Drugs, Health Technologies, Health Systems
Reimbursement Review
Exagamglogene Autotemcel (Casgevy)
Sponsor: Vertex Pharmaceuticals (Canada) Inc.
Therapeutic area: Sickle cell disease
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Ethics Review
Clinical Review
Abbreviations
AE
adverse event
ASCQ-Me
Adult Sickle Cell Quality of Life Measurement Information System
CanHaem
Canadian Hemoglobinopathy Association
CDA
Canada's Drug Agency
CRISPR–Cas9
clustered regularly interspaced short palindromic repeats–CRIPSR-associated protein 9
CTTC
Cell Therapy Transplant Canada
EMA
European Medicine Agency
FAS
full analysis set
GRADE
Grading of Recommendations Assessment, Development, and Evaluation
HRQoL
health-related quality of life
HSCT
hematopoietic stem cell transplant
MCID
minimum clinically important difference
PES
primary efficacy set
RCT
randomized controlled trial
SAE
serious adverse event
SD
standard deviation
VOC
vaso-occlusive crisis
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Exagamglogene autotemcel (Casgevy) Cell suspension in patient-specific vials, 4 to 13 × 106 cells/mL, for IV infusion |
Sponsor | Vertex Pharmaceuticals (Canada) Inc. |
Indication | For the treatment of patients 12 years of age and older with sickle cell disease with recurrent VOCs |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Priority review |
NOC date | September 23, 2024 |
Recommended dose | The minimum recommended dose is 3 × 106 viable CD34+ cells/kg |
NOC = Notice of Compliance; VOC = vaso-occlusive crisis.
Source: Exagamglogene autotemcel product monograph.1
Introduction
Sickle cell disease is a chronic, genetic, rare disease in which mutations in the beta-globin gene result in an increased production of sickle hemoglobin, giving the usually round red blood cells a sickle-like shape.2-5 Clinical manifestations arise as the sickle cells disrupt circulation in the small blood vessels.2,6 Vaso-occlusive crises (VOCs) are the hallmark clinical feature of sickle cell disease and involve the abrupt onset of severe, acute, and debilitating pain.5,7-9 The natural trajectory is generally poor.2 The clinical experts highlighted an unmet need in patients with severe manifestations of sickle cell disease, who typically present with recurrent VOCs, which are associated with ongoing organ damage, high health care use, and mortality,2,10 and can have a substantial impact on the daily lives of patients and their caregivers.
Prevalence data in Canada suggest that sickle cell disease affects 1 in 4,200 individuals.11 The current disease-modifying therapy for sickle cell disease includes hydroxyurea; it’s off-label use can reduce complications and mortality,2,10 and reduce transfusions, which are recommended for specific complications of sickle cell disease. Neither of these are curative therapies but, to date, they remain the only treatment options currently available for many patients. Hematopoietic stem cell transplant (HSCT) is a curative therapy that has the best overall and event-free survival outcomes in the few young patients who have a matched sibling donor who is available and willing to donate.2,10
Exagamglogene autotemcel (Casgevy) is approved by Health Canada for the treatment of patients aged 12 years and older with sickle cell disease and recurrent VOCs. Exagamglogene autotemcel is a cellular therapy that consists of autologous CD34+ hematopoietic stem cells and progenitor cells edited by clustered regularly interspaced short palindromic repeats (CRISPR)–CRISPR-associated protein 9 (Cas9) technology. Exagamglogene autotemcel is provided as a one-time single-dose suspension of CD34+ cells, administered by IV infusion.1 The minimum recommended dose, according to the product monograph, is 3 × 106 viable CD34+ cells/kg.1 The reimbursement request is per the indication.
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of the exagamglogene autotemcel cell suspension in patient-specific vials, for IV infusion, in the treatment of sickle cell disease in patients aged 12 years and older with recurrent VOCs.
Perspectives of Patients, Clinicians, and Drug Programs
The information in this section is a summary of input provided by the patient and clinician groups who responded to the CDA-AMC call for input and by clinical experts consulted by CDA-AMC for the purpose of this review.
Patient Input
CDA-AMC received patient group submissions from the Sickle Cell Awareness Group of Ontario (SCAGO), the Sickle Cell Disease Association of Canada (SCDAC), the Global Action Network for Sickle Cell & Other Inherited Blood Disorders (GANSID), and NotJustYou. Information-gathering methods included focus groups, one-on-one conversations, surveys of patients and caregivers, and a virtual webinar on gene therapy.
Patient groups highlighted the significant impact that sickle cell disease has on every aspect of an individual’s life. The multiple, unpredictable complications, such as severe painful attacks, fatigue, and organ damage, pose a substantial physical and mental burden. The clinical manifestations of the disease can be quite severe and may require frequent hospitalizations, leading to absenteeism from school or work and disruptions in family life. Social stigma, fertility issues, and the burden of managing a complex, painful condition have been emphasized as an important source of emotional suffering. Families also often face significant strain, which can be amplified in some instances by the financial burden of medical expenses. As such, patients placed a high value on avoiding VOCs and hospital visits, improving quality of life, facilitating access to treatment, and ensuring long-term safety.
Clinician Input
Input From Clinical Experts Consulted by CDA-AMC
The information in this section is based on input received from a panel of 3 clinical specialists consulted by CDA-AMC for the purpose of this review.
The clinical experts highlighted a significant unmet need in patients with severe manifestations of sickle cell disease. These patients typically present with recurrent pain crises, ongoing organ damage, and high health care use, which can have a substantial impact on the daily lives of patients and their caregivers. However, access to standard-of-care therapies can be limited and present a challenge in some regions of the country because of inconsistent coverage among jurisdictions and can be a challenge because of difficulties obtaining blood products for a lifetime of chronic transfusions, given that the Canadian blood donation pool is not always representative of most people living with sickle cell disease. Second-line and curative therapies include HSCT, which has the best outcomes in young patients who have a matched sibling donor available and willing to donate. According to the clinical experts, however, having a donor is a significant barrier for most patients, who are left with very limited therapeutic options despite substantial morbidity.
The clinical experts expect that exagamglogene autotemcel will be positioned as a second-line or later-line therapy for patients with severe manifestations of sickle cell disease for whom a matched sibling HSCT is not an option; for patients who have not had an optimal response or who have become resistant to hydroxyurea or red blood cell transfusions; for patients who cannot access these therapies because of a lack of coverage, the unavailability of a blood supply, or their distance from a tertiary centre; or in whom hydroxyurea or red blood cell transfusions are intolerable or contraindicated. These patients were identified by the clinical experts as those with the greatest unmet need.
Sickle cell disease is considered a rare disease; the prevalence of patients who would be considered candidates for exagamglogene autotemcel treatment is therefore limited. However, the clinical experts noted the limited health care resources and significant health care capacity issues at the time of this review. Individual patient prioritization is expected to be done by transplant experts, upon referral by the hemoglobinopathy specialist, as they have the necessary expertise to assess and identify the patients who are most likely to benefit from treatment and who have a sufficiently good health status to sustain the toxicities of myeloablative conditioning. The clinical experts indicated that socioeconomic factors often play an important role in the management of patients with sickle cell disease, and that nonclinical features could have a bearing on the selection of patients who receive exagamglogene autotemcel. These would include socioeconomic and geographic barriers, in addition to the psychological status of the patient and the patient’s support network.
Treatment with exagamglogene autotemcel requires an initial inpatient course, with hospital stays averaging 1 month. Patients should ideally be supported throughout hospitalization and follow-up by a multidisciplinary team that includes a pain specialist and a psychologist or social worker. Upon discharge, the treating hemoglobinopathy specialist and the multidisciplinary team would then switch to outpatient care, with additional follow-up by cell therapy specialists. The clinical experts emphasized that patients are expected to be very involved in discussions about the risks, benefits, and practicalities of exagamglogene autotemcel so that they can make an individualized and informed decision about treatment.
Clinician Group Input
CDA-AMC received submissions from 2 clinician groups: the Canadian Hemoglobinopathy Association (CanHaem) and Cell Therapy Transplant Canada (CTTC).
Both groups noted that sickle cell disease is the most common monogenetic rare disease, and currently affects more than 5,000 individuals in Canada. The input highlighted the severity of clinical manifestations, leading to significant morbidity and early death. Goals of therapy are to improve quality of life, decrease cumulative disease burden, and maximize life expectancy. Consequently, a clinically meaningful response to treatment, according to the input received, would include an absence of VOCs, improved quality of life, independence from transfusions, an absence of treatment-related neoplasms, and stable cardiovascular, renal, and pulmonary functioning.
Several unmet needs were identified from the input, including the fact that, despite the effectiveness of HSCT, most patients do not have access to this treatment because they do not have a matched sibling donor. Other available treatments do not consistently stop disease progression or ongoing organ damage, and all are associated with important toxicities. Considering the limited number of therapies available, additional therapeutic options are needed, the input stressed.
The place in therapy of exagamglogene autotemcel suggested by the 2 clinician groups was consistent with the input provided by the clinical experts consulted by CDA-AMC. Therapy must be delivered in the inpatient setting in specialized treatment centres that have experience in myeloablative therapy and/or cellular therapy and multidisciplinary teams that can provide specialty services.
The input noted that patients with sickle cell disease are at higher risk of myeloid malignancies, and that busulfan has been associated with myeloid malignancies and solid tumours in this patient population. The input also noted the need for equitable access, regardless of a patient’s geographic distance from a treatment centre, which can sometimes mean relocation. The clinician groups recognized the high risk of infertility and suggested that the cost of fertility preservation be included in price negotiations.
Drug Program Input
The drug programs provide input on each drug going through CDA-AMC reimbursement review processes by identifying issues that may affect their ability to implement a recommendation. For the CDA-AMC review of exagamglogene autotemcel, the drug plans raised issues pertaining to the eligibility criteria for initiation of therapy, assessment of long-term response, generalizability, and care provision. These questions were addressed by the clinical experts consulted by CDA-AMC for this review. The clinical experts’ responses have been included in Table 3.
Clinical Evidence
Systematic Review
Description of Studies
The 1 study reviewed — CLIMB-121 (63 patients enrolled and 30 patients analyzed) — was a single-arm, phase III, ongoing multicenter study designed to evaluate the efficacy and safety of exagamglogene autotemcel, administered after single-drug myeloablative conditioning chemotherapy, for the treatment of sickle cell disease in patients aged 12 to 35 years who have severe disease and recurrent VOCs (i.e., at least 2 protocol-defined severe VOC events per year in the 2 years before enrolment).
The primary outcome was the proportion of patients who did not experience any severe VOC events for at least 12 consecutive months from 60 days after the last red blood cell transfusion to up to 2 years after exagamglogene autotemcel infusion. Severe VOC was defined in the CLIMB-121 study as any of the following events: acute pain event that requires a visit to a medical facility and the administration of pain medications or red blood cell transfusions; acute chest syndrome; priapism lasting more than 2 hours and requiring a visit to a medical facility; or splenic sequestration. On-trial VOC events were adjudicated by an independent external end point adjudication committee.
Secondary outcomes in the study included hospitalizations and red blood cell transfusions, as well as health-related quality of life (HRQoL), which was assessed using the Adult Sickle Cell Quality of Life Measurement Information System (ASCQ-Me).12 ASCQ-Me is a disease-specific measurement system that enables adults to describe their functioning and well-being. Five question sets assess the impact of emotional functioning, social functioning, pain, stiffness, and sleep; higher scores indicate improved HRQoL. For pain-episode questions (which include pain-frequency and pain-severity scores) and the Sickle Cell Disease Medical History Checklist (SCD-MHC), lower scores indicate less severe pain. A reduction of 5 points in pain-episode scores and an increase of 5 points on impact subscales were considered minimum clinically important differences (MCIDs).13
The mean age at baseline was 22 years, and 6 patients (20%) were younger than 18 years. A total of 26 patients (87%) were Black or African American. The predominant genotype was betaS/betaS, which is considered a severe phenotype. In the 2 years before enrolment in the CLIMB-121 study, patients had a mean annualized rate of 3.9 severe VOCs (standard deviation [SD] = 2.1 severe VOCs). The mean annualized rate of inpatient hospitalizations for severe VOCs was 2.7 (SD = 2.0 hospitalizations), resulting in a mean annualized duration of hospitalizations of 17.1 days (SD = 14.3 days). The mean annual transfusion for a sickle cell disease–related indication was 8.4 units (SD = 14.9 units) of red blood cells.
Efficacy Results
The primary outcome — the absence of severe VOCs for at least 12 consecutive months — was considered the preferred clinical end point. In the CLIMB-121 study, 29 of 30 patients (96.7%) who were followed for at least 16 months after exagamglogene autotemcel infusion achieved the primary outcome and did not experience any severe VOCs for at least 12 consecutive months. In the 2 years preceding enrolment in the CLIMB-121 study, patients had a mean annualized rate of 3.9 severe VOCs (SD = 2.1). Results reached statistical significance against a prespecified but nonjustified sponsor-selected 50% response rate. The magnitude of the response was considered clinically meaningful by the clinical experts. There is, however, substantial uncertainty surrounding the severe VOC findings, given the limitations of the study and the fact that events of stroke were not included in the definition or captured in the trial, despite being considered a severe manifestation of sickle cell disease. Given the absence of comparative data, evidence for the effect of exagamglogene autotemcel on severe VOCs relative to any comparator is therefore very uncertain.
Secondary outcomes pertaining to health care use were hospitalizations and red blood cell transfusion, which is a highly resource-intensive treatment. These were deemed to be particularly relevant because they can have a substantial impact on the daily lives of patients and their caregivers. All 30 patients in the analysis achieved an absence of hospitalization for severe VOC for at least 12 consecutive months. In the 2 years preceding enrolment, patients had a mean annualized rate of 2.7 hospitalizations (SD = 2.0 hospitalizations). No patient received red blood cell transfusions for indications related to sickle cell disease during the 12-month period after exagamglogene autotemcel infusion. In the 2 years before enrolment, the mean annualized units of red blood cells transfused was 8.4 (SD = 14.9 units). The magnitude of the response for both outcomes (hospitalizations and transfusions) was considered clinically meaningful by the clinical experts. However, there is substantial uncertainty surrounding the findings. Given the absence of comparative data, evidence for the effect of exagamglogene autotemcel on health care use relative to any comparator is very uncertain.
Hematological outcomes were considered to be surrogate outcomes of efficacy; therefore, they are not as clinically meaningful for informing treatment decisions, according to the clinical experts. Results suggest that there was sufficient and stable allelic editing after exagamglogene autotemcel infusion to induce fetal hemoglobin levels above the 20% threshold in all 30 patients, significantly changing the phenotype. However, given the absence of comparative data, evidence for the effect of exagamglogene autotemcel on hematological outcomes relative to any comparator is very uncertain.
HRQoL was assessed using the disease-specific ASCQ-Me. The magnitude of the mean improvement from baseline through month 24 observed with exagamglogene autotemcel across the 7 subscales ranged from 3.3 points (SD = 13.3 points) to 21.0 points (SD = 7.7 points), which is considered clinically meaningful by the clinical experts, especially in regard to the impact of emotional functioning, social functioning, and pain. Substantial uncertainty, however, surrounds those findings, given the overall limitations of the trial and the subjectivity of the HRQoL assessments. And given the absence of comparative data, evidence for the effect of exagamglogene autotemcel on HRQoL outcomes relative to any comparator is very uncertain.
Gaps in the Evidence
The short follow-up duration of 20.1 months (SD = 10.37 months) in the trial was highlighted as a major evidence gap, as it does not address whether there could be a wanning of efficacy and whether that could lead to a loss of response over time. Limitations to generalizability include the fact that available evidence was insufficient to assess with certainty whether patients in the study had an adequate trial of first-line treatments, although exagamglogene autotemcel would be positioned as second-line or later-line therapy in clinical practice. In addition, patients whose health care use was consistent with that of patients with chronic pain were excluded from the study, although such patients may benefit from treatment that prevents further deterioration in their condition. However, the magnitude of the response to exagamglogene autotemcel in these patients is unknown.
Harms Results
All patients who received exagamglogene autotemcel in the CLIMB-121 study experienced at least 1 adverse event (AE). Serious adverse events (SAEs) were also relatively common; the safety profile of exagamglogene autotemcel was generally consistent with that associated with myeloablative busulfan conditioning and underlying disease, according to the clinical experts. A total of 6 patients discontinued the study due to inadequate cell collections. One death was due to respiratory failure after COVID-19 infection in a patient with preexisting lung disease and reported busulfan lung injury. Time to engraftment was an AE of special interest; however, although considered relatively long by the clinical experts, no association was reported between infection events and time to neutrophil engraftment or between bleeding events and time to platelet engraftment.
From the small number of patients, the short follow-up duration, and the very controlled setting of the clinical trial, the clinical experts indicated that the overall harms profile of the exagamglogene autotemcel treatment process in the CLIMB-121 study did not raise any particular safety signals.
Gaps in the Evidence
There are important evidence gaps in the safety assessment of exagamglogene autotemcel that limit interpretation of the findings. The short follow-up duration could not inform on longer-term toxicities, such as malignancies. These were highlighted as a significant concern by the clinical experts because of the increased baseline risk of leukemia in patients with sickle cell disease, the increased risk of developing secondary malignancies that are associated with busulfan, and the possibility of off-target editing.14 Although none of these notable harms were reported in the CLIMB-121 study, the follow-up duration was insufficient to assess the risk properly.
Critical Appraisal
Several limitations affect our confidence in the findings and lead to a risk of bias across all outcomes assessed in the trial. First is the absence of a control group, which precludes any conclusion from being drawn about the true effect of exagamglogene autotemcel relative to any comparator. With the Grading of Recommendations, Assessment, Development, and Evaluation (GRADE) approach, conclusions about relative efficacy in the absence of a comparator group cannot be drawn, and the certainty of the evidence is very low, as is typical for single-arm studies. Second is the lack of information regarding treatments received during the 2 years before enrolment (i.e., the baseline period), so the review team could not confirm whether patients in the study had an adequate trial of first-line treatments before receiving exagamglogene autotemcel. Therefore, what the baseline actually represents in terms of treatments received and compared is unknown. Third is the assessment of subjective outcomes, such as VOCs and HRQoL, in a single-arm trial, which can potentially influence the investigator’s assessment in favour of the drug. Finally, the review team noted that the sponsor made several changes to the conduct of the planned study once the trial was ongoing. This adds to the overall uncertainty; however, the impact on the results and on the risk of bias cannot be quantified.
As for generalizability, based on demographics and disease characteristics, the study population was considered to be mostly representative of patients with sickle cell disease seen in clinical practice who would be candidates for exagamglogene autotemcel.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For pivotal studies and randomized controlled trials (RCTs) identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered to be most relevant to CDA-AMC expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group.5,16
Although GRADE guidance is not available for noncomparative studies, the CDA-AMC review team assessed the pivotal single-arm trial for study limitations (which refer to internal validity or risk of bias), indirectness, and imprecision of effects to present these important considerations. Because the lack of a comparator arm does not allow for a conclusion to be drawn about the effect of the intervention relative to any comparator, the certainty of evidence for the single-arm study started at very low certainty with no opportunity to be rated up.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null. The target of the certainty of evidence assessment was defined as the presence or absence of an important effect, based on thresholds identified in the literature whenever possible or informed by the clinical experts consulted for this review.
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for exagamglogene autotemcel.
The selection of outcomes for the GRADE approach was based on the sponsor’s Summary of Clinical Evidence, consultation with clinical experts, and input received from patient groups, clinician groups, and public drug plans. The following list of outcomes was finalized in consultation with expert committee members:
Clinical outcomes of sickle cell disease (VOCs)
patients who have not experienced any severe VOCs for at least 12 consecutive months
Health care resource use
patients free from inpatient hospitalization for severe VOCs sustained for at least 12 months
reduction in units of red blood cell transfusion
Hematological outcomes
patients with sustained fetal hemoglobin of at least 20% for at least 12 consecutive months
the proportion of alleles with intended genetic modification present in CD34+ cells of the bone marrow
Patient-reported outcomes
change over time in ASCQ-Me score
Harms outcomes
patients with engraftment (neutrophil and platelet)
time to engraftment (neutrophil and platelet)
AEs and SAEs
mortality.
Table 2: Summary of Findings for Exagamglogene Autotemcel in Patients With Sickle Cell Disease
Outcome: follow-up at interim analysis data cut-off (June 14, 2023) | Patients (studies), N | Effect | Certaintya | What happens |
|---|---|---|---|---|
Clinical outcomes of sickle cell disease (VOCs) | ||||
No severe VOCs for ≥ 12 consecutive monthsb | N = 30, new drug (1 single-arm trial) | n = 29 (967 per 1,000 patients) Reduction from baseline (95% CI): 96.7% (82.8% to 99.9%) | Very lowc | The evidence is very uncertain about the effect of exagamglogene autotemcel on severe VOCs compared with any comparator, given the absence of comparative data |
Health care resource use | ||||
No inpatient hospitalization for severe VOCs sustained for ≥ 12 monthsb | N = 30, new drug (1 single-arm trial) | n = 30 (1,000 per 1,000 patients) Reduction from baseline (95% CI): 100.0% (88.4% to 100.0%) | Very lowd | The evidence is very uncertain about the effect of exagamglogene autotemcel on hospitalization for severe VOCs compared with any comparator, given the absence of comparative data |
Reduction in units of red blood cell transfusion | N = 30, new drug (1 single-arm trial) | Baseline mean (SD): 8.4 units (14.9 units) Reduction from baseline (95% CI): 100.0% (100.0% to 100.0%) | Very lowd | The evidence is very uncertain about the effect of exagamglogene autotemcel on red blood cell transfusions compared with any comparator, given the absence of comparative data |
Hematological outcomes | ||||
Sustained fetal hemoglobin of ≥ 20% for ≥ 12 consecutive months | N = 30, new drug (1 single-arm trial) | n = 30 (1,000 per 1,000 patients) | Very lowd | The evidence is very uncertain about the effect of exagamglogene autotemcel on fetal hemoglobin compared with any comparator, given the absence of comparative data |
Proportion of alleles with intended genetic modification present in CD34+ cells of the bone marrow | N = 30, new drug (1 single-arm trial) | Mean (SD) at: month 6, █████ ███████ month 12, █████ ████████ month 24, █████ █████████ | Very lowd | The evidence is very uncertain about the effect of exagamglogene autotemcel on intended allelic genetic modification compared with any comparator, given the absence of comparative data |
Patient-reported outcomes | ||||
Change over time in ASCQ-Me score, emotional impact subscale | N = 30, new drug (1 single-arm trial) | Change from baseline, mean score (SD) at:
| Very lowd | The evidence is very uncertain about the effect of exagamglogene autotemcel on ASCQ-Me emotional impact scores compared with any comparator, given the absence of comparative data |
Change over time in ASCQ-Me score, pain impact subscale | N = 30, new drug (1 single-arm trial) | Change from baseline, mean (SD) at:
| Very lowd | The evidence is very uncertain about the effect of exagamglogene autotemcel on ASCQ-Me pain impact scores compared with any comparator, given the absence of comparative data |
Change over time in ASCQ-Me score, social functioning impact subscale | N = 30, new drug (1 single-arm trial) | Change from baseline, mean (SD) at:
| Very lowd | The evidence is very uncertain about the effect of exagamglogene autotemcel on ASCQ-Me social functioning impact scores compared with any comparator, given the absence of comparative data |
Change over time in ASCQ-Me score, stiffness impact subscale | N = 30, new drug (1 single-arm trial) | Change from baseline, mean (SD) at:
| Very lowd | The evidence is very uncertain about the effect of exagamglogene autotemcel on ASCQ-Me stiffness impact scores compared with any comparator, given the absence of comparative data |
Change over time in ASCQ-Me score, sleep impact subscale | N = 30, new drug (1 single-arm trial) | Change from baseline, mean (SD) at:
| Very lowd | The evidence is very uncertain about the effect of exagamglogene autotemcel on ASCQ-Me sleep impact scores compared with any comparator, given the absence of comparative data |
Change over time in ASCQ-Me score, pain-episode frequency subscale | N = 30, new drug (1 single-arm trial) | Change from baseline, mean (SD) at:
| Very lowd | The evidence is very uncertain about the effect of exagamglogene autotemcel on ASCQ-Me pain-episode frequency scores compared with any comparator, given the absence of comparative data |
Change over time in ASCQ-Me score, pain-episode severity subscale | N = 30, new drug (1 single-arm trial) | Change from baseline, mean (SD) at:
| Very lowd | The evidence is very uncertain about the effect of exagamglogene autotemcel on ASCQ-Me pain-episode severity scores compared with any comparator, given the absence of comparative data |
Harms | ||||
Engraftment (neutrophil and platelet) | N = 44, new drug (1 single-arm trial) | Neutrophil: n = 44 (1,000 per 1,000 patients) Platelet: n = 43 (977 per 1,000 patients) | Very low | The evidence is very uncertain about the effect of exagamglogene autotemcel on neutrophil engraftment compared with any comparator, given the absence of comparative data |
Time to engraftment (neutrophil and platelet) | N = 44, new drug (1 single-arm trial) | Neutrophil, median (range): 27 days (15 to 40 days) Platelet, median (range): 35 days (23 to 126 days) | Very low | The evidence is very uncertain about the effect of exagamglogene autotemcel on neutrophil and platelet engraftment compared with any comparator, given the absence of comparative data |
AEs (in ≥ 25% of patients) and SAEs (in ≥ 2% of patients) | N = 44, new drug (1 single-arm trial) | AEs: n = 44 (1,000 per 1,000 patients) SAEs: n = 20 (455 per 1,000 patients) | Very low | The evidence is very uncertain about the effect of exagamglogene autotemcel on AEs, SAEs, and AEs of special interest compared with any comparator, given the absence of comparative data |
Mortality | N = 44, new drug (1 single-arm trial) | n = 1 (23 per 1,000 patients) | Very lowe | The evidence is very uncertain about the effect of exagamglogene autotemcel on mortality compared with any comparator, given the absence of comparative data |
AE = adverse event; ASCQ-Me = Adult Sickle Cell Quality of Life Measurement Information System; CI = confidence interval; SAE = serious adverse event; SD = standard deviation; VOCs = vaso-occlusive crisis.
Note: All serious concerns about study limitations (which refer to internal validity or risk of bias), indirectness, and imprecision of effects are documented in the table footnotes.
aIn the absence of a comparator group, conclusions about efficacy relative to any comparator cannot be drawn, and the certainty of evidence started at very low. None of the outcomes were rated up because of serious study limitations (refer to specific footnotes).
bStatistical testing for these outcomes was adjusted for multiplicity in the trial. Statistical testing for all other outcomes was not adjusted for multiplicity in the trial; therefore, findings for these other outcomes should be considered to be supportive evidence.
cSerious study limitations. The flexibility of the start and finish dates of patients who did not experience any severe VOC for at least 12 consecutive months during the 2-year follow-up period runs the risk of overestimating the treatment effect. Updates to the outcomes made to the study protocol after enrolment and with no rationale provided cause an unknown risk of bias.
dSerious study limitations. The interim analysis provided results only for the primary efficacy set, which is potentially a select sample, as it represents only patients who have completed a set follow-up time in the study to date, as opposed to the full enrolled sample. Information on the outcomes based on the full treatment experience is therefore lacking.
eSerious imprecision. The study captured a very small number of events, and the study duration is unlikely to be long enough to fully capture the outcome.
Sources: SCD Clinical Overview Addendum,17 sponsor’s Summary of Clinical Evidence.
Other Considerations
Sickle cell disease can be considered a rare disease for which a number of patients have a significant unmet need. The clinical experts emphasized that patients with severe manifestations of sickle cell disease typically present with recurrent pain crises, ongoing organ damage, and high health care use, which can have a substantial impact on the daily lives of patients and their caregivers. The natural trajectory is generally poor, as the disease has a substantial negative impact on life expectancy, and a limited number of effective therapeutic options are available.
According to the clinical experts, the drug under review could address this unmet need. They indicated that exagamglogene autotemcel is not for all patients with sickle cell disease; some patients respond well to standard first-line therapies, and these patients would not be candidates for this treatment. In clinical practice, exagamglogene autotemcel would likely be a second-line or later-line therapy in patients with severe manifestations of sickle cell disease for whom HSCT is not an option and who do not have an optimal response or who become resistant to hydroxyurea or red blood cell transfusions; in patients who cannot access these therapies because of a lack of coverage, the unavailability of a blood supply, or their distance from a tertiary centre; and in patients in whom hydroxyurea or red blood cell transfusions are intolerable or contraindicated.
Long-Term Extension Studies
Description of Studies
At the time of this review, 1 long-term extension study is in progress. CLIMB-131 is an ongoing prospective, multisite, observational study evaluating the long-term safety and efficacy of exagamglogene autotemcel in patients who received this treatment in the parent study, CLIMB-121. It is planned that patients be followed for up to 15 years after exagamglogene autotemcel infusion. The primary objective of the CLIMB-131 study is to evaluate the long-term safety of exagamglogene autotemcel. Because the CLIMB-121 study is ongoing, only a subset of patients with sickle cell disease has completed the parent study and enrolled in the CLIMB-131 study.
Efficacy Results
As of the data cut-off date (June 14, 2023), the median follow-up duration after exagamglogene autotemcel infusion in the CLIMB-121 and CLIMB-131 studies was 19.3 months (range, 0.8 to 48.1 months).
All patients in the primary efficacy set (PES) of the CLIMB-121 study who experienced — for at least 12 months during the available follow-up period — no severe VOC events, no inpatient hospitalization for severe VOC, or a fetal hemoglobin level of at least 20%, remained VOC-free, hospitalization-free, or above the minimal fetal hemoglobin threshold, respectively.
A total of 43 of 44 patients in the full analysis set (FAS) population had at least 60 days of follow-up after the last red blood cell transfusion and were included in the June 14, 2023, postaddendum analysis; of these, 6 patients had adjudicated VOCs and 3 patients had inpatient hospitalization for VOC during follow-up in the CLIMB-121 and CLIMB-131 studies. The proportion of total hemoglobin comprised of fetal hemoglobin (%) was maintained, generally, at least 40% from month 6 to the end of available follow-up.
Harms Results
A total of 17 of 44 patients (38.6%) had more than 24 months of follow-up and were included in the long-term extension for which harms results are reported. Of these, no deaths occurred during the CLIMB-131 study. ███ ███████ ███ ██ ███ ██ ███████████████ █████████ ████████ ██ ███ ████████████ ██ █████████ ██ █████████████ ██████████. No new malignancies, new or worsening hematologic disorders, or complications related to sickle cell disease occurred in patients who advanced from the CLIMB-121 study during the CLIMB-131 study.
Critical Appraisal
The study limitations regarding the single-arm and open-label nature of the CLIMB-121 study, as well as limitations related to generalizability, also apply to the CLIMB-131 long-term extension study. In addition, reporting of available data from the CLIMB-131 study is poor, and is limited because it is an interim analysis, which hampers the ability to draw definitive long-term conclusions before follow-up is complete. Furthermore, the PES population is potentially a select sample, not the full enrolled sample, and data reported so far in the CLIMB-131 study bring uncertainty regarding the true magnitude of the treatment effect. Finally, long-term data on HRQoL and complete harms reporting in the long-term extension are lacking.
Indirect Comparisons
No indirect treatment comparisons were submitted by the sponsor.
Studies Addressing Gaps in the Evidence From the Systematic Review
No studies addressing gaps in evidence were submitted by the sponsor.
Conclusions
Evidence from the ongoing, single-arm CLIMB-121 study (n = 30 patients in the interim analysis) is very uncertain for the effect of exagamglogene autotemcel on clinical efficacy and harms outcomes in patients with severe sickle cell disease who have recurrent VOCs relative to any comparator, given the absence of comparative data. The findings from the trial are consistent with a clinically meaningful prevention of VOCs, hospitalizations, and red blood cell transfusions, based clinical expert input. As well, clinically meaningful improvements in HRQoL, based on reported MCIDs, were observed. These clinical outcomes were consistent with the treatment goals of sickle cell disease in clinical practice, according to the experts (i.e., to prolong life, prevent end-organ toxicity, and reduce symptom severity). However, there is substantial uncertainty surrounding the evidence. The most important limitations include the use of a single-arm study design and uncertainty regarding the treatments that patients were receiving at baseline, which preclude definite conclusions from being drawn about the comparative efficacy of exagamglogene autotemcel. The available results come from a follow-up period of up to 2 years after exagamglogene autotemcel infusion, which is a relatively short period from which to determine longer-term effectiveness. Limitations with regard to outcome assessment introduce a risk of bias in favour the drug. Therefore, despite the magnitude of the response observed with exagamglogene autotemcel in the CLIMB-121 study, concerns remain as to whether the results present the true effect of the drug. A high proportion of patients experienced harms events, which were generally consistent with what is associated with the underlying disease and the notoriously difficult myeloablative busulfan conditioning. The study could not provide information on the issues of longer-term toxicities, such as loss of fertility and malignancies associated with the disease itself and with busulfan chemotherapy, or potential off-target gene editing. The clinical experts indicated that the overall harms profile of the exagamglogene autotemcel treatment process did not raise any particular safety signals, but the number of patients was small and the follow-up duration was short in the very controlled setting of the clinical trial.
Special consideration may be given to the fact that sickle cell disease is rare. Patients with severe manifestations present with recurrent pain crises, ongoing organ damage, and high health care use, which have a substantial negative impact on life quality and expectancy. The natural trajectory of the disease is generally poor, and only a limited number of effective therapies are available. In clinical practice, exagamglogene autotemcel would likely be reserved as a later-line therapy in select patients with severe disease for whom HSCT is not an option. The clinical experts emphasized that they are willing to tolerate a higher level of uncertainty in this patient population because of the magnitude of the unmet need. However, the risks should be weighed against the expected benefits in discussions with patients so they can make an individualized and informed decision.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of exagamglogene autotemcel cell suspension in patient-specific vials for IV infusion in the treatment of sickle cell disease in patients 12 years and older with recurrent VOCs.
Disease Background
The contents of this section have been informed by materials submitted by the sponsor and clinical expert input. The following information has been summarized and validated by the CDA-AMC review team.
Sickle cell disease is a chronic genetic rare disease in which mutations in the beta-globin gene result in an increased production of sickle hemoglobin, giving the usually round red blood cells a sickle-like shape.2-5 Clinical manifestations arise as the sickle cells disrupt circulation in the small blood vessels, resulting in unpredictable episodes of severe pain and widespread organ damage, both of which are associated with increased morbidity and mortality.2,6
More specifically, different combinations of the globin subunits give rise to multiple types of hemoglobin, which predominate at different stages of life.4 Hemoglobin A is the predominant type in adults, whereas fetal hemoglobin is the predominant hemoglobin type before birth and during the newborn phase.18 Shortly after birth, beta-globin levels increase and gamma-globin levels decline, leading to a switch from fetal hemoglobin to hemoglobin A.18 In individuals with sickle cell disease, mutations in the beta-globin gene result in an abnormal, reduced, or absent expression of hemoglobin A and an increased production of sickle hemoglobin with associated disease pathology.3-5 Sickle cell disease is characterized by the expression of abnormal sickle hemoglobin; the deoxygenated form of this polymerizes abnormally within red blood cells, giving them a characteristic sickle shape. As sickle hemoglobin polymers extend, they deform red blood cells and interfere with their flexibility, shape, and rheological and physical properties,4,19 which leads to a range of acute and chronic complications. Notably, an inverse relation exists between fetal hemoglobin levels and clinical manifestations of sickle cell disease.20
Although the disease presentation may vary greatly among patients, the natural trajectory is generally poor, as the disease has a substantial negative impact on life expectancy.2 The clinical experts emphasized that patients with severe manifestations of sickle cell disease typically present with recurrent pain crises, ongoing organ damage, and high health care use, which can have a substantial impact on the daily lives of patients and their caregivers. VOCs are the hallmark clinical feature of sickle cell disease, and involve the abrupt onset of severe, acute, and debilitating pain, which can lead to chronic pain and complications in patients and often require hospitalization.5,7-9 These events are caused by the cycle of blood vessel occlusion, impaired oxygen supply, and tissue injury from infarction and reperfusion, and are directly linked to end-organ damage and early mortality.5,19,21,22
Sickle cell disease is considered a rare disease. Prevalence data based on an analysis of health administrative databases in Ontario suggest that it affects 1 in 4,200 individuals in Canada.11 Screening for sickle cell disease is common practice as part of the newborn screening program in most regions of Canada.23 Once a diagnosis is confirmed by a physician, a life-long treatment program is initiated immediately. However, the clinical experts indicated that they often see patients, such as individuals newly arrived in Canada, who received a late diagnosis, or who have not received any treatment, and present with more severe manifestations of the disease.
Standards of Therapy
The contents of this section have been informed by materials submitted by the sponsor and clinical expert input. The following information has been summarized and validated by the CDA-AMC review team.
The clinical experts indicated that the most important treatment goals for patients with sickle cell disease are to prolong life, prevent end-organ toxicity, and reduce symptom severity. Both the medical literature2,10 and experience from clinical practice suggest that patients who experience frequent VOCs are at higher risk of complications and mortality. Additional goals of therapy include a reduction in health care use, which is typically high in patients with sickle cell disease. Improving quality of life, chronic pain, and working ability are also meaningful goals.
The consensus statement released by the Canadian Hemoglobinopathy Association2 serves as clinical practice guidelines in Canada. According to the statement, the current backbone of disease-modifying therapy in sickle cell disease includes the off-label use of hydroxyurea to reduce complications and mortality,2,10 and transfusions, which are recommended for specific complications of sickle cell disease; both are continued over a lifetime horizon. As such, neither of these are curative therapies and, to date, they remain the only treatment options available for many patients. The clinical experts indicated that they are effective in some patients for delaying complications, but they are associated with potential risks and require an ongoing commitment for continued benefit. For example, transfusions given to patients with stroke, acute chest syndrome, and recurrent VOCs are highly resource-intensive treatments that are accompanied by several risks and toxicities — when matching blood can be obtained — and are considered by some patients to be disruptive to life, at best, and to be an overwhelming burden by others. There remains a subset of patients with sickle cell disease and recurrent VOCs who receive no treatment.
HSCT is a curative therapy and has the best overall and event-free survival outcomes in patients younger than 12 years who have a matched sibling donor available and willing to donate, according to the clinical experts.2,10 According to the clinical practice guidelines,2 HSCT is indicated in patients younger than 16 years. In clinical practice, exagamglogene autotemcel would be positioned after HSCT in younger patients who have a matched sibling donor; the experts noted that matched sibling HSCT would remain the preferred treatment in these patients until further data become available regarding the long-term efficacy and safety of exagamglogene autotemcel. In patients for whom HSCT is not an option, the clinical experts explained that they are willing to tolerate a higher level of uncertainty because of the magnitude of the unmet need in this population. They indicated that the risks should be weighed against the expected benefits in discussions with patients so they can make an individualized decision about treatment.
Drug Under Review
Mechanism of Action
Exagamglogene autotemcel is a cellular therapy that consists of autologous CD34+ hematopoietic stems and progenitor cells edited by CRISPR–Cas9 technology. Further information on the CRISPR–Cas9 technology for gene editing can be found in a October 2024 CDA-AMC publication.14
Guide RNA enables CRISPR–Cas9 to make a precise DNA double-strand break at the critical transcription-factor binding site (GATA1) in the erythroid-specific enhancer region of the BCL11A gene.1 As a result of the editing, GATA1 binding is irreversibly disrupted and BCL11A gene expression is reduced, resulting in an increase in gamma-globin expression and fetal hemoglobin protein production in erythroid cells.1 This attempts to address the underlying cause of the disease with a single administration and curative intent. In patients with sickle cell disease, fetal hemoglobin expression reduces intracellular sickle hemoglobin concentration, preventing red blood cells from sickling and thereby eliminating VOCs.1
The editing is intended to be specific and, to date, the sponsor reported that no off-target editing has been observed in in vitro studies of exagamglogene autotemcel manufactured using either healthy donor cells or patient cells.
Health Canada Indication
The approved indication for exagamglogene autotemcel is for the treatment of patients 12 years and older with sickle cell disease with recurrent VOCs.1 The reimbursement request is aligned with the Health Canada indication.
Exagamglogene autotemcel has been approved in other jurisdictions, including the US,24 UK,25 and Europe.26
Dosing and Administration
Exagamglogene autotemcel is provided as a one-time single dose for IV infusion that contains a suspension of CD34+ cells in 1 or more vials, all of which must be administered.1 The minimum recommended dose is 3 × 106 viable CD34+ cells/kg.1 Exagamglogene autotemcel is for autologous use only, and can only be administered in an authorized treatment centre that has experience in stem cell transplants and in the treatment of patients with sickle cell disease, per the product monograph.1
Prescribing
The clinical experts confirmed that treatment requires both hematology and transplant expertise. Hematologists will need to have expertise in and the capacity to discuss treatment options, refer the appropriate patients for treatment, and provide advice on follow-up care. Hematologists should have expertise in identifying patients who would be best suited for exagamglogene autotemcel treatment that includes both clinical (e.g., eligible patients) and operational (e.g., which centres to refer to, how treatment and ancillary costs are managed) perspectives. Transplant physicians should have the capacity and expertise to finalize patient treatment plans, administer exagamglogene autotemcel treatment, and provide advice on follow-up care.
Given that access to these clinical specialists may differ across provinces and territories, the sponsor suggested that a referral network could be established. According to physicians consulted by the sponsor, such networks currently exist for other complex treatments, like HSCT and chimeric antigen receptor T-cell (CAR-T) therapy. However, the clinical experts consulted by CDA-AMC noted that most centres are geared toward treating patients with malignancies, and that very few centres have established funding sources for the treatment of nonmalignant disease or ancillary services. In addition, the clinical experts noted that there are limited health care resources and significant health care capacity issues at the time of this review.
The sponsor’s submission provided the anticipated exagamglogene autotemcel treatment journey, which involves multiple steps, and the health care resources that are expected to be involved based on clinical trial experience to date (Figure 1). Patients are required to undergo CD34+ hematopoietic stem cell and progenitor cell mobilization, followed by apheresis to isolate the CD34+ cells for medicinal product manufacturing. Before transplant of the edited cells, full myeloablative conditioning is required. After the final dose of myeloablative chemotherapy, the patient receives a single exagamglogene autotemcel infusion. After successful engraftment, the transplanted stem cells are nested in the bone marrow and begin to produce new blood cells. The treating physician will be a hematologist and/or a transplanter, depending on the step in the patient pathway.
Figure 1: Anticipated Exagamglogene Autotemcel Treatment Journey
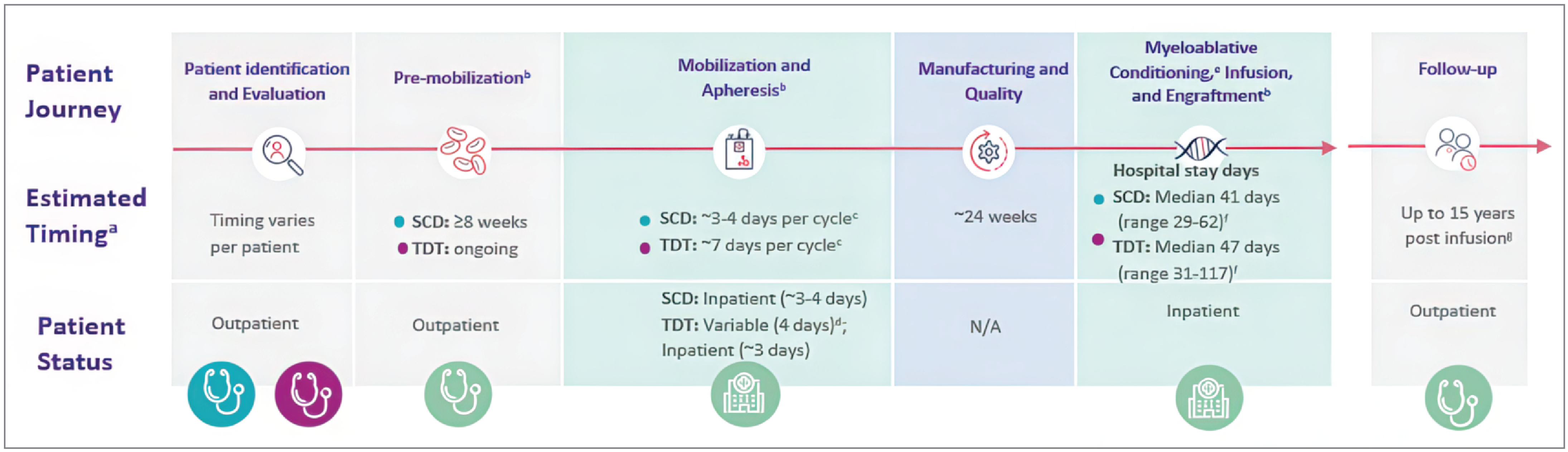
N/A = not applicable; SCD = sickle cell disease; TDT = transfusion-dependent beta-thalassemia.
Note: This figure represents the journey for patients with both SCD and TDT (exagamglogene autotemcel is also approved by Health Canada for the treatment of patients with TDT).
aThe time frame for each step of the exagamglogene autotemcel treatment journey is approximate and will vary by patient. The entire exa-cel treatment journey could take up to a year.
bTiming and patient status are based on clinical trials.
cThe median number of collection cycles for SCD and TDT was 2 (minimum to maximum, 1 to 6) and 1 (minimum to maximum, 1 to 4), respectively. Timing is reflective of a hospital admission on day 0 for SCD and day 4 for TDT.
dSubcutaneous mobilizing drugs can be self-administered at home, but certain circumstances and payer requirements mean that inpatient administration of the injection may be required.
eThe median total length of hospitalization for myeloablative conditioning and exagamglogene autotemcel infusion through discharge for SCD and TDT was approximately 6 weeks and 7 weeks, respectively.
fHospital stay days. It is recommended that patients with SCD or TDT be transfused for at least 8 weeks or for 60 days, respectively, before the initiation of myeloablative conditioning.
gTo be confirmed after Health Canada review.
Perspectives of Patients, Clinicians, and Drug Programs
The full patient and clinician group submissions received by CDA-AMC are available in the consolidated patient and clinician group input document for this review on the project website.
Patient Group Input
This section was prepared by the CDA-AMC review team based on the input provided by patient groups.
CDA-AMC received patient group submissions from the SCAGO, SCDAC, the Global Action Network for Sickle Cell & Other Inherited Blood Disorders, and NotJustYou.
Information-gathering methods included focus groups, one-on-one conversations, surveys of patients and caregivers, and a virtual webinar on gene therapy.
The patient groups emphasized that sickle cell disease has a significant impact not only on quality of life, but also on every aspect of an individual’s life. Patients indicated that multiple, unpredictable complications, such as severe painful attacks, fatigue, and organ damage, pose a substantial physical burden and take a toll on mental well-being. The clinical manifestations of the disease can be quite severe and may require frequent hospitalizations, leading to absenteeism from school or work and disruptions in family life. In addition, social stigma, fertility issues, and the burden of managing a complex, painful condition have been emphasized as sources of emotional suffering. Several patients reported that sickle cell disease affected educational and career opportunities, financial stability, and interpersonal relationships. The illness also has a profound impact on family members, who often face significant stress and emotional strain. These can be amplified in some instances by the financial burden of medical expenses.
Patients reported various experiences with currently available treatments, but a subset of individuals faces an unmet need. As such, patients placed a high value on avoiding VOCs and hospital visits, improving quality of life, facilitating access to treatment, and ensuring long-term safety. The patient input noted that the prospect of being able to modify the defective gene brings patients hope for a cure. Patients expressed the need for their voices to be taken into account during decision-making and the desire to access mental and reproductive health and financial support throughout the treatment and recovery process.
Clinician Input
Input From Clinical Experts Consulted by CDA-AMC
All CDA-AMC review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). In addition, as part of the review of exagamglogene autotemcel, a panel of 3 clinical experts from across Canada was convened to characterize unmet therapeutic needs, assist in identifying and communicating situations in which there are gaps in the evidence that could be addressed through the collection of additional data, promote the early identification of potential implementation challenges, gain further insight into the clinical management of patients living with a condition, and explore the potential place in therapy of the drug (e.g., potential reimbursement conditions). A summary of this panel discussion follows.
Unmet Needs
The clinical experts highlighted an unmet need in patients with severe manifestations of sickle cell disease. Such patients typically present with recurrent pain crises and high health care use, which can have a substantial impact on the daily lives of patients and their caregivers.
The clinical experts explained that severe disease may present when patients do not receive appropriate treatments, as access to standard-of-care therapies can be limited and present a challenge in some regions of the country. In their experience, coverage of first-line treatments, such as hydroxyurea and red blood cell transfusions, is not consistent among jurisdictions. Factoring into this is the difficulty obtaining blood products for lifetime and chronic transfusions, as the Canadian blood donation pool is not always representative of most people living with sickle cell disease.
Severe manifestations, such as ongoing organ damage, may also be observed in several patients despite an adequate trial of standard-of-care therapies. The clinical experts confirmed that there is a wide range of disease presentations, and emphasized the need for treatment options with different mechanisms of action to support patients who do not have an optimal response to or who become resistant to existing therapy.
Second-line and curative therapies include HSCT, which has the best outcomes in patients younger than 12 years who have a matched sibling donor available and willing to donate. According to the clinical experts, however, the conditioning regimen is associated with several meaningful toxicities. But most important the need for a donor is a significant barrier for some patients who are left with very limited therapeutic options despite substantial morbidity.
As novel drugs such as exagamglogene autotemcel gradually become available, their use may be limited by the fact that they can only be administered in tertiary centres, which are located mainly in urban and metropolitan settings; as a result, patients living in rural areas are required to travel a long way to receive treatment. In addition, the clinical experts noted significant health care capacity issues at the time of this review, even though gene therapy and transplant centres are required to expand their capacity to treat patients with these complex procedures and to not limit access to second-line and subsequent-line therapies for patients who need them.
Place in Therapy
According to the consensus statement released by the Canadian Hemoglobinopathy Association,2 which serves as clinical practice guidelines in Canada, the current backbone of disease-modifying therapy in sickle cell disease includes hydroxyurea and transfusions that are continued over a lifetime horizon. As such, neither of these are curative therapies. In some patients, they are effective in delaying complications, but they require an ongoing commitment for continued benefit, the clinical experts indicated. For example, transfusions given to patients with stroke, acute chest syndrome, and recurrent VOCs are highly resource-intensive treatments that are accompanied by several risks and toxicities — when matching blood can be obtained — and are considered by some patients to be disruptive to life, at best, and to be an overwhelming burden by others.
In this context, the clinical experts expect that exagamglogene autotemcel will cause a shift in the current treatment paradigm because it addresses the underlying disease process. However, they note that the drug will target a specific patient population (i.e., patients with severe manifestations of sickle cell disease). As such, exagamglogene autotemcel would be a stand-alone treatment and is likely to be positioned as a second-line or later-line therapy in patients who do not have an optimal response to or who become resistant to hydroxyurea or red blood cell transfusions; in patients who cannot access these therapies because of a lack of coverage, the unavailability of a blood supply, or their distance from a tertiary centre; or in whom hydroxyurea or red blood cell transfusions are intolerable or contraindicated.
In clinical practice, exagamglogene autotemcel would also be positioned after HSCT in younger patients who have a matched sibling donor; the experts mentioned that HSCT would remain the preferred treatment in these patients until further data become available on the long-term efficacy and safety of exagamglogene autotemcel. In patients for whom HSCT is not an option, the clinical experts said they are willing to tolerate a higher level of uncertainty because of the magnitude of the unmet need in this population. They indicated that the risks should be weighed against the expected benefits in discussions with patients so they can make an individualized decision about treatment.
Patient Population
Sickle cell disease is considered a rare disease; the prevalence of patients who would be considered candidates for exagamglogene autotemcel treatment, as previously discussed, is limited. Diagnosis is relatively straightforward and accessible in Canada, and no companion diagnostic test is required, other than perhaps human leucocyte antigen typing.
Patients enrolled in the CLIMB-121 study were, overall, representative of the target patient population, according to clinical experts. In clinical practice, there is substantial heterogeneity across patient characteristics, severity of disease manifestations, and complications. Some relevant categories of patients were excluded from the study, such as patients with chronic pain. This was deemed to be an important evidence gap. According to the clinical experts, it is not necessarily expected that treatment with exagamglogene autotemcel will have a meaningful impact on outcomes (such as chronic pain) or on preexisting complications (such as organ damage); nevertheless, patients experiencing these complications may benefit from a reduction in VOCs with exagamglogene autotemcel treatment and the prevention of further deterioration in their condition. Another significant exclusion criterion was patients with the betaS/betaC genotype, who account for approximately one-third of patients in clinical practice, according to the experts. These patients, however, typically present with milder manifestations of the disease and are not expected to be treated with exagamglogene autotemcel in the absence of efficacy and safety data. Finally, the definition used in the assessment of VOCs, both in the inclusion criteria and efficacy evaluation, did not include individual events of stroke, despite the fact that it is considered a severe manifestation of sickle cell disease in clinical practice. As it was not an exclusion criterion, however, patients with stroke could still be included in the trial if they had additional manifestations of the disease.
Socioeconomic factors often play an important role in the management of patients with sickle cell disease. As such, there are nonclinical features that can have a bearing on the selection of patients eligible to receive exagamglogene autotemcel. According to the experts, these would include socioeconomic and geographic barriers, as the treatment process requires a lengthy absence from work and/or caregiving, and may require geographic relocation, which can put additional economic strain on patients and their family. The psychological status of patients and members of their support networks are also important to consider, and includes issues such as identity, stigma, and expectations around treatment effects. The clinical experts emphasized that patients are expected to be very involved in discussions around risks, benefits, and the practicalities of exagamglogene autotemcel. Patients who have a religious belief that excludes blood transfusions and the collection of cells would not be suitable for this treatment.
In the context of limited health care resources, the clinical experts indicated that capacity building is important. For the time being, patients with sickle cell disease would likely be prioritized to receive exagamglogene autotemcel over patients with other indications, as the disease has a substantial negative impact on both life expectancy and quality of life, in addition to being associated with high health care resource use. Individual patient prioritization is expected to be done by transplant experts, upon referral by a hemoglobinopathy specialist. Transplant specialists have the necessary expertise to assess and identify patients who are most likely to benefit from treatment and who have a sufficiently good health status to sustain the toxicities of myeloablative conditioning.
Assessing the Response to Treatment
According to the experts, the disease trajectory in most patients with sickle cell disease is relatively constant. Minor improvements can sometimes be seen, for example, by changing habits, but nonpharmaceutical measures are not sufficient to stop the progression of the disease or related complications.
The clinical experts indicated that the most important treatment goals for patients with sickle cell disease are to prolong life, prevent end-organ toxicity, and reduce symptom severity. Both the medical literature2,10 and experience from clinical practice suggest that patients who experience frequent VOCs are at higher risk of complications and mortality. Therefore, the clinical experts indicated that the use of VOCs as the primary outcome in the CLIMB-121 study was appropriate. However, they noted that the identification of VOCs can be subjective, especially when the assessment is based on pain symptoms. They also noted that some patients may not consistently seek medical attention, which may result in an underestimation of the number of VOCs.
Additional goals of therapy include a reduction in health care use, which is typically high in patients with sickle cell disease. The clinical experts explained that preventing even a few VOCs or hospitalizations per year would be an important benefit to the patients they follow in their practice. Improvements in quality of life, chronic pain, and working ability would also be meaningful goals to achieve.
Prescribing Considerations
Patients with severe manifestations of sickle cell disease who may be candidates for exagamglogene autotemcel, according to their treating hemoglobinopathy specialists, are expected to be referred to an accredited transplant centre for assessment by a transplant physician. Treatment with exagamglogene autotemcel requires an initial inpatient course, with hospital stays averaging 1 month. Patients should ideally be supported throughout hospitalization and follow-up by a multidisciplinary team that includes a pain specialist and a psychologist or social worker. Upon discharge, the treating hemoglobinopathy specialist and the multidisciplinary team would then switch to outpatient care.
Clinician Group Input
This section was prepared by the CDA-AMC review team based on the input provided by clinician groups.
CDA-AMC received clinician group submissions from CanHaem and CTTC. CanHaem is a not-for-profit organization composed of health care providers that aims to provide multidisciplinary expertise and advance the quality of care to patients with hemoglobinopathies. CTTC is a member-led, national, multidisciplinary organization providing leadership and promoting excellence in patient care, research, and education in the field of HSCT and cell therapy.
Both groups noted that sickle cell disease is the most common monogenetic rare disease, and currently affects more than 5,000 individuals in Canada. The input highlighted the severity of clinical manifestations which can lead to significant morbidity and early death. Patients can suffer from chronic anemia, severe acute debilitating pain from VOCs, a higher risk of serious infection due to a compromised immune system, acute respiratory failure (acute chest syndrome), chronic pain, ischemic and hemorrhagic stroke, liver disease, nephropathy, and neurovascular disease. As a result, every organ in the body can be affected, including bones (osteoporosis, avascular necrosis of femoral and/or humeral heads), skin (retractable ulcers), heart (pulmonary hypertension, right heart failure), lungs (restrictive and obstructive lung defects), gastrointestinal system (chronic severe constipation, liver dysfunction), vision (retinopathy), and brain (progressive cognitive decline, ischemic and hemorrhagic stroke).
The goals of therapy are to improve quality of life, decrease cumulative disease burden, and maximize life expectancy. Specifically, these include preventing fatal complications such as infections, stroke, and acute chest crises, as well as minimizing painful crises and chronic pain and limiting end-organ damage. Consequently, a clinically meaningful response to treatment, according to the input received, would include the absence of VOCs, improved self-reported quality of life, engraftment of engineered cells with the persistence of genetically targeted stem cells, independence from transfusion, an absence of hemolysis, an absence of treatment-related neoplasms, and stability of cardiovascular, renal, and pulmonary function.
Several unmet needs were identified from the input, including the fact that despite the effectiveness of HSCT, most patients do not have access to this treatment, as they do not have a matched sibling donor. Other available treatments do not consistently stop disease progression or ongoing organ damage, and all are associated with important toxicities. The input highlighted the need for additional therapeutic options because of the limited number of therapies overall.
The 2 clinician groups indicated that exagamglogene autotemcel could be considered a first-line treatment for patients aged 12 years and older with severe sickle cell disease phenotypes, despite best supportive care measures, who do not have an available matched sibling donor (i.e., for whom HSCT is not an option), which is consistent with the input provided by the clinical experts consulted by CDA-AMC. Therapy must be delivered in the inpatient setting, in a specialized treatment centre with experience in myeloablative therapy and/or cellular therapy, and with specialty services from a multidisciplinary team. Monitoring would be best overseen by a hemoglobinopathy specialist.
The input noted that patients with sickle cell disease are at higher risk of myeloid malignancies, and that busulfan has been associated with myeloid malignancies and solid tumours in this patient population. Therefore, ongoing postmarket monitoring for treatment-related neoplasms is considered imperative. The input also noted the need for equitable access, regardless of a patient’s geographic distance from a treatment centre, which can sometimes mean relocation. In addition, clinician groups recognized that treatment is associated with a high risk of infertility and noted that the cost of fertility preservation should be included in price negotiations.
Drug Program Input
The drug programs provide input on each drug being reviewed through CDA-AMC reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CDA-AMC are summarized in Table 3.
Table 3: Summary of Drug Plan Input and Clinical Expert Responses
Drug program implementation questions | Clinical expert response |
|---|---|
Considerations for initiation of therapy | |
Eligibility criteria for the pivotal trial required patients to have:
Would the above criteria from the pivotal trial be appropriate for reimbursement purposes? Would any additional laboratory tests be required for reimbursement purposes, based on the pivotal trial inclusion and/or exclusion criteria? | The clinical experts considered these criteria to be fair. However, there are patients with severe phenotypes who would not be captured by the criteria. For example, the clinical experts highlighted patients who had severe VOCs but are now well controlled with chronic red blood cell transfusions. Considering the burden of transfusions for patients, caregivers, and the health care system, the clinical experts suggested that these patients not be excluded from the reimbursement criteria. The clinical experts noted that stroke is also considered a severe manifestation that may be included in the reimbursement criteria. Although the treatment is not entirely comparable to a bone marrow transplant, the clinical experts indicated that the selection criteria for a bone marrow transplant may be a benchmark against which to balance the risks and/or benefits of therapy regarding the conditioning risks associated with exagamglogene autotemcel. The clinical experts confirmed that no additional laboratory tests would be required for reimbursement purposes based on the pivotal trial selection criteria. |
Eligibility criteria for the pivotal trial required patients to be aged 12 to 35 years. The sponsor noted that “if patients with SCD or TDT who are over 35 years of age are deemed fit for treatment with exa-cel, there is no plausible biologic mechanism to limit access to exa-cel to those no older than 35 years.” Should patients older than 35 years be eligible to receive exagamglogene autotemcel? | The clinical experts indicated that patients older than 35 years should be eligible to receive, as several patients older than 35 years are likely to benefit from treatment. Therefore, the experts suggested that age should not be an absolute cut-off for reimbursement; rather, a patient’s fitness for treatment with exagamglogene autotemcel should be considered. |
The product is proposed as a “one-time treatment with potential for a functional cure.” Are there any instances in which a second dose would be considered appropriate? | The clinical experts considered it very unlikely that transplant specialists would recommend a second round of myeloablative conditioning chemotherapy. |
Considerations for continuation or renewal of therapy | |
Therapy would not be continued, per se, because exagamglogene autotemcel is a single-administration therapy. However, there may be a need to confirm long-term response. The sponsor notes that “Patients with SCD with recurrent VOCs who received exa-cel in CLIMB-121 were asked to enrol in the long-term follow-up study CLIMB-131 (NCT04208529), where they will be followed for up to 15 years post exa-cel infusion.” How should a clinically meaningful response be defined using objective parameters? How long should follow-up last to confirm that a clinically meaningful response is maintained? | The experts suggested that clinically meaningful responses be monitored by clinicians, based on routine evaluations. These would include mainly quality-of-life assessments and health care use in terms of emergency department visits and hospitalizations. Biochemical monitoring of treatment effects may also be performed by measuring hemoglobin and fetal hemoglobin percentages, which are objective measures that can be collected peripherally. However, the clinical experts indicated that these remain surrogate outcomes of lesser importance than clinical outcomes. The proportion of bone marrow genetically modified cells may theoretically inform on the maintenance of effect over time; the clinical experts mentioned, however, that there is no agreed-upon threshold to be reached. The clinical experts also noted that there is a current paucity of long-term data, and because complications from myeloablative conditioning may present late, the CLIMB-131 study has a 15-year follow-up. |
Generalizability | |
The pivotal trial listed numerous exclusion criteria, but there are no related contraindications, or warnings and/or precautions, to therapy listed in the product monograph for most of these. The sponsor noted that patients with an available HLA-matched related donor were excluded from the pivotal clinical trials due to ethical concerns around including patients with a viable treatment option in a trial for a treatment without proven efficacy or safety at the time. However, based on the results of the CLIMB-121 and CLIMB-111 studies, which demonstrated that exagamglogene autotemcel results in improved clinical outcomes (by significantly reducing VOCs in patients with SCD and by demonstrating transfusion independence in patients with TDT), this may no longer be a valid concern. Which, if any, of the pivotal trial exclusion criteria should be used for determining eligibility for treatment? | The clinical experts agreed that patients who were previously treated with HSCT should not be candidates for exagamglogene autotemcel, as having a second round of myeloablative conditioning chemotherapy would be contraindicated. In clinical practice, exagamglogene autotemcel would be positioned after HSCT in younger patients who have a matched sibling donor eligible and willing to donate, considering the lack of long-term efficacy and safety data. Therefore, the clinical experts indicated that these patients should not be eligible for exagamglogene autotemcel at the time of this review. The clinical experts also noted that patients who are ineligible for transplant or who present with unacceptable end-organ damage, at the discretion of the transplant physician, should not be candidates to receive exagamglogene autotemcel. |
Eligibility criteria for the pivotal trial required patients to be aged 12 to 35 years, and the product monograph states that “[n]o data in patients less than 12 years of age are available to Health Canada; therefore, Health Canada has not authorized an indication of pediatric use in patients less than 12 years of age.” Will there be interest in using exagamglogene autotemcel in those younger than 12 years? If so, should such patients be considered for reimbursement? | The clinical experts noted that there would likely be interest in using exagamglogene autotemcel in patients younger than 12 years. However, they also noted that there are several risks and uncertainty surrounding this treatment, which may limit the number of young patients to whom it may actually be offered. Some issues may resonate stronger in a younger population, such as the loss of fertility and the contraindication to receiving another gene therapy in the future. |
Care provision issues | |
The sponsor noted the following:
Patients not diagnosed through NBS could also have their blood drawn and sent to a laboratory for testing, with review by a hematopathologist (this is how most hemoglobinopathies are diagnosed later in life). Is the above accurate from a diagnostic standpoint? Is the blood spot screening test referenced by the sponsor widely available, in use in Canada and, most important, reliable and accurate? | The clinical experts agreed with the sponsor’s assessment of diagnosis testing in newborns. They indicated that newborn screening is an important diagnostic tool for identifying babies born in Canada with hemoglobinopathies. Newborn screening uses a spot screening test, which is widely available and tests for a number of conditions. Abnormal newborn screens suggestive of hemoglobinopathies are sent for confirmation with hemoglobin electrophoresis. If positive, genetic testing is often also performed. Screening and diagnosis of hemoglobinopathies would occur regardless of exagamglogene autotemcel eligibility. The sensitivity and specificity of blood spot testing are excellent for sickle cell disease. For adults, however, the clinical experts indicated that some people would not have had access to newborn screening, such as newcomers to Canada or those born before the implementation of newborn testing. These patients may be identified after they develop symptoms or during routine screening with hemoglobin electrophoresis, which is reviewed and interpreted by an expert (hematologist or hematopathologist). Genetic testing is often conducted to provide further information. Hemoglobin electrophoresis is widely available. |
The sponsor noted the following:
Is the above accurate from an implementation and/or resource standpoint? | The clinical experts did not agree with the sponsor’s assessment. The clinical experts highlighted that most centres are geared toward treating patients with malignancies, and that very few centres have established nonmalignant funding sources or ancillary services. Although the number of patients receiving exagamglogene autotemcel treatment is likely to be small, the source of funding for the use of resources, aside from the drug cost, is currently unclear. The clinical experts listed, for example, red blood cell exchange, stem cell collection, treatment with plerixafor, and admission to an inpatient ward for 1 month. |
ATC = authorized treatment centre; exa-cel = exagamglogene autotemcel; HLA = human leucocyte antigen; HSCT = hematopoietic stem cell transplant; NBS = newborn screening; SCD = sickle cell disease; TDT = transfusion-dependent beta-thalassemia; VOC = vaso-occlusive crisis.
Clinical Evidence
The objective of the CDA-AMC Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of exagamglogene autotemcel cell suspension in patient-specific vials for IV infusion in the treatment of sickle cell disease in patients 12 years and older with recurrent VOCs. The focus will be placed on comparing exagamglogene autotemcel to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of exagamglogene autotemcel is presented in 4 sections, with the CDA-AMC critical appraisal of the evidence at the end of each section. The first section, the Systematic Review, includes pivotal studies and RCTs that were selected in accordance with the sponsor’s systematic review protocol. The CDA-AMC’ assessment of the certainty of the evidence in this first section, using the GRADE approach, follows the critical appraisal of the evidence. The second section includes sponsor-submitted long-term extension studies. The third section includes indirect evidence from the sponsor; however, none was submitted. The fourth section includes additional studies that were considered by the sponsor to address important gaps in the systematic review evidence; however, none were included.
Included Studies
Clinical evidence from the following is included in the CDA-AMC review and appraised in this document:
1 pivotal study identified in the systematic review
1 long-term extension study.
Systematic Review
The contents of this section have been informed by materials submitted by the sponsor. The following information has been summarized and validated by the CDA-AMC review team.
Description of Studies
One study was identified and included in the systematic review. The ongoing CLIMB-121 study (n = 63 patients enrolled) is a single-arm, phase III, multicenter study designed to evaluate the efficacy and safety of exagamglogene autotemcel, administered after single-drug myeloablative conditioning chemotherapy, for the treatment of sickle cell disease in patients aged 12 and 35 years with severe disease who have recurrent VOCs (at least 2 protocol-defined severe VOC events per year in the 2 years before enrolment). The primary outcome was the proportion of patients who did not experience any severe VOC events for at least 12 consecutive months from 60 days after the last red blood cell transfusion to up to 2 years after the exagamglogene autotemcel infusion.
The study had a single-arm treatment design. According to the sponsor, this was based on the premise that allogeneic HSCT would not be considered a relevant comparator, due to ethical concerns around including patients with a viable treatment option in a trial for a treatment without proven efficacy or safety. Before being enrolled in the study, patients received standard-of-care treatments, such as off-label hydroxyurea and/or red blood cell transfusions with iron chelation therapy.
The CLIMB-121 study is ongoing. Results from the most recent interim analysis, with a data cut-off date of June 14, 2023, are presented in this report and constitute the main evidence informing the CDA-AMC review. Patients who complete the CLIMB-121 study are enrolled in the CLIMB-131 long-term extension study, and will be followed for up to 15 years after exagamglogene autotemcel infusion.
Characteristics of the included study are summarized in Table 4.
Table 4: Details of the Study Included in the Systematic Review
Detail | CLIMB-121 |
|---|---|
Design and populations | |
Study design | A phase I, II, and III single-arm, open-label, multisite, single-dose study (ongoing) |
Locations | 16 sites in 7 countries: Belgium, Canada (1 site at The Hospital for Sick Children in Toronto), France, Germany, Italy, UK, US |
Key dates | Start date: November 27, 2018 Data cut-off (interim analysis): June 14, 2023 |
Enrolled (N) | 63 patients enrolled (30 patients analyzed in the PES population) |
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Intervention | Single IV infusion of exa-cel at a minimum recommended dose of ≥ 3.0 × 106 CD34+ cells/kg administered in the hospital after myeloablative conditioning (minimum of 48 hours and maximum of 7 days after completion of myeloablative conditioning) |
Study duration | |
Premobilization | ≥ 8 weeks |
Mobilization | 3 to 4 days per cycle |
Manufacturing and quality control | Approximately 24 weeks |
Myeloablative conditioning, infusion, and engraftment | Median, 41 days (range, 29 to 62 days) |
Follow-up | 24 months (followed by entry into the LTE study for a total follow-up of up to 15 years) |
Outcomes | |
Primary end point | Proportion of patients who have not experienced any severe VOC event for ≥ 12 consecutive months from 60 days after the last RBC transfusion to up to 2 years after exa-cel infusion |
Secondary and exploratory end points | Key secondary
Secondary
Safety
|
Publication status | |
Publications | Frangoul et al. (2021)3 Frangoul et al. (2024)30 |
Clinical trial record number | NCT03745287 |
ACS = acute chest syndrome; AE = adverse event; AESI = adverse event of special interest; ASCQ-Me = Adult Sickle Cell Quality of Life Measurement Information System; EQ-5D-5L = 5-Level EQ-5D; EQ-5D-Y = EQ-5D Youth; exa-cel = exagamglogene autotemcel; FACT-BMT = Functional Assessment of Cancer Therapy-Bone Marrow Transplant; HLA = human leukocyte antigen; HSCT = hematopoietic stem cell transplant; LTE = long-term extension; PES = primary efficacy set; PRO = patient-reported outcome; RBC = red blood cell; SAE = serious adverse event; SCD = sickle cell disease; TRM = transplant-related mortality; VOC = vaso-occlusive crisis.
Sources: SCD Clinical Overview Addendum,17 Clinical Study Report for CLIMB-121,31 Clinicaltrials.gov. 2018,32 sponsor’s Summary of Clinical Evidence.
The administration of exagamglogene autotemcel is a multistep process, which is presented in Figure 2. Briefly, after screening, patients enrolled in the CLIMB-121 study underwent mobilization treatment to increase yields of CD34+ stem cells for collection. Then, patients started treatment with busulfan conditioning chemotherapy. This was followed by exagamglogene autotemcel infusion and, finally, by engraftment of the cells (the process by which the infused gene-edited stem cells travel through the blood to the bone marrow where they begin to produce gene-edited red blood cells, neutrophils, and platelets). Additional cells were collected during the CLIMB-121 study that could be used as backup for rescue therapy in the event of nonneutrophil engraftment with exagamglogene autotemcel; however, no patient required backup cells.
Populations
Inclusion and Exclusion Criteria
Patients aged 12 to 35 years were eligible for the trial if they had severe sickle cell disease, defined by the genotype, and a history of at least 2 protocol-defined severe VOC events per year in the 2 years before enrolment. Patients needed to have a good performance status and be eligible for autologous stem cell transplant. Patients were excluded from the trial if they had a prior HSCT, or if they had an available related donor that made HSCT possible. Other key exclusion criteria included clinically significant bacterial, viral, fungal, or parasitic infection; prior or current malignancy, myeloproliferative disorder, or significant immunodeficiency; white blood cell or platelet count below predefined levels and a fetal hemoglobin level above 15%; advanced liver, renal, lung, or cardiac disease, as specified; and unplanned hospitalizations or emergency department visits in the previous year that were related to sickle cell disease but consistent with significant chronic pain (rather than acute pain crises). Finally, prior treatment with gene therapy or an editing product was prohibited.
Figure 2: CLIMB-121 Study Design
Cond = busulfan conditioning; CRISPR/Cas9: clustered regularly interspaced short palindromic repeats–CRISPR-associated protein 9; Enroll = enrolment; exa-cel = exagamglogene autotemcel; hHSPCs = human hematopoietic stem and progenitor cells; M24 = month 24; Mob = mobilization; NE = neutrophil engraftment.
Note: Stage 1 = signing of the informed consent form to the start of mobilization; Period 2 = the start of the first mobilization cycle to 2 weeks after the end of the last mobilization cycle; Period 3 = 2 weeks after the end of the last mobilization cycle to the start of busulfan conditioning; Period 4 = the start of busulfan conditioning to the start of exagamglogene autotemcel infusion; Period 5 = the start of exagamglogene autotemcel infusion to month 24 (end-of-study visit).
aSafety analysis intervals: Enroll to <exa-cel = enrolment to the day before exagamglogene autotemcel infusion; Exa-cel to M24 = the day of exagamglogene autotemcel infusion to month 24 or the end-of-study visit; Enrol to M24 = enrolment to month 24 or the end-of-study visit.
bAdditional intervals: Enroll to <Mob = enrolment to the day before the first mobilization cycle; Mob to <Cond = the start of the first mobilization cycle to the day before the start of busulfan conditioning; Cond to NE = the start of busulfan conditioning to neutrophil engraftment; >NE to M24 = the day after neutrophil engraftment to month 24 or the end-of-study visit.
Source: Clinical Study Report for CLIMB-121.31
Interventions
The CLIMB-121 study evaluated the efficacy and safety of exagamglogene autotemcel using a single-arm treatment design. The following discussion points were provided by the sponsor to explain this decision:
HSCT was not considered a relevant comparator for exagamglogene autotemcel because of —
ethical concerns around including patients with a viable treatment option in a trial for a treatment without proven efficacy or safety at the time
a difference in patient populations (HSCT is best suited for very young patients, whereas exagamglogene autotemcel has been studied in patients 12 years and older, so it was assumed that most patients with a matched donor available would have received HSCT before they reached the age of eligibility for exagamglogene autotemcel).
Standard of care, such as off-label hydroxyurea and/or red blood cell transfusions with iron chelation therapy, was not considered a relevant comparator because patients would already be receiving these therapies at baseline, as the inclusion criteria required patients to experience VOC events while receiving best supportive care.
Placebo was not considered a relevant comparator, as the mobilization, myeloablation, and subsequent transplant procedures required before exagamglogene autotemcel treatment would be neither feasible nor ethical to perform as sham procedures in a placebo group.
In the CLIMB-121 study, all patients received a single IV infusion of exagamglogene autotemcel on day 1, at a minimum recommended dose of at least 3.0 × 106 CD34+ cells/kg. Exagamglogene autotemcel was administered in the hospital by trained professionals. Before infusion, patients were required to undergo a mobilization and apheresis process, as well as myeloablative conditioning chemotherapy, as follows:
During mobilization, patients received plerixafor at a dose of 0.24 mg/kg by subcutaneous injection 2 to 3 hours before the start of apheresis. Patients then underwent apheresis for 2 or 3 consecutive days to collect CD34+ hematopoietic stem cells and progenitor cells for exagamglogene autotemcel manufacturing.
For myeloablative conditioning, patients were hospitalized to receive busulfan IV, at a starting dose of 3.2 mg/kg per day, for 4 consecutive days. Exagamglogene autotemcel treatment had to be administered 2 to 7 days after the completion of myeloablative conditioning.
Outcomes
A list of efficacy end points assessed in this Clinical Review Report is provided in Table 5, followed by descriptions of the outcome measures. Summarized end points are based on outcomes included in the sponsor’s Summary of Clinical Evidence, as well as outcomes identified as important to this review by to the clinical experts consulted by CDA-AMC and from input from patient and clinician groups and public drug plans. Using the same considerations, the CDA-AMC review team selected end points that were considered to be most relevant to CDA-AMC expert committee deliberations, and finalized this list of end points in consultation with members of the expert committee. All summarized efficacy end points were assessed using the GRADE approach. Select notable harms outcomes considered important to CDA-AMC expert committee deliberations were also assessed using GRADE.
Table 5: Outcomes Summarized From the CLIMB-121 Study
Outcome measure | Time point | CLIMB-121 |
|---|---|---|
Clinical efficacy outcomes | ||
Proportion of patients who have not experienced any severe VOC event for at least 12 consecutive months | Any 12-month period, starting from 60 days after the last red blood cell transfusion to up to 2 years after exagamglogene autotemcel infusion | Primarya |
Health care use outcomes | ||
Proportion of patients free from inpatient hospitalization for severe VOCs sustained for at least 12 months | Any 12-month period, starting from 60 days after the last red blood cell transfusion to up to 2 years after exagamglogene autotemcel infusion | Key secondarya |
Reduction in units of red blood cell transfusions | Baseline to data cut-off | Secondary |
Hematological outcomes | ||
Proportion of patients with sustained fetal hemoglobin ≥ 20% for at least 12 consecutive months | Baseline to data cut-off | Secondary |
Proportion of alleles with intended genetic modification present in CD34+ cells of the bone marrow | Baseline to month 6, month 12, and month 24 | Secondary |
Patient-reported outcomes | ||
Change over time in ASCQ-Me score | Baseline to month 6, month 12, month 18, and month 24 | Secondary |
Safety outcomes | ||
Proportion of patients with neutrophil engraftment | Day 42 | Safety |
Time to engraftment (neutrophil and platelet) | Time to event | Safety |
AEs, SAEs, and AEs of special interest (e.g., malignancies) | Continuous | Safety |
Transplant-related mortality | Safety | |
All-cause mortality | Safety | |
AE = adverse event; ASCQ-ME = Adult Sickle Cell Quality of Life Measurement Information System; SAE = serious adverse event; VOC = vaso-occlusive crisis.
aStatistical testing for these end points was adjusted for multiple comparisons (e.g., hierarchal testing).
Sources: SCD Clinical Overview Addendum,17 Clinical Study Report for CLIMB-121,31 sponsor’s Summary of Clinical Evidence.
Vaso-Occlusive Crises
The primary outcome in the CLIMB-121 study was the proportion of patients who did not experienced any severe VOC events for at least 12 consecutive months from 60 days after the last red blood cell transfusion to up to 2 years after exagamglogene autotemcel infusion. On-trial VOC events were adjudicated by an independent external end point adjudication committee.
Severe VOC was defined in the CLIMB-121 study as any of the following:
acute pain event that requires a visit to a medical facility and administration of pain medications (opioids or IV nonsteroidal anti-inflammatory drugs) or red blood cell transfusions
acute chest syndrome, as indicated by the presence of a new pulmonary infiltrate associated with pneumonia-like symptoms, pain, or fever
priapism lasting more than 2 hours and requiring a visit to a medical facility
splenic sequestration, defined by an enlarged spleen, left upper quadrant pain, and an acute decrease in hemoglobin concentration of at least 2 g/dL.
Considering the consequences of severe VOCs on patient morbidity and quality of life, the clinical experts consulted by CDA-AMC emphasized that avoidance of these events is paramount in preventing patient suffering and mortality. In clinical practice, the determination of VOCs is considered a challenge, according to the clinical experts consulted, as it can be difficult to differentiate between VOCs and other diagnoses, especially with regard to chronic pain events. Assessment may be considered partly subjective and is based on a combination of patient-reported symptoms and clinical examination.
Hospitalizations
Hospitalizations were included in the study as a key secondary outcome, and assessed as the proportion of patients free from inpatient hospitalization for severe VOCs for at least 12 months, from 60 days after the last red blood cell transfusion to up to 2 years after exagamglogene autotemcel infusion. This outcome was deemed particularly relevant, according to the clinical experts, as reducing health care use is considered one of the main goals of sickle cell disease therapy.
Transfusions
Another secondary outcome in the study was the reduction in units of red blood cell transfusion; this was also considered an important treatment goal by the clinical experts, because of the particularly high burden associated with chronic transfusions for patients and for the health care system.
Hematological Outcomes
Biomarker end points of efficacy included the proportion of patients with sustained fetal hemoglobin of at least 20% for at least 12 consecutive months, as well as the proportion of alleles with intended genetic modification present in CD34+ cells of the bone marrow. These outcomes are consistent with the mechanism of action of exagamglogene autotemcel. The clinical experts indicated that the fetal hemoglobin threshold of at least 20% is considered to be the minimum level that leads to significant phenotypical modification. The experts also indicated that there was no known minimal clinically important threshold for alleles with intended genetic modification at the time of the review, and that the proportion needed to induce phenotypic changes is disease dependent.
Patient-Reported Outcomes
HRQoL was assessed as a secondary outcome in the study using ASCQ-Me,12 which is a disease-specific measurement system that enables adults to describe their functioning and well-being. Five question sets assess the impact of emotional functioning, social functioning, pain, stiffness, and sleep; higher scores indicate better HRQoL. For the pain-episode questions (which include pain-frequency and pain-severity scores) and the Sickle Cell Disease Medical History Checklist, lower scores indicate less severe pain.12 A reduction of 5 points for pain episodes and an increase of 5 points on impact subscales were considered MCIDs.13
The clinical experts indicated that HRQoL is considered an important outcome when it comes to treating patients with severe manifestation of sickle cell disease. Although the experts considered the use of ASCQ-Me to be appropriate, they indicated that it is not routinely used in clinical practice.
Safety Outcomes
The safety analysis included AEs, SAEs, and mortality. AEs of special interest included malignancies.
Neutrophil and platelet engraftment was assessed as a harms outcome in the safety population. Engraftment is the process by which the infused gene-edited stem cells travel through the blood to the bone marrow, where they begin to produce new red blood cells, neutrophils, and platelets. The clinical experts noted that patients who do not achieve engraftment, or for whom time to engraftment is particularly long, are at increased risk of bleeding events and infections in the period after myeloablative conditioning and exagamglogene autotemcel infusion. Although engraftment is an important milestone, it should not be considered a comprehensive measure of patient safety or overall treatment success. As such, engraftment does not provide any information on important AEs that could occur before or during the engraftment process; thus, the frequency and severity of AEs — such as infections (including febrile neutropenia), bleeding, veno-occlusive liver disease, and hemophagocytic lymphohistiocytosis — that can occur before engraftment is achieved should also be considered in the safety assessment.
Statistical Analysis
A summary of the statistical analyses used in the CLIMB-121 study is presented in Table 7. All efficacy analyses were performed in the PES (i.e., all enrolled patients who received exagamglogene autotemcel infusion, and who were followed for at least 16 months after infusion and for at least 14 months after completion of the red blood cell transfusions for posttransplant support or sickle cell disease management). Safety analyses were based on the safety analysis set (i.e., all enrolled patients who started the mobilization regimen).
Primary and Key Secondary End Point Analyses
The proportion of patients who did not experience any severe VOC for at least 12 consecutive months (primary efficacy end point) and the proportion of patients free from inpatient hospitalization for severe VOCs for at least 12 months (key secondary end point) were evaluated with a 1-sided P value against a 50% response rate, and the 2-sided 95% exact Clopper-Pearson confidence interval. According to the statistical analysis plan, if the prespecified efficacy boundary was crossed at any interim analysis, overwhelming efficacy was considered to be established for exagamglogene autotemcel.
Patients were considered responders if they met the response criteria any time during the response evaluation period. If a patient died or discontinued the study before achieving the end point for reasons other than exagamglogene autotemcel–related AEs, then the VOC-free status of that patient was carried forward for up to 24 months after infusion. Patients who died or discontinued the study due to exagamglogene autotemcel–related AEs before achieving the end point or who continuously received red blood cell transfusions for posttransplant support or sickle cell disease management after month 10 following infusion were considered to be nonresponders.
Other Secondary End Point Analyses
All secondary end points were summarized as continuous variables over time.
Sample Size and Power Calculation
With a planned total of 45 patients dosed, 3 interim analyses could be performed after a group sequential testing procedure in the study to allow for the evaluation of efficacy. This sample size provided at least 95% power to rule out a response rate of 50% when the true response rate is 80% for both the primary and key secondary efficacy end points, with 1-sided alpha of 2.5%. No rationale was provided by the sponsor to justify the use of a 50% response rate as the tested hypothesis; however, the clinical experts indicated that this was likely consistent with a meaningful improvement for patients with severe manifestations of sickle cell disease.
Statistical Testing
Multiplicity was considered with respect to testing the null hypothesis for the primary and key secondary efficacy end points in the 3 interim analyses and the final analysis. The familywise type I error rate was controlled with an alpha spending approach for tests at the interim and final analyses and a sequential testing of the primary and key secondary efficacy end points (i.e., the key secondary efficacy end point was to be tested only if the primary efficacy end point crossed the efficacy boundary and, thus, was statistically significant).
The efficacy boundaries for the primary and key secondary efficacy end points were specified to control the type I error at a 1-sided alpha of 2.5% across multiple looks, based on the exact binomial distribution. These are presented in Table 6. The first interim analysis was not conducted, but no rationale or justification was provided by the sponsor as to why. As a result of this, the primary end point and key secondary end point were considered to be statistically significant if the corresponding 1-sided P value was less than 0.0144. The subsequently tested secondary end point was considered to be statistically significant if the corresponding 1-sided P value was less than 0.025.
Table 6: Operating Characteristics of the Efficacy Boundaries for the Primary and Key Secondary Efficacy End Points
Analysis stage | Efficacy boundary (n/N) | Boundary in response rate, % (95% CI) | Probability of crossing efficacy boundaries under different response rates,a % | One-sided alpha spent at each IA or final analysis (assuming a response rate of 50%), % | ||
|---|---|---|---|---|---|---|
p1 = 80% | p1 = 85% | p1 = 90% | ||||
IA1 (N = 10) | 9/10 | 90.0 (55.5 to 99.7) | 37.6 | 54.4 | 73.6 | 1.074 |
IA2b (N = 17) | 14/17 | 82.4 (56.6 to 96.2) | 54.9 | 75.6 | 91.7 | 0.366 |
IA3 (N = 30) | 22/30 | 73.3 (54.1 to 87.7) | 87.1 | 97.2 | 99.8 | 0.540 |
Final analysis (N = 45) | 31/45 | 68.9 (53.4 to 81.8) | 97.5 | 99.8 | > 99.9 | 0.440 |
Overall power | 97.9 | 99.9 | > 99.9 | 2.420 | ||
IA = interim analysis; CI = confidence interval; p1 = probability of crossing efficacy boundaries under the specified response rate.
aMarginal probability of crossing the efficacy boundary at a specific interim or final analysis.
bIA1 was not conducted; hence, the alpha planned for IA1 was recovered for IA2. The primary end point and the key secondary end point of hospitalization were considered to be statistically significant if the corresponding 1-sided P value was < 0.0144.
Sources: Clinical Study Report for CLIMB-121,31 sponsor’s Summary of Clinical Evidence.
Subgroup Analyses
Subgroup analyses of the primary and the key secondary efficacy end point were performed with point estimates and 95% CIs for each subgroup, and were conducted using:
age at screening (≥ 12 to < 18 years and ≥ 18 to ≤ 35 years)
genotype (betaS/betaS and nonbetaS/betaS)
sex
race (Black or African American and other races)
at least 3 VOCs/year in the 2 years before baseline.
Prespecified subgroup analyses of age, genotype, sex, region, and race for selected AE summaries were also conducted.
The subgroups of age, genotype, and prior VOCs were considered relevant to the review, according to the clinical experts.
Table 7: Statistical Analysis of Efficacy End Points in the CLIMB-121 Study
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
Proportion of patients who achieved VF12 | 1-sided P value against a 50% response rate and 2-sided 95% exact Clopper-Pearson confidence interval | NR | If a patient died or discontinued the study before achieving VF12, the VOC-free status of that patient was carried forward for up to 24 months after exa-cel infusion | Proportion of patients who remained VOC-free until the end of the study (the data cut-off date) Summarized based on patients who had been followed for at least 18 months after exa-cel infusion |
Proportion of patients who achieved HF12 | If a patient died or discontinued the study before achieving HF12, the hospitalization-free status of that patient was carried forward for up to 24 months after exa-cel infusion | NR | ||
Proportion of patients who achieved VF9 | If a patient died or discontinued the study before achieving VF9, the VOC-free status of that patient was carried forward for up to 24 months after exa-cel infusion | NR | ||
Total hemoglobin concentration | Summarized as a continuous variable over time | NR | NR | NR |
Total fetal hemoglobin concentration | ||||
Proportion of patients with sustained fetal hemoglobin of ≥ 20% for at least 12 months | ||||
Relative reduction in units of red blood cell transfusion | ||||
Proportion of alleles with intended genetic modification present in the peripheral blood | ||||
Proportion of alleles with intended genetic modification present in CD34+ cells of the bone marrow | ||||
Change over time in weekly pain-scale score (11-point NRS) | Summarized as a continuous variable over time, and including domain score and total score (if applicable) | |||
Change over time in EQ-5D-5L score | ||||
Change over time in EQ-5D-Y score | ||||
Change over time in FACT-BMT score |
EQ-5D-5L = 5-Level EQ-5Q; EQ-5D-Y = EQ-5D Youth; exa-cel = exagamglogene autotemcel; FACT-BMT = Functional Assessment of Cancer Therapy-Bone Marrow Transplant; HF12 = no inpatient hospitalization for severe VOCs for at least 12 months; NR = not reported; NRS = numerical rating scale; VF12 = no severe VOC for at least 12 consecutive months; VF9 = no severe VOC for at least 9 consecutive months; VOC = vaso-occlusive crisis.
Sources: Clinical Study Report for CLIMB-121,31 sponsor’s Summary of Clinical Evidence.
Analysis Populations
A summary of the analysis populations used in the CLIMB-121 study is presented in Table 8. The PES is the main analysis population informing the baseline characteristics and efficacy assessments in this review.
Table 8: Analysis Populations of the CLIMB-121 Study
Population | Definition | Application |
|---|---|---|
Enrolled set (N = 63) | All enrolled patients who signed informed consent and met the eligibility criteria | Listings of the demographics and baseline characteristics |
FAS (N = 44) | A subset of the enrolled set that included patients who received an exa-cel infusion | Summary of demographics and baseline characteristics, and efficacy analyses where applicable Used in supportive analyses |
PES (N = 30) | A subset of the FAS that included all patients who were followed for at least 16 months after exa-cel infusion and for at least 14 months after the completion of RBC transfusions for posttransplant support or SCD management | Summary of demographics and baseline characteristics, and all efficacy analyses Used in the primary data analysis and supportive data analyses |
Early efficacy set (N = 32) | A subset of the FAS that included all patients who were followed for at least 12 months after exa-cel infusion and for at least 11 months after the completion of RBC transfusions for posttransplant support or SCD management | Summary of select demographic data, and efficacy analyses where applicable Used in supportive and subgroup analyses |
Safety analysis set (N = 58) | A subset of the enrolled set that included patients who started the mobilization regimen | Safety analyses |
exa-cel = exagamglogene autotemcel; FAS = full analysis set; PES = primary efficacy set; RBC = red blood cell; SCD = sickle cell disease.
Sources: SCD Clinical Overview Addendum,17 Clinical Study Report for CLIMB-121,31 sponsor’s Summary of Clinical Evidence.
Results
Patient Disposition
As of the clinical data cut-off date of June 14, 2023, 17 of the 63 enrolled patients (27%) completed the study, whereas ██ ████████ █████ were reported as continuing the study. A total of ██ ████████ █████ discontinued the study, almost exclusively before receiving exagamglogene autotemcel; of these, 6 patients (i.e.,10% of the 58 patients who started mobilization) discontinued due to inadequate cell collections. Details regarding patient disposition are presented in Table 9.
Table 9: Summary of Patient Disposition From the CLIMB-121 Study
Patient disposition | Exa-cel (N = 63) |
|---|---|
Enrolled | 63 |
Started mobilization | 58 |
Started the conditioning regimen | 44 |
Received exagamglogene autotemcel | 44 |
Patient disposition up to exagamglogene autotemcel infusion | |
Enrolled, n (%) | 63 (100.0) |
Discontinued the study before receiving exagamglogene autotemcel , n (%) | 16 (25.0) |
Before mobilization, n | 5 |
Reason for discontinuation | |
Withdrawal of consent | 3 |
Noncompliance | 1 |
Investigator decision | 1 |
After the start of mobilization but before myeloablative conditioning, n | 11 |
Reason for discontinuation, n | |
Inadequate cell collections | 6 |
Withdrawal of consent | ███ |
Patient decision | ███ |
Not meeting selection criteria | 1 |
Noncompliance | 1 |
Ongoing in CLIMB-121 but not yet dosed with exagamglogene autotemcel, n (%) | 3 (5.0) |
Received exagamglogene autotemcel infusion, n (%) | 44 (70.0) |
Patient disposition after exagamglogene autotemcel infusion | |
Dosed with exagamglogene autotemcel, n (%) | 44 (100.0) |
Ongoing in CLIMB-121 | ██ ██████ |
Completed CLIMB-121 | 17 (39.0) |
Discontinued the study after exagamglogene autotemcel infusion | █████ |
Reason for discontinuation, n | |
Death (respiratory failure after COVID-19 infection) | 1 |
Exa-cel = exagamglogene autotemcel.
Sources: SCD Clinical Overview Addendum,17 sponsor’s Summary of Clinical Evidence.
Baseline Characteristics
The baseline characteristics outlined in Table 10 are limited to those that are most relevant to this review or were felt to affect the outcomes or interpretation of the study results. For the 30 patients in the PES, which the efficacy analyses are based on, the mean age at baseline was 22 years; 6 patients (20%) were younger than 18 years. A total of 26 patients (87%) were Black or African American. The predominant genotype, in 29 patients (97%), was betaS/betaS, which is considered to be a severe phenotype.
In the 2 years before enrolment in the CLIMB-121 study, patients had a mean annualized rate of 3.9 severe VOC events (SD = 2.1 events). The mean annualized rate of inpatient hospitalizations for severe VOCs was 2.7 (SD = 2.0 hospitalizations), resulting in a mean annualized duration of hospitalizations of 17.1 days (SD = 14.3 days). Patients were transfused annually with a mean of 8.4 units (SD = 14.9 units) of red blood cells for an indication related to sickle cell disease.
According to the clinical experts, patients in the CLIMB-121 study were considered to be mostly representative of patients with sickle cell disease seen in clinical practice, although there can be a wide variety of disease presentation among patients.
Table 10: Summary of Baseline Characteristics From the CLIMB-121 Study
Patient demographics | FAS (N = 44) | PES (N = 30) |
|---|---|---|
Age at screening, years | ||
Mean (SD) | 21.2 (6.1) | 22.1 (6.0) |
Median (minimum to maximum) | 20.0 (12.0 to 34.0) | 21.0 (12.0 to 34.0) |
Age category at screening, n (%) | ||
≥ 12 years and < 18 years | 12 (27.3) | 6 (20.0) |
≥ 18 years and ≤ 35 years | 32 (72.7) | 24 (80.0) |
Sex, n (%) | ||
Male | 24 (54.5) | 16 (53.3) |
Female | 20 (45.5) | 14 (46.7) |
Childbearing potential, n (% of females) | ██ ███████ | ██ ███ |
Race, n (%) | ||
White | 3 (6.8) | 1 (3.3) |
Black or African American | 38 (86.4) | 26 (86.7) |
Asian | ██ █ | ██ █ |
American Indian or Alaska Native | ██ █ | ██ █ |
Native Hawaiian or other Pacific Islander | ██ █ | ██ █ |
Not collected, per local regulations | ██ █ | ██ █ |
Other | 3 (6.8) | 3 (10.0) |
Multiracial | 0 | 0 |
Ethnicity, n (%) | ||
Hispanic or Latino | 2 (4.5) | 2 (6.7) |
Not Hispanic or Latino | 41 (93.2) | 27 (90.0) |
Not collected, per local regulations | 1 (2.3) | 1 (3.3) |
Weight, kg | ||
Mean (SD) | 65.7 (17.3) | 65.5 (14.9) |
Median (minimum to maximum) | 67.0 (34.0 to 116.0) | 65.5 (43.0 to 95.0) |
Genotype, n (%) | ||
BetaS/betaS | 40 (90.9) | 29 (96.7) |
Nonbetas/betaS | ||
BetaS/beta0 | 3 (6.8) | 1 (3.3) |
BetaS/beta+ | 1 (2.3) | 0 |
Total Hb, g/dL | ||
Mean (SD) | 9.1 (1.6) | 9.0 (1.6) |
Median | 9.4 | 9.4 |
Minimum to maximum | 5.7 to 12.6 | 5.7 to 12.6 |
Fetal hemoglobin, % | ||
Mean (SD) | 5.4 (3.9) | 5.2 (3.8) |
Median | 5.0 | 5.3 |
Minimum to maximum | 0.0 to 14.7 | 0.0 to 14.7 |
Fetal hemoglobin, g/dL | ||
Mean (SD) | 0.5 (0.4) | 0.5 (0.4) |
Median | 0.4 | 0.4 |
Minimum to maximum | 0.0 to 1.5 | 0.0 to 1.5 |
Annualized rate of severe VOCs | ||
Mean (SD) | 4.1 (3.0) | 3.9 (2.1) |
Median | 3.5 | 3.3 |
Minimum to maximum | 2.0 to 18.5 | 2.0 to 9.5 |
Annualized rate of inpatient hospitalizations for severe VOCs | ||
Mean (SD) | 2.7 (2.0) | 2.7 (2.0) |
Median | 2.5 | 2.0 |
Minimum to maximum | 0.5 to 9.5 | 0.5 to 8.5 |
Annualized duration of inpatient hospitalizations for severe VOCs, days | ||
Mean (SD) | 19.7 (21.9) | 17.1 (14.3) |
Median | 14.0 | 12.3 |
Minimum to maximum | 2.0 to 136.5 | 2.0 to 64.6 |
Annualized units of red blood cells transfused for SCD-related indication | ||
Mean (SD) | 11.3 (18.4) | 8.4 (14.9) |
Median | 5.0 | 3.3 |
Minimum to maximum | 0.0 to 86.1 | 0.0 to 75.5 |
FAS = full analysis set; Hb = hemoglobin; PES = primary efficacy set; SCD = sickle cell disease; SD = standard deviation; VOC = vaso-occlusive crisis.
Notes: Baseline severe VOCs, inpatient hospitalizations for severe VOCs, and red blood cell transfusions were based on the 2 years before the most recent screening. Only severe VOCs adjudicated by an end point adjudication committee as meeting the protocol definition of severe VOCs were included.
Annualized rate = total number of events/number of years. Annualized duration = total duration of events/number of years. Annualized units = total units/number of years.
Sources: SCD Clinical Overview Addendum,17 sponsor’s Summary of Clinical Evidence.
Exposure to Study Treatments
Exposure to the study treatment is outlined in Table 11. A total of 44 patients received exagamglogene autotemcel infusion in the CLIMB-121 study. The mean dose was ██████████████ ███ █ █████.
As of the data cut-off date of June 14, 2023, the mean follow-up duration after exagamglogene autotemcel infusion was 20.1 months (SD = 10.37 months) in the analysis that included all patients who received the drug, irrespective of whether or not they met the minimum follow-up time to be included in the main PES analysis population.
Table 11: Summary of Patient Exposure From the CLIMB-121 Study
Exposure | Exa-cel (N = 44) |
|---|---|
Exa-cel dose (106 cells/kg) | |
Patients who received exa-cel infusion | 44 |
Mean (SD) | ███ ██████ |
Median | 4.0 |
Minimum to maximum | 2.9 to 14.4 |
Follow-up duration after exa-cel infusion, months | |
Mean (SD) | 20.1 (10.37) |
Median | 19.3 |
Minimum to maximum | 0.8 to 48.1 |
Follow-up duration after exa-cel infusion by interval, n (%) | |
≤ 3 months | 1 (2.3) |
> 3 months to ≤ 6 months | 2 (4.5) |
> 6 months to ≤ 12 months | 8 (18.2) |
> 12 months to ≤ 24 months | 16 (36.4) |
Exa-cel = exagamglogene autotemcel; SD = standard deviation.
Note: Follow-up duration after exa-cel infusion (months) = (Data cut-off date or end-of-study date whichever is earlier – exa-cel infusion date + 1)/30.
Sources: SCD Clinical Overview Addendum,17 sponsor’s Summary of Clinical Evidence.
Prior, Concomitant, and Subsequent Treatments
Prior medications were reported, but it is unclear whether the data were collected specifically for the 2-year period before patients were enrolled in the study. Of these, ██ █████ of 44 patients in the FAS received prior hydroxyurea (labelled as hydroxycarbamide). Data pertaining to red blood cell transfusions are available for the 2 years before screening and are reported in Table 10.
All patients used concomitant medications, which were defined in the study as a medication that is continued or newly received on or after exagamglogene autotemcel infusion until 24 months after infusion. The clinical experts confirmed that the most common concomitant medications were typical of those used for the management of patients during perimyeloablative and postmyeloablative conditioning and for sickle cell disease–related chronic pain. As such, a total of ██ ███████ patients received opioids.
Subsequent treatments received after study completion were not reported.
Efficacy
Results of the primary and key secondary end points are presented for the latest postaddendum analysis, with the data cut-off date of June 14, 2023. Results are presented in Table 12.
Vaso-Occlusive Crises
Of the 30 patients in the CLIMB-121 study who were followed for at least 16 months after exagamglogene autotemcel infusion and for at least 14 months after completion of posttransplant support (PES population), 29 patients (96.7%) achieved the primary outcome of the absence of any severe VOCs for at least 12 consecutive months. Testing against a prespecified 50% response rate showed statistical significance (P < 0.0001). The magnitude of the response from baseline was considered clinically meaningful by the clinical experts.
█████████ ██ ███ ████████ ███ ███████ ███ ███ ███ ███████ ███ ███████ ███████ ███████████ ██████ ████ ████ ███ ████████ ██ ████ █████████ ██ ███ █████████████ ███ ████ ████ ████████████ ███████████ ██ █████ ███ ███████ ████████ █ ███████ ██ ██████████ █████ █████ █████████ ███████ ████ ███████ ██ ██████ ████ ████████ █████████████ █████████ █████████ ███ ███████ █████████
Subgroup results show that all 6 patients (100.0%) aged 12 to younger than 18 years and 23 of 24 patients (95.8%) aged 18 to 35 years achieved the absence of any severe VOC for at least 12 consecutive months. A total of 16 of 17 patients (94.1%) who experienced at least 3 VOCs per year achieved the outcome. In the PES, 28 of 29 patients (96.6%) had the betaS/betaS genotype, so these subgroup data were not reported by the sponsor.
Hospitalizations
All patients in the PES population achieved the key secondary outcome of no inpatient hospitalizations for severe VOCs for at least 12 months. Testing against a prespecified 50% response rate showed statistical significance (P < 0.0001). The magnitude of the response from baseline was considered clinically meaningful by the clinical experts.
Red Blood Cell Transfusions
At baseline, patients had an annualized mean of 8.4 units (SD = 14.9 units) of red blood cells transfused; however, no patient received red blood cell transfusions for indications related to sickle cell disease after the 12-month period following the exagamglogene autotemcel infusion. This resulted in a 100% (SD = 0%) relative reduction from baseline in the mean number of annualized units of red blood cell transfusions. The magnitude of the response from baseline was considered clinically meaningful by the clinical experts.
Hematological Outcomes
All 30 patients (100.0%) in the PES had a sustained fetal hemoglobin level of at least 20% for at least 12 consecutive months starting 60 days after the last red blood cell transfusion.
The mean proportion of alleles with intended genetic modification in CD34+ cells of the bone marrow was █████ ███ █ █████ at month 6; results remained stable at month 12 ███████ ██ █ █████. After 24 months, results were still consistent, but based on fewer evaluable patients (██ ███ ██ ██ patients).
Table 12: Summary of Key Efficacy Results From the CLIMB-121 Study
Summary of key efficacy outcomes (PES) | Exa-cel (N = 30) |
|---|---|
Vaso-occlusive crises | |
Patients who achieved the absence of any severe VOC for at least 12 consecutive months | |
n | 29 |
%, 2-sided 95% CI | 96.7 (82.8 to 99.9) |
1-sided P value against a 50% response rate | < 0.0001 |
Hospitalizations | |
Patients who remained free from inpatient hospitalization for severe VOCs for at least 12 months | |
n | 30 |
%, 2-sided 95% CI | 100.0 (88.4 to 100.0) |
1-sided P value against a 50% response rate | < 0.0001 |
Red blood cell transfusions | |
Reduction in units of red blood cells transfused | |
Baseline number of annualized units of red blood cells transfused | |
n | 30 |
Mean (SD) | 8.4 (14.9) |
Median | 3.3 |
Minimum to maximum | 0.0 to 75.5 |
Number of annualized units of red blood cells transfused starting 12 months after exagamglogene autotemcel infusion | n = 30 |
Mean (SD) | 0.0 (0.0) |
Median | 0.0 |
Minimum to maximum | 0.0 to 0.0 |
Relative reduction from baseline in the number of annualized units of red blood cells transfused, % | n = 26 |
Mean (SD) | 100.0 (0.0) |
Median | 100.0 |
Minimum to maximum | 100.0 to 100.0 |
Hematological outcomes | |
Proportion of alleles with intended genetic modification in CD34+ cells of the bone marrow | |
Month 6 | |
n | ██ |
Mean (SD) | █████ ██████ |
Median | █████ |
Minimum to maximum | ██████ █████ |
Month 12 | |
n | ██ |
Mean (SD) | █████ ██████ |
Median | █████ |
Minimum to maximum | ██████ █████ |
Month 24 | |
n | ██ |
Mean (SD) | █████ ██████ |
Median | █████ |
Minimum to maximum | ██████ █████ |
Exa-cel = exagamglogene autotemcel; CI = confidence interval; PES = primary efficacy set; SD = standard deviation; VOC = vaso-occlusive crisis.
Sources: SCD Clinical Overview Addendum,17 sponsor’s Summary of Clinical Evidence.
Patient-Reported Outcomes
The mean changes from baseline on ASCQ-Me pain-episode and impact subscales are reported in Table 13.
According to the clinical experts consulted by CDA-AMC, the magnitude of the change from baseline through month 24 can be considered clinically meaningful for the emotional, pain, social-functioning, and stiffness impact subscales, as well as for the pain-episode frequency subscale.
Table 13: Summary of Patient-Reported Outcomes From the CLIMB-121 Study
Patients | ASCQ-Me Exa-cel (N = 24) | ||||||
|---|---|---|---|---|---|---|---|
Emotional impact | Pain impact | Social-functioning impact | Stiffness impact | Sleep impact | Pain-episode frequency | Pain-episode severity | |
Baseline | |||||||
n | 23 | 23 | 23 | 23 | 23 | 24 | 24 |
Mean (SD) | 51.9 (7.5) | 53.7 (8.8) | 50.2 (11.1) | 53.3 (8.4) | 47.6 (8.3) | 53.0 (6.2) | 52.6 (9.0) |
Change at month 6 | |||||||
n | 20 | 20 | 20 | 20 | 20 | 21 | 21 |
Mean (SD) | 8.6 (9.7) | 5.5 (8.8) | 11.2 (12.4) | 0.0 (11.5) | 4.2 (12.2) | −16.1 (9.1) | −0.6 (12.2) |
Change at month 12 | |||||||
n | 23 | 23 | 22 | 23 | 23 | 24 | 24 |
Mean (SD) | 9.4 (8.9) | 5.2 (8.6) | 13.7 (11.7) | 3.6 (10.5) | 4.4 (7.0) | −19.3 (8.1) | −3.6 (12.2) |
Change at month 18 | |||||||
n | 19 | 19 | 19 | 19 | 19 | 20 | 20 |
Mean (SD) | 9.7 (9.3) | 9.0 (9.2) | 14.0 (12.7) | 4.8 (8.3) | 2.9 (8.9) | −20.6 (8.8) | −1.9 (11.1) |
Change at month 24 | |||||||
n | 16 | 16 | 16 | 16 | 16 | 17 | 17 |
Mean (SD) | 10.3 (10.9) | 9.1 (10.5) | 16.4 (11.0) | 6.6 (10.5) | 4.7 (8.0) | −21.0 (7.7) | −3.3 (13.3) |
ASCQ-Me = Adult Sickle Cell Quality of Life Measurement Information System; Exa-cel = exagamglogene autotemcel; SD = standard deviation.
Note: The population includes 24 of 30 patients in the primary efficacy set who are aged 18 to 35 years.
Sources: Internal sponsor’s report,29 sponsor’s Summary of Clinical Evidence.
Harms
Results for the safety end points are from the postaddendum analysis, which had a data cut-off date of June 14, 2023, and are presented in Table 14. The sponsor indicated that the majority of AEs and SAEs occurred in the 6 months after exagamglogene autotemcel infusion, with most occurring in the first 3 months.
Adverse Events
In the CLIMB-121 study, all 44 patients in the safety analysis reported at least 1 AE. The most common AEs were nausea (70.5%), stomatitis (63.6%), vomiting (56.8%), febrile neutropenia (54.5%), abdominal pain (50.0%), headache (50.0%), and pruritus (50.0%). The time-adjusted AE rate for all AEs was █████ █████████████████████ during the period from exagamglogene autotemcel infusion to less than 6 months after infusion; █████ █████████████████████ from 6 months after infusion to less than 12 months after infusion; and █████ events/patient-months from 12 months after infusion to less than 18 months after infusion.
Serious Adverse Events
Overall, 45.5% of patients reported at least 1 SAE. The most common SAEs were cholelithiasis (9.1%), pneumonia (9.1%), abdominal pain (6.8%), constipation (6.8%), pyrexia (6.8%), and sickle cell anemia with crisis (6.8%), and there was a range of other events related to pain, which are outlined in Table 14. The time-adjusted SAE rate for all SAEs was █████ events/patient-months during the period from exagamglogene autotemcel infusion to less than 6 months after infusion; █████ events/patient-months from 6 months after infusion to less than 12 months after infusion; and █████ events/patient-months from 12 months after infusion to less than 18 months after infusion.
Mortality
One death was reported in the study; the cause was respiratory failure after COVID-19 infection. A potential contribution of busulfan lung injury was reported in this patient, as well as preexisting lung disease. The death was considered to be not related to exagamglogene autotemcel.
Notable Harms
No AEs of malignancies were reported by the sponsor as of the June 14, 2023, data cut-off date.
All 44 patients (100.0%) achieved neutrophil engraftment; the median time to neutrophil engraftment was 27.0 days (range, 15 to 40 days). A total of 43 (97.7%) patients achieved platelet engraftment; the median time to platelet engraftment was 35.0 days (range, 23 to 126 days). No association was reported between infection events and time to neutrophil engraftment, or between bleeding events and time to platelet engraftment.
Overall, 29 patients (65.9%) experienced at least 1 AE of infection, and 9 patients (20.5%) experienced at least 1 SAE of infection. A total of 24 (54.5%) patients reported febrile neutropenia. As for bleeding events, 21 patients (47.7%) experienced at least 1 AE, and 1 patient (2.3%) experienced at least 1 SAE. There were no clinically significant infusion-related reactions or anaphylaxis. There was 1 case (2.3%) of patient-reported veno-occlusive liver disease. No information was reported regarding hemophagocytic lymphohistiocytosis and engraftment syndrome.
Although planned by the sponsor, no detailed subgroup results were reported for any harms outcomes.
Table 14: Summary of Harms Results From the CLIMB-121 Study
Adverse events | Exa-cel (N = 44) |
|---|---|
Most common AEs (in ≥ 25% of patients), n (%) | |
Patients with any AEs | 44 (100.0) |
Nausea | 31 (70.5) |
Stomatitis | 28 (63.6) |
Vomiting | 25 (56.8) |
Febrile neutropenia | 24 (54.5) |
Abdominal pain | 22 (50.0) |
Headache | 22 (50.0) |
Pruritus | 22 (50.0) |
Decreased appetite | 21 (47.7) |
Decreased platelet count | 21 (47.7) |
Constipation | 20 (45.5) |
Pain in extremity | 20 (45.5) |
Arthralgia | 19 (43.2) |
Pyrexia | 18 (40.9) |
Diarrhea | 17 (38.6) |
Decreased neutrophil count | 17 (38.6) |
Anemia | 16 (36.4) |
Fatigue | 16 (36.4) |
Mucosal inflammation | 16 (36.4) |
Skin hyperpigmentation | 16 (36.4) |
Back pain | 15 (34.1) |
Hypokalemia | 15 (34.1) |
Neutropenia | 13 (29.5) |
Peripheral edema | 12 (27.3) |
Thrombocytopenia | 12 (27.3) |
Upper abdominal pain | 11 (25.0) |
Increased alanine aminotransferase | 11 (25.0) |
COVID-19 | 11 (25.0) |
Gastritis | 11 (25.0) |
Pain | 11 (25.0) |
Serious AEs (≥ 2 patients), n (%) | |
Patients with any SAEs | 20 (45.5) |
Cholelithiasis | 4 (9.1) |
Pneumonia | 4 (9.1) |
Abdominal pain | 3 (6.8) |
Constipation | 3 (6.8) |
Pyrexia | 3 (6.8) |
Sickle cell anemia with crisis | 3 (6.8) |
Upper abdominal pain | 2 (4.5) |
Noncardiac chest pain | 2 (4.5) |
Oropharyngeal pain | 2 (4.5) |
Pain | 2 (4.5) |
Sepsis | 2 (4.5) |
Patients who stopped treatment due to AEs, n (%) | |
Patients who stopped | 0 (0.0) |
Deaths, n (%) | |
Patients who died | 1 (2.4) |
Respiratory failure after COVID-19 | 1 (2.4) |
AEs of special interest, n (%) | |
Malignancy | 0 (0.0) |
Infections, AEs | 29 (65.9) |
Infections, SAEs | 9 (20.5) |
Febrile neutropenia | 24 (54.5) |
Bleeding, AEs | 21 (47.7) |
Bleeding, SAEs | 1 (2.3) |
Clinically significant infusion-related reactions | 0 (0.0) |
Anaphylaxis | 0 (0.0) |
Veno-occlusive liver disease | 1 (2.3) |
Hemophagocytic lymphohistiocytosis | NR |
Engraftment syndrome | NR |
AE = adverse event, Exa-cel = exagamglogene autotemcel; NR = not reported; SAE = serious adverse event.
Sources: SCD Clinical Overview Addendum,17 internal sponsor’s report,29 sponsor’s Summary of Clinical Evidence.
Critical Appraisal
Internal Validity
Study Design, Patient Population, and Interventions
Although well-designed RCTs allow for causal inferences to be drawn with greater certainty than any other study type, the CLIMB-121 study had a single-arm treatment design. This introduces a risk of bias because of the absence of a treatment comparison that includes proper randomization, allocation concealment, and blinding. The clinical experts acknowledged that there is a rationale behind the sponsor’s decision to perform a noncomparative trial, as there are several issues with potential comparators, such as HSCT or standard-of-care treatments (i.e., hydroxyurea and red blood cell transfusions), used for the first-line treatment of this rare disease. The experts indicated that exagamglogene autotemcel targets a different population than HSCT, which would remain the preferred treatment option in patients younger than 12 years who have a matched sibling donor eligible and willing to donate. They noted, however, that only approximately 10% of patients in their practices have a matched related donor, so HSCT is not being widely available or accessible.
As for standard-of-care treatments, it was assumed in the study that patients were already receiving appropriate supportive therapies. Considering the relatively constant natural trajectory of sickle cell disease, the clinical experts indicated that patients who do not respond to hydroxyurea and red blood cell transfusions, or who become resistant to these treatments, are unlikely to experience a sudden improvement in disease progression or related complications, supporting the use of the within-group change from baseline as a measure of efficacy. However, there is an important limitation with this assumption, in that there is uncertainty about the treatments patients were actually receiving during the 2-year period before being enrolled in the trial, which serves as the baseline from which to evaluate the efficacy of exagamglogene autotemcel. Despite additional data provided by the sponsor, the available evidence was insufficient to assess with certainty whether patients in the study actually received an appropriate treatment option, according to Canadian and international standards, without achieving a meaningful response, which could bias the results in favour of exagamglogene autotemcel.
As a result, the lack of a control group, in addition to the absence of information on treatments received during the 2-year baseline period, introduces uncertainty regarding the true effect of exagamglogene autotemcel compared to any comparator, which is why the certainty of evidence was graded as very low.
Outcome Measures
The primary outcome in the CLIMB-121 study was freedom from severe VOCs, which was considered the preferred clinical end point by the experts. The criteria used in the study for the VOC definition and assessment were considered, overall, generally adequate and representative of the way these are routinely assessed in clinical practice. However, events of stroke were not included in the definition, despite being considered a severe manifestation of sickle cell disease by the experts. This could potentially bias the results in favour of the drug, as individual events of stroke would not be captured under the trial’s VOC criteria. However, no severe neurologic events were reported as harms events.
According to the experts, the determination of VOCs is considered a challenge, as it can be difficult to differentiate between VOCs and other diagnoses, especially with regard to chronic pain events. Assessment may be considered partly subjective, and is based on a combination of patient-reported symptoms and clinical examination. The experts noted that patients who experienced VOCs related to chronic pain rather than acute pain could bias the results against the drug. However, the single-arm design of the CLIMB-121 study made it susceptible to assessment and reporting biases, because knowledge of the treatment received could influence an investigator’s assessment. Therefore, it is possible that this outcome could have been subject to detection bias that favoured exagamglogene autotemcel.
On-trial VOC events were adjudicated by an independent external end point adjudication committee. With a single-arm design, however, members of the end point adjudication committee were aware of which patients were receiving exagamglogene autotemcel. Considering the partly subjective nature of VOC assessment, the use of an independent external end point adjudication committee in this trial made the mitigation of bias related to outcome detection in favour of the drug less likely.
The primary and key secondary efficacy outcomes, which were defined as the proportion of patients free from severe VOCs and hospitalization, respectively, for at least 12 consecutive months, were assessed over a longer, 2-year time frame in the trial. The FDA statistical reviewer calculated that the flexible evaluation period would result in a 2-fold to 3-fold increase in the chance of observing a response when there is no treatment effect, compared to a fixed evaluation period (e.g., being event-free from month 7 to month 18).24 Therefore, this end point response window increases the likelihood that patients will achieve the outcome, potentially introducing bias in favour of exagamglogene autotemcel.
CDA-AMC reviewers noted that the protocol for the CLIMB-121 study went through several revisions to the design of the study (namely, it was upgraded from phase I and II study to a phase I, II, and III study) and, relatedly, to the primary outcome and key secondary outcomes, as well as the definitions, of these. The European Medicines Agency (EMA) also highlighted these repeated changes in their assessment of exagamglogene autotemcel. Although some of the revisions were identified as being necessary to address regulatory feedback, a key consideration is the timing of the amendments relative to study participant follow-up status and database analysis and, therefore, changes could be outcome (data) driven. According to the EMA European public assessment report,33 1 patient in the CLIMB-121 study had achieved the primary outcome at the time some of these key amendments were made. The EMA accepted that —although not ideal for internal validity — this likely had little effect on the overall validity or results of the study. Nonetheless, the number of important protocol and statistical analysis revisions for a study that does not have a true confirmatory phase III design adds to the very low certainty of the evidence.
Other efficacy outcomes included HRQoL, which was assessed using the appropriate tool. ASCQ-Me is a disease-specific instrument that is validated in patients with sickle cell disease.12 An MCID of 5 points has been used in the literature,13 although with very little rationale. Nevertheless, it was considered appropriate and consistent with clinical practice, according to the clinical experts. Assessment of these outcomes implies some level of subjectivity from the patients and investigators; as such, it is possible that knowledge of the treatment received may have favoured exagamglogene autotemcel.
Statistical Analysis
The CLIMB-121 study had sufficient power for the analysis of the primary and key secondary efficacy outcomes. However, the sample size is considered relatively small for drawing inferences with higher certainty. Moreover, the interim analysis provided results only for the PES (n = 30 evaluable patients of the 63 enrolled), which is potentially a select sample, as it represents only patients who have completed a set follow-up time in the study to date, as opposed to the full enrolled sample. Information on the outcomes based on the full treatment experience is, therefore, lacking.
No rationale was provided by the sponsor to justify the use of a 50% response rate as the null threshold for the primary testing hypothesis. The EMA and FDA24,33 also were concerned with the adequacy of this threshold for defining response. The sponsor provided references to the FDA to support the rationale for the threshold; however, the FDA’s assessment of the supportive literature and estimates for the null threshold indicated that there were important differences in end points and populations, among other considerations, that suggested the null threshold should have been higher than 50%, although an exact value was not suggested by the FDA. The EMA and FDA used the change from baseline and confidence intervals for the PES instead of the hypothesis test P value to determine treatment effects for their reviews. The clinical experts consulted by CDA-AMC indicated that, in patients with severe sickle cell disease, preventing even a few VOCs or hospitalizations per year would represent an important benefit for the patients they follow in their practices. Therefore, it was their opinion that a 50% response rate could be consistent with a meaningful improvement, given the expected later line of therapy for exagamglogene autotemcel and the severity of the disease, and considering the average annualized rate of severe VOC events of 3.9 (SD = 2.1 events) observed in the trial at baseline.
Analyses of the primary and key secondary efficacy end points were adjusted for multiplicity in the 3 interim analyses and the final analysis. The familywise type I error rate was controlled by an alpha spending approach for tests at the interim and final analyses and at sequential testing of the primary and key secondary efficacy end points. However, the first interim analysis was not conducted, and no rationale or justification was provided by the sponsor as to why this change was made to the prespecified statistical plan. As well, the alpha spend for each interim analysis appeared to be data driven, with alphas that varied by interim analysis so that there could be the possibility of both adjusting and not adjusting for type I errors, depending on the time point for the analysis. The statistical analysis for interim analysis 3 recycled the alpha from the previous unused alpha spend, but the appropriateness of this in terms of a planned approach to accounting for multiplicity is questionable and would not necessarily result in a sufficiently conservative threshold. Therefore, there are not only concerns about the validity of the hypothesis-testing approach, but also concerns about the overall study conduct, given the apparent lack of adherence to the analysis plan and protocol. In addition, the appropriateness of the methods used to maintain trial integrity with interim analyses could not be assessed.
The primary analysis was based on the last observation carried forward for missing data, which is unlikely to be an adequate strategy to handle intercurrent events and/or missing values for future analyses or estimating a causative estimand. Additional analyses were carried over after regulatory feedback, in which patients who died or discontinued the study because of exagamglogene autotemcel–related AEs before achieving the end point, or who continuously received red blood cell transfusions for posttransplant support of sickle cell disease beyond 10 months after exagamglogene autotemcel infusion, were considered nonresponders. Given that 29 of the 30 evaluable patients in the PES achieved the primary outcome, the overall impact on this analysis set is unlikely a major issue. However, a key gap in the evidence is the estimated effect on all enrolled patients who would likely go through the complete treatment, starting at stage 1. As mentioned, the PES (N = 30) is a subset of the FAS (N = 44), which is a subset of the enrolled population (N = 63). It is not clear if a final analysis will include all enrolled patients with appropriate approaches to handle intercurrent events. The PES may be more representative of real practice, but it represents a select population and may impact the treatment effect of exagamglogene autotemcel.
Subgroup analyses based on patient characteristics were specified a priori; however, no detailed subgroup results were reported for harms outcomes. Therefore, in addition to the relatively small sample sizes, it is difficult to assess the safety of exagamglogene autotemcel in these subpopulations.
External Validity
Patient Population
The inclusion and exclusion criteria were deemed to be clinically relevant and reasonable by the clinical experts. More important, baseline patient characteristics and disease histories were considered to be representative of patients with sickle cell disease seen in clinical practice who would be candidates for exagamglogene autotemcel. There is a wide variety of disease presentation among patients. For example, the clinical experts indicated that it is typical for patients to experience 2 to 10 VOCs per year; thus, the average annualized VOC rate of 4 in the study was deemed to be representative by the experts. According to the clinical experts, there is a rationale for patients who experience more VOCs to potentially get a greater benefit from exagamglogene autotemcel; although this treatment will not reverse existing damage, it may help prevent further damage that would have occurred at a high rate, as the number of annualized VOCs is unlikely to decrease over time.
However, an important limitation to generalizability is uncertainty about the use of standard-of-care therapies for sickle cell disease, other than red blood cell transfusions, in the 2 years before enrolment, which constitutes the baseline period for assessment of change over time. The sponsor reports that ██ █████ of 44 patients in the FAS population received prior hydroxyurea, but it is unclear if the data were collected specifically for the 2-year period before patients were enrolled in the study. Overall, the available evidence was insufficient to assess with certainty whether patients in the study had had an adequate trial of first-line treatments, as would be the case for patients who would be candidates for exagamglogene autotemcel in clinical practice (i.e., whether the treatments that patients received were appropriate in terms of dosage and duration, and whether they were consistent with current clinical practice in Canada). Indeed, the clinical experts noted that this drug would likely be a second-line or later-line therapy in patients with severe manifestations of sickle cell disease who did not have an optimal response or who became resistant to hydroxyurea or red blood cell transfusions; in patients who cannot access these therapies because of lack of coverage, the unavailability of a blood supply, or their distance from a tertiary centre; or in whom hydroxyurea or red blood cell transfusions are intolerable or contraindicated. Relevant information, such as start dates, adherence, or data on treatment optimization, would have been insightful but were not reported.
Treatment Regimen and Length of Follow-Up
The administration of exagamglogene autotemcel and busulfan myeloablative conditioning chemotherapy in the CLIMB-121 study was in line with the Health Canada–recommended dosages in this indication and with what is expected to be used in the reimbursement population.
The short follow-up of patients in the trial was highlighted as a major concern for both efficacy and safety assessments, especially in the context of a single-dose drug indicated for the treatment of a life-long disease. The mean follow-up after exagamglogene autotemcel infusion of 20.1 months (SD = 10.37 months) was not sufficient to provide information on the issues of the waning effect over time and of longer-term toxicities, such as the occurrence of malignancies.
Outcome Measures
The primary outcome of VOC prevention is consistent with the treatment goals of sickle cell disease in clinical practice, according to the clinical experts. Considering the established relations between VOCs and morbidity and/or mortality,2,10 avoidance of future VOCs is paramount in preventing complications, such as organ damage, in patients and in reducing mortality. The clinical experts highlighted that preventing even a few VOCs or hospitalizations per year would represent an important benefit for the patients they follow in their practice. Assessment of VOCs in the trial was considered to be performed in a manner that is similar enough to what is being done in clinical practice, according to the clinical experts, keeping in mind the challenges associated with ruling out differential diagnoses.
Among the secondary outcome measures, the use of ASCQ-Me was considered appropriate for assessing HRQoL, but the clinical experts noted that this assessment is not routinely performed in clinical practice. Improving HRQoL, chronic pain, and the ability to work were also presented as meaningful goals to achieve.
The patient groups that provided input for this review identified the outcomes assessed and reported in the CLIMB-121 study as being important, with a focus on preventing VOCs, reducing hospitalizations, maintaining HRQoL, and avoiding toxicities.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered most relevant to CDA-AMC expert committee deliberations, and a final certainty rating was determined, as outlined by the GRADE Working Group.15,16
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate. The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word likely for evidence of moderate certainty (e.g., X intervention likely results in Y outcome).
Low certainty: Our confidence in the effect estimate is limited. The true effect may be substantially different from the estimate of the effect. We use the word may for evidence of low certainty (e.g., X intervention may result in Y outcome).
Very low certainty: We have very little confidence in the effect estimate. The true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as very uncertain.
Although GRADE guidance is not available for noncomparative studies, the CDA-AMC review team assessed the pivotal single-arm trial for study limitations (which refer to internal validity or risk of bias), indirectness, and imprecision of effects to present these important considerations. Because the lack of a comparator arm does not allow for a conclusion to be drawn on the effect of the intervention relative to any comparator, the certainty of the evidence for the single-arm study was very low at the start, with no opportunity to be rated up.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null. The target of the certainty of evidence assessment was defined on the presence or absence of an important effect, based on thresholds identified in the literature, whenever possible, or informed by the clinical experts consulted for this review.
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for exagamglogene autotemcel.
Long-Term Extension Studies
The contents of this section have been informed by materials submitted by the sponsor. The following information has been summarized and validated by the CDA-AMC review team.
Description of Studies
At the time of this review, 1 long-term extension study is in progress. CLIMB-131 is an ongoing prospective, multisite, observational study designed to evaluate the long-term safety and efficacy of exagamglogene autotemcel in patients who received this treatment in the parent study, CLIMB-121. The CLIMB-131 study also enrolled patients with transfusion-dependent beta-thalassemia from the CLIMB-111 parent study. The planned follow-up of patients will be up to 15 years after exagamglogene autotemcel infusion (i.e., 2 years of follow-up in the parent study and up to 13 years of follow-up in the CLIMB-131 study).
The primary objective of the CLIMB-131 study is to evaluate the long-term safety of exagamglogene autotemcel. Because the CLIMB-121 study is ongoing, only a subset of patients with sickle cell disease has completed the parent study and enrolled in the CLIMB-131 study.
Populations
All patients who received an exagamglogene autotemcel infusion and completed or discontinued a parent study (CLIMB-121 or CLIMB-111) were asked to participate in the CLIMB-131 study. In addition, the CLIMB-131 study plans to include pediatric patients from the ongoing CLIMB-141 and CLIMB-151 parent studies (results are not yet available). There were no exclusion criteria beyond the eligibility described.
Interventions
Patients receive a one-time IV infusion of exagamglogene autotemcel in a parent study and do not receive any interventions in the long-term follow-up study.
Outcomes
There is no primary efficacy outcome in the CLIMB-131 study.
Safety evaluations included AEs, abbreviated physical examinations, clinical laboratory assessments, imaging assessments, and all-cause mortality. AEs, SAEs, AEs of special interest (malignancies), AEs of new or worsening hematologic disorders, and AEs of complications related to sickle cell disease were summarized according to the time periods and specifications.
Secondary efficacy outcomes for patients with sickle cell disease included the proportion of patients with the absence of any severe VOCs for at least 12 consecutive months, the proportion of patients with no inpatient hospitalizations for severe VOCs sustained for at least 12 months, and hematological outcomes. It is planned that patient-reported outcomes will be measured for up to 5 years in the CLIMB-131 study.
Statistical Analysis
Analysis sets for patients with sickle cell disease in the CLIMB-131 study were the same as for the CLIMB-121 parent study, and included the enrolled set, safety analysis set, FAS, and PES. The analysis sets included all patients from the parent study.
All efficacy analyses combined the data from the respective parent study and the extension study. The results of these combined analyses were considered to be the primary analyses because they represent the totality of the efficacy data from exagamglogene autotemcel infusion to the time of the analyses. Efficacy end points were analyzed based on the PES using pooled data from the CLIMB-121 and CLIMB-131 studies. As of the current data cut-off date of June 14, 2023, no patients had completed 5 or more years of follow-up after exagamglogene autotemcel infusion.
The percentage of patients who achieved the absence of any severe VOCs for at least 12 consecutive months was calculated relative to the number of patients in the PES. Only severe VOCs adjudicated by an end point adjudication committee as meeting the protocol definition of severe VOCs were included in the analysis. Efficacy end points were analyzed using the same statistical methods as in the CLIMB-121 study.
All safety analyses were conducted based on the respective safety analysis set by parent study (i.e., the CLIMB-121 study for patients with sickle cell disease). Only a descriptive analysis of safety was performed. For analysis purposes, AEs were summarized for defined periods (i.e., before exagamglogene autotemcel infusion, after exagamglogene autotemcel infusion up to no more than 2 years after infusion, from more than 2 years to 5 years after infusion, from more than 5 years to up to 15 years after infusion, and additional study intervals based on the parent study). Subgroup analyses by age, sex, region, race, and genotype were also performed for AE data.
Results
Patient Disposition
Baseline Characteristics
Demographic and other baseline characteristics for the 44 patients in the FAS and 30 patients in the PES were collected in the CLIMB-121 study and are presented in Table 10. As of the June 14, 2023, data cut-off date, 17 patients from the CLIMB-121 study had rolled over into the CLIMB-131 study; however, a summary of baseline demographics for this patient subset is currently unavailable.
Patient Disposition
As of the June 14, 2023, data cut-off date, 17 patients had completed the CLIMB-121 study and all 17 had enrolled in the CLIMB-131 study.
Exposure to Study Treatments
Study Treatments
The median follow-up duration after exagamglogene autotemcel infusion in the CLIMB-121 and CLIMB-131 studies was 19.3 months (range, 0.8 to 48.1 months), which corresponds to 73.5 patient-years (Table 15).
Table 15: Summary of Patient Exposure in the CLIMB-121 and CLIMB-131 Studies (FAS)
Exposure | Exa-cel (N = 44) |
|---|---|
Exposure after exa-cel infusion, patient-years | 73.5 |
Follow-up duration after exa-cel infusion, months | |
n | 44 |
Mean (SD) | 20.1 (10.37) |
Median | 19.3 |
Minimum to maximum | 0.8 to 48.1 |
Follow-up duration by interval after exa-cel infusion, n (%) | |
≤ 3 months | 1 (2.3) |
> 3 months to ≤ 6 months | 2 (4.5) |
> 6 months to ≤ 12 months | 8 (18.2) |
> 12 months to ≤ 24 months | 16 (36.4) |
> 24 months | 17 (38.6) |
Exa-cel = exagamglogene autotemcel; FAS = full analysis set; SD = standard deviation.
Sources: SCD Clinical Overview Addendum,17 Clinical Study Report for CLIMB-131.34
Concomitant Medications and Cointerventions
For patients whose first visit in the CLIMB-131 study did not correspond with their last visit in the parent study, concomitant medications and therapies received after the last visit in the parent study but before the first visit in the CLIMB-131 study were captured in the CLIMB-131 study; however, a summary of concomitant medications for this patient subset is currently unavailable.
Subsequent Treatment (If Applicable)
All patients in the CLIMB-131 study an received exagamglogene autotemcel infusion in a parent study; as such, no subsequent interventions were administered as part of the CLIMB-131 study.
Efficacy
Proportion of Patients Who Achieved the Absence of Any Severe VOCs for at Least 12 Consecutive Months
Patients Who Achieved the Primary Outcome in the CLIMB-121 Study
After infusion with exagamglogene autotemcel in the CLIMB-121 study, 29 of 30 patients (96.7%) in the PES achieved the absence of any severe VOCs for at least 12 consecutive months. No additional patients achieved that outcome in the CLIMB-131 study. The only patient who did not achieve the outcome was included in the 17 patients who rolled over to the CLIMB-131 study from the CLIMB-121 study. Of the 16 patients who achieved the outcome in the CLIMB-121 study and rolled over to the CLIMB-131 study, all (100.0%) remained VOC-free in the CLIMB-131 study.
As of the data cut-off date of 14 June 14, 2023, for the 29 patients who achieved the absence of any severe VOC for at least 12 consecutive months in the PES, the mean VOC-free duration was 22.4 months (SD = 7.2 months), including the follow-up in the CLIMB-131 study (range, 14.8 to 45.5 months). One patient who achieved the absence of any severe VOCs for at least 12 consecutive months had a single VOC during the CLIMB-121 study approximately 20.2 months after exagamglogene autotemcel infusion and has since had a VOC-free duration of approximately 12.3 months across the CLIMB-121 and CLIMB-131 studies.
Overall Evaluable Population (CLIMB-121 and CLIMB-131 Studies)
A total of 43 of 44 patients in the FAS population had at least 60 days of follow-up after the last red blood cell transfusion and were included in the June 14, 2023, postaddendum analysis; of these, 6 patients had adjudicated VOCs during the efficacy evaluation period.
Proportion of Patients Who Had No Inpatient Hospitalizations for Severe VOCs for at Least 12 Months
Patients Who Achieved the Outcome in the CLIMB-121 Study
After infusion with exagamglogene autotemcel, 100.0% of the 30 patients in the PES (CLIMB-121 study) had no inpatient hospitalizations for severe VOCs for at least 12 months. Of the 17 patients who rolled over from the CLIMB-121 study to the CLIMB-131 study (all of whom had achieved the outcome), all (100.0%) were free from hospitalizations for VOCs in the CLIMB-131 study.
Overall Evaluable Population (CLIMB-121 and CLIMB-131 Studies)
As of the data cut-off, 43 of 44 patients in the FAS population had at least 60 days of follow-up after the last red blood cell transfusion; of these, 40 patients were free from inpatient hospitalizations for VOCs for the duration of follow-up in the CLIMB-121 and CLIMB-131 studies, for a median of ████ ██████ ██████ ███ ██ 45.5 months).
Hematological Outcomes: Hemoglobin
Patients Who Achieved the Primary Outcome in the CLIMB-121 Study
In the CLIMB-121 study, all 30 patients (100.0%) in the PES had a sustained fetal hemoglobin level of at least 20% for at least 12 consecutive months starting 60 days after the last red blood cell transfusion, and a fetal hemoglobin level of at least 20% was maintained in all patients in the CLIMB-131 study as of the June 14, 2023, data cut-off date.
Overall Evaluable Population (CLIMB-121 and CLIMB-131 Studies)
In the FAS, increased mean hemoglobin levels and fetal hemoglobin levels were achieved by month 3 after exagamglogene autotemcel infusion, and were generally maintained over time from month 6 through to month 48. More specifically, the mean proportion of total hemoglobin comprised of fetal hemoglobin was 36.9% (SD = 9.0%) at month 3 and was maintained at, generally, at least 40% from month 6 over the duration of follow-up.
Patient-Reported Outcomes
Assessments conducted for patients with sickle cell disease included ASCQ-Me, 5-Level EQ-5D, EQ-5D Youth, Functional Assessment of Cancer Therapy-Bone Marrow Transplant (FACT-BMT), and the 11-point numerical rating scale. Due to a paucity of long-term data collected as of the June 14, 2023, data cut-off date, no results for patient-reported outcomes in the CLIMB-131 study were reported.
Harms
For safety evaluations in the CLIMB-131 study (i.e., beginning after the month 24 visit of the CLIMB-121 study), only AEs related or possibly related to exagamglogene autotemcel, SAEs, new malignancies, and new or worsening hematologic disorders were collected. Disorders related to sickle cell disease were recorded throughout the CLIMB-131 study. As of the June 14, 2023, data cut-off date, 17 of 44 patients (38.6%) had more than 24 months of follow-up, were include in the long-term extension, and had harms results reported in the June 14, 2023, postaddendum analysis. The patient with the longest follow-up after exagamglogene autotemcel infusion was followed for 48.1 months.
Of these 17 patients, no deaths occurred during the CLIMB-131 study. ███ ███████ ███ ██ ███ ██ ███████████████ █████████ █████ ███ █████ ██ ██████ █████ ███ ████████ ██ ███ ████████████ ██ █████████ ██ █████████████ ██████████ ███ ████████ ██████ █ █████. No new malignancies, new or worsening hematologic disorders, or complications related to sickle cell disease occurred during the CLIMB-131 study in patients from the CLIMB-121 study.
███████████ █████████ ███ ██████████ ██████ ████ ██████████ ████ ████ █████ ███ █████ ██ ██████ █████ ████ ██ ██████████ ████████ ██████ ██ █████ ██████████ ███████ █████ █████ ██ ████████ █████████████
Critical Appraisal
Internal Validity
The CLIMB-131 study was an open-label extension designed to evaluate the long-term efficacy and safety of exagamglogene autotemcel in patients with sickle cell disease. However, the same study limitations regarding the single-arm and open-label nature of the CLIMB-121 study also apply to the long-term extension. In addition, reporting of available data from the CLIMB-131 study has been poor and is limited because it is an interim analysis, which hampers the ability to draw definitive long-term conclusions until the follow-up is complete. Furthermore, the same limitations regarding the PES compares with the FAS apply to the long-term extension: results are only provided for the PES, which is potentially a select sample because it represents only patients who have completed a set follow-up time in the parent study to date, as opposed to the full enrolled sample. Although the efficacy of exagamglogene autotemcel appears to be maintained over the available follow-up duration, information on efficacy outcomes based on a larger population (FAS; n = 44) brings uncertainty regarding the magnitude of the treatment effect.
External Validity
The limitations regarding the external validity of the long-term extension identified in the CLIMB-121 study also apply to the CLIMB-131 study. It is reasonable to expect that the strengths and limitations related to generalizability also apply to the extension study. Additionally, HRQoL results for the CLIMB-131 period were not reported as of the data cut-off date, so long-term data on HRQoL are lacking. Another important limitation is the fact that no harms were reported after month 24 unless they were judged to be related to the study drug, which is an important limitation, as relation to the study drug can be a subjective measure, so complete harms reporting in the long-term extension is lacking.
Indirect Evidence
No indirect treatment comparisons were submitted by the sponsor.
Studies Addressing Gaps in the Systematic Review Evidence
No studies addressing gaps in the evidence were submitted by the sponsor.
Discussion
Summary of Available Evidence
One study was reviewed. CLIMB-121 (n = 63 patients enrolled and 30 patients analyzed) was a single-arm, phase III, ongoing multicenter study designed to evaluate the efficacy and safety of exagamglogene autotemcel, administered after single-drug myeloablative conditioning chemotherapy, for the treatment of sickle cell disease in patients aged 12 to 35 years with severe disease and recurrent VOCs (at least 2 protocol-defined severe VOC events per year for the 2 years before enrolment). The primary outcome was the proportion of patients who did not experience any severe VOCs for at least 12 consecutive months from 60 days after the last red blood cell transfusion to up to 2 years after exagamglogene autotemcel infusion. Based on demographics and disease characteristics, the study population was considered to be representative of patients with sickle cell disease seen in clinical practice who would be candidates for exagamglogene autotemcel; however, the sample size was small. In addition, the review team could not confirm whether patients in the study had received an adequate trial of first-line treatments before enrolment.
Interpretation of Results
Efficacy
The primary outcome in the CLIMB-121study pertained to the absence of severe VOCs for at least 12 consecutive months, which was considered the preferred clinical end point. Both the medical literature2,10 and experience from clinical practice suggest that patients who experience frequent VOCs are at higher risk of complications and mortality; therefore, VOC prevention is consistent with the treatment goals of sickle cell disease in clinical practice, according to the experts (i.e., to prolong life, prevent end-organ toxicity, and reduce symptom severity). However, assessment of VOCs is considered partly subjective and a challenge, according to the clinical experts, as it can be difficult to differentiate between VOCs and other diagnoses, especially with regard to chronic pain events.
In the CLIMB-121 study, 29 of 30 patients (96.7%; 95% confidence interval, 82.8% to 99.9%) who were followed for at least 16 months after exagamglogene autotemcel infusion achieved the primary outcome and did not experience any severe VOCs for at least 12 consecutive months in the PES. The lower limit of the confidence interval has higher than the prespecified sponsor-selected 50% null response rate. As mentioned, the appropriateness of this null threshold is unclear; nevertheless, the magnitude of the response was considered to be clinically meaningful by the clinical experts, who indicated that preventing even a few VOCs or hospitalizations per year would represent an important benefit for the patients they follow in their practices. In the 2 years preceding enrolment in the CLIMB-121 study, patients had a mean annualized rate of 3.9 severe VOC events (SD = 2.1 events) and, as the natural trajectory of sickle cell disease is relatively constant, the clinical experts indicated that patients would be unlikely to experience an unexplained sudden improvement in their disease manifestations.
However, substantial uncertainty surrounding those findings is related to several limitations in the evidence. Most limitations apply to all outcomes assessed in the study; these limitations and their impact on the interpretation of the findings are described here in detail. One limitation that is specific to the primary outcome is the definition and assessment of VOCs in the trial. Events of stroke were not included in the definition, despite being considered a severe manifestation of sickle cell disease by the experts, and, therefore, were not captured in the trial. It is unknown if any patients experienced an individual event of stroke that would have been counted as a VOC event, had it been included in the definition; however, no severe neurologic events were reported as harms events. It is also important to note that for patients to achieve the primary outcome, they could not experience severe VOCs for 12 consecutive months, but that could be any 12 consecutive months during a follow-up period that lasted up to 2 years after exagamglogene autotemcel infusion, which increases the likelihood that patients could achieve the outcome.
Patients with severe manifestations of sickle cell disease typically present with recurrent pain crises that are associated with high health care use. And, as the clinical experts and patient groups noted, reducing health care use is considered one of the main goals of therapy. The outcomes pertaining to hospitalization and red blood cell transfusion, which is a highly resource-intensive treatment, were therefore deemed to be particularly relevant, as they can have a substantial impact on the daily lives of patients and their caregivers. Results for the secondary outcome of the absence of hospitalizations for severe VOC for at least 12 consecutive months suggested the benefit of exagamglogene autotemcel, as all 30 patients in the analysis achieved the outcome. In the 2 years preceding enrolment, patients had a mean annualized rate of 2.7 hospitalizations (SD = 2.0 hospitalizations). In addition, no patient received a red blood cell transfusion for indications related to sickle cell disease during the 12-month period after the exagamglogene autotemcel infusion. The mean annualized units of red blood cells transfused was 8.4 (SD = 14.9 units) in the 2 years before enrolment. The magnitude of the response for both outcomes was considered to be clinically meaningful by the clinical experts. However, there is substantial uncertainty surrounding those findings, related to the limitations discussed in the subsequent Limitations section.
Hematological outcomes assessed in the CLIMB-121 study included the proportion of patients with a sustained fetal hemoglobin level of at least 20% for at least 12 consecutive months and the proportion of alleles with intended genetic modification present in CD34+ cells of the bone marrow. These surrogate outcomes of efficacy were, however, not considered to be clinically meaningful when making treatment decisions, according to the clinical experts. The findings are consistent with the mechanism of action of exagamglogene autotemcel, which increases fetal hemoglobin levels above the 20% threshold, which is considered in clinical practice to be the minimum level required for significant phenotypical modification. The results suggest that there was sufficient and stable allelic editing after exagamglogene autotemcel infusion to induce fetal hemoglobin levels above the 20% threshold in all 30 patients, significantly changing the phenotype.
The patient input and the clinical experts consulted highlighted the importance of improving HRQoL, which was assessed in the CLIMB-121 study using the disease-specific ASCQ-Me. According to the experts, the magnitude of the change from baseline through to month 24 observed with exagamglogene autotemcel can be considered clinically meaningful for the emotional-functioning, pain, social-functioning, and stiffness impact subscales, as well as for the pain-episode frequency subscale. The clinical experts emphasized that patients with severe manifestations of sickle cell disease often experience numerous comorbidities, such as mood disorders and anxiety; therefore, improvements in emotional functioning, social functioning, and pain are especially meaningful. Substantial uncertainty, however, surrounds those findings.
The efficacy of exagamglogene autotemcel appears to be maintained during the available follow-up in the long-term extension CLIMB-131 study. However, data on VOCs and hospitalizations based on the larger FAS population introduces uncertainty regarding the magnitude of the treatment effect with exagamglogene autotemcel because more patients experienced these events, although at a reduced frequency.
Limitations
Although results from the CLIMB-121 study were to be considered meaningful, several limitations affect our confidence in the findings and lead to a risk of bias for all outcomes assessed in the trial. First is the absence of a control group. Although the clinical experts acknowledged the rationale to perform a noncomparative trial by noting several important issues with potential comparators, such as HSCT, the single-arm design precludes any conclusion to be drawn regarding the true effect of exagamglogene autotemcel relative to any comparator. Second is the uncertainty regarding the treatments received during the 2 years before enrolment, which served as the baseline period. The available evidence is insufficient to assess whether patients had actually received an appropriate treatment option, in accordance with clinical practice standards, without achieving a meaningful response. Therefore, although findings from the CLIMB-121 study suggest that there is a reduction from baseline in the occurrence of severe VOCs after exagamglogene autotemcel infusion, what this baseline context truly represents in terms of treatments received and compared to is uncertain. The clinical experts noted, however, that the baseline disease severity in the CLIMB-121 study, based on VOC frequency, was consistent with that seen in clinical practice, and that not all patients receive appropriate standard-of-care therapies. Third is the assessment of subjective outcomes in the trial, such as VOCs and HRQoL. By having a single-arm design, the CLIMB-121 study was susceptible to biases because knowledge of the treatment received could influence an investigator’s assessment of this subjective outcome in favour of exagamglogene autotemcel. Finally, the review team noted that the sponsor made several changes to the planned study conduct once the trial was underway. This adds to the overall uncertainty; however, the impact on the results and on the risk of bias cannot be quantified.
In addition, there are important evidence gaps in the efficacy assessment of exagamglogene autotemcel that limits the interpretation of the findings. As of the latest interim analysis, the mean follow-up after exagamglogene autotemcel infusion was 20.1 months (SD = 10.37 months) in all patients who received the drug. The short follow-up of patients in the trial was highlighted as a major concern in the context of a single-dose drug indicated for the treatment of a life-long disease. Although there is no clear indication at this time from the CLIMB-121 study or the CLIMB-131 study that there will be a waning of efficacy, the question of response loss over time remains unanswered. The CLIMB-131 long-term extension study has the same limitation as its parent CLIMB-121 study, in addition to providing limited data because it is an interim analysis. An important limitation to generalizability is the lack of information to confirm whether patients in the study previously received an adequate trial of first-line treatments, as would be the case for patients who would be candidates for exagamglogene autotemcel in clinical practice. In addition, patients who had health care use that was consistent with chronic pain rather than acute pain crises were excluded from the study. According to the clinical experts, it is not necessarily expected that treatment with exagamglogene autotemcel will have a meaningful impact on outcomes such as chronic pain or on preexisting complications such as organ damage; nevertheless, patients experiencing these complications may benefit from a reduction in VOCs with exagamglogene autotemcel treatment and the prevention of further deterioration of their condition. Therefore, an evidence gap remains as to the magnitude of the response to exagamglogene autotemcel in these patients.
Harms
All patients who received exagamglogene autotemcel in the CLIMB-121 study experienced at least 1 AE. SAEs were also relatively common. Specific harms outcomes were not reported by time period, but rather for the entire study follow-up; however, the majority of AEs and SAEs occurred in the 6 months after exagamglogene autotemcel infusion. The clinical experts confirmed that the safety profile of exagamglogene autotemcel is generally consistent with that associated with myeloablative busulfan conditioning and the underlying disease. The clinical experts noted that myeloablative conditioning is a hard process for patients to go through, mainly because of tolerability issues, such as liver damage, lung disease, and infertility, which was highlighted as a serious concern for patients. The issue of loss of fertility with the exagamglogene autotemcel treatment process is discussed further in the CDA-AMC Exagamglogene Autotemcel Ethics Review Report.
Of the 63 enrolled patients, 16 (25%) discontinued the study before receiving exagamglogene autotemcel. Of the 58 patients who started the mobilization process, 6 patients (10%) discontinued due to inadequate cell collections. The clinical experts indicated that this is to be expected, as the manufacturing process of the drug — mainly the multiple mobilization cycles and stem cell collection — can be strenuous and burdensome for patients.
One death was reported during the study, which was due to respiratory failure after COVID-19 infection in a patient with preexisting lung disease and reported busulfan lung injury. All 44 patients achieved neutrophil engraftment, and 43 patients (97.7%) achieved platelet engraftment. The time to engraftment was considered relatively long by the clinical experts; infection and bleeding events were frequent, but were consistent with what would be expected in patients during the period of neutropenia and thrombocytopenia after myeloablative busulfan conditioning, and no association was reported between infection events and time to neutrophil engraftment, or between bleeding events and time to platelet engraftment. Other clinically important AEs of interest were infrequent and, according to the EMA European public assessment report, were consistent with those associated with HSCT; therefore, resource use was likely not greater with exagamglogene autotemcel than with standard therapy.33 The clinical experts indicated that, from the small number of patients and short follow-up duration, and in the very controlled setting of the clinical trial, the overall harms profile of the exagamglogene autotemcel treatment process in the CLIMB-121 study did not raise any particular safety signals.
There are, however, important evidence gaps in the safety assessment of exagamglogene autotemcel. The short follow-up duration in both the CLIMB-121 study and the long-term extension CLIMB-131 study was not sufficient to provide information on the issues of longer-term toxicities, such as the occurrence of malignancies, which was highlighted as a significant concern by the clinical experts. This is due to the increased baseline risk of leukemia in patients with sickle cell disease and to the increased risk of secondary malignancies associated with busulfan and the possibility of off-target gene editing (the process by which genes other than the intended target become altered).14 Although none of these notable harms were reported in the CLIMB-121 or CLIMB-131 studies, the follow-up time was likely insufficient to assess the risk properly; the clinical experts noted that these malignancies could reasonably be observed 5 to 10 years after infusion.
Other Considerations
Special consideration may be given to the fact that sickle cell disease can be considered a rare disease, but a number of patients have a significant unmet need. The clinical experts noted that patients with severe manifestations of sickle cell disease typically present with recurrent pain crises, ongoing organ damage, and high health care use, which can have a substantial impact on the daily lives of patients and their caregivers. The natural trajectory of the disease is generally poor, and patients are unlikely to improve spontaneously. The disease has a substantial negative impact on life expectancy, and the limited number of effective therapeutic options available require an ongoing commitment for continued benefit. Access can be very limited and present a challenge in some regions of the country; the clinical experts noted that coverage of first-line treatments, such as hydroxyurea and red blood cell transfusions, is not consistent among jurisdictions. The difficulty in obtaining blood products for a lifetime of chronic transfusions also has to be factored in, as the Canadian blood donation pool is not representative of most people living with sickle cell disease. When matching blood can be obtained, transfusions are a highly resource-intensive treatment associated with several risks and toxicities, and they are considered by some patients to be disruptive to life, at best, and to be an overwhelming burden by others. Second-line and subsequent-line therapies include HSCT, which is the preferred treatment for younger patients who have a matched sibling donor eligible and willing to donate. The clinical experts emphasized, however, that only approximately 10% of patients in their practices have a matched related donor, so HSCT is not considered widely available or accessible. Therefore, the lack of a donor is a significant barrier for patients who are left with very limited therapeutic options despite substantial morbidity.
According to the clinical experts, this unmet need may be met by the drug under review. They indicated that exagamglogene autotemcel is not for all patients with sickle cell disease; some patients respond well to standard first-line therapies, and these patients would not be candidates for this treatment. In clinical practice, exagamglogene autotemcel would likely be a second-line or later-line therapy in patients with severe manifestations of sickle cell disease for whom HSCT is not an option, and who do not have an optimal response or who become resistant to hydroxyurea or red blood cell transfusions; in patients who cannot access these therapies because of a lack of coverage, the unavailability of a blood supply, or their distance from a tertiary centre; or in whom hydroxyurea or red blood cell transfusions are intolerable or contraindicated. These patients were identified by the clinical experts as having the greatest unmet need.
In the context of limited health care resources, the clinical experts noted significant health care capacity issues at the time of this review. For the time being, patients with sickle cell disease would likely be prioritized to receive exagamglogene autotemcel over patients with other indications, as the disease has a substantial negative impact on both life expectancy and quality of life, in addition to being associated with a high health care resource use. Individual patient prioritization is expected to be done by transplant experts, upon referral by the hemoglobinopathy specialist, as they have the necessary expertise to assess and identify patients who are most likely to benefit from treatment and who have a sufficiently good health status to sustain the toxicities of myeloablative conditioning. Treatment with exagamglogene autotemcel requires an initial inpatient course, with hospital stays averaging 1 month. Patients should ideally be supported throughout hospitalization and follow-up by a multidisciplinary team, which would include a pain specialist and a psychologist or social worker. Upon discharge, the treating hemoglobinopathy specialist and the multidisciplinary team would then switch to outpatient care.
The clinical experts indicated that socioeconomic factors can often play an important role in the management of patients with sickle cell disease, and that nonclinical features can have a bearing in the selection of patients who receive exagamglogene autotemcel. These would include socioeconomic and geographic barriers, as the treatment process requires a lengthy absence from work and/or caregiving, and may require geographic relocation because it can only be administered in tertiary centres located mainly in urban or metropolitan settings, which can put an additional economic strain on the patient and family members. The psychological status of patients and members of their support network are also important to consider, and include issues such as identity, stigma, and expectations around treatment effects.
The clinical experts emphasized that patients are expected to be very involved in discussions about the risks, benefits, and practicalities of exagamglogene autotemcel. For patients who have severe phenotypes, high health care use, and no other effective therapeutic options, the balance between the known and unknown risks and benefits may be considered acceptable. The clinical experts indicated that they are willing to tolerate a higher level of uncertainty in this patient population because of the magnitude of the unmet need. However, the risks should be weighed against the expected benefits in discussions with patients so they can make an individualized and informed decision about treatment.
Conclusion
Evidence from the ongoing, single-arm CLIMB-121 study (n = 30 patients in the interim analysis) is very uncertain for the effect of exagamglogene autotemcel on clinical efficacy and harms outcomes in patients with severe sickle cell disease who have recurrent VOCs relative to any comparator, given the absence of comparative data. The findings from the trial are consistent with a clinically meaningful prevention of VOCs, hospitalizations, and red blood cell transfusions, based clinical expert input. As well, clinically meaningful improvements in HRQoL, based on reported MCIDs, were observed. These clinical outcomes were consistent with the treatment goals of sickle cell disease in clinical practice, according to the experts (i.e., to prolong life, prevent end-organ toxicity, and reduce symptom severity). However, there is substantial uncertainty surrounding the evidence. The most important limitations include the use of a single-arm study design and uncertainty regarding the treatments that patients were receiving at baseline, which preclude definite conclusions from being drawn about the comparative efficacy of exagamglogene autotemcel. The available results come from a follow-up period of up to 2 years after exagamglogene autotemcel infusion, which is a relatively short period from which to determine longer-term effectiveness. Limitations with regard to outcome assessment introduce a risk of bias in favour the drug. Therefore, despite the magnitude of the response observed with exagamglogene autotemcel in the CLIMB-121 study, concerns remain as to whether the results present the true effect of the drug. A high proportion of patients experienced harms events, which were generally consistent with what is associated with the underlying disease and the notoriously difficult myeloablative busulfan conditioning. The study could not provide information on the issues of longer-term toxicities, such as loss of fertility and malignancies associated with the disease itself and with busulfan chemotherapy, or potential off-target gene editing. The clinical experts indicated that the overall harms profile of the exagamglogene autotemcel treatment process did not raise any particular safety signals, but the number of patients was small and the follow-up duration was short in the very controlled setting of the clinical trial.
Special consideration may be given to the fact that sickle cell disease is rare. Patients with severe manifestations present with recurrent pain crises, ongoing organ damage, and high health care use, which have a substantial negative impact on life quality and expectancy. The natural trajectory of the disease is generally poor, and only a limited number of effective therapies are available. In clinical practice, exagamglogene autotemcel would likely be reserved as a later-line therapy in select patients with severe disease for whom HSCT is not an option. The clinical experts emphasized that they are willing to tolerate a higher level of uncertainty in this patient population because of the magnitude of the unmet need. However, the risks should be weighed against the expected benefits in discussions with patients so they can make an individualized and informed decision.
References
1.CASGEVY (exagamglogene autotemcel): Cell suspension in patient-specific vials, 4-13 × 106 cells/mL, for intravenous infusion [product monograph]. Toronto (ON): Vertex Pharmaceuticals (Canada) Incorporated; 2024.
2.CanHaem. Consensus statement on the Care of Patients with sickle Cell Disease in Canada. The Canadian Hemoglobinopathy Association; 2015: https://www.canhaem.org/wp-content/uploads/2018/05/Sickle-Cell-Consensus.pdf. Accessed 2024 Sep 27.
3.Frangoul H, Altshuler D, Cappellini MD, et al. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. N Engl J Med. 2021;384(3):252-260. PubMed
4.Kato GJ, Piel FB, Reid CD, et al. Sickle cell disease. Nat Rev Dis Primers. 2018;4:18010. PubMed
5.Ware RE, de Montalembert M, Tshilolo L, Abboud MR. Sickle cell disease. Lancet. 2017;390(10091):311-323. PubMed
6.Acute complications in children with sickle cell disease: Prevention and management. Ottawa (ON): Canadian Paediatric Scociety; 2024: https://cps.ca/en/documents/position/acute-complications-with-sickle-cell. Accessed 2024 Sep 27.
7.Brandow AM, Zappia KJ, Stucky CL. Sickle cell disease: a natural model of acute and chronic pain. Pain. 2017;158 Suppl 1(Suppl 1):S79-s84.
8.Kenney MO, Smith WR. Moving Toward a Multimodal Analgesic Regimen for Acute Sickle Cell Pain with Non-Opioid Analgesic Adjuncts: A Narrative Review. J Pain Res. 2022;Volume 15:879-894. PubMed
9.Osunkwo I, Manwani D, Kanter J. Current and novel therapies for the prevention of vaso-occlusive crisis in sickle cell disease. Ther Adv Hematol. 2020;11:2040620720955000. PubMed
10.Darbari DS, Sheehan VA, Ballas SK. The vaso-occlusive pain crisis in sickle cell disease: Definition, pathophysiology, and management. Eur J Haematol. 2020;105:237-246. PubMed
11.Pendergrast J, Ajayi TT, Kim E, Campitelli MA, Graves E. Sickle cell disease in Ontario, Canada: an epidemiologic profile based on health administrative data. CMAJ Open. 2023;11(4):E725-E733. PubMed
12.Keller SD, Yang M, Treadwell MJ, Werner EM, Hassell KL. Patient reports of health outcome for adults living with sickle cell disease: development and testing of the ASCQ-Me item banks. Health Qual Life Outcomes. 2014;12:125. PubMed
13.Esham KS, Rodday AM, Smith HP, et al. Assessment of health-related quality of life among adults hospitalized with sickle cell disease vaso-occlusive crisis. Blood Adv. 2020;4(1):19-27. PubMed
14.Canada’s Drug Agency. CRISPR Technologies for In Vivo and Ex Vivo Gene Editing. Ottawa (ON): CDA-AMC; 2024: https://www.cda-amc.ca/sites/default/files/pdf/htis/2024/EH0127_CRISPR_Final_Report_2.pdf. Accessed 2025 March11.
15.Balshem H, Helfand M, Schünemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-406. PubMed
16.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. PubMed
17.Exa-cel SCD Clinical Overview Addendum: Efficacy And Safety Update 14 June 2023 [internal sponsor’s report]. Toronto (ON): Vertex Pharmaceuticals, Inc.; 2023.
18.Sankaran VG, Orkin SH. The switch from fetal to adult hemoglobin. Cold Spring Harb Perspect Med. 2013;3(1):a011643-a011643. PubMed
19.Sundd P, Gladwin MT, Novelli EM. Pathophysiology of Sickle Cell Disease. Annu Rev Pathol. 2019;14:263-292. PubMed
20.Ngo DA, Aygun B, Akinsheye I, et al. Fetal haemoglobin levels and haematological characteristics of compound heterozygotes for haemoglobin S and deletional hereditary persistence of fetal haemoglobin. Br J Haematol. 2012;156(2):259-264. PubMed
21.Eltzschig HK, Eckle T. Ischemia and reperfusion—from mechanism to translation. Nat Med. 2011;17(11):1391-1401. PubMed
22.Ochocinski D, Dalal M, Black LV, et al. Life-Threatening Infectious Complications in Sickle Cell Disease: A Concise Narrative Review. Front Pediatr. 2020;8:38. PubMed
23.El-Haj N, Hoppe CC. Newborn Screening for SCD in the USA and Canada. Int J Neonatal Screen. 2018;4(4):36. PubMed
24.CASGEVY [sponsor supplied reference]. Silver Spring (MD): U.S. Food and Drug Administration; 2024: https://www.fda.gov/vaccines-blood-biologics/casgevy.
25.Medicines and Healthcare products Regulatory Agency. Decision Marketing authorisations granted in 2023. London (GB): Government of the United Kingdom; 2023: https://www.gov.uk/government/publications/marketing-authorisations-granted-in-2023. Accessed 2024 Sep 27.
26.Union Register of medicinal products for human use: Product information: Casgevy. Public Health - Union Register of medicinal products: European Commission; 2024: https://ec.europa.eu/health/documents/community-register/html/h1787.htm. Accessed 2024 Sep 27.
27.Vertex Pharmaceuticals Incorporated. Data on file. REF-15461 (v1.0) [sponsor supplied reference]. 2022.
28.Vertex Pharmaceuticals Incorporated. Data on file. REF-22852 (v3.0) [sponsor supplied reference]. 2023.
29.Data on file [internal sponsor’s report]. Toronto (ON): Vertex Pharmaceuticals, Inc.; 2024.
30.Frangoul H, Locatelli F, Sharma A, et al. Exagamglogene Autotemcel for Severe Sickle Cell Disease. N Engl J Med. 2024;Accepted for publication. PubMed
31.Interim Clinical Study Report: A Phase 1/2/3 Study to Evaluate the Safety and Efficacy of a Single Dose of Autologous CRISPR-Cas9 Modified CD34+ Human Hematopoietic Stem and Progenitor Cells (CTX001) in Subjects With Severe Sickle Cell Disease [internal sponsor’s report]. Toronto (ON): Vertex Pharmaceuticals, Inc.; 2023.
32.Clinicaltrials.gov. A Safety and Efficacy Study Evaluating CTX001 in Subjects With Severe Sickle Cell Disease (NCT03745287) [sponsor supplied reference]. 2018; https://clinicaltrials.gov/study/NCT03745287. Accessed November 6, 2023.
33.Committee for Medicinal Products for Human Use (CHMP). Assessment report: Casgevy. Procedure No. EMEA/H/C/005763/0000. Amsterdam (NL): European Medicines Agency; 2024: https://www.ema.europa.eu/en/documents/assessment-report/casgevy-epar-public-assessment-report_en.pdf. Accessed 2024 Sep 27.
34.Vertex Pharmaceuticals Incorporated. Interim Clinical Study Report: A Long-term Follow-up Study of Subjects With β-thalassemia or Sickle Cell Disease Treated with Autologous CRISPR-Cas9 Modified Hematopoietic Stem Cells (CTX001) [sponsor supplied reference]. 2023.
Pharmacoeconomic Review
Abbreviations
AE
adverse event
ATC
authorized treatment centre
BIA
budget impact analysis
CDA-AMC
Canada's Drug Agency
CIHI
Canadian Institute for Health Information
HR
hazard ratio
HRQoL
health-related quality of life
HSCT
hematopoietic stem cell transplant
ICER
incremental cost-effectiveness ratio
ICT
iron chelation therapy
LY
life-year
NICE
National Institute for Health and Care Excellence
OCCI
Ontario Case Costing Initiative
PES
primary efficacy set
QALY
quality-adjusted life-year
RBC
red blood cell
SCD
sickle cell disease
SOC
standard of care
VF12
reducing vaso-occlusive crises for at least 12 consecutive months
VOC
vaso-occlusive crisis
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Exagamglogene autotemcel (Casgevy), cell suspension for IV infusion |
Indication | For the treatment of patients 12 years of age and older with SCD with recurrent VOCs |
Health Canada approval status | NOC |
Health Canada review pathway | Priority review |
NOC date | September 23, 2024 |
Reimbursement request | As per indication |
Sponsor | Vertex Pharmaceuticals (Canada) Incorporated |
Submission history | Previously reviewed: Yes Indication: Transfusion-dependent beta-thalassemia Recommendation: Reimburse with conditions |
NOC = Notice of Compliance; SCD = sickle cell disease; VOC = vaso-occlusive crisis.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Markov model |
Target population | Patients aged 12 years and older with SCD with VOCs |
Treatments | Exagamglogene autotemcel |
Dose regimen | Single infusion of at least 3 × 106 CD34+ cells/kg |
Submitted price | Exagamglogene autotemcel, 4 × 106 cells/mL to 13 × 106 cells/mL: $2,800,000 per administration |
Submitted treatment cost | $2,800,000 per administration |
Comparator | SOC, composed of hydroxyurea, blood transfusions, and/or iron chelation therapy |
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | Lifetime (78 years) |
Key data source | Effectiveness of exagamglogene autotemcel informed by the CLIMB-121 study; effectiveness of SOC informed by data from the baseline period from the CLIMB-121 study |
Submitted results | ICER = $116,300 per QALY gained (incremental costs = $1,913,894; incremental QALYs = 16.46) |
Key limitations |
|
CDA-AMC reanalysis results |
|
CDA-AMC = Canada's Drug Agency; HSCT = hematopoietic stem cell transplant; ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year; SCD = sick cell disease; SOC = standard of care; VOC = vaso-occlusive crisis.
Conclusions
Evidence from the ongoing, single-arm CLIMB-121 study suggests that exagamglogene autotemcel may result in a clinically meaningful prevention of vaso-occlusive crises (VOCs), reductions in hospitalizations and red blood cell (RBC) transfusions, and improvements in health-related quality of life (HRQoL), compared to baseline, among patients with sick cell disease (SCD) aged 12 to 35 years with at least 2 severe VOCs per year. However, owing to the single-arm design of the study and uncertainty regarding the treatments received during the baseline period, the clinical efficacy of exagamglogene autotemcel compared to standard of care (SOC) is highly uncertain.
CDA-AMC was unable to address uncertainty related to comparative clinical data, including the magnitude and duration of benefit with exagamglogene autotemcel compared to SOC. Therefore, CDA-AMC was unable to provide a more reliable estimate of the cost-effectiveness of exagamglogene autotemcel.
Results of the sponsor’s base analysis suggest that exagamglogene autotemcel will prevent approximately 100 severe VOCs over a patient’s lifetime, reducing the number and duration of SCD-related complications and leading to $840,000 in cost savings from the avoidance of VOCs and other SCD-related complications. The sponsor anticipates that these savings will partially offset the acquisition cost of exagamglogene autotemcel ($2,800,000 per patient), resulting in an incremental cost-effectiveness ratio (ICER) of $116,300 (incremental cost of $1,913,894; incremental QALYs of 16.46; incremental life-years [LYs] of 13.33). If these results are realized in clinical practice, a 39% price reduction would be required for exagamglogene autotemcel to be considered cost-effective relative to SOC at a willingness-to-pay threshold of $50,000 per quality-adjusted life-year (QALY) gained. Given that 99% of the QALYs predicted by the sponsor’s model for exagamglogene autotemcel are based on extrapolation, and given the lack of robust long-term comparative data, the incremental gain in LYs and QALYs predicted by the sponsor’s model may be overestimated. Similarly, the cost savings that result from reduced VOCs and SCD-related complications may be overestimated, owing to the assumption that all events would be managed in an inpatient setting. If the difference in the magnitude of benefit between exagamglogene autotemcel and SOC is less than estimated by the sponsor, or if the costs of managing VOCs or SCD-related complications are lower than included in the sponsor’s model, a higher price reduction may be needed.
Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, clinician groups, and drug plans that participated in the CDA-AMC review process.
Patient input was received from 4 groups: the Sickle Cell Awareness Group of Ontario, the Sickle Cell Disease Association of Canada, NotJustYou, and the Global Action Network for Sickle Cell & Other Inherited Blood Disorders. Information was gathered through focus groups, one-on-one conversations, surveys, and a webinar. Patient input noted that SCD has a significant impact on quality of life because of its chronic nature and associated complications such as severe painful attacks, organ and/or bone damage, and fatigue. In addition to physical manifestations of the disease, patients described the associated emotional and financial burden of SCD. Patient group input indicated that there are only 2 treatment options available (i.e., RBC transfusion and hydroxyurea), neither of which are curative, and expressed a desire for additional treatments that could improve clinical outcomes with minimal adverse events (AEs). No respondents had experience with exagamglogene autotemcel.
Clinician group input was received from Cell Therapy Transplant Canada and the Canadian Hemoglobinopathy Association. Both groups noted that SCD is associated with a significant clinical burden, as it can lead to chronic anemia, severe acute debilitating pain (VOC), risk of serious infection, acute respiratory failure, chronic pain, ischemic and hemorrhagic stroke, liver disease, nephropathy, and neurovascular disease. Clinicians noted that allogenic hematopoietic stem cell transplant (HSCT), hydroxyurea, and blood transfusions are treatment options; however, many patients still experience a lack of effective therapy for their disease, which results in significant morbidity and early mortality. Clinician input noted that exagamglogene autotemcel may represent a promising first-line treatment for patients 12 years and older with severe SCD despite best supportive care, but note that in the absence of long-term data, it is unknown whether exagamglogene autotemcel is curative.
Drug plan input noted that public funding for hydroxyurea is widely available and that most jurisdictions fund at least 1 course of iron chelation therapy (ICT). Plans further noted that RBC transfusions are provided by Canadian Blood Services. The drug plans asked whether patients younger than 12 years or older than 35 years should be eligible to receive exagamglogene autotemcel. Drug plan feedback highlighted that exagamglogene autotemcel must be administered by trained personnel at an authorized treatment centre (ATC), and that there may be capacity constrains in the initial years after reimbursement is approved.
Several of these concerns were addressed in the sponsor’s model:
HRQoL was incorporated in the sponsor’s model, using data captured with the 5-Level EQ-5D questionnaire in the CLIMB-121 study.
Health care system capacity constraints associated with exagamglogene autotemcel administration were considered in the sponsor’s submitted budget impact analysis (BIA).
Acute and chronic complications were incorporated in the sponsor’s model, using data from an ICES database.
CDA-AMC was unable to address the following concerns raised from the input relevant to the economic review:
The uncertainty associated with the long-term efficacy of exagamglogene autotemcel could not be addressed due to a lack of long-term data.
Economic Review
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis to assess the cost-effectiveness of exagamglogene autotemcel for the treatment of patients aged 12 years and older with SCD and recurrent VOCs.1 In the model, the sponsor compared exagamglogene autotemcel to SOC, which consists of hydroxyurea, RBC transfusion, and ICT.1 The modelled population is in line with the Health Canada indication, and was based on patients enrolled in the CLIMB-121 study.1
Exagamglogene autotemcel is available as cell suspension for infusion (4 × 106 cells/mL to 13 × 106 cells/mL).2 The minimum recommended dose is 3 × 106 CD34+ cells/kg, infused as a single dose.2 Exagamglogene autotemcel must be administered between 48 hours and 7 days after the last dose of a myeloablative conditioning drug.2 The sponsor-submitted price for exagamglogene autotemcel is $2,800,000 per administration per patient, regardless of the number of vials required.1 The sponsor’s model estimated that the annual per-patient cost of SOC would be approximately $22,798 (assuming SOC comprises 68.2% hydroxyurea, 16% RBC transfusion, and 34.6% ICT).
The analysis was conducted from the perspective of the Canadian public health care payer. Cost and outcomes (QALYs, LYs) were estimated over a lifetime horizon (78 years; 1-month cycle length). Discounting (1.5% per annum) was applied for both costs and outcomes.
Model Structure
The sponsor submitted a Markov model with 4 health states: cured from SCD (patients who met the primary clinical outcome of the CLIMB-121 study: reducing VOCs for at least 12 consecutive months [VF12]), improved SCD (VF12 not achieved but VOCs reduced compared to baseline), severe SCD (no change in VOCs from baseline), and death (Figure 1).1 Each health state was associated with a risk of SCD-related complications; patients in the cured state were assumed to no longer be at risk of SCD-related complications, patients in the improved SCD state were assumed to have lower risk, and patients in the severe SCD state were assumed to have no change in risk. All patients entered the model in the severe SCD health state. Patients who received exagamglogene autotemcel were also assumed to receive SOC during the treatment phase (12 months) and to remain in the severe SCD state. After the treatment phase, patients who received exagamglogene autotemcel were assigned to a model health state based on efficacy from the CLIMB-121 study. The sponsor assumed that SOC alone has no clinical benefits and that patients receiving SOC maintain the same frequency of VOCs throughout the model horizon (i.e., remain in the severe health state for the remainder of their lifetime). The sponsor’s model included both acute and chronic complications, with the risk of each dependent on a patient’s health state. Chronic complications were considered to be independent and permanent once they developed (i.e., the risk of developing a chronic complication was independent of developing another complication, and each was assumed to last until death). Acute complications were assumed to occur at any time, last for 1 cycle per event, and not accumulate. An increased risk of infertility was incorporated for patients aged 13 to 52 years who received exagamglogene autotemcel. Death could occur from any health state.
Model Inputs
The baseline characteristics used to inform the model were based on the CLIMB-121 study,1 which enrolled patients 12 years and older with SCD and recurrent VOCs (defined as 2 or more VOCs per year that required medical attention in the 2 years preceding trial enrolment). The mean age of participants in the CLIMB-121 study was 22.1 years, and 46.7% of participants were female. In the CLIMB-121 study, of the 63 patients enrolled, 58 patients started mobilization. Based on the number of patients who discontinued the study (11 of the 58 patients who started mobilization), the sponsor assumed that treatment withdrawal was 19%. These patients were not included in the modelled cohort; however, pretreatment infusion costs were included for this group.
Clinical inputs for exagamglogene autotemcel were informed by data from the CLIMB-121 study. The sponsor assumed that 100% of patients who receive exagamglogene autotemcel would experience engraftment success, based on observations from the CLIMB-121 study. The efficacy of exagamglogene autotemcel was informed by data from the primary efficacy set (PES) of the CLIMB-121 study, which included 30 patients followed for at least 16 months after exagamglogene autotemcel infusion and for at least 14 months after the completion of posttransplant support. In the PES, 96.7% (29 of 30 patients in the PES) achieved the VF12 primary end point (absence of any severe VOCs for at least 12 consecutive months). In the model, the sponsor assumed that 96.7% of patients who received exagamglogene autotemcel would be cured, and thus experience no further VOCs or SCD-related complications. The remaining 3.3% of patients (i.e., those who did not reach VF12 in the CLIMB-121 study) were assumed by the sponsor to be nonresponders and to remain in the severe SCD health state.
General population mortality and SCD-related mortality was informed by Statistics Canada and a study by Bradt et al., respectively.3,4 The sponsor’s model included additional factors that could impact mortality, including cure status, exagamglogene autotemcel infusion, frequency of VOCs, and occurrence of complications and infusion-related events. Risk of death for patients in the cured SCD health state was estimated by applying a hazard ratio (HR) of 1.25 to age-specific and sex-specific general population mortality rates.5 The sponsor assumed that patients receiving exagamglogene autotemcel had no risk of infusion-related mortality, based on results from the CLIMB-121 study; however, patients who had an engraftment failure were assumed to have a 25% increased mortality risk after exagamglogene autotemcel engraftment failure. Patients with recurrent VOCs were assumed to have an increased risk of death, compared to patients without VOCs (HR, 1.56, informed by data from Shah et al., [2019]).6 The sponsor additionally assumed that the occurrence of complications (both acute and chronic) would impact mortality risk, with rates obtained from the literature.7-9
Rates of acute and chronic complications in the model were primarily informed by a retrospective cohort study that used ICES administrative database and assumptions. Acute complications included acute chest syndrome, acute renal failure, pulmonary embolism, gallstones, leg ulcers, and acute infection. Chronic complications included chronic kidney disease, pulmonary hypertension, avascular necrosis, heart failure, neurocognitive impairment, stroke, retinopathy, and liver complications. Rates of complications for patients in the severe SCD health state were informed by the subset of patients with SCD in the database who had recurrent VOCs and at least 2 VOCs per year in the follow-up period. Rates from the subset of patients with SCD in the database who had recurrent VOCs and fewer than 2 VOCs per year were applied to the improved SCD health state. Patients in the cured SCD response state were assumed to experience no further increased risk of complications related to SCD compared to the general population.1 The sponsor included the increased risk of infertility associated with myeloablative conditioning regimens administered before exagamglogene autotemcel. Infertility estimates associated with patients treated with SOC were assumed to reflect the prevalence of infertility in the general population, whereas the sponsor used infertility inputs from the National Institute for Health and Care Excellence (NICE) assessment of betibeglogene autotemcel in patients with transfusion-dependent beta-thalassemia to inform estimates for exagamglogene autotemcel.10-13
The model included grade 3 or higher treatment-related AEs for exagamglogene autotemcel (incorporated as a one-time event) and grade 3 or higher AEs for SOC (incorporated as recurring risk). Rates of AEs were obtained from the CLIMB-121 study for exagamglogene autotemcel and from the literature for SOC, based on the average rates of AEs in the placebo arms of trials of SCD treatment that included crizanlizumab, voxelotor, and L-glutamine.14-16 Disutility values were not included for AEs.
Health-state utility values in the model were informed by 5-Level EQ-5D health index scores from the CLIMB-121 trial for patients with uncomplicated SCD (average score for SCD in the absence of acute or chronic complications was 0.81) and for those with cured SCD (average score was 0.91).1 The uncomplicated SCD utility value was applied to patients in the severe SCD and improved SCD health states, with disutilities applied for those who experienced acute or chronic complications. Additional disutilities were incorporated for exagamglogene autotemcel (infusion, graft failure, and infertility), informed by published literature.12,13,17-27 A disutility of 0.18 was applied for each VOC event per month, based on the NICE assessment of crizanlizumab, in which a disutility for VOC was reported to be 0.46 per event for a duration of 12 days.
Costs included in the model consisted of treatment-acquisition costs, infusion-related costs, RBC transfusion and ICT costs, acute and chronic complication costs, disease-monitoring costs, AE costs, and terminal-care costs. Acquisition costs were based on the sponsor’s submitted price for exagamglogene autotemcel. SOC costs were informed by the Ontario or Saskatchewan drug formularies and previous reimbursement reviews published by CADTH.28-30 The number of RBC units and the frequency of transfusions were based on the assumptions used in the NICE assessment of crizanlizumab.17 Patients receiving exagamglogene autotemcel additionally incurred infusion-related costs, such as preinfusion costs, hospitalization and procedure costs, and postinfusion monitoring costs. The frequency and unit costs of these were informed by data from the CLIMB-121 study, clinical expert feedback received by the sponsor, and published literature. Routine disease-monitoring costs were included for patients with noncured SCD, such as lab tests and physician visits, where the frequency of each was informed by clinical expert feedback received by the sponsor, and costs were informed by the Ontario Schedule of Benefits for physician and laboratory services.31,32 The costs of complications were informed by the Ontario Case Costing Initiative (OCCI), the Canadian Institute for Health Information (CIHI) patient cost estimator, or the Ontario Schedule of Benefits for physician services.28,33,34 The cost of infertility was assumed to be associated with a one-time retrieval surgery and monthly storage costs. Last, a one-time cost of terminal care was informed by the average cost of palliative care from CIHI. All costs were reported in 2024 Canadian dollars.1
Summary of the Sponsor’s Economic Evaluation Results
All analyses were run probabilistically with 1,000 iterations. The deterministic and probabilistic results were similar. The probabilistic findings are presented here.
Base-Case Results
In the sponsor’s base case, exagamglogene autotemcel was associated with an estimated cost of $3,265,599 and 30.65 QALYs over the 78-year horizon, resulting in an ICER of $116,300 per QALY gained compared to SOC (Table 3). At a willingness-to-pay threshold of $50,000 per QALY gained, there was a 0% probability of exagamglogene autotemcel being cost-effective.
Results were driven by the acquisition cost of exagamglogene autotemcel ($2,800,000), the estimated gain in LYs (incremental LYs = 13.6) and QALYs (incremental QALYs = 16.46) associated with exagamglogene autotemcel, and the cost savings associated with reductions in VOCs and chronic complications (Table 9). The sponsors model predicted that patients who received exagamglogene autotemcel would experience approximately 100 fewer VOCs over their lifetime (Table 8), which was estimated to be associated with a cost savings of $624,569.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER vs. SOC ($/QALY) |
|---|---|---|---|---|---|
SOC | 1,351,615 | Reference | 14.19 | Reference | Reference |
Exagamglogene autotemcel | 3,265,599 | 1,913,984 | 30.65 | 16.46 | 116,300 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; SOC = standard of care; vs. = versus.
Source: Sponsor’s pharmacoeconomic submission.1
Sensitivity and Scenario Analyses Results
The sponsor conducted several scenario analyses that adopted alternative modelling assumptions (i.e., different discount rates and time horizons), as well as scenarios that adopted alternative assumptions related to the baseline number of VOCs, the baseline risk of complications, SCD-related mortality, and drug wastage. The ICERs for exagamglogene autotemcel versus SOC ranged from $56,787 to $197,093; the scenario assuming a 3% discount rate and the scenario examining fewer VOCs per year at baseline (2 VOCs per year, based on CLIMB-121 study eligibility criteria) had the largest impacts on the ICER.
The sponsor additionally conducted a scenario analysis from a societal perspective, which included indirect costs associated with patient productivity and caregiver burden. In this analysis, exagamglogene autotemcel was associated with an ICER of $59,485 per QALY gained (incremental costs = $1,034,406; incremental QALYs = 17.39) compared with SOC, which is lower than that in the sponsor’s base-case analysis that used a health care payer perspective.
CDA-AMC Appraisal of the Sponsor’s Economic Evaluation
CDA-AMC identified several key limitations of the sponsor’s analysis that have notable implications for the economic analysis:
The efficacy of exagamglogene autotemcel compared to SOC is highly uncertain. To inform efficacy in the pharmacoeconomic model (i.e., the proportion of patients who achieved VF12), the sponsor used data for exagamglogene autotemcel from the single-arm CLIMB-121 study. For SOC, the sponsor assumed that patients would experience no change in their baseline number of VOCs (3.9 VOCs per year) over their lifetime. As noted in the CDA-AMC clinical report, the absence of a comparator introduces uncertainty as to the true effect of exagamglogene autotemcel relative to any other treatment(s).
Clinical expert feedback received by CDA-AMC indicated that SOC comprises hydroxyurea, RBC transfusions, and ICT in Canada. However, as noted in the CDA-AMC clinical review, there was uncertainty regarding which treatments patients received during the baseline period and whether patients had received an adequate trial of first-line treatments. As such, it is unknown whether patients in the SOC group had received a treatment option in accordance with Canadian and international standards without achieving a meaningful response, which could bias the results in favour of exagamglogene autotemcel.
Given the absence of comparative clinical evidence and uncertainty as to whether patients in the CLIMB-121 study were receiving appropriate treatment during the baseline period, the magnitude of benefit associated with exagamglogene autotemcel versus SOC is highly uncertain. CDA-AMC was unable to address this limitation.
The cost-effectiveness of exagamglogene autotemcel versus allogeneic HSCT is unknown. HSCT is a potentially curative treatment for SCD; however, the sponsor excluded HSCT as a comparator from its analysis of the cost-effectiveness of exagamglogene autotemcel, assuming that only a small subset of patients would be eligible for HSCT, and that “most patients with willing matched sibling donors would likely have received HSCT at a young age, prior to reaching the age of eligibility for exagamglogene autotemcel.”1 Although CDA-AMC approved the sponsor’s request to exclude HSCT from its analysis, CDA-AMC notes that Canadian SCD guidelines indicate that HSCT may be considered for patients younger than 16 years.35 Clinical expert feedback received by CDA-AMC agreed with the sponsor that most eligible patients with willing matched sibling donors would likely have received an HSCT before they reached the age of eligibility for exagamglogene autotemcel (12 years); however, because of overlap in the age ranges in clinical practice guidelines, HSCT may be a treatment option for some patients in the indicated population. Clinical experts consulted by CDA-AMC noted that in clinical practice, exagamglogene autotemcel may be reserved for patients with severe disease for whom HSCT is not an option.
CDA-AMC was unable to address this limitation. The cost-effectiveness of exagamglogene autotemcel compared with allogeneic HSCT is unknown.
The long-term effectiveness of exagamglogene autotemcel is uncertain. In the pharmacoeconomic model, the sponsor assumed that patients who received exagamglogene autotemcel and were cured (VF12) would sustain this benefit for the duration of their lifetime. Approximately 99% of the incremental QALYs predicted by the sponsor’s model to be gained with exagamglogene autotemcel were derived on the basis of this assumption. However, evidence to support the duration and magnitude of benefit associated with exagamglogene autotemcel compared to SOC is unavailable. At the time of this review, the CLIMB-121 study is ongoing, with the most recent interim analysis based on a data cut-off date of June 14, 2023. CLIMB-131 is a long-term extension study designed to assess the long-term safety and efficacy in exagamglogene autotemcel in patients who received exagamglogene autotemcel in the CLIMB-121 study or the CLIMB-111 study, with up to 15 years of follow-up. As noted in the CDA-AMC clinical review, of the 16 patients who achieved VF12 in the CLIMB-121 study and enrolled in the CLIMB-131 study, all remain VOC-free in the CLIMB-131 study (a median of 16.8 months after exagamglogene autotemcel infusion; range, 0.6 to 45.5 months). However, in the absence of long-term follow-up data, there remains uncertainty as to the long-term efficacy and safety of exagamglogene autotemcel. Clinical expert feedback received by CDA-AMC for this review indicated that, in the absence of long-term data, the duration of benefit that patients receive from exagamglogene autotemcel is unknown.
CDA-AMC was unable to address this limitation. Additional data from a longer-duration follow-up will help reduce uncertainty about the duration of the effect, but will not reduce uncertainty about the magnitude of benefit relative to SOC due to the lack of a comparator group.
The survival benefit predicted for exagamglogene autotemcel is highly uncertain. In the sponsor’s base-case analysis, exagamglogene autotemcel was associated with an incremental gain of approximately 14 LYs compared to SOC. Clinical expert feedback received by CDA-AMC indicated that the frequency of VOCs and the occurrence of complications contribute to the risk of death in patients with SCD; however, mortality was not an outcome in the CLIMB-121 or CLIMB-131 studies, and there has been no comparison of survival between exagamglogene autotemcel and SOC. Thus, the magnitude of any survival benefit associated with exagamglogene autotemcel is uncertain.
The age-specific mortality rates for patients with SCD but no VOCs (cured SCD) and no SCD-related complications in the sponsor’s model were informed by annual mortality probabilities from an ICER report.4 Upon further investigation, CDA-AMC determined that these probabilities were derived using data from a retrospective study of SCD by Hassell et al.36 conducted in the US and based on data from 2008. CDA-AMC noted several limitations with these data, including the age of the data. Clinical expert feedback received by CDA-AMC indicated that, in Canada, there is a subset of younger patients with SCD whose disease has been well managed with hydroxyurea (i.e., SOC) from childhood, and that the expected survival of patients whose disease is well managed may approach 60 years.37 This is in contrast to older patients whose SCD was not well managed at a young age with historical SOC (i.e., before hydroxyurea was available). Should the life expectancy of patients receiving SOC in Canada be longer than that estimated by the sponsor’s model, the incremental LY and QALY difference between SOC and exagamglogene autotemcel may be smaller than predicted, which would increase the ICER.
CDA-AMC was unable to address this limitation. Although the clinical experts consulted by CDA-AMC agreed that a survival benefit for exagamglogene autotemcel is plausible (resulting from reductions in VOCs and SCD-related complications), the magnitude of this benefit is highly uncertain.
The cost of treating VOCs and SCD-related complications is uncertain. The sponsor’s analysis suggests that patients who receive exagamglogene autotemcel will have fewer acute and chronic complications, including approximately 100 fewer VOCs over their lifetime, resulting in cost savings of $840,606. In the sponsor’s base case, the cost of managing acute complications (including VOCs) and chronic complications associated with SCD were informed by values obtained from OCCI and CIHI.33,34 CDA-AMC noted that the use of OCCI and CIHI data to inform complication costs implies that all complications are treated in an inpatient setting. A recent study that assessed administration data for patients with SCD in Ontario showed that, over a 10-year study period (2007 to 2017), the mean number of emergency department visits was 6.69 (median, 2 visits; interquartile range, 1 to 7 visits) and the mean number of hospital admissions was 4.38 (median, 1 admission; interquartile range, 1 to 5 admissions) related to SCD.38 These data suggest that not all SCD-related events require admission to hospital. Clinical expert feedback received by CDA-AMC indicated that some events can be managed at home or in the outpatient setting, which is aligned with these data. The exclusive use of inpatient costs for managing SCD-related events in the model may overestimate costs, which would bias the results in favour of exagamglogene autotemcel.
CDA-AMC was unable to address this limitation. If the cost of managing VOCs or other SCD-related complications is lower than those incorporated in the sponsor’s model, the ICER for exagamglogene autotemcel compared to SOC may be higher.
Uncertainty in the efficacy of exagamglogene autotemcel was not modelled appropriately. The sponsor assumed that 96.7% of patients who receive exagamglogene autotemcel will experience a complete resolution of severe VOCs. This was based on the 29 of 30 patients in the PES who achieved VF12 in the CLIMB-121 study. The remaining patient (i.e., 1 of 30) was assumed by the sponsor to continue to have VOCs and to experience no improvement in frequency. This assumption is not aligned with data from CLIMB-121 study, in which 6 patients in the full analysis set (43 patients with at least 60 days of follow-up after their last RBC transfusion) continued to experience severe VOCs after receiving exagamglogene autotemcel, although at a lower frequency than during the baseline period. The sponsor additionally assumed 100% engraftment success after exagamglogene autotemcel administration, based on data from the CLIMB-121 study. It is unclear if engraftment success will be 100% in the clinical setting in Canada when more patients are infused. Thus, the sponsor’s model does not adequately reflect the uncertainty inherent in extrapolating clinical trial observations to whole populations.
CDA-AMC was unable to address these limitations, owing to the structure of the sponsor’s model.
Additionally, the key assumptions outlined in Table 4 were made by the sponsor and have been appraised by CDA-AMC.
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations in the Submission)
Sponsor’s key assumption | CDA-AMC comment |
|---|---|
The population enrolled in the CLIMB-121 trial was assumed to be representative of patients who will receive exa-cel in clinical practice. | Uncertain. CLIMB-121 enrolled patients aged 12 to 35 years with SCD and a history of 2 or more severe VOC episodes per year for the previous 2 years. CDA-AMC notes that the Health Canada indication for exa-cel does not specify the severity or frequency of VOCs. CDA-AMC approved a deviation request submitted by the sponsor to focus its pharmacoeconomic submission on the CLIMB-121 trial; however, CDA-AMC noted that patients in the CLIMB-121 trial experienced between 2 and 18 severe VOCs per year (annualized rate) in the full analysis set, and between 2 and 9.5 severe VOCs per year in the primary efficacy set (mean, 3.9 to 4.1 events per year). Although the clinical experts consulted by CDA-AMC deemed the CLIMB-121 population to be clinically relevant, a scenario analysis provided by the sponsor suggested that the ICER would be higher if the population that receives exa-cel in practice has fewer VOCs per year than the population enrolled in the CLIMB trial (2 VOCs per year = $137,192 per QALY gained [vs. $116,300 in the sponsor’s base case]). CDA-AMC additionally noted that, although the CLIMB-121 study included patients 12 years and older, the mean age of the modelled population was 22 years and, thus, may represent a group of patients with more severe disease than those eligible for exa-cel in clinical practice in Canada (starting at age 12). Clinical expert feedback received by CDA-AMC noted that older patients with SCD tend to have more severe disease because of the increased likelihood of organ damage and longer complication exposure. |
Health-state utility values for cured SCD and uncomplicated SCD were 0.91 and 0.81, respectively. Utility inputs were informed by EQ-5D-5L scores obtained from the CLIMB-121 study. | Uncertain. Utilities derived from the CLIMB-121 study suggest that patients who have SCD but no VOCs (cured SCD) will have higher HRQoL scores (0.91), and that patients who continue to have VOCs but no complications (uncomplicated SCD) will have utility values similar (0.810) to those in the general population (0.892 for people aged 20 to 24 years).39 The sponsor suggested that the higher utility values for cured patients may be due to an overall increased appreciation of life by patients. Clinical expert feedback received by CDA-AMC noted that, although the incremental difference between the health states may be reasonable, the numerical estimates may be overestimated. Because the increment between health states appears to be reasonable, the impact on the ICER is expected to be minimal. |
Patients receiving SOC experience no change in the number of VOCs over the modelled time horizon. | Uncertain. Clinical expert feedback received by CDA-AMC noted that patients with SCD likely experience little change in disease severity over time with SOC (if optimized); however, it is possible that patients with SCD may experience changes in their risk of VOCs and SCD-related complications. CDA-AMC was unable to explore this assumption, as the structure of the sponsor’s model does not allow patients to transition between health states. |
The cost of infertility was assumed to be associated with a one-time retrieval surgery and monthly storage costs informed by the Ottawa Fertility Clinic.40 | Uncertain. Clinical expert feedback received by CDA-AMC noted that many patients factor family planning into their treatment selection, and there are many factors to consider. CDA-AMC noted that the cost of retrieval surgery and monthly storage may vary by centre and that multiple rounds of retrieval surgery may be required. If the costs associated with fertility preservation are higher than those incorporated by the sponsor, the ICER associated with exagamglogene autotemcel may be higher than estimated. |
CDA-AMC = Canada's Drug Agency; EQ-5D-5L = 5-Level EQ-5D; exa-cel = exagamglogene autotemcel; HRQoL = health-related quality of life; ICER = incremental cost-effectiveness ratio ;QALY = quality-adjusted life-year; SCD = sickle cell disease, SOC = standard of care; VOC = vaso-occlusive crisis; vs. = versus.
CDA-AMC Reanalyses of the Economic Evaluation
Base-Case Results
CDA-AMC was unable to address the uncertainty related to the lack of robust comparative clinical data, including the magnitude and duration of benefit with exagamglogene autotemcel compared with SOC for the treatment of patients 12 years and older with SCD. CDA-AMC was additionally unable to resolve the uncertainty related to the predicted survival benefit associated with exagamglogene autotemcel, the modelled population, and inpatient complication costs. As such, CDA-AMC was unable to provide a more reliable estimate of the cost-effectiveness of exagamglogene autotemcel.
Results of the sponsor’s base case suggest that exagamglogene autotemcel will be more effective (an additional 16.46 QALYs) at an additional cost of $1,913,984 over a 78-year time horizon, resulting in an ICER of $116,300 compared to SOC. The sponsor’s base-case results further suggest that exagamglogene autotemcel would be associated with increased survival (incremental LYs of 13.33) and lower costs associated with blood transfusion, ICT, and complications (both acute and chronic). Based on the sponsor’s deterministic results, exagamglogene autotemcel is associated with reductions in the mean number VOCs over a person’s lifetime, the mean number of acute complications per person, and the mean number of years with a chronic complication per person.
Exploration of the sponsor’s model by CDA-AMC shows that approximately 99% of the predicated incremental gain in QALYs with exagamglogene autotemcel is expected to be accrued in the extrapolation period (i.e., after approximately 2 years). CDA-AMC noted that if the magnitude of benefit between exagamglogene autotemcel and SOC is less than estimated by the sponsor (i.e., exagamglogene autotemcel is associated a partial reduction in VOCs or complications), the ICER would be higher and further price reductions would be required to achieve cost-effectiveness.
Scenario Analysis Results
CDA-AMC conducted a price-reduction analysis on the sponsor’s base-case analysis (Table 9). Results of the price-reduction analysis suggest that exagamglogene autotemcel needs a price reduction of approximately 39% to be considered cost-effective relative to SOC at a willingness-to-pay threshold of $50,000 per QALY.
Table 5: CDA-AMC Price-Reduction Analyses
Price reduction | Unit drug cost ($) | ICERs for exagamglogene autotemcel vs. SOC ($/QALY), sponsor’s base case |
|---|---|---|
None | 2,800,000 | 116,300 |
10% | 2,520,000 | 99,286 |
20% | 2,240,000 | 82,273 |
30% | 1,960,000 | 65,259 |
40% | 1,680,000 | 50,000 |
50% | 1,400,000 | 48,245 |
60% | 1,120,000 | 31,231 |
70% | 840,000 | 14,218 |
80% | 560,000 | Dominant |
90% | 280,000 | Dominant |
CDA-AMC = Canada's Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; SOC = standard of care; vs. = versus.
Issues for Consideration
Exagamglogene autotemcel is currently undergoing review by CDA-AMC for the treatment of patients 12 years and older with transfusion-dependent beta-thalassemia.
Drug plan feedback and clinical expert input indicated that patients may face logistical issues trying to access specialized centres able to provide exagamglogene autotemcel. Financial support for travel and accommodation may be needed, particularly for patients who live in remote areas of the country.
Specialized centres will be needed to administer exagamglogene autotemcel, with training and accreditation provided by the sponsor. Obtaining and maintaining accreditation can result in a high resource burden, which includes the development of various protocols and supporting audits. In addition, this treatment has the added complexity of needing to coordinate patient care and product preparation with an external manufacturer. And because there will be patients receiving different treatments for different indications at specialized centres, there will be a need to manage various protocols for the preparation and delivery of each product type, which can increase the overall administrative burden.
Clinical expert feedback and patient input noted that the risk of infertility associated with myeloablative conditioning is of great concern to patients. Patients may opt to plan exagamglogene autotemcel treatment around their family planning, which may add an additional resource burden due to fertility preservation and emotional support.
Clinical expert feedback noted that life-long follow-up will be needed for patients who receive exagamglogene autotemcel, and emphasized the importance of alleviating the potential barriers associated with follow-up, including providing virtual care and community-based blood draws.
Overall Conclusions
Evidence from the ongoing single-arm CLIMB-121 study suggests that exagamglogene autotemcel may result in a clinically meaningful prevention of VOCs, reductions in hospitalizations and RBC transfusions, and improvements in HRQoL, compared to baseline, among patients aged 12 and 35 years with SCD and least 2 severe VOCs per year. However, owing to the single-arm study design and uncertainty regarding the treatments received during the baseline period, the clinical efficacy of exagamglogene autotemcel compared to SOC is highly uncertain.
CDA-AMC was unable to address the uncertainty related to comparative clinical data, including differences in the magnitude and duration of benefit between exagamglogene autotemcel and SOC. Therefore, CDA-AMC was unable to provide a more reliable estimate of the cost-effectiveness of exagamglogene autotemcel.
Results of the sponsor’s base analysis suggest that exagamglogene autotemcel will prevent approximately 100 severe VOCs over a patient’s lifetime, reducing the number and duration of SCD-related complications and leading to $840,000 in cost savings from the avoidance of VOCs and other SCD-related complications. The sponsor anticipates that these savings will partially offset the acquisition cost of exagamglogene autotemcel ($2,800,000 per patient), resulting in an ICER of $116,300 (incremental cost of $1,913,894 and incremental QALYs of 16.46). If these results are realized in clinical practice, a 39% price reduction would be required for exagamglogene autotemcel to be considered cost-effective relative to SOC at a willingness-to-pay threshold of $50,000 per QALY. Given that 99% of the QALYs predicted by the sponsor’s model for exagamglogene autotemcel are based on extrapolation, and given that there is a lack of robust long-term comparative data, the incremental QALYs predicted by the sponsor may be overestimated. Similarly, the cost savings related to reduced VOCs and SCD-related complications may be overestimated, owing to the assumption that all events would be managed in an inpatient setting. If the magnitude of benefit between exagamglogene autotemcel and SOC is less than that estimated by the sponsor, or if the costs of managing VOCs or SCD-related complications are lower than those included in the sponsor’s model, a higher price reduction may be needed.
References
1.Cost-effectiveness analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Exagamglogene Autotemcel (exa-cel). Toronto (ON): Vertex Pharmaceuticals (Canada) Incorporated; 2024 May 27.
2.Exa-cel Product Monograph (DRAFT) [sponsor supplied reference]. Toronto (ON): Vertex Pharmaceuticals (Canada), Inc; 2024.
3.Statistics Canada. Life Tables, Canada, Provinces and Territories 1980/1982 to 2020/2022 (three-year estimates), and 1980 to 2022 (single-year estimates) [sponsor supplied reference]. Ottawa (ON): Government of Canada; 2023: https://www150.statcan.gc.ca/n1/pub/84-537-x/84-537-x2023002-eng.htm.
4.Bradt P, Synnot PG, Chapman R, Beinfeld M, Rind DM, Pearson SD. Crizanlizumab, Voxelotor, and L-Glutamine for Sickle Cell Disease: Effectiveness and Value [sponsor supplied reference]. 2020.
5.Voskaridou E, Christoulas D, Bilalis A, et al. The effect of prolonged administration of hydroxyurea on morbidity and mortality in adult patients with sickle cell syndromes: results of a 17-year, single-center trial (LaSHS). Blood. 2010;115(12):2354-2363. PubMed
6.Shah N, Bhor M, Xie L, et al. Evaluation of Vaso-occlusive Crises in United States Sickle Cell Disease Patients: A Retrospective Claims-based Study. J Health Econ Outcomes Res. 2019;6(3):106-117. PubMed
7.Brunson A, Lei A, Rosenberg AS, White RH, Keegan T, Wun T. Increased incidence of VTE in sickle cell disease patients: risk factors, recurrence and impact on mortality. Br J Haematol. 2017;178(2):319-326. PubMed
8.Yeruva SL, Paul Y, Oneal P, Nouraie M. Renal Failure in Sickle Cell Disease: Prevalence, Predictors of Disease, Mortality and Effect on Length of Hospital Stay. Hemoglobin. 2016;40(5):295-299. PubMed
9.Elmariah H, Garrett ME, De Castro LM, et al. Factors associated with survival in a contemporary adult sickle cell disease cohort. Am J Hematol. 2014;89(5):530-535. PubMed
10.Datta J, Palmer MJ, Tanton C, et al. Prevalence of infertility and help seeking among 15 000 women and men. Hum Reprod. 2016;31(9):2108-2118. PubMed
11.Single Technology Appraisal: Betibeglogene autotemcel for treating transfusion-dependent beta-thalassaemia London (GB): National Institute for Health and Care Excellence; 2021: https://www.nice.org.uk/guidance/discontinued/gid-ta10334. Accessed 2024 Jul 12.
12.Costanian C, McCague H, Tamim H. Age at natural menopause and its associated factors in Canada: cross-sectional analyses from the Canadian Longitudinal Study on Aging. Menopause. 2018;25(3):265-272. PubMed
13.Al-Sahab B, Ardern CI, Hamadeh MJ, Tamim H. Age at menarche in Canada: results from the National Longitudinal Survey of Children & Youth. BMC Public Health. 2010;10:736. PubMed
14.Ataga KI, Kutlar A, Kanter J, et al. Crizanlizumab for the Prevention of Pain Crises in Sickle Cell Disease. N Engl J Med. 2017;376(5):429-439. PubMed
15.Vichinsky E, Hoppe CC, Ataga KI, et al. A Phase 3 Randomized Trial of Voxelotor in Sickle Cell Disease. N Engl J Med. 2019;381(6):509-519. PubMed
16.F. D. A. Oncologic Drugs Advisory Committee. Oral L-Glutamine Powder For The Treatment of Sickle Cell Disease: Sponsor Briefing Document [sponsor supplied reference]. Silver Spring (MD): U.S. Food and Drug Administration;2017.
17.Crizanlizumab for preventing sickle cell crises in sickle cell disease: Technology appraisal guidance [TA743]. London (GB): National Institute for Health and Care Excellence; 2021: https://www.nice.org.uk/guidance/ta743. Accessed 2024 Aug 2.
18.Matza LS, Paramore LC, Stewart KD, Karn H, Jobanputra M, Dietz AC. Health state utilities associated with treatment for transfusion-dependent β-thalassemia. Eur J Health Econ. 2020;21(3):397-407. PubMed
19.Stites SD, Harkins K, Rubright JD, Karlawish J. Relationships Between Cognitive Complaints and Quality of Life in Older Adults With Mild Cognitive Impairment, Mild Alzheimer Disease Dementia, and Normal Cognition. Alzheimer Dis Assoc Disord. 2018;32(4):276-283. PubMed
20.Cherry MG, Greenhalgh J, Osipenko L, et al. The clinical effectiveness and cost-effectiveness of primary stroke prevention in children with sickle cell disease: a systematic review and economic evaluation. Health Technol Assess. 2012;16(43):1-129. PubMed
21.Keogh AM, McNeil KD, Wlodarczyk J, Gabbay E, Williams TJ. Quality of life in pulmonary arterial hypertension: improvement and maintenance with bosentan. J Heart Lung Transplant. 2007;26(2):181-187. PubMed
22.Michaels JA, Campbell B, King B, Palfreyman SJ, Shackley P, Stevenson M. Randomized controlled trial and cost-effectiveness analysis of silver-donating antimicrobial dressings for venous leg ulcers (VULCAN trial). Br J Surg. 2009;96(10):1147-1156. PubMed
23.National Institute for H, Care E. Gallstone disease: diagnosis and management. Clinical guideline [CG188]. 2014.
24.Drabinski A, Williams G, Formica C. PID7: Observational evaluation of health state utilities among a cohort of sepsis patients Value Health. 2001;4(2):130.
25.Ojelabi AO, Bamgboye AE, Ling J. Preference-based measure of health-related quality of life and its determinants in sickle cell disease in Nigeria. PLoS One. 2019;14(11):e0223043. PubMed
26.Jiao B, Basu A, Ramsey S, et al. Health State Utilities for Sickle Cell Disease: A Catalog Prepared From a Systematic Review. Value Health. 2022;25(2):276-287. PubMed
27.Lloyd A, Price D, Brown R. The impact of asthma exacerbations on health-related quality of life in moderate to severe asthma patients in the UK. Prim Care Respir J. 2007;16(1):22-27. PubMed
28.Ontario Ministry of Health and Long-Term Care. Ontario drug benefit formulary/comparative drug index. 2024; https://www.formulary.health.gov.on.ca/formulary/. Accessed July 24, 2024.
29.Saskatchewan Drug Plan: search formulary. 2024; https://formulary.drugplan.ehealthsask.ca/SearchFormulary. Accessed Jul 24, 2024.
30.CADTH Reimbursement Review: Brexucabtagene Autoleucel (Tecartus). Can J Health Technol. 2021;1(11).
31.Schedule of benefits for physician services under the Health Insurance Act: (June 29, 2023 (effective July 24, 2023)). Toronto (ON): Ontario Ministry of Health; 2023: https://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master.pdf. Accessed 2024 Jul 24.
32.Schedule of benefits for laboratory services. (Effective July 24, 2023). Toronto (ON): Ontario Ministry of Health; 2023: https://www.health.gov.on.ca/en/pro/programs/ohip/sob/lab/sob_lab_2023.pdf. Accessed 2024 Jul 24.
33.Ontario Case Costing Initiative (OCCI). Toronto (ON): Ontario Health and Long-Term Care; 2017: https://data.ontario.ca/dataset/ontario-case-costing-initiative-occi. Accessed [CDA-AMC Jul 24, 2024; sponsor accessed 2023 May].
34.Patient cost estimator. Ottawa (ON): Canadian Institute for Health Information; 2024: https://www.cihi.ca/en/patient-cost-estimator. Accessed [CDA-AMC 2024 Jul 24; sponsor accessed May 2023].
35.Consensus Statement on the Care of Patients with Sickle Cell Disease in Canada. Version 2.0 [sponsor supplied reference]. Ottawa (ON): Canadian Haemoglobinopathy Association; 2015.
36.Hassell KL. Population Estimates of Sickle Cell Disease in the U.S. Am J Prev Med. 2010;38(4, Supplement):S512-S521. PubMed
37.Wailoo K. Sickle Cell Disease — A History of Progress and Peril. N Engl J Med. 2017;376(9):805-807. PubMed
38.Pendergrast J, Ajayi LT, Kim E, Campitelli MA, Graves E. Sickle cell disease in Ontario, Canada: an epidemiologic profile based on health administrative data. CMAJ Open. 2023;11(4):E725-e733. PubMed
39.Guertin JR, Feeny D, Tarride J-E. Age- and sex-specific Canadian utility norms, based on the 2013–2014 Canadian Community Health Survey. Can Med Assoc J. 2018;190(6):E155. PubMed
40.Ottawa Fertility Center Fees [sponsor supplied reference]. Ottawa Fertility Centre: https://conceive.ca/fees/.
41.Exceptional Access Program (EAP). Toronto (ON): Ontario Ministry of Health; Ontario Ministry of Long-Term Care; 2024: http://www.health.gov.on.ca/en/pro/programs/drugs/odbf/odbf_except_access.aspx. Accessed July 24, 2024.
42.Canadian Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Exagamglogene Autotemcel (exa-cel). Toronto (ON): Vertex Pharmaceuticals (Canada) Incorporated; 2024 May 27.
43.Epidemiology and Clinical Characteristics of SCD and B-Thal in Canada, Data on File [sponsor supplied reference]. Toronto (ON): Vertex Pharmaceuticals (Canada), Inc.; 2022.
44.Shah NR, Bhor M, Latremouille-Viau D, et al. Vaso-occlusive crises and costs of sickle cell disease in patients with commercial, Medicaid, and Medicare insurance - the perspective of private and public payers. J Med Econ. 2020;23(11):1345-1355. PubMed
45.Desai RJ, Mahesri M, Globe D, et al. Clinical outcomes and healthcare utilization in patients with sickle cell disease: a nationwide cohort study of Medicaid beneficiaries. Ann Hematol. 2020;99(11):2497-2505. PubMed
46.Statistics Canada. Population estimates, quarterly (Table 17-10-0009-01) [sponsor supplied reference]. Ottawa (ON): Government of Canada; 2024: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1710000901. Accessed March 27, 2024.
Appendix 1: Cost Comparison Table
Please note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical expert(s). Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 6: CDA-AMC Cost Comparison for Sickle Cell Anemia
Treatment | Strength / concentration | Form | Price | Recommended dosage | Daily cost ($) | Annual cost ($)a |
|---|---|---|---|---|---|---|
Exagamglogene autotemcel | 4 to 13 × 106 cells/mL | Cell suspension for IV infusion | 2,800,000b | Minimum recommended dose of 3 × 106 CD34+ cells/kg as a single dose for infusion | NA | 2,800,000 lifetime cost |
Antineoplastic drug | ||||||
Hydroxyurea | 500 mg | Cap | 1.0203 | 20 to 30 mg/kg once daily | 4.08 to 5.10 | 1,490 to 1,862 |
Iron chelation therapy | ||||||
Deferasirox | 90 mg 180 mg 360 mg 125 mg 250 mg 500 mg | Tablet | 2.6303 5.2610 10.5228 5.2408 10.4820 20.9649 | 7 to 14 mg/kg daily | 15.78 to 31.57 | 5,761 to 13,391 |
Deferiprone | 1,000 mg 100 mg/mL | Tablet Oral solution | 32.7452c 3.2766c | 25 to 33 mg/kg 3 times daily | 196.47 to 294.71 | 71,712 to 107,568 |
Deferoxamine mesylate | 95 mg/mL | 500 mg powder for SC injection 2 g powder for SC injection | 15.1700c 28.3500c | 20 to 60 mg/kg daily 4 to 7 times per week | 28.35 to 87.04 | 5,913 to 31,770 |
SC = subcutaneous.
Note: All prices are from the Ontario Drug Benefit Formulary or Ontario Exceptional Access Program Formulary (accessed August 2024) unless otherwise indicated, and do not include dispensing fees. Annual costs are based on 365 days per year.28,41
aRecommended dosages are from the respective product monographs. CDA-AMC assumed a patient weight of 76 kg.
bSponsor submitted price.1
cSaskatchewan drug benefit formulary (accessed August 2024).29
Appendix 2: Submission Quality
Please note that this appendix has not been copy-edited.
Description | Yes or No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | Refer to CDA-AMC critical appraisal. |
Model has been adequately programmed and has sufficient face validity | No | Refer to CDA-AMC critical appraisal. |
Model structure is adequate for decision problem | No | Refer to CDA-AMC critical appraisal. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | Yes | No comment. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | Yes | No comment. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | Yes | No comment. |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Please note that this appendix has not been copy-edited.
![Markov model with 4 health states: cured from SCD (patients who met the primary clinical outcome of the CLIMB-121 study: reducing VOCs for at least 12 consecutive months [VF12]), improved SCD (VF12 not achieved but VOCs reduced compared to baseline), severe SCD (no change in VOCs from baseline), and death. Each health state was associated with a risk of SCD-related complications; patients in the cured state were assumed to no longer be at risk of SCD-related complications, patients in the improved SCD state were assumed to have lower risk, and patients in the severe SCD state were assumed to have no change in risk. All patients entered the model in the severe SCD health state, and after the treatment phase, patients who received exagamglogene autotemcel were assigned to a model health state based on efficacy from the CLIMB-121 study.](https://canjhealthtechnol.ca/index.php/cjht/article/download/SG0830r/version/1113/2436/9565/SG0830-Pharmacoeconomic_Review-fig01.png)
SCD = sickle cell disease; SOC = standard of care; VOC = vaso-occlusive crisis.
Source: Sponsor’s pharmacoeconomic submission1
Detailed Results of the Sponsor’s Base Case
Table 8: Clinical Events Estimated by the Sponsor’s Economic Model
Parameter | Exagamglogene autotemcel | SOC |
|---|---|---|
Mean Number of Events Over a Lifetime, per Person | ||
VOC | 7.4 | 107.8 |
Acute chest syndrome | 0.08 | 1.11 |
Stroke | 0.02 | 0.31 |
Acute infection | 0.14 | 1.97 |
Acute kidney injury/failure | 0.16 | 2.41 |
Gallstones | 0.17 | 2.43 |
Pulmonary embolism | 0.13 | 1.89 |
Leg ulcers | 0.03 | 0.50 |
Mean Number of Years with Chronic Complication, per Person | ||
Chronic kidney disease | 1.63 | 3.40 |
Pulmonary hypertension | 0.61 | 3.83 |
Avascular necrosis | 17.35 | 17.34 |
Heart failure | 0.68 | 4.22 |
Neurocognitive impairment | 2.59 | 2.79 |
Poststroke | 0.24 | 1.43 |
Retinopathy | 4.27 | 6.31 |
Liver complications | 0.55 | 3.50 |
Patients Developing Chronic Complications by Age 99 (%) | ||
Chronic kidney disease | 3.5 | 18.8 |
Pulmonary hypertension | 1.7 | 22.7 |
Avascular necrosis | 34.2 | 77.5 |
Heart failure | 1.8 | 24.9 |
Neurocognitive impairment | 5.2 | 13.9 |
Post stroke | 0.6 | 7.8 |
Retinopathy | 8.7 | 32.7 |
Liver complications | 1.5 | 20.8 |
Exa-cel = exagamglogene autotemcel; SOC = standard of care; VOC = vaso-occlusive crisis.
Source: Sponsor’s pharmacoeconomic submission.1
Table 9: Disaggregated Summary of the Sponsor’s Economic Evaluation Probabilistic Results
Parameter | Exagamglogene autotemcel | SOC |
|---|---|---|
Discounted LYs | ||
Total LYs | 35.56 | 22.23 |
Discounted QALYs | ||
Total QALYs | 30.65 | 14.19 |
Cured / uncomplicated SCD | 32.21 | 18.02 |
Complication disutilitya | −1.57 | −3.84 |
Discounted costs ($) | ||
Total costs | 3,265,599 | 1,351,615 |
Treatment costs | 2,800,719 | 21,897 |
Mobilization, apheresis, conditioning, and pre-treatment lab costs | 54,286 | NA |
Additional infusion-related costs | 74,184 | NA |
Hospitalization costs for procedure | 67,606 | NA |
Postinfusion monitoring costs | 6,579 | NA |
Blood transfusion costs | 26,899 | 154,881 |
ICT costs | 1,385 | 18,228 |
Acute complication costs | 57,386 | 751,338 |
VOC costs | 51,974 | 680,488 |
Chronic complication costs | 236,190 | 382,844 |
Monitoring/lab costs | 917 | 11,953 |
AE management costs | 4,713 | 512 |
Infertility costs | 3,451 | 2,218 |
Terminal-care costs | 5,468 | 7,743 |
AE = adverse event, exa-cel = exagamglogene autotemcel; ICT = iron chelation therapy; LY = life-year; NA = not applicable; SCD = sickle cell disease; SOC = standard of care; QALY = quality-adjusted life-year; VOC = vaso-occlusive crisis.
aIncludes disutilites associated with VOCs, treatment-related adverse events, exagamglogene autotemcel infusion, and infertility (where appropriate).
Source: Sponsor’s pharmacoeconomic submission.1
Sensitivity and Scenario Analyses
The sponsor additionally conducted a distributional cost-effectiveness analysis. The methodology, and hence results, could not be validated by CDA-AMC.
Appendix 4: Additional Details on the CDA-AMC Reanalyses and Sensitivity Analyses of the Economic Evaluation
Please note that this appendix has not been copy-edited.
Given the identified limitations, CDA-AMC was unable to conduct any additional analyses to assess the relative cost-effectiveness of exagamglogene autotemcel for the treatment of patients with SCD with recurrent VOCs aged 12 years and older.
Appendix 5: Submitted BIA and CDA-AMC Appraisal
Please note that this appendix has not been copy-edited.
Table 10: Summary of Key Take-Aways
Key Take-aways of the budget impact analysis |
|---|
|
Summary of Sponsor’s BIA
The sponsor submitted a BIA to estimate the 3-year budget impact of reimbursing exagamglogene autotemcel for the treatment of patients aged 12 years and older with SCD with recurrent VOCs. The analysis was taken from the perspective of the Canadian public drug plan over a 3-year time horizon (2025 to 2027). The target population size was derived using an epidemiologic approach and included drug acquisition costs. The sponsor estimated province-specific prevalence rates using a linear regression model informed by published SCD prevalence estimates for Canada and the percentage of Black people in each jurisdiction during the study period.42 Age-specific prevalence estimates of SCD based on market research conducted by Bionest Partners were used to determine the number of eligible SCD patients.43 Further epidemiological criteria, such as proportion with more than 2 VOCs a year and proportion of patients fit for treatment, were derived from these studies.6,42-44 Key inputs to the BIA are documented in Table 12.
The sponsor compared a reference scenario in which patients received SOC (defined by the sponsor as a weighted average of hydroxyurea [alone or in combination with RBC transfusion and/or ICT], chronic RBC transfusions [alone or in combination], or no treatment) to a new drug scenario in which patients could receive exagamglogene autotemcel. Market share was informed by internal market estimates and clinical expert feedback obtained by the sponsor. Wastage and administration costs were not included.
Table 11: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3 if appropriate) |
|---|---|
Target Population | |
Number of patients with SCD (18+ years) Number of patients with SCD (12 to 17 years) Percentage of patients with SCD (18+) with ≥ 2 VOCs per year Percentage of patients with SCD (12 to 17) with ≥ 2 VOCs per year Percentage of patients (18+) fit for treatment Percentage of patients (12 to 17) fit for treatment Projected annual growth rate of SCD | 3,51242 87942 53%43 58%43 1.6%46 |
Number of patients eligible for drug under review | 1,086 / 1,103 / 1,121 |
Market Uptake (3 years) | |
Uptake (reference scenario) SOC | 100% / 100% / 100% |
Uptake (new drug scenario) Exagamglogene autotemcel SOC | 0%a / 0.5% / 1.9% 100% / 99.5% / 98.1% |
Cost of treatment (per patient, per year) | |
Exagamglogene autotemcel (one-time cost) SOC | $2,800,000b $8,002c |
SCD = sickle cell disease; SOC = standard of care; VOCs = vaso-occlusive crisis.
aSponsor assumed that no patients would receive exagamglogene autotemcel in year 1 due to the time needed to prep authorized treatment centres for exagamglogene autotemcel administration and that the process to receive exagamglogene autotemcel can take up to a year.
bApplied in the model as a one-time cost in the first year for when patients are treated.
cCalculated by the sponsor as a weighted average across all provinces as jurisdiction-specific treatment-acquisition costs were used in the pan-Canadian analysis. Annual SOC cost ranged from $7,991 to $8,667 depending on the jurisdiction.
Summary of the Sponsor’s BIA Results
The sponsor estimated the 3-year budget impact of reimbursing exagamglogene autotemcel for the treatment of patients aged 12 years and older with SCD with recurrent VOCs to be $59,337,641 (Year 1: $0; Year 2: $15,444,927; Year 3: $43,892,714).
CDA-AMC Appraisal of the Sponsor’s BIA
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
The number of patients eligible for exagamglogene autotemcel is uncertain. The number of eligible patients is uncertain for several reasons. First, the sponsor estimates that there are 4,391 patients in Canada (excluding Quebec) who have SCD, using a model informed by literature-derived prevalence, primary market research, and province-specific race- and ethnicity-based demographic data, which was then applied to province-specific demographic data to estimate the overall prevalence of SCD in CDA-AMC-participating jurisdictions.6,42,44,45 The results of these analyses were hard coded in the sponsor’s BIA model and could not be verified by CDA-AMC. Additionally, some of inputs used to derive the target population (i.e., the percentage of patients with SCD with ≥ 2 VOCs per year or proportion of patients fit to receive myeloablative treatment) was informed by proxy data from countries including the US and those in Europe. While CDA-AMC acknowledges that in the absence of Canadian data these estimates may be reasonable, differences in demographics between jurisdictions may increase uncertainty in the population estimate used in the BIA. Clinical expert opinion received by CDA-AMC additionally noted that the prevalence of SCD in Canada is increasing due to immigration from regions where the condition is more widely prevalent.
Second, the indication for exagamglogene autotemcel is for the treatment of patients with SCD with recurrent VOCs, without definition of “recurrent.” While the sponsor assumes “recurrent VOCs” means at least 2 severe VOCs (requiring hospitalization) per year, it is uncertain whether this threshold would be applied by all clinicians. If the number of people with SCD is higher than estimated by the sponsor or if a lower threshold for recurrent VOC is used in practice, the budget impact of reimbursing exagamglogene autotemcel will be higher than predicted by the sponsor’s model.
CDA-AMC explored uncertainty in the number of patients with recurrent VOCs in a scenario analysis.
The number of patients expected to receive exagamglogene autotemcel is uncertain. In the BIA, the sponsor has assumed that no patients will receive exagamglogene autotemcel for the treatment of SCD in the first year of reimbursement. The sponsor anticipates that exagamglogene autotemcel will be administered only in ATCs and that it will take 6 to 12 months before an ATC is “fully activated.” As per the sponsor’s implementation time, the sponsor has identified 4 ATCs in CDA-AMC-participating jurisdictions (2 each in Alberta and Ontario) that already have dedicated teams experienced with the steps required for delivery of exagamglogene autotemcel, and it is uncertain how long it would be before these centres are ready to administer exagamglogene autotemcel. The exact number of ATCs that will be ready to provide exagamglogene autotemcel in the first year of reimbursement is uncertain and if it is higher than anticipated by the sponsor, the budget impact of reimbursing exagamglogene autotemcel for the treatment of SCD will be higher than anticipated by the sponsor.
Additionally, in the sponsor’s base case, the anticipated market share of exagamglogene autotemcel for SCD was assumed to be 0% in Year 1, 0.5% in Year 2, and 1.9% in Year 3, resulting in 0, 6, and 21 patients receiving exagamglogene autotemcel in each year, respectively. These market share values were derived by the sponsor based on internal market estimates and clinical expert input received by the sponsor and incorporated the number of ATCs and bed capacity, with the latter being considered the most important factor. The sponsor thus calculated the total number of patients that they expect to receive exagamglogene autotemcel across both SCD and TDT indications, and then assumed that 2/3 of these patients have SCD and 1/3 would have TDT. While CDA-AMC acknowledges that bed capacity is an important consideration, the market share, derived in this way, may not accurately reflect the expected uptake of exagamglogene autotemcel. First, the number of ATCs, and hence bed capacity, is uncertain as noted in the previous appraisal point. Second, clinical expert feedback received by CDA-AMC noted that the exact proportion of patients with SCD to TDT is uncertain and that some clinicians may prioritize patients with SCD for receipt of exagamglogene autotemcel owing to the availability of other therapeutic options for TDT and the large health care resource burden associated with uncontrolled SCD. The specific distribution among the indications remains unknown.
CDA-AMC explored the impact of uncertainty in market uptake and ATC capacity in a scenario analysis.
Inclusion of blood product costs is inappropriate. As part of SOC the sponsor included the cost of RBC transfusions using a unit cost obtained from the CADTH reimbursement review of luspatercept in beta-thalassemia-associated anemia. As the budget impact is from the perspective of the Canadian drug plans and RBCs are funded by Canadian Blood Services, the inclusion of RBCs is not appropriate biasing BIA results in favour of exagamglogene autotemcel.
In the CDA-AMC base-case analysis, the cost of RBCs were excluded.
The price of drugs paid by public drug plans is uncertain: Both the sponsor and CDA-AMC analyses are based on publicly available list prices for all comparators. Actual costs paid by public drug plans are unknown.
CDA-AMC was unable to address this limitation.
CDA-AMC Reanalyses of the BIA
CDA-AMC revised the sponsor’s base case by adopting a public drug plan payer perspective. That is, costs associated with RBC were excluded from the CDA-AMC base case, as these costs are borne by Canadian Blood Services.
Table 12: CDA-AMC Revisions to the Submitted BIA
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Changes to derive the CDA-AMC base case | ||
1. Cost of RBC | Included | Excluded |
CDA-AMC base case | Reanalysis 1 | |
RBC = red blood cell.
The results of the CDA-AMC step-wise reanalysis are presented in summary format in Table 13 and a more detailed breakdown is presented in Table 14. All CDA-AMC reanalyses were based on publicly available prices of the comparator treatments.
In the CDA-AMC base case, the estimated incremental budget impact of reimbursing exagamglogene autotemcel is expected to be $59,373,150 (Year 1: $0; Year 2: $15,444,927; Year 3: $43,928,392).
Table 13: Summary of the CDA-AMC Reanalyses of the BIA
Stepped analysis | Three-year total ($) |
|---|---|
Submitted base case | 59,337,641 |
CDA-AMC reanalysis 1 | 59,373,150 |
CDA-AMC base case | 59,373,150 |
CDA-AMC conducted the following scenario analyses to address remaining uncertainty, using the CDA-AMC base case (results are provided in Table 14):
Increasing the number of patients with SCD with ≥ 2 VOCs per year by 10%.
Assuming that more patients will receive exagamglogene autotemcel (23 patients in year 1, 47 patients in year 2, and 65 patients in year 3), aligned with expert input obtained by the sponsor. This scenario considers the maximum possible anticipated uptake but does not consider the time from listing until treatment (e.g., time from listing until centre “activation,” time from listing to infusion).
Table 14: Detailed Breakdown of the CDA-AMC Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|---|
Submitted base case | Reference | 8,584,349 | 8,721,699 | 8,861,246 | 9,003,026 | 26,585,971 |
New drug | 8,584,349 | 8,721,699 | 24,306,173 | 52,895,740 | 85,923,612 | |
Budget impact | 0 | 0 | 15,444,927 | 43,892,714 | 59,337,641 | |
CDA-AMC base case | Reference | 1,780,619 | 1,809,109 | 1,838,055 | 1,867,464 | 5,514,627 |
New drug | 1,780,619 | 1,809,109 | 17,282,982 | 45,795,855 | 64,887,946 | |
Budget impact | 0 | 0 | 15,444,927 | 43,928,392 | 59,373,319 | |
CDA-AMC scenario analysis 1: Increased % of patients with ≥ 2 VOCs per year | Reference | 1,994,052 | 2,025,957 | 2,058,372 | 2,091,306 | 6,175,635 |
New drug | 1,994,052 | 2,025,957 | 19,047,294 | 50,410,935 | 71,484,187 | |
Budget impact | 0 | 0 | 16,988,922 | 48,319,629 | 65,308,551 | |
CDA-AMC scenario analysis 2: Increased number of patients who receive exagamglogene autotemcel a | Reference | 1,812,828 | 1,841,834 | 1,871,303 | 1,901,244 | 5,614,381 |
New drug | 1,812,828 | 74,809,994 | 60,517,115 | 48,895,629 | 184,222,738 | |
Budget impact | 0 | 72,968,160 | 58,645,812 | 46,994,385 | 178,608,358 |
ATC = authorized treatment centre, BIA = budget impact analysis; VOCs = vaso-occlusive crises.
aOwing to the structure of the sponsor’s model, assumptions about market share and number of ATCs were required to implement this scenario. Specifically, market uptake values for exagamglogene autotemcel in the new drug scenario were assumed to be 2.4% in year 1, 4.3% in Year 2, and 5.8% in Year 3 (i.e., equal to 7 ATCs), to result in 23, 47, and 65 patients who receive exagamglogene autotemcel in year 1, 2, and 3, respectively. This is aligned with the number of patients in the sponsor-submitted “high-end” uptake scenario.
Ethics Review
Abbreviations
allo-HSCT
allogenic hematopoietic stem cell transplant
exa-cel
exagamglogene autotemcel
GVHD
graft-versus-host disease
HRQoL
health-related quality of life
PES
primary efficacy set
RBC
red blood cell
SCD
sickle cell disease
VOC
vaso-occlusive crisis
Summary
Sickle cell disease (SCD) is a rare, progressive, life-limiting hereditary blood disorder that alters the shape and properties of red blood cells (RBCs). These altered RBCs result in acute and chronic multisystemic physical complications, including vaso-occlusive crises (VOCs) that are characterized by intense and debilitating pain episodes.
This report describes ethical considerations regarding the use of exagamglogene autotemcel (exa-cel), a gene therapy for patients aged 12 years and older with SCD and recurrent VOCs. Patient group, clinician group, clinical expert, and drug program input and relevant literature informed this review.
The ethical considerations identified include:
Diagnosis, treatment, and experiences of SCD: SCD and its treatment are physically and psychosocially burdensome. Existing disease-modifying and curative therapies have limitations in efficacy, present risks, and may be inaccessible or intolerable for some. For people with SCD who are ineligible for allogenic hemopoietic stem cell transplant (allo-HSCT) whose disease does not respond to, who do not tolerate, or who have difficulty accessing current therapies, there is an unmet need for effective treatments that reduce disease complications, decrease the burdens of long-term treatment, decrease health resource use, and increase quality of life. SCD disproportionately impacts people who are racialized, most commonly Black people. People impacted by intersecting factors related to race, disability, age, geography, income, immigration status, and opioid use may have more severe disease and a higher unmet need for novel treatment options because of the greater challenges they face in accessing and navigating standard care.
Evidence used in the evaluation of exa-cel: Findings from the ongoing, single-arm CLIMB-121 trial suggest that exa-cel demonstrates a potential clinically meaningful prevention of VOCs, hospitalizations, RBC transfusions, and improvements in health-related quality of life (HRQoL) in patients with SCD who have recurrent VOCs. Exa-cel displays a short-term safety profile consistent with a treatment requiring myeloablative conditioning. However, there is uncertainty about the true effect of the treatment because of methodological limitations of the CLIMB-121 trial; the efficacy and safety of exa-cel beyond the current trial follow-up of 24 months; and generalizability to groups that clinical experts suggested may benefit from treatment but were not included in the clinical trial (i.e., people with severe disease but fewer than 2 VOCs in the previous 2 years, people aged 35 years and older, and people with chronic pain). Additionally, there is no evidence on comparative effectiveness or safety. The trial could not provide information on longer-term toxicities important to patients, such as loss of fertility (a known risk of myeloablative conditioning), malignancies, and potential genotoxicities. Given that exa-cel has been proposed as a one-time treatment with potential for life-long effects, this evidentiary uncertainty highlights the importance of robust consent conversations to support informed, autonomous decision-making and establish reasonable expectations, including for people underrepresented in the trial. Evidentiary uncertainty also has implications for decision-making in health systems, as it presents challenges in the assessment of the value of exa-cel relative to the standard of care and understanding opportunity costs.
Clinical use and implementation of exa-cel: Based on available evidence, the clinical experts would consider exa-cel, given the high unmet treatment need, severe morbidity, and premature mortality for people experiencing severe complications of SCD, despite supportive care, and for whom allo-HSCT is not an option. As a gene therapy, exa-cel is associated with theoretical risks (e.g., genotoxicities) and known risks of myeloablative conditioning (e.g., secondary malignancy and infertility). Clinician groups and clinical experts suggested that providing access to fertility preservation (as is common for patients undergoing oncological treatments that present a risk of infertility) would help support equitable access to exa-cel and mitigate risks associated with infertility. Providers will need to facilitate thorough consent conversations to ensure that patients and their families are aware of the benefits, risks, and evidentiary uncertainty related to exa-cel and hold reasonable expectations. Managing expectations is especially important, considering that treatment with exa-cel may not cure SCD, will not reverse end-organ damage or related symptoms, and may preclude eligibility for re-treatment and future gene therapies. Addressing systemic racism and barriers to accessing standard SCD care may support equitable access to exa-cel. Equitable access may also be supported by addressing barriers to some elements of the exa-cel treatment journey, which includes care in specialized centres, prolonged hospitalization, and long-term follow-up.
Health systems: Uncertainty regarding exa-cel’s clinical effectiveness and safety and, in turn, cost-effectiveness, limits assessment of its value as a one-time therapy. Exa-cel has the potential to meet unmet needs for people with SCD, a historically underfunded and underresearched condition that disproportionately impacts groups experiencing health inequities. Treatment with exa-cel is resource-intensive, requires pre-treatment, a month-long hospitalization, and follow-up and administration by experienced personnel in authorized transplant and cell therapy centres. These factors, alongside current capacity constraints of health systems, will severely limit the number of eligible patients that can be treated each year and necessitate prioritizing patients for access. Clinical experts reported that, among people with SCD who are ineligible for allo-HSCT, they would prioritize those experiencing the most severe disease who were still fit and eligible for treatment with exa-cel. Because authorized treatment centres may only be situated in certain jurisdictions in Canada, consistent prioritization criteria and intrajurisdictional and interjurisdictional agreements are important for ensuring equitable access to exa-cel.
Objective and Research Questions
The objective of this ethics review is to identify and describe ethical considerations associated with the use of exa-cel for the treatment of patients 12 years and older with SCD and recurrent VOCs, including considerations related to the disease context, evidentiary basis, use of exa-cel, and impact on health systems.
To address this objective, this review addresses the following research questions:
What ethical considerations arise in the context of SCD, including considerations related to diagnosis, treatment, and outcomes?
What ethical considerations arise in relation to the evidence (e.g., clinical and economic data) used to evaluate exa-cel?
What ethical considerations arise in relation to the use of exa-cel for patients, their caregivers, and their clinicians?
What are the ethical considerations for health systems related to exa-cel?
Methods
Guiding questions identified in the EUnetHTA Core Model 3.0, Ethics Analysis Domain,1 and supplemented by relevant questions from the Equity Checklist for Health Technology Assessments (ECHTA),2 drive the identification of ethical considerations relevant to the use of exa-cel in the treatment of SCD in this ethics review. These guiding questions are organized to respond to the research questions and to investigate ethical considerations related to:
patients living with SCD and their caregivers (i.e., disparities in incidence, treatment, or outcomes; challenges or burdens related to diagnosis or clinical care; factors that might prevent patients from gaining access to therapies)
the evidence used to demonstrate the benefits, harms, and value of exa-cel (i.e., ethical considerations in relevant clinical trials, including their representativeness, the choice of outcome measures, the appropriateness of the analytical methods and models used for all population groups; ethical considerations related to the data or assumptions in the economic evaluation)
the use of exa-cel, including considerations related to benefits and harms to patients, relatives, caregivers, clinicians, and society, as well as considerations related to access to these therapies
the uptake of exa-cel in health systems, including considerations related to the distribution of health care resources.
Review of Project Inputs
A single reviewer collected and considered input from 7 main sources of data related to the ethical considerations relevant to the research questions guiding this ethics review. The reviewer considered the following sources:
evidence from a search of published literature
the sponsor submission, including noting relevant information and external references or sources relevant to each of the research questions driving this report
clinician group input received from Cell Therapy Transplant Canada and the Canadian Hemoglobinopathy Association
patient input received from the Global Action Network for Sickle Cell & Other Inherited Blood Disorders, NotJustYou, the Sickle Cell Awareness Group of Ontario, and the Sickle Cell Disease Association of Canada
drug program input received from drug programs participating in the reimbursement review process
discussion with clinical experts (n = 3) directly engaged over the course of this reimbursement review, including through 2 clinical and economic consultation meetings that involved 2 experts, and 1 panel meeting that involved 3 experts; during each of these meetings, the reviewer asked the clinical experts targeted questions related to ethical considerations corresponding to the research questions driving this report. All the clinical experts are practising hematologists with experience treating adult (n = 2) or pediatric (n = 1) patients with SCD in Canada; 1 had experience caring for patients who had received exa-cel
engagement with clinical and economic reviewers to identify domains of ethical interest arising from their respective reviews and to identify relevant questions and sources to further pursue in this report.
Details on the Published Literature Search
An information specialist conducted a literature search on key resources, including MEDLINE via Ovid, Philosopher’s Index via Ovid, and Scopus. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine Medical Subject Headings (MeSH), and keywords. To address the indication, the main search concept used was exagamglogene autotemcel, and retrieval was limited to documents published in English.
A focused search was performed to address the population (SCD), using MEDLINE and Philosopher’s Index, limiting the retrieval of English-language publications after January 1, 2019. Both searches were completed on June 21, 2024.
Search filters were applied to each search to limit retrieval to citations related to ethical concepts or considerations. Duplicates were removed by manual deduplication in EndNote. The search strategy is available on request.
Literature Screening and Selection
A single reviewer screened the literature in 2 stages. First, the reviewer screened the titles and abstracts of the retrieved citations and identified and retrieved articles for full-text review if their titles or abstracts identified ethical considerations, or provided normative analyses (i.e., focusing on “what ought to be” through argumentation), or empirical research (i.e., focusing on “what is” through observation) of ethical considerations related to the experience, incidence, diagnosis, treatment, or outcomes of patients with SCD, or related to evidence on the use of or the implications of exa-cel for patients with SCD. In the second stage, the same reviewer reviewed full-text publications categorized as “retrieve.” The reviewer included texts that had substantive information meeting the aforementioned criteria. Additionally, the reviewer retrieved and reviewed select sources drawn from relevant bibliographies, relevant key concepts, and consultations with experts or other reviewers using the selection criteria listed previously.
Data Analysis
The 4 research questions driving this review guided the collection, coding, and thematic analysis of data. The reviewer conducted 2 iterative cycles of coding and analysis to abstract, identify, and synthesize relevant ethical considerations from the literature and from relevant project inputs. In the initial coding phase, the reviewer read the publications and input sources for ethical content (e.g., claims related to potential harms, benefits, equity, justice, and resource allocation, as well as ethical issues in the evidentiary basis). The reviewer coded the identified claims related to ethical content using methods of qualitative description.3 In the second coding phase, the reviewer identified major themes and subcodes through repeated readings of the data,3 and summarized them into thematic categories within each guiding domain or research question. The reviewer noted if the ethical content did not fit into these categories or into the domains outlined in the research questions, or if there were discrepancies or conflicts between the ethical considerations or values identified between project sources or within thematic categories. The data analysis was iterative, and the reviewer used themes identified in the literature or in project inputs and during consultations with clinical experts to further refine and reinterpret the ethical considerations identified. Finally, the reviewer thematically organized and described the data according to the 4 research questions and domains driving this ethics review. The results, limitations, and conclusions of this analysis are described in the following sections.
Results
Key Ethical Considerations
Treatment and Experiences of SCD
SCD is a rare, progressive hereditary blood disorder characterized by a genetic mutation that causes the expression of abnormal hemoglobin S.4 Hemoglobin S polymerization causes RBCs to change their properties and shape (i.e., to a sickle shape), leading to blood vessel occlusion that prevents oxygen delivery to tissues.4 The hallmark manifestation of SCD are VOCs, which are characterized by intense and debilitating pain episodes.4,5 VOCs are directly linked to end-organ damage and early mortality6-9 and, as such, the frequency of VOCs inversely relates to life expectancy.4 As discussed in the clinical review report, SCD results in many additional acute and chronic multisystemic physical complications, which are associated with increased morbidity, mortality, and health use.4,5,10-13 In Canada, the average age of death of people living with SCD (although not necessarily with recurrent VOCs) is 55 years, according to Ontario-based data reported between 2007 and 2017.14 The same data suggest that SCD affects 1 in 4,200 individuals.14
Psychosocial Burdens of Living With SCD and Caring for Someone With SCD
SCD is both a physically and psychosocially burdensome condition.15-19 It can negatively impact quality of life due to associated medical complications, treatment burden, challenges in accessing treatment and care, stigmatization and discrimination, and lifestyle adjustments.15-19 These factors may negatively impact relationships, education, and employment opportunities.15,16,18,19 People with SCD may also experience fear of complications and a sense of fatalism related to their prognosis as their condition progresses.17,18 As 1 person living with SCD who provided patient input stated, “The constant vigilance and fear of crises robbed me of many of the simple joys of life.” SCD also impacts caregivers and families. Parents of children with SCD reported emotional distress, guilt related to the hereditary nature of the condition, and impact on relationships, employment, and education.5,15,19,20 Parental stress may be further associated with poorer health outcomes for children with SCD.20 Fear of SCD can also influence family-planning decisions, with some people forgoing having children or additional children, or choosing in vitro fertilization with preimplantation genetic testing or egg or sperm donation.21
Existing Treatment Options for SCD in Canada
Treatment for SCD in Canada includes disease-modifying therapies (hydroxyurea and RBC transfusions), curative therapy (allo-HSCT), prophylactic antibiotics, analgesics (including opioids), and surgery to address complications such as avascular necrosis.4 However, existing therapies have limitations in efficacy, present risks, and may be inaccessible, burdensome, or intolerable for some people with SCD in Canada. The only approved curative therapy for SCD is allo-HSCT, which involves using chemotherapy to weaken or destroy defective stem cells in the bone marrow (i.e., myeloablative conditioning) and replacing them with stem cells from a healthy donor.22 However, it is associated with risks such as organ damage, secondary malignancy, infertility, infection, graft rejection, and life-threatening graft-versus-host disease (GVHD).22 Moreover, as noted by clinical experts and in clinician group input, access to allo-HSCT is limited by donor availability (with only 10% to 20% of people with SCD having a matched sibling donor), age (with optimal outcomes for people younger than 12 years of age), and health system capacity.4,16
Hydroxyurea is an oral therapy that increases the production of fetal hemoglobin, which may protect against SCD complications such as VOCs.4,23 According to clinician group input and clinical experts, it can delay disease progression and prolong life. However, it is ineffective for some people with SCD, can cause myelosuppression that may prevent optimal dosing, and requires daily dosing and regular monitoring, which may limit adherence. Some patients may also wish to avoid using hydroxyurea until after childbearing because of its perceived association with teratogenicity and reduced fertility.4,10 The patient input and clinical experts also cited access challenges related to the limited availability of suspensions easily ingested by children in Canada.
RCB transfusions provide people with SCD with normal adult hemoglobin and suppress hemoglobin S to reduce SCD complications.4 However, chronic transfusions present risks such as iron overload (requiring iron chelation therapy), alloimmunization, and dangerous hemolytic transfusion reactions.4 Additionally, people who are racialized can have difficulty accessing blood with compatible antigens and may require access to blood through rare blood programs, as most blood donors in Canada are white.24 Clinical experts noted that people living far from specialized treatment centres may face barriers accessing exchange transfusions. Furthermore, chronic transfusion therapy is time-consuming and can interfere with social activities, education, and work, and even limit where people with SCD and their families can live, work, and travel.19 Ultimately, for people with SCD who are ineligible for allo-HSCT and whose disease does not respond to, who do not tolerate, or who have difficulty accessing current therapies, there is an unmet need for effective treatments. Specifically, there is an unmet need for treatment that reduces disease complications, decreases the burdens of long-term treatment, decreases health resource use, and increases HRQoL.
Inequities and Challenges Accessing Diagnosis, Treatment, and Care for SCD in Canada
Access to treatment and care for SCD is impacted by broader, intersecting factors related to race, disability, age, geography, income, immigration status, and opioid use.15-18,25-34 As detailed later in this report, groups impacted by these factors may experience increased disease severity due to limited access to standard care. They may also experience a higher unmet need for one-time disease-modifying therapies with the potential to reduce long-term treatment burdens. Simultaneously, however, they may experience disproportionate difficulty accessing such therapies.10,29,32,35 Although SCD does not occur exclusively in the Black population, Black people comprise 90% of those with SCD in the US.13,16 The clinical experts confirmed that this trend is transferable to Canada, where SCD impacts primarily people who are racialized and/or immigrants. Clinician group input reported that the prevalence of SCD is increasing in Canada because of immigration from regions where the condition is more widely prevelant.14,35 Black people with SCD often experience discrimination when interacting with the health system due to systemic racism compounded with the stigma associated with disability and opioid use.15-18,27-29 Health care providers often label people experiencing VOCs as drug-seeking, as overreporting pain, or as misusing opioids, which can lead to them being dismissed or ignored, harshly treated, and receiving delayed care, misdiagnoses, or undertreatment.15-17,29-32 Experiencing racial bias, discrimination, or pain perceived as unfair or unjust may lead people with SCD to have reduced physical and mental health, develop a mistrust of health care providers, avoid future medical care, and adhere less to treatment, all of which may have life-long impacts on health outcomes.27,32-34 Compared to rare diseases that primarily impact white people (e.g., cystic fibrosis and hemophilia), SCD is underresearched and underfunded, and care may be less available.16,29
The clinical experts reported that newborn screening to facilitate early SCD diagnosis and treatment is accessible Canada.22,36 However, they noted that immigrants with SCD born in regions without access to newborn screening or early treatment may have worse health status upon arrival in Canada. These groups may only obtain a diagnosis after acute complications.26 Clinical experts, patient input, and the published literature note that additional barriers to accessing and navigating treatment and care include administrative burdens, limited access to or difficulties contacting knowledgeable providers (especially in rural communities and provinces with fewer SCD cases), a lack of coordination between care providers, a lack of knowledge regarding where to access care, and being unable to afford out-of-pocket costs of treatment.31 Clinical experts also noted that people who are immigrants, have a low income, or lack employment-related insurance may find it particularly challenging to navigate these barriers. Furthermore, while transitioning from pediatric to adult care, patients are at an increased risk of health complications, morbidity, and early mortality, especially without transition-support programs.17 Clinical experts noted that access to such programs varies among jurisdictions.
Ethics of Evidence and Evaluation of Exa-cel
As detailed in the clinical review report, the clinical evidence for this review is drawn from the ongoing single-arm, phase III, open-label CLIMB-121 trial. The objective of the CLIMB-121 trial is to evaluate the safety and efficacy of exa-cel administered after single-drug myeloablative conditioning chemotherapy with busulfan for the treatment of SCD in patients 12 to 35 years who have recurrent VOCs. Participants in the CLIMB-121 trial were followed for at least 16 months after infusion and for at least 14 months after completion of the last RBC transfusion for posttransplant support or SCD management, as of the June 14, 2023, data cut-off date (i.e., the primary efficacy set [PES]). Participants who complete the CLIMB-121 trial (with a 2-year follow-up period) are enrolled in the CLIMB-131 long-term extension study (with a 13-year follow-up period), for a total of 15-years of follow-up.
The conclusions from the clinical review report were that interim findings of the CLIMB-121 trial are consistent with a clinically meaningful prevention of VOCs, hospitalizations, and RBC transfusions, and improvements in HRQoL. Of the 30 patients in the PES, 29 (96.7%) achieved absence of any severe VOCs for at least 12 consecutive months starting 60 days after their last RBC transfusion, which is the primary efficacy end point in the CLIMB-121 trial. Although the proportion of participants experiencing adverse events and serious adverse events was high, the clinical experts and clinician group input noted that this finding is generally consistent with harms associated with the underlying disease and myeloablative busulfan conditioning. However, there is uncertainty about whether results from the trial present the true effect of exa-cel because of important limitations in trial design, uncertainty regarding participants’ baseline treatments, and subjectivity in outcome assessments affect confidence in the findings. Additionally, there is uncertainty in the durability and long-term efficacy and safety of exa-cel beyond the current trial follow-up of 24 months, and there is no evidence on the comparative effectiveness and safety of exa-cel. The trial also cannot provide information on longer-term toxicities important to patients, such as loss of fertility, malignancies, and potential genotoxicities. Neither the CLIMB-121 trial nor the CLIMB-131 trial include infertility as a safety end point, despite this being a known risk of the conditioning required for treatment and a patient-important outcome.19,37 However, the clinical experts acknowledged that infertility would be challenging to measure in a trial and would be a known risk that they would discuss with patients.
There is also uncertainty in generalizability beyond the trial population. The clinical experts considered the trial population to be broadly generalizable in the Canadian context with respect to race. However, they cautioned that although reasonable for a trial context, the CLIMB-121 trial excluded people who might still benefit from exa-cel. This includes people who experienced fewer than 2 VOCs in the previous 2 years but who had other indications of severe disease, people 35 years and older, and those living with chronic pain. Clinical experts reported that VOC frequency may inadequately reflect disease severity, as it may be underestimated, and that there are additional indicators of severe disease. The experts also anticipated that people older than 35 years who are otherwise fit for treatment could benefit from exa-cel. Notably, they suggested that expanding the age of eligibility would increase people’s liberty in family planning, including choosing whether to delay treatment with exa-cel, which presents risk of infertility, until after having children. The clinical experts believed that people with chronic pain may still benefit from a treatment that has the potential to reduce VOCs and further organ damage, even if it does not alleviate preexisting pain.
Given that exa-cel has been proposed as a one-time treatment with the potential for life-long effects, evidentiary uncertainty highlights the importance of robust consent conversations to support informed, autonomous decision-making and establish reasonable expectations, including for people underrepresented in the trial. Evidentiary uncertainty also has implications for decision-making in health systems, as it presents challenges in the assessment of the value of exa-cel relative to the standard of care and understanding opportunity costs. Clinical experts and published literature emphasized the need for the life-long follow-up of people who have received exa-cel, and the collection and evaluation of long-term safety and efficacy data (e.g., through the CLIMB-131 trial and registries, the latter of which are not currently available in Canada).14,35,38 This follow-up and data would facilitate timely responses to harms and a better understanding of the benefits and risks of treatment. Clinical experts also noted that it will be important to ensure supports (e.g., virtual care and community-based blood draws) to alleviate potential barriers for people engaging in this continued monitoring.
Ethical Considerations in the Use of Exa-Cel
The use of exa-cel raises ethically salient considerations regarding the balancing of benefits and harms, risk of infertility, informed consent, and equitable access.
Balancing Benefits and Harms
The proposed value of exa-cel is its potential to be a one-time therapy that leads to sustained fetal hemoglobin expression over 20% to address the unmet need for an effective treatment for people with SCD and recurrent VOCs whose disease does not respond to standard care and who are not eligible for allo-HSCT. The clinical experts and clinician groups perceived the preliminary results of the CLIMB-121 trial to be promising for outcomes important to people with SCD and clinicians. These include preventing severe VOCs and related hospitalizations, reducing RBC transfusions, and increasing HRQoL. Furthermore, the clinical experts noted that the sustained expression of fetal hemoglobin over 20% could prevent or delay additional SCD complications. Despite evidentiary uncertainty, the clinical experts would consider exa-cel for patients experiencing recurrent VOCs despite supportive care and for whom allo-HSCT is not an option because of the severe morbidity, premature mortality, and high unmet need for effective, preventive therapy in this group. They noted, however, that they would reserve exa-cel for patients not eligible for allo-HSCT, whom they feel would better understand the benefits and risks at this time. However, when the evidence base for exa-cel grows, comparative safety and efficacy data for it and allo-HSCT may be warranted to inform clinical decision-making. Such data may be especially relevant because, unlike allo-HSCT, exa-cel also does not carry the risk of life-threatening GVHD.22
As a cellular therapy involving ex vivo gene editing using clustered regularly interspaced short palindromic repeats (CRISPR)–CRISPR-associated protein 9 (Cas9) technology, exa-cel is associated with theoretical and known risks. The theoretical risks include genotoxicities (i.e., unintended on-target and off-target effects), which could cause irreversible, unintended consequences or malignancy.22,23,35,38-40 The known risks include those related to myeloablative conditioning, which have common short-term adverse effects (e.g., neutropenia and thrombocytopenia) and long-term risks, such as organ damage, secondary malignancy, and infertility.22,35 The experts noted that a combined 15-year follow-up period (i.e., through the CLIMB-121 and CLIMB-131 trials) would be sufficient for detecting malignancies and potential off-target effects.
The clinical experts, patient input, and published literature reported that gene therapies offer hope for people with SCD who have no alternative treatment options and their caregivers, and that learning about ineligibility can be disappointing.19,37 Psychological support may be necessary to minimize harm related to unmet expectations for those who are ineligible, including those with irreversible organ damage related to SCD that prevents them from being fit for myeloablative condition. Of note, given that gene therapy is not associated with the risk of GVHD, some parents have expressed the desire to wait until their child with SCD meets the eligibility criteria for gene therapy before pursuing allo-HSCT.19
Risk of Infertility
Although infertility is not included as a safety outcome in the CLIMB-121 trial or the CLIMB-131 extension study, it is a known risk of myeloablative conditioning. As previously noted, people may experience reduced fertility or pregnancy-related complications due to SCD and its existing treatment options (e.g., hydroxyurea and allo-HSCT).4,10-12 The clinical experts did not consider infertility a reason to refrain from recommending exa-cel. However, the clinical experts and clinician groups suggested that offering and covering fertility preservation for patients receiving exa-cel (as is common for patients undergoing oncological treatments that present a risk of infertility) would help support equitable access and mitigate the risk of infertility. Of note, the clinical experts and literature reported that people with SCD already experience barriers to and inequities in accessing fertility preservation. These include inconsistent coverage across jurisdictions, funding being more widely available to people with oncological rather than hematological conditions, and out-of-pocket costs associated with collection and storage that are greater for people requiring ovum retrieval and preservation and may prevent some people with a lower income from being able to afford it.10,41 Some people with SCD consider infertility and possible secondary malignancy to be an unacceptable risk.10,37 This is even for treatments with curative intent that could prolong life expectancy (e.g., allo-HSCT), and even when educated about fertility preservation.10,37 Younger people may be especially likely to view gene therapy less positively when educated about these risks.37
Informed Consent
The clinical experts and literature emphasized the importance of carefully discussing the benefits, known and theoretical risks, and evidentiary uncertainty related to all treatment options, including exa-cel, to facilitate fully informed decision-making.17,22,36,38,42 It will be important for clinicians to establish reasonable expectations as part of informed consent processes to alleviate harms associated with false hope. This is especially important, given that people with SCD and their caregivers may initially expect and understand that gene therapy may be less invasive, lower risk, and easier to recover from than allo-HSCT.19,22,37 The clinical experts and the literature highlighted the need for clinicians to communicate that treatment with exa-cel may not cure SCD, will not reverse end-organ damage or related symptoms, such as chronic pain, and may preclude eligibility for re-treatment and future gene therapies.5,35,43,44 The clinical experts noted that consent to treatment with exa-cel is an ongoing process and may be withdrawn even after the collection of stem cells.
Promoting informed consent will also require considering peoples’ unique vulnerabilities and decision-making needs. For example, pediatric patients and young adults with limited decision-making experience or those with limited health or genetic literacy may require increased informational and decisional supports.17,45 Additionally, caregivers of children with SCD may be motivated to discuss higher-risk treatment options after their child experiences several debilitating VOCs, life-threatening complications, or medical recommendations for chronic transfusions, splenectomy, or surgery.19 This raises considerations regarding the vulnerability of people with life-limiting conditions and limited alternative treatment options. It also raises considerations about relational autonomy or the way social status and various forms of oppression influence an individual’s ability to act autonomously in medical decision-making.38
Equitable Access
The implementation of exa-cel raises challenges regarding equitable access and distributive justice.38 Equitable access requires attending to the previously discussed intersecting barriers to accessing care for SCD in Canada (e.g., related to systemic racism and discrimination, socioeconomic status, geography, and disability).35 It also requires addressing additional barriers related to accessing specialized treatment centres, undergoing a month-long hospitalization, and receiving long-term follow-up. Groups experiencing difficulty gaining timely access to the standard care for SCD may also be at a greater risk of not being healthy enough to undergo myeloablative conditioning and, thus, being deemed ineligible for treatment with exa-cel.35 People with severe disease who have developed a mistrust of the health system after experiences of racism or discrimination may also be less likely to access exa-cel.35 The literature suggested that addressing structural racism within the health system would help promote equity in access to care and treatment for SCD, including exa-cel.29,32 Treatment with exa-cel, paired with associated transportation, lodging costs, and loss of income, may be more financially and logistically burdensome for those who reside far from a specialized treatment centre and have a lower socioeconomic status.10 The clinical experts confirmed the importance of patient-support programs in mitigating barriers to equitable access. In the implementation plan submitted for this review, the sponsor reported that it is “exploring” funding and the implementation of such programs, should exa-cel be reimbursed.
Health Systems Considerations
The use of exa-cel for the treatment of SCD raises ethical considerations related to sustainable funding, health system capacity constraints, and prioritization among those eligible for treatment with exa-cel in Canada.
Sustainability of Funding Exa-Cel
The introduction of exa-cel, a highly expensive therapy, raises concerns regarding the ability of health systems to sustainably manage associated costs.35,40 It is hoped that one-time gene therapies like exa-cel with high upfront costs could result in long-term reductions in health care use.35,46 However, uncertainty regarding exa-cel’s clinical effectiveness and safety and, in turn, cost-effectiveness, limits assessments of its value, including as a one-time therapy.35,38,46 Clinician group input reported that consistent measures of outcomes related to exa-cel across treatment centres compared to best supportive care and allo-HSCT would be important for understanding the long-term benefits and risks and, in turn, cost-effectiveness and opportunity costs.
SCD has been underfunded, underresearched, and disproportionately impacts groups with ongoing health disparities related to systemic barriers to wellness.47 Given its high price and the number of potentially eligible recipients, reimbursement of exa-cel is associated with opportunity costs both within and outside of the health system, which may disproportionately impact equity-deserving groups.35,47 However, therapies reducing VOCs and hospitalizations could increase productivity and reduce caregiver burden, thus reducing income inequality for people with SCD who may already experience socioeconomic disadvantages due to structural racism and disability.25,46 The literature reports that the value of improved productivity, reduced caregiver burden, hope, and real option value (i.e., the potential to increase life expectancy to provide patients with the option to benefit from future therapies) may provide a rationale for higher price and cost-effectiveness thresholds for gene therapies for SCD.46 At the same time, using inequities to justify high prices may also reduce the availability of and access to treatment for equity-deserving groups.47
Health System Capacity Constraints and Priority Setting
The clinical experts anticipated that health system capacity will be the factor that will limit equitable access to exa-cel the most. This challenges deployment equity, or the need to ensure that innovations are accessible to and will benefit diverse populations, including those traditionally underserved.35 Exa-cel is a resource-intensive therapy that requires administration by highly trained personnel in authorized transplant and cellular therapy centres, which will severely limit the number of eligible patients that can be treated each year.23,35,40 The sponsor estimates that there will be 10 authorized treatment centres in 4 provinces by the third year of exa-cel implementation, if reimbursed. However, clinical experts reported that transplant and cellular therapy centres in Canada are currently “at or nearly at” capacity for delivering therapies to patients with oncological conditions for whom treatment is more time-sensitive. They noted that these centres currently lack the human and financial resources to treat patients with nonmalignant conditions, and many hemoglobinopathy clinics similarly lack the resources to assess and monitor patients receiving exa-cel.
In the context of capacity constraints, it will be necessary to prioritize access to exa-cel within the population with SCD, and between populations with SCD and transfusion-dependent thalassemia (the other condition for which Canada's Drug Agency (CDA-AMC) is reviewing exa-cel).48 Wesevich et al.,48 writing in a US context, proposed that the ethical principles of maximizing benefits, minimizing harm, equalizing concern, and prioritizing disadvantaged groups should underpin clear and transparent prioritization criteria created with input from key groups, including patient groups, clinician groups, and legislators.48 They proposed that these criteria could include factors such as age, disease intensity, the “number and severity of complications,” health care use, and measures “of patient vulnerability.”48 The clinical experts reported that they would prioritize people with SCD who had the highest unmet need, meaning those experiencing the most severe disease who are still fit for treatment with exa-cel but not allo-HSCT. However, they anticipated that people with the highest unmet need may also face geographic or socioeconomic barriers to accessing exa-cel. Considered together, these factors highlight the need for consistent prioritization criteria, intrajurisdictional and interjurisdictional agreements, and patient financial and social supports to ensure those with the greatest unmet need can access treatment.
Limitations
This review draws on published literature that discusses ethical considerations related to the use of exa-cel or gene therapy for the treatment of SCD. However, there may be additional ethical considerations in the context of exa-cel for SCD that are not captured in this review. Input received during this reimbursement review (i.e., patient group, clinician group, and drug program input; discussion with clinical experts; and engagement with the clinical and pharmacoeconomic review teams) provides a more comprehensive understanding of the ethical considerations related to the use of exa-cel for the treatment of SCD. It is possible that more direct engagement (e.g., through direct interviews) with patients, their caregivers, their family members, transplant specialists, and decision-makers on their specific experiences with SCD and/or exa-cel would offer additional relevant ethical considerations or domains of analysis.
Conclusion
Ethical considerations in the context of SCD highlighted the significant physical and psychosocial burdens of the life-limiting condition and its treatment. Existing disease-modifying and curative therapies have limitations in efficacy, present risks, and may be inaccessible, burdensome, or intolerable for some. There is an unmet need for effective therapies for people with SCD who are ineligible for allo-HSCT and whose disease does not respond to, who do not tolerate, or who have difficulty accessing current therapies. Specifically, there is an unmet need for effective treatments that reduce disease complications, decrease the burdens of long-term treatment, decrease health resource use, and increase HRQoL. People with SCD, a condition disproportionately experienced by people who are Black and impacted by intersecting factors related to race, disability, age, geography, income, immigration status, and opioid use, may have more severe disease and a higher unmet need for novel treatment options because of challenges in accessing and navigating standard care.
Clinical evidence suggests that exa-cel shows promise regarding a potentially clinically meaningful prevention of VOCs, hospitalizations, RBC transfusions, and improvements in HRQoL in patients with SCD who have recurrent VOCs. The evidence also suggests that exa-cel has a short-term safety profile consistent with a therapy requiring myeloablative conditioning. However, there is uncertainty about the true effect of the treatment due to methodological limitations of the CLIMB-121 trial, the efficacy and safety of exa-cel beyond the current trial follow-up of 24 month, and generalizability to groups that may benefit from exa-cel but were not included in the trial. Additionally, there is no evidence on the comparative effectiveness and safety of exa-cel, and the trial could not provide information on longer-term toxicities important to patients, such as loss of fertility, malignancies, and potential genotoxicities. Given that exa-cel has been proposed as a one-time treatment with the potential for life-long effects, this evidentiary uncertainty highlights the importance of robust consent conversations to support informed, autonomous decision-making and establish reasonable expectations, including for people underrepresented in the trial. Evidentiary uncertainty also has implications for decision-making by health systems, as it presents challenges in the assessment of the value of exa-cel relative to the standard of care and understanding opportunity costs. It will be necessary to address barriers to conducting and collecting long-term follow-up data to facilitate a better understanding of the benefits and risks of exa-cel.
The clinical experts would recommend exa-cel, based on the available evidence, for people with SCD experiencing recurrent VOCs despite supportive care and for whom allo-HSCT is not an option due to severe morbidity, premature mortality, and the high unmet treatment need in this group. However, as an ex vivo gene therapy, exa-cel’s use is associated with theoretical risks and known risks of myeloablative conditioning. Clinician groups and clinical experts suggested that providing access to fertility preservation would help support equitable access and mitigate risks associated with infertility. Additionally, providers will need to facilitate thorough consent processes to ensure that patients and their families understand the benefits, risks, and evidentiary uncertainty of exa-cel and have reasonable expectations. Managing expectations will be important to prevent harms related to false hope, as treatment with exa-cel may not cure SCD, will not reverse end-organ damage or related symptoms, and may preclude eligibility for re-treatment and future gene therapies. The clinical experts and literature suggested that addressing systemic racism and other barriers to accessing standard SCD care, as well as barriers to accessing or undergoing treatment with exa-cel, could support equitable access to exa-cel.
The use of exa-cel for the treatment of SCD raises ethical considerations related to sustainable funding, health system capacity constraints, and prioritization. Exa-cel has the potential to meet unmet needs for people with SCD, a historically underfunded and underresearched condition that disproportionately impacts groups experiencing health inequities. However, uncertainty regarding exa-cel’s clinical effectiveness and safety and, in turn, cost-effectiveness limits assessments of its value, including as a one-time therapy. Treatment with exa-cel is resource-intensive, requiring pretreatment, a month-long hospitalization, and follow-up and administration by experienced personnel in authorized transplant and cell therapy centres. These factors, alongside current capacity constraints in health systems, will severely limit the number of eligible patients that can be treated each year and necessitate prioritizing patients for access. The clinical experts reported that, among people with SCD, they would prioritize those experiencing the most severe disease despite supportive care who are still fit and eligible for treatment with exa-cel but not allo-HSCT. Authorized treatment centres may only be situated in certain jurisdictions, and people with highest unmet need may be living furthest from them and at the greatest socioeconomic disadvantage. These considerations highlight the need for consistent prioritization criteria, patient supports, and intrajurisdictional and interjurisdictional agreements to ensure equitable access to exa-cel and other therapies that require stem cell transplant resources in Canada.
References
1.EUnetHTA. EUnetHTA Joint Action 2 Work Package 8, HTA Core Model version 3.0. European Commission; 2016: http://www.htacoremodel.info/BrowseModel.aspx. Accessed 2024 Jun 27.
2.Benkhalti M, Espinoza M, Cookson R, Welch V, Tugwell P, Dagenais P. Development of a checklist to guide equity considerations in health technology assessment. Int J Technol Assess Health Care. 2021;37:e17. PubMed
3.Sandelowski M. Whatever happened to qualitative description? Res Nurs Health. 2000;23(4):334-340. PubMed
4.Kato GJ, Piel FB, Reid CD, et al. Sickle cell disease. Nat Rev Dis Primers. 2018;4(1):18010. PubMed
5.Locatelli F, Corbacioglu S, Hobbs W, Frangoul H, Walters MC. Defining curative endpoints for sickle cell disease in the era of gene therapy and gene editing. Am J Hematol. 2024;99(3):430-438. PubMed
6.Ware RE, de Montalembert M, Tshilolo L, Abboud MR. Sickle cell disease. Lancet. 2017;390(10091):311-323. PubMed
7.Sundd P, Gladwin MT, Novelli EM. Pathophysiology of Sickle Cell Disease. Annu Rev Pathol. 2019;14:263-292. PubMed
8.Eltzschig HK, Eckle T. Ischemia and reperfusion--from mechanism to translation. Nat Med. 2011;17(11):1391-1401. PubMed
9.Ochocinski D, Dalal M, Black LV, et al. Life-Threatening Infectious Complications in Sickle Cell Disease: A Concise Narrative Review. Front Pediatr. 2020;8:38. PubMed
10.Mishkin AD, Mapara MY, Barhaghi M, Reshef R. Fertility Concerns and Access to Care for Stem Cell Transplantation Candidates with Sickle Cell Disease. Biol Blood Marrow Transplant. 2020;26(8):e192-e197. PubMed
11.Boghossian NS, Greenberg LT, Saade GR, et al. Association of Sickle Cell Disease With Racial Disparities and Severe Maternal Morbidities in Black Individuals. JAMA Pediatr. 2023;177(8):808-817. PubMed
12.Early ML, Eke AC, Gemmill A, Lanzkron S, Pecker LH. Severe Maternal Morbidity and Mortality in Sickle Cell Disease in the National Inpatient Sample, 2012-2018. JAMA Netw Open. 2023;6(2):e2254552. PubMed
13.Lee L, Smith-Whitley K, Banks S, Puckrein G. Reducing Health Care Disparities in Sickle Cell Disease: A Review. Public Health Rep. 2019;134(6):599-607. PubMed
14.Pendergrast J, Ajayi LT, Kim E, Campitelli MA, Graves E. Sickle cell disease in Ontario, Canada: an epidemiologic profile based on health administrative data. CMAJ Open. 2023;11(4):E725-E733. PubMed
15.Essien EA, Winter-Eteng BF, Onukogu CU, Nkangha DD, Daniel FM. Psychosocial challenges of persons with sickle cell anemia: A narrative review. Medicine. 2023;102(47):e36147. PubMed
16.Anderson D, Lien K, Agwu C, Ang PS, Abou Baker N. The Bias of Medicine in Sickle Cell Disease. J Gen Intern Med. 2023;38(14):3247-3251. PubMed
17.Clayton-Jones D, Matthie N, Treadwell M, et al. Social and Psychological Factors Associated With Health Care Transition for Young Adults Living With Sickle Cell Disease. J Transcult Nurs. 2021;32(1):21-29. PubMed
18.Durgam N, Brion T, Lewis HB, et al. Patient and Caregiver Perspectives on Care-Seeking During a Vaso-Occlusive Crisis in Sickle Cell Disease: Results from Qualitative Interviews in Canada. Patient Prefer Adherence. 2023;17:41-49. PubMed
19.Sinha CB, Bakshi N, Ross D, Loewenstein G, Krishnamurti L. Primary caregiver decision-making in hematopoietic cell transplantation and gene therapy for sickle cell disease. Pediatr Blood Cancer. 2021;68(1):e28749. PubMed
20.Johnson YL, Woodward K, Dampier C, Cohen L, Sil S. Biopsychosocial Factors Associated with Parenting Stress in Pediatric Sickle Cell Disease. J Clin Psychol Med Settings. 2022;29(2):365-374. PubMed
21.Schultz CL, Tchume-Johnson T, Jackson T, Enninful-Eghan H, Schapira MM, Smith-Whitley K. Reproductive intentions in mothers of young children with sickle cell disease. Pediatr Blood Cancer. 2020;67(5):e28227. PubMed
22.Doerfler PA, Sharma A, Porter JS, Zheng Y, Tisdale JF, Weiss MJ. Genetic therapies for the first molecular disease. J Clin Invest. 2021;131(8):15. PubMed
23.Laurent M, Geoffroy M, Pavani G, Guiraud S. CRISPR-Based Gene Therapies: From Preclinical to Clinical Treatments. Cells. 2024;13(10):08.
24.Haw J, Walrond J, Jayachandran J, et al. Sickle cell disease and the need for blood: Barriers to donation for African, Caribbean, and Black young adults in Canada. Transfusion. 2023;63(7):1324-1332. PubMed
25.Graf M, Tuly R, Gallagher M, Sullivan J, Jena AB. Value of a cure for sickle cell disease in reducing economic disparities. Am J Hematol. 2022;97(8):E289-E291. PubMed
26.Monagel DA, Monteiro J, Thull-Freedman J, Ruzycki A, Leaker M, Steele M. Clinical features at diagnosis of sickle cell disease prior to universal newborn screening in Alberta. Paediatr Child Health. 2022;27(8):464-468. PubMed
27.Blakey AO, Lavarin C, Brochier A, et al. Effects of Experienced Discrimination in Pediatric Sickle Cell Disease: Caregiver and Provider Perspectives. J Racial Ethn Health Disparities. 2023;10(6):3095-3106. PubMed
28.Wahab S, Kelly K, Klingler M, et al. Impact of Race, Socioeconomic Status, and Geography on Healthcare Outcomes for Children With Sickle Cell Disease in the United States: A Scoping Review. Cureus. 2024;16(3):e56089. PubMed
29.Power-Hays A, McGann PT. When Actions Speak Louder Than Words - Racism and Sickle Cell Disease. N Engl J Med. 2020;383(20):1902-1903. PubMed
30.Srikanthan S. Contested Disability: Sickle Cell Disease. Health Soc Work. 2023;48(3):209-216. PubMed
31.Phillips S, Chen Y, Masese R, et al. Perspectives of individuals with sickle cell disease on barriers to care. PLoS One. 2022;17(3):e0265342. PubMed
32.Rosanwo TO, Kean LS, Archer NM. End the pain: Start with antiracism. Am J Hematol. 2021;96(1):4-6. PubMed
33.McGill LS, Hamilton KR, Letzen JE, et al. Depressive and Insomnia Symptoms Sequentially Mediate the Association Between Racism-Based Discrimination in Healthcare Settings and Clinical Pain Among Adults With Sickle Cell Disease. J Pain. 2023;24(4):643-654. PubMed
34.Miller MM, Rumble DD, Hirsh AT, et al. Pain-Related Injustice Appraisals in Youth with Sickle Cell Disease: A Preliminary Investigation. Pain Med. 2021;22(10):2207-2217. PubMed
35.Majumder MA, Fasipe T. Translational Research and Health Equity: Gene Therapies for Sickle Cell Disease as a Case Study. Ethics Hum Res. 2024;46(3):34-39. PubMed
36.Inusa BPD, Jacob E, Dogara L, Anie KA. Racial inequalities in access to care for young people living with pain due to sickle cell disease. Lancet Child Adolesc Health. 2021;5(1):7-9. PubMed
37.Strong H, Mitchell MJ, Goldstein-Leever A, Shook L, Malik P, Crosby LE. Patient Perspectives on Gene Transfer Therapy for Sickle Cell Disease. Adv Ther. 2017;34(8):2007-2021. PubMed
38.Kratzer K, Getz LJ, Peterlini T, Masson JY, Dellaire G. Addressing the dark matter of gene therapy: technical and ethical barriers to clinical application. Hum Genet. 2022;141(6):1175-1193. PubMed
39.Doudna JA. The promise and challenge of therapeutic genome editing. Nature. 2020;578(7794):229-236. PubMed
40.Urnov FD. The Cas9 Hammer-and Sickle: A Challenge for Genome Editors. CRISPR J. 2021;4(1):6-13. PubMed
41.Pecker LH, Oteng-Ntim E, Nero A, et al. Expecting more: the case for incorporating fertility services into comprehensive sickle cell disease care. Lancet Haematol. 2023;10(3):e225-e234. PubMed
42.Lugthart S, Ginete C, Kuona P, Brito M, Inusa BPD. An update review of new therapies in sickle cell disease: the prospects for drug combinations. Expert Opin Pharmacother. 2024;25(2):157-170. PubMed
43.Platt OS. Hydroxyurea for the Treatment of Sickle Cell Anemia. N Engl J Med. 2008;358(13):1362-1369. PubMed
44.Alsultan A, Alabdulaali MK, Griffin PJ, et al. Sickle cell disease in Saudi Arabia: the phenotype in adults with the Arab-Indian haplotype is not benign. Br J Haematol. 2014;164(4):597-604. PubMed
45.Zanchetta MS, Sarpong A, Osei-Boateng J, et al. Genetic literacy and experiential knowledge on sickle cell disease among Canadian- and foreign-born male and female Anglophone and Francophone youth in Canada. Int J Adolesc Med Health. 2023;35(6):443-455. PubMed
46.Quach D, Jiao B, Basu A, et al. A landscape analysis and discussion of value of gene therapies for sickle cell disease. Expert Rev Pharmacoecon Outcomes Res. 2022;22(6):891-911. PubMed
47.Tessema FA, Sarpatwari A, Rand LZ, Kesselheim AS. High-Priced Sickle Cell Gene Therapies Threaten to Exacerbate US Health Disparities and Establish New Pricing Precedents for Molecular Medicine. J Law Med Ethics. 2022;50(2):380-384. PubMed
48.Wesevich A, Peek ME, Ratain MJ. An Ethical and Financial Obligation for Sickle Cell Disease Gene Therapy in the United States. Ann Intern Med. 2024;177(1):85-86. PubMed
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.