Drugs, Health Technologies, Health Systems
Reimbursement Review
Toripalimab (Loqtorzi)
Sponsor: Apotex
Therapeutic area: Nasopharyngeal carcinoma
Summary
What Is Nasopharyngeal Carcinoma?
Nasopharyngeal carcinoma (NPC) is a rare cancer of the uppermost part of the throat that tends to be detected at a more advanced stage (stage III or IV) and therefore can be difficult to treat.
The incidence of NPC is 0.5 to 1 per 100,000, although it is more common in certain populations, including Inuit and people of Asian descent.
What Are the Treatment Goals and Current Treatment Options for Recurrent or Metastatic NPC?
The main goals of treatment are to prolong survival and minimize progression while maintaining health-related quality of life and minimizing drug toxicity.
The patient and clinician groups and clinical experts agreed the most important outcomes are overall survival (OS), progression-free survival (PFS), objective response rate, and duration of response.
There are no drugs currently approved for use in the second line or beyond for recurrent or metastatic NPC. Patients often seek out clinical trials as their best option, and other options include traditional chemotherapy.
What Is Loqtorzi and Why Did Canada’s Drug Agency Conduct This Review?
Loqtorzi is a drug that is administered by IV infusion. At the time this review was conducted, Health Canada was reviewing Loqtorzi for use as a single agent for the treatment of adults with recurrent unresectable or metastatic NPC with disease progression while on treatment or after treatment with a platinum-containing chemotherapy.
Canada’s Drug Agency (CDA-AMC) reviewed Loqtorzi to inform a recommendation for the participating public drug programs on whether it should be reimbursed for the indication under review by Health Canada.
How Did CDA-AMC Evaluate Loqtorzi?
CDA-AMC reviewed the clinical evidence on the beneficial and harmful effects of, as well as the economic evidence for, Loqtorzi versus other treatments used in Canada for use as a single drug for the treatment of adults with recurrent unresectable or metastatic NPC with disease progression while on treatment or after treatment with a platinum-containing chemotherapy. Cytotoxic chemotherapy drugs (taxanes, capecitabine) were considered relevant treatments to compare with toripalimab when reviewing the clinical evidence.
CDA-AMC identified equity and ethical considerations relevant to Loqtorzi and recurrent or metastatic NPC.
The review was informed by materials submitted by the sponsor.
The review was also informed by 3 patient group submissions and 1 clinician group submission in response to the CDA-AMC call for input, and by input from the participating public drug programs around issues that may impact their ability to implement a recommendation.
Three oncologists were consulted as part of the review process.
What Were the Findings?
Clinical Evidence
CDA-AMC reviewed the following clinical evidence:
One single-arm phase II trial (POLARIS-02) in which all 172 patients with recurrent or metastatic NPC received Loqtorzi.
Based on evidence without a comparison: The evidence is very uncertain about the efficacy or harms of toripalimab versus any comparator for outcomes such as OS, PFS, objective response rate, and duration of response. At a data cut-off of February 19, 2020, the median OS was 15.7 months (95% confidence interval [CI], 5.6 months to not estimable) and PFS was 1.9 months (95% CI, 1.8 to 3.5 months) in the platinum-treated analysis set. The objective response rate assessed by the independent review committee was 20.9% (95% CI, 15.1% to 27.8%) and the median duration of response was 14.9 months (95% CI, 10.3 months to not estimable).
There were no long-term extensions, studies addressing gaps, or indirect treatment comparison data included in this report.
Although there were no comparative data, the reported harms were consistent with those reported for other immune checkpoint inhibitors, namely, immune-related harms.
The key gap in the evidence for this review is a comparison of toripalimab versus any comparator; however, there are no drugs that are indicated for second-line or subsequent lines of therapy in this population.
Economic Evidence
Loqtorzi is available as a solution for IV injection (240 mg per 6 mL). At the submitted price of $8,800.00 per vial, the cost of Loqtorzi is expected to be $8,800 per patient per 14-day cycle, based on the Health Canada–recommended dosage. When considering a 28-day cost, the cost of Loqtorzi is expected to be $17,600 per patient.1,2
Clinical efficacy for Loqtorzi in the economic analysis was derived from the POLARIS-02 trial.3 Evidence submitted by the sponsor indicates that Loqtorzi is associated with significant uncertainty in terms of its impact on OS and PFS compared to any comparator among patients with recurrent or metastatic NPC with disease progression while on treatment or after treatment with a platinum-containing chemotherapy. Comparative evidence in the model was informed by naive comparison with data obtained from the KEYNOTE-122 (pembrolizumab and chemotherapy) and NCI-9742 (nivolumab) studies. As noted in the CDA-AMC clinical review, evidence about the efficacy or harms of toripalimab versus any comparator for outcomes such as OS and PFS is very uncertain due to the limitations of a single-arm trial and the absence of long-term extensions, studies addressing gaps, or indirect treatment comparison data.
The results of the CDA-AMC base case suggest:
Loqtorzi is predicted to be associated with higher costs to the health care system than chemotherapy (incremental costs = $89,608), driven primarily by increased costs associated with drug acquisition costs associated with Loqtorzi.
Loqtorzi is predicted to be associated with a gain of 0.54 life-years compared to chemotherapy and may result in a gain of 0.14 quality-adjusted life-years (QALYs) compared to chemotherapy.
The incremental cost-effectiveness ratio (ICER) of Loqtorzi compared to chemotherapy was $634,484 per QALY gained in the CDA-AMC base case. The estimated ICER was highly sensitive to the uncertainty in the comparative efficacy data, long-term survival estimates, and economic model structure. Given the identified limitations with the submitted model and comparative efficacy data, the economic analysis may not accurately assess the impact of Loqtorzi treatment on patient health and health care resources. If Loqtorzi were as equally effective as pembrolizumab, as demonstrated in the KEYNOTE-122 trial, then the ICER increases to $924,997 per QALY gained. Therefore, the cost-effectiveness estimates are uncertain, and higher price reductions may be required to achieve a given willingness-to-pay threshold.
CDA-AMC estimates that the incremental budget impact of reimbursing Loqtorzi for the treatment of patients with recurrent unresectable or metastatic NPC with disease progression while on treatment or after treatment with a platinum-containing chemotherapy will be approximately $3 million over the first 3 years of reimbursement compared to the amount currently spent on a comparator, with an estimated expenditure of $3.4 million on Loqtorzi over this period. The actual budget impact of reimbursing Loqtorzi will depend on the market uptake of Loqtorzi after the first year of its implementation, whether it is also available for first-line treatment, and the extent to which it will be used for patients previously exposed to toripalimab, pembrolizumab, or nivolumab.
Abbreviations
AE
adverse event
CCS
Canadian Cancer Society
CCSN
Canadian Cancer Survivor Network
CDA-AMC
Canada’s Drug Agency
CI
confidence interval
CORD
Canadian Organization for Rare Disorders
DOR
duration of response
EBV
Epstein-Barr virus
ECOG PS
Eastern Cooperative Oncology Group Performance Status
FAS
full analysis set
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HRQoL
health-related quality of life
ICER
incremental cost-effectiveness ratio
LTE
long-term extension
NPC
nasopharyngeal carcinoma
ORR
objective response rate
OS
overall survival
PFS
progression-free survival
PTAS
platinum-treated analysis set
QALY
quality-adjusted life-year
R/M
recurrent unresectable or metastatic
RCT
randomized controlled trial
Background
Introduction
Context for the Review
The objectives of this report are as follows:
Review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of toripalimab (Loqtorzi), 240 mg/6 mL (40 mg/mL) solution as a single agent, for the treatment of adults with recurrent unresectable or metastatic (R/M) nasopharyngeal carcinoma (NPC) with disease progression while on treatment or after treatment with after a platinum-containing chemotherapy. The focus will be placed on comparing toripalimab to relevant comparators in clinical practice in Canada and identifying gaps in the current evidence, and this focus is outlined in Table 1.
Review and critically appraise the economic information submitted by the sponsor, including a cost-effectiveness analysis and budget impact analysis. The focus of the economic review is aligned with the scope of the clinical review, unless otherwise stated. For most reviews, a Canada’s Drug Agency (CDA-AMC) base case is developed, informed by clinical expert input, the available clinical evidence, and the best interpretation of the economic evidence based on the information provided by the sponsor.
The application for review was submitted to CDA-AMC by the sponsor before its drug submission received a Notice of Compliance from Health Canada. This report therefore reflects the indication and recommended dosage for toripalimab that was anticipated during the initial CDA-AMC review period.
Table 1: Information on the Application Submitted for Review and on the CDA-AMC Review
Item | Description |
|---|---|
Information on the application submitted for review | |
Drug | Toripalimab (Loqtorzi), 240 mg/6 mL (40 mg/mL) solution for IV infusion |
Sponsor | Apotex |
Health Canada indication | Toripalimab is indicated:
|
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | October 17, 2025 |
Mechanism of action | PD-1 inhibitor |
Recommended dosage | Toripalimab 3 mg/kg body weight intravenously every 2 weeks. Administer until disease progression or unacceptable toxicity. |
Submission type | Initial |
Sponsor’s reimbursement request | Per indication |
Submitted price | The price of each single-dose vial is $8,800.00 |
Information on the CDA-AMC review | |
Review type | Complex |
Clinical review focusa | Population: Adults with recurrent unresectable or metastatic NPC with disease progression on or after a platinum-containing chemotherapy. Subgroups: None identified. Intervention: Toripalimab, per recommended dosage. Comparators: There are currently no drugs indicated for use in recurrent unresectable or metastatic NPC in the second-line setting or beyond. According to the clinical experts consulted on this review, other than clinical trials, options include conventional cytotoxic chemotherapy with drugs like taxanes or capecitabine. Outcomes: Overall survival, progression-free survival, objective response rate, duration of response, notable harms (immune related). |
CDA-AMC = Canada’s Drug Agency; NOC = Notice of Compliance; NPC = nasopharyngeal carcinoma.
aThe economic review aligns with the scope of the clinical review, unless otherwise stated.
Submission History for the Drug Under Review
CDA-AMC has not previously reviewed toripalimab through the reimbursement review process. Toripalimab in combination with cisplatin and gemcitabine is currently under review at CDA-AMC for the first-line treatment of adults with metastatic or recurrent locally advanced NPC.
Sources of Information
The contents of the Reimbursement Review report were informed by materials submitted by the sponsor, input received from interested parties (patient groups, clinician groups, and drug programs), and input from the clinical experts consulted for this review.
Calls for patient group and clinician group input are issued for each reimbursement review. Patient group submissions were received from 3 groups: the Canadian Organization for Rare Disorders (CORD), the Canadian Cancer Survivor Network (CCSN), and the Canadian Cancer Society (CCS). One clinician group submission was received from the Ontario Health (Cancer Care Ontario) Head and Neck Cancer Drug Advisory Committee. Information was gathered from patients via surveys of patients (12 respondents) and caregivers (3 respondents) and interviews with 3 patients. Clinician input was provided by 3 clinicians. The full submissions received are available on the project landing page in the consolidated input document. The drug programs provide input on each drug being reviewed through the reimbursement review process by identifying issues that may impact their ability to implement a recommendation.
Input from patient and clinician groups is considered throughout the review, including in the selection of outcomes to include in the clinical review and in the interpretation of the clinical and economic evidence. Relevant patient and clinician group input is summarized in the Disease Background, Current Management, and Unmet Needs and Existing Challenges sections of this report.
Each review team includes at least 1 clinical expert with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process. Three oncologists with expertise in the diagnosis and management of NPC participated as part of the review team.
Disease Background
NPC is a rare malignancy of the nasopharynx with distinct epidemiological, etiological, and histopathological characteristics, showing strong geographic and ethnic variation due to its association with Epstein-Barr virus (EBV) and other environmental and genetic risk factors.4 Diagnosis of NPC relies on histological confirmation via tissue biopsy, including EBV-related markers for nonkeratinizing subtypes and association with HPV in keratinizing subtypes, with further diagnostic staging supported by MRI, CT, CT in combination with PET imaging, and endoscopy.5-12
NPC is often diagnosed at a locally advanced stage (stages III to IV) due to its deep anatomic location, vague symptoms (e.g., hearing loss, nasal congestion), and early lymph node involvement, making early detection challenging.13-16 While Canadian data report a relatively high 5-year overall survival (OS) rate of 70% for all stages, up to 30% of patients still experience recurrence, with half of these occurring within 2 years of initial treatment.17 Patients with R/M NPC, especially those with de novo metastases, face poor long-term outcomes, with limited curative options and a 5-year disease-specific survival rate as low as 20%.18,19
Globally, NPC incidence is low (< 1 per 100,000 person-years), but rates are significantly higher in endemic regions such as Southeast Asia and among specific populations in Canada, including Inuit individuals and those of Asian descent.4,20-22 WHO classifies NPC into 3 histologic subtypes, with the EBV-associated nonkeratinizing subtype being the most prevalent, especially in both endemic regions and in Canada.23 In Canada, more than 80% of NPC cases are nonkeratinizing, reflecting the influence of immigration patterns and EBV prevalence; however, data remain limited due to the rare nature of the disease.17,24 Despite subtype differences, treatment approaches for both keratinizing and nonkeratinizing NPC are similar, with a poor prognosis in the R/M setting. In Canada, NPC is characterized by a remarkably low national level incidence rate of 0.5 to less than 1 per 100,000, according to reports from the Global Cancer Observatory (WHO International Agency for Research on Cancer) and Statistics Canada.20,21 A total of approximately 300 to 400 cases of NPC can be estimated annually in Canada, with approximately 120 (plus or minus 40) potentially being new cases of R/M NPC.20
Patient group input: CDA-AMC received patient group input from CORD, CCSN, and CCS. The 3 organizations distributed a joint survey targeting NPC patients through a patient panel, cancer connection forum, mailing list, newsletter, social media platforms, their member patient organizations, and other methods. Responses from 12 patients with NPC and 3 caregivers were collected between May 29, 2025, and June 13, 2025.
Patients described many symptoms (difficulty swallowing, changes in senses, nasal congestion, nosebleeds, hearing loss or ringing in ears, headaches, and neck swelling) as well as moderate to severe pain. They noted that NPC impaired their social interactions and quality of life and had negative impacts on sleep and emotional and mental well-being. A total of 50% of respondents reported moderate to severe pain and discomfort symptoms and adverse impacts on nutritional intake, likely due to difficulty with eating and drinking. Participation in work and routine activities was moderately to severely impacted in 50% of respondents, 70% experienced moderate to severe impact on sleep, and 60% experienced significant levels of anxiety or depression.
Current Management
Treatment Goals
Patient group input: Patients seek treatments with minimal additional toxicity, such as immunotherapy. No further input on treatment goals was received.
Clinician input: Input from 1 clinician group was received for this submission. The Ontario Health (Cancer Care Ontario) (OH [CCO]) Head and Neck Cancer Drug Advisory Committee provides timely evidence-based clinical and health system guidance on drug-related issues in support of Ontario Health’s mandate, including the Provincial Drug Reimbursement Programs and the Systemic Treatment Program. Information was gathered via email, and 3 clinicians provided their input.
According to the OH (CCO) Head and Neck Cancer Drug Advisory Committee, the incidence of NPC in Canada has increased. Although the initial response to the therapy is generally favourable, treatment of R/M disease remains a clinical challenge, given the restricted range of available therapies and limited options beyond first-line chemotherapy. Clinicians declared there is currently no immunotherapy that is funded for the management of R/M NPC in Canada. Therefore, NPC continues to represent a treatment challenge due to limited options. Clinicians agreed that improving OS is an important treatment goal. The prognosis for R/M NPC is generally quite poor, with 3- and 5-year OS rates of around 30% and 20%, respectively, with current chemotherapy options. Survival is generally less than 24 months. The OH (CCO) Head and Neck Cancer Drug Advisory Committee further noted that minimizing adverse events (AEs) and prolonging cancer control are other treatment goals for R/M NPC.
Current Treatment Options
There are no drugs approved for the second line or subsequent lines of treatment of patients with R/M NPC. According to the clinical experts consulted for this review, the best option for patients receiving a second or subsequent line of treatment is clinical trials. Otherwise, options include various cytotoxic chemotherapy including taxanes, capecitabine, and others. The clinical experts noted that although cytotoxic drugs do not target underlying disease mechanisms, they do tend to improve symptoms due to high objective response rates (ORRs) because NPC is a chemosensitive disease. Nivolumab and pembrolizumab may be prescribed off label; however, there are no reimbursement mechanisms available in jurisdictions in Canada for these drugs.
Key characteristics of toripalimab are summarized with other treatments available for NPC in the Supplemental Material document (available on the project landing page) in the Key Characteristics table in Appendix 1.
Unmet Needs and Existing Challenges
Patient group input: CCS, CORD, and CCSN highlighted that international treatment guidelines (European Society for Medical Oncology and National Comprehensive Cancer Network) suggest the use of PD-1 inhibitors in combination with gemcitabine plus cisplatin as the preferred first-line treatment for patients with R/M NPC. Furthermore, the latest guidelines (2021) from Alberta Health Services for managing NPC in Canada suggest that patients with R/M NPC should consider enrolling in clinical trials. This recommendation suggests there is a clinical unmet need due to limitations of current treatments for this patient group.
According to the input, the treatment regimen received most frequently was chemoradiation, reported by 7 participants (30%), followed by radiation therapy, chemotherapy, and surgery. Notably, only 1 patient had received immunotherapy (tislelizumab) during treatment. Some of the side effects of current treatments reported by patients with NPC included difficulty swallowing due to pain, alteration in sense of taste, pulmonary embolism, complicated neck surgery, incomplete recovery of sense of smell and persistent leaky eye, damaged nasal passages caused by radiation treatments, ringing in ear progressing to substantial hearing loss requiring hearing aids, neuropathy, loss of appetite, weight loss, radiation burns, brain fog, and hypothyroidism secondary to the radiation treatment. As observed by some patients, the standard of care for NPC can cause discomfort for some and, for others, serious side effects that impact quality of life during or after treatment. Some patients also reported long travel times to get to their treatment: 3 of 6 patients reported commutes of 30 minutes to 1 hour, and 1 patient reported a commute of 2 to 3 hours. Patients also reported the significant impact of their disease on quality of life, including social interactions, food intake, work or daily activities, sleep, emotional well-being, and mental health.
Clinician input: The clinician group noted that although NPC is responsive to chemotherapy, patients with relapsed or metastatic disease will eventually experience disease progression, even after multiple courses of chemotherapy. Therefore, there is a need for additional and better treatment options to improve outcomes. Moreover, there is no approved second-line treatment in those patients whose disease progresses either during or following treatment with a platinum-containing chemotherapy plus gemcitabine).
Considerations for Using the Drug Under Review
Contents within this section have been informed by input from the clinical experts consulted for the purpose of this review and from clinician groups, and by the reimbursement conditions proposed by the sponsor (refer to Table 2 in Appendix 1 of the Supplemental Material document). The implementation questions from the public drug programs and corresponding responses from the clinical experts consulted for this review are summarized in Table 3 in Appendix 1 of the Supplemental Material document. The following was summarized by the review team.
Place in Therapy
The clinical experts consulted for this review noted that the standard of care for patients who are eligible for second-line or subsequent lines of treatment varies and may include taxanes (paclitaxel or docetaxel), capecitabine, or others; however, clinical trials are the preferred option for patients who are eligible and interested. The clinician group agreed that the options for second-line or subsequent lines of treatment in NPC are very limited. The clinical experts also noted that these cytotoxic drugs are really aimed at relieving disease-related symptoms rather than targeting underlying disease mechanisms (better accomplished with targeted treatments or immunotherapies). The clinical experts and clinician group were of the opinion that toripalimab would change the treatment paradigm and would be used as monotherapy in the second-line setting in patients without prior exposure to immune checkpoint inhibitors.
Patient Population
The clinical experts and clinician groups believed that, given the data from the POLARIS-02 trial, it would be reasonable to offer single-drug toripalimab to any patients who are on their second or subsequent line of treatment and who had not been previously exposed to an immune checkpoint inhibitor. While NPC is considered a rare disease in Canada, the clinical experts noted that the risk of developing NPC is heavily concentrated in First Nations, Inuit, and Métis people, and people of Asian descent (e.g., Chinese). These groups may also experience additional barriers to care, including language, immigration-related factors, geographical isolation, potentially reduced health literacy, and low awareness of the disease, which can impact the outcomes of treatment. Misdiagnosis is unlikely at the R/M stage because patients are often seen at tertiary academic centres, but underdiagnosis and delayed diagnosis could be possible in patients who are from marginalized communities and who lack access to care.
With respect to whether a specific subpopulation might benefit most from toripalimab, the clinical experts believed that any patients in the second-line setting or beyond who are fit for treatment would be appropriate candidates, and the clinician groups agreed that even patients whose tumours have low PD-L1 expression may derive benefit. PD-L1 is not used as a selection biomarker in NPC because most NPC malignancies express PD-L1. The clinical experts added that, given that some patients who receive systemic therapy are long-term survivors, a watchful waiting approach, even if a patient has minimal or no symptoms, would not be appropriate. The clinical experts noted that the patients least suitable for treatment would be those with an absolute contraindication to anti-PD-1 antibodies, such as those who have received a solid organ transplant.
With respect to initiation criteria, the clinical experts believed that the sponsor’s initiation criteria for second-line or subsequent-line treatment are appropriate. One of the clinical experts raised a specific question as to whether patients who received standard-of-care induction of gemcitabine and cisplatin for 3 cycles followed by concurrent radiation with cisplatin for locoregionally advanced disease, and who subsequently experience disease recurrence or relapse within 1 year or less, should be rechallenged with the same chemotherapy (that is, gemcitabine and platinum-based therapies) with toripalimab added, or whether these patients should be considered to have disease that is refractory to platinum-containing therapy and therefore should be treated with toripalimab alone in the second or subsequent line. One of the clinical experts noted that most clinicians would consider disease recurring within 6 months of definitive therapy (i.e., induction gemcitabine plus cisplatin followed by chemoradiation with cisplatin) to be refractory to platinum; however, refractoriness is not as clearly defined in NPC as it is in other cancers.
Assessing the Response to Treatment
The clinical experts noted that, in practice, patients would routinely be assessed via their history and a physical examination, along with regular imaging (CT or MRI). Longer-term outcomes such as PFS and OS are not necessarily practical to assess on a visit-to-visit basis, according to the clinical experts, and tend to be more associated with the assessment of response in clinical trials. According to the clinical experts, health-related quality of life (HRQoL) is important but would be not typically be assessed using instruments in clinical practice; rather, an improvement in patient symptoms would be considered an indication of an improvement in HRQoL.
The clinical experts noted that depth of response (complete versus partial), duration of response (DOR), and an increase in ORR would be considered clinically meaningful. The clinical experts also noted that a transient response (duration of less than 4 weeks) with no or minimal improvement in disease-related symptoms and/or intolerable harms would not be considered a clinically meaningful benefit.
The clinical experts believed that patients treated for NPC would not be monitored as often as in a clinical trial because, for example, imaging (CT or MRI) for patients on systemic therapy would typically be performed every 9 to 12 weeks.
The sponsor did not propose renewal criteria for the second line or beyond.
Discontinuing Treatment
The clinical experts and clinician groups noted that toripalimab should be discontinued in patients with disease progression or intolerable toxicity, at the completion of the treatment course, or at patient request. The clinical experts agreed with the discontinuation criteria proposed by the sponsor, namely, patients with progressive disease or unacceptable toxicity.
Prescribing Considerations
The clinical experts noted that, because this is not the first PD-1 inhibitor, all cancer centres in Canada should be able to provide access to imaging or care infrastructure. The clinical experts noted, however, given the fairly low incidence of NPC in Canada, some community oncology centres may prefer to refer such patients to tertiary academic centres due to their lack of volume-driven experience and expertise in this disease. They added, however, that physicians in these centres should be able to reach out to more experienced physicians for advice and support. The clinical experts agreed with the prescribing conditions proposed by the sponsor, namely, that toripalimab be prescribed only by physicians with expertise in treating patients with R/M NPC.
Additional Considerations
The clinical experts noted there are a lack of data for toripalimab in R/M NPC in countries where the disease is not endemic (including the US and Canada) because most of the evidence had come from studies in China, Taiwan, and Singapore. One clinical expert added that there is an ongoing study (TRANSPARENT) with an accrual plan of 100 patients, with a primary objective to evaluate the efficacy of toripalimab in combination with chemotherapy (cisplatin and gemcitabine) in patients receiving first-line R/M NPC treatment, including those with NPC that is or is not associated with EBV. There are emerging cases of EBV-negative, HPV-positive squamous cell cancers of the nasopharynx in clinical practice. Until the TRANSPARENT study is completed, the allowance to use toripalimab for EBV-negative NPC (regardless of HPV status) should be clarified by drug plans.
Clinical Review
Methods
The review team considered the studies in the sponsor’s systematic review (pivotal studies and randomized controlled trials [RCTs]), sponsor-submitted long-term extension (LTE) studies, indirect treatment comparisons (ITCs), and studies addressing gaps in the evidence for inclusion. Eligible studies for the systematic review included published and unpublished pivotal studies and phase II, III, and IV RCTs or other designs, as relevant. Relevant patients and interventions were defined by the indication, that is, for use as a single drug for the treatment of adults with R/M NPC with disease progression on or after receiving a platinum-containing chemotherapy at the dosage recommended in the product monograph. Relevant comparators were drugs used in clinical practice in Canada to treat patients described in the indication under review. There are no drugs that are indicated for use in the second or a subsequent line of treatment for NPC; however, the clinical experts noted that chemotherapy is used in these patients. LTEs of the included pivotal studies and RCTs were to be included; however, none were available. ITCs and studies addressing gaps submitted by the sponsor were to be included when they filled an identified gap in the systematic review evidence (e.g., missing comparator, longer follow-up time); however, none were available.
The review team selected outcomes (and follow-up times) for review, considering the sponsor’s Summary of Clinical Evidence, clinical expert input, and patient and clinician group input. Included outcomes are those considered relevant to expert committee deliberations, and they were selected in consultation with committee members. Evidence from the systematic review for the most important outcomes was assessed using the Grading of Recommendations Assessment, Development and Evaluation (GRADE) approach. OS and PFS were selected for assessment with GRADE because these are important outcomes to patients and are standard outcomes for assessing drugs in oncology. ORR was chosen for assessment by GRADE because this is the outcome that is more likely to be used by clinicians to assess patients on a visit-by-visit basis. DOR was selected for GRADE because it was identified as important for the pharmacoeconomic review and because it is important to patients.
Methods for data extraction, the risk-of-bias appraisal, and the certainty-of-evidence assessment are in Appendix 2 of the Supplemental Material document.
Clinical Evidence
In this report, the following sources of evidence submitted by the sponsor are reviewed and appraised:
1 pivotal study included in the systematic review (POLARIS-02).
Systematic Review
Description of Studies
Study Characteristics
Characteristics of the included study are summarized in Table 2. Details pertaining to the eligibility criteria, interventions, and comparators and the relevant outcome measures are in Appendix 3 of the Supplemental Material document.
The POLARIS-02 trial was a phase II, multicentre, open-label, single-arm, activity estimation trial conducted in adults with different solid tumour malignancies, including a total of 8 different cohorts based on a basket design. The primary objective of the study was to evaluate the antitumour activity (measured as the ORR) of toripalimab for subsequent-line treatment in patients with R/M NPC. Secondary objectives were to evaluate the clinical activity of toripalimab, as determined by DOR, time to response, disease control rate, PFS, OS, 6-month PFS, 1-year OS, and its safety based on the incidence of AEs and laboratory abnormalities. Cohort 3 was conducted at 18 clinical study sites in China and assessed the efficacy and safety of toripalimab monotherapy in 190 adults with R/M NPC refractory to standard chemotherapy or who were intolerant to such therapy. The study was initiated on December 22, 2016, and the data cut-off date was February 19, 2020.
Protocol amendment 5 expanded the sample size and required more than 2 prior systemic therapies for inclusion in the study. According to the sponsor, this was enacted after advice from the Chinese regulator due to “substantial antitumour activity.”
Table 2: Characteristics of Studies Included in the Systematic Review
Study name, design, and sample size | Key inclusion criteria | Key exclusion criteria | Intervention and comparator | Relevant end points |
|---|---|---|---|---|
POLARIS-02 Phase II, multicentre, open-label study Total N = 190 |
|
| Toripalimab monotherapy: Toripalimab was administered by IV infusion at a recommended dosage of 3 mg/kg every 2 weeks for each 4-week cycle |
|
AE = adverse event; CTCAE = Common Terminology Criteria for Adverse Events; ECOG = Eastern Cooperative Oncology Group; NPC = nasopharyngeal carcinoma; ORR = overall response rate; OS = overall survival; PFS = progression-free survival; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1; TKI = tyrosine kinase inhibitor.
aIncluding, but not limited to, anticancer drug therapy or radiochemotherapy.
bAdjuvant or neoadjuvant chemoradiotherapy can be considered as 1 line of treatment if tumour recurrence or metastasis occurs within 6 months after the end of the definitive chemoradiotherapy treatment for nonmetastatic NPC.
cOnly lesions that progressed after radiation therapy were considered measurable disease.
dHowever, patients receiving local palliative radiotherapy for bone metastasis lesions could be included.
eExcluding alopecia and pigmentation changes, peripheral nerve toxicity that has improved to < CTCAE grade 2, and long-term toxicity related to radiotherapy that would likely not fully resolve, as judged by the investigator.
fTumour response evaluated by an independent review committee and investigator.
Source: Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Statistical Testing and Analysis Populations
The planned sample size for cohort 3 of the POLARIS-02 clinical trial was 180 patients. The sample size calculation was based on an anticipated ORR of 24% and the ability to exclude a lower-limit confidence interval (CI) for observed ORR of less than 15%. Enrolment of approximately 160 patients provided greater than 80% power and facilitated a lower-limit CI of no less than 15% using the Clopper-Pearson method.
Continuous data were summarized in terms of the mean, standard deviation, median, and range, unless otherwise stated. Categorical variables were summarized with descriptive statistics, including the number and proportion of patients and/or the number of events. If there were no events, the incidence was not calculated. The denominator used in the incidence calculations of efficacy variables was the total number of patients included in the analysis population, unless specified otherwise.
The full analysis set (FAS) and safety set both included patients who had received at least 1 dose of toripalimab. The platinum-treated analysis set (PTAS) included all patients with R/M NPC with disease progression on or after platinum-containing therapy who received at least 1 dose of toripalimab. The PTAS was used for all efficacy end points. The breakthrough therapy designation analysis set included all patients from the PTAS with nonkeratinizing NPC.
Patient Disposition
Patient disposition for the included study is summarized in Appendix 4 of the Supplemental Material document.
A total of 282 patients were screened and 190 were enrolled in the FAS population, including 172 patients in the PTAS. Following agreement with the FDA, the primary efficacy analyses were based on the 172 patients who had received prior platinum-based therapy. The most common reasons for the termination of study treatment were disease progression (70 of 172 patients; 41%) and second onset of disease progression (51 of 172 patients; 30%). The most common reason for the termination of study participation was death (97 of 172 patients; 56%). A total of 25 of 172 patients (15%) were lost to follow-up.
A total of 176 of 190 patients (93%) had at least 1 protocol deviation and 107 of 190 patients (56%) had at least 1 major protocol deviation. The most common major protocol deviations were related to “informed consent” (37 of 190 patients; 20%) and ”schedule criteria” (32 of 190 patients; 17%).
Baseline Characteristics
Table 3: Summary of Baseline Characteristics From Studies Included in the Systematic Review (PTAS)
Characteristics | Toripalimab (N = 172) |
|---|---|
Age, years mean (SD) | 46.4 (10.50) |
Sex, n (%) | |
Female | 14 (16.8) |
Male | 158 (83.2) |
Race, n (%) | |
Asian | 190 (100) |
ECOG PS at baseline, n (%) | |
0 | 66 (35) |
1 | 124 (65) |
PD-L1 tumour expression, n (%) | |
Negative | 130 (68) |
Positive | 52 (27) |
NA | 8 (4) |
Liver metastasis, n (%) | |
No | 75 (43.6) |
Yes | 97 (56.4) |
Extent of disease, n (%) | |
Metastatic | 170 (98.8) |
Recurrent | 2 (1.2) |
Pathological subtype, n (%) | |
Keratinizing | 5 (2.9) |
Nonkeratinizing | 164 (95.3) |
Missing | 3 (1.7) |
Median number of prior therapies (range) | 2 (1 to 13) |
Number of prior systemic therapies | |
1 | 56 (33) |
2 | 71 (41) |
3 or more | 45 (26) |
Prior therapies | |
Radiation therapy | 155 (90.1) |
Platinum agents | 172 (100) |
ECOG PS = Eastern Cooperative Oncology Group Performance Status; NA = not available; PTAS = platinum-treated analysis set; SD = standard deviation.
Note: Age is calculated from the informed consent date relative to the date of birth. Baseline is defined as the last nonmissing measurement taken before the reference start date (including unscheduled assessments).
Source: Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Treatment Exposure and Concomitant Medications
Details of patient treatment exposure, adherence, and use of concomitant medications in the included study are in Appendix 4 of the Supplemental Material document.
All of the patients in the POLARIS-02 trial received at least 1 dose of toripalimab. Throughout the study, the median number of infusions was 8.0 (range, 1.0 to 69.0) and the median number of completed cycles was 4.0 (range, 1.0 to 38.0). The mean dose of toripalimab per infusion was 179.8 mg (standard deviation of 34.1 mg).
Critical Appraisal
Internal Validity
The lack of a comparator prevents any conclusions from being drawn regarding the relative efficacy and harms of toripalimab versus any therapy. Although no drugs had received Health Canada approval of indications for the second-line treatment of NPC as of this writing, there are a number of drugs that are used for this purpose in clinical practice. As a result, there is also a lack of any hypothesis and statistical testing, which precludes assessment of the statistical significance of any observed effect and substantially reduces the certainty of the evidence. Furthermore, the lack of a comparator precludes any conclusion about the causality of the treatment effect. One cannot rule out that any observed outcome could be obtained by the natural evolution of the disease or simple chance.
The POLARIS-02 trial employed a basket design. Traditionally, 1 of the limitations of basket designs has been that several different populations are analyzed together; therefore, positive results for larger groups in the analysis may result in false-positive results for smaller groups. The POLARIS-02 trial does not appear to have this limitation because the NPC cohort was reported separately from the others. Additionally, no formal analysis was planned because this was a single-arm trial; therefore, the concern about analyses being driven by 1 group is not relevant. This exploratory and hypothesis-generating design of the POLARIS-02 trial is a consistent feature of basket designs.
The fact that all patients were aware of their treatment assignment may have biased the assessment of efficacy outcomes as well as harms. Although this bias tends to be most impactful for patient-reported outcomes, which were not assessed in the POLARIS-02 trial, it may still occur with the assessment of other efficacy outcomes. For example, investigator-assessed tumour end points such as ORR and PFS may be subject to bias in an open-label trial; however, this was apparently mitigated in the POLARIS-02 study through the use of an independent review committee. The use of an independent review committee for the primary analysis was noted in the Clinical Study Report but not in the protocol. Knowledge of assigned treatment may also impact patient adherence; however, this was reported as 100% in the trial. Reporting of harms can also be impacted because known harms of immune checkpoint inhibitors, such as immune-related harms, may be more frequently reported because patients are anticipating them. However, the most commonly reported immune-related harm reported was hypothyroidism, and this is a harm that should be objectively measured.
A relatively large number of patients (25 of 190 patients; 13%) who terminated the study were categorized as lost to follow-up. The most common reason for patients stopping the study was death (97 of 190 patients; 51%). There were similar numbers reported for missing data reported for assessment of ORR, with data missing for 20 of 172 patients (12%) in the PTAS population. All of these patients were counted in the denominator and were therefore assumed to be nonresponders. This is a conservative approach for accounting for missing data; therefore, it is possible the results for ORR could be underestimated. However, no sensitivity analyses were planned or conducted to test the robustness of the results for missing data.
External Validity
The POLARIS-02 study was conducted entirely in China, and although people of Chinese descent are at higher risk of NPC, the clinical experts noted that the population of patients with NPC in Canada is much more heterogeneous. According to the clinical experts, First Nations, Inuit, and Métis populations, particularly Inuit, are also at higher risk of developing NPC; therefore, this is a noteworthy gap in evidence relevant to Canada. The clinical experts also noted that several disease characteristics were more narrowly defined in the population enrolled in the POLARIS-02 trial than would be the case in clinical practice. For example, the POLARIS-02 trial restricted enrolment to patients with an Eastern Cooperative Oncology Group Performance Status (ECOG PS) of 0 or 1, while the clinical experts asserted they would treat patients with a worse performance status. Additionally, although the relatively small proportion of females in the study reflects the typical female-to-male ratio in this disease, only 14 females were enrolled in the study, which is a limited sample upon which to assess the drug.
The trial criteria did not specify that patients had to have not experienced a response to a prior platinum-based regimen; therefore, the PTAS that the sponsor used for its submission to CDA-AMC represents a subset (N = 172) of the FAS population (N = 190). The main publication for the POLARIS-02 trial, for example, reports efficacy data only for the FAS. The original enrolment criteria included patients who had received at least 1 prior line of therapy, which could include chemoradiation, and then this was changed under protocol amendment 5 to require patients to have received at least 2 prior lines of treatment, including (but not limited to) chemotherapy or chemoradiation. The sponsor stated that the reason for the protocol amendment was due to the positive response rates seen in the “original NPC cohort,” which prompted the Chinese regulatory body to also expand the sample size in the trial. There were 43 additional patients enrolled after the protocol amendment, which represents approximately one-quarter of the overall study population. Therefore, the POLARIS-02 trial included a mixture of 2 different populations, and this is a potential confounder when analyzing the data. Additionally, neither of these populations (the patients who were in the study before the amendment versus those after the amendment) in the POLARIS-02 trial matches the indication that was sought from Health Canada; thus, the proposed reimbursement criterion (patients with “…disease progression on or after a platinum-containing therapy”) matches the PTAS subset only.
The outcomes reported in the POLARIS-02 trial (OS, PFS, ORR, DOR) were key outcomes that are relevant to patients with NPC. HRQoL, which is an important outcome to patients, was not included as an outcome in that study.
Results
The key efficacy and harms results and findings from the GRADE assessment are presented in this section. Detailed efficacy and harms results can be found in Appendix 4 of the Supplemental Material document.
Efficacy
Key results include the following:
The median OS was 15.7 months (95% CI, 5.6 to not estimable) with toripalimab, and the median PFS was 1.9 months (95% CI, 1.8 to 3.5).
The ORR was 20.9% (95% CI, 15.1 to 27.8). Subgroups with differences in ORR were present in the subgroups based on previous radiotherapy (ORR of 21.9% for those who received prior radiotherapy versus 11.8% for those who did not), liver metastasis (16.5% for those who had liver metastasis versus 26.7% for those who did not), EBV DNA level (29.7% for those with less than 104 IU/mL versus 14.3% for those with ≥ 104 IU/mL), and lactate dehydrogenase levels (24.1% for those with < 2 × upper limit of normal [ULN] versus 8.8% for those with ≥ 2 × ULN).
The median DOR was 14.9 months (95% CI, 10.3 to not estimable).
Harms
A total of 181 of 190 patients (95%) reported at least 1 AE, and 46 of 190 patients (24%) had at least 1 serious adverse event (SAE).
The most common grade 3 or higher AE was hyponatremia (14 of 190 patients; 7%), followed by anemia (9 of 190 patients; 5%).
The most common SAE was lung infection (9 of 190 patients; 5%), followed by abnormal hepatic function (5 of 190 patients; 3%).
The most common immune-related harm was hypothyroidism (40 of 190 patients; 21%).
Summary of Findings and Certainty of the Evidence
Table 4 summarizes the findings from the POLARIS-02 study by the outcomes identified as most relevant to this reimbursement review of toripalimab. In accordance with the GRADE approach, evidence from nonrandomized studies without a comparator arm begins at very low certainty for each outcome due to inherent limitations in study design and risk of bias. No outcomes met the GRADE criteria for upgrading and, therefore, the certainty of evidence remained very low across all reported outcomes. Relatedly, no minimally important differences were sought as target thresholds for the outcomes of interest in this review. Refer to the summary of outcome measures in Appendix 3 of the Supplemental Material document.
Table 4: Summary of Findings for Toripalimab for Patients With R/M NPC
Outcome and follow-up | Patients, N (studies) | Effect | Certaintya | What happens |
|---|---|---|---|---|
Overall survival (PTAS) | ||||
Probabilityb of overall survival at 1 year (95% CI) Follow-up: 1 year | 172 (1 single-arm trial) | 56.7% (48.5% to 64.0%) | Very low | The evidence is very uncertain about the effects of toripalimab on overall survival when compared with any comparator |
PFS (PTAS) | ||||
Probabilityb of IRC-assessed PFS at 6 months (95% CI) Follow-up: 6 months | 172 (1 single-arm trial) | 29.1% (22.3% to 36.3%) | Very low | The evidence is very uncertain about the effects of toripalimab on PFS when compared with any comparator |
ORR (PTAS) | ||||
ORR, IRC assessed (95% CI) Follow-up: Data cut-off (February 19, 2020) | 172 (1 single-arm trial) | 20.9% (15.1% to 27.8%) | Very low | The evidence is very uncertain about the effects of toripalimab on ORR compared with any comparator |
DOR (PTAS) | ||||
Patients with a DOR > 12 months, n (%) Follow-up: 12 months | 172 (1 single-arm trial) | 14 (38.9) | Very low | The evidence is very uncertain about the effects of toripalimab on DOR compared with any comparator |
Harms (safety set) | ||||
Patients with pneumonitis, n (%) Follow-up: Data cut-off (February 19, 2020) | 190 (1 single-arm trial) | 1 (0.5) | Very low | The evidence is very uncertain about the effects of toripalimab on pneumonitis compared with any comparator |
CI = confidence interval; DOR = duration of response; GRADE = Grading of Recommendations Assessment, Development and Evaluation; IRC = independent review committee; NPC = nasopharyngeal carcinoma; ORR = objective response rate; PFS = progression-free survival; PTAS = platinum-treated analysis set; R/M = recurrent unresectable or metastatic.
Note: All serious concerns with study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, and publication bias are documented in the table footnotes.
aIn the absence of a comparator group, conclusions about efficacy relative to any comparator cannot be drawn. The certainty of evidence is therefore started at very low, and no factors were identified (in line with GRADE recommendations) that would support rating up.
bEstimated using the Kaplan-Meier method.
Source: Details included in the table are from the sponsor’s Summary of Clinical Evidence.
LTE Studies
The submission did not include any LTE studies.
Indirect Evidence
A review of the indirect evidence was merited because off-label comparators (pembrolizumab and nivolumab) were identified by the clinical experts as treatment options in the second-line setting and beyond, and naive comparisons of trials for toripalimab (POLARIS-02), pembrolizumab (KEYNOTE-122), and nivolumab (NCI-9742) were included in the pharmacoeconomic model.
Description of Indirect Treatment Comparison
Study Selection and Review Methods
At the time of submission, there was no direct comparative evidence of toripalimab relative to these comparators (pembrolizumab and nivolumab) for the treatment of adults with R/M NPC. The sponsor conducted a feasibility assessment to evaluate the clinical and methodological validity of conducting an ITC between toripalimab and pembrolizumab and between toripalimab and nivolumab in the second line or beyond, which consisted of a systematic literature review. Details of the search criteria and methods are in Table 5.
Table 5: Study Selection Criteria for the 2L+ Population ITC
Characteristics | Indirect comparison |
|---|---|
Population | Adults with recurrent unresectable or metastatic NPC with disease progression on or after treatment with a platinum-containing chemotherapy. |
Intervention | For the feasibility assessment, the following treatments were considered an intervention of interest:
|
Comparator | No restrictions were placed on comparators. |
Study designs | Prospective or retrospective clinical studies that could be used to inform an ITC. |
Publication characteristics | Published clinical studies. |
Exclusion criteria | Results were excluded if they were animal studies, conference abstracts, case reports, opinion articles, or review articles. |
Databases searched | PubMed. |
Selection process | Articles were screened by a researcher, and potentially relevant articles were subjected to full-text screening before a decision was made to include or exclude the study in the feasibility assessment. |
2L+ = second or subsequent line; ITC = indirect treatment comparison; NPC = nasopharyngeal carcinoma.
Source: Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Results
A total of 38 records were identified in the pembrolizumab literature search, and 32 records were identified in the nivolumab search. All of the studies identified were single-arm trials, and the sponsor elected not to conduct an unanchored matching-adjusted indirect comparison, citing limitations in the analysis and no access to individual patient data. The sponsor completed a naive comparison of toripalimab versus pembrolizumab and nivolumab for the second line or beyond in the pharmacoeconomic model, which was not included in the submission and not appraised in the clinical section of this report.
Studies Addressing Gaps in the Systematic Review Evidence
The submission did not include any studies addressing gaps in the systematic review evidence.
Discussion
Efficacy
There are no drugs approved in Canada for the treatment of R/M NPC in the second line or beyond; therefore, the clinical experts and clinician and patient groups consulted on this review believe that toripalimab potentially fills an important gap in the therapeutic landscape. However, with a lack of a concurrent control group or a comparator group in the POLARIS-02 trial, and in the absence of any other submitted comparative evidence, conclusions about the relative efficacy of toripalimab in the second line or beyond cannot be drawn. In the first-line setting, there is evidence from 1 RCT (JUPITER-02) demonstrating that toripalimab shows benefit as an add-on to gemcitabine plus cisplatin, suggesting activity in NPC. However, these results were in a treatment-naive population and the treatment regimens (combination versus monotherapy) differed between the 2 settings. This limits the generalizability of these results to the second-line population that is the focus of this review.
While cross-trial comparisons are inherently limited and not considered decision-level evidence in health technology assessment, the clinical experts consulted for this review suggested drawing from the KEYNOTE-122 study,25 which compared pembrolizumab monotherapy to nonplatinum chemotherapy (capecitabine, gemcitabine, or docetaxel) in patients with R/M NPC who had received at least 1 prior platinum-based regimen, which may provide some contextual insight into expected clinical outcomes. While pembrolizumab is not considered a valid comparator in this space because of a lack of approval and reimbursement, the data from the chemotherapy arm would be informative, according to the experts. The review team and sponsor did not identify any high-quality natural history data for the standard of care in the second-line setting and beyond. This supports the suggestion to consider the results from the chemotherapy arm of the KEYNOTE-122 trial.
Patients in the POLARIS-02 trial were slightly younger than patients in the chemotherapy group of the KEYNOTE-122 study (mean of 46 years versus median of 53 years, respectively); however, the population was predominantly male in both studies (80% male versus 20% female), approximately one-third of patients had an ECOG PS of 0 and the remaining patients had an ECOG PS of 1, and the majority had received 1 or 2 prior lines of therapy (74% in the POLARIS-02 trial; 62% in the KEYNOTE-122 trial). The clinical experts consulted by CDA-AMC agreed there are sufficient similarities between the 2 study populations to allow for a contextual comparison of outcomes, but they also acknowledged limitations with such a comparison. The median OS appeared comparable between the toripalimab group from the POLARIS-02 trial (15.7 months) and the chemotherapy group from the KEYNOTE-122 trial (15.3 months), and the clinical experts consulted on this review agreed with this observation. The ORR was also consistent between the toripalimab group in the POLARIS-02 trial (20.9%) and the chemotherapy group in the KEYNOTE-122 trial (23.3%); however, the median PFS in the POLARIS-02 trial was lower (1.9 months) than the median PFS in the chemotherapy group of the KEYNOTE-122 trial (5.5 months). The clinical experts noted challenges in interpreting PFS data in patients on immunotherapy because phenomena such as delayed responses and pseudoprogression can obscure treatment effects at earlier time points. Perhaps in support of the difficulties in interpreting PFS with immunotherapy (including toripalimab), despite OS being longer for pembrolizumab than chemotherapy in the KEYNOTE-122 trial (17.2 versus 15.3 months), the PFS was shorter for pembrolizumab than for chemotherapy (4.1 versus 5.5 months). In summary, this information suggests there may be a benefit in treating patients with toripalimab in the second-line setting or beyond at a scale similar to chemotherapy. However, whether the magnitude of the benefit is clinically important and transferable to clinical practice remains unclear. Furthermore, whether toripalimab provides similar efficacy for ORR and OS in the second line or beyond cannot be determined based on current evidence, and numerical similarities across trials should not be overinterpreted because these treatments have not been formally compared directly or indirectly. This information is provided only as contextual background and in the absence of decision-level comparative evidence.
HRQoL was not assessed in the POLARIS-02 trial. Given the lack of a control group and lack of blinding in the study, any assessment of HRQoL would have been at risk of bias. Nevertheless, the lack of HRQoL data is an important gap in the evidence, with the clinical experts, clinician groups and, most importantly, the patients who provided input to this review emphasizing the impact that this disease has on HRQoL. Patients describe symptoms such as difficulty swallowing and changes in senses, and moderate to severe pain was reported by half of the patients surveyed. These specific symptoms were also not assessed in the POLARIS-02 trial.
The clinical experts consulted on this review noted the differences in dosing between first-line toripalimab in the JUPITER-2 trial (flat dose) and the weight-based dosing in the POLARIS-02 trial in patients in the second line or beyond. The clinical experts all noted that weight-based dosing is more problematic from a practical perspective and may also result in more wastage if clinics are not able to share vials among patients. The clinical experts also noted that if toripalimab becomes widely used in the first-line setting, use of toripalimab in the second line may eventually fade to almost nothing because these patients would no longer be eligible for treatment with toripalimab.
Harms
The lack of a control group in the POLARIS-02 trial makes it difficult to place the findings related to harms into context. The clinical experts identified immune-related toxicities as being the most relevant harms when assessing PD-1 inhibitors such as toripalimab, and specifically noted the more serious harms, such as pneumonitis and myocarditis, rather than harms such as hypothyroidism, which are more common but less serious. Pneumonitis (grade 2) and myocarditis (grade 3) occurred only in 1 patient each; therefore, although we have nothing to compare these results to, in absolute terms, these more severe harms did not occur frequently over the median 1-year follow-up period.
Ethics and Equity Considerations
According to the clinical experts consulted on this review, in Canada, NPC tends to disproportionately impact First Nations, Inuit, and Métis populations, particularly Inuit, in addition to people of Asian descent, yet there were no Indigenous patients enrolled in the trial. These groups may also experience additional barriers to care, including language barriers, immigration-related factors, geographical isolation, potentially reduced health literacy, and low awareness of the disease, which can impact the outcomes of treatment. Misdiagnosis is unlikely at the R/M stage because patients are often seen at tertiary academic centres, but underdiagnosis and delayed diagnosis could be possible in patients who are from marginalized communities and who lack access to care.
Given the rarity of NPC, the clinical experts agreed there may be some challenges for patients trying to access toripalimab in rural and remote areas. Given that many First Nations, Inuit, and Métis people live in rural and remote areas, travel may be an important challenge for these patients to overcome. One of the clinical experts also noted the importance of having access to translators and other supports to reflect some of the relevant cultural sensitivities in Indigenous populations as well as among newcomers.
There were only 14 female patients in the POLARIS-02 trial, and this makes it challenging to draw conclusions about the efficacy and harms of toripalimab in this population.
The patient input suggested that NPC takes a significant emotional toll on patients and negatively impacts their mental health.
NPC is a rare disease; therefore, knowledge of this condition may be limited outside of academic centres, resulting in patients in rural areas having less access to timely care or being unduly burdened by travel to access care. This geographic isolation may also impose additional financial and caregiver burdens as well.
Currently, there is limited access to other immune checkpoint inhibitors or any other curative options in the second line or beyond.
Conclusion
The available evidence for the use of toripalimab in the second line or beyond for patients with R/M NPC is limited to 1 single-arm study (POLARIS-02) that enrolled patients who had previously experienced 1 or 2 prior therapies for this disease. The POLARIS-02 trial employed a basket design, and the focus of this CDA-AMC review was on a trial population comprising patients with R/M NPC who had previously received a platinum-containing regimen and were treated experimentally with toripalimab. As a result of the single-arm design, it is very uncertain whether toripalimab had any impact on OS, PFS, ORR, or DOR in this population, or on safety or tolerability, relative to any comparator. There were no studies addressing gaps in the literature or LTEs in this submission, and the sponsor determined it was not feasible to perform an ITC. Overall, there is no direct or indirect evidence upon which to assess the comparative efficacy or safety of toripalimab in previously treated NPC.
Economic Review
Methods
The review team appraised the pharmacoeconomic evidence submitted by the sponsor on the cost-effectiveness and budget impact of toripalimab compared to other relevant treatments for adults with R/M NPC with disease progression on or after treatment with a platinum-containing chemotherapy. The sponsor’s economic submission is summarized in Appendix 10 of the Supplemental Material document.
Summary of the Submitted Economic Evaluation
The sponsor submitted a cost-utility analysis to estimate the cost-effectiveness of toripalimab from the perspective of a public health care payer in Canada over a lifetime horizon (15 years).26 The modelled population comprised adults with R/M NPC with disease progression on or after treatment with a platinum-containing chemotherapy.26 This population was aligned with the Health Canada indication and was based on the participants in the POLARIS-02 trial.3,26 The sponsor’s base-case analysis included costs related to drug acquisition, administration, disease management, AEs, and end-of-life care.26
In the sponsor’s base case, toripalimab was associated with incremental costs of $93,510 and 0.39 incremental quality-adjusted life-years (QALYs) relative to chemotherapy.26 This resulted in an incremental cost-effectiveness ratio (ICER) of $242,215 per QALY gained. Compared to pembrolizumab and nivolumab, toripalimab was associated with an ICER of $90,056 (incremental costs = $35,707; incremental QALYs = 0.40) and $133,889 (incremental costs = $64,347; incremental QALYs = 0.48), respectively.26 Of the incremental benefit compared to chemotherapy (0.39 incremental QALYs), more than 96% of the benefit was predicted to be accrued after the treatment duration of the POLARIS-02 trial (median follow-up was 387.5 days).3,26 Additional information about the sponsor’s submission is summarized in Appendix 10 of the Supplemental Material document.
CDA-AMC identified several key issues with the sponsor’s analysis (refer to Table 6; full details are provided in Appendix 11 of the Supplemental Material document).
Table 6: Key Issues With the Sponsor’s Economic Submission
Issue | What evidence is there to inform this issue? | How was this issue addressed by CDA-AMC? | Did CDA-AMC explore uncertainty in a scenario analysis? |
|---|---|---|---|
The comparative clinical efficacy and safety of toripalimab is unknown. | In the absence of comparative evidence, the sponsor informed comparative clinical efficacy using a naive approach.26 Thus, the comparative results predicted from the model are associated with considerable uncertainty due to the limitations of a single-arm trial and the absence of long-term extensions, studies addressing gaps, and ITC data. | CDA-AMC could not address this issue owing to a lack of long-term comparative data. | A scenario analysis was conducted that assumed the efficacy of toripalimab is equal to that of pembrolizumab vs. chemotherapy from the KEYNOTE-122 trial. |
Nonrelevant comparators were included in the model. | The sponsor included chemotherapy, pembrolizumab, and nivolumab as comparators in its submitted model.26 The feedback received by CDA-AMC from the drugs plans and clinical experts noted that pembrolizumab and nivolumab are not publicly available across Canada in the clinical setting. | Only chemotherapy was maintained as a relevant comparator. | No scenario analysis was conducted. |
The cost of chemotherapy was uncertain. | Unit costs for carboplatin, cisplatin, docetaxel, and paclitaxel were informed by a medication list from L’Association québécoise des pharmaciens propriétaires.27 These costs were not aligned with the unit costs available in the IQVIA DeltaPA database.28 Additionally, the sponsor assumed a dose of 569 mg for carboplatin; however, this may underestimate the dose of carboplatin, which may be adjusted based on renal function and varies. | CDA-AMC updated the unit costs for carboplatin, cisplatin, docetaxel, and paclitaxel. Additionally, a full dose of carboplatin was assumed. | No scenario analysis was conducted. |
The health state utility values were uncertain and the model structure, which assumed no decrease in quality of life upon progression, lacks face validity. | The sponsor included only 1 health state to estimate LYs and QALYs and a single utility value derived from head and neck cancer.26 This implies that patients experience no change in quality of life if they experience progression after 2L+ treatment. The clinical expert feedback received by CDA-AMC noted that this lacks face validity and, at this stage of the disease and their expected frailty upon progression after 2L+ treatments, patients may experience an even further decline in quality of life than the decline observed upon progression to 1L treatments. Additionally, the preference would be to use utility values from patients with NPC. | CDA-AMC used a utility value of 0.57 at model entry and a 25% decrease in utility was assumed upon progression, as estimated by Yen et al. (2009)29 based on survey data obtained from oncologists and patients with recurrent NPC. | No scenario analysis was conducted. |
Treatment waning for toripalimab was excluded. | The sponsor’s model structure failed to allow for treatment-effect waning with toripalimab. The clinical expert feedback noted there is no evidence that may support the assumption of an indefinite treatment effect. In the absence of clinical evidence informing treatment-effect waning for toripalimab, the degree and magnitude of waning are unknown. | CDA-AMC could not address this issue owing to the structure of the sponsor’s model. | No scenario analysis was conducted. |
The model lacked transparency due to poor modelling practice. | The submitted model had numerous IFERROR statements that resulted in automated overwriting errors. | CDA-AMC could not address this issue owing to the structure of the sponsor’s model. | No scenario analysis was conducted. |
1L = first line; 2L+ = second or subsequent line; CDA-AMC = Canada’s Drug Agency; ITC = indirect treatment comparison; LY = life-year; NPC = nasopharyngeal carcinoma; PFS = progression-free survival; QALY = quality-adjusted life-year; vs. = versus.
Note: Full details of the issues identified by CDA-AMC are provided in Appendix 11 of the Supplemental Material document.
CDA-AMC Assessment of Cost-Effectiveness
The CDA-AMC base case was derived by making changes to model parameter values and assumptions (refer to Appendix 11, Table 15 of the Supplemental Material document), in consultation with clinical experts. Detailed information about the CDA-AMC base case is provided in Appendix 11 of the Supplemental Material document.
Impact on Health Care Costs
Toripalimab is predicted to be associated with additional health care costs compared to chemotherapy (incremental costs = $89,608). This increase in health care spending results primarily from the drug acquisition costs associated with toripalimab (refer to Figure 1).
Impact on Health
Relative to chemotherapy, toripalimab is predicted to extend OS by 0.54 years (refer to Figure 2). Considering the impact of treatment on both the quality and length of life, toripalimab is predicted to result in an additional 0.14 QALYs per patient compared to chemotherapy. More than 96% of the predicted incremental benefit was accrued on the basis of extrapolation.
Figure 1: Impact of Toripalimab vs. Chemotherapy on Health Care Costs
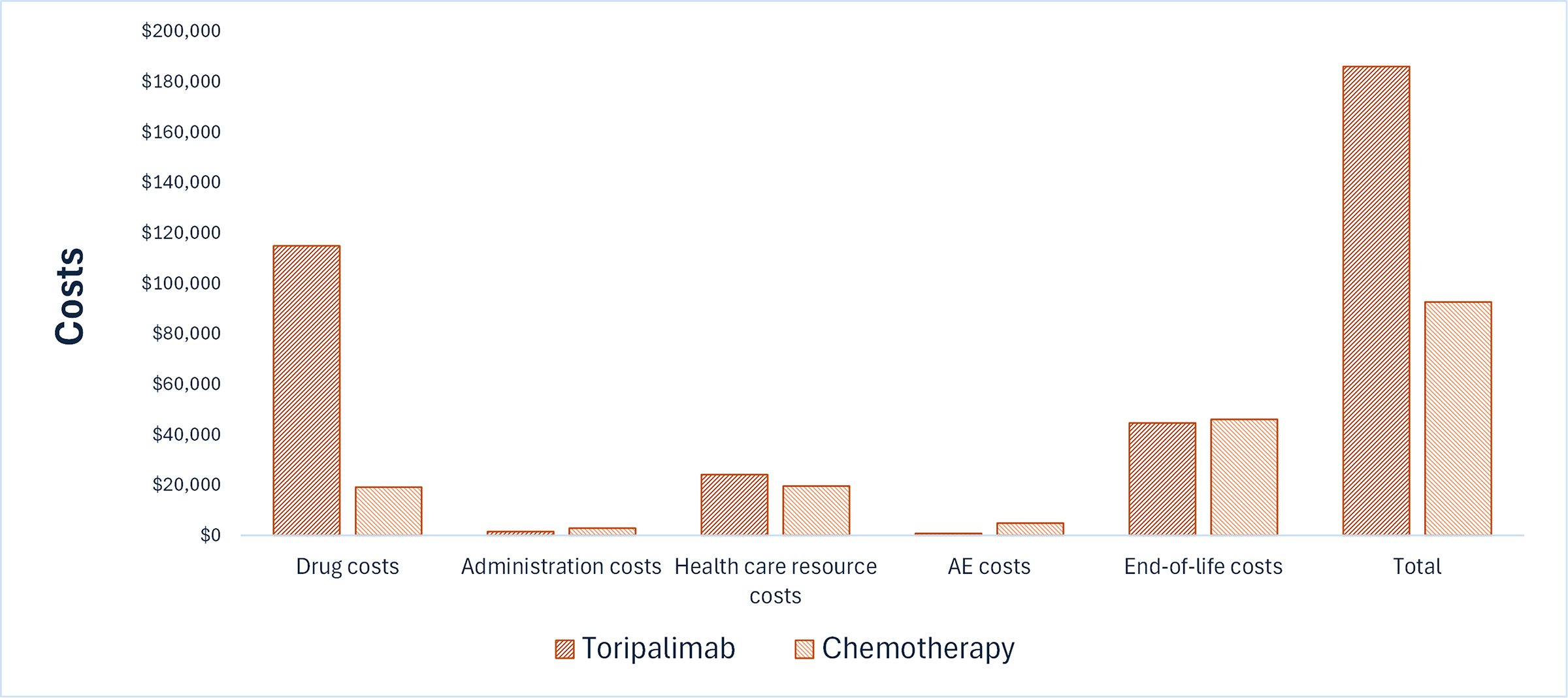
AE = adverse events; vs. = versus.
Figure 2: Impact of Toripalimab vs. Chemotherapy on Patient Health
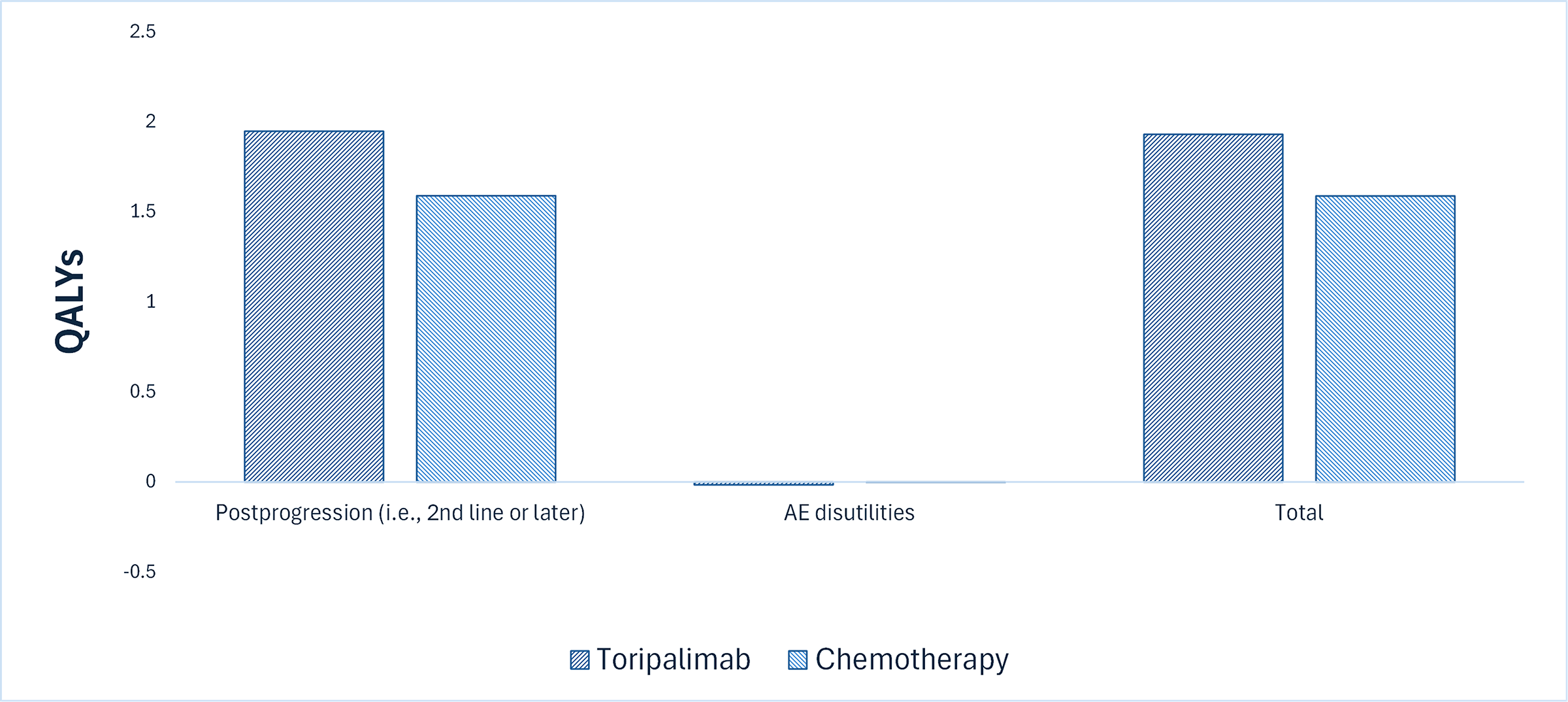
AE = adverse event; QALY = quality-adjusted life-year; vs. = versus.
Overall Results
The results of the CDA-AMC base case suggest an ICER of $634,484 per QALY gained for toripalimab compared to chemotherapy (refer to Table 7). Additional details on the CDA-AMC base case are available in Appendix 11 of the Supplemental Material document.
Table 7: Summary of CDA-AMC Economic Evaluation Results
Drug | Total costs ($) | Total LYs | Total QALYs | ICER vs. chemotherapy ($/QALY) |
|---|---|---|---|---|
Chemotherapy | 89,428 | 2.16 | 1.07 | Reference |
Toripalimab | 179,037 | 2.70 | 1.21 | 634,484 |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year; vs. = versus.
Note: Publicly available list prices were used for all comparators.
Uncertainty and Sensitivity
Uncertainty was explored in the scenario analyses outlined in Appendix 11 of the Supplemental Material document. There was uncertainty around the comparative efficacy against chemotherapy; thus, we assumed the efficacy of toripalimab was equal to that of pembrolizumab from the KEYNOTE-122 trial (refer to Appendix 11 of the Supplemental Material document).
Summary of the Budget Impact
The sponsor submitted a budget impact analysis to estimate the 3-year (2026 to 2028) budget impact of reimbursing toripalimab for the treatment of adults with R/M NPC with disease progression while on treatment or after treatment with a platinum-containing chemotherapy.2,29 The sponsor assumed that the payer would be the CDA-AMC–participating public drug plans and derived the size of the eligible population using an epidemiologic approach.29 The price of toripalimab was aligned with the price included in the sponsor’s economic evaluation, while the prices of comparators were based on the publicly available list prices.2 Additional information pertaining to the sponsor’s submission is provided in Appendix 12 of the Supplemental Material document.
CDA-AMC identified a number of issues with the sponsor’s estimated budget impact and made changes to model parameters and assumptions in consultation with the clinical experts to derive the CDA-AMC base case (Appendix 12 of the Supplemental Material document). CDA-AMC estimated that 86 patients would be eligible for treatment with toripalimab over a 3-year period (year 1 = 28; year 2 = 29; year 3 = 29), of whom 31 would be expected to receive toripalimab (year 1 = 22; year 2 = 6; year 3 = 3). The estimated incremental budget impact of reimbursing toripalimab is predicted to be approximately $3.0 million over the first 3 years, with an expected expenditure of $3.4 million on toripalimab. The actual budget impact will depend on the market uptake of Loqtorzi after the first year of its implementation, whether it is also available for first-line treatment, and the extent to which it will be used for patients previously exposed to toripalimab, pembrolizumab, or nivolumab.
Conclusion
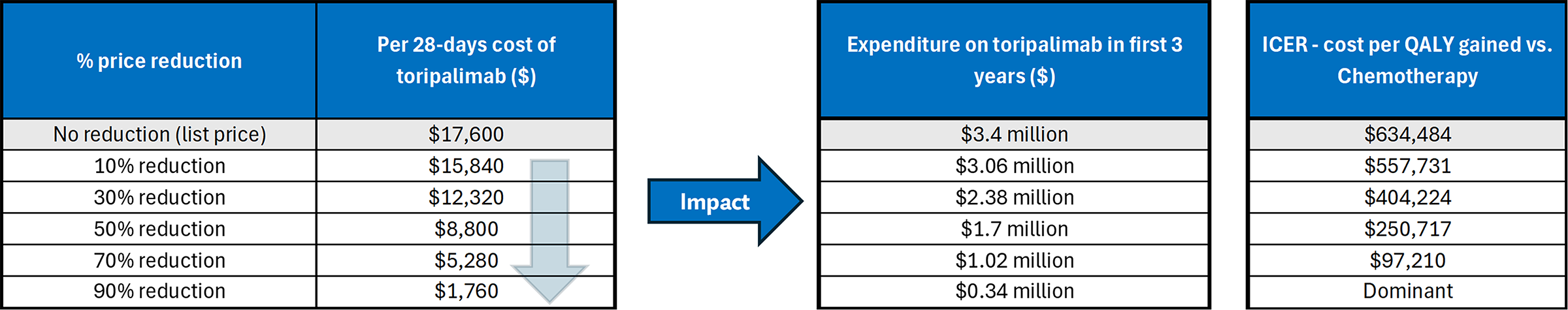
Based on the CDA-AMC base case, toripalimab would be considered cost-effective at the submitted price if the public health care system was willing to pay at least $634,484 for each additional QALY gained. If the public health care system is not willing to pay that amount, a price reduction should be considered (refer to Figure 3; full details of the impact of price reductions on cost-effectiveness are presented in Appendix 11, Table 18 of the Supplemental Material document). However, the estimated cost-effectiveness of toripalimab compared to chemotherapy is highly uncertain due to the lack of comparative evidence between toripalimab and chemotherapy, uncertainty in the long-term survival estimates, and the structure of the submitted economic analysis. Therefore, higher price reductions may be required.
The budget impact of reimbursing toripalimab to the public drug plans in the first 3 years is estimated to be approximately $3.0 million. The 3-year expenditure on toripalimab (i.e., not accounting for current expenditures on comparators) is estimated to be $3.4 million. The estimated budget impact is uncertain due to uncertainty associated with the market uptake of toripalimab after the first year of its implementation, whether it is also available for first-line treatment, and the extent to which it will be used for patients previously exposed to toripalimab, pembrolizumab, or nivolumab.
References
1.Apotex Inc. Loqtorzi (toripalimab solution for injection): Sterile solution, 240 mg/6 mL (40 mg/mL) for intravenous infusion after dilution [product monograph]. October 31, 2024.
2.Apotex Inc. Drug Reimbursement Review sponsor submission: Loqtorzi (toripalimab solution for injection), Sterile solution, 240 mg/6 mL (40 mg/mL) for intravenous infusion after dilution [internal sponsor's package]. July 16, 2025.
3.Shanghai Junshi Bioscience Co., Ltd. Clinical Study Report: JS00l-lb-CRP-1.0. A Multi-Center, Open-Label Phase lb/II Clinical Study to Evaluate JS00 1 in Patients With Advanced Gastric Adenocarcinoma, Esophageal Squamous Cell Carcinoma, Nasopharyngeal Carcinoma and Head and Neck Squamous Cell Carcinoma - Cohort 3 Nasopharyngeal Carcinoma [internal sponsor's report]. April 08, 2021.
4.Spreafico A, Winquist E, Ho C, et al. A Canadian Perspective on Systemic Therapy for Recurrent or Metastatic Nasopharyngeal Carcinoma. Curr Oncol. 2025;32(1):48. doi:10.3390/curroncol32010048 PubMed
5.Xu T, Tang J, Gu M, Liu L, Wei W, Yang H. Recurrent nasopharyngeal carcinoma: a clinical dilemma and challenge. Curr Oncol. 2013;20(5):e406-19. doi:10.3747/co.20.1456 PubMed
6.Lo AK, Dawson CW, Lung HL, Wong KL, Young LS. The Role of EBV-Encoded LMP1 in the NPC Tumor Microenvironment: From Function to Therapy. Front Oncol. 2021;11:640207. doi:10.3389/fonc.2021.640207 PubMed
7.Shah AB, Nagalli S. Nasopharyngeal Carcinoma [sponsor supplied reference]. StatPearls. 2025.
8.Pathology Outlines. Nasal cavity, paranasal sinuses, nasopharynx [sponsor supplied reference]. Updated December 15, 2020. Accessed June 13, 2025. https://www.pathologyoutlines.com/topic/nasalnasopharyngealgeneral.html
9.Zhao BY, Hirayama S, Goss D, Zhao Y, Faden DL. Human papillomavirus-associated nasopharyngeal carcinoma: A systematic review and meta-analysis. Oral Oncol. 2024;159:107057. doi:10.1016/j.oraloncology.2024.107057 PubMed
10.Wang WH, Lin YC, Chen WC, Chen MF, Chen CC, Lee KF. Detection of mucosal recurrent nasopharyngeal carcinomas after radiotherapy with narrow-band imaging endoscopy. Int J Radiat Oncol Biol Phys. 2012;83(4):1213-9. doi:10.1016/j.ijrobp.2011.09.034 PubMed
11.Ng SH, Chang JT, Ko SF, Wan YL, Tang LM, Chen WC. MRI in recurrent nasopharyngeal carcinoma. Neuroradiology. 1999;41(11):855-62. doi:10.1007/s002340050857 PubMed
12.Liu T, Xu W, Yan WL, Ye M, Bai YR, Huang G. FDG-PET, CT, MRI for diagnosis of local residual or recurrent nasopharyngeal carcinoma, which one is the best? A systematic review. Radiother Oncol. 2007;85(3):327-35. doi:10.1016/j.radonc.2007.11.002 PubMed
13.Cancer Care Alberta. Nasopharyngeal Cancer Treatment [sponsor supplied reference]. 2021. Accessed February 2025. https://www.albertahealthservices.ca/assets/info/hp/cancer/if-hp-cancer-guide-hn003-nasopharyngeal.pdf
14.Abdullah B, Alias A, Hassan S. Challenges in the management of nasopharyngeal carcinoma: a review. Malays J Med Sci. 2009;16(4):50-4. PubMed
15.Li AC, Xiao WW, Shen GZ, et al. Distant metastasis risk and patterns of nasopharyngeal carcinoma in the era of IMRT: long-term results and benefits of chemotherapy. Oncotarget. 2015;6(27):24511-21. doi:10.18632/oncotarget.4312 PubMed
16.Li JS, Blanchard P, Wong CHL, et al. International Recommendations on Postoperative Management for Potentially Resectable Locally Recurrent Nasopharyngeal Carcinoma. Int J Radiat Oncol Biol Phys. 2024;120(5):1294-1306. doi:10.1016/j.ijrobp.2024.07.2143 PubMed
17.Howlett J, Hamilton S, Ye A, et al. Treatment and outcomes of nasopharyngeal carcinoma in a unique non-endemic population. Oral Oncol. 2021;114:105182. doi:10.1016/j.oraloncology.2021.105182 PubMed
18.Liu Q, Li J, Ng WT, Lee AWM. Treatment strategy for de novo metastatic nasopharyngeal carcinoma: a literature review. Chin Clin Oncol. 2023;12(4):43. doi:10.21037/cco-23-32 PubMed
19.Lee AWM, Ng WT, Chan LK, et al. The strength/weakness of the AJCC/UICC staging system (7th edition) for nasopharyngeal cancer and suggestions for future improvement. Oral Oncol. 2012;48(10):1007-1013. doi:10.1016/j.oraloncology.2012.03.022
20.World Health Organization International Agency for Research on Cancer. Cancer Today [sponsor supplied reference]. Updated August 2, 2024. Accessed February 11, 2025. https://gco.iarc.who.int/today/en/dataviz/maps-heatmap?mode=population&cancers=4&zoom=3
21.Statistics Canada. Table 13-10-0747-01, Number of new cases and age-standardized rates of primary cancer, by cancer type and sex [sponsor supplied reference]. 2025. https://doi.org/10.25318/1310074701-eng
22.Young TK, Kelly JJ, Friborg J, Soininen L, Wong KO. Cancer among circumpolar populations: an emerging public health concern. Int J Circumpolar Health. 2016;75:29787. doi:10.3402/ijch.v75.29787 PubMed
23.Badoual C. Update from the 5th Edition of the World Health Organization Classification of Head and Neck Tumors: Oropharynx and Nasopharynx. Head Neck Pathol. 2022;16(1):19-30. doi:10.1007/s12105-022-01449-2
24.Peterson BR, Nelson BL. Nonkeratinizing undifferentiated nasopharyngeal carcinoma. Head Neck Pathol. 2013;7(1):73-5. doi:10.1007/s12105-012-0401-4 PubMed
25.Chan ATC, Lee VHF, Hong RL, et al. Pembrolizumab monotherapy versus chemotherapy in platinum-pretreated, recurrent or metastatic nasopharyngeal cancer (KEYNOTE-122): an open-label, randomized, phase III trial. Ann Oncol. 2023;34(3):251-261. doi:10.1016/j.annonc.2022.12.007 PubMed
26.Apotex Inc. Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Loqtorzi (toripalimab solution for injection), Sterile solution, 240 mg/6 mL (40 mg/mL) for intravenous infusion after dilution. July 16, 2025.
27.Association québécoise des pharmaciens propriétaires. AQPP’s Medication List [sponsor supplied reference]. 2024. https://www.monpharmacien.ca/en/aqpp/aqpps-medication-list/
28.IQVIA. DeltaPA. 2025. Accessed August 26, 2025. https://www.iqvia.com/
29.Apotex Inc. Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Loqtorzi (toripalimab solution for injection), Sterile solution, 240 mg/6 mL (40 mg/mL) for intravenous infusion after dilution. July 16, 2025.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.