CADTH Reimbursement Review
Fidanacogene Elaparvovec (Beqvez)
Sponsor: Pfizer Canada ULC
Therapeutic area: Hemophilia B
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Ethics Review
Stakeholder Input
Clinical Review
Abbreviations
AAV
adeno-associated virus
AAVrh74var
variant adeno-associated virus rh74 serotype
ABRjoint
annualized bleeding rate for treated and untreated joint bleeds
ABRtotal
annualized bleeding rate for treated and untreated bleeds
ABRtreat
annualized bleeding rate for treated bleeds
AE
adverse event
AHCDC
Association of Hemophilia Clinic Directors of Canada
AIR
annualized infusion rate
ALT
alanine transaminase
AST
aspartate aminotransferase
CANHC
Canadian Association of Nurses in Hemophilia Care
CFC
coagulation factor concentrate
CHS
Canadian Hemophilia Society
CI
confidence interval
EHL
extended half-life
FIX
coagulation factor IX
FIX:C
circulating coagulation factor IX
GRADE
Grading of Recommendations Assessment, Development and Evaluation
Haem-A-QoL
Haemophilia Quality of Life Questionnaire for Adults
HAL
Hemophilia Activities List
HJHS
Hemophilia Joint Health Score
HRQoL
health-related quality of life
nAb
neutralizing antibody
PROBE
Patient Reported Outcomes Burdens and Experiences
rFIX
recombinant coagulation factor IX
SAE
serious adverse event
SD
standard deviation
SHL
standard half-life
SLR
systematic literature review
TEAE
treatment-emergent adverse event
TESAE
treatment-emergent serious adverse event
ULN
upper limit of normal
Vg
vector genome
WFH
World Federation of Hemophilia
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Fidanacogene elaparvovec (Beqvez), concentrate solution for infusion, 1 × 1013 vg/mL, IV infusion |
Sponsor | Pfizer Canada ULC |
Indication | Fidanacogene elaparvovec is an AAV vector–based gene therapy indicated for the treatment of adults (aged 18 years or older) with moderately severe to severe hemophilia B (congenital factor IX deficiency) who test negative for neutralizing antibodies to variant AAV serotype rh74 |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Priority review |
NOC date | December 27, 2023 |
Recommended dose | 5 × 1011 vg/kg of body weight, administered as a single dose IV infusion |
AAV = adeno-associated virus; NOC = Notice of Compliance; vg = vector genome.
Sources: Sponsor’s Summary of Clinical Evidence1 and draft product monograph.2
Introduction
Hemophilia is a serious and lifelong genetic disorder linked to the X chromosome that leaves patients vulnerable to blood loss and organ damage due to impaired functioning of the coagulation cascade.3,4 Hemophilia B is the second most common type of hemophilia (after hemophilia A) and is characterized by an absence or shortage of coagulation factor IX (FIX) resulting from a mutation in the F9 gene.3,4 Moderate and severe hemophilia B cases are defined by the World Federation of Hemophilia (WFH) as those with 1% to 5% and less than 1% of normal enzymatic FIX activity, respectively.5 However, according to the clinical experts consulted by CADTH for this review, severity in clinical practice is defined by the patients' phenotype (i.e., tendency to bleed) and not simply their coagulation factor activity levels; the decision to initiate prophylaxis with clotting factor concentrates takes into the account both their clinical phenotype and factor activity levels, as well as lifestyle and professional activities. Individuals with moderately severe to severe hemophilia frequently experience bleeding and recurrent spontaneous bleeding into muscle, soft tissue, and joints (hemarthroses) starting from infancy and continuing through adulthood.4,6 Hemarthrosis is the most common manifestation of moderate and severe hemophilia B.4,5 As of 2021, there were 704 patients with hemophilia B (with recorded severity) in Canada, 535 of whom were adult males.
The treatment goal for hemophilia, as outlined by WFH guidelines,5 is to reduce or prevent bleeding while allowing patients to lead active lives and achieve a quality of life comparable to that of individuals not affected by the condition. Current management strategies for hemophilia B include on-demand treatment to stop bleeds as they occur and/or routine prophylaxis therapy to prevent bleeding, both involving the administration of exogenous FIX coagulation factor concentrates (CFCs) to treat the FIX deficiency.5 According to the clinical experts consulted by CADTH, routine FIX prophylaxis involving lifelong regular IV administration of FIX CFCs is currently the standard of care for patients with hemophilia B in Canada. Recombinant coagulation factor IX (rFIX) products are the mainstay of prophylactic treatments for hemophilia B.5,7,8 The clinical experts consulted by CADTH noted that the frequency of FIX injections varies from individual to individual depending on the type of FIX concentrate and the pharmacokinetics of individual patients. According to the clinical experts consulted by CADTH, plasma-derived FIX, such as factor IX concentrate (human) (Immunine), is also available in Canada but with very limited use.
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of fidanacogene elaparvovec (Beqvez) (concentrate solution for infusion, 1 × 1013 vector genomes [vg] per millilitre, IV infusion) for the treatment of moderately severe to severe hemophilia B in patients aged 18 years or older.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient and clinician groups who responded to CADTH’s call for input and from clinical experts consulted by CADTH for the purpose of this review.
Patient Input
The Canadian Hemophilia Society (CHS) provided input for the review of fidanacogene elaparvovec for the treatment of moderately severe to severe hemophilia B patients aged 18 years or older. Patient input was gathered from an online survey conducted between July 10 and July 31, 2023. In total 17 responses were gathered by the CHS. All respondents were affected by severe or moderately severe hemophilia B without inhibitors. In addition, in September 2022, a CHS online survey of Canadians with severe hemophilia A and B received 39 responses, of which 31 were from patients with hemophilia A, 7 were from those with hemophilia B, and 1 was from a patient whose hemophilia was not specified.
Joint damage, primarily to knees, ankles and elbows, caused by repeated internal hemarthroses was reported to be the primary physical health impact of hemophilia B. Regarding currently available treatments, 4 patients in the 2023 CHS survey reported being very satisfied, 7 were satisfied, 5 were neither satisfied nor dissatisfied, and 1 was very dissatisfied. Patients from this survey noted that treatments greatly complicate their everyday life, travel, and leisure activities. They also mentioned the infusion process was difficult due to vein visibility, poor vein issues, and side effects. Patients also reported facing socioeconomic problems due to the need for regular visits, missing work due to visits, travel and insurance issues, and accessing treatment.
When patients from the 2023 CHS survey were asked how gene therapy could potentially change their lives, all patients provided positive feedback. Patients hoped gene therapy would lead to fewer FIX infusions, minimal needle injections, less stress, less bleeding, and fewer restrictions on activities, and make it easier to travel. In addition, 63% of the respondents from the 2022 survey indicated they expected gene therapy to be effective in preventing bleeding for at least 10 years. The 2022 survey asked if people would be willing to receive gene therapy knowing that that there would be frequent blood draws in the weeks and months following administration, and they would need to be followed up in a registry for 10 to 20 years. In response, 66% answered yes, 10% answered no, and 24% indicated they did not know.
The CHS mentioned that a small number (likely close to 5) patients in Canada have undergone gene therapy for hemophilia B, but it had no information about their experience outside the preliminary data from the trials.
Clinician Input
Input From Clinical Experts Consulted by CADTH
The clinical experts consulted by CADTH noted that FIX prophylaxis requires frequent IV injections performed by the patients themselves, which poses a heavy burden for patients with hemophilia B and significantly undermines their ability to live normal lives. The clinical experts noted that poor adherence to FIX prophylaxis may result in reduced effectiveness and an increased risk of bleeding, and even patients who execute prophylaxis on the prescribed schedule (i.e., are adherent) can experience breakthrough bleeds, particularly in the days before the next infusion. According to the clinical experts consulted by CADTH, the key advantage of fidanacogene elaparvovec over an exogenous FIX prophylaxis regimen, if effective, would be avoiding fluctuations in FIX levels and eliminating the need for repeated CFC infusions. The clinical experts consulted by CADTH noted that fidanacogene elaparvovec could be a curative treatment if a steady high level of FIX is expressed and efficacy is maintained over the long-term. The clinical experts noted that it remains unclear whether the use of fidanacogene elaparvovec will lead to a shift in the treatment paradigm.
The clinical experts noted that all patients with hemophilia B who have a clinically severe phenotype regardless of FIX level are likely to benefit from treatment with fidanacogene elaparvovec in terms of reductions in burden of care, pain, and pain interference as well as improvement in mobility and quality of life. The clinical experts noted that those who would likely benefit the most from the treatment of fidanacogene elaparvovec would be patients without pre-existing joint damage due to hemophilia B, as well as younger patients who are usually more active. The clinical experts consulted by CADTH noted that identifying the patients best suited for treatment should involve clinical assessments and shared decision-making with patients. Based on the study design of the pivotal BeneGene-2 trial, the clinical experts indicated that testing for neutralizing antibodies (nAbs) against variant adeno-associated virus serotype rh74 (AAVrh74var) should be mandatory for patients to receive fidanacogene elaparvovec. The clinical experts noted that patients least suitable for fidanacogene elaparvovec include those with pre-existing nAbs against adeno-associated virus (AAV) and those who conclude that the benefit does not outweigh the risk associated with fidanacogene elaparvovec gene therapy, given that its long-term efficacy and safety remain unclear. In addition, some patients may not want to change their current treatment.
According to the clinical experts consulted by CADTH, the most important assessment of treatment response requires monitoring patients’ bleeding to determine whether fidanacogene elaparvovec prevents bleeding events and allows patients to live the lifestyle they want without concern about the risk of bleeding. The clinical experts agreed that the length of follow-up for hepatic function and FIX activity levels after fidanacogene elaparvovec infusion should be lifelong. The clinical experts noted that monitoring after fidanacogene elaparvovec infusion will be more frequent in the short-term and less frequent over time in the long-term. The clinical experts consulted by CADTH noted that it is reasonable to monitor FIX activity levels and liver function twice a week at the early stage postinfusion, although the production of FIX is unlikely to occur immediately postinfusion. The clinical experts noted that monitoring changes in the Hemophilia Joint Health Score (HJHS) as well as in quality-of-life–related end points following fidanacogene elaparvovec infusion (e.g., improvement in activities associated with daily living, physical activity, and functioning; decrease in development of disability; and improvement in psychosocial health and functioning) are also important. The clinical experts consulted by CADTH noted that treatment failure should be determined by the treating clinician on a case-by-case basis. The clinical experts consulted by CADTH noted that, if treatment with fidanacogene elaparvovec fails, patients may not be eligible for another gene therapy based on AAV vectors because they may present with cross-reactivity against most AAV vectors.
The clinical experts consulted by CADTH noted that fidanacogene elaparvovec should be prescribed based on the judgment of a multidisciplinary team at a comprehensive hemophilia treatment centre. The team may consist of specialists such as a hematologist with experience in treating hemophilia patients, a physiotherapist to assess joint function, a hepatologist for liver-related issues, a pharmacist, and an HIV specialist if the patient is HIV-positive. The clinical experts noted that administration of fidanacogene elaparvovec takes place on an outpatient basis, as do postinfusion follow-ups for most of the patients.
Clinician Group Input
Nine clinicians from the Association of Hemophilia Clinic Directors of Canada (AHCDC) and 3 nurses from Canadian Association of Nurses in Hemophilia Care (CANHC) provided input. Both the AHCDC and CANHC emphasized that currently available treatments in Canada do not modify or alter the underlying disease process, making persons with hemophilia B dependent for life on regular IV infusions of FIX to prevent and treat bleeding. In addition, the AHCDC noted that the frequent venipunctures required for prophylactic CFC replacement can pose challenges for patients with poor venous access. The group emphasized that all these factors lead to the need for persons with hemophilia B and a severe bleeding phenotype to restore coagulation factor levels to clinically effective levels without the need for frequent venipunctures on a regular basis throughout their lifespan. The AHCDC also mentioned the variability of the efficacy of prophylaxis with CFCs across individuals, which makes some patients susceptible to breakthrough bleeding into joints and muscles.
Both the AHCDC and the CANHC noted that fidanacogene elaparvovec would provide a 1-time treatment leading to sustained FIX production, which would address the underlying disease process and natural history, rather than provide symptomatic management. This would represent a paradigm shift in the treatment of hemophilia B. The AHCDC indicated that patients eligible for gene therapy include adults with hemophilia B and a clinically severe bleeding phenotype requiring prophylaxis, no history of inhibitory antibodies, no significant comorbidities, and no pre-existing anti-AAV nAbs.
Drug Program Input
Input was obtained from the drug programs that participate in the CADTH reimbursement review process. The following were identified as key factors that could affect the implementation of a CADTH recommendation for fidanacogene elaparvovec:
relevant comparators
consideration for initiation of therapy
consideration of continuation or renewal of therapy
consideration of discontinuation of therapy
consideration for prescribing of therapy
generalizability
care provision issues
system and economic issues.
Clinical Evidence
Systematic Review
Description of Studies
One phase III, single-arm, open-label clinical trial (BeneGene-2, N = 45) was included in the systematic literature review (SLR) conducted by the sponsor. The BeneGene-2 trial involved 45 participants from 27 centres across 13 countries and territories around the globe, including 3 centres in Canada.1 The BeneGene-2 trial enrolled adult male patients who had moderately severe to severe hemophilia B (defined as circulating coagulation factor IX [FIX:C] ≤ 2%). Patients were excluded if their anti-AAVrh74var nAb titre was equal to or greater than 1:1 or if they had a prior history of FIX inhibitors (i.e., nAbs against FIX) or a positive FIX inhibitor test result equal to greater than 0.6 Bethesda units.
The primary objective of the BeneGene-2 trial was to determine the noninferiority of fidanacogene elaparvovec relative to the standard of care in Canada (FIX prophylaxis), as measured by the annualized bleed rate for treated and untreated bleeds (ABRtotal) at week 12 to month 15 (denoted as year 1) following fidanacogene elaparvovec infusion. Other efficacy and safety end points were also examined in the BeneGene-2 trial, including number of patients without bleeds; annualized bleeding rate for treated bleeds (ABRtreat); annualized bleeding rate for treated and untreated joint bleeds (ABRjoint); annualized infusion rate (AIR); annualized FIX consumption; HJHS, Hemophilia Quality of Life Questionnaire for Adults (Haem-A-QoL), and Hemophilia Activities List (HAL) scores; withdrawals due to adverse events (AEs); treatment-emergent adverse events (TEAEs); treatment-emergent serious adverse events (TESAEs); deaths; and notable harms (e.g., increased alanine transaminase [ALT], abnormal hepatic function, increased aspartate aminotransferase [AST], increased hepatic enzyme, and increased transaminases). Tests of both noninferiority and superiority were also conducted, and a gatekeeping process was applied to control for multiplicity of testing multiple end points. For efficacy outcomes such as ABRtotal, ABRtreat, ABRjoint, AIR, and annualized FIX consumption, the 45 participants in the pivotal BeneGene-2 trial served as their own controls, using data collected from when they were on FIX prophylaxis during an open-label, noninvestigational, prospective, lead-in study (BeneGene-1, N = 102) for comparison.
Patients in the BeneGene-2 trial had a median age of 29 years, ranging from 18 to 62. The majority of patients were white (73.3%), followed by Asian (15.6%) as well as Black or African American (2.2%).
The BeneGene-2 trial is ongoing and expected to be completed in December 2029. Data gathered before the data cut-off date (November 16, 2022) were used to support the sponsor’s present submission to CADTH.9,10
Efficacy Results
As of the data cut-off date, the mean duration of follow-up in the BeneGene-2 trial was |||| ||||| (standard deviation [SD] = |||||) with a median of |||| ||||| ||||| ||||. For the lead-in BeneGene-1, the mean duration of follow-up in the lead-in BeneGene-1 trial was |||| ||||| ||||||| with a median of |||| ||||| ||||| |||||
Bleeding Outcomes
The model estimate of the difference in the ABRtotal between patients treated with fidanacogene elaparvovec during the BeneGene-2 trial versus the same patients treated with routine FIX prophylaxis during the lead-in BeneGene-1 study was −3.13 (95% CI, −5.44 to −0.81) at year 1 postinfusion, favouring fidanacogene elaparvovec. The difference in the ABRtotal from week 12 to the data cut-off date (overall) was ||||| |||||| || ||||||, in favour of fidanacogene elaparvovec. The analysis found that 64.4% (29 of 45) of the patients treated with fidanacogene elaparvovec and 28.9% (13 of 45) of the patients treated with routine FIX prophylaxis had no untreated and treated bleeds at year 1 postinfusion. From week 12 to the data cut-off date postinfusion, ||||| ||||||| of the patients treated with fidanacogene elaparvovec and ||||| ||||||| of the patients treated with routine FIX prophylaxis had no bleeds.
The estimated mean differences in the ABRtreat between patients treated with fidanacogene elaparvovec during the BeneGene-2 trial and the same patients treated with routine FIX prophylaxis during the lead-in BeneGene-1 trial were −2.62 (95% CI, −4.27 to −0.96) at year 1 postinfusion and ||||| |||||| || |||||| from week 12 to the data cut-off date, ||| ||||||||| fidanacogene elaparvovec. No treated bleeds at year 1 postinfusion were reported in 73.3% (33 of 45) of the patients treated with fidanacogene elaparvovec and 35.6% (16 of 45) of the patients treated with routine FIX prophylaxis. ||||| ||||||| of the patients treated with fidanacogene elaparvovec and ||||| ||||||| of the patients treated with routine FIX prophylaxis had no treated bleeds at year 1 postinfusion.
The estimated difference in the ABRjoint between patients treated with fidanacogene elaparvovec and the same patients treated with routine FIX prophylaxis during the lead-in BeneGene-1 trial was ||||| |||||| || |||||| at year 1 postinfusion, in favour of fidanacogene elaparvovec. From week 12 to the data cut-off date, the difference was ||||| |||||| || ||||||, |||| ||||||||| fidanacogene elaparvovec. No joint bleeds at year 1 postinfusion were reported in 68.9% (31 of 45) of the patients treated with fidanacogene elaparvovec and 44.4% (20 of 45) of the patients treated with routine FIX prophylaxis ||||| ||||||| of the patients treated with fidanacogene elaparvovec infusion and ||||| ||||||| of the patients treated with routine FIX prophylaxis had no joint bleeds.
Use of FIX Post–Fidanacogene Elaparvovec Infusion
The differences in the AIR between patients treated with fidanacogene elaparvovec during the BeneGene-2 trial and the same patients treated with routine FIX prophylaxis during the lead-in BeneGene-1 study were −54.37 (95% CI, −63.64 to −45.10) at year 1 postinfusion and |||||| ||||||| || ||||||| from week 12 to the data cut-off date, ||| ||||||||| fidanacogene elaparvovec. From week 12 to the data cut-off date, the difference in the annualized FIX consumption between patients treated with fidanacogene elaparvovec and the same patients treated with routine FIX prophylaxis during the lead-in BeneGene-1 study was |||||||| IU/kg ||||||||| || ||||||||), ||||||||| fidanacogene elaparvovec.
Patient-Reported Outcomes
Among patients treated with fidanacogene elaparvovec, change from baseline at week 52 or week 104 postinfusion |||||| |||||||||||| in the HJHS total score, Haem-A-QoL physical health domain, Haem-A-QoL total score, HAL complex lower extremity activities score, and HAL total score.
Harms Results
Treatment-emergent AEs were reported in 84.4% (38/45) of the safety population of the BeneGene-2 trial. The most commonly reported TEAE was increased ALT (26.7%), followed by nasopharyngitis (17.8%) and arthralgia (17.8%). Serious adverse events (SAEs) were reported in 7 patients (15.6%) in the BeneGene-2 trial. The most common SAE was anemia (4.4%). No patients in the BeneGene-2 trial discontinued the study due to AEs or died as of the data cut-off date of November 16, 2022.
In terms of notable harms, increased ALT and abnormal hepatic function occurred in 26.7% (12 of 45) and 13.3% (6 of 45) of the patients in the BeneGene-2 trial, respectively. Increased AST, increased hepatic enzyme, and increased transaminases occurred in 6.7% (3 of 45) of the patients in the BeneGene-2 trial.
Critical Appraisal
The only eligible study identified by the SLR, BeneGene-2, was a phase III, single-arm, open-label clinical trial that enrolled 45 patients. Although interpretation of the study results is limited due to the nonrandomized, open-label, single-arm design, the clinical experts consulted by CADTH for this review considered the discontinuity design appropriate for clinical studies of hemophilia B treatment. Participants in the BeneGene-2 trial were requested to suspend their FIX prophylaxis regimen following fidanacogene elaparvovec infusion but were allowed to resume FIX prophylaxis based on certain conditions. These conditions were considered generally appropriate by the clinical experts consulted by CADTH. Moreover, resumption of a FIX prophylaxis regimen postinfusion in the BeneGene-2 trial was not expected to modify treatment effects. This was supported by the “jump to reference” sensitivity analysis, in which participants who resumed FIX prophylaxis regimens following fidanacogene elaparvovec infusion were excluded and the difference in the ABRtotal was similar to that found in the primary analysis. The patients included in the pivotal BeneGene-2 trial were selected from the lead-in BeneGene-1 study. Of the 102 patients in the BeneGene-1 trial, only 45 were enrolled in the pivotal BeneGene-2 trial. CADTH determined that the potential selection bias due to a large number of patients being left out was not a serious concern because the data provided by the sponsor showed that outcomes reported in the majority of the patients who were left out (i.e., the 40 patients who were not enrolled in the BeneGene-2 trial because they had not completed the BeneGene-1 trial), such as the ABRtotal, ABRtreat, and AIR, were similar to the those at year 1 postinfusion among the 45 patients enrolled in the BeneGene-2 trial. The documentation of bleeding events in the BeneGene-2 trial relied on the use of an electronic diary by patients, and the determination of whether a bleed needed to be treated relied on physicians’ clinical decisions shared with patients. Despite the risk of bias likely being low, and based on information provided by the sponsor, CADTH determined that the potential risk of bias that may lead to exaggeration of treatment effects of fidanacogene elaparvovec (i.e., annualized bleeding rate [ABR] outcomes) could not be ruled out. Furthermore, due to a lack of comparative data for some end points and the open-label design, reliable assessments of patient-reported outcomes (e.g., health-related quality of life [HRQoL] end points) could not be made. CADTH concluded that the gatekeeping process used to control for multiplicity when testing multiple end points was appropriate. However, some concerns regarding the assumptions of the statistical models used in the BeneGene-2 trial were raised, which may make interpretation of the magnitude of the effect estimates of fidanacogene elaparvovec compared to FIX prophylaxis challenging.
CADTH identified several considerations related to the generalizability of the BeneGene-2 trial. First and most importantly, given the novelty of gene therapy as well as patients’ and clinicians’ expectation of long-lasting effects, it may not be possible to use evidence from the current follow-up period (|||| ||| |||||) in the BeneGene-2 trial to generalize about long-term efficacy and safety. Second, the indication includes patients with “moderately severe to severe” hemophilia B, and defining this description has implementation considerations. Whereas the BeneGene-2 trial defined “moderately severe to severe” as a FIX:C level less than or equal to 2%, the clinical experts consulted by CADTH noted that severity in clinical practice is defined by the patients' phenotype and not simply their factor activity levels. According to the clinical experts consulted by CADTH, hemophilia in some patients will be considered moderately severe to severe due to clinical symptoms despite a FIX level greater than 2%. Furthermore, the BeneGene-2 trial included only patients with an anti-AAVrh74var nAb titre of less than 1:1. According to the clinical experts consulted by CADTH, the efficacy of fidanacogene elaparvovec in patients with an anti-AAVrh74var nAb titre equal to or greater than 1:1 remains uncertain. Nonetheless, the clinical experts consulted by CADTH agreed that, if fidanacogene elaparvovec were to be publicly reimbursed, selection of eligible patients should follow the threshold used in the BeneGene-2 study.
GRADE Summary of Findings and Certainty of the Evidence
The selection of outcomes for Grading of Recommendations Assessment, Development and Evaluation (GRADE) was based on the sponsor’s Summary of Clinical Evidence, consultation with clinical experts, and input received from patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members: ABRtotal, ABRtreat, ABRjoint, percentage of patients without bleeds, AIR, annualized FIX consumption, HJHS, Haem-A-QoL (physical health and total scores), HAL (complex lower extremity activities and total scores), and harms. According to GRADE guidance, nonrandomized comparative evidence starts at low certainty and noncomparative evidence starts at very low certainty. The GRADE summary of findings is presented in Table 2 and Table 3.
Table 2: Summary of Findings for Fidanacogene Elaparvovec for Patients with Hemophilia B (Outcomes With Comparative Data)
Outcome and follow-up | Patients, N (studies) | Effect | Certainty | What happens |
|---|---|---|---|---|
Treated and untreated bleeds | ||||
ABRtotal Follow-up:
| 45 (1 single-arm study, with intrapatient comparison) | Year 1 postinfusion Number (%) of patients without any treated and untreated bleeds:
Mean ABRtotal estimate (95% CI):
Difference in ABRtotal, negative binomial estimate (95% CI):
Overall Number (%) of patients without any treated and untreated bleeds:
Mean ABRtotal estimate (95% CI):
Difference in ABRtotal, negative binomial estimate (95% CI):
| Lowa | Fidanacogene elaparvovec may result in a decrease in the ABR for treated and untreated bleeds when compared with FIX prophylaxis |
Treated bleeds | ||||
ABRtreat Follow-up: • Year 1 postinfusion of fidanacogene elaparvovec • Overall | 45 (1 single-arm study, with intrapatient comparison) | Year 1 postinfusion Number (%) of patients without any treated bleeds:
Mean ABRtreat estimate (95% CI)
Difference in ABRtreat, negative binomial estimate (95% CI)
Overall Number (%) of patients without any treated bleeds:
Mean ABRtreat estimate (95% CI)
Difference in ABRtreat, negative binomial estimate (95% CI):
| Lowa | Fidanacogene elaparvovec may result in a decrease in the ABR for treated bleeds when compared with FIX prophylaxis |
Treated and untreated joint bleeds | ||||
ABRjoint Follow-up:
| 45 (1 single-arm study, with intrapatient comparison) | Year 1 postinfusion Number (%) of patients without any treated or untreated joint bleeds:
Mean ABRjoint estimate (95% CI):
Difference in ABRjoint, negative binomial estimate (95% CI):
Overall Number (%) of patients without any treated or untreated joint bleeds:
Mean ABRjoint estimate (95% CI):
Difference in ABRjoint, negative binomial estimate (95% CI):
| Lowa | Fidanacogene elaparvovec may result in a decrease in the ABR for treated and untreated joint bleeds when compared with FIX prophylaxis |
Use of FIX following fidanacogene elaparvovec infusion | ||||
AIR Follow-up:
| 45 (1 single-arm study, with intrapatient comparison) | Year 1 postinfusion Mean AIR (SD):
Difference in AIR, estimate from paired t test (95% CI):
Overall Mean AIR (SD):
Difference in AIR, estimate from paired t test (95% CI):
| Lowa | Fidanacogene elaparvovec may result in a decrease in the AIR when compared with FIX prophylaxis |
Annualized FIX consumption (IU/kg) Follow-up:
| 45 (1 single-arm study, with intrapatient comparison) | Overall Mean annualized FIX consumption (SD):
Difference in annualized FIX consumption, estimate from paired t test (95% CI):
| Lowa | Fidanacogene elaparvovec may result in a decrease in total FIX consumption when compared with FIX prophylaxis |
ABR = annualized bleeding rate; ABRjoint = annualized bleeding rate for treated and untreated joint bleeds; ABRtotal = annualized bleeding rate for treated and untreated bleeds; ABRtreat = annualized bleeding rate for treated bleeds; AIR = annualized infusion rate; CI = confidence interval; FIX = coagulation factor IX; SD = standard deviation.
Note: Year 1 refers to the period between week 12 and month 15 following fidanacogene elaparvovec infusion. Overall refers to the period from week 12 following fidanacogene elaparvovec infusion to the data cut-off date of November 16, 2022. As of the data cut-off date, the mean duration of follow-up in the pivotal BeneGene-2 trial was |||| ||||| ||||||| with a median of |||| ||||| ||||| ||||. Week 52 and week 104’s baseline was defined as the last nonmissing measurement before the dosing date (day 1) in the pivotal study. The mean duration of follow-up in the lead-in BeneGene-1 study was |||| ||||| ||||||| with a median of |||| ||||| ||||| ||||.
aThe risk of bias was not rated down. According to the clinical experts consulted by CADTH, although not optimal, the study design adopted by the BeneGene-2 trial was considered to be of sufficiently low risk of confounding and sampling bias to not introduce serious risk of bias. Although there were differences between patients in the indication and patients in pivotal trial (e.g., definition of moderately severe to severe disease), the clinical experts consulted by CADTH did not consider them sufficient to result in important differences in the observed effect. Imprecision was not rated down as the improvement was considered clinically meaningful by the clinical experts consulted by CADTH.
Source: BeneGene-2 Clinical Study Report.9
Table 3: Summary of Findings for Fidanacogene Elaparvovec for Patients With Hemophilia B (Outcomes Without Comparative Data)
Outcome and follow-up | Patients, N (studies) | Effect | Certainty | What happens |
|---|---|---|---|---|
Joint health | ||||
HJHS (0 [best] to 124 [worst]) Follow-up:
| || (week 52) || (week 104) (1 single-arm study) | Week 52 postinfusion Mean HJHS score (SD):
Change from baseline, estimate from paired t test (95% CI):
Week 104 postinfusion Mean HJHS score (SD):
Change from baseline, estimate from paired t test (95% CI):
| Very lowa | The evidence is uncertain about the effect of fidanacogene elaparvovec on the HJHS |
HRQoL | ||||
Haem-A-QoL Physical health domain (5 [best] to 25 [worst]) Total score (0 [best] to 100 [worst]) Follow-up:
| 37 (physical health domain, week 52) || (physical health domain, week 104) 37 (total score, week 52) || (total score, week 104) (1 single-arm study) | Physical health domain, seek 52 postinfusion Mean Haem-A-QoL physical health score (SD):
Change from baseline, estimate from paired t test (95% CI):
Physical health domain, seek 104 postinfusion Mean Haem-A-QoL physical health score (SD):
Change from baseline, estimate from paired t test (95% CI):
Total score, week 52 postinfusion Mean Haem-A-QoL total score (SD):
Change from baseline, estimate from paired t test (95% CI):
Total score, week 104 postinfusion Mean Haem-A-QoL total score (SD):
Change from baseline, estimate from paired t test (95% CI):
| Very lowb, c, d | The evidence is uncertain about the effect of fidanacogene elaparvovec on the Haem-A-QoL physical health score or total score |
HAL Complex lower extremity activities (9 [worst] to 54 [best]) Total score (0 [worst] to 100 [best]) Follow-up:
| 37 (complex lower extremity activities, week 52) || (complex lower extremity activities, week 104) 37 (total score, week 52) || (total score, week 104) (1 single-arm study) | Complex lower extremity activities, week 52 postinfusion Mean HAL complex lower extremity activities score (SD):
Change from baseline, estimate from paired t test (95% CI):
Complex lower extremity activities, week 104 postinfusion Mean HAL complex lower extremity activities score (SD):
Change from baseline, estimate from paired t test (95% CI): ||||| ||||| || ||||||| Total score, week 52 postinfusion Mean HAL total score (SD):
Change from baseline, estimate from paired t test (95% CI):
Total score, week 104 postinfusion Mean HAL total score (SD):
Change from baseline, estimate from paired t test (95% CI):
| Very lowb,e | The evidence is uncertain about the effect of fidanacogene elaparvovec on the HAL complex lower extremity activities score or total score |
Harms | ||||
TESAEs Mortality Increased ALT Abnormal hepatic function Increased AST Increased hepatic enzyme Increased transaminases Follow-up:
| 45 (1 single-arm study) | TESAEs: 156 per 1,000 (most common: anemia [44 per 1,000]) Mortaliy: 0 ALT increased: 267 per 1,000 Abnormal hepatic function: 133 per 1000 AST increased: 67 per 1,000 Hepatic enzyme increased: 67 per 1,000 Transaminases increased: 67 per 1,000 | Very lowf | The evidence is uncertain about the effect of fidanacogene elaparvovec on TESAEs, mortality, increased ALT, abnormal hepatic function, increased AST, increased hepatic enzyme, increased transaminases |
ALT = alanine transaminase; AST = aspartate transaminase; CI = confidence interval; Haem-A-QoL = Hemophilia Quality of Life Questionnaire for Adults; HAL = Hemophilia Activities List; HJHS = Hemophilia Joint Health Score; HRQoL = health-related quality of life; MID = minimal important difference; SD = standard deviation; TESAE = treatment-emergent serious adverse event.
Note: Year 1 refers to the period between week 12 and month 15 following fidanacogene elaparvovec infusion. Overall refers to the period from week 12 following fidanacogene elaparvovec infusion to the data cut-off date of November 16, 2022. As of the data cut-off date, the mean duration of follow-up in the pivotal BeneGene-2 trial was |||| ||||| ||||||| with a median of |||| ||||| ||||| ||||. Week 52 and week 104’s baseline was defined as the last nonmissing measurement before the dosing date (day 1) in the pivotal study. The mean duration of follow-up in the lead-in the BeneGene-1 trial was |||| ||||| ||||||| with a median of |||| ||||| ||||| ||||.
aIn absence of a comparator arm, certainty of evidence started at very low. Although there were differences between patients in the indication and patients in the pivotal trial (e.g., definition of moderately severe to severe disease), the clinical experts consulted by CADTH did not consider them serious enough to result in important differences in the observed effect. No MID was identified. Imprecision was not rated down as the improvement was considered clinically meaningful by the clinical experts consulted by CADTH.
bIn absence of a comparator arm, certainty of evidence started at very low. Rated down 1 level for risk of bias due to potential for bias arising from the open-label nature of the study and the subjective nature of the outcome. Indirectness was not rated down. Although the Patient Reported Outcomes Burdens and Experiences instrument is more commonly used in Canada, this was not considered a serious generalizability issue by the clinical experts consulted by CADTH because all these HRQoL measurement instruments are closely aligned.
CRated down 1 level for imprecision. The meaningful within-patient change identified in the literature was 10.0 for Haem-A-QoL physical health domain, ||||| ||| ||||||| || ||| ||| ||| ||| ||||| ||| || ||| ||| || ||| ||| ||||| ||| || |||||| ||||.
dRated down 1 level for imprecision due to the small number of patients involved. The meaningful within-patient change identified in the literature was 7.1 for Haem-A-QoL total score, ||||| ||| ||| ||||||| || ||| ||| ||.
eRated down 1 level for imprecision. No MID was available, and the upper end of the 95% CI did not cross the no-effect line.
fIn absence of a comparator arm, certainty of evidence started at very low. Although there were differences between patients in the indication and patients in pivotal trial (e.g., definition of moderately severe to severe disease), the clinical experts consulted by CADTH did not consider them sufficient to result in important differences in the observed effect. Rated down 1 level for imprecision due to the small sample size, although the safety profile was considered acceptable by clinical experts consulted by CADTH.
Source: BeneGene-2 Clinical Study Report.9
Studies Addressing Gaps in the Evidence From the Systematic Review
The sponsor submitted 2 additional studies to address gaps in the pivotal trial evidence. Study C0371005 was submitted to address a gap in knowledge of the safety and kinetics of fidanacogene elaparvovec. Study C0371003 was a corresponding extension study submitted to address a gap in knowledge of the longer-term efficacy and safety of fidanacogene elaparvovec. Patients who completed Study C0371005 were encouraged to enrol in Study C0371003 to evaluate fidanacogene elaparvovec for up to an additional 5-year, longer-term follow-up.
Study C0371005
Description of Study
Study C0371005 (N = 15) was a phase I and IIa, open-label, nonrandomized, dose-escalation, multicentre study. The objective was to evaluate the safety, tolerability, and kinetics of a single IV infusion of fidanacogene elaparvovec (dose of 5 × 1011 vg/kg) in hemophilia B participants with endogenous FIX levels of less than of equal to 2%. Patients were followed for 52 weeks. No formal efficacy evaluations were performed. All efficacy analyses were exploratory in nature. The safety analysis set included 15 participants who received the infusion.
All 15 participants enrolled were male with a mean age of 38.6 years, ranging from 18 to 61 years. The majority of participants were white (80.0%). The majority of participants (80.0%) had no family history of FIX inhibitors and 66.7% had hemophilia B with a FIX:C level of less than 1%.
Efficacy Results
Bleeding outcomes: Among 15 treated participants, 12 participants (80.0%) did not experience any on-study bleeds. No traumatic bleeds were observed during the study, and all 3 participants who experienced bleeding episodes had spontaneous bleeds. The median ABR during the 52-week period preceding fidanacogene elaparvovec infusion (historical) was 4.00, ranging from 0.0 to 48.0. The median ABR decreased to 0.00 (range = 0.0 to 4.0) during the 52-week period following fidanacogene elaparvovec infusion (on study). The mean ABR decreased from 8.87 (SD = 14.040) to 0.40 (SD = 1.060).
The overall mean annualized FIX production consumption was |||| |||||||||| IU in all 15 participants, with a mean of |||| |||||||||| IU in the 11 participants previously on prophylaxis treatment and |||| |||||||| IU in the 4 participants previously receiving treatment on demand.
During the 52-week period preceding screening, the mean number of target joint bleeds was |||| ||||||| in 5 participants (4 previously on prophylactic treatment and 1 previously receiving treatment on demand). The mean number of target joint bleeds decreased from |||| ||||||| in 4 participants to ||| |||||| occurring in 2 participants previously on prophylactic treatment from 52 weeks preceding screening to the end of study.
Patient-Reported Outcomes: As the HJHS, HAL, and McGill pain questionnaire assessments were added in a protocol amendment, only the final | participants enrolled were evaluated for these assessments.
Regarding HJHS, || || participants had assessments done at baseline and end of study. In general, a |||||||| ||| |||||||| || ||| ||||||||| |||||||||||| |||| ||||| ||||| |||||| |||||| |||||| |||| |||||| ||||| |||||| |||| |||||| ||||| |||| ||| |||| ||||| |||| ||| |||| |||||| ||| ||| ||||| ||||| || |||| ||||| ||||||| ||||| ||||||| ||| ||||| ||||||.
A |||||||| || ||| ||||| ||| ||||| ||| ||| |||||| |||| |||| ||| |||||||| || |||| || participants who had assessments done at baseline and end of study. A |||||||| was also observed in |||||||||| ||||||| ||||| ||||| |||| ||||, as well as in the |||||| ||||| |||||| |||||| ||||||| ||||| |||||.
Harms Results
Fourteen out of 15 participants (93.3%) had at least 1 reported TEAE. A total of 81 TEAEs were reported in the study. The most commonly reported TEAEs were in the system organ class of infections and infestation (8 participants, or 53.3%), gastrointestinal disorders (7 participants, or 46.7%) and musculoskeletal and connective disorders (6 participants, or 40.0%). The majority of TEAEs (53 out of 81, or 65.4%) were mild in severity, and the other 28 (34.6%) were moderate in severity. No study drug discontinuation, study discontinuation, SAEs, or deaths were reported in the study.
Study C0371003
Description of Study
Study C0371003 (N = 17) is a phase IIa, open-label, nonrandomized, longer-term follow-up study designed to evaluate the safety and efficacy of previously administered fidanacogene elaparvovec at a dose of 5 × 1011 vg/kg for up to 6 years. Participants enrolled in this study either had been dosed with fidanacogene elaparvovec in Study C0371005 (summarized previously; N = 14) or received fidanacogene elaparvovec in a dose-escalation substudy (N = |) within this study. Results presented in this report are for the cohort of 14 patients from Study C0371005 who entered Study C0371003. The dose-escalation substudy is not covered in this report due to the small number of participants and the fact that the dose of fidanacogene elaparvovec used did not align with the recommended dose (patients received a dose of |||||| |||||| |||| || |||||||| |||).
The primary outcome measures for Study C0371003 were related to safety and immunogenicity, while secondary measures were related to efficacy. As the primary objective of this study was safety, no hypothesis testing was planned, and all summaries are descriptive.
At the data cut-off date (November 2, 2022), 2 patients had discontinued from the study, 5 patients had completed the longer-term follow-up, and 7 participants were continuing the study. The duration of follow-up at the data cut-off ranged from || |||||| || || |||||| following fidanacogene elaparvovec infusion.
The mean age of participants was 40.1 years, ranging from 18 to 61 years at the time of fidanacogene elaparvovec infusion. Most participants were aged 35 years or older (71.4%) and white (85.7%). There were 10 participants on FIX prophylaxis and 4 participants using on-demand regimens before fidanacogene elaparvovec infusion. All participants had FIX levels of 2% or lower.
Efficacy Results
Bleeding outcomes: The mean ABRtreat remained lower than 1.0 from year 2 through year 6 postinfusion, with || participants (|||||) having no bleeds during their entire time in the study. The mean treated ABRs were |||| |||||||, |||| |||||||, |||| |||||||, |||| |||||||, and |||| ||||||| during years 2, 3, 4, 5, and 6 postinfusion, respectively. |||| participants had treated bleeds from years 2 through 6.
The AIR generally decreased over the entire follow-up periods, from a mean of |||| in year 2 to |||| in year 6 following fidanacogene elaparvovec infusion. The mean AIRs were |||| |||||||, |||| |||||||, |||| |||||||, |||| |||||||, and |||| ||||||| during years 2, 3, 4, 5, and 6 postinfusion, respectively.
As of the data cut-off date, there were no prophylactic infusions in the study, and no participants had resumed prophylaxis. The median total factor consumption and annualized FIX consumption, excluding consumption required for surgery, was |||| for year 2 through year 6. |||| of the 14 participants have had no nonsurgical FIX consumption over the longer-term follow-up period.
From week 52 to week 130 following fidanacogene elaparvovec infusion, the number of participants with target joint bleeds decreased from || ||||||| to ||||||, based on responses to target joint assessment questionnaires. || ||||||||||| had target joint bleeding reported beyond week 130 as of the data cut-off (from weeks 156 to 312 or end of study).
Patient-Reported Outcomes: The HJHS, an exploratory end point, was added after most participants were dosed, resulting in a low number of assessments at baseline. The baseline HJHS score was the last nonmissing measurement before fidanacogene elaparvovec infusion in Study C0371005. The median HJHS total scores were |||| ||||| at baseline, |||| |||||| at week 156, |||| ||||| at week 208, |||| ||||| at week 260, and |||| ||||| at week 312 or end of study.
Haem-A-QoL total scores and domain scores |||||||| |||||||||| throughout the longer-term follow-up period (years 2 through 6). Median change from baseline in Haem-A-QoL total scores ranged from ||||| || |||||| over the longer-term follow-up (years 2 through 6).
Mean HAL domain scores |||||||| ||||| || and the total score |||||||| ||||| || at all following fidanacogene elaparvovec infusion visits over the longer-term follow-up period (years 2 through 6). HAL scores can range from 0 to 100, with higher scores indicating fewer functional limitations.
Harms Results
Of the 10 TEAEs reported, 5 were mild, 1 was moderate, and 4 were severe. These 10 TEAEs included 9 SAEs and 1 nonserious AE (back pain). The most frequently reported TEAEs regardless of severity were related to musculoskeletal and connective tissue disorders in 2 participants (14.3%).
Four of the 14 participants (28.6%) experienced a total of 9 SAEs. No participants discontinued from the study due to AEs. There were no deaths.
No participants experienced hypersensitivity reactions or another AE of special interest. During the longer-term follow-up period, 8 of 14 participants experienced increased ALT above the upper limit of normal (ULN), 3 of whom had increased AST above the ULN. None of these cases were managed with corticosteroids and, as of the data cut-off, all of these participants had ALT and AST levels back within normal limits, except for 1 patient who completed the study with an ALT level above the ULN. Regarding immunogenicity, all 14 participants remained negative for FIX inhibitors during the study.
Critical Appraisal
Internal Validity
Study C0371005 was an open-label, single-arm, multicentre, phase I and IIa study. All efficacy analyses were exploratory in nature and were presented using descriptive statistics. The absence of a comparator group limited the interpretation of results because causality could not be established. The open-label design may have biased the reporting of some end points because awareness of the study treatment received may have influenced the perception of improvement and/or harms by patients and clinicians, particularly for outcomes that are subjective in measurement and interpretation (e.g., patient-reported outcomes and subjective AEs). The follow-up period was only 1 year, which was insufficient to permit drawing any definite conclusions regarding long-term efficacy and safety outcomes. In addition to the general limitations of the study design, because the HJHS and HAL assessments were added to the study later during a protocol amendment, data were missing for most of the participants (only 4 patients contributed data to the analyses). As such, no conclusions can be drawn for these outcomes with certainty.
Study C0371003 provided a longer-term follow-up for 14 of the patients who had previously received fidanacogene elaparvovec in Study C0371005. As the primary objective of Study C0371003 was to evaluate safety, no hypothesis testing was planned. All efficacy and safety data were summarized descriptively, resulting in no statistical inferences. Data were missing for HJHS and HAL assessments in this study as well, for the reasons discussed for Study C0371005.
In Study C0371003, the duration of follow-up at the data cut-off ranged from || |||||| || || |||||| following fidanacogene elaparvovec infusion. Only 5 participants had completed the 6-year longer-term follow-up as of the data cut-off. According to the clinical experts consulted by CADTH, the data provided for up to 6 years of follow-up are limited but reasonable for the purposes of assessing safety and efficacy in the patient population. The clinical experts noted that a longer follow-up (20 to 25 years) involving more patients is warranted to make any definitive determinations on overall long-term safety and efficacy of fidanacogene elaparvovec. Although Study C0371003 provides the longest-term data available on the efficacy of fidanacogene elaparvovec, this evidence is inconclusive.
External Validity
The external validity was similar to that of the pivotal trial and its corresponding lead-in study. The dose of fidanacogene elaparvovec used in Study C0371005 aligns with the recommended dose in the draft product monograph. The majority of the patients enrolled were white (80.0% and 85.7% in Study C0371005 and Study C0371003, respectively), which, according to the clinical experts consulted by CADTH, was higher than what would be expected for the patient population in Canada. Both Study C0371005 and Study C0371003 enrolled only male patients, although the clinical experts noted this is likely not a serious generalizability issue because the treatment effects are not expected to differ between males and females due to the same underlying mechanism of disease, and female patients with moderately severe to severe hemophilia B are rare. One of the eligibility criteria in Study C0371005 was both hemophilia B with FIX activity less than or equal to 2% at screening and historical evidence or a documented genotype known to produce a clinically severe phenotype of hemophilia B. The clinical experts consulted by CADTH noted that severity in clinical practice is defined by the patients' phenotype and not simply their factor activity levels. Hemophilia in some patients will be considered moderately severe to severe due to clinical symptoms even for those with a FIX level greater than 2%, according to the clinical experts consulted by CADTH. Last, generalizability may be limited by the small sample size.
Conclusions
One phase III, single-arm, open-label trial (BeneGene-2) investigated the efficacy and safety of fidanacogene elaparvovec in 45 patients with moderately severe to severe hemophilia B (defined as FIX:C ≤ 2%). For efficacy outcomes regarding bleeding events and use of FIX following fidanacogene elaparvovec infusion, patients in the BeneGene-2 trial served as their own controls, using data collected from when these patients were on FIX prophylaxis during a lead-in study (BeneGene-1). Compared to FIX prophylaxis, fidanacogene elaparvovec may result in a decrease in the ABRtotal, ABRtreat, |||||| AIR, and |||||||||| ||| |||||||||||, and the effects observed for all of these outcomes were considered clinically relevant by the clinical experts consulted by CADTH. However, uncertainty associated with interpreting the clinical significance of the magnitude of the treatment differences remains due to limitations such as the nonrandomized comparative design, potential risk of bias in self-reporting bleeding events caused by the open-label design, and potential biases introduced by assumptions of the statistical models used to make the comparisons. The safety profile of fidanacogene elaparvovec during the follow-up period was considered acceptable by the clinical experts consulted by CADTH; however, the safety evidence is uncertain given the lack of comparative data, sample size, and limited duration of follow-up. To address the limited duration of follow-up in the BeneGene-2 study, evidence from a phase I and IIa, single-arm, open-label trial and a corresponding extension study that provided data for up to 6 years of follow-up was examined. However, the limitations of these supportive studies (e.g., a single arm and noncomparative design, descriptive analyses, small sample size, many patients ongoing, and missing data) preclude CADTH from drawing conclusions with certainty about the longer-term efficacy and safety of fidanacogene elaparvovec based on this evidence. Altogether, the long-term efficacy and safety of fidanacogene elaparvovec remains inconclusive.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of fidanacogene elaparvovec, 1 × 1013 vg/mL, supplied as a concentrate solution for IV infusion in the treatment of moderately severe to severe hemophilia B in patients aged 18 years or older.
Disease Background
Contents within this section were informed by materials submitted by the sponsor and clinical expert input. The following summary was validated by the CADTH review team.
Hemophilia is a serious X chromosome–linked, lifelong genetic disorder that leaves patients vulnerable to blood loss and organ damage due to impaired functioning of the coagulation cascade.3,4 Hemophilia B is the second most common type of hemophilia (after hemophilia A) and is characterized by an absence or shortage of FIX resulting from a mutation in the F9 gene.3,4 FIX is a vital component of the intrinsic coagulation cascade pathway, which is activated in response to vascular endothelium surface damage.11 Once initiated, the enzymes in the coagulation cascade activate in sequence until fibrin, a clot-forming protein, is produced.11,12 A FIX deficiency in hemophilia B prevents or reduces the ability of the coagulation cascade to produce fibrin.13
Moderate and severe hemophilia B cases are defined by the WFH as having 1% to 5% and less than 1% of normal enzymatic FIX activity, respectively.5 Moderately severe hemophilia has also been defined as factor levels of 1% to no more than 2% in previous clinical trials that have investigated treatment with prophylaxis.14 However, according to the clinical experts consulted by CADTH, severity in clinical practice is defined by the patients’ phenotype (i.e., tendency to bleed) and not simply their factor activity levels. The decision to initiate prophylaxis with clotting factor concentrates takes into the account both their clinical phenotype and factor activity levels, as well as lifestyle and professional activities.
Clinically, hemophilia B presents as a susceptibility to bruising and episodes of prolonged bleeding from surgery or trauma.4 In patients with moderate or severe hemophilia, spontaneous and internal serious and life-threatening bleeding into joints, muscles, and vital organs may also occur.4 The frequency of spontaneous bleeding episodes is variable in severe patients and bleeding may occur up to 20 or 30 times without an apparent cause or after minor trauma, each year.4,15 The majority of spontaneous bleeds occur in the joints (70% to 80%) and muscles (10% to 20%).5 Less than 5% of bleeds occur in the central nervous system (e.g., intracranial hemorrhage), but these can be particularly serious and debilitating, potentially leading to seizures, impaired motor function, or death in up to 20% of cases.5,16,17 Patients with hemophilia B are prone to prolonged bleeding after injury, surgery, or trauma, as well as nosebleeds and bleeding from the gums.4,18 Individuals with moderately severe to severe hemophilia frequently experience bleeding and recurrent spontaneous bleeding events into muscle, soft tissue, and joints (hemarthroses) throughout their entire lives.4,6 However, bleeds can occur in any organ, and other affected organs can include kidneys, stomach, intestines, and the brain.4,5,19 Hemarthrosis is the most common manifestation of moderate and severe hemophilia B.4,5
As of 2021, there were 704 patients with hemophilia B (with recorded severity) in Canada, 535 of whom were adult male patients. Of the adult male patients, 218 had moderate and 145 had severe hemophilia B.20 The mean prevalence per 100,000 males in Canada from 1998 to 2006 was 3.23.21 The estimated prevalences at birth per 100,000 males in Canada from 1991 to 2015 were 3.9 for all severities of hemophilia B and 1.3 for severe disease only.22
Standards of Therapy
Contents within this section were informed by materials submitted by the sponsor and clinical expert input. The following summary was validated by the CADTH review team.
The treatment goal for hemophilia, as outlined by WFH guidelines,5 is to reduce or prevent bleeding while allowing patients to lead active lives and achieve a quality of life comparable to that of individuals not affected by the condition. Current management strategies of hemophilia B for affected patients include on-demand treatment to stop bleeds as they occur and/or routine prophylaxis therapy to prevent bleeding, both involving the administration of exogenous FIX CFCs to treat a FIX deficiency.5
According to the clinical experts consulted by CADTH, routine FIX prophylaxis involving lifelong regular IV administration of FIX CFCs is currently the standard of care for patients with hemophilia B in Canada. The clinical experts consulted by CADTH noted that FIX prophylaxis should be based on clinical phenotype (e.g., presenting clinical bleeds) and not simply laboratory severity (FIX levels).
Prophylaxis aims to maintain hemostasis with the primary goal of preventing bleeds, especially into the joints, to avoid long-term joint damage and enable patients to live a full and active life.5,8,23 Products based on rFIX are the mainstay prophylactic treatments for hemophilia B.5,7,8 All rFIX products have either a standard half-life (SHL) and therefore must be administered once weekly at a minimum, and often 2 to 3 times per week, or an extended half-life (EHL), and therefore require administration either once weekly or once every 1 to 2 weeks.24-26 The clinical experts consulted by CADTH noted that the frequency of FIX injections varies from individual to individual depending on the type of FIX concentrate and the pharmacokinetics of individual patients. Preparations of rFIX CFCs for the treatment of hemophilia B are available in Canadian provinces and territories through Canadian Blood Services, excluding Québec, and include rFIX Fc fusion protein (Alprolix]), pegylated nonacog beta pegol (Rebinyn), and nonacog alfa (BeneFIX).1,27 According to the clinical experts consulted by CADTH, plasma-derived FIX, such as factor IX concentrate (human) (Immunine), is also available in Canada but with very limited use.
Drug Under Review
Key characteristics of fidanacogene elaparvovec and other treatments available for moderately severe to severe hemophilia B in patients aged 18 years of age and older are summarized in Table 4.
Fidanacogene elaparvovec is a gene therapy designed to introduce a functional copy of a high-activity variant of the F9 gene (FIX-R338L) in the transduced cells to address the monogenic root cause of hemophilia B. By providing an alternative active source of the FIX protein, which is secreted into the plasma, it is expected to restore hemostasis.2 Fidanacogene elaparvovec is a nonreplicating recombinant AAV vector that utilizes the AAVrh74var capsid to deliver a stable, fully functional human FIX transgene. The AAVrh74var capsid is derived from AAVrh74, which is not known to cause disease in humans. The AAVrh74var capsid is able to transduce hepatocytes, the natural site of FIX synthesis. The F9 gene present in fidanacogene elaparvovec is designed to reside predominately as episomal DNA within transduced cells. Expression of the transgene is driven by a liver-specific promoter, which results in tissue-specific, continuous, and sustained expression of the FIX protein.2
Table 4: Key Characteristics of Fidanacogene Elaparvovec, rFIXFc, Pegylated Nonacog Beta Pegol, and Nonacog Alfa
Characteristic | Fidanacogene elaparvovec | rFIXFc (Alprolix) | Pegylated nonacog beta pegol (Rebinyn) | Nonacog alfa (BeneFIX) |
|---|---|---|---|---|
Mechanism of action | Nonreplicating recombinant AAV vector that utilizes the AAVrh74var capsid to deliver a stable, fully functional human FIX transgene | Long-acting, fully recombinant, fusion protein comprising human coagulation FIX covalently linked to the Fc domain of human immunoglobulin G1 and produced by recombinant DNA technology | Upon activation, the peptide including the 40 kDa polyethylene-glycol moiety is cleaved off, leaving the native FIX molecule | Contains recombinant coagulation FIX (nonacog alfa); FIX is activated by factor VII or the tissue factor complex in the extrinsic pathway as well as factor XIa in the intrinsic coagulation pathway |
Indicationa | Anticipated for the treatment of moderately severe to severe hemophilia B in patients 18 years and older | Indicated in adults and children with hemophilia B (congenital FIX deficiency or Christmas disease) for:
| Indicated for adults and children with hemophilia B (congenital FIX deficiency or Christmas disease) for:
| Indicated for the control and prevention of hemorrhagic episodes and routine prophylaxis in patients with hemophilia B (congenital FIX deficiency or Christmas disease), including control and prevention of bleeding in surgical settings |
Route of administrationb | Single IV infusion over 1 hour | IV over several minutes after reconstitution | IV bolus injection over several minutes after reconstitution | IV infusion after reconstitution |
Recommended doseb | 5 × 1011 vg/kg of body weight | Starting regimens are either 50 IU/kg once weekly or 100 IU/kg once every 10 to 14 days | 40 IU/kg body weight once weekly | 40 IU/kg administered at intervals of 3 to 4 days Dosing regimens of 50 IU/kg twice weekly and 100 IU/kg once weekly have been demonstrated to be effective in clinical trials |
Serious adverse effects or safety issues | Theoretical risk of malignant transformation leading to cancer resulting from AAV-mediated integration into host cell DNA. Transient and asymptomatic elevation of transaminases; anti-AAVrh74var antibody formation can take place after exposure to a virus similar to the vector | Thromboembolic complications (e.g., pulmonary embolism, venous thrombosis, and arterial thrombosis). Inhibitors have been reported, including in previously untreated patients; allergic-type hypersensitivity reactions including anaphylactic reactions are possible | Similar to rFIXFc | Similar to rFIXFc |
AAV = adeno-associated virus; AAVrh74va = adeno-associated virus rh74 variant protein; FIX = coagulation factor IX; rFIXFc = recombinant factor IX Fc fusion protein; vg = vector genome.
aHealth Canada–approved indication.
bFor comparators, dose is for prophylaxis in adult patients.
Source: Pfizer (2023),2 Sanofi (2021),26 Novo Nordisk (2022),24 and Pfizer (2017).25
Stakeholder Perspectives
Patient Group Input
This section was prepared by the CADTH review team based on the input provided by patient groups. The full original patient inputs received by CADTH are included in the Stakeholder section of this report.
The CHS provided input for the review of fidanacogene elaparvovec for the treatment of moderately severe to severe hemophilia B patients who are 18 years of age and older. Patient input was gathered from an online survey, conducted between July 10 and July 31, 2023. In total 17 responses were gathered by the CHS. All respondents were affected by severe or moderately severe hemophilia B without inhibitors. In addition, in September 2022, the CHS conducted an online survey of patients in Canada with severe hemophilia A and B and received 39 responses, among which 31 were from patients with hemophilia A, 7 were from patients with hemophilia B, and 1 was from a patient whose hemophilia was not specified.
Joint damage, primarily to knees, ankles and elbows, caused by repeated internal hemarthroses, was reported to be the primary physical health impact of hemophilia B. Regarding currently available treatments, 4 patients in the 2023 CHS survey reported being very satisfied, 7 were satisfied, 5 were neither satisfied nor dissatisfied, and 1 was very dissatisfied. Patients from this survey noted that treatments greatly complicate their everyday life, travel, and leisure activities. They also mentioned the difficulty associated with infusions due to vein visibility, poor vein issues, and side effects, and reported dealing with socioeconomic problems due to the need for regular visits, missing work to attend visits, and travel and insurance issues, and accessing treatment.
When respondents to the 2023 CHS survey were asked how gene therapy could change their lives, all patients provided positive feedback. Patients hoped gene therapy would lead to fewer FIX infusions, minimal needle injections, less stress, less bleeding, and fewer restrictions on activities, and make it easier to travel. In addition, about 63% of the respondents from the 2022 survey indicated they expected gene therapy to be effective in preventing bleeding for at least 10 years. The 2022 survey asked if people would be willing to receive gene therapy knowing that that there would be frequent blood draws in the weeks and months following administration, and they would need to be followed up in a registry for 10 to 20 years. In response, 66% answered yes, 10% answered no, and 24% indicated they did not know.
The CHS mentioned that a small number of patients in Canada (likely close to 5) have undergone gene therapy for hemophilia B, but it had no information about their experiences beyond the preliminary trial data. The group also noted that, in the absence of peer-reviewed publications describing the results of phase III clinical trials for fidanacogene elaparvovec, it cannot comment on the relative benefits and risks compared to current therapies or other gene therapies for hemophilia B currently under review by Health Canada.
Clinician Input
Input From Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). In addition, as part of the review of fidanacogene elaparvovec, a panel of 4 clinical experts from across Canada was convened to characterize unmet therapeutic needs, help identify and describe gaps in the evidence that could be addressed through the collection of additional data, promote the early identification of potential implementation challenges, gain further insight into the clinical management of patients living with a condition, and explore the potential place in therapy of the drug (e.g., potential reimbursement conditions). A summary of this panel discussion follows.
Unmet Needs
The clinical experts consulted by CADTH noted several important goals of treatment for patients with hemophilia B, including allowing patients to have a normal life expectancy, improve their quality of life, and decrease the burden of disease by reducing or eliminating pain as well as preventing or reducing functional impairment; preventing or reducing bleeding such as joint bleeds and spontaneous bleeds; and preventing the development of chronic musculoskeletal complications due to recurrent bleeding, particularly chronic hemophilic arthropathy, and the resulting health system resource consumption of joint replacement.
The clinical experts consulted by CADTH noted that FIX prophylaxis requires frequent IV injections performed by the patients themselves. Patients who are on prophylactic therapy have to inject themselves at home, usually about 2 times per week (if on an SHL product) or once every 2 weeks (if on an EHL product). This poses a heavy burden for patients with hemophilia B and significantly affects patients’ ability to live a normal life. The clinical experts consulted by CADTH noted that poor adherence to FIX prophylaxis, which may result in reduced effectiveness of prophylaxis and increased risk of bleeding, has been a significant problem for patients with hemophilia B due to the frequency and difficulty associated with self-injections.
After infusion, the plasma FIX level varies over time. Patients must adapt their lifestyle to these continuous waves of FIX levels, limiting their freedom to enjoy activities (and productivity) to specific temporal windows. Furthermore, the decline in plasma FIX concentrations in the period between infusions is such that patients may have little or no protection from bleeding for variable periods of time. This is accentuated if an infusion is missed or delayed because of difficulty or misadventure in carrying out the infusion, or depletion of the home inventory (FIX concentrates are not delivered to the patient’s home but must be picked up at a designated hospital blood bank). Even patients who administer prophylaxis on the prescribed schedule (i.e., are adherent) can experience breakthrough bleeds, particularly in the days before the next infusion.
Place in Therapy
The clinical experts consulted by CADTH noted that the key advantage of fidanacogene elaparvovec over an exogenous FIX prophylaxis regimen is avoiding the fluctuations in FIX levels and eliminating the need for repeated CFC infusions. A stable level of FIX via a single infusion of fidanacogene elaparvovec is expected, while current FIX prophylaxis requires frequent self-injections of FIX to sustain what is still a fluctuating FIX activity level characterized by peaks and troughs. The clinical experts consulted by CADTH noted that fidanacogene elaparvovec could be a curative treatment if a steady and high level of FIX is expressed.
The clinical experts consulted by CADTH noted that a first or later line of treatment is not an appropriate framework to describe the relationship between fidanacogene elaparvovec and FIX prophylaxis. The adoption of fidanacogene elaparvovec or FIX prophylaxis depends on the situation. For instance, fidanacogene elaparvovec is unlikely to be offered to newborn patients with hemophilia B, and newborn patients may need FIX prophylaxis for years before they are considered for gene therapy such as fidanacogene elaparvovec. Additionally, fidanacogene elaparvovec can be offered to not only patients who respond to FIX prophylaxis but also those who cannot or will not perform the injections of FIX due to reasons such as unreliable venous access, elbow arthropathy limiting self-infusion, needle phobia, or unwillingness. The clinical experts consulted by CADTH noted that it remains uncertain whether the use of fidanacogene elaparvovec will cause a shift in treatment paradigm.
The clinical experts noted that patients who qualify for fidanacogene elaparvovec would have been exposed to FIX concentrates since early childhood and that there are potential situations in which clinicians may discuss other options with patients before initiating fidanacogene elaparvovec. For instance, prophylaxis with an available EHL FIX product may not have been attempted, or nonfactor therapies may become available in the future (currently no nonfactor therapies are licensed for the indicated population). However, the clinical experts consulted by CADTH noted that these situations do not necessarily mean that patients must try other options before fidanacogene elaparvovec. The clinical experts indicated that the selection of treatment options will involve a shared decision-making process between clinicians and patients.
Patient Population
The clinical experts consulted by CADTH noted that all patients with hemophilia B who have a clinically severe phenotype (regardless of FIX level) are likely to benefit from treatment with fidanacogene elaparvovec in terms of reductions in burden of care, pain, and pain interference as well as improvement in mobility and quality of life. The clinical experts noted that those who would gain the most from fidanacogene elaparvovec treatment would be patients without pre-existing joint damage due to hemophilia B in terms of preserving joint function, as well as younger patients who are usually more active and would enjoy physical activity and being able to practise sports in a safer way.
The clinical experts noted that other patients with hemophilia B who would also benefit from fidanacogene elaparvovec include those on FIX prophylaxis with an ABR of 0 as the burden of care would be reduced, those not on prophylaxis who experience bleeding (ABR > 0) as they are likely to achieve an ABR of 0, those who are unable to adhere to prophylactic therapy, those with pre-existing joint damage as fidanacogene elaparvovec may reduce the progression to arthropathy and eliminate daily pain and aches, and those with recurrent bleeding despite prophylactic therapy.
The clinical experts noted that the patients who would be best suited for treatment with fidanacogene elaparvovec will be identified primarily by clinical assessment and shared decision-making with patients. A misdiagnosis (a false-positive due to diagnosis of hemophilia B in a patient with another bleeding disorder) is unlikely to occur in practice, as the laboratory measurement of plasma factor IX is a relatively sensitive and specific test, and because it is standard practice in Canada to confirm the phenotypic diagnosis of hemophilia B with genotyping. Testing for nAbs against AAVrh74var should be mandatory as a companion diagnostic test.
The clinical experts consulted by CADTH noted that the patients least suitable for fidanacogene elaparvovec include those with pre-existing anti-AAV antibodies and those who conclude that the benefit does not outweigh the risk associated with fidanacogene elaparvovec gene therapy, given that its long-term efficacy and safety remain unclear. In addition, some patients may not want to change their current treatment.
Assessing the Response Treatment
The clinical experts consulted by CADTH noted that the most important assessment of treatment response is monitoring patients’ bleeding to determine whether fidanacogene elaparvovec prevents bleeding events and allows patients to live the lifestyle they want without concerns about the risk of bleeding. The clinical experts noted that FIX activity levels may also be monitored to assess response to treatment; this can allow clinicians to determine the degree to which the deficiency in FIX has been corrected by fidanacogene elaparvovec. The clinical experts consulted by CADTH noted that a higher FIX activity level is in general associated with better bleeding outcomes (e.g., no bleeding). However, in some cases, there can be a discrepancy between FIX activity levels and bleeding outcomes.
The clinical experts noted that follow-ups should focus on both efficacy and safety (e.g., checking patients’ bleeding events and joint status via phone or virtual check-ups) and lab tests (e.g., liver enzymes, FIX activity levels, liver ultrasounds to detect hepatocarcinomas). The length of follow-up for hepatic function and FIX activity levels following fidanacogene elaparvovec infusion should be lifelong. In terms of frequency, the clinical experts consulted by CADTH noted that monitoring after fidanacogene elaparvovec infusion will be more frequent in the short term (e.g., for the first 3 months postinfusion, lab tests mainly for liver enzymes and FIX levels twice a week, starting around week 3 postinfusion, or lab tests twice weekly initially and then once weekly) and less frequently over the long-term (e.g., after first 3 months, quarterly visits for the balance of the first year and then yearly visits lifelong, or monthly visit for the balance of the first year and then only as clinically indicated). The clinical experts consulted by CADTH noted that tests for FIX levels may not start immediately after fidanacogene elaparvovec infusion given that the production of FIX by fidanacogene elaparvovec is unlikely to happen immediately postinfusion, although it is reasonable to monitor FIX activity levels and liver function twice a week at the early stages postinfusion.
The clinical experts consulted by CADTH noted that monitoring changes in the HJHS as well as in quality-of-life–related end points following fidanacogene elaparvovec infusion (e.g., improvement in activities associated with daily living, physical activity, and functioning; decrease in development of disability; and improvement in psychosocial health and functioning) are also important. The clinical experts added that the PROBE tool is typically used to measure quality of life in patients with hemophilia B in the Canadian setting instead of Haem-A-QoL and HAL, although these latter instruments are closely aligned in measuring quality of life, and PROBE includes questions covering activities of daily life.
Discontinuing Treatment
The “discontinuation of treatment” concept is not applicable to gene therapy, which is a 1-time treatment. The clinical experts consulted by CADTH noted that determination of treatment failure should be made by the treating clinician on a case-by-case basis. Although the pivotal trial has provided some definitions of treatment failure, the clinical experts consulted by CADTH noted that determining treatment failure is more complicated in clinical practice than in the clinical trial setting. The clinical experts noted that, if fidanacogene elaparvovec fails, patients may not be eligible for another gene therapy based on AAV vectors because they may present cross-reactivity against most AAV vectors. However, the clinical experts added that patients may in the future try alternative approaches to a gene therapy based on other viral vectors or even nonviral vectors, although this is hypothetical because no such gene therapy is currently available.
Prescribing Considerations
The clinical experts consulted by CADTH noted that fidanacogene elaparvovec should be prescribed based on the judgment of a multidisciplinary team organized by a comprehensive hemophilia treatment centre, and the team may consist of specialists such as a hematologist with experience in treating hemophilia patients, a physiotherapist to assess joint function, a hepatologist for liver-related issues, a pharmacist, and an HIV specialist if the patient is HIV-positive. The clinical experts consulted by CADTH noted that the administration of fidanacogene elaparvovec is on an outpatient basis, as are follow-ups following fidanacogene elaparvovec infusion for most patients (some may occasionally need to be admitted for follow-up).
Clinician Group Input
This section was prepared by the CADTH review team based on the input provided by clinician groups. The full original clinician group inputs received by CADTH are included in the Stakeholder section of this report.
Nine clinicians from the AHCDC and 3 nurses from the CANHC provided input. Both the AHCDC and CANHC highlighted unmet needs for persons with hemophilia and a severe bleeding phenotype, and specifically hemophilia B. Both the AHCDC and CANHC mentioned that the treatment currently available in Canada does not modify or alter the underlying disease process, making persons with hemophilia B dependent for life on regular IV infusions of FIX to prevent and treat bleeding. In addition, the AHCDC emphasized the frequency of venipuncture required for prophylactic CFC replacement. The group noted that routine prophylaxis can be challenging for patients with poor venous access and the placement of a central venous catheter can lead to long-term complications, including risks of infection, bleeding, thromboembolism, and loss of function requiring removal. The group emphasized that all these factors lead to the need for persons with hemophilia B and a severe bleeding phenotype to restore coagulation factor levels to clinically effective levels without the need for frequent venipunctures on a regular basis throughout their lifespans. The AHCDC also discussed the variability of the efficacy of prophylaxis with CFCs across individuals, with some patients susceptible to breakthrough bleeding into joints and muscles. The group noted that these breakthrough bleeds result in pain, loss of function, absenteeism from work or school, reduced quality of life, and, more importantly, disability from progressive joint damage. Last, the AHCDC noted that the FIX trough levels associated with prophylaxis regimens are often insufficient to allow for safe anticoagulation or dual antiplatelet therapy.
Both the AHCDC and CANHC noted that fidanacogene elaparvovec would provide a 1-time treatment leading to sustained FIX production, addressing the underlying disease process and natural history rather than symptomatic management. This would represent a paradigm shift in the treatment of hemophilia B. The AHCDC also mentioned that, in contrast to patients with hemophilia A, who have the option of emicizumab, patients with hemophilia B have no current alternatives to CFCs outside of clinical trials, making the need for gene therapy greater for hemophilia B patients.
The AHCDC noted that candidates for gene therapy include adults with hemophilia B and a clinically severe bleeding phenotype requiring prophylaxis, no history of inhibitory antibodies, no significant comorbidities, and no pre-existing anti-AAV nAbs. The group also highlighted the difficulty involved in estimating the proportion of patients with hemophilia who would be eligible for gene therapy once it becomes commercially available due to the need for an anti-AAV antibody assay, detailed liver assessment, and assessment of the patient’s attitudes and perceptions.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may affect their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 5.
Table 5: Summary of Drug Plan Input and Clinical Expert Response
Implementation questions from the drug programs | Clinical panel response |
|---|---|
Relevant comparators | |
The current standard of care for those with moderately severe or severe hemophilia B is routine prophylaxis, involving the regular IV administration of FIX, e.g., nonacog alfa (BeneFIX), rFIXFc (Alprolix), pegylated nonacog beta pegol (Rebinyn), or factor IX concentrate (human) (Immunine). There is no direct gene therapy comparator product in the marketplace. | To inform expert committee deliberations. |
Are vendor-supplied real-world evidence and indirect treatment comparison studies appropriate to confirm better clinical outcomes for fidanacogene elaparvovec compared with available FIX? | The sponsor did not submit real-world evidence or an indirect treatment comparison for this review. The sponsor submitted a single-arm phase III pivotal trial that included comparisons to a lead-in study. The sponsor also submitted a phase II trial with an associated lead-in study to address the knowledge gap in longer-term impacts. |
The comparators in the sponsor’s submission are recombinant FIX products supplied by CBS for the management of moderately severe to severe hemophilia B in adults in Canada (excluding Québec). If fidanacogene elaparvovec is funded by the public drugs plans there would be need for coordination between the public drug plans and CBS (i.e., prophylactic dose of one of the comparators is given before infusion with fidanacogene elaparvovec). These treatments are provided at no cost to the patient (i.e., no deductibles or co-pays). If the comparators are considered under public drug plans, they would have to meet the eligibility requirements, which would also include co-pays in certain jurisdictions. In addition, there will likely be travel expenses. | To inform expert committee deliberations. |
What is the timing between prophylaxis and infusion of fidanacogene elaparvovec? What is the transition plan for patients moving from the comparator drug to this therapy? | It does not matter when the last FIX prophylaxis treatment before infusion of fidanacogene elaparvovec takes place. For a patient who is on FIX prophylaxis, the clinician can set a date for the patient to receive fidanacogene elaparvovec. Until that date, the patient can still follow a FIX prophylaxis regimen. After the infusion of fidanacogene elaparvovec, it will take a period of time (e.g., 1 to 4 weeks) for fidanacogene elaparvovec to start producing transgenic FIX. A FIX prophylaxis regimen should continue during this period to avoid bleeds and provide protection. |
Considerations for initiation of therapy | |
It was anticipated that the product monograph will include tests to confirm eligibility for fidanacogene elaparvovec and to ensure safety and effectiveness. Anticipated tests included a liver fibrosis test, liver function tests, FIX inhibitor assay, blood test for the presence of chronic infections (i.e., hepatitis B, hepatitis C, and HIV serology), and screening for nAb seropositivity against the specific AAVrh74var. In the event of a criteria-based recommendation for reimbursement, which marker(s) or criteria should be used to start therapy with fidanacogene elaparvovec? | Overall, many factors need to be considered before initiation of fidanacogene elaparvovec to identify patients who are likely to benefit from fidanacogene elaparvovec. The decision should be based on the judgment of the treating clinician via discussion with patients and their referring centres. The pivotal BeneGene-2 trial provided several criteria, including patients’ FIX levels, as well as the status of nAbs against AAVrh74var, nAbs against FIX, and liver function. The clinical experts noted that situations can be more complex in clinical practice. For example, in addition to the FIX level, clinicians must consider the clinical phenotype of the disease to determine its severity. |
Participants were excluded from the pivotal trial for reasons that may reduce the safety or efficacy of the infusion such as nAbs against AAVrh74var or a history of or presence of nAbs against FIX (i.e., FIX inhibitors). Testing for nAbs against AAVrh74var is expected to be required to confirm eligibility for fidanacogene elaparvovec. Should patients excluded from the pivotal study due to reasons such as being positive for nAbs against AAVrh74var or have a history of or presence of nAbs against FIX to be eligible for fidanacogene elaparvovec? Is a program needed to identify eligible patients? If nAb testing is required for eligibility, is this a test that is available in each jurisdiction (all provinces and territories)? (The sponsor indicated it is planning an optional patient support program, which would offer nAb testing.) | Overall, nAb testing should be required to select patients eligible for fidanacogene elaparvovec. In terms of testing for nAbs against FIX (i.e., FIX inhibitors), the clinical experts noted that it is a part of the standard of clinical practice in Canada. Clinicians will measure nAbs against FIX regularly. It is also reasonable to exclude a patient who has currently active nAbs against FIX, but these antibodies are rare in people with hemophilia B. Testing for nAbs against AAVrh74var should be a requirement for initiating fidanacogene elaparvovec. It is acceptable to exclude a patient who has an anti-AAVrh74var nAb titre ≥ 1:1, a criterion used in the pivotal BeneGene-2 trial, the evidence associated with the titre threshold (≥ 1:1) remains uncertain. The capacity for nAb testing against AAVrh74var in Canada remains unknown, and relevant issues (e.g., testing being done in the US through a support program offered by the sponsor, types of assays) should be discussed with the sponsor. No patients should be excluded based on a lack of access to nAb testing against AAVrh74var. |
The drug plans and CBS noted that patients eligible for the pivotal study would have already received rFIX therapy for hemophilia B and are seeking information on how long patients need to have received comparator drugs before starting this therapy. Should it be a requirement for the patient to be on FIX therapy to receive fidanacogene elaparvovec? If yes, what is the duration of time they should be on FIX therapy before receiving fidanacogene elaparvovec? | Because hemophilia B is a congenital disease, it is extremely unlikely that an adult candidate for gene therapy had never received FIX in their life. Prior lack of exposure may suggest that the patient’s clinical phenotype is so mild that FIX prophylaxis is not needed. It is more precise to state fidanacogene elaparvovec should be given to patients who need FIX prophylaxis, rather than to those who have been on a FIX prophylaxis regimen. |
Would there be a need to continue the comparator products after the 1-time IV infusion of fidanacogene elaparvovec? | The comparator products (FIX prophylaxis regimens) will be needed until fidanacogene elaparvovec begins to work (likely by 2 to 4 weeks postinfusion). In addition, comparator products may be needed when patients receive surgery following fidanacogene elaparvovec infusion. |
The indication specifies “moderately severe or severe hemophilia B.” How should this be defined? | Using FIX:C ≤ 2% as the definition is acceptable in a clinical trial. However, from the perspective of daily clinical practice, using this FIX level as a criterion for eligibility is not appropriate. Some patients’ disease may be clinically severe despite having a level of FIX > 2%. Therefore, disease severity should be determined through observation by clinicians in clinical practice. |
Considerations for continuation or renewal of therapy | |
Fidanacogene elaparvovec is indicated as a 1-time infusion. Would there be a situation where it would be needed or appropriate to administer a second treatment of fidanacogene elaparvovec? | It would not be appropriate to administer a second treatment of fidanacogene elaparvovec because nAbs against AAVrh74var will develop from the first treatment. |
What objective markers should be used to assess initial and ongoing response to treatment? What follow-up will be required for patients treated with fidanacogene elaparvovec? How long should patients be monitored for hepatic function and FIX activity levels following fidanacogene elaparvovec infusion? | The most important assessment for treatment response is to monitor patients’ bleeding. It can be considered a complete response if fidanacogene elaparvovec prevents bleeding and allows patients to live the lifestyle they want without concerns about the risk of bleeding. FIX activity levels should also be monitored to allow clinicians to determine whether the deficiency in FIX has been corrected by fidanacogene elaparvovec. In general, superior FIX activity level is associated with better bleeding outcomes (e.g., no bleeding). However, in some cases, there is a discrepancy between FIX activity levels and bleeding outcomes. There are also discrepancies in FIX levels measured using different assay methodologies. Follow-ups should focus on both efficacy and safety through clinical follow-ups (e.g., checking patients’ bleeding events and joint status via phone or virtual check-ups) and lab tests (e.g., liver enzymes, FIX activity levels, and liver ultrasounds to detect potential carcinomas). The follow-up period for hepatic function and FIX activity levels following fidanacogene elaparvovec infusion should be lifelong. In terms of frequency, monitoring after fidanacogene elaparvovec infusion will be more frequent in the short term (e.g., for the first 3 months postinfusion, lab tests mainly for liver enzymes and FIX levels twice a week, beginning at around week 3 postinfusion, or lab tests twice weekly initially and then once weekly) and less frequent over time in the long term (e.g., after the first 3 months, quarterly visits for the balance of the first year, and then yearly visits lifelong, or monthly visits for the balance of the first year and then only as clinically indicated). Tests of FIX levels may not begin immediately after fidanacogene elaparvovec infusion given that the production of FIX by fidanacogene elaparvovec is unlikely to happen immediately postinfusion, although it is reasonable to monitor FIX activity level and liver function tests twice a week at the early stage postinfusion. |
Considerations for discontinuation of therapy | |
In the pivotal BeneGene-2 trial, participants were asked to suspend their FIX prophylaxis regimen following fidanacogene elaparvovec infusion; however, FIX replacement was allowed as needed. The protocol contained guidance respecting when a participant could resume FIX prophylaxis if fidanacogene elaparvovec was not efficacious. In this study, this was defined as FIX activity of ≤ 2% after 12 weeks (in the absence of a confirmed FIX inhibitor) as determined by a central laboratory based on 2 consecutive samples collected within 2 weeks, and/or 2 or more spontaneous bleeds into a major joint and/or target joint over 4 weeks (in the absence of a confirmed FIX inhibitor) or 3 or more spontaneous bleeds (consisting of joint bleeds and/or significant soft tissue/muscle or other site bleeds) over 4 weeks (in the absence of a confirmed FIX inhibitor). | To inform expert committee deliberations. |
The drug plans noted that if treatment failure occurs, the patient may need to restart FIX therapy. How should treatment failure or refractory disease be defined? | The determination of treatment failure should be made by a treating clinician on a case-by-case basis, although the pivotal trial has provided some definitions of treatment failure. Determining treatment failure is more complicated in clinical practice than in the clinical trial setting. In general, the decision to restart factor concentrate prophylaxis should use the same criteria that are used for starting prophylaxis in a patient who did not receive gene therapy. |
If fidanacogene elaparvovec fails, can patients be treated with another gene therapy (e.g., a competitor product using different vector)? | If the other gene therapy uses an AAV vector, then the patients may not be eligible to be treated with the other product because anti-AAV nAbs will be positive to the companion test. Patients may try other products developed based on other viral vectors or even nonviral vectors, although this is hypothetical because no such gene therapy is currently available. |
Considerations for prescribing of therapy | |
The drugs plans and CBS noted the following considerations for prescribing of therapy:
| To inform expert committee deliberations. |
Does fidanacogene elaparvovec need to be prescribed by or in consultation with specialists who have expertise in the treatment of hemophilia B and/or gene therapy? If so, what specialists need to be involved in the initiation, administration, and follow-up? | Fidanacogene elaparvovec should be prescribed by or in consultation with specialists who have expertise in the treatment of hemophilia B and/or gene therapy. Fidanacogene elaparvovec should be prescribed based on the judgment of a multidisciplinary team, which is organized by a hemophilia comprehensive treatment centre and may consist of specialists such as a hematologist with experience in treating hemophilia patients, a physiotherapist to assess joint function, a hepatologist for liver-related issues, a pharmacist, and an HIV specialist if the patient is HIV-positive. |
What would be the most suitable setting for patients to receive the therapy: outpatient clinics at hospitals, specialized medical centres, or hemophilia treatment centres? Does that mean a designated infusion centre? Do the clinical experts anticipate there will be access issues regarding specialists and the infusion centres for patients in some regions? | As the administration of fidanacogene elaparvovec is on an outpatient basis, so is follow-up after fidanacogene elaparvovec infusion for most of the patients (some patients may need to be admitted for follow-up in the case of acute infusion reactions). There are hemophilia treatment centres or clinics across provinces in Canada, although these centres and clinics may not be evenly distributed within a province. In terms of infusion centres, while the situation is unclear, the number of such centres across Canada will likely remain low. As a result, patients from remote areas may face barriers in the form of travel and accommodation-related costs. |
Generalizability | |
The inclusion criteria of the pivotal trial stipulated a classification of “moderately severe” or severe, as defined by a FIX level of 2% or lower. Could individuals with moderate hemophilia having levels between 2% and 5% be considered eligible? Also, would individuals with “mild” hemophilia on regular prophylaxis be included? | A maximum FIX level of 2% was chosen as the inclusion criterion by the clinical trialists, but this does not correspond with the conventional definition of hemophilia severity. This question is partially addressed by with the response about how to define moderately severe to severe hemophilia B. Using FIX levels to define eligibility for fidanacogene elaparvovec is not appropriate in clinical practice (although it is acceptable in clinical trials). Disease severity sufficient to make a patient a candidate for gene therapy should be determined by clinicians based on clinical phenotype. Patients with moderate hemophilia having levels between 2% and 5% (or even > 5%) could be eligible for fidanacogene elaparvovec because these patients may have serious clinical phenotype. The correlation between clinical phenotype and baseline FIX level in hemophilia B can vary. With respect to whether patients with “mild” hemophilia on regular prophylaxis would be eligible for fidanacogene elaparvovec, few patients meet this description, and these “mild” patients who are on FIX prophylaxis likely require this because they need a high level of protection for their lifestyles (e.g., competitive or professional athlete). This scenario is more of an ethics issue and the experts were undecided. |
The indication restricts treatment to adults 18 years of age and older. Could fidanacogene elaparvovec be used in the pediatric population (< 18 years old)? | Fidanacogene elaparvovec should not be given to pediatric patients given the lack of evidence. |
Is there anticipation for any off-label use of the product for patients who do not strictly meet the criteria? | There should be no off-label use of fidanacogene elaparvovec based on current evidence. |
Do the clinical experts anticipate that the drug plans and/or CBS will experience an increase in the prescribing of medications outside parameters to prevent inhibitors, to ensure patients to maintain able to use this treatment in the future? | This is not expected to be an issue because cases described in the question are very rare and there is currently no such medication which can prevent the development of FIX inhibitors (i.e., nAbs against FIX). |
Care provision issues | |
Will fidanacogene elaparvovec be supplied directly to infusion clinics from the sponsor, and will there be an additional transportation fee? Are there any different storage conditions, or special equipment required for infusion not normally carried out by the clinic? | The sponsor provided information related to these implementation considerations. According to the sponsor, fidanacogene elaparvovec will be shipped directly from Pfizer’s manufacturing and packaging facility to the hospital where the infusion is to occur, and administration will be overseen by the associated hemophilia treatment centre. Shipping fees will be covered by Pfizer. Fidanacogene elaparvovec will not use specialty pharmacies to manage cold-chain supply and infusion. After a shipment is received, the product must be transferred, stored, and temperature-monitored in ultra-low-temperature environments (i.e., a −90°C to −60°C [−130°F to −76°F]) freezer. Original packages removed from frozen storage (−90°C to −60°C) may be at room temperature (up to 30°C) for up to 5 minutes for transfer between ultra-low-temperature environments. To ensure that gene infusion centres have all necessary processes in place to successfully order, receive, and unpack shipments as well as return thermal shippers and loggers, Pfizer is offering the option for gene infusion centres to order a dry-run test shipment. Fidanacogene elaparvovec contains genetically modified organisms and has special handling requirements. Recommendations in the safety data sheet as well as local regulations and practices for the handling of biohazardous agents must be followed. Personal protective equipment should be worn while preparing or administering fidanacogene elaparvovec. All handling and preparations of sterile and cytotoxic or hazardous products must be carried out in Class II, types A2, B1, or B2 and Class III biological safety cabinets as applicable under local regulations. Gene infusion centres are expected to have all necessary equipment on site required for storage, handling, dose preparation and administration of fidanacogene elaparvovec. No additional special equipment will be required. |
The plans noted the following considerations:
| To inform expert committee deliberations. |
The following considerations are related to additional supportive medications or other health interventions:
| To inform expert committee deliberations. |
System and economic issues | |
Additional gene therapies for hemophilia B are being reviewed by Health Canada. | To inform expert committee deliberations. |
There is a high 1-time cost of gene therapy, with unknown additional costs if patients need existing treatment options after gene therapy is administered. There is uncertainty regarding the duration of efficacy of the gene therapy. The drug plans and CBS noted concerns with affordability. The drug plans highlighted a need cost comparison between comparator drugs with this product before commencing. | To inform expert committee deliberations. |
Treatment sites may be limited. What are the parameters of the types of facilities that can manage the therapy, and who should make the determination? | The main parameter is the pharmacy’s capacity and willingness to store and reconstitute fidanacogene elaparvovec, and this is a primary parameter to determine whether fidanacogene elaparvovec can be given in a setting. The comfort of a hemophilia treatment centre in terms of infusing and dealing with immediate or short-term reaction following fidanacogene elaparvovec infusion can be a parameter of concern. The requirements in terms of a specific treating room and outpatient medical day unit should not be a major issue. |
The drug plans and CBS noted a need for long-term follow-up and data collection to the assess efficacy of gene therapy and the need for other products. There may be costs associated with data collection and gathering. In addition, they noted a need to monitor access to other therapies after gene therapy is administered. | To inform expert committee deliberations. |
Given the expected budget impacts and travel that will be required, the drug plans and CBS noted a need to consider funding some of these costs, co-pay assistance and travel assistance. | To inform expert committee deliberations. |
No specific program has been established for gene therapies. The mechanism of administration and funding are to be determined. | To inform expert committee deliberations. |
The drug plans noted that the sponsor included an option in its economic model to consider outcome-based arrangements. The drug plans indicated that drug plans may not be familiar with this funding model. | To inform expert committee deliberations. |
AAV = adeno-associated virus vector; AAVrh74var = adeno-associated virus rh74 variant protein; ALT = alanine transaminase; AST = aspartate transaminase; CBS = Canadian Blood Services; FIX = coagulation factor IX; FIX:C = circulating coagulation factor IX; nAb = neutralizing antibody; rFIX = recombinant coagulation factor IX; rFIXFc = recombinant factor IX Fc fusion protein; vg = vector genome.
Clinical Evidence
The objective of CADTH’s Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of fidanacogene elaparvovec, 1 × 1013 vg/mL, supplied as a concentrate solution for IV infusion in the treatment of moderately severe to severe hemophilia B in patients aged 18 years or older. The focus is on comparing fidanacogene elaparvovec to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of fidanacogene elaparvovec is presented in 2 sections, with CADTH’s critical appraisal of the evidence included at the end of each section. The first section, the systematic review, includes pivotal studies and randomized controlled trials that were selected according to the sponsor’s systematic review protocol. CADTH’s assessment of the certainty of the evidence in this first section using the GRADE approach follows the critical appraisal of the evidence. The second section includes additional studies that were considered by the sponsor to address important gaps in the systematic review evidence. No long-term extensions studies or indirect treatment comparisons were submitted by the sponsor.
Included Studies
Clinical evidence from the following is included in the CADTH review and appraised in this document:
One pivotal phase III, open-label, single-arm study (along with a lead-in study conducted before the pivotal study to provide a comparator) identified in the systematic review
One additional study (along with a lead-in study) addressing gaps in evidence.
Systematic Review
Contents within this section were informed by materials submitted by the sponsor. The following summary was validated by the CADTH review team.
Description of Studies
One study (BeneGene-2),1,9 which was conducted by the sponsor, met the inclusion criteria of the sponsor submitted SLR. Characteristics of BeneGene-2 are summarized in Table 6.
Table 6: Details of Studies Included in the Systematic Review
Detail | BeneGene-2 |
|---|---|
Designs and populations | |
Study design | Phase III, open-label, single-arm, multicentre study |
Locations | 28 sites across 27 centres in 13 countries or territories: US, Australia, Brazil, Canada, France, Germany, Greece, Japan, Saudi Arabia, Sweden, Taiwan, Turkey, and the UK |
Key dates | Start date: July 29, 2019 Actual primary completion date: November 16, 2022 Estimated study completion date: December 18, 2029 |
N | 45 |
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Intervention | Fidanacogene elaparvovec was administered as a single IV infusion over 1 hour on day 1 at a dose of 5 × 1011 vector genomes per kg of body weight. For participants with a BMI > 30 kg/m2, dose was calculated using a maximum permissible BMI of 30 kg/m2 |
Comparator(s) | The data collected throughout the lead-in study, BeneGene-1, were utilized as the FIX prophylaxis control data for comparison with post–fidanacogene elaparvovec infusion in the BeneGene-2 trial |
Study duration | |
Lead-in study | At least 6 months |
Screening phase | 6 weeks |
Treatment phase | Fidanacogene elaparvovec was administered as a single infusion |
Follow-up phase | Year 1 (1 year from week 12 to month 15 following infusion of fidanacogene elaparvovec) |
Long-term follow-up phase | Year 2 to year 6 following infusion of fidanacogene elaparvovec |
Outcomes | |
Primary end point | ABRtotal |
Secondary and exploratory end points | Secondary:
Exploratory:
|
Publication status | |
Publications | Clinical trial registry entry for BeneGene-2 trial:
|
AAVrh74var = adeno-associated virus rh74 variant protein; ABRl = annualized bleeding rate for treated and untreated bleeds; ABRtreat = annualized bleeding rate for treated bleeds; AIR = annualized infusion rate; ALP = alkaline phosphatase; ALT = alanine transaminase; AST = aspartate transaminase; BMI = body mass index; BU = Bethesda units; FIX = coagulation factor IX; FIX:C = circulating coagulation factor IX; Haem-A-QoL = Hemophilia Quality of Life Questionnaire for Adults; HAL = Hemophilia Activities List; HBsAg = hepatitis B surface antigen; HBV = hepatitis B virus; HCV = hepatitis C virus; HJHS = Hemophilia Joint Health Score; nAb = neutralizing antibody; ULN = upper limit of normal.
Sources: BeneGene-2 Clinical Study Report9 and the sponsor’s Summary of Clinical Evidence.1
BeneGene-2 is a phase III, open-label, single-arm study investigating the use of fidanacogene elaparvovec for the treatment of adult male patients with moderately severe to severe hemophilia B (defined as FIX:C ≤ 2%). BeneGene-2 was conducted in 45 participants from 27 centres across 13 countries or territories around the globe, including 3 centres in Canada.1 The primary objective was to demonstrate the noninferiority of fidanacogene elaparvovec as opposed to FIX prophylaxis replacement therapy via ABRtotal from week 12 to month 15 postinfusion.1,10 Superiority of fidanacogene elaparvovec via ABRtotal was a secondary objective if noninferiority was established. BeneGene-2 is ongoing and expected to be completed in December 2029. Data from the data cut-off date (November 16, 2022) were used to support the sponsor’s present submission to CADTH.9,10 As of the data cut-off date, the mean duration of follow-up in the BeneGene-2 trial was |||| ||||| ||||||| with a median of |||| ||||| ||||| ||||.
The 45 participants enrolled in the pivotal BeneGene-2 phase III trial were selected from patients who had completed at least 6 months of FIX prophylaxis during the BeneGene-1 study.28 The Bene-Gene-1 study was an open-label, noninvestigational-product, prospective, multicentre, lead-in study conducted before the BeneGene-2 trial to prospectively collect efficacy and safety data of current FIX prophylaxis replacement therapy in the usual care setting of adult male participants with moderately severe to severe hemophilia B (FIX:C ≤ 2%) (Figure 1). FIX prophylaxis replacement therapy was continued during the patient screening stage in the BeneGene-2 trial until the infusion of fidanacogene elaparvovec. The outcome data of the FIX prophylaxis replacement therapy during the BeneGene-1 trial as well as the stage of patient screening were used by the sponsor to serve as a comparator to the fidanacogene elaparvovec arm in the BeneGene-2 trial.
The duration of follow-up for participants in the lead-in BeneGene-1 trial was at least 6 months until the completion of the trial for patients with hemophilia B, which was determined when the number of treated participants required for the pivotal BeneGene-2 trial were met. The mean duration of follow-up in the lead-in BeneGene-1 trial was |||| ||||| ||||||| with a median of |||| |||||||||| ||||| Based on number of infusions, ||||| ||||||| of the patients in the BeneGene-1 trial were ≥ 80% compliant and |||| |||||| were < 70% compliant.
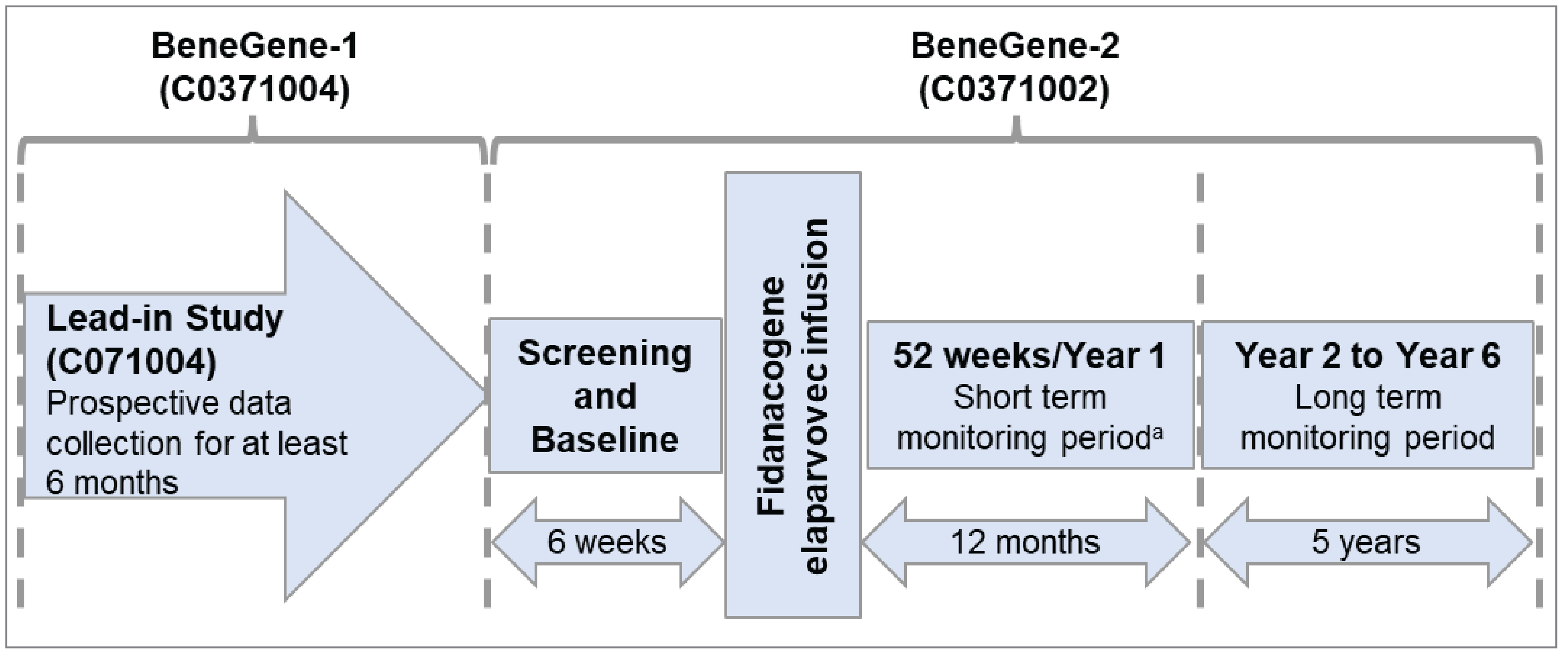
Populations
Inclusion and Exclusion Criteria
BeneGene-2 included male participants (≥ 18 years old) with moderately severe to severe hemophilia B (defined as FIX:C ≤ 2%), who must have completed 6 months or more of routine FIX prophylaxis therapy during the lead-in BeneGene-1 trial and agreed to suspend prophylactic FIX therapy following fidanacogene elaparvovec infusion. Patients with nAbs or a history of or presence of FIX inhibitors or with elevated LFTs or bilirubin, relevant unstable or significant liver disease, infection, or clinically relevant disease were excluded.
Interventions
In the pivotal BeneGene-2, fidanacogene elaparvovec was administered as a single IV infusion over 1 hour on day 1 at a dose of 5 × 1011 vg/kg of body weight. For participants with a body mass index greater than 30 kg/m2, the dose was calculated using a maximum permissible body mass index of 30 kg/m2.
Participants in the Benegene-2 trial were asked to suspend their FIX prophylaxis regimen following fidanacogene elaparvovec infusion. However, FIX replacement therapy was allowed as needed:
For a bleeding event, the trial investigator recommended an appropriate dose of FIX to treat the bleed because the dose of factor concentrate should include the recent steady-state fidanacogene elaparvovec–induced FIX activity levels to avoid overdosing resulting in a potential thrombotic event.
A participant might resume prophylaxis if fidanacogene elaparvovec was considered inefficacious, defined as FIX activity after 12 weeks of 2% or lower (in the absence of a confirmed FIX inhibitor) as determined by the central laboratory on 2 consecutive samples collected within a 2-week period, and/or over a 4-week period (in the absence of a confirmed FIX inhibitor) of 2 or more spontaneous bleeds into a major joint and/or target joint, or over a 4-week period (in the absence of a confirmed FIX inhibitor) of 3 or more spontaneous bleeds (consisting of joint bleeds and/or significant soft tissue/muscle or other site bleeds).
A tapering course of oral corticosteroids (i.e., prednisone or prednisolone) was the first consideration for the suppression of apparent immune hepatitis. Due to the importance of timely intervention of corticosteroids, decisions to begin treatment were based on local laboratory values. Approximately 60 mg to 100 mg of oral corticosteroids once a day for the first week was recommended as the starting dosage unless the investigator determined that a different regimen was preferable based on the patient’s medical history. The first-week dose could be extended for another week, according to the judgment of the investigator, if the patient experienced no adverse effects. Guidance given was that the subsequent prednisolone or prednisone taper should not be started until the ALT and/or AST levels had declined for at least 2 consecutive lab draws or returned to approximately baseline (pre-administration) levels and any decline in FIX:C activity had plateaued. Combined oral corticosteroids and IV corticosteroids (methylprednisolone) was recommended if there was no evidence of resolution of transaminase elevation while on oral corticosteroid treatment alone.
Outcomes
A list of efficacy end points assessed in this Clinical Review Report is provided in Table 7, followed by descriptions of the outcome measures. Summarized end points are based on outcomes included in the sponsor’s Summary of Clinical Evidence as well as any outcomes identified as important to this review by the clinical expert(s) consulted by CADTH, patient and clinician groups, and public drug plans. Using the same considerations, the CADTH review team selected end points that were considered to be most relevant to inform CADTH’s expert committee deliberations and finalized this list of end points in consultation with members of the expert committee. All summarized efficacy end points were assessed using GRADE. Select notable harms outcomes considered important for informing CADTH’s expert committee deliberations were also assessed using GRADE.
Descriptions of efficacy and safety outcomes presented in the BeneGene-2 trial and appraised in the CADTH Clinical Reviews are as described in the following section.10,29
Table 7: Outcomes Summarized from BeneGene-2
Outcome measure | Time point | BeneGene-2 |
|---|---|---|
ABRtotal (including information on percentage of patients without bleeds) | Year 1 following fidanacogene elaparvovec infusiona | Primary |
ABRtotal (including information on percentage of patients without bleeds) | Overallb | Secondary |
ABRtreat (including information on percentage of patients without bleeds) | Year 1 following fidanacogene elaparvovec infusiona | Key secondary |
ABRtreat (including information on percentage of patients without bleeds) | Overallb | Secondary |
ABRjoint (including information on percentage of patients without bleeds) | Year 1 following fidanacogene elaparvovec infusiona | Secondary |
ABRjoint (including information on percentage of patients without bleeds) | Overallb | Secondary |
AIR | Year 1 following fidanacogene elaparvovec infusiona | Key secondary |
AIR | Overallb | Secondary |
Annualized FIX consumption | Overallb | Secondary |
HJHS | Week 52 following fidanacogene elaparvovec infusion | Secondary |
HJHS | Week 104 following fidanacogene elaparvovec infusion | Exploratory |
Haem-A-QoL (physical health) | Week 52 following fidanacogene elaparvovec infusion | Secondary |
Haem-A-QoL (physical health) | Week 104 following fidanacogene elaparvovec infusion | Exploratory |
Haem-A-QoL (total score) | Week 52 following fidanacogene elaparvovec infusion | Exploratory |
Haem-A-QoL (total score) | Week 104 following fidanacogene elaparvovec infusion | Exploratory |
HAL (complex lower extremity activities) | Week 52 following fidanacogene elaparvovec infusion | Secondary |
HAL (complex lower extremity activities) | Week 104 following fidanacogene elaparvovec infusion | Exploratory |
HAL (total score) | Week 52 following fidanacogene elaparvovec infusion | Exploratory |
HAL (total score) | Week 104 following fidanacogene elaparvovec infusion | Exploratory |
ABRjoint = annualized bleeding rate for untreated and treated joint bleeds; ABRtotal = annualized bleeding rate for treated and untreated bleeds; ABRtreat = annualized bleeding rate for treated bleeds; AIR = annualized infusion rate; FIX = coagulation factor IX; Haem-A-QoL = Hemophilia Quality of Life Questionnaire for Adults; HAL = Hemophilia Activities List; HJHS = Hemophilia Joint Health Score.
aYear 1 refers to the period between week 12 and month 15 following fidanacogene elaparvovec infusion.
bOverall refers to the period between week 12 and the data cut-off date of November 16, 2022. The mean duration of follow-up in the pivotal BeneGene-2 was |||| ||||| ||||||| with a median of |||| ||||| ||||| |||||
Source: BeneGene-2 study protocol incorporating Amendment 3.10
Efficacy Outcomes
Annualized Bleeding Rate for Treated and Untreated Bleeds
The ABRtotal included treated bleeds (defined as an event that required FIX infusion within 72 hours of signs or symptoms of bleeding) and untreated bleeds (defined as a bleeding event that did not require FIX infusion within 72 hours of signs or symptoms of bleeding). Every occurrence of bleed events was counted as a separate bleed if occurring more than 72 hours after the previous bleed at the same site or more than 72 hours after stopping treatment.
The ABRtotal for each participant was calculated using the following formula:
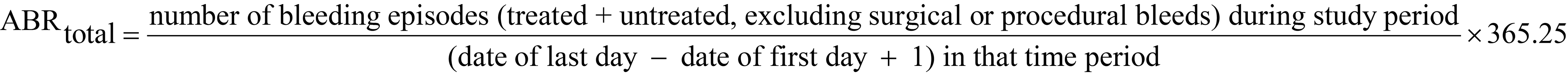
The ABRtotal excluded surgical or procedural bleeds (defined as a bleed related to a procedure or surgery such as hematomas or bruising resulting from any surgery or invasive procedure or invasive diagnostic procedure, and bleeds related to procedures or surgery not associated with any trauma except procedure- or surgery-induced trauma). If a prophylaxis FIX regimen was resumed for a participant after fidanacogene elaparvovec infusion, the time period following the resumption of the prophylaxis regimen was excluded from the ABR end point calculation, which means the bleeding events would be excluded and the time period of observation would be deducted as well.
To calculate the ABRtotal from the lead-in BeneGene-1 trial, the denominator of the formula presented earlier was the date of fidanacogene elaparvovec infusion minus the date of enrolment in the BeneGene-1 trial in the comparison between ABRtotal at year 1 following fidanacogene elaparvovec infusion versus the lead-in period. The same comparator group of the lead-in period was utilized in the comparison of ABRtotal (overall) versus the lead-in period. The time periods were the same across outcomes for the ABR, AIR, and annualized FIX consumption, except that the postresumption period for the ABR was not counted in the follow-up duration and bleeds occurring postresumption were excluded from the ABR calculation as well. However, the postresumption period was included for the AIR calculation.
In addition, a hand-held electronic diary was provided to all participants. The participants were required to enter any occurrence of hemophilic bleeding episodes (including date, time, location, and etiology) and any exogenous FIX replacement (including date, time, reason, and dose) required to treat the bleeds in the diary. If bleeding episodes or treatments were not entered in the diary during the appropriate time window, data were to be entered by the investigator (or appropriate site staff member) according to the process in place with appropriate source documentation in the participant’s medical record.
Annualized Bleeding Rate for Treated Bleeds and Annualized Bleeding Rate for Treated and Untreated Joint Bleeds
The ABRtreat involved treated bleeds only, while the ABRjoint involved joint bleeds only, which were defined as a bleeding episode characterized by rapid loss of range of motion compared with baseline that was associated with any combination of the following: pain or an unusual sensation in the joint, palpable swelling, and warmth of the skin over the joint. The calculation of an ABRtreat or ABRjoint used the same methods as for the ABRtotal, except only treated bleeds or joint bleeds were included.
Annualized Infusion Rate
The AIR included all FIX infusions during the observation period for any purpose, including treating bleeding, preventive purposes, perioperative purposes, or if a prophylaxis FIX regimen was resumed.
The AIR for each participant was calculated using the following formula:

Descriptions of HJHS, Haem-A-QoL, and HAL are shown in Table 8.
Table 8: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
HJHS | The HJHS version 2.1 comprises an assessment of specific features, or items, of the 6 index joints (elbows, knees, and ankles) as well as an assessment of global gait. For each of the 6 joints, the following items are scored:
The maximum score for an individual index joint is 20. Gait is scored 0 to 4 based on walking, stairs, running, and hopping on 1 leg. The total score is the sum of all joint and gait scores (range = 0 to 124), with a higher number indicating more severe joint damage.30,31 | Validity: In a multicentre international study containing hemophilia patients as well as healthy adults, HJHS total scores were highly correlated with the WFH Gilbert scores (Spearman correlation, rs = 0.95), which is the original WFH Orthopedic Joint Score, demonstrating convergent construct validity.30 Discriminant (known-groups) construct validity was evaluated by the Kruskal-Wallis nonparametric analysis of variance. The HJHS total score significantly differentiated between age groups (Kruskal-Wallis t = 35.02, P < 0.001) and disease severity in persons with hemophilia.30 Reliability: In a study consisting of male hemophilia patients in the US, the Cronbach alpha value was 0.97 for the HJHS total score, above the threshold of 0.70 established in previous studies, indicating sufficient internal consistency.32,33 All items on the HJHS had been reported to capture sufficient correlation with their respective joint total scores (r = 0.34 to 0.83, where r is the Pearson product moment correlation coefficient).32,34 In another study consisting of male hemophilia patients in the US, the HJHS Ankle domain reached a correlation of r > 0.5 for several domains and summary scores related to physical function, including scores specific to activity of the lower extremities (HAL lying/sitting/kneeling/standing, functions of the legs, use of transportation, complex lower extremity activities, and overall activity). HJHS total scores also demonstrated similar correlations for similar domains and summary scores, except use of transportation. However, HJHS global gait did not reach a correlation of r > 0.5 with any patient-reported outcome instrument domain or summary scores.31 In a multicentre international study of hemophilia patients and healthy adults, the HJHS 2.1 items demonstrated adequate internal reliability (Cronbach alpha = 0.88).33 Item scores were correlated with total scores, with almost all HJHS items (muscle atrophy, crepitus, flexion and extension loss, joint pain, and strength) being highly correlated (alpha > 0.70), except for swelling and duration of swelling, which were only moderately correlated.30 Responsiveness: The HJHS is more sensitive to early joint changes than the Gilbert score.35 It can reportedly distinguish between different prophylactic strategies in young adults with severe hemophilia,36 between severe and nonsevere hemophilia in children35,37 and is responsive to changes following physiotherapy treatment.38 However, it is so sensitive that it showed positive scores in 40% of unaffected young adults (total score ≤ 3 points).39,40 | No MID was identified in the sponsor’s literature search for this population. |
Haem-A-QoL | The Haem-A-QoL questionnaire assesses the quality of life of adults (aged ≥ 17 years) with hemophilia. It contains 46 items in 10 domains: physical health (5 items), feelings (4 items), view of self (5 items), sports and leisure (5 items), work and school (4 items), dealing with hemophilia (3 items), treatment (8 items), future (5 items), family planning (4 items), and partnership and sexuality (3 items); the total score is also considered. The physical health domain was considered the primary domain and was assessed as the secondary end point; other domains were considered exploratory. Each item was answered considering the last 4 weeks on the 5-point Likert scale ranging from “never” to “all of the time,” with several items having a “not applicable” option. Nonmissing scores were averaged for each domain then rescaled from 0 to 100; lower scores represent a higher quality of life.41-43 | Validity: In a severe hemophilia (A or B) population (aged ≥ 12 years), several Haem-A-QoL domains and “total score” demonstrated known-groups and convergent validity when compared with other trial measures, including the EQ-5D questionnaire (items and total scores) and joint impairment.42 Reliability: Internal consistency and reliability was previously reported to be sufficiently adequate (Cronbach alpha > 0.70)33 for 9 of the 10 Haem-A-QoL domains and for “total score” in a severe hemophilia (A or B) population (aged ≥ 12 years) at baseline.42 Responsiveness: Change in score correlations (baseline to 28 weeks) between the EQ-5D and the Haem-A-QoL total score, and physical health and feelings domains were moderate in magnitude (│r│ ≥ 0.33; P < 0.03), demonstrating sensitivity to change for these outcome measures in hemophilia A patients.42 | In a severe hemophilia (A or B) population (aged ≥ 12 years), the most indicative meaningful within-patient change was a reduction of 7.1 points for the total score and 10.0 points for physical health score over 6 months based on anchor- and distribution-based methods.43 In the same population, the Haem-A-QoL total-score threshold for meaningful change ranged from 3.5 to 8.1 points and the physical health domain threshold for meaningful change ranged from 8.0 to 11.9 points (distribution-based methods).43 These thresholds were consistent in other studies.41 |
HAL | HAL version 2 measures the impact of hemophilia on functional abilities in adults. It contains 42 items in 7 domains measuring aspects of physical function as experienced over the past month through a 0- to 100-point scale:31 lying/sitting/kneeling/standing (8 items), lower (leg) functioning (9), upper (arm) functioning (4), transportation (3), self-care (5), household tasks (6), and sports/leisure (7). An overall sum score and 3 component scores (upper extremity; basic lower extremity; and complex lower extremity activities) can also be calculated. The complex lower extremity activities component score was assessed as the secondary end point and other scores were considered exploratory. All individual items were rated on a 6-point scale from 1 (impossible) to 6 (never) describing difficulty due to hemophilia in the past month. Several items allowed a response of “not applicable.” Overall, component, individual domain, and total scores were calculated with higher values corresponding to a higher quality of life i.e., less functional limitations in performing tasks.44,45 | Validity: In a severe hemophilia (A or B) population, the HAL was correlated with 4 performance tests (Spearman rank correlation coefficient, r = 0.47 to 0.84), and was reported to have sufficient evidence of convergent validity.45 In another study of adults with mild to severe hemophilia B patients and caregivers of children with hemophilia B, construct validity was demonstrated as nearly all correlations showed satisfactory validity (Pearson product moment correlation coefficient, r > 0.37).46,47 Satisfactory construct validity was also demonstrated in other studies with similar populations.31,47 Reliability: In an adult (aged ≥ 18 years) mild to severe hemophilia (A or B) population, the HAL demonstrated acceptable reliability for the sum and component scores, with ICCs > 0.9.48,49 In another study of adults with mild to severe hemophilia B patients and caregivers of children with hemophilia B, adequate internal consistency (Cronbach alpha > 0.70)33 was demonstrated for all domains and component scores, except for HAL self‐care. The item‐total correlation generally was high for all items and was particularly high for self‐care (Pearson correlation range, r = 0.74 to 0.85) and basic lower extremity activities (r = 0.75 to 0.82).46 The internal consistency of the 3 components (upper extremity activities, basic lower extremity activities, and complex lower extremity activities) was high (Cronbach alpha = 0.93 to 0.95), as was internal consistency for the 7 domains of the HAL (alpha = 0.61 to 0.96).33,45 Test-retest reliability was also demonstrated in another study with adult male hemophilia patients.50 | No published thresholds based on anchor-based analyses were identified for the HAL. In an adult (aged ≥ 18 years) mild to severe hemophilia (A or B) population, the literature reports distribution-based estimates for meaningful within-patient change ranging from 4.85 (SEM) to 13.45 (SDC) for the HAL complex lower extremity activities score and 3.68 (SEM) to 10.20 (SDC) for the HAL total score. The basic lower extremity component score had the highest variation with SEM (6.0) and SDC value (16.7), the upper extremity component score had the lowest variation with SEM (3.3) and SDC value (9.2).48 |
Haem-A-QoL = Hemophilia Quality of Life Questionnaire for Adults; HAL = Hemophilia Activities List; HJHS = Hemophilia Joint Health Score; HJHS 2.1 = Hemophilia Joint Health Score version 2.1; ICC = intraclass correlation coefficient; MID = minimal important difference; SDC = smallest detectable change; SEM = standard error of measurement; WFH = World Federation of Hemophilia.
Harms Outcomes
The harms outcomes assessed in the BeneGene-2 trial included TEAEs, TESAEs, withdrawals due to AEs, mortality, and notable harms (e.g., increased ALT, increased AST, and abnormal hepatic function). AEs were coded using the Medical Dictionary for Regulatory Activities. TEAEs included any AEs that occurred on or after the infusion of fidanacogene elaparvovec.
Statistical Analysis
In the pivotal BeneGene-2 trial, all hypothesis testing was 2-sided, unless specified otherwise. The primary objective was to determine the efficacy of fidanacogene elaparvovec in adult males with moderately severe to severe hemophilia B (FIX:C ≤ 2%), quantified by the primary end point of an ABRtotal. The primary hypothesis was noninferiority pre- and posttreatment with fidanacogene elaparvovec on the ABRtotal. If noninferiority was demonstrated on the ABRtotal outcome, subsequent testing for superiority would be conducted. The test of superiority via an ABRtotal was considered a secondary analysis.
A gatekeeping process was applied to control for multiplicity when testing multiple end points at the primary analysis. The subsequent hypothesis testing was performed only after success on a previous hypothesis test, with each test performed at the type I error rate defined for the primary analysis (0.05). The analyses would cease when a failure occurred. The sequence of gatekeeping process of multiple hypothesis tests in the BeneGene-2 trial is shown below in the following order:
ABRtotal noninferior to FIX prophylaxis regimen (The upper bound of the confidence interval of the difference in ABRtotal and ABRtreat between pre– and post–fidanacogene elaparvovec infusion was compared to the noninferiority margin. All other hypothesis tests were 2-sided.).
ABRtreat noninferior to FIX prophylaxis regimen.
AIR superior to FIX prophylaxis regimen.
Steady state FIX:C > 5% (Steady-state FIX:C was analyzed using a 1-sided, 1-sample t test.).
FIX consumption superior to FIX prophylaxis regimen.
ABRtreat superior to FIX prophylaxis regimen.
ABRtotal superior to FIX prophylaxis regimen.
Haem-A-QoL Physical Health domain significantly improved from baseline.
HAL Complex Lower Extremity component significantly improved from baseline.
ABRtotal of specific bleed type noninferior to FIX prophylaxis regimen, and HJHS.
Participants who completed the lead-in BeneGene-1 study were screened into the BeneGene-2 trial to achieve a desired sample size of 40 eligible participants assigned to fidanacogene elaparvovec. The sample size could have exceeded 40 participants because all participants who completed the lead-in study and met other eligibility criteria were allowed to participate in this study. When all 40 participants had completed at least 15 months of follow-up after fidanacogene elaparvovec infusion, the number of observed ABRtotal events would provide at least 90% power (1-sided test with an alpha of 0.025) to demonstrate noninferiority of fidanacogene elaparvovec compared to prophylaxis treatment against a noninferiority margin of 3.0 bleeds per year under an assumed negative binomial regression with repeated measures.
The noninferiority margin for the ABRtotal was determined by the sponsor using the constancy assumption and the “95% to 95%” methods.51,52 The noninferiority margin was based on previously reported effects of prophylaxis over on-demand treatment and was expressed as the mean difference in the ABR in a single-arm trial, with a switch from on-demand to prophylaxis using a paired comparison. Based on the lower bound of the CI of an estimate for the ABR for treated bleeds (defined as an event that required FIX infusion within 72 hours of signs or symptoms of bleeding) in on-demand participants, the ABR for treated bleeds was assumed to be higher by at least 24.5 and was considered M1 in the noninferiority test setting for the ABR for treated bleeds. It was assumed that the treatment difference (on-demand – prophylaxis) in the ABRtotal was proportional to that in the ABR for treated bleeds. The ratio of the treatment difference (ABRtotal over ABRtreat) was estimated to be 1.17. The M1 for ABRtotal was therefore estimated to be 28.7 (1.17 × 24.5). Given the large effect size of prophylaxis treatment (over on-demand therapy), an appropriate value for M2 was considered to preserve a sufficiently large proportion of this effect. Simulations were conducted to assess preservation levels of 80%, 85%, and 90% of M1. These percentages corresponded to noninferiority margin values of 5.7, 4.3, and 2.9 bleeding events per year respectively, on an absolute scale. A value of 3.0 for M2 (approximately 89.5% of the M1 effect preserved) was proposed to be both clinically meaningful and yielded a reasonable sample size for establishing efficacy. In addition, the noninferiority margin of 3.0 was in line with differences observed in the ABR for treated bleeds in the real-world and clinical-trial settings. A superiority margin of 0 was used for the ABRtotal to demonstrate a better response for fidanacogene elaparvovec.
Details of the statistical analysis of efficacy end points in the BeneGene-2 trial are presented in Table 9.
Table 9: Statistical Analysis of Efficacy End Points in BeneGene-2
End point | Primary statistical model | Handling of missing data | Sensitivity/subgroup analyses |
|---|---|---|---|
ABRtotal |
| For participants with incomplete data for the analysis time period, or if a participant had not yet been followed for the full length of the analysis time period, individuals were censored at the time of last known follow-up. | Sensitivity analyses
Subgroup analyses
|
ABRtreat | As described for ABRtotal (a noninferiority test of treatment difference in ABRtreat was performed similarly as ABRtotal, with a noninferiority margin of 3.0). | Sensitivity analyses
| |
ABRjoint | ABRjoint was obtained using a similar approach of data collection and end point derivation as described for ABRtotal. | NA | |
AIR |
|
| Sensitivity analyses
|
Annualized FIX consumption | Annualized FIX consumption was obtained using a similar approach to data collection and end point derivation as described for AIR. | Sensitivity analyses
| |
HJHS |
| HJHS was not calculated if > 20% (> 10 of 49 items) items were missing or not evaluable. | NA |
Haem-A-QoL |
|
| NA |
HAL |
|
| NA |
ABRjoint = annualized bleeding rate for treated and untreated joint bleeds; ABRtotal = annualized bleeding rate for treated and untreated bleeds; ABRtreat = annualized bleeding rate for treated bleeds; AIR = annualized infusion rate; CI = confidence interval; FIX = coagulation factor IX; Haem-A-QoL = Hemophilia Quality of Life Questionnaire for Adults; HAL = Hemophilia Activities List; HJHS = Hemophilia Joint Health Score. NA = not applicable.
Sources: BeneGene-2 Study Protocol Incorporating Amendment 3,10 BeneGene-2 Statistical Analysis Plan version 629 and the sponsor’s Summary of Clinical Evidence.1
Analysis Populations
Analysis populations of the BeneGene-2 trial are summarized in Table 10.
Table 10: Analysis Populations of BeneGene-2
Population | Definition | Application |
|---|---|---|
Dosed | All participants enrolled in the study who received a fidanacogene elaparvovec infusion | Used for most primary analyses of efficacy outcomes |
Safety | All participants enrolled in the study who received a fidanacogene elaparvovec infusion | Same as the dosed population; used for analyses of safety outcomes |
Evaluable | All participants enrolled in the study who received a fidanacogene elaparvovec infusion and have no significant interruptiona of efficacy measurement | Additional analyses, such as sensitivity analyses |
aSignificant interruption was determined after discussion between the investigator and the medical monitor, e.g., if a participant required a major surgery, this would be a significant interruption of measurement.
Sources: BeneGene-2 study protocol incorporating Amendment 310 and the sponsor’s Summary of Clinical Evidence.1
Protocol Amendments and Deviations
Three amendments were made to the protocol. The original protocol and the protocol with Amendment 3 were issued on December 13, 2018, and June 29, 2022, respectively. In protocol Amendment 3, the primary end point was revised from the ABRtreat to the ABRtotal. In protocol amendments 2 and 3, the start point for outcome analysis was revised from day 1 following fidanacogene elaparvovec infusion to week 12 postinfusion to correspond with the estimated FIX:C steady-state onset.10
Common protocol deviations were related to specimens that could not be analyzed, lab work not done, procedures and or tests not done, procedures and/or tests not performed following protocol, procedures and or tests performed out of window, and electronic diary not completed.9
Results
Patient Disposition
The lead-in BeneGene-1 study enrolled 102 participants, of whom 59 (57.8%) completed the study and 3 (2.9%) discontinued. As of the data cut-off on November 2, 2022, 40 participants (39.2%) were continuing the study. Among 59 patients who completed the BeneGene-1 study, 8 decided not to continue onto the BeneGene-2 trial; as a result, 51 patients from the lead-in BeneGene-1 study entered the screening phase of the BeneGene-2 trial.
A summary of patient disposition in the BeneGene-2 trial is presented in Table 11. Of the 51 patients from the BeneGene-1 study, 5 (9.8%) discontinued due to screen failures, reasons for which included an nAb titre above the established threshold; the patient did not complete 6 months of routine FIX prophylaxis therapy during the lead-in study and had 50 or more lifetime exposure days to a FIX protein product; the patient was unable to comply with scheduled visits, treatment plans and laboratory tests and other study procedures for up to 6 years postinfusion; the patient had current unstable liver or biliary disease; and screening laboratory values for hemoglobin, platelets and creatinine were outside of acceptable range.28 In addition, 1 patient withdrew from screening due to the COVID-19 pandemic and hepatocellular carcinoma risk (classified under “Other” by the sponsor).
All 45 participants completed the 1-time fidanacogene elaparvovec IV infusion. Forty-three (84.3%) of the participants had completed follow-ups up to and including 52 weeks following fidanacogene elaparvovec IV infusion as of the data cut-off on November 16, 2022. During the long-term follow-up, 1 patient discontinued after losing motivation due to a self-reported “lack of efficacy.”28
Table 11: Summary of Patient Disposition from BeneGene-2
Patient disposition | BeneGene-2 |
|---|---|
Fidanacogene elaparvovec | |
Screening, N (%) | 51 (100.0) |
Completed, n (%) | 45 (88.2) |
Discontinued, n (%) | 6 (11.8) |
Screen failure | 5 (9.8) |
Withdrawal by participant | 0 |
Other, n (%) | 1 (2.0) |
Treatment, n (%) | 45 (88.2) |
Ongoing, n (%) | 0 |
Discontinued, n (%) | 0 |
Completed, n (%) | 45 (88.2) |
Follow-up (up to and including 52 weeks), n (%) | 45 (88.2) |
Ongoing, n (%) | 2 (3.9) |
Discontinued, n (%) | 0 |
Completed, n (%) | 43 (84.3) |
Long-term follow-up, n (%) | 43 (84.3) |
Ongoing, n (%) | 42 (82.4) |
Discontinued, n (%) | 1 (2.0) |
Withdrawal by participant | 1 (2.0)a |
Completed, n (%) | 0 |
Safety population, n (%) | 45 (88.2) |
FIX = coagulation factor IX.
Note: As of the data cut-off date of November 16, 2022.
aThe participant resumed FIX prophylaxis on day 365 following fidanacogene elaparvovec infusion and withdrew on day 910.
Sources: BeneGene-2 Clinical Study Report9 and the sponsor’s Summary of Clinical Evidence.1
Baseline Characteristics
The baseline characteristics listed in Table 12 are limited to those most relevant to this review or assumed to affect the outcomes or interpretation of the study results. Patients who completed the lead-in BeneGene-1 study and enrolled in the BeneGene-2 trial were aged between 18 and 62 years with a median of 29 years. The majority of patients were white (73.3%). All participants had a factor mutation, including 7 (15.6%) with a nonsense mutation, 20 (44.4%) with a missense mutation, 0 with an insertion, 9 (20%) with a deletion, 0 with an inversion, and 9 (20%) with other mutations. Thirteen participants (28.9%) had at least 1 target joint at baseline, which was defined as a major joint (e.g., hip, elbow, wrist, shoulder, knee, and ankle) into which repeated bleeds occur (3 or more spontaneous bleeds into a single joint within a consecutive 6-month period). Moreover, 84.4% of the 45 patients had a FIX level below 1%, while the remaining patients’ FIX levels were between 1% and 2% (inclusive). In addition, 53.3% of the patients had a family history of hemophilia.
Exposure to Study Treatments
Details on the extent of exposure to fidanacogene elaparvovec in the BeneGene-2 trial are summarized in Table 13. In the 45 participants, the median total dose of fidanacogene elaparvovec infusion was ||||||||||||||||||||||||||||||||||||||||||||||||||||| vg.
Prior and Concomitant Treatments
Prior or concomitant medications were reported in || ||||||| of the safety population of BeneGene-2 (N = 45).9 The most commonly reported prior medications included paracetamol in 7 participants (15.6%) and celecoxib in 6 participants (13.3%). The most commonly used concomitant medications included prednisone in || ||||||| participants, prednisolone in || ||||||| participants, and paracetamol in || ||||||| participants.
Table 12: Summary of Baseline Characteristics From BeneGene-2 (Safety Population)
Characteristic | BeneGene-2 |
|---|---|
Fidanacogene elaparvovec (N = 45) | |
Age (years), n (%) | |
n | 45 |
< 35 | 28 (62.2) |
≥ 35 | 17 (37.8) |
Mean (SD) | 33.18 (10.947) |
Median (minimum to maximum) | 29.0 (18.0, 62.0) |
Sex, n (%) | |
Male | 45 (100.0) |
Female | 0 |
Race, n (%) | |
White | 33 (73.3) |
Black or African American | 1 (2.2) |
Asian | 7 (15.6) |
American Indian or Alaska Native | 0 |
Native Hawaiian or other Pacific Islander | 0 |
Not reported | 4 (8.9) |
Region, n (%) | |
Asia and Pacific | 6 (13.3) |
Australia | 2 (4.4) |
Europe | 13 (28.9) |
Middle East | 9 (20.0) |
North America | 12 (26.7) |
South America | 3 (6.7) |
Weight (kg) | |
n | 45 |
Mean (SD) | 86.66 (16.988) |
Median (minimum to maximum) | 86.20 (53.4, 141.6) |
BMI (kg/m2) | |
n | 45 |
Mean (SD) | 27.85 (5.466) |
Median (minimum to maximum) | 27.7 (17.6, 48.4) |
Hepatitis C virus, n (%)a | 15 (33.3) |
Hepatitis B virus, n (%)a | 13 (28.9) |
HIV, n (%)a | 3 (6.7) |
Factor mutation, n (%) | |
Yes | 45 (100.0) |
Nonsense | 7 (15.6) |
Missense | 20 (44.4) |
Insertion | 0 |
Deletion | 9 (20.0) |
Inversion | 0 |
Other | 9 (20.0) |
No | 0 |
Number of participants with target joints,b n (%) | |
Yes | 13 (28.9) |
Right shoulder | 1 (2.2) |
Left shoulder | 0 |
Right elbow | 3 (6.7) |
Left elbow | 4 (8.9) |
Right wrist | 0 |
Left wrist | 0 |
Right hip | 0 |
Left hip | 1 (2.2) |
Right knee | 2 (4.4) |
Left knee | 2 (4.4) |
Right ankle | 6 (13.3) |
Left ankle | 6 (13.3) |
No | 32 (71.1) |
Medical history and concurrent illnesses, n (%) | |
Musculoskeletal and connective tissue disorders | 22 (48.9) |
Surgical and medical procedures | 21 (46.7) |
Prior FIX product exposure, n (%) | |
Standard half-life FIX | 16 (35.6) |
Plasma-derived FIX | 2 (4.4) |
Recombinant FIX | 14 (31.1) |
Extended half-life FIX | 29 (64.4) |
Disease severity | |
< 1% | 38 (84.4) |
1% to 2% (inclusive) | 7 (15.6) |
Family history of hemophilia | |
Yes | 24 (53.3) |
No | 20 (44.4) |
Unknown | 1 (2.2) |
Number of prior exposure days to FIX product | |
≥ 50 days | 45 (100.0) |
Unknown | 0 |
BMI = body mass index; FIX = coagulation factor IX; SD = standard deviation.
Note: As of the data cut-off date of November 16, 2022.
aCounts and percentages of subjects with positive laboratory results for the corresponding parameter.
bA target joint was defined as a major joint (e.g., hip, elbow, wrist, shoulder, knee, and ankle) into which repeated bleeds occur (3 or more spontaneous bleeds into a single joint within a consecutive 6-month period). A target joint is considered resolved when there are no more than 2 bleeds into the joint within a 12-month period.
Sources: BeneGene-2 Clinical Study Report9,53 and the sponsor’s Summary of Clinical Evidence.1
Table 13: Summary of Patient Exposure From BeneGene-2 (Dosed Population)
Exposure | BeneGene-2 |
|---|---|
Fidanacogene elaparvovec (N = 45) | |
Duration of infusion, (minutes) | |
n | 45 |
Mean (SD) | |||| |||||| |
Median (minimum to maximum) | |||| |||| |||| |
Total dose, (vg) | |
n | 45 |
Mean (SD) | |||| |||| |||||||| |||| |||| |
Median (minimum to maximum) | |||| |||| |||||||| |||| |||||||| |||| || |
Total dose infused, (vg/kg for fidanacogene elaparvovec) | |
n | 45 |
Mean (SD) | |||| |||| |||||||| |||| |||| |
Median (minimum to maximum) | |||| |||| |||||||| |||| |||||||| |||| || |
Actual dose was adjusted from planned,a n (%) | |
Yes | 0 |
Adverse events | 0 |
Insufficient clinical response | 0 |
Other | 0 |
No | 45 (100.0) |
SD = standard deviation; vg = vector genome.
Note: As of data cut-off date November 16, 2022.
aThe actual dose was adjusted from the planned dose when a participant’s body mass index was greater than 30 kg/m2.
Source: BeneGene-2 Clinical Study Report.9
Use of FIX Prophylaxis Post–Fidanacogene Elaparvovec Infusion
The summary of participants resuming FIX prophylaxis regimen after fidanacogene elaparvovec infusion in the BeneGene-2 trial is presented in Table 14. In total, 6 out of 45 (13.3%) participants had resumed prophylaxis therapy, with time to resumption ranging from 5.1 months to 20.5 months. All of the patients resumed FIX prophylaxis within 15 months after fidanacogene elaparvovec infusion except 1 patient who resumed after 15 months postinfusion.
Table 14: Summary of Participants Resuming FIX Prophylaxis Post–Fidanacogene Elaparvovec Infusion (Dosed Population)
Exposure | BeneGene-2 |
|---|---|
Fidanacogene elaparvovec (N = 45) | |
Participants resuming FIX prophylaxis, n (%)a | |
At 15 months postinfusion (45 patients at the start of the interval) | 5 (11.1) |
At year 2 postinfusion (39 patients at the start of the interval) | 1 (2.6) |
At year 3 postinfusion (24 patients at the start of the interval) | 0 (0) |
Overall (45 patients at the start of the interval) | 6 (13.3) |
FIX = coagulation factor IX.
Note: As of the data cut-off date of November 16, 2022. Overall includes from day 1 to data cut-off date.
aThe percentage was calculated using the number of participants who were in follow-up at the start of the interval as the denominator.
Source: BeneGene-2 Clinical Study Report.9
Efficacy
Key efficacy results in the dosed population of the BeneGene-2 trial are presented in Table 15. The data cut-off date was November 16, 2022.
Annualized Bleeding Rate for Treated and Untreated Bleeds
The model estimates of the mean difference in the ABRtotal between patients who were treated with fidanacogene elaparvovec during the BeneGene-2 trial and the ABRtotal from the same patients treated with routine FIX prophylaxis during the lead-in BeneGene-1 trial was −3.13 (95% CI, −5.44 to −0.81) at year 1 following fidanacogene elaparvovec infusion, favouring fidanacogene elaparvovec, which met the noninferiority margin (3.0 bleeds per year) as well as the criteria for subsequent hierarchical testing for superiority. The difference in the ABRtotal from week 12 to data cut-off date (overall) was ||||| |||||| || ||||||, in favour of fidanacogene elaparvovec.
Totals of 29 of 45 (64.4%) of the patients treated with fidanacogene elaparvovec during the BeneGene-2 trial and 13 of 45 (28.9%) of the patients treated with routine FIX prophylaxis during the lead-in BeneGene-1 trial had no bleeds (untreated and treated) at year 1 following fidanacogene elaparvovec infusion. From week 12 to the data cut-off date postinfusion, ||||| ||||||| of the patients treated with fidanacogene elaparvovec infusion during BeneGene-2 and ||||| ||||||| of the patients treated with routine FIX prophylaxis during the lead-in BeneGene-1 trial had no bleeds.
A “jump to reference” sensitivity analysis was conducted to assess the impact of participants who discontinued or resumed FIX prophylaxis before completing the 15 months of follow-up after fidanacogene elaparvovec infusion. In total, 5 patients were included in the sensitivity analysis, among them 4 who resumed FIX prophylaxis within 15 months after fidanacogene elaparvovec infusion as well as 1 who resumed FIX prophylaxis within 15 months after fidanacogene elaparvovec infusion but who withdrew on ||| ||| postinfusion. One other patient also resumed FIX prophylaxis but only after 15 months postinfusion, and was therefore excluded from the sensitivity analysis. Results |||| |||||||||| with the primary analyses: The difference in the ABRtotal was ||||| |||||| || ||||||, favouring fidanacogene elaparvovec, and the percentages of patients who had no treated and untreated bleeds were ||||| in patients who were treated with fidanacogene elaparvovec infusion during the BeneGene-2 trial versus ||||| the same patients who were treated with routine FIX prophylaxis during the lead-in BeneGene-1 trial (Appendix 1, Table 19).
Annualized Bleeding Rate for Treated Bleeds
The estimated mean differences in the ABRtreat between patients treated with fidanacogene elaparvovec during the BeneGene-2 trial and the same patients treated with routine FIX prophylaxis during the lead-in BeneGene-1 trial were −2.62 (−4.27 to −0.96) at year 1 following fidanacogene elaparvovec infusion and ||||| |||||| || |||||| from week 12 to the data cut-off date, all in favour of fidanacogene elaparvovec.
A comparison of the 2 studies found that 73.3% (33 of 45) of the patients treated with fidanacogene elaparvovec during the BeneGene-2 trial and 35.6% (16 of 45) of the patients treated with routine FIX prophylaxis during the lead-in BeneGene-1 trial had no treated bleeds at year 1 following fidanacogene elaparvovec infusion. ||||| ||||||| of the patients treated with fidanacogene elaparvovec infusion during the BeneGene-2 trial and ||||| ||||||| of the patients treated with routine FIX prophylaxis during the lead-in BeneGene-1 trial had no treated bleeds.
Annualized Bleeding Rate for Treated and Untreated Joint Bleeds
The estimated mean difference in the ABRjoint between patients treated with fidanacogene elaparvovec during the BeneGene-2 trial and the same patients treated with routine FIX prophylaxis during the lead-in BeneGene-1 trial was ||||| |||||| || |||||| at year 1 following fidanacogene elaparvovec infusion, ||||||||| fidanacogene elaparvovec. From week 12 to the data cut-off date, the difference was ||||| |||||| || ||||||| ||||||||| fidanacogene elaparvovec.
Comparing the 2 studies, 68.9% (31 of 45) of the patients treated with fidanacogene elaparvovec during the BeneGene-2 trial and 44.4% (20 of 45) of the patients treated with routine FIX prophylaxis during the lead-in BeneGene-1 trial had no joint bleeds at year 1 following fidanacogene elaparvovec infusion. ||||| ||||||| of the patients treated with fidanacogene elaparvovec infusion during BeneGene-2 and ||||| ||||||| of the patients treated with routine FIX prophylaxis during the lead-in BeneGene-1 trial had no joint bleeds.
Annualized Infusion Rate
The differences in the AIR between patients treated with fidanacogene elaparvovec during the BeneGene-2 trial and the same patients treated with routine FIX prophylaxis during the lead-in BeneGene-1 trial was −54.37 (−63.64 to −45.10) at year 1 following fidanacogene elaparvovec infusion and |||||| ||||||| || ||||||| from week 12 to data cut-off date, all favouring fidanacogene elaparvovec.
Annualized FIX Consumption
Overall (from week 12 to the data cut-off date following fidanacogene elaparvovec infusion), the difference in the annualized FIX consumption between patients treated with fidanacogene elaparvovec during the BeneGene-2 trial and the same patients treated with routine FIX prophylaxis during the lead-in BeneGene-1 trial was |||||||| ||||| ||||||||| || |||||||||| ||||||||| fidanacogene elaparvovec.
Hemophilia Joint Health Score
Change from baseline at week 52 or week 104 following fidanacogene elaparvovec infusion |||||| |||||||||||| in the HJHS total score in patients treated with fidanacogene elaparvovec.
Hemophilia Quality of Life Questionnaire for Adults
At week 52 or week 104 following fidanacogene elaparvovec infusion, the Haem-A-QoL physical health score and total score |||| ||||||||.
Hemophilia Activities List
|||||||||, at week 52 or week 104 following fidanacogene elaparvovec infusion, the score of HAL complex lower extremity activities and the HAL score |||||||||
Table 15: Summary of Key Efficacy Results From the Pivotal BeneGene-2 and Lead-in BeneGene-1 Trials (Dosed Population)
Outcome | Pivotal BeneGene-2 | Lead-in BeneGene-1 |
|---|---|---|
Fidanacogene elaparvovec (N = 45) | FIX prophylaxis (N = 45) | |
ABRtotal (year 1 following fidanacogene elaparvovec infusion) | ||
Number of participants who contributed to the analysisa | 45 | 45 |
Number of participants without any bleeds, n (%) | 29 (64.4) | 13 (28.9) |
Number of bleeds | ||
Mean (SD) | ||| |||||| | ||| |||||| |
Median (min to maximum) | ||| ||| ||| | ||| ||| ||| |
ABR | ||
Mean (SD) | |||| ||||||| | |||| ||||||| |
Median (minimum to maximum) | |||| ||||| ||||| | |||| ||||| ||||| |
Estimated ABRtotal (95% CI)b | 1.30 (0.59 to 2.02) | 4.43 (1.81 to 7.05) |
Treatment differencec | ||
Mean difference estimate (95% CI) | −3.13 (−5.44 to −0.81) | |
P value | |||||| | |
Reduction, %d | ||
Estimate (95% CI) | 70.63 (49.17 to 83.03) | |
P value | < 0.0001 | |
ABRtotal (overall) | ||
Number of participants who contributed to the analysisa | 45 | 45 |
Number of participants without any bleeds, n (%) | || |||||| | || |||||| |
Number of bleeds | ||
Mean (SD) | ||| |||||| | ||| |||||| |
Median (minimum to maximum) | ||| ||| ||| | ||| ||| ||| |
ABR | ||
Mean (SD) | |||| ||||||| | |||| ||||||| |
Median (minimum to maximum) | |||| ||||| |||| | |||| ||||| ||||| |
Estimated ABRtotal (95% CI)b | |||| ||||| || ||||| | |||| ||||| || ||||| |
Treatment differencec | ||
Mean difference estimate (95% CI) | ||||| |||||| || |||||| | |
P value | |||||| | |
Reduction, %d | ||
Estimate (95% CI) | ||||| |||||| || |||||| | |
P value | |||||| | |
ABRtreat (year 1 following fidanacogene elaparvovec infusion) | ||
Number of participants who contributed to the analysisa | 45 | 45 |
Number of participants without treated bleeds, n (%) | 33 (73.3) | 16 (35.6) |
Number of bleeds | ||
Mean (SD) | ||| |||||| | ||| |||||| |
Median (minimum to maximum) | ||| ||| || | ||| ||| ||| |
ABR | ||
Mean (SD) | |||| ||||||| | |||| ||||||| |
Median (minimum to maximum) | |||| ||||| |||| | |||| ||||| ||||| |
Estimated ABRtreat (95% CI)b | 0.73 (0.25 to 1.21) | 3.35 (1.71 to 4.98) |
Treatment differencec | ||
Mean difference estimate (95% CI) | −2.62 (−4.27 to −0.96) | |
P value | |||||| | |
Reduction, %d | ||
Estimate (95% CI) | 78.30 (52.75 to 90.03) | |
P value | |||||| | |
ABRtreat (overall) | ||
Number of participants who contributed to the analysisa | 45 | 45 |
Number of participants without treated bleeds, n (%) | || |||||| | || |||||| |
Number of bleeds | ||
Mean (SD) | ||| |||||| | ||| |||||| |
Median (minimum to maximum) | ||| ||| ||| | ||| ||| ||| |
ABR | ||
Mean (SD) | |||| ||||||| | |||| ||||||| |
Median (minimum to maximum) | |||| ||||| |||| | |||| ||||| ||||| |
Estimated ABRtreat (95% CI)b | |||| ||||| || ||||| | |||| ||||| || ||||| |
Treatment differencec | ||
Mean difference estimate (95% CI) | ||||| |||||| || |||||| | |
P value | |||||| | |
Reduction, %d | ||
Estimate (95% CI) | ||||| |||||| || |||||| | |
P value | |||||| | |
ABRjoint (year 1 following fidanacogene elaparvovec infusion) | ||
Number of participants who contributed to the analysisa | 45 | 45 |
Number of participants without any joint bleeds, n (%) | 31 (68.9) | 20 (44.4) |
Number of bleeds | ||
n | 45 | 45 |
Mean (SD) | ||| |||||| | ||| |||||| |
Median (minimum to maximum) | ||| ||| || | ||| ||| ||| |
ABR | ||
n | 45 | 45 |
Mean (SD) | |||| ||||||| | |||| ||||||| |
Median (minimum to maximum) | |||| ||||| |||| | |||| ||||| ||||| |
Estimated ABRjoint (95% CI)b | |||| ||||| || ||||| | |||| ||||| || ||||| |
Treatment differencec | ||
Mean difference estimate (95% CI) | ||||| |||||| || |||||| | |
P value | |||||| | |
Reduction, %d | ||
Estimate (95% CI) | ||||| |||||| || |||||| | |
P value | |||||| | |
ABRjoint (overall) | ||
Number of participants who contributed to the analysisa | 45 | 45 |
Number of participants without any joint bleeds, n (%) | || |||||| | || |||||| |
Number of bleeds | ||
Mean (SD) | ||| |||||| | ||| |||||| |
Median (minimum to maximum) | ||| ||| ||| | ||| ||| ||| |
ABR | ||
Mean (SD) | |||| ||||||| | |||| ||||||| |
Median (minimum to maximum) | |||| ||||| |||| | |||| ||||| ||||| |
Estimated ABRjoint (95% CI)b | |||| ||||| || ||||| | |||| ||||| || ||||| |
Treatment differencec | ||
Mean difference estimate (95% CI) | ||||| |||||| || |||||| | |
P value | |||||| | |
Reduction, %d | ||
Estimate (95% CI) | ||||| |||||| || |||||| | |
P value | |||||| | |
AIR (from tear 1 following fidanacogene elaparvovec infusion) | ||
Number of participants who contributed to the analysisa | 45 | 45 |
Number of participants without any infusions, n (%) | 29 (64.4) | 0 |
Number of infusions | ||
Mean (SD) | ||| ||||||| | |||| ||||||| |
Median (minimum to maximum) | ||| ||| ||| | |||| |||| |||| |
AIR | ||
Mean (SD) | 4.46 (10.028) | 58.83 (29.056) |
Median (minimum to maximum) | |||| ||||| ||||| | ||||| |||||| |||||| |
Treatment differencee | ||
Estimates (95% CI) | −54.37 (−63.64 to −45.10) | |
P value | < 0.0001 | |
Reduction in mean AIR, (%)f | 92.4 | |
AIR (overall) | ||
Number of participants who contributed to the analysisa | 45 | 45 |
Number of participants without any infusions, n (%) | || |||||| | || |||||| |
Number of infusions | ||
Mean (SD) | ||| ||||||| | |||| ||||||| |
Median (minimum to maximum) | ||| ||| |||| | |||| |||| |||| |
AIR | ||
Mean (SD) | |||| |||||||| | ||||| |||||||| |
Median (minimum to maximum) | |||| ||||| ||||| | ||||| |||||| |||||| |
Treatment differencee | ||
Estimates (95% CI) | |||||| ||||||| || ||||||| | |
P value | |||||| | |
Reduction in mean AIR, (%)f | |||| | |
Annualized FIX consumption (overall) | ||
Number of participants who contributed to the analysisa | 45 | 45 |
Total FIX consumption (IU/kg) | ||
Mean (SD) | |||||| |||||||||| | ||||||| |||||||||| |
Median (minimum to maximum) | |||| ||||| |||||||| | ||||||| ||||||| |||||||| |
Annualized total FIX consumption (IU/kg) | ||
Mean (SD) | |||||| |||||||||| | ||||||| |||||||||| |
Median (minimum to maximum) | |||| ||||| ||||||| | ||||||| |||||||| ||||||| |
Treatment differencee | ||
Estimates (95% CI) | |||||||| ||||||||| || ||||||||| | |
P value | |||||| | |
Reduction in mean, (%)g | |||| | |
HJHS | ||
Baseline | ||
n | || | — |
Mean (SD) | |||| ||||||| | — |
Median (minimum to maximum) | |||| ||| ||| | — |
Week 52 following fidanacogene elaparvovec infusion | ||
n | || | — |
Mean (SD) | |||| ||||||| | — |
Median (minimum to maximum) | |||| ||| ||| | — |
Change from baselineh | ||
n | || | — |
Mean (SD) [95% CI] | |||| |||||| ||||| || ||||| | — |
P value | |||||| | — |
Week 104 following fidanacogene elaparvovec infusion | ||
n | || | — |
Mean (SD) | |||| ||||||| | — |
Median (minimum to maximum) | ||| ||| ||| | — |
Change from baselineh | ||
n | || | — |
Mean (SD) [95% CI] | |||| |||||| ||||| || ||||| | — |
P value | |||||| | — |
Haem-A-QoL | ||
Haem-A-QoL physical health domain | ||
Baseline | ||
n | 40 | — |
Mean (SD) | 31.00 (24.967) | — |
Median (minimum to maximum) | ||||| ||||| ||||| | — |
Week 52 following fidanacogene elaparvovec infusion | ||
n | 42 | — |
Mean (SD) | 22.50 (23.511) | — |
Median (minimum to maximum) | ||||| ||||| |||||| | — |
Change from baselineh | ||
n | 37 | — |
Mean (SD) [95% CI] | −7.70 (15.750) ||||||| || |||||| | — |
P value | |||||| | — |
Week 104 following fidanacogene elaparvovec infusion | ||
n | || | — |
Mean (SD) | ||||| |||||||| | — |
Median (minimum to maximum) | ||||| ||||| ||||| | — |
Change from baselineh | ||
n | || | — |
Mean (SD) [95% CI] | |||||| |||||||| ||||||| || |||||| | — |
P value | |||||| | — |
Haem-A-QoL total score | ||
Baseline | ||
n | 40 | — |
Mean (SD) | 29.08 (14.891) | — |
Median (minimum to maximum) | ||||| ||||| ||||| | — |
Week 52 following fidanacogene elaparvovec infusion | ||
n | 42 | — |
Mean (SD) | 17.18 (13.535) | — |
Median (minimum to maximum) | ||||| ||||| ||||| | — |
Change from baselineh | ||
n | 37 | — |
Mean (SD) [95% CI] | −11.17 (9.145) ||||||| || |||||| | — |
P value | |||||| | — |
Week 104 following fidanacogene elaparvovec infusion | ||
n | || | — |
Mean (SD) | ||||| |||||||| | — |
Median (minimum to maximum) | ||||| ||||| ||||| | — |
Change from baselineh | ||
n | || | — |
Mean (SD) [95% CI] | |||||| ||||||| ||||||| || |||||| | — |
P value | |||||| | — |
HAL | ||
HAL complex lower extremity activities | ||
Baseline | ||
n | 40 | — |
Mean (SD) | 67.06 (24.887) | — |
Median (minimum to maximum) | ||||| |||||| |||||| | — |
Week 52 following fidanacogene elaparvovec infusion | ||
n | 42 | — |
Mean (SD) | 74.36 (24.668) | — |
Median (minimum to maximum) | ||||| |||||| |||||| | — |
Change from baselineh | ||
n | 37 | — |
mean (SD) [95% CI] | 7.59 (19.556) ||||| || |||||| | — |
P value | |||||| | — |
Week 104 following fidanacogene elaparvovec infusion | ||
n | || | — |
Mean (SD) | ||||| |||||||| | — |
Median (minimum to maximum) | ||||| |||||| |||||| | — |
Change from baselineh | ||
n | || | — |
Mean (SD) [95% CI] | ||||| |||||||| ||||| || |||||| | — |
P value | |||||| | — |
HAL total score | ||
Baseline | ||
n | 40 | — |
Mean (SD) | 81.02 (16.123) | — |
Median (minimum to maximum) | ||||| |||||| |||||| | — |
Week 52 following fidanacogene elaparvovec infusion | ||
n | 42 | — |
Mean (SD) | 88.34 (12.316) | — |
Median (minimum to maximum) | ||||| |||||| |||||| | — |
Change from baselineh | ||
n | 37 | — |
Mean (SD) [95% CI] | 6.66 (10.862) ||||| || |||||| | — |
P value | |||||| | — |
Week 104 following fidanacogene elaparvovec infusion | ||
n | || | — |
Mean (SD) | ||||| |||||||| | — |
Median (minimum to maximum) | ||||| |||||| |||||| | — |
Change from baselineh | ||
n | || | — |
mean (SD) [95% CI] | |||| |||||||| |||||| || |||||| | — |
P value | |||||| | — |
ABR = annualized bleeding rate; ABRjoint = annualized bleeding rate for treated and untreated joint bleeds; ABRtotal = annualized bleeding rate for treated and untreated bleeds; ABRtreat = annualized bleeding rate for treated bleeds; AIR = annualized infusion rate; CI = confidence interval; FIX = coagulation factor IX; Haem-A-QoL = Hemophilia Quality of Life Questionnaire for Adults; HAL = Hemophilia Activities List; HJHS = Hemophilia Joint Health Score; SD = standard deviation.
Note: Year 1 refers to the period between week 12 and month 15 following fidanacogene elaparvovec infusion. Overall refers to the period from week 12 following fidanacogene elaparvovec infusion to the data cut-off date of November 16, 2022. As of the data cut-off date, the mean duration of follow-up in the pivotal BeneGene-2 trial was |||| ||||| ||||||| with a median of |||| ||||| ||||| ||||| Week 52 and week 104’s baseline was defined as the last nonmissing measurement before the dosing date (day 1) in the pivotal study. The mean duration of follow-up in the lead-in BeneGene-1 trial was |||| ||||| ||||||| with a median of |||| ||||| ||||| ||||.
aNumber of patients who were in follow-up at the start of the period interval.
bDerived from a repeated measures generalized linear model with negative binomial distribution.
cThe treatment difference and P value were obtained from a repeated measures generalized linear model with negative binomial distribution and identity link function.
dThe percentage reduction and P value were obtained from a repeated measures generalized linear model with a negative binomial distribution and log link function.
eThe treatment difference estimate (95% CI) and P value were obtained from a paired t test.
fPercentage reduction in mean the AIR was calculated as (1 − mean AIR for FIX from week 12 to month 15 following fidanacogene elaparvovec infusion / mean AIR during standard of care FIX replacement regimen) × 100%.
gPercent reduction in mean annualized FIX consumption was calculated as (1 − mean annualized FIX consumption for FIX during the respective year interval following fidanacogene elaparvovec infusion / mean annualized FIX consumption during standard of care FIX replacement regimen) × 100%.
hThe change from baseline, 95% CI, and P value were obtained from a paired t test.
Source: BeneGene-2 Clinical Study Report.9
Harms
A summary of harms in the BeneGene-2 trial is shown in Table 16. The data cut-off date for the harms data was November 16, 2022.
Adverse Events
Treatment-emergent AEs were reported in 84.4% (38 of 45) of the safety population in the BeneGene-2 trial. The most commonly reported TEAE was increased ALT (26.7%), followed by nasopharyngitis (17.8%) and arthralgia (17.8%).
Serious Adverse Events
Serious adverse events were reported in 7 patients (15.6%) in the BeneGene-2 trial. The most common SAE was anemia (4.4%).
Withdrawal due to Adverse Events
No patients in the BeneGene-2 trial continued the study due to AEs as of the data cut-off date of November 16, 2022.
Mortality
No patients in the BeneGene-2 trial died as of the data cut-off date of November 16, 2022.
Notable Harms
Increased ALT and abnormal hepatic function occurred in 26.7% (12 of 45) and 13.3% (6 of 45) of the patients in the BeneGene-2 trial, respectively. Increased AST, increased hepatic enzyme, and increased transaminases occurred in 6.7% (3 of 45) of the patients in the BeneGene-2 trial, respectively.
Table 16: Summary of Harms Results from BeneGene-2 (Safety Population)
Adverse events | BeneGene-2 |
|---|---|
Fidanacogene elaparvovec (N = 45) | |
TEAE, n (%) | 38 (84.4) |
TEAEs that occurred in ≥ 10% of the safety population | |
Increased ALT | 12 (26.7) |
Nasopharyngitis | 8 (17.8) |
Arthralgia | 8 (17.8) |
Abnormal hepatic function | 6 (13.3) |
Headache | 6 (13.3) |
COVID-19 | 6 (13.3) |
TESAEs, n (%) | 7 (15.6) |
Anemia | 2 (4.4) |
Patients who discontinued due to adverse events, n (%) | 0 (0.0) |
Deaths, n (%) | 0 (0.0) |
Notable harms, n (%) | |
Increased ALT | 12 (26.7) |
Abnormal hepatic function | 6 (13.3) |
Increased AST | 3 (6.7) |
Increased hepatic enzyme | 3 (6.7) |
Increased transaminases | 3 (6.7) |
ALT = alanine transaminase; AST = aspartate transaminase; TEAE = treatment-emergent adverse event; TESAE = treatment-emergent serious adverse event
Note: Data cut-off date: November 16, 2022.
Source: BeneGene-2 Clinical Study Report.9
The harms data were reported in the 102 patients who participated in the lead-in to BeneGene-1 trial.1 Among them, a total of 16 (15.7%) participants had AEs, of whom 5 (4.9%) experienced injury, poisoning and procedural complications and 5 (4.9%) experienced musculoskeletal and connective tissue disorders. In terms of SAEs, out of 102 participants, 6 (5.9%) had SAEs, of whom 3 (2.9%) had gastrointestinal hemorrhage, joint injury, and arthropathy. No participants discontinued from the BeneGene-1 trial due to AEs, and no deaths occurred. In terms of notable harms, 2 patients (2.0%) reported hypersensitivity.
Critical Appraisal
Internal Validity
The BeneGene-2 trial, the only eligible study identified by the sponsor-conducted SLR, was a phase III, single-arm, open-label clinical trial that enrolled 45 patients. To determine the relative efficacy of fidanacogene elaparvovec and FIX prophylaxis on patient outcomes (i.e., the standard of care in Canada), patients in the BeneGene-2 trial served as their own controls for efficacy outcomes (e.g., ABRtotal, ABRtreat, ABRjoint, AIR, and annualized FIX consumption). Specifically, data collected from the same patients who were on FIX prophylaxis for at least 6 months during the lead-in phase (the BeneGene-1 trial) before the pivotal BeneGene-2 trial were used for comparison. Although interpretation of the study results is limited due to the nonrandomized, open-label, single-arm design, the discontinuity design was considered appropriate in the field of hemophilia B by the clinical experts consulted by CADTH for this review.
Participants in the BeneGene-2 trial were requested to suspend their FIX prophylaxis regimen after fidanacogene elaparvovec infusion but were allowed to resume FIX prophylaxis based on certain conditions. These conditions were considered generally appropriate by the clinical experts consulted by CADTH. Moreover, the resumption of FIX prophylaxis regimen postinfusion in the BeneGene-2 trial was not expected to modify treatment effects, ||||||||| || ||| ||||| || |||||||||| ||||||||||| |||||||| || ||||| |||||||||||| ||| ||||||| ||| ||||||||||| |||||||| |||| |||||||| || |||||||||||| ||||||||||| |||| |||||||| ||| ||| |||||||||| || || ||||||| || |||| |||| ||| ||||||| ||||||||.
To suppress immune hepatitis, patients can be treated with corticosteroids following fidanacogene elaparvovec infusion under certain circumstances, which the clinical experts consulted by CADTH determined were appropriate. Still, a relatively large proportion of patients (28 of 45) were given corticosteroids following fidanacogene elaparvovec infusion. Although a subgroup analysis based on corticosteroid use was carried out by the sponsor (data not shown), the effect of corticosteroid use following fidanacogene elaparvovec treatment remains unclear because the subgroup analysis was unadjusted for confounding variables, and the BeneGene-2 trial was not powered to perform a comparison based on corticosteroid use.
The clinical experts consulted by CADTH for this review noted that the inclusion and exclusion criteria in the BeneGene-2 trial were appropriate and reflective of patients they would have expected to experience in clinical practice. The patients included in the BeneGene-2 trial were selected from the lead-in BeneGene-1 trial according to an additional set of criteria on top of those specified in the BeneGene-1 trial. CADTH found that the purpose of the additional criteria specified in the BeneGene-2 trial was mainly to ensure that patients had acceptable conditions to receive the gene therapy of interest (fidanacogene elaparvovec). Of the 102 patients in the lead-in BeneGene-1 study, only 45 were enrolled in the pivotal BeneGene-2 trial, while 57 were not. Other than 8 who chose not to continue into the BeneGene-2 trial and 9 who failed the BeneGene-2 screening, the remaining and majority (40 patients) of the 57 patients did not enter the BeneGene-2 trial because they had not completed the BeneGene-1 study. It was determined by CADTH that the potential selection bias due to a large number of patients being left out was not a serious concern because the data provided by the sponsor showed that outcomes of the 40 patients, such as the ABRtotal, ABRtreat, and AIR, were similar to the those at year 1 postinfusion among the 45 patients enrolled in the BeneGene-2 trial.
No major concerns were associated with patient compliance to FIX prophylaxis in the BeneGene-1 trial, with ||| |||||||| || |||||||| |||||| |||||||| in the BeneGene-1 trial being 80% or more compliant based on number of infusions. The documentation of bleeding events in the BeneGene-2 trial relied on the use of electronic diaries by patients, and the determination of whether a bleed needs to be treated relied on physicians’ clinical decisions shared with patients. Despite the risk of bias likely being low, and based on information provided by the sponsor, CADTH determined that the possibility of bias that may exaggerate the treatment effects of fidanacogene elaparvovec (i.e., ABR outcomes) could not be ruled out. Furthermore, due to the single-arm, open-label design, reliable assessments of patient-reported outcomes (e.g., HRQoL end points) could not be made.
According to the clinical experts consulted by CADTH, the definitions of the efficacy outcomes such as the ABRtotal and AIR, the start point for outcome analysis (week 12 after fidanacogene elaparvovec infusion instead of immediately postinfusion), as well as the noninferiority margin for the ABRtotal were acceptable. In addition, CADTH determined that the gatekeeping process applied to control for multiplicity of testing multiple end points was appropriate. However, there were some concerns about the statistical models (assumptions) used to inform the comparative efficacy of fidanacogene elaparvovec relative to FIX prophylaxis. The first assumption, which was considered reasonable by the experts consulted by CADTH, was that rate of bleeding during FIX prophylaxis in the BeneGene-1 trial would be comparable to the bleeding rate during the BeneGene-2 trial if FIX prophylaxis had not been discontinued and fidanacogene elaparvovec not been given as an intervention. This assumption is required to interpret observed differences in bleeding rates pre– and post–fidanacogene elaparvovec treatment intervention in the BeneGene-2 trial. The second assumption of the negative binomial mixed model is that the cohort bleed rates were constant during the entire period of study. The challenge lies in interpreting the magnitude of the effect estimates of fidanacogene elaparvovec compared to FIX prophylaxis as the model describes a weighted average of the rate of bleeding over time that is dependent on the observed censoring mechanism.
External Validity
CADTH identified several considerations related to the generalizability of the BeneGene-2 trial. First and most importantly, given the novelty of gene therapy and the expectation of long-lasting effects, evidence from current follow-up period (|||| ||| |||||) in the BeneGene-2 trial may not be adequate to inform long-term efficacy and safety.
Second, the indication includes patients with “moderately severe to severe” hemophilia B. The BeneGene-2 trial defined “moderately severe to severe” as a FIX:C level less than or equal to 2%. However, the clinical experts consulted by CADTH noted that severity in clinical practice is defined by the patients’ phenotype and not simply their factor activity levels. In some patients, the disease will be considered moderately severe to severe due to clinical symptoms although the FIX level is greater than 2%, according to the clinical experts consulted by CADTH.
Third, the BeneGene-2 trial only included patients with an anti-AAVrh74var nAb titre of less than 1:1. According to the clinical experts consulted by CADTH, the efficacy of fidanacogene elaparvovec in patients with an anti-AAVrh74var nAb titre greater than or equal to 1:1 remains uncertain. However, the clinical experts consulted by CADTH agreed that selection of eligible patients should follow the threshold used in the BeneGene-2 trial.
Fourth, the indication does not specify sex (i.e., it includes both men and women) but the product monograph states that fidanacogene elaparvovec is not intended for administration in women. Following the protocol, BeneGene-2 only enrolled male patients.
Fifth, 73.3% (33 of 45) of the patients in the BeneGene-2 trial were white, which, according to the clinical experts consulted by CADTH, was higher than would be expected in the patient population in Canada.
Sixth, the clinical experts consulted by CADTH noted that PROBE is more commonly used in the Canadian settings to measure HRQoL, rather than Haem-A-QoL and HAL, which were used in the BeneGene-2 trial. However, this was not considered a serious generalizability issue because all these HRQoL measurement instruments are aligned.
Last, results from the lead-in BeneGene-1 trial were from patients treated with EHL FIX or SHL FIX. No supplemental analyses were submitted comparing fidanacogene elaparvovec with FIX prophylaxis based on the type of FIX concentrate (e.g., EHL versus SHL) used in prophylaxis. According to the clinical experts consulted by CADTH, the key difference between EHL and SHL FIX in treating patients is the frequency of infusion. Due to its half-life, EHL FIX may be associated with a lower ABR. However, the clinical experts consulted by CADTH raised no concerns about applying the mixed results to EHL FIX or SHL FIX.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For pivotal studies and randomized controlled trials identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered most relevant to inform CADTH’s expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group:54,55
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate — The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited — The true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate — The true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as “very uncertain.”
According to the GRADE guidance, nonrandomized comparative evidence starts at low certainty and noncomparative evidence starts at very low certainty. The CADTH review team carefully assessed the risk of selection bias and potential for unmeasured confounding of the pivotal intrapatient single-arm trial comparing bleeding pre- and postintervention. The GRADE report captures the study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias to present these important considerations.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null.
The reference points for the certainty of evidence assessment for the ABRtotal and ABRtreat was set according to the presence or absence of an important effect based on thresholds informed by the sponsor and agreed upon by clinical experts consulted by CADTH for this review. The target of the certainty of evidence assessment was the presence or absence of any (non-null) effect for the AIR and annualized FIX consumption due to the lack of a formal estimate of a minimally importance difference. The certainty of evidence was summarized narratively for the HJHS, Haem-A-QoL (physical health score and total score), and HAL (complex lower extremity activities score and total score), as well as harms outcomes due to lack of comparators.
Results of GRADE Assessments
Table 2 and Table 3 presents the GRADE summary of findings for fidanacogene elaparvovec versus FIX prophylaxis in adult patients (18 years and older) with moderately severe to severe hemophilia B.
Studies Addressing Gaps in the Systematic Review Evidence
Contents within this section were informed by materials submitted by the sponsor. The following summary was validated by the CADTH review team.
Two studies (C0371005 and C0371003) submitted by the sponsor to address gaps in the systematic review evidence are summarized in this section. Study C0371005 was submitted to address a gap in knowledge of the safety and kinetics of fidanacogene elaparvovec. Its corresponding extension study, C0371003, was submitted to address a gap in knowledge of the long-term efficacy and safety of fidanacogene elaparvovec.
Study C0371005
Study C037100556 was a phase I and IIa, open-label, single-arm, dose-escalation, multicentre study. The objective of this study was to evaluate the safety, tolerability, and kinetics of a single IV infusion of fidanacogene elaparvovec in hemophilia B participants with endogenous FIX levels less than or equal to 2%. Initially 5 participants were given an initial dose of 5 × 1011 vg/kg. Based on observed safety and efficacy data in the first 5 participants, an additional 10 participants were enrolled at the same dose level. A total of 15 participants completed the study. All dosed participants underwent safety observation for a total of 52 (± 2) weeks after infusion. At the end of study, all participants were encouraged to enrol in an extension study (Study C0371003) evaluating the long-term safety of fidanacogene elaparvovec for up to an additional 5-year long-term follow-up.
The objectives for Study C0371005 were:
to evaluate the safety and tolerability of a single IV infusion of fidanacogene elaparvovec in hemophilia B participants (primary)
to characterize the kinetics of fidanacogene elaparvovec. (secondary).
Key inclusion criteria of the participants were:56,57
male participants aged 18 years or older
have hemophilia B with a FIX activity of 2% or less of the normal value at screening, historical evidence, or a documented genotype known to produce a clinically severe phenotype of hemophilia B
nAbs to fidanacogene elaparvovec of less than 1:5 as determined by an in vitro transduction assay
have had 50 or more exposure days to any recombinant or plasma-derived FIX product
patients on prophylaxis treatment must have had documented bleeding events or FIX infusions in the previous 12 weeks
patients using on-demand treatment must have had 4 or more bleeding events in the previous 52 weeks and/or chronic hemophilic arthropathy in 1 or more joints
no prior history of hypersensitivity or anaphylaxis associated with FIX or IV immunoglobulin infusion
acceptable laboratory values for hemoglobin, platelets, AST, ALT, alkaline phosphatase, bilirubin, and creatinine
agree to use acceptable methods of contraception for the time required for 3 ejaculate samples negative for vector shedding.
Key exclusion criteria were:56,57
active hepatitis B or C; hepatitis B surface antigen, hepatitis B virus DNA positivity, or hepatitis C virus RNA positivity
currently on antiviral therapy for hepatitis B or C
a pre-existing diagnoses of significant liver disease including portal hypertension, splenomegaly, or hepatic encephalopathy
serological evidence of HIV-1 or HIV-2 with a CD4+ cell count less than or equal to 200 mm3
a history of chronic infection or other chronic disease deemed an unacceptable risk by the investigator
any concurrent clinically significant major disease or condition deemed unsuitable for participation by the investigator
previously dosed in a gene therapy trial within the last 52 weeks or in a clinical study with an investigational drug in the last 12 weeks.
The median duration of fidanacogene elaparvovec infusion was |||| minutes, ranging from || || || minutes. The median total dose of fidanacogene elaparvovec infused was |||||||| vg, ranging from |||||||| vg.
Results
Baseline Characteristics
A total of 22 participants signed the informed consent form: 15 of these participants received treatment and 7 did not participate due to screening failure. All 15 participants completed the study and were male, with a mean age of 38.6 years, ranging from 18 to 61 years. The majority of participants were aged 35 years or younger (66.7%) and white (80.0%). Among the 15 treated patients, 11 (73.3%) were prophylaxis participants and 4 (26.7%) were on-demand participants at baseline. The majority of participants had no family history of FIX inhibitors (80.0%) and had hemophilia B with a FIX:C level of less than 1% (66.7%). Among 15 participants, 14 (93.3%) had a medical or surgical history related to hemophilia. All 15 participants received at least 1 concomitant medication. The most commonly used concomitant medications included paracetamol in 10 participants (66.7%).56
Efficacy Outcomes
No formal efficacy evaluations were performed, and all analyses were exploratory in nature. The ABR (spontaneous and traumatic), annualized FIX consumption, number of target joints, changes in level of activity, HRQoL, and health-economic parameters were collected for exploratory efficacy evaluations. Vector-derived FIX:C levels were used as important indicators of efficacy for dose selection. The safety analysis set included 15 participants who received the infusion. The assessments of the HJHS, HAL and McGill pain questionnaire were added in protocol Amendment 5.1 (January 13, 2017). For this reason, only the final | participants enrolled under this amendment and subsequent versions of the protocol were evaluated for these assessments. All 15 participants were included for other efficacy, pharmacokinetic and vector shedding analyses.56
Annualized Bleeding Rate
Among the 15 treated participants, 12 (80.0%) experienced no on-study bleeds. No traumatic bleeds were observed during the study, and all 3 participants experiencing bleeding episodes had spontaneous bleeds. The total number of infusions ranged from 0 to 10 during the study. The median ABR during the 52-week period preceding fidanacogene elaparvovec infusion (historical) was 4.00, ranging from 0.0 to 48.0. The median ABR decreased to 0.00 (range = 0.0 to 4.0) during the 52-week period following fidanacogene elaparvovec infusion (on study). The mean ABR decreased from 8.87 (SD = 14.040) to 0.40 (SD = 1.060).56
Annualized FIX Consumption
The overall mean annualized FIX production consumption was |||| ||||||||| IU in all 15 participants, with a mean of |||| ||||||||| IU in the 11 participants previously on prophylaxis treatment and |||| |||||||| IU in the 4 participants previously receiving on-demand treatment.56
Target Joints Assessments
During the 52-week period preceding screening, the mean number of target joint bleeds was |||| ||||||| in a total of 5 participants (4 participants previously on prophylactic treatment and 1 participant previously receiving on-demand treatment). The mean number of target joint bleeds decreased from |||| ||||||| in 4 participants to ||| |||||| in 2 participants who were previously receiving prophylactic treatment.56
Hemophilia Joint Health Score
Regarding the HJHS, | participants were assessed at baseline and end of study. In general, a |||||||| ||| |||||||| || ||| ||||||||| |||||||||||| |||| ||||| ||||| |||||||| |||||| |||||| |||| |||||| ||||| |||||| |||| |||||| ||||| |||| ||| |||| ||||| |||| ||| |||| |||||| ||| ||| ||||| ||||| || |||| ||||| ||||||| ||||| |||||||| ||| ||||| ||||||.56
Hemophilia Activities List and Hemophilia Quality of Life Questionnaire for Adults
A |||||||| in HAL total sum scores and subscores over time was observed in the | participants who had assessments done at baseline and end of study. A |||||||| ||| |||| |||||||| in Haem-A-QoL overall total score over time, || |||| || in the domain total scores except dealing total score.56
Adverse Events
Fourteen of 15 participants (93.3%) reported 1 or more TEAE. No study drug discontinuation, study discontinuation, SAEs or deaths were reported in the study.56
A total of 81 TEAEs were reported in the study. The most commonly reported TEAEs were in the system organ class of infections and infestation (8 participants, or 53.3%), gastrointestinal disorders (7 participants, or 46.7%) and musculoskeletal and connective disorders (6 participants, or 40.0%). The most frequently reported TEAEs were upper respiratory tract infection (5 participants, or 33.3%), nasopharyngitis (3 participants, or 20.0%), back pain (3 participants, or 20.0%) and muscle strain (3 participants, or 20.0%). One participant (6.7%) reported experiencing arthralgia as a TEAE. Two drug-related TEAEs of increased transaminases (13.3%) were reported. The majority of TEAEs (53 of 81, or 65.4%) were mild in severity, and the other 28 TEAEs (34.6%) were moderate in severity. There were no severe or life-threatening TEAEs, or deaths.56
Study C0371003
Description of Studies
Study C037100358 (N = 17) is a phase IIa, open-label, nonrandomized, longer-term follow-up study designed to evaluate the safety and efficacy of previously administered fidanacogene elaparvovec at a dose of 5 × 1011 vg/kg for up to 6 years. Participants enrolled in this study either had been dosed with fidanacogene elaparvovec in Study C0371005 (summarized previously) (N = 14) or received fidanacogene elaparvovec in the dose-escalation substudy (N = |) within this study. Among the 15 participants who completed Study C0371005, a single participant did not provide informed consent for Study C0371003, resulting in 14 participants continuing to Study C0371003. Results presented in this report are for the cohort of 14 patients from Study C0371005 who entered Study C0371003.
The dose-escalation substudy is not summarized in this report because the dose of fidanacogene elaparvovec did not align with the recommended dose in the draft product monograph and the substudy’s small sample size. In the dose-escalation substudy, patients received a dose of |||||| |||||| |||| || ||||||| || ||. The original design of Study C0371003 included only the continuation of the patients previously dosed in Study C0371005; the dose-escalation study was added as a protocol amendment.
The objectives for Study C0371003 were:
to evaluate the long-term safety of a single fidanacogene elaparvovec infusion administered to participants in Study C0371005 (primary safety and immunogenicity)
to determine the durability of transgene expression of fidanacogene elaparvovec and to assess the effect of fidanacogene elaparvovec on clinical outcomes of FIX:C, ABR, AIR, FIX consumption, number of spontaneous or traumatic bleeding events, and HRQoL (secondary efficacy)
to evaluate and provide a descriptive analysis of other relevant outcomes including number of FIX infusions, joint assessments, activities and functioning, health care resource use, and productivity (tertiary/exploratory efficacy).
Study C0371003 included adult males with moderately severe to severe hemophilia B who received a 5 × 1011 vg/kg dose of fidanacogene elaparvovec in Study C0371005 and completed 1 year of follow-up.57,58 Patients were monitored for safety and efficacy outcomes from year 2 to year 6 in Figure 2.
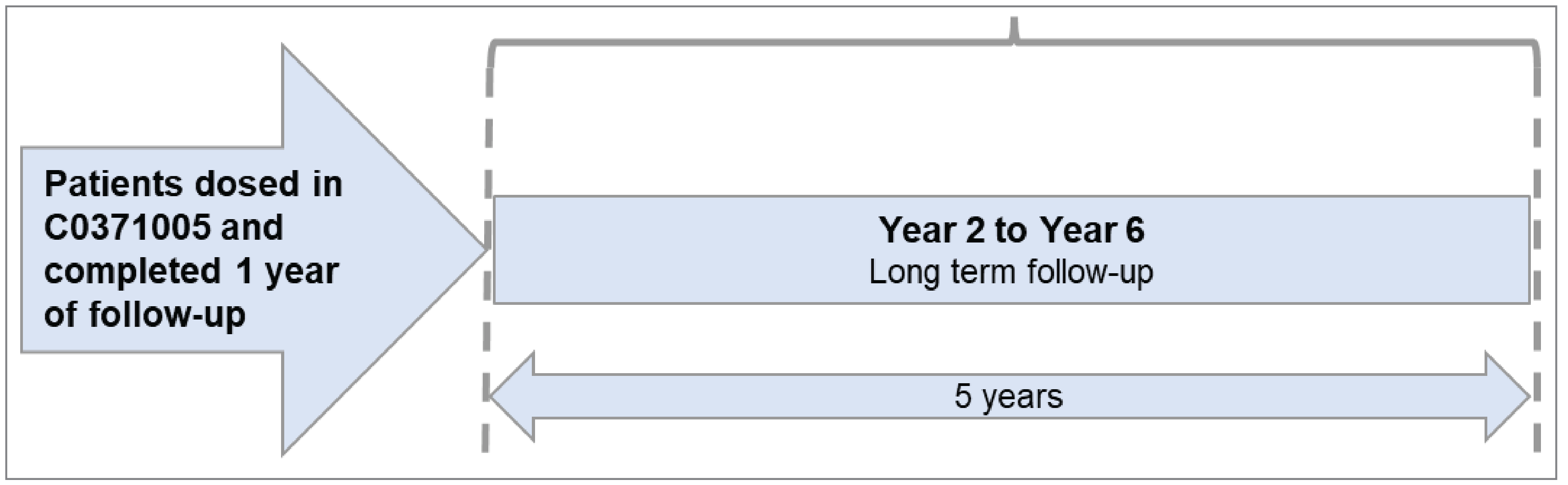
Populations
The longer-term follow-up study was conducted in the US and Australia; of the 15 participants dosed in C0371005, | were in Canada (||||| || ||| ||).58 Study C0371003 enrolled participants who had received a dose of 5 × 1011 vg/kg of fidanacogene elaparvovec and completed 1 year of follow-up in Study C0371005. The inclusion and exclusion criteria for Study C0371003 are the same as those for Study C0371005.
Interventions
There was no intervention in Study C0371003. Study participants were dosed in Study C0371005.
Outcomes
The primary outcome measures for Study C0371003 were related to safety and immunogenicity, while secondary measures were related to efficacy. The primary end point was the incidence of fidanacogene elaparvovec-related AEs. The secondary end points were efficacy and patient-reported outcome end points, including the ABR for treated bleeds, AIR, annualized FIX consumption, target joint assessments, HJHS, total number of bleeding events, and quality-of-life assessments.
Statistical Analysis
The primary analysis of Study C0371003 is planned to occur when the last patient completes 6 years of post–fidanacogene elaparvovec infusion follow-up or withdraws from the study.59 The results described here were part of an interim analysis based on the data available as of a data cut-off that occurred on November 2, 2022. No hypothesis testing was planned and all summaries are descriptive.59
The analyses of safety and efficacy in Study C0371003 were performed on the safety analysis set (i.e., all participants who received a single administration of fidanacogene elaparvovec in Study C0371005).59 Baseline data from Study C0371005 were used where possible.59 In general, descriptive statistics were reported each yearly period and for the duration of the study for the end points of FIX:C, AIR, annualized FIX consumption, number of target joints, HJHS, Haem-A-QoL, HAL, EQ-5D-5L, and safety.59 Missing data were not imputed for Study C0371003 unless specified for an individual end point.59
An exploratory analysis compared the mean ABRtreat by year to a historical ABRtotal obtained from Study C0371005 during the 52 weeks before screening.59 The historical ABR included spontaneous and traumatic bleeds, and excluded surgical and perioperative bleeds, but did not distinguish between treated and untreated bleeds; therefore, only the ABRtotal was available for the historical ABR.59
The ABRtreat and percentage reduction in ABRtreat by year were estimated using a negative binomial mixed model with repeated measures and a log link function.59 Treatment (pre- or postinfusion) was included as a factor, duration (elapsed time in years of each yearly treatment period) as numeric, and participants as a random effect.
Results
Patient Disposition
Fourteen of the 15 participants dosed in C0371005 entered into Study C0371003. Of these 14 patients, 2 discontinued from the longer-term follow-up (1 participant was lost to follow-up and 1 withdrew) and neither discontinuation was considered to be related to an AE. At the data cut-off on November 2, 2022, 5 participants had completed the longer-term follow-up and 7 were continuing the study. The duration of follow-up at the data cut-off ranged from || |||||| || || |||||| following fidanacogene elaparvovec infusion.58
Baseline Characteristics
The mean age of participants was 40.1 years, ranging from 18 to 61 years, at the time of fidanacogene elaparvovec infusion. Most participants were aged 35 years or older (71.4%) and white (85.7%). Ten participants were on FIX prophylaxis and 4 were using on-demand regimens before fidanacogene elaparvovec infusion. All participants had FIX levels below or equal to 2%.58
Concomitant Treatments
||||| ||||||| participants received at least 1 concomitant medication during the study period and 7 participants (50.0%) underwent at least 1 nondrug therapeutic procedure or diagnostic assessment. The most common concomitant medications included ||||||||||| (||| participants [|||||]), ||||||||||||||||||||||| (||| participants [|||||]). ||||| ||||||) participants underwent investigations including |||||||||||| ||||||| ||| |||||||||||| ||||||||||. ||||| ||||| participants underwent surgical procedures such as ||||||||||||||| |||||||||||| |||||| |||||| |||||||||||||| |||||||||||||||||||| |||||| ||||||| ||| ||||||||||||| |||||||||||||| |||| |||||||||| |||| |||||||||| |||||||||||| |||||| |||||| ||||||||| ||||| ||||||||||| ||| ||||||||||||.58
Efficacy
Annualized Bleeding Rate for Treated Bleeds
The ABRtreat by year and over time (year 2 to year 6), and the comparison with the historical ABR are presented in Table 17. The mean ABR |||||||| ||||| |||| ||| |||||| |||| year 2 through year 6 postinfusion with || participants (|||||) having 0 bleeds during their entire time in the study. The mean ABRstreat were |||| |||||||, |||| |||||||, |||| |||||||, |||| |||||||, and |||| ||||||| during years 2, 3, 4, 5, and 6 postinfusion, respectively. |||| participants had treated bleeds during years 2 through 6.58
Table 17: Summary of ABRtreat by Year Postinfusion, Safety Analysis Set
ABRtreat | Fidanacogene elaparvovec (N = 14) |
|---|---|
Historical | |
n | 14 |
ABRa | |
Mean (SD) | |||| |||||||| |
Median (minimum to maximum) | |||| ||||| ||||| |
Year 2 | |
n | 14 |
ABR | |
Mean (SD) | |||| |||||||| |
Median (minimum to maximum) | |||| ||||| |||||| |
Treatment difference estimate (95% CI)b | ||||| |||||||| ||||||| |
Reduction estimate, % (95% CI)b | ||||| ||||||| ||||||| |
Year 3 | |
n | 14 |
ABR | |
Mean (SD) | |||| ||||||| |
Median (minimum to maximum) | |||| ||||| |||| |
Treatment difference estimate (95% CI)b | ||||| |||||||| ||||||| |
Reduction estimate, % (95% CI)b | ||||| ||||||| ||||||| |
Year 4 | |
n | 14 |
ABR | |
Mean (SD) | |||| |||||||| |
Median (minimum to maximum) | |||| ||||| ||||| |
Treatment difference estimate (95% CI)b | ||||| |||||||| |||||| |
Reduction estimate, % (95% CI)b | ||||| ||||||| |||||| |
Year 5 | |
n | 13 |
ABR | |
Mean (SD) | |||| |||||||| |
Median (minimum to maximum) | |||| ||||| ||||| |
Treatment difference estimate (95% CI)b | |||||| |||||||| ||||||| |
Reduction estimate, % (95% CI)b | ||||| ||||||| ||||||| |
Year 6 | |
n | 8 |
ABR | |
Mean (SD) | |||| |||||||| |
Median (minimum to maximum) | |||| ||||| ||||| |
Treatment difference estimate (95% CI)b | |||||| |||||||| ||||||| |
Reduction estimate, % (95% CI)b | ||||| ||||||| ||||||| |
Over time | |
n | 14 |
ABR | |
Mean (SD) | |||| ||||||| |
Median (minimum to maximum) | |||| ||||| ||||| |
Treatment difference estimate (95% CI)b | ||||| |||||||| ||||||| |
Reduction estimate, % (95% CI)b | ||||| ||||||| ||||||| |
Participants with no bleeds, n (%) | || ||||||| |
Participants that resumed prophylaxis, n (%) | 0 |
ABR = annualized bleeding rate; ABRtreat = annualized bleeding rate for treated bleeds; CI = confidence interval; SD = standard deviation.
Notes: Traumatic and spontaneous bleedings are included. The historical ABR did not distinguish between treated and untreated bleeds; following fidanacogene elaparvovec infusion, only treated bleeding episodes were collected. Data cut-off was on November 2, 2022.
aThe reported number of bleeds in the 52 weeks before screening in Study C0371005 is used as a historical ABR.
bTreatment difference and P value were obtained from a repeated measures generalized linear model with negative binomial distribution and log link function comparing to a historical ABRtotal obtained from Study C0371005 during the 52 weeks before screening
Source: Pfizer (2023).58
Annualized Infusion Rate
The AIR by year and over time (year 2 to year 6) is presented in Table 18. The AIR generally decreased over the entire follow-up periods from a mean of |||| in year 2 to |||| in year 6 following fidanacogene elaparvovec infusion. The mean AIR was |||| |||||||, |||| |||||||, |||| |||||||, |||| ||||||| and |||| ||||||| during years 2, 3, 4, 5, and 6 postinfusion, respectively.58
Table 18: Summary of AIR by Year Postinfusion, Safety Analysis Set
AIR | Fidanacogene elaparvovec (N = 14) |
|---|---|
Year 2 | |
n | 14 |
||| | |
Mean (SD) | |||| |||||||| |
Median (minimum to maximum) | |||| ||||| |||||| |
Year 3 | |
n | 14 |
AIR | |
Mean (SD) | |||| |||||||| |
Median (minimum to maximum) | |||| ||||| ||||| |
Year 4 | |
n | 14 |
AIR | |
Mean (SD) | |||| |||||||| |
Median (minimum to maximum) | |||| ||||| ||||| |
Year 5 | |
n | 13 |
AIR | |
Mean (SD) | |||| |||||||| |
Median (minimum to maximum) | |||| ||||| ||||| |
Year 6 | |
n | 8 |
AIR | |
Mean (SD) | |||| |||||||| |
Median (minimum to maximum) | |||| ||||| ||||| |
Years 2 through 6 | |
n | 14 |
AIR | |
Mean (SD) | |||| |||||||| |
Median (minimum to maximum) | |||| ||||| ||||| |
AIR = annualized infusion rate; SD = standard deviation.
Note: Data cut-off on November 2, 2022.
Source: Pfizer (2023).58
Annualized FIX Consumption
As of the data cut-off date, there were no prophylactic infusions in the study, and no participants had resumed prophylaxis. Median total factor consumption and annualized FIX consumption, excluding consumption required for surgery, was |||| for year 2 through year 6. |||| of the 14 participants have had no nonsurgical FIX consumption over the longer-term follow-up period.58
Target Joints Assessments
From week 52 to week 130 following fidanacogene elaparvovec infusion, the number of participants with target joint bleeds ||||||||| from || ||||||| to ||||||, based on target joint assessment questionnaire results. || ||||||||||| had target joint bleeding reported beyond week 130 as of the data cut-off (from weeks 156 to 312 or end of study).58
Hemophilia Joint Health Score
The HJHS, an exploratory end point, was added after most participants were dosed, resulting in a low number of assessments at baseline. The median HJHS total scores were |||| ||||| at baseline, |||| |||||| at week 156, |||| ||||| at week 208, |||| ||||| at week 260, and |||| ||||| at week 312 or end of study. The baseline HJHS score was the last nonmissing measurement before fidanacogene elaparvovec infusion in Study C0371005.58
Hemophilia Quality of Life Questionnaire for Adults
Haem-A-QoL total scores and domain scores remained consistent throughout the longer-term follow-up period (years 2 through 6) in the 4 patients contributing to the analysis. Decreases from baseline (indicating a higher quality of life) were observed in the domains of physical health, feeling, view of yourself, sport and leisure, work and school, treatment, future, family planning, partnership and sexuality, as well as the total score, at all visits over the longer-term follow-up (years 2 through 6). Median change from baseline in Haem-A-QoL total scores ranged from ||||| || |||||| over the longer-term follow-up (years 2 through 6).58
Hemophilia Activities List
Mean HAL domain scores |||||||| ||||| || and the total score |||||||| ||||| || at all visits after fidanacogene elaparvovec infusion over the longer-term follow-up period (years 2 through 6) in the 4 patients contributing to the analysis.58
Harms
Four of the 14 participants (28.6%) experienced a total of 9 SAEs. No participants discontinued from the study due to AEs. No participants experienced hypersensitivity reactions or other AEs of special interest and there were no deaths.58
Of the 10 TEAEs reported, 5 were mild, 1 was moderate, and 4 were severe. These 10 TEAEs included 9 SAEs and 1 nonserious AE (back pain). The most frequently reported TEAEs regardless of severity were related to musculoskeletal and connective tissue disorders in 2 participants (14.3%).58
During the longer-term follow-up period, 8 of 14 participants experienced increased ALT levels above the ULN, 3 of whom had increased AST levels above the ULN. None of these cases were managed with corticosteroids and, as of the data cut-off, the ALT and AST levels of all 3 participants had returned to normal limits, except for 1 patient who completed the study with an ALT level above the ULN. Regarding immunogenicity, all 14 participants remained negative for FIX inhibitors during the study.58
Critical Appraisal
Internal Validity
Study C0371005
Study C0371005 is an open-label, single-arm, multicentre, phase I and IIa study. All efficacy analyses were exploratory in nature and were presented using descriptive statistics. The absence of a comparator group limited the interpretation of results because causality cannot be established. The open-label design may have biased the reporting of some end points because awareness of the study treatment received may have influenced patient and clinician perceptions of improvement and/or harms, particularly for outcomes that are subjective in measurement and interpretation (e.g., patient-reported outcomes and subjective AEs). Furthermore, the follow-up period was only 1 year, which is insufficient to draw any definitive conclusions regarding long-term efficacy and safety. In addition to the general limitations of the study design, data were missing for most of the participants (only 4 patients contributed to the analyses) because the HJHS and HAL were added later during a protocol amendment. No conclusions can be drawn for these outcomes with certainty.
Study C0371003
Study C0371003 provided a longer-term follow-up for the 14 patients previously given fidanacogene elaparvovec in Study C0371005. As the primary objective of C0371003 was safety, no hypothesis testing was planned, and all summaries were descriptive, resulting in no statistical inferences. Data were also missing for HJHS and HAL assessments for the reasons previously described.
The duration of follow-up at the data cut-off ranged from || |||||| || || |||||| following fidanacogene elaparvovec infusion, and only 5 participants had completed the 6-year longer-term follow-up as of the data cut-off date. According to the clinical experts consulted by CADTH, the data provided for the 6 years of follow-up are limited but reasonable for the purposes of assessing safety and efficacy in the patient population. The clinical experts consulted by CADTH noted that a longer follow-up period (20 to 25 years) involving more patients is warranted to make any definite determination about the overall long-term safety and efficacy of fidanacogene elaparvovec and may not be feasible in the clinical trial setting. The experts further noted that lifelong monitoring of liver health and carcinogenicity will be necessary as discussed previously. Altogether, the evidence with respect to the long-term efficacy and safety in C0371003 is inconclusive.
External Validity
The external validity of these supportive studies was similar to that of the pivotal trial and its corresponding lead-in study. The majority of the patients were white (80.0% and 85.7% in Study C0371005 and Study C0371003, respectively), which, according to the clinical experts consulted by CADTH, was higher than would be expected for a patient population in Canada. Both Study C0371005 and Study C0371003 enrolled only male patients, although the clinical experts noted this is likely not a serious generalizability issue because the treatment effects are not expected to differ between males and females due to the same underlying mechanism of disease, and female patients with moderately severe to severe hemophilia B are rare. One of the eligibility criteria in Study C0371005 was hemophilia B with FIX activity less than equal to 2% at screening and historical evidence or a documented genotype known to produce a clinically severe phenotype of hemophilia B. The clinical experts consulted by CADTH noted that severity in clinical practice is defined by the patient’s phenotype and not simply factor activity levels. According to the clinical experts consulted by CADTH, the disease will be considered moderately severe to severe in some patients due to clinical symptoms even with a FIX level greater than 2%. Last, generalizability may be limited by the small sample sizes of these studies.
Discussion
Summary of Available Evidence
One ongoing phase III, single-arm, open-label clinical trial (BeneGene-2, N = 45) was included in the SLR conducted by the sponsor. The primary objective of BeneGene-2 was to determine the noninferiority of fidanacogene elaparvovec relative to the standard of care FIX prophylaxis, as measured by the ABRtotal at year 1 following fidanacogene elaparvovec infusion. Other efficacy and safety end points were also examined, including the number of patients without bleeds, ABRtreat, ABRjoint, AIR, annualized FIX consumption, HJHS, Haem-A-QoL, HAL, and harms. For efficacy outcomes such as the ABRtotal, ABRtreat, ABRjoint, AIR, and annualized FIX consumption, the 45 participants in the pivotal BeneGene-2 trial served as their own controls, using data collected when these patients were on FIX prophylaxis during an open-label, noninvestigational, prospective, lead-in study (BeneGene-1, N = 102) for comparison. In addition to noninferiority, tests of superiority were also conducted, and a gatekeeping process was applied to control for multiplicity of testing multiple end points. The BeneGene-2 trial enrolled adult patients who had moderately severe to severe hemophilia B (defined as FIX:C ≤ 2%). Patients were excluded if their anti-AAVrh74var nAb titre was 1:1 or greater or if they had a prior history of FIX inhibitors or a positive FIX inhibitor test result of 0.6 Bethesda units or greater. Patients in the BeneGene-2 trial had a median age of 29 years, ranging from 18 to 62. The majority of patients were white (73.3%), followed by Black or African American (2.2%), Asian (15.6%), American Indian or Alaska Native (0), and Native Hawaiian or Other Pacific Islander (0). All of the participants had a factor mutation, of which 7 (15.6%) were nonsense, 20 (44.4%) were missense, 0 were insertions, 9 (20%) were deletions, 0 were inversions, and 9 (20%) were classified as “other.”
Two additional studies were submitted by the sponsor to address gaps in the pivotal trial evidence. Study C0371005 was a phase I and IIa, open-label, single-arm, dose-escalation, multicentre study that followed patients for 1 year. The objective was to evaluate the safety, tolerability, and pharmacokinetics of a single IV infusion of fidanacogene elaparvovec in male patients with hemophilia B and endogenous FIX levels less than or equal to 2%. Participants who completed Study C037105 were encouraged to enrol in an extension study (C0371003) evaluating the longer-term safety of fidanacogene elaparvovec for up to an additional 5-year follow-up.
Interpretation of Results
Efficacy
Overall, efficacy evidence from the pivotal BeneGene-2 trial favoured fidanacogene elaparvovec over FIX prophylaxis for the treatment of adult male patients with moderately severe to severe hemophilia B (FIX:C ≤ 2%) at the time of the interim analysis (median follow-up time was |||| |||||), although this evidence is associated with uncertainty. This conclusion was drawn after considering bleeding end points (e.g., ABRtotal, ABRtreat, ABRjoint, both mean reduction of bleeds and percentage of patients without bleeds) and end points relating to the use of FIX following fidanacogene elaparvovec infusion (e.g., AIR), as well as patient-reported outcomes (e.g., HJHS, Haem-A-QoL, HAL) examined in the CADTH report. These efficacy end points were selected based on input from stakeholders, including the clinical experts consulted by CADTH and patient and clinician groups. These efficacy end points were generally aligned with patients’ expectations of important outcomes. Patients placed importance on controlling bleeding (and joint bleeding caused by repeated internal hemarthroses in particular) and maintaining quality of life.
Patient group input reported that patients hoped gene therapy would lead to fewer FIX infusions, minimal needle injections, and less bleeding. Based on results of the BeneGene-2 trial, fidanacogene elaparvovec may result in a decrease in AIRs and total FIX consumption when compared with FIX prophylaxis. Furthermore, the BeneGene-2 trial results for bleeding end points overall indicated that treatment with fidanacogene elaparvovec may result in decreased bleeds when compared to FIX prophylaxis. Fidanacogene elaparvovec resulted in a decrease in the ABRtotal at both year 1 and from week 12 to the data cut-off date when compared with FIX prophylaxis received during the lead-in study, and the clinical experts consulted by CADTH determined that the magnitude of the effect size was a clinically relevant improvement. The improvement in the ABRtotal FIX activity levels measured by 2 single-stage assays and 1 chromogenic assay suggest that the steady-state FIX:C level of the majority of the patients was higher than the prespecified fixed threshold of 5% and remained stable (Appendix 1). Results from other bleeding outcomes (i.e., the ABRtreat, | |||||||||||| |||| ||| ||||||||| [Appendix 1]) were consistent with the ABRtotal, favouring fidanacogene elaparvovec compared to FIX prophylaxis during the lead-in study. The clinical experts consulted by CADTH considered the percentage of patients with no bleeds to be an efficacy end point that is more clinically relevant and informative compared with the ABR. Results at year 1 following fidanacogene elaparvovec infusion (i.e., 64.4% of patients treated with fidanacogene elaparvovec versus 28.9% of the patients treated with FIX prophylaxis during the lead-in period) suggest that there is a clinically meaningful improvement favouring fidanacogene elaparvovec, according to the clinical experts.
However, there was uncertainty in interpreting the positive findings in the bleeding outcomes. The comparative evidence is nonrandomized, which, according to GRADE guidance used by the CADTH review team, starts at low certainty. Furthermore, CADTH identified potential sources of bias. The magnitude of the observed effect may be biased due to assumptions of the models used to compare observations in the BeneGene-2 study to those in the lead-in BeneGene-1 study. In particular, the probability of no bleeds did not account for differences in follow-up times during the 2 studies, and this is expected to bias results, although the direction is unknown. The open-label design and self-reporting of bleeding events may also have led to an overestimate of the effect size. There was a potential risk of bias in the underestimation of the ABR or overestimation of percentage of patients with no bleeds among those treated with fidanacogene elaparvovec, given that the BeneGene-2 trial was open-label and the bleeding events were recorded by the patients themselves. Furthermore, according to the clinical experts consulted by CADTH, the ABRtreat can be misleading because the determination of whether a bleed needed treatment was a decision made by both clinicians and patients. The risk of bias could not be ruled out despite the sponsor’s decision to implement several measures to ensure patients correctly used electronic diaries. Similarly, given the open-label design of the BeneGene-2 trial, the subjective nature of outcomes and the lack of a comparator group, no valid inferences can be made on HRQoL outcomes despite improvement in change from baseline across all HRQoL outcomes examined in the CADTH report.
According to patient group input, more than 60% of the respondents expect a gene therapy to be effective in preventing bleeding for at least 10 years. In addition, the clinical experts consulted by CADTH noted that a longer follow-up period of 20 to 25 years is warranted to make any definite determinations on the overall long-term efficacy of fidanacogene elaparvovec. An important limitation in the efficacy results in the pivotal BeneGene-2 trial is the relatively short duration of follow-up (median: |||| ||||| with a minimum of ||| and a maximum of ||| |||||). To help address this gap in the pivotal trial evidence, the sponsor submitted results from a phase I and IIa trial (Study C0371005) and its corresponding extension study (Study C0371003), which provided evidence on the safety and efficacy of fidanacogene elaparvovec in 15 patients who were followed for up to 6 years. However, the limitations of these studies (e.g., a single-arm and noncomparative design, descriptive analyses, a small sample size, many patients ongoing, and missing data for some outcomes) preclude CADTH from using this evidence to draw conclusions about the longer-term efficacy of fidanacogene elaparvovec. Furthermore, 6 years of follow-up may not be considered sufficiently long-term, as patients and clinicians expect longer efficacy from a gene therapy.
Patient input emphasized that current treatments for hemophilia B can greatly complicate their everyday life, travel, and leisure activities. Patients indicated that they hoped gene therapy would lead to less stress and fewer restrictions on activities and make it easier to travel. Similarly, the clinical experts consulted by CADTH noted that monitoring changes in the HJHS as well as HRQoL end points following fidanacogene elaparvovec infusion (e.g., improvement in activity of daily living, physical activity and functioning, decrease in development of disability, and improvement in psychosocial health and functioning) are important for assessing treatment response. Although HRQoL was assessed in the BeneGene-2 trial using the Haem-A-QoL instrument, this evidence was noncomparative and therefore provided very low certainty according to GRADE guidance used by the CADTH review team. Similarly, effects on activities were assessed by the HAL tool, but the data were noncomparative. Although the Haem-A-QoL and HAL tools were also assessed in Study C0371005 and C0371003, both of which had only a single arm, data were available for only | patients, and the missing data further limits the interpretation of those results. As such, CADTH could draw no conclusions with certainty regarding the effect of fidanacogene elaparvovec on HRQoL and patient activities.
Harms
The safety profiles of patients treated with fidanacogene elaparvovec in the pivotal BeneGene-2 trial, were considered acceptable by the clinical experts consulted by CADTH. No deaths or study discontinuations due to AEs were reported in the trial. The most common SAE was anemia, and the most common notable harm was increased ALT (26.7%) followed by abnormal hepatic function (13.3%). As the available safety data are noncomparative, the evidence about the effect of fidanacogene elaparvovec on harms overall compared to current treatments (i.e., FIX prophylaxis) is uncertain. In addition, the evidence on safety was considered limited due to the small number of patients involved and the relatively short duration of the follow-up. Although Study C0371005 and C0371003 provide longer-term safety data, with up to 6 years of follow-up, the long-term safety of fidanacogene elaparvovec remains uncertain. Similar to the assessment of efficacy, the clinical experts consulted by CADTH noted that a longer follow-up period of 20 to 25 years involving more patients is warranted to make any definite determinations on the overall long-term safety of fidanacogene elaparvovec.
Conclusion
One phase III, single-arm, open-label trial (BeneGene-2) investigated the efficacy and safety of fidanacogene elaparvovec in 45 patients with moderately severe to severe hemophilia B (defined as FIX:C ≤ 2%). For efficacy outcomes regarding bleeding events and the use of FIX following fidanacogene elaparvovec infusion, patients in the BeneGene-2 trial served as their own controls, using data collected from when these patients were on FIX prophylaxis during a lead-in study (BeneGene-1). Compared to FIX prophylaxis, fidanacogene elaparvovec may result in a decrease in the ABRtotal, ABRtreat, ||||||||| AIR, and |||||||||| ||| |||||||||||| and the effects observed for all of these outcomes were considered clinically relevant by the clinical experts consulted by CADTH. However, uncertainty associated with interpreting the clinical significance of the magnitude of the treatment differences remains due to study limitations, such as the nonrandomized comparative design, potential risk of bias in self-reporting of bleeding events caused by the open-label design, and potential biases introduced by the assumptions of the statistical models used to make the comparisons. The safety profile of fidanacogene elaparvovec during the follow-up period was considered acceptable by the clinical experts consulted by CADTH; however, the safety evidence is also uncertain given the lack of comparative data, small sample size, and limited duration of follow-up. To address the limited duration of follow-up in the BeneGene-2 study, evidence from a phase I and IIa, single-arm, open-label trial and a corresponding extension study that provided data for up to 6 years of follow-up was examined. However, the limitations of these supportive studies (e.g., a single arm and noncomparative design, descriptive analyses, small sample size, many patients ongoing, and missing data) preclude CADTH from using this evidence to draw conclusions with certainty about the longer-term efficacy and safety of fidanacogene elaparvovec. Altogether, the long-term efficacy and safety of fidanacogene elaparvovec remains inconclusive.
References
1.Fidanacogene elaparvovec for the treatment of moderately severe to severe hemophilia B in patients 18 years of age and older [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: fidanacogene elaparvovec, 1 x 1013 vector genomes per mL, intravenous infusion. Kirkland (QC): Pfizer Canada ULC; 2023 Jul 31.
2.Fidanacogene elaparvovec: 1 x 1013 vector genomes per mL, intravenous infusion [product monograph]. Kirkland (QC): Pfizer Canada ULC; 2023 Dec 27.
3.Swystun LL, James P. Using genetic diagnostics in hemophilia and von Willebrand disease. Hematology Am Soc Hematol Educ Program. 2015;2015:152-159. PubMed
4.National Organization for Rare Disorders. Hemophilia B. Rare Disease Database 2023; https://rarediseases.org/rare-diseases/hemophilia-b/. Accessed 2023 Jun 29.
5.Srivastava A, Santagostino E, Dougall A, et al. WFH guidelines for the management of hemophilia, 3rd edition. Haemophilia. 2020;26(Suppl 6):1-158.
6.Konkle BA, Nakaya Fletcher S. GeneReviews® [Internet]: Hemophilia B. Seattle (WA): University of Washington, Seattle; 2023: https://www.ncbi.nlm.nih.gov/books/NBK1495/. Accessed 2023 Feb 9.
7.Document 276: MASAC Recommendations Concerning Products Licensed for the Treatment of Hemophilia and Selected Disorders of the Coagulation System. New York (NY): National Hemophilia Foundation; 2023: https://www.hemophilia.org/sites/default/files/document/files/MASACTreatment.pdf. Accessed 2023 Jun 29.
8.Rayment R, Chalmers E, Forsyth K, et al. Guidelines on the use of prophylactic factor replacement for children and adults with Haemophilia A and B. Br J Haematol. 2020;190(5):684-695. PubMed
9.Clinical Study Report: C0371002. Phase 3, Open Label, Single Arm Study to Evaluate Efficacy and Safety of FIX Gene Transfer With PF06838435 (rAAV-Spark100-hFIX-Padua) in Adult Male Participants With Moderately Severe to Severe Hemophilia B (FIX:C≤2%) (BeneGene-2) [internal sponsor's report]. Kirkland (QC): Pfizer Canada ULC; 2023 Feb 22.
10.Clinical Study Protocol: C0371002. Phase 3, Open-Label, Single-Arm Study to Evaluate Efficacy and Safety of FIX Gene Transfer with PF-06838435 (rAAV-Spark100-hFIX-R338L) in Adult Male Patients with Moderately Severe to Severe Hemophilia B (FIX:C≤2%) (BeneGene-2) [internal sponsor's report]. New York (NY): Pfizer, Inc.; 2021 Oct 12.
11.Grover SP, Mackman N. Intrinsic pathway of coagulation and thrombosis. Arterioscler Thromb Vasc Biol. 2019;39(3):331-338. PubMed
12.Perez-Pujol S, Aras O, Escolar G. Factor V Leiden and inflammation. Thrombosis. 2012;2012:594986. PubMed
13.Mann DM, Stafford KA, Poon MC, Matino D, Stafford DW. The Function of extravascular coagulation factor IX in haemostasis. Haemophilia. 2021;27(3):332-339. PubMed
14.Windyga J, Lissitchkov T, Stasyshyn O, et al. Pharmacokinetics, efficacy and safety of BAX326, a novel recombinant factor IX: a prospective, controlled, multicentre phase I/III study in previously treated patients with severe (FIX level <1%) or moderately severe (FIX level ≤2%) haemophilia B. Haemophilia. 2014;20(1):15-24. PubMed
15.Mannucci PM, Tuddenham EG. The hemophilias--from royal genes to gene therapy. N Engl J Med. 2001;344(23):1773-1779. PubMed
16.Witmer C, Presley R, Kulkarni R, Soucie JM, Manno CS, Raffini L. Associations between intracranial haemorrhage and prescribed prophylaxis in a large cohort of haemophilia patients in the United States. Br J Haematol. 2011;152(2):211-216. PubMed
17.Zanon E, Iorio A, Rocino A, et al. Intracranial haemorrhage in the Italian population of haemophilia patients with and without inhibitors. Haemophilia. 2012;18(1):39-45. PubMed
18.Kizilocak H, Young G. Diagnosis and treatment of hemophilia. Clin Adv Hematol Oncol. 2019;17(6):344-351. PubMed
19.Miesbach W, Reitter-Pfoertner SE, Klamroth R, et al. Co-morbidities and bleeding in elderly patients with haemophilia–a survey of the German, Austrian and Swiss Society of Thrombosis and Haemostasis Research (GTH). Haemophilia. 2017;23(5):721-727. PubMed
20.Association of Hemophilia Clinic Directors of Canada. CHR 2021 Data Summaries: Individuals with Factor IX Deficiency. 2023; https://ahcdc.ca/chr-2021. Accessed 2023 Jun 26.
21.Stonebraker JS, Bolton-Maggs PH, Michael Soucie J, Walker I, Brooker M. A study of variations in the reported haemophilia B prevalence around the world. Haemophilia. 2012;18(3):e91-94. PubMed
22.Iorio A, Stonebraker JS, Chambost H, et al. Establishing the prevalence and prevalence at birth of hemophilia in males: a meta-analytic approach using national registries. Ann Intern Med. 2019;171(8):540-546. PubMed
23.Konkle BA, Skinner M, Iorio A. Hemophilia trials in the twenty-first century: Defining patient important outcomes. Res Pract Thromb Haemost. 2019;3(2):184-192. PubMed
24.Rebinyn® Coagulation Factor IX (Recombinant), Pegylated nonacog beta pegol [product monograph]. Mississauga (ON): Novo Nordisk Canada, Inc; 2022: https://www.novonordisk.ca/content/dam/nncorp/ca/en/products/rebinyn-product-monograph.pdf. Accessed 2023 Jun 29.
25.BeneFIX® Coagulation Factor IX (Recombinant) [product monograph]. Kirkland (QC): Pfizer Canada Inc.; 2017: https://pdf.hres.ca/dpd_pm/00040290.PDF. Accessed by sponsor, no date provided.
26.Alprolix® Coagulation Factor IX (Recombinant), Fc Fusion Protein [product monograph]. Sanofi Aventis Canada, Inc: Laval (QC); 2020: https://pdf.hres.ca/dpd_pm/00058155.PDF. Accessed by sponsor, no date provided.
27.Canadian Blood Services. Plasma and the blood system supply chain. 2021; https://www.blood.ca/en/about-us/media/newsroom/plasma-and-blood-system-supply-chain. Accessed 2022 Sep 19.
28.Clinical Study Report: C0371004. Phase 3, An Open-Label, Non-Investigational Product, MultiCenter, Lead-in Study to Evaluate Prospective Efficacy and Selected Safety Data of Current Factor IX (FIX) or Factor VIII (FVIII) Prophylaxis Replacement Therapy in the Usual Care Setting of Moderately Severe to Severe Adult Hemophilia B Participants (FIX:C≤2%) Who are Negative for Neutralizing Antibodies to AdenoAssociated Virus Vector-Spark100 (Benegene-1) and Moderately Severe to Severe Hemophilia A Adult Participants (FVIII:C≤1%) Who are Negative for Neutralizing Antibodies to Adeno-Associated Virus Vector 6 (AAV6), Prior to the Respective Therapeutic Phase 3 Gene Therapy Studies [internal sponsor's report]. Kirkland (QC): Pfizer Canada ULC; 2023 Mar 17.
29.Clinical Study Report: C0371002. Phase 3, Open-Label, Single-Arm Study to Evaluate Efficacy and Safety of FIX Gene Transfer with PF-06838435 (rAAV-Spark100-hFIX-R338L) in Adult Male Patients with Moderately Severe to Severe Hemophilia B (FIX:C≤2%) (BeneGene-2) [internal sponsor's report]. Statistical Analysis Plan version 6 Kirkland (QC): Pfizer Canada ULC; 2022.
30.St-Louis J, Abad A, Funk S, et al. The Hemophilia Joint Health Score version 2.1 Validation in Adult Patients Study: A multicenter international study. Res Pract Thromb Haemost. 2022;6(2):e12690. PubMed
31.Batt K, Recht M, Cooper DL, Iyer NN, Kempton CL. Construct validity of patient-reported outcome instruments in US adults with hemophilia: results from the Pain, Functional Impairment, and Quality of life (P-FiQ) study. Patient Prefer Adherence. 2017;11:1369-1380. PubMed
32.Wang M, Batt K, Kessler C, et al. Internal consistency and item-total correlation of patient-reported outcome instruments and hemophilia joint health score v2.1 in US adult people with hemophilia: results from the Pain, Functional Impairment, and Quality of life (P-FiQ) study. Patient Prefer Adherence. 2017;11:1831-1839. PubMed
33.Nunnally JC. Psychometric Theory (2nd ed.). New York (NY): McGraw-Hill; 1978.
34.Kline P. A Handbook of Test Construction: Introduction to Psychometric Design. New York (NY): Methuen & Co; 1986.
35.Feldman BM, Funk SM, Bergstrom BM, et al. Validation of a new pediatric joint scoring system from the International Hemophilia Prophylaxis Study Group: validity of the hemophilia joint health score. Arthritis Care Res (Hoboken). 2011;63(2):223-230. PubMed
36.Fischer K, Steen Carlsson K, Petrini P, et al. Intermediate-dose versus high-dose prophylaxis for severe hemophilia: comparing outcome and costs since the 1970s. Blood. 2013;122(7):1129-1136. PubMed
37.Bladen M, Main E, Hubert N, Koutoumanou E, Liesner R, Khair K. Factors affecting the Haemophilia Joint Health Score in children with severe haemophilia. Haemophilia. 2013;19(4):626-631. PubMed
38.Groen W, van der Net J, Lacatusu AM, Serban M, Helders PJ, Fischer K. Functional limitations in Romanian children with haemophilia: further testing of psychometric properties of the Paediatric Haemophilia Activities List. Haemophilia. 2013;19(3):e116-125. PubMed
39.Fischer K, Poonnoose P, Dunn AL, et al. Choosing outcome assessment tools in haemophilia care and research: a multidisciplinary perspective. Haemophilia. 2017;23(1):11-24. PubMed
40.Sluiter D, Foppen W, de Kleijn P, Fischer K. Haemophilia Joint Health Score in healthy adults playing sports. Haemophilia. 2014;20(2):282-286. PubMed
41.von Mackensen S, Catalani O, Asikanius E, Paz-Priel I, Lehle M, Trask P. Determining meaningful health-related quality-of-life improvement in persons with haemophilia A using the Haemophilia Quality of Life Questionnaire for Adults (Haem-A-QoL). Haemophilia. 2020;26(6):1019-1030. PubMed
42.von Mackensen S, Eldar-Lissai A, Auguste P, et al. Measurement properties of the Haem-A-QoL in haemophilia clinical trials. Haemophilia. 2017;23(3):383-391. PubMed
43.Wyrwich KW, Krishnan S, Poon JL, et al. Interpreting important health-related quality of life change using the Haem-A-QoL. Haemophilia. 2015;21(5):578-584. PubMed
44.van Genderen FR, van Meeteren NL, van der Bom JG, et al. Functional consequences of haemophilia in adults: the development of the Haemophilia Activities List. Haemophilia. 2004;10(5):565-571. PubMed
45.van Genderen FR, Westers P, Heijnen L, et al. Measuring patients' perceptions on their functional abilities: validation of the Haemophilia Activities List. Haemophilia. 2006;12(1):36-46. PubMed
46.Buckner TW, Sidonio R, Jr., Guelcher C, et al. Reliability and validity of patient-reported outcome instruments in US adults with hemophilia B and caregivers in the B-HERO-S study. Eur J Haematol. 2018;101(6):781-790. PubMed
47.Cohen J. A power primer. Psychol Bull. 1992;112(1):155-159. PubMed
48.Kuijlaars IAR, Emst M, Net J, Timmer MA, Fischer K. Assessing the test–retest reliability and smallest detectable change of the Haemophilia Activities List. Haemophilia. 2020;27(1):108-112. PubMed
49.de Vet HCW, Terwee CB, Mokkink LB, Knol DL. Measurement in Medicine: A Practical Guide. Cambridge (UK): Cambridge University Press; 2011.
50.Kempton CL, Wang M, Recht M, et al. Reliability of patient-reported outcome instruments in US adults with hemophilia: the Pain, Functional Impairment and Quality of life (P-FiQ) study. Patient Prefer Adherence. 2017;11:1603-1612. PubMed
51.Non-Inferiority Clinical Trials to Establish Effectiveness: Guidance for Industry. Rockville (MD): U.S. Food and Drug Administration; 2016: https://www.fda.gov/media/78504/download. Accessed by sponsor, no date provided.
52.Guideline on the choice of the non-inferiority margin. London (UK): European Medicines Agency; 2005: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-choice-non-inferiority-margin_en.pdf. Accessed by sponsor, no date provided.
53.Drug Reimbursement Review sponsor submission: Fidanacogene elaparvovec: 1 x 1013 vector genomes per mL, intravenous infusion [internal sponsor's package]. Kirkland (QC): Pfizer Canada ULC; 1800.
54.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. Journal of clinical epidemiology. 2020;119:126-135. PubMed
55.Balshem H, Helfand M, Schünemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. Journal of clinical epidemiology. 2011;64(4):401-406. PubMed
56.Interim Clinical Study Report: C0371005. Gene Therapy, Open-Label, Dose-Escalation Study of SPK-9001 (Adeno-Associated Viral Vector With Human Factor IX Gene) in Subjects With Hemophilia B. Kirkland (QC): Pfizer Canada ULC; 2023.
57.George LA, Sullivan SK, Giermasz A, et al. Hemophilia B Gene Therapy with a High-Specific-Activity Factor IX Variant. N Engl J Med. 2017;377(23):2215-2227. PubMed
58.Interim Clinical Study Report: C0371003. A Factor IX (FIX) Gene Transfer, Multi-Center Evaluation of the Long-Term Safety and Efficacy Study of PF-06838435 and a Dose-Escalation Substudy in Individuals With Hemophilia B. Kirkland (QC): Pfizer Canada ULC; 2023.
59.Clinical Study Protocol: C0371003. A Factor IX (FIX) Gene Transfer, Multi-Center Evaluation of the Long-Term Safety and Efficacy Study of PF-06838435 and a Dose-Escalation Substudy in Individuals With Hemophilia B - Statistical Analysis Plan. Version 3. April, 29 2022. Kirkland (QC): Pfizer Canada ULC; 2022.
Appendix 1: Detailed Outcome Data
Note this appendix has not been copy-edited.
Table 19: Sensitivity Analysis — Jump to Reference of ABRtotal to Assess Impact of Discontinuation From BeneGene-2 Prior to Reaching 15 Months of Follow-Up (Dosed Population)
Outcome | Pivotal BeneGene-2 | Lead-in BeneGene-1 |
|---|---|---|
Fidanacogene elaparvovec (N = 45) | FIX prophylaxis (N = 45) | |
Jump to reference of ABRtotal (year 1 following fidanacogene elaparvovec infusion) | ||
Number of participants who contributed to the analysisa | 45 | 45 |
Number of participants without any bleeds, n (%) | || |||||| | || |||||| |
Number of bleeds | ||
Mean (SD) | ||| |||||| | ||| |||||| |
Median (minimum to maximum) | ||| ||| ||| | ||| ||| ||| |
ABR | ||
Mean (SD) | |||| ||||||| | |||| ||||||| |
Median (minimum to maximum) | |||| ||||| ||||| | |||| ||||| ||||| |
Model-derived ABRtotal estimate (95% CI) | |||| ||||| || ||||| | |||| ||||| || ||||| |
Treatment difference | ||
Negative binomial estimates (95% CI) | ||||| |||||| || |||||| | |
Negative binomial P value | |||||| | |
Percentage reduction, % | ||
Negative binomial estimate (95% CI) | ||||| ||||| || |||||| | |
Negative binomial P value | |||||| | |
ABRtotal = annualized bleeding rate for treated and untreated bleeds; CI = confidence interval; SD = standard deviation.
Note: Year 1 referred to the period between week 12 and month 15 following infusion of fidanacogene elaparvovec.
aNumber of patients who were in follow-up at the start of the period interval.
Source: BeneGene-2 Clinical Study Report.9
Table 20: Mean Steady-State FIX:C Between Week 12 and Month 15 Postinfusion of Fidanacogene Elaparvovec (Dosed Population)
Steady-state FIX:Ca | Fidanacogene elaparvovec (N = 45) |
|---|---|
One-stage assay with Actin-FSL reagent | |
Mean (SD) | 12.62 (8.92) |
P value | |||||| |
One-stage assay with SynthAsil reagent | |
Mean (SD) | 25.90 (16.89) |
P value | |||||| |
Chromogenic assay | |
Mean (SD) | 13.49 (10.40) |
P value | ||||||| |
FIX:C = circulating factor IX; SD = standard deviation.
Note: Data cut-off date November 16, 2022.
aBased on central laboratory data from 12 weeks to 15 months. Any samples taken within 7 days (14 days if EHL product is used) of exogenous FIX replacement therapy are not included in the assessment of steady-state FIX:C postinfusion.
bSignificantly higher than the fixed threshold of 5%
Sources: BeneGene-2 Clinical Study Report9 and the sponsor’s Summary of Clinical Evidence.1
Table 21: Results for the Analysis of Sustained FIX:C (Dosed Population)
Steady-state FIX:Ca | Month 3 (Week 12) | Month 15 (Week 65) | Month 24 (Week 104) |
|---|---|---|---|
One-stage assay with Actin-FSL reagent | |||
n | 43 | 34 | 22 |
Mean (SD) | 13.52 (8.134) | 13.10 (12.792) | 12.67 (11.884) |
One-stage assay with SynthAsil reagent | |||
n | 44 | 35 | 22 |
Mean (SD) | 27.79 (15.226) | 27.47 (25.739) | 25.00 (22.627) |
Chromogenic assay | |||
n | 44 | 35 | 22 |
Mean (SD) | 13.91 (9.302) | 15.82 (16.996) | 15.40 (18.829) |
FIX:C = circulating factor IX; SD = standard deviation.
Note: Data cut-off date November 16, 2022
aBased on central laboratory data from 12 weeks to 15 months. Any samples taken within 7 days (14 days if EHL product is used) of exogenous FIX replacement therapy are not included in the assessment of steady-state FIX:C postinfusion.
Sources: BeneGene-2 Clinical Study Report9 and the sponsor’s Summary of Clinical Evidence.1
Figure 3: Stacked Bar Plot for Proportions of Participants Within the FIX:C Category Over Time With Imputation by Assay (Dosed Population) [Redacted]

FIX:C = circulating coagulation factor IX; SD = standard deviation.
Notes: Analyses are based on central laboratory data and any samples taken within 7 days of exogenous FIX replacement therapy (or 14 days if EHL product used) are excluded; If a patient withdrew consent, discontinued or resumed FIX prophylaxis, then assessments for visits following withdrawal/discontinuation/FIX prophylaxis resumption were imputed as 1.9%
Source: BeneGene-2 Clinical Study Report9 and the sponsor’s Summary of Clinical Evidence.1
Figure 4: Box-Whisker Plot of FIX:C Over Time With Imputation by One-Stage Assay With Actin-FSL Reagent (Dosed Population) [Redacted]

EHL = extended half-life; FIX = coagulation factor IX; FIX:C = circulating factor IX; IQR = interquartile range.
Notes: Analyses are based on central laboratory data and any samples taken within 7 days of exogenous FIX replacement therapy (or 14 days if EHL product used) are excluded; If a patient withdrew consent, discontinued or resumed FIX prophylaxis, then assessments for visits following withdrawal/discontinuation/FIX prophylaxis resumption were imputed as 1.9%.
Box contains the 25th (Q1) and 75th (Q3) percentiles; median is denoted by the line inside the box; whiskers mark the 1.5*IQR; values beyond 1.5*IQR range are considered outliers and are denoted by open circles; mean values are denoted by filled diamonds.
Source: BeneGene-2 Clinical Study Report9 and the sponsor’s Summary of Clinical Evidence.1
Figure 5: Box-Whisker Plot of FIX:C Over Time with Imputation by One-Stage Assay with SynthASil Reagent (Dosed Population) [Redacted]

EHL = extended half-life; FIX = coagulation factor IX; FIX:C = circulating factor IX; IQR = interquartile range
Notes: Analyses are based on central laboratory data and any samples taken within 7 days of exogenous FIX replacement therapy (or 14 days if EHL product used) are excluded; If a patient withdrew consent, discontinued or resumed FIX prophylaxis, then assessments for visits following withdrawal/discontinuation/FIX prophylaxis resumption were imputed as 1.9% The box contains the 25th (Q1) and 75th (Q3) percentiles; median is denoted by the line inside the box; whiskers mark the 1.5*IQR; values beyond 1.5 × IQR range are considered outliers and are denoted by open circles; mean values are denoted by filled diamonds.
Sources: BeneGene-2 Clinical Study Report9 and the sponsor’s Summary of Clinical Evidence.1
Table 22: Analysis of Target Joints in the BeneGene-2 Trial (Dosed Population)
Outcome | Number of target jointsa | ||||
|---|---|---|---|---|---|
None | At least 1 | 1 | 2 | ≥ 3 | |
Number of participants with target joints, n (%) | |||||
Baseline | 32 (71.1) | 13 (28.9) | 7 (15.6) | 3 (6.7) | 3 (6.7) |
Day 1 to month 15 | || ||||||| | |||||| | |||||| | |||||| | |||||| |
Overallb | || ||||||| | |||||| | |||||| | |||||| | |||||| |
Number of participants whose baseline target joints resolved during time period, n (%) | |||||
Day 1 to month 15 | |||||| | |||||| | |||||| | |||||| | |||||| |
Overallb | |||||| | |||||| | |||||| | |||||| | |||||| |
Number of participants with newly developed target joints postbaseline, n (%) | |||||
Day 1 to month 15 | |||||| | |||||| | |||||| | |||||| | |||||| |
Overallb | |||||| | |||||| | |||||| | |||||| | |||||| |
Number of participants whose newly developed target joints postbaseline was resolved, n (%) | |||||
Day 1 to month 15 | |||||| | |||||| | |||||| | |||||| | |||||| |
Note: Data cut-off date November 16, 2022.
aTarget joint was defined as a major joint (e.g., hip, elbow, wrist, shoulder, knee, and ankle) into which repeated bleeds occur (≥ 3 spontaneous bleeds into a single joint within a consecutive 6-month period). A target joint is considered resolved when there are ≤ 2 bleeds into the joint within a 12-month period.
bOverall includes from day 1 to data cut-off date.
Sources: BeneGene-2 Clinical Study Report9 and the sponsor’s Summary of Clinical Evidence.1
Figure 6: Box-Whisker Plot of FIX:C Over Time With Imputation by Chromogenic Assay (Dosed Population) [Redacted]

EHL = extended half-life; FIX = coagulation factor IX; FIX:C = circulating factor IX; IQR = interquartile range.
Notes: Analyses are based on central laboratory data and any samples taken within 7 days of exogenous FIX replacement therapy (or 14 days if EHL product used) are excluded; If a patient withdrew consent, discontinued or resumed FIX prophylaxis, then assessments for visits following withdrawal/discontinuation/FIX prophylaxis resumption were imputed as 1.9% The box contains the 25th (Q1) and 75th (Q3) percentiles; median is denoted by the line inside the box; whiskers mark the 1.5*IQR; values beyond 1.5*IQR range are considered outliers and are denoted by open circles; mean values are denoted by filled diamonds.
Sources: BeneGene-2 Clinical Study Report9 and the sponsor’s Summary of Clinical Evidence.1
Table 23: ABR for Spontaneous Bleeding and Traumatic Bleeding in the BeneGene-2 Trial (Dosed Population)
Outcome | Pivotal BeneGene-2 | Lead-in BeneGene-1 |
|---|---|---|
Fidanacogene elaparvovec (N = 45) | FIX prophylaxis (N = 45) | |
ABR spontaneousa (year 1 following fidanacogene elaparvovec infusion) | ||
Number of participants who contributed to the analysisb | 45 | 45 |
Number of participants without any bleeds, n (%) | 35 (77.8) | 18 (40.0) |
Number of bleeds | ||
Mean (SD) | ||| |||||| | ||| |||||| |
Median (minimum to maximum) | ||| ||| || | ||| ||| ||| |
ABR | ||
Mean (SD) | |||| ||||||| | |||| |||||||| |
Median (minimum to maximum) | |||| ||||| |||| | |||| ||||| ||||| |
Model-derived ABRtotal estimate (95% CI) | |||| ||||| || ||||| | |||| ||||| || ||||| |
Treatment difference | ||
Negative binomial estimates (95% CI) | ||||| |||||| || |||||| | |
Negative binomial P value | |||||| | |
Percentage reduction, % | ||
Negative binomial estimate (95% CI) | ||||| |||||| || |||||| | |
ABR spontaneous (overall) | ||
Number of participants who contributed to the analysisb | 45 | 45 |
Number of participants without any bleeds, n (%) | || ||||||| | || ||||||| |
Number of bleeds | ||
Mean (SD) | ||| |||||| | ||| |||||| |
Median (minimum to maximum) | ||| ||| ||| | ||| ||| ||| |
ABR | ||
Mean (SD) | |||| ||||||| | |||| |||||||| |
Median (minimum to maximum) | |||| ||||| |||| | |||| ||||| ||||| |
Model-derived ABRtotal estimate (95% CI) | |||| ||||| || ||||| | |||| ||||| || ||||| |
Treatment difference | ||
Negative binomial estimates (95% CI) | ||||| |||||| || |||||| | |
Negative binomial P value | |||||| | |
Percentage reduction, % | ||
Negative binomial estimate (95% CI) | ||||| |||||| || |||||| | |
ABR traumaticc (year 1 following fidanacogene elaparvovec infusion) | ||
Number of participants who contributed to the analysisb | 45 | 45 |
Number of participants without any bleeds, n (%) | || ||||||| | || ||||||| |
Number of bleeds | ||
Mean (SD) | ||| |||||| | ||| |||||| |
Median (minimum to maximum) | ||| ||| || | ||| ||| ||| |
ABR | ||
Mean (SD) | |||| |||||| | |||| |||||| |
Median (minimum to maximum) | |||| ||||| |||| | |||| ||||| ||||| |
Model-derived ABRtotal estimate (95% CI) | |||| ||||| || ||||| | |||| ||||| || ||||| |
Treatment difference | ||
Negative binomial estimates (95% CI) | ||||| |||||| || ||||| | |
Negative binomial P value | |||||| | |
Percentage reduction, % | ||
Negative binomial estimate (95% CI) | ||||| |||||| || |||||| | |
ABR traumatic (overall) | ||
Number of participants who contributed to the analysisb | 45 | 45 |
Number of participants without any bleeds, n (%) | || |||||| | || |||||| |
Number of bleeds | ||
Mean (SD) | ||| |||||| | ||| |||||| |
Median (minimum to maximum) | ||| ||| || | ||| ||| ||| |
ABR | ||
Mean (SD) | |||| ||||||| | |||| ||||||| |
Median (minimum to maximum) | |||| ||||| |||| | |||| ||||| ||||| |
Model-derived ABRtotal estimate (95% CI) | |||| ||||| || ||||| | |||| ||||| ||||||| |
Treatment difference | ||
Negative binomial estimates (95% CI) | ||||| |||||| || ||||| | |
Negative binomial P value | |||||| | |
Percentage reduction, % | ||
Negative binomial estimate (95% CI) | ||||| |||||| || |||||| | |
ABR = annualized bleeding rate; CI = confidence interval; FIX = coagulation factor IX; SD = standard deviation
Note: Year 1 referred to the period between week 12 and month 15 following infusion of fidanacogene elaparvovec. Overall referred to the period from week 12 following fidanacogene elaparvovec infusion to the data cut-off date: November 16, 2022. As of the data cut-off date, the mean (SD) duration of follow-up in the pivotal BeneGene-2 trial was |||| ||||| ||||||| with a median (minimum to maximum) of |||| ||||| ||||| ||||. Week 52 and week 104’s baseline was defined as the last nonmissing measurement before the dosing date (day 1) in the pivotal study. The mean (SD) duration of follow-up in the lead-in BeneGene-1 was |||| ||||| ||||||| with a median (minimum to maximum) of |||| ||||| ||||| ||||.
aSpontaneous bleed was defined as a bleed that occurred for no apparent/known reason, particularly into the joints, muscles, and soft tissues.
bNumber of patients who were in follow-up at the start of the period interval.
cTraumatic bleed was defined as a bleed that occurred for an apparent/known reason.
Source: BeneGene-2 Clinical Study Report.9
Pharmacoeconomic Review
Abbreviations
ABR
annual bleed rate
AE
adverse event
BIA
budget impact analysis
CBS
Canadian Blood Services
EHL
extended half-life
FIX
factor IX
nAb
neutralizing antibody
QALY
quality-adjusted life-year
SHL
standard half-life
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Fidanacogene elaparvovec, concentrate solution of vector genomes, for infusion |
Submitted price | Fidanacogene elaparvovec, 1 × 1013 vector genomes per mL: $4,773,595.20 per administration |
Indication | For the treatment of adults (aged 18 years or older) with moderately severe to severe hemophilia B (congenital factor IX deficiency) who are negative for neutralizing antibodies to variant adeno-associated virus serotype Rh74 |
Health Canada approval status | NOC |
Health Canada review pathway | Priority review |
NOC date | December 27, 2023 |
Reimbursement request | As per indication |
Sponsor | Pfizer Canada ULC |
Submission history | Previously reviewed: No |
NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Markov model |
Target population | Adult patients (18 years and older) patients with moderately severe to severe hemophilia B |
Treatment | Fidanacogene elaparvovec |
Comparators | FIX prophylaxis treatments:
|
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, life-years |
Time horizon | Lifetime (77 years) |
Key data sources | Effectiveness of fidanacogene elaparvovec informed by the BeneGene-2 trial; effectiveness of FIX prophylaxis treatments informed by the BeneGene-1 trialc |
Submitted results | Fidanacogene elaparvovec was dominant (more effective and less costly) compared to EHL, SHL, and a weighted basket of SHL and EHL FIX prophylaxis treatments |
Key limitations |
|
CADTH reanalysis results |
|
EHL = extended half-life; FIX = coagulation factor IX; ICER = incremental cost-effectiveness ratio; nAb = neutralizing antibody; QALY = quality-adjusted life-year; SHL = standard half-life.
aComposed of FIX (recombinant) Fc fusion protein; and FIX (recombinant) and pegylated nonacog beta pegol.
bComposed of FIX (recombinant) and nonacog alfa.
cBased on data for the subgroup of patients in the BeneGene-1 trial who went on to receive fidanacogene elaparvovec infusion in the BeneGene-2 trial.
Conclusions
The CADTH clinical review concluded that fidanacogene elaparvovec may reduce bleeding events in adult patients with moderately severe to severe hemophilia B relative to treatment with coagulation factor IX (FIX) prophylaxis, based on observations from the single-arm BeneGene-2 trial for fidanacogene elaparvovec compared to observations from the lead-in BeneGene-1 trial for FIX prophylaxis. CADTH judged the certainty of the evidence to be low for most outcomes and noted that there is uncertainty in the magnitude of differences in bleeding outcomes between fidanacogene elaparvovec and FIX prophylaxis due to the open-label, single-arm study design and self-reporting of bleeding events. The long-term efficacy and safety of fidanacogene elaparvovec is highly uncertain due to limited long-term follow-up data beyond the BeneGene-2 trial period.
CADTH could not address uncertainty related to comparative clinical data, including the magnitude and duration of benefit for fidanacogene elaparvovec compared to FIX prophylaxis. CADTH was also unable to resolve the uncertainty related to the impact of adverse events (AEs), the model structure, the price of FIX prophylaxis, and the costs and consequences of neutralizing antibody (nAb) testing. As such, CADTH was unable to provide a more reliable estimate of the cost-effectiveness of fidanacogene elaparvovec.
Results of the sponsor’s base case suggest that fidanacogene elaparvovec will be more effective over a patient’s lifetime (i.e., 77 years) at reducing bleed events, which leads to a gain of approximately 1 quality-adjusted life-year (QALY), but no predicted change in life expectancy, when compared to FIX prophylaxis treatments (i.e., extended half-life [EHL], standard half-life [SHL], and a weighted basket of SHL and EHL). The sponsor also predicts that fidanacogene elaparvovec will be cost-saving due to the 1-time infusion of fidanacogene elaparvovec and reduced need for FIX prophylaxis and bleeding-related health care resource use. CADTH notes that most (93%) of the benefits associated with fidanacogene elaparvovec were accrued in the extrapolated period (i.e., after 2 years) and that, in the absence of robust long-term comparative data, the incremental gain in QALYs predicted by the sponsor may be overestimated. Similarly, the cost savings predicted by the sponsor’s model ($2,871,630 to $5,576,438) may be overestimated, as they rely on a sustained reduction in bleeding events for the first 25 years of the analysis. Based on the sponsor’s model, the acquisition cost of fidanacogene elaparvovec ($4,773,595 per administration) is predicted to be offset by the savings from reduced use of FIX prophylaxis and treatment of bleeding events after approximately 12 years. If the magnitude and duration of benefit between fidanacogene elaparvovec and FIX prophylaxis is less than estimated by the sponsor or if the actual cost of FIX prophylaxis treatments is lower than what is specified in the sponsor’s model, it will take longer for any potential savings to be realized.
Given the uncertainty in the clinical evidence base, including the magnitude and duration of benefit with fidanacogene elaparvovec compared to currently available FIX prophylaxis treatments, it is highly uncertain whether and to what extent the gains in QALYs and cost savings will be realized in clinical practice.
Stakeholder Input Relevant to the Economic Review
This section summarizes the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process.
Patient input was received from the Canadian Hemophilia Society, which conducted online surveys in July 2023 (17 respondents with moderately severe or severe hemophilia B) and September 2022 (31 respondents with hemophilia A, 7 with hemophilia B, and 1 not specified). Patients with hemophilia B noted that joint damage to the knees, ankles, and elbows caused by repeated internal hemarthroses is the primary physical health impact of hemophilia and that this affects their overall quality of life by reducing mobility, causing them to refrain from physical activity, and requiring joint replacements. Patients indicated that regular prophylactic IV infusions (1 to 3 times per week) were required to manage their hemophilia B and that the high frequency of trips to the clinic for infusions impacted their ability to work. Respondents expressed a desire for the effects of gene therapy to last for at least 10 years and noted concerns with short- and long-term side effects associated with gene therapy and that factor VIII or IX (FIX) levels would not be high enough or last long enough to prevent bleeding. No respondents had experience with fidanacogene elaparvovec.
Clinician input was received from members of the Novel Therapy Committee on behalf of the Association of Hemophilia Clinic Directors of Canada and from the Canadian Association of Nurses in Hemophilia Care. Clinicians noted that the current standard of care for hemophilia B in Canada is FIX concentrates for bleed management or prophylaxis and that approximately 80% of patients in Canada with clinically severe hemophilia B receive prophylaxis. Clinicians noted that current treatments do not modify the underlying disease and that patients with severe hemophilia B have an unmet need for effective prophylaxis. Clinician input noted that gene therapy is a promising long-term phenotypic treatment for patients with hemophilia, but that shared decision-making with patients and families is key in the patient selection process.
CADTH participating drug plans noted concerns about the implementation of fidanacogene elaparvovec, including coordination between Canadian Blood Services (CBS) and the drug plans for funding of products to treat hemophilia B, additional costs (i.e., travel), and the need for gene therapy–specific health care resources (e.g., testing and administration facilities). The drug plans commented on the requirement for nAb testing to confirm eligibility for fidanacogene elaparvovec and noted that long-term follow-ups and data collection are needed to assess the efficacy of gene therapy.
Two of these concerns were addressed in the sponsor’s model:
Costs related to FIX prophylaxis treatment were included; however, these costs may not reflect the true acquisition cost incurred by the public health care payer due to confidential pricing.
Health-related quality of life was incorporated in the sponsor’s model by use of EQ-5D questionnaire data captured in the BeneGene-2 trial. The impact of AEs was not included.
CADTH was unable to address 2 concerns raised in stakeholder input:
The uncertainty associated with the long-term efficacy of fidanacogene elaparvovec could not be addressed due to a lack of long-term comparative data.
Joint health and joint-related surgeries could not be addressed due to the structure of the submitted model.
Economic Review
The current review is for fidanacogene elaparvovec for the treatment of adults (aged 18 years or older) with moderately severe to severe hemophilia B (congenital FIX deficiency) who test negative for nAbs to variant adeno-associated virus serotype Rh74.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis to assess the cost-effectiveness of fidanacogene elaparvovec compared to FIX prophylaxis treatments for adult patients with moderately severe to severe hemophilia B.1 In the model, the sponsor compared fidanacogene elaparvovec to SHL FIX prophylaxis (i.e., FIX recombinant nonacog alfa), EHL FIX prophylaxis (i.e., FIX recombinant pegylated nonacog beta pegol) and FIX recombinant Fc fusion protein), and a weighted basket of SHL and EHL FIX prophylaxis (25% SHL and 75% EHL). The modelled population is in line with the Health Canada indication and was based on patients enrolled in the BeneGene-2 trial, although CADTH notes that women and those with a FIX:C level greater than 2% were excluded from the trial.2
Fidanacogene elaparvovec is available as a concentrate for solution for infusion (1 × 1013 vector genomes in 1 mL vials).3 The recommended dose of fidanacogene elaparvovec is a single IV infusion of 5 × 1011 vector genomes per kilogram of body weight.3 The sponsor submitted price for fidanacogene elaparvovec is $4,773,595.20 per administration per patient, regardless of the number of vials required.1 The sponsor estimated that the annual per-patient cost of SHL FIX prophylaxis and EHL FIX prophylaxis would be $321,734 and $417,964, respectively.
The analysis was conducted from the perspective of the Canadian public health care payer. Cost and clinical outcomes (QALYs and life-years) were estimated over a lifetime horizon of 77 years with a cycle length of 1 year. Discounting (1.5% per annum) was applied to both costs and outcomes.1
Model Structure
The sponsor submitted a Markov model consisting of 4 health states based on an annual number of bleeds (0 bleeds, > 0 to < 3 bleeds, ≥ 3 to < 5 bleeds, ≥ 5 bleeds) and death. Patients were distributed at model entry among the bleed-based health states based on observations from the BeneGene-2 trial for those who received fidanacogene elaparvovec and from the BeneGene-1 trial for those who received FIX prophylaxis.1 Patients who received fidanacogene elaparvovec were assumed to experience an immediate treatment benefit and remain in their initial health state until year 25, when patients were assumed to experience a 1-time 15% effect loss at the beginning of year 26 and have a higher risk of transitioning to the next more-severe health state.1 Of those, 4% were assumed to initiate FIX (SHL or EHL) prophylaxis each year. All fidanacogene elaparvovec patients were assumed to experience a loss of treatment effect by year 40. Patients on FIX infusion at baseline were assumed to remain in their initial bleed-based health state until death. In each cycle, a proportion of patients in all health states were at risk of death.
Model Inputs
The baseline population characteristics used to inform the model were based on the BeneGene-2 trial, which enrolled adult men (mean age 33 years, mean weight 86.66 kg) with moderately severe to severe hemophilia B, which was defined by the sponsor as a circulating FIX level of 2% or less. The BeneGene-2 trial enrolled 45 participants who had completed at least 6 months of FIX prophylaxis during the BeneGene-1 study. The baseline distribution of patients across the bleed-based health states was derived from the BeneGene-2 monitoring phase for the 45 patients who received fidanacogene elaparvovec and from the BeneGene-1 lead-in study for the same 45 patients (which reflects a subset of the overall BeneGene-1 study population). The sponsor assumed that the distributions would be the same for EHL and SHL.1
Mean annual bleed rate (ABR) values by health state were derived from patient-level data from the BeneGene-2 trial pooled across all time points. The sponsor assumed that a proportion of total bleeds would be untreated based on observations from the BeneGene-1 and BeneGene-2 trials.1 Specifically, the BeneGene-2 trial was used to inform the untreated-versus-treated bleed distribution for the 0-to-less-than-3-bleeds health state while the BeneGene-1 study was used to inform untreated-versus-treated bleeds for all other health states. The distribution of bleed type (i.e., target-joint bleeds, nontarget-joint bleeds, and nonjoint bleeds) for treated bleeds was informed by the period before FIX prophylaxis for all health states except for the 0-to-less-than-3-bleeds health state, which was informed by the period following fidanacogene elaparvovec infusion.1
Mortality in the model was informed inputs from Statistics Canada life tables, adjusted for the increased risk of mortality for hemophilia B patients described in a Dutch study that reported a standardized mortality rate of 2.4.4,5 AEs were not considered in the model.
Health-state utility values in the model were informed by 5-Level EQ-5D data collected from the BeneGene-2 trial and valued using UK tariffs. Values were stratified by bleed health states (no bleeds: 0.85; > 0 to < 3 bleeds: 0.74; ≥ 3 to < 5 bleeds: 0.74; ≥ 5 bleeds: 0.68). Disutilities associated with the type of bleed (i.e., target joint, nontarget-joint, and nonjoint) were obtained from the published literature.6,7 The sponsor assumed that initiation of fidanacogene elaparvovec treatment was associated with a 1-time utility decrement of 0.0164 lasting 1 year.
Costs included in the model consisted of acquisition costs for fidanacogene elaparvovec and FIX prophylaxis treatments, administration costs for FIX prophylaxis, health care resource use, and disease management costs.1 Acquisition costs were based on the sponsor’s submitted price for fidanacogene elaparvovec and a previous CADTH review for FIX recombinant nonacog alfa and FIX recombinant Fc fusion protein.1,8 The sponsor assumed that the costs for FIX recombinant nonacog alfa is representative for all available SHL products, whereas the cost of EHL was assumed to be a weighted average between FIX recombinant Fc fusion protein and FIX recombinant pegylated nonacog beta pegol (11.8% and 88.2%, respectively).1 The sponsor assumed that 10% of FIX prophylaxis treatments would be administered by health care professionals, based on expert opinion obtained by the sponsor, with the cost of administration assumed to be $769.90 when provided by a physician or $722.00 when provided by a nurse. Administration costs associated with fidanacogene elaparvovec were not included. The frequency of disease management and health care resource use were informed by clinical expert opinion obtained by the sponsor, with unit costs obtained from the Ontario Schedule of Benefits for physician and laboratory services, the Ontario Case Costing Initiative, and published literature.9-15 Clinical expert opinion was also used to inform the short-term resource use associated with gene therapies and acute bleeding events.1 The sponsor’s model included the ability to incorporate the cost of nAb testing to determine fidanacogene elaparvovec eligibility; however, the sponsor assumed that this cost would be covered by the sponsor and excluded it from the base-case analysis.
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically with 5,000 iterations. The deterministic results were aligned with the submitted probabilistic results. The probabilistic findings are presented in the following section. The submitted analysis was based on the submitted price for fidanacogene elaparvovec and prices from a previous CADTH report for FIX prophylaxis treatments.1,8
Base-Case Results
In the sponsor’s base-case analysis, fidanacogene elaparvovec was more effective and less costly (dominant) compared with SHL FIX prophylaxis, EHL FIX prophylaxis, and the basket of SHL and EHL FIX prophylaxis. Fidanacogene elaparvovec was estimated to be associated with a gain of approximately 1.08 QALYs relative to all comparators over the 77-year horizon, with incremental cost savings of $2,871,630 to $5,576,438 (Table 3). At a willingness-to-pay threshold of $50,000 per QALY gained, there was a 97% probability of fidanacogene elaparvovec being cost-effective.
Results were driven by the acquisition cost of fidanacogene elaparvovec ($4,773,595), as well as the predicted gain in QALYs with fidanacogene elaparvovec and the predicted cost savings resulting from reduced bleeds, the need for FIX prophylaxis, and health care resource use (Appendix 3). The acquisition costs of fidanacogene elaparvovec represent 90% of the total costs associated with fidanacogene elaparvovec. Fidanacogene elaparvovec was not associated with any life-year gains; however, the sponsor’s model predicts an incremental gain of 9.13 years spent in the “no bleeds” health state. Based on the deterministic results, approximately 93% of the predicted QALYs to be gained with fidanacogene elaparvovec accrued after the first 2 years of treatment (i.e., beyond the duration of the BeneGene-2 trial).
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Total QALYs | Sequential ICER ($ per QALY) |
|---|---|---|---|
Fidanacogene elaparvovec | 7,744,097 | 19.503 | Reference |
SHL FIX prophylaxis | 10,615,727 | 18.421 | Dominated by fidanacogene elaparvovec |
EHL FIX prophylaxis | 13,320,535 | 18.420 | Dominated by fidanacogene elaparvovec, SHL FIX prophylaxis, SHL/EHL FIX prophylaxis |
SHL/EHL FIX prophylaxisa | 12,644,333 | 18.420 | Dominated by fidanacogene elaparvovec, SHL FIX prophylaxis |
EHL = extended half-life, ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; SHL = standard half-life.
aAssumed to be composed of 25% SHL and 75% EHL FIX prophylaxis.
Source: Sponsor’s pharmacoeconomic submission.1
Sensitivity and Scenario Analysis Results
The sponsor conducted several scenario analyses that included adopting alternative modelling assumptions (i.e., a discount rate and treatment-effectiveness waning) as well as alternate assumptions related to treatment adherence for FIX prophylaxis, treatment of bleeds, and utility values. In all scenarios, fidanacogene elaparvovec remained dominant over FIX prophylaxis.
The sponsor also conducted 2 scenarios to explore the impact of adopting outcome-based agreements; in both, the outcome that triggers the outcome-based agreements is the addition of FIX prophylaxis infusions after fidanacogene elaparvovec. The first scenario considered annuity payments, in which the annual cost of fidanacogene elaparvovec is applied to each patient who has received fidanacogene elaparvovec but not initiated FIX prophylaxis infusion for 20 years. The second scenario considered lump-sum payments, in which the upfront cost of fidanacogene elaparvovec is applied to all patients who receive fidanacogene elaparvovec but a refund is applied if a patient switches to FIX infusion during an eligibility period (assumed by the sponsor to be 18 years) following treatment administration (refund percentage varies depending on how many years after fidanacogene elaparvovec patients switch to FIX infusions). In both outcome-based agreement scenarios, fidanacogene elaparvovec remained dominant over FIX prophylaxis.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations to the sponsor’s analysis that have notable implications for the economic analysis:
The comparative efficacy of fidanacogene elaparvovec to FIX treatments is uncertain. To inform efficacy in the pharmacoeconomic model (i.e., the number of annual bleeds), the sponsor used data from the BeneGene-2 trial for fidanacogene elaparvovec and from the lead-in study (BeneGene-1) for FIX prophylaxis. In the BeneGene-1 study, patients could receive SHL or EHL FIX prophylaxis, and data were not provided by FIX treatment received. Although the CADTH clinical review concluded that fidanacogene elaparvovec may result in a decrease in the number of bleeding events compared with FIX prophylaxis, the certainty of this finding is low due to the single-arm study design. Additionally, the magnitude of the difference between fidanacogene elaparvovec and FIX prophylaxis is highly uncertain due to the open-label study design and self-reporting of bleeding events. Data to support the equivalent efficacy of individual FIX treatments were not provided by the sponsor.
Given the limitations with the submitted clinical evidence, the magnitude of benefit (i.e., in reducing bleeds) associated with fidanacogene elaparvovec versus currently available SHL and EHL FIX prophylaxis treatments is highly uncertain. CADTH was unable to address this limitation in reanalysis.
The long-term effectiveness of fidanacogene elaparvovec is uncertain. Evidence to support the duration and magnitude of benefit associated with fidanacogene elaparvovec compared to FIX prophylaxis is unavailable. While the sponsor submitted Study C0371003 to support the long-term benefit of fidanacogene elaparvovec (in which 14 patients were followed up for a median of 5.8 years), the CADTH clinical review concluded that the long-term efficacy and safety of fidanacogene elaparvovec relative to FIX prophylaxis could not be determined due to the lack of a comparator group. In the pharmacoeconomic model, the sponsor assumed that patients who received fidanacogene elaparvovec would sustain the initial benefit for 25 years, after which effectiveness was assumed to begin to wane. Given that approximately 93% of the incremental QALYs predicted by the sponsor’s model to be gained with fidanacogene elaparvovec were derived from extrapolated findings rather than observed benefits, the lack of comparative long-term data introduces considerable uncertainty to the analysis. Clinical expert feedback received by CADTH for this review indicated that, in the absence of long-term data, the duration of benefit that patients will receive from fidanacogene elaparvovec is unknown.
CADTH was unable to address this limitation. The BeneGene-2 trial is ongoing and expected to be complete in December 2029. Additional data from longer-duration follow-up will help reduce uncertainty in the duration of effect but will not reduce uncertainty in the magnitude of benefit relative to FIX prophylaxis due to the lack of a comparator group.
The impact of AEs was not considered. Costs and consequences associated with AEs were not included in the model. As noted in the CADTH clinical review, treatment-emergent AEs were reported in 84% of the safety population of the BeneGene-2 trial, with the most common being increased alanine transaminase (27%), nasopharyngitis (18%), and arthralgia (18%), and serious AEs were reported in 16% of patients who received fidanacogene elaparvovec in the BeneGene-2 trial. Due to the lack of a comparator group in the BeneGene-2 trial, the CADTH clinical review concluded that the relative safety of fidanacogene elaparvovec compared to FIX prophylaxis cannot be determined.
CADTH was unable to address this limitation. Although the sponsor’s model included the option to include treatment-related grade 3 and higher AEs, this was based on a naive comparison of AEs across trials, and it is not possible to determine if any observed differences between the therapies are solely due to the treatment or, rather, to bias or confounding factors.
The model structure does not appropriately capture the current treatment experience of patients with hemophilia B. The sponsor submitted a Markov model with health states based on an annual number of bleeds (0 bleeds, > 0 to < 3 bleeds, ≥ 3 to < 5 bleeds, ≥ 5 bleeds). Patients on FIX prophylaxis treatment were assumed to experience no change in the ABR over time and therefore remained in the same model health state for the entire analysis horizon (or until death). A similar assumption was made for patients on fidanacogene elaparvovec except for the initiation of effectiveness-waning starting at year 26. Feedback received by clinical experts consulted by CADTH indicated that FIX prophylaxis treatment is highly individualized and that ABRs do not remain static over time as they depend on factors such as adherence to treatment and physical activity level. Clinical expert feedback additionally noted that patients who experience 3 or more bleeds in a year would undergo clinical assessment and treatment changes to address the issue. The assumption that patients will remain in their initial health state while on FIX prophylaxis or fidanacogene elaparvovec for the entirety of the time horizon is therefore likely not reflective of the treatment experience of hemophilia B patients in Canada.
In addition, while the sponsor implicitly assumed utility and resource use inputs accounted for joint health and joint-related surgeries, this simplifying approach does not appropriately capture the true impact of prophylaxis treatment on patients’ experiences with hemophilia B. Patient input received by CADTH for this review indicated that joint damage to the knees, ankles, and elbows results in the greatest physical health and quality-of-life impact of hemophilia B. Although patients with pre-existing joint damage may benefit from fidanacogene treatment, clinical expert feedback received by CADTH suggests that the patients who would benefit most from fidanacogene treatment would be those without pre-existing joint damage (e.g., to preserve joint function). The clinical efficacy of fidanacogene elaparvovec among those with or without joint damage is unknown.
CADTH was unable to address these limitations due to a lack of model flexibility in incorporating transitions in health state over time or joint health outcomes.
The status of nAb testing coverage is uncertain. The BeneGene-2 trial enrolled patients without nAbs to the adeno-associated virus–5 vector used for gene transfusion. This aligns with clinical expert input received by CADTH that indicates that only patients with a negative anti–adeno-associated virus Rh74 variant serotype nAb test result would be considered eligible to receive fidanacogene elaparvovec. In its submission, the sponsor excluded costs related to nAb testing, indicating that the sponsor would cover the costs. However, if the cost of nAb testing is not covered by the sponsor, the costs incurred by the public health care payer will be higher than anticipated by the sponsor. Based on the sponsor’s submission, the cost is anticipated to be $3,000 per patient tested. In its budget impact analysis (BIA), the sponsor estimated that 48% of patients tested will have nAbs and therefore be ineligible for fidanacogene elaparvovec, based on a retrospective cross-sectional study assessing the prevalence and titre level of 9 nAb serotypes.16 No data concerning the sensitivity or specificity of this test were provided by the sponsor. If a false-negative result is received (i.e., the test suggests that a patient does not have nAbs when they actually do), the patient would incur the cost of fidanacogene elaparvovec but likely derive no benefit.
CADTH was unable to address this limitation.
One additional limitation was identified but not considered to be a key limitation.
The cost of FIX administration was overestimated. The sponsor assumed the cost per IV administration of FIX drugs was $769.90 when provided by a physician or $722.00 when provided by a nurse. CADTH notes that these costs were obtained from Prince Edward Island Outpatient Charges Hospital Fees for International Patients. Overestimation of administration costs would bias results in favour of fidanacogene elaparvovec due to increased total costs associated with FIX prophylaxis. Based on 1 hour of nursing or physician time, administration costs for FIX prophylaxis treatments are likely to be approximately $40 to $87.90 per infusion.11,14
The following key assumptions were made by the sponsor and were appraised by CADTH (Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CADTH comment |
|---|---|
Women were excluded from the modelled population, in line with the patient population of BeneGene-2. | Inappropriate, as the Health Canada indication for fidanacogene elaparvovec does not exclude women. However, clinical expert feedback received by CADTH for this review noted that treatment effects are not expected to differ between men and women due to the underlying mechanism of disease. The experts also noted that the number of women with moderately severe to severe hemophilia B in Canada is low. The impact of this on the cost-effectiveness of fidanacogene elaparvovec is therefore unknown but not expected to be significant. |
The sponsor’s pharmacoeconomic analysis compared fidanacogene elaparvovec to SHL FIX prophylaxis, EHL FIX prophylaxis, and to a weighted average of SHL and EHL FIX prophylaxis. | Inappropriate. The sponsor modelled SHL FIX prophylaxis and EHL FIX prophylaxis as separate comparators, with the assumption that the efficacy of both SHL and EHL would be equal. Data to support this assumption were not provided, and data from the BeneGene-1 trial were not stratified by type of FIX prophylaxis received. For the weighted average of SHL and EHL FIX prophylaxis, the sponsor assumed that 25% of patients would receive SHL and 75% would receive EHL and weighted the costs and QALYs accordingly. This is not aligned with the distribution of treatments received in the BeneGene-1 trial, in which the distribution was 64.4% EHL and 35.6% SHL. |
Patients were assumed to immediately benefit from fidanacogene elaparvovec infusion. | Uncertain. Clinical expert feedback received by CADTH noted that there will likely be a delay after fidanacogene elaparvovec infusion before treatment benefit occurs. |
The sponsor assumed that 90% of patients will self-administer FIX prophylaxis. | Reasonable, as confirmed by clinical experts consulted by CADTH. |
The frequency of health care resource use (i.e., hospital admissions, ICU stays, surgeries requiring hospitalization, hematologist visits, physiotherapy, tests, FIX inhibitor monitoring) was based on clinical expert opinion obtained by the sponsor. | Uncertain. Feedback received by clinical experts consulted by CADTH noted that patients with hemophilia B on prophylaxis and able to self-administer treatment at home rarely require clinical and hospital visits and that health care resource use is more dependent on bleed severity than frequency. |
Patients experiencing > 0 to < 3 bleeds annually and ≥ 3 to < 5 bleeds annually were assumed to have the same health-state utility value. | Uncertain. Clinical expert feedback received by CADTH noted this may be a reasonable assumption as the described health states likely overlap; however, the experts indicated that higher frequencies of bleeds tend to be correlated with worse health-related quality of life. |
Utility decrements associated with bleed type (i.e., target-joint bleeds, nontarget-joint bleeds, and nonjoint bleeds) were included in the analysis. | Uncertain. While bleed type may have an impact on a patient’s quality of life, the inclusion of utility decrements associated with bleed type potentially leads to double counting of utility decrements when applied on top of health-state utilities based on number of bleeds, likely biasing the results in favour of fidanacogene elaparvovec. |
EHL = extended half-life; FIX = coagulation factor IX; QALY = quality-adjusted life-year; SHL = standard half-life.
CADTH Reanalyses of the Economic Evaluation
Base-Case Results
CADTH was unable to address uncertainty related to comparative clinical data, including the magnitude and duration of benefit for fidanacogene elaparvovec compared to FIX prophylaxis treatments. CADTH was additionally unable to resolve uncertainty related to the impact of AEs, the model structure, and the price of FIX prophylaxis treatments. As such, CADTH was unable to provide a more reliable estimate of the cost-effectiveness of fidanacogene elaparvovec.
Results of the sponsor’s base case suggest that fidanacogene elaparvovec will be more effective and less costly (dominant) over FIX prophylaxis treatments (i.e., EHL, SHL, and a weighted basket of SHL and EHL), with a gain of approximately 1.08 QALYs relative to all comparators over the 77-year horizon but no expectation of improvement in survival, and an incremental cost savings of $2,871,630 to $5,576,438 resulting from a reduction in the cost of FIX, administration costs, and a reduction in the average number of annual bleeds and cumulative bleeds (Table 7).
Exploration of the sponsor’s model by CADTH shows that 93% of the predicted incremental gain in QALYs with fidanacogene elaparvovec compared to FIX prophylaxis treatments is expected to accrue in the extrapolation period (i.e., after 2 years; the BeneGene-2 observation period). Similarly, approximately 97% of the predicted cost savings (i.e., resulting from a reduction in bleeding events, need for FIX prophylaxis, and health care resource use) with fidanacogene elaparvovec is expected to be realized beyond the BeneGene-2 observation period. Based on the sponsor’s model, the acquisition cost of fidanacogene elaparvovec ($4,773,595 per administration) is predicted to be offset by such savings after approximately 12 years. CADTH notes that if the magnitude of benefit between fidanacogene elaparvovec and FIX prophylaxis is less than the sponsor’s estimate or if the actual cost of FIX prophylaxis treatments is lower than what is incorporated in the sponsor’s model, it will take longer for any potential savings to be realized.
Issues for Consideration
The sponsor has indicated that it will cover costs related to nAb testing. Should this not be the case, the costs associated with fidanacogene elaparvovec will be higher than what is estimated in the sponsor’s submitted analysis.
The price of FIX prophylaxis in the sponsor’s submitted model was based on a previous CADTH review for FIX recombinant nonacog alfa and FIX recombinant Fc fusion protein1,8 and does not reflect any confidential pricing that may have been negotiated by CBS. The true acquisition costs paid by CBS may be lower than those included in the sponsor’s cost-effectiveness and BIA.
Etranacogene dezaparvovec is currently undergoing review by CADTH for the treatment of hemophilia B in adults.17 The cost-effectiveness of fidanacogene elaparvovec compared to etranacogene dezaparvovec is unknown.
Overall Conclusions
The CADTH clinical review concluded that fidanacogene elaparvovec may reduce the number of bleeding events in adult patients with moderately severe to severe hemophilia B relative to treatment with FIX prophylaxis, based on observations from the single-arm BeneGene-2 trial for fidanacogene elaparvovec compared to observations from the BeneGene-1 lead-in study of FIX prophylaxis. CADTH judged the certainty of the evidence to be low for most outcomes and notes that there is uncertainty in the magnitude of differences in bleeding outcomes between fidanacogene elaparvovec and FIX prophylaxis due to the open-label, single-arm study design and the self-reporting of bleeding events. The long-term efficacy and safety of fidanacogene elaparvovec is highly uncertain due to limited long-term follow-up data beyond the BeneGene-2 trial period.
CADTH was unable to address the uncertainty related to comparative clinical data, including the magnitude and duration of benefit for fidanacogene elaparvovec compared to FIX prophylaxis. CADTH was also unable to resolve the uncertainty related to the impact of AEs, the model structure, the price of FIX prophylaxis, and the costs and consequences of nAb testing. As such, CADTH was unable to provide a more reliable estimate of the cost-effectiveness of fidanacogene elaparvovec.
Results of the sponsor’s base case suggest that fidanacogene elaparvovec will be more effective over a patient’s lifetime (i.e., 77 years) at reducing bleed events, which leads to a gain of approximately 1 QALY but no predicted change in life expectancy, when compared to FIX prophylaxis treatments (i.e., EHL, SHL, and a weighted basket of SHL and EHL FIX). The sponsor also predicts that fidanacogene elaparvovec will be cost-saving due to the 1-time infusion of fidanacogene elaparvovec and a reduced need for FIX prophylaxis and bleed-related health care resource use. Most (93%) of the benefits associated with fidanacogene elaparvovec accrued in the extrapolated period (i.e., after 2 years) and, in the absence of robust long-term comparative data, the incremental QALYs predicted by the sponsor may be overestimated. Similarly, the cost savings predicted by the sponsor’s model ($2,871,630 to $5,576,438) may be overestimated, as they rely on a sustained reduction in bleeding events for the first 25 years of the analysis. Based on the sponsor’s model, the acquisition cost of fidanacogene elaparvovec ($4,773,595 per administration) is predicted to be offset by the savings from reduced use of FIX prophylaxis and treatment of bleeding events after approximately 12 years. If the magnitude and duration of benefit between fidanacogene elaparvovec and FIX prophylaxis are less than the sponsor’s estimates or if the actual cost of FIX prophylaxis treatments is lower than what is specified in the sponsor’s model, it will take longer for any potential savings to be realized.
Given the uncertainty in the clinical evidence base, including the magnitude and duration of benefit with fidanacogene elaparvovec compared to currently available FIX prophylaxis treatments, it is highly uncertain whether and to what extent the gains in QALYs and cost savings will be realized in clinical practice.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Fidanacogene elaparvovec. Concentrate for solution for infusion, 1 × 1013 vector genomes/mL. Kirkland (QC): Pfizer Canada ULC; 2023 Jul 31.
2.Reimbursement Review sponsor submission: Fidanacogene elaparvovec. Concentrate for solution for infusion, 1 × 1013 vector genomes/mL. Kirkland (QC): Pfizer Canada ULC; 2023 Jul 31.
3.BEQVEZ (Fidanacogene elaparvovec): concentrate for solution for infusion, 1 × 1013 vector genomes/mL [product monograph]. Kirkland (QC): Pfizer Canada ULC; 2023.
4.Alam AU, Karkhaneh M, Attia T, Wu C, Sun HL. All-cause mortality and causes of death in persons with haemophilia: A systematic review and meta-analysis. Haemophilia. 2021;27(6):897-910. PubMed
5.Table: 13-10-0114-01. Life expectancy and other elements of the life table, Canada, all provinces except Prince Edward Island. Ottawa (ON): Statistics Canada; 2022: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310011401. Accessed 2023 Sep 5.
6.Neufeld EJ, Recht M, Sabio H, et al. Effect of acute bleeding on daily quality of life assessments in patients with congenital hemophilia with inhibitors and their families: observations from the dosing observational study in hemophilia. Value Health. 2012;15(6):916-925. PubMed
7.O'Hara J, Walsh S, Camp C, et al. The impact of severe haemophilia and the presence of target joints on health-related quality-of-life. Health Qual Life Outcomes. 2018;16(1):84. PubMed
8.Coabulation factor IX FC fusion protein (Alprolix): treatment cost comparison and budget impact analysis. Ottawa (ON): CADTH; 2015: https://www.cadth.ca/sites/default/files/pdf/OB0003_Alprolix_Economic_Report_REV.pdf. Accessed 2023 Sep 26.
9.Ontario Case Costing Initiative (OCCI). Toronto (ON): Ontario Health and Long-Term Care; 2017: https://data.ontario.ca/dataset/ontario-case-costing-initiative-occi. Accessed 2023 Sep 5.
10.Schedule of benefits for laboratory services: effective July 1, 2020. Toronto (ON): Ontario Ministry of Health; 2020: http://www.health.gov.on.ca/en/pro/programs/ohip/sob/lab/lab_mn2020.pdf. Accessed 2023 Sep 5.
11.Schedule of benefits for physician services under the Health Insurance Act: (January 25, 2022 (effective July 1, 2022)). Toronto (ON): Ontario Ministry of Health; 2022: https://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master.pdf. Accessed 2023 Sep 5.
12.Ontario Ministry of H, Ontario Ministry of Long-Term C. Ontario drug benefit formulary/comparative drug index. 2022; https://www.formulary.health.gov.on.ca/formulary/. Accessed 2023 Sep 5.
13.Patient cost estimator. 2022; https://www.cihi.ca/en/patient-cost-estimator. Accessed 2023 Sep 5.
14.Job Bank. Registered Nurse (R.N.) hourly wages. 2023; https://www.jobbank.gc.ca/marketreport/wages-occupation/993/ON. Accessed 2023 Jul 5.
15.Job Bank. Median Physiotherapy Wage in Canada. 2023; https://www.jobbank.gc.ca/marketreport/wages-occupation/18214/ca. Accessed 2023 Jul 5.
16.Rasko PJ, Bashirians G, Petropoulos C, et al. Global Seroprevalence of Neutralizing Antibodies Against Adeno-Associated Virus (AAV) Serotypes of Relevance to Gene Therapy. Blood. 2022;140(Supplement 1):10668-10670.
17.etranacogene dezaparvovec. Ottawa (ON): CADTH; 2023: https://www.cadth.ca/etranacogene-dezaparvovec. Accessed 2023 Oct 3.
18.Clinical Study Report: C0371002 Phase 3, Open Label, Single Arm Study to Evaluate Efficacy and Safety of FIX Gene Transfer With PF06838435 (rAAV-Spark100-hFIX-Padua) in Adult Male Participants With Moderately Severe to Severe Hemophilia B (FIX:C≤2%) (BeneGene-2). New York (NY): Pfizer Inc.; 2023.
19.IDELVION (Coagulation Factor IX (Recombinant), Albumin Fusion Protein (rIX-FP)): 250, 500, 1000, 2000 and 3500 IU/vial For Intravenous Administration [product monograph]. Ottawa (ON): CSL Behring Canada, Inc.; 2020: https://labeling.cslbehring.ca/PM/CA/IDELVION/EN/IDELVION-Product-Monograph.pdf. Accessed 2023 Sep 26.
20.ALPROLIX (Coagulation Factor IX (Recombinant), Fc Fusion Protein): 250, 500, 1000, 2000, 3000 and 4000 IU/vial For Intravenous Administration [product monograph]. Laval (QC): Sanofi-Aventis Canada Inc.; 2020: https://pdf.hres.ca/dpd_pm/00058155.PDF. Accessed 2023 Sep 26.
21.BeneFIX (Coagulation Factor IX (Recombinant), Nonacog alfa): 250, 500, 1000, 1500, 2000 and 3000 IU per viall For Intravenous Administration [product monograph]. Kirkland (QC): Pfizer Canada Inc.,; 2020: https://www.pfizer.ca/files/BeneFIX_PM.pdf. Accessed 2023 Sep 26.
22.REBINYN (Coagulation Factor IX (Recombinant), pegylated nonacog beta pegol): 2500, 1000, 2000 and 3000 IU/vial, intravenous [product monograph]. Mississauga (ON): Novo Nordisk Canada, Inc.; 2020: https://www.novonordisk.ca/content/dam/nncorp/ca/en/products/rebinyn-product-monograph.pdf. Accessed 2023 Sep 26.
23.RIXUBIS (Recombinant Coagulation Factor IX (rFIX), Nonacog gamma): 250, 500, 1000, 2000 and 3000 International Units (IU) per vial for injection [product monograph]. Toronto (ON): Takeda Canada Inc.; 2020: https://assets-dam.takeda.com/raw/upload/v1662721804/legacy-dotcom/siteassets/en-ca/home/what-we-do/our-medicines/product-monographs/rixubis/rixubis-pm-en.pdf. Accessed 2023 Sep 26.
24.Association of Hemophilia Clinic Directors of Canada. Canadian Bleeding Disorders Registry (CBDR). 2023; https://www.ahcdc.ca/cbdr. Accessed 2023 Jun 2.
25.Factor FIX Deficiency - 2021. Ottawa (ON): Association of Hemophilia Clinic Directors of Canada; 2021: https://www.ahcdc.ca/storage/pdf/21/FIX-2021.pdf. Accessed by sponsor, no date provided.
26.Krumb E, Lambert C, Hermans C. Patient selection for hemophilia gene therapy: Real-life data from a single center. Res Pract Thromb Haemost. 2021;5(3):390-394. PubMed
27.Maor Y, Bashari D, Kenet G, et al. Non-invasive biomarkers of liver fibrosis in haemophilia patients with hepatitis C: can you avoid liver biopsy? Haemophilia. 2006;12(4):372-379. PubMed
28.Maor Y, Halfon P, Bashari D, et al. Fibrotest or Fibroscan for evaluation of liver fibrosis in haemophilia patients infected with hepatitis C. Haemophilia. 2010;16(1):148-154. PubMed
Appendix 1: Cost Comparison
Note this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical expert(s) and drug plan. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 5: CADTH Cost Comparison Table for Adult Patients with Moderately Severe and Severe Hemophilia B
Treatment | Strength / concentration | Form | Price ($)a | Recommended dosageb | Daily cost ($) | Average annual cost ($) |
|---|---|---|---|---|---|---|
Fidanacogene elaparvovec | 1 × 1013 vector genomes/mL | Solution for infusion | 4,773,595.2000 per patient | One-time infusion | NA | NA |
Factor IX treatments (extended half-life) | ||||||
Coagulation factor IX (recombinant), albumin fusion protein (Idelvion) | 250 IU 500 IU 1,000 IU 2,000 IU 3,500 IU | Powder for IV induction | 2.3124 per IU | 25 to 40 IU/kg every 7 days or 50 to 75 IU/kg every 14 days | 718 to 1,150 | 262,431 to 419,889 |
Coagulation factor IX (recombinant), Fc fusion protein (Alprolix) | 250 IU 500 IU 1,000 IU 2,000 IU 3,000 IU | Powder for IV injection | 2.0464 per IU | 50 IU/kg once weekly or 100 IU/kg once every 10 to 14 days | 1,272 to 1,780 | 464,485 to 650,279 |
Coagulation factor IX (recombinant), pegylated nonacog beta pegol (Rebinyn) | 500 IU 1,000 IU 2,000 IU | Powder for IV injection | 2.3124 per IU | 40 IU/kg once weekly | 1,150 | 419,889 |
Factor IX treatments (standard half-life) | ||||||
Coagulation factor IX (recombinant), nonacog alfa (BeneFIX) | 250 IU 500 IU 1,000 IU 1,500 IU 2,000 IU 3,000 IU | Powder for IV injection | 0.8834 per IU | 40 IU/kg every 3 to 4 days | 769 to 1,025 | 280,716 to 374,288 |
Recombinant coagulation factor IX, nonacog gamma for Injection (Rixubis) | 250 IU 500 IU 1,000 IU 2,000 IU 3,000 IU | Powder for IV injection | 0.7207 per IU | 40 to 60 IU/kg twice weekly | 717 to 1,075 | 261,731 to 392,597 |
Note: All prices do not include dispensing fees. Annual costs were calculated assuming patient weight of 87 kg and 365.25 days per year.18
aSponsor-submitted price for fidanacogene elaparvovec.1 Prices of FIX treatments are not available in CADTH participating drug formularies for factor IX treatments; as such, the price for FIX comparators was adopted from the sponsor’s submission (in which the price per IU for each FIX prophylaxis treatment was back calculated by the sponsor based on the weekly costs reported for coagulation factor IX (recombinant) nonacog alfa and coagulation factor IX (recombinant) Fc fusion protein.8 The sponsor assumed that the cost of coagulation factor IX (recombinant) Fc fusion protein is representative of all available EHLs and that the cost of coagulation factor IX (recombinant) nonacog alfa is representative of all SHL products).1
bRecommended dosages are informed by corresponding product monographs unless otherwise stated.19-23
Appendix 2: Submission Quality
Note this table has not been copy-edited.
Description | Yes/No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | Yes | While female patients with moderately severe to severe hemophilia B are captured in the reimbursement request, they were excluded from BeneGene-2. However, clinical expert feedback noted that treatment effects are not expected to differ between males and females due to the underlying mechanism of disease and the number of moderately severe to severe hemophilia B female patients in Canada is low. The impact of this on the cost-effectiveness of fidanacogene elaparvovec is not expected to be significant but is unknown. |
Model has been adequately programmed and has sufficient face validity | Yes | No comment. |
Model structure is adequate for decision problem | No | Refer to CADTH appraisal. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | Yes | No comment. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | Yes | No comment. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | No | CADTH identified discrepancies in the inputs used in the sponsor’s economic report and their submitted model. For example, although the submitted report stated that “the model assumes that the cost for coagulation factor IX (recombinant) Fc fusion protein is representative for all of the available EHLs,” the cost of EHL is the model was informed by a weighted average between coagulation factor IX (recombinant) Fc fusion protein and coagulation factor IX (recombinant) pegylated nonacog beta pegol. Furthermore, certain parameter inputs of the submitted model were not user friendly. For example, cells < N15:015 > on the < Results > sheet suggest user inputs for alternative distribution to inform the FIX prophylaxis SHL/EHL weighted average, however usage of these cells do not properly update the results. |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note this appendix has not been copy-edited.
Table 7: Number of Bleeds Predicted in the Sponsor’s Base Case
Component | FidaVec | FIX prophylaxis (SHL) | FIX prophylaxis (EHL) | FIX prophylaxis SHL/EHL weighted average |
|---|---|---|---|---|
Average annual number of bleeds | ||||
Target joint bleeds | 0.98 | 1.73 | 1.73 | 1.73 |
Non–target joint bleeds | 0.41 | 0.73 | 0.73 | 0.73 |
Nonjoint bleeds | 0.28 | 0.51 | 0.51 | 0.51 |
Untreated bleeds | 0.58 | 1.03 | 1.03 | 1.03 |
Total | 2.25 | 4.00 | 4.00 | 4.00 |
Cumulative number of bleeds | ||||
Target joint bleeds | 38.05 | 67.33 | 67.33 | 67.33 |
Non–target joint bleeds | 15.99 | 28.46 | 28.46 | 28.46 |
Nonjoint bleeds | 11.06 | 19.73 | 19.73 | 19.73 |
Untreated bleeds | 22.41 | 40.27 | 40.27 | 40.27 |
Total | 87.51 | 155.79 | 155.79 | 155.79 |
Note: Deterministic results; number of bleeds not available from the sponsor’s probabilistic analysis.
Source: Sponsor’s pharmacoeconomic submission.1
Table 8: Disaggregated Summary of the Sponsor’s Economic Evaluation Results
Component | Fidanacogene elaparvovec | FIX prophylaxis (SHL) | FIX prophylaxis (EHL) | FIX prophylaxis SHL/EHL weighted average |
|---|---|---|---|---|
Discounted LYs | ||||
No Bleeds | 17.40 | 8.27 | 8.27 | 8.27 |
0 < Bleeds < 3 | 6.60 | 12.92 | 12.90 | 12.90 |
3 ≤ Bleeds < 5 | 1.76 | 1.76 | 1.77 | 1.77 |
5 ≥ Bleeds | 2.82 | 5.63 | 5.64 | 5.64 |
Total | 28.58 | 28.58 | 28.58 | 28.58 |
Discounted QALYs | ||||
No Bleeds | 12.98 | 5.99 | 5.99 | 5.99 |
0 < Bleeds < 3 | 3.99 | 8.24 | 8.23 | 8.23 |
3 ≤ Bleeds < 5 | 1.12 | 1.12 | 1.13 | 1.13 |
5 ≥ Bleeds | 1.57 | 3.31 | 3.31 | 3.31 |
Target-Joint Bleeds Disutility | –0.09 | –0.17 | –0.17 | –0.17 |
Nontarget-Joint Bleeds Disutility | –0.02 | –0.04 | –0.04 | –0.04 |
Nonjoint Bleeds Disutility | –0.02 | –0.03 | –0.03 | –0.03 |
Disutility Associated with Treatment Initiation | –0.02 | 0.00 | 0.00 | 0.00 |
Total | 19.50 | 18.42 | 18.42 | 18.42 |
Discounted costs ($) | ||||
Treatment acquisition – Fidanacogene elaparvovec | 4,773,595 | 0.00 | 0.00 | 0.00 |
Treatment Acquisition – Other Therapies | 2,283,195 | 9,369,871 | 12,172,394 | 11,471,764 |
Treatment-Related Costs and Administration | 24,679 | 198,038 | 99,019 | 123,774 |
Acute Bleed Management | 465,264 | 869,258 | 870,133 | 869,915 |
Situational Prophylaxis Infusionsa | 101,426 | 0.00 | 0.00 | 0.00 |
Other Disease Medical Resource Utilizationb | 95,939 | 178,559 | 178,988 | 178,881 |
Total | 7,744,097 | 10,615,727 | 13,320,535 | 12,644,333 |
EHL = extended half-life, FIX = factor IX; QALY = quality-adjusted life-year; SHL = standard half-life.
aIncludes use of FIX infusion for patients receiving gene therapy under specific situations such as before high-risk physical activity or surgical procedures. Assumptions of use were informed by clinical expert feedback received by the sponsor.
bIncludes bleed-related hospital ward and ICU stays, surgery-related hospitalizations, visit to hematologists and other specialists, outpatient consultations (nurse and physiotherapy), test and procedures, and FIX inhibitor monitoring.
Appendix 4: Submitted BIA and CADTH Appraisal
Note this appendix has not been copy-edited.
Table 9: Summary of Key Take-Aways
Key Take-aways of the BIA |
|---|
|
Summary of Sponsor’s BIA
The sponsor submitted a BIA to estimate the three-year budget impact of reimbursing fidanacogene elaparvovec for the treatment of moderately severe to severe hemophilia B in patients 18 years and older. The BIA was undertaken over a three-year time horizon (2024 to 2026) using an epidemiological approach and included acquisition costs related to fidanacogene elaparvovec and FIX treatments (taken as prophylaxis, bleed episode management, and perioperative bleed management). Province-specific prevalence rates from the Canadian Bleeding Disorders registry (CBDR) were adopted for Alberta and Ontario, while regional prevalence rates were utilized for other jurisdictions.24,25 The sponsor excluded patients who had prior use of inhibitors (assumed to be 1% of eligible hemophilia B patients), those with liver disease, liver fibrosis/impaired liver function or status, and/or active HBV+HCV (20%), those with neutralizing antibodies (48%), and prior receipt of fidanacogene elaparvovec in a clinical trial program (9 patients).16,26-28 All patients were assumed to have public coverage. Data to inform the model were obtained from various sources, including the published literature, the sponsor’s internal data, and input from clinical experts consulted by the sponsor. Key inputs to the BIA are documented in Table 10.
The sponsor compared a reference scenario in which patients received EHL FIX or SHL FIX prophylaxis to a new drug scenario in which patients received fidanacogene elaparvovec. Market share of EHL and SHL FIX prophylaxis were estimated based on CBDR data. EHL was assumed to be comprised of 77% coagulation factor IX (recombinant) pegylated nonacog beta pegol and 23% coagulation factor IX (recombinant) Fc fusion protein, while SHL was assumed to be comprised solely of coagulation factor IX (recombinant), nonacog alfa. In the new drug scenario, uptake of fidanacogene elaparvovec was assumed to be with 12%, 28%, and 34% in year 1, year 2, and year 3, respectively, based on market share forecasting and expert opinion obtained by the sponsor. Wastage and administration costs were not included.
Table 10: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (Year 1 / Year 2 / Year 3) |
|---|---|
Target population | |
Number of patients with moderately severe to severe hemophilia B | 188 (baseline year) |
Number of patients eligible for drug under reviewa | 71 / 73 / 76 |
Market Uptake (3 years) | |
Uptake (reference scenario) | |
Fidanacogene elaparvovec | 0% / 0% / 0% |
Coagulation factor IX (recombinant) Fc fusion protein | 17.5% / 18.3% / 19.0% |
Coagulation factor IX (recombinant) pegylated nonacog beta pegol | 58.5% / 61.2% / 63.5% |
Coagulation factor IX (recombinant) nonacog alfa | 24.0% / 20.5% / 17.6% |
Uptake (new drug scenario) | |
Fidanacogene elaparvovec (cumulative intake) | 12.0% / 28.0% / 34.0% |
Coagulation factor IX (recombinant) Fc fusion protein | 15.4% / 13.5% / 13.1% |
Coagulation factor IX (recombinant) pegylated nonacog beta pegol | 51.6% / 45.0% / 43.8% |
Coagulation factor IX (recombinant) nonacog alfa | 21.0% / 13.5% / 9.15% |
Cost of treatment (per patient) | |
Fidanacogene elaparvovec (one-time costb) | $4,773,595 |
Coagulation factor IX (recombinant) Fc fusion protein (annual cost) | $417,964 |
Coagulation factor IX (recombinant) pegylated nonacog beta pegol (annual cost) | $381,946 |
Coagulation factor IX (recombinant) nonacog alfa (annual cost) | $321,734 |
HBV = hepatitis B virus; HCV = hepatitis C virus.
aAfter exclusion of patients with prior use of inhibitors; liver disease, liver fibrosis/impaired liver function or status, and/or active HBV+HCV; neutralizing antibodies; and prior receipt of fidanacogene elaparvovec.
bCost is assumed to be per infusion, regardless of the number of vials needed (based on patient weight).
Summary of the Sponsor’s BIA Results
The sponsor estimated the 3-year budget impact of reimbursing fidanacogene elaparvovec for the treatment of moderately severe to severe hemophilia B in patients 18 years and older to be $101,613,426 (year 1: $37,044,064; year 2: $50,213,749; year 3: $14,355,613).
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
The number of patients with moderately severe to severe hemophilia B is uncertain. The indication for fidanacogene elaparvovec is for the treatment of adults (aged 18 years or older) with moderately severe to severe hemophilia B (congenital factor IX deficiency) who are negative for neutralizing antibodies to variant adeno-associated virus serotype Rh74, without definition of what constitutes “moderately severe” or “severe” hemophilia B. Clinical expert feedback received by CADTH for this review indicated that clinical phenotype is the most important basis for clinicians to determine a patient’s disease severity and treatment, not a cut point of FIX:C of 2% which was used as an enrolment criteria in the BeneGene-2 trial. The clinical expert input noted that some patients will require FIX prophylaxis based on clinical symptoms despite a FIX level greater than 2%. In deriving the eligible population, the sponsor used prevalence estimates stratified by severity from CBDR, with the assumption that 40% of patients with moderate patients have moderately severe disease; no definition was provided for this distinction. Should the total number of patients with moderately severe to severe hemophilia B be larger than the 188 patients estimated by the sponsor, the budget impact of reimbursing fidanacogene elaparvovec will be greater than estimated by the sponsor.
CADTH explored uncertainty in the number of patients with moderately severe to severe hemophilia B in a scenario analysis.
The number of patients expected to receive fidanacogene elaparvovec in the next 3 years is uncertain. The sponsor assumed that the cumulative uptake of fidanacogene elaparvovec will be 34% over the first 3 years of reimbursement (2024 to 2026), based on clinical expert feedback received by the sponsor and internal market research. This uptake results in an estimated 26 patients receiving fidanacogene elaparvovec by the end of 2026. Clinical expert feedback received by CADTH indicated that uptake may be higher than estimated by the sponsor, with up to 50 patients anticipated to receive fidanacogene elaparvovec in the next 3 years.
CADTH explored uncertainty in the number of patients expected to receive fidanacogene elaparvovec in a scenario analysis.
Market share estimates of currently available FIX prophylaxis are not reflective of Canadian clinical practice. In the sponsor’s submitted BIA, market share estimates for FIX prophylaxis drugs in the reference scenario were informed by data from the Canadian Bleeding Disorder Registry, with market share over the analysis horizon based on the change rate between years 2020 and 2022. That is, the market share of coagulation factor IX (recombinant) Fc fusion protein and coagulation factor IX (recombinant) pegylated nonacog beta pegol were assumed to increase while the market share of coagulation factor IX (recombinant) nonacog alfa was assumed to decrease. Clinical expert feedback received by CADTH indicated that changes in the market share estimates for FIX prophylaxis treatments available for hemophilia B are expected to be minimal for the foreseeable future, given that these treatments have been available for many years.
In the CADTH scenario analysis adopting a health care system perspective, the market share distribution for FIX prophylaxis treatments in the reference scenario was assumed to be remain static in each analysis year.
Costing in the model is highly uncertain. The cost of FIX prophylaxis in the sponsor’s submitted model was based on a previous CADTH review for coagulation FIX recombinant nonacog alfa and coagulation FIX recombinant Fc fusion protein.1,8 These costs do not reflect any confidential pricing that may have been negotiated by CBS. As such, the estimated drug acquisition costs for FIX prophylaxis treatments are uncertain.
CADTH was unable to incorporate confidential pricing.
Neutralizing antibody testing coverage status is uncertain. The sponsor excluded costs related to nAb testing from the BIA, indicating that the sponsor would cover the costs. However, if the cost of nAb testing is not covered by the sponsor, the costs incurred by the payer will be higher than estimated by the sponsor.
CADTH conducted a scenario analysis in which the cost of nAb testing was included.
CADTH Reanalyses of the BIA
CADTH revised the sponsor’s base case by adopting a public drug plan payer perspective given feedback from the public payers (Table 11). That is, costs associated with FIX were excluded from the CADTH base case, as these costs are borne by CBS. The impact of reimbursing fidanacogene elaparvovec from the broader perspective of the Canadian health care system was explored in scenario analysis (Table 13).
Table 11: CADTH Revisions to the Submitted BIA
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
1. Fidanacogene elaparvovec cost | 4,773,595.1991 | 4,773,595.2000 |
2. Calculation error in year 3 for fidanacogene elaparvovec | Multiplied the cost of fidanacogene elaparvovec to a portion of the patients and subtracted this from the second prevalence population | Corrected bracket placement in the equation to calculate number of patients before multiplying by cost of fidanacogene elaparvovec |
Changes to derive the CADTH base case | ||
1. Perspective | CBS | Drug plan |
CADTH base case | Reanalysis 1 | |
CBS = Canadian Blood Services.
The results of the CADTH stepwise reanalysis are presented in summary format in Table 12 and a more detailed breakdown is presented in Table 13. All CADTH reanalysis were based on publicly available prices of the comparator treatments.
In the CADTH base case, the estimated incremental budget impact of reimbursing fidanacogene elaparvovec is expected to be $127,503,945 (year 1: $40,579,580; year 2: $58,746,280; year 3: $28,178,085).
Table 12: Summary of the CADTH Reanalyses of the BIA
Stepped analysis | 3-year total |
|---|---|
Submitted base casea | $101,613,426 |
Submitted base case (corrected)a | $104,731,331 |
CADTH base caseb | $127,503,945 |
aSponsor's submitted base case and the sponsor’s corrected base case adopt the perspective of the CBS. This analysis includes acquisition costs for fidanacogene elaparvovec and FIX prophylaxis treatment.
bCADTH’s base case adopts the perspective of CADTH participating drug plans. This analysis includes acquisition costs for fidanacogene elaparvovec. Acquisition costs for FIX are borne by the CBS and were excluded from this analysis.
CADTH conducted the following scenario analyses. Results are provided in Table 13.
Adopting the perspective of the public health care payer, in which costs related to treatment administration, health care resource use (i.e., bleed-related hospital visits, surgery-related hospitalizations, visits to hematologists and other specialists, outpatient consultations, tests and procedures, and FIX inhibitor monitoring, and AEs) were included.
Increasing the number of patients with moderately severe to severe hemophilia B by 10%.
Adopting higher uptake of fidanacogene elaparvovec, based on clinical expert feedback indicating that up to 50 patients could receive fidanacogene elaparvovec by the end of year 3.
Including costs related to nAb testing.
Table 13: Detailed Breakdown of the CADTH Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
Submitted base casea | Reference | $28,777,439 | $30,129,988 | $31,456,089 | $32,758,149 | $123,121,665 |
New drug | $28,777,439 | $67,174,053 | $81,669,838 | $47,113,762 | $224,735,091 | |
Budget impact | $0 | $37,044,064 | $50,213,749 | $14,355,613 | $101,613,426 | |
Submitted base case (corrected)a | Reference | $28,777,439 | $30,129,988 | $31,456,089 | $32,758,149 | $123,121,665 |
New drug | $28,777,439 | $67,174,053 | $81,669,838 | $50,231,666 | $227,852,996 | |
Budget impact | $0 | $37,044,064 | $50,213,749 | $17,473,518 | $104,731,331 | |
CADTH base caseb | Reference | $0 | $0 | $0 | $0 | $0 |
New drug | $0 | $40,579,580 | $58,746,280 | $28,178,085 | $127,503,945 | |
Budget impact | $0 | $40,579,580 | $58,746,280 | $28,178,085 | $127,503,945 | |
CADTH scenario 1: health care system perspectivec | Reference | $29,200,760 | $30,238,053 | $31,275,347 | $32,312,641 | $123,026,801 |
New drug | $29,200,760 | $67,270,789 | $81,447,608 | $49,717,716 | $227,636,873 | |
Budget impact | $0 | $37,044,064 | $50,213,749 | $17,473,518 | $104,731,331 | |
CADTH scenario 2: moderately severe to severe hemophilia B populationd,e | Reference | $0 | $0 | $0 | $0 | $0 |
New drug | $0 | $47,833,807 | $68,418,582 | $31,805,198 | $148,057,587 | |
Budget impact | $0 | $47,833,807 | $68,418,582 | $31,805,198 | $148,057,587 | |
CADTH scenario 3: fidanacogene elaparvovec uptaked,f | Reference | $0 | $0 | $0 | $0 | $0 |
New drug | $0 | $40,579,580 | $58,746,280 | $143,814,576 | $243,140,436 | |
Budget impact | $0 | $40,579,580 | $58,746,280 | $143,814,576 | $243,140,436 | |
CADTH scenario 4: nAb testing costsd,g | Reference | $0 | $0 | $0 | $0 | $0 |
New drug | $0 | $40,632,710 | $58,823,196 | $28,214,978 | $127,670,884 | |
Budget impact | $0 | $40,632,710 | $58,823,196 | $28,214,978 | $127,670,884 |
nAb = neutralizing antibody.
aSponsor's submitted base case and the sponsor’s corrected base case adopt the perspective of the CBS. This analysis includes acquisition costs for fidanacogene elaparvovec and FIX prophylaxis treatments.
bCADTH’s base case adopts the perspective of CADTH participating drug plans. This analysis includes acquisition costs for fidanacogene elaparvovec. Acquisition costs for FIX prophylaxis were excluded from this analysis.
cConducted from the perspective of the Canadian health care system. Costs related to treatment administration, health care resource use (i.e., bleed-related hospital visits, surgery-related hospitalizations, visits to hematologists and other specialists, outpatient consultations, tests and procedures, and FIX inhibitor monitoring, and AEs) were included. Administration costs for fidanacogene elaparvovec and FIX infusions conducted by physicians and nurses were assumed to be $87.90 and $40, respectively.11,14 Administration and health care resource use frequencies were informed by clinical expert feedback obtained by the sponsor. AE frequencies were informed by the US FDA prescribing information for serious treatment-related AEs experienced by at least 5% of patients.
dScenario based on the CADTH base case (drug plan perspective).
eAssumed 10% more patients with moderately severe to severe hemophilia B (i.e., 207 patients).
fA total of 50 patients were assumed to receive fidanacogene elaparvovec over the first 3 years of reimbursement, based on clinical expert opinion.
gCost of nAb testing provided by the sponsor: $3,000 per patient. Cost of testing was included for all patients screened, using the sponsor’s assumption that 48% of patients screened would have nAbs and thus be ineligible for fidanacogene elaparvovec.
Ethics Review
Abbreviations
AAV
adeno-associated virus
AAVrh74
adeno-associated virus rh74 serotype
ABRtotal
annualized bleeding rate for treated and untreated bleeds
APM
alternative payment model
FIX
coagulation factor IX
nAb
neutralizing antibody
Summary
Hemophilia B is a congenital, recessive bleeding disorder that is characterized by a deficiency of coagulation factor IX (FIX) and results in susceptibility to prolonged bleeding and subsequent organ and/or joint damage.
Patient group, clinician group, clinical expert, and drug program input gathered, as well as relevant literature, were reviewed to identify ethical considerations relevant to the use of fidanacogene elaparvovec for the treatment of moderately severe to severe hemophilia B in patients aged 18 years or older.
Ethical considerations identified in this review relate to:
Treatment, and experiences of hemophilia B: A significant burden is associated with the existing standard-of-care treatment, prophylactic FIX replacement therapy, for people with moderate to severe hemophilia B. Successful treatment with prophylactic FIX replacement therapy requires frequent IV infusions. People experience variable FIX activity levels due to waning of treatment effect despite high adherence. As a result, they remain susceptible to bleeds and, even when well-treated, people with hemophilia B may find it challenging to fully participate in some household, workplace, athletic, or other activities due to the elevated risk of bleeding. As an X chromosome–linked condition with infrequent occurrence in females, females with moderate to severe hemophilia B may experience inequitable access to existing care due to misdiagnosis or underdiagnosis.
Clinical and economic evidence use in the evaluation of fidanacogene elaparvovec: Clinical trial evidence indicated treatment with fidanacogene elaparvovec may result in a clinically relevant reduction in the primary end point of the annualized bleeding rate of treated and untreated bleeds during the median follow-up period of |||| |||||. However, interpretations of the magnitude of treatment effect are deemed uncertain due to the risk of bias in the statistical models used to inform the comparative efficacy of fidanacogene elaparvovec relative to FIX prophylaxis. Similarly, there is also uncertainty regarding the durability of effect and long-term safety. This uncertainty presents challenges for clinical and shared decision-making about the benefits and harms of fidanacogene elaparvovec, particularly as it is proposed as a 1-time therapy that is meant to remain effective over the duration of the patient’s life. This uncertainty may be further exacerbated for females, who were absent from the trial population, and nonwhite people, who were underrepresented in the trial. Limited long-term safety and efficacy data also limits the assessment of cost-effectiveness.
Clinical use and implementation of fidanacogene elaparvovec as a gene therapy: The use of fidanacogene elaparvovec as a gene therapy presents some known risks for patients, such as the development of transaminitis, and presently theoretical risks, such as the long-term possibility of genotoxicity leading to the development of cancer. As a result, it is important for clinicians to facilitate robust informed consent and shared decision-making processes with patients, particularly as there is no opportunity to discontinue this 1-time treatment. Further, due to the production of cross-reactive anti–adeno-associated virus (AAV) neutralizing antibodies (nAbs), some people may be rendered ineligible for additional gene therapies even if they experience limited to no clinical benefit after receiving fidanacogene elaparvovec. Even for those who experience benefits, transgene expression of the AAV vectors used in gene therapies is expected to diminish over time, leading to decreased efficacy and the need to return to FIX prophylaxis. Determining eligibility for fidanacogene elaparvovec may also present ethical challenges as it is presently unclear who is most likely to benefit from treatment. In addition, the absence or underrepresentation of some populations in trials (e.g., females and nonwhite people) may incidentally lead to inequitable access to treatment if access is prioritized for populations for whom some safety and efficacy data are available. As diagnosis and treatment with fidanacogene elaparvovec necessitates multidisciplinary care in specialized treatment centres, ensuring equitable access to this therapy requires addressing common geographic barriers to specialist care and monitoring.
Health systems: Implementation of fidanacogene elaparvovec presents ethical challenges associated with assessing opportunity costs and making funding and resource-allocation decisions for expensive drugs for rare diseases. Given the uncertainty around the durability of effect and safety of fidanacogene elaparvovec, alternative payment models (APMs) have been proposed to help mitigate the risks of paying for a highly expensive gene therapy (with a proposed lifelong efficacy) in the absence of long-term data. However, it is important to consider the concomitant challenges of building the data and clinical infrastructure needed to effectively execute the chosen APM. Similarly, it is important to consider that the design of an APM (e.g., the parameters of treatment success) may also affect how the benefits and burdens of risk-sharing are distributed among manufacturers, payers, patients, and the public. Clinical experts also noted the potential need to develop clear prioritization criteria should production shortages of the AAV vector used in fidanacogene elaparvovec (rh74 serotype [AAVrh74]) arise. Clinical experts also indicated there may be some geographic challenges to accessing treatment as not all treatment centre pharmacies may be able, or willing, to offer fidanacogene elaparvovec. As a result, some patients may need to travel out of the province to access fidanacogene elaparvovec, which can present challenges in determining which jurisdictions are responsible for reimbursing the therapy and other treatment-related costs.
Objective(s)
To identify and describe ethical considerations associated with the use of fidanacogene elaparvovec for the treatment of moderately severe to severe hemophilia B in patients ages 18 years or older, including considerations related to the context of hemophilia B, evidentiary basis, use of fidanacogene elaparvovec (as a gene therapy), and health systems.
Research Questions
This report addresses the following research questions:
What ethical considerations arise in the context of hemophilia B in adults?
What ethical considerations arise related to the evidence (e.g., clinical and economic data) used to evaluate fidanacogene elaparvovec?
What ethical considerations arise in the use of fidanacogene elaparvovec (as a gene therapy) for patients, their caregivers, and clinicians? What ethical considerations for health systems are involved in the context of fidanacogene elaparvovec as a gene therapy?
Methods
To identify ethical considerations relevant to the use of fidanacogene elaparvovec in the treatment of hemophilia B, this ethics report was driven by relevant questions identified in the EUnetHTA Core Model 3.0, Ethics Analysis Domain,1 supplemented by relevant questions from the Equity Checklist for Health Technology Assessment.2 These guiding questions were organized to respond to the research questions posed, and investigated ethical considerations related to:
adult patients living with hemophilia B and their caregivers (i.e., disparities in incidence, treatment, or outcomes; challenges related to diagnosis or clinical care; factors that might prevent patients from gaining access to therapies)
the evidence used to demonstrate the benefits, harms and value of fidanacogene elaparvovec (i.e., ethical considerations in relevant clinical trials, including their representativeness, choice of outcome measures, appropriateness of analytical methods and models to all population groups; ethical considerations related to the data or assumptions in the economic evaluation)
the use of fidanacogene elaparvovec (as a gene therapy), including considerations related to benefits and harms to patients, relatives, caregivers, clinicians or society, and considerations related to access to these therapies
the uptake of fidanacogene elaparvovec (as a gene therapy) in health systems, including considerations related to the distribution of health care resources.
Data Collection: Review of Project Inputs and Literature
Data to inform this ethics report drew from an identification of ethical considerations (e.g., values, norms, or implications related to the harms, benefits, and implications for equity, justice, resource allocation, and ethical considerations in the evidentiary basis) in the patient and clinician group, clinical expert, and drug program input collected by CADTH to inform this review, as well as a complementary search of the published literature. Ongoing collaboration and communication with CADTH reviewers working on the clinical and economic reviews for this submission also assisted in the clarification and identification of ethical considerations raised.
Review of Project Inputs
During this CADTH review, a single reviewer collected and considered input from 6 main sources for content related to ethical considerations relevant to addressing the research questions guiding this ethics report. In addition to published literature, this report considered several sources:
the sponsor submission, noting relevant information and external references or sources relevant to each of the research questions driving this report
clinician group input received by CADTH from the Association of Hemophilia Clinic Directors of Canada and the Canadian Association of Nurses in Hemophilia Care
patient input received by CADTH from the Canadian Hemophilia Society
drug program input received by CADTH from drug programs participating in the CADTH reimbursement review process
discussion with clinical experts who were practising hematologists with experience treating patients with hemophilia B (n = 4) directly engaged by CADTH over the course of this reimbursement review, including 2 clinical and economic consultation meetings involving 2 experts, and 1 panel meeting involving 3 experts; a fourth expert provided written responses to questions posed by the CADTH team in lieu of attending the panel discussion; during each of these meetings, clinical experts were asked targeted questions related to ethical considerations corresponding to the research questions driving this report
engagement with CADTH clinical and economic reviewers to identify domains of ethical interest arising from their respective reviews as well as relevant questions and sources to further pursue in this report.
Literature Search Methods
An information specialist conducted a literature search on key resources including MEDLINE via Ovid, Philosopher’s Index via Ovid, the Cumulative Index to Nursing and Allied Health Literature (via EBSCO, PsycInfo, and Scopus). A targeted Google Scholar search was also performed. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s Medical Subject Headings, and keywords. The main search concepts were fidanacogene elaparvovec and hemophilia B.
CADTH-developed search filters were applied to limit retrieval to citations related to ethical concepts or considerations, equity concepts or considerations, or qualitative studies. Duplicates were removed by manual deduplication in EndNote. Retrieval was limited to the English language. The search was completed on August 28, 2023. The search strategy is available on request.
Literature Screening and Selection
Literature retrieved according to the search and selection methods detailed earlier was screened in 2 stages. First, titles and abstracts of citations retrieved were screened for relevance by a single reviewer. Articles were identified and retrieved for full-text review by a single reviewer if their titles or abstracts identified ethical considerations, provided normative analysis (i.e., focusing on “what ought to be” through argumentation), or presented empirical research (i.e., focusing on “what is” through observation) of ethical considerations related to: the experiences, incidence, diagnosis, treatment, or outcomes of hemophilia B; or the evidence on, use of, or implications of fidanacogene elaparvovec (as a gene therapy) for patients with hemophilia B. In the second stage, full-text publications categorized as “retrieve” were reviewed by the same reviewer. Texts that included substantive information meeting the aforementioned criteria were included in the review, and reports that did not meet these criteria were excluded. As a parallel process, other sources drawn from relevant bibliographies, relevant key concepts, in consultation with experts or other CADTH reviewers, were retrieved and reviewed using the selection criteria listed described earlier.
Data Analysis
Data analysis was driven by the 4 research questions guiding this report and included the collection, coding, and thematic analysis of data drawn from the literature and project inputs. The reviewer conducted 2 iterative cycles of coding and analysis to abstract, identify, and synthesize relevant ethical considerations in the literature and from relevant project inputs.
In the initial coding phase, publications and input sources were reviewed for ethical content (e.g., claims related to potential harms, benefits, equity, justice, resource allocation, and ethical issues in the evidentiary basis). Once identified, claims related to ethical content were coded using methods of qualitative description.3 In the second coding phase, major themes and subcodes were identified through repeated readings of the data,3 and summarized into thematic categories within each guiding domain or research question. Ethical content that did not fit into these categories or domains outlined in the research questions was noted, as were discrepancies or conflicts between ethical considerations or values identified between project sources or within thematic categories. Data analysis was iterative, and themes identified in the literature, in project inputs, and during consultations with clinical experts were used to further refine and re-interpret ethical considerations identified.
Data collected and analyzed from these sources were organized thematically and described according to the 4 research questions and domains driving this report. The results of this analysis and its limitations and conclusions are described in the following section.
Results
Description of Included Sources
Data to inform this ethics report drew from patient group input, clinician group input, drug program input, and consultation with clinical experts engaged by CADTH for this review. All clinical experts were active in relevant clinical roles in Canada, and all had experience as hematologists treating patients with hemophilia B and some had experience treating patients with fidanacogene elaparvovec. A description and summary of these sources are included in the Clinical Review report.
The literature search identified 620 results and the grey literature search identified 22 additional results, for a total of 642 results. Following title and abstract screening, 617 citations were excluded and 25 potentially relevant publications from the electronic searches were retrieved for full-text review. Of the potentially relevant publications, 7 were excluded as they did not discuss ethical considerations of fidanacogene elaparvovec (as a gene therapy) or hemophilia B. A total of 18 publications meeting the inclusion criteria were included in this report. Eight additional publications were retrieved from backward searching of included publications’ reference lists or a manual search. A total of 26 publications were used to inform this report. Of these publications, 5 discussed ethical considerations in the context of hemophilia B, including those related to diagnosis and treatment; 2 discussed patient and/or family and caregiver experiences in the context of hemophilia B; and 12 discussed the use of the gene therapies (such as fidanacogene elaparvovec) for hemophilia. The remaining 7 publications were selected to provide a broader understanding of health systems considerations related to the costs of gene therapies, or other expensive treatment options, for rare diseases such as hemophilia B.
Key Ethical Considerations
Treatment and Experiences of Hemophilia B
Hemophilia B is a rare, congenital bleeding disorder that is characterized by a deficiency of FIX due to changes (variants or mutations) in the F9 gene on the X chromosome.4,5 As of 2021, there were 704 people living with hemophilia B in Canada.6 Of these patients, 535 were adult males and 43 were adult females.6 Of the 43 females, 3 had moderate hemophilia B, while of the 536 males, 145 had severe and 218 had moderate hemophilia B.6
Whether due to a shortage or complete absence of FIX activity, people living with hemophilia B of all severities are susceptible to prolonged bleeding episodes.4 In addition to trauma- or injury-related bleeding events, people with moderate to severe hemophilia B may also experience spontaneous and potentially life-threatening internal bleeding into joints, muscles, and organs.4 Although the frequency of bleeding episodes can vary significantly across patients, people with untreated severe hemophilia (A or B) experience, on average, upwards of 20 to 30 bleeding episodes annually.7 Frequent bleeding into joints (hemarthrosis) is not only painful in the short-term, but may lead to permanent joint damage and restricted mobility in the long-term.4
The World Federation of Hemophilia defines hemophilia B severity according to FIX activity levels, with “moderate” indicating 1% to 5% of normal and “severe” less than 1% of normal.5 Although not included in federation guidelines, clinical trials in hemophilia B (including the BeneGene-2 trial for fidanacogene elaparvovec) have used the term “moderately severe” (FIX activity 1% to 2% of normal) to further specify their target population. However, clinician input and the clinical experts engaged by CADTH suggested that clinical phenotype (i.e., tendency to bleed) is often used in clinical practice in conjunction with FIX activity levels to determine severity. This is noteworthy as the clinical experts suggested that, on rare occasions, some patients may have mild FIX activity levels (> 5% to 40% of normal) paired with a bleeding phenotype more typical of moderate to severe hemophilia B. Because severity, alongside lifestyle and professional activity considerations, is used as a key determinant of initiating FIX prophylaxis, this distinction can affect a person’s ability to receive preventive treatment (including gene therapies such as fidanacogene elaparvovec).
Current Treatment Options for Hemophilia B
According to clinician input and the clinical experts, standard of care for people living with moderate to severe hemophilia B (and a bleeding phenotype) in Canada is the use of prophylactic factor replacement therapy. The goal of prophylactic replacement therapy is to temporarily increase FIX activity so as to prevent, or reduce, bleeding events and help provide a quality of life comparable to that of the unaffected population.5 Factor replacement therapy involves IV injection of FIX coagulation factor concentrates at home by the patient or a caregiver.
Clinician input, patient input, and the clinical experts indicated that a limiting feature of FIX concentrates is the frequency at which the infusions must be administered. Depending on the FIX concentrate being used, people with hemophilia B undergo IV infusion as often as 2 to 3 times per week for products with a standard half-life or once every 1 to 2 weeks for those with an extended half-life. To this point, clinical experts indicated that FIX prophylaxis, while successful in raising FIX activity levels for most people with hemophilia B, is also a demanding, lifelong therapy that presents adherence challenges for some people. The clinical experts suggested that adherence may be particularly challenging for people with poor venous access. Given that these products only replace FIX for a limited amount of time, people with hemophilia B experience variable FIX activity levels, even when it is possible to adhere to a dosing regimen. As a result, people receiving FIX replacement therapy can remain susceptible to bleeds and may have to restrict their activity as FIX levels wane, and some remain at risk of developing joint damage due to internal bleeding. Although most participants in the patient group input indicated they were either very satisfied (4 of 17) or satisfied (7 of 17) with current treatment options, they also indicated that these options can greatly complicate their daily lives.
Experiences of Hemophilia B
Patient input, the clinical experts, and published literature all described the serious psychosocial and physical burden of living with or caring for someone with moderate to severe hemophilia (A or B).8 Patient input reported that a primary physical burden associated with hemophilia B is the joint damage caused by repeated episodes of internal bleeding. This is not only painful, but it was described as reducing people’s ability to participate in daily household and workplace activities.9 Furthermore, it contributed to an increased need for mobility support and joint-replacement procedures later in life. The ease with which bleeding episodes may occur was also described as a severe impact on quality of life because it can limit people’s ability to participate in sports or other daily activities. Patient input also indicated that the frequency of IV infusions of FIX prophylaxis could be challenging due to scarring and pain at injection sites as well as poor venous access.
As an X-linked condition, hemophilia (A or B) has historically been understood as a disease that primarily affects males and is carried (largely asymptomatically) by females.10 This understanding has fostered a medical research paradigm focused largely on the development and implementation of treatments for males with hemophilia (A or B) to the general exclusion of females.10 Although females have been recognized as carrying the potential to be diagnosed with hemophilia (A or B) since 2012, some authors have suggested it remains underrecognized and underdiagnosed.10 This fact highlights the possibility that females living with moderate to severe hemophilia B (although few in number) may experience disparities in access to care. While clinical experts and published literature indicated that psychological care is a standard offering in hemophilia treatment centres, it is also possible that access to this care may not be equitably distributed among all people potentially living with hemophilia, particularly females.8
Ethics of Evidence and Evaluation of Fidanacogene Elaparvovec.
The clinical evidence used to assess fidanacogene elaparvovec for the treatment of adult patients with hemophilia B is drawn from the pivotal, phase III, single-arm, open-label BeneGene-2 trial (n = 45). This trial is ongoing (with an expected completion in December 2029) and the data discussed in this larger Reimbursement Review reflects the November 16, 2022, data cut-off submitted by the sponsor. The primary objective of this trial is to determine whether fidanacogene elaparvovec can be considered noninferior to existing FIX prophylaxis as measured through the annualized bleeding rate for treated and untreated bleeds. To do this, BeneGene-2 trial participants were drawn from the noninvestigational BeneGene-1 lead-in study, in which participants were on FIX prophylaxis. Data collected from the BeneGene1 study (median follow-up of |||| |||||) was used to compare participant responses to FIX prophylaxis with responses to fidanacogene elaparvovec in the BeneGene-2 trial. Participants therefore served as their own controls. The details of the BeneGene-2 and BeneGene-1 trials are discussed further in the Clinical Review and Pharmacoeconomic Review reports.
Ethical Considerations in Trial Data
The clinical experts and clinician group input suggested that the preliminary results of the BeneGene-2 trial are promising, with a clinically relevant decrease in the annualized bleeding rate for treated and untreated bleeds. However, as described in the Clinical Review report, there is uncertainty in the interpretation of the magnitude of effect, as it may be biased due to assumptions in the statistical models used to inform the comparative efficacy of fidanacogene elaparvovec relative to FIX prophylaxis. Similarly, the durability of the effect and long-term safety of fidanacogene elaparvovec are presently uncertain, with a median follow-up time of only |||| |||||. The sponsor provided longer-term follow-up data (median of ||| |||||) for 14 participants enrolled in Study C0371003, a phase IIa, nonrandomized, open-label, longer-term follow-up study evaluating safety and efficacy for up to 6 years. However, the Clinical Review indicated that the efficacy and safety of fidanacogene elaparvovec relative to FIX prophylaxis could not be determined from this data due to the lack of a comparator group and the small sample size.
As an intervention that is meant to be effective over the course of a patient’s entire life, the uncertainty regarding durability of effect and long-term safety makes it challenging to accurately model and assess the cost-effectiveness of fidanacogene elaparvovec. This limitation presents challenges for assessing the opportunity costs — or forgone benefits — associated with reimbursing and resourcing a particular intervention over others, which are important for informing resource-allocation decisions at a health systems level.11 As discussed later in this report, this uncertainty can also make navigating the potential harms and benefits of fidanacogene elaparvovec challenging, which underscores the need for robust informed consent and shared decision-making.12
Ethical Considerations of Trial Representativeness
The clinical experts described the trial population as broadly generalizable to the Canadian context. However, it is important to note that eligibility was determined through severity according to FIX activity (i.e., ≤ 2%) and did not consider severity according to bleeding phenotype. As such, the clinical experts highlighted the possibility that people who would have been considered eligible in practice on the basis of clinical phenotype were excluded from the trial due to the presence of FIX activity greater that 2%. While the experts did not expect fidanacogene elaparvovec to act any differently in people with higher FIX activity, it is worth nothing that this exclusion criterion is misaligned with the current clinical practice in Canada described previously. As such, there is currently a lack of data for use in people who might otherwise be deemed eligible in clinical practice based on phenotypic severity.
Further, no females were included in the trial population for the BeneGene-2 study. Although cases of moderate to severe hemophilia B (defined using FIX activity levels) in females are rare, they are not altogether absent.10,13 However, the clinical experts did not anticipate that this would affect the generalizability of trial outcomes in practice as they expected the mechanism of action for fidanacogene elaparvovec to work similarly for males and females. The clinical experts also noted that the high number of white participants in the BeneGene-2 trial was not reflective of what they would expect to experience in clinical practice. Given the challenges in determining eligibility, it is possible that clinical decision-making regarding the offering of fidanacogene elaparvovec may favour those populations for whom some safety and efficacy data are available. This may be particularly challenging in light of ongoing uncertainty regarding who are most likely to benefit from fidanacogene elaparvovec. Additionally, the potential for AAV vector shortages may necessitate the formulation of criteria for prioritizing access to limited therapies.
Ethical Considerations in the Use of Fidanacogene Elaparvovec
Weighing Potential Harms and Potential Benefits
According to the sponsor’s submission, the value of fidanacogene elaparvovec lies in the possibility that a 1-time infusion can facilitate long-term endogenous FIX production and thereby eliminate, or reduce, the need for routine FIX prophylaxis as well as limit the frequency of spontaneous joint bleeds that can lead to long-term mobility challenges. Patient input reported that the possibility of eliminating IV infusions would be life-changing. Although their input also suggested a general degree of satisfaction with the clinical effectiveness of FIX prophylaxis, it was clear that the potential impact that gene therapies such as fidanacogene elaparvovec could have on their quality of life was desirable. For example, they emphasized the value of fewer infusions and fewer restrictions on activities such as athletics, physical labour, and travel. They also described the value of the potential to diminish developing long-term mobility challenges associated with frequent joint bleeds. This sentiment was echoed in clinician group input and the published literature, in which gene therapies such as fidanacogene elaparvovec were frequently described as paradigm-shifting due to their delivery as 1-time infusions, with the potential to deliver consistent, lifelong FIX production.14-16 However, the clinical experts consulted by CADTH noted that it is currently uncertain whether fidanacogene elaparvovec represents a shift in the current treatment paradigm.
While gene therapies (including fidanacogene elaparvovec) may offer novel therapeutic opportunities for people with hemophilia B, they involve both documented and theoretical risks that will need to be discussed as part of a robust informed-consent process.17,18 The Clinical Review report details adverse events identified in the BeneGene-2 trial, including increased alanine transaminase, abnormal hepatic function, increased aspartate transaminase, increased hepatic enzyme, increased transaminases, and anemia. Notably, navigating the risk of developing transaminitis (due to elevated alanine transaminase or aspartate transaminase levels) following gene therapy may be particularly challenging for patients who are considering gene therapy. As reported in 1 publication examining the experiences of early-phase hemophilia A or B gene-therapy trials, immunosuppressive therapy to treat, or prevent, transaminitis was frequently described as the most challenging aspect of undergoing gene therapy.16 One participant suggested that their experience was so difficult that, if they were to be hypothetically offered another gene therapy, they would only proceed if they could be guaranteed immunosuppressive therapy would not be required.16
Concerns about required postinfusion immunosuppressive therapy may remain a challenge if recommended for reimbursement, as 62.2% of participants in the BeneGene-2 trial underwent immunosuppressive therapy following infusion of fidanacogene elaparvovec. Although this may not prevent most people interested in gene therapy from pursuing fidanacogene elaparvovec, it will be important for clinical providers to ensure that patients are aware of the risk and understand both the potential duration and challenge of undergoing immunosuppressive therapy as part of the informed-consent process.
As previously noted, there are limited long-term efficacy and safety data on the use of fidanacogene elaparvovec. This means that there may be some theoretical risks that have yet to be demonstrated in gene therapy clinical trials (such as the BeneGene-2 trial for fidanacogene elaparvovec). As potential long-term risks, they are pertinent considerations for reimbursement decision-making. One such risk highlighted in the sponsor’s submission and published literature15,18-20 and by clinical experts was the theoretical risk that genotoxicity that could lead to the development of cancer following gene therapy. Although no cases of cancer were reported in the BeneGene-2 trial, clinical experts indicated that it would be important to continue postmarket monitoring of patients through patient registries and phase IV trials. To address this concern, clinical experts emphasized the potential challenge of losing track of patients during postmarket monitoring. This issue may become exacerbated if the gene therapy proves successful and allows patients to lead lives with few to no symptoms.
Overall, the clinical experts and clinician groups described being satisfied by the current safety profile of fidanacogene elaparvovec. They indicated that uncertainty regarding the durability of effect and long-term safety is not uncommon in gene therapies. They also suggested that this uncertainty would not prevent them from prescribing it to their patients who met eligibility criteria.
Determining Treatment Failure
Published literature14 and clinical experts reported that 1 post-infusion challenge would be how to determine treatment failure. Clarifying the parameters of treatment failure of a 1-time infusion with a potentially lifelong benefit will help with longer-term determinations of clinical benefit, use, and broader health systems value.
This is particularly important given that the transgene expression of AAV vectors is expected to diminish over time.15 This would result in a reduced treatment response and may require some patients to return to FIX prophylaxis.15 As some patients may be under the impression that gene therapies (such as fidanacogene elaparvovec) represent a lifelong cure,17-19 navigating potential treatment failure later in life could be challenging. While the sponsor has suggested that a proportion of patients may experience a gradual loss of therapeutic effect beginning in year 25 postinfusion, the clinical experts reported that it is currently impossible to know whether this will be observed in practice.
Furthermore, the clinical experts were hesitant to define what should qualify as treatment failure. However, they did state that treatment failure (or success) could be understood as happening in degrees rather than as a binary outcome. Additionally, the clinical experts suggested that evaluations of treatment response should be determined through a process of shared decision-making between clinical care teams and their patients. For example, literature suggests patients and clinicians may need to adjust how they understand and respond to potential bleeds.14 Prior to gene therapy, patients are encouraged to assume that all joint pain or worsened chronic pain is likely a new bleed.14 However, following gene therapy, there is the assumption that patients will experience fewer bleeds, which expands the possibility that novel joint pain, or aggravated chronic pain, may be related to something other than a new bleed.14 This points to a potential shift in the treatment paradigm and novel challenges that patients, their families, and their health care providers will need to navigate in the context of gene therapies such as fidanacogene elaparvovec.
Eligibility
Clinical experts highlighted 2 overarching ethically salient challenges associated with the establishment of eligibility criteria should fidanacogene elaparvovec be reimbursed. These include the need to consider clinical phenotype alongside FIX activity levels to determine severity, and considerations related to anti-AAV nAb testing.
FIX Activity and Phenotype
The BeneGene-2 trial determined eligibility according FIX activity levels (moderately severe to severe defined as FIX activity ≤ 2% of normal). However, the clinical experts expressed concern about the use of this criterion to determine treatment eligibility for fidanacogene elaparvovec. They emphasized how using FIX activity as the sole metric of severity would not align with clinical practice in Canada. This could also exclude people who present with a moderate to severe bleeding phenotype but FIX activity exceeding the moderately severe range. As most females with hemophilia B have mild FIX activity (5% to 40% of normal), the clinical experts suggested this would mean that females with a more severe bleeding phenotype may be unfairly excluded. Instead, they suggested that eligibility should be determined according to bleeding phenotype and whether patients require prophylactic FIX replacement therapy. This approach would better support equitable access for individuals with a potential to benefit from fidanacogene elaparvovec. Even with this broadened eligibility, it possible that not everyone eligible for treatment would pursue fidanacogene elaparvovec. Reasons for this include patient satisfaction with current treatment regimen or reluctance due to the lack of long-term data.19
Neutralizing Antibodies
Many people with hemophilia B will remain ineligible due to the presence of nAbs.18 Fidanacogene elaparvovec relies on successful hepatocyte transduction to deliver a stable, functional copy of a high-activity variant of the F9 gene (FIX-R338L) to liver cells. Cellular transduction is facilitated through a recombinant AAV vector — the rh74 serotype (AAVrh74) — and, as with other gene therapies, transduction may be prevented by the presence of anti-AAV nAbs.15 As a result, the presence of nAbs serves as an exclusion criterion in clinical trials for most gene therapies in development, including fidanacogene elaparvovec. Clinician input and clinical experts engaged by CADTH agreed with this exclusion criterion and emphasized that testing for the presence of anti-AAVrh74 nAbs would be a necessary component of determining patient eligibility for fidanacogene elaparvovec in clinical practice. However, they also reported the lack of standardization around nAb testing can make it challenging to interpret test results.21 Given the significance of nAb testing in determining eligibility for fidanacogene elaparvovec, the clinical experts emphasized the importance of accurate and complete reporting of test results, and suggested that it would be important to receive full results rather than a binary present-or-not determination.
The prevalence of pre-existing nAbs may be related to a number of factors, including serotype (e.g., AAVrh74 in the case of fidanacogene elaparvovec), types of antibodies being assessed, method of assessment, geographic location, and age.14,15,18,22 One publication investigating the use of different AAV serotypes for a novel gene therapy for Duchenne muscular dystrophy in the US reported that, while nAbs for AAVrh74 were less common that other AAV serotypes (e.g., AAV2 and AAV8), they were more frequently observed in Black, or African American, males.23 This may mean that Black males are more likely to test positive for anti-AAVrh74 nAbs and therefore have a higher chance of being ineligible for fidanacogene elaparvovec. Should other gene therapies for hemophilia B also use AAV vectors, this could present barriers for equitable access to gene therapy for people who are contraindicated due to presence of nAbs.
Although it is possible that Black males may be more likely to have anti-AAVrh74 nAbs, the presence of nAbs may be a barrier for many people with hemophilia B hoping to access gene therapies such as fidanacogene elaparvovec. According to the sponsor’s submission, roughly 60% of patients potentially eligible for the BeneGene-1 lead-in study were nAb-positive. In fact, of those excluded from the BeneGene-1 study, around 90% were nAb-positive. As such, it is important to recognize that many people may not be eligible for fidanacogene elaparvovec. This could be challenging in the context of the curative discourse surrounding gene therapies. Published literature has indicated there is a need to provide psychological support for those deemed ineligible for currently available gene therapies.17
Access
The clinical experts discussed the likelihood that geographic inequities in terms of access to fidanacogene elaparvovec (or other gene therapies) would exist due to limited infusion centres across Canada. According to the sponsor’s submission, there are ||| sites with experience serving as infusion centres for fidanacogene elaparvovec. There is also a ||||| with experience with referring patients to these ||| centres. Though the sponsor’s submission and the clinical experts indicated they expected the number of infusion sites to grow if fidanacogene elaparvovec is reimbursed, it is worth pointing out that this may take some time, and infusion sites may still not be equitably distributed across Canada. The clinical experts suggested that subsidy programs may help cover the costs of travel and accommodation for patients living in rural settings. They noted that this is already common practice for chimeric antigen receptor T-cell products and would likely be simpler to implement for fidanacogene elaparvovec (or other gene therapies) given the less-complex follow-up needs. Although patients would need to travel to designated infusion sites to receive fidanacogene elaparvovec, they could return home to be followed by their standard hematology treatment centre team.
Informed Consent
Weighing the potential benefits and harms of fidanacogene elaparvovec, as well as the challenges associated with determining treatment failure and eligibility, underscores the importance of robust informed consent and shared decision-making.12 Informed consent should not be considered a 1-time conversation, but an ongoing process, particularly in light of the reality that once administered, gene therapy cannot be discontinued.18 As the effectiveness of gene therapies (including fidanacogene elaparvovec) is expected to diminish over time,15 it is also important that clinical providers help establish reasonable expectations among their patients by clearly indicating that gene therapies (including fidanacogene elaparvovec) are not presently known to be curative.17-19
Furthermore, clinical experts and published literature noted that, following gene therapy, patients will develop nAbs for the AAV serotype serving as a vector for the therapy they received.22 This not only prevents people from being able to receive 2 infusions of the same gene therapy, or a gene therapy using the same AAV vector, but it may also prevent people from being eligible for future gene therapies. This is because nAbs are often cross-reactive to other AAV serotypes.21 This is important in the context of fidanacogene elaparvovec considering that 13.3% (n = 6) of the BeneGene-2 participants have resumed FIX prophylaxis as of the data cut-off. Having clear conversations about this possibility may help prevent patients from developing “buyer’s remorse” if another gene therapy is found to be more effective in the future.21
Clinical experts raised the importance of informed consent in the context of nAb testing. As there is currently no capacity in Canada, nAb testing will be conducted by US-based labs, which highlights the need for clinicians to discuss privacy with potential patients.
Health Systems Considerations
The use of fidanacogene elaparvovec as a gene therapy for hemophilia B raises several ethical considerations related to sustainable funding, resource allocation, and manufacturing and health system capacity for health systems in Canada.
Sustainability of Funding Gene Therapies
The introduction of gene therapies raises concerns regarding health care systems’ abilities to sustainably manage costs associated with these expensive, single-administration therapies.18,24 Fidanacogene elaparvovec’s long-term clinical effectiveness and cost-effectiveness are currently uncertain, which limits assessments of its long-term value and the opportunity costs of reimbursement. While this uncertainty will be familiar to decision-makers in the context of expensive drugs for rare diseases, it complicates reimbursement and resource-allocation decisions, particularly as fidanacogene elaparvovec has a proposed value as a 1-time therapy with a lifelong benefit.
Funding reforms for high-cost therapies with uncertain long-term evidence (e.g., gene therapy and chimeric antigen receptor T-cell therapy) have been suggested in the Canadian context, such as through some form of risk-sharing agreement.25-27 While risk-sharing agreements (also known as APMs) have been used in the US and some European jurisdictions, we are not aware of any existing APMs being used across Canadian jurisdictions at this time.25 Two overarching categories of APMs have been suggested within the hemophilia context: outcome-based and finance-based.24 As part of their pharmacoeconomic model in this review, the sponsor included 2 scenarios illustrating the potential impact of adopting an outcome-based agreement. In outcome-based models, payment is conditionally tied to meeting agreed-upon treatment outcomes (e.g., sustained FIX activity levels and limited breakthrough bleeds) and typically involves some form of delayed payment or full payment upfront with rebate options, depending on long-term success.24
From a health systems perspective, it is important to consider the clinical and data infrastructure that may be required to implement an APM. If choosing an outcomes-based model, for example, jurisdictions would need to agree on clear parameters of success (i.e., clinical outcome cut-offs) with manufacturers and establish postmarket surveillance mechanisms that could support data collection and management as well as value adjudication and contracting between parties.24,25 All parties would need to have confidence in the clinical data being collected.25 As such, the design of an outcome-based model has ethical implications for the distribution of the potential benefits and burdens associated with these arrangements.27 For example, how the parameters of success, or “value,” are defined has implications for how financial risks are distributed among manufacturers, payers, patients, and the public.27 In the context of hemophilia, it was suggested that some payers may find that the challenges of developing the necessary infrastructure for implementing these alternative funding arrangements outweigh their potential benefit for such a small population.24 However, as some authors have indicated the need to implement ongoing surveillance of long-term effectiveness and safety through extension studies and international registries,12,28 it is possible that some of the infrastructural requirements of a potential APM could be mitigated through shared administration of all postmarket surveillance.
Manufacturing and Health Systems Capacity
The clinical experts and published literature noted that manufacturing the AAV vectors used for AAV-mediated gene therapies is highly specialized and cumbersome.29 While some literature suggested that manufacturers have been able to keep up with demand for phase III and market-authorization trials,29 the clinical experts noted it is possible that supply shortages may become an issue as more gene therapies are made available in North American and European markets. Although they suggested shortages were less likely in the context of hemophilia B gene therapies (such as fidanacogene elaparvovec) given the small pool of potentially eligible patients, they expressed some concern that there is currently no guidance on how to prioritize access in the event of a shortage. However, they were wary of the development of strict prioritization criteria while it remains unclear who is most likely to benefit from fidanacogene elaparvovec. Without fair and transparent criteria for prioritizing access to limited therapy, there is a risk of inequitable access for patients with a limited capacity for self-advocacy. This issue could also affect those who lack support from proactive clinical providers, or those who reside in less-well-resourced jurisdictions, particularly in the event of AAVrh74 vector shortages.
Beyond potential manufacturing shortages, the clinical experts also indicated that some treatment centre pharmacies may either have a limited capacity, or desire, to store and reconstitute fidanacogene elaparvovec. This may limit the geographic distribution of infusion sites. It may also make accessing fidanacogene elaparvovec more challenging for people living outside the catchment areas of those treatment centres serving as infusion sites. Clinical experts noted that determining jurisdictional responsibility for reimbursement of the various costs associated with delivering fidanacogene elaparvovec may be complicated where patients may need to travel outside of their province or territory to access therapy.
Limitations
Little published literature discusses the ethical considerations related to the use of fidanacogene elaparvovec for the treatment of hemophilia B due to both the rarity of the disease and the novelty of the therapy under review. However, this does not imply an absence of ethical considerations in the context of fidanacogene elaparvovec (as a gene therapy) for hemophilia B, and this review of ethical considerations was augmented by drawing from additional resources collected in the course of the reimbursement review, including patient group, clinician group, and drug program input, and discussion with clinical experts, as well as engagement with CADTH clinical and pharmacoeconomic review teams, to provide a more comprehensive understanding of the ethical considerations related to the use of fidanacogene elaparvovec (as a gene therapy) for the treatment of hemophilia B. It is possible that more direct engagement with key stakeholders (e.g., direct interviews with patients, caregivers, family members, and decision-makers) on their specific experiences with hemophilia B and/or fidanacogene elaparvovec could offer additional relevant ethical considerations or domains of analysis.
Conclusion
Input from patient and clinician groups, drug programs, and relevant published literature were reviewed alongside direct engagement with clinical experts to identify and describe ethical considerations relevant to the use of fidanacogene elaparvovec for the treatment of adults living with hemophilia B. Ethical considerations in the context of hemophilia B include the significant treatment burdens associated with the existing standard of care for people with moderate to severe hemophilia B, which is FIX prophylaxis. While FIX prophylaxis is considered effective at elevating FIX activity levels, the effect is time-limited, it requires frequent IV infusions, and inevitably results in trough periods during which patients remain at risk for bleeds. As hemophilia B is an X-linked condition with infrequent occurrence in females, those females with moderate to severe hemophilia B may experience inequitable access to existing care due to misdiagnosis or underdiagnosis. While clinical trial evidence regarding the effectiveness and safety of fidanacogene elaparvovec is promising, the uncertainty regarding both the interpretation of preliminary trial results and the durability of effect and safety limits the ability to make definitive statements on the benefits and harms of fidanacogene elaparvovec. As a 1-time therapy that is meant to be effective for the duration of a person’s life, long-term data will be important in determining the realistic value of fidanacogene elaparvovec. Similarly, no females were included in the BeneGene-2 trial population and clinical experts suggested that white participants were overrepresented. Limited long-term data also limited the assessment of cost-effectiveness.
The use of fidanacogene elaparvovec as a gene therapy presents some known risks, such as developing transaminitis, and presently theoretical risks, such as genotoxicity, which may lead to the development of cancer. It is important to consider these risks in the context of fidanacogene elaparvovec as it is a 1-time infusion that cannot be discontinued. Additionally, due to the production of cross-reactive anti-AAV nAbs, some people may be rendered ineligible for future gene therapies even if they experience limited to no clinical benefit following fidanacogene elaparvovec. Further, transgene expression of the AAV vectors used in gene therapies is expected to diminish over time, leading to decreased efficacy and the need to return to FIX prophylaxis. If reimbursed, clinical experts suggested they would expect to experience a high uptake of fidanacogene elaparvovec in clinical practice. However, given the uncertainty surrounding the long-term safety and efficacy of fidanacogene elaparvovec, it will be important that clinical providers facilitate a robust informed consent and shared decision-making process. Determining eligibility for fidanacogene elaparvovec also presents ethical challenges as it is presently unclear who are most likely to benefit. With this in mind, the absence, or underrepresentation, of some populations (e.g., females and nonwhite people) may incidentally mean those included in the trial population are privileged for treatment-eligibility considerations. Further, it is possible that Black males are more likely to be living with anti-AAVrh74 nAbs, making them ineligible for fidanacogene elaparvovec. If other gene therapies for hemophilia B use AAV vectors that are more likely to be contraindicated in Black males due to the presence of nAbs, this may lead to fairness challenges. Equitable access to gene therapies such as fidanacogene elaparvovec will require addressing common geographic barriers to accessing specialist care and monitoring.
Ethical considerations for health systems related to fidanacogene elaparvovec include the challenges of funding decisions and assessments of opportunity costs for expensive drugs for rare diseases. Given the uncertainty around the durability of effect and safety of fidanacogene elaparvovec, APMs have been proposed as to help mitigate the risks of paying for an expensive gene therapy (with a proposed lifelong efficacy) in the absence of robust long-term data. Although the implementation of an APM may help mitigate risks, it is important to consider the concomitant challenges of building the data and clinical infrastructure needed to effectively execute the chosen APM. Similarly, it will be important to consider that the design of an APM (e.g., the parameters of treatment success) may affect how the benefits and burdens are distributed. It is also possible there will be production shortages of the AAV vector (AAVrh74) used in fidanacogene elaparvovec, which implies a need for clear prioritization criteria around which patients should be offered treatment first. Clinical experts also indicated there may be some geographic challenges to access, as not all treatment centre pharmacies will be able, or willing, to carry fidanacogene elaparvovec. As a result, some patients may need to travel out of province for treatment, and which province is responsible for which costs will have to be determined.
References
1.EUnetHTA Joint Action 2 Work Package 8. HTA Core Model ® version 3.0. Diemen (NL): European Network for Health Technology Assessment (EUnetHTA); 2016: https://www.htacoremodel.info/BrowseModel.aspx. Accessed 2022 Nov 18.
2.Benkhalti M, Espinoza M, Cookson R, Welch V, Tugwell P, Dagenais P. Development of a checklist to guide equity considerations in health technology assessment. Int J Technol Assess Health Care. 2021;37(1):e17. PubMed
3.Sandelowski M. Whatever happened to qualitative description? Res Nurs Health. 2000;23(4):334-340. PubMed
4.National Organization for Rare Disorders. Hemophilia B. Rare Disease Database 2023; https://rarediseases.org/rare-diseases/hemophilia-b/. Accessed June 29, 2023.
5.Srivastava A, Santagostino E, Dougall A, et al. WFH guidelines for the management of hemophilia, 3rd edition. Haemophilia. 2020;26(Suppl 6):1-158.
6.Association of Hemophilia Clinic Directors of Canada. Individuals with Factor IX Deficiency. 2021; https://ahcdc.ca/chr-2021. Accessed 2023 Oct 20.
7.Mannucci PM, Tuddenham EGD. The Hemophilias — From Royal Genes to Gene Therapy. N Engl J Med. 2001;344(23):1773-1779. PubMed
8.Arya S, Siad FM, Wilton P, et al. Invisible bleeds: Lived experiences and barriers to care for men with hemophilia. J Thromb Haemost. 2022;20(2):296-306. PubMed
9.Cutter S, Molter D, Dunn S, et al. Impact of mild to severe hemophilia on education and work by US men, women, and caregivers of children with hemophilia B: The Bridging Hemophilia B Experiences, Results and Opportunities into Solutions (B-HERO-S) study. Eur J Haematol. 2017;98 Suppl 86:18-24. PubMed
10.Weyand AC, James PD. Sexism in the management of bleeding disorders. Res Pract Thromb Haemost. 2021;5(1):51-54. PubMed
11.Kacetl J, Marešová P, Maskuriy R, Selamat A. Ethical Questions Linked to Rare Diseases and Orphan Drugs - A Systematic Review. Risk Manag Healthc Policy. 2020;13:2125-2148. PubMed
12.Valentino LA, Kaczmarek R, Pierce GF, et al. Hemophilia gene therapy: first, do no harm. J Thromb Haemost. 2023;21(9):2354-2361. PubMed
13.Miller CH, Soucie JM, Byams VR, et al. Women and girls with haemophilia receiving care at specialized haemophilia treatment centres in the United States. Haemophilia. 2021;27(6):1037-1044. PubMed
14.Yamaguti-Hayakawa GG, Ozelo MC. Gene therapy for hemophilia: looking beyond factor expression. Exp Biol Med (Maywood). 2022;247(24):2223-2232. PubMed
15.Ohmori T. Advances in gene therapy for hemophilia: basis, current status, and future perspectives. Int J Hematol. 2020;111(1):31-41. PubMed
16.Fletcher S, Jenner K, Pembroke L, Holland M, Khair K. The experiences of people with haemophilia and their families of gene therapy in a clinical trial setting: regaining control, the Exigency study. Orphanet J Rare Dis. 2022;17(1):155. PubMed
17.Fletcher S, Jenner K, Holland M, Khair K. Expectation and loss when gene therapy for haemophilia is not an option: An exigency sub-study. Haemophilia. 2023;29(3):776-783. PubMed
18.Baas L, van der Graaf R, van Hoorn ES, Bredenoord AL, Meijer K. The ethics of gene therapy for hemophilia: a narrative review. J Thromb Haemost. 2023;21(3):413-420. PubMed
19.Fletcher S, Jenner K, Holland M, Chaplin S, Khair K. An exploration of why men with severe haemophilia might not want gene therapy: The exigency study. Haemophilia. 2021;27(5):760-768. PubMed
20.Vasquez-Loarte TC, Lucas TL, Harris-Wai J, Bowen DJ. Beliefs and values about gene therapy and in-utero gene editing in patients with hemophilia and their relatives. The Patient: Patient-Centered Outcomes Research. 2020;13(5):633-642. PubMed
21.George LA. Hemophilia gene therapy: ushering in a new treatment paradigm? Hematology. 2021;2021(1):226-233. PubMed
22.Batty P, Lillicrap D. Gene therapy for hemophilia: Current status and laboratory consequences. Int J Lab Hematol. 2021;43 Suppl 1:117-123. PubMed
23.Verma S, Nwosu SN, Razdan R, et al. Seroprevalence of Adeno-Associated Virus Neutralizing Antibodies in Males with Duchenne Muscular Dystrophy. Hum Gene Ther. 2023;34(9-10):430-438. PubMed
24.Goodman C, Berntorp E, Wong O. Alternative payment models for durable and potentially curative therapies: The case of gene therapy for haemophilia A. Haemophilia. 2022;28(S2):27-34. PubMed
25.From Research to Reality. The Expert Panel on the Approval and Use of Somatic Gene Therapies in Canada. Ottawa (ON: Council of Canadian Academies; 2020: https://cca-reports.ca/wp-content/uploads/2019/08/Report-From-Research-to-Reality-EN.pdf. Accessed 2023 Sep 29.
26.Axicabtagene Ciloleucel (Yescarta). Ottawa (ON): CADTH; 2023: https://www.cadth.ca/sites/default/files/DRR/2023/PG0293-Yescarta.pdf. Accessed 2023 Sep 29.
27.Ciltacabtagene Autoleucel (Carvykti). Ottawa (ON): CADTH; 2023: https://www.cadth.ca/sites/default/files/DRR/2023/PG0302-Carvykti_combined.pdf. Accessed 2023 Sep 29.
28.Batty P, Lillicrap D. Advances and challenges for hemophilia gene therapy. Hum Mol Genet. 2019;28(R1):R95-R101. PubMed
29.Nathwani AC. Gene therapy for hemophilia. Hematology. 2022;2022(1):569-578. PubMed
Stakeholder Input
Patient Input
Canadian Hemophilia Society
About Canadian Hemophilia Society
Founded in 1953, the Canadian Hemophilia Society (CHS) is a national voluntary health charity. Its mission is to advocate to improve the health and quality of life for all people in Canada living with inherited bleeding disorder until cures are universally available. Its vision is a world free from the pain and suffering of inherited bleeding disorders.
The Canadian Hemophilia Society, whose national headquarters are in Montreal, is an organization that works at three levels: nationally, provincially and locally. We have ten provincial chapters across the country. Some of our chapters have additional local structures that we refer to as regions.
Its Board of Directors is made up of individuals with valuable skills and representing the organization’s ten provincial chapters. Each provincial chapter in turn is managed by its own Board of Directors. Many chapters are separately incorporated and have their own charitable registrations. Three provinces — Québec, Ontario and Manitoba — currently have offices with permanent staff. All chapters work in accordance with CHS by-law and conform to national policies. The national organization and its ten chapters share a common vision and mission. The CHS has approximately 300 active volunteers across the country.
The CHS is affiliated with the World Federation of Hemophilia which is officially recognized by the World Health Organization. We work in collaboration with health care providers in Canada’s 26 inherited bleeding disorder comprehensive care treatment centres, the blood system operators (Canadian Blood Services and Héma-Québec), the Network of Rare Blood Disorder Organizations, the rare disease community, and others who share our common interests.
Charitable Registration: 11883 3094 RR 0001
Website: www.hemophilia.ca
Information Gathering
The CHS gathers information on the patient perspective in a number of ways.
The CHS Blood Safety and Supply Committee (BSSC) is made up of a dozen patients, physicians and nurses. Meeting monthly, their role is to inform and advise the Board of Directors and the community on key issues pertaining to the safety, efficacy and availability of coagulation therapies for inherited bleeding disorders. Collectively, they have over 200 years of experience in this field. Members of the BSSC attended the latest Congresses of the International Society of Hemostasis and Thrombosis (Montréal, June 24-28, 2023) and the World Federation of Hemophilia (Montréal, May 8-11, 2022), where the latest research on novel therapies was presented.
Gene therapies for hemophilia B have been in clinical trials for more than ten years. The CHS and its BSSC have closely followed the results of this research by attending medical conferences where results are presented and reading peer-reviewed journal publications. Every two years, the CHS, in collaboration with the Association of Hemophilia Clinic Directors of Canada, organizes a three-day medical/scientific symposium where the latest research is presented and discussed. People with hemophilia B from Canada and abroad who have received gene therapy in clinical trials have presented their experience at these meetings. The latest symposium was held May 4 -7, 2023. A session, entitled GENE THERAPY: MANAGING EXPECTATIONS was dedicated to gene therapy, including patient perspectives, and can be viewed at …
“Hemophilia gene therapies: the current state of affairs”
Presented by DR. DAVID LILLICRAP, Kingston General Hospital, Ontario
"Why I said 'Yes' to gene therapy"
Presented by LUKE PEMBROKE, Greenwich, England, United Kingdom
"Why I said 'No' to gene therapy"
Presented by RICK WAINES, Victoria, British Columbia
“Updates on hemophilia gene therapies clinical trials in Canada”
Presented by DR. ALFONSO IORIO, Hamilton Health Sciences Centre, Ontario
“Hemophilia gene therapies roll-out: are HTCs ready?”
Presented by DR. ROY KHALIFÉ, The Ottawa Hospital, Ontario, and DR. JERRY TEITEL, St. Michael’s Hospital, Toronto, Ontario
Open discussion with panel Moderator: DR. ROY KHALIFÉ, AHCDC
Panelists: DR. ALFONSO IORIO, DR. DAVID LILLICRAP, DAVID PAGE, LUKE PEMBROKE, MARK SKINNER, DR. JERRY TEITEL, RICK WAINES
The CHS is in regular contact with its members through chapter meetings where current and future therapies of all types are discussed. In addition, members of the BSSC are in regular contact with their counterparts in hemophilia patient organizations around the world and the BSSC is represented on the World Federation of Hemophilia’s Coagulation Products Safety, Supply and Access Committee.
To collect specific perspectives from patients and caregivers with hemophilia B on the burden of disease and treatment, satisfaction with current treatment and the improvements people would like to see in a new treatment, the CHS in conducting an online survey launched on July 10, 2023. The survey was publicized via different CHS and chapter communication tools, including the CHS website, e-mail, Facebook, Twitter and Instagram. The questions asked are identical to those in the CADTH patient input template. We have received 17 responses up to July 31, 2023. All respondents are affected by severe or moderately severe hemophilia B without inhibitors. The results of that survey are presented below.
In addition, in September 2022, the CHS conducted an online survey of Canadians with severe hemophilia A and B to learn their hopes and expectations for gene therapy and received 39 responses. The results of that study are presented under the Improved Outcomes section.
Disease Experience
Joint damage, primarily to knees, ankles and elbows, caused by repeated internal hemarthroses, is the primary physical health impact of hemophilia B. Bleeding can be caused by very minor trauma. These impacts are clearly reflected in the survey results.
Overall Quality of Life
“Reduction in quality of life. Constant worry about injuries and bleeding, and the long recovery time. Twice-weekly treatments.”
“The exclusion from certain activities is a real factor in mental health. As an adult, not being able to participate in household duties, the chronic pain, knowing that I will have even more limitations in the future, not being able to contribute to savings for later invalidity, the worry that I will be a burden on loved ones; all these weigh on me.”
The Need to Refrain From Physical Activities and Sports
“As an adult, hemophilia has a big impact on my daily life. Many activities are chosen relative to my condition. I try to limit the risk of injuries. Even my career choice was affected by hemophilia.”
“My son has severe hemophilia B. There are certain activities he has to be careful doing or can't do at all. A small injury can easily become a trip to the hospital and weeks of recovery.”
“I take caution re activities, even benign sports.”
“There is constant worry that when a trauma happens there will be a delay in treatment and life-threatening response times.”
“We must make careful decisions about activities that come into play now that he is older.”
Reduced mobility
“My mobility, strength and endurance are significantly impacted on a daily basis.”
“I have joint pain and stiffness in knees and ankle that make walking painful and joint pain and stiffness in elbows that limit certain functions.”
“He is now too heavy for us to carry if he has a bleed that affects his walking. We have a wheelchair for him for these instances.”
“My joints are affected. Lots of pain, every day. I’ve had lots of surgeries and can’t function normally.”
Joint Replacements
“I have had several joint replacements and severe back pain due to the hemophilia.”
“I have had two knee replacements in the last five years.”
Experiences With Currently Available Treatments
The only currently approved products for the treatment of hemophilia B are clotting factor concentrates containing factor IX. Treatment for severe and moderately severe phenotypes of hemophilia B is for the vast majority of patients by regular prophylactic (preventative) intravenous infusions (IV), usually administered at home. Both recombinant and plasma-derived formulations are available in Canada. Recombinant forms can be either “standard half-life” preparations which require two to three IV infusions per week or “extended half-life” preparations, usually requiring only one infusion per week.
These treatments are prescribed through the Canadian network of 26 hemophilia treatment centres and are available at no direct cost to the patient through the Canadian Blood Services Plasma Protein and Alternative Products Formulary. Typically, patients/caregivers go to the treatment centre or hospital blood bank every one or two months to replenish their home inventory. In addition, they have more in-depth assessments by the interdisciplinary care team once or twice per year.
No alternatives to IV factor IX are currently approved. This is unlike hemophilia A where monoclonal antibodies (emicizumab) mimicking the function of factor VIII and injected subcutaneously are in widespread use. Subcutaneous non-factor IX replacement therapies to treat hemophilia B are in clinical trials. These include anti-tissue factor pathway inhibitors such as concizumab (licensed in Canada for those with inhibitors to factor IX) and marstacimab, anti-antithrombin therapies such as fitusiran, and anti-protein C therapies. It is difficult to predict if and when these products will get marketing approvals in Canada. Refer to www.hemophilia.ca/products-in-the-pipeline.
Early initiation of prophylaxis provides continued protection against joint damage throughout childhood compared with delayed initiation, but early prophylaxis is not sufficient to fully prevent damage. At the exit of the landmark Joint Outcome Continuation Study in hemophilia A, MR I osteochondral damage was found in 77% of those on secondary prophylaxis and 35% of those on primary prophylaxis. (Beth Boulden Warren, Marilyn J. Manco-Johnson et al. https://doi.org/10.1182/bloodadvances.2019001311, Blood Adv (2020) 4 (11): 2451–2459.)
While joint health research on hemophilia B lags behind that of hemophilia A because of the smaller numbers affected, there is little reason to believe that results are different. As long as factor levels fall below 10-15%, as is inevitable with factor replacement therapy, joint damage will occur in the long term. Only maintenance of higher levels will avoid this. I. E. M. Den UIJL, E. P. MAUSER BUNSCHOTEN, G. ROOSENDAAL, R. E. G. SCHUTGENS, D. H. BIESMA, D. E. GROBBEE, K. FISCHER
https://doi.org/10.1111/j.1365-2516.2011.02539.x
This is how the 17 respondents to the recent survey rated their satisfaction with current treatments.
Very satisfied: 4
Satisfied: 7
Neither satisfied nor dissatisfied: 5
Dissatisfied: 0
Very dissatisfied: 1
Patients and caregivers described their current treatments.
Safety and Efficacy
“Current factor concentrates protect well against most bleeding. I have approximately 2 to 3 joint bleeds per year despite prophylaxis.”
“While receiving his factor, my son is can run around like a normal kid, with minimal bleeds.”
“The support and care we get at the Children's Hospital are excellent. They are very knowledgeable and willing to help. I just wish my son didn't need to have so many needles all the time.”
“Bleeds seem to be controlled. We are very careful so this could be because of our efforts.”
“The concentrates are reasonably effective in protecting against bleeding.”
“With the EHL products, he doesn't bruise as easily now and has 1 or 2 minor bleeds per year, commonly in his ankle. No side effects or inhibitors.”
“Our current long-acting clotting factor works great. It is easy to use and infuse; however, so many pokes (IV infusions) every single year can be traumatic.”
“The current treatment regime had to be adjusted to be given within a shorter timeline as additional bleeds were happening.”
“We recently changed from an SHL product to an EHL. The number of treatments went from three times a week to one. That is a huge plus. Both medications have worked well.”
“We use an EHL FIX once a week through IV infusion. This is usually enough factor for him to get through a 7-day period without any bleeds. On this prophylaxis schedule, generally in one year we may need to take him to the Children's Hospital 1-2 times per year to treat a bleed.”
“He has missed some school with bleeds to improve healing.”
“The factor IX only lasts 24 hours in the bloodstream so if you have. A major trauma it means several days of infusions.”
The Burden of Treatments
“The treatments, even if they’re just IV infusions, greatly complicate everyday life, travel, and leisure activities.”
“Infusions can be difficult because the success of the needle getting in the vein is dependent on his vein visibility at the time of injection. A side effect would be that over the years he has complained about pain at the injection sites of his hands.”
“It is super hard, and I have always been hard to infuse.”
“Injection sites are scarred.”
“IV infusion every 5 days. I manage despite poor veins.”
“He has to have needles for factor replacement every week, and blood tests way more often than any other kid. A small injury can easily become a trip to the hospital and weeks of recovery. Then he gets more needles to add more factor IX to his blood to try to speed up his recovery.”
“Regular treatments are only a slight nuisance.”
“I get frequent phone calls from school due to cautious staff members not familiar with this disorder.”
“I give him weekly infusions, or more if injured, through a port.”
“The side effects are with my veins. I feel them getting used up. They are more and more discoloured. The aesthetic aspect bothers me.”
Socioeconomic Aspects
“There are a fair amount of trips to the clinic, so time off work for parents.”
“The clinic visits and follow-up are seemingly more difficult to access, and professional staff positions are not always filled.”
“The difficulties in accessing treatment are mostly time off work if something happens and we need to take him to the hospital. We live fairly close to the Children's Hospital. One of the reasons we live where we do is because of the access to the hospital.”
“Travel and insurance are an issue.”
“I take time off work to take my son to appointments. He needs frequent blood tests. He has pain at injection sites. Going to ER when clinic is closed is often a bad experience. Parking costs at hospitals are super high.”
“I have 6-8 clinic visits per year to pick up concentrates and have blood tests. I have to miss work.”
Improved Outcomes
In September 2022, the CHS conducted an online survey of Canadians with severe hemophilia A and B to learn their hopes and expectations for gene therapy and received 39 responses. The survey, whose answers were anonymous, was targeted at Canadian residents with severe hemophilia A or B, fourteen years of age or older, who represent the patient group that might consider taking gene therapy in the next five years.
The survey was publicized via the usual via CHS communication channels: website, Facebook, Twitter, and certain chapters’ social media, and was available in both English and French.
Thirty-nine people completed the survey, 31 with hemophilia A, seven with hemophilia B and one not specified. This accurately reflects the prevalence of severe hemophilia A and B in the population.
Fifty-four percent (54%) indicated they thought they would be eligible for gene therapy, 28% thought they would not be eligible and 18% said they didn’t know. Reasons for thinking themselves to be ineligible include a past history of inhibitors, pre-existing antibodies to the AAV vector used to deliver the gene for factor VIII or IX, age (under 18 or over 75) and other medical conditions, for example, active liver disease.
Respondents were asked the minimum level of factor VIII or IX expression predicted to be achieved that would make them want to have gene therapy. Answers varied widely, but 60% hoped for sustained expression of 30% or more.
Table 1: Minimum Factor VIII or IX Expression Desired
Minimum factor VIII or IX level desired (normal is 50-150%) | % of respondents |
|---|---|
5-10% | 7% |
10-20% | 17% |
20-30% | 7% |
30-40% | 17% |
40-100% | 43% |
I don’t know | 10% |
Respondents were also asked how long they would expect the factor level they chose in the question above to last for them to accept gene therapy. Again, answers varied widely, which is not surprising given that clinical trials for hemophilia gene therapy have given no clear answer to this question. It is worth noting that more than 6 out of 10 respondents (63%) indicated they expected gene therapy to be effective in preventing bleeding for at least 10 years.
Table 2: How Long Desired Factor Level Should Last to Be Acceptable
Time that gene therapy will be effective | % of respondents |
|---|---|
My whole life | 40% |
More than 10 years | 23% |
5-10 years | 10% |
3-5 years | 3% |
Less than 3 years | 7% |
I don’t know | 17% |
Respondents were asked how much certainty they needed as to the factor VIII or IX level they might achieve. More than half (56%) need to be quite sure or absolutely sure of the eventual factor level obtained. Unfortunately, clinical trial results, especially in hemophilia A, show a highly variable and unpredictable level of response from individual to individual, ranging from no response at all to levels above normal.
Table 3: Level of Certainty Needed Regarding Level and Duration of Factor Expression
Level of certainty needed regarding level and duration | % of respondents |
|---|---|
I need to be absolutely sure of the factor VIII or IX level I will get. | 13% |
I need to be quite sure of the factor VIII or IX level I will get. | 43% |
I am ready to accept some uncertainty. | 20% |
I realize my factor VIII or IX level could be much lower or higher than expected and I am ready to take a chance. | 17% |
I don’t know. | 7% |
Respondents were asked if they would accept gene therapy if they were told in advance that steroids would probably be needed in the period following administration. Thirty-eight percent (38%) said yes, 21% said no and 41% didn’t know. Many respondents commented that this was an important factor that would cause them to pause. Many others indicated they needed more information on this subject.
The survey asked if people would accept to receive gene therapy knowing that that there would be frequent blood draws in the weeks and months following administration, and they would need to be followed up in a registry for 10 to 20 years. Sixty-six percent (66%) answered yes, 10% answered no and 24% didn’t know.
Respondents were asked to express their overall attitude to gene therapy. (More than one answer was allowed.)
Table 4: Overall Attitudes to Gene Therapy
Overall Attitudes to Gene Therapy | % of respondents |
|---|---|
I am very interested in receiving gene therapy. | 45% |
I am not interested in receiving gene therapy at this time. | 14% |
I am concerned about short-term side effects. | 35% |
I am concerned about long-term side effects. | 52% |
I am concerned that FVIII or IX levels will not be high enough to prevent bleeding. | 44% |
I am concerned that FVIII or IX levels will not last long enough. | 55% |
I am waiting for more information. | 48% |
I intend to wait for future generations of gene therapy. | 31% |
I am ready to take a chance. | 10% |
I am not ready to take a chance. | 28% |
We asked respondents to indicate how knowledgeable they felt themselves to be.
Table 5: Level of Knowledge About Gene Therapy
Level of Knowledge About Gene Therapy | % of respondents |
|---|---|
Very knowledgeable | 13% |
Quite knowledgeable | 16% |
Somewhat knowledgeable | 38% |
Not very knowledgeable | 29% |
Not knowledgeable at all | 3% |
Respondents also indicated what they would like to know more about. Answers were:
Everything.
Nothing. I just wouldn't do it.
Nothing, I trust the science. I just want it.
How the therapy can be improved to provide better and more consistent results.
How to avoid the exclusion of those with HIV and/or HCV infection.
The complete working of it.
The experiences of those who have gone through the process.
Why and how it lasts for the amount of time that it does.
More about side effects.
If additional doses are possible if my levels drop, especially with future generations of gene therapy.
How it's being developed safely and securely.
Side effects, long term effectiveness.
Parallel information from other gene therapies for other disorders.
Trough levels, duration of levels, risks.
Cognitive/neurological risks.
Risks of comorbidities.
Will government be inclined to pay for gene therapy?
The kinds of support that would be available to a person in the first few weeks and months when there are numerous blood draws and appointments with medical personnel.
The lasting effects for those who went through clinical trials and received corticosteroids.
If I don’t respond, can I go back to my previous treatment with factor?
The long-term risks.
Patients and caregivers with hemophilia B, via the July 2023 survey undertaken for the review of fidanacogene elaparvovec, told us this about how gene therapy could potentially change their lives.
“How can it not? Nothing beats even a year of no infusions or bleeds.”
“Gene therapy would transform my life. I wouldn’t bleed. People who don’t have hemophilia cannot imagine the pain of a joint bleed; they have no idea.”
“I could imagine it being quite fantastic. Minimal needles, less stress and hopefully even fewer bleeds.”
“Gene therapy could revolutionize my daily functioning. It could optimize my current health state and improve my quality of life by reducing the amount of time and energy expended on treatments and preventing bleeding episodes.”
“Gene therapy would be life-changing for my son. He would go from having 52-75 needle pokes per year to only needing 2-5 needles with gene therapy. If his factor IX levels are consistently high from gene therapy, he can participate in activities that his doctors told us he can't because of physical contact. He tells me he always has to be aware of what dangers there are, even if they are minor. Something as minor as a hit with a ball or a bump against the wall can cause a bleed. Gene therapy could take some of his worries away if he doesn't have as high of a chance of a bleed. He wouldn't have bruises all over his body all the time. We would also have less trips to the hospital.”
“Confidence to travel and do physical work.”
“Less restrictions on activities. No weekly prophylaxis. No medications needed. A sense of safety knowing he has factors at all times in his body.”
“No more traumatic needles weekly. No more worry of injury response time. Ability to go out and take trips longer than a week without worry.”
“Gene therapy is a game changer. Going into teenage/adulthood, gene therapy would be huge for mental health and him feeling more “normal” and being able to enjoy life more fully.”
“Gene therapy has the potential to keep my factor IX at a level that would be very effective in preventing bleeding (i.e. 30-40%). I would no longer need IV infusions, except for surgeries or serious trauma.”
“Gene therapy would help my son dramatically without the fear of constant injuries. Mentally, removing his phobia around needles would improve his lifestyle incredibly.”
“I could travel more easily. Now, I limit my travel because of the difficulty of carrying bulky medication. Without 2 to 3 infusions per week, I’d have more time for my family and to do activities that improve my quality of life.”
“It would make the last years of my life so much easier.”
Experience With Drug Under Review
A small number (likely close to five) Canadians have undergone gene therapy for hemophilia B, but nothing is known to CHS about their experience outside the preliminary data for the full trials.
In early 2023, with the approach of gene therapies to the Canadian market, the Canadian Hemophilia Society produced All About Hemophilia Gene Therapy, A guide for patients and caregivers (bit.ly/AllAboutHemophiliaGeneTherapy).
This is an excerpt from the introduction to the booklet:
The hemophilia community has been waiting for gene therapy for years. Many have hoped it would be a cure. In the past few years, we have started to see promising results from the late stages of clinical trials for gene therapy in both hemophilia A and B.
With these results, however, we have learned that the reality of gene therapy differs from original hopes and expectations. The gene therapies that will be made available are promising new treatment options but are not full cures and are not for everyone.
Gene therapy is very different from the prophylaxis therapies we are used to. It is a one- time treatment that cannot be taken back and cannot be repeated. And we have also learned that we have a lot of work to do to ensure its safe and optimal introduction as a treatment option.
People with hemophilia (PwH) and their families must have all the information they need to make a fully informed decision as to whether or not to consider gene therapy.
PwH benefit from a number of therapeutic options, many of which have a long track record of safety and efficacy. Therefore, the benefits and risks of gene therapy must be seen in comparison to current treatments.
Ten key considerations that will be explored in this booklet include:
Gene therapy is not for everyone.
Many people are not eligible.
Predicting the outcome of gene therapy for an individual is not possible; however, for some, it can result in a significant improvement in quality of life.
Gene therapies for hemophilia A and B are different.
Decisions on moving ahead with gene therapy should be made only after a rigorous process of informed, shared decision-making.
Recipients of gene therapy must be ready for frequent blood draws and hospital visits in the first months after administration.
Most of those with hemophilia A and some with hemophilia B will require treatment with corticosteroids for up to many months after administration of gene therapy. These drugs can have significant side effects.
Reduction in consumption of alcohol may be recommended after gene therapy.
Clinicians monitoring people after gene therapy must be supported by a network of experienced experts.
All recipients of gene therapy must be enrolled in a registry. This registry will follow people for life.
Companion Diagnostic Test
Testing for antibodies to the AAV-5 vector is required before undergoing gene therapy. With regard to fidanacogene elaparvovec, those who test positive are deemed ineligible. Those who undergo gene therapy are required to have liver enzyme testing one to two times a week in the weeks and months following administration. A process needs to be in place to do the blood draws and send them to a laboratory for immediate analysis. Results must be analyzed very rapidly. Experts in hemophilia gene therapy must be available to advise physicians who are less experienced on when to initiate steroid treatment. Time is of the essence. If a rise in ALTs indicates a possible rejection of the vector, a course of steroids is started immediately and lasts for several months. A failure to act quickly can mean that expression of factor IX is permanently diminished or entirely eliminated. Side effects of the steroids, affecting both physical and mental health, can be significant. Patients and their families need to be adequately counselled regarding the potential need for steroids and their health impacts well in advance of any decision to receive gene therapy.
Anything Else?
In the absence of peer-reviewed publications describing the results of Phase III clinical trials for fidanacogene elaparvovec, the CHS is unable to comment on the relative benefits and risks compared to current therapies or other gene therapies for hemophilia B currently under review by Health Canada.
Conflict of Interest Declaration — Canadian Hemophilia Society
To maintain the objectivity and credibility of the CADTH CDR and pCODR programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission? If yes, please detail the help and who provided it.
The CHS received no help from outside our patient group to complete the submission.
Did you receive help from outside your patient group to collect or analyze data used in this submission? If yes, please detail the help and who provided it.
The CHS received no help from outside our patient group to collect or analyze data used in this submission.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review.
Table 6: Financial Disclosures for Canadian Hemophilia Society
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Bayer | — | — | — | X |
BioMarin | — | X | — | — |
CSL Behring | — | — | — | X |
Novo Nordisk | — | — | — | X |
Pfizer | — | — | — | X |
Roche | — | — | — | X |
Sanofi | — | — | — | X |
Takeda | — | — | — | X |
Clinician Input
The Association of Hemophilia Clinic Directors of Canada
About The Association of Hemophilia Clinic Directors of Canada
The Association of Hemophilia Clinic Directors of Canada (AHCDC) is a non-profit organization of Hemophilia Clinic Directors from across Canada. The goal of the AHCDC is to ensure excellent care for persons with bleeding disorders in Canada through clinical services, research and education. Our members are involved nationally and internationally in regulatory trials and research studies that investigate new factor replacement products or regimens, inhibitor development, prophylaxis, quality of life, women with bleeding disorders, genetic and clinical aspects of von Willebrand’s disease. In addition, our organization promotes clinical care through support of the National Inherited Bleeding Disorder Genotyping Lab. Prior to formal incorporation as the AHCDC in 1994, the organization was called the Canadian Hemophilia Clinic Directors Group (CHCDG) and ran from 1988-1994. The AHCDC is currently formed by 26 hemophilia treatment centers (HTC) and has 71 full members. The AHCDC members care for the totality of Canadian patients with hemophilia. AHCDC owns and manages the Canadian Bleeding Disorders Registry (CBDR, formerly CHARMS), a registry platform collecting demographics, clinical and quality of life data of all Canadian patients with hemophilia.
The organization’s website is: www.ahcdc.ca
Information Gathering
The information is gathered through a scoping literature review, expert presentations and member discussions through the AHCDC National Gene Therapy Learning Initiative (November 11, 2022), and drafted by members from the AHCDC Novel Therapy committee. It is circulated to AHCDC members for input and feedback before submitting the final version.
Current Treatments and Treatment Goals
Hemophilia B is an X-linked recessive bleeding disorder, affecting approximately 1 in 50,000 people, or about 600 Canadians [1]. The 2022 Canadian Hemophilia Registry reported a total of 553 adult males (318 years) registered in one of the 26 Canadian hemophilia treatment centres (HTCs), of whom 366 has moderate or severe hemophilia B [2]. Hemophilia B is classified as mild (baseline factor IX [FIX] activity 0.05-0.40 IU/ml), moderate (FIX 0.01-<0.05 IU/ml) and severe (FIX <0.01 IU/ml). Persons with severe hemophilia B and a proportion of moderate hemophilia B present with a clinically “severe” bleeding phenotype [3]. They suffer from recurrent bleeds into joints and muscles (spontaneous and traumatic), which may be mitigated but not eliminated by prophylactic treatment. Repeated bleeds into joints result in progressive joint damage (hemophilic arthropathy), chronic pain, loss of function, impairment in school and work productivity, and the need for early orthopedic interventions such as joint arthroplasties.
The current standard of care in Canada adheres to the World Federation of Hemophilia (WFH) Guidelines for the Management of Hemophilia. Persons with hemophilia (PWH) with a non-severe bleeding phenotype typically treat with episodic (also known as on-demand) clotting factor concentrates (CFCs) at the time of joint and muscle bleeds, and treat with tranexamic acid for mucocutaneous bleeding. For PWH with a severe bleeding phenotype, the WFH strongly recommends prophylactic replacement with CFCs or non-factor replacement therapy [3]. The goal of prophylaxis, the regular administration of therapeutic agents aimed at maintaining hemostasis, has evolved over the past decade. Historically, the primary goals of prophylaxis were to prevent repeated bleeding into joints and muscles which was supposedly achievable by maintaining FIX trough >0.01 IU/ml at all times, also preventing life-threatening bleeds such as intracranial hemorrhage, and prevent/slow down joint damage. Over time it became clear that prophylaxis targeting the 0.01 IU/ml threshold was only partially effective, and more so when associated with a careful avoidance of any moderate to intense physical activity, including the practice of most sports. Due to the very large variability in response to the infusion of CFCs, the administration of standardized doses of CFCs was producing higher trough levels in a sizeable minority of patients, showing the benefits of targeting higher trough level. For most patients, this would only be achievable very frequent administration of high dose of standard half-life concentrates which is both impractical and costly. Later on, higher trough levels became achievable for a larger majority of patient with the availability of extended half-life factor concentrates. More recently, the updated WFH Guidelines emphasized other important goals to aim for minimal bleeds and to empower PWH to lead healthy and active lives, and to participate fully in physical and social activities similar to the general population [3]. For PWH with breakthrough bleeds despite routine prophylaxis, the WFH recommends individualization and escalation of prophylaxis dose and/or frequency to prevent bleeding at all times. The current standard of care in Canada includes individualized or personalized prophylaxis, based on patient- and disease-related factors such as bleeding rates, joint health, physical activity and occupation, population pharmacokinetics profile on CFCs, and need for antiplatelet or anticoagulant therapy. Currently, approximately 80% of Canadian patients with clinically severe hemophilia B are receiving prophylaxis.
In Canada, CFCs are provided by the Canadian Blood Services (for provinces outside of Quebec) and Hema-Quebec (in the province of Quebec). Currently available Factor IX products include standard half-life factor CFCs (Benefix®, Rixubis®) and extended half-life CFCs (Alprolix ®, Rebinyn®). Non-factor replacement therapies are currently available only through clinical trials, although may eventually become available in the Canadian market within the next 2-5 years. These include RNA interference therapy targeting antithrombin (fitusiran), and monoclonal antibodies against tissue factor pathway inhibitors (anti-TFPI). One of the anti-TFPIs, concizumab, is recently licensed in Canada but only for hemophilia B with Factor IX inhibitors. Current treatments for hemophilia only target symptoms (prevention of bleeds or joint damage), without the ability to modify underlying disease mechanism, natural history, or provide potential cure. Prophylactic CFC replacement requires frequent intravenous (IV) infusions long-term, typically 1-2 times per week. The frequency of infusions, the consequences of breakthrough bleeds, and the need for IV access pose significant disease and treatment burden for patients, families, and caregivers. The latter barrier is particularly applicable to infants, children, and some older adults.
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
There are several unmet needs despite the currently available treatments in Canada for PWH with severe bleeding phenotype. First, prophylactic CFC replacement requires frequent venipuncture by patients and/or caregivers long-term, typically 1-2 intravenous infusions per week. Even with the advent of population pharmacokinetics and extended half-life FIX products, less than half of PWH are able to administer less than once weekly (e.g. every 10-14 days). Many individuals have poor venous access, posing a major challenge to routine prophylaxis. While placement of a central venous catheter (generally a Port-a-Catheter) is an option, it is associated with long-term complications including risks of infection, bleeding, thromboembolism, and loss of function requiring removal. Even among PWH with adequate venous access, non-adherence and/or treatment burden pose as key barriers to effective prophylaxis. For persons with hemophilia B and a severe bleeding phenotype, there is an unmet need to restore coagulation factor to clinically effective levels without the need for frequent venipunctures on a regular basis throughout one’s lifespan.
Second, the efficacy of prophylaxis with CFCs is variable across individuals. Given the half-life of CFCs, even with frequent administration of routine prophylaxis 1-2 times per week, PWH may experience low FIX trough levels (e.g. 0.01-0.05 IU/ml) prior to the next infusion. As a result, they are susceptible to breakthrough bleeds into joints and muscles, resulting in pain, loss of function, absenteeism from work or school, reduced quality of life, and more importantly disability from progressive joint damage. Even with the routine adoption of individualized, pharmacokinetics-guided prophylaxis (adjusting dose and/or frequency) in Canada, many PWH are still unable to achieve the goal of zero bleeds. For the period from January-December 2021, data from 149 severe hemophilia B patients on regular prophylaxis in Canada were available: of these patients, 55/149 (37%) had at least one hemarthroses in the calendar year. Additionally, 15 patients (10%) had a major bleeding episode.
Third, current treatments do not modify or alter the course of disease. Awareness of the progressive decline of trough factor IX levels following each factor concentrate infusion, many PWH live a restricted life, modifying their physical and social activities due to fear of bleeding and treatment burden. The impact on quality of life and participation varies among individuals, and may include (but not limited to): inability to pursue certain occupations, inability to participate in certain sports or physical activities, fear of bleeding or pain with sexual activities, mental health problems related to treatment burden, and chronic pain. The impact of hemophilia on quality of life has been highlighted in several studies [4-7].
Fourth, the factor IX trough levels associated with prophylaxis regimens are often insufficient to allow for safe anticoagulation or dual antiplatelet therapy. Historically, PWH had a shorter life expectancy than the general population due to life-threatening hemorrhages, as well as blood-borne pathogens such as human immunodeficiency virus (HIV) and hepatitis C from tainted blood products. As the life expectancy of PWH is approaching that of the general population, we observe a rise in the prevalence of cardiovascular and cerebrovascular diseases requiring antiplatelet or anticoagulation therapy. This provides a clinical conundrum, and is challenging to manage even with the use of aggressive prophylactic therapy.
Overall, there is a pressing need to provide effective therapy for a subgroup of PWH with a severe bleeding phenotype, who continue to experience breakthrough bleeds into joints/muscles despite routine prophylaxis. The ultimate goal, in keeping with the WFH treatment guidelines, is to minimize the number of bleeds to zero or near-zero, slow down the progression of hemophilic arthropathy, and minimize the adverse impact of recurrent bleeds on physical activity, physical and social function, and productivity loss. Among PWH with currently low rates of bleeding on prophylactic therapy, there is an unmet need to provide a therapy with curative potential, that would provide clinically adequate factor levels without long-term need for prophylactic replacement. This would improve health-related quality of life, reduce treatment burden, and save costs in the intermediate to long-term.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
As currently available hemophilia therapies (factor or non-factor replacement therapy) do not provide a curative option, gene therapy complements other available therapies by providing the possibility of long-term phenotypic cure for persons with hemophilia B. Hemophilia B is an X-linked monogenic disease leading to a single plasma protein deficiency. This pathology, along with the wide therapeutic margin of factor VIII and FIX levels have made hemophilia A and B recognized as ideal candidates for gene therapy for decades. Gene therapy provides a one-time treatment that inserts a functional factor IX gene into somatic cells, leading to sustained factor IX production. For the first time, we now have a treatment option that addresses the underlying disease process and natural history, rather than symptomatic management, representing a paradigm shift.
Until gene therapy can provide widely available, reliable long-term phenotypic normalization, prophylaxis with CFCs will remain the first-line treatment for PWH with a severe bleeding phenotype. Gene therapy is not currently studied or approved in the pediatric population under age 18 years. For adults with hemophilia who continue to experience breakthrough bleeds despite routine prophylaxis with CFCs or non-factor replacement (if available), who are unable to tolerate or adhere to prophylaxis, or who experience impaired health-related quality of life, impaired physical or social function related to hemophilia, gene therapy will be a very attractive therapeutic option.
In contrast to patients with hemophilia A, who have the option of emicizumab (a bi-specific monoclonal antibody mimicking the function of factor VIII, injected subcutaneously), patients with hemophilia B have no current alternatives to CFCs outside of clinical trials. This makes the need for gene therapy all the more pressing in hemophilia B.
Which patients would be best suited for treatment with the drug under review? Which patients would be least suitable for treatment with the drug under review?
Suitability and patient selection have been published in international guidelines and expert opinion pieces. A thorough assessment of eligibility is required and will be conducted by one or more specialists in hemophilia care (i.e. HTC Clinic Directors). Mechanisms will be in place to ensure equitable access to all eligible patients. Careful conversations and shared decision-making with patients and families are key aspects of the patient selection process, ensuring an individualized treatment decision based on eligibility, clinical and treatment factors including the potential need for corticosteroids after gene therapy, as well as patient’s values and preferences. This is especially relevant to gene therapy, as it is a one-time treatment at the present time without options for re-treatment. Patient identification includes:
Clinical examination and clinical judgment: annualized bleeding rate (all bleeds and bleeding into the index joints [ankles, knees, and elbows]), annualized spontaneous bleeding rate (all bleeds and bleeds into the index joints), annualized utilization of factor or non-factor replacement therapy (factor IX, or non-factor replacement therapies accessed through clinical trial), annualized number of factor IX infusions, adherence to prescribed prophylactic infusions, venous access, index joint scores using a standardized index joint examination using the Hemophilia Joint Health Score (the HJHS) performed by an experienced health care professional (generally a certified physical therapiest), and validated outcome measures of health-related quality of life (HRQoL), treatment burden and chronic pain and disability, and the ability to adhere to post-treatment laboratory tests
Laboratory tests: complete blood count and differential, liver enzymes, liver synthetic function, renal function, coagulation factor IX activity, factor IX inhibitor, assessment for human immunodeficiency virus (HIV), hepatitis B, and hepatitis C (serology and/or viral load, if relevant)
Imaging studies (if needed): abdominal ultrasound, +/- abdominal/liver ultrasound with elastography (i.e. Fibroscan)
Companion diagnostic tests: neutralizing antibody assay to identify pre-existing antibodies against AAV-5 vectors. As patients with positive AAV-5 antibodies are deemed ineligible for gene therapy at this time, this will be a first step in eligibility assessment. The AHCDC does not foresee any barriers or concerns with the adoption of centralized testing for AAV-5 antibodies.
Eligible candidates include adults with hemophilia B with clinically severe bleeding phenotype requiring prophylaxis, no history of inhibitory antibodies, no significant comorbidities, and no pre-existing anti-AAV neutralizing antibodies. For candidates with potential concerns on liver function (based on clinical history and laboratory evaluation), dedicated liver imaging with elastography and hepatology consultation are required. Given the possibility of corticosteroids use for adverse events, candidates should ideally have no absolute contraindications to corticosteroids such as severe psychiatric conditions, poorly controlled hypertension or diabetes, and severe osteoporosis. There are no concerns on misdiagnosis, under- or over-diagnosis in clinical practice. Current hemophilia gene therapies are not approved for patients under 18 years of age outside of clinical trials. While this may change in the future, individuals with hemophilia B under age 18 would not qualify for gene therapy.
At this time, it is unclear what are clinical or laboratory predictors of treatment response. This is an important area under study. However, the following factors will be considered to prioritize which candidates may benefit the most from gene therapy: severe bleeding phenotype (regardless of baseline factor IX activity) despite regular prophylaxis; poor venous access; non-adherence to routine prophylaxis resulting in recurrent bleeds; significant impairment in health-related quality of life and/or treatment burden from prophylaxis; need for a higher sustained FIX level (e.g. need for anticoagulants or dual antiplatelet therapy), ability to attend regular clinic follow-up and laboratory monitoring; and ability to abstain from alcohol for 6 months or longer post-infusion.
Given the need for anti-AAV antibody assay, detailed liver assessment, and assessment of PWH attitudes and perceptions, it is difficult to estimate the proportion of PWH eligible for gene therapy once it becomes commercially available. A recent single-centre study in Europe showed that most severe hemophilia A and B patients could not be enrolled due to eligibility criteria or lack of patient interest, only 8% of the patient population were eligible for a gene therapy trial [8].
AHCDC
A core outcome set for evaluating the efficacy, safety and value of gene therapy, incorporating both clinical outcomes and patient-reported outcomes, has been developed and proposed [9]. The core outcomes were selected based on a rigorous process including literature review, stakeholder engagement (PWH, clinicians, researchers, regulators and payers, including Health Canada) and adopting a Delphi methodology. The steering group included an AHCDC member. The core set has been endorsed by the World Federation of Hemophilia (WFH) gene therapy working group and included in the data collected by the Gene Therapy Registry. Its use has been endorsed as post-marketing surveillance system by all drug manufacturers, FDA and EMA (the latter official statement still pending after completed review). Of note, the CBDR has been recently updated by releasing a gene therapy module, colleting all the information included in the WFH Gene Therapy Registry. Canadian centers are ready to collect a thorough set of efficacy and safety outcomes.
The list of key outcome measures established by the WFH working group include:
Clinical outcomes
Frequency of bleeds: This is routinely collected by patients/ families on the MyCBDR platform and reviewed annually or more frequently by the HTC team.
Annualized frequency of bleeds: This is routinely collected by patients/ families on the MyCBDR platform and reviewed annually or more frequently by the HTC team. MyCBDR routinely collects detailed data on bleeding events, including spontaneous vs traumatic bleeds; index joint bleeds (knees, ankles, and elbows), bleeds into large joints not covered by the HJHS (e.g. shoulders, hips), treated vs non-treated bleeds.
Factor activity level: This will be regularly measured in a central coagulation laboratory. The frequency of Factor IX levels (one-stage, chromogenic) follows a schedule, based on the time from gene therapy infusion, at first weekly then with reduced frequency.
Duration of expression
Patient reported outcomes
Chronic pain
Mental health
Healthcare resource utilization: including hospitalization, emergency room visits, factor IX utilization, adjunctive medication (e.g. tranexamic acid, medications), homecare services, specialty consultations.
Safety outcomes
Mortality/ cause of death
Liver toxicity; short-term immune response to factor; thrombosis; vector integration into host genome; duration of vector-neutralizing response; other long-term adverse events
In addition, the following outcomes are crucial in assessing treatment response in routine clinical practice:
Ability to discontinue routine prophylaxis with minimal breakthrough bleeds: routine prophylaxis is defined as continuous replacement with standard CFCs or non-factor therapy for a minimum of 48 out of 52 weeks per week, with the intent to prevent bleeding. This information is routinely collected in the Canadian Bleeding Disorders Registry (CBDR) and updated annually or more frequently by the HTC.
Reduced annualized factor utilization: This includes factor utilization for routine prophylaxis, episodic treatment for acute bleeds and trauma, and surgical/situational prophylaxis. This information is routinely collected by patients/families on the MyCBDR portal, and available to clinicians and relevant stakeholders (e.g. AHCDC, Canadian Blood Services)
Improved joint health: presence of target joints (a single joint with 3 or more spontaneous bleeds in a 6-month period), hemophilic arthropathy as assessed by standardized instrument such as the HJHS score. Joint health is routinely assessed during annual comprehensive hemophilia assessments by physiotherapists. In addition to these routinely collected joint health data, imaging modalities such as point-of-care ultrasound of the index joints (performed by a trained healthcare professional) and magnetic resonance imaging (MRI) may be used as surveillance for subclinical joint disease resulting from clinical and subclinical bleeding. Surveillance point-of-care ultrasound is routinely performed by hemophilia clinic physiotherapists in many large centres in Canada.
Patient reported outcomes using validated generic and disease specific health-related quality of life, treatment burden, treatment satisfaction questionnaires; work and school absenteeism, and measures of physical activity. These measures are not routinely collected. Gene therapy participants and treating centres are strongly encouraged to enroll patients to the WFH gene therapy registry, and participate in the collection of the core outcome set including patient-reported outcomes in addition to routinely collected clinical information.
Treatment response including use of prophylaxis, annualized factor utilization, annualized bleeding rates, and joint health are formally assessed on an annual basis. However, as patients record their bleeding diaries and factor/ non-factor product utilization in real-time, any clinical changes will be flagged and reviewed sooner by members of the the hemophilia multidisciplinary healthcare teams. The factor IX activity will be measured regularly, the frequency based on timing from gene therapy infusion. The frequency will follow the protocol as per the clinical trial protocol and drug monograph, and also depends on the trend of serial factor IX activities.
A clinically meaningful response may include one or more of the following:
For patients on prophylactic replacement, the ability to discontinue routine prophylactic therapy. Of note, some patients may still require episodic prophylaxis during times of high-risk for bleeding, such as factor prophylaxis during times of major surgeries or trauma.
Reduction in the annualized utilization of CFCs or non-factor products
Low annualized bleeding rates: zero or near-zero spontaneous annualized bleeding rates including all bleeds and bleeds into the index joints.
Sustained expression of FIX activity: factor IX activity does not always correlate with the bleeding tendency or need for prophylactic therapy. While severe hemophilia is characterized by recurrent bleeds requiring routine prophylaxis, moderate hemophilia (0.01-<0.05 IU/ml) is a more heterogenous group. Some persons with moderate hemophilia (e.g. factor 0.01-0.02 IU/ml) may behave like those with severe hemophilia and require routine prophylactic replacement, while others may rarely bleed outside of traumatic or surgical settings. As a result, factor IX activity should only be used as a secondary outcome. For instance, for an individual with baseline factor IX of 0.01 IU/ml requiring long-term prophylaxis to prevent recurrent bleeds, the ability to discontinue prophylaxis with near-zero bleeds over the next 5-10 years would be considered a clinical meaningful response even if factor IX is only 0.04 IU/ml in year 10. The limitation of factor IX levels is further emphasized by substantial discrepancies in quantitating factor IX by different commonly used factor IX activity assays.
Improvement in patient reported outcomes including health-related quality of life, chronic pain and treatment burden
The magnitude of treatment response would not vary across multidisciplinary health care professionals involved in the assessment and care of PWH.
What factors should be considered when deciding to discontinue treatment with the drug under review?
Gene therapy is a one-time treatment. Criteria for treatment discontinuation is not relevant for the drug under review.
What settings are appropriate for treatment with [drug under review]? Is a specialist required to diagnose, treat, and monitor patients who might receive [drug under review]?
A “hub and spoke model” of gene therapy implementation and delivery has been proposed by a joint publication by the European Association for Haemophilia and Allied Disorders (EAHAD) and the European Haemophilia Consortium and adopted by Quebec with success [10, 11]. The current strategy from the AHCDC is to adopt the proposed “hub and spoke model” with appropriate adaptation to Canadian needs. Specifically, since we anticipate that not all HTC (Hemophilia Treatment Centres) in Canada may have the institutional facility to infuse the gene therapy product, “hub” centres will be those which can accommodate patients from other (“spoke”) HTC for the actual infusion. Once the infusion has been completed and the patient is discharged, the “spoke” centre will resume all responsibility for follow up care, and registry reporting. The model is characterized by a close collaboration and communication, between gene therapy dosing centres (“hubs”) and referral/follow-up centres (“spokes”). Empirically, the model has shown to perform efficiently and safely for enrollment of approximately 10 patients in gene therapy trials, whereby most patients were infused in a few clinical centers, and followed up in “spoke” centers. All referral/follow-up centres belong to one of the 26 HTCs. The local HTC clinic director will be responsible for the diagnosis, identification and screening of eligible patients, education and counselling, and post-infusion follow-up care. Once interested patients meet the eligibility criteria and are deemed a suitable candidate, they will be referred to one of the regional gene therapy dosing centres. The dosing centres include HTCs that have participated in or are selected to participate in gene therapy clinical trials as well as other HTCs with the resources and infrastructure to serve as an infusion site. The AHCDC, in collaboration with individual HTCs, is in the process of selecting a list of gene therapy dosing sites over the next year. Attention to geographic distribution will be paid to ensure equitable access to all Canadians.
Patients will travel to the closest regional gene therapy dosing site for infusion. Treatment will be delivered in a monitored setting in a tertiary hospital, which will ensure safe product storage and handling, safe area for containment, supplies and trained personnel for handling infusion reactions. Following initial treatment, patients will be monitored closely by their local HTC. The dosing site will continue to act as a consultant and provide expert advice, if needed, to support ongoing monitoring and treatment.
Other than hemophilia clinic directors, other specialties that may be involved in the care of patients undergoing gene therapy include:
Psychologists, psychiatrists, or counsellors: to provide psychosocial assessment and support throughout the patient journey pre- and post-infusion of gene therapy
Hepatologists: to assess baseline liver function to help determine eligibility (e.g. arrange liver ultrasound with elastography to assess baseline liver fibrosis or steatosis), assess and manage transaminitis that may occur following infusion.
Social workers/ occupational therapists: to assist with social, financial, and logistic support for patients and families throughout the journey.
Home care: to assist with sample collection in patients who live in rural/remote regions, and/or have barriers to frequent visits to the hospital laboratory for monitoring.
Additional Information
References
Iorio, A., Stonebraker, J. S., Chambost, H., Makris, M., Coffin, D., Herr, C., & Germini, F. Establishing the Prevalence and Prevalence at Birth of Hemophilia in Males. Annals of Internal Medicine. 2019; 171(8): 540. https://doi.org/10.7326/M19-1208
Canadian Bleeding Disorders Registry. “Factor FIX Deficiency-2022”. Unpublished data.
Srivastava A, Santagostino E, Dougall A, Kitchen S, Sutherland M, Pipe SW, Carcao M, Mahlangu J, Ragni MV, Windyga J, Llinás A, Goddard NJ, Mohan R, Poonnoose PM, Feldman BM, Lewis SZ, van den Berg HM, Pierce GF; WFH Guidelines for the Management of Hemophilia panelists and co-authors. WFH Guidelines for the Management of Hemophilia, 3rd edition. Haemophilia. 2020;26 Suppl 6:1-158.
Buckner TW, Witkop M, Guelcher C, Sidonio R, Kessler CM, Clark DB, Owens W, Frick N, Iyer NN, Cooper DL. Impact of hemophilia B on quality of life in affected men, women, and caregivers-Assessment of patient-reported outcomes in the B-HERO-S study. Eur J Haematol. 2018;100(6):592-602.
Steen Carlsson K, Winding B, Astermark J, Baghaei F, Brodin E, Funding E, Holmström M, Österholm K, Bergenstråle S, Lethagen S. High use of pain, depression, and anxiety drugs in hemophilia: more than 3000 people with hemophilia in an 11-year Nordic registry study. Res Pract Thromb Haemost. 2023 Jan 31;7(2):100061.
Nguyen NAT, Auquier P, Beltran Anzola A, d'Oiron R, Biron-Andréani C, Lienhart A, Rauch A, Baumstarck K, Boucekine M, Milien V, Rosso-Delsemme N, Tabele C, Giraud N, Sannié T, Chambost H, Resseguier N; INTHEMO Study Group. Occupational integration of adults with severe haemophilia (INTHEMO): A study based on the FranceCoag registry. Haemophilia. 2022;28(6):962-976.
Plug I, Peters M, Mauser-Bunschoten EP, de Goede-Bolder A, Heijnen L, Smit C, Willemse J, Rosendaal FR, van der Bom JG. Social participation of patients with hemophilia in the Netherlands. Blood. 2008;111(4):1811-5.
Krumb E, Lambert C, Hermans C. Patient selection for hemophilia gene therapy: real-life data from a single center. Res Pract Thromb Haemost 2021; 5: 390–394.
Iorio A, Skinner MW, Clearfield E, Messner D, Pierce GF, Witkop M, Tunis S; coreHEM panel. Core outcome set for gene therapy in haemophilia: Results of the coreHEM multistakeholder project. Haemophilia. 2018;24(4):e167-e172.
Miesbach W, Chowdary P, Coppens M, et al. Delivery of AAV‐based gene therapy through haemophilia centres‐A need for re‐evaluation of infrastructure and comprehensive care: a Joint publication of EAHAD and EHC. Haemophilia. 2021;27(6):967‐973.
Personal communications, Dr. Molly Warner.
Conflict of Interest Declarations — The Association of Hemophilia Clinic Directors of Canada
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please refer to the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
AHCDC received no help from outside our clinician group to complete the submission.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
AHCDC received no help from outside our clinician group to collect or analyze any information used in this submission.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician who contributed to the input.
Declaration for Clinician 1
Name: Dr. Haowei (Linda) Sun
Position: Chair, Novel Therapy Committee, AHCDC; Hemophilia Clinic Director, Northern Alberta Bleeding Disorders Program; Associate Professor, Division of Hematology, Department of Medicine, University of Alberta
Date: 01-07-2023
Table 7: COI Declaration for The Association of Hemophilia Clinic Directors of Canada — Clinician 1
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Pfizer | X | — | — | — |
Roche | X | — | — | — |
Sanofi | X | — | — | — |
Takeda/ Shire | X | — | — | — |
Declaration for Clinician 2
Name: Dr. Jerry Teitel
Position: Past president, AHCDC; Member, AHCDC Novel Therapy Committee; Professor, Division of Hematology, Department of Medicine, University of Toronto
Date: 08-08-2023
Table 8: COI Declaration for The Association of Hemophilia Clinic Directors of Canada — Clinician 2
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Pfizer | X | — | — | — |
Roche | X | — | — | — |
Sanofi | X | — | — | — |
Takeda | X | — | — | — |
Biomarin | — | X | — | — |
Vega Therapeutics | — | X | — | — |
Declaration for Clinician 3
Name: Davide Matino
Position: Assistant Professor, McMaster University; Member, AHCDC Novel Therapy Committee
Date: 07-08-2023
Table 9: COI Declaration for The Association of Hemophilia Clinic Directors of Canada — Clinician 3
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Novo Nordisk | X | — | — | — |
Sanofi | — | X | — | — |
Vega Therapeutics | X | — | — | — |
Pfizer | X | — | — | — |
Octapharma | X | — | — | — |
Bayer | X | — | — | — |
Precision Biosciences | X | — | — | — |
Declaration for Clinician 4
Name: Dr. Alfonso Iorio
Position: Professor, Health Research Methods, Evidence, and Impact, Faculty of Health Sciences, McMaster University; Co-chair of the World Federation of Haemophilia (WFH) World Bleeding Disorder Registry; Past Chair of the WFH Data and Demographics Committee
Date: Aug-14-2023
Table 10: COI Declaration for The Association of Hemophilia Clinic Directors of Canada — Clinician 4
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Pfizer | — | — | X | — |
Roche | — | — | — | X |
Takeda | — | — | — | X |
Spark | — | — | X | — |
Bayer | — | — | — | X |
Sanofi | — | X | — | — |
Sobi | — | — | — | X |
Note: No money was received personally by myself from any pharma company. The dollar ranges indicated below are for research contracts (e.g. conduct of clinical trials) or research service agreements (e.g. data analysis) paid from sponsors to Hamilton Health Sciences or McMaster University. All research relationships have been disclosed, irrespectively of the sponsor involvement with gene therapy trials or research.
Declaration for Clinician 5
Name: Dr. Adrienne Lee
Position: AHCDC executive board of directors; Member, AHCDC Novel Therapy Committee
Date: 11-08-2023
Table 11: COI Declaration for The Association of Hemophilia Clinic Directors of Canada — Clinician 5
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Takeda | X | — | — | — |
Pfizer | X | — | — | — |
Leo Pharma | X | — | — | — |
Declaration for Clinician 6
Name: Dr. Victor Blanchette
Position: Hemophilia Clinic Director, Hospital for Sick Children; Member, AHCDC Novel Therapy Committee; Professor, Department of Pediatrics, University of Toronto
Date: 12-08-2023
Table 12: COI Declaration for The Association of Hemophilia Clinic Directors of Canada — Clinician 6
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Takeda | — | X | — | — |
Sanofi | X | — | — | — |
Declaration for Clinician 7
Name: Dr. Mark Belletrutti
Position: Hemophilia Clinic Director, British Columbia Children’s Hospital; Member, AHCDC Novel Therapy Committee; AHCDC executive board of directors
Date: 10-08-2023
Table 13: COI Declaration for The Association of Hemophilia Clinic Directors of Canada — Clinician 7
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Roche Canada | — | X | — | — |
Takeda Canada | X | — | — | — |
Bayer Canada | X | — | — | — |
Sanofi Canada | X | — | — | — |
Octapharma Canada | X | — | — | — |
Declaration for Clinician 8
Name: Dr. Roy Khalife
Position: Member, AHCDC Novel Therapy Committee; AHCDC executive board of directors
Date: 11-08-2023
Table 14: COI Declaration for The Association of Hemophilia Clinic Directors of Canada — Clinician 8
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Pfizer Canada | X | — | — | — |
Takeda | X | — | — | — |
Novo Nordisk | X | — | — | — |
Bayer Canada | X | — | — | — |
Declaration for Clinician 9
Name: Dr. Roona Sinha
Position: President, AHCDC
Date: 13/08/2023
Table 15: COI Declaration for The Association of Hemophilia Clinic Directors of Canada — Clinician 9
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Thrombosis Canada | X | — | — | — |
Canadian Blood Services Board of Directors | X | — | — | — |
Bayer | X | — | — | — |
Takeda/ Shire | X | — | — | — |
Octapharm | X | — | — | — |
Canadian Association of Nurses in Hemophilia Care
About Canadian Association of Nurses in Hemophilia Care
CANHC is a group of Canadian nurses dedicated to the care of patients and families with bleeding disorders. The group engages in clinical, educational, and research activities, focused on improving the care of this population.
Information Gathering
Requested feedback from all CANHC members via email and collated the perspectives of the nurses.
Current Treatments and Treatment Goals
The current basic treatment to stop or prevent bleeding in people with hemophilia B is factor replacement therapy. This is the infusion (injection into the bloodstream) of factor IX concentrates to prevent or control bleeding. The factor products currently available in Canada include X recombinant products with a range in average ½ life of 18.1-115 hours and require intravenous infusion intervals ranging from daily to biweekly depending on the product for life in order to prevent bleeding. There is also one albumin containing recombinant FIX product available with a ½ life between 104 and 144 hours. For minor mucosal bleeding anti-fibrinolytic therapy may be useful as either standalone therapy or as add on therapy. While these therapies target the treatment of or prevention of bleeding, they do not modify the underlying disease process in any way. It is therapy that modifies the underlying disease process giving lasting increases to baseline FIX levels that is currently missing from the treatments available in Canada outside of a research setting. Permanently sustained elevated FIX levels decreases day to day bleeding in a reliable way without need for regularly self-administered intravenous products. Alleviating patients and families of the burden of regular infusions (which by human nature have variable compliance), joint and muscles bleeds that impact productivity (school and work) positively impacts quality of life.
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
As mentioned above the currently available treatments in Canada do not modify the underlying disease process and thus persons with Hemophilia B are dependent, life long, on regular IV infusions to prevent and treat bleeding. In addition all currently available treatments are administered intravenously.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
For people living with hemophilia B, once treated with Fidanacogene elaparvovec, would be able to produce FIX themselves via this one-time treatment rather than having to receive exogenous FIX. This would result in a complete shift in the treatment paradigm from one of ongoing optimization with regular infusions to treat once and modify the disease to a state where treatment to prevent bleeding (prophylaxis) would no longer be necessary. It would be a novel option for patients to consider along with the currently available therapies noted above.
Which patients would be best suited for treatment with the drug under review? Which patients would be least suitable for treatment with the drug under review?
Which patients are most likely to respond to treatment with drug under review? Persons with Moderate to Severe Hemophilia B.
Which patients are most in need of an intervention? Persons with Moderate to Severe Hemophilia B or those with Mild Hemophila B and an Annual bleed rate equal to or greater to 1.
Would this differ based on any disease characteristics (e.g., presence or absence of certain symptoms, stage of disease)? Annual bleed rate may be a factor to consider for those not currently receiving regular prophylaxis but should not be a consideration for those already on regular prophylaxis.
How would patients best suited for treatment with drug under review be identified (e.g., clinician examination/judgement, laboratory tests (specify), diagnostic tools (specify)) Multidisciplinary team discussion
Are there any issues related to diagnosis? No
Is a companion diagnostic test required? No
Is it likely that misdiagnosis occurs in clinical practice (e.g., underdiagnosis)? No
Is it possible to identify those patients who are most likely to exhibit a response to treatment with drug under review? Those with a high annual bleed rate, those not compliant with current prescribed or recommended treatment.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice? How often should treatment response be assessed?
Compliance with prescribed or recommended therapy, annual bleed rate, pain assessments and quality of life scales should be monitored on a 6 month, or yearly timeframe.
What factors should be considered when deciding to discontinue treatment with the drug under review?
Not applicable- Treatment is one time treatment and cannot be discontinued.
What settings are appropriate for treatment with [drug under review]? Is a specialist required to diagnose, treat, and monitor patients who might receive [drug under review]?
Patient must be registered with one of the 26 bleeding disorder centres across Canada with an interdisciplinary team of specialists including physiotherapy, social workers, hematologists, and nurses and nurse practitioners with specialty in the management of bleeding disorders. Final prescribing of the therapy would need to be done by a health care provider with prescription authority (Physician or Nurse Practitioner).
Additional Information
Is there any additional information you feel is pertinent to this review?
Not applicable.
Conflict of Interest Declarations — Canadian Association of Nurses in Hemophilia Care
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please refer to the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission?
No.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission?
No.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician who contributed to the input.
Declaration for Clinician 1
Name: Karen Sims
Position: Nurse Practitioner Adult Bleeding Disorder Program of BC and Yukon
Date: 24/07/2023
Table 16: COI Declaration for Canadian Association of Nurses in Hemophilia Care — Clinician 1
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 2
Name: Vanessa Bouskill
Position: Nurse Practitioner SickKids Hospital Toronto
Date: 25/07/2023
Table 17: COI Declaration for Canadian Association of Nurses in Hemophilia Care — Clinician 2
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 3
Name: Heather Bauman
Position: Registered Nurse
Date: 14-08-2023
Table 18: COI Declaration for Canadian Association of Nurses in Hemophilia Care — Clinician 3
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
Redactions: Confidential information in this document may be redacted at the request of the sponsor in accordance with the CADTH Drug Reimbursement Review Confidentiality Guidelines
Stakeholder Input: The views expressed in each submission are those of the submitting organization or individual; not necessarily the views of CADTH or of other organizations. As such, they are independent of CADTH and do not necessarily represent or reflect the view of CADTH. No endorsement by CADTH is intended or should be inferred. By filing with CADTH, the submitting organization or individual agrees to the full disclosure of the information. CADTH does not edit the content of the submissions.
CADTH does use reasonable care to prevent disclosure of personal information in posted material; however, it is ultimately the submitter’s responsibility to ensure no identifying personal information or personal health information is included in the submission. The name of the submitting organization or individual and all conflict of interest information are included in the submission; however, the name of the author, including the name of an individual patient or caregiver submitting the patient input, are not posted.
Accessibility: CADTH is committed to treating people with disabilities in a way that respects their dignity and independence, supports them in accessing material in a timely manner, and provides a robust feedback process to support continuous improvement. All materials prepared by CADTH are available in an accessible format. Where materials provided to CADTH by a submitting organization or individual are not available in an accessible format, CADTH will provide a summary document upon request. More details on CADTH’s accessibility policies can be found here.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.