Drugs, Health Technologies, Health Systems
Reimbursement Review
Atezolizumab (Tecentriq)
Sponsor: Hoffmann-La Roche Limited
Therapeutic area: Metastatic non–small cell lung cancer
Summary
What Is Non–Small Cell Lung Cancer?
Non–small cell lung cancer (NSCLC) is the most common type of lung cancer, accounting for nearly 90% of all lung cancers in Canada. In those with NSCLC, unusual growth of cells takes place inside the lungs or lining of the airways and forms into tumours. Cancer that is metastatic has spread to other parts of the body.
In 2024, it was estimated that there would be 32,100 new cases of lung cancer diagnosed and 20,700 deaths from lung cancer in Canada.1
What Are the Treatment Goals and Current Treatment Options for NSCLC?
The goals of first-line treatment of metastatic NSCLC are to improve survival, delay progression of cancer, decrease cancer-related symptoms, and improve quality of life.
In Canada, immunotherapy is considered the preferred first-line treatment of metastatic NSCLC without EGFR mutations or ALK aberrations and with high PD-L1 expression (PD-L1 expression on ≥ 50% of tumour cells [TCs]). Available options include pembrolizumab (funded by all participating drug programs) and cemiplimab (funded only in Ontario at the time of this review).2-4 Platinum-based chemotherapy alone is also a potential first-line treatment option; however, clinical experts consulted by Canada’s Drug Agency (CDA-AMC) noted that this would be reserved for patients with significant contraindications to immunotherapy and that very few patients would receive this option.
What Is Tecentriq and Why Did CDA-AMC Conduct This Review?
Tecentriq (atezolizumab) is a drug that is administered by IV infusion or subcutaneous (SC) injection. Health Canada has approved Tecentriq for use as monotherapy for the first-line treatment of patients with metastatic NSCLC whose tumours have high PD-L1 expression (PD-L1 staining in ≥ 50% of TCs or PD-L1-stained tumour-infiltrating immune cells [ICs] covering ≥ 10% of the tumour area), as determined by a validated test and who do not have EGFR or ALK genomic tumour aberrations.
CDA-AMC reviewed Tecentriq to inform a recommendation to the participating public drug programs on whether it should be reimbursed for use as monotherapy for the first-line treatment of patients with metastatic NSCLC whose tumours have high PD-L1 expression (PD-L1 staining in ≥ 50% of TCs or PD-L1 staining in tumour-infiltrating ICs covering ≥ 10% of the tumour area), as determined by a validated test and who do not have EGFR or ALK genomic tumour aberrations. The sponsor is seeking reimbursement for this patient population.
How Did CDA-AMC Evaluate Tecentriq?
Tecentriq was reviewed through a tailored review process that focuses on whether the drug under review has similar beneficial and harmful effects to the most appropriate treatments for comparison.
The review was informed by materials submitted by the sponsor, which included clinical and economic evidence.
The review was also informed by 1 patient group submission (jointly submitted by Lung Cancer Canada, the Lung Health Foundation, and the Canadian Cancer Survivor Network) and 2 clinician group submissions in response to our call for input and by input from the participating public drug programs around issues that may impact their ability to implement a recommendation.
Two clinical specialists with expertise in the management of NSCLC were consulted as part of the review process.
What Did CDA-AMC Find?
Clinical Evidence
We reviewed the following clinical evidence:
One randomized controlled phase III trial (IMpower110) compared atezolizumab with platinum-based chemotherapy in 572 patients (285 in the atezolizumab group and 287 in the chemotherapy group). The overall trial population of the IMpower110 trial was broader than the approved indication; therefore, the sponsor submitted subgroup data for patients with high PD-L1 expression (i.e., PD-L1 stained at least 50% of TCs or PD-L1 stained tumour-infiltrating ICs covering at least 10% of the tumour area) (107 in the atezolizumab group and 98 in the chemotherapy group). For the comparison of atezolizumab versus platinum-based chemotherapy, evidence from the IMpower110 trial demonstrated that atezolizumab results in clinically meaningful improvements compared with chemotherapy in outcomes that are relevant for the management of NSCLC in the first-line metastatic setting, including overall survival, progression-free survival, and objective response.
One network meta-analysis compared atezolizumab versus pembrolizumab, cemiplimab, and platinum-based chemotherapy. The estimates of effect from the sponsor’s network meta-analysis were limited by imprecision (i.e., wide credible intervals [CrIs]), making it challenging to evaluate if there are meaningful differences across the different PD-1 and PD-L1 inhibitors approved for use in the target population (i.e., atezolizumab, pembrolizumab, and cemiplimab). Although the evidence submitted by the sponsor does not support any definitive conclusions about the efficacy or safety of atezolizumab relative to pembrolizumab or cemiplimab, the clinical experts suggested that atezolizumab may present an additional treatment option for patients with metastatic NSCLC with high PD-L1 expression, particularly given the availability of a SC formulation. Based on their experience, they considered the adverse effects of different immune checkpoint inhibitors to be similar.
The clinical experts consulted by CDA-AMC, patient groups, and clinician groups all noted that reimbursement of atezolizumab would be beneficial as it would provide an SC treatment option for this patient population.
Economic Evidence
Tecentriq is available as a solution for IV infusion (submitted price: $4,743 per 840 mg vial or $6,776 per 1,200 mg vial) or SC injection (submitted price: $6,453 per 1,875 mg vial). Based on Health Canada–recommended dosages and sponsor-submitted prices, the 28-day cost of Tecentriq SC is expected to be $8,604 per patient and the 28-day cost of Tecentriq IV is expected to be $9,035 to $9,486 per patient, depending on dosage regimen.
Based on the sponsor’s submitted prices for atezolizumab IV and SC and publicly available list prices of all comparators, reimbursement of atezolizumab is expected to be associated with lower drug costs to the health care system versus cemiplimab and fixed-dose pembrolizumab (incremental savings: $1,447 to $3,129 per 28-day cycle) and higher or lower costs versus weight-based pembrolizumab (incremental savings: $313 to incremental cost: $569 per 28-day cycle).
CDA-AMC estimates that reimbursement of atezolizumab for use in the indicated population may result in cost savings for the public drug plans (approximately $3.5 million over the first 3 years of reimbursement) compared to the amount currently spent on comparators, with an estimated expenditure of $24 million on atezolizumab over this period. However, whether the reimbursement of atezolizumab is cost saving, and the extent of any savings, will depend on the number of people eligible for treatment, market uptake of atezolizumab, and confidentially negotiated prices for comparators. The magnitude of uncertainty in the budget impact must be addressed to ensure the feasibility of adoption, given the difference between the sponsor’s estimate and the CDA-AMC estimate.
Abbreviations
AE
adverse event
AESI
adverse event of special interest
CCOD
clinical cut-off date
CI
confidence interval
CrI
credible interval
ECOG
Eastern Cooperative Oncology Group
HR
hazard ratio
IC
tumour-infiltrating immune cell
IHC
Immunohistochemistry
ITC
indirect treatment comparison
KM
Kaplan-Meier
NMA
network meta-analysis
NPT
nonprotocol therapy
NSCLC
non–small cell lung cancer
ORR
objective response rate
OS
overall survival
PFS
progression-free survival
RPSFT
rank-preserving structural failure time
SAE
serious adverse event
SC
subcutaneous
TC
tumour cell
WT
wild type
Background
Sponsor’s Summary of Disease Background and Current Management
Application Summary
Table 1: Application Submitted for Review
Item | Description |
|---|---|
Drug (product) | Tecentriq (atezolizumab)
|
Sponsor | Hoffmann-La Roche Limited |
Health Canada indication | Tecentriq (atezolizumab) as monotherapy for the first-line treatment of patients with metastatic NSCLC whose tumours have high PD-L1 expression (PD-L1 stained ≥ 50% of TCs or PD-L1 stained tumour-infiltrating immune cells [ICs] covering ≥ 10% of the tumour area), as determined by a validated test and who do not have EGFR or ALK genomic tumour aberrations. |
Sponsor’s reimbursement request | Tecentriq (atezolizumab) as monotherapy for the first-line treatment of patients with metastatic NSCLC whose tumours have high PD-L1 expression (PD-L1 stained ≥ 50% of TCs or PD-L1 stained tumour-infiltrating immune cells [ICs] covering ≥ 10% of the tumour area), as determined by a validated test and who do not have EGFR or ALK genomic tumour aberrations. |
Health Canada approval status | Approved with NOC |
Health Canada review pathway | Standard review |
NOC date | Tecentriq (atezolizumab) IV on March 1, 2021 Tecentriq (atezolizumab) SC on March 13, 2024 |
ALK = anaplastic lymphoma kinase, EGFR = epidermal growth factor receptor, IC = immune cell, NOC = Notice of Compliance, NSCLC = non–small cell lung cancer, PACES = Pharmaceuticals With Anticipated Comparable Efficacy and Safety, PD-L1 = programmed death ligand 1, TC = tumour cell.
Disease Background
Lung cancer is one of the leading causes of cancer-associated mortality in Canada; it is estimated that lung cancer accounted for approximately one-quarter of all cancer-related deaths.5,6 In Canada, over half of NSCLC cases (53%) are metastatic (stage IV) which is no longer treatable with curative intent.7 The 5-year relative survival rate for metastatic NSCLC is only 3%.8,9
Lung cancer is classified based upon its histology, and is comprised of two main forms: non small-cell lung cancer (NSCLC) and small-cell lung cancer (SCLC).5 NSCLC is the predominant subtype, and can be further categorized into two major histologic subtypes: non-squamous carcinomas (including adenocarcinomas) and squamous cell carcinomas.10 In Canada, NSCLC accounts for approximately 88% of all lung cancer cases.5 Distinct subsets of NSCLC can also harbour driver mutations such as the activation mutation of Epidermal Growth Factor Receptor (EGFR) tyrosine kinase and the rearrangement of the anaplastic lymphoma kinase (ALK) or the c-ROS proto-oncogene 1 (ROS1) gene.11 High programmed death ligand 1 (PD-L1) expression on Tumour cells (TCs) or Immune cells (ICs) may indicate an increased magnitude of response to cancer immunotherapy.12-15 Approximately 1 in 3 patients have high PD-L1 expression.16
A definitive diagnosis of lung cancer can involve a chest x-ray and computed tomography (CT) scan.17 Tissue biopsy specimens can be taken from the lungs and mediastinal lymph nodes to enable more accurate diagnosis and staging, as well as histological and biomarker analyses.18 Determination of PD-L1 expression level in patients is identified using a validated immunohistochemical (IHC) assay in order to determine suitability for checkpoint inhibitor monotherapy.19 Molecular testing for oncogenic drivers is typically performed using a multigene panel.20 Both of these assays are broadly available in Canada. The diagnostic tests for lung cancer are well established and broadly available in Canada.
Current Management and Place in Therapy of the Drug Under Review
Current Treatment Options
The goal of lung cancer treatment in the metastatic setting is to prolong survival while also maintaining quality of life.21 For NSCLC patients undergoing first-line (1L) therapy with high PD-L1 expression and who do not harbour an oncogenic driver, the standard of care typically involves an anti PD-L1 monotherapy.11 CDA-AMC has recommended two 1L monotherapy options: pembrolizumab and cemiplimab. Currently, pembrolizumab represents the standard of care and cemiplimab is expected to be funded in the near future. In patients with significant contraindications to immunotherapy, platinum-based chemotherapy is a 1L treatment option.11
Impact of the Drug Under Review on Treatment Options
Anti-PD-L1 agents are an already established therapy for metastatic NSCLC adopted by many clinicians.11 Based on the results of the IMpower110 and indirect comparisons, the clinical experts believed that atezolizumab would offer a comparable clinical benefit, as well as safety profile to other recommended IO monotherapy options.22,23 Atezolizumab monotherapy is not expected to replace any existing treatments, but rather be added as an additional treatment option that may be easily adopted into the system – atezolizumab can be easily integrated into the current diagnosis and treatment of NSCLC. Overall, atezolizumab could present as a new first-line treatment option for all eligible patients with PD-L1 high metastatic NSCLC without EGFR or ALK driver mutations. Key characteristics of atezolizumab are summarized with other treatments available for 1L treatment of patients with metastatic NSCLC whose tumours have high PD-L1 expression and who do not have EGFR or ALK genomic tumour aberrations in the Supplemental Materials.
CDA-AMC Sources of Information and Summary of Input
This section was prepared by CDA-AMC based on materials submitted by the sponsor and input from interested parties.
The objective of the clinical review is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of Tecentriq (atezolizumab) IV (1,200 mg/20 mL and 840 mg/14 mL single use vials for IV infusion) and the subcutaneous (SC) formulation (1,875 mg/15 mL single use vial for SC injection) for use as monotherapy for the first-line treatment of patients with metastatic NSCLC whose tumours have high PD-L1 expression (PD-L1 staining in ≥ 50% of TCs or PD-L1 staining in tumour-infiltrating ICs covering ≥ 10% of the tumour area), as determined by a validated test, and who do not have EGFR or ALK genomic tumour aberrations.
The focus will be on comparing atezolizumab to relevant comparators in clinical practice in Canada and identifying gaps in the current evidence. This application has been submitted through the tailored review process; the focus of these reviews is whether the evidence supports that the drug under review demonstrates comparable clinical benefit and harms versus appropriate comparators, as well as a comparison of drug acquisition costs and budget impact analysis. The comparators considered relevant to this review were Keytruda (pembrolizumab) and Libtayo (cemiplimab).
Sources of Information
This report is informed by materials submitted by the sponsor and input received from interested parties. Calls for patient group and clinician group input are issued for each reimbursement review. We received 1 jointly submitted patient group submission from Lung Cancer Canada, the Lung Health Foundation, and the Canadian Cancer Survivor Network, and 2 clinician group submissions from the Lung Cancer Canada Medical Advisory Committee and the Ontario Health (Cancer Care Ontario) Lung Cancer Drug Advisory Committee.
The information submitted by the patient groups was collected through telephone interviews with patients and caregivers (5 interviews), an anonymous treatment-experience survey (74 respondents), and information from previous submissions to CDA-AMC and the Institut national d'excellence en santé et en services sociaux. For the patient interviews, 1 patient had previous experience with atezolizumab for the indication under review; the others had experience with atezolizumab for other indications (i.e., small cell lung cancer or adjuvant NSCLC).
The full submissions received are available on the project landing page in the consolidated input document. The drug programs provide input on each drug being reviewed through the reimbursement review process by identifying issues that may impact their ability to implement a recommendation.
Input from patient and clinician groups is considered throughout the review, including in the selection of outcomes to include in the clinical review and in the interpretation of the clinical evidence. Relevant patient and clinician group input is summarized in the Summary of Input section.
Each review team includes at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process. Two clinicians with expertise in the diagnosis and management of lung cancer participated as part of the review team, with representation from British Columbia and Ontario.
Summary of Input
Disease Background and Impacts of the Disease
The sponsor has provided an overview of the condition. Patient groups describe the diagnosis of metastatic lung cancer as a devastating and frightening experience for them and their loved ones. Patients describe a variety of troubling emotional and physical symptoms. Commonly reported physical symptoms included fatigue, shortness of breath, coughing, and pain. Patients reported negative and challenging impacts on their emotional well-being and family relationships. Feelings of isolation were common, as was the sense that they spend too much time at medical appointments.
Treatment Goals
Patients and clinicians describe the goals of treatment as extending survival, slowing disease progression, improving symptoms, and helping patients maintain their quality of life. Additional treatment options are valued as they may provide another option if alternative treatments are not effective or cannot be tolerated by the patient.
Treatment Options
There was consensus from the clinical specialists and clinician groups that monotherapy with pembrolizumab is the preferred first-line treatment option for the target patient population (recommended by CDA-AMC in August 2017; the pan-Canadian Pharmaceutical Alliance [pCPA] agreement was reached in November 2017). In June 2022, CDA-AMC delivered a recommendation in favour of reimbursing monotherapy with cemiplimab. An agreement was reached with the pCPA in March 2025, and 3 jurisdictions (Newfoundland and Labrador, Nova Scotia, and Ontario) are now reimbursing cemiplimab for the target population. Clinicians noted that platinum doublet chemotherapy and pembrolizumab, or platinum doublet chemotherapy and cemiplimab, can be considered for some patients, but these regimens were not considered to be comparators for this review, which is focused on the use of PD-1 or PD-L1 inhibitors as monotherapy.
Unmet Needs and Existing Challenges
The availability of a PD-1 or PD-L1 inhibitor that can be administered subcutaneously is a key unmet need identified by patients and clinicians that could be addressed by the reimbursement of atezolizumab for those living with metastatic NSCLC who are eligible for treatment in the first-line setting.
Considerations for Using the Drug Under Review
The contents within this section have been informed by input from the clinical experts consulted for the purpose of this review and from clinician groups. The implementation questions from the public drug programs and corresponding responses from the clinical experts consulted for this review are summarized in the Summary of Drug Program Input and Clinical Expert Responses table in Appendix 2 in the Supplemental Material document. The following has been summarized by CDA-AMC.
Place in Therapy
Atezolizumab is expected to be an additional option for those who receive first-line immuno-oncology monotherapy (the alternatives being pembrolizumab and cemiplimab). Given that atezolizumab is available as an SC formulation, it may have the potential to address an unmet need for patients who struggle to access IV infusion centres due to travel requirements or have challenges receiving or tolerating IV-administered therapies. The clinical experts consulted by CDA-AMC noted that many patients strongly prefer SC administration over IV administration when given the option. Some patients also prefer the shorter administration time with an SC formulation (though some noted the slower IV infusion was more tolerable).
Patient Population
As shown in Table 2, there is variation across the Health Canada–approved indications for atezolizumab, pembrolizumab, and cemiplimab with respect to the scope of the patient population, PD-L1 staining techniques used to assess eligibility, and exclusionary biomarkers.
Table 2: Comparison of Indicated Patient Populations
Characteristics | Atezolizumab | Pembrolizumab | Cemiplimab |
|---|---|---|---|
Disease stage |
|
|
|
Biomarkers (inclusion) | Patient must have:
| Patient must have:
| Patient must have:
|
Biomarkers (exclusion) | Patient must not have any:
| Patient must not have any:
| Patient must not have any:
|
IC = tumour-infiltrating immune cell; TC = tumour cell.
Disease Stage
There is variation across the Health Canada–approved indications for atezolizumab, pembrolizumab, and cemiplimab with respect to the scope of the patient population. The indication for atezolizumab is limited to patients with metastatic disease; whereas the indications for pembrolizumab and cemiplimab also include use in patients with stage III or locally advanced disease (respectively) who are not candidates for surgical resection or definitive chemoradiation. The scope of the CDA-AMC review is limited to the indication that has been approved by Health Canada and, therefore, does not include patients with stage III disease or locally advanced disease. However, the clinical experts consulted by CDA-AMC noted it could be considered appropriate to use atezolizumab for the same patient populations as currently reimbursed for pembrolizumab and cemiplimab for this disease setting.
Assessment of PD-L1 Status
There is variation in PD-L1 staining techniques across the approved indications for atezolizumab, pembrolizumab, and cemiplimab. All 3 indications state that those who have PD-L1 staining on at least 50% of TCs may be eligible for the treatment.24,25 However, the indication for atezolizumab allows eligibility if they have PD-L1 stained tumour-infiltrating immune cells covering at least 10% of the tumour area.26 The difference in indications appears to be due to the use of different PD-L1 testing kits applied in the clinical development programs — the IMpower110 trial (atezolizumab) used the Ventana (SP142) assay, whereas PD-L1 staining can be done on both TCs and tumour-infiltrating immune cells;26,27 and the pembrolizumab and cemiplimab pivotal trials (KEYNOTE-024 and EMPOWER-LUNG 1, respectively) both used the IHC 22C3 pharmDx assay, whereas PD-L1 staining is only done on TCs.24,25 The product monographs for all of the drugs do not mandate any particular test be used in the assessment of PD-L1 status, only that the assessment be done using a validated test. The clinical experts consulted by CDA-AMC emphasized that there is a preference to use PD-L1 staining on at least 50% of TCs to identify patients eligible for PD-L1 and PD-1 inhibitors, and that the PD-L1 IHC 22C3 pharmDx assay is generally the preferred test used in Canada. The experts noted that adoption of alternative testing strategies would pose implementation challenges.
Exclusion of EGFR, ALK, or ROS1 Aberrations
There is variation across the product monographs regarding the number of oncogenic mutations that are listed as exclusion criteria (i.e., atezolizumab and pembrolizumab list EGFR and ALK, and cemiplimab also lists ROS1).24-26 The clinical experts consulted by CDA-AMC noted that for patients with actionable oncogenic mutations, the use of targeted therapy is typically the preferred option. Consequently, although not excluded from the indication approved for atezolizumab, the clinical experts consulted by CDA-AMC noted patients with a ROS1 mutation would likely receive a targeted therapy as opposed to a PD-L1 or PD-1 inhibitor in the first-line metastatic setting (i.e., target population for this review).
Assessing Treatment Response
Previous CDA-AMC recommendations for pembrolizumab and cemiplimab have indicated that assessments should be performed based on clinical and radiographic evaluation every 3 to 4 months and that patients should be eligible to have reimbursement of the treatment renewed (i.e., continued), provided they demonstrate a continued response to treatment, defined as absence of disease progression.28,29 The clinical experts consulted by CDA-AMC noted that these reimbursement conditions would be appropriate for atezolizumab.
Discontinuing Treatment
There are differences between product monographs for atezolizumab, pembrolizumab, and cemiplimab with respect to the recommendations for the maximum number or cycles and maximum duration of therapy. The product monographs for atezolizumab, pembrolizumab, and cemiplimab all recommend discontinuation in the event of unacceptable toxicity.
Maximum Number of Cycles
The product monograph for pembrolizumab specifies a maximum number of cycles for those without disease progression (up to 24 months or 35 doses for 200 mg or 18 doses for 400 mg, whichever is longer).24 This is reflected in the CDA-AMC recommendation for the indication of interest for this (and others that have been issued for pembrolizumab). The product monograph for cemiplimab does not recommend a maximum number of cycles, but the recommendation from CDA-AMC specified a maximum of 108 weeks based on the upper limit of cycles administered in the EMPOWER-LUNG 1 trial.25,29 The product monograph for atezolizumab does not specify a maximum number of doses, and the IMpower110 clinical trial did not stop administration after a maximum number of cycles.27,30 The clinical experts consulted by CDA-AMC noted that continuation of treatment with atezolizumab beyond the maximum durations recommended for pembrolizumab and cemiplimab (e.g., 24 and 27 months, respectively) would be appropriate for those who are responding and continuing to benefit from the treatment in the opinion of the clinician.
Re-Treatment
Existing CDA-AMC recommendations for pembrolizumab and cemiplimab have included implementation guidance that patients could receive re-treatment with those products in 1 of the following scenarios:
patient discontinues treatment before completing the maximum number of cycles (e.g., 2 years of treatment) due to toxicity, but without relapse
patient experiences a relapse after discontinuing therapy following treatment with the maximum number of cycles
patient experiences a relapse after discontinuing therapy before reaching the maximum number of cycles earlier due to a confirmed complete response to the treatment.28,29
In the case of discontinuation due to toxicity, CDA-AMC has previously recommended that patients should be eligible to restart the treatment as long as the toxicity has resolved. In situations where a patient’s disease has relapsed following completion of the maximum number of cycles or earlier discontinuation due to complete response, CDA-AMC has previously recommended that patients should be eligible for re-treatment for up to an additional 1 year. As noted, there is no maximum treatment duration for atezolizumab; however, the clinical experts consulted by CDA-AMC noted that these reimbursement conditions would be appropriate for atezolizumab.
Discontinuation at the Time of Disease Progression
The product monographs for both pembrolizumab and cemiplimab state that treatment should be discontinued at the time of disease progression. In contrast, the product monograph for atezolizumab states that patients may be treated until “loss of clinical benefit,” which refers to the following description from the pivotal clinical trials for the use of atezolizumab as monotherapy (including IMpower110):
absence of symptoms and signs (including worsening of laboratory values [e.g., new or worsening hypercalcemia]) indicating unequivocal progression of disease
no decline in Eastern Cooperative Oncology Group (ECOG) performance status
absence of tumour progression at critical anatomic sites (e.g., leptomeningeal disease) that cannot be readily managed and stabilized by protocol-allowed medical interventions before repeat dosing
evidence of clinical benefit as assessed by the investigator.26
In their economic submission to CDA-AMC, the sponsor assumed that the treatment would be discontinued at the time of disease progression in the same manner as pembrolizumab and cemiplimab.
The clinical experts consulted by CDA-AMC noted that continuation of treatment with atezolizumab after disease progression could be appropriate for patients who are continuing to benefit, in the opinion of the clinician.
Prescribing Considerations
In line with previous CDA-AMC recommendations, atezolizumab should be prescribed by clinicians with expertise and experience in treating NSCLC and the treatment should be supervised and delivered in outpatient specialized oncology clinics with expertise in systemic therapy delivery and management of immunotherapy-related side effects.
These criteria were supported by input from the Lung Cancer Canada Medical Advisory Committee, which also noted that the SC formulation of atezolizumab would have the added benefit of being administered in infusion clinics outside of the hospital settings (particularly in rural communities), but the monitoring of patients would still be done by medical oncologists.
Sponsor’s Summary of the Systematic Review Evidence
Objective and Methods
The objective is to perform a systematic review of the beneficial and harmful effects of atezolizumab for first-line treatment of patients with metastatic NSCLC whose tumours have high PD-L1 expression as determined by a validated test and who do not have EGFR or ALK genomic tumour aberrations versus relevant comparators in clinical practice in Canada. Refer to the Supplemental Materials for details on the systematic review protocol, literature search strategy, study selection process and the list of excluded studies.
Included Studies
Table 3: Details of Included Studies
Item | IMpower110 |
|---|---|
Study design and population | |
Study design | Global, randomized, open-label, phase 3 trial |
Locations | Patients were recruited from 144 centers across 19 countries in North America, Europe and the Middle East, and Asia-Pacific. There were no Canadian sites. |
Patient enrolment Dates: | Start date: 21 July 2015 End date: 20 February 2018 |
Randomized (N) | 572 randomized patients in total (287 in the chemotherapy arm and 285 in the atezolizumab arm). |
Key inclusion criteria |
|
Key exclusion criteria |
|
Drugs | |
Intervention | Atezolizumab 1200 mg intravenously once every 3 weeks |
Comparator(s) | Platinum-based chemotherapy (4 or 6 cycles) once every 3 weeks
|
Duration | |
Treatment phase | Atezolizumab treatment was continued as long as patients experienced a clinical benefit as assessed by the investigator, or until unacceptable toxicity or death. During treatment, patients receiving atezolizumab and who showed evidence of clinical benefit were permitted to continue atezolizumab after RECIST version 1.1 for PD were met if they met specific criteria. The intended number of cycles planned for the platinum-based induction chemotherapy (i.e., four or six cycles) were specified by the investigator prior to study randomization. Treatment was continued until disease progression, unacceptable toxicity, or death. |
Follow-up phase | One interim efficacy analysis was planned for the primary endpoint of OS in the PD-L1 high (≥50% of TCs or ≥10% of tumour-infiltrating ICs; TC3 or IC3) WT, the high or intermediate PD-L1 (≥5% of TCs or ICs; TC2/3 or IC2/3) WT, and any PD-L1 expression (≥1% of TCs or ICs; intention-to-treat population; TC1/2/3 or IC1/2/3) WT populations, respectively. An interim analysis of OS in the PD-L1 high WT population was planned when both of the following criteria had been met: An approximately 45% event-patient ratio had been observed in the PD-L1 high WT subpopulation; Approximately 96 deaths had occurred in the PD-L1 high WT subpopulation. If the OS interim analysis in the PD-L1 high WT population was statistically significant, the OS analysis in the high or intermediate PD-L1 WT population would be tested under the overall type I error of 0.05. If the OS interim analysis in the PD-L1 high WT population was not statistically significant, the final analysis would be conducted when approximately 135 OS events had occurred in this population. If this final analysis was statistically significant, the OS in the high or intermediate PD-L1 WT and any PD-L1 WT populations would be tested at the planned interim and final analyses accordingly. The interim and final analyses were expected at approximately 40 and 55 months after the first patient was enrolled, respectively. |
Outcomes | |
Primary endpoint | The primary efficacy endpoint was OS in the PD-L1 high WT, high or intermediate PD-L1 WT, and the any PD-L1 WT populations |
Publication status | |
Publications | 1. Herbst et al., 202022 2. Jassem et al., 202123 |
ALK = anaplastic lymphoma kinase; AUC = area under the concentration-time curve; CNS = central nervous system; EGFR = epidermal growth factor receptor; IC = immune cells; IHC = immunohistochemistry; NSCLC = non-small cell lung cancer; OS = overall survival; PD-L1 = programmed death ligand 1; PD = progressive disease; RECIST = Response Evaluation Criteria in Solid Tumors; TC = tumour cells; WT = wild type.
Randomization was stratified according to sex (male vs. female), ECOG performance-status score (0 vs. 1), histologic type (non-squamous vs. squamous), and PD-L1 status (≥1% PD-L1 expression on tumour cells and any level of PD-L1 expression on tumour-infiltrating immune cells vs. <1% PD-L1 expression on tumour cells and ≥1% PD-L1 expression on tumour-infiltrating immune cells).31
To control for the overall type I error rate at a two-sided significance level of 0.05, the primary endpoint of overall survival was tested hierarchically in the population with EGFR and ALK wild-type tumours: high PD-L1 expression (≥50% of TC or ≥10% of IC), then high to intermediate PD-L1 expression (≥5% of TC or IC), and then any PD-L1 expression (≥1% of TC or IC; intention-to-treat population).31
Interventions
Patients on the atezolizumab arm received 1200 mg atezolizumab by IV infusion in a 250 mL 0.9% NaCl bag on Day 1 of every 21-day cycle. Atezolizumab was infused over 60 (±15) minutes for the first infusion, and if tolerated, subsequent infusions were administered over 30 (±10) minutes. No pre-medication was allowed in the first cycle. Afterwards, if patients experienced infusion-related reactions during any previous infusion, pre-medication with antihistamines may be administered for Cycles 2 and later at the discretion of the treating physician.31
Outcomes
The primary endpoint was overall survival (OS), defined as the time from randomization to death from any cause. Secondary endpoints included progression free survival (PFS).31 Detailed descriptions of relevant outcome measures are presented in the Supplemental Materials.
Sample Size and Power Calculation
Detailed descriptions of the sample size and power calculation are provided in the Supplemental Materials.
Statistical Testing
Detailed descriptions of statistical analysis methods are presented in the Supplemental Materials. For the primary comparison of OS, overall type I error rate for the two-sided tests was controlled at 0.05 for the PD-L1 high WT subpopulation, the high or intermediate PD-L1 WT subpopulation, and the any PD-L1 WT population, with all populations excluding patients with a sensitizing EGFR mutation or ALK translocation.31,32 The hierarchy of spending is specified in the Supplemental Materials.
Subgroup Analyses
The consistency of OS, investigator-assessed PFS and objective response rate (ORR) results in subgroups was examined in the PD-L1 high WT, the high or intermediate PD-L1 WT, and/or any PD-L1 WT populations where OS benefit had been demonstrated. The subgroups were defined by the following:
Demographics (age, sex, race/ethnicity)
Baseline disease characteristics (e.g., PD-L1 tumour expression status, histology, ECOG performance status, smoking history, number of metastatic sites, location of metastases, size of primary tumour, etc.)31
Analysis Populations
Table 4: Analysis Populations of IMpower110
Population | Definition | Application |
|---|---|---|
Efficacy Analysis Population: Randomized/ITT population | All randomized patients, regardless of receipt of the assigned treatment. | IMpower110 recruited 572 patients in total, including 18 with EGFR mutations or ALK rearrangements. Patients with EGFR mutations or ALK rearrangements were originally eligible for the study if they had received an appropriate targeted therapy. The protocol was then amended to exclude these patients, and the primary endpoint of the study is therefore analyzed in the ITT-WT population (patients without EGFR mutations or ALK rearrangements) consisting of 554 patients |
Efficacy Analysis Population: The wild-type population | All randomized patients excluding patients with a sensitizing EGFR mutation or ALK translocation. | The primary efficacy endpoint, OS, will be analyzed in three PD-L1 selected populations, the PD-L1 high subpopulation, the high or intermediate PD-L1 WT subpopulation and the any PD-L1 WT population.
|
Safety Population | Treated patients - randomized patients who received any amount of study treatment. | For the safety analyses, patients will be grouped according to whether any amount of atezolizumab was received, including when atezolizumab was received in error. Specifically for patients who were randomized to the control arm, if atezolizumab was received in error, patients will be grouped to the atezolizumab arm for the safety analyses. |
ALK = anaplastic lymphoma kinase; EGFR = epidermal growth factor receptor; IC = immune cells; ITT = intent-to-treat; PD-L1 = programmed death ligand 1; TC = tumour cells; WT = wild type.
Patient Population
Baseline Characteristics
Table 5: Summary of Baseline Characteristics of IMpower110
Characteristic | IMpower110 | |
|---|---|---|
PD-L1 high WT population | ||
Chemotherapy (N = 98) | Atezolizumab (N = 107) | |
Age (years) | ||
Mean (SD) | 64.2 (9.0) | 63.3 (9.1) |
Sex, n (%) | ||
Male | 64 (65.3%) | 79 (73.8%) |
Female | 34 (34.7%) | 28 (26.2%) |
Race, n (%) | ||
Asian | 15 (15.3%) | 20 (18.7%) |
White | 82 (83.7%) | 87 (81.3%) |
Baseline ECOG per eCRF, n (%) | ||
0 | 38 (38.8%) | 35 (32.7) |
1 | 60 (61.2%) | 72 (67.3) |
Tobacco use history, n (%) | ||
Never | 15 (15.3%) | 9 (8.4%) |
Current | 29 (29.6%) | 20 (18.7%) |
Previous | 54 (55.1%) | 78 (72.9%) |
Histology at diagnosis, n (%) | ||
Non-squamous | 75 (76.5%) | 80 (74.8%) |
Squamous | 12 (23.5%) | 27 (25.2%) |
ECOG = Eastern Cooperative Oncology Group; PD-L1 = programmed death ligand 1; SD = standard deviation; WT = wild type.
Patient Disposition
At the 2018 data cutoff, more patients on chemotherapy (███) had discontinued from the study compared with the atezolizumab arm (███), with the most common reason being death. In total, ██versus ██of patients in chemotherapy and atezolizumab arms, respectively, had study discontinuation reason: withdrawal by subject. At data cut-off in the “any PD-L1 population,” ███of patients in the atezolizumab arm had discontinued treatment, most commonly due to disease progression (███). In the chemotherapy arm, ██████████████of patients had completed the intended number of cycles of carboplatin, cisplatin, and gemcitabine, respectively. Treatment discontinuation due to AEs (█████████████████████████████████████████ ███████████████████████████████) was more common in the chemotherapy arm.
███████████████████████████████████████████████████████████████████████████████████████████████
Figure 1: Patient Disposition (CCOD: 10 September 2018)
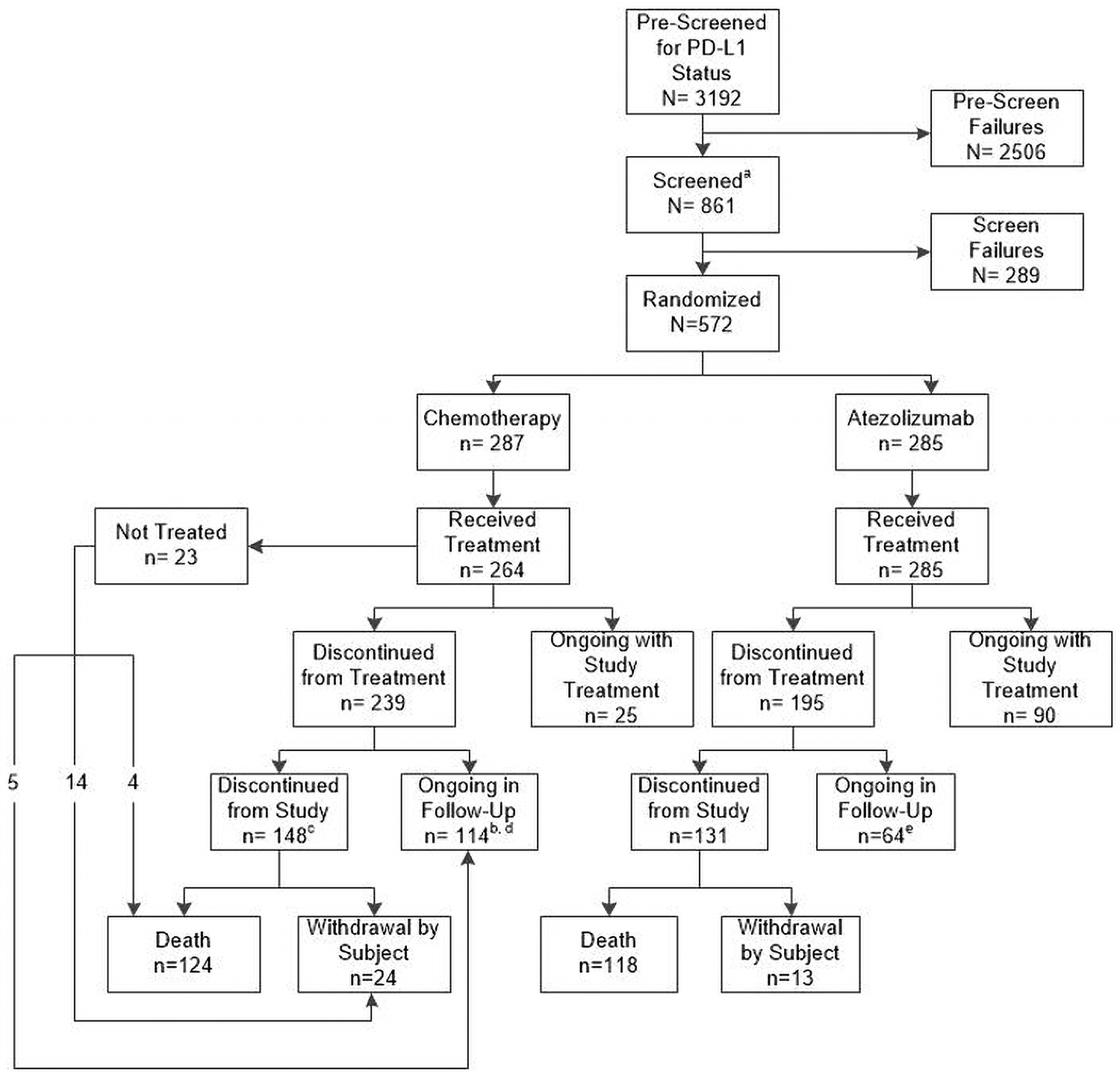
CCOD = clinical cut-off date; PD-L1 = programmed death-ligand 1.
aPre-screening for PD-L1 status was not required prior to patients entering screening. Therefore, total number of screened patients includes patients that were not pre-screened for PD-L1 status or that were re-screened.
bIncludes 5 patients who did not receive treatment.
cIncludes 18 patients who did not receive treatment.
dIncludes one patient that at the time of CCOD had survival follow up status lost to follow-up.
eIncludes two patients that at the time of CCOD had survival follow up status lost to follow-up.
Exposure to Interventions
Study Treatments
As of the CCOD of 04 February 2020, the median atezolizumab treatment duration was 5.3 months; median durations for cisplatin, carboplatin, gemcitabine, and pemetrexed were 2.1, 2.3, 2.6, and 3.5 months, respectively.
Table 6: Exposure to Study Treatment (Safety Evaluable Population, CCOD: 04 February 2020
Dosing information | Chemotherapy (n = 263) | Atezolizumab (n = 286) | |||
|---|---|---|---|---|---|
Cisplatin (n = 90) | Carboplatin (n = 177) | Gemcitabine (n = 75) | Pemetrexed (n = 191) | ||
Treatment duration (months) | |||||
Median | 2.1 | 2.3 | 2.6 | 3.5 | 5.3 |
Dose intensity (%) | |||||
Median | ████ | ████ | ████ | ████ | ████ |
Number of doses received | |||||
Median | ███ | ███ | ███ | ███ | ███ |
CCOD = clinical cut-off date.
Concomitant Medications and Co-Interventions
Concomitant medications that were allowed are described in the Supplemental Materials.
Results
Efficacy
Table 7: Summary of Key Efficacy Results in the PD-L1 High WT Population
Variable | Chemotherapy (N = 98) | Atezolizumab (N = 107) |
|---|---|---|
Overall survival (September 2018 CCOD) | ||
Number of patients | 98 | 107 |
Patients with event (%) | 57 (58.2%) | 44 (44.1%) |
Median duration of survival (95% CI) (Months) | 13.1 (7.4, 16.5) | 20.2 (16.5, NE) |
Stratified Hazard Ratio (95% CI) | 0.59 (0.40, 0.89) | |
p-value (Stratified log-rank) | 0.0106 | |
12-month OS rate | 50.6% | 64.9% |
24-month OS rate | 24.8% | 45.5% |
Overall survival (February 2020 CCOD) | ||
Number of patients | 98 | 107 |
Patients with event (%) | 64 (65.3%) | 64 (59.8%) |
Median duration of survival (95% CI) (Months) | 14.7 (7.4, 17.7) | 20.2 (17.2, 27.9) |
Stratified hazard ratio (95% CI) | 0.76 (0.54, 1.09) | |
p-value (stratified log-rank) | 0.1338* | |
12-month OS rate | 52.3% | 66.1% |
24-month OS rate | 34.1% | 47.1% |
Progression-free survival (September 2018 CCOD) | ||
Number of patients | 98 | 107 |
Patients with event (% | 79 (80.6%) | 67 (62.6%) |
Median duration of PFS-INV (95% CI) (months) | 5.0 (4.2, 5.7) | 8.1 (6.8, 11.0) |
Stratified hazard ratio (95% CI) | 0.63 (0.45, 0.88) | |
p-value (stratified log-rank) | 0.0070* | |
Progression-free survival (February 2020 CCOD) | ||
Number of patients | 98 | 107 |
Patients with event (% | 87 (88.8%) | 82 (76.6%) |
Median duration of PFS-INV (95% CI) (months) | 5.0 (4.2, 5.7) | 8.2 (6.8, 11.4) |
Stratified hazard ratio (95% CI) | 0.59 (0.43, 0.81) | |
p-value (stratified log-rank) | - | |
Objective response rate (September 2018 CCOD) | ||
Number of patients | 98 | 107 |
ORR (%) | 28.6% | 38.3% |
(95% CI) | (19.90, 38.58) | (29.08, 48.22) |
Objective response rate (February 2020 CCOD) | ||
Number of patients | 98 | 107 |
ORR (%) | 28.6% | 40.2% |
(95% CI) | (19.90, 38.58) | (30.82, 50.11) |
Duration of response (September 2018 CCOD) | ||
Number of patients | 28 | 41 |
Median DOR, months | 6.7 | NE |
(95% CI) | (5.5, 17.3) | (11.8, NE) |
Duration of response (February 2020 CCOD) | ||
Number of patients | 28 | 43 |
Median DOR, months | 8.3 | 38.9 |
(95% CI) | (5.6, 11.0) | (16.1, NE) |
CCOD = clinical cut-off date; CI = confidence interval; DOR = duration of response; NE = not estimable; ORR = objective response rate; OS = overall survival; WT = wild type.
*P value is descriptive only
At the interim analysis, the pre-specified OS interim analysis alpha boundary (α = 0.0413) was not crossed in the high or intermediate PD-L1-WT population (p = 0.0416). Therefore, OS in the “any PD-L1 WT population” was not formally tested.31 At the time of the updated clinical cutoff date (CCOD) (February 4th, 2020), the pre-specified OS final analysis alpha boundary was not crossed in the high or intermediate PD-L1 or any PD-L1 WT population. However, OS showed a numerical improvement with median OS of 19.9 months in the atezolizumab arm and 16.1 months in the chemotherapy arm. The stratified Cox hazard ratio (HR) was 0.87 (95% CI: 0.66, 1.14). OS was not formally tested in the “any PD-L1 WT population.” Nevertheless, OS also showed numerical improvement in the atezolizumab arm compared with the chemotherapy arm in this patient population.23,33 The focus of this report is on the PD-L1 high patients which aligns with the Health Canada marketing authorization.
Additional efficacy results are presented in the Supplemental Materials.
Overall Survival
September 2018 CCOD
At the CCOD of 10 September 2018, 101 death events were observed in the PD-L1 high WT population in the two arms combined; there were more deaths in the chemotherapy arm (58.2%) compared to the atezolizumab arm (41.1%).
The primary analysis endpoint was met as the pre-specified interim analysis alpha boundary (α = 0.0413) was crossed for OS in PD-L1 high WT population with a statistically significant and clinically meaningful OS benefit in the atezolizumab treatment arm. The stratified Cox HR was 0.59 (95% CI 0.40, 0.89; p = 0.0106), which corresponds to a 41% relative reduction in the risk of death associated with atezolizumab compared to chemotherapy.
The Kaplan-Meier (KM) estimated median OS was 7.1 months longer in the atezolizumab arm (20.2 months) compared to the chemotherapy arm (13.1 months). The minimum follow-up time at the time of CCOD was approximately 7 months (duration from last patient randomized date to CCOD) and the median follow-up was 15.7 months. The KM curves began to separate at approximately 4 to 5 months after randomization in favor of the atezolizumab arm; the benefit in the atezolizumab arm was maintained thereafter (Figure 2).31
Figure 2: Overall Survival in the PD-L1 High WT Population (CCOD: 10 September 2018)
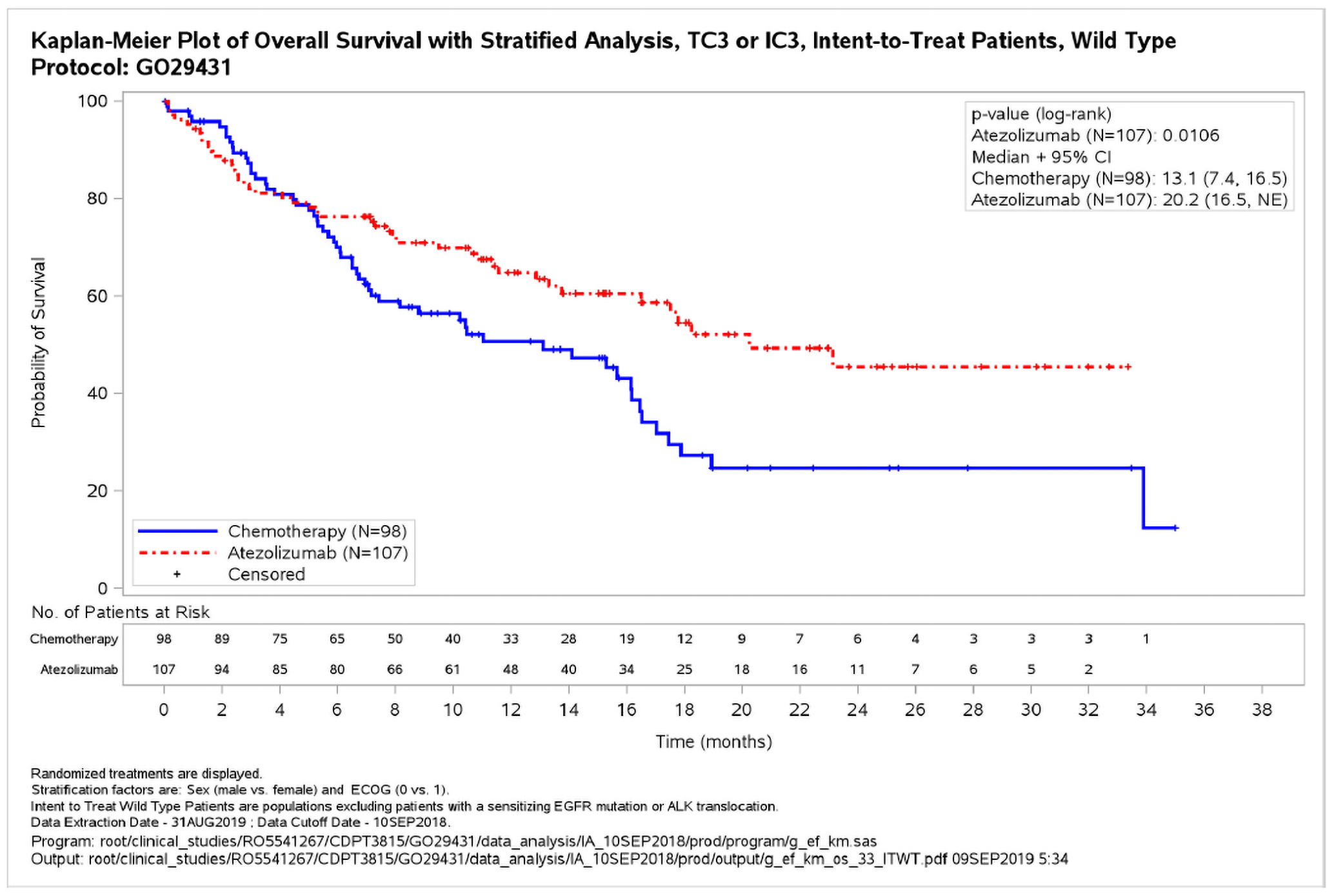
ALK = anaplastic lymphoma kinase; CCOD = clinical cutoff date; CI = confidence interval; ECOG = Eastern Cooperative Oncology Group; EGFR = epidermal growth factor receptor; IC = immune cell; NE = not estimable; PD-L1=programmed death-ligand 1; TC = tumour cell; WT = wild type.
February 2020 CCOD
As the OS benefit with atezolizumab in the high PD-L1 expression group crossed the boundary for significance at the interim analysis, that is considered the primary analysis for that population. Thus, the updated OS analysis for the high PD-L1 expression group presented here is exploratory and not formally tested.
At an additional 17 months of survival follow-up after the primary analysis, an exploratory OS analysis (CCOD: 04 February 2020) in the PD-L1 high WT population demonstrated continued clinically meaningful improvement, with a median OS of 20.2 months in the atezolizumab arm compared to 14.7 months in the chemotherapy arm (stratified HR 0.76; 95% CI, 0.54, 1.09). The median OS in the atezolizumab arm was maintained at this longer follow up.
The benefit in the atezolizumab arm was maintained after the separation of the KM curves, which again began at approximately 4 - 5 months. The median duration of survival follow-up at the time of this exploratory analysis was 31.3 months and minimum follow-up was 24 months; a total of 128 death events had occurred in the PD-L1 high WT population (Figure 3).23,33
Figure 3: Overall Survival in the PD-L1 High WT Population (CCOD: 04 February 2020)
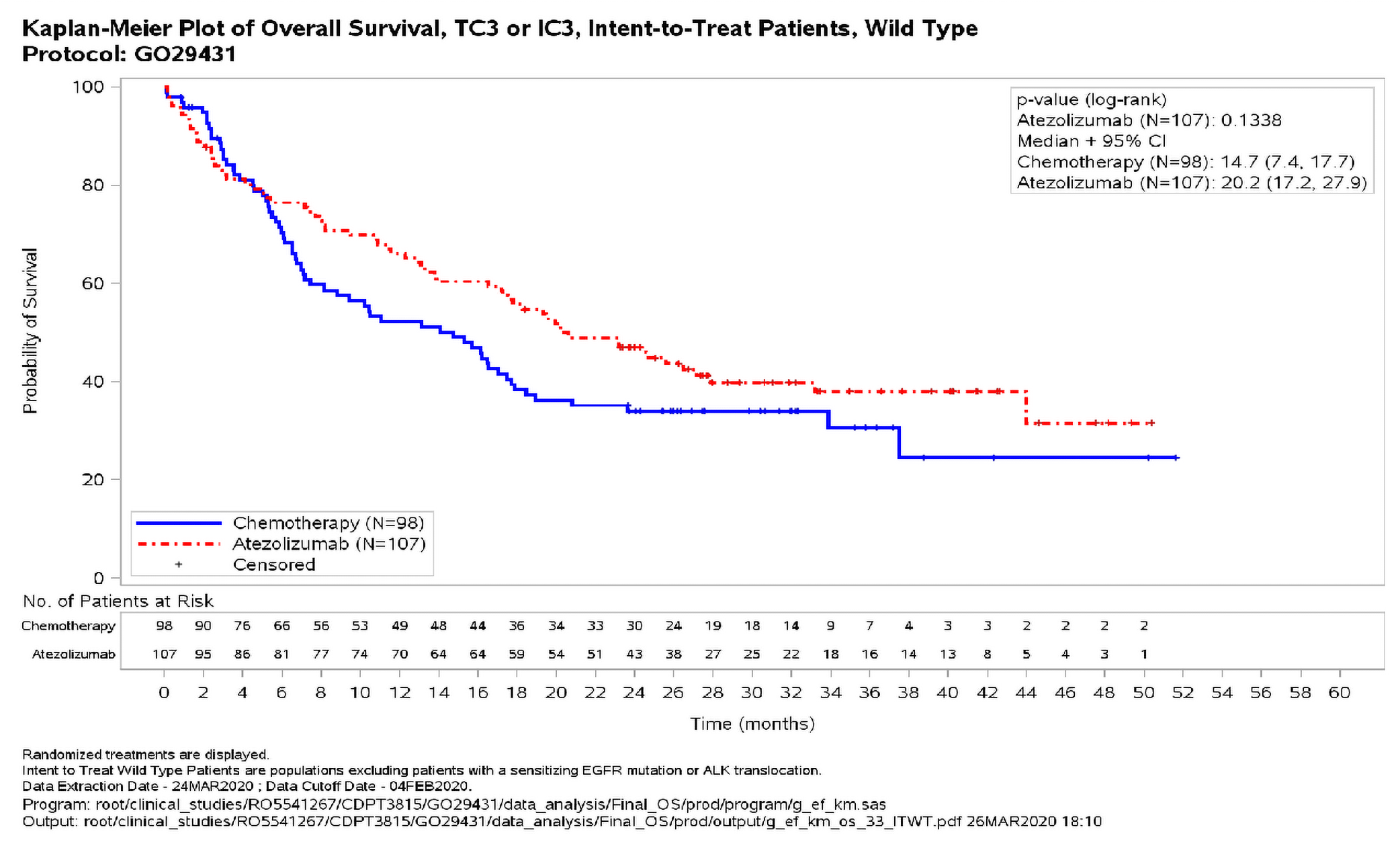
ALK = anaplastic lymphoma kinase; CCOD = clinical cutoff date; CI = confidence interval; ECOG = Eastern Cooperative Oncology Group; EGFR = epidermal growth factor receptor; IC = immune cell; NE = not estimable; PD-L1=programmed death-ligand 1; TC = tumour cell; WT = wild type.
Sensitivity Analyses of OS
The robustness of the treatment effect with atezolizumab on OS in the PD-L1 high WT population was confirmed in sensitivity analyses (CCOD of 10 September 2018). Despite the higher rate of 2L therapy use in the chemotherapy arm (47% chemotherapy vs. 24% atezolizumab), the treatment effect with atezolizumab remained robust, as demonstrated in a sensitivity analysis discounting survival duration for non-protocol-specified or follow-up anti-cancer therapy with 10% (HR, 0.59 [95% CI, 0.40 to 0.88]), 20% (HR, 0.58 [0.39 to 0.86]), or 30% reduction (HR, 0.58 [0.39 to 0.86]).33
At the updated exploratory OS analysis (CCOD: 04 February 2020) in the PD-L1 high WT population, a higher percentage of patients in the chemotherapy arm (54.1%) received at least one non-protocol specified or follow-up anti-cancer therapy (NPT) compared to the atezolizumab arm (35.5%). As expected, immunotherapies were reported at a much higher frequency in the chemotherapy arm (34.7%) compared to the atezolizumab arm (3.7%). A sensitivity analysis using the RPSFT (Rank Preserving Structural Failure Time) method was conducted at the updated analysis. Using this method, the median OS in the high PD-L1 expression WT group was 20.2 months in the atezolizumab arm and 13.0 months in the chemotherapy arm (HR: 0.69, 95% CI: 0.48–0.99), suggesting that NPT attenuated the treatment effect with atezolizumab (Figure 4).23
Figure 4: Overall Survival in the PD-L1 High WT Group, Adjusting for NPT Immunotherapy Using the RPSFT Method
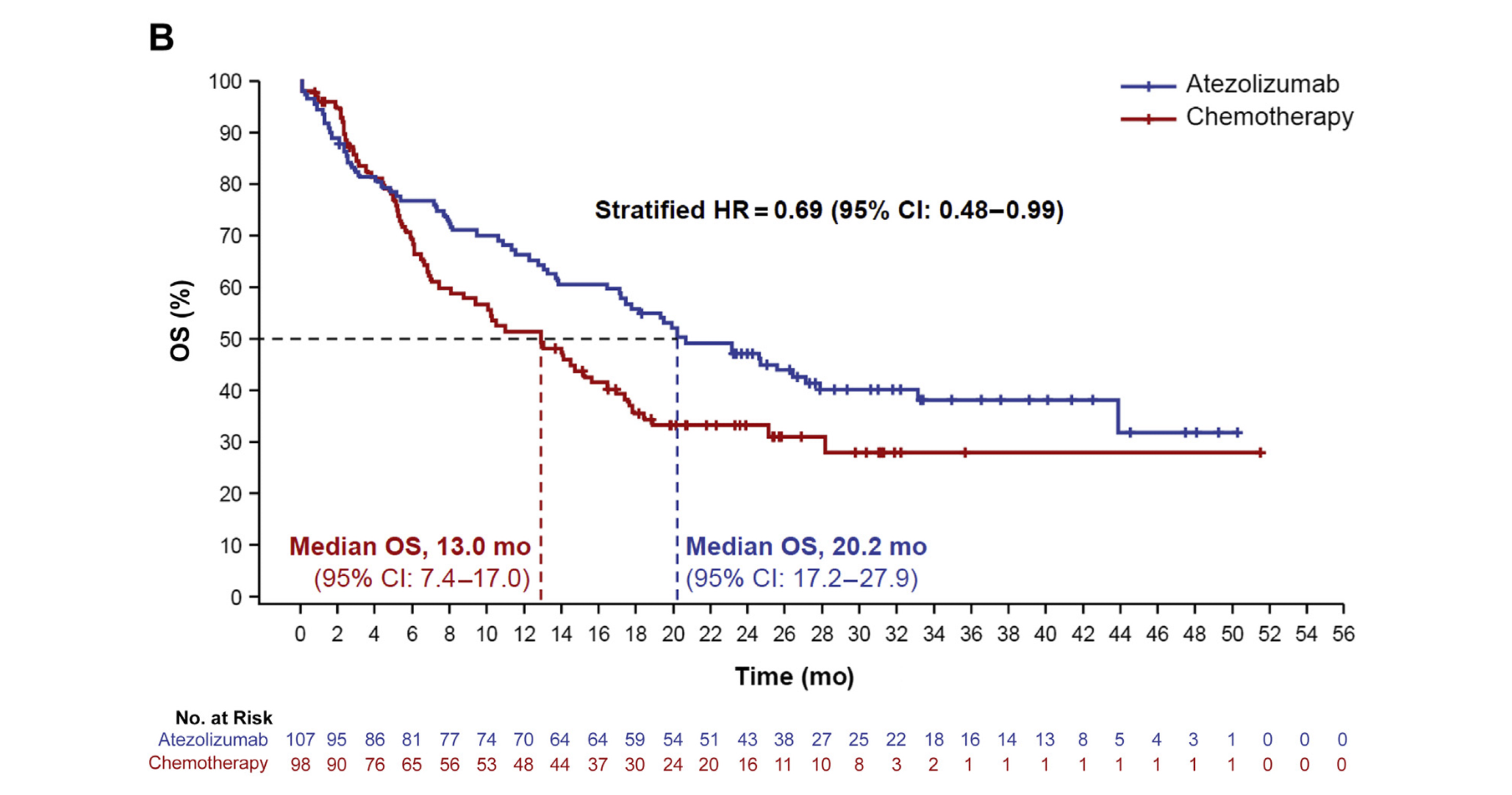
CI, confidence interval; HR, hazard ratio; IC, tumour-infiltrating immune cell; NPT, nonprotocol therapy; OS, overall survival; PD-L1, programmed death-ligand 1; RPSFT, rank-preserving structural failure time; TC, tumour cell; WT, wild type.
Harms
Overview of Safety
Detailed results for harms are presented in the Supplemental Materials.
At the CCOD of 10 September 2018, the median treatment duration was longer in the atezolizumab arm (5.3 months) compared with the chemotherapy arm (3.5 months pemetrexed, 2.1 months cisplatin, 2.3 months carboplatin, 2.6 months gemcitabine). The overall safety experience in the atezolizumab arm was consistent with the established safety profile of atezolizumab in a single-agent setting; being generally consistent with safety data from a pooled atezolizumab monotherapy population. No new safety signals were identified. Atezolizumab demonstrated better safety and tolerability than chemotherapy, as reflected by the lower incidence of treatment-related AEs, Grade 3-4 AEs, AEs leading to treatment discontinuation and AEs leading to dose modification/interruption. The incidence of serious adverse events (SAEs) was comparable between treatment arms. █████ ████ ██ ██████████ ████ ████████ ████ ███ ██████████ ██ █████████ ███████ █████31
Overall, the updated safety data at the CCOD of 04 February 2020 were consistent with the data from the 10 September 2018 analysis. Atezolizumab continued to demonstrate better safety and tolerability than chemotherapy. Although the frequency of overall SAEs was comparable between the two arms (31.8% for atezolizumab and 29.3% for chemotherapy), the frequency of treatment-related SAEs was lower in the atezolizumab arm (9.4% vs. 15.6% in the chemotherapy arm). There were no treatment related deaths in the atezolizumab arm. Adverse events of special interest (AESIs) reported in the atezolizumab arm (most commonly rash, hepatic lab abnormalities and hypothyroidism) were Grade 1-2 and manageable with treatment interruption, administration of systemic corticosteroids or hormone replacement therapy. The overall safety experience with atezolizumab at the updated analysis remained consistent with its well-established safety profile in the monotherapy setting. No new or unexpected safety signals were identified for atezolizumab (Table 8).23,31
At the last patient last visit (CCOD of 08 March 2022), safety data was also consistent with the previous two analyses, with no new safety signals identified. Atezolizumab was generally safe and tolerated compared to chemotherapy, as reflected by the lower incidence of treatment-related AEs (█████ ██ █████), Grade 3-4 AEs (█████ ██ █████), AEs leading to treatment discontinuation (████ ██ █████), and AEs leading to dose modification/interruption (█████ ██ █████). The incidence of Grade 5 AEs was comparable between treatment arms (████ ██ ████), and there were no treatment-related Grade 5 AEs reported in the atezolizumab arm. The incidence of SAEs was comparable between treatment arms (█████ ██ █████), with no significant SAEs reported with a 2% difference in incidence between arms. As expected, incidence of atezolizumab specific AESI was higher in the atezolizumab arm than chemotherapy arm. The majority of AESIs were Grade 1-2, with no Grade 5 AESIs reported.33 This final analysis only included updated safety findings not coupled with updated efficacy data. As such, only safety data from the first two cut-off dates is presented in the table below. For further details on safety data from the final analysis, please refer to the Clinical Study Report dated March 2022.
Table 8: Summary of Adverse Events
AE category | CCOD: 10 September 2018 | CCOD: 04 February 2020 | ||
|---|---|---|---|---|
Atezolizumab N = 286 n (%) | Chemo N = 263 n (%) | Atezolizumab N = 286 n (%) | Chemo N = 263 n (%) | |
Grade 3-4 AEsb | 86 (30.1) | 138 (52.5) | 97 (33.9) | 140 (53.2) |
Related Grade 3-4 AEsb | 37 (12.9) | 116 (44.1) | 41 (14.3) | 118 (44.9) |
Grade 5 AE | 11 (3.8) | 11 (4.2) | 12 (4.2) | 11 (4.2) |
Related Grade 5 AE | 0 | 1 (0.4) | 0 | 1 (0.4) |
AEs leading to any treatment withdrawal | 18 (6.3) | 43 (16.3) | 21 (7.3) | 45 (17.1) |
imAEs | 115 (40.2) | 44 (16.7) | 132 (46.2) | 48 (18.3) |
Grade 3-4 imAEsb | 19 (6.6) | 4 (1.5) | 25 (8.7) | 4 (1.5) |
imAEs requiring corticosteroid use | 30 (10.5) | 3 (1.1) | 38 (13.3) | 4 (1.5) |
AE, Adverse event; CCOD = clinical cutoff date; imAE, immune-related adverse event
b Patients with AEs with the highest grade of 3 or 4.
Additional Results From the Included Studies
The information in Table 9 was added by CDA-AMC to provide additional information regarding the OS and PFS endpoints.
Table 9: Summary of Event-Free Rates for OS and PFS at 12 and 24 Months
Variable | Chemotherapy (N = 98) | Atezolizumab (N = 107) |
|---|---|---|
Overall survival (September 2018 CCOD) | ||
12-month OS rate, % (95% CI) | 50.64 (40.00 to 61.27) | 64.90 (55.36 to 74.43) |
Difference | Reference | 14.3 (−0.03 to 28.55) |
24-month OS rate, % (95% CI) | █████ ██████ ██ ██████ | █████ ██████ ██ ██████ |
Difference | █████████ | █████ █████ ██ ██████ |
Overall survival (February 2020 CCOD) | ||
12-month OS rate, % (95% CI) | █████ ██████ ██ ██████ | █████ ██████ ██ ██████ |
Difference | █████████ | █████ █████ ██ ██████ |
24-month OS rate, % (95% CI) | █████ ██████ ██ ██████ | █████ ██████ ██ ██████ |
Difference | █████████ | █████ ██████ ██ ██████ |
Progression-free survival (September 2018 CCOD) | ||
12-month PFS rate, % (95% CI) | █████ ██████ ██ ██████ | █████ ██████ ██ ██████ |
Difference | █████████ | █████ █████ ██ ██████ |
24-month PFS rate, % (95% CI) | ████ █████ ██ ██████ | █████ ██████ ██ ██████ |
Difference | █████████ | █████ ██████ ██ ██████ |
Progression-free survival (February 2020 CCOD) | ||
12-month PFS rate, % (95% CI) | ██ | ██ |
Difference | █████████ | ██ |
24-month PFS rate, % (95% CI) | ██ | ██ |
Difference | █████████ | ██ |
CCOD = clinical cutoff date; CI = confidence interval; NR = not reported; OS = overall survival; PFS = progression-free survival
CDA-AMC Critical Appraisal of the Systematic Review Evidence
This section was prepared by CDA-AMC based on the sponsor’s summary of the systematic review evidence and other material submitted by the sponsor.
Internal Validity
Randomization was performed using an interactive web and voice response system for concealment and stratified by important prognostic factors (i.e., sex [female versus male]; ECOG performance status [0 versus 1]; histology [nonsquamous versus squamous]; and tumour tissue PD-L1 expression by IHC TC1/2/3 and any IC versus TC0 and IC1/2/3). The stratification factors were considered to be clinically relevant by regulatory authorities.34 The method of assigning patients to treatment groups was adequate to conceal allocation until the time of treatment assignment.
Patients were eligible for IMpower110 if they had tumour PD-L1 expression (1% PD-L1–expressing TCs and 1% of tumour area occupied by PD-L1–expressing ICs). This is broader than the indication for which the sponsor sought and obtained regulatory approval and the target population for this review, which is limited to patients with high PD-L1 expression (defined as ≥ 50% PD-L1 expression on TCs or ≥ 10% ICs) who did not have EGFR or ALK aberrations. As such, the submission to CDA-AMC and Health Canada is focused on the subgroup of patients with high PD-L1 expression. Randomization in the IMpower110 trial was stratified by PD-L1 status (≥ 1% PD-L1 expression on TCs and any level of PD-L1 expression on tumour-infiltrating immune cells versus < 1% PD-L1 expression on TCs and ≥ 1% PD-L1 expression on tumour-infiltrating immune cells), which is different than the thresholds for high PD-L1 that were ultimately used in efficacy analysis and reflected in the approved indication. Potential bias due to confounding due to the use of a subgroup population (that was not stratification factor at the time of randomization, and prognostic balance may not have been maintained across treatment groups) was investigated by Health Canada, which concluded that the sponsor’s approach was acceptable and supported by additional analyses (e.g., multivariable Cox regression and propensity score methods including all measured clinically relevant variables).35
The baseline demographic and disease characteristics for the relevant subpopulation were generally balanced between the atezolizumab and chemotherapy treatment groups. A notable exception was the higher proportion of nonsmokers in the chemotherapy group. Health Canada noted that a literature review has reported a more robust response to PD-1 inhibition in current or former smokers compared with those with no smoking history.35 In response to a request from Health Canada, the sponsor conducted an unstratified multivariate Cox regression adjusting for 6 baseline factors: PD-L1 status, age groups, sex, baseline ECOG, baseline sum of longest diameters, and tobacco use history.35 The results showed no significant interactions between each baseline factor and treatment. As noted previously, to better understand the impact of potential treatment imbalances, Health Canada requested that the sponsor reanalyze OS data using multivariable Cox regression and propensity score methods (inverse probability of treatment weight method), including all measured clinically relevant variables. The adjusted results were considered acceptable by Health Canada. The clinical experts consulted by CDA-AMC felt that the baseline characteristics were generally well-balanced across the treatment groups, and any imbalances were unlikely to result in a risk of bias in favour of or against atezolizumab.
Treatments were administered in an open-label manner in the IMpower110 trial. This is consistent with other phase III trials in the therapeutic space, given that blinding would not be feasible or appropriate with 1 treatment group receiving a cytotoxic chemotherapy regimen. Although patient blinding would not have been possible given the differences in the treatment regimens, detection and performance bias that may result from lack of blinding of patients and investigators to assigned study treatments cannot be ruled out, especially for subjective patient-reported outcomes, response end points (ORR, duration of response, PFS), and subjective harms.
The sponsor had a prespecified analysis hierarchy for evaluating OS in the 3 study populations (i.e., high PD-L1 tested first), and if significant, the testing continued to the broader patient populations in the trial (which were outside the scope of CDA-AMC review). The remaining end points may be at risk of type I error with no adjustment for multiple comparisons; however, the analyses were directionally aligned, and there were no significant concerns regarding the statistical approaches. The sponsor presented the results of subgroup analyses, and the direction of effect appears to differ for some subpopulations (e.g., people who have never smoked and older adults). These analyses are insufficient to inform credible conclusions of effect modification due to small sample sizes, wide confidence intervals (CIs), and lack of testing for treatment by subgroup interactions.
The protocol for the IMpower110 trial underwent several important amendments throughout the study, including revising the target population for the primary analysis to focus on patients with high PD-L1 expression (≥ 50% of TC or ≥ 10% of tumour-infiltrating ICs) and without a sensitizing EGFR mutation or ALK translocation. This is the population that would be submitted for regulatory review and reflected in the approved indications in Canada and internationally. Additionally, the timing of the primary end point evaluation was revised to focus on an earlier assessment of events accrued in the population of patients with high PD-L1 expression. The sponsor cited emerging evidence that external data suggested that PD-L1 inhibitors were more effective in those with high PD-L1 expression and that the events in this group may be sufficient to allow for an analysis of OS benefit with less confounding from subsequent therapies than an analysis based on a later time point with the overall study population.
Given the open-label study design, the nature and timing of the protocol amendments related to the study population and end point evaluation triggered a Good Clinical Practice inspection by the European Medicines Agency (EMA). The EMA accepted the sponsor’s explanation and rationale for the amendments, though the sponsor was criticized for the approach and advised to avoid this approach in future open-label clinical studies.34 Overall, regulatory authorities considered the analyses that focused on the subgroup of patients with high PD-L1 expression to be internally valid.34,35
There were imbalances in the proportion of patients who received subsequent therapies in the second-line setting (with more patients in the chemotherapy group receiving subsequent treatment). This included 34% of chemotherapy-treated patients receiving immunotherapy in the second-line setting, which could attenuate the treatment effect for atezolizumab for evaluating potential improvements in OS. The sponsor’s sensitivity analysis using the RPSFT method supported their interpretation that nonprotocol anticancer therapies (e.g., immunotherapies administered in the postprogression setting) attenuated the treatment effect with atezolizumab. CDA-AMC notes that the use of the RPSFT method to attempt to adjust for use of second-line immunotherapies in the chemotherapy group relies on untestable assumptions that may not be plausible.36
For the analysis of OS, there may be a violation of the proportional hazards assumption as evidenced by the early survival detriment with atezolizumab relative to placebo, though this is typical of immunotherapy, given the delayed treatment effect. However, the ratio of the hazard functions is not constant over time, and reliance on the HR to inform the OS benefit may be misleading. The between-group differences in KM-estimated probabilities of remaining event-free at clinically relevant follow-up times are not affected by this limitation. In addition, the sponsor reported analyses using piecewise HRs (estimated using weighted Cox regression models based on piecewise constant weight function) to understand the changes in treatment effect over time, and restricted mean survival time estimates the area under the KM survival curves up to a specific time point (i.e., the average of event-free survival time at 12 months, 18 months, and 24 months). Although the point estimates for the between-group differences in KM-estimated probabilities of OS at 12 and 24 months of follow-up suggested a clinically important benefit with atezolizumab, the 95% CIs included small differences that may not be considered clinically important.
The study was open-label, and PFS and ORR were assessed by the study investigators and not through a blinded independent central review. Given the open-label design, regulatory authorities noted that the robustness of the PFS result in the IMpower110 trial could have been greatly improved by an independent review of the scan results. For the time-to-event end points (OS and PFS), the sponsor did not report the proportions of patients censored in each group or the reason for censoring, so it was not possible to complete an appraisal of the risk of bias due to informative censoring. In the absence of this information, the potential for risk of bias due to informative censoring cannot be ruled out. For ORR, there is risk of bias due to missing outcome data because, at the time of the September 2018 data cut-off, ███ of patients in the chemotherapy group and ███ in the atezolizumab group were classified as “missing or unevaluable” and it was assumed that their disease had not responded to treatment. The plausibility of this assumption was not tested and the direction of bias cannot be predicted. Given that PFS or ORR were not prespecified in the hierarchy testing procedures, the statistical analyses for these outcomes were considered descriptive only. For the outcome of duration of response, which was evaluated only among patients with a response, the results are at risk of bias given that it is uncertain whether prognostic balance across treatment groups was maintained in this subpopulation of patients.
External Validity
According to the clinical experts consulted by CDA-AMC, the relevant subpopulation of the IMpower110 trial was a reasonable reflection of the target population in Canada. As is typically the case in phase III trials, the enrolled population was likely more fit than would be expected in the broader population encountered in routine clinical practice. The clinical experts consulted by CDA-AMC had no concerns regarding the generalizability of the IMpower110 trial to the setting in Canada. The clinical experts consulted by CDA-AMC noted that the patient disposition reported in the IMpower110 trial was reflective of what would have been expected in clinical practice (noting chemotherapy is no longer a preferred option for the target population).
The dosing of atezolizumab in the IMpower110 trial was 1,200 mg administered by IV infusion every 3 weeks, which reflects 1 of the approved dosing schedules for the IV formulation (the others being 840 mg every 2 weeks and 1,680 mg every 4 weeks). The clinical experts consulted by CDA-AMC noted that this is an appropriate regimen for IV-administered atezolizumab. Platinum-based chemotherapy is no longer the preferred first-line treatment option for the target patient population (i.e., metastatic NSCLC without actionable oncogenic mutations); however, it was considered an acceptable comparator at the time the IMpower110 trial was initiated in 2015.35 The clinical experts consulted by CDA-AMC similarly noted that the regimens were appropriate.
In total, 35.5% of patients in the atezolizumab group received treatment with at least 1 subsequent anticancer therapy after discontinuation of the study drug. The most common subsequent therapy for those in the atezolizumab group was platinum-based chemotherapy (30.8%), which is reflective of the current provisional funding algorithm from CDA-AMC for NSCLC patients without actionable oncogenic mutations who experience disease progression after immunotherapy in the first-line metastatic setting. The clinical experts noted that the proportion of patients receiving a second-line therapy in the atezolizumab group may be lower than would be expected in routine practice in the current environment. Immunotherapy was the most common subsequent therapy for those in the chemotherapy group (34.7%; nivolumab [14.3%], pembrolizumab [19.4%], atezolizumab [1.0%]) and 21.4% receiving treatment with chemotherapy. The clinical experts noted that more patients in the chemotherapy treatment group receiving a subsequent therapy is consistent with observations from the other phase III clinical trials in this therapeutic area. Overall, the clinical experts noted that the subsequent therapies were reasonable for the time the clinical trial was conducted.
Summary of Findings and Certainty of the Evidence
For a tailored review application, CDA-AMC only applies the Grading of Recommendations Assessment, Development and Evaluation (GRADE) approach when the sponsor submits direct evidence comparing the drug under review with a relevant comparator. The sponsor has submitted an indirect comparison to support the comparative effectiveness of atezolizumab versus the appropriate comparators; therefore, the GRADE approach has not been used.
Sponsor’s Summary of the Indirect Evidence
Description of Indirect Treatment Comparison(s)
Objectives
In the absence of head-to-head data, the objective of the ITC was to investigate the efficacy and safety of immunotherapies approved in Canada for the first-line monotherapy treatment of adult patients with stage IV squamous or non-squamous NSCLC and PD-L1 high expression.
Study Selection and Review Methods
Details on study selection criteria and review methods are presented in the Supplemental Materials. A systematic literature review was performed to identify published evidence regarding the clinical efficacy and safety of systemic therapies for stage IV squamous or non-squamous NSCLC from randomized controlled trials.
Indirect Comparison Analysis Methods
Details on analysis methods for the indirect comparisons are presented in the Supplemental Materials. A series of Bayesian network meta-analyses (NMAs) were conducted to produce comparative effectiveness estimates for atezolizumab versus existing treatments such as chemotherapy, cemiplimab, and pembrolizumab in Canada. The NMA was performed in accordance with best practice guidelines, including the National Institute for Health and Care Excellence (NICE) Decision Support Unit (DSU) guidelines.37
Results
Summary of Included Studies
The studies included in the systemic review included: IMpower110 (atezolizumab), KEYNOTE-024 (pembrolizumab), KEYNOTE-042 (pembrolizumab), and EMPOWER-LUNG 1 (cemiplimab).
Efficacy
The league table for all pairwise comparisons for OS for high PD-L1 expression groups is presented below. The results suggest insufficient evidence to conclude any superiority among the three immunotherapies.
CDA-AMC Note: Table 10 and Table 11 show the logHR (95% CrI) for OS and the logOR (95% CrI) for treatment-related adverse events. CDA-AMC requested the sponsor submit the data as HR (95% CrI) for ease of interpretation and consistency with prior applications. These are summarized in Table 12 for OS and in Table 13 for PFS.
Table 10: League Table for Overall Survival
Compared to | Atezolizumab | Cemiplimab | Chemotherapy | Pembrolizumab |
|---|---|---|---|---|
Atezolizumab | - | █████ ██████ ██ █████ | ████ ██████ ██ █████ | ████ ██████ ██ █████ |
Cemiplimab | ████ ██████ ██ █████ | - | ████ █████ ██ █████ | ████ ██████ ██ ████ |
Chemotherapy | █████ ██████ ██ █████ | █████ ██████ ██ ████ | - | █████ ██████ ██ █████ |
Pembrolizumab | █████ ██████ ██ █████ | █████ █████ ██ █████ | ████ ██████ ██ █████ | - |
Harms
The league table for all pairwise comparisons in TRAEs is presented below.
Table 11: League Table for Treatment-Related Adverse Events
Compared to | Atezolizumab | Cemiplimab | Chemotherapy | Pembrolizumab |
|---|---|---|---|---|
Atezolizumab | - | ████ █████ ██ ██████ | ████ ████ ██ ██████ | ████ █████ ██ ██████ |
Cemiplimab | ████ █████ ██ ██████ | - | ████ █████ ██ ██████ | ████ ████ ██ ██████ |
Chemotherapy | ████ █████ ██ █████ | ████ █████ ██ ████ | - | ████ █████ ██ █████ |
Pembrolizumab | ████ █████ ██ ██████ | ████ █████ ██ █████ | ███ █████ ██ ██████ | - |
CDA-AMC Critical Appraisal of the Indirect Evidence
This section was prepared by CDA-AMC based on the sponsor’s summary of the systematic review evidence and other material submitted by the sponsor.
Additional Results From the Indirect Treatment Comparison
CDA-AMC requested the sponsor to provide the between-group differences for OS and PFS from the NMA and corresponding HR with 95% CrI for each comparison. These are reported in Table 12 and Table 13 for OS and PFS, respectively.
Table 12: League Table and Corresponding HR (95% CrI) for OS
Compared to | Atezolizumab | Cemiplimab | Chemotherapy | Pembrolizumab |
|---|---|---|---|---|
Atezolizumab | — | ████ █████ ██ ██████ | ████ █████ ██ ██████ | ████ █████ ██ ██████ |
Cemiplimab | ████ █████ ██ ██████ | — | ████ █████ ██ ██████ | ████ █████ ██ ██████ |
Chemotherapy | ████ █████ ██ █████ | ████ █████ ██ █████ | — | ████ █████ ██ █████ |
Pembrolizumab | ████ █████ ██ █████ | ████ █████ ██ █████ | ████ █████ ██ █████ | — |
CrI = credible interval; HR = hazard ratio; OS = overall survival.
Table 13: League Table and Corresponding HR (95% CrI) for PFS
Compared to | Atezolizumab | Cemiplimab | Chemotherapy | Pembrolizumab |
|---|---|---|---|---|
Atezolizumab | — | ████████ | ████████ | ████████ |
Cemiplimab | ████████ | — | ████████ | ████████ |
Chemotherapy | ████████ | ████████ | — | ████████ |
Pembrolizumab | ████████ | ████████ | ████████ | — |
CrI = credible interval; HR = hazard ratio; PFS = progression-free survival.
Critical Appraisal
The sponsor-submitted NMA was informed by a systematic literature review that included searches of multiple databases and trial registries. The inclusion of studies was based on defined eligibility criteria (i.e., population, intervention, comparators, outcomes, study designs [PICOS]). The eligibility criteria used for study selection were appropriate and reflect the comparators that are relevant for the context in Canada. Study selection, data extraction, and risk of bias appraisal were undertaken by 2 reviewers with a third reviewer to adjudicate, limiting the risk of bias and errors in these processes. The risk of bias appraisals were undertaken using a validated tool; however, they were undertaken at the study level rather than at the level of the reported effects. This method ignores that risk of bias can vary depending on the effect estimate being evaluated, particularly for such domains as performance, detection, and reporting bias. As such, the risks of bias reported by the sponsor may not universally apply to all outcomes assessed. CDA-AMC is not aware of any relevant studies that were excluded from the systematic literature review. Upon request, the sponsor was unable to supply a protocol for the systematic review or a statistical analysis plan for the network meta-analysis. As such, there is an increased risk that the reported results were selected from among multiple analyses of the data, based on the direction and/or magnitude of the effects.
Base case analyses were conducted using random effects models, which assumes that there is between-study heterogeneity and that effect modifiers are not evenly distributed across all studies in the analysis. Sensitivity analyses using fixed effects models were conducted and submitted, for which the point estimates were consistent with the random effects base case, though the CrIs were more precise (though comparisons were only reported versus chemotherapy). However, this model assumes that there is a single true treatment effect across studies, potentially underestimating the uncertainty in the effect size (i.e., width of the 95% CrIs) when between-study variation in results exists. The credible intervals were wide for all of the NMAs comparing atezolizumab against pembrolizumab and cemiplimab, including the potential for both important benefit and harm, and no difference. Irrespective of other limitations, this imprecision rendered the NMA inconclusive for all outcomes analyzed. The evidence network was sparse, with only 1 or 2 studies per node, and the comparisons of interest (i.e., atezolizumab versus pembrolizumab and cemiplimab) relied entirely on indirect data. Due to the star-shaped evidence network, inconsistency (the statistical manifestation of intransitivity) could not be tested. The proportional hazards assumption may be violated for the IMpower110 trial due to delayed separation and crossing of the curves. It is unclear if the other trials included in the NMA have similar limitations, but this remains a potential source of bias with the indirect comparison (though the direction and magnitude of any potential bias is uncertain).
The sponsor did not report a comprehensive appraisal of potential treatment effect modifiers (e.g., a feasibility assessment with methods to identify and adjust for potential differences). Without such detail, it is not possible to make a comprehensive judgment of whether the assumptions underlying the NMA were upheld. CDA-AMC evaluated some of the key patient characteristics. These characteristics, and some potentially important differences, are as follows:
Study design: The IMpower110 (atezolizumab), KEYNOTE-024 and KEYNOTE-042 (pembrolizumab), and EMPOWER-LUNG 1 (cemiplimab) trials were generally similar in their overall design (i.e., phase III, open-label, trials comparing PD-1 and PD-L1 inhibitors versus chemotherapy in the first-line metastatic setting). The EMPOWER-LUNG 1 trial was stopped early because the prespecified OS efficacy criteria were met at the secondary interim analysis. All of the other trials continued to the final analysis of OS. This could potentially bias the comparative OS analysis against cemiplimab (i.e., in favour of atezolizumab) because those in the chemotherapy group could cross over to receive cemiplimab before disease progression (43% crossed over). None of the other trials included a crossover protocol.
Study locations: There were differences across study locations (e.g., the EMPOWER-LUNG 1 trial did not include any sites in North America due to the approval of pembrolizumab monotherapy for first-line treatment of metastatic NSCLC with high tumour PD-L1 expression, though CDA-AMC did not identify generalizability concerns with this study when reviewing cemiplimab for use as monotherapy in the first-line metastatic setting).
Open-label administration: Study treatments were administered in an open-label manner in all 4 trials. The NMA did not evaluate the patient-reported end points that could be most susceptible to bias because of open-label administration. As previously noted, in the IMpower110 trial, PFS was assessed by the investigator, which is at a greater risk of bias in an open-label trial.
Study treatments: Atezolizumab, pembrolizumab, and cemiplimab were all administered in accordance with a regimen that is recommended in the Canadian product monograph for the drugs. All of the trials used platinum-based chemotherapy as the comparator group, with different regimens applied for those with squamous or for those with nonsquamous disease. There was some variation across the chemotherapy regimens, but all were considered to be acceptable options by regulatory authorities when the trials were conducted.
Disease status: Patients in the KEYNOTE-024 and IMpower110 trials were required to have metastatic disease; whereas the EMPOWER-LUNG 1 and KEYNOTE-042 trials included patients with stage III or IV disease (though the sponsor reported that more than 80% of those enrolled had stage IV disease).
PD-L1 expression: The IMpower110 and KEYNOTE-042 trials enrolled patients with a PD-L1 expression of 1% or greater, but all 4 trials included analyses for patients with PD-L1 expression levels of 50% or greater (i.e., the target population for this review).
Squamous and nonsquamous: All 4 trials enrolled patients with both squamous and nonsquamous NSCLC. The proportion of patients with squamous NSCLC ranged from approximately 20% in the KEYNOTE-024 trial, approximately 30% in the IMpower110 trial, and approximately 40% in the KEYNOTE-042 and EMPOWER-LUNG 1 trials. The sponsor reported subgroup analysis based on squamous or nonsquamous disease, though the results are limited by considerable imprecision.
Subsequent therapy (intervention groups): In the IMpower110 trial, 35.5% of patients in the atezolizumab group received treatment with at least 1 subsequent anticancer therapy after discontinuation of the study drug. The most common subsequent therapy for those in the atezolizumab group was platinum-based chemotherapy (30.8%). In the EMPOWER-LUNG 1 trial, 19.9% of patients in the cemiplimab group received at least 1 subsequent anticancer therapy, with 14.3% continuing to receive cemiplimab plus chemotherapy and 6.5% receiving other systemic therapies (mainly chemotherapy).
Subsequent therapy (chemotherapy groups): In the IMpower110 trial, 54.1% of patients in the chemotherapy group received at least 1 subsequent anticancer therapy after discontinuation of the study drug (34.7% receiving immunotherapy and 21.4% receiving treatment with chemotherapy). In the EMPOWER-LUNG 1 trial, 44.1% of patients in the chemotherapy group of the cemiplimab trial received at least 1 subsequent anticancer therapy, with 43% receiving treatment with a PD-1 inhibitor (42.4% with cemiplimab and 0.6% with pembrolizumab). Complete details regarding subsequent anticancer therapies in the KEYNOTE-024 and KEYNOTE-042 trials were not reported by the sponsor and may not be available in the public domain.
Overall, CDA-AMC had no concerns regarding the decision to pool the trials based on the study design or patient characteristics; however, crossover in the EMPOWER-LUNG 1 trial may introduce bias for OS analysis.
Differences in subsequent therapy (particularly immunotherapy) across the trials make it challenging to assess comparability. Most notably, early stoppage of the EMPOWER-LUNG 1 trial due to a prespecified OS efficacy end point and widespread crossover may confound interpretation of the results. Lack of information in the public domain regarding subsequent therapies in the KEYNOTE-024 and KEYNOTE-042 trials makes it challenging to determine if the overall treatment course was comparable to the IMpower110 trial.
Follow-up times were different across the included studies, and the NMAs were not adjusted for these differences in exposure (IMpower110 [13.4 months], KEYNOTE-024 [25.2 months], EMPOWER-Lung 1 [10.8 months], and KEYNOTE-042 [12.8 months]). The proportion of patients with at least 1 treatment-related adverse event in the chemotherapy groups was similar across the studies (range: 85% to 90%).
Discontinuations due to adverse events are an end point for which the sponsor has claimed that atezolizumab offers improvements compared with cemiplimab and pembrolizumab. However, the proportion of patients in the chemotherapy group who withdrew because of adverse events (AEs) was considerably lower in the EMPOWER-LUNG 1 trial (4%) compared with the IMpower110 trial (16%) and KEYNOTE-024 and KEYNOTE-042 trials (11% and 14%, respectively).
CDA-AMC Clinical Review Discussion and Conclusion
This section was prepared by CDA-AMC based on the preceding sections.
Discussion
The evidence included in this review consisted of 1 pivotal study and an NMA. One randomized, open-label, phase III pivotal study (IMpower110) provided evidence for the efficacy and safety of atezolizumab compared to platinum chemotherapy in patients with stage IV NSCLC who have not received prior chemotherapy and who have tumour PD-L1 expression of 1% or more on PD-L1–expressing TCs or 1% of tumour area occupied by PD-L1–expressing ICs. The analysis submitted was based on a subpopulation of patients who were selected on the basis of high PD-L1 expression (≥ 50% of TC or ≥ 10% of IC). This subpopulation became the focus after a protocol amendment that revised the target population for the primary analysis of the IMpower110 trial to be those with high PD-L1 expression and without a sensitizing EGFR mutation or ALK translocation. The sponsor cited external data that suggested PD-L1 inhibitors were more effective in those with high PD-L1 expression and that the events in this group may be sufficient to allow for an analysis of OS benefit with less confounding from subsequent therapies than an analysis based on a later time point with the overall study population. This is the population that was submitted for regulatory review and reflected in the approved indications in Canada and internationally. Regulatory authorities examined the protocol amendment and ultimately concluded that analysis focusing on the subgroup of patients with high PD-L1 expression was internally valid and sufficient to warrant market authorization.34,35
In the absence of direct comparative evidence versus the relevant comparators of interest, the sponsor submitted an NMA comparing atezolizumab with pembrolizumab and cemiplimab for the outcomes of OS, PFS, and treatment-related adverse events.
The sponsor's application is seeking reimbursement for both IV and SC formulations of atezolizumab. There was no data for the SC formulation submitted for the target patient population because the SC formulation was evaluated in a different clinical development program. The SC formulation was approved by Health Canada in March 2024, and pCPA negotiations were completed for all indications in November 2024.
Efficacy
For patients who initiate immuno-oncology therapy, the product monograph for pembrolizumab specifies a maximum number of cycles for those without disease progression (i.e., up to 24 months or 35 doses for 200 mg or 18 doses for 400 mg, whichever is longer). The product monograph for cemiplimab does not recommend a maximum number of cycles, but the recommendation from CDA-AMC specified a maximum of 108 weeks based on the upper limit of cycles administered in the EMPOWER-LUNG 1 trial. The product monograph for atezolizumab does not specify a maximum number of doses, and patients in the IMpower110 trial could receive the treatment until disease progression or unacceptable toxicity. The patient and clinician groups who provided input to CDA-AMC did not comment on the absence of a specified maximum number of cycles in the product monograph, though the participating drug programs noted that it could increase expenditures relative to the comparators (particularly in the absence of clinical superiority). The review by NICE in the UK noted that the absence of a stopping rule could be valuable to people with untreated high PD-L1–expression metastatic NSCLC.38
The evidence for atezolizumab provided in this review was for the IV formulation, though a SC formulation has since been approved by Health Canada. The sponsor reported that the approval of the SC formulation was based on the IMscin001 trial results, which showed comparable pharmacokinetics and benefit-risk profiles to the IV formulation. Patient and clinician groups noted that the ability to administer atezolizumab SC would be beneficial for some patients, particularly those in rural communities who may need to travel extensively to receive IV administration of cancer therapies and those with poor IV access that would require central catheters that are prone to infection, dysfunction, and require constant maintenance. Some patients also prefer the shorter administration time with an SC formulation (though some noted the slower IV infusion was more tolerable). Clinician groups emphasized that SC administration can be beneficial for the health system and alleviate the burden on the infusion centres.
The IMpower110 trial met its primary end point with atezolizumab demonstrating a statistically significant improvement in OS compared with chemotherapy (HR: 0.59; 95% CI, 0.40 to 0.89) at the September 2018 CCOD (median follow-up of 15.7 months). The clinical experts consulted by CDA-AMC noted that the results from the IMpower110 trial suggested that treatment with atezolizumab monotherapy results in clinically meaningful improvements compared with chemotherapy, and the results were aligned with expectations for a PD-L1 inhibitor in this patient population. The sponsor’s updated OS analysis (February 2020 CCOD) for the target population (i.e., high PD-L1 expression) was considered exploratory and was not formally tested for statistical significance. At a median follow-up of 31.3 months (an additional 17 months from the interim analysis), the 95% CI for the HR crossed 1 (stratified HR, 0.76; 95% CI, 0.54 to 1.09), showing the point estimate for HR increased and the OS analysis was not nominally significant at this longer-term follow-up. In discussions with EMA, the sponsor noted that the change could be due to the subsequent use of immunotherapy by 34.7% of patients in the chemotherapy group following disease progression in the first-line setting (as the median OS increased only for the chemotherapy group [13.1 months versus 14.7 months at the updated analysis] and was maintained for the atezolizumab group). The EMA noted that further analyses substantiated the sponsor’s position but did not fully explain the improved response in the chemotherapy group. Overall, the EMA concluded that the failure to demonstrate nominal significance at 30.1 months of follow-up did not impact the benefit in OS shown for atezolizumab monotherapy, and regulatory approval was granted.34 As previously noted, the estimated between-group differences in OS rates at 12 and 24 months were imprecise (the 95% CIs included differences that are unlikely to be clinically meaningful) at both CCODs.
The sponsor submitted an NMA comparing atezolizumab versus pembrolizumab and cemiplimab for use as monotherapy in patients with metastatic NSCLC. Owing to multiple methodological concerns, the confidence of CDA-AMC in the results of the NMA is limited. Important limitations include the absence of a prespecified analysis plan or feasibility appraisal, risk of bias in the included trials, a sparse network, and uncertainty as to the plausibility of the transitivity assumption. Further, the between-group effect estimates for all relevant outcomes are critically imprecise; that is, they include the potential that atezolizumab may result in important benefit, important harm, or no difference compared with pembrolizumab or cemiplimab. This imprecision renders the NMA inconclusive, irrespective of other limitations.
Although the evidence submitted by the sponsor does not support any definitive conclusions about the efficacy of atezolizumab relative to pembrolizumab or cemiplimab, the clinical experts suggested that atezolizumab may present an additional treatment option for patients with metastatic NSCLC with high PD-L1 expression, particularly given the availability of a SC formulation. Based on their experience, they considered the adverse effects of different immune checkpoint inhibitors to be similar.
Reviews by NICE and the Scottish Medicines Consortium of atezolizumab only included pembrolizumab as a relevant comparator (cemiplimab was not approved or funded in the UK at the time of the review), and their expert committees concluded that the results from the sponsor’s NMA suggested no significant differences between atezolizumab and pembrolizumab.38
Harms
The product monographs for atezolizumab, pembrolizumab, and cemiplimab all include warnings regarding the risk of immune-mediated adverse reactions. The recommendations for monitoring patients receiving these therapies are similar in each of the product monographs and there is extensive clinical experience with PD-1 and PD-L1 inhibitors in the treatment of metastatic NSCLC. The clinical experts consulted by CDA-AMC noted that the adverse event profile of atezolizumab is similar to pembrolizumab and cemiplimab, and laboratory monitoring for patients receiving atezolizumab would generally be similar to the other regimens.
Indirect comparisons submitted by the sponsor included AEs and discontinuations due to AEs. The indirect estimates of effect were associated with very wide CrIs and lacked the precision to evaluate if there are meaningful differences across the treatments. The clinical experts consulted by CDA-AMC noted that clinical trial data from the IMpower110 trial do not suggest an increase or decrease in AEs compared with the alternative treatments. As with the assessment of comparative efficacy, the clinical experts consulted for this review consider the AEs of different immune checkpoint inhibitors to be similar based on their clinical experience.
Conclusion
Evidence from a pivotal phase III clinical trial (IMpower110) demonstrated that atezolizumab results in clinically meaningful improvements compared with chemotherapy in outcomes that are relevant for the management of NSCLC in the first-line metastatic setting, including OS, PFS, and objective response. The estimates of effect from the sponsor’s NMA for were limited by imprecision (i.e., wide CrIs), making it challenging to evaluate if there are meaningful differences across the different PD-1 and PD-L1 inhibitors approved for use in the target population (i.e., atezolizumab, pembrolizumab, and cemiplimab). However, the clinical experts consulted by CDA-AMC noted that in their experience, atezolizumab is associated with similar clinical benefits in comparison with pembrolizumab and cemiplimab (i.e., consistent with the sponsor’s claim of no added clinical benefit). Furthermore, the clinical experts consulted by CDA-AMC, patient groups, and clinician groups all noted that reimbursement of atezolizumab would be beneficial because it would provide a SC treatment option for this patient population. No data for the SC formulation were submitted in this application because the sponsor reported that the approval of the SC formulation was based on the IMscin001 trial results, which showed comparable pharmacokinetics and benefit-risk profiles to the IV formulation.
CDA-AMC Pharmacoeconomic Evaluation
The objective of the economic review undertaken by CDA-AMC is to review and critically appraise the pharmacoeconomic evidence submitted by the sponsor on the drug cost and budget impact of atezolizumab compared to pembrolizumab and cemiplimab for the Health Canada–indicated population. This review was submitted via the Pharmaceuticals With Anticipated Comparable Efficacy and Safety (PACES) Tailored Review pathway; therefore, the economic information provided within this report is limited to cost tables and budget impact.
Summary of the Submitted Cost Information
The sponsor submitted a cost comparison that included drug acquisition and health care resource costs associated with atezolizumab IV and SC versus pembrolizumab and cemiplimab. Drug costs were calculated by the sponsor using the submitted prices for atezolizumab IV and SC, assuming 95% of patients receive the SC form, and IQVIA DeltaPA (Delta Price Advisor) list prices for comparators.26,39 In their calculation of drug acquisition costs, the sponsor assumed that all patients will receive treatment for 5.3 months, which the sponsor equated to 7.7 cycles for treatments administered every 3 weeks (atezolizumab SC, cemiplimab), 5.8 cycles for treatments administered every 4 weeks (atezolizumab IV [1,680 mg every 4 weeks]), 3.8 cycles for treatments administered every 6 weeks (fixed-dose pembrolizumab [400 mg every 6 weeks]). The sponsor assumed that costs associated with genetic testing would not differ between atezolizumab and comparators.40
The sponsor assumed that the per-cycle costs associated with health care resource use would not differ between atezolizumab IV and comparators, whereas atezolizumab SC was assumed to require less preparation and administration time by nurses and less chemotherapy suite time). The sponsor’s calculations of costs associated with health care resource use is provided in Supplemental Materials, Appendix 10.
CDA-AMC Cost Comparison
CDA-AMC compared the drug acquisition costs for atezolizumab IV and SC to those for pembrolizumab and cemiplimab. Based on the sponsor’s submitted prices for atezolizumab IV and SC, and publicly available list prices of all comparators, reimbursement of atezolizumab is expected to be associated with lower drug costs to the health care system versus cemiplimab and fixed-dose pembrolizumab (incremental savings: $1,447 to $3,129 per 28-day cycle), and higher or lower cost versus weight-based pembrolizumab depending on the dosage of atezolizumab IV (incremental savings: $313 to incremental cost: $569 per 28-day cycle) (refer to Table 14 for cost differences between the SC and IV formulation of atezolizumab and comparators).
CDA-AMC notes that there may be health care resource use savings associated with the use of atezolizumab SC, given that SC administration may be associated with lower administration costs versus IV administration; however, the extent of cost savings is uncertain.
Table 14: CDA-AMC Drug Cost Comparison Table
Treatment | Strength or concentration | Form | Price ($) | Recommended dosage | 28-day drug cost ($) | Difference in 28-day cost ($) |
|---|---|---|---|---|---|---|
Atezolizumab (Tecentriq) | 840 mg 1,200 mg | 60 mg/mL vial for IV infusion | 4,743.2000a 6,776.0000a | 840 mg every 2 weeks, 1,200 mg every 3 weeks, or 1,680 mg every 4 weeks | 9,035 to 9,486 | Reference |
1,875 mg | 125 mg/mL solution for SC injection | 6,453.0000a | 1,875 mg every 3 weeks | 8,604 | ||
Comparators | ||||||
Cemiplimab (Libtayo) | 350 mg | 50 mg/mL vial for IV infusion | 8,200.0000 | 350 mg every 3 weeks | 10,934 | 1,447 to 2,329b |
Pembrolizumab (Keytruda) | 100 mg | 25 mg/mL vial for IV infusion | 4,400.0000 | Weight-based dosage: 2 mg/kg every 3 weeksc | 8,917 | −569 to 313b |
Fixed dosage: 200 mg every 3 weeks or 400 mg every 6 weeks | 11,733 | 2,247 to 3,129b | ||||
CDA-AMC = Canada’s Drug Agency; SC = subcutaneous.
Note: Prices are from the IQVIA DeltaPA (Delta Price Advisor) (accessed August 2025), unless otherwise indicated and do not include dispensing fees. All recommended dosages were obtained from the respective Health Canada product monographs. Cycle costs were calculated with an average patient weight of 76 kg.
aSponsor’s submitted price.40
bRange calculated using the lower and upper bounds of the expected atezolizumab 28-day drug cost.
cAssumes vial sharing.
Summary of the Budget Impact
The sponsor submitted a budget impact analysis to estimate the 3-year (2026 to 2028) budget impact of reimbursing atezolizumab for use in the Health Canada–indicated population.40 The sponsor assumed that the payer would be CDA-AMC–participating public drug plans and derived the size of the eligible population using an epidemiologic approach. The prices of atezolizumab IV and SC were aligned with the prices in the sponsor’s cost comparison, whereas the prices of comparators were based on the publicly available list prices.26,39 The sponsor assumed that all patients would undergo testing, with no difference in testing costs between treatments.40
CDA-AMC identified a number of issues with the sponsor’s estimated budget impact and made changes to model parameters and assumptions in consultation with clinical experts to derive the CDA-AMC base case (Supplemental Materials, Appendix 11). CDA-AMC estimated that 5,339 patients would be eligible for atezolizumab over a 3-year period (year 1 = 1,760; year 2 = 1,780; year 3 = 1,799), of whom 482 are expected to receive atezolizumab (year 1 = 88; year 2 = 178; year 3 = 216). CDA-AMC estimates that the reimbursement of atezolizumab for the Health Canada–indicated population may be cost saving for the public drug plans; however, the budget impact of reimbursing atezolizumab will depend on the number of eligible people, uptake of atezolizumab, and confidential prices for comparators.
Conclusion
At the submitted prices, the 28-day cycle cost per patient is expected to be $8,604 for atezolizumab SC and $9,035 to 9,486 for atezolizumab IV.
Using the sponsor’s submitted price for atezolizumab IV and SC and publicly available list prices for comparators, the 28-day drug cost of atezolizumab is expected to be lower than that of cemiplimab and fixed-dosage pembrolizumab and higher or lower than that of weight-based pembrolizumab.
CDA-AMC estimates that the reimbursement of atezolizumab for use in the Health Canada–indicated population may result in cost savings to the public drug plans; however, the extent of savings will depend on the number of people eligible for treatment, market uptake of atezolizumab and the proportion of patients who receive the IV versus SC formulation, and confidentially negotiated prices for comparators.
References
1.Canadian Cancer Society. Lung and bronchus cancer statistics [sponsor submitted reference]. 2024. https://cancer.ca/en/cancer-information/cancer-types/lung/statistics#:~:text=32%2C100%20Canadians%20will%20be%20diagnosed,and%20bronchus%20cancer%20every%20day.
2.Canada's Drug Agency. Provisional Funding Algorithm for Non-Small Lung Cancer without Actionable Oncogenic Alterations. June 2024. 2024.
3.Hendriks LE, Kerr KM, Menis J, et al. Non-oncogene-addicted metastatic non-small-cell lung cancer: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann Oncol. 2023;34(4):358-376. doi: 10.1016/j.annonc.2022.12.013 Medline PubMed
4.National Comprehensive Cancer N. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®). Non-Small Cell Lung Cancer Version 9.2024 — September 9, 2024. 2024.
5.Molina JR, Yang P, Cassivi SD, Schild SE, Adjei AA. Non-small cell lung cancer: epidemiology, risk factors, treatment, and survivorship. Mayo Clin Proc. 2008;83(5):584-94. doi: 10.4065/83.5.584 Medline PubMed
6.Howlader N, Noone A, Krapcho M, et al. SEER Cancer Statistics Review, 1975-2011 [sponsor submitted reference]. 2014. https://seer.cancer.gov/archive/csr/1975_2011/
7.Bradley SH, Kennedy MPT, Neal RD. Recognising Lung Cancer in Primary Care. Adv Ther. 2019;36(1):19-30. doi: 10.1007/s12325-018-0843-5 Medline PubMed
8.Canadian Cancer Statistics Advisory Committee, Canadian Cancer Society, Statistics Canada, Public Health Agency of Canada. Canadian cancer statistics 2023. Canadian Cancer Society; 2023. Accessed January 1, 1800. https://cdn.cancer.ca/-/media/files/cancer-information/resources/publications/canadian-cancer-statistics-2023/2023pdfen.pdf
9.Ellison LF, Saint-Jacques N. Five-year cancer survival by stage at diagnosis in Canada. Health Rep. 2023;34(1):3-15. doi: 10.25318/82-003-x202300100001-eng Medline PubMed
10.Grapatsas K, Leivaditis V, Tsilogianni Z, et al. Epidemiology, risk factors, symptomatology, TNM classification of Non Small Cell Lung Cancer. An overview while waiting the 8th TNM classification. Oncomedicine. 2017;2:14-23. doi: 10.7150/oncm.17097
11.Leighl NB, Ismaila N, Durm G, et al. Therapy for Stage IV Non-Small Cell Lung Cancer Without Driver Alterations: ASCO Living Guideline, Version 2024.3. J Clin Oncol. 2025;43(10):e17-e30. doi: 10.1200/JCO-24-02786 Medline PubMed
12.Borghaei H, Langer CJ, Gadgeel S, et al. Updated results from KEYNOTE-021 cohort G: A randomized, phase 2 study of pemetrexed and carboplatin (PC) with or without pembrolizumab (pembro) as first-line therapy for advanced nonsquamous NSCLC. Ann Oncol. 2017;28:v636-v637. doi: 10.1093/annonc/mdx440.052
13.Rittmeyer A, Barlesi F, Waterkamp D, et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. The Lancet. 2017;389(10066):255-265. doi: 10.1016/s0140-6736(16)32517-x Medline PubMed
14.Reck M, Rodriguez-Abreu D, Robinson AG, et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N Engl J Med. 2016;375(19):1823-1833. doi: 10.1056/NEJMoa1606774 Medline PubMed
15.Mok TSK, Wu Y-L, Kudaba I, et al. Pembrolizumab versus chemotherapy for previously untreated, PD-L1-expressing, locally advanced or metastatic non-small-cell lung cancer (KEYNOTE-042): a randomised, open-label, controlled, phase 3 trial. The Lancet. 2019;393(10183):1819-1830. doi: 10.1016/s0140-6736(18)32409-7 Medline PubMed
16.Hwang DM, Albaqer T, Santiago RC, et al. Prevalence and Heterogeneity of PD-L1 Expression by 22C3 Assay in Routine Population-Based and Reflexive Clinical Testing in Lung Cancer. J Thorac Oncol. 2021;16(9):1490-1500. doi: 10.1016/j.jtho.2021.03.028 Medline PubMed
17.Canadian Cancer Society. Diagnosis of lung cancer [sponsor submitted reference]. 2020. https://cancer.ca/en/cancer-information/cancer-types/lung/diagnosis
18.Pfister DG, Johnson DH, Azzoli CG, et al. American Society of Clinical Oncology treatment of unresectable non-small-cell lung cancer guideline: update 2003. J Clin Oncol. 2004;22(2):330-53. doi: 10.1200/JCO.2004.09.053 Medline PubMed
19.Grigg C, Rizvi NA. PD-L1 biomarker testing for non-small cell lung cancer: truth or fiction? J Immunother Cancer. 2016;4(1):48. doi: 10.1186/s40425-016-0153-x Medline PubMed
20.Sakaguchi T, Iketani A, Esumi S, et al. The Current Achievements of Multi-Gene Panel Tests in Clinical Settings for Patients with Non-Small-Cell Lung Cancer. Cancers (Basel). 2024;16(9):1670. doi: 10.3390/cancers16091670 Medline PubMed
21.Canadian Cancer Society. What is metastatic cancer? [sponsor submitted reference]. https://cancer.ca/en/cancer-information/cancer-types/metastatic/what-is-metastatic-cancer
22.Herbst RS, Giaccone G, de Marinis F, et al. Atezolizumab for First-Line Treatment of PD-L1-Selected Patients with NSCLC. N Engl J Med. 2020;383(14):1328-1339. doi: 10.1056/NEJMoa1917346 Medline PubMed
23.Jassem J, de Marinis F, Giaccone G, et al. Updated Overall Survival Analysis From IMpower110: Atezolizumab Versus Platinum-Based Chemotherapy in Treatment-Naive Programmed Death-Ligand 1-Selected NSCLC. J Thorac Oncol. 2021;16(11):1872-1882. doi: 10.1016/j.jtho.2021.06.019 Medline PubMed
24.Merck Canada. Keytruda Product Monograph [sponsor submitted reference]. 2025.
25.Regeneron Canada. Libtayo Product Monograph [sponsor submitted reference]. 2025.
26.Hoffmann-La Roche. Tecentriq SC Product Monograph [sponsor submitted reference]. 2025.
27.Hoffmann-La Roche. IMpower110 Protocol [sponsor submitted reference]. 2019.
28.CADTH. Drug Reimbursement Expert Review Committee final recommendation: pembrolizumab (Keytruda - Merck). August 23, 2017. Accessed August 13, 2025. https://www.cda-amc.ca/sites/default/files/pcodr/pcodr_pembrolizumab_keytruda_nsclc_1stln_fn_rec.pdf
29.Canada's Drug Agency. Reimbursement recommendation: cemiplimab (Libtayo). Can J Health Technol. 2022;2(6). doi: DOI.
30.Hoffmann-La Roche. Tecentriq Product Monograph [sponsor submitted reference]. 2025.
31.IMpower110 Primary Clinical Study Report. Hoffmann-La Roche.
32.Hoffmann-La Roche. IMpower110 Statistical Analysis Plan [sponsor submitted reference].
33.IMpower110 Final Clinical Study Report. Hoffmann-La Roche.
34.European Medicines Agency. Tecentriq (atezolizumab) CHMP extension of indication variation assessment report. 2021. https://www.ema.europa.eu/en/documents/variation-report/tecentriq-h-c-004143-ii-0033-epar-assessment-report-variation_en.pdf
35.Health Canada. Tecentriq (atezolizumab) Biologics Safety and Efficacy Assessment Report (BSEAR) [CONFIDENTIAL]. 2021.
36.Latimer N, Abrams K. NICE DSU Technical Support Document 16: Adjusting Survival Time Estimates in the Presence of Treatment Switching. Decision Support Unit, ScHARR, University of Sheffield; 2014. Accessed September 17, 2025. https://sheffield.ac.uk/sites/default/files/2022-02/TSD16_Treatment_Switching.pdf
37.McKesson Canada PDCI Market Access. Network Meta-analysis of Tecentriq and its relevant comparators for Canadian Submission for the treatment of stage IV squamous or non- squamous NSCLC and PD-L1 high-expression [sponsor submitted reference]. 2025.
38.National Institute for Health and Care Excellence. Atezolizumab monotherapy for untreated advanced non-small-cell lung cancer. 2021. https://www.nice.org.uk/guidance/ta705
39.IQVIA. Delta PA [sponsor submitted reference]. 2025. Accessed Aug 19, 2025.
40.Hoffmann-La Roche. Tecentriq Budget Impact Analysis (BIA) [sponsor submitted reference]. 2025.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.