CADTH Reimbursement Review
Axicabtagene Ciloleucel (Yescarta)
Sponsor: Gilead Sciences Canada Inc.
Therapeutic area: Large B-cell lymphoma
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Ethics Review
Stakeholder Input
Clinical Review
Abbreviations
AE
adverse event
allo-SCT
allogeneic stem cell transplant
ASCT
autologous stem cell transplant
axi-cel
axicabtagene ciloleucel
CAR
chimeric antigen receptor
CHOP
cyclophosphamide, hydroxydaunorubicin (doxorubicin), oncovin (vincristine), and prednisone
CI
confidence interval
CNS
central nervous system
CR
complete response
CRS
cytokine release syndrome
DLBCL
diffuse large B-cell lymphoma
DOR
duration of response
ECOG PS
Eastern Cooperative Oncology Group Performance Status
EFS
event-free survival
EORTC QLQ-C30
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30
EQ-5D-5L
5-Level EQ-5D
G-CSF
granulocyte colony-stimulating factor
HDT
high-dose therapy
HGBL
high-grade B-cell lymphoma
HR
hazard ratio
HRQoL
health-related quality of life
ICU
intensive care unit
IPCW
inverse probability of censoring weights
IxRS
Interactive Voice/Web (x) Response System
LBCL
large B-cell lymphoma
MID
minimal important difference
MMRM
mixed model for repeated measures
NHL
non-Hodgkin lymphoma
NOS
not otherwise specified
ORR
objective response rate
OS
overall survival
PD
progressive disease
PFS
progression-free survival
PMBCL
primary mediastinal B-cell lymphoma
PR
partial response
QoL
quality of life
R-CHOP
rituximab plus cyclophosphamide, hydroxydaunorubicin (doxorubicin), oncovin (vincristine), and prednisone
R-DHAP
rituximab plus dexamethasone, high-dose cytarabine, and cisplatin
R-DHAX
rituximab plus dexamethasone, high-dose cytarabine, and oxaliplatin
R-ESHAP
rituximab plus etoposide, solu-medrone (methylprednisolone), high-dose cytarabine, and cisplatin
R-GDP
rituximab plus gemcitabine, dexamethasone, and cisplatin
R-ICE
rituximab plus ifosfamide, carboplatin, and etoposide
RPSFT
rank-preserving structural failure time
sAAIPI
second-line age-adjusted International Prognostic Index
SAE
serious adverse event
SD
standard deviation
SOC
standard of care
TBI
total body irradiation
TEAE
treatment-emergent adverse event
VAS
visual analogue scale
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Item | Description |
|---|---|
Drug product | Axicabtagene ciloleucel (axi-cel; Yescarta), cell suspension in patient-specific single-infusion bag, target dose of 2 × 106 CAR-positive viable T cells/kg body weight to a maximum of 2 × 108 CAR-positive viable T cells, for IV infusion |
Indication | Treatment of adult patients with diffuse large B-cell lymphoma or high-grade B-cell lymphoma that is refractory to first-line chemoimmunotherapy or that relapses within 12 months of first-line chemoimmunotherapy |
Reimbursement request | Treatment of adult patients with relapsed or refractory large B-cell lymphoma, who are candidates for ASCT |
Health Canada approval status | NOC |
Health Canada review pathway | Standard review |
NOC date | December 6, 2022 |
Sponsor | Gilead Sciences Canada, Inc. |
ASCT = autologous stem cell transplant; CAR = chimeric antigen receptor; NOC = Notice of Compliance.
Introduction
Non-Hodgkin lymphoma (NHL) comprises a diverse group of closely related cancers of the lymphocytes.1 It is the most prevalent hematological malignancy and the fifth most common cancer diagnosed in Canada.1 In 2022 it was estimated that 11,400 people in Canada would be diagnosed with NHL and 3,000 would die from it.2 NHL can affect any organ in the body with a wide range of clinical presentations.3 Most patients with NHL present with painless enlarged lymph nodes with or without systemic symptoms (e.g., fevers, night sweats, weight loss, and fatigue). Large B-cell lymphoma (LBCL) is the most common subtype of NHL diagnosed in Canada, constituting 30% to 40% of all NHL cases in Canada.4 LBCL is an aggressive but potentially curable NHL, and is typically diagnosed at an advanced stage (stage III or IV).5 The 5-year progression-free survival (PFS) in patients with limited disease is 80% to 85%, whereas those with advanced disease have a 5-year PFS of approximately 50%.6
The standard of care (SOC) first-line treatment for patients with newly diagnosed LBCL is CHOP — cyclophosphamide, hydroxydaunorubicin (doxorubicin), oncovin (vincristine), and prednisone — often used in combination with rituximab (R-CHOP).4,7-10 However, 30% to 50% of patients are refractory to or relapse after first-line therapy.4,11 Overall survival (OS) in patients with primary refractory disease is very poor, with only 15% to 20% surviving at 5 years.12 Patients with partial response (PR) or complete response (CR) to first-line treatment also have poor survival at relapse, with 38% and 42% surviving at 5 years, respectively.12 In this population, 82% progressed within 12 months of first-line chemoimmunotherapy, and these patients had significantly worse 3-year OS rate compared with patients who relapsed after 12 months (10% versus 39%, respectively).13
For patients who relapse or whose disease is refractory to first-line chemoimmunotherapy, second-line treatment comprises salvage chemotherapy, and if responsive to salvage therapy, this is followed by high-dose therapy (HDT) and autologous stem cell transplant (ASCT). However, only about half of the patients with relapsed or refractory LBCL are fit enough for transplant (i.e., have adequate organ function with no major comorbidities), and only half of transplant-eligible patients respond to salvage chemotherapy and can proceed to ASCT. Treatment options for patients with relapsed or refractory LBCL who are ineligible for ASCT, do not respond to salvage chemotherapy, or relapse post-ASCT include palliative chemotherapy, radiotherapy, clinical trials, or third-line chimeric antigen receptor (CAR) T-cell therapy if the patient meets the eligibility criteria.
Axicabtagene ciloleucel (axi-cel) is a CD19 antigen–directed genetically modified autologous T-cell immunotherapy (i.e., CAR T-cell therapy). The objective of this review was to evaluate the efficacy and safety of axi-cel (IV infusion, target dose of 2 × 106 anti-CD19 CAR T cells/kg body weight) for the treatment of adult patients with relapsed or refractory LBCL. Of note, this CADTH reimbursement review was conducted before issuance of Health Canada Notice of Compliance (NOC) and the scope was based on the anticipated indication.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient groups who responded to CADTH’s call for patient input and from clinical expert(s) consulted by CADTH for the purpose of this review.
Patient Input
One patient group, Lymphoma Canada, submitted patient input for this review. This group gathered information from patients with diffuse large B-cell lymphoma (DLBCL) through 2 online surveys, 1 in 2018 and the other in 2022. Respondents from both surveys reported that fear of progression/relapse was the most common psychosocial impact (67%) affecting quality of life, followed by anxiety (37%), memory loss (37%), and concentration problems (36%). The majority of respondents (83%) were treated with CHOP with or without rituximab as their first line of treatment after diagnosis. The respondents reported that long-term treatment side effects (i.e., lasting longer than 2 years or appearing at least 2 years after the end of treatment) included fatigue (52%) and chemo brain (42%). Respondents from the first survey reported that their lymphoma treatment had the greatest negative impact on their work, travel, and other activities. Patients rated longer survival, longer remission, better quality of life, and fewer side effects as important outcomes expected from their treatment. About half of the patients stated that they would choose a treatment with potentially serious side effects if their doctor recommended it as the most effective option for DLBCL. Respondents also indicated that treatment choice was an important factor, and 91% of patients felt a need for more therapy options for patients with DLBCL.
Three patients reported receiving axi-cel as third-line therapy. All 3 patients were away from home for more than 3 months while receiving the treatment, pointing to the challenges in accessing CAR T-cell therapy. All 3 patients reported thrombocytopenia as a side effect of axi-cel treatment, with fever, anemia, nausea/vomiting, neutropenia, diarrhea, joint or muscle pain, and fatigue reported by 2 patients. Fear of progression/relapse and difficulty sleeping were reported by all 3 patients as psychosocial impacts related to their CAR T-cell therapy.
Clinician Input
Input From Clinical Experts Consulted by CADTH
Four clinical experts from across Canada contributed input to the CADTH review. The clinical experts consulted by CADTH noted the unmet treatment needs of patients who are refractory to or who relapsed after front-line therapy. While high-dose therapy and autologous stem cell transplant (HDT-ASCT) has curative potential for patients with relapsed or refractory LBCL, many are ineligible for ASCT or do not respond to salvage chemotherapy. The clinical experts indicated that axi-cel would fit well earlier in the treatment lines and could be used for most patients instead of ASCT. The clinical experts noted that patient outcomes are expected to improve if patients receive a potentially curative therapy earlier in the course of disease as some patients deteriorate rapidly and may be less likely to survive if definitive treatment is delayed.
The clinical experts noted that while the ZUMA-7 trial recruited only patients who were eligible for ASCT, in standard clinical practice there is no clinical rationale for restricting axi-cel only to those who are candidates for ASCT, and that any patient with adequate organ function and good performance status (Eastern Cooperative Oncology Group Performance Status [ECOG PS] score ≤ 2) who, based on the clinician’s judgment, can tolerate the known toxicities of CAR T-cell therapy (e.g., cytokine release syndrome [CRS]) would be suitable for axi-cel treatment. The clinical experts noted that axi-cel treatment can be provided by oncologists or hematologists in a hospital setting with adequate infrastructure for cell therapy and with access to highly specialized multidisciplinary clinical care including critical/intensive care and specialist care (e.g., neurology, nephrology) to manage toxicities, as well as laboratory support to handle and process samples. The clinical experts also pointed out that the 13-day median manufacturing time reported in the ZUMA-7 study is rapid and may not be reproducible outside the clinical trial setting; longer wait times may compromise patient outcomes.
Clinician Group Input
Clinician group input was received from 3 groups: Lymphoma Canada, Ontario Health (CCO) Hematology Cancer Drug Advisory Committee, and Cell Therapy Transplant Canada (CTTC). The clinician groups agreed that there are unmet needs in the current second-line treatment for patients with relapsed or refractory LBCL. The clinician groups indicated that there may be limited eligibility or tolerability to further salvage chemotherapy for some patients (e.g., patients with primary refractory disease or early relapse, and older patients). The clinician groups also noted that toxicities such as febrile neutropenia, bacteremia and other infections, gastrointestinal toxicity, mucositis, need for transfusion support, and secondary malignancies associated with ASCT treatment have made it unsuitable for patients who are high risk, who are refractory to treatment, or who relapse within 12 months of diagnosis.
Drug Program Input
Input was obtained from the drug programs that participate in the CADTH reimbursement review process. The following were identified as the key factors that could potentially impact the implementation of a CADTH recommendation for axi-cel:
Considerations for initiation of therapy
Considerations for prescribing of therapy
Care provision issues
System and economic issues
The clinical experts consulted by CADTH provided advice on the potential implementation issues raised by the drug programs.
Clinical Evidence
Pivotal Studies and Protocol-Selected Studies
Description of Studies
A single sponsor-submitted pivotal study was included in this review.14,15 The ZUMA-7 study is a phase III, multicentre, randomized, open-label study evaluating the efficacy of axi-cel compared with SOC (salvage chemoimmunotherapy followed by HDT-ASCT) as a second-line therapy in patients with relapsed or refractory LBCL after first-line rituximab and anthracycline-based chemotherapy. The trial was conducted in 14 countries; 20 patients from 8 centres were recruited in Canada. The first patient was enrolled (randomized) on January 25, 2018, and enrolment was completed on October 4, 2019; ZUMA-7 is currently ongoing. All patients had either primary refractory disease or relapse within 12 months of completing first-line therapy, were potentially eligible for ASCT, and had not yet received second-line treatment. The data cut-off date for the primary analysis was March 18, 2021. For patients in the axi-cel arm (N = 180), treatment consisted of lymphodepleting chemotherapy followed by a single IV infusion of axi-cel. Bridging therapy consisting of corticosteroids was allowed before lymphodepleting chemotherapy for patients with high disease burden, at the discretion of the investigator. For patients in the SOC arm (N = 179), treatment consisted of a single protocol-defined, platinum-based salvage chemotherapy regimen for 2 to 3 cycles as selected by the treating investigator. Patients who responded to salvage chemotherapy were to proceed to HDT followed by ASCT. The mean age of patients was 57 years (standard deviation [SD] = 12 years); 30% of the patients were aged 65 years or older. Overall, 74% of the study population had primary refractory disease and 26% had early relapse. Approximately one-quarter of patients in both treatment arms had achieved a best response of CR to first-line treatment.
Efficacy Results
Key efficacy results from the ZUMA-7 study are summarized in Table 2.
Overall Survival
OS was a key secondary outcome in the ZUMA-7 study. The OS data remain immature at the time of this review. At the time of the data cut-off date (March 18, 2021), 72 patients (40%) in the axi-cel arm had died and 81 patients (45%) in the SOC arm had died. The primary OS analysis will occur at approximately 210 deaths or 5 years after the first patient was enrolled. At the interim OS analysis, the hazard ratio (HR) for death was 0.730 (95% confidence interval [CI], 0.530 to 1.007; 1-sided stratified log-rank P value = 0.0270).
Event-Free Survival
Event-free survival (EFS) as determined by blinded central assessment was the primary outcome of the ZUMA-7 study. At the time of the data cut-off, 252 EFS events as determined by blinded central assessment had occurred for 108 patients (60%) in the axi-cel arm and for 144 patients (80%) in the SOC arm. The median EFS was 8.3 months (95% CI, 4.5 to 15.8 months) in the axi-cel arm and 2.0 months (95% CI, 1.6 to 2.8 months) in the SOC arm. The stratified HR for event or death was 0.398 (95% CI, 0.308 to 0.514; P < 0.0001).
Health-Related Quality of Life
HRQoL was assessed by changes from screening in the global health status scale and the physical functioning domain of the European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 (EORTC QLQ-C30) and the 5-Level EQ-5D (EQ-5D-5L) index and visual analogue scale (VAS) scores were secondary outcomes. There was a clinically meaningful difference (based on the trial-specified threshold of ± 10 points)16 in mean change of scores from baseline to study day 100 for the EORTC QLQ-C30 global health status and physical functioning, and EQ-5D-5L and VAS scores with axi-cel compared with SOC. However, the high attrition rate, which was imbalanced between groups at all follow-up time points, limits interpretation of these data.
For the EORTC QLQ-C30 global health status there was a clinically meaningful difference in mean change of scores from screening at study day 100 for patients treated with axi-cel compared with SOC (estimated difference = 18.1 points; 95% CI, 12.3 to 23.9; adjusted P < 0.0001). At study day 150, the estimated difference was 9.8 points (95% CI, 2.6 to 17.0; adjusted P = 0.0124).
For the EORTC QLQ-C30 physical functioning subscale, there was a clinically meaningful difference in mean change of scores from screening to study day 100 for patients treated with axi-cel compared with SOC (estimated difference = 13.1; 95% CI, 8.0 to 18.2; adjusted P < 0.0001).
For the EQ-5D-5L VAS there was a clinically meaningful difference in mean change of scores from screening in the axi-cel arm compared with SOC at study day 100 (estimated difference = 13.7; 95% CI, 8.5 to 18.8; adjusted P < 0.0001) and study day 150 (estimated difference = 11.3; 95% CI, 5.4 to 17.1; adjusted P = 0.0004).
Progression-Free Survival
PFS was a secondary outcome. At the data cut-off date, the median duration of PFS based on investigator disease assessments was 14.9 months (95% CI, 7.2 months to not estimable [NE]) in the axi-cel arm and 5.0 months (95% CI, 3.4 months to 8.5 months) in the SOC arm, with a stratified HR of 0.562 (95% CI, 0.414 to 0.762).
Objective Response Rate
Objective response rate (ORR) as determined by blinded central assessment was a key secondary outcome in ZUMA-7. The ORR (CR or PR) as determined by blinded central assessment was 83% in the axi-cel arm and 50% in the SOC arm (difference in ORR = 33.1%; 95% CI, 23.2% to 42.1%; P < 0.0001).
The CR rates in the axi-cel arm and the SOC arm were 65% (95% CI, 57.6% to 71.9%; n = 117) and 32% (95% CI, 25.6% to 39.8%; n = 58), respectively. The PR rates were 18% (95% CI, 13.0% to 24.8%; n = 33) in the axi-cel arm and 18% (95% CI, 12.6%, 24.3%; n = 32) in the SOC arm.
Duration of Response
Duration of response (DOR) was a secondary outcome. For the 150 patients in the axi-cel arm and the 90 patients in the SOC arm who achieved an objective response of CR or PR as determined by blinded central assessment, the Kaplan-Meier estimated median DOR was 26.9 months (95% CI, 13.6 months to NE) in the axi-cel arm compared with 8.9 months (95% CI, 5.7 months to NE) in the SOC arm (stratified HR = 0.736; 95% CI, 0.488 to 1.108).
The Kaplan-Meier estimates of the percentage of patients who remained in response at 12 and 24 months from first objective response were 60.9% (95% CI, 52.4% to 68.4%) and 54.0% (95% CI, 45.1% to 62.0%), respectively, in the axi-cel arm compared with 47.6% (95% CI, 35.2% to 58.9%) and 45.6% (95% CI, 33.2% to 57.1%), respectively, in the SOC arm.
Time to Next Treatment
Time to next treatment was an exploratory outcome. Time to next treatment events occurred for 99 patients (55%) in the axi-cel arm and 135 patients (75%) in the SOC arm. The median time to next treatment was 14.7 months (95% CI, 6.5 months to NE) and 3.4 months (95% CI, 3.1 months to 4.4 months), respectively (stratified HR = 0.430; 95% CI, 0.329 to 0.560).
Health Care Resource Utilization
A total of 42 patients (25%) in the axi-cel arm and 9 patients (5%) in the SOC arm were admitted to the intensive care unit (ICU). Median duration of ICU hospitalization was 5 days (range, 1 to 12 days) in the axi-cel arm and 3 days (range, 2 to 17 days) in the SOC arm. Median duration of hospitalization for axi-cel infusion was 16 days (range, 5 to 103 days); median duration of inpatient hospitalization for stem cell transplant in the SOC arm was 21 days (range, 1 to 53 days).
Table 2: Summary of Key Efficacy Results From the ZUMA-7 Study (Full Analysis Set)
Outcome | Axi-cel (N = 180) | SOC (N = 179) |
|---|---|---|
OS | ||
Death from any cause, n (%) | 72 (40) | 81 (45) |
Stratified 1-sided log-rank P valuea | 0.0270 | NA |
HR (95% CI), stratifiedb,c | 0.730 (0.530 to 1.007) | NA |
OS time (months), median (95% CI)d | NR (28.3 to NE) | 35.1 (18.5 to NE) |
EFS per central assessment | ||
Events, n (%) | 108 (60) | 144 (80) |
Stratified log-rank P valueb | < 0.0001 | NA |
Stratified HR (95% CI)b | 0.398 (0.308 to 0.514) | NA |
EFS time (months), median (95% CI)c | 8.3 (4.5 to 15.8) | 2.0 (1.6 to 2.8) |
PFS | ||
Events, n (%) | 93 (52) | 81 (45) |
Hazard ratio (95% CI), stratifiedb,e | 0.562 (0.414 to 0.762) | |
PFS (months), median (95% CI)c | 14.9 (7.2 to NE) | 5.0 (3.4 to 8.5) |
ORR | ||
Objective responders (CR + PR), n (%) | 150 (83) | 90 (50) |
95% CI for ORR, % | 77.1 to 88.5 | 42.7 to 57.8 |
Difference in ORR (95% CI), %f | 33.1 (23.2 to 42.1) | |
Stratified P valueb,g | < 0.0001 | |
CR, n (%) | 117 (65) | 58 (32) |
95% CI for response rate, %h | 57.6 to 71.9 | 25.6 to 39.8 |
PR, n (%) | 33 (18) | 32 (18) |
95% CI for response rate, %h | 13.0 to 24.8 | 12.6 to 24.3 |
DOR | ||
Number of objective responders (CR + PR), n | 150 | 90 |
Events, n (%) | 66 (44) | 37 (41) |
HR (95% CI), stratifiedb,e | 0.736 (0.488 to 1.108) | |
P value, stratifiedb,i | 0.0695j | |
DOR (months), median (95% CI)c | 26.9 (13.6 to NE) | 8.9 (5.7 to NE) |
Time to next treatment | ||
Events, n (%) | 99 (55) | 135 (75) |
HR (95% CI), stratifiedb,e | 0.430 (0.329 to 0.560) | |
P value, stratifiedb,i | < 0.0001j | |
axi-cel = axicabtagene ciloleucel; CI = confidence interval; CR = complete response; DOR = duration of response; EFS = event-free survival; NA = not applicable; NE = not estimable; NR = not reached; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; PR = partial response; SOC = standard of care.
aAlpha allocated to this analysis: 1-sided P = 0.004.
bThe stratification factors are responses to first-line therapy (primary refractory vs. relapse ≤ 6 months of first-line therapy vs. relapse > 6 and ≤ 12 months of first-line therapy) and second-line age-adjusted International Prognostic Index (0 to 1 vs. 2 to 3) as collected via interactive voice/web response system.
cEstimated using the Kaplan-Meier method.
dEstimated using the reverse Kaplan-Meier approach.
eHazard ratio and 95% CIs were estimated using Cox regression models for axi-cel relative to SOC. The Breslow method was used to handle ties for the Cox regression models.
f95% CI for the difference in ORR (with the SOC arm as reference group) was from the Wilson score method with continuity correction.
gOne-sided P value from the Cochran-Mantel-Haenszel test.
hEstimated using the Clopper-Pearson method.
iP value is based on the log-rank test.
jAnalysis not part of the statistical hierarchy.
Source: Clinical Study Report for Yescarta.15
Harms Results
Key harms results from the ZUMA-7 study are summarized in Table 3.
All patients in the axi-cel arm (170 patients; 100%) and the SOC arm (168; 100%) had at least 1 treatment-emergent adverse event (TEAE); 155 patients (91%) in the axi-cel arm and 140 patients (83%) in the SOC arm had grade 3 or higher TEAEs. The most frequently reported TEAEs of worst grade 3 or higher (reported in ≥ 20% of patients in either treatment arm) were neutropenia (73 patients; 43%); anemia (51 patients; 30%); neutrophil count decreased (49 patients; 29%); and white blood cell count decreased (43 patients; 25%) in the axi-cel arm, and anemia (65 patients; 39%); platelet count decreased (60 patients; 36%); decreased neutrophil count (47 patients; 28%); febrile neutropenia (46 patients; 27%); and thrombocytopenia (37 patients; 22%) in the SOC arm.
A total of 85 patients (50%) in the axi-cel arm and 77 patients (46%) in the SOC arm had at least 1 serious adverse event (SAE). The most frequently (in ≥ 5% of patients) reported SAEs of any grade in each treatment arm were pyrexia (27 patients; 16%), encephalopathy (17 patients; 10%), hypotension (15 patients; 9%), aphasia (9 patients; 5%), and pneumonia (8 patients; 5%) in the axi-cel arm, and febrile neutropenia (22 patients; 13%) and acute kidney injury and pyrexia (8 patients each; 5%) in the SOC arm.
No patient discontinued treatment due to TEAEs in the axi-cel arm. Two patients in the SOC arm discontinued treatment due to TEAEs of grade 4 acute kidney injury and grade 1 blood stem cell harvest failure.
In the axi-cel arm, 64 patients (38%) had died by the data cut-off date due to progressive disease (PD) (n = 47; 28%), TEAEs (n = 6; 4%), and other reasons (n = 10; 6%) and 1 patient (1%) had died from an event reported by the investigator as a “secondary malignancy” (lung adenocarcinoma). In the SOC arm, 78 patients (46%) had died by the data cut-off date due to PD (n = 64; 38%), TEAEs (n = 2; 1%), or other reasons (n = 12; 7%).
In total, 102 patients (60%) in the axi-cel arm and 33 patients (20%) in the SOC arm had at least 1 treatment-emergent neurologic event, including 36 patients (21%) and 1 patient (1%) in the axi-cel and SOC arms, respectively, with worst grade 3 or higher neurologic events. Of these, 10 patients (6%) in the axi-cel arm had worst grade 4 neurologic events; no patients in either treatment arm had a grade 5 neurologic event. Serious treatment-emergent neurologic events of any grade were reported for 34 patients (20%) in the axi-cel arm and 1 patient (1%) in the SOC arm, including 26 patients (15%) in the axi-cel arm with a serious worst grade 3 or higher neurologic event and 1 patient (1%) in the SOC arm with a serious worst grade 2 neurologic event.
CRS of any grade was reported for 157 patients (92%), including 11 patients (6%) who had worst grade 3 or higher CRS, in the axi-cel arm. No patient had grade 5 CRS.
The most common cytopenias of any grade reported in the axi-cel arm were thrombocytopenia (50 patients; 29%), neutropenia (122 patients; 72%), and anemia (73 patients; 43%). The most common cytopenias of any grade reported in the SOC arm were thrombocytopenia (101 patients; 60%), neutropenia (92 patients; 55%), and anemia (92 patients; 55%).
Seventy patients (41%) in the axi-cel arm and 51 patients (30%) in the SOC arm had at least 1 treatment-emergent infection, including 24 patients (14%) and 19 patients (11%), respectively, with worst grade 3 or higher infections. Three patients (2%) in the axi-cel arm and 6 patients (4%) in the SOC arm had worst grade 4 infections. Five patients (3%) in the axi-cel arm had a grade 5 TEAE of infection (2 patients with COVID-19, 1 patient with progressive multifocal leukoencephalopathy, 1 patient with hepatitis B reactivation, and 1 patient with sepsis). No patients in the SOC arm had a grade 5 TEAE of infection.
Table 3: Summary of Key Harms Results From the ZUMA-7 Study (Safety Analysis Set)
Harms, n (%) | Axi-cel (N = 170) | SOC (N = 168) |
|---|---|---|
Any TEAE | 170 (100) | 168 (100) |
Grade ≥ 3 | 155 (91) | 140 (83) |
Any serious TEAE | 85 (50) | 77 (46) |
Grade ≥ 3 | 72 (42) | 67 (40) |
Treatment-related TEAE | 163 (96) | 160 (95) |
Grade ≥ 3 | 112 (66) | 131 (78) |
Any treatment-emergent neurologic event | 102 (60) | 33 (20) |
Grade ≥ 3 | 36 (21) | 1 (1) |
Any treatment-emergent CRS | 157 (92) | NA |
Grade ≥ 3 | 11 (6) | NA |
Any treatment-emergent cytopenias | 136 (80) | 135 (80) |
Any treatment-emergent infections | 70 (41) | 51 (30) |
Any treatment-emergent hypogammaglobulinemia | 19 (11) | 1 (1) |
Withdrawal due to AEs | 0 (0) | 2 (1) |
Death due to an AE | 6 (4) | 2 (1) |
AE = adverse event; axi-cel = axicabtagene ciloleucel; CRS = cytokine release syndrome; NA = not applicable; TEAE = treatment-emergent adverse event; SOC = standard of care.
Critical Appraisal
The ZUMA-7 trial was open label. Despite the open-label design, there is low risk of bias in the measurement of outcomes such as EFS, PFS, and ORR, which were assessed via independent blinded radiologic review of disease response, or OS, which is an objective outcome. Although independent blinded radiologic review of disease response was performed as the primary analysis to minimize investigator bias, patients’ knowledge of their assigned treatment could impact HRQoL data and any subjective harm (which are particularly susceptible to bias from a lack of blinding of patients to their treatment). The HRQoL data were also at high risk of attrition bias because there were large amounts of missing data for all follow-up time points and the amount of missing data was not balanced across treatment arms. The HRQoL tools were not validated in patients with LBCL. The primary and secondary efficacy end points of EFS, ORR, and OS are considered appropriate for the disease setting. The OS data were immature and, because the results are based on an interim analysis, there is a risk that the effect of axi-cel compared with SOC on survival is overestimated.17,18 Although there is some evidence to suggest EFS is a robust surrogate end point for OS in hematological malignancies,19 it is unknown whether this could be extended to CAR T-cell therapies in relapsed or refractory LBCL.
The ZUMA-7 trial included patients with relapsed or refractory LBCL with a wide range of clinical presentations, but patients with HIV and those with an ECOG PS score of 2 or more were excluded. The clinical experts consulted by CADTH indicated that these patient groups should be eligible for CAR T-cell therapies including axi-cel. While the SOC treatment including salvage chemotherapy regimens used in the SOC arm of the ZUMA-7 trial are reflective of clinical practice in Canada, the clinical experts noted the challenges in reproducing the same treatment processes for axi-cel treatment in the real-world setting, notably the rapid manufacturing time (13 days in the ZUMA-7 trial). According to the clinical experts, delays in manufacturing and access times to axi-cel treatment would potentially compromise patient outcomes as the probability of disease progression or other complications increase with longer axi-cel manufacturing wait times. Data for all outcomes considered important to patients, clinicians, and drug plans, as per the systematic review protocol, were collected and reported; however, conclusions could not be drawn for effects of axi-cel compared with SOC on HRQoL due to limitations in the data.
Other Relevant Evidence
The sponsor provided long-term (≥ 4 year and ≥ 5 year) data from ZUMA-1, the pivotal multicentre, single-arm, registrational phase I and II study of axi-cel in patients with relapsed or refractory LBCL.20-22
In the 2-year analysis of ZUMA-1 (n = 101; median follow-up from axi-cel dosing to data cut-off = 27.1 months), the ORR was 83%; 58% of patients achieved a CR. The 2-year OS rate was 50.5%.
The most recently updated survival results from the phase II ZUMA-1 study showed a 5-year OS rate of 42.6% (95% CI, 32.8% to 51.9%) among patients treated with axi-cel after 5 years of follow-up. The 5-year OS rate among complete responders was 64.4% (95% CI, 50.8% to 75.1%). The median survival time among complete responders was not reached (95% CI, 63.4% to NE). Of 59 patients who achieved CR, 37 (63%) were alive at the 5-year data cut-off date. Since the 4-year data cut-off date, 1 death at month 63 (CR) and 1 PD at month 54 (PR) were observed.
Supportive safety data comparing axi-cel-treated populations in ZUMA-7 and ZUMA-1 suggest comparable TEAEs between the 2 trials.
Critical Appraisal
The ZUMA-1 trial was critically appraised in the 2019 CADTH Optimal Use Report report.23 ZUMA-1 was a single-arm, phase I and II study of axi-cel in patients with relapsed or refractory LBCL who had received at least 2 previous systemic therapies. The primary limitation of ZUMA-1 was the absence of a comparator group against which the treatment benefits and harms of axi-cel could be compared. As such, causal effects cannot be inferred. In addition, patients in ZUMA-1 received axi-cel as third- or later-line treatment. It is unknown whether results are generalizable to patients treated with axi-cel as second-line treatment, which is the indication under review.
Conclusions
Based on data from the ZUMA-7 trial, the anti-CD19 CAR T-cell axi-cel led to an improvement in EFS and PFS compared with SOC HDT-ASCT as second-line treatment in ASCT-eligible patients with relapsed or refractory LBCL within 12 months of first-line chemoimmunotherapy. It is not yet clear whether EFS benefits will translate to improved OS as the data remain immature and follow-up is ongoing. Because the OS results are based on an interim analysis, there is a risk that the effect of axi-cel compared with SOC on survival is overestimated. The risks associated with axi-cel treatment are consistent with its mechanism of action and acceptable given the high-risk patient population, but it must be administered in specialized centres equipped to manage CAR T-cell therapy–related toxicities. Although the most common toxicities were similar in the ZUMA-7 and ZUMA-1 studies, the lack of long-term safety and efficacy data from the ZUMA-7 trial remains an important limitation. A longer follow-up time is needed to better establish the survival benefit and long-term safety of axi-cel, as relapse of lymphoma and potential late toxicities of CAR T-cell therapy such as secondary malignancies may emerge with longer follow-up. Uncertainties remain regarding the implementation of CAR T-cell therapy and the support system needed to optimize timely access and deliverability of axi-cel in the real-world setting.
Introduction
Disease Background
NHL comprises a diverse group of at least 60 closely related cancers of the lymphocytes.1 About 85% to 90% of NHLs are derived from B lymphocytes; the remaining lymphomas are derived from T lymphocytes or natural killer cells.3 NHL is the most prevalent hematological malignancy and the fifth most common cancer diagnosed in Canada.1 An estimated 11,400 Canadians were diagnosed with NHL in 2022 and 3,000 died from it.2 Risk factors for NHL include immune disorders (e.g., rheumatoid arthritis, Sjögren syndrome, systemic lupus erythematosus), use of immunosuppressive therapies, bacterial and viral infections (e.g., Helicobacter pylori, Epstein-Barr virus, and hepatitis C virus), family history, and genetic, occupational, and lifestyle risk factors.3 NHL can affect any organ in the body and have a wide range of clinical presentations.3 Most patients with NHL present with painless enlarged lymph nodes with or without systemic symptoms (e.g., fevers, night sweats, weight loss, and fatigue); however, because NHL can involve any organ in the body, a wide range of presentations that mimic other conditions are possible.3 Diagnosis is established based on a biopsy sample with immunohistochemical and genetic testing, ideally from an excisional biopsy of an involved lymph node or a tumour in another organ, or a cutting-needle biopsy if an excisional biopsy is impractical.3 The differential diagnosis and classification of the many subtypes of NHL are complex and have evolved with advances in clinical science and understanding of their distinctive molecular and immunophenotypic features.
LBCL is the most prevalent subtype of NHL in Canada, constituting 30% to 40% of all NHL cases.4 LBCL comprises a group of cancers of B lymphocytes that develop and mature in the bone marrow and lymph nodes. The B-cell lineage-specific antigens CD19 and CD20 are highly and consistently expressed on the cell surface of most B-cell lymphomas. DLBCL not otherwise specified (NOS) is the most common subtype of LBCL, accounting for more than 80% of cases.24 DLBCL comprises a group of LBCL cases that remain biologically and clinically heterogeneous, for which there are no clear and accepted criteria for subclassification. Other subtypes of LBCL include DLBCL transformed from follicular lymphoma or chronic lymphocytic leukemia; high-grade B-cell lymphoma (HGBL); primary DLBCL of the central nervous system (CNS); primary cutaneous DLBCL, leg type; Epstein-Barr virus–positive DLBCL; DLBCL associated with chronic inflammation; and T-cell/histiocyte-rich LBCL.24-27 The median age at diagnosis of LBCL is 65 years, with 30% of patients older than 75 years. LBCL is slightly more common among males.5 LBCL often manifests as a rapidly enlarging painless mass at a nodal or extranodal site, and other general symptoms such as enlarged lymph nodes, fatigue, cough, itchy skin, and loss of appetite.5 In addition, 30% to 40% of patients present with “B symptoms,” including fevers, night sweats, and unexplained weight loss.5,24 LBCL is an aggressive but potentially curable NHL, and is typically diagnosed at an advanced stage (stage III or IV).5 The 5-year PFS in patients with limited disease is 80% to 85%, whereas those with advanced disease have a 5-year PFS of about 50%.6 Patients with LBCL experience a high symptom burden and poorer HRQoL compared with a general cancer reference population, with further decline in HRQoL observed with disease progression and subsequent lines of therapy.28,29
Patients with newly diagnosed LBCL are routinely treated with the chemotherapy regimen CHOP in combination with the anti-CD20 monoclonal antibody rituximab (R-CHOP). Treatment with R-CHOP is widely recognized as the first-line SOC treatment for LBCL based on Canadian and international clinical practice guidelines.4,7-10 Although many patients respond well to R-CHOP and achieve long-lasting remission, a large proportion (30% to 50%) will either be refractory to or relapse following front-line treatment.4,11 Most relapses occur within the 2 to 3 years after initial treatment.4 OS in patients with primary refractory disease is very low, with only 15% to 20% surviving at 5 years.12 Patients with partial or complete response to first-line treatment also have poor survival at relapse, with 38% and 42% surviving at 5 years, respectively.12 Patients with primary refractory disease or early relapsed disease (less than 12 months) have worse outcomes than patients who relapse more than 12 months after first-line treatment.30,31 Patients with relapsed or refractory LBCL who relapsed within 12 months of initial treatment had worse response rates to second-line salvage chemotherapy than patients who relapsed after 12 months.32 Another recent Canadian real-world study of patients with relapsed or refractory LBCL found a median OS of 5.1 months, with 15% of patients alive at 3 years. In this population, 82% progressed within 12 months of first-line chemoimmunotherapy, and these patients had significantly worse 3-year OS than those who relapsed after 12 months (10% versus 39%, respectively).13
Standards of Therapy
For patients who relapse or who have disease that is refractory to first-line chemoimmunotherapy, second-line treatment comprises salvage chemotherapy, followed by curative-intent HDT and ASCT, if the patients meet the eligibility criteria and have chemosensitive disease. The goal of salvage chemotherapy is to reduce tumour burden and demonstrate chemosensitivity before proceeding to HDT-ASCT. Achieving better response to salvage chemotherapy is associated with better outcomes following ASCT, but no salvage regimen has been shown to be superior to another.4,33 Commonly used salvage chemotherapy regimens in Canada include rituximab plus gemcitabine, dexamethasone, and cisplatin (R-GDP), rituximab plus ifosfamide, carboplatin, and etoposide (R-ICE), rituximab plus dexamethasone, high-dose cytarabine, and cisplatin (R-DHAP), and rituximab plus dexamethasone, cyclophosphamide, etoposide, and cisplatin (R-DICEP).4 Patients who have a CR or PR to salvage chemotherapy will then have their stem cells mobilized and collected by leukapheresis and be administered HDT consisting of a combination chemotherapy such as etoposide plus melphalan or carmustine (BCNU), etoposide, cytarabine (Ara-C), and melphalan (BEAM). The stem cells are then infused back into the patient as part of the ASCT. Although HDT-ASCT has curative potential, many patients are not fit enough for transplant, do not respond to salvage chemotherapy (i.e., do not have chemosensitive disease), or relapse following ASCT. Only about half of the patients with relapsed or refractory LBCL are fit enough for transplant (i.e., have adequate organ function with no major comorbidities), and only half of transplant-eligible patients respond to salvage chemotherapy and can proceed to ASCT.32,34 Of the patients who receive ASCT (25% of patients with relapsed or refractory LBCL), approximately half remain event-free 4 years after transplant.32,35
Patients with relapsed or refractory LBCL who are ineligible for ASCT, do not respond to salvage chemotherapy, or relapse post-ASCT have limited treatment options and a poor prognosis.4 Treatment options for these patients include palliative chemotherapy, radiotherapy, clinical trials, or third-line CAR T-cell therapy. Polatuzumab vedotin plus bendamustine and rituximab (Pola-BR) may provide a future treatment option for patients who are ineligible for ASCT as it is currently Health Canada–approved in this setting and has received a positive reimbursement recommendation from CADTH.4,36 The clinical experts consulted by CADTH indicated that provincial funding for CAR T-cell therapy currently varies and some patients are required to travel out of province or country to access it. For patients who are ineligible for CAR T-cell therapy or fail treatment, there is no standard treatment approach and further treatment depends on access to clinical trials.
Drug
Axi-cel (brand name Yescarta) is a CD19-directed genetically modified autologous T-cell immunotherapy that binds to CD19-expressing cancer cells and normal B cells.37 Following anti-CD19 CAR T-cell engagement with CD19-expressing target cells, the CD28 and CD3-zeta costimulatory domains activate downstream signalling cascades that lead to T-cell activation, proliferation, acquisition of effector functions, and secretion of inflammatory cytokines and chemokines. This sequence of events leads to killing of CD19-expressing cells.37 To prepare axi-cel, a patient’s own T cells are harvested and genetically modified ex vivo by retroviral transduction to express a CAR protein comprising a murine anti-CD19 single-chain variable fragment (scFV) linked to CD28 and CD3-zeta costimulatory domains. The anti-CD19 CAR T cells are expanded and infused back into the patient, where they recognize and eliminate CD19-expressing target cells.
Axi-cel was first approved by the FDA (FDA) in October 2017 for the treatment of adult patients with relapsed or refractory LBCL after 2 or more lines of systemic therapy, including DLBCL NOS, primary mediastinal B-cell lymphoma (PMBCL), HGBL, and DLBCL arising from follicular lymphoma.38 Approval by the European Medicines Agency followed in August 2018 as a treatment for adult patients with relapsed or refractory DLBCL and primary mediastinal B-cell lymphoma after 2 or more lines of systemic therapy.39
In Canada, axi-cel is currently approved for the treatment of patients with relapsed or refractory LBCL who have received at least 2 previous systemic therapies.37 Of note, this CADTH reimbursement review was conducted before issuance of Health Canada NOC and the scope was based on the anticipated indication. The anticipated indication during the time of this review was for the treatment of adult patients with LBCL that is refractory to first-line chemoimmunotherapy or that relapses within 12 months of first-line chemoimmunotherapy. The sponsor’s reimbursement request is the same as the anticipated Health Canada indication, except that it narrows the population to patients who are candidates for ASCT. NOC was received on December 6, 2022, and the final approved indication is for the treatment of adult patients with DLBCL or HGBL that is refractory to first-line chemoimmunotherapy or that relapses within 12 months of first-line chemoimmunotherapy. In 2019, CADTH published an Optimal Use Report evaluating the beneficial and harmful effects of axi-cel for eligible types of relapsed or refractory B-cell lymphomas in adult patients.23
Axi-cel is a single-dose, 1-time treatment in a patient-specific infusion bag. Axi-cel should be administered by experienced health professionals at specialized treatment centres. Each patient-specific, single-infusion bag contains a suspension of anti-CD19 CAR-positive viable T cells in approximately 60 mL for a target dose of 2 × 106 anti-CD19 CAR T cells/kg body weight (range, 1 × 106 cells/kg to 2.4 × 106 cells/kg), with a maximum of 2 × 108 anti-CD19 CAR T cells for patients weighing 100 kg and above.37
Stakeholder Perspectives
Patient Group Input
This section was prepared by CADTH staff based on the input provided by patient groups. The full original patient input(s) received by CADTH have been included in the stakeholder section at the end of this report.
One patient advocacy group, Lymphoma Canada, provided input on the treatment of adult patients with relapsed or refractory LBCL. This group gathered information from patients with DLBCL living in Canada through 2 online surveys, 1 in 2018 and another between April 26 and June 20, 2022. A total 97 patients with DLBCL completed the first survey and 23 patients with DLBCL completed the first and second survey, respectively. Patients from the second survey reported that fatigue (83%) was the most common physical symptom affecting their quality of life, followed by body aches and pains (67%), thrombocytopenia (67%), enlarged lymph nodes (58%), anemia (58%), neutropenia (58%) and night sweats (58%). Patients from both surveys reported that fear of progression/relapse was the most common psychosocial impact (67%) affecting quality of life, followed by anxiety (37%), memory loss (37%), and concentration problems (36%).
The majority of respondents (83%) were treated with CHOP with or without rituximab as their first-line treatment. When describing their experiences with their current or most recent therapy managing DLBCL symptoms, 6 out of 11 patients (55%) from the second survey agreed with the statement that it was able to manage their symptoms. While describing the side effects caused by the treatment, patients from both surveys reported that hair loss (93%) was the most common side effect, followed by fatigue (85%), neutropenia (70%), “chemo brain” or memory problems and confusion (67%), and nausea (60%). The most difficult-to-tolerate side effect was reported to be fatigue (40%). Long-term treatment side effects (lasting longer than 2 years or appearing 2 years or later after the end of treatment) were reported to be fatigue (52%) and chemo brain (42%). Patients from the first survey reported that their lymphoma treatment had the greatest negative impact on their work, travel, and other activities.
Regarding the importance of outcomes of new treatments on a scale of 1 (“not important”) to 5 (“very important”), patients from the first survey rated all of the following as important: longer survival (mean = 4.88), longer remission (mean = 4.84), better quality of life (mean = 4.64), and fewer side effects (mean = 4.12). Moreover, 51% of patients from both surveys stated that they would choose a treatment with potentially serious side effects if their doctor recommended it to be the most effective treatment option for DLBCL, whereas 46% of patients were not sure. Respondents from the first survey indicated treatment choice as important, with an average score of 4.54 out of 5. In addition, 91% patients from the second survey felt a need for more therapy options for patients with DLBCL.
Three patients from the second survey reported receiving axi-cel as third-line therapy. All 3 patients were away from home for more than 3 months while receiving the treatment, pointing to the challenges in accessing treatment currently. All 3 patients reported thrombocytopenia as a side effect, and 2 patients reported fever, anemia, nausea or vomiting, neutropenia, diarrhea, joint or muscle pain, and fatigue as side effects. Fear of progression/relapse and difficulty sleeping were reported by all 3 patients as psychosocial impacts related to their CAR T-cell therapy.
Clinician Input
Input From Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol; assisting in the critical appraisal of clinical evidence; interpreting the clinical relevance of the results; and providing guidance on the potential place in therapy). In addition, as part of the axi-cel review, a panel of 4 clinical experts from across Canada was convened to characterize unmet therapeutic needs, assist in identifying and communicating situations where there are gaps in the evidence that could be addressed through the collection of additional data, promote the early identification of potential implementation challenges, gain further insight into the clinical management of patients living with a condition, and explore the potential place in therapy of the drug (e.g., potential reimbursement conditions). A summary of this panel discussion is presented below.
Unmet Needs
The clinical experts highlighted that the most important goal of treatment for patients with LBCL is a cure. The clinical experts reported that about half of patients will be cured with front-line therapy. For patients who do not respond to front-line therapy or who relapse, intensive therapy with curative intent such as stem cell transplant is considered in second line. However, the clinical panel reported that only half of patients who relapse after front-line therapy or are refractory to front-line therapy will respond to salvage chemotherapy and proceed to transplant, per the clinical experts. The clinical experts noted that some patients do not tolerate or do not respond to salvage chemotherapy and therefore cannot continue to transplant. Others, who are responsive to salvage therapy may not continue to transplant for example those whose hematopoietic progenitor cells cannot be collected. Some patients may not be eligible for transplant, including patients with comorbidities or inadequate organ function. The clinical panel highlighted that patients with active CNS lymphoma and those who progress or relapse with CNS disease are a particularly difficult to treat population. In addition, patients with genetically high-risk disease (double-hit or triple-hit lymphoma) for whom the likelihood of responding to conventional chemotherapy is very low have limited treatment options, per the clinical experts. The clinical experts indicated that patients with relapsed or refractory LBCL who cannot tolerate or are ineligible for transplant have the largest unmet treatment need. The clinical panel noted that salvage chemotherapy and ASCT is an arduous treatment regimen and is associated with serious toxicities. The clinical panel indicated that more effective and safe treatment options are needed that are better tolerated than current conventional treatment and that can be used soon after failure of front-line therapy to improve patient outcomes.
Place in Therapy
The clinical experts noted that axi-cel is already in the current treatment paradigm as it is approved for the treatment of relapsed or refractory LBCL in third line. The clinical experts indicated that axi-cel fits well, earlier in the line of treatment and can move up to the second line and replace ASCT for most patients. The clinical experts noted that they expect patient outcomes to be better when the patients receive a potentially curative therapy earlier in the course of disease. The clinical experts noted that some patients deteriorate rapidly and thus may be less likely to survive if definitive treatment is delayed. The clinical panel thought that patients with primary refractory LBCL would benefit from CAR T-cell therapy in the second-line setting, as current salvage therapies can be ineffective and toxic in many patients, thereby preventing accessing or decreasing the efficacy of curative therapy. The clinical experts believed that a significant problem of the current treatment paradigm is that CAR T-cell therapy is only approved for third-line treatment, as this limits the number of patients with relapsed or refractory LBCL who can potentially benefit from a therapy that has demonstrated efficacy over SOC.
Patient Population
The clinical experts noted that although the ZUMA-7 trial recruited only patients who were eligible for ASCT, in clinical practice there is no clinical rationale for restricting axi-cel only to those who are candidates for ASCT. The clinical experts reported that there are patients who would be ineligible for ASCT based on fitness (i.e., comorbidities), whom the clinical experts thought should be eligible for CAR T-cell therapy. The clinical experts indicated that any patient with adequate organ function and good performance status (ECOG ≤ 2) who, based on the clinician’s judgment can tolerate the known toxicities of CAR T-cell therapy (e.g., CRS) would be suitable for axi-cel treatment. They also indicated that there are no uniformly implemented, easily reproducible or objective criteria for ASCT eligibility in Canada. Suitability for ASCT thus varies across provinces or even treatment centres depending on resources. For example, although some centres may use age as an eligibility criterion for ASCT, other centres may not exclude older patients if they have adequate organ function and performance status.
Assessing Response to Treatment
The clinical panel reported that response to treatment is typically assessed by CT scan or PET scan at 1 month and then at 3 months after initiation of therapy. The clinical panel noted that the 3-month assessment is important for assessing prognosis because patients who achieve CR at 3 months tend to have favourable outcomes, whereas those who have a PR or no response at the 3-month assessment tend to have poor long-term outcomes. The clinical experts highlighted that HRQoL is also important to assess to determine effectiveness of treatment.
Discontinuing Treatment
The clinical panel noted that in some cases patients may go through pretreatment (leukapheresis, bridging therapy, and conditioning chemotherapy), but do not receive axi-cel. These situations include lymphoma progression despite salvage and/or bridging therapy to the extent that performance status or vital organ function becomes a major concern (e.g., CNS disease, pulmonary disease), serious opportunistic infections or serious end-organ damage, and less commonly, therapy-related organ impairment (e.g., renal impairment) that limits patients’ predicted ability to tolerate CAR T-cell therapy. The clinical experts pointed out that the probability of disease progression or other complications increase with longer CAR T cell manufacturing wait times. The clinical experts noted that the 13-day manufacturing time reported in the ZUMA-7 study is rapid and may not be reproducible outside the clinical trial setting. According to the clinical experts, management of patients in such situations depends on patients’ disease-related characteristics; in most circumstances, lymphoma progression at this stage would require palliation and abandonment of axi-cel treatment. Organ toxicity could be managed, and reversed in rare circumstances, while infections could be managed for those who are amenable to treatment in the short term, as most patients are very immune deficient at this stage of the disease. The clinical experts reiterated that CAR T-cell therapy given as a second-line treatment (compared to third line or fourth line) would reduce the likelihood of patients progress or become too unwell after salvage treatment and before axi-cel infusion and are unable to complete CAR T-cell therapy.
For the 50% to 60% of patients who are expected to fail CAR T-cell therapy, subsequent treatment options currently include pola-BR (polatuzumab vedotin plus bendamustine and rituximab) or further chemotherapy, often in a palliative setting.
Prescribing Conditions
The clinical experts noted that axi-cel treatment can be provided by oncologists or hematologists in a hospital setting with adequate infrastructure for cell therapy and with access to highly specialized multidisciplinary clinical care including critical/intensive care and specialist care (e.g., neurology, nephrology) to manage toxicities, as well as laboratory support to handle and process samples.
Clinician Group Input
This section was prepared by CADTH staff based on the input provided by clinician groups. The full original clinician group input(s) received by CADTH have been included in the stakeholder section at the end of this report.
Clinician group input was received from 3 groups: Lymphoma Canada, Ontario Health (CCO) Hematology Cancer Drug Advisory Committee, and Cell Therapy Transplant Canada (CTTC).
The clinician groups agreed that, based on Canadian and international real-world evidence, there are unmet needs and challenges in the current second-line chemoimmunotherapy and ASCT treatment for patients with relapsed or refractory LBCL. The clinician groups indicated that there may be limited eligibility or tolerability to further salvage chemotherapy for some patients (e.g., patients with primary refractory disease or early relapse, and older patients). Moreover, toxicities such as febrile neutropenia, bacteremia and other infections, gastrointestinal toxicity, mucositis, need for transfusion support, and secondary malignancies associated with ASCT treatment have made it unsuitable for patients who are high risk, who are refractory to treatment or who relapse within 12 months of diagnosis.
The clinician groups also pointed out that they thought axi-cel could be expanded to specific groups of patients (e.g., older patients, patients with comorbidities, patients with ECOG PS score of 2) who may be unable to tolerate HDT and ASCT but could tolerate axi-cel. The clinician groups emphasized that CAR T-cell therapy needs to be prescribed and administered only by specialized, accredited centres for cell therapy programs, given the unique toxicities (notably CRS and immune effector cell-associated neurotoxicity syndrome [ICANS]) associated with this treatment.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
The ZUMA-7 trial comparator was platinum-chemoimmunotherapy (R-GDP, R-ICE, R-DHAP, R-ESHAP) followed by high-dose chemotherapy and then autologous stem cell transplant, which is aligned with the standard of care. | Comment from the drug programs to inform pERC deliberations. |
Considerations for initiation of therapy | |
For DLBCL arising from FL, do patients need to have a record of treatment for the diagnosis of DLBCL or is a biopsy-proven DLBCL sufficient (e.g., the patient only received treatment for FL and then transformed to DLBCL)? | The clinical experts indicated that in clinical settings, the diagnosis of transformation may be clinically driven, based on patient symptoms and signs, rather than pathologically driven. In some cases, biopsy is unavailable or risky to obtain. Therefore, a high clinical suspicion of transformation is sufficient and biopsy-proven DLBCL is not necessary to confirm transformation to DLBCL. The clinical experts indicated that, generally, once the diagnosis of transformation is made, line of therapy for the transformation (i.e., CAR T cell–eligible disease) starts at that point. However, the clinical experts noted that if a patient FL has already been given an active regimen for high-grade lymphoma (including DLBCL) that includes a rituximab-containing regimen with anthracycline (e.g., R-CHOP), especially when treatment is recent, the patients should be regarded as having failed first-line therapy and should be eligible for second-line CAR T-cell therapy. To be considered for second-line CAR T-cell therapy, the clinical experts noted that patients should have been exposed to a rituximab-containing regimen with an anthracycline, as in the ZUMA-7 trial (or etoposide if an anthracycline was unavailable), whether for DLBCL or FL. |
Can pERC clarify the definition of relapsed disease? In the ZUMA-7 trial, relapse was defined as relapse from complete remission no more than 12 months after the completion of first-line chemoimmunotherapy. | The clinical experts clarified that the definition used in the ZUMA-7 trial is reasonable and indicated this definition could be applied to eligibility criteria for axi-cel (i.e., relapse within 12 months from date of last exposure to active therapy). |
Should patients with the following be considered for axi-cel, as they were excluded from the ZUMA-7 trial?
| The clinical experts indicated the following:
|
ZUMA-7 only allowed bridging with corticosteroids. Should patients who are given other bridging therapies be considered for axi-cel? If yes, what other bridging therapies can be considered? | The clinical experts indicated that bridging therapies other than corticosteroids can be used. Any standard salvage chemotherapy regimen (e.g., R-GemOx, R-GDP, R-ICE, R-DHAP, R-ESHAP, pola-BR) could be used as bridging therapy. |
CAR T-cell therapy is funded in some jurisdictions for relapsed or refractory LBCL after 2 or more lines of systemic therapy. Is there evidence to support re-treatment with CAR T cells using either the same product, or a different product, or allogeneic CAR T? | The clinical experts reported that re-treatment is not based on robust clinical evidence and is not commonly practised in Canada. The clinical experts noted that a small cohort of patients in the ZUMA-1 study were re-treated (they needed to still be expressing CD19), and those who had a CR with the first treatment had a small chance of another CR following re-treatment with the same CAR T-cell product (i.e., axi-cel). The clinical experts indicated that the likelihood of patients benefiting from re-treatment with the same CAR T-cell product is low. Regarding allogeneic CAR T cell, the clinical experts noted that if a patient has relapsed post-allogenic stem cell transplant, they may still successfully undergo leukapheresis and CAR T-cell therapy using the patient’s T cells, even if donor derived. However, this situation will be rare as allo-SCT is no longer commonly used for DLBCL. Off-the-shelf allogeneic CARs (not autologous, created from donor cells) are still in clinical trials and are not ready for clinical consideration until further data on efficacy and safety are available, per the clinical experts. |
Considerations for prescribing of therapy | |
Axi-cel is a single-dose, 1-time treatment infused at a target dose of 2 × 106 CAR T cells/kg body weight. | Comment from the drug programs to inform pERC deliberations. |
Delivery must take place at specialized treatment centres that are accredited and certified by the manufacturer. There continues to be limited access to CAR T-cell services in Canada. Although access is expanding, interprovincial travel or out-of-country funding remains necessary in many parts of Canada. Due to geographical site limitations, patients may need to travel for treatment, requiring interprovincial agreements to ensure equitable access. | Comment from the drug programs to inform pERC deliberations. |
Special implementation issues | |
Should patients who recently started second-line platinum-chemoimmunotherapy be allowed to switch to CAR T-cell therapy provided all other criteria are met? | Depending on where the patient is in the course of treatment (e.g., completed salvage chemotherapy and a plan is in place for transplant), the clinical experts indicated they should be allowed to switch to CAR T-cell therapy. The decision of whether to switch should be left to the treating physician and the patient. |
Hospitalization for adverse events does occur and may include ICU admission. CRS is sometimes managed with tocilizumab. In the event of a tocilizumab shortage, is there another treatment that can be used to manage CRS? | The clinical experts reported that other treatments may be used to manage CRS. These include siltuximab, a next-generation IL-6 inhibitor, and steroids, if an IL-6 inhibitor is unavailable. |
Feasibility of adoption must be addressed. Given the anticipated patient volumes, PAG is concerned that existing capacity may not be able to meet demand. | Comment from the drug programs to inform pERC deliberations. |
Accessing CAR T-cell therapy may require interprovincial travel. A program to cover travel and accommodation expenses should be offered by the manufacturer until access across Canada is widespread. | Comment from the drug programs to inform pERC deliberations. |
There are patient privacy and patient cell ownership concerns because CAR T cells are manufactured by a US-based company outside of Canadian jurisdiction (this is also the case for the other CAR T-cell therapies that are publicly funded). | Comment from the drug programs to inform pERC deliberations. |
allo = allogeneic; ASCT = autologous stem cell transplant; axi-cel = axicabtagene ciloleucel; CAR = chimeric antigen receptor; CLL = chronic lymphocyte leukemia; CNS = central nervous system; CR = complete response; CRS = cytokine release syndrome; DLBCL = diffuse large B-cell lymphoma; ECOG = Eastern Cooperative Oncology Group; FL = follicular lymphoma; ICU = intensive care unit; IL = interleukin; LBCL = large B-cell lymphoma; PAG = provincial advisory group; pERC = CADTH pan-Canadian Oncology Drug Review Expert Review Committee; PMBCL = primary mediastinal B-cell lymphoma; R-CHOP = rituximab, cyclophosphamide, doxorubicin hydrochloride, vincristine, and prednisone; R-DHAP = rituximab, dexamethasone, cytarabine, and cisplatin; R-ESHAP = rituximab, etoposide, solu-medrone, high-dose cytarabine, and cisplatin; R-GDP = rituximab, gemcitabine, dexamethasone, and cisplatin; R-GemOx = rituximab, gemcitabine, and oxaliplatin; R-ICE = rituximab, ifosfamide, carboplatin, and etoposide; pola-BR = polatuzumab vedotin, bendamustine, and rituximab.
Clinical Evidence
The clinical evidence included in the review of axi-cel (Yescarta) is presented in 3 sections. The first section, the systematic review, includes pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those studies that were selected according to an a priori protocol. The second section includes indirect evidence from the sponsor (if submitted) and indirect evidence selected from the literature that met the selection criteria specified in the review. The third section includes sponsor-submitted long-term extension studies and additional relevant studies that were considered to address important gaps in the evidence included in the systematic review.
Systematic Review (Pivotal and Protocol-Selected Studies)
Objectives
To perform a systematic review of the efficacy and safety of axi-cel (IV infusion, target dose of 2 × 106 anti-CD19 CAR T cells/kg body weight) for the treatment of adult patients with relapsed or refractory LBCL.
Methods
Studies selected for inclusion in the systematic review included pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those meeting the selection criteria presented in Table 5. Outcomes included in the CADTH review protocol reflect outcomes considered to be important to patients, clinicians, and drug plans. Of note, the systematic review protocol presented below was established before the granting of a Notice of Compliance from Health Canada.
Table 5: Inclusion Criteria for the Systematic Review
Criteria | Description |
|---|---|
Population | Adult patients with relapsed or refractory large B-cell lymphoma Subgroups of interest:
|
Intervention | Axicabtagene ciloleucel as a single-dose, 1-time treatment (IV infusion containing a suspension of anti-CD19 CAR-positive viable T cells in approximately 60 mL for a target dose of 2 × 106 anti-CD19 CAR T cells/kg body weight [recommended dose range, 1 × 106 cells/kg to 2.4 × 106 cells/kg], with a maximum of 2 × 108 anti-CD19 CAR T cells for patients weighing 100 kg or more) |
Comparator | 1. Platinum-containing salvage chemoimmunotherapy:
2. Followed by high-dose therapy:
3. Followed by ASCT |
Outcomes | Efficacy and patient-reported outcomes:
Harms outcomes:
Notable harms:
|
Study designs | Published and unpublished phase III and IV randomized controlled trials |
ASCT = autologous stem cell transplant; CAR = chimeric antigen receptor; ECOG PS = Eastern Cooperative Oncology Group Performance Status; HRQoL = health-related quality of life; ICU = intensive care unit.
The literature search for clinical studies was performed by an information specialist using a peer-reviewed search strategy according to the PRESS Peer Review of Electronic Search Strategies checklist.40
Published literature was identified by searching the following bibliographic databases: MEDLINE All (1946—) via Ovid and Embase (1974—) via Ovid. All Ovid searches were run simultaneously as a multifile search. Duplicates were removed using Ovid deduplication for multifile searches, followed by manual deduplication in Endnote. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concept was Yescarta (axicabtagene ciloleucel). The following clinical trials registries were searched: the US National Institutes of Health’s clinicaltrials.gov, the WHO’s International Clinical Trials Registry Platform (ICTRP) search portal, Health Canada’s Clinical Trials Database, and the European Union Clinical Trials Register.
No filters were applied to limit the retrieval by study type. Retrieval was not limited by publication date or by language. Conference abstracts were excluded from the search results. Refer to Appendix 1 for the detailed search strategies.
The initial search was completed on July 7, 2022. Regular alerts updated the search until the meeting of the CADTH pan-Canadian Oncology Drug Review Expert Review Committee (pERC) on November 9, 2022.
Grey literature (literature that is not commercially published) was identified by searching relevant websites from the Grey Matters: A Practical Tool for Searching Health-Related Grey Literature checklist.41 Included in this search were the websites of regulatory agencies (US FDA and the European Medicines Agency). Google was used to search for additional internet-based materials. Refer to Appendix 1 for more information on the grey literature search strategy.
These searches were supplemented by reviewing bibliographies of key papers and through contacts with appropriate experts. In addition, the manufacturer of the drug was contacted for information about unpublished studies.
Two CADTH clinical reviewers independently selected studies for inclusion in the review based on titles and abstracts, according to the predetermined protocol. Full-text articles of all citations considered potentially relevant by at least 1 reviewer were acquired. Reviewers independently made the final selection of studies to be included in the review, and differences were resolved through discussion.
Findings From the Literature
A total of 484 records were identified via the searches (Figure 1). Of these, 483 records were excluded; 1 study was retained. An additional 2 reports provided by the sponsor were included, as was an FDA review report. In total, 4 reports presenting data from a single study, ZUMA-7, were included in this review.14,15,42 Details of the ZUMA-7 study are summarized in Table 6.
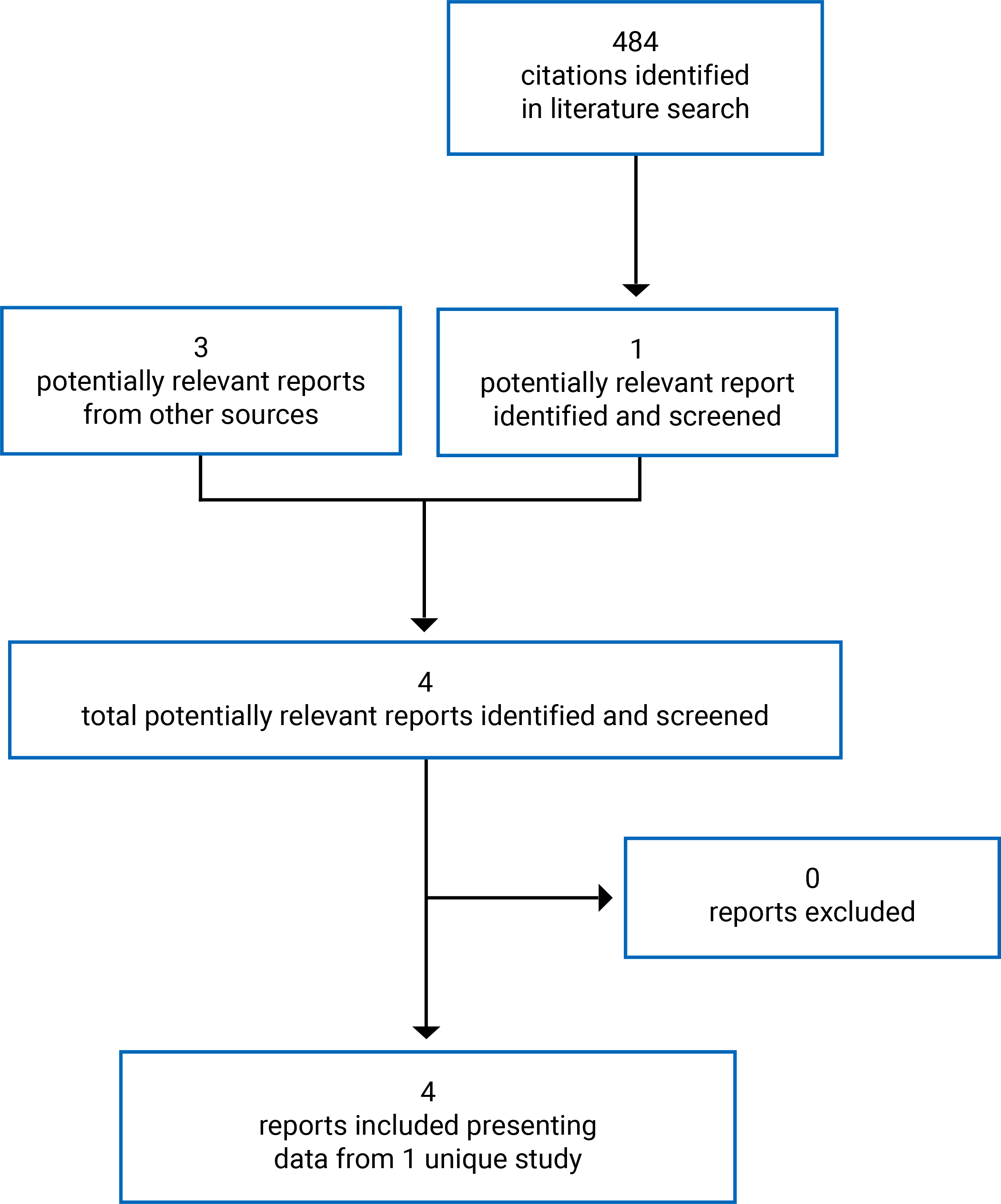
Table 6: Details of the ZUMA-7 Study
Study details | Description |
|---|---|
Designs and populations | |
Study design | Phase III, multicentre, open-label, randomized controlled trial |
Locations | 77 sites in 14 countries (Australia, Austria, Belgium, Canada [8 centres, 20 patients], France, Germany, Israel, Italy, Netherlands, Spain, Sweden, Switzerland, UK,US) |
Study duration | January 25, 2018, to ongoing |
Data cut-off date | March 18, 2021 |
No. of patients randomized (randomization ratio) | 359 (1:1) |
Main inclusion criteria |
|
Main exclusion criteria |
|
Drugs | |
Intervention | Axi-cel administered after a 3-day lymphodepleting chemotherapy regimen consisting of fludarabine 30 mg/m2/day and cyclophosphamide 500 mg/m2/day, followed by 2 rest days. A single infusion of axi-cel was administered intravenously at a target dose of 2 × 106 anti-CD19 CAR T cells/kg (minimum dose of 1 × 106 anti-CD19 CAR T cells/kg; for patients weighing ≥ 100 kg, the maximum flat dose was 2 × 108 anti-CD19 CAR T cells). |
Comparator | Protocol-defined salvage chemotherapy regimens: R-ICE, R-DHAP/R-DHAX, R-ESHAP, or R-GDP as selected by the treating investigator, administered every 2 to 3 weeks for 2 to 3 cycles. Patients responding to salvage chemotherapy after 2 or 3 cycles were to proceed with HDT-ASCT per institutional or regional standards. Patients not responding to salvage chemotherapy could receive additional treatment off-protocol. |
Duration | |
Phases | |
Screening | Within 14 days before randomization |
Open label | Axi-cel arm: lymphodepleting chemotherapy and axi-cel SOC arm: salvage chemotherapy, HDT, TBI (sometimes given as part of HDT), and ASCT |
Follow-up | Axi-cel arm: Up to 15 years SOC arm: Up to 5 years |
Outcomes | |
Primary end point | EFS per blinded central assessment |
Secondary and other end points | Key secondary end points
Other secondary end points
Exploratory end points
|
Safety end points |
|
Notes | |
Publications | Locke et al. (2022)14 |
ABC = activated B cell; AE = adverse event; ASCT = autologous stem cell transplant; axi-cel = axicabtagene ciloleucel; CAR = chimeric antigen receptor; CR = complete response; CRS = cytokine release syndrome; CNS = central nervous system; DLBCL = diffuse large B-cell lymphoma; DOR = duration of response; ECOG = Eastern Cooperative Oncology Group; EFS = event-free survival; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EQ-5D-5L = 5-Level EQ-5D; GCB = germinal centre B cell; HDT = high-dose therapy; HGBL = high-grade B-cell lymphoma; HRQoL = health-related quality of life; LBCL = large B-cell lymphoma; ORR = objective response rate; OS = overall survival; PCR = polymerase chain reaction; PD = progressive disease; PFS = progression-free survival; PR = partial response; R-DHAP/X = rituximab plus dexamethasone, high-dose cytarabine, and cisplatin/oxaliplatin; R-ESHAP = rituximab plus etoposide, methylprednisolone, cytarabine, and cisplatin; R-GDP = rituximab plus gemcitabine, dexamethasone, and cisplatin/carboplatin; R-ICE = rituximab plus ifosfamide, carboplatin, and etoposide; SD = stable disease; SOC = standard of care; TBI = total body irradiation; VAS = visual analogue scale; WDAE = withdrawal due to adverse event.
Description of the ZUMA-7 Trial
ZUMA-7 is a phase III, multicentre, randomized, open-label trial evaluating the efficacy of axi-cel compared with SOC (salvage chemoimmunotherapy followed by HDT-ASCT) as a second-line therapy in patients with relapsed or refractory LBCL after first-line rituximab and anthracycline-based chemotherapy (Figure 2). The trial was conducted in 14 countries; 20 patients were recruited in Canada. The first patient was enrolled (randomized) on January 25, 2018, and enrolment was completed on October 4, 2019. The ZUMA-7 is ongoing at the time of this review. The data cut-off date for the primary analysis was March 18, 2021.
For patients in the axi-cel arm, treatment consisted of lymphodepleting chemotherapy followed by a single IV infusion of axi-cel. Bridging therapy of corticosteroids was allowed before lymphodepleting chemotherapy for patients with high disease burden, at the discretion of the investigator. For patients in the SOC arm, treatment consisted of a single protocol-defined, platinum-based salvage chemotherapy regimen as selected by the treating investigator. Patients who responded to salvage chemotherapy were to proceed to HDT with or without total body irradiation (TBI), followed by ASCT.
Randomization and treatment allocation: After all screening activities were completed, an Interactive Voice/Web (x) Response System (IxRS) was used to randomly assign patients in a 1:1 ratio to receive axi-cel or SOC. Randomization was stratified by response to first-line therapy (primary refractory, relapse ≤ 6 months of first-line therapy, or relapse > 6 and ≤ 12 months of first-line therapy) and second-line age-adjusted International Prognostic Index (sAAIPI) (low: 0 to 1; or high: 2 to 3), as assessed at the time of screening.
Blinding: This was an open-label trial; patients were aware of their assigned treatment group and all staff at each investigative site involved in treating and caring for study patients had full knowledge of the patient’s treatment assignment. An open-label design was necessary due to the difference in the nature of treatment in the 2 arms. Radiologic evaluations of response (i.e., for EFS, PFS, ORR, and DOR) were performed both by an unblinded investigator and by an independent central reviewer who was unaware of the patient’s treatment arm.
Study phases: The screening phase started on the date the patient signed the informed consent form and continued through confirmation of eligibility and randomization into the study. Screening procedures were completed within 14 days before enrolment (i.e., randomization). Informed consent was obtained before completion of any study-specific procedures that were not SOC. To establish a baseline and to confirm eligibility, screening fluorodeoxyglucose (FDG)-PET from skull base to mid-thighs, diagnostic quality contrast-enhanced (unless contraindicated) CT from skull base through lesser trochanters (PET-CT), and the appropriate imaging of all other sites of disease were required to be performed within 28 days before randomization and as close to randomization as possible. Patients who failed to meet the eligibility criteria were allowed to rescreen once. The assessment that initially resulted in the failed screening was repeated, including any other procedures that fell outside of the designated screening window (i.e., laboratory assessments or PET-CT scans).
The treatment phase started with the first dose of treatment following randomization. In the axi-cel arm, treatment day 0 was defined as the day the patient received the first axi-cel infusion. End of study will occur when all patients in the axi-cel arm have been followed up for 15 years in the axi-cel arm and 5 years in the SOC arm after randomization, have withdrawn consent, have been lost to follow-up, or have died.
Protocol Amendments
The original protocol, dated May 22, 2017, was amended 6 times in the US; for all other regions, the protocol was amended 5 times. No patients were treated until the second amendment. Important protocol amendments included the following: the primary EFS analysis event trigger was modified from 270 to approximately 250 EFS events, with an acceptable lower limit for the observed total EFS events of 225, to maintain the power for the primary analysis to within 5% of the targeted 90%; the duration of follow-up for the primary analysis of EFS was increased from the study day 150 assessment to the month 9 assessment; and a second interim OS analysis and a sensitivity analysis of OS were added. The second interim analysis of OS was to occur when approximately 160 deaths were observed or no later than 4 years after the first patient was randomized. The sensitivity analyses were added to address the confounding effect from treatment switching (date of analysis June 25, 2020).
Some changes were made to the planned analyses after the finalization of the Statistical Analysis Plan. The primary EFS analysis had been planned to occur after 250 EFS events were observed in the study. Because the time to reach 250 EFS events was longer than estimated, the first interim OS analysis was conducted at 153 events instead of the planned 110 events. As a result, the interim OS analysis conducted at 153 events meets the criteria for both originally planned interim OS analyses at 110 and 160 events. The only subsequent planned OS analysis will be the primary (final) OS analysis, expected to occur when 210 events are observed or no later than 5 years after the first patient was randomized. Other minor changes included modifications within some categories of baseline characteristics and subgroup covariates and clarification to definitions and reporting of some end points.
Figure 2: ZUMA-7 Study Treatment Schema

Auto-SCT = autologous stem cell transplant; CAR = chimeric antigen receptor; HDT = high-dose therapy; R-ESHAP = rituximab+etoposide-methylprednisolone-cytarabine-cisplatin; R-GDP = rituximab+gemcitabine-dexamethasone-cisplatin/carboplatin; R-DHAP = rituximab+dexamethasone-cytarabine-cisplatin; R-ICE = rituximab+ifosfamide-carboplatin-etoposide; SOC = standard of care; SOCT = standard of care therapy.
Notes: Study day = number of days from the day of randomization; treatment day = number of days from the day of axicabtagene ciloleucel treatment.
a At the discretion of the investigator, corticosteroid bridging therapy could have been considered for patients with high disease burden at screening.
b Minimum observation period: 7 days unless otherwise required by country regulatory agencies (e.g., 10 days for patients treated in Germany, Switzerland, and France).
c Disease assessments were to be calculated from the date of randomization and not the date of dosing with axicabtagene ciloleucel or SOCT. Independent of the treatment arm, study procedures and disease assessments were to occur at the same protocol-defined time points.
Source: Clinical Study Report for Yescarta.15
Populations
Inclusion and Exclusion Criteria
Eligible patients were aged at least 18 years and had histologically confirmed LBCL, according to the WHO 2016 classification criteria,27 that was refractory to first-line treatment or that had relapsed from complete remission. Refractory disease was defined as a lack of CR to first-line therapy and relapsed disease was defined as biopsy-proven disease relapse occurring no more than 12 months after the completion of first-line chemoimmunotherapy. Patients must have received adequate first-line therapy, including at a minimum an anti-CD20 monoclonal antibody (unless their tumour was CD20 negative) and an anthracycline-containing chemotherapy regimen, and must have intended to proceed to HDT-ASCT if there was a response to salvage chemotherapy. Patients with a history of malignancy other than nonmelanoma skin cancer or carcinoma in situ (unless disease-free for at least 3 years), those with a history of Richter transformation of chronic lymphocytic leukemia or primary mediastinal B-cell lymphoma (PMBCL) were ineligible. Patients with a history of autologous or allogeneic stem cell transplant, or those who received prior CD19 targeted therapy, CAR T-cell therapy or other genetically modified T-cell therapy, or systemic immunostimulatory agents within 6 weeks or 5 half-lives of the drug before the first dose of the study drug were also excluded (Table 6).
Baseline Characteristics
The mean age of patients was 57 years (SD = 12); 30% of the patients were aged 65 years or older. The majority of patients were male (66%) and White (83%). Most patients were randomized in North America (75%), of whom the majority were in the US (70%). Approximately half of the patients had an sAAIPI score of 0 or 1 (52% and 56% in the axi-cel and SOC arms, respectively). Overall, 74% of the study population had primary refractory disease and 26% had early relapse. Based on central laboratory assessment, patients in the axi-cel and SOC arms were categorized as DLBCL NOS/without further classification possible (70% and 67%, respectively); HGBL with MYC, BCL2, and/or BCL6 rearrangements (17% and 14%, respectively); or HGBL, NOS, (1% in the SOC arm). The remaining patients were categorized under not confirmed, missing, or other. Approximately one-quarter of patients in both treatment arms had achieved a best response of CR to first-line treatment (Table 7). Overall, baseline characteristics were generally comparable between the 2 treatment arms, but a difference of at least 10 percentage points was observed between axi-cel and SOC arms for sex (61% versus 71% male, respectively) and extranodal disease (57% versus 67%, respectively).
Table 7: Summary of Baseline Characteristics — ZUMA-7 Study
Characteristic | Axi-cel (N = 180) | SOC (N = 179) |
|---|---|---|
Age (years), mean (SD) | 57.1 (12.0) | 57.4 (12.2) |
Sex | ||
Male n (%) | 110 (61) | 127 (71) |
Female n (%) | 70 (39) | 52 (29) |
Region, n (%) | ||
North America | 140 (78) | 130 (73) |
Europe | 34 (19) | 45 (25) |
Israel | 4 (2) | 2 (1) |
Australia | 2 (1) | 2 (1) |
Race and ethnicity, n (%) | ||
American Indian or Alaska Native | 0 (0) | 1 (1) |
Asian | 12 (7) | 10 (6) |
Black or African American | 11 (6) | 7 (4) |
Native Hawaiian or Other Pacific Islander | 2 (1) | 1 (1) |
White | 145 (81) | 152 (85) |
Other | 10 (6) | 8 (4) |
ECOG performance status, n (%) | ||
0 | 95 (53) | 100 (56) |
1 | 85 (47) | 79 (44) |
Disease stage, n (%) | ||
I | 10 (6) | 6 (3) |
II | 31 (17) | 27 (15) |
III | 35 (19) | 33 (18) |
IV | 104 (58) | 113 (63) |
Disease type per central laboratory, n (%) | ||
Diffuse large B-cell lymphomaa | 126 (70) | 120 (67) |
High-grade B-cell lymphoma, not otherwise specified | 0 (0) | 1 (1) |
High-grade B-cell lymphoma, including rearrangement of MYC with BCL or BCL6 or both | 31 (17) | 25 (14) |
Not confirmed or missing | 18 (10) | 28 (16) |
Other | 5 (3) | 5 (3) |
Molecular subgroup per central laboratory, n (%) | ||
Germinal centre B cell–like | 109 (61) | 99 (55) |
Activated B cell–like | 16 (9) | 9 (5) |
Unclassified | 17 (9) | 14 (8) |
Not applicable | 10 (6) | 16 (9) |
Missing | 28 (16) | 41 (23) |
CD19 IHC-positive per central laboratory at baseline, n (%)b | ||
Yes | 144 (80) | 134 (75) |
No | 13 (7) | 12 (7) |
Missingc | 23 (13) | 33 (18) |
CD19 H-Score, n (%) | ||
≤ 150 | 85 (47) | 67 (37) |
> 150 | 72 (40) | 79 (44) |
Missing | 23 (13) | 33 (18) |
Presence of B symptoms, n (%) | 21 (12) | 29 (16) |
Splenic involvement), n (%) | 19 (11) | 33 (18) |
Extranodal disease, n (%) | 103 (57) | 120 (67) |
Bulky disease, n (%) | 13 (7) | 16 (9) |
Bone marrow involvement, n (%)d | 17 (9) | 15 (8) |
Elevated lactate dehydrogenase level, n (%)e | 101 (56) | 94 (53) |
Tumour burden (mm2), median (range)f | 2,123 (181 to 22,538) | 2,069 (252 to 20,117) |
Prognostic marker per central laboratory, n (%) | ||
High-grade B-cell lymphoma, double hit | 25 (14) | 15 (8) |
High-grade B-cell lymphoma, triple hit | 6 (3) | 10 (6) |
Double expressor lymphoma | 57 (32) | 62 (35) |
MYC rearrangement | 15 (8) | 7 (4) |
Not applicable | 74 (41) | 70 (39) |
Missing | 3 (2) | 15 (8) |
Best response to first-line therapy, n (%) | ||
Complete response | 46 (26) | 47 (26) |
Partial response | 60 (33) | 62 (35) |
Stable disease | 11 (6) | 11 (6) |
Progressive disease | 63 (35) | 59 (33) |
Response to first-line therapy at randomization, n (%) | ||
Primary refractory | 133 (74) | 132 (74) |
Relapse ≤ 6 months of first-line therapy | 26 (14) | 22 (12) |
Relapse > 6 and ≤ 12 months of first-line therapy | 20 (11) | 24 (13) |
Missing | 1 (1) | 1 (1) |
Second-line age-adjusted International Prognostic Index total score, n (%) | ||
0 or 1 | 94 (52) | 100 (56) |
2 or 3 | 86 (48) | 79 (44) |
axi-cel = axicabtagene ciloleucel; ECOG = Eastern Cooperative Oncology Group; IHC = immunohistochemistry; SD = standard deviation; SOC = standard of care.
aThe definition of diffuse large B-cell lymphoma per central laboratory assessment included cases of incomplete evaluation due to inadequate sample amount or type for which further classification of the subtype was not possible.
bCD19 IHC-positive is defined as the H-score of staining ≥ 5.
cMissing CD19 H-scores are mainly due to quantity not sufficient, biopsy not available at central laboratory, CD19 negative, or tumour tissue not present in sample.
dBone marrow involvement is collected on the diagnosis history case report form.
eAn elevated lactate dehydrogenase level was defined as a level that was above the upper limit of the normal range according to the local laboratory.
fTumour burden was determined on the basis of the sum of product diameters of the target lesions, according to the Cheson criteria, and was assessed by the central laboratory.
Sources: Clinical Study Report for Yescarta15 and Locke et al. (2022).14
Interventions
Axi-Cel Arm
Bridging Therapy
Administration of bridging therapy was optional. Bridging therapy consisted of corticosteroids (e.g., dexamethasone at a dose of 20 mg to 40 mg or equivalent, either orally [PO] or IV daily for 1 to 4 days) administered after leukapheresis through 5 days before administration of axi-cel. Bridging therapy was sourced by the investigative site. Choice of corticosteroid and dosing could be adjusted for age, comorbidities, or per clinical judgment.
Lymphodepleting Chemotherapy
Lymphodepleting chemotherapy (conditioning chemotherapy) consisted of fludarabine and cyclophosphamide used for lymphodepletion before administration of axi-cel. Five days before the planned axi-cel infusion date, patients were treated with lymphodepleting chemotherapy. The 3-day lymphodepleting chemotherapy regimen consisted of fludarabine 30 mg/m2 per day and cyclophosphamide 500 mg/m2 per day on treatment days −5 to −3 followed by 2 rest days before axi-cel infusion on treatment day 0.
Axi-Cel
Any screening assessments or procedures that were repeated between confirmation of eligibility and the start of axi-cel infusion that had results outside the eligibility criteria were resolved before proceeding with the infusion (except for peripheral blood cell counts that had been impacted by lymphodepleting chemotherapy). The patient must not have shown any evidence or suspicion of an infection. If the axi-cel infusion was delayed for more than 2 weeks, protocol guidelines regarding the need for repeat lymphodepleting chemotherapy were to be followed.
Approximately 1 hour before the axi-cel infusion, the preinfusion medications acetaminophen (650 mg PO or equivalent) and diphenhydramine (12.5 mg PO or IV or equivalent) were administered. Axi-cel was administered intravenously as a single infusion of CAR-transduced autologous T cells. Axi-cel was supplied cryopreserved in cryostorage bags and the product was required to remain frozen until the patient was ready for treatment to assure viable live autologous T cells were administered. Axi-cel was administered at a target dose of 2 × 106 anti-CD19 CAR T cells/kg body weight but may have been dosed at a minimum of 1 × 106 anti-CD19 CAR T cells/kg. For patients weighing more than 100 kg, a maximum flat dose of axi-cel at 2 × 108 anti-CD19 CAR T cells was administered.
All patients received axi-cel infusion at a health care facility, followed by daily monitoring at a health care facility for at least 7 days to monitor for signs and symptoms of CRS and neurologic events. Alternatively, if deemed appropriate by the investigator, patients may have been hospitalized to receive their infusion and were observed for CRS and neurologic events in the hospital setting.
Re-Treatment
Axi-cel re-treatment was allowed based on experience from the ZUMA-1 study, in which 6 of 10 patients who were re-treated subsequently achieved a PR or CR.43 Patients in ZUMA-7 who achieved a PR or CR at the study day 50 disease assessment and subsequently experienced disease progression had the option to receive a second course of lymphodepleting chemotherapy and axi-cel if they met the following criteria: their disease was not known to be CD19 negative; they continued to meet the study eligibility criteria; they had no life-threatening toxicity related to axi-cel during the original course of treatment and any toxicities related to lymphodepleting chemotherapy (except for alopecia) had resolved to grade 1 or lower or baseline before re-treatment; they had not received subsequent therapy for lymphoma; and they did not have known axi-cel neutralizing antibodies. A maximum of 1 re-treatment per patient was permitted. Sites were strongly encouraged to collect and submit a biopsy to the central laboratory before initiating re-treatment for confirmation of disease progression and CD19 expression. The second dose of axi-cel could have been manufactured at the same time as the first axi-cel dose with existing cryopreserved peripheral blood mononuclear cells (PBMCs), or the patient could have undergone a second leukapheresis to collect T cells for manufacture of the second dose of axi-cel.
SOC Arm
Salvage Chemotherapy
Patients randomized to the SOC arm received a salvage chemotherapy regimen, and those who responded were to proceed to HDT-ASCT. The SOC treatment was sourced by and administered at the investigative site in a setting with emergency medical facilities. Multiple salvage chemotherapy regimens were allowed; dosing was recommended but not required due to regional and institutional differences. It was anticipated that patients who did not respond to salvage chemotherapy would receive subsequent, off-protocol treatment that could include commercially available CAR T-cell therapies.
Salvage chemotherapy consisted of 1 of the following platinum-based second-line combination chemotherapy regimens and recommended dosing:
R-ICE
rituximab: 375 mg/m2 before chemotherapy
ifosfamide: 5 g/m2 24-hour continuous infusion on cycle day 2 with mesna
carboplatin: area under the curve (AUC) 5 on cycle day 2, maximum dose 800 mg
etoposide: 100 mg/m2 per day on cycle days 1 to 3
R-ESHAP:
rituximab: 375 mg/m2 on cycle day 1
etoposide: 40 mg/m2/day IV on cycle days 1 to 4
methylprednisolone: 500 mg/day IV, on cycle days 1 to 4 or 5
cytarabine: 2 g/m2 on cycle day 5
cisplatin: 25 mg/m2/day continuous infusion, on cycle days 1 to 4
R-GDP:
rituximab: 375 mg/m2 on cycle day 1 (or day 8)
gemcitabine: 1 g/m2 on cycle days 1 and 8
dexamethasone: 40 mg on cycle days 1 to 4
cisplatin: 75 mg/m2 on cycle day 1 (or carboplatin, AUC = 5)
R-DHAP or R-DHAX:
rituximab: 375 mg/m2 before chemotherapy
dexamethasone: 40 mg/d on cycle days 1 to 4
high-dose cytarabine: 2 g/m2 every 12 hours for 2 doses on cycle day 2 following platinum treatment
cisplatin: 100 mg/m2 24-hour continuous infusion on cycle day 1 (or oxaliplatin 100 mg/m2)
Salvage chemotherapy was recommended to start within approximately 5 days after randomization. Patients received 2 or 3 cycles of a single permitted salvage chemotherapy regimen, with 1 cycle administered every 2 to 3 weeks. After salvage chemotherapy was administered, the patient received the same chemotherapy regimen for subsequent cycles. Dose modifications due to toxicity were allowed, but a change of regimen was noted as a new lymphoma therapy.
HDT and ASCT
If a patient was deemed transplant eligible (i.e., demonstrated adequate disease response with CR or PR after 2 or 3 cycles of salvage chemotherapy and collected a sufficient number of CD34+ stem cells), HDT and ASCT could be initiated. Given the lack of a standard high-dose regimen, HDT and ASCT procedures were performed according to regional and institutional standards. Before HDT, granulocyte colony-stimulating factor (G-CSF) was administered to mobilize stem cells from the bone marrow to the periphery, after which peripheral blood progenitor cells were collected by leukapheresis to a minimum target of 2 × 106 CD34+ hematopoietic stem cells/kg body weight.
The HDT conditioning regimen consisted of combination high-dose chemotherapy with or without TBI. Commonly used high-dose regimens include BEAM (carmustine, etoposide, ara-C, and melphalan) or cyclophosphamide-BCNU-etoposide (CBV). After HDT, the CD34+ hematopoietic stems cells were reinfused to rescue hematopoiesis.
Discontinuation
Patients could be removed from protocol-required investigational treatments or procedures due to AEs, disease progression, patient request or noncompliance, unavailability of product, loss to follow-up, death, or decision by sponsor.
Subsequent Therapy
Subsequent therapy that was administered after the protocol-specified study treatment (axi-cel or SOC) and was necessary to treat a patient’s PD, such as chemotherapy not specified in the study, immunotherapy, targeted agents, as well as allo-SCT or ASCT and radiation therapy, were to be recorded for all patients randomized until 1 of the following occurred: the patient completed the long-term follow-up period, was considered lost to follow-up, withdrew full consent, or died.
Prior and Concomitant Therapy
Investigators were allowed to prescribe any concomitant medications or treatment deemed necessary to provide adequate supportive care, including growth factor support (e.g., G-CSF) and routine antiemetic prophylaxis, with the following exceptions:
After treatment with axi-cel or SOC, lymphoma treatment such as chemotherapy, immunotherapy, targeted agents, radiation (TBI for HDT was allowed for the SOC arm), high-dose corticosteroids (other than those allowed in the protocol for either arm), and other investigational agents were prohibited, except as needed for the treatment of PD.
For patients in the axi-cel arm, corticosteroid therapy at a pharmacologic dose (≥ 5 mg/day of prednisone or equivalent doses of other corticosteroids) and other immunosuppressive drugs were to be avoided for 7 days before leukapheresis and 5 days before axi-cel administration.
Patients for whom CT scans with contrast are contraindicated (i.e., patients with contrast allergy or impaired renal clearance) were to undergo an MRI with contrast and noncontrast CT scans instead of premedicating with systemic corticosteroids.
For 3 months after axi-cel administration, corticosteroids and other immunosuppressive drugs were to be avoided unless used to manage axi-cel-related toxicities. Other medications that might have interfered with the evaluation of axi-cel, such as nonsteroidal anti-inflammatory agents, were also to be avoided for the same period unless medically necessary.
Outcomes
The efficacy end points identified in the CADTH review protocol that were assessed in the clinical trial included in this review are summarized below.
Efficacy
The primary end point was EFS according to blinded central assessment. The key secondary efficacy end points were ORR according to blinded central assessment, and OS. Other secondary end points included PFS based on investigator assessment, DOR according to blinded central assessment, and HRQoL. Time to next treatment was an exploratory end point. Health resource utilization was not measured as an efficacy end point but was reported supplementary to the assessment of safety.
Throughout the study, patients in both treatment groups were assessed for response and progression at the same times relative to randomization (study day 0), including study days 50, 100, and 150, month 9, and every 3 months thereafter until month 24, and then every 6 months from months 30 to 60. Patients were to have their first posttreatment planned PET-CT tumour assessment within the study day 50 assessment period (range, −7 to + 21 days, from study day 50). The patient’s disease was to be assessed at regular intervals through month 9 after randomization or until a change in lymphoma therapy or disease progression, whichever came first. If the patient’s disease had not progressed by month 9, disease assessments were to be evaluated by CT scans where a CR was suspected and by PET-CT scans where a PR was suspected. Patients with symptoms suggestive of disease progression were to be evaluated for progression at the time symptoms occurred. PET-CT scan could be performed at any time disease progression was suspected.
The fluorodeoxyglucose (FDG)-PET assessment was to take precedence over the CT assessment for the time points when both were available. In addition to the investigator’s assessment, PET-CT scans of all patients evaluated for disease response were to be submitted to and reviewed by an independent central reviewer who was blinded to the patient’s treatment arm. If PD was reported by the investigator in the absence of radiologic evidence, objective evidence of clinical progression (e.g., tumour tissue or bone marrow biopsy results, physical examination findings, lactate dehydrogenase elevation, weight loss, or fever) were to be submitted to and reviewed by an independent central reviewer. For patients who discontinued protocol therapy due to an assessment of PD but for whom there was no change in lymphoma therapy, any additional imaging data subsequent to the image in question were to be submitted to the central reviewer to confirm disease response. If the patient was eligible for re-treatment with axi-cel, the last scan before re-treatment was to be considered the baseline for the purpose of evaluating the response to re-treatment.
The primary end point of EFS was defined as the time from randomization to the earliest date of disease progression per the Lugano Classification,44 as determined by blinded central assessment; commencement of new lymphoma therapy (including stem cell transfer in the axi-cel arm without axi-cel–induced response or re-treatment of axi-cel); or death from any cause. Patients with established PR, CR, or stable disease who subsequently began a new lymphoma therapy in the absence of documented PD were to have an EFS event time defined as the time from randomization to the last evaluable disease assessment (or last time stable disease was established) before commencing the new therapy. For patients who started a new lymphoma therapy in the absence of an evaluable disease assessment, the EFS date was imputed as the randomization date. Patients with a best response of stable disease up to and including study day 150 were considered to have an EFS event, defined as the time from randomization to the time of the first establishment of stable disease.
The following criteria were used to define censoring times for EFS: (1) patients alive, in response, and with no new therapy were censored at the last evaluable disease assessment; (2) patients with no evaluable disease assessment by study day 150 were not considered to have an EFS event and event time was censored at the time of randomization; (3) for patients in the axi-cel arm who underwent ASCT without documented progression or new lymphoma therapy were censored at the time of ASCT; (4) for patients in the SOC arm, TBI, HDT, and ASCT that occurred while in response were not considered an EFS event; (5) at the time of the interim EFS analysis, patients who had not been followed for 150 days and who did not have an EFS event were censored at the last evaluable disease assessment.
ORR was defined as the incidence of either a CR or PR by the Lugano Classification44 as defined by blinded central assessment. Patients who did not meet the criteria for an objective response by the analysis cut-off date were considered as nonresponders.
OS was defined as the time from randomization to death from any cause. Patients who did not die by the analysis data cut-off date had survival time censored at their last date known to be alive. For patients alive or dead after the data cut-off date, survival time was to be censored at the data cut-off date.
PFS was defined as the time from randomization to disease progression per the Lugano Classification44 as determined by the investigator assessment or death from any cause. The following criteria were used to define censoring times for PFS: (1) patients who were alive and did not meet the criteria for progression at the analysis cut-off date had PFS censored a the last evaluable disease assessment, (2) patients who received subsequent lymphoma therapy (except HDT, TBI for HDT, and ASCT while in protocol therapy-induced response) in the absence of documented PD were censored at their last evaluable disease assessment date before starting the subsequent therapy, (3) the receipt of ASCT and allo-SCT while a patient was in response was not considered a PFS event and these patients were censored at the last evaluable disease assessment before ASCT or allo-SCT.
DOR was defined as the time from first response to disease progression per the Lugano Classification44 or death from any cause. DOR was derived only in patients who experienced an objective response per the Lugano Classification as determined by blinded central assessment. The following criteria were used to define censoring times for DOR: (1) patients not meeting criteria for progression or death by the analysis cut-off date were censored at the last evaluable disease assessment date; (2) patients who received subsequent new lymphoma therapy (except HDT, TBI for HDT, or ASCT while in protocol therapy-induced response) in the absence of documented PD were censored at the last evaluable disease assessment before starting the new therapy; (3) for the primary DOR analysis, DOR was censored at the last evaluable disease assessment date before ASCT or allo-SCT for patients undergoing ASCT or allo-SCT while in protocol-defined therapy response.
Time to next treatment was defined as the time from the randomization date to the start of the subsequent new lymphoma therapy (including re-treatment or subsequent stem cell transfer for patients in the axi-cel arm) or death from any cause. Patients who had not received subsequent new lymphoma therapy and were still alive were to be censored at the last contact date.
Patient-reported outcomes included HRQoL assessed using the EORTC QLQ-C30 version 3.0 and the EQ-5D-5L questionnaires. The EORTC QLQ-C30 is a 30-item cancer-specific questionnaire designed to provide a multidimensional assessment of HRQoL with a recall period of 1 week. The EORTC QLQ-C30 version 3.0 includes the following scales: 5 multi-item functional scales (assessing physical, role, emotional, cognitive, and social functioning) and 3 symptom scales (fatigue, nausea and vomiting, and pain); 6 single-item symptom scales (dyspnea, insomnia, appetite loss, constipation, diarrhea, and financial difficulties); and 2 global scales (global health status and HRQoL). No items are shared between the scales. The first 28 of 30 items have 4-level ordinal responses from 1 (“not at all”) to 4 (“very much”). The final 2 items, comprising the global health status and HRQoL scale, have 7-level ordinal responses from 1 (“very poor”) to 7 (“excellent”). Each scale is measured from 0 to 100 after a linear transformation. Higher scores for functioning scales and for the global health status or quality of life scales indicate a higher level of functioning and a better HRQoL, respectively, whereas higher scores in symptom scales represent a higher level of symptoms. The minimal important difference (MID) of the QLQ-C30 scores was 10 points. This MID was based on a study of patients with lung cancer.16
The EQ-5D-5L questionnaire is a generic and preference-weighted measure of health status captured on the day of assessment. Two components comprise the EQ-5D-5L: the EQ-5D descriptive system and the EQ VAS. The descriptive system has 5 dimensions (mobility, self-care, usual activities, pain/discomfort, and anxiety/depression) and is divided into 5 levels of severity: no problems, slight problems, moderate problems, severe problems, and extreme problems. The EQ VAS is a vertical visual analogue scale for recording self-rated HRQoL state reported from 0, described as “the worst health you can imagine,” to 100, described as “the best health you can imagine.” The MID was defined as a change of 0.06 points in the EQ-5D-5L index and a change of 7 points from screening in the EQ VAS score. This MID was based on a study of patients with general cancer.45
The HRQoL assessments were conducted at screening (within 14 days before randomization), 5 days after randomization, at study day 50, treatment period (day of transplant), and during posttreatment follow-up, that is, study day 100 and study day 150 (± 14 days).
Safety
Safety assessments included monitoring of AEs and concomitant medications, clinical laboratory analyses (serum chemistries, complete blood count with differential, alanine transaminase, aspartate transaminase, total bilirubin, lactate dehydrogenase, C-reactive protein, ferritin, urine/serum pregnancy tests, and viral serologic tests [for European sites only]), vital sign measurements, physical examinations, ECOG PS, neurologic examinations, cardiac function (echocardiography and electrocardiography), and testing for replication-competent virus in the blood, as well as for antibodies against the axi-cel CAR in the serum.
The severity of AEs and SAEs were graded using the National Cancer Institute’s Common Terminology Criteria for Adverse Events (CTCAE) version 4.03.46 The investigator was to assess and record whether the AE or SAE was possibly related to axi-cel, leukapheresis, or lymphodepleting chemotherapy (in the axi-cel arm); salvage chemotherapy, HDT, CD34+ leukapheresis, or CD34+ infusion (in the SOC arm); disease progression; concurrent disease; concomitant medication; or other possible reasons.
An AE was defined as any untoward medical occurrence in a clinical study experienced by a participant; the event did not necessarily have a relationship to the study treatment. Investigators were responsible for ensuring that any AEs observed by the investigator or reported by the patient were recorded in the patient’s medical record. The definition of an AE included the worsening of a pre-existing medical condition. Worsening indicated that the pre-existing medical condition had increased in severity, frequency, and/or duration or was associated with a worse outcome. When recording such events, descriptions that the pre-existing condition had changed (e.g., more frequent headaches for a patient with pre-existing headaches or increased blood pressure in a patient with pre-existing hypertension) were to be provided. A pre-existing condition that had not worsened during the study or involved an intervention, such as elective cosmetic surgery or a medical procedure while on study, was not to be considered an AE. Hospitalization for study treatment infusions or precautionary measures per institutional policy were not to be considered AEs. TEAEs were defined as any AE with onset on or after the axi-cel infusion for the axi-cel arm, and as any AE with onset on or after the first dose of salvage chemotherapy for the SOC arm.
An SAE was defined as an event that met at least 1 of the following: fatal; life-threatening; required inpatient hospitalization or prolongation of planned hospitalization; resulted in persistent or significant disability or incapacity; resulted in congenital anomaly or birth defect. Other medically important serious events could be classified as an SAE if an investigator considered them to be clinically important, but they did not meet any of the serious criteria.
AEs of special interest for axi-cel treatment included important identified risks (CRS, neurologic events [including cerebral edema], cytopenias [thrombocytopenia, neutropenia, and anemia], infections, and hypogammaglobulinemia) and important potential risks (secondary malignancies, immunogenicity, replication-competent virus, tumour lysis syndrome, and aggravation of graft-versus host disease). CRS is an identified risk for axi-cel but not for SOC; as such, CRS as a syndrome was only recorded for axi-cel. The term “disease progression” as assessed by measurement of malignant lesions on radiographs or other methods was not to be considered an AE. However, for situations when an AE or SAE was due to the disease under investigation, the signs and symptoms or worsening of signs and symptoms of the malignancy under study were considered to be AEs.
Investigators were to report all AEs observed by the investigator or reported by the patient and that occurred from enrolment (i.e., randomization) through the study day 150 visit or until a change in the patient’s lymphoma therapy, whichever occurred first. All SAEs that occurred from the date of the screening through the study day 150 visit or until initiation of new lymphoma therapy (whichever occurred first) were to be reported. Targeted SAEs (neurologic events, hematologic events, infections, autoimmune disorders, and secondary malignancies) were to be reported for up to 5 years for the SOC arm, 15 years for the axi-cel treatment arm, or until disease progression, whichever occurred first. SAEs assessed by the investigator as related to axi-cel were to be reported regardless of the time period. Patients who did not meet the screening criteria or who were enrolled but not dosed with axi-cel were to be followed for AEs and SAEs for 30 days after the last study-specific procedure (e.g., screening procedure, leukapheresis, or lymphodepleting chemotherapy).
Deaths were to be reported as SAEs if they occurred after randomization and before the study day 150 visit or until the initiation of new lymphoma therapy (whichever occurred first), regardless of attribution to treatment; the event or condition leading to death was to be recorded on the AE form. The term “disease progression” as assessed by measurement of malignant lesions on radiographs or other methods was not to be reported as an AE. Deaths attributed solely to progression of the underlying lymphoma and in the absence of signs and symptoms were to be recorded as SAEs and on the AE electronic case report form with the preferred term “B-cell lymphoma.” Deaths that occurred after the study day 150 visit during the long-term follow-up period or after initiation of new lymphoma therapy that were due to the underlying malignancy were to be recorded on the Survival Status electronic case report form and Death Summary Page.
Statistical Analysis
Sample Size Calculations
The anticipated enrolment in this study was approximately 350 patients. The primary analysis was planned to occur when all patients had had the opportunity to be followed for the month 9 disease assessment (i.e., the month 9 time point had passed for all patients) and 250 EFS events by blinded central assessment had been observed; the study was sized to achieve approximately 90% power at the 1-sided 2.5% significance level to detect a 50% improvement in EFS (the hypothesis for the study). The minimum effect size that could be determined to be statistically significant was an EFS HR of 0.79, or a 27% relative improvement in EFS. Assuming a concave accrual distribution with 50% of accrual in the last 33% of the accrual period of 24 months and a 10% rate of loss to follow-up (5% by month 1 and cumulative 10% by month 8) in the axi-cel arm and 15% rate of loss to follow-up (10% by month 1 and cumulative 15% by month 8) in the SOC arm, it was anticipated that the event goal would be achieved if 350 patients were randomized (175 patients per arm) and would occur approximately 31 months after the first patient was randomized.
Analyses of Outcomes
For the primary end point of EFS according to blinded central assessment, a stratified log-rank test was used for the primary comparison of EFS. Stratified Cox regression models were used to provide the estimated EFS HR and 2-sided 95% CIs for axi-cel relative to SOC. IxRS values for response to first-line therapy and sAAIPI were used as the stratification factors for the log-rank test and Cox regression model. The median EFS event time and event-free rates at 3-month intervals were calculated. The Breslow method and the Efron method were used to handle the ties for the Cox regression models. Nonproportionality among the treatment groups was assessed by comparing the standardized martingale residuals over time to a normal distribution at the 5% level. If the comparison of the standardized martingale residuals over time was significant, a piece-wise Cox model was used for the analysis. For the stratified piece-wise Cox model, 2 or more equal length intervals of 12 weeks was considered. This was to include 1 scheduled tumour assessment in each interval. These models allowed estimation of the overall as well as within-interval treatment HR. The overall log HR was calculated from the weighted average of the interval log HRs. The weight for each interval log HR estimate was to be inversely proportional to the variance of the interval log HR estimate. If there were intervals where 1 or more strata was too sparse, the stratified piece-wise Cox model was to be collapsed as specified for the primary analysis. Kaplan-Meier estimates and 2-sided 95% CIs were calculated for event-time quartiles, for event-free rates at 3-month intervals, and for the difference in event-free rates between treatment arms at these times.
For the analysis of ORR based on blinded central assessment, the incidence of objective response and best response was summarized. An exact binomial 2-sided 95% CI was generated for the ORR and best response rates for each treatment arm. The Wilson score method with continuity correction was used to calculate 95% CIs for the difference in ORRs between treatment arms.
The analysis of OS was conducted using the same methods as for the analysis of EFS. A stratified log-rank test was used for the primary comparison of OS. In addition, stratified Cox regression models were used to provide the estimated OS HR and 2-sided 95% CIs for the axi-cel arm relative to the SOC arm.
The analyses of PFS, DOR, and time to next treatment used the same methods as the analysis of EFS, with P values from the log-rank test descriptive.
Changes in the EORTC QLQ-C30 domains, EQ-5D-5L index, and EQ-5D-5L VAS scores were summarized with descriptive statistics. Each of the 3 HRQoL scores was examined via a series of mixed models for repeated measures (MMRM) for the individual change in HRQoL scores over time and systematic differences between treatment arms. The base MMRM was fit to each patient-reported outcome score with terms for treatment arm, time point (treated as discrete), and treatment by time interaction. An unstructured correlation structure was assumed for measurements collected across visits. In case the estimation procedure did not converge, the compound symmetry variance structure was considered. The fit of the model to the data was assessed via Akaike information criterion (AIC) and Bayesian information criterion (BIC). Using MMRM, planned comparisons were conducted between treatment arms at predetermined specific time points of interest (study days 100 and 150, and months 9, 12, 15, 18, 21, and 24). The MMRM was considered sufficient for handling missing data under the missing-at-random assumption. The base MMRM was run with variables for treatment, time, and treatment time by interaction (primary analysis) and controlling for Interactive voice/web (x) response IxRS values of response to first-line therapy and age-adjusted International Prognostic Index.
Descriptive statistics were used to summarize safety data. A summary of the patient incidence of key AEs (any, worst severity, serious, related, CRS, neurologic event) was provided. The incidence of AEs, SAEs, and grade 3 or higher AEs was provided for all TEAEs, all AEs related to lymphodepleting chemotherapy (axi-cel arm only), all TEAEs related to axi-cel (axi-cel arm only), all TEAEs related to salvage chemotherapy, HDT, or ASCT (SOC arm only), fatal AEs, and TEAEs of interest (including identified and potential risks). Summary statistics for the onset time, duration, and resolution of TEAEs of interest were reported.
Statistical Considerations for Multiple Comparisons
The study was planned to have an overall alpha (significance level) of 2.5% with 1-sided testing. To preserve the overall significance level, statistical testing of the primary and key secondary efficacy end points was to follow a hierarchical scheme: first, EFS was to be tested at the primary analysis. An EFS HR (test versus control arm) of 0.67 was hypothesized. Assuming an exponential distribution for EFS and a median EFS of 4 months in the SOC arm, this implied a 50% relative improvement in EFS, corresponding to a median EFS of 4 months versus 6 months (control arm versus test arm, respectively). A log-rank test stratified by randomization factors was to be used to test the null hypothesis of no difference in EFS using an overall 1-sided alpha level of 2.5%.
Conditional on a statistically significant improvement in EFS, ORR was to be tested at the 1-sided alpha level of 2.5% at the time of the primary EFS analysis. ORR was to be tested with a stratified Cochran-Mantel-Haenszel test using randomization factors. Conditional on a statistically significant improvement in EFS and ORR, OS was to be tested up to 3 times at an overall alpha level of 2.5%. The primary analysis of OS is to occur when approximately 210 deaths have been observed or no later than 5 years after the first patient was randomized. A first interim analysis of OS was to occur at the time of the primary EFS analysis and a second interim analysis when approximately 160 deaths have been observed or no later than 4 years after the first patient is randomized. A spending function of the rho family was to be used to allocate the alpha between the 2 interim analyses of OS and the primary analysis of OS. Log-rank tests stratified by randomization factors were to be used to test the null hypothesis of no difference in OS. If a statistically significant improvement in EFS was not demonstrated at the time of the primary EFS analysis, hierarchical testing of ORR and OS was not to occur. If a statistically significant improvement in EFS was demonstrated, but a statistically significant improvement in ORR was not demonstrated at the time of the primary EFS analysis, hierarchical testing of OS was not to occur.
One primary analysis of EFS was planned and was to be performed after 250 EFS events as determined by blinded central assessment had been observed and all patients had had the opportunity to be followed for 9 months (i.e., the month 9 disease assessment time point for all patients had passed at the time of the primary analysis, which allowed patients to be included in the analysis if they did not have their month 9 visit [e.g., due to death, disease progression, loss to follow-up, consent withdrawal, or the COVID-19 pandemic]). The primary analysis of ORR was to occur at the same time. The primary analysis for OS was planned to be performed after approximately 210 deaths had occurred or no later than 5 years after the first patient was randomized.
Interim Analyses
The study planned for 1 interim and 1 primary analysis for EFS. The interim analysis was to be a safety and futility analysis of EFS after 135 EFS events (by blinded central assessment) had occurred. A total of 135 EFS events occurred approximately 19 months after the first patient was randomized. Two interim analyses of OS were planned. A first interim analysis of OS was to occur at the time of the primary EFS analysis by when approximately 110 deaths were anticipated to occur, conditional upon statistically significant tests of EFS and ORR. A second interim analysis of OS was to occur when approximately 160 deaths had occurred or no later than 4 years after the first patient was randomized.
To allocate the type II error between the interim and primary EFS analyses, an O’Brien-Fleming spending function of the Lan-DeMets family was to be used. Under the null hypothesis, the probability of stopping for futility at the interim analysis was approximately 60%.
To allocate the alpha between the 2 interim analyses of OS and the primary analysis of OS, a spending function of the rho family with parameter (rho = 6) was to be used. The 1-sided alpha of 0.1% and 0.4% was to be allocated at the first and second interim analysis of OS, respectively.
Sensitivity Analyses
Sensitivity analyses of EFS were performed to assess ascertainment time bias in disease progression. An additional sensitivity analyses of EFS, PFS, DOR were performed in which patients in the axi-cel arm who underwent ASCT while in an axi-cel-induced response were imputed to have an event at the time the ASCT was performed.
Sensitivity analyses of OS were performed to address the confounding effects of treatment switching in the SOC arm (i.e., patients randomized to the SOC arm who did not respond or relapsed after SOC and subsequently received commercial or investigational cell therapy) using the rank-preserving structural failure time (RPSFT) model with g-estimation47 and inverse probability of censoring weights (IPCW) adjustment methods.48
Subgroup Analyses
The consistency of treatment effect on the EFS HR for axi-cel versus SOC was examined by estimating Cox model HRs within preplanned subgroups according to age, sex, ECOG PS, geographic region, race or ethnicity, response to first-line therapy, sAAIPI, prognostic marker (HGBL, double- or triple-hit and double expressor lymphoma), molecular subgroup (germinal centre B cell–like, activated B cell–like, unclassified), and disease type according to investigator and according to the central laboratory. ECOG PS score and response to first-line therapy were subgroups of interest identified in the CADTH systematic review protocol. A similar statistical analysis approach to the main EFS analysis was used. There was no multiplicity control. As such, all subgroup analyses are exploratory in nature.
Analysis Populations
The full analysis set (intention-to-treat population; N = 359) includes all randomized patients. Patients were analyzed by the protocol therapy to which they were randomized. The primary analysis of all efficacy end points, unless noted otherwise, was an intention-to-treat analysis conducted using the full analysis set. The safety analysis set (N = 338) was the subset of all randomized patients who received at least 1 dose of axi-cel as protocol therapy or SOC salvage chemotherapy as protocol therapy. The safety analysis set–ASCT (N = 62) comprised the subset of patients randomized to the SOC arm who underwent transplant as part of protocol therapy. The QoL analysis set (N = 296) comprised the subset of patients in the full analysis set who had a baseline and at least 1 completed postrandomization measurement through study day 150. The re-treatment analysis set (N = 9) comprised the subset of patients treated with axi-cel as study treatment who received any dose of axi-cel as re-treatment.
Results
Patient Disposition
Of the 437 patients screened, 359 (82%) patients met the inclusion criteria and underwent randomization (axi-cel arm: n = 180; SOC arm: n = 179). As of the data cut-off date of March 18, 2021, the median follow-up from randomization to the data cut-off date was 24.9 months (interquartile range [IQR], 21.0 to 28.7 months).
In the axi-cel arm, 178 of 180 randomized patients (99%) underwent leukapheresis. Of the 2 patients (1%) who did not undergo leukapheresis, 1 patient had rapid disease progression that required immediate antilymphoma therapy before the planned study treatment, and 1 patient was found to be ineligible after randomization due to cardiac lymphoma involvement. A total of 172 patients (96%) received lymphodepleting chemotherapy and 170 patients (94%) subsequently received axi-cel. Of the 178 patients who underwent leukapheresis, 6 patients (3%) received neither lymphodepleting chemotherapy nor axi-cel and 2 patients (1%) received lymphodepleting chemotherapy but not axi-cel due to AEs, disease progression or death (Table 8 and Figure 3). In the axi-cel arm, all 170 patients (100%) who received a single dose of axi-cel completed treatment. After axi-cel treatment, 9 patients who had had a response and later progressed were re-treated with axi-cel.
In the SOC arm, 168 of 179 randomized patients (94%) received at least 1 dose of salvage chemotherapy. In the SOC arm, completion of treatment was defined as completion of 2 or 3 cycles of salvage chemotherapy and either stable disease at study day 50 or PR or CR at study day 50 and completion of HDT-ASCT. Treatment was reported to be completed for 89 (53%) of the 168 patients who received at least 1 dose of salvage chemotherapy. Treatment was not completed for the remaining 79 (47%) patients for the following reasons: disease progression (71 patients), AEs (2 patients: 1 patient with grade 4 acute kidney injury related to SOC, and 1 patient with blood stem cell harvest failure), and other reasons (6 patients: 3 patients could not continue the initial salvage chemotherapy regimen, 1 patient was initiated on an alternative protocol, 1 patient achieved PR but the investigator deemed the response insufficient to proceed to ASCT, and 1 patient did not have a complete metabolic response and could not receive ASCT per institutional guidelines) (Table 8 and Figure 3).
According to the investigator assessment, 80 patients (45%) responded to salvage chemotherapy, of whom 69 patients (86%) underwent leukapheresis of CD34+ cells for ASCT. The other 11 patients (14%) who responded did not undergo leukapheresis of CD34+ cells for ASCT due to PD after the initial response (9 patients), an AE (1 patient; blood stem cell harvest failure), and investigator assessment of insufficient response to proceed to ASCT (1 patient). Another 5 patients underwent leukapheresis of CD34+ cells for ASCT but did not respond to salvage chemotherapy and could not proceed with ASCT. Thus, a total of 74 patients (41%) in the SOC arm underwent leukapheresis, of whom 69 patients responded, 64 patients (36%) received HDT and reached HDT-ASCT, and 62 patients (35%) subsequently received CD34+ stem cell rescue (ASCT) on-protocol. An additional 2 patients received CD34+ stem cell rescue off-protocol (1 patient was inadvertently started on an alternative protocol and 1 patient had disease progression 1 day after HDT was started and later received off-protocol HDT-ASCT).
At the data cut-off date, 66 patients (39%) in the axi-cel arm who received axi-cel and 86 patients (51%) in the SOC arm who received at least 1 dose of salvage chemotherapy had discontinued participation in the study. Reasons for discontinuation after receiving study therapy were death (64 patients in the axi-cel arm and 75 patients in the SOC arm), loss to follow-up (2 patients in each arm), withdrawal of consent (7 patients in the SOC arm), investigator decision (1 patient in the SOC arm), and other (1 patient in the SOC arm). In addition, 8 patients in the axi-cel arm who did not receive axi-cel were discontinued from the study due to death, and 7 patients in the SOC arm were discontinued from the study due to death (1 patient), full consent withdrawal (5 patients), or loss to follow-up (1 patient).
Table 8: Patient Disposition — ZUMA-7 Study (Full Analysis Set)
Disposition | Axi-cel (N = 180) n (%) | SOC (N = 179) n (%) |
|---|---|---|
Randomized | 180 (100) | 179 (100) |
Axi-cel | ||
Underwent leukapheresis | 178 (99) | NA |
Received bridging therapy | 65 (36) | NA |
Received conditioning chemotherapy | 172 (96) | NA |
Received axi-cel | 170 (94) | NA |
Received re-treatment axi-cel | 9 (5) | NA |
SOC therapy | ||
Received SOC salvage chemotherapy | NA | 168 (94) |
Underwent leukapheresis of CD34+ stem cells | NA | 74 (41) |
Received high-dose therapy | NA | 64 (36) |
Received CD34+ stem cell rescue | NA | 62 (35) |
Patients who did not receive conditioning chemotherapy, axi-cel, or SOC therapy | 8 (4) | 11 (6) |
Adverse event | 2 (1) | 0 (0) |
Death | 2 (1) | 0 (0) |
Disease progression | 2 (1) | 0 (0) |
Patient request | 0 (0) | 8 (4) |
Lost to follow-up | 0 (0) | 1 (1) |
Other | 2 (1) | 2 (1) |
Patients who received conditioning chemotherapy but not axi-cel | 2 (1) | NA |
Adverse event | 2 (1) | NA |
Patients who received axi-cel or SOC therapy | 170 (94) | 168 (94) |
Patients who received bridging therapy | 60 (33) | NA |
Patients who completed treatment | 170 (94) | 89 (50)a |
Patients who initiated but did not complete axi-cel infusion or SOC | 0 (0) | 79 (44) |
Adverse event | 0 (0) | 2 (1) |
Disease progression | 0 (0) | 71 (40) |
Other | 0 (0) | 6 (3) |
Primary reason for ending the study | ||
Patients who did not receive axi-cel or SOC therapy | 8 (4) | 7 (4) |
Death | 8 (4) | 1 (1) |
Lost to follow-up | 0 (0) | 1 (1) |
Full consent withdrawn | 0 (0) | 5 (3) |
Patients who received axi-cel or SOC therapy | 66 (37) | 86 (48) |
Death | 64 (36) | 75 (42) |
Due to COVID-19 | 4 (2) | 2 (1) |
Lost to follow-up | 2 (1) | 2 (1) |
Full consent withdrawn | 0 (0) | 7 (4) |
Investigator decision | 0 (0) | 1 (1) |
Other | 0 (0) | 1 (1) |
axi-cel = axicabtagene ciloleucel; NA = not applicable; SOC = standard of care.
Notes: In the SOC arm, patients who completed treatment are those who completed salvage chemotherapy (2 or 3 cycles), high-dose therapy, and stem cell transplant, or those who completed salvage chemotherapy and were assessed as stable disease at study day 50 without proceeding to stem cell transplant.
aFor 2 patients, the study day 50 disease assessments were updated by the clinical site to stable disease, but the corresponding end-of-treatment electronic case report form was not updated.
Source: Clinical Study Report for Yescarta.15
Figure 3: Patient Disposition — ZUMA-7
AE = adverse event; ASCT = autologous stem cell transplant; auto-SCT = autologous stem cell transplant; FAS = full analysis set; HDT = high-dose therapy; PD = progressive disease; SOCT = standard of care therapy; SD = stable disease.
a Patient was ineligible.
b One patient had an AE of increased alanine transaminase; 1 patient had an AE of hyperbilirubinemia.
c Patient in false progression at baseline; reassessment showed they were not progressing.
d One patient had an AE of cerebrovascular accident; 1 patient had an AE of small intestinal perforation.
e Three patients because of reasons related to insurance, 1 patient due to rapid progression, and 1 patient opted out.
f One patient had a negative disease biopsy; 1 patient had a false-positive PET-CT and no refractory double-hit lymphoma after R-EPOCH x 5 (rituximab+etoposide- prednisolone -vincristine-cyclophosphamide-doxorubicin).
g Withdrawals: 5 patients withdrew with full consent due to patient request. Patients are also included in the categories of reasons not received.
h Includes 4 patients with PD who were leukapheresed. PD represents best response to salvage chemotherapy.
i Includes 1 patient with SD who was leukapheresed. SD represents best response to salvage chemotherapy.
j Patient had an AE of acute kidney injury.
k Includes 1 patient with lack of response to salvage chemoimmunotherapy with rituximab+ifosfamide-carboplatin-etoposide (R-ICE); 1 patient who did not tolerate rituximab+gemcitabine-dexamethasone-cisplatin/carboplatin (R-GDP) and switched to R-ICE; 1 patient who changed treatment after 1 cycle of rituximab+dexamethasone-cytarabine-cisplatin (R-DHAP) due to renal impairment; and 1 patient with insufficient overall response to proceed to ASCT per investigator.
l Withdrawals: Patient withdrew with full consent; 4 patients completed therapy but no response; 3 patients with PD. Patients are also included in the categories of reasons for not proceeding.
m As determined by the investigator.
n PD represents disease progression after an initial response to salvage chemotherapy.
o Patient had an AE of blood stem cell harvest failure.
p As determined by the investigator.
q Patient was inadvertently enrolled on an alternative protocol.
Source: Clinical Study Report for Yescarta.15
Protocol Deviations
Important protocol deviations were reported for 56 patients (16%) during the study, including 31 patients (17%) in the axi-cel arm and 25 patients (14%) in the SOC arm. These included patients receiving prohibited investigational agents or radiation (2%), exclusion criteria (1%), inclusion criteria (4%), and missing data (8%). Each category of important protocol deviations occurred for less than 10% of patients overall. The most frequent important protocol deviation in both the axi-cel and SOC arms was missing data (20 patients [11%] and 10 patients [6%], respectively).
Exposure to Study Treatments
Axi-Cel Arm
Bridging Therapy
Of the 170 patients treated with axi-cel, 60 patients (35%) received bridging therapy, most of whom (48 patients, 80%) received at least 1 dose of dexamethasone.
Lymphodepleting Chemotherapy
Of the 180 patients randomized to the axi-cel arm, 172 (96%) received lymphodepleting chemotherapy. Of the 169 patients in the safety analysis set with available body surface area adjusted dose information, the planned total BSA-adjusted dose (± 10%) of cyclophosphamide (1,500 mg/m2) and fludarabine (90 mg/m2) was administered to 165 patients (98%) and 164 patients (97%), respectively. Six patients (3%) did not receive lymphodepleting chemotherapy, primarily due to AEs or disease progression.
Axi-Cel
Of the 180 patients randomized to the axi-cel arm, 170 patients (94%) received axi-cel infusion. The median weight-adjusted dose of axi-cel was 2.0 × 106 anti-CD19 CAR T cells/kg body weight (range, 1.0 × 106 cells/kg to 2.1 × 106 cells/kg). In total, 166 patients (98%) received within 10% of the target dose of 2 × 106 anti-CD19 CAR T cells/kg (or a flat dose of 200 × 106 anti-CD19 CAR T cells for patients weighing > 100 kg). The median total number of anti-CD19 CAR T cells in the axi-cel infusion was 170 × 106 cells (range, 58 × 106 cells to 200 × 106 cells), and the median total number of T cells infused was 301.5 × 106 cells (range, 88 × 106 cells to 633 × 106 cells).
Two patients who received lymphodepleting chemotherapy did not receive axi-cel due to AEs unrelated to leukapheresis or lymphodepleting chemotherapy. Manufacturing failure or delay in product availability was not reported as a limitation to any patient not receiving axi-cel. Nine patients (5%) who achieved a PR or CR at the study day 50 disease assessment and subsequently experienced disease progression were re-treated with axi-cel. The second dose of axi-cel could have been manufactured at the same time as the first axi-cel dose or manufactured later from existing cryopreserved peripheral blood mononuclear cells or a second leukapheresis.
Manufacturing Turnaround Time
For the 170 patients who received axi-cel ████ ████████ ██ █████ ███████ ██████ ███ ███ ███████ ███ ██ ████████ ██ ███ ████ ██ ███ █████ ██████ ███████ ███████ ███ ████████████ the median time from leukapheresis to the product’s release from the manufacturing site was 13 days (range, 10 to 24 days) overall, ██ ████ ███████ ██ ██ ██ █████ ███ █████ ███████ ███ ██ ████ ███████ ██ ██ ██ █████ ███ █████ ████████ Axi-cel was delivered to the study site after a median of 18 days (range, 13 to 49 days) overall, ██ ████ ███████ ██ ██ ██ █████ ███ █████ ████████ ███ ██ ████ ███████ ██ ██ ██ █████ ███ █████ ████████
███ ██████ ████ ████ █████████████ ██ █████████████ ███ ███ ████ ███████ █ ██ ██ ██████ The median time from leukapheresis to axi-cel administration was 26 days (range, 16 to 52 days) ████████ ██ ████ ███████ ██ ██ ██ █████ ███ ████████ ██ █████ ████████ ███ ██ ████ ███████ ██ ██ ██ █████ ███ ████████ ██ █████ ████████ ███ ████████ ██ ████ ████ █████████████ ██ ███████ ██████████████ ████████ ███████ █████████████ ███ ██████████ ██ ███████████████ █████████████ ████ █ ██████ ████ ██ ██ ████ ███████ ██ ██ ██ █████ ████████ ██ ████ ███████ ██ ██ ██ █████ ███ ████████ ██ █████ ████████ ███ ██ ████ ███████ ██ ██ ██ █████ ███ ████████ ██ ███ █████ ████████ ███ ████ ███████ ██████████ ██ ███████████████ ████████████ ███ ███████ ████████ ███ █ ████ ███ ███ ████████
Of the 170 patients who received axi-cel, 166 patients (98%) received within 10% of the planned dose of 2 × 106 anti-CD19 CAR T cells/kg body weight (or a flat dose of 200 × 106 anti-CD19 CAR T cells for patients weighing over 100 kg). For the 137 patients who received axi-cel and weighed 100 kg or less, the median weight-adjusted dose was 2 × 106 anti-CD19 CAR T cells/kg body weight (range, 1.0 × 106 cells/kg to 2.1 × 106 cells/kg). The 33 patients who received axi-cel and who weighed more than 100 kg received the planned flat dose of 200 × 106 cells. For all 170 patients, the median total number of anti-CD19 CAR T cells in the axi-cel infusion was 170 × 106 cells (range, 58 × 106 cells to 200 × 106 cells) and the median total number of T cells infused was 301.5 × 106 (range, 88 × 106 cells to 633 × 106 cells).
SOC Arm
Of the 168 patients who received SOC salvage chemotherapy, 84 patients (50%) received R-ICE, 42 patients (25%) received R-GDP, 37 patients (22%) received R-DHAP/R-DHAX, and 5 patients (3%) received rituximab+etoposide-methylprednisolone-cytarabine-cisplatin (R-ESHAP). Just over half of the patients (91 patients; 54%) received 2 cycles of salvage chemotherapy and 61 patients (36%) received 3 cycles; 16 patients (10%) received only 1 cycle. A total of 62 patients (37% of the SOC – safety analysis set) who achieved a CR or PR with SOC salvage chemotherapy went on to receive HDT-ASCT. Of the 62 patients who received HDT plus on-protocol ASCT, 42 achieved a response after 3 cycles and 20 patients achieved a response after 2 cycles.
Prior Therapy
All patients received chemotherapy with rituximab in first line. Exposure to prior chemotherapy regimens was generally balanced between the treatment arms. The majority of patients in both arms (59%) had previously been treated with CHOP or a CHOP-like regimen with rituximab only. Prior chemoimmunotherapy regimens are detailed in Table 9.
Table 9: Prior Therapy for Primary Study Disease — ZUMA-7 Study (Full Analysis Set)
Prior therapy | Axi-cel (N = 180) | SOC (N = 179) |
|---|---|---|
Patients with ≥ 1 prior therapy | 180 (100) | 179 (100) |
CHOP or CHOP-like regimen ± rituximab | ||
CHOP or CHOP-like regimen ± rituximab only | 107 (59) | 106 (59) |
With CNS prophylactic chemotherapy | 10 (6) | 8 (4) |
With other regimen | 8 (4) | 6 (3) |
With CNS prophylactic chemotherapy and other regimen | 3 (2) | 2 (1) |
Intensified regimen (e.g., EPOCH, HyperCVAD) ± rituximab | ||
Intensified regimen ± rituximab only | 26 (14) | 26 (15) |
With CNS prophylactic chemotherapy | 12 (7) | 6 (3) |
With other regimen | 2 (1) | 2 (1) |
With CNS prophylactic chemotherapy and other regimen | 0 (0) | 1 (1) |
CHOP or CHOP-like with intensified regimen ± rituximab | ||
CHOP or CHOP-like with intensified regimen ± rituximab only | 9 (5) | 8 (4) |
With CNS prophylactic chemotherapy | 2 (1) | 9 (5) |
With other regimen | 0 (0) | 2 (1) |
Other | 1 (1) | 2 (1) |
CNS prophylactic chemotherapy and other regimen | 0 (0) | 1 (1) |
Patients with ≥ 1 prior radiotherapy | 19 (11) | 16 (9) |
Intent | ||
Curative | 13 (7) | 10 (6) |
Palliative | 5 (3) | 5 (3) |
Other | 1 (1) | 1 (1) |
Body site | ||
Abdominal cavity | 3 (2) | 2 (1) |
Chest | 1 (1) | 0 (0) |
CNS or spine | 0 (0) | 1 (1) |
Gastrointestinal tract | 1 (1) | 0 (0) |
Lymph node | 2 (1) | 4 (2) |
Other | 12 (7) | 9 (5) |
axi-cel = axicabtagene ciloleucel; CHOP = cyclophosphamide+doxorubicin-vincristine-prednisone; CNS = central nervous system; EPOCH = etoposide-prednisone-vincristine-cyclophosphamide-doxorubicin; HyperCVAD = hyperfractionated therapy of cyclophosphamide-vincristine sulphate-doxorubicin (Adriamycin)-dexamethasone; SOC = standard of care.
Note: Multiple prior therapies within the same line are counted as 1 incidence.
Source: Clinical Study Report for Yescarta.15
Concomitant Medications and Procedures
Among patients who received axi-cel, 77 patients (45%) received corticosteroids (with or without tocilizumab), 112 patients (66%) were treated with tocilizumab (with or without corticosteroids), and 68 patients (40%) were treated with corticosteroids and tocilizumab. Reasons for corticosteroid treatment included neurologic events (54 patients; 32%), CRS (40 patients; 24%), for other AEs (18 patients; 11%), or other uses that included prophylaxis and medical history (36 patients; 21%). Reasons for tocilizumab treatment included CRS (111 patients; 65%), neurologic events (17 patients; 10%), for other AEs (11 patients, 6%), or for other uses (1 patient, 1%). Nineteen patients (11%) were treated with vasopressors, and 28 patients (16%) were treated with immunoglobulins.
Subsequent Anticancer Therapy
Although treatment switching between the treatment groups was not planned, patients who did not respond to SOC salvage chemotherapy or who relapsed after ASCT could receive cell therapy (including CAR T-cell therapy) outside the protocol as per local clinical decision.
Of the 70 patients (41%) in the axi-cel arm who received subsequent lymphoma therapy, none received a cell therapy. Of the 120 patients (71%) in the SOC arm who received subsequent lymphoma therapy, 75 patients (63%) received axi-cel, 16 patients (13%) received tisagenlecleucel, 5 patients (4%) were reported to receive a CAR T-cell therapy without further explanation of type, and 1 patient (0.8%) received CD19/CD22 CAR T-cell therapy.
Nineteen patients (11%) in the axi-cel arm received subsequent stem cell transfer, including 11 patients who received ASCT and 8 patients who received allo-SCT, and 11 patients (7%) in the SOC arm received subsequent stem cell transfer, including 5 patients who received ASCT, 5 patients who received allo-SCT, and 1 patient who received both.
Efficacy
Only those efficacy outcomes and analyses of subgroups identified in the review protocol are reported below.
All efficacy analyses reported below are based on the full analysis set (intention-to-treat population; N = 359).
Overall Survival
The OS data remain immature. The primary OS analysis will occur at approximately 210 deaths or 5 years after the first patient was enrolled, whichever is earlier. The interim analysis of OS occurred following demonstration of a statistically significant improvement in EFS and ORR for axi-cel compared with SOC. Based on the a priori alpha spending function, a 1-sided alpha of 0.004 was allocated for the interim analysis. The HR for death was 0.730 (95% CI, 0.530 to 1.007; 1-sided stratified log-rank P = 0.027) (Table 10 and Figure 4).
Table 10: Overall Survival — ZUMA-7 Study (Full Analysis Set)
Overall survival | Axi-cel (N = 180) | SOC (N = 179) |
|---|---|---|
Death from any cause, n (%) | 72 (40) | 81 (45) |
Alive, n (%) | 108 (60) | 98 (55) |
Full consent withdrawn | 0 (0) | 9 (5) |
Lost to follow-up | 2 (1) | 2 (1) |
End of study due to investigator decision | 0 (0) | 1 (1) |
End of study due to other reason | 0 (0) | 0 (0) |
Stratified 1-sided log-rank P valuea,b | 0.0270 | |
Hazard ratio (95% CI), stratifiedb,c | 0.730 (0.530 to 1.007) | |
Unstratified log-rank P value | 0.0442 | |
Hazard ratio (95% CI), unstratified | 0.759 (0.553 to 1.043) | |
OS time (months), median (95% CI) d | NR (28.3 to NE) | 35.1 (18.5 to NE) |
Survival rate, % (95% CI)d | ||
3 month | 96.7 (92.7 to 98.5) | 97.7 (93.9 to 99.1) |
6 month | 90.0 (84.6 to 93.6) | 87.1 (81.0 to 91.3) |
9 month | 83.9 (77.6 to 88.5) | 74.1 (66.9 to 80.1) |
12 month | 76.0 (69.1 to 81.6) | 64.7 (57.0 to 71.4) |
15 month | 67.6 (60.3 to 74.0) | 59.4 (51.6 to 66.3) |
18 month | 64.8 (57.3 to 71.3) | 58.2 (50.4 to 65.2) |
21 month | 63.6 (56.1 to 70.2) | 53.2 (45.2 to 60.5) |
24 month | 60.7 (52.8 to 67.7) | 52.1 (44.0 to 59.5) |
27 month | 59.4 (51.2 to 66.7) | 50.6 (42.2 to 58.3) |
30 month | 53.1 (43.1 to 62.2) | 50.6 (42.2 to 58.3) |
33 month | 53.1 (43.1 to 62.2) | 50.6 (42.2 to 58.3) |
36 month | 53.1 (43.1 to 62.2) | 33.7 (10.0 to 59.9) |
Follow-up time (months), median (95% CI) e | 24.7 (23.3 to 26.0) | 24.1 (22.1 to 25.1) |
axi-cel = axicabtagene ciloleucel; CI = confidence interval, NE = not estimable; NR = not reached; OS = overall survival; SOC = standard of care.
Note: Data cut-off date: March 18, 2021.
aA 1-sided alpha of 0.004 was allocated to this analysis.
bThe stratification factors are responses to first-line therapy (primary refractory vs. relapse ≤ 6 months of first-line therapy vs. relapse > 6 and ≤ 12 months of first-line therapy) and second-line age-adjusted International Prognostic Index (0 to 1 vs. 2 to 3).
cEstimated using Cox regression models.
dEstimated using Kaplan-Meier estimation.
eEstimated using the reverse Kaplan-Meier approach.
Source: Clinical Study Report for Yescarta.15
Figure 4: Kaplan-Meier Plot of Overall Survival (Full Analysis Set)
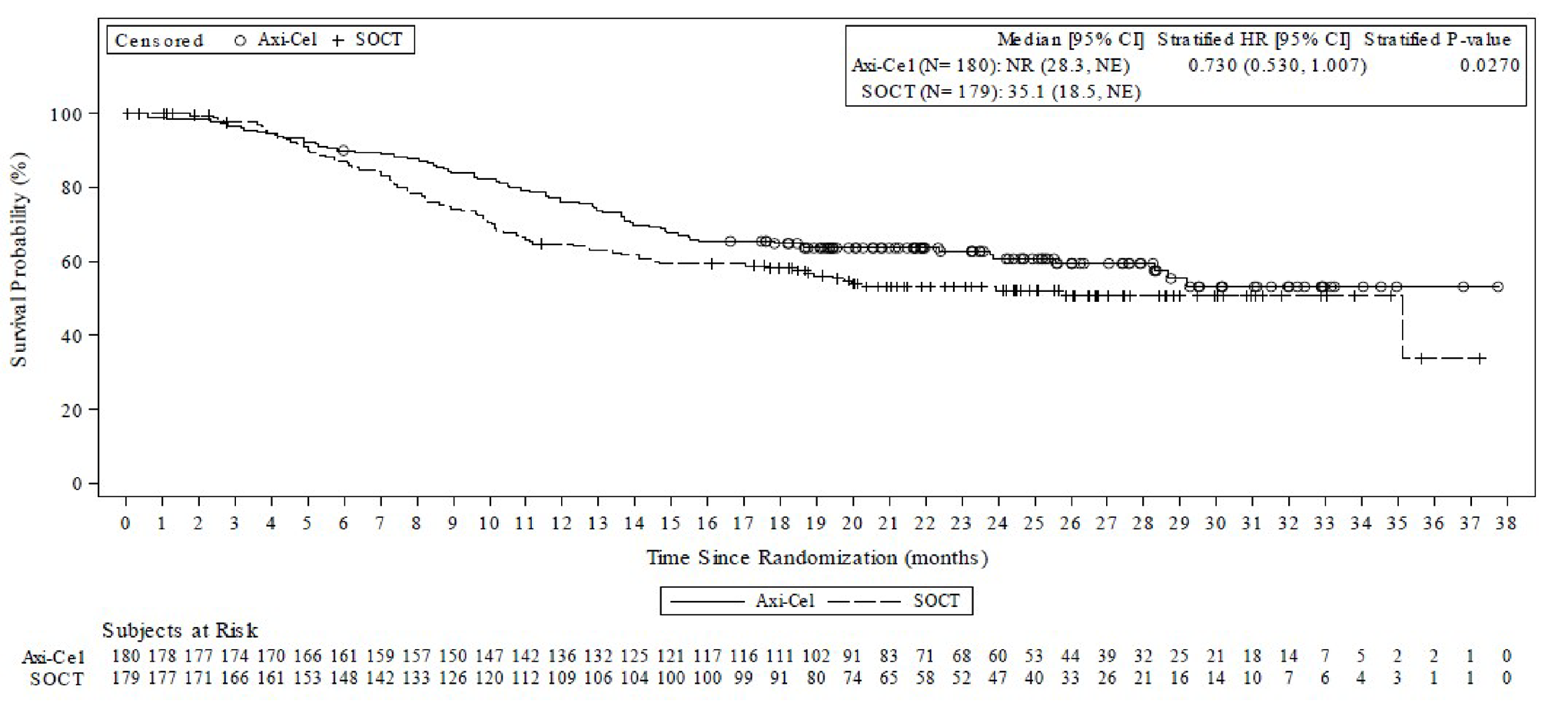
axi-cel = axicabtagene ciloleucel; CI = confidence interval; HR = hazard ratio; NE = not estimable; NR = not reached; OS = overall survival; SOC = standard of care; SOCT = standard of care therapy.
Notes: The stratification factors are responses to first-line therapy (primary refractory vs. relapse ≤ 6 months of first-line therapy vs. relapse > 6 and ≤ 12 months of first-line therapy) and second-line age-adjusted International Prognostic Index (0 to 1 vs. 2 to 3) as collected via interactive voice/web response system.
Stratified Cox regression models were used to provide the estimated HR and 2-sided 95% CIs for axi-cel relative to SOC. The Breslow method was used to handle the ties for the Cox regression models. One-sided P value from log-rank test is presented. A 1-sided alpha of 0.004 was allocated to this interim analysis.
Data cut-off date: March 18, 2021.
Source: Clinical Study Report for Yescarta.15
Addendum to OS Data
An addendum to the ZUMA-7 Clinical Study Report was made to provide data generated in response to a Health Authority request to obtain additional survival follow-up, including from public records, for patients discontinued from ZUMA-7 that was not available at the time of the interim OS analysis (which was conducted at the time of the primary EFS analysis) for completeness of the interim OS data.49
By the EFS primary analysis data cut-off date (March 18, 2021), 14 patients had discontinued from the study and were either lost to follow-up, had withdrawn consent, or had been withdrawn by the investigator. A subsequent search of public records identified additional survival data for 8 of the discontinued patients, including 4 patients (all in the SOC arm) who had died before the primary analysis data cut-off date, and 4 patients (3 in the SOC arm and 1 in the axi-cel arm) confirmed as being alive at the primary analysis cut-off date. Additional survival data for the remaining 6 patients (5 in the SOC and 1 in the axi-cel arm) could not be obtained. The interim OS analysis data were updated (with the same data cut-off date of March 18, 2021) to include these additional data.
By the primary analysis data cut-off date, 153 OS events (i.e., deaths) were observed in the full analysis set and included in the primary analysis. The additional deaths of 4 patients in the SOC arm that were identified after the primary analysis resulted in a total of 157 OS events (72 deaths in the axi-cel arm and 85 deaths in the SOC arm).
The update to the interim OS analysis with additional survival data were consistent with the OS interim analysis originally conducted. The stratified HR was 0.708 (95% CI, 0.515 to 0.972; P = 0.0159). While the median OS in the axi-cel arm was consistent with the median OS reported in the original interim OS analysis (not reached; 95% CI, 28.3 months to NE), the median OS in the SOC arm was reduced from 35.1 months (95% CI, 18.5 months to NE) in the original interim OS analysis to 25.7 months (95% CI, 17.6 months to NE) in the update to the interim OS analysis.
Sensitivity Analysis of OS
The results of the sensitivity analysis to address the confounding effects of treatment switching in the SOC arm were consistent with the original interim analysis of OS (RPSFT model stratified HR = 0.575 [95% CI, 0.413 to 0.800] and IPCW model stratified HR = 0.618 [95% CI, 0.417 to 0.916]).
Table 11: Overall Survival With Additional Survival Data for Discontinued Patients — ZUMA-7 Study (Full Analysis Set)
Overall survival | Axi-cel (N = 180) | SOC (N = 179) |
|---|---|---|
Death from any cause, n (%) | 72 (40) | 85 (47) |
Alive, n (%) | 108 (60) | 94 (53) |
Full consent withdrawn | 0 (0) | 5 (3) |
Lost to follow-up | 1 (1) | 0 (0) |
End of study due to investigator decision | 0 (0) | 0 (0) |
End of study due to other reason | 0 (0) | 0 (0) |
Stratified 1-sided log-rank P valuea | 0.0159 | |
Hazard ratio (95% CI), stratifieda,b | 0.708 (0.515 to 0.972) | |
Unstratified log-rank P value | 0.0275 | |
Hazard ratio (95% CI), unstratifiedb | 0.736 (0.538 to 1.008) | |
OS time (months), median (95% CI)c | NR (28.3 to NE) | 25.7 (17.6 to NE) |
Survival rate, % (95% CI)c | ||
3 month | 96.7 (92.7 to 98.5) | 97.7 (94.1 to 99.1) |
6 month | 90.0 (84.6 to 93.6) | 85.2 (79.0 to 89.6) |
9 month | 83.9 (77.6 to 88.5) | 72.6 (65.3 to 78.6) |
12 month | 76.0 (69.1 to 81.6) | 63.4 (55.8 to 70.1) |
15 month | 67.6 (60.3 to 74.0) | 58.3 (50.6 to 65.2) |
18 month | 64.8 (57.4 to 71.3) | 57.1 (49.4 to 64.1) |
21 month | 63.6 (56.1 to 70.2) | 52.4 (44.6 to 59.6) |
24 month | 60.7 (52.8 to 67.7) | 51.3 (43.4 to 58.7) |
27 month | 59.4 (51.3 to 66.7) | 49.9 (41.8 to 57.5) |
30 month | 53.2 (43.1 to 62.2) | 49.9 (41.8 to 57.5) |
33 month | 53.2 (43.1 to 62.2) | 49.9 (41.8 to 57.5) |
36 month | 53.2 (43.1 to 62.2) | 33.3 (9.9 to 59.2) |
Follow-up time (months), median (95% CI) d | 24.7 (23.3 to 26.0) | 24.4 (22.5 to 25.7) |
axi-cel = axicabtagene ciloleucel; CI = confidence interval, NA = not applicable; NE = not estimable; NR = not reached; OS = overall survival; SOC = standard of care.
Note: Data cut-off date: March 18, 2021.
aThe stratification factors are responses to first-line therapy (primary refractory vs. relapse ≤ 6 months of first-line therapy vs. relapse > 6 and ≤ 12 months of first-line therapy) and second-line age-adjusted International Prognostic Index (0 to 1 vs. 2 to 3).
bEstimated using Cox regression models.
cEstimated using the Kaplan-Meier estimation.
dEstimated using the reverse Kaplan-Meier approach.
Source: Addendum to the primary analysis clinical study report (internal sponsor’s report).49
Figure 5: Kaplan-Meier Plot of Overall Survival With Additional Survival Data for Discontinued Patients (Full Analysis Set)
axi-cel = axicabtagene ciloleucel; CI = confidence interval; HR = hazard ratio; NE = not estimable; NR = not reached; OS = overall survival; SOCT = standard of care therapy.
Notes: The stratification factors are responses to first-line therapy (primary refractory vs. relapse ≤ 6 months of first-line therapy vs. relapse > 6 and ≤ 12 months of first-line therapy) and second-line age-adjusted International Prognostic Index (0 to 1 vs. 2 to 3) as collected via interactive voice/web response system.
Stratified Cox regression models were used to provide the estimated HR and 2-sided 95% CIs for axi-cel relative to standard of care. The Breslow method was used to handle the ties for the Cox regression models. One-sided P value from log-rank test is presented.
Data cut-off date: March 18, 2021.
Source: Addendum to the primary analysis clinical study report (Sponsor’s internal report).49
OS Sensitivity Analyses
A prespecified sensitivity analysis of OS conducted to address the confounding effects of subsequent cell therapy in the SOC arm showed a stratified HR of 0.580 (95% CI, 0.416 to 0.809) using the RPSFT model and 0.695 (95% CI, 0.461 to 1.049) using the IPCW model. There was no multiplicity adjustment for these analyses.
Event-Free Survival
At the time of the data cut-off (March 18, 2021), 252 EFS events as determined by blinded central assessment had occurred for 108 patients (60%) in the axi-cel arm and 144 patients (80%) in the SOC arm. The median EFS was 8.3 months (95% CI, 4.5 to 15.8 months) in the axi-cel arm and 2.0 months (95% CI, 1.6 to 2.8 months) in the SOC arm. The stratified HR for event or death was 0.398 (95% CI, 0.308 to 0.514; P < 0.0001). The Kaplan-Meier estimates of the percentage of patients who remained event-free at 12 and 24 months after randomization in the axi-cel arm were 47.2% (95% CI, 39.8% to 54.3%) and 40.5% (95% CI, 33.2% to 47.7%), respectively, compared with 17.6% (95% CI, 12.3% to 23.6%) and 16.3% (95% CI, 11.1% to 22.2%), respectively, in the SOC arm (Table 12 and Figure 6).
The most common EFS events in either the axi-cel or SOC arm were disease progression (46% and 42%, respectively), new lymphoma therapy (5% and 35%, respectively), and death from any cause (6% and 3%, respectively).
Twelve patients (2 in the axi-cel and 10 in the SOC arm) initiated a new lymphoma therapy in the absence of an evaluable post-baseline disease assessment and had EFS event dates imputed as the randomization date as predefined in the statistical analysis plan. No patients in the axi-cel arm underwent ASCT while in response.
Table 12: Event-Free Survival Per Blinded Central Assessment (Full Analysis Set)
Event-free survival | Axi-cel (N = 180) | SOC (N = 179) |
|---|---|---|
Events, n (%) | 108 (60) | 144 (80) |
Censored, n (%) | 72 (40) | 35 (20) |
Stratified log-rank P valuea | < 0.0001 | NA |
Stratified HR (95% CI)a | 0.398 (0.308 to 0.514) | NA |
EFS time (months), median (95% CI) b | 8.3 (4.5 to 15.8) | 2.0 (1.6 to 2.8) |
Event, n (%) | ||
Disease progression | 82 (46) | 75 (42) |
Best response of stable disease up to and including day 150 assessment postrandomization | 4 (2) | 0 (0) |
New lymphoma therapy | 9 (5) | 63 (35) |
Axi-cel re-treatment | 2 (1) | 0 (0) |
Death from any cause | 11 (6) | 6 (3) |
Censoring reason, n (%) | ||
Response ongoing | 72 (40) | 28 (16) |
Response assessed but no disease at baseline and post baseline | 0 (0) | 3 (2) |
No post-baseline disease assessment | 0 (0) | 1 (1) |
Full withdrawal of consent | 0 (0) | 1 (1) |
Lost to follow-up | 0 (0) | 2 (1) |
Event-free rate,b % (95% CI) | ||
3 month | 80.6 (74.0 to 85.6) | 40.5 (33.2 to 47.8) |
6 month | 51.1 (43.6 to 58.1) | 26.6 (20.2 to 33.3) |
9 month | 49.4 (42.0 to 56.5) | 19.4 (13.8 to 25.6) |
12 month | 47.2 (39.8 to 54.3) | 17.6 (12.3 to 23.6) |
15 month | 43.9 (36.5 to 50.9) | 17.0 (11.8 to 23.0) |
18 month | 41.5 (34.2 to 48.6) | 17.0 (11.8 to 23.0) |
21 month | 41.5 (34.2 to 48.6) | 16.3 (11.1 to 22.2) |
24 month | 40.5 (33.2 to 47.7) | 16.3 (11.1 to 22.2) |
27 month | 40.5 (33.2 to 47.7) | 16.3 (11.1 to 22.2) |
30 month | 37.2 (28.0 to 46.3) | 16.3 (11.1 to 22.2) |
33 month | NE (NE to NE) | 16.3 (11.1 to 22.2) |
Follow-up time (months), median (95% CI)c | 23.0 (20.9 to 24.0) | 21.2 (20.4 to 23.7) |
axi-cel = axicabtagene ciloleucel; CI = confidence interval; EFS = event-free survival; HR = hazard ratio; NA = not applicable; NE = not estimable; SOC = standard of care.
Note: Data cut-off date: March 18, 2021.
aThe stratification factors are responses to first-line therapy (primary refractory vs. relapse ≤ 6 months of first-line therapy vs. relapse > 6 and ≤ 12 months of first-line therapy) and second-line age-adjusted International Prognostic Index (0 to 1 vs. 2 to 3) as collected via interactive voice/web response system.
bEstimated using the Kaplan-Meier method.
cEstimated using the reverse Kaplan-Meier approach.
Source: Clinical Study Report for Yescarta.15
Figure 6: Kaplan-Meier Plot of Event-Free Survival Per Central Assessment (Full Analysis Set)
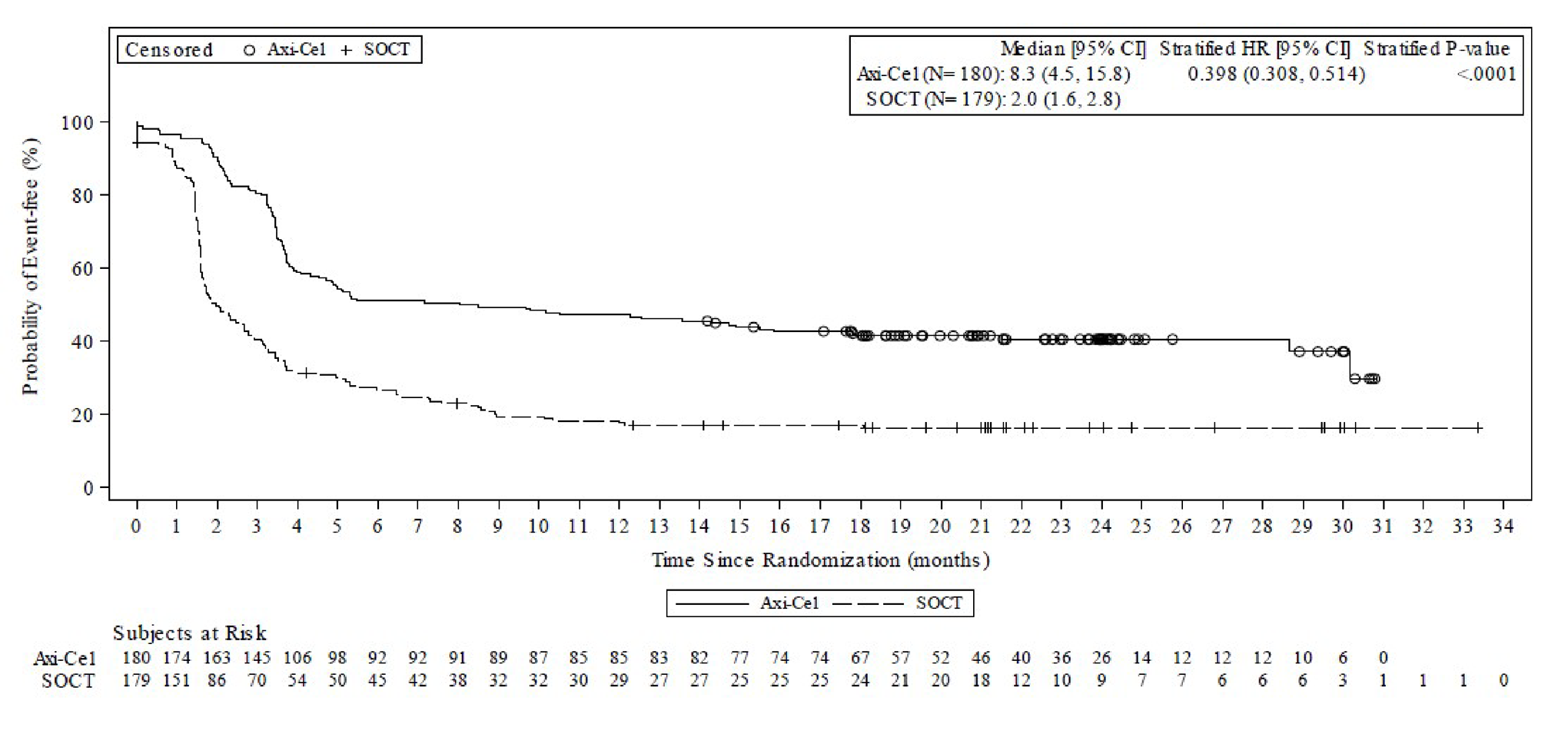
axi-cel = axicabtagene ciloleucel; CI = confidence interval; HR = hazard ratio; SOCT = standard of care therapy.
Note: Data cut-off date: March 18, 2021.
Source: Clinical Study Report for Yescarta.15
Subgroup Analysis
The improvement in EFS with axi-cel compared with SOC was consistent in the subgroups of interest (Table 13).
Table 13: Event-Free Survival Per Blinded Central Assessment by Subgroup
Subgroup | Events, n (%) | HR (95% CI) | |
|---|---|---|---|
Axi-cel | SOC | ||
ECOG Performance Status | |||
0 | 59 of 95 (62) | 76 of 100 (76) | 0.504 (0.356 to 0.713) |
1 | 49 of 85 (58) | 68 of 79 (86) | 0.292 (0.195 to 0.436) |
Response to first-line therapy | |||
Primary refractory | 85 of 133 (64) | 106 of 131 (81) | 0.426 (0.319 to 0.570) |
Relapse ≤ 12 months of the completion of the first-line therapy | 23 of 47 (49) | 38 of 48 (79) | 0.342 (0.202 to 0.579) |
axi-cel = axicabtagene ciloleucel; CI = confidence interval; ECOG = Eastern Cooperative Oncology Group; HR = hazard ratio; SOC = standard of care.
Note: Data cut-off date: March 18, 2021.
Source: Clinical Study Report for Yescarta.15
Sensitivity Analysis
A sensitivity analysis wherein patients who underwent ASCT while in axi-cel-induced response were imputed to have an EFS event at the time of ASCT was consistent with the main assessment of EFS (HR = 0.406; 95% CI, 0.313 to 0.525). An assessment of nonproportional hazards was also performed and the corresponding sensitivity analysis was consistent with the primary analysis (HR = 0.444; 95% CI, 0.333 to 0.590).
The FDA’s Assessment of EFS in Patients in the SOC Arm Who Underwent HDT-ASCT
To understand the outcome of the subgroup of patients that underwent ASCT, the FDA analyzed the EFS of the 62 patients who underwent transplant in the SOC arm. The objective of this exploratory subgroup analysis was not to compare the outcome of the transplant subgroup with the axi-cel arm given that this is not a randomized comparison and the 2 groups are not comparable in terms of baseline characteristics; patients who underwent ASCT were more likely than the patients in the axi-cel arm to have relapsed disease (37% versus 26%) and ECOG PS score of 0 (68% versus 53%). Rather, the purpose of the subgroup analysis was to evaluate the efficacy of ASCT in the subset of chemosensitive patients within the primary refractory and early relapsed population. Results of this exploratory analysis showed a median EFS of 12.1 months (95% CI, 8.5 months to NE) with a 1-year EFS of 51% (95% CI, 38% to 64%).42
Health-Related Quality of Life
Of the 359 patients enrolled in the ZUMA-7 study, 296 patients (165 [92%] patients in the axi-cel arm and 131 [73%] patients in the SOC arm) had baseline HRQoL data and at least 1 follow-up assessment and were included for analysis in the HRQoL Analysis set. By study day 100, data were available for 146 patients (81%) in the axi-cel arm and 62 patients (35%) in the SOC arm. By study day 150, data were available for 110 patients (61%) in the axi-cel arm and 56 patients (31%) in the SOC arm. Samples sizes continued to fall and by month 15, data were available for 67 patients (37%) in the axi-cel arm and 26 patients (15%) in the SOC arm.
EORTC QLQ-C30 Global Health Status/QoL
At screening, the mean EORTC QLQ-C30 global health status scores for evaluable patients were comparable in the axi-cel and SOC arms (mean = 68.6 [SD = 19.9] and mean = 70.1 [SD = 23.1], respectively). At study day 50, almost half of evaluable patients (41.1%) reported worsening (exceeding MID threshold of ± 10 points used in the trial) scores in both treatment arms (mean scores: 60.9 [95% CI, 57.7 to 64.2] and 61.3 [95% CI, 57.3 to 65.3], respectively). Starting at study day 100, scores in the axi-cel arm rebounded while those in the SOC arm declined, with 26.7% of evaluable patients who received axi-cel and 11.3% of patients who received SOC reporting improved scores compared with screening (mean scores: 70.4; [95% CI, 67.0 to 73.9] and 57.1 [95% CI, 51.6 to 62.7], respectively).
Figure 7 shows changes from screening in global health status/QoL. The summary of differences in change from screening is provided in Table 14.
There was a clinically meaningful difference in mean change of scores from screening at study day 100 (estimated difference = 18.1; 95% CI, 12.3 to 23.9; adjusted P < 0.0001) in favour of patients treated with axi-cel compared with patients treated with SOC. At study day 150, the estimated difference was 9.8 (95% CI, 2.6 to 17.0; adjusted P = 0.0124) in favour of patients treated with axi-cel compared with patients treated with SOC. The difference did not reach the trial-specified MID of ± 10 points. By study month 9, the estimated difference between treatment arms was 4.4 (95% CI, –3.3 to 12.0; adjusted P = 0.2655). At study months 12 and 15, the estimated differences between groups were –1.5 (95% CI, –9.6 to 6.6) and –4.9 (95% CI, –13.0 to 3.1), respectively. There was no statistical testing at either time point due to previous failure of the hierarchical testing procedure.
Figure 7: Change From Screening by Visit in EORTC QLQ-C30 Global Health Status/Quality of Life Scores (QoL Analysis Set)
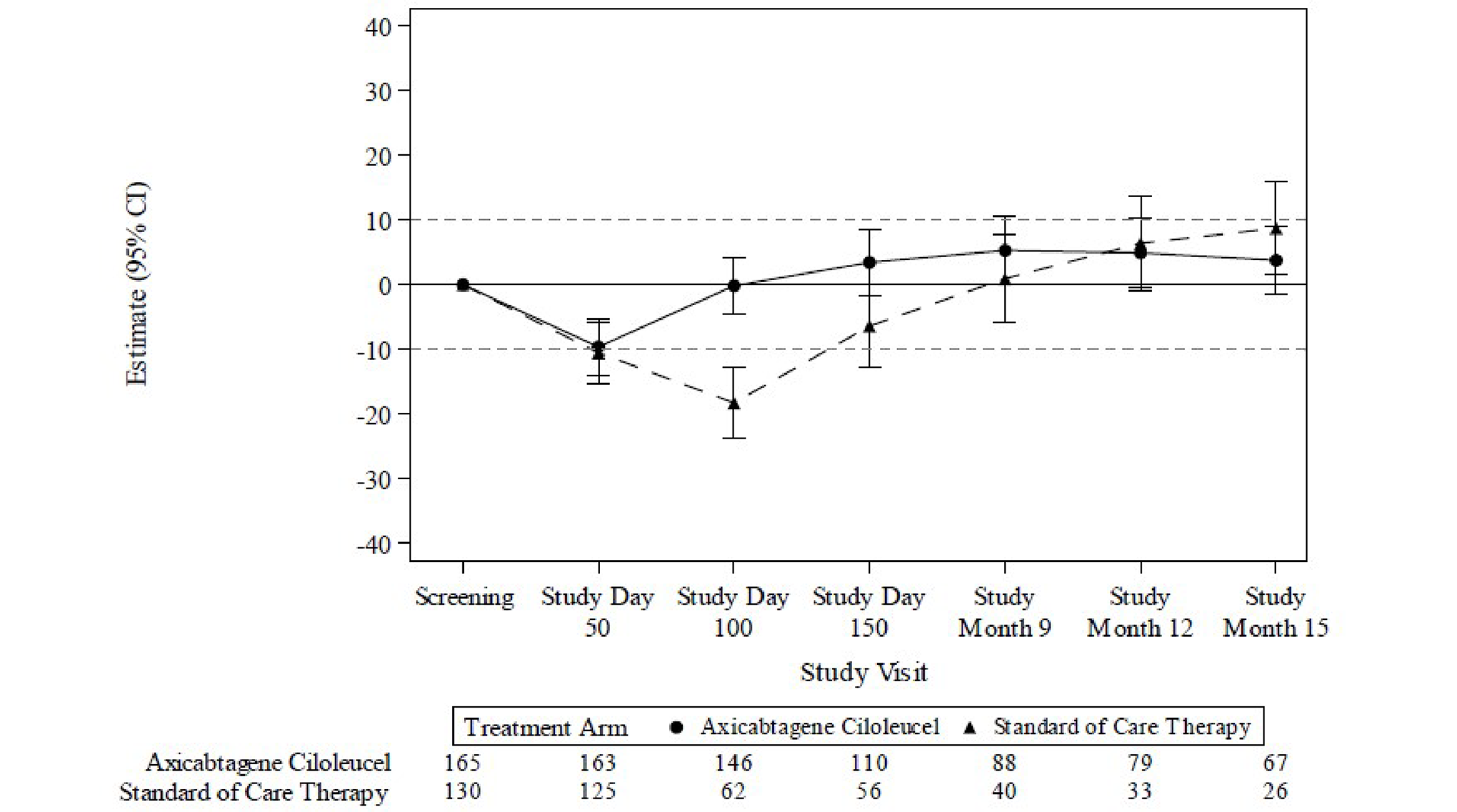
CI = confidence interval; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; QoL = quality of life.
Notes: Study day = number of days since the day of randomization.
Changes from screening in global health status/quality of life scores were estimated using mixed models for repeated measures.
Results populated only through month 15 due to lack of model convergence when using time points. Horizontal lines represent the minimal important difference thresholds for meaningful change and are provided for clarity of interpretation. Model included variables for treatment, time, and treatment by time interaction (primary analysis) and controlled for response to first-line therapy (primary refractory, relapse ≤ 6 months of first-line therapy vs. relapse > 6 and ≤ 12 months of first-line therapy) and age-adjusted International Prognostic Index (0 to 1 vs. 2 to 3) at time of screening.
Data cut-off date: March 18, 2021.
Source: Clinical Study Report for Yescarta.15
Table 14: Summary of Difference in Change From Screening in EORTC QLQ-C30 Global Health Status/QoL Scores (QoL Analysis Set)
Visit | Sample size, n | Estimate of difference between groups (95% CI)a | Unadjusted P valueb | Adjusted P valueb,c | |
|---|---|---|---|---|---|
Axi-cel | SOC | ||||
Day 100 | 146 | 62 | 18.1 (12.3 to 23.9) | < 0.0001 | < 0.0001 |
Day 150 | 110 | 56 | 9.8 (2.6 to 17.0) | 0.0077 | 0.0124 |
Month 9 | 88 | 40 | 4.4 (–3.3 to 12.0) | 0.2655 | 0.2655 |
Month 12 | 79 | 33 | –1.5 (–9.6 to 6.6) | NR | NR |
Month 15 | 67 | 26 | –4.9 (–3.0 to 3.1) | NR | NR |
axi-cel = axicabtagene ciloleucel; CI = confidence interval; EORTC QLC-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; NR = not reported; QoL = quality of life; SOC = standard of care.
Note: Data cut-off date: March 18, 2021.
aEstimated using mixed models for repeated measures. Between-group difference is between the axi-cel and the SOC arms.
bP values only presented for day 100 and only for subsequent visits when the previous visit was statistically significant (P < 0.05).
cAdjusted P values were calculated using the false discovery rate methodology.
Source: Clinical Study Report for Yescarta.15
EORTC QLQ-C30 Physical Functioning
At screening, the mean and median physical functioning scores for evaluable patients in the QoL analysis set were comparable in the axi-cel and SOC arms (mean [SD]: 83.5 [95% CI, 80.8 to 86.2] and 85.3 [95% CI, 82.0 to 88.6], respectively). Mean scores ranged from 70.7 (95% CI, 66.9 to 74.5) on study day 50 to 88.8 (95% CI, 83.0 to 94.6) at month 21 for the axi-cel and between 74.0 (95% CI, 68.7 to 79.4) at study day 100 to 97.2 (95% CI, 94.4 to 100.1) at month 24 in the SOC arm. At study day 50, 48.1% of evaluable patients in the axi-cel arm and 37.3% of patients in the SOC arm reported worsening scores. Starting at study day 100, scores for both treatment arms rebounded, with 19.9% of evaluable patients who received axi-cel and 4.7% of patients who received SOC reporting improved scores compared with screening. Both worsening and improved scores were based on a change of ± 10 points from screening (Figure 8).
For patients in the QoL analysis set treated with axi-cel versus SOC, there was a clinically meaningful difference in mean change of scores from screening to study day 100 (estimated difference = 13.1; 95% CI, 8.0 to 18.2; adjusted P < 0.0001) in favour of axi-cel. At day 150, the estimated difference between groups was 5.1 (95% CI, −0.9 to 11.0; adjusted P = 0.1253). At months 9, 12, and 15, the estimated differences between groups were 3.6 (95% CI, −2.7 to 9.8), −2.7 (95% CI, −9.8 to 4.5), and −2.9 (95% CI, −9.7 to 4.0), respectively. There was no statistical testing at these time points due to previous failure of the hierarchical testing procedure.
Figure 8: EORTC QLQ-C30 Physical Functioning Mixed Model With Repeated Measures for Change From Screening (QoL Analysis Set)
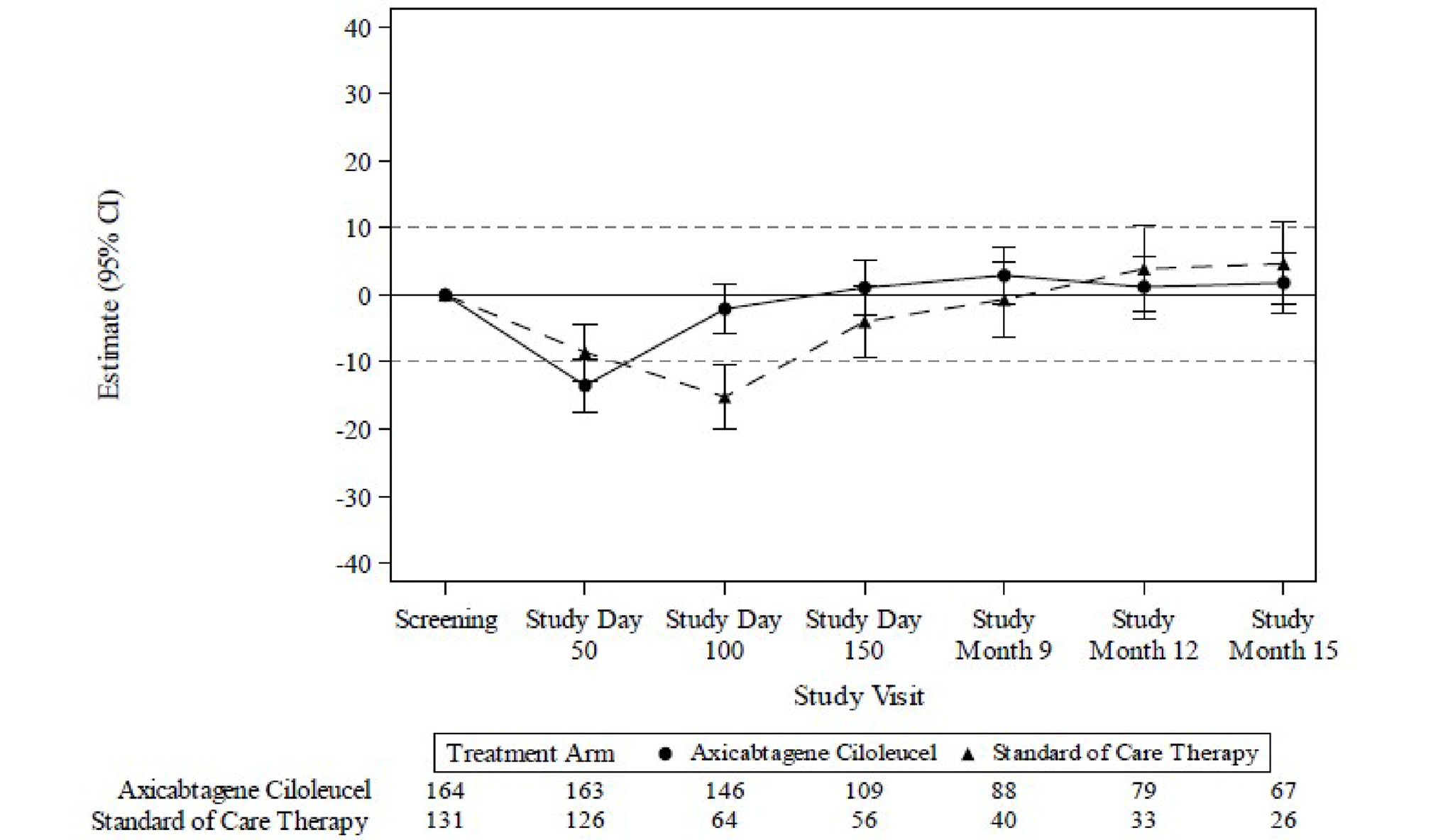
CI = confidence interval; EORTC QLC-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; MMRM = mixed model with repeated measures; QoL = quality of life.
Notes: Study day denotes number of days since the day of randomization. Results populated only through month 15 due to lack of model convergence when using time points. Horizontal lines represent the minimal important difference thresholds for meaningful change and are provided for clarity of interpretation. This model included variables for treatment, time, and treatment by time interaction (primary analysis) and controlled for response to first-line therapy (primary refractory, relapse ≤ 6 months of first-line therapy vs. relapse > 6 and ≤ 12 months of first-line therapy) and age-adjusted International Prognostic Index (0 to 1 vs. 2 to 3) at time of screening.
Data cut-off date: March 18, 2021.
Source: Clinical Study Report for Yescarta.15
EQ-5D-5L VAS
The mean EQ VAS score reported by evaluable patients in the axi-cel and SOC arms were comparable at screening (72.4 [95% CI, 69.5 to 75.2] and 74.4 [95% CI, 70.9 to 77.9], respectively). Based on the MMRM models, there was a clinically meaningful difference in mean change of scores for the EQ-5D-5L VAS from screening in the axi-cel arm compared with SOC at study day 100 (estimated difference = 13.7; 95% CI, 8.5 to 18.8; adjusted P < 0.0001) and study day 150 (estimated difference = 11.3, 95% CI, 5.4 to 17.1; adjusted P = 0.0004). At month 9, the estimated difference between groups was 3.8 (95% CI, –2.3 to 10.0; adjusted P = 0.2549). At months 12 and 15, the estimated differences between groups were 0.3 (95% CI, –6.3 to 6.8) and –3.4 (95% CI, –10.4 to 3.6), respectively. There was no statistical testing at these time points due to previous failure of the hierarchical testing procedure. Figure 9 shows changes from screening in EQ VAS. The summary of difference in change from screening is provided in Table 15.
Figure 9: Change From Screening by Visit in EQ-5D-5L VAS Scores (QoL Analysis Set)
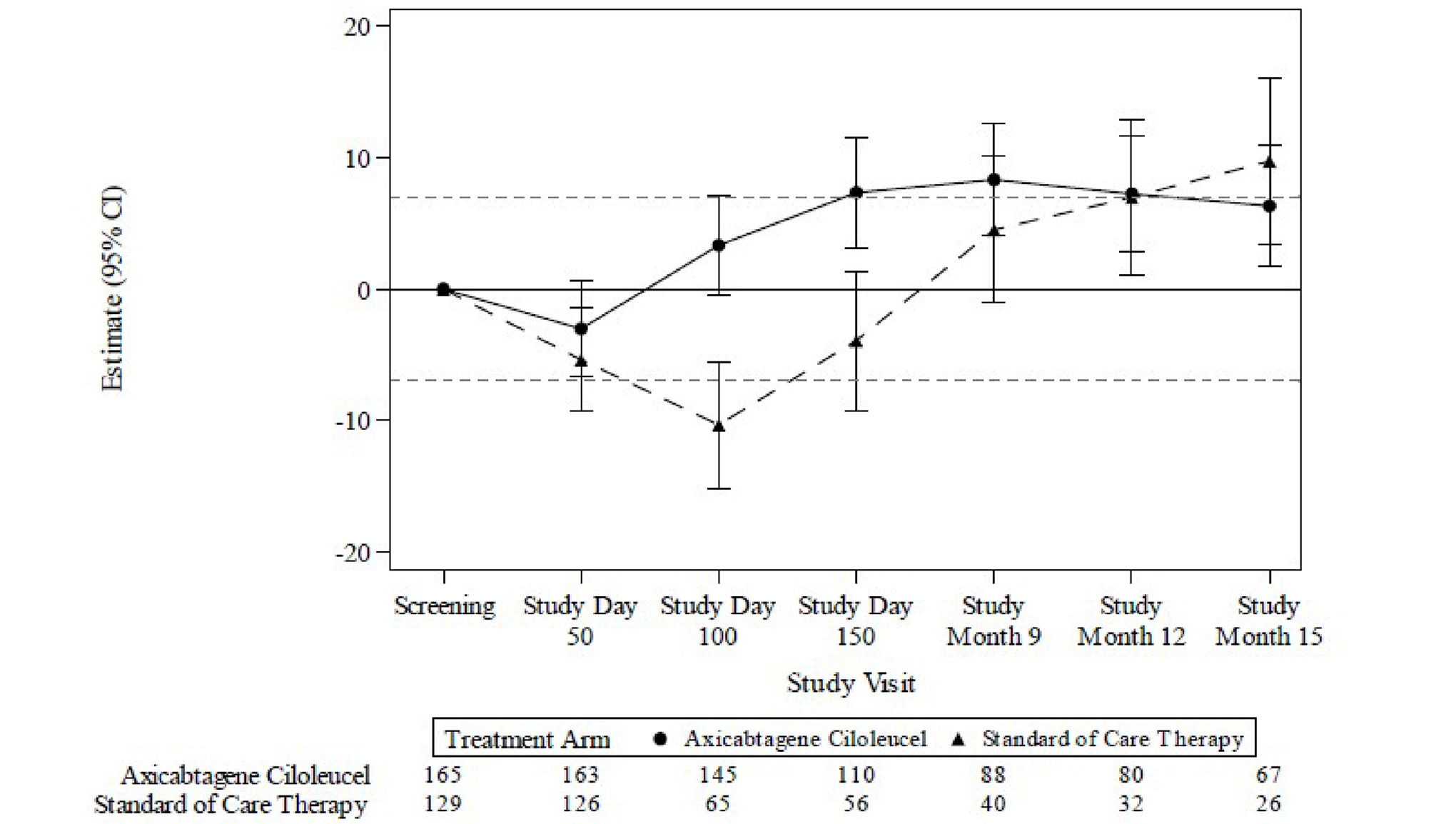
CI = confidence interval; EQ-5D-5L = 5-Level EQ-5D; QoL = quality of life; VAS = visual analogue scale.
Notes: Study day denotes number of days since the day of randomization.
Changes from screening in EQ-5D-5L VAS scores were estimated using mixed models for repeated measures.
Results populated only through month 15 due to lack of model convergence when using time points. Horizontal lines represent the minimal important difference thresholds for meaningful change and are provided for clarity of interpretation. Model included variables for treatment, time, and treatment by time interaction (primary analysis) and controlled for response to first-line therapy (primary refractory, relapse ≤ 6 months of first-line therapy vs. relapse > 6 and ≤ 12 months of first-line therapy) and age-adjusted International Prognostic Index (0 to 1 vs. 2 to 3) at time of screening.
Note: Data cut-off date: March 18, 2021.
Source: Clinical Study Report for Yescarta.15
Table 15: Summary of Difference in Change From Screening in EQ-5D-5L VAS Scores (QoL Analysis Set)
Visit | Sample size, n | Estimate of difference between groups (95% CI)a | Unadjusted P valueb | Adjusted P valueb,c | |
|---|---|---|---|---|---|
Axi-cel | SOC | ||||
Day 100 | 145 | 65 | 13.7 (8.5 to 18.8) | < 0.0001 | < 0.0001 |
Day 150 | 110 | 56 | 11.3 (5.4 to 17.1) | 0.0002 | 0.0004 |
Month 9 | 88 | 40 | 3.8 (–2.3 to 10.0) | 0.2230 | 0.2549 |
Month 12 | 80 | 32 | 0.3 (–6.3 to 6.8) | NR | NR |
Month 15 | 67 | 26 | –3.4 (–10.4 to 3.6) | NR | NR |
axi-cel = axicabtagene ciloleucel; CI = confidence interval; EQ-5D-5L = 5-Level EQ-5D; NR = not reported; QoL = quality of life; SOC = standard of care; VAS = visual analogue scale.
Note: Data cut-off date: March 18, 2021.
aEstimated using mixed models for repeated measures. Between-group difference is between the axi-cel and the SOC arms.
bP values only presented for study day 100 and only for subsequent visits when the previous visit was statistically significant (P < 0.05).
cAdjusted P values were calculated using the false discovery rate methodology.
Source: Clinical Study Report for Yescarta.15
Progression-Free Survival
At the data cut-off date, the median follow-up time was 21.6 months (95% CI, 20.7 to 23.7 months) in the axi-cel arm and 12.1 months (95% CI, 3.7 months to 18.3 months) in the SOC arm. The median duration of PFS was 14.9 months (95% CI, 7.2 months to NE) in the axi-cel arm and 5.0 months (95% CI, 3.4 to 8.5 months) in the SOC arm, with a stratified HR of 0.562 (95% CI, 0.414 to 0.762) (Table 16).
The Kaplan-Meier estimates of PFS rates at 12 and 24 months from randomization were 53.6% (95% CI, 45.8% to 60.7%) and 46.3% (95% CI, 38.5% to 53.7%), respectively, in the axi-cel arm and 32.3% (95% CI, 23.5% to 41.4%) and 30.9% (95% CI, 22.2% to 40.1%), respectively, in the SOC arm.
Table 16: Progression-Free Survival Per Central Assessment (Full Analysis Set)
Progression-free survival | Axi-cel (N = 180) | SOC (N = 179) |
|---|---|---|
Events, n (%) | 93 (52) | 81 (45) |
Censored, n (%) | 87 (48) | 98 (55) |
HR (95% CI), stratifieda,b | 0.562 (0.414 to 0.762) | |
PFS (months), median (95% CI)c | 14.9 (7.2 to NE) | 5.0 (3.4 to 8.5) |
Event, n (%) | ||
Disease progression | 82 (46) | 75 (42) |
Death from any cause | 11 (6) | 6 (3) |
Censoring reasons, n (%) | ||
Response ongoing | 76 (42) | 28 (16) |
New lymphoma therapy | 9 (5) | 61 (34) |
Subsequent stem cell transplant | 0 (0) | 2 (1) |
Axi-cel re-treatment | 2 (1) | 0 (0) |
Response assessed but no disease at baseline and post baseline | 0 (0) | 3 (2) |
No post-baseline disease assessment | 0 (0) | 1 (1) |
Full withdrawal of consent | 0 (0) | 1 (1) |
Lost to follow-up | 0 (0) | 2 (1) |
Follow-up time (months), median (95% CI)d | 21.6 (20.7 to 23.7) | 12.1 (3.7 to 18.3) |
axi-cel = axicabtagene ciloleucel; CI = confidence interval; HR = hazard ratio; NE = not estimable; PFS = progression-free survival; SOC = standard of care.
Note: Data cut-off date: March 18, 2021.
aThe stratification factors are responses to first-line therapy (primary refractory vs. relapse ≤ 6 months of first-line therapy vs. relapse > 6 and ≤ 12 months of first-line therapy) and second-line age-adjusted International Prognostic Index (0 to 1 vs. 2 to 3) as collected via interactive voice/web response system.
bHR and 95% CIs were estimated using Cox regression models for axi-cel relative to SOC. The Breslow method was used to handle ties for the Cox regression models.
cEstimated using the Kaplan-Meier method.
dEstimated using the reverse Kaplan-Meier approach.
Source: Clinical Study Report for Yescarta.15
Objective Response Rate
The difference in ORR between arms was tested statistically following demonstration of a significant improvement in EFS favouring axi-cel compared with SOC. Based on the a priori alpha spending function, an alpha of 0.025 was allocated to the analysis. The ORR (CR or PR) was 83% (95% CI, 77.1% to 88.5%) in the axi-cel arm and 50% (95% CI, 42.7% to 57.8%) in the SOC arm (difference in ORR = 33.1%; 95% CI, 23.2% to 42.1%; P < 0.0001).
The CR rates in the axi-cel arm and the SOC arm were 65% (95% CI, 57.6% to 71.9%; n = 117) and 32% (95% CI, 25.6% to 39.8%; n = 58), respectively. The PR rates were 18% (95% CI, 13.0% to 24.8%; n = 33) and 18% (95% CI, 12.6% to 24.3%; n = 32), respectively.
Table 17: Objective Response Rate and Best Overall Response Per Central Assessment (Full Analysis Set)
Objective response | Axi-cel (N = 180) | SOC (N = 179) |
|---|---|---|
Objective responders (CR + PR), n (%) | 150 (83) | 90 (50) |
95% CI for ORR, % | 77.1 to 88.5 | 42.7 to 57.8 |
Difference in ORR (95% CI), %a | 33.1 (23.2 to 42.1) | |
Stratified P valueb,c | < 0.0001 | |
CR, n (%) | 117 (65) | 58 (32) |
95% CI for response rate, %d | 57.6 to 71.9 | 25.6 to 39.8 |
PR, n (%) | 33 (18) | 32 (18) |
95% CI for response rate, %d | 13.0 to 24.8 | 12.6 to 24.3 |
Stable disease, n (%) | 5 (3) | 33 (18) |
95% CI for response rate, %d | 0.9 to 6.4 | 13.0 to 24.9 |
Progressive disease, n (%) | 21 (12) | 38 (21) |
95% CI for response rate, %d | 7.4 to 17.3 | 15.5 to 28.0 |
Undefined/no disease, n (%) | 0 (0) | 4 (2) |
95% CI for response rate, %d | 0.0 to 2.0 | 0.6 to 5.6 |
Not evaluable, n (%) | 0 (0) | 0 (0) |
95% CI for response rate, %d | 0.0 to 2.0 | 0.0 to 2.0 |
Not done, n (%) | 4 (2) | 14 (8) |
95% CI for response rate, %d | 0.6 to 5.6 | 4.3 to 12.8 |
axi-cel = axicabtagene ciloleucel; CI = confidence interval; CR = complete response; ORR = objective response rate; PR = partial response; SOC = standard of care.
Note: Data cut-off date: March 18, 2021.
a95% CI for the difference in ORR (with the SOC arm as reference group) was from the Wilson score method with continuity correction.
bThe stratification factors are responses to first-line therapy (primary refractory vs. relapse ≤ 6 months of first-line therapy vs. relapse > 6 and ≤ 12 months of first-line therapy) and second-line age-adjusted International Prognostic Index (0 to 1 vs. 2 to 3) as collected via interactive voice/web response system.
cOne-sided P value from the Cochran-Mantel-Haenszel test. An alpha of 0.025 was allocated to this analysis.
dEstimated using the Clopper-Pearson method.
Source: Clinical Study Report for Yescarta.15
Duration of Response
The median time from randomization to the first objective response for patients who achieved a CR or PR per central assessment in the axi-cel arm (150 of 180 patients) or the SOC arm (90 of 179 patients) was 2.04 months (range, 1.5 months to 18.0 months) and 1.58 months (range, 1.1 to 5.3 months), respectively.
DOR was analyzed for the 150 patients in the axi-cel arm and the 90 patients in the SOC arm who achieved an objective response of CR or PR. Median follow-up time was 19.5 months (95% CI, 18.2 to 21.7 months) in the axi-cel arm and 17.3 months (95% CI, 12.7 to 19.6 months) in the SOC arm. The Kaplan-Meier median DOR for the axi-cel arm was 26.9 months (95% CI, 13.6 months to NE) compared with 8.9 months (95% CI, 5.7 months to NE) for the SOC arm (stratified HR = 0.736; 95% CI, 0.488 to 1.108).
The Kaplan-Meier estimates of the percentage of patients who remained in response at 12 and 24 months from first objective response were 60.9% (95% CI, 52.4% to 68.4%) and 54.0% (95% CI, 45.1% to 62.0%), respectively, in the axi-cel arm compared with 47.6% (95% CI, 35.2% to 58.9%) and 45.6% (95% CI, 33.2% to 57.1%), respectively, in the SOC arm.
Table 18: Duration of Response Per Central Assessment (Full Analysis Set)
Duration of response | Axi-cel (N = 180) | SOC (N = 179) |
|---|---|---|
Number of objective responders (CR + PR), n | 150 | 90 |
Events, n (%) | 66 (44) | 37 (41) |
Censored, n (%) | 84 (56) | 53 (59) |
HR (95% CI), stratifieda,b | 0.736 (0.488 to 1.108) | |
P value, stratifieda,c | 0.0695 | |
DOR (months), median (95% CI)d | 26.9 (13.6 to NE) | 8.9 (5.7 to NE) |
Events, n (%) | ||
Disease progression | 58 (39) | 34 (38) |
Death from any cause | 8 (5) | 3 (3) |
Censoring reasons, n (%) | ||
Response ongoing | 76 (51) | 28 (31) |
New lymphoma therapy | 6 (4) | 23 (26) |
Subsequent stem cell transplant | 0 (0) | 1 (1) |
Axi-cel re-treatment | 2 (1) | 0 (0) |
Lost to follow-up | 0 (0) | 1 (1) |
Follow-up time (months), median (95% CI)e | 19.5 (18.2 to 21.7) | 17.3 (12.7 to 19.6) |
axi-cel = axicabtagene ciloleucel; CI = confidence interval; CR = complete response; DOR = duration of response; HR = hazard ratio; NE = not estimable; PR = partial response; SOC = standard of care.
Notes: Percentages are based on number of patients in the analysis set with objective response.
Data cut-off date: March 18, 2021.
aThe stratification factors are responses to first-line therapy (primary refractory vs. relapse ≤ 6 months of first-line therapy vs. relapse > 6 and ≤ 12 months of first-line therapy) and second-line age-adjusted International Prognostic Index (0 to 1 vs. 2 to 3) as collected via interactive voice/web response system.
bHR and 95% CIs were estimated using Cox regression models for axi-cel relative to SOC.
cOne-sided P value is based on log-rank test. No multiplicity adjustment was performed.
dEstimated using the Kaplan-Meier method.
eEstimated via the reverse Kaplan-Meier approach.
Source: Clinical Study Report for Yescarta.15
Time to Next Treatment
Time to next treatment events occurred for 99 patients (55%) in the axi-cel arm and 135 patients (75%) in the SOC arm. The median follow time was 24.6 months (95% CI, 22.0 to 25.4 months) in the axi-cel arm and 22.8 months (95% CI, 21.0 to 24.6 months) in the SOC arm. The median time to next treatment was 14.7 months (95% CI, 6.5 months to NE) in the axi-cel arm and 3.4 months (95% CI, 3.1 to 4.4 months) in the SOC arm (stratified HR = 0.430; 95% CI, 0.329 to 0.560; descriptive log-rank P value < 0.0001).
The Kaplan-Meier estimates of the percentage of patients with events 12 and 24 months from randomization were 51.7% (95% CI, 44.1% to 58.7%) and 45.0% (95% CI, 37.6% to 52.2%), respectively, in the axi-cel arm, and 24.2% (95% CI, 18.1% to 30.8%) and 21.1% (95% CI, 15.4% to 27.6%), respectively, in the SOC arm.
Table 19: Time to Next Treatment Per Central Assessment (Full Analysis Set)
Time to next treatment | Axi-cel (N = 180) | SOC (N = 179) |
|---|---|---|
Events, n (%) | 99 (55) | 135 (75) |
Censored, n (%) | 81 (45) | 44 (25) |
HR (95% CI), stratifieda,b | 0.430 (0.329 to 0.560) | |
P value, stratifieda,c | < 0.0001 | |
Time to next treatment (months), median (95% CI)d | 14.7 (6.5 to NE) | 3.4 (3.1 to 4.4) |
Event, n (%) | ||
Subsequent new lymphoma therapy | 75 (42) | 124 (69) |
Axi-cel re-treatment | 8 (4) | 0 (0) |
Subsequent stem cell transplant | 1 (1) | 3 (2) |
Death from any cause | 15 (8) | 8 (4) |
Censoring reasons, n (%) | ||
Did not receive subsequent therapy and still alive | 81 (45) | 35 (20) |
Full withdrawal of consent | 0 (0) | 6 (3) |
Lost to follow-up | 0 (0) | 2 (1) |
End of study due to investigator decision | 0 (0) | 1 (1) |
Follow-up time (months), median (95% CI)e | 24.6 (22.0 to 25.4) | 22.8 (21.0 to 24.6) |
axi-cel = axicabtagene ciloleucel; CI = confidence interval; HR = hazard ratio; NE = not estimable; SOC = standard of care.
Note: Data cut-off date: March 18, 2021.
aThe stratification factors are responses to first-line therapy (primary refractory vs. relapse ≤ 6 months of first-line therapy vs. relapse > 6 and ≤ 12 months of first-line therapy) and second-line age-adjusted International Prognostic Index (0 to 1 vs. 2 to 3) as collected via interactive voice/web response system.
bHR and 95% CIs were estimated using Cox regression models for axi-cel relative to SOC.
cBased on the log-rank test. Not adjusted for multiple comparisons.
dEstimated using the Kaplan-Meier method.
eEstimated via the reverse Kaplan-Meier approach.
Source: Clinical Study Report for Yescarta.15
Health Care Resource Utilization
A total of 42 patients (25%) in the axi-cel arm and 9 patients (5%) in the SOC arm were admitted to the ICU. Median duration of ICU hospitalization was 5 days (range, 1 to 12 days) in the axi-cel arm and 3 days (range, 2 to 17 days) in the SOC arm. Median duration of hospitalization for axi-cel infusion was 16 days (range, 5 to 103 days); median duration of inpatient hospitalization for stem cell transplant in the SOC arm was 21 days (range, 1 to 53 days).14
Harms
Adverse Events
All patients in the axi-cel arm (170 patients [100%]) and the SOC arm (168 patients [100%]) had at least 1 TEAE, and 155 patients (91%) in the axi-cel arm and 140 patients (83%) in the SOC arm had worst grade 3 or higher TEAEs.
TEAEs of any grade that were most frequently (in ≥ 30% of patients) reported in each treatment arm were as follows:
Axi-cel arm: pyrexia (158 patients; 93%); neutropenia and hypotension (75 patients each; 44%); anemia, fatigue, and diarrhea (71 patients each; 42%); headache (70 patients; 41%); nausea (69 patients; 41%), sinus tachycardia (58 patients; 34%); and neutrophil count decreased (52 patients; 31%)
SOC arm: nausea (116 patients; 69%); anemia (91 patients; 54%); fatigue (87 patients; 52%); diarrhea (66 patients; 39%); platelet count decreased (64 patients; 38%); constipation (58 patients; 35%); and vomiting (55 patients; 33%).
The most frequently reported TEAEs of worst grade 3 or higher (reported in ≥ 20% of patients in either treatment arm) were as follows:
Axi-cel arm: neutropenia (73 patients; 43%); anemia (51 patients; 30%); neutrophil count decreased (49 patients; 29%); and white blood cell count decreased (43 patients; 25%)
SOC arm: anemia (65 patients; 39%); platelet count decreased (60 patients; 36%); neutrophil count decreased (47 patients; 28%); febrile neutropenia (46 patients; 27%); and thrombocytopenia (37 patients; 22%).
Table 20: Summary of Adverse Events (Safety Analysis Set)
Eventa | Axi-cel (N = 170) n (%) | SOC (N = 168) n (%) |
|---|---|---|
Any TEAE | 170 (100) | 168 (100) |
Grade ≥ 3 | 155 (91) | 140 (83) |
Grade 5, excluding PD | 7 (4) | 2 (1) |
Any serious TEAE | 85 (50) | 77 (46) |
Grade ≥ 3 | 72 (42) | 67 (40) |
Grade 5, excluding PD | 7 (4) | 2 (1) |
Treatment-related TEAE | 163 (96) | 160 (95) |
Grade ≥ 3 | 112 (66) | 131 (78) |
Grade 5, excluding PD | 1 (1) | 2 (1) |
Any treatment-emergent neurologic event | 102 (60) | 33 (20) |
Grade ≥ 3 | 36 (21) | 1 (1) |
Any treatment-emergent CRS | 157 (92) | NA |
Grade ≥ 3 | 11 (6) | NA |
Any treatment-emergent cytopenias | 136 (80) | 135 (80) |
Any treatment-emergent infections | 70 (41) | 51 (30) |
Any treatment-emergent hypogammaglobulinemia | 19 (11) | 1 (1) |
Withdrawal due to AEs | 0 (0) | 2 (1) |
Death due to an AE | 6 (4) | 2 (1) |
AE = adverse event; axi-cel = axicabtagene ciloleucel; CRS = cytokine release syndrome; NA = not applicable; PD = progressive disease; SOC = standard of care; TEAE = treatment-emergent adverse event.
Note: TEAE includes all AEs with an onset on or after the axi-cel infusion date in the axi-cel arm or the first dose of salvage chemotherapy in the SOC arm.
aAEs are graded per Common Terminology Criteria for Adverse Events Version 4.03, and CRS events are graded according to a modified grading system proposed by Lee et al. (2014).50
Source: Clinical Study Report for Yescarta.15
Serious Adverse Events
A total of 85 patients (50%) in the axi-cel arm and 77 patients (46%) in the SOC arm had at least 1 SAE. The most frequently (in ≥ 5% of patients) reported SAEs of any grade in each treatment arm were as follows:
Axi-cel arm: pyrexia (27 patients; 16%), encephalopathy (17 patients; 10%), hypotension (15 patients; 9%), aphasia (9 patients; 5%), and pneumonia (8 patients; 5%)
SOC arm: febrile neutropenia (22 patients; 13%), and acute kidney injury and pyrexia (8 patients each; 5%)
Seventy-two patients (42%) in the axi-cel arm and 67 patients (40%) in the SOC arm had worst grade 3 or higher SAEs. The most frequently (in > 2% of patients) reported worst grade 3 or higher SAEs, excluding B-cell lymphoma, in each treatment arm were as follows:
Axi-cel arm: encephalopathy (15 patients; 9%), aphasia (8 patients; 5%), hypotension (7 patients; 4%), and pneumonia (6 patients, 4%)
SOC arm: febrile neutropenia (22 patients; 13%) and platelet count decreased (5 patients; 3%).
Withdrawals Due to Adverse Events
No patient discontinued treatment due to TEAEs in the axi-cel arm. Two patients in the SOC arm discontinued treatment due to TEAEs of grade 4 acute kidney injury and grade 1 blood stem cell harvest failure.
AEs Related to Either Leukapheresis or Lymphodepleting Chemotherapy (Axi-Cel Arm)
At least 1 AE related to leukapheresis (for T cell collection) was reported for 27 of 170 patients (16%) who later received axi-cel, including 5 patients (3%) with worst grade 3 or higher leukapheresis-related AEs. One patient (1%) had a worst grade 4 AE, and no patients had a grade 5 AE related to leukapheresis. At least 1 AE related to lymphodepleting chemotherapy was reported for 151 of 170 patients (89%) who later received axi-cel, including 130 patients (76%) with worst grade 3 or higher lymphodepleting chemotherapy-related AEs. Worst grade 4 lymphodepleting chemotherapy-related AEs were reported for 109 patients (64%), and 1 patient (1%) had a grade 5 AE of progressive multifocal leukoencephalopathy. The 3 most common AEs of any grade reported as related to lymphodepleting chemotherapy were nausea and neutropenia (63 patients each; 37%), and anemia (59 patients; 35%). The 3 most common worst grade 3 or higher lymphodepleting chemotherapy-related AEs were neutropenia (61 patients; 36%), anemia (45 patients; 26%), and neutrophil count decreased (42 patients; 25%).
TEAEs Related to Either Salvage Chemotherapy, HDT, or ASCT (SOC Arm)
At least 1 TEAE related to salvage chemotherapy was reported for 160 of 168 patients (95%) in the SOC arm, including 121 patients (72%) with a worst grade 3 or higher salvage chemotherapy-related TEAE. Worst grade 4 TEAEs were reported for 79 patients (47%), and no patients had a grade 5 TEAE. The 3 most common salvage chemotherapy-related TEAEs of any grade were nausea (95 patients; 57%), fatigue (73 patients; 43%), and anemia (70 patients; 42%). The 3 most common worst grade 3 or higher salvage chemotherapy-related TEAEs were anemia (50 patients; 30%), platelet count decreased (41 patients; 24%), and neutrophil count decreased (32 patients; 19%). Following salvage chemotherapy, 74 patients underwent leukapheresis for CD34+ stem cell collection. At least 1 TEAE related to leukapheresis was reported for 7 patients, including 2 patients with a worst grade 3 or higher leukapheresis-related TEAE. Worst grade 4 TEAEs were reported for 1 patient, and no patient had a grade 5 TEAE that was deemed related to leukapheresis for CD34+ stem cell collection.
Following leukapheresis, 64 patients received HDT. At least 1 HDT-related TEAE was reported for 58 patients, including 52 patients with a worst grade 3 or higher TEAE. Worst grade 4 HDT-related TEAEs were reported for 45 patients, and 2 patients had a grade 5 TEAE of cardiac arrest and acute respiratory distress syndrome. Following HDT, 62 patients received on-protocol CD34+ stem cell infusion. At least 1 CD34+ stem cell infusion–related TEAE was reported for 13 of the 62 patients (21%) who received ASCT in the SOC arm, including 6 patients (10%) with a worst grade 3 or higher TEAE. Worst grade 4 CD34+ stem cell infusion–related TEAEs were reported for 5 patients (8%). No patients had a grade 5 TEAE.
Mortality
In the axi-cel arm, 64 patients (38%) had died at the data cut-off date due to reasons including PD (n = 47; 28%), TEAEs (n = 6; 4%), and other reasons (n = 10; 6%) and 1 patient (1%) died from an event reported by the investigator as a “secondary malignancy” (lung adenocarcinoma) although deemed unrelated to axi-cel treatment (this event was reported as a grade 5 TEAE). In the SOC arm, 78 patients (46%) had died at the data cut-off date due to reasons including PD (n = 64; 38%), TEAEs (n = 2; 1%), and other reasons (n = 12; 7%).
Of the 64 patients in the axi-cel arm who died, 6 died more than 30 days but less than 3 months after the axi-cel infusion, and 58 died more than 3 months after the axi-cel infusion. Of the 78 patients in the SOC arm who died, 3 died more than 30 days but less than 3 months after the first dose of salvage chemotherapy, and 75 died more than 3 months after the first dose of salvage chemotherapy.
Six patients in the axi-cel arm and 2 patients in the SOC arm died due to TEAEs which included myocardial infarction, progressive multifocal leukoencephalopathy, hepatitis B reactivation, sepsis, and COVID19 in the axi-cel arm, and cardiac arrest and acute respiratory distress syndrome in the SOC arm (Table 22).
Table 21: Summary of Deaths (Safety Analysis Set)
Deaths | Axi-cel (N = 170) n (%) | SOC (N = 168) n (%) |
|---|---|---|
Patients who died | 64 (38) | 78 (46) |
Deaths that occurred ≤ 30 days from axi-cel infusion or first dose of SOC salvage chemotherapy | 0 (0) | 0 (0) |
Deaths that occurred > 30 days through 3 months (92 days) from axi-cel infusion or first dose of SOC salvage chemotherapy | 6 (4) | 3 (2) |
Deaths that occurred > 3 months (> 92 days) after axi-cel infusion or first dose of SOC salvage chemotherapy | 58 (34) | 75 (45) |
Primary cause of death | ||
Adverse event | 6 (4) | 2 (1) |
COVID-19 | 2 (1) | 0 (0) |
Progressive disease | 47 (28) | 64 (38) |
Secondary malignancya | 1 (1) | 0 (0) |
Other | 10 (6) | 12 (7) |
COVID-19 | 2 (1)b | 2 (1) |
axi-cel = axicabtagene ciloleucel; SOC = standard of care.
Note: Data cut-off date: March 18, 2021.
aOne death was reported by the investigator as a “secondary malignancy” although deemed unrelated to study therapy. The sponsor’s medical review considered the event to be a new malignancy unrelated to study therapy. (Note: this event was reported as a grade 5 treatment-emergent adverse event.)
bCOVID-19 infection was reported as an adverse event if it occurred during the adverse event reporting window and as “other” if it occurred outside the adverse event reporting window.
Source: Clinical Study Report for Yescarta.15
Table 22: Deaths Due to Adverse Events (Safety Analysis Set)
Treatment arm | Primary cause of death | Therapy day of death |
|---|---|---|
Axi-cel | Myocardial infarction | 53 |
PML | 207 | |
Hepatitis B reactivation | 422 | |
Sepsis | 442 | |
COVID-19 | 278 | |
COVID-19 | 275 | |
SOC | Cardiac arrest | 146 |
Acute respiratory distress syndrome | 161 |
axi-cel = axicabtagene ciloleucel; PML = progressive multifocal leukoencephalopathy; SOC = standard of care.
Notes: Deaths were to be reported as serious adverse events if they occurred after randomization and before the study day 150 visit or until the initiation of new lymphoma therapy (whichever occurred first), regardless of attribution to treatment. Targeted SAEs (neurologic events, hematologic events, infections, autoimmune disorders, and secondary malignancies) were to be reported for up to 5 years for the SOC arm and up to 15 years for the axi-cel arm, or until disease progression.
Data cut-off date: March 18, 2021.
Source: Clinical Study Report for Yescarta.15
Notable Harms
Neurologic Events
In the safety analysis set (N = 338), 102 patients (60%) in the axi-cel arm and 33 patients (20%) in the SOC arm had at least 1 treatment-emergent neurologic event, including 36 patients (21%) and 1 patient (1%), respectively, with worst grade 3 or higher neurologic events. Of these, 10 patients (6%) in the axi-cel arm had worst grade 4 neurologic events; no patients in either treatment arm had a grade 5 neurologic event. The most frequently (in ≥ 10% of patients) reported treatment-emergent neurologic events of any grade in the axi-cel arm were tremor (44 patients; 26%), confusional state (40 patients; 24%), aphasia (36 patients; 21%), encephalopathy (29 patients; 17%), and somnolence (19 patients; 11%). No neurologic events occurred with a patient incidence higher than 10% in the SOC arm; the most frequently (in ≥ 2% of patients) reported treatment-emergent neurologic events in the SOC arm were paresthesia (14 patients; 8%), delirium (5 patients; 3%), and confusional state (4 patients; 2%).The most frequently (in ≥ 5% of patients) reported worst grade 3 or higher treatment-emergent neurologic events in the axi-cel arm were encephalopathy (20 patients, 12%), aphasia (12 patients; 7%), and confusional state (9 patients, 5%). One patient (1%) in the SOC arm had a grade 3 or higher treatment-emergent neurologic event of delirium.
Serious treatment-emergent neurologic events of any grade were reported for 34 patients (20%) in the axi-cel arm and 1 patient (1%) in the SOC arm, including 26 patients (15%) in the axi-cel arm with a serious worst grade 3 or higher neurologic event and 1 patient (1%) in the SOC arm with a serious worst grade 2 neurologic event. The 3 most common serious treatment-emergent neurologic events of any grade in the axi-cel arm were encephalopathy (17 patients; 10%), aphasia (9 patients; 5%), and confusional state (6 patients; 4%). The only serious neurologic event in the SOC arm was encephalopathy.
Among patients who had a treatment-emergent neurologic event, the median time to onset was 7.0 days (range, 1 to 133 days) after the axi-cel infusion and 23.0 days (range, 1 to 108 days) after the first dose of SOC salvage chemotherapy. At the data cut-off date, neurologic events had resolved in 96 of the 102 patients in the axi-cel arm and in 32 of the 33 patients in the SOC arm; the median duration of neurologic events was 8.5 days (range, 1 to 817 days) for patients in the axi-cel arm and 23.0 days (range, 1 to 253 days) for the patients in the SOC arm.
A total of 6 patients in the axi-cel arm and 1 patient in the SOC arm had ongoing neurologic events at the data cut-off date or unresolved neurologic events at the time of death.
Cytokine Release Syndrome
All cases of CRS were considered related to axi-cel. CRS of any grade was reported for 157 patients (92%), including 11 patients (6%) who had worst grade 3 or higher CRS. No patient had grade 5 CRS. The most frequently reported CRS symptoms (in ≥ 30% of patients with CRS) were pyrexia (155 patients; 99%), hypotension (68 patients; 43%), and sinus tachycardia (49 patients; 31%). The most frequently reported worst grade 3 or higher CRS symptoms (in ≥ 5% of patients with CRS) were hypotension (18 patients; 11%), pyrexia (14 patients; 9%), and hypoxia (13 patients; 8%).
Cytopenias
The most common cytopenias of any grade reported in the axi-cel arm were thrombocytopenia (50 patients; 29%), neutropenia (122 patients; 72%), and anemia (73 patients; 43%). The most common cytopenias of any grade reported in the SOC arm were thrombocytopenia (101 patients; 60%), neutropenia (92 patients; 55%), and anemia (92 patients; 55%). Grade 3 or higher cytopenias reported in the axi-cel arm were thrombocytopenia (25 patients; 15%), neutropenia (119 patients; 70%), and anemia (51 patients; 30%). Grade 3 or higher cytopenias reported in the SOC arm were thrombocytopenia (95 patients; 57%), neutropenia (91 patients; 54%), and anemia (65 patients; 39%).
Infections
Seventy patients (41%) in the axi-cel arm and 51 patients (30%) in the SOC arm had at least 1 treatment-emergent infection, including 24 patients (14%) and 19 patients (11%), respectively, with worst grade 3 or higher infections. Three patients (2%) in the axi-cel arm and 6 patients (4%) in the SOC arm had worst grade 4 infections. Five patients (3%) in the axi-cel arm had a grade 5 TEAE of infection (2 patients with COVID-19, 1 patient with progressive multifocal leukoencephalopathy, 1 patient with hepatitis B reactivation, and 1 patient with sepsis). No patients in the SOC arm had a grade 5 TEAE of infection.
The most frequently (in ≥ 5% of patients) reported infection categories were unspecified (44 patients; 26%), viral infections (26 patients; 15%), bacterial infections (16 patients; 9%), upper respiratory tract infections (11 patients; 6%), and opportunistic infections (8 patients; 5%) in the axi-cel arm and unspecified (40 patients; 24%), bacterial infections (15 patients; 9%), and viral infections (8 patients; 5%) in the SOC arm.
The most frequently (in ≥ 2% of patients) reported worst grade 3 or higher treatment-emergent infections by preferred term (excluding COVID-19) were pneumonia (6 patients; 4%) and upper respiratory tract infection (3 patients; 2%) in the axi-cel arm, and pneumonia and sepsis (4 patients each; 2%) in the SOC arm.
Secondary Malignancies
No new malignancies were considered by the sponsor to be secondary to axi-cel treatment (new malignancies considered not to be secondary malignancies by the sponsor included events of the primary malignancy, nonmalignant events such as tumour pain, nonmelanoma skin cancer, events of treatment-related acute myeloid leukemia (t-AML) and treatment-related myelodysplastic syndrome (t-MDS) considered related to underlying genetic predisposition or prior chemotherapy [i.e., chemotherapy administered before axi-cel infusion or the first dose of SOC], and other new malignancies with alternative etiologies).
Critical Appraisal
Internal Validity
In ZUMA-7, treatment assignment was based on a central randomization scheme, which would ensure concealment of the randomized groups until allocation. Randomization was stratified according to response to first-line therapy and sAAIPI, which are important prognostic factors in this setting and ensured that the 2 treatment arms were balanced in the proportion of patients with primary refractory versus relapsed lymphoma and low versus high sAAIPI total score. Overall, the 2 treatment arms were balanced in terms of baseline demographic and disease characteristics. The 10% difference in the proportion of male patients between the 2 arms is unlikely to present a high risk of bias due to the randomization.
The trial was open label. Despite the open-label design, there is low risk of bias in the measurement of outcomes such as EFS, PFS, and ORR, which were assessed via independent blinded radiologic review of disease response, as well as OS, which is an objective outcome. For subjective outcomes like HRQoL and some harms outcomes, the patient’s knowledge of their assigned treatment could result in overestimation or underestimation. Of note, 4% of the enrolled patients in the SOC arm declined to proceed with the study compared to none in the axi-cel arm which could indicate patient bias against SOC therapy, possibly due to perceived lack of efficacy. In addition, although the EORTC QLQ-C30 and EQ-5D-5L are comprehensive and widely used instruments designed to measure HRQoL in the general population and in patient groups with diverse chronic diseases, both are currently not validated in patients with LBCL. Further, at most time points for the analysis of HRQoL, relatively few patients completed the assessment and the amount of missing data was imbalanced between the groups, so the results are at a high risk of attrition bias.
The study was powered (based on the full analysis set, N = 359) to test its primary end point, EFS. The statistical approach of sequentially testing the primary and secondary end points was acceptable to account for multiple testing across these analyses. Of note, the subgroup analyses, although prespecified, were not adjusted for multiple comparisons, there was no test of between subgroup differences, and the trial was not powered to detect subgroup differences. Strong conclusions should not be drawn from these data. The primary and secondary efficacy end points of EFS, ORR, and OS are considered appropriate for the disease setting. There is some evidence from the literature that EFS is a robust surrogate end point for OS in hematological malignancies; a systematic review of patients with DLBCL suggested that PFS and EFS could be regarded as earlier efficacy end points in patients with DLBCL primarily treated with rituximab-containing immunochemotherapy.19 However, whether this could be extended to CAR T-cell therapies for relapsed or refractory LBCL is unknown. For the purpose of regulatory decision-making, FDA has accepted EFS as primary end point in relapsed refractory lymphoma.42
OS results should be considered in the context of subsequent therapies as patients who did not respond to SOC could receive any subsequent treatment for lymphoma off-protocol deemed appropriate by the investigator (including non–study-specific chemotherapy, immunotherapy, targeted agents, and anti-CD19 CAR T-cell therapy). The treatment switching rate was more than 50%, with 56% of patients randomized to the SOC arm receiving new lymphoma therapy after SOC. Prespecified sensitivity analyses of OS were performed to address the confounding effects from subsequent cell therapy in the SOC arm. The sensitivity analysis results using RPSFT and IPCW were consistent with the main interim OS analysis. These sensitivity analyses, designed to address the confounding effect of subsequent cell therapy in the SOC arm on OS, were also updated to include the additional survival data for discontinued patients, and results also consistent with the main interim OS analysis. Since the OS data are immature and the current results are based on an interim analysis, there is a risk that the benefit of axi-cel compared with SOC is overestimated, although the degree to which this may be true is uncertain.17,18
External Validity
The ZUMA-7 trial included a heterogenous population of patients with relapsed or refractory LBCL. Although a wide range of clinical presentations were represented, a few patient groups were excluded from the trial. The clinical experts consulted by CADTH noted that patients with an ECOG PS score of 2 and those who are HIV positive should be eligible for treatment with CAR T-cell therapies including axi-cel. In addition, ZUMA-7 enrolled patients who were eligible for ASCT. Patients in need for urgent therapy due to tumour mass effect were excluded from the study limiting the applicability of the data in patients with bulky disease needing urgent treatment. It was also noted that the median age of 59 years is younger than the median age of patients with LBCL in Canada, reflecting the age of the study population that is transplant eligible.51 Therefore, efficacy results in the trial may differ from those in the real-world context. Most patients in the ZUMA-7 trial were treated with CHOP regimen and all patients received rituximab in first-line, which is the SOC in Canada. Thus, it can be assumed that patients were adequately treated in first line before being characterized as having relapsed or refractory LBCL. Although both the clinical experts and the clinician groups indicated that ASCT eligibility is highly variable in clinical practice and should not be used to determine eligibility for CAR T-cell therapy, given that the clinical evidence from ZUMA-7 is based solely on patients who are eligible for ASCT, generalizability of results to patients who are not considered eligible for ASCT remains uncertain.
The clinical experts consulted by CADTH indicated that the SOC treatment including salvage chemotherapy regimens used in the SOC arm of the ZUMA-7 trial are reflective of Canadian clinical practice. However, some challenges were noted for axi-cel treatment. The manufacturing turnaround time for axi-cel in the ZUMA-7 trial (i.e., the median time from leukapheresis to the product’s release from the manufacturing site) was reported as 13 days (range, 10 to 24 days). The clinical experts questioned whether the same highly standardized processes can be replicated in most centres across Canada and whether and to what extent delays and restrictions in cross-border transport may affect treatment delays in the real-world. In addition, outside the highly controlled clinical trial setting, circumstances encountered more often in the real world may complicate timely treatment of patients and lead to outcomes different than those observed in the trial. The clinical experts noted that while a 13-day manufacturing turnaround for axi-cel was rapid, it may not be reproducible outside the clinical trial setting because manufacturing will be conducted in the US according to the sponsor’s implementation plan. Longer delays from leukapheresis to axi-cel infusion could adversely affect patient outcomes if, for example, patients experience disease progression during this delay. In addition, in the trial setting of ZUMA-7, risk of life-threatening and fatal AEs attributed to axi-cel treatment was mitigated by stringent training of study site staff and investigators and careful monitoring to ensure timely detection and management of the most serious complications. In real-world settings, site preparation, patient and clinician education, and risk mitigation strategies with emphasis on early recognition and treatment of serious and life-threatening complications including CRS and neurologic toxicities will be crucial to ensure patient safety.
Data for all outcomes considered important to patients, clinicians, and drug plans, as outlined in the systematic review protocol, were collected and analyzed in the trial. However, data for HRQoL were collected using a tool not validated among patients with LBCL, and data were available for few patients at most time points, precluding strong conclusions for this outcome. There remains an evidence gap regarding the impact of treatment with axi-cel compared with SOC on the HRQoL of patients with relapsed or refractory LBCL.
Indirect Evidence
A focused literature search for indirect treatment comparisons dealing with LBCL was run in MEDLINE All (1946–) on July 7, 2022. No limits were applied to the search. The search retrieved 32 reports, but none met the eligibility criteria for the systematic review. No indirect treatment comparisons were submitted by the sponsor.
Other Relevant Evidence
Long-Term Data From the ZUMA-1 Study
The sponsor provided long-term (≥ 4 year and ≥ 5 year) data from ZUMA-1, the pivotal multicentre, single-arm, registrational phase I and II study of axi-cel in patients with relapsed or refractory LBCL who had received at least 2 previous systemic therapies. These data were presented as conference abstracts or posters.20-22
In the 2-year analysis of ZUMA-1 (n = 101; median follow-up from axi-cel dosing to data cut-off = 27.1 months; interquartile range [IQR], 25.7 to 28.8 months), the ORR was 83%; 58% of patients achieved a CR. The 2-year OS rate was 50.5%. The median OS was not reached).52
Year 420
As of August 11, 2020, median follow-up was 51.1 months. With more than 4 years of follow-up, median OS was 25.8 months, and 4-year OS rate was 44% . Median EFS in patients treated with axi-cel was 5.7 months (range NR), with a 12-month EFS rate of 43% (95% CI, 33% to 52%) and a 24-month EFS rate of 38% (95% CI, 28% to 47%).
Overall, 26 (26%) patients received subsequent anticancer therapy; median time to next therapy was 8.7 months (range, 0.3 to 53.8 months). Immunoglobulin therapy was administered to 38 (38%) patients.
Of the treated patients, 58 (57%) had died, primarily due to progressive disease (46%; n = 46), other reasons (7%; n = 7), AEs (4%; n = 4), and secondary malignancy (myelodysplastic syndrome, with onset on day 574) unrelated to axi-cel (1%; n = 1).
Year 522
The most recently updated survival results from phase II ZUMA-1, after 5 years of follow-up, showed a 5-year OS rate of 42.6% (95% CI, 32.8% to 51.9%) among patients treated with axi-cel. The 5-year OS rate among complete responders was 64.4% (95% CI, 50.8% to 75.1%). The median survival time among complete responders was not reached (95% CI, 63.4 months to NE). Of 59 patients who achieved CR, 37 (63%) were alive at the 5-year data cut-off date. Since the 4-year data cut-off date, 1 death at month 63 (CR) and 1 progressive disease at month 54 (PR) were observed.
Median time to next anticancer therapy from axi-cel infusion was 8.7 months (range, 0.3 to 63.4 months), unchanged from the previous (4-year) analysis. By the year 5 data cut-off, 34 patients (34%) were still alive and received no subsequent therapy (excluding stem cell transfer) or re-treatment with axi-cel. Compared with the year 4 data, 2 patients (2%) who had previously progressed received new anticancer therapy.
Exploratory (Post Hoc) Analysis of OS by EFS Status at 12 and 24 Months22
Among all treated patients, the 12-month and the 24-month EFS rates were 42.8% (95% CI, 33.0% to 52.3%) and 37.7% (95% CI, 28.3% to 47.2%), respectively (Figure 10).
Among patients with (n = 57) and without (n = 44) an EFS event by month 12, 5-year OS rates were 5.3% (95% CI, 1.4% to 13.2%) and 90.9% (95% CI, 77.6% to 96.5%), respectively.
Among patients with (n = 62) and without (n = 39) an EFS event by month 24, 5-year OS rates were 11.3% (95% CI, 5.0% to 20.5%) and 92.3% (95% CI, 78.0% to 97.5%), respectively.
Figure 10: Exploratory Analysis of Overall Survival by Event-Free Survival at 12 and 24 Months in the ZUMA-1 Study
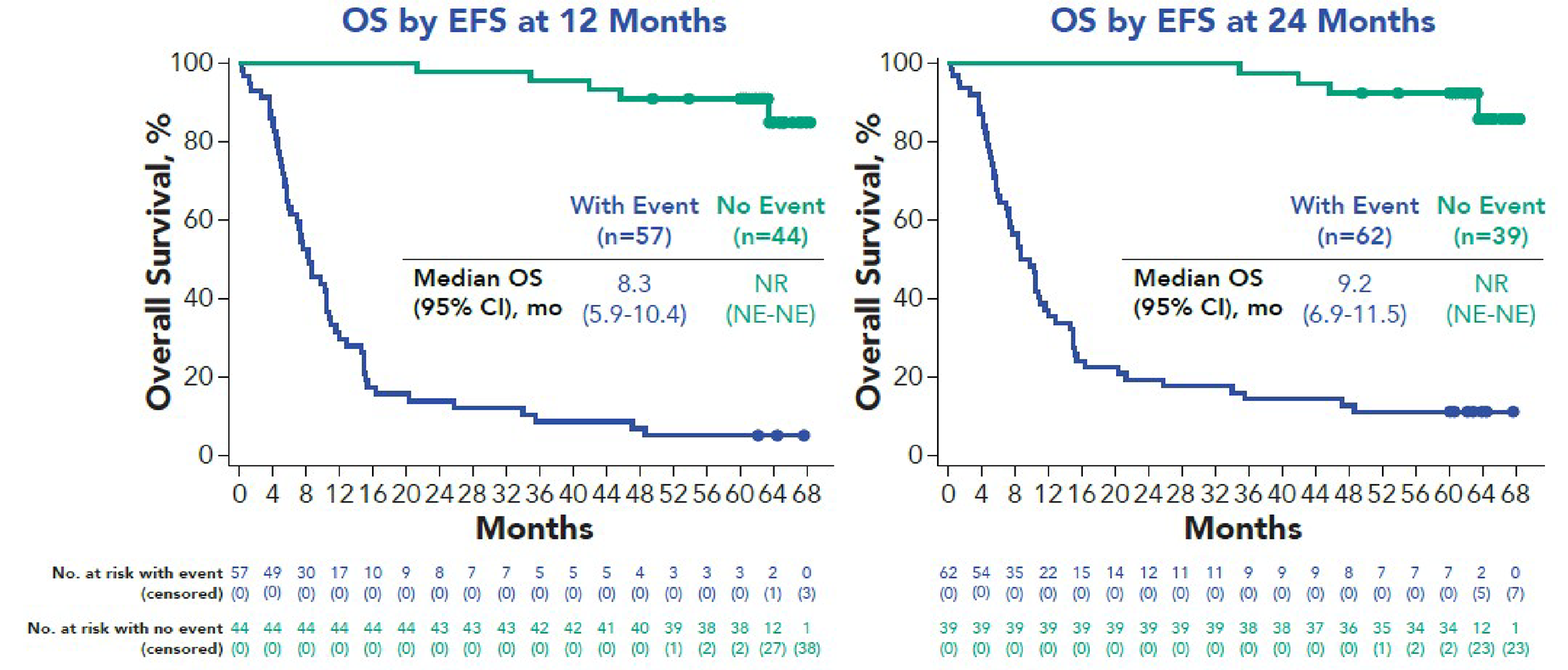
EFS = event-free survival; NE = not estimable; NR = not reached; OS = overall survival.
Source: Jacobson et al. (2021) (sponsor’s internal report).22
Supportive Safety Data: Comparison of Treated Populations Treated With Axi-Cel in the ZUMA-7 and ZUMA-1 Studies
In addition to data from the ZUMA-7 study, supportive safety data from the ongoing ZUMA-1 study (phase I and II) were included for consideration in the current CADTH review. Both studies treated patients with relapsed or refractory LBCL, including DLBCL and HGBL. The data cut-off date for both studies was March 18, 2021. TEAEs were comparable between patients treated with axi-cel in the ZUMA-1 and ZUMA-7 studies (Table 23). All patients in the ZUMA-7 and ZUMA-1 studies had at least 1 TEAE, including 91% and 96%, respectively, who had grade 3 or less TEAEs and 8% who had grade 5 TEAEs. Approximately half of the patients (50% in ZUMA-7 and 54% in ZUMA-1) had SAEs. The proportion of patients treated with axi-cel who experienced neurologic events or CRS was also similar in the 2 trials.
Table 23: Overall Summary of TEAEs — SOC From the ZUMA-7 Study and the Pooled Axi-Cel Population of the ZUMA-7 and ZUMA-1 Studies (Safety Analysis Set)
Event | SOC, n (%) | Axi-cel, n (%) | |
|---|---|---|---|
ZUMA-7 (N = 168) | ZUMA-7 (N = 170) | ZUMA-1 (N = 108) | |
Any TEAE | 168 (100) | 170 (100) | 108 (100) |
Worst grade ≥ 3 | 140 (83) | 155 (91) | 104 (96) |
Worst grade 5 | 7 (4) | 14 (8) | 9 (8) |
Due to disease progression | 5 (3) | 7 (4) | 5 (5) |
Any serious TEAE | 77 (46) | 85 (50) | 58 (54) |
Worst grade ≥ 3 | 67 (40) | 72 (42) | 53 (49) |
Worst grade 5 | 6 (4) | 14 (8) | 9 (8) |
Due to disease progression | 4 (2) | 7 (4) | 5 (5) |
Any axi-cel- or SOC-related TEAE | 160 (95) | 163 (96) | 107 (99) |
Worst grade ≥ 3 | 131 (78) | 112 (66) | 72 (67) |
Worst grade 5 | 2 (1) | 1 (1) | 2 (2) |
Any serious axi-cel- or SOC-related TEAE | 59 (35) | 63 (37) | 40 (37) |
Worst grade ≥ 3 | 51 (30) | 49 (29) | 36 (33) |
Worst grade 5 | 2 (1) | 1 (1) | 2 (2) |
Any CRS or neurologic event | NA | 159 (94) | 101 (94) |
Worst grade ≥ 3 | NA | 41 (24) | 39 (36) |
Worst grade 5 | NA | 0 (0) | 1 (1) |
Any CRS | NA | 157 (92) | 100 (93) |
Worst grade ≥ 3 | NA | 11 (6) | 12 (11) |
Worst grade 5 | NA | 0 (0) | 1 (1) |
Any serious CRS | NA | 29 (17) | – |
Worst grade ≥ 3 | NA | 10 (6) | – |
Worst grade 5 | NA | 0 (0) | – |
Any neurologic event | 33 (20) | 102 (60) | 71 (66) |
Worst grade ≥ 3 | 1 (1) | 36 (21) | 34 (31) |
Worst grade 5 | 0 (0) | 0 (0) | 0 (0) |
Any serious neurologic event | 1 (1) | 34 (20) | 27 (25) |
Worst grade ≥ 3 | 0 (0) | 26 (15) | 25 (23) |
Worst grade 5 | 0 (0) | 0 (0) | 0 (0) |
Any infection | 51 (30) | 70 (41) | 43 (40) |
Worst grade ≥ 3 | 19 (11) | 24 (14) | 29 (27) |
Worst grade 5 | 0 (0) | 5 (3) | 0 (0) |
Any cytopenias | 135 (80) | 136 (80) | 98 (91) |
Worst grade ≥ 3 | 126 (75) | 128 (75) | 89 (82) |
Worst grade 5 | 0 (0) | 0 (0) | 0 (0) |
Any hypogammaglobulinemia | 1 (1) | 19 (11) | 17 (16) |
Worst grade ≥ 3 | 0 (0) | 0 (0) | 0 (0) |
Worst grade 5 | 0 (0) | 0 (0) | 0 (0) |
axi-cel = axicabtagene ciloleucel; ASCT = autologous stem cell transplant; CRS = cytokine release syndrome; CTCAE = Common Terminology Criteria for Adverse Events; NA = not applicable; SOC = standard of care; TEAE = treatment-emergent adverse event.
Notes: Adverse events (AEs) are coded using Medical Dictionary for Regulatory Activities (MedDRA) Version 23.1. A TEAE is defined as any AE occurring on or after the first axi-cel infusion or the first dose of salvage chemotherapy in the SOC arm in ZUMA-7. TEAEs that occurred during the re-treatment period are excluded.
For patients treated with axi-cel, treatment-related TEAEs are those related to axi-cel. For the SOC arm of ZUMA-7, treatment-related TEAEs are those related to salvage chemotherapy, total body irradiation, high-dose therapy, or autologous stem cell transplant. CRS events were graded according to a modification of the criteria of Lee et al.50 Other events were graded according to Common Terminology Criteria for Adverse Events Version 4.03. Seriousness of CRS was not collected in ZUMA-1. Neurologic events are identified using a modified search strategy based on Topp et al. (2015). Multiple incidences of the same AE in 1 patient are counted once at the highest grade for each patient.
ZUMA-1 refers to ZUMA-1 phase I and phase II cohorts 1 and 2.
Data cut-off date is March 18, 2021.
Source: Clinical Summary Common Technical Document Section 2.7.4.53
Critical Appraisal
Internal Validity
The ZUMA-1 trial was critically appraised in the 2019 CADTH Optimal Use Report report.23 ZUMA-1 was a single-arm, phase I and II study of axi-cel in patients with relapsed or refractory LBCL who had received at least 2 previous systemic therapies. The primary limitation of ZUMA-1 was the absence of a comparator group against which the treatment benefits and harms of axi-cel could be compared. As such, causal effects cannot be inferred. Several limitations related to outcome assessment in the ZUMA-1 trial were also noted. These include the use of investigator disease assessment for the primary analysis (ORR based on independent review committee assessment, was reported as a secondary outcome), which can lead to detection bias particularly in open-label, single-arm trials, and inconsistency in censoring of patients throughout the analyses that could lead to more favourable estimates of the treatment effect of axi-cel. Notably, OS was not censored at the time of additional treatments (i.e., re-treatment with axi-cel, treatment with chemotherapy, or allo-SCT after relapse following axi-cel). Given the different study designs and patient populations in ZUMA-1 and ZUMA-7, the CADTH review team determined that data from the 2 trials cannot be reliably pooled. Therefore, the additional pooled safety data from the 2 trials were not interpreted for this review.
External Validity
ZUMA-1 was conducted in patients with relapsed or refractory LBCL who had received at least 2 previous systemic therapies (i.e., patients received axi-cel as third- or later-line treatment). It is unknown whether results are generalizable to patients treated with axi-cel as second-line treatment, which is the indication under review.
Discussion
Summary of Available Evidence
The ZUMA-7 trial that forms the main evidence base for this review is an ongoing phase III, open-label, randomized controlled trial that compares the efficacy and safety of axi-cel to SOC chemoimmunotherapy in patients with LBCL. Patients were those with disease that was refractory or had relapsed within 12 months of first-line chemoimmunotherapy, including an anti-CD20 monoclonal antibody and an anthracycline-containing chemotherapy regimen, who intended to proceed to HDT and ASCT. The primary end point was EFS by blinded central assessment. Secondary end points included ORR, OS, PFS, DOR, and HRQoL. Time to next treatment was an exploratory end point, and health care resource utilization was also reported. A total of 359 patients were enrolled; 180 were randomized to receive axi-cel, and 179 were randomized to receive SOC therapy consisting of salvage chemotherapy followed by HDT-ASCT. Axi-cel was manufactured for all 178 patients who underwent leukapheresis and administered to 170 (96%) patients. The primary efficacy analysis (data cut-off: March 18, 2021) was performed after 250 EFS events by blinded central assessment had been observed. The median follow-up was 23.0 months in the axi-cel arm and 21.2 months in the SOC arm. The mean age of patients was 57 years (SD = 12 years); 30% of the patients were aged 65 years or older. Overall, 74% of the study population had primary refractory disease and 26% had early relapse. Approximately one-quarter of patients in both treatment arms had achieved a best response of CR to first-line treatment.
The sponsor also provided long-term (≥ 4 year and ≥ 5 year) data from ZUMA-1, the pivotal multicentre, single-arm, registrational phase I and II study of axi-cel in patients with relapsed or refractory LBCL who had received at least 2 previous systemic therapies. Supportive safety data from ZUMA-1 were also considered.
Interpretation of Results
Efficacy
The ZUMA-7 trial demonstrated evidence of efficacy of axi-cel compared with SOC (salvage chemotherapy followed by HD-ASCT) in adult patients with primary refractory and early relapsed LBCL (< 12 months). EFS was improved for the axi-cel arm compared with the SOC arm. Secondary outcomes were supportive, with improvements observed for PFS and ORR. According to the clinical experts, the magnitude of the treatment effect is clinically meaningful. The interim analysis of OS did not reach statistical significance; the 95% CI was wide and included the possibility of appreciable OS benefit compared with SOC, as well as little-to-no difference. As the results are based on an interim analysis, there is the potential that the beneficial effect of axi-cel compared with SOC is overestimated. Results from the final analysis are required to determine with certainty the presence and magnitude of OS benefit. More mature OS data from the phase II cohort of the ZUMA-1 study for patients with LBCL treated with axi-cel in third line provide supporting evidence for the OS benefit of axi-cel in patients who had received at least 2 previous systemic therapies.21,22 However, the single-arm design of ZUMA-1 and different patient population precludes drawing causal inferences.
The clinical experts consulted by CADTH indicated that OS is the most important efficacy outcome to assess treatment effect between axi-cel and the SOC arms, and longer survival was an important priority indicated in the patient input. The interim analysis of OS is premature given the heavy censoring around 18 months. Overall, the difference in OS was not statistically significant. As the FDA review has also noted, while the difference in OS between the 2 arms is not statistically significant, the direction of the observed treatment effect is consistent with the EFS and PFS data.42 It is also noted that results of the OS analysis may be confounded by the fact that 55% of the patients in the SOC arm received autologous CD19-directed CAR T therapy after experiencing an event as crossover, which was not built into the main statistical analyses.42 As OS data are immature and the current results are based on an interim analysis, it is possible that the benefit of axi-cel compared with SOC is overestimated, although the degree to which this may be true is uncertain.17,18
HRQoL was also an outcome indicated as important by patients. However, findings were uncertain due to the large amount of missing data. It is thus unclear, based on these data, if axi-cel affords better QoL compared with SOC in patients with relapsed or refractory LBCL. Data on resource utilization, another important outcome as indicated by the clinical experts, were limited to acute care, and thus may not completely capture resource use associated with axi-cel.
Despite high initial response to first-line therapy, about 40% of patients with LBCL are refractory or will relapse and require second-line therapy.4 The current SOC for relapsed or refractory LBCL is salvage chemotherapy followed by HDT and ASCT if there is an adequate response to chemotherapy. However, a considerable proportion of patients do not benefit from current SOC second-line therapy. Many patients, even if fit enough for ASCT, ultimately cannot undergo HDT-ASCT for reasons including chemotherapy-resistant disease, treatment-emergent toxicities, or inability to mobilize stem cells. Poor outcomes have been observed in these patients, with 5-year survival below 50%. Although higher risk is associated with disease that is refractory or relapses within 12 months of first-line therapy, only 10% of all patients with relapsed or refractory LBCL are estimated to have long-term survival following ASCT.4 Therefore, there is an important need for effective alternative second-line therapies, including those with a mechanism of action independent of chemotherapy sensitivity, whether the patient relapses before or after 12 months of first-line therapy or is deemed eligible or ineligible to receive ASCT before starting salvage chemotherapy. In the ZUMA-7 trial, the most common reason for not proceeding with HD-ASCT in the SOC arm was due to the lack of response to salvage chemoimmunotherapy, indicating the chemorefractory nature of the study population. Axi-cel has a mechanism of action independent of chemotherapy sensitivity that may benefit patients with LBCL who require second-line therapy. No therapies are currently approved for the definitive management of LBCL in the second-line setting for potentially transplant-eligible patients who are either primary refractory or have early treatment failure to front-line therapy. The clinical experts consulted by CADTH also indicated that they thought axi-cel could fit well as a second-line treatment and replace ASCT for most patients. The clinical experts indicated that they would expect patient outcomes to be better when they receive this therapy earlier in the course of disease.
Of note, this CADTH Reimbursement Review was conducted before issuance of Health Canada NOC and the scope was based on the anticipated indication. The anticipated Health Canada indication when the submission was received by CADTH was for the treatment of adult patients with relapsed or refractory LBCL. The final approved indication is for the treatment of adult patients with DLBCL or HGBL that is refractory to first-line chemoimmunotherapy or that relapses within 12 months of first-line chemoimmunotherapy. The sponsor’s requested reimbursement criteria are for the treatment of adult patients with relapsed or refractory LBCL who are candidates for ASCT, which aligns with the trial population in ZUMA-7. However, the clinical experts expressed concern about using ASCT eligibility as a criterion for CAR T-cell therapy eligibility. The clinical experts noted that there are no standard criteria to determine ASCT eligibility; criteria vary widely across treatment centres depending on local clinical practices and resources. According to the clinical experts, only approximately 40% of patients are intended for ASCT. A recent Canadian study of real-world eligibility for second-line CAR T-cell therapy in patients with LBCL found that real-world eligibility for second-line CAR T-cell therapy may be as high as 65%.13 Although only 14% of patients met all the ZUMA-7 study inclusion criteria, as many as 65% of patients progressing within 12 months of chemoimmunotherapy had adequate performance status to be considered potentially eligible for second-line CAR T-cell therapy.13 Therefore, restricting axi-cel eligibility only to transplant-eligible patients would exclude a large proportion of patients. However, the proportion of patients who can be treated with axi-cel in real-world practice is uncertain and will likely depend on both patient-related and system-related factors, and although the definition of CAR T-cell therapy eligibility might differ from the definition of ASCT eligibility, not all patients would be considered suitable for axi-cel treatment in the second-line setting.
In the ZUMA-7 trial, only 35% of the patients randomized to the SOC arm underwent transplant. In comparison, almost 3 times as many patients (94%) randomized to the axi-cel arm underwent definitive treatment with CAR T-cell infusion. In addition to lack of chemotherapeutic response as a main reason for ineligibility to proceed with HDT-ASCT, the SOC regimen (preceded by salvage chemotherapy) is a multistep and arduous treatment regimen that most patients find difficult to tolerate. According to the clinical experts consulted by CADTH, single-infusion CAR T-cell therapy represents a less burdensome treatment option for patients and its use in the second line, particularly for patients with prognostic factors indicative of poor response to second-line SOC, could substantially increase the proportion of patients able to receive curative-intent treatment at first progression of LBCL.
Harms
All patients in the axi-cel and SOC arms had at least 1 TEAE. Serious TEAEs were reported in similar proportions of patients in the axi-cel and SOC arms. Grade 3 or higher TEAEs occurred in 91% and 83% of patients in the axi-cel and SOC arms, respectively, including 7 patients and 2 patients, respectively, with grade 5 TEAEs (excluding PD). Evidence for the safety of axi-cel was also reported from ZUMA-1. Safety data reported from ZUMA-1 were based on 108 patients treated with axi-cel; at the data cut-off date of March 18, 2021, patients in ZUMA-1 had the opportunity to be followed up for at least 54 months after their infusion of axi-cel. Nine patients (5%) in the axi-cel arm were re-treated. No new safety concerns were identified after re-treatment with axi-cel.
Differences in the TEAE profile between the 2 treatment arms were generally consistent with the AE profiles of the 2 treatments (e.g., CRS is a well-recognized toxicity of CAR T-cell therapies). TEAEs noted after axi-cel infusion in ZUMA-7 are consistent with those observed with the AE profile of other anti-CD19 CAR T therapies.54 Patients treated with CAR T-cell therapies commonly experience CRS, associated with the use of monoclonal antibodies and adoptive cell therapies that activate lymphocytes. The condition results from the release of cytokines from cells targeted by antibodies, immune effector cells recruited to the tumour area, and the patient’s immune cells activated during this process. CRS is characterized by high fevers, hemodynamic instability, multiorgan dysfunction, and neurologic events such as encephalopathy, delirium, seizures, and cerebral edema, with most patients requiring treatment in hospital with highly specialized and intensive care.54 In ZUMA-7, CRS occurred in 92% of patients (grade ≥ 3, 6%), and was generally manageable and resolved for all patients, with a median duration of 7.0 days. While the overall incidence of CRS observed in ZUMA-7 is similar to that observed in ZUMA-1 (92% and 94%, respectively), the incidence of grade 3 or greater CRS appeared lower in ZUMA-7 compared with ZUMA-1 (7% and 13%, respectively). This difference in the rate of grade 3 or greater CRS may be due to earlier intervention in ZUMA-7, or it may be related to differences in the study population (second-line versus third- or later-line population in the ZUMA-7 versus ZUMA-1 trials, respectively).42
Another AE of special interest was neurotoxicity. The ZUMA-7 trial reported 102 patients (60%) in the axi-cel arm and 33 patients (20%) in the SOC arm who had at least 1 treatment-emergent neurologic event, including 36 patients (21%) and 1 patient (1%), respectively, with grade 3 or higher neurologic events. However, the FDA reanalysis of the safety data found that 124 (74%) patients had 1 or more neurologic toxicity events considered to be related to axi-cel, and that 42 patients (25%) experienced grade 3 or higher events.42 Of the 124 patients who experienced neurotoxicity, neurologic toxicity occurred before CRS in 1 (0.8%) patient, during CRS in 110 (89%) patients, and after CRS in 11 (9%) patients. Two (2%) patients had neurotoxicity without CRS.42 Most of the neurotoxicity events reported in the trial were manageable and reversible. The clinical experts indicated that in clinical practice treating physicians would be vigilant to the early signs of neurotoxicity and can manage these in most cases.
Axi-cel is administered as a single infusion; no patients discontinued treatment due to TEAEs in the axi-cel arm. Two patients in the SOC arm discontinued treatment (due to TEAEs of grade 4 acute kidney injury and grade 1 blood stem cell harvest failure). Overall, 3 patients in the SOC arm were unable to tolerate protocol-specified chemotherapy due to toxicities, resulting in treatment discontinuation: 2 patients discontinued R-ICE and R-DHAP, respectively, due to renal impairment, and 1 patient was unable to tolerate R-GDP. One patient was unable to complete HSCT due to stem cell harvest failure. Similarly, because axi-cel is administered as a single infusion, no patients in the axi-cel arm had a dose interruption or reduction due to TEAEs. In the SOC arm, 30 patients had at least 1 TEAE that required dose interruption or reduction to salvage chemotherapy.
The patient groups indicated that having access to treatments with fewer side effects was very important. Although the safety profile of axi-cel is consistent with its known mechanism of action, the lack of long-term safety data from ZUMA-7 remains an important limitation as the risk of certain AEs that may develop over a longer time period after treatment, such as secondary malignancies, is not well-characterized.
Conclusions
Based on data from the ZUMA-7 trial, treatment with the anti-CD19 CAR T-cell axi-cel led to an improvement in EFS and PFS compared with SOC HDT-ASCT as second-line treatment in ASCT-eligible patients with relapsed or refractory LBCL within 12 months of first-line chemoimmunotherapy. It is not yet clear whether EFS benefits will translate to improved OS as the data remain immature, and follow-up is ongoing. As the OS results are based on an interim analysis, there is a risk that the effect of axi-cel compared with SOC on survival is overestimated. The risks associated with axi-cel treatment are consistent with its mechanism of action and acceptable given the high-risk patient population, but it must be administered in a specialized centres equipped to manage toxicities related to CAR T-cell therapy. Although the most common toxicities were similar in the ZUMA-7 and ZUMA-1 trials, the lack of long-term safety and efficacy data from the ZUMA-7 trial remains an important limitation. A longer follow-up time is needed to better establish the survival benefit and long-term safety of axi-cel as relapse of lymphoma and potential late toxicities of CAR T-cell therapy, such as secondary malignancies, may emerge with longer follow-up. Uncertainties remain regarding the implementation of CAR T-cell therapy and the support system needed to optimize timely access and deliverability of axi-cel in the real-world setting.
References
1.Canadian Cancer Society. Non-Hodgkin Lymphoma. 2022; https://www.lymphoma.ca/lymphoma/non-hodgkin-lymphoma/. Accessed 2022 Aug 12.
2.Canadian Cancer Society. Non-Hodgkin Lymphoma statistics. 2022; https://cancer.ca/en/cancer-information/cancer-types/non-hodgkin-lymphoma/statistics. Accessed 2022 Sep 1.
3.Armitage JO, Gascoyne RD, Lunning MA, Cavalli F. Non-Hodgkin lymphoma. Lancet. 2017;390(10091):298-310. PubMed
4.Shafey M, Savage KJ, Skrabek P, Elsawy M, Bosch M, Kuruvilla J. Canadian Evidence-based Guideline for the Treatment of Relapsed/Refractory Diffuse Large B-Cell Lymphoma. Mississauga (ON): Lymphoma Canada; 2021: https://www.lymphoma.ca/resources/healthcare-professionals/dlbcl-diffuse-large-b-cell-lymphoma/. Accessed 2022 Aug 15.
5.Lymphoma Canada. Diffuse Large B cell Lymphoma (DLBCL). 2022; https://www.lymphoma.ca/diffuse-large-b-cell-lymphoma/. Accessed 2022 Aug 12.
6.Martelli M, Ferreri AJ, Agostinelli C, Di Rocco A, Pfreundschuh M, Pileri SA. Diffuse large B-cell lymphoma. Crit Rev Oncol Hematol. 2013;87(2):146-171. PubMed
7.NCCN Guidelines for Patients: Diffuse Large B-cell Lymphoma. Plymouth Meeting (PA): National Comprehensive Cancer Network; 2020.
8.Lymphoma Clinical Practice Guidelines. Edmonton (AB): Alberta Health Services; 2021: https://www.albertahealthservices.ca/assets/info/hp/cancer/if-hp-cancer-guide-lyhe002-lymphoma.pdf. Accessed 2022 Aug 11.
9.Prica A, Baldassarre F, Hicks LK, et al. Rituximab in Lymphoma and Chronic Lymphocytic Leukemia: A Clinical Practice Guideline. (Guideline 6-8 Version 3). Toronto (ON): Cancer Care Ontario; 2015: https://www.cancercareontario.ca/en/guidelines-advice/types-of-cancer/721. Accessed 2022 Aug 15.
10.BC Cancer Protocol Summary for Treatment of Lymphoma with DOXOrubicin, Cyclophosphamide, vinCRIStine, predniSONE and riTUXimab (CHOP-R). [Victoria (BC)]: BC Cancer; 2022: http://www.bccancer.bc.ca/chemotherapy-protocols-site/Documents/Lymphoma-Myeloma/LYCHOPR_Protocol.pdf. Accessed 2022 Aug 15.
11.Coiffier B, Sarkozy C. Diffuse large B-cell lymphoma: R-CHOP failure-what to do? Hematology Am Soc Hematol Educ Program. 2016;2016(1):366-378. PubMed
12.Rovira J, Valera A, Colomo L, et al. Prognosis of patients with diffuse large B cell lymphoma not reaching complete response or relapsing after frontline chemotherapy or immunochemotherapy. Ann Hematol. 2015;94(5):803-812. PubMed
13.Puckrin R, Stewart DA, Shafey M. Real-World Eligibility for Second-Line Chimeric Antigen Receptor T Cell Therapy in Large B Cell Lymphoma: A Population-Based Analysis. Transplant Cell Ther. 2022;28(4):218.e211-218.e214. PubMed
14.Locke FL, Miklos DB, Jacobson CA, et al. Axicabtagene Ciloleucel as Second-Line Therapy for Large B-Cell Lymphoma. N Engl J Med. 2022;386(7):640-654. PubMed
15.Clinical Study Report: KTE-C19-107. A phase 3, randomized, open-label study evaluating the efficacy of axicabtagene ciloleucel versus standard of care therapy in subjects with relapsed/refractory diffuse large B-cell lymphoma (ZUMA-7) [internal sponsor's report]. Santa Monica (CA): Kite Pharma, Inc.; 2021 Sep 14.
16.Maringwa JT, Quinten C, King M, et al. Minimal important differences for interpreting health-related quality of life scores from the EORTC QLQ-C30 in lung cancer patients participating in randomized controlled trials. Support Care Cancer. 2011;19(11):1753-1760. PubMed
17.Guyatt GH, Briel M, Glasziou P, Bassler D, Montori VM. Problems of stopping trials early. BMJ. 2012;344:e3863. PubMed
18.Montori VM, Devereaux PJ, Adhikari NK, et al. Randomized trials stopped early for benefit: a systematic review. JAMA. 2005;294(17):2203-2209. PubMed
19.Zhu J, Yang Y, Tao J, et al. Association of progression-free or event-free survival with overall survival in diffuse large B-cell lymphoma after immunochemotherapy: a systematic review. Leukemia. 2020;34(10):2576-2591. PubMed
20.Jacobson C, Locke FL, Ghobadi A, et al. Long-Term (>4 Year and >5 Year) Overall Survival (OS) By 12- and 24-Month Event-Free Survival (EFS): An Updated Analysis of ZUMA-1, the Pivotal Study of Axicabtagene Ciloleucel (Axi-Cel) in Patients (Pts) with Refractory Large B-Cell Lymphoma (LBCL) [conference abstract]. Blood. 2021;138(Suppl 1):1764.
21.Jacobson C, Locke FL, Ghobadi A, et al. 10 - Long-Term (5 Year) Overall Survival in Zuma-1, the Pivotal Study of Axicabtagene Ciloleucel (Axi-Cel) in Patients with Refractory Large B-Cell Lymphoma (LBCL) [conference abstract]. Transplant Cell Ther. 2021;28(3S):S9-S10.
22.Jacobson C, Locke FL, Ghobadi A, et al. Long-Term (4- and 5- Year) Overall Survival in ZUMA-1, the Pivotal Study of Axicabtagene Ciloleucel in Patients with Refractory Large B-Cell Lymphoma [conference poster]. In: Drug Reimbursement Review sponsor submission: Yescarta® (axicabtagene ciloleucel), cell suspension in patient-specific single infusion bag, target of 2 × 106 chimeric antigen receptor (CAR)-positive viable T cells per kg body weight with a maximum of 2 × 108 CAR-positive viable T cells, for intravenous infusion. Mississauga (ON): Gilead Sciences Canada, Inc.; 2021.
23.Axicabtagene ciloleucel for large B-cell lymphoma: Clinical Report. (CADTH optimal use report vol. 9, no. 1c). Ottawa (ON): CADTH; 2019: https://www.cadth.ca/sites/default/files/pdf/car-t/ct0002-clinical-report-redacted.pdf. Accessed 2022 Sep 1.
24.Sehn LH, Salles G. Diffuse Large B-Cell Lymphoma. N Engl J Med. 2021;384(9):842-858. PubMed
25.Rosenwald A, Bens S, Advani R, et al. Prognostic Significance of MYC Rearrangement and Translocation Partner in Diffuse Large B-Cell Lymphoma: A Study by the Lunenburg Lymphoma Biomarker Consortium. J Clin Oncol. 2019;37(35):3359-3368. PubMed
26.Willenbacher E, Willenbacher W, Weger R, Dominik W, Manzl C, Brunner A. Patients with double/triple copy number gains on C-MYC, BCL2, and/or BCL6 treated with standard chemotherapy have a similarly poor prognosis than those with high-grade B cell lymphoma with C-MYC and BCL2 and/or BCL6 rearrangements: a single-center experience on a consecutive cohort of large B cell lymphomas. Ann Hematol. 2020;99(9):2125-2132. PubMed
27.Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127(20):2375-2390. PubMed
28.van der Poel MW, Oerlemans S, Schouten HC, et al. Quality of life more impaired in younger than in older diffuse large B cell lymphoma survivors compared to a normative population: a study from the population-based PROFILES registry. Ann Hematol. 2014;93(5):811-819. PubMed
29.Ma Q, Bailey A, Milloy N, Butcher J, Quek RG, Johnson PC. Real-World Health-Related Quality of Life in Patients with Diffuse Large B-Cell Lymphoma: Comparisons with Reference Populations and By Line of Therapy. Blood. 2021;138(S1):4111.
30.Gisselbrecht C, Glass B, Mounier N, et al. Salvage regimens with autologous transplantation for relapsed large B-cell lymphoma in the rituximab era. J Clin Oncol. 2010;28(27):4184-4190. PubMed
31.van Imhoff GW, McMillan A, Matasar MJ, et al. Ofatumumab Versus Rituximab Salvage Chemoimmunotherapy in Relapsed or Refractory Diffuse Large B-Cell Lymphoma: The ORCHARRD Study. J Clin Oncol. 2017;35(5):544-551. PubMed
32.Crump M, Kuruvilla J, Couban S, et al. Randomized comparison of gemcitabine, dexamethasone, and cisplatin versus dexamethasone, cytarabine, and cisplatin chemotherapy before autologous stem-cell transplantation for relapsed and refractory aggressive lymphomas: NCIC-CTG LY.12. J Clin Oncol. 2014;32(31):3490-3496. PubMed
33.Kondo E. Autologous Hematopoietic Stem Cell Transplantation for Diffuse Large B-Cell Lymphoma. J Clin Exp Hematop. 2016;56(2):100-108. PubMed
34.Friedberg JW. Relapsed/refractory diffuse large B-cell lymphoma. Hematology Am Soc Hematol Educ Program. 2011;2011:498-505. PubMed
35.Gisselbrecht C, Schmitz N, Mounier N, et al. Rituximab maintenance therapy after autologous stem-cell transplantation in patients with relapsed CD20(+) diffuse large B-cell lymphoma: final analysis of the collaborative trial in relapsed aggressive lymphoma. J Clin Oncol. 2012;30(36):4462-4469. PubMed
36.CADTH. Drug reimbursement review: Polatuzumab Vedotin (Polivy) for diffuse large B-cell lymphoma (DLBCL). 2021; https://www.cadth.ca/polatuzumab-vedotin-polivy-dlbcl-details. Accessed 2022 Sep 1.
37.Yescarta® (axicabtagene ciloleucel): cell suspension in patient-specific single infusion bag, target of 2 × 106 chimeric antigen receptor (CAR)-positive viable T cells per kg body weight with a maximum of 2 × 108 CAR-positive viable T cells, for intravenous infusion [product monograph]. Mississauga (ON): Gilead Sciences Canada, Inc.; 2021 Dec 15.
38.Summary Basis for Regulatory Action: Yescarta (axicabtagene ciloleucel). Silver Spring (MD): U.S. Food and Drug Administration; 2017: https://www.fda.gov/media/108788/download. Accessed 2022 Aug 10.
39.European Public Assessment Report (EPAR): Yescarta (axicabtagene ciloleucel). Amsterdam (NL): European Medicines Agency; 2022: https://www.ema.europa.eu/en/medicines/human/EPAR/yescarta. Accessed 2022 Aug 10.
40.McGowan J, Sampson M, Salzwedel DM, Cogo E, Foerster V, Lefebvre C. PRESS Peer Review of Electronic Search Strategies: 2015 guideline statement. J Clin Epidemiol. 2016;75:40-46. PubMed
41.Grey matters: a practical tool for searching health-related grey literature. Ottawa (ON): CADTH; 2019: https://www.cadth.ca/grey-matters. Accessed 2022 Jun 28.
42.BLA 125643/394 Clinical Review and Evaluation: Axicabtagene ciloleucel (Yescarta). Silver Spring (MD): U.S. Food and Drug Administration; 2022: https://fda.report/media/157687/April-1-2022-Clinical-Review-and-Evaluation-YESCART_0.pdf. Accessed 2022 Oct 18.
43.Neelapu SS, Locke FL, Bartlett NL, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med. 2017;377(26):2531-2544. PubMed
44.Cheson BD, Fisher RI, Barrington SF, et al. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: the Lugano classification. J Clin Oncol. 2014;32(27):3059-3068. PubMed
45.Pickard AS, Neary MP, Cella D. Estimation of minimally important differences in EQ-5D utility and VAS scores in cancer. Health Qual Life Outcomes. 2007;5:70. PubMed
46.National Cancer Institute Division of Cancer Treatment and Diagnosis. Common Terminology Criteria for Adverse Events (CTCAE) v4.0. [2010]; https://ctep.cancer.gov/protocoldevelopment/electronic_applications/ctc.htm#ctc_40. Accessed 2022 Sep 1.
47.Robins JM, Tsiatis AA. Correcting for Non-Compliance in Randomized Trials Using Rank Preserving Structural Failure Time Models. Commun Statist Theory Meth. 1991;20(8):22.
48.Robins JM, Finkelstein DM. Correcting for noncompliance and dependent censoring in an AIDS Clinical Trial with inverse probability of censoring weighted (IPCW) log-rank tests. Biometrics. 2000;56(3):779-788. PubMed
49.Addendum to the Primary Analysis Clinical Study Report: A phase 3, randomized, open-label study evaluating the efficacy of axicabtagene ciloleucel versus standard of care therapy in subjects with relapsed/refractory diffuse large B-cell lymphoma (ZUMA-7) (CSR KTE-C19-107) [internal sponsor's report]. Santa Monica (CA): Kite Pharma, Inc.; 2022 Mar 03.
50.Lee DW, Gardner R, Porter DL, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014;124(2):188-195. PubMed
51.Ye X, Mahmud S, Skrabek P, Lix L, Johnston JB. Long-term time trends in incidence, survival and mortality of lymphomas by subtype among adults in Manitoba, Canada: a population-based study using cancer registry data. BMJ Open. 2017;7(7):e015106. PubMed
52.Locke FL, Ghobadi A, Jacobson CA, et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1-2 trial. Lancet Oncol. 2019;20(1):31-42. PubMed
53.Section 2.7 Clinical Summary. Section 2.7.4: Summary of Clinical Safety Axicabtagene Ciloleucel Yescarta [internal sponsor's report]. Santa Monica (CA): Kite Pharma Inc.; 2021 Sep 23.
54.Neelapu SS. Managing the toxicities of CAR T-cell therapy. Hematol Oncol. 2019;37 Suppl 1:48-52. PubMed
55.EORTC Quality of Life. EORTC QLQ-C30 FAQ Categories: Scoring. https://qol.eortc.org/faq-category/scoring/. Accessed 2021 Oct 01.
56.Cocks K, King MT, Velikova G, et al. Evidence-based guidelines for interpreting change scores for the European Organisation for the Research and Treatment of Cancer Quality of Life Questionnaire Core 30. Eur J Cancer. 2012;48(11):1713-1721. PubMed
57.Osoba D, Rodrigues G, Myles J, Zee B, Pater J. Interpreting the significance of changes in health-related quality-of-life scores. J Clin Oncol. 1998;16(1):139-144. PubMed
58.van Reenen M, Janssen B, Oppe M, Kreimeier S, Greiner W, Stolk E. EQ-5D-Y User Guide. Version 2.0. 2020; https://euroqol.org/publications/user-guides. Accessed 2022 Mar 06.
59.McClure NS, Sayah FA, Xie F, Luo N, Johnson JA. Instrument-Defined Estimates of the Minimally Important Difference for EQ-5D-5L Index Scores. Value Health. 2017;20(4):644-650. PubMed
60.Elsawy M, Chavez JC, Avivi I, et al. Patient-reported outcomes in ZUMA-7, a phase 3 study of axicabtagene ciloleucel in second-line large B-cell lymphoma. Blood. 2022;15:15. PubMed
61.Shah J, Shacham S, Kauffman M, et al. Health-related quality of life and utility outcomes with selinexor in relapsed/refractory diffuse large B-cell lymphoma. Fut Oncol. 2021;17(11):1295-1310. PubMed
62.Giesinger JM, Kieffer JM, Fayers PM, et al. Replication and validation of higher order models demonstrated that a summary score for the EORTC QLQ-C30 is robust. J Clin Epidemiol. 2016;69:79-88. PubMed
63.Cochrane T, Enrico A, Gomez-Almaguer D, et al. Impact of venetoclax monotherapy on the quality of life of patients with relapsed or refractory chronic lymphocytic leukemia: results from the phase 3b VENICE II trial. Leuk Lymphoma. 2022;63(2):304-314. PubMed
Appendix 1: Literature Search Strategy
Note that this appendix has not been copy-edited.
Clinical Literature Search
Overview
Interface: Ovid
Databases:
MEDLINE All (1946 to present)
Embase (1974 to present)
Note: Subject headings and search fields have been customized for each database. Duplicates between databases were removed in Ovid.
Date of search: July 7, 2022
Alerts: Bi-weekly search updates until project completion
Search filters applied: No filters were applied to limit the retrieval by study type
Limits: Conference abstracts: excluded
Syntax | Description |
|---|---|
/ | At the end of a phrase, searches the phrase as a subject heading |
* | Before a word, indicates that the marked subject heading is a primary topic; or, after a word, a truncation symbol (wildcard) to retrieve plurals or varying endings |
.ti | Title |
.ot | Original title |
.ab | Abstract |
.hw | Heading word; usually includes subject headings and controlled vocabulary |
.kf | Keyword heading word |
.dq | Candidate term word (Embase) |
.pt | Publication type |
.rn | Registry number |
.nm | Name of substance word (MEDLINE) |
medall | Ovid database code: MEDLINE All, 1946 to present, updated daily |
oemezd | Ovid database code; Embase, 1974 to present, updated daily |
Multi-Database Strategy
(axicabtagene* or Yescarta* or axi-cel or axicel or KTEC19* or KTE-C19* or fkc 876* or fkc876* or U2I8T43Y7R).ti,ab,ot,kf,hw,nm,rn.
1 use medall
*Axicabtagene Ciloleucel/
(axicabtagene* or Yescarta* or axi-cel or axicel or KTEC19* or KTE-C19* or fkc 876* or fkc876*).ti,ab,kf,dq.
3 or 4
5 not (conference review or conference abstract).pt.
6 use oemezd
2 or 7
remove duplicates from 8
Clinical Trials Registries
ClinicalTrials.gov
Produced by the US National Library of Medicine. Targeted search used to capture registered clinical trials.
[Search -- axicabtagene OR axicel OR “axi cel” OR yescarta OR ktec19 OR “kte c19” OR “fkc 876” OR fkc876]
WHO ICTRP
International Clinical Trials Registry Platform, produced by the WHO. Targeted search used to capture registered clinical trials.
[Search terms -- axicabtagene* OR axicel* OR “axi cel” OR yescarta* OR ktec19 OR “kte c19” OR “fkc 876” OR fkc876]
Health Canada’s Clinical Trials Database
Produced by Health Canada. Targeted search used to capture registered clinical trials.
[Search terms – axicabtagene, axicel, “axi cel”, yescarta, ktec19, “kte c19”, “fkc 876”, fkc876]
EU Clinical Trials Register
European Union Clinical Trials Register, produced by the European Union. Targeted search used to capture registered clinical trials.
[Search terms -- axicabtagene* OR axicel* OR “axi cel” OR yescarta* OR ktec19 OR “kte c19” OR “fkc 876” OR fkc876]
Grey Literature
Search dates: June 24 to July 6, 2022
Keywords: Yescarta, axicabtagene, axi-cel, axicel, KTE-C19, KTEC19, CAR-T, chimeric antigen, lymphoma
Limits: None
Updated: No update
Relevant websites from the following sections of the CADTH grey literature checklist Grey Matters: A Practical Tool for Searching Health-Related Grey Literature were searched:
Health Technology Assessment Agencies
Health Economics
Clinical Practice Guidelines
Drug and Device Regulatory Approvals
Advisories and Warnings
Drug Class Reviews
Clinical Trials Registries
Databases (free)
Internet Search
Appendix 2: Description and Appraisal of Outcome Measures
Note this appendix has not been copy-edited.
Aim
To describe the following outcome measures and review their measurement properties (validity, reliability, responsiveness to change, and MID):
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 (EORTC QLQ-C30)
EQ-5D-5L
Findings
A focused literature search was conducted to identify the psychometric properties and minimally important difference (MID) of each of the stated outcome measures.
The findings about validity, reliability, responsiveness, and MID of each outcome measure are summarized in Table 25.
Table 25: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
EORTC QLQ-C30 | The EORTC QLQ-C30 is a standardized, patient self-administered questionnaire for evaluating the quality of life of patients with cancer. The questionnaire consists of 5 functional scales, 3 symptom scales, 6 single-item scales, and 1 global quality of life scale. Most questions have 4 response options, scored from 1 (“not at all”) to 4 (“very much”). Raw scores for each scale are computed as the average of the contributing items. These are converted to scores ranging from 0 to 100, with higher scores reflecting better function, more symptoms, or better quality of life.55 | Measurement properties of validity, reliability, and responsiveness have not been assessed in NHL patients. | For improvement and deterioration in patients with various types of cancers including hematological diseases, the following have been estimated as “small” mean differences56:
For HRQoL among patients with breast, ovarian, and lung cancer a 5-point change in the GHS/QoL subscale was estimated as “a little” change, whereas for other domains, a 5-point change from baseline and 10-point change in mean scores was estimated as “a little” change.56,57 In the ZUMA-7 trial clinically meaningful changes were defined as 10 points for QLQ-C30 involving large B-cell lymphoma patients.16 |
EQ-5D-5L | Generic, preference-based HRQoL instrument, consisting of an index score and VAS scale score. A higher score represents better HRQoL. The index score is based on 5 dimensions: mobility, self-care, usual activities, pain/discomfort, and anxiety/depression. Each dimension is rated on a scale of 1 (“no problems”) to 5 (“extreme problems”). EQ VAS scale ranges from 0 (worst health imaginable) to 100 (best health imaginable).58 | Measurement properties of validity, reliability, and responsiveness have not been assessed in NHL patients. | Simulation-based MID – Canadian general population (mean ± SD): 0.056 ± 0.01159 In the ZUMA-7 trial, clinically meaningful changes were defined as 7 points for EQ-5D-5L VAS score, and 0.06 points for the EQ-5D-5L index for patients with lymphoma.45,60 This was based on an estimated MID among patients with various cancers.59 Among patients with multiple myeloma the MID has been estimated as 0.08 for improvement and 0.10 for deterioration.61 |
EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EQ VAS = EuroQol-visual analogue scales; GHS = global health status; HRQoL = health-related quality of life; MCID = minimal clinically important difference; MID = minimal important difference; NHL = non-Hodgkin lymphoma; QoL = quality of life; SD = standard deviation; SF-36 = Short Form-36; VAS = visual analogue scale.
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire-C30
Description
The European Organization for Research and Treatment of Cancer (EORTC) Quality of Life Questionnaire Core 30, or EORTC QLQ-C30, is 1 of the most commonly used patient-reported outcomes (PRO) measures in oncology clinical trials.62 It is a multidimensional, cancer-specific, evaluative measure of health-related quality of life (HRQoL). It was designed specifically for the purpose of assessing changes in participants’ HRQoL in clinical trials, in response to treatment. The core questionnaire of the EORTC QLQ-C30 consists of 30 questions that are scored to create 5 multi-item functional scales, 3 multi-item symptom scales, 6 single-item symptom scales, and a 2-item quality of life (QoL) scale, as outlined in Table 26. Version 3.0 of the questionnaire is the most current version and has been in use since December 1997. It is available in 90 different languages and is intended for use in adult populations only. Notably, the global QoL scale is also known as the Global Health Status (GHS).55
Table 26: Scales of EORTC QLQ-C30
Functional scales (15 questions) | Symptom scales (7 questions) | Single-item symptom scales (6 questions) | Global quality of life (2 questions) |
|---|---|---|---|
Physical function (5) Role function (2) Cognitive function (2) Emotional function (4) Social function (2) | Fatigue (3) Pain (2) Nausea and vomiting (2) | Dyspnea (1) Insomnia (1) Appetite loss (1) Constipation (1) Diarrhea (1) Financial impact (1) | Global quality of life (2) |
EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30.
Scoring
The EORTC QLQ-C30 uses a 1-week recall period in assessing function and symptoms. Most questions have 4 response options (“not at all,” “a little,” “quite a bit,” “very much”), with scores on these items ranging from 1 to 4. For the 2 items form the global QoL scale, however, the response format is a 7-point Likert-type scale, with anchors between 1 (very poor) and 7 (excellent).55
Raw scores for each scale are computed as the average of the items that contribute to a particular scale. This scaling approach is based upon the assumption that it is appropriate to provide equal weighting to each item that comprises a scale. There is also an assumption that, for each item, the interval between response options is equal (for example, the difference in score between “not at all” and “a little” is the same as “a little” and “quite a bit,” at a value of 1 unit). All the scales and single-item measures range in score from 0 to 100. Higher score for the functioning scales and global health status denotes a better level of functioning (i.e., a better state of the patient), while higher scores on the symptom and single-item scales indicate a higher level of symptoms (i.e., a worse state of the patient). Each raw scale score is converted to a standardized score that ranges from 0 to 100 using a linear transformation, with a higher score reflecting better function on the function scales, higher symptoms on the symptom scales, and better quality of life (i.e., higher scores simply reflect higher levels of response on that scale). According to the EORTC QLQ-C30 scoring algorithm, if there are missing items for a scale (i.e., the participant did not provide a response), the score for the scale can still be computed if there are responses for at least one-half of the items. In calculating the scale score, the missing items are simply ignored — an approach that assumes that the missing items have values equal to the average of those items for what the respondent completed.55
Psychometric properties of the EORTC QLQ-C30 have not been assessed in patients with lymphoma, therefore its validity, reliability, and responsiveness to change in this patient population remain unknown.
Minimal Important Difference
Cocks et al.56 used a systematic review of the literature and experts’ opinion to evaluate meaningful differences and magnitude of change in the QLQ-C30 scores. In a meta-analysis of 118 relevant papers (13.6% from US and Canada, 5.1% about hematological diseases) with timescales ranging from 4 days to 5 years, authors estimated trivial, small, medium, and large size classes for meaningful change in the scales. Medium and large changes could not be estimated for all scales due to insufficient data and response shift (i.e., psychological adaption of patients to their changing health status). Small differences were defined as subtle but nevertheless clinically meaningful changes (Table 27).
Table 27: Small Mean Differences of EORTC QLQ-C30 Subscales
EORTC QLQ-C30 scales | Small mean differences for Improvement | Small mean differences for Deterioration |
|---|---|---|
Functional scales | ||
Physical function | 2 to 7 | −10 to −5 |
Role function | 6 to 12 | −14 to −7 |
Cognitive function | 3 to 7 | −7 to −1 |
Emotional function | 6 to 9 | −12 to −3 |
Social function | 3 to 8 | −11 to −6 |
Symptom scales | ||
Fatigue | −9 to −4 | –5 to 10 |
Pain | −9 to −5 | 3 to 11 |
Nausea and vomiting | −9 to −3 | 5 to 11 |
Single-item symptom scales | ||
Dyspnea | −9 to −2 | 5 to 11 |
Insomnia | −9 to −5 | 2 to 9 |
Appetite loss | −13 to −7 | 2 to 14 |
Constipation | −10 to −4 | 5 to 15 |
Diarrhea | −11 to −3 | 5 to 15 |
Financial impact | < −3 | 2 to 10 |
Global quality of life | ||
Global quality of life | 5 to 8 | −10 to −5 |
EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30.
Among patients with lung, ovarian, or breast cancer, a 5-point change in the GHS/QoL subscale has been estimated as a clinically meaningful difference, whereas for other domains, a 5-point change from baseline and 10-point change in mean scores were estimated as a clinically meaningful difference.57,63 In the ZUMA-7 trial, clinically meaningful changes were defined as 10 points for QLQ-C30.16,56,57,60
EuroQoL 5-Dimensions 5-Levels Questionnaire
The EQ-5D-5L is a generic preference-based HRQoL instrument that has been applied to a wide range of health conditions and treatments. The EQ-5D-5L was developed by the EuroQoL Group as an improvement to the EQ-5D 3 level (i.e., EQ-5D-3L) to measure small and medium health changes and reduce ceiling effects. The EQ-5D-5L consists of the EQ-5D descriptive system and the EQ visual analogue scale (EQ VAS). The descriptive system comprises 5 dimensions: mobility, self-care, usual activities, pain/discomfort, and anxiety/depression. Each dimension is rated on 5 levels: level 1 “no problems,” level 2 “slight problems,” level 3 “moderate problems,” level 4 “severe problems,” and level 5 “extreme problems” or “unable to perform.” A total of 3,125 unique health states are possible, with 55555 (extreme problems in all of the dimensions) representing the worst health state and 11111 (no problems in any of the dimensions) representing the best state. The corresponding scoring of EQ-5D-5L health states is based on a scoring algorithm that is derived from preference data obtained from interviews using choice-based techniques (e.g., time trade-off) and discrete choice experiment tasks. The lowest score varies depending on the scoring algorithm used. The anchors are 0 (dead) and 1 (full health); however, negative values are also allowed to represent health states that a society considers worse than death. As an example, a Canadian scoring algorithm results in a score of –0.148 for health state 55555 (worst health state).58
Another component of the EQ-5D-5L, the EQ VAS, asks respondents to rate their health on a visual analogue scale from 0 (worst health imaginable) to 100 (best health imaginable). The EQ VAS records the respondent’s self-rated health on a vertical VAS where the end points are labelled 0 and 100. The respondents are asked to mark an X on the point of the VAS that best represents their health on that day.
The EQ-5D-5L index and VAS scores can be summarized and analyzed as continuous data. Hence, the EQ-5D-5L produces 3 types of data for each respondent:
a profile indicating the extent of problems on each of the 5 dimensions represented by a 5-digit descriptor, such as 11121 or 21143
a population preference-weighted health index score based on the descriptive system
a self-reported assessment of health status based on the EQ VAS
The EQ-5D-5L has been extensively validated across countries around the world and in various conditions.58 However, the psychometric properties of the EQ-5D-5L have not been assessed in patients with lymphoma, therefore its validity, reliability, and responsiveness to change in this patient population remain unknown.
Minimal Important Difference
To estimate the MID values of the EQ-5D-5L for each country-specific scoring algorithm, a simulation-based approach based on instrument-defined single-level transitions has been used. The simulation-based instrument-defined generally accepted MID estimate (mean ± SD) for Canada is 0.056 ± 0.011.59 In the ZUMA-7 trial, clinically meaningful changes were defined as 7 points for EQ-5D-5L VAS score, and 0.06 points for the EQ-5D-5L index for patients with lymphoma, based on relevant literature.45,60 In a study of 239 patients with multiple myeloma, the MID was estimated as 0.08 for improvement and 0.10 for deterioration.61
Pharmacoeconomic Review
Abbreviations
AE
adverse event
axi-cel
axicabtagene ciloleucel
ASCT
autologous stem cell transplant
BEAM
carmustine, etoposide, cytarabine, and melphalan
CAR
chimeric antigen receptor
CRS
cytokine release syndrome
DLBCL
diffuse large B-cell lymphoma
EFS
event-free survival
EM
etoposide and melphalan
EQ-5D-5L
5-Level EQ-5D
HDT
high-dose therapy
HGBL
high-grade B-cell lymphoma
ICER
incremental cost-effectiveness ratio
LBCL
large B-cell lymphoma
MCM
mixture cure model
OS
overall survival
PSM
partitioned survival model
QALY
quality-adjusted life-year
R-DHAP
rituximab plus dexamethasone, cytarabine, and cisplatin
R-GDP
rituximab plus gemcitabine, dexamethasone, and cisplatin
R-ICE
rituximab plus ifosfamide, carboplatin, and etoposide
SOC
standard of care
SMR
standardized mortality ratio
WTP
willingness to pay
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Axicabtagene ciloleucel (Yescarta), cell suspension in infusion bag, for IV infusion |
Submitted price | Axicabtagene ciloleucel, suspension of anti-CD19 CAR-positive viable T cells in approximately 60 mL for a target dose of 2 × 106 anti-CD19 CAR T cells/kg body weight, with a maximum of 2 × 108 anti-CD19 CAR T cells for patients weighing 100 kg or more: $485,021 per administration |
Indication | Treatment of adult patients with diffuse large B-cell lymphoma or high-grade B-cell lymphoma that is refractory to first-line chemoimmunotherapy or that relapses within 12 months of first-line chemoimmunotherapy. |
Health Canada approval status | NOC |
Health Canada review pathway | Standard review |
NOC date | December 6, 2022 |
Reimbursement request | Treatment of adult patients with relapsed or refractory large B-cell lymphoma, who are candidates for autologous stem cell transplant. |
Sponsor | Gilead Sciences Canada Inc. |
Submission history | Previously reviewed: Yesa Indication: Treatment of adult patients with relapsed or refractory LBCL after 2 or more lines of systemic therapy Recommendation date: August 15, 2019 Recommendation: Recommended with clinical criteria and condition |
CAR = chimeric antigen receptor; NOC = Notice of Compliance.
aThe previous CAR T-cell therapy review of axicabtagene ciloleucel went through the interim CAR T-cell therapy review process in which recommendations were issued by the CADTH Health Technology Expert Review Panel (HTERP).
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis PSM |
Target population | Reimbursement request population: Adult patients with relapsed or refractory LBCL who are candidates for ASCT |
Treatment | Axicabtagene ciloleucel (axi-cel; Yescarta), followed by subsequent third-line therapy consisting of enrolment in clinical trials for investigational therapies (40%), salvage chemotherapy (20%), radiotherapy (20%), ASCT (10%), or no treatment (10%) |
Comparator | SOC: Salvage chemotherapy (defined as a basket of chemotherapy regimens including R-GDP, R-ICE, R-DHAP, followed by HDT (defined as a basket of drug regimens including EM and BEAM) and ASCT in responders. This is followed by third-line therapy consisting of CAR T-cell therapies (50%), radiotherapy (40%), or no treatment (10%) |
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, life-years |
Time horizon | Lifetime (50 years) |
Key data source | ZUMA-7 |
Submitted results | ICER = $141,757 per QALY compared with SOC (incremental costs = $232,469; incremental QALYs = 1.64) |
Key limitations |
|
CADTH reanalysis results |
|
axi-cel = axicabtagene ciloleucel; ASCT = autologous stem cell transplant; BEAM = carmustine, etoposide, cytarabine, and melphalan; CAR = chimeric antigen receptor; EM = etoposide and melphalan; HDT = high-dose therapy; ICER = incremental cost-effectiveness ratio; LBCL = large B-cell lymphoma; OS = overall survival; PSM = partitioned survival model; QALY = quality-adjusted life-year; R-DHAP = rituximab + dexamethasone, cytarabine, and cisplatin; R-GDP = rituximab + gemcitabine, dexamethasone, and cisplatin; R-ICE = rituximab + ifosfamide, carboplatin, and etoposide; SOC = standard of care.
Conclusions
Evidence from the ZUMA-7 trial suggests that axicabtagene ciloleucel (axi-cel) is associated with clinically meaningful improvements in event-free survival (EFS) compared to standard of care (SOC) as second-line treatment in autologous stem cell transplant (ASCT)-eligible patients with relapsed or refractory large B-cell lymphoma (LBCL) within 12 months of first-line chemoimmunotherapy. It is not yet clear whether this EFS benefit will translate to improved overall survival (OS) as the data remain immature and follow-up is ongoing. As noted in the CADTH Clinical Review, as OS results are based on an interim analysis, there is a risk that the effect of axi-cel compared with SOC on survival is overestimated. This uncertainty is propagated into the submitted model given that the sponsor’s partitioned survival model (PSM) is informed by OS curves. The clinical trial population upon which the economic analysis was based consists of patients who are relatively stable and may not be generalizable to patients who are less stable or who do not meet the specific inclusion criteria of the trial.
In addition to the aforementioned limitations with the clinical evidence, CADTH identified several limitations with the sponsor’s economic submission. As part of the base-case reanalysis, CADTH addressed the uncertainties associated with long-term treatment efficacy by selecting an alternative extrapolation curve to inform OS for axi-cel; revising the pre-event (for the first 149 days of treatment) and postevent utility values; adjusting the distribution of subsequent therapies in the axi-cel arm to exclude investigational therapies; and applying a higher hazard of death among long-term survivors based on North American data. In CADTH’s base-case reanalysis, the incremental cost-effectiveness ratio (ICER) of axi-cel is $404,418 per QALY (incremental costs: $235,146; incremental QALYs: 0.58) compared with SOC, assuming 50% of patients who fail SOC would be treated with CAR T-cell therapies in the third-line setting. A price reduction of 45% would be necessary for axi-cel to be considered cost-effective at a willingness-to-pay (WTP) threshold of $50,000 per QALY.
The cost-effectiveness of axi-cel was sensitive to the assumptions regarding the selected parametric model distribution used to extrapolate OS for axi-cel. The CADTH base-case reanalysis estimated a smaller OS benefit with axi-cel when compared to the sponsor’s base case (i.e., incremental life-years: 1.50 [sponsor’s base case] versus 0.24 [CADTH’s reanalysis]), and this translated to a smaller difference in QALYs between axi-cel and SOC. Nonetheless, similar to the sponsor’s analysis, the majority of the QALY gains (71%) conferred by axi-cel in the CADTH reanalysis were derived from the period beyond which there are observed trial data. Furthermore, clinical uncertainties in the extrapolation period could not be adequately explored due to the inflexible modelling approach. This is particularly concerning when considering the uncertainties in the distribution of subsequent therapy. The cost-effectiveness results from both the sponsor and CADTH’s base-case reanalysis are informed by underlying OS curves from ZUMA-7 trial in which 56% of patients on SOC therapy received subsequent CAR T-cell therapies in the third-line setting. It is uncertain whether this reflects the proportion of patients who will have access to CAR T-cell therapy for third-line treatment.
The cost-effectiveness of axi-cel as second-line treatment in patients who are ineligible for ASCT and in patients who may not meet the specific inclusion or exclusion criteria of the ZUMA-7 trial, which reflects a component of the Health Canada indication, is unknown.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process.
One patient group, Lymphoma Canada, provided input through data collected from 2 online patient surveys — the first gathered information from 97 patients with diffuse large B-cell lymphoma (DLBCL) in 2018 and the second gathered information from 23 patients with DLBCL in 2022. Overall, patients’ disease experience was influenced by the physical symptoms associated with LBCL (e.g., fatigue, body aches, thrombocytopenia, enlarged lymph nodes, neutropenia, and night sweats) and the psychosocial effect (e.g., fear of disease progression and relapse). The most important outcomes for patients included delaying disease progression and achieving long-term remission, with the ultimate objective of improving survival; reducing side effects from treatments; and maintaining quality of life. Of the 3 patients who had experience with the drug under review as third-line therapy, 1 had relapsed following treatment, 1 had been in remission for less than 6 months, and 1 had been in remission for 1 to 2 years. All 3 patients were away from home for more than 3 months to access treatment with axi-cel, and treatment access was described as a significant challenge. All 3 respondents reported thrombocytopenia as a treatment side effect, with 2 respondents reporting fever, neutropenia, anemia, nausea/vomiting, diarrhea, joint or muscle pain, and fatigue.
Registered clinician input was received from 3 groups: Lymphoma Canada, Ontario Health Hematology Cancer Drug Advisory Committee, and Cell Therapy Transplant Canada. According to clinician input, the current pathway of care for patients with relapsed or refractory LBCL is salvage chemotherapy, with an intent to proceed to ASCT in patients who are chemosensitive. The most common salvage chemotherapy regimen used in Canada is a combination of rituximab, gemcitabine, dexamethasone, and cisplatin (R-GDP). The most commonly used high-dose therapy (HDT) regimens were reported to be a combination of etoposide and melphalan (EM) or a combination of carmustine (BCNU), etoposide, cytarabine (Ara-C), and melphalan (BEAM). Clinicians noted that although ASCT is the best current curative option for patients with relapsed or refractory LBCL, it is an intensive intervention performed at a limited number of centres with known rates of relapse and multiple late toxicities. Clinicians indicated that axi-cel is currently available in Canada for patients with relapsed or refractory LBCL after 2 lines of systemic therapy and that ZUMA-7 explores the earlier use of axi-cel as second-line treatment. Across the feedback received, axi-cel was unanimously expected to shift the current treatment paradigm by replacing intensive chemoimmunotherapy and ASCT as the new preferred second-line treatment for patients with relapsed or refractory LBCL. Furthermore, it was noted that axi-cel should be eligible to the group of population (e.g., older age, comorbidities, Eastern Cooperative Oncology Group Performance Status [ECOG PS] score of 2) who might not be expected to tolerate ASCT. To assess treatment response to axi-cel, the clinician groups noted a variety of response outcomes may be used (e.g., overall response rate, complete response rate, EFS, progression-free survival, OS) and may involve restaging CT and PET/CT scans.
CADTH-participating drug plans highlighted several implementation and economic considerations for axi-cel. Plans noted concerns that the existing capacity may not be able to meet the anticipated demand in Canada. Given the requirement for specialized and accredited centres where the therapy can be administered, accessing CAR T-cell therapy may require interprovincial travel and, without full coverage of interprovincial reimbursement, may impact equitable access across Canada. The evidence surrounding CAR T-cell re-treatment was noted to be another area of uncertainty.
Several of these concerns were addressed in the sponsor’s model:
The PSM was informed by the EFS and OS curves, 2 outcomes that are valued by patients.
The impact of disease and treatment on patients’ quality of life was captured with utility values. Adverse events (AEs) were incorporated as disutilities within the analyses.
The SOC modelled by the sponsor reflected the current treatments available to patients with relapsed or refractory LBCL who undergo ASCT (including salvage chemotherapy and HDT regimens).
In addition, CADTH addressed some of these concerns as follows:
A scenario analysis exploring re-treatment based on the fact that 5% of patients who received axi-cel in the ZUMA-7 trial were re-treated.
CADTH was unable to address the following concerns raised from stakeholder input:
Clinical data were not available for the broader Health Canada–indicated population (i.e., not eligible for ASCT) who may benefit from CAR T-cell therapy. As such, the modelled population in the cost-effectiveness analysis and budget impact analysis (BIA) excludes this subgroup of patients.
Capacity is not explicitly considered in the model. The sponsor assumes all patients with relapsed or refractory LBCL have access to axi-cel if required and that the manufacturing time is similar to that observed in the trial.
Accessing axi-cel may require interprovincial travel. These costs were not considered in the analysis given heterogeneity across provinces in terms of their policy for interprovincial billings. Furthermore, given the public payer perspective, patient-borne interprovincial travel costs were not considered as it was considered outside the scope of this review’s perspective.
Economic Review
The current review is for axi-cel (Yescarta) for adult patients with relapsed or refractory LBCL who are candidates for ASCT.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
Axi-cel is indicated for the treatment of adult patients with DLBCL or high-grade B-cell lymphoma (HGBL) that is refractory to first-line chemoimmunotherapy or that relapses within 12 months of first-line chemoimmunotherapy. The sponsor submitted a cost-utility analysis of axi-cel compared with SOC in the treatment of adult patients with relapsed or refractory LBCL who are candidates for ASCT.1 The modelled population aligned with the reimbursement request criteria but differed from its Health Canada indication. SOC consisted of salvage chemoimmunotherapy followed by HDT and ASCT in responders. Salvage chemoimmunotherapy was defined as a basket of chemotherapy regimens including R-GDP, R-ICE (rituximab plus ifosfamide, carboplatin, and etoposide), and R-DHAP (rituximab plus dexamethasone, high-dose cytarabine, and cisplatin. HDT was defined as a basket of drug regimens including EM and BEAM.1 For patients who failed their second-line treatment, the sponsor assumed 90% would proceed to subsequent therapy. For patients who received axi-cel, subsequent treatment consisted of enrolment in a clinical trial for investigational treatment (40%), salvage chemoimmunotherapy (i.e., R-GDP, R-DHAP, and R-ICE; 20%), radiotherapy (20%), or ASCT (10%), whereas patients who failed SOC proceeded to CAR T-cell therapies (50%) or radiotherapy (40%).
Axi-cel is a CD19-directed genetically modified autologous T-cell immunotherapy individually prepared from patients’ peripheral blood mononuclear cells. It is available as a cell suspension for infusion containing a target dose of 2 × 106 CAR T cells/kg body weight (range, 1 × 106 cells/kg to 2.4 × 106 cells/kg), with a maximum of 2 × 108 CAR T cells for patients weighing 100 kg or more.2 It is provided as a single-dose, one-time treatment.2 The sponsor’s submitted price of axi-cel is $485,021 per infusion,1 not including costs associated with pretreatment (i.e., leukapheresis, bridging therapy, conditioning chemotherapy) and postinfusion management (i.e., ICU and non-ICU inpatient stay). The comparator for this analysis consisted of SOC, with treatment costs divided into 3 phases: salvage chemoimmunotherapy, HDT, and ASCT. Based on the proportion of patients receiving R-GDP (80%), R-ICE (10%), and R-DHAP (10%), the drug cost of salvage chemotherapy over a mean of 2.3 treatment cycles was estimated by the sponsor to be $10,011. The drug cost of HDT, derived from either 1 cycle of EM (80%) or 1 cycle of BEAM (20%), was estimated to be $3,529.1 The cost of ASCT was $77,956 based on the Ontario interprovincial billing rates for an adult ASCT with a hospital stay longer than 72 hours, which includes all facility costs including inpatient and diagnostic costs.3
The outcomes of interest were QALYs and life-years. The analysis takes the perspective of a public health care payer. The time horizon in the base case was specified by the sponsor as a lifetime time horizon (50 years) with monthly cycles. The discount rate for costs and outcomes was 1.5% per year.1
Model Structure
The sponsor used a PSM to capture all costs and outcomes associated with axi-cel and SOC. The model included 3 health states: event-free, postevent, and death (Figure 1).1 The model considered the pretreatment period with patients who are considering second-line treatment entering the PSM in the event-free health state. The proportion of patients with event-free, postevent, and death were estimated over time based on the OS and EFS curves, informed by the ZUMA-7 trial.1 The proportion of patients with progressed disease (i.e., postevent state) was estimated as the difference between the proportion of patients who are living (estimated from the OS curve) and the proportion of patients who are event-free (estimated from the EFS curve).1 In other words, from the event-free state, patients can transition to the death state or progress to the postevent state where they can remain until transitioning to the death state (i.e., patients cannot go back to the event-free health state). Events were defined as disease progression per Lugano Classification, commencement of new lymphoma therapy (including stem cell transplant in the axi-cel arm without axi-cel–induced response or re-treatment of axi-cel), or death from any cause.4 Patients who experienced an event that was not death may transition to third-line therapy as determined by the time-to-next-treatment curve derived from the ZUMA-7 trial data.1 If patients were in the EFS state for more than 5 years, they were assumed to be in long-term remission.1
Model Inputs
Baseline patient characteristics were derived from ZUMA-7, a phase III, randomized, open-label multicentre clinical trial evaluating the efficacy of axi-cel versus SOC in patients with relapsed or refractory LBCL (i.e., age = 57 years, weight = 84 kg; proportion male = 66%).1
The clinical outcomes informing the model (i.e., OS, EFS) were those reported by the ZUMA-7 trial, based on the intention-to-treat population. Survival data from the ZUMA-7 trial (data cut-off date: March 18, 2021) were extrapolated to derive long-term survival estimates of OS, EFS, and time to next treatment to inform the economic model.1 The data were fitted by different statistical methods (i.e., parametric curve, mixture cure model [MCM], spline model) and different parametric functions. Under the assumption that both axi-cel treatment and ASCT may be curative, the MCM was selected for use in the sponsor’s base case. This statistical method estimates a likelihood of “cure” (i.e., cure fraction) in which the survival outcomes for this group, compared to the group who does not achieve long-term remission, would be different. The OS for the “cured” group was informed by adjusting pre-2020 age- and sex-matched Canadian mortality rates,1 with an additional risk of excess mortality related to the disease (hazard ratio [HR] = 1.09).5 The survival function for the noncured group was evaluated with a diversity of parametric models with the statistical model selected based on clinical plausibility and statistical fit. The sponsor used a generalized gamma hazard to model the OS for the uncured fraction (i.e., patients who do not achieve long-term remission) in both the axi-cel and SOC arm. An MCM was also selected to estimate EFS in which, rather than estimating the cure fraction, the model predicted the likelihood of remaining event-free (i.e., event-free fraction). In the sponsor’s base case, log-logistic and exponential distributions were selected for the EFS for axi-cel and SOC, respectively.1
Utility data for the event-free health state were derived directly from the EQ-5D-5L index data collected in the ZUMA-7 trial with Canadian tariffs applied.1 Treatment-specific utilities were applied to the event-free state during the first month for axi-cel and the first 3 months for SOC. The sponsor assumed the utility for the postevent state would be equal to the utility derived from the JULIET trial.6 Moreover, the sponsor assumed that the utility values for patients who survived for at least 5 years would be equal to the sex-matched general population values, with an age-related decrement incorporated.1,7 Refer to Table 11 for the health state utility values used in the sponsor’s base case.
The model included AEs grade 3 or higher as well as those graded 2 or higher for AEs of special interest, including cytokine release syndrome (CRS), neurologic events, and hypogammaglobulinemia.1 Treatment-specific AE rates were informed from the ZUMA-7 trial.8 The value of the utility decrements was informed from published literature and applied as a one-time decrement to the first model cycle.9-12 It was assumed that AEs would have no further impact on costs as these would be managed during the initial hospitalization period.
Costs captured in the model included drug acquisition, administration, monitoring, disease management, subsequent therapy, and end-of-life care. Drug acquisition costs were sourced from the Ontario Drug Benefit Formulary,13 CADTH’s previous pharmacoeconomic reviews,14-19 and the Canadian Association of Provincial Cancer Agencies,20 with dosing schedules based on the chemotherapy regimen monographs from Cancer Care Ontario21 and the Alberta Health Services clinical practice guideline on lymphoma.22 Costs specific to axi-cel included leukapheresis, bridging therapy, conditioning chemotherapy, and drug acquisition and administration (Table 12). The sponsor considered the costs typically associated with the ongoing monitoring, which were obtained from the Canadian Institute for Health Information’s Patient Cost Estimator for an inpatient hospitalization for malignant lymphoma.23 For patients receiving SOC, treatment costs included salvage chemoimmunotherapy, HDT, and ASCT (Table 12). The salvage chemotherapy and HDT regimen were assumed to be administered on an outpatient basis. The total weighted costs of subsequent therapy differed by prior treatment (e.g., those who received axi-cel or SOC as second line) and were applied as a one-off cost at the time of initiation of third-line therapy based on the TTNT curve (Table 14). Treatment monitoring costs and health care resource use costs were sourced from the Ontario Ministry of Health Schedule of Benefits for laboratory and physician services,24 whereas end-of-life costs were based on the average cost of care for patients with terminal lymphoma from the Ontario Cancer Registry.25
Summary of Sponsor’s Economic Evaluation Results
The sponsor conducted a probabilistic analysis based on 1,000 simulations. Discrepancies were observed between the deterministic (Table 13) and probabilistic results (Table 3). This discrepancy was due to a divergence in the predicted life-years as the deterministic analysis estimated fewer life-years associated with SOC compared to the probabilistic analysis (deterministic analysis: 9.96 versus probabilistic analysis: 10.28). Upon further investigation, CADTH noted that the source of this discrepancy lay in the definition of the probabilistic distributions associated with the OS parametric distributions. The probabilistic findings are presented below.
Base-Case Results
In the sponsor’s requested reimbursement population, axi-cel was associated with an incremental QALY gain of 1.64 and an incremental cost of $232,469, resulting in an ICER of $141,757 per QALY compared with SOC (Table 3) under the assumption that 50% of patients who fail SOC would be treated with CAR T-cell therapies in third line.
The sponsor’s analysis predicted that axi-cel was associated with a longer duration of life than SOC (i.e., incremental life-years: 1.50). Given the duration of the ZUMA-7 trial (i.e., 37 months) in contrast to the model’s time horizon (i.e., 50 years), it is important to note that the majority of the QALY benefit (87%) realized by patients receiving axi-cel was derived from the period beyond which there are observed trial data (i.e., extrapolated period). Most of the QALYs gained by patients receiving axi-cel were realized in the event-free state (74%), whereas patients receiving SOC realized most of their QALY gains in the postevent state (60%). The key cost driver among patients receiving axi-cel was the cost of axi-cel acquisition, accounting for 91% of the total cost. Given the sponsor’s assumption that patients treated with SOC would be receiving CAR T-cell therapies in subsequent treatment lines (≥ third line), the main cost driver in the SOC arm that accounted for 85% of its total expected cost was the acquisition cost of CAR T-cell therapy. The sponsor’s submitted analysis is based on the publicly available prices for all drug treatments.
Table 3: Summary of the Sponsor’s Economic Evaluation Results (Probabilistic)
Drug | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER vs. SOC ($/QALY) |
|---|---|---|---|---|---|
SOC | 336,588 | Reference | 7.71 | Reference | Reference |
Axi-cel | 569,057 | 232,469 | 9.35 | 1.64 | 141,757 |
axi-cel = axicabtagene ciloleucel; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; SOC = standard of care.
Source: Sponsor’s pharmacoeconomic submission.1
Additional results and calculations from the sponsor’s submitted economic evaluation base case are presented in Appendix 3.
Scenario Analysis Results
The sponsor assessed several model parameters and assumptions in probabilistic scenario analyses. The sponsor’s findings were generally sensitive to the scenario analyses tested. When adjusting the model for treatment switching to cell immunotherapy in the subsequent line of SOC, the ICER decreased to $40,554 per QALY. In contrast, when selecting the log-logistic distribution to extrapolate the OS curves for axi-cel and SOC, the ICER increased to $514,797 per QALY. All other scenarios resulted in ICERs between $58,898 and $402,350 per QALY.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis:
Exclusion of ASCT-ineligible patient population: The sponsor’s economic evaluation was specifically conducted using adult patients with relapsed or refractory LBCL who are candidates for ASCT, which is a subset of the Health Canada indication. The sponsor submitted a request for deviation to focus their analysis on this subpopulation (i.e., their reimbursement request). During the review process, the clinical expert panel convened by CADTH noted that the expected place in therapy for axi-cel would align more closely with its Health Canada indication (i.e., second-line treatment of adult patients with DLBCL or HGBL that is refractory to first-line chemoimmunotherapy or that relapses within 12 months of first-line chemoimmunotherapy, regardless of ASCT eligibility). From their perspective, patients who could potentially benefit from CAR T-cell therapy should not be excluded on the basis of ASCT eligibility. This is because the current SOC requires chemosensitivity which cannot be established up front; as such, patients must start salvage chemotherapy with the intent of receiving ASCT before a response can be observed, and those without a response would switch to receive CAR T cells in the third-line setting. They further noted challenges in implementing an initiation criterion based on ASCT eligibility (refer to the Issue for Consideration section for more information). The clinical experts indicated that any patient with adequate organ function and good performance status (i.e., Eastern Cooperative Oncology Group Performance Status [ECOG PS] ≤ 2) who could tolerate the known toxicities of CAR T-cell therapy (e.g., CRS) would be suitable for axi-cel treatment in real-world clinical practice. However, the clinical evidence informing the economic evaluation was based on the ZUMA-7 trial in which ASCT eligibility was an inclusion criterion. To evaluate the broader Health Canada population, clinical evidence on the population ineligible for ASCT would be required to inform the clinical effectiveness inputs within the economic model and alternative comparators would need to be considered (e.g., salvage chemotherapy, polatuzumab in combination with bendamustine and rituximab [Pola-BR]).
CADTH could not address this limitation given the lack of clinical data on the efficacy of axi-cel as second-line treatment in ASCT-ineligible patients. As such, the cost-effectiveness of axi-cel in this population is unknown.
Inflexible modelling approach: The sponsor used a PSM to capture all costs and outcomes associated with axi-cel and SOC. Although the PSM has routinely been used to model oncology treatments, there are numerous issues documented with its use.26,27 As there is no explicit modelling of transitions between states, subsequent therapy had no impact on OS within the submitted model. This is concerning given access to CAR T-cell therapy varies across jurisdictions. The OS curves are informed by the ZUMA-7 trial in which 56% of patients in the SOC group received subsequent CAR T-cell therapy in the third-line setting, while the sponsor assumed that 50% of patients who fail SOC would incur the cost of CAR T-cell therapy. Although it was possible to alter the distribution of subsequent therapies, doing so would only impact the cost of subsequent therapy and not of the underlying OS estimates, with the sole exception of the scenario analysis that incorporated the crossover adjustment (i.e., this would address a scenario whereby no patients receiving SOC would receive subsequent third-line CAR T-cell therapies). The clinical effects for alternate distribution of CAR T cells in third-line treatment is unknown.
The PSM approach further introduces structural assumptions about the relationship between EFS and OS (i.e., non–mutually exclusive curves), which is potentially problematic since EFS and OS are likely dependent outcomes. Clinical expert opinion suggested that survival is linked to the occurrence of an event and thus the transition probability to death should vary for patients within the event-free state compared to those in the postevent state.
CADTH could not fully address this limitation given the structure of the model.
Both axi-cel and tisagenlecleucel are indicated in the third-line setting in Canada, although access to CAR T-cell therapy varies across jurisdictions. To reflect the jurisdictions that do not presently fund CAR T-cell therapy in the third-line setting, CADTH conducted scenario analyses. These scenario analyses made the following adjustment: (1) incorporated the sponsor’s crossover adjustments (i.e., applied an HR to adjust the OS curve for SOC); and (2) reassigned the proportion of patients who were expected to be on subsequent third-line therapy with CAR T-cell therapy to chemotherapy (i.e., excluded CAR T-cell therapies).
Uncertainty in the extrapolation of OS for axi-cel: The sponsor selected an MCM based on a study showing the long-term OS of axi-cel in ZUMA-1 (a phase II, single-arm study of patients with LBCL [N = 101] treated with axi-cel in the third line), as well as a study that compared the fit of mixture cure and standard parametric models to the 12-month data cut-off of ZUMA-1.1,28 Although the clinical expert feedback sought by CADTH noted the plausibility of cure in both interventions, there remains uncertainty in the sponsor’s predicted cure rate for axi-cel. The sponsor’s base case estimated a 53% cure fraction at 5 years for axi-cel. This is highly uncertain given the limited follow-up time of the ZUMA-7 trial. As noted in the CADTH clinical report, the sponsor's OS results are immature and, because it is based on an interim analysis, there is a risk that the effect of axi-cel compared with SOC on survival is overestimated. Indeed, considering that the OS data were based on an interim analysis with large confidence intervals that included appreciable benefit as well as little-to-no difference, longer follow-up is required to ascertain the presence and magnitude of OS benefit. The ZUMA-1 trial reported a cure fraction of 52% at year 2, and existing real-world evidence have reported a 5-year OS for CAR T-cell therapies of between 30% and 50%.29 Although these estimates are derived in a later line of therapy (third line), CADTH clinical expert feedback noted that any estimates of more than 50% would be considered highly optimistic.
There is further uncertainty regarding the expected OS benefit in a Canadian setting following the implementation of axi-cel to a broader population that goes beyond the selective patient population recruited within the clinical trial. ZUMA-7 consisted exclusively of patients with an ECOG PS of 1 or less with a mean age of 57 years. The clinical experts noted that the average age for patients with relapsed or refractory LBCL is expected to be higher in Canada. Indeed, a Canadian real-world population-based study of a cancer registry estimated the median age of patients with incident LBCL to be 67 years in males and 71 in females.30 As such, if axi-cel were to become available in a clinical practice where patients are likely to have more diverse clinical and demographic profiles, there remains uncertainty about the presence and the magnitude of the OS benefit in a real-world setting.
In the sponsor’s base case, a generalized gamma hazard was used as the parametric distribution of OS for both axi-cel and SOC. According to the sponsor, their model selection was based on several factors including best statistical fit, clinical expert opinion and the requirement that long-term survivors in both arms should follow the same functional form for the remainder of the model time horizon. However, the generalized gamma distribution selected by the sponsor to extrapolate OS for axi-cel yielded the worst goodness of fit, based on Bayesian information criterion (BIC), of the available distributions and lacked face validity as noted above when CADTH sought clinical expert feedback on the OS extrapolations. The sponsor’s model allowed exploration of several parametric distributions. However, significant uncertainty remains around these and their impact on the extrapolation of overall comparative costs and effectiveness. The appropriateness of any extrapolation depends on the quantity of observed data available. Given the lack of long-term evidence and the high amount of censoring observed in the interim analysis of OS at 18 months within the ZUMA-7 trial, it is not possible to conclude that axi-cel is a curative therapy. Thus, although an OS benefit with axi-cel was deemed plausible, the magnitude of such a benefit was uncertain in the absence of more robust long-term evidence.
This limitation is particularly concerning giving that the majority of QALY benefit (87%) realized by patients receiving axi-cel in the sponsor’s base-case analysis was derived from the period beyond which there are observed trial data (i.e., extrapolated period). To address some of the uncertainties in long-term OS estimates, CADTH changed the distributional assumption to log-logistic to derive a more plausible OS curve for axi-cel. This distribution had the best statistical fit of the 7 parametric distributions provided by the sponsor (BIC = 714.9 [generalized gamma] versus 709.6 [log-logistic], respectively). According to clinical expert feedback, this change further produced a more plausible OS curve in the absence of long-term evidence (i.e., 5-year cure fraction = 44%), while still conferring a survival benefit with axi-cel.
Utility values associated with uncertainty: The sponsor derived utility estimates for the on- and off-treatment event-free states directly from the raw EQ-5D index data collected in the ZUMA-7 trial and transformed them to utility weights based on Canadian population tariffs. However, in selecting this approach, patients preparing to receive and receiving the axi-cel infusion (in the first month) or undergoing SOC including ASCT (in the first 3 months) would have similar or better quality of life than the reported general age- and sex-adjusted Canadian population (index score = 0.827).7 Clinical expert feedback sought by CADTH further noted that time-dependent utilities may better reflect the impact of treatment in the short-term given that patients are expected to undergo an array of taxing treatments with varying timelines as part of axi-cel infusion or SOC. However, after 5 months posttreatment, the quality of life would be similar for patients in the event-free state, regardless of the second-line treatment received.
In addition, the sponsor sourced the utility value associated with the postevent state from the JULIET trial data for tisagenlecleucel in patients with relapsed or refractory LBCL following 2 or more lines of systemic therapy. In doing so, the sponsor assumed that the postevent utility for this patient population (i.e., progressed disease following axi-cel or SOC in second line) would be equal to the postevent utility derived from the JULIET trial (i.e., further progressed LBCL following failure of CAR T-cell therapy in the third-line setting). The clinical experts consulted by CADTH indicated that the quality of life among patients who progress following CAR T-cell therapy in second line would not be equivalent to that of patients who progress following CAR T-cell therapy in later lines because this reflects a more biologically aggressive disease.
To address these limitations, CADTH applied time-dependent utility estimates based on shorter 50-day intervals, based on the ZUMA-7 trial, for the first 149 days. Thereafter, CADTH assumed that utility values did not exceed age-adjusted population utility norms. Utilities based on shorter time intervals were considered more valid (i.e., utility estimates did not exceed age- and sex-matched Canadian utility estimates) and yielded higher sensitivity to detect differences in the quality-of-life impacts between therapies. For the postevent health state, in line with clinical expert opinion, a utility value of 0.73 was assumed. This value had improved face validity as it remained lower than the utility estimates in the pre-event health states but was higher than the original postevent estimates derived from the JULIET trial.
CADTH acknowledges that the trial-based values may overestimate the event-free utilities giving that the ZUMA-7 population was biologically fitter by design (refer to Key Limitations in Table 2). In light of the uncertainties in the utility estimates, CADTH conducted a scenario analysis sourcing utilities from alternate sources.31-33 The utility value assigned to patients while on treatment with axi-cel in the first month (0.74) was sourced from an ad hoc analysis from a phase I/II safety management study of axi-cel in combination with atezolizumab (ZUMA-6) for the treatment of patients with refractory LBCL,33,34 whereas the utility estimate for patients while on treatment with SOC in the first 3 months (0.673) was sourced from a published study.31 Finally, the event-free utility assigned to patients following treatment with either axi-cel or SOC and before an event (0.823) was sourced from a study reporting preference-based EQ-5D index scores for chronic conditions in the US.32
Inconsistent modelling of subsequent treatment mix: CADTH noted that the estimated treatment mix for subsequent therapies varied substantially between patients receiving axi-cel and patients receiving SOC. Of note, 40% of patients receiving the axi-cel infusion were assumed to seek subsequent treatment through clinical trials for investigational therapies (at no additional cost to drug plans), while this assumption was extended to zero patients in the SOC arm. This implicitly assumes patients receiving axi-cel would have access to other treatment options that are not available to those on SOC, which lacks face validity. Furthermore, by assuming that patients receiving axi-cel were disproportionately more likely than patients receiving SOC to seek clinical trials, the sponsor effectively reduced the costs associated with subsequent treatment in the axi-cel arm.
In light of this limitation, CADTH conducted a reanalysis assuming that zero patients in the axi-cel arm receive subsequent therapies through clinical trials. The proportion of axi-cel clinical trial recipients was redistributed to chemotherapy, radiotherapy, and no active treatment based on clinical expert feedback.
Mortality of long-term survivors: The sponsor applied a standardized mortality ratio (SMR) of 1.09 to increase the hazard of death relative to the general population for those who are assumed to achieve long-term remission (i.e., the cure fraction). The sponsor relied on a previous study of newly diagnosed DLBCL that reported the SMR of patients in a French cohort (N = 820) who were event-free at 24 months following immunochemotherapy.5 The same study reported higher excess mortality (SMR = 1.18) in the American cohort of patients with DLBCL (N = 767). CADTH considers that the higher estimate would better capture the excess mortality of an adult cohort with relapsed or refractory LBCL who have received extensive prior treatments including first-line chemotherapy followed by second-line axi-cel or ASCT. A higher risk of mortality is expected due to potential late toxicities of therapy. CADTH sought clinical expert feedback that supported the use of the higher SMR in the long term surviving population, and further noted that it would be more reasonable to align the potential excess mortality experienced by a Canadian population of patients to estimates derived from a North American population rather than a European population.
To address this limitation, CADTH performed a reanalysis that adjusted the hazard of death to 1.18%.
In addition, the following key assumptions were made by the sponsor and have been appraised by CADTH (refer to Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation
Sponsor’s key assumption | CADTH comment |
|---|---|
Long-term survivors (i.e., patients who were alive and free of progression at 5 years and beyond) were assumed to have utility values equal to those of the age-matched and sex-matched general Canadian population for the remainder of the model’s lifetime horizon. | CADTH sought clinical expert feedback who concurred that, notwithstanding the existing data gap, it is broadly recognized that once patients reach the 5-year posttreatment point, the clinical expectation is that their quality of life begins to match that of the general population. However, it remains uncertain whether long-term survivors would attain a health state utility in line with the general population while experiencing an elevated mortality risk. |
All patients expected to receive a CAR T-cell infusion will receive leukapheresis, bridging therapy, and conditioning chemotherapy. | Conservative. In ZUMA-7, the proportions of patients who received leukapheresis (98.9%), bridging therapy (36.1%), and conditioning chemotherapy (95.6%) were lower. |
94% of patients will receive a CAR T-cell infusion. | Uncertain. Based on clinical expert feedback, the rates of CAR T-cell infusion may be impacted by both patient characteristics and implementation consideration. Time to manufacture and infuse CAR T cells beyond that observed in the ZUMA-7 trial may affect the proportion of patients who could receive a CAR T-cell infusion. |
Chemotherapy drug acquisition and administration costs for patients who receive SOC reflect Canadian practice. Equal efficacy was assumed across regimens. | Reasonable. |
Cost of in-hospital care for axi-cel and SOC included posttreatment AE monitoring and care. | Appropriate. In the ZUMA-7 trial, CRS, neurologic events, and hypogammaglobulinemia were monitored during the first 7 days after infusion with axi-cel. It appears that the most concerning AEs occurred in the initial days after infusion. Because the model assumed 14 days of hospitalization, the assumption that AE costs would already be captured as part of their initial hospitalization stay is appropriate. |
AE = adverse event; axi-cel = axicabtagene ciloleucel; CAR = chimeric antigen receptor; CRS = cytokine release syndrome; SOC = standard of care.
CADTH Reanalyses of the Economic Evaluation
Base-Case Results
CADTH’s reanalysis addressed several limitations within the submitted economic model. The CADTH base case was derived by making changes in the model parameter values and assumptions, in consultation with clinical experts. The following changes were applied: adjusting the parametric distribution to log-logistic to derive a more plausible OS curve for axi-cel; employing time-dependent utility estimates based on shorter 50-day intervals, based on the ZUMA-7 trial, for the first 149 days, while assuming that utility values did not exceed age- and sex-adjusted population utility norms thereafter; revising the postevent utility value; assuming that zero patients in the axi-cel group receive subsequent therapies through clinical trials; and adjusting the hazard of death among long-term survivors. These changes are summarized in Table 5.
Table 5: CADTH Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Changes to derive the CADTH base case | ||
1.Inappropriate modelling and distributional assumptions in estimating OS (axi-cel) |
(5-year OS: 51%; cure fraction: 53%) |
(5-year OS: 46%; cure fraction: 44%) |
2.Utility values | Axi-cel (event-free)a = 0.833 SOC (event-free)a = 0.826 Off treatment (event-free) = 0.837 Postevent = 0.710 | Pre-event
Postevent = 0.73 |
3.Subsequent treatment mix (axi-cel) | Chemotherapy: 20% Radiotherapy: 20% ASCT: 10% Clinical trial: 40% Not seeking active treatment: 10% | Chemotherapy: 50% Radiotherapy: 20% ASCT: 10% Clinical trial: 0 Not seeking active treatment: 20% |
4.Excess mortality of “cure” group, SMR (SE) | 1.09 (0.04) | 1.18 (0.17) |
CADTH base case | 1 + 2 + 3 + 4 | |
ASCT = autologous stem cell transplant; axi-cel = axicabtagene ciloleucel; MCM = mixture cure model; OS = overall survival; SE = standard error; SMR = standardized mortality ratio.
aTreatment-specific utilities were applied to the event-free state during the first month for axi-cel and the first 3 months for SOC.
bCADTH assumed that beyond 149 days, utility values did not exceed age- and sex-adjusted utility norms. For instance, the age-adjusted utility values for the age range of 55 years to 64 years is 0.830 for men and 0.820 for women.
In the reimbursement requested population, CADTH’s base-case reanalysis estimated that axi-cel was $235,146 more expensive and yielded 0.58 more QALYs when compared to SOC for adult patients with relapsed or refractory LBCL who are candidates for ASCT. This resulted in an ICER for axi-cel of $404,418 per QALY when compared to SOC (Table 6). The probability that axi-cel was cost-effective at a WTP threshold of $50,000 per QALY was 0%. A detailed breakdown of the disaggregated results is available in Table 15. It is important to note that, similar to the sponsor’s analysis, the CADTH reanalysis was based on the assumption that 50% of patients who fail SOC would be treated with CAR T-cell therapies in the third-line setting.
The estimated ICER was higher than the sponsor’s base-case value, driven primarily by adjusting the parametric distribution of OS to log-logistic. This produced a more plausible OS curve for axi-cel in the absence of long-term evidence, while still conferring a survival benefit with axi-cel. All of axi-cel’s QALY gain compared with SOC was found to be derived in the event-free health state. Furthermore, 71% of the QALY benefit was derived from the extrapolated period in which there are no OS data to confirm this modelled benefit.
The majority of the total costs in the axi-cel group (80%) and the SOC group (68%) were similarly related to CAR T-cell treatment acquisition costs.
Table 6: Summary of the Stepped Analysis of the CADTH Reanalysis Results
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
Sponsor’s base case (probabilistic) | SOC | 336,588 | 7.71 | Reference |
Axi-cel | 569,057 | 9.35 | 141,757 | |
Sponsor’s base case (deterministic) | SOC | 336,857 | 7.47 | Reference |
Axi-cel | 569,098 | 9.31 | 126,046 | |
CADTH reanalysis 1 | SOC | 336,857 | 7.47 | Reference |
Axi-cel | 569,547 | 8.35 | 265,051 | |
CADTH reanalysis 2 | SOC | 336,857 | 7.55 | Reference |
Axi-cel | 569,098 | 9.28 | 134,444 | |
CADTH reanalysis 3 | SOC | 336,857 | 7.47 | Reference |
Axi-cel | 571,537 | 9.31 | 127,369 | |
CADTH reanalysis 4 | SOC | 337,280 | 7.33 | Reference |
Axi-cel | 569,229 | 9.13 | 128,635 | |
CADTH base case 1 + 2 + 3 + 4 (deterministic) | SOC | 337,280 | 7.41 | Reference |
Axi-cel | 572,112 | 8.13 | 323,802 | |
CADTH base case 1 + 2 + 3 + 4 (probabilistic) | SOC | 336,966 | 7.67 | Reference |
Axi-cel | 572,112 | 8.25 | 404,418 |
axi-cel = axicabtagene ciloleucel; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; SOC = standard of care.
Scenario Analysis Results
CADTH undertook price reduction analyses based on the CADTH base case. These analyses demonstrated that a price reduction of 45% would be necessary to achieve cost-effectiveness at a WTP threshold of $50,000 per QALY in the reimbursement request population (Table 7).
Table 7: CADTH Price Reduction Analyses
Price reduction analysis | ICER for axi-cel vs. SOC ($/QALY) | |
|---|---|---|
Sponsor base case | CADTH reanalysis | |
No price reduction | $141,757 | $405,424 |
10% | $113,949 | $326,817 |
20% | $86,149 | $248,210 |
30% | $58,349 | $169,603 |
40% | Dominant | $90,997 |
45.2% | NA | $50,000 |
50% | Dominant | Dominant |
axi-cel = axicabtagene ciloleucel; ICER = incremental cost-effectiveness ratio; NA = not applicable; QALY = quality-adjusted life-year; SOC = standard of care.
In addition, CADTH conducted a series of scenario analyses to determine the impact of alternative assumptions on the cost-effectiveness of axi-cel, including adopting alternative health state utilities values based on the literature and assuming no access to CAR T-cell therapies in the third-line setting.
In a scenario in which patients do not have access to CAR T-cell therapy in the third-line setting (i.e., incorporate crossover adjustment and redistribute CAR T cells in the third-line setting to chemotherapy), the deterministic ICER ranged from $85,299 to $163,890 per QALY for axi-cel compared with SOC based on the statistical method taken to conduct the crossover adjustment. Although these scenarios are especially relevant for drug plans that do not currently fund CAR T-cell therapies for third-line treatment, these results should be interpreted with caution given the sensitivity to the statistical approaches taken to make the crossover adjustments.
Exploratory analyses were further conducted to assess the possibility of re-treatment (i.e., assuming 5% of patients who receive axi-cel are re-treated in third line, as reported in the ZUMA-7 trial); increased mean patient age (67 years); and other proportions of patients on axi-cel receiving pretreatment (i.e., leukapheresis [98.9%]), bridging chemotherapy [36.1%], and conditioning chemotherapy [95.6%], based on the ZUMA-7 trial). Further, there is uncertainty around the pricing negotiations of axi-cel. The details of potential negotiations between the sponsor and the payers, and whether value-based pricing or performance pricing is to be employed, remain unclear. This has been the case in private arrangements in the US, whereby the public funder would only pay the drug acquisition costs for patients who have a response.35,36 CADTH adopted an outcome-based agreement in which reimbursement of axi-cel would be based on OS rate at 12 months (e.g., 76% of patients receiving axi-cel), based on clinician input.
The results of the scenario and exploratory analyses are presented in Table 16.
Issues for Consideration
ASCT eligibility: The clinical expert feedback noted significant challenges in implementing an initiation criterion for axi-cel based on ASCT eligibility. Clinicians highlighted the differences that exist across jurisdictions, as well as between institutions and transplanters, with regard to the set of criteria employed to determine ASCT eligibility. Indeed, clinical expert feedback indicated that there are no uniformly implemented, easily reproducible, or objective criteria for ASCT eligibility in Canada. For instance, while some centres may use age as an eligibility criterion for ASCT, other centres might not exclude older patients if they have adequate organ function and performance status.
Long-term clinical impacts: The evidence for the effectiveness of CAR T-cell therapy is still in its early stages and evidence is emerging about the rate of late treatment-related toxicities, duration-of-treatment effect, and what comprises follow-up for patients receiving CAR T-cell therapy in Canada. Furthermore, if the therapy is curative as claimed, the expectation is that patients with relapsed or refractory LBCL would live a longer life, and as such, may incur additional costs to the health system.
Shortage of drugs to manage CRS: CRS is sometimes managed with tocilizumab. Tocilizumab has been on the Drug Shortages Canada website list due to its use in COVID-19 treatment. Health Canada has previously declared a “Tier 3” shortage of tocilizumab, a designation reserved for shortages with the greatest potential impact on Canada’s health care system.37 The use of siltuximab has been considered by some clinicians if there is a severe shortage of tocilizumab, but this treatment is currently only publicly funded via the Alberta drug formulary.38 Shortage of treatments for CRS may impact axi-cel use because of the risk of CRS associated with CAR T-cell therapy. This is especially relevant because 92% of patients who received axi-cel in the ZUMA-7 trial experienced a CRS event, and 52% had a CRS event of grade 2 or higher.1 Indeed, real-world evidence corroborates the high incidence of CRS among patients with relapsed or refractory LBCL treated with CAR T-cell therapy. A recent study conducted in the US based on 3 commercial claims databases revealed that an estimated 75% to 84% of patients experienced any-grade CRS, and between 15% to 32% of patients experienced severe CRS.39
Sequential use of CAR T-cell products: Axi-cel alongside other CAR T-cell therapies (e.g., tisagenlecleucel) is reimbursed in some jurisdictions for the treatment of adult patients with relapsed or refractory LBCL after 2 or more lines of systemic therapy. As axi-cel is now coming in at an earlier line of treatment (second line), it is unclear whether sequential use of CAR T-cell therapy will be permitted. Evidence for the sequential use of CAR T-cell therapies does not exist and, thus the cost-effectiveness and budget impact of sequential use of CAR T cells is unknown.
Therapies to augment residual effect of CAR T-cell therapies: Clinical expert feedback indicated that immunosuppressive treatments such as pembrolizumab or nivolumab may be considered for use following CAR T-cell treatment failure to augment the residual effects of the CAR T-cell therapy. These treatments were not included in the sponsor’s submission.
Future treatments: Clinical expert feedback indicated that, in the future, other CAR T-cell therapies, such as tisagenlecleucel and lisocabtagene maraleucel, may also pursue the indication for treatment of patients with relapsed or refractory LBCL in second line given there are clinical trial programs currently under way.40
Travel-associated costs: Travel costs and the requirement for time spent away from work was not considered. The sponsor’s implementation plan indicated that not all provinces and territories will have a site to provide axi-cel.41 For jurisdictions that do not currently have a site to provide axi-cel, patients will need to travel out of province or out of country for treatment. Furthermore, it was noted by clinical experts that some provinces do not even have capacity to assess patients’ eligibility for CAR T-cell therapy. The implementation plan suggests that the sponsor will coordinate travel and lodging logistics for the patient and their caregiver who arrive to receive treatment and who need to remain within close proximity of the qualified treatment centre for at least 4 weeks following infusion.41 The sponsor stated that the program is intended to support adherence to monitoring requirements for axi-cel by providing financial support to cover transportation-related expenses and lodging costs for the patient and their caregiver during the pretreatment and treatment periods and for at least the 4 weeks when they are required to stay within close proximity of the qualified treatment centre. If this patient support program is not operationalizable, and travel expenses (e.g., travel, lodging, food) have to be absorbed by the patient or the public payer, this may impact access to axi-cel. Disparities in funding and treatment access may vary depending on the province or territory, and the requirement for access to a tertiary care centre for delivery of axi-cel may have equity of access implications, which were not substantively considered in the economic submission.
Manufacturing delays: The sponsor’s implementation plan indicated that, in the ZUMA-7 trial, the median time from apheresis collection to release from the manufacturing site was 13 days, and the median time to final product delivery (i.e., time axi-cel is ready to be infused back into the patient) was 18 days. A recent real-world study conducted in the US based on 3 commercial claims databases revealed that the median time from leukapheresis to CAR T-cell infusion was 26 to 27 days.39 However, CADTH clinical expert feedback noted the potential for greater variability in manufacturing time in the real-world setting given apheresis collection would be conducted across a broad network of pan-Canadian sites with manufacturing conducted across the border in the US.41
Manufacturing failures: Issues pertaining to manufacturing are important to the successful delivery of CAR T-cell therapies. Manufacturing failure may occur due to an inadequate number of T cells in the apheresed product, poor selection of T cells on day zero of manufacturing, or irreversibly impaired T cells (i.e., no response to stimulation in culture), microbial contamination, equipment-related cell loss, high endotoxin levels, and accidents. The sponsor noted that the axi-cel manufacturing success rate in the ZUMA-7 trial was 99% and the impacts of manufacturing failure were not considered in the submitted economic model. However, manufacturing failure of CAR T-cell therapies is not uncommon and has been previously observed in trials for axi-cel19 and other CAR T-cell products.42 There may be additional costs associated with manufacturing failure, including increased hospital stay while a second sample is prepared, if possible. In addition, manufacturing failure may impact patient outcomes due to treatment delays or compromised doses.
Workplace productivity: The patient input noted workplace productivity to be an outcome of interest. ZUMA-7 explored this outcome using the Work Productivity Activity Impairment (WPAI) questionnaire at different time points (i.e., screening, day 100, day 150, month 9, month 12, and month 15).8 Given that mean age of participants in the ZUMA-7 trial was 57 years, data were reported for only a small proportion of patients. As such, the trial was not powered to detect a difference between treatment arms for this outcome. There was insufficient evidence to show a difference in mean overall work impairment at day 100. Thereafter, differences in work impairment between patients who received axi-cel compared with those who received SOC were not tested statistically due to nonsignificance at day 100.
Capacity constraints: The sponsor’s implementation plan indicated the capacity for production of axi-cel for patients living in Canada annually. The sponsor stated that it currently has the capacity to produce therapy for 4,500 patients per year, with capacity increasing to 7,100 patients per year by December 2023. The sponsor did not consider potential capacity constraints within the submitted economic evaluation (i.e., those considered eligible for treatment would not have adverse clinical outcomes or additional costs arising from treatment delays due to capacity issues).
Previous CADTH review: Axi-cel was reviewed by CADTH and was given a conditional positive recommendation with substantial price reductions by CADTH’s Health Technology Expert Review Panel (HTERP). The rationale for the price conditions was based on CADTH reanalyses indicating that a price reduction of 83% was required to achieve an ICER of $50,000 per QALY for axi-cel. Further, it was noted in the recommendation that these results should be interpreted with caution given the uncertainty associated with the clinical evidence.
Overall Conclusions
Evidence from the ZUMA-7 trial suggests that axi-cel is associated with clinically meaningful improvements in EFS compared to SOC as second-line treatment in ASCT-eligible patients with relapsed or refractory LBCL within 12 months of first-line chemoimmunotherapy. It is not yet clear whether this EFS benefit will translate to improved OS as the data remain immature and follow-up is ongoing. As noted in the CADTH Clinical Review, because the OS results are based on an interim analysis, there is a risk that the effect of axi-cel compared with SOC on survival is overestimated. This uncertainty is propagated into the submitted model given the sponsor’s PSM is informed by these OS curves. The clinical trial population, upon which the economic analysis was based, consists of relatively stable patients that may not be generalizable to patients who are less stable or who do not meet the specific inclusion criteria of the trial.
In addition to the aforementioned limitations with the clinical evidence, CADTH identified several limitations with the sponsor’s economic submission. These limitations included the following: the sponsor’s request for deviation to focus their analysis on adult patients with relapsed or refractory LBCL who are candidates for ASCT, which does not align with the drug’s expected place in therapy; the use of a model structure that lacks flexibility, which is particularly concerning given uncertainties in the distribution of subsequent therapy and its impact on OS; the choice of an MCM and the uncertainties in the predicted cure rate for axi-cel given the immaturity of the OS data in the ZUMA-7 trial; the assumption that the postevent utility for patients with progressed disease would be equal to that derived from a population with further progressed disease following CAR T-cell therapy in third line; the assumption that patients receiving axi-cel would exclusively seek subsequent treatment through clinical trials for investigational therapies at no additional cost to drug plans; and the use of a mortality hazard estimate for long-term survivors that was deemed to be too low to capture the excess mortality of this patient population whom have received extensive prior treatments that are associated with late toxicities.
As part of the base-case reanalysis, CADTH addressed the uncertainties associated with long-term treatment efficacy by selecting an alternative extrapolation curve to inform OS for axi-cel; revising the pre-event (for the first 149 days of treatment) and postevent utility values; adjusting the distribution of subsequent therapies in the axi-cel arm to exclude investigational therapies; and applying a higher hazard of death among long-term survivors based on North American data.
In CADTH’s base-case reanalysis, the ICER of axi-cel is $404,418 per QALY (incremental costs: $235,146; incremental QALYs: 0.58) compared with SOC. This is based on the assumption that 50% of patients who fail SOC would be treated with CAR T-cell therapies in the third-line setting. Adjusting the distributional assumption to log-logistic to derive a more plausible OS curve for axi-cel in the absence of long-term evidence, while still conferring a survival benefit to axi-cel, resulted in the largest change to the sponsor’s base case. A price reduction of 45% would be necessary for axi-cel to be considered cost-effective at a WTP threshold of $50,000 per QALY.
The cost-effectiveness of axi-cel was sensitive to the assumptions regarding the selected parametric model distribution used to extrapolate OS. The CADTH base-case reanalysis estimated a smaller OS benefit with axi-cel when compared to the sponsor’s base case (i.e., incremental life-years: 1.50 [sponsor’s base case] versus 0.24 [CADTH’s reanalysis]), and this translated to a smaller difference in QALYs between axi-cel and SOC. Nonetheless, similar to the sponsor’s analysis, the majority of the QALY gains (71%) conferred by axi-cel in the CADTH reanalysis were derived from the period beyond which there are observed trial data. Furthermore, clinical uncertainties in the extrapolation period could not be adequately explored due to the inflexible modelling approach. This is particularly concerning when considering the uncertainties in the distribution of subsequent therapy. The cost-effectiveness results from both the sponsor and CADTH’s base-case reanalysis are informed by underlying OS curves from the ZUMA-7 trial in which 56% of the patients in the SOC arm received subsequent CAR T-cell therapies in the third-line setting. It remains uncertain whether it is feasible to expect more than half of the patients receiving SOC to subsequently receive CAR T cells as third-line therapy in clinical practice.
The cost-effectiveness of axi-cel as second-line treatment in patients who are ineligible for ASCT and in patients who may not meet the specific inclusion or exclusion criteria of the ZUMA-7 trial, which reflects a component of the Health Canada indication, is unknown.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Yescarta® (axicabtagene ciloleucel), cell suspension in patient-specific single infusion bag, target of 2 × 106 chimeric antigen receptor (CAR)-positive viable T cells per kg body weight with a maximum of 2 × 108 CAR-positive viable T cells, for intravenous infusion. Mississauga (ON): Gilead Sciences Canada, Inc.; 2022 Jun 9.
2.Yescarta® (axicabtagene ciloleucel): cell suspension in patient-specific single infusion bag, target of 2 × 106 chimeric antigen receptor (CAR)-positive viable T cells per kg body weight with a maximum of 2 × 108 CAR-positive viable T cells, for intravenous infusion [product monograph]. Mississauga (ON): Gilead Sciences Canada, Inc.; 2021 Dec 15.
3.Interprovincial Health Insurance Agreements Coordinating Committee (IHIACC). Final - Interprovincial Billing Rates for Designated High Cost Transplants Effective for Discharges on or After April 1, 2022. Organ Transplants. Toronto (ON): Ontario Ministry of Health, Ontario Ministry of Long-Term Care; 2022: https://www.health.gov.on.ca/en/pro/programs/ohip/bulletins/docs/interprovincia220408/2022-2023_rates_high_cost_procedures.pdf. Accessed 2022 Oct 18.
4.Cheson BD, Fisher RI, Barrington SF, et al. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: the Lugano classification. J Clin Oncol. 2014;32(27):3059-3068. PubMed
5.Maurer MJ, Ghesquieres H, Jais JP, et al. Event-free survival at 24 months is a robust end point for disease-related outcome in diffuse large B-cell lymphoma treated with immunochemotherapy. J Clin Oncol. 2014;32(10):1066-1073. PubMed
6.National Institute for Health and Care Excellence. Tisagenlecleucel for treating relapsed or refractory diffuse large B-cell lymphoma after 2 or more systemic therapies. (Technology appraisal guidance TA567) 2019; https://www.nice.org.uk/guidance/ta567. Accessed 2022 Aug 15.
7.Alberta PROMs & EQ-5D Research & Support Unit (APERSU). Alberta Population Norms for EQ-5D-5L. Vol 2022. Edmonton (AB): School of Public Health, University of Alberta; 2018: https://apersu.ca/wp-content/uploads/2020/10/Alberta-Norms-Report_APERSU-1.pdf. Accessed 2022 Sep 5.
8.Clinical Study Report: KTE-C19-107. A phase 3, randomized, open-label study evaluating the efficacy of axicabtagene ciloleucel versus standard of care therapy in subjects with relapsed/refractory diffuse large B-cell lymphoma (ZUMA-7) [internal sponsor's report]. Santa Monica (CA): Kite Pharma, Inc.; 2021 Sep 14.
9.Howell TA, Matza LS, Jun MP, Garcia J, Powers A, Maloney DG. Health State Utilities for Adverse Events Associated with Chimeric Antigen Receptor T-Cell Therapy in Large B-Cell Lymphoma. Pharmacoecon Open. 2022;6(3):367-376. PubMed
10.Beusterien KM, Davies J, Leach M, et al. Population preference values for treatment outcomes in chronic lymphocytic leukaemia: a cross-sectional utility study. Health Qual Life Outcomes. 2010;8:50. PubMed
11.Nafees B, Stafford M, Gavriel S, Bhalla S, Watkins J. Health state utilities for non small cell lung cancer. Health Qual Life Outcomes. 2008;6:84. PubMed
12.Tolley K, Goad C, Yi Y, Maroudas P, Haiderali A, Thompson G. Utility elicitation study in the UK general public for late-stage chronic lymphocytic leukaemia. Eur J Health Econ. 2013;14(5):749-759. PubMed
13.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario drug benefit formulary/comparative drug index. 2022; https://www.formulary.health.gov.on.ca/formulary/. Accessed 2022 Aug 1.
14.CADTH. Drug reimbursement review: brexucabtagene autoleucel (Tecartus) for mantle cell lymphoma. 2021; https://www.cadth.ca/brexucabtagene-autoleucel. Accessed 2022 Aug 31.
15.CADTH. Drug reimbursement review: nivolumab-ipilimumab (Opdivo-Yervoy) for malignant pleural mesothelioma. 2021; https://www.cadth.ca/nivolumab-ipilimumab. Accessed 2022 Aug 31.
16.CADTH. Drug reimbursement review: larotrectinib (Vitrakvi) for solid tumours with NTRK gene fusion. 2021; https://www.cadth.ca/larotrectinib. Accessed 2022 Aug 31.
17.CADTH. Drug reimbursement review: durvalumab (Imfinzi) for extensive-stage small cell lung cancer. 2021; https://www.cadth.ca/durvalumab. Accessed 2022 Aug 31.
18.CADTH. Drug reimbursement review: daratumumab (Darzalex) for multiple myeloma. 2021; https://www.cadth.ca/daratumumab-darzalex-multiple-myeloma. Accessed 2022 Aug 31.
19.Axicabtagene ciloleucel for diffuse large B-cell lymphoma: economic report. (CADTH optimal use report vol. 9, no. 1d). Ottawa (ON): CADTH; 2019: https://www.cadth.ca/sites/default/files/pdf/car-t/ct0002-axicabtagene-ciloleucel-economic-report-redacted.pdf. Accessed 2022 Aug 18.
20.Logan H, Livingstone S. Subject: Patented Medicine Prices Review Board (PMPRB) Guidelines Modernization. Toronto (ON): Canadian Association of Provincial Cancer Agencies (CAPCA); 2016: http://www.pmprb-cepmb.gc.ca/CMFiles/Consultations/Rethinking_the_Guidelines_2016/Submission_CAPCA_Oct_2016.pdf. Accessed 2022 Aug 18.
21.Cancer Care Ontario. Drug Formulary. GDP - Provider Monograph. 2019; https://www.cancercareontario.ca/en/drugformulary/regimens/monograph/46166. Accessed 2022 Aug 15.
22.Guidelines Resource Unit. Lymphoma Clinical Practice Guideline. (Clinical Practice Guideline LYHE-002 V16). Edmonton (AB): Cancer Care Alberta; 2021: https://www.albertahealthservices.ca/assets/info/hp/cancer/if-hp-cancer-guide-lyhe002-lymphoma.pdf. Accessed 2021 Oct 14.
23.Canadian Institute for Health Information (CIHI). Patient Cost Estimator. 2022; https://www.cihi.ca/en/patient-cost-estimator. Accessed 2022 Mar 10.
24.Schedule of benefits for physician services under the Health Insurance Act: effective October 1, 2021. Toronto (ON): Ontario Ministry of Health; 2022: https://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master.pdf. Accessed 2022 Aug 1.
25.de Oliveira C, Pataky R, Bremner KE, et al. Phase-specific and lifetime costs of cancer care in Ontario, Canada. BMC Cancer. 2016;16(1):809. PubMed
26.Woods B, Sideris E, Palmer S, Latimer N, Soares M. Partitioned survival analysis for decision modelling in health care: a critical review. (NICE DSU technical support document 19). Sheffield (GB): Decision Support Unit, ScHARR University of Sheffield; 2017: https://www.sheffield.ac.uk/nice-dsu/tsds/partitioned-survival-analysis. Accessed 2022 Oct 18.
27.Woods BS, Sideris E, Palmer S, Latimer N, Soares M. Partitioned Survival and State Transition Models for Healthcare Decision Making in Oncology: Where Are We Now? Value Health. 2020;23(12):1613-1621. PubMed
28.Jacobson C, Locke FL, Ghobadi A, Miklos DB. Long-Term (=4 Year and =5 Year) Overall Survival (OS) By 12- and 24-Month Event-Free Survival (EFS): An Updated Analysis of ZUMA-1, the Pivotal Study of Axicabtagene Ciloleucel (Axi-Cel) in Patients (Pts) with Refractory Large B-Cell Lymphoma (LBCL) [conference abstract]. Blood. 2021;138(Suppl 1):1764.
29.Chong EA, Ruella M, Schuster SJ, Lymphoma Program Investigators at the University of Pennsylvania. Five-Year Outcomes for Refractory B-Cell Lymphomas with CAR T-Cell Therapy. N Engl J Med. 2021;384(7):673-674. PubMed
30.Ye X, Mahmud S, Skrabek P, Lix L, Johnston JB. Long-term time trends in incidence, survival and mortality of lymphomas by subtype among adults in Manitoba, Canada: a population-based study using cancer registry data. BMJ Open. 2017;7(7):e015106. PubMed
31.Huntington SF, Svoboda J, Doshi JA. Cost-effectiveness analysis of routine surveillance imaging of patients with diffuse large B-cell lymphoma in first remission. J Clin Oncol. 2015;33(13):1467-1474. PubMed
32.Sullivan PW, Ghushchyan V. Preference-Based EQ-5D index scores for chronic conditions in the United States. Med Decis Making. 2006;26(4):410-420. PubMed
33.Lin VW, Jiang Y, Chuang LH, Navale L, Cheng P, Purdum A. P889 Health Utilities for Patients with Relapsed or Refractory Large B-Cell Lymphoma (R/R-LBCL): Ad Hoc Analysis From an Axicabtagene Ciloleucel (Axi-cel) Safety Management Study [conference poster]. Bone Marrow Transplant. 2019;53(Suppl 1):879.
34.Locke FL, Westin JR, Miklos DB. Phase 1 results from ZUMA-6: axicabtagene ciloleucel (axi-cel; KTE-c19) in combination with atezolizumab for the treatment of patients with refractory diffuse large B cell lymphoma (DLBCL) [conference abstract]. Blood. 2017;130(Suppl 1):2826.
35.Bach PB, Giralt SA, Saltz LB. FDA Approval of Tisagenlecleucel: Promise and Complexities of a $475000 Cancer Drug. JAMA. 2017;318(19):1861-1862. PubMed
36.Karlin-Smith S, Pittman D. CMS quit test of pricey cancer treatment amid concerns over industry role. Politico 2018 Jul 9; https://www.politico.com/story/2018/07/09/cms-quit-test-of-pricey-cancer-treatment-amid-concerns-over-industry-role-674086. Accessed 2022 Sep 6.
37.Drug Shortages Canada. Drug Shortage Report for Actembra. 2022; https://www.drugshortagescanada.ca/shortage/157625. Accessed 2022 Sep 1.
38.DeltaPA. Ottawa (ON): IQVIA; 2021: https://www.iqvia.com/. Accessed 2022 Feb 28.
39.Keating SJ, Gu T, Jun MP, McBride A. Health Care Resource Utilization and Total Costs of Care Among Patients with Diffuse Large B Cell Lymphoma Treated with Chimeric Antigen Receptor T Cell Therapy in the United States. Transplant Cell Ther. 2022;28(7):404 e401-404 e406.
40.Juno Therapeutics. NCT03483103: Lisocabtagene Maraleucel (JCAR017) as Second-Line Therapy (TRANSCEND-PILOT-017006). ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine; 2022: https://clinicaltrials.gov/ct2/show/NCT03483103. Accessed 2022 Sep 2.
41.CADTH Reimbursement Review Implementation Planning for Cell or Gene Therapy [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Yescarta® (axicabtagene ciloleucel), cell suspension in patient-specific single infusion bag, target of 2 × 106 chimeric antigen receptor (CAR)-positive viable T cells per kg body weight with a maximum of 2 × 108 CAR-positive viable T cells, for intravenous infusion. Mississauga (ON): Gilead Sciences Canada, Inc.; 2022 Jun 9.
42.CADTH Reimbursement review: lisocabtagene maraleucel (Breyanzi) for the treatment of relapsed or refractory large B-cell lymphoma. Can J Health Technol. 2022;2(10). https://www.cadth.ca/sites/default/files/DRR/2022/PG0258-Breyanzi.pdf. Accessed 2022 Aug 20.
43.PrRinvoq® (upadacitinib): 15 mg, 30 mg extended-release oral tablets [product monograph]. St-Laurent (QC): AbbVie; 2021 Sep 30: https://www.abbvie.ca/content/dam/abbvie-dotcom/ca/en/documents/products/RINVOQ_PM_EN.pdf. Accessed 2022 Oct 1.
44.Agathou S. To BeEAM or to R-BEAM prior to ASCT in B-cell lymphomas? LymphomaHub 2018; https://lymphomahub.com/medical-information/to-beeam-or-to-r-beam-prior-to-asct-in-b-cell-lymphomas. Accessed 2022 Aug 20.
45.pan-Canadian Oncology Drug Review (pCODR) Initial Economic Guidance Report - Gemtuzumab Ozogamicin (Mylotarg) for Acute Myeloid Leukemia. Ottawa (ON): CADTH; 2020: https://www.cadth.ca/sites/default/files/pcodr/Reviews2020/10190GemtuzumabOzogamicinAML_inEGR_NOREDACT-ABBREV_Post_30Jan2020_final.pdf. Accessed 2022 Aug 20.
46.Ellis K. Cost-effectiveness of Chimeric Antigen Receptor T-cell Therapy for Treating Large B-cell Lymphoma Patients in Canada [thesis]. Waterloo (ON): University of Waterloo; 2019: https://uwspace.uwaterloo.ca/bitstream/handle/10012/15515/Ellis_Kristina.pdf. Accessed 2022 Aug 15.
47.Leighl NB, Shepherd FA, Kwong R, Burkes RL, Feld R, Goodwin PJ. Economic analysis of the TAX 317 trial: docetaxel versus best supportive care as second-line therapy of advanced non-small-cell lung cancer. J Clin Oncol. 2002;20(5):1344-1352. PubMed
48.Zheng B, Reardon PM, Fernando SM, et al. Costs and Outcomes of Patients Admitted to the Intensive Care Unit With Cancer. J Intensive Care Med. 2021;36(2):203-210. PubMed
49.Wagner-Johnston ND, Link BK, Byrtek M, et al. Outcomes of transformed follicular lymphoma in the modern era: a report from the National LymphoCare Study (NLCS). Blood. 2015;126(7):851-857. PubMed
50.Armitage JO, Gascoyne RD, Lunning MA, Cavalli F. Non-Hodgkin lymphoma. Lancet. 2017;390(10091):298-310. PubMed
51.Canadian Cancer Statistics Advisory Committee in collaboration with the Canadian Cancer Society, Statistics Canada, the Public Health Agency of Canada. Canadian cancer statistics 2021. Toronto (ON): Canadian Cancer Society; 2021: https://cdn.cancer.ca/-/media/files/research/cancer-statistics/2021-statistics/2021-pdf-en-final.pdf. Accessed 2022 Aug 15.
52.Cancer-specific stats 2020: Snapshot of incidence, mortality and survival estimates by cancer type. Toronto (ON): Canadian Cancer Society; 2020: https://cdn.cancer.ca/-/media/files/research/cancer-statistics/2020-statistics/canadian-cancer-statistics/2020-resources/res-cancerstatistics-canadiancancerstatistics-2020_cancer-specific-stats.pdf. Accessed 2022 Aug 15.
53.Drug Reimbursement Review sponsor submission: Yescarta® (axicabtagene ciloleucel), cell suspension in patient-specific single infusion bag, target of 2 × 106 chimeric antigen receptor (CAR)-positive viable T cells per kg body weight with a maximum of 2 × 108 CAR-positive viable T cells, for intravenous infusion [internal sponsor's package]. Mississauga (ON): Gilead Sciences Canada, Inc.; 2022 Jun 9.
54.Coiffier B, Sarkozy C. Diffuse large B-cell lymphoma: R-CHOP failure-what to do? Hematology Am Soc Hematol Educ Program. 2016;2016(1):366-378. PubMed
Appendix 1: Cost Comparison Table
Note this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical expert(s) and drug plans. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 8: CADTH Cost Comparison Table for Relapsed or Refractory LBCL (Gene Therapy)
Treatment | Strength / concentration | Form | Price ($) | Recommended dosage | Cost per cycle or 1-time use ($) | Cost per 28 days ($) |
|---|---|---|---|---|---|---|
CAR T-cell therapy | ||||||
Axicabtagene ciloleucel (Yescarta) | Refer to dosage | Suspension for IV infusion | 485,021.0000a | Target of 2 × 106 anti-CD19 CAR T cells/kg body weight (range, 1 × 106 to 2.4 × 106 cells/kg) to a maximum of 2 × 108 anti-CD19 CAR T cells | 485,021 | NA |
CAR = chimeric antigen receptor; LBCL = large B-cell lymphoma; NA = not applicable.
aSponsor’s submitted price.1
Table 9: CADTH Cost Comparison Table for Relapsed or Refractory LBCL (Salvage Chemotherapy, HDT, ASCT)
Treatment | Strength / concentration | Form | Price ($) | Recommended dosage | Cost per cycle or 1-time use ($) | Cost per 28 days ($) |
|---|---|---|---|---|---|---|
Salvage chemotherapy | ||||||
R-GDP | ||||||
Rituximab (biosimilars) | 100 mg vial | IV infusion | 297.0000a | 21-day cycles: 375 mg/m2 on day 1b | 2,079 | 2,772 |
Gemcitabine (generic) | 1,000 mg 2,000 mg | Lyophilized powder | 270.0000 540.0000 | 21-day cycles: 1,000 mg/m2 days 1 and 8b | 1,080 | 1,440 |
Dexamethasone (generics) | 4 mg | Tablet | 0.6112 | 21-day cycles: 40 mg days 1 to 4c | 24 | 33 |
Cisplatin (generic) | 50 mg vial 100 mg vial | 1 mg/mL solution for injection | 135.0000 270.0000 | 21-day cycles: 75 mg/m2 on day 1b | 405 | 540 |
R-GDP regimen cost (21-day cycle) | — | — | — | — | 3,588 | 4,785 |
R-ICE | ||||||
Rituximab (biosimilars) | 100 mg vial | IV infusion | 297.0000a | 21-day cycles: 375 mg/m2 on day 1b | 2,079 | 2,772 |
Ifosfamide (Ifex) | 1,000 mg vial 3,000 mg vial | Powder for solution | 131.4900 403.4900 | 21-day cycles: 1,667 mg/m2 on days 1 to 3b | 1,183 | 1,578 |
Carboplatin (generic) | 50 mg 150 mg 450 mg 600 mg | 10 mg/mL vial for injection | 70.0000 210.0000 600.0000 775.0020 | 21-day cycles: AUC 5 on day 1; maximum dose for AUC 5 is 750 mgb | Max: 1,050 | 1,400 |
Etoposide (generic) | 100 mg | 20 mg/mL vial for injection | 75.0000 | 21-day cycles: 100 mg/m2 on days 1 to 3b | 450 | 600 |
R-ICE regimen cost (21-day cycle) | — | — | — | — | 4,762 | 6,350 |
R-DHAP | ||||||
Rituximab (biosimilars) | 100 mg vial | IV | 297.0000a | 21- or 28-day cycles: 375 mg/m2 on day 1b | 2,079 | 2,079 to 2,772 |
Dexamethasone (generics) | 4 mg | Tablet | 0.6112 | 21-day cycles: 40 mg days 1 to 4b | 24 | 33 |
Cytarabine (generic) | 500 mg vial 2,000 mg vial | 100 mg/mL IV solution | 76.8500 306.5000 | 21- or 28-day cycles: 2,000 mg/m2 every 12 hours on day 2b | 1,230 | 1,230 to 1,639 |
Cisplatin (generic) | 50 mg vial 100 mg vial | 1 mg/mL IV solution | 135.0000 270.0000 | 21- or 28-day cycles: 100 mg/m2 on day 1b | 540 | 540 to 720 |
R-DHAP regimen cost (21- or 28-day cycle) | — | — | — | — | 3,873 | 3,873 to 5,164 |
Salvage chemotherapy costs | — | — | — | — | 3,588 to 4,762 | 3,873 to 6,350 |
High-dose therapy | ||||||
EM | ||||||
Etoposide (generic) | 100 mg vial 200 mg vial 500 mg vial 1,000 mg vial | 20 mg/mL injection | 75.0000 150.0000 375.0000 750.0000 | 60 mg/kg on day –4c | 3,375 | NA |
Melphalan (generic) | 50 mg vial | 50 mg/mL injection | 88.0700 | 180 mg/m2 on day –3c | 616.49 | NA |
EM regimen cost | — | — | — | — | 3,991 | NA |
BEAM | ||||||
Carmustine (generic) | 100 mg vial | 100 mg/mL injection | 4,965.14d | 300 mg/m2 on day –2 or –6e | 29,791 | NA |
Etoposide (generic) | 100 mg vial 200 mg vial 500 mg vial 1,000 mg vial | 20 mg/mL injection | 75.0000 150.0000 375.0000 750.0000 | 150 mg/m2 to 200 mg/m2 twice daily, on day –5 to –2e | 1,800 to 2,400 | NA |
Cytarabine (generic) | 100 mg vial 500 mg vial 1,000 mg vial 2,000 mg vial | 100 mg/mL IV solution | 5.0900g 76.8500 153.2500 306.5000 | 200 mg/m2 to 400 mg/m2 twice daily on days –5 to –2e | 162.88 to 325.76 | NA |
Melphalan (generic) | 50 mg vial | 50 mg/mL injection | 88.0700 | 140 mg/m2 on day –1e | 528.42 | NA |
BEAM regimen cost | — | — | — | — | 32,282 to 33,045 | NA |
High-dose therapy costs | — | — | — | — | 3,991 to 33,045 | NA |
Autologous stem cell transplant | ||||||
Autologous stem cell transplant | Adult autologous stem cell transplant (> 72 hours) includes all facility costs including inpatient and diagnostic costs | 77,956g per transplant | NA | |||
BEAM = carmustine, etoposide, cytarabine, and melphalan; CAR = chimeric antigen receptor; EM = etoposide and melphalan; LBCL = large B-cell lymphoma; NA = not applicable; R-DHAP = rituximab + dexamethasone, cytarabine, and cisplatin; R-GDP = rituximab + gemcitabine, dexamethasone, and cisplatin; R-ICE = rituximab + ifosfamide, carboplatin, and etoposide.
Note: All prices are wholesale from IQVIA Delta PA (accessed October 2021), unless otherwise indicated, and do not include dispensing fees. Calculations assume a patient body weight of 75 kg and a body surface area of 1.8 m2.
aOntario Drug Benefit Formulary or Exceptional Access Program list price13 (accessed August 2022).
bCancer Care Ontario Drug Formulary: Regimens database.43
cRegimen as developed by Princess Margaret Hospital and validated by clinical experts consulted by CADTH.
dPatented Medicine Prices Review Board (PMPRB) Guidelines Modernization.20
eRegimen as cited in Lymphoma Hub44 and validated by clinical experts consulted by CADTH.
fpCODR Final Economic Guidance Report for Gemtuzumab Ozogamicin (Mylotarg) for Acute Myeloid Leukemia.45
gInterprovincial Billing Rates for Designated High Cost Transplants Effective for Discharges on or After 1, 2022.3 The cost includes all facility costs associated with a single transplant episode including inpatient and diagnostic costs, with a maximum length of stay of 16 days.
Appendix 2: Submission Quality
Note this appendix has not been copy-edited.
Description | Yes/No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | Refer to CADTH appraisal regarding the generalizability of the patient population. |
Model has been adequately programmed and has sufficient face validity | Yes | No comment. |
Model structure is adequate for decision problem | No | Refer to CADTH appraisal regarding the inflexible modelling approach. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | Yes | No comment. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | No | Refer to CADTH appraisal. Discrepancies were observed between the deterministic and probabilistic results. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | Yes | No comment. |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note this appendix has not been copy-edited.
3L = third line; EFS = event-free survival; OS = overall survival; TTNT = time to next treatment.
Source: Sponsor’s pharmacoeconomic submission.1
Table 11: Health State Utility Values
Health state | Utility value | Source |
|---|---|---|
Axicabtagene ciloleucel, event freea | 0.833 | ZUMA-7 EQ-5D-5L Index (Canadian value set) |
SOC, event free1 | 0.826 | ZUMA-7 EQ-5D-5L Index (Canadian value set) |
Off treatment, event-free | 0.837 | ZUMA-7 EQ-5D-5L Index (Canadian value set) |
Post event | 0.710 | JULIET as per NICE (TA567)6 |
aTreatment-specific utilities were applied to the event-free state during the first month for axi-cel and the first 3 months for SOC.
Source: Sponsor’s pharmacoeconomic submission.1
Table 12: Weighted Acquisition and Administration Costs
Treatment | Cost ($) | % Receiving | Weighted cost ($) |
|---|---|---|---|
Axi-cel | |||
Leukapheresis46 | $2,760 | 100% | $2,760 |
Bridging therapy | — | — | $506.35 |
Dexamethasone14 | $4.57 | 100% | $4.57 |
Radiotherapy47 | $1,572 | 20% | $314.46 |
GDP21 | $1,873 | 10% | $187.32 |
Conditioning chemotherapy | — | — | $2,703a |
Fludarabine19 | $1,530 | 100% | $1,530 |
Cyclophosphamide14 | $496.56 | 100% | $496.56 |
Axi-cel | — | — | $487,108 |
Acquisition1 | $485,021 | 94% | $455,920 |
$33,179b | 94% | $31,188 | |
Total cost | — | — | $493,076 |
SOC | |||
Salvage chemotherapy | — | 100% | $13,217c |
$4,249 | 80% | $7,920d | |
$5,056 | 10% | $1,138d | |
$4,293 | 10% | $953.09d | |
HDT | — | 35.8% | $3,912e |
$4,530 | 80% | $1,296 | |
$31,227 | 20% | $2,233 | |
ASCT3 | $67,621 | 34.6% | $23,422 |
Total costs | — | — | $40,551 |
ASCT = autologous stem cell transplant; axi-cel = axicabtagene ciloleucel; BEAM = carmustine, etoposide, cytarabine, and melphalan; EM = etoposide and melphalan; R-GDP = rituximab + gemcitabine, dexamethasone, and cisplatin; HDT = high-dose therapy; R-DHAP = rituximab + dexamethasone, cytarabine-, and cisplatin; R-GDP = rituximab + gemcitabine, dexamethasone, and cisplatin; R-ICE = rituximab + carboplatin, etoposide, and ifosfamide; SOC = standard of care.
aIncludes the administration cost of $676.43, based on the cost of administering a standard chemotherapy agent plus the cost of an additional chemotherapy agent for a total of 3 hours of chair time.
bSponsor assumed an inpatient length of stay of 14 days, and 28% of patients would be admitted to the intensive care unit for a median of 5 days (based on ZUMA-7).
cIncludes weighted administration cost of $3,206 based on the cost of complex chemotherapy administration from the Ontario Schedule of Benefits for Physician Services plus the cost of each additional chemotherapy agent in the regimen.24
dNumber of cycles was based on the average number of treatment cycles (~2.3) from the ZUMA-7 trial.
eIncludes weighted administration cost of $383.08 based on the cost of administering myeloablative therapy from the Ontario Schedule of Benefits and Physician Services.24
Source: Sponsor’s pharmacoeconomic submission.1
Table 13: Summary of the Sponsor’s Economic Evaluation Results (Deterministic)
Drug | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER vs. SOC ($/QALY) |
|---|---|---|---|---|---|
SOC | $336,857 | Reference | 7.47 | Reference | Reference |
Axi-cel | $569,098 | $232,241 | 9.31 | 1.84 | $126,046 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Source: Sponsor’s pharmacoeconomic submission.1
Table 14: Subsequent Therapy Use and Costs
Subsequent therapy | Axi-cel | SOC | ||
|---|---|---|---|---|
% receiving | Weighted cost | % receiving | Weighted cost | |
Chemotherapy | 20% | $1,895.40 | 0 | $0 |
Radiotherapy | 20% | $314.46 | 40% | $628.92 |
ASCT | 10% | $7,856.14 | 0 | $0 |
Allo-SCT | 0 | $0 | 0 | $0 |
Axi-cel | 0 | $0 | 25% | $130,961.84 |
Tisagenlecleucel (Kymriah) | 0 | $0 | 25% | $122,286.82 |
Clinical trial | 40% | $0 | 0 | $0 |
Patients not seeking active treatment | 10% | $0 | 10% | $0 |
Total costs | — | $6,729.95 | — | $259,119.33 |
allo-SCT = allogenic stem cell transplant; ASCT = autologous stem cell transplant; SOC = standard of care.
Source: Sponsor’s pharmacoeconomic submission.1
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
This appendix has not been copy-edited.
Detailed Results of CADTH Base Case
Table 15: Disaggregated Summary of CADTH’s Economic Evaluation Results
Parameter | Axi-cel | SOC | Incremental |
|---|---|---|---|
Discounted LYs | |||
Total | 10.34 | 10.11 | 0.24 |
Event-free | 8.06 | 3.63 | 4.43 |
Post event | 2.28 | 6.47 | –4.19 |
Discounted QALYs | |||
Total | 8.25 | 7.67 | 0.58 |
Event-free | 6.59 | 2.97 | 3.62 |
Post event | 1.65 | 4.69 | –3.04 |
Discounted costs ($) | |||
Total | $572,112 | $336,966 | $235,146 |
Second-line treatment costs | $493,048 | $40,456 | $452,592 |
Axi-cel | $493,048 | $0 | $493,048 |
Drug acquisition | $455,920 | $0 | $455,920 |
Leukapheresis | $2,759 | $0 | $2,759 |
Drug administration | $31,160 | $0 | $31,160 |
Conditioning chemotherapy | $3,209 | $0 | $3,209 |
Salvage chemotherapy | $0 | $13,199 | –$13,199 |
ASCT | $0 | $27,256 | –$27,256 |
Third-line treatment costs | $11,220 | $229,505 | –$218,285 |
CAR T cells | $0 | $228,918 | –$228,918 |
Salvage chemotherapy | $4,375 | $586 | $3,789 |
Allo-SCT | $0 | $0 | $0 |
ASCT | $6,844 | $0 | $6,844 |
Disease management | $9,827 | $8,746 | $1,081 |
Terminal care | $58,018 | $58,259 | –$242 |
ICER ($/QALY) | $404,418 | ||
allo-SCT = allogenic stem cell transplant; ASCT = autologous stem cell transplant; axi-cel = axicabtagene ciloleucel; CAR = chimeric antigen receptor; ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year; SOC = standard of care.
Scenario Analyses
Table 16: Summary of Probabilistic Scenario Analyses Conducted on CADTH Base Case
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
CADTH base case | SOC | $336,966 | 7.67 | Reference |
Axi-cel | $572,112 | 8.25 | $404,418 | |
Scenario 1: Health state utilities based on literature | SOC | $336,966 | 7.64 | Reference |
Axi-cel | $572,112 | 8.25 | $386,914 | |
Scenario 2: Assuming 5% of patients are re-treated with axi-cel in third line | SOC | $336,966 | 7.67 | Reference |
Axi-cel | $591,611 | 8.25 | $437,953 | |
Scenario 3: Assuming mean age of 67 years | SOC | $342,468 | 5.68 | Reference |
Axi-cel | $573,731 | 6.16 | $486,906 | |
Scenario 4: Assuming trial-based pretreatment proportions for patients receiving axi-cel | SOC | $336,778 | 7.67 | Reference |
Axi-cel | $571,631 | 8.25 | $403,913 | |
Scenario 5: Value-based pricing (76% OS rate at 12 months among patients receiving axi-cel) | SOC | $311,451 | 7.67 | Reference |
Axi-cel | $462,691 | 8.25 | $260,112 | |
Scenario 6: Crossover adjustment based on RPSFTM and exclusion of CAR T-cell therapy in third line for SOC (Deterministic)a | SOC | $114,310 | 2.77 | Reference |
Axi-cel | $572,112 | 8.13 | $85,299 | |
Scenario 7: Crossover adjustment based on IPCW and exclusion of CAR T-cell therapy in third line for SOC (Deterministic)a | SOC | $113,432 | 5.33 | Reference |
Axi-cel | $572,112 | 8.13 | $163,890 |
ICER = incremental cost-effectiveness ratio; IPCW = inverse probability of censoring weights model; QALY = quality-adjusted life-year; OS = overall survival; RPSFTM = rank-preserving structural failure time model; SOC = standard of care.
aScenario presented deterministically as the probabilistic scenario ran with errors. Specifically, the model could not compute SOC costs probabilistically after redistributing treatments in 3L SOC CAR T-cell therapy recipients to other treatments.
Appendix 5: Submitted BIA and CADTH Appraisal
Note this appendix has not been copy-edited.
Table 17: Summary of Key Take-Aways
Key take-aways of the BIA |
|---|
|
Summary of Sponsor’s BIA
The sponsor submitted a BIA to estimate the incremental 3-year budget impact of reimbursing axi-cel for the treatment of adult patients with relapsed or refractory LBCL, who are candidates for ASCT. The analysis was performed from the perspective of the Canadian public drug plan with a scenario analysis based on the health care system perspective. The sponsor estimated the budget impact by comparing 2 scenarios: a reference scenario that estimated the total costs associated with SOC for the treatment of adults with relapsed or refractory LBCL who are candidates for ASCT, with a new drug scenario, where axi-cel is funded in the second-line setting as per its reimbursement requested criteria. SOC included salvage chemotherapy regimens (i.e., R-ICE, R-GDP, and R-DHAP) and HDT (i.e., EM and BEAM) followed by ASCT in cases of adequate disease response.
The sponsor estimated the eligible population using an epidemiology-based approach, leveraging data from multiple sources in the scientific literature and assumptions based on clinical expert input. The sponsor included second- and third-line drug acquisition costs associated with axi-cel and SOC. Key inputs are documented in Table 18.
Key assumptions made by the sponsor include: Treatment regimens and the proportion of patients receiving each therapy (i.e., conditioning chemotherapy, salvage chemotherapy, HDT) before axi-cel infusion or ASCT were based on the ZUMA-7 clinical trial and assumed reflective of Canadian clinical practice.
Costs associated with axi-cel and SOC would be paid by the province in which the patients reside (i.e., no patients assumed covered by Non-Insured Health Benefit [NIHB] program).
Table 18: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3 if appropriate) |
|---|---|
Target population | |
General Canadian population, sum of included jurisdictions in base year | ~29,758,786 |
10.54 | |
Percentage incident patients with LBCL requiring first-line treatment53 | 95% |
Percentage LBCL patients with primary refractory disease54 | 20% |
Percentage eligible for ASCT after first-line treatment | 50% |
Percentage proceeding to treatment53 | 80% |
Percentage LBCL patients who relapse after first-line treatment54 | 20% |
Percentage who relapse within 12 months | 67% |
Percentage eligible for ASCT after relapse | 50% |
Percentage proceeding to treatment53 | 80% |
Number of patients eligible for drug under review | 402 / 407 / 412 |
Market uptake (3 years) | |
Uptake (reference scenario) SOC | 100% / 100% / 100% |
Uptake (new drug scenario) Axi-cel SOC | ████% / ████% / ████% ████% / ████% / ████% |
Cost of treatment (per patient) | |
Axi-cel (1 time) Acquisition Leukapheresis Bridging therapy (weighted) Conditioning chemotherapy (weighted) | $485,021 $2,759 $506 $2,027 |
SOC Salvage chemotherapy (weighted) HDT (weighted) ASCT | $4,334 (per cycle) $3,529 (1 time) $67,621 (1 time) |
ASCT = autologous stem cell transplant; HDT = high-dose therapy; LBCL = large B-cell lymphoma; SOC = standard of care.
Note: Table 14 presents the estimated treatment mix for subsequent treatment therapies (and proportion of patients assumed to receive each therapy) in third line, following axi-cel and SOC that also informed the sponsor’s budget impact analysis.
Summary of the Sponsor’s BIA Results
Results of the sponsor’s base-case BIA suggest that the incremental expenditures associated with the reimbursement of axi-cel for the reimbursement request population would be $7,302,910 in year 1, $14,705,537 in year 2, and $27,063,656 in year 3, for a 3-year cumulative total of $49,072,104, under the drug plan perspective. When considering a health care system perspective, the sponsor’s base case estimated a budgetary impact of $7,214,321 in year 1, $14,545,375 in year 2, and $26,770,766 in year 3, for a 3-year cumulative total of $48,530,462.
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Exclusion of ASCT-ineligible patient population: As noted above, CADTH received clinical expert feedback that the expected place in therapy for axi-cel would align more closely with its Health Canada indication (i.e., second-line treatment of adult patients with DLBCL or HGBL that is refractory to first-line chemoimmunotherapy or that relapses within 12 months of first-line chemoimmunotherapy, regardless of ASCT eligibility). They further noted challenges in implementing an initiation criterion based on ASCT eligibility (refer to Issue for Consideration). Hence, it is expected that, if funded, axi-cel may be offered to the broader Health Canada indication – and not solely to the subpopulation considered by the sponsor in its reimbursement request.
CADTH conducted a scenario analysis that considered the broader Health Canada–indicated population by removing the ASCT eligibility criterion from the calculation of the total number of patients eligible for axi-cel. This approach, however, is not without limitations, considering that (1) it is not feasible to adequately explore the comparators in this broader population of patients with relapsed or refractory LBCL who are ASCT-ineligible, and (2) the treatment mix in the reference and new drug scenario did not differ from the CADTH reanalysis (e.g., SOC remains as ASCT rather than chemotherapy). Given this limitation, the estimated magnitude of the impact may be underestimated given the costs of ASCT are assumed for those on SOC.
Projected market share of axi-cel is underestimated: The sponsor assumed that axi-cel would have a market share of ███%, ████%, and ████% in years 1, 2, and 3, respectively. The clinical expert feedback emphasized that the sponsor’s market share projections were significantly lower than they would anticipate observing in practice once a therapy like axi-cel is funded in second line. This aligned with the feedback received from registered clinician groups in Canada who noted that axi-cel was unanimously expected to shift the current treatment paradigm by replacing intensive chemoimmunotherapy and ASCT as the new preferred second-line treatment for patients with relapsed or refractory LBCL.
CADTH conducted a reanalysis by adjusting the projected market share of axi-cel to 77.4%, 87.6%, and 93.8% in years 1, 2, and 3, respectively, based on feedback sought from CADTH clinical experts.
CADTH conducted a scenario analysis assuming axi-cel’s market shares to be 67.2%, 77.4%, and 87.6% in years 1, 2, and 3, respectively, to show how a more conservative projection may impact the budget.
CADTH conducted a scenario analysis assuming axi-cel’s market shares to be 40%, 50%, and 60% in years 1, 2, and 3, respectively to show how a more conservative projection may impact the budget.
Projected market size is underestimated: The sponsor assumed that 80% of patients with relapsed or refractory LBCL who are eligible for ASCT would proceed to second-line treatment. Clinical expert feedback highlighted that there is uncertainty associated with this number and that it may be higher in real-practice.
CADTH conducted a reanalysis by assuming that 90% of patients with relapsed or refractory LBCL who are eligible for ASCT would proceed to second-line treatment.
Inconsistent modelling of subsequent treatment mix: As noted above, the estimated treatment mix for subsequent therapies included in the BIA varied substantially between patients receiving axi-cel and SOC. Specifically, 40% of patients receiving axi-cel were assumed to seek subsequent treatment through clinical trials for investigational therapies (at no additional cost to drug plans), while this assumption was extended to zero patients in the SOC arm. By assuming that patients receiving axi-cel will exclusively be able to seek investigational therapies through clinical trials relative to patients receiving SOC, the sponsor effectively reduced the costs associated with subsequent treatment in the axi-cel arm.
CADTH conducted a reanalysis redistributing the subsequent therapy mix for patients in the axi-cel arm such that none would be receiving clinical trials (i.e., distributions reflected the revisions CADTH made to derive the CADTH reanalysis within the economic evaluation).
CADTH Reanalyses of the BIA
CADTH conducted reanalyses of the BIA for the reimbursement request population by revising the projected market share of axi-cel, increasing the proportion of patients eligible for ASCT who would proceed to treatment with axi-cel in the second line, and redistributing subsequent therapies in the axi-cel arm to reflect the distribution assumed in the CADTH base-case reanalysis within the economic evaluation.
Table 19: CADTH Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
None | — | — |
Changes to derive the CADTH base case | ||
1. Projected market share of axi-cel | Year 1: ███% Year 2: ████% Year 3: ████% | Year 1: 77.4% Year 2: 87.6% Year 3: 93.8% |
2. Projected market size (i.e., ASCT-eligible population proceeding to treatment) | 80% | 90% |
3. Subsequent treatment mix (axi-cel) | Chemotherapy: 20% Radiotherapy: 20% ASCT: 10% Clinical trial: 40% Not seeking active treatment: 10% | Chemotherapy: 50% Radiotherapy: 20% ASCT: 10% Clinical trial: 0% Not seeking active treatment: 20% |
CADTH base case | 1 + 2 + 3 | |
ASCT = autologous stem cell transplant.
The results of the CADTH step-wise reanalysis are presented in summary format in Table 20. Based on the CADTH base case, the budget impact in the population of patients with relapsed or refractory LBCL who are candidates for ASCT is expected to be $103,063,855 in year 1, $117,507,525 in year 2, and $127,069,602 in year 3, with a 3-year total of $347,640,982. It was estimated that 350, 401, and 434 patients would be treated with axi-cel in years 1, 2, and 3 of the analysis, respectively. The submitted analysis is based on the publicly available prices of the comparator treatments.
Table 20: Summary of the CADTH Reanalyses of the BIA
Stepped analysis | Three-year total |
|---|---|
Submitted base case | $49,072,104 |
CADTH reanalysis 1 | $307,382,244 |
CADTH reanalysis 2 | $55,206,117 |
CADTH reanalysis 3 | $49,337,923 |
CADTH base case | $347,640,982 |
BIA = budget impact analysis.
CADTH conducted additional exploratory analyses on the CADTH base case (Table 21). These included: assuming axi-cel’s market shares to be lower; considering the broader Health Canada–indicated population by removing the ASCT eligibility criterion from the calculation of the total number of patients eligible for axi-cel, assuming a value-based pricing scheme based on 76% OS rate at 12 months among patients treated with axi-cel; assuming the health care payer perspective; excluding CAR T cells from the subsequent therapy mix for patients receiving SOC; and assuming a 45% price reduction in the reimbursement request population.
Of note, the scenario that considered the broader Health Canada–indicated population, by removing the ASCT eligibility criterion from the calculation of the total number of patients eligible for axi-cel, resulted in an estimated 3-year total budget impact of $695,281,964. The scenario that excluded CAR T cells from the subsequent therapy mix for patients receiving SOC, which is informative for drug plans that do not currently fund CAR T-cell therapy in third line, resulted in an estimated 3-year total budget impact of $565,340,233. All other scenarios explored resulted in 3-year total budget impacts ranging from $139,382,205 to $343,756,318.
Table 21: Detailed Breakdown of the CADTH Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
Submitted base case | Reference | $3,984,270 | $79,158,744 | $80,629,559 | $81,734,951 | $241,523,254 |
New drug | $3,984,270 | $86,461,654 | $95,335,096 | $108,798,607 | $290,595,357 | |
Budget impact | $0 | $7,302,910 | $14,705,537 | $27,063,656 | $49,072,104 | |
CADTH base case | Reference | $4,482,304 | $89,053,586 | $90,708,254 | $91,951,820 | $271,713,660 |
New drug | $4,482,304 | $192,117,441 | $208,215,778 | $219,021,422 | $619,354,642 | |
Budget impact | $0 | $103,063,855 | $117,507,525 | $127,069,602 | $347,640,982 | |
CADTH scenario analysis 1: No ASCT eligibility criterion | Reference | $8,964,607 | $178,107,173 | $181,416,508 | $183,903,640 | $543,427,321 |
New drug | $8,964,607 | $384,234,883 | $416,431,557 | $438,042,845 | $1,238,709,285 | |
Budget impact | $0 | $206,127,710 | $235,015,049 | $254,139,205 | $695,281,964 | |
CADTH scenario analysis 2: Alternative market share 1 | Reference | $4,482,304 | $89,053,586 | $90,708,254 | $91,951,820 | $271,713,660 |
New drug | $4,482,304 | $178,535,383 | $194,533,396 | $210,622,366 | $583,691,144 | |
Budget impact | $0 | $89,481,797 | $103,825,142 | $118,670,546 | $311,977,484 | |
CADTH scenario analysis 3: Alternative market share 2 | Reference | $4,482,304 | $89,053,586 | $90,708,254 | $91,951,820 | $271,713,660 |
New drug | $4,482,304 | $142,316,561 | $157,778,759 | $173,233,016 | $473,328,335 | |
Budget impact | $0 | $53,262,974 | $67,070,505 | $81,281,196 | $201,614,675 | |
CADTH scenario analysis 4: Value-based pricing (76% OS rate among patients on axi-cel) | Reference | $4,482,304 | $78,630,819 | $80,087,755 | $81,184,357 | $239,902,931 |
New drug | $4,482,304 | $148,966,840 | $160,205,955 | $167,791,237 | $476,964,032 | |
Budget impact | $0 | $70,336,022 | $80,118,199 | $86,606,880 | $237,061,101 | |
CADTH scenario analysis 5: Excluding CAR T-cell therapy from subsequent therapy mix for SOC | Reference | $4,482,304 | $6,455,322 | $6,543,014 | $6,621,921 | $19,620,258 |
New drug | $4,482,304 | $173,450,234 | $197,779,289 | $213,730,969 | $584,960,491 | |
Budget impact | $0 | $166,994,911 | $191,236,275 | $207,109,047 | $565,340,233 | |
CADTH scenario analysis 6: Assuming 5% of patients are re-treated with axi-cel in 3L | Reference | $4,482.304 | $89,053,586 | $90,708,254 | $91,951,820 | $271,713,660 |
New drug | $4,482.304 | $192,117,441 | $208,215,778 | $219,021,422 | $619,354,642 | |
Budget impact | $0 | $103,063,855 | $117,507,525 | $127,069,602 | $347,640,982 | |
CADTH scenario analysis 7: Health care payer perspective | Reference | $18,922,852 | $110,331,311 | $112,676,942 | $114,494,716 | $337,502,969 |
New drug | $18,922,852 | $212,150,978 | $228,911,567 | $240,196,742 | $681,259,287 | |
Budget impact | $0 | $101,819,666 | $116,234,625 | $125,702,027 | $343,756,318 | |
CADTH scenario analysis 8: 45.2% price reduction | Reference | $4,482,304 | $69,424,041 | $70,706,315 | $71,673,099 | $211,803,454 |
New drug | $4,482,304 | $110,850,476 | $117,797,277 | $122,537,907 | $351,185,660 | |
Budget impact | $0 | $41,426,436 | $47,090,962 | $50,864,808 | $139,382,205 |
ASCT = autologous stem cell transplant; BIA = budget impact analysis; CAR = chimeric antigen receptor; OS = overall survival; SOC = standard of care.
a67.2%, 77.4%, and 87.6% in years 1, 2, and 3, respectively.
Ethics Review
Abbreviations
ASCT
autologous stem cell transplant
axi-cel
axicabtagene ciloleucel
CAR
chimeric antigen receptor
LBCL
large B-cell lymphoma
Summary
Patient and clinician group, clinical expert, and drug program input gathered in the course of this CADTH review, as well as relevant literature, were reviewed to identify ethical considerations relevant to the use of axicabtagene ciloleucel (axi-cel) for the treatment of adult patients with relapsed or refractory large B-cell lymphoma (LBCL).
Ethical considerations arising in the context of LBCL highlight the impacts on patients and the disparities in the incidence, treatment, and outcomes of LBCL, especially as they affect racialized or other groups that are underserved. The diagnosis of relapsed or refractory disease, as well as determinations of eligibility for autologous stem cell transplant, were cited as complex barriers to treatment.
Ethical considerations arising in the evidence used to evaluate axi-cel indicated some limitations in clinical trial data, including limitations on included groups and long-term data on safety and effectiveness. As well, budget forecasting may underestimate the overall budget impact of these therapies if implemented fairly and as needed.
Several access considerations arise in the context of chimeric antigen receptor (CAR) T-cell therapies in Canada, including those related to geographic access, especially because they may disproportionately affect racialized or underserved groups, as well as inequities that might emerge in the process of patient referral. Considerations also arise in the context of cell and tissue use and ownership during CAR T-cell manufacture and disposal as do considerations related to informed consent and balanced communication about CAR T-cell therapies.
Ethical considerations for health systems related to implementing axi-cel therapy include challenges to implementing and scaling CAR T-cell treatment sites across Canada as well as the other considerations related to high-cost therapies.
Objective
The aim of this review is to identify and describe ethical considerations associated with the use of axicabtagene ciloleucel (axi-cel) for the treatment of adult patients with relapsed or refractory large B-cell lymphoma (LBCL), including those related to the context of LBCL, evidentiary basis, and use of axi-cel as a chimeric antigen receptor (CAR) T-cell therapy, and considerations relevant to health systems.
Research Questions
This report addresses the following research questions:
What ethical considerations arise in the context of LBCL in adult patients, including those related to diagnosis, treatment, and outcomes?
What ethical considerations arise related to the evidence (e.g., clinical and economic data) related to the validation and approval of axi-cel?
What ethical considerations arise in the use of CAR T-cell therapies for clinicians, patients, and their caregivers?
What ethical considerations for health systems are involved in the context of CAR T-cell therapies?
Methods
To identify ethical considerations relevant to the use of axi-cel in the treatment of relapsed or refractory LBCL, this ethics report was driven by relevant questions identified from the EUnetHTA HTA Core Model Version 3.0 (ethical analysis domain)1 and supplemented with relevant questions from the equity checklist for health technology assessment (ECHTA).2 These guiding questions were organized to respond to the research questions posed, and ethical considerations were investigated relating to the following:
Patients with LBCL and their caregivers (i.e., disparities in incidence, treatment, or outcomes; challenges related to diagnosis or clinical care; factors that might prevent patients from gaining access to therapies).
The evidence used to demonstrate the benefits, harms, and value of axi-cel (i.e., ethical considerations in relevant clinical trials, including their representativeness, choice of outcome measures, appropriateness of analytical methods, and models to all population groups; ethical considerations related to the data or assumptions in the economic evaluation).
The use of axi-cel as a CAR T-cell therapy, including considerations related to benefits and harms to patients, relatives, caregivers, clinicians, or society, and considerations related to access to these therapies.
The uptake of CAR T-cell therapies in health systems, including considerations related to the distribution of health care resources.
Data Collection: Review of Project Inputs and Literature
Data to inform this ethics report drew from an identification of ethical considerations (e.g., values, norms, or implications related to the harms, benefits, and implications for equity, justice, resource allocation, and ethical considerations in the evidentiary basis) in the patient and clinician group, clinical expert, and drug program input collected in the course of this review, as well as a complimentary search of the published literature. Ongoing collaboration and communication with the CADTH review team also assisted in the clarification and identification of ethical considerations raised.
Review of Project Inputs
Six main sources of inputs collected in the course of this CADTH review were reviewed by a single reviewer to inform the ethics report:
The sponsor submission was reviewed for content related to ethical considerations (e.g., values, norms, or implications related to the harms, benefits, and implications for equity, justice, or resource allocation), noting relevant information and external references or sources relevant to each of the research questions driving this report.
Clinician group input received by CADTH from Lymphoma Canada, Ontario Health Hematology Cancer Drug Advisory Committee, and Cell Therapy Transplant Canada was reviewed for content related to ethical considerations, relevant to each of the research questions driving this report.
Patient input received by CADTH from Lymphoma Canada was reviewed for content related to ethical considerations relevant to each of the research questions driving this report.
Drug program input received by CADTH was reviewed for content related to ethical considerations relevant to each of the research questions driving this report.
Clinical experts (n = 4) were engaged by CADTH during this Reimbursement Review through 2 teleconference discussions and 1 panel discussion. These clinical experts were active in relevant clinical roles in Canada, and all had experience treating patients with LBCL and using CAR T-cell therapies. During each of the 3 interactions, the clinical experts were asked targeted questions related to ethical considerations corresponding to the research questions driving this report.
Collaboration with the CADTH clinical and economic reviewers identified domains of ethical interest arising in their reviews and relevant questions and sources pursued in this report.
Literature Search and Selection Methods
The literature search strategies used in this ethics report are updates of those developed for 2 previous CADTH reports.3,4 The original searches were as follows: a search run on November 26, 2018, with the main search concept of axicabtagene ciloleucel (Yescarta), using the databases MEDLINE All via Ovid, PsycInfo via Ovid, PubMed, and the Cumulative Index to Nursing and Allied Health Literature (CINAHL) via EBSCO3 and a search run on August 19, 2021, with the main search concepts of large B-cell lymphoma or CAR T-cell therapy using the databases MEDLINE All via Ovid, Philosopher’s Index via Ovid, CINAHL via EBSCO, and Scopus.4 The searches for axicabtagene ciloleucel were restricted to articles published from January 1, 2008, to the search date and were limited to citations published in English or French; the searches for large B-cell lymphoma or CAR T-cell therapy were not restricted by date and were limited to citations published in English. CADTH-developed search filters were applied to both searches to limit retrieval to publications related to empirical and normative ethical considerations.
For the current report, 2 literature searches were conducted by an information specialist using the following databases: MEDLINE All (1946‒) via Ovid, Philosopher’s Index via Ovid, CINAHL via EBSCO, and Scopus. The database searches were rerun on July 5, 2022, to capture any articles published or made available since the initial search dates. Because Ovid Philosopher's Index and Scopus were not searched during the initial axicabtagene ciloleucel search, no date limits were used for that concept in these databases. Duplicates were removed by manual deduplication in Endnote. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concepts were axicabtagene ciloleucel (Yescarta), large B-cell lymphoma, or CAR T-cell therapy.
CADTH-developed search filters were applied to limit retrieval to citations related to empirical and normative ethical considerations. Retrieval was limited to citations published in the English language.
Grey literature (literature that is not commercially published) was identified by searching sources listed in the ethics section of the Grey Matters: A Practical Tool For Searching Health-Related Grey Literature checklist.5 The grey literature search for ethical considerations was conducted on July 5, 2022. The main search concepts were Yescarta (axicabtagene ciloleucel), large B-cell lymphoma, or CAR T-cell therapy. Search results were not limited by publication date but were limited to articles published in the English language. Google was used to search for additional internet-based materials. These searches were supplemented by reviewing bibliographies of key papers and through contacts with experts, as appropriate.
Literature Screening and Selection
The ethics review sections of 2 previous CADTH reports3,4 served as the foundations for this review, and the literature retrieved from the search and selection methods (described in the Literature Search and Selection Methods section) provided updated information. The titles and abstracts of citations retrieved were screened for relevance by a single reviewer. Articles were retrieved for full-text review if they identified or provided normative analysis (i.e., focusing on “what ought to be” through argumentation) or presented empirical research (i.e., focusing on “what is” through observation) of ethical considerations related to the incidence, diagnosis, treatment, or outcomes of LBCL or arising in the evidence used to support, use, or implications of axi-cel as a CAR T-cell therapy. Full-text publications categorized as “retrieve” were then reviewed by the same reviewer. Reports meeting the above criteria were included in the review, and reports that did not meet these criteria were excluded.
In a parallel process, the grey literature and other sources drawn from relevant bibliographies or in consultation with experts or other CADTH reviewers were retrieved and reviewed following the selection criteria listed previously.
Data Analysis
Data analysis included the collection, coding, and thematic analysis of data drawn from the literature and project inputs, driven by the 4 research questions guiding this ethics review report. The reviewer conducted 3 iterative cycles of coding and analysis to abstract, identify, and synthesize relevant ethical considerations in the literature and from relevant project inputs.
In the initial coding phase, the main ethical considerations posed and discussed in the 2 previous CADTH ethics reports were abstracted. These considerations were used to guide questions asked of the clinical experts, and the reviews of project inputs and additional literature sources. In the second coding phase, publications retrieved from the updated search as well as project input sources were reviewed for ethical content (e.g., claims related to potential harms, benefits, equity, justice, resource allocation, and ethical issues in the evidentiary basis). Once identified, claims related to ethical content were coded using qualitative description methods.6 In the final coding phase, major themes and subcodes were identified through repeated readings of the data6 and through comparisons with the initial themes and considerations that emerged in the previous CADTH reviews. These were then summarized into thematic categories within each domain or research question. Where ethical content emerged that did not fit into these categories or domains outlined in the research questions, this was noted, as were discrepancies within or conflicts between ethical considerations or values identified between project sources or within thematic categories. Data analysis was iterative, and themes identified in the literature, in project inputs, and during consultations with clinical experts were used to further refine and reinterpret the ethical considerations identified.
Results
Description of Included Sources
Two previous CADTH reports of ethical considerations in the context of CAR T-cell therapies provided the foundation for the literature search strategy. These 2 reports were reviews of relevant ethical considerations in the use of axi-cel for LBCL3 and lisocabtagene maraleucel for LBCL.4 The search strategies used in these reports were updated to identify any recent relevant publications. From these updated searches, a total of 244 citations were identified. Following screening of titles and abstracts, 208 citations were excluded and 36 potentially relevant publications from the electronic search were retrieved for full-text review. Of the potentially relevant publications, 24 publications were excluded because they did not include ethical considerations of axi-cel or LBCL (n = 19) or they were not relevant to the Canadian context (n = 5). Twelve publications met the inclusion criteria and were included in this report. In addition, 2 relevant publications were retrieved from other sources, including the grey literature search.
In addition to the 2 previous CADTH reviews, a total of 14 other publications were used to inform this report: 11 publications discussed ethical considerations in the context of access, use, or implementation of CAR T-cell therapies; 2 publications discussed considerations in the implementation of CAR T-cell or gene therapies in Canada; and 1 publication discussed disparities in LBCL treatment. The characteristics of included publications are listed in Table 1.
In addition to sources from the published literature, data used to inform this ethics report drew from a review of the patient group input, clinician group input, drug program input, and consultation with clinical experts engaged by CADTH for this review. A description and summary of these sources are included in the Clinical Review Report.
Key Ethical Considerations
Diagnosis of and Experiences With LBCL
Patient Experiences With LBCL
Patient input received by CADTH identified how patients with LBCL face both physical symptoms and the psychosocial impacts of their conditions, including fear of relapse and becoming dependent on others. Patients reported that their therapy was able to manage many of their symptoms, although they tended to prioritize long-term health outcomes over short-term improvements in quality of life. Patients also valued the opportunity to exercise their autonomy through choice of treatment and felt there was a need for more therapeutic options for LBCL. A previous CADTH review of ethical issues related to LBCL found that patients may underestimate the severity of their illness and tend not to document their care preferences or participate in advance care planning.4 This has highlighted a need to expand the availability and accessibility of patient information about LBCL, and for these materials to reflect a diversity of lived experiences.
Disparities in LBCL Incidence, Treatment, and Outcomes
A previous CADTH review of ethical issues in LBCL found significant disparities reported in the incidence of LBCL where these cancers are more prevalent in certain racialized groups, particularly among Black people in the US.4 Disparities in treatment have also disproportionately affected many groups; women and patients who are white had longer survival outcomes compared to other racialized groups, those of higher socioeconomic status experience superior treatment and survival outcomes, and patients living in lower income neighbourhoods have worse treatment and survival outcomes.4 Reported rationales for disparities in treatment and outcomes include differential access to health care, challenges in patient-physician relationships, and socioeconomic status. Therefore, equity-deserving patients may require more assistance navigating treatment pathways for LBCL to attempt to mitigate these disparities.4
When discussing disparities in the incidence, treatment, and outcomes of LBCL in Canada, the clinical experts engaged by CADTH noted that there have been few evaluations of the experiences and outcomes of Black patients in Canada with LBCL; most studies have been based in the US and the results may not be directly comparable. The clinical experts noted that Indigenous Peoples in Canada can also face many barriers to care; some Indigenous patients may live far away from cancer treatment centres, have language barriers, or encounter systemic biases in health care. They discussed that all patients with cancer are vulnerable, but these vulnerabilities are amplified in these groups.
Diagnosis and Treatment of Relapsed or Refractory LBCL
Clinical experts engaged by CADTH discussed how the classification of lymphomas can be complex, and how the subset of patients with relapsed or refractory LBCL can be a difficult-to-treat population. Although frontline therapy tends to be somewhat standard across patients, they discussed how decisions around second-line therapy are more varied and complicated. Patients who have relapsed or who have disease that is refractory to first-line therapies may be referred for high-dose chemotherapy followed by autologous stem cell transplant (ASCT). However, the clinical experts discussed how the eligibility criteria for ASCT can be nebulous and can vary across the country, leading to fairness and equity challenges. The clinical experts noted that responses vary at the time of relapse and that many patients may not make it to transplant. Input from clinician groups also indicated that the majority of patients do not achieve durable remission with the current standard of care of second-line therapy or ASCT, and they indicated that as few as 10% of patients achieve durable remissions after ASCT. In addition, the clinician groups identified ASCT as intensive therapy associated with significant toxicities, and eligibility for this treatment is limited to younger, medically fit patients. In many patients, sufficient stem cells cannot be collected, and they cannot proceed to transplant.
Ethics of Evidence and Evaluation of Axi-Cel
Ethical Considerations in Clinical Trial Data
The clinical evidence used to evaluate the use of axi-cel for relapsed or refractory LBCL is drawn from the ZUMA-7 trial. In considering the populations included in this trial, the clinical experts noted that patients who were HIV-positive were noticeably excluded from this trial, despite some types of lymphoma being associated with HIV. The clinical experts also noted that patients with an Eastern Cooperative Oncology Group Performance Status (ECOG PS) score greater than 1 were not included in this trial, and that these exclusions may not be reflective of patients seen in regular clinical practice in Canada. In addition, the median age of participants in the ZUMA-7 trial participants was 59 years, which is younger than the median age of patients with LBCL in Canada, which may affect the applicability to older patients with LBCL, especially those who may not be eligible for ASCT. The choice of participants included in the trial may raise considerations about the applicability of the trial results to the broader patient population in Canada who have LBCL and are eligible for CAR T-cell therapy.
The clinical experts also discussed the challenges related to CAR T-cell manufacture time and noted that, in the ZUMA-7 trial, cells would be returned to patients by the manufacturer in 13 days, but this time may be longer in practice. They commented that the trial data presented an ideal but not likely realistic representation of the manufacture and return time. Clinical experts also noted that there may be circumstances when manufacturing fails or the cells do not meet the criteria for release, and that these circumstances may be more common in the real world than in clinical trials.
In international settings, it has been noted that Black patients tend to be underrepresented in clinical trials of CAR T-cell therapies internationally, and this lack of access to clinical trials may further widen existing disparities.4,7 Further, enrolment in clinical trials tends not to be matched to disease prevalence in racialized or other underserved populations, and that these groups are often not reported in clinical trial data (e.g., are included in an “other” category).7 In the ZUMA-7 trial, 81% of patients in the treatment arm, and 85% of patients in the standard of care arm were white. In addition to concerns about the limited inclusion of Black patients in CAR T-cell therapy clinical trials, young adults and older adults, and other racialized patients also tend to be underrepresented in these trials, potentially limiting the applicability of the results to these populations.4 Given the limited treatment options for patients with relapsed or refractory LBCL, demand for access to clinical trials on CAR T-cell therapies is high, and concerns have been expressed that this imbalance in demand versus availability may privilege some groups over others.8
Despite advances in clinical trial data for CAR T-cell therapies and 15 years of data on patients, there is a lack of long-term safety and efficacy data for CAR T-cell therapies which has been widely discussed by clinical experts and in the published literature.3,4,8
Ethical Considerations in Economic Models
Given the high costs and complexity of CAR T-cell therapies, and the opportunity costs posed by these therapies in the Canadian health care system, there have been calls to include diverse stakeholders in health economic modelling.9 It is not clear the extent to which the input of stakeholders, beyond clinical experts (e.g., patients, the public, payers, industry), have been included in the economic modelling used to support axi-cel.
In considering the budget impact analysis, the clinical experts engaged by CADTH for this review questioned the low projected uptake at year 3 of availability. They indicated that the projected uptake would not be sufficient to meet demand and would require clinicians to choose which patients would have access and which would not, raising ethical considerations around fairness and resource allocation. Although there may be rationales for limiting the availability of axi-cel, and therefore its budget impact, limited access raises ethical considerations for clinicians who must determine criteria for access or deny access to certain patients, and for patients who may lack access despite meeting eligibility criteria.
Ethical Considerations in the Use of CAR T-Cell Therapies
Access to CAR T-Cell Therapies
Access to CAR T-cell therapies has been a long-standing concern in the literature, raising considerations around equity, fairness, resource allocation, and distributive justice. Although there has been much discussion of costs as a barrier in non-Canadian health care systems,4 there are several other access barriers that have been discussed. Drug program input noted that there is limited access to CAR T-cell services in Canada, and the drug programs suggested the need for mechanisms to ensure equitable access. The clinical experts engaged by CADTH also discussed challenges with the ability of facilities in Canada to meet the demand that would be present for CAR T-cell therapy and the challenges for physicians in making equitable access decisions regarding patients when treatment centres in Canada will not have the capacity to treat all eligible patients.
Input from the clinician groups and drug programs highlighted how all CAR T-cell therapies, including axi-cel, are resource intensive to deliver and can only be administered in specialized, accredited treatment centres with certified laboratories with access to highly trained personnel. This resource-intensive nature of CAR T-cell delivery and support is also widely discussed in the literature.3,4 The complexities, and related expenses, of establishing CAR T-cell treatment centres have limited their availability to certain geographic locations. Geographic access challenges thus exist for patients who live far from treatment sites and who face costs related to travel, lodging, and absence from work as well as the psychological burdens of having to be away from families and communities.3,4,10,11
In the delivery of axi-cel, it is expected that patients must remain within proximity of a specialized treatment centre for at least 4 weeks following their infusion. The clinical experts described how before CAR T-cell infusion, patients must undergo leukapheresis, bridging therapy, and conditioning chemotherapy, so time that must be spent in proximity to a treatment centre may be significantly longer than this. This time required to be in close proximity to treatment centres places similar geographic and support barriers and limitations on patients who must travel far from home to access CAR T-cell therapies, as well as all patients and their caregivers who must disrupt their lives and livelihoods to receive therapy.
However, not all patients can access CAR T-cell therapy at distant treatment centres equally. It has been demonstrated that patients with diffuse LBCL are more likely to choose treatments offered in distant cancer centres if follow-up care can be provided locally.12 For Black patients with DLBCL in the US, travel to treatment sites was a significant negative factor in choosing treatment at a distant cancer centre.12 Although all patients face challenges with continuity of care when accessing therapies far from home,3 histories and experiences of structural racism in the health care system have made seeking care far from home or from familiar care providers a barrier for some Black patients.12 Similar considerations may be relevant for Indigenous people and other underserved populations in Canada who face barriers to care far from home as well as histories and experiences of racism and discrimination in the health care system. Collaborative care between specialized centres and local oncologists and reductions in travel burden are important considerations for all patients, but additional barriers to travel and transfers in care for structurally marginalized patients are important contextual considerations around access to CAR T-cell therapies.
Much like disparities in access to clinical trials or LBCL therapies, racial disparities have been reported in access to CAR T-cell therapies in the US, especially for Black and Hispanic populations, with Black patients less likely to receive CAR T-cell therapies.13,14 Neighbourhood disparities in the US have also been described; most CAR T-cell therapy recipients have been residents of metropolitan areas with access to teaching hospitals14 and few CAR T-cell recipients are from lower socioeconomic neighbourhoods13 or from rural areas.15 In the Canadian context, similar race-based disparities in access should be considered because they could impact Indigenous people or other underserved people or groups.
The manufacturer of axi-cel has proposed a travel and lodging support program for patients who meet certain criteria for financial support, including being a Canadian resident, having consented to receive treatment, being enrolled in their product management software, having a primary residence within 2 hours or 200 km from the treatment centre, and not having sufficient coverage for travel and lodging. However, it has been suggested that similar assistance programs provided by manufacturers may not sufficiently cover all costs borne by patients in travelling to treatment centres,15 and the role of manufacturers as “gatekeepers” to travel assistance and compassionate access programs has been questioned.8
To support access to and receipt of CAR T-cell therapy, caregiver support is often needed,4,16 but not all patients have access to a caregiver. Clinical experts noted the challenges for patients who lack caregiver support and the significant financial burdens of accessing nonfamilial, formal caregivers. Caregivers must also be trained to identify signs and symptoms of severe adverse events that might arise from a patient’s CAR T-cell therapy, posing additional burdens and challenges for these caregivers.
Clinical Judgment and Equity in Referral
Another access barrier has been noted at the point of patient referral to CAR T-cell therapy.3,4 There are finite time points when patients can benefit most from the administration of CAR T-cell therapies, and the value of these therapies can be lost with treatment delays.4 Authors have called for the education of clinicians about CAR T-cell therapies so that timely referral can occur,16 including calls for improved communication between referring oncologists and treatment centres, for training of referring physicians in working through the logistics of CAR T-cell referral,11 and for clear criteria for prioritizing patients and transparency in this process.3
As noted previously, the clinical experts consulted by CADTH discussed the difficulties in defining eligibility criteria for ASCT and they noted differences across jurisdictions as well as clinician subjectivity in making eligibility distinctions and decisions. The experts questioned why the reimbursement criteria requested by the sponsor of this submission was restricted to ASCT eligibility given these challenges in defining this population. They noted that although this restriction in reimbursement criteria may align with the data of the ZUMA-7 trial, it may exclude some patients who may benefit from access to axi-cel. They indicated that this restriction would raise questions about biases in clinical assessments of patient suitability, and considerations around fairness and transparency in clinical decision-making. The clinical experts called for broader access to axi-cel treatment, noting that there would be patients who would be considered eligible for CAR T-cell therapy but not ASCT in clinical practice, including older patients and those with primary refractory disease or early relapse. Clinician group input indicated that fitness for CAR T-cell therapy should be assessed on an individual basis and that fitness for ASCT should not necessarily be used to define fitness for CAR T-cell therapy in clinical practice.
Differences in access to CAR T-cell therapies across jurisdictions may also pose challenges for patients who feel that they are “missing out”3 and for those with unequal access across regions.17 The clinical experts discussed the need to develop equitable, transparent, and standardized criteria for CAR T-cell therapy eligibility across the country, and indicated that patients and citizens should be involved in developing and setting these criteria. The clinical experts also indicated that differences in access to CAR T-cell therapies across jurisdictions in Canada should be diminished, and that clinicians and patients should have access to all approved CAR T-cell products so they can make decisions about them based on clinical presentations and the available evidence.
Potential Harms in the Use and Delivery of CAR T-Cell Therapies
CAR T-cell therapies have unique and frequent toxicities to patients, notably cytokine release syndrome. Adverse events related to axi-cel are detailed in the Clinical Review. The potential for these toxicities can be barriers for some individuals or families seeking CAR T-cell treatment.11 Adverse events related to CAR T-cell therapies are becoming better understood and managed, yet the overall unknown safety risks of CAR T-cell therapies have been positioned as an ethical issue.4 Clinician group inputs point to the unique toxicities associated with CAR T-cell therapies and the importance of administering these by well-trained medical teams at well-resourced centres.
The potential for harm to patients may also lie in delayed access to CAR T-cell therapies. Clinician group input suggested that patients with relapsed or refractory LBCL who may not benefit from current second-line therapies and their associated toxicities would potentially benefit from earlier access to CAR T-cell therapy. The clinical experts engaged by CADTH discussed the potential benefits of moving access to a CAR T-cell therapy to earlier in the course of treatment, to be considered as soon as patients are relapsed or refractory, such as in the case of axi-cel. They spoke about the risks and harms of patients going through additional chemotherapy regimens to meet eligibility criteria when they could potentially benefit from CAR T-cell therapy, and the importance of second-line access to CAR T-cell therapy. The potential harms in delayed access to CAR T cells were characterized as direct harms to patients because of exposure to the toxicities of potentially ineffective therapies, possibly causing them to be less fit and more frail at the point of access to CAR T-cell therapy. Clinical experts also spoke about financial harms and costs to the health care system if patients are using potentially ineffective therapies to reach the eligibility criteria for CAR T-cell therapy access.
Cell and Tissue Ownership
The collection and storage of patients’ cells in the course of CAR T-cell manufacture may raise patient privacy and cell ownership Drug plan input questioned how these issues would be managed, given that CAR T cells are currently manufactured by a US-based company and, therefore, outside of Canadian jurisdiction. The clinical experts engaged by CADTH felt that many of the issues related to patient data and privacy were adequately covered in patient consent forms. However, although informed consent processes may address some of these considerations, it has been noted that tissue and genetic materials are valued differently by different cultural groups, and consent processes need to have clear details about cell processing and ownership, as well as how remaining cells that are produced but not infused will be handled or disposed of.18 To this end, consultation with diverse groups has been suggested as essential to CAR T-cell research and delivery to ensure that cell handling and disposal practices are sensitive to the needs and values of patients and communities so the educational and consent materials will resonate with them.3,18 These considerations of tissue ownership and tissue retention and disposal need to be considered in the context of how the benefits of cell ownership accrue to different actors (e.g., patients, the public, and manufacturers) and the challenges that may arise regarding public trust and the legitimacy of these initiatives.3
Considerations for Informed Consent
In addition to the unique potential for toxicities and uncertainties related to cell ownership, considerations about informed consent to CAR T-cell therapies may also arise. It has been argued that attention should also be paid to the “hype” surrounding CAR T-cell therapies.3 Many patients position CAR T cells as a “cure” despite treatment and access uncertainties.3,19 This “hype” may come from physicians, industry, or media releases.8,19 To address this, the risks and benefits of CAR T-cell therapies to patients as well as the public should be described and communicated in a robust, iterative, and balanced way, taking into account the unique vulnerability of patients with cancer who face few therapeutic options.3,4
Health System and Funding Considerations
The sponsor’s implementation plan indicates that there are currently 6 locations in Canada that will be able to supply axi-cel, with an additional 6 locations proposed. However, there are no current plans for treatment centres in New Brunswick, Newfoundland and Labrador, Prince Edward Island, the Northwest Territories, Nunavut, or Yukon, leaving substantial gaps in treatment for populations in these provinces and territories as well as for people living far from treatment sites. The clinical experts were hopeful that geographic access considerations would diminish as more centres become available, although they cautioned that growth in the system would be necessary as eligibility criteria evolve for CAR T-cell because our current health system may be ill-equipped to meet future needs. It has also been noted that rapid scaling of CAR T-cell therapies availability can be challenging because CAR T-cell therapies are produced through complex and individualized processes,20 increased access through more treatment sites needs to be balanced with safety and quality of treatment centres,4,16 and geographic allocation of CAR T-cell therapy centres should explicitly consider regional, rural-urban, and sociodemographic equity.10
Concerns have been raised about the Canadian health care system not being able to meet the costs of additional CAR T-cell therapies being approved.9 Funding reforms for high-cost therapies have been suggested in the Canadian context, such as the use of performance-based or outcome-based agreements to share the risk associated with funding high-cost therapies, such as CAR T-cell therapies.3,17 These funding reforms might be accompanied by postmarket surveillance mechanisms to inform regulatory and economic reassessments, which would require collaboration across provinces in developing and maintaining these registries.17 In the context of CAR T-cell therapies, it has been noted, however, that price negotiations can only reduce costs so much, and the total costs of these therapies will remain high because the price of the therapy is only 1 component of its high cost.3 Further, it has been argued that the full costs of CAR T-cell therapies may be unknown to payers and there have been calls for full transparency around total costs of CAR T-cell therapy, including those associated with preinfusions and postinfusions, treatment of adverse events, and other clinical costs.4 Clinical experts noted that therapies to manage adverse events related to CAR T-cell therapies (such as tocilizumab in the treatment of cytokine release syndrome) should also be available across provinces, as a matter of equity and fairness, and the high costs of managing toxicities associated with CAR T-cell therapies should be understood in the context of these therapies.8
As is relevant with many high-cost therapies, clinical experts discussed the opportunity costs of CAR T-cell therapies and the challenges of funding these therapies in the context of fixed budgets. A prior review of ethical considerations in CAR T-cell therapy conducted by CADTH discussed how funding of CAR T-cell therapies can impact the sustainability of health systems, and fair and just funding allocation with fair distribution of risks and benefits of innovations should be considered.4 These considerations become amplified as CAR T-cell therapies apply to more cancer subtypes, and the need to manage health care system costs and improve access persists.4 These considerations call for clarity and transparency in justifications for policy decisions about expanding access to CAR T-cell therapies in the context of considering the long-term sustainability of the Canadian health care system.3
Limitations
This review is limited by the paucity of published literature examining ethical considerations directly relevant to the use of axi-cel for the treatment of adult patients with LBCL in Canada. Although there are no directly applied ethical analyses published, that does not mean that there are no ethical considerations present. Many ethical considerations in the context of LBCL more broadly, or those related to CAR T-cell therapies in general, are also relevant in this context. Augmenting the somewhat limited literature available with inputs from patient and clinician groups, drug programs, and clinical experts collected during the course of this Reimbursement Review has provided a more comprehensive overview of the ethical considerations in the context of axi-cel as treatment of relapsed or refractory LBCL.
Although this ethics report drew on and extracted from patient group, clinician group, clinical expert, and drug program inputs, it is possible that more directed engagement (such as through direct interviews with patients, caregivers, family members, or health system payers) would have yielded more relevant domains of analysis. Given the proposed high cost of this therapy in the context of the limited resources of the Canadian health care system, it is also possible that inputs from community members may have identified additional relevant considerations or trade-offs relevant to the funding and implementation of axi-cel.
Conclusion
Inputs from patient and clinician groups, clinical experts, drug programs, and relevant published literature were reviewed for ethical considerations relevant to the use of axi-cel for the treatment of relapsed or refractory LBCL in adult patients. Ethical considerations in the context of LBCL highlighted the impacts on patients and disparities in the incidence, treatment, and outcomes for racialized or other underserved populations. Regarding the evidence used to evaluate axi-cel, evidence from clinical trials may not have included all relevant populations, and economic models may have underrepresented the budget impact of implementation. The implementation of axi-cel involves several access considerations given the limited and resource-intensive nature of delivery, and these access considerations may disproportionately affect underserved persons or groups or be challenged by differential applications of eligibility criteria for treatment. There are unique toxicities involved in the delivery of CAR T-cell therapies, and there is the potential for harm in delayed access to therapy. In addition, the manufacture and storage of cellular tissues used in the course of therapy raises ethical considerations, as do issues related to informed consent and balanced communication about the risks and benefits of CAR T-cell therapies. Finally, the implementation of axi-cel raises ethical considerations for health systems related to the challenges and high costs of scaling CAR T-cell therapy infrastructure and trade-offs in the payment of high-cost therapies.
Overall, axi-cel for treatment of LBCL poses ethical considerations related to access challenges and the disproportionate effects on certain persons or groups. Considerations about the implementation and use of axi-cel should thus take into account the context of these challenges of differential and delayed access to CAR T-cell therapy; limited data for effectiveness in certain populations, including long-term safety and efficacy data; and high costs for the implementation of this therapy.
References
1.EUnetHTA Joint Action 2 Work Package 8. HTA Core Model ® version 3.0. 2016; https://www.htacoremodel.info/BrowseModel.aspx. Accessed 2021 Dec 17.
2.Benkhalti M, Espinoza M, Cookson R, Welch V, Tugwell P, Dagenais P. Development of a checklist to guide equity considerations in health technology assessment. Int J Technol Assess Health Care. 2021;37(1):e17. PubMed
3.Axicabtagene ciloleucel for large B-cell lymphoma: ethics and implementation report. (CADTH optimal use report vol. 9, no. 1e). Ottawa (ON): CADTH; 2019: https://www.cadth.ca/sites/default/files/pdf/car-t/ct0002-ipe-report.pdf. Accessed 2022 Jul 7.
4.CADTH Reimbursement Review: lisocabtagene maraleucel (Breyanzi) for relapsed or refractory large B-cell lymphoma. Can J Health Technol. 2022;2(10). https://www.cadth.ca/sites/default/files/DRR/2022/PG0258-Breyanzi.pdf. Accessed 2022 Oct 3.
5.Grey matters: a practical tool for searching health-related grey literature. Ottawa (ON): CADTH; 2019: https://www.cadth.ca/grey-matters. Accessed 2022 Jun 28.
6.Sandelowski M. Whatever happened to qualitative description? Res Nurs Health. 2000;23(4):334-340. PubMed
7.Al Hadidi S, Schinke C, Thanendrarajan S, Zangari M, van Rhee F. Enrollment of Black Participants in Pivotal Clinical Trials Supporting US Food and Drug Administration Approval of Chimeric Antigen Receptor-T Cell Therapy for Hematological Malignant Neoplasms. JAMA Netw Open. 2022;5(4):e228161. PubMed
8.Imbach KJ, Patel A, Levine AD. Ethical considerations in the translation of CAR-T cell therapies. Cell Gene Ther Insights. 2018;4(4):295-307.
9.Wilson M, Thavorn K, Hawrysh T, et al. Stakeholder engagement in economic evaluation: Protocol for using the nominal group technique to elicit patient, healthcare provider, and health system stakeholder input in the development of an early economic evaluation model of chimeric antigen receptor T-cell therapy. BMJ Open. 2021;11(8):e046707. PubMed
10.Snyder S, Chung KC, Jun MP, Gitlin M. Access to Chimeric Antigen Receptor T Cell Therapy for Diffuse Large B Cell Lymphoma. Adv Ther. 2021;38(9):4659-4674. PubMed
11.Schneider ME. CAR T-Cell Therapy Emerges as Option in Follicular Lymphoma. Oncology Times. 2021;43(18):1,8-8.
12.Frosch ZAK, Namoglu EC, Mitra N, et al. Willingness to Travel for Cellular Therapy: The Influence of Follow-Up Care Location, Oncologist Continuity, and Race. JCO Oncol Pract. 2022;18(1):e193-e203. PubMed
13.Ahmed N, Shahzad M, Shippey E, et al. Socioeconomic and Racial Disparity in Chimeric Antigen Receptor T Cell Therapy Access. Transplant Cell Ther. 2022;28(7):358-364. PubMed
14.Emole J, Lawal O, Lupak O, Dias A, Shune L, Yusuf K. Demographic differences among patients treated with chimeric antigen receptor T-cell therapy in the United States. Cancer Med. 2022;08:08.
15.Snyder S, Albertson T, Garcia J, Gitlin M, Jun MP. Travel-Related Economic Burden of Chimeric Antigen Receptor T Cell Therapy Administration by Site of Care. Adv Ther. 2021;38(8):4541-4555. PubMed
16.Kansagra A, Farnia S, Majhail N. Expanding Access to Chimeric Antigen Receptor T-Cell Therapies: Challenges and Opportunities. Am Soc Clin Oncol Educ Book. 2020;40:1-8. PubMed
17.Council of Canadian Academies. From Research to Reality. Ottawa (ON): The Expert Panel on the Approval and Use of Somatic Gene Therapies in Canada, Council of Canadian Academies; 2020: https://cca-reports.ca/reports/somatic-gene-and-engineered-cell-therapies/. Accessed 2022 Jul 18.
18.Weinkove R, George P, Ruka M, Haira TH, Giunti G. Chimeric antigen receptor T-cells in New Zealand: challenges and opportunities. N Z Med J. 2021;134(1542):96-108. PubMed
19.Jenei K, Burgess M, Peacock S, Raymakers AJN. Experiences and perspectives of individuals accessing CAR-T cell therapy: A qualitative analysis of online Reddit discussions. J Cancer Policy. 2021;30:100303. PubMed
20.Thornton Snider J, Brauer M, Kee R, et al. The potential impact of CAR T-cell treatment delays on society. Am J Manag Care. 2019;25(8):379-386. PubMed
Appendix 1: Details of Included Publications
Table 1: Details of Included Publications
First author, year | Publication type | Objective | Key ethical considerations | Funding source |
|---|---|---|---|---|
Ahmed (2022)13 | Database analysis | To determine patterns of racial and ethnic distribution, socioeconomic status, insurance coverage, and travel time of CAR T-cell recipients |
| None declared |
Al Hadidi (2022)7 | Cross-sectional study | To examine the enrolment of Black participants in clinical trials that resulted in a subsequent FDA approval of CAR T-cell products in hematologic malignant neoplasms |
| None declared |
CADTH (2019)3 | Ethics review report | To discuss the major ethical issues raised by the implementation of axicabtagene ciloleucel for adults with relapsed or refractory non-Hodgkin lymphoma |
| Canada’s federal, provincial, and territorial governments, with the exception of Quebec |
CADTH (2022)4 | Ethics review report | To identify and describe ethical considerations raised in the literature associated with the use of lisocabtagene maraleucel for treatment of relapsed or refractory LBCL |
| Canada’s federal, provincial, and territorial governments, with the exception of Quebec |
Council of Canadian Academies (2020)17 | Report and recommendations | To report on the findings of the Expert Panel on the Approval and Use of Somatic Gene Therapies in Canada |
| National Research Council Canada |
Emole (2022)14 | Database analysis | To evaluate if demographic differences existed among adult patients who received CAR T-cell therapy and to assess predictors of CAR T-cell treatment outcomes |
| None declared |
Frosch (2022)12 | Survey study | To determine the relative value patients place on clinical factors, oncologist continuity, and travel time under different posttreatment follow-up arrangements |
| National Institutes of Health; University of Pennsylvania |
Imbach (2018)8 | Commentary | To articulate key ethical challenges for the field of CAR T cells and suggest some strategies to help navigate these challenges |
| None declared |
Jenei (2021)19 | Reddit analysis | To explore how patients and their families understand novel treatments such as CAR T-cell therapies and their associated uncertainties |
| Michael Smith Foundation for Health Research |
Kansagra (2020)16 | Review | To review opportunities for centres, manufacturers, payers, and policy-makers to address barriers to care in CAR T-cell therapies |
| None declared |
Schneider (2021)11 | Review | To discuss opportunities and barriers to the use of CAR T-cell therapies |
| None declared |
Snyder (2021a)15 | Original research (GIS mapping) | To estimate the travel-related economic burden associated with site-of-care options among patients with relapsed or refractory DLBCL |
| Bristol Myers Squibb |
Snyder (2021b)10 | Original research (GIS mapping) | To examine how expanding access to CAR T-cell therapy administration sites impacts patient travel distances and times |
| Bristol Myers Squibb |
Thornton Snider (2019)20 | Economic evaluation | To measure the social value of CAR T-cell therapy for relapsed or refractory DLBCL in the US and quantify social value lost due to treatment delays |
| None declared |
Weinkove (2021)18 | Review | To outline CAR T-cell manufacturing and logistic considerations, with a focus on New Zealand’s environment for personalized cell and gene therapy |
| Health Research Council of New Zealand; Ministry of Business Innovations and Employment; LifeBlood Trust; Freemasons New Zealand |
Wilson (2021)9 | Protocol | To outline a specific methodology for stakeholder engagement in economic evaluations to inform decision-making for resource allocation for CAR T-cell therapies |
| Joint OICR-BioCanRx Health Services Competition |
CAR = chimeric antigen receptor; DLBCL = diffuse large B-cell lymphoma; GIS = geographic information system; LBCL = large B-cell lymphoma.
Note that this appendix has not been copy-edited.
Stakeholder Input
Patient Input
Lymphoma Canada
About Lymphoma Canada
Lymphoma Canada is a national Canadian registered charity that empowers the lymphoma community through education, support, advocacy, and research. Based out of Mississauga (ON), we collaborate with patients, caregivers, healthcare professionals, and other organizations and stakeholders, to promote early detection, find new and better treatments for lymphoma patients, help patients access those treatments, learn about the causes of lymphoma, and work together to find a cure. Resources are provided in both English and French. www.lymphoma.ca
Information Gathering
Lymphoma Canada (LC) collected the data for this submission from two online patient surveys. Links to these surveys were sent via e-mail to people registered through the LC database. The survey was also made available via social media outlets, including Twitter, Instagram and Facebook accounts, and was sent to healthcare professionals across Canada to share with their patients. The survey had a combination of multiple choice, rating and open‐ended questions. Open-ended responses to surveys that reflected the sentiment of a majority are included to provide a deeper understanding of patient perspectives.
The first survey of diffuse large B-cell lymphoma (DLBCL) patients was conducted in 2018 to support a previous CADTH submission. A total of 97 DLBCL patients completed the survey. These results are used in the next three sections.
A second survey of DLBCL patients with and without Yescarta experience was conducted between April 26 and June 20, 2022. A total of 23 patients completed this survey. Of these respondents, 3 had treatment experience with Yescarta. The former patients will be included in the results for the next three sections; the latter patients will be profiled in the Experience With Drug Under Review section.
Disease Experience
Respondents to the most recent survey were asked which symptoms of DLBCL were affecting their current quality of life on a scale from 1 (no impact) to 5 (significantly negative impact). Fatigue was the most commonly cited symptom (83%, n=12), followed by body aches and pains (67%), thrombocytopenia (67%), enlarged lymph nodes (58%), anemia (58%), neutropenia (58%) and night sweats (58%). However, symptoms were not described as having a significantly negative impact– frequent infections was the only symptom for which a majority of respondents (60%, n=5) gave a score of 4 or 5.
Apart from their physical symptoms, there are a number of psychosocial impacts that can affects the quality of life for DLBCL patients. Fear of progression/relapse was by far the most commonly cited impact by respondents to both surveys (67%, n=107). Anxiety (37%), memory loss (37%), and concentration problems (36%) were also cited by at least one third of respondents.
Respondents made numerous comments about the impact of DLBCL on their day-to-day lives:
“It changed my life and ability to enjoy activities of daily living because I now have to depend on others.”
“Stress/Anxiety/Fear of relapse, recurrence or new cancer a daily concern. Memory/Focus has been affected to large degree post treatment, which affects my daily functions at work/home. However, I've been able to manage my obligations overall.”
“When I had lymphoma, I was unable to work, I had incredible fatigue, difficulty with cognition and a hard time doing very much on a day-to-day basis. My fatigue lasted for almost two years following treatment and was a very difficult side effect to manage.”
“My immunocompromised situation restricts my ability to be in social settings, particularly with Covid-19 still being a major concern.”
“I am lucky in the sense that I can almost do the same as before, but at times I lack energy or I should say that I crash for a couple of days, I then need time to rest, I sleep a lot and am back in shape in less than a week. I always trained hard, now I cannot do as much and need to adjust my mind to this new reality.”
“I manage the day-to-day fine. It’s the constant fear of recurrence that haunts me the most.”
Experiences With Currently Available Treatments
107 respondents from both surveys provided information about the treatments that they have received since their diagnosis.
Table 1: Treatments Respondents Received Since Diagnosis
Treatments Received | n | Treatments Received | n |
|---|---|---|---|
CHOP +/- R | 94 | Mini-CHOP | 2 |
Radiation therapy | 22 | ICE +/- R | 2 |
Stem cell transplant | 15 | Ibrutinib | 2 |
Rituximab | 9 | R-CVP | 2 |
Methotrexate | 4 | DCA | 1 |
Unspecified CAR-T therapy | 4 | CUPR | 1 |
COP-R | 3 | MXT | 1 |
GDP +/- R | 3 | BEAM | 1 |
Bendamustine | 3 | Mini-Beam | 1 |
DHAP +/- R | 3 | Obinutuzumab | 1 |
EZH2 HST Inhibitor | 2 | Leucovorin | 1 |
Polatuzumab | 2 | ESHAP | 1 |
CEPD +/- R | 2 | Panobinostat | 1 |
EPOCH +/- R | 2 | CMC-544 | 1 |
A large majority of respondents (83%, n=103) to both surveys received CHOP +\- R (cyclophosphamide, doxorubicin, vincristine, prednisone, with or without rituximab) as their first line of treatment; eight more respondents received it as a subsequent line of treatment. There was no consistent pattern for later lines of treatment and only three forms of treatment (radiation therapy, stem cell transplant and rituximab) were reported by more than 4 people.
Respondents to the most recent survey generally felt that their therapy was able to manage their DLBCL symptoms. When asked to rate their agreement with the statement – “My current therapy (or most recent therapy) was able to manage my Diffuse Large B-Cell Lymphoma symptoms.” – on a scale of 1 (strongly disagree) to 5 (strongly agree), the average score was 3.8 (n=11) and six of the eleven respondents gave a rating of 5.
Patients from both surveys were asked to identify the side effects that they had experienced as a result of lymphoma treatment. Hair loss was the most commonly cited side effect (93%, n=103), followed by fatigue (85%), neutropenia (70%), chemo-brain - memory problems and confusion (67%) and nausea (60%). Fatigue was most commonly cited as the most difficult-to-tolerate side effect (40%, n=87). Over 40% of respondents to the 2018 survey reported that they had experienced fatigue (52%, n=91) and chemo brain (42%) as a long-term treatment side effect, meaning side effects that lasted longer than 2 years or appeared two years or later after the end of treatment.
When asked if their lymphoma treatment had a negative impact on different aspects of their quality of life on a scale of 1 (no impact) to 5 (significant negative impact), respondents to the 2018 survey indicated that the greatest negative impact was to their work, travel and other activities.
Table 2: Impact of Treatment on Quality of Life
Impact on Quality of Life | n | Average |
|---|---|---|
Work | 73 | 4.04 |
Travel | 82 | 3.87 |
Activities | 87 | 3.85 |
Intimate relations | 76 | 3.25 |
Family | 84 | 2.90 |
Friendships | 85 | 2.42 |
School | 22 | 2.00 |
Improved Outcomes
Lymphoma Canada asked respondents to the 2018 survey to evaluate the importance of different aspects of new treatments on a scale of 1 (not important) to 5 (very important).
Table 3: Importance of Different Aspects of New Treatments
Importance of Outcome | Average (n = 89) |
|---|---|
Longer survival than current therapies | 4.88 |
Longer remission than current therapies | 4.84 |
Better quality of life than current therapies | 4.64 |
Fewer side effects than current therapies | 4.12 |
All of these outcomes were rated as important. However, longer survival and longer remission received higher scores than better quality of life and fewer side effects. This suggests that patient values prioritize long-term health outcomes over short-term improvements to quality of life. This conclusion is reinforced by other results.
When patients from both surveys were asked if they would choose a treatment with potentially serious side effects if their doctor recommended that it was the best and most effective option for DLBCL, 51% (n=100) said that they would and 46% said that they were unsure. Only 3% said that they would not.
Comments included:
“Overall, long term health effects and potential future risks is primary deciding factor.”
“It would have to be a drug that would give me very good chances of surviving.”
“Yes, if there are potential long-term benefits.”
“I would be in favour of any new therapy that would increase the chances of DLBCL being cured.”
Treatment choice was also identified as an important value for DLBCL patients. When respondents to the 2018 survey were asked about the importance of choice in deciding on treatment on a scale from 1 (not important) to 5 (very important), the average score was 4.54 (n=89) and 68 respondents gave a score of 5. As one person commented:
“I would like for patients to have more treatment options where they can honestly weigh the risks of side effects with the benefits of disease management.”
In the most recent survey, 91% of respondents (n=11) answered Yes to the question - “Do you feel there is currently a need for more therapy options for patients with DLBCL?”
Experience With Drug Under Review
Three DLBCL patients with Yescarta treatment experience completed the most recent survey. All three respondents are Canadian and all three received Yescarta as third-line therapy:
Patient A is a woman, aged 45-54. She was diagnosed 3-5 years ago. She received Yescarta following CHOP- R and an unspecified second-line chemotherapy. She accessed Yescarta via a clinical trial in the United States because it was unavailable in her province. She relapsed following treatment.
Patient B is a woman, aged 45-54. She was diagnosed 3-5 years ago. She received Yescarta following CHOP- R and GDBP. She also received polituzamub following Yescarta. She accessed Yescarta via a clinical trial because CAR-T therapy was not available at her local cancer centre and was not covered by insurance. She has been in remission for 1-2 years.
Patient C is a man, aged 55-64. He was diagnosed less than 1 year ago. He received Yescarta following CHOP-R and a stem cell transplant that had to be aborted due to high tumour load. He accessed Yescarta through the public system. He has been in remission for less than six months.
All three respondents were away from home for more than three months for treatment with Yescarta. Access to treatment was described as a significant challenge to these patients as the therapy was not available in their community and most had to travel to receive it.
All three respondents reported thrombocytopenia as a side effect of Yescarta. Two respondents reported fever, neutropenia, anemia, nausea/vomiting, diarrhea, joint or muscle pain and fatigue.
All three respondents reported fear of progression/relapse and difficulty sleeping as psychosocial impacts related to their CAR-T therapy. Two respondents reported anxiety/worry, loss of sexual desire, isolation and challenges with support (family/friends).
When asked about the effect of different aspects of CAR-T therapy on their life on a scale of 1 (no negative impact on my life) to 5 (significant negative impact on my life), respondents gave the following scores:
Table 4: Effect of Different Aspects of CAR-T Therapy on Life
Importance of Outcome | Average (n=3) |
|---|---|
Number of clinic visits | 4.67 |
Monitoring for side-effects post-infusion (hospitalization) | 4.67 |
Long-term side effects | 4.67 |
Travel to treatment centre | 4.67 |
Away from family/friends | 4.67 |
Inability to attend work/school/volunteer | 4.67 |
Inability to perform daily activities | 4.67 |
CAR-T cell re-infusion process | 4.33 |
Short-term side effects | 4.33 |
Financial cost of treatment | 3.00 |
Extraction of blood(leukapheresis) | 2.33 |
All three patients reported their overall experience with Yescarta therapy as good (on a scale of poor, satisfactory, good and very good).
When asked if they would recommend Yescarta to other patients with relapsed/refractory DLBCL, all three respondents with Yescarta said that they would.
Comments include:
“It saved my life.”
“Overall, the treatment helped my symptoms, but did not cure the disease.”
“I am now living a better life.”
Companion Diagnostic Test
Not applicable.
Anything Else?
These are the key points made by survey respondents:
Current DLBCL therapies cause significant side effects, including long-term side effects.
Patient values prioritize long-term health outcomes over short-term improvements to quality of life.
Treatment choice was also identified as an important patient value.
Patients are required to make significant logistical efforts to access Yescarta under the current treatment regime.
Every respondent that received Yescarta said that they would recommend it to other patients as treatment for relapsed/refractory DLBCL.
Conflict of Interest Declaration — Lymphoma Canada
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission? If yes, please detail the help and who provided it.
Adam Waiser, an independent consultant, completed this submission with assistance and oversight from Lymphoma Canada staff. The surveys were created and promoted by Lymphoma Canada staff.
Did you receive help from outside your patient group to collect or analyze data used in this submission? If yes, please detail the help and who provided it.
The data was collected by Lymphoma Canada. Adam Waiser, an independent consultant, analyzed the data for this submission with assistance and oversight from Lymphoma Canada staff.
List any companies or organizations that have provided your group with financial payment over the past 2 years AND who may have direct or indirect interest in the drug under review.
Table 5: Financial Disclosures for Lymphoma Canada
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Gilead | — | — | — | X |
Bristol Myers Squibb | — | — | — | X |
Novartis | — | — | X | — |
Clinician Input
Lymphoma Canada
About Lymphoma Canada
Lymphoma Canada is a national organization dedicated to research, education, and raising awareness to benefit patients with lymphoma across Canada. (Home - Lymphoma Canada)
Information Gathering
We conducted a literature search of PubMed for published clinical trials of axicabtagene ciloleucel and other chimeric antigen receptor (CAR) T cell products used for the treatment of large B-cell lymphoma (LBCL). In addition, we referred to pivotal clinical trials of second line treatment for LBCL including LY.12, a Canadian study of second line chemoimmunotherapy and autologous stem cell transplant (ASCT), as well as the Canadian Lymphoma Treatment Guidelines (DLBCL - Diffuse Large B Cell Lymphoma - Lymphoma Canada).
Current Treatments and Treatment Goals
Diffuse large B-cell lymphoma (DLBCL) is an aggressive lymphoid malignancy which represents approximately 30% of all non-Hodgkin lymphomas. First line treatment for fit patients is multiagent chemoimmunotherapy with R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone) which may cure up to 60-70% of patients (1). However, 30-40% of patients will relapse or be refractory to initial therapy and experience poor outcomes. The current standard of care for fit patients with relapsed/refractory LBCL is to administer intensive second line chemoimmunotherapy, with responding patients proceeding to high-dose chemotherapy and ASCT which resulted in long-term remissions in 46% of transplanted patients in the PARMA trial (2).
In Canada, the most common second line chemoimmunotherapy regimen is R-GDP (rituximab, gemcitabine, dexamethasone, cisplatin) based on the multicenter Canadian randomized trial LY.12 (3). In this trial, 2-3 cycles of R-GDP or R-DHAP (rituximab, dexamethasone, cytarabine, cisplatin) were administered with the aim of proceeding to ASCT if a response was achieved. However, a significant proportion of patients had insufficient response to second line chemoimmunotherapy, and only 52% of patients randomized to R-GDP and 49% to R-DHAP were able to undergo ASCT. In addition, relapse occurred frequently among patients after ASCT, and as a result, the 4-year event-free survival for all patients intended for ASCT in this trial was only 26%.
Although the goal of treatment in relapsed/refractory LBCL is to prolong life by curing the patient of their lymphoma, the majority of patients do not achieve durable remissions with the current standard of care second line chemoimmunotherapy and ASCT. Accordingly, the prognosis of patients with relapsed/refractory LBCL is dismal with overall survival rates of only 20-30% (4-6). However, the recent introduction of anti-CD19 CAR-T cell therapy as third line treatment for patients with relapsed/refractory LBCL has resulted in significant improvements in patient outcomes, with long-term remissions achieved in 30-40% of infused patients. In Canada, axicabtagene ciloleucel, tisagenlecleucel, and recently lisocabtagene maraleucel have been reviewed by CADTH with recommendation to support funding for this indication based on the ZUMA-1, JULIET, and TRANSCEND trials, respectively (7-9). As a result, CAR-T cell therapy has become the current standard of care for patients with relapsed/refractory LBCL after 2 lines of systemic therapy who are unable to proceed to ASCT due to chemorefractory disease.
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
Although second line chemoimmunotherapy and ASCT is effective in a subset of patients with relapsed/refractory LBCL, real-world evidence from Canada and other countries show that just 40-50% of patients are considered fit enough to receive intensive second line chemoimmunotherapy, only 20-25% undergo ASCT, and as few as 10% achieve durable remissions after ASCT (5, 6, 10). In addition, ASCT is an intensive therapy associated with significant toxicities, including febrile neutropenia, bacteremia and other infections, need for transfusion support, gastrointestinal toxicity, mucositis, and secondary malignancies. As a result, only younger, medically fit patients less than 65-70 years old are typically considered eligible for ASCT, and a significant proportion of these patients are unable to proceed to ASCT due to chemorefractory disease or inability to adequately mobilize and collect stem cells. This is especially true for high-risk patients who are refractory to or relapse within 12 months of diagnosis, with only 20% of these patients achieving durable remissions with intensive second line chemoimmunotherapy and ASCT in the CORAL trial (11). These findings show that a new treatment paradigm is greatly needed for the second line treatment of LBCL.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
CAR-T cell therapy has a novel targeted immune-based mechanism of action which enables it to effectively treat lymphoma, including in chemorefractory cases which do not respond to other conventional therapies. CAR-T cell therapy is currently only available in Canada for patients with relapsed/refractory LBCL after 2 lines of systemic therapy. However, the phase III randomized controlled trial ZUMA-7 was designed to determine if earlier use of axicabtagene ciloleucel in second line improves outcomes compared to the current standard of care of salvage chemoimmunotherapy +/- ASCT. The trial included 359 patients with LBCL refractory to or relapsing within 12 months of first line chemoimmunotherapy. Because this group of patients is at high risk of chemoresistance, a significantly higher proportion of patients were able to receive their intended definitive therapy in the CAR-T group compared to the ASCT group (94% versus 36%). In addition, axicabtagene ciloleucel achieved significant improvements in important clinical outcomes including complete response rates (65% versus 32%), 2-year event-free survival (41% versus 16%), and 2-year progression-free survival (46% versus 27%). The interim analysis of this trial also demonstrated a trend to superior 2-year overall survival with axicabtagene ciloleucel (61% versus 52%) which requires confirmation with further follow-up. It is noteworthy that >50% of patients in the standard of care arm ultimately went off trial to receive CAR-T cell therapy in third line, highlighting that the majority of high-risk patients with relapsed DLBCL ultimately require CAR-T cell therapy and do not benefit from intensive second line chemoimmunotherapy and its associated toxicities. The results of this trial demonstrate that second line CAR-T cell therapy is more effective than the current standard of care and will likely result in a paradigm shift, with axicabtagene ciloleucel replacing intensive chemoimmunotherapy and ASCT as the new preferred second line treatment for high-risk patients with LBCL refractory to or relapsing within 12 months of first line chemoimmunotherapy.
Which patients would be best suited for treatment with the drug under review? Which patients would be least suitable for treatment with the drug under review?
Patients with the greatest need for more effective second line therapies are those with primary refractory LBCL and those relapsing within 12 months of first line therapy, who have superior outcomes with second line CAR-T cell therapy compared to the current standard of care. Patients eligible for treatment with second line axicabtagene ciloleucel would be identified by the Hematologist/Oncologist according to the following criteria: relapsed/refractory LBCL within 12 months of completion of first line chemoimmunotherapy, adequate performance status and vital organ function (as defined per institutional guidelines), no prior CAR-T cell therapy, and sufficient clinical stability to be expected to tolerate the 3–4-week manufacturing period for CAR-T cell therapy. Patients not meeting these criteria would not be suitable for axicabtagene ciloleucel as they would be unlikely to successfully respond to or tolerate the potential toxicities of CAR-T cell therapy.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice? How often should treatment response be assessed?
The outcomes used in clinical practice and clinical trials to determine response to CAR-T cell therapy include overall response rate, complete response rate, progression-free survival, and overall survival. The time to response after axicabtagene ciloleucel infusion is usually rapid (median 1 month) although some patients may have delayed responses. Response assessment varies according to institutional guidelines and may include restaging CT or PET/CT scans at 1 month and 3 months after CAR-T cell infusion. The majority of patients with ongoing responses 6-12 months after CAR-T cell therapy will achieve long-lasting remissions.
What factors should be considered when deciding to discontinue treatment with the drug under review?
This question is not applicable to CAR-T cell therapy because it is administered as a one-time infusion of a cellular therapy product.
What settings are appropriate for treatment with axicabtagene ciloleucel? Is a specialist required to diagnose, treat, and monitor patients who might receive axicabtagene ciloleucel?
CAR-T cell therapy must be administered by a specialized, accredited center with a certified laboratory for handling cellular therapy products. In addition, Hematologists/Oncologists with expertise in cellular therapy are required to monitor and manage adverse events occurring after CAR-T cell infusion, including cytokine release syndrome, neurotoxicity, cytopenias, infections, and hypogammaglobulinemia. Intensive care unit support must be available for patients experiencing severe toxicities. Over the past several years, a growing number of treatment centers in Canada have developed expertise in the administration of CAR-T cell therapy including axicabtagene ciloleucel.
Additional Information
The proposed indication submitted to CADTH is "For the treatment of adult patients with relapsed or refractory large B-cell lymphoma (LBCL), who are candidates for autologous stem cell transplant (ASCT)". This differs from the indication adopted by the US FDA which was also the key enrolment criteria in the ZUMA-7 trial: "For the treatment of adult patients with large B-cell lymphoma that is refractory to first-line chemoimmunotherapy or that relapses within 12 months of first-line chemoimmunotherapy”. This is a subtle difference, but it is potentially important as the wording submitted to CADTH implies that patients must be eligible for ASCT to receive axicabtagene ciloleucel. Given the significant differences in the toxicity profile of CAR-T cell therapy versus ASCT, we feel it is important to note that a patient’s fitness and candidacy for CAR-T cell therapy should be assessed on an individual basis, and that fitness for ASCT is not necessarily used to define fitness for CAR-T cell therapy in clinical practice. We feel that revising the proposed CADTH indication to “adult patients with LBCL refractory to or relapsing within 12 months of first line chemoimmunotherapy” would better define the high-risk patient population who would benefit from second line axicabtagene ciloleucel in Canada and prevent the needless exclusion of patients who may be sufficiently fit for CAR-T cell therapy but not ASCT.
Conflict of Interest Declarations — Lymphoma Canada
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please refer to the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission?
No.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission?
No.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician who contributed to the input.
Declaration for Clinician 1
Name: Robert Puckrin
Position: Cellular Therapy Fellow, University of Calgary
Date: 15-6-2022
Table 6: COI Declaration for Lymphoma Canada — Clinician 1
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Beigene | X | — | — | — |
Declaration for Clinician 2
Name: Pamela Skrabek
Position: Associate Professor, Department of Internal Medicine, Max Rady College of Medicine, Winnipeg Manitoba
Date: 17-06-2022
Table 7: COI Declaration for Lymphoma Canada — Clinician 2
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Incyte Biosciences Canada | X | — | — | — |
Bristol-Myers Squibb | X | — | — | — |
Novartis | X | — | — | — |
Declaration for Clinician 3
Name: John Kuruvilla
Position: Hematologist, Princess Margaret Cancer Centre, Chair Lymphoma Canada Scientific Advisory Board
Date: 28-06-2022
Table 8: COI Declaration for Lymphoma Canada — Clinician 3
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Kite | — | — | X | — |
Novartis | — | — | X | — |
BMS | — | X | — | — |
Declaration for Clinician 4
Name: Carolyn Owen
Position: Assistant Professor, Division of Hematology & Hematological Malignancies, University of Calgary
Date: 21-06-2022
Table 9: COI Declaration for Lymphoma Canada — Clinician 4
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Janssen | X | — | — | — |
AbbVie | — | — | X | — |
Astrazeneca | — | — | X | — |
Beigene | — | — | X | — |
Novartis | X | — | — | — |
Incyte Biosciences Canada | X | — | — | — |
Declaration for Clinician 5
Name: Mahmoud Elsawy
Position: Assistant Professor/Hematologist QEII Halifax
Date: 27/06/2022
Table 10: COI Declaration for Lymphoma Canada — Clinician 5
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Kite/Gilead | X | — | — | — |
Novartis | X | — | — | — |
BMS | X | — | — | — |
Declaration for Clinician 6
Name: Mona Shafey
Position: Clinical Associate Professor, Depts of Medicine and Oncology, University of Calgary
Date: 20-Jun-2022
Table 11: COI Declaration for Lymphoma Canada — Clinician 6
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Kite/Gilead | X | — | — | — |
Novartis | X | — | — | — |
BMS | X | — | — | — |
Ontario Health Cancer Care Ontario Hematology Cancer Drug Advisory Committee
About the Ontario Health Cancer Care Ontario Hematology Cancer Drug Advisory Committee
OH-CCO’s Drug Advisory Committees provide timely evidence-based clinical and health system guidance on drug-related issues in support of CCO’s mandate, including the Provincial Drug Reimbursement Programs (PDRP) and the Systemic Treatment Program.
Information Gathering
The information was discussed jointly at a DAC meeting.
Current Treatments and Treatment Goals
Axicabtagene ciloleucel would be a potentially curative therapy for DLBCL.
Treatment goals for axicabtagene ciloleucel would be cure of relapse DLBCL.
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
Chemo-refractory DLBCL is difficult to salvage with chemotherapy prior to considering CAR-T as third line therapy (current indication and funding). There may be benefits to having a CAR-T option as second line.
There are patients who would be considered for CAR-T and not ASCT, specifically older patients, primary refractory patients and early relapse patients. These patients may not tolerate or respond well to further salvage chemotherapy so having a CAR-T option as second line would be preferred.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
Second line auto transplant is the current standard but depends on chemo sensitivity, which can be challenging for this patient population. For patients without autologous stem cell transplant (ie, chemo-refractory), this would be the second line option.
Which patients would be best suited for treatment with the drug under review? Which patients would be least suitable for treatment with the drug under review?
Patients best suited would align with the inclusion criteria and least suited would align with the exclusion criteria of the clinical trial (ie. Refractory patients or early chemo failure). Allowing for any second line patient is beyond what the trial demonstrated. Also, for patients relapsing late (ie, post-1 year) ASCT results are good.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice? How often should treatment response be assessed?
Standard lymphoma response measures including CT and PET CT.
What factors should be considered when deciding to discontinue treatment with the drug under review?
Challenges often rise during the time of T-Cell collection and processing if the underlying lymphoma is unstable and patients may not be fit enough to proceed with CAR-T. In such situations, the plan for CAR-T can be discontinued.
What settings are appropriate for treatment with [drug under review]? Is a specialist required to diagnose, treat, and monitor patients who might receive [drug under review]?
CAR-T cell therapy is only available in selected centers and is resource intensive.
Additional Information
The trial eligibility for CAR-T was more limited than this CADTH indication. There are DLBCL subgroups who may not do well even with a ASCT beyond 1 year of initial therapy (ie, double/triple hit lymphoma). These patients may be better suited for CAR-T even as second line therapy given the dismal results with ASCT. As per the clinical trial, CAR-T cell therapy in second line for trial eligible patients would be preferred. This would spare patients additional chemotherapy toxicity if CAR-T therapy was only available as third line therapy.
It is unlikely or unknown whether there could be therapeutic value in ASCT as third line therapy following CAR-T as second line therapy. Novel treatment options/clinical trials would be preferred in this patient population. Approximately 10% of patients in the study received ASCT.
Conflict of Interest Declarations — Ontario Health Cancer Care Ontario Hematology Cancer Drug Advisory Committee
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please refer to the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
Yes, OH-CCO provided secretariat functions to the DAC.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission?
No.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician who contributed to the input.
Declaration for Clinician 1
Name: Dr. Tom Kouroukis
Position: Ontario Health (CCO) Hematology Cancer Drug Advisory Committee Lead
Date: 09-06-2022
Table 12: COI Declaration for OH-CCO Hematology Cancer Drug Advisory Committee — Clinician 1
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 2
Name: Dr. Jordan Herst
Position: Ontario Health (CCO) Hematology Cancer Drug Advisory Committee Member
Date: 09-06-2022
Table 13: COI Declaration for OH-CCO Hematology Cancer Drug Advisory Committee — Clinician 2
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 3
Name: Dr. Anca Prica
Position: Ontario Health (CCO) Hematology Cancer Drug Advisory Committee Member
Date: 09-06-2022
Table 14: COI Declaration for OH-CCO Hematology Cancer Drug Advisory Committee — Clinician 3
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Cell Therapy Transplant Canada
About Cell Therapy Transplant Canada
Cell Therapy Transplant Canada (CTTC; www.cttcanada.org) is a member-led, national, multidisciplinary organization providing leadership and promoting excellence in patient care, research and education in the field of hematopoietic stem cell transplant and cell therapy.
We are the professional society representing the stem cell transplant community in Canada, including physician, nursing, laboratory, and allied health professionals, along with an active family and caregiver group.
Information Gathering
The proposed submission was discussed by two CTTC committees – our Board of Directors, and our standing committee of program directors, representing the cell therapy and stem cell transplant programs across Canada. These two committees were provided an opportunity to review this report and provide input.
Current Treatments and Treatment Goals
The current treatment for this population is salvage chemotherapy, with an intent to proceed to autologous stem cell transplant in those patients who are chemosensitive (achieve at least a partial response). Salvage chemotherapy is typically administered by the primary hematologist/oncologist, with the most common regimen used in Canada being a combination of rituximab, gemcitabine, cisplatin, and dexamethasone (R-GDP). Patients who respond are referred for autologous stem cell transplant.
Autologous stem cell transplant is an intensive procedure, performed at a limited number of centres in Canada. Hematopoietic stem cells are mobilized from the bone marrow into the peripheral blood using a combination of growth factor (G-CSF or grastofil) and chemotherapy, and then collected using peripheral blood apheresis, and cryopreserved. Several weeks later, patients are admitted to hospital, and given high dose chemotherapy, followed by re-infusion of thawed stem cells. A variety of chemotherapy regimens are used in Canada. The most commonly used regimens for patients with DLBCL are a combination of melphalan and etoposide (Mel/Etop) or a combination of bendamustine, cytarabine, melphalan, and etoposide (Benda-EAM). These medications are generally used off label, without specific Health Canada approval for this indication. The administration of high dose chemotherapy and subsequent recovery period is often completed as an inpatient, with the typical length of stay in hospital of around 3 weeks. The role for autologous stem cell transplant was established by the PARMA trial several decades ago (Phillip et al, NEJM, 1995), which showed the superiority of high dose chemotherapy/stem cell transplant compared to conventional chemotherapy alone.
Patients that do not respond to salvage chemotherapy may be given CAR-T therapy as third line therapy. Patients who do not receive CAR-T therapy as third line therapy may receive best supportive care, or additional salvage chemotherapy, but outcomes in the third line setting are poor.
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
Despite the known superiority of autologous transplant over conventional chemotherapy, several significant challenges remain.
Autologous transplant is an intensive procedure, associated with very high short-term morbidity, and multiple known late toxicities. While some centres in Canada do perform autologous transplants as outpatients, the majority of patients even at these centres will require hospitalization at some point due to toxicities such as severe mucositis or febrile neutropenia. In the majority of centres that perform autologous transplant as an inpatient, the typical stay in hospital is 3-4 weeks. There is significant short-term morbidity associated with autologous transplant, with patients frequently requiring parenteral analgesia and nutritional support. As mentioned, rates of febrile neutropenia are very high. The treatment related mortality associated with autologous transplant in this population is 3-5%, depending on the patient group being studied, typically due to infection, but rarely due to organ failure. Due to the exposure to high dose cytotoxic chemotherapy, the risk of a late second malignancy is approximately 10%.
There remains a significant risk of relapse following autologous transplant. While autologous transplant can cure approximately 50% of patients with relapsed/refractory DLBCL, disease relapse/progression after transplant remains the most common cause of treatment failure.
Thus, while autologous transplant is the best current curative option for patients with relapsed DLBCL, it is an incredibly toxic intervention, with very high failure rates due to relapse, and multiple known late toxicities.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
This treatment would replace salvage chemotherapy and autologous stem cell transplant as the preferred salvage therapy (second line therapy overall) for patients with relapsed or refractory DLBCL, and would be done with curative intent.
Based on the ZUMA-7 trial, a significantly higher portion of patients will be cured with axicabtagene ciloleucel therapy than with standard of care chemotherapy/transplant. In that sense, it will cause a shift in the current treatment paradigm.
In response to the template questions, it would not be appropriate to reserve this treatment for patients who are not tolerant to other treatments, as other treatments have been shown to be inferior. Similarly, it would not be appropriate to recommend that patients try other treatments before axicabtagene ciloleucel, as other treatments have been shown to be inferior. The results of ZUMA-7 have clearly outlined the role of axicabtagene ciloleucel and the ideal line of therapy in which it should be used.
Which patients would be best suited for treatment with the drug under review? Which patients would be least suitable for treatment with the drug under review?
The patients best suited for this this therapy are largely identified by the ZUMA-7 trial – namely, patients with large B-cell lymphoma that was refractory to or had relapsed no more than 12 months after first-line chemoimmunotherapy.
These patients are easily identified and are currently managed in a limited number of specialized centres, by experienced hematologists and oncologists. They are currently referred to stem cell transplant teams, which in most centres, are the teams that will be delivering Yescarta. Thus, no changes in the existing pathway for treatment will be required. There are no issues related to diagnosis, no companion diagnostic testing required, and no issues with misdiagnosis.
Experience to date suggests that axicabtagene ciloleucel is better tolerated than autologous stem cell transplant, and thus the potential exists to expand curative based salvage therapy to a broader group of patients. There does exist a small patient population (older age, comorbidities, ECOG 2) who might not be expected to tolerate autologous transplant but likely could tolerate axicabtagene ciloleucel. Given the known superiority of axicabtagene ciloleucel, these patients should be eligible for this important therapy.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice? How often should treatment response be assessed?
Outcomes used in clinical practice mirror those used in clinical trial – diagnostic imaging (CT scan, PET scan), in combination with clinical evaluation by hematologists/medical oncologists.
The ZUMA-7 trial demonstrated superior event free survival with axicabtagene ciloleucel compared to salvage chemotherapy/autologous stem cell transplant. Additional clinically meaningful endpoints known to be superior with axicabtagene ciloleucel compared to standard of care chemotherapy/transplant include higher overall response rates, and improved quality of life.
What factors should be considered when deciding to discontinue treatment with the drug under review?
Given that axicabtagene ciloleucel is a one-time therapy, there is no need to consider when to discontinue treatment.
What settings are appropriate for treatment with [drug under review]? Is a specialist required to diagnose, treat, and monitor patients who might receive [drug under review]?
This therapy should only be prescribed for this indication by specialists working in a clinic associated with a cell therapy program. In general, these are located in cancer centres associated with tertiary care hospitals in Canada. There are very unique toxicities associated with axicabtagene ciloleucel, and it is critical that these therapies are only administered by well-trained medical teams.
Cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS) are well known effects of CAR-T therapies such as axicabtagene ciloleucel. In the ZUMA-7 trial, these were seen at rates comparable to that in other studies (6% rate of grade 3 or higher CRS, 21% rate of grade 3 or higher neurotoxicity). These toxicities, while significant, are usually manageable if treated by well-trained and experienced teams.
At this point, only a limited number of centres in Canada are delivering CAR-T therapy. It is critical that additional centres offer this therapy, so that patients can be treated closer to home, without delays that might necessitate bridging therapy.
Additional Information
No additional information is pertinent.
Conflict of Interest Declarations — Cell Therapy Transplant Canada
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please refer to the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission?
No additional help was provided.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission?
Not applicable.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician who contributed to the input.
Declaration for Clinician 1
Name: Kristjan Paulson
Position: Past President, CTTC; Hematologist, CancerCare Manitoba; Assistant Professor, University of Manitoba
Date: 24-06-2022
Table 15: COI Declaration for Cell Therapy Transplant Canada — Clinician 1
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 2
Name: Mona Shafey
Position: Medical Director, Alberta Blood & Marrow Transplant Program
Date: 28-06-2022
Table 16: COI Declaration for Cell Therapy Transplant Canada — Clinician 2
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Kite/Gilead | X | — | — | — |
Novartis | X | — | — | — |
Bristol Myers Squibb | X | — | — | — |
Declaration for Clinician 3
Name: Nicole Prokopishyn
Position: Director at Large, Regulatory and Quality, CTTC; Cellular Therapy Lab Director, Alberta Precision Labs, Alberta
Date: 30-06-2022
Table 17: COI Declaration for Cell Therapy Transplant Canada — Clinician 3
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 4
Name: Kevin Song
Position: Interim Medical Director, Leukemia/BMT Program of BC; Director, Hematology Research Program - VGH
Date: 30-06-2022
Table 18: COI Declaration for Cell Therapy Transplant Canada — Clinician 4
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Gilead | X | — | — | — |
Kite – Gilead | X | — | — | — |
Declaration for Clinician 5
Name: Guy Cantin
Position: Hémato-oncologist et Medical Director, Transplantation de cellules souches hématopoïétiques, CHU de Québec, Université Laval.
Date: 30-06-2022
Table 19: COI Declaration for Cell Therapy Transplant Canada — Clinician 5
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 6
Name: Mohamed Elemary
Position: Secretary, CTTC, Hematologist, Saskatoon Cancer Center, Professor, University of Saskatchewan
Date: 04-07-2022
Table 20: COI Declaration for Cell Therapy Transplant Canada — Clinician 6
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Jazz Pharmaceuticals | X | — | — | — |
AbbVie pharmaceuticals | X | — | — | — |
Bristol Myers Squibb | X | — | — | — |
Paladin Labs Inc. | X | — | — | — |
AstraZeneca | X | — | — | — |
Pfizer | X | — | — | — |
Novartis | X | — | — | — |
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
Redactions: Confidential information in this document may be redacted at the request of the sponsor in accordance with the CADTH Drug Reimbursement Review Confidentiality Guidelines.
Stakeholder Input: The views expressed in each submission are those of the submitting organization or individual; not necessarily the views of CADTH or of other organizations. As such, they are independent of CADTH and do not necessarily represent or reflect the view of CADTH. No endorsement by CADTH is intended or should be inferred. By filing with CADTH, the submitting organization or individual agrees to the full disclosure of the information. CADTH does not edit the content of the submissions.
CADTH does use reasonable care to prevent disclosure of personal information in posted material; however, it is ultimately the submitter’s responsibility to ensure no identifying personal information or personal health information is included in the submission. The name of the submitting organization or individual and all conflict of interest information are included in the submission; however, the name of the author, including the name of an individual patient or caregiver submitting the patient input, are not posted.
Accessibility: CADTH is committed to treating people with disabilities in a way that respects their dignity and independence, supports them in accessing material in a timely manner, and provides a robust feedback process to support continuous improvement. All materials prepared by CADTH are available in an accessible format. Where materials provided to CADTH by a submitting organization or individual are not available in an accessible format, CADTH will provide a summary document upon request. More details on CADTH’s accessibility policies can be found here.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.