Drugs, Health Technologies, Health Systems
Reimbursement Review
Talazoparib (Talzenna)
Sponsor: Pfizer Canada ULC
Therapeutic area: Metastatic castration-resistant prostate cancer
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
AAP
abiraterone acetate plus prednisone
ADT
androgen deprivation therapy
AE
adverse event
ARPI
androgen receptor pathway inhibitor
BICR
blinded independent central review
BPI-SF
Brief Pain Inventory–Short Form
CCS
Canadian Cancer Society
CDA-AMC
Canada’s Drug Agency
CI
confidence interval
CRPC
castration-resistant prostate cancer
CSPC
castration-sensitive prostate cancer
ECOG PS
Eastern Cooperative Oncology Group Performance Status
EORTC QLQ-PR25
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Prostate Cancer
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HR
hazard ratio
HRQoL
health-related quality of life
HRR
homologous recombination repair
ITT
intention to treat
MAIC
matching-adjusted indirect comparison
mCRPC
metastatic castration-resistant prostate cancer
MID
minimal important difference
NGS
next-generation sequencing
NHT
novel hormonal therapy
OH (CCO)
Ontario Health (Cancer Care Ontario)
OS
overall survival
PARP
poly(adenosine diphosphate-ribose) polymerase
PSA
prostate-specific antigen
RCT
randomized controlled trial
RECIST
Response Evaluation Criteria in Solid Tumours
rPFS
radiographic progression-free survival
TEAE
treatment-emergent adverse event
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Talazoparib (Talzenna), 0.1 mg and 0.25 mg oral capsules |
Sponsor | Pfizer Canada ULC |
Indication | Talzenna (talazoparib) in combination with enzalutamide is indicated for the treatment of adult patients with homologous recombination repair gene-mutated metastatic castration-resistant prostate cancer |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | January 30, 2025 |
Recommended dose | Talazoparib 0.5 mg orally once daily in combination with enzalutamide 160 mg orally once daily, until disease progression or unacceptable toxicity. Patients receiving talazoparib plus enzalutamide should also receive a gonadotropin-releasing hormone analogue concurrently or should have had bilateral orchiectomy. |
NOC = Notice of Compliance.
Introduction
Castration-resistant prostate cancer (CRPC) is defined by disease progression despite castrate levels of testosterone and may present as either a continuous rise in serum prostate-specific antigen (PSA) levels, the progression of preexisting disease, and/or the appearance of new metastases. A patient may progress to metastatic CRPC (mCRPC) from metastatic castration-sensitive prostate cancer (CSPC) based on biochemical recurrence (characterized by rising PSA levels despite medical or surgical castration) or from nonmetastatic CRPC based on presentation of metastases (assessed radiographically). Progressing to mCRPC is characterized by increased symptomatic burden and reduced health-related quality of life (HRQoL). In Canada, the estimated prevalence of mCRPC is 1.2% to 2.1% of total prostate cancer cases. The expected 5-year survival for males diagnosed with prostate cancer is 91% for all stages combined, and for metastatic disease, the 5-year survival rate is approximately 28%.
According to the clinical experts consulted by Canada’s Drug Agency (CDA-AMC), the main treatment goals for patients with mCRPC are to prolong survival, delay disease progression, improve symptoms, and maintain HRQoL. Systemic therapies for the treatment of patients with mCRPC, and the sequencing of these treatments, depends on patient and disease factors, prior treatments used in the metastatic CSPC setting, and access, which varies across Canada. Docetaxel, cabazitaxel, abiraterone acetate, enzalutamide, radium-223 (for patients with bone-only metastatic disease), lutetium vipivotide tetraxetan, and olaparib, olaparib plus abiraterone acetate plus prednisone (AAP), or niraparib-abiraterone plus prednisone for patients with BRCA1, BRCA2, and/or ATM mutations, are all Health Canada approved, and most are available across the jurisdictions in Canada.
Talazoparib is a potent inhibitor of poly(adenosine diphosphate-ribose) polymerase (PARP) enzymes, which are involved in the homologous recombination repair (HRR) pathway. The approved Health Canada indication for talazoparib is in combination with enzalutamide for the treatment of adult patients with HRR gene-mutated mCRPC. The reimbursement request is aligned with the Health Canada–approved indication. The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of talazoparib 0.5 mg plus enzalutamide 160 mg, taken orally once daily in the treatment of adults with HRR gene-mutated mCRPC.
Perspectives of Patients, Clinicians, and Drug Programs
The information in this section is a summary of input provided by the patient and clinician groups who responded to our call for input and from clinical experts consulted by CDA-AMC for the purpose of this review.
Patient Input
CDA-AMC received input from 2 patient groups, PROCURE and the Canadian Cancer Society (CCS). According to these inputs, the main concerns of patients included distress and treatment decisions, rising PSA levels after treatment, recurrence, hormone therapy and its side effects, as well as metastases. mCRPC mostly impacted patients in sexual activity, followed by the ability to work, maintaining a positive mental health, concentration, travel, exercise, or conducting household chores. Both patient groups clarified that patients' expectations with regard to new treatments included cancer control with fewer side effects, longer-lasting effects, delaying the onset or elimination of metastases, decreasing or maintaining PSA levels over a long period, convenient treatment regimens, prolonging life, and improving quality of life. Patients also expected treatments to be affordable, have better accessibility, better follow-up for long-term issues, and to be heard and taken seriously.
Clinician Input
Input From Clinical Experts Consulted for This Review
The clinical experts indicated that, because mCRPC is a terminal phase of prostate cancer, the unmet needs of patients would be new treatments that would prolong survival and maintain or improve quality of life, while exposing patients to minimal toxicity. Both clinical experts highlighted that the balance between treatment efficacy and quality of life would be important. The clinical experts noted that it remains unclear whether talazoparib plus enzalutamide would lead to a shift in the current treatment paradigm. This uncertainty stems from the increased use of androgen receptor pathway inhibitors (ARPIs) in patients with metastatic CSPC and nonmetastatic CRPC in recent years. They noted that many medical oncologists currently favour prescribing chemotherapy as first-line mCRPC treatment in patients who have previously progressed on an ARPI in the metastatic CSPC and nonmetastatic CRPC settings. They noted that if a treatment was used in metastatic CSPC, it is not likely for the patient to receive it again for first-line mCRPC (with the occasional exception of docetaxel if given at least 1 year prior). The clinical experts noted that talazoparib plus enzalutamide may have a limited role as a first-line treatment in the mCRPC setting due to the decreasing number of patients who are ARPI naive and the few patients who would be clinically ineligible for docetaxel; however, it may have a role in subsequent lines of therapy. The clinical experts indicated that patients best suited for talazoparib plus enzalutamide would be those who match the eligibility criteria of the TALAPRO-2 trial, which included first-line treatment for HRR gene-mutated mCRPC, and no contraindications to talazoparib or enzalutamide. The experts indicated that in clinical practice, a combination of radiographic, biochemical (e.g., PSA), and clinical parameters (i.e., decrease in disease-related symptoms) is used to determine whether a patient with mCRPC is responding or progressing while receiving treatment. They noted that, at the very least, assessments should be performed at 3-month intervals. The experts indicated that treatment with talazoparib plus enzalutamide should be discontinued if patients experience disease progression (as defined radiologically or clinically), treatment is intolerable, or if this is the patient’s preference. The experts noted that PARP inhibitors such as talazoparib have the potential to be toxic; therefore, patients receiving talazoparib plus enzalutamide should be under the care of a medical oncologist who can manage toxicity associated with the therapy.
Clinician Group Input
CDA-AMC received 1 input from the Ontario Health (Cancer Care Ontario) (OH [CCO]) Genitourinary Cancer Drug Advisory Committee. According to the clinician group input, the treatment goal is prolonging life and improving quality of life. The group noted that there are no cures currently available for metastatic prostate cancer and there is a need for treatments that prolong life. The group noted that talazoparib in combination with enzalutamide should be used in patients with treatment-naive mCRPC and that treatment with chemotherapy or an ARPI in the metastatic CSPC setting should not preclude eligibility for treatment with talazoparib plus enzalutamide, as per the TALAPRO-2 trial. The group noted that PSA and serial radiographic imaging would be used to monitor response to therapy and that significant side effects and progression of disease on imaging are among the factors for considering discontinuation of the treatment. The clinician group noted that medical oncologists, radiation oncologists, and urologists specialized in prostate cancer care are among the specialists or prescribers that would be required for prescribing the treatment. The OH (CCO) Genitourinary Cancer Drug Advisory Committee added that access to this (and other) combinations in this setting would also require ongoing efforts to ensure equitable, timely access to genomic testing of relevant alterations for all eligible males with prostate cancer living in Canada.
Drug Program Input
Input was obtained from the drug programs that participate in the CDA-AMC reimbursement review process. The following were identified as key factors that could potentially impact the implementation of a CDA-AMC recommendation for talazoparib plus enzalutamide:
relevant comparators
considerations for initiation of therapy
considerations for discontinuation of therapy
considerations for prescribing of therapy
generalizability
funding algorithm
care provision issues.
The clinical experts consulted by CDA-AMC provided advice on the potential implementation issues raised by the drug programs (refer to Table 4).
Testing Procedure Considerations
According to the clinical experts consulted by CDA-AMC, public funding status and clinical practices for somatic or germline HRR mutation testing in patients with mCRPC are not consistent across Canada. Next-generation sequencing (NGS) panels for mutations in BRCA1, BRCA2, and ATM are generally available in jurisdictions. However, there are several other genes that are involved in the HRR pathway, the testing for which is not available or accessible to all patients. Therefore, a small proportion of patients who have mutations in genes other than BRCA1, BRCA2, or ATM may not be identified for eligibility for talazoparib depending on what testing panels are available in their region. There are existing implementation concerns related to testing within health systems, patients, and costs; however, minimal additional impact is anticipated if talazoparib were to be reimbursed.
Clinical Evidence
Systematic Review
Description of Studies
The TALAPRO-2 trial (cohort 2, N = 399) met the inclusion criteria for the systematic review conducted by the sponsor. An objective of the trial was to assess the efficacy and safety of talazoparib 0.5 mg plus enzalutamide 160 mg, taken orally once daily, or matched placebo plus enzalutamide in adult patients with HRR-deficient mCRPC. The trial enrolled adults with asymptomatic or mildly symptomatic mCRPC who had not started systemic cancer treatment after the diagnosis of CRPC (metastatic or nonmetastatic), with the exception of androgen deprivation therapy (ADT) and first-generation antiandrogen drugs. Patients were allowed to have previously received abiraterone acetate or docetaxel for CSPC but were ineligible to participate if they had received any prior treatment with second-generation ARPIs androgen receptor inhibitors (enzalutamide, apalutamide, and darolutamide), a PARP inhibitor, cyclophosphamide, or mitoxantrone for prostate cancer. Patients were required to have had an Eastern Cooperative Oncology Group Performance Status (ECOG PS) score of 0 or 1, and progressive disease at study entry. The approved Health Canada indication and reimbursement request aligned with the trial’s HRR-deficient population. The outcomes relevant to this review included the primary outcome of radiographic progression-free survival (rPFS), key secondary outcome of overall survival (OS), and HRQoL outcomes of time to deterioration of pain symptoms measured via the Brief Pain Inventory–Short Form (BPI-SF) and the European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Prostate Cancer (EORTC QLQ-PR25) functional and symptom scales and safety. The rPFS data are based on the primary analysis data cut-off date of October 3, 2022, and supportive data from the data cut-off date of September 3, 2024. All other outcomes are based on the data cut-off date of September 3, 2024. Overall, key baseline characteristics were generally balanced between treatment groups. The trial population was predominately white (68%), with a median age of 70 years. Most patients had an ECOG PS score of 0 (approximately 62%), indicating good overall performance; normal or mild renal impairment (approximately 90%); bone and soft tissue disease-site metastasis (approximately 84%); a Gleason score greater than or equal to 8 (approximately 74%); and had prior therapy with surgery or biopsy ██████████████ ████ and first-generation antiandrogens ██████████████ █████ In both groups, the most common detected HRR gene alteration was BRCA2 (33.8%), followed by ATM (21.6%), CDK12 (18.8%), and CHEK2 (17.8%). The talazoparib plus enzalutamide group had a ██████ ████ ████████ █████ ███ █████ ██████ █████ ███ █ ██████ ██████████ ██ ████████ ███ ███ ████████ ██████ █████████ ██████ ██████ ██████. These imbalances were likely due to chance, as ███ █████ ████████ ███████████████ ██ ████████ ████████ ████████ ███████ ███████
Efficacy Results
Only those efficacy outcomes and analyses of subgroups identified as important to this review are reported. The main findings for the efficacy outcomes for the TALAPRO-2 trial are from the data cut-off dates of October 3, 2022, and September 3, 2024. The boundary for statistical significance for the primary outcome of rPFS was met at the data cut-off date of October 3, 2022; therefore, rPFS data were reported descriptively at the September 3, 2024, data cut-off (i.e., not controlled for type I error). All other outcomes are based on the data cut-off date of September 3, 2024.
rPFS by Blinded Independent Central Review
In total, 170 events had occurred in both groups by the data cut-off date of October 3, 2022. The median duration of follow-up for rPFS was 17.5 months (range not reported) for the talazoparib plus enzalutamide group and 16.8 months (range not reported) for the placebo plus enzalutamide group. The median rPFS was not reached (95% confidence interval [CI], 21.9 months to not reached) in the talazoparib plus enzalutamide group and 13.8 months (95% CI, 11.0 months to 16.7 months) for the placebo plus enzalutamide group (log-rank test P < 0.0001), with a between-group hazard ratio (HR) of 0.45 (95% CI, 0.33 to 0.61) in favour of talazoparib plus enzalutamide. The Kaplan-Meier–estimated probability of rPFS at 12 months was ████ █ █████████████ ██████████ ██ █████ ████ ███ ████ ██ ██████ in the talazoparib plus enzalutamide and placebo plus enzalutamide groups, respectively, ████ █ █████████████ ██████████ ██ █████ ████ ███ ████ ██ ██████ The results of sensitivity analyses were consistent with the primary analysis. At the second data cut-off date of September 3, 2024, rPFS descriptive results were consistent with the first data cut-off date. The Kaplan-Meier–estimated probability of rPFS at 48 months was █████ ████ ███ ████ ██ █████ ██████ █████ ████ ███ ███ ██ █████ in the talazoparib plus enzalutamide and placebo plus enzalutamide groups, respectively, ████ █ █████████████ ██████████ ██ █████ ████ ███ ████ ██ █████. The rPFS results were consistent across the subgroup analyses of interest by BRCA alteration status (with or without) and prior treatment with novel hormonal therapy (NHT) or taxane therapy in favour of talazoparib plus enzalutamide.
Overall Survival
In total, 219 events had occurred in both groups by the data cut-off date of September 3, 2024. The median follow-up for OS was 44.2 months for the talazoparib plus enzalutamide group and ████ ██████ for the placebo plus enzalutamide group. The median OS was 45.1 months (95% CI, 35.4 months to not reached) for the talazoparib plus enzalutamide group and 31.1 months (95% CI, 27.3 months to 35.4 months) in the placebo plus enzalutamide group (log-rank test P < 0.0001), with a between-group HR of 0.62 (95% CI, 0.48 to 0.81) in favour of talazoparib plus enzalutamide. The results of sensitivity analyses were consistent with the primary analysis. The Kaplan-Meier–estimated probability of OS at 12 months was █████ ████ ███ ████ ██ █████ ██████ █████ ████ ███ ████ ██ █████ ███████ █████ ███████████ ████ ████ ███ ████ ██ █████; and OS at 48 months was █████ ████ ███ ████ ██ █████ ██████ █████ ████ ███ ████ ██ █████ █████████████ ███████████ █████ ████ ███ ████ ██ ██████ in the talazoparib plus enzalutamide and placebo plus enzalutamide groups, respectively.
Time to Deterioration of Pain by BPI-SF
At the data cut-off date of September 3, 2024, 58 events had occurred in both groups, and the median time to deterioration of pain was not reached in either group. The stratified between-group HR was 0.55 (95% CI, 0.33 to 0.94) in favour of talazoparib plus enzalutamide. The Kaplan-Meier–estimated probability of being free of pain progression at 12 months was █████ ████ ██ ███ █████████ ██████ █████ ████ ██ ███ █████████ █████████████ ███████████ ████ ████ ███ ████ ██ ██████, and at 48 months it ███ █████ ████ ██ ███ █████████ ██████ █████ ████ ██ ███ █████████ █████████████ ███████████ █████ ████ ███ ███ ██ ██████ in the talazoparib plus enzalutamide and placebo plus enzalutamide groups, respectively.
HRQoL by EORTC QLQ-PR25
At the data cut-off date of September 3, 2024, ██ ███ ██ ███ patients in the talazoparib plus enzalutamide group and ██ ███ ██ ███ patients in the placebo plus enzalutamide group contributed to almost all the EORTC QLQ-PR25 functional and symptom scale score analyses up to week 109; the incontinence aid symptoms score was informed by ██ ███ ██ patients, respectively. No comparison was made for the sexual functioning score due to limited data. The sexual activity score and the symptom scale scores were generally maintained and similar in both treatment groups up to week 109. Improvements in urinary and bowel symptoms favoured the talazoparib plus enzalutamide group, with urinary symptoms reaching the minimal important difference (MID) threshold of greater or equal to 5 points. All other scale scores did not reach the clinically important MID threshold.
Harms Results
Harms data reported in this section are from the data cut-off date of September 3, 2024. Almost all patients in both treatment groups reported at least 1 treatment-emergent adverse event (TEAE) (99.5% with talazoparib plus enzalutamide and 97.5% with placebo plus enzalutamide). The most frequently reported TEAEs in the talazoparib plus enzalutamide group were anemia (66.7% versus 18.6% with placebo plus enzalutamide), fatigue (34.8% versus 28.1% with placebo plus enzalutamide), and decreased neutrophil count (34.8% versus 7.0% with placebo plus enzalutamide); a higher proportion of patients in the talazoparib plus enzalutamide group reported these TEAEs than the placebo plus enzalutamide group. The most frequently reported TEAEs in the placebo plus enzalutamide group were fatigue (28.1% versus 34.8% with talazoparib plus enzalutamide), arthralgia (24.6% versus 16.7% with talazoparib plus enzalutamide), and back pain (23.1% versus 24.2% with talazoparib plus enzalutamide). A higher proportion of patients in the talazoparib plus enzalutamide group experienced at least 1 grade 3 or grade 4 TEAE ███████ versus the placebo plus enzalutamide group ████████ The incidence of serious TEAEs was higher in the talazoparib plus enzalutamide group (█████) versus the placebo plus enzalutamide group (██████ In both groups, ██████ █████ ████████ ███ ███████ █████ █████████ █████ ██ █████ were the most frequently reported serious TEAEs. A higher proportion of TEAEs that led to study treatment discontinuation was reported in the talazoparib plus enzalutamide group ██████ versus the placebo plus enzalutamide group ███████ with anemia being the most common reason █████ ███ ██████ Enzalutamide-only treatment discontinuation was reported for ████ of patients in the talazoparib plus enzalutamide group versus ████ in the placebo plus enzalutamide group. A lower proportion of deaths was reported in the talazoparib plus enzalutamide group (46.5%) versus in the placebo plus enzalutamide group (63.3%), with ███████ ███████████ being the primary reason for death in both groups ██████ ██████ ███████ The incidence of notable harms in both treatment groups was comparable and infrequent. Second primary malignancies (other than hematologic) occurred in ████ ███ ████ of patients, and embolic and thrombotic events occurred in 5.6% and 1.0% of patients in the talazoparib plus enzalutamide and placebo plus enzalutamide groups, respectively. There was 1 case of pneumonitis in each group.
Critical Appraisal
TALAPRO-2 was a randomized, double-blind, phase III trial. The patients and investigators were blinded to talazoparib or placebo, but enzalutamide was open label. This design was appropriate because different dosing and oral tablets across the 2 treatment groups may have made blinding to enzalutamide impractical. Randomization procedures, including stratification by prior NHT or taxane therapy for CSPC and HRR gene alteration status (deficient versus nondeficient or unknown), were appropriate and conducted by interactive response technology. The talazoparib plus enzalutamide group had a ██████ ████ ████████ █████ ███ █████ ██████ █████ ███ █ ██████ ██████████ ██ ████████ ███ ███ ████████ ██████ █████████ ██████ ██████ ██████. These imbalances were likely due to chance, as ███ █████ ████████ ███████████████ ██ ████████ ████████ ████████ ███████ ██████ and based on clinical expert feedback, unlikely to have confounded the effect between treatment and outcomes. Sample size and power calculations were based on the primary outcome of rPFS, and the trial was powered to detect significant differences for rPFS, but it was underpowered for the secondary outcome of OS. The analyses were preplanned with adequately justified stopping boundaries and the prespecified analyses of rPFS and OS were appropriately controlled for multiple comparisons. All other analyses were descriptive (i.e., not controlled for type I error). To minimize the risk of bias in the measurement of rPFS, the trial performed tumour assessments using Response Evaluation Criteria in Solid Tumours Version 1.1 (RECIST 1.1) criteria and radiographic scans were assessed by blinded independent central review (BICR). In addition, the findings of the sensitivity analyses for rPFS were consistent with the primary analysis. Patients were permitted to receive posttreatment anticancer medications after study treatment had been discontinued (36.9% in the talazoparib plus enzalutamide group versus 56.8% in the placebo plus enzalutamide group), which may influence the assessment of OS. Because no sensitivity analyses were performed to test treatment policy strategy for OS (e.g., exclude the effect of subsequent therapies), the estimated effect would be a combination of treatment with talazoparib plus enzalutamide versus placebo plus enzalutamide, plus subsequent treatments. Therefore, survival results might be partially attributable to treatments administered after disease progression rather than the study treatment itself, although without the necessary analysis, the direction and magnitude of bias are unclear. This is a relevant comparison; however, it is reflective of how the intervention and comparator and subsequent therapies would be used in practice in Canada. Based on the Clinical Study Report and statistical analysis plan, the proportional hazards assumption was not assessed or discussed. Despite the absence of these results, visual inspection of the Kaplan-Meier curves for rPFS and OS appear to indicate a clear separation (at approximately 3 months and 8 months, respectively), after which there appeared to be sustained proportionality throughout study treatment. HRQoL was assessed by the BPI-SF and EORTC QLQ-PR25 questionnaires, which have been validated in patients with metastatic prostate cancer with evidence of reliability, responsiveness, and MID. The result of these outcomes was subject to potential bias due to ████ ████ ███ ██ ███████ ████ ██ ████ ██████ ████ █ ██████ ██████████ ██ ███████ ████ ██ ███ ████████████████████ ██████ which could have biased the results in favour of talazoparib plus enzalutamide. For the EORTC QLQ-PR25, no models other than the mixed model for repeated measures were tested that applied alternative imputation methods or sensitivity analyses to assess the impact of missing data.
In general, the population requested for the reimbursement aligns with the approved Health Canada indication, and the dosing and administration of talazoparib plus enzalutamide were consistent with the approved product monograph. However, the trial provided talazoparib plus enzalutamide as first-line treatment only (i.e., patients had not received prior systemic therapy for mCRPC) while the approved Health Canada indication is line agnostic. Therefore, there is no direct comparative evidence for the use of talazoparib plus enzalutamide in later-line settings. The clinical experts consulted by CDA-AMC noted that many medical oncologists currently favour prescribing chemotherapy for the first-line treatment of mCRPC in patients who have previously progressed on an ARPI in the metastatic CSPC and nonmetastatic CRPC settings. Therefore, the clinical experts indicated that most clinicians would likely prescribe talazoparib plus enzalutamide as a second-line and beyond treatment due to the decreasing number of patients who are ARPI naive in the first-line mCRPC setting. According to the clinical experts, the eligibility criteria and baseline characteristics of the TALAPRO-2 trial are generalizable to adult patients with mCRPC in Canada, except that the trial did not include patients with an ECOG PS of greater than 1. The clinical experts indicated that patients with good ECOG PS or a score of 0 to 2 should be eligible for talazoparib plus enzalutamide, if they are able to tolerate the therapy. The experts noted that although enzalutamide was an appropriate comparator when the TALAPRO-2 trial was designed and executed, the current treatment paradigm has shifted since then toward the use of chemotherapy as the most common first-line treatment for patients with mCRPC. The evidence submitted to CDA-AMC did not include head-to-head comparisons between talazoparib plus enzalutamide and chemotherapy, which represents a gap in the available direct evidence given the potential shared place in therapy when used as first-line treatments for mCRPC.
GRADE Summary of Findings and Certainty of the Evidence
For pivotal studies and randomized controlled trials (RCTs) identified in the sponsor’s systematic review, Grading of Recommendations, Assessment, Development and Evaluations (GRADE) was used to assess the certainty of the evidence for outcomes considered most relevant to inform CDA-AMC expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group.
Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null.
The reference points for the certainty of evidence assessment for rPFS and OS were set according to the presence or absence of an important effect based on thresholds informed by the clinical experts consulted for this review. A threshold could not be determined for time to deterioration of pain by BPI-SF; therefore, the target of certainty appraisal was any effect. The reference point for the certainty of the evidence assessment for EORTC QLQ-PR25 functional and symptom scale scores was set according to the presence or absence of an important effect based on a threshold informed by the literature.
The selection of outcomes for GRADE assessment was based on the sponsor’s Summary of Clinical Evidence, consultation with clinical experts, and input received from patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members:
survival outcomes (rPFS, OS)
HRQoL outcomes (time to deterioration of pain by BPI-SF, EORTC QLQ-PR25 functional and symptom scale scores).
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for talazoparib plus enzalutamide versus placebo plus enzalutamide.
Table 2: Summary of Findings for Talazoparib Plus Enzalutamide vs. Placebo Plus Enzalutamide for Patients With HRR Gene-Mutated mCRPC
Outcome and follow-up | Patients (studies) N | Relative effect (95% CI) | Absolute effects (95% CI) | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
Placebo + enzalutamide | Talazoparib + enzalutamide | Difference | |||||
rPFS — ITT population | |||||||
Probability of rPFS at 12 months Median follow-up: 17.5 months for talazoparib+ enzalutamide and 16.8 months for placebo + enzalutamide; data cut-off date of October 3, 2022 | 399 (1 RCT) | NA | ███ ███ █████ | ███ ███ █████ ████ ██ ████ | ███ ████ ███ █████ ████ ████ ██ ███ █████ | Moderatea | Talazoparib + enzalutamide likely results in a clinically important higher probability of rPFS at 12 months when compared with placebo + enzalutamide. |
Probability of rPFS at 48 months Median follow-up: 38.0 months for talazoparib + enzalutamide and 30.8 months for placebo + enzalutamide; data cut-off date of September 3, 2024 | 399 (1 RCT) | NA | ███ ███ █████ | ███ ███ █████ ████ ██ ████ | ███ ████ ███ █████ ████ ████ ██ ███ █████ | Moderateb | Talazoparib + enzalutamide likely results in a clinically important higher probability of rPFS at 48 months when compared with placebo + enzalutamide. |
OS — ITT population, data cut-off date of September 3, 2024 | |||||||
Probability of survival at 12 months Median follow-up: 44.2 months for talazoparib + enzalutamide and ████ ██████ for placebo + enzalutamide | 399 (1 RCT) | NA | ███ ███ █████ | ███ ███ █████ ████ ██ ████ | ██ ████ ███ █████ ███ █████ ██ ██ █████ | Highc | Talazoparib + enzalutamide results in little to no clinically important difference in the probability of survival at 12 months when compared with placebo + enzalutamide. |
Probability of survival at 48 months Median follow-up: 44.2 months for talazoparib + enzalutamide and ████ ██████ for placebo + enzalutamide | 399 (1 RCT) | NA | ███ ███ █████ | ███ ███ █████ ████ ██ ████ | ███ ████ ███ █████ ████ ████ ██ ███ █████ | Highd | Talazoparib + enzalutamide results in a clinically important higher probability of survival at 48 months when compared with placebo + enzalutamide. |
Time to deterioration of pain by BPI-SF — ITT subset population, data cut-off date of September 3, 2024 | |||||||
Probability of being free of pain progression at 12 months Median follow-up: NR | 394 (1 RCT) | NA | ███ ███ █████ | ███ ███ █████ ████ | ██ ████ ███ █████ ███ █████ ██ ███ █████ | Lowe | Talazoparib + enzalutamide may result in a higher probability of being free of pain progression at 12 months when compared with placebo + enzalutamide. The clinical importance of the increase is uncertain. |
Probability of being free of pain progression at 48 months Median follow-up: NR | 394 (1 RCT) | NA | ███ ███ █████ | ███ ███ █████ ████ | ███ ████ ███ █████ ███ ████ ██ ███ █████ | Lowf | Talazoparib + enzalutamide may result in a higher probability of being free of pain progression at 48 months when compared with placebo + enzalutamide. The clinical importance of the increase is uncertain. |
EORTC QLQ-PR25 functional scale — ITT subset population, data cut-off date of September 3, 2024 | |||||||
Mean change from baseline in sexual activity score; scores range from 0 to 100, with higher scores indicating better function Time point: up to 109 weeks | ██ (1 RCT) | NA | ████ | ████ █████ ██ █████ | ████ █████ ██ ████ | Lowg | Talazoparib + enzalutamide may result in little to no clinically important difference in sexual activity up to 109 weeks when compared with placebo + enzalutamide. |
Mean change from baseline in EORTC QLQ-PR25 sexual functioning score; scores range from 0 to 100, with higher scores indicating better function Time point: up to 109 weeks | || (1 RCT) | NA | ██ | ██ | ██ | NE | NE |
EORTC QLQ-PR25 symptom scale — ITT subset population, data cut-off date of September 3, 2024 | |||||||
Mean change from baseline in urinary symptoms score; scores range from 0 to 100, with higher scores indicating worsened symptoms Time point: up to 109 weeks | ██ (1 RCT) | NA | ████ | ████ █████ ██ █████ | −5.0 (−7.6 to −2.4) | Very lowh | The evidence is very uncertain about the effect of talazoparib + enzalutamide on urinary symptoms up to 109 weeks when compared with placebo + enzalutamide. |
Mean change from baseline in bowel symptoms score; scores range from 0 to 100, with higher scores indicating worsened symptoms Time point: up to 109 weeks | ██ (1 RCT) | NA | ███ | ███ ████ ██ ████ | −1.8 (−3.3 to −0.4) | Lowg | Talazoparib + enzalutamide may result in little to no clinically important difference in bowel symptoms up to 109 weeks when compared with placebo + enzalutamide. |
Mean change from baseline in hormonal treatment-related symptoms score; scores range from 0 to 100, with higher scores indicating worsened symptoms Time point: up to 109 weeks | ██ (1 RCT) | NA | ████ | ████ █████ ██ █████ | −0.8 (−2.5 to 1.0) | Lowg | Talazoparib + enzalutamide may result in little to no clinically important difference in hormonal treatment-related symptoms up to 109 weeks when compared with placebo + enzalutamide. |
Mean change from baseline in incontinence aid symptoms score; scores range from 0 to 100, with higher scores indicating worsened symptoms Time point: up to109 weeks | ██ (1 RCT) | NA | ████ | ████ ████ ██ █████ | 0.2 (−4.8 to 5.3) | Very lowi | The evidence is very uncertain about the effect of talazoparib + enzalutamide on incontinence aid symptoms up to 109 weeks when compared with placebo + enzalutamide. |
BPI-SF = Brief Pain Inventory–Short Form; CI = confidence interval; EORTC QLQ-PR25 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Prostate Cancer; HRR = homologous recombination repair; ITT = intention to treat; mCRPC = metastatic castration-resistant prostate cancer; NA = not applicable; NE = not estimable; NR = not reported; OS = overall survival; RCT = randomized controlled trial; rPFS = radiographic progression-free survival; vs. = versus.
Note: Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aRated down 1 level for serious imprecision due to the 95% CI for the between-group difference including the possibility of both important benefit and trivial effect; a between-group absolute risk difference of 20% (200 fewer or more events per 1,000 patients) at 12 months was clinically significant according to the clinical experts.
bRated down 1 level for serious risk of bias due to few patients at risk at 48 months. A between-group absolute risk difference of 5% (50 fewer or more events per 1,000 patients) at 48 months was clinically significant according to the clinical experts. The point estimate and entire CI exceeded the threshold.
cA between-group absolute risk difference of 10% (100 fewer or more events per 1,000 patients) at 12 months was clinically significant according to the clinical experts. There is no imprecision in the estimate (the point estimate and entire 95% CI for the between-group difference shows little to no difference).
dA between-group absolute risk difference of 5% (50 fewer or more events per 1,000 patients) at 48 months was clinically significant according to the clinical experts. The point estimate and entire CI exceeded the threshold.
eRated down 2 levels for very serious imprecision due to the 95% CI for the between-group absolute risk difference including the possibility of benefit and harm. No known minimal important difference so target of certainty appraisal was any effect.
fRated down 2 levels for very serious risk of bias due to missing outcome data. No known minimal important difference so target of certainty appraisal was any effect. The point estimate and entire CI exceeded the null.
gRated down 2 levels for very serious risk of bias due to missing outcome data. There is no imprecision in the estimate (the point estimate and entire 95% CI for the between-group difference shows little to no difference); based on literature, a 5-point change from baseline score was considered clinically important.
hRated down 1 level for serious imprecision due to the 95% CI for the between-group difference including the possibility of both important benefit and little to no difference; based on literature, a 5-point change from baseline score was considered clinically important. Rated down 2 levels for very serious risk of bias due to missing outcome data.
iRated down 1 level for serious imprecision due to the 95% CI for the between-group difference including the possibility of both important harm and little to no difference; based on literature, a 5-point change from baseline score was considered clinically important. Rated down 2 levels for very serious risk of bias due to missing outcome data.
Sources: TALAPRO-2 HRR-deficient Clinical Study Report.1 Details included in the table are from the sponsor’s Summary of Clinical Evidence and additional information provided in the submission.
Long-Term Extension Studies
No long-term extension studies were submitted by the sponsor.
Indirect Comparisons
One sponsor-submitted matching-adjusted indirect comparison (MAIC) was submitted to inform the pharmacoeconomic model and fill gaps in the comparative evidence for other treatments of interest for adults with HRR gene-mutated mCRPC.
Description of Studies
A systematic literature review identified 49 unique RCTs, of which 42 had published results that met the inclusion criteria for first-line treatments in adults with mCRPC. Three trials (TALAPRO-2, PROpel, and MAGNITUDE) reported on survival data for patients with HRR deficiencies receiving relevant first-line treatment for mCRPC, and were included in a feasibility assessment. The TALAPRO-2 study compared talazoparib plus enzalutamide versus enzalutamide, the PROpel study compared olaparib plus AAP versus AAP, and the MAGNITUDE study compared niraparib-abiraterone plus prednisone versus AAP. The feasibility assessment explored potential sources of heterogeneity that included study design, patient eligibility criteria, baseline patient characteristics, outcome characteristics, and efficacy or safety results. ███ ███████ ██ ███ ██████████ ██████████ ████ █████████████ ██████ ███ ███████ but the sponsor assessed them as sufficiently similar to conduct MAICs between the TALAPRO-2 study and each of the PROpel and MAGNITUDE trials. There was no common treatment comparator between the TALAPRO-2 trial and comparator trials; therefore, unanchored MAICs were conducted using individual patient data from the TALAPRO-2 study and summary-level data from the comparator trials. Key treatment effect modifiers and prognostic factors for adjustment were identified and ranked in order of importance based on published analyses on prognostic strength in mCRPC and refined based on external clinical input from a practising clinician experienced in treating mCRPC. Individual patient data from the TALAPRO-2 trial HRR-deficient population were adjusted to match the marginal distribution (e.g., mean, variance) of clinical factors of patients for each comparison between the PROpel (olaparib plus AAP versus AAP) and MAGNITUDE (niraparib-abiraterone plus prednisone versus AAP) trials. Point estimates for rPFS and OS were reported as HRs with 95% CIs.
Efficacy Results
The rPFS HR point estimates and 95% CIs ████████ ████████████████████████ ██████ ████████ ████ ████ █████████ ████ ███ ███ ███ ██████ ███ ███ ███ ███████ ████████ ████████████████████████ ██████ █████████ ████ ███ ███ ███ ██████ ███ ██ █████ ████████ ████████ ████████████████████████ ██████ ████████ ████ ███ ███ ███ ███ ██ ████████ ███ ███████████ ██ ██ ██████████ ██ ████ ███ ██████████ ███ ████████ ██████ ███████ ███ ██████ ███ ████████████ ███████ ██████████ ███ ███ ████████ ██ █████████. The rPFS and OS primary analyses for the MAICs for the PROpel and MAGNITUDE studies adjusted for 5 and 8 treatment effect modifiers and prognostic factors, respectively. However, not all factors could be matched or adjusted; therefore, imbalances remained.
Harms Results
The MAIC did not include harms, and therefore no conclusions could be drawn on the relative safety of talazoparib plus enzalutamide versus relevant comparators.
Critical Appraisal
The methods used to conduct the systematic literature review for the MAIC were a priori registered, and used appropriate criteria to search databases, select studies, extract data, and assess risk of bias of the included studies. The MAIC did not include comparisons between talazoparib plus enzalutamide and chemotherapy, which represents a gap in the available indirect evidence given the shared place in therapy for mCRPC. The MAIC included relevant outcomes identified by the CDA-AMC team (rPFS and OS); however, clinically relevant and patient-relevant outcomes such as pain, HRQoL, and harms were not included in the comparisons. To account for between-study differences in patient baseline characteristics, several potentially relevant treatment effect modifiers and prognostic factors (i.e., clinical factors) were matched in the weighting process for each comparison between the TALAPRO-2 study and comparator trials. The methods used to identify and rank the clinical factors were considered appropriate. For both the PROpel and MAGNITUDE trials, adjustments were limited by how, and if, these variables were reported in both trials, and the highest-ranking factor could not be included in the adjustment for either study. The TALAPRO-2 study stratified by prior NHT or taxane therapy per interactive web response system as a single variable; whereas the PROpel and MAGNITUDE studies reported these variables separately. The PROpel study used the interactive web response system value, whereas the MAGNITUDE study did not specify interactive web response system or electronic data capture. Because interactive web response system values were combined in the TALAPRO-2 study but electronic data capture values were presented separately, electronic data capture values were used to align with reporting of comparator trials. Further, the TALAPRO-2 study stratified by prior therapy in the CSPC stage. The PROpel study reported the proportion of patients who received NHT and taxane-based therapy but did not specify the disease stage. The MAGNITUDE study reported the proportion of patients who received NHT and taxane-based therapy in the metastatic CSPC or nonmetastatic CRPC stage into a single variable. There were important differences in eligibility criteria between the MAGNITUDE and TALAPRO-2 studies. The MAGNITUDE study allowed the use of AAP in the mCRPC setting for 4 months or less, whereas all patients were treatment naive in the mCRPC setting within the TALAPRO-2 trial. Because only summary-level data were available for the MAGNITUDE study, patients who were not truly treatment naive could not be removed when performing analyses. It is possible that these patients had different disease characteristics compared to the rest of the trial population. Overall, the magnitude and direction of potential bias due to imbalances for the rPFS and OS estimates cannot be predicted. Because the unanchored nature of the MAIC requires a stronger assumption (than an anchored MAIC) that all effect modifiers and prognostic factors have been included in the analysis, which was not possible, the effects of talazoparib plus enzalutamide on rPFS and OS compared with relevant comparators are uncertain and definitive conclusions based on these results are not recommended.
Studies Addressing Gaps in the Evidence From the Systematic Review
No additional studies were submitted by the sponsor.
Conclusions
Evidence from 1 phase III, randomized, double-blind trial (TALAPRO-2) reported on outcomes that were important to both patients and clinicians. The trial showed moderate certainty of evidence that treatment with talazoparib plus enzalutamide results in a clinically important increase in rPFS at 12 months and 48 months compared to placebo plus enzalutamide in adults with HRR-deficient mCRPC in the first-line setting. The trial showed high certainty of evidence of a clinically important increase in OS at 48 months, in favour of talazoparib plus enzalutamide. There was low certainty of evidence for a higher probability of being free of pain progression at 12 months and 48 months in favour of talazoparib plus enzalutamide. Up to 109 weeks, no definitive conclusions can be drawn on other HRQoL outcomes due to concerns of imprecision and missing outcome data. There were no new safety signals identified, and the safety of talazoparib plus enzalutamide was consistent with the known safety profiles of the individual drugs, although the trial showed a higher proportion of TEAEs and serious TEAEs when compared with placebo plus enzalutamide. Due to limitations of the indirect treatment comparison, mostly attributed to the heterogeneity across studies and lack of safety assessment, no conclusions can be drawn on the relative efficacy and safety of talazoparib plus enzalutamide compared to relevant comparators, which included olaparib plus AAP, niraparib-abiraterone plus prednisone, and AAP alone. The evidence submitted to CDA-AMC did not include direct or indirect comparisons between talazoparib plus enzalutamide and chemotherapy, which represents a gap in the available evidence given the potential shared place in therapy for HRR-deficient mCRPC when used as first-line treatments. The TALAPRO-2 trial and submitted indirect treatment comparison evaluated talazoparib plus enzalutamide as a first-line treatment only; therefore, the efficacy and safety of talazoparib plus enzalutamide as a second-line or later-line treatment for HRR-deficient mCRPC represents a gap in the evidence.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of talazoparib 0.5 mg plus enzalutamide 160 mg, taken orally once daily in the treatment of adult patients with HRR gene-mutated mCRPC.
Disease Background
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following have been summarized and validated by the CDA-AMC review team.
The stages of prostate cancer are classified in terms of localized, locally advanced, or metastatic disease, with further subcategorization according to hormone therapy status, whether hormone naive, hormone sensitive, or mCRPC.2 CRPC is defined by disease progression despite castrate levels of testosterone and may present as either a continuous rise in serum PSA levels, the progression of pre-existing disease, and/or the appearance of new metastases.3 Nonmetastatic CRPC has not spread to other parts of the body (based on bone scans and CT scans); whereas, mCRPC has spread to lymph nodes or other parts of the body, such as the bones.4 A patient may progress from metastatic CSPC to mCRPC based on biochemical recurrence (characterized by rising PSA levels despite medical or surgical castration) or from nonmetastatic CRPC based on presentation of metastases (assessed radiographically).5,6 The mechanisms driving progression from androgen-dependent (hormone sensitive or castration sensitive) prostate cancer to CRPC are still largely unclear, although continued androgen receptor signalling, despite depletion of circulating androgens and androgen receptor blockade, is thought by many to be central to the development of CRPC.7
Patients with metastatic prostate cancer typically have declining urinary, sexual, and bowel functions (due to the primary tumour or its treatment) as well as signs and symptoms that are related to the location of the metastasis, which can include bone pain and pathologic fractures, hepatic disorders, neurologic symptoms, weight loss, and fatigue.3,8 Approximately 90% of mCRPC cases involve bone metastases that may cause significant morbidity.3 Progressing to mCRPC is characterized by increased symptomatic burden and reduced HRQoL.9-11
Prostate cancer is the most common cancer among males living in Canada, accounting for 11% of cancer-related deaths.12 It was estimated that in 2022, 24,600 males in Canada were diagnosed with prostate cancer based on an incidence of 117.8 cases per 100,000 population.13 As an advanced form of prostate cancer, the prevalence of mCRPC was estimated as 1.2% to 2.1% of total prostate cancer cases.14 Even though the expected 5-year survival for males diagnosed with prostate cancer in Canada is 91% for all stages combined,12 for metastatic disease, the 5-year survival rate reduces to approximately 28%.15
Standards of Therapy
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following have been summarized and validated by the review team.
According to the clinical experts consulted by CDA-AMC, the main treatment goals for patients with mCRPC are to prolong survival, delay disease progression, improve symptoms, and maintain HRQoL. Systemic therapies for the treatment of patients with mCRPC, and the sequencing of these treatments, depend on patient and disease factors, prior treatments used in the metastatic CSPC setting, and access, which varies across Canada. Docetaxel, cabazitaxel, abiraterone acetate, enzalutamide, radium-223 (for patients with bone-only metastatic disease), lutetium vipivotide tetraxetan, and olaparib, olaparib plus AAP, or niraparib-abiraterone plus prednisone for patients with BRCA1, BRCA2, and/or ATM mutations, are all Health Canada approved, and most are available across Canada. According to the Canadian Urological Association–Canadian Uro-Oncology Group guideline for the management of mCRPC3 and clinical experts consulted by CDA-AMC, abiraterone acetate, enzalutamide, and docetaxel are treatment options in all lines of therapy for mCRPC. Additionally, cabazitaxel, radium-223, lutetium vipivotide tetraxetan, olaparib, and niraparib are treatment options as second-line, third-line, and later lines of therapy, with olaparib and niraparib for patients with BRCA-mutated or ATM-mutated mCRPC.
Drug Under Review
Key characteristics of talazoparib and the comparators are summarized in Table 3.
Talazoparib is indicated in combination with enzalutamide for the treatment of adult patients with HRR gene-mutated mCRPC. The recommended dose is talazoparib 0.5 mg orally once daily in combination with enzalutamide 160 mg orally once daily, until disease progression or unacceptable toxicity. The 0.1 mg and 0.25 mg capsules are available for dose reduction. Talazoparib is also approved for the treatment of breast cancer. Talazoparib has not been previously reviewed by CDA-AMC.
Talazoparib is an inhibitor of the PARP enzymes, PARP1 and PARP2. PARP enzymes are involved in cellular DNA damage response signalling pathways such as DNA repair, gene transcription, cell cycle regulation, and cell death. PARP inhibitors exert cytotoxic effects on cancer cells by 2 mechanisms, inhibition of PARP catalytic activity and by PARP trapping, whereby a PARP protein bound to a PARP inhibitor does not readily dissociate from a DNA lesion, thus preventing DNA repair, replication, and transcription and ultimately leading to apoptosis and/or cell death.
The reimbursement request is aligned with the Health Canada–approved indication.
The drug underwent the standard review pathway at Health Canada.
Table 3: Key Characteristics of Talazoparib, Enzalutamide, Abiraterone Acetate, Docetaxel, Olaparib, and Niraparib-Abiraterone Acetate
Characteristic | Talazoparib16,a | Enzalutamide17 | Abiraterone acetate18,b | Docetaxel19,c | Olaparib20 | Niraparib-abiraterone acetate21 |
|---|---|---|---|---|---|---|
Mechanism of action | Potent inhibitor of PARP enzymes, which are involved in the HRR pathway | Androgen receptor inhibitor that acts on several steps in the androgen receptor signalling pathway | Converted in vivo to abiraterone, an androgen biosynthesis inhibitor. Abiraterone selectively inhibits the enzyme CYP17 | Antineoplastic drug, which acts by disrupting the microtubular network in cells that is essential for vital mitotic and interphase cellular functions | Selective inhibitor of human PARP enzymes, which are involved in normal cellular functions, such as DNA transcription and DNA repair | Niraparib is an inhibitor of PARP enzymes, which play a role in DNA repair. Abiraterone acetate is converted in vivo to abiraterone, an androgen biosynthesis inhibitor. This combination targets 2 oncogenic dependencies in patients with mCRPC and HRR gene alterations. |
Indicationd | In combination with enzalutamide for the treatment of adult patients with homologous recombination repair (HRR) gene-mutated metastatic castration-resistant prostate cancer (mCRPC). | In the setting of medical or surgical castration for the treatment of metastatic castration-resistant prostate cancer (CRPC) in patients who:
| In combination with prednisone for the treatment of metastatic prostate cancer (castration-resistant prostate cancer, mCRPC) in patients who:
| In combination with prednisone or prednisolone for the treatment of patients with androgen-independent (hormone-refractory) metastatic prostate cancer. | As monotherapy for the treatment of adult patients with deleterious or suspected deleterious germline and/or somatic BRCA or ATM-mutated mCRPC who have progressed following prior treatment with a new hormonal agent. | Niraparib-abiraterone acetate is indicated with prednisone or prednisolone for the treatment of adult patients with deleterious or suspected deleterious BRCA-mutated (germline and/or somatic) mCRPC, who are asymptomatic/mildly symptomatic, and in whom chemotherapy is not clinically indicated. |
Route of administration | Oral | Oral | Oral | IV | Oral | Oral |
Recommended dose | 0.5 mg administered once daily | 160 mg (four 40 mg tablets) administered once daily | 1 g (two 500 mg tablets or four 250 mg tablets) as a single daily dose | 75 mg/m2 administered as a 1-hour IV infusion every 3 weeks | Daily dose of 600 mg, taken as two 150 mg tablets twice daily | 200 mg niraparib and 1,000 mg abiraterone acetate (two 100 mg + 500 mg tablets), as a single daily dose that must be taken on an empty stomach at approximately the same time every day |
Serious adverse effects or safety issues | Myelodysplastic syndrome or AML has been reported in patients exposed to this drug; can cause fetal harm when administered to a person who is pregnant. | The following are clinically significant adverse events: seizures and posterior reversible encephalopathy syndrome. | May cause hypertension, hypokalemia, and fluid retention due to mineralocorticoid excess. Hepatotoxicity, including fatal cases, has been observed. | Higher risk of developing toxic myelodysplastic syndrome or AML, death, and fatal gastrointestinal hemorrhage in patients with hepatic impairment. Fatal cases of enterocolitis, including ischemic colitis, colitis, and neutropenic enterocolitis have been reported. Treatment-related AML may occur. | Myelodysplastic syndrome or AML, pneumonitis, fetal harm, venous thromboembolic events including pulmonary embolism, hematological toxicity, and hepatotoxicity Coadministration of olaparib with strong or moderate CYP3A inhibitors is not recommended. | May cause hypertension, hypokalemia, and fluid retention, should be used with caution in patients with a history of cardiovascular disease, and should not be given to patients with moderate to severe hepatic impairment. Myelodysplastic syndrome or AML, venous thromboembolic events including pulmonary embolism, hematological adverse reaction, anaphylactic reactions |
AML = acute myeloid leukemia; HRR = homologous recombination repair; mCRPC = metastatic castration-resistant prostate cancer; PARP = poly(adenosine diphosphate-ribose) polymerase.
aGiven in combination with enzalutamide.
bGiven in combination with prednisone.
cGiven in combination with prednisone or prednisolone.
dHealth Canada–approved indication.
Sources: Product monographs for talazoparib, abiraterone acetate, docetaxel, and enzalutamide.16,18,19,22
Testing Procedure Considerations
Somatic or germline mutations occurring in the genes involved in DNA damage repair through the HRR pathway have been of interest for targeted treatments in prostate cancer.23 Published literature suggests that alterations in HRR genes are present in approximately 25% to 30% of patients with mCRPC.23-27 The clinical experts consulted for this review stated that, due to the occurrence of driving mutations based on prior therapy, the rate of somatic HRR mutations in this population is likely increasing over time. The presence of HRR gene mutations is associated with early onset of disease, aggressive tumours, higher recurrence rates, and worse prognosis.23,28,29 BRCA1 and BRCA2 mutations, in particular, are associated with poor prognosis compared to other HRR mutations in the mCRPC population.27
There are at least 18 genes involved in the HRR pathway that could harbour mutations associated with mCRPC.23,30 Among them, participants in the TALAPRO-2 trial had mutations in 1 or more of the following 12 genes that are directly or indirectly involved in the HRR pathway: ATM, ATR, BRCA1, BRCA2, CDK12, CHEK2, FANCA, MLH1, MRE11A, NBN, PALB2, and RAD51C.31,32 In this trial, the most commonly mutated HRR genes in the combined intervention and placebo groups of the population with HRR deficiencies were BRCA2 (33.8%), ATM (21.6%), CDK12 (18.8%), CHEK2 (17.8%), and BRCA1 (5.8%).32 The most prevalent HRR gene mutations detected in a real-world cohort of individuals with mCRPC using both somatic and germline testing methods were in BRCA2 (13.4%), CDK12 (7.7%), and ATM (6.2%).30 Mutations in the other genes in the HRR pathway are less common and the prevalence varies across the published literature.27,28,33,34 The clinical experts noted that, in the rare occurrence where a patient is identified with an HRR gene mutation other than 1 of the 12 genes included in the TALAPRO-2 study, treatment with talazoparib would still be considered.
Germline, or hereditary, mutations are identified using NGS from a saliva or blood sample.3 In Canada, patients can typically access this test by referral to specialized hereditary cancer clinic services.3 In some jurisdictions, such as British Columbia, germline testing can be accessed through “mainstreaming,” where the medical oncologist can directly order the test.35 The patient would then be referred to a hereditary cancer clinic with a geneticist and genetic counselling services only if a pathogenic, likely pathogenic, or variant of uncertain significance was found.3,35-37 Somatic mutations are also identified through NGS, usually using fresh or archival tumour biopsy tissue or circulating tumour DNA from a blood sample.3 NGS testing on tumour tissue can identify mutations of both germline and somatic origins, although it is unable to differentiate between them.3,36,37
The 2023 Canadian Urological Association guideline recommends testing patients with metastatic prostate cancer for hereditary mutations in ATM, BRCA1, BRCA2, CHEK2, MLH1, and PALB2.36 Similarly, the Canadian Urological Association recommends that patients with mCRPC should receive tumour genomic profiling to inform the selection of therapy. At minimum, this should include mutations in ATM, BRCA1, and BRCA2; however, panels should align with germline panels as much as possible, and ideally also include CHEK2, MLH1, PALB2, and CDK12.36 To make the most efficient use of available resources and timelines when determining eligibility for a targeted treatment in patients with metastatic prostate cancer, the Canadian Urological Association suggests starting with somatic mutation testing of archived primary or metastatic tumour tissue and following with germline testing only if variants associated with hereditary cancer risk are identified.36 If no mutations are identified using a tumour NGS panel, germline testing may still be indicated if the panel did not include the recommended genes for hereditary mutation testing.36 According to the clinical experts consulted for this review, practice in Canada does not always reflect the Canadian Urological Association recommendations. There is considerable variability between and within jurisdictions in the timing of testing, who orders the test, the type of test offered, and the genes covered.
We considered the potential impacts of HRR gene mutation testing to ascertain eligibility for talazoparib for the treatment of adult patients with mCRPC, including to health systems, patients (including families and caregivers), and costs. There are existing concerns related to testing within health systems, impacting patients, and costs. However, if talazoparib were to be funded, minimal additional impact is anticipated. A small proportion of patients who have mutations in genes other than BRCA1, BRCA2, or ATM may not be identified for eligibility for talazoparib depending on what testing panels are available in their jurisdiction. Key considerations and relevant information available from materials submitted by the sponsor, input from the clinical experts consulted by the review team, and sources from the literature were validated by the review team when possible and are summarized in Table 4.
Table 4: Considerations for HRR Gene Mutation Testing for Establishing Treatment Eligibility With Talazoparib in mCRPC
Consideration | Criterion | Available information |
|---|---|---|
Health system related | Number of individuals in Canada expected to require the test (e.g., per year) | According to sponsor-submitted materials, approximately 2,500 individuals in Canada are diagnosed with mCRPC annually.38 The sponsor assumed that approximately 1,600 of these patients (65%) would likely be tested for HRR gene mutation status.38 The clinical experts agree that this is an acceptable estimate. According to the clinical experts, the number of individuals undergoing testing would likely not change if talazoparib were to be funded. There are already funded HRR mutation-targeted treatment options available for this population, such as olaparib, that require eligibility determination by tumour or germline NGS. |
Availability and reimbursement status of the testing procedure in jurisdictions across Canada | The clinical experts described the availability of HRR mutation testing as heterogeneous within and between jurisdictions. While NGS panels for mCRPC are generally available across Canada, there is variability in the genes included in each panel.39-41 According to the sponsor, panels for mutations in BRCA1, BRCA2, and ATM may be available in most provinces, although it is unclear if this refers to somatic or germline testing.32 It was unclear whether an NGS panel covering all HRR genes is available in Canada. As well, it is unclear if there are any tumour NGS panels available in Canada that provide ATR, FANCA, or MRE11A gene coverage.39-41 Provincial hereditary cancer programs are established in Nova Scotia, Ontario, Saskatchewan, Alberta, and British Columbia, where, if certain eligibility criteria are met, patients with mCRPC can obtain germline testing for a select number of genes.35,37,42-44 Individuals in Prince Edward Island and New Brunswick can access germline testing through out-of-province referral to Nova Scotia.42 Manitoba has a hereditary cancer program with germline testing; however, it is unclear if patients with mCRPC would be able to access this program.45 It is unclear if the remaining provinces and territories have hereditary cancer programs with germline genetic testing available. The provinces with known hereditary cancer programs test for germline mutations in at least 4 HRR genes, BRCA1, BRCA2, CHEK2, and MLH1.37,46-50 Depending on the panel used in each province, germline testing for ATM, NBN, PALB2, and RAD51C mutations may also be available.37,46-50 Tumour NGS testing in patients with mCRPC is publicly funded in Nova Scotia, Ontario, and British Columbia. For example, in Ontario, funded testing of the BRCA1, BRCA2, ATM, PALB2, and MLH1 genes is available to individuals with newly diagnosed advanced prostate cancer.39 Funding status in the other provinces is unknown. Somatic HRR mutation testing using ctDNA is rarely done in Canada and is available and publicly funded in Alberta and British Columbia. Patients in other provinces may be able to access ctDNA testing through other avenues, such as clinical trials. Germline NGS testing offered through provincial programs is funded for patients who have a medical or family history suggestive of hereditary cancer.35,37,42-44 | |
Testing procedure as part of routine care | CUA guidelines recommend routine somatic and germline testing for HRR mutations in patients with mCRPC in Canada.36 However, according to the clinical experts, real-world testing practices are heterogenous across the country and may not reflect the guideline recommendations. | |
Repeat testing requirements | According to the clinical experts, testing in most patients would be done on primary tumour tissue obtained at the initial diagnostic work up. They noted that somatic HRR alterations could develop later in the clinical course. At present, CUA does not provide any guidance on repeat NGS testing of tumour tissue after disease progression to mCRPC.14 As well, in some cases, tumour testing may miss germline variants.36 Even with negative tissue results, additional germline testing in patients with a personal or family history strongly suggestive of hereditary cancer may be helpful.36,51 The clinical experts noted that if talazoparib were to be funded, repeat testing requirements will likely not change. Because germline mutations are hereditary and remain throughout an individual's lifetime, such testing can be done at any time and repeat testing is not necessary.3,36 | |
Impacts on human and other health care resources by provision of the testing procedure | The clinical experts remarked how genomic testing practice in the population with prostate cancer is not standardized across Canada. A 2022 Environmental Scan listed several barriers to the implementation of widespread genomic testing practices for patients with prostate cancer in the country. These included unclear processes for requisitioning, lack of standardization of clinical reporting, insufficient laboratory funding, and lack of a formal priority process to manage the expected volume of testing requests.51 As well, to align with Canadian guidelines, some jurisdictions may need to expand existing NGS panels.36 If talazoparib is reimbursed, jurisdictions may need to add even more genes to existing panels. There are also existing concerns specifically regarding germline genomic testing. For example, the clinical experts mentioned that there already are large backlogs in the hereditary cancer clinics.52 A 2023 scenario analysis conducted in Canada concluded that there may not be enough clinical genetics professionals, including geneticists and genetic counsellors, to meet the projected demand for hereditary cancer services in Canada.52 To provide consistent service across the country, upscaling of existing testing infrastructure, workflows, and technologies may be needed to meet an increase in testing.3,51,52 In addition, there may be a need for formal education strategies directed toward health care workers, such as primary care physicians, medical oncologists, and geneticists, to increase awareness and knowledge regarding precision medicine options and genomic testing in patients with mCPRC.3,51-53 According to the clinical experts, these human and other health care resources concerns related to genomic testing in patients with mCRPC already exist and minimal additional impact is anticipated if talazoparib is reimbursed. | |
Patient related | Accessibility of the testing procedure in jurisdictions across Canada | As noted previously, the availability of tumour and germline NGS testing for specific HRR mutations is highly variable across jurisdictions. According to the sponsor, most individuals with mCRPC in Canada should be able to access either somatic or germline testing for at least BRCA1, BRCA2, and ATM mutations.32 Mutations in these 3 genes were present in up to approximately 60% of patients with HRR mutations in the TALAPRO-2 study.32 Patients with a mutation in 1 of the other HRR genes may not be identified for eligibility with talazoparib depending on what testing panels are available in their region. According to the clinical experts, this could mean, in rare instances, some patients may be at risk of undergoing a less optimal course of treatment. |
Expected turnaround times for the testing procedure | Turnaround time for tumour NGS testing is approximately 3 weeks after the receipt of the specimen in the testing laboratory.40,41 Turnaround time for germline testing ranges from 4 weeks (in urgent cases) to 16 weeks after specimen receipt.48-50 This may be prolonged in areas without mainstream referral options, as wait times to see a medical geneticist or genetic counsellor can be weeks to years long.3 The clinical experts mentioned that they often have to make treatment decisions in the absence of genetic testing results due to prolonged wait times. However, they also noted that these turnaround time concerns for testing already exist in the population with mCRPC, and no additional impact is anticipated if talazoparib is reimbursed. | |
Burden associated with the testing procedure for patients, families, and/or caregivers | Patients who receive germline testing may have to undergo pretest and/or posttest genetic counselling.3,36 If a pathogenic or likely pathogenic variant is found, this could trigger cascade testing in family members.36 Patients or their families may experience emotional burden, such as feeling guilt toward their children if a hereditary cancer gene is found.3 According to the clinical experts, these potential burdens associated with testing already exist in the mCRPC population and no additional impact is anticipated if talazoparib is reimbursed. The ability of NGS testing to generate results decreases with increasing sample age due to DNA degradation.36,54,55 Patients may need a repeat biopsy if testing using archival tissue fails.3,36 This could pose additional burden on patients and caregivers in a heterogeneous testing landscape with inconsistent access to funded testing. | |
Clinical | Clinical utility and validity of the testing procedure | There is evidence showing analytical and clinical validity and clinical utility of NGS testing to detect mutations in HRR genes.56,57,a Accordingly, both germline and somatic NGS testing methods for HRR mutations show high sensitivity and specificity (> 95%).57 As well, there is good analytical concordance between the tumour NGS panel used in the TALAPRO-2 study and an externally validated NGS assay.56 A cohort study found that tumour NGS testing was unable to detect approximately 6.8% (95% CI, 3.5% to 12.8%) of pathogenic hereditary HRR gene variants in prostate tumours.58 |
Risks of harm associated with the testing procedure | There is minimal clinical risk of harm or adverse events involved with the testing procedure itself, beyond those associated with biopsy or blood collection. The Genetic Non-Discrimination Act, upheld by the Canadian Supreme Court in 2020, prohibits both the mandatory disclosure of genetic results and the collection and use of those results without written consent.3,59 However, there may be personal or financial risks to patients and their families, such as workplace discrimination or exclusion from life insurance, if they are found to have a pathogenic or likely pathogenic hereditary variant.3,59 According to the clinical experts, these risks associated with HRR testing already exist in the mCRPC population and no additional impact is anticipated if talazoparib is reimbursed. | |
Cost | Projected cost of the testing procedure | The sponsor estimated the unit cost of tumour NGS testing to be $1,919.38 There could potentially be additional costs related to adding HRR genes to current panels or switching to a comprehensive genomic profiling assay. However, these cost-related concerns already exist. One of the clinical experts consulted for this review mentioned that it is relatively inexpensive to add genes to an NGS panel. |
CDA-AMC = Canada's Drug Agency; CI = confidence interval; ctDNA = circulating tumour DNA; CUA = Canadian Urological Association; HRR = homologous recombination repair; mCRPC = metastatic castration-resistant prostate cancer; NGS = next-generation sequencing.
aCDA-AMC have not evaluated or critically appraised this evidence to determine its validity or reliability.
Perspectives of Patients, Clinicians, and Drug Programs
The full patient and clinician group submissions received are available in the consolidated patient and clinician group input document for this review on the project website.
Patient Group Input
This section was prepared by the review team based on the input provided by patient groups.
CDA-AMC received 2 patient group inputs from PROCURE and CCS. PROCURE is a charitable organization which promotes and contributes to the financing of research and educates, supports, and informs patients affected by prostate cancer. PROCURE gathered information from more than 3,500 patients with localized, locally advanced, metastatic, recurrent, hormone sensitive, or CRPC, with or without metastases through phone calls, and conducting 2 online surveys — the Canadian survey in 2022 with 263 patients who responded and the Quebec survey in 2018 (the total number of patients who responded was not reported). CCS supports patients living with cancer in Canada through research, transformative advocacy, and compassionate support. CCS conducted a survey to gather perspectives on disease experience from 21 patients with mCRPC and 3 caregivers of patients with mCRPC from April to May 2023.
According to PROCURE’s input, in addition to distress and treatment decisions, rising PSA levels after treatment, recurrence, hormone therapy and its side effects, as well as metastases are among the main concerns of patients. Additionally, 50% of the patients who responded to the survey in Canada reported that the main challenges posed by treatment included managing side effects, living with uncertainty, and maintaining a positive attitude. PROCURE also noted that patients who responded to the survey in Quebec hope that future treatments for prostate cancer slow down the progression of cancer (94%), extend life expectancy (94%), improve the quality of life (98%), help manage or diminish side effects (93%), and decrease PSA levels (91%).
Based on the CCS input, mCRPC mostly impacted the sexual activity of patients, followed by their ability to work, maintain positive mental health, concentrate, travel, exercise, or conduct household chores. CCS added that some of the barriers the patients faced when receiving treatment included transportation costs to appointments, long wait times to receive tests or treatments, and lack of familiarity with the health care system. According to the CCS input, the majority of patients who responded to the survey (67%) reported receiving 3 or more lines of treatment and 8% indicated they had received 1 line of treatment, while 13% reported having received 2 lines of treatment. CCS noted that the most common current treatments included hormone therapies, external beam radiation, surgical procedures, chemotherapy, and corticosteroids. Notably, no patients who responded to the survey reported using targeted therapy. Some of the side effects of current treatments reported by CCS included fatigue, hot flashes, dizziness, peripheral neuropathy, pain, nausea, vomiting, changes in libido, sexual function or fertility, and problems in the mouth, tongue, and throat. CCS stated that many patients are highly motivated to extend survival, despite potential side effects.
PROCURE noted that prostate cancer treatments have physical and psychological impacts. Physical impacts included side effects (incontinence, erectile dysfunction, and bowel issues), effects associated with hormone therapies (hot flashes, loss of libido, weight gain, cardiovascular risks, osteoporosis, and so forth), and pain related to metastases; psychological impacts included anxiety, depression, loss of self confidence, fatigue, and fear of cancer returning which causes stress and uncertainty, and affecting sleep, appetite, and concentration. PROCURE emphasized the importance of emotional support in addition to medical care and added that access to health care professionals (sexologists, psychologists, physiotherapists, kinesiologists) is often limited due to cost, lack of available services, or long wait times, leaving many patients without adequate support.
Regarding patients’ partners, family members, and caregivers, PROCURE explained that caregiving is a demanding role and comes with stress, anxiety, and depression. Intimate relationships are affected due to loss of libido, fatigue, and changes in masculine characteristics often lead males to avoid intimacy with their partners, resulting in significant consequences. PROCURE added that if a father carries a genetic mutation and has prostate cancer, both sons and daughters are at an increased risk of developing certain cancers, such as prostate, breast, and ovarian cancer, if they inherit the altered gene, thus ███████ ██████████ ███ ███████ ███████ █████████ ███ █████████ ██ ██████ ███ █████ ███ █████ ██ ███ ███████ ██████ Children may experience anxiety due to the family history and concerns about their own risk of developing cancer. CCS also noted the negative impact of disease on caregivers and children such as transportation costs, caregiving costs, loss of income due to absence from work, and lack of familiarity with the health care system.
Based on the PROCURE input, while current treatments are often effective, they eventually face resistance over time, limiting their long-term benefits. PROCURE added that standard treatments for metastatic cancer, such as hormone therapy and chemotherapy, are systemic and nontargeted, exposing the entire body to often severe side effects. Therapeutic options targeting genetic mutations in prostate cancer remain limited to the BRCA gene and are only available when this mutation is confirmed through tumour biopsy or genetic testing. This restriction reduces treatment alternatives for many patients, highlighting the need for new therapeutic approaches. PROCURE highlighted that patients face both emotional and practical concerns regarding treatment efficacy, cancer progression, long-term health impact, and overall life expectancy.
Both patient groups clarified that patients' expectations with regards to new treatments included cancer control with fewer side effects, longer-lasting effects, delayed onset or elimination of metastases, decreased or maintained PSA levels over a long period, convenient treatment regimens, prolonged life, and improved quality of life. Patients also expected treatments to be affordable, have better accessibility, better follow-up for long-term issues, and to be heard and taken seriously.
In terms of experience with the drug under review, the patient groups explained that they did not have access to patients who have experience with the drug under review. PROCURE clarified that there are 2 distinct indications for this treatment, depending on its use in North America versus Europe. The European indication, which is broader, allows the use of this combination in patients with mCRPC, with or without a genetic mutation, when chemotherapy is not clinically indicated. In contrast, in the US and Canada, the approach remains more conservative, restricting the use of this treatment to patients with HRR gene alterations. CCS highlighted the importance of companion diagnostic genetic testing.
Clinician Input
Input From Clinical Experts Consulted for This Review
All CDA-AMC review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of mCRPC.
Unmet Needs
The clinical experts indicated that because mCRPC is a terminal phase of prostate cancer, the unmet needs of patients would be new treatments that would prolong survival and improve quality of life, while exposing patients to minimal toxicity. Both clinical experts highlighted that the balance between treatment efficacy and quality of life would be important.
Place in Therapy
Clinical experts noted that it remains unclear whether talazoparib plus enzalutamide would lead to a shift in the current treatment paradigm. This uncertainty stems from the increased use of ARPIs in patients with metastatic CSPC and nonmetastatic CRPC. They noted that many medical oncologists would favour a change to chemotherapy in patients progressing on an ARPI in the metastatic CSPC and nonmetastatic CRPC settings. They noted that if a treatment was used in metastatic CSPC, it is not likely the patient will receive it again for first-line mCRPC (with the occasional exception of docetaxel if given at least 1 year prior). The clinical experts noted that talazoparib plus enzalutamide may have a limited role as a first-line treatment in the mCRPC setting due to the decreasing number of patients who are ARPI naive and the few patients who would be clinically ineligible for docetaxel; however, it may have a role in subsequent lines of therapy.
Patient Population
The clinical experts indicated that patients best suited for talazoparib plus enzalutamide would be those who match the eligibility criteria of the TALAPRO-2 trial, which included first-line treatment for HRR gene-mutated mCRPC, with no prior ARPI in the metastatic CSPC setting (with the exception of abiraterone acetate) and no contraindications to talazoparib or enzalutamide. They did not indicate any issues or challenges related to diagnosis or misdiagnosis of mCRPC and noted that there are no predictive biomarkers to guide treatment.
Assessing the Response Treatment
The clinical experts indicated that in clinical practice, a combination of radiographic, biochemical (e.g., PSA), and clinical parameters (i.e., decrease in disease-related symptoms) is used to determine whether a patient with mCRPC is responding or progressing while receiving treatment. They noted that the frequency of evaluation can be variable from patient to patient, but at the very least, assessments should be performed at 3-month intervals.
Discontinuing Treatment
The clinical experts indicated that treatment with talazoparib plus enzalutamide should be discontinued if patients experience disease progression (as defined radiologically or clinically), treatment is intolerable, or if it is the patient’s preference.
Prescribing Considerations
The clinical experts noted that PARP inhibitors such as talazoparib have the potential to be toxic; therefore, patients receiving talazoparib plus enzalutamide should be under the care of a medical oncologist who can manage toxicity associated with therapy, in the community.
Clinician Group Input
This section was prepared by the review team based on the input provided by clinician groups.
CDA-AMC received 1 input from the OH (CCO) Genitourinary Cancer Drug Advisory Committee for this submission. The OH (CCO) Drug Advisory Committees provide timely, evidence-based clinical and health system guidance on drug-related issues in support of the OH (CCO) mandate, including the Provincial Drug Reimbursement Programs and the Systemic Treatment Program. The OH (CCO) Genitourinary Cancer Drug Advisory Committee gathered information from 5 clinicians.
According to the clinician group input, the treatment goal is prolonging life and improving quality of life. The current treatments for mCRPC include abiraterone acetate, enzalutamide, docetaxel, and radium-223 (in patients who are ineligible to receive docetaxel), all given concurrently with ADT. Cabazitaxel is another option after docetaxel use in the mCRPC setting. Similarly, olaparib monotherapy might be a treatment option after treatment with an ARPI for mCRPC in males with BRCA2, BRCA1, or ATM aberrations. Olaparib plus abiraterone acetate is also available (while niraparib-abiraterone acetate is under funding consideration) for those who have not been treated with ARPI in the metastatic CSPC setting or nonmetastatic CRPC setting.
The clinician group explained that there are no cures currently available for metastatic prostate cancer and there is a need for treatments that prolong life.
Based on input from a clinical expert consulted by CDA-AMC, the place in therapy of talazoparib in combination with enzalutamide is for the treatment of patients with mCRPC who are treatment naive. The group noted that treatment with chemotherapy or ARPI in the metastatic CSPC setting should not preclude eligibility for treatment with talazoparib plus enzalutamide, as per the TALAPRO-2 trial. The group noted that PSA and serial radiographic imaging would be used to monitor response to therapy and that significant side effects and progression of disease on imaging are among the factors for considering discontinuation of the treatment. The clinician group noted that medical oncologists, radiation oncologists, and urologists specialized in prostate cancer care are among the specialists or prescribers that would be required for prescribing the treatment. The OH (CCO) Genitourinary Cancer Drug Advisory Committee added that access to this (and other) combinations in this setting would also require ongoing efforts to ensure equitable, timely access to genomic testing of relevant alterations for all eligible males with prostate cancer living in Canada.
Drug Program Input
The drug programs provide input on each drug being reviewed through the reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CDA-AMC for this review are summarized in Table 5.
Table 5: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
The comparator in the TALAPRO-2 study was enzalutamide and placebo. This is a reasonable comparator, but is there any evidence comparing talazoparib with enzalutamide to other funded PARP inhibitors, either alone (i.e., olaparib) or in combination with an androgen receptor inhibitor (i.e., olaparib plus AAP or niraparib-abiraterone plus prednisone)? | The CDA-AMC team noted that the comparison between talazoparib + enzalutamide and relevant comparators is to be addressed in the Clinical Review report. The clinical experts noted that, to their knowledge, there is no direct comparative efficacy and safety between talazoparib + enzalutamide and other funded PARP inhibitors, either alone or in combination with an androgen receptor inhibitor. |
Considerations for initiation of therapy | |
In the trial, mutation status of HRR genes was determined prospectively using solid tumour tissue or circulating tumour DNA–based next-generation sequencing assays. Patients were required to have a mutation in at least 1 of 12 genes involved directly or indirectly in the HRR pathway (ATM, ATR, BRCA1, BRCA2, CDK12, CHEK2, FANCA, MLH1, MRE11A, NBN, PALB2, or RAD51C). Not all jurisdictions have access to testing for all HRR pathway genes. | Comment from the drug plans to inform pERC deliberations. |
Should consider alignment with other PARP inhibitor or androgen receptor pathway inhibitors where possible. | Comment from the drug plans to inform pERC deliberations. |
Considerations for discontinuation of therapy | |
In the trial, study treatment could continue after radiographic PFS if the investigator determined benefit was still being derived. What parameters should be used to determine when a patient should discontinue treatment? | The clinical experts indicated that in clinical practice, a combination of radiographic, biochemical, and clinical parameters (e.g., progressing radiological disease, worsening symptoms), as well as tolerability are used to determine whether a patient should discontinue treatment. |
Should consider alignment with other PARP inhibitor or androgen receptor pathway inhibitors either in monotherapy or combination therapy as several have been evaluated previously by CDA-AMC. | Comment from the drug plans to inform pERC deliberations. |
Considerations for prescribing of therapy | |
Should patients continue on talazoparib monotherapy if enzalutamide requires discontinuation for reasons other than progression? Are there relevant dosing considerations (i.e., dose escalations)? | The clinical experts indicated that it would be reasonable for patients to continue on talazoparib monotherapy if enzalutamide requires discontinuation for reasons other than disease progression. The experts noted that they would not consider dose escalation, but dose reduction if patients experience toxicities. |
Generalizability | |
Patients with ECOG PS > 1 were not eligible for the trial. Should the eligibility criteria be expanded to PS > 1? | The clinical experts indicated that patients with good performance status should be eligible for talazoparib + enzalutamide, if they are able to tolerate the therapy as determined by the treating physician. |
Should patients currently receiving monotherapy with enzalutamide (or abiraterone acetate) be able to switch to talazoparib + enzalutamide? And if yes, what are the parameters for this? | The clinical experts indicated that there are no data on the impact of switching patients currently receiving monotherapy with enzalutamide (or abiraterone acetate) to talazoparib + enzalutamide; however, they considered the switch to be reasonable within 3 months to 6 months of starting enzalutamide or abiraterone acetate without disease progression. |
Funding algorithm | |
An update to the algorithm to incorporate the recommendation, if positive. | Comment from the drug plans to inform pERC deliberations. |
Under what clinical circumstances would talazoparib + enzalutamide be preferred over olaparib-abiraterone acetate or niraparib-abiraterone acetate? | The clinical experts noted that there is no clear approach because there is no head-to-head comparative evidence in favour of one treatment over another. The clinical experts also noted that talazoparib + enzalutamide could be considered in patients with HRR gene alterations beyond BRCA, as it is currently the only combination treatment with an indication covering a broader range of HRR mutations. |
Care provision issues | |
There is a statement in the product monograph, that talazoparib is maintained in the original bottle to protect from light. Is this applicable only in the pharmacy or does this include when dispensed to the patient? Would the usual light-blocking medication vials be appropriate to use? Otherwise, there will need to be consideration regarding drug wastage, depending on how the drug is packaged (i.e., how many capsules are in the original container). Talazoparib is noted in the product monograph to be available in several strengths: 0.1 mg, 0.25 mg, 0.35 mg, 0.5 mg, and 1 mg. The starting dose for mCRPC is 0.5 mg daily with or without food. | Comment from the drug plans to inform pERC deliberations. The CDA-AMC review team noted that in Canada, only the 0.1 mg and 0.25 mg capsules of talazoparib will be made available by the sponsor. |
The TALAPRO-2 trial shows a higher rate of adverse effects, particularly anemia and neutropenia, and this may result in dose modification. | Comment from the drug plans to inform pERC deliberations. |
System and economic issues | |
Enzalutamide, olaparib, and niraparib-abiraterone acetate have confidential pricing. Abiraterone acetate is available in a generic form. | Comment from the drug plans to inform pERC deliberations. |
AAP = abiraterone acetate plus prednisone; CDA-AMC = Canada's Drug Agency; ECOG PS = Eastern Cooperative Oncology Group Performance Status; HRR = homologous recombination repair; mCRPC = metastatic castration-resistant prostate cancer; PARP = poly(adenosine diphosphate-ribose) polymerase; pERC = pan-Canadian Oncology Drug Review Expert Review Committee; PFS = progression-free survival.
Clinical Evidence
The objective of this Clinical Review report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of talazoparib 0.5 mg plus enzalutamide 160 mg, taken orally once daily in the treatment of adult patients with HRR gene-mutated mCRPC. The focus will be placed on comparing talazoparib plus enzalutamide to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of talazoparib plus enzalutamide is presented in 4 sections with our critical appraisal of the evidence included at the end of each section. The first section, the systematic review, includes pivotal studies and RCTs that were selected according to the sponsor’s systematic review protocol. Our assessment of the certainty of the evidence in this first section using the GRADE approach follows the critical appraisal of the evidence. The second section typically includes sponsor-submitted long-term extension studies; however, none were submitted by the sponsor. The third section includes indirect evidence from the sponsor. The fourth section typically includes additional studies that were considered by the sponsor to address important gaps in the systematic review evidence; however, none were submitted by the sponsor.
Included Studies
Clinical evidence from the following are included in the review and appraised in this document:
1 pivotal trial identified in the systematic review
1 indirect treatment comparison.
Systematic Review
Contents within this section have been informed by materials submitted by the sponsor. The following have been summarized and validated by the review team.
Description of Studies
Characteristics of the included studies are summarized in Table 6.
Table 6: Details of Studies Included in the Systematic Review
Detail | TALAPRO-2 |
|---|---|
Designs and populations | |
Study design | Phase III, randomized, double-blind, placebo-controlled trial
Note: The focus of this review is cohort 2 (i.e., population with HRR deficiencies). |
Locations | 287 study centres in 26 countries: Argentina, Australia, Belgium, Brazil, Canada, Chile, China, Czech Republic, Finland, France, Germany, Hungary, Israel, Italy, Japan, New Zealand, Norway, Peru, Poland, Portugal, Republic of Korea, South Africa, Spain, Sweden, UK, US ██ ████████ ████ ██████████ across 6 sites in Canada; ██ ████████ ████ ██████ were included in cohort 2 |
Key dates | Study initiation date: August 8, 2017 Analyses data cut-off of September 3, 2024, for the population with HRR deficiencies |
Randomized (N) | Cohort 2 enrolled 399 patients with HRR deficiencies, including 169 patients with HRR deficiencies from cohort 1 and 230 patients subsequently enrolled after cohort 1 enrolment was completed. |
Key inclusion criteria |
|
Key exclusion criteria |
|
Intervention | Talazoparib (orally, 0.5 mg per day) + enzalutamide (orally, 160 mg per day) |
Comparator(s) | Matched placebo + enzalutamide (orally, 160 mg per day) |
Screening phase | Up to 42 days before first treatment dose |
Treatment phase | From time of first dose until discontinuation criteria were met |
Follow-up phase | Through 28 days after last dose of study treatment for safety follow-up. Follows imaging schedule for long-term follow-up. |
Primary end point | rPFS by BICR per RECIST 1.1 (soft tissue disease) and PCWG3 (bone disease) in patients with mCRPC harbouring HRR deficiencies |
Secondary and exploratory end points | Key secondary (alpha protected):
Secondary (not alpha protected):
Exploratory:
|
Publications | Agarwal et al. (2022)60 Agarwal et al. (2023)31 Fizazi et al. (2024)61 Lloyd et al. (2024)62 McKay et al. (2024)63 Azad et al. (2024)64 |
ADT = androgen deprivation therapy; AE = adverse event; AR = androgen receptor; BICR = blinded independent central review; BPI-SF = Brief Pain Inventory–Short Form; CRPC = castration-resistant prostate cancer; CSPC = castration-sensitive prostate cancer; ECOG PS = Eastern Cooperative Oncology Group Performance Status; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-PR25 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Prostate Cancer; GnRH = gonadotropin-releasing hormone; HRR = homologous recombination repair; mCRPC = metastatic castration-resistant prostate cancer; mCSPC = metastatic castration-sensitive prostate cancer; OS = overall survival; PARP = poly(adenosine diphosphate-ribose) polymerase; PCWG3 = Prostate Cancer Clinical Trials Working Group 3; PFS = progression-free survival; PK = pharmacokinetics; PSA = prostate-specific antigen; QoL = quality of life; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1; rPFS = radiographic progression-free survival.
Note: PFS2 was defined as the time from the date of randomization to the date of investigator documented disease progression on the first subsequent antineoplastic therapy for prostate cancer, or death from any cause, whichever occurs first.
Sources: TALAPRO-2 HRR-deficient Clinical Study Report.1 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
TALAPRO-21 is a phase III, randomized, double-blind, placebo-controlled, multicentre trial that aimed to assess the efficacy and safety of talazoparib 0.5 mg plus enzalutamide 160 mg, taken orally once daily, or matched placebo plus enzalutamide in adult patients with mCRPC (Figure 1). The trial enrolled adult patients with asymptomatic or mildly symptomatic mCRPC who had not started systemic cancer treatment after the diagnosis of CRPC (metastatic or nonmetastatic), with the exception of ADT and first-generation antiandrogen drugs. The trial included 2 prespecified populations. Enrolment began in cohort 1, which included patients with and without HRR gene alterations. Once enrolment was complete in cohort 1, enrolment continued but was restricted to patients with HRR gene alterations. Cohort 2 (population with HRR deficiencies) consisted of all patients with HRR deficiencies enrolled in the trial and is the focus of this review as it aligns with the Health Canada–approved indication and the sponsor’s reimbursement request.
Patients who were enrolled in cohort 2 were randomly assigned 1:1 using a centralized interactive web response system (N = 399, at 287 sites across 26 countries) to talazoparib plus enzalutamide (n = 200) or placebo plus enzalutamide (n = 199). In total, 10 patients from Canada were included. Randomization was stratified by previous treatment with NHT (abiraterone acetate, abiraterone, or orteronel) or taxane-based chemotherapy (docetaxel) for CSPC (yes or no), and HRR gene alteration status (deficient versus nondeficient or unknown). The patients and investigators were blinded to talazoparib or placebo, while enzalutamide was open label.
The trial included a screening period up to 42 days, a treatment period until discontinuation criteria were met, which included radiographic progression by BICR, an adverse event (AE) leading to permanent discontinuation, patient decision to discontinue treatment, or death. All patients were followed during a posttreatment phase after discontinuation criteria were met, which included the 28 days after the last dose of study treatment. Patients were subsequently followed during a long-term follow-up phase with imaging and PSA measurements.
The outcomes relevant to this review included the primary outcome of rPFS, and secondary outcomes of OS, patient-reported pain symptoms measured via BPI-SF, HRQoL measured via EORTC QLQ-PR25 functional and symptom scales, and safety. The rPFS data are based on the primary analysis data cut-off date of October 3, 2022, and supported by the data cut-off date of September 3, 2024. All other outcomes are based on the data cut-off date of September 3, 2024.
Populations
Inclusion and Exclusion Criteria
Patients who were eligible were asymptomatic or mildly symptomatic adult patients with mCRPC who had not started systemic cancer treatment after the diagnosis of CRPC (metastatic or nonmetastatic), with the exception of ADT and first-generation antiandrogen drugs. Patients were allowed to have previously received abiraterone acetate or docetaxel for CSPC but were ineligible to participate if they had received any prior treatment with second-generation androgen receptor inhibitors (enzalutamide, apalutamide, and darolutamide), a PARP inhibitor, cyclophosphamide, or mitoxantrone for prostate cancer. Patients were required to have had an ECOG PS score of 0 or 1, and progressive disease at study entry, which was based on at least 1 of the following criteria during ADT: PSA progression, soft tissue disease progression defined by RECIST 1.1, or bone disease progression defined by the Prostate Cancer Clinical Trials Working Group 3.
Interventions
Patients received talazoparib 0.5 mg as two 0.25 mg capsules plus enzalutamide 160 mg as four 40 mg capsules, taken orally once daily, or matched placebo plus enzalutamide. Placebo capsules were identical in appearance to talazoparib capsules. Participants with moderate renal impairment would receive talazoparib 0.35 mg once a day plus enzalutamide 160 mg once a day to account for the lower talazoparib clearance in this subpopulation. Patients were to continue treatment until BICR-determined radiographic progression, no longer clinically benefiting in the opinion of the investigator, unacceptable toxicity, withdrawal of consent, or death. After radiographic progression, treatment could be continued if the investigator determined benefit was still being derived.
The following concomitant treatments were permitted during the trial: bisphosphonates or denosumab, ADT with a gonadotropin-releasing hormone agonist or antagonist, hematopoietic growth factors, red blood cell transfusions, erythropoietin and erythropoiesis-stimulating drugs, thrombopoietin analogues and/or platelet transfusions, COVID-19 vaccines, and analgesics. The following treatments were prohibited during the trial: prednisone, cytotoxic chemotherapy, hormonal therapy (e.g., bicalutamide, nilutamide, flutamide, estrogens, 5-alpha reductase inhibitors), NHT with the exception of enzalutamide (e.g., abiraterone acetate, apalutamide, darolutamide), biologic therapy, radionuclide therapy, or other PARP inhibitors.
Outcomes
A list of efficacy end points assessed in this Clinical Review report is provided in Table 7, followed by descriptions of the outcome measures. Summarized end points are based on outcomes included in the sponsor’s Summary of Clinical Evidence as well as any outcomes identified as important to this review according to the clinical experts consulted for this review and input from patient and clinician groups and public drug plans. Using the same considerations, we selected end points that were considered to be most relevant to inform expert committee deliberations and finalized this list of end points in consultation with members of the expert committee. All summarized efficacy end points were assessed using GRADE.
Table 7: Outcomes Summarized From the Studies Included in the Systematic Review
Outcome measure | Time point | TALAPRO-2 trial |
|---|---|---|
rPFS by BICR | At 12 months and 48 months | Primarya |
OS | At 12 months and 48 months | Key secondarya |
Time to deterioration of pain via BPI-SF | At 12 months and 48 months | Secondary |
EORTC QLQ-PR25 functional and symptom scales | At up to 109 weeks | Secondary |
BICR = blinded independent central review; BPI-SF = Brief Pain Inventory–Short Form; EORTC QLQ-PR25 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Prostate Cancer; OS = overall survival; rPFS = radiographic progression-free survival.
aStatistical testing for these outcomes were adjusted for multiple comparisons (e.g., hierarchal testing).
Sources: TALAPRO-2 HRR-deficient Clinical Study Report.1 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Radiographic Progression-Free Survival
The primary outcome of rPFS assessed by BICR was defined as the time from the date of the randomization to first objective evidence of radiographic progression as assessed in soft tissue per RECIST 1.1 or in bone (upon subsequent confirmation) per Prostate Cancer Clinical Trials Working Group 3, or death due to any cause, whichever occurred first. Tumour assessments were done with CT or MRI of chest, abdomen, and pelvis, and whole-body bone scan every 8 weeks through to week 25 and every 12 weeks thereafter. Soft tissue responses were confirmed by a follow-up radiographic assessment at least 4 weeks later with no evidence of confirmed bone disease progression on repeated bone scans at least 6 weeks apart per Prostate Cancer Clinical Trials Working Group 3.
Overall Survival
The secondary outcome of OS was defined as the time from randomization to the date of death due to any cause. Patients last known to be alive were censored at the date of last contact.
Time to Deterioration of Pain
The secondary outcome of time to deterioration in patient-reported pain symptoms was measured using question 3 from the BPI-SF: “Please rate your pain by marking the box beside the number that best describes your pain at its worst in the last 24 hours.” A summary of its measurement properties is in Table 8. The BPI-SF is a 9-item instrument that uses a self-reported scale assessing level of pain, its effect on activities of daily living, and analgesic medication use. Question 3 uses a scale of 0 (no pain) to 10 (pain as bad as you can imagine), with lower scores indicating less pain. The MID for the BPI-SF and for question 3 is considered greater or equal to 2 points in patients with various cancers, including metastatic prostate cancer.65,66 Time to this event was defined as the time from randomization to onset of pain progression, where pain progression was defined as a 2-point or more increase from baseline for 2 consecutive visit periods at least 4 weeks apart without a decrease in WHO analgesic usage score. Patients without observed pain progression at the time of analysis were censored at the date of last BPI-SF assessment. Pain and analgesic assessments were completed via pain and analgesic logs, respectively, for 7 consecutive days before study visits, and pain score averages during the period of reporting were calculated. Assessments were performed at baseline, every 4 weeks through week 53, every 8 weeks after week 53, and then every 12 weeks until the end of the study.
Health-Related Quality of Life
The secondary outcome of HRQoL was measured by change in baseline in patient-reported disease-specific functioning and symptom scales using EORTC QLQ-PR25 (Table 8). The EORTC QLQ-PR25 is a prostate cancer-specific module of the European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire that assesses the functioning and symptoms of patients with prostate cancer. Patients self-rate their current state of pain as it relates to urination, ease and frequency of urination, bowel, and other symptoms and signs during the past week. Patients also answer 5 questions about weight loss or gain and sexual interest and 4 questions about sexual activity during the past 4 weeks and choose 1 of 4 possible responses that record level of intensity (not at all, a little, quite a bit, very much) within each dimension. Scores are linearly transformed onto a scale of 0 to 100, with higher scores indicating worse symptoms or better functioning. A MID of 5 points has been used for the EORTC QLQ-PR25 in patients from Taiwan with various stages of prostate cancer and treatments.67 Assessments were performed at baseline, every 4 weeks through week 53, every 8 weeks after week 53, and then every 12 weeks until end of study.
Safety Outcomes
The assessment of safety was based on the incidence of TEAEs, serious TEAEs, notable TEAEs, TEAEs leading to discontinuation, TEAEs leading to dose modification, and deaths. AEs were reported at each study visit and coded to preferred term and system organ class using the Medical Dictionary for Regulatory Activities and classified by severity using the National Cancer Institute Common Terminology Criteria for Adverse Events version 4.0. Safety assessments were done at screening and every 2 weeks up to week 17, every 4 weeks up to week 53, every 8 weeks thereafter while on study drug, then 28 days after discontinuation of all study treatments or before initiation of a new antineoplastic or investigational therapy, whichever occurred first.
Table 8: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
BPI-SF | Patient-reported generic questionnaire for pain intensity and impact. It is a validated 9-item instrument that uses a self-reported scale assessing level of pain, its effect on activities of daily living, and analgesic medication use. Tracking analgesic use is of particular importance to ensure that the delay in pain progression observed is the result of the treatment being studied rather than the result of increased analgesic use.68-70 Each item is scored on an 11-point scale from 0 to 10, where 0 is no pain or no interference and 10 is the worst pain or complete interference.68 The BPI-SF is available as a long and short version, the latter of which has a 24-hour recall period for both worst pain and least pain items.71 Score reductions indicate improvement in the patient’s perception of pain.68-70 A composite of the 4 pain items (a mean severity score) can be presented and pain interference is typically scored as the mean of the 7 interference items.68 | Validity: Strong correlations between worst pain and average pain items (r = 0.79) and between the worst pain item and the present pain intensity (r = 0.52).71 Support for content validity via in-depth interviews for worst pain item in a study of patients with CRPC and bone metastases.72 Reliability: Good internal consistency reliability in study of patients with mCRPC with alpha ≥ 0.89 and good internal consistency reliability with ICC values ≥ 0.73.71 Responsiveness: Not assessed in indicated population. | An MID estimate of ≥ 2 points or 30% change in pain intensity items from baseline was previously used in studies in patients with mCRPC.65,66 |
EORTC QLQ-PR25 | The EORTC QLQ-PR25 is a prostate cancer-specific module of the EORTC questionnaire that assesses the functioning and symptoms of patients with prostate cancer. Patients self-rate their current state of pain as it relates to urination, ease and frequency of urination, and bowel and hormonal treatment-related symptoms and signs during the past week.73 Patients also answer 5 questions about weight loss or gain and sexual interest and 4 questions about sexual activity during the past 4 weeks. There are 4 possible responses within each question to reflect intensity: “not at all,” “a little,” “quite a bit,” and “very much.”67 Questions are grouped into symptom and functional scales. Raw scores from each scale are linearly transformed onto a scale of 0 to 100, with higher scores indicating worse symptoms (urinary, bowel, hormonal treatment-related symptoms) or better functioning (sexual activity, sexual functioning).67,73 | Validity: The EORTC QLQ-PR25 questionnaire demonstrated discrimination between clinically distinct patient subgroups (curative vs. palliative treatment intent and higher vs. lower Karnofsky performance status scores), indicating known-group validity for patients with prostate cancer.73 Reliability: Internal consistency reliability (Cronbach coefficient alpha) was between 0.70 and 0.86 for the urinary symptoms and sexual function scales, but < 0.70 for the bowel function and side effects of hormonal treatment scales for patients with prostate cancer.73 In another study consisting of patients with prostate cancer, ICCs ranged from 0.45 to 0.78 for all the domains in the EORTC QLQ-PR25, indicating moderate to good agreement for each domain.67 Responsiveness: Responsiveness of the EORTC QLQ-PR25 scale to changes in health status over time was evaluated in patients with prostate cancer by testing for significant changes in questionnaire scores as a function of observed changes in performance status (defined as a shift of at least 1 level upwards or downwards on the Karnofsky scale). Statistically significant differences over time in the expected direction were observed for the urinary symptom scale, the bowel symptom scale, and the sexual functioning scale, but not for the hormonal treatment-related symptom scale or the sexual activity scale.73 | A between-group MID of 5 points has been used for each of the domains of the EORTC QLQ-PR25 in patients from Taiwan with various stages of prostate cancer and treatment.67 |
BPI-SF = Brief Pain Inventory–Short Form; CRPC = castration-resistant prostate cancer; EORTC = European Organisation for Research and Treatment of Cancer; EORTC QLQ-PR25 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Prostate Cancer; ICC = intraclass correlation coefficient; mCRPC = metastatic castration-resistant prostate cancer; MID = minimal important difference; vs. = versus.
Statistical Analysis
A summary of the statistical analysis of efficacy outcomes is provided in Table 9.
Sample Size and Power Calculation
Sample size and power calculations were based on a stratified log-rank test. The following assumptions were used to determine the sample size for the primary outcome of rPFS:
1:1 randomization of patients to talazoparib plus enzalutamide versus placebo plus enzalutamide
15% discontinuation rate at 12 months for rPFS and 6% dropout at 20 months for OS
median rPFS assumed to be 16 months for placebo plus enzalutamide and the median rPFS for talazoparib plus enzalutamide assumed to be 23 months, with an HR of 0.696
median OS assumed to be 35 months for placebo plus enzalutamide and the median OS for talazoparib plus enzalutamide assumed to be 46.7 months, with an HR of 0.75 under the exponential model assumption
approximately 15% of the all-comers population were assumed to harbour HRR deficiencies
nonuniform patient accrual over 16.3 months in the all-comers population followed by an additional 13.5 months to enrol additional patients with HRR-deficient mCRPC.
For the primary comparison of rPFS in the population with HRR deficiencies (i.e., combining patients with HRR deficiencies from cohort 1 and cohort 2), 224 rPFS events based on BICR assessment would provide 85% power to detect an HR of 0.64 using a 1-sided stratified log-rank test at a significance level of 0.0125, and 2 interim analyses using a Lan DeMets alpha spending and a Lan DeMets beta spending function to determine the nonbinding boundaries and preserve the overall error rate. It was estimated that 380 patients with HRR deficiencies would be needed to observe the 224 events.
The study will be underpowered for OS in the population with HRR deficiencies. The final analysis for OS in the population with DNA damage response deficiency will occur at the time of the final analysis of OS in the all-comers population. It was estimated that 173 OS events in the population with HRR deficiencies will have occurred at this time, providing 36% power to detect an HR of 0.75 using a 1-sided log-rank test at a significance level of 0.0125.
Statistical Testing
rPFS was analyzed using a 1-sided log-rank test, stratified by randomization stratification factors, and the Kaplan-Meier method. To maintain overall type I error at or below a 1-sided alpha of 0.025, alphas were split equally (1-sided alpha = 0.0125) between cohorts 1 and 2. The HR and associated 95% CI were estimated using a Cox proportional hazards model, with a 2-sided 95% CI and corresponding P value estimated based on the Brookmeyer-Crowley method. The distribution of time-to-event rPFS, including median rPFS and event-free rates at selected time points, was estimated using the Kaplan-Meier method for each treatment group.
For OS, a hierarchical stepwise gatekeeping testing procedure was used to preserve the overall type I error only if the difference in rPFS was significant, using a 1-sided alpha of 0.0125. Time to deterioration of pain was estimated by the Kaplan-Meier method, and 2-sided 95% CIs were based on the Brookmeyer-Crowley method. Analysis for EORTC QLQ-PR25 consisted of mean change from baseline and overall change from baseline using a longitudinal mixed effects model. The pain and HRQoL outcomes were not controlled for multiplicity. Safety data were analyzed descriptively.
Subgroup Analysis
For the prespecified subgroups of interest, the stratified HR of rPFS were performed by BRCA alteration status and prior treatment with NHT or taxane therapy. These analyses were not adjusted for multiplicity.
Censoring and Data Imputation Methods
For rPFS, the following criteria and hierarchy were applied to censoring: no adequate baseline assessment, start of new antineoplastic therapy, event after missing assessments, withdrawal of consent, lost to follow-up, no adequate postbaseline tumour assessment, and ongoing without an event. For OS, the following criteria and hierarchy were applied to censoring: withdrawal of consent, lost to follow-up, and ongoing without an event. For all outcomes, missing data were not imputed.
Sensitivity Analyses
Sensitivity analyses are summarized in Table 9.
Table 9: Statistical Analysis of Efficacy End Points — TALAPRO-2 Trial
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
rPFS by BICR | Stratified log-rank test; Cox proportional hazard analysis | Stratified according to:
| Missing data were not imputed |
|
OS | Stratified log-rank test; Cox proportional hazard analysis | Same as rPFS | Missing data were not imputed | None |
Time to deterioration of pain via BPI-SF | Kaplan-Meier method | NR | NR | NR |
EORTC QLQ-PR25 functional and symptom scales | Descriptive; longitudinal mixed effects model | NR | NR | NR |
BICR = blinded independent central review; BPI-SF = Brief Pain Inventory–Short Form; CSPC = castration-sensitive prostate cancer; EORTC QLQ-PR25 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Prostate Cancer; HRR = homologous recombination repair; NHT = novel hormonal therapy; NR = not reported; OS = overall survival; rPFS = radiographic progression-free survival; vs. = versus.
Sources: TALAPRO-2 HRR-deficient Clinical Study Report.1 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Analysis Populations
The efficacy outcomes were analyzed based on the intention-to-treat population (ITT), regardless of whether treatment was administered. For the pain and HRQoL outcomes, only patients with baseline and at least 1 postbaseline assessment were analyzed. The safety outcomes were analyzed using the safety population, defined as patients who received at least 1 dose of any study medication.
Results
Patient Disposition
A summary of patient disposition is in Figure 1.
In total, 2,896 patients were screened, and 200 were randomized to the talazoparib plus enzalutamide and 199 to placebo plus enzalutamide groups; 169 patients were included from cohort 1 and 230 patients from cohort 2. Two patients in the talazoparib plus enzalutamide group were not treated with either study drug and 1 patient was treated with only enzalutamide. At the data cut-off date of September 3, 2024, 74.7% (n = 148) of patients in the talazoparib plus enzalutamide group discontinued treatment with talazoparib, and 89.9% (n = 179) of patients in the placebo plus enzalutamide group discontinued placebo. The most common reason for discontinuation in both groups was disease progression (talazoparib plus enzalutamide = 28.3%; placebo plus enzalutamide = 41.7%), followed by global deterioration of health status (talazoparib plus enzalutamide, n = 35; placebo plus enzalutamide, n = 44), and AEs (talazoparib plus enzalutamide, n = 25; placebo plus enzalutamide, n = 15). Discontinuations from enzalutamide were reported for 75.8% (n = 150) of patients in the talazoparib plus enzalutamide group and 89.9% (n = 179) of patients in the placebo plus enzalutamide group, with disease progression as the most frequently reported reason for discontinuation in both groups (28.8% and 41.2%, respectively).
Figure 1: Summary of Patient Disposition — Population With HRR Deficiencies, TALAPRO-2 Trial
CSR = Clinical Study Report; DDR = DNA damage response (described as HRR in this figure); HRR = homologous recombination repair.
Sources: TALAPRO-2 HRR-deficient Clinical Study Report.1 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Baseline Characteristics
A summary of baseline patient characteristics of the ITT population is in Table 10. The baseline characteristics outlined in Table 11 are limited to those that are most relevant to this review or were felt to affect the outcomes or interpretation of the study results. Overall, key baseline characteristics were generally balanced between treatment groups, except the talazoparib plus enzalutamide group had a ██████ ████ ████████ █████ ███ █████ ██████ █████ ███ █ ██████ ██████████ ██ ████████ ███ ███ ████████ ██████ █████████ ██████ ██████ ██████. The trial population was predominately white (68%), with a median age of 70 years. Most patients had an ECOG PS score of 0 (approximately 62%), indicating good overall performance; normal or mild renal impairment (approximately 90%); bone and soft tissue disease-site metastasis (approximately 84%); a Gleason score greater than or equal to 8 (approximately 74%); and had █████ ███████ ████ ███████ ██ ██████ ██████████████ ████ ███ ████████████████ ██████████████ ██████████████ █████ In both groups, the most commonly detected HRR gene alteration was BRCA2 (33.8%), followed by ATM (21.6%), CDK12 (18.8%), and CHEK2 (17.8%).
Table 10: Summary of Baseline Characteristics — Population With HRR Deficiencies, TALAPRO-2 Trial
Characteristic | Talazoparib + enzalutamide n = 200 | Placebo + enzalutamide n = 199 |
|---|---|---|
Age (years) | ||
Mean (SD) | █████ ██████ | █████ ██████ |
Median (range) | 70 (41 to 90) | 71 (44 to 90) |
Race, n (%) | ||
Asian | 45 (22.5) | 39 (19.6) |
Black or African American | 6 (3.0) | 5 (2.5) |
Multiracial | 0 | 1 (0.5) |
Native Hawaiian or Other Pacific Islander | 1 (0.5) | 1 (0.5) |
Not reported | 10 (5.0) | 17 (8.5) |
White | 137 (68.5) | 136 (68.3) |
ECOG PS, n (%) | ||
0 | 128 (64.0) | 118 (59.3) |
1 | 72 (36.0) | 81 (40.7) |
Renal impairment at baseline (mL/min/1.73 m2), n (%) | ||
Mild (60 to 89) | ██ ██████ | ██ ██████ |
Moderate (30 to 59) | ██ (10.0) | ██ ██████ |
Normal (≥ 90) | ██ ██████ | ██ ██████ |
Gleason score, n (%) | ||
< 8 | 42 (21.0) | 53 (26.6) |
≥ 8 | 152 (76.0) | 142 (71.4) |
███ ████████ | | █████ | | █████ |
Baseline serum PSA (ng/mL) | ||
Patients, n | 199 | 199 |
Mean (SD) | █████ ████████ | █████ ████████ |
Median (range) | 19.65 (0.22 to 3,412.00) | 18.00 (0.04 to 1,055.00) |
Previous anticancer treatment, n (%) | ||
██████ █████████ | ██ ██████ | ██ ██████ |
██████████████ | ███ ██████ | ███ ██████ |
Docetaxel | 56 (28.3) | 60 (30.2) |
████████████████ ██████████████ ██████████████ ██████████ ███████████ ████████████ ███████████ ████████ | ███ ██████ | ███ ██████ |
Novel hormonal drugs (abiraterone, abiraterone acetate, orteronel) | 17 (8.6) | 17 (8.5) |
Distribution of disease at screening, n (%) | ||
Bone (includes bone with soft tissue component) | 176 (88.0) | 158 (79.4) |
Lymph node | 81 (40.5) | 93 (46.7) |
Visceral disease (lung or liver) | ██ ██████ | ██ ██████ |
Visceral disease (lung) | 23 (11.5) | 26 (13.1) |
Visceral disease (liver) | 10 (5.0) | 6 (3.0) |
Other soft tissue | 23 (11.5) | 20 (10.1) |
HRR gene alterations, n (%) | ||
≥ 1 alteration in HRR genes | 198 (99.0) | 197 (99.0) |
ATM | 47 (23.5) | 39 (19.6) |
ATR | 3 (1.5) | 12 (6.0) |
BRCA1 | 11 (5.5) | 12 (6.0) |
BRCA2 | 62 (31.0) | 73 (36.7) |
CDK12 | 36 (18.0) | 39 (19.6) |
CHEK2 | 34 (17.0) | 37 (18.6) |
FANCA | 4 (2.0) | 5 (2.5) |
MLH1 | 9 (4.5) | 1 (0.5) |
MRE11A | 1 (0.5) | 2 (1.0) |
NBN | 8 (4.0) | 3 (1.5) |
PALB2 | 9 (4.5) | 8 (4.0) |
RAD51C | 2 (1.0) | 2 (1.0) |
ECOG PS = Eastern Cooperative Oncology Group Performance Status; HRR = homologous recombination repair; PSA = prostate-specific antigen; SD = standard deviation.
Note: Racial categories used in the table are as reported in the source and may not align with Canada's Drug Agency inclusive language guidelines.
Sources: TALAPRO-2 HRR-deficient Clinical Study Report.1 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Exposure to Study Treatments
The median treatment durations for talazoparib and placebo were 88.3 weeks in the talazoparib plus enzalutamide group (relative dose intensity = 78.3%) and 60.0 weeks in the placebo plus enzalutamide group (relative dose intensity = 100.00%). The median treatment durations for enzalutamide were 90.5 weeks (relative dose intensity = 99.83%) in the talazoparib plus enzalutamide group and 60 weeks (relative dose intensity = 100.00%) in the placebo plus enzalutamide group. The average daily dose administered for talazoparib was ████ ██ and ████ ██ for placebo. The average daily dose administered for enzalutamide in the talazoparib plus enzalutamide group was █████ ██ and █████ ██ in the placebo plus enzalutamide.
There were more dose interruptions and reductions due to AEs in the talazoparib plus enzalutamide group than in the placebo plus enzalutamide group. Dose interruptions of talazoparib due to AEs were reported in █████ ███████ ██ █████████ ███ █████ ███ ███ of patients taking placebo. In both groups, the most common AEs leading to dose interruptions were anemia, neutrophil count decreased, and platelet count (percent not reported). Dose reductions of talazoparib due to AEs were reported in 54.5% (n = 108) of patients taking talazoparib and 5.0% (n = 10) of patients taking placebo. In both groups, the most common AE leading to dose reductions was anemia (percent not reported).
████ ████████ █████ █ ██ ██████ received at least 1 concomitant medication during the trial ███ ████ ████ ███ ██████████ ███████ ██████. In both groups, the most common classes of concomitant medications were ██████████ ██████████████ ████ and ████████████████ ██████ ██ ██████. Most concomitant medications were used consistently between the 2 treatment groups, except ███████████ ████████████ were more commonly used in the talazoparib plus enzalutamide group than placebo plus enzalutamide ██████ ███ ███████
Subsequent Anticancer Therapy
Fewer patients (36.9%) in the talazoparib plus enzalutamide group received subsequent anticancer therapy than in the placebo plus enzalutamide group (56.8%), with the most common subsequent anticancer therapy being taxane-based therapy (26.3% versus 39.2%, respectively).
Efficacy
Only those efficacy outcomes and analyses of subgroups identified as important to this review are reported. The main findings for the efficacy outcomes for the TALAPRO-2 trial are from the data cut-off dates of October 3, 2022, and September 3, 2024. The boundary for statistical significance for the primary outcome of rPFS was met at the data cut-off date of October 3, 2022; therefore, rPFS data were reported descriptively at the September 3, 2024, data cut-off date (i.e., not controlled for type I error). All other outcomes are based on the data cut-off date of September 3, 2024.
rPFS by BICR
Table 11 provides a summary of results for rPFS by BICR. In total, 170 events had occurred in both groups by the data cut-off date of October 3, 2022. The median duration of follow-up for rPFS was 17.5 months (range not reported) for the talazoparib plus enzalutamide group and 16.8 months (range not reported) for the placebo plus enzalutamide group. The median rPFS was not reached (95% CI, 21.9 months to not reached) in the talazoparib plus enzalutamide group and 13.8 months (95% CI, 11.0 months to 16.7 months) for the placebo plus enzalutamide group (log-rank test P < 0.0001), with a between-group HR of 0.45 (95% CI, 0.33 to 0.61) in favour of talazoparib plus enzalutamide. The Kaplan-Meier–estimated probability of rPFS at 12 months was █████ ████ ███ ████ ██ █████ ██████ █████ ████ ███ ████ ██ █████ in the talazoparib plus enzalutamide and placebo plus enzalutamide groups, respectively, with a between-group difference of █████ ████ ███ ████ ██ ██████ The results of sensitivity analyses were consistent with the primary analysis. At the second data cut-off date of September 3, 2024, rPFS descriptive results were consistent with the first data cut-off date (Table 10). The median rPFS was 30.7 months (95% CI, 24.3 months to 38.5 months) in the talazoparib plus enzalutamide group and 12.3 months (95% CI, 11.0 months to 16.5 months) in the placebo plus enzalutamide group. The Kaplan-Meier–estimated probability of rPFS at 48 months was █████ ████ ███ ████ ██ █████ ██████ █████ ████ ███ ███ ██ █████ in the talazoparib plus enzalutamide and placebo plus enzalutamide groups, respectively, with a between-group difference of █████ ████ ███ ████ ██ ██████
rPFS Subgroup Analyses
The rPFS results were consistent across the subgroup analyses of interest by BRCA alteration status (with or without) and prior treatment with NHT or taxane therapy in favour of talazoparib plus enzalutamide. Based on the data cut-off date of September 3, 2024, the stratified HR for patients with BRCA alterations was 0.26 (95% CI, 0.16 to 0.42), and without BRCA alterations was 0.65 (95% CI, 0.47 to 0.91). For patients with prior NHT or taxane exposure, the rPFS HR was ████ ████ ███ ████ ██ █████, and for patients with no prior NHT or taxane exposure, the HR was ████ ████ ███ ████ ██ ██████
Table 11: rPFS by BICR — ITT Population With HRR Deficiencies, TALAPRO-2 Trial
rPFS by BICR | Talazoparib + enzalutamide n = 200 | Placebo + enzalutamide n = 199 |
|---|---|---|
Data cut-off date of October 3, 2022 | ||
Patients with events, n (%) | ||
Total | 66 (33.0) | 104 (52.3) |
Progressive disease | ██ ██████ | ██ ██████ |
Death | ██ █████ | ██ █████ |
Patients censored | 134 (67.0) | 95 (47.7) |
Reason for censoring, n (%) | ||
███████ ███████ ██ █████ | ███ ██████ | ██ ██████ |
█████ ██ ███ ██████████ █████ ██████ ███████ | ██ █████ | ██ ██████ |
██████████ ██ ███████ | █████ | ██ █████ |
█████ █████ ██ ███████ ██ ██████████ █████████████ ███████████ | █████ | █████ |
████ ██ ██████ ██ | █████ | || |
██ ████████ ██████████ | █████ | || |
██ █████████████ ██████ ██████████ | ███ | ███ |
rPFS (months) | ||
Median (95% CI)a | NE (21.9 to NE) | 13.8 (11.0 to 16.7) |
Hazard ratio (95% CI) | 0.45 (0.33 to 0.61) | |
Log-rank test P value, 1-sidedb | < 0.0001 | |
Probability of being event free, % (95% CI) | ||
At 12 monthsa | ████ █████ ██ █████ | ████ █████ ██ █████ |
Absolute between-group difference, % (95% CI) | ████ █████ ██ █████ | |
Data cut-off date of September 3, 2024 | ||
Patients with events, n (%) | ||
Total | 99 (49.5) | 127 (63.8) |
Progressive disease | ██ ██████ | ███ ██████ |
Death | ██ █████ | ██ █████ |
Patients censored | 101 (50.5) | 72 (36.2) |
Reason for censoring, n (%) | ||
███████ ███████ ██ █████ | ██ ██████ | ██ ██████ |
█████ ██ ███ ██████████ █████ ██████ ███████ | ██ ██████ | ██ ██████ |
██████████ ██ ███████ | █████ | █████ |
█████ █████ ██ ███████ ██ ██████████ █████████████ ███████████ | █████ | █████ |
████ ██ ██████ ██ | █████ | || |
██ ████████ ██████████ | █████ | || |
██ █████████████ ██████ ██████████ | || | || |
rPFS (months) | ||
Median (95% CI)a | 30.7 (24.3 to 38.5) | 12.3 (11.0 to 16.5) |
Hazard ratio (95% CI) | 0.47 (0.36 to 0.61) | |
Probability of being event free, % (95% CI) | ||
At 12 monthsa | ████ █████ ██ █████ | ████ █████ ██ █████ |
Absolute between-group difference, % (95% CI) | ████ █████ ██ █████ | |
At 48 monthsa | ████ █████ ██ █████ | ████ ████ ██ █████ |
Absolute between-group difference, % (95% CI) | ████ █████ ██ █████ | |
BICR = blinded independent central review; CI = confidence interval; HRR = homologous recombination repair; ITT = intention to treat; NE = not estimable; rPFS = radiographic progression-free survival.
aBased on Kaplan-Meier estimates.
bP values were tested within a hierarchical testing strategy to control for multiple comparisons.
Sources: TALAPRO-2 HRR-deficient Clinical Study Report.1 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Figure 2: Kaplan-Meier Plot of rPFS by BICR — ITT Population With HRR Deficiencies, TALAPRO-2 Trial
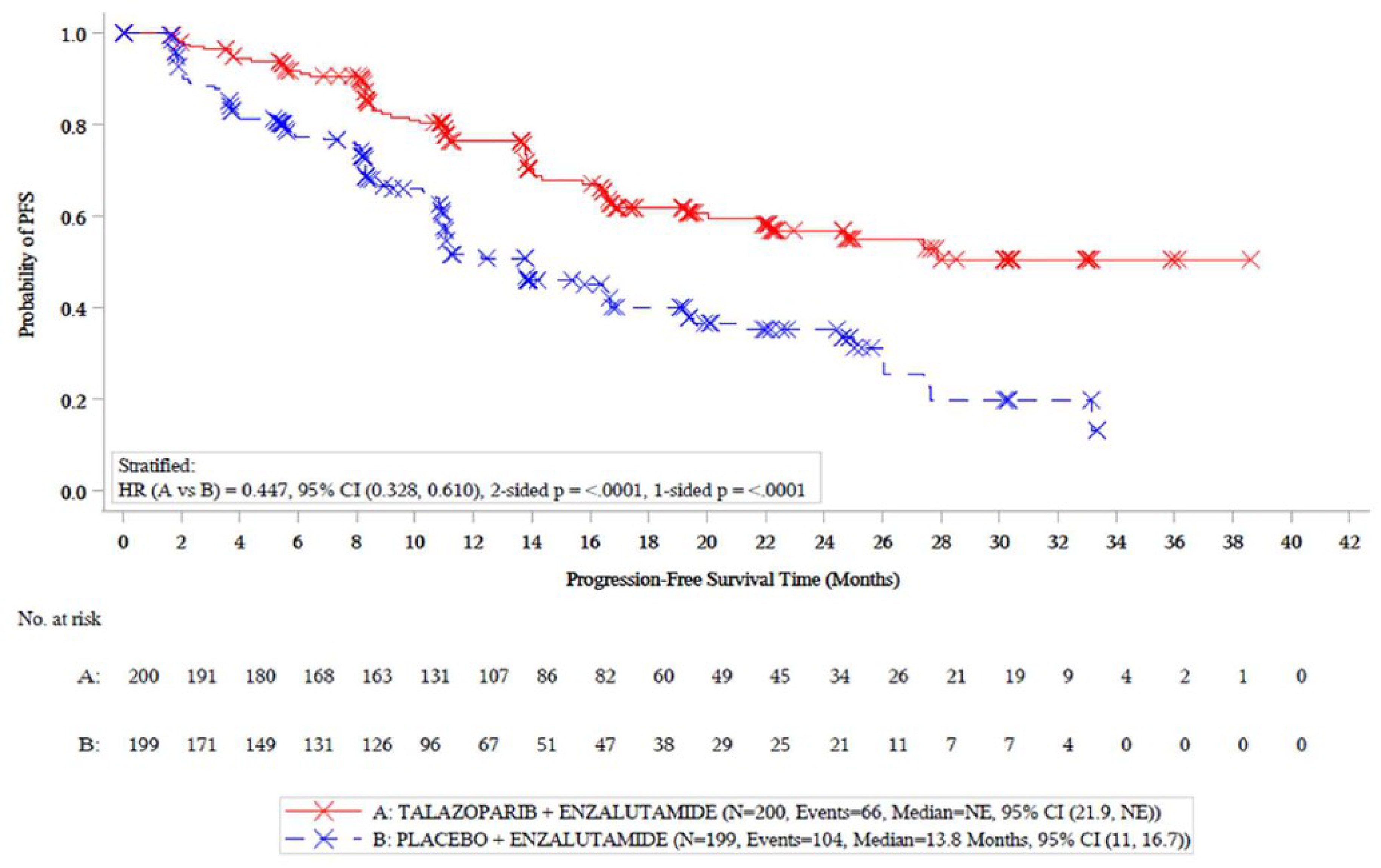
BICR = blinded independent central review; CI = confidence interval; HR = hazard ratio; HRR = homologous recombination repair; ITT = intention to treat; NE = not estimable; PFS = progression-free survival; rPFS = radiographic progression-free survival.
Sources: TALAPRO-2 HRR-deficient Clinical Study Report.1 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Overall Survival
Table 12 provides a summary of results for OS. In total, 219 events had occurred in both groups by the data cut-off date of September 3, 2024. The median follow-up for OS was 44.2 months (range not reported) for the talazoparib plus enzalutamide group and ████ ██████ (range not reported) for the placebo plus enzalutamide group (Figure 3). The median OS was 45.1 months (95% CI, 35.4 months to not reached) for the talazoparib plus enzalutamide group and 31.1 months (95% CI, 27.3 months to 35.4 months) in the placebo plus enzalutamide group (log-rank test P < 0.0001), with a between-group HR of 0.62 (95% CI, 0.48 to 0.81) in favour of talazoparib plus enzalutamide. The results of sensitivity analyses were consistent with the primary analysis. The Kaplan-Meier–estimated probability of OS at 12 months was █████ ████ ███ ████ ██ █████ ██████ █████ ████ ███ ████ ██ ████ ██ ███ ████████████████████████ ███ ████████████████████ ███████ █████████████ ███████ █████ ███████████ ████ ████ ███ ████ ██ ██████ The Kaplan-Meier–estimated probability of OS at 48 months was █████ ████ ███ ████ ██ █████ ██████ █████ ████ ███ ████ ██ █████ █████████████ ███████████ █████ ████ ███ ████ ██ ██████ ██ ███ ████████████████████████ ███ ████████████████████ ███████ █████████████
Table 12: Overall Survival — ITT Population With HRR Deficiencies, TALAPRO-2 Trial
Overall survival | Talazoparib + enzalutamide n = 200 | Placebo + enzalutamide n = 199 |
|---|---|---|
Data cut-off date of September 3, 2024 | ||
Deaths, n (%) | 93 (46.5) | 126 (63.3) |
Patients censored | ███ ██████ | ██ ██████ |
Reason for censoring, n (%) | ||
█████ | ██ ██████ | ██ ██████ |
██████████ ██ ███████ | ██ █████ | ██ █████ |
████ ██ ██████ ██ | █████ | █████ |
Overall survival (months) | ||
Median (95% CI)a | 45.1 (35.4 to NE) | 31.1 (27.3 to 35.4) |
Hazard ratio (95% CI) | 0.62 (0.48 to 0.81) | |
Log-rank test P value, 1 sidedb | 0.0002 | |
Probability of being event free, % (95% CI) | ||
At 12 monthsa | 91.4 █████ ██ █████ | 88.5 █████ ██ █████ |
Absolute between-group difference, % (95% CI) | 2.9 █████ ██ ████ | |
At 48 monthsa | 48.1 █████ ██ █████ | 28.5 █████ ██ █████ |
Absolute between-group difference, % (95% CI) | 19.6 █████ ██ █████ | |
CI = confidence interval; HRR = homologous recombination repair; ITT = intention to treat; NE = not estimable.
aBased on Kaplan-Meier estimates.
bP values were tested within a hierarchical testing strategy to control for multiple comparisons.
Sources: TALAPRO-2 HRR-deficient Clinical Study Report.1 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Figure 3: Kaplan-Meier Plot of OS by BICR — ITT Population With HRR Deficiencies, TALAPRO-2 Trial
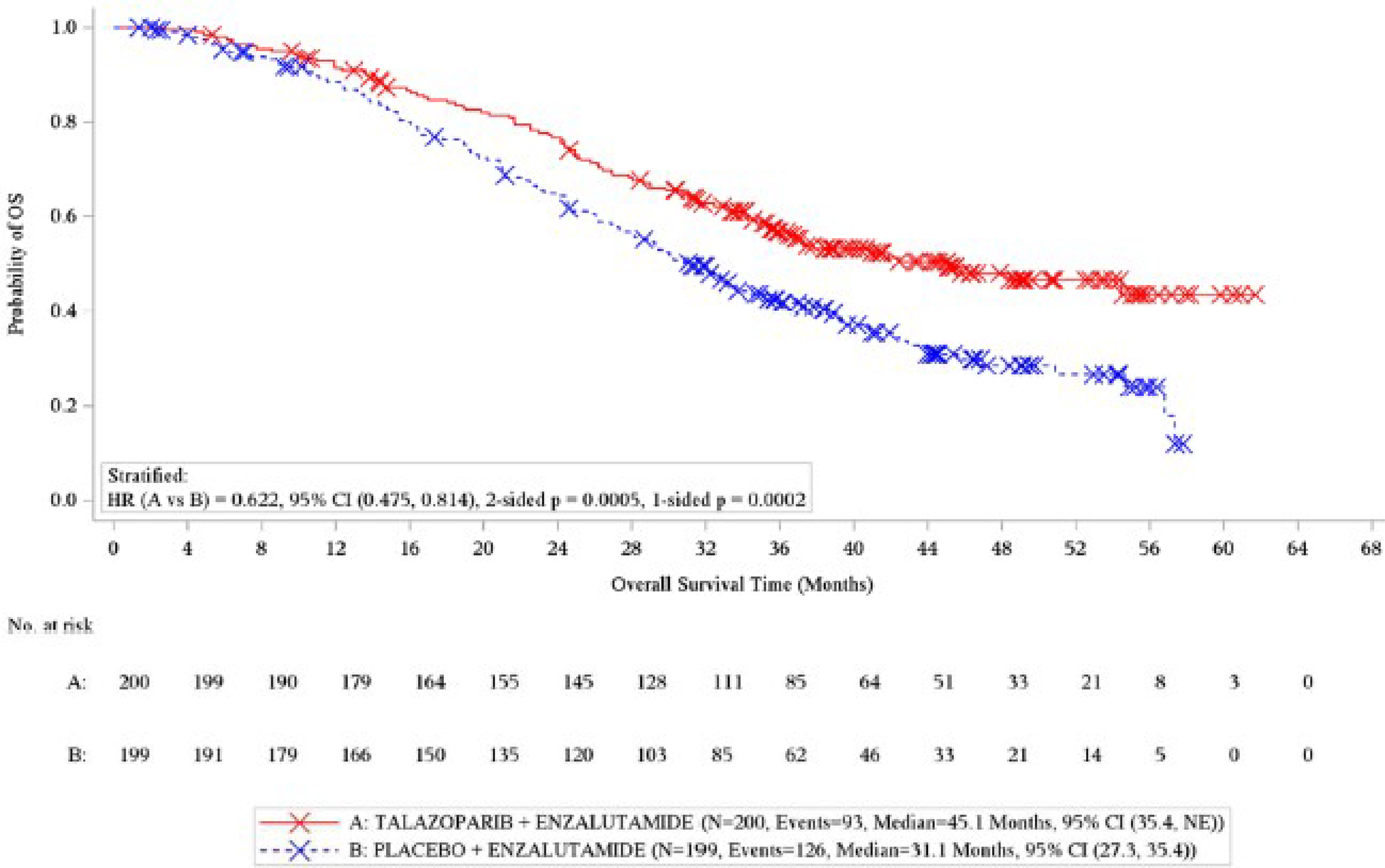
BICR = blinded independent central review; CI = confidence interval; HR = hazard ratio; HRR = homologous recombination repair; ITT = intention to treat; NE = not estimable; OS = overall survival.
Sources: TALAPRO-2 HRR-deficient Clinical Study Report.1 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Time to Deterioration of Pain by BPI-SF
At the data cut-off date of September 3, 2024, 58 events had occurred in both groups, and the median time to deterioration of pain was not reached in either group (Figure 4). The stratified between-group HR was 0.55 (95% CI, 0.33 to 0.94) in favour of talazoparib plus enzalutamide. ███ ████████████ ███████████ ██ █████ ████ ██ ████ ███████████ ██ ██ ██████ ███ █████ ████ ██ ███ █████████ ██████ █████ ████ ██ ███ █████████ █████████████ ███████████ ████ ████ ███ ████ ██ ███████ ███ ██ ██ ██████ ███ █████ ████ ██ ███ █████████ ██████ █████ ████ ██ ███ █████████ █████████████ ███████████ █████ ████ ███ ███ ██ ██████ ██ ███ ████████████████████████ ███ ████████████████████ ███████ █████████████
Figure 4: Kaplan-Meier Plot of Time to Deterioration in Pain Symptoms by BPI-SF — ITT Subset Population With HRR Deficiencies, TALAPRO-2 Trial
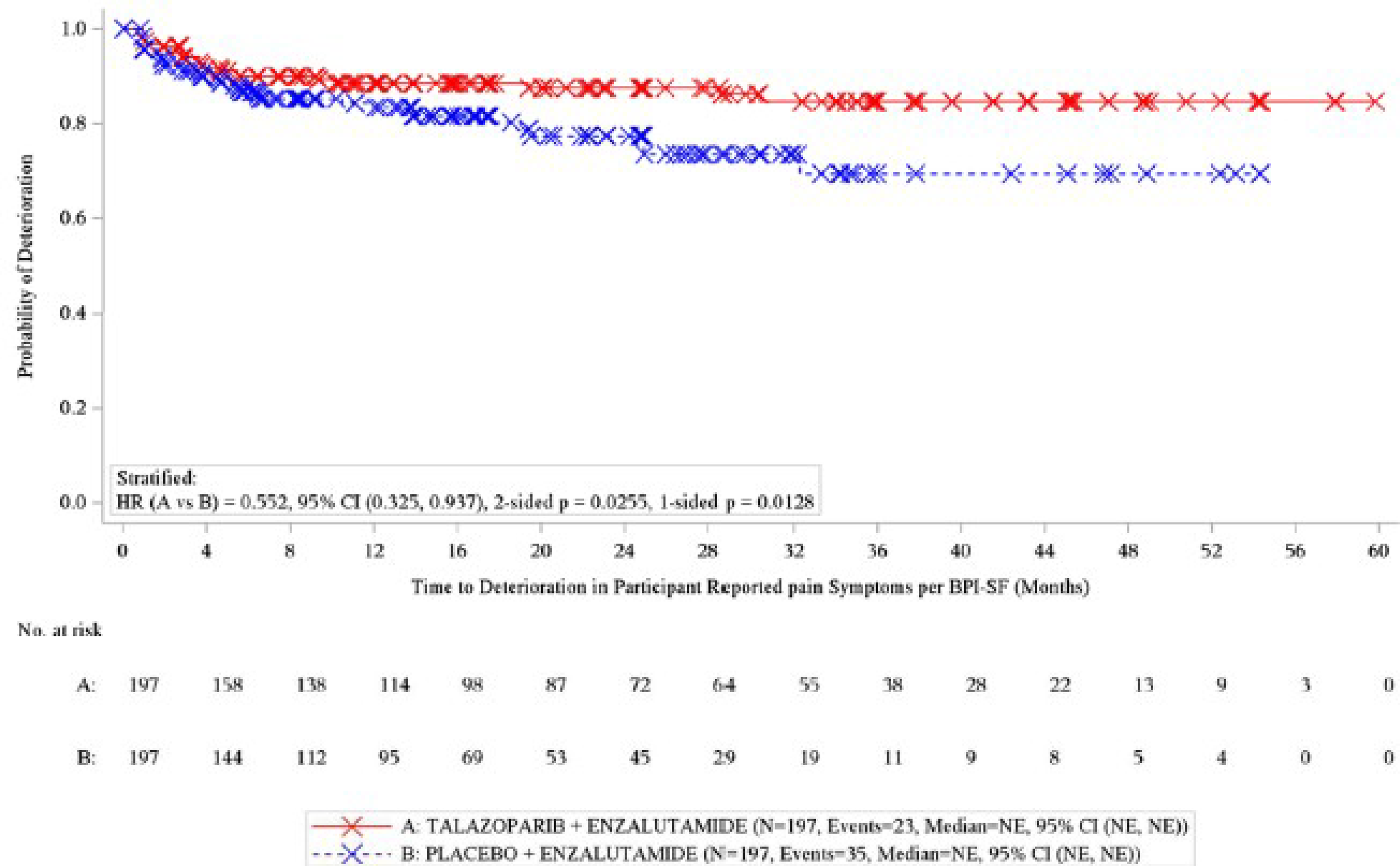
BPI-SF = Brief Pain Inventory–Short Form; CI = confidence interval; HR = hazard ratio; HRR = homologous recombination repair; ITT = intention to treat; NE = not estimable.
Sources: TALAPRO-2 HRR-deficient Clinical Study Report.1 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
HRQoL by EORTC QLQ-PR25
The EORTC QLQ-PR25 functional and symptom scale scores at the data cut-off date of September 3, 2024, are provided in Figure 5 and Figure 6, respectively. Of the ITT population, ██ ███ ██ ███ patients in the talazoparib plus enzalutamide group and ██ ███ ██ ███ patients in the placebo plus enzalutamide group contributed to the analyses at week 109, except for the incontinence aid symptoms score, which was informed ██ ██ ███ ██ patients, respectively. No comparison was made for the sexual functioning score due to limited data. The sexual activity score and the symptom scale scores were generally maintained and similar in both treatment groups up to week 109. Improvements in urinary and bowel symptoms favoured the talazoparib plus enzalutamide group, with urinary symptoms reaching the MID threshold of greater or equal to 5 points, suggesting a clinically important improvement. All other scale scores did not reach the clinically important MID threshold.

Note: Figure 5 contained confidential information and was removed at the request of the sponsor.
Figure 6: Forest Plot of Estimated Treatment Difference in EORTC QLQ-PR25 Symptom Scales — ITT Population With HRR Deficiencies, TALAPRO-2 Trial
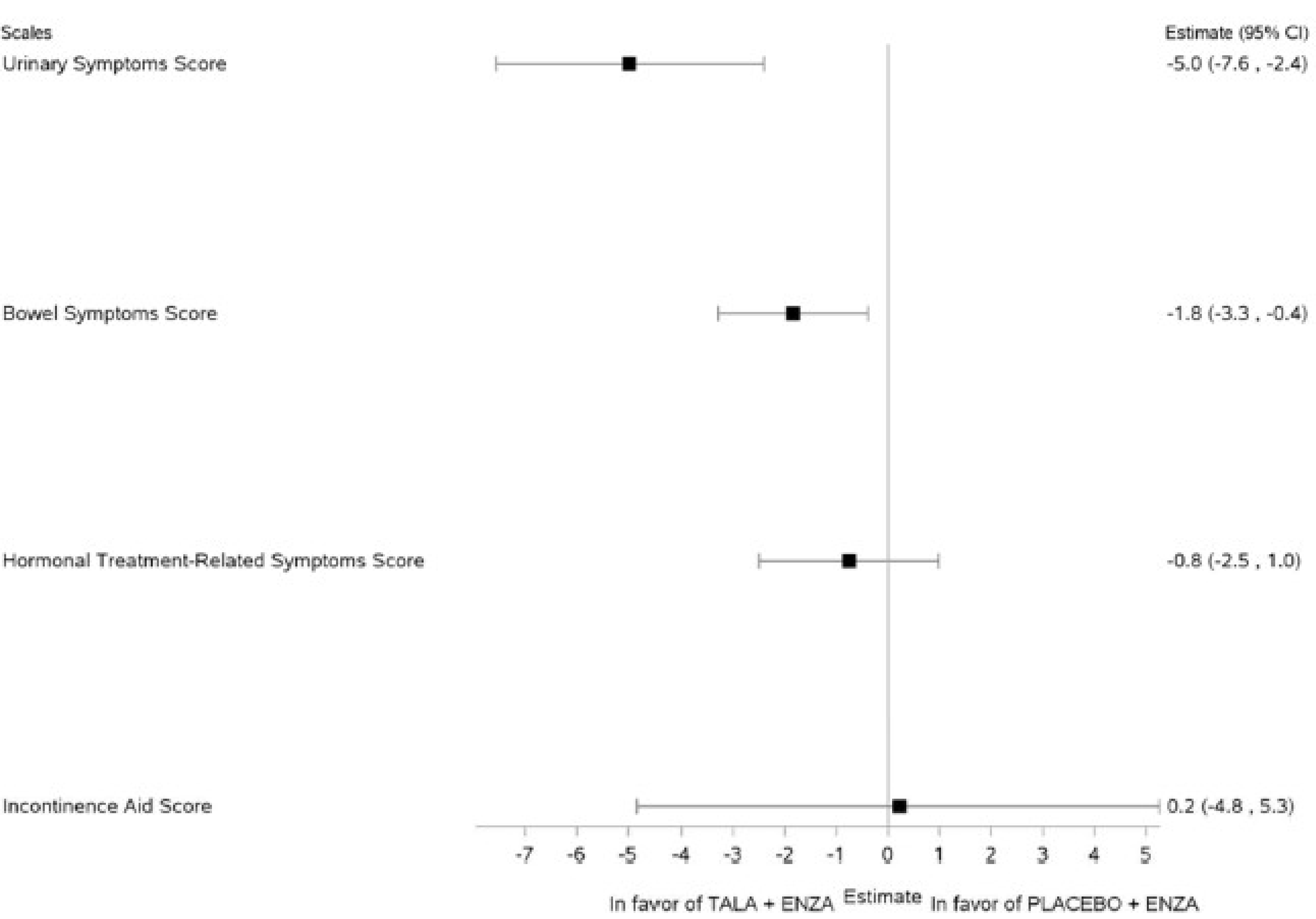
CI = confidence interval; ENZA = enzalutamide; EORTC QLQ-PR25 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Prostate Cancer; HRR = homologous recombination repair; ITT = intention to treat; TALA = talazoparib.
Note: Results are based on the data cut-off date of September 3, 2024.
Sources: TALAPRO-2 HRR-deficient Clinical Study Report.1 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Harms
Harms data reported in this section are from the data cut-off date of September 3, 2024, and summarized in Table 13.
Treatment-Emergent Adverse Events
Almost all patients in both treatment groups reported at least 1 TEAE (99.5% with talazoparib plus enzalutamide and 97.5% with placebo plus enzalutamide). The most frequently reported TEAEs in the talazoparib plus enzalutamide group were anemia (66.7% versus 18.6% with placebo plus enzalutamide), fatigue (34.8% versus 28.1% with placebo plus enzalutamide), and decreased neutrophil count (34.8% versus 7.0% with placebo plus enzalutamide); a higher proportion of patients in the talazoparib plus enzalutamide group reported these TEAEs than the placebo plus enzalutamide group. The most frequently reported TEAEs in the placebo plus enzalutamide group were fatigue (28.1% versus 34.8% with talazoparib plus enzalutamide), arthralgia (24.6% versus 16.7% with talazoparib plus enzalutamide), and back pain (23.1% versus 24.2% with talazoparib plus enzalutamide). A higher proportion of patients in the talazoparib plus enzalutamide group experienced at least 1 grade 3 or grade 4 TEAE ███████ versus the placebo plus enzalutamide group ████████
Serious TEAEs
The incidence of serious TEAES were higher in the talazoparib plus enzalutamide group ███████ versus the placebo plus enzalutamide group ███████ In both groups, ██████ █████ ████████ ███ ███████ █████ █████████ █████ ██ █████ were the most frequently reported serious TEAEs.
Withdrawals Due to TEAES
A higher proportion of TEAEs that led to study treatment discontinuation were reported in the talazoparib plus enzalutamide group (████) versus the placebo plus enzalutamide group (████), with anemia being the most common reason (████ ███ ████). Enzalutamide-only treatment discontinuation was reported for ████ of patients in the talazoparib plus enzalutamide group versus ████ in the placebo plus enzalutamide group.
Mortality
A lower proportion of deaths were reported in the talazoparib plus enzalutamide group (46.5%) versus placebo plus enzalutamide (63.3%), with ███████ ███████████ being the primary reason for death in both groups ██████ ██████ ███████
Notable Harms
The incidence of notable harms in both treatment groups was comparable and infrequent. Second primary malignancies (other than hematologic) occurred in ████ ███ ████ of patients, and embolic and thrombotic events occurred in 5.6% and 1.0% of patients in the talazoparib plus enzalutamide and placebo plus enzalutamide groups, respectively. There was 1 case of pneumonitis in each group.
Table 13: Summary of Harms Results — Safety Population With HRR Deficiencies, TALAPRO-2 Trial
Harms | Talazoparib + enzalutamide n = 198 | Placebo + enzalutamide n = 199 |
|---|---|---|
≥ 1 TEAE, n (%) | 197 (99.5) | 194 (97.5) |
Patients with grade 3 or 4 TEAEs, n (%) | ███ ██████ | ██ ██████ |
Most common any grade TEAEs ≥ 10% in either treatment group, n (%) | ||
Anemia | 132 (66.7) | 37 (18.6) |
Fatigue | 69 (34.8) | 56 (28.1) |
Decreased neutrophil count | 69 (34.8) | 14 (7.0) |
Decreased platelet count | 51 (25.8) | 5 (2.5) |
Back pain | 48 (24.2) | 46 (23.1) |
Decreased appetite | 46 (23.2) | 31 (15.6) |
Hypertension | 44 (22.2) | 39 (19.6) |
Nausea | 43 (21.7) | 36 (18.1) |
Decreased WBCs | 43 (21.7) | 15 (7.5) |
Fall | 39 (19.7) | 28 (14.1) |
Asthenia | 34 (17.2) | 33 (16.6) |
Arthralgia | 33 (16.7) | 49 (24.6) |
Constipation | 32 (16.2) | 41 (20.6) |
Diarrhea | 27 (13.6) | 24 (12.1) |
Hot flush | 24 (12.1) | 33 (16.6) |
Pyrexia | 22 (11.1) | 4 (2.0) |
Dizziness | 21 (10.6) | 16 (8.0) |
Weight decreased | 21 (10.6) | 18 (9.0) |
Dyspnea | 20 (10.1) | 11 (5.5) |
Headache | 14 (7.1) | 24 (12.1) |
Any grade SAEs, n (%) | ||
Patients with ≥ 1 SAE | ██ ██████ | ██ █████ |
██████ | ██ █████ | █████ |
███████ █████ █████████ | █████ | █████ |
█████████ | █████ | █████ |
█████████ ████████ | █████ | █████ |
███████ | █████ | █████ |
Patients who discontinued treatment due to TEAEs, n (%)a | ||
Patients who permanently discontinued both talazoparib or placebo and enzalutamide | ██ █████ | ██ █████ |
Reasons for discontinuation in ≥ 1% of either group | ||
Anemia | █████ | █████ |
Patients who permanently discontinued only talazoparib or placebo | ██ █████ | █████ |
Reasons for discontinuation in ≥ 1.5% of either group | ||
Anemia | █████ | || |
Patients who permanently discontinued only enzalutamide (only 1 case per preferred term) | █████ | █████ |
█████████ | █████ | || |
Deaths, n (%) | ||
Patients who died | 93 (46.5) | 126 (63.3) |
Cause of death | ||
███████ ███████████ | ██ ██████ | ██ ██████ |
███████ █████ ███ ███████ ██ █████ █████████ | █████ | █████ |
█████ | █████ | ██ █████ |
███████ | ██ █████ | ██ ██████ |
Any grade notable AEs related to talazoparib, n (%) | ||
Patients with ≥ 1 notable AEs related to talazoparib | ██ ██████ | ██ █████ |
██████ ███████ ████████████ ██████ ████ ████████████ | ██ █████ | █████ |
Embolic and thrombotic events, venous | 11 (5.6) | 2 (1.0) |
Pneumonitis | 1 (0.5) | 1 (0.5) |
AE = adverse event; HRR = homologous recombination repair; SAE = serious adverse event; TEAE = treatment-emergent adverse event; WBC = white blood cell.
aOnly included events which occurred in > 2 patients.
Sources: TALAPRO-2 HRR-deficient Clinical Study Report.1 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Critical Appraisal
Internal Validity
TALAPRO-2 was a randomized, double-blind, phase III trial. The patients and investigators were blinded to talazoparib or placebo but enzalutamide was open label. This design was appropriate because different dosing and oral tablets across the 2 treatment groups may have made blinding to enzalutamide impractical. Randomization procedures, including stratification by prior NHT or taxane therapy for CSPC and HRR gene alteration status (deficient versus nondeficient or unknown) were appropriate and conducted by interactive response technology. The talazoparib plus enzalutamide group had a ██████ ████ ████████ █████ ███ █████ ██████ █████ ███ █ ██████ ██████████ ██ ████████ ███ ███ ████████ ██████ █████████ ██████ ██████ ██████. These imbalances were likely due to chance, as ███ █████ ████████ ███████████████ ██ ████████ ████████ ████████ ███████ ██████ and based on clinical expert feedback, unlikely to have confounded the effect between treatment and outcomes.
Sample size and power calculations were based on the primary outcome of rPFS, and the trial was powered to detect significant differences for rPFS, but it was underpowered for the secondary outcome of OS. The interim analyses were preplanned with adequately justified stopping boundaries and the prespecified analyses of rPFS and OS were appropriately controlled for multiple comparisons. All other analyses were descriptive (i.e., not controlled for type I error), including the pain and HRQoL outcomes, which were deemed clinically important outcomes for the disease. The sample size for the prespecified subgroup analyses of interest for rPFS was small; the trial may not have been powered to detect subgroup differences.
Although patients and investigators were blinded to treatment assignment (enzalutamide was open label), there was a potential for unblinding due to the imbalances in TEAEs, particularly anemia across treatment groups (i.e., higher in the talazoparib plus enzalutamide group). Knowledge of treatment assignment could increase the risk of bias in the measurement of subjective outcomes, including HRQoL and subjective harms, although the extent and direction of bias cannot be predicted. To minimize the risk of bias in the measurement of rPFS, the trial performed tumour assessments using RECIST 1.1 criteria and radiographic scans were assessed by BICR. In addition, the findings of the sensitivity analyses for rPFS were consistent with the primary analysis.
Patients were permitted to receive posttreatment anticancer medications after study treatment had been discontinued (36.9% in the talazoparib plus enzalutamide group versus 56.8% in the placebo plus enzalutamide group), which may influence the assessment of OS. Because no sensitivity analyses were performed to test treatment policy strategy for OS (e.g., exclude the effect of subsequent therapies), the estimated effect would be a combination of treatment with talazoparib plus enzalutamide versus placebo plus enzalutamide, plus subsequent treatments. Therefore, survival results might be partially attributable to treatments administered after disease progression rather than the study treatment itself, although without the necessary analysis the direction and magnitude of bias is unclear. However, this is a relevant comparison, as it is reflective of how the intervention and comparator and subsequent therapies would be used in practice in Canada.
Based on the Clinical Study Report and statistical analysis plan, the proportional hazards assumption was not assessed or discussed. Despite the absence of these results, visual inspection of the Kaplan-Meier curves for rPFS and OS appears to indicate a clear separation (at approximately 3 months and 8 months, respectively), after which there appeared to be sustained proportionality throughout study treatment.
HRQoL was assessed by the BPI-SF and EORTC QLQ-PR25 questionnaires, which have been validated in patients with metastatic prostate cancer with evidence of reliability, responsiveness, and MID. The result of these outcomes was subject to potential bias due to more than 50% of missing data in each group, with a higher proportion of missing data in the placebo plus enzalutamide group, which could have biased the results in favour of talazoparib plus enzalutamide. For the EORTC QLQ-PR25, no models other than the mixed model for repeated measures were tested that applied alternative imputation methods or sensitivity analyses to assess the impact of missing data. The results were also subject to bias potentially due to differential incidence of notable harms such as anemia, although the direction and extent of bias are unclear.
External Validity
In general, the population requested for the reimbursement aligns with the approved Health Canada–approved indication, and the dosing and administration of talazoparib plus enzalutamide were consistent with the approved product monograph. However, the trial provided talazoparib plus enzalutamide as first-line treatment only (i.e., patients had not received prior systemic therapy for mCRPC) while the approved Health Canada indication is line agnostic. Therefore, there is no direct comparative evidence for the use of talazoparib plus enzalutamide in the second-line, third-line, and later-line settings. The clinical experts consulted by CDA-AMC noted that many medical oncologists currently favour prescribing chemotherapy for the first-line treatment of mCRPC in patients who have previously progressed on an ARPI in the metastatic CSPC and nonmetastatic CRPC settings. Therefore, the experts indicated that most clinicians would likely prescribe talazoparib plus enzalutamide as a second-line and beyond treatment due to the decreasing number of patients who are ARPI naive in the first-line mCRPC setting.
According to the clinical experts consulted by CDA-AMC, the eligibility criteria and baseline characteristics of the TALAPRO-2 trial are generalizable to adult patients with mCRPC in the setting in Canada, except that the trial did not include patients with an ECOG PS of greater than 1. The clinical experts indicated that patients with good ECOG PS or a score of 0 to 2 should be eligible for talazoparib plus enzalutamide, if they are able to tolerate the therapy. The clinical experts noted that although enzalutamide was an appropriate comparator when the TALAPRO-2 trial was designed and executed, the current treatment paradigm has shifted since then toward the use of chemotherapy as the most common first-line treatment for patients with mCRPC. The evidence submitted to CDA-AMC did not include head-to-head comparisons between talazoparib plus enzalutamide and chemotherapy, which represents a gap in the available direct evidence given the potential shared place in therapy when used as first-line treatments for mCRPC.
Dose adjustments were allowed in the trial and the methods were outlined in the protocol. The clinical experts noted that dose adjustments or modifications are anticipated in a clinical practice setting to manage AEs while maintaining drug benefit.
The trial included outcomes that were important to patients. The patient group indicated that stopping disease progression, prolonging life, improving symptoms and HRQoL, and reducing treatment side effects are important to them.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered most relevant to inform expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group.74,75
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate — the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited — the true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate — the true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as “very uncertain.”
Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null.
The reference points for the certainty of evidence assessment for rPFS and OS were set according to the presence or absence of an important effect based on thresholds informed by the clinical experts consulted for this review. A threshold could not be determined for time to deterioration of pain by BPI-SF; therefore, the target of certainty appraisal was any effect. The reference points for the certainty of the evidence assessment for EORTC QLQ-PR25 functional and symptom scale scores were set according to the presence or absence of an important effect based on a threshold informed by the literature.
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for talazoparib plus enzalutamide versus placebo plus enzalutamide.
Long-Term Extension Studies
No long-term extension studies were submitted by the sponsor.
Indirect Evidence
Contents within this section have been informed by materials submitted by the sponsor. The following have been summarized and validated by the review team.
Objectives for the Summary of Indirect Evidence
The aim of this section is to summarize and critically appraise 1 sponsor-submitted MAIC76 to inform the pharmacoeconomic model and fill gaps in the comparative evidence for other treatments of interest for adults with HRR gene-mutated mCRPC.
Description of Indirect Comparison
The study selection criteria and methods for the MAIC are summarized in Table 14. The protocol for the systematic literature review was a priori registered with PROSPERO (registration number CRD42021283512).
Table 14: Study Selection Criteria for Indirect Treatment Comparisons Submitted by the Sponsor
Characteristics | Indirect comparison |
|---|---|
Population | Males (aged ≥ 18 years old) with asymptomatic or mildly symptomatic mCRPC |
Intervention | Talazoparib (orally, 0.5 mg per day) + enzalutamide (orally, 160 mg per day) |
Comparator | Relevant comparators:
|
Outcome |
|
Study designs | Phase II and phase III randomized controlled trials |
Publication characteristics | Comparator studies were identified via published literature (published reports and conference abstracts). TALAPRO-2 study data (protocol, individual patient data, and Clinical Study Report) were provided by the sponsor. |
Exclusion criteria | Population:
Study and report types:
|
Databases searched | Published reports (no date restrictions):
Conference abstracts (2019 onward):
|
Selection process | Records were independently screened by 2 reviewers and any discrepancies were resolved by consensus; a third independent reviewer was used to resolve any differences. |
Data extraction process | Data were extracted using standardized data extraction templates created using Microsoft Excel. A single reviewer performed the data extraction and then a second reviewer verified the data accuracy; a third independent reviewer was used to resolve any differences. |
Quality assessment | Risk of bias assessments were performed for full-text reports using NICE Single Technology Appraisal Evidence Submission checklist. A single reviewer performed the assessments, and a second independent reviewer verified the content; a third independent reviewer was used to resolve any differences. |
CDA-AMC = Canada’s Drug Agency; CRPC = castration-resistant prostate cancer; ITC = indirect treatment comparison; mCRPC = metastatic castration-resistant prostate cancer; NICE = National Institute for Health and Care Excellence; ORR = objective response rate; OS = overall survival; PSA = prostate-specific antigen; rPFS = radiographic progression-free survival.
Sources: Matching-adjusted indirect comparison technical report.76 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
MAIC Design
Objectives
The objective of the MAIC was to assess the treatment effects of talazoparib plus enzalutamide versus relevant comparators of interest, which included enzalutamide monotherapy, AAP, niraparib-abiraterone plus prednisone, and olaparib plus AAP in adults with HRR-gene-mutated mCRPC.
███ ████ ███ █████████ ██ ████ ██ █ ██████ ███████ ███ ████████ █ █████ █████ ██ ███████████. The comparators previously listed were considered relevant to public payers in Canada by the sponsor and the comparisons are summarized in this section.
Study Selection Methods
A systematic literature review was initially conducted on September 9, 2021, and updated on October 3, 2022, and August 8, 2024. The review included RCTs assessing first-line treatments in adults with asymptomatic or mildly symptomatic mCRPC and reported on relevant outcomes (Table 13). Identified trials were assessed for eligibility against predefined inclusion and exclusion criteria and further narrowed to only include trials that reported relevant outcomes for patients with HRR gene-mutated alterations. Two reviewers independently screened relevant citations for inclusion, and discrepancies were resolved through consensus or by a third reviewer. Data extraction and quality assessment of included studies were performed by 1 reviewer and validated by a second reviewer, and discrepancies were resolved by a third reviewer. Risk of bias assessment of included studies was performed using the National Institute for Health and Care Excellence: Single Technology Appraisal Evidence Submission checklist for assessment of risk of bias in RCTs.
Search Results and Feasibility Assessment
The literature review identified 49 unique RCTs, of which 42 had published results that met the inclusion criteria for first-line treatments in adults with mCRPC. Three trials (TALAPRO-2, PROpel, and MAGNITUDE) reported data on patients receiving relevant first-line treatment for mCRPC who had HRR gene-mutated alterations and data on survival outcomes, and were included in the feasibility assessment. One trial (BRCAAway) that compared olaparib plus abiraterone acetate to olaparib alone and abiraterone acetate alone among patients with HRR gene-mutated mCRPC was excluded because it did not report on rPFS or OS. The TALAPRO-2 study compared talazoparib plus enzalutamide versus enzalutamide, the PROpel study compared olaparib plus AAP versus AAP, and the MAGNITUDE study compared niraparib-abiraterone plus prednisone versus AAP.
A feasibility assessment was undertaken to determine the extent of heterogeneity across the included 3 trials, and if indirect treatment comparisons would be appropriate. The following potential sources of heterogeneity were explored: study design, patient eligibility criteria, baseline patient characteristics, outcome characteristics, and efficacy or safety results. ███ ███████ ██ ███ ██████████ ██████████ ████ █████████████ ██████ ███ ███████ ███ ███ ███████ ████████ ████ ██ ████████████ ███████ ██ ███████ ████ ███████ ███ █████████ ███ ████ ██ ██████ ███ █████████ ██████ ███ ████ ███ ██ ██ ███ ███ ████████████ ███████████ There was no common treatment comparator between the TALAPRO-2, PROpel, and MAGNITUDE studies; therefore, unanchored MAICs were chosen using individual patient data from the TALAPRO-2 study and summary-level data from the comparator trials. The authors noted that a network meta-analysis was considered but determined unfeasible due to the lack of a common comparator between the TALAPRO-2, PROpel, and MAGNITUDE trials.
MAIC Analysis Methods
Unanchored MAICs were performed by matching and adjusting patients from the TALAPRO-2 trial to match the marginal distribution (e.g., mean and variance) of covariates in patients who received the comparator treatment. Key factors for adjustment were identified and ranked in order of importance based on published analyses77 on prognostic strength in mCRPC and refined based on clinical input from a practising clinician experienced in treating mCRPC consulted by the sponsor. Presence of BRCA1, BRCA2, or PALB2 HRR gene alterations was also considered for adjustment based on external clinical input as an exploratory analysis. The final list of 12 factors, ranked from 1 (most important) to 12 (least important) is in Table 15.
After completing the matching phase of the MAIC, patients remaining from the TALAPRO-2 study were weighted using logistic regression and method of moments. Trial stratification factors at randomization by HRR alternation status and prior NHT or taxane-based therapy in the CSPC setting, which were reported in all 3 trials, were adjusted where necessary. Following the population adjustment of the TALAPRO-2 study’s population with HRR deficiencies to the comparator trials, rPFS and OS estimates of the comparative efficacy of talazoparib plus enzalutamide versus comparator treatments were derived by digitizing published Kaplan-Meier curves and applying the Guyot method. A dataset combining weighted individual patient data and pseudoindividual patient data were then used to fit a weighted Cox proportional hazards model with a binary treatment indicator. The estimated regression coefficient for the treatment indicator was used to represent the log HR for talazoparib plus enzalutamide versus the comparator treatment, and the corresponding variance was estimated using a robust sandwich estimator. Effect estimates were exponentiated and reported as HRs with 95% CIs for the population with HRR gene mutations. The effective sample size was calculated to reflect the sample size of the weighted population, and population differences between the TALAPRO-2 study and the comparator trials were assessed using standardized mean differences; a standardized mean difference between 0 and 0.1 was considered a small difference, a standardized mean difference of greater than 0.1 and less than or equal to 0.2 was a moderate difference, and a standardized mean difference of greater than 0.2 was a substantial difference. Scenario analyses were conducted to investigate the impact on the treatment effect estimates, effective sample size, and standardized mean difference when adjusting for additional covariates in the analyses. These analyses were conducted sequentially, adjusting incrementally for the remaining factors in order of importance, until the final model contained all available factors.
Table 15: Factors for Adjustment in the MAICs
Rank | Identified factor | Available for TALAPRO-2 trial | Adjusted in PROpel trial | Adjusted in MAGNITUDE trial |
|---|---|---|---|---|
1 | Time to mCRPC from continuous ADT (ADT alone or in combination) | No; NR | No; NR | No; NR |
2 | Presence of liver metastases | Yes | Yes; adjusted using visceral metastases (e.g., lung or liver) | Yes |
3 | Number of bone metastases (< 10 vs. > 10) | Yes | Yes; adjusted using bone-only metastases | Yes; adjusted using presence or absence of bone metastases |
4 | ECOG PS (0 to 1 vs. 2, 3) | Yes; all patients had ECOG PS of 0 or 1 | Yes; all patients had ECOG PS of 0 or 1 | Yes; all patients had ECOG PS of 0 or 1 |
5 | BPI-SF | Yes | No | No |
6 | PSA kinetics or PSA levels in absence of kinetics data | Yes | Yes | Yes |
7 | Gleason score | Yes | Yes | Yes |
8 | Hemoglobin level | Yes | No; NR | Yes |
9 | Lactate dehydrogenase level | Yes | No; NR | Yes |
10 | Albumin level | Yes | No; NR | No; NR |
11 | Alkaline phosphatase level | Yes | No; NR | Yes |
12 | Neutrophil to lymphocyte ratio | Yes | No; NR | No; NR |
Exploratory | BRCA1 | Yes | Yes | Yes |
Exploratory | BRCA2 | Yes | Yes | Yes |
ADT = androgen deprivation therapy; BPI-SF = Brief Pain Index–Short Form; ECOG PS = Eastern Cooperative Oncology Group Performance Status; MAIC = matching-adjusted indirect comparison; mCRPC = metastatic castration-resistant prostate cancer; NR = not reported in the trial; PSA = prostate-specific antigen; vs. = versus.
Sources: MAIC technical report.76 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Results of Indirect Treatment Comparison
Summary of Included Studies
Trial characteristics such as phase, study design, blinding, setting, and number of patients randomized, and outcome reporting for rPFS and OS were generally similar between the TALAPRO-2, PROpel, and MAGNITUDE trials. In the assessment of patient eligibility criteria, the included trials generally recruited adult patients with confirmed adenocarcinoma of the prostate, who had asymptomatic or mildly symptomatic disease, were surgically or medically castrated, had metastatic disease, and did not receive prior cytotoxic chemotherapy. Substantial heterogeneity was noted for the MAGNITUDE study, which had broader eligibility criteria than the TALAPRO-2 study by permitting the prior use of AAP in the mCRPC state. Although only 4 months or less of AAP was allowed, approximately 25% of the patients were not truly naive, whereas all patients were treatment naive in the mCRPC state in the TALAPRO-2 trial. Therefore, matching was not possible for this criterion. The PROpel trial was broader than the TALAPRO-2 study for the pain criteria measured by BPI-SF. The PROpel study had no eligibility restrictions based on BPI-SF, whereas the TALAPRO-2 study required patients to have a score of less than or equal to 3 on question 3 of the BPI-SF (worst pain in the last 24 hours). In the PROpel study, 25.8% of patients in the olaparib plus AAP group were considered symptomatic (defined as those with a BPI-SF score ≥ 4 and/or opiate use). Therefore, matching was not possible on this criterion.
The authors noted that it was not possible to account for all differences between the trial populations. Analyses between the TALAPRO-2 study and comparator trials were limited by reporting of baseline characteristics of interest in the comparator trials. Additionally, the ability to account for intertrial differences was further limited by those characteristics for which eligibility criteria were broader in the aggregate comparator trials than in the TALAPRO-2 study. Both comparator trials included patients with the HRR gene mutations BRCA1 and BRCA2.
Results
The unadjusted and adjusted baseline characteristics for the population with HRR deficiencies for the TALAPRO-2 study versus the PROpel study and the TALAPRO-2 study versus the MAGNITUDE study are summarized in Figure 7 and Figure 8, respectively. The primary analysis for the PROpel and MAGNITUDE studies adjusted MAICs for 5 factors and 8 factors, respectively. ███ ████ ██████ ███ ██████████ ███████████ ███████ █████████ ███ ███ ██████████ ██████ ██████ ████ ██ ████ ████ ████ ██████████ █ █████ ██████████. For the TALAPRO-2 trial, the rPFS and OS data are based on the data cut-off date of September 3, 2024. A summary of the primary rPFS and OS results in the population with HRR deficiencies is in Table 16.

Note: Figure 7 contained confidential information and was removed at the request of the sponsor.

Note: Figure 8 contained confidential information and was removed at the request of the sponsor.
Radiographic Progression-Free Survival
Talazoparib Plus Enzalutamide Versus Olaparib Plus AAP (PROpel Trial)
In the primary analysis, the result ████████ ████████████████████████ ██████ ████████ ████ ████ ███ ███████ ████ ██████████ ███████ ███ ███████ ████████ ███ ███ ████████ ████████ ██████ ████████ █████ ██████ ███ ████ ███ ██████ ███████████████ ███ ███████████ ████████ █████████ ███ ████ ████ ████████████
Talazoparib Plus Enzalutamide Versus Niraparib-Abiraterone Plus Prednisone (MAGNITUDE Trial)
In the primary analysis, the result ████████ ████████████████████████ ██████ █████████ ████ ████ ███ ███████ ████ ██████████ ███████ ███ ███████ ████████ ███ ███ ████████ ████████ ██████ ████████ █████ ██████ ███ ████ ███ ██████ ███████████████ ███ ███████████ ████████ █████████ ███ ████ ████ ████████████
Talazoparib Plus Enzalutamide Versus AAP
In the primary analysis for each comparison between the TALAPRO-2 study versus the PROpel and MAGNITUDE studies, the results ████████ ████████████████████████ ██████ ████ ███ ███████ ████ ██████████ ███████ ███ ███████ ████████ ███ ███ ████████ ████████ ██████ ████████ █████ ██████ ███ ████ ███ ██████ ███████████████ ███ ███████████ ████████ █████████ ███ ████ ████ ████████████
Overall Survival
Talazoparib Plus Enzalutamide Versus Olaparib Plus AAP
In the primary analysis, the point estimate for the between-group HR ████████ ████████████████████████ ██████ ████████ ████ ███ ███ ███ ███ ██ ████████ ███ ███████████ ██ ██ ██████████ ██ ████ ███ ██████████ ███ ████████ ██████ ███████ ███ ██████ ███ ███████ ████ ██████████ ███████ ███ ███████ ████████ ███ ███ ████████ ████████ ██████ ████████ █████ ██████ ███ ████ ███ ██████ ███████████████ ███ ███████████ ████████ █████████ ███ ████ ████ ████████████
Talazoparib Plus Enzalutamide Versus Niraparib-Abiraterone Plus Prednisone
In the primary analysis (adjusted for 8 factors), the result ████████ ████████████████████████ ██████ █████████ ████ ████ ███ ███████ ████ ██████████ ███████ ███ ███████ ████████ ███ ███ ████████ ████████ ██████ ████████ █████ ██████ ███ ████ ███ ██████ ███████████████ ███ ███████████ ████████ █████████ ███ ████ ████ ████████████
Talazoparib Plus Enzalutamide Versus AAP
In the primary analysis for each comparison between the TALAPRO-2 study versus the PROpel and MAGNITUDE studies, the ███████ ████████ ████████████████████████ ██████ ████ ███ ███████ ████ ██████████ ███████ ███ ███████ ████████ ███ ███ ████████ ████████ ██████ ████████ █████ ██████ ███ ████ ███ ██████ ███████████████ ███ ███████████ ████████ █████████ ███ ████ ████ ███████████.
Table 16: Summary of MAIC Results for rPFS and OS — Population With HRR Gene Mutations
Outcome | Talazoparib + enzalutamide vs. olaparib + AAP | Talazoparib + enzalutamide vs. niraparib-abiraterone plus prednisone | Talazoparib + enzalutamide vs. AAP | |||||
|---|---|---|---|---|---|---|---|---|
TALAPRO-2 trial (cohort 2) vs. PROpel trial HR (95% CI) | TALAPRO-2 trial (cohort 2) vs. MAGNITUDE trial HR (95% CI) | TALAPRO-2 trial (cohort 2) vs. PROpel trial HR (95% CI) | TALAPRO-2 (cohort 2) trial vs. MAGNITUDE trial HR (95% CI) | |||||
rPFS (BICR) | ████ █████ ██ █████ | ████ █████ ██ █████ | ████ █████ ██ █████ | ████ █████ ██ █████ | ||||
OS | ████ █████ ██ █████ | ████ █████ ██ █████ | ████ █████ ██ █████ | ████ █████ ██ █████ | ||||
AAP = abiraterone acetate plus prednisone; BICR = blinded independent central review; CI = confidence interval; HR = hazard ratio; HRR = homologous recombination repair; MAIC = matching-adjusted indirect comparison; OS = overall survival; rPFS = radiographic progression-free survival; vs. = versus.
Note: An HR below 1.0 indicates an improved outcome for talazoparib plus enzalutamide relative to olaparib plus AAP, niraparib-abiraterone plus prednisone, or AAP.
Sources: MAIC technical report.76 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Critical Appraisal of MAIC
The methods used to conduct the systematic literature review for the MAIC were a priori registered, and used appropriate criteria to search databases, select studies, extract data, and assess risk of bias of the included studies. The MAIC did not include comparisons between talazoparib plus enzalutamide and chemotherapy, which represents a gap in the available indirect evidence given the shared place in therapy for mCRPC. The MAIC included relevant outcomes identified by the CDA-AMC team (rPFS and OS); however, clinically relevant and patient-relevant outcomes such as pain, HRQoL, and harms were not included in the comparisons. Due to a lack of a common comparator group between trials, unanchored MAICs were conducted, and attempts were made to adjust for important prognostic factors and treatment effect modifiers (i.e., clinical factors).
To account for between-study differences in patient baseline characteristics, several potentially relevant clinical factors were matched in the weighting process for each comparison between the TALAPRO-2 study and comparator trials. The methods used to identify and rank the clinical factors were considered appropriate. The process involved identifying and ranking clinical factors in order of importance based on published analyses for mCRPC and refined based on clinical input from a practising clinician experienced in treating patients with mCRPC consulted by the sponsor. For both the PROpel and MAGNITUDE trials, adjustments were limited by how, and if, these variables were reported in both trials. The TALAPRO-2 study stratified by prior NHT or taxane therapy per interactive web response system as a single variable whereas the PROpel and MAGNITUDE studies reported these variables separately. The PROpel study used the interactive web response system value, whereas the MAGNITUDE study did not specify interactive web response system or electronic data capture. Because interactive web response system values were combined in the TALAPRO-2 study but electronic data capture values were presented separately, electronic data capture values were used to align with reporting of comparator trials. Further, the TALAPRO-2 study stratified by prior therapy in the CSPC stage. The PROpel study reported the proportion of patients who received NHT and taxane-based therapy but did not specify the disease stage. The MAGNITUDE study reported the proportion of patients who received NHT and taxane-based therapy in the metastatic CSPC or nonmetastatic CRPC stage into a single variable. There were important differences in eligibility criteria between the MAGNITUDE and TALAPRO-2 studies. The MAGNITUDE study allowed the use of AAP in the mCRPC setting for 4 months or less whereas all patients were treatment naive in the mCRPC setting within the TALAPRO-2 trial. Because only summary-level data were available for the MAGNITUDE study, patients who were not truly treatment naive could not be removed when performing analyses. It is possible that these patients had different disease characteristics compared to the rest of the trial population. Overall, the magnitude and direction of potential bias due to imbalances for the rPFS and OS estimates cannot be predicted. The proportional hazards assumption was also not assessed or discussed. Because the unanchored nature of the MAIC requires a stronger assumption (than an anchored MAIC) that all effect modifiers and prognostic factors have been included in the analysis, which was not possible, definitive conclusions based on these results are not recommended. Further, the sponsor’s highest-ranked (most important) factor for adjustment in the MAIC, time to mCRPC from continuous ADT, could not be adjusted for in the analysis.
Additionally, following the weighting process after adjusting for 8 clinical factors, the effective sample size of the talazoparib cohort for the primary analysis declined by more than 50% of the original sample size for the comparison with niraparib-abiraterone plus prednisone. A significant reduction in sample size can contribute to imprecision, increasing uncertainty of the results. A notable reduction in effective sample size also suggests that results may be heavily influenced by a subset of the sample in the trials that may not be representative of the full sample. Given that the population of these analyses were weighted to the TALARPO-2 study, the results may be driven by a subset of patients in the talazoparib group, limiting generalizability to the full population presented by the TALAPRO-2 study. Due to the limitations in the MAICs, the effects of talazoparib plus enzalutamide on PFS and OS compared with comparators in the MAICs are uncertain.
Studies Addressing Gaps in the Systematic Review Evidence
No additional studies were submitted by the sponsor.
Discussion
Summary of Available Evidence
One pivotal phase III, randomized, double-blind trial and 1 indirect treatment comparison submitted by the sponsor were summarized in this report.
The TALAPRO-2 trial (cohort 2, N = 399) met the inclusion criteria for the systematic review conducted by the sponsor. An objective of the trial was to assess the efficacy and safety of talazoparib 0.5 mg plus enzalutamide 160 mg, taken orally once daily, or matched placebo plus enzalutamide in adult patients with HRR-deficient mCRPC. The trial enrolled adults with asymptomatic or mildly symptomatic mCRPC who had not started systemic cancer treatment after the diagnosis of CRPC (metastatic or nonmetastatic), with the exception of ADT and first-generation antiandrogen drugs. Patients were allowed to have previously received abiraterone acetate or docetaxel for CSPC but were ineligible to participate if they had received any prior treatment with second-generation androgen receptor inhibitors (enzalutamide, apalutamide, and darolutamide), a PARP inhibitor, cyclophosphamide, or mitoxantrone for prostate cancer. Patients were required to have had an ECOG PS score of 0 or 1, and progressive disease at study entry. The approved Health Canada indication and reimbursement request aligned with the trial’s population with HRR deficiencies. The outcomes relevant to this review included the primary outcome of rPFS, the key secondary outcome of OS and HRQoL outcomes of time to deterioration of pain symptoms measured via BPI-SF and EORTC QLQ-PR25 functional and symptom scales, and safety. The rPFS data are based on the primary analysis data cut-off date of October 3, 2022, and supportive data from the data cut-off date of September 3, 2024. All other outcomes were based on the data cut-off date of September 3, 2024. Overall, key baseline characteristics were generally balanced between treatment groups. The trial population was predominately white (68%), with a median age of 70 years. Most patients had an ECOG PS score of 0 (approximately 62%), indicating good overall performance; normal or mild renal impairment (approximately 90%); bone and soft tissue disease-site metastasis (approximately 84%); a Gleason score greater than or equal to 8 (approximately 74%); and had prior therapy with surgery or biopsy (█████████████ ████ and first-generation antiandrogens (█████████████ ███). In both groups, the most commonly detected HRR gene alteration was BRCA2 (33.8%), followed by ATM (21.6%), CDK12 (18.8%), and CHEK2 (17.8%). ███ ████████████████████████ █████ ███ █ ██████ ████ ████████ █████ ███ █████ ██████ █████ ███ █ ██████ ██████████ ██ ████████ ███ ███ ████████ ██████ █████████ ██████ ██████ ██████. These imbalances were likely due to chance, as ███ █████ ████████ ███████████████ ██ ████████ ████████ ████████ ███████ ██████.
To inform the pharmacoeconomic model and fill gaps in the comparative evidence for other treatments of interest for adults with HRR gene-mutated mCRPC, a MAIC was conducted by the sponsor. The objective of the MAIC was to assess the treatment effects of talazoparib plus enzalutamide versus relevant comparators, which included enzalutamide monotherapy, AAP, niraparib-abiraterone plus prednisone, and olaparib plus AAP in adults with HRR gene-mutated mCRPC. Outcomes of interest included rPFS and OS. Key clinical factors for adjustment were identified and ranked in order of importance based on published analyses on prognostic strength in mCRPC and refined based on clinical input received by the sponsor from a practising clinician experienced in treating mCRPC. Individual patient data from the TALAPRO-2 trial population with HRR deficiencies were adjusted to match the marginal distribution (e.g., mean, variance) of clinical factors of patients for each comparison between the PROpel (olaparib plus AAP versus AAP) and MAGNITUDE (niraparib-abiraterone plus prednisone versus AAP) trials. Because there was no common treatment comparator between the TALAPRO-2 trial and the comparator trials, unanchored MAICs were conducted. Point estimates for rPFS and OS were reported as HRs with 95% CIs.
Interpretation of Results
The evidence from the pivotal trial, the TALAPRO-2 study, addressed treatment outcomes noted to be important by both patients and clinicians. The patient group input indicated that stopping disease progression, prolonging life, improving symptoms and HRQoL, and reducing treatment side effects are important to them. Similarly, the clinical experts consulted by the review team indicated that because mCRPC is a terminal phase of prostate cancer, the unmet needs of patients would be new treatments that would prolong survival and improve quality of life, while exposing patients to minimal toxicity. Both clinical experts highlighted that the balance between treatment efficacy and quality of life would be important.
Efficacy
The TALAPRO-2 trial supported a clinically meaningful improvement of talazoparib plus enzalutamide over placebo plus enzalutamide for rPFS and OS in adults with HRR gene-mutated mCRPC. At the data cut-off date of October 3, 2022, the trial met its primary outcome of rPFS, with a median duration of follow-up of 17.5 months for the talazoparib plus enzalutamide group and 16.8 months for the placebo plus enzalutamide group. The median rPFS was not reached (95% CI, 21.9 months to not reached) in the talazoparib plus enzalutamide group and was 13.8 months (95% CI, 11.0 months to 16.7 months) for the placebo plus enzalutamide group. At the data cut-off date of September 3, 2024, the median rPFS was 30.7 months (95% CI, 24.3 months to 38.5 months) in the talazoparib plus enzalutamide group and 12.3 months (95% CI, 11.0 months to 16.5 months) in the placebo plus enzalutamide group. The between-group difference in probability of rPFS at 12 months (data cut-off date of October 3, 2022) and 48 months (data cut-off date of September 3, 2024) was █████ ████ ███ ████ ██ █████ ███ █████ ████ ███ ████ ██ ██████ ████████████. For the GRADE assessment, the clinical experts consulted by CDA-AMC suggested a clinically important threshold of 20% and 5% for between-group absolute risk difference at 12 months and 48 months, respectively. Based on these thresholds, there was moderate certainty of evidence for a clinically important increase in the probability of rPFS at 12 months and 48 months, respectively. The rPFS findings were generally consistent across the subgroup analyses by BRCA alteration status (with or without) and prior treatment with NHT or taxane therapy in favour of talazoparib plus enzalutamide. The HR for the BRCA alterations subgroup was larger than the subgroup without BRCA alteration, in favour of talazoparib plus enzalutamide. The sample size for the prespecified subgroup analyses of interest was small and was not powered to detect subgroup differences. At the data cut-off date of September 3, 2024, the median duration of follow-up for OS was 44.2 months in the talazoparib plus enzalutamide group and ████ ██████ for the placebo plus enzalutamide group. The median OS was 45.1 months (95% CI, 35.4 months to not reached) for the talazoparib plus enzalutamide group and 31.1 months (95% CI, 27.3 months to 35.4 months) for the placebo plus enzalutamide group. The between-group difference in probability of OS at 12 months and 48 months was ████ ████ ███ ████ ██ ████ ███ █████ ████ ███ ████ ██ ██████ ████████████. For the OS GRADE assessment, the clinical experts suggested a clinically important threshold of 10% and 5% for between-group absolute risk difference at 12 months and 48 months, respectively. Based on these thresholds, there was high certainty of evidence for little to no clinically important difference in the probability of OS at 12 months, and clinically important increase in the probability of OS at 48 months. Because patients were permitted to receive posttreatment anticancer therapy after study treatment had been discontinued (36.9% in the talazoparib plus enzalutamide group versus 56.8% in the placebo plus enzalutamide group), the potential treatment benefit on OS would have been subject to a degree of uncertainty.
At the data cut-off date of September 3, 2024, the median time to deterioration of pain symptoms was not reached in either group. For the GRADE assessment, clinically important thresholds at 12 months and 48 months could not be determined; therefore, the target of certainty was any effect. There was low certainty of evidence for a higher probability of being free of pain progression at 12 months and 48 months, respectively, in favour of talazoparib plus enzalutamide. The low certainty of evidence at 12 months was attributed to very serious imprecision due to the 95% CI for the between-group difference including both the possibility of benefit and harm, and at 48 months was attributed to risk of bias due to missing outcome data. For the EORTC QLQ-PR25 functional and symptom scale scores at week 109, there was low certainty of evidence in little to no clinically important difference in functional or symptom scores, based on an MID estimate of 5 points, or the evidence was very uncertain about the effect compared to placebo plus enzalutamide. The low certainty of evidence was attributed to risk of bias due to missing outcome data. The very low certainty of evidence was attributed to serious imprecision due to the 95% CI for the between-group difference including the possibility of important harm, little to no difference or important benefit, and risk of bias due to missing outcome data. No data were available for the sexual functioning score.
The CDA-AMC reviewers and the clinical experts noted some potential gaps and implementation challenges in applying the evidence from the trial. The CDA-AMC reviewers noted that because the trial and sponsor-submitted MAIC evaluated talazoparib plus enzalutamide as first-line treatment only (i.e., patients had not received prior systemic therapy for mCRPC) and the approved Health Canada indication is line agnostic, there is no direct or indirect comparative evidence for the use of talazoparib plus enzalutamide in later-line settings.
The clinical experts noted that enzalutamide was an appropriate comparator when the TALAPRO-2 trial was designed and executed; however, the current treatment paradigm has shifted since then toward the use of chemotherapy as the most common first-line treatment for patients with HRR-deficient mCRPC. The evidence submitted to CDA-AMC did not include head-to-head comparisons between talazoparib plus enzalutamide and chemotherapy, which represents a gap in the available direct evidence given the potential shared place in therapy for mCRPC when used as first-line treatments. As such, the clinical experts indicated that most clinicians would likely prescribe talazoparib plus enzalutamide as a second-line and beyond treatment due to the decreasing number of patients who are ARPI naive in the first-line mCRPC setting.
Based on the sponsor-submitted unanchored MAICs, the rPFS HR point estimates and 95% CIs ████████ ████████████████████████ ██████ ████████ ████ ████ █████████ ████ ████ ███ ███ ██████ ███ ███ ███ ███████ ████████ ████████████████████████ ██████ █████████ ████ ███ ███ ███ ██████ ███ ██ █████ ████████ ████████ ████████████████████████ ██████ ████████ ████ ███ ███ ███ ███ ██ ████████ ███ ███████████ ██ ██ ██████████ ██ ████ ███ ██████████ ███ ████████ ██████ ███████ ███ ██████ ███ ████████████ ███████ ██████████ ███ ███ ████████ ██ █████████. The rPFS and OS primary analyses for the MAICs in the PROpel and MAGNITUDE studies adjusted for 5 and 8 treatment effect modifiers and prognostic factors, respectively. However, not all factors could be matched or adjusted, including the factor rated of highest importance; therefore, imbalances remained. There were also differences in trial eligibility criteria that could not be controlled for by matching. Because the unanchored nature of the MAIC requires a stronger assumption (than an anchored MAIC) that all effect modifiers and prognostic factors have been included in the analysis, which was not possible, the effects of talazoparib plus enzalutamide on rPFS and OS compared with the comparator drugs in the MAICs are uncertain and conclusions based on these results are not recommended.
Harms
At the data cut-off date of September 3, 2024, almost all patients in both treatment groups reported at least 1 TEAE. The most frequently reported TEAEs in the talazoparib plus enzalutamide group were anemia, fatigue, and decreased neutrophil count, and occurred more frequently than in the placebo plus enzalutamide group. A higher proportion of patients in the talazoparib plus enzalutamide group also experienced at least 1 grade 3 or grade 4 TEAE. The incidence of serious TEAEs was higher in the talazoparib plus enzalutamide group, with ██████ ███ ███████ █████ ██████████ being the most frequently reported. A higher proportion of TEAEs that led to study treatment discontinuation was reported in the talazoparib plus enzalutamide group, with anemia being the most common reason. The incidence of death was lower in the talazoparib plus enzalutamide group, with ███████ ███████████ being the primary reason for death in both groups. The incidence of notable harms, which included embolic and thrombotic events and pneumonitis, was comparable and infrequent in both treatment groups. The clinical experts indicated that the incidence of these TEAEs is expected with talazoparib plus enzalutamide, and that with appropriate care, the TEAEs would be manageable for many patients. The patient group input for this review highlighted a need for alternative tolerable treatments. However, it is uncertain whether talazoparib plus enzalutamide is a more tolerable treatment compared to placebo plus enzalutamide because a higher incidence of TEAEs, particularly anemia, and serious TEAEs were reported with this treatment regimen.
The sponsor-submitted MAIC did not include harms; therefore, no conclusions could be drawn on the relative safety of talazoparib plus enzalutamide versus other relevant comparators.
Conclusion
Evidence from 1 phase III, randomized, double-blind trial (TALAPRO-2) reported on outcomes that were important to both patients and clinicians. The trial showed moderate certainty of evidence that treatment with talazoparib plus enzalutamide results in a clinically important increase in rPFS at 12 months and 48 months, respectively, compared to placebo plus enzalutamide in adults with HRR-deficient mCRPC in the first-line setting. The trial showed high certainty of evidence of a clinically important increase in OS at 48 months in favour of talazoparib plus enzalutamide. There was low certainty of evidence for a higher probability of being free of pain progression at 12 months and 48 months, respectively, in favour of talazoparib plus enzalutamide. Up to 109 weeks, no definitive conclusions can be drawn on other HRQoL outcomes due to concerns of imprecision and missing outcome data. There were no new safety signals identified, and the safety of talazoparib plus enzalutamide was consistent with the known safety profiles of the individual drugs, although the trial showed a higher proportion of TEAEs and serious TEAEs when compared with placebo plus enzalutamide. Due to limitations of the indirect treatment comparison, mostly attributed to the heterogeneity across studies and lack of safety assessment, no conclusions can be drawn on the relative efficacy and safety of talazoparib plus enzalutamide compared to relevant comparators, which included olaparib plus AAP, niraparib-abiraterone plus prednisone, and AAP alone. The evidence submitted to CDA-AMC did not include direct or indirect comparisons between talazoparib plus enzalutamide and chemotherapy, which represents a gap in the available evidence given the potential shared place in therapy for HRR-deficient mCRPC when used as first-line treatments. The TALAPRO-2 trial and submitted indirect treatment comparison evaluated talazoparib plus enzalutamide as a first-line treatment only; therefore, the efficacy and safety of talazoparib plus enzalutamide as a second-line or later-line treatment for HRR-deficient mCRPC represents a gap in the evidence.
References
1.Pfizer Inc. P. Clinical Study Report: TALAPRO-2 (C3441021): A phase 3, randomized, double-blind, placebo-controlled study of talazoparib with enzalutamide in metastatic castration-resistant prostate cancer; Cohort 2 HRR-deficient [internal sponsor's report]. 2024.
2.Scher HI, Morris MJ, Stadler WM, et al. Trial Design and Objectives for Castration-Resistant Prostate Cancer: Updated Recommendations From the Prostate Cancer Clinical Trials Working Group 3. J Clin Oncol. 2016;34(12):1402-1418. PubMed
3.Kolinsky MP, Niederhoffer KY, Kwan EM, et al. Considerations on the identification and management of metastatic prostate cancer patients with DNA repair gene alterations in the Canadian context. Can Urol Assoc J. 2022;16(4):132-143. PubMed
4.Canadian Cancer S. Treatments for castration-resistant prostate cancer. 2021; https://cancer.ca/en/cancer-information/cancer-types/prostate/treatment/castration-resistant-prostate-cancer. Accessed 29 September, 2023.
5.Malone S, Shayegan B, Basappa NS, et al. Management algorithms for metastatic prostate cancer. Canadian Urological Association journal = Journal de l'Association des urologues du Canada. 2019;14(2):50-60. PubMed
6.So AI, Chi KN, Danielson B, et al. Update: Canadian Urological Association-Canadian Urologic Oncology Group guideline on metastatic castration-naive and castration-sensitive prostate cancer [Draft]. Accessed at: https://www.cua.org/system/files/Guideline-Files/8148_mCNPC_mCSPC%20guideline%20update.pdf. Can Urol Assoc J. 2022.
7.Dawson NA, MD, Leger, Paul, MD. Overview of the treatment of castration-resistant prostate cancer (CRPC). In: TW P, ed. UpToDate. Waltham (MA): UpToDate; 2023: https://www.uptodate.com/contents/overview-of-the-treatment-of-castration-resistant-prostate-cancer-crpc. Accessed Oct 12, 2023.
8.Rebello RJ, Oing C, Knudsen KE, et al. Prostate cancer. Nature Reviews Disease Primers. 2021;7(1). PubMed
9.Fizazi K, Jenkins C, Tannock IF. Should docetaxel be standard of care for patients with metastatic hormone-sensitive prostate cancer? Pro and contra. Ann Oncol. 2015;26(8):1660-1667. PubMed
10.Jenkins V, Solis-Trapala I, Payne H, et al. Treatment Experiences, Information Needs, Pain and Quality of Life in Men with Metastatic Castrate-resistant Prostate Cancer: Results from the EXTREQOL Study. Clinical Oncology. 2019;31(2):99-107. PubMed
11.Saad F, Ivanescu C, Phung D, et al. Skeletal-related events significantly impact health-related quality of life in metastatic castration-resistant prostate cancer: data from PREVAIL and AFFIRM trials. Prostate Cancer Prostatic Dis. 2017;20(1):110-116. PubMed
12.Canadian Cancer S. Survival statistics for prostate cancer. https://www.cancer.ca/en/cancer-information/cancer-type/prostate/prognosis-and-survival/survival-statistics/?region=on.
13.Brenner DR, Poirier A, Woods RR, et al. Projected estimates of cancer in Canada in 2022. Canadian Medical Association Journal. 2022;194(17):E601-E607. PubMed
14.Shore N, Oliver L, Shui I, et al. Systematic literature review of the epidemiology of advanced prostate cancer and associated homologous recombination repair gene alterations. J Urol. 2021;205(4):977-986. PubMed
15.Sheen D. Towards the Long-Term Protection for Prostate Cancer Undergraduate Journal of Experimental Microbiology and Immunology (UJEMI) 2020;4:1-10 (September).
16.Pfizer C. Product Monograph: TALZENNA (talazoparib). 2022.
17.Astellas Pharma Canada I. XTANDI® (enzalutamide) Product Monograph. January 14 2019.
18.Janssen. Product Monograph: ZYTIGA (abiraterone acetate). 2018.
19.Sanofi. Product Monograph: TAXOTERE (docetaxel). 2020.
20.AstraZeneca. Product Monograph: LYNPARZA (olaparib). 2024.
21.Janssen. Product Monograph: AKEEGA (niraparib/abiraterone acetate). 2023.
22.Astellas. Product Monograph: XTANDI (enzalutamide). 2019.
23.Scott RJ, Mehta A, Macedo GS, Borisov PS, Kanesvaran R, El Metnawy W. Genetic testing for homologous recombination repair (HRR) in metastatic castration-resistant prostate cancer (mCRPC): challenges and solutions. Oncotarget. 2021;12(16):1600-1614. PubMed
24.Shui IM, Burcu M, Shao C, et al. Real-world prevalence of homologous recombination repair mutations in advanced prostate cancer: an analysis of two clinico-genomic databases. Prostate Cancer Prostatic Dis. 2024;27(4):728-735. PubMed
25.Bilen MA, Khilfeh I, Rossi C, et al. Homologous Recombination Repair Testing Patterns and Outcomes in mCRPC by Alteration Status and Race. Clinicoecon Outcomes Res. 2024;16:657-674. PubMed
26.Fan Y, Liu Z, Chen Y, He Z. Homologous Recombination Repair Gene Mutations in Prostate Cancer: Prevalence and Clinical Value. Adv Ther. 2024;41(6):2196-2216. PubMed
27.Olmos D, Lorente D, Alameda D, et al. Treatment patterns and outcomes in metastatic castration-resistant prostate cancer patients with and without somatic or germline alterations in homologous recombination repair genes. Ann Oncol. 2024;35(5):458-472. PubMed
28.Kwon WA. PARP Inhibitors in the Treatment of Prostate Cancer: From Scientific Rationale to Clinical Development. World J Mens Health. 2024;42(2):290-303. PubMed
29.Serritella AV, Taylor A, Haffner MC, et al. Therapeutic implications of homologous repair deficiency testing in patients with prostate cancer (Part 2 of 2). Prostate Cancer Prostatic Dis. 2024. PubMed
30.Shore N, Ionescu-Ittu R, Yang L, et al. Real-world genetic testing patterns in metastatic castration-resistant prostate cancer. Future Oncol. 2021;17(22):2907-2921. PubMed
31.Agarwal N, Azad AA, Carles J, et al. Talazoparib plus enzalutamide in men with first-line metastatic castration-resistant prostate cancer (TALAPRO-2): a randomised, placebo-controlled, phase 3 trial. The Lancet. 2023;402(10398):291-303. PubMed
32.Pfizer. Clinical Evidence [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: talazoparib, 0.1 mg and 0.25 mg, oral capsules. 2025.
33.Lang S, Swift S, White H, Misso K, Kleijnen J, Quek R. A systematic review of the prevalence of DNA damage response gene mutations in prostate cancer. International Journal of Oncology. 2019. PubMed
34.Xia M, Guo Z, Hu Z. The Role of PARP Inhibitors in the Treatment of Prostate Cancer: Recent Advances in Clinical Trials. Biomolecules. 2021;11(5). PubMed
35.BC Cancer. Mainstreamed Hereditary Cancer Testing. https://cancergeneticslab.ca/hereditary/mainstreamed-testing/. Accessed May 01, 2025.
36.Rendon RA, Selvarajah S, Wyatt AW, et al. 2023 Canadian Urological Association guideline: Genetic testing in prostate cancer. Can Urol Assoc J. 2023;17(10):314-325. PubMed
37.Cancer Care Ontario. Hereditary Cancer Testing Eligibility Criteria: Version 3.1. 2024: https://www.cancercareontario.ca/en/guidelines-advice/types-of-cancer/70161. Accessed May 01, 2025.
38.Pfizer. Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: talazoparib, 0.1 mg and 0.25 mg, oral capsules. 2025.
39.Cancer Care Ontario. Comprehensive Cancer Biomarker Testing Program - As of April 1, 2025. 2025: https://www.cancercareontario.ca/en/guidelines-advice/treatment-modality/pathology-laboratory-testing/genetic-testing-resources/comprehensive-cancer-biomarker-testing-program. Accessed May 01, 2025.
40.Alberta Precision Laboratories. Cancer Biomarker Comprehensive DNA Panel, Tumor. 2024: https://www.albertahealthservices.ca/webapps/labservices/indexAPL.asp?id=9362&tests=&zoneid=1&details=true. Accessed May 01, 2025.
41.BC Cancer. Prostate Cancer. https://cancergeneticslab.ca/solid-tumour/prostate-cancer/. Accessed May 01, 2025.
42.IWK Health. Maritime Medical Genetics Service. https://iwkhealth.ca/clinics-programs-services/maritime-medical-genetics-service. Accessed May 01, 2025.
43.Saskatchewan Health Authority. Roy Romanow Provincial Laboratory Molecular Diagnostics - Hereditary Cancer Requisition. 2023: https://www.saskhealthauthority.ca/system/files/2023-09/Form-LabMed-Hereditary-Cancer-Requisition-Fillable.PDF. Accessed May 01, 2025.
44.Alberta Health Services. Genetic testing for hereditary cancer: Breast/Ovarian/Prostate cancer panel. 2024: https://myhealth.alberta.ca/health/AfterCareInformation/pages/conditions.aspx?hwid=custom.ab_genetictest_hered_cancer_bop_inst. Accessed May 01, 2025.
45.Winnipeg Regional Health Authority. Hereditary Cancer Service. https://wrha.mb.ca/genetics-and-metabolism/hereditary-cancer-service/. Accessed May 01, 2025.
46.Saskatchewan Health Authority. Hereditary Cancer Gene Panels. 2023: https://rrpl-testviewer.ehealthsask.ca/SCI/Requisitions/Requisitions%20for%20Molecular%20Diagnostics%20-%20Human%20Molecular%20Genetics/Hereditary%20Cancer%20Gene%20Panels%20Version%20May%202022.pdf. Accessed May 01, 2025.
47.IWK Health. IWK CGL Test Menu. 2024: https://iwkhealth.ca/sites/default/files/2024-08/IWK%20CGL%20Test%20Menu%20-%20with%20restrictions%20and%20gene%20content.pdf. Accessed May 01, 2025.
48.Alberta Precision Laboratories. Breast/Ovarian/Prostate Cancer Panel. 2024: https://www.albertahealthservices.ca/webapps/labservices/indexAPL.asp?id=9431&tests=&zoneid=1&details=true. Accessed May 01, 2025.
49.BC Cancer. Hereditary Cancer Panel. https://cancergeneticslab.ca/genes/hereditary-cancer-panel/. Accessed May 01, 2025.
50.Shared Health Manitoba. Manitoba Health Lab Information Manual 2.9.2: Hereditary Cancer Panel https://apps.sbgh.mb.ca/labmanual/test/view?seedId=43680. Accessed May 01, 2025.
51.Selvarajah S, Schrader KA, Kolinsky MP, et al. Recommendations for the implementation of genetic testing for metastatic prostate cancer patients in Canada. Can Urol Assoc J. 2022;16(10):321-332. PubMed
52.Dragojlovic N, Borle K, Kopac N, et al. Workforce Implications of Increased Referrals to Hereditary Cancer Services in Canada: A Scenario-Based Analysis. Current Oncology. 2023;30(8):7241-7251. PubMed
53.Snow S, Brezden-Masley C, Carter MD, et al. Barriers and Unequal Access to Timely Molecular Testing Results: Addressing the Inequities in Cancer Care Delays across Canada. Curr Oncol. 2024;31(3):1359-1375. PubMed
54.Schostak M, Bradbury A, Briganti A, et al. Practical Guidance on Establishing a Molecular Testing Pathway for Alterations in Homologous Recombination Repair Genes in Clinical Practice for Patients with Metastatic Prostate Cancer. Eur Urol Oncol. 2024;7(3):344-354. PubMed
55.Armstrong AJ, Taylor A, Haffner MC, et al. Germline and somatic testing for homologous repair deficiency in patients with prostate cancer (part 1 of 2). Prostate Cancer Prostatic Dis. 2024. PubMed
56.Milbury CA, Creeden J, Yip WK, et al. Clinical and analytical validation of FoundationOne®CDx, a comprehensive genomic profiling assay for solid tumors. PLoS One. 2022;17(3):e0264138. PubMed
57.Pfizer. Pfizer Inc. response to Canada's Drug Agency request for additional information regarding Talzenna review on May 26, 2025 [internal additional sponsor's information]. 2025.
58.Terraf P, Pareja F, Brown DN, et al. Comprehensive assessment of germline pathogenic variant detection in tumor-only sequencing. Ann Oncol. 2022;33(4):426-433. PubMed
59.Cowan JS, Kagedan BL, Graham GE, Heim-Myers B, Bombard Y. Health care implications of the Genetic Non-Discrimination Act. Canadian Family Physician. 2022;68(9):643. PubMed
60.Agarwal N, Azad A, Shore ND, et al. Talazoparib plus enzalutamide in metastatic castration-resistant prostate cancer: TALAPRO-2 phase III study design. Future Oncol. 2022;18(4):425-436. PubMed
61.Fizazi K, Azad AA, Matsubara N, et al. First-line talazoparib with enzalutamide in HRR-deficient metastatic castration-resistant prostate cancer: the phase 3 TALAPRO-2 trial. Nature Medicine. 2024;30(1):257-264. PubMed
62.Lloyd J, Zomorodian N, Devgan G, Batten J. Talazoparib Plus Enzalutamide in Patients With HRR-Deficient mCRPC: Practical Implementation Steps for Oncology Nurses and Advanced Practice Providers. Clinical journal of oncology nursing. 2024;28(5):483-491. PubMed
63.McKay RR, Morgans AK, Shore ND, Dunshee C, Devgan G, Agarwal N. First-line combination treatment with PARP and androgen receptor-signaling inhibitors in HRR-deficient mCRPC: Applying clinical study findings to clinical practice in the United States. Cancer Treat Rev. 2024;126:102726. PubMed
64.Azad AA, Fizazi K, Matsubara N, et al. Talazoparib plus enzalutamide in metastatic castration-resistant prostate cancer: Safety analyses from the randomized, placebo-controlled, phase III TALAPRO-2 study. Eur J Cancer. 2024;213:115078. PubMed
65.Logothetis CJ, Basch E, Molina A, et al. Effect of abiraterone acetate and prednisone compared with placebo and prednisone on pain control and skeletal-related events in patients with metastatic castration-resistant prostate cancer: exploratory analysis of data from the COU-AA-301 randomised trial. Lancet Oncol. 2012;13(12):1210-1217. PubMed
66.Kuppen MCP, Westgeest HM, van den Eertwegh AJM, et al. Health-related Quality of Life and Pain in a Real-world Castration-resistant Prostate Cancer Population: Results From the PRO-CAPRI Study in the Netherlands. Clin Genitourin Cancer. 2020;18(3):e233-e253. PubMed
67.Chang Y-J, Chang C-H, Peng C-L, et al. Measurement equivalence and feasibility of the EORTC QLQ-PR25: paper-and-pencil versus touch-screen administration. Health and Quality of Life Outcomes. 2014;12(1). PubMed
68.Cleeland CS, Ryan KM. Pain assessment: global use of the Brief Pain Inventory. Ann Acad Med Singap. 1994;23(2):129-138. PubMed
69.Atkinson TM, Mendoza TR, Sit L, et al. The Brief Pain Inventory and Its “Pain At Its Worst in the Last 24 Hours” Item: Clinical Trial Endpoint Considerations. Pain Medicine. 2010;11(3):337-346. PubMed
70.Stenzl A, Dunshee C, De Giorgi U, et al. Effect of Enzalutamide plus Androgen Deprivation Therapy on Health-related Quality of Life in Patients with Metastatic Hormone-sensitive Prostate Cancer: An Analysis of the ARCHES Randomised, Placebo-controlled, Phase 3 Study. Eur Urol. 2020;78(4):603-614. PubMed
71.Robinson DW, Jr., Zhao N, Dawkins F, Qi M, Revicki D. Pain questionnaire performance in advanced prostate cancer: comparative results from two international clinical trials. Qual Life Res. 2013;22(10):2777-2786. PubMed
72.Gater A, Abetz-Webb L, Battersby C, et al. Pain in castration-resistant prostate cancer with bone metastases: a qualitative study. Health Qual Life Outcomes. 2011;9:88. PubMed
73.van Andel G, Bottomley A, Fosså SD, et al. An international field study of the EORTC QLQ-PR25: a questionnaire for assessing the health-related quality of life of patients with prostate cancer. Eur J Cancer. 2008;44(16):2418-2424. PubMed
74.Balshem H, Helfand M, Schünemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. Journal of clinical epidemiology. 2011;64(4):401-406. PubMed
75.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. Journal of clinical epidemiology. 2020;119:126-135. PubMed
76.Inc. P. Talazoparib in combination with enzalutamide for the first-line treatment of adult patients with metastatic castration-resistant prostate cancer: updated matching-adjusted indirect comparison [internal sponsor’s report]. In: Drug Reimbursement Review sponsor submission: Talzenna (talazoparib); 0.5 mg oral tablet. 2025.
77.Armstrong AJ, Lin P, Higano CS, et al. Development and validation of a prognostic model for overall survival in chemotherapy-naïve men with metastatic castration-resistant prostate cancer. Ann Oncol. 2018;29(11):2200-2207. PubMed
Pharmacoeconomic Review
Abbreviations
AAP
abiraterone acetate plus prednisone
ARPI
androgen receptor pathway inhibitor
BIA
budget impact analysis
CDA-AMC
Canada’s Drug Agency
HRR
homologous recombination repair
ICER
incremental cost-effectiveness ratio
MAIC
matching-adjusted indirect comparison
mCRPC
metastatic castration-resistant prostate cancer
OS
overall survival
PARP
poly(adenosine diphosphate-ribose) polymerase
QALY
quality-adjusted life-year
RDI
relative dose intensity
rPFS
radiographic progression-free survival
TTD
time to treatment discontinuation
Economic Review
The objective of the economic review undertaken by Canada’s Drug Agency (CDA-AMC) is to review and critically appraise the pharmacoeconomic evidence submitted by the sponsor on the cost-effectiveness and budget impact of talazoparib plus enzalutamide compared to abiraterone acetate plus prednisone (AAP), enzalutamide, niraparib-abiraterone plus prednisone, and olaparib plus AAP for treatment of adult patients with homologous recombination repair (HRR) gene-mutated metastatic castration-resistant prostate cancer (mCRPC).
Item | Description |
|---|---|
Drug product | Talazoparib (Talzenna), 0.1 mg and 0.25 mg oral capsules |
Indication | In combination with enzalutamide for the treatment of adult patients with homologous recombination repair (HRR) gene-mutated metastatic castration-resistant prostate cancer (mCRPC) |
Submitted price | 0.1 mg capsule: $99.35 0.25 mg capsule: $99.35 |
Health Canada approval status | NOC |
Health Canada review pathway | Standard review pathway |
NOC date | January 30, 2025 |
Reimbursement request | Per indication |
Sponsor | Pfizer Canada ULC |
Submission history | Previously reviewed: No |
HRR = homologous recombination repair; mCRPC = metastatic castration-resistant prostate cancer; NOC = Notice of Compliance.
Note: While 0.1 mg, 0.25 mg, 0.35 mg, 0.5 mg, and 1 mg capsules are listed in the product monograph, the sponsor noted that 0.35 mg, 0.5 mg, and 1 mg capsules are not currently available in Canada.
Summary
Talazoparib is available as 0.1 mg and 0.25 mg capsules.1 At the submitted price of $99.35 per capsule, the 28-day cost of talazoparib is expected to be $5,564 per patient, based on the Health Canada–recommended dosage.1,2 When used in combination with enzalutamide, the 28-day cost is expected to be $8,833 per patient.
Clinical efficacy in the economic analysis for talazoparib plus enzalutamide compared to enzalutamide was derived from the TALAPRO-2 trial. Evidence submitted by the sponsor indicates that talazoparib plus enzalutamide is likely to improve radiographic progression-free survival (rPFS) and overall survival (OS) compared with enzalutamide among patients with HRR gene-mutated mCRPC. Clinical efficacy for all other comparators was informed by a sponsor-submitted matching-adjusted indirect comparison (MAIC). As not all effect modifiers and prognostic factors could be matched or adjusted for, the results of the MAIC are uncertain and conclusions could not be drawn on the relative efficacy of talazoparib plus enzalutamide with comparators included in the MAIC (olaparib plus AAP, niraparib-abiraterone plus prednisone, and AAP).
The results of the CDA-AMC base case suggest the following.
Talazoparib plus enzalutamide will be associated with higher costs to the health care system than enzalutamide (incremental costs = $183,217), primarily driven by increased costs associated with drug acquisition.
Talazoparib plus enzalutamide may be associated with a gain of 0.87 life-years compared to enzalutamide. When the impact on health-related quality of life is also considered, talazoparib plus enzalutamide may result in a gain of 0.76 quality-adjusted life-years (QALYs) compared to enzalutamide.
The incremental cost-effectiveness ratio (ICER) of talazoparib plus enzalutamide compared to enzalutamide was $242,571 per QALY gained in the CDA-AMC base case. Approximately 49% of the incremental benefit of talazoparib plus enzalutamide versus enzalutamide was gained in the extrapolated period (median OS follow-up = 44.2 months; maximum follow-up not reported). CDA-AMC was unable to adequately explore uncertainty due to limitations with the available evidence (the absence of data to support sustained treatment effect and use beyond first-line treatment), and issues with the model structure.
Due to significant uncertainty associated with the sponsor’s submitted MAIC, comparative effectiveness between talazoparib plus enzalutamide and olaparib plus AAP, niraparib-abiraterone plus prednisone, and AAP is too uncertain to determine. Therefore, there is no robust evidence to support a price premium of talazoparib plus enzalutamide in relation to other poly(adenosine diphosphate-ribose) polymerase (PARP) inhibitors plus androgen receptor pathway inhibitor (ARPI) combinations (i.e., olaparib plus AAP and niraparib-abiraterone plus prednisone).
Clinical expert input sought for this review noted that chemotherapy (e.g., docetaxel) and radium-223 may also be considered relevant comparators for talazoparib plus enzalutamide in the indicated population. Neither of these treatments were included in the analysis.
CDA-AMC estimates that the budget impact of reimbursing talazoparib plus enzalutamide for the indicated population will be approximately $21.8 million over the first 3 years of reimbursement compared to the amount currently spent on enzalutamide, AAP, olaparib plus AAP, and niraparib-abiraterone plus prednisone, with an estimated expenditure of $23.4 million on talazoparib over this period (talazoparib plus enzalutamide = $37.2 million). The actual budget impact of reimbursing talazoparib plus enzalutamide will depend on the distribution of market shares for patients with HRR gene-mutated mCRPC, the proportion of patients with HRR BRCA mutations, and the uptake of talazoparib plus enzalutamide.
Summary of the Submitted Economic Evaluation
The sponsor submitted a cost-utility analysis to estimate the cost-effectiveness of talazoparib plus enzalutamide from the perspective of a public drug plan payer in Canada over a lifetime horizon (20 years).3 The modelled population comprised adult patients with HRR gene-mutated mCRPC, which is aligned with the Health Canada–approved indication and was based on a subset of the participants in the TALAPRO-2 trial. The sponsor’s base-case analysis included costs related to drug acquisition, adverse events, disease management, subsequent therapies, and end of life.
In the sponsor’s base case, talazoparib plus enzalutamide was associated with incremental costs of $155,649 and 0.93 incremental QALYs relative to enzalutamide. This resulted in an ICER of $168,095 per QALY gained. Of the incremental benefit compared to enzalutamide (0.93 incremental QALYs), approximately 57% of the benefit was predicted to be accrued after the treatment duration of the TALAPRO-2 trial (median OS follow-up = 44 months). Additional information about the sponsor’s submission is summarized in Appendix 3.
CDA-AMC identified several key issues with the sponsor’s analysis (refer to Table 2; full details are provided in Appendix 4). A revised base case was therefore developed.
Table 2: Key Issues With the Sponsor’s Economic Submission
Issue | What evidence is there to inform this issue? | How was this issue addressed by CDA-AMC? | Did CDA-AMC explore uncertainty in a scenario analysis? |
|---|---|---|---|
The effectiveness and cost-effectiveness of TALA + ENZA in subsequent lines of treatment is unknown. | The TALAPRO-2 trial was restricted to patients receiving their first-line treatment while the approved Health Canada indication allows for use in any line of treatment. There is no direct comparative evidence for the use of TALA + ENZA in the second-line, third-line, and later-line settings. The clinical expert input sought by CDA-AMC indicated that TALA + ENZA would be used in second-line and later-line treatment. | This issue could not be addressed. The analysis therefore focuses on TALA + ENZA in the first-line setting only. | No scenario analysis was conducted given the lack of available evidence. |
Comparative effectiveness of TALA + ENZA vs. OLA + AAP, NIRA-ABI, and AAP is uncertain. | The MAIC submitted by the sponsor has several limitations that deemed the comparative effectiveness between TALA + ENZA and OLA + AAP, NIRA-ABI, and AAP in this population too uncertain to determine. | This issue could not be addressed. | No scenario analysis was conducted given the lack of available evidence. |
The long-term treatment effect of TALA is uncertain. | Based on the sponsor’s rPFS extrapolation, the proportion of patients who remain progression free at 10 years were considered overestimated according to clinical expert input. There is no evidence that TALA + ENZA will sustain clinical benefit for the entire model time horizon, which includes approximately 15 years of extrapolated data. Clinical expert input indicated that the benefit accrued with TALA treatment is not expected to be sustained indefinitely. | CDA-AMC revised the waning of treatment effect assumption to start waning gradually for 12 months after the TALAPRO-2 trial. | To explore uncertainty around this issue, CDA-AMC conducted 2 scenario analysis in which treatment effect is lost immediately after the TALAPRO-2 trial or treatment effect is sustained indefinitely. |
TTD for patients receiving TALA + ENZA is uncertain and underestimates costs for TALA + ENZA. | TTD for patients receiving TALA + ENZA was informed by the TALAPRO-2 trial. TTD estimated by the sponsor lacks face validity as patients continue to experience rPFS benefits after treatment is discontinued (patients accrue benefits while not incurring any treatment costs). Also, only a small percentage of patients are expected to remain progression free without any treatment. | This issue was associated with high uncertainty and it was addressed in a scenario analysis. | To explore uncertainty, CDA-AMC aligned TTD with rPFS curves because based on clinical expert input, this is more aligned with clinical practice. |
Use of RDI underestimated drug acquisition costs. | In the sponsor’s base-case analysis, the mean RDIs observed in the TALAPRO-2 trial and literature were used to derive the drug acquisition cost for all therapies. For oral therapies, pharmacies in Canada are likely to fill and dispense prescriptions in full. Without evidence to suggest that patients will delay filling prescriptions, it is not certain that unused tablets will result in lower drug costs. | The CDA-AMC base case does not incorporate RDI. | No scenario analysis was conducted. |
Exclusion of docetaxel and radium-223, relevant comparators to the indicated population, was inappropriate. | Clinical expert input noted that, currently, given patients who progress to mCRPC have largely already received ARPI treatment and are unlikely to be treated with an ARPI again, docetaxel and other treatments such as radium-223 are relevant comparators for TALA + ENZA in the reimbursement request population. | This issue could not be addressed due to lack of comparative effectiveness between TALA + ENZA and docetaxel or radium-223. | No scenario analysis was conducted. |
The model structure does not reflect clinical practice. | The submitted model only accounted for 1 line of subsequent treatment, although clinical expert input indicated that patients with HRR gene-mutated mCRPC are generally treated with an average of 3 lines of therapy in clinical practice. Additionally, CDA-AMC noted a coding error in the subsequent treatment costs for OLA + AAP and NIRA-ABI resulting in those treatments having zero subsequent treatment costs. | CDA-AMC was unable to address issues related to the model structure. CDA-AMC fixed a coding error to reflect the actual weighted average cost of second-line treatment for OLA + AAP and NIRA-ABI. | No scenario analysis was conducted. |
AAP = abiraterone acetate plus prednisone; ABI = abiraterone acetate; ARPI = androgen receptor pathway inhibitor; CDA-AMC = Canada’s Drug Agency; ENZA = enzalutamide; HRR = homologous recombination repair; HRR = homologous recombination repair; MAIC = matching-adjusted indirect comparison; mCRPC = metastatic castration-resistant prostate cancer; NIRA = niraparib; OLA = olaparib; RDI = relative dose intensity; rPFS = radiographic progression-free survival; TALA = talazoparib; TTD = time to treatment discontinuation; vs. = versus.
Note: Full details of the issues identified by CDA-AMC are provided in Appendix 3.
CDA-AMC Assessment of Cost-Effectiveness
The CDA-AMC base case was derived by making changes to model parameter values and assumptions (refer to Table 8), in consultation with clinical experts. Due to the limitations identified with the MAIC that preclude conclusions regarding the relative effects of talazoparib plus enzalutamide compared with olaparib plus AAP, niraparib-abiraterone plus prednisone, and AAP, the results in this section will be presented for talazoparib plus enzalutamide versus enzalutamide only. Detailed information about the comparison of talazoparib plus enzalutamide versus enzalutamide only is provided in Appendix 4.
Impact on Health Care Costs
Talazoparib plus enzalutamide is expected to be associated with additional health care costs compared to enzalutamide (incremental costs = $183,217). This increase in health care spending results from drug acquisition costs associated with talazoparib plus enzalutamide in the first-line setting (refer to Figure 1). All other costs to the health system are expected to be largely similar between talazoparib plus enzalutamide versus enzalutamide.
Figure 1: Impact of Talazoparib Plus Enzalutamide Versus Enzalutamide on Health Care Costs
AAP = abiraterone acetate plus prednisone; ENZA = enzalutamide; TALA = talazoparib; vs. = versus.
Notes: Drug-related costs include drug acquisition costs, administration, premedication, drug monitoring, and adverse event management.
Results for olaparib plus AAP, niraparib-abiraterone plus prednisone, and AAP are not presented in this figure. Refer to Appendix 4 for full results.
Impact on Health
Relative to enzalutamide, talazoparib plus enzalutamide is expected to increase the amount of time a patient remains progression free by approximately 0.87 years (refer to Figure 2). Considering the impact of treatment on both quality and length of life, talazoparib plus enzalutamide is expected to result in 0.76 additional QALYs per patient compared to enzalutamide.
Figure 2: Impact of Talazoparib Plus Enzalutamide Versus Enzalutamide on Patient Health
AAP = abiraterone acetate plus prednisone; ENZA = enzalutamide; QALY = quality-adjusted life-year; TALA = talazoparib vs. = versus.
Note: Results for olaparib plus AAP, niraparib-abiraterone plus prednisone, and AAP are not presented in this figure. Refer to Appendix 4 for full results.
Overall Results
The results of the CDA-AMC base case suggest an ICER of $242,571 per QALY gained for talazoparib plus enzalutamide compared to enzalutamide (refer to Table 3). Additional details on the CDA-AMC base case are available in Appendix 4.
Table 3: Summary of CDA-AMC Economic Evaluation Results
Drug | Total costs ($) | Total QALYs | ICER vs. ENZA ($/QALY) |
|---|---|---|---|
ENZA | 106,076 | 2.04 | Reference |
TALA + ENZA | 289,294 | 2.79 | 242,571 |
CDA-AMC = Canada's Drug Agency; ENZA = enzalutamide; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; TALA = talazoparib; vs. = versus.
Note: Publicly available list prices were used for all comparators.
Uncertainty and Sensitivity
Due to uncertainty in the comparative clinical evidence, including the lack of long-term evidence and lack of data to inform treatment effectiveness waning over time, the predicted costs and QALYs in the CDA-AMC base case are uncertain.
Uncertainty was able to be explored for the scenario analyses outlined in Table 2. Assumptions on sustaining treatment effect indefinitely had the largest impact on the cost-effectiveness conclusions (refer to Table 12, Appendix 4).
HRR gene mutations need to be confirmed before talazoparib plus enzalutamide treatment is initiated. The clinical experts described the availability of HRR mutation testing as very heterogeneous within and between jurisdictions, including variability in the genes included in each panel. If talazoparib is reimbursed, jurisdictions may need to add more genes to existing panels. Based on clinical expert feedback, it is unclear how or if this would impact health care resources.
CDA-AMC noted that the proposed Health Canada indication for talazoparib plus enzalutamide allows for use in any line of treatment. Therefore, there is no direct comparative evidence for the use of talazoparib plus enzalutamide in the second-line, third-line, and later-line settings. The cost-effectiveness of talazoparib plus enzalutamide in later lines of treatment remains unknown.
Summary of the Budget Impact
The sponsor submitted a budget impact analysis (BIA) to estimate the 3-year (2026 to 2028) budget impact of reimbursing talazoparib plus enzalutamide for use in adult patients with HRR gene-mutated mCRPC. The sponsor assumed that the payers would be CDA-AMC–participating public drug plans and derived the size of the eligible population using an epidemiologic approach based on incident (new) patients only. The price of talazoparib was aligned with the price included in the sponsor’s economic evaluation, while the prices of comparators were based on the publicly available list prices. Additional information pertaining to the sponsor’s submission is provided in Appendix 5.
CDA-AMC identified a number of issues with the sponsor’s estimated budget impact and made changes to model parameters and assumptions in consultation with clinical experts to derive the CDA-AMC base case (Appendix 5). CDA-AMC estimated that 790 individual patients would be eligible for treatment with talazoparib plus enzalutamide over a 3-year period, of whom 204 are expected to receive talazoparib plus enzalutamide. The estimated incremental budget impact of reimbursing talazoparib plus enzalutamide is expected to be approximately $21.8 million over the first 3 years, with an expected expenditure of $23.4 million on talazoparib (the 3-year expenditure on talazoparib plus enzalutamide regimen is estimated to be $37.2 million). The actual budget impact of reimbursing talazoparib plus enzalutamide will depend on the number of people eligible for treatment, the proportion of patients with BRCA mutations, and the uptake of talazoparib plus enzalutamide.
Conclusion
Based on the CDA-AMC base case, talazoparib plus enzalutamide would be considered cost-effective at the submitted price if the public health care system was willing to pay at least $242,571 for each additional QALY gained. If the public health care system is not willing to pay that amount, a price reduction should be considered (refer to Figure 3; full details of the impact of price reductions on cost-effectiveness are presented in Table 11). The estimated cost-effectiveness is uncertain due to different comparators within subgroups of the indication, limitations with the clinical evidence, exclusion of a relevant comparator, and issues with the model structure. Based on available information on indirect comparative effectiveness, there is no robust evidence to support a price premium of talazoparib plus enzalutamide in relation to olaparib plus AAP and niraparib-abiraterone plus prednisone.
The budget impact of reimbursing talazoparib plus enzalutamide to the public drug plans in the first 3 years is estimated to be approximately $21.8 million. The 3-year expenditure on talazoparib alone (i.e., not accounting for current expenditure on the rest of the regimen or comparators) is estimated to be $23.4 million (the 3-year expenditure on the talazoparib plus enzalutamide regimen is estimated to be $37.2 million). The estimated budget impact is uncertain due to uncertainty in the distribution of market shares for patients with HRR gene-mutated mCRPC, in the proportion of patients with BRCA mutations, and in the uptake of talazoparib plus enzalutamide.
Figure 3: Summary of the CDA-AMC Economic Analysis and Price Reduction
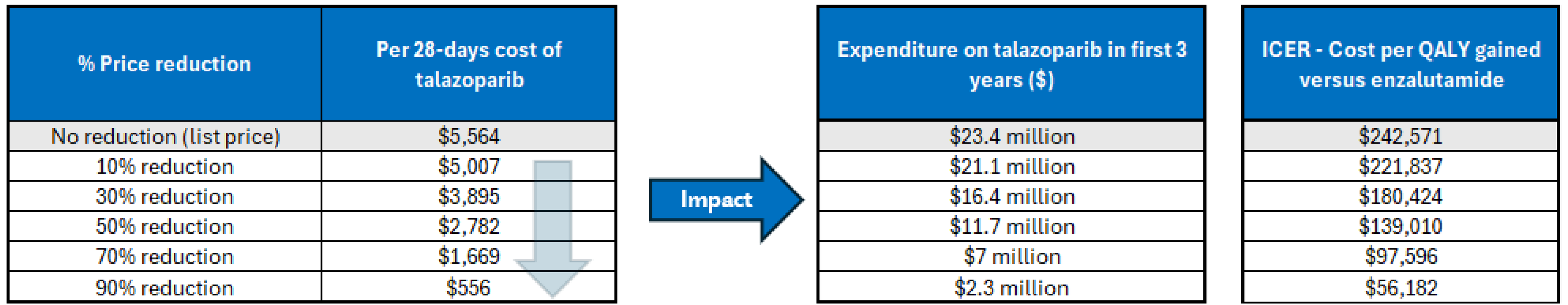
AAP = abiraterone acetate plus prednisone; CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: “Expenditure” includes only the drug cost of talazoparib. The price reductions in this table are applied only to talazoparib. Results for olaparib plus AAP, niraparib-abiraterone plus prednisone, and AAP are not presented in this figure.
References
1.Pfizer Canada. Drug Reimbursement Review sponsor submission: TALZENNA® (talazoparib), Capsules, 0.1 mg, 0.25 mg, 0.35 mg, 0.5 mg and 1 mg talazoparib (as talazoparib tosylate) [internal sponsor's package]. 2025.
2.Pfizer C. Product Monograph: TALZENNA (talazoparib). 2025. Accessed January 30, 2025. https://webfiles.pfizer.com/file/471a94d8-92af-4ea2-bf98-a6f1ecb1013b?referrer=ccb731e5-4f2d-4f4a-b2dc-e5e912145fc6
3.Pfizer Canada. Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: TALZENNA® (talazoparib), Capsules, 0.1 mg, 0.25 mg, 0.35 mg, 0.5 mg and 1 mg talazoparib (as talazoparib tosylate). 2025.
4.Ontario Ministry of H. Ontario drug benefit formulary/comparative drug index. Accessed January 1, 1800. https://www.formulary.health.gov.on.ca/formulary/
5.Ontario Ministry of H. Exceptional Access Program (EAP). Accessed January 1, 1800. http://www.health.gov.on.ca/en/pro/programs/drugs/odbf/odbf_except_access.aspx
6.Woon DTS, Chandrasekar T, Aaron L, et al. Disparity in public funding of therapies for metastatic castrate-resistant prostate cancer across Canadian provinces. Can Urol Assoc J. 2018;12(10):328-336. doi: 10.5489/cuaj.5378 PubMed
7.Nice. Enzalutamide for treating metastatic hormone-relapsed prostate cancer [TA377]. 2015. Accessed January 2025. https://www.nice.org.uk/guidance/ta377/documents/final-appraisal-determination-document
8.Nice. Daratumumab with Bortezomib and Dexamethasone for Previously Treated Multiple Myeloma [TA573]. 2023. Accessed February 6, 2025. https://www.nice.org.uk/guidance/ta897/evidence/committee-papers-pdf-13069187965
9.Nice. Enzalutamide for treating nonmetastatic hormone-relapsed prostate cancer [ID1359]. 2019. Accessed February 6, 2025. https://www.nice.org.uk/guidance/ta580/documents/committee-papers
10.Diaby V, Adunlin G, Ali AA, et al. Cost-effectiveness analysis of 1st through 3rd line sequential targeted therapy in HER2-positive metastatic breast cancer in the United States. Breast Cancer Res Treat. 2016;160(1):187-196. doi: 10.1007/s10549-016-3978-6 PubMed
11.Ontario Ministry of H, Long-Term C. Ontario Drug Benefit Formulary. Updated 2025. Accessed February 2025. https://www.formulary.health.gov.on.ca/formulary/
12.Scotia GoN. Nova Scotia Formulary. Updated March 2025. Accessed March 2025. https://novascotia.ca/dhw/pharmacare/documents/formulary.pdf
13.Ontario Ministry of H, Long-Term C. Schedule of Benefits Physician Services Under the Health Insurance Act. 2025. Accessed March 2025. https://www.ontario.ca/files/2025-03/moh-schedule-benefit-2025-03-19.pdf
14.Ontario Ministry of H, Long-Term C. Schedule of Benefits for Laboratory Services. 2025. Accessed March 2025. https://www.ontario.ca/files/2025-03/moh-ohip-schedule-of-benefits-laboratory-services-2025-03-03.pdf
15.Latimer N. NICE DSU Technical Support Document 14: Undertaking survival analysis for economic evaluations alongside clinical trials - extrapolation with patient-level data. 2011. Accessed February 2023. https://www.ncbi.nlm.nih.gov/books/NBK395885/pdf/Bookshelf_NBK395885.pdf
16.Authors, Coyle D, Haines A, Lee K. CADTH Health Technology Review. Extrapolating Clinical Evidence Within Economic Evaluations: CADTH Methods and Guidelines. Canadian Agency for Drugs and Technologies in Health; 2023.
17.Pfizer. Clinical Study Report: TALAPRO-2 Interim HRR-deficient CSR (Cohort 2 Final Overall Survival), Version 1.0 (C3441021, data cut-off: 03Sep2024). 2024.
18.Olmos D, Lorente D, Alameda D, et al. Treatment patterns and outcomes in metastatic castration-resistant prostate cancer patients with and without somatic or germline alterations in homologous recombination repair genes. Annals of Oncology. 2024;35(5):458-472. doi: 10.1016/j.annonc.2024.01.011 PubMed
19.Brenner DR, Gillis J, Demers AA, et al. Projected estimates of cancer in Canada in 2024. CMAJ. 2024;196(18):E615-E623. doi: 10.1503/cmaj.240095 PubMed
20.Kirby M, Hirst C, Crawford ED. Characterising the castration-resistant prostate cancer population: a systematic review. Int J Clin Pract. 2011;65(11):1180-92. doi: 10.1111/j.1742-1241.2011.02799.x PubMed
21.Pfizer. Clinical Study Report: TALAPRO-2 Cohort 1, Version 2.0 (C3441021, data cut-off: 16Aug2022). 2022.
22.Shayegan B, Wallis CJD, Malone S, et al. Real-world use of systemic therapies in men with metastatic castration resistant prostate cancer (mCRPC) in Canada. 2022;(1873-2496 (Electronic)).
23.ONCO-CAPPS physician chart audit. Pfizer internal files.
24.Shore N, Oliver L, Shui I, et al. Systematic Literature Review of the Epidemiology of Advanced Prostate Cancer and Associated Homologous Recombination Repair Gene Alterations. J Urol. 2021;205(4):977-986. doi: 10.1097/ju.0000000000001570 PubMed
25.Canadian Cancer Society. Canadian Cancer Statistics: A 2022 special report on cancer prevalence. 2023:49. Accessed June 27, 2025.
26.Government of Canada. Alignment Among Public Formularies in Canada, Part 2: Oncology Medicines. 2021. Accessed June 27, 2025. https://www.canada.ca/en/patented-medicine-prices-review/services/npduis/analytical-studies/formularies-part2-oncology-medicines.html
27.Sutherland G, Dihn T. Understanding the gap: a pan-Canadian analysis of prescription drug insurance coverage. The Conference Board of Canada; 2017. Accessed January 1, 1800. https://www.conferenceboard.ca/e-library/abstract.aspx?did=9326
28.Fay AP, Fizazi K, Matsubara N, et al. First-line talazoparib plus enzalutamide versus placebo plus enzalutamide in men with metastatic castration-resistant prostate cancer and homologous recombination repair gene alterations: patient-reported outcomes from the randomised, double-blind, placebo-controlled, phase 3 TALAPRO-2 trial. The Lancet Oncology. 2025;26(4):481-490. doi: 10.1016/s1470-2045%2825%2900031-2 PubMed
29.Chi KN, Sandhu S, Smith MR, et al. Niraparib plus abiraterone acetate with prednisone in patients with metastatic castration-resistant prostate cancer and homologous recombination repair gene alterations: second interim analysis of the randomized phase III MAGNITUDE trial. Ann Oncol. 2023;34(9):772-782. doi: 10.1016/j.annonc.2023.06.009 PubMed
30.Ryan CJ, Small EJ, Smith MR, et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. New England Journal of Medicine. 2013;368(2):138-148. doi: 10.1056/NEJMoa1209096 PubMed
31.Clarke Noel W, Armstrong Andrew J, Thiery-Vuillemin A, et al. Abiraterone and Olaparib for Metastatic Castration-Resistant Prostate Cancer. NEJM Evidence. 2022;1(9):EVIDoa2200043. doi: 10.1056/EVIDoa2200043
32.Chi KN, Rathkopf D, Smith MR, et al. Niraparib and Abiraterone Acetate for Metastatic Castration-Resistant Prostate Cancer. J Clin Oncol. 2023;41(18):3339-3351. doi: 10.1200/JCO.22.01649 PubMed
Appendix 1: Cost Comparison Table
Please note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical experts and CDA-AMC–participating public drug plans. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 4: Cost Comparison for Treatments for Metastatic Castration-Resistant Prostate Cancer
Treatment | Strength and/or concentration | Form | Price ($) | Recommended dosage | Daily cost ($) | 28- day cost ($) |
|---|---|---|---|---|---|---|
Talazoparib (Talzenna) | 0.25 mg 0.10 mg | Capsule | 99.3500a 99.3500a | 0.50 mg daily | 198.70 | 5,564 |
Enzalutamide (Xtandi) | 40 mg | Capsule | 29.1954b | 160 mg daily | 116.78 | 3,270 |
Talazoparib plus enzalutamide | — | — | — | — | 315.48 | 8,833 |
Enzalutamide | ||||||
Enzalutamide (Xtandi) | 40 mg | Capsule | 29.1954b | 160 mg daily | 116.78 | 3,270 |
Abiraterone plus prednisone | ||||||
Abiraterone (generic) | 250 mg 500 mg | Tablet | 7.6563 15.3125 | 1,000 mg daily | 30.63 | 858 |
Prednisone (generic) | 5 mg 50mg | Tablet | 0.0220 0.1735 | 10 mg daily | 0.04 | 1 |
Abiraterone plus prednisone | — | — | — | — | 30.67 | 859 |
Olaparib plus abiraterone | ||||||
Olaparib (Lynparza) | 100 mg 150 mg | Tablet | 69.9482b 69.9482b | 600 mg daily | 279.79 | 7,834 |
Abiraterone (generic) | 250 mg 500 mg | Tablet | 7.6563 15.3125 | 1,000 mg daily | 30.63 | 858 |
Prednisone (generic) | 5 mg 50mg | Tablet | 0.0220 0.1735 | 10 mg daily | 0.04 | 1 |
Olaparib plus abiraterone plus prednisone | — | — | — | — | 310.46 | 8,693 |
Niraparib plus abiraterone | ||||||
Niraparib (Zejula) | 100 mg | Capsule | 133.2346b | 200 mg daily | 266.47 | 7,461 |
Abiraterone (generic) | 250 mg 500 mg | Tablet | 7.6563 15.3125 | 1,000 mg daily | 30.63 | 858 |
Niraparib-abiraterone (Akeega) | 50 mg / 500 mg 100 mg / 500 mg | Tablet | 149.3065 149.3065 | 200 mg of niraparib daily 1,000 mg of abiraterone daily | 298.61 | 8,361 |
Prednisone (generic) | 5 mg 50mg | Tablet | 0.0220 0.1735 | 10 mg daily | 0.04 | 1 |
Niraparib plus abiraterone plus prednisone | — | — | — | — | 297.14 | 8,320 |
Niraparib-abiraterone plus prednisone | — | — | — | — | 298.65 | 8,362 |
Note: All prices are from the Ontario Drug Benefit Formulary (accessed May 2025),4 unless otherwise indicated, and do not include dispensing fees. Recommended dosages are based on product monographs, unless otherwise indicated.
aSponsor’s submitted price and recommended dosage.3
bPrices from Ontario Exceptional Access Program (accessed May 2025).5
Table 5: Cost Comparison for Additional Treatments for Metastatic Castration-Resistant Prostate Cancer
Treatment | Strength and/or concentration | Form | Price ($) | Recommended dosage | Daily cost ($) | 28- day cost ($) |
|---|---|---|---|---|---|---|
Docetaxel | ||||||
Docetaxel (generic) | 10 mg/mL 20 mg/mL | 8 mL Vial 16 mL Vial 4 mL Vial | 970.2000 1,940.4000 497.0000 | 75 mg/m2 IV every 3 weeksa | 85.76 | 2,401 |
Prednisone (generic) | 5 mg 50mg | Tablet | 0.0220 0.1735 | 10 mg daily | 0.04 | 1 |
Docetaxel plus prednisone | — | — | — | — | 85.78 | 2,402 |
Radium-223 | ||||||
Radium-223 (Xofigo) | 1,100 kBq/mL | Vial | 5,640.0000b | 55 kBq per kg, given at 4-week intervals for a total of 6 injections | 5,640.00 | 33,840 |
Note: All prices are from the IQVIA Delta PA database (accessed May 2025), unless otherwise indicated, and do not include dispensing fees. Recommended dosages are based on product monographs, unless otherwise indicated.
aBased on an assumed body surface area of 1.98 m2.
bPrice sourced from Woon et al. (2018).6
Appendix 2: Input Relevant to the Economic Review
Please note that this appendix has not been copy-edited.
This section is a summary of the input received from the patient groups, clinician groups, and drug plans that participated in the CDA-AMC review process.
Patient input was received from Canadian Cancer Society and PROCURE. Both groups conducted surveys with patients living in Canada (PROCURE surveyed 263 patients living in Canada with prostate cancer in 2018 and 2022, and the Canadian Cancer Society surveyed 21 individuals with mCRPC and 3 caregivers). Patient input indicates that treatment goals include slowing down the progression of cancer and extending OS without introducing additional severe side effects than standard therapies. Moreover, patient input noted that talazoparib plus enzalutamide allows patients to refrain from taking corticosteroids as part of their treatment regimen, which can be beneficial in reducing the side effects associated with long-term steroid use (e.g., weight gain, osteoporosis, and increased risk of infections) and reducing the number of pills required per day. Regarding HRR testing, patient input noted that while most provinces have access to publicly funded genetic testing programs, the availability and scope of these tests can vary significantly, which can be a barrier to timely access to treatment. Regarding current treatment options, most patients who responded to the survey reported receiving 3 or more lines of treatment, with the most commonly being hormone therapies (i.e., luteinizing hormone-releasing hormone agonists, antagonists, and antiandrogen drugs). Patient input also noted many severe adverse events associated with current treatment options (e.g., nausea, vomiting, change in sexual function, fertility) with significant impact on patient’s quality of life. No survey respondents had experience with the drug under review.
Clinician group input was received from Ontario Health (Cancer Care Ontario) Genitourinary Cancer Drug. The current pathway of care for patients with mCRPC includes abiraterone, enzalutamide, docetaxel, radium-223 (in docetaxel ineligible patients) all given concurrently with androgen deprivation therapy. In addition, clinical input indicates that cabazitaxel is another option after docetaxel intensification, as well as olaparib-abiraterone and niraparib-abiraterone for those who have not intensified with ARPIs. Clinical input noted that treatment goals include prolonging life and maximizing quality of life, as there currently remains no cure for these patients. Finally, clinician input indicates that talazoparib plus enzalutamide will be best suited for patients with mCRPC who are treatment naive.
Input from CDA-AMC–participating drug plans noted that there is a statement in the product monograph that the talazoparib should maintained in the original bottle to protect the capsules from the light. However, it is unclear whether the capsules need to be maintained in the original packaging when dispensed to the patients, and if so, consideration regarding drug wastage, depending on how the drug is packaged, needs to be taken into account. Drug plan input indicates that not all jurisdictions have access to testing for all HRR pathway genes. Finally, plans questioned whether patients currently receiving monotherapy with enzalutamide (or abiraterone) be able to switch to talazoparib plus enzalutamide.
Several of these concerns were addressed in the sponsor’s model:
Treatment goals of OS, as well as adverse events, were modelled.
CDA-AMC was unable to address the following concerns:
Comparative effectiveness between talazoparib plus enzalutamide and other relevant comparators such as docetaxel, cabazitaxel, and radium-223.
Appendix 3: Summary of the Sponsor’s Submission
Please note that this appendix has not been copy-edited.
Summary of the Sponsor’s Economic Evaluation
For the pharmaceutical reviews program, clinical and economic information is submitted to CDA-AMC by the sponsor. The CDA-AMC health economics team reviews the submitted economic information and appraises the information in collaboration with clinical experts and the clinical review team to evaluate key assumptions, influential parameters, and the overall rigour of the economic submission. Based on what the team learns through this process, adjustments may be made to the sponsor’s model to produce the CDA-AMC base case. The CDA-AMC base case represents the team’s current understanding of the clinical condition, clinical evidence currently available, and best interpretation of the economic evidence based on the information provided.
For the review of talazoparib plus enzalutamide, the sponsor provided a cost-utility analysis and a BIA. The sponsor’s economic submission is summarized in Table 6.
Table 6: Key Components of the Sponsor’s Economic Evaluation
Component | Description |
|---|---|
Treatment information | |
Drug under review | Talazoparib (Talzenna), oral capsules (0.1 mg and 0.25 mg) |
Submitted price of drug under review | TALA: 0.25 mg capsule: $99.35 0.1 mg capsule: $99.35 |
Regimen | 0.5 mg of TALA administered orally once daily in combination with ENZA 160 mg orally once daily, until disease progression or unacceptable toxicity occurs |
Per 28-day cycle cost of drug under reviewa | TALA: $4,509 per patient per 28-day cycle TALA + ENZA: $7,779 per patient per 28-day cycle |
Model information | |
Type of economic evaluation | Cost-utility analysis, PSM |
Treatment | TALA + ENZA |
Included comparator(s) |
|
Perspective | Publicly funded health care payer perspective |
Time horizon | Lifetime (20 years) |
Cycle length | One month |
Modelled population(s) | Patients aged 18 years and older with asymptomatic or mildly symptomatic HRR gene-mutated metastatic CRPC who have not started systemic cancer treatment after diagnosis of CRPC (metastatic or nonmetastatic), with the exception of androgen deprivation therapy and first-generation antiandrogen drugs. |
Characteristics of modelled population | Derived from the TALAPRO-2 trial (mean age: 70.6 years; BSA: 1.98 mg/m2) |
Model health states |
|
Data sources | |
Comparative efficacy |
|
Natural history and/or clinical pathway |
|
Health-related utilities and disutilities | |
Costs |
|
Summary of the submitted results | |
Base case results |
|
Scenario analysis resultsc |
|
AAP = abiraterone acetate plus prednisone; ABI = abiraterone acetate; AE = adverse event; BSA = body surface area; CRPC = castration-resistant prostate cancer; ENZA = enzalutamide; ICER = incremental cost-effectiveness ratio; HRR = homologous recombination repair; MAIC = matching-adjusted indirect comparisons; NIRA = niraparib; OLA = olaparib; OS = overall survival; NICE = National Institute for Health and Care Excellence; PD = progressed disease; PSM = partitional survival model; QALY = quality-adjusted life-year; rPFS = radiographic progression-free survival; TALA = talazoparib; TTD = time to treatment discontinuation.
aSponsor assumed relative dose intensity of 81.05% for talazoparib and 100% for enzalutamide.3
bThe sponsor’s rationale for this methodologic choice was that it allows the use of real-world evidence to adjust ENZA’s survival curves, although this approach was not part of the sponsor’s base case. Additionally, the long-term OS evidence for ENZA was not used to inform the MAIC.
cResults of scenario analyses that had a meaningful impact on the estimated ICER compared to the sponsor’s base case. Additional scenarios were submitted that had no meaningful impact on the estimated ICER included time horizon of 10 years, discount rates of 0% and 3%, health states utilities for PD from TALAPRO-2, alternate parametric functions for OS, and inclusion of genetic testing.
Table 7: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Total QALYs | TALA + ENZA vs. comparators ICER ($/ QALY) |
|---|---|---|---|
TALA + ENZA vs. ENZA | |||
ENZA | 105,317 | 2.05 | Reference |
TALA + ENZA | 260,966 | 2.97 | 168,095 |
TALA + ENZA vs. OLA + AAP | |||
OLA + AAP | 263,538 | 2.73 | Reference |
TALA + ENZA | 260,966 | 2.97 | Dominant |
TALA + ENZA vs. NIRA-ABI | |||
NIRA-ABI | 211,496 | 2.09 | Reference |
TALA + ENZA | 260,966 | 2.97 | 55,971 |
TALA + ENZA vs. AAP | |||
AAP | 58,115 | 1.78 | Reference |
TALA + ENZA | 260,966 | 2.97 | 170,727 |
AAP = abiraterone acetate plus prednisone; ABI = abiraterone acetate; ENZA = enzalutamide; ICER = incremental cost-effectiveness ratio; NIRA = niraparib; OLA = olaparib; QALY = quality-adjusted life-year; TALA = talazoparib; vs. = versus.
Appendix 4: Additional Details of CDA-AMC Reanalyses
Please note that this appendix has not been copy-edited.
Clinical Data in the Economic Model
The key clinical efficacy data used to inform the economic model (i.e., rPFS, OS, and time to treatment discontinuation [TTD]) were derived from the TALAPRO-2 trial (data cut: September 2024) for talazoparib plus enzalutamide and enzalutamide. The sponsor submitted a model that used an alternative definition for rPFS definition that incorporates the progression or death event into the rPFS end point for patients previously censored, instead of the primary rPFS definition from the TALAPRO-2 trial. The rationale for using an alternate rPFS definition is that the primary rPFS definition led patients to be censored before experiencing radiographic progression or death – due to either missing assessments or the timing of events – which can lead to an inflation of rPFS and cause the rPFS Kaplan-Meier curve to cross the OS Kaplan-Meier curve.3 The sponsor did not apply the same censoring adjustment to the OS curve. The relative efficacy between enzalutamide and talazoparib plus enzalutamide appeared to be similar when comparing both rPFS definitions, with the key difference being a reduction in proportion in rPFS in both treatment arms. Based on the TALAPRO-2 trial, the CDA-AMC Clinical Review found that talazoparib plus enzalutamide may improve rPFS and OS when compared to enzalutamide monotherapy, with the potential treatment benefit on OS subjected to a degree of uncertainty due to differences in the proportion of patients permitted to receive posttreatment anticancer therapy after study treatment had been discontinued.
Estimates of relative efficacy for the other comparators in economic evaluation were obtained from the sponsor’s MAIC. The outcome of interest from the MAIC, as it relates to the economic evaluation, was rPFS and OS. The CDA-AMC Clinical Review noted that although rPFS and OS were adjusted some effect modifiers and prognostic factors in the MAIC, not all factors could be matched or adjusted, therefore imbalances remained. Overall, the magnitude and direction of potential bias due to imbalances for the rPFS and OS estimates cannot be predicted and definitive conclusions based on these results are not recommended. Furthermore, the MAIC used to inform the economic model matched patients from the enzalutamide monotherapy arm from the TALAPRO-2 trial to olaparib plus AAP, niraparib-abiraterone plus prednisone, and AAP, instead of patients from the talazoparib plus enzalutamide arm, the drug under review. The hazard ratio derived from this analysis was then applied to the enzalutamide monotherapy rPFS and OS curves. Thus, in the analysis talazoparib plus enzalutamide was not compared to olaparib plus AAP, niraparib-abiraterone plus prednisone, and AAP. Finally, the proportional hazards assumption was also not assessed or discussed.
Key Issues of the Submitted Economic Evaluation
CDA-AMC identified the following key issues with the sponsor’s analysis:
The submitted model does not align with the indicated population. The proposed indication for talazoparib plus enzalutamide is for all adult patients with HRR gene-mutated mCRPC, irrespective of the line of treatment. The TALAPRO-2 trial was restricted to patients receiving their first-line treatment in the mCRPC setting (i.e., patients had not received prior systemic therapy for mCRPC) while the approved Health Canada indication allows for use in any line of treatment. Therefore, there is no direct comparative evidence for the use of talazoparib plus enzalutamide in the second-line, third-line, and later-line settings. The clinical expert input indicated that talazoparib plus enzalutamide would likely be used as second-line and beyond treatment in clinical practice in Canada. The cost-effectiveness of talazoparib plus enzalutamide as later lines of treatment remains unknown.
CDA-AMC was unable to address this limitation associated with the model structure and the lack of clinical data in later lines of therapy.
Comparative effectiveness estimates derived from the MAIC are highly uncertain. Given there is no head-to-head comparison between talazoparib plus enzalutamide and olaparib plus AAP, niraparib-abiraterone plus prednisone, and AAP, the sponsor submitted an unanchored MAIC as source of comparative effectiveness to inform rPFS and OS in the economic model. CDA-AMC noted that olaparib plus AAP and niraparib-abiraterone plus prednisone are both indicated to be used in first-line treatment of patients with deleterious or suspected deleterious germline and/or somatic BRCA-mutated mCRPC, which represent only part of the population eligible for talazoparib plus enzalutamide based on Health Canada indication (i.e., any HRR gene mutations, line agnostic). Based on TALAPRO-2 trial distribution of mutations, BRCA mutations are expected to be between 30% to 40% of all HRR mutations in this population. Therefore, olaparib plus AAP and niraparib-abiraterone plus prednisone would not be relevant comparators for between 60% to 70% of patients receiving first-line therapy. Olaparib and niraparib are available as second-line or later-line treatments in patients with BRCA and/or ATM mutations (based on the TALAPRO-2 trial, BRCA and ATM mutations account for up to 60% of all HRR mutations), so if talazoparib plus enzalutamide is used in subsequent lines of therapy there are various potential comparators. Moreover, as stated in the Clinical Data in the Economic Model section, the MAIC has several limitations that preclude any conclusions regarding the comparative effectiveness between talazoparib plus enzalutamide and olaparib plus AAP, niraparib-abiraterone plus prednisone, and AAP in this population.
Additionally, in the MAIC, the sponsor matched patients from the enzalutamide monotherapy arm from the TALAPRO-2 trial to patients from the PROpel study (olaparib plus AAP) and MAGNITUDE study (niraparib-abiraterone plus prednisone and AAP), instead of patients from the talazoparib plus enzalutamide arm, the drug under review. The hazard ratio derived from the MAIC were then applied to enzalutamide monotherapy arm to derive progression-free survival and OS curves for olaparib plus AAP, niraparib-abiraterone plus prednisone, and AAP in the economic model. Therefore, there is no analysis comparing talazoparib plus enzalutamide with olaparib plus AAP, niraparib-abiraterone plus prednisone, and AAP, and the use of the MAIC estimates that uses enzalutamide as the reference treatment to inform the efficacy of talazoparib plus enzalutamide relative to the other comparators is methodologically inappropriate. Any conclusions regarding comparative effectiveness remain highly uncertain given the modelling choice made by the sponsor (i.e., all comparators were modelled against enzalutamide monotherapy). In addition to these concerns, the CDA-AMC Clinical Review identified limitations with the MAICs, such that the effects of talazoparib plus enzalutamide on rPFS and OS compared with olaparib plus AAP, niraparib-abiraterone plus prednisone, and AAP are uncertain and definitive conclusions based on these results are not recommended. Therefore, based on available information, there is no robust evidence to support a price premium of talazoparib plus enzalutamide in relation to olaparib plus AAP and niraparib-abiraterone plus prednisone. The clinical expert input sought by CDA-AMC for this review noted that AAP is in the same class of treatments (ARPIs) as enzalutamide, and these treatments are generally considered to have similar effects.
This issue could not be addressed due to issues related to the MAIC.
The long-term treatment effect of talazoparib is uncertain. In the base-case analysis submitted by the sponsor, the clinical evidence was based on the TALAPRO-2 trial (median OS follow-up of 44.2 months) which were extrapolated for the sponsor’s lifetime time horizon. Talazoparib plus enzalutamide’s treatment effects on rPFS and OS were maintained for the entire time horizon, which includes approximately 15 years of extrapolated data. For instance, CDA-AMC noted that 0.4% and 2.2% of patients who received enzalutamide and talazoparib plus enzalutamide, respectively, were projected to be progression free after 10 years in the sponsor’s base case. These proportions were considered significantly overestimated according to clinical expert input sought by CDA-AMC during this review. Based on clinical expert input, the proportion of patients who were progression free would likely approach zero after 3 years. Moreover, clinical expert input sought by CDA-AMC for this review indicated that the benefit accrued while receiving talazoparib treatment is not expected to be sustained indefinitely, with no known mechanism of action to support the sustained effect of talazoparib over time, especially after disease progression. In circumstances where either treatment will be delivered for a time horizon longer than the trial duration, or that treatment is assumed to have a continued effect on event rates postdelivery, assumptions relating to the continuation of long-term treatment effect must be considered carefully.15,16
In addition to issues related to the plausibility of sustained treatment effect, CDA-AMC highlights that the use of different definitions of rPFS in the TALAPRO-2 trial and the economic model introduce additional uncertainty in the analysis. CDA-AMC maintained the use of alternative rPFS definition as the relative efficacy between enzalutamide and talazoparib plus enzalutamide remained similar and programming issues prevented CDA-AMC to use the original rPFS definition.
Finally, CDA-AMC noticed that the sponsor assumed the end of follow-up for OS was 67 months. The submitted Kaplan-Meier graph for OS showed no patients at risk of an event after 64 months and the sponsor did not provide the maximum follow-up for OS upon request.
CDA-AMC revised the waning of treatment effect assumption to start waning gradually for 12 months after the trial. It is important to clarify that the CDA-AMC revised assumption does not imply a complete absence of benefits from talazoparib plus enzalutamide versus enzalutamide monotherapy. Instead, it still shows some level of sustained separation between the rPFS and OS curves. However, the relative advantages of talazoparib plus enzalutamide versus enzalutamide gradually decline. CDA-AMC conducted 2 scenario analyses to explore the impact of the waning treatment effect of talazoparib plus enzalutamide: treatment effect is lost immediately after the TALAPRO-2 trial and treatment effect is sustained indefinitely. In addition, CDA-AMC changed the maximum follow-up of OS to 64 months.
TTD is uncertain. In the sponsor’s base case, time on treatment for patients receiving talazoparib plus enzalutamide was informed by parametric distributions fitted to patient-level data on TTD of talazoparib plus enzalutamide and enzalutamide from the TALAPRO-2 trial. Based on the TALAPRO-2 trial, patients continued treatment until at least 1 of the following events occurred: radiographic progression and the patient is no longer clinically benefiting in the opinion of the investigator, an adverse event leading to permanent discontinuation, patient decision to discontinue study treatment, or death.17 The sponsor selected the gamma distribution for the base-case analysis, stating that it provided a more clinically plausible extrapolation. CDA-AMC notes that the TTD estimated in the sponsor’s base case resulted in 24% and 7% of patients still receiving talazoparib plus enzalutamide treatment at year 3 and 5 posttreatment initiation. In contrast, the sponsor’s extrapolation of rPFS estimated that 38% and 17% of patients treated with talazoparib plus enzalutamide would be progression free at 3-years and 5-years posttreatment initiation. That is, within this period between 10% and 14% of patients receiving talazoparib plus enzalutamide would continue to experience rPFS benefit after treatment with talazoparib plus enzalutamide is discontinued. In addition, based on the sponsor’s choice of extrapolation, the proportion of patients expected to remain on treatment with talazoparib plus enzalutamide only reaches 0% at approximately 16 years, and the proportion of patients would be progression free only reaches 0% at 19 years, showing a gap of 3 years where patients would remain progression free without receiving any treatment. The gap between rPFS and TTD results in patients accruing benefits while not incurring any treatment costs. Clinical expert input indicated that TTD should be better aligned with time to radiological progression among patients treated with talazoparib plus enzalutamide, as patients were not expected to remain progression free while not on treatment. Additionally, only a small percentage of patients are expected to remain without receiving any treatment. Clinical expert input indicated that the long-term extrapolation proposed by the sponsor lacked face validity considering the discrepancy generated between TTD and rPFS extrapolations. Furthermore, clinical expert input remarked that if it is assumed that rPFS is durable over time, then it should also be reasonable to assume that time on treatment is maintained for a comparable period of time. Because the sponsor estimated that 0.01% of patients receiving talazoparib plus enzalutamide remain radiologically progression free at the end of the model’s 20-year lifetime horizon, a TTD of 0.1% at the same time point was deemed more clinically plausible than patients remaining 3 years in progression free without any treatment. In comparison with enzalutamide monotherapy, the gap between TTD and rPFS of patients receiving talazoparib plus enzalutamide is much wider, underestimating the treatment costs for talazoparib plus enzalutamide, and resulting in bias that favour talazoparib plus enzalutamide.
CDA-AMC conducted a scenario analysis where TTD was set to be equal to rPFS for all treatments, which based on clinical expert input is more reflective of what is observed in clinical practice.
Use of relative dose intensity (RDI) underestimated drug acquisition costs. In the sponsor’s base-case analysis, the mean RDI observed in the TALAPRO-2 trial and literature were used to derive the drug acquisition cost for all therapies (i.e., expected versus observed doses; talazoparib plus enzalutamide = 81%, enzalutamide = 100%, olaparib = 98.2%, niraparib = 99.4%). The inclusion of RDI may underestimate the total cost of talazoparib in real-world clinical practice as the dose received by patients may be different from the planned dose for several reasons (i.e., missed, delayed, or de-escalated doses). CDA-AMC notes that, when considering wastage, each reason determining a reduction in RDI may have a different impact on drug costs. Likewise, it is unclear how treatment discontinuation influences RDI. Furthermore, for oral therapies, pharmacies in Canada are likely to fill and dispense prescriptions in full. Without evidence to suggest that patients will delay filling prescriptions, it is not certain that unused tablets will result in lower drug costs. Additionally, the sponsor did not provide justification as to why compliance in the real-world setting would be expected to be different across treatments included in the model.
In the CDA-AMC base case, RDI was assumed to be 100% for all treatments.
Exclusion of potentially relevant comparators. The sponsor did not include chemotherapy (e.g., docetaxel) or radium-223 as relevant comparators from the economic analysis. Clinical expert input sought by CDA-AMC for this review noted that there have been changes in the treatment landscape for prostate cancer patients since the TALAPRO-2 trial was conducted). Clinical expert input obtained by CDA-AMC indicated that in many treatment settings in Canada, patients with HRR gene-mutated metastatic castration-sensitive prostate cancer are receiving treatment intensification with ARPIs (including AAP and enzalutamide) earlier in the disease management (i.e., during the castration-sensitive phase). As a result, it is expected that patients who progress to mCRPC may have already received ARPI treatment and were unlikely to be treated with them again. Clinical expert input sought by CDA-AMC for this review noted that docetaxel is indicated in combination with prednisone or prednisolone for the treatment of patients with androgen-independent (hormone-refractory) mCRPC, and radium-223 is indicated for patients with mCRPC with symptomatic bone metastases and no known visceral metastatic disease; both treatments were considered relevant comparator for the current review of talazoparib plus enzalutamide. No direct or indirect evidence was provided to inform the comparative effectiveness of these treatments in the shared place in therapy for HRR gene-mutated mCRPC. Therefore, the cost-effectiveness of talazoparib plus enzalutamide in relation to docetaxel and radium-223 is unknown.
CDA-AMC was unable to address this limitation given lack of clinical comparatives effectiveness data between talazoparib plus enzalutamide and docetaxel or radium-223.
The model structure does not reflect clinical practice. The model submitted by the sponsor only accounted for 1 line of subsequent treatment after first-line therapy and assumed that patients would remain in palliative care after the failure of second-line treatment. For patients receiving talazoparib plus enzalutamide as first-line treatment, this assumption resulted in patients spending 30% of their time in the progressed disease state receiving subsequent treatment, and the remaining time (70%) in palliative treatment. Feedback from clinical experts consulted by CDA-AMC indicated that patients with HRR gene-mutated mCRPC are generally treated with an average of 3 lines of therapy in clinical practice. Patients are expected to spend most of their time on subsequent lines of treatment, instead of palliative care.
Additionally, CDA-AMC noted a coding error in the subsequent treatment costs for olaparib plus AAP and niraparib-abiraterone plus prednisone resulting in those treatments having zero subsequent treatment costs.
CDA-AMC was unable to address issues related to the model structure. CDA-AMC fixed the coding error to reflect the actual weighted average cost of second-line treatment for olaparib plus AAP and niraparib-abiraterone plus prednisone.
CDA-AMC Reanalysis of the Economic Evaluation
The CDA-AMC base case was derived by making changes in model parameter values and assumptions, in consultation with clinical experts (refer to Table 8). The impact of these changes, individually and collectively, is presented in Table 9. Due to high uncertainty with the MAIC results, Table 9 and Table 11 will be presented for talazoparib plus enzalutamide versus enzalutamide only, while Table 10 will include all comparators.
Table 8: Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
1. Treatment effects of talazoparib sustained indefinitely | Treatment effect sustained for the remainder of the time horizon | Waning effect gradually over 12 months after trial (42 months for rPFS and 64 months for OS) |
2. Inclusion of RDI | Derived from the TALAPRO-2 trial and literature | Assumed to be 100% for all therapies |
3. Subsequent treatment costs for OLA + AAP and NIRA-ABI | $0 | Fixed to reflect the weighted average cost of second-line treatment for OLA + AAP and NIRA-ABI |
CDA-AMC base case (health care payer perspective) | ― | Reanalysis 1 + 2 + 3 |
AAP = abiraterone acetate plus prednisone; ABI = abiraterone acetate; CDA-AMC = Canada’s Drug Agency; NIRA = niraparib; OLA = olaparib; OS = overall survival; RDI = relative drug intensity; rPFS = radiographic progression-free survival.
Note: CDA-AMC was unable to resolve the issues with uncertain cost-effectiveness of TALA + ENZA in later lines of therapy, lack of clinical plausibility of rPFS estimates, uncertainty in comparative effectiveness between TALA + ENZA and OLA + AAP, NIRA-ABI, and AAP, and exclusion of docetaxel as comparator.
Table 9: Summary of the Stepped Analysis
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
Sponsor base case | ENZA | 104,688 | 2.02 | Reference |
TALA + ENZA | 254,852 | 2.95 | 161,523 | |
CDA-AMC reanalysis 1 | ENZA | 104,688 | 2.02 | Reference |
TALA + ENZA | 255,477 | 2.78 | 198,717 | |
CDA-AMC reanalysis 2 | ENZA | 105,091 | 2.02 | Reference |
TALA + ENZA | 284,064 | 2.95 | 192,511 | |
CDA-AMC reanalysis 3 | ENZA | 104,688 | 2.02 | Reference |
TALA + ENZA | 254,852 | 2.95 | 161,523 | |
CDA-AMC base case Reanalysis 1 + 2 + 3 (deterministic) | ENZA | 105,091 | 2.02 | Reference |
TALA + ENZA | 283,051 | 2.78 | 236,619 | |
CDA-AMC base case Reanalysis 1 + 2 + 3 (probabilistic) | ENZA | 106,076 | 2.04 | Reference |
TALA + ENZA | 289,294 | 2.79 | 242,571 |
CDA-AMC = Canada’s Drug Agency; ENZA = enzalutamide; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; TALA = talazoparib.
Note: The CDA-AMC reanalysis is based on the publicly available prices of the comparator treatments.
Table 10: Disaggregated Results of the CDA-AMC Base Case
Parameter | TALA + ENZA | ENZA | OLA + AAP | NIRA-ABI | AAP |
|---|---|---|---|---|---|
Discounted LYs | |||||
Total | 3.83 | 2.96 | 3.82 | 2.77 | 2.57 |
By health state | |||||
Progression free | 2.58 | 1.55 | 2.39 | 2.02 | 1.40 |
Progressed (second-line) | 0.47 | 0.49 | 0.39 | 0.32 | 0.44 |
Progressed (palliative) | 0.78 | 0.92 | 1.03 | 0.43 | 0.73 |
Discounted QALYs | |||||
Total | 2.79 | 2.04 | 2.72 | 2.06 | 1.79 |
By health state | |||||
Progression free | 2.10 | 1.26 | 1.95 | 1.64 | 1.14 |
Progressed (second-line) | 0.31 | 0.32 | 0.26 | 0.21 | 0.29 |
Progressed (palliative) | 0.39 | 0.46 | 0.51 | 0.21 | 0.36 |
AEs | −0.008 | −0.005 | −0.005 | −0.007 | −0.006 |
Discounted costs ($) | |||||
Total | 289,294 | 106,076 | 279,933 | 222,084 | 59,494 |
Progression free | |||||
Drug acquisition | 240,042 | 57,556 | 227,307 | 186,563 | 13,512 |
Drug monitoring | 2,446 | 1,549 | 2,417 | 2,102 | 2,487 |
AE management | 1,167 | 210 | 360 | 842 | 336 |
Disease monitoring | 10,295 | 6,187 | 9,549 | 8,049 | 5,609 |
Progressed disease | |||||
Drug acquisition | 14,064 | 16,274 | 12,547 | 10,294 | 17,760 |
Drug monitoring | 540 | 567 | 1,878 | 1,673 | 372 |
AE management | 318 | 343 | 380 | 392 | 400 |
Disease monitoring | 1,878 | 1,964 | 1,575 | 1,292 | 1,761 |
Palliative care | 16,856 | 19,715 | 22,232 | 9,161 | 15,536 |
End of life | 1,688 | 1,711 | 1,688 | 1,716 | 1,721 |
AAP = abiraterone acetate plus prednisone; ABI = abiraterone; AE = adverse event; ABI = abiraterone acetate; CDA-AMC = Canada’s Drug Agency; ENZA = enzalutamide; LY = life-year; NIRA = niraparib; OLA = olaparib; QALY = quality-adjusted life-year; TALA = talazoparib.
Note: The model results for OLA + AAP, NIRA-ABI, and AAP are uncertain, as the evidence informing these treatments is associated with substantial uncertainty.
Price Reduction Analysis
CDA-AMC conducted price reduction analyses using the sponsor’s base case and the CDA-AMC base case (refer to Table 11).
Table 11: Results of the Price Reduction Analysis for Talazoparib
Price reduction | Unit drug cost ($) | Cost per 28 days ($) | ICERs for TALA + ENZA vs. ENZA ($/QALY) | |
|---|---|---|---|---|
Sponsor base case | CDA-AMC base case | |||
No price reduction | 99.35a | 5,564 | 168,095 | 242,571 |
10% | 89.42 | 5,007 | 154,727 | 221,837 |
20% | 79.48 | 4,451 | 141,349 | 201,131 |
30% | 69.55 | 3,895 | 127,971 | 180,424 |
40% | 59.61 | 3,338 | 114,592 | 159,717 |
50% | 49.68 | 2,782 | 101,214 | 139,010 |
60% | 39.74 | 2,225 | 87,836 | 118,303 |
70% | 29.81 | 1,669 | 74,457 | 97,596 |
80% | 19.87 | 1,113 | 61,079 | 76,889 |
90% | 9.94 | 556 | 47,701 | 56,182 |
CDA-AMC = Canada’s Drug Agency; ENZA = enzalutamide; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; TALA = talazoparib; vs. = versus.
aSponsor’s submitted price for talazoparib.3
Assessment of Uncertainty
CDA-AMC used the CDA-AMC base case to conduct scenario analyses to address uncertainty within the economic evaluation. The results are provided in Table 12.
Talazoparib plus enzalutamide treatment effect was assumed to be sustained indefinitely throughout the entire time horizon (10 years).
Talazoparib plus enzalutamide treatment effect was assumed to wane immediately after the trial (42 months for rPFS and 64 months for OS).
TTD was assumed to be equal to progression-free survival for talazoparib plus enzalutamide and comparators.
Table 12: Results of CDA-AMC Scenario Analyses
Analysisa | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
CDA-AMC base case | ENZA | 105,091 | 2.02 | Reference |
TALA + ENZA | 284,641 | 2.78 | 236,619 | |
CDA-AMC scenario 1: Treatment effect sustained indefinitely | ENZA | 105,091 | 2.02 | Reference |
TALA + ENZA | 284,641 | 2.95 | 192,511 | |
CDA-AMC scenario 2: Treatment effect wanes after trial | ENZA | 105,091 | 2.02 | Reference |
TALA + ENZA | 284,641 | 2.74 | 247,744 | |
CDA-AMC scenario 3: TTD equal to PFS | ENZA | 113,432 | 2.02 | Reference |
TALA + ENZA | 340,044 | 2.78 | 298,639 |
CDA-AMC = Canada’s Drug Agency; ENZA = enzalutamide; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; TALA = talazoparib; TTD = time to treatment discontinuation.
aDeterministic analyses.
Issues for Consideration
Talazoparib plus enzalutamide is indicated for the treatment of adult patients with HRR gene mutations. HRR gene mutations need to be confirmed before talazoparib plus enzalutamide treatment is initiated. The clinical experts described the availability of HRR mutation testing as very heterogeneous within and between jurisdictions. While next generation sequencing panels for mCRPC are generally available across Canada, there is variability in the genes included in each panel. There are also concerns regarding large backlogs in the hereditary cancer clinics for germline genomic testing based on clinical expert input sought by CDA-AMC for this review. If talazoparib is reimbursed, jurisdictions may need to add more genes to existing panels. It is unclear how or if this would impact human and other health care resources. To provide consistent service across the country, upscaling existing testing infrastructure, workflows, and technologies may be needed. In addition, there may be a need for formal education strategies directed toward health care workers, such as primary care physicians, medical oncologists, and geneticists, to increase awareness and knowledge regarding precision medicine options and genomic testing in patients with mCRPC. However, according to clinical expert input, the structure needed for genomic testing already exists, and no to minimal additional impact is anticipated if talazoparib is reimbursed. The sponsor submitted a scenario analysis that suggested incorporation of genetic testing considerations (current cost of next generation sequencing and number of patients needed to test to identify 1 individual with HRR mutation) would have a minimal impact on the ICER was considered. The impact of the potential need for upscaling testing capacity or expanding testing panels were not considered in the cost-effectiveness analysis.
CDA-AMC noted that olaparib plus AAP and niraparib-abiraterone plus prednisone are both indicated to be used in first-line treatment of patients with deleterious or suspected deleterious germline and/or somatic BRCA-mutated mCRPC, which represent only between 30% and 40% of all HRR mutations in this population based on the TALAPRO-2 trial. Available evidence suggests patients with HRR BRCA mutations may have worse treatment outcomes in comparison with patients with non-BRCA HRR gene mutations.18
The sponsor indicated that the smallest dispensable unit is per capsule, which indicates that the original package does not need to be maintained. The product monograph for talazoparib states that it should be maintained in the original bottle to protect the capsules from light. If individual capsules are not dispensed, the total costs and wastage associated with talazoparib are underestimated in the analysis, and the cost-effectiveness is underestimated.
The flat pricing of talazoparib ($99.35 for 0.25 mg, and 0.1 mg presentation forms) may be an issue for consideration if there are supply chain issues to deliver the standard dose presentation in Canada. This can lead to a significant cost increase when the same treatment dose needs to be dispensed using lower-dose tablets. For example, a shortage in the 0.25 mg tablet could lead to the need to dispense 5 tablets of 0.1 mg instead of 2 tablets of 0.25 mg, increasing the drug costs by 2 and a half times.
Appendix 5: Budget Impact Analysis
Please note that this appendix has not been copy-edited.
Summary of the Submitted BIA
The sponsor submitted a BIA that estimated the expected incremental budgetary impact of reimbursing talazoparib for the treatment of adult patients with HRR gene-mutated mCRPC.
The BIA was conducted from the perspective of public drug plan payers over a 3-year time horizon (2026 to 2028), with 2025 as the base year. The sponsor’s estimate reflects the aggregated results from the jurisdictional provincial budgets (excluding Quebec) as well as the Non-Insured Health Benefits Program. The sponsor estimated the eligible population using an epidemiological approach incorporating incident (i.e., new) patients only. It was assumed that the uptake of talazoparib would displace all the other comparators by the same proportion (10%), compared to a scenario without reimbursement of talazoparib. The sponsor’s base case included drug acquisition costs. The market uptake for talazoparib was estimated using the sponsor’s internal market research and interviews with clinicians in Canada. The key inputs to the BIA are documented in Table 13. The sponsor assumed no patients died or switched therapies within the BIA.
The sponsor estimated the 3-year incremental budget impact associated with reimbursing talazoparib for the treatment of adult patients with HRR gene-mutated mCRPC would be $20,002,109 (year 1 = $1,971,265; year 2 = $6,231,893; year 3 = $11,798,951).
Table 13: Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3 if appropriate) |
|---|---|
Target population | |
Incidence of disease | 119.7 per 100,000 males19 |
Incident patients with PC in 2025 | 20,031a |
Proportion of patients with PC progressing to CRPC | 15%20 |
Proportion of patients with CRPC with mCRPC | 84%20 |
Proportion of patients with mCRPC and HRR mutations | 21%21 |
Proportion of patients eligible for treatment with TALA | 94.7% |
Proportion of patients exposed to ENZA, OLA, or NIRA (derived through the following steps) | 5.3% |
Proportion of patients who were ARAT naive | 86%b |
Percentage of patients requiring subsequent therapy | 28.5%22 |
Percentage of patients requiring subsequent therapy eligible for treatment with TALA in 2L+ | 6%23 |
Proportion of patients exposed to ABI | 4.0%b |
Proportion of patients exposed to chemotherapy | 3.0%b |
Percentage of patients covered by public drug plan | NL, PEI, NS = 41%; NB = 41%; ON = 64%; MB = 87%; SK, AB = 88%; BC = 83%; NIHB = 70% |
Number of patients eligible for drug under review | 365 / 373 / 381 |
Market shares (reference scenario) | |
TALA + ENZA | 0% / 0% / 0% |
ENZA | 40.0% / 33.0% / 31.0% |
AAP | 35.0% / 29.0% / 25.0% |
OLA + AAP | 15.0% / 20.0% / 22.0% |
NIRA-ABI | 10.0% / 18.0% / 22.0% |
Market shares (new drug scenario) | |
TALA + ENZA | 10.0% / 20.0% / 30.0% |
ENZA | 36.0% / 26.4% / 21.7% |
AAP | 31.5% / 23.2% / 17.5% |
OLA + AAP | 13.5% / 16.0% / 15.4% |
NIRA-ABI | 9.0% / 14.4% / 15.4% |
Cost of treatment (per patient per day) | |
TALA + ENZAc | $271.77 |
ENZA | $116.78 |
AAP | $30.67 |
OLA + AAP | $248.91 |
NIRA-ABId | $291.30 |
2L = second line; AAP = abiraterone acetate plus prednisone; AB = Alberta; ABI = abiraterone acetate plus prednisone; ARAT = androgen receptor axis-targeted therapy; BC = British Columbia; CRPC = castration-resistant prostate cancer; ENZA = enzalutamide; HRR = homologous recombination repair; MB = Manitoba; mCRPC = metastatic castration-resistant prostate cancer; NB = New Brunswick; NIHB = Non-Insured Health Benefit; NIRA = niraparib; NL = Newfoundland and Labrador; NS = Nova Scotia; OLA = olaparib; ON = Ontario; PC = prostate cancer; PEI = Prince Edward Island; SK = Saskatchewan; TALA = talazoparib.
aCalculated based on the population of Canada excluding Quebec, incorporating an incidence of 119.7 per 100,000.19
bSource: Clinicians in Canada.
cSponsor included a RDI of 78%. No source was provided for this assumption and it did not align with the RDI in the cost-utility analysis.
dSponsor included a RDI of 99%. No source was provided for this assumption and it did not align with the RDI in the cost-utility analysis.
Note: Time on treatment (in months) for the treatments were assumed as follows: TALA + ENZA = 30.7; ENZA = 12.3; AAP = 16.5; OLA + AAP = 27.6; NIRA-ABI = 16.7
Key Issues of the Submitted BIA
CDA-AMC identified several key issues to the sponsor’s analysis that have notable implications on the results of the BIA:
The eligible population is uncertain: Clinical experts consulted by CDA-AMC noted changes in the treatment landscape for patients with prostate cancer in recent years. In many treatment settings in Canada, patients with metastatic castration-sensitive prostate cancer are receiving treatment intensification with ARPIs including abiraterone acetate and enzalutamide. As a result, most patients who progress to mCRPC have already received ARPI treatment and are unlikely to be treated with them again. Clinical experts consulted by CDA-AMC indicated that approximately 40% of patients might be deemed ineligible for talazoparib plus enzalutamide due to prior exposure to enzalutamide, niraparib, or olaparib.
CDA-AMC assumed that approximately 61.7% of patients with mCRPC would be considered eligible for talazoparib plus enzalutamide based on the sponsor’s model structure (e.g., ARPI naive: 10%, abiraterone exposed: 40%; chemotherapy exposed: 10%; subsequent treatment: 1.7%).
Assumption that existing (prevalent) patients would not be eligible for treatment with talazoparib plus enzalutamide does not align with clinical expectations: The sponsor used the incidence of prostate cancer to calculate the population size and assumed that by the time of reimbursement, existing (prevalent) patients with HRR gene-mutated mCRPC would not be eligible for treatment with talazoparib plus enzalutamide as they would likely have had exposure to enzalutamide, niraparib-abiraterone plus prednisone, or olaparib plus AAP by that time. However, the sponsor also assumed that 1.7% of new patients would be eligible for talazoparib plus enzalutamide as second-line or later-line treatment, which indicates misalignment in their assumptions. Clinical experts consulted for this review confirmed that the proportion of existing patients that would likely be eligible for treatment with talazoparib plus enzalutamide is likely to be small.
CDA-AMC undertook a scenario analysis using an alternate approach that incorporated prevalence to explore possible implications of this limitation. Estimates of eligible population were obtained by applying the proportion of patients with mCRPC obtained from literature (1.65%)24 to the prevalence of patients with prostate cancer (993.66 per 100,000).25
Underestimated public drug coverage: The sponsor used the percentage of public coverage from a Patented Medicine Prices Review Board report on oncology medicine funding for take home medications26 as a proxy for public drug coverage for all CDA-AMC-participating jurisdictions. Given that the majority of patients eligible for treatment with talazoparib plus enzalutamide are aged 65 years or older, the treatment will be covered by most of CDA-AMC-participating drug plans.
CDA-AMC estimated that the public coverage for eligible patients living in CDA-AMC-participating drug plans would be 100%, except for the provinces of Newfoundland and Labrador (50.8%) and New Brunswick (84.9%).27
Exclusion of potentially relevant comparators to the indicated population: The sponsor did not include docetaxel and radium-223 as relevant comparators in the BIA. Clinical expert input sought by CDA-AMC for this review noted that chemotherapy (docetaxel) and radium-223 are indicated for first and subsequent lines of treatment of patients with HRR gene-mutated mCRPC, and that clinical practice is changing such that chemotherapy is more commonly being used as a first-line treatment for patients with HRR gene-mutated mCRPC, given patients are likely to have prior exposure to an ARPI and/or PARP inhibitor. Therefore, they are potentially relevant comparators to the reimbursement request population, which represents a gap in the available direct evidence given the shared place in therapy for HRR gene-mutated mCRPC. Therefore, the budget impact of talazoparib plus enzalutamide in relation to docetaxel or radium-223 is unknown.
CDA-AMC was unable to address this limitation. A supplementary cost comparison table with treatment costs of docetaxel and radium-223 can be found in Appendix 1. Docetaxel and radium-223 are expected to have per-patient costs of $2,402 and $33,840 per 28-day cycle, respectively.
Use of RDI underestimated drug acquisition costs: The sponsor’s BIA incorporated RDIs into talazoparib and comparators based on data from the TALAPRO-228 and the MAGNITUDE29 trials. This consideration of RDI is problematic as this parameter can be influenced by several factors. The dose received by a patient may differ from the full planned dose of the drug due to dose delays, missed doses, dose reductions to manage toxicity, or subsequent dose re-escalation; each of these have differing impacts on drug costs.
Furthermore, prescriptions for talazoparib may be filled and reimbursed regardless of treatment adherence.
In the CDA-AMC reanalysis, RDI for talazoparib and comparators was set to be 100%.
TTD is uncertain: The sponsor noted that treatment duration estimates for talazoparib plus enzalutamide and enzalutamide were informed by the TALAPRO-2 trial, while treatment duration for AAP,30 olaparib plus AAP,31 and niraparib-abiraterone plus prednisone32 were informed by the literature. TTD from the TALAPRO-2 trial was noted to be associated with uncertainty in Appendix 4. Furthermore, given that the indirect treatment comparisons conducted by the sponsor were associated with limitations, which preclude CDA-AMC from reaching conclusions regarding comparative effect, there is no robust evidence to suggest that talazoparib plus enzalutamide is more effective than niraparib-abiraterone plus prednisone, or olaparib plus AAP. The clinical experts consulted for the review indicated that they would expect the treatment duration for enzalutamide and AAP to be similar, and treatment duration of talazoparib plus enzalutamide, olaparib plus AAP, and niraparib-abiraterone plus prednisone to be similar.
CDA-AMC undertook a scenario analysis assuming that treatment duration for olaparib plus AAP and niraparib-abiraterone plus prednisone would align with talazoparib plus enzalutamide (30.7 months), and that AAP would align with enzalutamide (12.3 months).
The price of drugs paid by public drug plans is uncertain: Both the sponsor’s analysis and the analysis by CDA-AMC are based on publicly available list prices for all comparators. The actual costs paid by public drug plans are unknown.
CDA-AMC was unable to address this limitation in reanalysis.
The market share estimates in the reference scenario do not align with clinical expectations: In the sponsor’s submitted BIA, the market shares in the reference scenario were derived from interviews with clinicians in Canada. CDA-AMC obtained clinical expert feedback that suggested the distribution of market shares in the current treatment landscape is likely different, and dependent on 2 subgroups of patients with HRR gene-mutated mCRPC. Patients with non-BRCA mutations are eligible for enzalutamide and AAP, while those who have mutations on BRCA genes are also eligible for olaparib plus AAP and niraparib-abiraterone plus prednisone. Clinical experts did not believe there would be a change in market shares over the next 3 years other than the possible reimbursement of talazoparib plus enzalutamide. Clinical experts provided 2 possible scenarios for market shares among patients with BRCA mutations, without reimbursement of talazoparib plus enzalutamide: 1 in which 50% of the market share are ARPIs (AAP or enzalutamide), 15% are PARP inhibitors (olaparib or niraparib), and 35% are other treatments (predominantly chemotherapy) (scenario 2), and another in which 10% are ARPIs, 15% are PARP inhibitors, and 75% are other treatments (predominantly chemotherapy) (scenario 3). Given that the sponsor model did not stratify their BIA into patients with HRR non-BRCA mutations and patients with HRR BRCA mutations, the market shares estimates obtained by CDA-AMC were weighted to exclude other treatments that were not incorporated in the model, and then combined into 1, considering the proportion of each subgroup among the population with HRR gene-mutated mCRPC. Based on data from the TALAPRO-2 trial,28 the proportion of patients with BRCA1 or BRCA2 was 39.6% of the group with HRR gene-mutated mCRPC.
CDA-AMC addressed this limitation by conducting scenario analyses (scenarios 2 and 3) that revised the market shares of the treatment(s) in the reference scenario to reflect the distribution of treatments expected based on clinical expert feedback.
The market uptake of talazoparib is overestimated: The sponsor’s submitted BIA indicated that talazoparib would result in a market uptake of 10.0% in year 1, 20.0% in year 2, and 30.0% in year 3 based on the sponsor’s internal market research and interviews with clinicians in Canada. However, CDA-AMC obtained clinical expert feedback indicating that the market uptake of 30% in year 3 is reasonable only among the subgroup of patients with non-BRCA mutations, and that for patients with BRCA mutations the uptake of talazoparib plus enzalutamide would be lower, around 5% or 12%, given that patients with non-BRCA mutations are eligible for enzalutamide and AAP only, while those who have mutations on BRCA genes are also eligible for olaparib and niraparib. As previously noted, the sponsor did not explicitly consider these 2 subgroups in the model. When taking into account the different uptake in these subgroups, talazoparib plus enzalutamide would have 20% or 23% of the market shares in year 3.
To address this limitation, CDA-AMC undertook scenario analyses by revising the market shares for talazoparib in the new drug scenario to 20% (scenario 2) or 23% (scenario 3) in year 3. For patients with BRCA mutations, in scenario 2 talazoparib plus enzalutamide was estimated to have a market share of 5% in year 3, displacing both PARP inhibitors (olaparib and niraparib). In scenario 3, talazoparib plus enzalutamide was estimated to have a market share of 12% in year 3, displacing market shares from olaparib only. Among patients with non-BRCA mutations, it was assumed for both scenarios that talazoparib plus enzalutamide would have a market share of 30% in year 3, displacing 20% from enzalutamide and 10% from AAP.
The Non-Insured Health Benefit population was inappropriately calculated: The sponsor calculated the total population of CDA-AMC-participating drug plans by adding the population of the provinces, excluding Quebec, to the population of clients of the Non-Insured Health Benefit. Clients of the Non-Insured Health Benefit living within the borders of a province are counted within provincial population data as reported by Statistics Canada, thus the Non-Insured Health Benefit population was double counted in the sponsor’s analysis. Additionally, the provinces of Saskatchewan and Alberta fund all oncology products for patients residing within their borders, including those who would otherwise be reimbursed by the Non-Insured Health Benefit, as does Ontario for patients aged younger than 25 years or 65 years and older. Clients of the Non-Insured Health Benefit of the appropriate ages who are living within the borders of these 3 jurisdictions should therefore be considered clients of the provincial plan for the purposes of modelling the budget impact of reimbursing talazoparib.
CDA-AMC did not adjust for this limitation in reanalysis. The impact on pan-Canadian model results is expected to be minimal.
CDA-AMC Reanalyses of the BIA
CDA-AMC revised the sponsor’s submitted analyses by making changes in model parameter values and assumptions, in consultation with clinical experts, as outlined in Table 14.
Table 14: Revisions to the Submitted BIA
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
1. Proportion of patients with HRR gene-mutated mCRPC eligible for TALA | 94.7% of patients with HRR gene-mutated mCRPC clinically eligible for treatment with TALA ARPI naive = 86% Abiraterone exposed = 4% Chemotherapy exposed = 3% Subsequent treatment: 1.7% | 61.7% of patients with HRR gene-mutated mCRPC clinically eligible for treatment with TALA: ARPI naive = 10% Abiraterone exposed = 40% Chemotherapy exposed = 10% Subsequent treatment: 1.7% |
2. RDI for talazoparib and comparators | TALA, OLA = 78% NIRA = 99% | All drugs = 100% |
3. Public coverage | NL, PEI, NS, NB = 41% ON = 64% MB = 87% SK, AB = 88% BC = 83% NIHB = 70% | 100% for all provinces/plans, except: NL = 50.8% NB = 84.9% |
CDA-AMC base case | ― | Reanalysis 1 + 2 + 3 |
AB = Alberta ; ARPI = androgen receptor pathway inhibitor; BC = British Columbia; BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency; ENZA = enzalutamide; HRR = homologous recombination repair; MB = Manitoba; mCRPC = metastatic castration-resistant prostate cancer; NB = New Brunswick; NIHB = Non-Insured Health Benefit; NIRA = niraparib; NL = Newfoundland and Labrador; NS = Nova Scotia; OLA = olaparib; RDI = relative dose intensity; ON = Ontario; PEI = Prince Edward Island; SK = Saskatchewan; TALA = talazoparib.
Note: CDA-AMC was unable to resolve the issues with the calculation of the NIHB population.
The results of the CDA-AMC step-wise reanalysis are presented in summary format in Table 15 and a more detailed breakdown is presented in Table 16. In the CDA-AMC base case, the 3-year budget impact of reimbursing talazoparib plus enzalutamide for the treatment of adult patients with HRR gene-mutated mCRPC was $21,840,671 (year 1 = $2,196,847; year 2 = $6,817,135; year 3 = $12,826,688).
CDA-AMC used the CDA-AMC base case to conduct scenario analyses to explore uncertainty in the estimated budget impact of reimbursing talazoparib plus enzalutamide. The results are provided in Table 16.
Table 15: Summary of the Stepped Analysis of the CDA-AMC Base Case
Stepped analysis | Three-year total ($) |
|---|---|
Submitted base case | $20,002,109 |
CDA-AMC reanalysis 1 | $13,031,997 |
CDA-AMC reanalysis 2 | $24,128,592 |
CDA-AMC reanalysis 3 | $27,789,087 |
CDA-AMC base case: Reanalysis 1 + 2 + 3 | $21,840,671 |
CDA-AMC = Canada’s Drug Agency.
Note: The CDA-AMC reanalysis is based on publicly available prices of the comparator treatments
Table 16: Disaggregated Summary of the BIA
Stepped analysis | Scenario | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|---|
Submitted base case | Reference total | $12,347,517 | $20,567,915 | $28,554,159 | $34,304,767 | $83,426,842 |
TALA + ENZA | 0 | 0 | 0 | 0 | 0 | |
All other comparators | $12,347,517 | $20,567,915 | $28,554,159 | $34,304,767 | $83,426,842 | |
New drug total | $12,347,517 | $22,539,180 | $34,786,052 | $46,103,718 | $103,428,950 | |
TALA + ENZA | 0 | $3,623,331 | $11,028,842 | $20,775,077 | $35,427,251 | |
All other comparators | $12,347,517 | $18,915,849 | $23,757,210 | $25,328,641 | $68,001,699 | |
Budget Impact | 0 | $1,971,265 | $6,231,893 | $11,798,951 | $20,002,109 | |
CDA-AMC base case | Reference total | $11,902,443 | $20,493,156 | $28,776,090 | $34,718,350 | $83,987,596 |
TALA + ENZA | 0 | 0 | 0 | 0 | 0 | |
All other comparators | $11,902,443 | $20,493,156 | $28,776,090 | $34,718,350 | $83,987,596 | |
New drug total | $11,902,443 | $22,690,004 | $35,593,224 | $47,545,039 | $105,828,267 | |
TALA + ENZA | $0 | $3,807,189 | $11,588,754 | $21,830,374 | $37,226,316 | |
All other comparators | $11,902,443 | $18,882,815 | $24,004,471 | $25,714,665 | $68,601,951 | |
Budget Impact | $0 | $2,196,847 | $6,817,135 | $12,826,688 | $21,840,671 | |
CDA-AMC scenario analyses | ||||||
Scenario 1: Existing patients with HRR gene-mutated mCRPC eligible for treatment with TALA (Prevalence model) | Reference total | $12,938,775 | $22,277,472 | $31,281,591 | $37,741,237 | $91,300,300 |
New drug total | $12,938,775 | $24,665,596 | $38,692,286 | $51,684,731 | $115,042,613 | |
Budget Impact | $0 | $2,388,125 | $7,410,695 | $13,943,494 | $23,742,313 | |
Scenario 2: Updated market shares and TALA + ENZA market uptake | Reference total | $10,193,490 | $13,899,651 | $14,866,140 | $15,187,853 | $43,953,644 |
New drug total | $10,193,490 | $15,562,064 | $20,614,089 | $26,519,306 | $62,695,460 | |
Budget Impact | $0 | $1,662,413 | $5,747,949 | $11,331,453 | $18,741,816 | |
Scenario 3: Updated market shares and TALA + ENZA market uptake | Reference total | $14,568,210 | $21,632,296 | $23,429,758 | $23,937,671 | $68,999,726 |
New drug total | $14,568,210 | $23,290,982 | $29,068,355 | $35,203,638 | $87,562,974 | |
Budget Impact | $0 | $1,658,685 | $5,638,596 | $11,265,967 | $18,563,249 | |
Scenario 4: Alternative treatment durations for OLA + AAP, NIRA-ABI, and ABI | Reference total | $11,902,443 | $19,973,711 | $31,429,457 | $41,742,441 | $93,145,609 |
New drug total | $11,902,443 | $22,170,558 | $38,075,917 | $53,507,751 | $113,754,226 | |
Budget Impact | $0 | $2,196,847 | $6,646,460 | $11,765,310 | $20,608,618 | |
AAP = abiraterone acetate plus prednisone; ABI = abiraterone acetate; BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency ENZA = enzalutamide; HRR = homologous recombination repair; mCRPC = metastatic castration-resistant prostate cancer; NIRA = niraparib; OLA = olaparib; TALA = talazoparib.
Note: The CDA-AMC reanalysis is based on the publicly available prices of the comparator treatments.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.