Drugs, Health Technologies, Health Systems
Reimbursement Review
Pembrolizumab (Keytruda)
Sponsor: Merck Canada Inc.
Therapeutic area: Head and neck squamous cell carcinoma
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
AE
adverse event
AEOSI
adverse event of special interest
APaT
all participants as treated
BICR
blinded independent central review
CCSN
Canadian Cancer Survivor Network
CI
confidence interval
CPS
combined positive score
CRT
chemoradiotherapy
DMFS
distant metastasis-free survival
ECOG
Eastern Cooperative Oncology Group
EFS
event-free survival
EORTC QLQ-C30
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30
EORTC QLQ-H&N35
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Head and Neck Module 35
FAS
full analysis set
GHS
global health status
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HNSCC
head and neck squamous cell carcinoma
HR
hazard ratio
HRQoL
health-related quality of life
IA1
first interim analysis
IA2
second interim analysis
ICI
immune checkpoint inhibitor
ITT
intention to treat
KM
Kaplan-Meier
LRC
local regional control
LS
least squares
LSTN
Life-Saving Therapies Network
mPR
major pathological response
NR
not reported
OH (CCO)
Ontario Health (Cancer Care Ontario)
OS
overall survival
pCR
pathological complete response
PRO
patient-reported outcome
PS
Performance Status
QoL
quality of life
RCT
randomized controlled trial
RECIST
Response Evaluation Criteria in Solid Tumours
RT
radiotherapy
SAE
serious adverse event
SCC
squamous cell carcinoma
SOC
standard of care
TPS
tumour proportion score
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Pembrolizumab (Keytruda), 100 mg/4 mL vial, solution for infusion |
Sponsor | Merck Canada Inc. |
Indication | For the treatment of adult patients with resectable locally advanced HNSCC whose tumours express PD-L1 (CPS ≥ 1), as determined by a validated test, as neoadjuvant treatment as monotherapy, continued as adjuvant treatment in combination with RT with or without cisplatin and then as monotherapy |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Priority review, ORBIS |
NOC date | August 11, 2025 |
Recommended dose | For neoadjuvant treatment, pembrolizumab should be given as monotherapy for 2 doses of 200 mg every 3 weeks, or 1 dose of 400 mg every 6 weeks, or until disease progression that precludes definitive surgery or unacceptable toxicity. For adjuvant treatment, pembrolizumab should be given in combination with RT with or without cisplatin for 3 doses of 200 mg every 3 weeks or 2 doses of 400 mg every 6 weeks, followed by 12 doses of pembrolizumab as monotherapy at 200 mg every 3 weeks or 6 doses of 400 mg every 6 weeks, or until disease recurrence or unacceptable toxicity. |
CPS = combined positive score; HNSCC = head and neck squamous cell carcinoma; NOC = Notice of Compliance; RT = radiotherapy.
Introduction
Head and neck cancers encompass malignancies that originate in the mouth, throat, sinuses, and salivary glands.1 In Canada, an estimated 8,100 new patients were diagnosed and 2,100 deaths were attributable to head and neck cancers in 2024.1,2 More than 90% of head and neck cancers begin in squamous cells and are termed head and neck squamous cell carcinomas (HNSCCs).2 Patients with HNSCC experience symptoms that affect basic physiological functions (e.g., chewing, swallowing, and breathing), reduce senses (e.g., taste, smell, and hearing), and impact human characteristics (e.g., appearance and voice).3 Treatment guidelines in Canada support a multimodal approach tailored to patient and tumour-specific factors, to achieve optimal tumour control and survival outcomes while minimizing treatment-related morbidity for resectable, locally advanced HNSCC.4-9 For stage III or stage IVA disease, surgical resection of the primary tumour and involved lymph nodes is the main treatment, followed by adjuvant radiotherapy (RT) or chemoradiotherapy (CRT) based on pathologic risk features such as positive surgical margins or extranodal extension.4,5,7,10 High-risk cases typically receive high-dose cisplatin (100 mg/m2 on days 1, 22, and 43) with RT.4,6,11,12 For those unable to tolerate cisplatin, alternatives such as carboplatin with or without 5-fluorouracil or cetuximab are available but may be limited by access and uncertainties in strength of their supporting evidence.9,13,14 In certain cases and anatomic sites, definitive CRT or RT alone may be appropriate, especially for patients unfit for surgery or seeking organ preservation.5 Patients with PD-L1 combined positive score (CPS)-negative HNSCC generally have a poor prognosis and may respond less favourably to immune checkpoint inhibitors (ICIs).15-18 The complex anatomy of the head and neck often results in lasting disabilities for HNSCC survivors, with more than half unable to return to work posttreatment.19 These challenges contribute to psychological distress, reduced health-related quality of life (HRQoL), and increased suicide risk.20,21
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of neoadjuvant pembrolizumab 100 mg/4 mL, administered as an IV infusion (either 2 doses of 200 mg every 3 weeks or 1 dose of 400 mg every 6 weeks or until disease progression or unacceptable toxicity), continued as adjuvant pembrolizumab in combination with RT with or without cisplatin (3 doses of 200 mg every 3 weeks or 2 doses of 400 mg every 6 weeks), followed by 12 doses of 200 mg every 3 weeks or 6 doses of 400 mg every 6 weeks as monotherapy or until disease recurrence or unacceptable toxicity, for the treatment of adults with resectable locally advanced HNSCC whose tumours express PD-L1 (CPS ≥ 1).
Perspectives of Patients, Clinicians, and Drug Programs
The information in this section is a summary of input provided by the patient and clinician groups who responded to our call for input and from clinical experts consulted by for the purpose of this review.
Patient Input
The CDA-AMC received a joint patient group input from the Canadian Cancer Survivor Network (CCSN) and Life-Saving Therapies Network (LSTN), with their information collected via an online survey between February 24 and March 17, 2025, and follow-up interviews. Eight participants (5 survivors, 1 patient in active treatment, and 2 caregivers) contributed, all based in Canada. Head and neck cancer significantly affects daily life, causing pain, difficulty with speech, eating, and social activities, and leading to reduced self-esteem. Caregivers are also heavily impacted due to treatment burdens and emotional strain. Patients prioritize treatments that minimize pain, functional impairment, and disfigurement, while preserving quality of life (QoL) and dignity. They value manageable side effects and the potential for less invasive treatment options. Patient feedback suggests pembrolizumab may offer better tolerability and QoL compared to traditional treatments. Its use could reduce surgical invasiveness and improve psychological well-being, making it a meaningful option in the Canadian context, especially for recurrent or metastatic disease.
Clinician Input
Input From Clinical Experts Consulted for This Review
The clinical experts consulted for this review emphasized that treatment of resectable HNSCC aims for cure and long-term disease control while minimizing toxicity. According to the clinical experts, current standard of care (SOC) involves surgery followed by adjuvant RT with or without chemotherapy (generally, cisplatin), or primary RT with or without chemotherapy for organ preservation. Locoregional control is critical, but recurrence (especially in p16-negative cases) is common, and salvage options like surgery or re-irradiation are often limited. Comorbidities may also restrict treatment choices. The clinical experts noted that the KEYNOTE-689 study may shift practice; however, it excluded concurrent chemotherapy with neoadjuvant pembrolizumab, leaving limited evidence to guide this approach. Nonetheless, the clinical experts suggested chemotherapy could be added in neoadjuvant settings for eligible, fit patients. The clinical experts indicated that pembrolizumab may be best suited for patients with resectable, locally advanced HNSCC not requiring urgent surgery who are medically fit. The clinical experts noted that CPS testing may be needed, particularly if chemotherapy is considered in the neoadjuvant phase, especially for CPS-negative or high-burden, symptomatic patients. While overall survival (OS) and HRQoL remain primary goals, improvements in event-free survival (EFS) are also meaningful given the disease’s morbidity. Response is assessed through clinical exam, endoscopy, and imaging. The clinical experts noted that treatment should be stopped if there is disease progression or severe or persistent adverse events (AEs). According to the clinical experts, prescribing pembrolizumab would require a multidisciplinary team at a tertiary cancer centre, with a medical oncologist leading treatment and monitoring.
Clinician Group Input
One clinician group, the Ontario Health (Cancer Care Ontario) (OH [CCO]) Head and Neck Cancer Drug Advisory Committee, submitted input. The clinician group had similar input to the clinical experts consulted for this review. They agreed the SOC for resectable, locally advanced HNSCC is surgery followed by adjuvant radiation (60 Gy to 66 Gy) with risk-adapted cisplatin chemotherapy (weekly 40 mg/m2 or high-dose therapy of 100 mg/m2 every 3 weeks) for high-risk patients. The clinician group emphasized the long-term QoL impacts of surgery, radiation, and cisplatin, along with high recurrence rates and limited salvage options, underscoring the need for new treatments to improve survival and reduce recurrence. While the clinician group felt it was premature to define which patients would benefit most from pembrolizumab without further data, they highlighted the potential role of PD-L1 testing to guide use. The clinician group also noted the need to determine if results from the KEYNOTE-689 study apply to cisplatin-ineligible patients, because the trial only included those who were cisplatin eligible. The group agreed that treatment should occur in multidisciplinary clinics to reduce risks of disease progression, acknowledged the increased visit burden from the adjuvant pembrolizumab component, and emphasized considering resource implications. For monitoring, the clinician group recommended clinical assessments before each pembrolizumab cycle and surgery, along with cross-sectional imaging (CT or MRI) for restaging. They supported discontinuing pembrolizumab upon disease progression or significant toxicity.
Drug Program Input
Input was obtained from the drug programs that participate in the reimbursement review process. The following were identified as key factors that could potentially impact the implementation of a recommendation for pembrolizumab:
relevant comparators
considerations for initiation of therapy
considerations for prescribing of therapy
generalizability
system and economic issues.
The clinical experts consulted for this review provided advice on the potential implementation issues raised by the drug programs. Refer to Table 4 for more details.
Clinical Evidence
Systematic Review
Description of Studies
One ongoing, multicentre, open-label, phase III randomized controlled trial (RCT), the KEYNOTE-689 study (N = 714) submitted by the sponsor was included for this review. The KEYNOTE-689 study investigated the efficacy and safety of pembrolizumab as neoadjuvant therapy, followed by postsurgical (adjuvant) treatment combining pembrolizumab and SOC RT (with or without cisplatin) followed by pembrolizumab monotherapy in treatment-naive patients with resectable locally advanced HNSCC. Eligible patients were adults with newly diagnosed, histologically confirmed locally advanced (stage III or stage IVA) HNSCC amenable to primary surgery. Eligible patients had an Eastern Cooperative Oncology Group (ECOG) Performance Status (PS) of 0 or 1, adequate organ function, available HPV (p16) results for oropharyngeal tumours, and provided a tumour sample for PD-L1 testing. Patients who had received prior therapy with an anti-programmed cell death 1 protein (PD-1), anti-PD-L1, anti–PD-L2 drug, or with a drug directed to another co-inhibitory T-cell receptor, or who had received prior RT treatment or systemic anticancer therapy including investigational agents for the head and neck cancer under study before randomization, were excluded. The outcomes relevant to this review included OS, EFS assessed by blinded independent central review (BICR), HRQoL, and safety.
The overall intention-to-treat (ITT) population of the KEYNOTE-689 study had a median age of 60 years. The proportion of male patients (79%) was higher than that of female patients (21%). Most patients were white (77% to 78%), followed by Asian (13% to 14%). Most patients were current or former smokers (78%) and alcohol users (68%). In the overall ITT population, 65% of the patients had a PD-L1 CPS of 10 or greater, and 95.5% of the patients had a PD-L1 CPS of 1 or greater. More patients had an ECOG PS of 0 (57%) compared to those with an ECOG PS of 1 (43%). Most patients had a negative HPV status (96%) and stage IVA disease (74%). Most patients had their primary tumour located in the oral cavity (61%), followed by the larynx (22%), oropharynx (10%), and hypopharynx (8%).
Efficacy Results
The efficacy and harms outcomes of the KEYNOTE-689 study reported in this review were based on the protocol-prespecified first interim analysis (IA1) for which the data cut-off date was July 25, 2024. For the overall ITT population (N = 714), the median durations of follow-up were 30.0 months (range, 0.8 months to 64.9 months) in the pembrolizumab plus SOC group, and 23.4 months (range, 0.5 months to 66.5 months) in the SOC group.
Overall Survival
The median OS was not reached (95% confidence interval [CI], 61.9 months to not reached) in the pembrolizumab plus SOC group, and 61.8 months (95% CI, 50.1 months to not reached) in the SOC group. The hazard ratio (HR) was 0.76 (95% CI, 0.59 to 0.98). This was not formally tested. According to the multiple testing strategy of the KEYNOTE-689 study, formal OS testing was not conducted at IA1, as the statistical significance boundary was not met in the PD-L1 population with a CPS of 10 or greater (earlier in the hierarchy). The Kaplan-Meier (KM) estimates of the probability of OS in the pembrolizumab plus SOC and SOC alone groups were 86.7% (95% CI, 82.7% to 89.8%) versus 77.9% (95% CI, 73.2% to 81.9%) at 12 months, and 63.6% (95% CI, 57.4% to 69.1%) versus 58.0% (95% CI, 51.6% to 63.9%) at 48 months, respectively.
EFS by BICR
In the overall ITT population, the median EFS was 51.8 months (95% CI, 37.5 months to not reached) in the pembrolizumab plus SOC group, and 30.4 months (95% CI, 21.8 months to 50.1 months) in the SOC group. The HR was 0.73 (95% CI, 0.58 to 0.92; P = 0.00411). The KM estimates of the probability of EFS in the pembrolizumab plus SOC and SOC alone groups were 75.1% (95% CI, 70.0% to 79.4%) versus 62.5% (95% CI, 56.9% to 67.5%) at 12 months, and 52.0% (95% CI, 45.1% to 58.4%) versus 44.2% (95% CI, 37.5% to 50.8%) at 48 months, respectively.
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 Global Health Status/QoL Score
The between-group differences in least squares (LS) mean change from baseline to postadjuvant CRT or RT at week 25 and week 51 in the European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 (EORTC QLQ-C30) global health status (GHS)/QoL scores were −1.29 (95% CI, −4.78 to 2.20; P = 0.4671) in a total of 350 patients, and 0.39 (95% CI, −3.35 to 4.13; P = 0.8379) in a total of 280 patients, respectively.
Harms Results
Of note, the pembrolizumab plus SOC group had a longer treatment duration as per the protocol design, with a median of 9.1 months for pembrolizumab plus SOC compared to 2.9 months for SOC alone.
The harms outcomes were available among the all participants as treated (APaT) population in the KEYNOTE-689 study (n = 676). AEs were reported in 96% and 97% of patients in the pembrolizumab plus SOC and SOC alone groups, respectively. The most common AEs were stomatitis (42% in the pembrolizumab plus SOC group and 55% in the SOC group), and radiation skin injury (40% and 48%). There were similar proportions of patients between the pembrolizumab plus SOC and the SOC groups for grade 3 to grade 5 AEs (76% and 74%, respectively), and AEs resulting in death (7% and 8%, respectively). There were higher proportions of patients in the pembrolizumab plus SOC group than in the SOC group for serious AE (SAEs) (50% and 37%), AEs leading to any study treatment discontinuation (24% and 14%), and AEs of special interest (AEOSIs, i.e., immune-mediated events and infusion-related reactions; 44% and 11%). The most frequently reported AEOSIs in the pembrolizumab plus SOC group and the SOC group were hypothyroidism (25% and 5%), hyperthyroidism (9% and 3%), pneumonitis (5.5% and 0), colitis (2.5% and 0.3%), and hepatitis (2% and 0.3%).
Critical Appraisal
The KEYNOTE-689 trial ensured allocation concealment through a central interactive voice-response or web-response system. Stratification by key prognostic factors such as tumour site, disease stage, and PD-L1 tumour proportion score (TPS) contributed to ensuring comparability between treatment groups at baseline. Overall, patient characteristics were well balanced, although a slight imbalance in smoking status (never smoker: 17.6% in the pembrolizumab plus SOC group versus 23.1% in the SOC group) was observed, which clinical experts considered unlikely to influence results. The trial also benefited from the use of centralized, blinded reviews for imaging, pathology, and PD-L1 status, mitigating assessment bias despite the open-label design. Compared to the SOC group, more patients in the pembrolizumab plus SOC group discontinued treatment (54% in the pembrolizumab group versus 17% in the SOC group), mainly due to AEs and disease progression. The longer treatment duration (per protocol) in the pembrolizumab group (median of 9.1 months in the pembrolizumab group versus 2.9 months in the SOC group) likely contributed to this difference. Additionally, more patients in the SOC group were randomized but not treated (0.8 % in the pembrolizumab group versus 10% in the SOC group), ██████ ███ ██ ███████████ ███████████ ██████████ ██████████ ██ █████ █████████████ ██ ██ ██████████ █████. This introduces some concern for risk of bias due to deviations from the intended intervention in the SOC group that arose because of the trial context, though the direction and magnitude are unclear. Furthermore, while efficacy outcomes like EFS and OS were objectively assessed, patient-reported HRQoL measures may be more susceptible to bias under open-label conditions. The handling of missing HRQoL data — assumed to be missing at random by the study investigators — is also a notable concern, as there were no sensitivity analyses provided to confirm the robustness of the results to different plausible assumptions about the missing data. Lastly, the trial's interim OS and EFS analyses introduce uncertainty, as HRs may be overestimated, and longer follow-up is needed for robust OS conclusions.
The clinical experts noted that the study population and eligibility criteria in the KEYNOTE-689 study is reflective of patients seen in clinical practice in Canada. Patients were enrolled from multiple countries, including Canada, and had demographic characteristics consistent with real-world populations. The inclusion of only patients with ECOG PS 0 to 1 may limit generalizability slightly, though experts noted that pembrolizumab could be beneficial in patients with lower PS and an ECOG score of 2 as well. Treatment regimens used in the trial align with clinical practice in Canada, supporting generalizability of the trial results. However, the small proportion of PD-L1 CPS-negative patients in the study (approximately 4% in each group) limits the certainty of findings for this subgroup. Clinical experts advised caution in using pembrolizumab as neoadjuvant monotherapy in CPS-negative patients. Given the limited representation of CPS-negative patients in the KEYNOTE-689 study and the associated prognostic challenges, further evidence is needed before extending the findings to this population. Moreover, nearly one-third of screened patients were excluded due to ineligibility, although experts felt this did not compromise generalizability. The end points used in the study and reported in the main body of this report, including OS, EFS, HRQoL, and safety, were considered clinically relevant and appropriate for decision-making. The use of EFS as a surrogate for OS is supported by good correlations between treatment effects on EFS and OS reported in previous HNSCC meta-analyses, indicating improvements in EFS likely predict OS improvements.22 Nonetheless, uncertainties remain regarding the applicability of these findings to the KEYNOTE-689 study, as it is unclear how closely the patient populations, treatment modalities (e.g., inclusion of pembrolizumab, specifics of SOC), and EFS definitions in the KEYNOTE-689 study align with those in the meta-analyses.22,23 Therefore, caution should be exercised when extrapolating EFS findings to long-term survival benefit, especially given that OS data remain immature.
GRADE Summary of Findings and Certainty of the Evidence
For pivotal studies and RCTs identified in the sponsor’s systematic review, Grading of Recommendations, Assessment, Development and Evaluations (GRADE) was used to assess the certainty of the evidence for outcomes considered most relevant to inform expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group.24,25 Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), indirectness, imprecision of effects, and publication bias.
The selection of outcomes for GRADE assessment was based on the sponsor’s summary of clinical evidence, consultation with clinical experts, and input received from patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members:
probability of OS at 12 months and 60 months
probability of EFS at 12 months, 36 months, and 48 months
EORTC QLQ-C30 GHS/QoL at postadjuvant CRT or RT weeks 25 and 51
grade 3 to grade 5 AEs.
Table 2 presents the GRADE summary of findings for pembrolizumab plus SOC versus SOC alone for adult patients with resectable locoregionally advanced HNSCC.
Table 2: Summary of Findings for Pembrolizumab Plus SOC vs. SOC Alone for Adult Patients With Resectable Locoregionally Advanced HNSCC
Outcome and follow-up | Patients (studies), N | Relative effect (95% CI) | Absolute effects (95% CI) | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
SOC | Pembrolizumab plus SOC | Difference | |||||
OS | |||||||
Probability of survival at 12 months Median (range) follow-up: 27.1 months (0.5 months to 66.5 months) | 714 (1 RCT) | NR | ███ | ███ | ███ | Moderatea | Pembrolizumab plus SOC likely results in a clinically important increase in OS compared to SOC at 12 months. |
Probability of survival at 60 months Median (range) follow-up: 27.1 months (0.5 months to 66.5 months) | 714 (1 RCT) | NR | ███ | ███ | ███ | Lowb | Pembrolizumab plus SOC may result in a clinically important improvement in OS compared to SOC at 60 months. |
EFS by BICR | |||||||
Probability of EFS at 12 months Median (range) follow-up: 27.1 months (0.5 months to 66.5 months) | 714 (1 RCT) | NR | ███ | ███ | ███ | Moderatec | Pembrolizumab plus SOC likely results in a clinically important increase in EFS compared to SOC at 12 months. |
Probability of EFS at 36 months Median (range) follow-up: 27.1 months (0.5 months to 66.5 months) | 714 (1 RCT) | NR | ███ | ███ | ███ | Moderated | Pembrolizumab plus SOC likely results in a clinically important increase in EFS compared to SOC at 36 months. |
Probability of EFS at 48 months Median (range) follow-up: 27.1 months (0.5 months to 66.5 months) | 714 (1 RCT) | NR | ███ | ███ | ███ | Lowe | Pembrolizumab plus SOC may result in little to no clinically important difference in EFS compared to SOC at 48 months. |
HRQoL (measured with EORTC QLQ-C30 global health status/QoL) | |||||||
LS mean change from baseline in EORTC QLQ-C30 global health status/QoL score (0 [worst] to 100 [best]) Follow-up: at week 25 postadjuvant CRT or RT | 350 (1 RCT) | NR | ███ | ███ | ███ | Lowf | Pembrolizumab plus SOC may result in little to no clinically important difference on the EORTC QLQ-C30 global health status/QoL compared to SOC at week 25 postadjuvant CRT or RT. |
LS mean change from baseline in EORTC QLQ-C30 global health status/QoL score (0 [worst] to 100 [best]) Follow-up: at week 51 postadjuvant CRT or RT | 280 (1 RCT) | NR | ███ | ███ | ███ | Lowg | Pembrolizumab plus SOC may result in little to no clinically important difference on the EORTC QLQ-C30 global health status/QoL compared to SOC at week 51 postadjuvant CRT or RT. |
Harms | |||||||
Grade 3 to grade 5 AEs Median (range) follow-up: 27.7 months (0.6 months to 66.5 months) | 676 (1 RCT) | NR | ███ | ███ | ███ | Moderateh | Pembrolizumab plus SOC likely results in little to no clinically important difference in grade 3 to grade 5 AEs compared to SOC. |
AE = adverse event; BICR = blinded independent central review; CDA-AMC = Canada's Drug Agency; CI = confidence interval; CRT = chemoradiotherapy; EFS = event-free survival; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; FAS = full analysis set; HNSCC = head and neck squamous cell carcinoma; HRQoL = health-related quality of life; LS = least squares; MID = minimal important difference; NR = not reported; OS = overall survival; PRO = patient-reported outcome; QoL = quality of life; RCT = randomized controlled trial; RT = radiotherapy; SOC = standard of care; vs. = versus.
Note: Data presented in this table were based on analyses at the clinical cut-off date of July 25, 2024. Of note, the median OS data had not been reached in the pembrolizumab plus SOC group as of July 25, 2024. As per the protocol design, the pembrolizumab plus SOC group had a longer treatment duration, with a median of 9.1 months for pembrolizumab plus SOC compared to 2.9 months for SOC alone. The between-group difference for grade 3 to grade 5 AEs in this table was requested from the sponsor to aid in interpretation and was not part of the sponsor’s analysis plan. Study limitations (which refer to internal validity or risk of bias), indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aRated down 1 level for serious imprecision. At the data cut-off date, there was relatively short follow-up and the median OS had not been reached in the pembrolizumab plus SOC group. A difference of 5% between the groups was identified by the clinical expert consulted by CDA-AMC as a threshold of clinical importance for this outcome. At 12 months, the 95% CI of the absolute effect includes the possibility of little to no difference and a clinically important increase in OS.
bRated down 2 levels for serious study limitations and imprecision. At the data cut-off date, there was relatively short follow-up and the median OS had not been reached in the pembrolizumab plus SOC group. A difference of 5% between the groups was identified by the clinical expert consulted by CDA-AMC as a threshold of clinical importance for this outcome. At 60 months, the 95% CI of the absolute effect includes the possibility of little to no difference and a clinically important benefit; the lower bound of the CI is approaching a threshold of clinically important harm. The estimate is from the tail of the KM curve where there is a high level of censoring and few patients left at risk.
cRated down 1 level for serious imprecision. A difference of 10% between the groups was identified by the clinical expert consulted by CDA-AMC as a threshold of clinical importance for this outcome. At 12 months, the 95% CIs of the absolute effect include the potential for little to no difference and a clinically important increase in EFS.
dRated down 1 level for serious imprecision. A difference of 10% between the groups was identified by the clinical expert consulted by CDA-AMC as a threshold of clinical importance for this outcome. At 36 months, the 95% CIs of the absolute effect include the potential for little to no difference and a clinically important increase in EFS.
eRated down 2 levels for serious study limitations and imprecision. A difference of 10% between the groups was identified by the clinical expert consulted by CDA-AMC as a threshold of clinical importance for this outcome. At 48 months, the 95% CI of the absolute effect included the potential for little to no difference and clinically important benefit. The estimate is from the tail of the KM curve where there is a high level of censoring and few patients left at risk.
fRated down 2 levels for very serious risk bias of due to the open-label nature of the study and the subjective nature of the outcome. A total of 47.1% of the total PRO FAS population, or 43.5% of the patients with this score assessed at baseline, are missing some data. In the absence of sensitivity analyses, risk of bias cannot be excluded.
gRated down 2 levels for very serious risk bias of due to the open-label nature of the study and the subjective nature of the outcome. A total of 57.7% of the total PRO FAS population, or 54.8% of the patients with this score assessed at baseline, are missing some data. In the absence of sensitivity analyses, risk of bias cannot be excluded.
hRated down 1 level for serious imprecision. The review team was unable to identify the MID to assess a between-group difference from the literature or the clinical experts consulted for this review; therefore, the null was used to assess certainty. The 95% CI of the absolute effect includes the potential for both benefit (reduced AEs) and harm (increased AEs). The CDA-AMC review team assessed that lower boundary of the 95% CI was not a source of serious imprecision, although the upper lower boundary of the 95% CI may indicate an important difference.
Sources: Clinical Study Report for the KEYNOTE-689 study26 and sponsor’s submissions.27,28 Details included in the table are from the sponsor’s summary of clinical evidence.29
Long-Term Extension Studies
No long-term extension studies were submitted for this review as the KEYNOTE-689 study is still ongoing for OS follow-up.
Indirect Comparisons
No studies with indirect evidence were submitted for this review.
Studies Addressing Gaps in the Evidence From the Systematic Review
No studies addressing gaps in the pivotal and RCT evidence were submitted for this review.
Conclusions
One ongoing, open-label, phase III RCT comparing efficacy and safety between neoadjuvant and adjuvant pembrolizumab plus SOC and SOC alone in treatment-naive, adult patients with stage III or stage IVA resectable locally advanced HNSCC contributed to this review. As of the data cut-off date (July 25, 2024), OS was not formally tested and the median OS was not reached in the pembrolizumab plus SOC group, with OS events observed in 31% and 37% of patients in the pembrolizumab plus SOC and SOC groups, respectively. The evidence suggests that there is likely a clinically meaningful benefit of pembrolizumab plus SOC for OS at 12 months which appeared to extend throughout the available follow-up but is less certain at longer time points (60 months). Pembrolizumab plus SOC likely results in a clinically important improvement in EFS at 12 months and 36 months. The results suggest low-certainty evidence of little to no difference in EFS at 48 months; however, there is increased uncertainty for all time points beyond 36 months due to few patients remaining at risk and imprecision. Patients who received pembrolizumab plus SOC may have comparable HRQoL to the patients who received SOC more than 51 weeks after surgery. According to the clinical experts consulted for this review, the safety profile of pembrolizumab was consistent with their expectations for this drug. Given the small proportion of patients with CPS-negative disease, representing less than 4% of patients in the KEYNOTE-689 study, the clinical experts emphasized that there is insufficient evidence to support or oppose pembrolizumab monotherapy in this subgroup and advised caution in its use.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of neoadjuvant pembrolizumab 100 mg/4 mL, administered as an IV infusion (either 2 doses of 200 mg every 3 weeks or 1 dose of 400 mg every 6 weeks or until disease progression or unacceptable toxicity), continued as adjuvant pembrolizumab in combination with RT with or without cisplatin (3 doses of 200 mg every 3 weeks or 2 doses of 400 mg every 6 weeks), followed by 12 doses of 200 mg every 3 weeks or 6 doses of 400 mg every 6 weeks as monotherapy or until disease recurrence or unacceptable toxicity, for the treatment of adults with resectable locally advanced HNSCC whose tumours express PD-L1 (CPS ≥ 1).
Disease Background
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the review team.
Head and neck cancers encompass malignancies that originate in the mouth, throat, sinuses, and salivary glands.1 In Canada, these cancers represent the 10th most common type of malignancy, with an estimated 8,100 new patients diagnosed and 2,100 deaths attributable to head and neck cancers in 2024.1,2 The estimated 5-year net survival in Canada from the date of head and neck cancer diagnosis is 64%.30 First Nations adults face an estimated 15% to 22% lower five-year survival despite similar incidence rates, underscoring the disparities faced by Indigenous Peoples in Canda.31
More than 90% of head and neck cancers begin in squamous cells and are termed HNSCCs.2 HNSCC is more prevalent in men than women and typically affect adults aged older than 50 years.32 The primary risk factors for HNSCCs include tobacco use, alcohol consumption, chewing betel quid or areca nut, HPV infection (particularly for oropharyngeal cancers), and Epstein–Barr virus infection.32
HNSCC diagnosis typically begins with a comprehensive medical history, an endoscopy, and standard tests including HPV testing. Imaging techniques such as X-rays, CT scans, PET scans, and MRIs are often used to support diagnosis and staging along with a tissue biopsy that confirms the diagnosis.21,33 Of note, the clinical experts consulted for this review pointed out that tissue biopsy may be performed before imaging in some cases, as biopsy is not always preceded by imaging. To determine the prognosis and best course of treatment, HNSCC is staged using the American Joint Committee on Cancer staging criteria, which involves tumour-node-metastasis classification based on the size and spread of the primary tumour, lymph node involvement, and distant metastasis.33 Patients with PD-L1 CPS-negative (CPS < 1) HNSCC generally have a poor prognosis and may respond less favourably to ICIs, such as pembrolizumab or nivolumab, compared to patients with a CPS of 1 or more.15-18
Those diagnosed with early-stage HNSCC (i.e., stage I and stage II) generally have a good prognosis with 80% to 90% of patients achieving a cure following a single treatment modality, either surgery or RT alone.34 However, in Canada almost half (46%) of patients present with locally advanced HNSCC (defined as stage III or stage IVA).2 Locally advanced cases require more intensive multimodality treatment, typically combining surgery with RT and potentially chemotherapy as a radiosensitizer.34 Those with locally advanced HNSCC have a high risk of local recurrence (18% to 33%) and distant metastasis (approximately 20% to 25%) leading to a 5-year OS of 40% to 53%.10,35-41
Patients with HNSCC experience a variety of symptoms including compromised basic physiological functions (e.g., chewing, swallowing, and breathing), reduced senses (e.g., taste, smell, and hearing), and impacted human characteristics (e.g., appearance and voice).3 Treatment sequelae encompass physical (e.g., dysphagia, radionecrosis, mucositis, and xerostomia), emotional (e.g., disfigurement and depression), functional (e.g., pain, voice or speech impairment, and masticatory and dental problems), social, and occupational (e.g., delayed return to work) dysfunction, affecting both patients and their family members.21 The anatomic complexity of the head and neck region leads to significant disabilities in HNSCC survivors with more than half of patients unable to return to work after completing treatment.19 These multifaceted challenges contribute to psychological distress and compromised HRQoL, and have led to increased suicide ideation and rates among HNSCC survivors.20,21
Standards of Therapy
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the review team.
The treatment goals for resectable, locally advanced HNSCC are to achieve optimal tumour control and survival outcomes while minimizing treatment-related morbidity. For patients with stage III or stage IVA disease, surgical resection of the primary tumour and involved lymph nodes is often a key component of treatment, typically followed by adjuvant RT or CRT based on pathologic risk features such as positive surgical margins or extranodal extension.4,5,7,10 Furthermore, the clinical experts consulted for this review noted that surgery is not always the cornerstone for all patients in this group. For example, primary CRT may be preferred for HPV-related oropharyngeal, nasopharyngeal, and select laryngeal or hypopharyngeal cancers. The clinical experts noted that even minor tumour volume reductions can notably lessen symptom burden due to the tight anatomic space in the head and neck region. In certain cases and anatomic sites, definitive CRT or RT alone may be appropriate, particularly for patients who are ineligible for surgery, seek larynx preservation, or would face unacceptable functional impairment from surgery.5 While surgery plays an important role in treatment of most resectable cases, especially oral cavity cancers, CRT may be used as an alternative depending on tumour site, functional considerations, and patient preferences.4-6,8,10
Treatment guidelines in Canada — including those from the National Comprehensive Cancer Network; the European Head and Neck Society, European Society for Medical Oncology, and European SocieTy for Radiotherapy and Oncology (EHNS-ESMO-ESTRO); and Cancer Care Alberta — support a multimodal approach tailored to patient and tumour-specific factors.4-9 The standard regimen for high-risk postoperative cases is high-dose cisplatin (100 mg/m2 on days 1, 22, and 43) concurrent with RT.4,6,11,12 The clinical experts noted that low-dose cisplatin (40 mg/m2 weekly) with RT may be used as an alternative regimen.42 For patients ineligible for cisplatin, alternatives such as carboplatin with or without 5-fluorouracil or cetuximab are available, but their use may be limited by access restrictions and uncertainties regarding the strength of supporting evidence in this setting.9,13,14 A multidisciplinary team is essential to optimize treatment decisions and support patient care throughout the therapeutic process.5
Drug Under Review
Key characteristics of pembrolizumab and other treatments for adults with resectable locally advanced HNSCC are summarized in Table 3. Pembrolizumab is a high-affinity anti–PD-1 antibody that exerts dual ligand blockade of the PD-1 pathway, including PD-L1 and PD-L2, on antigen-presenting or tumour cells. This dual blockade prevents tumour cells and antigen-presenting cells from suppressing immune responses, ultimately restoring the antitumour activity of cytotoxic T lymphocytes within the tumour microenvironment.43
The recommended dosage of pembrolizumab for the treatment of HNSCC in adults with resectable locally advanced HNSCC is either 200 mg every 3 weeks or 400 mg every 6 weeks. Pembrolizumab is administered as an IV infusion over 30 minutes. In the neoadjuvant phase, patients are treated with 400 mg of pembrolizumab (2 doses of 200 mg every 3 weeks or 1 dose of 400 mg every 6 weeks) until disease progression that precludes definitive surgery or unacceptable toxicity. Pembrolizumab is continued as adjuvant treatment in combination with RT with or without cisplatin for 3 doses of 200 mg every 3 weeks or 2 doses of 400 mg every 6 weeks, followed by 12 doses of 200 mg every 3 weeks or 6 doses of 400 mg every 6 weeks as monotherapy or until disease recurrence or unacceptable toxicity. Pembrolizumab should be administered before CRT or before RT when given on the same day. The sponsor’s reimbursement request is the same as the proposed Health Canada–approved indication.
███████████ ██████████████ ████████ ██ ████████ █████████████ ██ ███████████ ████ ██ ████ ██ ███████ ████████████ ████████ ██ █████████████ ██ █ ███████████ ██ █████████ █████ ██████ ██ ███ ███ ███ ███ ███ █████████.
Table 3: Key Characteristics of Pembrolizumab, Cisplatin, Carboplatin, 5-Fluorouracil, and Cetuximab
Drug | Mechanism of action | Indicationa | Route of administration | Recommended dose | Serious AEs of safety issues |
|---|---|---|---|---|---|
Pembrolizumab with adjuvant RT with or without cisplatin44 | A high-affinity anti–PD‑1 antibody that exerts dual ligand blockade of the PD‑1 pathway, including PD‑L1 and PD‑L2, on antigen-presenting or tumour cells. | Treatment of adult patients with resectable, locally advanced HNSCC whose tumours express PD‑L1 (CPS ≥ 1), as neoadjuvant treatment, continued as adjuvant treatment in combination with RT with or without cisplatin and then as monotherapy. | IV | Neoadjuvant: 200 mg q.3.w for 2 cycles or 400 mg for 1 cycle. Adjuvant: 200 mg q.3.w. for 15 cycles or 400 mg q.6.w. for 8 cycles. | Hepatic, biliary, or pancreatic impairment; immune AEs; renal impairment; and teratogenic risk. |
RT with or without cisplatin or carboplatinb | Antineoplastic agents form crosslinks with the DNA in cancer cells, preventing proper DNA replication and transcription, triggering apoptosis (cell death) and inhibiting tumour growth. | Not approved. | IV | Adjuvant cisplatin: 100 mg/m2 q.3.w. for 3 cycles or 40 mg/m2 q.w. for 7 cycles concurrently with RT. Adjuvant carboplatin: AUC 5 q.3.w. for 3 cycles or AUC 2 q.w. for 7 cycles concurrently with RT. | Nephrotoxicity, neurotoxicity, ocular toxicity or retinopathy, ototoxicity, severe nausea and vomiting, gonadotoxicity, and myelosuppression. |
RT with or without carboplatin and 5‑fluorouracilb,45 | A fluorinated pyrimidine antimetabolite that is metabolized intracellularly to its active form, fluorouridine monophosphate, which then inhibits DNA synthesis by inhibiting thymidylate synthetase and the normal production of thymidine. | In the treatment of head and neck cancer with or without carboplatin. | IV | Adjuvant: carboplatin 70 mg/m2/day for 4 days with 5‑fluorouracil 600 mg/m2/day for 4 days, q.3.w. for 3 cycles concurrently with RT. | Contraindicated in patients with known complete absence of dihydropyrimidine dehydrogenase activity. Teratogenic risk, increased risk of infection, and cardiac events. |
Cetuximab with RTb,46 | Monoclonal antibody that binds specifically to the extracellular domain of the human EGFR with high affinity. | In combination with radiation therapy for the initial treatment of locally or regionally advanced HNSCC. | IV | Adjuvant: 400 mg/m2 1 week before initiation of RT. Subsequent weekly dose is 250 mg/m2 for the duration of RT (6 weeks to 7 weeks). | Infusion reactions and cardiopulmonary arrest. |
AE = adverse event; AUC = area under the curve; CPS = combined positive score; EGFR = epidermal growth factor receptor; HNSCC = head and neck squamous cell carcinoma; q.w. = every week; q.3.w. = every 3 weeks; q.6.w. = every 6 weeks; RT = radiotherapy.
aHealth Canada–approved indication.
bThe treatments for HNSCC were based on those recommended by BC Cancer and validated by the clinical expert consulted for this review.47
Sources: Draft product monograph for Keytruda (pembrolizumab),44 product monograph for fluorouracil injection,45 and product monograph for Erbitux (cetuximab).46
Perspectives of Patients, Clinicians, and Drug Programs
The full patient and clinician group submissions received are available in the consolidated patient and clinician group input document for this review on the project website.
Patient Group Input
This section was prepared by the review team based on the input provided by patient groups.
A joint submission was received from the CCSN and LSTN in response to the CDA-AMC call for patient input on the current review of pembrolizumab. They collected information via an online survey between February 24 and March 17, 2025. The LSTN also conducted interviews with head and neck cancer survivors who had past or current experience with pembrolizumab.
A total of 8 respondents completed the survey. All respondents were from Canada. One respondent was in active treatment, 5 were head and neck cancer survivors, and 2 were caregivers to someone with head and neck cancer.
Respondents were asked to rate on a 5-point scale how head and neck cancer has impacted their ability to perform daily activities (0 = no impact and 5 = severe impact). The 3 most affected areas were participating in hobbies or leisurely activities (3.25 of 5), exercising (3.13 of 5), and working (3.00 of 5). Through open-ended answers, respondents noted substantial pain and discomfort, including difficulty talking, eating or swallowing, and laughing, as well as impacted self-esteem related to disfigurement.
Respondents felt currently available treatments, including radiation and cisplatin, were associated with substantial side effects that impacted both the patient and caregivers. Additionally, a common theme among respondents was reduced self-esteem after surgery either due to visual disfigurement or impairments that must be overcome, such as a speech impediment.
Four of the respondents had experience with pembrolizumab (either previous or current) and participated in LSTN’s interview. Interviewees often felt currently available treatments were more physically and psychologically demanding than pembrolizumab. The LSTN noted interviewees indicated that pembrolizumab was associated with improved QoL over currently available treatments. Interviewees were asked to rate on a 5-point scale the severity of side effects associated with pembrolizumab (0 = no effects and 5 = severe or intolerable effects). The most severe side effects experienced were muscle weakness, pain, or cramps (2.00 of 5), a stiff neck (1.60 of 5), and tingling or numbness in the arms and legs (0.80 of 5). Interviewees felt the side effects were manageable and the benefits of pembrolizumab outweighed the side effects.
The CCSN added that despite the relatively limited number of respondents participating in the survey and interview, results showed the benefits of pembrolizumab over currently available treatment options. The CCSN noted that pembrolizumab offers patients with head and neck cancer hope and reassurance that there are treatment options, which is especially impactful for patients whose cancer has become metastatic or recurrent. Particularly for this indication under review, the CCSN felt pembrolizumab may provide an opportunity to shrink the HNSCC tumour, thereby limiting the invasiveness of surgery and reducing discomfort and disfigurement.
Clinician Input
Input From Clinical Experts Consulted for This Review
All CDA-AMC review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of HNSCC.
Unmet Needs
The clinical experts consulted for this review emphasized that in the treatment of resectable HNSCC, the SOC aims for curative intent and durable long-term disease control while minimizing disease morbidity and long-term toxicities. The clinical experts pointed out that the current SOC for resectable HNSCC involves either surgery followed by adjuvant RT with or without chemotherapy, or primary RT with or without concurrent chemotherapy, particularly for patients seeking organ preservation. Concurrent chemotherapy is generally cisplatin, administered either as 100 mg/m2 every 3 weeks or 40 mg/m2 weekly during RT, with the weekly regimen being the most commonly used in some provinces, and this often consists of 7 weekly cycles or, alternatively, 3 cycles of high-dose cisplatin, depending on regional practice patterns. The clinical experts indicated that locoregional control is of significant importance due to the morbidity associated with poor disease control in the head and neck region. According to the clinical experts, despite advances in the management of head and neck cancers, some patients frequently experience incomplete responses to initial treatment and are at high risk of recurrence, particularly those with p16-negative tumours. Additionally, the clinical experts noted that recurrences are often not amenable to surgical salvage, and curative re-irradiation is frequently not a viable option due to cumulative toxicity. Moreover, patients with multiple comorbidities cannot tolerate the standard treatment regimens that combine surgery with adjuvant CRT.
Place in Therapy
The clinical experts noted that the KEYNOTE-689 trial could be practice-changing depending on forthcoming data. However, the clinical experts highlighted several considerations. First, the KEYNOTE-689 study included patients with CPS-negative HNSCC, who are often excluded from ICI studies, but prior evidence (the KEYNOTE-048 trial) suggested pembrolizumab monotherapy may be inferior to chemotherapy (EXTREME regimen) in this subgroup in the setting of unresectable locally advanced HNSCC.17,48 Additionally, the KEYNOTE-689 study did not allow for the combination of pembrolizumab with chemotherapy, which the clinical experts felt could limit its benefit in patients with CPS-negative tumours, particularly for symptomatic patients with a high tumour burden, who may derive greater benefit from chemo-immunotherapy combinations.
The clinical experts suggested that depending on the KEYNOTE-689 study results, offering an option for additional chemotherapy in neoadjuvant settings for patients with locoregionally advanced HNSCC should be considered. Such an approach may be appropriate for selected patients who are fit and meet inclusion criteria (e.g., ECOG PS, surgical eligibility) and are willing to undergo a more prolonged treatment course. The clinical experts noted that currently, pembrolizumab is not used in the curative-intent upfront setting, so its introduction could represent a significant shift in the treatment paradigm. The clinical experts also raised questions about whether the EFS results are primarily driven by neoadjuvant, adjuvant, or a combination of these pembrolizumab treatments, noting previous perioperative ICI trials — either as a radiosensitizer along with chemotherapy and RT or in the consolidative setting after CRT — have failed to show meaningful benefit. However, the clinical experts pointed out that an updated KEYNOTE-412 study with an additional 2 years of follow-up, suggested pembrolizumab plus CRT appeared to have clinically meaningful EFS, when compared to placebo plus CRT (HR = 0.79; 95% CI, 0.65 to 0.96).16 The clinical experts added that a meta-analysis of the KEYNOTE-412, JAVALIN Head and Neck 100, and NRG-HN004 studies (N = 1,687) suggested chemotherapy and RT with concurrent adjuvant ICIs was associated with increased progression-free survival in patients with PD-L1–positive, locally advanced HNSCC (HR = 0.78; 95% CI, 0.63 to 0.97) but not in the PD-L1–negative subgroup (HR = 1.31; 95% CI, 0.99 to 1.75).15 Cost, patient burden, and treatment toxicity implications must also be considered.
Patient Population
The clinical experts noted that the patients most suited for treatment with pembrolizumab are those with resectable locoregionally advanced HNSCC. These patients should be fit to undergo a prolonged and intensive treatment course and should not require urgent surgical intervention (e.g., due to impending airway obstruction or bleeding). The clinical experts noted that CPS testing may be necessary as a companion diagnostic, particularly if chemotherapy is to be optionally added to pembrolizumab in the neoadjuvant setting, especially for CPS-negative patients or those with high tumour burden and significant symptoms.
Assessing the Response Treatment
The clinical experts noted that although OS and HRQoL remain the primary goals for treatment, improvements in EFS or progression-free survival without a corresponding OS benefit may still be considered clinically meaningful, given the significant morbidity associated with HNSCC. The clinical experts indicated that in routine clinical practice, treatment response is typically assessed through a combination of clinical evaluation (including physical examination and endoscopy) and radiographic imaging such as CT, MRI, or PET scans. The clinical experts noted that while these methods are similar to those used in clinical trials, trials typically apply Response Evaluation Criteria in Solid Tumours (RECIST) criteria to objectively and consistently measure tumour burden. The clinical experts pointed out that in the setting of curative intent for HNSCC, achieving a complete response or no evidence of disease is the ultimate goal of treatment.
Discontinuing Treatment
The clinical experts noted that treatment should be discontinued in cases of disease progression, the occurrence of intolerable AEs, or based on patient preference. Examples of disease progression may include clinically significant deterioration, inadequate response, or the development of new metastatic lesions. AEs warranting discontinuation would include immune-related toxicities that are severe (e.g., ≥ grade 3) or persistent despite appropriate management. Specific to the regimen used in the KEYNOTE-689 study, the clinical experts expressed skepticism regarding the benefit of adjuvant pembrolizumab, noting that previous trials evaluating immunotherapy either as a radiosensitizer during chemotherapy or RT or as consolidative therapy after CRT had failed to meet their primary end points.
Prescribing Considerations
The clinical experts indicated that a specialty head and neck outpatient clinic, most likely within a tertiary cancer centre, would be needed for pembrolizumab prescription, due to the involvement of ear, nose, and throat specialists, medical oncologists, radiation oncologists, and allied health professionals. The clinical experts noted that medical oncology would be responsible for treating and monitoring patients receiving pembrolizumab.
Clinician Group Input
This section was prepared by the review team based on the input provided by clinician groups.
One clinician group submitted input to CDA-AMC, the OH (CCO) Head and Neck Cancer Drug Advisory Committee, providing input from 3 clinicians.
The clinician group was generally in agreement with the feedback received from the clinical experts consulted for this review. They agreed that the current standard treatment for resectable locally advanced HNSCC typically involves surgery followed by adjuvant radiation (60 Gy to 66 Gy in 30 to 33 fractions). Additionally, risk-adapted concurrent cisplatin chemotherapy (either weekly at 40 mg/m2 or high-dose therapy at 100 mg/m2 every 3 weeks) is recommended for patients with positive surgical margin with or without extranodal extension.
The OH (CCO) Head and Neck Cancer Drug Advisory Committee highlighted that extensive surgery, radiation, and cisplatin toxicities have detrimental impacts on QoL for patients with HNSCC. The clinician group noted the substantial risk of recurrence in patients treated with current conventional treatment options, with only a minority of these patients eligible for salvage therapy (either further surgery or radio therapy), emphasizing the need for additional treatments options to improve survival, recurrence rates, and QoL.
The clinician group felt it was difficult to comment on which patients would be best suited for treatment with pembrolizumab without access to current data, but noted an important consideration is if PD-L1 testing will be required to either select patients or determine risk of disease progression or toxicities that may hinder surgery. The trial restricted enrolment to cisplatin-eligible patients and the OH (CCO) Head and Neck Cancer Drug Advisory Committee noted the importance of determining if findings can be generalized to patients who are ineligible for cisplatin.
The OH (CCO) Head and Neck Cancer Drug Advisory Committee noted treatment should be administered in multidisciplinary clinics involving surgical, medical, and radiation oncologists to mitigate progression risks. The clinician group noted the adjuvant component of pembrolizumab would increase the number of visits and felt the impact on resourcing needs should be considered.
According to input from the OH (CCO) Head and Neck Cancer Drug Advisory Committee, clinical assessments should be performed before each cycle in the neoadjuvant and adjuvant setting and before surgery. The clinician group also felt patients should be restaged using cross-sectional imaging (CT or MRI) before surgery. The clinician group agreed that pembrolizumab should be discontinued in the event of disease progression or significant toxicities.
Drug Program Input
The drug programs provide input on each drug being reviewed through the reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted for this review are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
1. Issues with the choice of comparator in the submitted trial For CRT, the preferred chemotherapy regimen is high-dose cisplatin (100 mg/m2) on days 1, 22, and 43 concurrent with RT. For cisplatin-ineligible patients, alternatives include carboplatin with 5-fluorouracil or cetuximab concurrent with RT, as well as hyperfractionated or accelerated RT without chemotherapy. Note: In Canada, cetuximab is only funded for patients with a medical contraindication to cisplatin and carboplatin or 5-fluorouracil. This restricted funding positions its use outside the scope of the KEYNOTE-689 study. | This is a comment from the drug plans to inform pERC deliberations. The clinical experts consulted for this review agree with the statement. Additionally, the clinical experts note that low-dose cisplatin (40 mg/m2 weekly) concurrent with RT may be used as an alternative regimen in clinical practice in Canada. |
Considerations for initiation of therapy | |
2. Other patient characteristics for eligibility The KEYNOTE-689 study excluded previously treated patients (recurrent cases). Question to the clinical experts: Should these patients be eligible? | The clinical experts consulted for this review generally agreed that patients with recurrent disease could be eligible for treatment with pembrolizumab, depending on their prior therapy history. Specifically, if a patient's initial treatment occurred a long time ago (e.g., > 1 year ago), and they have not received prior systemic therapy, they should be considered eligible. The clinical experts noted that pembrolizumab should be reserved for systemic therapy–naive patients, or patients who recurred over 6 months after prior systemic therapy. The clinical experts indicated that because there is currently no approved indication for immunotherapy outside of the noncurative setting, these patients — if previously treated only with local therapies — could appropriately be included in trials like the KEYNOTE-689 study. |
3. Eligibility for re-treatment Question to the clinical experts: Would patients be eligible for downstream immunotherapy in the following situations? 3.1. The patient’s disease progresses during neoadjuvant pembrolizumab. 3.2. The patient receives neoadjuvant pembrolizumab but is not able to proceed to surgery. 3.3. The patient’s disease progresses during or recurs shortly after adjuvant pembrolizumab. 3.4. The patient has started but is not able to complete adjuvant pembrolizumab for reasons other than disease progression. Question to the clinical experts: Should patients who complete 1 year of treatment and experience disease progression or recurrence off of pembrolizumab treatment be eligible for up to 1 year (18 cycles) of pembrolizumab re-treatment? | The clinical experts generally agreed that patients would be eligible for downstream immunotherapy in scenarios 3.1, 3.2, and 3.4 for the following reasons. 3.1. Disease progression during neoadjuvant pembrolizumab: Yes. The clinical experts noted that because only 2 cycles are typically given up front and pseudoprogression occurs in approximately 20% of patients, continuing treatment and restaging may be appropriate. 3.2. Inability to proceed to surgery after neoadjuvant pembrolizumab: Yes. The clinical experts indicated that in such cases, treatment would shift toward a palliative approach. 3.4. Incomplete adjuvant pembrolizumab due to reasons other than progression: Yes. For scenario 3.3, disease progression during or shortly after adjuvant pembrolizumab, the clinical experts expressed hesitation, suggesting these patients may be considered PD-1 refractory, making further immunotherapy less appropriate. pERC acknowledged that pembrolizumab may be reasonable if disease progress or recurs at least 6 months after completion of the originally planned adjuvant treatment with pembrolizumab. In response to the question of whether patients who complete 1 year of pembrolizumab treatment and subsequently experience disease progression or recurrence off treatment should be eligible for up to 1 additional year (18 cycles) of pembrolizumab re-treatment, the clinical experts noted that evidence supporting re-treatment is currently lacking, as the treatment duration in the KEYNOTE-689 study was limited to approximately 1 year. However, the clinical experts acknowledged that if recurrence occurs > 6 months after completing pembrolizumab, the disease may not reflect true PD-1 resistance, and re-treatment with a PD-1–based regimen could be reconsidered on a case-by-case basis. |
Considerations for prescribing of therapy | |
4. Dosing, schedule and frequency, dose intensity Neoadjuvant pembrolizumab (2 cycles of 200 mg q.3.w. or 1 cycle of 400 mg). Patients receiving neoadjuvant pembrolizumab would continue pembrolizumab postsurgery as adjuvant treatment in combination with standard RT or CRT, depending on the risk factors, and then as monotherapy for a total of 1 year. The OWG would like to inform pERC that most jurisdictions use weight-based dosing up to a maximum dose for pembrolizumab (2 mg/kg up to 200 mg q.3.w. or 4 mg/kg up to 400 mg every 6 weeks). | This is a comment from the drug plans to inform pERC deliberations. The clinical experts agree with the statement. |
5. Concerns related to combination usage Question to the clinical experts: Should pembrolizumab be given with an alternative systemic regimen if the patient cannot receive or tolerate cisplatin? | The clinical experts agreed that pembrolizumab can be given with an alternative systemic regimen if a patient cannot receive or tolerate cisplatin. The clinical experts indicated a previous study suggesting that using a radiosensitizer, regardless of the specific agent, is more beneficial than omitting it entirely. |
Generalizability | |
6. Populations of interest matching the indication but with insufficient data Questions to the clinical experts: 6.1. Should patients with ECOG > 1 be eligible? 6.2. Should the ability to continue to surgery following neoadjuvant therapy be a consideration? | 6.1. The clinical experts indicated that patients with an ECOG Performance Status > 1 (e.g., ECOG = 2) could still be eligible for treatment. The clinical experts noted that in clinical practice in Canada, if the surgeon deems the patient fit for surgery, inclusion of patients with ECOG > 1 in the treatment protocol remains reasonable. 6.2. The clinical experts agreed that the ability to proceed to surgery after neoadjuvant therapy should be a consideration. The clinical experts emphasized that patients must be fit enough to undergo surgery for neoadjuvant treatment to be appropriate. |
System and economic issues | |
7. Concerns regarding the anticipated budget impact and sustainability There is potential for significant budget impact. | This is a comment from the drug plans to inform pERC deliberations. |
CRT = chemoradiotherapy; ECOG = Eastern Cooperative Oncology Group; OWG = Oncology Working Group; pERC = pan-Canadian Oncology Drug Review Expert Review Committee; q.3.w. = every 3 weeks; RT = radiotherapy.
Clinical Evidence
The objective of this clinical review report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of neoadjuvant pembrolizumab (either 2 doses of 200 mg every 3 weeks or 1 dose of 400 mg every 6 weeks or until disease progression or unacceptable toxicity), continued as adjuvant pembrolizumab in combination with RT with or without cisplatin (3 doses of 200 mg every 3 weeks or 2 doses of 400 mg every 6 weeks), followed by 12 doses of 200 mg every 3 weeks or 6 doses of 400 mg every 6 weeks as monotherapy or until disease recurrence or unacceptable toxicity, administered as an IV infusion, for the treatment of adults with resectable locally advanced HNSCC whose tumour expresses PD-L1 (CPS ≥ 1). The focus will be placed on comparing pembrolizumab to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of pembrolizumab is presented in 4 sections with our critical appraisal of the evidence included at the end of each section. The first section, the systematic review, includes pivotal studies and RCTs that were selected according to the sponsor’s systematic review protocol. Our assessment of the certainty of the evidence in this first section using the GRADE approach follows the critical appraisal of the evidence. The second section includes sponsor-submitted long-term extension studies. The third section includes indirect evidence from the sponsor. The fourth section includes additional studies that were considered by the sponsor to address important gaps in the systematic review evidence. There were no long-term extension studies (section 2), indirect evidence (section 3), or additional studies to address important gaps in the systematic review evidence (section 4) submitted by the sponsor.
Included Studies
Clinical evidence from the following is included in the review and appraised in this document:
1 pivotal RCT identified in the sponsor-conducted systematic review (the KEYNOTE-689 study).
Systematic Review
Contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
Description of Study
Characteristics of the included study are summarized in Table 5.
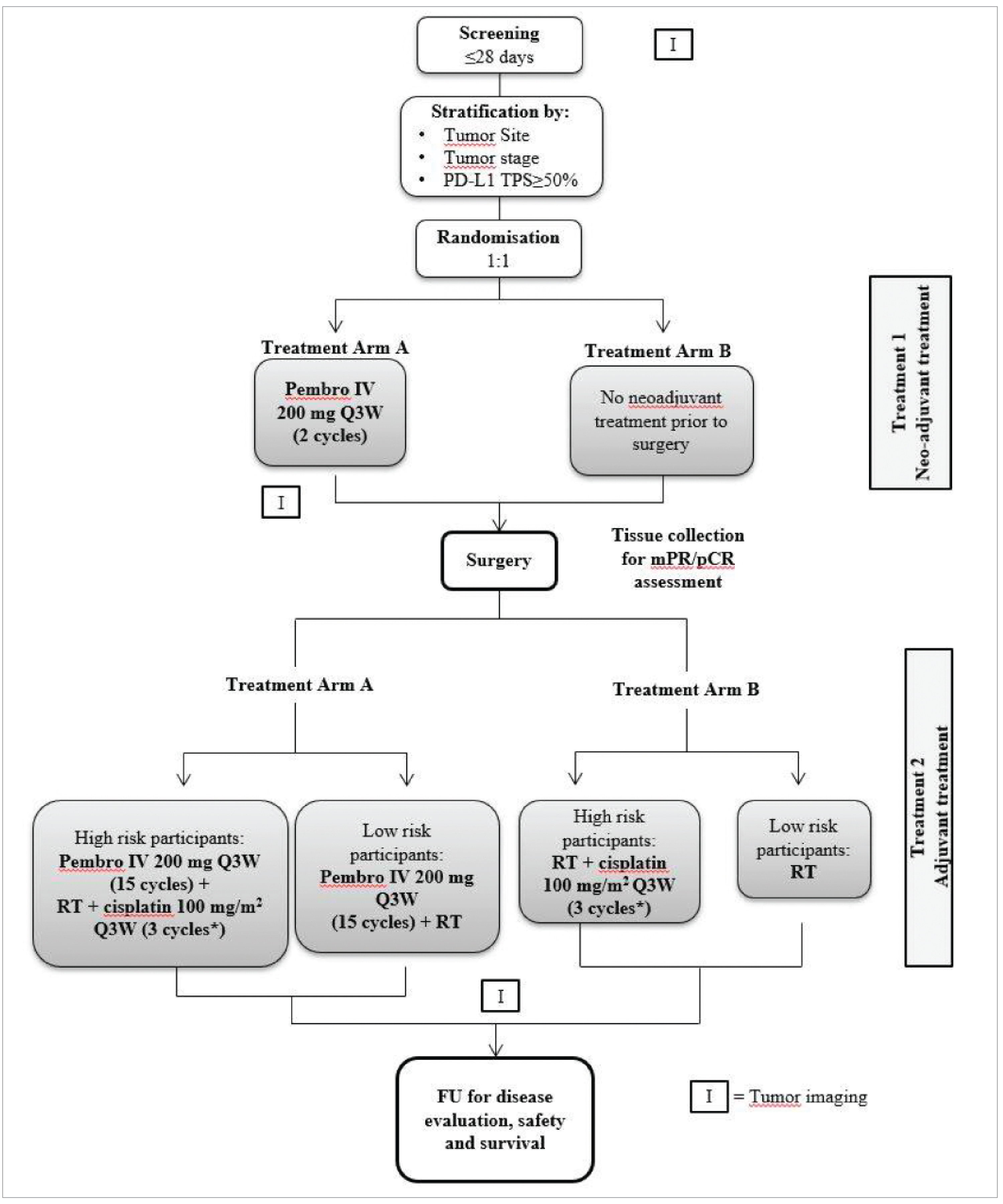
One pivotal, multicentre, open-label, active-controlled randomized phase III trial (the KEYNOTE-689 trial) met the inclusion criteria for the sponsor’s systematic review, in which a total of 192 sites randomized 714 patients (Table 5). The KEYNOTE-689 study investigated the efficacy and safety of pembrolizumab as neoadjuvant therapy, followed by postsurgical (adjuvant) treatment combining pembrolizumab and SOC RT (with or without cisplatin) followed by pembrolizumab monotherapy in treatment-naive patients with newly diagnosed stage III or stage IVA, resectable locally advanced HNSCC (Figure 1). Eligible patients were randomized in a 1:1 ratio to pembrolizumab plus SOC (arm A) and SOC (arm B). Randomization was stratified by primary tumour site (oropharynx or oral cavity versus larynx versus hypopharynx), tumour stage (III versus IVA), and PD-L1 status (TPS ≥ 50% versus TPS < 50%).26,49 The study consisted of the following study periods.49
Screening period (approximately 28 days): Patients were assessed for study eligibility.
Visits during treatment periods: Only patients in the pembrolizumab plus SOC group (arm A) received neoadjuvant pembrolizumab. In arm A, presurgery neoadjuvant treatment (pembrolizumab) began within 3 days of randomization. The recommended time frame for surgery (after the presurgery visit) was within 6 weeks (± 10 days) after randomization. Patients in arm A were allowed to go to surgery outside of this time frame if delays were due to an AE. Patients in the SOC alone group (arm B) proceeded directly to surgery within 4 weeks (± 10 days) after randomization. The postsurgery safety follow-up visit was approximately 7 days to 30 days postsurgery. The postsurgery adjuvant treatment started when patients had recovered adequately from the morbidity and/or complications from the surgery and with RT with or without cisplatin commencing at a recommended minimum range of within 6 weeks after definitive surgery. If the start of RT was delayed, up to 4 doses of pembrolizumab might be given. The timing of this dose of pembrolizumab could be adjusted based on timing of surgery after discussion with the sponsor. The first dose of pembrolizumab given during RT should be given such that pembrolizumab continued to be given every 21 days (± 3 days).
End-of-treatment and safety follow-up: The end-of-treatment visit was at the time study treatment was discontinued for any reason, or 30 days from the last dose of study treatment. The mandatory safety follow-up visit was approximately 30 days after the last dose of study treatment or before the initiation of a new anticancer treatment, whichever came first.
Posttreatment follow-up: Patients who completed study treatment or discontinued study treatment for a reason other than an event, disease progression, or recurrence moved into the posttreatment follow-up phase and were assessed in office every 3 months (91 days ± 7 days) until the end of year 3 (measured from the date of randomization) to monitor disease status. In year 4 and year 5, patients were assessed in office every 6 months (182 days ± 14 days) to monitor disease status. After year 5, patients moved into the survival follow-up.
Survival follow-up: Patients who completed the posttreatment follow-up or who experienced an event or disease progression or recurrence verified by BICR moved into the survival follow-up phase and were contacted by telephone every 3 months (91 days ± 14 days) to assess for survival status until death, withdrawal of consent, or the end of the study, whichever occurs first.
The KEYNOTE-689 study is ongoing, and the results reported in this review are based on the protocol-prespecified IA1 of EFS and OS for which the data cut-off date was July 25, 2024.26,49
Table 5: Details of Studies Included in the Systematic Review
Detail | KEYNOTE-689 study |
|---|---|
Designs and populations | |
Study design | Phase III, open-label, active-controlled RCT |
Locations | 192 centres in 24 countries from continents of Australia, the Americas (Canada, 5 sites with 13 patients), Asia, and Europe |
Patient enrolment dates | Start date: December 17, 2018 End date: July 25, 2024 |
Data cut-off date | For this review: July 25, 2024 |
Randomized (N) | Total N = 714
|
Inclusion criteria | Male and female patients at least 18 years of age were eligible to enrol in the study if they:
|
Exclusion criteria |
|
Drugs | |
Intervention | Pembrolizumab IV 200 mg q.3.w. (2 cycles as neoadjuvant therapy), followed by surgical resection, followed by adjuvant therapy with pembrolizumab IV 200 mg q.3.w. (3 cycles) plus SOC RT with or without cisplatin IV 100 mg/m2 q.3.w. (3 doses) based on the presence of high-risk pathologic features of extranodal extension or positive margins, followed by pembrolizumab monotherapy (12 cycles) as maintenance. |
Comparator | Surgical resection followed by adjuvant therapy with SOC RT with or without cisplatin IV 100 mg/m2 q.3.w. (3 doses) based on the presence of high-risk pathologic features of extranodal extension or positive margins. |
Study duration | |
Screening phase | Approximately 28 days before treatment randomization |
Treatment phase | Intervention arm: 2 cycles of pembrolizumab (q.3.w.) followed by surgical resection followed by 3 cycles of pembrolizumab (q.3.w.) concurrently with 6 weeks to 6.5 weeks of RT (to start within 6 weeks of the surgery) with or without concurrent cisplatin q.3.w. (3 doses), followed by pembrolizumab monotherapy (12 cycles) as maintenance Comparator arm: Surgical resection followed by 6 weeks to 6.5 weeks of RT (to start within 6 weeks of the surgery) with or without concurrent cisplatin q.3.w. (3 doses) |
Follow-up phase | Until an event or disease progression, pregnancy, withdrawal of consent, death, or the end of the study, whichever occurs first. |
Outcomes | |
Primary end point | Event-free survival |
Secondary and exploratory end points | Secondary:
Exploratory:
|
Publication status | |
Publication | Clinicaltrials.gov identifier: NCT03765918 |
ECOG = Eastern Cooperative Oncology Group; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-H&N35 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Head and Neck Module 35; FNA = fine needle aspiration; HNSCC = head and neck squamous cell carcinoma; IHC = immunohistochemistry; LA = locally advanced; q.3.w. = every 3 weeks; RCT = randomized controlled trial; RECIST = Response Evaluation Criteria in Solid Tumours Version 1.1; RT = radiotherapy; SOC = standard of care.
Source: Clinical Study Report for the KEYNOTE-689 study.26 Details included in the table are from the sponsor’s summary of clinical evidence.29
Populations
Inclusion and Exclusion Criteria
Eligible patients were aged 18 years or older with newly diagnosed, histologically confirmed locally advanced (stage III or stage IVA) HNSCC amenable to primary surgery. Patients had an ECOG PS of 0 or 1, adequate organ function, available HPV (p16) results for oropharyngeal tumours, and provided a tumour sample for PD-L1 testing. Patients who had received prior therapy with an anti–PD-1, anti–PD-L1, or anti–PD-L2 drug, or with a drug directed to another co-inhibitory T-cell receptor, or prior RT treatment or systemic anticancer therapy including investigational agents for the head and neck cancer under study before randomization, were excluded.
In the KEYNOTE-689 study, data were available for patients whose tumours express PD-L1 with a CPS of 1 or more, 10 or more, or 50 or more, and in all patients regardless of CPS status.26 The current clinical report focuses on the overall ITT population, as this aligns with the scope of the reimbursement request.
Interventions
Treatment flow in the neoadjuvant and adjuvant phases in both study arms is described in Figure 1. Pembrolizumab (200 mg) was administered via 30-minute IV infusion on day 1 of each 3-week cycle, after completing all scheduled assessments.
In the pembrolizumab plus SOC group (arm A), patients received 2 cycles of pembrolizumab (initiated within 3 days of randomization) presurgery during neoadjuvant treatment and underwent surgery within 6 weeks (± 10 days) after randomization. In arm A, delays for surgery due to an AE were allowed. Patients in the SOC group (arm B) proceeded directly to surgery within 4 weeks (± 10 days) after randomization.
Figure 1: KEYNOTE-689 Clinical Trial Design
FU = follow-up; mPR = major pathological response; pCR = pathological complete response; pembro = pembrolizumab; Q3W = every 3 weeks; RT = radiotherapy; TPS = tumour proportion score.
Note: Randomization was stratified by primary tumour site (oropharynx or oral cavity versus larynx versus hypopharynx), tumour stage (III versus IVA), and PD-L1 status (TPS ≥ 50% versus TPS < 50%).
*If there are delays in cisplatin treatment, cisplatin may continue for up to 1 week following completion of RT.
Sources: Clinical Study Report for the KEYNOTE-689 study26 and study protocol for the KEYNOTE-689 study.49
After surgical resection, patients were pathologically categorized as either high or low risk, based on assessment of the surgically resected specimens from the primary tumour site and neck lymph nodes. High-risk patients, defined as those with positive margins or extranodal extension following surgical resection, received SOC RT and cisplatin (100 mg/m2 of body surface area every 3 weeks for duration of RT). Low-risk patients, defined as having clear margins and no extranodal extension following surgical resection, received SOC RT.49
In the pembrolizumab plus SOC group (arm A), adjuvant pembrolizumab began after adequate surgery recovery. Postsurgery, patients in arm A could receive up to 4 cycles of pembrolizumab (every 3 weeks) before RT with or without cisplatin, with timing adjusted for RT delays. Pembrolizumab continued during RT with or without cisplatin, then as monotherapy for a total of 15 infusions postsurgery (17 cycles in total). If surgical recovery prevented pembrolizumab reinitiation within 3 weeks of surgery, treatment began concurrently with RT with or without cisplatin. During the combination portion of the study, treatments were sequenced as pembrolizumab, cisplatin (high-risk only), then RT.49
Adjuvant RT with or without cisplatin was planned to start within 6 weeks after surgery. All patients in the study were treated with 6 MV to 10 MV photons. Low-risk patients received RT at a dose of 2 Gy per day in 30 fractions for a total of 60 Gy; high-risk patients received cisplatin 100 mg/m2 of body surface area every 3 weeks for 3 cycles and RT at a dose of 2 Gy per day in 33 fractions for a total of 66 Gy; the RT was increased to 35 fractions for a total of 70 Gy in patients with gross residual disease. RT treatment for patients who underwent surgery was delivered over 6 weeks to 6.5 weeks, depending on risk of recurrence, with 5 fractions per week (5 fractions over 5 days [1 fraction per day], 2 days off). Patients who did not undergo planned surgery, for whatever reason, proceeded directly to salvage CRT where they received an RT dose of 70 Gy (total of 35 fractions) over 6.5 weeks to 7 weeks.49
Supportive care for pembrolizumab included oral or IV treatment with corticosteroids, and additional anti-inflammatory drugs if symptoms did not improve with administration of corticosteroids. Medications or vaccinations specifically prohibited in the exclusion criteria were not allowed during study treatment through follow-up. Antineoplastic systemic chemotherapy or biologic therapy was not allowed during the study. The study could be terminated early if the extent (incidence and/or severity) of emerging effects or clinical end points was such that the risk–benefit ratio to the study population as a whole was unacceptable. Pembrolizumab dose reductions were not permitted. Per protocol, pembrolizumab treatment could be interrupted or discontinued due to toxicity. Patients could have a maximum of 2 dose modifications throughout the course of the study for toxicities.49
Outcomes
A list of efficacy end points assessed in this clinical review report is provided in Table 6, followed by descriptions of the outcome measures. Summarized end points are based on outcomes included in the sponsor’s summary of clinical evidence as well as any outcomes identified as important to this review according to the clinical experts consulted for this review and input from patient and clinician groups and public drug plans. Using the same considerations, we selected end points that were considered to be most relevant to inform expert committee deliberations and finalized this list of end points in consultation with members of the expert committee. All summarized efficacy end points were assessed using GRADE. Select notable harms outcomes considered important for informing expert committee deliberations were also assessed using GRADE.
Considerations that informed the selection of outcomes to be summarized and assessed using GRADE include the following.
OS in the overall ITT population was identified by the patient and clinician groups’ input, and the clinical experts consulted for this review. Probability of OS at months 12 and months 60 were assessed using the GRADE approach.
EFS in the overall ITT population was identified by the patient and clinician groups’ input, and the clinical experts consulted for this review. Probability of EFS at months 12, months 36, and months 48 were assessed using the GRADE approach.
HRQoL outcomes were identified by the patient and clinician groups’ input, and specified by the clinical experts to include change from baseline in the EORTC QLQ-C30 GHS/QoL score at postadjuvant CRT or RT weeks 25 and 51 in the overall ITT population.
Harms are considered important outcomes according to input from the patient group and the clinical experts. Grade 3 to grade 5 AEs were assessed using the GRADE approach.
Additional outcome data are provided in the Appendix for end points highlighted by clinical experts as important, including EFS in patients with a CPS of 10 or more, major pathological response (mPR), pathological complete response (pCR), distant metastasis-free survival (DMFS), and local regional control (LRC).
Table 6: Outcomes Summarized From the KEYNOTE-689 Study
Outcome measure | Time point | KEYNOTE-689 study |
|---|---|---|
OS | The HR and medians were presented up to the DCO of July 25, 2024. Probabilities at 6, 12, 24, 36, 48, and 60 months were reported. | Key secondary outcomea |
EFS by BICR | The HR and medians were presented up to the DCO of July 25, 2024. Probabilities at 6, 12, 24, 36, and 48 months were reported. | Primary outcomea |
EORTC QLQ-C30 global health status/QoL scores | Change from baseline to postadjuvant CRT or RT (weeks 25 and 51). | Secondary outcome |
AEs, SAEs, AEs leading to treatment discontinuation | Results were based on a DCO of July 25, 2024, in the APaT population. | Secondary outcome |
AE = adverse event; APaT = all participants as treated; BICR = blinded independent central review; CRT = chemoradiotherapy; DCO = data cut-off; EFS = event-free survival; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; HR = hazard ratio; OS = overall survival; QoL = quality of life; RT = radiotherapy; SAE = serious adverse event.
aStatistical testing for these end points was adjusted for multiple comparisons using the graphical method of Maurer and Bretz.50
Source: Clinical Study Report for the KEYNOTE-689 study.26 Details included in the table are from the sponsor’s summary of clinical evidence.29
In the KEYNOTE-689 study, the primary efficacy end point was EFS. Secondary efficacy outcomes were mPR, OS, and pCR. Both mPR and pCR were assessed by blinded central pathologists. OS and mPR were key secondary outcomes. LRC and DMFS, based on BICR, were some of the other exploratory efficacy end points. Other secondary end points were patient-reported outcomes (PROs) and safety. PROs included change from baseline in GHS/QoL and physical functioning scores from the EORTC QLQ-C30, as well as swallowing, speech, and pain symptoms based on the European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Head and Neck Module 35 (EORTC QLQ-H&N35). Safety end points were AEs, SAEs, AEOSIs, changes in vital signs and laboratory values, and discontinuation of study treatment because of AEs.26,49
Overall Survival
OS was defined as the time from randomization to death due to any cause. Patients without an event at the data cut-off date were censored at the date that they were last known to be alive.
Event-Free Survival
EFS was defined as the time from randomization to the first of the following events.
Radiographic disease progression per RECIST Version 1.1 by BICR — participants who undergo a definitive biopsy of the progressed lesion and are found to have no histologic evidence of invasive cancer do not meet criteria for an event.
Radiographic disease progression during neoadjuvant phase that precludes surgery by BICR.
Local or distant progression or recurrence (as assessed with imaging or biopsy as indicated) by BICR.
Death due to any cause.
A secondary malignancy was not considered an EFS event. Patients without an event at the data cut-off date were censored at their last disease assessment, regardless of whether a subsequent anticancer therapy was initiated.
Major Pathological Response
mPR was defined as having 10% or less residual invasive squamous cell carcinoma (SCC) in the resected primary tumour specimen and all sampled regional lymph nodes.
Pathological Complete Response
pCR was defined as absence of residual invasive SCC in the resected primary tumour specimen and all sampled regional lymph nodes.
Local Regional Control
LRC was defined as time from date of randomization to the date of first record of radiographic disease progression (the same exceptions as for the EFS outcome) or local recurrence as assessed with imaging or biopsy as indicated. Distant metastases diagnosed before any locoregional failure and death in absence of locoregional failures were not considered events, but as competing risks, were censored at the date of documented disease metastasis or death, whichever occurred first.
Distant Metastasis-Free Survival
DMFS was defined as time from date of randomization to the date of first record of appearance of distant metastasis or death due to any cause. Patients without documented metastatic disease diagnosis at the data cut-off date were censored at the date of their last disease assessment.
Health-Related Qualify of Life
The summaries of the EORTC QLQ-C30 and EORTC QLQ-H&N35 are shown in Table 7. These PRO questionnaires were administered by trained site personnel and completed electronically by patients, before drug administration, AE evaluation, and disease status notification.
Harms
Definitions of the safety end points in the KEYNOTE-689 study are as follows.
AEs: AEs, irrespective of causality, were reported from the time of treatment randomization through 30 days after the last dose of study treatment. AE severity was graded as defined by the Common Terminology Criteria for Adverse Events Version 4.03.49
SAEs: SAEs included those reported from the time of treatment randomization through 90 days after the last dose of study treatment or 30 days following discontinuation of study treatment if the patient initiates a new anticancer treatment, whichever occurred first. SAEs are AEs resulting in death; that are life-threatening; that require inpatient hospitalization or prolongation of existing hospitalization; that result in persistent or significant disability or incapacity; congenital anomalies or birth defects; or other important medical events.
Death due to AE: Grade 5 AEs (AEs leading to death) were reported.
Discontinuation of study treatment due to AE: These include permanent discontinuation of any study treatment due to any study intervention-related toxicity specified as a reason for permanent discontinuation as defined in the guidelines for dose modification due to AEs.
AEOSI: AEOSIs were immune-mediated events and infusion-related reactions associated with pembrolizumab, regardless of causality as reported by investigators. AEOSIs were reported from randomization through 90 days after the last dose of study treatment or 30 days following discontinuation of study treatment if the patient initiated a new anticancer treatment, whichever occurred first.
Table 7: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
EORTC QLQ-C30 global health status/QoL | Cancer-specific self-reported measure of HRQoL. A 30-item questionnaire, consisting of 5 functional scales (physical, role, emotional, cognitive, and social); 3 multi-item symptom scales (fatigue, pain, and nausea and vomiting); and 7 single items (i.e., dyspnea, appetite loss, sleep disturbance, constipation, and diarrhea); these are measured on a 4-point response scale. The instrument also contains an item assessing the perceived financial impact of the disease and treatment and two 7-point response scales pertaining to global health and QoL. A higher score for global health status represents better functioning ability or HRQoL. | In studies with patients with HNC:
| A recent study estimated MIDs for between-group difference in change over time for the EORTC QLQ-C30 using data from 2 phase III trials involving 808 patients with HNC.56 The anchor-based MIDs for global health status were 5 points for improvement and −7 points for deterioration.56 |
EORTC QLQ-H&N35 (pain, swallowing, and speech symptom score) | Head and neck cancer-specific self-reported module of the EORTC QLQ-C30, which measures HRQoL in HNC.57,58 Consists of 7 multi-item scales (pain in the mouth, problems with swallowing, senses, speech, social eating, social contact, and sexuality), and 11 single-item scales (problems with teeth, mouth opening, dry mouth, sticky saliva, coughing, feeling ill, use of analgesics, use of nutritional supplements, use of feeding tube, weight gain, and weigh loss); these are measured on a 4-point response scale except for questions related to treatments and weight which are measured on a 2-point yes or no scale. A higher score represents worse symptoms or more frequent medication or treatment use.57,58 | In studies with patients with HNC:
| MID for the setting of the current review is not known. |
EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-H&N35 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Head and Neck Module 35; HNC = head and neck cancer; HRQoL = health-related quality of life; MID = minimal important difference; QoL = quality of life.
Statistical Analysis
Clinical Trial End Points
The primary hypotheses for EFS and key secondary hypotheses for OS were evaluated by comparing the pembrolizumab plus SOC group to the SOC group using a stratified log-rank test. Estimations of the HRs were performed by using a stratified Cox regression model with Efron’s tie-handling method. The nonparametric KM method was used to estimate the survival curves.26
The key secondary hypotheses for mPR were evaluated by comparing the pembrolizumab plus SOC group to the SOC group with respect to the rate of mPR using the stratified Miettinen and Nurminen method with strata weighting by sample size. The same method was used to evaluate the pCR rate.26 The nonparametric KM method was used to estimate the DMFS curves. A stratified Cox proportional hazard model with Efron’s method of tie handling was used to estimate the magnitude of the treatment difference (i.e., HR) between the groups for DMFS.59 The nonparametric cumulative incidence estimator was used to estimate the LRC curve.59-61
The LS mean changes and 95% CIs from baseline in the EORTC QLQ-C30 GHS/QoL and EORTC QLQ-H&N35 swallowing, speech, and pain scores were modelled using a constrained longitudinal data analysis model with the PRO score as the response variable and treatment, time, treatment by time interaction, and stratification factors as covariates.
Details about the statistical model, adjustment factors, handling of missing data, and sensitivity analyses of efficacy end points analyses are described in Table 8.
Sample Size and Power Calculation
The study intended to enrol approximately 704 patients. The actual number of patients enrolled was 714, which was expected to result in approximately 65% (approximately 462) patients whose tumours express PD-L1 with a CPS of 10 or more and approximately 95% (approximately 680) patients whose tumours express PD-L1 with a CPS of 1 or more.49 The EFS hypothesis testing strategy was designed with an alpha of 0.025 (1-sided) and power of 94.9% to detect a HR of 0.62 with 232 EFS events for patients whose tumours express PD-L1 with a CPS of 10 or more at the second interim analysis (IA2) (the final EFS analysis). In addition, at an alpha of 0.025 (1-sided), the EFS hypothesis for patients whose tumours express PD-L1 with a CPS of 1 or more yields 91.2% power to detect a HR of 0.70 with 354 events at IA2; the EFS hypothesis for all patients yields 92.5% power to detect a HR of 0.70 with 371 events at IA2.59 The duration of EFS in the SOC arm was assumed to follow a Poisson mixture model with approximately 34% of patients achieving excellent long-term results (i.e., cure rate of 34%), with a cumulative EFS rate of 90% at 4 months and 42% at 34 months based on historical data.38,59 The enrolment duration was assumed to be 58 months, and the yearly dropout rate was assumed to be 5% for both arms.59
If the underlying mPR rate in the SOC arm is 2%, based on 462 patients whose tumours express PD-L1 with a CPS of 10 or more, the study has approximately greater than 99.9% power to detect a true mPR rate difference of 25 percentage points, at an alpha of 0.0005 (1-sided). Based on 680 patients whose tumours express PD-L1 with a CPS of 1 or more, the study has approximately greater than 99.9% power to detect a true mPR rate difference of 20 percentage points, at an alpha of 0.0005 (1-sided). Under the same assumptions, the power to detect a true mPR rate difference of 20 percentage in 714 patients irrespective of PD-L1 CPS is higher. At an alpha of 0.0245 (1-sided), the final OS hypothesis for patients whose tumours express PD-L1 with a CPS of 10 or more yields 84.9% power to detect a HR of 0.65 with 201 deaths observed. The final OS hypothesis for patients whose tumours express PD-L1 with a CPS of 1 or more yields 69.7% power to detect a HR of 0.75 with 309 events. The final OS hypothesis for all patients yields 71.9% power to detect a HR of 0.75 with 324 deaths observed.59
Statistical Testing
All efficacy analyses were performed in patients whose tumours express PD-L1 with a CPS of 10 or more, a CPS of 1 or more, and in all patients regardless of CPS score.49 Multiplicity was controlled across the primary efficacy hypothesis regarding EFS, and the 2 key secondary efficacy hypotheses regarding mPR and OS, as well as across the 3 PD-L1 CPS populations.49,59 The overall type I error across the EFS, mPR, and OS end points and across PD-L1 CPS populations was strongly controlled at an alpha of 2.5% (1-sided), with an alpha of 2.5% initially allocated to the hypothesis for EFS in patients whose tumours express PD-L1 with a CPS of 10 or more (hypothesis 1 [H1]). Alpha could be reallocated using the graphical approach of Mauer and Bretz.50 Group sequential methods (Lan-DeMets O’Brien-Fleming) were planned to be used to allocate alpha among the interim and final analyses for EFS and OS based on the information fraction. Figure 4 in the Appendix shows the 1-sided alpha allocation for each hypothesis in the ellipse representing the hypothesis. The weights for reallocation from each hypothesis to the others are represented in the boxes on the lines connecting hypotheses.49,59
If the EFS hypotheses (H1, H2, and H3) were rejected at any analysis, the alpha of 0.025 (1-sided) was split, and the alpha of 0.0005 (1-sided) would be rolled over to the mPR test for patients whose tumours express PD-L1 with a CPS of 10 or more (H4).49 The mPR rate hypothesis for patients whose tumours express PD-L1 with a CPS of 10 or more (H4) would be tested at an alpha of 0.0005 (1-sided) if all EFS null hypotheses (H1, H2, and H3) were rejected, or at an alpha of 0.025 (1-sided) if all EFS (H1, H2, and H3) and OS hypotheses (H7, H8, and H9) were rejected. OS would be tested only if all EFS null hypotheses (H1, H2, and H3) were rejected. The OS hypothesis for patients whose tumours express PD-L1 with a CPS of 10 or more (H7) would be tested at an alpha of 0.0245 (1-sided) if all EFS null hypotheses (H1, H2, and H3) were rejected, or at an alpha of 0.025 (1-sided) if all EFS (H1, H2, and H3) and mPR rate hypotheses (H4, H5, and H6) were rejected.49
Interim Analyses
Two interim analyses and 1 final analysis were planned in this study.26,49 This submission summarizes the results of the IA1, which was planned to be performed when approximately 207 EFS events occurred in patients whose tumours express PD-L1 with a CPS of 10 or more and approximately 6 months after the last patient had been randomized. Per protocol, if the EFS events in patients with a CPS of 10 or more accrued slower than expected, the analysis could be delayed up to 3 months after the projected timing.26,49 Based upon the event accrual rate, IA1 was performed approximately 9 months after the last patient had been randomized. There were 192 EFS events in the patient population with a CPS of 10 or more as of the IA1 data cut-off date of July 25, 2024.26 IA1 was planned to be the first interim analysis of EFS and OS and the final analysis of mPR.26,59
Subgroup Analyses
To determine whether the treatment effect is consistent across various subgroups, the estimate of the between-group treatment effect (with 95% CI) for the primary end point (i.e., EFS) and OS were estimated and plotted within each category of the following classification variables: primary tumour site (oropharynx or oral cavity versus larynx versus hypopharynx), tumour stage (III versus IVA), PD-L1 status (TPS ≥ 50% versus TPS < 50%), age (aged < 65 years versus aged ≥ 65 years), sex (female versus male), race (white versus all others), smoking status (never smoker versus former smoker versus current smoker), and geographical regions (North America versus European Union versus rest of the world). The consistency of the treatment effect was assessed descriptively via summary statistics by category for these subgroup analysis variables, using unstratified methods. In addition, a forest plot was produced, which provides the estimated point estimates and CIs for the treatment effect across the subgroups categories. If the number of patients in a category of a subgroup variable was less than 10% of the analysis population, the subgroup analysis was not performed for this category of the subgroup variable.49
Sensitivity Analyses
Sensitivity analyses for EFS are summarized in Table 8.
Analysis Populations
Analyses of primary and secondary efficacy end points were conducted on all randomized patients according to the ITT principle. Safety analyses were conducted in the APaT population, which consists of all randomized participants who received study treatment. PRO analyses were conducted in the PRO full analysis set (FAS) population, defined as participants who have received treatment and have at least 1 PRO assessment available. The analyses populations are defined in Table 9.
Table 8: Statistical Analysis of Efficacy End Points for the KEYNOTE-689 Study
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
Overall survival |
|
| Censored at the last date participant was known to be alive. | None |
EFS |
|
| Censored at the last disease assessment. |
|
HRQoL (EORTC QLQ-C30 GHS/QoL score, EORTC QLQ-C30 physical functioning score, EORTC QLQ-H&N35 swallowing, speech, and pain symptom score) | Constrained longitudinal data analysis model | Treatment, time, the treatment by time interaction and stratification factors as covariates | Assumed missing at random. | None |
CI = confidence interval; EFS = event-free survival; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-H&N35 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Head and Neck Module 35; HR = hazard ratio; HRQoL = health-related quality of life; TPS = tumour proportion score; vs. = versus.
Sources: Clinical Study Report for the KEYNOTE-689 study26 and study protocol for the KEYNOTE-689 study.49 Details included in the table are from the sponsor’s summary of clinical evidence.29
Table 9: Analysis Populations of the KEYNOTE-689 Study
Population | Definition | Application |
|---|---|---|
ITT | All randomized participants. Patients were analyzed in the treatment group to which they were randomized. | Analysis of all efficacy outcomes |
APaT | All randomized participants who received study treatment (note that surgery was considered part of study treatment). Participants were included in the treatment group corresponding to the study treatment they actually received. | Analysis of safety outcomes and treatment exposure |
PRO FAS | Participants who have received treatment and have at least 1 PRO assessment available. | Analysis of PRO |
APaT = all participants as treated; FAS = full analysis set; ITT = intention to treat; PRO = patient-reported outcome.
Source: Clinical Study Report for the KEYNOTE-689 study.26 Details included in the table are from the sponsor’s summary of clinical evidence.29
Results
Patient Disposition
A total of 1,044 patients were screened for eligibility and 714 participants were randomized to receive pembrolizumab plus SOC (n = 363) or SOC alone (n = 351). These patients were included in the ITT population. All nonrandomized participants (n = 330) were screen failures, including 327 patients who did not meet the eligibility criteria (Table 10).
In the neoadjuvant phase, 360 of 363 (97%) patients in the pembrolizumab plus SOC group started study treatment (i.e., pembrolizumab), and of the 360 patients, 341 (94.7%) completed neoadjuvant study treatment (Table 10). In the SOC group, there was no neoadjuvant treatment; however, 2 patients received neoadjuvant treatment mistakenly. One of these 2 patients received 1 cycle of neoadjuvant treatment and was returned to SOC treatment for the remainder of the study, while the other patient received 2 neoadjuvant cycles of pembrolizumab, underwent surgery, and was discontinued from study treatment; this patient did not receive any adjuvant treatment.26
Of the overall ITT population, 322 (88.7%) in the pembrolizumab plus SOC group and 308 (87.7%) in the SOC group underwent in-study surgery (Table 10), while 41 patients in the pembrolizumab plus SOC group and 43 patients in the SOC group did not undergo in-study surgery. Reasons for in-study surgery not being performed included receiving adjuvant treatment without surgery (8 patients in each group; 2.2% in the pembrolizumab group and 2.3% in the SOC group) and discontinuation of treatment before surgery (30 patient [8.3%] in the pembrolizumab group) and randomized but not treated (35 patients [10.0%] in the SOC group). The most common reason for discontinuing treatment before surgery was due to progressive disease in the pembrolizumab group (15 patients [4.1%]).26
A total of 54 patients in the pembrolizumab group and 41 in the SOC group completed surgery but did not receive adjuvant treatment. The most common reasons in the pembrolizumab group were progressive disease (27 patients [50.0%]), withdrawal by patient (8 patients [14.8%]), and AEs (9 patients [16.7%]). The most common reasons in the SOC group were AEs (11 patients [26.8%]), withdrawal by patient (10 patients [24.4%]), physician decision (9 patients [22.0%]), and progressive disease (7 patients [17.1%]).
Among patients who underwent surgery (323 in the pembrolizumab plus SOC group and 307 in the SOC group, as treated), 266 (82.4%) patients in the pembrolizumab plus SOC group and 267 (87.0%) patients in the SOC group received in-study surgery and adjuvant RT; 100 (31.0%) participants in the pembrolizumab plus SOC group and 132 (43.0%) participants in the SOC group received in-study surgery and adjuvant RT plus cisplatin.26,29
The adjuvant phase following surgery was longer in the pembrolizumab plus SOC group (15 cycles: 3 cycles of adjuvant pembrolizumab in combination with SOC RT with or without 3 cycles of cisplatin, followed by 12 cycles of pembrolizumab monotherapy as maintenance) than the SOC group (RT with or without 3 cycles of cisplatin). In the adjuvant phase following surgery, 275 patients in the pembrolizumab plus SOC group started study treatment of which 155 (56.4%) completed study treatment. In these 275 patients, 255 (92.7%) received adjuvant pembrolizumab, 36 (14.1%) completed 3 or fewer cycles of pembrolizumab, 219 (85.9%) completed more than 3 cycles of pembrolizumab, and 20 (7.3%) patients received RT with or without cisplatin only. In the SOC group, 275 patients started study treatment, and 261 (94.9%) completed study treatment consisting of RT with or without 3 cycles of cisplatin.
The proportion of patients who discontinued overall study treatment was greater in the pembrolizumab plus SOC group than in the SOC group (53.9% in the pembrolizumab plus SOC group and 17.4% in the SOC group). The most common reasons for discontinuation of all study treatment were progressive disease (21.9%) and AEs (18.1%) in the pembrolizumab plus SOC group and AEs (5.1%), withdrawal by patients (4.1%), and progressive disease (3.2%) in the SOC group. As of the IA1 data cut-off date (July 25, 2024), 11 (3.1%) patients in the pembrolizumab plus SOC group and no patients in the SOC group were still receiving study treatment.
Table 10: Summary of Patient Disposition From the KEYNOTE-689 Study
Patient disposition | Pembrolizumab plus SOC n = 363 | SOC n = 351 |
|---|---|---|
Number of patients screened | 1,044 | |
Number of patients not randomized | 330 | |
Screen failure | 330 | |
Did not meet eligibility criteria | 327 | |
Number of participants randomized (ITT) | 363 | 351 |
Status for neoadjuvant study treatment | ||
Started, n | 360 | 2 |
Completed, n (%) | 341 (94.7) | 0 |
Discontinued, n (%) | 19 (5.3) | 0 |
Adverse event | 6 (1.7) | 0 |
Clinical progression | 4 (1.1) | 0 |
Lost to follow-up | 1 (0.3) | 0 |
Physician decision | 2 (0.6) | 0 |
Associated with COVID-19 | 1 (0.3) | 0 |
Progressive disease | 3 (0.8) | 0 |
Withdrawal by patient | 3 (0.8) | 0 |
Status not recorded, n (%) | 0 | 2 (100.0) |
Status for in-study surgery | ||
Started in-study surgery, n | 322 | 308 |
Without in-study surgery (reasons for in-study surgery not performed listed in the following rows), n | 41 | 43 |
Adjuvant treatment without surgerya, subtotal, n (%) | 8 (2.2) | 8 (2.3) |
Adverse events | 1 (0.3) | 1 (0.3) |
Physician decision | 2 (0.6) | 2 (0.6) |
Tumour found to be surgically unresectable | 2 (0.6) | 0 |
Withdrawal by patient | 3 (0.8) | 5 (1.4) |
Discontinued treatment before surgeryb, subtotal, n (%) | 30 (8.3) | 0 |
Adverse events | 4 (1.1) | 0 |
Lost to follow-up | 1 (0.3) | 0 |
Nonstudy anticancer therapy | 1 (0.3) | 0 |
Physician decision | 2 (0.6) | 0 |
Progressive disease | 15 (4.1) | 0 |
Withdrawal by patient | 7 (1.9) | 0 |
Randomized but not treatedc, subtotal, n (%) | 3 (0.8) | 35 (10.0) |
████████ ███████████ | █████ | █████ |
████████ ███████████ | █████ | █████ |
████████ ███████████ | █████ | █████ |
████████ ███████████ | █████ | █████ |
████████ ███████████ | █████ | █████ |
████████ ███████████ | █████ | █████ |
Completed, n (%) | 321 (99.7) | 308 (100.0) |
Discontinued, n (%) | 1 (0.3) | 0 |
Tumour found to be surgically unresectable | 1 (0.3) | 0 |
Patients who completed surgery but did not receive adjuvant therapy, n (%) | 54 (14.9) | 41 (11.7) |
Adverse event | 9 (2.5) | 11 (3.1) |
Clinical progression | 2 (0.6) | 1 (0.3) |
Noncompliance with study drug | 1 (0.3) | 2 (0.6) |
Nonstudy anticancer therapy | 1 (0.3) | 1 (0.3) |
Physician decision | 6 (1.7) | 9 (2.6) |
Progressive disease | 27 (7.4) | 7 (2.0) |
Withdrawal by patient | 8 (2.2) | 10 (2.8) |
Status for adjuvant study treatment | ||
Started, n | 275 | 275 |
Completed, n (%) | 155 (56.4) | 261 (94.9) |
Discontinued, n (%) | 109 (39.6) | 14 (5.1) |
Adverse event | 52 (18.9) | 5 (1.8) |
Associated with COVID-19 | 1 (0.4) | 0 |
Clinical progression | 1 (0.4) | 0 |
Noncompliance with study drug | 1 (0.4) | 1 (0.4) |
Nonstudy anticancer therapy | 2 (0.7) | 2 (0.7) |
Physician decision | 5 (1.8) | 0 |
Associated with COVID-19 | 1 (0.4) | 0 |
Progressive disease | 36 (13.1) | 3 (1.1) |
Withdrawal by patient | 12 (4.4) | 3 (1.1) |
Patients ongoing | 11 (4.0) | 0 |
Status for study treatment (overall) | ||
Started, n | 360 | 316 |
Completed, n (%) | 155 (43.1) | 261 (82.6) |
Discontinued, n (%) | 194 (53.9) | 55 (17.4) |
Adverse event | 65 (18.1) | 16 (5.1) |
Associated with COVID-19 | 2 (0.6) | 0 |
Clinical progression | 3 (0.8) | 1 (0.3) |
Lost to follow-up | 1 (0.3) | 0 |
Noncompliance with study drug | 2 (0.6) | 3 (0.9) |
Nonstudy anticancer therapy | 4 (1.1) | 3 (0.9) |
Physician decision | 13 (3.6) | 9 (2.8) |
Associated with COVID-19 | 2 (0.6) | 0 |
Progressive disease | 79 (21.9) | 10 (3.2) |
Withdrawal by patient | 27 (7.5) | 13 (4.1) |
Patients ongoing (receiving study treatment) | 11 (3.1) | 0 |
Status for trial | ||
Discontinued, n (%) | 123 (33.9) | 146 (41.6) |
Death | 109 (30.0) | 127 (36.2) |
Associated with COVID-19 | 2 (0.6) | 4 (1.1) |
Lost to follow-up | 2 (0.6) | 3 (0.9) |
Withdrawal by patient | 12 (3.3) | 16 (4.6) |
COVID-19 association unspecified, subsequently died | 4 (1.1) | 4 (1.1) |
Patients ongoing | 240 (66.1) | 205 (58.4) |
PRO FAS, N | 356 | 306 |
Safety population (APaT), N | 361 | 315 |
APaT = all participants as treated; FAS = full analysis set; ITT = intention to treat; PRO = patient-reported outcome; SOC = standard of care.
Note: Data presented in this table were based on analyses at the clinical cut-off date of July 25, 2024.
aPatients receiving SOC in this category were eligible to receive radiation ± cisplatin. Patients receiving pembrolizumab plus SOC were eligible to receive pembrolizumab ± radiation ± cisplatin.
bPatients in this category received at least 1 dose of pembrolizumab and discontinued treatment before surgery.
cPatients in this category did not continue to any phase of treatment (including surgery) following randomization.
Source: Clinical Study Report for the KEYNOTE-689 study.26 Details included in the table are from the sponsor’s summary of clinical evidence.29
Baseline Characteristics
The baseline characteristics outlined in Table 11 are limited to those that are most relevant to this review or were felt to affect the outcomes or interpretation of the study results.
Of the 714 patients enrolled in the study, 465 (65.1%) had PD-L1 with a CPS of 10 or more and 682 (95.5%) had PD-L1 with a CPS of 1 or more. Overall, demographic and baseline disease characteristics of the ITT population were generally similar between the treatment groups. In the KEYNOTE-689 study, the proportion of male patients (78.9%) was higher than female patients (21.1%). Patients had a median age of 60.0 years (range, 22 years to 87 years). In the overall ITT population, 60.5% of patients had their primary tumour located in the oral cavity, 73.9% had stage IVA disease, and 57.1% had an ECOG PS of 0 at baseline. Most patients were current or former smokers (78.4%) and alcohol users (68.3%).26,29
Table 11: Summary of Baseline Characteristics From the KEYNOTE-689 Study (Overall ITT Population)
Characteristic | Pembrolizumab plus SOC n = 363 | SOC n = 351 |
|---|---|---|
Demographic characteristics | ||
Age (years) | ||
Mean (SD) | 59.4 (10.0) | 60.6 (10.0) |
Median (range) | 60.0 (29 to 82) | 61.0 (22 to 87) |
< 65 years, n (%) | 249 (68.6) | 227 (64.7) |
≥ 65 years, n (%) | 114 (31.4) | 124 (35.3) |
Sex, n (%) | ||
Female | 77 (21.2) | 74 (21.1) |
Male | 286 (78.8) | 277 (78.9) |
Race, n (%) | ||
American Indian or Alaska Native [wording from original source] | 0 | 1 (0.3) |
Asian | 51 (14.0) | 44 (12.5) |
Black or African American | 9 (2.5) | 9 (2.6) |
Multiple | 9 (2.5) | 20 (5.7) |
Native Hawaiian or Other Pacific Islander | 1 (0.3) | 1 (0.3) |
White | 284 (78.2) | 270 (76.9) |
Missing | 9 (2.5) | 6 (1.7) |
Smoking status, n (%) | ||
Never smoker | 64 (17.6) | 81 (23.1) |
Former smoker | 176 (48.5) | 144 (41.0) |
Current smoker | 117 (32.2) | 123 (35.0) |
Unknown | 6 (1.7) | 3 (0.9) |
Cigarette use (packs per year), n (%) | ||
0 (no cigarette use) | 64 (17.6) | 81 (23.1) |
≤ 10 | 67 (18.5) | 73 (20.8) |
> 10 | 216 (59.5) | 186 (53.0) |
Unknown | 16 (4.4) | 11 (3.1) |
Alcohol use, n (%) | ||
Yes | 250 (68.9) | 238 (67.8) |
No | 107 (29.5) | 110 (31.3) |
Unknown | 6 (1.7) | 3 (0.9) |
Disease characteristics | ||
PD-L1 status by TPS score, n (%) | ||
TPS < 50% | 257 (70.8) | 242 (68.9) |
TPS ≥ 50% | 103 (28.4) | 107 (30.5) |
Missing | 3 (0.8) | 2 (0.6) |
PD-L1 status by CPS score, n (%) | ||
CPS < 50 | 244 (67.2) | 224 (63.8) |
CPS ≥ 50 | 116 (32.0) | 125 (35.6) |
Missing | 3 (0.8) | 2 (0.6) |
PD-L1 status by CPS score, n (%) | ||
CPS < 10 | 126 (34.7) | 118 (33.6) |
CPS ≥ 10 | 234 (64.5) | 231 (65.8) |
Missing | 3 (0.8) | 2 (0.6) |
PD-L1 status by CPS score, n (%) | ||
CPS < 1 | 13 (3.6) | 14 (4.0) |
CPS ≥ 1 | 347 (95.6) | 335 (95.4) |
Missing | 3 (0.6) | 2 (0.6) |
Primary tumour site, n (%) | ||
Oropharynx | 35 (9.6) | 38 (10.8) |
Oral cavity | 219 (60.3) | 213 (60.7) |
Larynx | 81 (22.3) | 73 (20.8) |
Hypopharynx | 28 (7.7) | 26 (7.4) |
Missing | 0 | 1 (0.3) |
ECOG PS, n (%) | ||
0 | 199 (54.8) | 209 (59.5) |
1 | 164 (45.2) | 142 (40.5) |
HPV status, n (%) | ||
Positive | 12 (3.3) | 15 (4.3) |
Negative | 351 (96.7) | 335 (95.4) |
Unknown | 0 | 1 (0.3) |
Overall cancer staging at baseline, n (%) | ||
III | 90 (24.8) | 93 (26.5) |
IVA | 271 (74.7) | 257 (73.2) |
IVB | 2 (0.6) | 0 |
Missing | 0 | 1 (0.3) |
Cancer T staging at baseline, n (%) | ||
T1 | 9 (2.5) | 4 (1.1) |
T2 | 49 (13.5) | 47 (13.4) |
T3 | 110 (30.3) | 113 (32.2) |
T4 | 11 (3.0) | 15 (4.3) |
T4A | 184 (50.7) | 170 (48.4) |
T4B | 0 | 1 (0.3) |
Missing | 0 | 1 (0.3) |
Cancer N staging at baseline, n (%) | ||
N0 | 94 (25.9) | 88 (25.1) |
N1 | 81 (22.3) | 72 (20.5) |
N2 | 32 (8.8) | 31 (8.8) |
N2A | 12 (3.3) | 7 (2.0) |
N2B | 83 (22.9) | 88 (25.1) |
N2C | 58 (16.0) | 64 (18.2) |
N3 | 1 (0.3) | 0 |
N3B | 2 (0.6) | 0 |
Missing | 0 | 1 (0.3) |
Cancer M staging at baseline, n (%) | ||
M0 | 363 (100.0) | 350 (99.7) |
Missing | 0 | 1 (0.3) |
CPS = combined positive score; ECOG PS = Eastern Cooperative Oncology Group Performance Status; ITT = intention to treat; SD = standard deviation; SOC = standard of care; TPS = tumour proportion score.
Note: Data presented in this table were based on analyses at the clinical cut-off date of July 25, 2024.
Source: Clinical Study Report for the KEYNOTE-689 study.26 Details included in the table are from the sponsor’s summary of clinical evidence.29
Exposure to Study Treatments
Study Treatments
Overall, the median duration of therapy was longer in the pembrolizumab plus SOC group compared with the SOC group (9.1 months; range, 0.03 months to 22.3 months versus 2.9 months; range, 0.03 months to 7.2 months). The proportion of participants on therapy for 1 months or longer, 3 months or longer, 6 months or longer, 9 months or longer, 12 months or longer, and 15 months or longer was higher in the pembrolizumab plus SOC group (Table 12).
In the neoadjuvant phase, patients in the pembrolizumab plus SOC arm received treatment for a median duration of 0.7 months (2.0 cycles), with 360 of 361 participants completing all doses of pembrolizumab within 1 month of initiation. The adjuvant phase showed the expected difference between groups — the pembrolizumab plus SOC group had a median treatment duration of 9.7 months (with a median of 15.0 cycles of pembrolizumab administered) compared to 1.5 months in the SOC group. Cisplatin exposure was similar in both arms, with a median duration of 1.4 months (3.0 cycles). Radiation therapy delivery was similar between groups, with median doses of 60 Gy in the pembrolizumab plus SOC group (n = 274) and 66 Gy in the SOC group (n = 275).26
Table 12: Summary of Patient Exposure From the KEYNOTE-689 Study (APaT Population)
Treatment exposure | Pembrolizumab plus SOC n = 361 | SOC n = 315 |
|---|---|---|
Study duration on therapya (months) | ||
Mean (SD) | 8.0 (5.1) | 2.7 (1.2) |
Median (range) | 9.1 (0.03 to 22.3) | 2.9 (0.03 to 7.2) |
Duration of exposure, n (%), person-years | ||
> 0 month | 361 (100.0), 240.5 | 315 (100.0), 70.9 |
≥ 1 month | 326 (90.3), 238.7 | 274 (87.0), 70.7 |
≥ 3 months | 272 (75.3), 231.9 | 125 (39.7), 37.1 |
≥ 6 months | 216 (59.8), 211.4 | 2 (0.6), 1.2 |
≥ 9 months | 182 (50.4), 191.2 | 0 (0.0), 0.0 |
≥ 12 months | 142 (39.3), 155.0 | 0 (0.0), 0.0 |
≥ 15 months | 5 (1.4), 7.1 | 0 (0.0), 0.0 |
≥ 18 months | 1 (0.3), 1.9 | 0 (0.0), 0.0 |
APaT = all participants as treated; SD = standard deviation; SOC = standard of care.
Note: Data presented in this table were based on analyses at the clinical cut-off date of July 25, 2024.
aTherapy includes study drugs, in-study surgery, and on-study radiotherapy.
Source: Clinical Study Report for the KEYNOTE-689 study.26 Details included in the table are from the sponsor’s summary of clinical evidence.29
Concomitant Medications and Co-Interventions
Most patients received 1 or more concomitant medications (98.6% in the pembrolizumab plus SOC group versus 98.4% in the SOC group) during the study period of the KEYNOTE-689 study. Classes of concomitant medications used by more than 20% of patients in either treatment group and the most commonly used drugs are shown in Table 13. There were higher proportions of patients in the pembrolizumab plus SOC group compared with the SOC group who were on levothyroxine sodium (32.4% versus 15.9%), ibuprofen (25.5% versus 19.4%), and fentanyl (23.5% versus 16.8%), among the others. On the contrary, there were lower proportion of patients in the pembrolizumab plus SOC group compared with the SOC group who were on dexamethasone (27.4% versus 32.7%), metamizole sodium (21.6% versus 27.9%), and tramadol hydrochloride (20.2% versus 25.1%), among the others. There is no notable between-group difference in the rest of the concomitant medications.
Table 13: Concomitant Medications in the KEYNOTE-689 Study (APaT Population)
Concomitant medicationsa, n (%) | Pembrolizumab plus SOC n = 361 | SOC n = 315 |
|---|---|---|
Patients with ≥ 1 concomitant medications | 356 (98.6) | 310 (98.4) |
Alimentary tract and metabolism | ||
Antidiarrheals, intestinal anti-inflammatory, or anti-infective drugs | 206 (57.1) | 154 (48.9) |
Antiemetics and antinauseants | 185 (51.2) | 181 (57.5) |
Bile and liver therapy | 95 (26.3) | 71 (22.5) |
Drugs for acid-related disorders | 223 (61.8) | 215 (68.3) |
Drugs for constipation | 207 (57.3) | 178 (56.5) |
Drugs for functional gastrointestinal disorders | 136 (37.7) | 129 (41.0) |
Mineral supplements | 186 (51.5) | 172 (54.6) |
Other alimentary tract and metabolism products | 110 (30.5) | 72 (22.9) |
Stomatological preparations | 296 (82.0) | 269 (85.4) |
Vitamins | 106 (29.4) | 86 (27.3) |
Anti-infectives for systemic use | ||
Antibacterials for systemic use | 260 (72.0) | 221 (70.2) |
Antimycotics for systemic use | 127 (35.2) | 105 (33.3) |
Blood and blood-forming organs | ||
Antithrombotic drugs | 182 (50.4) | 160 (50.8) |
Blood substitutes and perfusion solutions | 232 (64.3) | 204 (64.8) |
Cardiovascular system | ||
Drugs acting on the renin-angiotensin system | 97 (26.9) | 96 (30.5) |
Antihypertensives | 63 (17.5) | 67 (21.3) |
Beta-blocking drugs | 71 (19.7) | 65 (20.6) |
Calcium channel blockers | 74 (20.5) | 63 (20.0) |
Cardiac therapy | 193 (53.5) | 157 (49.8) |
Diuretics | 83 (23.0) | 79 (25.1) |
Lipid-modifying drugs | 82 (22.7) | 79 (25.1) |
Vasoprotectives | 219 (60.7) | 188 (59.7) |
Dermatologicals | ||
Antiacne preparations | 162 (44.9) | 121 (38.4) |
Antibiotics and chemotherapeutics for dermatological use | 161 (44.6) | 122 (38.7) |
Antifungals for dermatological use | 127 (35.2) | 108 (34.3) |
Antipruritics, including antihistamines, anesthetics, and so forth | 146 (40.4) | 101 (32.1) |
Antiseptics and disinfectants | 78 (21.6) | 60 (19.0) |
Corticosteroids, dermatological preparations | 213 (59.0) | 169 (53.7) |
Emollients and protectives | 80 (22.2) | 58 (18.4) |
Medicated dressings | 147 (40.7) | 128 (40.6) |
Other dermatological preparations | 222 (61.5) | 195 (61.9) |
Preparations for treatment of wounds and ulcers | 138 (38.2) | 127 (40.3) |
Genitourinary system and sex hormones | ||
Gynecological anti-infectives and antiseptics | 190 (52.6) | 175 (55.6) |
Other gynecologicals | 169 (46.8) | 127 (40.3) |
Urologicals | 167 (46.3) | 96 (30.5) |
Musculoskeletal system | ||
Anti-inflammatory and antirheumatic products | 219 (60.7) | 184 (58.4) |
Topical products for joint and muscular pain | 236 (65.4) | 205 (65.1) |
Nervous system | ||
Analgesics | 303 (83.9) | 262 (83.2) |
Paracetamol (acetaminophen) | 193 (53.5) | 172 (54.6) |
Metamizole sodium | 78 (21.6) | 88 (27.9) |
Tramadol hydrochloride | 73 (20.2) | 79 (25.1) |
Anesthetics | 149 (41.3) | 106 (33.7) |
Fentanyl | 85 (23.5) | 53 (16.8) |
Fentanyl citrate | 22 (6.1) | 12 (3.8) |
Antiepileptics | 145 (40.2) | 126 (40.0) |
Other nervous system drugs | 106 (29.4) | 75 (23.8) |
Gabapentin | 39 (10.8) | 26 (8.3) |
Psychoanaleptics | 96 (26.6) | 59 (18.7) |
Psycholeptics | 217 (60.1) | 179 (56.8) |
Respiratory system | ||
Antihistamines for systemic use | 105 (29.1) | 66 (21.0) |
Cough and cold preparations | 170 (47.1) | 159 (50.5) |
Drugs for obstructive airway diseases | 177 (49.0) | 159 (50.5) |
Nasal preparations | 236 (65.4) | 203 (64.4) |
Throat preparations | 233 (64.5) | 184 (58.4) |
Ibuprofen | 92 (25.5) | 61 (19.4) |
Sensory organs | ||
Ophthalmological and ontological preparations | 200 (55.4) | 186 (59.0) |
Ophthalmologicals | 303 (83.9) | 274 (87.0) |
Otologicals | 255 (70.6) | 223 (70.8) |
Systemic hormonal preparations, excluding sex hormones and insulins | ||
Corticosteroids for systemic use | 200 (55.4) | 156 (49.5) |
Dexamethasone | 99 (27.4) | 103 (32.7) |
Prednisolone | 42 (11.6) | 12 (3.8) |
Prednisone | 27 (7.5) | 5 (1.6) |
Hydrocortisone | 25 (6.9) | 6 (1.9) |
Thyroid therapies | 124 (34.3) | 59 (18.7) |
Levothyroxine sodium | 117 (32.4) | 50 (15.9) |
APaT = all participants as treated; SOC = standard of care.
Note: Data presented in this table were based on analyses at the clinical cut-off date of July 25, 2024.
aOnly the concomitant medication classes received by at least 20% of the patients in either group are presented in this table.
Source: Clinical Study Report for the KEYNOTE-689 study.26 Details included in the table are from the sponsor’s summary of clinical evidence.29
Subsequent Treatment
Subsequent oncologic therapy was reported in 19.3% and 22.2% of patients in the pembrolizumab plus SOC group and SOC group, respectively (Table 14). The most common (> 10% of participants) subsequent oncologic therapy was cisplatin (11.0% in the pembrolizumab plus SOC group versus 10.8% in the SOC group). A higher proportion of patients in the SOC group (7.7%) received fluorouracil compared with that in the pembrolizumab plus SOC group (3.9%). There is no notable between-group difference in the rest of the subsequent oncology therapies listed in Table 14.
Efficacy
Results of the key efficacy outcomes (OS and EFS) are presented in Table 15. EFS in patients with a CPS of 1 or more and additional efficacy outcomes (mPR, pCR, DMFS, LRC, and EORTC QLQ-H&N35 symptom scores for pain, swallowing, and speech) that were also highlighted by the clinical experts as important are presented in the Appendix. As of the data cut-off date of July 25, 2024, the median durations of follow-up were 30.0 months (range, 0.8 months to 64.9 months) in the pembrolizumab plus SOC group, 23.4 months (range, 0.5 months to 66.5 months) in the SOC group, and 27.1 months (range, 0.5 months to 66.5 months) in the overall ITT population.
Table 14: Summary of Subsequent Oncology Therapies From the KEYNOTE-689 Study (Overall ITT Population)
Oncologic therapiesa, n (%) | Pembrolizumab plus SOC n = 363 | SOC n = 351 |
|---|---|---|
Participants in population with ≥ 1 subsequent therapies | 70 (19.3) | 78 (22.2) |
Antineoplastic and immunomodulating agents | ||
Antineoplastic drugs | 70 (19.3) | 78 (22.2) |
Carboplatin | 20 (5.5) | 19 (5.4) |
Cetuximab | 20 (5.5) | 19 (5.4) |
Cisplatin | 40 (11.0) | 38 (10.8) |
Fluorouracil | 14 (3.9) | 27 (7.7) |
Paclitaxel | 20 (5.5) | 19 (5.4) |
Pembrolizumab | 8 (2.2) | 32 (9.1) |
ITT = intention to treat; SOC = standard of care.
Note: Data presented in this table were based on analyses at the clinical cut-off date of July 25, 2024.
aOnly the subsequent therapies received by at least 5% of the patients in either group are presented in this table.
Source: Clinical Study Report for the KEYNOTE-689 study.26 Details included in the table are from the sponsor’s summary of clinical evidence.29
Survival
Overall Survival
At the data cut-off date of the IA1 and in the overall ITT population (N = 714), the median OS was not reached (95% CI, 61.9 months to not reached; OS events observed in 31.1% of patients) in the pembrolizumab plus SOC group, and 61.8 months (95% CI, 50.1 months to not reached; OS events observed in 37.3% of patients) in the SOC group. The HR was 0.76 (95% CI, 0.59 to 0.98). The KM estimates of the probability of OS in the pembrolizumab plus SOC and SOC alone groups were 86.7% (95% CI, 82.7% to 89.8%) versus 77.9% (95% CI, 73.2% to 81.9%) at 12 months, and 63.6% (95% CI, 57.4% to 69.1%) versus 58.0% (95% CI, 51.6% to 63.9%) at 48 months, respectively (Figure 2).
According to the prespecified multiplicity strategy, formal OS testing was not conducted in the populations with a CPS of 1 or more and overall ITT populations at IA1, as the statistical significance boundary was not met in the population with PD-L1 with a CPS of 10 or more.49,59
The OS results were consistent for point estimates of HR across most of the subgroups with point estimates that favoured the pembrolizumab plus SOC group and a 95% CI that often crossed the null (refer to Figure 5 in the Appendix). For patients with the hypopharynx as the primary tumour site, the HR was 2.23 with a wide 95% CI (0.69 to 7.20) due to the small sample size of the hypopharynx category and the small number of events in the SOC group; whereas, the results for the primary tumour site of the oropharynx or oral cavity (HR = 0.70; 95% CI, 0.52 to 0.95) and larynx (HR = 0.71; 95% CI, 0.42 to 1.21) had point estimates favouring pembrolizumab. In female patients, the point estimate approached null (HR = 0.94; 95% CI, 0.51 to 1.73) compared with the male patients (HR = 0.72; 95% CI, 0.55 to 0.95). In patients with the tumour stage of IVA, the 95% CI crossed the null (HR = 0.84; 95% CI, 0.63 to 1.11) compared with patients with the tumour stage of III (HR = 0.52; 95% CI, 0.30 to 0.92).
Table 15: OS and EFS Results From the KEYNOTE-689 Study (Overall ITT Population)
Efficacy outcome | Pembrolizumab plus SOC n = 363 | SOC n = 351 |
|---|---|---|
Follow-up duration (months), median (range) | 30.0 (0.8 to 64.9) | 23.4 (0.5 to 66.5) |
OS | ||
Number of events, n (%) | 113 (31.1) | 131 (37.3) |
OS (months), median (95% CI)a | Not reached (61.9 to not reached) | 61.8 (50.1 to not reached) |
HR (95% CI)b | 0.76 (0.59 to 0.98) | Reference |
1-sided P valuec | 0.01529d | |
OS probability (%) at 6 months (95% CI) | 94.2 (91.2 to 96.2) | 88.6 (84.8 to 91.5) |
Difference in OS probability (%) (95% CI) | ███ ████ ██ ████ | |
OS probability (%) at 12 months (95% CI) | 86.7 (82.7 to 89.8) | 77.9 (73.2 to 81.9) |
Difference in OS probability (%) (95% CI) | ███ ████ ██ █████ | |
OS probability (%) at 24 months (95% CI) | 75.9 (70.9 to 80.1) | 67.9 (62.5 to, 72.7) |
Difference in OS probability (%) (95% CI) | ███ ████ ██ █████ | |
OS probability (%) at 36 months (95% CI) | 68.4 (62.9 to 73.3) | 61.1 (55.1 to 66.5) |
Difference in OS probability (%) (95% CI) | ███ █████ ██ █████ | |
OS probability (%) at 48 months (95% CI) | 63.6 (57.4 to 69.1) | 58.0 (51.6 to 63.9) |
Difference in OS probability (%) (95% CI) | ███ █████ ██ █████ | |
OS probability (%) at 60 months (95% CI) | 59.8 (52.4 to 66.4) | 52.9 (45.2 to 60.0) |
Difference in OS probability (%) (95% CI) | ███ █████ ██ █████ | |
EFS by BICR | ||
Number of events, n (%) | 136 (37.5) | 159 (45.3) |
Death | 67 (18.5) | 64 (18.2) |
Distant progressive disease | 26 (7.2) | 51 (14.5) |
Local and distant progressive disease | 4 (1.1) | 7 (2.0) |
Local progressive disease or recurrence | 39 (10.7) | 37 (10.5) |
Number censored (%) | 227 (62.5) | 192 (54.7) |
Last assessment showing no progression | 224 (61.7) | 181 (51.6) |
Randomizatione | 3 (0.8) | 11 (3.1) |
EFS (months), median (95% CI)a | 51.8 (37.5 to not reached) | 30.4 (21.8 to 50.1) |
HR (95% CI)b | 0.73 (0.58 to 0.92) | Reference |
One-sided P valuec | 0.00411 | |
EFS probability (%) at 6 months (95% CI) | 86.3 (82.1 to 89.5) | 80.3 (75.5 to 84.2) |
Difference in EFS probability (%) (95% CI) | ███ ████ ██ █████ | |
EFS probability (%) at 12 months (95% CI) | 75.1 (70.0 to 79.4) | 62.5 (56.9 to 67.5) |
Difference in EFS probability (%) (95% CI) | ████ ████ ██ █████ | |
EFS probability (%) at 24 months (95% CI) | 65.0 (59.4 to 70.1) | 54.6 (48.7 to 60.1) |
Difference in EFS probability (%) (95% CI) | ████ ████ ██ █████ | |
EFS probability (%) at 36 months (95% CI) | 57.6 (51.5 to 63.3) | 46.4 (40.0 to 52.5) |
Difference in EFS probability (%) (95% CI) | ████ ████ ██ █████ | |
EFS probability (%) at 48 months (95% CI) | 52.0 (45.1 to 58.4) | 44.2 (37.5 to 50.8) |
Difference in EFS probability (%) (95% CI) | ███ █████ ██ █████ | |
BICR = blinded independent central review; CI = confidence interval; CPS = combined positive score; EFS = event-free survival; HR = hazard ratio; IA1 = first interim analysis; ITT = intention to treat; OS = overall survival; SOC = standard of care; sSAP = supplementary statistical analysis plan; vs. = versus.
Note: Data presented in this table were based on analyses at the clinical cut-off date of July 25, 2024.
aFrom product-limit (Kaplan-Meier) method for censored data.
bBased on Cox regression model with Efron’s method of tie handling with treatment as a covariate stratified by primary tumour site (oropharynx or oral cavity vs. larynx vs. hypopharynx) and tumour stage (III vs. IVA) with small strata collapsed as prespecified in the sSAP.
cBased on log-rank test stratified by primary tumour site (oropharynx or oral cavity vs. larynx vs. hypopharynx) and tumour stage (III vs. IVA) with small strata collapsed as prespecified in the sSAP. Statistical testing for this end point was adjusted for multiple comparisons using the graphical method of Maurer and Bretz.50
dThe test for OS is not considered statistically significant. According to the multiple testing strategy, formal OS testing was not conducted at IA1, as the statistical significance boundary was not met in the PD-L1 CPS ≥ 10 population (earlier in the hierarchy).
eThe first assessment for patients was screening or randomization because at randomization patients were confirmed to not have progressive disease or recurrence. Patients who did not have any postrandomization radiographic disease assessments or on-study surgery were censored at their last assessment showing no progression which occurs at randomization. For specificity, the label “randomization” is used within the EFS analyses tables to distinguish from individuals with postrandomization assessments.
Source: Clinical Study Report for the KEYNOTE-689 study.26 Details included in the table are from the sponsor’s summary of clinical evidence.29
Figure 2: Kaplan-Meier Plot of Overall Survival in the KEYNOTE-689 Study (Overall ITT Population)
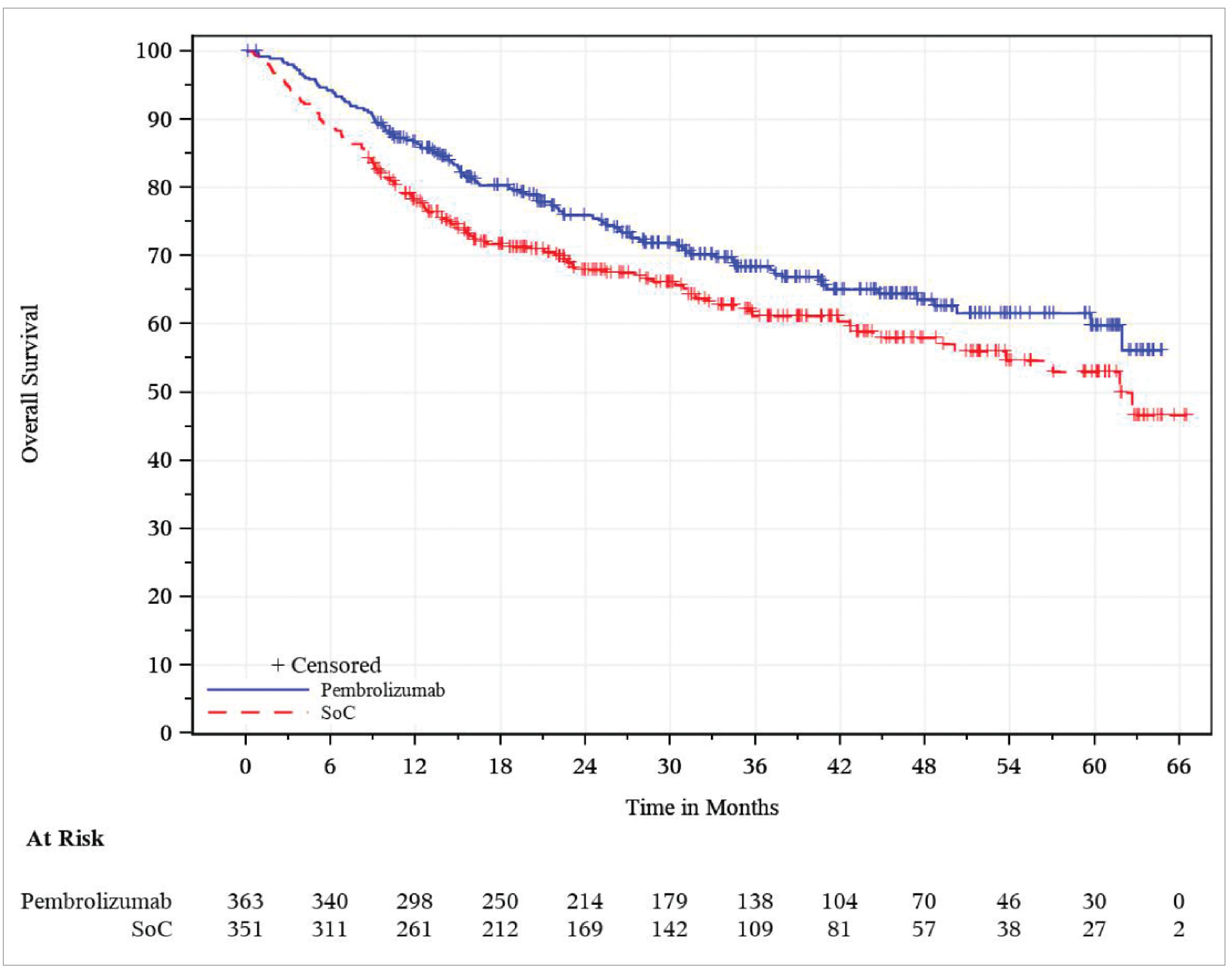
ITT = intention to treat; SoC = standard of care.
Note: This figure was based on analyses at the clinical cut-off date of July 25, 2024.
Source: Clinical Study Report for the KEYNOTE-689 study.26
EFS by BICR
At the data cut-off date of the IA1, the median EFS was 51.8 months (95% CI, 37.5 months to not reached; EFS events observed in 37.5% of patients) in the pembrolizumab plus SOC group, and 30.4 months (95% CI, 21.8 months to 50.1 months; EFS events observed in 45.3% of patients) in the SOC group. The HR was 0.73 (95% CI, 0.58 to 0.92; P = 0.00411). The KM estimates of the probability of EFS in the pembrolizumab plus SOC and SOC alone groups were 75.1% (95% CI, 70.0% to 79.4%) versus 62.5% (95% CI, 56.9% to 67.5%) at 12 months, and 52.0% (95% CI, 45.1% to 58.4%) versus 44.2% (95% CI, 37.5% to 50.8%) at 48 months, respectively.
The KM curve for the pembrolizumab plus SOC group separated from the SOC curve at approximately 3 months and maintained this separation throughout the evaluation period (Figure 3).
EFS as assessed by BICR in the sensitivity analysis based on different censoring rules outlined in the study protocol49 (HR = 0.70; 95% CI, 0.54 to 0.91; P = 0.00349) was consistent with EFS analysis results as assessed by BICR in the primary analysis. The EFS results based on investigator assessment with the primary censoring rule (HR = 0.72; 95% CI, 0.58 to 0.91; P = 0.00265) and in the sensitivity analysis (HR = 0.73; 95% CI, 0.57 to 0.93; P = 0.00577) were consistent with EFS analysis results as assessed by BICR in the primary analysis.26
The EFS results were consistent for point estimates of the HR across most of the subgroups in favour of the pembrolizumab plus SOC group; in many cases the 95% CIs cross the null (refer to Figure 6 in the Appendix). For patients with a primary tumour site of the hypopharynx, the HR was 2.55 with a wide 95% CI (0.91 to 7.17, due to the small sample size of this category and the small number of events in the SOC group), whereas the results for the primary tumour site of the oropharynx or oral cavity (HR = 0.66; 95% CI, 0.51 to 0.87) and larynx (HR = 0.68; 95% CI, 0.41 to 1.14) had point estimates favouring pembrolizumab. For female patients, the point estimate was null with a wide 95% CI (0.57 to 1.76). For male patients (HR = 0.67; 95% CI, 0.52 to 0.87) pembrolizumab was favoured. In patients with the tumour stage of IVA, the 95% CI crossed the null (HR = 0.85; 95% CI, 0.66 to 1.10) compared with patients with the tumour stage of III (HR = 0.42; 95% CI, 0.25 to 0.71).
Figure 3: Kaplan-Meier Plot of EFS Based on BICR (Primary Censoring Rule) in the KEYNOTE-689 Study (Overall ITT Population)
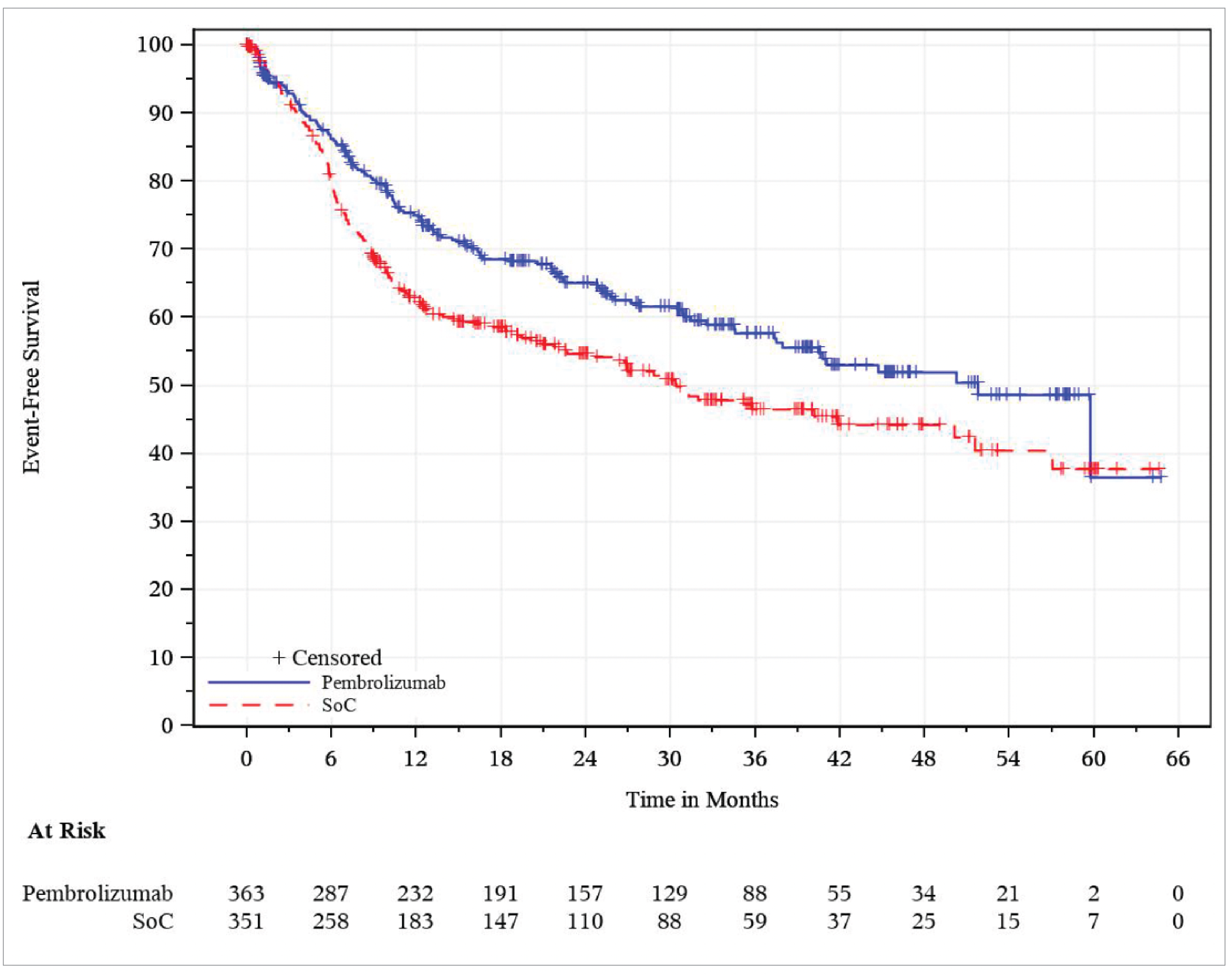
BICR = blinded independent central review; EFS = event-free survival; ITT = intention to treat; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1; SoC = standard of care.
Note: This figure was based on analyses at the clinical cut-off date of July 25, 2024.
Source: Clinical Study Report for the KEYNOTE-689 study.26
Health-Related Quality of Life
Change From Baseline in EORTC QLQ-C30 GHS/QoL Score
The between-group differences in LS mean change from baseline to postadjuvant CRT or RT at week 25 and week 51 in the EORTC QLQ-C30 GHS/QoL score were −1.29 (95% CI, −4.78 to 2.20; P = 0.4671) and 0.39 (95% CI, −3.35 to 4.13; P = 0.8379), respectively (Table 16; also refer to Figure 7 in the Appendix).
No subgroup or sensitivity analyses were included in the sponsor’s reimbursement review submission for this review.
Table 16: EORTC QLQ-C30 Global Health Status/QoL Results From the KEYNOTE-689 Study (PRO FAS Population)
EORTC QLQ-C30 global health status/QoL | Pembrolizumab plus SOC n = 356 | SOC n = 306 |
|---|---|---|
Number of patients contributing to the analysis at baselinea | 340 | 280 |
Baseline score, mean (SD) | 63.73 (20.31) | 62.71 (21.50) |
Postadjuvant CRT or RT at week 25 | ||
Number of patients at postadjuvant CRT or RT at week 25a | 178 | 172 |
Score at postadjuvant CRT or RT at week 25, mean (SD) | 69.34 (18.59) | 70.40 (18.63) |
Change from baseline to postadjuvant CRT or RT at week 25, LS mean (95% CI)b | 3.69 (1.03 to 6.34) | 4.98 (2.26 to 7.70) |
Treatment group difference vs. control at week 25, LS mean (95% CI)b | −1.29 (−4.78 to 2.20) | |
P valueb | 0.4671 | |
Postadjuvant CRT or RT at week 51 | ||
Number of patients at week 51a | 146 | 134 |
Score at postadjuvant CRT or RT at week 51, mean (SD) | 71.00 (18.08) | 70.52 (19.05) |
Change from baseline to postadjuvant CRT or RT at week 51, LS mean (95% CI)b | 4.44 (1.64 to 7.23) | 4.05 (1.13 to 6.97) |
Treatment group difference vs. control at week 51, LS mean (95% CI)b | 0.39 (−3.35 to 4.13) | |
P valueb | 0.8379 | |
CI = confidence interval; cLDA = constrained longitudinal data analysis; CRT = chemoradiotherapy; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; FAS = full analysis set; LS = least squares; PRO = patient-reported outcome; QoL = quality of life; RT = radiotherapy; SD = standard deviation; SOC = standard of care; sSAP = supplementary statistical analysis plan; TPS = tumour proportion score; vs. = versus.
Note: Data presented in this table were based on analyses at the clinical cut-off date of July 25, 2024.
aNumber of patients in each treatment group with nonmissing assessments at the specific time point.
bBased on a cLDA model with the PRO scores as the response variable with covariates for treatment by study visit interaction, and stratification factors by primary tumour site (oropharynx or oral cavity vs. larynx vs. hypopharynx), tumour stage (III vs. IVA), and PD-L1 status (TPS ≥ 50% vs. TPS < 50%) as prespecified in the sSAP.
Source: Clinical Study Report for the KEYNOTE-689 study.26 Details included in the table are from the sponsor’s summary of clinical evidence.29
Harms
The harms observed among the APaT population in the KEYNOTE-689 study (data cut-off date of July 25, 2024) are summarized in Table 17. As per the protocol design, the pembrolizumab plus SOC group had a longer treatment duration, with a median of 9.1 months for pembrolizumab plus SOC compared to 2.9 months for SOC alone.26
Adverse Events
In the KEYNOTE-689 study, there were similar proportions of patients with any AEs between the pembrolizumab plus SOC (96.4%) and the SOC (96.8%) groups. The most frequently reported AEs were stomatitis (41.6% in the pembrolizumab plus SOC group versus 54.6% in the SOC group), radiation skin injury (39.9% versus 47.9%), weight decrease (35.7% versus 27.3%), anemia (34.1% versus 33.3%), and dysphagia (29.1% versus 31.7%).
Grade 3 to Grade 5 AEs
Grade 3 to grade 5 AEs were reported in 76.2% and 74.0% of patients in the pembrolizumab plus SOC and SOC groups, respectively. The most frequently reported grade 3 to grade 5 AEs were weight decrease (13.9% in the pembrolizumab plus SOC group versus 9.5% in the SOC group), stomatitis (12.5% versus 13.3%), dysphagia (12.2% versus 10.8%), and anemia (10.5% versus 11.1%).
Serious AEs
SAEs were reported by 49.6% of patients in the pembrolizumab plus SOC group and 36.8% of patients in the SOC group. SAEs reported in at least 2% of patients in either group were pneumonia (3.6% in the pembrolizumab plus SOC group versus 5.7% in the SOC group), postoperative wound infection (1.4% versus 2.9%), as well as pneumonia aspiration (2.2% versus 0.6%), and wound dehiscence (2.2% versus 0.6%; Table 17).
Withdrawals Due to AEs
The proportion of patients with AEs leading to any study treatment discontinuation was 24.4% in the pembrolizumab plus SOC group and 14.3% in the SOC group, respectively. The most frequently (in at least 1% of patients) reported AEs resulting in discontinuation of any study treatments (i.e., either pembrolizumab, RT, cisplatin, or any combination of these) were neutrophil count decrease (1.9% in the pembrolizumab plus SOC group versus 3.2% in the SOC group), pneumonitis (1.9% versus 0%), colitis (1.7% versus 0%), acute kidney injury (1.1% versus 2.2%), immune-mediated hepatitis (1.1% versus 0%), stomatitis (1.1% versus 1.3%), and white blood cell count decrease (1.1% versus 0.3%).26
Mortality
There were similar proportions of patients with AEs resulting in death in the pembrolizumab plus SOC (6.9%) and the SOC (7.6%) groups. The most common AEs resulting in death were “death” (3 patients [0.8%] in the pembrolizumab plus SOC group versus 2 patients [0.6%] in the SOC group), and pneumonia and septic shock (2 patients [0.6%] in the pembrolizumab plus SOC group versus 3 patients [1.0%] in the SOC group). The 3 patients in the pembrolizumab plus SOC group for whom the term “death” was reported included a patient with cardiovascular failure as a result of electrolyte disorder, a patient with excessive alcohol consumption, and a patient with no cause cited for the death.26
Notable Harms
The proportion of patients with an AEOSI, considered to be medically equivalent to the immune-mediated events and infusion-related reactions, was higher in the pembrolizumab plus SOC group (43.8%) compared with the SOC group (10.8%). The most frequently reported AEOSIs (in at least 2% of patients) were hypothyroidism (24.7% in the pembrolizumab plus SOC group versus 5.4% in the SOC group), hyperthyroidism (8.9% versus 3.2%), pneumonitis (5.5% versus 0%), colitis (2.5% versus 0.3%), and hepatitis (2.2% versus 0.3%). The majority of AEOSIs were managed by standard clinical practice, including supportive care, treatment interruption, and administration of corticosteroids and/or hormone replacement therapy.26
Table 17: Summary of Harms Results From the KEYNOTE-689 Study (APaT Population)
AEs | Pembrolizumab plus SOC n = 361 | SOC n = 315 |
|---|---|---|
AEs reported in ≥ 20% of patients in any treatment group, n (%) | ||
Patients with ≥ 1 AE | 348 (96.4) | 305 (96.8) |
Stomatitis | 150 (41.6) | 172 (54.6) |
Radiation skin injury | 144 (39.9) | 151 (47.9) |
Weight decreased | 129 (35.7) | 86 (27.3) |
Anemia | 123 (34.1) | 105 (33.3) |
Dysphagia | 105 (29.1) | 100 (31.7) |
Constipation | 97 (26.9) | 70 (22.2) |
Hypothyroidism | 89 (24.7) | 17 (5.4) |
Nausea | 88 (24.4) | 87 (27.6) |
Fatigue | 85 (23.5) | 52 (16.5) |
Dry mouth | 81 (22.4) | 81 (25.7) |
Diarrhea | 78 (21.6) | 33 (10.5) |
Grade 3 to grade 5 AEs reported in ≥ 5% of patients in any treatment group | ||
Patients with ≥ 1 grade 3 to grade 5a AE, n (%) | 275 (76.2) | 233 (74.0) |
Risk difference vs. SOC alone, % (95% CI) | ███ █████ ██ ████ | Reference |
Weight decreased, n (%) | 50 (13.9) | 30 (9.5) |
Stomatitis, n (%) | 45 (12.5) | 42 (13.3) |
Dysphagia, n (%) | 44 (12.2) | 34 (10.8) |
Anemia, n (%) | 38 (10.5) | 35 (11.1) |
Lymphocyte count decreased, n (%) | 33 (9.1) | 36 (11.4) |
Hyponatremia, n (%) | 28 (7.8) | 16 (5.1) |
Neutrophil count decreased, n (%) | 20 (5.5) | 37 (11.7) |
Hypokalemia, n (%) | 19 (5.3) | 17 (5.4) |
White blood cell count decreased, n (%) | 17 (4.7) | 29 (9.2) |
Radiation skin injury, n (%) | 15 (4.2) | 18 (5.7) |
Pneumonia, n (%) | 14 (3.9) | 22 (7.0) |
SAEs reported in ≥ 2% of patients in any treatment group, n (%) | ||
Patients with ≥ 1 SAE | 179 (49.6) | 116 (36.8) |
Pneumonia | 13 (3.6) | 18 (5.7) |
Pneumonia aspiration | 8 (2.2) | 2 (0.6) |
Postoperative wound infection | 5 (1.4) | 9 (2.9) |
Wound dehiscence | 8 (2.2) | 2 (0.6) |
Patients who discontinued treatment due to AEs, number of events in the total number of patients contributing to the analysis (%) | ||
Patients who stopped any drug | 88 of 361 (24.4) | 45 of 315 (14.3) |
Patients who stopped pembrolizumab | 66 of 361 (18.3) | Not applicable |
Patients who stopped cisplatinb | 30 of 107 (28.0)b | 41 of 139 (29.5)b |
Patients who stopped radiation therapyc | 8 of 275 (2.9) | 4 of 275 (1.5) |
Deaths, n (%) | ||
Patients who died | 25 (6.9) | 24 (7.6) |
AEOSIs, n (%) | ||
Patients with ≥ 1 AEOSI | 158 (43.8) | 34 (10.8) |
Adrenal Insufficiency | 7 (1.9) | 0 |
Colitis | 9 (2.5) | 1 (0.3) |
Gastritis | 5 (1.4) | 1 (0.3) |
Hepatitis | 8 (2.2) | 1 (0.3) |
Immune-mediated hepatitis | 5 (1.4) | 0 |
Hyperthyroidism | 32 (8.9) | 10 (3.2) |
Hypoparathyroidism | 1 (0.3) | 1 (0.3) |
Hypophysitis | 2 (0.6) | 1 (0.3) |
Hypothyroidism | 89 (24.7) | 17 (5.4) |
Infusion reactions | 4 (1.1) | 2 (0.6) |
Myocarditis | 1 (0.3) | 0 |
Myositis | 1 (0.3) | 0 |
Nephritis | 3 (0.8) | 0 |
Immune-mediated nephritis | 1 (0.3) | 0 |
Pancreatitis | 5 (1.4) | 0 |
Pneumonitis | 20 (5.5) | 0 |
Severe skin reactions | 10 (2.8) | 2 (0.6) |
Thyroiditis | 2 (0.6) | 1 (0.3) |
Uveitis | 1 (0.3) | 0 |
AE = adverse event; AEOSI = adverse events of special interest; APaT = all participants as treated; NCI CTCAE = National Cancer Institute Common Terminology Criteria for Adverse Events; SAE = serious adverse event; SOC = standard of care.; vs. = versus.
Note: Data presented in this table were based on analyses at the clinical cut-off date of July 25, 2024.
aAEs were graded in severity according to the guideline outlined in the NCI CTCAE Version 4.03.
bReported among the 107 and 139 patients who received cisplatin in the pembrolizumab plus SOC and the SOC treatment groups, respectively.
cReported among the 275 patients who received adjuvant treatment in each treatment group.
Sources: Clinical Study Report for the KEYNOTE-689 study26 and sponsor’s submission.28 Details included in the table are from the sponsor’s summary of clinical evidence.29
Critical Appraisal
Internal Validity
The KEYNOTE-689 study is a randomized, multicentre, parallel-group, active-controlled, open-label phase III trial. The pembrolizumab regimen included both neoadjuvant and adjuvant treatment added to SOC, in addition to pembrolizumab monotherapy as maintenance. Due to the study design, it is not possible to isolate the contribution of neoadjuvant and adjuvant components of the treatment to the overall efficacy. Treatment allocation (randomization) was performed using an appropriate methodology with adequate allocation concealment (i.e., central interactive voice-response system or integrated web-response system). Randomization stratification was prespecified and was based on relevant prognostic factors of patients including primary tumour site, disease stage, and PD-L1 status defined by TPS. The patient demographic and disease characteristics generally appeared balanced between the treatment groups in the overall ITT population. However, the review team noted that smoking status was slightly unbalanced between the 2 groups (17.6% were never smokers in the pembrolizumab plus SOC group versus 23.1% in the SOC group). The extent to which this imbalance could bias the results is unknown; however, the clinical experts consulted for this review did not express significant concerns, considering the imbalance unlikely to meaningfully affect the efficacy and safety outcomes.
Compared to the SOC group, a greater proportion of patients in the pembrolizumab plus SOC group discontinued the study treatment (54% in the pembrolizumab plus SOC group versus 17% in the SOC alone group), mainly due to AEs and progressive disease. Of note, per study protocol, the overall maximum duration of pembrolizumab treatment was planned to be 1 year (i.e., a total of 17 cycles of pembrolizumab) in the pembrolizumab plus SOC group, while the overall maximum duration of SOC treatment was planned to be 6 weeks to 7 weeks (i.e., the duration of treatment in the SOC alone group). As a result, the duration of therapy was longer in the pembrolizumab plus SOC group compared with the SOC group (median = 9.1 months; range, 0.03 months to 22.3 months versus 2.9 months; range, 0.03 months to 7.2 months). The review team and the clinical experts considered that it is reasonable that the longer duration of exposure in the pembrolizumab plus SOC group could play a role in the difference in discontinuation rates and the most common reasons for treatment discontinuation between the treatment groups. Additionally, there was a higher rate of patients who were randomized but not treated in the SOC group compared to the pembrolizumab plus SOC group (3 patients [0.8%] in the pembrolizumab plus SOC group versus 35 patients [10.0% of the overall ITT population] in the SOC group). █████ ████ ██ ███ ███████ ███ ████████ █████ ██████████ ███ ███ ███████ ██ ███ ███ █████ ███ ██ ██████████ ██████████ ██████ ████████████ ████ ███ ██████████ ███████ ███ █ ██████ ███ █████████ ██████ ██ ███████ ██████████ ██████████████ ████ ████ █ ████████ ██████ ███ ████ ██ ████████ ████████ ████ ███████████ ███ ██ ██████████ ██ ███ ██████████ █████ ███████ ██ ████ ████████ █████████ ██ ███ ███ █████ ███ ████ ████ █████████ ██ █████ ██████ ██ █████████████ ████ ███ ██████ ██ ███ █████████████ ████████ █████ ███████. This introduces some concern for risk of bias due to deviations from the intended intervention in the SOC group that arose because of the trial context. It is challenging to determine the direction of the bias.
In the APaT population of the KEYNOTE-689 study (n = 676), nearly all the study patients (98.5%) received at least 1 concomitant medication. The proportion of patients with the use of most medications was similar between treatment groups. However, the review team noted some imbalances between the treatment groups, with a higher proportion of certain concomitant medications observed in the pembrolizumab plus SOC group compared to the SOC alone group. For instance, the use of levothyroxine sodium (32% versus 16%), ibuprofen (25.5% versus 19%), and fentanyl (23.5% versus 17%) was more common in the pembrolizumab plus SOC group versus the SOC alone group. The clinical experts noted that the higher use of thyroid treatments in the pembrolizumab group is reasonable, given the immune-related AEs associated with pembrolizumab. Conversely, a lower proportion of patients in the pembrolizumab group versus the SOC alone group received dexamethasone (27% versus 33%), metamizole sodium (22% versus 28%), and tramadol hydrochloride (20% versus 25%). In the overall ITT population (N = 714), the proportion of patients with subsequent oncologic therapy during the trial was lower in the pembrolizumab plus SOC group (19%) than in the SOC group (22%), mainly driven by fluorouracil which was received by 3.9% in the pembrolizumab plus SOC group versus 7.7% in the SOC group. The review team and the clinical experts did not regard these between-group imbalances in the concomitant and subsequent medications would impact the validity of study results, as their use was allowed per study protocol, and aligns with the real-world practice that patients would necessarily also receive these treatments.
The KEYNOTE-689 study is an open-label trial; therefore, the sponsor, investigators, and patients were aware of the study treatment administered. Imaging and pathology data for the planned study analyses were centrally reviewed by independent radiologists and pathologists, respectively, without knowledge of patient treatment assignment. Additionally, the patient-level PD-L1 biomarker results were masked in the database to the investigator. Being an objective outcome, the open-label study would not have affected the evaluation of treatment effect on the OS. EFS and the other efficacy outcomes including mPR, pCR, DMFS, and LRC were assessed by BICR or blinded independent pathologist review; thus, assessment bias associated with the open-label design for these outcomes is likely low. However, the open-label design may introduce a potential risk of bias due to more unscheduled assessments, which could alter the timing of events, though it is unclear whether this has actually occurred. Additionally, the open-label design introduces a risk of bias in the subjective assessment of PROs (i.e., EORTC QLQ-C30 GHS/QoL and EORTC QLQ-H&N35 pain, swallowing, and speech symptoms), whereas the direction and magnitude of this potential bias on HRQoL outcomes remain uncertain. On one hand, patients receiving a new drug may perceive an improvement in their HRQoL simply due to expectations. On the other hand, the longer treatment duration associated with the drug may lead some patients to perceive a decline in their QoL.
No significant protocol deviations were noted in either group in the KEYNOTE-689 study. The sensitivity analyses using alternative censoring rules (which censored patients after ≥ 2 missed disease assessments or the initiation of new anticancer therapy; Table 8) were conducted and supported the results in the primary analyses for EFS.
Overall, the statistical methods used in the KEYNOTE-689 study are appropriate. Losses to follow-up for both OS and EFS were limited. The review team noted that OS and EFS results were based on interim analyses, which may have overestimated the treatment effect (HR) estimates for both OS and EFS.62,63 The presence and extent of any overestimation that may have been introduced could not be determined. The follow-up for OS is relatively short, such that the median was not yet reached in the pembrolizumab plus SOC group. The median in the SOC group occurred at the tail of the KM curve where few patients remained at risk; therefore, there is uncertainty associated with the estimate. The effect of pembrolizumab plus SOC on OS was not formally tested due to earlier failure of the testing hierarchy. Therefore, despite the exclusion of the null, the result is not considered statistically significant. Longer follow-up for OS will be needed to provide insight on the potential longer-term benefits on OS. Meta-analyses of patients with locally advanced HNSCC in the study by Michiels and colleagues demonstrated strong correlations between treatment effects on EFS and OS with an r of 0.86 for concomitant chemotherapy, 0.79 for induction chemotherapy, and 0.93 for adjuvant chemotherapy at the trial level.22 These findings support the use of EFS as a surrogate end point for OS in RCTs involving chemotherapy, as improvements in EFS are likely to predict OS improvements.22 However, uncertainties remain regarding the applicability of these findings to the KEYNOTE-689 study, as it is unclear how closely the patient populations, treatment modalities (e.g., inclusion of pembrolizumab, specifics of SOC), and EFS definitions in the KEYNOTE-689 study align with those in the meta-analyses.64 For the HRQoL assessed with EORTC QLQ-C30 and EORTC QLQ-H&N35 (secondary outcome) at postadjuvant CRT or RT week 25 and week 51, respectively, outcome data were available for approximately half and more than half of the total PRO FAS population or those with their scores assessed at baseline. As is common in oncology trials, the analysis of HRQoL in the KEYNOTE-689 study assumed that the data were missing at random.59 While this is acceptable as a primary analysis method, patients’ outcome data may have been missing for a variety of reasons (including death or a terminal event), which might not be compatible with this assumption. Given that the validity of the assumption cannot be verified using the observed data, sensitivity analyses making different plausible assumptions about the missing data would have been required to rule out bias.65,66 Although some categories in prespecified subgroup analyses results showed different magnitude or direction of HR estimates from the primary analysis for OS and EFS, in particular the subgroups by primary tumour site (hypopharynx versus other sites) and the findings that females had wider 95% CI s and smaller effects than males, these results should not be used to draw conclusions regarding effect modification.23 The intent of these analyses was to demonstrate consistency, not to test for subgroup differences.
External Validity
Patients in the KEYNOTE-689 study were recruited from multiple countries, including Canada. Approximately 30% of the screened patients were not randomized due to not meeting the eligibility criteria. The clinical experts did not regard this as a factor that might substantially influence the generalizability of the study’s results. The clinical experts considered that the eligibility criteria of patients in the KEYNOTE-689 study are appropriate and the demographic characteristics of the patients from the diversity aspect in the study were mostly in line with the patients seen in clinical practice in Canada. The clinical experts noted that even though only patients with an ECOG PS of 0 to 1 were enrolled in the KEYNOTE-689 study per the inclusion criteria, patients with an ECOG PS of 2 might gain benefit from pembrolizumab. The clinical experts commented that pembrolizumab can be prescribed to patients up to ECOG PS of 2 in clinical practice in Canada.
In the KEYNOTE-689 study, most patients (95.5%) had PD-L1 with a CPS of 1 or more. A total of 13 patients (3.6%) in the pembrolizumab plus SOC group and 14 patients (4.0%) in the SOC group had a CPS of less than 1 (i.e., PD-L1 CPS-negative). The clinical experts consulted for this review expressed concerns about the generalizability of the KEYNOTE-689 study in CPS-negative patients, particularly as neoadjuvant monotherapy. The clinical experts pointed out that in clinical practice, clinicians are generally hesitant to use pembrolizumab monotherapy in CPS-negative patients with such an aggressive disease, due to the risk of disease progression rendering tumours unresectable. In such cases, RT might be considered upfront instead. According to the clinical experts, given the small proportion of CPS-negative patients enrolled in the KEYNOTE-689 study, and the fact that CPS-negative status is associated with a poorer prognosis, there is uncertainty regarding the benefit of pembrolizumab, particularly as neoadjuvant monotherapy, in this subgroup. There is currently a lack of evidence to either support or oppose its use in CPS-negative patients. Therefore, the clinical experts advised caution when considering pembrolizumab monotherapy in CPS-negative patients in the neoadjuvant setting, due to the potential risk of delaying surgery or contributing to disease progression. The clinical experts did not suggest excluding CPS-negative patients from receiving immunotherapy like pembrolizumab, but proposed a potential approach of granting preliminary approval for adding chemotherapy to immunotherapy in the neoadjuvant setting, with the condition that efficacy and safety data be collected through a phase IV trial in Canada.
The clinical experts noted that the KEYNOTE-689 study excluded certain HNSCC patient groups (e.g., those with nasopharyngeal, sinonasal, unknown primary SCC), underscoring the ongoing unmet needs and limited salvage options for these populations.
The clinical experts consulted for this review noted that the dosing and schedule of pembrolizumab in the KEYNOTE-689 study as well as that specified in the drug’s product monograph (neoadjuvant pembrolizumab as 2 doses of 200 mg every 3 weeks or 1 dose of 400 mg every 6 weeks or until disease progression that precludes definitive surgery or unacceptable toxicity, continued as adjuvant pembrolizumab in combination with RT with or without cisplatin for 3 doses of 200 mg every 3 weeks or 2 doses of 400 mg every 6 weeks, followed by 12 doses of 200 mg every 3 weeks or 6 doses of 400 mg every 6 weeks as monotherapy or until disease recurrence or unacceptable toxicity) are appropriate and align with the clinical practice in Canada.
The KEYNOTE-689 study included outcomes that were important to patients. The outcomes of OS, EFS, HRQoL, and safety were considered appropriate by the clinical experts and the clinician group. The use of EFS as a surrogate end point for OS is accepted by the FDA for both accelerated and regular approval.67 Strong correlations between treatment effects on EFS and OS were reported in locally advanced HNSCC.22,67 The clinical experts and the clinician group also noted that EFS is generally considered the most common surrogate survival end point for HNSCC treatments and is appropriate to be an end point for evidence in the current review.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered most relevant to inform expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group.24,25
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate — the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited — the true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate — the true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as “very uncertain.”
Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null. For this review, the target of the certainty of evidence assessment was based on presence or absence of an important effect based on thresholds informed by the clinical experts consulted for this review for OS and EFS. The target of the certainty of evidence assessment was based on presence or absence of an important effect based on a threshold identified in the literature for EORTC QLQ-C30 GHS/QoL. Due to the lack of a formal minimal important differences estimate, the target of certainty of evidence assessment was the presence or absence of any (nonnull) effect for grade 3 to grade 5 AEs.
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for pembrolizumab plus SOC versus SOC alone for adult patients with resectable locoregionally advanced HNSCC.
Long-Term Extension Studies
No long-term extension studies were submitted for this review as the KEYNOTE-689 study is still ongoing for OS follow-up and there are no other long-term extension studies currently ongoing or completed.
Indirect Evidence
No studies with indirect evidence were submitted for this review.
Studies Addressing Gaps in the Systematic Review Evidence
No studies addressing gaps in the pivotal and RCT evidence were submitted for this review.
Discussion
This report summarizes the evidence for neoadjuvant and adjuvant pembrolizumab in combination of SOC compared with SOC alone in adult patients with resectable locally advanced HNSCC.
Summary of Available Evidence
This review included 1 sponsor-conducted study (the KEYNOTE-689 study).
The KEYNOTE-689 study is an ongoing, multicentre, open-label, phase III RCT (N = 714) that investigated the efficacy and safety of pembrolizumab as neoadjuvant therapy, followed by postsurgical (adjuvant) treatment combining pembrolizumab and SOC RT (with or without cisplatin) followed by pembrolizumab monotherapy in treatment-naive patients with resectable locally advanced HNSCC. Eligible patients were adults with newly diagnosed, histologically confirmed locally advanced (stage III or stage IVA) HNSCC amenable to primary surgery. Eligible patients had an ECOG PS of 0 or 1, adequate organ function, available HPV (p16) results for oropharyngeal tumours, and provided a tumour sample for PD-L1 testing. Patients who had received prior therapy with an anti–PD-1, anti–PD-L1, anti–PD-L2 drug, or with a drug directed to another co-inhibitory T-cell receptor, or prior RT treatment or systemic anticancer therapy including investigational agents for the head and neck cancer under study before randomization, were excluded. Patients were randomized to receive pembrolizumab as neoadjuvant therapy (for 2 cycles), followed by postsurgical (adjuvant) treatment combining pembrolizumab (for 15 cycles) and SOC (RT with or without cisplatin) followed by pembrolizumab monotherapy (the pembrolizumab plus SOC arm; n = 363) or SOC alone (the SOC arm; n = 351). The outcomes relevant to this review included OS, EFS, EORTC QLQ-C30 GHS/QoL, and safety. Additional efficacy outcomes including mPR, pCR, LRC, DMFS, and EORTC QLQ-H&N35 symptom scores (pain, swallowing, and speech) are presented in the Appendix of this report.
The overall ITT population of the KEYNOTE-689 study had a median age of 60 years. The proportion of male patients (79%) was higher than that of female patients (21%). Most patients were white (77% to 78%), followed by Asian (13% to 14%), among the others. Most patients were current or former smokers (78%) and alcohol users (68%). In the overall ITT population, 65% of the patients had a PD-L1 CPS of 10 or greater, and 95.5% of the patients had a PD-L1 CPS of 1 or greater. More patients had an ECOG PS of 0 (57%) compared to those with an ECOG PS of 1 (43%). Most patients had a negative HPV status (96%), and stage IVA disease (74%). Most patients had their primary tumour located in the oral cavity (61%), followed by the larynx (22%), oropharynx (10%), and hypopharynx (8%).
No long-term extension studies, indirect comparative evidence, and other studies addressing gaps in the systematic review evidence were submitted for this review.
Interpretation of Results
Efficacy
OS is the standard end point for phase III oncology studies and has been recognized as the gold standard for demonstrating superiority of new antineoplastic therapies in RCTs.49,67 OS may be affected by subsequent therapies such as PD-1 inhibitors, particularly with their widespread use in recurrent or metastatic HNSCC. In the overall ITT population in the KEYNOTE-689 study, OS was not formally tested at IA1 due to earlier failure of the statistical hierarchy. Nevertheless, the KM OS curves demonstrated early separation within the first few months that persisted throughout the follow-up period. The follow-up for OS is relatively short, with OS events observed in 31.1% of patients in the pembrolizumab plus SOC group and in 37.3% of patients in the SOC group as of the data cut-off date for the IA1. The median was not yet reached in the pembrolizumab plus SOC group. The median in the SOC group occurred at the tail of the KM curve where few patients remained at risk; therefore, there is uncertainty associated with the estimate. The available data show a likely clinically important increase in OS at 12 months with pembrolizumab plus SOC compared to SOC alone that appears to extend throughout follow-up, though longer-term estimates become more uncertain due to few patients remaining at risk. Longer follow-up is needed to draw more definitive conclusions regarding the potential for long-term benefits of pembrolizumab plus SOC on OS.
As the primary end point of the KEYNOTE-689 study, EFS is a commonly used surrogate end point for OS in evaluating the efficacy of neoadjuvant and adjuvant cancer therapies. EFS is recognized by the FDA for regulatory approvals.67 Meta-analyses in locally advanced HNSCC support the validity of EFS as a surrogate for OS in chemotherapy trials,22,67 a view also shared by clinical experts and the clinician group. However, the applicability of these findings to the KEYNOTE-689 study is uncertain, given potential differences in patient population, treatment and comparison (e.g., inclusion of pembrolizumab and SOC components), and EFS definition.23 In the KEYNOTE-689 study, EFS does not incorporate a pathological component; instead, it defines a presurgical radiographic event as disease progression that precludes surgery. This approach aligns with the design of ongoing and recently completed neoadjuvant trials across various cancer types, which similarly do not include pathologic criteria in their definitions of an event.49,68-70 EFS is considered a clinically meaningful end point for patients with HNSCC, given the significant morbidity associated with recurrence and its potentially severe impact on essential functions such as eating, swallowing, speaking, and breathing.49 The clinical experts consulted for this review noted EFS as an important and appropriate efficacy outcome. In the overall ITT population, the trial’s statistical success criterion for EFS was met at IA1. The main analysis was supported by sensitivity analyses showing similar results. The available data show a likely clinically important increase in EFS at 12 months and 36 months. However, the results at longer follow-up become less certain. The data suggest that there may be little to no difference at 48 months but there was increased uncertainty because of imprecision and few patients remaining at risk at this time point. Crossing of the KM curves at 60 months postrandomization should not be overinterpreted, as the very few patients (n = 9) at risk at this time point results in estimates that are highly unreliable and likely to change with additional follow-up (Figure 3). The clinical experts noted that uncertainty remains on whether EFS gains stem from the neoadjuvant, adjuvant, or combined approach, and considerations include cost, toxicity, and patient burden.
Data from multiple studies (e.g., the KEYNOTE-040,71 KEYNOTE-048,17,48 and KEYNOTE-412 studies16) consistently show that pembrolizumab provides greater benefit (for outcomes including response rates, OS, and EFS) in patients with relapsed or metastatic HNSCC with higher PD-L1 expression, supporting a biomarker-enriched analysis. Amendments to the KEYNOTE-689 study protocol included the addition of efficacy analyses in subpopulations with a CPS of 1 or more, 10 or more, and 50 or more, in addition to the primary analyses in the overall ITT population. The OS and EFS results in these CPS-defined subpopulations were generally consistent with those observed in the overall, CPS unselected study population (EFS outcome in patients with CPS ≥ 10 is presented in Table 15; other data not shown).26
Of note, the clinical experts consulted for this review expressed concerns about the generalizability of the KEYNOTE-689 study results to CPS-negative patients, particularly in the context of neoadjuvant monotherapy. They noted that clinicians are typically hesitant to use pembrolizumab monotherapy in CPS-negative patients with aggressive disease due to the risk of progression that could render tumours unresectable, prompting consideration of RT instead. In the KEYNOTE-689 study, only a small proportion of patients were PD-L1 CPS-negative (CPS < 1) at baseline: 13 patients (3.6%) in the pembrolizumab plus SOC group and 14 patients (4.0%) in the SOC group. The CDA-AMC review team assessed that no conclusions can be made to the efficacy and safety results for this subgroup due to the small sample size. Given the small proportion (representing < 4% of the overall ITT population in the KEYNOTE-689 study) and poorer prognosis of CPS-negative patients, experts emphasized that there is insufficient evidence to support or oppose pembrolizumab monotherapy in this subgroup and advised caution in its use, particularly in the neoadjuvant setting where delays in surgery could be detrimental.
Other secondary outcomes, specifically mPR and pCR, showed results favouring pembrolizumab plus SOC (refer to Table 19 in the Appendix). The clinical experts also considered DMFS and LRC (both were exploratory end points) to be of interest. For example, LRC may help determine whether the improved EFS in the pembrolizumab plus SOC group is attributable to radiosensitization effects or the treatment of micrometastatic disease. As of the data cut-off date, the median duration of DMFS was longer in the pembrolizumab plus SOC group compared with SOC group (51.8 months in the pembrolizumab plus SOC group and 35.7 months in the SOC group; HR = 0.71; 95% CI, 0.56 to 0.90; P = 0.0025). However, the 95% CI of HR crossed the null for LRC (HR = 0.92; 95% CI, 0.61 to 1.41; P = 0.3539), though the reason for this remains unclear.
As all high-risk patients in the KEYNOTE-689 trial received adjuvant cisplatin per study protocol, the benefit of combining pembrolizumab with alternative chemotherapies remains unknown due to a lack of clinical evidence.
The patient and clinician groups also regarded HRQoL as an outcome of importance. The clinical experts indicated that both overall QoL and specific symptom domains are important. LS mean change from baseline in EORTC QLQ-H&N35 symptom domains, specifically pain, swallowing, and speech, are presented in the Appendix (Figure 8 and Figure 9). The EORTC QLQ-C30 and EORTC QLQ- H&N35 are validated tools commonly used to measure the HRQoL among patients with HNSCC. At postadjuvant weeks 25 and 51, the between-group difference in the EORTC QLQ-C30 GHS/QoL change was not regarded as clinically meaningful. The substantial proportion of missing data (47% to 58% of the total PRO FAS population) for HRQoL outcomes and open-label nature of the trial introduces considerable uncertainty to any conclusion drawn. Nevertheless, the available evidence suggest that patients who received pembrolizumab plus SOC experienced HRQoL outcomes that may be comparable to patients who received SOC alone at postadjuvant CRT or RT weeks 25 and 51.
Harms
In the overall APaT population of the KEYNOTE-689 study, there were comparable proportions of patients between the 2 treatment groups who experienced any AEs (96% of the pembrolizumab plus SOC group versus 97% of the SOC group), grade 3 to grade 5 AEs (76% of the pembrolizumab plus SOC group versus 74% of the SOC group), and AEs resulting in death (7% of the pembrolizumab plus SOC group versus 8% of the SOC group). There were higher proportions of patients in the pembrolizumab plus SOC group compared with the SOC group for SAEs (50% versus 37%, respectively), and AEs leading to any study treatment discontinuation (24% versus 14%, respectively). Of note, the pembrolizumab plus SOC group had a longer treatment duration per the protocol design, with a median of 9.1 months for pembrolizumab plus SOC compared to 2.9 months for SOC alone.
As expected, based on the drug’s mechanism of action, the proportion of patients with an AEOSI, considered to be medically equivalent to the immune-mediated events and infusion-related reactions, was higher in the pembrolizumab plus SOC group (44%) compared with the SOC group (11%), and the greater proportion of AEOSIs in the pembrolizumab plus SOC group was driven by hypothyroidism (25% in the pembrolizumab plus SOC group and 5% in the SOC group). The clinical experts noted that these most frequently reported AEOSIs are consistent with the known AEs identified for pembrolizumab in the HNSCC perioperative and postoperative settings. Most AEOSIs were grade 1 or grade 2 in severity, nonserious, and generally consistent with the known safety profile of pembrolizumab.
Conclusion
One ongoing, open-label, phase III RCT comparing efficacy and safety between neoadjuvant and adjuvant pembrolizumab plus SOC and SOC alone in treatment-naive, adult patients with stage III or stage IVA resectable locally advanced HNSCC contributed to this review. As of the data cut-off date (July 25, 2024), OS was not formally tested and the median OS was not reached in the pembrolizumab plus SOC group, with OS events observed in 31% and 37% of patients in the pembrolizumab plus SOC and SOC groups, respectively. The evidence suggests that there is likely a clinically meaningful benefit of pembrolizumab plus SOC for OS at 12 months which appeared to extend throughout the available follow-up but is less certain at longer time points (60 months). Pembrolizumab plus SOC likely results in a clinically important improvement in EFS at 12 months and 36 months. The results suggest low-certainty evidence of little to no difference in EFS at 48 months; however, there is increased uncertainty for all time points beyond 36 months due to few patients remaining at risk and imprecision. Patients who received pembrolizumab plus SOC may have comparable HRQoL compared to the patients who received SOC more than 51 weeks after surgery. According to the clinical experts consulted for this review, the safety profile of pembrolizumab was consistent with their expectations for this drug. Given the small proportion of patients with CPS-negative disease, representing less than 4% of patients in the KEYNOTE-689 study, the clinical experts emphasized that there is insufficient evidence to support or oppose pembrolizumab monotherapy in this subgroup and advised caution in its use.
The sponsor’s application was filed on a pre-Notice of Compliance basis, and the main body of this report reflects the originally proposed indication to Health Canada and CDA-AMC. The updated indication is highlighted in Table 1. Data for patients with PD-L1 with a CPS of less than 1 (i.e., PD-L1 CPS-negative), were not included in the sponsor’s submission for the KEYNOTE-689 study. This subgroup comprises 13 patients (3.6%) in the pembrolizumab plus SOC group and 14 patients (4.0%) in the SOC group. On the other hand, data for patients with PD-L1 with a CPS of 1 or more were available in the sponsor’s Clinical Study Report. Safety outcomes were available only for the overall study population. Between-group balance in the subpopulation with PD-L1 with a CPS of 1 or more for baseline characteristics were consistent with those observed in the overall population. Based on efficacy and baseline data for this subgroup (refer to Table 20 and Table 21 in the Appendix), the CDA-AMC review team assessed that results were generally aligned with those of the overall population.
██ █████ ██ ████████ ███████ ███ █████████ ██ ███ ███████ ████████ ████ █████ ███████ ██████ ███████████ ███ ████████████ ██████ █ ████ ██ █████████ ███████ ████ █████████████ ██ ████████ ████ █████████ ██ ██████████ █████ ███ ████ █████ ███ ████ ████ ██ ████ ███████████ ██ ██████████ ████ ███ █████████ ██ ██████ ██ ██████████████ █ █████████████ █████████ ████████ ████ ██████ ████ █████ ███ █████ ███████ ██ ██████████████████ ███ █████ █████.
In summary, the CDA-AMC review team assessed that, given the small proportion of patients with PD-L1 with a CPS of less than 1 and the similarity of data of patients with PD-L1 with a CPS of 1 or more to the overall population in the KEYNOTE-689 study, the conclusions of the review remain unaffected. The final indication should be based on the narrowed Health Canada–approved indication.
References
1.Centers for Disease Control and Prevention. Head and Neck Cancers Basics. 2023. Accessed March 25, 2025. https://www.cdc.gov/head-neck-cancer/about/index.html
2.Byrne K, Hallworth P, Monfared AAT, Moshyk A, Shaw JW. Real-world systemic therapy treatment patterns for squamous cell carcinoma of the head and neck in Canada. Curr Oncol. 2019;26(2):e167-e174. doi: 10.3747/co.26.3946 PubMed
3.Howren MB, Christensen AJ, Karnell LH, Funk GF. Psychological factors associated with head and neck cancer treatment and survivorship: evidence and opportunities for behavioral medicine. J Consult Clin Psychol. 2013;81(2):299-317. doi: 10.1037/a0029940 PubMed
4.Machiels JP, René Leemans C, Golusinski W, Grau C, Licitra L, Gregoire V. Squamous cell carcinoma of the oral cavity, larynx, oropharynx and hypopharynx: EHNS-ESMO-ESTRO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2020;31(11):1462-1475. doi: 10.1016/j.annonc.2020.07.011 PubMed
5.National Comprehensive Cancer N. NCCN Clinical Practice Guidelines: Head and Neck Cancers. Version 1. 2025. Accessed December 3. https://www.nccn.org/professionals/physician_gls/pdf/head-and-neck.pdf
6.Alberta Cancer C. Clinical Practice Guideline: Oropharyngeal Cancer. 2019. Accessed December 4. https://www.albertahealthservices.ca/assets/info/hp/cancer/if-hp-cancer-guide-hn004-oropharyngeal.pdf
7.Alberta Cancer C. Clinical Practice Guideline: Oral Cavity Cancer. 2023. Accessed December 4. https://www.albertahealthservices.ca/assets/info/hp/cancer/if-hp-cancer-guide-hn002-oral-cavity.pdf
8.Alberta Cancer C. Clinical Practice Guideline: Laryngeal Cancer. 2022. Accessed December 4. https://www.albertahealthservices.ca/assets/info/hp/cancer/if-hp-cancer-guide-hn009-laryngeal.pdf
9.Cancer Care O. Drug Formulary. Accessed August 5, 2024. https://www.cancercareontario.ca/en/drugformulary/search
10.Chow LQM. Head and Neck Cancer. N Engl J Med. 2020;382(1):60-72. doi: 10.1056/NEJMra1715715 PubMed
11.Cancer BC. BC Cancer Protocol Summary for Treatment of Locally Advanced Squamous Cell Carcinoma of the Head and Neck with Concurrent 3-Weekly CARBOplatin and Radiation. Protocol Code: HNLACART3. Accessed February 6. https://www.bccancer.bc.ca/chemotherapy-protocols-site/Documents/Head%20and%20Neck/HNLACART3_Protocol.pdf
12.Cancer BC. BC Cancer Protocol Summary for Combined Chemotherapy CISplatin and Radiation Treatment for Locally Advanced Squamous Cell Carcinoma of the Head and Neck. Protocol Code: HNLAPRT. Accessed February 6. https://www.bccancer.bc.ca/chemotherapy-protocols-site/Documents/Head%20and%20Neck/HNLAPRT_Protocol.pdf
13.Drug I. ONCO-CAPPS. Locally Advanced Resectable Head & Neck Cancer Market Landscape. Report prepared for Merck Canada. 2025.
14.Survey Conducted Among Eight Canadian Key Opinion Leaders (KOLs) at Ad-Board. 2025.
15.Aboaid H KT, Jones D, Nanda R, Ta J, Srinivasmurthy R, Nguyen K, Fama K, Aamer S, Myat Y, Bigcas J-L, Thein K. Efficacy of definitive radiotherapy with concurrent and adjuvant immune checkpoint inhibitors in patients with locally advanced head and neck squamous cell carcinoma (LA HNSCC). Presented at: 2024 ASCO Annual Meeting; May 31–June 4, 2024; Chicago, IL. Abstract 250338.
16.Machiels JP, Tao Y, Licitra L, et al. Pembrolizumab plus concurrent chemoradiotherapy versus placebo plus concurrent chemoradiotherapy in patients with locally advanced squamous cell carcinoma of the head and neck (KEYNOTE-412): a randomised, double-blind, phase 3 trial. Lancet Oncol. 2024;25(5):572-587. doi: 10.1016/s1470-2045(24)00100-1 PubMed
17.Burtness B, Harrington KJ, Greil R, et al. Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-048): a randomised, open-label, phase 3 study. Lancet. 2019;394(10212):1915-1928. doi: 10.1016/s0140-6736(19)32591-7 PubMed
18.Ferris RL, Blumenschein G, Jr., Fayette J, et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N Engl J Med. 2016;375(19):1856-1867. doi: 10.1056/NEJMoa1602252 PubMed
19.Taylor JC, Terrell JE, Ronis DL, et al. Disability in patients with head and neck cancer. Arch Otolaryngol Head Neck Surg. 2004;130(6):764-9. doi: 10.1001/archotol.130.6.764 PubMed
20.Henry M, Rosberger Z, Bertrand L, et al. Prevalence and Risk Factors of Suicidal Ideation among Patients with Head and Neck Cancer: Longitudinal Study. Otolaryngol Head Neck Surg. 2018;159(5):843-852. doi: 10.1177/0194599818776873 PubMed
21.Johnson DE, Burtness B, Leemans CR, Lui VWY, Bauman JE, Grandis JR. Head and neck squamous cell carcinoma. Nat Rev Dis Primers. 2020;6(1):92. doi: 10.1038/s41572-020-00224-3 PubMed
22.Michiels S, Le Maitre A, Buyse M, et al. Surrogate endpoints for overall survival in locally advanced head and neck cancer: meta-analyses of individual patient data. Lancet Oncol. 2009;10(4):341-50. doi: 10.1016/S1470-2045(09)70023-3 PubMed
23.Schandelmaier S, Briel M, Varadhan R, et al. Development of the Instrument to assess the Credibility of Effect Modification Analyses (ICEMAN) in randomized controlled trials and meta-analyses. CMAJ. 2020;192(32):E901-e906. doi: 10.1503/cmaj.200077 PubMed
24.Balshem H, Helfand M, Schünemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-6. doi: 10.1016/j.jclinepi.2010.07.015 PubMed
25.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. doi: 10.1016/j.jclinepi.2019.10.014 PubMed
26.Merck Sharp & Dohme L. L. C. Clinical Study Report: P689V01MK3475 (Data Cutoff Date 25-JUL-2024). A Phase III, Randomized, Open-label Study to Evaluate Pembrolizumab as Neoadjuvant Therapy and in Combination With Standard of Care as Adjuvant Therapy for Stage III-IVA Resectable Locoregionally Advanced Head and Neck Squamous Cell Carcinoma (LA HNSCC) [internal sponsor's report]. 2024 Dec 9.
27.Merck Canada Inc. Merck Canada Inc. response to Canada's Drug Agency request for additional information regarding Keytruda (pembrolizumab) HNSCC review on May 9, 2025 [internal additional sponsor's information]. Merck Canada Inc.; May 12, 2025.
28.Merck Canada Inc. Merck Canada Inc. response to Canada's Drug Agency request for additional information regarding Keytruda (pembrolizumab) HNSCC review on April 24, 2025 [internal additional sponsor's information]. Merck Canada Inc.; April 30, 2025.
29.Merck Canada Inc. Pembrolizumab as Neoadjuvant Therapy and in Combination With Standard of Care as Adjuvant Therapy for Stage III-IVA Resectable Locoregionally Advanced Head and Neck Squamous Cell Carcinoma [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Keytruda (pembrolizumab), solution for infusion 100 mg/4 mL vial. 2025.
30.Canadian Cancer Statistics Advisory Committee in collaboration with the Canadian Cancer S, Statistics C, and the Public Health Agency of C. Canadian Cancer Statistics 2024. Toronto, ON: Canadian Cancer Society. 2024. Accessed July 17, 2024. https://cdn.cancer.ca/-/media/files/research/cancer-statistics/2024-statistics/2024-cmaj/2024_cancer-specific-stats.pdf
31.Chiefs of Ontario CCO, and Institute for Clinical Evaluative Sciences, Cancer in First Nations People in Ontario: Incidence, Mortality, Survival and Prevalence. 2017. https://www.cancercareontario.ca/sites/ccocancercare/files/assets/CancerInFirstNationsPeopleInOntario.pdf
32.Barsouk A, Aluru JS, Rawla P, Saginala K, Barsouk A. Epidemiology, Risk Factors, and Prevention of Head and Neck Squamous Cell Carcinoma. Med Sci (Basel). 2023;11(2). doi: 10.3390/medsci11020042 PubMed
33.National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines: Head and Neck Cancers. Version 2.2025. 2025. https://www.nccn.org/professionals/physician_gls/pdf/head-and-neck.pdf
34.Koh J, Walsh P, D'Costa I, Bhatti O. Head and neck squamous cell carcinoma survivorship care. Aust J Gen Pract. 2019;48(12):846-848. doi: 10.31128/ajgp-08-19-5032 PubMed
35.National Cancer I. SEER Program. Cancer Stat Facts: Laryngeal Cancer. Accessed December 4. https://seer.cancer.gov/statfacts/html/laryn.html
36.National Cancer I. SEER Program. Cancer Stat Facts: Oral Cavity and Pharynx Cancer. Accessed December 4. https://seer.cancer.gov/statfacts/html/oralcav.html
37.Bernier J, Domenge C, Ozsahin M, et al. Postoperative irradiation with or without concomitant chemotherapy for locally advanced head and neck cancer. N Engl J Med. 2004;350(19):1945-52. doi: 10.1056/NEJMoa032641 PubMed
38.Cooper JS, Pajak TF, Forastiere AA, et al. Postoperative concurrent radiotherapy and chemotherapy for high-risk squamous-cell carcinoma of the head and neck. N Engl J Med. 2004;350(19):1937-44. doi: 10.1056/NEJMoa032646 PubMed
39.Black CM, Kong A, Chiu G, et al. Treatment patterns and clinical outcomes of newly diagnosed locally advanced head and neck squamous cell carcinoma (LA HNSCC): A retrospective cohort analysis of national real-world data in England. J Clin Oncol. 2023;41(16_suppl):e18075-e18075. doi: 10.1200/JCO.2023.41.16_suppl.e18075
40.Braakhuis BJ, Brakenhoff RH, Leemans CR. Treatment choice for locally advanced head and neck cancers on the basis of risk factors: biological risk factors. Ann Oncol. 2012;23 Suppl 10:x173-7. doi: 10.1093/annonc/mds299 PubMed
41.Bernier J, Cooper JS, Pajak TF, et al. Defining risk levels in locally advanced head and neck cancers: a comparative analysis of concurrent postoperative radiation plus chemotherapy trials of the EORTC (#22931) and RTOG (# 9501). Head Neck. 2005;27(10):843-50. doi: 10.1002/hed.20279 PubMed
42.Kiyota N, Tahara M, Mizusawa J, et al. Weekly Cisplatin Plus Radiation for Postoperative Head and Neck Cancer (JCOG1008): A Multicenter, Noninferiority, Phase II/III Randomized Controlled Trial. J Clin Oncol. 2022;40(18):1980-1990. doi: 10.1200/jco.21.01293 PubMed
43.Product Monograph: KEYTRUDA (pembrolizumab) - draft. January 23, 2025.
44.Merck Canada Inc. Keytruda (pembrolizumab), solution for infusion 100 mg/4 mL vial [draft product monograph]. 2025.
45.Biolyse Pharma Corp. Product monograph: Fluorouracil Injection 2024. Accessed May 20, 2025. https://pdf.hres.ca/dpd_pm/00077457.PDF
46.Eli Lilly Canada Inc. Product monograph: Erbitux (cetuximab). 2018. Accessed May 20, 2025. https://pdf.hres.ca/dpd_pm/00043071.PDF
47.B.C. Cancer. Head & Neck Protocols: Squamous Cell Carcinoma. 2025. Accessed May 20, 2025. http://www.bccancer.bc.ca/health-professionals/clinical-resources/chemotherapy-protocols/head-neck#Squamous
48.Harrington KJ, Burtness B, Greil R, et al. Pembrolizumab With or Without Chemotherapy in Recurrent or Metastatic Head and Neck Squamous Cell Carcinoma: Updated Results of the Phase III KEYNOTE-048 Study. J Clin Oncol. 2023;41(4):790-802. doi: 10.1200/jco.21.02508 PubMed
49.Merck Sharp & Dohme L. L. C. Protocol No. 689-09 MK3475: A Phase III, Randomized, Open-label Study to Evaluate Pembrolizumab as Neoadjuvant Therapy and in Combination With Standard of Care as Adjuvant Therapy for Stage III-IVA Resectable Locoregionally Advanced Head and Neck Squamous Cell Carcinoma (LA HNSCC) [internal sponsor's report]. 2024.
50.Maurer W, Bretz F. Multiple testing in group sequential trials using graphical approaches. Stat Biopharm Res. 2013;5(4):311-320.
51.Arraras JI, Arias F, Tejedor M, et al. The EORTC QLQ-C30 (version 3.0) Quality of Life questionnaire: validation study for Spain with head and neck cancer patients. Psychooncology. 2002;11(3):249-56. doi: 10.1002/pon.555 PubMed
52.Chaukar DA, Das AK, Deshpande MS, et al. Quality of life of head and neck cancer patient: validation of the European organization for research and treatment of cancer QLQ-C30 and European organization for research and treatment of cancer QLQ-H&N 35 in Indian patients. Indian J Cancer. 2005;42(4):178-84. PubMed
53.Singer S, Amdal CD, Hammerlid E, et al. International validation of the revised European Organisation for Research and Treatment of Cancer Head and Neck Cancer Module, the EORTC QLQ-HN43: Phase IV. Head Neck. 2019;41(6):1725-1737. doi: 10.1002/hed.25609 PubMed
54.Singer S, Wollbrück D, Wulke C, et al. Validation of the EORTC QLQ-C30 and EORTC QLQ-H&N35 in patients with laryngeal cancer after surgery. Head Neck. 2009;31(1):64-76. doi: 10.1002/hed.20938 PubMed
55.Kyrgidis A, Printza A, Vitkos E, et al. Minimal Clinically Important Differences in the Cancer Quality of Life Questionnaires in Patients with Head and Neck Cancer. Clin Pract. 2024;14(6):2329-2340. doi: 10.3390/clinpract14060182 PubMed
56.Musoro JZ, Coens C, Sprangers MAG, et al. Minimally important differences for interpreting EORTC QLQ-C30 change scores over time: A synthesis across 21 clinical trials involving nine different cancer types. Eur J Cancer. 2023;188:171-182. doi: 10.1016/j.ejca.2023.04.027 PubMed
57.Bjordal K, Ahlner-Elmqvist M, Tollesson E, et al. Development of a European Organization for Research and Treatment of Cancer (EORTC) questionnaire module to be used in quality of life assessments in head and neck cancer patients. EORTC Quality of Life Study Group. Acta Oncol. 1994;33(8):879-85. doi: 10.3109/02841869409098450 PubMed
58.Bjordal K, de Graeff A, Fayers PM, et al. A 12 country field study of the EORTC QLQ-C30 (version 3.0) and the head and neck cancer specific module (EORTC QLQ-H&N35) in head and neck patients. EORTC Quality of Life Group. Eur J Cancer. 2000;36(14):1796-807. doi: 10.1016/s0959-8049(00)00186-6 PubMed
59.Merck Sharp & Dohme L. L. C., ed. Supplemental SAP Protocol No. 689-09 MK3475: A Phase III, Randomized, Open-label Study to Evaluate Pembrolizumab as Neoadjuvant Therapy and in Combination With Standard of Care as Adjuvant Therapy for Stage III-IVA Resectable Locoregionally Advanced Head and Neck Squamous Cell Carcinoma (LA HNSCC) [internal sponsor's report]. 2024.
60.Gray RJ. A Class of K-Sample Tests for Comparing the Cumulative Incidence of a Competing Risk. The Annals of Statistics. 1988;16(3):1141-1154.
61.Prentice RL, Kalbfleisch JD, Peterson AV, Jr., Flournoy N, Farewell VT, Breslow NE. The analysis of failure times in the presence of competing risks. Biometrics. 1978;34(4):541-54. PubMed
62.Bassler D, Briel M, Montori VM, et al. Stopping randomized trials early for benefit and estimation of treatment effects: systematic review and meta-regression analysis. JAMA. 2010;303(12):1180-7. doi: 10.1001/jama.2010.310 PubMed
63.Guyatt GH, Briel M, Glasziou P, Bassler D, Montori VM. Problems of stopping trials early. BMJ. 2012;344:e3863. doi: 10.1136/bmj.e3863 PubMed
64.Ciani O, Buyse M, Drummond M, Rasi G, Saad ED, Taylor RS. Time to Review the Role of Surrogate End Points in Health Policy: State of the Art and the Way Forward. Value Health. 2017;20(3):487-495. doi: 10.1016/j.jval.2016.10.011 PubMed
65.Little RJ, D'Agostino R, Cohen ML, et al. The prevention and treatment of missing data in clinical trials. N Engl J Med. 2012;367(14):1355-60. doi: 10.1056/NEJMsr1203730 PubMed
66.Sterne JAC, Savović J, Page MJ, et al. RoB 2: a revised tool for assessing risk of bias in randomised trials. BMJ. 2019;366:l4898. doi: 10.1136/bmj.l4898 PubMed
67.Clinical trial endpoints for the approval of cancer drugs and biologics: Guidance for industry. Silver Spring (MD): U.S. Food and Drug Administration; 2018: https://www.fda.gov/media/71195/download. Accessed May 8, 2025.
68.Forde PM, Spicer J, Lu S, et al. Neoadjuvant Nivolumab plus Chemotherapy in Resectable Lung Cancer. N Engl J Med. 2022;386(21):1973-1985. doi: 10.1056/NEJMoa2202170 PubMed
69.Patel SP, Othus M, Chen Y, et al. Neoadjuvant-Adjuvant or Adjuvant-Only Pembrolizumab in Advanced Melanoma. N Engl J Med. 2023;388(9):813-823. doi: 10.1056/NEJMoa2211437 PubMed
70.Schmid P, Cortes J, Pusztai L, et al. Pembrolizumab for Early Triple-Negative Breast Cancer. N Engl J Med. 2020;382(9):810-821. doi: 10.1056/NEJMoa1910549 PubMed
71.Cohen EEW, Soulières D, Le Tourneau C, et al. Pembrolizumab versus methotrexate, docetaxel, or cetuximab for recurrent or metastatic head-and-neck squamous cell carcinoma (KEYNOTE-040): a randomised, open-label, phase 3 study. Lancet. 2019;393(10167):156-167. doi: 10.1016/s0140-6736(18)31999-8 PubMed
Appendix 1: Detailed Outcome Data
Please note that this appendix has not been copy-edited.
Additional information and data from the KEYNOTE-689 study are presented as follows.
Figure 4: Multiplicity Graph for Type I Error Control of Study Hypothesis in the KEYNOTE-689 Study
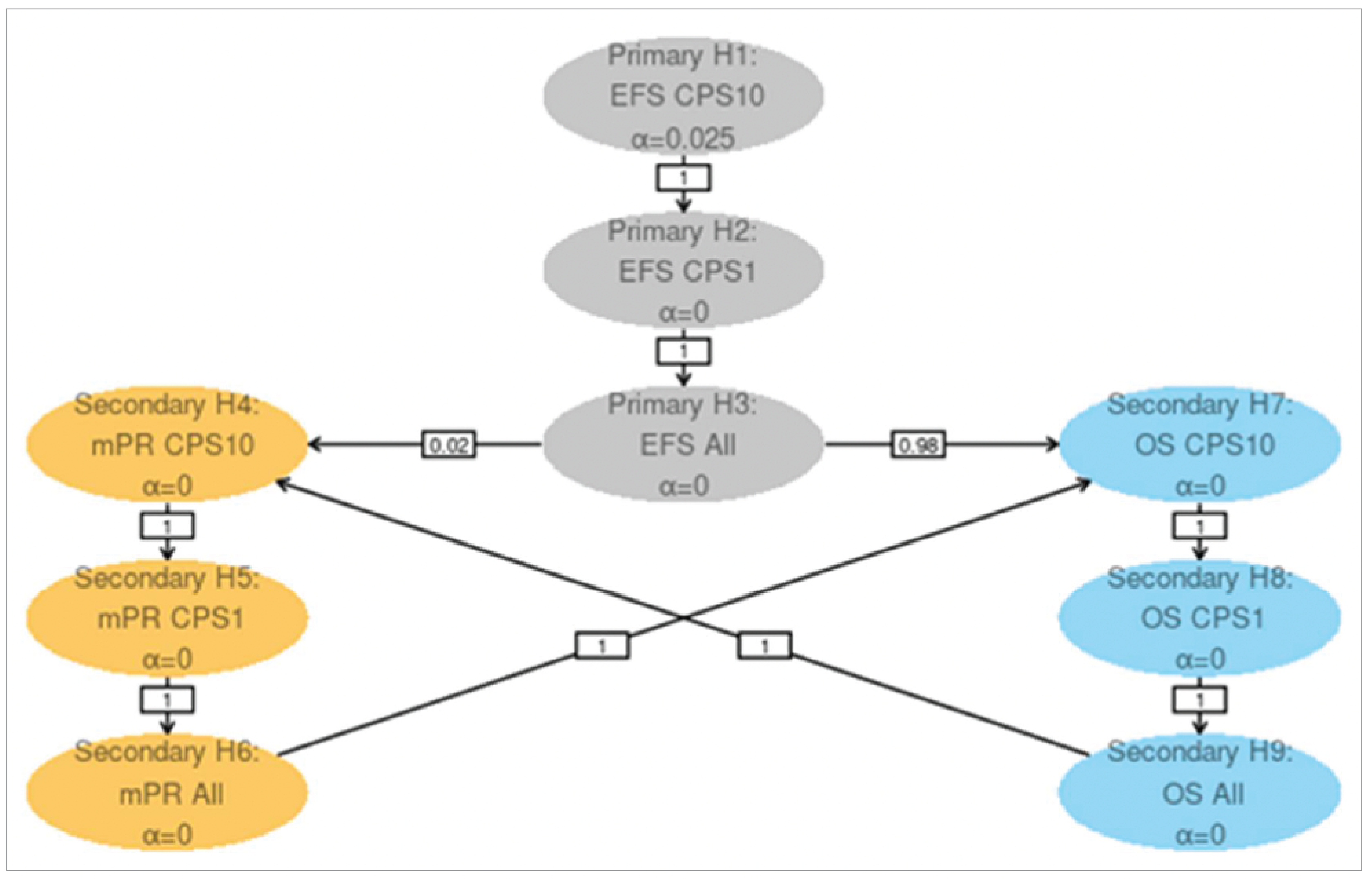
CPS = combined positive score; EFS = event-free survival; H1-H9 = hypothesis 1 to 9; mPR = major pathologic response; OS = overall survival.
Source: Study protocol for the KEYNOTE-689 study.49
Figure 5: Forest Plot of OS HR by Subgroup Factors in the KEYNOTE-689 Study (Overall ITT Population)
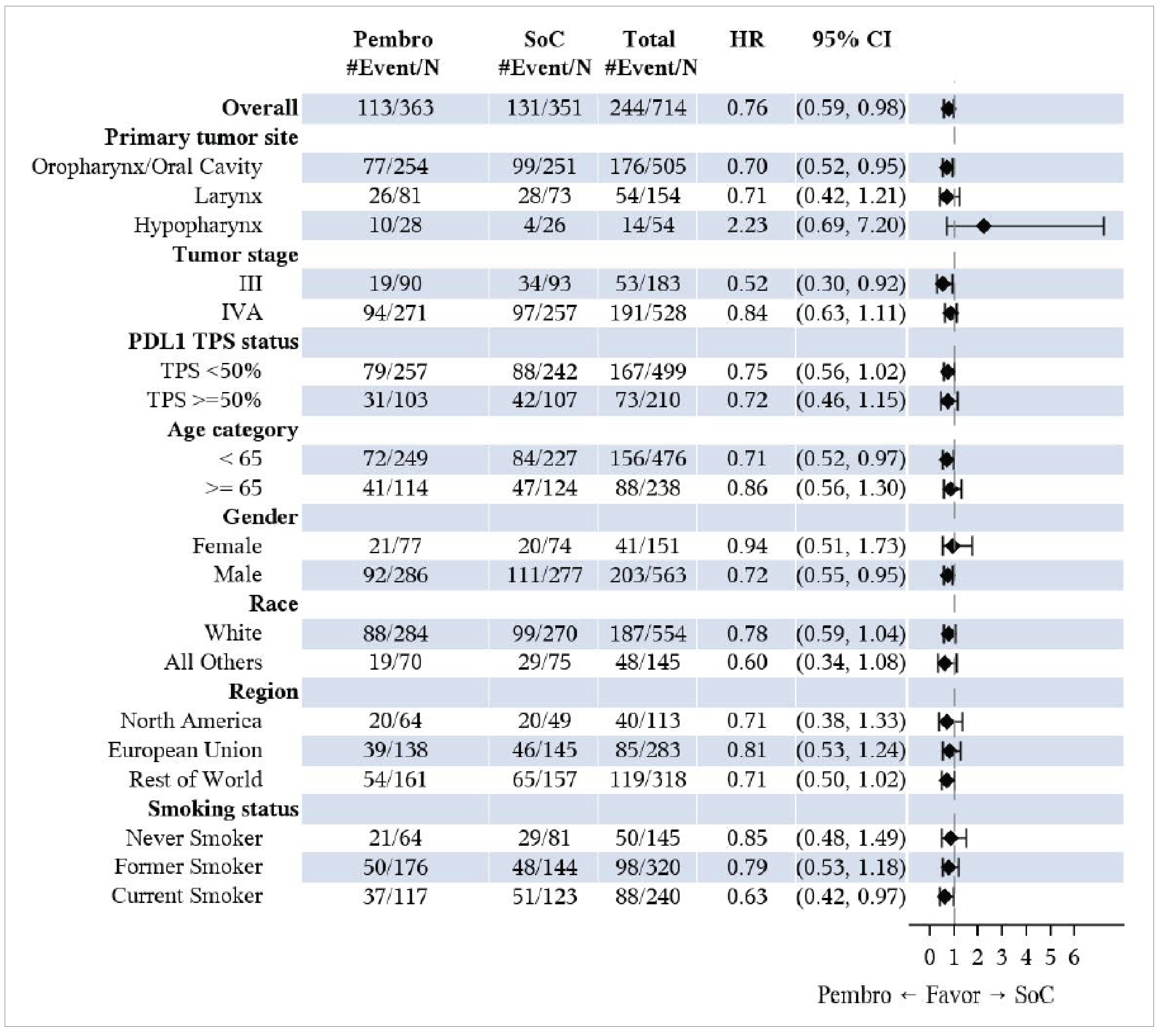
CI = confidence interval; CPS = combined positive score; HR = hazard ratio; ITT = intention to treat; Pembro = pembrolizumab; OS = overall survival; SOC = standard of care; TPS = tumour proportion score.
Note: For subgroups, analysis was based on unstratified Cox regression model with treatment as a covariate. If total number of patients in one level of a subgroup was < 10% of the CPS ≥ 10 population, that particular level would not be displayed. European Union category in this graph includes European Economic Area, UK, and Switzerland. Terminology used in this figure (i.e., “gender” rather than “sex”) reflects the original source and has been reproduced without modification. Database cut-off date was July 25, 2024.
Source: Clinical Study Report for the KEYNOTE-689 study.26
Figure 6: Forest Plot of EFS HR by Subgroup Factors in the KEYNOTE-689 Study (Overall ITT Population)
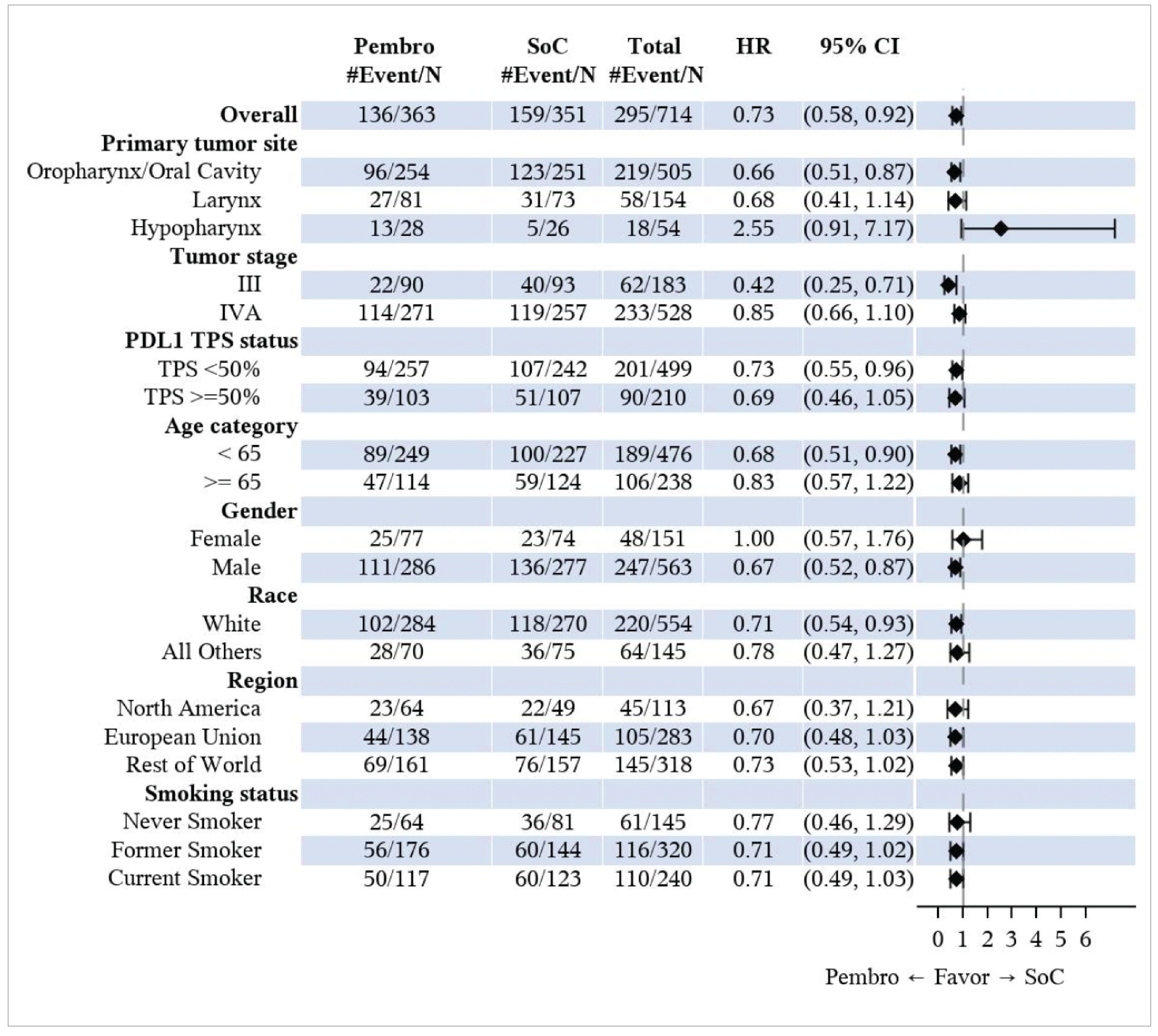
BICR = blinded independent central review; CI = confidence interval; CPS = combined positive score; EFS = event-free survival; HR = hazard ratio; ITT = intention to treat; Pembro = pembrolizumab; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours version 1.1; SOC = standard of care; TPS = tumour proportion score.
Note: The EFS assessment was based on BICR per RECIST 1.1 (primary censoring rule). For subgroups, analysis was based on unstratified Cox regression model with treatment as a covariate. If total number of patients in one level of a subgroup was < 10% of the CPS ≥ 10 population, that particular level would not be displayed. European Union category in this graph includes European Economic Area, UK, and Switzerland. Terminology used in this figure (i.e., “gender” rather than “sex”) reflects the original source and has been reproduced without modification. Database cut-off date was July 25, 2024.
Source: Clinical Study Report for the KEYNOTE-689 Study.26
EFS by BICR in the Patients With CPS Greater Than or Equal to 10 (Total N = 465)
In the patient population with CPS ≥ 10 at baseline, the median durations of follow-up were 31.1 months (range, 0.8 to 64.9) in the pembrolizumab plus SOC group, and 24.3 months (range, 0.6 to 66.5) in the SOC group. For patients with CPS ≥ 10, the median EFS in the pembrolizumab plus SOC group was 59.7 months (95% CI, 41.1 months to not reached), while the median EFS in the SOC alone group was 26.9 months (95% CI, 18.3 to 51.5). The HR was 0.66 (95% CI, 0.49 to 0.88; P = 0.00217). The KM estimates of the probability of EFS in the pembrolizumab plus SOC and SOC alone groups were 74.0% (95% CI, 67.6% to 79.3%) versus 60.0% (95% CI, 53.0% to 66.3%) at 12 months; and 65.1% (95% CI, 58.2% to 71.2%) versus 53.2% (95% CI, 45.9% to 59.9%) at 24 months, respectively (Table 18).
Table 18: Event-Free Survival Results From the KEYNOTE-689 Study (Patients With CPS Greater Than or Equal to 10 at Baseline)
Efficacy outcome | Pembrolizumab plus SOC (N = 234) | SOC (N = 231) |
|---|---|---|
EFS by BICR (patients with PD-L1 CPS ≥ 10, total N = 465) | ||
Follow-up duration (months), median (range) | 31.1 (0.8 to 64.9) | 24.3 (0.6 to 66.5) |
Number of patients contributing to the analysis | 234 | 231 |
Number of events, n (%) | 85 (36.3) | 107 (46.3) |
Death | 43 (18.4) | 37 (16.0) |
Distant progressive disease | 15 (6.4) | 39 (16.9) |
Local and distant progressive disease | 2 (0.9) | 2 (0.9) |
Local progressive disease/recurrence | 25 (10.7) | 29 (12.6) |
Number censored, n (%) | 149 (63.7) | 124 (53.7) |
Last assessment showing no progression | 148 (63.2) | 116 (50.2) |
Randomizationa | 1 (0.4) | 8 (3.5) |
EFS (months), median (95% CI)b | 59.7 (41.1 to not reached) | 26.9 (18.3 to 51.5) |
HR (95% CI)c | 0.66 (0.49 to 0.88) | Reference |
One-sided P valued | 0.00217 | |
EFS probability (%) at 6 months (95% CI) | 85.7 (80.4 to 89.7) | 79.4 (73.3 to 84.2) |
EFS probability (%) at 12 months (95% CI) | 74.0 (67.6 to 79.3) | 60.0 (53.0 to 66.3) |
Difference in EFS probability (%) (95% CI) | ████ ████ ██ █████ | |
EFS probability (%) at 24 months (95% CI) | 65.1 (58.2 to 71.2) | 53.2 (45.9 to 59.9) |
Difference in EFS probability (%) (95% CI) | ████ ████ ██ █████ | |
EFS probability (%) at 36 months (95% CI) | 59.8 (52.3 to 66.5) | 45.9 (38.0 to 53.4) |
EFS probability (%) at 48 months (95% CI) | 55.9 (47.7 to 63.4) | 44.4 (36.3 to 52.2) |
BICR = blinded independent central review; CI = confidence interval; CPS = combined positive score; EFS = even-free survival; HR = hazard ratio; SOC = standard of care; sSAP = supplementary statistical analysis plan.
Note: Data presented in this table were based on analyses at clinical cut-off date of July 25, 2024.
aThe first assessment for patients was screening/randomization because at randomization patients were confirmed to not have progressive disease/recurrence. Patients who did not have any postrandomization radiographic disease assessments or on-study surgery were censored at their last assessment showing no progression which occurs at randomization. For specificity, the label “randomization” is used within the EFS analyses tables to distinguish from individuals with postrandomization assessments.
bFrom product-limit (Kaplan-Meier) method for censored data.
cBased on Cox regression model with Efron’s method of tie handling with treatment as a covariate stratified by primary tumour site (oropharynx/oral cavity vs larynx vs hypopharynx) and tumour stage (III vs IVA) with small strata collapsed as prespecified in the sSAP.
dBased on log-rank test stratified by primary tumour site (oropharynx/oral cavity vs larynx vs hypopharynx) and tumour stage (III vs IVA) with small strata collapsed as prespecified in the sSAP. Statistical testing for this end point was adjusted for multiple comparisons using the graphical method of Maurer and Bretz (2013).50
Source: Clinical Study Report for the KEYNOTE-689 study,26 sponsor’s submissions.27,28
EFS as assessed by BICR in the sensitivity analysis based with different censoring rules (HR = 0.62; 95% CI, 0.45 to 0.86; P = 0.00172) was consistent with EFS analysis results as assessed by BICR in the primary analysis. The EFS results based on investigator assessment on the primary censoring rule (HR = 0.64; 95% CI, 0.48 to 0.85; P = 0.001) and in the sensitivity analysis (HR = 0.64; 95% CI, 0.47 to 0.87; P = 0.00196) were consistent with EFS analysis results as assessed by BICR in the primary analysis.26 The EFS results were generally consistent across the prespecified subgroups in favour of pembrolizumab plus SOC group, but 95% CI of the HR for some categories of patients included the null.26
Table 19: Additional Efficacy Outcomes in the KEYNOTE-689 Study (Overall ITT Population)
Efficacy outcome | Pembrolizumab plus SOC (N = 363) | SOC (N = 351) |
|---|---|---|
Major pathological response (mPR) based on BIPR | ||
Number of patients with mPR | 34 | 0 |
Proportion of patients with mPR (%) (95% CI) | 9.4 (6.6 to 12.8) | 0 (0.0 to 1.0) |
Difference in % vs. SOC (95% CI)a | 9.3 (6.7 to 12.8) | Reference |
One-sided P valueb | < 0.00001 | |
Pathological complete response (pCR) based on BIPR | ||
Number of patients with pCR | 11 | 0 |
Proportion of patients with pCR (%) (95% CI) | 3.0 (1.5 to 5.4) | 0 (0.0 to 1.0) |
Difference in % vs. SOC (95% CI)a | 3.0 (1.5 to 5.3) | Reference |
One-sided P valueb | 0.0006 | |
Distant metastasis-free survival (DMFS) based on BICR per RECIST 1.1 | ||
Number of events, n (%) | 127 (35.0) | 148 (42.2) |
DMFS (months), median (95% CI)c | 51.8 (37.9 to not reached) | 35.7 (26.3 to 57.0) |
HR (95% CI)d | 0.71 (0.56 to 0.90) | Reference |
One-sided P valuee | 0.00252 | |
DMFS probability (%) at 6 months (95% CI) | 92.0 (88.5 to 94.4) | 84.2 (79.7 to 87.7) |
DMFS probability (%) at 12 months (95% CI) | 81.9 (77.3 to 85.7) | 66.5 (61.0 to 71.5) |
DMFS probability (%) at 24 months (95% CI) | 68.3 (62.6 to 73.2) | 56.6 (50.7 to 62.1) |
DMFS probability (%) at 36 months (95% CI) | 59.1 (52.8 to 64.8) | 49.0 (42.5 to 55.3) |
DMFS probability (%) at 48 months (95% CI) | 52.6 (45.6 to 59.2) | 45.4 (38.1 to 52.4) |
Local regional control (LRC) based on BICR per RECIST 1.1 | ||
Number of events, n (%) | 136 (37.5) | 159 (45.3) |
Number of competing eventsf, n (%) | 93 (25.6) | 115 (32.8) |
HR (95% CI)d | 0.92 (0.61 to 1.41) | Reference |
One-sided P valueg | 0.3539 | |
Event cumulative incidence (%) at 6 months (95% CI)h | 6.9 (4.6 to 9.9) | 7.4 (4.9 to 10.6) |
Event cumulative incidence (%) at 12 months (95% CI)h | 10.7 (7.6 to 14.2) | 11.5 (8.3 to 15.3) |
Event cumulative incidence (%) at 18 months (95% CI)h | 12.0 (8.8 to 15.8) | 11.5 (8.3 to 15.3) |
Event cumulative incidence (%) at 24 months (95% CI)h | 12.4 (9.1 to 16.2) | 11.5 (8.3 to 15.3) |
Event cumulative incidence (%) at 36 months (95% CI)h | 13.4 (9.9 to 17.5) | 14.3 (10.4 to 18.7) |
Event cumulative incidence (%) at 48 months (95% CI)h | 13.4 (9.9 to 17.5) | 15.3 (11.1 to 20.1) |
BICR = blinded independent central review; BIPR = blinded independent pathologist review; CI = confidence interval; DMFS = distant metastasis-free survival; EFS = even-free survival; HR = hazard ratio; ITT = intention to treat; LRC = local regional control; mPR = major pathological response; pCR = pathological complete response; RECIST v1.1 = Response Evaluation Criteria in Solid Tumours version 1.1; SOC = standard of care; sSAP = supplementary statistical analysis plan.
Note: Database cut-off date was July 25, 2024. Definitions of outcomes in this table are as follows: mPR: ≤ 10% of invasive SCC in the resected primary tumour specimen and all sampled regional lymph nodes. pCR: absence of residual invasive SCC in the resected primary tumour specimen and all sampled regional lymph nodes. DMFS: time from date of randomization to the date of first record of appearance of distant metastasis or death due to any cause. LRC: time from date of randomization to the date of first record of radiographic disease progression (same exceptions as for the EFS outcome), local recurrence as assessed with imaging or biopsy as indicated.
aBased on Miettinen and Nurminen method stratified by primary tumour site (oropharynx/oral cavity vs larynx vs hypopharynx) and tumour stage (III vs IVA) with small strata collapsed as prespecified in the sSAP.
bH0: difference in % = 0 vs. H1: difference in % > 0. Statistical testing for this end point was adjusted for multiple comparisons using the graphical method of Maurer and Bretz (2013).50
cFrom product-limit (Kaplan-Meier) method for censored data.
dBased on Cox regression model with Efron’s method of tie handling with treatment as a covariate stratified by primary tumour site (oropharynx/oral cavity vs larynx vs hypopharynx) and tumour stage (III vs IVA) with small strata collapsed as prespecified in the sSAP.
eBased on log-rank test stratified by primary tumour site (oropharynx/oral cavity vs larynx vs hypopharynx) and tumour stage (III vs IVA) with small strata collapsed as prespecified in the sSAP.
fDistant metastases diagnosed before any locoregional failure and death in the absence of locoregional failures are not considered events of interest, but as competing risk events.
gBased on Gray’s test stratified by primary tumour site (oropharynx/oral cavity vs larynx vs hypopharynx) and tumour stage (III vs IVA) with small strata collapsed as prespecified in the sSAP.
hCumulative incidence at specified time points is based on nonparametric estimation of cumulative incidence of the event of interest accounting for competing risk events.
Figure 7: Line Plot of Empirical Mean Change From Baseline and 95% CI for the EORTC QLQ-C30 GHS/QoL Over Time by Treatment Group in the KEYNOTE-689 Study (PRO FAS Population)
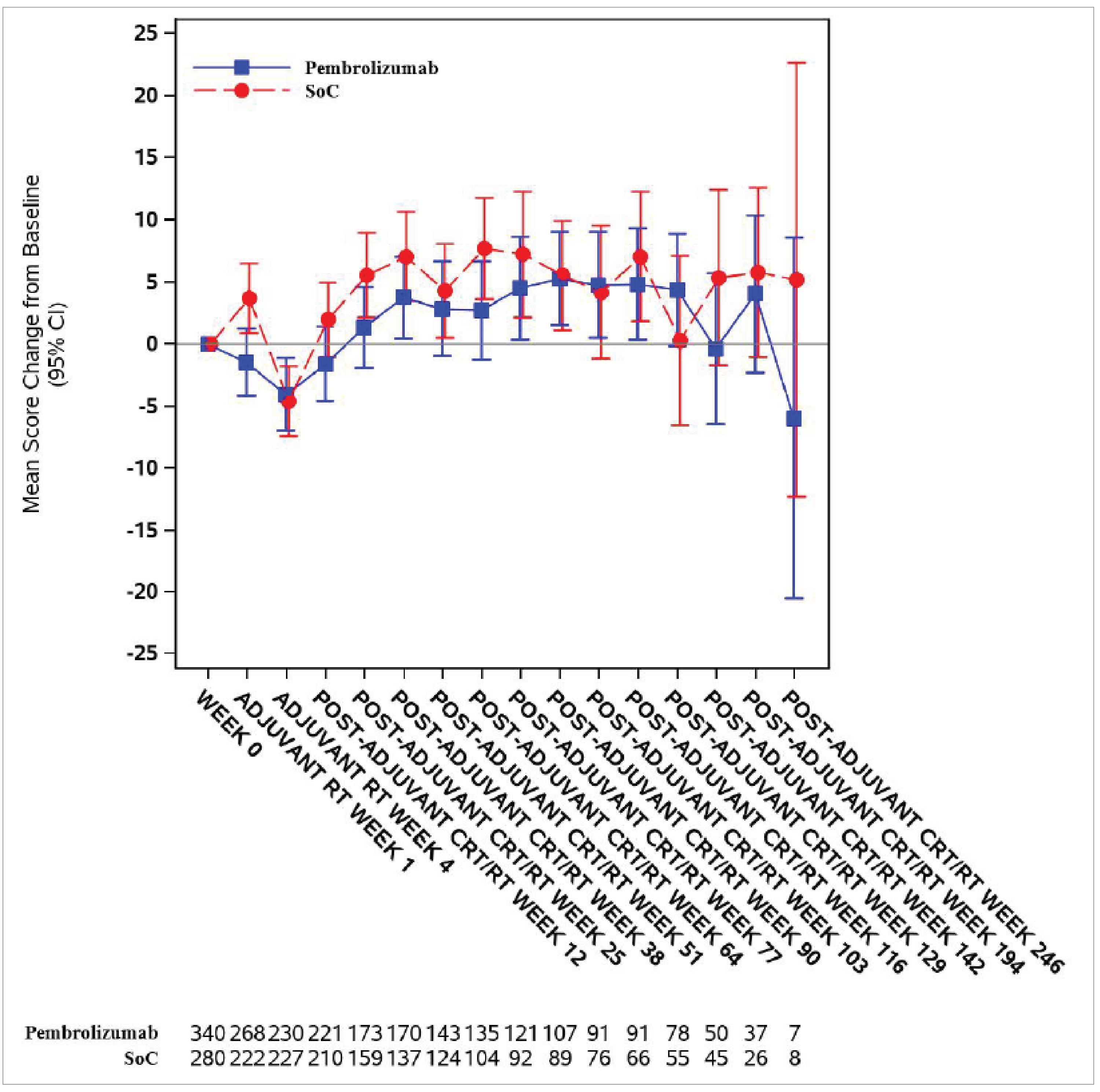
CI = confidence interval; CRT = chemoradiotherapy; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core-30; FAS = full analysis set; GHS = global health status; PRO = patient-reported outcome; QoL = quality of life; RT = radiotherapy; SOC = standard of care.
Note: Database cut-off date was July 25, 2024.
Source: Clinical Study Report for the KEYNOTE-689 study.26
Figure 8: Bar Plot of LS Mean Change From Baseline to Postadjuvant CRT/RT Week 25 and 95% CI in EORTC QLQ-H&N35 Pain/Swallowing/Speech Symptoms (PRO FAS Population)
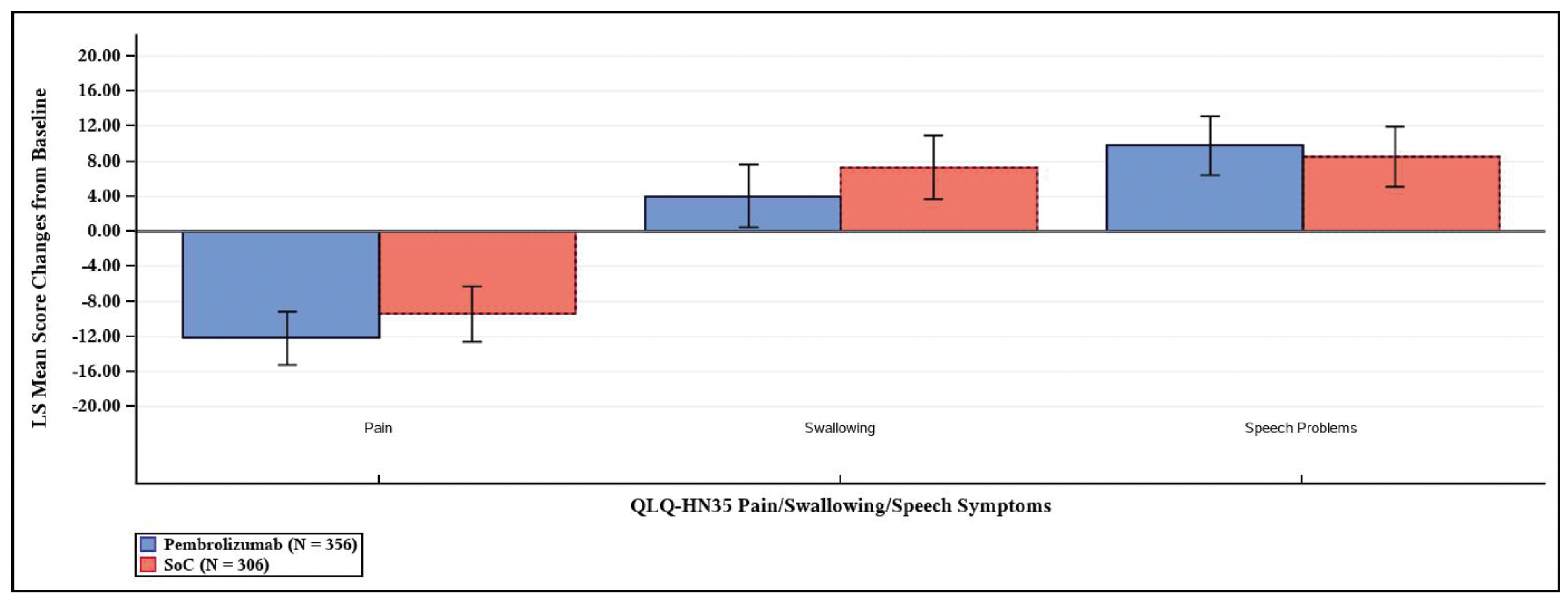
CI = confidence interval; CRT = chemoradiotherapy; EORTC QLQ-H&N35 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Head and Neck cancer module 35; FAS = full analysis set; LS = least squares; PRO = patient-reported outcome; RT = radiotherapy; SOC = standard of care.
Note: Data presented in this figure were based on analyses at clinical cut-off date of July 25, 2024.
Source: Clinical Study Report for the KEYNOTE-689 study.26
Figure 9: Bar Plot of LS Mean Change From Baseline to Postadjuvant CRT/RT Week 51 and 95% CI in EORTC QLQ-H&N35 Pain/Swallowing/Speech Symptoms (PRO FAS Population)
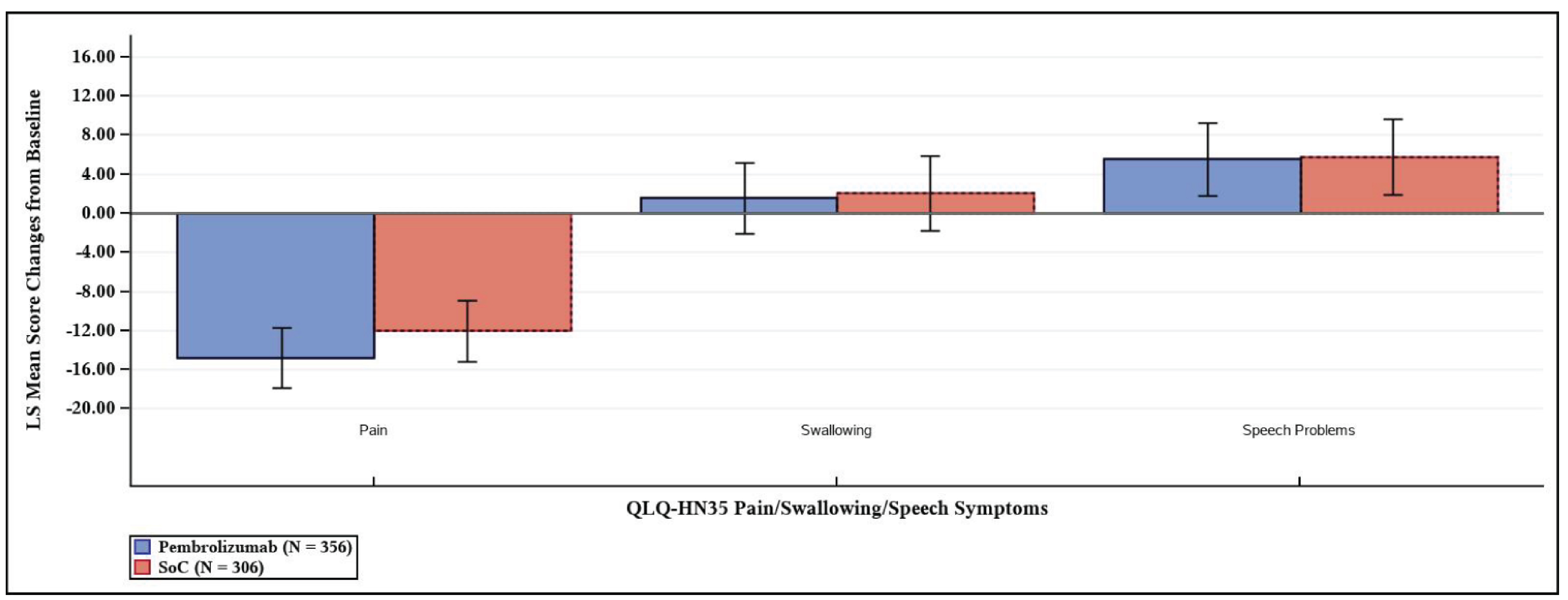
CI = confidence interval; CRT = chemoradiotherapy; EORTC QLQ-H&N35 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Head and Neck cancer module 35; FAS = full analysis set; LS = least squares; PRO = patient-reported outcome; RT = radiotherapy; SOC = standard of care.
Note: Data presented in this figure were based on analyses at clinical cut-off date of July 25, 2024.
Source: Clinical Study Report for the KEYNOTE-689 study.26
Table 20: Summary of Baseline Characteristics From the KEYNOTE-689 Study (Patients With PD-L1 CPS ≥ 1)
Characteristic | Pembrolizumab plus SOC (N = 347) | SOC (N = 335) |
|---|---|---|
Demographic characteristics | ||
Age, years | ||
Mean (SD) | 59.5 (10.0) | 60.4 (10.0) |
Median (range) | 60.0 (29 to 82) | 61.0 (22 to 87) |
< 65 years, n (%) | 237 (68.3) | 220 (65.7) |
≥ 65 years, n (%) | 110 (31.7) | 115 (34.3) |
Sex, n (%) | ||
Male | 272 (78.4) | 265 (79.1) |
Female | 75 (21.6) | 70 (20.9) |
Race, n (%) | ||
American Indian or Alaska Native [wording from original source] | 0 | 1 (0.3) |
Asian | 47 (13.5) | 42 (12.5) |
Black or African American | 8 (2.3) | 9 (2.7) |
Multiple | 9 (2.6) | 16 (4.8) |
Native Hawaiian or Other Pacific Islander | 1 (0.3) | 1 (0.3) |
White | 273 (78.7) | 260 (77.6) |
Missing | 9 (2.6) | 6 (1.8) |
Smoking status, n (%) | ||
Never smoker | 61 (17.6) | 76 (22.7) |
Former smoker | 170 (49.0) | 135 (40.3) |
Current smoker | 110 (31.7) | 121 (36.1) |
Unknown | 6 (1.7) | 3 (0.9) |
Cigarette use (packs per year), n (%) | ||
0 (No cigarette use) | 61 (17.6) | 76 (22.7) |
≤ 10 | 64 (18.4) | 68 (20.3) |
> 10 | 206 (59.4) | 180 (53.7) |
Unknown | 16 (4.6) | 11 (3.3) |
Alcohol use, n (%) | ||
Yes | 238 (68.6) | 228 (68.1) |
No | 103 (29.7) | 104 (31.0) |
Unknown | 6 (1.7) | 3 (0.9) |
Disease characteristics | ||
PD-L1 status by TPS score, n (%) | ||
TPS < 50% | 244 (70.3) | 228 (68.1) |
TPS ≥ 50% | 103 (29.7) | 107 (31.9) |
Primary tumour site, n (%) | ||
Oropharynx | 34 (9.8) | 37 (11.0) |
Oral Cavity | 210 (60.5) | 205 (61.2) |
Larynx | 77 (22.2) | 67 (20.0) |
Hypopharynx | 26 (7.5) | 25 (7.5) |
Missing | 0 | 1 (0.3) |
ECOG PS, n (%) | ||
0 | 187 (53.9) | 201 (60.0) |
1 | 160 (46.1) | 134 (40.0) |
HPV status, n (%) | ||
Positive | 11 (3.2) | 14 (4.2) |
Negative | 336 (96.8) | 320 (95.5) |
Unknown | 0 | 1 (0.3) |
Overall cancer staging at baseline, n (%) | ||
III | 86 (24.8) | 88 (26.3) |
IVA | 259 (74.6) | 246 (73.4) |
IVB | 2 (0.6) | 0 |
Missing | 0 | 1 (0.3) |
Cancer T staging at baseline, n (%) | ||
T1 | 8 (2.3) | 4 (1.2) |
T2 | 48 (13.8) | 47 (14.0) |
T3 | 107 (30.8) | 109 (32.5) |
T4 | 10 (2.9) | 14 (4.2) |
T4A | 174 (50.1) | 159 (47.5) |
T4B | 0 | 1 (0.3) |
Missing | 0 | 1 (0.3) |
Cancer N staging at baseline, n (%) | ||
N0 | 90 (25.9) | 80 (23.9) |
N1 | 74 (21.3) | 71 (21.2) |
N2 | 31 (8.9) | 29 (8.7) |
N2A | 12 (3.5) | 7 (2.1) |
N2B | 80 (23.1) | 87 (26.0) |
N2C | 57 (16.4) | 60 (17.9) |
N3 | 1 (0.3) | 0 |
N3B | 2 (0.6) | 0 |
Missing | 0 | 1 (0.3) |
Cancer M staging at baseline, n (%) | ||
M0 | 347 (100.0) | 334 (99.7) |
Missing | 0 | 1 (0.3) |
CPS = combined positive score; ECOG = Eastern Cooperative Oncology Group; PS = Performance Status; SD = standard deviation; SOC = standard of care; TPS = tumour proportion score.
Note: Data presented in this table were based on analyses at clinical cut-off date of July 25, 2024.
Source: Clinical Study Report for the KEYNOTE-689 study.26
Table 21: OS and Event-Free Survival Results From the KEYNOTE-689 Study (Patients With PD-L1 CPS Greater Than or Equal to 1)
Efficacy outcome | Pembrolizumab plus SOC (N = 347) | SOC (N = 335) |
|---|---|---|
Follow-up duration (months), median (range) | 30.0 (0.8 to 64.9) | 23.4 (0.5 to 66.5) |
OS | ||
Number of events, n (%) | 106 (30.5) | 128 (38.2) |
OS (months), median (95% CI)a | Not reached (not reached to not reached) | 61.8 (49.2 to not reached) |
HR (95% CI)b | 0.72 (0.56 to 0.94) | Reference |
One-sided P valuec | 0.00666 | |
OS probability (%) at 6 months (95% CI) | 94.2 (91.2 to 96.2) | 88.4 (84.4 to 91.4) |
OS probability (%) at 12 months (95% CI) | 86.3 (82.2 to 89.5) | 77.5 (72.6 to 81.6) |
OS probability (%) at 24 months (95% CI) | 75.3 (70.2 to 79.7) | 67.3 (61.7 to, 72.2) |
OS probability (%) at 36 months (95% CI) | 69.0 (63.3 to 74.0) | 60.2 (54.1 to 65.8) |
OS probability (%) at 48 months (95% CI) | 64.0 (57.6 to 69.6) | 57.0 (50.4 to 63.0) |
OS probability (%) at 60 months (95% CI) | 60.1 (52.4 to 66.8) | 51.7 (43.8 to 59.1) |
EFS by BICR | ||
Number of events, n (%) | 128 (36.9) | 156 (46.6) |
Death | 63 (18.2) | 62 (18.5) |
Distant progressive disease | 24 (6.9) | 51 (15.2) |
Local and distant progressive disease | 4 (1.2) | 6 (1.8) |
Local progressive disease/recurrence | 37 (10.7) | 37 (11.0) |
Number censored (%) | 219 (63.1) | 179 (53.4) |
Last assessment showing no progression | 216 (62.2) | 168 (50.1) |
Randomizationd | 3 (0.9) | 11 (3.3) |
EFS (months), median (95% CI)a | 59.7 (37.9 to not reached) | 29.6 (19.5 to 41.9) |
HR (95% CI)b | 0.70 (0.55 to 0.89) | Reference |
One-sided P valuec | 0.00140 | |
EFS probability (%) at 6 months (95% CI) | 86.2 (82.0 to 89.5) | 80.0 (75.1 to 84.0) |
EFS probability (%) at 12 months (95% CI) | 74.8 (69.6 to 79.2) | 61.3 (55.5 to 66.5) |
EFS probability (%) at 24 months (95% CI) | 64.6 (58.8 to 69.8) | 53.4 (47.4 to 59.1) |
EFS probability (%) at 36 months (95% CI) | 58.2 (51.9 to 64.0) | 44.9 (38.4 to 51.2) |
EFS probability (%) at 48 months (95% CI) | 52.1 (45.0 to 58.8) | 42.8 (35.9 to 49.4) |
EORTC QLQ-C30 global health status/QoL | ||
Number of patients contributing to the analysis at baselinee | 324 | 265 |
Baseline score, mean (SD) | 63.73 (20.36) | 62.52 (21.69) |
Postadjuvant CRT/RT Week 25 | ||
Number of patients at postadjuvant CRT/RT Week 25e | 170 | 162 |
Score at postadjuvant CRT/RT Week 25, mean (SD) | 68.97 (18.42) | 70.32 (18.81) |
Change from baseline to postadjuvant CRT/RT Week 25, LS mean (95% CI)f | 3.62 (0.90 to 6.34) | 5.03 (2.23 to 7.83) |
Treatment group difference vs. control at Week 25, LS mean (95% CI)f | −1.41 (−4.96 to 2.15) | |
P valuef | 0.4374 | |
Postadjuvant CRT/RT Week 51 | ||
Number of patients at postadjuvant CRT/RT Week 25e | 139 | 125 |
Score at postadjuvant CRT/RT Week 25, mean (SD) | 70.44 (18.18) | 70.93 (18.83) |
Change from baseline to postadjuvant CRT/RT Week 25, LS mean (95% CI)f | 4.00 (1.14 to 6.86) | 4.29 (1.29 to 7.29) |
Treatment group difference vs. control at Week 25, LS mean (95% CI)f | −0.29 (−4.12 to 3.54) | |
P valuef | 0.8813 | |
BICR = blinded independent central review; CI = confidence interval; cLDA = constrained longitudinal data analysis; CPS = combined positive score; CRT = chemoradiotherapy; EFS = even-free survival; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; HR = hazard ratio; LS = least squares; OS = overall survival; PRO = patient-reported outcome; RT = radiotherapy; SD = standard deviation; SOC = standard of care; sSAP = supplementary statistical analysis plan; TPS = tumour proportion score.
Note: Data presented in this table were based on analyses at clinical cut-off date of July 25, 2024.
aFrom product-limit (Kaplan-Meier) method for censored data.
bBased on Cox regression model with Efron’s method of tie handling with treatment as a covariate stratified by primary tumour site (oropharynx/oral cavity vs larynx vs hypopharynx) and tumour stage (III vs IVA) with small strata collapsed as prespecified in the sSAP.
cBased on log-rank test stratified by primary tumour site (oropharynx/oral cavity vs larynx vs hypopharynx) and tumour stage (III vs IVA) with small strata collapsed as prespecified in the sSAP. Statistical testing for this end point was adjusted for multiple comparisons using the graphical method of Maurer and Bretz (2013).50
dThe first assessment for patients was screening/randomization because at randomization patients were confirmed to not have progressive disease/recurrence. Patients who did not have any postrandomization radiographic disease assessments or on-study surgery were censored at their last assessment showing no progression which occurs at randomization. For specificity, the label “randomization” is used within the EFS analyses tables to distinguish from individuals with postrandomization assessments.
eNumber of patients in each treatment group with nonmissing assessments at the specific time point.
fBased on a cLDA model with the PRO scores as the response variable with covariates for treatment by study visit interaction, and stratification factors by primary tumour site (oropharynx/oral cavity vs. larynx vs. hypopharynx), Tumour Stage (III vs. IVA) and PD-L1 status (TPS ≥ 50% vs. TPS < 50%) as prespecified in the sSAP.
Source: Clinical Study Report for the KEYNOTE-689 study.26
Pharmacoeconomic Review
Abbreviations
AE
adverse event
BIA
budget impact analysis
CDA-AMC
Canada’s Drug Agency
EF
event-free
EFS
event-free survival
HNSCC
head and neck squamous cell carcinoma
HR
hazard ratio
HRQoL
health-related quality of life
ICER
incremental cost-effectiveness ratio
IRP
incurable recurrence or progression
LA
locally advanced
LR
local recurrence
LY
life-year
OS
overall survival
QALY
quality-adjusted life-year
SOC
standard of care
Economic Review
The objective of the economic review undertaken by Canada’s Drug Agency (CDA-AMC) is to review and critically appraise the pharmacoeconomic evidence submitted by the sponsor on the cost-effectiveness and budget impact of neoadjuvant monotherapy pembrolizumab, adjuvant pembrolizumab with standard of care (SOC) radiotherapy, and then monotherapy pembrolizumab (subsequently referred to as pembrolizumab plus SOC) compared to radiotherapy SOC (subsequently referred to as SOC) for treatment of adult patients with resectable locally advanced (LA) head and neck squamous cell carcinoma (HNSCC). In high-risk patients, neoadjuvant monotherapy pembrolizumab, adjuvant pembrolizumab with SOC radiotherapy plus cisplatin, and then monotherapy pembrolizumab was compared with SOC radiotherapy plus cisplatin.
Note: The sponsor’s application was filed on a pre-Notice of Compliance basis and the pharmacoeconomic submission and budget impact analysis (BIA) are reflective of the indication that was initially submitted to Health Canada and CDA-AMC. The updated indication is not expected to materially impact conclusions on cost-effectiveness. As the updated indication is narrower than the submitted indication, the budget impact presented is slightly overestimated.
Item | Description |
|---|---|
Drug product | Pembrolizumab (Keytruda), 100 mg/4 mL vial, solution for infusion |
Indication | For the treatment of adult patients with resectable locally advanced HNSCC whose tumours express PD-L1 (CPS ≥ 1), as determined by a validated test, as neoadjuvant treatment as monotherapy, continued as adjuvant treatment in combination with RT with or without cisplatin and then as monotherapy |
Submitted price | Pembrolizumab: $4,400.00 per 100 mg/4 mL vial |
Health Canada approval status | NOC |
Health Canada review pathway | Priority review, ORBIS |
NOC date | August 11, 2025 |
Reimbursement request | Per indication |
Sponsor | Merck Canada Inc. |
Submission history | Previously reviewed: Yes Indication: First-line treatment of metastatic or unresectable recurrent HNSCC as monotherapy, in adult patients whose tumours have PD-L1 expression (CPS ≥ 1) as determined by a validated test. First-line treatment of metastatic or unresectable recurrent HNSCC in combination with platinum and fluorouracil chemotherapy, in adult patients Recommendation date: December 22, 2020 Recommendation: Reimburse with clinical criteria and/or conditions |
CPS = combined positive score; HNSCC = head and neck squamous cell carcinoma; NOC = Notice of Compliance; RT = radiotherapy.
Pagebreak
Key Messages
Pembrolizumab is available as a solution for infusion (100 mg/4 mL vial).1 At the submitted price of $4,400.00 per 100 mg/4 mL vial, the per 28-day cycle cost of pembrolizumab is expected to be $11,733 per patient using a fixed dosage (200 mg once every 3 weeks) and $8,917 using a weight-based dosage (2 mg/kg [up to 200 mg] once every 3 weeks).1,2 When pembrolizumab is used in combination with cisplatin, the expected 28-day cost is $12,399 per patient using fixed dosage and $9,583 per patient using weight-based dosage.
Clinical efficacy in the economic analysis was derived from the KEYNOTE-689 trial (with a data cut-off date of July 25, 2024), which compared pembrolizumab plus SOC with SOC.3 Evidence submitted by the sponsor indicates that pembrolizumab plus SOC likely results in a clinically important benefit in overall survival (OS) and event-free survival (EFS) compared with SOC among adult patients with LA HNSCC.
The results of the CDA-AMC base case suggest:
Pembrolizumab plus SOC is predicted to be associated with higher costs to the health care system than SOC (incremental costs = $46,655), primarily driven by increased costs associated with drug acquisition.
Pembrolizumab plus SOC is predicted to be associated with a gain of 1.48 life-years (LYs) compared to SOC and may result in a gain of 1.23 quality-adjusted life-years (QALYs) compared to SOC.
The incremental cost-effectiveness ratio (ICER) of neoadjuvant pembrolizumab and adjuvant pembrolizumab plus SOC compared to SOC is $37,888 per QALY gained. The estimated ICER was sensitive to long-term extrapolation of EFS. Approximately 75% of the incremental QALYs relative to SOC were gained in the extrapolated period (i.e., after 282 weeks). Given the lack of evidence beyond this time point, the QALYs gained for patients receiving pembrolizumab plus SOC predicted in the CDA-AMC base case are uncertain.
CDA-AMC estimates that the budget impact of reimbursing pembrolizumab plus SOC for the treatment of resectable LA HNSCC is predicted to be approximately $72 million over the first 3 years of reimbursement compared to the amount currently spent on SOC, with an estimated expenditure of $85 million on pembrolizumab over this period. The actual budget impact of reimbursing pembrolizumab plus SOC will depend on the number of people eligible for treatment and its uptake.
Summary of the Submitted Economic Evaluation
The sponsor submitted a cost-utility analysis to estimate the cost-effectiveness of pembrolizumab plus SOC from the perspective of a public health care payer in Canada over a lifetime horizon (40 years).4 The modelled population comprised adult patients with resectable LA HNSCC, which is aligned with the Health Canada indication and the reimbursement request and was based on the participants in the KEYNOTE-689 trial.3 The sponsor’s base-case analysis included costs related to neoadjuvant or adjuvant drug acquisition and administration, initial surgery and adjuvant radiotherapy in the event-free (EF) state, salvage surgery and radiotherapy in the local recurrence (LR) state, subsequent drug acquisition and administration in the incurable recurrence or progression (IRP) state, adverse events (AEs), nondrug disease management in each health state, and terminal care.
In the sponsor’s base case, pembrolizumab plus SOC was associated with incremental costs of $61,121 and 1.38 incremental QALYs relative to SOC. This resulted in an ICER of $44,163 per QALY gained. Of the incremental benefit compared to SOC (1.33 incremental QALYs in the deterministic analysis), approximately 76% of QALYs were predicted to be accrued after the treatment duration of the KEYNOTE-689 trial (maximum trial observation period = 282 weeks). Additional information about the sponsor’s submission is summarized in Appendix 3.
CDA-AMC identified several key issues with the sponsor’s analysis (refer to Table 2; full details are provided in Appendix 4).
Table 2: Key Issues With the Sponsor’s Economic Submission
Issue | What evidence is there to inform this issue? | How was this issue addressed by CDA-AMC? | Did CDA-AMC explore uncertainty in a scenario analysis? |
|---|---|---|---|
Impact of pembrolizumab plus SOC on long-term OS is uncertain. | OS results were based on interim analyses that may have overestimated the treatment effect. The efficacy of pembrolizumab plus SOC beyond 60 months is unknown. The sponsor extrapolated KM curves in the KEYNOTE-689 study by using parametric functions to inform the model with time horizon of 40 years, introducing uncertainty to the results. | CDA-AMC could not address this issue in the base case due to lack of robust clinical evidence. | No scenario analysis was conducted. |
Long-term EFS is uncertain. | The long-term impact of pembrolizumab plus SOC on EFS is unknown in the absence of evidence and parametric modelling was required to extrapolate benefit across the model time horizon. | CDA-AMC retained the sponsor’s extrapolation in the base case because, although alternative extrapolations of EFS were available, all were considered clinically plausible. | To explore the uncertainty, CDA-AMC conducted a scenario analysis by selecting alternative distributions for transitions from EF to LR states and EF to IRP states in the pembrolizumab plus SOC arm. |
The assumption for treatment effect waning is uncertain. | No additional treatment effect waning for transitions from the EF state was assumed for pembrolizumab plus SOC, with no long-term supporting clinical evidence. Additionally, the implied EFS HRs demonstrated initial treatment waning, then a regained treatment effect for pembrolizumab, which is unlikely to be clinically plausible. | CDA-AMC applied a treatment waning rule which produced more clinically plausible long-term treatment effects. | No scenario analysis was conducted. |
The approach to model the transitions from LR and IRP is uncertain. | Constant transition probabilities over time were assumed for the LR to IRP states, LR to death, and IRP to death. Clinical experts noted the risk of death and recurrence for patients in these health states would change over time. | CDA-AMC could not address these issues in the base case due to lack of robust clinical evidence and the inflexibility of the model. | No scenario analysis was conducted. |
The censoring rule applied in the model introduced bias. | The sponsor used the EFS censoring rule (at last disease assessment) for OS, which would introduce downward bias for OS in both groups. | CDA-AMC could not address these issues in the base case due to lack of robust clinical evidence. | CDA-AMC conducted a scenario analysis by applying the OS censoring rule for both EFS and OS. |
The dosing regimen of pembrolizumab does not reflect the clinical practice in Canada. | Fixed dosing (200 mg IV once every 3 weeks) for pembrolizumab was used in the model. However, clinical experts and drug plan input noted that weight-based dosing (2 mg/kg IV once every 3 weeks) is commonly used in clinical practice. | CDA-AMC applied weigh-based dosing for pembrolizumab. | No scenario analysis was conducted. |
CDA-AMC = Canada’s Drug Agency; EF = event-free; EFS = event-free survival; HR = hazard ratio; IRP = incurable recurrence or progression; KM = Kaplan-Meier; LR = local recurrence; OS = overall survival; SOC = standard of care.
Note: Full details of the CDA-AMC identified issues are provided in Appendix 3.
CDA-AMC Assessment of Cost-Effectiveness
The CDA-AMC base case was derived by making changes to model parameter values and assumptions (refer to Table 7), in consultation with clinical experts. Detailed information about the base case is provided in Appendix 4.
Impact on Health Care Costs
Pembrolizumab plus SOC is predicted to be associated with additional health care costs compared to SOC (incremental costs = $46,655). This increase in health care spending results from drug acquisition costs associated with pembrolizumab (refer to Figure 1).
Figure 1: Impact of Pembrolizumab Plus SOC vs. SOC on Health Care Costs
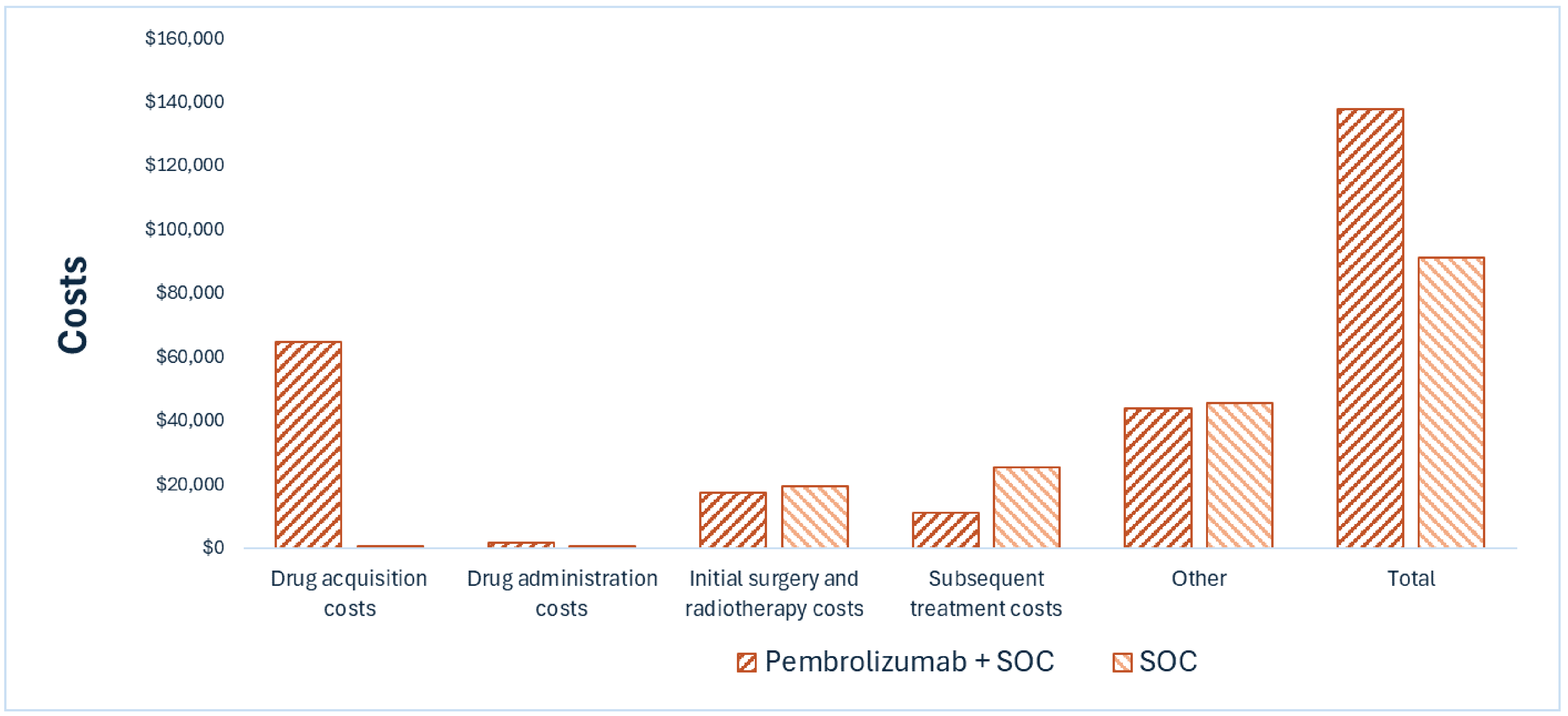
SOC = standard of care; vs. = versus.
Impact on Health
Relative to SOC, pembrolizumab plus SOC is predicted to increase the amount of time a patient remains in the EF health state by approximately 1.52 years and extend OS by 1.48 years. Considering the impact of treatment on both quality and length of life, pembrolizumab plus SOC is predicted to result in 1.23 additional QALYs per patient compared to SOC (refer to Figure 2). Approximately 75% of the predicted incremental benefit was accrued on the basis of extrapolation.
Figure 2: Impact of Pembrolizumab Plus SOC vs. SOC on Patient Health
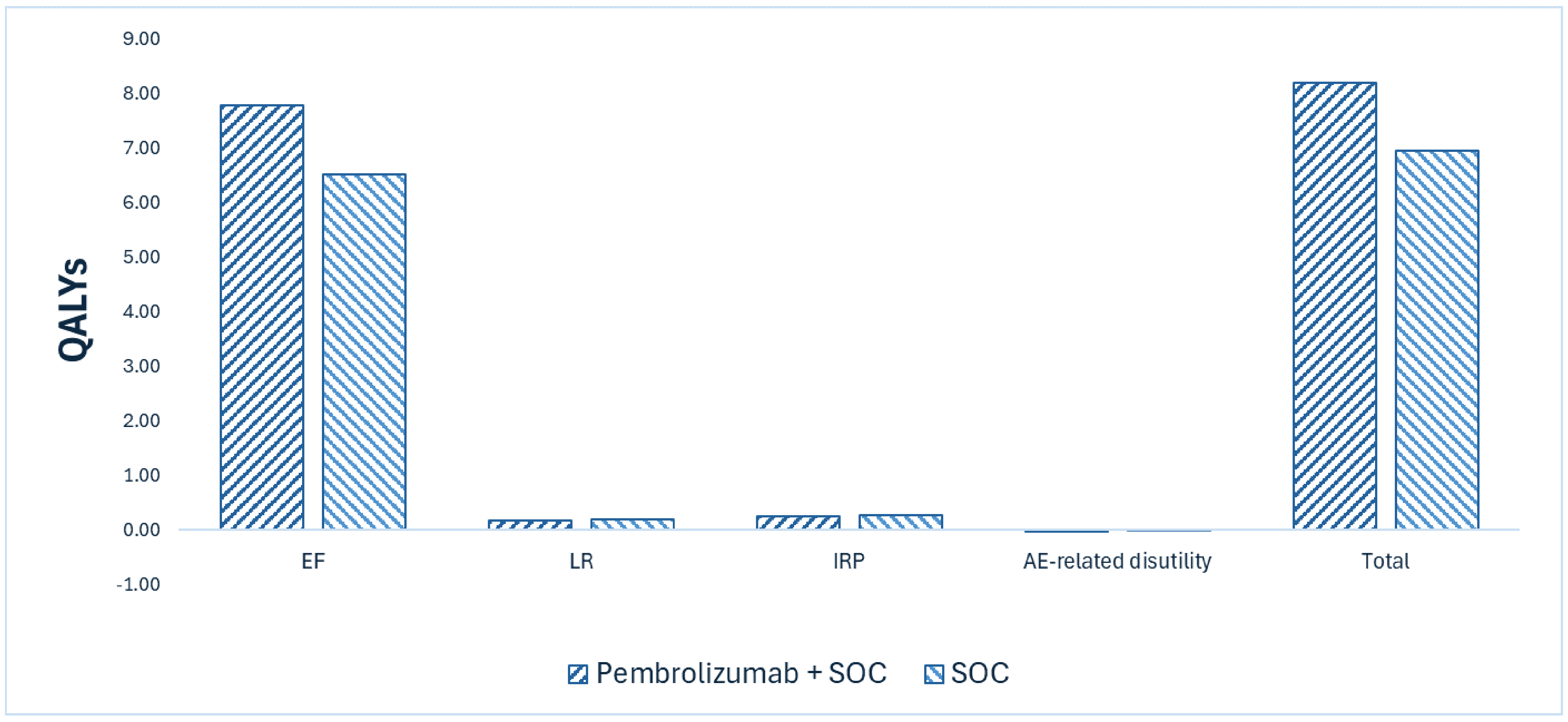
AE = adverse event; EF = event-free; IRP = incurable recurrence or progression; LR = local recurrence; QALY = quality-adjusted life-year; SOC = standard of care; vs. = versus.
Overall Results
The results of the CDA-AMC base case suggest an ICER of $37,888 per QALY gained for pembrolizumab plus SOC compared to SOC (refer to Table 3). Additional details on the CDA-AMC base case are available in Appendix 4.
Table 3: Summary of CDA-AMC Economic Evaluation Results
Drug | Total costs ($) | Total LYs | Total QALYs | ICER vs. SOC ($/QALY) |
|---|---|---|---|---|
SOC | 91,132 | 8.42 | 6.95 | Reference |
Pembrolizumab plus SOC | 137,787 | 9.90 | 8.18 | 37,888 |
CDA-AMC = Canada's Drug Agency; ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year; SOC = standard of care; vs. = versus.
Note: Publicly available list prices were used for all comparators.
Uncertainty and Sensitivity
Due to immature OS data and uncertainty regarding long-term EFS, extrapolation of transitions from the EF health state were uncertain. The impact of uncertainty on long-term EFS was explored in a scenario analysis. which increased ICER to $45,586 per QALY gained compared to SOC (refer to Table 11 in Appendix 4).
CDA-AMC also conducted a scenario analysis to explore the uncertainty pertaining to the censoring rule, which increased the ICER for pembrolizumab plus SOC to $41,890 per QALY gained compared to SOC.
Summary of the Budget Impact
The sponsor submitted a BIA to estimate the 3-year (2026–2028) budget impact of pembrolizumab plus SOC for the treatment of adult patients with resectable LA HNSCC.5 The sponsor assumed that the payer would be CDA-AMC–participating public drug plans and derived the size of the eligible population using an epidemiologic approach. The price of pembrolizumab was aligned with the price included in the sponsor’s economic evaluation, while the prices of other drugs were sourced from IQVIA DeltaPA.6 Additional information pertaining to the sponsor’s submission is provided in Appendix 5.
CDA-AMC identified a number of issues with the sponsor’s estimated budget impact and made changes to model parameters and assumptions in consultation with clinical experts to derive the CDA-AMC base case (Appendix 5). CDA-AMC estimated that 2,530 patients would be eligible for treatment with pembrolizumab over a 3-year period (year 1 = 817; year 2 = 843; year 3 = 870), of whom 1,733 are expected to receive pembrolizumab plus SOC (year 1 = 251; year 2 = 699; year 3 = 783). The estimated incremental budget impact of reimbursing pembrolizumab plus SOC is predicted to be approximately $72 million over the first 3 years, with a predicted expenditure of $85 million on pembrolizumab. The actual budget impact will depend on the number of people eligible for treatment and its uptake.
Conclusion
Based on the CDA-AMC base case, pembrolizumab plus SOC would be considered cost-effective at the submitted price if the public health care system was willing to pay at least $37,888 per QALY gained for each additional QALY gained. If the public health care system is not willing to pay that amount, a price reduction should be considered (refer to Figure 3; full details in Table 10). The estimated cost-effectiveness of pembrolizumab plus SOC compared to SOC is uncertain due to the lack of long-term clinical evidence.
The budget impact of reimbursing pembrolizumab plus SOC to the public drug plans in the first 3 years is estimated to be approximately $72 million. The 3-year expenditure on pembrolizumab is estimated to be $85 million.
Figure 3: Summary of the CDA-AMC Economic Analysis and Price Reduction
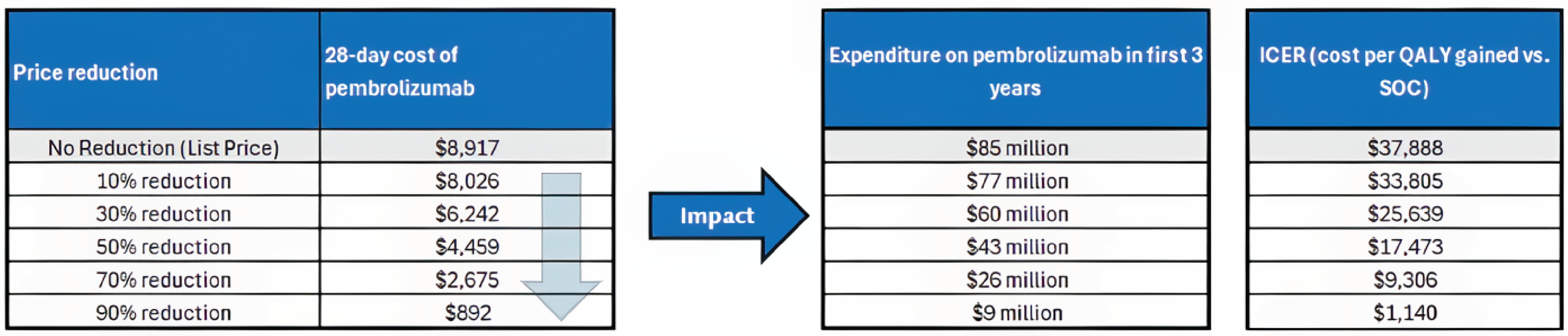
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; SOC = standard of care; vs. = versus.
Note: The per-cycle cost of pembrolizumab plus SOC includes only the drug cost and excludes the cost of the radiosensitizer used in high-risk patients. The expenditure reflects only the neoadjuvant and adjuvant costs of pembrolizumab plus SOC, as the sponsor did not report subsequent therapy costs by treatment arm.
References
1.Merck Canada Inc. Product monograph: Keytruda (pembrolizumab) solution for infusion 100 mg/4 mL vial. 2025. https://pdf.hres.ca/dpd_pm/00078714.PDF
2.INESSS. INESSS Submission Guide for Drugs, Blood System Products and Medical Devices Related to the Administration of Drugs. 2024. https://www.inesss.qc.ca/fileadmin/doc/INESSS/Inscription_medicaments/Fiches_inscription/en/Submission_guidance_document.pdf
3.Merck Sharp & Dohme L. L. C. Clinical Study Report: P689V01MK3475 (Data Cutoff Date 25-JUL-2024). A Phase III, Randomized, Open-label Study to Evaluate Pembrolizumab as Neoadjuvant Therapy and in Combination With Standard of Care as Adjuvant Therapy for Stage III-IVA Resectable Locoregionally Advanced Head and Neck Squamous Cell Carcinoma (LA HNSCC) [internal sponsor's report]. 2024 Dec 9.
4.Merck Canada Inc. Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Keytruda (pembrolizumab), solution for infusion 100 mg/4 mL vial. 2024.
5.Merck Canada Inc. Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Keytruda (pembrolizumab), solution for infusion 100 mg/4 mL vial. 2024.
6.IQVIA. DeltaPA. 2025. https://www.iqvia.com/
7.Merck, Co. Structured Literature Review on Patients Diagnosed with Locally Advanced Head and Neck Squamous Cell Carcinoma: Treatment Pattern/Guidelines Review Report. Jan 2025.
8.Hannouf MB, Zaric GS, Blanchette P, et al. Cost-effectiveness analysis of multigene expression profiling assays to guide adjuvant therapy decisions in women with invasive early-stage breast cancer. The Pharmacoeconomics Journal. 2020;20:27-46. PubMed
9.Ontario Health Insurance Plan. Schedule of Benefits: Physician Services Under the Health Insurance Act (Feb 20, 2024 (effective Apr 1, 2024)). https://www.ontario.ca/files/2024-08/moh-schedule-benefit-2024-08-30.pdf
10.Mittmann NLN, Cheng SY, Seung SJ, et al. Health system costs for cancer medications and radiation treatment in Ontario for the 4 most common cancers: a retrospective cohort study. Canadian Medical Association Open Access Journal. 2020;1(8):E191-8. PubMed
11.Ministry of Health and Long-Term Care. Ontario Case Costing Initiative (OCCI) Acute Inpatient Database for fiscal year 2016/17.
12.Canadian Institute for Health Information. Patient Cost Estimator Results. Ottawa, ON: CIHI; 2024. https://www.cihi.ca/en/patient-cost-estimator
13.National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology: Head and Neck Cancers. Version 3.2024.
14.Enomoto LM, Schaefer EW, Goldenberg D, et al. The Cost of Hospice Services in Terminally Ill Patients With Head and Neck Cancer. JAMA Otolaryngol Head Neck Surg. 2015;14(12):1066-74. PubMed
15.Eggermont AMM, Blank CU, Mandalà M, et al. Adjuvant pembrolizumab versus placebo in resected stage III melanoma (EORTC 1325-MG/KEYNOTE-054): distant metastasis-free survival results from a double-blind, randomised, controlled, phase 3 trial. The Lancet Oncology. 2021;22(5):643-654. PubMed
16.Schmid P, Cortes J, Dent R, et al. Pembrolizumab or placebo + chemotherapy followed by pembrolizumab or placebo for early-stage triple-negative breast cancer: Updated event-free survival results from the phase 3 KEYNOTE-522 study. presented at: European Society for Medical Oncology (ESMO) Congress; 2023; Madrid, Spain.
17.Schmid P, Cortes J, Dent R, et al. Event-free Survival with Pembrolizumab in Early Triple-Negative Breast Cancer. N Engl J Med. 2022;386(6):556-567. doi:10.1056/NEJMoa2112651 PubMed
18.Merck S, Dohme C. Protocol: A Phase III, Randomized, Open-label Study to Evaluate Pembrolizumab as Neoadjuvant Therapy and in Combination With Standard of Care as Adjuvant Therapy for Stage III-IVA Resectable Locoregionally Advanced Head and Neck Squamous Cell Carcinoma (KEYNOTE-689). 2023.
19.CDA-AMC. Pembrolizumab (Keytruda) for HNSCC. 2020. https://www.cda-amc.ca/pembrolizumab-keytruda-hnscc-details
20.pan-Canadian Pharmaceutical Alliance. Keytruda (pembrolizumab). 2021. https://www.pcpacanada.ca/negotiation/21386
21.Canadian Cancer Statistics Advisory Committee in collaboration with the Canadian Cancer S, Statistics C, and the Public Health Agency of C. Canadian Cancer Statistics 2024. Toronto, ON: Canadian Cancer Society. 2024. Accessed July 17, 2024. https://cdn.cancer.ca/-/media/files/research/cancer-statistics/2024-statistics/2024-cmaj/2024_cancer-specific-stats.pdf
22.Byrne K, Hallworth P, Monfared AAT, Moshyk A, Shaw JW. Real-world systemic therapy treatment patterns for squamous cell carcinoma of the head and neck in Canada. Curr Oncol. 2019;26(2):e167-e174. doi:10.3747/co.26.3946 PubMed
23.Drug I. ONCO-CAPPS: 1L NSCLC New Start Treatment Dynamics Q4 2023. 2024.
24.Oracle Life Sciences Cancer M. Treatment Architecture Head and Neck Cancer, United-States. 2024.
Appendix 1: Cost Comparison Table
Please note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical experts and CDA-AMC–participating public drug plans. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 4: Cost Comparison for the Treatment of Adult Patients With Resectable LA HNSCC
Treatment | Strength and/or concentration | Form | Price ($) | Recommended dosage | Average daily cost ($) | Average 28‑day cost ($) |
|---|---|---|---|---|---|---|
Neoadjuvant pembrolizumab and adjuvant pembrolizumab plus SOC | ||||||
Pembrolizumab (Keytruda) | 100 mg/4 mL | 4 mL vial | 4,400.0000a | Fixed dosage: 200 mg once every 3 weeks, or 400 mg once every 6 weeks | 419.05 | 11,733 |
Weight-based dosage: 2 mg/kg (up to 200 mg) once every 3 weeks or 4 mg/kg (up to 400 mg) once every 6 weeksb | 318.48 | 8,917 | ||||
Cisplatin | 1 mg/mL | 50 mL vial 100 mL vial | 135.0000 270.0000 | 100 mg/m2 IV every 3 weeks for up to 3 cycles | 23.79 | 666 |
Pembrolizumab (fixed dosage, neoadjuvant therapy or adjuvant therapy for low-risk patientsc) | 419.05 | 11,733 | ||||
Pembrolizumab (weight-based dosage, neoadjuvant therapy or adjuvant therapy for low-risk patientsc) | 318.48 | 8,917 | ||||
Pembrolizumab plus cisplatin (fixed dosage, adjuvant therapy for high-risk patientsd) | 442.84 | 12,399 | ||||
Pembrolizumab plus cisplatin (weight-based dosage, adjuvant therapy for high-risk patientsd) | 342.26 | 9,583 | ||||
SOC (adjuvant therapy) | ||||||
Cisplatin | 1 mg/mL | 50 mL vial 100 mL vial | 135.0000 270.0000 | 100 mg/m2 IV once every 3 weeks up to 3 cycles | 23.79 | 666 |
LA = locally advanced; HNSCC = head and neck squamous cell carcinoma; SOC = standard of care.
aSponsor’s submitted price.4
bInput from clinical experts and participating drug plans indicated that weight-based dosage may be used for pembrolizumab. Weight-based dosage assumes a mean weight of 76 kg2 and vial sharing. BSA based dosed assumed a BSA of 1.85 m2 and vial sharing.2
cLow risk is defined by the absence of extranodal extension and/or positive margins of resection.
dHigh risk is defined by the presence of extranodal extension and/or positive margins of resection.
Note: All prices are from IQVIA DeltaPA (accessed June 2025),6 unless otherwise indicated, and do not include dispensing fees. Wastage was not included in costs. Recommended dosages are based on Cancer Care Ontario monographs, unless otherwise indicated.
Appendix 2: Input Relevant to the Economic Review
Please note that this appendix has not been copy-edited.
This section is a summary of the input received from the patient groups, clinician groups, and drug plans that participated in the CDA-AMC review process.
Patient input was received from Canadian Cancer Survivor Network and Life-Saving Therapies Network, through an online survey, with all respondents based in Canada. Respondents included 1 patient currently in treatment, 2 caregivers of individuals with head and neck cancer, and 5 survivors, all residing in Canada. Patients reported difficult experiences with current treatments, including the harsh side effects of radiation, such as fatigue, and the toxicities of cisplatin, such as nausea. Patients noted a significant impact on self-esteem following surgery, due to disfigurement or functional impairments like speech difficulties. A few respondents had direct experience with pembrolizumab, describing it as less intense than radiation or chemotherapy and generally more tolerable. Side effects such as skin rashes and mild discomfort were noted among patients who received pembrolizumab, however, patients noted that these did not significantly interfere with daily functioning.
Clinician group input was received from Ontario Health (Cancer Care Ontario) Head and Neck Cancer Drug Advisory. The current treatment pathway for patients with LA HNSCC was noted to be upfront surgery, followed by adjuvant radiation and, for high-risk patients (positive margins and/or extranodal extension), concurrent cisplatin-based chemotherapy. Despite this approach, recurrence rates remain high, and many patients experience lasting toxicities from treatments such as radiation and chemotherapy, which can affect quality of life. Clinicians highlighted the need for therapies that improve oncologic outcomes (relapse-free survival and OS) while reducing treatment-related harm. Clinicians raised key considerations for treatment with pembrolizumab, including the need for biomarker-based patient selection, the risk of disease progression during before surgery, and whether response to neoadjuvant pembrolizumab could downstage patients from high to low risk. Clinicians also noted that the adjuvant component may increase clinical workload and resource needs (e.g., administration costs).
Input from CDA-AMC–participating drug plans noted that alternative treatments for high-risk patients include carboplatin with 5-fluorouracil or cetuximab concurrent with radiotherapy, and hyperfractionated or accelerated radiotherapy without chemotherapy. Drug plans also noted that most jurisdictions use weight-based dosing up to a cap for pembrolizumab instead of fixed dose. Plans raised questions about patient eligibility for treatment with pembrolizumab, including among those with Eastern Cooperative Oncology Group less than 1, those unable to proceed to surgery, and those with disease progression or incomplete treatment. Drug plans also had questions regarding whether patients would be eligible for re-treatment after recurrence and for previously treated patients excluded from the KEYNOTE-689 study.
Several of these concerns were addressed in the sponsor’s model:
The impact of disease and the side effects of treatments (i.e., radiotherapy, surgery, pembrolizumab, chemotherapy) on patient’s quality of life was captured with utility values.
The impact of neoadjuvant pembrolizumab on proportion of high-risk patients in adjuvant therapy was captured in the model.
The impact of treatment on EFS and OS were included in the model.
Administration costs of receiving pembrolizumab in neoadjuvant/adjuvant therapy were included.
CDA-AMC addressed some of these concerns as follows:
The weight-based dosing was applied in CDA-AMC base case.
CDA-AMC was unable to address the following concerns:
The impact of alternative chemotherapies other than cisplatin for high-risk patients on clinical benefits and costs is unknown due to the lack of clinical evidence.
Based on patient eligibility criteria of the KEYNOTE-689 study, the efficacy and safety of pembrolizumab in patients with poor performance status, and the impact of treatment interruptions on study outcomes, is unknown due to the lack of clinical evidence.
Appendix 3: Summary of the Sponsor’s Submission
Please note that this appendix has not been copy-edited.
Summary of the Sponsor’s Economic Evaluation
For the pharmaceutical reviews program, clinical and economic information is submitted to CDA-AMC by the sponsor. The CDA-AMC health economics team reviews the submitted economic information and appraises the information in collaboration with clinical experts and the clinical review team to evaluate key assumptions, influential parameters, and the overall rigour of the economic submission. Based on what the team learns through this process, adjustments may be made to the sponsor’s model to produce the CDA-AMC base case. The CDA-AMC base case represents the team’s current understanding of the clinical condition, clinical evidence currently available, and best interpretation of the economic evidence based on the information provided.
For the review of neoadjuvant pembrolizumab and adjuvant pembrolizumab plus SOC, the sponsor provided a cost-utility analysis and a BIA. The sponsor’s economic submission is summarized in Table 5.
Table 5: Key Components of the Sponsor’s Economic Evaluation
Component | Description |
|---|---|
Treatment information | |
Drug under review | Pembrolizumab (Keytruda), IV solution (100 mg/4 mL) |
Submitted price of drug under review | Pembrolizumab: $4,400.00 per 100 mg/4 mL vial |
Regimen | Pembrolizumab: 200 mg IV once every 3 weeks for up to 2 cycles as neoadjuvant therapy; 200 mg IV once every 3 weeks for up to 15 cycles as adjuvant therapy after surgery; For high-risk patients, adjuvant pembrolizumab is given in combination with cisplatin 100 mg/m2 IV once every 3 weeks for up to 3 cycles. |
28-day cost of drug under review | $11,733 for pembrolizumab neoadjuvant and monotherapy treatment (fixed dose) $12,399 for high-risk patients for adjuvant pembrolizumab plus cisplatin (fixed dose) |
Model information | |
Type of economic evaluation | Cost-utility analysis Markov model |
Treatment | Neoadjuvant monotherapy pembrolizumab followed by adjuvant pembrolizumab with SOC radiotherapy and then monotherapy pembrolizumab. High-risk patients only: neoadjuvant monotherapy pembrolizumab followed by adjuvant pembrolizumab in combination with up to 3 cycles of cisplatin with SOC radiotherapy and then monotherapy pembrolizumab |
Included comparator | SOC radiotherapy alone; No neoadjuvant therapy High-risk patients only: SOC radiotherapy plus up to 3 cycles of cisplatin; No neoadjuvant therapy |
Perspective | Publicly funded health care payer perspective |
Time horizon | Lifetime (40 years) |
Cycle length | 1 week |
Modelled population | Adult patients (ages 18 years or older) who have newly diagnosed, treatment-naive resectable LA HNSCC |
Characteristics of modelled population | Derived from the KEYNOTE-689 trial (mean age = 60 years; sex = 21.1% female; baseline weight = 73 kg, BSA = 1.8 m2) |
Model health states |
For additional information, refer to Model Structure |
Data sources | |
Comparative efficacy |
|
Natural history and/or clinical pathway |
|
|
|
|
|
Summary of the submitted results | |
Base case results | ICER = $44,163 per QALY gained (incremental costs = $61,121; incremental QALYs = 1.38) |
Scenario analysis results |
|
AE = adverse event; BSA = body surface area; CIHI = Canadian Institute for Health Information; EF = event-free; HNSCC = head and neck squamous cell carcinoma; ICER = incremental cost-effectiveness ratio; IRP = incurable recurrence or progression; KM = Kaplan-Meier; LA = locally advanced; LR = local recurrence; OHIP = Ontario Health Insurance Plan; QALY = quality-adjusted life-year; SOC = standard of care; ToT = time-on-treatment.
Model Structure
The sponsor submitted a Markov model with 4 health states: EF, LR, IRP, and death. Two types of recurrence (either LR or IRP) were expected to have different implications for patients’ prognosis, and therefore result in different health outcomes and costs. All patients enter the model in the EF state at the start of neoadjuvant therapy.5 From the EF state, patients may remain EF or transition to LR, IRP, or death, depending on treatment effectiveness and background mortality. Cause specific transitions from the EF state were based on parametric models fitted to trial data from the KEYNOTE-689 study with each specific type of EF transition being treated as a censoring event (for e.g., to estimate EF to IRP, all patients who experienced LR or death before IRP were censored). Upon transition to the LR state may receive salvage surgery or radiotherapy and either remain in this state or progress to IRP or death, depending on treatment response and disease progression. Patients in the IRP state are assumed to receive first-line therapy for recurrent/metastatic HNSCC, with the possibility of receiving second-line treatment, and either remain in the IRP state or transition to death. The risk of death from IRP was determined by parametric survival models fitted to treatment arm–specific patient-level data from the KEYNOTE-689 study. The distribution of individuals across health states is used in conjunction with state-specific costs and utility values to estimate LYs, QALYs, and costs. A figure of the sponsor’s model structure is available in Appendix 3 (Figure 4).
Table 6: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER vs. ($/QALY) |
|---|---|---|---|---|---|
SOC | 96,867 | Reference | 6.95 | Reference | Reference |
Neoadjuvant pembrolizumab and adjuvant pembrolizumab plus SOC | 157,988 | 61,121 | 8.33 | 1.38 | 44,163 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; SOC = standard of care; vs. = versus.
Figure 5: Implied EFS HRs — Sponsor’s Base Case
EFS = event-free survival; HR = hazard ratio.
Source: Sponsor’s pharmacoeconomic submission.4
Appendix 4: Additional Details of CDA-AMC Reanalyses
Please note that this appendix has not been copy-edited.
Clinical Data in the Economic Model
The clinical review by CDA-AMC found that pembrolizumab plus SOC likely results in a clinically important increase in OS and EFS at 12 months when compared to SOC. Pembrolizumab plus SOC may result in clinically important improvement in OS compared to SOC at 60 months, but little to no clinically important difference in EFS compared to SOC at 48 months. The CDA-AMC clinical review also found that pembrolizumab plus SOC may result in little to no clinically important difference in health-related quality of life (HRQoL) compared to SOC at post adjuvant chemoradiotherapy or radiotherapy (RT), and it likely results in little to no clinically important difference in grade 3 to grade 5 AEs compared to SOC. The clinical review team noted that the OS results at longer follow-up become less certain. The EFS results suggest that there may be little to no difference at 48 months but there was increased uncertainty because of imprecision and few patients remaining at risk at this time point. Moreover, there is a risk of bias due to missing outcome data, because the method of handling the missing data for HRQoL (assumed missing at random) is not plausible.
In the economic model submitted by the sponsor, transition probabilities for pembrolizumab plus SOC and SOC were extrapolated beyond the KEYNOTE-689 trial period using survival analysis. As a result, 76% of the incremental benefit associated with pembrolizumab plus SOC in the sponsor’s model was accrued beyond the observed trial follow-up, contributing to additional uncertainty in the projected LYs and QALYs gained. Because the long-term clinical efficacy of pembrolizumab plus SOC compared to SOC is uncertain, the assumptions pertaining to the extrapolated period introduce uncertainty to the overall conclusions regarding cost-effectiveness. For example, the sponsor assumed no waning of treatment effect of pembrolizumab plus SOC over the 40-year lifetime time horizon, despite a lack of evidence to support this beyond the trial period. Additionally, clinical experts consulted by CDA-AMC noted that the risk of death and recurrence for patients in LR and IRP states would change over time. Therefore, the assumption of constant transition probabilities over time for these transitions was uncertain Consequently, the CDA-AMC reanalysis is subject to uncertainty due to the lack of long-term comparative efficacy data and the assumptions underpinning long-term survival benefit for pembrolizumab plus SOC versus SOC. Taken together, these limitations contribute to uncertainty in the estimated cost-effectiveness of pembrolizumab plus SOC relative to SOC for adult patients with newly diagnosed, treatment-naive resectable LA HNSCC.
Key Issues of the Submitted Economic Evaluation
CDA-AMC identified the following key issues with the sponsor’s analysis:
Impact of pembrolizumab plus SOC on long-term OS is uncertain. The sponsor’s base case predicts a survival advantage for pembrolizumab plus SOC over SOC (incremental LYs = 1.65). However, at the data cut-off date of July 25, 2024, median OS was not reached and OS was not formally tested, indicating OS data immaturity. According to the Clinical Review of the KEYNOTE-689 trial, the certainty of the OS evidence is low at 60 months due to study limitations and imprecision, with absolute effect includes the possibility of little to no difference and a clinically important benefit. Additionally, the CDA-AMC clinical report noted that OS results were based on interim analyses, which may have overestimated the treatment effect (hazard ratio [HR]) estimates. The presence and extent of any overestimation that may have been introduced could not be determined.
Given the limited follow-up time of the KEYNOTE-689 trial (median study observation time: 166.6 weeks), parametric distributions were used to extrapolate the Kaplan-Meier data in the KEYNOTE-689 trial to inform the economic model with 40-year time horizon.
CDA-AMC could not address this limitation in base case owing to the lack of clinical data. Approximately 75% of the modelled incremental survival benefit gained by patients treated with pembrolizumab plus SOC were accrued beyond the trial’s maximum follow-up, representing model-generated outcomes rather than trial-based evidence.
The sponsor’s analysis is aligned with OS outcomes observed in the trial. When looking at predictions beyond the trial, 1 of the following scenarios would have to occur for OS curves to merge faster than what is predicted by the model:
Patients who received pembrolizumab will start to progress faster than those who did not receive pembrolizumab
The risk of death is higher in those who remain EF after receiving pembrolizumab than those who did not.
No evidence from the trial or external sources could currently support either of the scenarios occurring. Given the absence of evidence, the sponsors approach to estimating OS was deemed appropriate based on the available data.
Long-term EFS is uncertain. The sponsor used parametric modelling to extrapolate long-term EFS beyond the observable time points in the KEYNOTE-689 trial (median study observation time: 166.6 weeks) to a lifetime horizon of 40 years. Parametric functions were fitted by treatment strategy to each of the 3 transitions originating from the “EF” health state — transitioning to “LR,” “IRP,” or “Death.” As a result, the projected long-term EFS curve in each treatment strategy was influenced by the combination of distributions applied to these transitions.
The sponsor evaluated 343 potential combinations of parametric functions for transitions from the “EF” health state by fitting distributions to each treatment arm separately. The following distributions for both interventions were selected: Gompertz for the transitions from “EF” to “LR” and from “EF” to “IRP,” and generalized gamma for the transition from “EF” to “Death.”
According to the CDA-AMC clinical review, there is moderate certainty that pembrolizumab plus SOC results in a clinically important benefit in EFS compared with SOC as 12 and 36 months; however, at 48 months, there is a chance that pembrolizumab plus SOC results in little to no difference in EFS. Due to uncertain evidence at 48-months and unknown long-term effects of EFS due to the absence of evidence, the long-term extrapolated EFS is uncertain.
CDA-AMC conducted a scenario analysis by selecting generalized gamma distribution for transitions of EF to LR and EF to IRP in pembrolizumab plus SOC arm to explore the uncertainty, which showed best statistical fit among the distributions that were generated by separately fitting parametric models to each treatment arm and retained based on visual fit.
The assumption for treatment effect waning is uncertain. In the sponsor’s base case, no treatment effect waning for transitions from the EF state was explicitly assumed. According to the sponsor, the implied HRs for EFS were close to or approaching 1, which the sponsor claims was consistent with an assumption of treatment waning.4 However, as demonstrated in Figure 5, while the implied HRs in the sponsor’s base case initially approach (and exceed) 1, consistent with a treatment waning effect, there is then an additional treatment benefit for pembrolizumab plus SOC observed at around 10 years, after which there is a second episode of treatment waning. This is inconsistent with what would be expected clinically. According to clinical expert feedback received for this review it is very unlikely for patients to experience recurrence after 5 years of being in the EF state. Therefore, a renewed treatment benefit for pembrolizumab at 10 years is not proven or clinically expected.
The sponsor stated that there is clinical evidence showing sustained benefit with pembrolizumab for up to 5 years; however, this follow-up period is substantially shorter than the 40-year time horizon used in the model.15-17 Therefore, whether the approach to treatment waning chosen by the sponsor (i.e., no explicit incorporation of treatment waning beyond the selected parametric functions) will reflect that actual clinical impact of pembrolizumab beyond 5 years is unknown in the absence of clinical evidence.
In reanalyses, CDA-AMC applied a treatment effect waning from the EF state, starting in year 5 and ending in year 10, which produced more clinically plausible long-term treatment effects (Figure 6).
The censoring rule applied in the model introduced bias. The censoring rules for EFS and OS in the KEYNOTE-689 trial were as follows: for EFS, patients without any disease recurrence or death were censored at the last disease assessment; for OS, patients without death were censored at the last date that the patient was known to be alive. In the sponsor’s base case, the sponsor selected the EFS rule to align the censoring rules for EFS and OS to inform transitions from the EF state. However, either approach could introduce bias: the EFS censoring rule (used by the sponsor) may lead to downward bias in OS, while the OS censoring rule may lead to upward bias in EFS. As a result, the sponsor’s approach introduces uncertainty in the transition estimates by potentially underestimating the treatment benefit of both arms. Therefore, the approach applied by the sponsor to reconcile the censoring rules used for EFS and OS introduced uncertainty the results of the model.
CDA-AMC conducted a scenario analysis applying the alternative censoring rule, in which patients without death were censored at the last date they were known to be alive for both EFS and OS
Dosing regimen of pembrolizumab does not reflect public drug plans’ implementation strategy. Fixed dosing was assumed for pembrolizumab (200 mg IV once every 3 weeks) in sponsor’s base case, which was aligned with dosing in the KEYNOTE-689 trial. However, according to drug plan input and clinical expert feedback received by CDA-AMC indicated that a weight-based dosing approach for pembrolizumab (2 mg/kg up to 200 mg once every 3 weeks or 4 mg/kg up to 400 mg once every 6 weeks) is likely to be adopted. CDA-AMC notes that using a weight-based dosage approach would reduce drug acquisition costs for pembrolizumab.
In the CDA-AMC base case, weight-based dosing (2 mg/kg once every 3 weeks) for pembrolizumab was applied. CDA-AMC was unable to fully address this limitation, given the uncertainty around the impact of different dosage regimens on treatment efficacy.
The approach to model the transitions from LR and IRP is uncertain. To model the transitions from LR (i.e., LR to IRP and LR to death) and IRP (i.e., IRP to death), exponential models fitted to patient-level data from the KEYNOTE-689 study were used. This approach is based on the assumption that the hazard rate is constant and does not depend on time since entry into the health state. However, the clinical experts consulted by CDA-AMC noted that the hazard is expected to be higher in the first 2 years and lower after 5 years. Therefore, there is uncertainty pertaining to the sponsor’s approach to modelling the transition probabilities from LR and IRP.
CDA-AMC could not address these issues in base case due to inflexibility of the model. Given that a greater proportion of patients receiving SOC transition to LR and IRP compared with pembrolizumab plus SOC, assuming a constant rate of transition for those in those health states likely overestimates long-term mortality risk for those with LR and IRP. This is because these rates are expected to decrease over time and transitions from the KEYNOTE-689 study would represent transitions happening earlier in progression to these states. This therefore will bias the results in favour of pembrolizumab as it reduces the frequency of progression events. However, the magnitude of the bias is uncertain.
The intervention and comparator treatment regimens in the model are not reflective of clinical practice in Canada. In the intervention arm, all patients were assumed to receive pembrolizumab monotherapy for neoadjuvant therapy, which was aligned with its use in the KEYNOTE-689 trial.3 However, clinical expert input obtained by CDA-AMC noted that for the PD-L1 combined positive score (CPS)-negative patients (approximately 4% in each group) with more aggressive disease, pembrolizumab in combination with chemotherapy may be considered for neoadjuvant therapy. As patients in the pembrolizumab arm of the KEYNOTE-689 study were prohibited from using combination therapy, and given the small proportion of this subgroup in the study, the clinical efficacy of pembrolizumab-chemotherapy combination therapy in CPS-negative patients is unknown.
Additionally, the sponsor assumed that all high-risk patients would receive cisplatin as their radiosensitizer during adjuvant therapy. Based on drug plan input and clinical expert feedback, approximately 20 to 40% of high-risk patients are ineligible to receive cisplatin. These patients would receive other chemotherapy (i.e., carboplatin with or without 5-fluorouracil, cetuximab) as radiosensitizer during adjuvant therapy. As all high-risk patients in the KEYNOTE-689 trial received adjuvant cisplatin, the treatment effect of pembrolizumab in combination of other chemotherapies than cisplatin is unknown.18 Therefore, the cost-effectiveness of pembrolizumab plus SOC compared to SOC in high-risk cisplatin-ineligible patients is unknown. Taken together, the limitation of the design for the KEYNOTE-689 trial introduces uncertainty and may limit the generalizability of the economic results to real-world clinical practice.
CDA-AMC could not address these issues in base case due to lack of clinical evidence. The cost-effectiveness of pembrolizumab combination therapy in CPS-negative patients and the cost-effectiveness of alternative radiosensitizers in high-risk patients is therefore unknown.
Age-related disutility is not incorporated in the model. The sponsor’s base case did not incorporate age-related disutilities, which is a critical omission in a cohort model with a lifetime horizon for an aging population. Age-related disutilities reflect the natural decline in HRQoL due to aging, including the cumulative impact of comorbidities and reduced physical and cognitive functioning. Omitting these factors can lead to overestimation of QALYs, particularly in older patients, as utility values remain unrealistically high over time. This may result in an inflated perception of pembrolizumab’s long-term effectiveness and bias the incremental QALYs in its favour, making the treatment appear more cost-effective than it may be in practice. Incorporating age-related disutilities would allow for a more accurate assessment of patient outcomes and cost-effectiveness in a progressively aging population.
CDA-AMC could not address these issues in base case due to the inflexibility of the model.
CDA-AMC Reanalysis of the Economic Evaluation
The CDA-AMC base case was derived by making changes in model parameter values and assumptions, in consultation with clinical experts (refer to Table 7). The impact of these changes, individually and collectively, is presented in Table 8.
Table 7: Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
1. Dosing regimen of pembrolizumab | Fixed dosing (200 mg IV once every 3 weeks) | Weight-based dosing (2 mg/kg IV once every 3 weeks) |
2. Treatment effect waning of pembrolizumab | No additional treatment effect waning assumed for pembrolizumab | Assumed treatment effect waning starting in year 5 and ending in year 10 |
CDA-AMC base case (health care payer perspective) | ― | Reanalysis 1 + 2 |
CDA-AMC = Canada’s Drug Agency.
Note: CDA-AMC was unable to resolve the issues with long-term comparative efficacy, modelling approaches, treatment regimen in the model, and the age-related utility due to lack of clinical evidence and inflexibility of the model.
Table 8: Summary of the Stepped Analysis
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
Sponsor’s base case | SOC | 94,416 | 7.14 | Reference |
Pembrolizumab plus SOC | 155,903 | 8.47 | 46,246 | |
CDA-AMC reanalysis 1: dosing regimen of pembrolizumab | SOC | 88,931 | 7.14 | Reference |
Pembrolizumab plus SOC | 131,844 | 8.47 | 32,276 | |
CDA-AMC reanalysis 2: treatment effect waning of pembrolizumab | SOC | 94,416 | 7.14 | Reference |
Pembrolizumab plus SOC | 158,692 | 8.41 | 50,726 | |
CDA-AMC base case (Reanalysis 1 + 2) (deterministic) | SOC | 88,931 | 7.14 | Reference |
Pembrolizumab plus SOC | 134,612 | 8.41 | 36,050 | |
CDA-AMC base case (probabilistic) | SOC | 91,132 | 6.95 | Reference |
Pembrolizumab plus SOC | 137,787 | 8.18 | 37,888 |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; SOC = standard of care.
Note: The CDA-AMC reanalysis is based on the publicly available prices of the comparator treatments. Deterministic results are presented, unless otherwise indicated.
Table 9: Disaggregated Results of the CDA-AMC Base Case
Parameter | Pembrolizumab plus SOC | SOC |
|---|---|---|
Discounted LYs | ||
Total | 9.90 | 8.42 |
EF | 9.34 | 7.81 |
LR | 0.22 | 0.24 |
IRP | 0.34 | 0.37 |
Discounted QALYs | ||
Total | 8.18 | 6.95 |
EF | 7.78 | 6.51 |
LR | 0.17 | 0.19 |
IRP | 0.26 | 0.28 |
AE-related disutility | −0.02 | −0.02 |
Discounted costs ($) | ||
Total | 137,787 | 91,132 |
Neoadjuvant/adjuvant treatment costs | 66,109 | 1,030 |
Initial surgery and radiotherapy costs | 17,154 | 19,459 |
Subsequent treatment costs in LR state | 1,341 | 1,575 |
Subsequent treatment costs in IRP state | 9,636 | 23,623 |
AE costs | 910 | 1,377 |
Disease management costs | 21,039 | 21,898 |
Terminal care costs | 21,598 | 22,169 |
AE = adverse event; CDA-AMC = Canada’s Drug Agency; EF = event-free; IRP = incurable recurrence or progression; LR = local recurrence; LY = life-year; QALY = quality-adjusted life-year; SOC = standard of care.
Price Reduction Analysis
Table 10: Results of the Price Reduction Analysis
Price reduction | Unit drug cost ($) | Cost per 28 days ($) | ICERs for pembrolizumab plus SOC vs. SOC ($/QALY) | |
|---|---|---|---|---|
Sponsor base case | CDA-AMC base case | |||
No price reduction | 4,400a | 8,917 | 44,163 | 37,888 |
10% | 3,960 | 8,026 | 39,192 | 33,805 |
20% | 3,520 | 7,134 | 34,221 | 29,722 |
30% | 3,080 | 6,242 | 29,251 | 25,639 |
40% | 2,640 | 5,350 | 24,280 | 21,556 |
50% | 2,200 | 4,459 | 19,310 | 17,473 |
60% | 1,760 | 3,567 | 14,339 | 13,390 |
70% | 1,320 | 2,675 | 9,368 | 9,306 |
80% | 880 | 1,783 | 4,398 | 5,223 |
90% | 440 | 892 | Dominant | 1,140 |
100% | 0 | 0 | Dominant | Dominant |
CDA-AMC = Canada’s Drug Agency; SOC = standard of care; vs. = versus.
aSponsor’s submitted price for pembrolizumab.
Assessment of Uncertainty
CDA-AMC used the CDA-AMC base case to conduct scenario analyses to address uncertainty within the economic evaluation. The results are provided in Table 11.
Scenario 1: Used alternative parametric distribution (generalized gamma) for transitions of EF to LR and EF to IRP in pembrolizumab plus SOC arm, which showed best statistical fit among the distributions that were generated by separately fitting parametric models to each treatment arm and retained based on visual fit.
Scenario 2: Assumed that the censoring rule for OS in protocol of the KEYNOTE-689 trial is applied for both EFS and OS. Specifically, patients without death were censored at the last date they were known to be alive.
Table 11: Results of CDA-AMC Scenario Analyses
Analysisa | Drug | Total costs ($) | Total QALYs | ICER ($/QALYs) |
|---|---|---|---|---|
CDA-AMC base case | SOC | 91,132 | 6.95 | Reference |
Pembrolizumab + SOC | 137,787 | 8.18 | 37,888 | |
CDA-AMC scenario 1: Distribution for EF to LR and EF to IRP in pembrolizumab + SOC arm | SOC | 91,132 | 6.95 | Reference |
Pembrolizumab + SOC | 139,325 | 8.01 | 45,586 | |
CDA-AMC scenario 2: Censoring rule | SOC | 83,461 | 8.60 | Reference |
Pembrolizumab + SOC | 131,069 | 9.74 | 41,890 |
CDA-AMC = Canada’s Drug Agency; EF = event-free; ICER = incremental cost-effectiveness ratio; IRP = incurable recurrence or progression; LR = local recurrence; QALY = quality-adjusted life-year; SOC = standard of care.
aProbabilistic analyses.
Issues for Consideration
CDA-AMC has previously reviewed pembrolizumab for the first-line treatment of metastatic or unresectable recurrent HNSCC in 2 settings: (1) as monotherapy in adult patients whose tumours express PD-L1 (CPS ≥ 1), as determined by a validated test; and (2) in combination with platinum and fluorouracil chemotherapy in adult patients.19 The cost-effectiveness results of these evaluations may not be directly comparable to those in the current review, owing to differences in target population, model structure, clinical effectiveness parameters, health state utility values, and cost inputs. CDA-AMC further notes that the pan-Canadian Pharmaceutical Alliance (pCPA) concluded negotiations with a letter of intent for pembrolizumab for this indication.20 As such, pembrolizumab has a confidential negotiated price, and is currently funded by jurisdictional cancer formularies. The CDA-AMC reanalyses are based on the publicly available price of pembrolizumab, which may be different than the confidential price and may influence the results of the cost-effectiveness and budget impact analyses.
Appendix 5: Budget Impact Analysis
Please note that this appendix has not been copy-edited.
Summary of the Submitted BIA
The sponsor submitted a BIA that estimated the expected incremental budgetary impact of reimbursing neoadjuvant pembrolizumab and adjuvant pembrolizumab plus SOC for the treatment of adult patients with resectable LA HNSCC.
The BIA was conducted from the perspective of public drug plan payers over a 3-year time horizon (2026 to 2028), with 2025 as the base year. The sponsor’s estimate reflects the aggregated results from the jurisdictional provincial budgets (excluding Quebec). The sponsor estimated the eligible population using an epidemiological approach. The sponsor’s base case included drug acquisition costs. All patients receiving adjuvant therapy were assumed to be high-risk. Subsequent treatment costs were included based on therapies used in Canada, with market shares informed by the KEYNOTE-689 trial.3 The market uptake for pembrolizumab was estimated using data from internal market research. The key inputs to the BIA are documented in Table 12.
The sponsor estimated the 3-year incremental budget impact associated with reimbursing pembrolizumab plus SOC would be $101,513,522 (year 1 = $7,475,589; year 2 = $38,831,374; year 3 = $55,206,559).
Table 12: Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3 if appropriate) |
|---|---|
Target population | |
Number of incident patients with head and neck cancer | 6,35121 |
Percent with squamous cell carcinoma | 90%22 |
Percent with locally advanced HNSCC | Stage III: 29%22 Stage IVA: 17%22 |
Percent referred to medical oncologists | 63%23 |
Percent who are operable patients | 52%24 |
Systemic treatment rate by medical oncologists | 95%a |
Number of patients eligible for drug under review | 817 / 843 / 870 |
Market shares (reference scenario) | |
Pembrolizumab plus SOC | 0% / 0% / 0% |
SOC | 100% / 100% / 100% |
Market shares (new drug scenario) | |
Pembrolizumab plus SOC | 31% / 83% / 90% |
SOC | 69% / 17% / 10% |
Cost of treatment (per patient per 21-day cycle) | |
Pembrolizumab plus SOC (neoadjuvant therapy or adjuvant therapy for low-risk patients) | $8,800 |
Pembrolizumab plus SOC (adjuvant therapy for high-risk patients) | $9,286 |
SOC (cisplatin adjuvant therapy for high-risk patients) | $486 |
HNSCC = head and neck cancer incidence squamous cell carcinoma; SOC = standard of care.
aBased on assumption.
Key Issues of the Submitted BIA
CDA-AMC identified several key issues to the sponsor’s analysis that have notable implications on the results of the BIA:
Dosing regimen of pembrolizumab does not reflect the clinical practice in Canada. Fixed dosing was assumed for pembrolizumab (200 mg IV once every 3 weeks) in sponsor’s BIA. As noted in earlier section of this report, according to drug plan input and the feedback from clinical experts consulted by CDA-AMC, weight-based dosing (2 mg/kg up to 200 mg once every 3 weeks or 4 mg/kg up to 400 mg once every 6 weeks) for pembrolizumab is likely to be adopted.
CDA-AMC conducted a reanalysis base applying weight-based dosing (2 mg/kg once every 3 weeks) for pembrolizumab. CDA‑AMC explored the impact of fixed dosage for pembrolizumab (200 mg every 3 weeks) in scenario analysis.
Proportion of high-risk patients is not aligned with economic model. In the sponsor’s BIA, all patients who receive adjuvant therapy were assumed to be at high-risk, meaning 100% of patients receiving adjuvant therapy received cisplatin. However, clinical expert feedback obtained by CDA-AMC noted that some patients receiving adjuvant therapy are not high risk, which was also aligned with the KEYNOTE-689 trial. Therefore, the sponsor’s assumption does not reflect the clinical practice and is not aligned with the assumptions in sponsor’s economic model, where 29.6% in pembrolizumab plus SOC arm and 44.1% in SOC arm received adjuvant cisplatin (note this appears to be slightly different than the proportion of patients who are high risk in the KEYNOTE-689 study, which was 32.5% and 44.4% of patients who received pembrolizumab plus SOC and SOC, respectively).4
CDA-ACM conducted a reanalysis assuming the proportion of high-risk patients receiving adjuvant therapy was equal to that in the SOC arm of the economic model (i.e., 44.1%). A scenario analysis was also conducted assuming the proportion was equal to that in the pembrolizumab plus SOC arm (i.e., 29.6%).
Assumptions for market share of pembrolizumab plus SOC are uncertain. The sponsor assumed that, following reimbursement, pembrolizumab plus SOC would capture a peak market share of 90% among patients with resectable LA HNSCC, based on internal market research. Clinical expert feedback obtained by CDA-AMC noted that this could be an overestimate, as some patients may not be able to tolerate the long treatment course with pembrolizumab and therefore may not uptake the new regimen.
CDA-AMC could not address these issues in base case due to lack of robust evidence for the market uptake of after pembrolizumab is reimbursed for treatment of adult patients who have newly diagnosed, treatment-naive resectable LA HNSCC.
CDA-AMC Reanalyses of the BIA
CDA-AMC revised the sponsor’s submitted analyses by making changes in model parameter values and assumptions, in consultation with clinical experts, as outlined in Table 13.
Table 13: Revisions to the Submitted BIA
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
1. Dosing regimen of pembrolizumab | Fixed dosing (200 mg IV once every 3 weeks) | Weight-based dosing (2 mg/kg up once every 3 weeks) |
2. Proportion of patients who are high-risk | 100% | 44.1% |
CDA-AMC base case | ― | Reanalysis 1 + 2 |
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency.
The results of the CDA-AMC stepwise reanalysis are presented in summary format in Table 14 and a more detailed breakdown is presented in Table 15. In the CDA-AMC base case, the 3-year budget impact of reimbursing pembrolizumab plus SOC for the treatment of adult patients with resectable LA HNSCC as neoadjuvant treatment, continued as adjuvant treatment in combination with radiotherapy with or without platinum-containing chemotherapy and then as monotherapy was $72,260,881 (year 1 = $5,324,091; year 2 = $27,650,404; year 3 = $39,306,387).
Table 14: Summary of the Stepped Analysis of the CDA-AMC Base Case
Stepped analysis | Three-year total ($) |
|---|---|
Submitted base case | 101,513,522 |
CDA-AMC reanalysis 1 | 72,094,147 |
CDA-AMC reanalysis 2 | 101,680,256 |
CDA-AMC base case: (Reanalysis 1 + 2) | 72,260,881 |
CDA-AMC = Canada’s Drug Agency.
Note: The CDA-AMC reanalysis is based on publicly available prices of the comparator treatments.
CDA-AMC used the CDA-AMC base case to conduct scenario analyses to explore uncertainty in the estimated budget impact of reimbursing pembrolizumab plus SOC. The results are provided in Table 15.
Table 15: Disaggregated Summary of the BIA
Stepped analysis | Scenario | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|---|
Submitted base case | Reference total | 3,788,853 | 12,775,406 | 19,204,457 | 23,006,979 | 54,986,842 |
Pembrolizumab | 0 | 0 | 0 | 0 | 0 | |
Cisplatin (used with pembrolizumab) | 0 | 0 | 0 | 0 | 0 | |
SOC | 343,064 | 421,836 | 435,387 | 449,374 | 1,306,597 | |
Subsequent therapies | 3,445,789 | 12,353,570 | 18,769,070 | 22,557,605 | 53,680,245 | |
New drug total | 3,788,853 | 20,250,995 | 58,035,831 | 78,213,538 | 156,500,364 | |
Pembrolizumab | 0 | 8,092,745 | 43,907,377 | 66,945,818 | 118,945,940 | |
Cisplatin (used with pembrolizumab) | 0 | 46,340 | 213,535 | 274,297 | 534,173 | |
SOC | 343,064 | 330,206 | 99,007 | 44,937 | 474,151 | |
Subsequent therapies | 3,445,789 | 11,781,703 | 13,815,912 | 10,948,485 | 36,546,100 | |
Budget Impact | 0 | 7,475,589 | 38,831,374 | 55,206,559 | 101,513,522 | |
CDA-AMC base case | Reference total | 2,732,011 | 9,324,113 | 14,010,957 | 16,784,489 | 40,119,559 |
Pembrolizumab | 0 | 0 | 0 | 0 | 0 | |
Cisplatin (used with pembrolizumab) | 0 | 0 | 0 | 0 | 0 | |
SOC | 151,291 | 186,029 | 192,006 | 198,174 | 576,209 | |
Subsequent therapies | 2,580,720 | 9,138,084 | 13,818,951 | 16,586,315 | 39,543,350 | |
New drug total | 2,732,011 | 14,648,204 | 41,641,361 | 56,090,875 | 112,380,440 | |
Pembrolizumab | 0 | 5,759,404 | 31,247,782 | 47,643,665 | 84,650,852 | |
Cisplatin (used with pembrolizumab) | 0 | 20,436 | 94,169 | 120,965 | 235,570 | |
SOC | 151,291 | 145,621 | 43,662 | 19,817 | 209,101 | |
Subsequent therapies | 2,580,720 | 8,722,743 | 10,255,747 | 8,306,428 | 27,284,918 | |
Budget Impact | 0 | 5,324,091 | 27,630,404 | 39,306,387 | 72,260,881 | |
CDA-AMC scenario analyses | ||||||
Scenario 1: Fixed dosage of pembrolizumab | Reference total | 3,597,080 | 12,539,600 | 18,961,076 | 22,755,779 | 54,256,455 |
New drug total | 3,597,080 | 20,040,505 | 57,861,120 | 78,035,086 | 155,936,711 | |
Budget Impact | 0 | 7,500,905 | 38,900,044 | 55,279,307 | 101,680,256 | |
Scenario 2: Alternative proportion of high-risk patients | Reference total | 2,682,267 | 9,262,947 | 13,947,826 | 16,719,329 | 39,930,102 |
New drug total | 2,682,267 | 14,593,605 | 41,596,042 | 56,044,586 | 112,234,233 | |
Budget Impact | 0 | 5,330,658 | 27,648,216 | 39,325,257 | 72,304,131 | |
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency; SOC = standard of care.
Note: The CDA-AMC reanalysis is based on the publicly available prices of the comparator treatments.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.