Drugs, Health Technologies, Health Systems
Reimbursement Review
Durvalumab (Imfinzi)
Sponsor: AstraZeneca Canada Inc.
Therapeutic area: Muscle invasive bladder cancer
Summary
What Is Muscle Invasive Bladder Cancer?
Muscle invasive bladder cancer (MIBC) is a serious type of bladder cancer in which the tumour has grown into the muscle layer of the bladder. This makes it more likely to spread to other parts of the body.1
About 12,300 new cases of bladder cancer were expected in Canada in 2024, and roughly 25% of these were expected to be MIBC at the time of diagnosis.1,2
What Are the Treatment Goals and Current Treatment Options for MIBC?
Clinician groups noted that the most important treatment goals for MIBC are to stop the cancer from growing or coming back and to help patients maintain their quality of life. Other important outcomes include the cancer no longer being detectable after the patient has received treatment (complete response) and the patient staying free of cancer events (decreasing the risk of recurrence or spread).
The patient groups noted that key goals of treatment are to control the disease and help patients live longer.
Radical cystectomy, hereafter referred to as surgery, is a procedure that typically involves removal of the bladder and prostate gland in males or removal of the bladder, urethra, uterus, and the anterior vaginal wall in females.3,4
Clinician group input stated that standard treatments in Canada include platinum-based chemotherapy administered before (neoadjuvant) or after (adjuvant) surgery and nivolumab administered after surgery.
The clinical experts noted that in academic hospitals in Canada, up to 60% of patients receive chemotherapy before surgery. The choice of chemotherapy is usually gemcitabine-cisplatin, or, more infrequently, dose-dense methotrexate, vinblastine, Adriamycin (doxorubicin), and cisplatin (ddMVAC).
What Is Imfinzi and Why Did Canada’s Drug Agency Conduct This Review?
Imfinzi is a drug that is administered by IV infusion. It is a member of a class of drugs called PD-L1 inhibitors, which help the immune system target and destroy tumour cells.
Health Canada has approved Imfinzi combination with gemcitabine-cisplatin chemotherapy before surgery, followed by Imfinzi alone after surgery, for patients with MIBC.
Canada’s Drug Agency (CDA-AMC) reviewed Imfinzi so that it could make a recommendation to participating public drug programs on whether it should be reimbursed for use in combination with gemcitabine-cisplatin as neoadjuvant treatment, followed by Imfinzi as monotherapy adjuvant treatment after surgery, for the treatment of patients with MIBC.
How Did CDA-AMC Evaluate Imfinzi?
CDA-AMC reviewed clinical evidence on the beneficial and harmful effects, as well as the economic evidence, of Imfinzi compared with other treatments used in Canada for patients with MIBC. Gemcitabine-cisplatin and adjuvant nivolumab were considered to be relevant treatments to compare with Imfinzi when the clinical evidence was reviewed.
CDA-AMC identified equity and ethical considerations relevant to Imfinzi and MIBC.
The review was informed by materials submitted by the sponsor, which included clinical and economic evidence.
The review was also informed by 1 patient group submission and 2 clinician group submissions in response to the CDA-AMC call for input, and by input from participating public drug programs related to issues that may impact their ability to implement a recommendation.
Two clinical oncologists, who represented Ontario, were consulted as part of the review process.
What Were the Findings?
Clinical Evidence
CDA-AMC reviewed the following clinical evidence:
One phase III clinical trial (the NIAGARA study) compared Imfinzi with gemcitabine-cisplatin in 1,063 patients with MIBC. Patients were assigned to 1 of 2 groups:
Those in the Imfinzi group received Imfinzi with chemotherapy (gemcitabine-cisplatin) before surgery, followed by Imfinzi alone after surgery.
Those in the gemcitabine-cisplatin group received gemcitabine-cisplatin before surgery and no study treatment after surgery.
The sponsor’s feasibility assessment evaluated whether available clinical trials of patients with MIBC provided sufficiently comparable evidence to support the conduct of an indirect treatment comparison (ITC).
For the comparison:
Treatment with Imfinzi plus gemcitabine-cisplatin before surgery followed by Imfinzi alone after surgery likely results in a clinically important improvement in pathologic complete response compared with gemcitabine-cisplatin alone before surgery.
Treatment with Imfinzi plus gemcitabine-cisplatin before surgery followed by Imfinzi alone after surgery likely results in a clinically important improvement in event-free survival at 24 months compared with gemcitabine-cisplatin alone before surgery.
Treatment with Imfinzi plus gemcitabine-cisplatin before surgery followed by Imfinzi alone after surgery likely results in a clinically important improvement in overall survival (OS) at 36 months compared with gemcitabine-cisplatin before surgery.
Treatment with Imfinzi plus gemcitabine-cisplatin before surgery followed by Imfinzi alone after surgery may result in little-to-no clinically important difference in health-related quality of life compared with gemcitabine-cisplatin before surgery (measured using European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 [EORTC QLQ-C30] global health status over a median follow-up of 42.3 months). Certainty of the evidence was low, owing to the risk of bias.
In the overall study period, treatment with Imfinzi plus gemcitabine-cisplatin before surgery followed by Imfinzi alone after surgery likely results in an increase in the proportion of patients who experience serious adverse events (SAEs) compared with gemcitabine-cisplatin before surgery. Although there was a slight increase in SAEs observed with Imfinzi plus gemcitabine-cisplatin during the neoadjuvant phase, most of the difference in overall SAEs is attributable to the adjuvant treatment phase, during which patients in the gemcitabine-cisplatin group were not receiving postsurgical therapy.
Based on the NIAGARA study, harms were consistent with the known safety profile of cisplatin-based chemotherapy and PD-L1 inhibitors, with no new safety signals identified.
Other considerations:
Based on the provided evidence, the efficacy and safety of perioperative Imfinzi added to the ddMVAC regimen are unknown.
Based on the provided evidence, the efficacy of perioperative Imfinzi added to gemcitabine-cisplatin compared to neoadjuvant gemcitabine-cisplatin followed by adjuvant nivolumab in patients at high risk is not known because these are different patient populations.
A subset of patients in the NIAGARA study did not undergo surgery. The efficacy and safety of Imfinzi in patients receiving neoadjuvant gemcitabine-cisplatin followed by bladder-sparing therapy are not well understood.
Because the NIAGARA study looked at the effectiveness of the whole treatment process with Imfinzi (both before and after surgery), it is unclear how much benefit comes from each of the Imfinzi presurgery and postsurgery treatment periods.
Economic Evidence
Imfinzi is available as a solution for IV infusion (50 mg/mL).5 At the submitted price of $938.67 per 2.4 mL vial and $3,911.11 per 10 mL vial,6 the cost of Imfinzi is expected to range from $11,733 (adjuvant) to $15,644 (neoadjuvant) per patient per 28-day cycle, based on the Health Canada–recommended dosage.5 The cost of neoadjuvant Imfinzi in combination with gemcitabine-cisplatin is expected to be $17,624 per patient per 28-day course for the first 4 cycles of treatment.
Clinical efficacy in the economic analysis was derived from the NIAGARA trial, which compared neoadjuvant Imfinzi in combination with gemcitabine-cisplatin followed by adjuvant Imfinzi monotherapy after surgery with neoadjuvant gemcitabine-cisplatin.7 Evidence submitted by the sponsor indicates that the perioperative Imfinzi regimen is likely to improve event-free survival and OS compared with neoadjuvant gemcitabine-cisplatin, among patients with MIBC. An ITC was not submitted by the sponsor, as a sponsor-submitted feasibility assessment concluded that a valid ITC could not be conducted; thus, the efficacy and safety of the Imfinzi regimen compared with ddMVAC and adjuvant nivolumab is unknown.6
The results of the CDA-AMC base case suggest:
A perioperative Imfinzi regimen is predicted to be associated with higher costs to the health care system than neoadjuvant gemcitabine-cisplatin (incremental costs = $98,565), primarily driven by increased costs associated with drug acquisition.
A perioperative Imfinzi regimen is predicted to be associated with a gain of 1.83 life-years compared to neoadjuvant gemcitabine-cisplatin, and may result in a gain of 1.51 quality-adjusted life-years (QALYs) compared to gemcitabine-cisplatin.
The incremental cost-effectiveness ratio (ICER) of the perioperative Imfinzi regimen compared to neoadjuvant gemcitabine-cisplatin was $65,194 per QALY gained in the CDA-AMC base case. The estimated ICER was highly sensitive to time-to-event outcome extrapolations. Approximately 92% of the incremental QALYs were estimated based on extrapolations outside the trial period.
Neoadjuvant gemcitabine plus cisplatin was the only comparator included in the cost-utility analysis. The cost-effectiveness of perioperative Imfinzi versus ddMVAC or adjuvant nivolumab is unknown. If decision-makers determine that there are no differences in health between perioperative Imfinzi and ddMVAC or adjuvant nivolumab, then the cost of perioperative Imfinzi should not exceed that of these comparators.
CDA-AMC estimates that the budget impact of reimbursing the perioperative Imfinzi regimen for the treatment of patients with MIBC will be approximately $111 million over the first 3 years of reimbursement, compared to the amount currently spent on comparators, with an estimated expenditure of $170 million on Imfinzi over this period. The actual budget impact of reimbursing the perioperative Imfinzi regimen will depend on the potential continuation of Imfinzi treatment if surgery is refused and the cost of subsequent treatment. The incremental budget impact of reimbursing Imfinzi is predicted to be greater than $40 million in year 3, and the economic feasibility of adoption must be addressed.
Abbreviations
AE
adverse event
BCC
Bladder Cancer Canada
BICR
blinded independent central review
CDA-AMC
Canada’s Drug Agency
CI
confidence interval
CrCl
creatinine clearance
CUA
Canadian Urologic Association
DCO
data cut-off
ddMVAC
dose-dense methotrexate, vinblastine, Adriamycin (doxorubicin), cisplatin
ECOG PS
Eastern Cooperative Oncology Group Performance Status
EFS
event-free survival
EORTC QLQ-C30
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30
GHS
global health status
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HR
hazard ratio
HRQoL
health-related quality of life
HTA
health technology assessment
IA2
second interim analysis
ICER
incremental cost-effectiveness ratio
IDMC
independent data monitoring committee
ITC
indirect treatment comparison
ITT
intention to treat
M
metastasis staging
MIBC
muscle invasive bladder cancer
N
lymph node staging
OH (CCO)
Ontario Health Cancer Care Ontario
OR
odds ratio
OS
overall survival
pCR
pathologic complete response
QALY
quality-adjusted life-year
RCT
randomized controlled trial
RECIST 1.1
Response Evaluation Criteria in Solid Tumours Version 1.1
SAE
serious adverse event
SLR
systematic literature review
T
tumour staging
TMT
trimodal therapy
TURBT
transurethral resection of bladder tumour
UC
urothelial carcinoma
Background
Context for the Review
This is the first review by CDA-AMC for MIBC. The objectives of this report are as follows:
Review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of durvalumab (Imfinzi), 50 mg/mL, concentrate for IV infusion in the treatment of MIBC in adult patients. The focus will be on comparing durvalumab to relevant comparators used in clinical practice in Canada and identifying gaps in the current evidence. The focus of the review is outlined in Table 1.
Review and critically appraise the economic information submitted by the sponsor, including a cost-effectiveness analysis and budget impact analysis. The focus of the Economic Review is aligned with the scope of the Clinical Review, unless otherwise stated. For most reviews, a CDA-AMC base case is developed, informed by clinical expert input, the available clinical evidence, and the best interpretation of the economic evidence based on the information provided by the sponsor.
Table 1: Information on the Application Submitted for Review and on the CDA-AMC Review
Item | Description |
|---|---|
Information on the application submitted for review | |
Drug | Durvalumab (Imfinzi), 50 mg/mL, concentrate for IV infusion |
Sponsor | AstraZeneca Canada Inc. |
Health Canada indication | For the treatment of patients with resectable muscle invasive bladder cancer (MIBC)a in combination with gemcitabine and cisplatin as neoadjuvant treatment, followed by adjuvant Imfinzi monotherapy treatment after radical cystectomy |
Health Canada approval status | NOC |
Health Canada review pathway | Priority review |
NOC date | July 17, 2025 |
Mechanism of action | PD-L1 inhibitor |
Recommended dosage | Weight > 30 kg
Weight ≤ 30 kg
|
Submission type | Initial |
Sponsor’s reimbursement request | Per indication |
Submitted price | Durvalumab: $938.67 per 120 mg/2.4 mL single-use vial for IV infusion Durvalumab: $3,911.11 per 500 mg/10 mL single-use vial for IV infusion |
Information on the CDA-AMC review | |
Review type | Standard |
Clinical review focusb | Population: as defined in the Health Canada indication Subgroups: node-positive disease (N0 vs. N1), tumour stage (T2N0 vs. > T2N0) Intervention: per recommended dosage Comparators:
Outcomes: EFS, pCR, OS, MFS, DSS, proportion of patients who achieve < P2 (stages T0, Ta, T1, and carcinoma in situ) at the time of cystectomy, EFS at 24 months, OS at 5 years, PFS2, DFS, HRQoL, proportion of patients who undergo cystectomy, AEs, SAEs |
AE = adverse event; CDA-AMC = Canada’s Drug Agency; ddMVAC = dose-dense methotrexate, vinblastine, Adriamycin (doxorubicin), cisplatin; DFS = disease-free survival; DSS = disease-specific survival; EFS = event-free survival; HRQoL = health-related quality of life; MFS = metastasis-free survival; MIBC = muscle invasive bladder cancer; N = lymph node staging; NOC = Notice of Compliance; OS = overall survival; P = pathologic staging; pCR = pathologic complete response; PFS2 = time from randomization to subsequent progression or recurrence after an EFS event; q.3.w. = every 3 weeks; q.4.w. = every 4 weeks; SAE = serious adverse event; T = tumour staging; vs. = versus.
aAdjuvant durvalumab would only be available for patients who received neoadjuvant durvalumab plus cisplatin-based chemotherapy, in alignment with the Health Canada indication.
bThe Economic Review aligns with the scope of the Clinical Review, unless otherwise stated.
Sources: NIAGARA Clinical Study Report and sponsor’s Summary of Clinical Evidence.8,9 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Submission History for the Drug Under Review
CDA-AMC previously reviewed durvalumab through the Reimbursement Review process for biliary tract cancer, endometrial cancer that is mismatch repair deficient, extensive-stage small cell lung cancer, unresectable hepatocellular carcinoma, and unresectable non–small cell lung cancer. Each of these indications were issued a recommendation of reimburse with conditions.10-14
Sources of Information
The contents of the Reimbursement Review report are informed by materials submitted by the sponsor, input received from interested parties (patient groups, clinician groups, and drug programs), and input from clinical experts consulted for this review.
Calls for patient group and clinician group input are issued for each Reimbursement Review. One patient group submission from Bladder Cancer Canada (BCC) and 2 clinician group submissions from BCC and Ontario Health Cancer Care Ontario (OH [CCO]), Genitourinary Cancer Drug Advisory Committees were received. The patient group input gathered information for this submission through online surveys conducted by BCC between May 16 and June 20, 2025. The input from BCC consolidated feedback from Association Cancer Vessie France, which is a patient group based in France. The feedback from Association Cancer Vessie France was gathered through an online survey conducted by Association Cancer Vessie France (in French) in March 2025. The survey conducted by BCC received 4 responses, all from respondents who had experience with MIBC. The French survey received 26 responses: 23 from patients with MIBC and 3 from caregivers. For the clinician group input, BCC gathered information through an online survey conducted between May 8 and May 24, 2025. A total of 3 clinicians completed the survey. The OH (CCO) gathered information for this input from 4 clinicians by email. The full submissions received are available on the project landing page in the consolidated input document. The drug programs provide input on each drug being reviewed through the Reimbursement Review process by identifying issues that may impact their ability to implement a recommendation.
Input from patient and clinician groups is considered throughout the review, including in the selection of outcomes included in the Clinical Review and in the interpretation of the clinical and economic evidence. Relevant patient and clinician group input is summarized in the Disease Background, Current Management, and Unmet Needs and Existing Challenges sections.
Each review team includes at least 1 clinical expert with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process. Two medical oncologists with expertise in the diagnosis and management of MIBC, representing Ontario, participated as part of the review team.
Disease Background
Bladder cancer is the most common malignancy of the urinary system and originates in cells lining the interior of the urinary tract.15 The most common histologic form of bladder cancer is urothelial carcinoma (UC). UC was previously referred to as transitional cell carcinoma and represents more than 90% of all cases of bladder cancer.16,17 Other histologic subtypes, including squamous cell and adenocarcinoma of bladder, are uncommon and account for less than 5% of all cases.18 Clinically, UC is categorized as non–muscle invasive bladder carcinoma, MIBC, and metastatic UC, which are differentiated based on the extent of tumour growth into the bladder wall and the detection of distal metastases.19,20 Compared to non-MIBC, MIBC permeates the deeper (detrusor) muscle layers of the bladder and is associated with a high rate of recurrence and poor overall prognosis, despite aggressive local and systemic therapies.19 Patients with MIBC often present with increased urinary frequency, urgency, and dysuria; pain secondary to lymphadenopathy or ureteric obstruction; weight loss; fatigue; anorexia; poor health-related quality of life (HRQoL); and poor ability to perform activities of daily living.19,21 The patient group input noted that common symptoms of MIBC include problems with urination, such as frequent urination, painful urination, blood in the urine, and poor HRQoL. The stage at which bladder cancer is diagnosed is a key predictor of patient outcome.22 The clinical experts consulted for this review noted that patients living in rural areas are often far from specialized medical care. As such, patients in rural locations may face greater barriers to accessing care, which can result in delayed diagnoses, more advanced disease at presentation, and worse cancer-related outcomes.22 Information was not available regarding whether MIBC disproportionately affects any other specific subpopulations in Canada. International guidelines recommend detailed history-taking and physical examination (which can include bimanual palpation; cystoscopy to look inside the bladder; urinary cytology to look for cancer cells in the urine; cross sectional CT imaging of the chest, abdomen, and pelvis; and transurethral resection of invasive bladder tumours [TURBT]) to establish the diagnosis of MIBC and determine appropriate treatment and prognosis.23 Cystectomy samples need to be assessed by a pathologist to determine the presence and extent of muscle invasion, as well as the presence of mutations or alterations in the FGFR gene, which may impact treatment selection in the metastatic setting.
In 2024, about 12,300 people in Canada were diagnosed with bladder cancer; the age-standardized incidence rate was projected to be 24.6 per 100,000 and the age-standardized mortality rate was projected to be 5.1 per 100,000.24 In 2022, the Canadian Cancer Statistics special report indicated that the number of people living with bladder cancer in the previous 2 years was 17,595 and the 5-year prevalence was 37,315.25 Among individuals diagnosed with bladder cancer, 25% have MIBC.25 In 2019, the 5-year mortality rate for MIBC was approximately 40% to 50%,26 imposing a significant burden on the Canadian health care system.
Current Management
Treatment Goals
The patient group input indicated that the most important treatment goals for patients are controlling disease progression and improving OS. Based on a score of 1 to 10, where 1 indicates no willingness to tolerate side effects and 10 indicates a high willingness to tolerate significant side effects, patients reported an average score of 7.5, suggesting a strong willingness to accept new side effects from drugs that can control disease progression or improve OS.
Input from the 2 clinician groups noted that the main treatment goals for patients with MIBC are to control the disease progression, prevent disease recurrence, and maintain HRQoL. Both clinician groups noted that improvements in outcomes such as pathologic complete response (pCR) and event-free survival (EFS), as well as the prevention of disease recurrence, are clinically meaningful responses to treatment. According to the OH (CCO) clinician group, outcomes should be assessed using CT and/or MRI imaging at diagnosis (preoperatively), followed by repeat CT and/or MRI imaging and bloodwork every 3 months to 6 months after cystectomy or nephroureterectomy for 2 years, then every 6 months for 2 years to 3 years, and annually thereafter. According to both clinician groups, factors to consider for discontinuing treatment are disease progression, severity, and recurrence of immune-related side effects.
Current Treatment Options
According to the 2025 Canadian Urologic Association (CUA) guideline for MIBC, eligible patients (clinical tumour staging of T2 to T4a, and lymph node and metastasis staging, respectively, of N0M0) should receive neoadjuvant chemotherapy with gemcitabine-cisplatin or ddMVAC before radical cystectomy.27 Based on a change in staging, patients with N1 disease are also considered for neoadjuvant chemotherapy. Eligibility for neoadjuvant chemotherapy, has recently been redefined by Jiang et al.28 and allows patients with impaired renal function to receive cisplatin-based neoadjuvant chemotherapy using the split-dose regimen. In this regimen, the dose is divided and given on day 1 and day 8. After neoadjuvant chemotherapy, patients need to be evaluated for optimal local therapy, either radical cystectomy or bladder-sparing therapy.
Patients with contraindications to cisplatin-based neoadjuvant chemotherapy, according to criteria outlined by Jiang et al.,28 should proceed directly to radical local therapy. This can either be radical cystectomy or trimodal therapy (TMT), which consists of TURBT followed by external beam radiotherapy, with concurrent chemotherapy.29 TMT can be offered to carefully selected patients wishing to preserve their bladder, those unfit for cystectomy, or those who refuse cystectomy.27 Radical cystectomy or TMT should occur 4 weeks to 6 weeks after the completion of neoadjuvant chemotherapy and no later than 10 weeks after the last treatment dose.27
After surgery, patients who received neoadjuvant chemotherapy but had residual T2 to T4 and/or node-positive disease and patients who did not receive neoadjuvant chemotherapy but had pathologic T3/T4 and/or node-positive disease and would not be candidates for adjuvant platinum-based chemotherapy should be offered adjuvant nivolumab (checkpoint inhibitor) for a year to prevent recurrence.30 Patients who do not receive neoadjuvant chemotherapy but who would be eligible for adjuvant chemotherapy should receive platinum-based adjuvant chemotherapy.31
A clinical expert consulted for this review noted that about 60% of eligible patients in Canada receive neoadjuvant chemotherapy within a multidisciplinary care framework that involves urologists and radiation oncologists. After evaluation and the review of scans, patients are assessed for either bladder-sparing therapy (TMT) or cystectomy, with roughly a 50:50 distribution between the 2 options in current clinical practice, according to the clinical expert. However, the expert noted that in more remote settings, cystectomy is more common, mainly because of the limited expertise with concurrent radiation approaches.
Key characteristics of durvalumab are summarized with other treatments available for MIBC in the Key Characteristics table in Appendix 1 of the Supplemental Material document.
Unmet Needs and Existing Challenges
According to the patient group input, the side effects of available treatments can include fatigue, nausea, and sometimes hospitalization, highlighting the fact that available treatment options can be detrimental to the lives of patients. When patients were asked whether the side effects of available treatments affected their daily lives, 35% reported a very strong impact, 46% reported a strong impact, 15% reported a medium impact, and 4% reported a weak impact. Overall, patients noted that they are willing to tolerate new side effects from drugs that can control their disease progression and improve OS.
Input from the OH (CCO) noted that current treatment with adjuvant nivolumab has not demonstrated a statistically significant OS benefit and that the availability of platinum-based chemotherapy with an immune checkpoint inhibitor (durvalumab) may increase the use of neoadjuvant systemic therapy. The OH (CCO) also noted that durvalumab in combination with platinum-based chemotherapy is expected to replace platinum-based chemotherapy alone as the preferred neoadjuvant treatment, resulting in a reduction in the use of chemotherapy or nivolumab in the adjuvant setting.
The clinician group input from BCC noted that there is a need for treatments that spare the patient’s bladder, as well as need for curative treatments and improvements in response rates, as disease relapse is common. According to the clinical experts consulted, only approximately 25% of patients with MIBC currently achieve pCR after neoadjuvant cisplatin-based chemotherapy. The experts stated that about 40% to 60% of eligible patients in Canada are not referred for neoadjuvant chemotherapy, reflecting variability in referral practices among urologists. Consequently, these patients may proceed directly to surgery without preoperative chemotherapy, and if the disease is subsequently found to be more advanced, the opportunity to benefit from neoadjuvant therapy is lost. Furthermore, the experts stated that some patients may be unable to tolerate the standard dose of neoadjuvant gemcitabine-cisplatin and may require a split-dose treatment approach. The clinical experts stated that bladder-sparing treatments (such as TMT) are also limited to certain centres, so rural and remote populations are less likely to have access to the expertise necessary to provide bladder-sparring approaches. However, the experts noted that if more patients achieve pCR upon completion of neoadjuvant treatment, bladder preservation may become an option for a greater number of patients. The clinical experts stated that although the main treatment goal is to cure all patients, variabilities in treatment access, tolerability, and response to treatment continue to pose challenges in clinical practice in Canada.
Considerations for Using the Drug Under Review
Contents within this section have been informed by input from the clinical experts consulted for the purpose of this review and from clinician groups, as well as the reimbursement conditions proposed by the sponsor (refer to the Initiation, Renewal, Discontinuation, and Prescribing Conditions Proposed by the Sponsor table in Appendix 1 of the Supplemental Material document, available on the project landing page). The implementation questions from the public drug programs and corresponding responses from the clinical experts consulted for this review are summarized in the Summary of Drug Program Input and Clinical Expert Responses table in Appendix 1 of the Supplemental Material document. The following has been summarized by the review team.
Place in Therapy
The clinical experts stated that durvalumab would fit well within a perioperative treatment framework as a first-line treatment, administered as neoadjuvant therapy alongside cisplatin-based chemotherapy and continued as adjuvant treatment after cystectomy. The clinical experts also noted that it is worth considering whether it could also be offered to patients who do not undergo radical cystectomy, particularly in cases where bladder preservation is pursued or surgery is not feasible, as these patients were also included in the trial. The sponsor noted that perioperative durvalumab in combination with gemcitabine-cisplatin is primarily expected to displace neoadjuvant cisplatin-based chemotherapy (primarily gemcitabine-cisplatin and minimal ddMVAC displacement). In addition, the sponsor noted that there may be displacement of adjuvant treatment options, namely adjuvant nivolumab, as the physician and/or patient choice of perioperative durvalumab plus gemcitabine-cisplatin may reduce the number of patients eligible for adjuvant therapy, such as nivolumab or other cisplatin-based chemotherapy.
Patient Population
The sponsor noted that durvalumab in combination with neoadjuvant gemcitabine-cisplatin will be the first immunotherapy-based perioperative regimen indicated for the treatment of all patients with T2N0/1M0 to T4aN0/1M0 MIBC whose tumours are resectable. The clinical experts agreed that the full patient population included in the indication may be eligible for treatment with the drug under review, including patients with baseline renal impairment (creatinine clearance [CrCl], 40 mL/min to 60 mL/min). The clinical experts suggested that patients who have an excellent response to neoadjuvant treatment with durvalumab in combination with gemcitabine-cisplatin, and who wish to spare their bladders, could also experience benefits with adjuvant durvalumab. The experts noted that patients with contraindications to immunotherapy, such as those with severe autoimmune conditions that cannot be adequately managed, and individuals with organ transplants may not be suitable candidates for perioperative durvalumab. These situations should be assessed on a case-by-case basis, according to the clinical experts.
According to the clinical experts, standard staging for MIBC includes TURBT and CT imaging. For patients unable to undergo CT, MRI is a reasonable alternative. Currently, there are no validated biomarkers to identify the subgroups of patients most likely to respond to treatment, and no companion diagnostic is required. The experts noted that although clinical understaging can occasionally occur, offering durvalumab with neoadjuvant chemotherapy broadly to eligible patients may help mitigate this issue.
Assessing the Response to Treatment
According to the clinical experts, in clinical practice, response to treatment is typically assessed using staging scans and symptom improvement, such as resolution of hematuria. These measures are aligned with outcomes used in the NIAGARA study, including pCR, EFS, and OS. A clinically meaningful response, according to the experts, would be a between-group difference in pCR of at least 8% to 10%, or a between-group difference of 5% to 10% in EFS or OS. Although interpretations may vary slightly among physicians, these thresholds are generally viewed as meaningful. The experts stated that treatment response is usually assessed after neoadjuvant chemotherapy, followed by routine monitoring with scans and bloodwork every 4 months for 2 years, every 6 months for the subsequent 2 years, and annually thereafter. The clinical experts consulted for this review noted that patients living in rural areas are often far from specialized medical care. As such, patients in rural locations may face greater barriers to accessing care, which can result in delayed diagnoses, more advanced disease at presentation, and worse cancer-related outcomes.22
Discontinuing Treatment
According to the clinical experts consulted, treatment should be discontinued in cases of disease progression or unacceptable toxicity, per the sponsor’s proposed discontinuation criteria. The experts noted that these discontinuation criteria are appropriate, align with current practice, and can be readily implemented without major challenges.
Prescribing Considerations
The clinical experts noted that durvalumab should be prescribed and managed by medical oncologists with expertise in managing patients with bladder cancer.
Clinical Review
Methods
The review team considered studies in the sponsor’s systematic review (pivotal studies and randomized controlled trials [RCTs]) in the evidence for inclusion. Eligible studies for the systematic review included published and unpublished pivotal studies and phase III and phase IV RCTs. Relevant patients and interventions were defined by the indication and the recommended dosage in the product monograph. Disease staging (T2N0 versus > T2N0) was considered to be potentially important for informing the reimbursement recommendation. Relevant comparators were drugs used in clinical practice in Canada to treat patients described in the indication under review. These included neoadjuvant ddMVAC and adjuvant nivolumab. Long-term extensions of included pivotal studies and RCTs were eligible, regardless of whether there was a comparison group. ITCs and studies addressing gaps submitted by the sponsor were eligible if they filled an identified gap in the systematic review evidence (e.g., missing comparator, longer follow-up time). The sponsor did not submit any long-term extensions or ITCs for inclusion.
The review team selected outcomes (and follow-up times) for review after consideration of the sponsor’s Summary of Clinical Evidence, clinical expert input, and patient and clinician group input. Included outcomes are those considered relevant to expert committee deliberations, and they were selected in consultation with committee members. Evidence from the systematic review for the most important outcomes was assessed using the Grading of Recommendations Assessment, Development and Evaluation (GRADE) approach.
Considerations that informed the selection of efficacy outcomes to be summarized and assessed using GRADE include the following:
pCR was identified as important by the clinical experts consulted for this review, as it is an outcome that is routinely used in clinical practice and is used as a surrogate end point in clinical trials.
Survival outcomes were identified by patient and clinician group input and specified by the clinical experts consulted for this review. EFS was a key input for the pharmacoeconomic model and was deemed relevant by the clinical experts consulted for this review. The clinical experts noted that EFS at 24 months is a relevant time point for GRADE, as recurrence rates are highest during the first 2 years of treatment. According to the experts, OS with the longest follow-up was critical for informing the long-term benefits and harms of treatment; as such, OS at 36 months was selected for GRADE.
The ability to maintain HRQoL while receiving treatment was identified by the patient and clinician groups as an important outcome, and the clinical experts consulted for this review specified the EORTC QLQ-C30 global health status (GHS) as the most informative scale.
Patient and clinical group input highlighted safety and toxicity as important outcomes, with clinical experts identifying SAEs as a key measure with which to evaluate the safety profile, owing to the risk of immune-related toxicities. Methods for data extraction, risk of bias appraisal, and certainty of evidence assessment are described in Appendix 2 of the Supplemental Material document.
Clinical Evidence
In this report, the following sources of evidence submitted by the sponsor are reviewed and appraised:
1 pivotal study — NIAGARA — which was included in the systematic review
1 feasibility assessment, which was used to evaluate whether available evidence supports the conduct of an ITC.
Systematic Review
Description of Studies
Study Characteristics
The characteristics of the included study are summarized in Table 2. Details pertaining to the eligibility criteria, study design, and relevant outcome measures are presented in Appendix 3 of the Supplemental Material document.
NIAGARA is a phase III, randomized, open-label, multicentre, global study conducted to evaluate the efficacy and safety of durvalumab in adult patients with resectable MIBC. The study was conducted in Australia and in 21 countries across Asia, Europe, North America, and South America, with 7 sites in Canada. The perioperative study design consisted of 3 phases: neoadjuvant treatment, radical cystectomy, and adjuvant treatment. Randomization (1:1) was stratified by clinical tumour stage (T2N0 versus > T2N0), renal function (CrCl, ≥ 40 mL/min to < 60 mL/min versus ≥ 60 mL/min), and PD-L1 expression (high versus low/negative). Patients randomized to the 2 treatment groups were treated according to their renal function. The recruitment of patients with borderline renal function was limited to 20% of the global study population. The enrolment of patients with T2N0 disease was capped at approximately 40% across both treatment groups.
Intervention and Comparator
Patients were randomized to 1 of 2 treatment groups.
Patients in the durvalumab group (n = 533) received:
neoadjuvant durvalumab plus gemcitabine-cisplatin (every 3 weeks for 4 cycles),
subsequent radical cystectomy, and
adjuvant durvalumab monotherapy (every 4 weeks up to 8 cycles).
Patients in the gemcitabine-cisplatin group (n = 530) received:
neoadjuvant gemcitabine-cisplatin (every 3 weeks for 4 cycles),
subsequent radical cystectomy, and
no adjuvant treatment.
Neoadjuvant Phase
During the neoadjuvant phase, patients with Response Evaluation Criteria in Solid Tumours Version 1.1 (RECIST 1.1)-defined progression could proceed to the adjuvant phase if the progression did not preclude radical cystectomy (i.e., progression was local or limited to regional lymph nodes). Patients with progression that prevented surgery (i.e., distant metastases) were transitioned to follow-up and monitored for OS.
Cystectomy and Noncystectomy Extension Phase
It was recommended that surgery take place from 14 to 56 days after the last neoadjuvant dose. In the event that progression (distant metastases) precluded the patient from undergoing radical cystectomy, the patient proceeded to follow-up, with no additional tumour assessments, and was followed for OS.
Patients who underwent partial cystectomy based on clinical assessment could continue to the adjuvant phase, per their treatment assignment. Patients in either treatment group who refused to undergo cystectomy and had a complete clinical response could enter a noncystectomy extension phase instead of the follow-up phase after consultation with and approval by the sponsor. Patients enrolled into the durvalumab group who entered the noncystectomy extension phase could be administered durvalumab 1,500 mg (as monotherapy) every 28 days for a maximum of 8 doses.
Adjuvant Phase
It was recommended that adjuvant therapy begin from 42 to 120 days after radical cystectomy. In the adjuvant phase, treatment was discontinued upon RECIST-defined progression, and patients with confirmed recurrence after surgery proceeded to follow-up.
Table 2: Characteristics of the Study Included in the Systematic Review
Study | Key inclusion criteria | Key exclusion criteria | Intervention and comparator | Relevant end points |
|---|---|---|---|---|
NIAGARA, a phase III, randomized, open-label, multicenter, global study Total N = 1,063 |
|
| Intervention:
Comparator:
| Dual primary end points:
Key secondary end point:
Other secondary end points:
|
BCG = Bacillus Calmette-Guérin; CrCl = creatinine clearance; DFS = disease-free survival; DSS = disease-specific survival; ECOG PS = European Cooperative Oncology Group Performance Status; EFS = event-free survival; HRQoL = health-related quality of life; M = metastasis staging; MFS = metastasis-free survival; MIBC = muscle invasive bladder cancer; N = node staging; OS = overall survival; P = pathologic staging; pCR = pathologic complete response; PFS2 = time from randomization to subsequent progression or recurrence after an EFS event; q.3.w. = every 3 weeks; q.4.w. = every 4 weeks; T = tumour staging; UC = urothelial carcinoma.
aIncluding but not limited to other anti-CTLA-4, anti-PD-1, anti-PD-L1, or anti-PD-L2 antibodies.
bExceptions include intranasal, inhaled, topical, or local steroid injections (e.g., intra-articular injection); systemic corticosteroids at physiologic doses not exceeding 10 mg/day of prednisone or its equivalent; steroids as premedication for hypersensitivity reactions (e.g., CT scan premedication).
cIntervention group: on day 1, patients received durvalumab (1,500 mg IV), cisplatin (70 mg/m2) and gemcitabine (1,000 mg/m2); on day 8, patients received gemcitabine 1,000 mg/m2 q.3.w.; for 4 cycles. This was followed by radical cystectomy and up to 8 cycles of adjuvant durvalumab (1,500 mg IV) q.4.w.
dPatients in both treatment groups with borderline renal function (CrCl ≥ 40 mL/min to < 60 mL/min) received split-dose cisplatin: 35 mg/m2 IV on day 1 and day 8, q.3.w.
ePatients received the same neoadjuvant gemcitabine-cisplatin regimen as in the intervention group, followed by radical cystectomy, with no adjuvant treatment.
fEvaluated by blinded independent central review (BICR) or central pathology review (if a biopsy was required for a suspected new lesion).
gEvaluated by BICR.
hPer investigator assessment or local biopsy review.
Sources: NIAGARA Clinical Study Report and sponsor’s Summary of Clinical Evidence.8,9 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
The final pCR analysis is based on the data cut-off (DCO) of January 14, 2022, and all other reported efficacy analyses are based on results from the DCO of April 29, 2024, corresponding to the second interim analysis (IA2). Interim analyses were prespecified for both EFS and OS, with type I error controlled through a Lan-DeMets alpha-spending function. An independent data monitoring committee (IDMC) provided oversight, reviewing interim safety and efficacy data at regular intervals and issuing recommendations to continue, modify, or stop the trial without unblinding the results. The final analyses for EFS and OS were initially expected to occur in June 2025 and June 2026, respectively. However, according to the sponsor, because the IDMC indicated that EFS and OS had reached clinical significance at IA2, these were considered the final analyses, and the sponsor proceeded to unblind the study. Subsequent EFS and OS analyses will be event-driven and descriptive in nature.
The outcome of pCR rate was assessed with a central pathology review of the radical cystectomy sample, and EFS was assessed by blinded independent central review (BICR) of imaging scans, according to RECIST 1.1 or by central pathology review if a biopsy was required for a suspected new lesion.
Substantial protocol amendments dated June 23, 2023, were made by the sponsor in response to published evidence that most MIBC events occur in the 12 months after surgery and slow after 2 years.32-34 Key protocol amendments included reducing the target number of EFS events for the final analysis from 509 to 451 and adjusting statistical power from 93% to 90%. Calendar-based DCOs were introduced for interim and final EFS analyses to address potential delays, and an interim OS analysis was added at EFS IA2, contingent on EFS reaching statistical significance. A substantial amendment dated January 29, 2024, included the removal of the censoring rule for 2 consecutive missed visits for the primary EFS end point to minimize the loss of clinically relevant events.
Statistical Testing and Analysis Populations
The NIAGARA study was powered to detect differences in the dual primary end points: pCR and EFS. With 525 patients planned per treatment group, the study had at least 95% power to detect a statistically significant difference at a 2-sided alpha of 0.1%, assuming that the pCR rate was 50% and 35% for patients in the intention-to-treat (ITT) population in each of the durvalumab and gemcitabine-cisplatin groups, respectively. The final pCR analysis was planned at approximately 6 months after randomization of the last patient in. EFS was event-driven, with the final analysis initially planned after approximately 451 EFS events (43% maturity) or by June 2025, providing at least 90% power to detect a hazard ratio (HR) of 0.733 at a 2-sided alpha of 4.9%. Two interim analyses were planned for EFS at approximately 67% and 91% of target events. As EFS was statistically significant at IA2, this was considered the final analysis by the sponsor. Subsequent EFS analyses will be descriptive in nature, event-driven, and are expected to mature in 2026.
OS was modelled assuming an exponential distribution, with an expected HR of █████ favouring the durvalumab group. Median OS was assumed to be ███ █████ ██████ ██ █ ██████ in the durvalumab group and ███ █████ ██████ ██ █ ██████ in the gemcitabine-cisplatin group. The 5-year OS analysis, based on █████ ███ ██████ ████ █████████, was planned 5 years after the last patient was randomized (≥ 80% power; 2-sided alpha of 4.9%). OS interim analyses were planned a ███ ███ ███ of target events and tested only if EFS was positive, per the multiple testing procedure. As OS was statistically significant at IA2, this was considered the final analysis by the sponsor. Subsequent OS analyses will be descriptive in nature, event-driven, and expected to mature in 2027.
A multiple testing procedure (described in Appendix 3 of the Supplemental Material document) with an alpha-exhaustive recycling strategy controlled the overall 2-sided error rate at 5%, with an initial split between pCR (0.1%) and EFS (4.9%). The Lan-DeMets spending function, approximating an O’Brien-Fleming boundary, was used to allocate alpha at the interim and final analyses of pCR, EFS, and OS, based on the proportion of information available. The other prespecified secondary analyses, apart from OS at 5 years after randomization, were not included in the multiple testing procedure and, therefore, are nonconfirmatory.
Efficacy end points were analyzed in the ITT population. Disease-free survival was assessed in the cystectomy set (patients who underwent partial or radical cystectomy). Safety was evaluated in the safety analysis set (patients who received ≥ 1 dose of study treatment).
Patient Disposition
Patient disposition for the included study is summarized in Appendix 4 of the Supplemental Material document.
In the NIAGARA study, of the 1,530 patients screened, 1,063 (69.5%) were randomized and 1,056 (69.0%) were treated. Among the 428 patients who were screened out of the study, the most common reasons were the presence of lymph node or metastatic disease ████████ lack of confirmed MIBC ████████ and inadequate organ or marrow function ███████. As of IA2 █████ of patients in durvalumab group █████████ ███ █████ of patients in the gemcitabine-cisplatin group █████████ discontinued neoadjuvant treatment. Discontinuation of neoadjuvant treatment related to patient decision occurred ██ ████ and ████ of patients in the durvalumab and gemcitabine-cisplatin groups, respectively.
Before the IA2 DCO, a total of 470 (88.2%) and 446 (84.2%) randomized patients underwent cystectomy in the durvalumab group and gemcitabine-cisplatin group, respectively. Six patients (1.1%) in the durvalumab group and 0 patients (0.0%) in the gemcitabine-cisplatin group entered the noncystectomy extension phase.
At IA2, a total of 288 (54.3%) patients in the durvalumab group completed all 8 cycles of adjuvant treatment. Ninety-five (17.9%) patients discontinued adjuvant treatment, mainly because of adverse events (AEs) or disease relapse █████ █████ as well as patient decision ███████
A total of ███ ███████ patients in the durvalumab group and ███ ███████ patients in the gemcitabine-cisplatin group terminated the study because of death ██████ ███ ██████ and patient decision █████ ███ ██████
At IA2, 84 of randomized patients (7.9%) had at least 1 important protocol deviation, 47 (8.8%) in the durvalumab group and 37 (7.0%) in the gemcitabine-cisplatin group. The most common deviations were incorrect tumour staging entered into the interactive voice response system, causing patients to be stratified incorrectly █████ ███ █████ and missed baseline patient-reported outcome assessments or compliance below ███ █████ ███ ██████.
Baseline Characteristics
Baseline characteristics of patients in the NIAGARA study are summarized in Table 3.
Table 3: Summary of Baseline Characteristics From the NIAGARA Study
Characteristic | Durvalumab group (N = 533) | Gemcitabine-cisplatin group (N = 530) |
|---|---|---|
Age, years, median (range) | 65 (34 to 84) | 66 (32 to 83) |
Sex, n (%) | ||
Female | 96 (18.0) | 97 (18.3) |
Male | 437 (82.0) | 433 (81.7) |
Race, n (%) | ||
Asian | 152 (28.5) | 145 (27.4) |
Black | 6 (1.1) | 4 (0.8) |
White | 354 (66.4) | 358 (67.5) |
Missing or other | 21 (3.9) | 23 (4.4) |
Region, n (%) | ||
Asia | 151 (28.3) | 143 (27.0) |
Europe | 265 (49.7) | 287 (54.2) |
North America and Australia | 66 (12.4) | 62 (11.7) |
South America | 51 (9.6) | 38 (7.2) |
ECOG PS score, n (%) | ||
0 | 418 (78.4) | 415 (78.3) |
1 | 115 (21.6) | 115 (21.7) |
Smoking status, n (%) | ||
Current | 122 (22.9) | 130 (24.5) |
Former | 255 (47.8) | 269 (50.8) |
Never | 144 (27.0) | 120 (22.6) |
Missing | 12 (2.3) | 11 (2.1) |
Histologic type, n (%) | ||
Invasive UC, not otherwise specified | 457 (85.7) | 441 (83.2) |
UC with squamous differentiation | 38 (7.1) | 49 (9.2) |
UC with glandular differentiation | 10 (1.9) | 15 (2.8) |
Tumour stage, n (%) | ||
T2N0 | 215 (40.3) | 213 (40.2) |
> T2N0 | 318 (59.7) | 317 (59.8) |
Regional lymph nodes, n (%) | ||
N0 | 505 (94.7) | 500 (94.3) |
N1 | 28 (5.3) | 30 (5.7) |
Renal function, per eCRF, n (%)a,b | ||
Adequate | 431 (80.9) | 431 (81.3) |
Borderline | 102 (19.1) | 99 (18.7) |
PD-L1 status, per laboratory data, n (%)c,d | ||
High | 388 (72.8) | 388 (73.2) |
Low | 144 (27.0) | 142 (26.8) |
ECOG PS = Eastern Cooperative Oncology Group Performance Status; eCRF = electronic Case Report Form; N = node; T = tumour; UC = urothelial carcinoma.
aIn the interactive voice report system (IVRS), 1 patient in the durvalumab group was mis-stratified to adequate renal function and 1 patient in the gemcitabine-cisplatin group was mis-stratified to borderline renal function; both classifications were adjusted in the eCRF.
bCreatinine clearance (CrCl) for adequate renal function: ≥ 60 mL/min; CrCl for borderline renal function: ≥ 40 mL/min to < 60 mL/min.
cOne patient in the durvalumab group was mis-stratified to the PD-L1 high group, according to IVRS, and should belong in the PD-L1 low group, per central laboratory data.
dPD-L1 high status was considered high if any of the following were met: ≥ 25% of tumour cells exhibit membrane staining; immune cells present > 1% and immune cell+ ≥ 25%; immune cells present = 1% and immune cell+ = 100%. PD-L1 low/negative status was considered low/negative if none of the criteria for PD-L1 high status were met.
Sources: NIAGARA Clinical Study Report and sponsor’s Summary of Clinical Evidence.8,9 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Treatment and Concomitant Medications
Details of patients’ treatment exposure, use of concomitant medications, and subsequent treatments in the included study are presented in Appendix 4 of the Supplemental Material document.
Exposure to Durvalumab
At IA2, the median total duration of durvalumab exposure was 44.0 weeks, overall (range, 1 to 84 weeks), with a median of 12.1 weeks in the neoadjuvant period and 32.0 weeks in the adjuvant period. A total of 53.8% (287/533) of patients completed all 8 adjuvant cycles of durvalumab. As allowed in the protocol, 6 patients in the durvalumab group who declined cystectomy and had a complete clinical response entered the noncystectomy extension phase, during which they received an additional 8 cycles of durvalumab after neoadjuvant therapy. This treatment was included as part of the neoadjuvant period.
Exposure to Gemcitabine-Cisplatin
Note, in scenarios in which patients were unable to complete the intended 4 cycles of chemotherapy before radical cystectomy, patients were permitted to receive fewer than 4 cycles of chemotherapy at the discretion of the investigator and upon discussion with the sponsor.
At IA2, the median total duration of exposure to gemcitabine-cisplatin for the overall treatment period was ██ █████ in both the durvalumab group ██████ ██ ██████ and the gemcitabine-cisplatin group ██████ ██ ███████ In the safety analysis set, as of IA2, █████ █████████ of patients in the durvalumab group and █████ ████████) of patients in the gemcitabine-cisplatin group had a cumulative exposure of at least ██ █████ to gemcitabine-cisplatin in the neoadjuvant period.
Concomitant Medications
In the ITT population, at IA2, almost all patients had received at least 1 allowed concomitant medication during the study; █████ █████████ in the durvalumab group and █████ █████████ in the gemcitabine-cisplatin group. Common concomitant medications included steroids ██████ ███ ██████ and antiemetics ██████ and ███████Fluoroquinolones and combinations of penicillin were used more frequently in the durvalumab group ██████ ███ ██████ respectively, than in the gemcitabine-cisplatin group ██████ and ██████respectively).
Subsequent Treatments
In the ITT population, at IA2, 9.9% (53 of 533) of patients in the durvalumab group and 17.5% (93 of 530) in the gemcitabine-cisplatin group received subsequent anticancer therapy after study treatment discontinuation. The most common subsequent anticancer therapies administered were immunotherapy █████ in the durvalumab group versus █████ in the gemcitabine-cisplatin group), cytotoxic chemotherapy-based regimens █████ ███ ██████ and radiotherapy █████ ███ ██████
Critical Appraisal
Internal Validity
NIAGARA is a phase III, randomized, open-label, multicentre, global study. The study used a computer-generated blocked randomization scheme to stratify patients by clinical tumour stage (T2N0 versus > T2N0), renal function (adequate versus borderline), and PD-L1 status (high and low/negative baseline characteristics were generally balanced, suggesting that randomization was successful). More patients in the durvalumab group than in the gemcitabine-cisplatin group continued to surgery after neoadjuvant treatment (88.2% versus 84.2%). Reasons for not continuing with surgery were generally balanced between treatment groups. However, a higher proportion of patients in the gemcitabine-cisplatin group did not proceed with cystectomy because of study discontinuation than in the durvalumab group (12 versus 3).
Patients and treating physicians were unblinded, and end points were subject to unblinded review by an IDMC to allow for safety and efficacy oversight. The IDMC reviewed interim efficacy and safety data and provided recommendations to the sponsor regarding continuation, modification, and unblinding. The sponsor remained blinded to treatment assignment until the IDMC confirmed that the predefined threshold for statistical significance at IA2 was reached. Several protocol amendments were implemented during the NIAGARA study, resulting in changes to the study population, the required number of events for the final analysis, and censoring rules. In Amendment 5 (June 2023), the planned number of EFS events was revised from 509 to 451, based on external evidence indicating slower event accumulation after 2 years. These modifications were not expected to have meaningfully affected the study results.32-34 Importantly, the study team remained sponsor-blinded until after the IA2 DCO. This structure limited potential bias in study conduct, data collection, and interpretation of results, thereby supporting internal validity.
Although immature data and a limited number of events may raise the possibility that early statistical significance could reflect random fluctuation rather than a true effect that is not sustained when the data are mature, this risk was addressed through the prespecified analysis plan. The risk of bias from early termination was mitigated through preplanned interim analyses with multiplicity control, using a Lan-DeMets alpha-spending function.
Outcome validity was supported by the use of standard oncology measures. Imaging was performed per protocol and assessed according to RECIST 1.1, and both imaging and pathology assessments were centrally read and blinded. Quality-of-life outcomes were measured using the validated EORTC QLQ-C30.
Efficacy outcomes were reported for the entire perioperative treatment course, which limits the ability to distinguish the relative contributions of the neoadjuvant and adjuvant phases to the overall treatment effect. To limit bias, both primary end points were assessed by independent reviewers who were blinded to treatment assignment; pCR was reviewed by central pathology and EFS by BICR. The sponsor was only unblinded after the IDMC confirmed that the EFS results at IA2 met the predefined threshold for statistical significance. A programming error in the initial pCR analysis led to the misclassification of certain patients as nonresponders, as the date of central assessment was incorrectly applied in place of the date of surgery. This issue was subsequently identified and corrected, with the revised analysis appropriately incorporating the actual outcomes. This amendment was handled transparently and does not raise concerns regarding bias or the overall validity of the efficacy analyses.
The definition of EFS included patients who experienced progression and were precluded from a radical cystectomy (1.7% of patients in each group), patients who refused to undergo a radical cystectomy, and patients with residual disease who did not undergo a radical cystectomy (████ in the durvalumab group versus 11.3% in the gemcitabine-cisplatin group). Although recurrence, progression, and death are objective end points, the classification of surgery refusal or omission as an event introduces subjectivity, as these decisions may be influenced by patient preference or physician judgment in an open-label setting. This component of the EFS definition, therefore, increases the risk of performance and detection bias. Furthermore, patients who refused cystectomy or did not undergo cystectomy despite residual disease were assigned an EFS event at the time of their expected surgery date. This conservative approach counts these patients as events without waiting for progression or death, but introduces some uncertainty, as the expected surgery date may not precisely correspond to each patient’s actual clinical course. However, in a post hoc sensitivity analysis in which EFS was censored for patients who refused or did not undergo radical cystectomy, the HR remained consistent with the primary results. This suggests that any imbalance related to this factor did not account for the observed EFS benefit.
The observed median EFS in the gemcitabine-cisplatin group █████ ██████ at IA2) exceeded the protocol assumption of ████ ███████ This improved performance in the gemcitabine-cisplatin group did not undermine the study’s internal validity, but did result in smaller absolute and relative effect sizes than originally anticipated. Upon visual inspection, EFS Kaplan-Meier curves separated after approximately 2 months and remained durably apart thereafter in favour of the durvalumab group. At 24 months, absolute EFS was higher in the durvalumab group than in the gemcitabine-cisplatin group (67.8% versus 59.8%), with median EFS not reached the durvalumab group and 46.1 months for the gemcitabine-cisplatin group. For OS, the Kaplan-Meier curves were immature (28.7% maturity) but showed a similar pattern of progressive and sustained separation after approximately 6 months. Median OS was not reached in either group, but follow-up was adequate, based on clinical expert input (40 months to 42 months), and showed a consistent treatment effect over time. Numbers at risk and censoring were balanced, and the shape of the curves suggests approximate proportional hazards, with consistent separation maintained after the early period (as shown in Appendix 4 of the Supplemental Material document).
In terms of relevant subgroups, the clinical experts consulted noted that patients with a more advanced stage of MIBC (i.e., > stage T2N0) may experience worse outcomes. In patients with T2N0 disease at baseline, EFS and OS outcomes presented 95% confidence intervals (CIs) that included the null value, indicating a lack of statistical precision (Appendix 4 of the Supplemental Material document). Although OS subgroup analyses were not specified as a priori, the results suggest that most of the observed benefits with durvalumab may come from patients with more advanced disease (> T2N0). These subgroup findings should be interpreted with caution, given the small sample sizes, which limit the ability to draw definitive conclusions regarding treatment effect. Furthermore, as multiple subgroups were considered without control for multiplicity, apparent differences observed may be due to chance.
Patients who had a pCR and did not undergo surgery were counted as nonresponders, which is a conservative approach that may slightly underestimate the treatment effect but does not compromise internal validity. Of the patients in the durvalumab group who initiated neoadjuvant treatment, 27.7% (147 of 530) did not start adjuvant durvalumab, including 18.5% (87 of 470) of those who underwent cystectomy. The reasons for not continuing adjuvant durvalumab were not collected in the NIAGARA study, which introduces some uncertainty in our understanding of treatment adherence and the effects of the full treatment regimen.
A limitation in the safety analysis is the potential underestimation of AEs in the neoadjuvant period due to overlapping study phases. Events occurring near the time of surgery may have been attributed to the adjuvant phase, even if they were related to neoadjuvant treatment. Additionally, ongoing AEs that began in the neoadjuvant phase may not have been consistently counted unless they worsened, and patients who did not proceed to adjuvant treatment may have had limited follow-up. This overlap makes it difficult to fully isolate and interpret harms associated with each treatment phase.
The coprimary outcomes (pCR, EFS) and key secondary outcomes (OS) were adjusted for multiplicity. Secondary and exploratory outcomes were not controlled for multiplicity. As previously described, the post hoc analysis of the primary end point of pCR was descriptive in nature and was not controlled for multiplicity, increasing the risk of type I error. However, the revised analysis aligns with the planned approach and maintains methodological integrity.
Baseline compliance with the EORTC QLQ-C30 was 69.8% in the durvalumab group and 72.8% in the gemcitabine-cisplatin group. Compliance with the questionnaire generally decreased in both groups throughout the study, with ████ and ████ compliance in the durvalumab group and the gemcitabine-cisplatin group at week 25, respectively. Because there were no data imputations in these analyses, there is a risk of bias due to missing outcomes data.
External Validity
In consultation with the clinical experts on this review, the NIAGARA study population was considered broadly representative of patients seen in routine clinical practice in Canada; however, some generalizability issues were identified. The caps on enrolment for patients with borderline renal function (20%) and T2N0 disease (approximately 40%) restrict the representativeness of the study population, thereby limiting the generalizability of the findings to the broader MIBC population encountered in routine clinical practice. Although patients enrolled in the study were slightly younger than typical patients presenting in clinical practice (mean age, 64.4 years), the female-to-male ratio (approximately 20:80), racial distribution (28% Asian, 1% Black, 67% white), and Eastern Cooperative Oncology Group Performance Status (ECOG PS) (predominantly 0 or 1) are consistent with clinical experience, the clinical experts report. It should be noted that this patient population may limit the generalizability of the results to diverse populations in Canada. The experts noted that patients with an ECOG PS of 2 would be considered eligible for treatment with durvalumab in practice, as long as they were deemed fit to receive treatment. Further, there were no issues with generalizing the evidence to patients with an ECOG PS of 2. Smoking history was also reflective of real-world patterns; roughly 60% of patients were current or former smokers and 25% were never smokers. Histologic subtypes, including predominant squamous differentiation, aligned with expectations, although rare subtypes, such as small cell or pure adenocarcinoma, were not included, which limits the generalizability of the study results to these patient populations that may experience different prognosis and treatment responsiveness. The experts also noted that PD-L1 status is not measured in this patient population in clinical practice in Canada.
Experts stated that patient preferences and clinical context may influence the study’s applicability to certain patients. The clinical experts noted that an increasing number of patients, particularly those achieving pCR after neoadjuvant treatment (about 25% of patients in current clinical practice in Canada), may opt for bladder-sparing approaches, such as TMT. An expert noted that in high-volume treatment centres, decisions are approximately 50:50 between bladder preservation and cystectomy, whereas patients in remote centres are more likely to undergo cystectomy, owing to limited resources. According to the clinical experts, about 40% to 60% of eligible patients are referred to neoadjuvant chemotherapy in clinical practice in Canada and can readily adopt this regimen. The clinical experts also stated that adjuvant completion rates are likely to be slightly higher in clinical practice than in the NIAGARA study, which may affect the generalizability of the study results. The clinical experts noted that the comparator of gemcitabine-cisplatin was appropriate. The experts also noted that the study design allowed for use of split-dose gemcitabine-cisplatin in patients with compromised renal function (CrCl, 40 mL/min to 60 mL/min), which is commonly used in clinical practice.
Results
The key efficacy and harms results and findings from the GRADE approach are presented in this section. Detailed efficacy and harms results can be found in Appendix 4 of the Supplemental Material document.
Efficacy
Key results include the following.
Pathologic Complete Response
In the ITT population, at the time of the final (original) pCR analysis (DCO: January 14, 2022), the pCR end point was not met, with an odds ratio (OR) of 1.49 (95% CI, 1.14 to 1.96; P = 0.0038; threshold for significance, P = 0.001).
A post hoc reanalysis (after correction of a postprogramming error in which 59 evaluable patients were classified as nonresponders) showed a pCR rate of 37.3% in the durvalumab group and of 27.5% in the gemcitabine-cisplatin group, and the OR was 1.60 (95% CI, 1.22 to 2.08; P = 0.0005 [nominal]) in favour of the durvalumab group.
Event-Free Survival
The median duration of follow-up for all patients for EFS was 34.7 months (range = ████ ██ █████) in the durvalumab group and 27.7 months (range = 0.03 to 61.27 months) in the gemcitabine-cisplatin group.
In the ITT population, at the IA2 DCO, the HR for EFS was 0.68 (95% CI, 0.558 to 0.817; P < 0.0001) in favour of the durvalumab group, indicating a 32% overall reduction in the risk of experiencing an EFS event (43% maturity; 451 events).
The Kaplan-Meier–estimated probability of being alive and event-free at 24 months was 67.8% (95% CI, 63.6% to 71.7%) in the durvalumab group and 59.8% (55.4% to 64.0%) in the gemcitabine-cisplatin group. Refer to the EFS Kaplan-Meier Plot, per BICR or by Central Pathology Review (ITT Population; IA2) figure in the Supplemental Material document.
Median EFS was not reached in the durvalumab group and was 46.1 months in the gemcitabine-cisplatin group.
Investigator-assessed EFS results were consistent with the primary analysis.
Prespecified subgroup and sensitivity analyses supported the primary EFS results, with a between-group HR of less than 1 across all subgroups, and a consistent treatment effect observed (HR range = 0.67 to 0.70).
Overall Survival
The median duration of follow-up of all patients for OS was 42.3 months (range = 0.26 to 64.62 months) in the durvalumab group and 39.6 months (range = 0.03 to 64.66 months) in the gemcitabine-cisplatin group.
In the ITT population, at the IA2 DCO, the interim analysis of OS resulted in an HR of 0.75 (95% CI, 0.59 to 0.93; P = 0.0106) in favour of the durvalumab group, based on 305 events (28.7% maturity).
The Kaplan-Meier-estimated probability of being alive at 36 months and 60 months was 76.6% (95% CI, █████ ██ ██████ and █████(95% CI, █████ ██ ██████ in the durvalumab group versus █████ 95% CI, █████ ██ ██████ ███ █████ (95% CI, ████ ██ ██████ in the gemcitabine-cisplatin group, respectively. Refer to the OS Kaplan-Meier Plot, (ITT Population; IA-2) figure in the Supplemental Material document.
Median OS was not reached in either group at the time of analysis.
The OS treatment effect was generally consistent across all subgroups.
Health-Related Quality of Life
A trend toward deterioration from baseline was observed in the first 25 weeks for EORTC QLQ-C30 scales of GHS, physical functioning, fatigue, and pain in both treatment groups, followed by a decrease in deterioration or a return to baseline levels thereafter.
Overall change from baseline favoured the durvalumab group in the GHS, physical functioning, fatigue, and pain domains across all visits. Refer to the Change From Baseline in EORTC QLQ-C30 GHS by MMRM Analysis (ITT Population) figure in the Supplemental Material document.
Other Secondary End Points
The following secondary end points were directionally consistent with the EFS primary end point:
The proportion of patients who achieved less than P2 (the proportion of patients whose pathologic staging at radical cystectomy was P0 [T0N0M0]/Pa/P1/carcinoma in situ, assessed per local pathology review using specimens obtained from radical cystectomy after the neoadjuvant treatment) was 265 (49.7%) in the durvalumab group and 215 (40.6%) in the gemcitabine-cisplatin group, with a between-group OR of 1.47 (95% CI, 1.15 to 1.88)
Disease-free survival with a between-group HR of ████(95% CI, ████ ██ █████
Time from randomization to subsequent progression or recurrence after an EFS event (PFS2), with a between-group HR of ████95% CI, ████ ██ █████
Harms
Key results include the following:
Adverse Events
Any AEs were reported in 99.4% (n = 527 of 530) of patients in the durvalumab group versus 99.8% (n = 525 of 526) of patients in the gemcitabine-cisplatin group. AE rates in the durvalumab group versus the gemcitabine-cisplatin group were high during the neoadjuvant period (98.1% versus 97.9%) and lower in the adjuvant period (86.4% versus 71.3%).
Overall, the most common AEs occurring in at least 15% of patients in either group (durvalumab group versus gemcitabine-cisplatin group) included nausea (53.6% versus 48.5%), anemia (38.7% versus 40.5%), constipation (38.7% versus 38.6%), fatigue (36.0% versus 32.1%), urinary tract infection (30.0% versus 29.1%), decreased appetite (26.6% versus 24.9%), neutropenia (25.8% versus 31.4%), pyrexia (20.8% versus 16.5%), diarrhea (20.6% versus 14.1%), vomiting (19.2% versus 18.4%), increased blood creatinine (18.5% versus 14.6%), asthenia (17.5% versus 18.3%), decreased neutrophil count (15.3% versus 14.1%), and pruritus (15.1% versus 7.2%).
The most common AEs occurring in at least 15% of patients in either group (durvalumab group versus gemcitabine-cisplatin group) in the neoadjuvant period included nausea (50.6% versus 45.6%), fatigue (33.4% versus 30.0%), constipation (30.8% versus 31.6%), anemia (27.4% versus 31.7%), neutropenia (25.7% versus 31.2%), decreased appetite (21.9% versus 22.6%), decreased neutrophil count (15.3% versus 13.9%), vomiting (15.3% versus 14.3%), and asthenia (14.5% versus 15.2%).
The most common AEs in the adjuvant phase were urinary tract infection (18.0% versus 17.0%) and pruritus (12.8% versus 2.6%).
In the overall study period, other AEs with a greater than 5% difference between treatment groups (durvalumab group versus gemcitabine-cisplatin group) in either direction included rash (12.6% versus 5.7%), abdominal pain (12.5% versus 7.8%), hypothyroidism (11.5% versus 2.1%), and dyspnea (9.6% versus 3.8%).
Serious Adverse Events
In the overall study period, SAEs were reported in 61.5% versus 54.6% of patients in each group (durvalumab group versus gemcitabine-cisplatin group); the most common SAEs included urinary tract infection (11.1% versus 13.1%) and prostate cancer (6.6% versus 5.1%).
In the neoadjuvant phase, SAEs were reported in 23.6% and 22.4% of patients in the durvalumab and gemcitabine-cisplatin groups, respectively; the most common SAEs included urinary tract infection (1.3% versus 2.7%) and anemia (0.8% versus 2.9%).
In the adjuvant period, SAEs were reported in 26.4% and 22.2% of patients in the durvalumab and gemcitabine-cisplatin groups, respectively; the most common SAEs included urinary tract infection (6.0% versus 7.0%), acute kidney injury (2.9% versus 2.1%), pyelonephritis (2.1% versus 2.1%), and hydronephrosis (2.1% versus 1.6%).
In the overall study period, SAEs assessed by the investigator to be possibly related to any study treatment were reported for 16.2% of patients in the durvalumab group and 12.0% of patients in the gemcitabine-cisplatin group.
Withdrawals Due to Adverse Events
In the overall study, AEs leading to study treatment discontinuation affected 21.1% and 15.2% of patients in the durvalumab and gemcitabine-cisplatin groups, respectively; the most common events included neutropenia (9 patients versus 11 patients), anemia (7 patients versus 7 patients), increased blood creatinine (7 patients versus 4 patients), chronic kidney disease (6 patients versus 3 patients), and acute kidney injury (5 patients versus 2 patients).
In the neoadjuvant phase, AEs leading to the discontinuation of any neoadjuvant study treatment were reported in 14.9% and 15.2% of patients in the durvalumab and gemcitabine-cisplatin groups, respectively; the most common events included neutropenia (9 patients versus 11 patients), anemia (6 patients versus 7 patients), and increased blood creatinine (6 patients versus 4 patients).
AEs leading to the discontinuation of durvalumab in the adjuvant phase were reported in 7.8% of patients, the most common of which included pneumonitis, acute kidney injury, diarrhea, nephritis, fatigue, decreased appetite, and chronic kidney disease (2 patients for each).
Mortality
AEs with an outcome of death were reported in 27 (5.1%) patients versus 29 (5.5%) patients in each group (durvalumab group versus gemcitabine-cisplatin group). Deaths due to AEs occurred most frequently within the system organ class of infections and infestations, affecting 1.9% (10 of 530) of patients in the durvalumab group and 1.3% (7 of 526) in the gemcitabine-cisplatin group.
In the neoadjuvant phase, AEs with an outcome of death were reported in 6 (1.1%) patients versus 10 (1.9%) patients in each group (durvalumab groups versus gemcitabine-cisplatin group). There were no AEs with outcome of death by preferred term in at least 2 patients in either group.
In the adjuvant phase, AEs with an outcome of death were reported in 7 (1.8%) patients versus 6 (1.6%) patients in the durvalumab and gemcitabine-cisplatin groups, respectively. Overall, the most common fatal events in the durvalumab group (≥ 2 patients) included sepsis, cardiorespiratory arrest, death, COVID-19, and pulmonary embolism. In the gemcitabine-cisplatin group, the most common fatal events were recorded as death, septic shock, sepsis, and multiple organ dysfunction syndrome.
Notable Harms
In the overall study period, the proportion of patients with immune-mediated AEs was higher in the durvalumab group (20.9%) than the gemcitabine-cisplatin group (3.0%). Immune-mediated AEs were also higher in the durvalumab group than the gemcitabine-cisplatin group in the neoadjuvant period (8.3% versus 1.5%) and the adjuvant period (13.1% versus 2.1%).
In the overall period, infusion and hypersensitivity reactions were low in both the durvalumab and gemcitabine-cisplatin groups (3.2% versus 2.7%).
Surgery Delayed or Not Performed Due to AEs
In the neoadjuvant phase, AEs leading to surgery not being performed were reported in 1.1% and 1.3% of patients in the durvalumab and gemcitabine-cisplatin groups, respectively, and surgery delays were reported in 1.7% and 1.1% of patients, respectively.
Summary of Findings and Certainty of the Evidence
In the absence of literature-based minimal important difference estimates, thresholds suggested by the clinical experts were used for the following outcomes: pCR (threshold of 8% to 10%), EFS (threshold of 8% to 10%), and OS (threshold of 5% to 10%). In the absence of a known threshold, the certainty in the presence of a nonnull effect was rated for SAEs.
Table 4: Summary of Findings for Perioperative Durvalumab Plus Neoadjuvant Gemcitabine-Cisplatin vs. Neoadjuvant Gemcitabine-Cisplatin for Patients With MIBC
Outcome and follow-up | Patients (studies), N | Relative effect (95% CI) | Absolute effects (95% CI)a | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
Neoadjuvant gemcitabine-cisplatin | Perioperative durvalumab + neoadjuvant gemcitabine-cisplatin | Difference | |||||
Complete response | |||||||
pCRb Evaluated upon surgical resection approximately 6 months after the last patient was randomized | 1,063 (1 RCT) | ██████ ████ ██████ █████ ████ | ██████ ████ ██████ █████ ████ | ██████ ████ ██████ █████ ████ | ██████ ████ ██████ █████ ████ | Moderatec | Perioperative durvalumab plus neoadjuvant gemcitabine-cisplatin likely results in an increased probability of pCR compared to neoadjuvant gemcitabine-cisplatin |
Event-free survival | |||||||
Probability of being alive and event-free at 24 months Median follow-up: 34.7 months (durvalumab group), 27.7 months (gemcitabine-cisplatin group) | 1,063 (1 RCT) | ██ | █████ ████ █████ ████ ███ | █████ ████ █████ ████ ███ | █████ ████ █████ ████ ███ | Moderated | Perioperative durvalumab plus neoadjuvant gemcitabine-cisplatin likely results in an increase probability of event-free survival at 24 months compared to neoadjuvant gemcitabine-cisplatin |
Overall survival | |||||||
Probability of being alive at 36 months Median follow-up: 42.3 months (durvalumab group), 39.6 months (gemcitabine-cisplatin group) | 1,063 (1 RCT) | ██ | █████ ████ █████ ████ ███ | █████ ████ █████ ████ ███ | █████ ████ █████ ████ ███ | Moderatee | Perioperative durvalumab plus neoadjuvant gemcitabine-cisplatin likely results in an increase in overall survival at 36 months compared to neoadjuvant gemcitabine-cisplatin |
HRQoL EORTC QLQ-C30 (GHS) | |||||||
GHS score, change from baseline (95% CI) Median follow-up: 42.3 months (durvalumab group), 39.6 months (gemcitabine-cisplatin group) | 1,063 (1 RCT) | ██ | █████ ████ █████ ███ ██ █ █████ ██ ██ █████ █████ █████ ██████ █████ | Lowf | Perioperative durvalumab plus neoadjuvant gemcitabine-cisplatin may result in little-to-no difference in GHS score compared to neoadjuvant gemcitabine-cisplatin | ||
Harms | |||||||
Proportion of patients with SAEs Median follow-up: 42.3 months (durvalumab group), 39.6 months (gemcitabine-cisplatin group) | 1,056 (1 RCT) | ██ | █████ ████ █████ ████ | █████████ █████████ ███ | █████ ████ █████ ████ | Moderateg | Perioperative durvalumab plus neoadjuvant gemcitabine-cisplatin likely results in an increase in the proportion of patients with SAEs compared to neoadjuvant gemcitabine-cisplatin |
CI = confidence interval; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; GHS = global health status; HRQoL = health-related quality of life; MIBC = muscle invasive bladder cancer; NR = not reported; pCR = pathologic complete response; RCT = randomized controlled trial; SAE = serious adverse event; vs. = versus.
Note: Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aThe 95% CI is reported for absolute effects unless otherwise noted.
bA descriptive reanalysis of pCR was conducted at the second interim analysis (IA2) to incorporate the results from 59 assessments that were excluded from the final pCR analysis because of a programming error. Statistical testing for this outcome was not adjusted for multiplicity and should be considered supportive evidence.
cRated down 1 level for imprecision. The point estimate suggests a clinically important difference in pCR based on a between-group difference threshold of 8% to 10% (80 per 1,000 to 100 per 1,000) suggested by clinical experts; however, the lower bound of the 95% CI crosses this threshold.
dRated down 1 level for imprecision. The point estimate suggests a clinically important difference in event-free survival (EFS) based on a between-group difference threshold of 8% to 10% (80 per 1,000 to 100 per 1,000) suggested by clinical experts; however, the lower bound of the 95.877% CI crosses this threshold. The between-group difference in the probability of EFS was requested from the sponsor to aid in the interpretation of the results for this end point. In the analysis, EFS included patients who refused or did not undergo surgery, which could introduce bias in an open-label setting. A post hoc analysis censoring these patients produced consistent results, mitigating but not eliminating this concern. Patient and investigator decisions to not proceed with surgery were infrequent, balanced between treatment groups, and did not warrant a downgrade in the certainty of evidence.
eRated down 1 level for imprecision. The point estimate suggests a clinically important difference in overall survival (OS) based on a between-group difference threshold of 5% to 10% (50 per 1,000 to 100 per 1,000) suggested by clinical experts; however, the lower bound of the 98.457% CI crosses this threshold. Although OS data may be considered immature at 28.7%, statistical significance was already achieved at IA2; therefore, any subsequent OS results would be descriptive in nature. The between-group difference in survival probability was requested from the sponsor to aid in the interpretation of the results for this end point.
fRated down 2 levels because of missing data in both treatment groups; 30.2% were missing in the perioperative durvalumab plus neoadjuvant gemcitabine-cisplatin group and 27.2% were missing in the neoadjuvant gemcitabine-cisplatin group at baseline, with missing data generally increasing in both treatment groups throughout the study (≥ 50% missing in each treatment group at week 25). Statistical testing for this outcome was not adjusted for multiplicity and should be considered supportive evidence.
gRated down 1 level for serious imprecision. A minimally important difference was not identified, so the target of certainty appraisal was any effect; the 95% CI around the point estimate contains the potential for an increase in SAEs. The between-group difference was requested from the sponsor to aid in the interpretation of the results.
Sources: NIAGARA Clinical Study Report and sponsor’s Summary of Clinical Evidence.8,9 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Long-Term Extension Studies
The submission did not include any long-term extension studies.
Indirect Evidence
In the absence of direct head-to-head trials, indirect evidence can be useful to inform the clinical effectiveness of durvalumab relative to the sponsor-identified relevant comparators. The sponsor provided a feasibility assessment of the possibility of an ITC comparing durvalumab with potential comparator therapies in the perioperative and/or neoadjuvant setting and in the adjuvant setting. After this feasibility assessment, the sponsor concluded that an ITC was not appropriate.
Description of ITCs
The aims of the ITC were to determine the relative efficacy of durvalumab in combination with chemotherapy as neoadjuvant treatment, followed by durvalumab as adjuvant monotherapy, in patients with resectable MIBC (T2N0/1M0 to T4aN0/1M0) compared with other treatments licensed in both neoadjuvant and adjuvant settings (perioperative), other treatments licensed and/or guideline-recommended in the neoadjuvant setting only, and other treatments licensed and/or guideline-recommended in an adjuvant setting only.
Study Selection and Review Methods
The sponsor’s feasibility assessment evaluated whether available clinical trials of patients with MIBC provided sufficiently comparable evidence to support the conduct of an ITC. To do this, a systematic literature review (SLR) was conducted on July 22, 2024, to identify and summarize published evidence from RCTs on the clinical efficacy and safety of existing treatments for adult patients with MIBC. The SLR was conducted according to the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guideline.35 The eligibility criteria were developed using the Population, Intervention, Comparison, Outcomes and Study (PICOS) framework. Briefly, the SLR focused on adults (18 years or older) with untreated MIBC, or with muscle invasive UC if at least 75% of patients had bladder cancer, per baseline data, using perioperative regimens, in accordance with the experimental NIAGARA trial arm or just neoadjuvant or adjuvant treatments.36 Further information on the SLR methods can be found in sponsor-submitted report.37
Summary of Included Studies
A total of 104 publications describing 52 RCTs that reported efficacy and/or safety end points of interest for MIBC interventions were retrieved in the systematic review. The NIAGARA trial, the pivotal trial assessing the efficacy and safety of perioperative durvalumab in combination with neoadjuvant chemotherapy, was not identified in the SLR because it was not published at the time the database was searched.36 The study was then added to the 52 RCTs identified in the SLR. Of the 53 RCTs, 32 were not included in the ITC feasibility assessment because they evaluated different protocols for radical cystectomy alone (3 studies), evaluated 2 or more bladder-preservation regimens (14 studies), assessed neoadjuvant radiotherapy followed by radical cystectomy or assessed adjuvant radiotherapy (5 studies), or assessed adjuvant therapies that are not currently licensed and/or guideline-recommended (10 studies). Briefly, the studies considered in the feasibility assessment included 2 RCTs of perioperative regimens,36,38 17 RCTs of neoadjuvant regimens,39-54 and 3 RCTs evaluating either adjuvant nivolumab or ddMVAC.5,32,50,55
Critical Appraisal of the Sponsor’s Feasibility Assessment
This section appraises the sponsor’s rationale for not conducting an ITC.
Comparison of Perioperative Durvalumab and Adjuvant Nivolumab
The sponsor noted that a standard anchored indirect comparison of perioperative durvalumab to adjuvant nivolumab was infeasible because the timing of randomization in the CheckMate 274 trial differed from that in the NIAGARA trial, selection bias due to differences in randomization criteria, baseline differences in the risk of recurrence, and bias related to the relative efficacy and/or safety of prior immunotherapy. Randomization in the CheckMate 274 trial occurred only after patients had successfully undergone surgery and achieved disease-free status, which excluded those who became ineligible, refused surgery, or had residual disease. This meant that patients had a guaranteed survival period before randomization. In contrast, the NIAGARA trial randomized patients before surgery, meaning that patients with poorer prognoses remained in the study, which could make adjuvant nivolumab appear more effective in comparison. Regarding the baseline risk of recurrence bias, the CheckMate 274 trial recruited only high-risk patients at adjuvant baseline, whereas the NIAGARA trial enrolled patients before neoadjuvant treatment, independent of recurrence risk, resulting in fewer high-stage tumours (pT3 to pT4a or pN+). The sponsor noted that in the NIAGARA trial, durvalumab increased pCR rates and reduced progression to high-risk disease in the neoadjuvant phase, creating a lower-risk tumour stage distribution at adjuvant baseline compared to the CheckMate 274 trial. The clinical experts agreed that comparing perioperative durvalumab to adjuvant nivolumab is not feasible, mainly because of population differences rather than trial timing.
Comparison of Adjuvant Durvalumab and Adjuvant Nivolumab
The sponsor’s feasibility assessment noted that a comparison between adjuvant durvalumab (as part of the perioperative regimen in the NIAGARA trial) and adjuvant nivolumab (in the CheckMate 274 trial) is not feasible because of limited population overlap and the impact of prior immunotherapy exposure in the NIAGARA trial. Several differences in postsurgical baseline characteristics were cited, including the higher proportion of patients with pN0 and pT0 to pT2 disease in the NIAGARA trial and differences in histologic subtypes. Notably, only about 27.5% of patients enrolled in the NIAGARA trial met the high-risk inclusion criteria of the CheckMate 274 trial, compared to more than 90% of patients in the CheckMate 274 trial. Both clinical experts agreed that a comparison between adjuvant durvalumab and adjuvant nivolumab may not be feasible, given the differences in population characteristics.
Comparison of Perioperative Durvalumab and Neoadjuvant ddMVAC
According to the sponsor’s feasibility assessment, multiple methodological limitations that violated the transitivity assumption render an indirect comparison between perioperative durvalumab and neoadjuvant ddMVAC infeasible. For efficacy outcomes, only the VESPER trial could be linked to the NIAGARA trial for pCR, but differences in follow-up durations, nonproportional hazards for OS, and inconsistent definitions of EFS undermined comparability. In addition, substantial baseline imbalances in tumour stage, nodal involvement, and histology, along with missing or inconsistently reported key effect modifiers across trials, made population-adjusted analyses to correct for these biases impossible. As such, the clinical experts agreed that any indirect comparison would likely yield unreliable and potentially misleading estimates of relative treatment effects.
In a recently published systematic review and network meta-analysis (NMA) that included perioperative durvalumab (NIAGARA trial) and ddMVAC (VESPER trial), leveraging gemcitabine-cisplatin as a common comparator to assess relative efficacy,56 the authors applied standard random-effects NMA methodology and reported no statistically significant difference in OS. The analysis showed significantly higher pCR rates with immune checkpoint inhibitors plus chemotherapy than with chemotherapy alone (OR = 2.90; 95% CI, 1.43 to 5.87). A second NMA synthesized data from 7 RCTs, including the NIAGARA and VESPER trials, and used gemcitabine-cisplatin as a common comparator to assess comparative efficacy and safety.57 The findings showed that durvalumab plus gemcitabine-cisplatin had significantly higher pCR rates than gemcitabine-cisplatin (OR = 1.5; 95% CI, 1.1 to 1.9) and numerically superior OS (OR = 0.61; 95% CI, 0.48 to 0.78). However, the clinical experts indicated that it is not possible to make clinical decisions based on these analyses, and agreed with the sponsor that the NMAs did not adequately account for trial heterogeneity or population overlap.
Comparison of Perioperative Durvalumab and Adjuvant ddMVAC
According to the sponsor, an indirect comparison between perioperative durvalumab (NIAGARA trial) and adjuvant ddMVAC (EORTC 30994 and VESPER trials) was not feasible because of incomplete time-to-event data and a lack of common outcomes. In addition to data limitations, the NIAGARA trial evaluated a perioperative treatment that combines neoadjuvant and adjuvant immunotherapy with chemotherapy, whereas the EORTC 30994 trial and the adjuvant cohort in the VESPER trial focused exclusively on postsurgical cytotoxic treatment. The clinical experts noted that without clear baseline stratification, adjustment for key effect modifiers is unreliable, making a comparison of perioperative durvalumab and adjuvant ddMVAC likely infeasible.
Summary
In summary, the sponsor-submitted feasibility assessment concluded that a valid ITC could not be conducted between perioperative durvalumab and relevant comparators because of baseline population differences, a lack of clear stratification of outcomes, and trial heterogeneity, which would make any estimates unreliable and misleading for clinical and health technology assessment (HTA) decision-making. However, this represents an evidence gap, as the absence of such comparisons affects both HTA assessment decisions and clinical guidance.
Studies Addressing Gaps in the Systematic Review Evidence
The submission did not include any studies addressing gaps in the systematic review evidence.
Discussion
This report summarizes the evidence for perioperative durvalumab plus neoadjuvant gemcitabine-cisplatin compared to neoadjuvant gemcitabine-cisplatin in adult patients with MIBC. Eligible patients had a tumour stage of T2 to T4aN0/1M0 and were deemed medically fit to receive neoadjuvant chemotherapy and radical cystectomy. The evidence appraisal was based on the results from the final analysis of pCR (DCO of January 14, 2022) and the IA2 of EFS and OS (DCO of April 29, 2024) from a randomized, open-label, multicentre, global study (NIAGARA; N = 1,063). Outcomes of key interest to patients and clinicians were pCR, EFS, OS, and HRQoL.
The report also summarized a sponsor-submitted report on a feasibility assessment for conducting an ITC between durvalumab in the perioperative or neoadjuvant setting and the adjuvant setting versus potential comparator therapies.
Of note, in March 2025, perioperative treatment with durvalumab, cisplatin-based neoadjuvant chemotherapy, and cystectomy was added as a category 1 recommendation in the National Comprehensive Cancer Network (NCCN) guidelines for patients with resectable MIBC.58
Efficacy
Input from 1 patient group collected for this review highlighted the key treatment goals for patients: to control disease progression, prevent recurrence, improve OS, maintain HRQoL, and improve treatment tolerability. The effect of perioperative durvalumab with neoadjuvant chemotherapy was assessed in the NIAGARA trial by the outcomes of pCR, EFS, OS, EORTC QLQ-C30 GHS, and SAEs.
The Society for Immunotherapy of Cancer and the International Bladder Cancer Group note that pCR is associated with improved OS but is not yet validated as a surrogate in MIBC trials, leading the joint consensus panel to recommend pairing pCR with a coprimary end point such as EFS in phase III trials.59 The clinical experts consulted for this review considered pCR, EFS, and OS to be clinically important outcomes. They noted that a minimum between-group difference of 5% to 10% in OS and of 8% to 10% for each of pCR and EFS would be considered clinically meaningful. Based on these thresholds, perioperative durvalumab with neoadjuvant chemotherapy likely results in an increased probability of the achievement of pCR and EFS at 24 months and OS at 36 months compared with neoadjuvant gemcitabine-cisplatin alone. At IA2, median OS had not yet been reached in either treatment group (immature at 28.7%), with 305 deaths observed in the ITT population. The results for EFS and OS at IA2 were both statistically significant and considered clinically meaningful by the experts; any subsequent data on EFS and OS from the study are descriptive in nature.
A key limitation of the current evidence in the NIAGARA study is that the relative contributions of the neoadjuvant and adjuvant components of perioperative durvalumab treatment are unknown. It remains uncertain how best to manage patients with varying pathologic responses after neoadjuvant immunotherapy treatment plus chemotherapy. For instance, in patients who achieve pCR after neoadjuvant durvalumab plus chemotherapy, the necessity, duration, and potential benefit of continuing with adjuvant immunotherapy is unclear. Conversely, for patients who do not respond to neoadjuvant treatment, it is unknown whether continuing immunotherapy in the adjuvant setting offers any additional benefit or whether it potentially exposes patients to unnecessary toxicity.
In the NIAGARA study, surgery timing was not meaningfully delayed when durvalumab was added in the neoadjuvant setting. The proportion of patients who underwent radical cystectomy was comparable between treatment groups, with a nearly identical median time to surgery of approximately 16 weeks. Another important gap in the NIAGARA study is the lack of information related to patients who did not undergo cystectomy. It is unclear whether these patients received bladder-sparing treatments, such as TMT, or whether they discontinued treatment altogether. Their long-term outcomes, including disease control and survival, remain unknown. This is particularly relevant for patients who may wish to avoid cystectomy in favour of preserving their bladder, which the clinical experts suggested is becoming more common in clinical practice in Canada. According to the 2025 CUA guideline for MIBC, although radical cystectomy has long been the standard treatment for localized MIBC, modern care emphasizes a multidisciplinary approach that includes consideration of bladder preservation with TMT.27 TMT includes maximal TURBT, external beam radiotherapy, and concurrent chemotherapy, and may be offered to patients wishing to retain their bladder, those unfit for cystectomy, or those who decline surgery.27 The guideline states that ideal candidates typically have small (< 5 cm), unifocal tumours without extensive carcinoma in situ, minimal hydronephrosis, good bladder function, and are motivated to participate in regular cystoscopy follow-up.27 The 2025 CUA guideline suggests that TMT is a viable option, not only for those unfit for surgery, but also for any suitable patient, provided treatment decisions are made within a multidisciplinary, shared decision-making framework. Studies show that, in carefully selected patients, TMT offers cancer-specific survival rates comparable to radical cystectomy, with a 5-year disease-specific survival rate of 77% for TMT versus 73% for radical cystectomy, and similar rates of metastasis-free survival.27,29 The guideline notes that only about 30% of surgically fit patients typically meet the criteria for bladder preservation with TMT.27 According to the clinical experts consulted on this review, patients generally have a strong preference to preserve their bladder whenever possible. The experts noted that patients choosing bladder-sparing approaches typically do not receive additional systemic therapy. In their view, TMT alone may be insufficient for disease control, which could disadvantage these patients and unintentionally encourage cystectomy over bladder preservation. Although some concerns exist regarding unbalanced censoring and potential informative dropout in the NIAGARA study, particularly because of the higher proportion of patients in the gemcitabine-cisplatin group who did not proceed to surgery, the study addressed these limitations through sensitivity analyses. In the primary EFS analysis, patients who did not undergo radical cystectomy were conservatively counted as having experienced an event. Sensitivity analyses were then conducted by censoring these patients instead, and the results remained consistent, supporting the robustness of the main findings. Health Canada requested a sensitivity analysis for OS that excluded patients who did not undergo radical cystectomy, which resulted in a wide 95% CI that crosses the null, indicating uncertainty in the results. Although sensitivity analyses offer additional context, excluding patients after randomization may disrupt the balance between treatment groups.
The NIAGARA study did not provide comparative data against other therapies used in clinical practice in Canada, such as adjuvant nivolumab or ddMVAC. The sponsor-submitted ITC feasibility assessment concluded that comparisons between perioperative durvalumab and potential comparators in the perioperative, neoadjuvant, or adjuvant settings were not feasible because of multiple limitations, including immortal time bias, selection bias, differences in recurrence risk, and substantial baseline population differences across trials. The clinical experts consulted for the review agreed that differences in patient characteristics render an ITC between perioperative durvalumab to adjuvant nivolumab infeasible and comparisons with adjuvant nivolumab or adjuvant ddMVAC unreliable because of the lack of a clear stratification of outcomes at adjuvant baseline. According to the clinical experts, existing NMAs did not adequately address trial heterogeneity or population overlap. The clinical experts concluded that no valid ITC could be conducted to inform clinical or HTA decision-making in this context.
Harms
Durvalumab was generally well tolerated in the NIAGARA study, and no new safety signals were observed in the neoadjuvant and adjuvant settings (median duration of 41 months of AE follow-up). A balanced proportion of patients completed 4 neoadjuvant chemotherapy cycles in the NIAGARA study, indicating that the addition of durvalumab did not impact a patient’s ability to receive all cycles of neoadjuvant chemotherapy.
In the study, the addition of neoadjuvant durvalumab to gemcitabine-cisplatin did not lead to a noticeable increase in overall AEs, AEs of grade 3 or 4, infusion reactions, surgery delays, or treatment discontinuations during the neoadjuvant phase. However, there was a slightly greater proportion of patients with SAEs in the neoadjuvant period. AEs of grade 3 or 4, SAEs, discontinuation due to AEs, and immune-related AEs were more frequent during the adjuvant period in the durvalumab group, given that patients in the gemcitabine-cisplatin group did not receive postsurgery treatment. The clinical experts observed that some reported AEs were more likely attributable to the underlying disease or surgery rather than to durvalumab or gemcitabine-cisplatin treatment. Importantly, in the NIAGARA study, SAEs deemed by investigators to be possibly related to the study treatment were comparable between groups. The clinical experts agreed that there was no clinically meaningful difference in treatment-related SAEs.
The sponsor-submitted ITC feasibility assessment also identified limitations in the evidence base for safety outcomes, noting that only the NIAGARA and SWOG S1314 trials were connected for any-grade treatment-related AEs and that no connected data existed for grade 3 or 4 treatment-related AEs across any relevant trials, preventing meaningful indirect comparisons of tolerability.
Ethics and Equity Considerations
According to the clinical experts consulted, a key ethical concern is that some patients in clinical practice in Canada, particularly those treated at nonacademic centres, may miss the opportunity to receive neoadjuvant chemotherapy because of inconsistent referral practices among urologists before surgery. If these patients are later found to have more advanced disease, the optimal treatment window for neoadjuvant chemotherapy has often passed. Establishing a standardized, approved neoadjuvant treatment approach could help ensure that more patients receive guideline-recommended care, according to the experts.
Evidence notes that radical cystectomy often leads to permanent changes in urinary function (including the need for urinary diversion), alterations in body image and sexual function, and associated emotional and/or psychological burdens.60 The CUA guideline suggests that contemporary care should involve a multidisciplinary approach, including a discussion about bladder preservation with TMT. However, access to bladder-sparing strategies such as TMT remains limited in certain centres in Canada, raising equity concerns. The experts noted that patients in remote or less specialized settings are less likely to receive TMT and more likely to undergo cystectomy because of a lack of local expertise. Experts emphasized that patients from urban marginalized populations may encounter structural or socioeconomic barriers that limit their ability to attend the daily radiation treatments required for bladder-sparing approaches. These barriers may reduce the acceptability or accessibility of such treatments, thereby influencing the likelihood that therapy will be completed. The experts noted that improving pCR rates through neoadjuvant treatment could potentially expand the number of patients eligible for bladder preservation.
Conclusion
One phase III, open-label, multicentre RCT (NIAGARA) provided evidence for the efficacy and safety of perioperative durvalumab in combination with neoadjuvant gemcitabine-cisplatin in patients with resectable MIBC. According to the experts, the study population was broadly representative of patients seen in clinical practice in Canada, including those with impaired renal function (CrCl, 40 mL/min to 60 mL/min) and node-positive disease. The NIAGARA study demonstrated a likely clinically important increase in pCR, EFS at 24 months, and OS at 36 months compared to gemcitabine-cisplatin alone. Harms were consistent with the known safety profile of cisplatin-based chemotherapy and PD-L1 inhibitors, and no new safety signals were identified. These results are derived from an interim analysis of immature data; long-term follow-up data will be forthcoming. Uncertainties remain regarding the relative contribution of the neoadjuvant versus adjuvant components of therapy and the applicability of the findings to patients undergoing bladder-sparing approaches. The absence of direct or indirect comparative evidence against other relevant systemic therapies, such as ddMVAC or adjuvant nivolumab, is a noted limitation. The sponsor-submitted feasibility assessment concluded that a valid ITC could not be conducted between perioperative durvalumab and relevant comparators because of substantial methodological and clinical limitations, including baseline population differences, a lack of clear stratification of outcomes, and trial heterogeneity. As a result, the sponsor noted that any effect estimates from such comparisons would be unreliable and could mislead both clinical and HTA decision-making. Nonetheless, this represents an evidence gap, as the efficacy of durvalumab relative to other available treatments remains unknown.
Economic Review
Methods
The review team appraised the pharmacoeconomic evidence submitted by the sponsor on the cost-effectiveness and budget impact of neoadjuvant durvalumab in combination with gemcitabine-cisplatin followed by adjuvant durvalumab monotherapy after radical cystectomy (henceforth referred to as the perioperative durvalumab regimen) compared to relevant comparators for the treatment of patients with MIBC.
Summary of the Submitted Economic Evaluation
The sponsor submitted a cost-utility analysis to estimate the cost-effectiveness of the perioperative durvalumab regimen from the perspective of a public health care payer in Canada over a lifetime horizon (35.6 years). The modelled population comprised cisplatin-eligible patients with MIBC who are considered medically fit to undergo radical cystectomy, which is aligned with the Health Canada indication and was based on participants in the NIAGARA trial.61 The sponsor’s base-case analysis included costs related to drug acquisition, administration, AEs, subsequent treatment, disease management, terminal care, and radical cystectomy.61
In the sponsor’s base case, the perioperative durvalumab regimen was associated with incremental costs of $89,724 and 1.7 incremental QALYs relative to neoadjuvant gemcitabine-cisplatin. This resulted in an ICER of $52,316 per QALY gained. Of the incremental benefit compared to neoadjuvant gemcitabine-cisplatin (1.74 incremental QALYs), approximately 92% of benefit was predicted to be accrued after the treatment duration of the NIAGARA trial7 (median follow-up period of 42.3 months). Additional information about the sponsor’s submission is summarized in the Supplemental Materials document, Appendix 10.
CDA-AMC identified several key issues with the sponsor’s analysis (refer to Table 5; full details are provided in the Supplemental Materials document, Appendix 11).
Table 5: Key Issues With the Sponsor’s Economic Submission
Issue | What evidence is there to inform this issue? | How was this issue addressed by CDA-AMC? | Did CDA-AMC explore uncertainty in a scenario analysis? |
|---|---|---|---|
Relevant comparators were omitted. | The sponsor did not submit indirect evidence for ddMVAC or adjuvant nivolumab, both relevant comparators.6 The cost-effectiveness of perioperative durvalumab vs. these or any comparator other than neoadjuvant gemcitabine-cisplatin is unknown. | CDA-AMC could not address this issue in the base case, as no indirect evidence was provided. | No scenario analysis was conducted. |
Uncertainty in long-term efficacy. | The sponsor’s extrapolations suggested ongoing recurrence or death with gemcitabine-cisplatin beyond 5 years,61 whereas the clinical experts noted patients may be cured after this time. | CDA-AMC aligned gemcitabine-cisplatin and durvalumab TTR and TTD (pre recurrence) curves after 7 years. | Because of limited long-term data, CDA-AMC tested scenarios with alignment at 5 years and 10 years. |
The ToT estimate did not align with clinical expectations, leading to an underestimation of durvalumab costs. | In the NIAGARA trial, 54% of patients completed durvalumab and 81% completed gemcitabine-cisplatin;7 the experts expect higher completion rates in practice. | CDA-AMC assumed that at least 70% of patients would complete the perioperative durvalumab regimen. | A scenario analysis was conducted in which the observed ToT from the NIAGARA trial was maintained. |
Patient outcomes associated with subsequent treatments are uncertain. | The model assumes 47.5% of patients receive pembrolizumab and 47.5% receive enfortumab vedotin; these rates are higher than in the NIAGARA trial,61 which adds costs without extending post recurrence survival.7 | CDA-AMC was unable to address this limitation because of a lack of post recurrence survival evidence. | No scenario analysis was conducted. |
The model may overestimate the proportion of patients undergoing RC, inflating costs. | In the NIAGARA trial, 83% to 88% of patients underwent RC,7 but experts expect only 50% to 75% would in practice because of bladder-sparing preferences. | CDA-AMC could not address this limitation, given the uncertainty of outcomes for patients who decline RC. | A scenario analysis was conducted in which 50% of patients declined RC. |
Health-state utilities exceeded those in the age-matched general population. | Model utilities from the NIAGARA trial exceeded age-adjusted norms,7,62 although experts did not expect patients with EF or RD MIBC to have a higher QoL than those in the general population. | CDA-AMC was unable to address this limitation because of a lack of alternative estimates. | Two scenarios were tested: EF utilities were capped at population norms while the NIAGARA trial’s relative difference was retained; and EF was capped and RD was reduced by 15%. |
Resource costs were underestimated for neoadjuvant gemcitabine-cisplatin because they did not reflect clinical practice. | The clinical experts indicated a higher frequency of oncology-nurse encounters and medical-oncology consultations in clinical practice.61 | CDA-AMC adjusted the frequency of oncology-nurse encounters and medical-oncology consultations to reflect clinical practice. | No scenario analysis was conducted. |
CDA-AMC = Canada’s Drug Agency; ddMVAC = dose-dense methotrexate, vinblastine, Adriamycin (doxorubicin), and cisplatin; EF = event-free; MIBC = muscle invasive bladder cancer; QoL = quality of life; RC = radical cystectomy; RD = recurrent disease; ToT = time on treatment; TTD = time to death; TTR = time to recurrence; vs. = versus.
Note: Full details of the issues identified by CDA-AMC are provided in the Supplemental Materials document, Appendix 11.
CDA-AMC Assessment of Cost-Effectiveness
The CDA-AMC base case was derived by making changes to model parameter values and assumptions (refer to the Supplemental Materials document, Appendix 11, Table 17), in consultation with clinical experts. Detailed information about the CDA-AMC base case is provided in the Supplemental Materials document, Appendix 11.
Impact on Health Care Costs
The perioperative durvalumab regimen is predicted to be associated with additional health care costs compared to neoadjuvant gemcitabine-cisplatin (incremental costs = $98,565). This increase in health care spending results from drug-acquisition costs associated with durvalumab (refer to Figure 1).
Figure 1: Impact of a Perioperative Durvalumab Regimen vs. Neoadjuvant Gemcitabine and Cisplatin on Health Care Costs
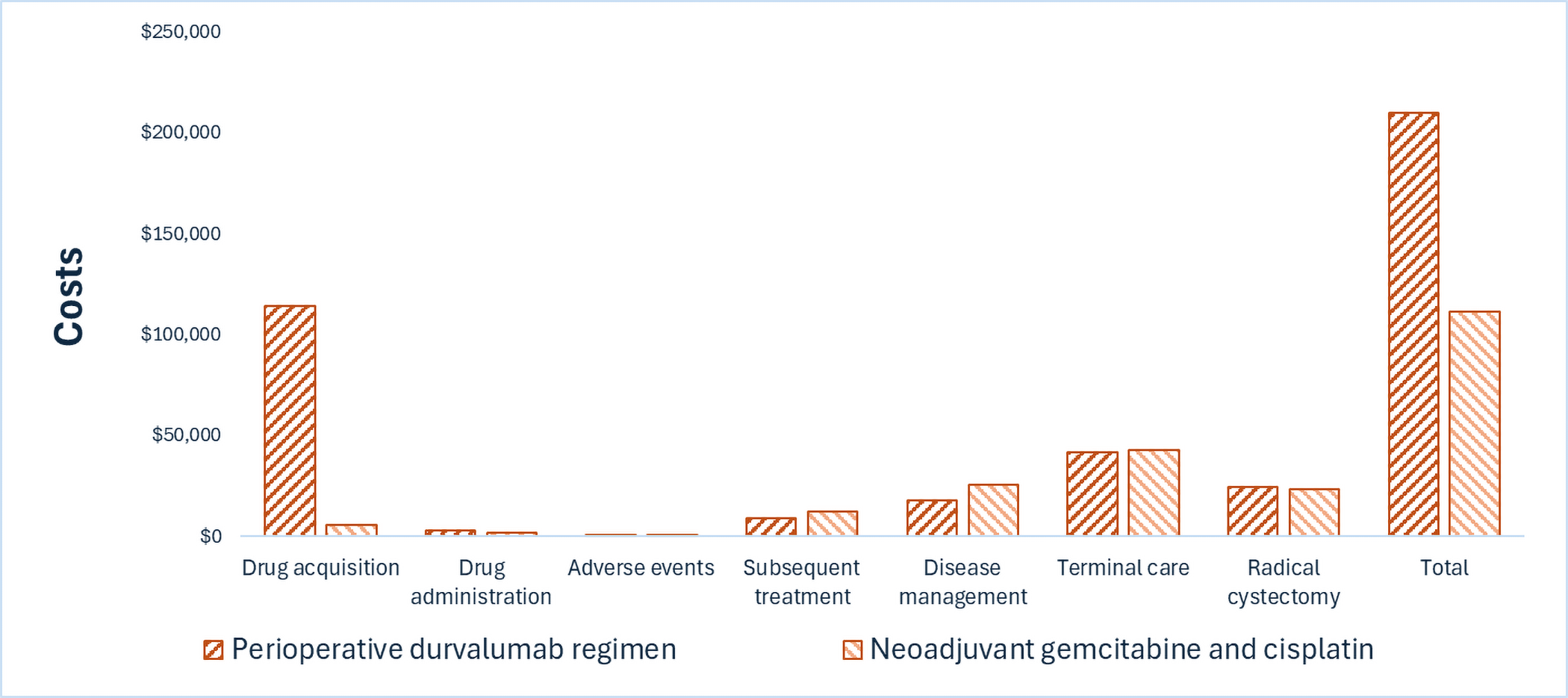
Impact on Health
Relative to neoadjuvant gemcitabine-cisplatin, the perioperative durvalumab regimen is predicted to increase the amount of time a patient remains in the event-free health state by approximately 2 years and to extend OS by 1.8 years (refer to Figure 2). Considering the impact of treatment on both quality and length of life, the perioperative durvalumab regimen is predicted to result in 1.51 additional QALYs per patient compared to neoadjuvant gemcitabine-cisplatin. Approximately 92% of the predicted incremental benefit was accrued on the basis of extrapolation.
Figure 2: Impact of a Perioperative Durvalumab Regimen vs. Neoadjuvant Gemcitabine and Cisplatin on Patient Health
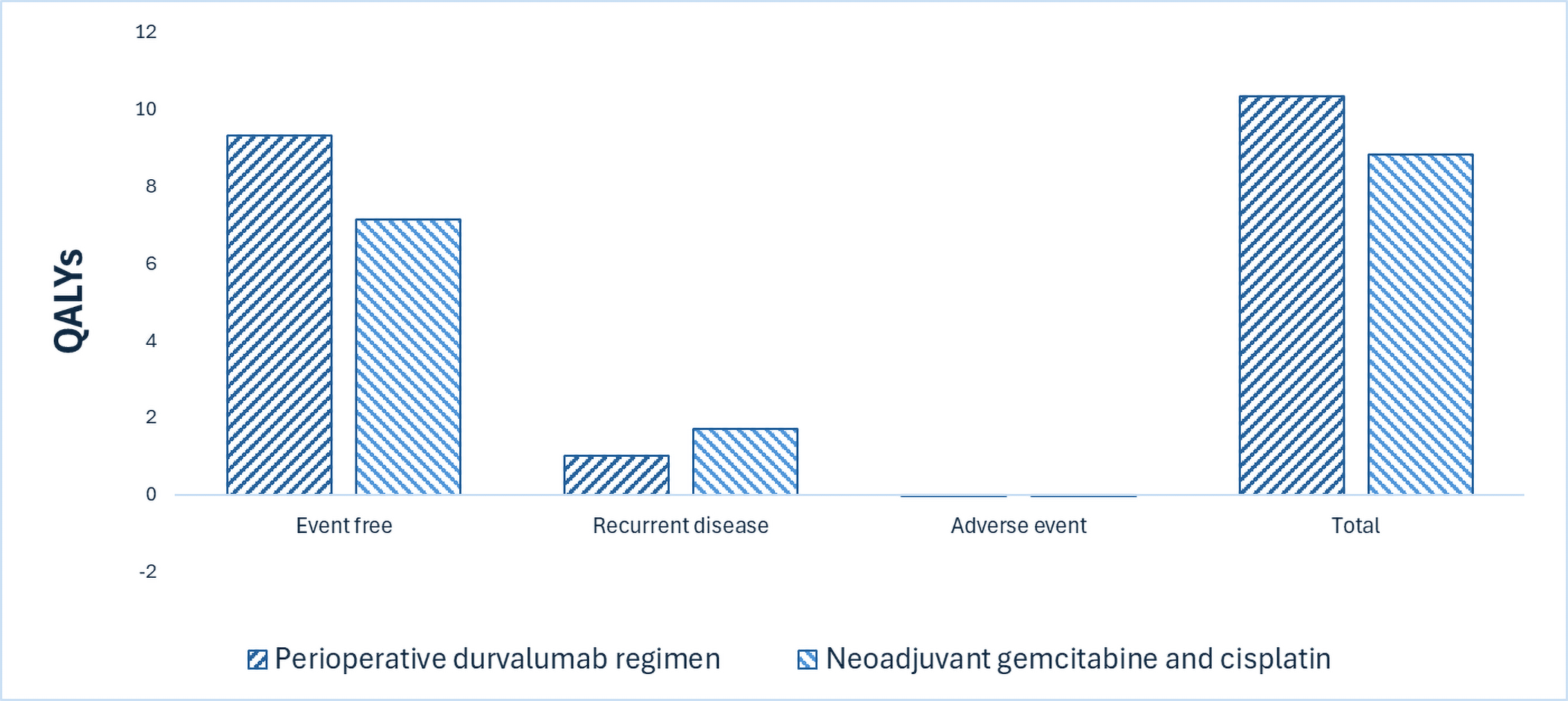
QALY = quality-adjusted life-year.
Overall Results
The results of the CDA-AMC base case suggest an ICER of $65,194 per QALY gained for the perioperative durvalumab regimen compared to neoadjuvant gemcitabine-cisplatin (refer to Table 6). Additional details on the CDA-AMC base case are available in the Supplemental Materials document, Appendix 11.
Table 6: Summary of CDA-AMC Economic Evaluation Results
Drug | Total costs ($) | Total LYs | Total QALYs | ICER vs. neoadjuvant gemcitabine-cisplatin ($/QALY) |
|---|---|---|---|---|
Neoadjuvant gemcitabine-cisplatin | 111,152 | 10.63 | 8.81 | Reference |
Perioperative durvalumab regimen | 209,717 | 12.46 | 10.32 | 65,194 |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year; vs. = versus.
Note: Publicly available list prices were used for all comparators.
Uncertainty and Sensitivity
Treatment waning at the 5-year and 10-year time points was applied, in which the hazard of pre recurrence death and progression for neoadjuvant gemcitabine-cisplatin was assumed to be equal to that for the perioperative durvalumab regimen.
The proportion of patients who undergo radical cystectomy was assumed to be 50%.
Two scenarios regarding health-state utilities were tested. In the first, event-free utilities were capped at population norms while the NIAGARA trial’s relative difference was retained; in the second, event-free utilities were capped and recurrent-disease utilities were reduced by 15%.
The time on treatment observed in the NIAGARA trial was maintained.
Summary of the Budget Impact
The sponsor submitted a budget impact analysis to estimate the 3-year (2026 to 2028) budget impact of reimbursing neoadjuvant durvalumab in combination with gemcitabine-cisplatin, followed by adjuvant durvalumab monotherapy, for use in the Health Canada–indicated population. The sponsor assumed that the payer would be CDA-AMC–participating public drug plans, and derived the size of the eligible population using an epidemiologic approach. The price of durvalumab was aligned with the price included in the sponsor’s economic evaluation, while the prices of comparators were based on the publicly available list prices. Additional information pertaining to the sponsor’s submission is provided in the Supplemental Materials document, Appendix 12.
CDA-AMC identified a number of issues with the sponsor’s estimated budget impact and made changes to model parameters and assumptions in consultation with clinical experts to derive the CDA-AMC base case (Supplemental Materials document, Appendix 12). CDA-AMC estimated that 2,384 patients would be eligible for treatment with the perioperative durvalumab regimen over a 3-year period (year 1 = 788; year 2 = 793; year 3 = 802), of whom 1,269 are expected to receive durvalumab (year 1 = 331; year 2 = 420; year 3 = 518). The estimated incremental budget impact of reimbursing durvalumab is predicted to be approximately $111 million over the first 3 years, with an expected expenditure of $170 million on durvalumab. The actual budget impact will depend on the number of eligible patients and the cost of subsequent treatment.
Conclusion
Based on the CDA-AMC base case, the perioperative durvalumab regimen would be considered cost-effective at the submitted price if the public health care system was willing to pay at least $65,194 for each additional QALY gained. If the public health care system is not willing to pay that amount, a price reduction should be considered (refer to Figure 3; full details of the impact of price reductions on cost-effectiveness are presented in the Supplemental Materials document, Appendix 11, Table 20).
The budget impact of reimbursing the perioperative durvalumab regimen on the public drug plans in the first 3 years is estimated to be approximately $111 million. The 3-year expenditure on durvalumab (i.e., not accounting for current expenditure on comparators) is estimated to be $170 million. The estimated budget impact is uncertain because of potential continuation of durvalumab treatment if surgery is refused and because of the cost of subsequent treatment.
Figure 3: Summary of the CDA-AMC Economic Analysis and Price Reduction
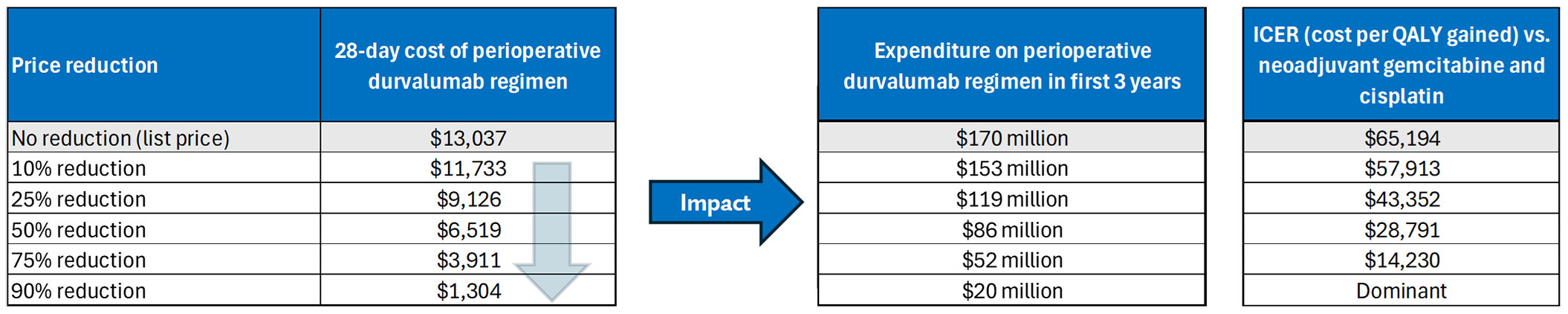
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Note: Expenditure includes only the drug cost of durvalumab. The term dominant indicates that a drug costs less and provides more QALYs than the comparator.
References
1.Kulkarni GS, Black PC, Sridhar SS, et al. Canadian Urological Association Guideline: Muscle-invasive bladder cancer. Canadian Urological Association Journal. 2019;13(8):230-238. PubMed
2.Canadian Cancer Society. Cancer statistics at a glance. 2024. Accessed May 28, 2025. https://cancer.ca/en/research/cancer-statistics/cancer-statistics-at-a-glance
3.Bree KK, Hensley PJ, Westerman ME, et al. Contemporary Rates of Gynecologic Organ Involvement in Females with Muscle Invasive Bladder Cancer: A Retrospective Review of Women Undergoing Radical Cystectomy following Neoadjuvant Chemotherapy. J Urol. 2021;206(3):577-585. doi:10.1097/JU.0000000000001784 PubMed
4.Royal-Preyra B, Cury FL, Katib Y, et al. The Rate of Prostatic Involvement in Men Treated With Radical Cystectomy for Muscle Invasive Bladder Cancer. Pract Radiat Oncol. 2023;13(1):e68-e72. doi:10.1016/j.prro.2022.06.001 PubMed
5.AstraZeneca Canada Inc. Imfinzi (durvalumab for injection): concentrate for solution for infusion, 50 mg / mL, intravenous [product monograph]. November 03, 2017. Updated January 17, 2025. Accessed July 21, 2025. https://pdf.hres.ca/dpd_pm/00078342.PDF
6.AstraZeneca Canada Inc. Drug Reimbursement Review sponsor submission: Imfinzi (durvalumab for injection), concentrate for solution for infusion, 50 mg / mL, intravenous [internal sponsor's package]. June 13, 2025.
7.NIAGARA Clinical Study Report - AstraZeneca. Data on file [sponsor supplied reference]. 2024.
8.AstraZeneca Canada Inc. Clinical Study Report: D933RC00001. A Phase III, Randomized, Open-Label, Multi-Center, Global Study to Determine the Efficacy and Safety of Durvalumab in Combination with Gemcitabine+Cisplatin for Neoadjuvant Treatment Followed by Durvalumab Alone for Adjuvant Treatment in Patients with Muscle-Invasive Bladder Cancer (NIAGARA) [internal sponsor's report]. September 16, 2024.
9.CDA-AMC Sponsor Summary of Clinical Evidence Template [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Imfinzi (durvalumab), Concentrate for solution for infusion, 50 mg/mL, Intravenous. AstraZeneca Canada Inc.; 2025.
10.CADTH Reimbursement Recommendation: Durvalumab (Imfinzi). Can J Health Technol 2021;1(7). doi:10.51731/cjht.2021.109
11.CADTH Reimbursement Recommendation Durvalumab (Imfinzi). Can J Health Technol. 2023;3(2). doi:10.51731/cjht.2023.635
12.pCODR Expert Review Committee (pERC) final recommendation: Durvalumab (Imfinzi). CADTH. 2019. Accessed July 21, 2025. https://www.cda-amc.ca/sites/default/files/pcodr/Reviews2019/10131DurvalumabNSCLC_fnRec_02May2019_approvedbyChair_Post_03May2019_final.pdf
13.CADTH Reimbursement Recommendation: Tremelimumab (Imjudo) in Combination With Durvalumab (Imfinzi). Can J Health Technol. 2023;3(11). doi:10.51731/cjht.2023.788
14.CDA-AMC Reimbursement Recommendation: Durvalumab (Imfinzi). Can J Health Technol. 2025;5(5). doi:10.51731/cjht.2025.1133
15.Sung H, Ferlay J, Siegel RL, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021;71(3):209-249. doi:10.3322/caac.21660 PubMed
16.Dawood EE, Awadalla A, Hashem A, Shokeir AA, Abdel-Aziz AF. Evaluation of molecular signatures in the urinary bladder and upper tract urothelial carcinomas: a prospective controlled clinical study. J Egypt Natl Canc Inst. 2022;34(1):47. doi:10.1186/s43046-022-00148-x PubMed
17.Govindasamy R, Selvi SK, Srivani S, Ivan EA. Rare histological types of bladder carcinoma from a tertiary care hospital: Case reports. Journal of Medical and Scientific Research. 2023;11(1):60-63. doi:10.17727/jmsr.2023/11-12
18.American Cancer Society: What Is Bladder Cancer? [sponsor supplied reference]. March 2024; https://www.cancer.org/cancer/types/bladder-cancer/about/what-is-bladder-cancer.html. Accessed on 31 Jan 2025.
19.Park JC, Citrin DE, Agarwal PK, Apolo AB. Multimodal management of muscle-invasive bladder cancer. Curr Probl Cancer. 2014;38(3):80-108. doi:10.1016/j.currproblcancer.2014.06.001 PubMed
20.Krishna SR, Konety BR. Current concepts in the management of muscle invasive bladder cancer. Indian J Surg Oncol. 2017;8(1):74-81. doi:10.1007/s13193-016-0586-1 PubMed
21.Jakus D, Solic I, Juric I, Borovac J, Situm M. The impact of the initial clinical presentation of bladder cancer on histopathological and morphological tumor characteristics. J Clin Med. 2023;12(13):4259. doi:10.3390/jcm12134259. PubMed
22.Morra M, Chung D, Kassouf W, et al. MP08-15 BLADDER CANCER STAGE AT DIAGNOSIS IN RURAL VERSUS URBAN PATIENTS IN CANADA: RESULTS FROM THE CANADIAN BLADDER CANCER INFORMATION SYSTEM (CBCIS). J Urol. 2025;213(5S):e248.
23.Alfred Witjes J, Max Bruins H, Carrion A, et al. European Association of Urology guidelines on muscle-invasive and metastatic bladder cancer: Summary of the 2023 guidelines. Eur Urol. 2024;85(1):17-31. doi:10.1016/j.eururo.2023.08.016 PubMed
24.Brenner DR, Gillis J, Demers AA, et al. Projected estimates of cancer in Canada in 2024. CMAJ. 2024;196(18):E615-E623. doi:10.1503/cmaj.240095 PubMed
25.CCSAC. Canadian cancer statistics: A 2024 special report on the economic impact of cancer in Canada. Canadian Cancer Society; 2024. Accessed July 21, 2025. https://cdn.cancer.ca/-/media/files/research/cancer-statistics/2024-statistics/2024-special-report/2024_pdf_en.pdf
26.Kulkarni GS, Urbach D, Austin P, Fleshner N, Laupacis A. Higher surgeon and hospital volume improves long‐term survival after radical cystectomy. Cancer 2013;119 (19):3546-3554. PubMed
27.Kulkarni GS, Black PC, Sridhar SS, et al. 2025 Canadian Urological Association Expert Report: Muscle-invasive bladder cancer. Can Urol Assoc J. 2025;19(1):E1-E16. doi:10.5489/cuaj.9096 PubMed
28.Jiang DM, Gupta S, Kitchlu A, et al. Defining cisplatin eligibility in patients with muscle-invasive bladder cancer. Nat Rev Urol. 2021;18(2):104-114. doi:10.1038/s41585-020-00404-6 PubMed
29.Zlotta AR, Ballas LK, Niemierko A, et al. Radical cystectomy versus trimodality therapy for muscle-invasive bladder cancer: a multi-institutional propensity score matched and weighted analysis. Lancet Oncol. 2023;24(6):669-681. doi:10.1016/S1470-2045(23)00170-5 PubMed
30.Galsky MD, Witjes JA, Gschwend JE, et al. Adjuvant Nivolumab in High-Risk Muscle-Invasive Urothelial Carcinoma: Expanded Efficacy From CheckMate 274. J Clin Oncol. 2025;43(1):15-21. doi:10.1200/JCO.24.00340 PubMed
31.Vale CL. Neoadjuvant chemotherapy in invasive bladder cancer: update of a systematic review and meta-analysis of individual patient data: advanced bladder cancer (ABC) meta-analysis collaboration. Eur Urol. 2005;48(2):202-206. PubMed
32.Bajorin DF, Witjes JA, Gschwend JE, et al. Adjuvant Nivolumab versus Placebo in Muscle-Invasive Urothelial Carcinoma. N Engl J Med. 2021;384(22):2102-2114. doi:10.1056/NEJMoa2034442 PubMed
33.Bellmunt J, Hussain M, Gschwend JE, et al. Adjuvant atezolizumab versus observation in muscle-invasive urothelial carcinoma (IMvigor010): a multicentre, open-label, randomised, phase 3 trial. . Lancet Oncol. 2021;22(4):525-537. doi:10.1016/S1470-2045(21)00004-8 PubMed
34.Cathomas R, Rothschild SI, Hayoz S, et al. Perioperative chemoimmunotherapy with durvalumab for muscle-invasive urothelial carcinoma: Primary analysis of the single-arm Phase II trial SAKK 06/17. J Clin Oncol 2023;41(33):5131-5139. doi:10.1200/JCO.23.00363 PubMed
35.Page MJ, McKenzie JE, Bossuyt PM, et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ. 2021;372. PubMed
36.Powles T, Catto JWF, Galsky MD, et al. Perioperative durvalumab with neoadjuvant chemotherapy in operable bladder cancer. N Engl J Med. 2024;391(19):1773-1786. doi:10.1056/NEJMoa2408154 PubMed
37.IQVIA. Systematic Literature Review (SLR) report of durvalumab in MIBC [sponsor supplied reference]. 2024.
38.NCT04209114. A study of nivolumab plus bempegaldesleukin (bempeg/NKTR-214) vs nivolumab alone vs standard of care in participants with bladder cancer that may have invaded the muscle wall of the bladder and who cannot get cisplatin, a type of medicine given to treat bladder cancer [sponsor provided reference]. https://clinicaltrials.gov/study/NCT04209114
39.Martinez Chanza N, Carnot A, Barthelemy P, et al. Avelumab as the basis of neoadjuvant regimen in platinum-eligible and-ineligible patients with nonmetastatic muscle-invasive bladder cancer: AURA (Oncodistinct-004) trial. American Society of Clinical Oncology; 2022.
40.Cooke PW, Dunn JA, Latief T, Bathers S, James N, Wallace D. Long–Term Risk of Salvage Cystectomy after Radiotherapy for Muscle–Invasive Bladder Cancer. Eur Urol. 2000;38(3):279-286. PubMed
41.Flaig TW, Tangen CM, Daneshmand S, et al. A randomized phase II study of coexpression extrapolation (COXEN) with neoadjuvant chemotherapy for bladder cancer (SWOG S1314; NCT02177695). Clin Cancer Res. 2021;27(9):2435-2441. PubMed
42.Galsky MD, Sfakianos JP, Ye D, et al. Oral APL-1202 in combination with tislelizumab as neoadjuvant therapy in patients with muscle-invasive bladder cancer (MIBC): Interim analysis of ANTICIPATE phase II trial. American Society of Clinical Oncology; 2024.
43.Grossman H. Neoadjuvant chemotherapy plus cystectomy compared with cystectomy alone for locally advanced bladder cancer (vol 349, pg 859, 2003). N Engl J Med. 2003;349(19):1880-1880.
44.Huddart RA, Birtle A, Maynard L, et al. Clinical and patient‐reported outcomes of SPARE–a randomised feasibility study of selective bladder preservation versus radical cystectomy. BJU Int. 2017;120(5):639-650. PubMed
45.Griffiths G, R; Raghavan, D; Parmar M; Hall R;. International phase III trial assessing neoadjuvant cisplatin, methotrexate, and vinblastine chemotherapy for muscle invasive bladder cancer: Long-term results of the BA06 30894 trial [sponsor provided reference]. J Clin Oncol. 2011;29(16):2171-2177. PubMed
46.Khaled HM, Shafik HE, Zabhloul M, et al. Gemcitabine and cisplatin as neoadjuvant chemotherapy for invasive transitional and squamous cell carcinoma of the bladder: effect on survival and bladder preservation. Clin Genitourin Cancer. 2014;12(5):e233-e240. PubMed
47.Kitamura H, Tsukamoto T, Shibata T, et al. Randomised phase III study of neoadjuvant chemotherapy with methotrexate, doxorubicin, vinblastine and cisplatin followed by radical cystectomy compared with radical cystectomy alone for muscle-invasive bladder cancer: Japan Clinical Oncology Group Study JCOG0209. Ann Oncol. 2014;25(6):1192-1198. PubMed
48.Martinez-Piñeiro JA, Martin MG, Arocena F, et al. Neoadjuvant cisplatin chemotherapy before radical cystectomy in invasive transitional cell carcinoma of the bladder: a prospective randomized phase III study. The Journal of urology. 1995;153(3 Pt 2):964-973. PubMed
49.Osman MA, Gabr AM, Elkady MS. Neoadjuvant chemotherapy versus cystectomy in management of stages II, and III urinary bladder cancer. Archivio Italiano di Urologia e Andrologia. 2014;86(4):278-283. PubMed
50.Pfister C, Gravis G, Flechon A, et al. Dose-Dense Methotrexate, Vinblastine, Doxorubicin, and Cisplatin or Gemcitabine and Cisplatin as Perioperative Chemotherapy for Patients With Nonmetastatic Muscle-Invasive Bladder Cancer: Results of the GETUG-AFU V05 VESPER Trial. J Clin Oncol. 2022;40(18):2013-2022. doi:10.1200/JCO.21.02051 PubMed
51.Sengeløv L, Maase HVD, Lundbeck F, et al. Neoadjuvant chemotherapy with cisplatin and methotrexate in patients with muscle-invasive bladder tumours. Acta Oncol. 2002;41(5):447-456. PubMed
52.Sherif A, Rintala E, Mestad O, et al. Neoadjuvant cisplatin-methotrexate chemotherapy for invasive bladder cancer-Nordic cystectomy trial 2. Scand J Urol Nephrol. 2002;36(6):419-425. PubMed
53.Thibault C, Bennamoun M, Flechon A, et al. 2364MO Durvalumab (D)+/−tremelimumab (T) in combination with dose-dense MVAC (ddMVAC) as neoadjuvant treatment in patients with muscle-invasive bladder carcinoma (MIBC): Results of NEMIO, a randomized phase I-II trial. Ann Oncol. 2023;34:S1202.
54.Wallace DM, Raghavan D, Kelly K, et al. Neoadjuvant (preemptive) cisplatin therapy in invasive transitional cell carcinoma of the bladder. Br J Urol. 1991;67(6):608-615. PubMed
55.Sternberg C, N Skoneczna I, Kerst JM, et al. Immediate versus deferred chemotherapy after radical cystectomy in patients with pT3-pT4 or N+ M0 urothelial carcinoma of the bladder (EORTC 30994): an intergroup, open-label, randomised phase 3 trial. Lancet Oncol. 2015;16(1):76-86. doi:10.1016/S1470-2045(14)71160-X PubMed
56.Matsukawa A,Cormio A, Miszczyk M, et al. Impact of Immune Checkpoint Inhibitors as Neoadjuvant Therapy for Muscle-invasive Bladder Cancer: A Systematic Review, Meta-analysis, and Network Meta-analysis. Eur Urol Oncol 2025:S2588-9311: 1673-1684. doi:10.1016/j.euo.2025.02.009 PubMed
57.Noelle P, Joseph F, Tsao A, et al. Efficacy and Safety of Neoadjuvant Chemotherapy for Muscle-Invasive Bladder Cancer: A Systematic Review and Network Meta-Analysis of RCTs. Journal of Clinical Question. 2025;2(2):e62. doi:10.69854/jcq.2025.0001
58.NCCN. Guidelines for Patients: Bladder Cancer Version 1.2025 [sponsor supplied reference]. 2025. NCCN. Guidelines for Patients: Bladder Cancer.(https://www.nccn.org/guidelines/guidelines-detail?category=1&id=1417).
59.Kamat AM, Apolo AB, Babjuk M, et al. Definitions, End Points, and Clinical Trial Designs for Bladder Cancer: Recommendations From the Society for Immunotherapy of Cancer and the International Bladder Cancer Group. J Clin Oncol 2023;41(35):5437-5447. doi:10.1200/JCO.23.00307 PubMed
60.Canadian Cancer Society. Supportive care for bladder cancer. 2024. Accessed September 16, 2025. https://cancer.ca/en/cancer-information/cancer-types/bladder/supportive-care
61.AstraZeneca Canada Inc. Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Imfinzi (durvalumab for injection), concentrate for solution for infusion, 50 mg / mL, intravenous. June 13, 2025.
62.Yan J, Xie S, Johnson JA, et al. Canada population norms for the EQ-5D-5L. Eur J Health Econ. 2024;25(1):147-155. doi:10.1007/s10198-023-01570-1 PubMed
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.