Drugs, Health Technologies, Health Systems
Reimbursement Review
Lisocabtagene Maraleucel (Breyanzi)
Sponsor: Bristol Myers Squibb Canada
Therapeutic area: Relapsed or refractory large B-cell lymphoma
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Ethics Report
Clinical Review
Abbreviations
AE
adverse event
ALC
absolute lymphocyte count
ASCT
autologous stem cell transplant
axi-cel
axicabtagene ciloleucel
CAR
chimeric antigen receptor
CD
cluster of differentiation
CDA-AMC
Canada’s Drug Agency
CI
confidence interval
CNS
central nervous system
COO
cell of origin
CRR
complete response rate
CRS
cytokine release syndrome
DA-EPOCH-R
dose-adjusted etoposide, prednisone, vincristine sulfate (Oncovin), cyclophosphamide, and doxorubicin hydrochloride (hydroxydaunomycin) plus rituximab
DLBCL
diffuse large B-cell lymphoma
ECOG
Eastern Cooperative Oncology Group
EFS
event-free survival
EORTC QLQ-C30
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30
ESS
effective sample size
FACT-Lym
Functional Assessment of Cancer Therapy–Lymphoma
FACT-LymS
Functional Assessment of Cancer Therapy–Lymphoma “additional concerns” subscale
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HGBCL
high-grade B-cell lymphoma
HR
hazard ratio
HRQoL
health-related quality of life
HSCT
hematopoietic stem cell transplant
IHC
immunohistochemistry
IPD
individual patient data
IQR
interquartile range
IRC
independent review committee
ITC
indirect treatment comparison
ITT
intention to treat
KM
Kaplan-Meier
LBCL
large B-cell lymphoma
LC
Lymphoma Canada
LDC
lymphodepleting chemotherapy
liso-cel
lisocabtagene maraleucel
LS
least squares
LVEF
left ventricular ejection fraction
MAIC
matching-adjusted indirect comparison
MID
minimal important difference
NE
not estimable
NHL
non-Hodgkin lymphoma
NOS
not otherwise specified
OS
overall survival
pERC
pan-Canadian Oncology Drug Review Expert Review Committee
PET
positron emission tomography
PFS
progression-free survival
PMBCL
primary mediastinal large B-cell lymphoma
R-CHOP
rituximab, cyclophosphamide, doxorubicin hydrochloride (hydroxydaunomycin), vincristine sulfate (Oncovin), and prednisone
R-DHAP
rituximab, dexamethasone, cytarabine, and cisplatin
R-GDP
rituximab, gemcitabine, dexamethasone, and cisplatin
R-ICE
rituximab, ifosfamide, carboplatin, and etoposide
RCT
randomized controlled trial
sAAIPI
secondary Age-Adjusted International Prognostic Index
SCT
stem cell transplant
SMD
standardized mean difference
SOC
standard of care
SPD
sum of the product diameters
TEAE
treatment-emergent adverse event
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Lisocabtagene maraleucel (liso-cel; Breyanzi), 60 × 106 to 120 × 106 CAR-positive viable T cells, presented as a cell suspension in patient-specific single-dose vials, for IV infusion (1-time infusion, non–weight-based dose) |
Sponsor | Bristol Myers Squibb Canada |
Indication | For the treatment of adult patients with DLBCL NOS, PMBCL, HGBCL, and DLBCL arising from follicular lymphoma, who have refractory disease to first-line chemoimmunotherapy or relapse within 12 months of first-line chemoimmunotherapy, and who are candidates for autologous HSCT. |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Standard review |
NOC date | September 25, 2024 |
CAR = chimeric antigen receptor; DLBCL = diffuse large B-cell lymphoma; HGBCL = high-grade B-cell lymphoma; HSCT = hematopoietic stem cell transplant; liso-cel = lisocabtagene maraleucel; NOC = Notice of Compliance; NOS = not otherwise specified; PMBCL = primary mediastinal large B-cell lymphoma.
Introduction
Non-Hodgkin lymphoma (NHL) is the most common type of blood cancer that originates from lymphocytes, a type of white blood cell crucial to the immune system, and represents 90% of all lymphomas.1 The Canadian Cancer Society estimated that, in 2024, 11,700 people in Canada would be diagnosed with NHL and 3,100 would die from NHL.2 The 2023 Canadian Cancer Statistics reported an age-standardized incidence rate of 24.0 per 100,000 people in Canada for NHL.3 Large B-cell lymphoma (LBCL), a diverse and aggressive NHL type, prominently features large lymphoid cells expressing B-cell antigens such as cluster of differentiation (CD) 19 and CD20.4 The most common subtype of LBCL is diffuse large B-cell lymphoma (DLBCL), accounting for 30% to 40% of NHL cases.5 The median age of diagnosis for DLBCL is the mid-60s, with men more commonly affected.5-7
Patients with DLBCL typically present with enlarged lymph nodes and systemic issues like fever, weight loss, and night sweats.8 Most individuals with DLBCL have a type that remains biologically and clinically heterogeneous for which there are no clear and accepted criteria for subclassification. This type is known as not otherwise specified (NOS) and constitutes 80% to 85% of all DLBCL cases.8,9 Other subtypes of DLBCL include primary mediastinal large B-cell lymphoma (PMBCL), a rare subtype of DLBCL5 that occurs in the thymus or in lymph nodes in the mediastinum (centre of the chest) and represents approximately 10% of all DLBCLs.10
The diagnostic process for LBCL comprises a complete physical exam,11 imaging, and laboratory testing.11 The diagnosis of LBCL is confirmed through an excisional or core biopsy.6 Once the biopsy is obtained, cytomorphology and subclassification are determined using immunohistochemistry (IHC) and/or flow cytometry.12,13 Confirmation of the cell of origin (COO) is then assessed using gene expression profiling, immunophenotyping, or IHC algorithms.11,13,14
Lisocabtagene maraleucel (liso-cel) is a genetically modified autologous cell immunotherapy targeting CD19. It specifically binds to CD19, a protein expressed on the surface of B-cell precursors and malignant B-cells in DLBCL and other lymphomas. By binding to CD19, liso-cel activates and proliferates the chimeric antigen receptor (CAR) T cells, resulting in the release of proinflammatory cytokines and cytotoxic agents that destroy the targeted cancer cells.
Liso-cel has been approved by Health Canada for the treatment of adult patients with DLBCL NOS, PMBCL, high-grade B-cell lymphoma (HGBCL), or DLBCL arising from follicular lymphoma that is refractory to first-line chemoimmunotherapy or relapses within 12 months of first-line chemoimmunotherapy and who are candidates for autologous hematopoietic stem cell transplant (HSCT). The sponsor reimbursement request is as per the indication.
Canada’s Drug Agency (CDA-AMC) has reviewed liso-cel previously for the treatment of adult patients with relapsed or refractory LBCL after 2 or more lines of systemic therapy, including DLBCL NOS, HGBCL, DLBCL arising from follicular lymphoma, PMBCL, and grade 3B follicular lymphoma after at least 2 prior therapies. The pan-Canadian Oncology Drug Review Expert Review Committee (pERC) recommended the medication be reimbursed with conditions (June 29, 2022).15
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of a 1-time infusion of liso-cel 60 × 106 to 120 × 106 CAR-positive viable T cells for the treatment of adults with relapsed or refractory LBCL.
Patient Group and Clinician Group Perspectives
The information in this section is a summary of input provided by the patient and clinician groups that responded to a call for input and from the clinical experts consulted by CDA-AMC for the purpose of this review.
Patient Input
One patient group, Lymphoma Canada (LC), responded to a call for patient input on the current review of liso-cel. LC conducted an anonymous online survey from March 18 to May 13, 2024. The survey included responses from 90 patients with LBCL, primarily in Canada. Of these, 23 patients had experience with liso-cel in the third line or later and 5 had experience with this therapy in the second line (2 males and 3 females aged 25 to 44 years).
The majority of LC survey respondents lived in Canada (66%), were aged 25 to 44 years (30%) or 35 to 44 years (21%), and many (38%) were diagnosed with DLBCL NOS. They reported significant physical impacts, including fatigue, enlarged lymph nodes, body aches, swelling, and night sweats. Psychosocial effects included stress, difficulty sleeping, fear of disease progression, trouble with daily activities, concentration problems, and depression. LC survey respondents indicated that these challenges severely impacted daily life, with many struggling to travel and manage work or family obligations.
LC survey respondents reported receiving 1 or 2 lines of treatment for LBCL, with satisfaction decreasing from first-line to third-line treatments. Common treatments included rituximab, cyclophosphamide, doxorubicin hydrochloride (hydroxydaunomycin), vincristine sulphate (Oncovin), and prednisone (R-CHOP); dose-adjusted etoposide, prednisone, vincristine sulphate (Oncovin), cyclophosphamide, and doxorubicin hydrochloride (hydroxydaunomycin), and rituximab (DA-EPOCH-R); radiation; and various salvage therapies. Patients reported that difficult side effects like fatigue, hair loss, and nausea significantly impacted quality of life. Patients reported that access to treatment was challenging for many, with barriers such as local availability and financial burdens from drug costs and travel expenses.
Most patients with LBCL in the LC survey stressed the need for more treatment options. They prioritized longer remission, survival, improved quality of life, and normalizing blood counts. Additionally, they indicated they were willing to tolerate short-term, nonsevere side effects for new treatments, emphasizing the desire for options with fewer side effects and effective disease control. Based on the input, 5 patients, including 1 residing in Canada, reported receiving liso-cel as second-line treatment and are currently in remission. The main side effects observed were decreased appetite, nausea or vomiting, and fever. All patients are experiencing positive outcomes and unanimously recommended liso-cel for relapsed or refractory LBCL.
Clinician Input
Input From the Clinical Experts Consulted by CDA-AMC
The clinical experts indicated that the treatment goal for fit patients with relapsed or refractory LBCL is cure and long-term survival. The experts noted that fit patients typically receive salvage platinum-containing chemotherapy as second-line treatment, followed by an autologous hematopoietic stem cell transplant (HSCT) if they are eligible and respond to salvage chemotherapy. The experts also noted that many patients are not eligible for stem cell transplant (SCT) due to age (e.g., older than 70 to 75 years), comorbidities (e.g., related to liver, pulmonary, or cardiac function), chemorefractory disease, or an inability to mobilize stem cells, and these criteria vary across treatment centres. The experts noted that patients whose disease relapses soon after treatment or does not respond to first-line therapy typically have chemotherapy-refractory disease and are unlikely to benefit from ASCT. As such, the experts indicated that the unmet needs of patients would be new treatments that would prevent progression, prolong overall survival (OS), and improve quality of life while exposing patients to reduced toxicity. The clinical experts agreed that liso-cel would be used in the second-line setting for patients with DLBCL that is refractory to or relapses within 12 months of the end of first-line therapy. Since axicabtagene ciloleucel (axi-cel) is approved for the same indication, liso-cel would be in direct competition with axi-cel. The experts believe it is important for more than 1 CAR T product to be available for this indication, given differences in product availability, manufacturing technique, and safety profile. The clinical experts noted that the patients most likely to benefit from second-line liso-cel would be those with characteristics similar to those of the patients in the TRANSFORM trial (e.g., refractory to or relapse within 12 months of first-line therapy with adequate performance status and organ function); patients would not be suitable for treatment with second-line liso-cel if they have later relapses. The clinical experts indicated there should be some leeway with age, Eastern Cooperative Oncology Group (ECOG) Performance Status, and organ function parameters. The clinical experts indicated that, in clinical practice, response rates on imaging beyond 30 to 90 days and clinical symptoms are used to determine whether a patient’s disease is responding or progressing on treatment. The clinical experts indicated that patients receiving liso-cel should be under the care of a clinician (e.g., hematologist or medical oncologist) who can manage the toxicity associated with the therapy within centres that have cellular therapy experience. The experts also noted that patients should have access to an intensive care unit in case of rare high-grade toxicities and to consultative support from an infectious disease specialist or neurologist, if needed.
Clinician Group Input
Three clinician groups, LC (3 clinicians contributed to the input), Ontario Health, Cancer Care Ontario (OH-CCO) Hematology Cancer Drug Advisory Committee (7 clinicians contributed to the input), and Leukemia & Lymphoma Society of Canada Nurses Network (5 clinicians contributed to the input) responded to a call for clinician group input. Overall, the input aligned with that provided by the clinical experts consulted by CDA-AMC.
The clinician groups stated that the primary treatment goals are prolonging life, slowing disease progression, and enhancing quality of life for patients with DLBCL, aiming to minimize the need for additional treatments and toxic chemotherapy. As per the clinician groups, available treatments for DLBCL are effective for some patients, but there are unmet needs, particularly for high-risk patients such as those with primary refractory disease or early relapse. Second-line chemoimmunotherapy and autologous HSCT are successful in a subset of patients, but only about 20% achieve durable remission with this approach. CAR T-cell therapy in the second line could address this gap by offering more effective treatment earlier in the disease course, potentially leading to more cures and reducing the need for other salvage strategies.
According to the clinician groups, liso-cel is best suited over salvage chemotherapy and autologous HSCT for those with high-risk disease. Patients with a low tumour burden or other DLBCL subtypes may also benefit. Fitness for treatment will be determined by primary hematologists or oncologists based on institutional guidelines, considering factors like performance status and organ function. Clinical practice and trials utilize various outcomes, including overall response rate and complete response rates (CRR), progression-free survival (PFS), and OS, employing the Lugano criteria for remission confirmation. Response assessment involves restaging CT or PET scans at 1, 3, and 6 months post infusion, with patients sustaining responses beyond 6 to 12 months typically experiencing long-lasting remissions.
The clinician groups agreed that liso-cel is administered as a single infusion; the concept of discontinuation does not typically apply in the context of CAR T treatment. Liso-cel should only be administered in established CAR T-cell therapy programs approved to deliver this therapy.
Drug Program Input
Input was obtained from the drug programs that participate in the CDA-AMC Reimbursement Review process. The following were identified as key factors that could potentially impact the implementation of a CDA-AMC recommendation for liso-cel:
relevant comparators
consideration for initiation of therapy
considerations for prescribing of therapy
generalizability.
The clinical expert consulted by CDA-AMC provided advice on the potential implementation issues raised by the drug programs. Refer to Table 4.
Clinical Evidence
Systematic Review
Description of Studies
One trial, TRANSFORM (N = 184), met the inclusion criteria for the systematic review conducted by the sponsor. The objective of TRANSFORM is to assess the efficacy and safety of a 1-time IV infusion of liso-cel 100 × 106 CAR T cells compared with standard of care (SOC), defined as 3 cycles with 1 of 3 prespecified salvage immunochemotherapy regimens followed, depending on response, by 1 cycle of high-dose chemotherapy and an autologous HSCT in adult patients with relapsed or refractory LBCL. During liso-cel manufacturing, patients in the liso-cel group could receive bridging therapy with 1 of the 3 defined salvage immunochemotherapy regimens allowed in the SOC group, if needed. The trial enrolled patients who had LBCL that was refractory to or relapsed within 12 months after initial response to first-line therapy (including an anthracycline and an anti-CD20 monoclonal antibody), were considered candidates for autologous HSCT, and had an ECOG score of 1 or less. The approved Health Canada indication and reimbursement request aligned with the trial population. The outcomes most relevant to the CDA-AMC review included the primary outcome of event-free survival (EFS) per independent review committee (IRC), and secondary outcomes of CRR, PFS, OS, health-related quality of life (HRQoL) and safety. The HRQoL outcomes included the European Organisation for the Research and Treatment of Cancer Quality of Life Questionnaire Core 30 (EORTC QLQ-C30) global health status score and Functional Assessment of Cancer Therapy–Lymphoma (FACT-Lym) “additional concerns” subscale (FACT-LymS) total score. The trial population was predominately white (approximately 59%) and male (57%), with a median age of 59 years (range, 20 to 75 years). Most patients had an ECOG Performance Status of 0 (52%) or 1 (47%), indicating good overall performance; a secondary Age-Adjusted International Prognostic Index (sAAIPI) of 0 or 1 (60%); had disease that had relapsed or was refractory to their last therapy (26% and 74%, respectively); and had LBCL subtype DLBCL NOS (56%), followed by HGBCL (23%), PMBCL (9%), or DLBCL from transformed indolent lymphoma (8%). Compared with the SOC group, the liso-cel group had a lower proportion of male patients (47.8% versus 66.3%) and a higher proportion of patients with chemorefractory disease (28.3% versus 19.6%).
Efficacy Results
Only those efficacy outcomes and analyses of subgroups identified as important to this review are reported. Efficacy and safety data were evaluated at the second interim analysis (data cut-off date of March 8, 2021), the primary analysis (data cut-off date of May 13, 2022), and the final analysis (data cut-off date of October 23, 2023). The primary efficacy outcome of EFS was met at the second interim analysis and was presented descriptively (i.e., not included in the hierarchical testing strategy) in the primary analysis. Therefore, hypothesis testing on the key secondary outcomes of CRR, PFS, and OS was performed hierarchically in the primary analysis.
Event-Free Survival
At the time of the second interim analysis, the median duration of follow-up for all patients was 6.2 months (interquartile range [IQR], 4.4 to 11.5 months), and EFS events had been reported for 35 patients (38.0%) in the liso-cel group and 63 patients (68.5%) in the SOC group. The median EFS was 10.1 months (95% confidence interval [CI], 6.1 months to not estimable [NE]) in the liso-cel group versus 2.3 months (95% CI, 2.2 to 4.3 months) in the SOC group (1-sided P value < 0.0001), with a between-group hazard ratio (HR) of 0.35 (95% CI, 0.23 to 0.53). The results of sensitivity analyses were consistent with the primary analysis. The Kaplan-Meier (KM)–estimated probability of EFS at 12 months was 44.5% (95% CI, 29.4% to 59.6%) for the liso-cel group versus 23.7% (95% CI, 13.4% to 34.1%) for the SOC group, with a between-group difference of 20.8% (95% CI, 2.5% to 39.1%).
At the time of the primary efficacy analysis, the median duration of follow-up for all patients was 17.5 months (IQR, 0.9 to 37 months), and EFS events had been reported in 44 patients (47.8%) in the liso-cel group and 71 patients (77.2%) in the SOC group. The median EFS was not reached (EFS = NE; 95% CI, 9.5 to NE) in the liso-cel group versus 2.4 months (95% CI, 2.2 to 4.9 months) in the SOC group (HR = 0.36; 95% CI, 0.24 to 0.52). The KM-estimated probability of EFS at 12 months was consistent with the second interim analysis, with a between-group difference of 34.6% (95% CI, 21.2% to 48.0%).
At the final analysis, the median duration of follow-up of 33.9 months (IQR, 11.6 to 39.2 months) and EFS events had been reported in ██ ███████ patients in the liso-cel group and █████ patients in the SOC group. The median EFS was 29.5 months (95% CI, 9.5 months to NE) in the liso-cel group and 2.4 months (95% CI, 2.2 to 4.9 months) in the SOC group, with a between-group HR of 0.38 (95% CI, 0.259 to 0.542). The KM-estimated probability of EFS at 12 months was consistent with the interim and primary analyses and, at 36 months, was 45.8% (95% CI, 35.2% to 56.5%) for the liso-cel group versus 19.1% (95% CI, 11.0% to 27.3%) for the SOC group, with a between-group difference of 26.7% (95% CI, 13.3% to 40.1%).
At the second interim analysis, the efficacy results for EFS were consistent across the subgroup analyses by histological subtype, use of bridging therapy (data not shown), and prior response status in favour of liso-cel. In general, the results of the subgroup analyses were consistent across all data cut-offs.
Complete Response Rate
At the time of the primary analysis, the CRR in the liso-cel group was 73.9% (95% CI, 63.7% to 82.5%) versus 43.5% (95% CI, 33.2% to 54.2%; stratified 1-sided P < 0.0001), with a between-group difference of 29.3% (95% CI, 16.4% to 42.2%). The CRR remained consistent at the final analysis. The results of the sensitivity analyses were consistent with the primary analysis.
Progression-Free Survival
At the time of the primary analysis, PFS events had been reported for 37 patients (40.2%) in the liso-cel group and 52 patients (56.5%) in the SOC group. The median PFS was not reached (95% CI, 12.6 to NE) in the liso-cel group versus 6.2 months (95% CI, 4.3 to 8.6 months) in the SOC group (1-sided P < 0.0001), with a between-group HR of 0.40 (95% CI, 0.26 to 0.62). The results of sensitivity analyses were consistent with the primary analysis. The KM-estimated probability of PFS at 12 months was 63.1% (95% CI, 53.0% to 73.3%) in the liso-cel group versus 31.2% (95% CI, 20.2% to 42.3%) in the SOC group, with a between-group difference of 31.9% (95% CI, 16.9% to 46.9%).
At the time of the final analysis, PFS events had been reported in ██ ███████ patients in the liso-cel group and ██ ███████ patients in the SOC group. The median PFS was not reached (95% CI, 12.6 to NE) in the liso-cel group and 6.2 months (95% CI, 4.3 to 8.6 months) in the SOC group. The KM-estimated probability of PFS at 12 months was consistent with the primary analysis and, at 36 months, was 50.9% (95% CI, 39.9% to 62.0%) for the liso-cel group versus 26.5% (95% CI, 15.9% to 37.1%) in the SOC group, with a between-group difference of 24.4% (95% CI, 9.1% to 39.7%).
Overall Survival
By the primary analysis, there were 28 ███████ deaths in the liso-cel group and 38 ███████ deaths in the SOC group. The median OS was not reached (95% CI, 29.5 to NE) in the liso-cel group versus 29.9 months (95% CI, 17.9 to NE) months in the SOC group (1-sided P = 0.0987), with a between-group HR of 0.72 (95% CI, 0.44 to 1.18). The KM-estimated probability of being alive at 12 months was 83.4% (95% CI, 75.7% to 91.1%) in the liso-cel group versus 72.0% (95% CI, 62.7% to 81.3%) in the SOC group, with a between-group difference of 11.4% (95% CI, −0.7% to 23.5%).
At the time of final analysis, there were 34 ███████ deaths in the liso-cel group and 42 ███████ deaths in the SOC group. The median OS was not reached for either treatment group (liso-cel: 95% CI, 42.8 to NE; SOC: 95% CI, 18.2 to NE), with a between-group HR of 0.76 (95% CI, 0.481 to 1.19). The KM-estimated probability of OS at 12 months was consistent with the primary analysis and, at 36 months, was 62.8% (95% CI, 52.7% to 72.9%) in the liso-cel group versus 51.8% (95% CI, 41.2% to 62.4%) in the SOC group, with a between-group difference of 11.0% (95% CI, −3.7% to 25.7%)
Health-Related Quality of Life
At baseline, EORTC QLQ-C30 global health status scores were similar between treatment groups and there were clinically meaningful changes observed (defined by the sponsor as a change of ≥ 5 points in the score from baseline) in both groups at 6 months. The between-group least squares (LS) mean difference in change from baseline was ████ ████ ███ █████ ██ ██████. At baseline, total FACT-LymS scores were similar between groups. At 6 months, there was no clinically meaningful change (defined by the sponsor as a change of ≥ 3 points in the score from baseline) observed in the liso-cel group, while a clinically meaningful change was observed in the SOC group. The between-group LS mean difference in change from baseline was █████ ████ ███ █████ ██ █████.
Harms Results
Harms data reported in this section are from the primary analysis (data cut-off date of May 13, 2022). There were no significant changes in the incidence of treatment-emergent adverse events (TEAEs) from the time of the interim analysis to the time of the primary analysis. Almost all patients in the trial reported at least 1 TEAE (liso-cel: 100%; SOC: 98.9%). The most frequently reported TEAEs of any grade in both treatment groups were neutropenia (liso-cel: 82.6%; SOC: 54.9%), anemia (liso-cel: 67.4%; SOC: 68.1%), thrombocytopenia (liso-cel: 59.8%; SOC: 72.5%), and nausea (liso-cel: 53.3%; SOC: 58.2%). Of these TEAEs, a numerically higher proportion of neutropenia was reported in patients taking liso-cel, while a higher proportion of thrombocytopenia was reported in patients taking SOC. Most patients in both groups reported at least 1 grade 3 or 4 TEAE (liso-cel: 92.4%; SOC: 89.0%). The incidence of grade 3 or 4 neutropenia (liso-cel: 81.5%; SOC: 51.6%) and lymphopenia (liso-cel: 26.1%; SOC: 9.9%) was numerically higher in the liso-cel group versus the SOC group. The incidence of serious TEAEs was similar between groups (liso-cel: 47.8%; SOC: 48.4%). The most frequently reported serious TEAEs were cytokine release syndrome (CRS) (liso-cel: 13%; SOC: 0%), febrile neutropenia (liso-cel: 7.6%; SOC: 9.9%), pyrexia (liso-cel: 6.5%; SOC: 7.7%), and neutropenia (liso-cel: 7.6%; SOC: 4.4%). The frequency of these TEAEs was similar between groups, except a higher proportion of CRS was reported in patients taking liso-cel. Four patients (4.4%) in the SOC group experienced TEAEs leading to treatment withdrawal. No patients in the liso-cel group had a TEAE that led to the withdrawal of the study drug (including bridging therapy and lymphodepleting chemotherapy [LDC]). Deaths were reported in 14.1% of patients in the liso-cel group and 8.8% of patients in the SOC group. The majority of deaths in both groups were attributed to disease progression (liso-cel: 7.6%; SOC: 4.4%), followed by TEAEs (liso-cel: 2.2%; SOC: 4.4%). A numerically higher proportion of notable TEAEs were reported in patients taking liso-cel (90.2%) than SOC (75.8%). The most frequently reported notable harms of any grade were neurologic toxicity (liso-cel: 64.1%; SOC: 62.6%), CRS (liso-cel: 48.9%; SOC: 0.0%), prolonged cytopenia (liso-cel: 43.5%; SOC: 3.3%), and investigator-identified neurologic toxicity (liso-cel: 10.9%; SOC: not applicable). These events occurred more frequently in patients taking liso-cel, except for neurologic toxicity, which was similar in both groups.
Critical Appraisal
The TRANSFORM trial randomization procedures, including stratification by response to first-line therapy (relapsed versus refractory) and sAAIPI (0 to 1 versus 2 to 3) were appropriate and conducted by interactive response technology. The liso-cel group had a lower proportion of male patients (47.8% versus 66.3%), and a higher proportion of patients with chemorefractory disease (28.3% versus 19.6%) compared with the SOC group. According to the clinical experts consulted by CDA-AMC, it was unlikely that these imbalances confounded the effect between treatment and outcomes. Treatment period discontinuation was numerically higher in the SOC group (59.8%) versus the liso-cel group (12.0%), with lack of efficacy being the most common reason (SOC: 30%; liso-cel = 0%). The open-label design introduces a potential bias in the assessment of efficacy for EFS, CRR, and PFS, as well as a potential reporting bias of the subjective outcomes of HRQoL and safety, although, this bias was mitigated by use of an IRC for EFS, CRR, and PFS. To minimize the risk of differential measurement error, the trial performed tumour assessments using the Lugano criteria and radiographic scans were assessed by IRC. For the HRQoL and safety outcomes, the source of bias could overestimate the efficacy of liso-cel. Sample size and power calculations were based on EFS, and the trial was powered to detect significant differences between groups for EFS. The prespecified analysis of EFS, CRR, PFS, and OS were appropriately controlled for multiple comparisons; all other analyses were descriptive. This included the HRQoL outcomes EORTC QLQ-C30 and FACT-LymS, which were deemed clinically important outcomes for the disease. The sample size for most subgroup analyses of interest appeared large enough to detect subgroup differences for EFS, except for DLBCL transformed from indolent NHL, which may not have been powered to detect subgroup differences. The findings of the sensitivity analyses for the primary outcome of EFS were consistent with the primary analysis. The proportional hazards assumption was assessed via inspection of Schoenfeld residuals, and the trial authors stated there was no evidence of a violation of this assumption. The median OS was not reached in either treatment group at the primary and final analyses due to the small number of OS events. As such, longer follow-up is needed to inform the true effect of liso-cel on survival compared with SOC. In addition, patients were permitted to receive posttreatment anticancer medications after study treatment had been discontinued (liso-cel: 34.8%; SOC: 70.7% [65 patients]), of which 61 patients were approved to cross over to liso-cel. The proportion of patients permitted to receive posttreatment anticancer medications was not balanced between groups and may have influenced the assessment of OS due to crossover bias. The certainty of evidence for the HRQoL outcomes was limited because of the risk of bias due to missing outcomes data, both at baseline and at the selected follow-up times, and imprecision due to the 95% CI for the between-group difference including the possibility of both benefit and little to no difference. However, the direction and extent of bias is unclear and, as such, the potential differences on patients’ HRQoL remains very uncertain.
The population requested for reimbursement aligns with the approved Health Canada indication. The dosing and administration of liso-cel was consistent with the approved product monograph. According to the clinical experts consulted by CDA-AMC, the eligibility criteria and baseline characteristics of the TRANSFORM trial were generalizable to adults with R/R LBCL in the Canadian setting, although, the trial did not include patients with a poor ECOG Performance Status. The clinical experts noted that enrolling patients with an ECOG score of only 0 and 1 is not entirely representative of patients with R/R LBCL in Canada, as they expect to have patients with higher ECOG scores in their practice. The clinical experts also noted that autologous HSCT eligibility is highly variable in clinical practice across Canada, as there are no standardized criteria to identify patients. The trial included outcomes that were important to patients and clinicians. The patient group indicated that stopping disease progression, prolonging life, improving HRQoL, and reducing treatment side effects are important to them.
GRADE Summary of Findings and Certainty of the Evidence
For pivotal studies and randomized controlled trials (RCTs) identified in the sponsor’s systematic review, Grading of Recommendations Assessment, Development and Evaluation (GRADE) was used to assess the certainty of the evidence for outcomes considered most relevant to inform CDA-AMC’s expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group.
Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty-of-evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null.
The reference points for the certainty-of-evidence assessment for EFS, CRR, PFS, OS and EORTC QLQ-C30 global health status score were set according to the presence or absence of an important effect based on thresholds informed by the clinical experts consulted for this review, and ranges identified in the literature for the EORTC QLQ-C30. The reference point for the certainty-of-evidence assessment for the FACT-LymS total score was set according to the presence or absence of an important effect based on a threshold suggested by the sponsor that was informed by the literature.
The selection of outcomes for the GRADE assessment was based on the sponsor’s summary of clinical evidence, consultation with clinical experts, and the input received from patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members:
survival outcomes (EFS, PFS, and OS)
complete response
HRQoL outcomes (EORTC QLQ-C30 global health status and FACT-LymS scores).
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for liso-cel versus SOC.
Long-Term Extension Studies
No long-term extension studies were submitted by the sponsor.
Indirect Comparisons
One sponsor-submitted matching-adjusted indirect comparison (MAIC)17 was submitted by the sponsor to fill gaps in the comparative evidence for other treatments of interest for relapsed or refractory LBCL, and 1 indirect treatment comparison (ITC) using the Bucher approach was conducted to inform the pharmacoeconomic model. The MAIC is the focus of this report. The authors did not report a systematic literature search or describe the methods for study selection, data extraction, and quality assessment for both ITCs.
Table 2: Summary of Findings for Liso-Cel Versus SOC for Patients With Relapsed or Refractory Large B-Cell Lymphoma — TRANSFORM Trial
Outcome and follow-up | Patients (studies), N | Relative effect (95% CI) | Absolute effects (95% CI) | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
SOC | Liso-cel | Difference | |||||
EFS — ITT set, second interim analysis data cut-off date of March 8, 2021 | |||||||
Probability of EFS at 12 months. Median follow-up for all patients: 6.2 months. | 184 (1 RCT) | NA | 237 per 1,000 | 445 per 1,000 (294 to 596) | 208 per 1,000 (25 more to 391 more) | Higha | Liso-cel results in a clinically important increase in the probability of EFS at 12 months when compared with SOC. |
EFS — ITT set, final analysis data cut-off date of October 23, 2023 | |||||||
Probability of EFS at 36 months. Median follow-up for all patients: 33.9 months. | 184 (1 RCT) | NA | 191 per 1,000 | 458 per 1,000 (352 to 565) | 267 per 1,000 (133 more to 401 more) | Higha | Liso-cel results in a clinically important increase in the probability of EFS at 36 months when compared with SOC. |
CRR — ITT set, primary analysis data cut-off date of May 13, 2022 | |||||||
Complete response rate. Median follow-up for all patients: 17.5 months. | 184 (1 RCT) | NR | 435 per 1,000 | 739 per 1,000 (637 to 825) | 293 more per 1,000 (164 more to 422 more) | Highb | Liso-cel results in a clinically important increase in the proportion of patients who experience a complete response when compared with SOC. |
PFS — ITT set, primary analysis data cut-off date of May 13, 2022 | |||||||
Probability of PFS at 12 months. Median follow-up for all patients: 17.5 months. | 184 (1 RCT) | NA | 312 per 1,000 | 631 per 1,000 (530 to 733) | 319 per 1,000 (169 more to 469 more) | Higha | Liso-cel results in a clinically important increase in the probability of PFS at 12 months when compared with SOC. |
PFS — ITT set, final analysis data cut-off date of October 23, 2023 | |||||||
Probability of PFS at 36 months. Median follow-up for all patients: 33.9 months. | 184 (1 RCT) | NA | 265 per 1,000 | 509 per 1,000 (399 to 620) | 244 per 1,000 (91 more to 397 more) | Moderatec | Liso-cel likely results in a clinically important increase in the probability of PFS at 36 months when compared with SOC. |
OS — ITT set, primary analysis data cut-off date of May 13, 2022 | |||||||
Probability of survival at 12 months. Median follow-up for all patients: 17.5 months. | 184 (1 RCT) | NA | 720 per 1,000 | 834 per 1,000 (757 to 911) | 114 per 1,000 (7 fewer to 235 more) | Moderated | Liso-cel likely results in a clinically important increase in the probability of survival at 12 months when compared with SOC. |
OS — ITT set, final analysis data cut-off date of October 23, 2023 | |||||||
Probability of survival at 36 months. Median follow-up for all patients: 33.9 months. | 184 (1 RCT) | NA | 518 per 1,000 | 628 per 1,000 (527 to 729) | 110 per 1,000 (37 fewer to 257 more) | Moderated | Liso-cel likely results in a clinically important increase in the probability of survival at 36 months when compared with SOC. |
EORTC QLQ-C30 global health status score — HRQoL set, primary analysis data cut-off date of May 13, 2022 | |||||||
LS mean change from baseline in global health status; scores range from 0 to 100, with higher scores indicating better health status. Time point: 6 months. | 36 (1 RCT) | NA | █████ | ████ ███████ ██ █████ | ████ ███████ ██ ██████ | Very lowe | The evidence is very uncertain about the effect of liso-cel on global health status at 6 months when compared with SOC. |
FACT-LymS total score — HRQoL set, primary analysis data cut-off date of May 13, 2022 | |||||||
LS mean change from baseline in symptoms score; scores range from 0 to 60, with higher scores indicating lower levels of symptoms. Time point: 6 months. | 37 (1 RCT) | NA | ████ | ████ ███████ ██ █████ | █████ ███████ ██ █████ | Very lowf | The evidence is very uncertain about the effect of liso-cel on symptoms at 6 months when compared with SOC. |
CI = confidence interval; CDA-AMC = Canada’s Drug Agency; CRR = complete response rate; EFS = event-free survival; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; FACT-LymS = Functional Assessment of Cancer Therapy–Lymphoma “additional concerns” subscale; GRADE = Grading of Recommendations Assessment, Development and Evaluation; HRQoL = health-related quality of life; ITT = intention to treat; liso-cel = lisocabtagene maraleucel; LS = least squares; NA = not applicable; OS = overall survival; PFS = progression-free survival; RCT = randomized controlled trial; SOC = standard of care.
Note: Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes. The between-group absolute effects for EFS, CRR, PFS, and OS at the time points included in the table were requested by CDA-AMC from the sponsor to facilitate the GRADE assessment.
aA between-group absolute risk difference of 15% and 10% (150 and 100 fewer or more events per 1,000 patients) at 12 and 36 months, respectively, was clinically important, according to the clinical experts. The point estimate and entire CI exceeded the threshold.
bA between-group absolute risk difference of 15% (150 fewer or more events per 1,000 patients) was clinically important, according to the clinical experts. The point estimate and entire CI exceeded the threshold.
cRated down 1 level for serious imprecision due to the 95% CI for the between-group absolute risk difference included the possibility of both an important benefit and a trivial effect when compared with SOC; a between-group absolute risk difference of 10% (100 fewer or more events per 1,000 patients) at 36 months was clinically important, according to the clinical experts.
dRated down 1 level for serious imprecision due to the 95% CI for the between-group absolute risk difference included the possibility of both an important benefit and little to no difference when compared with SOC; a between-group absolute risk difference of 10% and 5% (100 and 50 fewer or more events per 1,000 patients) at 12 and 36 months, respectively, was clinically important, according to the clinical experts.
eRated down 1 level for serious imprecision due to the 95% CI for the between-group difference included the possibility of both benefit and little to no difference when compared with SOC; based on the ranges identified in the literature and suggested by the clinical experts, a 10-point change from baseline in EORTC QLQ-C30 scale score was considered clinically important. Rated down 2 levels for risk of bias due to missing outcome data.
fRated down 1 level for serious imprecision due to the 95% CI for the between-group difference included the possibility of both harm and little to no difference when compared with SOC; based on the sponsor’s suggestion that was informed by ranges identified in the literature, a 3-point change from baseline in FACT-LymS total score was considered clinically important. Rated down 2 levels for risk of bias due to missing outcome data.
Sources: TRANSFORM Clinical Study Report.16 Details included in the table are from the sponsor’s summary of clinical evidence and the sponsor’s response to a request for additional information.
Description of Studies
A feasibility assessment using study design, eligibility criteria, baseline characteristics, and outcomes was performed and determined the TRANSFORM and ZUMA-7 trials to be comparable enough to allow for the indirect comparison between liso-cel and axi-cel on key efficacy and safety outcomes through unadjusted (Bucher) or population-adjusted ITC methods. MAIC and simulated treatment comparison approaches were considered feasible to minimize potential sources of bias while comparing these therapies, and the MAIC approach was preferred by the investigators over simulated treatment comparison. The MAIC assessed the comparative efficacy and safety of liso-cel and axi-cel using individual patient data (IPD) from the TRANSFORM trial (for liso-cel) and summary-level data from the ZUMA-7 trial in patients with relapsed or refractory LBCL who were intended for transplant. Outcome of interests included EFS, CRRs, PFS, OS, and TEAEs. The IPD from the TRANSFORM trial were adjusted to match the marginal distribution (e.g., mean, variance) of clinical factors among patients from the ZUMA-7 trial. For EFS, PFS, and OS, an anchored MAIC was performed using Cox proportional hazards models to estimate HRs for liso-cel versus axi-cel. Generalized linear models for CRR were used to estimate ORs. For the unanchored analysis of safety outcomes, weighted log odds for liso-cel were estimated in the TRANSFORM trial by fitting an intercept-only logistic regression model with MAIC adjustment weights. Point estimates (HRs or ORs) and 95% CIs were reported for all analyses. The Bucher ITC used a mixture cure modelling framework to derive relative efficacy estimates for EFS and OS, which were reported as ORs and 95% CIs.
Efficacy Results
Based on the anchored MAIC, the results did not support a clinically meaningful difference between liso-cel and axi-cel for EFS, CRR, PFS, or OS. The Bucher ITC results for EFS and OS were consistent with the MAIC findings.
Harms Results
Based on the unanchored MAIC, the results showed no difference between liso-cel and axi-cel for grade 3 or greater TEAE events, prolonged cytopenia, severe infections, and hypogammaglobulinemia. The results for CRS, neurologic toxicity, pyrexia, and encephalopathy favoured liso-cel.
Critical Appraisal
For the MAIC and Bucher ITC, the authors did not report a systematic literature search, describe their methods for data extraction, or conduct a quality assessment of the TRANSFORM and ZUMA-7 trials. The MAIC included relevant outcomes identified by the CDA-AMC team (EFS, CRR, PFS, OS, and safety). However, because data for several safety outcomes (e.g., CRS) were very limited or not available for the SOC arm in both the TRANSFORM and ZUMA-7 trials, an anchored MAIC was not feasible. Therefore, an unanchored MAIC was conducted for the safety outcomes. The Bucher ITC assessed only EFS and OS. For the MAIC, to account for between-study differences in patient baseline characteristics, several potentially relevant clinical factors (i.e., treatment effect modifiers) were matched in the weighting process. The methods used to identify and rank the clinical factors were considered appropriate. The process involved a systematic literature review and a panel of 3 clinical experts to validate the selection and ranking of the treatment effect modifiers based on their strength to influence the specific outcomes under study for patients with relapsed or refractory LBCL. For the anchored MAIC efficacy analysis, 10 clinical factors were adjusted for and sensitivity scenarios, which included all relevant clinical factors available (total = 14), were conducted to test robustness of the findings. After matching and adjusting for the 11 factors in the primary efficacy analyses, imbalances remained for ECOG Performance Status and COO, although the clinical experts consulted by CDA-AMC did not think these imbalances could bias the results. The authors noted that although the definition of EFS was similar between trials, some EFS events between randomization and treatment were treated differently. Among randomized patients who did not receive treatment in the ZUMA-7 trial, the majority were assigned an event immediately (i.e., at time 0 or day 1 of the KM curves) for the intention-to treat (ITT) analysis given commencement of new lymphoma therapy and lack of evaluable disease assessment. This was considered a potential source of bias favouring axi-cel. Whereas the date of imaging that served as the basis of starting new antineoplastic therapy was used in the TRANSFORM trial, rather than day 1, which was used in the ZUMA-7 trial. Overall, the magnitude and direction of potential bias due to imbalances in the efficacy estimates could not be predicted. Among the 17 identified clinical factors for the unanchored MAIC safety analyses, 2 factors (prior HSCT and number of prior lines of therapy) were excluded because they were not relevant to the second line–approved indication, 6 factors (bulky disease, metabolic tumour volume, serum albumin, interleukin-6, fibrinogen level, and C-reactive protein) were not considered due to lack of reporting in the ZUMA-7 trial, and lactate dehydrogenase at baseline and bridging therapy were excluded due to differences in definitions between the TRANSFORM and ZUMA-7 trials. ECOG Performance Status was also not included in the MAIC, which was considered by the clinical experts consulted by CDA-AMC to be an important potential effect modifier. In addition, the TEAE reporting window differed between trials. Events from bridging therapy were included only for the TRANSFORM trial, thus potentially biasing the safety comparison against liso-cel. Following the weighting process, the effective sample size (ESS) for all the efficacy outcomes declined by more than 50% from the size of the original sample in comparison with axi-cel. This percent declined further for the adjusted safety analyses and sensitivity analyses, which included all relevant clinical factors available. These reductions in the ESS meant the final matched patient population was highly selective when compared with the original patient population and may have led to uncertainty in the estimated treatment effects. Since there were no major generalizability issues in the axi-cel population compared with the liso-cel population, the concern for bias due to influential subgroups is less of a concern. Overall, the relative efficacy and safety estimates were subject to uncertainty due to imprecision, and the unanchored safety analysis was also subject to imbalances in potential effect modifiers and prognostic factors.
The Bucher ITC, which was used to inform the economic model, showed similar results to the MAIC for EFS and OS. The main limitation of this approach was that the ITC estimates did not adjust for between-study differences in patient baseline characteristics. Since there were notable baseline differences between the TRANSFORM and ZUMA-7 trials, as described in the MAIC approach, drawing definitive conclusions based on the Bucher results is not recommended.
Studies Addressing Gaps in the Evidence From the Systematic Review
One study that was submitted by the sponsor was excluded because it did not match the patient population of the approved Health Canada indication.
Conclusions
Evidence from 1 phase III open-label RCT (TRANSFORM) reported on outcomes that were important to both patients and clinicians. The trial showed a high certainty of evidence that treatment with liso-cel results in a clinically meaningful increase in EFS at 12 months and in CRR at 36 months compared with SOC in adults with relapsed or refractory LBCL. The trial also showed a high certainty of evidence at 12 months and a moderate certainty of evidence at 36 months that liso-cel results in a clinically meaningful increase in PFS. At the time of the final analysis, median OS had not been reached in either group, and no definitive conclusions could be drawn for HRQoL due to concerns of imprecision and missing outcome data. There were no new safety signals identified and the safety of liso-cel was consistent with the known safety profile of the drug. The results of the ITC did not support a clinically meaningful difference between liso-cel and axi-cel for EFS, CRR, PFS, or OS, but were suggestive of a more favourable safety profile for liso-cel; however, these estimates were subject to uncertainty due to imprecision and imbalances in potential effect modifiers and prognostic factors.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of liso-cel (Breyanzi) as 1-time infusion of CD8 and CD4 CAR T cells at a total target dose of 100 × 106 CAR T cells for the treatment of adults with relapsed or refractory LBCL.
Disease Background
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the CDA-AMC review team.
NHL is the most common type of blood cancer that comprises a range of closely related cancers that originates from lymphocytes and represents 90% of all lymphomas.1 The Canadian Cancer Society estimated that, in 2024, 11,700 people in Canada would have been diagnosed with NHL (6,600 men and 5,100 women) and 3,100 would have died from NHL (1,800 men and 1,300 women).3 The 2023 Canadian Cancer Statistics reported an age-standardized incidence rate of 24.0 per 100,000 Canadians for NHL.3 LBCL is a diverse and aggressive type of NHL. This malignancy is marked by the presence of large lymphoid cells that are significantly larger than macrophages or at least twice the size of normal lymphocytes. The most common subtype of LBCL is DLBCL, accounting for 30% to 40% of NHL cases.5 DLBCL is a heterogenous group of aggressive B-cell malignancies. The median age at diagnosis of DLBCL is in the mid-60s; 30% of patients are older than 75 years of age.6,7 Men are more likely to develop the disease than women.5 Patients with DLBCL typically present with enlarged symptomatic masses in the lymph nodes, typically in the neck, chest, or abdomen. However, widespread DLBCL can also arise in tissues outside the lymph nodes (i.e., extranodal involvement) in the bone marrow, bones, brain, and gastrointestinal tract, among others. DLBCL can also cause systemic B symptoms (i.e., unexplained fever, weight loss, night sweats).8
Most individuals with DLBCL (80% to 85%) have a type that remains biologically and clinically heterogeneous, for which there are no clear and accepted criteria for subclassification, that is known as DLBCL NOS.8,9 PMBCL is a rare subtype of DLBCL that occurs in the thymus or in lymph nodes in the mediastinum (centre of the chest) and represents approximately 10% of all DLBCLs.10 Both DLBCL NOS and PMBCL have a similar course and treatment regimen.18 DLBCL transformed from follicular lymphoma or DLBCL arising from indolent lymphoma are additional DLBCL subtypes that are all initially slow-growing types of B-cell lymphomas that transform into DLBCL.5
The diagnostic process for LBCL comprises a complete physical exam, including screening for B symptoms and assessing the size of the liver and spleen.11 Laboratory testing is also completed during this initial evaluation.11 The diagnosis of LBCL is confirmed through an excisional biopsy.6 Once the biopsy is obtained, cytomorphology and subclassification are determined using IHC and/or flow cytometry.12,13 Confirmation of the COO is then assessed using gene expression profiling, immunophenotyping, or IHC algorithms according to WHO classification, identifying whether it is germinal centre B cell–like (GCB) or non-GCB.11,13,14 Cytogenetic fluorescence in situ hybridization testing may also be conducted to determine COO molecular classification and to check for MYC, BCL2, and/or BCL6 rearrangements.19,20 Staging and response assessment should follow the Ann Arbor staging and Lugano classification criteria.6
Standards of Therapy
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following was summarized and validated by the CDA-AMC review team.
Treatment priorities for patients with relapsed or refractory LBCL are curing disease when possible, prolonging survival, improving HRQoL, and minimizing treatment adverse events (AEs). The goals of treatment are dependent on the stage and extent of disease, the age of the patient, the presence of comorbidities, and prognostic factors. The current SOC for first-line treatment is R-CHOP. Further treatment of LBCL that is refractory or relapsing following first-line therapy will depend on patient eligibility for further intensive therapy (i.e., SCT or CAR T-cell therapies). Approximately 40% of patients will have refractory or relapsed disease and, of these, approximately 50% will be eligible for SCT.6 Patients eligible for SCT can further receive therapy with platinum-based salvage therapy as second-line treatment. Salvage therapy regimens could include gemcitabine, dexamethasone, and cisplatin, with or without rituximab. Other options include rituximab plus ifosfamide, carboplatin, and etoposide (R-ICE); rituximab plus dexamethasone, cytarabine, and cisplatin (R-DHAP); and dexamethasone, ifosfamide, cyclophosphamide, etoposide, and procarbazine, with existing variation in some regimens based on funding in specific jurisdictions across Canada. Patients with a partial or complete response to these regimens can receive high-dose chemotherapy plus SCT. If, despite these treatments, there is relapse and the patient is eligible for intensive therapy, then CAR T-cell therapy is an option. From the outset, patients who are not fit for intensive therapy or ineligible for SCT can be considered for salvage chemotherapy as second line. Eligible CAR T-cell therapies (axi-cel, tisagenlecleucel) are considered third-line therapy. Some patients who are fit for intensive therapy and eligible for SCT may still be unable to undergo the transplant due to unsuccessful stem cell collection (which occurs in approximately 10% of attempts).
Drug Under Review
Liso-cel is a genetically modified autologous cell immunotherapy targeting CD19, also known as CAR T-cell therapy. It binds specifically to CD19, a protein expressed on the surface of B-cell precursors and malignant B cells in DLBCL and other lymphomas. This binding initiates a series of intracellular signalling events that activate and proliferate the CAR T cells, leading to the release of proinflammatory cytokines and cytotoxic agents that destroy the targeted cancer cells.21
Liso-cel is approved by Health Canada for the treatment of adult patients with DLBCL NOS, PMBCL, HGBCL, or DLBCL arising from follicular lymphoma that is refractory to first-line chemoimmunotherapy or relapsed within 12 months of first-line chemoimmunotherapy and who are candidates for autologous HSCT.
Liso-cel is provided as a single-dose, 1-time treatment.21,22 A single dose of liso-cel contains 60 × 106 to 120 × 106 CAR-positive viable T cells (consisting of CD4 and CD8 components at a ratio range of 0.8 to 1.2), with each component supplied separately in 1 to 4 single-dose vials. According to the product monograph,21 liso-cel should be administered at a qualified treatment centre by health care professionals trained in its administration and patient management, including monitoring and handling of CRS and neurotoxicity.
CDA-AMC has reviewed liso-cel previously for the treatment of adult patients with relapsed or refractory LBCL after 2 or more lines of systemic therapy, including those with DLBCL NOS, PMBCL, HGBCL, DLBCL arising from follicular lymphoma, or grade 3B follicular lymphoma after at least 2 prior therapies. pERC the medication be reimbursed with conditions (June 29, 2022).15
Key characteristics of liso-cel and relevant comparators are summarized in Table 3.
Table 3: Key Characteristics of Liso-Cel, Axi-Cel, and Rituximab-Based Chemotherapies
Characteristic | Liso-cel | Axi-cel | Rituximab-based chemotherapy (e.g., R-GDP, R-ICE, R-DHAP) |
|---|---|---|---|
Mechanism of action | CD19-directed genetically modified autologous T-cell immunotherapy. | CD19-directed genetically modified autologous T-cell immunotherapy. | Rituximab is a chimeric mAb that binds to the antigen CD20, a transmembrane protein found on the surface of normal and malignant B lymphocytes. CD20 regulates an early step in the activation of cell cycle initiation and differentiation. |
Indication | For the treatment of adult patients with LBCL, including those with DLBCL NOS, PMBCL, HGBCL, or DLBCL arising from follicular lymphoma that is refractory or has relapsed within 12 months of first-line therapy and who are candidates for autologous HSCT. | For the treatment of adult patients with DLBCL or HGBL that is refractory to first-line chemoimmunotherapy or that relapses within 12 months of first-line chemoimmunotherapy. | Not approved in Canada in the relapsed or refractory setting for DLBCL. |
Route of administration | IV infusion. | IV infusion. | IV infusion. |
Recommended dose | A single-dose, 1-time treatment of liso-cel is recommended. A single dose contains 60 × 106 to 120 × 106 CAR-positive viable T cells (consisting of CD4 and CD8 components at a ratio range from 0.8 to 1.2), with each component supplied separately in 1 to 4 single-dose vials with extractable total volume of 4.6 mL of each CD8 and CD4 component. | A single-dose, 1-time treatment in a patient-specific infusion bag. Each single infusion bag of axi-cel contains a suspension of anti-CD19 CAR T cells in approximately 68 mL. The target dose is 2 × 106 CAR-positive viable T cells per kg body weight (range, 1 × 106 cells/kg to 2.4 × 106 cells/kg), with a maximum of 2 × 108 CAR T cells for patients ≥ 100 kg. | Indication in the relapsed or refractory setting for DLBCL was not identified in the product monograph. |
Serious adverse effects or safety issues | CRS, neurologic toxicities, secondary malignancies, hypogammaglobulinemia, prolonged cytopenias, infections, febrile neutropenia, and TLS have been observed. | CRS, TLS, neurologic toxicities, secondary malignancies, hypogammaglobulinemia, prolonged cytopenias, infections, febrile neutropenia. | Rituximab: Infusion reactions, PML, TLS, HBV reactivation, mucocutaneous reactions, infections, cardiovascular events. |
Other | Must be administered in a qualified treatment centre under the supervision of health care professionals experienced in the treatment of hematological malignancies. | Must be administered in a qualified treatment centre under the supervision of health care professionals experienced in the treatment of hematological malignancies. | NA |
ALL = acute lymphoblastic leukemia; axi-cel = axicabtagene ciloleucel; CAR = chimeric antigen receptor; CD = cluster of differentiation; CRS = cytokine release syndrome; DLBCL = diffuse large B-cell lymphoma; FLG3b = follicular lymphoma grade 3B; HBV = hepatitis B virus; HGBCL = high-grade B-cell lymphoma; LBCL = large B-cell lymphoma; liso-cel = lisocabtagene maraleucel; mAb = monoclonal antibody; NA = not applicable; NOS = not otherwise specified; PMBCL = primary mediastinal large B-cell lymphoma; PML = progressive multifocal leukoencephalopathy; R-DHAP = rituximab, dexamethasone, cytarabine, and cisplatin; R-GDP = rituximab plus gemcitabine, dexamethasone, and cisplatin; R-ICE = rituximab, ifosfamide, carboplatin, etoposide; TLS = tumour lysis syndrome.
Sources: Product monographs for liso-cel (Breyanzi) for injection,21 axi-cel (Yescarta) for injection,23 and rituximab (Rituxan) for injection.24
Patient Group and Clinician Group Perspectives
Patient Group Input
This section was prepared by the CDA-AMC review team based on the input provided by patient groups. The full patient group submissions received are available in the consolidated patient and clinician group input document for this review on the project website publicly accessible here.
One patient group, LC, responded to CDA-AMC’s call for patient input on the current review of liso-cel. LC is a national charity that empowers the lymphoma community through education, support, advocacy, and research, promoting early detection, improving treatments, and facilitating patient access to treatments. Information for this submission was collected via an anonymous online patient survey conducted by LC from March 18 to May 13, 2024. LC reached out to clinicians and researchers involved in relevant trials to share the survey with their patients. Ninety responses from patients with LBCL were collected, including 23 with experience with liso-cel in the third line or later and 5 with experience with this therapy in the second line (2 males and 3 females aged 25 to 44 years). The majority of patients in the survey lived in Canada (66%), were aged 25 to 34 years (30%) or 35 to 44 years (21%), were female (53%), and were diagnosed 1 to 2 years ago (38%), 3 to 5 years ago (33%), or less than a year ago (18%). Among the survey respondents, 38% were diagnosed with DLBCL NOS.
Respondents rated physical symptoms affecting their quality of life on a scale of 1 (no impact) to 5 (significant impact). The symptoms most commonly rated as a 5 were fatigue (24%), enlarged lymph nodes (22%), indigestion or abdominal pain (14%), body aches and pains (13%), bodily swelling (13%), and night sweats (13%). The psychosocial impacts of LBCL included stress or worry (68%), difficulty sleeping (56%), fear of progression (49%), not being able to continue daily activities (43%), problems concentrating (33%), depression (31%), and fear of not being able to attend school or work (30%). Patients currently continue to experience physical symptoms like indigestion and fatigue alongside mental health challenges such as fear of relapse (66%) and anxiety (58%). Daily life is notably affected, with 31% reporting an impaired ability to travel and 29% struggling with work or family obligations.
LC survey respondents reported receiving 1 (42%) or 2 (33%) lines of treatment for LBCL, with varying satisfaction levels across treatment stages. In the first-line setting, 42 patients received R-CHOP; 19 received DA-EPOCH-R; and 8 received radiation. Second-line treatments included salvage therapy with autologous HSCT (n = 15); radiation (n = 10); R-ICE (n = 7); R-DHAP (n = 7); and rituximab, gemcitabine, dexamethasone, and cisplatin (R-GDP) (n = 6). Third-line treatments included polatuzumab vedotin, bendamustine, and rituximab (Pola-BR) (n = 14), CAR T-cell therapy (n = 13), and glofitamab (n = 9). First-line treatment options were generally well received (65% satisfaction), but satisfaction decreased for second-line (44%) and third-line treatments (43%). Based on these results, LC indicated a need for more second- and third-line treatment options. Commonly reported difficult side effects included fatigue, hair loss, and nausea, impacting quality of life. Of 77 patients surveyed, 26 found it easy to access LBCL treatment, while 22 had some difficulty and 3 had significant difficulty. Main barriers included lack of availability at local cancer centres (56%), living in a community without a cancer centre (10%), and residing in a province where treatment was not available (6%). Patients reported that accessing treatment posed challenges leading to financial burdens such as drug costs and travel expenses for many patients.
Most (66%) patients with LBCL who completed the LC survey emphasized the need for more therapy options. They indicated that factors such as longer remission (61%), survival (60%), improved quality of life and symptom control (55%), and normalizing blood counts (54%) were very important to them. Additionally, 67% were willing to tolerate short-term, nonsevere side effects for new treatments. Patients highlighted the desire for treatments with fewer side effects and effective disease control.
Five patients, including 1 residing in Canada, reported receiving liso-cel as second-line treatment and being currently in remission. The main side effects observed were decreased appetite, nausea or vomiting, and fever. All patients reported they are experiencing positive outcomes and unanimously recommended liso-cel for relapsed or refractory LBCL.
Clinician Input
Input From the Clinical Experts Consulted by CDA-AMC
All CDA-AMC review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). In addition, as part of the review of liso-cel, a panel of 3 clinical experts from across Canada was convened to characterize unmet therapeutic needs, assist in identifying and communicating situations where there are gaps in the evidence that could be addressed through the collection of additional data, promote the early identification of potential implementation challenges, gain further insight into the clinical management of patients living with the condition, and explore the potential place in therapy of the drug (e.g., potential reimbursement conditions). A summary of this panel discussion follows.
Unmet Needs
The clinical experts indicated that the treatment goal for fit patients with relapsed or refractory LBCL is cure and long-term survival. The experts noted that fit patients typically receive salvage platinum-containing chemotherapy as second-line treatment, followed by an autologous HSCT if they are eligible and respond to salvage chemotherapy. The experts also noted that many patients are not eligible for SCT due to age (e.g., older than 70 to 75 years of age), comorbidities (e.g., related to liver, pulmonary, or cardiac function), chemorefractory disease, or an inability to mobilize stem cells, and these criteria vary across treatment centres. The experts noted that patients whose disease relapses soon after treatment who do not experience a response to first-line therapy typically have chemotherapy-refractory disease and are unlikely to benefit from autologous HSCT. As such, the experts indicated that the unmet needs of patients would be new treatments that would prevent progression, prolong OS, and improve quality of life while exposing patients to reduced toxicity.
Place in Therapy
The clinical experts agreed that liso-cel would be used in the second-line setting for patients with DLBCL that is refractory to or that relapses within 12 months of the end of first-line therapy. Since axi-cel is approved for the same indication, liso-cel would be in direct competition with axi-cel. The experts believe it is important for more than 1 CAR T product to be available for this indication, given differences in product availability, manufacturing technique, and safety profile.
Patient Population
The clinical experts noted that the patients most likely to benefit from second-line liso-cel would be those with characteristics similar to those of the patients in the TRANSFORM trial (e.g., with disease that is refractory to or that relapses within 12 months of first-line therapy with an adequate performance status and organ function), and patients who would not be suitable for treatment with second-line liso-cel are those who experience later relapses. The clinical experts indicated there should be some leeway with age, ECOG Performance Status, organ function parameters, and whether or not patients are required to be candidates for an autologous HSCT.
Assessing the Response to Treatment
The clinical experts indicated that in clinical practice, the rate of response that is evident on imaging 30 to 90 days after treatment and/or the presence of clinical symptoms are used to determine whether a patient’s disease is responding or progressing on treatment.
Discontinuing Treatment
The clinical experts noted that discontinuation of liso-cel would not be relevant, as it is a single-dose therapy. However, the experts noted that between the time of undergoing leukapheresis and liso-cel infusion, some patients may experience significant disease progression or become clinically unstable and, at that point, it may be necessary to discontinue treatment with liso-cel, at the discretion of the treating clinician.
Prescribing Considerations
The clinical experts indicated that patients receiving liso-cel should be under the care of a clinician (e.g., hematologist or medical oncologist) who can manage toxicity associated with the therapy within centres that have cellular therapy experience. The experts also noted that patients should have access to an intensive care unit in case of rare high-grade toxicities, and consultative support from an infectious disease specialist or neurologist if needed.
Clinician Group Input
This section was prepared by the CDA-AMC review team based on the input provided by clinician groups. The full clinician group submissions received are available in the consolidated patient and clinician group input document for this review on the project website publicly accessible here.
Three clinician groups comprising a total of 15 clinicians provided input for this review: LC (3 clinicians contributed to the input), OH (CCO) Hematology Cancer Drug Advisory Committee (7 clinicians contributed to the input), and Leukemia & Lymphoma Society of Canada Nurses Network (5 clinicians contributed to the input). Overall, the input was aligned with that provided by the clinical experts consulted by CDA-AMC.
According to the clinician groups, first-line treatment for patients with DLBCL involves multiagent chemotherapy with R-CHOP, curing approximately 60% of patients. The remaining patients have disease that either relapses or is refractory and, in Canada, the second-line treatment is salvage chemoimmunotherapy, most commonly R-GDP followed by autologous HSCT. LC added that only about half of patients experience a response that is sufficient to undergo autologous HSCT, and relapses are common even after this procedure. Those who do not experience a sufficient response are now offered CAR T-cell therapy in the third-line (or later) setting. CAR T-cell therapy, now approved in the second line with axi-cel, is awaiting provincial funding for broader availability.
The clinician groups stated that the primary treatment goals are to prolong life, slow disease progression, improve quality of life, avoid toxic chemotherapy protocols and, ideally, prevent the need for further treatments. Available treatments for DLBCL are effective for some patients, but there are unmet needs, particularly for high-risk patients such as those with primary refractory disease or early relapse. Second-line chemoimmunotherapy and autologous HSCT are successful in a subset of patients, but only about 20% achieve durable remission with this approach. If effective, CAR T-cell therapy in the second line could address this gap earlier in the disease course, potentially leading to more cures and reducing the need for other salvage strategies.
The clinician groups indicated that liso-cel is best suited over salvage chemotherapy and autologous HSCT for those with high-risk diseases, such as those that are primary refractory or relapse within 12 months of first-line therapy. The groups suggested it has potential benefits for older patients and those with moderate comorbidities; patients with a low tumour burden or other DLBCL subtypes may also benefit. The clinician groups noted that fitness for treatment will be determined by the primary hematologists or oncologists based on institutional guidelines, considering factors like performance status and organ function.
According to the clinician groups, clinical practice and trials assess various outcomes, including overall response rate and CRR, PFS, and OS, employing the Lugano criteria for remission confirmation. Response assessment involves a restaging CT or a combined PET-CT scan at 1, 3, and 6 months post infusion, with patients sustaining responses beyond 6 to 12 months typically experiencing long-lasting remissions.
The clinician groups agreed that liso-cel is administered as a single infusion; thus, the concept of discontinuation does not typically apply in the context of CAR T treatment. However, the clinician groups noted that decisions to withhold infusion post manufacturing are patient-specific, typically due to rapidly progressive disease with organ failure. The clinician groups stated that liso-cel should be administered only in established CAR T therapy programs approved to deliver this therapy and that encompass certified laboratories, specialized hematologists or oncologists, and include access to subspecialists for managing potential AEs.
Drug Program Input
The drug programs provide input on each drug being reviewed through the Reimbursement Review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CDA-AMC are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
The TRANSFORM comparator was salvage chemoimmunotherapy (R-DHAP, R-ICE, R-GDP), depending on response, followed by HDCT and HSCT. This is aligned with standard of care. Yescarta (axi-cel) received a positive CDA-AMC review for the same indication. | Comment from the drug plans to inform pERC deliberations. |
Considerations for initiation of therapy | |
For DLBCL arising from follicular lymphoma, do patients need to have a record of treatment for the diagnosis of DLBCL or is a biopsy-proven DLBCL sufficient (e.g., the patient only received treatment for follicular lymphoma that then transformed to DLBCL)? | The clinical experts indicated that biopsy-proven DLBCL is sufficient to qualify for liso-cel (if the patient received R-CHOP or similar treatment for follicular lymphoma that then transformed to DLBCL within 12 months). |
Patients in the TRANSFORM trial were aged 18 to 75 years; had an ECOG of 1 or less; had PET-positive disease per Lugano 2014 criteria; had DLBCL (transformed from indolent NHL); had B-cell lymphoma with MYC and either BCL2, BCL6, or both with DLBCL histology; or had primary mediastinal B-cell lymphoma, T-cell LBCL, or follicular lymphoma grade 3B. Patients with secondary CNS lymphoma were allowed. Can pERC clarify relapsed or refractory disease? Should patients with the following, who were excluded from the trial, be considered for liso-cel:
| The clinical experts noted there is variability among definitions across trials but, generally, refractory disease refers to patients who do not achieve a complete response by the end of first-line therapy, while relapsed disease refers to patients experience an initial response but then experience disease progression. The clinical experts noted that eligibility for CAR T-cell therapy is based on provincial or program guidelines. The experts indicated that patients with an ECOG Performance Status of 0 to 2 and rare subtypes of DLBCL, including Richter transformation, should be considered for liso-cel. Burkitt lymphoma is a separate entity that is managed differently. The experts also noted there are limited data if patients with prior gene- or CD19-targeted therapy should be considered for liso-cel treatment. |
TRANSFORM allowed immunochemotherapy as bridging while awaiting liso-cel with R-DHAP, R-ICE, or R-GDP. Could patients be bridged with corticosteroid or other treatments? | The clinical experts noted that patients could receive bridging therapy, if needed to control symptoms or disease burden, with corticosteroids or other treatments before receiving liso-cel treatment. |
Considerations for prescribing of therapy | |
Liso-cel is a single-dose, 1-time treatment infused at a target dose of 60 × 106 to 120 × 106 CAR-positive viable T cells. | Comment from the drug plans to inform pERC deliberations. |
In the trial, one-fifth of the patients in the liso-cel arm received liso-cel in the outpatient setting. Is it safe to administer liso-cel in the outpatient setting and what criteria can be used to determine outpatient eligibility? | The clinical experts noted that most provinces have outpatient CAR T treatment standards and, in general, it would be safe to administer liso-cel in the outpatient setting if patients are able to reside close to the treatment centre, have a reliable caregiver, and do not have significant comorbidities or uncontrolled disease burden. |
Delivery must take place at specialized treatment centres that are accredited and certified by the manufacturer. There continues to be limited access to CAR T-cell services in Canada. While access is expanding, interprovincial travel or out-of-country funding remains necessary in many parts of Canada. Due to geographical site limitations, patients may need to travel for treatment requiring interprovincial agreements to ensure equitable access. | Comment from the drug plans to inform pERC deliberations. |
Consider alignment with prescribing criteria for axi-cel. | Comment from the drug plans to inform pERC deliberations. |
Generalizability | |
Can patients who recently started second-line chemotherapy (SOC) be allowed to switch to CAR T provided all criteria are met? | The clinical experts noted that patients who recently started second-line chemotherapy could be allowed to switch to CAR T therapy, at the discretion of the physician and patient, and provided they meet the eligibility requirements for second-line CAR T. |
Funding algorithm (oncology only) | |
Complex therapeutic space with multiple lines of therapy, subpopulations, or competing products. | Comment from the drug plans to inform pERC deliberations. |
Care provision issues | |
Patients will require hospitalization for adverse events and possible intensive care unit admission. CRS may be managed by tocilizumab or siltuximab and steroids. | Comment from the drug plans to inform pERC deliberations. |
System and economic issues | |
The feasibility of adoption must be addressed. Given the anticipated patient volumes, PAG is concerned that existing capacity may not be able to meet demand. | Comment from the drug plans to inform pERC deliberations. |
Liso-cel will require specialized facility eligible for CAR T therapy and patients may require interprovincial travel. This therapy requires facilities that are not available in all provinces. Drug plans (or manufacturer) may need to cover travel expenses for eligible patients. | Comment from the drug plans to inform pERC deliberations. |
At the time of PAG input, axi-cel is undergoing pCPA negotiations for the same indication as liso-cel. | Comment from the drug plans to inform pERC deliberations. |
There are patient privacy and patient cell ownership concerns due to the fact that CAR T is manufactured by a US-based company outside of Canadian jurisdiction (this is also the case for the other CAR T therapies that are publicly funded). | Comment from the drug plans to inform pERC deliberations. |
axi-cel = axicabtagene ciloleucel; CAR T = chimeric antigen receptor T cell; CD19 = cluster of differentiation 19; CDA-AMC = Canada’s Drug Agency; CLL = chronic lymphocytic leukemia; CNS = central nervous system; CRS = cytokine release syndrome; ECOG = Eastern Cooperative Oncology Group; DLBCL = diffuse large B-cell lymphoma; HDCT = high-dose chemotherapy; HSCT = hematopoietic stem cell transplant; LBCL = large B-cell lymphoma; liso-cel = lisocabtagene maraleucel; NHL = non-Hodgkin lymphoma; PAG = Provincial Advisory Group; pCPA = pan-Canadian Pharmaceutical Alliance; pERC = pan-Canadian Oncology Drug Review Expert Review Committee; R-CHOP = rituximab, cyclophosphamide, doxorubicin hydrochloride (hydroxydaunomycin), vincristine sulphate (Oncovin), and prednisone; R-DHAP = rituximab, dexamethasone, cytarabine, and cisplatin; R-GDP = rituximab, dexamethasone, gemcitabine, and cisplatin; R-ICE = rituximab, ifosfamide, carboplatin, and etoposide; SLL = small lymphocytic lymphoma; SOC = standard of care.
Clinical Evidence
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of liso-cel as 1-time IV infusion of 100 × 106 CAR T cells for the treatment of adults with LBCL, including DLBCL NOS, PMBCL, HGBCL, and DLBCL arising from follicular lymphoma that is refractory or has relapsed within 12 months of first-line therapy and who are candidates for autologous HSCT. The focus will be placed on comparing liso-cel with relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of liso-cel is presented in 4 sections, with a CDA-AMC critical appraisal of the evidence included at the end of each section. The first section, the systematic review, includes pivotal studies and RCTs that were selected according to the sponsor’s systematic review protocol. The CDA-AMC assessment of the certainty of the evidence in this first section using the GRADE approach follows the critical appraisal of the evidence. The second section would include long-term extension studies; however, none were submitted by the sponsor. The third section includes indirect evidence from the sponsor. The fourth section would include additional studies to address important gaps in the systematic review evidence; however, the 1 study that was submitted by the sponsor was excluded because it did not match the patient population for the approved Health Canada indication.
Included Studies
Clinical evidence from the following is included in the CDA-AMC review and appraised in this document:
1 pivotal trial identified in systematic review
2 ITCs.
Systematic Review
Contents within this section have been informed by materials submitted by the sponsor. The following was summarized and validated by the CDA-AMC review team.
Description of Studies
Characteristics of the included studies are summarized in Table 5.
TRANSFORM was a phase III, randomized, parallel-group, open-label, multicentre trial that aimed to assess the efficacy and safety of 1-time IV infusion of liso-cel 100 × 106 CAR T cells compared with SOC, defined as 3 cycles with 1 of 3 prespecified salvage immunochemotherapy regimens followed, depending on response, by 1 cycle of high-dose chemotherapy and autologous HSCT) in adult patients with relapsed or refractory LBCL. Patients in the liso-cel group could receive bridging therapy with 1 of the 3 defined salvage immunochemotherapy regimens allowed in the SOC group during liso-cel manufacturing, if needed.16 The trial enrolled patients who had LBCL that was refractory to or had relapsed within 12 months after initial response to first-line therapy, including an anthracycline and an anti-CD20 monoclonal antibody and were considered candidates for autologous HSCT with an ECOG Performance Status score of 1 or less. The focus of the approved Health Canada indication and reimbursement request is aligned with the trial population.
Enrolled patients were randomly assigned via interactive response system in a 1:1 ratio (N = 184 from 47 sites) to receive liso-cel (n = 92) or SOC (n = 92). No patients from Canada were included in the trial. Randomization was stratified by response to first-line therapy (relapsed versus refractory) and sAAIPI (0 to 1 versus 2 to 3). The patients and investigators were not blinded to treatments, although the IRC, which was responsible for reviewing data related to disease response assessments during the study and to determine remission and relapse for the primary analysis, was blinded to treatment groups.
Table 5: Details of Studies Included in the Systematic Review
Detail | TRANSFORM Trial |
|---|---|
Design and population | |
Study design | Phase III, open-label, parallel-group, randomized trial |
Locations | 47 sites in 11 countries (Belgium, France, Germany, Italy, Japan, Netherlands, Spain, Sweden, Switzerland, the UK, and the US) |
Patient enrolment dates | Start date: October 23, 2018 (first patient signed an ICF) End date: December 8, 2020 (last patient randomized) |
Randomized (N) | Total: N = 184:
|
Key inclusion criteria |
|
Key exclusion criteria |
|
Drugs | |
Intervention | Liso-cel at a dose of 100 × 106 CAR+ viable T cells (CAR+ T cells) on day 29 (2 to 7 days after completion of LDC), presented as a cell suspension in patient-specific single-dose vials, for IV infusion |
Comparator(s) | SOC followed by HDCT and HSCT for patients responding to SOC (CR or PR) Randomized patients were administered 1 of the following 3 SOC regimens, as per the investigator’s decision, followed, depending on dose, by HDCT and a HSCT:
All patients randomized received 3 cycles of SOC salvage therapy (R-DHAP, R-ICE, or R-GDP) as per physician’s choice, during which peripheral blood hematopoietic stem cells for HSCT were harvested. After 3 cycles, response was evaluated by PET-CT. Patients responding to SOC (CR and PR) were to proceed to HDCT and HSCT. |
Study duration | |
Screening phase | Screening (days −28 to −1): consisted of screening assessments to determine eligibility for randomization and unstimulated leukapheresis for all eligible patients before randomization (irrespective of treatment arm). |
Treatment phase | Treatment (days 1 [± 3 days] to 126 [± 7 days]): randomization to either arm A (SOC followed by HDCT and HSCT) or arm B (bridging therapy [if needed], LDC followed by liso-cel infusion day 29 ± 7 days [2 to 7 days after completion of LDC]). The first response evaluations were performed at week 9 (after 3 cycles of SOC for arm A and 5 weeks after liso-cel infusion for arm B) and week 18 (8 weeks after the start of HDCT for arm A and 14 weeks after liso-cel infusion for arm B). |
Follow-up phase |
|
Outcomes | |
Primary outcome | EFS, per IRC, defined as time from randomization to death from any cause, PD, failure to achieve CR or PR by 9 weeks post randomization or the start of new antineoplastic therapy due to efficacy concerns, whichever occurred first. Failure to achieve CR or PR was evaluated after 3 cycles of SOC (expected 9 weeks post randomization) and 5 weeks after the liso-cel infusion. |
Secondary and exploratory outcomes | Key secondary:
Secondary:
Exploratory:
|
Publication status | |
Publications | ClinicalTrials.gov number NCT03575351 Kamdar et al. (2022) Abramson et al. (2023) |
CAR+ = chimeric antigen receptor–positive; CD = cluster of differentiation; CNS = central nervous system; CR = complete response; CRR = complete response rate; DHL = double-hit lymphoma; DLBCL = diffuse large B-cell lymphoma; DoR = duration of response; ECOG = Eastern Cooperative Oncology Group; EFS = event-free survival; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EOS = end of study; FL3B = follicular lymphoma grade 3B; HDCT = high-dose chemotherapy; HRQoL = health-related quality of life; HSCT = hematopoietic stem cell transplant; ICF = informed consent form; ICU = intensive care unit; IRC = independent review committee; LBCL = large B-cell lymphoma; LDC = lymphodepleting chemotherapy; liso-cel = lisocabtagene maraleucel; LTFU = long-term follow-up; NHL = non-Hodgkin lymphoma; NOS = not otherwise specified; ORR = overall response rate; OS = overall survival; PD = progressive disease; PET-CT = combined PET and CT scan; PFS = progression-free survival; PMBCL = primary mediastinal large B-cell lymphoma; PR = partial response; R-DHAP = rituximab, dexamethasone, cytarabine, and cisplatin; R-GDP = rituximab, gemcitabine, dexamethasone, and cisplatin; R-ICE = rituximab, carboplatin, etoposide, and ifosfamide with mesna; SAE = serious adverse event; SD = stable disease; SOC = standard of care; THL = triple-hit lymphoma; THRBCL = T-cell or histiocyte-rich large B-cell lymphoma.
Sources: TRANSFORM Clinical Study Report.16 Details included in the table are from the sponsor’s summary of clinical evidence.
The trial included a screening phase 1 to 2 weeks before leukapheresis. If eligible, patients underwent leukapheresis to enable liso-cel product generation followed by a pretreatment evaluation that included reconfirmation of study eligibility before LDC and liso-cel administration. The treatment period was defined as the date on which LDC was first administered until day 29 after liso-cel infusion, and the posttreatment follow-up period was defined as day 30 after liso-cel infusion to end of study at 24 months. After completion of the posttreatment follow-up period or a premature withdrawal, all patients who received liso-cel were monitored (if they consented) for long-term safety and efficacy under a separate protocol for up to 15 years.
The outcomes relevant to this review include the primary outcome of EFS per IRC, and secondary outcomes of CRR, PFS, OS, and safety. HRQoL measured using the EORTC QLQ-C30 and FACT-LymS, a secondary outcome in the trial, was also considered relevant. Efficacy and safety data were evaluated at the data cut-off dates of March 8, 2021 (interim analysis), May 13, 2022 (primary analysis), and October 23, 2023 (final analysis).
Populations
Inclusion and Exclusion Criteria
A detailed description of the key inclusion and exclusion criteria for the TRANSFORM trial is in Table 5. Eligible patients were adults aged 18 to 75 years, considered candidates for autologous HSCT and had LBCL that was refractory to or relapsed within 12 months after initial response to first-line therapy, including an anthracycline and an anti-CD20 monoclonal antibody. Patients had to have an ECOG Performance Status score of 1 or less, adequate organ function, and PET-positive disease per Lugano (2014) criteria before randomization. Histologies were determined locally and subsequently confirmed by a central laboratory and included DLBCL NOS, DLBCL transformed from indolent NHL, HGBCL, PMBCL, T-cell or histiocyte-rich LBCL, and grade 3B follicular lymphoma. Patients with secondary central nervous system lymphoma were allowed.
Figure 1: Study Design of TRANSFORM Trial
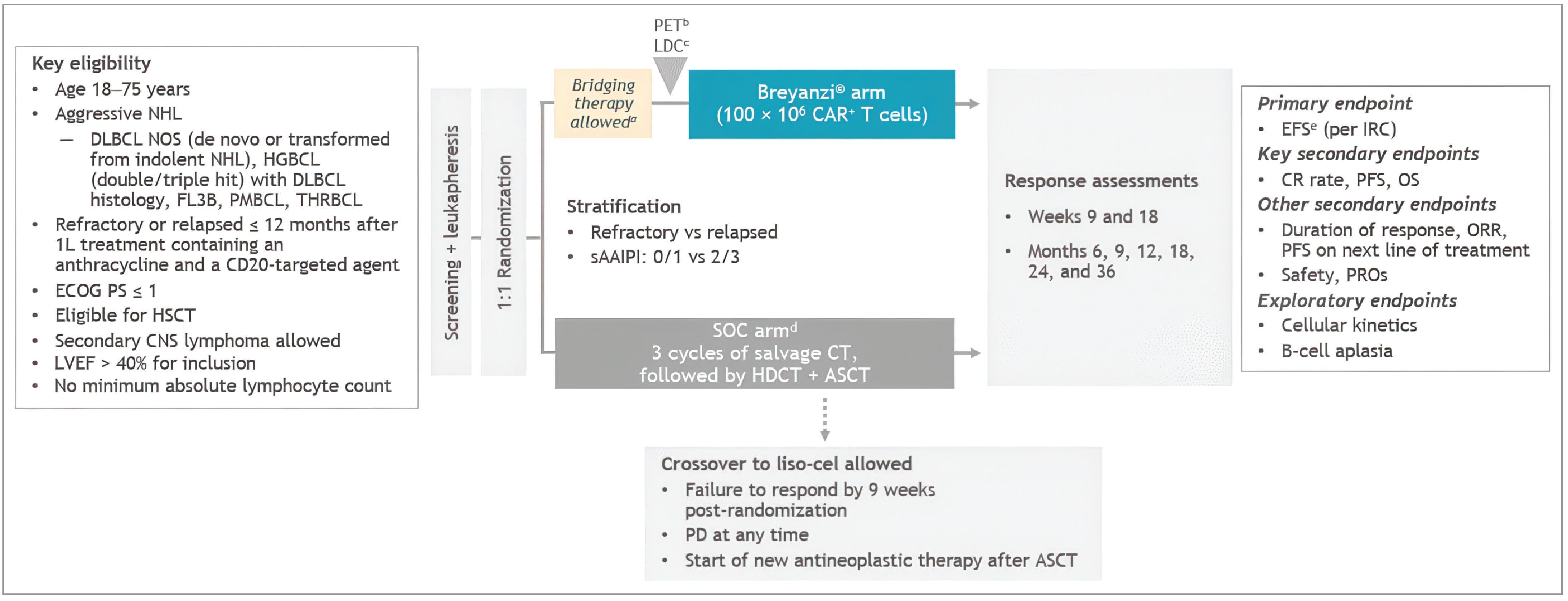
ASCT = autologous stem cell transplant; CAR = chimeric antigen receptor; CD = cluster of differentiation; CNS = central nervous system; CR = complete response; DLBCL NOS = diffuse large B-cell lymphoma not otherwise specified; ECOG PS = Eastern Cooperative Oncology Group Performance Status; EFS = event-free survival; FL3B = follicular lymphoma grade 3B; HDCT = high-dose chemotherapy; HGBCL = high-grade B-cell lymphoma; HSCT = hematopoietic stem cell transplant; IRC = independent review committee; LDC = lymphodepleting chemotherapy; liso-cel = lisocabtagene maraleucel; LVEF = left ventricular ejection fraction; NHL = non-Hodgkin lymphoma; ORR = overall response rate; OS = overall survival; PD = progressive disease; PFS = progression-free survival; PMBCL = primary mediastinal large B-cell lymphoma; PR = partial response; PRO = patient-reported outcome; R-DHAP = rituximab, dexamethasone, high-dose cytarabine, and cisplatin; R-GDP = rituximab, gemcitabine, dexamethasone, and cisplatin; R-ICE = rituximab, ifosfamide, carboplatin, and etoposide; sAAIPI = secondary Age-Adjusted International Prognostic Index; SOC = standard of care; THRBCL = T-cell or histiocyte-rich large B-cell lymphoma; vs = versus.
aPatients may have received a protocol-defined SOC regimen to stabilize their disease during liso-cel manufacturing.
bOnly for the 58 patients (63.0%) who received bridging therapy.
cLymphodepletion with fludarabine 30 mg/m2 and cyclophosphamide 300 mg/m2 for 3 days.
dSOC was defined as physician’s choice of R-DHAP, R-ICE, or R-GDP.
eEFS was defined as time from randomization to death due to any cause; PD was defined as failure to achieve a CR or PR by 9 weeks post randomization, or the start of a new antineoplastic therapy, whichever occurred first.
Sources: TRANSFORM Clinical Study Report.16 Details included in the table are from the sponsor’s summary of clinical evidence.
Interventions
Patients randomized to the liso-cel arm received LDC with IV fludarabine (30 mg/m2 per day for 3 days) plus cyclophosphamide IV (300 mg/m2 per day for 3 days) concurrently followed at least 2 days later by a liso-cel infusion (100 × 106 CAR T cells). Patients were permitted to receive bridging therapy with 1 cycle of an SOC regimen to stabilize their disease during liso-cel manufacturing, if deemed necessary by the investigator (i.e., after leukapheresis and before LDC). Bridging therapy given after randomization must have been stopped at least 7 days before LDC. Patients who received bridging chemotherapy were to have a PET scan before the start of LDC. Only fludarabine dose modification was allowed per protocol, based on renal function.
Patients randomized to the SOC arm received 3 cycles of 1 of the following SOC salvage regimens:
R-DHAP (rituximab 375 mg/m2 on day 1, dexamethasone 40 mg on days 1 to 4, cytarabine 2 × 2,000 mg/m2 on day 2, and cisplatin 100 mg/m2 on day 1)
R-ICE (rituximab 375 mg/m2 on day 1, ifosfamide 5,000 mg/m2 on day 2, etoposide 100 mg/m2 on days 1 to 3, and carboplatin area under the curve 5 [maximum dose of 800 mg] on day 2)
R-GDP (rituximab 375 mg/m2 on day 1, dexamethasone 40 mg on days 1 to 4, gemcitabine 1,000 mg/m2 on days 1 and 8, and cisplatin 75 mg/m2 on day 1).
Patients experiencing a complete or partial response were to proceed to 1 cycle of high-dose chemotherapy (IV carmustine 300 mg/m2 on day 1, etoposide 200 mg/m2 on days 2 to 5, cytarabine 200 mg/m2 on days 2 to 5, and melphalan 140 mg/m2 on day 6) and autologous HSCT. Choice of the 3 SOC regimens, schedule of the regimen, dose adjustment for toxicities, and the administration of premedication were done as per the site standard, local label, and investigator’s decision. Investigators could switch a patient’s regimen within 1 of the 3 protocol-defined salvage regimens in case of toxicity or unsatisfactory response. A switch within 1 of the 3 defined regimens was allowed to maximize a patient’s chance to receive the full 3 cycles of salvage immunochemotherapy before declaring failure of the treatment, which was not considered an EFS event.
Patients could be discontinued from treatment with the investigational products due to AEs, withdrawal by patient, disease relapse, death, lost to follow-up, physician decision, or other reason (e.g., COVID-19). If requested by the investigator, patients randomized to SOC were allowed to cross over to receive liso-cel upon central confirmation of 1 of the following criteria: failure to achieve a complete response or partial response by 9 weeks post randomization (after 3 cycles of SOC), disease progression at any time, and the need to start a new antineoplastic therapy due to efficacy concerns (absence of complete response) after 18 weeks post randomization. These patients were referred to as the crossover subgroup and were not considered part of the liso-cel group. Because the crossover group would be receiving liso-cel as third-line therapy, and the approved Health Canada indication is for second-line therapy only, the results for this group are not reported.
Outcomes
A list of efficacy end points assessed in this Clinical Review Report is provided in Table 6 followed by descriptions of the outcome measures. Summarized end points are based on outcomes included in the sponsor’s summary of clinical evidence as well as any outcomes identified as important to this review, according to the clinical experts consulted by CDA-AMC and the input from patient and clinician groups and public drug plans. Using the same considerations, the CDA-AMC review team selected end points that were considered to be most relevant to inform CDA-AMC’s expert committee deliberations and finalized this list of end points in consultation with members of the expert committee. All summarized efficacy end points were assessed using GRADE.
Event-Free Survival
The primary outcome of the TRANSFORM trial was EFS, per IRC. EFS was defined as time from randomization to death from any cause, progressive disease, failure to achieve complete response or partial response by 9 weeks post randomization, or the start of a new antineoplastic therapy due to efficacy concerns, whichever occurred first. Assessments were done according to the Lugano 2014 criteria, based on radiographic tumour evaluation by diagnostic-quality CT or MRI scans (chest, neck, abdomen, and pelvis) and PET scans, at weeks 9 (5 weeks after liso-cel infusion and after 3 cycles of immunochemotherapy) and 18 (14 weeks after liso-cel infusion and 8 weeks after the start of high-dose chemotherapy for the SOC group) and months 6, 9, 12, 18, 24, and 36 from randomization.
Table 6: Efficacy Outcomes Summarized From the TRANSFORM Trial
Outcome measure | Time point | TRANSFORM trial |
|---|---|---|
Event-free survival | At 12 and 36 months | Primarya |
Complete response rate | Up to 3 years after randomization | Key secondarya |
Progression-free survival | At 12 and 36 months | Key secondarya |
Overall survival | At 12 and 36 months | Key secondarya |
EORTC QLQ-C30 global health status | At 6 months | Secondary |
FACT-LymS | At 6 months | Secondary |
EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Cancer Questionnaire Core 30; FACT-LymS = Functional Assessment of Cancer Therapy–Lymphoma “additional concerns” subscale.
aStatistical testing for these end points was performed hierarchically.
Sources: TRANSFORM Clinical Study Report.16 Details included in the table are from the sponsor’s summary of clinical evidence.
Complete Response Rate
The secondary outcome of CRR per IRC was defined as the proportion of patients achieving a complete response from randomization up to 3 years post randomization. CRR and EFS assessments were conducted in the same way.
Progression-Free Survival
The secondary outcome of PFS per IRC was defined as the time from randomization to death from any cause or progressive disease, whichever occurred first. PFS and EFS assessments were conducted in the same way using the Lugano (2014) criteria.
Overall Survival
The secondary outcome of OS was defined as the time from randomization to death due to any cause. OS and EFS assessments were conducted in the same way.
Health-Related Quality of Life
The secondary outcome of HRQoL was measured by the change in baseline in patient-reported global health status and “additional concerns” scales, using the EORTC QLQ-C30 and FACT-Lym questionnaires, respectively. The EORTC QLQ-C30 consists of 30 questions that can be combined to produce 5 functional domains (physical, role, cognitive, emotional, and social), 3 symptom scales (fatigue, pain, and nausea or vomiting), and a 2-item global health status/QoL scale along with 5 individual item symptom scores (appetite loss, dyspnea, insomnia, constipation, and diarrhea). An outcome variable consisting of a score from 0 to 100 was derived for the global measure of health status scale, with higher scores indicating better health status. The FACT-Lym questionnaire consists of a general scale and a 15-item lymphoma-specific “additional concerns” subscale. This subscale (FACT-LymS) addresses symptoms and functional limitations that are important to lymphoma patients. The scale items are scored on a 0 (not at all) to 4 (very much) response scale, and items were aggregated to a single score on a 0 to 60 scale. The validity, reliability, and minimal important difference (MID) of both instruments is summarized in Table 7. For both questionnaires, no data were identified in the literature for MID in patients with relapsed or refractory LBCL. For both the EORTC QLQ-C30 global health status and FACT-LymS, the sponsor suggested an absolute change greater than or equal to 3 points from baseline, which was informed by the literature, to define a clinically meaningful change. The CDA-AMC team, in consultation with the clinical experts and based on ranges identified in the literature for the EORTC QLQ-C30 global health status score, determined an absolute change greater than or equal to 10 points from baseline is a more clinically meaningful threshold.
Safety Outcomes
The assessment of safety was based on the proportion of patients experiencing 1 or more TEAEs, serious TEAEs (SAEs), notable AEs, AEs leading to discontinuation, AEs leading to dose modification, and deaths. AEs were reported at each study visit and coded using the Medical Dictionary for Regulatory Activities (MedDRA) version 23.0. An independent data and safety monitoring committee assessed the progress of the trial.
Table 7: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
EORTC QLQ-C30 | The EORTC QLQ-C30 is a standardized patient self-administered questionnaire designed to assess HRQoL of adult patients with cancer.25 It consists of 30 items divided into several domains and scales:
Each item is scored on a 4-point Likert scale ranging from “not at all” to “very much,” except for the GHS (QoL) scale, which uses a 7-point scale. Scores are then converted to a 0 to 100 scale, with higher scores indicating better functioning or worse symptoms, or better quality of life, depending on the scale.25-27 | Measurement properties of validity, reliability, and responsiveness have not been assessed in patients with NHL. | No MID has been established for patients with NHL. In patients with various types of cancers, including hematological diseases, the following have been estimated as “small” mean differences:28
For HRQoL among patients with breast, ovarian, and lung cancer a 5-point change in the GHS (QoL) subscale was estimated as “a little” change; whereas, for other domains, a 5-point change from baseline and 10-point change in mean scores was estimated as “a little” change.28,29 |
FACT-Lym30 | This patient-reported questionnaire is used to assess HRQoL, specifically, disease-specific symptoms and concerns relevant to patients with lymphoma. The FACT-Lym questionnaire can be separated into the 27 items of the FACT-G (the general core questionnaire used for all cancer types, which consists of physical, social [including family], emotional, and functional well-being subscales) and the 15 lymphoma-specific subscales.30 Each item is typically rated on a 5-point Likert scale, where patients indicate how true each statement has been for them over the past 7 days. The response options range from “not at all” to “very much.” Higher scores indicate better HRQoL.30 | In a study,30 84 patients with NHL had measurements taken at baseline, 3 to 7 days, and 8 to 12 weeks. Validity: Construct validity was demonstrated by the FACT-LymS score, which differentiated between patients with an ECOG PS of 0, 1, or 2, and between patients on or off active treatment (e.g., radiation and chemotherapy), but did not differentiate between patient groups defined by their NHL grade.30 Concurrent validity was demonstrated based on correlations between the FACT-LymS and SF-36 PCS (r = 0.62), and the MCS (r = 0.48) and POMS total score (r = 0.60).30 Divergent validity was demonstrated based on the near-zero association between FACT-LymS and Marlowe-Crowne Social Desirability Scale–Short Form (r = 0.15).30 Reliability: The FACT-LymS demonstrated good internal consistency, with the Cronbach alpha ranging from 0.79 to 0.85 at each assessment time point.30 FACT-LymS demonstrated good test–retest reliability based on an ICC of 0.84 (retested at 3 to 7 days from baseline; n = 74).30 Responsiveness: FACT-Lym subscale scores were sensitive to change in patient’s PS over a 3-month period (worse, unchanged, better), with effect sizes > 0.5.30 | Using distribution- and anchor-based methods, the investigators suggested the likely MID range for the FACT-LymS in patients with NHL is approximately 3 to 5 points or 5% to 8% of the scale range (0 to 60).30 |
ECOG PS = Eastern Cooperative Oncology Group Performance Status; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; FACT-G = Functional Assessment of Cancer Therapy–General; FACT-Lym = Functional Assessment of Cancer Therapy–Lymphoma; FACT-LymS = Functional Assessment of Cancer Therapy–Lymphoma “additional concerns” subscale; GHS = global health status; HRQoL = health-related quality of life; ICC = intraclass correlation coefficient; MCS = Mental Component Summary; MID = minimal important difference; NHL = non-Hodgkin lymphoma; PCS = Physical Component Summary; POMS = Profile of Mood States; PS = performance status; QoL = quality of life; SF-36 = Short Form (36) Health Survey Version 1.
Statistical Analysis
A summary of the statistical analysis of efficacy outcomes is provided in Table 8.
Sample Size and Power Calculation
The TRANSFORM trial planned to randomize approximately 182 patients to treatment, given an expected randomization rate of up to 12 patients per month, a 20% dropout rate before first response assessment, and a yearly dropout rate of 10% (30% cumulative). Assuming a screening failure rate of 15%, 215 patients were planned to be screened. The trial was designed to have a power of 90% at a 1-sided 2.5% significance level to identify an approximate 81% increase in median EFS for the liso-cel group compared with the SOC group, equivalent to an HR of 0.55, and a median EFS of 5.455 months with liso-cel and 3 months with SOC. The primary analysis was planned to be conducted when 119 EFS events would have occurred.
Interim, Primary, and Final Analyses
Two interim efficacy analyses, at 60% and 80% of the planned EFS information fraction, were performed in the trial, and an O’Brien-Fleming boundary alpha spending function was used to control the type I error. The purpose of the first interim analysis was to stop for futility in case of no efficacy signal based on the CRR at 9 weeks post randomization after 3 cycles of salvage immunochemotherapy for the SOC group and 5 weeks after liso-cel infusion for the liso-cel group. This analysis was to be performed when approximately 30 evaluable patients, approximately 15 patients per group, had received their assigned treatment, had their week 9 response assessment, or had been confirmed with disease progression before this time point. For the second prespecified interim analysis (data cut-off date of March 8, 2021) planned at 80% and performed at 82% of the 119 events needed for the primary analysis, the null hypothesis was to be rejected if the P value associated to the test was less than 0.012. If the null hypothesis was rejected for EFS, hypothesis testing on CRR, PFS, and OS was performed hierarchically to control the overall type I error rate. At the time of the primary analysis (data cut-off date of May 13, 2022), 115 EFS events were accrued (96.6% of the information fraction) out of the 119 targeted events. The primary efficacy outcome of EFS was met at the interim analysis and was presented descriptively (i.e., not included in the hierarchical testing strategy) in the primary analysis. Therefore, only the key secondary outcomes were retested in the primary analysis. At the time of the interim analysis, the null hypothesis for OS was not rejected. If the null hypothesis was not rejected for any 1 of the key secondary outcomes, they were to be re-evaluated in the primary analysis. Therefore, hypothesis testing on the key secondary outcome of CRR and, subsequently, on PFS and OS, was performed hierarchically in the primary analysis. The significance threshold to reject the null hypothesis for the key secondary outcomes was a P value of 0.021 or less (adjusted for the actual number of events available for the primary analysis and the alpha spent at the previous interim analyses). The final analysis reported the preplanned final efficacy analysis results following the last patient visit milestone (data cut-off date of October 23, 2023) at a median follow-up of 33.9 months.
Statistical Testing
For EFS, PFS, and OS, the KM product limit method was used to extract summary information, including time-to-event rates and median survival times together with 2-sided 95% CIs. A Greenwood formula was used to estimate the 95% CIs for time-to-event rates. HR and P value were estimated from a stratified Cox proportional hazards model. The stratification factors used in the model correspond to the stratification at the time of randomization, and the model included treatment as the only covariate. The proportional hazard assumption was evaluated via inspection of Schoenfeld residuals. For CRR, a Cochran-Mantel-Haenszel test with stratification factors as strata was used for the analysis and calculation of P values. An O’Brien-Fleming boundary and a hierarchical testing strategy were used to control the familywise type I error rate. If the null hypothesis was rejected for EFS, the testing was to be performed on the CRR and subsequently on PFS and OS. An O’Brien-Fleming boundary alpha spending function was used to adjust for multiplicity for the interim analyses for efficacy and the primary analysis.
For the HRQoL outcomes of the EORTC QLQ-C30 and FACT-LymS, analyses were descriptive and included patients with an evaluable baseline assessment and at least 1 evaluable postbaseline assessment and were reported as a change from baseline. A linear mixed-effects regression model for repeated measures for randomized patients with an evaluable baseline assessment and at least 1 evaluable postbaseline assessment was performed. Analyses were not controlled for multiplicity. Safety data were summarized descriptively.
Data Imputation Methods and Censoring
If patients were event-free, EFS was censored based on the following criteria: no baseline or no postbaseline response assessment and no death; start of a new antineoplastic therapy for reasons other than efficacy concern; failure to proceed to high-dose chemotherapy and HSCT; or no death, no progressive disease, no failure to achieve a complete response or partial response by 9 weeks post randomization (after 3 cycles of SOC and 5 weeks after the liso-cel infusion), and no start of new antineoplastic therapy due to efficacy concerns. For CRR, patients with an unknown or missing response were counted as nonresponders in the analysis. Any responses after the start of a new antineoplastic therapy taken for efficacy concerns were not considered. For PFS, patients were censored based on the following criteria: no baseline or postbaseline response assessment and no death, start of a new antineoplastic therapy before death or progressive disease, or no death or no progressive disease. For OS, patients alive or lost to follow-up at the time of analysis were censored at the last date the patient was known to be alive. For the HRQoL outcomes, missing values were addressed according to questionnaire guidelines; no other details were reported.
Subgroup Analyses
For the prespecified subgroups of interest, the stratified HR and 95% CI for EFS, CRR, PFS, and OS were performed by histological subtype, use of bridging therapy, and prior response status. Subgroup analysis was conducted using a Cox proportional hazards model and presented as forest plots.
Sensitivity Analyses
Sensitivity analyses for the primary and key secondary outcomes are summarized in Table 8. No sensitivity analysis was performed for the HRQoL outcomes.
Analysis Populations
The analysis populations of the TRANSFORM trial are provided in Table 9. The primary and key secondary outcomes were analyzed based on the ITT population. The per-protocol population, defined as all patients from the ITT population with minimal exposure to treatment, a baseline assessment, and at least 1 postbaseline response assessment without important protocol deviations was used for sensitivity analyses. The HRQoL outcomes were analyzed based on the HRQoL population, defined as all patients from the ITT population who completed a baseline and at least 1 postbaseline assessment for each of the included instruments. The safety outcomes were analyzed using the safety population, defined as patients who received at least 1 dose of any study medication.
Table 8: Statistical Analysis of Efficacy Outcomes
Outcome | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
EFS | Stratified Cox PH model if the PH assumption holds (unstratified Cox PH model as supportive analysis). The number and percentage of patients with EFS events and censored were provided per treatment group using the KM product limit along with the SEs and associated 2-sided 95% CIs (Greenwood formula). The 25th percentile, median, and 75th percentile along with CIs were extracted from KM curves (using log-log transformation) and tabulated per treatment group. | The stratification factors at the time of randomization; in addition, the model included treatment as the only covariate for analysis. | Based on the following censoring rules:
| The PH assumption was to be evaluated through the inspection of Schoenfeld residuals. If non-PH are observed to the extent that an HR will not reliably represent the differences between treatment arms, then a restricted mean survival approach or piecewise stratified Cox PH model was also to be investigated as a sensitivity analysis. A sensitivity analysis was also be performed on:
|
CRR | A CMH test with stratification factors as strata was to be used for analysis and calculation of P values. | Randomization was to be stratified by best overall response to first-line therapy:
| Patients with an unknown or missing response were to be counted as nonevaluable in the analysis. Any responses after a start of a new antineoplastic therapy taken for efficacy concerns were not to be considered. All new antineoplastic therapies were to be considered as being started for efficacy concerns, unless explicitly recorded as not been given for efficacy concerns in the appropriate eCRF form. |
|
PFS | Refer to OS (next row) | None | Based on the following censoring rules: no baseline, or no postbaseline response assessment and no death; death; progressive disease; start of a new antineoplastic therapy before death or progressive disease; or no death or no progressive disease. | RPSFT method and IPCW method. |
OS | As patients from arm A could possibly cross over to lis-cel, a 2-stage Weibull approach, (primary analysis) was used. For PFS instead, patients who crossed over to liso-cel without progression or death were censored. In addition, the rank preserving structural failure time method and inverse probability of censoring weighting method were investigated for OS as supportive analyses and for PFS as sensitivity analyses, if applicable, depending on the rate of patients crossing over to liso-cel without previous disease progression. | In the 2-stage Weibull model, confirmation of EFS was to be considered as secondary baseline for all control group patients. Post-EFS survival in the control group was compared between those patients who switched and those who did not switch using an accelerated failure time model, adjusting for prognostic characteristics measured at baseline and at the time of the confirmation of EFS. An acceleration factor obtained from this model was used to adjust survival times observed in switching patients to derive counterfactual survival times. A stratified Cox proportional hazards regression model was then fitted to the observed liso-cel group survival times and the counterfactual control group survival times to estimate a treatment switching-adjusted HR. The following covariates measured at baseline and at the time of secondary baseline (when available) were considered in the model, as applicable: age, height, weight, BSA, sex, BMI, FEV1, oxygen saturation, HCT-CI, LVEF, MMSE, AAIPI, best response on the first line of treatment (relapse versus refractory), type of B-cell NHL, histological and molecular subtype for B-cell NHL, CNS involvement, Ann Arbor stage, bone marrow involvement, presence of B symptoms. In addition, the following variables collected at the secondary baseline were considered for inclusion in the models:
| Patients alive or lost to follow-up at the time of analysis will be censored at the last date the patient was known to be alive. |
|
HRQoL outcomes | Descriptive | None | None | None |
AAIPI = Age-Adjusted International Prognostic Index; BMI = body mass index; BSA = body surface area; CI = confidence interval; CMH = Cochran-Mantel-Haenszel; CNS = central nervous system; CR = complete response; CRR = complete response rate; DoR = duration of response; ECOG = Eastern Cooperative Oncology Group; eCRF = electronic case report form; EFS = event-free survival; FEV1 = forced expiratory volume in 1 second; HCT-CI = Hematopoietic Cell Transplant–Specific Comorbidity Index; HDCT = high-dose chemotherapy; HR = hazard ratio; HSCT = hematopoietic stem cell transplant; IPCW = inverse probability of censoring weighting; ITT = intention to treat; KM = Kaplan-Meier; liso-cel = lisocabtagene maraleucel; LVEF = left ventricular ejection fraction; MMSE = Mini-Mental State Examination; ORR = overall response rate; OS = overall survival; PD = progressive disease; PFS = progression-free survival; PH = proportional hazards; PR = partial response; RPSFT = rank preserving structural failure time; sAAIPI = secondary Age-Adjusted International Prognostic Index; SD = stable disease; SE = standard error; SOC = standard of care.
Sources: TRANSFORM Clinical Study Report.16 Details included in the table are from the sponsor’s summary of clinical evidence.
Table 9: Analysis Populations of the TRANSFORM Trial
Population | Definition | Application |
|---|---|---|
ITT population | All randomized patients, regardless of whether they received study treatment | EFS, CRR, PFS, OS analyses |
Per-protocol population | All patients from the ITT population with minimal exposure to treatment (liso-cel group: dose of conforming liso-cel; SOC group: 1 cycle of SOC), and a baseline assessment and at least 1 postbaseline response assessment without important protocol deviations | Sensitivity analyses |
Safety population | All patients who received at least 1 dose of study treatment | Safety and sensitivity analyses |
HRQoL population | All patients from the ITT population who completed a baseline and at least 1 postbaseline assessment for each of the included instruments | EORTC QLQ-C30 and FACT-LymS analyses |
CRR = complete response rate; EFS = event-free survival; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; FACT-Lym = Functional Assessment of Cancer Therapy–Lymphoma; FACT-LymS = Functional Assessment of Cancer Therapy–Lymphoma “additional concerns” subscale; HRQoL = health-related quality of life; ITT = intention to treat; liso-cel = lisocabtagene maraleucel; OS = overall survival; PFS = progression-free survival; SOC = standard of care.
Sources: TRANSFORM Clinical Study Report.16 Details included in the table are from the sponsor’s summary of clinical evidence.
Results
Patient Disposition
A summary of patient disposition for the primary analysis (data cut-off date of May 13, 2022) is in Table 10. At this time point, study enrolment was complete, and patients were either in the posttreatment follow-up period or the survival follow-up period. In total, 232 patients were screened, of which 184 patients were randomized to either liso-cel (n = 92) or SOC (n = 92). For the 48 patients (20.7%) who were screened out, the most common reason was not meeting 1 or more of the eligibility criteria (n = 31). In the liso-cel group, 89 patients (96.7%) received liso-cel and 1 patient (1.1%) received a nonconforming product. A total of 81 patients (88.0%) completed the treatment period, and 11 patients (12%) discontinued the treatment period after receiving liso-cel due to disease relapse (6.5%) or death (3.3%). Most patients in the liso-cel group received LDC (98%) and bridging therapy (63.0%), with the most common reason for bridging therapy being high tumour burden (28 patients; 30.4%) and rapid progression (23 patients; 25.0%), per investigator assessment. In the SOC group, 91 patients (98.9%) started SOC treatment, with 43 patients (46.7%) receiving high-dose chemotherapy and 43 patients (46.7%) receiving an autologous HSCT. A total of 37 patients (40.2%) completed the treatment period and 55 patients (59.8%) discontinued treatment. The most common reason for treatment discontinuation was lack of efficacy (30.4%) and disease relapse (16.3%).
Table 10: Summary of Patient Disposition — TRANSFORM Trial (Primary Analysis)
Patient disposition | TRANSFORM trial | |
|---|---|---|
Liso-cel | SOC | |
Screened, N | 232 | |
Failed screening | 48 | |
Reason for screening failure, n (%) | ||
Failure to meet eligibility criteria | 31 | |
COVID-19 | 7 | |
No apheresis slots provided by the sponsor | ██ | |
Randomized, N (%) | 92 (100) | 92 (100) |
Underwent leukapheresis, N (%) | 92 (100) | 92 (100) |
Liso-cel group, n (%) | ||
Received bridging therapy | 58 (63.0) | NA |
Received lymphodepleting chemotherapy | ██ ██████ | NA |
Received liso-cel | 89 (96.7) | NA |
SOC group, n (%) | ||
Received SOC salvage chemotherapy | NA | 91 (98.9) |
Received high-dose chemotherapy | NA | 43 (46.7) |
Received autologous stem cell transplant | NA | ██ ██████ |
Completed treatment, N (%) | ██ ██████ | ██ ██████ |
Discontinued from study, n (%) | 11 (12.0) | 55 (59.8) |
Reason for discontinuation, n (%) | ||
Adverse events | 0 (0) | 1 (1.1) |
Death | 3 (3.3) | 2 (2.2) |
COVID-19 | 1 (1.1) | 0 (0) |
Lack of efficacy | 0 (0) | 28 (30.4) |
Withdrawal by patient | 1 (1.1) | 1 (1.1) |
Physician decision | 0 (0) | 3 (3.3) |
Disease relapse | 6 (6.5) | 15 (16.3) |
Study drug manufacturing failure | 1 (1.1) | 0 (0) |
Other | 0 (0) | 5 (5.4) |
ITT, N | 92 | 92 |
PP, N | ██ | ██ |
Safety, N | 92 | 91 |
ITT = intention to treat; liso-cel = lisocabtagene maraleucel; NA = not applicable; PP = per protocol; SOC = standard of care.
Sources: TRANSFORM Clinical Study Report.16 Details included in the table are from the sponsor’s summary of clinical evidence.
Baseline Characteristics
A summary of the baseline patient demographic and disease characteristics of the ITT population are in Table 11. The baseline characteristics outlined in the table are limited to those that are most relevant to this review or were felt to affect the outcomes or interpretation of the study results. Overall, key baseline characteristics were generally balanced between treatment groups. The trial population was predominately white (approximately 59%), male (57%), with a median age of 59 years (range, 20 to 75 years). Most patients had an ECOG Performance Status at baseline of 0 (approximately 52%) or 1 (approximately 47%), indicating good overall performance; had an sAAIPI of 0 or 1 (60%); had disease that was refractory to or relapsed after their last therapy (74% and 26%, respectively); and had LBCL subtype DLBCL NOS (56%) followed by HGBCL (23%), PMBCL (9%), and DLBCL from transformed indolent lymphoma (8%). The liso-cel group had a lower proportion of male patients (47.8% versus 66.3%) and a higher proportion of patients whose condition was chemorefractory (28.3% versus 19.6%), compared with the SOC group.
Table 11: Summary of Baseline Characteristics — TRANSFORM Trial (Primary Analysis)
Characteristic | TRANSFORM Trial | |
|---|---|---|
Liso-cel (N = 92) | SOC (N = 92) | |
Male sex, n (%) | 44 (47.8) | 61 (66.3) |
Age (years) | ||
Median (IQR) | 60.0 (53.5 to 67.5) | 58.0 (42.0 to 65.0) |
Range | 20 to 74 | 26 to 75 |
Age group (years), n (%) | ||
< 65 | 56 (60.9) | 67 (72.8) |
≥ 65 to < 75 | 36 (39.1) | 23 (25.0) |
≥ 75 | 0 | 2 (2.2) |
Race, n (%) | ||
White | 54 (59) | 55 (60) |
Not collected or reported | 22 (24) | 25 (27) |
ECOG PS score at baseline, n (%) | ||
0 | 46 (50.0) | 49 (53.3) |
1 | 45 (48.9) | 41 (44.6) |
2 | 1 (1.1) | 2 (2.2) |
HCT-CI | ||
n | 86 | 90 |
Median | 1.0 | 1.0 |
Range | 0 to 7 | 0 to 5 |
LVEF | ||
Median | 61.00 | 60.00 |
Range | ██████ █████ | ██████ █████ |
Creatinine clearance at screening (mL/min) | ||
n | ██ | ██ |
Median | 107.10 | 113.40 |
Range | █████ █████ | █████ █████ |
Large B-cell lymphoma subtypes, n (%) | ||
DLBCL NOS | 53 (57.6) | 50 (54.3) |
DLBCL from transformed indolent lymphoma | 7 (7.6) | 8 (8.7) |
FL3B | 1 (1.1) | 0 |
HGBL | 22 (23.9) | 21 (22.8) |
PMBCL | 8 (8.7) | 9 (9.8) |
THRBCL | 1 (1.1) | 4 (4.3) |
sAAIPI at screening, n (%) | ||
0 or 1 | 56 (60.9) | 55 (59.8) |
2 or 3 | 36 (39.1) | 37 (40.2) |
Prior response status (only first-line complete response), n (%)a | ||
Refractory | 67 (72.8) | 70 (76.1) |
Relapse | 25 (27.2) | 22 (23.9) |
Prior chemotherapy response status (only first-line stable disease or partial response), n (%)b | ||
Chemorefractory | 26 (28.3) | 18 (19.6) |
Chemosensitive | 66 (71.7) | 74 (80.4) |
Secondary CNS lymphoma, n (%) | 1 (1.1) | 3 (3.3) |
CNS = central nervous system; ECOG PS = Eastern Cooperative Oncology Group Performance Status; DLBCL = diffuse large B-cell lymphoma; FL3B = follicular lymphoma grade 3B; HCT-CI = hematopoietic cell transplant–specific comorbidity index; HGBL = high-grade B-cell lymphoma; IQR = interquartile range; ITT = intention to treat; liso-cel = lisocabtagene maraleucel; LVEF = left ventricular ejection fraction; NOS = not otherwise specified; PMBCL = primary mediastinal large B-cell lymphoma; sAAIPI = secondary Age-Adjusted International Prognostic Index; SOC = standard of care; THRBCL = T-cell or histiocyte-rich large B-cell lymphoma.
Note: Baseline values were defined as the last value on the randomization date (+ 3 days) or before the date and time of randomization (date only if time not collected).
aOnly patients with a best response of complete response during first-line treatment are included.
bThe status is chemorefractory if a patient achieved stable disease or experienced a partial response to the last chemotherapy-containing regimen; otherwise, the status is chemosensitive.
Sources: TRANSFORM Clinical Study Report.16 Details included in the table are from the sponsor’s summary of clinical evidence.
Exposure to Study Treatments
By the primary analysis data cut-off date of May 13, 2022, the median total liso-cel dose was 99.92 × 106 cells; the median CD8 and CD4 doses were 49.97 × 106 cells and 49.89 × 106 cells, respectively. One patient (1.1%) received nonconforming product because the purity of the CD4 component was outside specifications.
In the SOC group, the most frequently used starting regimen was R-ICE (63.7%; median of 3 cycles), followed by R-GDP (19.8%; median of 3 cycles) and R-DHAP (16.5%; median of 2 cycles). Twelve patients (13.2%) were switched to SOC salvage therapy. The most frequently reported reason for switching to SOC salvage therapy was suboptimal response (5 patients; 5.5%) followed by AE (4 patients; 4.4%).
No patients in either group discontinued treatment due to toxicity, and no patients in the liso-cel group had dose modifications for toxicity. In the SOC group, 4 patients switched salvage regimens due to an AE, and a dose modification for toxicity was reported in 3 patients who received R-DHAP, 5 patients who received R-ICE, 3 patients who received R-GDP, and 1 patient who received high-dose chemotherapy.
All patients in the safety population received 1 or more concomitant medications. In the liso-cel group, 12 patients (13%) received immunoglobulins, 11 (12.0%) received corticosteroids, and 21 (22.8%) were treated with tocilizumab for the management of CRS. For the management of neurologic events, 6 patients (7.0%) received corticosteroids and 1 patient (1.1%) was treated with tocilizumab.
Subsequent Treatment
In the ITT population, ██ ███████ ████████ ██ ███ ████████ █████ ███ ██ ███████ ██ ███ ███ █████ ████████ ██████████ ███████████ ████████ ██ ███ ██ ████████ ███████ ██ ███ ███ █████ ███ ████████ ██ █████ ███ ██████████ ███████████ ████████ █ ████████ ████████ █ ████ ████ ██████████ and 61 patients were approved to cross over to liso-cel as part of an SOC crossover arm. Of those patients, 58 were infused, of which 57 (98.3%) received liso-cel and 1 (1.7%) received a nonconforming product.
Efficacy
Only those efficacy outcomes and analyses of subgroups identified as important to this review are reported. The main findings for the efficacy outcomes for the TRANSFORM trial are from the second interim analysis (data cut-off date of March 8, 2021), primary analysis (data cut-off date of May 13, 2022), and final analysis (data cut-off date of October 23, 2023). The primary efficacy outcome of EFS was met at the second interim analysis and was presented descriptively (i.e., not included in the hierarchical testing strategy) in the primary analysis. Therefore, hypothesis testing on the key secondary outcomes of CRR, PFS, and OS was performed hierarchically in the primary analysis.
Event-Free Survival by IRC
Table 12 provides a summary of results for EFS by IRC. At the time of the second interim analysis, the median duration of follow-up in all patients was 6.2 months (IQR, 4.4 to 11.5 months), and EFS events had been reported for 35 patients (38.0%) in the liso-cel group and 63 patients (68.5%) in the SOC group. The median EFS was 10.1 months (95% CI, 6.1 months to NE) in the liso-cel group versus 2.3 months (95% CI, 2.2 to 4.3 months) in the SOC group (1-sided P value < 0.0001), with a between-group HR of 0.35 (95% CI, 0.23 to 0.53). The results of sensitivity analyses were consistent with the primary analysis. The KM-estimated probability of EFS at 12 months was 44.5% (95% CI, 29.4% to 59.6%) for the liso-cel group versus 23.7% (95% CI, 13.4% to 34.1%) for the SOC group, with a between-group difference of 20.8% (95% CI, 2.5% to 39.1%).
At the time of the primary efficacy analysis, the median duration of follow-up in all patients was 17.5 months (IQR, 0.9 to 37 months), and EFS events had been reported in 44 patients (47.8%) in the liso-cel group and 71 patients (77.2%) in the SOC group. The median EFS was not reached (NE; 95% CI, 9.5 to NE) in the liso-cel group versus 2.4 months (95% CI, 2.2 to 4.9 months) in the SOC group (HR = 0.36; 95% CI, 0.24 to 0.52). The KM-estimated probability of EFS at 12 months was consistent with the second interim analysis, with a between-group difference of 34.6% (95% CI, 21.2% to 48.0%).
At the final analysis, the median duration of follow-up of 33.9 months (IQR, 11.6 to 39.2 months), and EFS events had been reported in ██ ███████ patients in the liso-cel group and ██ ███████ patients in the SOC group. The median EFS was 29.5 months (95% CI, 9.5 to NE) in the liso-cel group and 2.4 months (95% CI, 2.2 to 4.9 months) in the SOC group, with a between-group HR of 0.38 (95% CI, 0.259 to 0.542). The KM-estimated probability of EFS at 12 months was consistent with the interim and primary analyses and, at 36 months, was 45.8% (95% CI, 35.2% to 56.5%) for the liso-cel group versus 19.1% (95% CI, 11.0% to 27.3%) for the SOC group, with a between-group difference of 26.7% (95% CI, 13.3% to 40.1%) (Figure 2).
EFS Subgroup Analyses
At the second interim analysis, the efficacy results for EFS were consistent across the subgroup analyses by histological subtypes, use of bridging therapy (data not shown), and prior response status in favour of liso-cel (Figure 3). In general, the results of the subgroup analyses were consistent across all data cut-offs.
Table 12: Event-Free Survival — ITT Population, TRANSFORM Trial
EFS by IRC | TRANSFORM trial | |
|---|---|---|
Liso-cel (N = 92) | SOC (N = 92) | |
Second interim analysis data cut-off date of March 8, 2021 | ||
Patients with events, n (%) | ||
Total | 35 (38.0) | 63 (68.5) |
Death | 2 (2.2) | 2 (2.2) |
PD | 26 (28.3) | 39 (42.4) |
Failure to achieve CR or PR by 9 weeks post randomization | 4 (4.3) | 17 (18.5) |
Start a new anticancer therapy due to efficacy concerns | 3 (3.3) | 5 (5.4) |
Patients censored | 57 (62.0) | 29 (31.5) |
Median EFS, months (95% CI) | 10.1 (6.1 to NE) | 2.3 (2.2 to 4.3) |
Stratified HR (95% CI)a,b | 0.35 (0.23 to 0.53) | |
One-sided P valueb | < 0.0001 | |
KM probability of being event-free at 12 months, % (95% CI) | 44.5 (29.4 to 59.6) | 23.7% (13.4 to 34.1) |
Between-group difference, % (95% CI) | 20.8 (2.5 to 39.1) | |
Primary analysis data cut-off date of May 13, 2022 | ||
Patients with events, n (%) | ||
Total | 44 (47.8) | 71 (77.2) |
Death | █████ | █████ |
PD | ██ ██████ | ██ ██████ |
Failure to achieve CR or PR by 9 weeks post randomization | █████ | ██ ██████ |
Start a new anticancer therapy due to efficacy concerns | █████ | █████ |
Patients censored | 48 (52.2) | 21 (22.8) |
Median EFS, months (95% CI) | NE (9.5 to NE) | 2.4 (2.2 to 4.9) |
Stratified HR (95% CI)a,b,c | 0.36 (0.24 to 0.52) | |
KM probability of being event-free at 12 months, % (95% CI) | 57.1 (47.0 to 67.3) | 22.5 (13.9 to 31.2) |
Between-group difference, % (95% CI) | 34.6 (21.2 to 48.0) | |
Final analysis data cut-off date of October 23, 2023 | ||
Patients with events, n (%) | ||
Total | ██ ██████ | ██ ██████ |
Death | █████ | █████ |
PD | ██ ██████ | ██ ██████ |
Failure to achieve CR or PR by 9 weeks post randomization | █████ | ██ ██████ |
Start a new anticancer therapy due to efficacy concerns | █████ | █████ |
Patients censored | ██ ██████ | ██ ██████ |
Median EFS, months (95% CI) | 29.5 (9.5 to NE) | 2.4 (2.2 to 4.9) |
Stratified HR (95% CI)a,b,c | 0.38 (0.26 to 0.54) | |
KM probability of being event-free at 12 months, % (95% CI) | 57.0 (46.8 to 67.2) | 22.6 (13.9 to 31.3) |
Between-group difference, % (95% CI) | 34.4 (21.0 to 47.8) | |
KM probability of being event-free at 36 months, % (95% CI) | 45.8 (35.2 to 56.5) | 19.1 (11.0 to 27.3) |
Between-group difference, % (95% CI) | 26.7 (13.3 to 40.1) | |
CI = confidence interval; CR = complete response; EFS = event-free survival; HR = hazard ratio; IRC = independent review committee; ITT = intention to treat; KM = Kaplan-Meier; liso-cel = lisocabtagene maraleucel; NE = not estimable; NR = not reported; PD = progressive disease; PR = partial response; SOC = standard of care.
Note: EFS was defined as the time from randomization to death due to any cause; PD was defined as failure to achieve a CR or PR by 9 weeks post randomization, or the start of a new antineoplastic therapy due efficacy concerns, whichever occurred first.
aGreenwood formula.
bBased on a stratified Cox proportional hazards model.
cThe analysis was descriptive.
Sources: TRANSFORM Clinical Study Report.16 Details included in the table are from the sponsor’s summary of clinical evidence.
Figure 2: Kaplan-Meier Plot of Event-Free Survival — ITT Population, TRANSFORM Trial (Final Analysis) — Redacted

Sources: TRANSFORM Clinical Study Report.16 Details included in the table are from the sponsor’s summary of clinical evidence.
Complete Response Rate by IRC
At the time of the primary analysis, the CRR in the liso-cel group was 73.9% (95% CI, 63.7% to 82.5%) versus 43.5% (95% CI, 33.2% to 54.2%; stratified 1-sided P < 0.0001) with a between-group difference of 29.3% (95% CI, 16.4% to 42.2%). The CRR remained consistent at the final analysis. The results of the sensitivity analyses were consistent with the primary analysis.
Progression-Free Survival by IRC
Table 13 provides a summary of results for PFS by IRC. At the time of the primary analysis, PFS events had been reported for 37 patients (40.2%) in the liso-cel group and 52 patients (56.5%) in the SOC group. The median PFS was not reached (95% CI, 12.6 to NE) in the liso-cel group versus 6.2 months (95% CI, 4.3 to 8.6 months) in the SOC group (1-sided P < 0.0001), with a between-group HR of 0.40 (95% CI, 0.26 to 0.62). The results of the sensitivity analyses were consistent with the primary analysis. The KM-estimated probability of PFS at 12 months was 63.1% (95% CI, 53.0% to 73.3%) in the liso-cel group versus 31.2% (20.2 to 42.3) in the SOC group, with a between-group difference of 31.9% (95% CI, 16.9% to 46.9%).
At the time of the final analysis, PFS events had been reported in ██ ███████ patients in the liso-cel group and ██ ███████ patients in the SOC group. The median PFS was not reached (95% CI, 12.6 to NE) in the liso-cel group and was 6.2 months (95% CI, 4.3 to 8.6 months) in the SOC group. The KM-estimated probability of PFS at 12 months was consistent with the primary analysis and, at 36 months, was 50.9% (95% CI, 39.9% to 62.0%) for the liso-cel group versus 26.5% (95% CI, 15.9% to 37.1%) in the SOC group, with a between-group difference of 24.4% (95% CI, 9.1% to 39.7%) (Figure 4).
Figure 3: Forest Plot of EFS by Subgroups — ITT Population, TRANSFORM Trial (Second Interim Analysis)
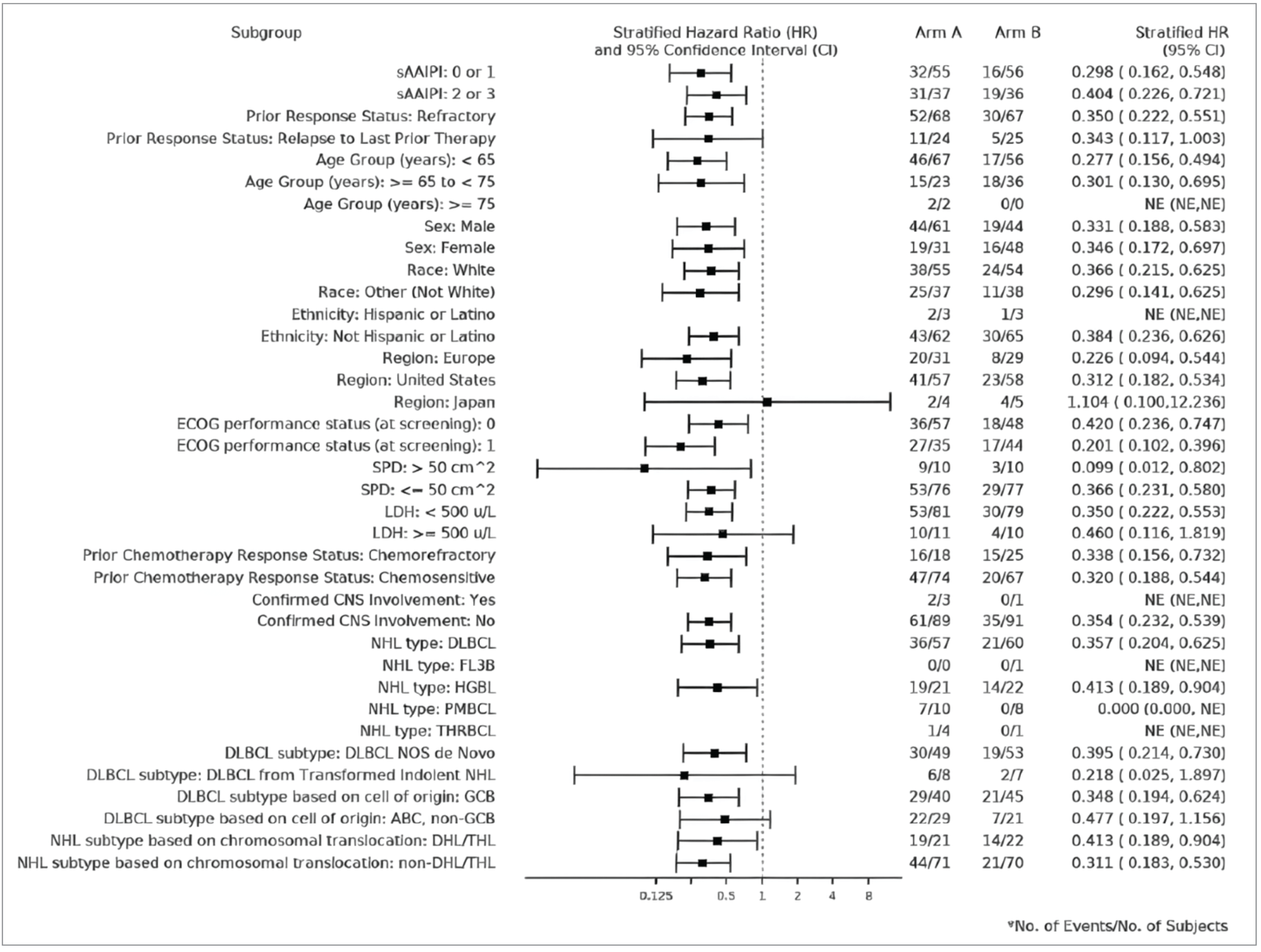
ABC = activated B cell; CNS = central nervous system; CI = confidence interval; DHL = double-hit lymphoma; DLBCL = diffuse large B-cell lymphoma; ECOG = Eastern Cooperative Oncology Group; EFS = event-free survival; FL3B = follicular lymphoma grade 3B; GCB = germinal centre B cell; HGBL = high-grade B-cell lymphoma; HR = hazard ratio; ITT = intention to treat; LDH = lactate dehydrogenase; liso-cel = lisocabtagene maraleucel; NE = not estimable; NHL = non-Hodgkin lymphoma; NOS = not otherwise specified; PMBCL = primary mediastinal B-cell lymphoma; sAAIPI = secondary Age-Adjusted International Prognostic Index; SOC = standard of care; SPD = sum of the product diameters; THL = triple-hit lymphoma; THRBCL = T-cell or histiocyte-rich large B-cell lymphoma.
Note: Arm A was the SOC group; arm B was the liso-cel group.
Sources: TRANSFORM Clinical Study Report.16 Details included in the table are from the sponsor’s summary of clinical evidence.
Table 13: Progression-Free Survival — ITT Population, TRANSFORM Trial
PFS by IRC | TRANSFORM trial | |
|---|---|---|
Liso-cel (N = 92) | SOC (N = 92) | |
Primary analysis data cut-off date of May 13, 2022 | ||
Patients with events, n (%) | ||
Total | 37 (40.2) | 52 (56.5) |
Death | █████ | █████ |
PD | ██ ██████ | ██ ██████ |
Patients censored | 55 (59.8) | 40 (43.5) |
Median PFS, months (95% CI) | NE (12.6 to NE) | 6.2 (4.3 to 8.6) |
Stratified HR (95% CI)a,b | 0.40 (0.26 to 0.62) | |
One-sided P valueb | < 0.0001 | |
KM probability of being event-free at 12 months, % (95% CI) | 63.1 (53.0 to 73.3) | 31.2 (20.2 to 42.3) |
Between-group difference, % (95% CI) | 31.9 (16.9 to 46.9) | |
Final analysis data cut-off date of October 23, 2023 | ||
Patients with events, n (%) | ||
Total | 41 (44.6) | 54 (58.7) |
Death | █████ | █████ |
PD | ██ ██████ | ██ ██████ |
Patients censored | 51 (55.4) | 38 (41.3) |
Median PFS, months (95% CI) | NE (12.6 to NE) | 6.2 (4.3 to 8.6) |
Stratified HR (95% CI)a,b | 0.42 (0.28 to 0.64) | |
One-sided P valueb,c | < 0.0001 | |
Difference, % (95% CI) | 31.9 (16.9 to 46.9) | |
KM probability of being event-free at 12 months, % (95% CI) | 63.0 (52.8 to 73.2) | 31.3 (20.3 to 42.4) |
Between-group difference, % (95% CI) | 31.7 (16.7 to 46.7) | |
KM probability of being event-free at 36 months, % (95% CI) | 50.9 (39.9 to 62.0) | 26.5 (15.9 to 37.1) |
Between-group difference, % (95% CI) | 24.4 (9.1 to 39.7) | |
CI = confidence interval; EFS = event-free survival; HR = hazard ratio; IRC = independent review committee; ITT = intention to treat; KM = Kaplan-Meier; liso-cel = lisocabtagene maraleucel; NE = not estimable; NR = not reported; PD = progressive disease; PFS = progression-free survival; SOC = standard of care.
Note: EFS was defined as the time from randomization to death due to any cause; PD was defined as failure to achieve a complete response or a partial response by 9 weeks post randomization, or the start of a new antineoplastic therapy due to efficacy concerns, whichever occurred first.
Data cut-off date: May 13, 2022 (primary analysis).16
aGreenwood formula.
bBased on a stratified Cox proportional hazards model.
cNot included in the hierarchical testing strategy.
Sources: TRANSFORM Clinical Study Report.16 Details included in the table are from the sponsor’s summary of clinical evidence.
Figure 4: Kaplan-Meier Plot of Progression-Free Survival — ITT Population, TRANSFORM Trial (Final Analysis) — Redacted

Sources: TRANSFORM Clinical Study Report.16 Details included in the table are from the sponsor’s summary of clinical evidence.
Overall Survival
Table 14 provides a summary of OS findings for the primary and final analyses. By the primary analysis, there were 28 ███████ deaths in the liso-cel group and 38 ███████ deaths in the SOC group. The median OS was not reached (95% CI, 29.5 to NE) in the liso-cel group versus 29.9 months (95% CI, 17.9 to NE) months in the SOC group (1-sided P = 0.0987), with a between-group HR of 0.72 (95% CI, 0.44 to 1.18). The KM-estimated probability of being alive at 12 months was 83.4% (95% CI, 75.7 to 91.1) in the liso-cel group versus 72.0% (95% CI, 62.7 to 81.3) in the SOC group, with a between-group difference of 11.4% (95% CI, −0.7 to 23.5).
At the time of the final analysis, there were 34 ███████ deaths in the liso-cel group and 42 ███████ deaths in the SOC group. The median OS was not reached for either treatment group (liso-cel: 95% CI, 42.8 to NE; SOC: 95% CI, 18.2 to NE), with a between-group HR of 0.76 (95% CI, 0.481 to 1.19). The KM-estimated probability of OS at 12 months was consistent with the primary analysis and, at 36 months, was 62.8% (95% CI, 52.7 to 72.9) in the liso-cel group versus 51.8% (95% CI, 41.2 to 62.4) in the SOC group, with a between-group difference of 11.0% (95% CI, −3.7 to 25.7) (Figure 5).
Table 14: Overall Survival — ITT Population, TRANSFORM Trial
OS | TRANSFORM Trial | |
|---|---|---|
Liso-cel (N = 92) | SOC (N = 92) | |
Primary analysis data cut-off date of May 13, 2022 | ||
Death, N (%) | 28 (30.4) | 38 (41.3) |
Patients censored | 64 (69.6) | 54 (58.7) |
Median OS, months (95% CI) | NE (29.5 to NE) | 29.9 (17.9 to NE) |
Stratified HR (95% CI)a,b | 0.72 (0.44 to 1.18) | |
One-sided P valueb | 0.0987 | |
KM probability of being event-free at 12 months, % (95% CI) | 83.4 (75.7 to 91.1) | 72.0 (62.7 to 81.3) |
Between-group difference, % (95% CI) | 11.4 (−0.7 to 23.5) | |
Final analysis data cut-off date of October 23, 2023 | ||
Death, N (%) | 34 ██████ | 42 ██████ |
Patients censored | ██ ██████ | ██ ██████ |
Median OS, months (95% CI) | NE (42.8 to NE) | NR (18.2 to NE) |
Stratified HR (95% CI)a,b,c | 0.76 (0.48 to 1.19) | |
KM probability of being event-free at 12 months, % (95% CI) | ████ ████ | ████ ██ |
Between-group difference, % (95% CI) | ████ █████ ██ █████ | |
KM probability of being event-free at 36 months, % (95% CI) | 62.8 (52.7 to 72.9) | 51.8 (41.2 to 62.4) |
Between-group difference, % (95% CI) | 11.0 (−3.7 to 25.7) | |
CI = confidence interval; HR = hazard ratio; ITT = intention to treat; KM = Kaplan-Meier; liso-cel = lisocabtagene maraleucel; NE = not estimable; NR = not reported; OS = overall survival; SOC = standard of care.
Note: EFS was defined as the time from randomization to death due to any cause, progressive disease, failure to achieve a complete response or a partial response by 9 weeks post randomization, or the start of a new antineoplastic therapy due efficacy concerns, whichever occurred first.
aGreenwood formula.
bBased on a stratified Cox proportional hazards model.
cThe analysis was descriptive.
Sources: TRANSFORM Clinical Study Report.16 Details included in the table are from the sponsor’s summary of clinical evidence.
Figure 5: Kaplan-Meier Plot of Overall Survival — ITT Population, TRANSFORM Trial (Final Analysis) — Redacted

Sources: TRANSFORM Clinical Study Report.16 Details included in the table are from the sponsor’s summary of clinical evidence.
HRQoL by EORTC QLQ-C30 and FACT-LymS
The EORTC QLQ-C30 global health status and FACT-LymS scores are summarized in Table 15. Of the ITT population, ██ ███████ patients in the liso-cel group and ██ ███████ patients in the SOC group were included in the EORTC QLQ-C30 analysis set. For the FACT-LymS analysis set, ██ ███████ and ██ ███████ patients were included, respectively. At baseline, global health status scores were similar between treatment groups and there were clinically meaningful changes (defined by the sponsor as change in the score from baseline of ≥ 5 points) observed in both groups at 6 months. The between-group LS mean difference in change from baseline was ████ ████ ███ █████ ██ ██████. At baseline, total FACT-LymS scores were similar between groups. At 6 months, there was no clinically meaningful change (defined by the sponsor as change in the score from baseline of ≥ 3 points) observed in the liso-cel group, while a clinically meaningful change was observed in the SOC group. The between-group LS mean difference in change from baseline was █████ ████ ███ █████ ██ █████.
Harms
The harms data reported in this section are from the primary analysis (data cut-off date of May 13, 2022). There were no significant changes in the incidence of TEAEs from the time of the interim analysis to the time of the primary analysis. The key harm results for the safety analysis population are summarized in Table 16.
Table 15: Least Squares Mean Changes in EORTC QLQ-C30 and FACT-LymS Scores — HRQoL Population, TRANSFORM Trial (Primary Analysis)
Scales (data cut-off date of May 13, 2022) | TRANSFORM trial | |
|---|---|---|
Liso-cel (██) | SOC (██) | |
EORTC QLQ-C30 global health status score (higher score indicates better health status) | ||
Baseline, mean (95% CI), n | ████ █████ ██ ███ | ████ █████ ██ ███ |
At 6 months, mean (95% CI), n | ████ █████ ██ ███ | ████ █████ ██ ███ |
Change from baseline, LS mean (95% CI)a | ████ █████ ██ ███ | ████ █████ ██ ███ |
Between-group difference, LS mean (95% CI) | ████ █████ ██ ███ | |
FACT-LymS score (higher score indicates lower levels of symptoms) | ||
Baseline, mean (95% CI), n | ████ █████ ██ ███ | ████ █████ ██ ███ |
At 6 months, mean (95% CI), n | ████ █████ ██ ███ | ████ █████ ██ ███ |
Change from baseline, LS mean (95% CI) | ████ █████ ██ ███ | ████ █████ ██ ███ |
Between-group difference, LS mean (95% CI) | ████ █████ ██ ███ | |
CI = confidence interval; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; FACT-LymS = Functional Assessment of Cancer Therapy–Lymphoma “additional concerns” subscale; HRQoL = health-related quality of life; liso-cel = lisocabtagene maraleucel; LS = least squares; sAAIPI = secondary Age-Adjusted International Prognostic Index; SOC = standard of care.
aThe analysis was performed using all HRQoL data, and the visits were truncated at month 6. Changes from baseline are used as the dependent variable. The mixed effects with repeated measures model assumes an unstructured covariance and includes a random intercept or slope and fixed effects, including treatment group, time (i.e., visit as a categorical variable), stratification factors (i.e., the best overall response to first-line therapy [refractory versus relapse] and sAAIPI [0 or 1 versus 2 or 3]), baseline score, and treatment group by time interaction. An evaluable EORTC QLQ-C30 assessment was defined as having at least 1 of the 15 domains answered at a given assessment. An evaluable FACT-LymS assessment was defined as having at least 8 of the 15 items answered at a given assessment visit.
Sources: TRANSFORM Clinical Study Report.16 Details included in the table are from the sponsor’s TRANSFORM PRO Tables. Data cut-off May 13, 2022.
Adverse Events
Almost all patients in the trial reported at least 1 TEAE (liso-cel: 100%; SOC: 98.9%). The most frequently reported TEAEs of any grade in both treatment groups were neutropenia (liso-cel: 82.6%; SOC: 54.9%), anemia (liso-cel: 67.4%; SOC: 68.1%), thrombocytopenia (liso-cel: 59.8%; SOC: 72.5%), and nausea (liso-cel: 53.3%; SOC: 58.2%). Of these TEAEs, a numerically higher proportion of neutropenia was reported in patients taking liso-cel and a higher proportion of thrombocytopenia was reported in patients taking SOC. Most patients in both groups reported at least 1 grade 3 or 4 TEAE (liso-cel: 92.4%; SOC: 89.0%). The incidence of grade 3 or 4 neutropenia (liso-cel: 81.5%; SOC: 51.6%) and lymphopenia (liso-cel: 26.1%; SOC: 9.9%) was numerically higher in the liso-cel group versus the SOC group.
Serious Adverse Events
The incidence of serious TEAEs was similar between groups (liso-cel: 47.8%; SOC: 48.4%). The most frequently reported serious TEAEs were CRS (liso-cel: 13%; SOC: 0%), febrile neutropenia (liso-cel: 7.6%; SOC: 9.9), pyrexia (liso-cel: 6.5%; SOC: 7.7), and neutropenia (liso-cel: 7.6; SOC: 4.4%). The frequency of these TEAEs was similar between groups, except a higher proportion of CRS was reported in patients taking liso-cel.
Withdrawals Due to Adverse Events
Four patients (4.4%) in the SOC group experienced TEAEs leading to treatment withdrawal. No patients in the liso-cel group had a TEAE that led to the withdrawal of the study drug (including bridging therapy and LDC).
Mortality
██████████ ████ ███ ██████ ██ deaths were reported in ███ of patients in the liso-cel group and ███ of patients in the SOC group. The majority of deaths in both groups were attributed to disease progression (█████████ ██) followed by TEAEs (█████████ █████).
Notable Harms
A numerically higher proportion of notable TEAEs was reported in patients taking liso-cel (90.2%) versus SOC (75.8%). The most frequently reported notable harms of any grade were neurologic toxicity (liso-cel: 64.1%; SOC: 62.6%), CRS (liso-cel: 48.9%; SOC: 0.0%), prolonged cytopenia (liso-cel: 43.5%; SOC: 3.3%), and investigator-identified neurologic toxicity (liso-cel: 10.9; SOC: not applicable). These events occurred more frequently in patients taking liso-cel, except for neurologic toxicity, which was similar in both groups.
Table 16: Summary of Harms — Safety Population, TRANSFORM Trial (Primary Analysis)
Harms (data cut-off date of May 13, 2022) | Liso-cel (N = 92) | SOC (N = 91) | ||
|---|---|---|---|---|
Any grade | Grade 3 or 4 | Any grade | Grade 3 or 4 | |
Primary analysis data cut-off date of May 13, 2022 | ||||
TEAEs, n (%) | 92 (100) | 85 (92.4) | 90 (98.9) | 81 (89.0) |
Most common TEAEs (≥ 20% of any grade in either treatment group), n (%) | ||||
Thrombocytopenia | 55 (59.8) | 46 (50.0) | 66 (72.5) | 62 (68.1) |
Anemia | 62 (67.4) | 48 (52.2) | 62 (68.1) | 51 (56.0) |
Nausea | 49 (53.3) | 3 (3.3) | 53 (58.2) | 4 (4.4) |
Neutropenia | 76 (82.6) | 75 (81.5) | 50 (54.9) | 47 (51.6) |
Diarrhea | 23 (25.0) | 0 (0) | 39 (42.9) | 3 (3.3) |
Fatigue | 37 (40.2) | 0 (0) | 37 (40.7) | 2 (2.2) |
Decreased appetite | 21 (22.8) | 1 (1.1) | 32 (35.2) | 4 (4.4) |
Vomiting | 18 (19.6) | 1 (1.1) | 27 (29.7) | 2 (2.2) |
Febrile neutropenia | 15 (16.3) | 11 (12.0) | 24 (26.4) | 21 (23.1) |
Constipation | 30 (32.6) | 2 (2.2) | 24 (26.4) | 0 (0) |
Pyrexia | 28 (30.4) | 0 (0) | 23 (25.3) | 0 (0) |
Hypokalemia | 21 (22.8) | 4 (4.3) | 22 (24.2) | 4 (4.4) |
Hypomagnesemia | 15 (16.3) | 0 (0) | 21 (23.1) | 1 (1.1) |
Headache | 40 (43.5) | 4 (4.3) | 21 (23.1) | 1 (1.1) |
Dizziness | 22 (23.9) | 0 (0) | 13 (14.3) | 0 (0) |
Lymphopenia | 25 (27.2) | 24 (26.1) | 11 (12.1) | 9 (9.9) |
Insomnia | 19 (20.7) | 0 (0) | 10 (11.0) | 0 (0) |
Hypotension | 19 (20.7) | 3 (3.3) | 6 (6.6) | 0 (0) |
Cytokine release syndrome | 45 (48.9) | 1 (1.1) | 0 (0) | 0 (0) |
Any serious TEAEs, n (%) | 44 (47.8) | — | 44 (48.4) | — |
Serious TEAEs reported in ≥ 2% of patients in either group, n (%) | ||||
Cytokine release syndrome | ██ ██████ | — | █████ | — |
Febrile neutropenia | █████ | — | █████ | — |
Pyrexia | █████ | — | █████ | — |
Neutropenia | █████ | — | █████ | — |
Acute kidney injury | █████ | — | █████ | — |
Thrombocytopenia | █████ | — | █████ | — |
Anemia | █████ | — | █████ | — |
Pneumonia | █████ | — | █████ | — |
Sepsis | █████ | — | █████ | — |
COVID-19 | █████ | — | █████ | — |
Escherichia sepsis | █████ | — | █████ | — |
Peripheral swelling | █████ | — | █████ | — |
Headache | █████ | — | █████ | — |
Aphasia | █████ | — | █████ | — |
Pulmonary embolism | █████ | — | █████ | — |
Confusional state | █████ | — | █████ | — |
All-cause TEAEs leading to treatment discontinuation, n (%) | █████ | — | █████ | — |
Deaths, n (%) | ██ ██████ | — | █████ | — |
Disease progression | █████ | — | █████ | — |
Due to AE | █████ | — | █████ | — |
Other | █████ | — | █████ | — |
Unknown | █████ | — | █████ | — |
Notable harms, n (%) | ||||
Neurologic toxicity | ██ ██████ | ██ ██████ | ██ ██████ | █████ |
Cytokine release syndrome | 45 (48.9) | 0 (0) | █████ | █████ |
Prolonged cytopenia | 40 (43.5) | ██ ██████ | 3 (3.3) | █████ |
iiNT | 11 (10.9) | NA | NA | NA |
Severe infections (grade ≥ 3) | 14 (15.2) | 14 (15.2) | 19 (20.9) | 19 (20.9) |
Hypogammaglobulinemia | 10 (10.9) | █████ | 3 (3.3) | █████ |
Infusion-related reaction | █████ | █████ | █████ | █████ |
COVID-19 | █████ | █████ | █████ | █████ |
Second primary malignancy | █████ | █████ | █████ | █████ |
Tumour lysis syndrome | █████ | █████ | █████ | █████ |
Macrophage activation syndrome | █████ | █████ | █████ | █████ |
AE = adverse event; iiNT = investigator-identified neurologic toxicity; liso-cel = lisocabtagene maraleucel; NA = not applicable; SOC = standard of care; TEAE = treatment-emergent adverse event.
Sources: TRANSFORM Clinical Study Report.16 Details included in the table are from the sponsor’s summary of clinical evidence.
Critical Appraisal
Internal Validity
The TRANSFORM trial was a randomized, open-label, phase III trial. Randomization procedures, including stratification by response to first-line therapy (relapsed versus refractory) and sAAIPI (0 to 1 versus 2 to 3) were appropriate and conducted by interactive response technology. The liso-cel group had a lower proportion of male patients (47.8% versus 66.3%) and a higher proportion of patients with chemorefractory disease (28.3% versus 19.6%) compared with the SOC group. These imbalances were likely due to chance, as all other baseline patient characteristics appeared balanced between groups and, based on clinical expert feedback, were unlikely to have confounded the effect between treatment and outcomes.
Treatment period discontinuation was numerically higher in the SOC (59.8%) versus the liso-cel (12.0%) group, with lack of efficacy being the most common reason (SOC: 30%; liso-cel: 0%). Important protocol deviations were similar between groups (liso-cel: 13%; SOC: 14.1%) and were due primarily to failure to report serious AEs or suspected unexpected serious adverse reactions per regulations. Since the type and frequency of these deviations were comparable between the treatment groups, the presence, magnitude, and direction of potential bias are unclear.
The open-label design introduces potential bias in the assessment of efficacy for EFS, CRR, and PFS and introduces a potential reporting bias for the subjective outcomes of HRQoL and safety, although this bias was mitigated by the use of an IRC for EFS, CRR, and PFS. To minimize the risk of differential measurement error, the trial investigators performed tumour assessments using Lugano criteria, and radiographic scans were assessed by IRC. For the HRQoL and safety outcomes, the extent and direction of bias could overestimate the efficacy of liso-cel. Objective outcomes such as OS are unlikely to be affected by bias due to the open-label design of the trial.
Sample size and power calculations were based on EFS, and the trial was powered to detect significant differences between groups for EFS. Prespecified analysis of EFS, CRR, PFS, and OS were appropriately controlled for multiple comparisons. All other analyses were descriptive. This included the HRQoL outcomes of EORTC QLQ-C30 and FACT-LymS, which were deemed clinically important outcomes for the disease. The sample size for most subgroup analyses of interest appeared large enough to detect subgroup differences, except for DLBCL transformed from indolent NHL, which may not have been powered to detect subgroup differences. The findings of the sensitivity analyses for the primary outcome of EFS were consistent with the primary analysis. The proportional hazards assumption was assessed via inspection of Schoenfeld residuals, and the trial authors stated there was no evidence of a violation of this assumption.
The median OS was not reached in either treatment group at the primary and final analyses due to the small number of OS events. As such, longer follow-up is needed to inform the true effect of liso-cel compared with SOC on survival. In addition, patients were permitted to receive posttreatment anticancer medications after study treatment had been discontinued (liso-cel: 34.8%; SOC: 70.7%; 65 patients, of which 61 patients were approved to cross over to liso-cel); such medications were not balanced between groups, which may have influenced the assessment of OS.
The EORTC QLQ-C30 and FACT-LymS questionnaires have been validated in patients with cancer and lymphoma, respectively, with evidence of reliability and no established MIDs, although ranges for clinically meaningful change thresholds for both instruments have been identified in the literature. Based on the clinically meaningful change thresholds identified in the literature and feedback from the clinical experts consulted by CDA-AMC, a 10-point change from baseline score was used as a clinically meaningful change for EORTC QLQ-C30. For the FACT-LymS score, the sponsor suggested a 3-point change from baseline score as a clinically meaningful change, which was considered reasonable by the CDA-AMC team. No evidence was identified in the literature for responsiveness for both instruments. The certainty of evidence for the HRQoL outcomes was limited due to risk of bias due to missing outcomes data, both at baseline and at the selected follow-up times, and due to imprecision because the 95% CI for the between-group difference included the possibility of both benefit and little to no difference; however, the direction and extent of bias is unclear and, as such, the potential differences on patients’ HRQoL remains very uncertain.
External Validity
The population requested for reimbursement aligns with the approved Health Canada indication. The dosing and administration of liso-cel was consistent with the approved product monograph.
According to the clinical experts consulted by CDA-AMC, the eligibility criteria and baseline characteristics of the TRANSFORM trial were generalizable to adults with relapsed or refractory LBCL in the Canadian setting. The trial did not include patients with a poor ECOG Performance Status, and the clinical experts noted that only enrolling patients with an ECOG score of 0 and 1 is not entirely representative of patients with relapsed or refractory LBCL in Canada, as they expect to have patients with higher ECOG scores in their practice. The clinical experts also noted that autologous HSCT eligibility is highly variable in clinical practice across Canada, as there is no standardized criteria to identify patients.
The trial included outcomes that were important to patients and clinicians. The patient group indicated that stopping disease progression, prolonging life, improving HRQoL, and reducing treatment side effects are important to them.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For the pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered most relevant to inform CDA-AMC’s expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group:31,32
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate. The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited. The true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate. The true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as “very uncertain.”
Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty-of-evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null.
The reference points for the certainty-of-evidence assessment for the EFS, CRR, PFS, OS, and EORTC QLQ-C30 global health status scores were set according to the presence or absence of an important effect based on thresholds informed by the clinical experts consulted for this review and ranges identified in the literature for the EORTC QLQ-C30. The reference point for the certainty-of-evidence assessment for the FACT-LymS total score was set according to the presence or absence of an important effect based on a threshold suggested by the sponsor that was informed by the literature.
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for liso-cel versus SOC.
Long-Term Extension Studies
No long-term extension studies were submitted by the sponsor.
Indirect Evidence
Contents within this section have been informed by materials submitted by the sponsor. The following was summarized and validated by the CDA-AMC review team.
Objectives for the Summary of Indirect Evidence
The aim of this section is to summarize and critically appraise 1 sponsor-submitted MAIC17 to fill gaps in the comparative evidence for other treatments of interest for relapsed or refractory LBCL, and to briefly appraise 1 sponsor-submitted ITC (i.e., Bucher ITC)33 that was used to inform the pharmacoeconomic model.
Description of MAIC
The study selection criteria and methods for the MAIC are summarized in Table 17. The sponsor did not report a systematic literature search or describe the methods for study selection, data extraction, and quality assessment.
Table 17: Study Selection Criteria and Methods for MAIC Submitted by the Sponsor
Characteristics | Indirect comparison |
|---|---|
Population | The study populations for the TRANSFORM and ZUMA-7 trials were compared in terms of eligibility criteria and baseline characteristics to determine whether imbalances existed and to what extent they might affect the indirect comparison or could be dealt with quantitatively via population-adjustment ITC approaches. |
Intervention | Liso-cel per TRANSFORM trial dosing regimen; patients received liso-cel as 2 sequential IV infusions of CD8+ and CD4+ CAR+ T cells at a total target dose of 100 × 106 CAR+ T cells. |
Comparator | Axi-cel per ZUMA-7 trial dosing regimen; patients received a single infusion of axi-cel with a target dose of 2 × 106 CAR T cells per kilogram of body weight. |
Outcome | Efficacy outcomes evaluated were EFS, PFS, OS, ORR, CRR, and safety. The median study follow-up time was shorter in the TRANSFORM trial (defined as time from randomization to last known alive date; 33.86 months) than in the ZUMA-7 trial (defined as time from randomization to data cut-off; 47.2 months). For this updated ITC analysis, IRC-assessed ORR and CRR as well as OS were reported in both trials and therefore compared between liso-cel and axi-cel. For PFS, the ZUMA-7 trial presented only investigator-assessed data; therefore, investigator-assessed PFS was compared between liso-cel and axi-cel. For EFS, IRC-assessed EFS was not reported in Westin et al. (2023); therefore, IRC-assessed EFS was compared using the 2-year data for axi-cel and investigator-assessed EFS was compared using the long-term follow-up data for axi-cel versus TRANSFORM IPD. |
Study designs | Both the TRANSFORM and ZUMA-7 studies were phase III, open-label, multicentre randomized control trials with 1:1 randomization. The majority of centres were located in North America and Europe. Both trials had a similar induction period of 3-week cycles of salvage chemotherapy for the SOC arm. Both trials included R-DHAP, R-GDP, and R-ICE as salvage chemotherapy options per National Comprehensive Cancer Network guidelines. The ZUMA-7 trial also included R-ESHAP as an additional option. Responders to the salvage chemotherapy induction therapy proceeded to transplant for the SOC arm in both trials. Bridging chemotherapy was allowed only for patients receiving liso-cel in the TRANSFORM trial, which permitted the use of protocol-defined SOC chemotherapy regimens to stabilize disease. The ZUMA-7 trial permitted the use of corticosteroids only for patients with high disease burden at screening. Due to the difference in the use of bridging therapy between the trials, adjustment for this characteristic would not be feasible in the MAIC. The effect of bridging therapy on outcomes is complex. While it may improve disease control before CAR T-cell infusion, providing a response benefit and decreased severe toxicity, it has been suggested that patients who require bridging therapy may have inferior outcomes compared with those who do not, as these patients tend to have bulkier, more rapidly progressive or symptomatic disease compared with patients who do not receive bridging therapy. Patients in the SOC arm of both trials could receive subsequent CAR T-cell therapy. Specifically, the TRANSFORM trial permitted crossover for SOC patients with progression or lack of response. Although the ZUMA-7 trial did not plan for crossover, patients who progressed or did not respond to SOC could receive CAR T-cell therapy off protocol. The percentage of patients who crossed over or received subsequent CAR T-cell therapy was higher in the TRANSFORM trial (66.3%) than in the ZUMA-7 trial (56%). |
Publication characteristics | The manuscripts of both the TRANSFORM and ZUMA-7 studies have been published in peer-reviewed journals with supplements detailing the study protocols. The primary source of data for axi-cel used to inform the current analyses was the peer-reviewed publication for the ZUMA-7 trial (data cut-off: March 18, 2021) by Locke et al. (2021) and the longer-term follow-up data from the Westin (2023) publication. For liso-cel, the primary data source used to inform the current analysis was IPD from the TRANSFORM final analysis (data cut-off: October 23, 2023). |
Exclusion criteria | This MAIC focused on the pivotal trials for liso-cel (TRANSFORM) and axi-cel (ZUMA-7) used for second-line treatment of patients with high-risk, relapsed or refractory LBCL who are intended to receive a stem cell transplant. |
Databases searched | The systematic review identified the TRANSFORM trial with liso-cel and ZUMA-7 trial with axi-cel as appropriate supportive evidence to address this question. These 2 trials included the patient population targeted by the indication under review at CDA-AMC. As a result, additional database searches were not required to conduct this ITC. |
Selection process | NR |
Data extraction process | NR |
Quality assessment | NR |
axi-cel = axicabtagene ciloleucel; CAR = chimeric antigen receptor; CD = cluster of differentiation; CRR = complete response rate; CRS = cytokine release syndrome; EFS = event-free survival; IPD = individual patient data; IRC = independent review committee; ITC = indirect treatment comparison; LBCL = large B-cell lymphoma; liso-cel = lisocabtagene maraleucel; MAIC = matched-adjusted indirect comparison; NR = not reported; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; PR = partial response; R-DHAP = rituximab, dexamethasone, cytarabine, and cisplatin; R-ESHAP = rituximab, etoposide, methylprednisolone sodium succinate, cytarabine, and cisplatin; R-GDP = rituximab, gemcitabine, dexamethasone, and cisplatin; R-ICE = rituximab, ifosfamide, carboplatin, and etoposide; SOC = standard of care; TEAE = treatment-emergent adverse event.
Sources: MAIC technical report.17 Details included in the table are from the sponsor’s summary of clinical evidence.
MAIC Design
Objectives
The objective of the MAIC was to assess the comparative efficacy and safety of liso-cel and axi-cel using IPD from the TRANSFORM trial16 (for liso-cel) and summary-level data from the ZUMA-7 trial34 in patients with relapsed or refractory LBCL who are intended for transplant.
MAIC Analysis Methods
A feasibility assessment using study design, eligibility criteria, baseline characteristics, and outcomes was performed and determined the TRANSFORM and ZUMA-7 trials to be comparable enough to allow for the indirect comparison between liso-cel and axi-cel on key efficacy and safety outcomes through unadjusted (Bucher) or population-adjusted ITC methods. MAIC and simulated treatment comparison approaches were considered feasible to minimize potential sources of bias while comparing these therapies, and the MAIC approach was preferred by the investigators over a simulated treatment comparison.
A treatment effect modifier identification and ranking process involving eligibility criteria and baseline characteristics that were deemed most likely to modify treatment effect were prioritized, and interviews with clinical experts were undertaken to support the prioritization of the adjustment for the most relevant effect-modifying factors (hereafter referred as clinical factors). These clinical factors were identified based on variables available in both studies and a prior ranking exercise for CAR T-cell therapies. A data-driven ranking for each outcome was initially performed using the Bayesian additive regression trees method using IPD from the TRANSFORM trial. The clinical factors were ranked by assessing the interaction effect between treatment (i.e., liso-cel versus SOC) and the clinical factors in predicting the efficacy outcomes in a statistical model. The rankings were then reviewed and revised by a panel of 3 external clinicals experts to create the final ranking of clinical factors to be considered for analysis. The clinical factors for the efficacy and safety analyses are in Table 18.
Matching consisted of aligning trials on inclusion and exclusion criteria. Point estimates representing an ITC for liso-cel versus axi-cel were derived as the difference between this prediction and the estimated outcome using published summary-level data from the comparator trial. Given there was a common comparator group between the TRANSFORM and ZUMA-7 trials (the SOC group), anchored MAICs were conducted for the efficacy analyses. For the safety outcomes, the authors performed unanchored MAICs for 2 reasons: SOC arm data were not available or very limited for several safety outcomes, and the authors deemed it may not be clinically valid to use the SOC arm as the common comparator due to the fundamental differences in treatment modalities and toxicity profiles between CAR T-cell therapy and chemotherapies.
Outcome of interests included EFS, PFS, CRR, OS, and TEAEs. IPD from the TRANSFORM trial were adjusted to match the marginal distribution (e.g., mean, variance) of clinical factors among patients from the ZUMA-7 trial. As the TRANSFORM trial included a broader patient population than the ZUMA-7 trial, patients from the TRANSFORM trial were excluded from the analyses if they did not satisfy the eligibility criteria specified in the ZUMA-7 trial. Data for patients who remained in the TRANSFORM trial were weighted using a method of moments approximation to a propensity score model. Baseline characteristics and outcome definitions were compared and aligned with patients in the ZUMA-7 trial, when feasible. For the anchored efficacy analyses, patients from the TRANSFORM trial were weighted so they matched the marginal distribution of baseline characteristics from the combined population of axi-cel and SOC groups from the ZUMA-7 trial. For the unanchored safety analyses, patients from the TRANSFORM trial’s liso-cel group were weighted so they matched the marginal distribution of baseline characteristics from the axi-cel group from the ZUMA-7 trial. Clinically relevant factors (i.e., eligibility criteria and baseline characteristics suspected to be treatment effect modifiers) were adjusted collectively and in order of by ranked importance. Primary scenarios were selected to capture the impact of most clinically important effect modifiers while maintaining a reasonable ESS. Sensitivity scenarios, which include all relevant effect modifiers available, were conducted to test robustness of the findings. Scenario selections were validated with clinical experts.
For EFS, PFS, and OS, Cox proportional hazards models were used to estimate HRs for liso-cel versus axi-cel. Proportionality was assessed through visual inspections of the KM curves, plots of log cumulative hazards, and a Grambsch-Therneau test on the slope of the Schoenfeld residuals. Generalized linear models for binary outcomes (e.g., CRR) were used to estimate odds ratios (ORs) for liso-cel versus axi-cel. For the unanchored analysis of safety outcomes, weighted log odds for liso-cel were estimated in the TRANSFORM trial by fitting an intercept-only logistic regression model with MAIC adjustment weights. Point estimates (HRs or ORs) and 95% CIs were reported for all analyses.
Table 18: Clinical Factors Included in Efficacy and Safety Analyses Comparing Liso-Cel With Axi-Cel
Clinical factors | Efficacy analyses | Safety analysis | |
|---|---|---|---|
Primary analysis | Sensitivity analysis | ||
Factors that were matched on |
|
|
|
Factors that were adjusted for |
|
|
|
ALC = absolute lymphocyte count; axi-cel = axicabtagene ciloleucel; CNS = central nervous system; ECOG PS = Eastern Cooperative Oncology Group Performance Status; liso-cel = lisocabtagene maraleucel; LVEF = left ventricular ejection fraction; MAIC = matching-adjusted indirect comparison; sAAIPI = secondary Age-Adjusted International Prognostic Index; SPD = sum of the product diameters.
Sources: MAIC technical report.17 Details included in the table are from the sponsor’s summary of clinical evidence.
Results of MAIC
Summary of Included Studies
A summary of key study characteristics for the TRANSFORM and ZUMA-7 trials is in Table 19. The TRANSFORM trial enrolled a broader patient population than the ZUMA-7 trial in terms of bone marrow function, secondary central nervous system (CNS) involvement, and comorbidities. The TRANSFORM trial permitted the enrolment of patients with secondary CNS involvement (not allowed in the ZUMA-7 trial), more impaired organ functions, a left ventricular ejection fraction (LVEF) of 40% or greater (versus ≥ 50% in the ZUMA-7 trial), an alanine aminotransferase level of 5 times the upper limit of normal (ULN) or greater (versus ≤ 2.5 × ULN in the ZUMA-7 trial), a total bilirubin of less than 2 mg/dL (versus ≤ 1.5 mg/dL in the ZUMA-7 trial), and a creatinine clearance by Cockcroft Gault estimation greater than 45 mL/min (versus ≥ 60 mL/min in the ZUMA-7 trial). The TRANSFORM trial also had a lower enrolment threshold for platelet count of 50,000 cells/μL or greater (versus ≥ 75,000 cells/μL in the ZUMA-7 trial), with no requirement for absolute lymphocyte count (ALC). Both trials enrolled adult patients with an ECOG Performance Status of 0 or 1, 1 prior line of therapy, and disease that had relapsed or was refractory to the first-line treatment. To be enrolled, patients also needed to be eligible for an autologous HSCT with no prior autologous or allogeneic HSCT. Both studies enrolled patients with DLBCL NOS, transformed follicular lymphoma, HGBCL, and T-cell or histiocyte-rich LBCL. However, patients with PMBCL, grade 3B follicular lymphoma, and DLCBL transformed from indolent lymphoma (other than transformed follicular lymphoma) were eligible only in the TRANSFORM trial, while patients with Epstein-Barr virus plus DLBCL and primary cutaneous DLBCL (leg type) were permitted only in the ZUMA-7 trial. Other criteria such as prior regimen requirement, infections, history or presence of nonmalignant CNS disorders, infections, and cardiovascular conditions or clinically significant cardiac disease were similar across the trials.
Table 19: Comparison of Trial Design Features — TRANSFORM and ZUMA-7 Trials
Key trial design features | TRANSFORM (liso-cel) | ZUMA-7 (axi-cel) |
|---|---|---|
Study structure | 1:1 randomization, open-label, multicentre, phase III | 1:1 randomization, open-label, multicentre, phase III |
SOC induction period | Three 3-week cycles of salvage chemotherapy | Three 3-week cycles of salvage chemotherapy |
SOC regimen options | R-DHAP, R-GDP, R-ICE | R-DHAP, R-GDP, R-ICE, R-ESHAP |
Crossover for SOC patients | SOC patients could cross over if 1 of the following criteria were met: failure to achieve CR or PR after 3 cycles of SOC, progression at any time, or need to start a new antineoplastic therapy due to efficacy concerns after 18 weeks post randomization | No crossover planned; however, patients who had progression or lack of responsea to SOC could receive commercial CAR T-cell therapy off protocol |
Transplant requirements for SOC patients | Responders with a CR or PR to induction therapy (evaluated by a PET-CT); 3 cycles of SOC | Responders to induction therapy (CR or PR); 2 or 3 cycles of SOC |
Bridging chemotherapy for patients in the treatment arm | Patients in the treatment arm could receive bridging chemotherapy with a protocol-defined SOC regimen to stabilize their disease during liso-cel manufacturing | Not allowed; only corticosteroid was allowed for patients in the treatment arm with a high disease burden at screening |
Study locations | Belgium, France, Germany, Italy, Japan, Netherlands, Spain, Sweden, Switzerland, UK, US | Australia, Austria, Belgium, Canada, France, Germany, Israel, Italy, Netherlands, Spain, Sweden, Switzerland, UK, US |
axi-cel = axicabtagene ciloleucel; CAR T = chimeric antigen receptor T cells; CR = complete response; liso-cel = lisocabtagene maraleucel; MAIC = matching-adjusted indirect comparison; PET-CT = combined PET and CT scan; PR = partial response; R-DHAP = rituximab dexamethasone cytarabine cisplatin; R-ESHAP = rituximab, etoposide, and methylprednisolone sodium succinate, cytarabine, cisplatin; R-GDP = rituximab, gemcitabine, cisplatin, and dexamethasone; R-ICE = rituximab, ifosfamide, carboplatin, and etoposide; SOC = standard of care.
aBased on the definition for tumour response in the ZUMA-7 trial, it was assumed that response refers to CR and PR for this context.
Sources: MAIC technical report.17 Details included in the table are from the sponsor’s summary of clinical evidence.
Efficacy Results
Clinical factors from the ITT set in both trials were used to inform the efficacy analyses. The baseline values after redefinition or recategorization are summarized in Table 20. Comparisons of the ranked clinical factors between the 2 trials at baseline before or after the matching or adjusting of the ITT population are in Table 21. The results demonstrated that some factors were similar (i.e., standardized mean difference [SMD] of < 0.1) between liso-cel and axi-cel. Notable differences (i.e., SMDs ≥ 0.2) were observed for age, region, sum of the product diameters (SPD), disease histology, COO, bone marrow involvement, and secondary CNS lymphoma. After matching and adjusting for 11 factors in the primary analysis, the proportion of ranked clinical factors achieving an SMD of less than 0.2 increased to 85.7%. Factors with an SMD of less than 0.2 after MAIC included age, sex, region, sAAIPI score, SPD, relapsed or refractory status, double- or triple-hit lymphoma, disease histology, bone marrow involvement, disease stage, ALC, and secondary CNS lymphoma. Imbalances remained for ECOG Performance Status and COO. Similar trends in balance reduction after matching and adjusting were observed when using a more conservative SMD threshold of 0.1 (64.3% factors with an SMD of < 0.1). The sensitivity analysis, which matched and adjusted for all ranked factors, showed improvements in balance, with 100% of included factors achieving SMDs of less than 0.1.
Table 20: Comparison of Clinical Factors at Baseline After Definition and Categorization Alignment Between the TRANSFORM and ZUMA-7 Trials — Efficacy Analyses
Baseline clinical factors | ZUMA-7 (axi-cel) trial | TRANSFORM (liso-cel) trial | ||
|---|---|---|---|---|
Axi-cel | SOC | Liso-cel | SOC | |
N | 180 | 179 | 92 | 92 |
Age, mean (SD)a | ██ ██████ | ████ ██ | ████ ██ | █████ |
Sex, n (%) | ||||
Male | 70 (38.9) | 52 (29.1) | 44 (47.8) | 61 (66.3) |
Female | 110 (61.1) | 127 (70.9) | 48 (52.2) | 31 (33.7) |
Region, n (%)b | ||||
Europe | ██ ██████ | ██ ████ | ██ ████ | ██ ██ |
North America | ███ ██████ | ███ ███ | ██ ████ | ██ ████ |
Other | █████ | █████ | █████ | █████ |
sAAIPI score, n (%) | ||||
0 to 1 | 98 (54.4) | 100 (55.9) | 56 (60.9) | 55 (59.8) |
2 to 3 | 82 (45.6) | 79 (44.1) | 36 (39.1) | 37 (40.2) |
SPD (mm2), mean (SD) | ████ ████████ | █████ | █████ | █████ |
Relapse or refractory status, per ZUMA-7 trial definition, n (%) | ||||
Refractory | ███ ██████ | ███ ███ | ██ █████ | █████ |
Relapsed | ██ ██████ | ██ █████ | ██ █████ | █████ |
Double or triple hit, n (%) | ||||
Not applicable | ███ ██████ | ███ ███ | ██ █████ | █████ |
Unknown | █████ | ██ █████ | █████ | █████ |
Yes | ██ ██████ | ██ ████ | ██ █████ | █████ |
Histology, n (%) | ||||
DLBCL transformed from FL | ██ ██████ | ██ ████ | █████ | █████ |
DLBCL NOS | ███ ████ | ███ ████ | ██ ████ | █████ |
HGBCL | ██ ██████ | ██ ████ | ██ █████ | █████ |
FL3B | █████ | █████ | █████ | █████ |
PMBCL | █████ | █████ | █████ | █████ |
DLBCL transformed from indolent lymphoma (other than FL) | █████ | █████ | █████ | █████ |
Otherc | █████ | █████ | █████ | █████ |
Cell of origin, n (%) | ||||
ABC, non-GCB | █████ | █████ | █████ | █████ |
GCB | █████ | █████ | █████ | █████ |
Unknown | █████ | █████ | █████ | █████ |
Bone marrow involvement, n (%) | ||||
No | █████ | █████ | █████ | █████ |
Unknown | █████ | █████ | █████ | █████ |
Yes | █████ | █████ | █████ | █████ |
ECOG PS at baseline, n (%) | ||||
0 | 95 (52.8) | 100 (55.9) | 46 (50) | 49 (53.3) |
1 | 85 (47.2) | 79 (44.1) | 45 (48.9) | 41 (44.6) |
2 | 0 (0) | 0 (0) | 1 (1.1) | 2 (2.2) |
Disease stage, n (%) | ||||
Stage I or II | █████ | █████ | █████ | █████ |
Stage III or IV | █████ | █████ | █████ | █████ |
ALC, n (%) | ||||
< 100/uL | █████ | █████ | █████ | █████ |
Secondary CNS lymphoma, n (%) | █████ | █████ | █████ | █████ |
ABC = activated B cell; ALC = absolute lymphocyte count; axi-cel = axicabtagene ciloleucel; CNS = central nervous system; DLBCL = diffuse large B-cell lymphoma; EBV+ = Epstein-Barr virus–positive; ECOG PS = Eastern Cooperative Oncology Group Performance Status; FL = follicular lymphoma; FL3B = follicular lymphoma grade 3B; GCB = germinal centre B cell; HGBCL = high-grade B-cell lymphoma; ITT = intention to treat; liso-cel = lisocabtagene maraleucel; MAIC = matching-adjusted indirect comparison; NOS = not otherwise specified; PMBCL = primary mediastinal large B-cell lymphoma; sAAIPI = secondary Age-Adjusted International Prognostic Index; SD = standard deviation; SOC = standard of care; SPD = sum of the product diameters; THRBCL = T-cell or histiocyte-rich LBCL.
aThe mean and SD were estimated from the reported median and range using the methods proposed in the study by McGrath et al.
bRegion was reported in the FDA assessment of the ZUMA-7 trial.
cThe category included THRBCL, EBV+ DLBCL, and primary cutaneous leg-type DLBCL.
Sources: MAIC technical report.17 Details included in the table are from the sponsor’s summary of clinical evidence.
Table 21: Clinical Factor Balance Between Liso-Cel and Axi-Cel Before and After an MAIC — Efficacy Analyses
Clinical factor | ZUMA-7 ITT | TRANSFORM ITT | |||
|---|---|---|---|---|---|
Before MAIC | After MAIC (Primarya) | ||||
███ | ███ | ███ █ ██ | |||
Statistics | Statistics | SMD | Statistics | SMD | |
Age, mean (SD)b | ████ █ | ████ █ | ████ █ | ████ | ████ |
Sex, % | |||||
Female | ████ █ | ████ █ | ████ █ | ████ | ████ |
Region,c % | |||||
Europe | ████ █ | ████ █ | ████ █ | ████ | ████ |
North America | ████ | ████ | — | ████ | — |
Other | ███ | ███ | — | ███ | — |
sAAIPI score, % | |||||
0 to 1 | ████ | ████ | █████ | ████ | ████ |
2 to 3 | ████ | ████ | — | ████ | — |
SPD, mean (SD) | ████ | ████ | ████ | ████ | ████ |
Relapsed or refractory status, % | |||||
Refractory | ████ | ████ | ██████ | ████ | ████ |
Relapsed | ████ | ████ | — | ████ | — |
Double- or triple-hit lymphoma, % | |||||
Not applicable | ████ | ██ | ██████ | ████ | ████ |
Yes | ████ | ██ | — | ████ | — |
Histology, % | |||||
DLBCL transformed from FL | ████ | ████ | ██████ | ████ | █████ |
DLBCL NOS | ████ | ████ | — | ████ | — |
HGBCL | ████ | ████ | — | ████ | — |
FL3B | ████ | ████ | — | ████ | — |
PMBCL | ████ | ████ | — | ████ | — |
DLBCL transformed from indolent lymphoma other than FL | ████ | ████ | — | ████ | — |
Otherd | ████ | ████ | — | ████ | — |
Cell of origin, % | |||||
ABC, non-GCB | ████ | ████ | ██████ | ████ | █████ |
GCB | ████ | ████ | — | ████ | — |
Unknown | ████ | ████ | — | ████ | — |
Bone marrow involvement, % | |||||
No | ████ | ████ | ██████ | ████ | █████ |
Unknown | ████ | ████ | — | ███ | — |
Yes | ███ | ██ | — | ████ | — |
ECOG PS, % | |||||
0 | ████ | ████ | ██████ | ████ | █████ |
1 | ████ | ████ | — | ████ | — |
2 | ████ | ███ | — | ████ | — |
Disease stage, % | |||||
Stage I or II | ████ | ████ | ██████ | ████ | █████ |
Stage III or IV | ████ | ████ | — | ████ | — |
ALC, % | |||||
< 100/uL | ████ | ████ | ████ | ████ | ████ |
≥ 100/uL | ███ | ████ | — | ███ | — |
Secondary CNS lymphoma, % | |||||
No | ███ | ████ | ███ | ███ | ███ |
Yes | ███ | ███ | — | ███ | — |
Statistics | |||||
SMD < 0.2, % | ███ | ███ | ███ | ███ | ███ |
SMD < 0.1, % | ███ | ███ | ███ | ███ | ███ |
ABC = activated B cell; ALC = absolute lymphocyte count; axi-cel = axicabtagene ciloleucel; CNS = central nervous system; DLBCL = diffuse large B-cell lymphoma; EBV+ = Epstein-Barr virus–positive; ECOG PS = Eastern Cooperative Oncology Group Performance Status; ESS = effective sample size; FL = follicular lymphoma; FL3B = follicular lymphoma grade 3B; GCB = germinal centre B cell; HGBCL = high-grade B-cell lymphoma; ITT = intention to treat; liso-cel = lisocabtagene maraleucel; MAIC = matching-adjusted indirect comparison; NOS = not otherwise specified; PMBCL = primary mediastinal large B-cell lymphoma; sAAIPI = secondary Age-Adjusted International Prognostic Index; SD = standard deviation; SMD = standardized mean difference; SPD = sum of the product diameters; THRBCL = T-cell or histiocyte-rich large B-cell lymphoma.
aThe primary MAIC scenario matched and adjusted for 11 factors (histology, secondary CNS involvement, ALC, age, sex, region, sAAIPI score, SPD, relapse or refractory status, double or triple hit, and histology). This table displays all factors considered, including the 11 adjusted for.
bThe mean and SD were estimated from the reported median and range using methods proposed in the study by McGrath et al.143
cRegion was reported in the FDA assessment of the ZUMA-7 trial.122
dThe category included THRBCL, EBV+ DLBCL, and primary cutaneous leg-type DLBCL.
Sources: MAIC technical report.17 Details included in the table are from the sponsor’s summary of clinical evidence.
Event-Free Survival
In the primary scenario that matched and adjusted for 10 factors (secondary CNS involvement, ALC, age, sex, region, sAAIPI score, SPD, relapse or refractory status, double or triple hit, and histology), the TRANSFORM trial retained an ESS of ██. The median investigator-assessed EFS was not reached for liso-cel (95% CI, ███ ██ ███ ███████). The point estimate for the HR between liso-cel and axi-cel favoured liso-cel (███ █████ ███ ███ ██), but the 95% CI included the possibility of no difference or that the comparator was favoured (i.e., crossed the null). The results of the sensitivity scenario, adjusting for all clinical factors, was consistent with the primary scenario (███ █████ ███ ███ ██). Visual inspections of the KM curves and log cumulative hazard plots (data not shown) were generally parallel across study groups, supportive of the proportional hazards assumption.
Complete Response Rate
In the primary scenario that matched and adjusted for 10 factors, the TRANSFORM trial retained an ESS of ███. The point estimate for the odds of complete response between liso-cel and axi-cel favoured axi-cel (███ █████ ███ ██████ ██), but the 95% CI included the possibility of no difference or that the comparator was favoured (i.e., crossed the null). In the sensitivity scenario adjusting for all clinical factors, the CRR was █████ for liso-cel, and the odds of complete response were consistent with the primary scenario (███ █████ ███ ██).
Progression-Free Survival
In the primary scenario that matched and adjusted for 10 factors, the TRANSFORM trial retained an ESS of ██. The median PFS was not reached for liso-cel (95% CI, ████ ██ ███ ███████). The point estimate for the HR between liso-cel and axi-cel favoured liso-cel (████████ ███ ███ ██), but the 95% CI included the possibility of no difference or that the comparator was favoured (i.e., crossed the null). The results of the sensitivity scenario, adjusting for all clinical factors, was consistent with the primary scenario (███ █████ ███ █ ████). The results were supportive of the proportional hazards assumption.
Overall Survival
In the primary scenario that matched and adjusted for 10 factors, the TRANSFORM trial retained an ESS of ██. The median OS for liso-cel was not reached (95% CI, █████ ██ ███ ███████). The point estimate for the HR between liso-cel and axi-cel favoured liso-cel (███ █████ ███ ███ ████), but the 95% CI included the possibility of no difference or that the comparator was favoured (i.e., crossed the null). The results of the sensitivity scenario, adjusting for all clinical factors, were consistent with the primary scenario (███ █████ ███ ███ ██). The results were supportive of the proportional hazards assumption.
Harms
Clinical factors from the safety population in the TRANSFORM trial and the ITT set in the ZUMA-7 trial were used to inform the safety analyses. The baseline values after alignment are summarized in Table 22. Comparisons of the ranked clinical factors between the 2 trials at baseline before and after the matching or adjusting of the ITT population are in Table 23. The results demonstrated that notable differences (i.e., SMDs ≥ 0.2) were observed for age, SPD, disease histology, COO, and LVEF at screening. The matching and adjusting of 4 factors for patients from the TRANSFORM trial matched to the population of the ZUMA-7 trial produced improvements in the balance of clinical factors between studies. The proportion of ranked clinical factors achieving ███ ████ ████ ███. Factors with an SMD of less than 0.2 after matching and adjusting included age, ECOG at baseline, sAAIPI score, LVEF at screening, and bilirubin at screening. Similar trends in balance reduction after matching and adjusting were observed when using a more conservative SMD threshold of 0.1 (█████ ███████ ██).
Table 22: Comparison of Clinical Factors at Baseline After Definition and Categorization Alignment Between the TRANSFORM and ZUMA-7 Trials — Safety Analyses
Clinical factor | ZUMA-7 trial ITT population | TRANSFORM trial safety analysis set |
|---|---|---|
Age, mean (SD) | ██ ██████ | ████ ██████ |
sAAIPI score, % | ||
0 to 1 | 98 (54.4) | 56 (60.9) |
2 to 3 | 82 (45.6) | 36 (39.1) |
SPD (mm2), mean (SD), | ████ ████████ | ██████ ██████ |
Cell of origin, % | ||
ABC, non-GCB | 16 (8.9) | 21 (22.8) |
GCB | 109 (60.6) | 45 (48.9) |
Unknown | 55 (30.6) | 26 (28.3) |
ECOG at baseline, % | ||
0 | 95 (52.8) | ██ ████ |
1 | 85 (47.2) | ██ ██████ |
2 | 0 (0) | █████ |
LVEF at screening, % | ||
< 50% | █████ | █████ |
≥ 50% | ███ █████ | ██ ██████ |
Bilirubin at screening, % | ||
≤ 1.5 mg/dL | ███ █████ | ██ █████ |
> 1.5 mg/dL | █████ | █████ |
ABC = activated B cell; axi-cel = axicabtagene ciloleucel; ECOG = Eastern Cooperative Oncology Group; ESS = effective sample size; GCB = germinal centre B cell; ITT = intention to treat; liso-cel = lisocabtagene maraleucel; LVEF = left ventricular ejection fraction; MAIC = matching-adjusted indirect comparison; sAAIPI = secondary Age-Adjusted International Prognostic Index; SD = standard deviation; SMD = standardized mean difference; SOC = standard of care; SPD = sum of the product diameters.
Sources: MAIC technical report.17 Details included in the table are from the sponsor’s summary of clinical evidence.
Table 23: Clinical Factor Balance Before and After the MAIC Between Liso-Cel and Axi-Cel — Safety Analyses
Clinical factor | ZUMA-7 ITT N = 180 | Liso-cel (TRANSFORM) | |||
|---|---|---|---|---|---|
Before MAIC (unmatched and unadjusted) liso-cel arm N = 92 | After MAIC (Primarya) ESS = █████ | ||||
Statistics | Statistics | SMD | Statistics | SMD | |
Age, mean (SD) | ██ ██ | ████ █ | ██████ | ██ █ | ███ |
sAAIPI score, % | |||||
0 to 1 | 54.4 | 60.9 | ██████ | ████ | ███ |
2 to 3 | 45.6 | 39.1 | — | ████ | — |
SPD, mean (SD) | ████ ██ | ██████ | ██████ | █████ | ████ |
Cell of origin, % | |||||
ABC, non-GCB | 8.9 | 22.8 | ██████ | ████ | ████ |
GCB | 60.6 | 48.9 | — | ████ | — |
Unknown | 30.6 | 28.3 | — | ████ | — |
ECOG at baseline, % | |||||
0 | 52.8 | ████ | ██████ | ████ | ████ |
1 | 47.2 | ████ | — | ████ | — |
2 | 0 | ███ | — | ███ | — |
LVEF at screening, % | |||||
< 50% | ███ | ███ | ███ | ███ | ███ |
≥ 50% | ███ | ████ | — | ███ | — |
Bilirubin at screening, % | |||||
≤ 1.5 mg/dL | ███ | ███ | ███ | ███ | ███ |
> 1.5 mg/dL | ███ | ███ | — | ███ | — |
Statistics | |||||
SMD < 0.2, % | ██ | ████ | ██ | ████ | ██ |
SMD < 0.1, % | ██ | ████ | ██ | ████ | ██ |
ABC = activated B cell; axi-cel = axicabtagene ciloleucel; ECOG = Eastern Cooperative Oncology Group; ESS = effective sample size; GCB = germinal centre B cell; ITT = intention to treat; liso-cel = lisocabtagene maraleucel; LVEF = left ventricular ejection fraction; MAIC = matching-adjusted indirect comparison; NA = not applicable; sAAIPI = secondary Age-Adjusted International Prognostic Index; SD = standard deviation; SMD = standardized mean difference; SOC = standard of care; SPD = sum of the product diameters.
aThe primary MAIC scenario matched and adjusted for 4 factors (LVEF at screening, bilirubin at screening, age, and sAAIPI score).
Sources: MAIC technical report.17 Details included in the table are from the sponsor’s summary of clinical evidence.
Any TEAEs
The unmatched and unadjusted rate of any TEAEs was 100% for both liso-cel and axi-cel. An OR was therefore NE and an MAIC was not performed. The unmatched and unadjusted rate of any grade 3 or greater TEAE was similar between liso-cel (█████) and axi-cel (█████), and the odds of a grade 3 or greater TEAE were higher with liso-cel than axi-cel (███ █████ ███ ███ ████ ), but the 95% CI included the possibility of no difference or that the comparator had higher odds of an event (i.e., crossed the null). After MAIC, liso-cel had an event rate of 95.5%, and the odds of a grade 3 or greater TEAE were consistent with the unmatched and unadjusted comparison (███ █████ ███ ███).
Cytokine Release Syndrome
The unmatched and unadjusted rate of any-grade CRS was lower for liso-cel (█████) than for axi-cel (█████) and corresponded to lower odds of an event for liso-cel (███ █████ ███ ███ ████). After MAIC, liso-cel had an event rate of ██████. The odds of an any-grade CRS event were lower for liso-cel than for axi-cel (OR = 0.09; 95% CI, 0.04 to 0.18; P < 0.0001). Results for grade 3 or higher CRS were consistent with any-grade CRS.
Neurologic Toxicity
The unmatched and unadjusted rate of any-grade study-defined neurologic toxicity was lower for liso-cel (██████) than for axi-cel (███), and corresponded to lower odds of an event for liso-cel than for axi-cel (███ █████ ███ ███ ████). After MAIC, liso-cel had an event rate of █████, and the odds of any-grade neurologic toxicity were lower for liso-cel than for axi-cel (OR = 0.08; 95% CI, 0.03 to 0.18; P < 0.0001). Results for grade 3 or higher neurologic toxicity were consistent with any-grade neurologic toxicity.
Prolonged Cytopenia
The unmatched and unadjusted rate of grade 3 or greater prolonged cytopenia by laboratory assessment was higher in liso-cel than axi-cel (█████ █) and corresponded to higher odds of an event for liso-cel (███ █████ ███ ███ ████), but the 95% CI included the possibility of no difference or that the comparator had higher odds of an event (i.e., crossed the null). After MAIC, liso-cel had an event rate of █████, and the odds of a grade 3 or greater infection event were consistent with the unmatched and unadjusted comparison (███ █████ ███ ███ █).
Severe Infections
The unmatched and unadjusted rate of severe (i.e., grade ≥ 3) infections was similar between liso-cel (█████) and axi-cel (███), and the odds of an event were not different between treatments (███ █████ ███ ███ ████). After MAIC, liso-cel had an event rate of ██████ and the odds of a grade 3 or greater infection event remained consistent with the unmatched and unadjusted comparison (OR = 0.93; 95% CI, 0.41 to 2.13; ██████).
Hypogammaglobulinemia
The unmatched and unadjusted rates of any-grade hypogammaglobulinemia were similar between liso-cel (█████) and axi-cel (█████), and the odds of an event were not different between treatments (███ █████ ███ ███ ████). After MAIC, the event rate for liso-cel was █████ and the odds of an any-grade hypogammaglobulinemia event were consistent with the unmatched and unadjusted comparison (OR = 0.79; 95% CI, 0.31 to 2.01; ██████).
Serious AEs Occurring in at Least 10% of the Population
The unmatched and unadjusted rates of individual treatment-emergent serious AEs of any grade that occurred in 10% or more of patients were lower for liso-cel than axi-cel for pyrexia (████ ████) and encephalopathy (████ ████), and the odds of an event favoured liso-cel versus axi-cel (████████ ██ █ █████ ███ ███ ████). After MAIC, ████████ ███ █████ █████ ████ ████ ███ ███████ ███ ████ ███ ███████████████ ███ ███ ████ ██ ███ █████ █████ ████████ ████████ ███ ███████ ████ █████ ███ ███ ████ ██ ███████████ ███ ██████████████ ████ █████ ███ ███ ████ ██ ███████████ ██████ ███████.
Bucher ITC
Objectives
To inform the economic model, a Bucher ITC33 using a mixture cure modelling framework was conducted to assess the comparative efficacy and safety of liso-cel and axi-cel using IPD from the TRANSFORM trial16 (for liso-cel) and summary-level data from the ZUMA-7 trial34 in patients with relapsed or refractory LBCL who were intended for transplant.
Bucher Analysis Methods
Using the same methodology as the MAIC, a feasibility assessment was performed and determined the TRANSFORM and ZUMA-7 trials to be comparable enough to allow for the indirect comparison between liso-cel and axi-cel. MAIC and simulated treatment comparison approaches were considered feasible to minimize potential sources of bias while comparing these therapies, and the MAIC approach was preferred by the investigators over a simulated treatment comparison. ████████ ███ ███████ █████ ████ ███ ██████ ███ ███ ████ ██ ██████ ███ ████████ █████ ███████ ████ ███ █████████ ███████ ███████ ████ ███ ███████ ████ ████ ███████ ██ ███ ████████ █████████ █████ ██ ███ ██████████████████ ██████ ███ ███████ ██████ ██████████████ █████ ███ █████████ ██ ███ ███ ████ ███████ ██ ███ █████ ███████████ ██ ███ ████████ ███████████ █ ███ ███ ██████ ██████ ███ ██████████ ██ ██████ ██ ████ ███████████ ████ ███████████ ██ ████████ ███ █████████████ ███████ ███████ ████ ███ █████ ██ ███ ███
A 3-step process was followed for deriving relative efficacy for the cost-effectiveness model. First, a mixture cure modelling framework was jointly fit for liso-cel and SOC to derive the associated cure fractions and survival function for the noncured population based on the ZUMA-7 trial’s pseudo patient-level data. For the ZUMA-7 trial, pseudo patient-level data were first derived for the digitized OS and EFS for SOC and axi-cel. Then, a joint mixture curing model was fitted for the ZUMA-7 trial to derive the associated cure fractions and survival function for the noncured population. In the second step, a 2-dimensional treatment effect for liso-cel versus SOC was estimated from the jointly fitted mixture cure model using TRANSFORM trial IPD. Similarly, a 2-dimensional treatment effect for axi-cel versus SOC was estimated from a jointly fitted mixture cure model using patient-level data from the ZUMA-7 trial. The treatment effect on the cure fraction was estimated using a binary logistic regression model. In the third step, because the SOC arm of each trial made a connected network possible, the 2-dimensional treatment effect estimates from each trial were then combined using a Bucher approach to make an anchored ITC of liso-cel versus axi-cel. The efficacy outcomes for comparisons included EFS and OS.
Results
For EFS, the estimated odds ratio for the base case and 95% CI for the cure fraction (███ █████ ███ ███ ████) and noncured (███ █████ ███ ███ ████) with liso-cel versus axi-cel were not statistically significant. For OS, the estimated odds ratio for the base case and 95% CI for the cure fraction (███ █████ ███ ███ ████) and noncured (███ █████ ███ ███ ████) with liso-cel versus axi-cel were not statistically significant. These results were consistent with the MAIC estimates, albeit the point estimates were more conservative.
Critical Appraisal of MAIC and Bucher ITC
For the MAIC and Bucher ITC, the authors did not report a systematic literature search, describe their methods for data extraction, or conduct a quality assessment of the TRANSFORM and ZUMA-7 trials. The MAIC included relevant outcomes identified by the CDA-AMC team (EFS, CRR, PFS, OS, and safety). However, because data for several safety outcomes (e.g., CRS) were very limited or not available for the SOC arm in either the TRANSFORM or ZUMA-7 trials, an anchored MAIC was not feasible. Therefore, an unanchored MAIC was conducted for the safety outcomes. The Bucher ITC assessed only EFS and OS.
For the MAIC, to account for between-study differences in patient baseline characteristics, several potentially relevant clinical factors (i.e., treatment effect modifiers) were matched in the weighting process. The methods used to identify and rank the clinical factors were considered appropriate. The process involved a systematic literature review and a panel of 3 clinical experts to validate the selection and ranking of the treatment effect modifiers based on their strength to influence the specific outcomes under study for patients with relapsed or refractory LBCL. For the anchored MAIC efficacy analysis, 10 clinical factors were adjusted for and sensitivity scenarios, which included all relevant clinical factors available (total = 14), were conducted to test the robustness of the findings. After matching and adjusting for the 11 factors in the primary efficacy analyses, imbalances remained for ECOG Performance Status and COO, although the clinical experts consulted by CDA-AMC did not think these imbalances could bias the results. The authors noted that although the definition of EFS was similar between trials, some EFS events between randomization and treatment were treated differently. Among randomized patients who did not receive treatment in the ZUMA-7 trial, the majority were assigned an event immediately (i.e., at time 0, or day 1, of the KM curves) for the ITT analysis, given commencement of new lymphoma therapy and lack of evaluable disease assessment. This was considered to be a potential source of bias favouring axi-cel. Also, in the TRANSFORM trial, the date of imaging served as the basis for starting new antineoplastic therapy rather than day 1, which was used for the ZUMA-7 trial. Overall, the magnitude and direction of potential bias due to imbalances in the efficacy estimates could not be predicted.
For the unanchored MAIC safety analyses, the authors used the ITT population from the ZUMA-7 trial and the safety population from the TRANSFORM trial. Since the safety analysis set comprised 94.2% of the ITT population in the ZUMA-7 trial, the potential risk of selection bias was low. Among the 17 clinical factors identified for the safety analyses, 2 factors (prior HSCT and number of prior lines of therapy) were excluded because they were not relevant to the second line–approved indication, 6 factors (bulky disease, metabolic tumour volume, serum albumin, interleukin-6, fibrinogen level, and C-reactive protein) were not considered due to lack of reporting in the ZUMA-7 trial, and lactate dehydrogenase at baseline and bridging therapy were excluded due to differences in definitions between the TRANSFORM and ZUMA-7 trials. ECOG Performance Status was also not included in the MAIC, which was considered by the clinical experts consulted by CDA-AMC to be an important potential effect modifier. In addition, the TEAE reporting window differed between trials. Events from bridging therapy were included only for the TRANSFORM trial, thus potentially biasing the safety comparison against liso-cel. It should also be noted that the unanchored nature of the MAIC requires a stronger assumption (than an anchored MAIC) that all effect modifiers and prognostic factors have been included in the analysis, which is unlikely. Given these limitations, drawing definitive conclusions based on these results is not recommended.
Following the weighting process, the ESS for all of the efficacy outcomes declined by more than 50% from the size of the original sample in comparison with axi-cel. This proportion declined further for the adjusted safety analyses and sensitivity analyses, which included all relevant clinical factors available. These reductions in the ESS meant the final matched patient population was highly selective when compared with the original patient population and could lead to uncertainty in the estimated treatment effects. Since there were no major generalizability issues in the axi-cel population compared with the liso-cel population, the concern for bias due to influential subgroups is less of a concern.
The Bucher ITC, which was used to inform the economic model, showed results similar to the MAIC for EFS and OS. The main limitation of this approach was that the ITC estimates did not adjust for between-study differences in patient baseline characteristics. Since there were notable baseline differences between the TRANSFORM and ZUMA-7 trials, as described in the MAIC approach, drawing definitive conclusions based on the Bucher results is not recommended.
Studies Addressing Gaps in the Systematic Review Evidence
One study that was submitted by the sponsor was excluded because it did not match the patient population of the approved Health Canada indication.
Discussion
Summary of Available Evidence
One pivotal phase III open-label RCT and 2 ITCs submitted by the sponsor are summarized in this report.
One trial, TRANSFORM (N = 184), met the inclusion criteria for the systematic review conducted by the sponsor. The objective of the TRANSFORM trial was to assess the efficacy and safety of liso-cel 100 × 106 CAR T-cells, IV infusion, compared with SOC per investigator discretion that included 1 cycle of bridging therapy with 1 of 3 prespecified salvage immunochemotherapy regimens (R-DHAP, R-ICE or R-GDP) in adults with relapsed or refractory LBCL. The trial enrolled patients who had LBCL that was refractory to or relapsed within 12 months after initial response to first-line therapy (including an anthracycline and an anti-CD20 monoclonal antibody), were considered candidates for autologous HSCT, and had an ECOG score of 1 or less. The approved Health Canada indication and reimbursement request aligned with the trial population. The outcomes most relevant to the CDA-AMC review included the primary outcome of EFS per IRC, and secondary outcomes of CRR, PFS, OS, HRQoL, and safety. The HRQoL outcomes included EORTC QLQ-C30 global health status score and FACT-LymS total score. The trial population was predominately white (approximately 59%), male (57%), with a median age of 59 years (range, 20 to 75 years). Most patients had an ECOG Performance Status at baseline of 0 (52%) or 1 (47%), indicating good overall performance, an sAAIPI of 0 or 1 (60%), had disease that was refractory to or relapsed after their last therapy (74%; 26% respectively), and had LBCL subtype DLBCL NOS (56%), followed by HGBCL (23%), PMBCL (9%), or DLBCL from transformed indolent lymphoma (8%). Compared with the SOC group, the liso-cel group had a lower proportion of male patients (47.8% versus 66.3%) and a higher proportion of patients with chemorefractory disease (28.3% versus 19.6%).
In the absence of direct comparative evidence for liso-cel versus axi-cel, an MAIC was conducted by the sponsor. The objective of the MAIC was to assess the comparative efficacy and safety of liso-cel and axi-cel using IPD from the TRANSFORM trial (for liso-cel) and summary-level data from the ZUMA-7 trial (for axi-cel) in patients with relapsed or refractory LBCL who were intended for transplant. Outcome of interests included EFS, CRR, PFS, OS, and TEAEs. IPD from the TRANSFORM trial were adjusted to match the marginal distribution (e.g., mean, variance) of clinical factors among patients from the ZUMA-7 trial. For EFS, PFS, and OS, an anchored MAIC was performed using Cox proportional hazards models to estimate HRs for liso-cel versus axi-cel. Generalized linear models for CRR were used to estimate ORs. For the unanchored analysis of safety outcomes, weighted log odds for liso-cel were estimated in the TRANSFORM trial by fitting an intercept-only logistic regression model with MAIC adjustment weights. Point estimates (HRs or ORs) and 95% CIs were reported for all analyses. The sponsor also conducted a Bucher ITC to inform the pharmacoeconomic model; however, since it reported findings similar to the MAIC and was not the focus of this Clinical Review Report, it is not further discussed here.
Interpretation of Results
The evidence from the pivotal trial, TRANSFORM, addressed treatment outcomes noted to be important by both patients and clinicians. The patient group input indicated that stopping disease progression, prolonging life, improving HRQoL, and reducing treatment side effects are important to them. Similarly, the clinical experts consulted by CDA-AMC indicated that since the treatment goal for patients is cure or long-term disease control, the unmet needs of patients would be new treatments that would delay progression, prolong OS, and improve quality of life, while exposing patients to minimal toxicity.
Efficacy
The TRANSFORM trial supported a clinically meaningful improvement with liso-cel versus SOC for EFS, CRR, and PFS in adults with relapsed or refractory LBCL whose disease is refractory or has relapsed within 12 months of first-line therapy and who are candidates for autologous HSCT based on the second interim (data cut-off date of March 18, 2021) and final (data cut-off date of October 23, 2023) analyses. The between-group difference in probabilities of EFS at 12 and 36 months were 20.8% (95% CI, 2.5% to 39.1%) and 26.7% (95% CI, 13.3% to 40.1%), respectively. For the GRADE assessment, the clinical experts consulted by CDA-AMC suggested a clinically important threshold of 15% and 10% for between-group absolute risk difference for 12 and 36 months, respectively. Based on these thresholds, there was a high certainty of evidence for a clinically important increase in the probability of EFS at 12 and 36 months. The EFS findings were consistent across the subgroup analyses by histological subtypes, use of bridging therapy, and prior response status, although the analyses were exploratory and potentially not powered to detect differences between groups. At the primary analysis data cut-off date of May 13, 2022, the between-group difference in CRR was 29.3% (95% CI, 16.4% to 42.2%). For the GRADE assessment, the clinical experts suggested a clinically important threshold of 15% for between-group absolute risk difference. Based on this threshold, there was high-certainty evidence for a clinically important increase in CRR. The between-group difference in probabilities for PFS at 12 and 36 months were 31.9% (95% CI, 16.9% to 46.9%) and 24.4% (95% CI, 9.1% to 39.7%), respectively. For the GRADE assessment, the clinical experts suggested the same thresholds as EFS and, based on these thresholds, there was a high and moderate certainty of evidence for a clinically important increase in the probability of PFS at 12 and 36 months, respectively. The moderate certainty of evidence was attributed to serious imprecision due to the 95% CI including the possibility of an important benefit and a trivial effect when compared with SOC.
By the primary (data cut-off date of May 13, 2022) and final (data cut-off date of October 23, 2023) analyses, the median OS was not reached in either treatment group. The between-group difference in probabilities of OS at 12 and 36 months were 11.4% (95% CI, −0.7% to 23.5%) and 11.0% (95% CI, −3.7% to 25.7%), respectively. For the GRADE assessment, the clinical experts suggested a clinically important threshold of 10% and 5% for between-group absolute risk difference for 12 and 36 months, respectively. Thus, there was a moderate certainty of evidence for a clinically important increase in the probability of OS at 12 and 36 months when compared with SOC. The moderate certainty of evidence was attributed to serious imprecision due to the 95% CI including the possibility of an important benefit and little to no difference when compared with SOC. Since patients were permitted to receive posttreatment anticancer therapy (liso-cel: 34.8%; SOC: 70.7%), of which most patients in the SOC group (61 out of 65) crossed over to liso-cel treatment, the potential treatment benefit on OS would have been subjected to a degree of uncertainty.
For the HRQoL outcomes of EORTC QLQ-C30 global health status and FACT-LymS scores, the certainty of evidence was very low for the effect of liso-cel at 6 months when compared with SOC. The very low certainty of evidence was attributed to serious imprecision because the 95% CI for the between-group difference included the possibility of either benefit, little to no difference, or harm, and because of the risk of bias due to missing outcome data.
Based on the sponsor-submitted anchored MAIC, the HR point estimates for EFS, CRR, PFS, and OS between liso-cel and axi-cel favoured liso-cel; however, the 95% CI did not support the conclusion of a clinically meaningful difference. The results were supportive of the proportional hazards assumption. Ten clinical factors were adjusted for and sensitivity scenarios, which included all relevant clinical factors available (total = 14), were conducted to test the robustness of the findings. After matching and adjusting for the 11 factors in the primary efficacy analyses, imbalances remained for ECOG Performance Status and COO. The magnitude and direction of potential bias due to imbalances in the efficacy estimates could not be predicted. Additionally, the MAIC authors noted that EFS events between randomization and treatment were treated differently in the TRANSFORM and ZUMA-7 trials, which was considered a potential source of bias favouring axi-cel. Following the weighting process, the ESS for all of the efficacy outcomes declined by more that 50% from the original sample size in the comparison with axi-cel. These reductions in the ESS meant the final matched patient population was highly selective compared with the original patient population and could lead to uncertainty in the estimated treatment effects. Overall, the efficacy results suggest there are no meaningful differences between liso-cel and axi-cel; however, the estimates are subject to uncertainty due to imprecision.
Harms
Almost all patients in the trial reported at least 1 TEAE. The most frequently reported TEAEs of any grade in both treatment groups were neutropenia, anemia, thrombocytopenia, and nausea. Of these TEAEs, a numerically higher proportion of neutropenia was reported in patients taking liso-cel and a higher proportion of thrombocytopenia was reported in patients taking SOC. Most patients in both groups reported at least 1 grade 3 or 4 TEAE. The incidence of grade 3 or 4 neutropenia and lymphopenia was numerically higher in the liso-cel group versus SOC. The incidence of serious TEAEs was similar between groups, with the most frequently reported being CRS, febrile neutropenia, pyrexia, and neutropenia. The frequency of these serious TEAEs was similar between groups, except a higher proportion of CRS was reported in patients taking liso-cel. The clinical experts indicated that the incidence of these TEAEs is expected with liso-cel and that, with appropriate care, the AEs would be manageable for many patients. No patients in the liso-cel group had a TEAE that led to the withdrawal of the study drug (including bridging therapy and LDC), and 4 patients in the SOC group experienced TEAEs leading to treatment withdrawal. The incidence of death was higher in patients in the liso-cel group versus SOC, with the majority of deaths in both groups attributed to disease progression, followed by TEAEs. A numerically higher proportion of notable TEAEs of any grade were reported in patients taking liso-cel, with the most frequently reported being neurologic toxicity, CRS, prolonged cytopenia, and investigator-identified neurologic toxicity. These events occurred more frequently in patients taking liso-cel, except for neurologic toxicity, which was similar in both groups. Based on the sponsor-submitted unanchored MAIC comparing liso-cel with axi-cel, the odds of a grade 3 or greater TEAE event, prolonged cytopenia, severe infections, and hypogammaglobulinemia were either not different between groups or higher with liso-cel, but the 95% CI did not support the conclusion of a clinically meaningful difference. For CRS, neurologic toxicity, pyrexia, and encephalopathy, the odds of an any-grade event favoured liso-cel versus axi-cel. Among the 17 identified clinical factors for the safety analyses, 2 factors (prior HSCT and number of prior lines of therapy) were excluded because they were not relevant to the second line–approved indication, 6 factors (bulky disease, metabolic tumour volume, serum albumin, interleukin-6, fibrinogen level, and C-reactive protein) were not considered due to lack of reporting in the ZUMA-7 trial, and lactate dehydrogenase at baseline and bridging therapy were excluded due to differences in definitions between the TRANSFORM and ZUMA-7 trials. ECOG Performance Status was also not included in the MAIC, which was considered by the clinical experts consulted by CDA-AMC to be an important potential effect modifier. In addition, the TEAE reporting window differed between trials. Events from bridging therapy were included only for the TRANSFORM trial, thus potentially biasing the safety comparison against liso-cel. As such, the safety results are subject to uncertainty due to imprecision and imbalances in potential effect modifiers and prognostic factors.
Conclusion
Evidence from 1 phase III open-label RCT (TRANSFORM) reported on outcomes that were important to both patients and clinicians. The trial showed high certainty of evidence that treatment with liso-cel results in a clinically meaningful increase in EFS at 12 months and in CRR at 36 months compared with SOC in adults with relapsed or refractory LBCL. The trial also showed high and moderate certainty of evidence that liso-cel results in a clinically meaningful increase in PFS at 12 and 36 months, respectively. At the time of the final analysis, median OS had not been reached in either group, and no definitive conclusions could be drawn on HRQoL due to concerns of imprecision and missing outcome data. No new safety signals were identified, and the safety of liso-cel was consistent with the known safety profile for the drug. The results of the ITC did not support a clinically meaningful difference between liso-cel and axi-cel for EFS, CRR, PFS, and OS, but were suggestive of a more favourable safety profile for liso-cel; however, these estimates were subject to uncertainty due to imprecision and imbalances in potential effect modifiers and prognostic factors.
References
1.Jamil A, Mukkamalla SKR. Lymphoma. Treasure Island (FL): StatPearls Publishing; 2024: https://www.ncbi.nlm.nih.gov/books/NBK560826/. Accessed 2024 Jun.
2.Lymphoma Canada. Non-Hodgkin lymphoma. 2023; https://www.lymphoma.ca/lymphoma/non-hodgkin-lymphoma/. Accessed 2023 Nov 24.
3.Canadian Cancer Statistics Advisory Committee, Canadian Cancer Society, Statistics Canada, Public Health Agency of Canada. Canadian Cancer Statistics 2023. Toronto (ON): Canadian Cancer Society; 2023: https://cdn.cancer.ca/-/media/files/research/cancer-statistics/2023-statistics/2023_PDF_EN.pdf. Accessed 2023 Nov 24.
4.Kurz KS, Ott M, Kalmbach S, et al. Large B-Cell Lymphomas in the 5th Edition of the WHO-Classification of Haematolymphoid Neoplasms-Updated Classification and New Concepts. Cancers (Basel). 2023;15(8):2285.
5.Understanding NHL: a patient's guide to non-Hodgkin lymphoma. Mississauga (ON): Lymphoma Canada; 2020: https://www.lymphoma.ca/wp-content/uploads/2020/05/2020430-NHL-Interior-WEB.pdf. Accessed 2023 Nov 24.
6.Sehn LH, Salles G. Diffuse Large B-Cell Lymphoma. N Engl J Med. 2021;384(9):842-858. PubMed
7.Leukemia and Lymphoma Society of Canada. About blood cancers: facts and statistics. 2020; https://www.bloodcancers.ca/. Accessed 2021 May 9.
8.Armitage JO, Gascoyne RD, Lunning MA, Cavalli F. Non-Hodgkin lymphoma. Lancet. 2017;390(10091):298-310. PubMed
9.Martelli M, Ferreri AJ, Agostinelli C, Di Rocco A, Pfreundschuh M, Pileri SA. Diffuse large B-cell lymphoma. Crit Rev Oncol Hematol. 2013;87(2):146-171. PubMed
10.Vitolo U, Seymour JF, Martelli M, et al. Extranodal diffuse large B-cell lymphoma (DLBCL) and primary mediastinal B-cell lymphoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2016;27(suppl 5):v91-v102. PubMed
11.NCCN Guidelines. B-Cell Lymphomas, version 1.2024. Plymouth Meeting (PA): National Comprehensive Cancer Network (NCCN); 2024: https://www.nccn.org/home. Accessed 2024 Feb 20.
12.Kroft SH, Sever CE, Bagg A, et al. Laboratory Workup of Lymphoma in Adults: Guideline From the American Society for Clinical Pathology and the College of American Pathologists. Arch Pathol Lab Med. 2021;145(3):269-290. PubMed
13.Tilly H, Gomes da Silva M, Vitolo U, et al. Diffuse large B-cell lymphoma (DLBCL): ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2015;26 Suppl 5:v116-125. PubMed
14.Alaggio R, Amador C, Anagnostopoulos I, et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia. 2022;36(7):1720-1748.
15.Drug Reimbursement Expert Review Committee final recommendation: lisocabtagene maraleucel (Breyanzi - Celgene Inc., a Bristol Myers Squibb company). Can J Health Technol. 2022. https://www.canjhealthtechnol.ca/index.php/cjht/article/view/PG0258/PG0258. Accessed 2024 Jun 13.
16.Clinical Study Report: JCAR017-BCM-003, Addendum 01. A global randomized multicenter phase 3 trial to compare the efficacy and safety of JCAR017 to standard of care in adult subjects with highrisk, transplant-eligible relapsed or refractory aggressive b-cell non-Hodgkin lymphomas (TRANSFORM) [internal sponsor's report]. Princeton (NJ): Bristol-Myers Squibb Company; 2022 Oct 26.
17.Indirect treatment comparison of lisocabtagene maraleucel (liso-cel) and axicabtagene ciloleucel (axicel) for second-line treatment of transplant intended patients with relapsed or refractory large b-cell lymphoma [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Breyanzi (lisocabtagene maraleucel), cell suspension in patient-specific single-dose vials for intravenous infusion. Saint-Laurent (QC): Bristol Myers Squibb; 2024 Apr 17.
18.Giulino-Roth L. How I treat primary mediastinal B-cell lymphoma. Blood. 2018;132(8):782-790. PubMed
19.Rimsza L, Pittaluga S, Dirnhofer S, et al. The clinicopathologic spectrum of mature aggressive B cell lymphomas. Virchows Arch. 2017;471(4):453-466. PubMed
20.Di Napoli A, Remotti D, Agostinelli C, et al. A practical algorithmic approach to mature aggressive B cell lymphoma diagnosis in the double/triple hit era: selecting cases, matching clinical benefit: A position paper from the Italian Group of Haematopathology (G.I.E.). Virchows Arch. 2019;475(4):513-518. PubMed
21.Breyanzi (lisocabtagene maraleucel): cell suspension in patient-specific single-dose vials for intravenous infusion [product monograph]. Saint-Laurent (QC): Bristol Myers Squibb Canada; 2024 Sep 25.
22.Breyanzi (lisocabtagene maraleucel): cell suspension in patient-specific single-dose vials for intravenous infusion [product monograph]. Saint-Laurent (QC): Celgene Inc., a Bristol Myers Squibb company; 2022 May 6.
23.Yescarta (axicabtagene ciloleucel): cell suspension in patient-specific single infusion bag [product monograph]. Mississauga (ON): Gilead Sciences Canada, Inc.; 2022 Oct 13: https://pdf.hres.ca/dpd_pm/00067754.PDF. Accessed 2024 Aug 13.
24.Rituxan (rituximab for injection): 10 mg/mL intravenous infusion [product monograph]. Mississauga (ON): Hoffmann-La Roche Limited; 2023 Jun 2: https://pdf.hres.ca/dpd_pm/00071131.PDF. Accessed 2024 Aug 13.
25.Aaronson NK, Ahmedzai S, Bergman B, et al. The European Organization for Research and Treatment of Cancer QLQ-C30: a quality-of-life instrument for use in international clinical trials in oncology. J Natl Cancer Inst. 1993;85(5):365-376. PubMed
26.Abramson JS, Johnston PB, Kamdar M, et al. Health-related quality of life with lisocabtagene maraleucel vs standard of care in relapsed or refractory LBCL. Blood Adv. 2022;6(23):5969-5979. PubMed
27.Davda J, Kibet H, Achieng E, Atundo L, Komen T. Assessing the acceptability, reliability, and validity of the EORTC Quality of Life Questionnaire (QLQ-C30) in Kenyan cancer patients: a cross-sectional study. J Patient Rep Outcomes. 2021;5(1):4. PubMed
28.Cocks K, King MT, Velikova G, et al. Evidence-based guidelines for interpreting change scores for the European Organisation for the Research and Treatment of Cancer Quality of Life Questionnaire Core 30. Eur J Cancer. 2012;48(11):1713-1721. PubMed
29.Osoba D, Rodrigues G, Myles J, Zee B, Pater J. Interpreting the significance of changes in health-related quality-of-life scores. J Clin Oncol. 1998;16(1):139-144. PubMed
30.Hlubocky FJ, Webster K, Beaumont J, et al. A preliminary study of a health related quality of life assessment of priority symptoms in advanced lymphoma: the National Comprehensive Cancer Network-Functional Assessment of Cancer Therapy - Lymphoma Symptom Index. Leuk Lymphoma. 2013;54(9):1942-1946. PubMed
31.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. PubMed
32.Balshem H, Helfand M, Schünemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-406. PubMed
33.Summary of Clinical Evidence: Breyanzi (lisocabtagene maraleucel, liso-cel, JCAR 017) for the treatment of adult patients with large B-cell lymphoma (LBCL), who are refractory or have relapsed within 12 months of first-line therapy and are candidates for autologous haematopoeitic stem cell transplant (HSCT) [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Breyanzi (lisocabtagene maraleucel), cell suspension in patient-specific single-dose vials for intravenous infusion Saint-Laurent (QC): Bristol Myers Squibb; 2024 May.
34.Locke FL, Miklos DB, Jacobson CA, et al. Axicabtagene Ciloleucel as Second-Line Therapy for Large B-Cell Lymphoma. N Engl J Med. 2022;386(7):640-654. PubMed
Pharmacoeconomic Review
Abbreviations
AE
adverse event
BIA
budget impact analysis
CAR
chimeric antigen receptor
CDA-AMC
Canada’s Drug Agency
CRS
cytokine release syndrome
DLBCL
diffuse large B-cell lymphoma
EFS
event-free survival
HDCT
high-dose chemotherapy
HRQoL
health-related quality of life
HSCT
hematopoietic stem cell transplant
ITC
indirect treatment comparison
LBCL
large B-cell lymphoma
liso-cel
lisocabtagene maraleucel
LY
life-year
MAIC
matching-adjusted indirect comparison
OS
overall survival
pCPA
pan-Canadian Pharmaceutical Alliance
PSM
partitioned survival model
R-GDP
rituximab, gemcitabine, dexamethasone, and cisplatin
SOC
standard of care
tisa-cel
tisagenlecleucel
QALY
quality-adjusted life-year
WTP
willingness to pay
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Lisocabtagene maraleucel (Breyanzi), cell suspension for IV infusion |
Indication | Treatment of adult patients with diffuse large B-cell lymphoma (DLBCL) not otherwise specified, primary mediastinal large B-cell lymphoma (PMBCL), high-grade B-cell lymphoma, and DLBCL arising from follicular lymphoma, who have refractory disease to first-line chemoimmunotherapy or relapse within 12 months of first-line chemoimmunotherapy, and who are candidates for autologous hematopoietic stem cell transplant. |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | September 25, 2024 |
Reimbursement request | As per indication |
Sponsor | Bristol Myers Squibb Canada |
Submission history | Previously reviewed: Yes Indication: For the treatment of adult patients with relapsed or refractory large B-cell lymphoma after 2 or more lines of systemic therapy, including DLBCL not otherwise specified, PMBCL, high-grade B-cell lymphoma, and DLBCL arising from follicular lymphoma, after at least 2 prior therapies. Recommendation date: June 29, 2022 Recommendation: Recommended with clinical criteria and/or conditions. |
DLBCL = diffuse large B-cell lymphoma; NOC = Notice of Compliance; PMBCL = primary mediastinal large B-cell lymphoma.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis PSM |
Target population | Adults with second-line LBCL who are transplant eligible whose disease is refractory to first-line immunochemotherapy or has relapsed within 12 months |
Treatment | Liso-cel |
Dose regimen | Single infusion containing 60 × 106 to 120 × 106 CAR-positive viable T cells |
Submitted price | $501,900.00 per patient per infusion |
Submitted treatment cost | $508,934a per patient per infusion |
Comparators |
|
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | Lifetime (50 years) |
Key data sources | Efficacy (EFS, OS) of liso-cel and SOC informed by the TRANSFORM trial; efficacy of axi-cel informed by Bucher ITCsb |
Submitted results | In a sequential analysis, liso-cel was associated with an ICER of $137,251 per QALY gained compared with SOC (incremental cost: $264,234; incremental QALYs: 1.93). Axi-cel was extendedly dominated through a combination of liso-cel and SOC. |
Key limitations |
|
CDA-AMC reanalysis results |
|
axi-cel = axicabtagene ciloleucel; CAR = chimeric antigen receptor; CDA-AMC = Canada’s Drug Agency: EFS = event-free survival; HSCT = hematopoietic stem cell transplant; ICER = incremental cost-effectiveness ratio; ITC = indirect treatment comparison; LBCL = large B-cell lymphoma; liso-cel = lisocabtagene maraleucel; LY = life-year; MAIC = matching-adjusted indirect comparison; OS = overall survival; PSM = partitioned survival model; QALY = quality-adjusted life-year; SOC = standard of care.
aCosts included by the sponsor: liso-cel acquisition, leukapheresis, bridging therapy, lymphodepleting chemotherapy.1
bIn its ITC, the sponsor utilized reconstructed patient data from ZUMA-7. The sponsor additionally submitted an MAIC as part of its clinical evidence package. Data from the MAIC were not used in the sponsor’s economic evaluation.
Conclusions
The Canada’s Drug Agency (CDA-AMC) Clinical Review found that lisocabtagene maraleucel (liso-cel) likely results in a clinically meaningful increase in event-free survival (EFS) at 12 and 36 months compared with standard of care (SOC) in adults with relapsed or refractory large B-cell lymphoma (LBCL). At the time of the submitted analysis, median overall survival (OS) had not been reached in either group, and no definitive conclusions could be drawn as to the impact of liso-cel on health-related quality of life (HRQoL) because of concerns about imprecision and missing outcome data. There have been no head-to-head trials of liso-cel versus axicabtagene ciloleucel (axi-cel), and the results of both indirect treatment comparisons (ITCs) submitted by the sponsor (matching-adjusted indirect comparison [MAIC] and Bucher ITC) suggest there may be no differences between liso-cel and axi-cel for EFS or OS. The sponsor-submitted MAIC suggested there may be lower odds of some adverse events (AEs) with liso-cel versus axi-cel, including cytokine release syndrome (CRS), neurologic toxicity, pyrexia, and encephalopathy; however, these findings were subject to uncertainty due to imprecision and imbalances in potential effect modifiers.
CDA-AMC identified important limitations with the sponsor’s economic evaluation that precluded reanalysis. Notably, CDA-AMC was unable to address uncertainty related to comparative clinical data, including the magnitude and duration of benefit for liso-cel compared with axi-cel, limitations with the model structure, and uncertainty in the impact of liso-cel on HRQoL. CDA-AMC was additionally unable to resolve uncertainty related to the impact of AEs on the cost-effectiveness of liso-cel. Although the sponsor conducted a MAIC to estimate the risk of AEs in the indicated population, these data were not incorporated in the economic model, and the impact of AEs on the cost-effectiveness of liso-cel was based on naive comparison of data from clinical trials. Thus, it is not possible to determine if any differences between the therapies are due to the treatment or due to bias or confounding factors. CDA-AMC was unable to provide a more reliable estimate of the cost-effectiveness of liso-cel owing to these identified limitations.
Given the limitations in the indirect clinical evidence and the submitted economic model, there is insufficient evidence to suggest that liso-cel should be priced higher than axi-cel. Should negotiations with the pan-Canadian Pharmaceutical Alliance (pCPA) for the use of axi-cel in the second line conclude without a letter of intent, a price reduction of at least 35% for liso-cel would be required for liso-cel to be considered cost-effective relative to SOC at a willingness-to-pay (WTP) threshold of $50,000 per quality-adjusted life-year (QALY) gained.
Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, clinician groups, and drug plans that participated in the CDA-AMC review process (specifically, information that pertains to the economic submission).
Patient group input was received from Lymphoma Canada based on a survey conducted in 2024. Respondents were from Canada and other jurisdictions and included those with diffuse large B-cell lymphoma (DLBCL), primary mediastinal B-cell lymphoma, DLBCL arising from follicular lymphoma, high-grade B-cell lymphoma, and other types of DLBCL. Respondents indicated that LBCL affects their quality of life, with symptoms of the disease leading to anxiety and stress, fatigue, enlarged lymph nodes, indigestion, abdominal pain or bloating, body aches and pains, and limits their ability to continue their daily activities (e.g., travel, attending work or school). Respondents indicated that current treatment options include chemotherapy, radiation therapy, salvage therapy plus autologous hematopoietic stem cell transplant (HSCT), and chimeric antigen receptor (CAR) T-cell therapy, or treatments offered via clinical trials. Most respondents had received 1 line or 2 lines of treatment and noted that current treatments were associated with AEs including fatigue, hair loss, nausea, loss of appetite and/or weight loss, constipation, joint pain, bodily aches, neuropathy, mouth sores, muscle weakness, and diarrhea. Patients reported challenges with accessing treatment locally, as well as financial implications of treatment (e.g., costs of drugs to treat AEs, travel costs, medical supply costs, drug costs, absence from work). Patients expressed a desire for treatments that improve survival, extend remission, and improve quality of life with fewer AEs. Patients who had experience with liso-cel reported having experienced decreased appetite, nausea/vomiting, and fever.
Input was received from 3 clinician groups: Leukemia & Lymphoma Society of Canada Nurses Network, the Lymphoma Canada Clinician Group, and the Ontario Health–Cancer Care Ontario Hematology Cancer Drug Advisory Committee. Clinicians indicated that current treatment options include chemotherapy with rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) and salvage chemoimmunotherapy with rituximab, gemcitabine, dexamethasone, and cisplatin (R-GDP) followed with autologous HSCT. Clinicians noted that liso-cel may provide an alternative treatment option for patients with relapsed or refractory LBCL who are transplant eligible to bypass aggressive chemotherapy protocols. Challenges noted by clinicians included timing CAR T-cell therapy when patients are well enough to receive treatment and the limited availability of CAR T-cell therapy at select centres. Clinicians noted that clinically meaningful outcomes would include lasting remission, overall response rate, complete response rate, progression-free survival, and extending survival.
The drug plan input noted that axi-cel had previously received a positive recommendation from the pan-Canadian Oncology Drug Review Expert Review Committee (pERC) and is currently undergoing negotiation with pCPA for this indication. Plans noted that patients who experience an AE likely require admission to hospital, which may include care in an intensive care unit. Plans asked whether patients who recently started SOC should be allowed to receive to liso-cel if it is approved. Plans noted that, at present, there is limited access to CAR T services in Canada and that interprovincial travel or out-of-country funding is necessary for patients in many parts of Canada. Plans additionally expressed concerns about the feasibility of adoption, of liso-cel and noted that existing capacity may be insufficient to meet demand.
Several of these concerns were addressed in the sponsor’s model:
Axi-cel and SOC were considered comparators in the sponsor model. Should pCPA negotiations for axi-cel in the second line conclude without a letter of intent, SOC would be the most relevant comparator.
Quality of life was incorporated in the sponsor’s model, with health state utility values obtained from the literature.
The cost of intensive care unit stays was incorporated for patients who experienced grade 3 or higher CRS or neurotoxicity.
CDA-AMC was unable to address the following concern raised from the input:
CDA-AMC was unable to consider costs associated with interprovincial or out-of-country travel necessary for patients to access CAR T-cell therapy in specialized centres.
CDA-AMC was unable to fully consider the capacity of the public health care system to meet the demand for CAR T-cell therapy. In the budget impact analysis (BIA), the sponsor assumed that liso-cel would take market share only from axi-cel and thus not affect overall demand for CAR T-cell therapy. However, if patients who would have otherwise received SOC are considered candidates for liso-cel, the overall demand for CAR T-cell therapy will be increased. This was assessed by the sponsor in a scenario analysis that suggests liso-cel will no longer be cost-saving to the public health care payer under these conditions.
Economic Review
The current review is for liso-cel (Breyanzi) for the treatment of adult patients with LBCL, including DLBCL not otherwise specified, primary mediastinal B-cell lymphoma, high-grade B-cell lymphoma, and DLBCL arising from follicular lymphoma that is refractory to first-line therapy or relapsed within 12 months of first-line treatment, and who are eligible for transplant.1
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis comparing liso-cel with axi-cel and SOC, which the sponsor defined as salvage chemotherapy therapy with platinum-based chemoimmunotherapy regimens (including R-GDP and rituximab, ifosfamide, carboplatin, and etoposide [R-ICE]), that may or may not be followed by high-dose chemotherapy (HDCT) plus autologous HSCT.1 The modelled population is in line with the Health Canada indication and was based on patients enrolled in the TRANSFORM trial.
Liso-cel is a CD19-directed CAR T-cell immunotherapy available as a single-dose cell suspension for IV administration (60 × 106 to 120 × 106 CAR-positive viable T cells).2 The sponsor adjusted the submitted price of liso-cel ($501,900 per administration per patient) in the model by the proportion of patients who received liso-cel in the TRANSFORM trial (96.74%). Similarly, the sponsor adjusted the price of axi-cel ($485,021 per administration per patient) by the proportion of patients who received axi-cel in the ZUMA-7 trial. Costs associated with pretreatment for liso-cel and axi-cel (leukapheresis, bridging therapy, and lymphodepleting chemotherapy) as well as administration costs related to infusion for all treatments were incorporated separately.2 As part of SOC, the sponsor estimated the per-patient cost of salvage chemotherapy to be $4,834 per treatment cycle1 and the cost of autologous HSCT to be $82,555.3
The analysis was conducted from the perspective of the Canadian public health care payer. Costs and outcomes (QALYs, life-years [LYs]) were estimated over a lifetime time horizon (50 years). Weekly cycle lengths were used in the first 5 years of the model, with yearly cycles thereafter. An annual discount rate of 1.5% was applied to costs and outcomes.
Model Structure
The sponsor submitted a partitioned survival model (PSM) with 3 health states (event free, progressed disease, dead) (Figure 1).1 Patients entered the model in the event-free state (represented by “2L” in Figure 1), and were assumed to progress to third-line treatment following disease progression, failure to achieve a complete response or partial response, or the start of a new antineoplastic therapy (represented by “3L” in Figure 1). The proportion of patients who were event free in the second line, had progressed disease, or were dead was estimated over time based on EFS and OS curves informed by the TRANSFORM trial data. Health state membership was determined using an area under the curve approach.1 The sponsor assumed that patients who received SOC in the second line would be eligible to receive CAR T-cell therapy in the third line; patients who received CAR T in the second line were considered ineligible for CAR T in subsequent lines. Patients who remained in the event-free state for more than 2 years were assumed by the sponsor to “cured,” with the OS of cured patients assumed to be the same as the age- and sex-adjusted mortality for the general population, with adjustment using a standardized mortality ratio to account for excess mortality risk. Alternative assumptions about the timing of the cure were tested in scenario analyses. The survival of patients who were not cured was extrapolated based on parametric functions.
Model Inputs
The baseline patient characteristics used to inform the model were obtained from the TRANSFORM trial, which randomized patients with relapsed or refractory LBCL to receive liso-cel or SOC (mean age = 56 years; 43% female, 57% male).4
Clinical efficacy inputs for the model (OS, EFS) were obtained from a sponsor-conducted Bucher ITC, with the efficacy of liso-cel informed by data from the TRANSFORM trial (data cut-off date: October 23, 2023; median of 33.9 months of follow-up).1 The sponsor used a mixture cure model, which allowed the treatment effect of liso-cel and comparators to differ between patients considered to be “cured” (cured fraction) and those who were not cured (noncured fraction). The sponsor assumed that those who remained in the EFS health state at 2 years (i.e., patients who had not experienced an event in the first 2 years of treatment) comprise the cured fraction. The sponsor fit joint distributions to patient data from the TRANSFORM trial and reconstructed patient data from the ZUMA-7 trial to estimate treatment effects (i.e., the cure fraction and survival functions for the noncured fraction) of liso-cel versus SOC and axi-cel versus SOC, respectively. The treatment effects from each trial were combined using a Bucher approach to produce an anchored ITC of liso-cel versus axi-cel. The sponsor chose the gamma and lognormal distributions as the best-fitting survival curves for OS and EFS, respectively.
To estimate the risk of death, the sponsor adopted a standardized mortality ratio of 1.40 compared with the general population for the first 2 years, based on Statistics Canada life tables.5 After 2 years, the sponsor assumed that the risk of death for the cure fraction would be equivalent to the risk in the age- and sex-adjusted general population, while the survival of patients in the noncured fraction was predicted with parametric distributions based on the mixture cure model equation.
Health state utility values (EFS = 0.83; progressed disease = 0.71) were sourced from the Norwegian Medicines Agency’s health technology assessment report evaluating tisagenlecleucel (tisa-cel) for the treatment of relapsed or refractory DLBCL after 2 or more lines of chemotherapy (median prior therapies = 3; range, 1 to 6).6
Costs and consequences (utility decrements) were included in the model for grade 3 and 4 AEs, as well as costs of managing grade 1 and 2 AEs. Utility decrements were sourced from the published literature7 and the TRANSFORM trial, with the probability of AEs sourced from the TRANSFORM trial (for liso-cel and SOC) and Westin et al. (2023)8 (for axi-cel).
Costs captured in the model included those related to CAR T (acquisition, leukapheresis, bridging therapy, administration), HSCT, chemotherapy (bridging therapy, lymphodepletion, SOC, HDCT), subsequent treatment, CAR T–specific health care resource use (post–CAR T-cell therapy hospital stays, office visits), health care resource (e.g., outpatient visits, oncology visits, monitoring), management of AEs, and end-of-life care. Acquisition costs for liso-cel and axi-cel were assumed to be a 1-time cost, based on the sponsor’s submitted price and a prior CADTH review, respectively.9 Costs associated with chemotherapy were calculated by the sponsor as a function of unit drug price, dosing schedules, and the proportion of patients who received each treatment. Treatment costs for SOC were divided into 3 phases: salvage chemotherapy, HDCT, and autologous HSCT. The sponsor assumed that 36% of patients would receive R-GDP and 64% would receive R-ICE as part of salvage chemotherapy. The cost of HSCT was based on the Interprovincial Billing Rates for Designated High Cost Transplants (2023).3 The sponsor assumed that 90% of patients who progress on second-line therapy would receive subsequent therapy in the third line. For patients who received liso-cel or axi-cel in the second line, subsequent treatment was assumed to consist of allogeneic HSCT (5%), SOC (85%), and enrolment in a clinical trial (10%). For those who received SOC in the second line, subsequent treatment was assumed to consist of CAR T-cell therapy (60%), allogeneic HSCT (5%), SOC (20%), or enrolment in a clinical trial (15%). The proportion of patients receiving each type of subsequent treatment was based on expert opinion obtained by the sponsor, and subsequent therapy costs were applied as a 1-time cost. Other drug acquisition costs were obtained from the Ontario Drug Benefit Formulary10,11 and IQVIA Delta PA. Dosing schedules were obtained from Cancer Care Ontario,12 the TRANSFORM trial,13 the National Health Service Chemotherapy Protocol,14 Locke et al. (2021),15 and the polatuzumab vedotin product summary from the European Medicines Agency.16 Where applicable, costs were based on mean patient weight and body surface area from the TRANSFORM trial. The cost of managing AEs was obtained from the Ontario Physician Schedule of Benefits for grade 1 and 2 AEs (assumed to require 1 general practitioner visit) or Canadian Institute for Health Information costing (CIHI) for AEs of grade 3 or higher, with the exception of CRS, neurotoxicity, and hypogammaglobulinemia (microcosting approach). Resource use included inpatient stay after CAR T-cell administration and post-transplant resource use after discharge from the hospital, and resource use by health state (includes routine monitoring). Patients who remained in the EFS health state after 2 years were assumed to incur reduced monitoring costs. The cost of end-of-life care was obtained from the literature.17
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically (5,000). The deterministic and probabilistic results were similar. The probabilistic findings are presented subsequently. The submitted analysis was based on the submitted price for liso-cel and publicly available prices for comparators. Additional results from the sponsor’s submitted economic evaluation base case are presented in Appendix 3.
Base-Case Results
In the sponsor’s base case, liso-cel was associated with an estimated cost of $639,948 and 10.79 QALYs over a lifetime horizon (Table 3). In the sponsor’s sequential analysis, liso-cel was associated with an incremental cost-effectiveness ratio of $137,251 per QALY gained compared with SOC (incremental cost: $264,234; incremental QALYs: 1.92). At a WTP threshold of $50,000 per QALY gained, liso-cel had a 2% probability of being considered the optimal treatment.
Results were driven by the acquisition cost of liso-cel (submitted price: $501,900; cost incorporated in the model after adjustment for the proportion of patients who received planned treatment in the TRANSFORM trial for liso-cel [97%]: $482,284), as well as the predicted gain in LYs (incremental LYs: 0.41 versus axi-cel; 1.82 versus SOC) and QALYs (incremental QALYs: 0.48 versus axi-cel; 1.92 versus SOC). Of the 10.79 QALYs estimated to be gained with liso-cel, approximately 83% were accrued on the basis of extrapolation after the first 34 months (i.e., beyond the median follow-up period of the TRANSFORM trial). The acquisition costs of liso-cel represent approximately 75% of the total costs associated with liso-cel.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
SOC | 375,714 | 8.87 | Reference |
Liso-cel | 639,948 | 10.79 | 137,251 versus SOC |
Dominated treatments | |||
Axi-cel | 634,299 | 10.31 | Extendedly dominated |
axi-cel = axicabtagene ciloleucel; ICER = incremental cost-effectiveness ratio; liso-cel = lisocabtagene maraleucel; QALY = quality-adjusted life-year; SOC = standard of care.
Source: Sponsor’s pharmacoeconomic submission.1
Sensitivity and Scenario Analysis Results
The sponsor conducted several sensitivity analyses that explored, for example, alternative inputs or assumptions related to discount rate, horizon, wastage, efficacy, mortality, utility values, and perspective. However, only pairwise analyses were provided, limiting interpretation of the results. In a pairwise analysis adopting a societal perspective (i.e., including additional costs associated with transportation and lodging required for CAR T-cell therapy, and productivity losses by patients and caregivers), liso-cel was dominant over axi-cel and SOC; however, disaggregated costs and QALYs were not provided.
CDA-AMC Appraisal of the Sponsor’s Economic Evaluation
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis:
The comparative efficacy of liso-cel versus axi-cel is highly uncertain: To inform the economic model, the sponsor adopted efficacy inputs (EFS, OS) for liso-cel and SOC from the TRANSFORM trial. Because there have been no head-to-head trials comparing liso-cel with axi-cel, the sponsor undertook ITCs to inform the efficacy of axi-cel in the model. The sponsor first conducted a MAIC, which adjusted for differences in population characteristics between the TRANSFORM and ZUMA-7 trials; however, the sponsor deemed that the MAIC results could not be used in the economic evaluation “because ████ ████ ███ █████ ██ ██████ for the OS outcomes.” The sponsor additionally conducted a naive Bucher ITC (i.e., unadjusted for differences in patient characteristics), which was then used to inform the economic evaluation. As noted in the CDA-AMC Clinical Review, the results of the Bucher ITC were largely comparable with those of the MAIC; that is, in both analyses, there were no statistically significant differences in EFS or OS between liso-cel and axi-cel. However, given that the Bucher ITC estimates are not adjusted for between-study differences in patient baseline characteristics and notable baseline differences between the TRANFORM and ZUMA-7 trials (e.g., age, region, SPD, disease histology, cell of origin, bone marrow involvement and secondary central nervous system lymphoma) were identified, there is uncertainty as to the relative treatment effects of liso-cel compared with axi-cel.
Given the lack of direct evidence for liso-cel compared with axi-cel and limitations with the sponsor’s ITC, it remains highly uncertain whether liso-cel provides a net clinical benefit relative to axi-cel.
The comparative safety of liso-cel versus axi-cel is uncertain: In the economic model, the sponsor included costs associated with treatment-related AEs (grade 1 to 4), as well as utility decrements for grade 3 and 4 treatment-related AEs. Data to inform the rate of these AEs were incorporated in the model via naive comparison, without adjustment or accounting for differences in patient characteristics. Owing to the direct use of clinical trial data, it is not possible to determine if any observed differences between the therapies are solely due to the treatment or, rather, due to bias or confounding factors. As noted in the previous limitation, there were notable differences in the TRANSFORM and ZUMA-7 patient populations at baseline for liso-cel and axi-cel, respectively. Although the sponsor conducted MAICs for several harm outcomes (including treatment-related AEs, CRS and neurotoxicity, prolonged cytopenia, severe infections, and hypogammaglobulinemia), data from these analyses were not used in the economic evaluation.
The sponsor asserts that liso-cel “can offer a more favourable benefit-risk profile compared with axi-cel.”18 The CDA-AMC Clinical Review concluded that liso-cel may have a favourable safety profile, but this is subject to uncertainty due to imprecision and imbalances in potential effect modifiers. The sponsor’s economic model predicts that liso-cel will be associated with a marginal difference in QALYs related to treatment-related AEs (incremental QALYs = −0.04) but higher costs (incremental costs = $1,148) compared with axi-cel.
CDA-AMC was unable to address this limitation. Owing to the use of naive comparison, the economic model is unable to provide a sufficiently rigorous estimate of incremental QALYs related to AEs.
The long-term effectiveness of liso-cel is uncertain. Evidence to support the duration and magnitude of benefit associated with liso-cel compared with SOC is limited (median follow-up = 33.9 months). Although Study GC-LTFU-001, a long-term follow-up study of the efficacy and safety of liso-cel and other gene-modified T-cell therapies, is ongoing with an estimated completion date of 2036. No preliminary data were submitted for review.
The sponsor’s economic model suggests that liso-cel will be associated with an incremental gain in LYs and QALYs relative to axi-cel and SOC. In addition to the limitations noted in the previous bullets, CDA-AMC notes that the OS data from the TRANSFORM trial is immature, which introduces additional uncertainty into the long-term extrapolation. Given that approximately 83% of the QALYs predicted to be gained with liso-cel by the sponsor’s model were derived on the basis of extrapolated findings rather than observed benefit, the lack of comparative long-term data introduces considerable uncertainty into the findings.
CDA-AMC was unable to address this limitation owing to a lack of long-term data for liso-cel and comparators. The direction and magnitude of the impact of this limitation is unknown.
The model structure was inappropriate for the decision problem. The sponsor used a PSM to estimate the costs and outcomes associated with second-line treatment. Although PSMs are routinely used in the economic evaluation of oncology treatments, this approach is not suitable for a decision problem in which the primary goal of treatments is to achieve a cure. Although the sponsor’s model did account for patients who achieved cure in the second-line setting, the model did not to account for patients who experienced long-term remission (i.e., were cured) with subsequent lines of therapy, and did not capture changes in OS or HRQoL for such patients (i.e., in line with the general population). In the model, for patients who initially received SOC, the efficacy of CAR T-cell therapy received in the third line was informed by data from the SOC arm of the TRANSFORM trial, in which 63% of patients went on to receive liso-cel in the third line (i.e., after disease progression on SOC). Observations from the TRANSCEND study for liso-cel and the ZUMA-7 trial for axi-cel suggest that approximately 38% to 41% of patients who receive CAR T-cell therapy in the third line remained event free at 2 years (the time at which the sponsor considered patients to be cured).19,20 Thus, the magnitude of the benefit in the extrapolated period was inaccurately estimated, as the model did not allow for the possibility of future cures and related change in OS and HRQoL. A more appropriate model for this decision problem would have been a Markov model with a long-term remission state, which could have incorporated different survival assumptions based on subsequent therapy received.
The clinical expert input received by CDA-AMC for this review indicated that the assumption of a cure at 2 years may capture the majority of patients; however, approximately 5% of patients who are event free at 2 years may experience an event between year 2 and year 5. In practice, patients who are event free after 2 years would continue to be seen by an oncologist and undergo monitoring for progression until year 5. As such, the use of a 2-year cut-off for the definition of “cure” is not consistent with clinical practice.
CDA-AMC was unable to address this limitation owing to the structure of the sponsor’s model.
The impact of liso-cel on HRQoL is uncertain. The sponsor’s base case predicts an incremental gain of 1.92 QALYs with liso-cel compared with SOC over a lifetime horizon. As noted in the CDA-AMC Clinical Review, there was no significant between-group difference observed in the TRANSFORM trial for either European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 (EORTC QLQ-30) global health status and Functional Assessment of Cancer Therapy–Lymphoma “additional concerns” subscale (FACT-LymS) total scores for liso-cel and SOC at 6 months (May 13, 2022, data cut), and the certainty of evidence about the effect of liso-cel on HRQoL was considered to be very low owing to imprecision and concerns about missing data. Thus, whether liso-cel will lead to improved quality of life for patients in clinical practice is highly uncertain.
Although the sponsor provided health state utility values based on EQ-5D-5L observations from the TRANSFORM trial, these values were not utilized in the sponsor’s economic evaluation, as they deemed that the utility estimates for the event-free and progressed disease health states were higher that the utility norms reported for the general population in Canada for the age group of 55 to 59 years (men: 0.85; women: 0.83). Instead, the sponsor adopted utility values from the literature,6 which were based on data from the JULIET trial for tisa-cel in patients with relapsed or refractory LBCL following 2 or more lines of systemic therapy, which were mapped from Short-Form (36) Health Survey (SF-36) and valued using UK EQ-5D tariffs. Thus, the sponsor has implicitly made the assumption that the utility value for patients who are event free in the second or later line (i.e., 0.83 from the tisa-cel submission with a median number of 3 prior lines; range, 1 to 6) would be equivalent to the utility value for the patients who are event free in the second line in the indicated population, which lacks face validity. CADTH previously noted this issue with the use of utilities derived from the JULIET trial for CAR T-cell therapies in the second line.9
CDA-AMC was unable to address this limitation.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CDA-AMC (Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CDA-AMC comment |
|---|---|
The sponsor assumed that salvage chemotherapy (as part of SOC) would comprise R-ICE (64%) and R-GDP (36%), based on clinical expert opinion. | Uncertain. In the TRANSFORM trial, patients received R-DHAP, R-ICE, or R-GDP; however, the sponsor did not include R-DHAP in the model. Clinical expert input received by CDA-AMC for this review indicated that approximately 80% of patients receive R-GDP in Canadian clinical practice as part of salvage chemotherapy, with the remainder receiving R-DHAP or R-ICE. Including R-DHAP in the model would have increased the total cost of SOC. |
Salvage chemotherapy was assumed to be followed by HDCT (BEAM with or without carmustine) and HSCT for 47% of patients who receive SOC. | Uncertain. Clinical expert input noted that between 40% and 80% of patients who receive SOC will likely go on to receive HDCT and that most of those patients will go on to receive transplant (i.e., those who respond to salvage chemotherapy). |
The sponsor assumed that the time from apheresis to infusion would vary between liso-cel and axi-cel based on trial data and literature. | Uncertain. Clinical expert input noted that vein-to-vein time is dependent on company-specific manufacturing time, capacity, and shipping. |
The sponsor estimated that 79% and 97% of patients would receive liso-cel and axi-cel in an inpatient setting, respectively. | Reasonable, according to clinical expert input. |
The sponsor assumed that patients who received SOC in the second line would receive third-line CAR T (60%), allogeneic HSCT (5%), third-line or later SOC (20%), or an experimental treatment via a clinical trial (15%). | Inappropriate. While some patients may enter a clinical trial in the third line, the cost of such investigational therapies would not be covered by the public health care payer and thus should not be included in analyses using a health care payer perspective. Clinical expert input additionally indicated that patients are unlikely to receive allogeneic HSCT in the third line (i.e., after failure of second-line SOC) and that up to 80% of patients may receive CAR T-cell therapy in the third line. |
axi-cel = axicabtagene ciloleucel; BEAM = carmustine, etoposide, cytarabine, and melphalan; CAR T = chimeric antigen receptor T cells; CDA-AMC = Canada’s Drug Agency; HDCT = high-dose chemotherapy; HSCT = hematopoietic stem cell transplant; liso-cel = lisocabtagene maraleucel; R-DHAP = rituximab, dexamethasone, cytarabine, and cisplatin; R-GDP = rituximab, gemcitabine, dexamethasone, and cisplatin; R-ICE = rituximab, ifosfamide, carboplatin, and etoposide; SOC = standard of care.
CDA-AMC Reanalyses of the Economic Evaluation
Base-Case Results
CDA-AMC was unable to address uncertainty related to the comparative clinical data, including the magnitude and duration of benefit for liso-cel. CDA-AMC was additionally unable to resolve uncertainty related to the impact of comparative safety and health state utility values, as well as the limitations with the chosen model structure. As such, CDA-AMC was unable to provide a more reliable estimate of the cost-effectiveness of liso-cel.
Results of the sponsor’s base case suggest that liso-cel will be more effective and more costly compared with axi-cel (incremental QALYs: 0.48; incremental cost: $5,649) over a 50-year horizon, as well as more effective and more costly compared with SOC (Table 3). In the sponsor’s sequential analyses, liso-cel was associated with an incremental cost-effectiveness ratio of $137,251 per QALY gained compared with SOC. The incremental costs compared with SOC are driven by the drug acquisition costs of liso-cel (approximately 75% of the total costs associated with liso-cel predicted by the sponsor’s model). Exploration of the sponsor’s model shows that 85% of the predicted incremental gain in QALYs with liso-cel compared with axi-cel is expected to be accrued in the extrapolation period (i.e., after 34 months; the median length of follow-up in the TRANSFORM trial). Whether these gains are realized in clinical practice is highly uncertain owing to uncertainty in the comparative clinical data and lack of long-term data.
The sponsor asserts that liso-cel can offer a more favourable benefit-risk profile compared with axi-cel.1 While the CDA-AMC Clinical Review concluded that there may be a favourable safety profile for liso-cel, this is subject to uncertainty due to imprecision and imbalances in potential effect modifiers. The sponsor’s economic model predicts there will be a marginal difference in QALYs related to the management of treatment-related AEs with liso-cel compared with axi-cel (incremental QALYs due to AEs: −0.04), as well as higher costs related to treatment-related AEs for liso-cel compared with axi-cel (incremental $1,148). However, given that the rate of AEs in the model was based on a naive comparison that did not control for any potential confounding, the evidence is insufficiently robust to provide a rigorous estimate of incremental QALYs and costs due to AEs. As a result, CDA-AMC cannot draw conclusions about the economic impact of any differences in AEs between liso-cel and axi-cel.
Scenario Analysis Results
CDA-AMC undertook a price reduction analysis using the sponsor’s base case to explore the price reduction that would be required for liso-cel to be considered cost-effective compared with SOC at a WTP threshold of $50,000 per QALY gained. Although the clinical experts identified axi-cel as the most relevant comparator for this indication, axi-cel is currently under negotiation with pCPA for use in the second line. If axi-cel does not become reimbursed on public drug formularies, SOC would be the most relevant comparator.
Based on the sponsor’s base case, which is subject to the limitations identified in the CDA-AMC appraisal of the sponsor’s submitted economic evaluation, the price of liso-cel would need to be reduced by approximately 35% for liso-cel to be considered cost-effective at a WTP threshold of $50,000 per QALY gained (Table 5). CDA-AMC notes that this price reduction assumes there will be a 1.8-year gain in LYs (as per the sponsor’s base case). If this magnitude of gain is not realized, a greater price reduction would be required to achieve cost-effectiveness.
Table 5: CDA-AMC Price Reduction Analyses
Price reduction | Unit drug cost ($) | ICERs for liso-cel versus SOC ($/QALY)a,b sponsor base case |
|---|---|---|
No price reduction | 501,900 | 137,622 |
10% | 451,710 | 112,371 |
20% | 401,520 | 87,120 |
30% | 351,330 | 61,869 |
40% | 301,140 | 36,618 |
50% | 250,950 | 11,367 |
60% | 200,760 | Dominant |
CDA-AMC = Canada’s Drug Agency; HSCT = hematopoietic stem cell transplant; ICER = incremental cost-effectiveness ratio; liso-cel = lisocabtagene maraleucel; QALY = quality-adjusted life-year; SOC = standard of care.
aPrice reduction analyses were conducted using the sponsor’s base case and are subject to the limitations identified in the CDA-AMC appraisal of the sponsor’s economic evaluation.
bSOC was assumed by the sponsor to include salvage chemotherapy therapy with platinum-based chemoimmunotherapy, which may be followed by high-dose chemotherapy and autologous HSCT.
Issues for Consideration
Liso-cel has successfully concluded negotiation with pCPA with a letter of intent for use as third- or later-line therapy (i.e., for the treatment of patients with LBCL that is refractory or relapsed after 2 or more lines of systemic therapy). It is therefore likely that liso-cel is reimbursed by jurisdictional drug plans at confidential prices that are less than the sponsor’s submitted price for use in the second line.
Axi-cel is currently under negotiation with pCPA for this indication. Should negotiations conclude with a letter of intent, the price of axi-cel paid by the drug plans may be lower than incorporated in the sponsor’s model, which was based on the submitted price in the CDA-AMC review of axi-cel.9
Liso-cel and axi-cel, as well as tisa-cel, are reimbursed in some jurisdictions for the treatment of LBCL in the third- or later-line setting, and it is unclear whether sequential use of CAR T-cell therapy will be reimbursed. The cost-effectiveness and budget impact of sequential use of CAR T-cell therapies is unknown.
Clinical expert input received by CDA-AMC indicated differences between jurisdictions and institutions as to the criteria used to determine eligibility for HSCT, with no uniformly implemented, easily reproducible, or objective criteria for HSCT eligibility in Canada. For example, some centres may use age as an eligibility criterion for HSCT, while other centres may not exclude older patients if they have adequate organ function and performance status.
In the sponsor’s economic model, the median time from leukapheresis to liso-cel infusion was assumed to be 36 days based on trial data.13,15,21 Clinical expert input received for this review indicates variability in manufacturing time in practice, given that apheresis collection would be conducted across pan-Canadian sites and manufacturing is conducted in limited sites in Canada and the US.
Issues pertaining to manufacturing are important to the successful delivery of CAR T-cell therapies. Manufacturing failure may occur for a variety of reasons, including an inadequate number of T cells in the apheresed product, poor selection of T cells on day zero of manufacturing, irreversibly impaired T cells (i.e., no response to stimulation in culture), microbial contamination, equipment-related cell loss, and high endotoxin level. In its economic model, the sponsor estimated that 96.7% of patients receiving liso-cel and 95.51% of patients receiving axi-cel received planned treatment.13,15,22 Reasons for not receiving planned CAR T-cell therapy included manufacturing failure, not having measurable disease, AEs related to conditioning chemotherapy, death before CAR T-cell infusion, and receipt of CAR T cells that are “out of spec.” Additional costs associated with manufacturing failures not included in the sponsor’s model may include a longer hospital stay, while a second sample is prepared, if possible, as well as the potential impact on clinical outcomes.
The sponsor assumed in the BIA that liso-cel would not affect overall demand for CAR T-cell therapy. However, if patients who would have otherwise received SOC are considered candidates for liso-cel, the overall demand for CAR T-cell therapy will be higher than anticipated by the sponsor and may have important capacity implications.
The evidence for the effectiveness of CAR T-cell therapy is still in its early stages and evidence is emerging about the rate of late treatment-related toxicities, duration of treatment effect, and what comprises follow-up for patients receiving CAR T-cell therapy in Canada. If the therapy is curative, patients with relapsed or refractory LBCL would be expected to live a longer life and, as such, may incur additional costs to the health system.
To be able to treat patients with CAR T-cell therapy, personnel at specialized treatment centres need to be trained by the manufacturer. For sites already administering liso-cel in the third-line setting, the sponsor anticipates that no additional start-up activities will be required. Since there will likely be multiple CAR T-cell therapies administered by the specialized treatment centres, various protocols for preparation will need to be managed along with delivery of each product type, which would increase the overall administrative burden. Patient care and product preparation will additionally need to be coordinated with the manufacturer.
The sponsor’s implementation plan indicated that it will initially target the 7 provinces that have sites able to administer CAR T-cell therapies. For other jurisdictions, patients will need to travel out of province or out of country for treatment. Travel will also be required for patients in rural areas. The sponsor’s implementation plan describes patient support programs to provide travel assistance to those living at least 2 hours or 200 km from an infusion site. These cut-offs could leave gaps in coverage for some patients or provide insufficient support.
Clinical experts consulted by CDA-AMC noted that they would offer liso-cel in the outpatient setting on a case-by-case basis (e.g., for those who live closer to treatment centres, have caregiver support, and who do not have significant comorbidities). The clinical experts noted that outpatient delivery may expand CAR T-cell therapy availability, which is currently limited due to inpatient capacity (i.e., hospital beds, intensive care unit capacity, and apheresis facilities).
Overall Conclusions
The CDA-AMC Clinical Review found that liso-cel likely results in a clinically meaningful increase in EFS at 12 and 36 months compared with SOC in adults with relapsed or refractory LBCL. At the time of the submitted analysis, median OS had not been reached in either group, and no definitive conclusions could be drawn as to the impact of liso-cel on HRQoL because of concerns about imprecision and missing outcome data. There have been no head-to-head trials of liso-cel versus axi-cel, and the results of both ITCs submitted by the sponsor (MAIC, Bucher ITC) suggest there may be no differences between liso-cel and axi-cel for EFS or OS. The sponsor-submitted MAIC suggested there may be lower odds of CRS, neurologic toxicity, pyrexia, and encephalopathy with liso-cel versus axi-cel; however, these findings were subject to uncertainty due to imprecision and imbalances in potential effect modifiers.
CDA-AMC identified important limitations with the sponsor’s economic evaluation that precluded reanalysis. Notably, CDA-AMC was unable to address uncertainty related to comparative clinical data, including the magnitude and duration of benefit for liso-cel compared with axi-cel, and the model structure, which was insufficient to reflect the impact of subsequent therapies on health outcomes in later lines of therapy.
CDA-AMC was additionally unable to resolve uncertainty related to the impact of AEs on the cost-effectiveness of liso-cel. The sponsor asserts that liso-cel “can offer a more favourable benefit-risk profile compared to axi-cel.” While the CDA-AMC Clinical Review concluded that there may be a favourable safety profile for liso-cel, this is subject to uncertainty due to imprecision and imbalances in potential effect modifiers and because data were incorporated in the model via naive comparison of clinical trial data. It is thus not possible to determine if any differences between the therapies are solely due to the treatment or, rather, due to bias or confounding factors. The sponsor’s base case predicts there will be a marginal difference in QALYs related to the management of treatment-related AEs with liso-cel compared with axi-cel (incremental QALYs: −0.04), as well as higher costs related to treatment-related AEs for liso-cel compared with axi-cel (incremental cost: $1,148). However, given that the rate of AEs in the model was based on a naive comparison that did not control for any potential confounding, the evidence is insufficiently robust to provide a rigorous estimate of incremental QALYs. As a result, CDA-AMC cannot draw conclusions about the economic impact of any differences in AEs between liso-cel and axi-cel.
Given the limitations in the indirect clinical evidence and uncertainties with the submitted economic model, there is insufficient evidence to suggest that liso-cel should be priced higher than axi-cel. Should negotiations with pCPA for the use of axi-cel in the second line conclude without a letter of intent, a price reduction of liso-cel of at least 35% would be required for liso-cel to be considered cost-effective relative to SOC at a WTP threshold of $50,000 per QALY gained.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Breyanzi (lisocabtagene maraleucel), cell suspension in patient-specific single-dose vials for intravenous infusion. Saint-Laurent (QC): Bristol Myers Squibb; 2024 May 8.
2.Breyanzi (lisocabtagene maraleucel): cell suspension in patient-specific single-dose vials for intravenous infusion [product monograph]. Saint-Laurent (QC): Bristol Myers Squibb company; 2024 Sep 25.
3.Interprovincial Health Insurance Agreements Coordinating Committee (IHIACC). Interprovincial billing rates for designated high cost transplants effective for discharges on or after April 1, 2023. Toronto (ON): Government of Ontario; 2023: https://files.ontario.ca/moh-2023-2024-rates-high-cost-procedures-en-2023-05-09.pdf. Accessed by sponsor, no date provided.
4.Abramson JS, Solomon SR, Arnason J, et al. Lisocabtagene maraleucel as second-line therapy for large B-cell lymphoma: primary analysis of the phase 3 TRANSFORM study. Blood. 2023;141(14):1675-1684. PubMed
5.Table: 13-10-0837-01. Life expectancy and other elements of the complete life table, single-year estimates, Canada, all provinces except Prince Edward Island. Ottawa (ON): Statistics Canada; 2023: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310083701. Accessed 2024 Apr 24.
6.ID2019_141: tisagenlecleucel (Kymriah) for the treatment of second or later relapsed/refractory diffuse large B cell lymphoma (DLBCL): update of the previous health economic evaluation. Oslo (NO): Norwegian Medicines Agency; 2022: https://nyemetoder.no/Documents/Rapporter/ID2019_141_Tisagenlecleucel_Kymriah_DLBCL-subgruppe%20-%20hurtig%20metodevudering%20_kun%20offentlig%20versjon.pdf. Accessed by sponsor, no date provided.
7.Howell TA, Matza LS, Jun MP, Garcia J, Powers A, Maloney DG. Health State Utilities for Adverse Events Associated with Chimeric Antigen Receptor T-Cell Therapy in Large B-Cell Lymphoma. Pharmacoecon Open. 2022;6(3):367-376. PubMed
8.Westin JR, Oluwole OO, Kersten MJ, et al. Survival with Axicabtagene Ciloleucel in Large B-Cell Lymphoma. N Engl J Med. 2023;389(2):148-157. PubMed
9.Drug Reimbursement Review clinical, pharmacoeconomic, and ethics report: axicabtagene ciloleucel (Yescarta) for large B-cell lymphoma. Can J Health Technol. 2023;3(6). https://www.canjhealthtechnol.ca/index.php/cjht/article/view/PG0293r/PG0293r. Accessed by sponsor, no date provided.
10.Ontario Drug Benefit Formulary/Comparative Drug Index, effective from March 28, 2024. 43 ed. Toronto (ON): Ontario Ministry of Health; 2024: https://www.ontario.ca/files/2024-05/moh-formulary-edition-43-2024-05-02.pdf. Accessed 2024 Apr.
11.IQVIA Delta PA Ontario. Accessed 2023 Dec 5 [sponsor provided reference].
12.Cancer Care Ontario: funded evidence-informed regimens. 2024; https://www.cancercareontario.ca/en/drugformulary/regimens. Accessed by sponsor, no date provided.
13.Clinical Study Report: JCAR017-BCM-003, Addendum 01. A global randomized multicenter phase 3 trial to compare the efficacy and safety of JCAR017 to standard of care in adult subjects with highrisk, transplant-eligible relapsed or refractory aggressive b-cell non-Hodgkin lymphomas (TRANSFORM) [internal sponsor's report]. Princeton (NJ): Bristol-Myers Squibb Company; 2022 Oct 26.
14.Chemotherapy Protocol: lymphoma: cyclophosphamide-etoposide oral. London (UK): NHS England; 2015: https://www.uhs.nhs.uk/Media/UHS-website-2019/Docs/Chemotherapy-SOPs1/Lymphoma/Cyclophosphamide-Etoposide-PO-Ver-1.1.pdf. Accessed 2024 Apr 2.
15.Locke FL, Miklos DB, Jacobson CA, et al. Axicabtagene Ciloleucel as Second-Line Therapy for Large B-Cell Lymphoma. N Engl J Med. 2022;386(7):640-654. PubMed
16.Product information: Polivy (polatuzumab vedotin). (European public assessment report). Amsterdam (NL): European Medicines Agency; 2023: https://www.ema.europa.eu/en/documents/product-information/polivy-epar-product-information_en.pdf. Accessed 2024 May 2.
17.de Oliveira C, Pataky R, Bremner KE, et al. Phase-specific and lifetime costs of cancer care in Ontario, Canada. BMC Cancer. 2016;16(1):809. PubMed
18.Drug Reimbursement Review sponsor submission: Breyanzi (lisocabtagene maraleucel), cell suspension in patient-specific single-dose vials for intravenous infusion [internal sponsor's package]. Saint-Laurent (QC): Bristol-Myers Squibb Canada; 2024 May 8.
19.Abramson JS, Palomba ML, Gordon LI, et al. Two-Year Follow-up of Transcend NHL 001, a Multicenter Phase 1 Study of Lisocabtagene Maraleucel (liso-cel) in Relapsed or Refractory (R/R) Large B-Cell Lymphomas (LBCL). Blood. 2021;138(Supplement 1):2840.
20.Jacobson C, Locke FL, Ghobadi A, et al. Long-Term (≥4 Year and ≥5 Year) Overall Survival (OS) By 12- and 24-Month Event-Free Survival (EFS): An Updated Analysis of ZUMA-1, the Pivotal Study of Axicabtagene Ciloleucel (Axi-Cel) in Patients (Pts) with Refractory Large B-Cell Lymphoma (LBCL). Blood. 2021;138(Supplement 1):1764.
21.Schuster SJ, Bishop MR, Tam CS, et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N Engl J Med. 2019;380(1):45-56. PubMed
22.Abramson JS, Siddiqi T, Garcia J, et al. Cytokine release syndrome and neurological event costs in lisocabtagene maraleucel-treated patients in the TRANSCEND NHL 001 trial. Blood Adv. 2021;5(6):1695-1705. PubMed
23.Drug Reimbursement Expert Review Committee final recommendation: axicabtagene ciloleucel (Yescarta - Gilead Sciences Canada Inc.). Can J Health Technol. 2023;3(2). https://www.canjhealthtechnol.ca/index.php/cjht/article/view/PG0293/PG0293. Accessed by sponsor, no date provided.
24.Drug Reimbursement Review clinical, pharmacoeconomic, and ethics report: tisagenlecleucel (Kymriah) for relapsed or refractory follicular lymphoma. Can J Health Technol. 2023;3(11). https://www.canjhealthtechnol.ca/index.php/cjht/article/view/PG0306r/PG0306r. Accessed 2024 Apr 2.
25.eviQ. Non-Hodgkin lymphoma R-DHAOx (rituximab dexamethasone cytarabine oxaliplatin). 2023; https://www.eviq.org.au/haematology-and-bmt/lymphoma/other-b-cell-lymphoma/1674-r-dhaox-rituximab-dexamethasone-cytarabine-o#collapse139255. Accessed 2024 Aug 7.
26.Agathou S. To BeEAM or to R-BEAM prior to ASCT in B-cell lymphomas. LymphomaHub 2018; https://lymphomahub.com/medical-information/to-beeam-or-to-r-beam-prior-to-asct-in-b-cell-lymphomas. Accessed 2024 Jul 22.
27.Table: 17-10-0057-01. Projected population, by projection scenario, age and gender, as of July 1 (x 1,000). Ottawa (ON): Statistics Canada; 2022: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1710005701. Accessed 2024 May 2.
28.Canadian Cancer Statistics Advisory Committee, Canadian Cancer Society, Statistics Canada, Public Health Agency of Canada. Canadian Cancer Statistics 2023. Toronto (ON): Canadian Cancer Society; 2023: https://cdn.cancer.ca/-/media/files/research/cancer-statistics/2023-statistics/2023_PDF_EN.pdf. Accessed by sponsor, no date provided.
29.Raut LS, Chakrabarti PP. Management of relapsed-refractory diffuse large B cell lymphoma. South Asian J Cancer. 2014;3(1):66-70. PubMed
30.Sehn LH, Salles G. Diffuse Large B-Cell Lymphoma. N Engl J Med. 2021;384(9):842-858. PubMed
31.ONCO-CAPPS Patient Treatment Dynamics Relapsed/Refractory Diffuse Large B-Cell Lymphoma (R/R DLBCL): Q4 2021 Update and Trends [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Breyanzi (lisocabtagene maraleucel), cell suspension in patient-specific single-dose vials for intravenous infusion. Saint-Laurent (QC): Bristol Myers Squibb; 2022 Jan.
32.Westin J, Sehn LH. CAR T cells as a second-line therapy for large B-cell lymphoma: a paradigm shift? Blood. 2022;139(18):2737-2746. PubMed
Appendix 1: Cost Comparison Table
Table 6: CDA-AMC Cost Comparison Table for Refractory or Relapsed LBCL
Treatment | Strength / concentration | Form | Price ($) | Recommended dosage | Daily cost ($) | Cost per 28 days or 1-time cost ($) |
|---|---|---|---|---|---|---|
CAR T-cell therapy | ||||||
Lisocabtagene maraleucel (Breyanzi) | 60 × 106 to 120 × 106 CAR-positive viable T cells | Suspension for IV infusion | 501,900.0000a | One-time dose | NA | One-time cost: 501,900 |
Axicabtagene ciloleucel (Yescarta) | Target of 2 × 106 anti-CD19 CAR T cells/kg body weight (range, 1 × 106 cells/kg to 2.4 × 106 cells/kg) to a maximum of 2 × 108 anti-CD19 CAR T-cells | Suspension for IV infusion | 485,021.0000b | One-time dose | NA | One-time cost: 485,021 |
Salvage chemotherapy | ||||||
R-GDP | ||||||
Rituximab (biosimilars) | 100 mg vial | IV infusion | 297.0000d | 21-day cycles: 375 mg/m2 on day 1e | 99.00 | 2,772 |
Gemcitabine (generic) | 1,000 mg 2,000 mg | Powder for solution | 270.0000 540.0000 | 21-day cycles: 1,000 mg/m2 on days 1 and 8e | 51.43 | 1,440 |
Dexamethasone (generics) | 4 mg | Tablet | 0.6112d | 21-day cycles: 40 mg on days 1 to 4e | 1.18 | 33 |
Cisplatin (generic) | 50 mg vial 100 mg vial | 1 mg/mL solution for injection | 135.0000 270.0000 | 21-day cycles: 75 mg/m2 on day 1e | 19.29 | 540 |
R-GDP regimen cost (21-day cycle) | 170.90 | 4,785 | ||||
R-ICE | ||||||
Rituximab (biosimilars) | 100 mg vial | IV infusion | 297.0000d | 21-day cycles: 375 mg/m2 on day1e | 99.00 | 2,772 |
Ifosfamide (Ifex) | 1,000 mg vial 3,000 mg vial | Powder for solution | 143.8700 440.5899 | 21-day cycles:1,667 mg/m2 on days 1 to 3c | 61.66 | 1,726 |
Carboplatin (generic) | 50 mg 150 mg 450 mg 600 mg | 10 mg/mL vial for injection | 70.0000 210.0000 600.0000 775.0020 | 21-day cycles: AUC 5 on day 1; maximum dose for AUC 5 is 750 mge | 50.00 | 1,400 |
Etoposide (generic) | 100 mg | 20 mg/mL injection | 75.0000d | 21-day cycles: 100 mg/m2 on days 1 to 3e | 21.43 | 600 |
R-ICE regimen cost (21-day cycle) | 232.09 | 6,498 | ||||
R-DHAP | ||||||
Rituximab (biosimilars) | 100 mg vial | IV infusion | 297.0000d | 21- or 28-day cycles: 375 mg/m2 on day 1e | 74.25 to 99.00 | 2,079 to 2,772 |
Dexamethasone (generics) | 4 mg | Tablet | 0.6112d | 21-day cycles: 40 mg on days 1 to 4e | 1.18 | 33 |
Cytarabine (generic) | 100 mg vial 500 mg vial 1,000 mg vial 2,000 mg vial | 100 mg/mL IV solution | 5.0900 76.8500 153.2500 306.5000 | 21- or 28-day cycles: 2,000mg/m2 twice daily on day 2 | 58.57 to 58.54 | 1,230 to 1,639 |
Cisplatin (generic) | 50 mg vial 100 mg vial | 1 mg/mL solution for injection | 135.0000 270.0000 | 21- or 28-day cycles: 100 mg/m2 on day 1e | 25.71 to 25.71 | 540 to 720 |
R-DHAP regimen cost | 159.71 to 184.43 | 3,873 to 5,164 | ||||
R-DHAX | ||||||
Rituximab (biosimilars) | 100 mg vial | IV infusion | 297.0000d | 21-day cycle: 375 mg/m2 on day 1f | 99.00 | 2,772 |
Dexamethasone (generics) | 4 mg | Tablet | 0.6112d | 21-day cycle: 40 mg on days 1 to 4f | 1.18 | 33 |
Cytarabine (generic) | 100 mg vial 500 mg vial 1,000 mg vial 2,000 mg vial | 100 mg/mL IV solution | 5.0900 76.8500 153.2500 306.5000 | 21-day cycle: 2,000mg/m2 on day 2 and 3f | 43.93 | 1,230 |
Oxaliplatin (generics) | 100 mg vial 200 mg vial | Vial for injection | 90.0000 180.0000 | 21-day cycle: 130 mg/m2 on day 1f | 12.86 | 360 |
R-DHAX regimen cost | 156.97 | 4,395 | ||||
High-dose therapy | ||||||
BEAM | ||||||
Carmustine (generic) | 100 mg vial | 100 mg/mL injection | 4,965.1400e | 300 mg/m2 on day −2 or −6g | NA | One-time cost: 29,791 |
Etoposide (generic) | 100 mg vial 200 mg vial 500 mg vial 1,000 mg vial | 20 mg/mL injection | 75.0000d 150.0000d 375.0000d 750.0000d | 150 to 200 mg/m2 twice daily, on day −5 to −2g | NA | One-time cost: 1,800 to 2,400 |
Cytarabine (generic) | 100 mg vial 500 mg vial 1,000 mg vial 2,000 mg vial | 100 mg/mL IV solution | 5.0900 76.8500 153.2500 306.5000 | 200 to 400 mg/m2 twice daily on days −5 to −2g | NA | One-time cost: 163 to 326 |
Melphalan (generic) | 50 mg vial | 50 mg/mL injection | 88.0700 | 140 mg/m2 on day −1g | NA | One-time cost: 440 |
BEAM regimen cost | NA | One-time cost: 32,194 to 32,957 | ||||
EM | ||||||
Etoposide (generic) | 100 mg vial 200 mg vial 500 mg vial 1,000 mg vial | 20 mg/mL injection | 75.0000d 150.0000d 375.0000d 750.0000d | 60 mg/kg, on day −4 | NA | 3,375 |
Melphalan (generic) | 50 mg vial | 50 mg/mL injection | 88.0700 | 180 mg/m2 on day −3 | NA | 616 |
EM regimen cost | NA | 3,991 | ||||
High-dose therapy costs | ||||||
Autologous HSCT | Adult autologous stem cell transplant (< 72 hours)- includes all facility costs including inpatient and diagnostic costs | NA | 82,555h | |||
Allogeneic HSCT | Adult allogeneic stem cell transplant: includes all facility costs including inpatient and diagnostic costs, excludes matched unrelated donor patients | NA | 189,976h | |||
BEAM = carmustine, etoposide, cytarabine, and melphalan; CAR t = chimeric antigen receptor T-cells; EM = etoposide and melphalan; NA = not applicable; R-GDP = rituximab, gemcitabine, dexamethasone, and cisplatin; R-ICE = rituximab, ifosfamide, carboplatin, and etoposide.
Notes: This table has not been copy edited.
The comparators presented in the above table have been deemed to be appropriate based on feedback from clinical expert(s). Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
All prices are wholesale from IQVIA Delta PA (accessed June 2024), unless otherwise indicated, and do not include dispensing fees. Calculations assume a patient body weight of 75 kg and a body surface area of 1.8 m2.
aSponsor’s submitted price.1
bCADTH Reimbursement Review for axicabtagene ciloleucel.23
cCADTH Reimbursement Review for tisagenlecleucel.24
dOntario Drug Benefit Formulary or Exceptional Access Program list price (accessed June 2024).
eCancer Care Ontario Formulary: Regimens database.
fRegimen as cited in Cancer Institute NSW.25
gRegimen as cited in Lymphoma Hub26 and validated by clinical experts consulted by CDA-AMC.
hInterprovincial Billing Rates for Designated High Cost Transplants Effective for Discharges on or After April 1, 2023. The cost includes all facility costs associated with a single transplant episode including inpatient and diagnostic costs, with a maximum length of stay of 16 days.
Appendix 2: Submission Quality
Please note that this appendix has not been copy-edited.
Description | Yes or No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | Yes | No comment. |
Model has been adequately programmed and has sufficient face validity | No | Refer to key limitation on long-term effectiveness of liso-cel uncertain. |
Model structure is adequate for decision problem | No | Refer to key limitation on long-term effectiveness of liso-cel uncertain and model structure. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | Yes | No comment. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | Yes | No comment. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | Yes | No comment. |
liso-cel = lisocabtagene maraleucel.
Appendix 3: Additional Information on the Submitted Economic Evaluation
Please note that this appendix has not been copy-edited.
2L = second line; 3L+ = third line or later; EFS = event-free survival; OS = overall survival.
Note: Patients enter the model in the 2L state and are assumed to be event free. After an event, patients progress to the 3L+ state. In the right-hand panel, the solid line reflects the OS curve; dashed line represents the EFS curve. The proportion of patients under the EFS curve is assumed to be event free, while the proportion of patients between the EFS and OS lines are assumed to have progressed to third-line treatment.
Source: Sponsor’s pharmacoeconomic submission.1
Detailed Results of the Sponsor’s Base Case
Table 8: Disaggregated Summary of the Sponsor’s Economic Evaluation Results
Parameter | Liso-cel | Axi-cel | SOC |
|---|---|---|---|
Discounted LYs | |||
Total | 13.92 | 13.51 | 12.10 |
By health state | |||
Event free | 11.88 | 10.55 | 6.00 |
Progressed disease | 2.04 | 2.96 | 6.11 |
Discounted QALYs | |||
Total | 10.79 | 10.31 | 8.87 |
By health state | |||
Event free | 9.37 | 8.30 | 4.63 |
Progressed disease | 1.44 | 2.07 | 4.25 |
Treatment-related AEs (2L) | −0.02 | −0.06 | −0.01 |
Discounted costs ($) | |||
Total | 639,948 | 634,299 | 375,714 |
Pre-treatment | 7,017 | 6,947 | Not applicable |
Treatment acquisition (2L) | 482,284a | 462,300a | 61,523 |
Subsequent treatment (3L+) | 28,358 | 36,133 | 190,365 |
Treatment-related AEs | 28,614 | 27,466 | 21,978 |
Resource use | 41,268 | 48,610 | 47,548 |
End-of-life care | 52,407 | 52,843 | 54,301 |
2L = second-line treatment; 3L+ = third- or later-line treatment; AE = adverse event; axi-cel = axicabtagene ciloleucel; liso-cel = lisocabtagene maraleucel; LY = life-year; QALY = quality-adjusted life-year; SOC = standard of care.
aTreatment acquisition costs for liso-cel and axi-cel were adjusted by the sponsor by proportion of patients who received planned treatment in the TRANSFORM trial for liso-cel (96.74%) and the ZUMA-7 trial for axi-cel (95.51%). Reasons for not receiving planned CAR T-cell therapy included those who underwent apheresis but not infusion due to manufacturing failure, not having measurable disease, or AEs related to conditioning chemotherapy; death before CAR T infusion, and receipt of CAR T “out of spec.”
Source: Sponsor’s pharmacoeconomic submission.1
Appendix 4: Additional Details on the CDA-AMC Reanalyses and Sensitivity Analyses of the Economic Evaluation
Please note that this appendix has not been copy-edited.
Given the identified limitations within the submitted pharmacoeconomic model, CDA-AMC was unable to conduct any additional analyses to assess the relative cost-effectiveness of liso-cel for the indicated population.
Appendix 5: Submitted Budget Impact Analysis and CDA-AMC Appraisal
Please note that this appendix has not been copy-edited.
Table 9: Summary of Key Take-Aways
Key take-aways of the budget impact analysis |
|---|
|
AE = adverse event; axi-cel = axicabtagene ciloleucel; BIA = budget impact analysis; CAR = chimeric antigen receptor; CDA-AMC = Canada’s Drug Agency; HSCT = hematopoietic stem cell transplant; liso-cel = lisocabtagene maraleucel; pCPA = pan-Canadian Pharmaceutical Alliance.
Summary of Sponsor’s Budget Impact Analysis
The sponsor submitted a BIA estimating the budgetary impact of reimbursing liso-cel for the treatment of adult patients in the indicated population. The BIA was performed from the perspective of the Canadian public drug plans over a 3-year time horizon (2026 to 2028). The sponsor estimated the eligible population using an epidemiologic approach, with inputs obtained from the literature and clinical expert opinion. The sponsor’s pan-Canadian estimates reflect the aggregated results from provincial budgets, excluding Quebec and the territories. The sponsor assumed that no patients would be covered by the Non-Insured Health Benefits Program (NIHB). The sponsor’s base case included drug acquisition costs, as well costs related to the management of AEs and subsequent treatment. Costs of CAR T provision included pretreatment (i.e., leukapheresis, bridging, and lymphodepleting therapies). Cost of subsequent treatment was incorporated as a 1-time cost, based on EFS (assumed to be a proxy for time to next treatment) from the TRANSFORM trial (for liso-cel and SOC) or ZUMA-7 trial (for axi-cel). Subsequent treatment included CAR T-cell therapies, third-line chemotherapy (including a basket of salvage chemotherapy]), allogeneic HSCT, and clinical trials, with the distribution informed by clinical expert opinion.
The BIA compared a reference scenario in which axi-cel and SOC are reimbursed for the indicated population in the second line with a new drug scenario in which liso-cel is also funded in this setting. SOC was assumed by the sponsor to include salvage chemotherapy, HDCT, and HSCT. In the reference scenario, the market shares for axi-cel and SOC were based on the sponsor’s forecasts for treatments used in the second line, as well as IQVIA sales data for CAR T-cell therapies used in the third line. Market shares and uptake of liso-cel were estimated based on sponsor’s internal forecasts. The sponsor assumed that liso-cel would displace only axi-cel and thus not increase the number of patients who receive CAR T.
In scenario analyses, the sponsor explored the impact of adopting a broader health systems perspective (e.g., incorporating hospitalization costs related to the provision of CAR T, routine monitoring costs, nondrug health care costs associated with managing and monitoring AEs), as well as assuming market expansion with the reimbursement of liso-cel (i.e., that the reimbursement of liso-cel would result increase the number of patients receiving CAR T).
Table 10: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3 if appropriate) |
|---|---|
Target population | |
Canadian population (base year, excluding Quebec and territories) | 31,470,90027 |
Annual incidence of non-Hodgkin lymphoma | 29.68 per 100,00028 |
Percentage of patients with LBCL | 35%29 |
Percentage of patients with LBCL who receive first-line systemic therapy | 90%30 |
Percentage of patients refractory or relapsed after first-line therapy | 45% (35% relapsing, 10% refractory)29 |
Percentage of patients refractory or relapsed within 12 months | 66%31 |
Percentage of patients eligible for autologous HSCT as second-line therapy | 50%30 |
Percentage of patients eligible for CAR T-cell therapy | 100%32 |
Number of patients eligible for drug under review | 394 / 394 / 394 |
Market Uptake (3 years) | |
Uptake (reference scenario) Axi-cel SOC | 31.00% / 37.00% / 38.00% 69.00% / 63.00% / 62.00% |
Uptake (new drug scenario) Liso-cel Axi-cel SOC | 6.20% / 15.17% / 18.62% 24.80% / 21.83% / 19.38% 69.00% / 63.00% / 62.00% |
Cost of second-line treatment (per patient) | |
Liso-cel (one-time cost) Axi-cel (one-time cost) SOC (maximum cost if all cycles completed) | $501,900 $485,021 $15,066 |
axi-cel = axicabtagene ciloleucel; CAR = chimeric antigen receptor; HSCT = hematopoietic stem cell transplant; LBCL = large B-Cell lymphoma; liso-cel = lisocabtagene maraleucel; SOC = standard of care.
Summary of the Sponsor’s BIA Results
Results of the sponsor’s analysis suggest that the reimbursement of liso-cel for the indicated population will be associated with incremental cost savings of $4,996,903 (year 1: $931,721; year 2: $2,279,710; year 3: $1,785,472) under the drug plan perspective.
When a health care system perspective is adopted, the sponsor estimated an incremental cost savings of $4,385,053 (year 1: $882,479; year 2: $2,086,965; year 3: 1,415,608).
The sponsor submitted a scenario analysis in which liso-cel cel was assumed to take market share from SOC (i.e., increasing the total number of patients who receive CAR T). In this scenario, liso-cel was no longer cost-saving to the public health care payer (incremental costs: $13,909,409 over 3 years).
CDA-AMC Appraisal of the Sponsor’s BIA
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Cost related to the management of AEs and subsequent therapy are highly uncertain. In the base case, the sponsor included drug costs related to subsequent therapy and AE management. The sponsor’s results suggest that, although the list price of liso-cel is higher than that of axi-cel, reimbursement of liso-cel will result in savings of more than $8 million in subsequent therapy costs and $77 thousand in AE management costs. As noted in the CDA-AMC Appraisal of the Sponsor’s Economic Evaluation, there is considerable uncertainty associated with these inputs, including the use of naive comparison to inform the rates of AE and the distribution of subsequent therapy. It is thus uncertain if the cost savings estimated by the sponsor for subsequent treatment and AE management will be realized in clinical practice.
In the CDA-AMC base case, drug costs associated with AE management and subsequent therapy were excluded. The impact of this was explored in scenario analyses.
The market share estimates in the reference scenario do not align with clinical expectations: The sponsor assumed that, in the reference scenario, axi-cel will capture 31% of the market share in year 1, 37% in year 2, to 38% in year 3, with the remainder receiving SOC. Clinical expert opinion received by CDA-AMC for this review indicated that the market share of axi-cel is expected to be higher than estimated by the sponsor (i.e., that the uptake of axi-cel in the indicated population would be closer to what the sponsor estimated for SOC). If the number of patients who receive axi-cel is higher than estimated by the sponsor, this will also increase the number of patients expected to receive liso-cel (because the sponsor assumed displacement only of axi-cel).
CDA-AMC explored uncertainty in the market share of axi-cel in scenario analysis.
The market uptake of liso-cel does not align with clinical expectations: The sponsor assumed that liso-cel would capture approximately 6% of the eligible market in year 1, 15% in year 2, and 19% in year 3 based on internal forecasts and expert input. However, clinical expert input received by CDA-AMC suggests that the uptake of liso-cel will be higher than estimated by the sponsor. The underestimation of liso-cel market share leads to an underestimation of the budget impact of liso-cel.
CDA-AMC explored uncertainty in the market uptake of liso-cel in scenario analysis.
Markups, dispensing fees, and copayment discounts were included in the BIA: The BIA included markups, dispensing fees, and copayment discounts that were drug- and province-specific. However, markups, dispensing fees, and copayment discounts may have different criteria dependent on the drug plan and subject to change. Hence, the BIA base case should not include these estimates.
In the CDA-AMC base case, markups, dispensing fees, and copayment discounts were excluded.
The price of drugs paid by public drug plans is uncertain: The prices for CAR T and chemotherapy regimens were based on publicly available list prices and may not reflect the actual prices paid by public drug plans. Any potential confidential rebates are not reflected in this analysis. Axi-cel is currently being negotiated with pCPA, and any potential (future) confidential rebates are not reflected in this analysis.
CDA-AMC could not address this limitation.
The sponsor’s submitted model is not user-friendly and unnecessarily complicated: Several of the model inputs and assumptions in the sponsor’s submitted BIA model were difficult to test or modify with alternate inputs or assumptions due to unnecessary complexity (i.e., nonfunctioning cells). Many cost values were hard coded into the results tables (e.g., AE costs) and created difficulty in validating the calculations.
CDA-AMC could not address this limitation.
CDA-AMC Reanalyses of the BIA
CDA-AMC revised the sponsor’s base case by removing drug costs related to AE management and subsequent therapy, as well as markups, dispensing fees, and copayment discounts. The changes made to derive the CDA-AMC base case are described in Table 11.
Table 11: CDA-AMC Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Changes to derive the CDA-AMC base case | ||
1. Subsequent treatment costs | Included | Excluded |
2. Drug costs related to management of adverse events | Included | Excluded |
3. Markups, dispensing fees, and copayment discounts | Included | Excluded |
CDA-AMC base case | Reanalysis 1 + 2 + 3 | |
The results of the CDA-AMC reanalyses are presented in summary format in Table 12 and a more detailed breakdown is presented in Table 13. In the CDA-AMC base case, the 3-year budget impact is expected to be $3,488,830 (year 1: $540,904; year 2: 1,323,470; year 3: $1,624,457) should liso-cel be reimbursed as per Health Canada’s indication for use in the second-line setting.
Table 12: Summary of the CDA-AMC Reanalyses of the Budget Impact Analysis
Stepped analysis | Three-year total ($) |
|---|---|
Submitted base case | −4,996,903 |
CDA-AMC reanalysis 1 | 3,411,928 |
CDA-AMC reanalysis 2 | −4,920,855 |
CDA-AMC reanalysis 3 | −4,990,792 |
CDA-AMC base case | 3,488,830 |
CDA-AMC conducted the following scenario analyses to explore uncertainty in the market share assumptions for liso-cel and axi-cel.
Aligning market share estimates for axi-cel in the Reference Scenario to align with clinical input received by CDA-AMC (62% in year 1, 63% in year 2, and 69% in year 3).
Assuming higher market uptake of axi-cel (as described in scenario 1) and assuming that liso-cel will displace half of axi-cel’s market share in the new drug scenario (year 1: 31.0%; year 2: 31.5%; year 3: 34.5%).
The disaggregated results of the submitted BIA and CDA-AMC scenario analyses are provided in Table 13.
Table 13: Detailed Breakdown of the CDA-AMC Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|---|
Submitted base case | Reference | 120,537,206 | 137,604,969 | 144,005,381 | 145,072,116 | 426,682,467 |
New drug | 120,537,206 | 136,673,249 | 141,725,671 | 143,286,644 | 421,685,564 | |
Budget impact | 0 | −931,721 | −2,279,710 | −1,785,472 | −4,996,903 | |
CDA-AMC base case | Reference | 35,109,402 | 63,266,245 | 73,825,061 | 75,584,864 | 212,676,169 |
New drug | 35,109,402 | 63,807,149 | 75,148,531 | 77,209,320 | 216,165,000 | |
Budget impact | 0 | 540,904 | 1,323,470 | 1,624,457 | 3,488,830 | |
CDA-AMC scenario analysis 1: Increased market share of axi-cel | Reference | 35,109,402 | 117,820,128 | 119,579,930 | 130,138,746 | 367,538,804 |
New drug | 35,109,402 | 118,901,935 | 121,833,406 | 133,088,418 | 373,823,759 | |
Budget impact | 0 | 1,081,808 | 2,253,476 | 2,949,671 | 6,284,955 | |
CDA-AMC scenario analysis 2: Increased market share of liso-cela | Reference | 35,109,402 | 117,820,128 | 119,579,930 | 130,138,746 | 367,538,804 |
New drug | 35,109,402 | 120,524,647 | 122,328,071 | 133,148,615 | 376,001,333 | |
Budget impact | 0 | 2,704,520 | 2,748,141 | 3,009,869 | 8,462,529 |
BIA = budget impact analysis.
aIn addition to the increased market share of axi-cel as described in scenario 1.
Ethics Report
Abbreviations
ALL
acute lymphoblastic leukemia
axi-cel
axicabtagene ciloleucel
CAR
chimeric antigen receptor
CRS
cytokine release syndrome
DLBCL
diffuse large B-cell lymphoma
HSCT
hematopoietic stem cell transplant
ICU
intensive care unit
liso-cel
lisocabtagene maraleucel
LBCL
large B-cell lymphoma
MCL
mantle cell lymphoma
MM
multiple myeloma
MAIC
matching-adjusted indirect comparison
SCT
stem cell transplant
SOC
standard of care
Supplementary Ethical Considerations: Lisocabtagene Maraleucel for Relapsed or Refractory Large B-Cell Lymphoma
Ethical considerations relevant to all chimeric antigen receptor (CAR) T-cell therapies in the treatment of hematological cancers are described in the Summary Report: Ethical Considerations in the Use of CAR T-Cell Therapies for Hematological Cancers. This review, outlined in the first portion of this report, highlights ethical considerations specific to the use of lisocabtagene maraleucel (liso-cel; Breyanzi) for the treatment of adults with the indication of large B-cell lymphoma (LBCL), including diffuse large B-cell lymphoma (DLBCL) not otherwise specified, primary mediastinal large B-cell lymphoma (PMBCL), high-grade B-cell lymphoma, and DLBCL arising from follicular lymphoma who have disease that is refractory to first-line chemoimmunotherapy or relapses within 12 months of first-line chemoimmunotherapy and who are candidates for autologous hematopoietic stem cell transplant (HSCT). This supplement draws on patient and clinician group and drug program input as well as consultation with clinical experts and clinical and economic reviewers. Liso-cel was previously reviewed for the treatment of adult patients with relapsed or refractory LBCL after 2 or more lines of systemic therapy, including DLBCL not otherwise specified, high-grade B-cell lymphoma, and DLBCL arising from follicular lymphoma, PMBCL, and grade 3B follicular lymphoma after at least 2 prior therapies.
Treatment options for LBCL and patient experiences: LBCL is a heterogeneous and aggressive subtype of non-Hodgkin lymphoma, which is the most common type of blood cancer. The burdens of this disease are further exacerbated by geographic barriers to accessing treatment and variability in reimbursement and financial supports across jurisdictions. Approximately 30% to 50% of patients with LBCL have disease that is refractory to or relapses following first-line treatment with standard of care (SOC) chemotherapy. Approximately 50% of those with relapsed or refractory LBCL are eligible for autologous HSCT in the second line. Patients with disease that is refractory to or relapses after 2 or more lines of therapy may be eligible for 3 CAR T-cell therapies currently approved for use in the third line, which include liso-cel, axicabtagene ciloleucel (axi-cel), and tisagenlecleucel. Additionally, axi-cel is under negotiation with the pan-Canadian Pharmaceutical Alliance for a similar indication in the same line of therapy currently under review. The existing treatments options for LBCL are physically, psychosocially, and financially burdensome for patients and their caregivers. The patient and clinician group input received for this review identified that patients’ goals include the desire for more treatment options, including in earlier lines of therapy, that offer advantages such as longer remission and survival (including cure) as well as improved quality of life and fewer side effects.
Evidentiary uncertainties related to liso-cel for relapsed or refractory LBCL: The safety and efficacy of liso-cel compared with SOC (including 1 cycle of bridging therapy with 1 of 3 prespecified salvage immunochemotherapy regimens followed by 1 cycle of high-dose chemotherapy and autologous HSCT) in adult patients with relapsed or refractory LBCL was evaluated in the pivotal, phase III, open-label, randomized TRANSFORM trial. The conclusions from the Clinical Review of the TRANSFORM trial were that treatment with liso-cel resulted in a clinically meaningful increase in the primary end point of event-free survival at 12 and 36 months and in complete response rate compared with SOC, with a moderate and high certainty of evidence, respectively. At the time of the final analysis, median overall survival had not been reached in either group, and no definitive conclusions could be drawn on health-related quality of life due to concerns of imprecision and missing outcome data. No new safety signals were reported. The sponsor also submitted comparative evidence in the form of a matching-adjusted indirect comparison (MAIC) and an indirect treatment comparison used to inform the pharmacoeconomic model, with the Clinical Review focusing on the MAIC. The MAIC assessed the comparative efficacy and safety of liso-cel (from the TRANSFORM trial) with that of axi-cel (from the ZUMA-7 trial). The Clinical Review concluded that the results of the MAIC did not demonstrate a clinically meaningful difference between liso-cel and axi-cel for efficacy end points, including event-free survival, complete response rate, progression-free survival, and overall survival. However, the results of the MAIC were suggestive of a more favourable safety profile for liso-cel, including lower rates of serious adverse events such as cytokine release syndrome (CRS) and immune effector cell–associated neurotoxicity syndrome. However, the Clinical Review reported there is uncertainty in these estimates due to imprecision and imbalances in potential effect modifiers and prognostic factors. The TRANSFORM trial did not yield long-term safety and efficacy data. The clinical experts consulted stated they were comfortable with recommending liso-cel in the second line based on currently available evidence but noted the need for long-term data on safety, efficacy, and comparative effectiveness (especially with axi-cel) to further inform clinical decision-making. Together, the uncertainties in comparative effectiveness and long-term effectiveness and safety have ethical implications for informed consent. As described in the Pharmacoeconomic Review report, uncertainty about long-term safety, efficacy, and comparative effectiveness also presents challenges for pharmacoeconomic assessments. This presents challenges for understanding the opportunity costs of implementing liso-cel and associated resource allocation decisions at the health system level.
Risk of secondary T-cell lymphomas: As a class, CAR T-cell therapies, including liso-cel, pose a rare risk of secondary malignancy in the form of CAR-positive T-cell lymphoma. Although the development of CAR-positive T-cell lymphoma was not observed in the TRANSFORM trial, the clinical experts acknowledged the possibility of this risk with liso-cel. However, the clinical experts suggested that, based on currently available evidence, this risk would not alter their decision-making regarding liso-cel given the rarity of the risk, survival advantage offered by liso-cel over SOC in second line, and lack of available alternatives in third line. They also noted that currently available second-line treatments, including HSCT, also pose a risk of secondary malignancies. Additionally, the clinical experts noted the importance of informing patients of this risk during consent conversations because, as described in the product monograph, it requires life-long monitoring.
Increasing therapeutic options and access: The input from the clinical experts and clinician groups noted that offering liso-cel in the second line could increase access to more effective therapy earlier in the disease course compared with SOC for patients with LBCL. Notably, they discussed how patients who do not experience a response to second-line therapy may no longer be sufficiently fit to be eligible to receive and withstand the intensity of CAR T-cell therapy in the third line. They noted that offering liso-cel in earlier lines of therapy could potentially help address some geographic barriers to accessing CAR T-cell therapy for patients who reside far from treatment centres, since patients have to be sufficiently well to endure more significant travel, including potential interjurisdictional travel. The input from the clinical experts and clinician groups described that CAR T-cell therapy in the second line has advantages over autologous HSCT, as it tends to be less burdensome for patients and eliminates the need for salvage chemotherapy and stem cell collection. They also noted the benefit of having liso-cel in the second line for patients who are transplant-ineligible but otherwise fit for CAR T-cell therapy. The clinical experts noted that having access to multiple CAR T-cell therapies for relapsed or refractory LBCL in the second line would be preferable, as liso-cel and axi-cel have different toxicity profiles and would thus allow the choice of therapy to be tailored to a patient’s needs, increase access to additional options, and reduce the need for resources (e.g., intensive care unit [ICU] care, hospitalizations) to address severe toxicities. They noted that liso-cel may be a preferable option for patients who are older or have comorbidities that would make them less likely to tolerate axi-cel.
Outpatient delivery of liso-cel: The clinical experts noted they would offer liso-cel in the outpatient setting (if available) because of what they perceive to be a relatively favourable safety profile (characterized by lower rates of CRS and immune effector cell–associated neurotoxicity syndrome) compared with axi-cel. However, a more favourable safety profile for liso-cel was not supported by the Clinical Review assessment of the MAIC, given uncertainty due to methodological limitations. The clinical experts suggested that outpatient delivery could improve quality of life for patients and help expand capacity to deliver CAR T-cell therapy by requiring fewer institutional resources. They described that the choice of outpatient treatment should be determined on a case-by-case basis, with patients who live close to treatment centres, have a reliable caregiver, and do not have significant comorbidities or uncontrolled tumour burden being the most likely to be recommended for outpatient treatment. The clinical experts emphasized the importance of having a reliable caregiver to facilitate safe outpatient delivery and recovery. However, they also highlighted the importance of continuing to offer inpatient treatment with CAR T-cell therapy, since exclusive outpatient delivery would disadvantage patients without reliable caregiver support or those who would otherwise be ineligible for outpatient treatment. They noted that, in the future, outpatient delivery might also permit the expansion of CAR T-cell therapy delivery to some additional centres that are currently unable to offer CAR T-cell therapy due to capacity constraints (e.g., bed shortages).
Health system considerations associated with the use of liso-cel in earlier lines of therapy for LBCL: The clinical experts reiterated that Canada still lacks sufficient health system capacity to deliver CAR T-cell therapy to all eligible patients, given the resource-, personnel-, and infrastructure-intensive nature of CAR T-cell therapy. The ethical, equity, and access challenges arising from existing limitations in manufacturing and delivery capacity for CAR T-cell therapy, and the need for fair and equitable prioritization of access to limited therapy, are detailed further in the summary report that follows. However, the clinical experts noted that the use of liso-cel for the treatment of LBCL in the second line may reduce demand for CAR T-cell therapy in the third line. They also noted that, as the use of liso-cel in the second line is meant to replace autologous HSCT, the use of health system resources (e.g., infrastructure and personnel) may be attenuated by the resources that will be saved by not conducting transplants. Nonetheless, they cautioned that the high cost of liso-cel and CAR T-cell therapies raised ethical considerations about fair and equitable resource allocation and opportunity costs within and beyond the health care system. The clinical experts also reiterated the importance of offering support for patients and caregivers who reside in rural or remote communities to reduce geographic and financial barriers to equitable access to liso-cel, which is currently expected to be offered at █████ specialized treatment centres in ████ jurisdictions. The sponsor’s implementation plan indicates a plan to offer a travel assistance program to cover travel and accommodation expenses for patients and caregivers for a period before and following administration of liso-cel.
Summary Report: Ethical Considerations in the Use of CAR T-Cell Therapies for Hematological Cancers
Summary
Normative and empirical literature on CAR T-cell therapies, as well as past CADTH ethics reports of CAR T-cell therapies for hematological cancers, were reviewed to summarize the ethical considerations associated with the use of CAR T-cell therapies for the treatment of hematological cancers.
Ethical considerations arising in the context of hematological cancers include the unmet need for durable, life-prolonging treatment for patients with relapsed or refractory disease, as well as disparities in the incidence, diagnosis, treatment, and outcomes in hematological cancers, especially the way these affect patients from groups that are racialized, marginalized, or have low socioeconomic status, and those residing in rural areas.
Ethical considerations arising in the evidence used to evaluate CAR T-cell therapies indicate limitations in the representativeness of clinical trial populations, the absence of long-term safety and efficacy data, and the absence of comparative effectiveness data. Uncertainty about the magnitude of clinical benefit presents challenges for the pharmacoeconomic assessment of CAR T-cell therapies and the assessment of opportunity costs and may expose payers to greater financial risks. Budget forecasting may underestimate the overall budget impact of reimbursing CAR T-cell therapies if they are implemented fairly and as needed.
Ethical considerations arise with respect to the potential benefits and harms related to the use and delivery of CAR T-cell therapies. Several access considerations arise in the context of CAR T-cell therapies in Canada, including those related to geographical access, especially as they may disproportionately impact groups that are racialized, marginalized, or have low socioeconomic status and patients lacking caregiver support, as well as inequities that may arise during referral or treatment. Considerations related to privacy and culturally sensitive practices also arise in the context of cell and tissue ownership, as do considerations related to informed consent, shared decision-making, and balanced communication related to CAR T-cell therapies.
Ethical considerations for health systems include challenges associated with the capacity to manufacture and deliver CAR T-cell therapy and scale CAR T-cell centres across Canada due to the complex infrastructure and personnel requirements. Fair priority-setting criteria are required if demand for therapy exceeds manufacturing or delivery capacity. The reimbursement of high-cost, resource-intensive therapies such as CAR T-cell therapies presents opportunity costs for health systems within and beyond the hematological-oncological cancer space. Resources for health information infrastructure may be required to support postmarket surveillance, the collection of real-world evidence, and the implementation of alternative pricing or financing models.
Objectives
This report summarizes the ethical considerations common to the use of CAR T-cell therapies for the treatment of children and adults with hematological cancers in Canada, as identified in the normative and empirical literature on CAR T-cell therapies and informed by previous CADTH ethics reports of CAR T-cell therapies for hematological cancers. These reports addressed ethical considerations related to CAR T-cell therapies in the context of acute lymphoblastic leukemia (ALL), DLBCL, follicular lymphoma, mantle cell lymphoma (MCL), and multiple myeloma (MM).1-8 Past CADTH reports drew upon published literature, consultation with clinical experts, consideration of input from patient groups, clinician groups, and drug programs, and collaboration with the organization’s clinical and Pharmacoeconomic Review teams. Domains of interest in this summary report include ethical considerations related to the therapeutic context of hematological cancers, the evidentiary basis and use of CAR T-cell therapies, and health systems. In the context of this report, any reference to CAR T-cell therapy refers to CAR T-cell therapies used to treat hematological cancers.
Key Ethical Considerations
Therapeutic Context: Hematological Cancers
Patient and caregiver experiences, as well as diagnostic and treatment pathways, vary across the different hematological cancers for which CAR T-cell therapies are available or are under development (e.g., ALL, DLBCL, follicular lymphoma, MCL, and MM). Nonetheless, common ethical considerations are reported across indications, including those related to the high unmet needs of the patient population and equity issues related to disparities in diagnosis, treatment, and outcomes of these cancers. Presently, CAR T-cell therapies are reimbursed, or are under consideration for reimbursement, as second-line, third-line, and fourth-line therapies for patients with relapsed or refractory disease for whom there are few or no available alternative treatments or for whom alternative treatments have failed. As a result, patients eligible for CAR T-cell therapy are usually characterized as having a high unmet need for durable, life-prolonging therapy.
Published literature, which is largely reported from the US, indicates that there are disparities in diagnosis, treatment, and outcomes across hematological cancers, especially for groups that are racialized, marginalized, or have low socioeconomic status and patients residing in rural areas or far from tertiary care centres, and sometimes across age groups.1,2,5-8 Published literature concerning the distribution, incidence, treatment, and outcomes of hematological cancers in Canada is more limited, in part due to gaps in the collection of age-, sex-, and race-related demographic data in Canadian health information databases.9,10 This may limit a contextualized understanding of cancer-related disparities observed in Canada and its subnational jurisdictions.1
The clinical experts consulted during previous reimbursement reviews indicated that geography (residence in rural areas and/or far from tertiary centres) and socioeconomic status could impact the distribution of diagnosis, treatment, and outcomes for hematological cancers in Canada.1,2 They noted that disparities are more likely to be observed in access to primary care before diagnosis than once a patient is actively followed in the cancer care system. However, even in cancer care, requirements to travel and leave one’s support system and costs associated with travel, time off work, or childcare, as well as inconsistent funding and support across Canadian jurisdictions, can differentially impact patients’ and caregivers’ decision-making about treatment and care, including for CAR T-cell therapies, as will be discussed later. Disparities in outcomes between age groups have also been reported in Canada, as adults older than 70 years may have fewer therapeutic options if they are considered ineligible for common second-line or third-line treatments for hematological cancers, including allogenic stem cell transplant (SCT) and autologous SCT.2
Evidence and Evaluation of CAR T-Cell Therapies
Ethical Considerations in Clinical Trial Data
During reimbursement reviews, CAR T-cell therapies have usually been evaluated with phase I and II or phase II, single-arm, open-label trials that offer only limited certainty about short-term therapeutic safety and efficacy and lack head-to-head comparative effectiveness and long-term safety, efficacy, and survival data.1-8 Uncertainty about the magnitude and duration of clinical benefit presents challenges for the assessment of clinical benefits and harms.11 The clinical experts consulted during previous reimbursement reviews of CAR T-cell therapies noted that the risks associated with evidentiary uncertainty for particular therapies are partially mitigated by the growing body of evidence on CAR T-cell therapies as a therapeutic class, which facilitates earlier identification and response to adverse events.1,2 Evidence-generating measures, such as active postmarket surveillance, are required to better understand the risk-benefit profile and cost-effectiveness of CAR T-cell therapies in practice,12 and to inform the clinical and policy decision-making that serves the interests of patients and the public.11,13,14
The extent to which participants in CAR T-cell therapy trials are representative of patients in clinical practice in Canada varies. CAR T-cell therapy trials have generally tended to exclude patients with an Eastern Cooperative Oncology Group Performance Status (ECOG PS) greater than 1, which may not be reflective of clinical practice.1,2,6 ECOG PS is a measure of a patient’s level of functioning in terms of their physical ability and ability to care for themselves and perform daily activities, with a lower score (0 or 1) denoting the highest levels of functioning and a score of 5 denoting death. Further, trials tend to include patients with a median age that is lower than that observed in practice, which may present challenges for the applicability of results to patients who are older and may exclude patients with HIV or hepatitis B.1,6 CAR T-cell therapy trials also tend to include disproportionately higher rates of patients who are white than from other racial or ethnic groups, irrespective of disease incidence within the patient population.1,2,6 Indeed, racial and socioeconomic disparities in access to, and inclusion in, clinical trials have been reported in clinical trials for CAR T-cell therapies in the US (where most CAR T-cell trials are conducted).15,16 For example, participants who are African American or Black were underrepresented in clinical trials of 5 CAR T-cell products across 7 indications for hematological cancers, and are often underrepresented in clinical trials for cancer therapies across hematological indications more generally.1,6-8,15 This may potentially exacerbate the existing health disparities observed in these populations15 and lead to a limited understanding of, and hinder efforts to eliminate, the racial and ethnic disparities observed in disease outcomes for these populations.17
The underrepresentation of racial, ethnic, and other marginalized groups, as well as women, in clinical trials has been identified as a common issue in clinical trials more generally. Underrepresentation in trial participation is ethically concerning, as diverse clinical trial participation contributes to building trust in medical research and institutions (which can impact a patient’s willingness to pursue treatment), promotes fairness for potential participants and their communities, and produces higher-quality biomedical knowledge.18 The clinical experts consulted in a previous Reimbursement Review were uncertain about the clinical implications of the underrepresentation of racial or ethnic groups in CAR T-cell trials.1 However, demographically representative clinical trial data for CAR T-cell therapies may help determine whether therapeutic efficacy varies between subgroups and whether nontherapeutic factors (such as caregiver support or socioeconomic status) have an impact on effectiveness and clinical outcomes in the real world.1,19 Greater support is required to facilitate equitable access to clinical trial participation and to CAR T-cell treatment centres,15,18 and it is important to consider how trial participant selection may privilege certain groups and disadvantage others where demand for CAR T-cell therapy and trial participation exceeds supply.11,20
Ethical Considerations in Economic Models
The lack of long-term safety, efficacy, and survival data, as well as head-to-head comparative effectiveness data, at the time of a reimbursement review has implications for the pharmacoeconomic assessment of CAR T-cell therapies, as it limits the ability to accurately model and assess cost-effectiveness.1,21,22 Uncertainty about pharmacoeconomic assessments, which are used to support the ethical principles of stewardship and public accountability in resource allocation,3 has implications for resource allocation at a health systems level because it hinders assessments of the opportunity costs (or forgone benefits) associated with the reimbursement and resourcing of CAR T-cell therapies compared with other resources.1,6,23 Data collection for long-term safety, efficacy, and comparative effectiveness may support the more robust pharmacoeconomic assessments used to inform reimbursement recommendations and decisions.23
Concerns about evidentiary limitations in pharmacoeconomic assessments and health system sustainability have prompted consideration of alternative pricing and reimbursement models (e.g., value-based agreements, outcome-based pricing) as potential risk-sharing mechanisms that could possibly help mitigate the risks that payers face when reimbursing high-cost therapies, including CAR T-cell therapies, based on uncertain clinical and pharmacoeconomic evidence.6,23-28 Although not currently used in Canada for the reimbursement of CAR T-cell therapies, risk-sharing payment models have been used in other jurisdictions (especially in Europe).24 However, the way such financial arrangements are designed has ethical implications for the distribution of their potential benefits and burdens (e.g., for patients, the public, patients, payers, and manufacturers).28 For example, the way the value of a drug is defined, such as which surrogate outcomes are selected to evaluate efficacy, impacts how financial risks are distributed between manufacturers and payers.
The budget impact of implementing a CAR T-cell therapy may be underestimated if the estimated uptake does not reflect expected demand by patients and clinicians. In the absence of challenges related to manufacturing and delivery capacity, which will be discussed later, CAR T-cell therapies that are reimbursed are expected to be widely adopted by clinicians and patients, resulting in a high expected budget impact.1 Higher budget impacts may present challenges for health systems with respect to the consideration of opportunity costs and fair resource allocation within and beyond the reimbursement of hematological-oncological therapies.6
Use of CAR T-Cell Therapies
Potential Benefits and Harms in the Use and Delivery of CAR T-Cell Therapies
CAR T-cell therapies have the potential to expand access to therapeutic options for patients without alternatives, including those who are ineligible for SCT (e.g., patients who are still sufficiently healthy to receive CAR T-cell therapy but not to undergo SCT, patients who could not find a suitable match for an allogeneic SCT, and patients who exceed the age cut-offs for SCT). As a result, CAR T-cell therapies may offer equity-related advantages by expanding therapeutic options for older patients and for patients who are Black, Indigenous, and racialized, who may be underrepresented in SCT registries and thus unable to find adequate matches for allogeneic SCT in a timely manner.2,29 CAR T-cell therapies may offer additional practical advantages over existing therapies, especially for patients residing in rural or remote regions or with mobility issues, as they require a single infusion and treatment period; as a durable therapy, CAR T-cell therapies may offer the first treatment-free window for patients with some cancers (e.g., MM).1,30,31
Nonetheless, most CAR T-cell therapies lack long-term safety and efficacy data at the time of this Reimbursement Review, which limits the assessment of clinical benefits and harms. In practice, the balance of potential risks and benefits associated with CAR T-cell therapy is assessed relative to available alternative therapeutic options and to a patient’s condition (which, in the case of relapsed or refractory cancer, may have a poor prognosis).1,11,32,33 CAR T-cell therapies bear the risk of severe toxicities, including CRS and other adverse events. Moreover, shortages or inconsistent availability of treatments (e.g., tocilizumab) used to treat patients who develop adverse events (e.g., CRS) after CAR T-cell therapy could impact the safe administration of these therapies.4
The evidence base for CAR T-cell therapies continues to evolve, especially as more therapies are introduced and used in real-world settings. For example, the US FDA announced a safety signal in November 2023 and subsequently issued a boxed warning regarding a class-level risk of secondary malignancy based on postmarketing adverse event and clinical trial reports.53,54 CAR T-cell therapies that use an integrating (retroviral or lentiviral) vector are considered to pose a risk of developing T-cell lymphoma, which may develop as soon as weeks following infusion.54 While incidence is currently expected to be low, it may be underestimated, as sequencing may not have been conducted when a subsequent T-cell lymphoma was observed.53 The clinical experts noted that this risk will need to be considered in the context of disease severity and the availability, or lack thereof, of other therapeutic options, which may also carry risks of secondary malignancy. Patients should be informed of the risk of secondary malignancy in consent conversations for CAR T-cell therapy and provide acknowledgement.53 The risk of secondary malignancy also highlights the importance of randomized clinical trial data to inform comparative clinical risk-benefit assessments, especially as CAR T-cell therapies are offered earlier in the disease course when other therapeutic options may be available.53
Although the long-term safety of CAR T-cell therapies remains uncertain, the clinical experts consulted in previous reimbursement reviews noted that the safety of CAR T-cell therapies has improved as clinicians have become more experienced at administering treatment and identifying and responding to adverse events.1,2 This suggests that the safety of CAR T-cell therapies is context-dependent, where safety and efficacy may be impacted by the level of experience of the treating team and centre and the availability of supportive resources.12 The collection of postmarket data and real-world evidence related to the use of novel CAR T-cell therapies could contribute to a more robust understanding of the real-world safety and efficacy of CAR T-cell therapies, and the balance of risks and benefits, in diverse clinical practice settings and communities.
Equitable Access to CAR T-Cell Therapies
The safe and effective administration of CAR T-cell therapies presently requires administration in a limited number of accredited treatment centres equipped with specialized infrastructure and highly trained providers, which are currently localized in large urban centres in Canada. As a result, access to CAR T-cell therapies may be moderated by geographic and financial barriers. Patients residing far from treatment centres (including in other provinces or territories) must travel to access treatment and spend more than a month near the treatment centre for preinfusion and postinfusion treatment and care.1-3 The financial and psychosocial burdens resulting from geographic distance may impact patients’ therapeutic decision-making (e.g., patients opting for noncurative or inferior treatments to avoid leaving their communities or spending an extended time in hospital).1
Disparities in access to CAR T-cell therapies have been widely reported in the US context, including across race, geography (residence), and socioeconomic status.34,35 Geographic disparities in access to CAR T-cell therapies are especially salient in Canada, and especially for populations residing in rural and Northern communities or in provinces and territories without CAR T-cell centres, given Canada’s vast geography and the limited number of established and proposed CAR T-cell centres.1,2 In the Canadian context, race-based disparities in access should be considered, as they impact First Nations, Inuit, and Métis Peoples — especially in light of their disproportionately higher representation in rural and Northern communities — as well as other marginalized people or groups.1,2 At the same time, CAR T-cell therapies may offer access-related advantages over, and be less burdensome than, existing treatments, as they only require a single treatment period.1,31 Ensuring equitable access to high-quality care across Canada may also require considering what, if anything, might be owed to patients who are eligible for, but opt not to pursue, effective therapeutic options such as CAR T-cell therapy due to geographic or other barriers.1
Presently in Canada, most jurisdictions provide some support for accommodation and/or food-related expenses for people who reside a certain distance from an infusion centre, whereas fewer provide support for travel costs.1 CAR T-cell manufacturers may offer programs for financial and/or accommodation support for required travel, but often include distance-related eligibility cut-offs, which could leave gaps in coverage for some patients or provide insufficient support to cover all costs borne by patients and caregivers.1,2,6,36 Adequate financial support for patients and caregivers may be important for facilitating equitable access to CAR T-cell therapies by mitigating cost-related barriers that are exacerbated by geography (e.g., costs associated with travel, accommodations, and lost income for patients and caregivers who reside outside of cities with CAR T-cell treatment facilities).1,6
Referral practices can also impact access to CAR T-cell therapies in Canada.6,12,37,38 Not only do patients require access to primary care to be referred for CAR T-cell therapy, physicians must be aware of available therapies, eligibility criteria, and the processes involved in making a referral to a treatment centre (which could be located in a different jurisdiction).1,2 Providers less confident in their knowledge about CAR T-cell therapies might be less likely to refer,37 and racial and ethnic disparities observed in the distribution of patients receiving CAR T-cell therapy may be, in part, explained by disparities in referral patterns in primary care rather than in treatment practices in cancer care.38 Accordingly, it is important to have clear and equitable referral practices, educate clinicians about CAR T-cell therapies and referral processes, facilitate communication between clinicians and treatment centres, and provide systems-level supports for clinicians practising outside the large metropolitan centres where CAR T-cell centres are located.1,2 Eligibility for CAR T-cell therapy presently requires patients to have already undergone and failed several lines of therapy. However, not all patients may have had access to, or been eligible for, earlier lines of therapy for reasons outside of their or their providers’ control; this may present a barrier to access to CAR T-cell therapy for a subset of patients.1,31
Cell Ownership
The collection and storage of patients’ cells during CAR T-cell manufacturing may raise questions related to patient privacy and cell ownership, particularly when manufacturers are outside of Canadian jurisdictions.1,6,39 It is important to recognize that tissue and genetic materials are valued differently by different cultural groups (e.g., Indigenous groups internationally), and that informed consent processes need to clearly detail cell processing and ownership, as well as how remaining cells that are not infused will be handled or disposed of.40 Consultation with diverse groups has been identified as essential to CAR T-cell research and implementation to ensure that cell handling and disposal practices, as well as educational and consent materials, are sensitive to the needs and values of diverse patients and communities.6,39,40 In the Canadian context, attention should be paid to understanding Indigenous communities’ values and practices with respect to cell and tissue ownership and governance (e.g., with reference to guidance such as the First Nations principles of OCAP [ownership, control, access, possession]).41
Considerations for Informed Consent
Processes should be in place to ensure that patients (and caregivers) are apprised of the unique risks of, and evidentiary uncertainties related to, CAR T-cell therapies to support robust and ongoing, iterative informed consent, including as patients transition between care settings.6,42-45,53 Robust consent processes should recognize the unique vulnerabilities of patients with cancer who have limited or no alternative therapeutic options and who may be exposed to hype or the underreporting of treatment-related harms or uncertainties related to CAR T-cell therapies, and also recognize their autonomous decision-making capacity.4,6,8 The term “cure” should be avoided in discussions to avoid misleading or promoting false hope for therapies for which long-term clinical effectiveness remains unknown.46 The balance of potential risks and benefits associated with CAR T-cell therapy should be assessed in a process of shared decision-making by patients, providers, and caregivers. For CAR T-cell therapies approved for use in pediatric populations, it is important to recognize the unique vulnerability of children who are reliant on parents or caregivers for decision-making, as well as broader support. Depending on age or determined level of competency, minors may have a more active role in consent or assent to treatment, supported by age-appropriate educational materials about the potential benefits and harms of CAR T-cell therapy to facilitate family-based discussions.43,45 Discussions related to the preservation of fertility may also be important for adolescents and young adults considering CAR T-cell therapy.2 Studying and considering patient reported outcomes and patient experiences may better facilitate shared decision-making about the use of CAR T-cell therapies.12 Additional resources, including the use of translators and the provision of age-appropriate and language-appropriate educational materials for patients and caregivers, may be required to support patient decision-making.45
Health Systems
Manufacturing and Health Systems Capacity
There are at least 2 challenges related to CAR T-cell therapy delivery in Canada: manufacturing and health systems capacity.12 The first concerns the capacity to manufacture and supply CAR T-cell therapies and the timely coordination between manufacturers and CAR T-cell centres for limited manufacturing slots and a multiweek preparatory and manufacturing period (e.g., stabilizing patients’ conditions before apheresis, manufacturing and treatment, coordinating bridging therapy, apheresis, and the transport of cells). As each step in the complex sequence of manufacturing and delivery requirements for CAR T-cell therapy represents an opportunity for disruption or delay, it may be important to consider the development of contingency plans to ensure a stable supply.1,47 Patients may be harmed by delays in access to therapy because they have to be in sufficiently stable and good health to remain eligible for, and be able to withstand, treatment.1,31 The proliferation of CAR T-cell therapies also presents a growing administrative burden for centres, which must maintain resource-intensive accreditations and manage multiple protocols for the preparation and delivery of a growing number of therapies.1 The possibility of domestic, local CAR T-cell manufacturers in hospital and research settings is currently under investigation in the CLIC-01 clinical trial in British Columbia.48 Although still nascent, the potential use of a local CAR T-cell manufacturer in the future may expedite access to CAR T-cell therapies for patients (including eliminating the time required to transport cells to and from international manufacturing facilities) and is expected to be less costly and more cost-effective than CAR T-cell therapies produced by pharmaceutical manufacturers.48
The second challenge concerns the health systems capacity required to meet the therapeutic demand for CAR T-cell therapies in Canada due to the complex infrastructure and personnel requirements.6,39 For example, implementation requires tertiary medical centres with specialized expertise; specialized training for staff; infrastructure modifications; close interactions among experienced inpatient, ICU, outpatient, and emergency personnel and facilities; and the identification of and planning for patients before and after treatment. The implementation of an increasing number of CAR T-cell therapies for a growing number of indications may exacerbate existing health systems capacity challenges. Presently, there are a limited number of pediatric and adult CAR T-cell centres in Canada, which are localized in large urban centres in only some provinces. Although access in provinces and territories lacking CAR T-cell centres is managed through interjurisdictional agreements, the distribution of CAR T-cell centres in Canada could present a barrier for access to treatment for patients residing far from, or in jurisdictions without, CAR T-cell facilities. As a result, it is important to consider the allocation of CAR T-cell centres in a way that reflects regional, rural-urban, and sociodemographic equity.6,49
Although not currently used, outpatient delivery of CAR T-cell therapies has been suggested as a potential mechanism to address capacity limitations and expand access to a greater number of patients by circumventing limitations in inpatient capacity (e.g., health human resources, hospital beds, ICU capacity, apheresis facilities) and to reduce health systems costs.1,49 However, outpatient delivery would increase the need for patients to have access to social supports and a reliable caregiver, because the responsibility for care would be shifted largely onto patients and caregivers and away from trained health care personnel and health systems.1 Thus, a shift to outpatient delivery could potentially exacerbate burdens and the resulting inequities associated with accessing CAR T-cell therapies for patients and caregivers in lower socioeconomic strata and residing far from CAR T-cell centres, as is already observed in the context of SCTs.1 Outpatient delivery would still require significant health systems resources to deliver safe follow-up care for patients presenting with severe side effects or requiring ongoing care, emphasizing the need to invest in the infrastructure required to implement CAR T-cell therapies.6,39
Resource Allocation in the Context of Capacity Limitations
Insufficient supply or capacity to deliver CAR T-cell therapies raises ethical questions related to distributive justice (e.g., Who should be prioritized for access to a particular CAR-T-cell therapy, and why?), as well as procedural justice (e.g., Who should decide how to allocate limited resources and capacity? What constitutes a fair allocation process?).1,3,20,47,50 Fair decision-making processes and priority-setting criteria are required to inform the prioritization of patients for access to CAR T-cell therapies within and across indications to facilitate the equitable allocation of limited resources in Canada.1-8 Indeed, as multiple CAR T-cell therapies become available for single indications, criteria may also be required to determine whether to use 1 therapy over another,31 or whether patients would be eligible (and if so, under what conditions) for re-treatment with CAR T-cell therapy. The development of pan-Canadian priority-setting criteria for prioritizing access to CAR T-cell therapies and/or pan-Canadian coordination could facilitate fair resource allocation processes, accountability in decision-making, equitable pan-Canadian access to CAR T-cell therapies, reduce decision-making burden for clinicians, and reduce inefficiencies as a result of duplicated efforts.1,3,50,55 Consideration of manufacturing and health systems capacity implications may be required if CAR T-cell therapies demonstrate long-term curative potential, which could prompt the use of CAR T-cell therapy in earlier lines of treatment and, thus, for a greater number of patients.11
The introduction of multiple CAR T-cell therapies in Canada and abroad has heightened attention on the importance of fair allocation of scarce CAR T-cell therapies.55-58 In Canada, allocation occurs largely on an ad hoc basis at jurisdictional or institutional levels, which may result in inequitable and inefficient allocation of limited CAR T-cell therapy.55 One of Ontario’s CAR T-cell centres has published a framework to facilitate the systematized, fair allocation of CAR T-cell therapy.55 The framework describes the need for prioritization both at the disease-site level (including prioritization within each disease site) and cell therapy review committee level (requiring prioritization across disease sites).55 Prioritization occurs in a 3-step process:
assessment of patients for medical benefit
evaluation of functional and psychosocial challenges that may limit optimal CAR T-cell tolerance and caregiver availability
consensus review and prioritization by a multidisciplinary team requiring clear, transparent documentation of rationales on the basis of 9 prioritization criteria: medical need or acuity, likelihood of benefit and/or ability to tolerate therapy, safety and risk of complications, adherence to treatment regimen (compliance), social or caregiver support, impact on other resources, length of wait, first-come first-served, exhausted all other treatment options.
Given the context-dependent nature of the prioritization criteria, the authors of the framework note the importance of understanding and providing support to address underlying factors that might disadvantage a patient and deprioritize them for access (e.g., availability of caregiver support or potential challenges in adhering to treatment).55 Additionally, they emphasize the importance of pan-Canadian coordination to ensure equitable access within and across jurisdictions.55 Experience in the US also indicates the importance of explicitly attending to the fair allocation of CAR T-cell therapy to mitigate inequities arising from structural and systemic factors such as socioeconomic status, geography, and race.56-58 For example, the importance of addressing inequities faced by populations who are marginalized, including patients who are Black and those with lower socioeconomic status, may require reconsidering existing allocation practices such as “first-come, first-served,” which inadvertently prioritize those who are wealthier and have better and earlier access to health care.56
Funding, Opportunity Costs, and Data Infrastructure
The reimbursement and implementation of CAR T-cell therapies, which are highly expensive and resource intensive, raises concerns about the sustainability of the Canadian health care system1,6,12 and stewardship and the responsible use of health resources based on available evidence.3 Reimbursing and implementing CAR T-cell therapies presents opportunity costs (or forgone benefits for other treatments or health care services) for fixed health care budgets in which not all services or therapies can be reimbursed, both within hematological and oncological therapies and in other therapeutic classes.12,14,23,42,51,52 Additionally, it presents opportunity costs for health systems resources (e.g., hospital beds, ICU capacity, access to clinical specialists) due to the resource-intensive nature of CAR T-cell therapies.1,3 As discussed previously, uncertainty in the clinical evidence and pharmacoeconomic models used to evaluate CAR T-cell therapies limits the ability to accurately assess the magnitude of benefit of CAR T-cell therapies relative to other treatments or services and thus to inform an understanding of whether the benefits and burdens associated with funding some therapies or services but not others are distributed fairly.23 Clear and transparent decisions about the expansion of access to CAR T-cell therapies in the context of existing systems constraints, competing health care priorities, and long-term health systems sustainability are required to support fair decision-making and sustain patient and public trust.1,11,26,42 Although, as discussed previously, alternative pricing and reimbursement models may potentially help attenuate the risks faced by payers reimbursing therapies based on uncertain clinical and pharmacoeconomic evidence, it is still important to recognize that CAR T-cell therapies would still remain very expensive and resource intensive from a health systems perspective.1
From a health systems perspective, it is also important to consider the clinical and health informatics infrastructure and resources required to collect the data needed to implement novel funding models and postmarket surveillance.14,39
Conclusion
CAR T-cell therapies are being introduced as second-line, third-line, and fourth-line therapies for the treatment of various hematological cancers (e.g., ALL, DLBCL, follicular lymphoma, MCL, MM). Published empirical and normative literature, as well as past ethics reviews of CAR T-cell therapies, were reviewed to identify the ethical considerations relevant to the use of CAR T-cell therapies for the treatment of hematological cancers. Ethical considerations in the context of hematological cancers include the need for an effective, durable treatment that prolongs life, as well as existing disparities in the incidence, diagnosis, treatment, and outcomes for groups that are racialized, marginalized, or have low socioeconomic status, although more data are required to inform a greater understanding of disparities in the Canadian context. Clinical trials assessing CAR T-cell therapies may not be fully representative of the patient population in Canada (e.g., across race, age, and functional status) and lack long-term safety and efficacy data and comparative effectiveness data. The lack of long-term and comparative clinical data limits the certainty of pharmacoeconomic assessments, which poses challenges for the assessment of opportunity costs and may expose payers to greater financial risks. The way alternate pricing or funding arrangements are designed has implications for the distribution of the potential benefits and risks associated with the reimbursement of high-cost therapies that are based on uncertain clinical and pharmacoeconomic evidence. Underestimates in the demand for CAR T-cell therapy can lead to underestimates in the total budget impact of reimbursing and implementing CAR T-cell therapies.
The implementation of CAR T-cell therapies to clinical practice raises several access-related considerations, given a limited delivery capacity and resulting geographic barriers to access; notably, barriers to access may disproportionately impact groups that are racialized, marginalized, or have low socioeconomic status, as well as patients lacking caregiver support. The implementation and reimbursement of an increasing number of CAR T-cell therapies raises several ethical considerations for health systems, including challenges associated with scaling CAR T-cell delivery across Canada due to the complex and resource-intensive infrastructure and personnel requirements. A possible shift to outpatient delivery in the future may expand access to CAR T-cell therapies, but may also shift responsibility for care onto patients and caregivers and may disproportionately burden patients without robust caregiver support. The development of fair, consistent criteria to prioritize access to CAR T-cell therapy would facilitate equitable access across Canada, especially if demand exceeds manufacturing or delivery capacity (e.g., the growing number of CAR T-cell therapies and use in earlier lines of therapy if CAR T-cell therapies demonstrate curative potential may exacerbate demand). Additionally, the high cost of implementing CAR T-cell therapies presents a challenge for health care budgets and raises questions about the systems-level opportunity costs (both within and beyond the oncological space) of reimbursing CAR T-cell therapies.
The absence of long-term and comparative evidence for the safety and efficacy of CAR T-cell therapies necessitates robust postmarket surveillance to better understand the risk-benefit profile and cost-effectiveness of CAR T-cell therapies in practice. Moreover, where possible, postmarket surveillance and the use of real-world evidence may contribute to a better understanding of how the safety and efficacy of CAR T-cell therapies in clinical practice may be impacted by nonclinical factors, and whether this has an impact on how the benefits and burdens associated with the use of this therapy are distributed fairly across diverse demographic subgroups of patients with hematological cancers in Canada.
References
1.Drug Reimbursement Review clinical, pharmacoeconomic, and ethics reports: ciltacabtagene autoleucel (Carvykti) for the treatment of adult patients with multiple myeloma, who have received at least three prior lines of therapy, including aproteasome inhibitor, an immunomodulatory agent and ananti-CD38 antibody, and who are refractory to their last treatment. Can J Health Technol. 2023;3(8). 10.51731/cjht.2023.706. Accessed 2023 Aug 24.
2.Drug Reimbursement Review clinical, pharmacoeconomic, and ethics reports: brexucabtagene autoleucel (Tecartus) for the treatment of adult patients with relapsed or refractory B-cell precursor acute lymphoblastic leukemia (ALL). Can J Health Technol. 2023;3(6). 10.51731/cjht.2023.668. Accessed 2023 Aug 24.
3.Tisagenlecleucel for acute lymphoblastic leukemia and diffuse large b-cell lymphoma: ethics and implementation report. (CADTH optimal use report; vol.8, no.3d). Ottawa (ON): CADTH; 2019: https://www.cadth.ca/sites/default/files/pdf/car-t/op0538-tisagenlecleucel-ethics-and-implementation-jan2019.pdf. Accessed 2023 Aug 24.
4.Axicabtagene ciloleucel for large B-cell lymphoma: ethics and implementation report. (CADTH optimal use report; vol. 9, no. 1e). Ottawa (ON): CADTH; 2019: https://www.cadth.ca/sites/default/files/pdf/car-t/ct0002-ipe-report.pdf. Accessed 2023 Aug 24.
5.Drug Reimbursement Review clinical, pharmacoeconomic, and ethics reports: brexucabtagene autoleucel (Tecartus) for mantle cell lymphoma. Can J Health Technol. 2021;1(11). 10.51731/cjht.2021.187. Accessed 2023 Aug 24.
6.Drug Reimbursement Review clinical, pharmacoeconomic, and ethics reports: axicabtagene ciloleucel (Yescarta) for treatment of adult patients with relapsed or refractory large B-cell lymphoma (LBCL), who are candidates for autologous stem cell transplant (ASCT). Can J Health Technol. 2023;3(6). 10.51731/cjht.2023.667. Accessed 2023 Aug 24.
7.Drug Reimbursement Review clinical, pharmacoeconomic, ethics, and stakeholder input reports: lisocabtagene maraleucel (Breyanzi) for relapsed or refractory large B-cell lymphoma. Can J Health Technol. 2022;2(10). 10.51731/cjht.2022.461. Accessed 2023 Aug 24.
8.Drug Reimbursement Review clinical, pharmacoeconomic, and ethics reports: idecabtagene vicleucel (Abecma) for multiple myeloma. Can J Health Technol. 2022;2(2). 10.51731/cjht.2022.250. Accessed 2023 Aug 24.
9.Tsang M, Le M, Ghazawi FM, et al. Multiple myeloma epidemiology and patient geographic distribution in Canada: a population study. Cancer. 2019;125(14):2435-2444. PubMed
10.Mohyuddin GR, Sinnarajah A, Gayowsky A, Chan KKW, Seow H, Mian H. Quality of end-of-life care in multiple myeloma: a 13-year analysis of a population-based cohort in Ontario, Canada. Br J Haematol. 2022;199(5):688-695. PubMed
11.Imbach KJ, Patel A, Levine AD. Ethical considerations in the translation of CAR-T cell therapies. Cell Gene Ther Insights. 2018;4(4):295-307.
12.Gagelmann N, Sureda A, Montoto S, et al. Access to and affordability of CAR T-cell therapy in multiple myeloma: an EBMT position paper. Lancet Haematol. 2022;9(10):e786-e795. PubMed
13.Buitrago J, Adkins S, Hawkins M, Iyamu K, Oort T. Adult survivorship: considerations following CAR T-Cell therapy. Clin J Oncol Nurs. 2019;23(2):42-48. PubMed
14.Atilla E, Kilic P, Gurman G. Cellular therapies: day by day, all the way. Transfus Apher Sci. 2018;57(2):187-196. PubMed
15.Al Hadidi S, Schinke C, Thanendrarajan S, Zangari M, van Rhee F. Enrollment of Black participants in pivotal clinical trials supporting US Food and Drug Administration approval of chimeric antigen receptor-T cell therapy for hematological malignant neoplasms. JAMA Netw Open. 2022;5(4):e228161. PubMed
16.Alqazaqi R, Schinke C, Thanendrarajan S, et al. Geographic and racial disparities in access to chimeric antigen receptor-T cells and bispecific antibodies trials for multiple myeloma. JAMA Netw Open. 2022;5(8):e2228877. PubMed
17.Marinac CR, Ghobrial IM, Birmann BM, Soiffer J, Rebbeck TR. Dissecting racial disparities in multiple myeloma. Blood Cancer J.2020;10(2):19. PubMed
18.Schwartz AL, Alsan M, Morris AA, Halpern SD. Why diverse clinical trial participation matters. N Engl J Med. 2023;388(14):1252-1254. PubMed
19.Knight JM, Hackett E, Szabo A, et al. Associations between socioeconomic status and bispecific LV20.19 CAR T-cell therapy outcomes. Haematologica. 2023;108(2):588-593. PubMed
20.Jecker NS, Wightman AG, Rosenberg AR, Diekema DS. From protection to entitlement: selecting research subjects for early phase clinical trials involving breakthrough therapies. J Med Ethics. 2017;43(6):391-400. PubMed
21.CAR T-cell therapy: perceived need versus actual evidence. Lancet Oncol. 2018;19(10):1259 PubMed
22.Jonsson B, Hampson G, Michaels J, Towse A, von der Schulenburg JG, Wong O. Advanced therapy medicinal products and health technology assessment principles and practices for value-based and sustainable healthcare. Eur J Health Econ. 2019;20(3):427-438. PubMed
23.Choi G, Shin G, Bae S. Price and prejudice? The value of chimeric antigen receptor (CAR) T-cell therapy. Int J Environ Res Public Health. 2022;19(19):12366. PubMed
24.Goncalves E. Value-based pricing for advanced therapy medicinal products: emerging affordability solutions. Eur J Health Econ.2022;23(2):155-163. PubMed
25.Holmes H. Barriers to patient access to CAR-T therapy in the community setting. Journal of Clinical Pathways. 2018; https://www.texasoncology.com/who-we-are/media-center/in-the-news/news-articles/2018/june/barriers-to-patient-access-to-car-t-therapy-in-the. Accessed 2023 Aug 24.
26.de Lima Lopes G, Nahas GR. Chimeric antigen receptor T cells, a savior with a high price. Chin Clin Oncol. 2018;7(2):21. PubMed
27.Brigand T, Prokupkova A, Muller G, ECL Access to Medicines Task Force. CAR-T cell therapies: how much for survival? Brussels (BE): Association of European Cancer Leagues; 2018: https://www.cancer.eu/wp-content/uploads/2018/06/CAR-T-ECL-Article_Final_20062018.pdf. Accessed 2023 Aug 24.
28.Kannarkat JT, Good CB, Parekh N. Value-based pharmaceutical contracts: value for whom? Value Health. 2020;23(2):154-156. PubMed
29.Apostolidou E, Lachowiez C, Juneja HS, et al. Clinical outcomes of patients with newly diagnosed acute lymphoblastic leukemia in a county hospital system. Clin Lymphoma Myeloma Leuk. 2021;21(11):e895-e902. PubMed
30.Cohen AD, Hari P, Htut M, et al. Patient perceptions regarding ciltacabtagene autoleucel treatment: qualitative evidence from interviews with patients with relapsed/refractory multiple myeloma in the CARTITUDE-1 study. Clin Lymphoma Myeloma Leuk.2023;23(1):68-77. PubMed
31.Goldfinger M, Shah N, Cooper DL. Chimeric antigen receptor T-cell therapy for patients with multiple myeloma-a call for equal opportunity. JAMA Oncol. 2023;9(3):297-298. PubMed
32.Kuehn BM. The promise and challenges of CAR-T gene therapy. JAMA. 2017;318(22):2167-2169. PubMed
33.Chimeric antigen receptor T-cell therapy for B-cell cancers: effectiveness and value - final evidence report. Boston (MA): Institute for Clinical and Economic Review (ICER); 2018: https://icer.org/wp-content/uploads/2020/10/ICER_CAR_T_Final_Evidence_Report_032318.pdf. Accessed 2023 Aug 24.
34.Ahmed N, Shahzad M, Shippey E, et al. Socioeconomic and racial disparity in chimeric antigen receptor T cell therapy access. Transplant Cell Ther. 2022;28(7):358-364. PubMed
35.Emole J, Lawal O, Lupak O, Dias A, Shune L, Yusuf K. Demographic differences among patients treated with chimeric antigen receptor T-cell therapy in the United States. Cancer Med. 2022;08:08.
36.Snyder S, Albertson T, Garcia J, Gitlin M, Jun MP. Travel-related economic burden of chimeric antigen receptor T cell therapy administration by site of care. Adv Ther. 2021;38(8):4541-4555. PubMed
37.Blue B. Socioeconomic and racial disparity in chimeric antigen receptor T cell (CART) therapy access. Transplant Cell Ther. 2022;28(7):345-346. PubMed
38.Molina A, Muffly L. CT-095 disparities and incomplete capture of race/ethnicity data in adults with hematologic malignancies referred for cellular therapies. Clin Lymphoma Myeloma Leuk. 2022;22(Suppl 2):S433-S434.
39.Lamprecht M, Dansereau C. CAR T-cell therapy: update on the state of the science. 2019;23(2):6-12.
40.Weinkove R, George P, Ruka M, Haira TH, Giunti G. Chimeric antigen receptor T-cells in New Zealand: challenges and opportunities. N Z Med J. 2021;134(1542):96-108. PubMed
41.First Nations Information Governance Centre (FNIGC). The First Nations Principles of OCAP®. https://fnigc.ca/ocap-training/. Accessed 2023 Aug 24.
42.Cossu G, Birchall M, Brown T, et al. Lancet Commission: stem cells and regenerative medicine. Lancet. 2018;391(10123):883-910. PubMed
43.Mahadeo KM, Khazal SJ, Abdel-Azim H, et al. Management guidelines for paediatric patients receiving chimeric antigen receptor T cell therapy. Nat Rev Clin Oncol. 2019;16(1):45-63. PubMed
44.Darrow JJ, Sarpatwari A, Avorn J, Kesselheim AS. Practical, legal, and ethical issues in expanded access to investigational drugs. N Engl J Med. 2015;372(3):279-286. PubMed
45.McConville H, Harvey M, Callahan C, Motley L, Difilippo H, White C. CAR T-cell therapy effects: review of procedures and patient education. Clin J Oncol Nurs. 2017;21(3):E79-E86.
46.Bach PB, Giralt SA, Saltz LB. FDA approval of tisagenlecleucel: promise and complexities of a $475 000 cancer drug. JAMA. 2017;318(19):1861-1862. PubMed
47.Couzin-Frankel J. For experimental cancer therapy, a struggle to ensure supply keeps up with demand. Science 2017; https://www.sciencemag.org/news/2017/06/experimental-cancer-therapy-struggle-ensure-supply-keeps-demand. Accessed 2023 Aug 24.
48.Kekre N, Hay KA, Webb JR, et al. CLIC-01: Manufacture and distribution of non-cryopreserved CAR-T cells for patients with CD19 positive hematologic malignancies. Front Immunol. 2022;13:1074740. PubMed
49.Snyder S, Chung KC, Jun MP, Gitlin M. Access to chimeric antigen receptor T cell therapy for diffuse large B cell lymphoma. Adv Ther. 2021;38(9):4659-4674. PubMed
50.Unguru Y, Fernandez CV, Bernhardt B, et al. An ethical framework for allocating scarce life-saving chemotherapy and supportive care drugs for childhood cancer. J Natl Cancer Inst. 2016;108(6):djv392. PubMed
51.Gumer JM. A new frontier in the fight against cancer (that is, for those who can afford it): the approval and cost of CAR T-cell therapies. Voices Bioeth. 2018;4. https://journals.library.columbia.edu/index.php/bioethics/article/view/6024/2987. Accessed 2023 Aug 24.
52.Gonzalez-Vicent M, Sanz J, Fuster JL, et al. Autoimmune hemolytic anemia (AIHA) following allogeneic hematopoietic stem cell transplantation (HSCT): a retrospective analysis and a proposal of treatment on behalf of the Grupo Espanol De Trasplante de Medula Osea en Ninos (GETMON) and the Grupo Espanol de Trasplante Hematopoyetico (GETH). Transfus Med Rev.2018;32(3):179-185. PubMed
53.Prasad V. T-Cell lymphoma from CAR T-cell therapy—a new safety notice. JAMA. 2024;331(5):389-390. PubMed
54.U.S. Food and Drug Administration (FDA). FDA requires boxed warning for T cell malignancies following treatment with BCMA-directed or CD19-directed autologous chimeric antigen receptor (CAR) T cell immunotherapies. 2024; https://www.fda.gov/vaccines-blood-biologics/safety-availability-biologics/fda-requires-boxed-warning-t-cell-malignancies-following-treatment-bcma-directed-or-cd19-directed. Accessed 2024 Jul 8.
55.Bell J, Jeffries G, Chen C. Mitigating inequity: ethically prioritizing patients to CAR T-cell therapy. Blood. 2023;142(15):1263-1270. PubMed
56.Derman BA, Parker WF. Fair allocation of scarce CAR T-cell therapies for relapsed/refractory multiple myeloma. JAMA. 2023;330(8):687-688. PubMed
57.Goldfinger M, Shah N, Cooper DL. Chimeric antigen receptor T-cell therapy for patients with multiple myeloma—a call for equal opportunity. JAMA Oncology. 2023;9(3):297-298. PubMed
58.Kuipers MT, Kersten MJ. CAR Traffic jam: who can use the fast lane? Blood. 2023;142(15):1257-1258. PubMed
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.