CADTH Reimbursement Review
Brexucabtagene Autoleucel (Tecartus)
Sponsor: Gilead Sciences Canada Inc.
Therapeutic area: Acute lymphoblastic leukemia
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Ethics Review
Stakeholder Input
Clinical Review
Abbreviations
allo-SCT
allogeneic stem cell transplant
AE
adverse event
ALL
acute lymphoblastic leukemia
Brexu-cel
brexucabtagene autoleucel
CAR T
chimeric antigen receptor T
CI
confidence interval
CNS
central nervous system
CR
complete remission
CRh
complete remission with partial hematologic recovery
CRi
complete remission response with incomplete hematologic recovery
CRS
cytokine release syndrome
CSF
cerebrospinal fluid
CTTC
Cell Therapy Transplant Canada
DOR
duration of remission
ECOG
Eastern Cooperative Oncology Group
EFS
event-free survival
ESS
effective sample size
FAS
full analysis set
HR
hazard ratio
HRQoL
health-related quality of life
ITC
indirect treatment comparison
ITT
intention to treat
KM
Kaplan-Meier
LLSC
Leukemia & Lymphoma Society of Canada
MAIC
matching adjusted indirect comparison
mITT
modified intention to treat
MRD
minimal residual disease
NE
not estimable
OCR
overall complete remission
OH-CCO
Ontario Health – Cancer Care Ontario
OS
overall survival
Ph
Philadelphia chromosome
RCT
randomized controlled trial
RFS
relapse-free survival
R/R
relapsed or refractory
SAE
serious adverse event
SCA
synthetic control arm
SCT
stem cell therapy
SMD
standardized mean difference
SOC
standard of care
TKI
tyrosine kinase inhibitor
VAS
visual analogue scale
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Item | Description |
|---|---|
Drug product | Brexucabtagene autoleucel (Tecartus) cell suspension in a patient-specific single-infusion bag for IV use at a target dose of 1 × 106 chimeric antigen receptor T-cells per kilogram |
Indication | For the treatment of adult patients with relapsed or refractory B-cell precursor ALL |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Priority review |
NOC date | November 16, 2022 |
Sponsor | Gilead Sciences Canada Inc. |
ALL = acute lymphoblastic leukemia; NOC = Notice of Compliance.
Introduction
Acute lymphoblastic leukemia (ALL) is a rare form of leukemia in adults.1 It accounts for approximately 5% of all adult leukemia cases in Canada. Among these ALL cases, 80% are of B-cell lineage and the B-cell precursor ALL is found in 75% of adult ALL. About 50% of the patients who have B-cell precursor ALL have relapsed or refractory (R/R) disease. The estimated prevalence and incidence of R/R B-cell precursor ALL is 1,148 and 58 people, respectively, based on an estimated population in 2021 in Canada.2 Typical clinical presentations of ALL are associated with anemia, neutropenia, and/or thrombocytopenia due to bone marrow involvement. Although more than 80% of adult patients with newly diagnosed ALL will achieve a complete remission (CR) with intensive induction chemotherapy, the majority of these patients will ultimately relapse and the prognosis is poor.3
For patients with R/R B-cell precursor ALL, treatment options include cytotoxic chemotherapy regimens, targeted therapies, allogeneic stem cell transplant (allo-SCT) and the emerging chimeric antigen receptor T (CAR T)-cell therapy.4,5 CAR T-cell therapy is a treatment in which T lymphocytes are removed from a patient via apheresis, transduced ex vivo with a gene rendering them immunogenic against certain cancer cells, grown, and subsequently reinfused in to the patient. The activated T-cells then circulate, attack, and kill the targeted cancer cells.5 After the induction therapy, the patients should proceed to allo-SCT as soon as possible if they are eligible, to consolidate the treatment effect obtained from the initial induction therapy.6
Brexucabtagene autoleucel (brexu-cel; brand name Tecartus) is a CD19-directed genetically modified autologous T-cell immunotherapy that binds to CD19-expressing cancer cells and normal B-cells.7 On November 16, 2022, brexu-cel was approved by Health Canada for the treatment of adult patients with R/R B-cell precursor ALL.7 The sponsor’s reimbursement request is the same as the Health Canada indication. Brexu-cel is a single-dose, one-time treatment in a patient-specific infusion bag. Each patient-specific, single-infusion bag of brexu-cel contains a suspension of anti-CD19 CAR-positive viable T-cells in approximately 68 mL for a target dose of 1 × 106 CAR-positive viable T-cells per kg of body weight, with a maximum of 1 × 108 CAR-positive viable T-cells for patients weighing 100 kg or more.7
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient groups who responded to CADTH’s call for patient input and from clinical experts consulted by CADTH for the purpose of this review.
Patient Input
Patient input for the review of brexu-cel was provided by the Leukemia and Lymphoma Society of Canada (LLSC). An online survey was distributed by the LLSC between August 15 and September 21, 2022. A total of 22 individuals across Canada responded to the survey. Two respondents reported experience with brexu-cel .
The majority of the survey respondents indicated that typical symptoms include fatigue or weakness, followed by loss of appetite or weight loss, bone or joint pain, headaches, blurred vision, nausea, or vomiting, which had a significant impact on their ability to work, exercise, and continue everyday activities. This was followed by the ability to travel and pursue activities and hobbies, and to maintain intimate relationships. The majority of the survey responses indicated that the interruption of life goals and accomplishments (e.g., career and schooling) was a psychological and social factor of the disease that had a significant impact on their quality of life. This was followed by stress, anxiety, and worry; feelings of isolation; problems concentrating; loss of sexual desire; and financial impacts.
The outcomes that were considered most important to patients when making decisions related to treatment were the degree of certainty that ALL will respond to treatment and improve quality of life. These outcomes were followed by insurance or drug plan coverage and prolonged survival. Of note, the LLSC indicated that reduced side effects and easier accessibility were frequently mentioned as an improvement that respondents would like to see in any new treatment for ALL.
Clinician Input
Input From Clinical Experts Consulted by CADTH
The clinical experts indicated that for many adult patients with ALL, the most important treatment goal is to cure the disease and improve their health-related quality of life (HRQoL). The clinical experts indicated that the prognosis for these patients is poor. Once targeted therapy of blinatumomab or inotuzumab, or SCT, has been used and has failed, or if such treatments cannot be used, options available to the patients in this situation are limited, which represents an unmet need for effective treatments for B-cell precursor ALL.
The experts stated that brexu-cel can be used in patients who are ineligible for treatment with inotuzumab or blinatumomab, or who have relapsed once or twice after prior treatment with inotuzumab or blinatumomab. The clinical experts indicated that it would be beneficial if all these treatments were available for the patients with R/R ALL, which is a difficult-to-treat disease. The experts also agreed that brexu-cel is expected to cause a shift in the current treatment paradigm if it is approved and reimbursed, particularly for patients aged 25 years and older. It was noted that another CAR T-cell therapy, tisagenlecleucel, has been approved by Health Canada for the treatment of pediatric and young adult patients aged 3 to 25 years with B-cell precursor ALL who are refractory, have relapsed after SCT or are otherwise ineligible for SCT, or have experienced second or later relapse.8
Per the clinical experts, patients with a higher percentage of blasts in bone marrow at baseline or the presence of central nervous system (CNS) leukemia may have poor response to CAR T-cell therapy. The clinical experts noted that more clinical evidence is needed to identify the subsets of patients with R/R B-cell precursor ALL who would be best suited for treatment with brexu-cel .
The experts indicated that in clinical practice, patients are evaluated and followed in a similar manner described in the ZUMA-3 study. Bone marrow biopsies, the level of remission, and complete blood counts are routinely conducted to assess treatment response. In practice, complete blood counts are assessed during a patient’s routine visits, while bone marrow biopsy is less frequently performed, unless unusual results from other examinations are observed, or when brexu-cel is used as a bridge to an eventual allo-SCT and the clinician wants to know if remission has been achieved at the end of the treatment.
The clinical experts reported that meaningful responses to treatment with brexu-cel include prolonged overall survival (OS), minimal residual disease (MRD)–negative rate, improved HRQoL, better performance status, and the durability of treatment response.
The experts indicated that treatment with brexu-cel needs to be provided by hematologists and/or oncologists, who have experience in treating leukemia and in treating leukemia with cellular therapy or SCT.
Clinician Group Input
Two clinician groups provided input for the review of brexu-cel: Cell Therapy Transplant Canada (CTTC), which was represented by 4 clinicians; and the Ontario Health – Cancer Care Ontario (OH-CCO) Complex Malignant Hematology Group, which was represented by 2 clinicians. The OH-CCO Drug Advisory Committees provide evidence-based clinical and health system guidance on drug-related issues.
The clinician group input is consistent with the input provided by the experts consulted by CADTH for the brexu-cel review. They also pointed out that with the currently available targeted therapies, such as blinatumomab or inotuzumab, few patients have a long-term remission with these therapies alone, and allo-SCT has remained the only curative option for patients with R/R disease, but not every patient is eligible to receive this treatment. The clinician group suggested that the patients who are best suited for brexu-cel are adult patients with R/R B-cell precursor ALL with morphological disease in the bone marrow (greater than 5% blasts).
Drug Program Input
Input was obtained from the drug programs that participate in the CADTH reimbursement review process. The following were identified as key factors that could potentially affect the implementation of a CADTH recommendation for brexu-cel:
Considerations for initiation of therapy
Considerations for prescribing of therapy
Generalizability
Funding algorithm
System and economic issues
The clinical experts consulted by CADTH provided advice on the potential implementation issues raised by the drug programs.
Clinical Evidence
Pivotal Studies and Protocol Selected Studies
Description of Studies
One clinical study (the ZUMA-3 study) is included the CADTH systematic review. The ZUMA-3 study9 (N = 71) is an ongoing phase I/II, open-label, single-arm study that evaluated the efficacy and safety of brexu-cel in patients with R/R B-cell ALL. The primary end point was overall complete remission (OCR) rate (defined as CR plus CR response with incomplete hematologic recovery [CRi]) by central assessment. Secondary end points included OS, relapse-free survival (RFS), duration of remission (DOR), MRD-negative rate, subsequent allo-SCT rate, and HRQoL. A total of 55 of the 71 patients enrolled received brexu-cel and were included in the primary efficacy and safety analyses. Data up to 21 months of follow-up were available at the time of this review, with a data cut-off date of July 23, 2021. For patients treated in phase II of the ZUMA-3 study, the median actual follow-up time from the brexu-cel infusion was 20.5 months (range, 0.3 to 32.6 months), and the median potential follow-up time from the brexu-cel infusion was 26.8 months (range, 20.7 to 32.6 months). The mean age of patients was 42 years. The majority of patients were male (60%), white (67%), and had an Eastern Cooperative Oncology Group (ECOG) performance status of 1 (71%). Overall, 27% of patients were Philadelphia chromosome (Ph) positive. Overall, 45%, 22%, and 42% of the patients had prior blinatumomab, inotuzumab, or prior allo-SCT, respectively; 33% of the study population had primary refractory disease; and 78% had R/R to second-line or greater therapy. The mean percentage of blasts in bone marrow at baseline was 33%. Extramedullary disease at baseline was reported in 11% of the patients. All patients had CNS-1 disease (no detectable leukemia in the cerebrospinal fluid [CSF]) before entering the study.
Efficacy Results
At the data cut-off of July 23, 2021, based on the 21-month follow-up data in phase II of the ZUMA-3 study, the median OS measured with the Kaplan-Meier (KM) method was 25.4 months (95% confidence interval [CI]: 16.2 months to not estimable [NE]) in the overall patient population. The median OS was 26.0 months (95% CI, 21.9 months to NE) for those who had achieved CR or CRi, and was 2.4 months (95% CI, 0.7 months to NE) for all other patients who did not achieve CR or CRi. The median OS was not reached (95% CI, 25.4 months to NE) for patients with CR.
Another survival outcome, RFS, was defined as the length of time from the brexu-cel infusion date to the date of disease relapse or death from any cause. The median RFS was 11.6 months (95% CI, 2.7 to 20.5 months) in the overall population. Among patients with CR or CRi, the median RFS was 15.5 months (95% CI, 11.6 months to NE). The median RFS was 22.1 months (95% CI, 11.6 months to NE) for patients with CR and 11.7 months (95% CI, 1.8 months to NE) for those with CRi.
The OCR rate (including CR and CRi) per central assessment was the primary outcome of the ZUMA-3 study. For patients in the phase II modified intention to treat (mITT) analysis set in the ZUMA-3 study, the OCR rate was 70.9% (39 of 55 patients, 95% CI, 57% to 82%), with a CR rate of 56.4% (31 of 55 patients, 95% CI, 42% to 70%), which was higher than a prespecified historical overall response rate of 40% identified for adult patients with ALL who received standard of care (SOC) treatment.
Eleven patients (20%) received subsequent allo-SCT. Among them, 10 (18%) achieved OCR and 8 (15%) achieved CR.
The median EQ-5D visual analogue scale (VAS) score was 70.0 (range, 5 to 100; n = 51) at screening and increased over time: 80.0 (range, 20 to 100; n = 41) at day 28, 80.0 (range, 50 to 100; n = 26) at month 3, 85.0 (range, 40 to 100; n = 25) at month 6, 87.5 (range, 70 to 100; n = 14) at Month 12, ██ ███████ ██ ██ ███ ████ ██ █████ ███ ███ ████ ███████ ██ ██ ████ ████ ██ █████ ███ ████ ████ ████████ ██ █████ ███ ████████ ██ ███ █████ ███ █████ ████ █████ ██ ██████████
Harms Results
At the data cut-off date of July 23, 2021, all 55 patients in the safety analysis set in phase II component of the study ZUMA-3 reported at least 1 adverse event (AE). The most commonly reported AEs included pyrexia (95%), hypotension (67%), anemia (53%), nausea (38%), sinus tachycardia (38%), headache (36%), chills (33%), and decreased platelet count (33%). Serious adverse events (SAEs) were reported in 41 patients (75%). The most commonly reported SAEs were hypotension (29%), pyrexia (27%), and hypoxia (13%). In total, 25 of 55 patients (45%) had died as of the data cut-off date. Eleven patients (20%) had died due to AEs, including 4 (7%) who died due to disease progression within 3 months after the brexu-cel and 7 (1%) who died due to AEs other than disease progression. Brexu-cel is administered as a one-time single infusion; no patients discontinued treatment due to treatment-emergent AEs in the ZUMA-3 study.
In terms of notable harms, cytokine release syndrome (CRS) was the most commonly reported notable harm in the study population. A total of 49 patients (89%) had CRS, and 13 (24%) had worst grade 3 or higher CRS. No patient had grade 5 CRS. Pyrexia, hypotension, sinus tachycardia, chills, and hypoxia were typically reported notable harms. Thirty-three patients (60%) had at least 1 neurologic AE. Frequently reported neurologic AEs in the study population were tremor, confusional state, and encephalopathy.
According to the clinical experts consulted by CADTH, the safety profile of brexu-cel is consistent with other CAR T-cell therapy, and no unexpected safety signals are observed from the included studies.
Table 2: Summary of Key Results from the ZUMA-3 Study
Results | ZUMA-3 study (phase II) |
|---|---|
Efficacy (mITT set, N = 55) | |
OS by central assessment | |
Death, n (%) | 25 (45.5) |
KM median OS, months (95% CI), range | 25.4 (16.2 to NE), 0.30 to 32.56+ |
For patients with CR or CRi | 26.0 (21.9 to NE) |
For patients with CR | Not reached (25.4 to NE) |
For patients with all other responses | 2.4 (0.7 to NE) |
RFS by central assessment | |
Events, n (%) | 33 (60.0) |
KM median RFS, months (95% CI), range | 11.6 (2.7 to 20.5), 0.03 to 26.02 |
For patients with CR or CRi | 15.5 (11.6 to NE) |
For patients with CR | 22.1 (11.6 to NE) |
For patients with CRi | 11.7 (1.8 to NE) |
Patients with overall response by central assessment (CR + CRi), n (%) | 39 (70.9) |
95% CI (Clopper-Pearson method) | 57 to 82 |
Patients with CR by central assessment, n (%) | 31 (56.4) |
95% CI (Clopper-Pearson method) | 42 to 70 |
Patients with CRi by central assessment, n (%) | 8 (14.5) |
95% CI (Clopper-Pearson method) | 6 to 27 |
Patients with MRD-negative remission by central assessment, n (%) | 42 (76) |
95% CI (Clopper-Pearson method) | 63 to 87 |
DOR by central assessment, months, KM median (95% CI), range | 14.6 (9.4 to NE); 0.03+ to 24.08 |
Incidence of subsequent allo-SCT | |
Patients with allo-SCT posttreatment, n (%) | 11 (20) |
Patients with allo-SCT and achieved CR or CRi, n (%) | 10 (18) |
Safety (safety analysis set, N = 55) | |
Patients with ≥ 1 AE, n (%) | 55 (100) |
Patients with ≥ 1 SAE, n (%) | 41 (75) |
Death, n (%) | 25 (45) |
Notable harms, n (%) | |
Any CRS | 49 (89) |
Neurologic AEs | 33 (60) |
Anaphylaxis | NR |
Cytopenias (thrombocytopenia, neutropenia, and anemia) | 42 (76) |
Hypogammaglobulinemia | 4 (7) |
Serious infection | 14 (25) had worst grade 3 or higher AEs |
Potential secondary malignancies | 2 (4) |
Antibrexu-cel antibodies | NR |
AE = adverse event; allo-SCT = allogeneic stem cell therapy; CI = confidence interval; CR = complete remission; CRi = complete remission response with incomplete hematologic recovery; CRS = cytokine release syndrome; DOR = duration of remission; KM = Kaplan-Meier; mITT = modified intention to treat; MRD = minimal residual disease; NE = not estimable; NR = not reported; OS = overall survival; RFS = relapse-free survival; SAE = serious adverse event.
“+” indicated censoring.
Note: data cut-off of July 23, 2021.
Source: Clinical Study Report for the ZUMA-3 study.9
Critical Appraisal
The single-arm, noncomparative study design for the ZUMA-3 study is 1 of the key limitations of this evidence. Although the primary efficacy outcome, OCR per central assessment in the mITT analysis set (70.9%), was higher than the prespecified historical control rate of 40%, without a control arm, it is not possible to assess the relative efficacy and safety of brexu-cel versus currently available treatments for patients with R/R B-cell precursor ALL based on the results of the ZUMA-3 study. As well, the study design increases the possibility for bias in the estimation of treatment effects due to the potential for confounding related to selection bias, fluctuations in health status, and unidentified prognostic factors.
Another limitation of the ZUMA-3 study is the relatively small sample size and a selective study population. Although 71 patients were enrolled, only 55 patients received treatment with brexu-cel and were included in the primary analyses. Furthermore, 18 patients (33%) had an important protocol deviation. This as-treated population potentially introduces selection bias because it deviates from the ITT principle, which could bias the effect estimate away from the null hypothesis favouring brexu-cel . It is not possible to determine the magnitude of the potential overestimation of the treatment effect based on the available data and conducted analyses from this lone study.
Follow-up time was likely sufficient for assessing response and safety outcomes associated with brexu-cel . Although the median OS was estimable, the upper limit of the 95% CI was not, suggesting that the follow-up duration was not long enough to fully capture the effects on OS, and thus these results are considered immature.
No conclusion can be drawn for HRQoL outcomes because the analyses on EQ-5D VAS scores had considerable missing data throughout the study time points; ██ ████████ ███ █████████ ████████ ████ █████ ██ █████████ ████ █ ████████ ████.
After the infusion of brexu-cel, 20% of the patients received subsequent allo-SCT. Some patients may have received other subsequent treatments, such as chemotherapy or tyrosine kinase inhibitors (TKIs), for the purpose of consolidating the treatment effect from CAR T-cell therapy. Data on subsequent treatments other than allo-SCT were not reported. The survival results (OS, RFS) should be considered in the context of subsequent treatments, since it may be difficult to tell which treatment has more impact on a patient’s survival, especially when there is a lack of comparative data in the ZUMA-3 study.
According to the clinical experts consulted by CADTH, the study population of the ZUMA-3 study generally represents the patients living in Canada with R/R B-cell precursor ALL that would be receiving brexu-cel . However, the clinical experts noted that patients seen in clinical practice would include those with poorer performance status (the ZUMA-3 study only included patients with ECOG performance status of 0 or 1) and have more comorbidities.
According to the clinical experts consulted by CADTH, the efficacy outcomes used in this study are clinically relevant and important for the clinical trials of leukemia. Because the ZUMA-3 study was an open-label trial, all patients knew about the treatment they received. This would have some impact on patient-reported outcomes such as HRQoL, but is less likely to affect the objective outcomes such as OS and remission rate.
Indirect Comparisons
Description of Studies
The sponsor-submitted indirect treatment comparison (ITC)10 included a systematic literature review and an unanchored matching adjusted indirect comparison (MAIC) that compared brexu-cel to targeted therapies (blinatumomab and inotuzumab) or chemotherapy in patients with R/R B-cell precursor ALL. Three studies were included in this ITC: the ZUMA-3 study, the INO-VATE study, and the TOWER study. The outcomes assessed in the ITC were OS and event-free survival (EFS).
Efficacy Results
The results from the sponsor-submitted ITC suggested that the median OS was longer for brexu-cel compared to the comparators. The estimated hazard ratios (HRs) for OS ranged from ████ ██ █████ ████ ████ ██ █████ ████ ████ ██ ████ ███ ████ ████ ██ ████ for the comparisons to inotuzumab, blinatumomab, chemotherapy, and pooled chemotherapy, respectively. The upper limit of the 95% CI for the HRs ██████████ █████ ██████ for the ZUMA-3 phase II trial’s mITT population for the comparisons with inotuzumab and blinatumomab.
The median EFS was longer for brexu-cel compared to the comparators. The estimated HRs for EFS ranged from ████ ██ ████ ████ ████ ██ █████ ████ ████ ██ ████ ███ ████ ████ ██ ████ for the comparisons to inotuzumab, blinatumomab, chemotherapy, and pooled chemotherapy, respectively. Statistical significance depended on the study population for which the HRs were estimated for (e.g., mITT).
Harms Results
Harm outcomes were not assessed in this ITC.
Critical Appraisal
The authors of the ITC conducted a thorough review of the study design, inclusion and exclusion criteria, patient population characteristics, and outcomes measured in the included clinical trials and identified a number of differences in study design and patients’ baseline characteristics across studies that could potentially threaten the validity of an ITC. The rationale for conducting a MAIC was provided. A limitation of the MAIC is that MAIC can only adjust for heterogeneity that is directly related to differences in baseline patient characteristics. It is out of scope for a MAIC to account for differences between studies other than patient characteristics, such as those related to differences in study design, definitions of study outcomes, or changes in the management of support of patients over time.
When conducting an unanchored MAIC, identifying all effect modifiers and prognostic factors that could influence the results of the analysis is essential. The technical report indicated that 9 prognostic factors were identified and confirmed by the clinical experts consulted by the sponsor before the ITC analysis. However, not all factors could be used in the ITC because the complete list prevented the models from converging.
Considerable reductions in the effective sample size (ESS) were observed during the weighting process. For example, when comparing brexu-cel with inotuzumab for the outcome of OS, the sample size in the phase II ZUMA-3 study’s mITT population reduced ████ ██ ██ █████ ██ █████████ ██ ████. This suggests significant heterogeneity between the ZUMA-3 study and the comparator trials, and could lead to greater uncertainty of the validity of the comparison as well as poor precision. The results for comparisons with major reductions in ESS may not be reliable.
The outcome measures (OS and EFS) assessed in this study were clinically important. Other clinically relevant outcomes were not included in this report, such as treatment response rate, HRQoL, and safety outcomes. Outcome definitions varied across the studies as well, requiring modifications to the outcome definitions from the ZUMA-3 study to increase similarity with those used in the comparator studies.
Other Relevant Evidence
This section includes a summary of 1 additional relevant study, the SCHOLAR-3 study,11 included in the sponsor’s submission to CADTH that was considered to provide additional comparative effectiveness data for brexu-cel from the ZUMA-3 study versus matched historical cohorts.
Description of Studies
The SCHOLAR-3 study was a retrospective matched cohort study including adult patients with R/R B-cell precursor ALL that compared the patients who received brexu-cel in the mITT analysis set from the ZUMA-312 phase II study with 2 propensity score-matched historical cohorts of patients (N = 89). The comparator regimens were blinatumomab, inotuzumab ozogamicin, or SOC chemotherapy regimens.
Historical clinical trials were identified within the Medidata Enterprise Data Store database. Eligible historical clinical trials were phase I/II, II, or III multicentre, multinational, open-label, single-arm, or parallel assignment and randomized trials that were conducted between 2010 and 2017.11
Two cohorts were created to account for relevant previous treatment experience: patients previously treatment naive to blinatumomab and inotuzumab at enrolment (synthetic control arm 1 [SCA-1]) and SCA-2, which consisted of patients experienced with blinatumomab or inotuzumab.11
The inclusion criteria used in the SCHOLAR-3 study were generally consistent with the inclusion criteria used in the ZUMA-3 study. Overall, a relatively broader patient population was enrolled in the SCHOLAR-3 study compared to the ZUMA-3 study. Although response definitions across all historical clinical trials were harmonized to the same definitions used in the ZUMA-3 study, definitions for allo-SCT rate, RFS, and OS in the SCHOLAR-3 study were not completely aligned with the corresponding definitions used in the ZUMA-3 study.11
For the ZUMA-3 study, the data cut-off date for the primary analysis was September 9, 2020, and the data cut-off date for the 21-month follow-up analyses and sensitivity analysis that used the full analysis set (FAS) was July 23, 2021. The method for creating the matched historical cohorts of patients to the FAS population was consistent with the method described previously.13 The effectiveness outcomes of OCR, RFS, and OS were analyzed; OCR is designated as the primary outcome for the comparison with SCA-1, while only OS was analyzed for SCA-2 limited data availability. The secondary effectiveness outcome results were considered as supportive evidence (refer to Appendix 2).
Effectiveness Results
The primary outcome analyses were the focus for this review given the limitations of the SCHOLAR-3 study, including the lack of adjustment for multiplicity. For OCR at week 24, the estimated difference in the percentage of patients in the ZUMA-3 study’s mITT population (17 of 20 patients) compared with patients in SCA-1 (7 of 20 patients) was 50% (95% CI, 17.9 to 73.7; odds ratio = 10.5; 95% CI, 2.3 to 48.7). The comparison of OS between the ZUMA-3 study’s mITT (N = 29; median follow-up = 24 months) and SCA-2 (N = 20; median follow-up = 24 months) populations suggested that the ZUMA-3 study’s patients had a longer median OS (15.90 [95% CI, 3.19 to 26.02] months versus 4.76 [95% CI, 2.66 to 12.35] months; HR = 0.55; 95% CI, 0.26 to 1.13). The results from the sensitivity analysis were generally consistent with this 21-month follow-up updated analysis.13
Harms Results
Safety outcomes were not evaluated in the SCHOLAR-3 study.
Critical Appraisal
Internal Validity
As with the MAIC approach used for the ITC, ensuring homogeneity and accounting for potential confounding, effect modifiers, and prognostic factors is key to the validity of comparisons using external comparison groups. It was noted that duration of first remission of less than 12 months and complex karyotype were not included as factors used in the propensity matching in the SCHOLAR-3 study but were considered valid in the ITC by the clinical experts consulted by the sponsor and by CADTH for this review. Additionally, the heterogeneity of the historical clinical study designs that were included, and the dissimilar baseline characteristics between the ZUMA-3 study’s population and that of the historical studies, highlights that there is likely confounding of the treatment-effect estimates due to known and unknown confounders that could not be adjusted for. It should also be noted that a sensitivity analysis using a matching method other than the primary matching method was not conducted, and as such, the reliability and validity of the results were reduced.
The interpretation of the comparative effectiveness results, specifically the secondary outcomes, in the SCHOLAR-3 study is limited by the sampling approach that was used in the construction of the SCAs. In particular, the data pool for SCA-2 (treatment experienced with blinatumomab or inotuzumab) included patients who were previously treatment naive to blinatumomab and inotuzumab and had an on-study treatment switch from blinatumomab or inotuzumab to other SOC treatments. The baseline for these patients was redefined as the first day of the new treatment. Although the number of prior lines of therapy was a prognostic factor used in the propensity score matching, the data pool for SCA-2 was a heterogeneous population because patients entered the data pool with different treatment histories (i.e., it included both historical patients who were and were not truly treatment experienced with blinatumomab or inotuzumab). Moreover, the data pool for SCA-1 (treatment naive to blinatumomab and inotuzumab) did not include all eligible historical patients who were treatment naive to blinatumomab and inotuzumab; the impact, if any, of this sampling approach on the results is unknown. The interpretation of the comparative effectiveness results is further limited by the recruitment of patients from both the active and control arms of historical clinical trials that reflected approved SOC treatments in the European Union.
There was no formal hypothesis stated (e.g., superiority), no power or sample size considerations, and no adjustments for multiple comparisons. As such, the statistical inference from the results of this study has low reliability and validity. Additionally, a relatively small numbers of patients were included in the analysis sets; according to the preliminary feasibility assessments, it was anticipated that approximately 490 patients were eligible to participate in the study, yet a total of 89 patients formed the primary ZUMA-3 study’s mITT versus SCA-1 and SCA-2 comparisons.
External Validity
In SCA-1, 45% of patients were treated with blinatumomab and 55% of patients were treated with SOC chemotherapy; no patients received inotuzumab, which was identified as a relevant comparator by the clinical experts consulted by CADTH for this review.
In SCA-2, the majority (90%) of patients were treated with SOC chemotherapy. The clinical experts consulted by CADTH for this review indicated that there is no backbone chemotherapy identified because many options are available, depending on previous treatment experience; moreover, most regimens have been stable since 2010.
Conclusions
Evidence from a single-arm study (the ZUMA-3 study) suggests that treatment with brexu-cel may be associated with benefits in OS and RFS based on the clinical experts’ experience and expectations of the natural progression of the disease in adult patients with R/R B-cell precursor ALL. However, because the OS data are immature, analyses were based on a select patient population and there was no comparator arm in the ZUMA-3 study; therefore, it is possible that the effect of brexu-cel on survival is overestimated in the ZUMA-3 study. It is unclear if treatment with brexu-cel would improve a patient’s quality of life. Data from a retrospective matched cohort (the SCHOLAR-3 study) suggest that the response rate (e.g., CR) in patients treated with brexu-cel was higher than the rate observed in patients who received SOC in historical trials; however, the study was considered by CADTH reviewers to have poor internal validity and the findings were associated with a high degree of uncertainty. Findings from an ITC analysis suggest favourable survival benefits associated with brexu-cel treatment; however, definitive conclusions on survival benefits cannot be made due to the significant uncertainties in the indirect comparison. The harms associated with brexu-cel infusion are consistent with its mechanism of action and there were no unexpected safety signals observed. The single-arm study design of the ZUMA-3 study and the lack of long-term data are key limitations of the evidence; therefore, uncertainties remain regarding the magnitude of the clinical benefit from treatment with brexu-cel .
Introduction
Disease Background
ALL is a rare form of leukemia in adults.1 It accounts for approximately 5% of all adult leukemia cases in Canada. It is estimated that the age-standardized incidence rate of ALL is 0.79 cases per 100,000 person-years, while the prevalence rate is 15.7 cases per 100,000 persons.2 The lymphoblastic neoplasms are classified based on B-cell versus T-cell lineage.14 Among these ALL cases, 80% are of B-cell lineage and the B-cell precursor ALL is found in 75% of adult cases of ALL. Furthermore, about 50% of the patients who have B-cell precursor ALL have R/R disease. The estimated prevalence and incidence of R/R B-cell precursor ALL is 1,148 and 58 people, respectively, based on an estimated population in 2021 in Canada.2
B-cell precursor ALL is primarily a hematological malignancy of children, with 3/4 of cases occurring in children who are aged younger than 6 years. There is a second peak of incidence in adults aged older than 60 years.14 Typical clinical presentations of ALL are associated with anemia, neutropenia, and/or thrombocytopenia due to bone marrow involvement. Symptoms of ALL include fatigue, infections, and easy or spontaneous bruising or bleeding. Patients may complain about bone pain, arthralgias, and mild constitutional symptoms such as fever, night sweats, or unintentional weight loss. Hepatomegaly, splenomegaly, and/or lymphadenopathy are present in up to 50% of adult patients. CNS involvement may manifest as cranial neuropathies or meningeal symptoms.5,14
B-cell ALL or lymphoma is usually suspected in a child or adult with circulating lymphoblasts or painless lymphadenopathy, or when the following are observed: unexplained cytopenias, fatigue, infections, easy or spontaneous bruising or bleeding, constitutional symptoms, bone pain, or hepatomegaly or splenomegaly. A comprehensive diagnosis requires the study of cell morphology, immunophenotype, genetics and cytogenetics, and genomics. Diagnosis of B-cell precursor ALL requires the detection of lymphoblasts with the characteristic immunophenotype in peripheral blood, bone marrow, or other involved tissue.14
More than 80% of adult patients with newly diagnosed ALL will achieve a CR with intensive induction chemotherapy.3 However, after further consolidation therapy and maintenance chemotherapy, the majority of these patients will ultimately relapse.3 Older age (e.g., aged older than 60 years), reduced tolerability to treatments, higher white blood cell count on presentation (e.g., greater than 30 × 109/L), duration of first CR, refractoriness to prior therapy, and subsequent allo-SCT are generally recognized as prognostic factors for poorer prognosis, such as inferior OS, lower CR rate, and shorter CR duration.1,4,5 Relapsed disease is defined as the reappearance of leukemia cells in the bone marrow or peripheral blood after the achievement of a CR. Refractory disease is defined as those patients who fail to obtain a CR with induction therapy (e.g., failure to eliminate all detectable leukemia cells from the bone marrow and blood with subsequent restoration of normal hematopoiesis).3
Standards of Therapy
According to the patient groups that provided input to this review and a panel of clinicians consulted by CADTH, for patients who are relapsed or become refractory to the upfront treatments, the most important treatment goals are to improve patients’ HRQoL and prolong treatment response. After initial therapy for ALL, patients are followed at routine intervals to monitor for treatment-related complications and disease progression.3 For patients with R/R B-cell precursor ALL, induction of a CR is the first goal. Treatment options in R/R disease include cytotoxic chemotherapy regimens, targeted therapies, allo-SCT, and CAR T-cell therapy.4,5 Results of previous clinical trials have demonstrated that treatment with targeted therapy such as blinatumomab or inotuzumab ozogamicin was associated with superior rates of CR, OS, and EFS compared with conventional chemotherapy for R/R ALL; however, they do not induce long-term remission.15,16 They can be offered to patients with Ph-positive or Ph-negative ALL. For patients with R/R Ph-positive ALL, TKI with or without chemotherapy would be an option.1,17 For patients with R/R Ph-negative ALL, salvage therapy using a combination of targeted therapy and chemotherapy (e.g., cytarabine-based or others) were related to sustained treatment effect: 80% overall response rate and 57% to 80% CR rate were reported.18,19 CAR T-cell therapy is a treatment in which T lymphocytes are removed from a patient via apheresis, transduced ex vivo with a gene rendering them immunogenic against certain cancer cells, grown, and subsequently reinfused into the patient. The activated T-cells then circulate, attack, and kill the cancer cells.5 Treatment with CAR T-cell therapy was reported to be related to a higher risk of CRS (a severe systemic response to the activation and proliferation of CAR T-cell that includes high fever, flu-like symptoms, hypotension and mental status changes) and neurologic toxicities.3,4 Tisagenlecleucel is an autologous anti-CD19 CAR T-cell therapy that was approved for the treatment of R/R B-cell precursor ALL in pediatric and young adult patients aged up to 25 years.3,8
After the induction therapy, the patients should proceed to allo-SCT as soon as possible if they are eligible, to consolidate the treatment effect obtained from the initial induction therapy. Allo-SCT can be given to patients during their first CR or following relapse if a partial or complete remission can be reached.6 In a randomized study that enrolled 1,929 adult patients with ALL, patients assigned to the allo-SCT group showed improved 5-year OS compared to those assigned to the chemotherapy and autologous SCT (53% versus 45%).20 Common complications related to SCT include serious infections, risk of bleeding and transfusions, interstitial pneumonitis and other lung problems, graft-versus-host disease, graft failure, organ damage, relapsed disease, or secondary malignancies.6 Although allo-SCT may be the only curative approach for patients with R/R ALL, it can be performed in less than 50% of adult patients with ALL.21
Drug
Brexu-cel (brand name Tecartus) is a CD19-directed genetically modified autologous T-cell immunotherapy that binds to CD19-expressing cancer cells and normal B-cells.7 Following anti-CD19 CAR T-cell engagement with CD19-expressing target cells, the CD28 and CD3-zeta co-stimulatory domains activate downstream signalling cascades that lead to T-cell activation, proliferation, acquisition of effector functions, and secretion of inflammatory cytokines and chemokines. This sequence of events leads to killing of CD19-expressing cells.7 To prepare brexu-cel, a patient’s own T-cells are harvested and genetically modified ex vivo by retroviral transduction to express a CAR comprising a murine anti-CD19 single chain variable fragment linked to CD28 and CD3-zeta co-stimulatory domains. The anti-CD19 CAR T-cells are expanded and infused back into the patient, where they can recognize and eliminate CD19-expressing target cells.7
On November 16, 2022, brexu-cel was approved by Health Canada for the treatment of adult patients with R/R B-cell precursor ALL.7 The sponsor’s reimbursement request is the same as the Health Canada indication.
Brexu-cel is a single-dose, one-time treatment in a patient-specific infusion bag. The product monograph of brexu-cel notes that it should be administered by experienced health professionals at specialized treatment centres.7 Each patient-specific, single-infusion bag of brexu-cel contains a suspension of anti-CD19 CAR-positive viable T-cells in approximately 68 mL for a target dose of 1 × 106 CAR-positive viable T-cells per kg of body weight, with a maximum of 1 × 108 CAR-positive viable T-cells for patients weighing 100 kg or more.7 Prior to the infusion of brexu-cel and during the recovery period, 4 doses of tocilizumab and access to emergency equipment should be made available.
The key characteristics of commonly used targeted therapies and CAR T-cell therapies are presented in Table 3.
Table 3: Key Characteristics of Brexucabtagene Autoleucel, Blinatumomab, Inotuzumab, and Tisagenlecleucel
Detail | Brexucabtagene autoleucel | Blinatumomab | Inotuzumab ozogamicin | Tisagenlecleucel |
|---|---|---|---|---|
Mechanism of action | CD19-directed, genetically modified, autologous T-cell immunotherapy | Bispecific T-cell engager antibody construct | CD22-directed antibody-drug conjugate | CD19-directed, genetically modified, autologous T-cell immunotherapy |
Indicationa | Adult patients with R/R B-cell precursor ALL | Adult patients with R/R B-cell precursor ALL | Adults with R/R CD22-positive B-cell precursor ALL | Pediatric and young adults aged 3 to 25 years with B-cell ALL who are refractory, have relapsed after allo-SCT or are otherwise ineligible for SCT, or have experienced second or later relapse |
Route of administration | IV infusion | |||
Recommended dose | Target dose is 1 × 106 anti-CD19 CAR-positive viable T-cells per kg of body weight, with a maximum of 1 × 108 CAR-positive viable T-cells for patients weighing 100 kg or more. | ≥ 45 kg: Cycle 1: Days 1 to 7: 9 mcg/day. Days 8 to 28: 28 mcg/day. Days 29 to 42: treatment-free Subsequent cycles: Days 1 to 28: 28 mcg/day Days 29 to 42: treatment-free < 45 kg: Cycle 1: Days 1 to 7: 5 mcg/m2/day Days 8 to 28: 15 mcg/m2/day Subsequent cycles: Days 1 to 28: 15 mcg/m2/day. | Cycle 1: Total dose is 1.8 mg/m2 administered as 3 divided doses on days 1 (0.8 mg/m2), 8 (0.5 mg/m2), and 15 (0.5 mg/m2). Cycle 1 is 3 weeks in duration, but may be extended to 4 weeks if the patient achieves a CR or a CRi, and/or to allow recovery from toxicity. Subsequent cycles: Total dose is 1.5 mg/m2 per cycle, administered as 3 divided doses on days 1 (0.5 mg/m2), 8 (0.5 mg/m2), and 15 (0.5 mg/m2) for patients who achieve a CR or CRi or 1.8 mg/m2 per cycle given as 3 divided doses on days 1 (0.8 mg/m2), 8 (0.5 mg/m2), and 15 (0.5 mg/m2) for patients who do not achieve a CR or CRi. Subsequent cycles are 4 weeks in duration. | For patients weighing 50 kg and less: 0.2 to 5.0 × 106 CAR-positive viable T-cells per kg of body weight For patients weighing more than 50 kg: 0.1 to 2.5 × 108 CAR-positive viable T-cells (nonweight based). |
Serious AEs or safety Issues |
|
|
|
|
AE = adverse event; ALL = acute lymphoblastic leukemia; CAR = chimeric antigen receptor; CRS = cytokine release syndrome; R/R = relapsed or refractory; SCT = stem cell therapy; TLS = tumour lysis syndrome.
Source: Product monographs for brexucabtagene autoleucel,7 blinatumomab,22 inotuzumab ozogamicin,23 and tisagenlecleucel.8
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient and clinician groups that responded to CADTH’s call for patient and clinician input, respectively, and from clinical experts consulted by CADTH for the purpose of this review.
Patient Group Input
This section was prepared by CADTH staff based on the input provided by patient groups. The full original patient input(s) received by CADTH has been included in the stakeholder section at the end of this report.
Patient Input
Patient input for the review of brexu-cel was provided by the LLSC. The LLSC is a national charitable-status organization dedicated to finding a cure for blood cancers and improving the quality of life of people affected by blood cancers and their families by funding life-enhancing research and providing educational resources, services, and support. An online survey was distributed by the LLSC between August 15 and September 21, 2022. A total of 22 individuals across Canada responded to the survey (Quebec, 14; British Columbia, 5; Ontario, 2; Alberta, 1). Two respondents reported experience with brexu-cel ; their age ranges were 25 to 34 years and 65 to 74 years. Among the 20 respondents who did not have experience with brexu-cel, most respondents (n = 6) were within the age group of 35 to 44 years (range, 18 years or younger to 75 years or older).
The majority of the survey respondents (73%) indicated that fatigue or weakness was a symptom that had a significant impact on their quality of life. This was followed by loss of appetite or weight loss, bone or joint pain, headaches, blurred vision, nausea, or vomiting. The majority of the survey respondents (73%) indicated that their symptoms had a significant impact on their ability to work, exercise, and continue everyday activities. This was followed by the ability to travel and pursue activities and hobbies, and to maintain intimate relationships. The majority of the survey respondents (77%) indicated that interruption of life goals and accomplishments (e.g., career and schooling) was a psychological/social factor of the disease that had a significant impact on their quality of life. This was followed by stress, anxiety, and worry; feelings of isolation; problems concentrating; loss of sexual desire; and financial impacts.
For previous treatments received following their diagnosis with ALL, respondents reported experience with chemotherapy (n = 18), chemotherapy with SCT (n = 9), radiation therapy (n = 7), and targeted therapy (n = 2); 8 of which reported experience with 5 or more lines of treatment since diagnosis. Half of the respondents indicated that they have incurred more than 5 hospital visits per month for ALL-related reasons (e.g., treatment, scans, follow-ups, and emergency department visits). According to respondents, the impact of travelling to receive care included the costs of travel and accommodations, emotional hardships, being away from their support system for extended periods of time, and impact to daily activities and routines. For 82%, 73%, and 50% of respondents, weakness, fatigue, and nausea was identified as a treatment-related side effect that had a significant impact on their everyday lives, respectively. Finally, the LLSC indicated that the majority of the survey respondents reported a positive experience with ease of access to treatments and treatment results.
Two respondents reported experience with brexu-cel ; both accessed the drug via a clinical trial. The first respondent was aged between 25 and 34 years and previously received chemotherapy with SCT plus whole-body irradiation. This respondent reported an overall positive experience with brexu-cel, such that the respondent was able to achieve CR and reported fewer side effects compared to an allo-SCT. This respondent reported manageable or minor side effects, other than nausea and loss of appetite, which they considered to be a serious side effect. This respondent also indicated that brexu-cel had a positive impact on their quality of life (e.g., relationships with friends and family, mental health, ability to travel and perform everyday activities). Of note, this respondent was able to return to work and resume normal activities since receiving brexu-cel .
The second respondent who reported experience with brexu-cel was aged between 65 and 74 years and previously received chemotherapy and targeted therapy. This respondent reported very serious but somewhat manageable side effects including slurred speech, fever, chills, cough, or other signs of infection; feeling tired or lightheaded; fast or irregular heartbeat; headache; muscle or joint pain; diarrhea and/or constipation; nausea; loss of appetite, and insomnia. This respondent reported a strong negative impact of the treatment on their quality of life, including the ability to work, attend school, or volunteer, as well as on their mental health and ability to perform daily activities. Of note, this respondent was unable to return to work or resume normal activities since receiving brexu-cel ; their ALL responded partially to the treatment. Finally, both respondents indicated that they would recommend brexu-cel to other patients and would receive brexu-cel again if their doctor recommended it as the best choice.
The outcomes that were considered most important to patients when making decisions related to treatment were the degree of certainty that their ALL will respond to treatment and improve their quality of life. These outcomes were followed by coverage by insurance or drug plans and improved length of survival. Of note, the LLSC indicated that reduced side effects and easier accessibility were frequently mentioned as an improvement that respondents would like to see in any new treatment for ALL.
Clinician Input
Input from Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). In addition, as part of the brexu-cel review, a panel of 4 clinical experts from across Canada was convened to characterize unmet therapeutic needs, assist in identifying and communicating situations where there are gaps in the evidence that could be addressed through the collection of additional data, promote the early identification of potential implementation challenges, gain further insight into the clinical management of patients living with a condition, and explore the potential place in therapy of the drug (e.g., potential reimbursement conditions). A summary of this panel discussion is presented below.
Unmet Needs
The clinical panel indicated that for many adult patients with ALL, the most important treatment goals are to cure the disease. However, for the frail patients who receive palliative care, the patients who relapse or become refractory to the upfront treatments, or those ineligible for SCT, improving the patient’s quality of life as long as possible and prolonging the duration of treatment response are also important treatment goals. The clinical panel reported that the prognosis for these patients is poor (i.e., the chance of achieving a cure in these patients is diminished). Once targeted therapy (i.e., blinatumomab or inotuzumab) or SCT has been used and has failed, or if such treatments cannot be used, options available are limited, which is an unmet need for effective treatments for B-cell precursor ALL.
One of the treatment gaps identified by the Panel is the limited differential strategies and funded therapies available to adult patients with R/R B-cell precursor ALL. For example, within the publicly funded sector, a CAR T-cell therapy usually would not be available to patients with ALL who are aged 25 years or older per the clinical panel. Note that another CAR T-cell therapy, tisagenlecleucel, was approved by Health Canada for the treatment of pediatric and young adult patients aged 3 to 25 years with B-cell ALL who are refractory, have relapsed after allo-SCT or are otherwise ineligible for SCT, or have experienced second or later relapse.8 As well, if a patient has been treated with potent agents (such as ponatinib) but relapses thereafter, the clinical panel indicated that it would be difficult to choose another treatment. Another unmet need identified by the Panel is that patients in certain subgroups (e.g., aged 70 years or older or frail due to comorbidities) are not considered eligible for allo-SCT in some of the transplant centres in Canada; however, these patients may still be eligible to receive CAR T-cell therapy. The clinical panel noted a potential benefit from CAR T-cell therapy is that the patients would need only 1 single infusion, and long-term treatment is not required.
Place in Therapy
The panel stated that brexu-cel can be used in patients who are ineligible for treatment with inotuzumab or blinatumomab or who have relapsed once or twice after prior treatment with inotuzumab or blinatumomab. The clinical panel indicated that it would be beneficial if all these treatments were available for the patients with R/R ALL, which is a difficult-to-treat disease. The panel suggested that brexu-cel be used in patients who fail on at least 1 potent prior treatment (e.g., blinatumomab, inotuzumab or allo-SCT), those who are ineligible for allo-SCT, or as a bridging therapy to allo-SCT in the future. The panel also agreed that brexu-cel is expected to cause a shift in the current treatment paradigm if it is approved and reimbursed, particularly for patients aged 25 years and older.
Patient Population
The panel identified potential subgroups of patients who may respond differently to brexu-cel . Per the clinical panel, patients with a higher percentage of blasts in bone marrow at baseline or the presence of CNS leukemia may have poor response to CAR T-cell therapy. Although refractory CNS disease is considered a contraindication to CAR T-cell therapy, the Panel indicated that patients with previous CNS disease who responded to prior treatment and who currently have negative CNS disease may still be candidates for CAR T-cell therapy. The clinical panel noted that more clinical evidence is needed to identify the subsets of patients with R/R B-cell precursor ALL who would be best suited for treatment with brexu-cel .
Assessing Response to Treatment
The panel indicated that, in clinical practice, patients are evaluated and followed in a similar manner described in the ZUMA-3 study. Bone marrow biopsies, the level of remission, and complete blood counts are routinely conducted to assess treatment response. In terms of the timing of assessment, the clinical panel noted that complete blood counts are assessed during a patient’s routine visits, while bone marrow biopsy is less frequently performed unless unusual results from other examinations are observed or when brexu-cel is used as a bridge to an eventual allo-SCT and the clinician wants to know if remission has been achieved at the end of the treatment.
The clinical panel reported that meaningful responses to treatment with brexu-cel include prolonged OS, MRD-negative rate, improved HRQoL, better performance status, and the durability of treatment response.
Patients Who Go Through Pretreatment but Do Not Receive Brexu-cel
The panel noted that a small proportion of patients may go through pretreatment, such as leukapheresis and bridging therapy, but do not receive brexu-cel . The panel reported that this can be due to manufacturing failures or clinical reasons such as opportunistic infections and fungal infections resulting from the bone marrow failure and immunologic failure related to the disease, occurrence of active CNS disease during bridging therapy, or withdrawal of consent by the patient.
Subsequent Therapy
The panel indicated that after infusion with brexu-cel, allo-SCT can be offered to patients who are young and physically fit and who have an eligible donor. Patients who are not eligible for subsequent SCT will be monitored clinically, while repeated bone marrow biopsies are discouraged. If clinical relapses occur, the patients can be given the targeted therapy agents (blinatumomab or inotuzumab) that haven’t been used before their treatment with brexu-cel, or palliative chemotherapy.
Prescribing Conditions
The panel noted that the infrastructure and resources for many of the procedures (e.g., apheresis, bone marrow transplant programs, and the cellular therapy labs) required to facilitate treatment with CAR T-cell therapy are located in highly specialized tertiary care institutions. The panel indicated that treatment with brexu-cel needs to be provided by hematologists or oncologists who not only have the experience in treating leukemia but also experience in treating leukemia with cellular therapy or SCT.
Clinician Group Input
This section was prepared by CADTH staff based on the input provided by clinician groups. The full original clinician group input(s) received by CADTH has been included in the stakeholder section at the end of this report.
Two clinician groups provided input for the review of brexu-cel: CTTC, which was represented by 4 clinicians, and the OH-CCO Complex Malignant Hematology Group, which was represented by 2 clinicians.
The CTTC indicated that the current treatment options are immunotherapy with blinatumomab or inotuzumab; the goal is to bridge eligible patients who achieve remission to allo-SCT. Patients who relapse after allo-SCT can receive immunotherapy followed by supportive care alone when the disease no longer responds to therapy. Patients whose disease responds to immunotherapy but are not a candidate for allo-SCT will typically receive 5 to 6 cycles of 1 drug. If relapse occurs, patients will typically receive the alternative immunotherapy followed by supportive care alone or supportive care and low-dose chemotherapy when the disease no longer responds to therapy. The OH-CCO Complex Malignant Hematology Group indicated that tisagenlecleucel is available to patients with relapsed/refractory ALL who are aged 26 years or younger, while older patients could be treated with blinatumomab, inotuzumab, allogeneic transplant, or combination chemotherapy.
The treatment gaps identified by the clinician groups were consistent with the clinical panel, including a cure for the disease, the fact that few patients have a long-term remission with blinatumomab and inotuzumab, limited therapeutic options after relapse following transplant, and aged 26 years or older. Additionally, the CTTC indicated that blinatumomab and inotuzumab provide a short median OS of less than 8 months. The goals of therapy identified by the CTTC were consistent with the clinical panel, including prolonging life, improving quality of life, and delaying disease progression.
Similar to the clinical panel, the CTTC advocated for brexu-cel to be given to fit patients with CD19+ B-cell ALL who have relapsed disease (after allo-SCT, or those who are not candidates for allo-SCT), or who have refractory disease. In fit patients with refractory disease who have not undergone allo-SCT but have a donor available, the CTTC advocated for brexu-cel to be given before allo-SCT. The CTTC also advocated for brexu-cel to be used instead of (or in sequence with) blinatumomab and inotuzumab, and in patients who are ineligible for allo-SCT.
The outcomes identified by the CTTC that are used to determine response to treatment were consistent with the clinical panel, including peripheral blood counts and bone marrow biopsies. Meaningful responses to treatment with brexu-cel identified by the CTTC were consistent with the clinical panel, including prolonged OS and MRD-negative rate.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may affect their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
Relevant comparators for brexucabtagene autoleucel include blinatumomab, inotuzumab, and salvage multidrug chemotherapy. Patients with Ph-positive disease are treated with TKIs (e.g., dasatinib and ponatinib). For patients aged between 18 and 25 years, tisagenlecleucel may be another comparator, but it is only available in some jurisdictions across Canada. Tisagenlecleucel is funded for patients who are refractory, have relapsed after allo-SCT or are otherwise ineligible for SCT, or have experienced second or later relapse. | Comment from the drug programs to inform pERC deliberations. |
Considerations for initiation of therapy | |
Per the ZUMA-3 study eligibility criteria:
If brexucabtagene autoleucel is recommended for reimbursement, should patients be required to be ineligible for allo-SCT and/or other therapies? | The clinical experts indicated that patients with R/R Ph-positive ALL may be eligible to receive brexucabtagene autoleucel if they have not failed 2 different TKIs. The clinical experts also indicated that being ineligible for allo-SCT and/or other therapies should not be included as a criterion for patients to be treated with brexucabtagene autoleucel. The experts noted that treatment selection in patients with R/R ALL should be individualized. The clinicians and the patients need to engage in a dialogue and decide which treatment regimen fits the patient best. The clinical experts underlined the importance of allowing some flexibility in providing the optimal treatment(s) to patients. |
Is there sufficient evidence to support re-treatment with brexucabtagene autoleucel in case of disease relapse in the future? | The clinical experts noted that there is no evidence to support re-treatment with brexucabtagene autoleucel in case of disease relapse in the future. |
Which exclusion criteria from the ZUMA-3 study should be applied in determining eligibility for brexucabtagene autoleucel, if recommended for reimbursement? | The clinical experts indicated that some of the exclusion criteria from the ZUMA-3 study should be applied, such as patients with inadequate renal, hepatic, pulmonary, or cardiac function, so that the patients who receive brexucabtagene autoleucel can tolerate the treatment. For patients with HIV infection or hepatitis B, if the viremia is undetectable and the patients can restart their antiviral therapy quickly or stay on antiviral therapy throughout the brexucabtagene autoleucel therapy, it is reasonable for these patients to be considered eligible. The experts also indicated that hepatitis C infection should not be considered an exclusion criterion when this is a potentially curable disease. The experts noted that patients with prior CD19-targeted therapy could still be eligible for the treatment with brexucabtagene autoleucel. The experts agreed that patients with active or uncontrolled CNS disease should be excluded because CAR T-cell therapy would be excessively risky for these patients. The experts noted that in clinical practice, whether a patient should be given a particular treatment or not depends on the assessment of the risks and benefits associated with that treatment. This is different from the criteria used in clinical trials. |
Considerations for prescribing of therapy | |
Access would be limited to jurisdictional capacity. Although the manufacturer is planning to roll out additional centres across Canada, there are current capacity limitations (e.g., health human resources, bed limitations). As more CAR T-cell products are implemented, it is anticipated that the capacity may not be able to meet the demand. Out-of-province or out-of-country care may still be needed. There may be issues with access and prolonged stay in (or near) specialized centres, especially for patients from remote areas. Financial support for travel and accommodation would be needed. | Comment from the drug programs to inform pERC deliberations. |
The ZUMA-3 study noted that patients who had complete remission could resume TKIs 2 months after brexucabtagene autoleucel infusion and that these patients contributed to the derivation of duration of remission. To what extent did the use of TKIs contribute to the remission? | The clinical experts suggested that the added contribution to maintaining remission from the subsequent TKIs after brexucabtagene autoleucel infusion would likely have been small. However, the rationale to use TKIs in this way for patients with Ph-positive B-cell ALL is understandable, and is in line with the current guidance on the management of this subtype of B-cell precursor ALL. The clinical experts noted that subgroup analyses based on subsequent TKI therapy (with vs. without) on survival and remission rates may address this issue. |
Generalizability | |
Should brexucabtagene autoleucel be used in patients with ECOG performance status > 1? | The clinical experts noted that in clinical practice, patients with poorer performance status, such as ECOG performance status of 2, may be treated with brexucabtagene autoleucel. The clinical experts noted that in a clinical trial, patients who could better tolerate the treatment are more likely to be recruited, to avoid confounding effects from certain patient characteristics such as serious comorbid conditions that can affect performance status. |
Funding algorithm (oncology only) | |
Complex therapeutic space with multiple lines of therapy, subpopulations, or competing products. | Comment from the drug programs to inform pERC deliberations. |
For patients aged between 18 and 25 years, under what clinical circumstances would brexucabtagene autoleucel be preferred over tisagenlecleucel, and vice versa? | The clinical experts noted that there is a lack of direct evidence to answer this question. Longer-term data are available for tisagenlecleucel; data for brexucabtagene are limited in the ZUMA-3 study, which is a small, single-arm study. |
Care provision issues | |
There will be significant resource use for patient preparation, including leukapheresis, cell processing, and the use of bridging and lymphodepleting chemotherapy. Specialized centres need to be trained and accredited by the manufacturer. There is a high resource burden to obtain and maintain certification (including developing various protocols and supporting yearly audits). There is a need to coordinate patient care and product preparation with an external manufacturer. There are now multiple CAR T-cell therapies being administered by specialized centres; managing various protocols for preparation and deliver of each product type poses an administrative burden. | Comment from the drug programs to inform pERC deliberations. |
System and economic issues | |
There is a need for data collection to understand long-term outcomes. What outcomes should be measured, what constitutes treatment success, and what stopping rules should be considered? | The clinical experts indicated that overall survival and durability of the treatment response are outcomes reflecting treatment success. Health-related quality of life is also helpful in determining treatment effect. The clinical experts noted that the currently available data are still immature. |
Travel expenses for eligible patients. In some jurisdictions, the cost of CAR T-cell therapy may be through other departments in each province’s Ministry of Health rather than the drug programs. High upfront costs of this gene therapy may require special payment arrangements. Patient privacy and patient cell ownership concerns due to the fact that CAR T-cell is manufactured by a US-based company, which is outside of Canada. This is also the case for the other CAR T-cell therapies that are publicly funded. | Comment from the drug programs to inform pERC deliberations. |
ALL = acute lymphoblastic leukemia; allo-SCT = allogeneic stem cell therapy; CAR t = chimeric antigen receptor T; CNS = central nervous system; ECOG = Eastern Cooperative Oncology Group; pERC = pan-Canadian Oncology Drug Review Expert Review Committee; Ph = Philadelphia chromosome; R/R = relapsed or refractory; SCT = stem cell transplant; TKI = tyrosine kinase inhibitor.
Clinical Evidence
The clinical evidence included in the review of brexu-cel (Tecartus) is presented in 3 sections. The first section, the Systematic Review, includes pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those studies that were selected according to an a priori protocol. The second section includes indirect evidence from the sponsor and indirect evidence selected from the literature that met the selection criteria specified in the review. The third section includes sponsor-submitted additional relevant studies that were considered to address important gaps in the evidence included in the systematic review.
Systematic Review (Pivotal and Protocol Selected Studies)
Objectives
To perform a systematic review of the beneficial and harmful effects of brexu-cel for the treatment of adult patients with R/R B-cell precursor ALL.
Methods
Studies selected for inclusion in the systematic review will include pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those meeting the selection criteria presented in Table 5. Outcomes included in the CADTH review protocol reflect outcomes considered to be important to patients, clinicians, and drug plans.
Table 5: Inclusion Criteria for the Systematic Review
Criteria | Description |
|---|---|
Population | Adult patients with relapsed or refractory B-cell precursor ALL. Subgroups:
|
Intervention | Brexucabtagene autoleucel, IV Target dose: 1 × 106 anti-CD19 CAR-positive viable T-cells per kg of body weight Maximum dose: 1 × 108 CAR-positive viable T-cells |
Comparator | CAR T-cell therapy (e.g., tisagenlecleucel) For Ph-positive ALL:
For Ph-negative ALL:
|
Outcomes | Efficacy outcomes:
Harms outcomes:
|
Study designs | Published and unpublished phase I, II, III, and IV RCTs |
AE = adverse event; ALL = acute lymphoblastic leukemia; allo-SCT = allogeneic stem cell transplant; CAR = chimeric antigen receptor; CNS = central nervous system; CR = complete remission; CRi = complete remission response with incomplete hematologic recovery; CRS = cytokine release syndrome; HRQoL = health-related quality of life; ICU = intensive care unit; MRD = minimal residual disease; Ph = Philadelphia chromosome; RCT = randomized controlled trial; SAE = serious adverse event; TKI = tyrosine kinase inhibitor; WDAE = withdrawal due to adverse event.
aFor the outcome of event-free survival, “event” may include death, relapse, and relapse requiring treatment, according to the clinical expert consulted by CADTH.
The literature search for clinical studies was performed by an information specialist using a peer-reviewed search strategy according to the PRESS Peer Review of Electronic Search Strategies checklist.24
Published literature was identified by searching the following bibliographic databases: MEDLINE All (1946—) via Ovid and Embase (1974—) via Ovid. All Ovid searches were run simultaneously as a multifile search. Duplicates were removed using Ovid deduplication for multifile searches, followed by manual deduplication in Endnote. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concept was Tecartus (brexucabtagene autoleucel). Clinical trials registries were searched: the US National Institutes of Health’s clinicaltrials.gov, WHO’s International Clinical Trials Registry Platform (ICTRP) search portal, Health Canada’s Clinical Trials Database, and the European Union Clinical Trials Register.
No filters were applied to limit the retrieval by study type. Retrieval was not limited by publication date or by language. Conference abstracts were excluded from the search results. Refer to Appendix 1 for the detailed search strategies.
The initial search was completed on October 17, 2022. Regular alerts updated the search until the meeting of the CADTH pan-Canadian Oncology Drug Review Expert Committee (pERC) on February 8, 2023.
Grey literature (literature that is not commercially published) was identified by searching relevant websites from the Grey Matters: A Practical Tool For Searching Health-Related Grey Literature checklist.25 Included in this search were the websites of regulatory agencies (US FDA and European Medicines Agency). Google was used to search for additional internet-based materials. Refer to Appendix 1 for more information on the grey literature search strategy.
These searches were supplemented by reviewing bibliographies of key papers. In addition, the manufacturer of the drug was contacted for information regarding unpublished studies.
Two CADTH clinical reviewers independently selected studies for inclusion in the review based on titles and abstracts, according to the predetermined protocol. Full-text articles of all citations considered potentially relevant by at least 1 reviewer were acquired. Reviewers independently made the final selection of studies to be included in the review, and differences were resolved through discussion.
Findings from the Literature
One study was identified for inclusion in the systematic review (Figure 1). The included study is summarized in Table 6.
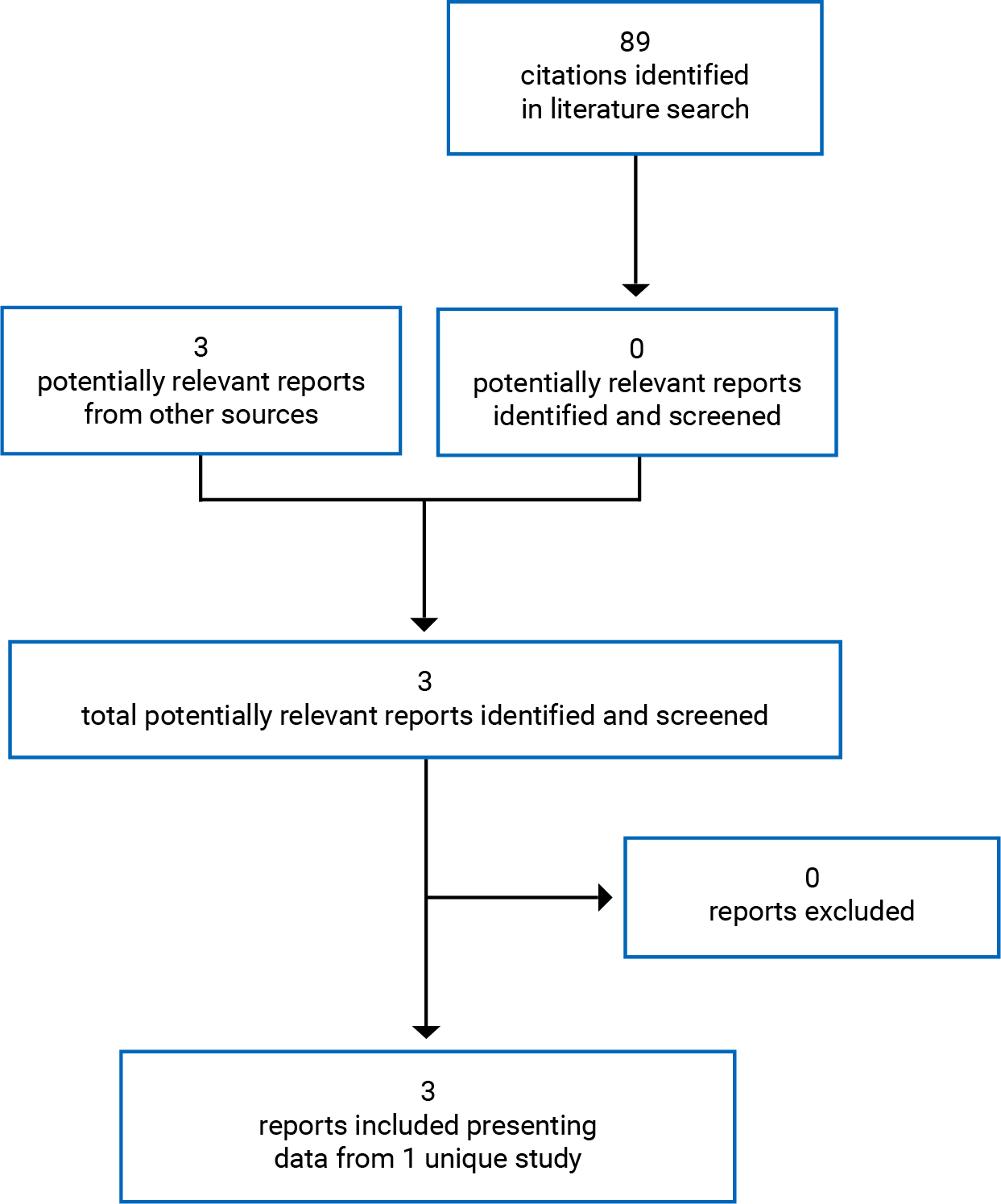
Table 6: Details of Included Studies
Characteristic | ZUMA-3 study |
|---|---|
Designs and populations | |
Study design | Phase I/II, multicentre, open-label, single-arm study |
Locations | 35 sites in Europe and the US |
Patient enrolment date | March 7, 2016 |
Enrolled (N) | 71 patients enrolled in phase II |
Inclusion criteria | Aged 18 years or older Relapsed or refractory B precursor ALL defined as one of the following:
Morphological disease in the bone marrow (> 5% blasts) Patients with Ph-positive disease were eligible if they were intolerant to TKI therapy or if they had relapsed or refractory disease despite treatment with at least 2 different TKIs ECOG performance status = 0 or 1 ANC ≥ 500/μL unless in the opinion of the PI cytopenia was due to underlying leukemia and was potentially reversible with leukemia therapy Platelet count ≥ 50,000/μL unless in the opinion of the PI cytopenia was due to underlying leukemia and was potentially reversible with leukemia therapy Absolute lymphocyte count ≥ 100/μL Adequate renal, hepatic, pulmonary, and cardiac function |
Exclusion criteria | Diagnosis of Burkitt leukemia/lymphoma according to WHO classification or chronic myelogenous leukemia lymphoid blast crisis History of malignancy other than nonmelanoma skin cancer or carcinoma in situ (e.g., cervix, bladder, breast) unless disease free for at least 3 years History of severe hypersensitivity reaction to aminoglycosides or any of the drugs used in this study CNS abnormalitiesa History of concomitant genetic syndrome associated with bone marrow failure such as Fanconi anemia, Kostmann syndrome, or Shwachman-Diamond syndrome History of myocardial infarction, cardiac angioplasty or stenting, unstable angina, or other clinically significant cardiac disease within 12 months of enrolment History of symptomatic deep vein thrombosis or pulmonary embolism within 6 months of enrolment Primary immunodeficiency Known infection with HIV, hepatitis B virus, or hepatitis C virus. A history of hepatitis B or hepatitis C is permitted if the viral load is undetectable per quantitative PCR and/or nucleic acid testing Presence of fungal, bacterial, viral, or other infection that is uncontrolled or requiring antimicrobials for management Prior medication:
Presence of any indwelling line or drain Acute GVHD grade II-IV by Glucksberg criteria or severity B-D by IBMTR index; acute or chronic GVHD requiring systemic treatment within 4 weeks before enrolment Live vaccine ≤ 4 weeks before enrolment History of autoimmune disease resulting in end organ injury or requiring systemic immunosuppression/systemic disease-modifying drugs within the past 2 years |
Drugs | |
Intervention | Brexu-cel at a target dose of 1 × 106 anti-CD19 CAR T-cells per kg, with a maximum dose of 1 × 108 anti-CD19 CAR T-cells for patients weighing ≥ 100 kg as a single bag for IV infusion |
Comparator(s) | None |
Duration | |
Phase | |
Screening | 28 days |
Treatment conditioning |
|
Treatment period | Single infusion at the start of the study period |
Follow-up | All enrolled patients were followed in the long term follow-up period for survival and disease status if possible Patients began the long-term follow-up period after they had completed the month 3 visit of the posttreatment assessment period (whether they have responded to treatment or went straight to the month 3 visit due to disease progression):
|
Outcomes | |
Primary end point | Overall complete remission rate (defined as CR + CRi) per independent review |
Secondary and exploratory end points | Secondary:
Exploratory:
|
Notes | |
Publications | Shah et al., 202126 |
AE = adverse event; ALL = acute lymphoblastic leukemia; allo-SCT = allogeneic stem cell transplant; ANC = absolute neutrophil count; brexu-cel = brexucabtagene autoleucel; CAR = chimeric antigen receptor; CNS = central nervous system; CR = complete remission; CRh = complete remission with partial hematologic recovery; CRi = complete remission response with partial hematologic recovery; CRS = cytokine release syndrome; CSF = cerebrospinal fluid; CTCAE = Common Terminology Criteria for Adverse Events; DOR = duration of remission; ECOG = Eastern Cooperative Oncology Group; GVHD = graft-vs.-host disease; HRQoL = health-related qualify of life; IBMTR = International Bone Marrow Transplant Registry; MRD = minimal residual disease; OS = overall survival; PCR = polymerase chain reaction; PH = Philadelphia chromosome; PI = primary immunodeficiency; PR = partial remission; RFS = relapse-free survival; SCT = stem cell therapy; TKI = tyrosine kinase inhibitor.
aCNS abnormalities: presence of CNS-3 disease, defined as detectable cerebrospinal blast cells in a sample of CSF with 5 or more white blood cells per mm3 with or without neurologic changes, or presence of CNS-2 disease, defined as detectable cerebrospinal blast cells in a sample of CSF with less than 5 white blood cells per mm3 with neurologic changes.
Note: Two additional reports were included: Sponsor’s submission2 and FDA Medical Review.27
Source: Clinical Study Report for the ZUMA-3 study.12
Description of Studies
ZUMA-3 is a phase I/II, multicenter, open-label study evaluating the safety and efficacy of brexu-cel in adult patients with R/R B precursor ALL. It is a single-arm study without a comparator group. One patient from Canada was enrolled. Phase I of the ZUMA-3 study was a dose-exploration phase. The Health Canada review of brexu-cel was based on the study’s phase II component; therefore, only the results of phase II of the ZUMA-3 study are presented in this review. The primary objective of phase II of the ZUMA-3 study was to evaluate the efficacy of brexu-cel, as measured by the OCR rate, defined as the combined rate of CR and CRi in adult patients with R/R B-cell precursor ALL.
The ZUMA-3 study is ongoing at the time of this review. The data cut-off date for the primary analysis was September 9, 2020, and the data cut-off date for the 21-month follow-up analyses was July 23, 2021.
Populations
Inclusion and Exclusion Criteria
Eligible patients in the ZUMA-3 study were aged 18 years and older and had R/R B precursor ALL. In this study, R/R was defined as 1 of the following: primary refractory, first relapse following a remission lasting less than 12 months, R/R after second-line or higher therapy, and R/R after allo-SCT (provided the transplant occurred 100 days or more before enrolment and that no immunosuppressive medications were taken within 4 weeks before enrolment). Patients with Ph-positive disease were eligible if they were intolerant to TKI therapy or if they had R/R disease despite treatment with at least 2 different TKIs. Patients were required to have an ECOG performance status of 0 or 1.
Patients who received prior CD19 targeted therapy other than blinatumomab; had severe CRS related to prior CD19-directed therapy; had been treated with medications within certain periods of time before study enrolment; had known infection with HIV, hepatitis B, or hepatitis C; had CNS abnormalities; or had acute graft-versus-host disease were excluded from this study.
Details of inclusion and exclusion criteria used in the ZUMA-3 study are provided in Table 6.
Baseline Characteristics
The mean age of patients was 42 years (standard deviation = 16). The majority of patients were male (60%) and white (67%). The majority of the patients (71%) had an ECOG performance status of 1. Ph-positive presented in 27% of the enrolled patients. Overall, 45%, 22%, and 42% of the patients had prior blinatumomab, inotuzumab, or prior allo-SCT, respectively; 33% of the study population had primary refractory disease and 78% had R/R to second-line or greater therapy. The mean percentage of blasts in bone marrow at baseline was 54% (standard deviation = 33.1). Extramedullary disease at baseline was reported in 11% of the patients. All patients reported CNS disease before entering the study (Table 7).
Table 7: Summary of Baseline Characteristics (Safety Analysis Set)
Characteristic | ZUMA-3 study (phase II) N = 55 |
|---|---|
Age, years, mean (SD) | 42.2 (16.1) |
Sex, n (%) | |
Male | 33 (60) |
Female | 22 (40) |
Race, n (%) | |
American Indian or Alaska Native | 1 (2) |
Asia | 3 (5) |
Black or African American | 1 (2) |
White | 37 (67) |
Other | 9 (16) |
Missing | 4 (7) |
ECOG performance status, n (%) | |
0 | 16 (29) |
1 | 39 (71) |
Philadelphia chromosome t(9;22) mutation, n (%) | |
Yes | 15 (27) |
No | 40 (73) |
Prior blinatumomab, n (%) | 25 (45) |
Prior inotuzumab, n (%) | 12 (22) |
Prior allo-SCT, n (%) | 23 (42) |
Prior autologous SCT, n (%) | 2 (4) |
Prior radiotherapy, n (%) | 13 (24) |
Number of lines of therapy, n (%) | |
1 | 10 (18) |
2 | 19 (35) |
3 | 14 (25) |
4 | 10 (18) |
5 | 1 (2) |
8 | 1 (2) |
Median (range) | 2.0 (1 to 8) |
Primary refractory, n (%) | 18 (33) |
Relapsed or refractory to second-line or greater therapy, n (%) | 43 (78) |
Relapsed or refractory disease after allo-SCT, n (%) | 24 (44) |
First relapse with first remission ≤ 12 months, n (%) | 16 (29) |
Response to the last prior therapy, n (%) | |
CR | 16 (29) |
CRi | 1 (2) |
PR | 2 (4) |
NR | 20 (36) |
PD | 10 (18) |
Not evaluated | 6 (11) |
% blasts in bone marrow at baseline | |
Mean (SD) | 54.0 (33.1) |
Median (range) | 60.0 (0 to 98) |
% blasts in bone marrow after bridging chemotherapy | |
Mean (SD) | 53.3 (32.8) |
Median (range) | 59.0 (0 to 98) |
Extramedullary disease at screening, n (%) | 6 (11) |
CNS disease at baseline, n (%) | CNS-1 55 (100) |
CNS = central nervous system; CR = complete remission; CRi = complete remission with incomplete hematologic recovery; ECOG = Eastern Cooperative Oncology Group; NR = no response; PD = progressive disease; PR = partial remission; SCT = stem cell therapy; SD = standard deviation.
Source: Clinical Study Report for the ZUMA-3 study.12
Interventions
Treatment in this study consisted of leukapheresis, bridging chemotherapy, and CSF prophylaxis, followed by a single IV infusion of brexu-cel.
Patients underwent leukapheresis to obtain leukocytes for the manufacture of brexu-cel . When patients commenced leukapheresis, they were considered to be enrolled in the study.
Bridging therapy could be administered after leukapheresis and before lymphodepleting chemotherapy at the discretion of the investigator. Bridging chemotherapy was recommended for all patients, particularly those with high disease burden at baseline (higher than 25% leukemic blasts in bone marrow or more than 1,000 blasts/mm3 in the peripheral circulation). If prescribed, bridging chemotherapy was to be administered after leukapheresis and completed at least 7 days or 5 half-lives, whichever was shorter, before initiating lymphodepleting chemotherapy.
All patients received CSF prophylaxis, consisting of an intrathecal regimen according to institutional or national guidelines. CSF prophylaxis was supplied any time during screening through 7 days before brexu-cel infusion.
Patients also received lymphodepleting chemotherapy (consisting of fludarabine 25 mg/m2/day administered IV over 30 minutes on day −4, day −3, and day −2, and cyclophosphamide 900 mg/m2/day administered IV over 60 minutes on day −2 before the administration of brexu-cel) to induce lymphocyte depletion and create an optimal environment for expansion of anti-CD19 CAR T-cells in vivo.
For all patients in ZUMA-3, brexu-cel was manufactured in the US. Brexu-cel was administered to the patient a median of 28.0 days (range, 20 to 56 days) after leukapheresis for patients in the US and 37.0 days (range, 28 to 60 days) after leukapheresis for patients in Europe. Patients were required to be hospitalized to receive the infusion of brexu-cel, followed by a minimum 7-day observation period.
Brexu-cel re-treatment was allowed if patients achieved remission of leukemia (CR, CR with partial hematologic recovery [CRh], or CRi) after the initial brexu-cel infusion at month 3 or later disease assessment and subsequently progressed (more than 5% bone marrow blasts or progression of extramedullary disease per local assessment).
Outcomes
A list of efficacy end points identified in the CADTH review protocol that were assessed in the clinical trials included in this review is provided in Table 8. These end points are further summarized in the following section. A detailed discussion and critical appraisal of the outcome measures is provided in Appendix 3.
OCR rate (defined as CR + CRi) is as per the independent review.
Table 8: Summary of Outcomes of Interest Identified in the CADTH Review Protocol
Outcome measure | ZUMA-3 study |
|---|---|
Survival including OS and RFS | Secondary |
Treatment response: OCR (defined as CR + CRi) MRD-negative rate DOR | Primary Secondary Secondary |
HRQoL | Secondary |
Allo-SCT rate | Secondary |
ICU admission | Not assessed |
Safety | Secondary |
allo-SCT = allogeneic stem cell therapy; CR = complete remission; CRi = complete remission response with incomplete hematologic recovery; DOR = duration of remission; HRQoL = health-related quality of life; ICU = intensive care unit; OCR = overall complete remission; OS = overall survival; RFS = relapse-free survival.
Efficacy Outcomes
OCR rate (CR and CRi): The primary study end point in the ZUMA-3 study was the OCR rate, which included complete remission or complete remission with incomplete hematological recovery (CR + CRi), by central assessment.
CR was reached when 1) the percentage of blasts was equal to or less than 5%, 2) absolute neutrophil count (ANC) was equal to or higher than 1,000/µL in peripheral blood, platelet count was equal to or higher than 100,000/µL in peripheral blood, and 3) CNS-1 (no detectable leukemia in the CSF).
CRi was defined when 1) the percentage of blasts was equal to or fewer than 5%, 2) ANC was equal to or higher than 1,000/µL and platelet count was lower than 100,000/µL, or ANC was equal to or fewer than 1,000/µL and platelet count was higher than 100,000/µL in peripheral blood, and 3) CNS-1 (no detectable leukemia in the CSF).
All patients who did not meet the criteria for CR or CRi by the analysis data cut-off date were considered nonresponders for the OCR rate evaluation.
MRD-negative rate: MRD-negative was defined as lower than 10-4 lymphoblasts in the bone marrow per the standard assessment. Not all patients who achieve a morphological CR achieved an MRD-negative remission. Previous studies have shown that the achievement of an MRD-negative response with ALL treatment is associated with prolonged leukemia remission in the study population.1
OS was defined as the time from brexu-cel infusion date to the date of death from any cause in the mITT analysis. Patients who had not died by the analysis data cut-off date were censored at their last contact date before the data cut-off date, with the exception that patients known to be alive or determined to have died after the data cut-off date were censored at the data cut-off date. In the FAS, OS was defined as the time from enrolment to the date of death from any cause. This was the secondary efficacy end point in ZUMA-3.
RFS was defined as the time from the brexu-cel infusion date to the date of disease relapse or death from any cause. RFS for all enrolled patients on the FAS was defined as the time from enrolment to the date of disease relapse or death from any cause. This was a secondary efficacy outcome in ZUMA-3.
DOR was defined as the time from the first CR (CR or CRi) to relapse or death from any cause in the absence of documented relapse.
HRQoL was assessed in the ZUMA-3 study using the EQ-5D-5L. The EQ-5D-5L questionnaire is a generic and preference-weighted measure of health status captured on the day of assessment. It comprises 2 components: the EQ-5D descriptive system and the EQ VAS. The descriptive system comprises 5 dimensions (mobility, self-care, usual activities, pain/discomfort, and anxiety/depression) and is divided into 5 levels of severity: no problems, slight problems, moderate problems, severe problems, and extreme problems. The EQ-5D-5L VAS is a vertical VAS for recording self-rated HRQoL state, which is reported from 0, described as “the worst health you can imagine,” to 100, described as “the best health you can imagine.” A MID for the VAS score was not identified in the literature for patients with ALL. For more information on the properties of the EQ-5D-5L, refer to Appendix 3.
Allogeneic stem cell transplant rate was defined as the incidence of transplant among transplant-eligible patients with an available donor.
Harms Outcomes
The safety analysis included the incidence and severity of AEs occurring throughout the study period. The occurrence of AEs was assessed at all assessment time points. The severity of AEs and SAEs was graded using the National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE) version 4.03.
Statistical Analysis
Sample Size Calculation
The ZUMA-3 study used a single-arm design to test for an improvement in OCR rate compared to a prespecified, historical control rate of 40% or less. The sponsor noted that the rationale for the 40% threshold was informed by rates observed in published studies of second-line or later chemotherapy and SCT regimens and in pivotal studies of blinatumomab. For the test of efficacy, this study has approximately 93% power to distinguish between brexu-cel with a 65% true OCR rate from a therapy with an OCR rate of 40% or less with a 1-sided alpha level of 0.025. In phase II of this study, approximately 50 patients treated with brexu-cel were needed.
Statistical Analysis for Efficacy Outcomes
The primary analysis was conducted when the overall study enrolment had been completed and the last treated patient in the mITT analysis set had had the opportunity to complete the month 6 disease assessment. A final analysis will be conducted when all patients have completed the study.
Primary Outcome
For the primary efficacy end point, the incidence of OCR rate and exact 2-sided 95% CIs were generated using an exact binomial test.
This study assumes that the underlying response rate (in the absence of treatment with investigational therapy) is 40% and that an improvement in the response rate to 65% provides clinically meaningful benefit.
Secondary Outcomes
DOR: KM estimates and 2-sided 95% CIs were generated for DOR. Patients who did not meet the criteria for relapse and who have not died were censored at the last evaluable disease assessment or disease status follow-up assessment. Disease assessments obtained after new anticancer therapies (including allo-SCT) did not contribute to the derivation of DOR. The DOR for patients who underwent allo-SCT while in remission was censored at the date of the allo-SCT; the DOR for patients who underwent other new anticancer therapies in the absence of relapse was censored at the last evaluable disease assessment before the new anticancer therapies.
MRD-negative rate: The incidence of MRD-negative rate and exact 2-sided 95% CIs were generated.
OS and RFS: KM estimates and 2-sided 95% CIs were generated for OS and RFS. Estimates of the proportion of patients alive at 3-month intervals were provided. For the outcome of OS, the following were censored: death after data cut-off date for analysis, known to be alive after data cut-off date for analysis, alive up through data cut-off date and no further information available after data cut-off date, or full withdrawal of consent or lost to follow-up before data cut-off date. For RFS, the following were considered censored: remained in remission and alive without new anticancer therapy (including allo-SCT but excluding resumption of TKI), patient has a CR or CRi and subsequently initiated new anticancer therapy (including allo-SCT but excluding resumption of TKI) before documented relapse or death, remained in remission without new anticancer therapy (including allo-SCT but excluding resumption of TKI) until withdrawal of consent or loss to follow-up, or enrolled and treated with brexu-cel but the disease assessment has not been done and the patient is still alive and has not received any new anticancer therapy.
Incidence of subsequent allo-SCT: The incidence of allo-SCT in the mITT set and 2-sided 95% CIs was generated.
HRQoL: The changes in the EQ-5D scale score and EQ-5D VAS score at each assessment time compared to baseline were presented.
Subgroup Analyses
To examine the consistency of treatment effect on selected efficacy outcomes (e.g., OCR and AEs), subgroup analyses were planned if there were at least 10% observations in 2 or more subcategories of the covariate, based on the following baseline covariates: ECOG performance status at baseline (0 versus 1), age at baseline (aged less than 65 years versus 65 years or older), sex (male versus female), race, region, R/R subgroup (primary refractory versus first relapse if duration of first remission was shorter than 12 months versus R/R to second-line or greater therapy versus R/R post–allo-SCT), prior blinatumomab treatment (yes versus no), prior inotuzumab treatment (yes versus no), prior allo-SCT (yes versus no), extramedullary disease (yes versus no), lines of prior therapies (1, 2, greater than 2), percentage of bone marrow blasts at screening (less than 50% versus 50% or more), peripheral blasts (0, greater than 0 to 1,000, greater than 1,000 blasts/mm3), normal karyotype, CD19 expression based on central read (positive, negative), or Ph t(9;22) (yes versus no).
Sensitivity Analyses
Sensitivity analyses of OS, RFS, OCR, and DOR were performed in the FAS. Sensitivity analyses were also performed in which disease assessment was obtained after allo-SCT was included in the derivation of DOR or RFS. Other sensitivity analyses were performed in which the DOR or RFS for patients undergoing TKI was censored at the last disease assessment before the resumption of TKI therapy.
Missing Data Handling
Imputing rules for partial or missing data were provided for the outcomes of AEs, deaths, concomitant medication, and subsequent anticancer therapy.
Analysis Populations
The mITT analysis set consisted of all patients enrolled and treated with brexu-cel in phase II of the ZUMA-3 study. This analysis set was used for all efficacy analyses unless specified otherwise and for hypothesis testing of the primary end point of OCR rate.
The safety analysis set was defined as all patients treated with any dose of brexu-cel .
The FAS consisted of all enrolled (leukapheresed) patients and was used for the summary of patient disposition, patient listings of deaths, and sensitivity analyses of efficacy.
Results
Patient Disposition
In phase II of the ZUMA-3 study, 71 patients were enrolled and underwent leukapheresis as of the data cut-off on July 23, 2021. Of the 57 patients who received lymphodepleting chemotherapy, 55 received brexu-cel (including 2 who received re-treatment with brexu-cel), and 2 did not receive brexu-cel due to AEs (1 experienced AEs of bacteremia and neutropenic fever that precluded further treatment, and 1 deteriorated after lymphodepleting chemotherapy and no longer met eligibility criteria). Fourteen patients received neither lymphodepleting chemotherapy nor brexu-cel after leukapheresis: 7 due to AEs (including 2 patients whose product was not successfully manufactured from the initial leukapheresis), 3 not meeting eligibility criteria after leukapheresis, and 4 due to manufacturing failures. In total, in the phase II component of the ZUMA-3 study, products were not successfully manufactured for 6 patients. Both the safety analysis set and mITT analysis set comprised 55 patients.
Disposition | ZUMA-3 study (phase II) |
|---|---|
Enrolled, N | 71 |
Patients treated with lymphodepleting chemotherapy, N (%) | 57 (80.3) |
Patients treated with brexucabtagene autoleucel, N (%) | 55 (77.5) |
Reasons for not treated with lymphodepleting chemotherapy or brexucabtagene autoleucel, N (%) | 14 (19.7) |
Adverse events | 7 (9.9) (including 2 patients whose product was not successfully manufactured from the initial leukapheresis) |
Not meeting eligibility criteria after leukapheresis | 3 (4.2) |
Manufacturing failures | 4 (5.6) |
FAS, N (%) | 71 (100) |
mITT, N (%) | 55 (77.5) |
SAS, N (%) | 55 (77.5) |
FAS = full analysis set (defined as all enrolled [leukapheresed] patients); mITT = modified intention to treat (defined as all patients enrolled and treated with brexucabtagene autoleucel in phase II); SAS = safety analysis set (defined as all patients treated with any dose of brexucabtagene autoleucel).
Note: Data cut-off date of July 23, 2021.
Source: Clinical Study Report for the ZUMA-3 study.9
Exposure to Study Treatments
For patients treated in phase II of the ZUMA-3 study, the median actual follow-up time from brexu-cel infusion was 20.5 months (range, 0.3 to 32.6 months), and the median potential follow-up time from the brexu-cel infusion was 26.8 months (range, 20.7 to 32.6 months); all patients had more than 18 months of potential follow-up, and 42 of 55 patients (76%) had at least 24 months of potential follow-up.
After leukapheresis, 51 patients (93%) received bridging therapy. The most commonly administered therapies were dexamethasone (███ ███), nonliposomal vincristine (███ ███), and cytarabine (███ ███) in the safety analysis set.
For lymphodepleting chemotherapy, all patients received the planned total body surface area–adjusted dose of cyclophosphamide (900 mg/m2). Patients received a median total body surface area–adjusted dose of fludarabine of 75 mg/m2 (range, 71 to 75 mg/m2), and all received within 10% of the planned total dose.
In phase II of the ZUMA-3 study, the median weight-adjusted dose of brexu-cel was 1.0 × 106 anti-CD19 CAR T-cells per kg (range, 0.5 × 106 to 1.0 × 106 cells per kg). The median total number of anti-CD19 CAR T-cells in the brexu-cel infusion was 75.7 × 106 cells (range, 39.3 × 106 to 101.0 × 106 cells), and the median total number of T-cells infused was 128.4 × 106 cells (range, 65.5 × 106 to 277.8 × 106 cells) (Table 10).
Table 10: Summary of Study Treatment Exposure in the ZUMA-3 Study (SAS set)
Exposure | ZUMA-3 study, phase II |
|---|---|
Brexucabtagene autoleucel (N = 55) | |
Exposure to bridging therapy, n (%) | 51 (93) |
Exposure to lymphodepleting chemotherapy, n (%) | 55 (100) |
Exposure to brexucabtagene autoleucel | |
Weight-adjusted brexucabtagene autoleucel dose received (x 106 anti-CD19 CAR T-cells per kg) | Mean (SD): 1.0 (0.1) Median (range): 1.0 (0.5 to 1.0) |
Total number of CAR T-cells (x 106) | Mean (SD): 77.4 (16.8) Median (range): 75.7 (39.3 to 101.0) |
CAR = chimeric antigen receptor; SD = standard deviation; SAS = safety analysis set.
Note: Data cut-off of July 23, 2021.
Source: Clinical Study Report for the ZUMA-3 study.9
Efficacy
Only those efficacy outcomes and analyses of subgroups identified in the review protocol are reported in the following section.
Survival
Overall Survival
At data cut-off of July 23, 2021, the KM estimates of OS at 12, 18, and 24 months were 71.7% (95% CI, 57.5% to 81.9%), 63.9% (95% CI, 49.4% to 75.3%), and 55.7% (95% CI, 41.2% to 68.1%), respectively. The KM median OS was 25.4 months (95% CI, 16.2 months to NE); OS was 24.0 months (95% CI, 23.3 to 24.6 months) using a reverse KM approach.
The KM median OS was 26.0 months (95% CI, 21.9 months to NE) for patients with CR or CRi and was 2.4 months (95% CI, 0.7 months to NE) for all other patients in the mITT analysis set. The KM median OS was NE (95% CI, 25.4 months to NE) for patients with CR (Table 11).
The KM estimate of OS at month 24 in the mITT analysis set was examined in subgroups based on prior treatment. The following OS rates were reported for patients who had been previously treated with blinatumomab (████ █ █ ██), inotuzumab (████ █ █ ██), SCT (████ █ █ ██), SCT and blinatumomab (████ █ █ ██), and SCT and inotuzumab (████ █ █ █).
Results of sensitivity analyses in the FAS set were similar to those in the mITT analysis set.
Relapse-Free Survival
Overall, 22 patients were censored: 6 were in ongoing remission as of the data cut-off date, 10 had an allo-SCT, and 6 started new anticancer therapy. Fourteen relapsed, 3 died, and 16 did not have a best response of CR or CRi.
KM estimates of RFS rates at 6, 12, and 18 months were 57.6% (95% CI, 42.6%, 69.9%), 45.4% (95% CI, 30.0%, 59.6%), and 35.4% (95% CI, 20.5%, 50.6%), respectively. The KM median RFS was 11.6 months (95% CI, 2.7 to 20.5 months), with a reverse KM median follow-up time for RFS of 17.8 months (95% CI, 3.2 to 24.0 months).
Among patients with CR or CRi, the KM median RFS was 15.5 months (95% CI, 11.6 months to NE). The KM median RFS was 22.1 months (95% CI, 11.6 months to NE) for those with CR and 11.7 months (95% CI, 1.8 months to NE) for those with Cri (Table 11).
Table 11: Summary of Survival (OS and RFS), mITT and FAS Populations
Survival | ZUMA-3 study (phase II) N = 55, mITT | ZUMA-3 study (phase II) N = 71, FAS |
|---|---|---|
OS by central assessment | ||
Death, n (%) | 25 (45.5) | ██ ██████ |
Censored, n (%) | 30 (54.5) | ██ ██████ |
Alive on or after data cut-off, n (%) | ██ ██████ | ██ ██████ |
Full withdrawal of consent, n (%) | █ █████ | █ █████ |
KM median OS, months (95% CI), range | 25.4 (16.2 to NE), 0.30 to 32.56+ | 23.1 (10.4 to NE), 0.69+ to 33.51+ |
For patients with CR or CRi | 26.0 (21.9 to NE) | ████ ██████ ███ |
For patients with CR | Not reached (25.4 to NE) | ███ █████ ████ ███ |
For patients with all other responses | 2.4 (0.7 to NE) | ███ █████ █████ |
RFS by central assessment | ||
Events, n (%) | 33 (60.0) | ██ ██████ |
Relapse | 14 (25.5) | ██ ██████ |
Death | 3 (5.5) | █ █████ |
Patient’s best overall response not CR or CRi | 16 (29.1) | ██ ██████ |
Censored, n (%) | 22 (40.0) | ██ ██████ |
Ongoing remission | 6 (10.9) | 6 (8.5) |
Allo-SCT | 10 (18.2) | 10 (14.1) |
Started new anticancer therapy | 6 (10.9) | 6 (8.5) |
Lost to follow-up | 0 | 0 |
Withdrawal of consent | 0 | 0 |
KM median RFS, months (95% CI), range | 11.6 (2.7 to 20.5), 0.03 to 26.02 | 3.7 (0.0 to 12.9), 0.03 to 26.84 |
For patients with CR or CRi | 15.5 (11.6 to NE) | NR |
For patients with CR | 22.1 (11.6 to NE) | NR |
For patients with CRi | 11.7 (1.8 to NE) | NR |
allo-SCT = allogeneic stem cell therapy; CI = confidence interval; CR = complete remission; CRi = complete remission response with incomplete hematologic recovery; FAS = full analysis set; KM = Kaplan-Meier; mITT = modified intention to treat; NE = not estimable; NR = not reported; OS = overall survival; RFS = relapse-free survival. “+” indicated censoring.
Note: Data cut-off of July 23, 2021.
Source: Clinical Study Report for the ZUMA-3 study.9
Treatment Response
Response (CR Plus CRi)
Among the 55 patients who were treated in phase II of the ZUMA-3 study, the OCR rate was 70.9% (39 of 55 patients, 95% CIs, 57% to 82%) in the mITT analysis set (Table 12).
In subgroup analyses, OCR rates were 90% for patients with only 1 prior line of therapy (N = 10), 60% for patients who had received prior blinatumomab (N = 25), 67% for those with prior inotuzumab (N = 12), 70% for those with prior SCT (N = 23), 67% for those with prior blinatumomab and inotuzumab (N = 6), ███ for those with prior SCT and blinatumomab ██ █ ████ ███ for those with prior SCT and inotuzumab ██ █ ███ and ████ for those with prior SCT, blinatumomab, and inotuzumab ██ █ █).
Complete Remission
Among the 55 patients who were treated in phase II, the CR rate was 56.4% (31 of 55 patients, 95% CI, 42% to 70%) in the mITT analysis set (Table 12).
In the subgroup analyses, CR rates were ███ for patients with only 1 prior line of therapy ██ █ ████ ███ for patients who had received prior blinatumomab ██ █ ████ ███ for patients with prior inotuzumab ██ █ ████ ███ for patients with prior SCT ██ █ ████ ███ for patients with prior blinatumomab and inotuzumab ██ █ ███ ███ for patients with prior SCT and blinatumomab ██ █ ████ ███ for patients with prior SCT and inotuzumab ██ █ ███ and ███ for patients with prior SCT, blinatumomab, and inotuzumab ██ █ ██.
MRD-Negative Remission
The overall MRD-negative rate was 76% (42 of 55 patients; 95% CI, 63% to 87%). Among patients with CR or CRi, the MRD-negative rate was 97% (38 of 39 patients; 95% CI, 87% to 100%). One patient who achieved a CR did not have samples sent to the central laboratory for MRD assessment (Table 12).
Duration of Remission
In the main analysis of DOR, patients were censored at their last evaluable disease assessment before initiation of a new anticancer therapy (excluding resumption of a TKI) or allo-SCT.
Among the 39 patients in the mITT analysis set who achieved a CR or CRi, the KM median DOR was 14.6 months (95% CI, 9.4 months to NE). Overall, 22 patients were censored: 6 were in ongoing remission as of the data cut-off date, 10 had an allo-SCT, and 6 started new anticancer therapy (Table 12).
Re-treatment
In ZUMA-3, patients were allowed to receive 1 additional brexu-cel infusion provided they achieved remission of leukemia (CR, CRh, or CRi) after the initial brexu-cel infusion at month 3 or later disease assessment and subsequently progressed. There were 2 patients who received re-treatment with brexu-cel in phase II of this study. Both patients did not respond to the re-treatment (1 died), and neither patient received transplant after the second dose of brexu-cel.
Table 12: Summary of Treatment Response, mITT and FAS Populations
Treatment response | ZUMA-3 study (phase II) N = 55, mITT | ZUMA-3 study (phase II) N = 71, FAS |
|---|---|---|
Patients with overall response by central assessment (CR + CRi), n (%) | 39 (70.9) | 39 (54.9) |
95% CI (Clopper-Pearson method) | 57 to 82 | 43 to 67 |
Patients with overall response by investigator assessment (CR + CRi), n (%) | 40 (72.7) | ██ ██████ |
95% CI (Clopper-Pearson method) | 59 to 84 | NR |
Patients with CR by central assessment, n (%) | 31 (56.4) | ██ ██████ |
95% CI (Clopper-Pearson method) | 42 to 70 | 32 to 56 |
Patients with CR by investigator assessment, n (%) | 33 (60) | 33 (46.5) |
95% CI (Clopper-Pearson method) | 46 to 73 | NR |
Patients with CRi by central assessment, n (%) | 8 (14.5) | █ ██████ |
95% CI (Clopper-Pearson method) | 6 to 27 | NR |
Patients with MRD-negative remission by central assessment, n (%) | 42 (76) | 42 (59) |
95% CI (Clopper-Pearson method) | 63 to 87 | 47 to 71 |
DOR by central assessment, months, KM median (95% CI), range | 14.6 (9.4 to NE); 0.03+ to 24.08 | DOR was defined only for patients achieving an OCR (CR or CRi); results of DOR analysis in FAS were identical to that in the mITT analysis set |
CI = confidence interval; CR = complete remission; CRi = complete remission response with incomplete hematologic recovery; DOR = duration of remission; FAS = full analysis set; KM = Kaplan-Meier; mITT = modified intention to treat; MRD = minimal residual disease; NE = not estimable; NR = not reported; OCR = overall complete remission.
“+” indicates censoring.
Source: Clinical Study Report for the ZUMA-3 study.9
Health-Related Quality of Life
In phase II of the ZUMA-3 study, the median EQ-5D VAS score was 70.0 (range, 5 to 100; n = 51) at screening and increased over time, with higher median scores of 80.0 (range, 20 to 100; n = 41) at day 28, 80.0 (range, 50 to 100; n = 26) at month 3, 85.0 (range, 40 to 100; n = 25) at month 6, 87.5 (range, 70 to 100; n = 14) at month 12, ██ ███████ ██ ██ ███ ████ ██ █████ ███ ███ ████ ███████ ██ ██ ████ ████ ██ █████ ██ ██ ███ ██████ ████████ ████ ████ ████ ████████ ██ █████ ███ ████████ ██ ███ █████ ███ █████ ████ █████ ██ █ █████████.
Allo-SCT Rate
The ZUMA-3 study did not specify the criteria that were used to determine which patients were eligible for a subsequent allo-SCT after infusion of brexu-cel.
Eleven of 55 patients (20%) in the mITT analysis set received subsequent allo-SCT after the initial brexu-cel infusion; of these, 8 had achieved a CR and 2 had achieved a CRi to brexu-cel treatment based on the central assessment of disease response. Overall, the median time from brexu-cel infusion to allo-SCT was 101 days (range, 60 to 390 days) (Table 13).
Table 13: Summary of Incidence of Subsequent Allo-SCT after Treatment with Brexucabtagene Autoleucel, mITT Population
Subsequent allo-SCT | ZUMA-3 study (phase II) N = 55, mITT |
|---|---|
Patients with allo-SCT posttreatment, n (%) | 11 (20) |
Patients with allo-SCT and achieved CR or CRi, n (%) | 10 (18) |
95% CI | 0.0908, 0.3090 |
Patients with allo-SCT and achieved CR, n (%) | 8 (15) |
95% CI | 0.0650, 0.2666 |
Patients with allo-SCT and achieved CRi, n (%) | 2 (4) |
95% CI | 0.0044, 0.1253 |
allo-SCT = allogeneic stem cell therapy; CI = confidence interval; CR = complete remission; CRi = complete remission response with incomplete hematologic recovery; mITT = modified intention-to-t; SCT = stem cell transplant.
“+” indicates censoring.
Note: Only transplants received while in remission after brexucabtagene autoleucel infusion and before re-treatment were included. Transplants that were received after subsequent anticancer therapy were not included. Response of CR or CRi was based on central assessment.
aThe overall patient incidence includes 1 patient who had CRi per investigator assessment but was assessed as blast-free hypoplastic or aplastic bone marrow per central assessment.
Source: Clinical Study Report for the ZUMA-3 study.9
ICU Admission
ICU admission was not assessed in the ZUMA-3 study.
Harms
Only those harms identified in the review protocol are reported below. See Table 14 for detailed harms data.
Adverse Events
At the data cut-off date of July 23, 2021, all (100%) of the 55 patients treated in phase II of the ZUMA-3 study had at least 1 AE. The most commonly reported AEs included pyrexia (95%), hypotension (67%), anemia (53%), nausea (38%), sinus tachycardia (38%), headache (36%), chills (33%), and decreased platelet count (33%).
Serious AEs
SAEs were reported in 41 patients (75%). Hypotension (29%), pyrexia (27%), and hypoxia (13%) were the most commonly reported SAEs.
Withdrawal Due to AEs
Brexu-cel is administered as a single infusion; no patients discontinued treatment due to treatment-emergent AEs in the ZUMA-3 study. AEs that led to study discontinuation were not reported in the ZUMA-3 study.
Mortality
As of the data cut-off date of July 23, 2021, 25 of 55 patients (45%) had died. Eleven patients (20%) had died due to AEs, including 4 (7%) who died due to disease progression within 3 months after the brexu-cel infusion (reported as grade 5 ALL) and 7 (1%) who died due to AEs other than disease progression.
Of the 25 patients in phase II who died, 4 died within 30 days after the brexu-cel infusion, 5 died between 30 days and 3 months after the infusion, and 16 died more than 3 months after the infusion.
Notable Harms
CRS was the most commonly reported notable harm in the study population. A total of 49 patients (89%) had CRS, and 13 (24%) had worst grade 3 or higher CRS. No patient had grade 5 CRS. Pyrexia, hypotension, sinus tachycardia, chills, and hypoxia were frequently reported symptoms of CRS. Thirty-three patients (60%) had at least 1 neurologic AE; 14 (25%) had worst grade 3 or higher neurologic AEs, and 14 (25%) had serious neurologic AEs. One patient had a grade 5 neurologic AE of brain herniation. Frequently reported neurologic AEs in the study population were tremor, confusional state, and encephalopathy. Prolonged cytopenias (n = 42, 75%), prolonged hypogammaglobulinemia (n = 4, 7%), and serious infection (grade 3 or higher; n = 14, 25%) were also reported. Two (4%) patients had potential secondary malignancies.
There were no reports of anaphylaxis or antidrug antibodies in phase II of the ZUMA-3 study.
Table 14: Summary of Harms, SAS
Harms | ZUMA-3 study (phase II) N = 55 |
|---|---|
Patients with ≥ 1 AE | |
n (%) | 55 (100) |
Most common events,a n (%) | |
Pyrexia | 52 (95) |
Hypotension | 37 (67) |
Anemia | 29 (53) |
Nausea | 21 (38) |
Sinus tachycardia | 21 (38) |
Headache | 20 (36) |
Chills | 18 (33) |
Platelet count decreased | 18 (33) |
Hypoxia | 16 (29) |
Fatigue | 15 (27) |
Hypokalemia | 15 (27) |
Hypophosphatemia | 15 (27) |
Neutrophil count decreased | 15 (27) |
Tremor | 15 (27) |
Confusional state | 14 (25) |
Tachycardia | 14 (25) |
White blood cell count decreased | 14 (25) |
Alanine aminotransferase increased | 12 (22) |
Diarrhea | 12 (22) |
Encephalopathy | 12 (22) |
Hypomagnesemia | 12 (22) |
Patients with ≥ 1 SAE | |
n (%) | 41 (75) |
Most common events,a n (%) | |
Hypotension | 16 (29) |
Pyrexia | 15 (27) |
Hypoxia | 7 (13) |
Patients who stopped treatment due to AEs | |
n (%) | NA |
Deaths | |
n (%) | 25 (45) |
Causes of death, n (%) | |
AEb | 7 (13) |
Progressive disease | 14 (25) |
Otherc | 4 (7) |
Notable harms, n (%) | |
Any CRS that included the following symptoms: | 49 (89) |
Pyrexia | 46 (94) |
Hypotension | 33 (67) |
Sinus tachycardia | 18 (37) |
Chills | 14 (29) |
Hypoxia | 14 (29) |
Tachycardia | 12 (24) |
Fatigue | 10 (20) |
Headache | 10 (20) |
Neurologic AEs | 33 (60) |
Tremor | 15 (27) |
Confusional state | 14 (25) |
Encephalopathy | 12 (22) |
Aphasia | 9 (16) |
Agitation | 7 (13) |
Anaphylaxis | NR |
Cytopenias (thrombocytopenia, neutropenia, and anemia) | 42 (76) |
Hypogammaglobulinemia | 4 (7) |
Serious infection | 14 (25) had worst grade 3 or higher AEs |
Potential secondary malignancies | 2 (4) |
Anti-brexucabtagene autoleucel antibodies | NR |
AE = adverse event; CRS = cytokine release syndrome; NA = not applicable; NR = not reported; SAE = serious adverse event; SAS = safety analysis set.
aFrequency is more than 10%.
bIncluded 7 patients who had adverse events other than grade 5 acute lymphoblastic leukemia: graft-vs.-host disease (GVHD), brain herniation, septic shock, pneumonia, fungal pneumonia, sepsis, and respiratory failure.
cIncluded COVID-19, multiorgan failure due to infection and GVHD, cardiopulmonary arrest, hemorrhagic shock secondary to a gastrointestinal bleed, and disseminated intravascular coagulation.
Note: Data cut-off date of July 23, 2021.
Source: Clinical Study Report for the ZUMA-3 study.9
Critical Appraisal
Internal Validity
ALL is a rare hematological malignancy in adult patients; therefore, it is challenging in terms of recruiting patients to an RCT. Nonetheless, the single-arm, noncomparative study design for the ZUMA-3 study is 1 of the key limitations of this evidence. Although the primary efficacy outcome, OCR per central assessment in the mITT analysis set (70.9%), was higher than the prespecified historical control rate of 40% (informed by rates observed in published studies of second-line or later chemotherapy and SCT regimens, as well as in pivotal studies of blinatumomab), without a control arm, it is not possible to assess the relative efficacy and safety of brexu-cel versus currently available treatments for patients with R/R B-cell precursor ALL based on the results of the ZUMA-3 study. As well, the study design increases the possibility for bias in the estimation of treatment effects due to the potential for confounding related to selection bias, fluctuations in health status, and unidentified prognostic factors.
Another limitation for the ZUMA-3 study is the relatively small sample size and selective study population. Although 71 patients were enrolled, only 55 patients received treatment with brexu-cel and were included in the primary analyses. Furthermore, 18 patients (33%) had an important protocol deviation. In phase II of the ZUMA-3 study, a mITT population was used in the primary efficacy analysis. This as-treated population potentially introduces selection bias because it deviates from the ITT principle. The FAS was used in the sensitivity analyses and the results were different from those obtained in the mITT set — for example, between the median RFS estimates (11.6 months in the mITT analysis set versus 3.7 months in the FAS), which suggested that the analysis population had an impact on the results, at least for certain outcomes. If the study reflects the treatment protocol, then the analyses based on all patients enrolled after screening is important to assess the effect of all the procedures and co-interventions involved once it has been decided to start the treatment; this includes the manufacturing process, depleting chemotherapy, bridging therapy, and all possible consequences of these steps. The analysis based on the mITT population could bias the effect estimate away from the null hypothesis favouring brexu-cel . It is not possible to determine the magnitude of the potential overestimation of the treatment effect based on the available data and conducted analyses from this lone study.
Follow-up time was likely sufficient for assessing response and safety outcomes associated with brexu-cel . Although the median OS was estimable, the upper limit of the 95% CI was not, suggesting that the follow-up duration was not long enough to fully capture the effects on OS, and thus these results are considered immature.
Relatedly, the study presented HRQoL data at 24 months; however, no conclusion can be drawn for this outcome because the analyses on EQ-5D VAS scores had considerable missing data throughout the study time points; ██ █████████ ███ ██████████ ████████ ████ █████ ██ █ ████████ ████.
After the infusion of brexu-cel, 20% of the patients received subsequent allo-SCT. Some patients may have received other subsequent treatments, such as chemotherapy or TKIs, for the purpose of consolidating the treatment effect from CAR T-cell therapy. Data on subsequent treatments other than allo-SCT were not reported. The survival results (OS, RFS) should be considered in the context of subsequent treatments, because it may be difficult to tell which treatment has more impact on a patient’s survival, especially when there is a lack of comparative data in the ZUMA-3 study.
External Validity
According to the clinical experts consulted by CADTH, the study population of the ZUMA-3 study generally represents the patients living in Canada with R/R B-cell precursor ALL who would be receiving brexu-cel . However, the clinical experts noted that patients seen in clinical practice would include those with poorer performance status (the ZUMA-3 study only included patients with ECOG performance status of 0 or 1) and have more comorbidities.
The clinical experts noted that the ZUMA-3 study allowed re-treatment with brexu-cel, which they indicated is not reflective of the treatment approach in current clinical practice.
According to the clinical experts consulted by CADTH, the efficacy outcomes used in this study are clinically relevant and important for the clinical trials in precursor B-cell ALL. Because the ZUMA-3 study was an open-label trial, all patients knew about the treatment they received. This would have some impact on patient-reported outcomes such as HRQoL, but be less likely to affect the objective outcomes such as OS and remission rate.
In addition, lack of long-term data on patients’ survival and response rate is another limitation.
Indirect Evidence
Objectives and Methods for the Summary of Indirect Evidence
Because there was no direct evidence comparing brexu-cel to other active therapies for the treatment of adult patients with R/R B-cell precursor ALL, a review of indirect evidence was undertaken. In addition to reviewing the sponsor’s submission, CADTH conducted a literature search to identify potentially relevant indirect treatment comparisons (ITCs) in patients with R/R ALL. A focused literature search for ITCs dealing with brexu-cel or ALL was run in MEDLINE All (1946-) on October 17, 2022. No limits were applied to the search to limit results. Titles, abstracts, and full-text articles were screened for inclusion by 1 reviewer based on the population, intervention, comparator, and outcome criteria outlined in Table 5.
No potentially relevant ITCs were identified in the literature search. One sponsor-submitted ITC was included in this review.10
Description of Indirect Comparison
The sponsor-submitted ITC included a systematic literature review and an unanchored MAIC that compared brexucabtagene autoleucel to blinatumomab, inotuzumab, or chemotherapy in patients with R/R B-cell precursor ALL.
Inclusion and exclusion criteria for the clinical studies for the ITC are presented in Table 15.
Table 15: Study Selection Criteria and Methods for ITC
Criteria | Sponsor-submitted ITC |
|---|---|
Population | Adult patients with B-cell precursor R/R ALL |
Intervention | Brexucabtagene autoleucel at 1 × 106 dose |
Comparators | CAR T-cell therapy Blinatumomab Inotuzumab ozogamicin TKIs Chemotherapy |
Outcome | OS RFS EFS Overall response rate Complete response rate Partial response rate Duration of response Duration of remission MRD Progressive disease Allo-SCT rate Discontinuation rates |
Study design | RCT or non-RCT |
Exclusion criteria | B-cell precursor ALL that is not relapsed/refractory Burkitt leukemia or lymphoma Nonhuman study Other indications not included under inclusion criteria Biomarker or genetic studies Studies that investigated SCT only |
Databases searched | Various databases (MEDLINE, Embase, Cochrane Database of Systematic Reviews, Database of Abstracts of Reviews of Effects, Cochrane Central Register of Controlled Trials), conference proceedings, and clinical trial registries. The search was conducted in June 2019 and updated in November 2020. |
Selection process | Articles screened independently by 2 researchers |
Data extraction process | Unclear |
Quality assessment | Unclear |
ALL = acute lymphoblastic leukemia; allo-SCT = allogeneic stem cell therapy; CAR = chimeric antigen receptor; EFS = event-free survival; ITC = indirect treatment comparison; MRD = minimal residual disease; OS = overall survival; RCT = randomized controlled trial; RFS = relapse-free survival; R/R = relapsed or refractory; SCT = stem cell therapy; TKI = tyrosine kinase inhibitor.
Source: Sponsor-submitted indirect treatment comparison.10
Methods of ITC
Objectives
The objective of the sponsor-submitted ITC was to assess the relative treatment-effect estimates for OS and RFS/EFS of brexucabtagene autoleucel versus interventions considered to be SOC for patients with R/R ALL by means of a MAIC.
Study Selection Methods
The randomized controlled trials (RCTs) and nonrandomized trials that were used to inform the ITC were identified through a systematic literature search conducted by the ITC authors. Multiple databases were searched to identify clinical trials that evaluated the efficacy and safety of drug therapies for R/R ALL. Two reviewers independently screened and selected studies. It was unclear if data extraction was performed by 2 reviewers. Quality of the included studies was not assessed in the report.
ITC Analysis Methods
A feasibility assessment was conducted to determine if it was possible to perform a network meta-analysis or an anchored MAIC for the outcomes of OS and RFS. The study design, patient population, intervention, comparator, and outcome definitions were examined to determine the comparability of the included studies for analyses. The authors of the ITC found that in the absence of an RCT featuring brexu-cel that can be connected to other RCTs of relevant comparators in a network, it was not feasible to perform a network meta-analysis or an anchored MAIC. Instead, unanchored indirect comparisons were used to estimate the relative treatment effects between brexu-cel and the SOC interventions given the nonrandomized design of ZUMA-3. Individual patient data from brexu-cel –treated patients from the ZUMA-3 study were assigned statistical weights to adjust for between-study differences in patient characteristics relative to that observed in the comparison studies (based on study-level aggregate data) using propensity score methods.
Variables that were considered as treatment-effect modifiers and prognostic factors were selected by examining baseline patient characteristics reported in the studies, fitting univariate and multivariable logistic regression models to identify potentially important variables, and consulting an external clinical expert. A logistic propensity score model was created to estimate weights for the individual patients in the ZUMA-3 study. The degree of similarity after weighting between the individual patient data and the aggregate data populations was evaluated based on the ESS. After the weighting procedure was conducted and weights derived, efficacy outcomes were compared between balanced treatment groups.
Out of 14 variables identified as potentially relevant effect modifiers or prognostic factors, 9 variables were kept and ranked by importance based on input from clinicians consulted for the sponsor’s ITC. The factors were primary refractory disease, duration of first remission shorter than 12 months, prior allo-SCT, age at baseline, performance status at baseline, lines of prior therapies/salvage status, bone marrow blast at screening, complex karyotype, and Ph status. If models did not converge, variables were removed using a stepwise approach to achieve the model with the most factors that allowed for convergence.
The feasibility assessment identified important differences in the outcome measures (type of outcome measured, variation in definition of similar outcomes, index date definitions, how events and end dates were defined). Therefore, a decision matrix was used to align or convert outcomes in the ZUMA-3 study to those in the comparator studies.
In ZUMA-3, 4 different populations were utilized for each comparison:
The mITT phase II population (all treated patients in phase II).
ITT phase II population (all enrolled patients in phase II).
The mITT phase I and II population (all patients who were treated in phase I and II).
ITT phase I and II population (all enrolled patients in phase I and II).
The relative effect of brexu-cel versus each comparator was calculated as the adjusted HRs in the MAIC based on Cox proportional hazards models. The technical report for the MAIC states that the assumption of proportional hazards was assessed by visual inspection of plots of the log cumulative hazards and Schoenfeld residuals, as well as with the Grambsch and Therneau test.
Results of ITC
Summary of Included Studies
Of the 12 studies included in the sponsor’s systematic literature review, 2 comparator studies (the INO-VATE and TOWER studies) were determined to be relevant for the ITC. These studies investigated inotuzumab or blinatumomab versus chemotherapy regimens in adult patients with R/R B-cell precursor ALL and assessed the treatment effect on patient’s survival (Table 16). Both the INO-VATE and TOWER studies were phase III, open-label RCTs. OS and RFS or EFS were measured in the 2 studies.
Table 16: Summary of Trials Included in the MAIC
Study | Study design | N | Population | Interventions and comparators | |
|---|---|---|---|---|---|
ZUMA-3 | Phase I/II single-arm | mITT phase I: 23 mITT phase II: 55 | Adult patients with R/R B-cell ALL | Brexucabtagene autoleucel 2, 1, or 0.5 × 106 cells/kg | No comparator |
INO-VATE | Phase III open-label RCT | 164 vs. 143 | Inotuzumab 1.8 mg/m2 | SOC intensive chemotherapy (investigator choice of FLAG; MXN/Ara-C or HiDAC) | |
TOWER | Phase III open-label RCT | 267 vs. 109 | Blinatumomab 28 mcg/day | SOC chemotherapy (investigator choice of FLAG ± anthracycline; HiDAC based; high-dose methotrexate-based; or clofarabine based) | |
Ara-C = brand name of cytarabine; ALL = acute lymphoblastic leukemia; FLAG = fludarabine, cytarabine and granulocyte colony-stimulating factor; FLAG-IDA = fludarabine, cytarabine, idarubicin, filgrastim; HiDAC = high-dose cytarabine; MAIC = matching adjusted indirect comparison; mITT = modified intention to treat; MXN = cytarabine and mitoxantrone; RCT = randomized controlled trial; R/R = relapsed or refractory; SOC = standard of care; vs. = versus.
Source: sponsor-submitted ITC10
Results
Comparisons were performed for each of the 4 analysis population sets of the ZUMA-3 study (mITT phase II, mITT phase I+II, ITT phase II, ITT phase I and II) with inotuzumab in the INO-VATE study, blinatumomab in the TOWER study, chemotherapy in the INO-VATE study, and the pooled chemotherapy arms in the INO-VATE and TOWER studies for OS and EFS.
After exploring different models and examining the ESS for the 4 analysis populations, the most inclusive model that achieved convergence was selected for the MAIC comparisons. The base case MAIC with the INO-VATE study used duration of first remission less than 12 months, prior stem cell transplant, age, ECOG performance status (0 versus 1, 2 or 3), salvage status, bone marrow blast at screening, complex karyotype, and Ph status. For the TOWER study, the MAIC used primary refractory, duration of first remission more than 12 months, prior stem cell transplant, age, ECOG performance status (0 versus rest), salvage status, bone marrow blast at screening, and Ph. Covariates reported by both trials (the INO-VATE and TOWER trials) were matched for the pooled comparison with chemotherapy. The ESS for the 4 ZUMA-3 study populations is presented in Table 17 for comparisons to inotuzumab, blinatumomab, and chemotherapy. Across the different ZUMA-3 study populations and comparisons, high reductions in ESS ███████ ███████ were observed for comparisons with inotuzumab and chemotherapy after matching to the characteristics in the INO-VATE study. Reductions in ESS were slightly lower when matched to the TOWER study ███████ █████████.
Table 17: ESS for Comparisons of the ZUMA-3 Study Versus Comparator Studies
ZUMA-3 study | Sample size | ESS | % reduction in ESS |
|---|---|---|---|
INO-VATE study (inotuzumab) | |||
mITT phase II, 3-level salvage | 55 | █████ | █████ |
mITT phase I + II, 3-level salvage | ██ | █████ | █████ |
ITT phase II, 3-level salvage | 71 | █████ | █████ |
ITT phase I + II, 3-level salvage | ██ | █████ | █████ |
TOWER study (blinatumomab) | |||
mITT phase II, 3-level salvage | 55 | █████ | █████ |
mITT phase I + II, 3-level salvage | ██ | █████ | █████ |
ITT phase II, 3-level salvage | 71 | █████ | █████ |
ITT phase I + II, 3-level salvage | ██ | █████ | █████ |
INO-VATE study (chemotherapy) | |||
mITT phase II, 3-level salvage | 55 | █████ | █████ |
mITT phase I + II, 3-level salvage | ██ | █████ | █████ |
ITT phase II, 3-level salvage | 71 | █████ | █████ |
ITT phase I +II, 3-level salvage | ██ | █████ | █████ |
INO-VATE and TOWER studies (chemotherapy) | |||
mITT phase II, 3-level salvage | 55 | █████ | █████ |
mITT phase I + II, 3-level salvage | ██ | █████ | █████ |
ITT phase II, 3-level salvage | 71 | █████ | █████ |
ITT phase I + II, 3-level salvage | ██ | █████ | █████ |
ESS = effect sample size; mITT = modified intention to treat; ITT = intention to treat.
Source: Sponsor-submitted indirect treatment comparison.10
As a result of differences in the outcome measures used in the trials, the outcomes were aligned for the MAICs and were reported as OS (with the index date from the ZUMA-3 study converted from date of infusion to study enrolment, except for the mITT population) and EFS (converted from RFS in the ZUMA-3 study and progression-free survival in the INO-VATE trial).
Overall Survival
For the ZUMA-3 study mITT populations, OS was calculated from date of infusion, whereas for ITT populations it was calculated from date of trial enrolment. The OS data were immature at the time of analysis.
In general, following adjustment, the median OS was reported to be longer for brexu-cel compared to the comparators (Table 18). The estimated HRs ranged from ████ ██ █████ ████ ████ ██ █████ ████ ████ ██ ████ ███ ████ ████ ██ █████ for the comparisons to inotuzumab, blinatumomab, chemotherapy, and pooled chemotherapy, respectively. The upper limit of the 95% CI for the HRs ██████████ █████ ███████ for the ZUMA-3 phase II trial mITT population for the comparisons with inotuzumab and blinatumomab.
Event-Free Survival
For the ZUMA-3 study mITT populations, EFS was calculated from date of infusion, whereas for ITT populations it was calculated from date of trial enrolment.
In general, following adjustment, the median EFS was longer for brexu-cel compared to the comparators (Table 18). The estimated HRs ranged from ████ ██ ████ ████ ████ ██ █████ ████ ████ ██ ████ ███ ████ ████ ██ █████ for the comparisons to inotuzumab, blinatumomab, chemotherapy, and pooled chemotherapy, respectively. Statistical significance depended on the study population for which the HRs were estimated.
Table 18: Brexucabtagene Autoleucel Versus Comparators (MAIC Results for Survival)
ZUMA-3 study | Brexu-cel | Comparator | OS HR (95% CI) | Brexu-cel | Comparator | EFS HR (95% CI) |
|---|---|---|---|---|---|---|
Median OS | Median OS | Median EFS | Median EFS | |||
INO-VATE study (inotuzumab) | ||||||
mITT phase II | ████ ███ | ███ ████ | ████ ███ | ███ ████ | ███████ | ████ ███ |
mITT phase I + II | ████ ███ | ███████ | ███████ | ███████ | ███ ████ | ███ ████ |
ITT phase II | ████ ███ | ███ ████ | ███████ | ███████ | ███ ████ | ███ ████ |
ITT phase I + II | ████ ███ | ███ ████ | ███████ | ████ ███ | ███████ | ████ ███ |
TOWER study (blinatumomab) | ||||||
mITT phase II | ████ ███ | ████ ███ | ████ ███ | ███ ████ | ███ ████ | ███ ████ |
mITT phase I + II | ████ ███ | ████ ███ | ████ ███ | ███████ | ███████ | ███ ████ |
ITT phase II | ████ ███ | ████ ███ | ████ ███ | ███████ | ███████ | ███ ████ |
ITT phase I + II | ██ ██ ███ | ████ ███ | ███████ | ███████ | ████ ███ | ███ ████ |
INO-VATE study (chemotherapy) | ||||||
mITT phase II | ████ ███ | ████ ███ | ████ ███ | ███ █ ███ | ███ ████ | ███ ████ |
mITT phase I + II | █████ ██ | ████ ███ | ███████ | ██ ██ ███ | ███ ████ | ████ ███ |
ITT phase II | ███████ | ████ ███ | ████ ███ | ██ ██ ███ | ███ ████ | ███ ████ |
ITT phase I + II | █████ ██ | ████ ███ | ████ ███ | ██ ██ ███ | ███ ████ | ████ ███ |
INO-VATE and TOWER studies (chemotherapy pooled) | ||||||
mITT phase II | ████ ███ | ████ ███ | ███████ | ████ ███ | ███ ████ | ████ ███ |
mITT phase I + II | █████ ██ | ████ ███ | ████ ███ | ███ ████ | ███ ████ | ███ ████ |
ITT phase II | ████ ███ | ████ ███ | ████ ███ | ███████ | ███ ████ | ███ ████ |
ITT phase I + II | █████ ██ | ████ ███ | ████ ███ | ████ ███ | ████ ███ | ████ ███ |
brexu-cel = brexucabtagene autoleucel; CI = confidence interval; EFS = even-free survival; HR = hazard ratio; ITT = intention to treat; MAIC = matching adjusted indirect comparison; mITT = modified intention to treat; NR = not reached; OS = overall survival.
Source: Sponsor-submitted indirect treatment comparison.10
Critical Appraisal of ITC
In this ITC, studies were identified and selected using a systematic review approach — for example, multiple databases were searched, and 2 independent reviewers performed study selection. It was unclear whether data extraction was conducted by 2 reviewers independently. Quality assessment of the included studies was not performed.
The authors conducted a thorough review of the study design, inclusion and exclusion criteria, patient population characteristics, and outcomes measured in the included clinical trials and identified a number of differences in study design and patients’ baseline characteristics across studies that could potentially threaten the validity of an ITC. The rationale for conducting a MAIC instead of an network meta-analysis (NMA) was provided and consistent with usual practices (i.e., NMA techniques were not appropriate due to lack of a common comparator between the studies, and individual patient data were available for the ZUMA-3 study). The methods used for the MAIC followed the recommendations from the National Institute for Health and Care Excellence (NICE) technical guidance for population-adjusted indirect comparisons.28
The technical report identified differences in study design, enrolment criteria, patient baseline characteristics, treatments, and outcomes across included studies. A limitation of the MAIC is that it can only adjust for heterogeneity that is directly related to differences in baseline patient characteristics. It is out of scope for a MAIC to account for differences between studies other than patient characteristics, such as those related to differences in study design, definitions of study outcomes, or changes in the management of support of patients over time. As a result, these differences could not be adjusted for in the analyses conducted.
The analyses were unable to account for key sources of heterogeneity. Identifying all effect modifiers and prognostic factors that could influence the results of the analysis is essential when conducting an unanchored MAIC. The technical report indicated that 9 prognostic factors were identified and confirmed by the clinical experts consulted by the sponsor before the ITC analysis. However, not all factors could be used in the ITC because the complete list prevented the models from converging. Therefore, the base case adjustment factors were the following:
The base case MAIC with the INO-VATE study used duration of first remission less than 12 months, prior stem cell transplant, age, ECOG performance status (0 versus rest), salvage status, bone marrow blast at screening, complex karyotype, and Ph status.
For the TOWER study, the MAIC used on primary refractory, duration of first remission less than 12 months, prior stem cell transplant, age, ECOG performance status (0 versus rest), salvage status, bone marrow blast at screening, and Ph.
According to the clinical experts consulted by CADTH, these are all important factors that need to be adjusted for. The clinical experts indicated that another important prognostic factor is the presence of extramedullary disease at baseline. However, as per the author of this ITC, extramedullary disease could not be adjusted for because there were limited data in the comparator studies available for this factor.
Considerable reductions in the ESS were observed during the weighting process. For example, when comparing brexu-cel with inotuzumab for the outcome of OS, the sample size in the phase II ZUMA-3 study mITT population reduced ████ ██ ██ █████ ██ █████████ ██ ████. This suggests significant heterogeneity between the ZUMA-3 study and the comparator trials, and could lead to greater uncertainty of the validity of the comparison as well as poor precision. The results for comparisons with major reductions in ESS may not be reliable.
The outcome measures (OS and EFS) assessed in this study were clinically important. Other clinically relevant outcomes were not included in this report, such as treatment response rate, HRQoL, and safety outcomes. Outcome definitions varied across the studies as well, requiring modifications to the outcome definitions from the ZUMA-3 study to increase similarity with those used in the comparator studies.
The authors of this MAIC analysis acknowledged that the proportional hazards assumption was violated in some comparisons (such as the comparisons with chemotherapy for EFS), which further reduces the certainty in these results.
Other Relevant Evidence
This section includes a summary of 1 additional relevant study, the SCHOLAR-3 study, included in the sponsor’s submission to CADTH that was considered to provide additional comparative effectiveness data for brexu-cel from the ZUMA-3 study versus matched historical cohorts.
According to the sponsor, the ZUMA-3 study met all conditions outlined by regulatory guidelines for when an external control study that utilizes individual patient-level data from historical clinical trials would be appropriate to contextualize the investigational trial results for regulatory decision-making. These conditions were that 1) the disease is rare, leading to recruitment challenges in a randomized study design; 2) there is a high unmet medical need with suboptimal outcomes being achieved with current SOC; and 3) the therapy under investigation is expected to have a meaningful treatment effect in the context of magnitude.11
The sponsor-provided rationale for using historical clinical trials was fourfold based on the following assumptions: 1) the quality of the data in the context of accuracy and completeness was expected to be high because most trials will have undergone source data verification; 2) comparative inference was expected to be completed under strong assumptions versus real-world data because clinical trials will have prespecified response assessments and relatively well harmonized end point definitions across trials; 3) the recruitment of a congruent population to the ZUMA-3 study was expected to be made with a high degree of confidence because trials will have been selective of their populations and have descriptions of the eligibility criteria; and 4) the interpretation and comparison of response assessments were expected to be done with fewer assumptions because these will be done centrally in most trials.11
SCHOLAR-3 Study
Methods
The SCHOLAR-3 study11 was a noninterventional, retrospective matched cohort study of adult patients with R/R B-cell precursor ALL. It was conducted to compare the patients who received brexu-cel in the mITT analysis set from the ZUMA-312 phase II study with 2 propensity score-matched historical cohorts of patients (N = 89). The comparator regimens were blinatumomab, inotuzumab ozogamicin, or SOC, defined as high-dose cytarabine, fludarabine, cytarabine and filgrastim, mitoxantrone, methotrexate, or clofarabine regimens.
The first phase of the SCHOLAR-3 study identified appropriate historical clinical trials within the Medidata Enterprise Data Store database from which patients would be sampled. These trials were conducted by research institutes and the pharmaceutical industry for the purposes of drug development. Eligible historical clinical trials were phase I/II, II, or III, multicentre, multinational, open-label, single-arm or parallel assignment and randomized trials that were conducted between 2010 and 2017.11 Details of the ZUMA-3 study are presented in the Clinical Evidence section of the Systematic Review.
The second phase of the SCHOLAR-3 study constructed 2 propensity score-matched historical cohorts of patients to the ZUMA-3 study using patient-level data from the historical clinical trials identified in the first phase. Two cohorts were created to account for relevant previous treatment experience because prior treatment with blinatumomab or inotuzumab may be a predictive factor for response to CAR T-cell therapy in the patient population currently under review.11 The first cohort consisted of patients previously treatment naive to blinatumomab and inotuzumab at enrolment (SCA-1) and the second cohort consisted of patients experienced with blinatumomab or inotuzumab (SCA-2).11
Of the eligible historical cohort patients who were treatment naive to blinatumomab and inotuzumab, the SCA-1 sampling pool consisted of 1) all patients who received SOC and 2) the randomly selected 20% of patients who received blinatumomab or inotuzumab in the historical clinical trials. Of note, the number of eligible historical patients who were treatment experienced with blinatumomab or inotuzumab at baseline was fewer relative to the corresponding mITT population in the ZUMA-3 study. Hence, the SCA-2 sampling pool consisted of 1) all patients who received blinatumomab or inotuzumab before the start of the historical clinical trial and 2) patients from the remaining 80% in the previously noted group for SCA-1 who switched to other SOCs after completing the planned blinatumomab or inotuzumab treatment or becoming R/R to on-study blinatumomab or inotuzumab treatment and were continued to be followed for survival (baseline for these patients was redefined as the first day of the new treatment). Patients were recruited from both the active and control arms of appropriate trials that reflected approved therapies in the European Union, which constituted the SOC.11 The sampling approach used in the construction of the SCAs is presented in Figure 2.
Figure 2: Flow Diagram of the Sampling Approach Used in the Construction of SCA (mITT Population)
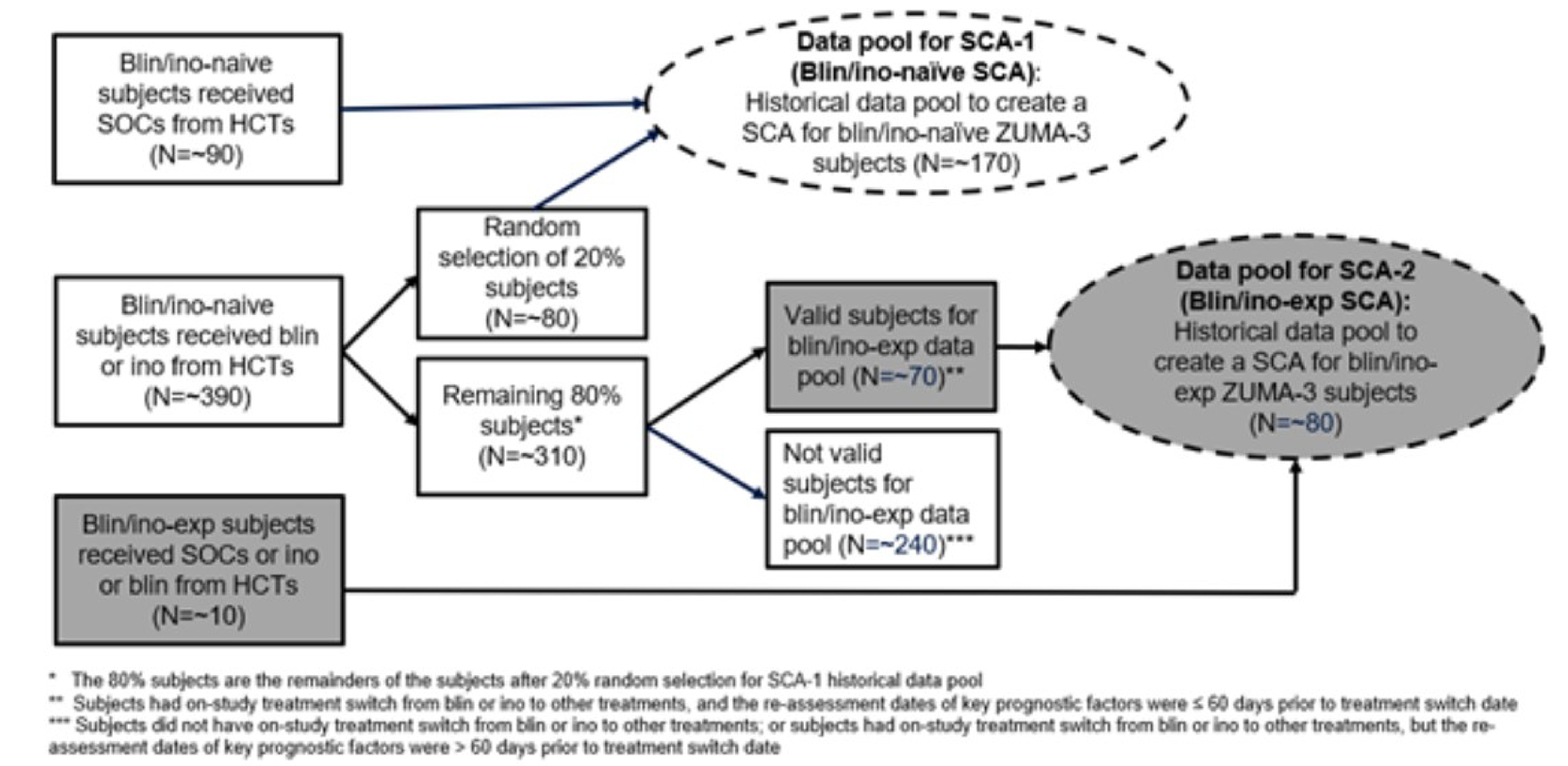
Blin = blinatumomab; exp = experienced; HCT = historical control; Ino = inotuzumab; SCA = synthetic control arm; SOC = standard of care.
Source: Clinical Study Report of the SCHOLAR-3 study.11
The matched historical cohorts of patients were created through a propensity score–based matching method; the propensity score was defined as the probability of a patient belonging to the ZUMA-3 study and receiving brexu-cel, conditional on the baseline characteristics using logistic regression. The following variables were used for propensity score matching: age at baseline, sex, ECOG performance, Ph status, percentage of bone marrow blasts at baseline, presence of extramedullary disease, primary refractory status, number of lines of prior therapy, and prior allo-SCT; depending on the final patient numbers, the last 2 prognostic factors used exact matching. After a propensity score was estimated for each patient included in the analysis, the ZUMA-3 study patients in the mITT population were randomly ordered and then sequentially matched (1 to 1) using the greedy nearest-neighbour matching without replacement algorithm and a caliper width of 0.25 of the pooled standard deviation of logit of the propensity score from the groups used. To ensure the arms were appropriately matched, the standardized mean difference (SMD) should be less than 0.25. Of note, SCA-1 and SCA-2 were constructed separately using the same method previously described.11
For the ZUMA-3 study, the data cut-off date for the primary analysis was September 9, 2020, and the data cut-off date for the 21-month follow-up analyses and sensitivity analysis that used the FAS was July 23, 2021. The method for creating the matched historical cohorts of patients to the FAS population was consistent with the method previously described.13
Populations
Inclusion and Exclusion Criteria
The inclusion criteria used in the SCHOLAR-3 study were generally consistent with the inclusion criteria used in the ZUMA-3 study. The SCHOLAR-3 study included adult patients (18 years or older) with R/R B-cell precursor ALL (definition was consistent with the ZUMA-3 study), the presence of morphological disease in the bone marrow (greater than 5% blasts), and an ECOG performance status of 0 or 1. Patients with Ph-positive disease were eligible if they were intolerant to TKI therapy or if they have R/R disease despite treatment with at least 2 different TKIs.11
Overall, a relatively broader patient population was enrolled in the SCHOLAR-3 study compared to the ZUMA-3 study. Of note, a history of severe hypersensitivity reaction to aminoglycosides or any of the drugs used in the ZUMA-3 study was not an exclusion criterion in the SCHOLAR-3 study. For prior medications, only prior CD19-directed therapy other than blinatumomab was an exclusion criterion in the SCHOLAR-3 study.11 Additionally, patients with the following medical histories were not excluded in the SCHOLAR-3 study but were excluded in the ZUMA-3 study (list is not exhaustive): clinically significant cardiac disease within 12 months of enrolment, symptomatic deep vein thrombosis or pulmonary embolism within 6 months of enrolment, and primary immunodeficiency.
Baseline Characteristics
The baseline characteristics of matched ZUMA-3 study (target group) and SCA-1 patients in the mITT and FAS population are presented in Table 19. For the mITT population, the characteristics were the same after propensity score matching for sex, ECOG performance status, and extramedullary disease status; exact matching was achieved for number of prior lines of therapy and prior allo-SCT status. In comparison to SCA-1, patients in the target group had a higher percentage of bone marrow blasts at baseline. Age, Ph status, and primary refractory status were similar between groups.11
Of note, matching was considered successful by the investigators based on the SMD of less than 0.25 between the target group and SCA-1 in the mITT and FAS population.11,13 The propensity score distributions of matched patients in the ZUMA-3 study mITT population and SCA-1 before and after matching are presented in Figure 3.
Table 19: Baseline Characteristics of Matched Patients in the ZUMA-3 Study and SCA-1
Characteristic | ZUMA-3 study mITT | ZUMA-3 study FAS | ||
|---|---|---|---|---|
Target group N = 20 | SCA-1 N = 20 | Target group N = 25 | SCA-1 N = 25 | |
Age (years), mean (SD) | 42.5 (15.3) | 44.8 (16.9) | ████ ████ | ████ ████ |
Sex, n (%) | ||||
Male | 12 (60.0) | 12 (60.0) | ██ ██████ | ██ ██████ |
Female | 8 (40.0) | 8 (40.0) | ██ ██████ | █ ██████ |
ECOG performance status, n (%) | ||||
0 | 7 (35.0) | 7 (35.0) | █ ██████ | █ ██████ |
1/Unknown | 13 (65.0) | 13 (65.0) | ██ ██████ | ██ ██████ |
Philadelphia chromosome status, n (%) | ||||
Positive | 4 (20.0) | 3 (15.0) | █ ██████ | █ ██████ |
Negative/unknown | 16 (80.0) | 17 (85.0) | ██ ██████ | ██ ██████ |
Bone marrow blasts (%), mean (SD) | 48.2 (31.6) | 41.6 (30.2) | ████ ████ | ████ ████ |
Prior lines of therapy, n (%) | ||||
≤ 2 | 16 (80.0) | 16 (80.0) | ██ ██████ | ██ ██████ |
> 2 | 4 (20.0) | 4 (20.0) | █ ██████ | █ ██████ |
Presence of extramedullary disease, n (%) | ||||
Yes | 1 (5.0) | 1 (5.0) | █ █████ | █ |
No/unknown | 19 (95.0) | 19 (95.0) | ██ ██████ | ██ ██████ |
Prior allo-SCT, n (%) | ||||
Yes | 7 (35.0) | 7 (35.0) | █ ██████ | █ ██████ |
No/unknown | 13 (65.0) | 13 (65.0) | ██ ██████ | ██ ██████ |
Primary refractory status, n (%) | ||||
Yes | 7 (35.0) | 6 (30.0) | █ ██████ | █ ██████ |
No/unknown | 13 (65.0) | 14 (70.0) | ██ ██████ | ██ ██████ |
allo-SCT = allogeneic stem cell transplant; ECOG = Eastern Cooperative Oncology Group; FAS = full analysis set; mITT = modified intention to treat; SCA = synthetic control arm; SD = standard deviation.
Source: Clinical Study Report of the SCHOLAR-3 study11 and 21-month update.13
Figure 3: Boxplot of Propensity Score Distribution of Matched Patients in the ZUMA-3 Study and SCA-1 (MITT Population)
![The propensity score distributions of patients in the SCHOLAR-3 study, before and after matching, are presented. Based on the propensity score distributions, the baseline characteristics (i.e., selected prognostic factors) between the patients in the target group (patients in the ZUMA-3 study mITT population who were naive to blinatumomab and inotuzumab) and the historical data (data pool for synthetic control arm 1 [SCA-1]) were observed to be dissimilar before matching. After matching, the characteristics between the patients in the target group and the SCA-1 were observed to be similar.](https://canjhealthtechnol.ca/index.php/cjht/article/download/PG0304r/version/669/1420/5229/PG0304CL-fig03.png)
SCA = synthetic control arm.
Source: Clinical Study Report of the SCHOLAR-3 study.11
The baseline characteristics of the matched ZUMA-3 study (target group) and SCA-2 patients in the mITT and FAS population are presented in Table 20. Due to the exclusion of 9 patients with protocol deviations in SCA-2, certain variables were no longer similar between groups in the mITT population. Specifically, a greater proportion of patients in the target group compared to SCA-2 were male, had an ECOG performance status of 1, had a negative or unknown Ph status, had 2 or fewer prior lines of therapy, had no or unknown extramedullary disease, had prior allo-SCT, and had no or unknown primary refractory disease. Although age was similar, patients in the target group had a lower mean percentage of bone marrow blasts compared to patients in SCA-2.11
Assessment of the SMD between groups confirmed the percentage bone marrow blasts and number of prior lines of therapy at baseline were not similar (SMD greater than 0.25); these 2 variables were adjusted for in the outcome analysis.11 Matching was considered successful based on the SMD between the target group and SCA-2 in the FAS population for the other charcteristics.13 The propensity score distributions of matched patients in the ZUMA-3 study mITT population and SCA-2 before and after matching are presented in Figure 4; however, this was before the exclusion of the 9 patients described previously.
Table 20: Baseline Characteristics of Matched Patients in the ZUMA-3 Study and SCA-2
Detail | mITT | FAS | ||
|---|---|---|---|---|
Target group N = 29 | SCA-2 N = 20 | Target group N = 40 | SCA-2 N = 40 | |
Age (years), mean (SD) | 40.9 (16.9) | ████ ████ | ████ █████ | ███ █████ |
Sex, n (%) | ||||
Male | 19 (65.5) | ██ █████ | ████ █████ | ████████ |
Female | 10 (34.5) | █ ██████ | ████ █████ | ████████ |
ECOG performance status, n (%) | ||||
0 | 9 (31.0) | █ ██████ | ████ █████ | ████ ████ |
1 | 20 (69.0) | ██ ██████ | ████ █████ | ████ ████ |
Philadelphia chromosome status, n (%) | ||||
Positive | 6 (20.7) | █ ██████ | ███ ██████ | ███ █████ |
Negative/unknown | 23 (79.3) | ██ █████ | ████ █████ | ████ ████ |
Bone marrow blasts (%), mean (SD) | 59.3 (32.2) | ███ ████ | ████ █████ | ████ ████ |
Prior lines of therapy, n (%) | ||||
≤ 2 | 11 (37.9) | █ ██████ | ████ █████ | ████ ████ |
> 2 | 18 (62.1) | ██ █████ | ████ █████ | ████ ████ |
Presence of extramedullary disease, n (%) | ||||
Yes | 5 (17.2) | █ ██████ | ███ ██████ | ███ █████ |
No/unknown | 24 (82.8) | ██ █████ | ████ █████ | ████ ████ |
Prior allo-SCT, n (%) | ||||
Yes | 13 (44.8) | █ ██████ | ████ █████ | ████ ████ |
No/Unknown | 16 (55.2) | ██ █████ | ████ █████ | ████ ████ |
Primary refractory status, n (%) | ||||
Yes | 8 (27.6) | █ ██████ | ████ █████ | ███ ████ |
No/unknown | 21 (72.4) | ██ █████ | ████ █████ | ████ ███ |
allo-SCT = allogeneic stem cell transplant; ECOG = Eastern Cooperative Oncology Group; FAS = full analysis set; mITT = modified intention to treat; SCA = synthetic control arm; SD = standard deviation.
Source: Clinical Study Report of the SCHOLAR-3 study11 and 21-month update.13
Figure 4: Boxplot of Propensity Score Distribution of Matched Patients in the ZUMA-3 study and SCA-2 (MITT Population)
![The propensity score distributions of patients in the SCHOLAR-3 study, before and after matching, are presented. Based on the propensity score distributions, the baseline characteristics (i.e., selected prognostic factors) between the patients in the target group (patients in the ZUMA-3 study modified intention-to-treat population who were experienced with blinatumomab or inotuzumab) and the historical data (data pool for synthetic control arm 2 [SCA-2]) were observed to be dissimilar before matching. After matching, the characteristics between the patients in the target group and SCA-2 were observed to be similar. Note that this was before the exclusion of the 9 patients who had protocol deviations.](https://canjhealthtechnol.ca/index.php/cjht/article/download/PG0304r/version/669/1420/5230/PG0304CL-fig04.png)
SCA = synthetic control arm.
Source: Clinical Study Report of the SCHOLAR-3 study.11
Interventions
The mITT population consisted of patients enrolled in the ZUMA-312 phase II study and who received brexu-cel at any dose. The FAS population consisted of patients enrolled in the ZUMA-3 study and leukapheresed. Details of the ZUMA-3 study are presented in the Clinical Evidence section of the Systematic Review.
The comparator regimens from the historical clinical trials were blinatumomab, inotuzumab ozogamicin, or SOC, defined as high-dose cytarabine, fludarabine, cytarabine and filgrastim, mitoxantrone, methotrexate, or clofarabine regimens.11
Outcomes
The primary objective was to describe the OCR rate, defined as CR and CRi, in the matched cohort of patients sampled from the historical clinical trials who were previously treatment naive to blinatumomab or inotuzumab ozogamicin.11
The following secondary objectives were assessed in the SCHOLAR-3 study and identified in the CADTH review protocol11:
Compare OCR between the matched ZUMA-3 study and SCA-1 patients.
Describe and compare CR in the matched ZUMA-3 study and SCA-1 patients.
Describe and compare the allo-SCT rate in the matched ZUMA-3 study and SCA-1 patients.
Describe and compare the OS in the matched ZUMA-3 study and SCA-1 patients.
Describe and compare the RFS in the matched ZUMA-3 study and SCA-1 patients.
Describe and compare the OS in the matched ZUMA-3 study and SCA-2 patients.
Compare the OS between all matched ZUMA-3 study and historical patients.
Of note, OS was the only effectiveness end point analyzed for SCA-2 due to the limited historical data that were available for this subset.
Safety outcomes were not evaluated in the SCHOLAR-3 study.
Statistical Analysis
All statistical analyses were performed by Medidata and matching was performed by a biostatistician and/or epidemiologist independent of the sponsor with access to the baseline data from the ZUMA-3 study only. The sponsor was blinded to all matching, treatment assignments, and outcomes until the results of the study were complete. All statistical tests were 2-sided at the alpha level of 0.05.11 Of note, an additional sensitivity analysis that used the ZUMA-3 study FAS was included.13
For the primary effectiveness end point, the number and proportion of patients with either CR or CRi per independent or investigator review before subsequent anticancer therapy and allo-SCT were described at week 24.11
For the secondary effectiveness end point of response rate, including OCR rate, CR rate, and allo-SCT rate, the crude incidence rates in each group with an exact binomial 95% CI were presented. The odds ratio (OR) together with the associated 95% CI and 2-sided P value were estimated from a logistic regression model with treatment group (ZUMA-3 study versus SCA) as a single covariate. Response definitions across all historical clinical trials were harmonized to the same definitions in the ZUMA-3 study. The index date was defined as the date on which a patient met all eligibility criteria and started therapy. The follow-up time to estimate the allo-SCT rate was defined as the period between the index date and initiation of allo-SCT, death, loss to follow-up, or initiation of a new line of therapy other than allo-SCT, whichever came first.11 These definitions were similar to the ZUMA-3 study. The definitions of the corresponding outcomes used in the ZUMA-3 study are presented in the Clinical Evidence section of the Systematic Review.
For the time-to-event end point, including RFS and OS, survival curves and rates were estimated using the KM method. A 2-sided log-rank test was used for testing the difference between ZUMA-3 study matched patients and the corresponding SCA group. The HR between the 2 comparison groups and the 95% CIs were estimated using a Cox proportional hazard regression model. RFS was defined as the time from the index date to the date of disease relapse or death from any cause. Patients who did not meet the criteria for relapse by the analysis data cut-off date were censored at their last evaluable disease assessment date. Patients who did not achieve a complete response (i.e., CR and CRi) at the analysis data cut-off date were evaluated as having an RFS event on day 0. The follow-up time to estimate OS was defined as the period between the index date and death or loss to follow-up, whichever came first.11 These definitions were similar to the definitions for RFS and OS used in the ZUMA-3 study.
For the propensity score model, only baseline variables with limited or no missingness (i.e., less than 15%) were included. According to the sponsor, the 15% threshold was selected to balance the benefits of incorporating as many historical patients as possible and the data that are recorded for those patients to avoid inaccuracies that could be induced by imputation of missing information. For baseline categorical variables, missing values were coded as an unknown category. For baseline continuous variables, missing values were imputed by a single regression imputation approach.11
There was no prespecified hypothesis (e.g., superiority), a priori sample size, or power calculation, and no control for type I error.
Patient Disposition
An approximate total of 490 eligible historical clinical trial patients were identified as potential external control arm candidates (sampling set). A total of 40 patients previously treatment naive to blinatumomab and inotuzumab were included in the SCHOLAR-3 study; 20 patients from the ZUMA-3 study were matched to 20 patients in SCA-1. A total of 49 patients experienced with blinatumomab were also included in the study; 29 patients from the ZUMA-3 study were matched to 20 patients in SCA-2. Of note, 29 patients were originally included in SCA-2; however, 9 patients from the historical clinical trials were found to have important protocol deviations (i.e., did not have documented relapse before starting a subsequent therapy) and were thus excluded. No matches from the historical clinical trials were found for 6 patients from the ZUMA-3 study mITT population; these 6 patients were excluded from the study.11
For the sensitivity analyses using the FAS population █ █ █████ ██ ██ ████████ ██████████ █████████ █████ ██ ████████████ ███ ██████████ ████ █████████ ██ ████████ ████ ██████ ███ █████ ██ ████████ ████ ███ ███████ ████ ██████████████ ███████████ ████ ███████ ██ ██ ████████ ██ ██████ █ █████ ██ ██ ████████ ███████████ ████ ████████████ ██ ██████████ ████ ████ ████████ ██ ███ ███████████ █████████ ██ ████████ ████ ██████ ███ █████ ██ ████████ ████ ███ ███████ ████ ██████████████ ███████████ ████ ███████ ██ ██ ████████ ██ ██████13
Exposure to Study Treatments
In the mITT population █ ███ █████████ ██████████ ██ █████ ███ ███ ██ ████████ ████ ███████ ████ ████████████ ███ ███ ██ ████████ ████ ███████ ████ ███ █████████████ ███ █████████ ██████████ ██ █████ ███ ██ ██ ████████ ████ ███████ ████ █████████████ ██ ██ ████████ ████ ███████ ████ ███████████ ███ ███ ██ ████████ ████ ███████ ████ ███ █████████████11
Effectiveness
Primary Analyses
The primary outcome analyses were the focus of this review. For OCR at week 24, the estimated difference in the percentage of patients in the ZUMA-3 mITT population (17 of 20 patients) compared with patients in SCA-1 (7 of 20 patients) was 50% (95% CI, 17.9 to 73.7; OR = 10.5, 95% CI, 2.3 to 48.7). The comparison of OS between the ZUMA-3 study mITT (N = 29; median follow-up = 24 months) and SCA-2 (N = 20; median follow-up = 24 months) populations suggested that ZUMA-3 study patients had a longer median OS (15.90 [95% CI, 3.19 to 26.02] months versus 4.76 [95% CI, 2.66 to 12.35] months; HR = 0.55; 95% CI, 0.26 to 1.13). The results from the sensitivity analysis were generally consistent with the 21-month follow-up updated analysis (refer to Appendix 2).13
Detailed effectiveness outcomes are presented in Appendix 2.
Harms
Safety outcomes were not assessed in the SCHOLAR-3 study.
Critical Appraisal
Internal Validity
In the absence of a comparator arm in the ZUMA-3 study (phase I/II, single-arm, open-label study), the SCHOLAR-3 study (a noninterventional, retrospective matched cohort study) was conducted to contextualize the results from the ZUMA-3 study. Eligible historical clinical trials included phase I/II, II, or III, multicentre, multinational, open-label, single-arm, or parallel assignment and randomized trials that were conducted between 2010 and 2017. Although the ZUMA-3 study enrolled patients from 2016, the clinical experts consulted by CADTH for this review did not express concerns about any time-related confounders because most of the regimens, including supportive care, and the diagnosis of ALL have been stable since 2010. Furthermore, the prognostic variables used for the propensity score matching (age at baseline, sex, ECOG performance status, Ph status, percentage bone marrow blasts at baseline, presence of extramedullary disease, primary refractory status, number of lines of prior therapy, and prior allo-SCT) were considered to be valid by the clinical experts consulted by CADTH for this review. However, it should be noted that duration of first remission of less than 12 months and complex karyotype were not included as prognostic factors in the SCHOLAR-3 study but were considered valid in the ITC by the clinical experts consulted by CADTH for this review. Additionally, the eligible historical clinical study designs were different (and those selected were not summarized and therefore cannot be appraised), and the dissimilar baseline characteristics before matching between the ZUMA-3 study population and that of the historical studies indicates that there was considerable heterogeneity between the comparison groups; therefore, it is likely that there is confounding of the treatment-effect estimates due to known and unknown confounders that were not adjusted for. Although matching was considered successful by the investigators based on the SMDs for the matched factors being within the prespecified threshold between the target group and SCA-1, it should be noted that the percentage bone marrow blasts and prior lines of therapy at baseline were not considered similar between the target group and SCA-2 postweighting and were adjusted for in the statistical analyses. It should also be noted that sensitivity analyses (i.e., using a matching method other than the primary matching method) were not conducted, and as such, the reliability and validity of the results were reduced. As well, the propensity score matching may make the patient characteristics between the populations similar, but it does not account for heterogeneity in study designs, conduct, analysis, or duration, nor does it adjust for limitations associated with the eligibility criteria.
The interpretation of the comparative effectiveness results, specifically the secondary outcomes, in the SCHOLAR-3 study is limited by the sampling approach that was used in the construction of the SCAs. In particular, the data pool for SCA-2 (treatment experienced with blinatumomab or inotuzumab) included patients who were previously treatment naive to blinatumomab and inotuzumab and had an on-study treatment switch from blinatumomab or inotuzumab to other SOC treatments. The baseline for these patients was redefined as the first day of the new treatment. Moreover, these patients who switched to other SOC treatments could have either completed the planned blinatumomab or inotuzumab treatment or became R/R to on-study blinatumomab or inotuzumab treatment. Although the number of prior lines of therapy was a prognostic factor used in the propensity score matching, the data pool for SCA-2 was a heterogeneous population because patients entered the data pool with different treatment histories (i.e., it included both historical patients who were and were not truly treatment experienced with blinatumomab or inotuzumab). Moreover, the data pool for SCA-1 (treatment naive to blinatumomab and inotuzumab) did not include all eligible historical patients who were treatment naive to blinatumomab and inotuzumab; the impact, if any, of this sampling approach on the results is unknown. The interpretation of the comparative effectiveness results is further limited by the recruitment of patients from both the active and control arms of historical clinical trials that reflected approved SOC treatments in the European Union.
All statistical analyses were performed by Medidata and matching was performed by a biostatistician and/or epidemiologist independent of the sponsor with access to the baseline data from the ZUMA-3 study only. The sponsor was blinded to all matching, treatment assignments, and outcomes until the results of the study were complete. However, for the primary effectiveness end point, the number and proportion of patients with either CR or CRi per independent or investigator review before subsequent anticancer therapy and allo-SCT were described. Therefore, there was a potential risk of bias in the measurement of outcomes; however, the magnitude and direction of this bias are unknown.
There was no formal hypothesis statement, no power or sample size considerations, and no adjustments for multiple comparisons. As such, the statistical inference from the results of this study has low reliability and validity. Additionally, relatively small numbers of patients were included in the analysis sets; according to the preliminary feasibility assessments, it was anticipated that approximately 490 patients were eligible to participate in the study, yet a total of 89 patients formed the primary ZUMA-3 study mITT versus SCA-1 and SCA-2 comparisons. Moreover, 6 patients from the ZUMA-3 study mITT population were excluded from the study because no matches from the historical clinical trials were found; the impact, if any, of this exclusion on the results is unknown. A total of 9 historical patients were found to have important protocol deviations and were thus excluded from SCA-2, and as a result, the baseline characteristics of matched ZUMA-3 study and SCA-2 patients were no longer well balanced, further adding to the uncertainty of the results.
External Validity
The inclusion criteria used in the SCHOLAR-3 study were generally consistent with the inclusion criteria used in the ZUMA-3 study. Overall, a relatively broader patient population was enrolled in the SCHOLAR-3 study compared to the ZUMA-3 study. For example, patients with a history of severe hypersensitivity reaction to aminoglycosides or any of the drugs used in the ZUMA-3 study were not excluded in the SCHOLAR-3 study. Additionally, only prior CD19-directed therapy other than blinatumomab was an exclusion criterion in the SCHOLAR-3 study.
In SCA-1, 45% of patients were treated with blinatumomab and 55% of patients were treated with SOC chemotherapy; no patients received inotuzumab, which was identified as a relevant comparator by the clinical experts consulted by CADTH for this review.
In SCA-2, 5% of patients were treated with blinatumomab, 5% of patients were treated with inotuzumab, and the majority (90%) of patients were treated with SOC chemotherapy. SOC was defined as high-dose cytarabine, fludarabine, cytarabine and filgrastim, mitoxantrone, methotrexate, or clofarabine regimens. The clinical experts consulted by CADTH for this review indicated that there is no backbone chemotherapy identified because many options are available, depending on previous treatment experience; moreover, most regimens have been stable since 2010.
Discussion
Summary of Available Evidence
One clinical study, the ZUMA-3 study, was included in the systematic review. The ZUMA-3 study is an ongoing phase I/II, open-label, single-arm study that evaluated the efficacy and safety of brexu-cel in patients with R/R B-cell precursor ALL. The primary end point was OCR rate (defined as CR plus CRi) by central assessment. Secondary end points included OS, RFS, DOR, MRD-negative rate, subsequent allo-SCT rate, and HRQoL. A total of 55 of the 71 patients enrolled received brexu-cel and were included in the primary efficacy and safety analyses. Data up to 21 months of follow-up were available at the time of this review (data cut-off date of July 23, 2021). For patients treated in phase II of the ZUMA-3 study, the median actual follow-up time from brexu-cel infusion was 20.5 months (range, 0.3 to 32.6 months), and the median potential follow-up time from the brexu-cel infusion was 26.8 months (range, 20.7 to 32.6 months). The mean age of patients was 42 years. The majority of patients were male (60%), white (67%), and had an ECOG performance status of 1 (71%). Ph-positive presented in 27% of the enrolled patients. Overall, 45%, 22%, and 42% of the patients had prior blinatumomab, inotuzumab, and prior allo-SCT, respectively; 33% of the study population had primary refractory disease and 78% had R/R to second-line or greater therapy. The mean percentage of blasts in bone marrow at baseline was 33%. Extramedullary disease at baseline was reported in 11% of the patients. All patients reported CNS-1 disease before entering the study.
One sponsor-submitted ITC was summarized and critically appraised. The sponsor performed an unanchored MAIC analysis to estimate the comparative effectiveness and safety of brexu-cel in patients with R/R B-cell precursor ALL relative to other targeted therapy (blinatumomab, inotuzumab) and chemotherapy. The outcomes assessed in the ITC were OS and EFS.
The sponsor also provided an additional study, the SCHOLAR-3 study, which was a retrospective matched cohort study conducted to provide comparative efficacy data for brexu-cel between the ZUMA-3 study and matched historical cohorts.
Interpretation of Results
Efficacy
OS was indicated by the clinical experts consulted by CADTH to be the most important efficacy outcome to assess treatment effect in patients with refractory and relapsed ALL. This is also an important outcome indicated in the patient group input. At the data cut-off of July 23, 2021, based on the 21-month data in phase II of the ZUMA-3 study, the median OS measured with the KM method was 25.4 months (95% CI, 16.2 months to estimable) in a modified ITT population, which included the patients who received treatment with brexu-cel . Another survival outcome, RFS, measures the length of time from the brexu-cel infusion date to the date of disease relapse or death from any cause. The median RFS was 11.6 months (95% CI, 2.7 to 20.5 months) in the mITT population. According to the clinical experts consulted by CADTH, the survival data look promising based on the experts’ experience treating patients with R/R B-cell precursor ALL. It is notable that over the 21-month follow-up, 45% of the patients had died, suggesting that brexu-cel is not curative. Given that the ZUMA-3 study is ongoing and the currently available OS data are immature (21 months of data thus far), it is still too early to assess the impact of brexu-cel on patients’ long-term survival.
The survival results were supported by the response rates, which are clinically relevant outcomes in the clinical trials of leukemia as well as in clinical practice. The OCR rate (including CR and CRi) per central assessment for patients in the phase II mITT analysis set in the ZUMA-3 study was 70.9% (39 of 55 patients, 95% CI, 57% to 82%), with a CR rate of 56.4% (31 of 55 patients, 95% CI, 42% to 70%). The primary end point of OCR was higher than a prespecified historical overall response rate of 40% identified for adult patients with ALL. The clinical experts indicated that the response rate results were favourable based on their clinical experience treating patients with R/R B-cell precursor ALL.
While these results indicate that brexu-cel has a tumour effect that is beneficial, the magnitude of the effect is difficult to determine because of the noncomparative design of the ZUMA-3 study and focus on the mITT population for the analyses. It is notable that the median OS and median RFS benefits were smaller in the FAS population than in the mITT population (23.1 versus 25.4 months and 3.7 versus 11.6 months, respectively). This is not surprising given that the index date for the FAS was closer to a true ITT population (all enrolled patients with leukapheresis) and included the full CAR T-cell protocol period versus the mITT index date set at the date of infusion. The mITT therefore represents a select population and the results may not be generalizable to clinical practice. An FAS analysis was not reported for the response outcomes.
If brexu-cel were reimbursed, some patients might be eligible to receive subsequent allo-SCT therapy to consolidate the effect from CAR T-cell therapy. Eleven patients (20%) received subsequent allo-SCT. Among them, 10 (18%) achieved OCR and 8 (15%) achieved CR. It is unknown how the subsequent treatment with allo-SCT and other cancer treatment would affect the patient’s survival or response to brexu-cel treatment.
HRQoL was an outcome indicated as important by patients with ALL and by clinicians. HRQoL is also useful in judging patient-centred effects of a treatment and to help evaluate the impacts of other outcomes, such as survival. However, findings related to HRQoL based on the EQ-5D VAS from the ZUMA-3 study could not be interpreted due to the large amount of missing data ███ ███ ███ ██████████ █████ ████ ████ ████ █████████ ██ ██ ██ ███ ████ ███████████. It is unclear if treatment with brexu-cel is associated with improved HRQoL.
The prognosis of R/R ALL is poor: The 5-year survival is approximately 20% in this population. For the patients with R/R disease, the treatment options are cytotoxic chemotherapy, targeted therapies (e.g., blinatumomab), allo-SCT, and, for some patients, enrolment in a clinical trial for an investigational therapy. Even though allo-SCT would be effective for some patients, not all are eligible to receive this treatment, especially older patients or those who are too frail to tolerate the procedure. CADTH consulted a clinical panel of cancer hematologists who indicated that patients may benefit from brexu-cel based on the promising, albeit relatively short-term, data and increasing experience with CAR T-cell therapies in hematologic cancers, including pediatric patients and a subset of younger adult patients with ALL. Brexu-cel is provided as a single-dose, one-time treatment per the Health Canada product monograph. In the ZUMA-3 study, re-treatment was allowed and 2 patients received a second treatment with brexu-cel, which differs from the expected use of brexu-cel in clinical practice. The data from the ZUMA-3 study did not show additional benefit from a second infusion with brexu-cel . However, given that the main evidence for the benefit comes from a single phase I/II noncomparative study that enrolled a range of patients with 1, 2, or more than 2 prior lines of therapy, the exact place in therapy based on the available evidence is difficult to determine.
Due to a lack of direct comparative evidence, data from an ITC and a retrospective matched cohort study (the SCHOLAR-3 study) were considered. The results from the sponsor-submitted ITC suggested that OS and EFS may be prolonged; this is related to the treatment with brexu-cel in the study population compared to blinatumomab, inotuzumab, or chemotherapy; however, definitive conclusions related to the survival benefits of brexu-cel cannot be drawn from this ITC analysis. The key limitation for the ITC is significant heterogeneity across the included RCTs and associated uncertainty in result interpretation. The heterogeneity existing in the included trials cannot be adequately addressed by the MAIC approach. Results of the MAIC suggested longer OS and EFS, and reduced risk of death associated with treatment with brexu-cel, compared to other treatments including blinatumomab, inotuzumab, and chemotherapy. However, given the limitations of these data (no effect modifiers identified and it’s likely that not all prognostic factors have been identified, as well as small sample size and small evidence base), there is a substantial risk of bias in the MAIC results.
In the SCHOLAR-3 study, the results suggested that brexu-cel was associated with improved OCR and OS compared with other standard treatments. However, interpretation of the comparative results is limited by the potential for selection bias and unaccounted for confounding despite the propensity score matching approach. Therefore, the statistical inference from the study findings has low reliability and validity.
A key gap in the evidence is the lack of comparison between brexu-cel and tisagenlecleucel. The latter has been approved for the treatment of R/R B-cell ALL in patients aged 18 to 25 years; it therefore overlaps with a proportion of the population indicated for brexu-cel .
Harms
At data cut-off date of July 23, 2021, all 55 patients in phase II of the ZUMA-3 study reported at least 1 AE. The most commonly reported AEs included pyrexia, hypotension, and anemia. Most of these AEs are symptoms of CRS, which is a usual but severe AE associated with treatment with CAR T-cell therapy. SAEs were reported in most patients (75%), with the most common SAEs being hypotension, pyrexia, and hypoxia. In total, 25 of 55 patients (45%) had died as of the data cut-off date. Eleven patients (20%) had died due to AEs, including 4 (7%) who died due to disease progression within 3 months after the brexu-cel infusion and 7 (1%) who died due to AEs other than disease progression. Brexu-cel is administered as a single infusion; no patients discontinued treatment due to treatment-emergent AEs in the ZUMA-3 study.
The Health Canada–approved product monograph of brexu-cel contains serious warnings for CRS and neurologic AEs, and states that brexu-cel should be administered by experienced health professionals at specialized treatment centres.
In terms of notable harms, CRS was the most commonly reported notable harm in the study population. A total of 49 patients (89%) had CRS, and 13 (24%) had worst grade 3 or higher CRS. No patient had grade 5 CRS. Pyrexia, hypotension, sinus tachycardia, chills, and hypoxia were the most commonly reported symptoms of CRS. Thirty-three patients (60%) had at least 1 neurologic AE. Frequently reported neurologic AEs in the study population were tremor, confusional state, and encephalopathy. Prolonged cytopenias, prolonged hypogammaglobulinemia (lower than grade 3 cases were reported), and serious infection were also reported in the study population.
According to the clinical experts consulted by CADTH, the safety profile of brexu-cel is consistent with other CAR T-cell therapy, and no unexpected safety signals are observed from the included study.
The ITC and the SCHOLAR-3 study did not assess harms; therefore, the safety of brexu-cel compared to other treatments for R/R B-cell precursor ALL remains unknown.
Conclusions
Evidence from a single-arm study (the ZUMA-3 study) suggests that treatment with brexu-cel may be associated with benefits in OS and RFS based on the clinical experts’ experience and expectations of the natural progression of the disease in adult patients with R/R B-cell precursor ALL. However, because the OS data are immature, analyses were based on a select patient population, and there was no comparator arm in the ZUMA-3 study, it is possible that the effect of brexu-cel on survival is overestimated in the ZUMA-3 study. It is unclear if treatment with brexu-cel would improve patients’ quality of life. Data from a retrospective matched cohort (the SCHOLAR-3 study) suggest that the response rate (e.g., CR) in patients treated with brexu-cel was higher than those observed in patients who received SOC in historical trials; however, the study was considered by CADTH reviewers to have poor internal validity and the findings were associated with a high degree of uncertainty. Findings from an ITC analysis suggest favourable survival benefits associated with brexu-cel treatment; however, definitive conclusions on survival benefits cannot be made due to the significant uncertainties in the indirect comparison. The harms associated with brexu-cel infusion are consistent with its mechanism of action and there were no unexpected safety signals observed. The single-arm study design of the ZUMA-3 study and lack of long-term data are key limitations of the evidence; therefore, uncertainties remain regarding the magnitude of the clinical benefit from treatment with brexu-cel.
References
1.Hoelzer D, Bassan R, Dombret H, et al. Acute lymphoblastic leukaemia in adult patients: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2016;27(suppl 5):v69-v82. PubMed
2.Drug Reimbursement Review sponsor submission: Tecartus (brexucabtagene autoleucel), target of 1 × 106 chimeric antigen receptor (CAR)-positive viable T cells per kg body weight with a maximum of 1 × 108 CAR-positive viable T cells, cell suspension in patient-specific single infusion bag for intravenous infusion [internal sponsor's package]. Mississauga (ON): Gilead Sciences Canada, Inc.; 2022 Oct 03.
3.Larson RA. Treatment of relapsed or refractory acute lymphoblastic leukemia in adults. In: Post TW, ed. UpToDate. Waltham (MA): UptoDate; 2022: www.uptodate.com. Accessed 2022 Oct 04.
4.Bouchkouj N, Lin X, Wang X, et al. FDA Approval Summary: Brexucabtagene Autoleucel for Treatment of Adults With Relapsed or Refractory B-Cell Precursor Acute Lymphoblastic Leukemia. Oncologist. 2022;27(10):892-899. PubMed
5.Brandwein J, Tilley D, Cancer Care Alberta. Acute lymphoblastic leukemia in adults, version 2. Edmonton (AB): Alberta Health Services; 2022: https://www.albertahealthservices.ca/assets/info/hp/cancer/if-hp-cancer-guide-lyhe005-all.pdf. Accessed 2022 Nov 30.
6.Stem cell transplant for acute lymphocytic leukemia. Toronto (ON): Canadian Cancer Society; 2022: https://cancer.ca/en/cancer-information/cancer-types/acute-lymphocytic-leukemia-all/treatment/stem-cell-transplant. Accessed 2022 Nov 30.
7.Tecartus (brexucabtagene autoleucel): target of 2 × 106 chimeric antigen receptor (CAR)-positive viable T cells per kg body weight with a maximum of 2 × 108 anti-CD19 CAR-positive viable T cells, cell suspension in patient-specific single infusion bag for intravenous infusion; or Tecartus (brexucabtagene autoleucel): target of 1 × 106 chimeric antigen receptor (CAR)-positive viable T cells per kg body weight with a maximum of 1 × 108 anti-CD19 CAR-positive viable T cells, cell suspension in patient-specific single infusion bag for intravenous infusion [product monograph]. Mississauga (ON): Gilead Sciences Canada, Inc.; 2022 Nov 16.
8.Kymriah (tisagenlecleucel): 2.0 x 106 to 6.0 x 108 CAR-positive viable T cells, cell suspension in infusion bag for intravenous use [product monograph]. Dorval (QC): Novartis Pharmaceuticals Canada Inc.; 2018 Sep 05: https://pdf.hres.ca/dpd_pm/00047188.PDF. Accessed 2022 Nov 30.
9.Clinical Study Report: KTE-C19-103 (ZUMA-3) 21-month follow-up. A phase 1/2 multi-center study evaluating the safety and efficacy of KTE-X19 in adult subjects with relapsed/refractory B-precursor acute lymphoblastic leukemia (r/r ALL) [internal sponsor's report]. Santa Monica (CA): Kite Pharma, Inc.; 2022 Jan 11.
10.Updated matching-adjusted indirect comparison for adult patients with relapsed/refractory B-precursor acute lymphoblastic leukemia: technical report, version 1 [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Tecartus (brexucabtagene autoleucel), target of 1 × 106 chimeric antigen receptor (CAR)-positive viable T cells per kg body weight with a maximum of 1 × 108 CAR-positive viable T cells, cell suspension in patient-specific single infusion bag for intravenous infusion. Mississauga (ON): Gilead Sciences Canada, Inc.; 2022 Mar 08.
11.Clinical Study Report: SCHOLAR-3. A retrospective cohort study of adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia sampled from historical clinical trials (SCHOLAR-3) [internal sponsor's report]. Santa Monica (CA): Kite Pharma, Inc.; 2021 May 07.
12.Clinical Study Report: KTE-C19-103 (ZUMA-3). A phase 1/2 multi-center study evaluating the safety and efficacy of KTE-X19 in adult subjects with relapsed/refractory B-precursor acute lymphoblastic leukemia (r/r ALL) [internal sponsor's report]. Santa Monica (CA): Kite Pharma, Inc.; 2021 Feb 09.
13.Clinical Study Report: SCHOLAR-3, addendum 21-month. A retrospective cohort study of adult subjects with relapsed or refractory B precursor acute lymphoblastic leukaemia sampled from historical clinical trials (SCHOLAR-3) [internal sponsor's report]. Santa Monica (CA): Kite Pharma, Inc.; 2021 Nov 30.
14.Advani AJ, Aster JC. Clinical manifestations, pathologic features, and diagnosis of B cell acute lymphoblastic leukemia/lymphoma. In: Post TW, ed. UpToDate. Waltham (MA): UpToDate; 2022: www.uptodate.com. Accessed 2022 Oct 04.
15.Kantarjian H, Stein A, Gokbuget N, et al. Blinatumomab versus Chemotherapy for Advanced Acute Lymphoblastic Leukemia. N Engl J Med. 2017;376(9):836-847. PubMed
16.Kantarjian HM, DeAngelo DJ, Stelljes M, et al. Inotuzumab Ozogamicin versus Standard Therapy for Acute Lymphoblastic Leukemia. N Engl J Med. 2016;375(8):740-753. PubMed
17.Fielding A, Nicholson E, Wrench B, Palanicawandar R, Easdale S, Kirschke S. Pan-London Haemato-Oncology Clinical Guidelines: Acute Leukaemias and Myeloid Neoplasms Part 1: Acute Lymphoblastic Leukaemia. London (UK): RM Partners, South East London Cancer Alliance Network, North Central and East London Cancer Alliance; 2020: https://www.kingshealthpartners.org/assets/000/003/339/Pan_London_ALL_Guidelines_Jan_2020_original.pdf Accessed 2022 Nov 30.
18.Jabbour E, Sasaki K, Short NJ, et al. Long-term follow-up of salvage therapy using a combination of inotuzumab ozogamicin and mini-hyper-CVD with or without blinatumomab in relapsed/refractory Philadelphia chromosome-negative acute lymphoblastic leukemia. Cancer. 2021;127(12):2025-2038. PubMed
19.Kunz T, Hauswirth AW, Hetzenauer G, Rudzki J, Nachbaur D, Steiner N. Changing Landscape in the Treatment of Adult Acute Lymphoblastic Leukemia (ALL). Cancers (Basel). 2022;14(17):4290. PubMed
20.Goldstone AH, Richards SM, Lazarus HM, et al. In adults with standard-risk acute lymphoblastic leukemia, the greatest benefit is achieved from a matched sibling allogeneic transplantation in first complete remission, and an autologous transplantation is less effective than conventional consolidation/maintenance chemotherapy in all patients: final results of the International ALL Trial (MRC UKALL XII/ECOG E2993). Blood. 2008;111(4):1827-1833. PubMed
21.Gokbuget N, Stanze D, Beck J, et al. Outcome of relapsed adult lymphoblastic leukemia depends on response to salvage chemotherapy, prognostic factors, and performance of stem cell transplantation. Blood. 2012;120(10):2032-2041. PubMed
22.Blincyto (blinatumomab): 38.5 mcg lyophilized powder for solution for intravenous infusion [product monograph]. Mississauga: Amgen Canada Inc.; 2018 Jun 08: https://pdf.hres.ca/dpd_pm/00045873.PDF. Accessed 2022 Nov 30.
23.Besponsa (inotuzumab ozogamicin for injection): 0.9 mg single-dose vial, lyophilized powder for solution for intravenous infusion [product monograph]. Kirkland (QC): Pfizer Canada Inc.; 2018 Mar 15: https://www.pfizer.ca/files/BESPONSA_PM_204077_15March2018_E.pdf. Accessed 2022 Nov 30.
24.McGowan J, Sampson M, Salzwedel DM, Cogo E, Foerster V, Lefebvre C. PRESS Peer Review of Electronic Search Strategies: 2015 guideline statement. J Clin Epidemiol. 2016;75:40-46. PubMed
25.Grey matters: a practical tool for searching health-related grey literature. Ottawa (ON): CADTH; 2019: https://www.cadth.ca/grey-matters. Accessed 2022 Oct 03.
26.Shah BD, Ghobadi A, Oluwole OO, et al. KTE-X19 for relapsed or refractory adult B-cell acute lymphoblastic leukaemia: phase 2 results of the single-arm, open-label, multicentre ZUMA-3 study. Lancet. 2021;398(10299):491-502. PubMed
27.Center for Drug Evaluation Research. Medical review(s). Tecartus (brexucabtagene autoleucel) intravenous infusion. Company: Kite Pharma, Inc. Application No.:BL 125703. Approval date: 10/01/2021 (FDA approval package). Silver Spring (MD): U.S. Food and Drug Administration (FDA); 2022: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/tecartus-brexucabtagene-autoleucel Accessed 2022 Nov 30.
28.Phillippo DM, Ades AE, Dias S, Palmer S, Abrams KR, Welton NJ. NICE DSU Technical Support Document 18: Methods for population-adjusted indirect comparisons in submission to NICE. Sheffield (UK): NICE Decision Support Unit; 2016: https://research-information.bris.ac.uk/ws/portalfiles/portal/94868463/Population_adjustment_TSD_FINAL.pdf. Accessed 2023 Jan 11.
29.van Reenen M, Janssen B, Stolk E, et al. EQ-5D-5L User Guide, v.3. Rotterdam (NL): EuroQol Research Foundation; 2019: https://euroqol.org/publications/user-guides. Accessed 2022 Nov 04.
30.van Dongen-Leunis A, Redekop WK, Uyl-de Groot CA. Which Questionnaire Should Be Used to Measure Quality-of-Life Utilities in Patients with Acute Leukemia? An Evaluation of the Validity and Interpretability of the EQ-5D-5L and Preference-Based Questionnaires Derived from the EORTC QLQ-C30. Value Health. 2016;19(6):834-843. PubMed
Appendix 1: Literature Search Strategy
Note that this appendix has not been copy-edited.
Clinical Literature Search
Overview
Interface: Ovid
Databases:
MEDLINE All (1946-present)
Embase (1974-present)
Note: Subject headings and search fields have been customized for each database. Duplicates between databases were removed in Ovid.
Date of Search: October 17, 2022
Alerts: Bi-weekly search updates until project completion
Search filters applied: No filters were applied to limit the retrieval by study type.
Limits:
Publication date limit: none
Language limit: none
Conference abstracts: excluded
Syntax | Description |
|---|---|
/ | At the end of a phrase, searches the phrase as a subject heading |
MeSH | Medical Subject Heading |
* | Before a word, indicates that the marked subject heading is a primary topic; or, after a word, a truncation symbol (wildcard) to retrieve plurals or varying endings |
.ti | Title |
.ot | Original title |
.ab | Abstract |
.hw | Heading word; usually includes subject headings and controlled vocabulary |
.kf | Author keyword heading word (MEDLINE) |
.dq | Candidate term word (Embase) |
.pt | Publication type |
.rn | Registry number |
.nm | Name of substance word (MEDLINE) |
medall | Ovid database code: MEDLINE All, 1946 to present, updated daily |
oemezd | Ovid database code; Embase, 1974 to present, updated daily |
Multidatabase Strategy
(tecartus* or brexucabtagene autoleucel* or brexu cel* or brexucel* or kte x19* or ktex19* or 4MD2J2T8SJ).ti,ab,kf,ot,hw,nm,rn
(autologous* and anti-CD19* and transduced and CD3*).ti,ab,kf,ot,hw,nm,rn
or/1-2
3 use medall
*brexucabtagene autoleucel/
(tecartus* or brexucabtagene autoleucel* or brexu cel* or brexucel* or kte x19* or ktex19*).ti,ab,kf,dq
(autologous* and anti-CD19* and transduced and CD3*).ti,ab,kf,dq
or/5-7
8 use oemezd
9 not (conference abstract or conference review).pt.
4 or 10
remove duplicates from 11
Clinical Trials Registries
ClinicalTrials.gov
Produced by the U.S. National Library of Medicine. Targeted search used to capture registered clinical trials.
[Search -- tecartus OR brexucabtagene autoleucel OR brexu cel OR brexucel OR kte x19 OR ktex19 OR autologous anti-CD19 transduced CD3+]
WHO ICTRP
International Clinical Trials Registry Platform, produced by the World Health Organization. Targeted search used to capture registered clinical trials.
[Search terms -- tecartus OR brexucabtagene autoleucel OR brexu cel OR brexucel OR kte x19 OR ktex19 OR autologous anti-CD19 transduced CD3]
Health Canada’s Clinical Trials Database
Produced by Health Canada. Targeted search used to capture registered clinical trials.
[Search terms -- tecartus OR brexucabtagene autoleucel OR brexu cel OR brexucel OR kte x19 OR ktex19 OR autologous anti-CD19 transduced CD3]
EU Clinical Trials Register
European Union Clinical Trials Register, produced by the European Union. Targeted search used to capture registered clinical trials.
[Search terms -- tecartus OR brexucabtagene autoleucel OR brexu cel OR brexucel OR kte x19 OR ktex19 OR autologous anti-CD19 transduced CD3]
Grey Literature
Search dates: October 03, 2022 – October 17, 2022
Keywords: [tecartus, brexucabtagene autoleucel, brexu cel, brexucel, kte x19, ktex19, autologous anti-CD19 transduced CD3, lymphocytic leukemia, lymphoblastic leukemia, lymphoid leukemia, lymphatic leukemia, lymphocyte leukemia, BCell leukemia, B-cell leukemia, lymphocytic leukaemia, lymphoblastic leukaemia, lymphoid leukaemia, lymphatic laeukemia, lymphocyte leukaemia, BCell leukaemia, B-cell leukaemia, or lymphoma]
Limits: Publication years: 2017-present for guidelines, no limits for other sections
Relevant websites from the following sections of the CADTH grey literature checklist Grey Matters: A Practical Tool for Searching Health-Related Grey Literature were searched:
Health Technology Assessment Agencies
Health Economics
Clinical Practice Guidelines
Drug and Device Regulatory Approvals
Advisories and Warnings
Drug Class Reviews
Clinical Trials Registries
Databases (free)
Internet Search.
Appendix 2: Efficacy Outcomes in SCHOLAR-3
Note that this appendix has not been copy-edited.
Survival
For the fourth secondary objective, the comparison of OS between matched ZUMA-3 and SCA-1 patients, the median OS was not evaluable (NE) (95% CI, 18.20 to NE) in the target group compared to 5.49 months (95% CI, 1.94 to 12.09) in SCA-1 (HR = 0.15; 95% CI, 0.05 to 0.45) (Table 22). In the sensitivity analysis, the median OS was NE in the target group compared to 8.53 months (95% CI, 4.21 to 20.27) in SCA (HR = 0.21; 95% CI, 0.08 to 0.55).13
For the fifth secondary objective, the comparison of RFS between matched ZUMA-3 and SCA-1 patients, the median RFS was 20.47 months (95% CI, 2.79 to NE) in the target group compared to 0.03 months (95% CI, 0.03 to 4.63) in the SCA-1 (HR = 0.18; 95% CI, 0.06 to 0.52) (Table 22). In the sensitivity analysis, the median RFS was 11.53 months (95% CI, 2.99 to NE) in the target group compared to 0.03 months (95% CI, 0.03 to 4.63) in the SCA-1 (HR = 0.45; 95% CI, 0.21 to 0.95).13
Table 22: Summary of Survival in Matched ZUMA-3 and SCA-1 Patients
Detail | mITT | FAS | ||
|---|---|---|---|---|
Target group N = 20 | SCA-1 N = 20 | Target group N = 25 | SCA-1 N = 25 | |
OS | ||||
Death, n (%) | █ ██████ | ██ █████ | █ ██████ | ██ █████ |
Censored, n (%) | ██ █████ | █ ██████ | ██ █████ | █ ██████ |
Alive, n (%) | ██ █████ | █ ██████ | ██ █████ | █ ██████ |
Withdrawal of consent, n (%) | █ | █ | █ | █ █████ |
Reverse KM median follow-up time, months (95%) | ███ ██ ██ | ███ ██ ██ | ███ ██ ██ | ███ ██ ██ |
KM median OS, months (95% CI) | NE (18.20 to NE) | 5.49 (1.94 to 12.09) | NE (NE) | 8.53 (4.21 to 20.27) |
Range, months | 0.72 to 29.83+ | 0.49 to 24.94+ | 1.02 to 30.98+ | 0.03 to 24.94+ |
P-valuea | 0.0001 | 0.0006 | ||
Hazard ratiob (95% CI) | 0.15 (0.05 to 0.45) | reference | 0.21 (0.08 to 0.55) | reference |
RFS | ||||
Events, n (%) | NR | NR | NR | NR |
Censored, n (%) | NR | NR | NR | NR |
Reverse KM median follow-up time, months (95%) | NR | NR | NR | NR |
KM median RFS, months (95% CI) | 20.47 (2.79 to NE) | 0.03 (0.03 to 4.63) | 11.53 (2.99 to NE) | 0.03 (0.03 to 4.63) |
Range, months | ██ ██ ███ | ██ ██ ███ | ██ ███ █ | ██ ████ █ |
P-valuea | 0.0004 | 0.0337 | ||
Hazard ratiob (95% CI) | 0.18 (0.06 to 0.52) | reference | 0.45 (0.21 to 0.95) | reference |
CI = confidence interval; FAS = full analysis set; KM = Kaplan-Meier method; mITT = modified intent to treat; NE = not evaluable; NR = not reported; OS = overall survival; RFS = relapse-free survival; SCA = synthetic control arm.
aFor time-to-event end points, a 2-sided log-rank test was used for testing the difference between ZUMA-3 matched patients and the corresponding SCA group. Note, P-values are descriptive only as there was no control for type I error rate.
bHazard ratios between the 2 comparison groups and the 95% CIs were estimated using a Cox proportional hazard regression model.
Source: Clinical Study Report of SCHOLAR-3, 21-month Update13
The comparison of OS between matched ZUMA-3 and SCA-2 patients was the only analysis conducted between these groups. The median OS was 15.90 months (95% CI, 3.19 to 26.02) in the target group compared to 4.76 months (95% CI, 2.66 to 12.35) in SCA-2 (adjusted HR = 0.55; 95% CI, 0.26 to 1.13) (Table 23). In the sensitivity analysis, the median OS was 9.72 (95% CI, 4.11 to 18.99) in the target group compared to 4.73 months (95% CI, 3.52 to 6.83) in SCA-2 (adjusted HR = 0.66; 95% CI, 0.37 to 1.17).13
Table 23: Summary of Overall Survival in Matched ZUMA-3 and SCA-2 Patients
Detail | mITT | FAS | ||
|---|---|---|---|---|
Target group N = 29 | SCA-2 N = 20 | Target group N = 40 | SCA-2 N = 40 | |
Death, n (%) | ██ █████ | ██ █████ | ██ █████ | ██ ████ |
Censored, n (%) | ██ █████ | █ ██████ | ██ █████ | █ █████ |
Alive on or after data cut-off, n (%) | █ ██████ | █ ██████ | ██ █████ | ███ ███ |
Full withdrawal of consent, n (%) | █ █████ | █ | █ █████ | █ |
Lost to follow-up, n (%) | █ | █ | █ | ███ ███ |
Reverse KM median follow-up time, months (95%) | ██ ██ ███ | ██ ██ ███ | ██ ██ ███ | ██ █ ██ |
KM median OS, months (95% CI) | 15.90 (3.19 to 26.02) | 4.76 (2.66 to 12.35) | 9.72 (4.11 to 18.99) | 4.73 (3.52 to 6.83) |
Range, months | ██ ██ ███ | ██ █ ████ | ██ ██ ███ | ██ █ ███ |
P-valueb | NRc | NRc | ||
Adjusted hazard ratiod (95% CI) | 0.55 (0.26 to 1.13) | reference | 0.66 (0.37 to 1.17) | reference |
CI = confidence interval; FAS = full analysis set; KM = Kaplan-Meier; mITT = modified intent to treat; NR = not reported; OS = overall survival; SCA = synthetic control arm.
aThe full matching approach was used in the sensitivity analysis using FAS to achieve balanced baseline covariates due to the limited sample size. One ZUMA-3 patient was allowed to be matched with multiple SCA patients, which resulted in weights for SCA patients to be presented as decimals.
bFor time-to-event end points, a 2-sided log-rank test was used for testing the difference between ZUMA-3 matched patients and the corresponding SCA group. Note, P-values are descriptive only as there was no control for type I error rate.
cDid not achieve statistical significance.
dHazard ratios between the 2 comparison groups and the 95% CIs were estimated using a Cox proportional hazard regression model adjusted for percentage bone marrow blasts and prior lines of therapy at baseline.
Source: Clinical Study Report of SCHOLAR-3, 21-month Update13
For the final secondary objective, the comparison of OS between all matched ZUMA-3 and SCA patients, the median OS was 25.43 months (95% CI, 15.90 to NE) in the target group compared to 5.49 months (95% CI, 3.32 to 9.23) in SCA (HR = 0.32; 95% CI, 0.18 to 0.58) (Table 24). In the sensitivity analysis, the median OS was 23.06 months (95% CI, 9.92 to NE) in the target group compared to 5.95 months (95% CI, 4.21 to 7.26) in SCA (HR = 0.47; 95% CI, 0.29 to 0.76).13
Of note, the results for the OS in all matched patients should be interpreted with caution as the propensity score matching was based on the 2 separate cohorts (i.e., patients who were either naive to or experienced with blinatumomab and/or inotuzumab) and as such, may have dissimilar distributions of characteristics.
Table 24: Summary of Overall Survival in All Matched Patients
Detail | mITT | FAS | ||
|---|---|---|---|---|
Target group N = 49 | SCA N = 40 | Target group N = 65 | SCA N = 65 | |
Death, n (%) | ██ █████ | ██ █████ | ██ █████ | ██ █████ |
Censored, n (%) | ██ █████ | ██ █████ | ██ █████ | ██ █████ |
Alive on or after data cut-off, n (%) | ██ █████ | ██ █████ | ██ █████ | ████ ███ |
Full withdrawal of consent, n (%) | █ █████ | █ | █ █████ | █ █████ |
Lost to follow-up, n (%) | █ | █ | █ | ███████ |
Reverse KM median follow-up time, months (95%) | ██ █ ███ | ██ █████ | ██ █ ███ | ██ █████ |
KM median OS, months (95% CI) | 25.43 (15.90 to NE) | 5.49 (3.32 to 9.23) | 23.06 (9.92 to NE) | 5.95 (4.21 to 7.26) |
Range, months | ██ █ ███ | ██ █ ███ | ██ █████ | ██ █ ███ |
P-valueb | 0.0001 | 0.0011 | ||
Hazard ratioc (95% CI) | 0.32 (0.18 to 0.58) | reference | 0.47 (0.29 to 0.76) | reference |
CI = confidence interval; FAS = full analysis set; KM = Kaplan-Meier; mITT = modified intent to treat; NE = not evaluable; OS = overall survival; SCA = synthetic control arm.
aThe full matching approach was used in the sensitivity analysis using FAS to achieve balanced baseline covariates due to the limited sample size. One ZUMA-3 patient was allowed to be matched with multiple SCA patients, which resulted in weights for SCA patients to be presented as decimals.
bFor time-to-event end points, a 2-sided log-rank test was used for testing the difference between ZUMA-3 matched patients and the corresponding SCA group. Note, P-values are descriptive only as there was no control for type I error rate.
cHazard ratios between the 2 comparison groups and the 95% CIs were estimated using a Cox proportional hazard regression model.
Source: Clinical Study Report of SCHOLAR-3, 21-month Update13
Treatment Response
The comparison of OCR rate between matched ZUMA-3 and SCA-1 patients was the primary efficacy outcome analysis. The estimated proportion of patients who achieved OCR at week 24 was 85% (95% CI, 62.1 to 96.8) in the target group compared to 35% (95% CI, 15.4 to 59.2) in SCA-1 (OR = 10.5; 95% CI, 2.3 to 48.7) (Table 25). In the sensitivity analysis, the estimated proportion of patients who achieved OCR at week 24 was 72% (95% CI, 50.6 to 87.9) in the target group compared to 36% (95% CI, 18.0 to 57.5) in SCA-1 (OR = 4.6; 95% CI, 1.4 to 15.1).13
For the second secondary objective, the comparison of CR rate between matched ZUMA-3 and SCA-1 patients, the estimated proportion of patients who achieved CR at week 24 was 75% (95% CI, 50.9 to 91.3) in the target group compared to 30% (95% CI, 11.9 to 54.3) in SCA-1 (OR = 7.0; 95% CI, 1.7 to 28.2) (Table 25). In the sensitivity analysis, the estimated proportion of patients who achieved ██ ██ ████ ██ ███ ███ ████ ███ ████ ██ █████ ██ ███ ██████ █████ ████████ ██ ███ ████ ███ ████ ██ █████ ██ █████ ███ █ ████ ███ ███ ███ ██ ████████
Table 25: Summary of Treatment Response in Matched ZUMA-3 and SCA-1 Patients
Response category | mITT | FAS | ||
|---|---|---|---|---|
Target group N = 20 | SCA-1 N = 20 | Target group N = 25 | SCA-1 N = 25 | |
OCR | ||||
Patients with OCR at week 24, n (%) | 17 (85.0) | 7 (35.0) | 18 (72.0) | 9 (36.0) |
95% CI | 62.1 to 96.8 | 15.4 to 59.2 | 50.6 to 87.9 | 18.0 to 57.5 |
Rate difference (95% CI) | 50.0 (17.9 to 73.7) | reference | 36.0 (7.4, 60.4) | reference |
Odds ratioa (95% CI) | 10.5 (2.3 to 48.7) | reference | 4.6 (1.4 to 15.1) | reference |
P-valueb | 0.0031 | 0.0222 | ||
CR | ||||
Patients with CR at week 24, n (%) | 15 (75.0) | 6 (30.0) | 16 (64.0) | 8 (32.0) |
95% CI | 50.9 to 91.3 | 11.9 to 54.3 | 42.5 to 82.0 | 14.9 to 53.5 |
Rate difference (95% CI) | 45.0 (12.2 to 70.8) | reference | 32.0 (2.5 to 57.6) | reference |
Odds ratioa (95% CI) | 7.0 (1.7 to 28.2) | reference | 3.8 (1.2 to 12.2) | reference |
P-valueb | 0.0104 | 0.0465 | ||
CI = confidence interval; CR = complete remission; FAS = full analysis set; mITT = modified intent to treat; OCR = overall complete remission; SCA = synthetic control arm.
aThe odds ratio and associated 95% CI and 2-sided P value were estimated from a logistic regression model with treatment group (ZUMA-3 versus SCA) as a single covariate. Note, P-values are descriptive only as there was no control for type I error rate.
Source: Clinical Study Report of SCHOLAR-3, 21-month Update13
Health-Related Quality of Life
Health-related quality of life was not assessed in SCHOLAR-3.
Allogeneic Stem Cell Transplant Rate
For the third secondary objective, the comparison of allo-SCT rate between the matched ZUMA-3 and SCA-1 patients, the estimated proportion of patients who had an allo-SCT was 35% (95% CI, 15.4 to 59.2) in the target group compared to 20% (95% CI, 5.7 to 43.7) in SCA-1 (OR = 2.2; 95% CI, 0.5 to 9.0) (Table 26). In the sensitivity analysis, the estimated proportion of patients who had an allo-SCT was 24% (95% CI, 9.4 to 45.1) in the target group compared to 32% (95% CI, 14.9 to 53.5) in SCA-1 (OR = 0.7; 95% CI, 0.2 to 2.3).13
Table 26: Summary of Allo-SCT in Matched ZUMA-3 and SCA-1 Patients
Detail | mITT | FAS | ||
|---|---|---|---|---|
Target group N = 20 | SCA-1 N = 20 | Target group N = 25 | SCA-1 N = 25 | |
Patients who had an allo-SCT, n (%) | 7 (35.0) | 4 (20.0) | 6 (24.0) | 8 (32.0) |
95% CI | 15.4 to 59.2 | 5.7 to 43.7 | 9.4 to 45.1 | 14.9 to 53.5 |
Rate difference (95% CI) | 15.0 (−13.7 to 42.4) | reference | −8.0 (−33.0 to 17.7) | reference |
Odds ratioa (95% CI) | 2.2 (0.5 to 9.0) | reference | 0.7 (0.2 to 2.3) | reference |
P-valueb | 0.4801 | 0.7536 | ||
allo-SCT = allogeneic stem cell transplant; CI = confidence interval; FAS = full analysis set; mITT = modified intent to treat; SCA = synthetic control arm.
aThe odds ratio and associated 95% CI and 2-sided P value were estimated from a logistic regression model with treatment group (ZUMA-3 versus SCA) as a single covariate.
bP-values are descriptive only as there was no control for type I error rate.
Source: Clinical Study Report of SCHOLAR-3, 21-month Update13
Intensive Care Unit Admission
Intensive care unit admission was not assessed in SCHOLAR-3.
Appendix 3: Description and Appraisal of Outcome Measures
Note that this appendix has not been copy-edited.
Aim
To describe the outcome measure, EQ-5D-5L, and review its measurement properties, including validity, reliability, responsiveness to change, and the minimal important difference.
Findings
Table 27: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
EQ-5D-5L | A generic measure of health status comprised of 2 parts. The descriptive system assesses health in 5 dimensions (mobility, self-care, usual activities, pain/discomfort and anxiety/depression).29 Each dimension has 5 increasing levels of severity/response (no problems, slight problems, moderate problems, severe problems and unable to/extreme problems). A unique health state profile is generated as a 5-digit code (e.g., 12345 indicates no problems with mobility, slight problems with self-care, moderate problems with usual activities, severe pain or discomfort, and extreme anxiety or depression).29 The health state can be converted to a summary index score based on societal (countries/regions) preference weights for the health state. Index scores range from less than 0 (negative values represent worse than dead, which is represented by 0) to 1 (full health), with higher scores representing higher health utility.29 Patient’s perceived health status on that day is also rated using the VAS, ranging from 0 (worst imaginable health) to 100 (best imaginable health).29 | Validity: The validity and interpretability of the EQ-5D-5L, QLQ-PBM, and EORTC-8D were evaluated in patients with acute leukemia who received first-line treatment in the HOVON clinical trials in the Netherlands between 1999 and 2011 (N = 111).a The mean age was 51 years (SD = 13.4), and 19 patients (17%) had ALL; the remaining 92 patients (83%) had acute myeloid leukemia.30 For the purposes of this review, only the results for the EQ-5D-5L are reported here. For content validity, all EQ-5D-5L dimensions, with the exception of self-care, demonstrated a strong correlation (Spearman correlation coefficients > 0.50) with certain domains of the QLQ-PBM and EORTC-8D.30 For construct validity, the EQ-5D-5L utility score demonstrated strong correlation (Spearman correlation coefficient > 0.50) with the VAS and QL scale. The EQ-5D-5L was able to distinguish between patients with different health statuses according to the VAS and QL scale (subgroups were determined using the quartile scores observed in this patient population). All standardized effect sizes were moderate to strong (Cohen’s d > 0.50), with the exception of detecting differences between the second and third quartile of the VAS.30 Note, the authors acknowledged that the lack of cancer-relevant domains, such as fatigue and cognitive functioning, is a limitation of the EQ-5D-5L. A high ceiling effect was reported for the EQ-5D-5L in patients with acute leukemia (30% of patients reported full health on the EQ-5D-5L vs. 14% and 16% on the QLQ-PBM and EORTC-8D, respectively); the largest ceiling effect was reported for the self-care dimension.30 Reliability: Not identified in patients with ALL. Responsiveness to change: Not identified in patients with ALL. | Not identified in patients with ALL |
ALL = acute lymphoblastic leukemia; EORTC-8D = European Organization of Randomized Controlled Trials 8 Dimension; HOVON = Hemato-Oncologie voor Volwassenen Nederland (the Haemato Oncology Foundation for Adults in the Netherlands); MID = minimal important difference; QL = global quality of life scale of the QLQ-C30; QLQ-C30 = Quality of Life Questionnaire for Cancer; QLQ-PBM = Quality of Life Questionnaire Preference-Based Measure; SD = standard deviation; VAS = visual analogue scale.
aNote, the EQ-5D-5L utilities were derived from the UK and Dutch tariffs; the QLQ-PBM was valued by the Dutch general public and the EORTC-8D was valued by the UK general public.
Pharmacoeconomic Review
Abbreviations
AE
adverse event
ALL
acute lymphoblastic leukemia
allo-SCT
allogeneic stem cell transplant
brexu-cel
brexucabtagene autoleucel
CAR T
chimeric antigen receptor T
CI
confidence interval
EFS
event-free survival
ICER
incremental cost-effectiveness ratio
ICU
intensive care unit
LY
life-year
mITT
modified intention to treat
OS
overall survival
PSM
partitioned survival model
QALY
quality-adjusted life-year
R/R
relapsed or refractory
tisa-cel
tisagenlecleucel
TKI
tyrosine kinase inhibitor
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Brexucabtagene autoleucel (Tecartus), cell suspension in 68 mL infusion bag, for single IV infusion |
Submitted price | Brexucabtagene autoleucel: $533,523 per administration |
Indication | Treatment of adult patients with relapsed or refractory (R/R) B-cell precursor acute lymphoblastic leukemia (ALL) |
Health Canada approval status | NOC |
Health Canada review pathway | Priority review |
NOC date | November 16, 2022 |
Reimbursement request | As per indication |
Sponsor | Gilead Sciences Canada Inc. |
Submission history | Yes Indication: Adult patients with relapsed or refractory (R/R) mantle cell lymphoma who have received treatment with a Bruton tyrosine kinase inhibitor Recommendation date: August 24, 2021 Recommendation: Reimburse with clinical criteria and/or conditions |
ALL = acute lymphoblastic leukemia; NOC = Notice of Compliance; R/R = relapsed or refractory.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Decision tree followed by PSM |
Target population | Adult patients with R/R B-cell precursor ALL |
Treatment | Brexucabtagene autoleucel (Tecartus) |
Comparators | Blinatomumab ± TKIs (dasatinib or ponatinib) Inotuzumab ± TKIs (dasatinib or ponatinib) Salvage chemotherapy (FLAG-IDA or hyper CVAD) |
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | Lifetime (59 years) |
Key data sources | ZUMA-3 study, INO-VATE trial, and TOWER trial |
Submitted results |
|
Key limitations |
|
CADTH reanalysis results |
|
ALL = acute lymphoblastic leukemia; allo-SCT = allogeneic stem cell transplant; brexu-cel = brexucabtagene autoleucel; EFS = event-free survival; FLAG-IDA = fludarabine, cytarabine, idarubicin, filgrastim; hyper CVAD = alternating courses of cyclophosphamide, vincristine, doxorubicin, and dexamethasone with courses of methotrexate and cytarabine; ICER = incremental cost-effectiveness ratio; LY = life-year; OS = overall survival; PSM = partitioned survival model; QALY = quality-adjusted life-year; R/R = relapsed or refractory; TKI = tyrosine kinase inhibitor.
aExtendedly dominated = in a sequential analysis, when 1 intervention has a higher ICER than the next more costly comparator.
Conclusions
Evidence from the ZUMA-3 study suggests that brexucabtagene autoleucel (brexu-cel) may be associated with clinically meaningful event-free survival (EFS) (11.6 months; 95% confidence interval [CI], 2.7 to 20.5 months) and overall survival (OS) (25.4 months; 95% CI, 16.2 months to not estimable) for the treatment of patients with relapsed or refractory (R/R) B-cell acute lymphoblastic leukemia (ALL). As noted in the CADTH Clinical Review, this benefit was based on 21 months of follow-up; therefore, the long-term benefit of brexu-cel treatment is currently unknown. However, because the ZUMA-3 study was a single-arm trial, comparative efficacy of brexu-cel to inotuzumab plus or minus tyrosine kinase inhibitors (TKIs), blinatumomab plus or minus TKIs, and salvage chemotherapy is unknown. The CADTH Clinical Review concluded that inferences regarding the efficacy of brexu-cel could not be made due to data immaturity and that there is insufficient evidence to support brexu-cel as a curative treatment. Therefore, uncertainties remain regarding the magnitude of the clinical benefit from the treatment with brexu-cel. No conclusions could be made regarding health-related quality of life due to missing data.
In addition to the aforementioned limitations with the clinical evidence, CADTH identified several limitations with the sponsor’s economic submission, including the use of naive indirect comparisons to derive comparative clinical efficacy for brexu-cel, the overestimation of overall survival for brexu-cel, the lack of face validity of long-term OS for comparators, the overestimation of postprogression effects of brexu-cel, the use of a model structure that assumes independence of EFS and OS, the assumption of a cure point of 2 years, and the lack of a consistent approach when incorporating comparator treatment durations.
Given the uncertainty associated with the comparative treatment effects and the limitations with the modelling approach, CADTH could not estimate a robust single base-case estimate of cost-effectiveness for brexu-cel; instead, 2 CADTH reanalyses were conducted based on the possible range of OS benefits (Figure 4, Appendix 4). If brexu-cel is associated with a 5-year and 25-year OS of 26% and 6%, respectively (reanalysis 1; log normal distribution), the incremental cost-effectiveness ratio (ICER) for brexu-cel relative to salvage chemotherapy was estimated to be $164,545 per quality-adjusted life-year (QALY). In this reanalysis, blinatumomab plus or minus TKIs and inotuzumab plus or minus TKIs did not lie in the cost-effectiveness frontier because they were extendedly dominated by brexu-cel. A price reduction of 71% would be required for brexu-cel to be considered cost-effective at a $50,000-per-QALY threshold. If brexu-cel’s OS benefit is reduced to a 5-year and 25-year OS of 21% and 0%, respectively (reanalysis 2; Weibull distribution), the ICER increases to $679,053 per QALY relative to inotuzumab plus or minus TKIs. In this reanalysis, blinatumomab plus or minus TKIs did not lie on the cost-effectiveness frontier because it was extendedly dominated by inotuzumab plus or minus TKI. A price reduction of 88% would be required to achieve cost-effectiveness at a $50,000 per QALY threshold.
CADTH explored the next most optimistic OS extrapolation (generalized gamma distribution) in a scenario analysis, which resulted in a 5-year and 25-year OS of 36% and 21%, respectively. However, this scenario analysis lacked face validity because it overestimated the proportion of patients alive at the 40-year and 50-year time horizon (13% and 4%, respectively). Furthermore, it inflated brexu-cel’s postprogression survival benefit, which was fivefold higher than salvage chemotherapy (5.46 versus 1.02 life-years (LYs) in the progressed disease state for brexu-cel and salvage chemotherapy, respectively).
Although the CADTH reanalyses attempted to address the identified limitations of the sponsor’s economic submission, significant uncertainty still exists due to a lack of comparative and long-term efficacy data, uncertainties in the extrapolation period, and an overestimation of postprogression benefit for brexu-cel. Therefore, CADTH’s estimates of cost-effectiveness were likely biased in favour of brexu-cel.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process.
Patient input was received from the Leukemia and Lymphoma Society of Canada. Input from this group was based on an online survey completed by 22 patients from Canada. Patient input highlighted that the symptoms that most affected patients’ daily quality of life were fatigue or weakness, loss of appetite or weight loss, bone or joint pain, headaches, blurred vision, nausea, or vomiting. A total of 73% of patients noted that ALL significantly affected their ability to work, exercise, and continue everyday activities. ALL interrupted patients’ life goals, such as their career and schooling, and had a significant burden on patients’ psychological health through stress, anxiety, and overall worry levels. Half of the patients surveyed indicated that ALL-related issues required more than 5 visits to the hospital per month, with 3 patients having to travel more than 100 km to access care. Patients highlighted that the need to travel had a negative impact on their lives by incurring extensive expenses from accommodations, disrupting daily activities and routines, and causing emotional hardship, such as being away from their support systems for extended periods of time. Patient input highlighted the following criteria they would use to consider treatment options: certainty of ALL response to treatment, improvement in quality of life, coverage by insurance, and improved length of life. In addition, patients were concerned about the potential side effects of new therapies. A total of 2 patients had experience with brexu-cel: 1 patient had a positive overall experience with manageable side effects and overall improvement in quality of life, and the other patient reported that brexu-cel negatively affected their quality of life due to serious side effects including slurred speech, fever, chills, cough, other signs of infection, feeling tired or lightheaded, fast or irregular heartbeat, and the inability to resume normal activities or return to work.
Clinician input was received from the Ontario Health – Cancer Care Ontario Complex Malignant Hematology Group and Cell Therapy Transplant Canada. Clinician feedback highlighted that existing chimeric antigen receptor T (CAR T) products in Canada for ALL have limited inclusion criteria based on age (only available for patients aged younger than 26 years). Clinician input indicated that current treatment in this population included blinatumomab or inotuzumab, the clinical goal of which was to bridge eligible patients who achieve a remission to allogeneic stem cell transplant (allo-SCT). For patients who were not eligible for an allo-SCT, the treatment goals were to prolong life and delay disease relapse.
CADTH participating drug plans highlighted several implementation and economic considerations, including concerns with access to brexu-cel and concerns with the financial burden incurred by patients and their families due to travelling and accommodation. Drug plan input highlighted concerns associated with the need for additional resources (e.g., nursing, hospital bed, intensive care unit) to treat adverse events (AEs), and issues with jurisdictional capacity. In addition, they indicated concerns about whether there is sufficient evidence to support re-treatment with brexu-cel and whether brexu-cel could be used in patients with an Eastern Cooperative Oncology Group (ECOG) performance status of less than 1. For patients aged between 18 and 25 years, drug plans questioned what criteria would be used to choose between brexu-cel and tisagenlecleucel. Finally, drug plans highlighted potential issues with patient privacy and patient cell ownership due to the fact that CAR T is manufactured by a US-based company, which is outside of Canadian jurisdiction.
Some of these concerns were addressed in the sponsor’s model:
The sponsor’s submitted model accounted for quality of life and length of life by using QALYs as the primary outcome.
The sponsor considered costs and disutilities associated with grade 3 AEs, which occurred in more than 5% of patients.
CADTH was unable to address the following concerns raised from stakeholder input:
The burden of out-of-pocket costs incurred to receive treatment (travelling and accommodation).
The need for additional resources to treat adverse reactions.
Access and capacity constraints associated with the number of centres specialized and accredited to provide CAR T-cell treatment.
Economic Review
The current review is for brexucabtagene autoleucel (Tecartus) for adult patients with R/R B-cell precursor ALL.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
Brexu-cel is indicated for the treatment of adult patients with R/R B-cell precursor ALL. The sponsor submitted a cost-utility analysis comparing brexu-cel to blinatumomab plus or minus TKIs, inotuzumab plus or minus TKIs, and salvage chemotherapy in the base-case analysis.1 In addition, tisagenlecleucel (tisa-cel), which is recommended for the treatment of children and young adults (aged 3 to 25 years) with R/R B-cell ALL, was considered as a comparator in a scenario analysis. The use of TKIs (dasatinib and ponatinib) for treatment was dictated by whether patients were Philadelphia chromosome positive. The salvage chemotherapy comparator was defined as a weighted basket of 2 regimens, which included FLAG-IDA (fludarabine, cytarabine, filgrastim, idarubicin and hyper CVAD [alternating courses of cyclophosphamide, vincristine, doxorubicin, and dexamethasone with courses of methotrexate and cytarabine]). The modelled population aligned with the Health Canada indication and the sponsor’s reimbursement request.
Brexu-cel is a CD19-directed genetically modified autologous T-cell immunotherapy individually prepared from patients’ peripheral blood mononuclear cells.2 It is available as a cell suspension for infusion containing a target dose of 1 × 106 CAR T-cells per kg of body weight, with a maximum of 1 × 108 CAR T-cells for patients weighing 100 kg or more. It is provided as a single-dose, one-time treatment. The sponsor’s submitted price of brexu-cel was $533,523 per infusion, not including costs associated with pretreatment (i.e., leukapheresis, bridging therapy, conditioning chemotherapy) and postinfusion management (i.e., intensive care unit and non- intensive care unit inpatient stay).
The comparator for this analysis consisted of blinatumomab plus or minus TKIs, inotuzumab plus or minus TKIs, and salvage chemotherapy. The sponsor estimated that the 28-day drug acquisition cost was $62,542 for blinatumomab and $115,247 for inotuzumab. Based on a 50% weighted cost between dasatinib and ponatinib, the sponsor estimated that the 28-day TKI cost was $6,672. The drug cost of salvage chemotherapy over 28 days, considering a 50% weighted cost between FLAG-IDA and hyper CVAD, was $1,544. In addition, tisa-cel costs were estimated to be $450,000 per one-time treatment. The sponsor did not incorporate vial sharing in the calculation of drug acquisition costs.
The outcomes of interest were QALYs and LYs. The analysis took the perspective of a Canadian publicly funded health care payer. The time horizon in the base case was specified by the sponsor as a lifetime time horizon (59 years) with weekly cycles. The discount rate for costs and outcomes was 1.5% annually.
Model Structure
The sponsor submitted a partitioned survival model (PSM), including 3 health states: event-free, progressed disease, and death (Figure 1, Appendix 3). For brexu-cel, the sponsor used a decision tree to account for patients who were eligible to receive brexu-cel, but ultimately fail to receive it due to a range of reasons, including AEs, death, and other reasons (e.g., consent withdrawn, lack of product availability, eligibility not met, and patient clinical deterioration after the product was not successfully manufactured from leukapheresis attempts) (Figure 2, Appendix 3). The decision tree was then linked to the PSM. The sponsor claimed that patients who failed to receive brexu-cel were assumed to be equally distributed across the other comparator treatments and incur the same survival outcomes as that comparator.
The proportion of patients who were event-free, experienced disease progression, or dead at any time over the model’s time horizon was derived from nonmutually exclusive survival curves. All patients entered the model in the event-free state. The proportion of patients in the progressed disease state was calculated as the proportion alive (based on the OS curve) minus the proportion of patients alive and event-free (based on the EFS curve). In the model, EFS was capped by OS. Patients in the event-free state were assumed to receive treatment for a defined number of treatment cycles. After disease progression, patients were assumed to receive subsequent treatments.
Model Inputs
The modelled cohort’s characteristics were based on the ZUMA-3 study (mean age 43 years, 1.92 m2 body surface area, 64% male), a single-arm trial that enrolled 71 patients with R/R B-cell precursor ALL. The decision tree for the brexu-cel treatment was informed by data from the ZUMA-3 study, where 55 out of 71 (77.4%) patients were ultimately treated with brexu-cel. Reasons for not receiving brexu-cel included AEs, not meeting eligibility criteria after leukapheresis, and manufacturing failure. EFS and OS estimates for brexu-cel were also obtained from the ZUMA-3 study.3
EFS and OS estimates for blinatumomab and inotuzumab were obtained from the TOWER trial and the INO-VATE trial, respectively.4,5 The proportion of patients receiving TKIs in addition to their underlying treatment (27%) was based on literature.6,7 The addition of TKIs to blinatumomab and inotuzumab was assumed to incur additional treatment costs but to have no impact on treatment efficacy (i.e., efficacy inputs reflected monotherapy blinatumomab or inotuzumab). EFS and OS estimates for salvage chemotherapy were obtained from the INO-VATE trial, in which most patients received a FLAG-IDA regimen (69.7% received FLAG-IDA, 19.1% received cytarabine plus mitoxantrone, and 11.2% received high-dose cytarabine).4 The sponsor assumed that the hyper CVAD regimen had the same efficacy as the FLAG-IDA regimen (salvage chemotherapy consisted of both regimens in a 50/50 split).
The sponsor conducted a naive indirect comparison between brexu-cel’s EFS and OS and blinatumomab, inotuzumab, and salvage chemotherapy’s EFS and OS. For brexu-cel, Kaplan-Meier estimates of EFS and OS from the ZUMA-3 study were used to fit parametric survival curves to extrapolate the observed data beyond the trial period (maximum observation: 26 months).3 The same method was used to fit parametric distributions for each of the comparators using EFS and OS data from the TOWER trial for blinatumomab (approximate median follow-up for OS: 12 months) and the INO-VATE trial for inotuzumab and salvage chemotherapy (median follow-up: 29.6 months).4,5 The choices between curves were based on Akaike Information Criteria, Bayesian Information Criteria, expert opinion, and visual inspection. For the scenario including tisa-cel, its OS and EFS were assumed to be the same as for brexu-cel.
The sponsor incorporated a cure point at the 2-year time point of the model. Patients who were event-free at the cure time point were deemed functionally cured and incurred an age-adjusted and sex-adjusted background mortality, sourced from Statistics Canada life tables.8 However, that background mortality was further increased by fourfold based on literature, to represent the patient population cured of B-cell precursor ALL.9
The duration of treatment for the comparators was obtained from the literature. The sponsor assumed an average of 2.24 cycles of therapy for blinatumomab, a maximum of 5 28-day cycles and 1 21-day cycle for inotuzumab, a maximum of 4 cycles for salvage chemotherapy, and 90 days for TKIs. In the base-case analysis, the sponsor used the maximum treatment cycles for inotuzumab, and salvage chemotherapy.
In addition to brexu-cel acquisition costs, the model also included the acquisition costs of bridging and conditioning chemotherapy, which were used to stabilize the patient’s condition while brexu-cel was being manufactured and for lymphodepletion, respectively. Bridging therapy was incorporated as a weighted average of bridging chemotherapy regimens observed in the ZUMA-3 study, while conditioning therapy was included as an average of 3 chemotherapy doses based on data from the ZUMA-3 study and a market information database. The sponsor assumed that 100% and 35% of patients would receive the bridging and conditioning chemotherapy in the outpatient setting, respectively.
For patients who fail treatment, the sponsor assumed that 41.8% would proceed to subsequent therapy, based on the frequency observed in the ZUMA-3 study.3 Subsequent treatment consisted of inotuzumab plus or minus ponatinib, cyclophosphamide and dexamethasone, or blinatumomab. The distribution of each subsequent therapy’s frequency depended on prior treatment and was based on the ZUMA-3 study and the sponsor’s assumption. In addition, the sponsor assumed that patients could receive subsequent allo-SCT. The proportion of patients undergoing allo-SCT by treatment arm was informed by literature.3-5
Health state utility values were obtained for preinfusion, event-free, and the progressed state from the ZUMA-3 study, based on EQ-5D-5L data valued by using Canadian preference weights. Health state utility values for cured patients was obtained from a blinatumomab submission to the Scottish Medicine Consortium and based on an unpublished study.
The incidence of AEs for each comparator was taken from each individual clinical trial as follows: for brexu-cel, data on grade 3 or higher AEs occurring in 5% or more of the population, including AEs occurring pretreatment (i.e., after conditioning chemotherapy and leukapheresis), were obtained from the ZUMA-3 study; for blinatumomab, data on grade or higher 3 AEs occurring in 5% or more of the population in the first cycle of therapy were obtained from the TOWER trial; for inotuzumab, data on grade 3 or higher AEs occurring in 2% or more of the population were obtained from the INO-VATE trial; for salvage therapy, data on AEs were pooled from the standard of care treatment arms of the INO-VATE and TOWER trials; and for ponatinib, data of any grade AE occurring in 20% or more of the total population were taken from the phase II PACE trial. Disutility decrements associated with AEs were multiplied by the duration of each event and applied to the first cycle of the model. The duration of AEs was obtained from the ZUMA-1 trial. The disutility values were obtained from multiple sources, including the literature and assumptions.
The model included costs related to drug acquisition and administration, subsequent treatment after disease progression, AEs and associated treatment costs, health care resource use, and mortality costs. Drug acquisition costs for brexu-cel were based on the sponsor’s submitted price, while the drug acquisition costs of comparators were retrieved from previous CADTH reviews and the Ontario Drug Benefit Formulary.10 Administration costs were obtained from the Ontario Schedule of Benefits, with the duration of administration for each treatment taken from the literature. The cost of drug acquisition for subsequent treatments was also obtained from the Ontario Drug Benefit Formulary.10 The cost of treating AEs was assumed to be equal to a doctor’s visit, which was obtained from the Ontario Case Costing Initiative.11
Health care resource use was assumed to include consultant visits, imaging (e.g., echocardiogram, electrocardiogram), and laboratory diagnostics, with the frequency of use assumed to vary by whether the patient was on treatment with brexu-cel or comparators, and their specific health state. Estimates of frequency of use were obtained from clinical expert input, and costs were obtained from the Ontario Schedule of Benefits. End-of-life costs were derived from the literature and were applied as a 1-off cost.12
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically (1,000 iterations for the base case and scenario analyses). The deterministic and probabilistic results were similar.
Base-Case Results
In the sponsor’s base-case analysis, treatment with brexu-cel was associated with incremental costs of $81,581 and a gain of 4.11 QALYs compared to salvage chemotherapy over the lifetime (59-year) time horizon, resulting in an ICER of $58,177 per QALY gained (Table 3). Treatment with brexu-cel was both more costly and produced more QALYs than all comparators. The probability of brexu-cel being cost-effective at a willingness-to-pay threshold of $50,000 per QALY compared to salvage chemotherapy was 38%. In the sponsor’s base case, 87% of predicted brexu-cel’s QALYs were generated through extrapolation beyond available ZUMA-3 study data (maximum observation of 26 months). At the end of the 59-year time horizon, more than 1% of brexu-cel patients remained alive. The sponsor also conducted a scenario analysis to determine the cost-effectiveness of brexu-cel when tisa-cel was included as comparator (Table 4).
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Total QALYs | Sequential ICER ($ per QALY) |
|---|---|---|---|
Salvage chemotherapy | $173,407 | 0.71 | Reference |
Blinatumomab ± TKIs | $357,243 | 2.36 | Extendedly dominated |
Inotuzumab ± TKIs | $506,087 | 3.72 | Extendedly dominated |
Brexu-cel | $587,667 | 7.83 | $58,177 vs. salvage chemotherapy |
brexu-cel = brexucabtagene autoleucel; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; TKI = tyrosine kinase inhibitor; vs. = versus.
Source: Sponsor’s pharmacoeconomic submission.
Table 4: Summary of the Sponsor’s Scenario Analysis Results, Including Tisa-cel
Drug | Total costs ($) | Total QALYs | Sequential ICER ($ per QALY) |
|---|---|---|---|
Salvage chemotherapy | $172,265 | 0.71 | Reference |
Blinatumomab ± TKIs | $356,525 | 2.28 | Extendedly dominated |
Inotuzumab ± TKIs | $504,866 | 3.73 | Extendedly dominated |
Tisa-cel | $511,635 | 7.38 | $50,884 vs. salvage chemotherapy |
Brexu-cel | $587,871 | 7.38 | Dominated by tisa-cel |
brexu-cel = brexucabtagene autoleucel; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; tisa-cel = tisagenlecleucel; TKI = tyrosine kinase inhibitor; vs. = versus.
Sensitivity and Scenario Analysis Results
The sponsor provided several pairwise scenario and sensitivity analyses, including adopting a shorter time horizon (i.e., 30 years), a change in discount rates (i.e., 0 and 3%), an alternative patient population (i.e., intention to treat, modified intention to treat [mITT], phase II, phase I and II), an alternative cure point (i.e., 4 years), and including tisa-cel as a comparator. The majority of scenarios included by the sponsor had little impact on the ICER, with the exception being changing the population to mITT (100% eligibility for brexu-cel), which increased the ICER to $100,494 versus inotuzumab plus or minus TKI. Further, when tisa-cel was included as a comparator, brexu-cel was dominated by tisa-cel, which was assumed to have the same efficacy but was also less costly.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis:
Uncertainty in the clinical efficacy of brexu-cel: The ZUMA-3 study, which was used to inform the efficacy of brexu-cel, was a phase II trial that consisted of 71 patients who were enrolled between October 2018 and October 2019. Although 71 patients were enrolled, only 55 patients received treatment with brexu-cel and were included in the primary analyses. Due to the small sample size, the CADTH Clinical Review Report noted that it was difficult to determine if an observed improvement would be a true finding. Also, as noted in the CADTH Clinical Review Report, the median OS and median relapse-free survival for brexu-cel were smaller in the full analysis set (n = 77) than the mITT population (n = 55). One of the reasons for this difference was that the index date for the full analysis set was defined as all enrolled patients (who were leukopharesed) and included the full CAR T protocol period, which represented more closely a true intention-to-treat population, while the mITT index date was set at the date of infusion. The mITT therefore characterized a select population and the results may not be generalizable to clinical practice.
As of September 2020, the median follow-up time was 12.4 months; however, based on the most recent data cut-off (July 2021), the median potential follow-up was 20.5 months. As of the July 2021 data cut-off, 50% of patients had been censored. The benefit provided by brexu-cel beyond the study duration was informed by extrapolation. The appropriateness and quality of any extrapolation is dependent on the quantity of observed data available. As noted in the CADTH Clinical Review Report, although follow-up time was considered appropriate to assess response to treatment, it was considered to be immature for assessing survival outcomes. The small sample, the short duration of follow-up, and the high amount of censoring in the trial resulted in considerable uncertainty in the clinical efficacy of brexu-cel.
The sponsor’s projection of long-term clinical benefit of brexu-cel lacked face validity. First, the sponsor’s choice for brexu-cel’s OS extrapolation curve resulted in 6% of patients remaining alive after a time horizon of 50 years (mean age: 93 years). Clinical experts consulted during this review considered this an overestimate and highlighted the high degree of uncertainty around OS estimates. Second, in the sponsor’s choice for extrapolating brexu-cel’s OS, the mortality risk decreased substantially after 5 years, at which time point approximately 40% of patients were alive (Figure 3, Appendix 3). However, the EFS curve estimated that approximately 23% of patients would be cured by brexu-cel at 5 years. This difference between EFS and OS implied that for those 17% of patients whose disease progressed, the mortality risk would be greatly decreased and similar to the mortality rate of cured patients, which lacks face validity. Finally, the OS benefit may be influenced by subsequent treatments that were received, the timing of when they were received, the duration of treatment, and the presence of treatment with allo-SCT, which all add to the uncertainty in extrapolating OS estimates.
Given the overestimate in OS extrapolation, CADTH revised the parametric distribution for brexu-cel. However, due to the uncertainty associated with brexu-cel’s efficacy, CADTH could not derive a single, robust base-case estimate for the cost-effectiveness of brexu-cel. Instead, the CADTH reanalysis explored 2 OS extrapolations: Reanalysis 1 used a log normal distribution, which resulted in brexu-cel having a 5-year and 25-year OS of 26% and 6%, respectively; and reanalysis 2 used a Weibull distribution, which generated a OS extrapolation curve with a 5-year and 25-year OS of 21% and 0%, respectively (Figure 4, Appendix 4). These distributions were aligned with clinical experts’ expectations of plausible OS over time.
Given the uncertainty around brexu-cel’s long-term OS estimates, CADTH conducted scenario analyses to assess its impact on the cost-effectiveness of brexu-cel by altering the time horizon to 5 years, which corresponds to the cure point used in the CADTH reanalysis. CADTH also conducted a scenario analysis exploring the next most optimistic extrapolation of brexu-cel’s OS, using a generalized gamma distribution (5-year and 25-year OS of 36% and 21%, respectively).
Comparative clinical efficacy of brexu-cel is unknown. There have been no head-to-head trials of brexu-cel to any of the comparators included in the economic model. Given the nonrandomized, single-arm design of the ZUMA-3 study, the interpretation of all outcomes was hampered by the lack of a control group, which makes the relative magnitude of any benefits to a comparator highly uncertain. The sponsor provided a matching-adjusted indirect comparison as part of its submission to CADTH; however, it was not utilized in the pharmacoeconomic submission because the sponsor deemed the results of the naive indirect comparison more clinically plausible than those of the matching-adjusted indirect comparison. Therefore, treatment comparators in the pharmacoeconomic submission were modelled as a naive indirect comparison of treatments, in which treatment efficacy data (i.e., OS, EFS) and safety data (i.e., AEs) of brexu-cel or comparators were obtained directly from clinical trials without adjustment or accounting for differences in patient characteristics. The use of a naive indirect comparison is subject to strong, untestable assumptions, such as the concept of conditional constancy (the requirement that there are no prognostic variables or effect modifiers imbalance between the 2 populations), which introduces substantial uncertainty into the determination of comparative efficacy and the magnitude of any relative efficacy benefits associated with brexu-cel.
CADTH notes that, owing to the direct use of clinical trial data from various sources, it was not possible to determine if any observed differences in EFS, OS, proportion of patients receiving allo-SCT, or AEs between therapies were solely due to the treatment or, rather, due to bias or confounding (e.g., differences in study populations, definitions of outcomes, study designs). Together, this reinforces the significant uncertainty in using naive indirect estimates to inform clinical inputs. As such, the incremental gains in QALYs and LYs predicted by the sponsor’s model for brexu-cel relative to comparators should be considered exploratory and interpreted with a high degree of uncertainty.
CADTH was unable to address the lack of comparative data for brexu-cel versus all model comparators.
Given the lack of direct evidence and the use of naive indirect comparisons to inform the model, CADTH could not estimate a robust single base case for assessing brexu-cel’s cost-effectiveness.
Long-term extrapolation of comparators’ OS lacked face validity: Clinical experts consulted for this review indicated that long-term extrapolation of OS for brexu-cel’s comparators was likely underestimated. Particularly, the OS estimate for salvage chemotherapy resulted in less than 1% of patients alive at year 9 and 1% of patients cured at year 2 (using the sponsor’s cure time point assumption). For blinatumomab plus or minus TKIs, when the cure point was changed to 5 years, the model estimated that 0% of patients would be alive at year 5. Based on feedback from experts consulted for this review, neither of those extrapolations were consistent with clinical practice. The sponsor indicated that parametric curves were chosen based on statistical approaches (i.e., Akaike Information Criteria, Bayesian Information Criteria) and then validated by experts. However, statistical fit speaks only to the fit of the predicted data to the observed data within the trial period, not to the validity of the predicted data to the extrapolated period. This issue is compounded by the trial data for the comparators, which had a relatively short follow-up (median of 29 months for the INO-VATE trial, and 12 months for the TOWER trial).
In addition, CADTH noted that in the sponsor’s base case, inotuzumab’s OS estimates were higher than blinatumomab’s OS estimates. Studies evaluating the indirect comparisons between blinatumomab and inotuzumab revealed no statistical difference in OS between the 2 comparators.13,14
The EFS and OS extrapolation curves for comparators were modified based on feedback from clinical experts consulted for this review and the matching-adjusted indirect comparisons evaluating inotuzumab and blinatumomab.
Model structure may overestimate comparative efficacy: Results from the sponsor’s model suggested that brexu-cel and inotuzumab were associated with longer survival after relapse (e.g., LY gain for brexu-cel in the progressed disease health state was fourfold to sevenfold higher compared to blinatumomab and salvage chemotherapy). While the pivotal trials for these drugs showed their impact on EFS and OS estimates, there was no clear mechanism by which brexu-cel or inotuzumab would continue to provide clinical benefit after relapse versus other comparators. The sponsor’s use of a PSM introduces structural assumptions about the relationship between EFS and OS that likely do not accurately reflect causal relationships within the disease pathway. These assumptions may produce a biased postrelapse survival benefit that favours brexu-cel. Due to the assumed independence between OS and EFS end points in a PSM, extrapolations for each end point may reflect within-trial trends in the rates of relapse and death.
The clinical experts consulted by CADTH concluded that there was insufficient evidence to explain the postrelapse survival difference between comparators in the sponsor’s model. CADTH was unable to determine the extent to which the implied postrelapse benefit was due to the effect of treatment or to structural bias within the PSM. Changes in the comparators’ OS distribution substantially reduced inotuzumab postprogression benefit. However, this biased benefit for brexu-cel could not be fully addressed in the CADTH reanalysis.
Treatment duration of comparators was modelled using distinct approaches: The duration of treatment for each of the comparators was incorporated in the model using distinct approaches: for blinatumomab, the sponsor modelled treatment duration using the number of average treatment cycles patients received in the TOWER trial, while for inotuzumab and salvage chemotherapy, treatment duration was assumed to be the maximum number of cycles for each therapy (five 28-day cycles for inotuzumab, and 4 28-day cycles for salvage chemotherapy). For ponatinib, the sponsor assumed a treatment duration of 90 days. According to the clinical experts consulted for this review, patients can remain on TKIs for longer than 90 days, but the experts agreed that 90 days could be considered an average treatment duration. The use of distinct approaches to account for comparators’ treatment duration hindered the comparability of drug acquisition costs and likely overestimated the cost of inotuzumab and salvage chemotherapy.
CADTH addressed this issue by changing inotuzumab and salvage chemotherapy maximum treatment duration to the median number of days from the INO-VATE trial (77 days and 28 days for inotuzumab and salvage chemotherapy, respectively).15
Concerns with the modelled comparator: Tisa-cel is a CAR T-cell technology recommended for the treatment of children and young adults (aged 3 to 25 years) with R/R B-cell ALL. Input provided by clinical experts consulted for this review indicated that tisa-cel would be considered as a comparator for the young adult population because there is an overlap in target populations between both CAR T technologies (young adults aged between 18 and 25 years). However, the sponsor only considered tisa-cel in a scenario analysis, and it assumed that tisa-cel’s OS and EFS were the same as brexu-cel, resulting in tisa-cel being cost-saving due to its lower costs.
CADTH conducted scenario analyses including tisa-cel as a comparator.
Re-treatment with brexu-cel was not incorporated in the model: Although a total of 2 patients (4%, 2 of 55 patients who ultimately received brexu-cel) were re-treated with brexu-cel in the ZUMA-3 study, the sponsors did not consider the costs of re-treatment with brexu-cel in subsequent treatment options. Their omission underestimated subsequent treatment drug acquisition costs, thus favouring brexu-cel.
CADTH addressed this issue in a scenario analysis using an adjustment weight for brexu-cel’s drug acquisition costs, to account for re-treatment with brexu-cel.
The 2-year cure point was considered underestimated: In the base-case analysis, the sponsor assumed that patients who remained event-free at year 2 were considered functionally cured. Although clinical experts consulted by CADTH agreed that a relationship between time without relapse and cure exists, this relationship was not linear, and the probabilities of relapse at 2 years was not zero. According to clinical experts consulted for this review, a 5-year cure point would be more appropriate.
CADTH addressed this limitation by modifying the cure point to 5 years in the CADTH reanalysis.
Lack of transparency and flexibility in the model: Several limitations were observed in the submitted model, including numerous errors in discount calculations, errors in the calculations of the treatment duration with TKIs, errors in the sequential cost-effectiveness calculator, lack of differentiation between extendedly dominated and dominated interventions, hidden Excel spreadsheets, poorly reported details in the pharmacoeconomic report, and many other limitations, which made validation of the model difficult. Additionally, the sponsor used numerous IFERROR statements in their model. IFERROR statements lead to situations in which the parameter value is overwritten with an alternative value without alerting the user to the automatized overwriting. The systematic use of IFERROR statements makes thorough auditing of the sponsor’s model challenging, because it remains unclear whether the model is running inappropriately by overriding errors. Best programming practices are such that any errors alert the user to a specific error. Overall, the submitted model was not transparent or flexible to changes.
CADTH was able to address only some of these deficiencies. CADTH cautions that results from submitted economic model could not be fully validated.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CADTH (Table 5).
Table 5: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CADTH comment |
|---|---|
Patients receiving TKIs incur the relevant costs of TKIs, but have no impact on efficacy estimates. | Uncertain. The sponsor assumed that adding TKIs to blinatumomab and inotuzumab for Ph-positive patients would not increase treatment efficacy in comparison with treatment with blinatumomab and inotuzumab as monotherapy. Feedback from clinical experts consulted for this review indicated that adding TKIs to blinatumomab or inotuzumab would be expected to increase the efficacy of treatment in this population. However, CADTH was unaware of any studies quantifying the increase in efficacy of adding TKIs to blinatumomab or inotuzumab. The magnitude of the impact of this assumption is uncertain, but it favours the cost-effectiveness of brexu-cel by not accounting for a potential increase in the efficacy of its comparators. |
Hyper CVAD’s efficacy is assumed to be equal to FLAG-IDA’s efficacy. | Reasonable. According to clinical experts consulted for this review, FLAG-IDA and hyper CVAD were expected to have similar efficacy. |
Relevant costs and disutilities were related to grade 3 or higher AEs with an incidence between 2% and 5% for brexu-cel, blinatumomab, inotuzumab, and salvage chemotherapy, obtained from the ZUMA-3, TOWER, and INO-VATE trials, respectively, while for ponatinib, data of any grade AE occurring in ≥ 20% of the total population were taken from the phase II PACE trial. | Inappropriate. The sponsor selected arbitrary thresholds to capture the impact of treatment-related AEs rather than selecting the most clinically meaningful AEs to include within the model. In addition, the AE threshold was distinct among the therapies, thus hindering comparability. CADTH guidelines recommend that all AEs that have clinical or cost significance should be included in the model. |
All AE costs were assumed to be the cost of a physician visit. | AEs were costed as a physician visit, which was not representative of the true cost. Because brexu-cel had the highest AE frequencies, the model likely underestimated the true costs of its AEs, and thus the overall cost of brexu-cel. |
AE = adverse event; FLAG-IDA = fludarabine, cytarabine, idarubicin, filgrastim; hyper CVAD = alternating courses of cyclophosphamide, vincristine, doxorubicin, and dexamethasone with courses of methotrexate and cytarabine; Ph = Philadelphia chromosome; TKI = tyrosine kinase inhibitor.
CADTH Reanalyses of the Economic Evaluation
Base-Case Results
Given the uncertainty associated with the comparative treatment effects and the limitations with the modelling approach, CADTH could not estimate a single robust base-case estimate of cost-effectiveness for brexu-cel compared to inotuzumab plus or minus TKIs, blinatumomab plus or minus TKIs, and salvage chemotherapy in the Canadian setting. The CADTH reanalyses were derived by making changes in model parameter values and assumptions, in consultation with clinical experts.
Table 6 details each change made to derive the CADTH revised reanalysis, which was conducted in a stepwise approach to highlight the impact of each change. The summary of results from the stepped reanalysis is presented in Table 7 and Table 15, Appendix 4.
Table 6: CADTH Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
1. Incorporate discount rate for drug acquisition costs | Error in applying discount rate to drug acquisition costs by multiplying the discount rate | Drug acquisition costs were divided by the discount rate |
2. Maximum treatment duration for TKIs | Error in the cell input text resulting in no maximum treatment duration | 90 days |
3. Correct drug acquisition price for salvage chemotherapy | Error in calculating drug cost for idarubicin (cost equal to zero) and cyclophosphamide (use of oral cyclophosphamide prices instead of IV) | Drug acquisition costs were fixed in the model |
Changes to derive the CADTH reanalysis | ||
1. OS and EFS extrapolation curves for brexu-cel (log normal; 5-year and 25-year OS of 26% and 6%, respectively) | EFS: Log logistic OS: Gompertz | EFS: Log logistic OS: Log normal |
2. OS and EFS extrapolation curves for brexu-cel (Weibull; 5-year and 25-year OS of 21% and 0%, respectively) | EFS: Log logistic OS: Gompertz | EFS: Log logistic OS: Weibull |
3. OS and EFS extrapolation curves for comparators | Salvage chemotherapy: EFS: Gompertz OS: Generalized gamma Inotuzumab: EFS: Log normal OS: Log logistic Blinatumomab: EFS: Exponential OS: Exponential | Salvage chemotherapy: EFS: Log logistic OS: Gompertz Inotuzumab: EFS: Gompertz OS: Log normal Blinatumomab: EFS: Log logistic OS: Log normal |
4. Duration of inotuzumab treatment | Maximum of 161 days | Median 3 cycles (77 days) |
5. Duration of salvage chemotherapy | Maximum of 112 days | Median 1 cycle (28 days) |
6. Acquisition costs for re-treatment with brexu-cel | Not incorporated in the model | Incorporated into brexu-cel drug acquisition cost considering re-treatment rate of 4% based on the ZUMA-3 study |
7. Cure point | 2 years | 5 years |
CADTH reanalysis 1 | 1 + 3 + 4 + 5 + 6 + 7 | |
CADTH reanalysis 2 | 2 + 3 + 4 + 5 + 6 + 7 | |
OS = overall survival; EFS = event-free survival; TKI = tyrosine kinase inhibitor.
The CADTH reanalyses were based on publicly available prices of the comparator and subsequent therapies.
In the first CADTH reanalysis, brexu-cel was estimated to have a 5-year and 25-year OS of 26% and 6%, respectively. Brexu-cel was associated with a total cost of $593,180 and 3.77 QALYs compared to $156,957 and 1.12 QALYs for patients receiving salvage chemotherapy. Blinatumomab plus or minus TKIs and inotuzumab plus or minus TKIs were extendedly dominated by brexu-cel in this reanalysis. The ICER for brexu-cel in the sequential analysis was $164,545 per QALY gained relative to salvage chemotherapy. The postprogression benefit for brexu-cel was twofold higher than salvage chemotherapy in this reanalysis (1.99 versus 1.01 LYs for brexu-cel and salvage chemotherapy, respectively). Of note, of the 2.71 incremental QALYs associated with brexu-cel, 21% were accrued during the trial period (maximum observation of 26 months).
In the second CADTH reanalysis, brexu-cel was estimated to have a 5-year and 25-year OS of 21% and 0%, respectively. Blinatumomab plus or minus TKIs was extendedly dominated by inotuzumab plus or minus TKIs in this reanalysis. The ICER increased to $679,053 per QALY relative to inotuzumab plus or minus TKIs, with approximately 47% of incremental QALYs accrued during the trial period. In this reanalysis, the postprogression benefit for brexu-cel was similar among comparators. Brexu-cel had a 0% probability of being cost-effective at a willingness-to-pay threshold of $50,000 per QALY in all of CADTH’s reanalyses.
Because the sponsor used a PSM, which assumed that EFS and OS are independent and fitted separately (i.e., no explicit relationship between EFS and OS), changes in the distributions used to extrapolate EFS and OS resulted in misalignment of the curves. In reanalysis 1, the long-term EFS (i.e., consistent with survival of cured patients) was not aligned with OS, resulting in a proportion of patients with disease progression after 35 years. Likewise, in reanalysis 2, the Weibull distribution increased the mortality rate monotonically, resulting in a shorter long-term survival compared to the other comparators. However, brexu-cel still sustained an OS benefit greater than the comparators for the first 10 years of the analysis. Ultimately, CADTH could not address these model limitations due to the lack of flexibility with long-term extrapolations, and due to the model structure, which failed to consider the explicit relationship between EFS and OS.
Detailed information and disaggregated results are presented in Appendix 4.
Table 7: Summary of the CADTH Reanalysis Results
Drug | Total costs ($) | Total QALYs | Total LYs | ICER vs. salvage chemotherapy ($ per QALY) | Sequential ICER ($ per QALY) |
|---|---|---|---|---|---|
Sponsor-corrected base case | |||||
Salvage chemotherapy | $173,221 | 0.71 | 1.13 | Reference | Reference |
Blinatumomab ± TKIs | $300,207 | 2.37 | 3.30 | $79,721 | Extendedly dominated by brexu-cel |
Inotuzumab ± TKIs | $436,627 | 3.73 | 6.35 | $87,155 | Extendedly dominated by brexu-cel |
Brexu-cel | $588,001 | 7.63 | 10.50 | $59,987 | $59,987 |
CADTH reanalysis 1 | |||||
Salvage chemotherapy | $156,957 | 1.12 | 1.69 | Reference | Reference |
Blinatumomab ± TKIs | $301,451 | 1.83 | 2.58 | $202,450 | Extendedly dominated by brexu-cel |
Inotuzumab ± TKIs | $316,929 | 1.92 | 2.61 | $199,396 | Extendedly dominated by brexu-cel |
Brexu-cel | $593,180 | 3.77 | 5.04 | $164,545 | $164,545 |
CADTH reanalysis 2 | |||||
Salvage chemotherapy | $156,764 | 1.10 | 1.67 | Reference | Reference |
Blinatumomab ± TKIs | $300,717 | 1.82 | 2.58 | $202,073 | Extendedly dominated by inotuzumab ± TKIs |
Inotuzumab ± TKIs | $316,248 | 1.94 | 2.59 | $190,979 | $190,979 |
Brexu-cel | $592,920 | 2.35 | 3.12 | $351,024 | $679,053 |
brexu-cel = brexucabtagene autoleucel; ICER = incremental cost-effectiveness ratio; LY = life-years; QALY = quality-adjusted life-years; TKI = tyrosine kinase inhibitor; vs. = versus.
Scenario Analysis Results
CADTH undertook price reduction analyses based on the sponsor base case and CADTH’s reanalyses (Table 8). These analyses demonstrated that a price reduction of 71% for reanalysis 1, and 88% for reanalysis 2, would be necessary to achieve cost-effectiveness at a willingness-to-pay threshold of $50,000 per QALY. Detailed results of the sequential analysis’ price reduction are presented in Table 17, Appendix 4.
Table 8: CADTH Price Reduction Analyses
Analysis | ICERs for brexu-cel vs. salvage chemotherapy ($ per QALY) | ||
|---|---|---|---|
Price reduction | Sponsor base case | CADTH reanalysis 1 | CADTH reanalysis 2 |
No price reduction | 57,702 | 164,545 | 351,032 |
10% | 52,048 | 148,584 | 316,929 |
20% | 46,394 | 132,563 | 282,827 |
30% | 40,740 | 116,541 | 248,725 |
40% | 35,087 | 100,520 | 214,623 |
50% | 29,433 | 84,498 | 180,520 |
60% | 23,779 | 68,477 | 146,418 |
70% | 18,125 | 52,544 | 112,316 |
80% | 12,471 | 36,434 | 78,214 |
90% | 6,818 | 20,412 | 44,111 |
100% | 1,164 | 4,390 | 10,009 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-years; vs. = versus.
In addition, CADTH conducted a series of scenario analyses to determine the impact of alternative assumptions on the cost-effectiveness of brexu-cel, which are outlined as follows:
Time horizon of 5 years
Inclusion of tisa-cel as a comparator
No re-treatment with brexu-cel
OS extrapolation curve for brexu-cel using generalized gamma
Results are described in Table 18, Appendix 4. CADTH considered a scenario analysis to explore the substantial uncertainty associated with OS benefits by reducing the time horizon of the analysis to 5 years and another scenario analysis to explore the impact of including tisa-cel as 1 of the comparators. In the scenario analysis in which the time horizon was changed to 5 years, the ICER increased substantially for all CADTH reanalyses. When tisa-cel was included as a comparator, it dominated brexu-cel in all analyses because it was assumed to have the same QALY gains as brexu-cel, but a lower drug acquisition cost. However, given that the list price of tisa-cel is unknown, no conclusions on the incremental cost-effectiveness can be drawn when comparing tisa-cel to brexu-cel.
CADTH conducted a scenario analysis that explored the next most optimistic OS extrapolation (generalized gamma distribution), in which the 5-year and 25-year OS was 36% and 21%, respectively. In this reanalysis, both blinatumomab plus or minus TKIs and inotuzumab plus or minus TKIs were extendedly dominated by brexu-cel. Brexu-cel’s ICER was $85,559 per QALY relative to salvage chemotherapy, with 10% of incremental QALYs accrued during the trial period. CADTH also notes that this scenario analysis generated an OS curve that lacks face validity, due to it both overestimating the proportion of patients alive at the 40-year and 50-year time horizon (13% and 4%, respectively) and inflating the postprogression survival benefit of brexu-cel, which was fivefold higher than salvage chemotherapy (5.46 versus 1.02 LYs in the progressed disease state for brexu-cel and salvage chemotherapy, respectively). This postprogression benefit is outlined in Figure 5, Appendix 4, in which brexu-cel’s OS remained proportional to the EFS curves and the OS and EFS curves did not converge; therefore, in this analysis, the OS benefits were maintained in approximately 30% of patients treated with brexu-cel despite the model assumption that only approximately 12% of brexu-cel patients were cured.
Issues for Consideration
Travel costs for patients (and their families) and the requirement for time spent away from work were not included in the sponsor’s submissions to CADTH. The drug plan input indicated that not all provinces and territories will have a local site to provide brexu-cel. For jurisdictions that do not currently have a site to provide brexu-cel, there will be a need for patients to travel out of province or out of country for treatment. The sponsor did not conduct a societal perspective, so these travel costs were not accounted for, and these costs were not applicable in the Canadian health care payer perspective. However, to mitigate this issue, the sponsor indicated that they offer a travel support program that assists in coverage for related travel and lodging expenses.
Disparities in funding and treatment access may vary depending on the province or territory, and the requirement for access to a tertiary care centre for delivery of brexu-cel may have equity-of-access implications, which were not substantively considered in the economic submission.
To be able to treat patients with brexu-cel, specialized centres need to be trained and accredited by the manufacturer. Both obtaining and maintaining this accreditation process can result in a high resource burden, including the development of various protocols and supporting yearly audits. In addition, this treatment has the added complexity of needing to coordinate patient care and product preparation with an external manufacturer. Because there are likely multiple CAR T therapies being administered by specialized centres, there will be a need to manage various protocols for the preparation and delivery of each product type, which can increase the overall administrative burden. Out-of-province or out-of-country care may still be needed.
Issues pertaining to the manufacturing process are important factors in the successful delivery of CAR T therapies. Manufacturing failure was observed in 8% of patients enrolled in the ZUMA-3 study (6 out of 71 enrolled). In cases of manufacturing failure, it’s expected that jurisdictions will not pay for the cost of the failed product. However, this does not account for the costs associated with an increased hospital stay while a second sample is prepared, if possible and required, alternate treatment if initiated, or the impact on patient outcomes due to treatment delays or compromised doses. Manufacturing failure is likely to increase the ICER because patients may require continued health care services while waiting for their dose of brexu-cel or they may experience disease progression, which requires intensive formal and informal care, including hiring paid help and family or partner help, respectively.
Although the budget impact analysis assumes that public drug programs will be paying for CAR T therapy, it remains unclear if other departments in each province’s ministry of health would be paying for this therapy. This may vary by jurisdiction.
Overall Conclusions
Evidence from the ZUMA-3 study suggests that brexu-cel was associated with clinically meaningful EFS (11.6 months; 95% CI, 2.7 to 20.5 months) and OS (25.4 months; 95% CI, 16.2 months to not estimable) for the treatment of patients with R/R B-cell ALL. As noted in the CADTH Clinical Review Report, this benefit was based on 21 months of follow-up; therefore, the long-term benefit of brexu-cel treatment is currently unknown. However, because the ZUMA-3 study was a single-arm trial, comparative efficacy of brexu-cel to inotuzumab plus or minus TKIs, blinatumomab plus or minus TKIs, and salvage chemotherapy is unknown. The CADTH Clinical Review Report concluded that inferences regarding the efficacy of brexu-cel could not be made due to data immaturity and that there is insufficient evidence to support brexu-cel as a curative treatment. No conclusions could be made regarding health-related quality of life due to missing data.
In addition to the aforementioned limitations with the clinical evidence, CADTH identified several limitations with the sponsor’s economic submission. These limitations included the uncertainty associated with the clinical efficacy of brexu-cel, the overestimation of brexu-cel overall survival, the use of naive indirect comparisons to derive comparative clinical efficacy for brexu-cel, the lack of face validity of long-term OS for comparators, the overestimation of postprogression effects of brexu-cel, the use of a model structure that assumed EFS and OS were independent, the assumption of a cure point of 2 years, a decision tree that failed to consider the QALY implications of not receiving brexu-cel, and the lack of a consistent approach when incorporating comparator’s treatment duration.
Given the uncertainty associated with the comparative treatment effects and the limitations with the modelling approach, CADTH could not estimate a single robust base-case estimate for brexu-cel, and instead conducted a series of CADTH reanalyses. Given the absence of long-term clinical evidence, CADTH reanalyses explored the uncertainties associated with long-term treatment efficacy by selecting 2 alternative extrapolation curves to inform the OS for brexu-cel.
If brexu-cel is associated with a 5-year and 25-year OS of 26% and 6%, respectively (reanalysis 1), then the ICER for brexu-cel relative to inotuzumab plus or minus TKI was estimated to be $164,545 per QALY. In this analysis, blinatumomab plus or minus TKIs and inotuzumab plus or minus TKI did not lie in the cost-effectiveness frontier, because they were extendedly dominated by brexu-cel. A price reduction of 71% would be required for brexu-cel to be considered cost-effective at a $50,000-per-QALY threshold. If brexu-cel’s OS benefit is reduced to a 5-year and 25-year OS of 21% and 0%, respectively (reanalysis 2), then the ICER increases to $679,053 per QALY relative to inotuzumab plus or minus TKIs. In this analysis, blinatumomab plus or minus TKIs did not lie on the cost-effectiveness frontier, because it was extendedly dominated by inotuzumab plus or minus TKI. A price reduction of 88% would be required to achieve cost-effectiveness at a $50,000-per-QALY threshold.
CADTH also explored the next most optimistic OS extrapolation (generalized gamma distribution) in a scenario analysis, which resulted in a 5-year and 25-year OS of 36% and 21%, respectively. However, this scenario analysis lacked face validity because it overestimated the proportion of patients alive at the 40-year and 50-year time horizon (13% and 4%, respectively). Furthermore, it inflated brexu-cel’s postprogression survival benefit, which was fivefold higher than salvage chemotherapy (5.46 versus 1.02 LYs in the progressed disease state for brexu-cel and salvage chemotherapy, respectively). This postprogression benefit is outlined in Figure 5, Appendix 4, in which brexu-cel’s OS remained proportional to the EFS curves and the OS and EFS curves did not converge; therefore, in this analysis, the OS benefits were maintained in approximately 30% of patients treated with brexu-cel despite the model assumption that only approximately 12% of brexu-cel patients were cured.
Although the CADTH reanalyses attempted to address the identified limitations of the sponsor’s economic submission, significant uncertainty still exists due to a lack of comparative and long-term efficacy data. As such, the cost-effectiveness of brexu-cel in comparison to other treatments is unknown. Given the absence of comparative data, naive indirect comparisons were conducted, and the cost-effectiveness of brexu-cel was sensitive to the parametric distribution used to extrapolate brexu-cel’s OS. In both CADTH reanalyses, OS benefits for brexu-cel were smaller than in the sponsor’s base-case (e.g., incremental LYs for brexu-cel versus salvage chemotherapy was 9.67 in the sponsor’s base case versus 3.35 in CADTH reanalysis 1) and this translated to a smaller difference in QALYs between brexu-cel and the other comparators. In addition, CADTH noted that a lower proportion of incremental QALYs accrued by brexu-cel were derived from the extrapolation period beyond the observed trial in the CADTH reanalyses (53% and 79%), contrary to the sponsor’s analysis, in which 92% of incremental QALYs were accrued beyond the trial’s observation period. Finally, although not able to fully address this limitation, CADTH reanalyses reduced the postprogression benefit from approximately 5 LY between brexu-cel and salvage chemotherapy in the sponsor’s base case to 1 LY in CADTH reanalysis 1 and 0.15 LY in CADTH reanalysis 2.
Although the CADTH reanalyses attempted to address the identified limitations of the sponsor’s economic submission, significant uncertainty still exists due to a lack of comparative and long-term efficacy data, uncertainties in the extrapolation period, and an overestimation of postprogression benefit for brexu-cel. Therefore, CADTH’s estimates of cost-effectiveness were likely biased in favour of brexu-cel.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Tecartus (brexucabtagene autoleucel), target of 1 × 106 chimeric antigen receptor (CAR)-positive viable T cells per kg body weight with a maximum of 1 × 108 CAR-positive viable T cells, cell suspension in patient-specific single infusion bag for intravenous infusion. Mississauga (ON): Gilead Sciences Canada, Inc.; 2022 Oct 03.
2.Tecartus (brexucabtagene autoleucel): target of 2 × 106 chimeric antigen receptor (CAR)-positive viable T cells per kg body weight with a maximum of 2 × 108 anti-CD19 CAR-positive viable T cells, cell suspension in patient-specific single infusion bag for intravenous infusion; or Tecartus (brexucabtagene autoleucel): target of 1 × 106 chimeric antigen receptor (CAR)-positive viable T cells per kg body weight with a maximum of 1 × 108 anti-CD19 CAR-positive viable T cells, cell suspension in patient-specific single infusion bag for intravenous infusion [product monograph]. Mississauga (ON): Gilead Sciences Canada, Inc.; 2022 Nov 16.
3.Clinical Study Report: KTE-C19-103 (ZUMA-3). A phase 1/2 multi-center study evaluating the safety and efficacy of KTE-X19 in adult subjects with relapsed/refractory B-precursor acute lymphoblastic leukemia (r/r ALL) [internal sponsor's report]. Santa Monica (CA): Kite Pharma, Inc.; 2021 Feb 09.
4.Kantarjian HM, DeAngelo DJ, Stelljes M, et al. Inotuzumab ozogamicin versus standard of care in relapsed or refractory acute lymphoblastic leukemia: Final report and long-term survival follow-up from the randomized, phase 3 INO-VATE study. Cancer. 2019;125(14):2474-2487. PubMed
5.Kantarjian H, Stein A, Gokbuget N, et al. Blinatumomab versus Chemotherapy for Advanced Acute Lymphoblastic Leukemia. N Engl J Med. 2017;376(9):836-847. PubMed
6.Gokbuget N, Dombret H, Ribera JM, et al. International reference analysis of outcomes in adults with B-precursor Ph-negative relapsed/refractory acute lymphoblastic leukemia. Haematologica. 2016;101(12):1524-1533. PubMed
7.Katz AJ, Chia VM, Schoonen WM, Kelsh MA. Acute lymphoblastic leukemia: an assessment of international incidence, survival, and disease burden. Cancer Causes Control. 2015;26(11):1627-1642. PubMed
8.Table: 13-10-0114-01. Life expectancy and other elements of the life table, Canada, all provinces except Prince Edward Island. Ottawa (ON): Statistics Canada; 2022: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310011401. Accessed 2022 Sep 01.
9.Martin PJ, Counts GW, Jr., Appelbaum FR, et al. Life expectancy in patients surviving more than 5 years after hematopoietic cell transplantation. J Clin Oncol. 2010;28(6):1011-1016. PubMed
10.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario drug benefit formulary/comparative drug index. 2022; https://www.formulary.health.gov.on.ca/formulary/. Accessed 2022 Sep 01.
11.Ontario Case Costing Initiative (OCCI). Toronto (ON): Ontario Health and Long-Term Care; 2017: https://data.ontario.ca/dataset/ontario-case-costing-initiative-occi. Accessed 2022 Sep 01.
12.Walker H, Anderson M, Farahati F, et al. Resource use and costs of end-of-Life/palliative care: Ontario adult cancer patients dying during 2002 and 2003. J Palliat Care. 2011;27(2):79-88. PubMed
13.Song J, Ma Q, Gao W, et al. Matching-Adjusted Indirect Comparison of Blinatumomab vs. Inotuzumab Ozogamicin for Adults with Relapsed/Refractory Acute Lymphoblastic Leukemia. Adv Ther. 2019;36(4):950-961. PubMed
14.Proskorovsky I, Su Y, Fahrbach K, et al. Indirect Treatment Comparison of Inotuzumab Ozogamicin Versus Blinatumomab for Relapsed or Refractory Acute Lymphoblastic Leukemia. Adv Ther. 2019;36(8):2147-2160. PubMed
15.Kantarjian HM, DeAngelo DJ, Stelljes M, et al. Inotuzumab Ozogamicin versus Standard Therapy for Acute Lymphoblastic Leukemia. N Engl J Med. 2016;375(8):740-753. PubMed
16.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Tecartus (brexucabtagene autoleucel), target of 1 × 106 chimeric antigen receptor (CAR)-positive viable T cells per kg body weight with a maximum of 1 × 108 CAR-positive viable T cells, cell suspension in patient-specific single infusion bag for intravenous. Mississauga (ON): Gilead Sciences Canada, Inc.; 2022 Oct 03.
17.pan-Canadian Oncology Drug Reimbursment Review clinical guidance report: inotuzumab ozogamicin (Bespona) for acute lymphoblastic leukemia. Ottawa (ON): CADTH; 2018 Jul 06: https://www.cadth.ca/sites/default/files/pcodr/pcodr_inotuzumab_ozogamicin_besponsa_all_fn_cgr.pdf. Accessed 2022 Dec 12.
18.Drug Reimbursement Review clinical and pharmacoeconomic reviews: daunorubicin and cytarabine (Vyxeos) for acute myeloid leukemia. Can J Health Technol. 2021;1(10). 10.51731/cjht.2021.183. Accessed 2022 Dec 12.
Appendix 1: Cost Comparison Table
Table 9: CADTH Cost Comparison Table for R/R B-cell ALL
Treatment | Strength/concentration | Form | Price ($) | Recommended dosage | Daily cost ($) | 28-day course cost ($) |
|---|---|---|---|---|---|---|
Brexucabtagene autoleucel | 2*106 CAR + viable T-cells per kg body weight to a maximum of 2*108 cells for patients 100 kg and above | Cell suspension in patient-specific single infusion bag | 533,523.1000 | One-time dosea | NA | NA |
CAR T | ||||||
Tisagenlecleucel | 2*106 CAR + viable T-cells per kg body weight for patients 50kg and below and 2*108 cells for patients 50 kg and above | Cell suspension in patient-specific single infusion bag | 450,000.000b | One-time dosea | NA | NA |
Monoclonal antibody ± TKIs | ||||||
Blinatumomab | 0.0385 mg in 10 mL | Injectable solution | 77.3600 per mgc | 0.028 mg/day from day 1 to 28 (for patients with weight > 45kg); followed by a 14-day interval | 1,985.51 | 55,594 |
Inotuzumab | 0.009 mg in 20 mL | Injectable solution | 14,405.8500 per vialc | First cycle: 0.8 mg/m2 on day 1, and 0.5 mg/m2 on day 8 and 15; Subsequent cycles: 0.5 mg/m2 on day 1,8, and 15. | 1,543.48 | 43,218 |
Ponatinib | 15 mg 45 mg | Tablet | 157.0815d 351.0267d | 45 mg daily | 351.03 | 9,829 |
Dasatinib | 20 mg 50 mg 70 mg 100 mg | Tablet | 32.8823c 66.1782c 72.9336c 132.2671c | 140 mg daily | 198.03 | 5,545 |
Blinatumomab + ponatinib | 2,336.53 | 65,423 | ||||
Blinatumomab + dasatinib | 2,183.54 | 61,139 | ||||
Inotuzumab + ponatinib | 1,894.51 | 53,046 | ||||
Inotuzumab + dasatinib | 1,741.52 | 48,762 | ||||
Hyper CVAD | ||||||
Cyclophosphamide (Proxytox) | 20 mg/mL | 500 mg 1,000 mg Powder for IV infusion | 97.8000c 177.2700c | 300 mg/m2 on Day 1 to 3 of a 21-day cycle | 50.65 | 1,064 |
Vincristine (generic) | 1 mg/mL (1 mL vial) 1 mg/mL (2 mL vial) 1 mg/mL (5 mL vial) | Injectable solution | 30.6000 mg per mL | 1.45 mg/m2 day 4 and 11 of a 21-day cycle | 5.83 | 163 |
Doxorubicin | 2 mg/mL | 5 mL vial 25 mL vial 50 mL vial 100 mL vial Injectable solution | 10.000 per mL (5 mL vial)c 26.0800 per mL (50 mL vial)c 10.2000 per mL (2 5 mL vial)c 7.7000 per mL (100 mL vial)c | 50 mg/m2 on Day 4 of a 21-day cycle | 17.16 | 480 |
Methotrexate | 25 mg/ mL (2mL vial) 10 mg/ mL (2 mL vial) | Injectable solution | 8.9200 per vial 12.5000 per vial | 1,000 mg/m2 Day 1 of a 21-day cycle | 15.29 | 428 |
Cytarabine | 100 mg/mL (20 mL vial) 100 mg/mL (5 mL vial) | Injectable solution | 306.5000 (15.3250 per mL)c 76.8500 (15.3700 per mL)c | 3,000 mg/m2 Days 2 and 3 of a 21-day-cycle | 73.02 | 2,045 |
Hyper CVAD | 161.95 | 4,535 | ||||
FLAG-IDA | ||||||
Filgrastim | 0.48 mg/1 mL vial 0.30 mg/1 mL vial 0.30 mg/1 mL pack 0.48 mg/0.8 mL prefilled syringe 0.30 mg/0.5 mL prefilled syringe | Injectable solution | 281.8120 176.1330 144.3100 230.9000 144.3135 | 0.3 mg days 1 to 4 of a 28-day-cycle | 20.62 | 577 |
Idarubicin | 1 mg/mL vial | IV solution (5 mL vial) | 211.5200 (42.304 per mL in 5 mL vial)c | 10 mg/m2 days 1 and 2 of a 28-day-cycle | 60.43 | 1,692 |
Fludarabine | 10 mg | Tablet | 40.5167 | 30 mg/m2 days 1 to 4 of a 28-day-cycle | 34.73 | 972 |
Cytarabine | 100 mg/mL (20 mL vial) 100 mg/mL (5 mL vial) | Injectable solution | 306.5000 (15.3250 per mL)c 76.8500 (15.3700 per mL)c | 2,000 mg/m2 days 1 to 4 of a 28-day-cycle | 87.57 | 2,452 |
FLAG-IDA | 203.35 | 5,694 | ||||
Note: All prices are from the Ontario Drug Benefit Formulary (accessed October 2022), unless otherwise indicated, and do not include dispensing fees. Calculations assume a patient body weight of 80.97 kg and a body surface area of 1.92 m2
aBrexucabtagene autoleucel and tisagenlecleucel are delivered as a one-time dose. Daily and annual costs were not calculated.
bNo public price available. Price listed was submitted by sponsor as part of submitted model; CADTH was unable to confirm accuracy
cCosts from IQVIA Delta PA database (accessed October 2022)
dOntario Drug Benefit Formulary or Exceptional Access Program list price (accessed October 2022)
Note that this appendix has not been copy-edited.
The comparators presented in this table have been deemed to be appropriate based on feedback from clinical expert(s). Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Appendix 2: Submission Quality
Note that this appendix has not been copy-edited.
Description | Yes/no | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | Yes | No comment |
Model has been adequately programmed and has sufficient face validity | No | As per CADTH appraisal, the model used naive indirect comparisons to assess the cost-effectiveness of brexu-cel, which lacks face validity. In addition, brexu-cel long-term OS extrapolation was overestimated according to clinical experts consulted for this review. |
Model structure is adequate for decision problem | Yes | No comment |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | No | As per CADTH appraisal, different approaches for treatment duration were used for comparators, and re-treatment with brexu-cel was not considered in the analysis. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | Yes | No comment |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | No | As per CADTH appraisal, the submission lacked details and the model had multiple errors and hidden spreadsheets that hindered the validation of results. |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note that this appendix of figures and tables has not been copy-edited.
Figure 3: Sponsor’s Choice of EFS and OS extrapolations
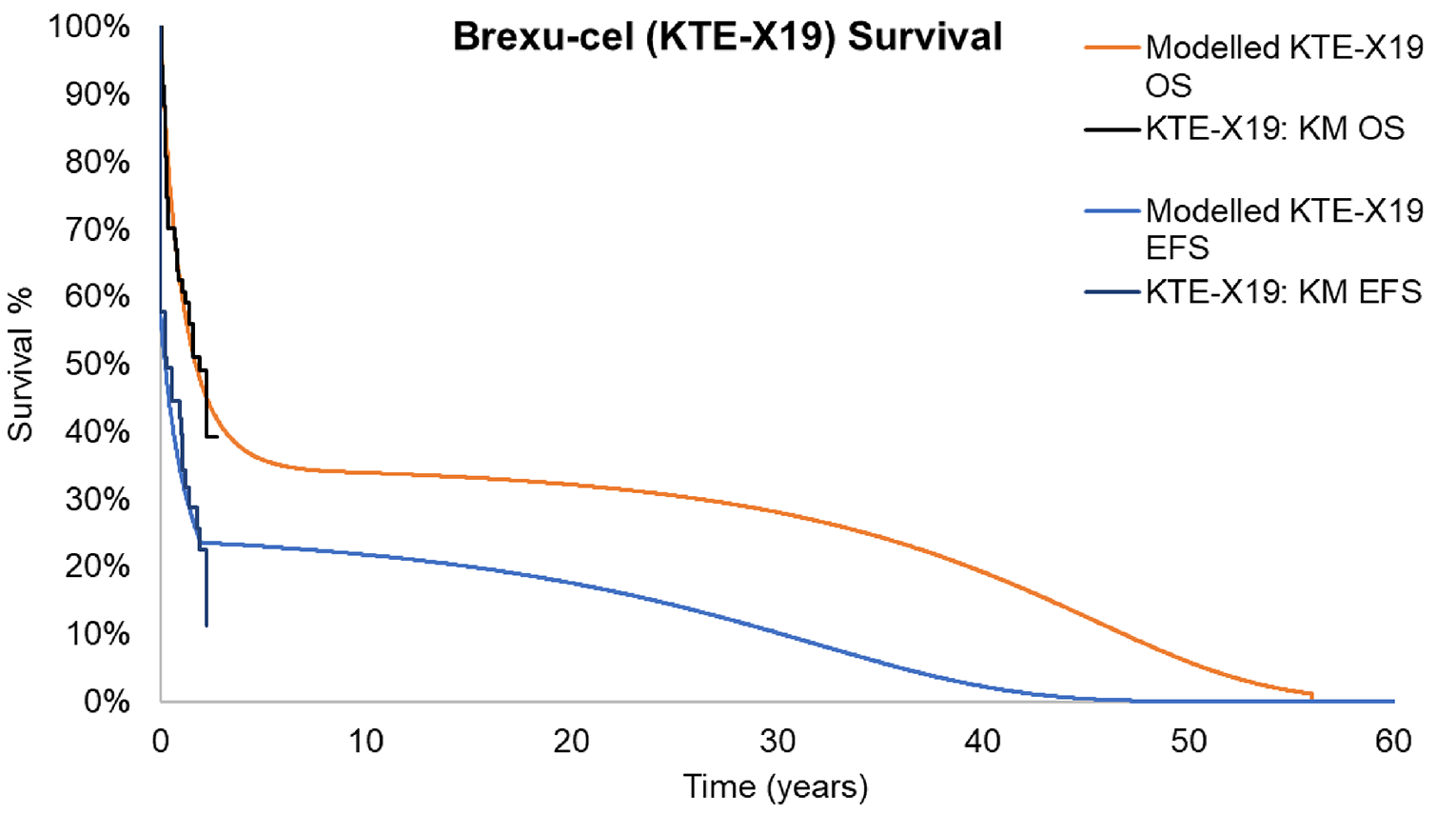
Source: Sponsor’s Pharmacoeconomic Submission.1
Detailed Results of the Sponsor’s Base Case
Table 11: Sponsor’s Economic Evaluation Results (Probabilistic)
Drug | Total costs | Total QALYs | Sequential ICER ($ per QALY) |
|---|---|---|---|
Salvage chemotherapy | $173,407.14 | 0.71 | Ref. |
Blinatumomab ± TKIs | $357,242.92 | 2.36 | Extendedly Dominated |
Inotuzumab ± TKI | $506,086.68 | 3.72 | Extendedly Dominated |
Brexu-cel | $587,667.51 | 7.83 | $58,177.29 |
TKI = tyrosine kinase inhibitor.
Table 12: Sponsor’s Economic Evaluation Life-Years and QALYs by Health State
Drug | EFS | PD | Total |
|---|---|---|---|
Life-years | |||
Salvage chemotherapy | 0.39 | 0.74 | 1.13 |
Blinatumomab ± TKIs | 1.71 | 1.59 | 3.30 |
Inotuzumab ± TKI | 2.17 | 4.18 | 6.35 |
Brexu-cel | 4.98 | 5.82 | 10.80 |
QALYs | |||
Salvage chemotherapy | 0.32 | 0.55 | 0.87 |
Blinatumomab ± TKIs | 1.38 | 1.13 | 2.51 |
Inotuzumab ± TKI | 1.06 | 2.94 | 4.00 |
Brexu-cel | 4.01 | 3.39 | 7.40 |
EFS = event-free survival; PD = progressed disease; TKI = tyrosine kinase inhibitor.
Table 13: Sponsor’s Economic Evaluation Disaggregated Costs
Drug | Salvage chemotherapy | Blinatumomab ± TKIs | Inotuzumab ± TKI | Brexu-cel |
|---|---|---|---|---|
Drug costs | $673 | $201,045 | $385,189 | $430,461 |
Administration costs | $38,724 | $14,294 | $14,562 | $24,716 |
Monitoring costs (EFS) | $451 | $999 | $1,501 | $2,582 |
Monitoring costs (PD) | $1,618 | $3,496 | $9,108 | $12,641 |
Subsequent treatment acquisition costs | $67,582 | $73,818 | $15,126 | $62,851 |
Subsequent treatment administration costs | $3,913 | $3,311 | $2,563 | $3,644 |
Allo-SCT cost (one-off) | $18,645 | $19,806 | $39,560 | $14,764 |
AE costs | $96 | $102 | $31 | $332 |
End-of-life costs | $41,705 | $40,372 | $38,445 | $35,676 |
Total | $173,407 | $357,243 | $506,087 | $587,668 |
EFS = event-free survival; PD = progressed disease; TKI = tyrosine kinase inhibitor; allo-SCT = allogeneic stem cell transplant; AE = adverse events.
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Note that this appendix of figures and tables has not been copy-edited.
Detailed Results of CADTH Base Case
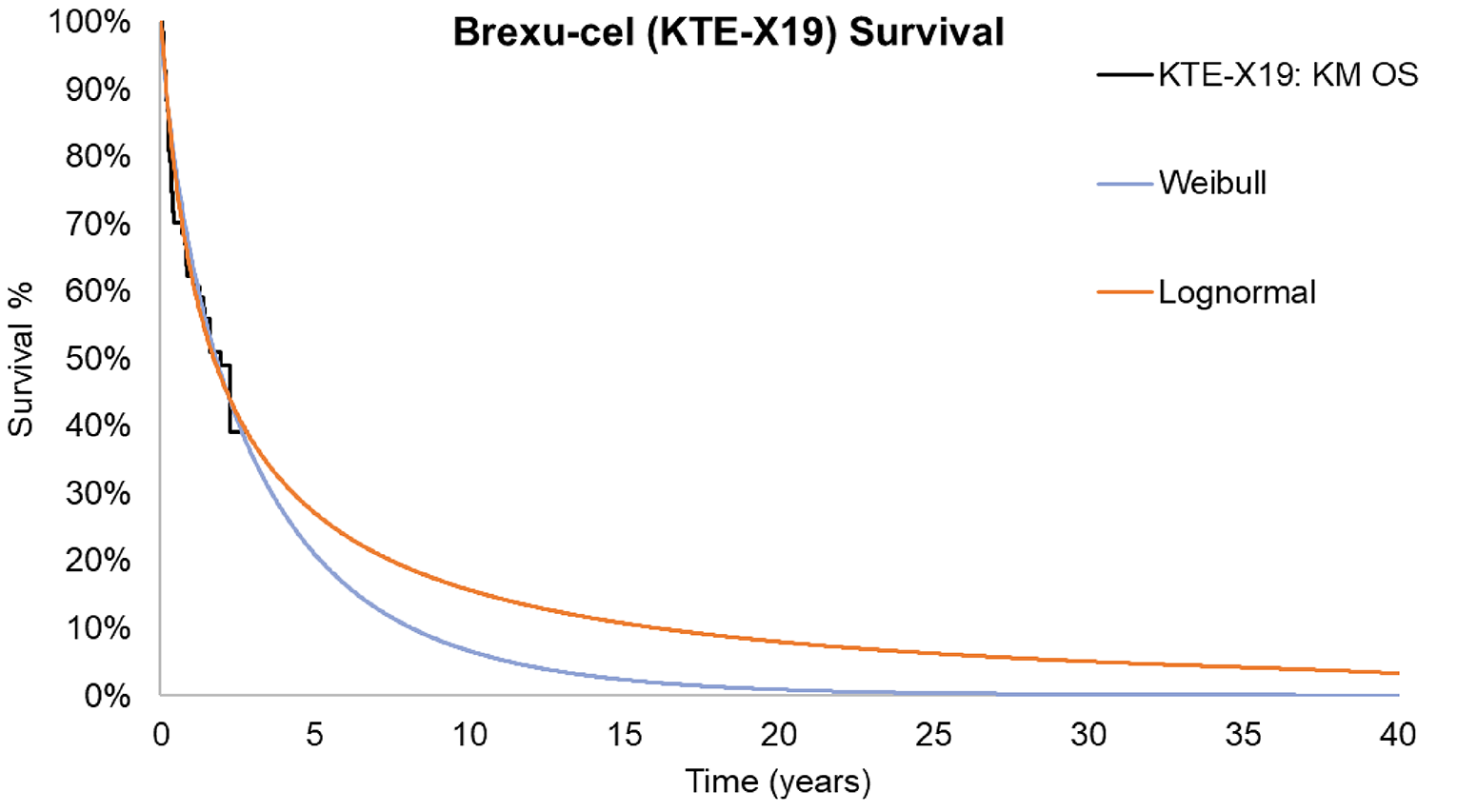
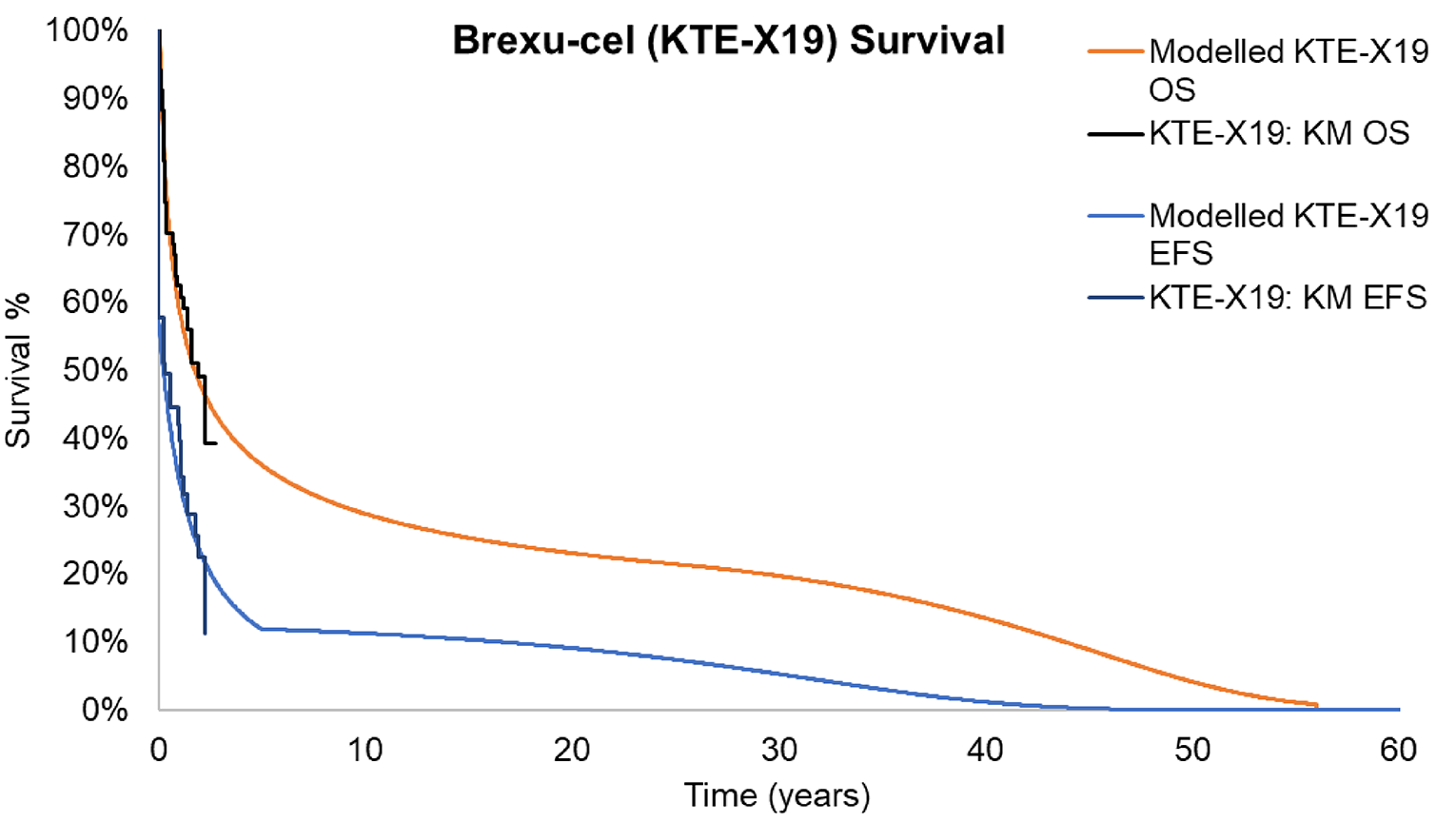
Table 14: Summary of the Stepped Analysis of the CADTH Reanalysis Results
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER vs. salvage chemotherapy ($/QALY) | Sequential ICER ($/QALY) |
|---|---|---|---|---|---|
Sponsor-corrected base case (probabilistic) | Salvage chemotherapy | $173,221 | 0.71 | Ref | Ref. |
Blinatumomab ± TKIs | $300,207 | 2.37 | $79,721 | Extendedly dominated by brexu-cel | |
Inotuzumab ± TKIs | $436,627 | 3.73 | $87,155 | Extendedly dominated by brexu-cel | |
Brexu-cel | $588,001 | 7.63 | $59,987 | $59,987 | |
Sponsor-corrected base case (deterministic) | Salvage chemotherapy | $173,481 | 0.68 | Ref. | Ref. |
Blinatumomab ± TKIs | $300,281 | 2.20 | $83,280 | Extendedly dominated by brexu-cel | |
Inotuzumab ± TKIs | $436,790 | 3.73 | $86,197 | Extendedly dominated by brexu-cel | |
Brexu-cel | $595,291 | 7.99 | $57,674 | $57,674 | |
1. OS and EFS extrapolation curves for brexu-cel (log normal; 5- and 25-year OS of 26% and 6%, respectively) | Salvage chemotherapy | $173,481 | 0.68 | Ref. | Ref. |
Blinatumomab ± TKIs | $300,281 | 2.20 | $83,280 | $83,280 | |
Inotuzumab ± TKIs | $436,790 | 3.73 | $86,197 | $89,096 | |
Brexu-cel | $590,591 | 3.83 | $132,353 | $1,589,015 | |
2. OS and EFS extrapolation curves for brexu-cel (Weibull; 5- and 25-year OS of 21% and 0%, respectively) | Salvage chemotherapy | $173,481 | 0.68 | Ref. | Ref. |
Blinatumomab ± TKIs | $300,281 | 2.20 | $83,280 | $83,280 | |
Inotuzumab ± TKIs | $436,790 | 3.73 | $86,197 | $89,096 | |
Brexu-cel | $592,923 | 2.37 | $247,789 | Dominated by inotuzumab ± TKIs | |
3. OS and EFS extrapolation curves for comparators | Salvage chemotherapy | $171,119 | 1.08 | Ref. | Ref. |
Blinatumomab ± TKIs | $301,321 | 3.58 | $52,284 | $52,284 | |
Inotuzumab ± TKIs | $409,595 | 2.42 | $178,994 | Extendedly dominated by brexu-cel | |
Brexu-cel | $595,291 | 7.99 | $61,410 | 66,555 | |
4. Duration of inotuzumab treatment | Salvage chemotherapy | $173,481 | 0.68 | Ref. | Ref. |
Blinatumomab ± TKIs | $300,281 | 2.20 | $83,280 | Extendedly dominated by inotuzumab ± TKIs | |
Inotuzumab ± TKIs | $332,372 | 3.73 | $52,015 | $52,015 | |
Brexu-cel | $591,660 | 7.99 | $57,177 | $60,879 | |
5. Duration of salvage chemotherapy | Salvage chemotherapy | $156,878 | 0.68 | Ref. | Ref. |
Blinatumomab ± TKIs | $300,281 | 2.20 | $94,185 | Extendedly dominated by brexu-cel | |
Inotuzumab ± TKIs | $436,790 | 3.73 | $91,633 | Extendedly dominated by brexu-cel | |
Brexu-cel | $590,101 | 7.99 | $59,234 | $59,234 | |
6. Acquisition costs for re-treatment with brexu-cel | Salvage chemotherapy | $173,481 | 0.68 | Ref. | Ref. |
Blinatumomab ± TKIs | $300,281 | 2.20 | $83,280 | Extendedly dominated by brexu-cel | |
Inotuzumab ± TKIs | $436,790 | 3.73 | $86,197 | Extendedly dominated by brexu-cel | |
Brexu-cel | $611,823 | 7.99 | $59,934 | $59,934 | |
7. Cure point | Salvage chemotherapy | $173,997 | 0.66 | Ref. | Ref. |
Blinatumomab ± TKIs | $301,103 | 0.62 | Dominated | Dominated by salvage chemotherapy | |
Inotuzumab ± TKIs | $435,891 | 3.03 | $110,746 | Extendedly dominated by brexu-cel | |
Brexu-cel | $601,794 | 7.76 | $60,262 | $60,262 | |
CADTH reanalysis 1 (log normal; 5- and 25-year OS of 26% and 6%, respectively) | Salvage chemotherapy | $156,957 | 1.12 | Ref. | Ref. |
Blinatumomab ± TKIs | $301,451 | 1.83 | $202,450 | Extendedly dominated by brexu-cel | |
Inotuzumab ± TKIs | $316,929 | 1.92 | $199,396 | Extendedly dominated by brexu-cel | |
Brexu-cel | $593,180 | 3.77 | $164,545 | $164,545 | |
CADTH reanalysis 2 (Weibull; 5- and 25-year OS of 21% and 0%, respectively) | Salvage chemotherapy | $156,764 | 1.10 | Ref. | Ref. |
Blinatumomab ± TKIs | $300,717 | 1.82 | $202,073 | Extendedly dominated by inotuzumab ± TKIs | |
Inotuzumab ± TKIs | $316,248 | 1.94 | $190,979 | $190,979 | |
Brexu-cel | $592,920 | 2.35 | $351,024 | $679,053 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-years; Ref. = reference; vs. = versus; brexu-cel = brexucabtagene autoleucel; TKIs = tyrosine kinase inhibitors.
Note: All steps were reported as deterministic analysis for comparability, while the results of the probabilistic analysis were reported for CADTH reanalysis 1 and 2.
Table 15: Disaggregated Summary of CADTH’s Economic Evaluation Results for Reanalysis 1
Treatment | Component | Value | Incremental (vs. reference)a | Incremental (sequential)a |
|---|---|---|---|---|
Discounted LYs | ||||
Salvage chemotherapy | Event-free | 0.68 | Ref. | Ref. |
Progressed disease | 1.01 | Ref. | Ref. | |
Total | 1.69 | Ref. | Ref. | |
Blinatumomab ± TKIs | Event-free | 1.30 | 0.62 | NA |
Progressed disease | 1.31 | 0.29 | NA | |
Total | 2.61 | 0.92 | NA | |
Inotuzumab ± TKI | Event-free | 1.46 | 0.78 | 0.78 |
Progressed disease | 1.12 | 0.11 | 0.11 | |
Total | 2.58 | 0.89 | 0.89 | |
Brexu-cel | Event-free | 3.05 | 2.38 | 1.60 |
Progressed disease | 1.99 | 0.97 | 0.87 | |
Total | 5.04 | 3.35 | 2.47 | |
Discounted QALYs | ||||
Salvage chemotherapy | Event-free | 0.38 | Ref. | Ref. |
Progressed disease | 0.74 | Ref. | Ref. | |
Total | 1.12 | Ref. | Ref. | |
Blinatumomab ± TKIs | Event-free | 0.89 | 0.51 | NA |
Progressed disease | 0.94 | 0.21 | NA | |
Total | 1.83 | 0.71 | NA | |
Inotuzumab ± TKI | Event-free | 1.12 | 0.73 | 0.73 |
Progressed disease | 0.80 | 0.07 | 0.07 | |
Total | 1.92 | 0.80 | 0.80 | |
Brexu-cel | Event-free | 2.34 | 1.96 | 1.22 |
Progressed disease | 1.43 | 0.69 | 0.63 | |
Total | 3.77 | 2.65 | 1.85 | |
Discounted costs ($) | ||||
Salvage chemotherapy | Acquisition | $512 | Ref. | Ref. |
Administration | $22,761 | Ref. | Ref. | |
Monitoring - EFS | $518 | Ref. | Ref. | |
Monitoring - PD | $2,195 | Ref. | Ref. | |
Subsequent treatment - drug acquisition | $66,941 | Ref. | Ref. | |
Subsequent treatment - administration | $3,893 | Ref. | Ref. | |
Allo-SCT | $18,704 | Ref. | Ref. | |
AEs | $96 | Ref. | Ref. | |
End of life | $41,353 | Ref. | Ref. | |
Total | $156,974 | Ref. | Ref. | |
Blinatumomab ± TKIs | Acquisition | $144,826 | $144,313 | NA |
Administration | $14,283 | -$8,478 | NA | |
Monitoring - EFS | $915 | $397 | NA | |
Monitoring - PD | $2,829 | $634 | NA | |
Subsequent treatment - drug acquisition | $74,517 | $7,575 | NA | |
Subsequent treatment - administration | $3,354 | -$539 | NA | |
Allo-SCT | $19,843 | $1,139 | NA | |
AEs | $102 | $6 | NA | |
End of life | $40,781 | -$571 | NA | |
Total | $301,451 | $144,476 | NA | |
Inotuzumab ± TKI | Acquisition | $204,781 | $204,269 | $204,269 |
Administration | $9,429 | -$13,332 | -$13,332 | |
Monitoring - EFS | $1,342 | $823 | $823 | |
Monitoring - PD | $2,432 | $236 | $236 | |
Subsequent treatment - drug acquisition | $15,827 | -$51,114 | -$51,114 | |
Subsequent treatment - administration | $2,607 | -$1,286 | -$1,286 | |
Allo-SCT | $39,649 | $20,944 | $20,944 | |
AEs | $31 | -$66 | -$66 | |
End of life | $40,830 | -$522 | -$522 | |
Total | $316,929 | $159,954 | $159,954 | |
Brexu-cel | Acquisition | $443,540 | $443,028 | $238,758 |
Administration | $19,839 | -$2,923 | $10,409 | |
Monitoring - EFS | $2,239 | $1,721 | $898 | |
Monitoring - PD | $4,304 | $2,108 | $1,872 | |
Subsequent treatment - drug acquisition | $64,997 | -$1,945 | $49,169 | |
Subsequent treatment - administration | $3,778 | -$115 | $1,171 | |
Allo-SCT | $14,855 | -$3,849 | -$24,793 | |
AEs | $335 | $238 | $304 | |
End of life | $39,294 | -$2,059 | -$1,537 | |
Total | $593,180 | $436,206 | $276,251 | |
Treatment | ICER vs. reference ($/QALY) | Sequential ICER ($/QALY) | ||
Salvage chemotherapy | Ref. | Ref. | ||
Blinatumomab ± TKIs | $202,450 | Extendedly dominated by brexu-cel | ||
Inotuzumab ± TKI | $199,396 | Extendedly dominated by brexu-cel | ||
Brexu-cel | $164,545 | $164,545 | ||
ICER = incremental cost-effectiveness ratio; LY = life-year; NA = not applicable; QALY = quality-adjusted life-year; Ref. = reference; vs. = versus.
aBoth dominated/extendedly dominated comparators were excluded from the sequential incremental results.
Table 16: Disaggregated Summary of CADTH’s Economic Evaluation Results for Reanalysis 2
Treatment | Component | Value | Incremental (vs. reference)a | Incremental (sequential)a |
|---|---|---|---|---|
Discounted LYs | ||||
Salvage chemotherapy | Event-free | 0.67 | Ref. | Ref. |
Progressed disease | 1.00 | Ref. | Ref. | |
Total | 1.67 | Ref. | Ref. | |
Blinatumomab ± TKIs | Event-free | 1.30 | 0.63 | NA |
Progressed disease | 1.28 | 0.28 | NA | |
Total | 2.58 | 0.91 | NA | |
Inotuzumab ± TKI | Event-free | 1.43 | 0.76 | 0.76 |
Progressed disease | 1.15 | 0.15 | 0.15 | |
Total | 2.59 | 0.92 | 0.92 | |
Brexu-cel | Event-free | 1.95 | 1.28 | 0.51 |
Progressed disease | 1.17 | 0.17 | 0.02 | |
Total | 3.12 | 1.45 | 0.53 | |
Discounted QALYs | ||||
Salvage chemotherapy | Event-free | 0.38 | Ref. | Ref. |
Progressed disease | 0.72 | Ref. | Ref. | |
Total | 1.10 | Ref. | Ref. | |
Blinatumomab ± TKIs | Event-free | 0.90 | 0.51 | NA |
Progressed disease | 0.92 | 0.20 | NA | |
Total | 1.82 | 0.71 | NA | |
Inotuzumab ± TKI | Event-free | 1.11 | 0.73 | 0.73 |
Progressed disease | 0.82 | 0.10 | 0.10 | |
Total | 1.94 | 0.84 | 0.84 | |
Brexu-cel | Event-free | 1.48 | 1.10 | 0.37 |
Progressed disease | 0.87 | 0.15 | 0.04 | |
Total | 2.35 | 1.24 | 0.41 | |
Discounted costs ($) | ||||
Salvage chemotherapy | Acquisition | $512 | Ref. | Ref. |
Administration | $23,003 | Ref. | Ref. | |
Monitoring - EFS | $513 | Ref. | Ref. | |
Monitoring - PD | $2,159 | Ref. | Ref. | |
Subsequent treatment - drug acquisition | $66,684 | Ref. | Ref. | |
Subsequent treatment - administration | $3,896 | Ref. | Ref. | |
Allo-SCT | $18,534 | Ref. | Ref. | |
AEs | $96 | Ref. | Ref. | |
End of life | $41,366 | Ref. | Ref. | |
Total | $156,764 | Ref. | Ref. | |
Blinatumomab ± TKIs | Acquisition | $144,485 | $143,973 | NA |
Administration | $14,367 | -$8,635 | NA | |
Monitoring - EFS | $907 | $395 | NA | |
Monitoring - PD | $2,784 | $625 | NA | |
Subsequent treatment - drug acquisition | $74,276 | $7,592 | NA | |
Subsequent treatment - administration | $3,351 | -$545 | NA | |
Allo-SCT | $19,646 | $1,112 | NA | |
AEs | $104 | $7 | NA | |
End of life | $40,797 | -$569 | NA | |
Total | $300,717 | $143,953 | NA | |
Inotuzumab ± TKI | Acquisition | $204,582 | $204,070 | $204,070 |
Administration | $9,462 | -$13,541 | -$13,541 | |
Monitoring - EFS | $1,328 | $815 | $815 | |
Monitoring - PD | $2,500 | $341 | $341 | |
Subsequent treatment - drug acquisition | $15,706 | -$50,978 | -$50,978 | |
Subsequent treatment - administration | $2,610 | -$1,286 | -$1,286 | |
Allo-SCT | $39,205 | $20,671 | $20,671 | |
AEs | $31 | -$65 | -$65 | |
End of life | $40,825 | -$542 | -$542 | |
Total | $316,248 | $159,485 | $159,485 | |
Brexu-cel | Acquisition | $443,643 | $443,130 | $239,061 |
Administration | $19,899 | -$3,104 | $10,437 | |
Monitoring - EFS | $2,026 | $1,514 | $699 | |
Monitoring - PD | $2,524 | $365 | $25 | |
Subsequent treatment - drug acquisition | $65,401 | -$1,283 | $49,695 | |
Subsequent treatment - administration | $3,816 | -$81 | $1,206 | |
Allo-SCT | $14,772 | -$3,762 | -$24,433 | |
AEs | $337 | $241 | $306 | |
End of life | $40,503 | -$863 | -$322 | |
Total | $592,920 | $436,157 | $276,672 | |
Treatment | ICER vs. reference ($/QALY) | Sequential ICER ($/QALY) | ||
Salvage chemotherapy | Ref. | Ref. | ||
Blinatumomab ± TKIs | $202,073 | Extendedly dominated by inotuzumab ± TKIs | ||
Inotuzumab ± TKI | $190,979 | $190,979 | ||
Brexu-cel | $351,024 | $679,053 | ||
ICER = incremental cost-effectiveness ratio; LY = life-year; NA = not applicable; QALY = quality-adjusted life-year; Ref. = reference; vs. = versus.
aBoth dominated/extendedly dominated comparators were excluded from the sequential incremental results.
Table 17: CADTH Price Reduction Analyses
Analysis | ICERs for brexu-cel vs. comparators ($/QALY) | ||
|---|---|---|---|
Price reduction | Sponsor base case | CADTH reanalysis 1 | CADTH reanalysis 2 |
No price reduction | λ < 57,702: salvage chemotherapy λ ≥ 57,702: brexu-cel | λ < 164,959: salvage chemotherapy λ ≥ 164,959: brexu-cel | λ < 190,977: salvage chemotherapy 190,977 > λ < 263,093: inotuzumab ± TKIs λ ≥ 263,093: brexu-cel |
10% | λ < 52,048: salvage chemotherapy λ ≥ 52,048: brexu-cel | λ < 148,584: salvage chemotherapy λ ≥ 148,584: brexu-cel | |
20% | λ < 46,394: salvage chemotherapy λ ≥ 46,394: brexu-cel | λ < 132,563: salvage chemotherapy λ ≥ 132,563: brexu-cel | |
30% | λ < 40,740: salvage chemotherapy λ ≥ 40,740: brexu-cel | λ < 116,541: salvage chemotherapy λ ≥ 116,541: brexu-cel | |
40% | λ < 35,087: salvage chemotherapy λ ≥ 35,087: brexu-cel | λ < 100,520: salvage chemotherapy λ ≥ 100,520: brexu-cel | |
50% | λ < 29,433: salvage chemotherapy λ ≥ 29,433: brexu-cel | λ < 84,498: salvage chemotherapy λ ≥ 84,498: brexu-cel | λ < 180,520: salvage chemotherapy λ ≥ 180,520: brexu-cel |
60% | λ < 23,779: salvage chemotherapy λ ≥ 23,779: brexu-cel | λ < 68,477: salvage chemotherapy λ ≥ 68,477: brexu-cel | λ < 146,418: salvage chemotherapy λ ≥ 146,418: brexu-cel |
70% | λ < 18,125: salvage chemotherapy λ ≥ 18,125: brexu-cel | λ < 52,455: salvage chemotherapy λ ≥ 52,455: brexu-cel | λ < 112,316: salvage chemotherapy λ ≥ 112,316: brexu-cel |
80% | λ < 12,471: salvage chemotherapy λ ≥ 12,471: brexu-cel | λ < 36,434: salvage chemotherapy λ ≥ 36,434: brexu-cel | λ < 78,214: salvage chemotherapy λ ≥ 78,214: brexu-cel |
90% | λ < 6,818: salvage chemotherapy λ ≥ 6,818: brexu-cel | λ < 20,412: salvage chemotherapy λ ≥ 20,412: brexu-cel | Λ < 44,111: salvage chemotherapy λ ≥ 44,111: brexu-cel |
100% | λ < 1,164: salvage chemotherapy λ ≥ 1,164: brexu-cel | λ < 4,390: salvage chemotherapy λ ≥ 4,390: brexu-cel | λ < 10,009: salvage chemotherapy λ ≥ 10,009: brexu-cel |
λ = willingness-to-pay threshold; ICER = incremental cost-effectiveness ratio; vs. = versus.
Note: This table presents the willingness-to-pay threshold at which each treatment is cost-effective for a given price reduction. Only nondominated strategies are presented.
Scenario analysis | Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|---|
Time horizon: 5 years (reanalysis 1) | Salvage chemotherapy | $153,502 | 0.54 | Ref. |
Blinatumomab ± TKI | $294,857 | 0.79 | Extendedly dominated by Inotuzumab ± TKI | |
Inotuzumab ± TKI | $312,209 | 1.48 | $169,280 | |
Brexu-cel | $574,838 | 1.70 | $1,172,608 | |
Time horizon: 5 years (reanalysis 2) | Salvage chemotherapy | $152,885 | 0.55 | Ref. |
Blinatumomab ± TKI | $295,154 | 0.79 | Extendedly dominated by Inotuzumab ± TKI | |
Inotuzumab ± TKI | $311,873 | 1.49 | $169,880 | |
Brexu-cel | $578,155 | 1.63 | $1,856,886 | |
Tisa-cel included as a comparator (reanalysis 1) | Salvage chemotherapy | $156,698 | 1.12 | Ref. |
Blinatumomab ± TKI | $300,700 | 1.83 | Extendedly dominated by tisa-cel | |
Inotuzumab ± TKI | $316,480 | 1.92 | Extendedly dominated by tisa-cel | |
Tisa-cel | $505,542 | 5.08 | $88,092 | |
Brexu-cel | $593,023 | 5.08 | Dominated | |
Tisa-cel included as a comparator (reanalysis 2) | Salvage chemotherapy | $157,162 | 1.15 | Ref. |
Blinatumomab ± TKI | $301,596 | 1.82 | Extendedly dominated by tisa-cel | |
Inotuzumab ± TKI | $317,034 | 1.92 | Extendedly dominated by tisa-cel | |
Tisa-cel | $503,142 | 3.15 | $172,990 | |
Brexu-cel | $593,259 | 3.15 | Dominated | |
No re-treatment with brexu-cel (reanalysis 1) | Salvage chemotherapy | $156,888 | 1.15 | Ref. |
Blinatumomab ± TKIs | $301,699 | 1.82 | Extendedly dominated by brexu-cel | |
Inotuzumab ± TKIs | $316,916 | 1.92 | Extendedly dominated by brexu-cel | |
Brexu-cel | $576,381 | 3.75 | $161,370 | |
No re-treatment with brexu-cel (reanalysis 2) | Salvage chemotherapy | $157,221 | 1.13 | Ref. |
Blinatumomab ± TKIs | $301,624 | 1.82 | Extendedly dominated by Inotuzumab ± TKI | |
Inotuzumab ± TKIs | $316,251 | 1.92 | $200,623 | |
Brexu-cel | $577,537 | 2.31 | $677,600 | |
OS extrapolation curve for brexu-cel using generalized gamma | Salvage chemotherapy | $157,693 | 1.12 | Ref. |
Blinatumomab ± TKIs | $300,576 | 1.83 | Extendedly dominated by brexu-cel | |
Inotuzumab ± TKIs | $316,407 | 1.92 | Extendedly dominated by brexu-cel | |
Brexu-cel | $597,712 | 6.26 | $85,559 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; Ref. = reference.
Appendix 5: Submitted Budget Impact Analysis (BIA) and CADTH Appraisal
Note that this appendix has not been copy-edited.
Table 19: Summary of Key Takeaways
Key Takeaways of the BIA |
|---|
|
Summary of Sponsor’s BIA
The sponsor submitted a BIA estimating the budget impact of brexu-cel for treating adult patients with R/R B-cell precursor acute lymphoblastic leukemia (ALL). The BIA’s base case was undertaken from a publicly funded drug plan perspective, considering drug costs over a 3-year time horizon. The sponsor also provided a scenario analysis from the public health care payer perspective, which included drug acquisition costs and health care resource use (i.e., laboratory tests, physician’s visits, and terminal care costs).
The analytic framework, which used an epidemiology-based approach, leveraged data from multiple sources in the literature and from assumptions to determine the estimated population size (Figure 6). The sponsor compared a reference scenario where brexu-cel is not reimbursed, with a new drug scenario where brexu-cel is publicly funded as per the Health Canada indication. Treatments available in the reference scenario included blinatumomab ± TKI, inotuzumab ± TKI, salvage chemotherapy, TKI monotherapy, and tisa-cel. The TKIs included in the analysis were ponatinib and dasatinib, while salvage chemotherapy regimens included FLAG-IDA (fludarabine, cytarabine, idarubicin, filgrastim) and hyper CVAD (alternating courses of cyclophosphamide, vincristine, doxorubicin, and dexamethasone with courses of methotrexate, and cytarabine).
Brexu-cel costs were obtained from the sponsor but did not include drug costs for brexu-cel conditioning and bridging chemotherapy. The calculation of costs for blinatumomab ± TKI and inotuzumab ± TKI assumed that 27% of patients were Philadelphia chromosome positive, and were treated with a 50/50 weighted bucket of ponatinib and dasatinib.6,7 The sponsor also assumed that patients treated with TKIs would receive a maximum 90-day course of therapy.16 Regimen costs like salvage chemotherapy were obtained from previous CADTH reports and calculated as a 50/50 weighted bucked of FLAG-IDA and hyper CVAD.17,18 Annual drug acquisition costs for comparators were obtained from the pharmacoeconomic report and previous CADTH reports.1,17,18 Costs of subsequent therapies were not included in the analysis.
The sponsor estimated that brexu-cel will reach a market share of ██% after 3 years. They also assumed that 0% of patients would receive TKI monotherapy or tisa-cel. Key inputs to the BIA are documented in Table 20.
Figure 6: Sponsor’s Estimation of the Size of the Eligible Population
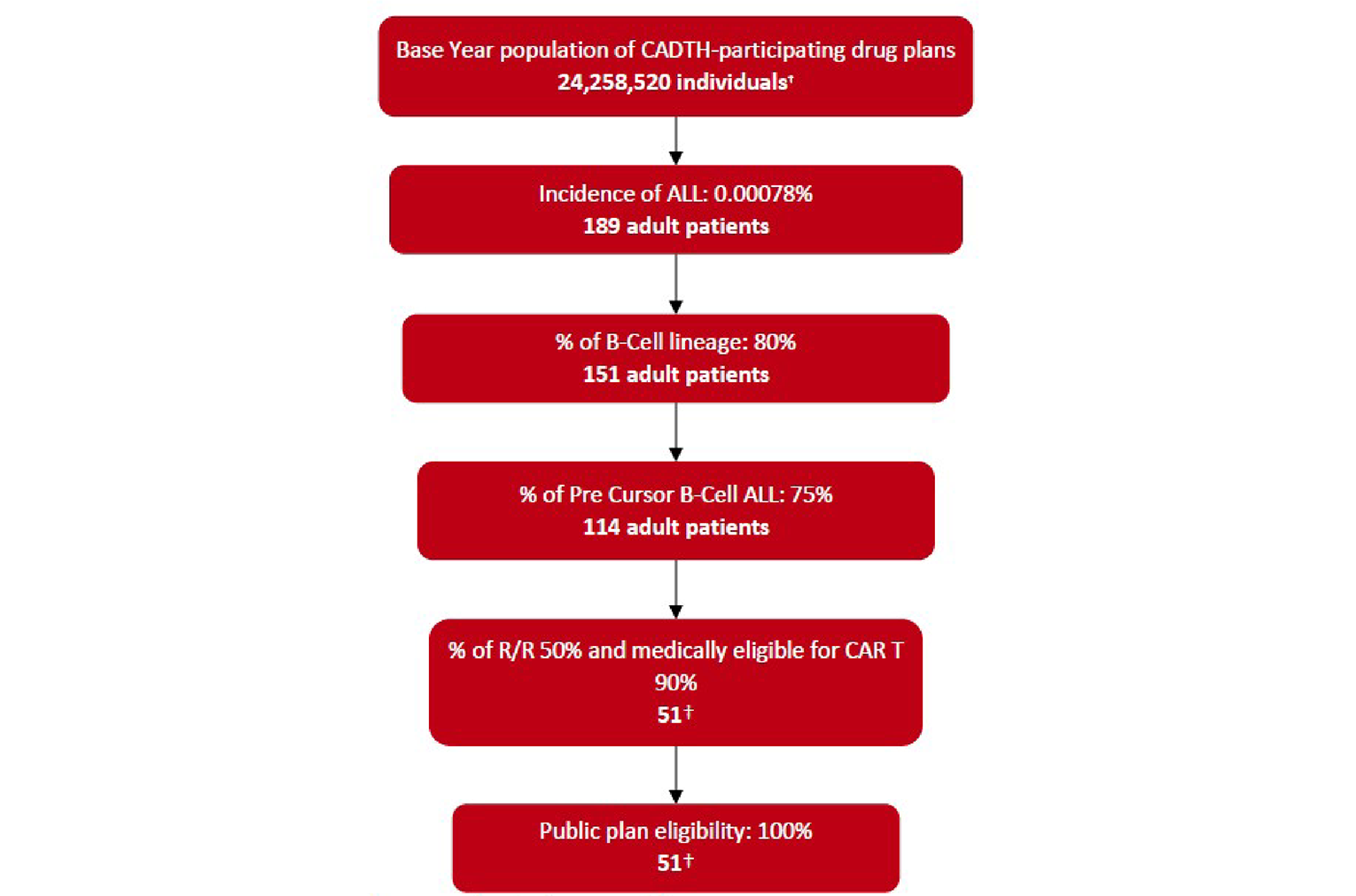
Source: Sponsor’s Budget Impact Submission.16
Table 20: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1/year 2/year 3 if appropriate) |
|---|---|
Target population | |
Number of patients eligible for drug under review | 53 / 54 / 55 |
Market Uptake (3 years) | |
Uptake (reference scenario) | |
Blinatumomab ± TKI | 40% / 40% / 40% |
Inotuzumab ± TKI | 40% / 40% / 40% |
Salvage therapy | 20% / 20% / 20% |
TKI monotherapy | 0% / 0% / 0% |
Tisagenlecleucel | 0% / 0% / 0% |
Uptake (new drug scenario) | |
Brexu-cel | ██% / ██% / ██% |
Blinatumomab ± TKI | ██% / ██% / ██% |
Inotuzumab ± TKI | ██% / ██% / ██% |
Salvage therapy | ██% / ██% / ██% |
TKI monotherapy | █% / █% / █% |
Tisagenlecleucel | █% / █% / █% |
Cost of treatment (per patient) | |
Cost of treatment over | |
Brexu-cel | $533,523 |
Blinatumomab ± TKI | $121,520 |
Inotuzumab ± TKI | $154,377 |
Salvage therapy | $6,015 |
TKI monotherapy | $19,881 |
Tisagenlecleucel | $533,523 |
Brexu-cel = brexucabtagene autoleucel; TKI = tyrosine kinase inhibitor.
Summary of the Sponsor’s BIA Results
The estimated budget impact of funding brexu-cel for the adjuvant treatment of adult patients with R/R B-cell precursor ALL from the drug plan perspective was $4,730,216, $6,396,738, and $7,647,071 for years 1, 2, and 3, respectively. The 3-year total budget impact was $18,774,025.
In the health care payer perspective, the estimated budget impact was $4,662,904, $6,306,653, and $7,539,891 for years 1, 2, and 3, respectively. The 3-year total budget impact was $18,509,448.
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Treatment duration was overestimated and inconsistently calculated for comparators: The drug acquisition costs for the BIA were obtained from 2 sources: the pharmacoeconomic cost-utility analysis (CUA) model and CADTH reports. In the CUA, the duration of treatment for each of the comparators was incorporated in the model using distinct approaches, as previously discussed. For blinatumomab, the sponsor modelled treatment duration using the number of average treatment cycles patients received treatment during the TOWER trial, while for inotuzumab and salvage chemotherapy treatment duration was assumed to be the maximum number of cycles for each therapy (5x 28-day cycles for inotuzumab, and 4x 28-day cycles for salvage chemotherapy). While for ponatinib, the sponsor assumed a treatment duration of 90 days. According to clinical experts consulted for this review, patients can remain on TKIs for longer than 90 days but agreed that 90 days would be an average treatment duration. However, the use of distinct approaches to account for comparators’ treatment duration hindered comparability of drug acquisition costs and overestimated the cost of inotuzumab and salvage chemotherapy.
For consistency, CADTH derived all acquisition costs from the CADTH’s CUA reanalysis estimates, where CADTH standardized the approach to calculating treatment duration by changing inotuzumab and salvage chemotherapy maximum treatment duration to their median number of days of treatment from the INO-VATE trial.15
In addition, CADTH also conducted a scenario analysis using the CADTH CUA reanalysis’ estimates for the health care payer perspective (e.g., administration costs, AEs costs, end-of-life costs).
Subsequent treatment costs, including re-treatment with brexu-cel, were omitted: The sponsor did not consider the drug acquisition costs of subsequent treatments in the BIA. No justification regarding their exclusion were provided. In addition, the sponsors did not consider the costs of re-treatment with brexu-cel, which occurred in a total of 4% of patients (2 out of 55 patients who ultimately received brexu-cel) in the ZUMA-3 study.
CADTH could not address the issue of excluding subsequent treatment costs from the analysis. In addition, not considering re-treatment with brexu-cel underestimate treatment costs, thus favouring brexu-cel.
Bridging and consolidating chemotherapy costs for patients receiving brexu-cel were omitted: The sponsor did not include the drug acquisition costs for bridging and consolidating chemotherapy. However, bridging and consolidating chemotherapy were pivotal to the successful administration of brexu-cel. Although bridging and consolidating chemotherapy acquisition costs were not main drivers in the CUA analysis, all relevant treatments should be included in the BIA, and excluding costs of the evaluated intervention favours brexu-cel results.
CADTH included the drug acquisition costs of bridging and consolidating therapies obtained from the CADTH’s CUA reanalysis’ estimates in the BIA.
Additional limitations were identified but were not considered to be key limitations:
Market shares for tisa-cel: The submitted BIA assumed that tisa-cel would have a market share of 0% in the analysis. Input provided by clinical experts consulted for this review indicated that tisa-cel would be expected to account for 10% of the current market share. The inclusion of tisa-cel in the BIA would increase the total costs of treatment for patients with R/R B-cell ALL. However, given tisa-cel’s market share would remain unchanged in both the reference and new drug scenarios, it is anticipated to not have an impact on the estimated incremental budget impact of reimbursing brexu-cel.
CADTH Reanalyses of the BIA
CADTH’s base-case case revised the costs of comparators, and costs of bridging and consolidation chemotherapy.
Table 21: CADTH Revisions to the Submitted BIA
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Correctionsa to sponsor’s base case (None) | ||
Changes to derive the CADTH base case | ||
1. Change in treatment costs for comparators | Blinatumomab: $116,152.14 Inotuzumab: $149,008.68 Salvage chemotherapy: $6,014.66 TKIs: $19,881.14 | Blinatumomab: $142,152.35 Inotuzumab: $200,903.85 Salvage chemotherapy: $386.06 TKIs: $16.617.93 |
2. Incorporate bridging and consolidation drug acquisition costs | No | Yes |
CADTH base case | 1 + 2 | |
AE = adverse events; BIA = budget impact analysis.
The results of the CADTH stepwise reanalysis are presented in summary format in Table 22 and a more detailed breakdown is presented in Table 23.
Based on CADTH’s base-case and using a drug plan perspective, the expected budget impact for funding brexu-cel as a treatment for patients with R/R B-cell ALL was expected to be $4,408,819 in Year 1, $5,962,938 in Year 2, and $7,128,931 in Year 3, with a 3-year budget impact of $17,500,689.
Table 22: Summary of the CADTH Reanalyses of the BIA
Stepped analysis | Three-year total ($) |
|---|---|
Submitted base case | 18,774,025 |
CADTH reanalysis 1 | 17,463,853 |
CADTH reanalysis 2 | 18,810,861 |
CADTH base case | 17,500,689 |
BIA = budget impact analysis.
CADTH also conducted additional scenario analyses to address remaining uncertainty. Results are provided in Table 23:
Price reduction of 75% in brexu-cel cost.
Price reduction of 89% in brexu-cel cost.
Updated values for administration, monitoring, AEs, allo-SCT costs obtained from CADTH’s CUA reanalysis for the health care payer perspective analysis.
Results of CADTH’s scenario analyses demonstrate that CADTH’s base case was sensitive to changes in brexu-cel’s price.
Table 23: Detailed Breakdown of the CADTH Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
Submitted base case | Reference | $5,868,183 | $5,952,519 | $6,038,159 | $6,125,123 | $18,115,801 |
New drug | $5,868,183 | $10,682,736 | $12,434,897 | $13,772,193 | $36,889,826 | |
Budget impact | $0 | $4,730,216 | $6,396,738 | $7,647,071 | $18,774,025 | |
CADTH base case | Reference | $7,410,825 | $7,517,332 | $7,625,484 | $7,735,309 | $22,878,126 |
New drug | $7,410,825 | $11,926,151 | $13,588,422 | $14,864,241 | $40,378,814 | |
Budget impact | $0 | $4,408,819 | $5,962,938 | $7,128,931 | $17,500,689 | |
CADTH scenario analysis 1: 75% price reduction | Reference | $7,410,825 | $7,517,332 | $7,625,484 | $7,735,309 | $22,878,126 |
New drug | $7,410,825 | $7,442,623 | $7,524,378 | $7,614,399 | $22,581,400 | |
Budget impact | $0 | -$74,709 | -$101,106 | -$120,911 | -$296,726 | |
CADTH scenario analysis 2: 89% price reduction | Reference | $7,410,825 | $7,517,332 | $7,625,484 | $7,735,309 | $22,878,126 |
New drug | $7,410,825 | $6,605,698 | $6,392,423 | $6,261,095 | $19,259,216 | |
Budget impact | $0 | -$911,634 | -$1,233,061 | -$1,474,214 | -$3,618,910 | |
CADTH scenario analysis 3: updated values for health care payer perspective | Reference | $13,419,423 | $13,612,283 | $13,808,124 | $14,006,994 | $41,427,402 |
New drug | $13,419,423 | $17,973,286 | $19,706,451 | $21,058,713 | $58,738,450 | |
Budget impact | $0 | $4,361,003 | $5,898,327 | $7,051,719 | $17,311,048 |
BIA = budget impact analysis.
Ethics Review
Abbreviations
ALL
acute lymphoblastic lymphoma
allo-SCT
allogeneic stem cell transplant
AYA
adolescent and young adult
Brexu-cel
brexucabtagene autoleuce
CAR T
chimeric antigen receptor T
R/R
relapsed or refractory
Summary
To identify ethical considerations relevant to the use of brexucabtagene autoleucel (brexu-cel ) for the treatment of adult patients with relapsed or refractory B-cell precursor acute lymphoblastic leukemia (ALL), input provided by patient groups, clinician groups, and provincial drug programs was reviewed along with information collected through direct engagement with clinical experts and relevant literature.
Ethical considerations arising in the context of relapsed or refractory (R/R) ALL highlight impacts on patients as well as disparities in diagnosis, treatment, and treatment outcomes among racialized or socioeconomically disadvantaged populations. Challenges associated with accessing and enduring current second-line treatments, particularly allogeneic stem cell transplant (allo-SCT), were noted potential barriers to treatment.
Ethical considerations arising in the evidence used to evaluate brexu-cel highlight limitations related to the absence of long-term effectiveness and safety data and representativeness of trial participants in the ZUMA-3 study.
Ethical considerations related to the use of brexucabtagene autoleucel, as with other chimeric antigen receptor T (CAR T) therapies, highlight challenges related to the location of specialized CAR T treatment centres and the geographic, financial, and referral barriers faced by patients who do not live near these treatment centres. Cell use, ownership, and challenges around informed consent were also highlighted as considerations in CAR T manufacturing and delivery.
Ethical considerations for health systems related to the implementation of brexu-cel involve challenges of sustainability to the health care system due to the high cost of CAR T-cell therapies and the related challenge of navigating limited capacity (e.g., resources and costs to the health care system) to keep up with demand for new CAR T-cell therapies.
Objective
To identify and describe ethical considerations associated with the use of brexu-cel for the treatment of adult patients with R/R B-cell precursor ALL. These considerations include those associated with the broader context of ALL, the evidentiary base and use of brexu-cel as a CAR T-cell therapy, and other considerations relevant to health care systems.
Research Questions
This report will address the following research questions:
What ethical considerations arise in the context of R/R ALL in adult patients?
What ethical considerations arise related to the evidence (e.g., clinical trial data and economic models) used to evaluate brexucabtagene autoleucel?
What ethical considerations arise in the use of brexucabtagene autoleucel as a CAR T-cell therapy, for clinicians, patients, and their caregivers?
What ethical considerations for health care systems are involved in the context of brexu-cel as a CAR T-cell therapy?
Methods
Overview
To identify ethical considerations relevant to the use of brexu-cel in the treatment of adults with R/R ALL, this Ethics Review Report was driven by relevant questions identified in the European Network for Health Technology Assessment (EUnetHTA)Core Model 3.0, Ethics Analysis Domain,1 and supplemented by relevant questions from the Equity Checklist for Health Technology Assessment.2 These guiding questions were organized to respond to the 4 research questions posed. In response to each of these 4 questions, this report investigated ethical considerations in 4 domains of interest:
People living with R/R ALL and their caregivers (i.e., disparities in incidence, treatment, or outcomes; challenges related to diagnosis or clinical care; factors that might prevent patients from gaining access to therapies).
The evidence used to demonstrate the benefits, harms, and value of brexu-cel (i.e., ethical considerations in relevant clinical trials, including their representativeness, choice of outcome measures, appropriateness of analytical methods and models to all population groups; and ethical considerations related to the data or assumptions in the economic evaluation).
The use of brexu-cel as a CAR T-cell therapy, including considerations related to benefits and harms to patients, relatives, caregivers, clinicians, or society; and considerations related to access to these therapies.
The uptake of CAR T-cell therapies, including brexucabtagene autoleucel, in health care systems, including considerations related to the distribution of health care resources.
These domains were explored through a review and synthesis of project inputs and relevant literature to highlight ethical considerations across each of them.
Data Collection Approach: Review of Project Inputs and Literature
Data to inform this Ethics Review Report drew from an identification of ethical considerations (e.g., claims related to potential harms, benefits, equity, justice, resource allocation, and ethical issues in the evidentiary basis) in the patient and clinician group, clinical expert, and drug program input collected in the course of this review, as well as a complementary review of the published literature. Ongoing collaboration and communication with the CADTH review team also assisted in the clarification and identification of ethical considerations raised.
Review of Project Inputs
Over the course of this CADTH review, a single reviewer collected and considered input from a variety of sources to inform this Ethics Review Report. All input was reviewed for content related to ethical considerations (e.g., claims related to potential harms, benefits, equity, justice, resource allocation, and ethical issues in an evidentiary basis) relevant to the research questions driving this report. In addition to the published literature, this report considered the following sources:
sponsor’s submission
clinician group input that was coordinated by Cell Therapy Transplant Canada
patient group input that was gathered by the Leukemia and Lymphoma Society of Canada
drug program input that was submitted to CADTH
discussion with a panel of clinical experts (n = 4) directly engaged by CADTH over the course of this reimbursement review. This discussion came through 2 teleconferences with 2 clinical experts and a panel discussion consisting of all 4 experts
CADTH clinical and health economics reviewers, who were engaged to identify domains of ethical interest arising from their reviews and identify further questions or sources to pursue in the Ethics Review Report
Literature Search Methods
Three literature searches were conducted by an information specialist on key resources, including MEDLINE All (1946—) via Ovid and Philosopher’s Index via Ovid, the Cumulative Index to Nursing and Allied Health Literature via EBSCO, and Scopus. Two search strategies made use of strategies developed for previous CADTH reports and were updated for this review.3,4 Additionally, 1 strategy was created de novo for this review. Duplicates were removed by manual deduplication in EndNote.
All 3 search strategies comprised both controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concepts were relapsed/refractory ALL, Tecartus (brexucabtagene autoleucel), and CAR T-cell therapies.
Updated searches were conducted for the concepts of Tecartus (brexucabtagene autoleucel), which made use of a previous search completed on December 8, 2020,4 and CAR T-cell therapies, which made use of a previous search completed on July 05, 2022.3 For the current report, database searches for Tecartus (brexucabtagene autoleucel) and CAR T-cell therapies were rerun to capture any articles published or made available since the initial search date. A de novo search was completed for the concept of relapsed/refractory ALL; results were not limited by publication date. The updated searches and the de novo search were completed on October 21, 2022, and limited to English-language documents. CADTH-developed search filters were applied to limit retrieval to citations related to ethical concepts or considerations.
Grey literature (literature that is not commercially published) was identified by searching sources listed in relevant sections of the Grey Matters: A Practical Tool For Searching Health-Related Grey Literature checklist.5 The grey literature search for ethical considerations was conducted on October 25, 2022. The main search concepts were ALL, Tecartus (brexucabtagene autoleucel), and CAR T-cell therapies. The search results for ALL and Tecartus (brexucabtagene autoleucel) were not limited by publication date, because grey literature searches on these concepts had not previously been completed. The search results for CAR T-cell therapies were limited to documents published since July 5, 2022, when a grey literature search for the concept was last completed.3 All search results were limited to English-language documents. Google was used to search for additional internet-based materials. These searches were supplemented by reviewing bibliographies of key papers and through contacts with experts, as appropriate.
Literature Screening and Selection
The ethics review reports for 4 previous CADTH reviews on CAR T-cell therapies3,4,6,7 served as foundational sources for this Ethics Review Report. Additional literature retrieved according to the search methods detailed in the previous section were screened and selected across 2 stages. In the first stage, titles and abstracts of citations retrieved were screened for relevance by a single reviewer. Articles were retrieved for full-text review if they identified ethical considerations or provided normative analysis (i.e., focusing on “what ought to be” through argumentation) or empirical research (i.e., focusing on “what is” through observation) of ethical considerations related to:
living with, or treating, R/R ALL
the evidence on, use of, or implications of brexu-cel as a CAR T-cell therapy for adults with R/R ALL
the implications, or use, of CAR T-cell therapies generally.
In the second stage, full-text publications categorized as “retrieve” were then reviewed by the same reviewer. Reports meeting the above criteria were included in the review, and reports that did not meet these criteria were excluded.
As a parallel process, grey literatures, and other sources drawn from relevant bibliographies or in consultation with experts or other CADTH reviewers were retrieved and reviewed following the selection criteria listed previously.
Data Analysis
Data analysis for this Ethics Review Report included the collection, coding, and thematic analysis of data drawn from the literature and project inputs, driven by the 4 research questions guiding this report. The reviewer conducted 3 iterative cycles of coding and analysis to abstract, identify, and synthesize relevant ethical considerations in the literature and from relevant project inputs.
In the initial coding phase, the main ethical considerations posed and discussed in the 4 previous CADTH ethics review reports were abstracted. These considerations were used to guide questions asked of the clinical experts and the reviews of project inputs and additional literature sources. In the second coding phase, publications retrieved from the updated search, as well as project input sources, were reviewed for ethical content (e.g., claims related to potential harms, benefits, equity, justice, resource allocation, and ethical issues in the evidentiary basis). Once identified, claims related to ethical content were coded using methods of qualitative description.8 In the final coding phase, major themes and subcodes were identified through repeated readings of the data8 and comparisons to initial themes and considerations that emerged in the previous CADTH reviews. These were then summarized into thematic categories within each domain or research question. When ethical content emerged that did not fit into these categories or the domains outlined in the research questions, this was noted, as were discrepancies or conflicts between ethical considerations or values identified between project sources or within thematic categories. Data analysis was iterative, and themes identified in the literature, in project inputs, and during consultations with clinical experts were used to further refine and re-interpret ethical considerations identified.
Data collected and analyzed from these sources were thematically organized and described according to the 4 research questions and domains driving this report. The results of this analysis and its limitations and conclusions are described in the following section.
Results
Description of Included Sources
Four previous CADTH reports of ethical considerations in the context of CAR T-cell therapies provided the foundation for both the literature search strategy and thematic analysis of ethical considerations. These 4 reports had undertaken reviews of relevant ethical considerations in the use of:
tisagenlecleucel for adults with R/R diffuse large B-cell lymphoma or children and young adults with R/R ALL6
brexu-cel for adults with R/R mantle cell lymphoma4
axicabtagene ciloleucel for adults with R/R large B-cell lymphoma.3,7
The literature search identified a total of 425 results. Following title and abstract screening, 394 citations were excluded and 31 potentially relevant publications from the electronic search were retrieved for full-text review. Of these potentially relevant publications, 21 were excluded because they did not discuss ethical considerations of brexu-cel or R/R ALL in adult populations (n = 17) or they were not relevant to the context of those living in Canada (n = 4). Ten publications met the inclusion criteria and were included in this report. Four additional studies were retrieved from backward searching of included publications’ reference lists.
In addition to the 4 previous CADTH reviews, 14 publications were used to inform this report. Of these publications, 6 discussed ethical considerations in the context of access, use, or implementation of CAR T-cell therapies; 1 discussed challenges to the ZUMA-3 study; 3 discussed disparities in ALL treatment; and the remaining 4 discussed challenges around ALL more broadly. Details regarding the characteristics of included publications are reported in Table 1.
In addition to sources from the published literature, data to inform this Ethics Review Report drew from a review of the patient group input, clinician group input, drug program input, and consultation with clinical experts (all hematologists practising in cancer centres across Canada) engaged by CADTH for this review. A description and summary of these sources are included in the Clinical Review Report.
Key Ethical Considerations
Diagnosis and Experiences of ALL
Disparities in Incidence, Treatment, and Outcomes of ALL
Literature has suggested that there are disparities in incidence of ALL where it has been identified as more prevalent among Hispanic/Latinx populations in the US.9 Disparities in access to treatment and specialized care for ALL have also been identified for Hispanic and non-Hispanic Black populations in the US.10 These studies have described how disparities in access to care and treatment are further exacerbated for those living in socioeconomically deprived areas that are less likely to host specialized treatment centres.10 Patients with limited resources or social support may also face challenges in accessing treatment given the costs of travel, childcare, and missing work.10 These studies point toward the importance of attending to social determinants of health by addressing socioeconomic and institutional barriers of access for disadvantaged populations in the context of those living in Canada.
Clinical experts and published literature also indicated that there are disparities in outcomes across demographics related to age and race.9-11 Significant disparities have been noted regarding 5-year survival outcomes between pediatric, adolescent and young adult (AYA), and adult populations.9 According to US-based data, while 5-year survival for children has been noted to be upward of 90%, for AYAs this figure drops to 60% to 85%, and it falls even further, to 30%, for adults, who make up about 54% of ALL deaths.9,11 In Canada, clinical experts suggested that much older patients (those aged 70 years of older) may expect even poorer outcomes because they are generally considered ineligible for a common second-line treatment, allo-SCT.
Patient Experiences of ALL
Patient group input received by CADTH has highlighted several physical and psychosocial impacts of living with ALL. Some of the more commonly identified physical symptoms included fatigue, weakness, loss of appetite, weight loss, and bone or joint pain. Dealing with these symptoms, and the effects they could have on people’s ability to work, travel, exercise, or complete daily tasks, were all described as interrupting life goals and contributing to feelings of isolation, depression, stress, and anxiety.
Broadly speaking, patient group input reported that patients want more effective treatment options that have a greater degree of certainty that their ALL would respond. Desired outcomes included improved overall survival and better quality of life. Some individual respondents in the patient group input indicated that the amount of time travelling to and spent in hospital for current (or former) treatments (e.g., chemotherapy and stem cell transplant) was challenging.
Both clinical experts and reports in the published literature have suggested that ALL may be particularly disruptive for AYA populations because this is often a period of transition involving critical physical and psychological development.11 Educational attainment, career advancement, and financial stability were all identified as being negatively affected for AYA patients.11 AYAs may also be more likely to experience challenges to their self-esteem due to their reliance on others for support and dramatic shifts in physical appearance due to treatment side effects.11 Clinical experts noted that this population may also have limited outside support because they are transitioning from childhood families or homes and may not have had the opportunity to develop new relationships or support systems yet. This age group may also face serious challenges regarding preserving fertility because some may hope to build a family in the future.11 Authors have suggested that the psychological distress associated with these challenges belies the importance of having multidisciplinary teams that can attend to the unique challenges for this population.11,12
Authors have also suggested that older patients may be more likely to experience hopelessness in relation to their care and treatment for acute leukemia broadly.13 This highlights the importance of fostering a strong relationship between providers and their patients as 1 way to mitigate the likelihood of developing hopelessness or depression for this population.12,13
Treatment of Relapsed and Refractory ALL
In its submission, the sponsor has indicated that complete remission is achieved in more than 80% of adult patients with newly diagnosed ALL after first-line chemotherapy. However, after further consolidation therapy and maintenance chemotherapy, the majority of these patients will ultimately relapse. For patients with R/R B-cell precursor ALL, induction of a complete remission is the first goal. Treatment options for remission induction therapy in R/R disease include cytotoxic chemotherapy regimens, targeted therapies (e.g., blinatumomab, inotuzumab ozogamicin), allo-SCT, and CAR T-cell therapy. For people who have Philadelphia chromosome–positive subtype ALL, tyrosine kinase inhibitors with or without chemotherapy may also be considered.
Clinical experts suggested that 2 of these therapies, blinatumomab and allo-SCT, were particularly challenging treatment options to access and endure. Blinatumomab needs to be delivered as an inpatient treatment and can involve a number of challenges associated with the expenses of travel, lodging, and time outside of 1’s community.10 Clinical experts described how allo-SCT eligibility varies across provinces, particularly in relation to age, which could lead to fairness challenges. Because allo-SCT also requires that a suitable match be found, clinical experts and authors suggested that there may be some disparities in access because transplant registries are often not representative of racialized populations.10 Clinician group input has indicated that allo-SCT requires that patients stay in hospital for 4 weeks and are closely followed for another 100 days. As such, allo-SCT can be a very intimidating and burdensome treatment to undergo, and patients may live in fear of remission.14
Ethics of Evidence and Evaluation of Brexucabtagene Autoleucel
The primary clinical evidence used to evaluate brexu-cel for R/R ALL is drawn from the ongoing phase I/II, open-label, single arm ZUMA-3 study. While the CADTH clinical report for this reimbursement review has only focused on the phase II component of the ZUMA-3 study in its review, it has described the ZUMA-3 study as being a noncomparative study with a small sample size and relatively short-term data represent limitations to the interpretation of trial results.
When considering the populations included in this trial, clinical experts noted that the exclusion of hepatitis B or C and HIV-positive patients from this trial may have been unnecessary. Given the availability of antivirals for hepatitis B and C, and antiretrovirals for HIV that help to make viral loads undetectable, experts suggested that the exclusion of these populations from the ZUMA-3 study could be considered discriminatory. Similarly, clinical experts were concerned that the exclusion of patients with an Eastern Cooperative Oncology Group (ECOG) performance status greater than 1 may not be reflective of clinical practice. They clarified by noting that many patients with R/R B-cell precursor ALL have a higher (or worse) ECOG performance status.
Previous CADTH reviews have identified ongoing underrepresentation of Black patients in clinical trials for CAR T-cell therapies.3In the ZUMA-3 study, 72% of patients in the intention-to-treat set were white, and 67% of those who actually received treatment were white. Given disparities in treatment access and outcomes noted for Black and Hispanic patients with R/R ALL noted previously,10,15 limited access to clinical trials may only serve to widen disparities. In addition to concerns about the limited inclusion of Black patients in CAR T clinical trials, young adult, older adults, and other nonwhite patients tend to be underrepresented in these trials, potentially limiting the applicability of the results to these populations.3 The mean age of the ZUMA-3 study participants who received treatment was 42.2 years, with 15% of participants being identified as aged 65 years or older.
Despite advances in clinical trial data for CAR T-cell therapies and 15 years of data on patients, the lack of long-term safety and efficacy data for CAR T-cell therapies has been widely discussed by clinical experts and in the published literature.3,6,7 However, while limited long-term safety and effectiveness data are a challenge for brexu-cel for R/R ALL, clinical experts suggested that data from other CAR T-cell therapies targeted toward B-cells could guide clinical practice.
Ethical Considerations in the Use of Brexucabtagene Autoleucel
Access to CAR T-Cell Therapies
Literature and project inputs have a identified several access challenges related to CAR T-cell therapies, including brexucabtagene autoleucel, that raise considerations of equity, fairness, resource allocation, and distributive justice. Outside of the health care system in Canada, costs have often been described as a primary barrier to accessing CAR T-cell therapies.3 This is not to suggest that costs are not a burden in the health care system in Canada, but that those living in Canada who bear that burden may be different.
Geographic and Financial Challenges
A substantial challenge described in the literature and through clinician group and drug program input relates to the extensive resourcing needs associated with the delivery of CAR T-cell therapies. As unique biologic therapies, CAR T products can only be delivered in accredited treatment centres with specialized infrastructures and highly trained providers.3,6,7 Drug program input has highlighted how challenging it can be for treatment centres to obtain and maintain these certifications as well as how resource intense the delivery of CAR T-cell therapies can be inside of these treatment centres (e.g., patient preparation preinfusion involves multiple steps, professionals, and technologies). While the certification process and need for highly trained providers are all mechanisms that could help to ensure the safe delivery of CAR T-cell therapies, the high costs and resource-intensive nature of providing CAR T-cell therapies has, at present, limited their availability to more metropolitan locations. This has had the subsequent effect of limiting access to CAR T-cell therapies for people who do not live near 1 of these treatment centres.3,6,7,10,15,16 For those who do access CAR T-cell therapies far from home, this adds costs associated with transportation, lodging, and the need to take time off work, and could also be psychologically and emotionally difficult given the need to spend time away from family and community.3,6,7
This time away from family and community is not insignificant. While CAR T-cell therapies such as brexu-cel are delivered as a one-off infusion, there are substantial time commitments associated with preinfusion and postinfusion activities as well. Patients must first undergo leukapheresis to harvest the cells to send to the manufacturer for modification. Depending on their health status, some patients may need to undergo bridging therapy while they wait for their modified T-cells to be returned from the manufacturer for infusion. Once the final product is ready, patients undergo conditioning chemotherapy for 4 days leading up to CAR T-cell infusion. Postinfusion, patients are required to remain onsite or near the treatment centre for another 4 weeks to monitor for adverse events such as cytokine release syndrome. Clinical experts described how challenging this process could be for people who lived several hours away from the treatment centre because it further compounds financial and caregiver support challenges experienced throughout the course of their treatment.
To mitigate some of these challenges with travel, clinician group input has suggested that there is a critical need for additional treatment centres that allow patients to be treated closer to home. Literature has additionally suggested that CAR T-cell therapies may be offered in outpatient settings.3 The sponsor has provided an implementation plan that details both which specialist centres they have already, or will be, training toward accreditation, and the supports they are willing to offer patients who need to travel for care. These include financial supports for residents of Canada who need to travel and stay at a treatment centre that is 2 hours or 200 km from their primary residence, who are enrolled in their product management software, who have consented to treatment, and who could not afford travel or lodging otherwise. While clinical experts suggested that this was a positive step, when considering similar programming for other CAR T-cell therapies, authors have questioned whether these steps provide enough financial support to cover all costs for patients travelling to treatment centres17 and whether manufacturers ought to take on this gatekeeping role.18
Disparities for Racialized Populations
In addition to challenges for patients who do not live nearby treatment centres, authors have noted how Black and Hispanic populations are much less likely to have access to and receive CAR T-cell therapy in the US context.10,15,19 This disparity in access to CAR T-cell therapies has also been identified along socioeconomic lines, with people who are poorer being less likely to have access.10,15,17 Of note, clinical experts suggested that CAR T-cell therapies may have some accessibility benefits over stem cell transplant for Black, Indigenous, or other racialized persons. They indicated, in accordance with the literature,10 that these populations are persistently underrepresented in transplant registries, which can result in people not finding matches for allo-SCT in a timely fashion. Because CAR T-cell therapies modify the individual’s own biologic material, it was suggested that this underrepresentation may be somewhat mitigated for CAR T-cell therapies. However, clinical experts did note that, unlike in the US, they were unaware of any directed work being done in Canada to remediate inequities in access to CAR T-cell therapies, so disparities in access for Black (and other racialized populations) are likely to persist.
Caregiver Support
Clinical experts and literature also pointed to the importance of having strong caregiver and/or family support when engaging with CAR T-cell therapies such as brexucabtagene autoleucel.3 However, not all patients will have this support and clinical experts suggested that patients who do not have access to caregivers should be considered as a particularly vulnerable population in the context of R/R ALL and CAR T-cell therapies. Without these supports already available, it could be a barrier to accessing CAR T-cell therapies because there may be additional costs associated with hiring formal caregivers.3
Clinical Judgment and Equity in Referral
Previous CADTH reviews of CAR T-cell therapies have also highlighted timing challenges for referral to CAR T-cell therapy.3,6,7 Given the finite windows in which CAR T is most likely to provide benefit, it is important that providers be adequately educated about CAR T-cell therapies so that a timely referral can be made.3 This may be challenging because clinical experts and literature both suggested that it is possible that referring oncologists from nonspecialized centres may not be familiar enough with referral and administrative processes for CAR T-cell therapies, including when and how to refer.19
Given the limited resources and unequal distribution of CAR T-cell therapies across Canada, it is also important that there are clear and transparent criteria for how to prioritize patients in this process.3,6,7
Potential Harms in the Use and Delivery of CAR T-Cell Therapies
As has been well described in previous CADTH reviews and across the literature,3,6,7 CAR T-cell therapy is often accompanied by unique toxicities with extensive effects on patients (e.g., cytokine release syndrome). The clinical review has detailed the adverse events specific to brexucabtagene autoleucel. While there has been a growth in understanding and management techniques for these toxicities over the past several years, there remains uncertainty around the long-term safety and risks of CAR T-cell therapies.3,20 Drug program and clinician group input for brexu-cel reiterated the importance of CAR T-cell therapies being delivered by well-trained medical teams who have competency and capacity in how to manage these toxicities.
Clinical experts also suggested that delayed access to CAR T-cell therapies may potentially harm patients with R/R ALL. Any delay may lead to patients being more frail at the time of infusion, which could have harmful consequences. Clinical experts also spoke about financial harms and costs to the health care system in patients using potentially ineffective therapies for the purpose of meeting eligibility criteria for CAR T-cell therapy access.
Cell and Tissue Ownership
Previous CADTH ethics review reports have identified challenges around cell ownership in the context of CAR T-cell therapies.6,7 Because CAR T-cell therapies involve the modification of patients’ T-cells using proprietary methods, there are questions around who owns the modified cells (e.g., patient, manufacturer, payer) and, if not the patient, at what point ownership might be transferred.6 Relatedly, if the patient is not considered the owner of the modified cells, there are also questions regarding when transfer of ownership happens and whether consent forms clearly articulate the amount of control patients have over their modified cells.6 Once manufactured, there has also been the question of what happens to the modified T-cells if the patient dies before delivery.6
Drug plans indicated that there remains uncertainty about how these challenges around ownership and patient privacy would be managed for brexu-cel given that it would currently be manufactured outside of Canada. While clinical experts felt that these concerns are largely resolved through consent forms, authors have indicated that different cultural groups understand and value tissue and genetic material differently, which implies the need to clearly detail and attend to cell processing and ownership and what will be done with remaining modified cells postproduction.21 Consulting with diverse groups on how to handle, deliver, and dispose of CAR T-cell products in ways that are sensitive to the needs and values of diverse patients would be an important step in making sure consent forms are appropriate across this diversity.7,21 This is important to consider because the benefits of cell ownership may differently accrue across actors (e.g., manufacturers, public, patients).6,7
Considerations for Informed Consent
There have also been challenges noted around the “hype” surrounding CAR T-cell therapies.4,6,7 There may be some confusion for patients who may understand CAR T-cell therapy as a “cure” despite uncertainties around long-term effectiveness and their ability to access CAR T-cell therapy.6,7 The risks and benefits of CAR T-cell therapies will need to be described and communicated to patients in a balanced way that accounts for their vulnerability as cancer patients with limited therapeutic options.3,4,6,7
Health System and Funding Considerations
In the submission, the sponsor has indicated that there are 12 treatment centres that are, or are in the process of becoming (in 2023), authorized to deliver brexu-cel in Canada. All of these sites are located in major metropolitan locations across Canada, with 3 each in Quebec and Ontario, 2 in Alberta, and 1 each in British Columbia, Manitoba, Saskatchewan, and Nova Scotia. This leaves substantial gaps in geographic availability and will require that some patients travel outside of their communities to receive treatment. Given the limited or lack of availability in some provinces (or territories), drug programs have highlighted the likelihood that some patients may even need to travel out of province, or potentially out of country, to access brexucabtagene autoleucel.
As previously described, clinician group input and clinical experts suggested that there is need to continue expanding sites capable of delivering CAR T-cell therapies. Drug program input has highlighted that current treatment centres are facing capacity challenges and may struggle to keep up with demand as new CAR T-cell therapies are approved and funded. Similarly, clinical experts did note some concern that it could be challenging for health care systems to meet growing needs for different CAR T-cell therapies that operate on different systems and involve different manufacturers. Growth needs to be matched with safety and quality standards across all new treatment centres,6,7 and the geographic dispersion of new centres should consider regional rural, urban, and sociodemographic equity.
One of the primary challenges affecting access to CAR T-cell therapies identified in the literature is the high costs of these therapies to the health care system.3,6,20 This has become a particularly salient challenge as more CAR T-cell therapies have been approved by regulatory agencies and are being considered for reimbursement in Canada. Drug program input and literature have suggested that some of the cost challenges could be mitigated by special payment arrangements (e.g., outcomes-based risk sharing) with the manufacturer that involve long-term follow-up and economic reassessment.7,20
It has been noted, however, that price negotiations for CAR T-cell therapies can only reduce costs so much, and that the total costs of these therapies will remain high because the price of the therapy itself is only 1 component of its high cost.6,7 Authors have argued that the full costs of CAR T-cell therapies may be unknown to payers and have called for full transparency around total costs of CAR T, including those associated with preinfusions and postinfusions, treatment of adverse events, and other clinical costs.3 Drug program input and clinical experts noted that therapies to manage adverse events related to CAR T-cell therapies (such as tociluzimab in the treatment of cytokine release syndrome) should also be available across provinces, as a matter of equity and fairness, and that the high costs of managing toxicities associated with CAR T-cell therapies should be understood in the context of these therapies.3
As is relevant with many high-cost therapies, clinical experts discussed the opportunity costs of CAR T-cell therapies and the challenges of funding these therapies in the context of fixed budgets. A prior review of ethical considerations in CAR T conducted by CADTH discussed how funding of CAR T-cell therapies can affect the sustainability of health systems. As such, fair and just funding allocation, with fair distribution of risks and benefits of innovations, should be considered alongside fair and transparent criteria for patient prioritization and allocation to CAR T-cell therapies.3 These considerations become amplified as CAR T-cell therapies apply to more cancer subtypes and the need to manage health care system costs and improve access persists.3 These considerations call for clarity and transparency in justifications for policy decisions about expanding access to CAR T-cell therapies in the context of considering the long-term sustainability of the health care systems in Canada.6,7
Limitations
This review is limited by the paucity of published literature examining ethical considerations directly relevant to the use of brexu-cel for adults with R/R ALL in Canada. The absence of directly applied published ethical analyses does not indicate that ethical considerations are not present, and many ethical considerations in the context of ALL more broadly, or those related to CAR T-cell therapies in general, are also salient in this context. Augmenting this somewhat limited literature with inputs from patient and clinician groups, drug programs, and clinical experts collected in the course of this reimbursement review provided a more fulsome picture of ethical considerations in the context of brexu-cel for the treatment of adults with R/R ALL.
Though this Ethics Review Report drew and extracted from patient group, clinician group, clinical expert, and drug program inputs, it is possible that more directed engagement (such as through direct interviews with patients, caregivers, family members, or health system payers) would have yielded more relevant domains of analysis. Given the proposed high cost of this therapy in the context of the limited resources of the health care system in Canada, is it also possible that inputs from citizens may have identified additional relevant considerations or trade-offs relevant to the funding and implementation of brexucabtagene autoleucel. This is particularly salient in a context in which much of the data available through published literature are developed outside of Canada. Without data derived from Canada, it can be challenging to negotiate the applicability of what is being seen in other health care systems to health care systems in Canada, particularly when the data that are available come from private-pay systems.
Conclusion
Inputs from patient and clinician groups, clinical experts, drug programs, and relevant published literatures were reviewed for ethical considerations relevant to the use of brexu-cel for the treatment of R/R B-cell precursor ALL in adult patients. Ethical considerations in the context of ALL highlighted the impacts on patients and disparities in incidence, treatment, and outcomes across racialized and marginalized groups as well as across ages. Ethical considerations related to the evidence used to evaluate brexu-cel identified discrepancies between eligibility criteria for trial participants and those who might be considered for brexu-cel in clinical practice. The implementation of brexu-cel involves several access considerations given the limited and resource-intensive nature of delivery. These access considerations involve substantial geographic disparities given the limited dispersion of specialized centres across Canada with appropriate accreditation to provide brexu-cel and may disproportionately affect marginalized persons or groups. There are unique toxicities involved in the delivery of CAR T-cell therapies and the potential for harm in delayed access to therapy. In addition, the manufacture and storage of cellular tissues used in the course of therapy raises ethical considerations, as do issues related to informed consent and balanced communication about the risks and benefits of CAR T-cell therapies. Finally, the implementation of brexu-cel raises ethical considerations for health care systems related to the challenges and high costs of scaling CAR T infrastructure and trade-offs in the payment of high-cost therapies. These considerations, challenges, and uncertainties should be understood alongside the potential for benefits that the introduction of brexu-cel may entail for some R/R ALL patients.
Because adults living with R/R B-cell precursor ALL are a population with a particularly poor prognosis and current treatment options that present their own access challenges (e.g., limited representation in transplant registries and age cut-offs for allo-SCT), brexu-cel has been proposed as a treatment option for these patients. Given the favourable overall survival and relapse-free survival outcomes apparent in early data from the ZUMA-3 study (and detailed in the clinical report of this review), it is possible that brexu-cel may meet patients’ desire to have more effective treatment options with a higher likelihood of benefit. However, the relatively short follow-up period of only 2 years and the absence of any head-to-head comparative data make it challenging to determine whether these preliminary outcomes will persist long-term. Paired with the numerous access disparities of CAR T-cell therapies broadly and implementation challenges for health care systems, questions remain around how to introduce the use of autoleucel brexucabtagene equitably and fairly for adults with R/R ALL in Canada.
Table 1: Details of Included Publications
First author, year | Publication type | Objective | Key ethical considerations | Funding source |
|---|---|---|---|---|
Ahmed, 202215 | Database analysis | To explore the impacts of race/ethnicity, socioeconomic–status, insurance coverage, travel time to CAR T-cell therapy centres, and participation in clinical trials on people’s likelihood of receiving CAR T-cell therapy | Universal access to CAR T-cell therapy is limited There are disparities in access to commercial and clinical trial CAR T-cell therapy for minorities, especially Black and Hispanic populations Majority of patients in clinical trials are white; Black patients are less likely to receive CAR T-cell therapy Few CAR T-cell therapy recipients are from low socioeconomic–status neighbourhoods | None reported |
Blue, 202219 | Commentary | To highlight similarities in social barriers to access between stem cell transplant and novel CAR T-cell therapies | CAR T-cell therapies are not universally accessible, particularly for “minority” populations or those living outside geographic catchment areas of specialized treatment centres Disparities in access to CAR T-cell therapies across racial and socioeconomic factors are reflective of those in stem cell transplant Community oncologists may have limited familiarity with CAR T-cell therapies, which could result in their patients not being referred to tertiary cancer centres; education is necessary | None reported |
Bouchkouj, 202222 | Review | To summarize the FDA clinical review and regulatory considerations regarding the licensing application for brexu-cel for treatment of adult patients with R/R ALL | Challenging to establish optimal sequencing of brexu-cel because duration of response is unknown Given that R/R B-ALL is life-threatening, adverse reactions of CRS and neurologic toxicity are considered acceptable from a “benefit-risk perspective in the intended population” | None reported |
CADTH, 20223 | Ethics review report | To identify and describe ethical considerations associated with the use of axicabtagene ciloleucel for the treatment of adults with relapsed or refractory LBCL | There are disparities in the incidence, treatment, and outcomes of patients with LBCL Barriers to access for CAR T-cell therapies include those related to costs, geography, and patient selection Resource allocation considerations call for fair prioritization processes, opportunities to expand access, and implications for health care systems Need to balance risks and benefits of CAR T-cell therapies and provide informed consent and balanced communication to patients | Canada’s federal, provincial, and territorial governments, with the exception of Quebec |
Choi, 202220 | Commentary | To describe the challenges of providing a reliable, cost-effectiveness analysis of CAR T-cell therapies | High costs of CAR T-cell therapies are a barrier to access Serious adverse events are a barrier of access to CAR T-cell therapies Limited long-term data for clinical effectiveness and safety data are barriers to access because they make it challenging to produce accurate cost-effectiveness analyses | National Research Foundation of Korea |
Apostolidou, 202110 | Retrospective study | To evaluate outcome disparities in a cohort of adult patients with newly diagnosed ALL treated in a US county with wide socioeconomic diversity, predominantly underserved by health care, and with limited access to specialized treatment centres | People identified as Hispanic have higher incidence and mortality rates of ALL Important to pay attention to how social determinants of health may affect ALL treatment and outcomes Given that their treatment occurred outside of specialized cancer centres, patients had limited access to stem cell transplant and other advanced therapies such as monoclonal antibodies (e.g., blinatumomab) and CAR T-cell therapies due to limited experience or infrastructure to deliver these therapies Lack of childcare or transportation can be barriers to follow-up care for some people undergoing treatment for ALL | None reported |
CADTH, 20214 | Ethics review report | To describe and summarize the ethical considerations raised explicitly in the literature associated with the use of brexu-cel for the treatment of adult patients with relapsed or refractory MCL | There are disparities in the incidence, treatment, and outcomes of patients with MCL Need to balance risks and benefits of CAR T-cell therapies and provide informed consent and balanced communication to patients | Canada’s federal, provincial, and territorial governments, with the exception of Quebec |
Snyder, 202117 | Original research (geographic information system mapping) | To estimate the travel-related economic burden associated with site-of-care options among patients with R/R DLBCL | Travel costs to CAR T-cell therapy centres could be decreased if access were to be expanded to nonacademic hospitals and specialty oncology centres. Patients living below the poverty line and those in rural areas are particularly disadvantaged with regards to CAR T-cell therapy access Assistance provided by pharmaceutical companies related to travel costs may not sufficiently cover all costs borne by patients to travel to treatment centres | Bristol Myers Squibb |
Weinkove, 202121 | Review | To outline CAR T-cell manufacturing and logistical considerations, with a focus on New Zealand’s environment for personalized cell and gene therapy | CAR T-cells are manufactured in a limited number of sites; shipping of cells can pose challenges Tissue and genetic material valued differently by different cultural groups (e.g., Maori). Details of processing and ownership must be made clear in consent processes Consultation with diverse groups (e.g., Maori) is essential in CAR T-cell research and delivery, including development of educational material for these groups and their clinicians | Health Research Council of New Zealand; Ministry of Business, Innovation and Employment; LifeBlood Trust; Freemasons New Zealand |
Thiele, 202014 | Survey study | To assess the prevalence of “fear of progression” and other relevant correlates before acute leukemia patients undergoing stem cell transplant | Acute leukemia patients undergoing stem cell transplant are at a heightened risk for “fear of progression” and may need extra psychological support | None reported |
Wang, 202011 | Review | To describe new insights in the biology of B-cell ALL and identify emergent treatment options that may help to close the disparity in survival outcomes for AYAs compared to pediatric populations | There is a disparity in survival outcomes between pediatric and AYA (ages 15 to 39) populations with ALL Living with ALL as an AYA can carry unique challenges associated with physical and psychological development, which could result in substantial psychological distress Multidisciplinary teams that can address the complexity of psychosocial, treatment-related, or supportive care needs for this population are necessary | None reported |
CADTH, 20196 | Ethics review report | To discuss the major ethical issues raised by the implementation of tisagenlecleucel for children or young adults with relapsed or refractory ALL and adults with relapsed or refractory DLBCL | The long-term risks and benefits of CAR T-cell therapies remain unknown There are several access challenges in the context of CAR T-cell therapies, including those related to geography, supply, and patient selection Need to consider the context of “hype” around CAR T-cell therapies and implications for informed consent Considerations of ownership of genetic materials on the context of CAR T-cell therapy | Canada’s federal, provincial, and territorial governments, with the exception of Quebec |
CADTH, 20197 | Ethics review report | To discuss the major ethical issues raised by the implementation of axicabtagene ciloleucel for adults with relapsed or refractory non-Hodgkin lymphoma | The long-term risks and benefits of CAR T-cell therapies remain unknown There are several access challenges in the context of CAR T-cell therapies, including those related to geography, supply, and patient selection Need to consider the context of “hype” around CAR T-cell therapies and implications for informed consent Considerations of ownership of genetic materials in the context of CAR T-cell therapy | Canada’s federal, provincial, and territorial governments, with the exception of Quebec |
Imbach, 201818 | Commentary | To articulate key ethical challenges for the field of CAR T-cell therapy and suggest some strategies to help navigate these challenges | Uncertainty about long-term benefits and risks of CAR T-cell therapies “Hype” and role of stakeholders in accurately communicating risks and benefits Manufacturers serve as a gatekeeper to compassionate access requests Demand for access to clinical trials exceeds capacity. This may privilege some groups over others | None declared |
Huguet, 201716 | Commentary | To walk through growth of biologic therapeutics in ALL and describe trial results for various monocloncal antibodies and CAR T-cells | Growth in biologic therapeutics available, or being tested, in certain subtypes of ALL has made it more challenging to determine optimal sequencing Production issues and costs of monoclonal antibodies and CAR T-cell therapies for ALL are likely to deepen existing inequalities in access to care | None reported |
Gheihman, 201613 | Survey study | To assess the prevalence and correlates of depression and hopelessness in patients with some form of acute leukemia | Older patients may be more likely to experience hopelessness around care and treatment of acute leukemia Fostering strong communication between providers and their patients may help limit the likelihood that patients develop depression or feelings of hopelessness | Canadian Institutes of Health Research; Ontario Ministry of Health and Long-Term Care; Princess Margaret Cancer Foundation Hertz Centre Fund |
Rodin, 201312 | Survey study | To examine the prevalence and correlates of post-traumatic stress syndrome in people with some form of acute leukemia | Poor communication between providers and patients may lead to a greater likelihood of developing post-traumatic stress syndrome Psychological supports should be available early in treatment for acute leukemia to reduce the risk of developing post-traumatic stress syndrome | Canadian Institutes of Health Research; Ontario Ministry of Health and Long-Term Care |
ALL = acute lymphoblastic lymphoma; AYA = adolescent and young adult; CAR t = chimeric antigen receptor T; CRS = cytokine release syndrome; DLBCL = diffuse large B-cell lymphoma; LBCL = large B-cell lymphoma; MCL = mantle cell lymphoma; R/R = relapsed or refractory.
References
1.EUnetHTA Joint Action 2 Work Package 8. HTA Core Model ® version 3.0. Diemen (NL): European Network for Health Technology Assessment (EUnetHTA); 2016: https://www.htacoremodel.info/BrowseModel.aspx. Accessed 2022 Nov 18.
2.Benkhalti M, Espinoza M, Cookson R, Welch V, Tugwell P, Dagenais P. Development of a checklist to guide equity considerations in health technology assessment. Int J Technol Assess Health Care. 2021;37(1):e17. PubMed
3.Drug Reimbursement Review: axicabtagene ciloleucel (Yescarta) for treatment of adult patients with relapsed or refractory large B-cell lymphoma (LBCL), who are candidates for autologous stem cell transplant (ASCT) [in progress]. Ottawa (ON): CADTH; 2022: https://www.cadth.ca/axicabtagene-ciloleucel. Accessed 2022 Nov 30.
4.Drug Reimbursement Review clinical, pharmacoeconomic, and ethics reports: brexucabtagene autoleucel (Tecartus) for mantle cell lymphoma. Can J Health Technol. 2021;1(11). https://www.cadth.ca/sites/default/files/DRR/2021/PG0219-tecartus-combined-report-meta.pdf. Accessed 2022 Nov 30.
5.Grey matters: a practical tool for searching health-related grey literature. Ottawa (ON): CADTH; 2019: https://www.cadth.ca/grey-matters. Accessed 2022 Oct 25.
6.Tisagenlecleucel for acute lymphoblastic leukemia and diffuse large b-cell lymphoma: Ethics and implementation report. (CADTH optimal use report; vol.8, no.3d). Ottawa (ON): CADTH; 2019: https://www.cadth.ca/sites/default/files/pdf/car-t/op0538-tisagenlecleucel-ethics-and-implementation-jan2019.pdf. Accessed 2022 Nov 30.
7.Axicabtagene ciloleucel for large B-cell lymphoma: ethics and implementation report. (CADTH optimal use report; vol. 9, no. 1e). Ottawa (ON): CADTH; 2019: https://www.cadth.ca/sites/default/files/pdf/car-t/ct0002-ipe-report.pdf. Accessed 2022 Nov 30.
8.Sandelowski M. Whatever happened to qualitative description? Res Nurs Health. 2000;23(4):334-340. PubMed
9.Xu K, Feng Q, Wiemels JL, de Smith AJ. Disparities in acute lymphoblastic leukemia risk and survival across the lifespan in the United States of America. J Transl Genet Genom. 2021;5(3):218-239.
10.Apostolidou E, Lachowiez C, Juneja HS, et al. Clinical Outcomes of Patients With Newly Diagnosed Acute Lymphoblastic Leukemia in a County Hospital System. Clin Lymphoma Myeloma Leuk. 2021;21(11):e895-e902. PubMed
11.Wang AY, Muffly LS, Stock W. Philadelphia Chromosome-Negative B-Cell Acute Lymphoblastic Leukemia in Adolescents and Young Adults. JCO Oncol Pract. 2020;16(5):231-238. PubMed
12.Rodin G, Yuen D, Mischitelle A, et al. Traumatic stress in acute leukemia. Psychooncology. 2013;22(2):299-307. PubMed
13.Gheihman G, Zimmermann C, Deckert A, et al. Depression and hopelessness in patients with acute leukemia: the psychological impact of an acute and life-threatening disorder. Psychooncology. 2016;25(8):979-989. PubMed
14.Thiele S, Goebel S, Kroger N, Pedersen A. Fear of disease progression and relevant correlates in acute leukemia patients prior to allogeneic hematopoietic stem cell transplantation. Psychooncology. 2020;29(8):1248-1254. PubMed
15.Ahmed N, Shahzad M, Shippey E, et al. Socioeconomic and Racial Disparity in Chimeric Antigen Receptor T Cell Therapy Access. Transplant Cell Ther. 2022;28(7):358-364. PubMed
16.Huguet F, Tavitian S. Emerging biological therapies to treat acute lymphoblastic leukemia. Expert Opin Emerg Drugs. 2017;22(1):107-121. PubMed
17.Snyder S, Albertson T, Garcia J, Gitlin M, Jun MP. Travel-Related Economic Burden of Chimeric Antigen Receptor T Cell Therapy Administration by Site of Care. Adv Ther. 2021;38(8):4541-4555. PubMed
18.Imbach KJ, Patel A, Levine AD. Ethical considerations in the translation of CAR-T cell therapies. Cell Gene Ther Insights. 2018;4(4):295-307.
19.Blue B. Socioeconomic and Racial Disparity in Chimeric antigen receptor T cell (CART)Therapy Access. Transplant Cell Ther. 2022;28(7):345-346. PubMed
20.Choi G, Shin G, Bae S. Price and Prejudice? The Value of Chimeric Antigen Receptor (CAR) T-Cell Therapy. Int J Environ Res Public Health. 2022;19(19):12366. PubMed
21.Weinkove R, George P, Ruka M, Haira TH, Giunti G. Chimeric antigen receptor T-cells in New Zealand: challenges and opportunities. N Z Med J. 2021;134(1542):96-108. PubMed
22.Bouchkouj N, Lin X, Wang X, et al. FDA Approval Summary: Brexucabtagene Autoleucel for Treatment of Adults With Relapsed or Refractory B-Cell Precursor Acute Lymphoblastic Leukemia. Oncologist. 2022;27(9):892-899. PubMed
Stakeholder Input
Patient Input
The Leukemia & Lymphoma Society of Canada
About The Leukemia & Lymphoma Society of Canada
The Leukemia and Lymphoma Society of Canada is a national charitable status organization dedicated to finding a cure for blood cancers and its ability to improve the quality of life of people affected by blood cancers and their families by funding life-enhancing research and providing educational resources, services, and support. The Leukemia and Lymphoma Society of Canada is the largest charitable organization in Canada dedicated to blood cancer, our focus includes:
Funding research from bench to bedside.
Rethinking how a person navigates their blood cancer experience
Providing targeted blood cancer information
Offering tools for psychological and emotional support
Empowering Canadians to take charge of their blood cancer experience through practical support and advocacy
To learn more, visit: bloodcancers.ca
Information Gathering
The LLSC created an online survey, that was distributed through social media networks [Facebook, Twitter, Instagram] and by email, between August 15 to September 21, 2022, in both English and French. The survey uses multiple choice, open-ended and rating questions, and uses skipping logic to allow respondents to pass on questions not relevant to them. Open-ended responses to surveys that reflected the sentiment of a majority are included verbatim to provide a deeper understanding of patient perspectives.
There were 22 respondents to the surveys, of which 15 are from Quebec, 5 from British Columbia, 2 from Ontario, and 1 from Alberta. Of all 22 respondents, only two had experience with Tecartus. The remaining 20 respondents were able to share their experience with ALL. Refer to Table 1 for breakdown of respondents by age.
Table 1: Age of Survey Respondents
Respondents | Age range | |||||||
|---|---|---|---|---|---|---|---|---|
<18 | 18-24 | 25-34 | 35-44 | 45-54 | 55-64 | 65-74 | 75 + | |
Patients WITHOUT Tecartus Experience | 2 | 1 | 4 | 6 | 1 | 3 | 2 | 1 |
Patients WITH Tecartus experience | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 |
Disease Experience
Respondents were asked to rate on a scale of 1 (no impact) to 5 (extremely significant impact), the impact that the disease symptoms had affected their quality of life. Refer to Table 2 for breakdown of responses. 73% of the responses listed fatigue or weakness as having the most significant impacts on their quality of life. These were followed by loss of appetite or weight loss (45%), bone or joint pain (41%) and headaches, blurred vision, nausea, or vomiting (41%).
Table 2: Disease Symptoms’ Effects on Quality of Life
Side Effect | Significant Impact (4-5) |
|---|---|
Fatigue or weakness | 16 |
Fever | 5 |
Loss of appetite or weight loss | 10 |
Night sweats | 5 |
Spots under the skin (petechiae) | 2 |
Stomach pain | 7 |
Infections | 6 |
Dizziness or lightheadedness | 8 |
Feeling cold | 1 |
Shortness of breath | 7 |
Frequent or severe bleeding (nosebleeds, gums bleeding, unusual bleeding from minor cuts) or bruising | 3 |
Enlarged lymph nodes | 0 |
Bone or Joint pain | 9 |
Headaches, blurred vision, nausea, or vomiting | 9 |
When asked how the disease impacted their day-to-day lives, the ability to work (73%), ability to exercise (73%), and ability to continue everyday activities (73%) were indicated as the most significantly impacted by the disease. Refer to Table 3 for the breakdown of responses.
Table 3: Disease Symptoms’ Effects on Day-to-Day Life Following Diagnosis
Effect | Significant Impact (4-5) |
|---|---|
Ability to work | 16 |
Ability to travel | 15 |
Ability to exercise | 16 |
Personal image | 11 |
Ability to spend time with family and friends | 11 |
Intimate relationships | 14 |
Ability to continue daily activities | 16 |
Ability to concentrate | 12 |
Mental health | 8 |
Ability to manage family responsibilities | 13 |
Ability to pursue activities or hobbies | 14 |
Most notably, were the psychological/social factors of the disease that have significantly impacted their quality of life. Refer to Table 4. Interruption of life goals/accomplishments such as careers, schooling and such had the most impact on the respondent’s quality of life after diagnosis (77%). This was followed closely by stress, anxiety and worry (68%).
Table 4: Psychological/Social Factors’ Effects on Quality-of-Life Following Diagnosis
Psychological/social factors | Significant Impact (4-5) |
|---|---|
Stress/anxiety/worry | 15 |
Depression | 7 |
Difficulty sleeping | 9 |
Feeling isolated | 10 |
Lack of support | 4 |
Difficulty with friend or family relationships | 3 |
Problems concentrating | 10 |
Loss of sexual desire | 10 |
Interruption of life goals/accomplishments (career, schooling, etc.) | 17 |
Financial impacts (cost of travel, inability to work, etc.) | 10 |
Loss of appetite | 5 |
One respondent said she felt a real need to speak to someone who had the same experience as she did. Although she had a support system, she felt isolated: “I was 38 years old and had three young children, including a one-year-old, and despite all the support of those around me, I felt like I was going through something so big that there were no words strong enough or understandable enough for those around me to understand how I felt. Plus, I had to deal with their own pain.”
Experiences With Currently Available Treatments
We asked the respondents which previous treatments they had received following their diagnosis. 18 indicated chemotherapy, 9 received chemotherapy with stem cell transplant, 7 received radiation therapy and 2 received targeted therapy. Of those who responded, 8 mentioned having had 5 or more lines of treatment since diagnosis.
We also inquired how many hospital visits were incurred for ALL related issues (i.e., Treatment, scans, follow ups, ER visits, etc.): 50% of the respondents indicated more than 5 hospital visits per month. Table 5 shows the number of years the respondents have been in treatment and the number of kms travels to access care (4 respondents skipped this question).
Table 5: Years in Treatment Versus Distance to Travel to Receive Care
Years in treatment | less than 100 kms | 100 to 199 kms | Over 400 kms |
|---|---|---|---|
1 year | 1 | 1 | — |
2 years | 7 | 1 | — |
3 years | — | — | — |
4 years | 3 | — | — |
5 years | — | — | 1 |
More than 5 years | 4 | — | — |
We asked the respondents what the impact of having to travel receive care and treatment were: extensive cost of travel and accommodations, emotional hardship, being away from support system for extended periods of time, and Impact to daily activities/routine were indicated as having significant impact. One respondent said, “Needed to fly to a major city, live with a family member and rent an apartment close to the hospital because treatment was longer than expected.”
When asked to rate the impact of treatment related side-effects, 82% felt weakness, 73% felt fatigue, and 50% felt nausea that significantly impacted their everyday lives. Some shared further details of the impact the treatments had on their quality of life:
“Avascular necrosis of the hip joints was most significant outcome in regard to the quality of my life.”
“The chemotherapy protocol is long and is extremely tiring.”
“Very difficult protocol of chemotherapy.”
“Intense and aggressive chemo protocol, difficult to tolerate, include intrathecal chemo, followed by allogenic stem cell transplant (2016). Very difficult, recovery over 2 years, major chicken pox affliction at 1.5 years from transplant, severely ill. Today I am doing well.”
“I developed Avascular necrosis as a result of the steroids taken during chemotherapy.”
“I had a hard time getting a diagnosis of ALL even though I had all the symptoms visible to the naked eye, an internist made a joke about pulses when my husband mentioned that my scalp was itchy, in addition to the other symptoms including my spleen hemorrhaging. Once I was diagnosed with ALL after 1 month, the Gatineau hospital transferred me to the Ottawa hospital where I was able to consider living again and received exceptional care. They were able to get me into pre-transplant remission despite the 95% leukemia cells in my marrow at the time of my transfer. The sad thing is that if my husband had not fought for further testing, I probably would have died before I could have been transferred to Ottawa. I am forever grateful to the Ottawa Hospital and its caregivers.”
“I stayed in the hospital for my entire chemo and will return there 3 times a week for week for transfusions.”
Majority of the respondents indicated that ease of access to treatments and treatment results were positive experiences for them:
“I was lucky enough to live close to Princess Margaret and get treatment there.”
“I had quick access to health care professionals and appropriate treatment.”
“Very good medical follow-up.”
Improved Outcomes
Respondents were asked to rate the most key factors or outcomes considered when deciding about taking a new treatment. Of the 18 responses received, the majority agreed that the degree of certainty that ALL will respond to treatment (17) and improved quality of life (16) are important or significantly important outcomes considered when making decisions about treatment options. Coverage by insurance/drug plan (15) and Improved length of survival (15) were the third most crucial factors. 4 people did not provide an answer to this question.
Although, reduced side effects and easier accessibility were not rated as highly as the above-mentioned factors, these were frequently mentioned in the open-ended question “What kinds of improvement would you like to see in any new treatment for ALL?”
“At home accessibility, remote nurses able to travel to homes or provide treatment in centers closer with less travel time”
“Less side effects and adverse reactions.”
“Something more gentle than a BMT.”
“Accessibility without hospitalization.”
“Fewer side effects during and after treatment.”
“Reduce dependence on chemo. Find ways to counteract the immense fatigue/weakness and nausea.”
Experience With Drug Under Review
The two respondents who had experience with Tacartus accessed the drug via clinical trial. Both received the treatment in centers close to their homes, had no issues access the treatment and neither had any out-of-pocket costs associated with this treatment.
The first respondent is between the ages of 25-34 years and had previously been treated with “Chemotherapy with Stem Cell Transplant PLUS Whole-Body Irradiation.” Regarding Tecartus, they reported an overall positive experience with the drug in question. Although the side-effects were explained, this has no impact on their decision to take this treatment. This person had no difficulties accessing this treatment and claims that their ALL responded completely to the drug. This person experienced manageable or minor sides effects other than nausea and loss of appetite which they considered a serious side effect. This person also reported Tecartus having a very positive impact on their quality of life: relationships with friends and family, metal health, ability to travel, ability to perform everyday activities and so forth. This respondent weas able to return to work and resume normal activities since taking Tecartus. When asked what their overall experience with Tecartus was like, they responded with “Very positive. I was able to achieve complete remission while only suffering a fraction of the side effects of an allogeneic stem cell transplant.”
The second respondent is between the ages of 65 to 74 and had previously been treated with “chemotherapy and targeted therapy,” No further details of the specific targeted therapy received. Unlike the first respondent, this person had different experiences with Tecartus. This person was also explained the potential side effects and claims this influenced the decision to take this treatment. The respondent reported experiencing very serious side effects: slurred speech, fever, chills, cough, or other signs of infection, feeling tired or lightheaded, fast or irregular heartbeat, headache, muscle or joint pain, diarrhea and/or constipation, nausea, loss of appetite and insomnia. Although these side effects were very serious, the respondent reported that these were somewhat manageable. This person also reported a strong negative impact of the treatment on certain aspects of their quality of life: Ability to work/go to school/volunteer, mental health, and ability to perform daily activities. The respondent report that they have not been able to resume normal activities or return to work since taking the drug under review. However, their ALL responded partially to the treatment. No comments were left by this respondent when asked about their overall experience with the drug.
Significant side effects are reported with genetically modified autologous T cell immunotherapies in general and the ones experience by these individuals are consistent with those reports. It is also important to note that these side effects are very individual and that there is a wide range of experiences. Clinicians generally believe that the side effects can be managed. It is significant to note that the data shows that response rate to this treatment is very high. Importantly, no matter the difference in their experiences, both patients with experience with Tecartus said that they would recommend this treatment to other patients, and both would take this treatment again if their doctor recommended it as the best choice.
Companion Diagnostic Test
Not applicable.
Anything Else?
Not at this time.
Conflict of Interest Declaration — The Leukemia & Lymphoma Society of Canada
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission?
No.
Did you receive help from outside your patient group to collect or analyze data used in this submission?
No.
List any companies or organizations that have provided your group with financial payment over the past 2 years AND who may have direct or indirect interest in the drug under review.
Table 6: Financial Disclosures for The Leukemia & Lymphoma Society of Canada
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Gilead | — | — | — | X |
Clinician Input
Ontario Health (Cancer Care Ontario) Complex Malignant Hematology Group
About Ontario Health (Cancer Care Ontario) Complex Malignant Hematology Group
OH-CCO’s Drug Advisory Committees provide timely evidence-based clinical and health system guidance on drug-related issues in support of CCO’s mandate, including the Provincial Drug Reimbursement Programs (PDRP) and the Systemic Treatment Program.
Information Gathering
Information was jointly discussed via email and meetings.
Current Treatments and Treatment Goals
Current treatments in Canada have limited inclusion criteria based on age limit for existing CAR-T product (KYMRIAH). Kymriah is available for RR ALL up to the age of 26. Older patients with RR ALL could be treated with blinatumamab, inotuzumab, allogeneic transplant, or combination chemotherapy.
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
Older patients (greater than 26) with RR ALL do not have access to CAR-T products outside of clinical trials. Allogeneic transplant can be associated with short and long-term adverse events. In the setting where CAR-T is not available and patient has already has an allogeneic transplant, combination chemotherapy would not be curative.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
As per ZUMA-3 or Health Canada indication (pending).
Which patients would be best suited for treatment with the drug under review? Which patients would be least suitable for treatment with the drug under review?
The applicability of tecartus likely includes patients who would not have been eligible for the the ZUMA 3 trial. This is similar to the expanded applicability compared to the clinical trial inclusion/exclusion criteria that we saw with the two CD19 CAR T products for 3rd line DLCL.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice? How often should treatment response be assessed?
Usual ALL response criteria. Treatment response should be assessed as per standard of care.
What factors should be considered when deciding to discontinue treatment with the drug under review?
Not relevant as this is a single treatment.
What settings are appropriate for treatment with [drug under review]? Is a specialist required to diagnose, treat, and monitor patients who might receive [drug under review]?
There are limited number of CAR-T treatment centers in Canada. The treatment should be in a CAR-T center with an expert in CAR-T. In Canada, these centers are closely linked with acute leukemia programs and expertise.
Additional Information
Not applicable.
Conflict of Interest Declarations — Ontario Health (Cancer Care Ontario) Complex Malignant Hematology Group
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please refer to the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
Yes, Ontario Health provided secretariat function to the DAC.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission?
No.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician who contributed to the input — please add more tables as needed (copy and paste). It is preferred for all declarations to be included in a single document.
Declaration for Clinician 1
Name: Dr. Tom Kouroukis
Position: Lead, Ontario Health CCO Hematology Cancer Drug Advisory Committee
Date: 28-09-2022
Table 7: COI Declaration for OH-CCO Complex Malignant Hematology Group — Clinician 1
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 2
Name: Dr. Chris Bredeson
Position: Lead, Ontario Health CCO Complex Malignant Hematology Group
Date: 09-28-2022
Table 8: COI Declaration for OH-CCO Complex Malignant Hematology Group — Clinician 2
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Cell Therapy Transplant Canada
About Cell Therapy Transplant Canada
Cell Therapy Transplant Canada (CTTC; www.cttcanada.org) is a member-led, national, multidisciplinary organization providing leadership and promoting excellence in patient care, research and education in the field of hematopoietic stem cell transplant and cell therapy.
We are the professional society representing the stem cell transplant community in Canada, including physician, nursing, laboratory, and allied health professionals, along with an active family and caregiver group.
Information Gathering
The proposed submission was discussed by two CTTC committees – our Board of Directors, and our standing committee of program directors, representing the cell therapy and stem cell transplant programs across Canada. These two committees were provided an opportunity to review this report and provide input.
Current Treatments and Treatment Goals
Approximately half (40-50%) of all adult patients with B-cell acute lymphoblastic leukemia (ALL) will relapse after or be refractory to 1-2 lines of standard intensive chemotherapy. Outcomes in this patient population are poor, and the current treatment in this population is immunotherapy with blinatumomab or inotuzumab ozogamicin with a goal to bridge eligible patients who achieve a remission to allogeneic hematopoietic stem cell transplantation (allo-HSCT). Typically, patients receive 1-2 cycles of blinatumomab or inotuzumab prior to allo-HSCT, and occasionally patients receive both of these therapies in sequence prior to the allo-HSCT procedure.
Allo-HSCT is an intensive procedure, performed at a limited number of centres in Canada. In this procedure, the patient’s immune marker genes (human leukocyte antigen – HLA) are stringently matched to a healthy human donor, typically a sibling/family member or a volunteer donor identified through an international database (BeTheMatch, Canadian Blood Services). Hematopoietic stem cells are mobilized from the bone marrow of the healthy donor into the peripheral blood using a combination of growth factor (G-CSF or grastofil) and chemotherapy, and then collected using peripheral blood apheresis, and the product is transferred to the patient’s hospital. Meanwhile, the patient is admitted to hospital and given high dose chemotherapy and/or total body irradiation, followed by infusion of the hematopoietic stem cell product. The subsequent recovery period is prolonged, with a length of stay in hospital of around 4 weeks and close follow-up at a transplant centre for approximately the first 100 days after the stem cell infusion. All patients require immunosuppressive therapy to prevent graft versus host disease and require lifelong follow-up for complications. Treatment related mortality with allo-HSCT is significant at 10-35%, even when restricted to patients with a closely matched donor and good overall fitness level without significant co-morbidities.
Patients whose disease responds to blinatumomab or inotuzumab but are unable to receive allo-HSCT typically receive 5-6 cycles of one of these medications with a goal of prolonging life and delaying disease relapse. When relapse occurs, patients will typically receive the alternative immunotherapy (inotuzumab if received blinatumomab the first time). When the disease no longer responds to one of these agents, patients receive either supportive care alone (i.e. blood transfusion, antibiotics for infection), or supportive care and low dose chemotherapy with a goal to improve quality of life. Patients who relapse after allo-HSCT can also receive immunotherapy with a goal of prolonging life, followed by supportive care alone when the disease no longer responds to therapy.
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
Although blinatumomab and inotuzumab have improved survival in patients with B-cell ALL compared to chemotherapy alone (based on the results of the TOWER and INO-VATE trials, respectively), median overall survival is still short at less than 8 months. Furthermore, few patients have a long-term remission with these therapies alone, and allo-HSCT has remained the only curative option for patients with relapsed/refractory disease.
Allo-HSCT is an intensive procedure, associated with very high short-term morbidity, and multiple known late toxicities. There is significant short-term morbidity associated with allo-HSCT, with patients frequently requiring parenteral analgesia and nutritional support, high rates of febrile neutropenia, and treatment related mortality of 10-35%, depending on the patient’s co-morbid conditions and the degree of mismatch between patient and donor. Due to the exposure to high dose cytotoxic chemotherapy, the risk of a late second malignancy is approximately 10%. There still also remains a significant risk of relapse following transplant, with no good therapy options.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
Brexucabtagene autoleucel would be given to fit patients with CD19+ B-ALL who have relapsed disease (after allo-HSCT, or those not a candidate for allo-HSCT), or who have refractory disease. The goal of therapy would be to prolong life, improve quality of life, and delay disease progression. Fit patients with refractory disease who have not already undergone allo-HSCT and have a donor available may proceed to allo-HSCT after receiving brexucabtagene autoleucel. This treatment may be used instead of (or in sequence with) blinatumomab and inotuzumab, and in patients who have received either of these agents that still have disease that responds to brexucabtagene autoleucel.
In response to the template questions, it would not be appropriate to reserve this treatment for patients who are not tolerant to other treatments, as other treatments have been shown to have similar or inferior outcomes.
Which patients would be best suited for treatment with the drug under review? Which patients would be least suitable for treatment with the drug under review?
The patients best suited for this this therapy are largely identified by the ZUMA-3 trial – namely, adult patients with relapsed or refractory B-cell ALL with morphological disease in the bone marrow (>5% blasts). Relapsed or refractory disease was defined in the study as “primary refractory, first relapse with remission of 12 months or less, relapsed or refractory after at least two previous lines of systemic therapy, or relapsed or refractory after allo-HSCT.” This are the B-ALL patients who are most in need of this intervention.
These patients are easily identified and are currently managed in a limited number of specialized centres, by experienced hematologists and oncologists, who have close links to the stem cell transplant teams, which in most centres, are the teams that will be delivering brexucabtagene autoleucel. Thus, no changes in the existing pathway for treatment will be required. There are no issues related to diagnosis, no companion diagnostic testing required, and no issues with misdiagnosis.
There does exist a small patient population (older age, comorbidities) who might not be expected to tolerate allo-HSCT but likely could tolerate brexucabtagene autoleucel. Given that brexucabtagene autoleucel provides the longest leukemia-free survival for these patients of all the non-transplant therapies, these patients should be eligible for this important therapy.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice? How often should treatment response be assessed?
Outcomes used in clinical practice mirror those used in clinical trial – monitoring of peripheral blood counts and bone marrow biopsies with measurable residual disease (MRD) testing by flow cytometry (and/or next-generation sequencing if available), in combination with clinical evaluation by hematologists/medical oncologists. Biopsies for disease response assessment should be performed in all patients on day 28 after the cell infusion and on an as needed basis thereafter based on clinical evaluation including blood cell count results.
The ZUMA-3 trial demonstrated 71% of treated patients achieved a complete remission (CR) or complete remission with incomplete hematologic recovery (CRi) following brexucabtagene autoleucel. Among the responders, 97% had no MRD detected. Median overall survival was 1.5 years, and notably in patients who responded to therapy the median overall survival was not yet reached.
In relapsed/refractory patients who have a donor available and whose only barrier to allo-HSCT is the need to obtain disease control, the high rate of MRD negativity achieved with this therapy is a clinically meaningful outcome as allo-HSCT outcomes are significantly improved in patients with no evidence of MRD at time of transplant.
In patients who have already undergone allo-HSCT or are ineligible for allo-HSCT, the median overall survival of 1.5 years is, so far, the best-known outcome in this patient population.
What factors should be considered when deciding to discontinue treatment with the drug under review?
Given that brexucabtagene autoleucel is a one-time therapy, there is no need to consider when to discontinue treatment.
What settings are appropriate for treatment with [drug under review]? Is a specialist required to diagnose, treat, and monitor patients who might receive [drug under review]?
This therapy should only be prescribed for this indication by specialists working in a clinic associated with a cell therapy program. In general, these are located in cancer centres associated with tertiary care hospitals in Canada. There are very unique toxicities associated with brexucabtagene autoleucel, and it is critical that these therapies are only administered by well-trained medical teams.
Cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS) are well known effects of CAR-T therapies. In the ZUMA-3 trial, these were seen at rates comparable to that in other studies of patients with B-ALL (24% rate of grade 3 or higher CRS, 25% rate of grade 3 or higher neurotoxicity). These toxicities, while significant, are usually manageable if treated by well-trained and experienced teams.
At this point, only a limited number of centres in Canada are delivering CAR-T therapy. It is critical that additional centres offer this therapy, so that patients can be treated closer to home, without delays that might necessitate bridging therapy.
Additional Information
No additional information is pertinent.
Conflict of Interest Declarations — Cell Therapy Transplant Canada
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please refer to the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
No additional help was provided.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
Not applicable.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician who contributed to the input — please add more tables as needed (copy and paste). It is preferred for all declarations to be included in a single document.
Declaration for Clinician 1
Name: Kevin Hay
Position: Assistant Professor, Department of Medicine, University of British Columbia
Date: 25-09-2022
Table 9: COI Declaration for Cell Therapy Transplant Canada — Clinician 1
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Kite/Gilead | X | — | — | — |
Novartis | X | — | — | — |
BMS | X | — | — | — |
Jazz Pharmaceuticals | X | — | — | — |
Janssen | X | — | — | — |
Declaration for Clinician 2
Name: Christopher Bredeson
Position: Head, Malignant Hematology, Transplant and Cellular Therapy, The Ottawa Hospital; Professor, University of Ottawa
Date: 26-Sept-2022
Table 10: COI Declaration for Cell Therapy Transplant Canada — Clinician 2
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Kite/Gilead | X | — | — | — |
Novartis | X | — | — | — |
Declaration for Clinician 3
Name: Terrance Comeau
Position: Director of New Brunswick Stem Cell Transplant Program, Horizon Health, Saint John, NB
Date: 28-09-2022
Table 11: COI Declaration for Cell Therapy Transplant Canada — Clinician 3
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 4
Name: Kevin Song
Position: Interim Medical Director, Leukemia/BMT Program of BC, Vancouver General Hospital
Date: 29-09-2022
Table 12: COI Declaration for Cell Therapy Transplant Canada — Clinician 4
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
Redactions: Confidential information in this document may be redacted at the request of the sponsor in accordance with the CADTH Drug Reimbursement Review Confidentiality Guidelines.
Stakeholder Input: The views expressed in each submission are those of the submitting organization or individual; not necessarily the views of CADTH or of other organizations. As such, they are independent of CADTH and do not necessarily represent or reflect the view of CADTH. No endorsement by CADTH is intended or should be inferred. By filing with CADTH, the submitting organization or individual agrees to the full disclosure of the information. CADTH does not edit the content of the submissions.
CADTH does use reasonable care to prevent disclosure of personal information in posted material; however, it is ultimately the submitter’s responsibility to ensure no identifying personal information or personal health information is included in the submission. The name of the submitting organization or individual and all conflict of interest information are included in the submission; however, the name of the author, including the name of an individual patient or caregiver submitting the patient input, are not posted.
Accessibility: CADTH is committed to treating people with disabilities in a way that respects their dignity and independence, supports them in accessing material in a timely manner, and provides a robust feedback process to support continuous improvement. All materials prepared by CADTH are available in an accessible format. Where materials provided to CADTH by a submitting organization or individual are not available in an accessible format, CADTH will provide a summary document upon request. More details on CADTH’s accessibility policies can be found here.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.