Drugs, Health Technologies, Health Systems
Reimbursement Review
Glofitamab (Columvi)
Sponsor: Hoffmann-La Roche Limited
Therapeutic area: Relapsed or refractory diffuse large B-cell lymphoma
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
AE
adverse event
ASCT
autologous stem cell transplant
CAR
chimeric antigen receptor
CDA-AMC
Canada’s Drug Agency
CI
confidence interval
CR
complete response
CRS
cytokine release syndrome
DAC
Drug Advisory Committee
DLBCL
diffuse large B-cell lymphoma
DOR
duration of objective response
ECOG PS
Eastern Cooperative Oncology Group Performance Status
EORTC QLQ-C30
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30
ESS
effective sample size
FACT-Lym
Functional Assessment of Cancer Therapy–Lymphoma
Glofit-GemOx
glofitamab in combination with gemcitabine and oxaliplatin
GRADE
Grading of Recommendations Assessment, Development, and Evaluation
HGBCL
high-grade B-cell lymphoma
HR
hazard ratio
HRQoL
health-related quality of life
ICANS
immun effector cell-associated neurotoxicity syndrome
iDMC
independent data monitoring committee
IPD
individual patient-level data
IPI
International Prognostic Index
IPTW
inverse probability of treatment weighting
IRC
independent review committee
IRR
infusion-related reaction
ITC
indirect treatment comparison
ITT
intention to treat
LBCL
large B-cell lymphoma
LC
Lymphoma Canada
LDH
lactate dehydrogenase
LymS
lymphoma subscale
MAIC
matching adjusted indirect comparison
NALT
new antilymphoma therapy
NE
not estimable
NHL
non-Hodgkin lymphoma
NOS
not otherwise specified
OH (CCO)
Ontario Health (Cancer Care Ontario)
OR
odds ratio
ORR
objective response rate
OS
overall survival
PFS
progression-free survival
PMBCL
primary mediastinal B-cell lymphoma
pola-BR
polatuzumab vedotin plus bendamustine and rituximab
PSA
propensity score analysis
QoL
quality of life
r/r
relapsed or refractory
R-CHOP
rituximab in combination with cyclophosphamide, doxorubicin, vincristine, and prednisone
RCT
randomized controlled trial
R-GemOx
rituximab in combination with gemcitabine and oxaliplatin
R-GDP
rituximab in combination with gemcitabine, dexamethasone, and cisplatin
SAE
serious adverse event
SD
standard deviation
tFL
transformed follicular lymphoma
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Glofitamab, 1 mg/mL, concentrate for solution for IV infusion |
Sponsor | Hoffmann-La Roche Limited |
Indication | COLUMVI (glofitamab for injection), in combination with gemcitabine and oxaliplatin, is indicated for the treatment of adult patients with relapsed or refractory diffuse large B‑cell lymphoma not otherwise specified (DLBCL NOS) who are not candidates for autologous stem cell transplant (ASCT). |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | August 13, 2025 |
Recommended dose | Recommended dose and dosage adjustments for notable subpopulations per the product monograph Cycle 1:
Cycle 2:
Cycles 3 to 8:
Cycles 9 to 12:
|
NOC = Notice of Compliance.
Note: Each treatment cycle is 21 days.
aCycles 2 to 8: Glofitamab, gemcitabine, and oxaliplatin can be administered in any order. Gemcitabine and oxaliplatin may be given on day 1 or 2.
bInfusion time may be shortened to 2 hours at the discretion of the treating physician, if the previous infusion was well tolerated. If the patient experienced cytokine release syndrome with the previous dose, the duration of infusion should be maintained at 4 hours.
Introduction
Diffuse large B-cell lymphoma (DLBCL) is the most common type of non-Hodgkin lymphoma (NHL) in North America, accounting for approximately 30% to 40% of all NHL cases in Canada. DLBCLs are a heterogeneous group of aggressive B-cell malignancies that differ in clinical presentation, molecular features, prognosis, and treatment options. The disease often presents with rapidly enlarging lymph nodes, elevated serum lactate dehydrogenase (LDH) and systemic “B symptoms,” such as fever, night sweats, and unexplained weight loss. In 2024, it was estimated that approximately 11,700 new cases of NHL would occur in Canada. Up to 40% of patients with DLBCL will have refractory or relapsed (r/r) disease, with most patients experiencing rapid disease progression. While the 5-year overall survival (OS) rate for DLBCL is approximately 60% to 70%, it is estimated to be as low as 6.6 months in patients with r/r DLBCL, particularly those who are ineligible for autologous stem cell transplant (ASCT) or who have experienced relapse after second-line therapy.
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of glofitamab (1 mg/mL concentrate solution for IV infusion) in combination with gemcitabine and oxaliplatin in the treatment of adult patients with r/r DLBCL not otherwise specified (NOS). In February 2024, glofitamab received a recommendation to reimburse with clinical criteria and/or conditions from Canada’s Drug Agency (CDA-AMC) for the treatment of adult patients with r/r DLBCL NOS, transformed follicular lymphoma (tFL), or primary mediastinal B-cell lymphoma (PMBCL) who have received 2 or more lines of systemic therapy and are ineligible to receive or cannot receive chimeric antigen receptor (CAR) T-cell therapy or have previously received CAR T-cell therapy.
Perspectives of Patients, Clinicians, and Drug Programs
The information in this section is a summary of input provided by the patient and clinician groups who responded to a call for input and from the clinical experts consulted by for the purpose of this review.
Patient Input
One patient group, Lymphoma Canada (LC) provided input for this submission. LC is a national Canadian registered charity whose mission is to empower patients and the lymphoma community through education, support, advocacy, and research. Data for this submission were gathered from an online anonymous patient survey conducted from February 24 to March 23, 2025. A total of 41 responses were collected and used to identify the main areas of concern for patients with DLBCL. Two patients, a 45-year-old and 54-year-old, living in the US had experience with glofitamab in combination with gemcitabine and oxaliplatin (Glofit-GemOx) for DLBCL.
Of the respondents who answered the demographic questions, 80% lived in Canada, 40% were between the ages of 65 and 74, and 60% were male. Six respondents were diagnosed 1 to 2 years previously, 5 respondents were diagnosed less than a year previously, 5 were diagnosed 3 to 5 years previously, and 7 respondents were diagnosed 5 to 10 years previously. The subtype of large B-cell lymphoma (LBCL) was identified as DLBCL NOS (n = 13); DLBCL arising from follicular lymphoma (n = 7); and DLBCL arising from PMBCL, Richter’s transformation, and germinal centre B-cell–like lymphoma (1 respondent each).
The respondents highlighted the following symptoms impacting quality of life (QoL): fatigue or lack of energy, neutropenia, enlarged lymph nodes, night sweats, shortness of breath, bodily aches and pains, reduced appetite, and weight loss. Regarding psychosocial impacts, respondents (n = 23) noted anxiety, fear of progression, difficulty sleeping, stress of diagnosis, problems concentrating, and inability to continue daily activities. On a scale of 1 (no impact) to 5 (significant impact), respondents assigned the ability to work, school and volunteer, ability to contribute financially to household expenses, and ability to travel a value of 4 or higher. Respondents reported that fatigue, nausea, vomiting, infections, a reduced white blood cell count, bone pain, neutropenia, and hair loss were the most difficult side effects of treatment to tolerate.
Among 11 respondents, 36% indicated they had received 1 line of treatment, another 36% reported receiving 2 lines of treatment, and 27% reported receiving 3 or more lines of treatment. The majority of the respondents (63%) experienced a relapse and needed treatment past the front-line setting. In the front-line setting, 10 respondents indicated that they received rituximab in combination with cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) and 1 received R-CHOP and rituximab in combination with etoposide, prednisone, vincristine, cyclophosphamide, and doxorubicin. In the second-line setting, 2 respondents each received rituximab, cisplatin, dexamethasone and gemcitabine (R-GDP) and salvage therapy plus ASCT, 3 received radiation, and 4 participated in a clinical trial. In the third-line setting, 3 respondents received CAR T-cell therapy, 1 received polatuzumab vedotin plus bendamustine and rituximab (pola-BR), 2 received glofitamab, and 5 participated in a clinical trial. The patient group noted that 54% of respondents indicated they were very satisfied with their front-line treatment options compared to 20% of respondents in the r/r setting. Eight of the 11 respondents had no or little difficulty accessing treatment, while 3 respondents had some difficulty. Common financial implications reported by respondents for treatment for DLBCL were absence from work, drug costs, supplementary drug costs for side effects and travelling costs. Respondents provided positive feedback on the care and expertise provided by the health care team treating them.
Eleven respondents considered longer disease remission, longer survival, control of disease symptoms, normalization of blood counts, and improved QoL that allowed them to perform daily activities as important outcomes for new treatments. Seven respondents indicated they were willing to tolerate side effects to access new treatments. Six respondents indicated that treatment choice based on knowledge of side effects and expected outcomes was an important factor.
Two respondents indicated they had experience with Glofit-GemOx. One respondent was treated less than 6 months previously, while the other was treated 1 to 2 years previously. Respondents indicated they were in remission upon receiving the therapy. Side effects such as decreased appetite, nausea, vomiting, and fatigue were noted by both respondents. One respondent also reported experiencing cytokine release syndrome (CRS), fever, neutropenia, and a low platelet count as side effects. Both respondents experienced financial impacts due to absence from work and the need to travel. The patient group noted that the respondents rated their overall experience with the therapy as good and satisfactory, and that they would recommend the therapy to other patients with DLBCL.
Clinician Input
Input From Clinical Experts Consulted by CDA-AMC for This Review
Unmet Needs
The clinical experts consulted for this review noted that the goal of treatment in transplant-ineligible patients with second-line and beyond r/r DLBCL is generally palliative despite the treatments available today, requiring access to new treatment options (e.g., new immunotherapies and cellular therapies). In the absence of a cure, the goal is to prevent disease progression, prolong life, and improve QoL, determined by a combination of validated QoL scales, patient reports, and clinical assessments. Furthermore, the side effects of treatment, including cytopenia, infections, and adverse events (AEs) of special interest for immunotherapies (e.g., CRS and immune effector cell-associated neurotoxicity syndrome [ICANS]) are serious conditions. Last, health care system resources are required to monitor new interventions and the risk of serious adverse events (SAEs) (e.g., CRS and ICANS) can be a burden on the health care system; requiring additional inpatient capacity to deliver and monitor patients on these treatments.
Place in Therapy
According to the clinical experts, because glofitamab works through mechanisms that are different from the current standard cytotoxic drugs, it is expected to be active even in very resistant DLBCLs. Furthermore, its combination with gemcitabine and oxaliplatin is believed to enhance T-cell activity, and is likely to be a promising option in patients with r/r DLBCL.
As patients have more courses of cytotoxic drugs (e.g., in the second line and beyond and the third-line and beyond), their immune system may not respond as well to combinations such as Glofit-GemOx. As such, it is likely that this combination will be more active earlier in the disease course as immune therapies in general require an intact immune system. Even so, it is not yet clear what the efficacy and safety of gemcitabine and oxaliplatin compared with other types of immunotherapies used in practice, such as cellular therapies (e.g., CAR T cell), or what the longer-term outcomes for this combination will be.
With regard to toxicity, 1 clinical expert consulted for this review stated that, while Glofit-GemOx is considered to have a worse safety profile than competing treatments that contain noncytotoxic agents (e.g., polatuzumab vedotin, tafasitamab, lenalidomide), the anticipated efficacy probably favours the use of Glofit-GemOx. There are still concerns about the toxicity, potential for CRS, ICANS, and treatment-related deaths from this combination. The clinical experts consulted for this review anticipated that the future evolution of treatment with Glofit-GemOx may include a reduced need for hospitalization when administering and monitoring these drugs, which would minimize health disparities between provinces and prove advantageous for rural patients who do not live in an urban environment.
Patient Population
According to the clinical experts, the patient population included in the STARGLO trial was relatively young (e.g., aged approximately 65 years) and in good general health (e.g., an Eastern Cooperative Oncology Performance Status [ECOG PS] of 0 to 2). The clinical experts stated that a patient with a poor functional status may not be able to tolerate the toxicities. In addition, the trial excluded important DLBCL groups (e.g., patients who had transformed disease and those with double-hit genetics), and patients with aggressive forms of DLBCL (e.g., those with a high International Prognostic Index [IPI] score, bulky disease, and non–germinal centre subtypes) had disease that did not respond as well to therapy. Furthermore, in the STARGLO trial, 1 of the most common reasons for transplant ineligibility was patient choice. According to the clinical experts, this is not expected to be a common reason among patients in Canada who are otherwise eligible.
Assessing the Response Treatment
According to the clinical experts, traditional measures of response (e.g., progression-free survival [PFS], OS, and duration of response [DOR]) can be used to determine the effectiveness of this treatment. Improvement in QoL and disease-related symptoms would be important patient-reported outcomes to assess as well.
Discontinuing Treatment
According to the clinical experts, disease progression and/or permanent toxicity (e.g., significantly impaired renal dysfunction, worsening neuropathy, and other toxicities with damage to major organs) would be causes for discontinuing treatment.
Prescribing Considerations
The clinical experts noted that treatment would require inpatient admission so that detailed and specialized management algorithms can be followed, similar to other T-cell–engaging therapies. This includes premedications, careful monitoring of the step-up dose where the risk of CRS or ICANS is greatest, and ensuring emergency facilities are available for acute complications. Managing CRS requires experienced care in the proper settings, thus restricting the use and availability of glofitamab in Canada.
Clinician Group Input
The following is a summary of the input received from clinician groups, and not the clinical experts consulted by CDA-AMC for this review.
Two clinician groups, LC and the Ontario Health (Cancer Care Ontario) (OH [CCO]) Hematology Cancer Drug Advisory Committee (DAC) provided input for this submission. Information was gathered from a literature search of published clinical trials of Glofit-GemOx as well as other treatments for r/r DLBCL. Four clinicians provided their input to LC. OH-CCO gathered information via a teleconference and included the input of 7 clinicians.
The LC clinician group noted that, for eligible patients, second-line treatment may include CAR T-cell therapy, if they experience a relapse within 12 months of R-CHOP. Chemoimmunotherapy and ASCT may be options if a patient experiences a relapse more than 12 months after R-CHOP. Those with disease that does not respond to second-line chemoimmunotherapy or who experience relapse after ASCT may receive third-line CAR T-cell therapy.
Both clinician groups noted that the goals of therapy were to prolong survival and improve QoL. In addition, the OH-CCO Hematology Cancer DAC noted an improvement in disease response and disease-related symptoms, and the LC clinician group noted a cure for lymphoma as important goals. The LC clinician group highlighted that most patients with r/r DLBCL would receive pola-BR or rituximab in combination with gemcitabine and oxaliplatin (R-GemOx) or monotherapy in the form of glofitamab or epcoritamab. They noted an unmet need for effective therapies and well-tolerated treatments among patients ineligible for, or who are unable to receive or have already received, intensive treatments (i.e., ASCT or CAR T cell). The OH-CCO noted poor response rates, PFS, and OS with available therapies.
The LC clinician group mentioned that, once funded, Glofit-GemOx would replace R-GemOx or pola-BR as the preferred treatment in the second-line setting for many patients who are ineligible for or do not wish to receive therapies such as ASCT or CAR T-cell therapy. They also noted that Glofit-GemOx would be an appropriate third-line treatment option for r/r DLBCL. However, this choice would depend on various factors, such as patient age, comorbidities, values and preferences, treatment history, prior response to chemotherapy, tumour burden, and cytopenias.
The LC clinician group mentioned that patients with r/r DLBCL who have received 1 or more lines of therapy and are ineligible for, are unable to receive, or have declined intensive therapies, would be best suited for treatment with Glofit-GemOx. They also highlighted that patients would be expected to have adequate performance status, hematopoietic reserve, and organ function to tolerate both components of Glofit-GemOx. The OH-CCO noted that patients not eligible for high-dose chemotherapy due to comorbidities would be best suited to receive Glofit-GemOx. They also mentioned that patients eligible for CAR T-cell therapy in the second line should proceed along that route.
The LC clinician group noted that Glofit-GemOx would not be recommended for patients whose disease is refractory to CD20xCD3 bispecific antibodies or to gemcitabine and oxaliplatin chemotherapy. Other patient factors that may make candidates inappropriate for this regimen were poor performance status, inadequate organ function, active uncontrolled infections, active central nervous system lymphoma, or eligibility to receive intensive therapies (e.g., ASCT). The LC clinician group noted that other histologic subtypes of DLBCL were likely to benefit from Glofit-GemOx. The OH-CCO noted that patients whose disease is not refractory to second-line glofitamab should be eligible for re-treatment with bispecific antibodies in later lines.
With regard to response assessment, the LC clinician group noted PET or CT scans were appropriate after cycle 4 of the treatment. Repeat imaging may be considered after cycle 8 and at the end of the treatment, depending on the initial depth of response and physician discretion. Both clinician groups agreed that treatment should be discontinued if disease progression or unacceptable toxicities occurred.
According to the clinical experts consulted for this review, glofitamab should be administered by a hematologist or oncologist with experience managing the potential AEs of bispecific antibodies (including CRS, neurotoxicity, cytopenias, tumour lysis syndrome, and tumour flares) and cytotoxic chemotherapy. Both clinician groups agreed that initial treatment with glofitamab requires inpatient monitoring. The LC clinician group also noted that patients should receive the first 3 doses of glofitamab at a facility with access to tocilizumab and an intensive care unit (to treat high-grade CRS).
Drug Program Input
The Provincial Advisory Group identified jurisdictional implementation issues related to relevant comparators, considerations for initiation of therapy, considerations for prescribing of therapy, generalizability, considerations for a funding algorithm, care provision, and systemic and economic issues. The clinical experts consulted by CDA-AMC weighed evidence from the STARGLO trial and other clinical considerations to provide responses to the Provincial Advisory Group’s implementation questions.
Clinical Evidence
Systematic Review
Description of Studies
The systematic review included 2 reports of 1 pivotal trial (STARGLO). The STARGLO study is a phase III, ongoing, prospective, international (no Canadian sites), multicentre, open-label, parallel-group randomized controlled trial (RCT) designed to assess Glofit-GemOx compared with R-GemOx in patients with r/r DLBCL NOS who have failed 1 line of therapy and are not candidates for transplant, as well as those patients who have failed at least 2 lines of therapy. Patients were randomized in a 2:1 ratio to receive either Glofit-GemOx or R-GemOx. Randomization was stratified by the number of previous lines of systemic therapy for DLBCL (1 versus 2 or more) and the outcome of the last systemic therapy (relapsed versus refractory). Patients in the investigational arm received an IV dose of obinutuzumab pretreatment 7 days before the first dose of glofitamab, then up to 8 cycles of Glofit-GemOx, followed by up to 4 cycles of glofitamab monotherapy to complete up to 12 cycles of glofitamab, with each cycle being 21 days (i.e., every 3 weeks). Patients in the control arm received R-GemOx for up to 8 cycles with each cycle being 21 days (i.e., every 3 weeks). Patients permanently discontinued study treatment if they experienced any of the following: any medical condition that could have jeopardized the safety of patients who continued to receive study treatment, documented infection with SARS-CoV-2 during the study, pregnancy, use of an anticancer therapy not required by protocol, and confirmed disease progression (as assessed by the investigator according to the 2014 Lugano response criteria for malignant lymphoma) and in case of unacceptable toxicity. The primary outcome was OS, and the key secondary outcomes included PFS, complete response (CR) rate, and duration of CR as assessed by an independent review committee (IRC). Harms, including SAEs and notable harms, were also measured and reported. Patient-reported outcomes (e.g., health-related quality of life [HRQoL]) were also measured.
A total of 274 patients underwent randomization (Glofit-GemOx = 183; R-GemOx = 91). At the updated analysis data cut-off (February 16, 2024), 86 patients (47.0%) in the Glofit-GemOx arm and 28 patients (30.8%) in the R-GemOx arm were still in the study. Patients were aged an average of 65.0 years (standard deviation [SD] = 13.0), 57.7% were male, and 50.0% were Asian. Most patients (89.9%) had an ECOG PS of 0 or 1, had 1 previous line of therapy (62.8%), and had refractory disease to any prior line of therapy (66.4%). Discontinuation from the study occurred in 97 patients (53.0%) in the Glofit-GemOx arm and 63 patients (69.2%) in the R-GemOx arm. The most frequent reason for study discontinuation in both treatment arms was death (accounting for 43.7% and 57.1% in the Glofit-GemOx and R-GemOx populations, respectively).
Efficacy Results
Overall Survival
For OS, at the time of the primary analysis, with median durations of follow-up of 12.0 months (95% confidence interval [CI], 10.2 to 13.2) in the Glofit-GemOx arm and 9.6 months (95% CI, 7.9 to 12.0) in the R-GemOx arm, the median OS was not reached for the Glofit-GemOx arm (95% CI, 13.8 to not estimable [NE]) and was 9.0 months (95% CI, 7.3 to 14.4) in the R-GemOx arm. These findings for OS favoured Glofit-GemOx (hazard ratio [HR] = 0.59; 95% CI, 0.40 to 0.89).
At the time of the updated analysis, with median durations of follow-up of 22.5 months (95% CI, 20.0 to 24.5) in the Glofit-GemOx arm and 19.7 months (95% CI, 18.0 to 23.1) in the R-GemOx arm, the median OS was 25.5 months (95% CI, 18.3 to NE) for the Glofit-GemOx arm and 12.9 months (95% CI, 7.9 to 18.5) for the R-GemOx arm. These findings for OS favoured Glofit-GemOx (HR = 0.62; 95% CI, 0.43 to 0.88).
IRC-Assessed Progression-Free Survival
For PFS, at the time of the primary analysis, with median durations of follow-up of 9.0 months (95% CI, 6.2 to 9.7) in the Glofit-GemOx arm and 6.1 months (95% CI, 3.4 to 8.8) in the R-GemOx arm, the median PFS was 12.1 months (95% CI, 6.8 to 18.3) for the Glofit-GemOx and 3.3 months (95% CI, 2.5 to 5.6) in the R-GemOx arm. These findings for PFS favoured Glofit-GemOx (HR = 0.37, CI, 0.25 to 0.55).
At the time of the updated analysis, with median durations of follow-up of 16.3 months (95% CI, 15.3 to 20.1) in the Glofit-GemOx arm and 8.6 months (95% CI, 5.9 to 14.6) in the R-GemOx arm, the median PFS was 13.8 months (95% CI, 8.7 to 20.5) for the Glofit-GemOx arm and 3.6 months (95% CI, 2.5 to 7.1) for the R-GemOx arm. These findings for PFS favoured Glofit-GemOx (HR = 0.40; 95% CI, 0.28 to 0.57).
IRC-Assessed Complete Response
For CR, at the time of the primary analysis, the rates were 50.3% (95% CI, 42.8 to 57.7) for Glofit-GemOx and 22.0% (95% CI, 14.0 to 31.9) for R-GemOx; the between-group difference of 28.3% (95% CI, 16.3 to 40.3) favoured Glofit-GemOx.
At the time of the updated analysis, the CR rates were 58.5% (95% CI, 51.0 to 65.7) for Glofit-GemOx and 25.3% (95% CI, 16.8 to 35.5) in the R-GemOx arm; the between-group difference of 33.2% (95% CI, 20.9 to 45.5) favoured Glofit-GemOx.
IRC-Assessed Duration of Objective Response
For DOR at the time of the primary analysis, with median durations of follow-up of 6.9 months (95% CI, 6.3 to 7.6) in the Glofit-GemOx arm and 5.8 months (95% CI, 2.8 to 6.1) in the R-GemOx arm, the median DORs in patients were 15.4 months (95% CI, 14.4 to NE) for Glofit-GemOx and 9.1 months (95% CI, 5.3 to NE) for R-GemOx. There was no evidence of a statistically significant difference in the DOR between the 2 groups (unstratified HR = 0.58; 95% CI, 0.26 to 1.30).
At the time of the updated analysis, with median durations of follow-up of 14.3 months (95% CI, 13.0 to 18.0) in the Glofit-GemOx arm and 5.8 months (95% CI, 3.0 to 12.5) in the R-GemOx arm, the median DOR for the Glofit-GemOx arm was not reached (NE months; 95% CI, 17.6 to NE) and it was 10.3 months (95% CI, 6.5 to NE) in the R-GemOx arm. There was no evidence of a statistically significant difference in the DOR between the 2 groups (unstratified HR = 0.57; 95% CI, 0.30 to 1.10).
Health-Related Quality of Life
For HRQoL, descriptive summary statistics and changes from baseline were to be calculated by treatment arm at each assessment. The mean difference between groups in change from baseline was not tested statistically.
Change From Baseline in EORTC QLQ-C30 Scores
At baseline, the European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 (EORTC QLQ-C30) fatigue scores for Glofit-GemOx and R-GemOx were 32.98 (SD = 25.20) and 32.18 (SD = 21.17), respectively. At the time of the primary analysis, the mean changes from baseline scores were 2.42 (SD = 23.10) in the Glofit-GemOx arm and 4.98 (SD = 22.43) in the R-GemOx arm at the end of treatment (either completion or discontinuation). At the time of the updated analyses, the mean changes from baseline in EORTC QLQ-C30 fatigue scores were −1.24 (SD = 25.64) in the Glofit-GemOx arm and 4.59 (SD = 21.64) in the R-GemOx arm at the end of treatment (either completion or discontinuation).
At baseline, the EORTC QLQ-C30 physical functioning scores for Glofit-GemOX and R-GemOx were 77.14 (SD = 20.01) and 76.63 (SD = 18.72), respectively. At the time of primary analysis, the mean changes from baseline in the EORTC QLQ-C30 physical functioning subscale were −1.65 (SD = 19.97) in the Glofit-GemOx arm and −4.25 (SD = 17.82) in the R-GemOx arm at the end of treatment (either completion or discontinuation). At the time of the updated analyses, the mean changes from baseline in the physical functioning subscale were 0.36 (SD = 20.29) in the Glofit-GemOx arm and −4.13 (SD = 17.12) in the R-GemOx arm at the end of treatment (either completion or discontinuation).
Change From Baseline in FACT-Lym LymS
At baseline, the Functional Assessment of Cancer Therapy–Lymphoma (FACT-Lym) lymphoma subscale (LymS) scores for Glofit-GemOX and R-GemOx were 45.38 (SD = 10.60) and 43.90 (SD = 10.43), respectively. At the time of primary analysis, the mean changes from baseline in FACT-Lym LymS scores were 1.71 (SD = 10.58) in the Glofit-GemOx arm and 0.41 (SD = 8.00) in the R-GemOx arm at the end of treatment (either completion or discontinuation). At the time of the updated analyses, the mean changes from baseline in LymS scores were 2.32 (SD = 10.05) in the Glofit-GemOx arm and 0.64 (SD = 7.82) in the R-GemOx arm at the end of treatment (either completion or discontinuation).
Harms Results
Adverse Events
At the updated analyses, the majority of patients in both the Glofit-GemOx (172 of 172 [100%]) and R-GemOx (84 of 88 [95.5%]) treatment arms experienced at least 1 AE. The most frequently reported AEs in the Glofit-GemOx arm were CRS (76 patients [44.2%]), nausea (71 patients [41.3%]), and anemia (71 patients [41.3%]). The most frequently reported AEs in the R-GemOx arm were nausea (35 patients [39.8%]), platelet count reduction (27 patients [30.7%]) and diarrhea (24 patients [27.3%]).
Serious Adverse Events
At the time of the updated analyses, SAEs were reported in a greater proportion of patients exposed to any treatment in the Glofit-GemOx population (98 patients [54.4%]), compared with the R-GemOx population (15 patients [17.0%]). The most common SAEs with an incidence rate equal to or higher than 5% in the Glofit-GemOx and R-GemOx populations were CRS (20.3% versus 0.0%, respectively), pyrexia (6.4% versus 1.1%, respectively), and pneumonia (5.8% versus 4.5%, respectively).
Withdrawals due to Adverse Events
At the updated analyses, withdrawal of any study treatment was reported in a greater proportion of patients exposed to any treatment in the Glofit-GemOx arm (48 patients [26.7%]), compared with the R-GemOx population (11 patients [12.5%]).
Notable Harms: Cytokine Release Syndrome
At the time of the updated analysis, 76 patients (42.2%) and 0 patients had experienced CRS events of any grade in the Glofit-GemOx (any treatment exposed) and R-GemOx arm, respectively. Of those in the Glofit-GemOx (any treatment exposed) arm who experienced CRS, 54 (30.0%) reported a grade 1 even, 18 (10.0%) a grade 2 event, and 4 (2.2%) a grade 3 event.
Critical Appraisal
The STARGLO trial was planned with adequate power to meet the goals of the study’s primary outcome of OS, randomizing 274 patients in a 2:1 ratio to Glofit-GemOx or R-GemOx. The assumptions made when performing power calculations were considered reasonable. The randomization method and allocation concealment appeared to be adequate. Further, the randomization was stratified based on important prognostic factors to minimize important differences in baseline characteristics between groups and to allow for maintenance of randomization for exploratory subgroup analyses based on these baseline characteristics. The use of IRC assessments for the important outcomes provides objectivity to the outcome assessments. To minimize bias, the IRC remained blinded to treatment assignment and the sponsor did not have access to efficacy and safety summaries, which compared treatment arms before the formal reporting of study results, with the exception that the randomization code may have been made available to facilitate the analysis of pharmacokinetic samples. However, some limitations and potential sources of bias are outlined in this section.
The results reported to date were based on an interim analysis at the clinical cut-off date of March 29, 2023, and a follow-up analysis was conducted at the subsequent clinical cut-off date of February 16, 2024. The final data will not be available for several years. Interim analyses are typically at risk of overestimating the true magnitude of any benefit.
As the STARGLO trial was open-label, the lack of blinding may bias results, particularly for subjective patient-reported outcomes. HRQoL self-reporting and reporting of more subjective AEs may also be influenced by a lack of blinding, as patients may anticipate known adverse effects and thus may be more likely to report them when they do occur. Knowledge of a patient’s assigned treatment may also affect the way physicians manage their patients, and patient knowledge of their assigned treatment may make them more or less likely to remain in the study. An IRC was used to evaluate the study end points of PFS, CR, and DOR in a blinded manner as it would be less likely to be influenced by the lack of blinding. Although the median OS was not reached at the time of the primary analysis, and the OS data were immature with respect to OS, the median OS was reached at the time of the updated analysis. Moreover, estimates of the median DOR were not yet reached at the time of the primary analysis or the updated analysis, remaining immature.
Multiplicity was controlled in the study by use of a hierarchical testing procedure. The procedure appeared to appropriate for the primary and key secondary outcomes (alpha spending between primary and updated analyses). Because statistical significance was reached for the primary analyses, the subsequent updated clinical cut-off analyses were considered descriptive. Moreover, the subgroup analyses were not adjusted for multiplicity nor powered to detect effect modification. However, the results of most sensitivity and subgroup analyses for OS were generally consistent with the primary analysis, supporting the consistency of the effect across subgroups.
Additionally, for OS, new antilymphoma therapy (NALT) was allowed in patients who completed study treatment or discontinued trial medications based on investigator assessment. This can potentially bias the true effect of the interventions on survival and can vary across settings as the decision on whether to use an NALT was not systematically applied across all the enrolling centres. Patients who started on an NALT were not censored at the time of initiation for the OS end point. Moreover, patients who discontinued therapy remained in the OS analysis. This was also the case with the analysis of PFS, CR, and DOR; however, additional sensitivity analyses that explored the potential impact of NALTs on treatment differences were supportive of the overall findings. However, at the time of the evidence review for OS by CDA-AMC, these sensitivity analyses were not available for review; thus, we could not be certain of the magnitude and direction of the potential bias.
For both OS and PFS, patients could be censored for several reasons, including missed assessments. The amount of censoring in both arms was high, which can introduce bias and reduce the validity of the assessments. At the time of the evidence review by CDA-AMC, sensitivity analysis addressing each reason for censoring data and the direction of potential bias, was not available for review; thus, we could not be certain of the magnitude and direction of the potential bias.
At the time of the second interim analysis, most patients had discontinued treatment, due primarily to disease progression or death. This large level of treatment discontinuation may influence any potential comparative evaluation and interpretation of HRQoL and harms outcomes. Also, the number of patients remaining to complete HRQoL assessments declined over time, resulting in a risk of bias due to missing outcome data. As such, HRQL was likely biased due to the large number of participants who did not complete the assessments during the follow-up period.
GRADE Summary of Findings and Certainty of the Evidence
The selection of outcomes for Grading of Recommendations Assessment, Development, and Evaluation (GRADE) was based on the sponsor’s Summary of Clinical Evidence, consultation with clinical experts, and input received from patient and clinician groups and public drug plans (Table 2). The following list of outcomes was finalized in consultation with expert committee members:
OS at 6, 12, and 18 months
PFS at 6, 12, and 18 months
HRQoL using the FACT-Lym LymS: change from baseline at 6, 12, and 18 months
individuals with SAEs: up to the data cut-off.
Table 2: Summary of Findings for Glofit-GemOx vs. R-GemOx for Patients With DLBCL NOS
Outcome and follow-up | Patients, N (studies) | Absolute effects (95% CI) | Certainty | Interpretation | ||
|---|---|---|---|---|---|---|
R-GemOx | Glofit-GemOx | Difference | ||||
Survival outcomes | ||||||
Probability of OS by IRC at 6 months | 274 (1 RCT) | 687 per 1,000 (587 to 787 per 1,000) | 780 per 1,000 (719 to 841 per 1,000) | 93 per 1,000 (25 less to 201 more per 1,000) | Moderatea | Glofit-GemOx likely results in an improvement in OS compared to R‑GemOx |
Probability of OS by IRC at 12 months | 274 (1 RCT) | 525 per 1,000 (416 to 6,334 per 1,000) | 629 per 1,000 (557 to 701 per 1,000) | 104 per 1,000 (26 less to 234 more per 1,000) | Moderatea | Glofit-GemOx likely results in an improvement in OS compared to R‑GemOx |
Probability of OS by IRC at 18 months | 274 (1 RCT) | 397 per 1,000 (287 to 507 per 1,000) | 576 per 1,000 (502 to 651 per 1,000) | 180 per 1,000 (47 to 312 more per 1,000) | Highb | Glofit-GemOx likely results in an improvement in OS compared to R‑GemOx |
Probability of PFS by IRC at 6 months | 274 (1 RCT) | 390 per 1,000 (276 to 504 per 1,000) | 659 per 1,000 (587 to 730 per 1,000) | 269 per 1,000 (134 to 403 more per 1,000) | Highb | Glofit-GemOx likely results in an improvement in PFS compared to R‑GemOx |
Probability of PFS by IRC at 12 months | 274 (1 RCT) | 252 per 1,000 (136 to 369 per 1,000) | 517 per 1,000 (440 to 594 per 1,000) | 265 per 1,000 (125 to 405 more per 1,000) | Highb | Glofit-GemOx likely results in an improvement in PFS compared to R‑GemOx |
Probability of PFS by IRC at 18 months | 274 (1 RCT) | 221 per 1,000 (104 to 338 per 1,000) | 454 per 1,000 (373 to 535 per 1,000) | 233 per 1,000 (91 to 376 more per 1,000) | Highb | Glofit-GemOx likely results in an improvement in PFS compared to R‑GemOx |
Patient-reported outcomes (HRQoL) | ||||||
FACT-Lym LymS at 6 months | 274 (1 RCT) | 4.30 points lower (SD = 8.35) | 2.80 points lower (SD = 9.42) | The mean difference between groups in change from baseline was not tested statistically | Very lowc | Glofit-GemOx may result in little to no difference in FACT‑Lym LymS scores compared to R‑GemOx |
FACT-Lym LymS at 12 months | 274 (1 RCT) | 5.81 points lower (SD = 11.47) | 3.30 points lower (SD = 10.66) | The mean difference between groups in change from baseline was not tested statistically | Very lowc | Glofit-GemOx may result in little to no difference in FACT‑Lym LymS scores compared to R-GemOx |
FACT-Lym LymS at 18 months | 274 (1 RCT) | 5.43 points lower (SD = 11.44) | 6.60 points lower (SD = 8.00) | The mean difference between groups in change from baseline was not tested statistically | Very lowc | Glofit-GemOx may result in little to no difference in FACT‑Lym LymS scores compared to R-GemOx |
Safety outcomes (treatment-emergent SAEs) | ||||||
SAEs Follow-up: up to data cut-off | 260 (1 RCT) | 150 per 1,000 | 544 per 1,000 | The absolute difference between groups was not tested statistically | Highb | Glofit-GemOx may result in an increase in SAEs compared to R‑GemOx |
CI = confidence interval; FACT-Lym = Functional Assessment of Cancer Therapy–Lymphoma; Glofit-GemOx = glofitamab in combination with gemcitabine and oxaliplatin; HRQoL = health-related quality of life; LymS = lymphoma subscale; OS = overall survival; PFS = progression-free survival; R-GemOx = rituximab in combination with gemcitabine and oxaliplatin; RCT = randomized controlled trial; SAE = serious adverse event; SD = standard deviation; vs. = versus.
Note: Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aNo threshold of clinical importance could be established; effects were appraised using the null. Rated down 1 level for imprecision; there is the possibility for both benefit and harm.
bNo threshold of clinical importance could be established; effects were appraised using the null.
cRated down 3 levels for study limitations; there is risk of bias due to (a) lack of blinding and a subjective outcome, (b) substantial missing outcome data, and (c) imprecision; there is the possibility for both benefit and harm.
Sources: Details included in the table are from the sponsor’s Summary of Clinical Evidence and additional information provided by the sponsor.
Long-Term Extension Studies
There is no long-term extension phase planned for the STARGLO trial at this time.
Indirect Comparisons
Description of Studies
The sponsor submitted an indirect treatment comparison (ITC) report with 2 comparisons considered relevant to the current CDA-AMC review. These analyses used individual patient-level data (IPD) from the pivotal STARGLO trial (GO41944) and 2 other trials (the GO29365 study for pola-BR and the NP30179 study for glofitamab monotherapy). Propensity score analyses (PSAs) were performed to provide an estimate of treatment effects after accounting for differences in covariates considered to be potential prognostic factors or treatment-effect modifiers across treatment groups. The analyzed outcomes were OS, PFS, DOR, objective response rate (ORR), and discontinuation due to AEs.
Efficacy Results
Overall Survival
The HR for OS between Glofit-GemOx and pola-BR was ████ ████ ███ ████ ██ █████. As the CIs crossed 1, there was no evidence of a statistically significant difference between the 2 treatments. These results were confirmed in the sensitivity analyses.
The HR for OS between Glofit-GemOx and glofitamab monotherapy was ████ ████ ███ ████ ██ █████. As the CIs crossed 1, there was no evidence of a statistically significant difference between the 2 treatments. These results were confirmed in the sensitivity analyses.
IRC-Assessed Progression-Free Survival
The HR for PFS between Glofit-GemOx and pola-BR was ████ ████ ███ ████ ██ █████. As the CIs crossed 1, there was no evidence of a statistically significant difference between the 2 treatments. These results were confirmed in the sensitivity analyses.
Tumour Response Variables
For IRC-assessed CR rates, the odds ratio (OR) between Glofit-GemOx and pola-BR was ████ ████ ███ █████ █████. As the CIs crossed 1, there was no evidence of a statistically significant difference between the 2 treatments. These results were confirmed in the full matching analysis, but inverse probability of treatment weighting (IPTW) with multiple imputation analysis showed a more favourable benefit with Glofit-GemOx.
For IRC-assessed CR rates, the OR between Glofit-GemOx and glofitamab was ████ ████ ███ ████ ██ █████. As the CIs crossed 1, there was no evidence of a statistically significant difference between the 2 treatments. These results were confirmed in the sensitivity analyses.
For IRC-assessed DOR, the OR between Glofit-GemOx and pola-BR was ████ █████ ██ █████. As the CIs crossed 1, there was no evidence of a statistically significant difference between the 2 treatments. These results were confirmed in the sensitivity analyses.
For IRC-assessed DOR, the OR between Glofit-GemOx and glofitamab was ████ █████ ██ █████. As the CIs crossed 1, there was no evidence of a statistically significant difference between the 2 treatments. These results were confirmed in the sensitivity analyses.
Harms Results
Treatment Discontinuation due to AEs
The OR between Glofit-GemOx and pola-BR for treatment discontinuation due to an AE was ████ █████ ██ █████. As the OR crossed 1, there was no evidence of a statistically significant difference between the 2 treatments. These results were confirmed in the sensitivity analyses.
The OR between Glofit-GemOx and glofitamab for treatment discontinuation due to an AE was ████ █████ ██ █████. As the OR CIs were wide and crossed 1, there was no evidence of a statistically significant difference between the 2 treatments. These results were confirmed in the sensitivity analyses.
Critical Appraisal
No major issues were identified with regard to the systematic search for identifying relevant studies for the ITC. The ITC analyses were preceded by a feasibility assessment, with a predefined hierarchy of evidence to guide study inclusion. In addition, of all the indirect comparisons submitted by the sponsor, only 2 PSAs were considered relevant to the current CDA-AMC review.
The choice of the matching factors was based on input from external clinical experts on an advisory board run by the sponsor. For the glofitamab monotherapy comparison, only high- and medium-priority variables were included due to the limitations of the adjusted model, with the Glofit-GemOx arm exhibiting a small effective sample size (ESS) of less than 30 when including high-, medium-, and low-priority variables, which adds uncertainty to these analyses.
While the ITC evaluated important clinical outcomes such as OS, PFS, CR rate and/or ORR, DOR, and treatment discontinuation due to AEs, other important outcomes for decision-making, such as HRQoL and SAEs, were not included in the analyses. This limits the ability to evaluate the true balance of comparative benefits and harms. Even for the evaluated outcomes, in most cases the CIs were wide and crossed the null, introducing further imprecision and uncertainty in the comparative effects.
Before the ITC adjustments, notable differences were observed between the trial patient characteristics. Following adjustment, the populations were balanced. Even so, adjustment methods cannot overcome methodological or design differences (e.g., study design, region or setting, length of follow-up, outcome definitions [event and censoring rules, schedule and method of assessments], cointerventions, and subsequent treatments) across the comparators, all of which can introduce bias. For example, the study designs for the 3 trials differed with the GO29365 trial being a phase Ib and II study, the NP30179 trial a phase II study, and the STARGLO trial a phase III study. In the study, GO29365, patients who received pola-BR had different histologies, such as DLBCL NOS, high-grade B-cell lymphoma (HGBCL), PMBCL, and tFL. However, the NP30179 trial of glofitamab monotherapy included patients with DLBCL NOS, tFL, PMBCL, and HGBCL, and the STARGLO trial only included patients with DLBCL NOS. Further, the NP30179 trial only included individuals with third-line and beyond r/r DLBCL (including only DLBCL NOS and 2, 3, and 4 prior treatment lines). As these factors could not be accounted for in the PSA models, differences between the study samples would be expected to introduce bias into the estimates, although the magnitude and direction of any bias is unclear.
The matching and/or adjustments conducted as part of the PSAs did sometimes result in residual imbalances for multiple prognostic factors. Although these are further controlled for in subsequent outcome analyses (following a doubly robust approach), it is important to note that the second adjustment could only be performed for summary statistics (HR or OR) but not for Kaplan-Meier curves.
Generalizability may be an issue due to the small sample size remaining after the exclusions and weighting in some of the analyses. For example, after subsetting the STARGLO and GO29365 patient cohorts used for the comparisons, there were 170 patients in the Glofit-GemOx arm and 132 patients in the pola-BR arm. Following IPTW adjustment, the ESS for Glofit-GemOx was 132.60 and the ESS for pola-BR was 107.36, which could affect the power of the analysis, resulting in wide CIs and a lack of ability to find true differences.
Studies Addressing Gaps in the Evidence From the Systematic Review
No additional studies were submitted by the sponsor.
Conclusions
One multinational, ongoing, open-label, phase III RCT (STARGLO), was included in the CDA-AMC review, along with ITCs (PSAs) comparing Glofit-GemOx to pola-BR and glofitamab monotherapy. The RCT included a population of 274 patients with DLBCL NOS, for whom Glofit-GemOx resulted in better clinical outcomes compared with those who received R-GemOx. While the trial is still ongoing, from the latest available evidence, data for most outcomes are mature, and the evidence suggests that Glofit-GemOx is significantly superior to R-GemOx. The clinical experts consulted by the review team considered the findings clinically meaningful. HRQoL appeared to be similar between groups, but due to the lack of blinding and the high attrition rate as the trial progressed, the certainty in the effect estimates is unclear. Data censoring in the OS and PFS analysis decreased the certainty in the results due to the high level of censoring. Treatment with Glofit-GemOx was associated with a substantially higher incidence of SAEs and notable harms compared with R-GemOx; however, the experts noted that these were known and manageable in the proper inpatient setting.
Using PSAs, Glofit-GemOx was indirectly compared to pola-BR and glofitamab monotherapies. After adjustments, the results generally did not provide evidence of a more favourable benefit of 1 treatment over another for all the evaluated outcomes (OR, PFS, CR rate, DOR, and treatment discontinuation due to AEs), with the CIs crossing the null. Furthermore, the wide CIs led to substantial uncertainty in many of the end points.
In conclusion, Glofit-GemOx was generally more efficacious than R-GemOx, providing a benefit in OS and PFS, especially on longer follow-ups. The QoL in patients who remained alive was uncertain due to a large loss to follow-up and the open-label nature of the trial. SAEs were more common with Glofit-GemOx and should be monitored closely. The evidence did not show that Glofit-GemOx had a conclusive benefit over pola-BR or glofitamab monotherapy.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of glofitamab (1 mg/mL concentrate solution for IV infusion) in combination with gemcitabine and oxaliplatin in the treatment of adult patients with r/r diffuse DLBCL NOS.
Disease Background
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following have been summarized and validated by the review team.
DLBCL is the most common type of NHL in North America, accounting for approximately 30% to 40% of all NHL cases in Canada.1 DLBCLs are a heterogeneous group of aggressive B-cell malignancies that differ in clinical presentation, molecular features, prognosis, and treatment options. Some types of indolent B-cell lymphomas (e.g., follicular lymphoma) can transform into DLBCL.1,2 The disease often presents with rapidly enlarging lymph nodes, elevated serum LDH and systemic “B symptoms,” such as fever, night sweats, and unexplained weight loss.3,4
In 2024, it was estimated that approximately 11,700 new cases of NHL would occur in Canada.5 Incidence increases with age, with a median age at presentation of 64 years.3 Up to 40% of patients with DLBCL will have r/r disease, with most patients experiencing rapid disease progression.9 The 5-year OS rate for DLBCL is approximately 60% to 70%.6
Prognosis is influenced by several factors, including the molecular subtype, with the germinal centre B-cell–like subtype having superior outcomes compared to the activated B-cell subtype.7 Factors such as age, disease stage, performance status, LDH levels, and extranodal involvement are important considerations when evaluating overall risk in patients with DLBCL. Prognosis is particularly poor for patients who experience r/r DLBCL, particularly those who are ineligible for ASCT, or who have experienced a relapse after second-line therapy.8 The median OS in patients with refractory or relapsed DLBCL has been previously reported to be only 6.6 months.9 As such, rapid diagnosis and initiation of treatment are essential.
Diagnosis of DLBCL includes an assessment of disease stage, medical history, and physical examination (cardiac function tests and blood tests), imaging (PET, CT, and MRI), biopsy, and IPI score.10 A morphological diagnosis of DLBCL should be confirmed by immunohistochemistry with essential markers, including CD20, CD79a, BCL2, BCL6, and MYC among others.11 The clinical experts consulted by CDA-AMC noted that fluorescence in situ hybridization evaluation looking for evidence of high-risk translocations (i.e., BCL2, MYC, BCL6) should be performed on tumour tissue from all patients at diagnosis. Disease staging is a crucial component for determining the extent of the disease, and the classification into 1 of 4 disease stages is according to the Ann Arbor and/or Lugano classification systems.11-13
Standards of Therapy
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following have been summarized and validated by the review team.
Pharmacotherapy regimens for DLBCL are complex and evolving rapidly as novel agents and multidrug combination regimens of various drug classes are increasingly being incorporated into clinical practice. The standard approach for DLBCL patients in the second line has been salvage chemotherapy (typically R-GDP in Canada) followed by high-dose chemotherapy and ASCT.1,11,52-54 However, this approach has several limitations. First, only approximately 50% of patients are suitable for this intensive approach, based on age or comorbidities. In addition, less than 50% of patients are reported to experience prolonged PFS with second-line therapies and the 3-year PFS of those patients that undergo ASCT is only 53%.55 Finally, only approximately 40% of patients who proceed to ASCT (representing approximately 10% of all patients with r/r) will ultimately be cured.55,56
The introduction of CAR T-cell therapy in this setting has demonstrated improved PFS (the 4-year PFS is approximately 40%) and OS compared to salvage therapy and ASCT (the 4-year PFS is 24%) for patients with primary refractory or early relapsing LBCL (i.e., within 12 months of therapy).55,57 Thus, second-line CAR T-cell therapy, specifically with axicabtagene ciloleucel (Yescarta), has recently been approved and is now funded in Canada for transplant-eligible patients with primary and/or refractory or early relapsing disease.24,58 Considering this, the decision between the use of salvage chemotherapy and ASCT versus CAR T-cell therapy is largely influenced by the timing of the relapse: CAR T-cell therapy for eligible patients whose disease is refractory to or who experience relapse within 1 year of first-line therapy, and ASCT for eligible patients with LBCL whose disease is refractory to or who experience relapse beyond 1 year of first-line therapy.59 However, many patients will not be able to receive CAR T-cell therapy due to logistical, geographical, or resource constraints.60
In ASCT-ineligible patients, after failure of CRS (second-line, transplant-ineligible), the goal of treatment is to prolong life, reduce symptoms, and provide a good QoL. Palliative approaches and mild single-drug chemotherapy may be used to control disease as treatments in this patient population are not thought to be curative, with life expectancy generally not longer than a year. The 2025 National Comprehensive Cancer Network guidelines suggest that, for those who experience relapse within 12 months, the preferred regimens include CAR T-cell therapy, Glofit-GemOx, epcoritamab in combination with gemcitabine and oxaliplatin, polatuzumab-mosunetuzumab, pola-BR, and tafasitamab-lenalidomide.52 Beyond CAR T-cell therapy, the only off-the-shelf regimens from this list that are currently approved and/or funded include pola-BR (outside of Quebec).61 Additional palliative-in-intent treatment options include rituximab-based chemotherapy.53,62 Tafasitamab-lenalidomide has also been approved in Canada,22,53 but is not publicly reimbursed outside of Quebec.63
In the third-line and beyond setting, other than CAR T-cell therapy in transplant-ineligible patients, other drugs such as pola-BR (excluding Quebec)61,62 and tafasitamab-lenalidomide (Quebec only)22,63 are approved for use for LBCL patients in Canada. Bispecific therapy has been approved and is funded in Canada for patients who previously received or are unable to receive transplant or CAR T-cell therapy. Finally, rituximab-based chemotherapies, such as R-GemOx or R-GDP, may also be considered with limited utility.43,44,64
Drug Under Review
Key characteristics of glofitamab are summarized in Table 3 with other treatments available for DLBCL.
Per the product monograph,14 glofitamab is a bispecific monoclonal antibody that binds bivalently (with high avidity) to CD20 expressed on the surface of B cells and monovalently to CD3 in the T-cell receptor complex expressed on the surface of T cells. By simultaneous binding to CD20 on the B cell and CD3 on the T cell, glofitamab mediates the formation of an immunological synapse with subsequent potent T-cell activation and proliferation, secretion of cytokines, and release of cytolytic proteins, resulting in the lysis of of CD20-expressing B cells.
Premedication with glofitamab is intended to reduce the risk of CRS and should be administered in well-hydrated patients. Glofitamab must be administered as an IV infusion according to the dose step-up schedule leading to the recommended dosage of 30 mg, after completion of pretreatment with obinutuzumab on day 1 of cycle 1. Each treatment cycle is 21 days, with a fixed duration of therapy of up to 12 cycles.
All patients must receive a single 1,000 mg dose of obinutuzumab on day 1 of cycle 1 (7 days before initiation of glofitamab treatment).14 The purpose of obinutuzumab pretreatment is to deplete circulating and lymphoid-tissue B cells and minimize the risk of CRS. Obinutuzumab should be administered as an IV infusion at 50 mg/h. The rate of infusion can be escalated in 50 mg/h increments every 30 minutes to a maximum of 400 mg/h. The recommended doses are 1,000 mg/m2 for gemcitabine and 100 mg/m2 for oxaliplatin, administered in any order. The recommended doses for glofitamab for days 8 and 15 of cycle 1 are 2.5 mg and 10 mg, respectively, with a duration of infusion of 4 hours. If patients have experienced CRS during premedication, the duration of infusion may be extended up to 8 hours. The recommended dose for glofitamab from cycles 2 to 12 is 30 mg.
Treatment with Glofit-GemOx is recommended for a fixed duration of treatment of 8 cycles, followed by 4 cycles of glofitamab monotherapy for a maximum of 12 cycles in total or until disease progression or unmanageable toxicity, whichever occurs first.14 For cycles 3 to 12, if the previous infusion with glofitamab was well tolerated, the infusion time may be shortened to 2 hours at the discretion of the treating physician. If the patient experienced CRS with the previous dose, the duration of infusion must be maintained at 4 hours. All patients must be monitored for signs and symptoms of CRS and/or neurologic toxicity, including ICANS, following glofitamab administration.
The Health Canada indication for Glofit-GemOx is for the treatment of adult patients with r/r DLBCL NOS who are not candidates for ASCT.14 The sponsor noted that the STARGLO trial (NCT04408638) included patients who were both transplant-ineligible and transplant-eligible in the third-line and beyond setting; however, the reimbursement request for transplant-ineligibility at the second line and beyond is aligned with the Health Canada indication.
Glofitamab has been previously reviewed by CDA-AMC for the treatment of adult patients with r/r DLBCL NOS, tFL, or PMBCL who have received 2 or more lines of systemic therapy and are ineligible to receive or cannot receive CAR T-cell therapy or have previously received CAR T-cell therapy, following pretreatment with obinutuzumab.15 A recommendation to reimburse with clinical criteria and/or conditions was issued on February 2, 2024.16
Table 3: Key Characteristics of Glofitamab, Polatuzumab Vedotin, Rituximab, Tafasitamab, Epicoritamab, Axicabtagene Ciloleucel, Tisagenlecleucel, and Lisocabtagene Maraleucel
Drug | Indicationa | Mechanism of action | Route of administration | Recommended dose | Serious adverse effects or safety issues |
|---|---|---|---|---|---|
Glofitamab | Glofitamab (for injection), in combination with gemcitabine and oxaliplatin, is indicated for the treatment of adult patients with r/r DLBCL NOS who are not candidates for ASCT. As monotherapy, glofitamab is indicated for the treatment of adult patients with r/r DLBCL NOS, DLBCL tFL, or PMBCL who have received 2 or more lines of systemic therapy and are ineligible to receive or cannot receive CAR T-cell therapy or have previously received CAR T‑cell therapy. | Binds to CD20 and CD3, mediating the formation of an immunological synapse with subsequent potent T-cell activation and proliferation, secretion of cytokines, and release of cytolytic proteins that results in the lysis of CD20‑expressing B cells. | IV infusion | Premedication
Subsequent infusions
Combination
Monotherapy
| Obinutuzumab
Glofitamab
Gemcitabine
Oxaliplatin
|
Polatuzumab vedotin | In combination with bendamustine and rituximab; indicated for the treatment of adult patients with r/r DLBCL NOS who are not eligible for ASCT and have received at least 1 prior therapy. | Polatuzumab vedotin is a CD79b-targeted antibody drug conjugate that preferentially delivers an antimitotic drug, resulting in lysis of malignant B cells. The polatuzumab vedotin molecule consists of monomethyl auristatin E, which binds to microtubules and kills dividing cells by inhibiting cell division and inducing apoptosis. | IV infusion | Polatuzumab vedotin, bendamustine, and rituximab can be administered in any order on day 1 of each cycle.
|
|
Rituximab-based chemotherapy | In combination with chemotherapy (CHOP) for induction treatment of CD20 positive, DLBCL. | Chimeric monoclonal antibody; binds to the transmembrane antigen CD20. | IV infusion, SC injection | Rituxan should be administered on day 1 of each chemotherapy cycle (CHOP), after the administration of the glucocorticoid component of the chemotherapy, for up to 8 cycles.
|
|
Tafasitamab | In combination with lenalidomide is indicated for the treatment of adult patients with r/r DLBCL NOS, including DLBCL arising from low-grade lymphoma who are not eligible for ASCT. | Tafasitamab is an Fc‑enhanced monoclonal antibody that targets the CD19 antigen expressed on the surface of pre-B and mature B lymphocytes and on several B‑cell malignancies. Upon binding to CD19, tafasitamab mediates B-cell lysis through apoptosis and immune-effector mechanisms including antibody-dependent cellular cytotoxicity and antibody-dependent cellular phagocytosis. In combination with lenalidomide, associated with greater cytotoxicity. | IV | The recommended dose is 12 mg of tafasitamab per kilogram of body weight administered as an IV infusion according to the following schedule:
Tafasitamab is to be administered with lenalidomide for up to 12 cycles. Patients should self-administer lenalidomide capsules at the recommended starting dose of 25 mg daily on days 1 to 21 of each cycle; the starting dose and subsequent dosing should be adjusted, as necessary, according to the lenalidomide product monograph. After a maximum of 12 cycles of combination therapy, stop treatment with lenalidomide and continue to administer tafasitamab infusions on day 1 and 15 of each 28‑day cycle, until disease progression or unacceptable toxicity. |
|
Epicoritamab | For the treatment of adult patients with r/r DLBCL NOS, DLBCL transformed from indolent lymphoma, HGBCL, primary mediastinal B-cell lymphoma or follicular lymphoma grade 3B after 2 or more lines of systemic therapy and who have previously received or are unable to receive CAR T-cell therapy. | An immunoglobin G1 bispecific antibody that binds to CD20 and CD3 cells, inducing specific T-cell activation and T-cell mediated killing of CD20 expressing cells. | SC injection | Cycle 1 (weekly dosing schedule):
Cycle 2 and 3 (weekly dosing schedule): Days 1, 8, 15 and 22: 48 mg of epcoritamab Cycles 4 to 9 (biweekly dosing schedule): Days 1 and 15: 48 mg of epcoritamab Cycles 10+ (dosing schedule of every 4 weeks): Day 1: 48 mg of epcoritamab |
|
Axicabtagene ciloleucel | Indicated for the treatment of adult patients with r/r large B‑cell lymphoma after 2 or more lines of systemic therapy, including DLBCL NOS, PMBCL, HGBL, and DLBCL arising from follicular lymphoma. Also, for second-line treatment in patients with progression within 12 months of first-line therapy (Zuma 7). | A CD19-directed genetically modified autologous T‑cell immunotherapy; binds to CD19-expressing cancer cells and normal B cells. Following anti-CD19 CAR T‑cell engagement with CD19-expressing target cells, the CD28 and CD3‑zeta costimulatory domains activate downstream signalling cascades that lead to T‑cell activation, proliferation, acquisition of effector functions and secretion of inflammatory cytokines and chemokines; this sequence of events leads to elimination of CD19-expressing cells. | IV | Provided as a single-dose, one-time treatment in a patient-specific infusion bag. Each single-infusion bag contains a suspension of anti-CD19 CAR-positive T cells in approximately 68 mL. The target dose is 2 × 106 CAR-positive viable T cells per kg of body weight (range: 1 × 106 cells/kg to 2.4 × 106 cells/kg), with a maximum of 2 × 108 CAR‑positive viable T cells for patients weighing 100 kg or more. |
|
Tisagen-lecleucel | Indicated for the treatment of adult patients with r/r large B‑cell lymphoma after 2 or more lines of systemic therapy including DLBCL NOS, HGBCL and DLBCL arising from follicular lymphoma | An autologous, immunocellular cancer therapy, involved in reprogramming of T cells with a transgene encoding a chimeric antigen receptor to identify and eliminate CD19-expressing malignant and normal cells. | IV | Provided as a single-dose, one-time treatment, in a patient- specific infusion bag(s). Adult r/r DLBCL and follicular lymphoma: 0.6 to 6.0 × 108 CAR-positive viable T cells (non–weight‑based). |
|
Lisocabtagene maraleucel | For the treatment of adult patients with r/r large B‑cell lymphoma after 2 or more lines of systemic therapy, including DLBCL NOS, PMBCL, HGBCL, and DLBCL arising from follicular lymphoma | A CD19‑directed genetically modified autologous cellular immunotherapy. CAR binding to CD19 expressed on the cell surface of tumour and normal B cells induces activation and proliferation of CAR T cells, release of proinflammatory cytokines, and cytotoxic killing of target cells. | IV | Provided as a single-dose, one-time treatment. A single dose contains 60 × 106 to 120 × 106 CAR‑positive viable T cells (consisting of CD4 and CD8 components at a ratio ranging from 0.8 to 1.2), with each component supplied separately in 1 to 4 single-dose vials. |
|
ASCT = autologous stem cell transplant; CAR = chimeric antigen receptor; CHOP = cyclophosphamide-doxorubicin-vincristine-prednisone; CRS = cytokine release syndrome; DLBCL = diffuse large B-cell lymphoma; HGBCL = high-grade B-cell lymphoma; ICANS = immune effector cell-associated neurotoxicity syndrome; NOS = not otherwise specified; PMBCL = primary mediastinal B-cell lymphoma; SC = subcutaneous; tFL = transformed follicular lymphoma.
Note: Examples of rituximab-based chemotherapy include rituximab plus gemcitabine, dexamethasone, and cisplatin; rituximab plus ifosfamide, carboplatin, and etoposide; and rituximab in combination with gemcitabine and oxaliplatin.
aHealth Canada–approved indication.
Source: Product monographs of glofitamab (draft),14 glofitamab,17 obinutuzumab,18 polatuzumab vedotin,19 rituximab,20 lisocabtagene maraleucel,21 tafasitamab,22 epicoritamab,23 axicabtagene ciloleucel,24 and tisagenlecleucel.25
Perspectives of Patients, Clinicians, and Drug Programs
The full patient and clinician group submissions received are available in the consolidated patient and clinician group input document for this review on the project website.
Patient Group Input
This section was prepared by the review team based on the input provided by patient groups.
One patient group (LC), provided input for this submission. LC is a national Canadian registered charity whose mission is to empower patients and the lymphoma community through education, support, advocacy, and research. Data for this submission were gathered from an online anonymous patient survey conducted from February 24, 2025, to March 23, 2025. A total of 41 collected responses were used to identify the main areas of concern for patients with DLBCL. Two patients living in the US (aged 45 to 54 years) had experience with Glofit-GemOx for the treatment of DLBCL.
Of the respondents who answered the demographic questions, 80% lived in Canada, 40% were between the age of 65 and 74, and 60% were male. Six respondents were diagnosed 1 to 2 years previously, 5 respondents each were diagnosed less than a year previously or 3 to 5 years previously, and 7 respondents were diagnosed 5 to 10 years previously. The subtypes of LBCL were identified as DLBCL NOS (n = 13); DLBCL arising from follicular lymphoma (n = 7); and DLBCL arising from PMBCL, Richter’s transformation, or germinal centre B-cell–like lymphoma (1 respondent each).
The respondents highlighted the following symptoms affecting QoL: fatigue or lack of energy, neutropenia, enlarged lymph nodes, night sweats, shortness of breath, bodily aches and pains, reduced appetite, and weight loss. Regarding psychosocial impacts, respondents (n = 23) noted anxiety, fear of progression, difficulty sleeping, stress of diagnosis, problems concentrating, and inability to continue daily activities. On a scale of 1 (no impact) to 5 (significant impact), respondents rated the ability to work, attend school, and volunteer; ability to contribute financially to household expenses; and ability to travel as 4 or higher. Respondents highlighted fatigue, nausea, vomiting, infections, low white blood cell counts, bone pain, neutropenia, and hair loss as treatment side effects that were the most difficult to tolerate.
Among 11 respondents, 36% indicated they had received 1 line of treatment, another 36% reported receiving 2 lines of treatment, and 27% reported receiving 3 or more lines of treatment. A majority of the respondents (63%) experienced a relapse and needed treatment past the front-line setting. In the front-line setting, 10 respondents indicated that they received R-CHOP, and 1 received R-CHOP and rituximab in combination with etoposide, prednisone, vincristine, cyclophosphamide and doxorubicin. In the second-line setting, 2 respondents each received R-GDP and salvage therapy plus ASCT, 3 received radiation, and 4 participated in a clinical trial. In the third-line setting, 3 respondents received CAR T-cell therapy, 1 received pola-BR, 2 received glofitamab, and 5 participated in a clinical trial. The patient group noted that 54% of respondents indicated they were very satisfied with their front-line treatment options compared to 20% of respondents in the r/r setting. Eight of the 11 respondents had no difficulty or little difficulty accessing treatment, while 3 respondents had some difficulty. Common financial implications of treatment for DLBCL reported by respondents were absence from work, drug costs, supplementary drug costs for side effects, and travelling costs. Respondents provided positive feedback on the care and expertise provided by the health care team treating them.
Respondents (n = 11) considered longer disease remission, longer survival, control of disease symptoms, normalization of blood counts, and an improved QoL that allows them to perform daily activities as important outcomes for new treatments. Seven respondents indicated they were willing to tolerate side effects to access new treatments. Six respondents indicated treatment choice based on knowledge of side effects and expected outcomes was an important factor.
Two respondents indicated they had experience with Glofit-GemOx. One respondent was treated less than 6 months ago, while the other was treated 1 to 2 years ago. Respondents indicated they were in remission upon receiving the therapy. Side effects, including decreased appetite, nausea, vomiting, and fatigue, were noted by both respondents. One respondent also noted CRS, fever, neutropenia, and low platelet count as side effects. Both respondents experienced financial impacts due to absence from work and travel. The patient group noted that the respondents rated their overall experience with the therapy as good and satisfactory, and that they would recommend the therapy to other patients with DLBCL.
Clinician Input
This section was prepared by the review team based on the input provided by clinician groups.
Input From Clinical Experts Consulted for This Review
All CDA-AMC review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of adult patients with r/r DLBCL NOS.
Unmet Needs
The clinical experts noted that the goal of treatment in transplant-ineligible patients with r/r DLBCL in the second-line and beyond is generally palliative despite the treatments available today requiring access to new treatment options (e.g., new immunotherapies and cellular therapies). In absence of a cure, the goal is to delay disease progression, prolong life, and improve QoL. It should be noted that QoL in clinical practice may be determined by a combination of validated QoL scales, patient reports, and clinical determination. Furthermore, the side effects and toxicity of treatment, including cytopenia, infections, and AEs of special interest for immunotherapies (e.g., CRS and ICANS) are serious conditions. Last, both newer treatment options and the risk of SAEs (e.g., CRS and ICANS) can be a burden on the health care system, and additional inpatient capacity may be needed to deliver treatments and monitor patients.
Place in Therapy
According to the clinical experts, because glofitamab works through mechanisms that are different from the current standard cytotoxic drugs, it is expected to be active even in very resistant DLBCLs. Furthermore, its combination with gemcitabine and oxaliplatin is believed to enhance T-cell activity, and is likely to be a promising option in patients with r/r DLBCL.
As patients have more courses of cytotoxic drugs (e.g., in the second line and beyond as well as the third line and beyond), their immune systems may not respond as well to combinations such as Glofit-GemOx. As such, it is likely that this combination will be more active earlier in the disease course as immune therapies in general require an intact immune system. It is not yet clear what the efficacy and safety of Glofit-GemOx compared with other types of immunotherapies used in practice such as cellular therapies (e.g., CAR T cell) or what the longer-term outcomes for this combination will be.
With regard to toxicity, while Glofit-GemOx is considered to have an inferior safety profile than competing treatments that contain noncytotoxic drugs (e.g., polatuzumab vedotin, tafasitamab, and lenalidomide), the anticipated efficacy likely favours the use of Glofit-GemOx. There are still concerns about the toxicity, potential for CRS, ICANS, and treatment-related deaths from this combination. The clinical experts noted that, in the future, the evolution of treatment with gemcitabine and oxaliplatin may include less need for hospitalization for administration and monitoring of these drugs, minimizing health disparities between provinces and improving access for patients from rural and/or remote areas.
Patient Population
The patients included in the STARGLO trial were relatively young (e.g., aged approximately 65 years) and in good general health (e.g., an ECOG PS of 0 to 2). Patient with a poor functional status may not be able to tolerate the toxicities. In addition, the trial excluded important DLBCL groups (e.g., patients who had transformed disease and those with double-hit genetics), and patients with aggressive forms of DLBCL (e.g., those with a high IPI, bulky disease, and non–germinal centre subtypes) did not respond as well to therapy. Furthermore, in the STARGLO trial, 1 of the most common reasons for transplant ineligibility was patient choice. This is not expected to be a common reason in a typical patient in Canada among those otherwise eligible.
Assessing the Response Treatment
Traditional measures of response (e.g., PFS, OS, and DOR) can be used to determine the effectiveness of this treatment. Improvement in QoL and disease-related symptoms would be important patient-reported outcomes to assess as well.
Discontinuing Treatment
Disease progression and permanent toxicity (e.g., significantly impaired renal dysfunction, worsening neuropathy, and other toxicities with damage major organs) would be causes for discontinuing treatment.
Prescribing Considerations
The clinical experts consulted by CDA-AMC noted that treatment would require inpatient admission so that detailed and specialized management algorithms can be followed, similar to other T-cell–engaging therapies. This includes managing premedications, careful monitoring of the step-up dose (where the risk of CRS or ICANS is greatest), and ensuring emergency facilities are available for acute complications. Because CRS requires experienced care in proper settings, this restricts the use and availability of glofitamab in Canada.
Clinician Group Input
This section was prepared by the review team based on the input provided by clinician groups.
Two clinician groups, LC and the OH-CCO Hematology Cancer DAC, provided input for this submission. LC is a national organization dedicated to research, education, and raising awareness for the benefit of patients with lymphoma across Canada. Information was gathered from a literature search of published clinical trials of Glofit-GemOx as well as other treatments for r/r DLBCL. Four clinicians provided their input for this submission. The OH-CCO Hematology Cancer DAC provides evidence-based clinical and health system guidance on drug-related issues in support of CCO’s mandate, including the Provincial Drug Reimbursement Programs and the Systemic Treatment Program. Information was gathered via a teleconference and included input from 7 clinicians.
The LC clinician group noted that, for eligible patients, second-line treatment may include CAR T-cell therapy, if patients experience relapse within 12 months of R-CHOP. Chemoimmunotherapy and ASCT may be options if a patient experiences relapses more than 12 months after R-CHOP. Those not responding to second-line chemoimmunotherapy or who experience relapse after ASCT may receive third-line CAR T-cell therapy.
Both clinician groups noted the goals of therapy were to prolong survival and improve QoL. In addition, the OH-CCO Hematology Cancer DAC noted that an improvement in disease response and disease-related symptoms, and the LC clinician group noted that a cure for lymphoma were important goals. The LC clinician group highlighted that most patients with r/r DLBCL would receive pola-BR or R-GemOx or monotherapy with glofitamab or epcoritamab. They noted that an unmet need exists for effective therapies and well-tolerated treatments among patients ineligible for or unable to receive, or those who have already received, intensive treatments (i.e., ASCT or CAR T-cell). The OH-CCO noted poor PFS and OS with available therapies.
The LC clinician group mentioned that, once it is funded, Glofit-GemOx would replace R-GemOx or pola-BR as the preferred treatment in the second-line setting for many patients ineligible for or who do not wish to receive therapies such as ASCT or CAR T-cell therapy. They also noted that Glofit-GemOx would be an appropriate third-line treatment option for r/r DLBCL. However, this choice would depend on various factors, such as patient age, comorbidities, values and preferences, treatment history, prior response to chemotherapy, tumour burden, cytopenias.
The LC clinician group mentioned that patients with r/r DLBCL after 1 or more lines of therapy who are ineligible for or unable to receive intensive therapies, or those who have declined them, would be best suited for treatment with Glofit-GemOx. They also highlighted patients would be expected to have an adequate performance status, hematopoietic reserve, and organ function to tolerate both components of glofitamab and GemOx treatment. The OH-CCO noted patients not eligible for high-dose chemotherapy related to comorbidities would be best suited to receive Glofit-GemOx. They also mentioned that patients eligible for CAR T-cell therapy in the second line should proceed along that route.
The LC clinician group noted that Glofit-GemOx would not be recommended for patients whose disease is refractory to a CD20xCD3 bispecific antibody or to chemotherapy consisting of gemcitabine and oxaliplatin. Other factors that would make patients inappropriate candidates for this regimen were a poor performance status, inadequate organ function, active uncontrolled infections, active central nervous system lymphoma, or eligibility to receive intensive therapies.
The LC clinician group noted that other histologic subtypes of DLBCL were likely to benefit from Glofit-GemOx. The OH-CCO noted that patients whose disease is not refractory to second-line glofitamab should be eligible for re-treatment with bispecific antibodies in later lines.
With regard to response assessment, the LC clinician group noted PET or CT scans after cycle 4 of the treatment would be appropriate. Repeat imaging may be considered after cycle 8 and at the end of the treatment, depending on the initial depth of response and physician discretion. Both clinician groups agreed that treatment should be discontinued if disease progression or unacceptable toxicities occur.
Glofitamab should be administered by a hematologist or oncologist familiar with managing the potential AEs of bispecific antibodies (including CRS, neurotoxicity, cytopenias, tumour lysis syndrome, and tumour flares) and cytotoxic chemotherapy. Both clinician groups agreed that treatment with glofitamab required inpatient monitoring. The LC clinician group further noted that patients should receive the first 3 doses of glofitamab at a facility with access to tocilizumab and an intensive care unit (to treat high-grade CRS).
Drug Program Input
The drug programs provide input on each drug being reviewed through the Reimbursement Review processes by identifying issues that may affect their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted for this review are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
Other comparators for r/r DLBCL include pola-BR and R-chemo. In the third line and later, other comparators would include CAR T-cell therapy, pola-BR, and glofitamab or epcoritamab monotherapy. CAR T-cell therapy is publicly funding in most jurisdictions for transplant-ineligible r/r DLBCL after 2 prior therapies. Is there evidence comparing Glofit-GemOx to these therapies? | No such evidence is available. |
Considerations for initiation of therapy | |
Are there circumstances for patients who only had 1 prior line of therapy, in which this ASCT ineligibility criteria falls outside of the preceding list? | Ineligibility for transplant usually included age, or a comorbid illnesses. Geographic issues may also prevent ASCT use. |
Considerations for continuation or renewal of therapy | |
Should patients treated with Glofit-GemOx be eligible for downstream glofitamab or epcoritamab? If so, should there be a minimum treatment interval between the last cycle of glofitamab and re-treatment? | If patients experienced a relapse or progress on treatment, then they should not receive glofitamab or epcoritamab downstream. If the treatment is topped and they experienced a relapse afterward, they can still be candidates for the individual drugs, particularly if the relapse is more than 6 months to 1 year after stopping the original treatment. |
Considerations for discontinuation of therapy | |
If 1 of the agents has to be discontinued due to intolerance, can the remaining agents be continued? | Yes. |
Generalizability | |
Should the following patients be considered for glofitamab?
| Yes, to all these specific populations. |
Should patients on existing regimens be switched to Glofit-GemOx? | — |
System and economic issues | |
Is there any guidance regarding which patients will need to be admitted vs. potentially some receiving the initial dose in the outpatient setting? | Patients with a high tumour burden or very kinetically active tumours (those with high Ki-67 indexes). |
ASCT = autologous stem cell transplant; CAR = chimeric antigen receptor; DLBLC = diffuse large B-cell lymphoma; Glofit-GemOx = glofitamab in combination with gemcitabine and oxaliplatin; HGBLC = high-grade B-cell lymphoma; NOS = not otherwise specified; pola-BR = polatuzumab vedotin plus bendamustine and rituximab; R-chemo = rituximab-based chemotherapy; vs. = versus.
Clinical Evidence
The objective of this Clinical Review report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of glofitamab (1 mg/mL concentrate solution for IV infusion) in combination with gemcitabine and oxaliplatin in the treatment of adult patients with r/r diffuse DLBCL NOS. The focus will be placed on comparing glofitamab to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of glofitamab is presented in 2 sections, with a critical appraisal of the evidence included at the end of each section. The first section, the systematic review, includes pivotal studies and RCTs that were selected according to the sponsor’s systematic review protocol. Our assessment of the certainty of the evidence in this first section using the GRADE approach follows a critical appraisal of the evidence. The second section includes indirect evidence from the sponsor.
Included Studies
Clinical evidence from 1 RCT identified in the systematic review and 1 ITC is included in the review and appraised in this document.
Systematic Review
Contents within this section have been informed by materials submitted by the sponsor. The following have been summarized and validated by the review team.
Description of Studies
Characteristics of the included studies are summarized in Table 5.
Table 5: Details of Studies Included in the Systematic Review
Characteristics | STARGLO (GO41944) |
|---|---|
Designs and populations | |
Study design | Phase III, open-label, multicentre RCT |
Locations | 62 centres across 13 countries: Australia (6 centres), Belgium (2 centres), Switzerland (2 centres), China (8 centres), Germany (3 centres), Denmark (2 centres), Spain (5 centres), France (5 centres), UK (5 centres), Republic of Korea (6 centres), Poland (5 centres), Taiwan (3 centres), and the US (10 centres) |
Patient enrolment dates | Start date: February 23, 2021 End date: Study ongoing |
Randomized (N) | Intention-to-treat population: N = 274
|
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Intervention | Glofit-GemOx
|
Comparator(s) | R-GemOx
|
Study duration | |
Screening phase | Within 28 days before treatment initiation |
Treatment phase | Glofit-GemOx arm: 12 cycles (8.3 months) R-GemOx arm: 8 cycles (5.5 months) |
Follow-up phase | Approximately 5 years after last patient enrolled |
Outcomes | |
Primary end point | OS |
Secondary and exploratory end points | Secondary
Safety
Exploratory
|
Publication status | |
Publications | Abramson et al. (2024)26 Hertzberg et al. (2021)27 GO41944; NCT04408638 (2021)28 |
AE = adverse event; ASCT = autologous stem cell transplant; CAR = chimeric antigen receptor; CNS = central nervous system; CR = complete response; DLBCL = diffuse large B-cell lymphoma; DOR = duration of response; ECOG PS = Eastern Cooperative Oncology Group Performance Status; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; FACT-Lym = Functional Assessment of Cancer Therapy−Lymphoma; Glofit-GemOx = glofitamab in combination with gemcitabine and oxaliplatin; HSCT = hematopoietic stem cell transplant; INV = investigator; IRC = independent review committee; LymS = lymphoma subscale; NOS = not otherwise specified; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; PRO = patient-reported outcome; R-GemOx = rituximab in combination with gemcitabine and oxaliplatin; RCT = randomized controlled trial; r/r = relapsed or refractory.
aAll response assessments were based on the 2014 Lugano response criteria.12
Sources: Sponsor’s Summary of Clinical Evidence and STARGLO (GO41944) Clinical Study Report.29
The STARGLO (GO41944) trial is an ongoing, phase III, open-label, multicentre study that aims to assess the efficacy and safety of Glofit-GemO) compared with R-GemOx in patients with r/r DLBCL NOS who have failed 1 line of therapy and are not candidates for transplant, as well as those patients who have failed at least 2 lines of therapy. The study design of the trial is summarized in Figure 1.
The study was conducted at 62 centres, none in Canada, that enrolled patients: Australia (6 centres), Belgium (2 centres), Switzerland (2 centres), China (8 centres), Germany (3 centres), Denmark (2 centres), Spain (5 centres), France (5 centres), UK (5 centres), Republic of Korea (6 centres), Poland (5 centres), Taiwan (3 centres), and the US (10 centres).
In total, 274 patients were randomized in the trial (91 in the R-GemOx arm and 183 in the Glofit-GemOx arm). Patients in the investigational arm received an IV dose of obinutuzumab pretreatment 7 days before the first dose of glofitamab, then up to 8 cycles of Glofit-GemOx, followed by up to 4 cycles of glofitamab monotherapy to complete up to 12 cycles of glofitamab, with each cycle being 21 days (i.e., every 3 weeks). Patients in the control arm received R-GemOx for up to 8 cycles with each cycle being 21 days (i.e., every 3 weeks). Patients were randomized via an interactive voice or web-based response system in a 2:1 ratio to receive either Glofit-GemOx or R-GemOx. The randomization was stratified according to the number of previous lines of systemic therapy for DLBCL (1 versus equal to or higher than 2) and the outcome of the last systemic therapy (relapsed versus refractory).
The study began enrolling patients on February 23, 2021, and is currently ongoing but not actively recruiting. The prespecified interim analysis was conducted at the clinical cut-off date of March 29, 2023, and a follow-up analysis was conducted at the subsequent clinical cut-off date of February 16, 2024.
Figure 1: STARGLO (GO41944) Study Design
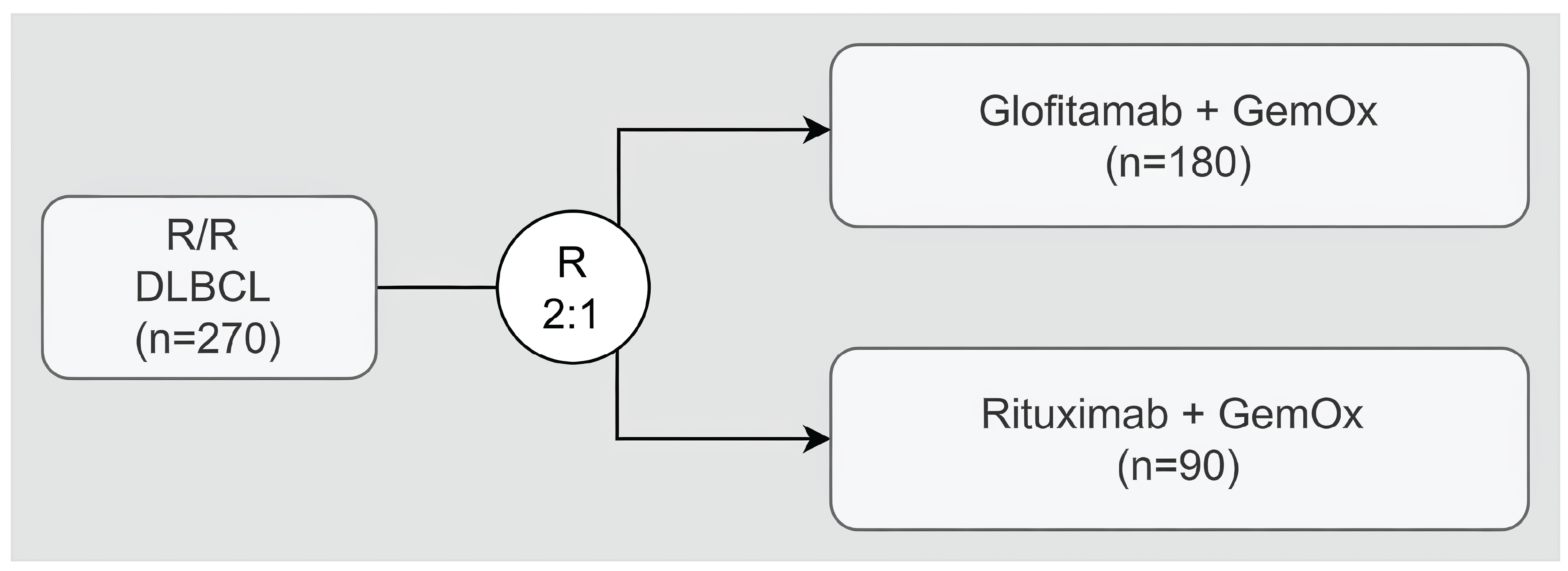
DLBCL = diffuse large B-cell lymphoma; GemOx = gemcitabine and oxaliplatin; R = ratio; R/R = relapsed or refractory.
Note: The planned enrolment was 270 patients, while the actual number of patients enrolled was 274.
Sources: STARGLO (GO41944) Clinical Study Report and updated Clinical Study Report.29
Populations
Inclusion and Exclusion Criteria
The STARGLO study included adult patients with histologically confirmed DLBCL NOS who had r/r disease after 1 or more previous treatments (Table 5). CAR T-cell and bridging therapies were counted as 1 line of therapy, while local therapies (e.g., radiotherapy) were not considered lines of therapy. Patients who had failed only 1 prior line of therapy had to be ASCT-ineligible, based on 1 of the following criteria: aged 70 years or older, ECOG PS of 2 or higher, left ventricular ejection fraction lower than or equal to 40%, creatinine clearance or glomerular filtration rate lower than or equal to 45 mL/min, refusal to receive high-dose chemotherapy and/or ASCT, insufficient response to pre-ASCT chemotherapy to be able to proceed with transplant, and comorbidities or criteria that precluded ASCT.
Patients who had failed only 1 prior line of therapy and were candidates for stem cell transplant were excluded from the trial. Moreover, key exclusion criteria of the trial covered the following: patients with DLBCL transformed from indolent lymphoma; HGBCL with MYC and BCL2 and/or BCL6 rearrangements and HGBCL NOS; PMBCL; prior treatment with glofitamab, other bispecific antibodies, R-GemOx or GemOx; current or previous primary or secondary central nervous system lymphoma; current or history of CNS disease; or any history of other malignancy.
Interventions
Patients in the Glofit-GemOx arm received a single IV dose of obinutuzumab pretreatment 7 days before the first dose of glofitamab, then up to 8 cycles of Glofit-GemOx, followed by up to 4 cycles of glofitamab monotherapy, to complete up to a total of 12 cycles of glofitamab. The treatment regimen and planned dosing are summarized in Figure 2.
Figure 2: Regimen in the Study Treatment Group — Glofit-GemOx Arm

Glofit-GemOx = glofitamab in combination with gemcitabine and oxaliplatin.
Note: For cycles 1 to 8, gemcitabine should have been administered before oxaliplatin. For cycles 2 to 8, glofitamab should have been given before gemcitabine and oxaliplatin.
Source: Clinical study protocol for STARGLO (GO41944), Version 7.29
Patients in the R-GemOx arm received rituximab in combination with gemcitabine plus oxaliplatin for up to 8 cycles. The treatment regimen and planned dosing are summarized in Figure 3.
Figure 3: Regimen in the Control Group — R-GemOx Arm
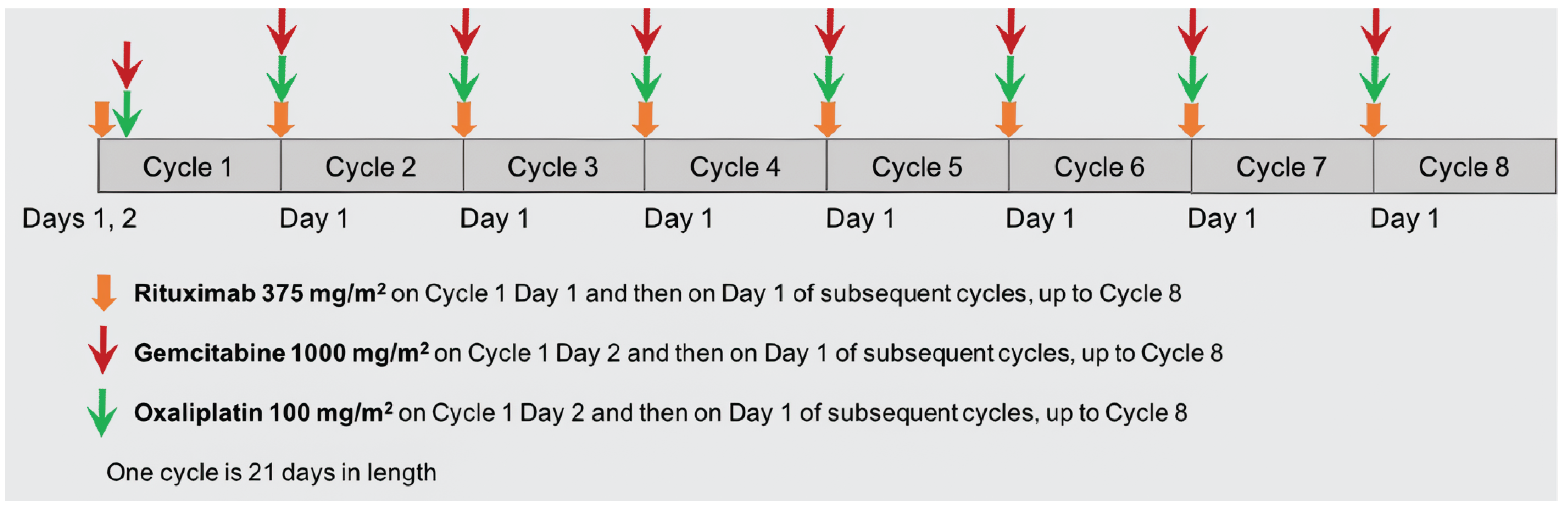
R-GemOx = rituximab in combination with gemcitabine and oxaliplatin.
Note: For cycles 1 to 8, gemcitabine should have been administered before oxaliplatin. For cycles 2 to 8, rituximab should have been given before gemcitabine and then oxaliplatin.
Source: Clinical study protocol for STARGLO (GO41944), Version 7.29
Premedication
The required pretreatment regimen and planned dosing are summarized in Table 6.
Table 6: Premedications Before Obinutuzumab and Glofitamab Infusions
Time point | Patients requiring premedication | Premedication | Administration |
|---|---|---|---|
Obinutuzumaba Cycle 1, day 1 | All patients | Hydration | Fluid intake of approximately 2 L to 3 L per day starting 24 to 48 hours before the first dose of study treatment |
IV glucocorticoidsb | At least 60 minutes before obinutuzumab infusion | ||
Oral or IV analgesic or antipyretic medicine | At least 30 minutes before obinutuzumab infusion | ||
Oral or IV antihistaminec | At least 30 minutes before obinutuzumab infusion | ||
Obinutuzumaba Cycle 1, day 1 (continued) | Patients at risk of TLS (e.g., because of bulky disease or renal impairment creatinine clearance < 70 mL/min) | Allopurinol or suitable alternative, such as rasburicase, along with adequate hydration | 48 to 72 hours before study treatment |
Glofitamabd,e Cycle 1, day 8 and onward; all doses | All patients | IV dexamethasoned,f | At least 60 minutes before the first dose of study treatment |
Oral or IV analgesic and/or antipyretic medication | At least 30 minutes before glofitamab infusion | ||
Oral or IV antihistaminec | At least 30 minutes before glofitamab infusion | ||
Patients at risk of TLS (e.g., because of bulky disease or renal impairment [creatinine clearance < 70 mL/min]) | Allopurinol or suitable alternative, such as rasburicase, along with adequate hydration | 48 to 72 hours before study treatment |
CRS = cytokine release syndrome; PO = orally; TLS = tumour lysis syndrome.
aClosely monitor patients during the entire infusion. Infusion reactions within 24 hours of receiving obinutuzumab have occurred. Obinutuzumab should be administered to well-hydrated patients.
bIV glucocorticoid before obinutuzumab: dexamethasone 20 mg IV or methylprednisolone 80 mg IV.
cFor example, 50 mg diphenhydramine (unless contraindicated).
dOptional at later cycles based on investigator’s assessment for patients that have tolerated two 30 mg doses of glofitamab without experiencing CRS.
eAll glofitamab doses should be administered to well-hydrated patients. Closely monitor patients during the entire infusion.
fAdminister dexamethasone 20 mg IV before glofitamab. If dexamethasone is not available or the patient has an intolerance to dexamethasone, an alternate corticosteroid (methylprednisolone 80 mg IV, prednisone 100 mg PO, or prednisolone 100 mg PO or IV) may be used for glofitamab premedication, after consulting with the medical monitor.
Source: Clinical study protocol for STARGLO (GO41944), Version 7.29
Obinutuzumab
Obinutuzumab pretreatment was to be administered by IV infusion 7 days before the first administration of glofitamab as an absolute (flat) dose of 1,000 mg on day 1 of cycle 1 for patients in the Glofit-GemOx Arm. The obinutuzumab infusion may have been split over 2 days if a patient was at increased risk for an immune-related response (IRR) (e.g., because of high tumour burden or a high peripheral lymphocyte count) or if an AE occurred during the infusion. Obinutuzumab must have been administered in a clinic or hospital equipped for systemic (IV) cancer treatment.29
Glofitamab
Patients randomized to the Glofit-GemOx treatment arm received glofitamab in a step-up dosing schedule starting on day 8 of cycle 1 (2.5 mg), day 15 of cycle 1 (10 mg), followed by 30 mg on day 1 of cycles 2 to 12, with each cycle being 21 days in length (i.e., every 3 weeks). Glofitamab was administered by IV infusion in a setting with immediate access to trained critical care personnel and facilities equipped to respond to and manage medical emergencies.
Initially, glofitamab was administered over 4 hours (± 15 minutes) on days 8 and 15 of cycle 1 and on day 1 of cycle 2. Following each glofitamab dose, patients were observed for at least 90 minutes for fever, chills, rigours, hypotension, hypoxia, nausea, or other signs and symptoms of CRS. At cycle 3 and beyond, if the patient had not experienced CRS during previous cycles, and had tolerated the preceding glofitamab infusion with no signs or symptoms of CRS, the window of observation may have been shortened at the discretion of the investigator. For patients who developed CRS with onset of associated signs or symptoms during glofitamab infusion, the infusion was discontinued immediately. In the absence of infusion-related AEs or CRS with their previous dose of glofitamab, the infusion time of glofitamab in cycles 3 and beyond could be reduced to 2 hours (± 15 minutes), at the discretion of the investigator. Alternatively, patients who were, in the investigator’s judgment, at an increased risk of recurrent CRS with subsequent doses could have had the time of infusion extended to up to 8 hours.
Premedication with dexamethasone 20 mg IV was administered 1 hour before the administration of glofitamab; premedication with acetaminophen or paracetamol (500 to 1,000 mg) and an antihistamine, such as diphenhydramine (50 mg), was administered approximately 30 minutes before the start of the infusion (Table 6).
Dexamethasone premedication was optional at later cycles based on investigator’s assessment for patients who have tolerated two 30 mg doses of glofitamab without experiencing CRS. However, if the patient experienced CRS, premedication with dexamethasone was required for subsequent glofitamab doses until no additional CRS events were observed. Patients were hospitalized at least overnight after the infusion of glofitamab on day 8 of cycle 1. Patients with a CRS event of grade 2 or higher associated with the preceding dose of glofitamab were hospitalized at least overnight for the next glofitamab dose. Investigators could choose to hospitalize a patient following any glofitamab dose for close monitoring if indicated and based on their clinical judgment. Hospitalization was considered following the second dose of glofitamab for patients who lacked support for close monitoring at home.29
Rituximab
Rituximab was administered by IV infusion at a dose of 375 mg/m2 in patients randomized to the R-GemOx arm. During the treatment period, rituximab was administered to patients in a setting where full emergency resuscitation facilities were immediately available.
On day 1 of each 21-day cycle, rituximab was administered to patients for 8 cycles. Only rituximab was administered on day 1 of cycle 1, followed by gemcitabine and oxaliplatin on day 2 of cycle 1. Rituximab was administered before gemcitabine and oxaliplatin on the same day during cycles 2 to 8. In the event of a late or prolonged infusion for rituximab because of starting late in the day, or slowed infusion rate because of an IRR, GemOx could be administered the following day. Once the rituximab infusion was completed, patients were observed for 30 minutes before the start of the other infusions.
If a dose of rituximab was split over 2 days, both infusions occurred with appropriate premedication (including prednisone) and at the first infusion rate. All rituximab infusions was administered to patients after premedication with acetaminophen (e.g., 650 to 1,000 mg) and an antihistamine such as diphenhydramine hydrochloride (50 to 100 mg) 30 to 60 minutes before starting each infusion (unless contraindicated). A dose of a glucocorticoid (methylprednisolone 80 mg IV or equivalent dose of dexamethasone [20 mg IV], prednisone [100 mg], or prednisolone [100 mg]) was allowed at the investigator’s discretion. For patients who did not experience infusion-related symptoms with their previous infusion, premedication at subsequent infusions could be omitted at the investigator’s discretion. If a patient tolerated the first cycle of rituximab without significant IRRs, rituximab was administered as a rapid infusion (over 60 to 90 minutes).29
Gemcitabine
Gemcitabine was administered by IV infusion to patients in both treatment arms (Glofit-GemOx and R-GemOx) at 1,000 mg/m2 on day 2 of cycle 1. Starting at cycle 2, gemcitabine was given on day 1 or 2 (following local practice) of each 21-day cycle (cycles 2 to 8). Gemcitabine was administered before oxaliplatin on the same day. Gemcitabine was to be administered in accordance with local institutional guidelines and prescribing information.29
Oxaliplatin
Oxaliplatin was administered by IV infusion to patients in both treatment arms (Glofit-GemOx or R-GemOx) at 100 mg/m2 on day 2 of cycle 1. Starting at cycle 2, oxaliplatin was given on day 1 or 2 (following local practice) of each 21-day cycle (cycles 2 to 8). Oxaliplatin was administered after gemcitabine on the same day. Oxaliplatin was to be administered in accordance with local institutional guidelines and prescribing information.29
Rescue Medication — Tocilizumab
Tocilizumab was not administered to all patients but only to those who experienced a CRS event for which tocilizumab was indicated as a rescue medicine. Before infusing glofitamab, the health care facility must have had onsite access to tocilizumab and ensured that a minimum of 2 doses of tocilizumab were available for the patient, if needed to treat CRS.
For the required treatment of CRS, patients received tocilizumab by IV infusion. Patients who weighed 30 kg or more received 8 mg/kg tocilizumab and patients who weighed less than 30 kg received 12 mg/kg tocilizumab. Doses exceeding 800 mg per infusion were not recommended. Treatment could be repeated every 8 hours as necessary (for a maximum of 4 doses).29
Concomitant Medication
Per the study protocol, concomitant therapy consisted of any medication used by a patient in addition to study treatment from 7 days before initiation of study treatment until the treatment-discontinuation visit.29
Patients were permitted to use the following therapies during the treatment period:
oral contraceptives with a failure rate of 1% per year
hormone-replacement therapy
treatment of hemophagocytic lymphohistiocytosis according to published recommendations and/or institutional practice
if dexamethasone is not available when dexamethasone is required in the protocol, or the patient has an intolerance to dexamethasone, then methylprednisolone, prednisone, or prednisolone may be used after consultation with the medical monitor.
Use of the following therapies was prohibited during the trial: investigational, unlicensed, or unapproved drugs, live vaccines, cytotoxic chemotherapy other than study treatments intended for lymphoma treatment, CNS prophylaxis, radiotherapy for lymphoma treatment, immunotherapy other than study treatments for lymphoma, immunosuppressive therapy (except medications indicated per protocol, including corticosteroids and tocilizumab), hormone therapy (other than contraceptives, hormone-replacement therapy, or megestrol acetate, and adjuvant endocrine therapy for nonmetastatic hormone receptor–positive breast cancer), biologic or targeted agents for lymphoma treatment, herbal therapies intended as treatment of lymphoma, and any therapies intended for lymphoma treatment whether approved by local regulatory authorities or investigational.29
NALTs, including radiotherapy and/or systemic therapies, were not permitted during study therapy but could be administered after patients had discontinued or completed study treatment. The decision to use an NALT was at the discretion of the investigator.
Dose Modifications
No dose modifications of glofitamab, obinutuzumab, rituximab, or gemcitabine were permitted. Oxaliplatin could be reduced per the management of oxaliplatin neuropathy guidelines provided by the sponsor.
If a cycle of therapy was delayed for more than 21 days for reasons other than toxicity, reinitiation of treatment was allowed, per investigators’ judgment. For patients receiving glofitamab, if a dose delay resulted in a treatment-free interval of 6 weeks or longer, obinutuzumab pretreatment needed to be reinitiated 7 days before resuming glofitamab treatment, and step-up dosing of glofitamab was required for the first cycle after the dose delay.29
Treatment Discontinuation
Patients permanently discontinued study treatment if they experienced any of the following: any medical condition that could have jeopardized the safety of a patient who continues to receive study treatment; documented SARS-CoV-2 infection during the study; pregnancy; use of an anticancer therapy not required per protocol; confirmed disease progression (per investigator assessment according to the 2014 Lugano response criteria for malignant lymphoma); and unacceptable toxicity.29 Patients needed to return for a treatment-discontinuation visit 6 weeks (± 14 days) after the final dose of study treatment, after which data on survival follow-up and new anticancer therapy were collected (via telephone calls, patient medical records, and/or clinic visits) approximately every 90 days (± 14 days) until death (unless the patient withdrew from the study).29
Study Discontinuation
Patients could voluntarily withdraw from the study at any time for any reason. Reasons for patient discontinuation from the study included, but were not limited to, the following: patient withdrawal of consent, study termination or site closure, patient noncompliance, and loss to follow-up. Every effort needed to be made to obtain a reason for patient discontinuation from the study. In case of patient study withdrawal, the study staff may have used public information sources (e.g., county records), when permissible, to obtain information about survival status.29
Outcomes
A list of efficacy end points assessed in this Clinical Review is provided in Table 7, followed by descriptions of the outcome measures. Summarized end points are based on outcomes included in the sponsor’s Summary of Clinical Evidence as well as any outcomes identified as important to this review by the clinical experts consulted for this review and input from patient and clinician groups and public drug plans. Using the same considerations, end points considered the most relevant to inform expert committee deliberations were selected and finalized in consultation with members of the expert committee. Key efficacy and safety end points were assessed using GRADE. Select notable harms outcomes considered important for informing expert committee deliberations were also assessed using GRADE.
Table 7: Outcomes Summarized From the Studies Included in the Systematic Review
Outcome measure | Definition | Time point | STARGLO (GO41944) |
|---|---|---|---|
OS | OS is defined as the time from randomization to date of death from any cause. | 6 months, 12 months, 18 months | Primarya |
PFS | The time from randomization to the first occurrence of disease progression, or death due to any cause, whichever occurs first. | 6 months, 12 months, 18 months | Key secondary (IRC-assesseda) |
CR | The proportion of patients whose BOR was a CR based on IRC assessment of PET and/or CT scans using the Lugano criteria. | 6 months, 12 months, 18 months | Key secondary (IRC-assesseda) |
DOR | The time from the initial occurrence of a documented objective response (PR or CR) until documented disease progression or death, whichever occurs first. | 6 months, 12 months, 18 months | Secondary |
EORTC QLQ-C30 | Time to deterioration in physical functioning and fatigue is defined as the time from randomization to the first documentation of a 10-point or more decrease or increase, respectively. | 6 months, 12 months, 18 months | Secondary |
FACT-Lym LymS | Time to deterioration in lymphoma-specific symptoms is defined as the time from randomization to the first documentation of a decrease of 3 or more points. | 6 months, 12 months, 18 months | Secondary |
EORTC QLQ-C30 | Change from baseline analyses. | 6 months, 12 months, 18 months | Exploratory |
FACT-Lym LymS | Change from baseline analyses. | 6 months, 12 months, 18 months | Exploratory |
SAEs | The proportion of patients with serious adverse events. | Up to the data cut-off | |
Notable harms (CRS) | The proportion of patients with CRS. | Up to the data cut-off |
BOR = best objective response; CR = complete response; CRS = cytokine release syndrome; DOR = duration of response; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; FACT-Lym = Functional Assessment of Cancer Therapy–Lymphoma; IRC = independent review committee; LymS = lymphoma subscale; OS = overall survival; PFS = progression-free survival; SAE = serious adverse event.
aStatistical testing for these end points, assessed by the IRC, was adjusted for multiple comparisons (e.g., hierarchal testing).
Source: Details included in the table are from the sponsor’s Summary of Clinical Evidence.
EORTC QLQ-C30
The EORTC QLQ-C30 is a validated, self-reported measure30,31 with a recall period of the previous week and consisting of 30 questions that assess the following: 5 aspects of patient functioning (physical, emotional, role, cognitive, and social), 3 symptom scales (fatigue, nausea and vomiting, and pain), global health/QoL, and 6 single items (dyspnea, insomnia, appetite loss, constipation, diarrhea, and financial difficulties) (Table 8). Scale scores can be obtained for the multi-item scales. The first 28 items are scored on a 4-point scale that ranges from “not at all” to “very much,” and the last 2 items are scores on a 7-point scale that ranges from “very poor” to “excellent.” Scores are then converted to a 0-to-100 scale, with higher scores indicating higher response levels (i.e., higher HRQoL and higher symptom severity).
No minimal important difference has been established, but the sponsor indicated that, for EORTC QLQ-C30 physical functioning and fatigue, a clinically meaningful change was defined as a 10-point decrease and increase, respectively, from baseline, based on the published literature.32,33
FACT-Lym LymS
The FACT-Lym is a validated, self-reported, disease-specific measure of HRQoL for lymphoma patients (Table 8).34 The full measure consists of the Functional Assessment of Cancer Therapy–General physical, social and family, emotional, and functional well-being scales (27 items), as well as the LymS (15 items). In the STARGLO trial, only the items in the LymS were administered to patients. Each item is rated on a 5-point response scale that ranges from “not at all” to “very much.” Scores are then converted to a 0-to-60 scale, with higher scores indicative of better HRQoL. The sponsor indicated that, for the FACT-Lym LymS, a clinically meaningful change was defined as a 3-point decrease from baseline, based on the published literature.35,36
Safety Outcomes
AEs and SAEs were defined according to the International Council for Harmonisation Guideline for Good Clinical Practice.29
AEs of special interest specific for glofitamab included the following: grade 2 or higher CRS, grade 2 or higher neurologic AEs; any suspected hemophagocytic lymphohistiocytosis tumour lysis syndrome (minimum grade 3 by definition); febrile neutropenia (minimum grade 3 by definition); grade 2 or higher aspartate transaminase, alanine transaminase, or total bilirubin elevation; any grade of disseminated intravascular coagulation (minimum grade 2 by definition); grade 2 or higher tumour flare (e.g., manifestation of signs or symptoms associated with an increase in the size of known nodal or extranodal lesions by clinical or radiographic assessment); any grade pneumonitis or interstitial lung disease (excluding pneumonia of infectious etiology); and colitis of any grade (excluding infectious etiology).29 Of these events, the clinical expects consulted noted that CRS was the most important in the context of this review.
Table 8: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
EORTC QLQ-C30 | The EORTC QLQ-C30 is a standardized patient self-administered questionnaire designed to assess the HRQoL of adult patients with cancer.37 The core questionnaire consists of 30 items divided into several domains and scales:
Patients complete the questionnaire based on a 1-week recall period by rating most items on a 4-point Likert scale (1 = not at all; 2 = a little; 3 = quite a bit; 4 = very much). For the 2 items in the global QoL scale, the response format is a 7-point Likert-type scale (1 = very poor; 7 = excellent).38 Scores are then converted to a 0-to-100 scale, with higher scores indicating better functioning or worse symptoms, or better QoL, depending on the scale.39,40 | Measurement properties of validity, reliability, and responsiveness have not been assessed in patients with NHL. | No MID has been established for patients with NHL. |
FACT-Lym LymS | The FACT-Lym consists of the 27-item FACT-G and the 15-item LymS.35 The FACT-G questionnaire assesses 4 dimensions of HRQoL: physical, social/family, emotional, and functional well-being. The LymS is an NHL-specific, patient-reported questionnaire used to assess HRQoL in terms of disease-specific symptoms and concerns. Items are rated on a 5-point Likert scale, with higher scores indicating better HRQoL. | In a study35 of 84 adult patients with NHL, measurements were taken at baseline, at 3 to 7 days, and at 8 to 12 weeks. Validity: Known-groups (construct) validity was demonstrated by the LymS score, which differentiated between patients with an ECOG PS of 0, 1, or 2 and between patients on and off active treatment (e.g., radiation and chemotherapy) but did not differentiate between patient groups defined by their NHL grade. Concurrent validity was demonstrated based on correlations between LymS and SF-36 PCS (r = 0.62) and MCS (r = 0.48) scores and the POMS total score (r = 0.60). Divergent validity was demonstrated based on the near-zero association between LymS and the Marlowe-Crowne Social Desirability Scale–Short Form (r = 0.15). Reliability: LymS demonstrated good internal consistency, with the Cronbach alpha ranging from 0.79 to 0.85 at each assessment time point. LymS demonstrated good test-retest reliability based on an ICC of 0.84 (retested at 3 to 7 days from baseline; n = 74). Responsiveness: FACT-LymS was able to differentiate patients in each of the 3 groups (worse, unchanged, better) defined by the patients’ retrospective ratings of change at the final assessment, and by change over 3 months in performance status (effect sizes > 0.50). | Using distribution-based and anchor-based methods, the investigators suggested the likely MID range for the LymS in patients with NHL is approximately 3 to 5 points, or 5% to 8% of the scale range (0 to 60).35 |
ECOG PS = Eastern Cooperative Oncology Group Performance Status; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; FACT-G = Functional Assessment of Cancer Therapy–General; FACT-Lym = Functional Assessment of Cancer Therapy–Lymphoma; GHS = global health status; HRQoL = health-related quality of life; ICC = intraclass correlation coefficient; LymS = lymphoma subscale; MCS = mental component summary; MID = minimal important difference; NHL = non-Hodgkin lymphoma; PCS = physical component summary; POMS = Profile of Mood States; QoL = quality of life; SF-36 = Short Form (36) Health Survey.
Statistical Analysis
Analyses Timing and Interim Analyses
Approximately 270 eligible patients were planned to be randomized in a 2:1 ratio to receive either Glofit-GemOx or R-GemOx. Recruitment for the study was expected to occur over approximately 17 months.
According to the statistical analyses plan, up to 2 analyses of the data had alpha-spending methodologies applied:
the interim analysis, which was planned to occur when 70% of the OS events have been documented (i.e., 97 events will trigger the interim analysis), and when all patients are randomized (expected to occur at a similar time, around 20 months after the first patient was randomized)
subsequent final analysis, which would be performed if the study does not stop for positive efficacy at the interim analysis; the analyses would be conducted once 138 OS events are documented (expected to occur around 26 months after the first patient was randomized).
One additional exploratory follow-up analysis will be conducted following a positive read-out at the interim analysis approximately 11 months after the last patient enrolled. In this analyses, no adjustment would have been made for alpha spending, given the primary end point would have been met at the interim analysis.29,41 These results are not yet available for review.
Multiplicity
To control the overall type I error rate at a 2-sided 0.05 level of significance, a hierarchical testing procedure was used to adjust for multiple statistical testing of the primary and key secondary efficacy end points. The following key secondary end points were formally tested:
PFS by IRC
best overall CR rate (based on response including PET and/or CT data) by IRC
duration of CR by IRC.
Overall survival was tested at the significance level determined using the Lan-DeMets spending function with an O’Brien-Fleming boundary such that the overall 2-sided type I error rate would be maintained at the 0.05 level. Based on the planned numbers of OS events, the O’Brien-Fleming boundary for statistical significance at the interim analysis was P = 0.0148.
The independent data monitoring committee (iDMC) evaluated efficacy data at the interim analysis during the study and, should the boundary for statistical significance be achieved and supported by other efficacy and safety results, would recommend that the study results be released without further hypothesis tests on the primary and key secondary end points after interim analysis. In this case, the interim analysis would be considered the primary analysis for this study and further analyses will be descriptive only. Otherwise, interim analysis results would be held by the iDMC until the final analysis is performed after 138 events. The boundary for efficacy at the final analysis was adjusted to incorporate the alpha spent at the interim analysis, such that the overall 2-sided type I error rate would be maintained at 0.05.29,41
A nonbinding futility analysis consisting in an observed HR for OS of 1.2 or greater would be performed at the same time as the interim efficacy analysis. If the HR for OS was 1.2 or greater, then the iDMC may consider the study as futile.29,41
At both the interim and final OS analyses, key secondary end points were evaluated in the order specified previously for statistical significance only if the study’s primary efficacy end point of OS was statistically significant at the appropriate boundary level. For key secondary end points, the boundaries for statistical significance were based on a Pocock alpha-spending function (i.e., 0.03244). In the case that the null hypothesis could not be rejected for any key secondary end point, the results for that end point and all following end points were considered not statistically significant.29,41
Sample Size and Power Calculation
Sample size calculation considered the following assumptions:
a median OS of 11 months in the R-GemOx arm based on the median OS reported in the largest multisite phase II study of R-GemOx44 and other similar references42,43
a randomization ratio of 2:1
an interim analysis for efficacy when 70% of events have been documented.
Taking into account these assumptions, 138 events were required to detect a between-group difference of 7.3 months in median OS (HR = 0.6), assuming an exponential distribution of OS using a log-rank test with 80% power and a 2-sided alpha of 0.05.
Based on these statistical assumptions and anticipating a recruitment period of approximately 17 months and follow-up of 9 months after the last patient is randomized, approximately 270 patients needed to be randomized in the global enrolment phase of this study (assuming an estimated annual dropout rate of 2%).
The clinical cut-off for the primary OS analysis was expected to be 26 months after the first patient was enrolled once the targeted number of mortality events (138 deaths) had occurred.41
Statistical Testing
OS was defined as the time from randomization to date of death from any cause. The main analytical approach and sensitivity analyses for the primary end point are described in Table 9. Censoring rules for OS are reported in Table 10. Assessment of the proportional hazards assumption on OS may have been examined using both graphical and analytical methods (if hazards were not proportional). Briefly, if the hazards were not proportional, supportive analyses may have been conducted using the restricted mean survival time, which would be computed for OS using the area under the curve from baseline to several time points for each treatment arm. The difference and its 95% CI (by the Greenwood method) and P values (by a z test) will be provided for descriptive purpose.41
PFS was defined as the time from randomization to the first occurrence of disease progression, or death due to any cause, whichever occurs first. The main analytical approach and sensitivity analyses for PFS end point are described in Table 9. Censoring rules for PFS are reported in Table 10. Assessment of proportional hazards followed the same approach used for the primary end point.
The CR rate is defined as the proportion of patients whose best overall response is a CR on a PET and/or CT scan during the study, according to the 2014 Lugano response criteria, as determined by the IRC and investigator. The main analytical approach for CR end points is described in Table 9.
The DOR is defined as the time from the date of the first occurrence of an objective response (partial response or CR) until the first date on which progressive disease or death is documented, whichever occurs first. The main analytical approach for the DOR end point is described in Table 9. The same PFS censoring rules as described in Table 10 were applied to the DOR, except the first scenario because all responders would have at least 1 baseline assessment.
For the patient-reported outcome questionnaires (EORTC QLQ-C30 and FACT-Lym LymS), analyses of time-to-deterioration analyses (secondary end points) and change from baseline (exploratory end points) were performed (details available in Table 9). For the EORTC QLQ-C30, time to deterioration in physical functioning and/or fatigue was defined as the time from randomization to the first documentation of a 10-point or more decrease. For the FACT-Lym LymS, time to deterioration in lymphoma-specific symptoms was defined as the time from randomization to the first documentation of a 3-point or more decrease. The FACT-Lym LymS and EORTC QLQ-C30 analysis was completed on evaluations of patient-reported outcomes.
Table 9: Statistical Analysis of Efficacy End Points
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
OS | The Kaplan-Meier estimate was used to estimate the median OS, if reached, and OS distribution for each treatment arm. The Brookmeyer-Crowley method was used to construct the 95% CI for the median OS for each treatment arm. Cox proportional hazards models were used to estimate the stratified hazard ratio and its 95% CI. A treatment comparison was made using a 2-sided 0.05-level stratified log-rank test. | The randomization stratification factors to be used in the efficacy analyses are the number of previous lines of systemic therapy for DLBCL (1 vs. ≥ 2) and the outcome of the last systemic therapy (relapsed versus refractory). | For OS, data from patients who did not have death documented will be censored on the last date they were known to be alive. Patients without information after baseline will be censored at the date of randomization. | The ITT population is the primary population for all efficacy measures and will be the only population examined in all sensitivity analyses. Sensitivity analyses was performed to study the impact on the analysis of OS for missing data and /or assessments due to COVID-19, and any stratification discrepancies between the eCRF and IxRS. |
PFS | The Kaplan-Meier estimate was used to estimate the median PFS, if reached, and PFS distribution for each treatment arm. The Brookmeyer-Crowley method was used to construct the 95% CI for the median PFS for each treatment arm. Cox proportional hazards models were used to estimate the stratified hazard ratio and its 95% CI. A treatment comparison was made using a 2-sided 0.05‑level stratified log-rank test. | The randomization stratification factors to be used in the efficacy analyses are the number of previous lines of systemic therapy for DLBCL (1 vs. ≥ 2) and the outcome of the last systemic therapy (relapsed versus refractory). | Patients who have neither progressed nor died at the time of analysis and patients who are lost to follow-up were censored according to the prespecified censoring rules. Patients who did not undergo a postbaseline tumour assessment were censored at the time of randomization. | Sensitivity analyses were performed to study the impact on the analysis of PFS for missing data and/or assessments due to COVID-19, and any loss to follow-up or discontinuation of assessments of PFS not due to an event. Additional sensitivity analyses will also be performed on PFS without censoring for new NALTs and censoring for NALTs, except for HSCT. |
CR | An estimate of the CR rate and its 95% CI were calculated using the Clopper-Pearson method for each treatment arm. The CR rate was compared between treatment arms using the Cochran-Mantel-Haenszel test stratified by randomization stratification factors. | The Cochran-Mantel-Haenszel test was stratified by the randomization stratification factors. | For the response end points, patients with no response assessments (for any reason) will be considered nonresponders. | NA |
DOR | The Kaplan-Meier estimate was used to estimate the median DOR, for each treatment arm. The Brookmeyer-Crowley method was used to construct the 95% CI for the median DOR for each treatment arm. Treatment comparison was made using a 2‑sided 0.05‑level stratified log-rank test. | The analyses were not stratified. | Patients who have neither progressed nor died at the time of analysis and patients who are lost to follow-up were censored according to the prespecified censoring rules. Patients who did not undergo a postbaseline tumour assessment were censored at the time of randomization. | NA |
EORTC QLQ-C30 and FACT-Lym LymS | Time-to-deterioration analysis. Change from baseline analyses. | Patients without an observed deterioration at the time of data cut-off will be censored at the last non-missing assessment date. Patients without a postbaseline assessment will be censored at randomization. For EORTC QLQ‑C30 subscales with 50% or more of the constituent items completed, a prorated score will be computed consistent with the scoring manuals and validation papers. For subscales with less than 50% of the items completed, the subscale will be considered as missing. | NA |
CI = confidence interval; CR = complete response; DLBCL = diffuse large B-cell lymphoma; DOR = duration of response; eCRF = electronic case report form; EORTC = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; FACT-Lym = Functional Assessment of Cancer Therapy–Lymphoma; HSCT = hematopoietic stem cell transplant; ITT = intention to treat; IxRS = interactive voice or web-based response system; LymS = lymphoma subscale; NA = not applicable; NALT = new antilymphoma therapy; OS = overall survival; PFS = progression-free survival; vs. = versus.
Source: Sponsor’s Summary of Clinical Evidence.
Table 10: Censoring Rules Analysis of OS (Primary End Point) and PFS (Secondary End Point)
Situation | Date of event or censoring | Outcome |
|---|---|---|
OS | ||
Death | Death date | Event |
No death | Last known alive datea before data cut-off | Censored |
No death and no postbaseline survival information available | Randomization date | Censored |
PFS | ||
No baseline disease assessments | Date of randomization | Censored |
New antilymphoma therapy started before documentation of disease progression or death | Date of last adequate disease assessment before start of new antilymphoma therapy | Censored |
Alive and without disease progression documentation | Date of last adequateb disease assessment | Censored |
Patients had 2 or more consecutive missed response assessments | Date of last adequate disease assessment before the missing assessments with documented nonprogression | Censored |
Death or disease progression between planned disease assessments | Date of death or first disease assessment showing disease progression, whichever occurs first | Event |
Death after first on-treatment disease assessment | Date of death | Event |
OS = overall survival; PFS = progression-free survival.
aLast known alive date is defined as the last date for which documented clinical data show the patient is alive. Scenarios considered in this definition may include last survival follow-up date with patient status of “alive,” date of last tumour assessment with a valid response, date of last treatment administration with a valid dose, date of last lab assessment with valid results, and date of last update of adverse event information.
bTo be considered adequate, a response assessment not including PET scans should have complete response, partial response, stable disease, or progressive disease as an outcome; and/or a response assessment including PET and CT scans should have complete metabolic response, partial metabolic response, no metabolic response, or partial metabolic disease using Lugano criteria. Assessments that are “unevaluable” and “not done” are considered not adequate.
Source: STARGLO Statistical Analyses Plan (version 6).41
Subgroup Analyses
Per version 6 of the STARGLO statistical analysis plan, subgroup analyses were conducted for the primary end point only, based on the variables listed in Table 11. The subgroup analyses were not to be adjusted for multiplicity, and all subgroup analyses were exploratory.
Table 11: Subgroups for Subgroup Analysis of STARGLO (GO41944)
Subgroup | Grouping |
|---|---|
ECOG Performance Status | 0, 1, or 2 |
Relapsed or refractory to last line of therapy (IxRS) | Relapsed, refractory |
Prior autologous stem cell transplant | Yes, No |
Double expresser (MYC and BCL2 overexpression) | Yes, No |
IPI score at study entry (CRF and derived) | 0, 1, 2, 3, 4, or 5 |
Prior CAR T-cell therapy | Yes, No |
Cell of origin (grouped data by IHC and gene expression) | Activated B-cell–like, germinal centre B-cell–like, unclassified |
Enrolment by geographic region | North America, Europe, and the rest of the world |
CAR = chimeric antigen receptor; CRF = case report form; ECOG = Eastern Cooperative Oncology Group; IPI = International Prognostic Index.
Source: STARGLO Statistical Analyses Plan (version 6).41
Analysis Populations
The populations analyzed in the STARGLO (GO41944) study are defined in Table 12. All patients randomized, whether or not they received the assigned treatment, were included in the efficacy analyses as part of the intention to treat (ITT) population. Any patient who received any amount of any study treatment was included in the safety-evaluable population. For all safety analyses, patients were grouped according to the treatment actually received (patients with any dose of glofitamab or obinutuzumab were analyzed in the Glofit-GemOx arm).
Table 12: Analysis Populations of STARGLO (GO41944)
Population | Definition | R-GemOx (N = 91) | Glofit-GemOx (N = 183) | All patients (N = 274) | |||
|---|---|---|---|---|---|---|---|
Included | Excluded | Included | Excluded | Included | Excluded | ||
Intention to treat | All randomized patients, whether or not the patient received the assigned treatment | 91 (100%) | — | 183 (100%) | — | 274 (100%) | — |
Safety-evaluable | Patients who receive any amount of any study treatment | 88 (96.7%) | 3 (3.3%) | 180 (98.4%) | 3 (1.6%) | 268 (94.5%) | 6 (5.5%) |
Modified safety-evaluable | Patients who receive at least 1 dose of rituximab in the R-GemOx arm and patients who receive at least 1 dose of obinutuzumab plus at least 1 dose of glofitamab in the Glofit-GemOx arm | 88 (96.7%) | 3 (3.3%) | 172 (94.0%) | 11 (6.0%) | 260 (94.9%) | 14 (5.1%) |
Glofit-GemOx = glofitamab in combination with gemcitabine and oxaliplatin; R-GemOx = rituximab in combination with gemcitabine and oxaliplatin.
Note: For all efficacy analyses (including patient-reported outcomes), patients will be grouped according to the treatment assigned at randomization. For all safety analyses, patients will be grouped according to the treatment actually received (patients with any dose of glofitamab or obinutuzumab will be analyzed in the Glofit-GemOx arm).
Source: STARGLO (GO41944) Clinical Study Report29 at the clinical cut-off date of March 29, 2023.
Results
Patient Disposition
The patient disposition of the ITT populations at the primary analysis of March 29, 2023, is summarized in Figure 4. Of the 274 patients enrolled in the STARGLO study (comprising the ITT population), 183 patients were randomized to receive Glofit-GemOx and 91 patients to receive R-GemOx. At the primary analysis data cut-off (March 29, 2023), 112 patients (61.2%) in the Glofit-GemOx arm and 43 patients (47.3%) in the R-GemOx arm were still in the study. Discontinuation from the study occurred in 71 and 48 patients (38.8% and 52.7%, respectively) in the Glofit-GemOx and R-GemOx arms, respectively. The most frequent reason for study discontinuation in both treatment arms was death (accounting for 33.3% and 42.9% in the Glofit-GemOx and R-GemOx populations, respectively). Discontinuation of treatment occurred in 95 patients (51.9%) and 62 patients (68.1%) in the Glofit-GemOx and R-GemOx arms, respectively. In the Glofit-GemOx arm, the main reasons for treatment discontinuation were progressive disease or disease relapse (21.3%), AEs (13.1%) and death (8.7%). In the R-GemOx arm, the main reasons for treatment discontinuation were progressive disease or disease relapse (39.6%), AEs (7.7%), and withdrawal by subject (6.6%).
Figure 4: STARGLO (GO41944) Disposition of Patients (Primary Analysis Data Cut-Off, March 29, 2023)
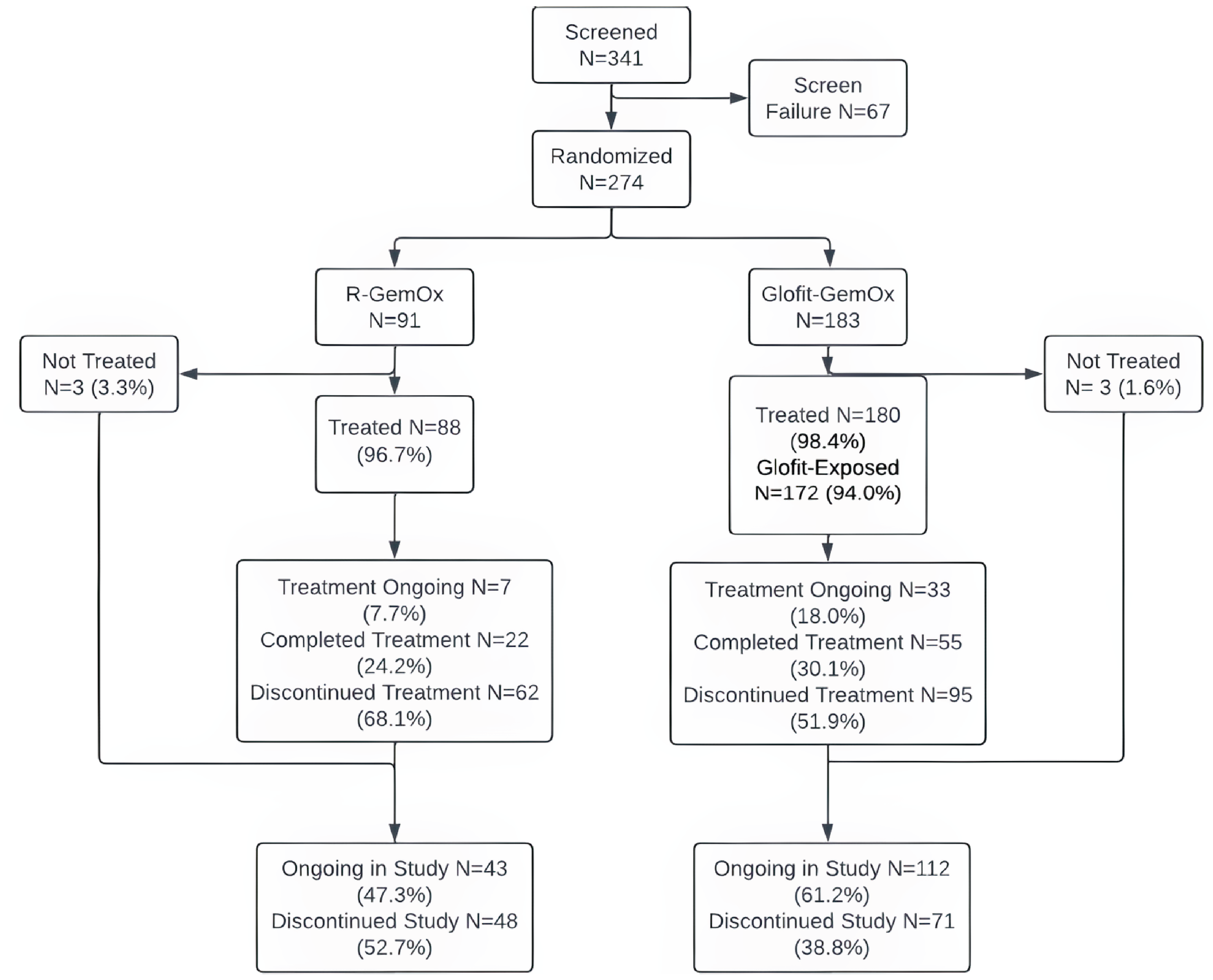
Glofit-GemOx = glofitamab in combination with gemcitabine and oxaliplatin; R-GemOx = rituximab in combination with gemcitabine and oxaliplatin.
Source: STARGLO (GO41944) Clinical Study Report29 at the clinical cut-off date of March 29, 2023.
At the updated analysis data cut-off (February 16, 2024), 86 patients (47.0%) in the Glofit-GemOx arm and 28 patients (30.8%) in the R-GemOx arm were still in the study. Discontinuation from the study occurred in 97 and 63 patients (53.0% and 69.2%, respectively) in the Glofit-GemOx and R-GemOx arms, respectively. The most frequent reason for study discontinuation in both treatment arms was death (accounting for 43.7% and 57.1% in the Glofit-GemOx and R-GemOx populations, respectively). Discontinuation of treatment occurred in 106 patients (57.9%) and 65 patients (71.4%) of patients in Glofit-GemOx and R-GemOx arms, respectively. In the Glofit-GemOx study arm, the main reasons for treatment discontinuation were progressive disease or disease relapse (22.4%), AEs (15.3%), and death (8.2%). In the R-GemOx arm, the main reasons for treatment discontinuation were progressive disease or disease relapse (41.8%), AEs (7.7%), and withdrawal by subject (7.7%).
Baseline Characteristics
As the study had completed enrolment at the time of the primary analysis (March 29, 2023), patient demographics and other baseline characteristics remained unchanged at the time of the updated analysis (February 16, 2024). The baseline demographic data and disease characteristics of the patients included in the ITT population are summarized in Table 13.
In the STARGLO trial, there were 42.6% (N = 78) of female patients in the Glofit-GemOx arm compared to 41.8% (N = 38) female patients in the R-GemOx arm. The mean age of patients in the Glofit-GemOx arm was 65.7 (SD = 12.3), and the mean age of patients in the R-GemOx arm was 63.8 (SD = 14.10). The majority of participants in the 2 treatment groups were Asian (Glofit-GemOx versus R-GemOx: 47.0% versus 56.0%) and white (Glofit-GemOx versus R-GemOx: 44.8% versus 36.3%).
In the Glofit-GemOx and R-GemOx treatment arms, 89.4% and 90.9% of patients had an ECOG PS of 0 or 1, respectively. An ECOG PS of 2 was reported in 10.6% of patients in the Glofit-GemOx arm and in 9.1% of patients in the R-GemOx arm.
Over 60% of the patients in the 2 treatment groups had received 1 previous line of systemic therapy for r/r DLBCL (Glofit-GemOx versus R-GemOx: 62.8% versus 62.6%). The majority of patients had advanced-stage disease, with an Ann Arbor stage of III reported in 12.6% and in 8.8% of patients in the Glofit-GemOx and R-GemOx arms, respectively, and an Ann Arbor stage of IV reported in 54.6% and 68.1% of patients in Glofit-GemOx and R-GemOx arms, respectively.
Regarding the proportions of patients who experienced relapse or had disease that was refractory to previous lines of therapies, the following data were reported: r/r to the first line of therapy (Glofit-GemOx versus R-GemOx: 57.4% versus 51.6%), and r/r to the last line of therapy (Glofit-GemOx versus R-GemOx: 61.2% versus 59.3%).
In the Glofit-GemOx arm, 67.8% of patients had disease that was refractory and 32.2% of patients experienced relapse after any prior therapy. In the R-GemOx arm, 63.7% of patients had disease that was refractory and 36.3% of patients experienced a relapse to any prior therapy.
Table 13: Summary of Baseline Characteristics of the STARGLO Study — ITT Population
Characteristic | Glofit-GemOx population (N = 183) | R-GemOx population (N = 91) | Total ITT population (N = 274) |
|---|---|---|---|
Age, years | |||
Mean (SD) | 65.7 (12.3) | 63.8 (14.1) | 65.0 (13.0) |
Median | 68.0 | 68.0 | 68.0 |
Minimum to maximum | 20 to 84 | 22 to 88 | 20 to 88 |
< 65, n (%) | 67 (36.6) | 35 (38.5) | 102 (37.2) |
≥ 65, n (%) | 116 (63.4) | 56 (61.5) | 172 (62.8) |
Sex, n (%) | |||
Female | 78 (42.6) | 38 (41.8) | 116 (42.3) |
Male | 105 (57.4) | 53 (58.2) | 158 (57.7) |
Race, n (%) | |||
Asian | 86 (47.0) | 51 (56.0) | 137 (50.0) |
Black or African American | 2 (1.1) | 1 (1.1) | 3 (1.1) |
White | 82 (44.8) | 33 (36.3) | 115 (42.0) |
Unknown | 13 (7.1) | 6 (6.6) | 19 (6.9) |
ECOG PS at baseline, n (%) | |||
0 | 72 (40.0) | 44 (50.0) | 116 (43.3) |
1 | 89 (49.4) | 36 (40.9) | 125 (46.6) |
2 | 19 (10.6) | 8 (9.1) | 27 (10.1) |
Geographic region, n (%) | |||
Europe | 62 (33.9) | 26 (28.6) | 88 (32.1) |
North America | 15 (8.2) | 10 (11.0) | 25 (9.1) |
Rest of the world | 106 (57.9) | 55 (60.4) | 161 (58.8) |
Previous lines of systemic therapy for DLBCL (IxRS), n (%) | |||
1 | 115 (62.8) | 57 (62.6) | 172 (62.8) |
≥ 2 | 68 (37.2) | 34 (37.4) | 102 (37.2) |
Relapsed or refractory to last line of therapy (IxRs), n (%) | |||
Refractory | 112 (61.2) | 54 (59.3) | 166 (60.6) |
Relapsed | 71 (38.8) | 37 (40.7) | 108 (39.4) |
Relapse or refractory to any prior therapy, n (%) | |||
Refractory | 124 (67.8) | 58 (63.7) | 182 (66.4) |
Relapsed | 59 (32.2) | 33 (36.3) | 92 (33.6) |
Relapse or refractory to first line of prior therapy, n (%) | |||
Refractory | 105 (57.4) | 47 (51.6) | 152 (55.5) |
Relapsed | 78 (42.6) | 44 (48.4) | 122 (44.5) |
Early relapse from ASCT (PD ≤ 12 months from completion), n (%) | |||
No | 4 (2.2) | 1 (1.1) | 5 (1.8) |
Yes | 4 (2.2) | 2 (2.2) | 6 (2.2) |
No prior ASCT | 175 (95.6) | 88 (96.7) | 263 (96.0) |
Ann Arbor staging at study entry, n (%) | |||
n | 182 | 90 | 272 |
Stage I | 19 (10.4) | 8 (8.8) | 27 (9.9) |
Stage II | 40 (21.9) | 12 (13.2) | 52 (19.0) |
Stage III | 23 (12.6) | 8 (8.8) | 31 (11.3) |
Stage IV | 100 (54.6) | 62 (68.1) | 162 (59.1) |
Patients who received prior ASCT | 8 (4.4) | 3 (3.3) | 11 (4.0) |
Patients who received prior CAR T-cell therapy, n (%) | 14 (7.7) | 8 (8.8) | 22 (8.0) |
Cell of origin, n (%) | |||
GCB | 56 (30.6) | 28 (30.8) | 84 (30.7) |
Non-GCB (by IHC plus non-GCB unclassified) | 102 (55.7) | 49 (53.8) | 151 (55.1) |
ABC | 3 (1.6) | 2 (2.2) | 5 (1.8) |
Unknown | 22 (12.0) | 12 (13.2) | 34 (12.4) |
Patients who have received anti-CD20 monoclonal antibody therapy, n (%) | 179 (97.8) | 89 (97.8) | 267 (97.8) |
ASCT = autologous stem cell transplant; CAR = chimeric antigen receptor; DLBCL = diffuse large B-cell lymphoma; ECOG PS = Eastern Cooperative Oncology Group Performance Status; GCB = germinal centre B-cell–like; Glofit-GemOx = glofitamab in combination with gemcitabine and oxaliplatin; IHC = immunohistochemistry; ITT = intention to treat; IxRS = interactive voice or web-based response system; PD = progressive disease; R-GemOx = rituximab in combination with gemcitabine and oxaliplatin; SD = standard deviation.
Sources: Sponsor’s Summary of Clinical Evidence and STARGLO (GO41944) Clinical Study Report29 at clinical cut-off dates of March 29, 2023, and February 16, 2024.
Prior ASCT had been given to 4.4% and 3.3% of patients in the Glofit-GemOx and R-GemOx arms, respectively. The most common reasons for ASCT ineligibility were patient refusing transplant (Glofit-GemOx: 35.0%; R-GemOx: 33.0%), age (Glofit-GemOx: 34.4%; R-GemOx: 27.5%), aged 70 years or older (Glofit-GemOx: 8.2%; R-GemOx: 14.3%), and insufficient response to salvage therapy (Glofit-GemOx: 8.2%; R-GemOx: 12.1%). Similarly, of 115 patients in Glofit-GemOx arm and 57 patients in R-GemOx arm with 1 prior line of therapy, the most common reason for not being a candidate for transplant was age (64 patients [55.7%] and 34 patients [59.6%], respectively) and patient refusal (39 patients [33.9%] and 19 patients [33.3%], respectively).29
CAR T-cell therapy had been given to 7.7% of patients in the Glofit-GemOx arm and to 8.8% of patients in the R-GemOx arm. Most patients (Glofit-GemOx: 97.8%; R-GemOx: 97.8%) had received anti-CD20 monoclonal antibody therapy.
Exposure to Study Treatments
The exposure of the primary safety-evaluable population to glofitamab, obinutuzumab, rituximab, gemcitabine, oxaliplatin, and tocilizumab at the primary analysis (data cut-off: March 29, 2023) and the updated analyses (data cut-off: February 16, 2024) are summarized in Table 14.
Subsequent Antilymphoma Treatment
After documented disease progression, patients were followed for survival or NALT.
In the ITT population at the time of the primary analyses, 32 of patients (17.5%) in the Glofit-GemOx arm and 35 patients (38.5%) in the R-GemOx arm received at least 1 NALT. Among patients who received an NALT, the majority received an NALT after documented investigator-assessed disease progression (24 [13.1%] in the Glofit-GemOx arm versus 28 [30.8%] in the R-GemOx arm). A list of the most frequently received subsequent antilymphoma treatments is reported in Table 14.
Table 14: Summary of Patient Exposure From the STARGLO Study Included in the Systematic Review (Primary Safety-Evaluable Population)
Treatment | Primary analysis data cut-off March 29, 2023 | Updated analysis data cut-off February 16, 2024 | ||
|---|---|---|---|---|
Glofit-GemOx (Glofit-exposed) (N = 172) | R-GemOx (N = 88) | Glofit-GemOx (Glofit-exposed) (N = 172) | R-GemOx (N = 88) | |
Gemcitabine | ||||
Median treatment duration,a days (range) | 147.0 (1 to 241) | 53.0 (1 to 183) | 147.0 (1 to 241) | 63.0 (1 to 183) |
Median number of treatment cycles, n (range) | 8.0 (1 to 8) | 3.5 (1 to 8) | 8.0 (1 to 9) | 4.0 (1 to 8) |
Median dose intensity,b % (range) | 100.0 (46.5 to 105.2) | 100.0 (92.9 to 111.0) | 100.0 (46.5 to 105.2) | 100.0 (92.7 to 111.0) |
Median total cumulative dose, mg/m2 (range) | 7,739.32 (465.1 to 8,415.9) | 3,494.26 (1,000.0 to 8,314.7) | 7,882.8 (465.1 to 9,000.0) | 39,97.7 (1,000.0 to 8,314.7) |
Median number of dose reductions,c n (range) | 0.0 (0.0 to 0.3) | 0.0 (0.0 to 0.1) | 0.0 (0.0 to 4.0) | 0.0 (0.0 to 1.0) |
Median number of dose delays, n (range) | 0.0 (0.0 to 5.0) | 0.0 (0.0 to 2.0) | 0.0 (0.0 to 5.0) | 0.0 (0.0 to 3.0) |
Glofitamab | ||||
Median treatment duration,a days (range) | 162.0 (1 to 296) | NE (NE to NE) | 218.0 (1 to 296) | NE (NE to NE) |
Median number of treatment cycles, n (range) | 8.0 (1 to 12) | NE (NE to NE) | 11.0 (1 to 13) | NE (NE to NE) |
Median dose intensity,b % (range) | 100.0 (96.1 to 116.9) | NE (NE to NE) | 100.0 (96.1 to 116.4) | NE (NE to NE) |
Median total cumulative dose, mg (range) | 222.5 (2.5 to 345.0) | NE (NE to NE) | 303.8 (2.5 to 355.0) | NE (NE to NE) |
Median number of dose reductions,c n (range) | 0.0 (0.0 to 1.0) | NE (NE to NE) | 0.0 (0.0 to 1.0) | NE (NE to NE) |
Median number of dose delays, n (range) | 0.0 (0.0 to 4.0) | NE (NE to NE) | 1.0 (0.0 to 6.0) | NE (NE to NE) |
Obinutuzumab | ||||
Median treatment duration,a days (range) | 1.0 (1 to 241) | NE (NE to NE) | 1.0 (1 to 241) | NE (NE to NE) |
Median number of treatment cycles, n (range) | 1.0 (1 to 2) | NE (NE to NE) | 1.0 (1 to 2) | NE (NE to NE) |
Median dose intensity,b % (range) | 100.0 (10.0 to 200.0) | NE (NE to NE) | 100.0 (10.0 to 200.0) | NE (NE to NE) |
Median total cumulative dose, mg (range) | 1,000.0 (100.0 to 2,000.0) | NE (NE to NE) | 1,000.0 (100.0 to 2,000.0) | NE (NE to NE) |
Median number of dose reductions,c n (range) | 0.0 (0.0 to 1.0) | NE (NE to NE) | 0.0 (0.0 to 1.0) | NE (NE to NE) |
Median number of dose delays, n (range) | 0.0 (0.0 to 1.0) | NE (NE to NE) | 0.0 (0.0 to 1.0) | NE (NE to NE) |
Oxaliplatin | ||||
Median treatment duration,a days (range) | 146.0 (1 to 241) | 53.0 (1 to 183) | 147.0 (1 to 241) | 63.0 (1 to 183) |
Median number of treatment cycles, n (range) | 8.0 (1 to 8) | 3.0 (1 to 8) | 8.0 (1 to 9) | 4.0 (1 to 8) |
Median dose intensity,b % (range) | 100.0 (88.0 to 311.7) | 100.0 (73.6 to 104.0) | 100.0 (85.0 to 103.6) | 100.0 (73.6 to 104.0) |
Median total cumulative dose, mg/m2 (range) | 777.4 (97.8 to 1,703.4) | 300.3 (99.0 to 810.5) | 788.5 (97.8 to 900.0) | 396.2 (99.0 to 810.5) |
Median number of dose reductions,c n (range) | 0.0 (0.0 to 2.0) | 0.0 (0.0 to 4.0) | 0.0 (0.0 to 4.0) | 0.0 (0.0 to 4.0) |
Median number of dose delays, n (range) | 0.0 (0.0 to 5.0) | 0.0 (0.0 to 2.0) | 0.0 (0.0 to 5.0) | 0.0 (0.0 to 3.0) |
Rituximab | ||||
Median treatment duration,a days (range) | NE (NE to NE) | 54.0 (1 to 183) | NE (NE to NE) | 64.0 (1 to 183) |
Median number of treatment cycles, n (range) | NE (NE to NE) | 3.5 (1 to 8) | NE (NE to NE) | 4.0 (1 to 8) |
Median relative dose intensity,b % (range) | NE (NE to NE) | 100.0 (93.2 to 125.4) | NE (NE to NE) | 100.0 (95.2 to 125.4) |
Median total cumulative dose, mg/m2 (range) | NE (NE to NE) | 1,287.5 (366.7 to 3,379.0) | NE (NE to NE) | 1,488.2 (366.7 to 3,379.0) |
Median number of dose reductions,c n (range) | NE (NE to NE) | 0.0 (0.0 to 1.0) | NE (NE to NE) | 0.0 (0.0 to 1.0) |
Median number of dose delays, n (range) | NE (NE to NE) | 0.0 (0.0 to 3.0) | NE (NE to NE) | 0.0 (0.0 to 3.0) |
Tocilizumab | ||||
Median treatment duration,a days (range) | 1.0 (1 to 58) | NE (NE to NE) | 1.0 (1 to 58) | NE (NE to NE) |
Median number of treatment cycles, n (range) | 1.0 (1 to 1) | NE (NE to NE) | 1.0 (1 to 1) | NE (NE to NE) |
Median dose intensity,b % (range) | 97.8 (50.1 to 114.3) | NE (NE to NE) | 97.7 (50.1 to 114.3) | NE (NE to NE) |
Median total cumulative dose, mg/kg (range) | 8.0 (4.0 to 31.9) | NE (NE to NE) | 8.0 (4.0 to 31.9) | NE (NE to NE) |
Median number of dose reductions,c n (range) | 0.0 (0.0 to 1.0) | NE (NE to NE) | 0.0 (0.0 to 1.0) | NE (NE to NE) |
Median number of dose delays, n (range) | 0.0 (0.0 to 0.0) | NE (NE to NE) | 0.0 (0.0 to 0.0) | NE (NE to NE) |
Glofit-GemOx = glofitamab in combination with gemcitabine and oxaliplatin; NE = not estimable; R-GemOx = rituximab in combination with gemcitabine and oxaliplatin.
aTreatment duration is the date of the last dose of study medication minus the date of the first dose plus 1 day.
bDose intensity is the total dose actually received divided by the expected total dose.
cA dose reduction is considered a dose that is less than 80% of the planned dose.
Sources: Details included in the table are from the sponsor’s Summary of Clinical Evidence and the STARGLO (GO41944) Clinical Study Report29 at the clinical cut-off dates of March 29, 2023, and February 16, 2024.
In the ITT population at the time of the updated analyses, 46 of patients (25.1%) in the Glofit-GemOx arm and 52 patients (57.1%) in the R-GemOx arm received at least 1 NALT. Among patients who received an NALT, the majority received an NALT after documented investigator-assessed disease progression (36 [19.7%] in the Glofit-GemOx arm versus 41 [45.1%] in the R-GemOx arm).
Efficacy
Overall Survival
The results for OS at the March 29, 2023, primary analyses in the overall ITT population are summarized in Table 15 and displayed in Figure 5. Sixty-one patients (33.3%) in the Glofit-GemOx arm and 40 patients (44.0%) in the R-GemOx arm had died. At the time of the primary analysis, with median durations of follow-up of 12.0 months (95% CI, 10.2 to 13.2) in the Glofit-GemOx arm and 9.6 months (95% CI, 7.9 to 12.0) in the R-GemOx arm, the median OS was not reached for the Glofit-GemOx arm (95% CI, 13.8 to NE) and was 9.0 months (95% CI, 7.3 to 14.4) in the R-GemOx arm. These findings for OS favoured Glofit-GemOx (HR = 0.59; 95% CI, 0.40 to 0.89).
The sponsor noted that, during the initial 90-day study period, there were 30 OS events (early deaths) in the ITT population — 18 in the Glofit-GemOx arm and 12 in the R-GemOx arm — which accounted for a transient inversion of the separation in the OS curve in favour of the R-GemOx arm.29 The majority of deaths during this period were attributable to progressive disease, all of which occurred in patients who entered the study with refractory DLBCL (as compared with those who enrolled with relapsed DLBCL after initial benefit with prior therapy). One patient who had progressive disease on study day 42 received an NALT on study day 50 and died on study day 57.
Figure 5: Kaplan-Meier Plot of Overall Survival in the STARGLO Trial (ITT Population, Primary Analysis, March 29, 2023)
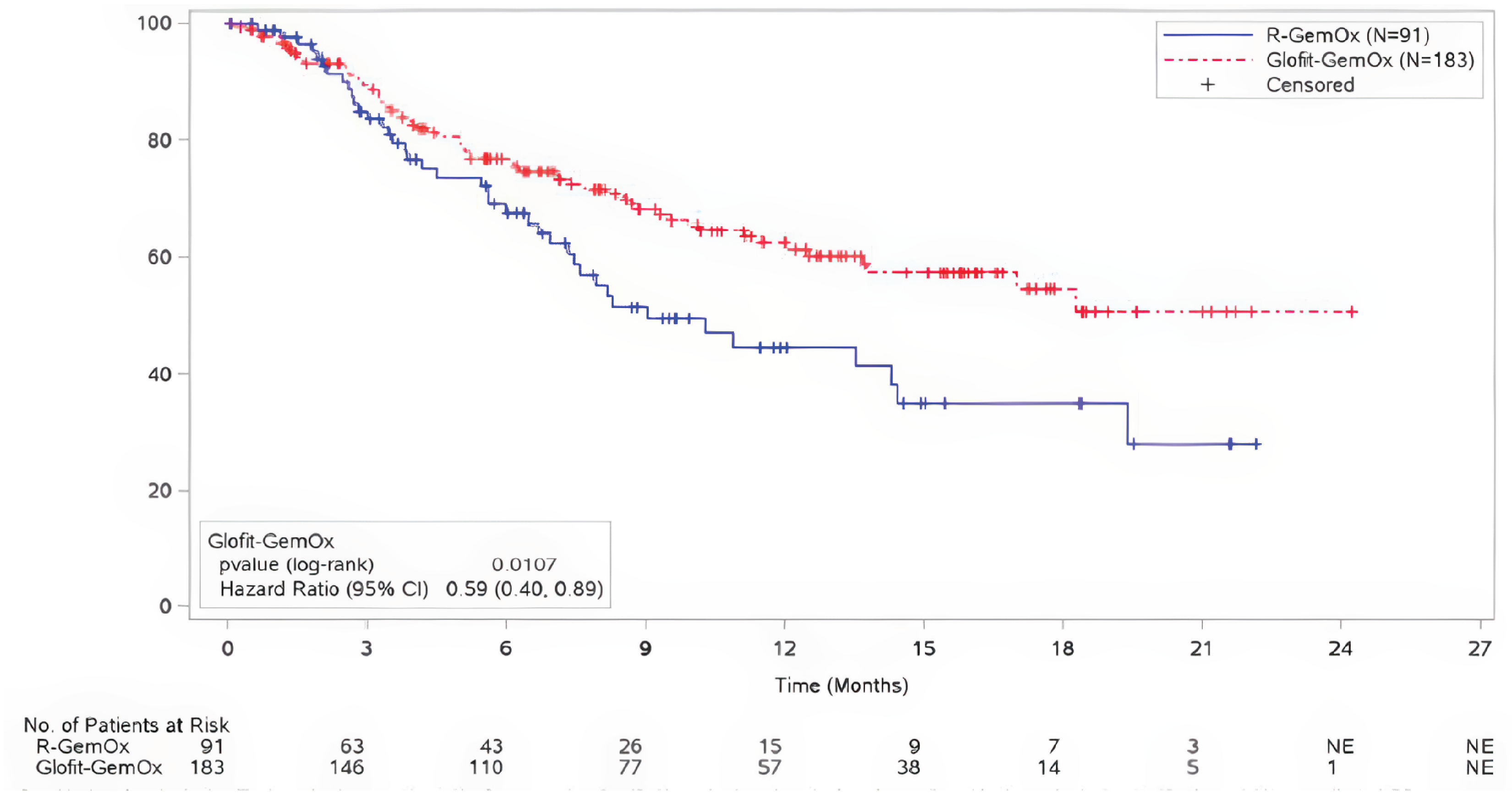
CI = confidence interval; Glofit-GemOx = glofitamab in combination with gemcitabine and oxaliplatin; ITT = intention to treat; NE = not estimable; R-GemOx = rituximab in combination with gemcitabine and oxaliplatin.
Source: STARGLO (GO41944) Clinical Study Report29 at the clinical cut-off dates of March 29, 2023, and February 16, 2024.
The results for OS at the February 16, 2024, updated analyses, which provided additional median durations of follow-ups of 10.5 months in the Glofit-GemOx arm and 10.1 months in the R-GemOx arm compared with the primary analysis, are summarized in Table 15 and displayed in Figure 6. Eighty patients (43.7%) in the Glofit-GemOx arm and 52 patients (57.1%) in the R-GemOx arm had died. At the time of the updated analysis, with median durations of follow-ups of 22.5 months (95% CI, 20.0 to 24.5) in the Glofit-GemOx arm and 19.7 months (95% CI, 18.0 to 23.1) in the R-GemOx arm, the median OS was 25.5 months (95% CI, 18.3 to NE) in the Glofit-GemOx arm and was 12.9 months (95% CI, 7.9 to 18.5) in the R-GemOx arm. These findings for OS favoured Glofit-GemOx (HR = 0.62; 95% CI, 0.43 to 0.88).
As with the primary analyses, the sponsor noted that, in the first 90 days of study participation, there were 31 OS events (early deaths) in the ITT population (18 in the Glofit-GemOx arm and 13 in the R-GemOx arm), which accounted for a transient inversion of the separation in the OS curve in favour of the R-GemOx arm before day 60.45 The majority of deaths in the initial 90-day period were attributable to progressive disease, and all patients entered the study with refractory DLBCL (as opposed to relapsed DLBCL after initial benefit from therapy). Of the early deaths, 2 were attributable to COVID-19, and both occurred in patients in the Glofit-GemOx arm. At the time of the updated analyses, more COVID-related deaths were recorded in the Glofit-GemOx arm than in the R-GemOx arm.
Figure 6: Kaplan-Meier Plot of Overall Survival in the STARGLO Trial (ITT Population, Updated Analysis, February 16, 2024)
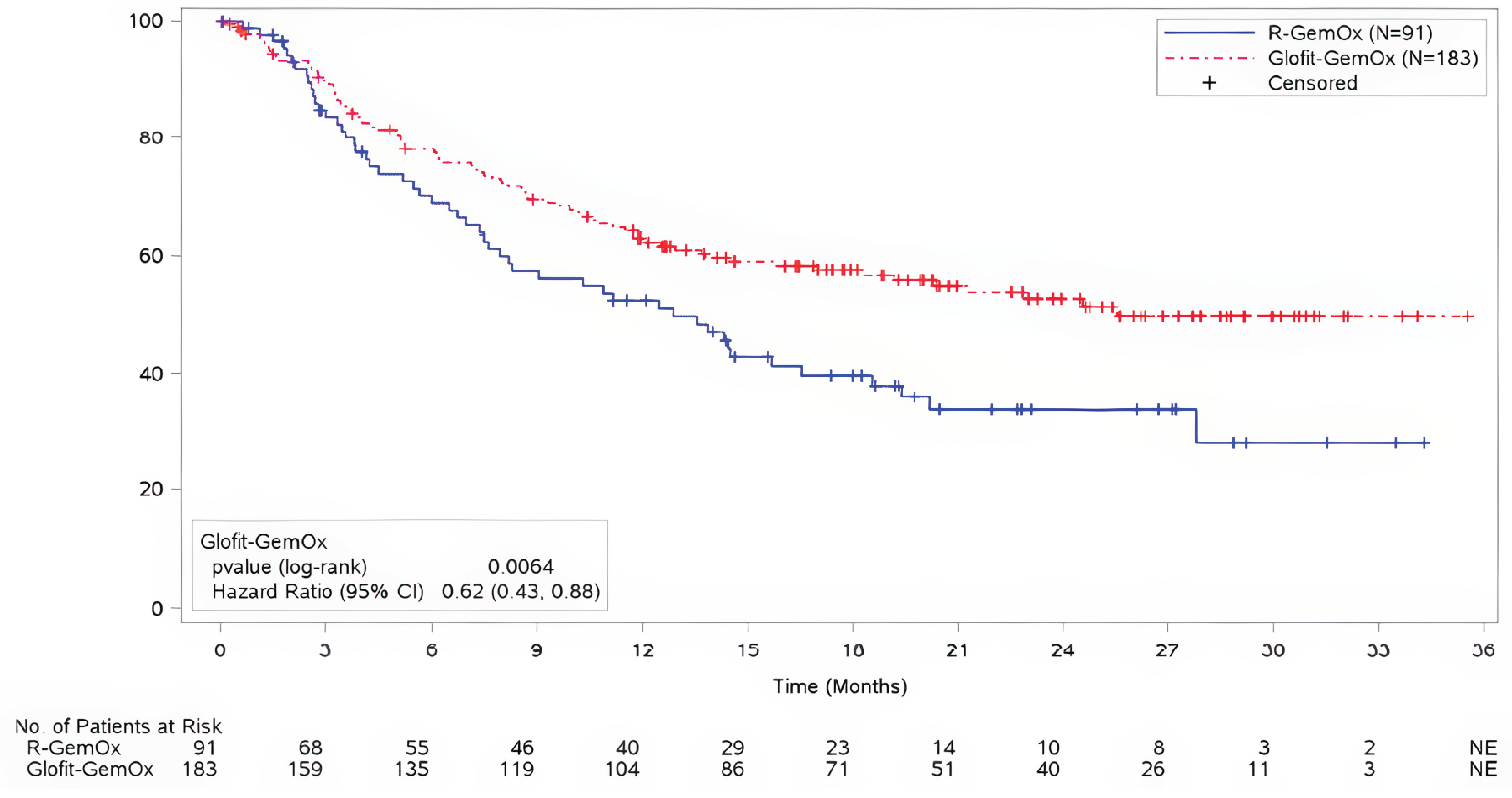
CI = confidence interval; Glofit-GemOx = glofitamab in combination with gemcitabine and oxaliplatin; ITT = intention to treat; NE = not estimable; R-GemOx = rituximab in combination with gemcitabine and oxaliplatin.
Source: STARGLO (GO41944) Clinical Study Report29 at the clinical cut-off dates of March 29, 2023, and February 16, 2024.
Pagebreak
Subgroup Analyses on Overall Survival
The following estimates were obtained in the subgroup analyses for the OS at the time of the primary analyses.
Table 15: Estimates in the Subgroup Analyses for OS at the Time of Primary Analyses
Subgroup | Point estimate |
|---|---|
ECOG PS 0 | HR = 0.56 (95% CI, 0.29 to 1.06) |
ECOG PS 1 | HR = 0.46 (95% CI, 0.25 to 0.84) |
ECOG PS 2 | HR = 1.67 (95% CI, 0.48 to 5.87) |
Refractory to the last line of therapy | HR = 0.62 (95% CI, 0.39 to 0.99) |
Relapsed to the last line of therapy | HR = 0.49 (95% CI, 0.21 to 1.16) |
Refractory to any line of therapy | HR = 0.64 (95% CI, 0.42 to 1.00) |
Relapsed to any line of therapy | HR = 0.43 (95% CI, 0.15 to 1.24) |
Cell of origin — ABC | HR = 1.41 (95% CI, 0.12 to 15.84) |
Cell of origin — GCB | HR = 0.36 (95% CI, 0.17 to 0.78) |
Cell of origin — non-GCB | HR = 0.70 (95% CI, 0.42 to 1.16) |
Prior CAR T-cell therapy | HR = 0.63 (95% CI, 0.11 to 3.53) |
No prior CAR T-cell therapy | HR = 0.61 (95% CI, 0.40 to 0.92) |
Prior SCT | HR > 999.99 (95% CI, 0 to not estimable) |
No prior SCT | HR = 0.60 (95% CI, 0.40 to 0.89) |
ABC = activated B-cell–like; CAR = chimeric antigen receptor; CI = confidence interval; ECOG PS = Eastern Cooperative Oncology Group Performance Status; HR = hazard ratio; GCB = germinal centre B-cell–like; OS = overall survival; SCT = stem cell transplant.
The following estimates were obtained in the subgroup analyses for the OS at the time of the updated analyses.
Table 16: Estimates in the Subgroup Analyses for OS at the Time of the Updated Analyses
Subgroup | Point estimate |
|---|---|
ECOG PS 0 | HR = 0.60, (95% CI, 0.35 to 1.05) |
ECOG PS 1 | HR = 0.46 (95% CI, 0.27 to 0.77) |
ECOG PS 2 | HR = 1.61 (95% CI, 0.53 to 4.89) |
Refractory to the last line of therapy | HR = 0.65 (95% CI, 0.43 to 0.99) |
Relapsed to the last line of therapy | HR = 0.51 (95% CI, 0.26 to 0.98) |
Refractory to any line of therapy | HR = 0.64 (95% CI, 0.42 to 1.00) |
Relapsed to any line of therapy | HR = 0.43 (95% CI, 0.15 to 1.24) |
Cell of origin — ABC | HR = 0.69 (95% CI, 0.18 to 2.60) |
Cell of origin — GCB | HR = 0.66 (95% CI, 0.35 to 1.25) |
Cell of origin — non-GCB | HR = 0.44 (95% CI, 0.17 to 1.17) |
Prior CAR T-cell therapy | HR = 0.84 (95% CI, 0.23 to 3.01) |
No prior CAR T-cell therapy | HR = 0.62 (95% CI, 0.43 to 0.89) |
Prior SCT | HR = 0.62 (95% CI, 0.09 to 4.50) |
No prior SCT | HR = 0.63 (95% CI, 0.44 to 0.90) |
ABC = activated B-cell–like; CAR = chimeric antigen receptor; CI = confidence interval; ECOG PS = Eastern Cooperative Oncology Group Performance Status; HR = hazard ratio; GCB = germinal centre B-cell–like; OS = overall survival; SCT = stem cell transplant.
IRC-Assessed Progression-Free Survival
The results for PFS at the March 29, 2023, primary analyses in the overall ITT population are summarized in Table 17 and displayed in Figure 7. Sixty-eight patients (37.2%) in the Glofit-GemOx arm and 44 patients (48.4%) in the R-GemOx arm experienced an event (death or disease progression). At the time of the primary analysis, with median durations of follow-ups of 9.0 months (95% CI, 6.2 to 9.7) in the Glofit-GemOx arm and 6.1 months (95% CI, 3.4 to 8.8) in the R-GemOx arm, the median PFSs were 12.1 months (95% CI, 6.8 to 18.3) for the Glofit-GemOx and 3.3 months (95% CI, 2.5 to 5.6) for the R-GemOx arm. These findings for PFS favoured Glofit-GemOx (HR = 0.37, CI, 0.25 to 0.55). Post hoc sensitivity analyses of IRC-assessed PFS in the ITT population without censoring for NALTs were consistent with the results of the IRC-assessed OS analysis in the ITT population censored before NALTs.
Figure 7: Kaplan-Meier Plot of IRC-Assessed PFS in the STARGLO Study (ITT Population, Primary Analysis, March 29, 2023)
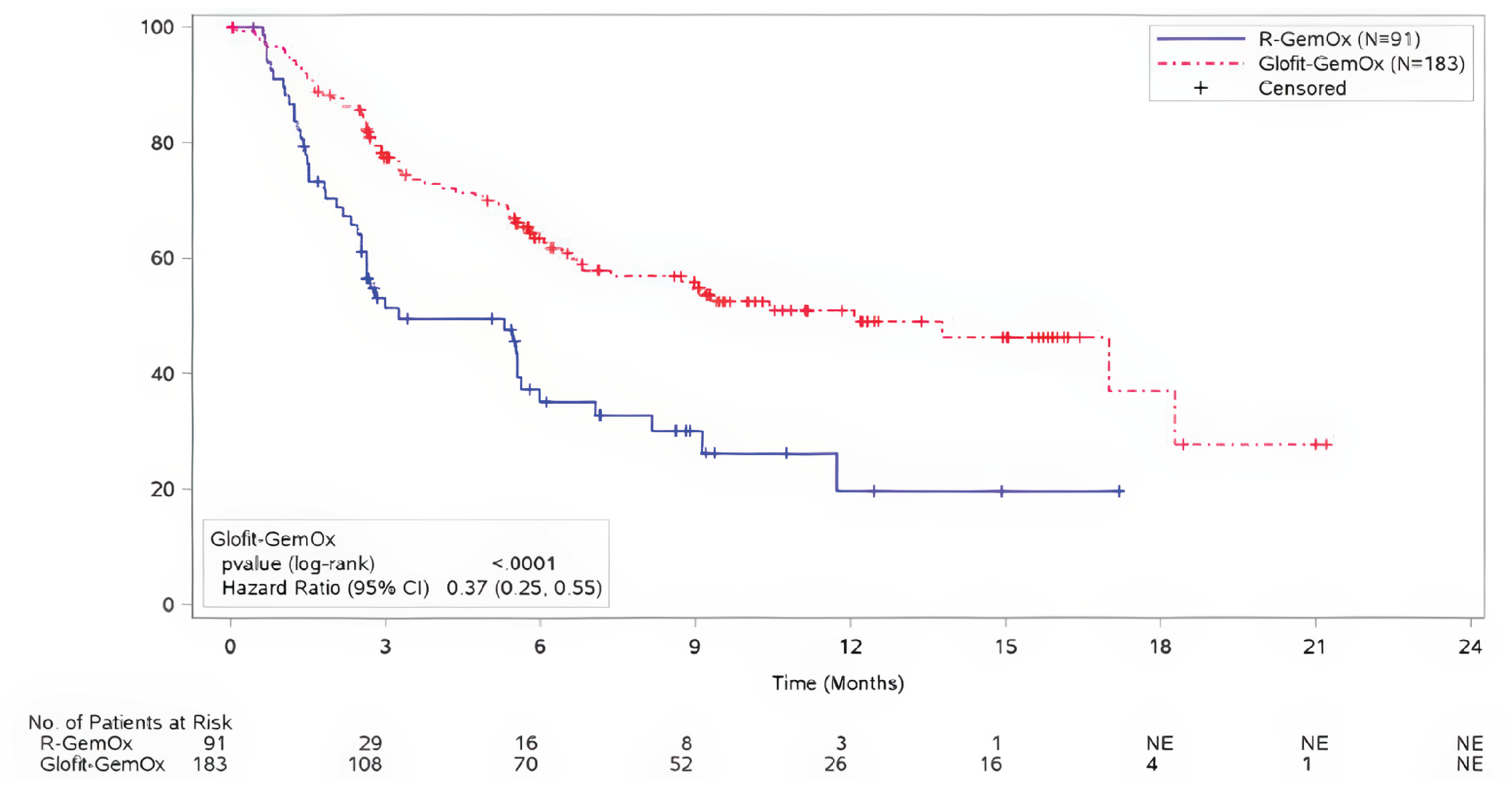
CI = confidence interval; Glofit-GemOx = glofitamab in combination with gemcitabine plus oxaliplatin; IRC = independent review committee; ITT = intention to treat; NE = not estimable; PFS = progression-free survival ; R-GemOx = rituximab in combination with gemcitabine and oxaliplatin.
Note: Censored before new antilymphoma treatment.
Source: STARGLO (GO41944) Clinical Study Report29 at the clinical cut-off dates of March 29, 2023, and February 16, 2024.
The results for PFS at the February 16, 2024, updated analyses, are summarized in Table 17 and displayed in Figure 8. Ninety patients (49.2%) in the Glofit-GemOx arm and 54 patients (59.3%) in the R-GemOx arm had a PFS event. At the time of the updated analysis, with median durations of follow-ups of 16.3 months (95% CI, 15.3 to 20.1) in the Glofit-GemOx arm and 8.6 months (95% CI, 5.9 to 14.6) in the R-GemOx arm, the median PFSs were 13.8 months (95% CI, 8.7 to 20.5) for the Glofit-GemOx arm (95% CI, 13.8 to NE) and 3.6 months (95% CI, 2.5 to 7.1) in the R-GemOx arm. These findings for PFS favoured Glofit-GemOx (HR = 0.40; 95% CI, 0.28 to 0.57). Post hoc sensitivity analyses of the IRC-assessed PFS in the ITT population without censoring for NALT were consistent with the results of the IRC-assessed PFS analysis in the ITT population censored before NALT.
Figure 8: Kaplan-Meier Plot of IRC-Assessed PFS in the STARGLO Study (ITT Population, Updated Analysis, February 16, 2024)
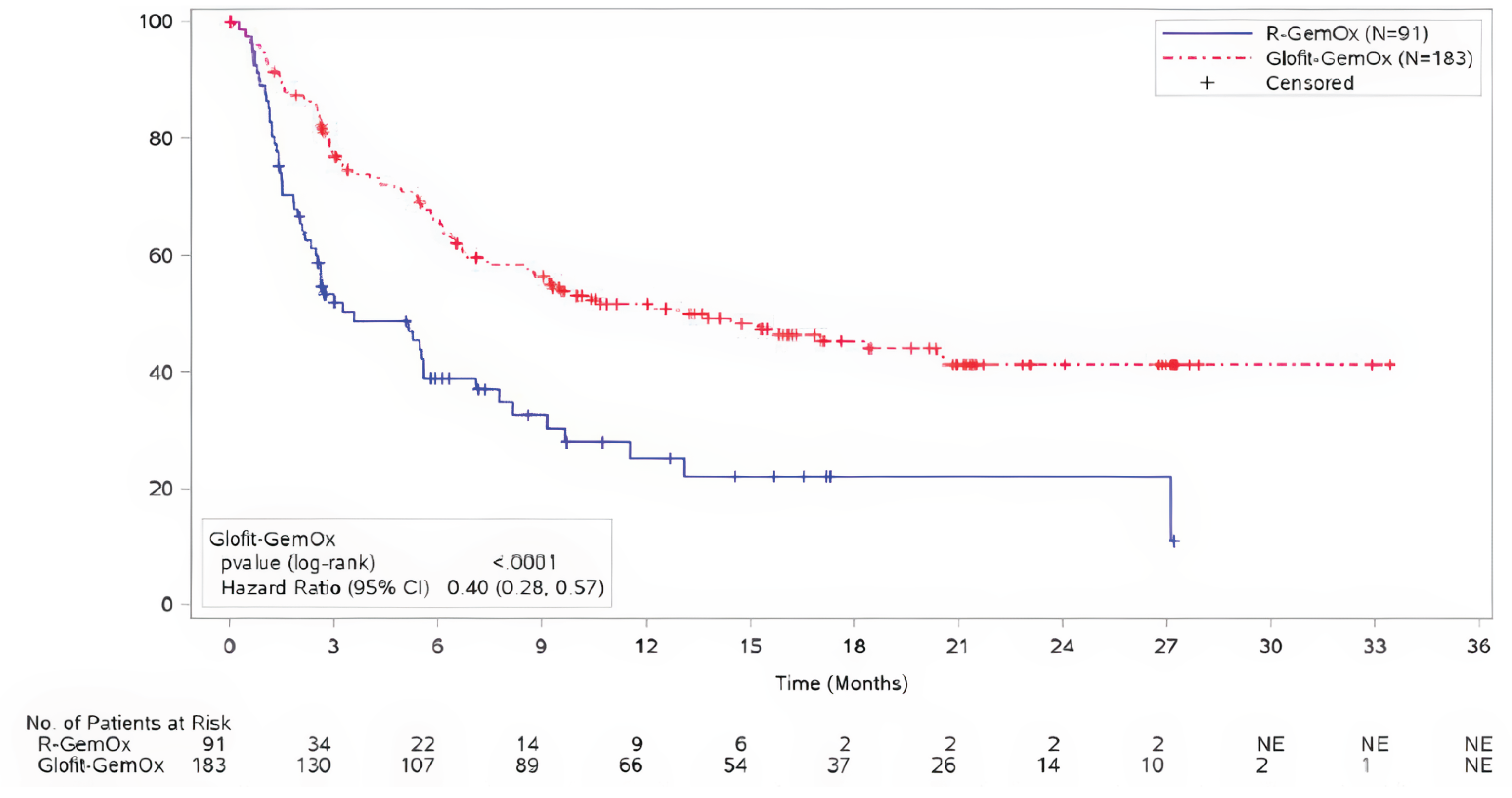
CI = confidence interval; Glofit-GemOx = glofitamab in combination with gemcitabine plus oxaliplatin; IRC = independent review committee; ITT = intention to treat; NE = not estimable; PFS = progression-free survival; R-GemOx, rituximab in combination with gemcitabine plus oxaliplatin.
Note: PFS was censored before new antilymphoma treatment.
Source: STARGLO (GO41944) Clinical Study Report29 at the clinical cut-off dates of March 29, 2023, and February 16, 2024.
IRC-Assessed Complete Response
The results for CR rate at the March 29, 2023, primary analyses in the overall ITT population are summarized in Table 17. The CR rates were 50.3% (95% CI, 42.8 to 57.7) and 22.0% (95% CI, 14.0 to 31.9) in the Glofit-GemOx (N = 183) and R-GemOx (N = 91) arms, respectively, with a between-group difference of 28.3% (95% CI, 16.3 to 40.3; P < 0.0001) in favour of Glofit-GemOx.
At the February 16, 2024, updated analyses of the ITT population, the CR rates were 58.5% (95% CI, 51.0 to 65.7) and 25.3% (95% CI, 16.8 to 35.5) in the Glofit-GemOx (N = 183) and R-GemOx (N = 91) arms, respectively, with a between-group difference of 33.2% (95% CI, 20.9 to 45.5; P < 0.0001) in favour of Glofit-GemOx (Table 17).
IRC-Assessed Duration of Objective Response
The results for IRC-assessed DOR at the March 29, 2023, primary analyses in the overall ITT population, with median durations of follow-ups of 6.9 months (95% CI, 6.3 to 7.6) in the Glofit-GemOx arm and 5.8 months (95% CI, 2.8 to 6.1) in the R-GemOx arm, are summarized in Table 17 and displayed in Figure 9. In the Glofit-GemOx arm, of the 110 patients (60.1%) who achieved an IRC-assessed response, 26 (23.6%) had subsequent disease progression or died by the time of the data cut-off. In the R-GemOx arm, of the 29 patients (31.9%) who achieved an IRC-assessed response, 8 (27.6%) had subsequent disease progression or died by the time of the data cut-off. The IRC-assessed median DORs in patients were 15.4 months (95% CI, 14.4 to NE) and 9.1 months (95% CI, 5.3 to NE) in the Glofit-GemOx and R-GemOx arms, respectively.
Figure 9: Kaplan-Meier Plot of IRC-Assessed DOR in the STARGLO Study (ITT Population, Primary Analysis, March 29, 2023)
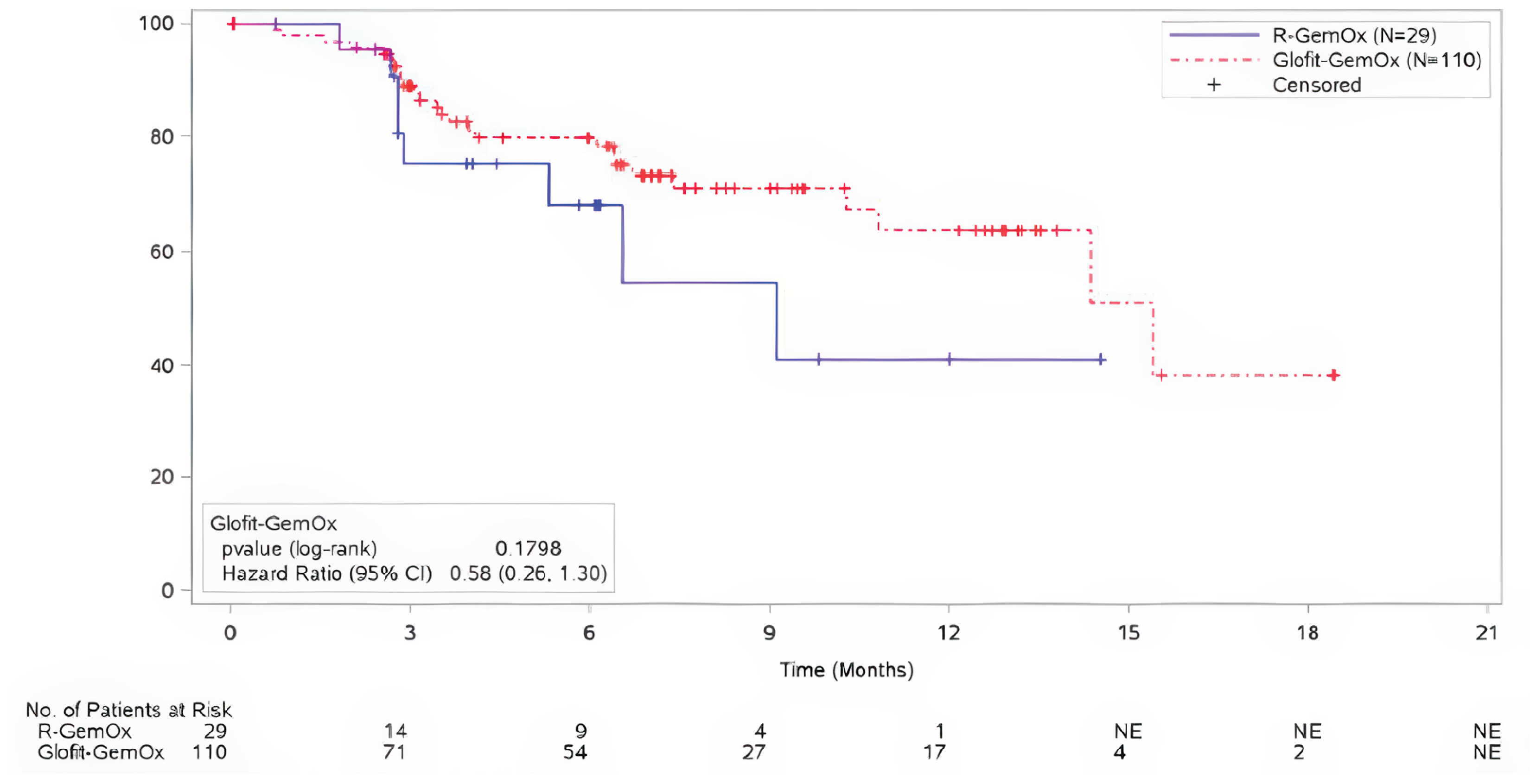
CI = confidence interval; DOR = duration of response; Glofit-GemOx = glofitamab in combination with gemcitabine plus oxaliplatin; IRC = independent review committee; ITT = intention to treat; NE = not estimable; R-GemOx = rituximab in combination with gemcitabine plus oxaliplatin.
Source: STARGLO (GO41944) Clinical Study Report29 at the clinical cut-off date of March 29, 2023, and February 16, 2024.
The results for IRC-assessed DOR at the February 16, 2024, updated analyses in the overall ITT population are summarized in Table 17 and displayed in Figure 10. With median durations of follow-up of 14.3 months (95% CI, 13.0 to 18.0) in the Glofit-GemOx arm and 5.8 months (95% CI, 3.0 to 12.5) in the R-GemOx arm, of the 125 patients (68.3%) in the Glofit-GemOx arm who achieved an IRC-assessed response, 42 (33.6%) had subsequent disease progression or died by the time of the data cut-off. Of the 37 patients (40.7%) in the R-GemOx arm who achieved an IRC-assessed response, 12 (32.4%) had subsequent disease progression or died by the time of the data cut-off. The IRC-assessed median DORs were not reached (NE months; 95% CI, 17.6 to NE) in the Glofit-GemOx arm and 10.3 months (95% CI, 6.5 to NE) in the R-GemOx arm.
Figure 10: Kaplan-Meier Plot of IRC-Assessed DOR in the STARGLO Study (ITT Population, Updated Analysis, February 16, 2024)
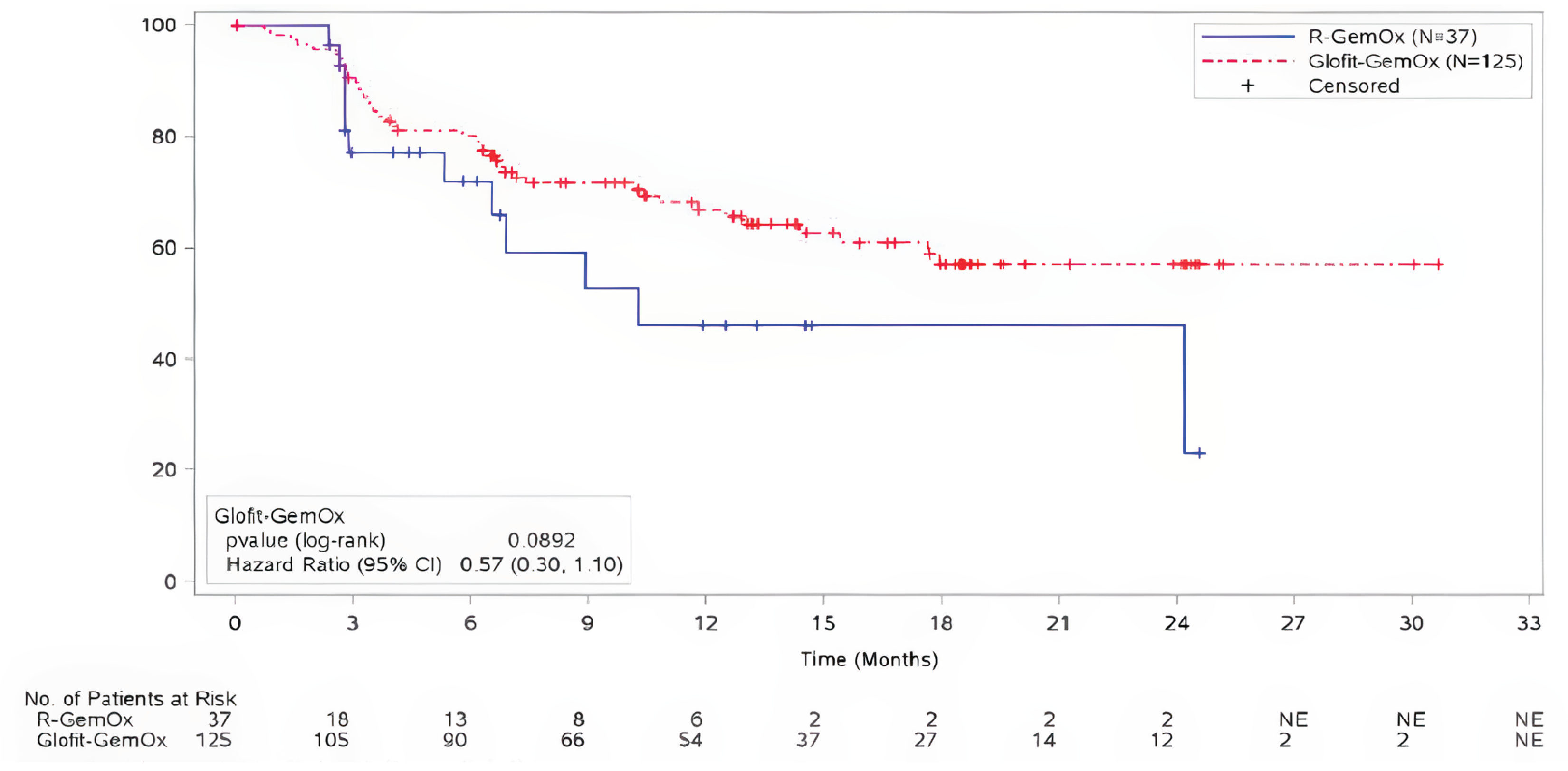
CI = confidence interval; DOR = duration of response; Glofit-GemOx = glofitamab in combination with gemcitabine plus oxaliplatin; IRC = independent review committee; ITT = intention to treat; NE = not estimable; R-GemOx = rituximab in combination with gemcitabine plus oxaliplatin.
Source: STARGLO (GO41944) Clinical Study Report29 at the clinical cut-off dates of March 29, 2023, and February 16, 2024.
Change From Baseline in EORTC QLQ-C30 Fatigue
At baseline, the EORTC QLQ-C30 fatigue scores for Glofit-GemOX and R-GemOx were 32.98 (SD = 25.20) and 32.18 (SD = 21.17), respectively. The results for the EORTC QLQ-C30 fatigue subscale at the March 29, 2023, primary analyses and in the February 16, 2024, updated analyses in the overall ITT population are summarized in Table 17.
At baseline, the EORTC QLQ-C30 physical functioning scores for Glofit-GemOX and R-GemOx were 77.14 (SD = 20.01) and 76.63 (SD = 18.72), respectively. At baseline, the mean EORTC QLQ-C30 fatigue score for the 175 patients in Glofit-GemOx arm was 32.98 (SD = 25.20) and for the 87 patients in the R-GemOx arm it was 32.18 (SD = 21.17).
In the primary analyses, the mean changes from baseline in EORTC QLQ-C30 fatigue scores were 2.42 (SD = 23.10) in the Glofit-GemOx arm and 4.98 (SD = 22.43) in the R-GemOx arm at the end of treatment (either completion or discontinuation).
In the February 16, 2024, updated analyses, the mean changes from baseline in EORTC QLQ-C30 fatigue scores were −1.24 (SD = 25.64) in the Glofit-GemOx arm and 4.59 (SD = 21.64) in the R-GemOx arm at the end of treatment (either completion or discontinuation).
Change From Baseline in EORTC QLQ-C30 Physical Functioning
The results for the EORTC QLQ-C30 physical functioning subscale at the March 29, 2023, primary analyses and in the February 16, 2024, updated analyses in the overall ITT population are summarized in Table 17.
At baseline, the mean EORTC QLQ-C30 physical functioning score for the 175 patients in the Glofit-GemOx arm was 77.14 (SD = 20.01) and for the 87 patients in the R-GemOx arm it was 76.63 (18.72) in the primary analyses.
In the primary analyses, the mean changes from baseline in EORTC QLQ-C30 physical functioning scores were −1.65 (SD = 19.97) in the Glofit-GemOx arm and −4.25 (SD = 17.82) in the R-GemOx arm at the end of treatment (either completion or discontinuation).
In the updated analyses, the mean changes from baseline in EORTC QLQ-C30 physical functioning score were 0.36 (SD = 20.29) in the Glofit-GemOx arm and −4.13 (SD = 17.12) in the R-GemOx arm at the end of treatment (either completion or discontinuation).
Change From Baseline in FACT-Lym LymS
The results for the FACT-Lym LymS at the March 29, 2023, primary analyses and in the February 16, 2024, updated analyses in the overall ITT population are summarized in Table 17. Time-to-deterioration analyses are available in Appendix 1 for additional HRQoL context.
At baseline, the mean FACT-Lym LymS score for the 175 patients in Glofit-GemOx arm was 45.38 (SD = 10.60) and for the 86 patients in the R-GemOx arm it was 43.90 (SD = 10.43).
In the primary analyses, the mean changes from baseline in the FACT-Lym LymS score were 1.71 (SD = 10.58) in the Glofit-GemOx arm and 0.41 (SD = 8.00) in the R-GemOx arm at the end of treatment (either completion or discontinuation).
In the updated analyses, the mean changes from baseline in the FACT-Lym LymS score were 2.32 (SD = 10.05) in the Glofit-GemOx arm and 0.64 (SD = 7.82) in the R-GemOx arm at the end of treatment (either completion or discontinuation).
Table 17: Summary of Key Efficacy Results From Studies Included in the Systematic Review (ITT Population)
Variable | Primary analysis data cut-off March 29, 2023 | Updated analysis data cut-off February 16, 2024 | ||
|---|---|---|---|---|
Glofit-GemOx (N = 183) | R-GemOx (N = 91) | Glofit-GemOx (N = 183) | R-GemOx (N = 91) | |
Overall survival | ||||
Patients alive, n (%) | 122 (66.7%) | 51 (56.0%) | 103 (56.3%) | 39 (42.9%) |
Deaths, n (%) | 61 (33.3) | 40 (44.0) | 80 (43.7) | 52 (57.1) |
Median time to event in months (95% CI) | NE (13.8 to NE) | 9.0 (7.3 to 14.4) | 25.5 (18.3 to NE) | 12.9 (7.9 to 18.5) |
Stratified HR (95% CI) | 0.59 (0.40 to 0.89) | 0.62 (0.43 to 0.88) | ||
Log-rank P value (stratified) | 0.010706 | 0.006366 | ||
6-month survival ratea (95% CI) | N = 110 (70.3 to 83.3) | N = 43 (56.7 to 78.3) | N = 135 (71.9 to 84.1) | N = 55 (58.7 to 78.7) |
Difference in survival rate (95% CI) | 9.29 (–3.3 to 21.9) | 9.27 (–2.5 to 21.0) | ||
12-month survival ratea (95% CI) | N = 57 (54.4 to 70.7) | N = 15 (31.6 to 57.6) | N = 104 (55.7 to 70.1) | N = 40 (41.6 to 63.3) |
Difference in survival rate (95% CI) | 17.9 (2.6 to 33.2) | 10.4 (–2.6 to 23.4) | ||
18-month survival ratea (95% CI) | N = 14 (44.7 to 64.6) | N = 7 (21.1 to 49.0) | N = 71 (50.2 to 65.1) | N = 23 (28.7 to 50.7) |
Difference in survival rate (95% CI) | 19.6 (2.4 to 36.8) | 18.0 (4.7 to 31.2) | ||
Progression-free survival (IRC assessed) | ||||
Patients with event, n (%) | 68 (37.2) | 44 (48.4) | 90 (49.2) | 54 (59.3) |
Median, months (95% CI) | 12.1 (6.8 to 18.3) | 3.3 (2.5 to 5.6) | 13.8 (8.7 to 20.5) | 3.6 (2.5 to 7.1) |
Stratified HR (95% CI) | 0.37 (0.25 to 0.55) | 0.40 (0.28 to 0.57) | ||
P value (log rank) | < 0.000001 | < 0.000001a | ||
6-month event-free rate (95% CI) | N = 70 (55.6 to 71.6) | N = 16 (22.6 to 47.7) | N = 107 (58.7 to 73.0) | N = 22 (27.6 to 50.4) |
Difference in survival rate (95% CI) | NR | 26.85 (13.40 to 40.31) | ||
12-month event-free rate (95% CI) | N = 26 (41.8 to 60.1) | N = 3 (4.9 to 34.6) | N = 66 (44.0 to 59.4) | N = 9 (13.6 to 36.9) |
Difference in survival rate (95% CI) | NR | 26.46 (12.48 to 40.45) | ||
18-month event-free rate (95% CI) | N = 4 (18.8 to 55.3) | NE (NE to NE) | N = 37 (37.3 to 53.5) | N = 2 (10.4 to 33.8) |
Difference in survival rate (95% CI) | NR | 23.31 (9.05 to 37.56) | ||
Complete response rate (IRC-assessed) | ||||
Complete responders, n (%) | 92 (50.3) | 20 (22.0) | 107 (58.5) | 23 (25.3) |
95% CI | (42.8 to 57.7) | (14.0 to 31.9) | (51.0 to 65.7) | (16.8 to 35.5) |
Difference in CR rate, (95% CI) | 28.3% (16.3 to 40.3) | 33.2% (20.9 to 45.5) | ||
P value (CMH) | < 0.0001 | < 0.0001a | ||
Duration of objective response | ||||
Responders, n | 110 (54.6) | 29 (31.9) | 125 (68.3) | 37 (40.7) |
Patients with event, n (%) | 26 (23.6) | 8 (27.6) | 42 (33.6) | 12 (32.4) |
Median, months (95% CI) | 15.4 (14.4 to NE) | 9.1 (5.3 to NE) | NE (17.6 to NE) | 10.3 (6.5 to NE) |
Unstratified HR (95% CI) | 0.58 (0.26 to 1.30) | 0.57 (0.30 to 1.10) | ||
P value (log rank) | 0.1798 | 0.0892 | ||
6-month event-free rate (95% CI) | N = 54 (71.1 to 88.5) | N = 9 (47.0 to 89.8) | N = 90 (72.9 to 87.4) | N = 13 (54.0 to 89.9) |
12-month event-free rate (95% CI) | N = 17 (50.1 to 77.4) | N = 1 (9.0 to 73.1) | N = 54 (57.9 to 75.9) | N = 6 (22.8 to 69.5) |
18-month event-free rate (95% CI) | N = 2 (9.7 to 66.8) | N = NE (NE to NE) | N = 27 (46.4 to 67.8) | N = 2 (22.8 to 69.5) |
Change from baseline in EORTC QLQ-C30 fatigue | ||||
Baseline, n | 175 | 87 | 175 | 87 |
Score, mean (SD) | 32.98 (25.20) | 32.18 (21.17) | 32.98 (25.20) | 32.18 (21.17) |
Long-term follow-up at month 6, n (%) | 36 (20.6%) | 12 (13.8%) | 75 (42.9%) | 28 (32.2%) |
Change from baseline, mean (SD) | −3.24 (22.16) | 4.63 (23.91) | −1.70 (24.83) | 1.19 (23.68) |
Long-term follow-up at month 12, n (%) | 8 (4.6%) | 6 (6.9%) | 52 (29.7%) | 21 (24.1%) |
Change from baseline, mean (SD) | −1.39 (27.50) | −7.41 (24.00) | −0.75 (22.00) | −2.65 (25.07) |
Long-term follow-up at month 18, n (%) | 1 (0.6%) | 0 (0%) | 21 (12.0%) | 7 (8.0%) |
Change from baseline, mean (SD) | 33.33 (NE) | NE (NE) | −7.94 (16.53) | 12.70 (26.78) |
Change from baseline in EORTC QLQ-C30 physical functioning | ||||
Baseline, n | 175 | 87 | 175 | 87 |
Score, mean (SD) | 77.14 (20.01) | 76.63 (18.72) | 77.30 (20.03) | 76.63 (18.72) |
Long-term follow-up at month 6, n (%) | 36 (20.6%) | 12 (13.8%) | 75 (42.9%) | 28 (32.2%) |
Change from baseline, mean (SD) | −1.11 (17.49) | −2.78 (24.03) | 0.98 (18.97) | −0.95 (22.20) |
Long-term follow-up at month 12, n (%) | 8 (4.6%) | 6 (6.9%) | 52 (29.7%) | 12 (13.8%) |
Change from baseline, mean (SD) | −2.50 (21.06) | 14.44 (12.23) | −3.72 (14.90) | 3.17 (18.57) |
Long-term follow-up at month 18, n (%) | 1 (0.6%) | 0 (0%) | 21 (12.0%) | 7 (8.0%) |
Change from baseline in FACT-Lym LymS | ||||
Baseline, n | 175 | 86 | 175 | 86 |
Score, mean (SD) | 45.38 (10.60) | 43.90 (10.43) | 45.38 (10.60) | 43.90 (10.43) |
Long-term follow-up at month 6, n (%) | 36 (20.6%) | 12 (13.8%) | 75 (42.9%) | 28 (32.2%) |
Change from baseline, mean (SD) | 2.58 (10.70) | 5.25 (9.70) | 2.80 (9.42) | 4.30 (8.35) |
Long-term follow-up at month 12, n (%) | 7 (4.0%) | 6 (6.9%) | 52 | 21 |
Change from baseline, mean (SD) | 4.86 (7.80) | 10.50 (11.31) | 3.30 (10.66) | 5.81 (11.47) |
Long-term follow-up at month 18, n (%) | 1 (0.6%) | 0 (0%) | 21 (12.0%) | 7 (8.0%) |
CI = confidence interval; CMH = Cochran-Mantel-Haenszel; CR = complete response; DOR = duration of response; EORTC QLQ-C30 European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; Glofit-GemOx = glofitamab in combination with gemcitabine and oxaliplatin; FACT-Lym = Functional Assessment of Cancer Therapy–Lymphoma; HR = hazard ratio; IRC = independent review committee; ITT = intention to treat; LymS = lymphoma subscale; NE = not estimable; NR = not reported; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; R-GemOx = rituximab in combination with gemcitabine and oxaliplatin; SD = standard deviation.
Note: IRC-assessed response rates were assessed using the Lugano classification.12 The randomization stratification factors to be used in the efficacy analyses are number of previous lines of systemic therapy for diffuse large B-cell lymphoma (1 versus ≥ 2) and outcome of last systemic therapy (relapsed versus refractory).
aEstimated by Kaplan-Meier analysis (OS, PFS, DOR); estimated by Cochran-Mantel-Haenszel test (CR, ORR).
Source: STARGLO (GO41944) Clinical Study Report29 at the clinical cut-off dates of March 29, 2023, and February 16, 2024.
Harms
Adverse Events
At the primary and updated analyses, the majority of patients in the safety-evaluable populations of both the Glofit-GemOx and R-GemOx treatment arms (i.e., patients who receive any amount of any study treatment) experienced at least 1 AE (Table 18). A total of 171 patients (99.4%) and 172 patients (100%) treated with Glofit-GemOx experienced at least 1 AE at the primary and updated analyses, respectively. Eighty-four patients (95.5%) treated with R-GemOx experienced at least 1 AE at both time points.
The most frequently reported AEs in the Glofit-GemOx arm were CRS (primary analyses: 73 patients [42.4%]; updated analyses: 76 patients [44.2%]), nausea (primary analyses: 70 patients [40.7%]; updated analyses: 71 patients [41.3%]), and anemia (primary analyses: 69 patients [40.1%]; updated analyses: 71 patients [41.3%]).
The most frequently reported AEs in the R-GemOx arm were nausea (primary analyses: 34 patients [38.6%]; updated analyses: 35 patients [39.8%]), reduced platelet count (primary analyses: 25 patients [28.4%]; updated analyses: 27 patients [30.7%]) and diarrhea (primary analyses: 23 patients [26.1%]; updated analyses: 24 patients [27.3%]).
Serious Adverse Events
SAEs were reported in a greater proportion of patients in the Glofit-GemOx population exposed to any treatment (primary analyses: 89 patients [51.7%]; updated analyses: 98 patients [54.4%]), compared with the R-GemOx population (primary analyses: 15 patients [17.0%]; updated analyses; 15 patients [17.0%]).
At the primary analyses, the most common SAEs with an incidence rate equal to or higher than 5% in the Glofit-GemOx and R-GemOx populations were CRS (19.4% versus 0.0%, respectively), pneumonia (6.7% versus 3.4%, respectively), COVID-19 (5.0% versus 2.3%, respectively), and pyrexia (5.6% versus 0%, respectively).29
At the updated analyses, the most common SAEs with an incidence rate equal to or higher than 5% in the Glofit-GemOx and R-GemOx populations were CRS (20.3% versus 0.0%, respectively), pyrexia (6.4% versus 1.1%, respectively), and pneumonia (5.8% versus 4.5%, respectively).29
A greater proportion of patients in the Glofit-GemOx arm than in the R-GemOx arm experienced SAEs that led to discontinuation from glofitamab or rituximab (primary analyses: 11.0% versus 4.5%, respectively); updated analyses: 12.2% versus 6.8%, respectively).
Withdrawals due to Adverse Events
Withdrawal of any study treatment was reported in a greater proportion of patients in the Glofit-GemOx population exposed to any treatment (primary analyses: 46 patients [25.6%]; updated analyses; 48 patients [26.7%]), compared with the R-GemOx population (primary analyses: 9 patients [10.2%]; updated analyses; 11 patients [12.5%]). AEs leading to the withdrawal of specific treatment, dose interruptions, or dose modifications are reported in Table 18.
Notable Harms
Cytokine Release Syndrome
All CRS AEs met the criteria for an SAE due to hospitalization or prolonged hospitalization. At the time of the primary analysis (March 29, 2023), 73 patients (40.6%) and 0 patients exposed to any treatment had experienced CRS events of any grade in the Glofit-GemOx and R-GemOx arms, respectively. At the time of the updated analysis (February 16, 2024), 76 patients (42.2%) and 0 patients exposed to any treatment had experienced CRS events of any grade in the Glofit-GemOx and R-GemOx arms, respectively. In patients receiving Glofit-GemOx, the majority of CRS AEs were low grade, with grade 1 CRS events accounting for 30.2% of AEs and grade 2 CRS events accounting for 30.2% at the primary analysis, compared with 31.4% for grade 1 events and 10.5% for grade 2 events at the updated analysis. A total of 35 of 172 patients (20.3%) experienced a serious CRS event at both analyses time points (primary analysis: grade 1 event reported in 18 patients [10.5%], grade 2 events reported in 14 patients [8.1%], and grade 3 events reported in 3 patients [1.7%];29 updated analysis: grade 1 events reported in 17 patients [9.9%], grade 2 events reported in 15 patients [8.7%], and grade 3 events reported in 3 patients [1.7%]).
In the primary analysis, 21 of 172 patients (12.2%) experienced a CRS of grade 2 or higher, with grade 3 CRS events reported in 4 patients (2.3%) and no grade 4 or grade 5 CRS reported. In the updated analysis, 1 additional patient experienced a grade 2 CRS AE. Three patients (1.7%) had their dose interrupted because of a CRS AE of grade 2 or higher at both analyses. At the primary analysis, 1 patient (0.6%) had glofitamab treatment discontinued for the same reason, and CRS AEs of grade 2 or higher were unresolved for 1 patient (0.6%). At the updated analysis, all CRS AEs of grade 2 or higher were resolved.29
Table 18: Summary of Harms Results From Studies Included in the Systematic Review (Safety-Evaluable Population)
AE profile | Primary analysis (March 29, 2023) | Updated analysis (February 16, 2024) | ||
|---|---|---|---|---|
Glofit-GemOx (Glofit-exposed) (N = 172) | R-GemOx | Glofit-GemOx (Glofit-exposed) (N = 172) | R-GemOx | |
Total number of patients with at least 1 AE (%) | 171 (99.4) | 84 (95.5) | 172 (100) | 84 (95.5) |
Total number of AEs | 3,099 | 811 | 3,299 | 868 |
Total number of patients with at least 1 SAE | 89 (51.7%) | 15 (17.0%) | 98 (54.4%) | 15 (17.0%) |
Total numbers of deaths (%) | 55 (32.0) | 39 (44.3) | 74 (43.0) | 51 (58.0) |
Total number of patients discontinued from any treatment because of an AE (%) | 41 (23.8) | 9 (10.2) | 43 (25.0) | 11 (12.5) |
Total number of patients with an AE with fatal outcome (%) | 12 (7.0) | 3 (3.4) | 12 (7.0) | 4 (4.5) |
AEs leading to treatment discontinuation of glofitamab or rituximab, n (%) | 34 (19.8) | 9 (10.2) | 36 (20.9) | 11 (12.5) |
AEs leading to treatment discontinuation of obinutuzumab, n (%) | 2 (1.2) | 0 | 1 (0.6) | 0 |
AEs leading to treatment discontinuation of gemcitabine, n (%) | 23 (13.4) | 9 (10.2) | 26 (15.1) | 11 (12.5) |
AEs leading to treatment discontinuation of oxaliplatin, n (%) | 25 (14.5) | 9 (10.2) | 29 (16.9) | 11 (12.5) |
AEs leading to dose interruption of glofitamab or rituximab, n (%) | 71 (41.3) | 12 (13.6) | 75 (43.6) | 14 (15.9) |
AE leading to dose interruption of obinutuzumab, n (%) | 10 (5.8) | 1 (1.1) | 8 (4.7) | 0 |
AEs leading to dose interruption of gemcitabine, n (%) | 56 (32.6) | 12 (13.6) | 61 (35.5) | 14 (15.9) |
AEs leading to dose interruption of oxaliplatin, n (%) | 55 (32.0) | 12 (13.6) | 60 (34.9) | 14 (15.9) |
AEs leading to dose modification of gemcitabine, n (%) | 2 (1.2) | 0 | 2 (1.2) | 0 |
AEs leading to dose modification of oxaliplatin, n (%) | 5 (2.9) | 5 (5.7) | 5 (2.9) | 6 (6.8) |
SAEs leading to discontinuation from glofitamab or rituximab | 19 (11.0) | 4 (4.5) | 21 (12.2) | 6 (6.8) |
SAEs leading to dose interruption of glofitamab or rituximab | 37 (21.5) | 4 (4.5) | 35 (20.3) | 5 (5.7) |
AEs related to glofitamab or rituximab leading to discontinuation from glofitamab or rituximab | 13 (7.6) | 3 (3.4) | 13 (7.6) | 3 (3.4) |
AEs related to glofitamab or rituximab leading to dose interruption from glofitamab of rituximab | 39 (22.7) | 7 (8.0) | 43 (25.0) | 9 (10.2) |
Total number of patients with at least 1 AE (%) | 170 (98.8) | 82 (93.2) | 170 (98.8) | 83 (94.3) |
Total number of AEs | 1,920 | 502 | 2,081 | 555 |
CRS,a n (%) | 73 (42.4)a | 0 | 76 (44.2)a | 0 |
Nausea, n (%) | 70 (40.7) | 34 (38.6) | 71 (41.3) | 35 (39.8) |
Anemia, n (%) | 69 (40.1) | 19 (21.6) | 71 (41.3) | 19 (21.6) |
Reduced platelet count, n (%) | 64 (37.2) | 25 (28.4) | 66 (38.4) | 27 (30.7) |
Increased aspartate transaminase, n (%) | 57 (33.1) | 16 (18.2) | 59 (34.3) | 17 (19.3) |
Diarrhea, n (%) | 56 (32.6) | 23 (26.1) | 60 (34.9) | 24 (27.3) |
Increased alanine transaminase, n (%) | 54 (31.4) | 18 (20.5) | 56 (32.6) | 19 (21.6) |
Reduced neutrophil count, n (%) | 50 (29.1) | 15 (17.0) | 51 (29.7) | 18 (20.5) |
Decreased appetite, n (%) | 45 (26.2) | 22 (25.0) | 50 (29.1) | 24 (27.3) |
Pyrexia, n (%) | 44 (25.6) | 4 (4.5) | 42 (24.4) | 5 (5.7) |
Vomiting, n (%) | 40 (23.3) | 17 (19.3) | 41 (23.8) | 19 (21.6) |
Fatigue, n (%) | 38 (22.1) | 17 (19.3) | 38 (22.1) | 19 (21.6) |
Reduced lymphocyte count, n (%) | 37 (21.5) | 12 (13.6) | 37 (21.5) | 12 (13.6) |
Reduced white blood cell count, n (%) | 35 (20.3) | 11 (12.5) | 37 (21.5) | 12 (13.6) |
Hypokalemia, n (%) | 30 (17.4) | 7 (8.0) | 32 (18.6) | 6 (6.8) |
Constipation, n (%) | 29 (16.9) | 13 (14.8) | 32 (18.6) | 14 (15.9) |
Increased blood alkaline phosphatase, n (%) | 27 (15.7) | 16 (18.2) | 31 (18.0) | 16 (18.2) |
Thrombocytopenia, n (%) | 27 (15.7) | 14 (15.9) | 26 (15.1) | 15 (17.0) |
COVID-19, n (%) | 27 (15.7) | 7 (8.0) | 28 (16.3) | 8 (9.1) |
Increased serum ferritin, n (%) | 26 (15.1) | 9 (10.2) | 29 (16.9) | 10 (11.4) |
Neutropenia, n (%) | 24 (14.0) | 10 (11.4) | 29 (16.9) | 9 (10.2) |
Neuropathy peripheral, n (%) | 22 (12.8) | 6 (6.8) | 20 (11.6) | 6 (6.8) |
Peripheral sensory neuropathy, n (%) | 20 (11.6) | 7 (8.0) | 27 (15.7) | 8 (9.1) |
Rash, n (%) | 22 (12.8) | 5 (5.7) | 23 (13.4) | 6 (6.8) |
AE = adverse event; CRS = cytokine release syndrome; Glofit-GemOx = glofitamab combined with gemcitabine and oxaliplatin; R-GemOx = rituximab combined with gemcitabine and oxaliplatin; SAE = serious adverse event.
Note: Investigator text for AEs encoded using the Medical Dictionary for Regulatory Activities, version 26.1. Percentages are based on N in the column headings. Only treatment-emergent AEs are displayed. Multiple occurrences of the same AE in 1 individual are counted only once except for the “Total number of AEs” row, in which multiple occurrences of the same AE are counted separately. Tocilizumab is not included as a study drug. A gemcitabine dose is considered modified if the accompanying oxaliplatin dose was modified, the AE is related to gemcitabine, and the dose is less than 80% of the planned dose.
aAccording to the American Society for Transplantation and Cellular Therapy 2019 grading criteria.
Source: STARGLO (GO41944) Clinical Study Report at the clinical cut-off dates of March 29, 2023,29 and February 16, 2024.45
Critical Appraisal
Internal Validity
The STARGLO trial was planned with adequate power to meet the goals of the study’s primary outcome (OS), randomizing 274 patients in a 2:1 ratio to Glofit-GemOx or R-GemOx. The assumptions made when performing power calculations were considered reasonable, and the randomization method, and allocation concealment, appeared adequate. Further, the randomization was stratified based on important prognostic factors to minimize important differences in baseline characteristics between groups and to allow for maintenance of randomization for exploratory subgroup analyses based on these baseline characteristics. The use of IRC assessments for the important outcomes (PFS and CR) provides objectivity to the outcome assessments. Furthermore, to minimize bias, the IRC remained blinded to treatment assignment and the sponsor did not have access to efficacy and safety summaries that compare treatment arms before the formal reporting of study results, with the exception that the randomization code may be made available to facilitate the analysis of pharmacokinetic samples. There were, however, some limitations and potential sources of bias are outlined in this section.
The reported results to date were based on an interim analysis at the clinical cut-off date of March 29, 2023, and a follow-up analysis was conducted at the subsequent clinical cut-off date of February 16, 2024. The final data will not be available for several years. At time of the primary analysis, the median OS was not yet reached, and the OS data were immature. Subsequently the median OS was reached at the time of the updated analysis. Moreover, estimates of the median DOR were not yet reached at the time of the primary analysis or the updated analysis, remaining immature. This is not unexpected as interim analyses are typically at risk of overestimating the true magnitude of benefit.
The STARGLO trial was open-label, and the lack of blinding may bias results, particularly for subjective, patient-reported outcomes. HRQoL self-reporting and reporting of more subjective AEs may also be influenced by a lack of blinding, as patients may anticipate known adverse effects and thus may be more likely to report them when they do occur. Physician knowledge of a patient’s assigned treatment may also affect the way they manage their patient, and patient knowledge of their assigned treatment may make them more or less likely to remain in the study. Objective clinical outcomes such as OS, PFS, and CR are less likely to be influenced by a lack of blinding, especially when they are evaluated by a blinded IRC, as in this case.
Multiplicity was controlled in the study by use of a hierarchical testing procedure. The procedure seems appropriate for the primary and key secondary outcomes (alpha spending between primary and updated analyses). Statistical significance was reached for the primary analyses, the subsequent clinical cut-off analyses were considered descriptive. Additionally, the trial was not stratified for most subgroup analyses, and imbalances may exist within the subgroups. Moreover, the subgroup analyses were not adjusted for multiplicity nor powered to detect effect modification. However, the results of most sensitivity and subgroup analyses for OS were generally consistent with the primary analysis supporting the consistency of the effect across subgroups.
Additionally, patients who completed study treatment or discontinued trial medications were allowed NALT based on investigator assessment; the use of NALTs was common in the study (32 of 183 patients [17.5%] in the Glofit-GemOx arm and 35 of 91 [38.5%] in the R-GemOx arm). This is expected to affect estimates of OS, although the direction and magnitude are uncertain.
For both OS and PFS, patients could be censored for several reasons, including missed assessments. The amount of censoring in both arms was very high, which can introduce bias and uncertainty in the estimates or bias the estimates, but the direction and magnitude are uncertain. There was no sensitivity analysis addressing each reason for censoring data, and the amount and direction of potential bias cannot be ascertained. The presented sensitivity analyses for the impact on the analysis of PFS for missing data and assessments due to the COVID-19 pandemic, and loss to follow-up and discontinuation of assessments of PFS not due to an event, PFS without censoring for new NALTs, and censoring for NALTs except for HSCT were supportive of the primary analysis.
At the time of the second interim analysis, most patients had discontinued treatment, mainly due to progressive disease or death. This large level of treatment discontinuation may influence any potential comparative evaluation and interpretation of HRQoL and harms outcomes. Also, the number of patients remaining to complete HRQoL assessments substantially declined over time, resulting in a risk of bias due to missing outcome data. As such, HRQoL was likely biased due to the large number of participants that did not complete the assessments during the follow-up period, with only those who were still alive and remaining in the trial providing most of the assessments.
External Validity
The STARGLO trial included patients who were relatively young (e.g., aged approximately 65 years) and in good general health (e.g., an ECOG PS of 0 to 2). Patients with a poor functional status may not be able to tolerate the toxicities. In addition, the trial excluded important DLBCL groups (e.g., those with a history of transformation of indolent disease to DLBCL, PMBCL, HGBCL with MYC and BCL2 and/or BCL6 rearrangements, and HGBCL NOS). Furthermore, 1 of the most common reasons for transplant ineligibility was patient choice. This is not expected to be a common reason in a typical patient in Canada who is otherwise eligible.
Furthermore, treatment would require inpatient admission so that detailed and specialized management algorithms can be followed. This restricts the use, and availability, of Glofit-GemOx in Canada, and may be a barrier for implementation, especially for individuals from rural communities. Because patients must be hospitalized and monitored for SAEs while receiving treatment, delays in beginning treatment may occur, even in communities with the required health care resources.
Last, while the study was conducted in 62 centres across 13 countries, none were in Canada. Therefore, the generalizability of the findings to the Canadian context is unclear, given the role of investigator discretion, potential for differences in practice patterns, or the resources used that might affect the results.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered most relevant to inform expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group:46,47
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate — The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited — The true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate — The true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as “very uncertain.”
Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null. Certainty in the effects for OS, PFS, and SAEs was based on the presence or absence of any effect, as a threshold of clinical importance could not be established. A literature-based minimal important difference was available for HRQoL; however the difference between groups was not tested statistically. Therefore, a narrative description of the findings was used.
The selection of outcomes for GRADE assessment was based on the sponsor’s Summary of Clinical Evidence, consultation with clinical experts, and input received from patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members:
OS at 6, 12, and 18 months
PFS at 6, 12, and 18 months
HRQoL using the FACT-Lym LymS: change from baseline at 6, 12, and 18 months
individuals with SAEs: up to the data cut-off.
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for Glofit-GemOx versus R-GemOx in patients with DBLCL NOS.
Long-Term Extension Studies
There is no long-term extension phase planned for the STARGLO trial at this time.
Indirect Evidence
Contents within this section have been informed by materials submitted by the sponsor. The following have been summarized and validated by the review team.
Objectives for the Summary of Indirect Evidence
The pivotal STARGLO trial provided a head-to-head comparison of Glofit-GemOx and R-GemOx among patients with r/r DLBCL. However, no direct evidence was included in the submission to support comparisons of the efficacy or safety of Glofit-GemOx with other available treatments for the patient population under review in Canada. Hence, an ITC is warranted to address this evidence gap.
Description of Indirect Comparisons
The sponsor submitted an ITC report with 2 analyses comparing Glofit-GemOx to relevant treatment comparators.
One of these was a matched adjusted indirect comparison (MAIC) analyses, in which IPD from the STARGLO (GO41944) study were weighted to match the prognostic factors and effect modifiers reported in each of the comparator studies (i.e., axicabtagene ciloleucel from the ALYCANTE trial; lisocabtagene maraleucel from the PILOT trial; and tafasitamab-lenalidomide from the L-MIND trial). However, the sponsor specified that, for the purposes of the current CDA-AMC review, these comparators were excluded from the pharmacoeconomic analysis due to the following reasons:
Axicabtagene ciloleucel (in the ALYCANTE trial) and lisocabtagene maraleucel (in the PILOT trial) included a second-line transplant-ineligible r/r DLBCL patient population which is not approved or funded for indications in Canada.
Tafasitamab-lenalidomide (in the L-MIND trial) is funded in Quebec, but not funded in rest of Canada.
As such, these analyses were not considered relevant to the current CDA-AMC review.
Other analyses presented in the sponsor’s ITC that were deemed relevant to this review used IPD from the pivotal STARGLO (GO41944) trial and 2 other trials (the GO29365 study for pola-BR and the NP30179 study for glofitamab monotherapy). PSAs were performed to provide an estimate of treatment effects after accounting for differences in covariates considered to be potential prognostic factors or treatment-effect modifiers across treatment groups.
Indirect Treatment Comparison Design
Objectives
The objectives of the sponsor-submitted ITC evidence were to conduct a systematic literature review to gather evidence on standard of care treatments and emerging therapies for managing r/r DLBCL, conduct an ITC feasibility assessment based on available studies and reported outcomes for comparing Glofit-GemOx with other treatments, and conduct MAICs and PSAs.
Study Selection Methods
The sponsor conducted a literature review to identify studies on adult patients (aged 18 years and older) with r/r DLBCL treated at the second line and beyond and a feasibility assessment to understand the appropriateness of conducting ITCs of Glofit-GemOx against commonly used regimens. For the purposes of this CDA-AMC review, only comparators relevant to, and available in, Canada were considered relevant (i.e., pola-BR and glofitamab monotherapy).
The review analyzed data from RCTs, nonrandomized clinical studies, and real-world observational studies in a clinical setting that involved at least 1 systemic line of therapy.
Feasibility assessment included the evaluation and selection of studies, based on the hierarchical criteria to minimize bias, identified by the sponsor and guided by internal medical and scientific feedback:
Prospective studies were prioritized over retrospective studies (with a view to revisiting them only if they were not feasible due to insufficient baseline characteristics for adjustment).
Studies conducted in a clinical setting were prioritized over observational study designs (e.g., registries).
Larger cohort studies with 40 or more patients were prioritized over those with smaller cohorts.
Patients receiving second-line treatment were prioritized if patients were ineligible for ASCT.
Studies were prioritized if they included patients whose disease was not refractory to rituximab exposure, particularly those studies initiated before 2010 or those in which it was reasonable to assume that rituximab was not received.
Following the feasibility assessment, from 304 unique studies identified by the SLR, 8 studies provided data for 3 MAIC comparators (axicabtagene ciloleucel from ALYCANTE trial; lisocabtagene maraleucel from the PILOT trial; and tafasitamab-lenalidomide from L-MIND trial), based on ESS and overlap in study and population characteristics, and 2 studies with IPD were eligible for the PSA.
A summary of the studies selected for inclusion in PSAs for each feasible comparator and the study end points measured in each comparison study are presented in Table 19. For end points that were both IRC- and investigator-assessed, IRC end points were used.
Table 19: Summary of Studies, Treatments, and Outcome Measures Involved in PSA Analyses
Comparator | Comparator study details | Comparator study outcomes | ||||||
|---|---|---|---|---|---|---|---|---|
Study identifier | Population | OS | PFS (IRC-assessed) | CR rate/ORR (IRC-assessed; based on response including PET and CT data) | DOR (IRC-assessed) | Treatment discontinuation due to AEs | Tumour assessment criteria | |
pola-BR | GO29365 (N = 142; ITT) | Second-line and beyond r/r DLBCL (including only DLBCL NOS, without prior CAR T-cell therapy patients, and 1, 2, 3 and 4 prior treatment lines only) | — | — | — | — | — | Lugano 2014 |
Glofitamab monotherapy | NP30179 (N = 100; ITT) | Third-line and beyond r/r DLBCL (including only DLBCL NOS and 2, 3 and 4 prior treatment lines only) | — | — | — | — | — | Lugano 2014 |
AE = adverse event; CAR = chimeric antigen receptor; DLBCL = diffuse large B-cell lymphoma; DOR = duration of response; Glofit-GemOx = glofitamab in combination with gemcitabine and oxaliplatin; IRC = independent review committee; ITT = intention to treat; NOS = not otherwise specified; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; pola-BR = polatuzumab vedotin plus bendamustine and rituximab; PSA = propensity score analysis; r/r = relapsed or refractory.
Source: Details included in the table are from the sponsor’s Summary of Clinical Evidence.
ITC Analysis Methods
Prognostic baseline characteristics and effect modifiers for adjustment in the analyses were identified and ranked in order of priority, based on input from independent clinical experts as well as the sponsor’s internal clinical experts and an advisory board:
High priority: Age, ECOG PS (0, 1, or 2), Ann Arbor stage (III to IV), high LDH levels (above-normal serum LDH level), presence of extranodal disease (greater than or equal to 2 sites), IPI (3 to 5), refractory to first line of treatment, refractory to last line of treatment, number of prior treatment lines (1, 2, 3, or 4)
Medium priority: Refractory to chemotherapy (i.e., any prior platinum therapy or any prior anti-CD20 and anthracycline-based regimen, either alone or in combination), refractoriness to rituximab (i.e., any prior anti-CD20 therapy), bulky disease (definitions can vary across studies), time since completion of last-line therapy to first study treatment
Low priority: Sex, cell type of origin (germinal centre B cell, non–germinal centre B cell, or activated B cell), bone marrow involvement, prior ASCT, prior CAR T-cell therapy
To better align the baseline characteristic of the population included in the STARGLO trial to the comparator trials, subsetting of the STARGLO trial was used, as detailed in Table 20.
Table 20: STARGLO Subset Based on Comparator Studies
Subsetting IPD study (treatment) | Comparator study (treatment) | Subsetting (include only) |
|---|---|---|
STARGLO (Glofit-GemOx) | GO29365 (pola-BR and BR) | Patients without prior CAR T-cell therapy |
GO29365 (pola-BR and BR) | STARGLO (Glofit-GemOx) | Patients without prior CART-cell therapy; only DLBCL NOS histology; and 1, 2, 3, and 4 prior treatment lines |
STARGLO (Glofit-GemOx) | NP30179 (Glofit monotherapy) | Patients with 2 or more prior lines of therapy without ECOG PS 2 |
NP30179 (Glofit monotherapy) | STARGLO (Glofit-GemOx) | Only DLBCL NOS histology |
BR = bendamustine and rituximab; CAR = chimeric antigen receptor; DLBCL = diffuse large B-cell lymphoma; Glofit-GemOx = glofitamab in combination with gemcitabine and oxaliplatin; ITC = indirect treatmemt comparison; NOS = not otherwise specified; pola-BR = polatuzumab vedotin plus bendamustine and rituximab.
Source: Details included in the table are from the sponsor’s ITC technical report.
The summary of baseline characteristics for the unweighted and weighted after IPTW samples for Glofit-GemOx, polar-BR, and glofitamab monotherapy are presented in (Table 21). The end points used in the ITC analysis are listed in Table 19. The STARGLO data cut-off date that was used for analysis is February 16, 2024.
The primary estimand was average treatment effect, which was computed after the necessary subselections of the study populations were made, according to the criteria detailed in Table 21.48
The sponsor presented the distribution balance on each of the individual matched covariates before and after adjustment. IPTW was the main analysis for PSAs. In addition, the sponsor reported the unweighted analyses (before IPTW) and a sensitivity analysis (IPTW with multiple imputations) to illustrate the impact of the matching adjustment. They also presented the results of a second sensitivity analysis using the double-robust method by further adjusting for matched covariates (full matching analyses).
IPTW Analyses
Weights for the IPTW analysis were computed that denote the probability of receiving the actual treatment assigned. These weights were applied in Cox proportional hazards or logistic regression models to estimate treatment effects. For propensity score weighting, bootstrapping was used to compute standard errors and CIs after weighting.
IPTW Analyses With Multiple Imputation
Five imputed datasets for multiple imputation were used as part of a sensitivity analysis in the IPTW. As recommended, outcome variables were included in the imputation model.49 For each imputed dataset, treatment effects were estimated and then combined using the Rubin rule.50
Full Matching Analyses
Full matching assigned every treated and control unit in the sample to 1 subclass each, with each subclass containing 1 treated unit and 1 or more control units, or 1 control unit and 1 or more treated units.51 Cluster bootstrap standard errors were used, and the 2.5 and 97.5 percentiles were used as the 95% CI boundaries. A total of 2,000 bootstrap samples were used in this study. Similar to IPTW, when an imbalance remained after matching, all baseline covariates were weighted for in the outcome regression after full matching was performed.
Estimation of the Propensity Score
Logistic regression methods were adopted to estimate propensity scores, with treatment as the dependent variable and baseline covariates as explanatory variables. The variables that were preselected for the regression model were used in the propensity score model.
Results of ITC 1
Summary of Included Studies
Comparisons Between Glofit-GemOx and pola-BR
After subsetting the STARGLO and GO29365 patient cohorts used for the comparisons, there were 170 patients in the Glofit-GemOx arm and 132 patients in the pola-BR arm. As there were many missing values for cell type of origin and bone marrow involvement in the GO29365 study, the main analysis was performed without adjusting for these factors. In both the main and full matching analyses, the missing values of other covariates were set to be equal to the mean or mode of each covariate to ensure that patients were not dropped from the analysis. In the IPTW with multiple imputation analysis, imputation was used to estimate values for cell type of origin, bone marrow involvement, and other covariates.
Baseline covariates for populations in the STARGLO and GO29365 trials, pre- and post-IPTW adjustments are summarized in Table 21. Before adjustment, there were numerous pairwise imbalances (absolute standardized mean difference > 0.1) in baseline characteristics between the STARGLO and GO29365 trials (age, ECOG PS, disease stage, LDH, prognostic index, refractoriness to first or last line of therapy, number of previous lines of therapies, and previous ASCT). After adjustment, there were no imbalances with an absolute standardized mean difference of less 0.1 with regard to baseline variables used in the main analyses. Following IPTW adjustment, the ESS of Glofit-GemOx was 132.60 and the ESS for pola-BR was 107.36.
Table 21: Overview of Patient Characteristics Before and After IPTW Weighting (Glofit-GemOx and pola-BR Populations)
Variable | Unweighted analyses | After IPTW | ||||
|---|---|---|---|---|---|---|
Glofit-GemOx (n = 170), mean (SD) | pola-BR (n = 132), mean (SD) | SMD | Glofit-GemOx (ESS = 132.60), mean (SD) | pola-BR (ESS = 107.36), mean (SD) | SMD | |
Age, years | 65.88 (12.46) | 67.52 (11.50) | −0.14 | 67.15 (11.79) | 67.62 (12.77) | −0.04 |
ECOG PS 1, % | 51 (50) | 58 (49) | −0.16 | 54 (50) | 56 (50) | −0.03 |
ECOG PS 2, % | 11 (31) | 14 (35) | −0.12 | 12 (33) | 12 (32) | 0.02 |
Ann Arbor stage III to IV, % | 68 (47) | 80 (40) | −0.29 | 74 (44) | 76 (43) | −0.04 |
High LDH, % | 53 (50) | 58 (49) | −0.11 | 56 (50) | 59 (49) | −0.06 |
Extranodal disease, % | 62 (49) | 64 (48) | −0.04 | 66 (47) | 66 (47) | 0.01 |
IPI 3 to 5, % | 50 (50) | 62 (49) | −0.25 | 57 (49) | 60 (49) | −0.05 |
Refractory in the first line, % | 57 (50) | 62 (49) | −0.10 | 60 (49) | 58 (49) | 0.04 |
Refractory in the last line, % | 59 (49) | 74 (44) | −0.32 | 65 (48) | 66 (47) | −0.02 |
Two prior lines of therapy, % | 19 (39) | 30 (46) | −0.25 | 22 (41) | 25 (43) | −0.07 |
Three prior lines of therapy, % | 8 (28) | 21 (41) | −0.37 | 12 (33) | 14 (34) | −0.04 |
Four prior lines of therapy, % | 5 (21) | 14 (34) | −0.31 | 12 (33) | 9 (29) | 0.10 |
Bulky disease | 59.70 (33.44) | 60.70 (39.37) | −0.03 | 59.58 (33.07) | 60.49 (38.25) | −0.03 |
Time since last treatment to first study treatment, months | 14.49 (27.39) | 12.29 (27.42) | 0.08 | 13.96 (27.59) | 14.34 (27.80) | −0.01 |
Male sex, % | 56 (50) | 58 (49) | −0.04 | 61 (49) | 60 (49) | 0.02 |
Prior ASCT, % | 4 (20) | 14 (34) | −0.34 | 8 (27) | 8 (27) | −0.00 |
ASCT = autologous stem cell transplant; ECOG PS = Eastern Cooperative Oncology Group Performance Status; Glofit-GemOx = glofitamab in combination with gemcitabine and oxaliplatin; IPI = International Prognostic Index; IPTW = inverse probability of treatment weighting; ITC = indirect treatment comparison; LDH = lactate dehydrogenase; pola-BR = polatuzumab vedotin plus bendamustine and rituximab; SD = standard deviation; SMD = standardized mean difference.
Source: Details included in the table are from the sponsor’s ITC technical report.
Comparisons Between Glofit-GemOx and Glofitamab Monotherapy
After subsetting the STARGLO and NP30179 patient cohorts used for the comparisons, there were 61 patients in the Glofit-GemOx arm and 100 patients in the glofitamab monotherapy arm.
In the weighted model, the Glofit-GemOx arm had an ESS smaller than 30 when including high-, medium-, and low-priority variables. To address the low ESS when all priority variables were included, the sponsor included only high- and medium-priority variables as covariates, which resulted in an ESS of 61 for of Glofit-GemOx and an ESS of 100 for pola-BR. After limiting the covariates, there were no variables with significant missing data; as such, multiple imputations with IPTW were not performed, and only the full matching method was performed.
Baseline covariates for populations in the STARGLO and NP30179 trials, pre- and post-IPTW adjustments are summarized in Table 22. Before adjustment, there were numerous pairwise imbalances (standardized mean difference > 0.1) in baseline characteristics between the STARGLO and NP30179 trials (age, ECOG PS, disease stage, LDH, refractoriness to first or last line of therapy). After adjustment, there were no imbalances with a standardized mean difference greater than 0.1 for baseline variables used in the main analyses. Following IPTW adjustment, the ESSs of Glofit-GemOx and glofitamab monotherapy were the same as in the subsetted sample.
Table 22: Overview of Patient Characteristics Before and After IPTW Weighting (Glofit-GemOx and Glofitamab Monotherapy Populations)
Variable | Unweighted analyses | After IPTW | ||||
|---|---|---|---|---|---|---|
Glofit-GemOx (n = 61), mean (SD) | Glofitamab monotherapy (n = 100), mean (SD) | SMD | Glofit-GemOx (n = 61), mean (SD) | Glofitamab monotherapy (n = 100), mean (SD) | SMD | |
Age, years | 60.36 (12.26) | 64.03 (13.91) | −0.28 | 63.62 (12.81) | 62.90 (13.98) | −0.04 |
ECOG PS 1, % | 57 (50) | 46 (50) | 0.23 | 51 (50) | 50 (50) | −0.03 |
Ann Arbor stage III to IV, % | 67 (47) | 78 (42) | −0.24 | 75 (44) | 74 (44) | −0.04 |
High LDH, % | 59 (50) | 69 (46) | −0.21 | 63 (48) | 65 (48) | −0.06 |
Extranodal disease, % | 67 (47) | 65 (48) | 0.05 | 69 (46) | 67 (47) | 0.01 |
IPI 3 to 5, % | 57 (50) | 56 (50) | 0.03 | 58 (50) | 56 (50) | −0.05 |
Refractory in the first line, % | 56 (50) | 60 (49) | −0.09 | 58 (50) | 59 (49) | 0.04 |
Refractory in the last line, % | 75 (43) | 81 (39) | −0.14 | 81 (40) | 79 (41) | −0.02 |
Three prior lines of therapy, % | 34 (48) | 30 (46) | 0.09 | 33 (47) | 33 (47) | −0.04 |
Four prior lines of therapy, % | 16 (37) | 20 (46) | −0.09 | 21 (41) | 18 (39) | 0.10 |
Bulky disease | 11 (32) | 13 (34) | −0.05 | 12 (33) | 13 (34) | −0.03 |
Time since last treatment to first study treatment, months | 7.59 (13.52) | 8.07 (18.48) | −0.03 | 6.55 (12.67) | 7.72 (16.25) | −0.01 |
ECOG PS = Eastern Cooperative Oncology Group Performance Status; Glofit-GemOx = glofitamab in combination with gemcitabine and oxaliplatin; IPI = International Prognostic Index; IPTW = inverse probability of treatment weighting; ITC = indirect treatment comparison; LDH = lactate dehydrogenase; SD = standard deviation; SMD = standardized mean difference.
Source: Details included in the table are from the sponsor’s ITC technical report.
Results
The results of the main ITC analyses comparing the OS, PFS, CR rare, DOR and discontinuation due to AEs estimates for Glofit-GemOx versus comparators (pola-BR and glofitamab monotherapy) are presented in the following section. The results for the unweighted analyses (before IPTW), IPTW analyses, and sensitivity analyses (IPTW with multiple imputations and full matching analyses) are available in Table 23.
Overall Survival
The HR for OS between Glofit-GemOx and pola-BR was ████ ████ ███ ████ ██ █████. As the CIs crossed 1, there was no evidence of a statistically significant difference between the 2 treatments. These results were confirmed in the sensitivity analyses.
The HR for OS between Glofit-GemOx and glofitamab monotherapy was ████ ████ ███ ████ ██ █████. As the CIs crossed 1, there was no evidence of a statistically significant difference between the 2 treatments. These results were confirmed in the sensitivity analyses.
Progression-Free Survival
The HR for PFS between Glofit-GemOx and pola-BR was ████ ████ ███ ████ ██ █████. As the CIs crossed 1, there was no evidence of a statistically significant difference between the 2 treatments. These results were confirmed in the sensitivity analyses.
Tumour Response Variables
For CR rate, the OR between Glofit-GemOx and pola-BR was ████ ████ ███ █████ █████. As the CIs crossed 1, there no evidence of a statistically significant difference between the 2 treatments. These results were confirmed in the full matching analysis, but IPTW with multiple imputation analysis showed a more favourable benefit with Glofit-GemOx.
For CR rate, the OR between Glofit-GemOx and glofitamab was ████ ████ ███ ████ ██ █████. As the CIs crossed 1, there was no evidence of a statistically significant difference between the 2 treatments. These results were confirmed in the sensitivity analyses.
For DOR, the OR between Glofit-GemOx and pola-BR was ████ █████ ██ █████. As the CIs crossed 1, there was no evidence of a statistically significant difference between the 2 treatments. These results were confirmed in the sensitivity analyses.
For DOR, the OR between Glofit-GemOx and glofitamab was ████ █████ ██ █████. As the CIs crossed 1, there was no evidence of a statistically significant difference between the 2 treatments. These results were confirmed in the sensitivity analyses.
Harms Results
Treatment Discontinuation due to an AE
The OR between Glofit-GemOx and pola-BR for treatment discontinuation due to an AE was ████ █████ ██ █████. These results were confirmed in the sensitivity analyses.
The OR between Glofit-GemOx and glofitamab for treatment discontinuation due to an AE was ████ █████ ██ █████. These results were confirmed in the sensitivity analyses.
Table 23: ITC Comparing Glofit-GemOx and pola-BR
Comparison (Glofit-GemOx vs. comparator) | pola-BR | Glofitamab monotherapy |
|---|---|---|
OS, HR (95% CI) | ||
Unweighted | 0.71 (0.51 to 0.97) | 0.74 (0.47 to 1.15) |
IPTWa | ████ █████ █ | ████ █████ ██ |
IPTW with multiple imputation | 0.92 (0.65 to 1.31) | NA |
Full matching | 0.97 (0.65 to 1.49) | 0.75 (0.48 to 1.11) |
PFS IRC-assessed HR (95% CI) | ||
Unweighted | 0.62 (0.46 to 0.83) | 0.73 (0.48 to 1.12) |
IPTWa | ████ █████ ██ | 0.79 (0.48 to 1.18) |
IPTW with multiple imputation | 0.83 (0.58 to 1.20) | NA |
Full matching | ████ █████ ██ | 0.77 (0.51 to 1.13) |
CRR, IRC-assessed OR (95% CI) | ||
Unweighted | 1.09 (0.69 to 1.72) | 1.89 (0.99 to 3.60) |
IPTWa | 0.73 (0.47, 1.17) | ████ █████ ██ |
IPTW with multiple imputation | 0.59 (0.34, 0.94) | NA |
Full matching | 0.86 (0.70, 1.05) | 1.26 (0.95 to 1.69) |
DOR, IRC-assessed HR (95% CI) | ||
Unweighted | 0.60 (0.39 to 0.93) | 1.08 (0.56 to 2.09) |
IPTWa | ████ █████ ██ | ████ █████ ██ |
IPTW with multiple imputation | 0.64 (0.23 to 1.41) | NA |
Full matching | 0.87 (0.50 to 1.49) | 1.30 (0.65 to 2.38) |
Treatment discontinuation due to AE, OR (95% CI) | ||
Unweighted | 0.62 (0.33 to 1.13) | 2.48 (0.98 to 6.28) |
IPTW | ████ █████ ██ █ | ████ █████ ██ |
IPTW with multiple imputation | 0.59 (0.26 to 1.31) | NA |
Full matching | 0.78 (0.37 to 1.62) | 2.01 (0.71 to 5.78) |
CI = confidence interval; Glofit-GemOx = glofitamab in combination with gemcitabine and oxaliplatin; HR = hazard ratio; IPTW = inverse probability of treatment weighting; IRC = independent review committee; IPTW = inverse probability of treatment weighting; ITC = indirect treatment comparison; NA = not applicable; pola-BR = polatuzumab vedotin plus bendamustine and rituximab; vs. = versus.
aThe IPTW was used for the main analysis.
Source: Details included in the table are from the sponsor’s ITC technical report.
Critical Appraisal of ITC 1
No major issues were identified with regard to the systematic search for identifying relevant studies for the ITC. The ITC analyses were preceded by a feasibility assessment, with a predefined hierarchy of evidence to guide study inclusion with 2 PSAs considered relevant to the current CDA-AMC review.
The choice of the matching factors was based on input from external clinical experts on an advisory board run by the sponsor. For the glofitamab monotherapy comparison, only high- and medium-priority variables were included due to the limitations of the weighted model, in which the Glofit-GemOx arm had a very small ESS (< 30) when including high-, medium-, and low- priority variables which adds uncertainty to these analyses.
While the ITC evaluated important clinical outcomes such as OS, PFS, CR rate and/or DOR, and treatment discontinuation due to AEs, other important outcomes for decision-making, such as HRQoL and SAEs, were not included in the analyses. This limits the ability to evaluate the true balance of comparative benefits and harms. Even for the evaluated outcomes, in most cases the CIs were wide and crossed the null, introducing further imprecision and uncertainty to the comparative effects.
Before the ITC adjustments, there were notable observed differences between the trial patient characteristics. Following adjustment, the populations were balanced. Even so, adjustment methods cannot overcome methodological or design differences (e.g., study design, region or setting, length of follow-up, outcome definitions [event and censoring rules, schedule and method of assessments], cointerventions, and subsequent treatments) across the comparators, which can introduce bias. For example, the study designs for the 3 trials differed, with the GO29365 trial being a phase Ib and II study, the NP30179 trial a phase II study, and the STARGLO trial a phase III study. In study GO29365, patients who received pola-BR had different histologies, such as DLBCL NOS, HGBCL, PMBCL, and tFL. However, the NP30179 trial (of glofitamab monotherapy) included patients with DLBCL NOS, tFL, PMBCL, and HGBCL, while the STARGLO trial only includes patients with DLBCL NOS. Further, the NP30179 trial only included individuals with third-line and beyond r/r DLBCL (including only DLBCL NOS and 2, 3, and 4 prior treatment lines). As these factors could not be accounted for in the PSA models, differences between the study samples would be expected to introduce bias into the estimates, although the magnitude and direction are unclear.
The matching and/or adjustments conducted as part of the PSAs did sometimes result in residual imbalances for multiple prognostic factors. Although these are further controlled for in subsequent sensitivity analyses (following a doubly robust approach), generalizability may be an issue due to the small sample size remaining after the exclusions and weighting in some of the analyses. For example, after subsetting the STARGLO and GO29365 patient cohorts used for the comparisons, there were 170 patients in the Glofit-GemOx arm and 132 patients in the pola-BR arm. Following IPTW adjustment, the ESS of the Glofit-GemOx arm was 132.60 and the ESS for the pola-BR arm was 107.36, which could affect the power of the analysis and result in wide CIs and a lack of ability to find true differences.
Summary
The sponsor-submitted ITC report of PSAs did not show a conclusive benefit of Glofit-GemOx over pola-BR or glofitamab monotherapy with regard to OS, PFS, DOR, ORR, and discontinuation due to AEs.
Studies Addressing Gaps in the Systematic Review Evidence
No additional studies were submitted by the sponsor.
Discussion
Summary of Available Evidence
The systematic review included 2 reports of 1 pivotal trial (STARGLO). The STARGLO trial is a phase III, ongoing, prospective, international (no Canadian sites), multicentre, open-label, parallel-group RCT to assess Glofit-GemOx compared with R-GemOx in patients with r/r DLBCL NOS who have failed 1 line of therapy and are not candidates for transplant, as well as those patients who have failed at least 2 lines of therapy. Patients were randomized in a 2:1 ratio to receive either Glofit-GemOx or R-GemOx. Randomization was stratified by the number of previous lines of systemic therapy for DLBCL (1 versus equal to or higher than 2) and the outcome of the last systemic therapy (relapsed versus refractory). The primary outcome was OS, and the key secondary outcomes included IRC-assessed PFS and IRC-assessed CR rates. Harms including SAEs, and notable harms were also measured and reported. Patient-reported outcomes (e.g., HRQoL) were also measured. All results presented were from the primary analysis data cut-off (March 29, 2023) and the updated analysis data cut-off (February 16, 2024).
A total of 274 patients underwent randomization (Glofit-GemOx = 183; R-GemOx = 91). At the time of the primary analysis, the median duration of follow-up for OS in the overall ITT population was 11.3 months (95% CI, 9.6 to 12.7): Glofit-GemOx (12.0 months [95% CI, 10.2 to 13.2; N = 183]); R-GemOx (9.6 months [95% CI, 7.9 to 12.0; N = 91]). The median durations of follow-up for OS in the updated analysis were 22.5 months (95% CI, 20.0 to 24.5) in the Glofit-GemOx arm (N = 183) and 19.7 months (95% CI, 18.0 to 23.1) in the R-GemOx arm (N = 91) (Appendix 1, Table 24), corresponding to follow-ups 10.5 months and 10.1 months longer than in the primary analysis.
At the updated analysis data cut-off, 86 patients (47.0%) in the Glofit-GemOx arm and 28 patients (30.8%) in the R-GemOx arm were still in the study. Patients were aged an average of 65.0 years (SD = 13.0), 57.7% were male, and 50.0% were Asian. Most patients (89.9%) had an ECOG PS of 0 or 1, 1 previous line of therapy (62.8%), and refractory disease (66.4%). Discontinuation from the study occurred in 97 patients (53.0%) in the Glofit-GemOx arm and 63 patients (69.2%) in the R-GemOx arm. The most frequent reason for study discontinuation in both treatment arms was death (accounting for 43.7% and 57.1% in the Glofit-GemOx and R-GemOx populations, respectively).
The sponsor submitted an ITC report with 2 comparisons considered relevant to the current CDA-AMC review. These analyses used IPD from the pivotal STARGLO (GO41944) trial and 2 trials (the GO29365 study for pola-BR and the NP30179 study for glofitamab monotherapy). PSAs were performed to provide an estimate of treatment effects after accounting for differences in covariates considered to be potential prognostic factors or treatment-effect modifiers across treatment groups. The analyzed outcomes were OS, PFS, DOR, ORR, and discontinuation due to AEs.
Interpretation of Results
Efficacy
Patient and clinician input received by CDA-AMC for this review indicated that outcomes such as OS, PFS, HRQoL, and safety outcomes are highly valued. The clinical experts consulted by the review team confirmed that the goal of treatment is generally palliative despite the treatments available today, reflecting an unmet clinical need for new treatment options. The experts also concurred that most patients will approach prioritizing survival and PFS over AEs that are manageable as a critical factor when making treatment decisions, particularly given the poor prognosis associated with this condition. According to the experts, even modest improvements in survival time will potentially influence patients' daily lives and decision-making processes.
The clinical experts consulted by CDA-AMC and other interest holders considered OS critical outcome for decision-making. The body of evidence from the pivotal study showed that Glofit-GemOx results in a clinically important increase in median survival and PFS when compared with R-GemOx, particularly on longer follow-ups. At the longest follow-up, the findings for OS favoured Glofit-GemOx, with median durations of follow-up of 22.5 months (95% CI, 20.0 to 24.5) in the Glofit-GemOx arm and 19.7 months (95% CI, 18.0 to, 23.1) in the R-GemOx arm (HR = 0.62; 95% CI, 0.43 to 0.88) and PFS, with median durations of follow-up of 16.3 months (95% CI, 15.3 to 20.1) in the Glofit-GemOx arm and 8.6 months (95% CI, 5.9, 14.6) in the R-GemOx arm (HR = 0.37, CI, 0.25, 0.55). The clinical experts emphasized that the results were encouraging, but that there was uncertainty as the trial is still ongoing. Similar results for patients with CR rate were reported in favour of Glofit-GemOx, with a between-group difference of 33.2% (95% CI, 20.9 to 45.5), compared to GemOx; however, no statistical differences in DOR were noted (HR = 0.59; 95% CI, 0.25 to 1.35). With regard to HRQoL, the results are uncertain because of the large number of patients who did not complete assessments over the longer periods and the open-label design of the trial.
The sponsor submitted an ITC report with 2 comparisons considered relevant to the current CDA-AMC review. These analyses used IPD from the pivotal STARGLO (GO41944) trial and 2 other trials (the GO29365 study for pola-BR and the NP30179 study for glofitamab monotherapy). PSAs were performed to provide an estimate of treatment effects after accounting for differences in covariates considered to be potential prognostic factors or treatment-effect modifiers across treatment groups. The analyzed outcomes were OS, PFS, DOR, ORR, and discontinuation due to AEs. After adjustments, the results generally did not provide evidence of a more favourable benefit of 1 treatment over another, with wide CIs crossing the null.
Harms
In both treatment arms, almost all patients reported at least 1 AE (100% of patients receiving Glofit-GemOx and 95.5% of those receiving R-GemOx), and more than half the patients receiving Glofit-GemOx had an SAE compared to less than 20% of patients receiving R-GemOx. Further, all cases of CRS (any grade) occurred in the Glofit-GemOx arm. The most frequently reported AEs in the Glofit-GemOx arm were CRS events, with 76 patients (44.2%); nausea, with 71 patients (41.3%); and anemia, with 71 patients (41.3%). The most frequently reported AEs in the R-GemOx arm were nausea (35 patients [39.8%]), platelet count reduction (27 patients [30.7%]), and diarrhea (24 patients [27.3%]). The most common SAEs with an incidence rate equal to or higher than 5% in the Glofit-GemOx and R-GemOx populations were CRS (20.3% versus 0.0%, respectively), pyrexia (6.4% versus 1.1%, respectively), and pneumonia (5.8% versus 4.5%, respectively). There were 76 patients (42.2%) and 0 patients experiencing CRS events of any grade in the Glofit-GemOx (any treatment exposed) and R-GemOx arms, respectively.
Other Considerations
Glofit-GemOx requires strict protocols in specialized centres to be administered safely. This can affect the real-world results of patients who may be candidates for this intervention but must wait for health care resources to become available.
Conclusion
One multinational, ongoing, open-label, phase III RCT (STARGLO), was included in the CDA-AMC review, along with ITCs (PSAs) comparing Glofit-GemOx to pola-BR and glofitamab monotherapy. The RCT included a population of 274 patients with DLBCL NOS, Glofit-GemOx resulted in superior clinical outcomes compared with R-GemOx. While the trial is still ongoing, from the latest available evidence, data for most outcomes are mature and the evidence suggests that Glofit-GemOx is significantly superior to R-GemOx. The clinical experts consulted by the review team considered the findings clinically meaningful. HRQoL appeared to be similar between groups, but due to a lack of blinding, and a high attrition rate as the trial progressed, the certainty in the effect estimates is unclear. Data censoring in the OS and PFS analysis decreased the certainty in the results. Treatment with Glofit-GemOx was associated with a substantially higher incidence of SAEs and notable harms than with R-GemOx; however, the experts noted that these were known and manageable in the proper inpatient setting.
Using a PSA, Glofit-GemOx was indirectly compared to pola-BR and glofitamab monotherapy. After adjustments, the results generally did not provide evidence of a more favourable benefit of 1 treatment over another for the evaluated outcomes (OR, PFS, CR rate, DOR, and treatment discontinuation due to AEs), with the CIs crossing the null. Furthermore, the wide CIs led to substantial uncertainty in many of the end points.
In conclusion, Glofit-GemOx was generally more efficacious than R-GemOx, with a benefit in OS and PFS, especially on longer follow-ups. The QoL in patients who remained alive was uncertain due to a large loss to follow-up and the open-label nature of the trial. SAEs were more common with Glofit-GemOx and should be monitored closely. The evidence did not show that Glofit-GemOx had a conclusive benefit over pola-BR or glofitamab monotherapy.
References
1.Shafey M, Savage KJ, Skrabek P, Elsawy M, Bosch M, Kuruvilla J. Canadian Evidence-Based Guideline for the Treatment of Relapsed/Refractory Diffuse Large B-Cell Lymphoma. Lymphoma Canada; 2021. Accessed sponsor ref, no date provided. https://www.lymphoma.ca/wp-content/uploads/2021/09/LymphomaCanada_Guideline_Relapsed_Refractory_DLBCL_VF_Digital.pdf
2.Canadian Cancer Society. Diffuse large B-cell lymphoma. 2023. Accessed May 14, 2025. https://cancer.ca/en/cancer-information/cancer-types/non-hodgkin-lymphoma/treatment/treatment-by-type/diffuse-large-b-cell-lymphoma
3.Freedman AS, Aster JC. Epidemiology, clinical manifestations, pathologic features, and diagnosis of diffuse large B cell lymphoma. UpToDate; July 29, 2022. Accessed March 10, 2025. http://www.uptodate.com
4.Goh SC, Ngai S. The clinical challenge of relapsed diffuse large B-cell lymphoma with disseminated infection. Radiol Case Rep. 2023;18(9):3046-3053. doi: 10.1016/j.radcr.2023.05.068 PubMed
5.Brenner DR, Gillis J, Demers AA, et al. Projected estimates of cancer in Canada in 2024. CMAJ. 2024;196(18):E615-E623. doi: 10.1503/cmaj.240095 PubMed
6.Crump M, Neelapu SS, Farooq U, et al. Outcomes in refractory diffuse large B-cell lymphoma: results from the international SCHOLAR-1 study. Blood. 2017;130(16):1800-1808. doi: 10.1182/blood-2017-03-769620 PubMed
7.Roschewski M, Staudt LM, Wilson WH. Diffuse large B-cell lymphoma-treatment approaches in the molecular era. Nat Rev Clin Oncol. 2014;11(1):12-23. doi: 10.1038/nrclinonc.2013.197 PubMed
8.Gisselbrecht C, Van Den Neste E. How I manage patients with relapsed/refractory diffuse large B cell lymphoma. Br J Haematol. 2018;182(5):633-643. doi: 10.1111/bjh.15412 PubMed
9.Harrysson S, Eloranta S, Ekberg S, et al. Outcomes of relapsed/refractory diffuse large B‐cell lymphoma and influence of chimaeric antigen receptor T trial eligibility criteria in second line—A population‐based study of 736 patients. Br J Haematol. 2022;198(2):267-277. doi: 10.1111/bjh.18197 PubMed
10.Freedman AS, Friedberg JW. Initial treatment of advanced-stage diffuse large B cell lymphoma. UpToDate; February 19, 2024. Accessed March 10, 2025. http://www.uptodate.com
11.Tilly H, Gomes da Silva M, Vitolo U, et al. Diffuse large B-cell lymphoma (DLBCL): ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2015;26 Suppl 5:v116-25. doi: 10.1093/annonc/mdv304 PubMed
12.Cheson BD, Fisher RI, Barrington SF, et al. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: the Lugano classification. J Clin Oncol. 2014;32(27):3059-68. doi: 10.1200/JCO.2013.54.8800 PubMed
13.Lister TA, Crowther D, Sutcliffe SB, et al. Report of a committee convened to discuss the evaluation and staging of patients with Hodgkin's disease: Cotswolds meeting. J Clin Oncol. 1989;7(11):1630-6. doi: 10.1200/JCO.1989.7.11.1630 PubMed
14.Hoffmann-La Roche Limited. Columvi (glofitamab for injection): 1mg/mL, concentrate for solution for intravenous infusion [draft product monograph; sponsor supplied reference]. 2024.
15.CADTH. Reimbursement review: glofitamab (Columvi). Can J Health Technol. 2024;4(4). doi: 10.51731/cjht.2024.873
16.CADTH. Reimbursement recommendation: glofitamab (Columvi). Can J Health Technol. 2024;4(2). doi: 10.51731/cjht.2024.837
17.Hoffmann-La Roche Limited. Columvi (glofitamab for injection): 2.5 mg/2.5 mL or 10 mg/10 mL, Concentrate for solution for intravenous infusion, in single-use glass vial [product monograph]. 2023. Accessed by sponsor, no date provided. https://assets.roche.com/f/173850/x/f54500f377/columvi_pm_e.pdf
18.Hoffmann-La Roche Limited. Gazyva (obinutuzumab): 25 mg/mL, concentrate for solution for intravenous infusion [product monograph]. November 25, 2014. Updated July 25, 2022. Accessed by sponsor, no date provided. https://assets.roche.com/f/173850/x/0c07cb9986/gazyva_pm_e.pdf
19.Hoffmann-La Roche Limited. Polivy (polatuzumab vedotin): 30 mg or 140 mg, lyophilized powder for solution for intravenous infusion, in single-use vial [product monograph]. July 9, 2020. Updated January 20, 2023. Accessed by sponsor, no date provided. https://assets.roche.com/f/173850/x/dbbfa021ce/polivy_pm_e.pdf
20.Hoffmann-La Roche Ltd. Rituxan (rituximab injection): 120 mg/mL, solution for subcutaneous injection [product monograph]. September 9, 2016. Updated June 2, 2023. Accessed September 23, 2025. https://pdf.hres.ca/dpd_pm/00071132.PDF
21.Bristol Myers Squibb Canada. Breyanzi (lisocabtagene maraleucel): cell suspension in patient-specific single-dose vials for intravenous infusion [product monograph]. May 6, 2022. Updated September 25, 2024. Accessed by sponsor, no date provided. https://pdf.hres.ca/dpd_pm/00077215.PDF
22.Innomar Strategies. Minjuvi (tafasitamab): 200 mg, lyophilized powder for solution for infusion, in single-use vial [product monograph]. August 19, 2021. Accessed by sponsor, no date provided. https://pdf.hres.ca/dpd_pm/00062585.PDF
23.AbbVie Corporation. Epkinly (epcoritamab for injection): 5 mg/mL, concentrate for solution, for subcutaneous injection; Epkinly (epcoritamab injection): 60 mg/mL, solution, for subcutaneous injection [product monograph]. October 13, 2023. Updated April 23, 2024. Accessed by sponsor, no date provided. https://pdf.hres.ca/dpd_pm/00075345.PDF
24.Gilead Sciences Canada Inc. Yescarta (axicabtagene ciloleucel): cell suspension in patient-specific single infusion bag, for intravenous infusion [product monograph]. February 13, 2019. Updated May 17, 2024. Accessed by sponsor, no date provided. https://pdf.hres.ca/dpd_pm/00075646.PDF
25.Novartis Pharmaceuticals Canada Inc. Kymriah (tisagenlecleucel): cell suspension in infusion bag, 1.2 x 106 to 6.0 x 108 CAR-positive viable T cells for intravenous use [product monograph]. September 5, 2018. Updated October 29, 2024. Accessed by sponsor, no date provided. https://pdf.hres.ca/dpd_pm/00077617.PDF
26.Abramson JS, Ku M, Hertzberg M, et al. Glofitamab plus gemcitabine and oxaliplatin (GemOx) versus rituximab-GemOx for relapsed or refractory diffuse large B-cell lymphoma (STARGLO): a global phase 3, randomised, open-label trial. Lancet. 2024;404(10466):1940-1954. doi: 10.1016/S0140-6736(24)01774-4 PubMed
27.Hertzberg M, Ku M, Catalani O, Althaus B, Simko S, Gregory GP. A phase III trial evaluating glofitamab in combination with gemcitabine plus oxaliplatin versus rituximab in combination with gemcitabine and oxaliplatin in patients with relapsed/refractory (R/R) diffuse large B-cell lymphoma (DLBCL). J Clin Oncol. 2021;39(15_suppl):TPS7575-TPS7575. doi: 10.1200/jco.2021.39.15_suppl.tps7575
28.Hoffmann-La Roche. NCT04408638: A Phase III Study Evaluating Glofitamab in Combination With Gemcitabine + Oxaliplatin vs Rituximab in Combination With Gemcitabine + Oxaliplatin in Participants With Relapsed/Refractory Diffuse Large B-Cell Lymphoma. ClinicalTrials.gov. Accessed by sponsor, no date provided. https://www.clinicaltrials.gov/study/NCT04408638
29.F. Hoffmann-La Roche Ltd. Clinical Study Report: GO41944, primary. A phase III, open-label, multicenter, randomized study evaluating the efficacy and safety of glofitamab in combination with gemcitabine plus oxaliplatin versus rituximab in combination with gemcitabine and oxaliplatin in patients with relapsed/refractory diffuse large B cell lymphoma [internal sponsor's report]. July 15, 2024.
30.Fitzsimmons D, Johnson CD, George S, et al. Development of a disease specific quality of life (QoL) questionnaire module to supplement the EORTC core cancer QoL questionnaire, the QLQ-C30 in patients with pancreatic cancer. EORTC Study Group on Quality of Life. Eur J Cancer. 1999;35(6):939-41. doi: 10.1016/s0959-8049(99)00047-7 PubMed
31.Aaronson NK, Ahmedzai S, Bergman B, et al. The European Organization for Research and Treatment of Cancer QLQ-C30: a quality-of-life instrument for use in international clinical trials in oncology. J Natl Cancer Inst. 1993;85(5):365-76. doi: 10.1093/jnci/85.5.365 PubMed
32.Cocks K, King MT, Velikova G, et al. Evidence-based guidelines for interpreting change scores for the European Organisation for the Research and Treatment of Cancer Quality of Life Questionnaire Core 30. Eur J Cancer. 2012;48(11):1713-21. doi: 10.1016/j.ejca.2012.02.059 PubMed
33.Osoba D, Rodrigues G, Myles J, Zee B, Pater J. Interpreting the significance of changes in health-related quality-of-life scores. J Clin Oncol. 1998;16(1):139-44. doi: 10.1200/JCO.1998.16.1.139 PubMed
34.Hlubocky FJ, Webster K, Beaumont J, et al. A preliminary study of a health related quality of life assessment of priority symptoms in advanced lymphoma: the National Comprehensive Cancer Network-Functional Assessment of Cancer Therapy - Lymphoma Symptom Index. Leuk Lymphoma. 2013;54(9):1942-6. doi: 10.3109/10428194.2012.762977 PubMed
35.Hlubocky FJ, Webster K, Cashy J, Beaumont J, Cella D. The Development and Validation of a Measure of Health-Related Quality of Life for Non-Hodgkin’s Lymphoma: The Functional Assessment of Cancer Therapy—Lymphoma (FACT-Lym). Lymphoma. 2013;2013(1):147176. doi: https://doi.org/10.1155/2013/147176
36.Carter GC, Liepa AM, Zimmermann AH, Morschhauser F. Validation of the Functional Assessment of Cancer Therapy–Lymphoma (FACT-LYM) in Patients with Relapsed/Refractory Mantle Cell Lymphoma. Blood. 2008;112(11):2376-2376. doi: 10.1182/blood.V112.11.2376.2376
37.Giesinger JM, Kieffer JM, Fayers PM, et al. Replication and validation of higher order models demonstrated that a summary score for the EORTC QLQ-C30 is robust. J Clin Epidemiol. 2016;69:79-88. doi: 10.1016/j.jclinepi.2015.08.007 PubMed
38.Fayers P, Bottomley A. Quality of life research within the EORTC—the EORTC QLQ-C30. Eur J Cancer. 2002;38:125-133. doi: 10.1016/S0959-8049(01)00448-8 PubMed
39.de Lima Lopes G, Nahas GR. Chimeric antigen receptor T cells, a savior with a high price. Chin Clin Oncol. 2018;7(2):21. doi: 10.21037/cco.2018.04.02 PubMed
40.Brigand T, Prokupkova A, Muller G. CAR-T Cell Therapies: How much for survival? Association of European Cancer Leagues; 2018. Accessed March 20, 2025. https://www.cancer.eu/wp-content/uploads/2018/06/CAR-T-ECL-Article_Final_20062018.pdf
41.F. Hoffmann-La Roche Ltd. Statistical Analysis Plan: GO41944, version 6. A phase III, open-label, multicenter, randomized study evaluating the efficacy and safety of glofitamab in combination with gemcitabine plus oxaliplatin versus rituximab in combination with gemcitabine and oxaliplatin in patients with relapsed/refractory diffuse large B cell lymphoma [internal sponsor's report]. July 16, 2024.
42.López A, Gutiérrez A, Palacios A, et al. GEMOX-R regimen is a highly effective salvage regimen in patients with refractory/relapsing diffuse large-cell lymphoma: a phase II study. Eur J Haematol. 2008;80(2):127-32. doi: 10.1111/j.1600-0609.2007.00996.x PubMed
43.Corazzelli G, Capobianco G, Arcamone M, et al. Long-term results of gemcitabine plus oxaliplatin with and without rituximab as salvage treatment for transplant-ineligible patients with refractory/relapsing B-cell lymphoma. Cancer Chemother Pharmacol. 2009;64(5):907-16. doi: 10.1007/s00280-009-0941-9 PubMed
44.Mounier N, El Gnaoui T, Tilly H, et al. Rituximab plus gemcitabine and oxaliplatin in patients with refractory/relapsed diffuse large B-cell lymphoma who are not candidates for high-dose therapy. A phase II Lymphoma Study Association trial. Haematologica. 2013;98(11):1726-31. doi: 10.3324/haematol.2013.090597 PubMed
45.F. Hoffmann-La Roche Ltd. Clinical Study Report: GO41944, update. A phase iii, open-label, multicenter, randomized study evaluating the efficacy and safety of glofitamab in combination with gemcitabine plus oxaliplatin versus rituximab in combination with gemcitabine and oxaliplatin in patients with relapsed/refractory diffuse large B cell lymphoma [internal sponsor's report]. July 16, 2024.
46.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. doi: 10.1016/j.jclinepi.2019.10.014 PubMed
47.Balshem H, Helfand M, Schünemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-406. doi: 10.1016/j.jclinepi.2010.07.015 PubMed
48.Faria R, Hernandez Alava M, Manca A, Wailoo AJ. NICE DSU Technical Support Document 17: The Use of Observational Data to Inform Estimates of Treatment Effectiveness in Technology Appraisal: Methods for Comparative Individual Patient Data. Decision Support Unit, ScHARR, University of Sheffield; 2015. Accessed September 23, 2025. https://www.sheffield.ac.uk/sites/default/files/2022-02/TSD17-DSU-Observational-data-FINAL.pdf
49.Austin PC, White IR, Lee DS, van Buuren S. Missing Data in Clinical Research: A Tutorial on Multiple Imputation. Can J Cardiol. 2021;37(9):1322-1331. doi: 10.1016/j.cjca.2020.11.010 PubMed
50.Leyrat C, Seaman SR, White IR, et al. Propensity score analysis with partially observed covariates: How should multiple imputation be used? Stat Methods Med Res. 2019;28(1):3-19. doi: 10.1177/0962280217713032 PubMed
51.Hansen BB. Full Matching in an Observational Study of Coaching for the SAT. J Am Stat Assoc. 2004;99(467):609-618. doi: 10.1198/016214504000000647
52.Zelenetz AD, Gordon LI, Abramson JS et al. NCCN Guidelines: B-Cell Lymphomas. J Natl Compr Canc Netw. 2025;3. https://www.nccn.org/guidelines/guidelines-detail?category=1&id=1480 [sponsor supplied reference] PubMed
53.Alberta Cancer Care. Lymphoma Clinical Practice Guideline. Published online 2024. Accessed November 28, 2024. https://www.albertahealthservices.ca/assets/info/hp/cancer/if-hp-cancer-guide-lyhe002-lymphoma.pdf [sponsor supplied reference]
54.Philip T, Guglielmi C, Hagenbeek A, et al. Autologous Bone Marrow Transplantation as Compared with Salvage Chemotherapy in Relapses of Chemotherapy-Sensitive Non-Hodgkin’s Lymphoma. N Engl J Med. 1995;333(23):1540-1545. doi:10.1056/nejm199512073332305 [sponsor supplied reference] PubMed
55.Gisselbrecht C, Glass B, Mounier N, et al. Salvage Regimens With Autologous Transplantation for Relapsed Large B-Cell Lymphoma in the Rituximab Era. J Clin Oncol. 2010;28(27):4184-4190. doi:10.1200/jco.2010.28.1618 PubMed
56.Seyfarth B, Josting A, Dreyling M, Schmitz N. Relapse in common lymphoma subtypes: salvage treatment options for follicular lymphoma, diffuse large cell lymphoma and Hodgkin disease. Br J Haematol. 2006;133(1):3-18. doi:10.1111/j.1365-2141.2006.05975.x [sponsor supplied reference] PubMed
57.Friedberg JW. Relapsed/Refractory Diffuse Large B-Cell Lymphoma. Hematology. 2011; 1; 498-505. Doi: 10.1182/asheducation-2011.1.498 [sponsor supplied reference] PubMed
58.CADTH. CADTH Reimbursement Recommendation: Axicabtagene Ciloleucel (Yescarta). Can J of Health Technol. 2023;3(2). https://www.cda-amc.ca/sites/default/files/DRR/2023/PG0293REC-Yescarta-meta.pdf [sponsor supplied reference]
59.Westin J, Sehn LH. CAR T cells as a second-line therapy for large B-cell lymphoma: a paradigm shift? Blood. 2022;139(18):2737-2746. doi:10.1182/blood.2022015789 [sponsor supplied reference] PubMed
60.Shi S, Laverdure E, Cohen SRA, et al. Real-world barriers to CAR-T access: A Canadian referral center perspective. J Clin Oncol. 2024;42(16_suppl):e19002-e19002. doi:10.1200/jco.2024.42.16_suppl.e19002 [sponsor supplied reference]
61.Sehn LH, Herrera AF, Flowers CR, et al. Polatuzumab Vedotin in Relapsed or Refractory Diffuse Large B-Cell Lymphoma. J Clin Oncol. 2020;38(2):155-165. doi:10.1200/jco.19.00172 [sponsor supplied reference] PubMed
62.CADTH. CADTH Reimbursement Review: Provisional Funding Algorithm for Indication - Large B-cell lymphoma. 2024. Accessed October 25, 2024. https://www.cda-amc.ca/sites/default/files/DRR/2024/PH0048_Rapid_Algorithm_Report_LBCL_Final.pdf [sponsor supplied reference]
63.INESSS. Extract notice to the minister: Minjuvi. 2022. Accessed November 14, 2024. https://www.inesss.qc.ca/fileadmin/doc/INESSS/Inscription_medicaments/Avis_au_ministre/Novembre_2022/Novembre_2022__avis_31_octobre_/Minjuvi_2022_10.pdf [sponsor supplied reference]
64.Crump M, Leppä S, Fayad L, et al. Randomized, Double-Blind, Phase III Trial of Enzastaurin Versus Placebo in Patients Achieving Remission After First-Line Therapy for High-Risk Diffuse Large B-Cell Lymphoma. J Clin Oncol. 2016;34(21):2484-2492. doi:10.1200/jco.2015.65.7171 [sponsor supplied reference] PubMed
Appendix 1: Detailed Outcome Data
Please note that this appendix has not been copy-edited.
Table 24: Sensitivity Analyses of Overall Survival (STARGLO Study, Primary Analysis, March 29, 2023, and Updated Analysis, February 16, 2024)
Adverse events | Primary analysis data cut-off; March 29, 2023 | Updated analysis data cut-off; February 16, 2024 | ||
|---|---|---|---|---|
Glofit-GemOx (N = 183) | R-GemOx (N = 91) | Glofit-GemOx (N = 183) | R-GemOx (N = 91) | |
Primary analysis (ITT Population)a | ||||
Median, months (95% CI) | NE (13.8, NE) | 9.0 (7.3, 14.4) | 25.5 (18.3, NE) | 12.9 (7.9, 18.5) |
Stratified HR (95% CI) | 0.59 (0.40, 0.89) | — | 0.62 (0.43, 0.88) | — |
P value (log rank) | 0.010706 | — | 0.006366b | — |
Stratification according to eCRF (ITT Population) | ||||
Median, months (95% CI) | NE (13.8, NE) | 9.0 (7.3, 14.4) | 25.5 (18.3, NE) | 12.9 (7.9, 18.5) |
Stratified HR (95% CI) | 0.60 (0.40, 0.90) | — | 0.61 (0.43, 0.87) | — |
P value (log rank) | 0.0128 | — | 0.0053b | — |
Stratification according to eCRF (SE Population) | ||||
Median, months (95% CI) | NE (13.8, NE) | 10.3 (7.3, 14.4) | NE (18.3, NE) | 13.5 (7.9, 18.5) |
Stratified HR (95% CI) | 0.60 (0.40, 0.91) | — | 0.60 (0.42, 0.86) | — |
P value (log rank) | 0.0145 | — | 0.0051b | — |
Censoring for deaths due to COVID-19 infection within 3 months of treatment discontinuation/completion on date of treatment discontinuation/completion | ||||
Median, months (95% CI) | NE (17.0, NE) | 10.3 (7.3, 19.4) | NE (21.2, NE) | 13.5 (8.2, 18.5) |
Stratified HR (95% CI) | 0.55 (0.37, 0.84) | — | 0.57 (0.40, 0.82) | — |
P value (log rank) | 0.004887 | — | 0.002242b | — |
Censoring for death due to COVID-19 infection on date of death | ||||
Median, months (95% CI) | NE (17.0, NE) | 10.3 (7.3, 19.4) | NE (24.5, NE) | 13.8 (8.3, 19.4) |
Stratified HR (95% CI) | 0.55 (0.36, 0.84) | — | 0.56 (0.39, 0.81) | — |
P value (log rank) | 0.004856 | — | 0.001775 b | — |
Censoring for treatment discontinuation due to COVID-19 AE | ||||
Median, months (95% CI) | 10.3 (7.5, 19.4) | 10.3 (7.5, 19.4) | NE (19.2, NE) | 13.5 (8.2, 19.4) |
Stratified HR (95% CI) | 0.54 (0.36, 0.82) | — | 0.60 (0.42, 0.88) | — |
P value (log rank) | 0.003801 | — | 0.007737b | — |
eCRF = electronic case report form; ITT = intent-to-treat; NE = not evaluable; SE = safety evaluable.
aUsing IxRS-recorded stratification factors.
bP values are descriptive.
Source: STARGLO (GO41944) Clinical Study Report at the clinical cut-off dates of March 29, 2023,29 and February 16, 2024.45
HRQoL Time to Deterioration Analysis
Time-to-deterioration analysis of EORTC QLQ-C30 (fatigue and physical functioning subscale) and FACT-Lym LymS were the secondary end points in the STARGLO trial. For the EORTC QLQ-C30, time to deterioration in fatigue and physical functioning was defined as the time from randomization to the first documentation of a 10-point or more increase or decrease, respectively. For the FACT-Lym LymS, time to deterioration in lymphoma-specific symptoms was defined as the time from randomization to the first documentation of a 3-point or more decrease in mean score.
Figure 11: Kaplan-Meier Plot of Time From Randomization to a Clinically Meaningful Deterioration in Fatigue, EORTC QLQ-C30 (ITT Population) — A (Primary Analysis Data Cut-Off; March 29, 2023); B (Updated Analysis Data Cut-Off; February 16, 2024)
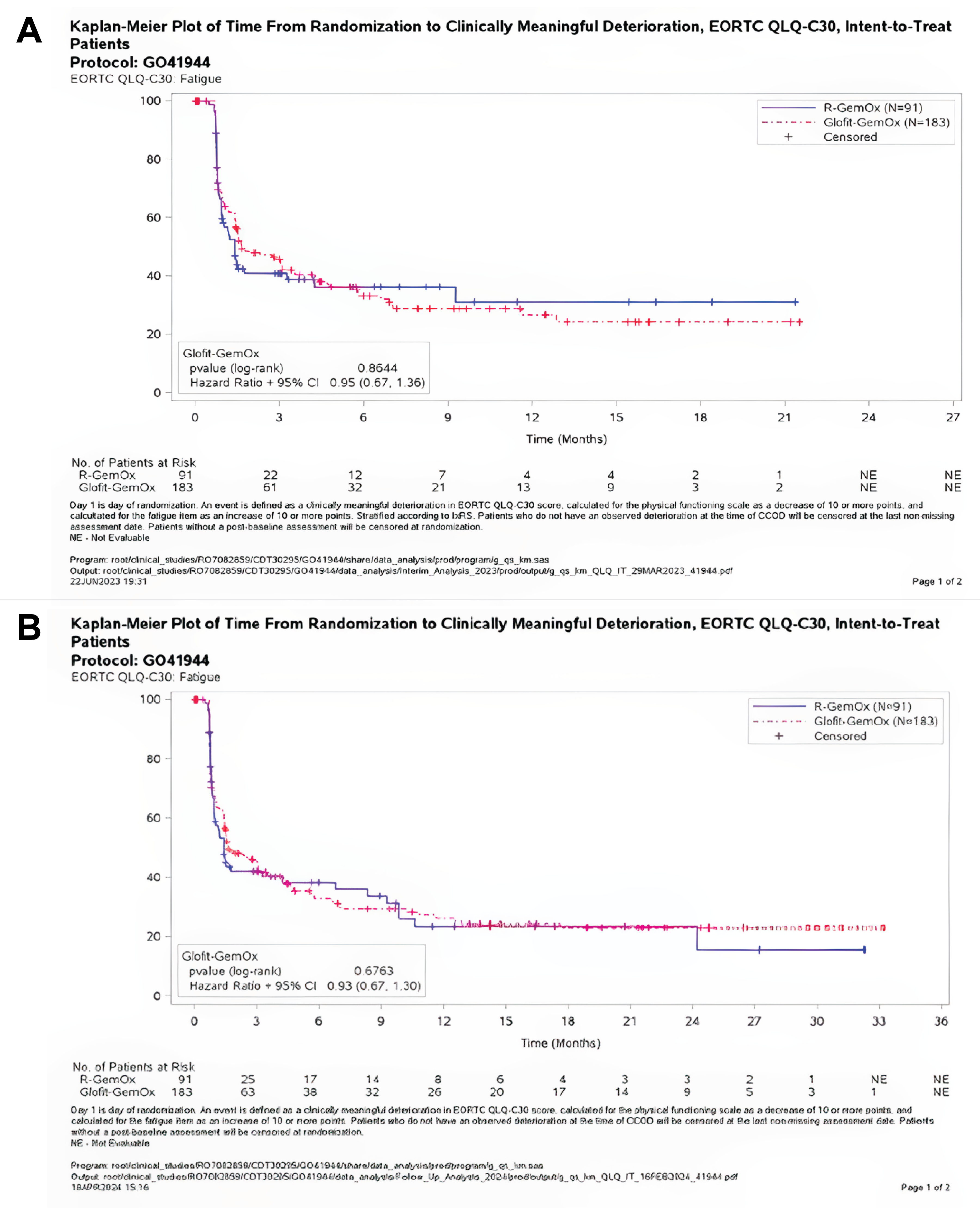
Source: STARGLO (GO41944) Clinical Study Report at the clinical cut-off dates of March 29, 2023,29 and February 16, 2024.45
Figure 12: Kaplan-Meier Plot of Time From Randomization to a Clinically Meaningful Deterioration in Physical Functioning, EORTC QLQ-C30 (ITT Population) — A (Primary Analysis Data Cut-Off; March 29, 2023); B (Updated Analysis Data Cut-Off; February 16, 2024)
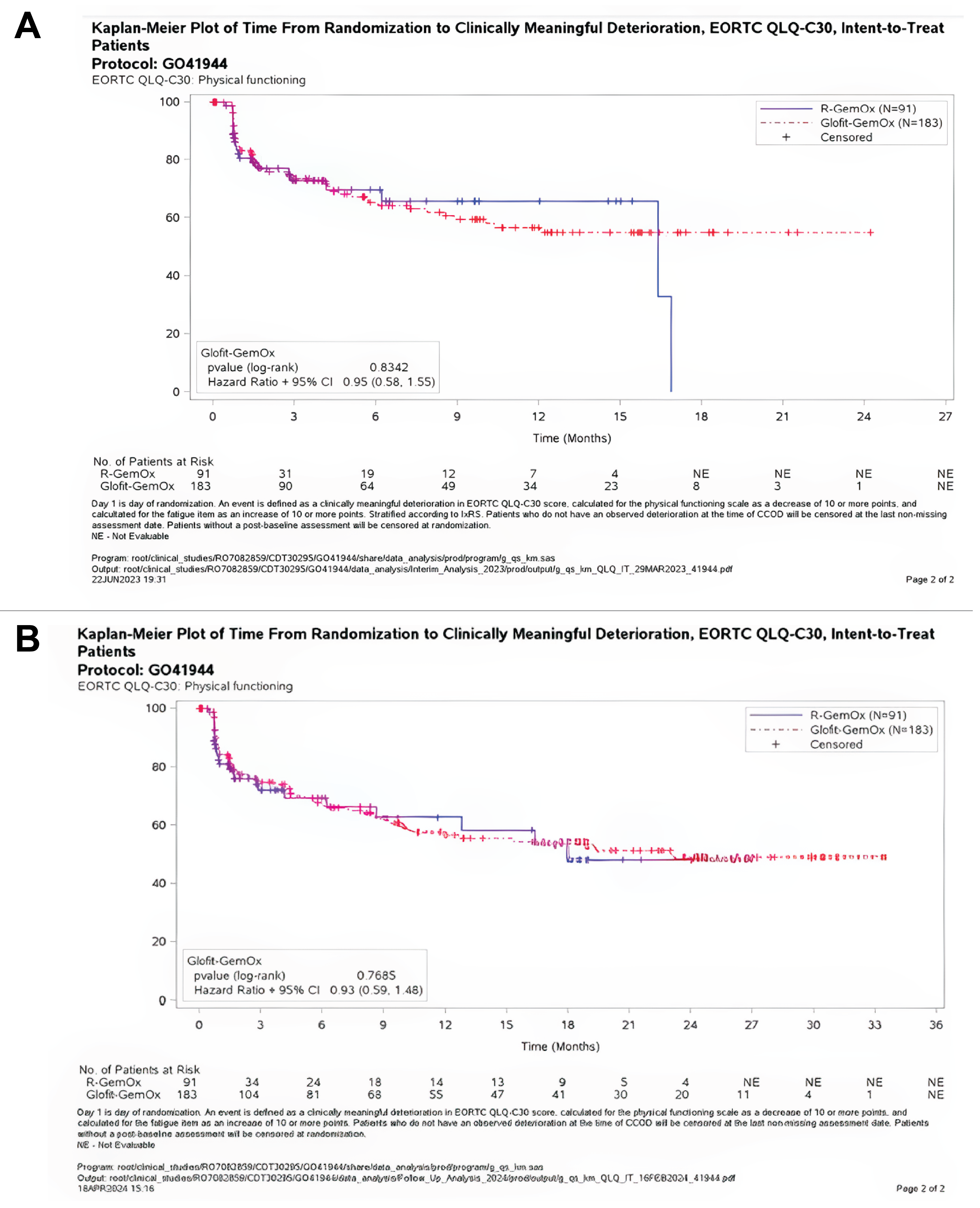
Source: STARGLO (GO41944) Clinical Study Report at the clinical cut-off dates of March 29, 2023,29 and February 16, 2024.45
Figure 13: Kaplan-Meier Plot of Time From Randomization to a Clinically Meaningful Deterioration in FACT-Lym LymS (ITT Population) — A (Primary Analysis Data Cut-Off of March 29, 2023); B (Updated Analysis Data Cut-Off of February 16, 2024)
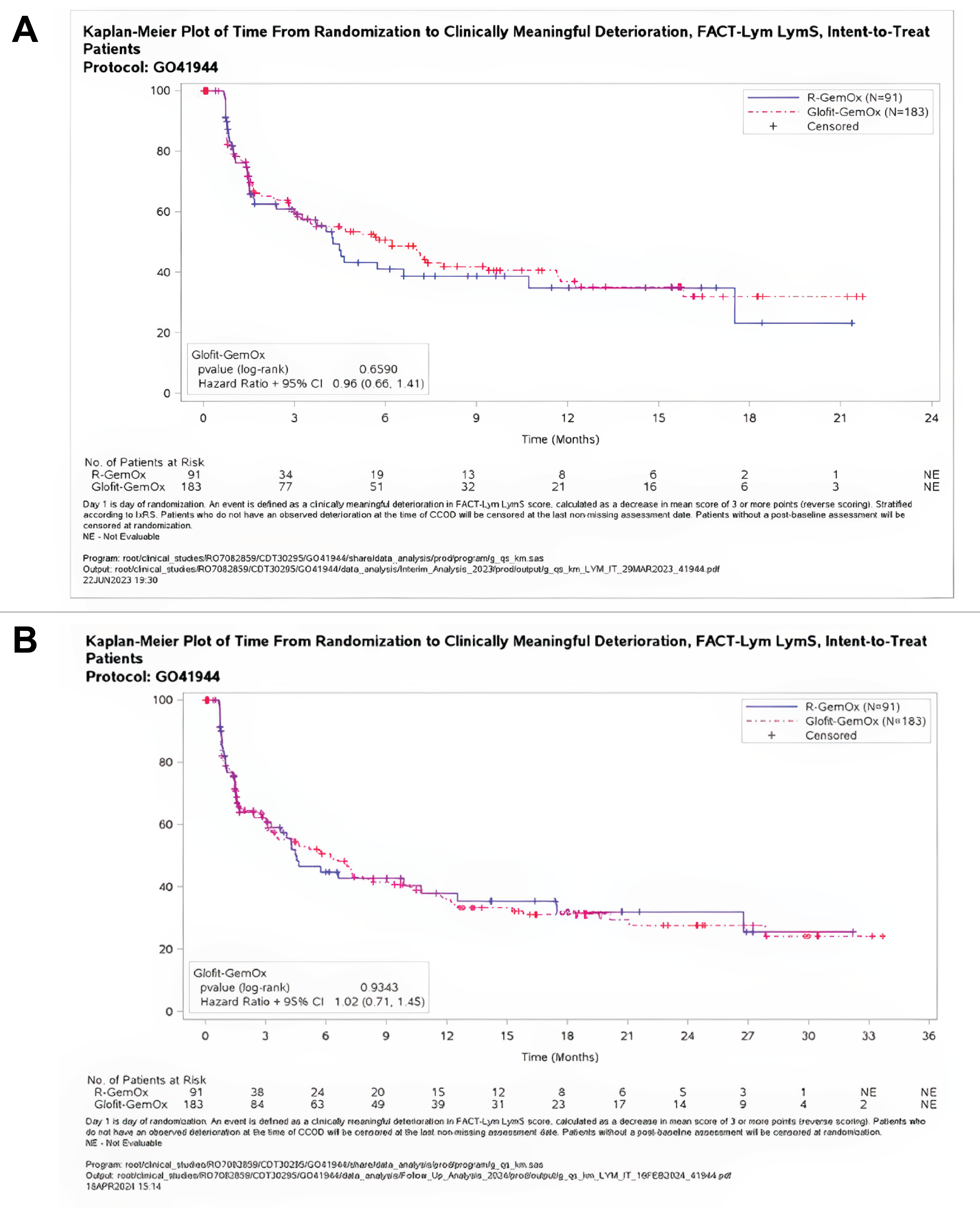
Source: STARGLO (GO41944) Clinical Study Report at the clinical cut-off dates of March 29, 2023,29 and February 16, 2024.45
Pharmacoeconomic Review
Abbreviations
AE
adverse event
ASCT
autologous stem cell transplant
BIA
budget impact analysis
CDA-AMC
Canada’s Drug Agency
CAR
chimeric antigen receptor
DLBCL
diffuse large B-cell lymphoma
Glofit-GemOx
glofitamab in combination with gemcitabine and oxaliplatin
HRQoL
health-related quality of life
ICER
incremental cost-effectiveness ratio
ITC
indirect treatment comparison
LY
life-year
NOS
not otherwise specified
OS
overall survival
PFS
progression-free survival
pola-BR
polatuzumab vedotin plus bendamustine and rituximab
QALY
quality-adjusted life-year
R-GemOx
rituximab in combination with gemcitabine and oxaliplatin
Economic Review
The objective of the economic review undertaken by Canada’s Drug Agency (CDA-AMC) is to review and critically appraise the pharmacoeconomic evidence submitted by the sponsor on the cost-effectiveness and budget impact of glofitamab in combination with gemcitabine and oxaliplatin (Glofit-GemOx) compared to rituximab in combination with gemcitabine and oxaliplatin (R-GemOx), polatuzumab vedotin plus bendamustine and rituximab (pola-BR), and glofitamab monotherapy for the treatment of patients with relapsed or refractory diffuse large B-cell lymphoma (DLBLC) not otherwise specified (NOS) in adults who are not candidates for autologous stem cell transplant (ASCT). The sponsor-submitted comparison to R-GemOx and pola-BR is aligned with the Health Canada indication (i.e., use in the second line or later), with the comparison to glofitamab monotherapy reflecting usage in the third line or later, which is a subset of the indicated population.
Item | Description |
|---|---|
Drug product | Golfitamab (Columvi), 1 mg/mL for IV infusion |
Indication | Proposed: Glofitamab in combination with gemcitabine and oxaliplatin (Glofit-GemOx) for the treatment of adult patients with relapsed or refractory diffuse large B-cell lymphoma not otherwise specified (DLBCL NOS) who are not candidates for autologous stem cell transplant (ASCT) |
Submitted price | $1,040.00 per 2.5 mg vial $4,160.00 per 10 mg vial |
Health Canada approval status | Under review (pre-NOC) |
Health Canada review pathway | Standard |
NOC date | Anticipated: August 17, 2025 |
Reimbursement request | Per indication |
Sponsor | Hoffmann-La Roche Ltd. |
Submission history | Yes Indication: treatment of adult patients with relapsed or refractory DLBCL NOS, DLBCL arising from follicular lymphoma, or primary mediastinal B-cell lymphoma, who have received 2 or more lines of systemic therapy and are ineligible to receive or cannot receive CAR T-cell therapy or have previously received CAR-T-cell therapy, following obinutuzumab pre-treatment. Recommendation date: February 2, 2024 Recommendation: Reimburse with clinical criteria and/or conditions |
ASCT = autologous stem cell transplant; CAR = chimeric antigen receptor; DLBCL = diffuse large B-cell lymphoma; Glofit-GemOx = glofitamab in combination with gemcitabine and oxaliplatin; NOC = Notice of Compliance; NOS = not otherwise specified.
Pagebreak
Summary
Glofitamab is available as 2.5 mL and 10 mL vials (1 mg/mL) of solution for IV infusion. At the submitted price of $1,040 per 25 mL vial (2.5 mg glofitamab) and $4,160 per 10 mL vial (10 mg glofitamab), the cost of glofitamab per 21-day cycle is expected to be $5,200 per patient in the first cycle of treatment and $12,480 in subsequent cycles, based on the Health Canada–recommended dosage. When used in combination with gemcitabine and oxaliplatin, the 21-day costs are expected to be $11,672, $13,200, and $12,480 per patient in cycle 1, cycles 2 to 8, and cycles 9 to 12, respectively.
Clinical efficacy in the economic analysis for Glofit-GemOx versus R-GemOx was derived from the STARGLO trial. Evidence submitted by the sponsor suggests that Glofit-GemOx likely results in an improvement in overall survival (OS) and progression-free survival (PFS) compared with R-GemOx among patients with relapsed or refractory DLBCL NOS who are not candidates for ASCT. Comparative clinical efficacy for Glofit-GemOx versus pola-BR and versus glofitamab monotherapy was informed by sponsor-submitted indirect treatment comparisons (ITCs), which did not show a conclusive benefit of Glofit-GemOx for efficacy (i.e., OS or PFS) or discontinuation due to adverse events (AEs). Health-related quality of life (HRQoL) and AEs were not included in the sponsor’s ITC.
The results of the CDA-AMC base case suggest:
Glofit-GemOx is predicted to be associated with higher costs to the health care system compared with R-GemOx (incremental costs = $165,684) and pola-BR (incremental costs = $49,252), primarily driven by increased drug acquisition costs associated with Glofit-GemOx.
Glofit-GemOx is predicted to be associated with a gain of 2.96 life-years (LYs) compared to R-GemOx and 0.43 LYs compared to pola-BR. When the impact on HRQoL is also considered, Glofit-GemOx is predicted to be associated with gains of 2.07 and 0.35 quality-adjusted life-years (QALYs) compared to R-GemOx and pola-BR, respectively.
The incremental cost-effectiveness ratio (ICER) of Glofit-GemOx compared to R-GemOx was $79,881 per QALY gained in the CDA-AMC base case. Compared to pola-BR, the ICER of Glofit-GemOx was $141,006 per QALY gained. The estimated ICER is uncertain because of the uncertainty in the comparative efficacy, long-term survival estimates, and economic model structure. Given the identified limitations with the submitted model structure and the uncertainty in the long-term efficacy data, the economic analysis may not accurately assess the impact on patient health and health care resources. The cost-effectiveness estimates are therefore uncertain, and greater price reductions may be required to achieve a given willingness-to-pay threshold.
CDA-AMC estimates that the budget impact of reimbursing Glofit-GemOx for the Health Canada–indicated population will be approximately $43 million over the first 3 years of reimbursement compared to the amount currently spent on comparators, with an estimated expenditure of $127 million on Glofit-GemOx over this period (expenditure on glofitamab alone: $108 million). The actual budget impact of reimbursing Glofit-GemOx will depend on the number of patients eligible for treatment, the market uptake of Glofit-GemOx, and the treatment duration of therapies in clinical practice.
Summary of the Submitted Economic Evaluation
The sponsor submitted a cost-utility analysis to estimate the cost-effectiveness of Glofit-GemOx from the perspective of a public health care payer in Canada over a lifetime horizon (35 years). The modelled population comprised adult patients with DLBCL NOS who are not candidates for ASCT, which is aligned with the Health Canada indication and was based on data gathered from participants in the STARGLO trial. The sponsor’s base-case analysis included costs related to drug acquisition (submitted price for glofitamab and public list prices for other drugs), administration, supportive care, subsequent treatment, AEs, and health care resource use. In the sponsor’s base case, Glofit-GemOx was associated with incremental costs of $49,633 and 2.44 incremental QALYs relative to R-GemOx, resulting in an ICER of $20,376 per QALY gained. Compared to pola-BR and glofitamab monotherapy, Glofit-GemOx was associated with ICERs of $20,387 per QALY gained (incremental costs = $5,345; incremental QALYs = 0.26) and $134,829 per QALY gained (incremental costs = $65,421; incremental QALYs = 0.49), respectively. Additional information about the sponsor’s submission is summarized in Appendix 3.
CDA-AMC identified several key issues with the sponsor’s analysis (refer to Table 2; full details are provided in Appendix 4).
Table 2: Key Issues With the Sponsor’s Economic Submission
Issue | What evidence is there to inform this issue? | How was this issue addressed by CDA-AMC? | Did CDA-AMC explore uncertainty in a scenario analysis? |
|---|---|---|---|
The comparative efficacy of Glofit-GemOx is uncertain. | The magnitude of OS and PFS benefits with Glofit-GemOx vs. R‑GemOx is uncertain because of data censoring. There is no head-to-head direct evidence comparing Glofit-GemOx to pola-BR or glofitamab monotherapy. The indirect evidence submitted by the sponsor did not show a conclusive benefit of Glofit-GemOx vs. pola-BR or glofitamab monotherapy. | CDA-AMC could not address uncertainty in the comparative efficacy data. | CDA-AMC conducted a scenario analysis assuming equal efficacy for Glofit-GemOx and pola-BR. |
The sponsor’s pharmacoeconomic analysis does not adequately reflect the clinical pathway or cost-effectiveness by line of therapy. | The comparative efficacy estimates used by the sponsor, and therefore the resulting cost-effectiveness estimates, reflect the comparison of Glofit-GemOx vs. R-GemOx and pola-BR in the second or later line and vs. glofitamab monotherapy in the third or later line. The care pathway for relapsed or refractory DLBCL depends on prior treatment received as well as eligibility for ASCT and CAR T-cell therapy and cannot be adequately reflected in a partitioned survival model. | CDA-AMC was unable to address the structural limitations of the sponsor’s modelling approach. The CDA-AMC base case compared Glofit-GemOx to R-GemOx and pola-BR in the second or later line, which is aligned with the draft Health Canada indication. | The cost-effectiveness of Glofit-GemOx vs. glofitamab monotherapy, which reflects usage in the third or later line, was explored in a scenario analysis. |
The impact of Glofit-GemOx on long-term survival is uncertain. | The sponsor’s analysis predicts an incremental gain of LYs for Glofit-GemOx compared to R-GemOx, pola-BR, and glofitamab monotherapy. While a gain in LYs vs. R-GemOx is supported by OS data from the STARGLO trial, the magnitude of gain is uncertain because of uncertainty in the OS data from STARGLO, and the submitted indirect evidence did not show a conclusive benefit for OS between Glofit-GemOx and pola-BR or glofitamab monotherapy. The sponsor’s assumption that patients who remain progression-free for 3 years are no longer at risk of disease progression introduces additional uncertainty. | CDA-AMC was unable to fully address uncertainty in the impact of Glofit-GemOx on long-term survival. CDA-AMC removed the sponsor’s cure assumption from the base case. | CDA-AMC explored a 5‑year cure assumption in a scenario analysis. |
The costs associated with subsequent therapy are highly uncertain. | The sponsor incorporated the cost of subsequent therapy as a one-time cost for all patients who transition out of the PFS health state, without accounting for the proportion of PFS events that are deaths. Use of a one-time cost assumes that the duration and intensity of treatment is the same for all patients and therefore does not account for variability in treatment regimens. Clinical expert input obtained by CDA-AMC indicated that the distribution of subsequent therapies in the model is not aligned with clinical practice in Canada. | CDA-AMC could not fully address this issue because of the structure of the sponsor’s model. | No scenario analyses were conducted. |
Treatment costs are uncertain. | All patients in the model received the same number of treatment cycles based on the mean number of treatment cycles in the STARGLO trial for Glofit-GemOx and from GO29365 and NP30179 studies for comparators. This approach fails to incorporate variability in treatment exposure. | CDA-AMC could not fully address this issue because of the structure of the sponsor’s model. | CDA-AMC conducted a scenario analysis in which treatment was received until disease progression. |
Treatment effectiveness waning was not considered. | The sponsor assumed that patients would maintain treatment benefit even if off treatment. In the absence of long-term data, the impact of waning effectiveness on patients who discontinue or complete a course of treatment is unknown. | CDA-AMC could not fully address this issue because of a lack of long-term data. | CDA-AMC explored the impact of waning treatment effectiveness in a scenario analysis. |
The impact of adverse events is uncertain. | Cytokine release syndrome, identified as a notable harm by clinical experts consulted by CDA-AMC, was not included in the model, despite a reported incidence of 2.2% in the Glofit-GemOx group of the STARGLO trial. The sponsor assumed that the impact of AEs on health-related quality of life would be captured through health state utility values. | CDA-AMC could not address this issue because of the structure of the sponsor’s model. | No scenario analyses were conducted. |
ASCT = autologous stem cell transplant; CAR = chimeric antigen receptor; CDA-AMC = Canada’s Drug Agency; DLBCL = diffuse large B-cell lymphoma; Glofit-GemOx = glofitamab in combination with gemcitabine and oxaliplatin; LY = life-years; OS = overall survival; PFS = progression-free survival; pola-BR = polatuzumab vedotin plus bendamustine and rituximab; R-GemOx = rituximab in combination with gemcitabine and oxaliplatin; vs. = versus.
Note: Full details of the issues identified by CDA-AMC are provided in Appendix 3.
CDA-AMC Assessment of Cost-Effectiveness
The CDA-AMC base case was derived by making changes to model parameter values and assumptions (refer to Table 6), in consultation with clinical experts. Detailed information about the base case is provided in Appendix 4.
Impact on Health Care Costs
Glofit-GemOx is expected to be associated with additional health care costs compared to R-GemOx (incremental costs = $165,684). Compared to pola-BR, Glofit-GemOx is expected to be associated with additional health care costs (incremental costs = $49,252). This increase in health care spending primarily results from drug acquisition and supportive care costs associated with Glofit-GemOx (refer to Figure 1 and Figure 2).
Figure 1: Impact of Glofit-GemOx vs. R-GemOx on Health Care Costs
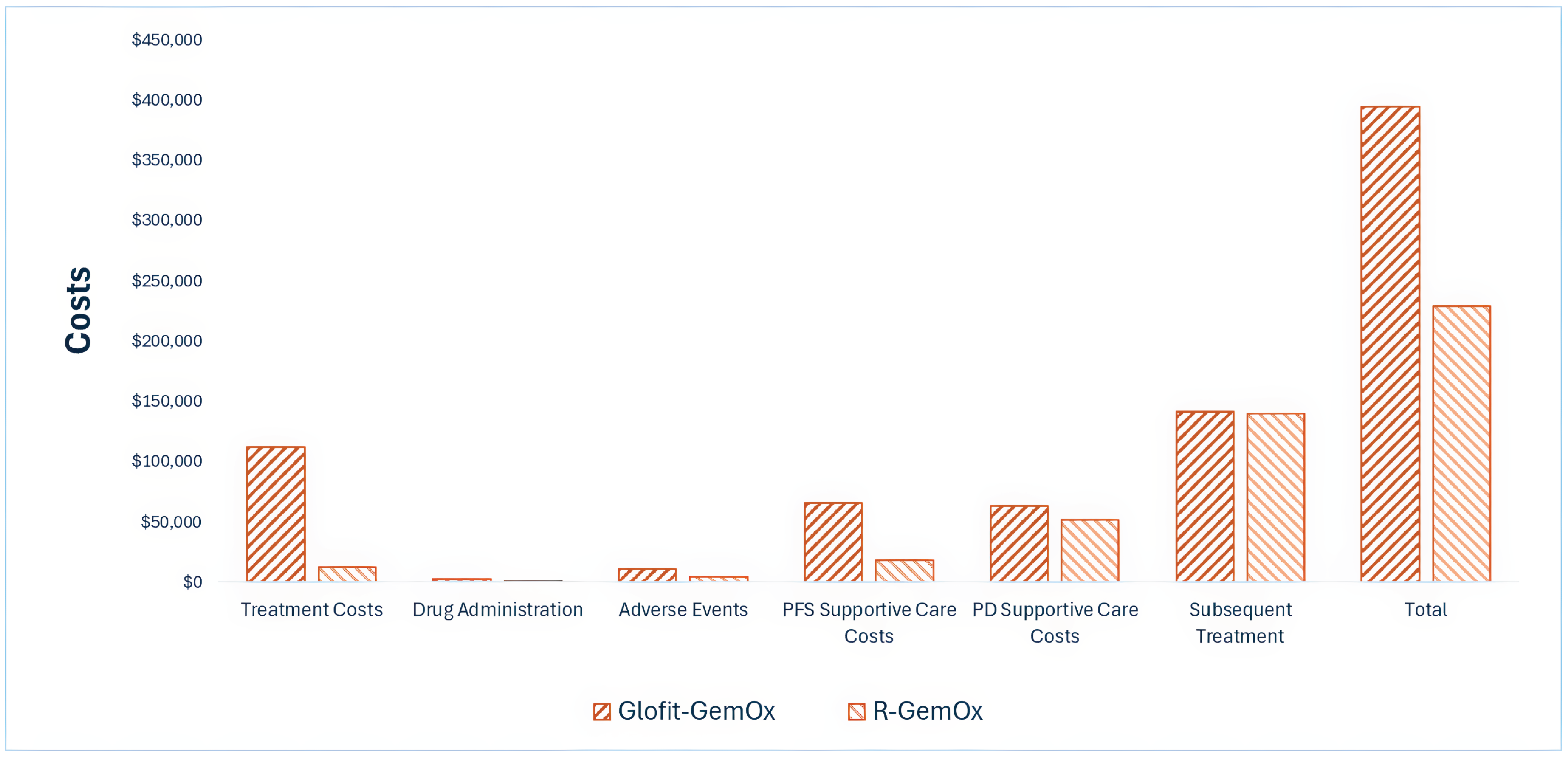
Glofit-GemOx = glofitamab in combination with gemcitabine and oxaliplatin; PD = progressed disease; PFS = progression-free survival; R-GemOx = rituximab in combination with gemcitabine and oxaliplatin; vs. = versus.
Figure 2: Impact of Glofit-GemOx vs. pola-BR on Health Care Costs
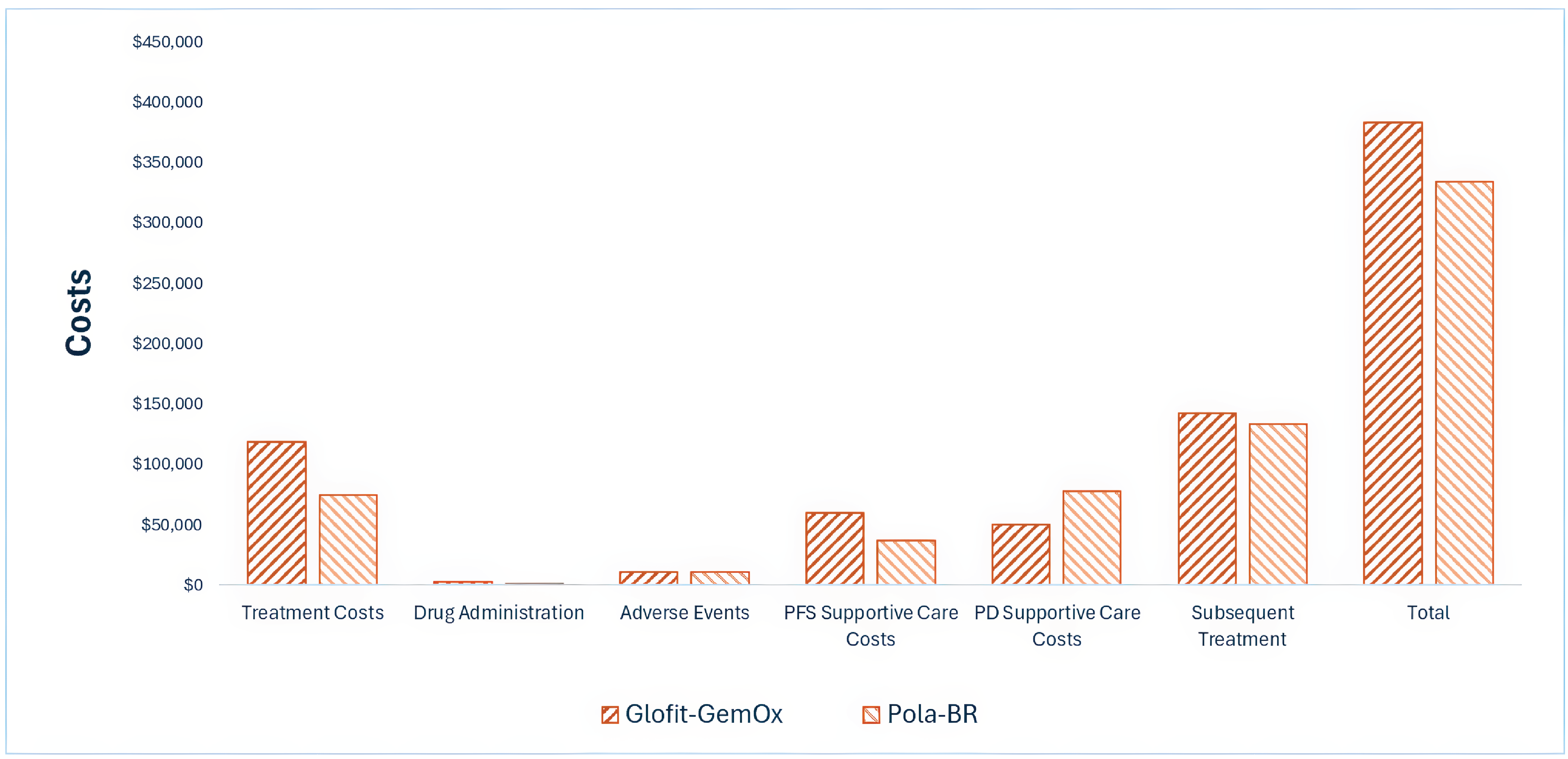
Glofit-GemOx = glofitamab in combination with gemcitabine and oxaliplatin; pola-BR = polatuzumab vedotin plus bendamustine and rituximab; vs. = versus.
Impact on Health
Relative to R-GemOx, Glofit-GemOx is expected to increase the amount of time a patient remains in the PFS health state by approximately 2.67 years and to extend OS by 2.95 years (refer to Appendix 4). Considering the impact of treatment on both quality and length of life, Glofit-GemOx is expected to result in a gain of 2.07 QALYs compared to R-GemOx.
Compared to pola-BR, Glofit-GemOx is expected to increase the amount of time a patient remains in the PFS health state by 1.15 years. Considering the impact on both quality and length of life, Glofit-GemOx is expected to result in an additional 0.35 QALYs per patient compared to pola-BR.
Overall Results
The results of the CDA-AMC base case suggest an ICER of $79,881 per QALY gained for Glofit-GemOx compared to R-GemOx (refer to Table 3). Compared to pola-BR, the ICER of Glofit-GemOx was $141,006. Additional details on the CDA-AMC base case are available in Appendix 4.
Table 3: Summary of CDA-AMC Economic Evaluation Results
Drug | Total costs ($) | Total QALYs | ICER vs. reference ($ per QALY) |
|---|---|---|---|
Glofit-GemOx vs. R-GemOx | |||
R-GemOx | 228,624 | 1.54 | Reference |
Glofit-GemOx | 394,308 | 3.62 | 79,881 |
Glofit-GemOx vs. pola-BR | |||
Pola-BR | 334,305 | 2.81 | Reference |
Glofit-GemOx | 383,557 | 3.16 | 141,006 |
Glofit-GemOx = glofitamab in combination with gemcitabine and oxaliplatin; ICER = incremental cost-effectiveness ratio; pola-BR = polatuzumab vedotin plus bendamustine and rituximab; QALY = quality-adjusted life-year; R-GemOx = rituximab in combination with gemcitabine and oxaliplatin; vs. = versus.
Note: Publicly available list prices were used for all comparators.
Uncertainty and Sensitivity
Uncertainty was explored in the scenario analyses outlined in Table 13 and Table 14. Assumptions influencing OS (i.e., including a cure assumption, assuming equal efficacy compared to pola-BR) had the largest impact on cost-effectiveness (refer to Appendix 4).
Summary of the Budget Impact
The sponsor submitted a budget impact analysis (BIA) to estimate the 3-year (2026 to 2028) budget impact of reimbursing glofitamab for use in combination with gemcitabine and oxaliplatin for the treatment of relapsed or refractory DLBCS NOS in adults who are not candidates for ASCT. The sponsor assumed that the payer would be CDA-AMC–participating public drug plans and derived the size of the eligible population using an epidemiologic approach. The price of glofitamab was aligned with the price included in the sponsor’s economic evaluation, while the prices of gemcitabine and oxaliplatin, as well as those of comparators, were based on the publicly available list prices. Additional information pertaining to the sponsor’s submission is provided in Appendix 5.
CDA-AMC identified a number of issues with the sponsor’s estimated budget impact. The CDA-AMC was derived by correcting a coding error in the submitted BIA model. CDA-AMC estimated that 3,502 patients will be eligible for treatment with Glofit-GemOx over a 3-year period (year 1 = 1,178; year 2 = 1,162; year 3 = 1,162), of whom 833 are expected to receive Glofit-GemOx (year 1 = 173; year 2 = 292; year 3 = 833). The estimated incremental budget impact of reimbursing Glofit-GemOx is expected to be approximately $43 million over the first 3 years, with an expected expenditure of $127 million on Glofit-GemOx (expenditure on glofitamab alone: $108 million). The actual budget impact of reimbursing Glofit-GemOx will depend on the number of people eligible for treatment, the market uptake of Glofit-GemOx, and the duration of treatment in clinical practice.
Conclusion
Results of the CDA-AMC base case suggest that Glofit-GemOx would be considered cost-effective at the submitted price if the public health care system is willing to pay at least $79,881 for each additional QALY gained. If the public health care system is not willing to pay that amount, a price reduction should be considered (refer to Figure 3; full details of the impact of price reductions on cost-effectiveness are presented in Table 10). The estimated cost-effectiveness of Glofit-GemOx compared to R-GemOx is uncertain due to the uncertainty in the comparative efficacy, long-term survival estimates, and structure of the submitted economic model.
The budget impact of reimbursing Glofit-GemOx to the public drug plans in the first 3 years is estimated to be approximately $43 million. The 3-year expenditure on Glofit-GemOx (i.e., not accounting for current expenditure on comparators) is estimated to be $127 million. The estimated budget impact is uncertain due to the uncertainty in the number of patients eligible for treatment, the market uptake of Glofit-GemOx, and the duration of treatment in clinical practice.
Figure 3: Summary of the CDA-AMC Economic Analysis and Price Reduction

CDA-AMC = Canada’s Drug Agency; QALY = quality-adjusted life-year; pola-BR = polatuzumab vedotin plus bendamustine and rituximab; R-GemOx = rituximab in combination with gemcitabine and oxaliplatin; vs. = versus.
Note: Expenditure includes only the drug cost of glofitamab (as part of Glofit-GemOx).
References
1.Cancer Care Ontario (CCO). Drug Formulary: GEMOX+RITU regimen (Gemcitabine-Oxaliplatin-riTUXimab). 2025. Accessed May 23, 2025. https://www.cancercareontario.ca/en/drugformulary/regimens/monograph/67996
2.Cancer Care Ontario (CCO). Drug Formulary: BEND+POLA+RITU regimen (Bendamustine-Polatuzumab-Rituximab). 2025. Accessed May 23, 2025. https://www.cancercareontario.ca/en/drugformulary/regimens/monograph/62866
3.Hoffmann-La Roche Limited. Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Columvi (glofitamab for injection), 1 mg/mL concentrate for solution for intravenous infusion. March 7, 2025.
4.IQVIA. DeltaPA. 2023. Accessed May 24, 2025. https://www.iqvia.com/
5.Hoffmann-La Roche Limited. Columvi (glofitamab for injection): 2.5 mg/2.5 mL or 10 mg/10 mL, concentrate for solution for intravenous infusion, in single-use glass vial [product monograph]. August 13, 2025.
6.Maurer MJ, Ghesquieres H, Jais JP, et al. Event-free survival at 24 months is a robust end point for disease-related outcome in diffuse large B-cell lymphoma treated with immunochemotherapy. J Clin Oncol. 2014;32(10):1066-73. doi: 10.1200/JCO.2013.51.5866 PubMed
7.Ara R, Brazier JE. Populating an economic model with health state utility values: moving toward better practice. Value Health. 2010;13(5):509-18. doi: 10.1111/j.1524-4733.2010.00700.x PubMed
8.Hernandez Alava M, Pudney S, Wailoo A. Estimating the Relationship Between EQ-5D-5L and EQ-5D-3L: Results from a UK Population Study. Pharmacoeconomics. 2023;41(2):199-207. doi: 10.1007/s40273-022-01218-7 PubMed
9.Ontario Ministry of Health. Schedule of benefits for physician services under the Health Insurance Act: (February 15, 2025 (effective March 3, 2025)). 2025. Accessed June 2, 2025. https://www.ontario.ca/files/2025-03/moh-schedule-benefit-2025-03-19.pdf
10.Ontario Ministry of Health. Schedule of benefits for laboratory services: February 14, 2025 (effective March 3, 2025). 2025. Accessed June 2, 2025. https://www.ontario.ca/files/2025-03/moh-ohip-schedule-of-benefits-laboratory-services-2025-03-03.pdf
11.Canada's Drug Agency. Provisional Funding Algorithm: large B-cell lymphoma. 2025. Accessed June 12, 2025. https://www.cda-amc.ca/sites/default/files/DRR/2025/PH0064-Rapid_Algorithm_Report_LBCL.pdf
12.Brenner DR, Gillis J, Demers AA, et al. Projected estimates of cancer in Canada in 2024. CMAJ. 2024;196(18):E615-E623. doi: 10.1503/cmaj.240095 PubMed
13.Shafey M, Savage KJ, Skrabek P, Elsawy M, Bosch M, Kuruvilla J. Canadian Evidence-Based Guideline for the Treatment of Relapsed/Refractory Diffuse Large B-Cell Lymphoma. Lymphoma Canada; 2021. Accessed by sponsor, no date provided. https://www.lymphoma.ca/wp-content/uploads/2021/09/LymphomaCanada_Guideline_Relapsed_Refractory_DLBCL_VF_Digital.pdf
14.CADTH. Reimbursement recommendation: glofitamab (Columvi). Can J Health Technol. 2024;4(2). doi: 10.51731/cjht.2024.837
15.INESSS. Notice to the Minister: Minjuvi (diffuse large B-cell lymphoma). 2022. Accessed by sponsor, no date provided. https://www.inesss.qc.ca/fileadmin/doc/INESSS/Inscription_medicaments/Avis_au_ministre/Novembre_2022/Novembre_2022__avis_31_octobre_/Minjuvi_2022_10.pdf
16.Raut LS, Chakrabarti PP. Management of relapsed-refractory diffuse large B cell lymphoma. South Asian J Cancer. 2014;3(1):66-70. doi: 10.4103/2278-330X.126531 PubMed
17.Sehn LH, Salles G. Diffuse Large B-Cell Lymphoma. N Engl J Med. 2021;384(9):842-858. doi: 10.1056/NEJMra2027612 PubMed
18.CADTH. Reimbursement recommendation: axicabtagene ciloleucel (Yescarta). Can J Health Technol. 2023;3(11). doi: 10.51731/cjht.2023.787
19.Locke FL, Miklos DB, Jacobson CA, et al. Axicabtagene Ciloleucel as Second-Line Therapy for Large B-Cell Lymphoma. N Engl J Med. 2022;386(7):640-654. doi: 10.1056/NEJMoa2116133 PubMed
Appendix 1: Cost Comparison Table
Please note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical experts and CDA-AMC–participating public drug plans. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 4: Cost Comparison for Relapsed or Refractory DLBCL NOS Who Are Not Candidates for ASCT
Treatment | Strength | Form | Price | Recommended dosage | Daily cost ($) | 21-day cycle cost ($)a |
|---|---|---|---|---|---|---|
Glofitamab | 2.5 mg/ 2.5 mL 10 mg/ 10mL | Vial for injection | 1,040.0000a 4,160.0000 | Cycle 1: 2.5 mg on day 8 and 10 mg on day 15 Cycle 2 to 12: 30 mg on day 1 | Cycle 1: 247.62 Cycle 2 to 12: 594.29 | Cycle 1: 5,200 Cycle 2 to 12: 12,480 |
Gemcitabine | 1,000 mg/ 25 mL 2,000 mg/ 50 mL | Vial for injection | 270.0000b 540.0000b | Cycle 1: 1,000 mg/m2 on day 2 Cycle 2 to 8: 1,000 mg/m2 on day 1 | 25.71 | 540 |
Oxaliplatin | 100 mg/ 20 mL 200 mg/ 40 mL | Vial for injection | 90.0000b 180.0000b | Cycle 1: 100 mg/m2 on day 2 Cycle 2 to 8: 100 mg/m2 on day 1 | 8.57 | 180 |
Obinutuzumab (Gazyva) | 1,000 mg/ 40 mL | Solution for infusion | 5,751.7320b | Cycle 1 (Premedication): 1,000 mg dose on day 1 | 273.89 | 5,752 |
Glofit-GemOx | — | — | — | — | Cycle 1: 555.80 Cycle 2 to 8: 628.57 Cycle 9 to 12: 594.29 | Cycle 1: 11,672 Cycle 2 to 8: 13,200 Cycle 9 to 12: 12,480 |
R-GemOx | ||||||
Gemcitabine (generics) | 1,000 mg/ 25 mL 2,000 mg/ 50 mL | Vial for injection | 270.0000b 540.0000b | Cycle 1 (14-day cycle): 1,000 mg/m2 on day 2 Cycle 2 to 4 (14-day cycles): 1,000 mg/m2 on day 1 | 38.57 | 810 |
Oxaliplatin (generics) | 100 mg/ 20 mL 200 mg/ 40 mL | Vial for injection | 90.0000b 180.0000b | Cycle 1 (14-day cycle): 100 mg/m2 on day 2 Cycle 2 to 4 (14-day cycles): 100 mg/m2 on day 1 | 12.86 | 270 |
Rituximab (biosimilars) | 100 mg/ 10 mL | IV infusion | 297.0000 | Cycle 1 to 4 (14-day cycles): 375 mg/m2 on day 1 | 148.50 | 3,119 |
R-GemOx | — | — | — | — | 199.93 | 4,199 |
pola-BR | ||||||
Polatuzumab vedotin (Polivy) | 140 mg/ 20 mL | Lyophilized powder | 14,750.0000b | Cycle 1: 1.8 mg/m2 on day 2 Cycle 2 to 6: 1.8 mg/m2 on day 1 | 702.38 | 14,750 |
Bendamustine (generic) | 25 mg/ 8 mL 100 mg/ 20 mL | Lyophilized powder | 250.0000b 1,000.0000b | Cycle 1: 90 mg/m2 on day 2 and 3 Cycle 2 to 6: 90 mg/m2 on days 1 and 2 | 166.67 | 3,500 |
Rituximab (biosimilars) | 100 mg/ 10 mL 500 mg/ 50 mL | IV infusion | 297.0000 | Cycle 1 to 6: 375 mg/m2 on day 1 | 99.00 | 2,079 |
pola-BR | — | — | — | — | 968.05 | 20,329 |
ASCT = autologous stem cell transplant; DLBCL = diffuse large B-cell lymphoma; Glofit-GemOx = glofitamab in combination with gemcitabine and oxaliplatin; NOS = not otherwise specified; pola-BR = polatuzumab vedotin plus bendamustine and rituximab; R-GemOx = rituximab in combination with gemcitabine and oxaliplatin.
Note: All prices are from the Ontario Drug Benefit Formulary (accessed May 2025), unless otherwise indicated, and do not include dispensing fees. All cycle regimens have a cycle length of 21 days, unless otherwise stated. Recommended dosing is informed by Cancer Care Ontario regimen monographs unless otherwise stated.1,2
aSponsor submitted price.3
bObtained from IQVIA DeltaPA database accessed May 2025.4
Appendix 2: Input Relevant to the Economic Review
Please note that this appendix has not been copy-edited.
This section is a summary of the input received from the patient groups, clinician groups, and drug plans that participated in the CDA-AMC review process.
Patient input was received from Lymphoma Canada, with input gathered via an online survey. Respondents with DLBCL noted that it has a significant impact on their physical and psychosocial well-being. Respondents noted that travelling and financial implications were a concern with currently available treatment and indicated a desire for new treatments that extend disease remission and improve survival. Two respondents with glofitamab experience (in combination with gemcitabine and oxaliplatin [Glofit-GemOx]; both from the US) indicated that their experience was good but that they had experienced side effects including decreased appetite, nausea, fatigue, and cytokine release syndrome (CRS).
Clinician group input was received from Lymphoma Canada and Ontario Health (Cancer Care Ontario) Hematology Cancer Drug Advisory Committee. Clinician group input noted that the current standard of care for patients with relapsed or refractory DLBCL who are ineligible for treatments with curative intent (i.e., ASCT or chimeric antigen receptor (CAR) T-cell therapy) or who relapse after such treatments includes chemotherapies such as rituximab gemcitabine oxaliplatin (R-GemOx) or polatuzumab vedotin-bendamustine-rituximab (pola-BR), which are given with palliative intent. Clinicians indicated that patients may experience a temporary response with these chemotherapies; however, most will experience relapse and require additional treatment, therefore there is a need for effective and well-tolerated treatments remains. Clinician input noted that if reimbursed, Glofit-GemOx may replace R-GemOx or pola-BR as the preferred second-line regimen; however, they noted there is an absence of clinical trials directly comparing Glofit-GemOx with pola-BR.
Input from CDA-AMC–participating drug plans noted that the comparator included in the STARGLO trial (i.e., R-GemOx) was not reflective of all relevant comparators in Canada. They further inquired about the eligibility of patients for re-treatment with Glofit-GemOx. Drug plan input noted that patients receiving their first dose of glofitamab should be monitored for AEs such as CRS. Plans additionally notes that confidential prices exist for glofitamab, polatuzumab vedotin, rituximab (brand and biosimilar).
Several of these concerns were addressed in the sponsor’s model:
HRQoL was included in the model, by use of EQ-5D-5L data from the STARGLO.
The sponsor included R-GemOx, pola-BR, and glofitamab monotherapy as comparators in their analysis.
Costs associated with monitoring for CRS were included for Glofit-GemOx.
Pagebreak
CDA-AMC was unable to address the following concerns:
Confidential pricing noted by the drug plans could not be incorporated into the model.
Costs associated with the management of CRS were not included in the sponsor’s model.
Appendix 3: Summary of the Sponsor’s Submission
Please note that this appendix has not been copy-edited.
Summary of the Sponsor’s Economic Evaluation
For the pharmaceutical reviews program, clinical and economic information is submitted to CDA-AMC by the sponsor. The CDA-AMC health economics team reviews the submitted economic information and appraises the information in collaboration with clinical experts and the clinical review team to evaluate key assumptions, influential parameters, and the overall rigour of the economic submission. Based on what the team learns through this process, adjustments may be made to the sponsor’s model to produce the CDA-AMC base case. The CDA-AMC base case represents the team’s current understanding of the clinical condition, clinical evidence currently available, and best interpretation of the economic evidence based on the information provided.
For the review of glofitamab for use in combination with gemcitabine and oxaliplatin, the sponsor provided a cost-utility analysis and a BIA. The sponsor’s economic submission is summarized in Table 5.
Table 5: Key Components of the Sponsor’s Economic Evaluation
Component | Description |
|---|---|
Treatment information | |
Drug under review | Glofitamab (Columvi), solution for IV infusion (1 mg/mL) |
Submitted price of drug under review | $1,040.00 per 2.5 mL vial $4,160.00 per 10 mL vial |
Regimen5 | Cycle 1 (21 days):
Cycle 2 to 8 (21 days): Day 1: 30 mg glofitamab, 1,000 mg/m2 gemcitabine, and 100 mg/m2 oxaliplatin Cycle 9 to 12 (21 days): Day 1: 30 mg glofitamab |
21-day course cost of drug under review | Glofitamab: $5,200 (Cycle 1), $12,480 (Cycle 2 to 12) Glofitamab in combination with gemcitabine and oxaliplatin (Glofit-GemOx): $11,672 (Cycle 1), $13,200 (Cycle 2 to 8), $12,480 (Cycle 9 to 12) |
Model information | |
Type of economic evaluation | Cost-utility analysis Partitioned survival model |
Treatment | Glofit-GemOx |
Included comparators |
|
Perspective | Publicly funded health care payer perspective |
Time horizon | Lifetime (35 years) |
Cycle length | 7 days |
Modelled populations | Adult patients with relapsed or refractory DLBCL NOS who are not candidates for ASCT |
Characteristics of modelled population | Derived from the STARGLO trial (mean age: 65 years, mean weight: 69 kg, 57.3% men, 42.7% women) |
Model health states3 |
|
Data sources | |
Comparative efficacy |
|
Natural history and/or clinical pathway |
|
Health-related utilities and disutilities |
|
Costs included in the model |
|
Summary of the submitted results | |
Base case results |
|
Scenario analysis results |
Additional scenarios were submitted that had no meaningful impact on the estimated ICER, including adopting alternative dosing scenarios, utility values, background mortality risk, and parametric distributions for PFS or OS. |
AE = adverse events; ASCT = autologous stem cell transplant; CRS = cytokine release syndrome; DLBCL = diffuse large B-cell lymphoma; Glofit-GemOx = glofitamab in combination with gemcitabine and oxaliplatin; HR = hazard ratio; ICER = incremental cost-effectiveness ratio; LY = life-year; NOS = not otherwise specified; OS = overall survival; PFS = progression-free survival; pola-BR = polatuzumab vedotin plus bendamustine and rituximab; PSM = partitioned survival model; QALY = quality-adjusted life-years; R-GemOx = rituximab in combination with gemcitabine and oxaliplatin; TTOT = time to off-treatment; vs. = versus; 3L+ = third line plus
aNo cure assumption, treatment effectiveness waning assumed to begin at 3 years until null at 5.
Appendix 4: Additional Details of CDA-AMC Reanalyses
Please note that this appendix has not been copy-edited.
Clinical Data in the Economic Model
The key clinical efficacy data in the model (OS, PFS, HRQoL) were derived from the STARGLO trial (data cut-off February 16, 2024) for Glofit-GemOx versus R-GemOx and from the sponsor’s submitted ITC for Glofit-GemOx versus pola-BR and Glofit-GemOx versus glofitamab monotherapy. Evidence from the STARGLO trial suggests that Glofit-GemOx likely results in an improvement in PFS and OS compared to R-GemOx; however, HRQoL was similar between groups, but due to lack of blinding and high attrition rates, the certainty in the effect estimates is unclear. Glofit-GemOx was associated with a substantially higher incidence of serious AEs and notable harms than with R-GemOx. Clinical expert feedback received by CDA-AMC indicated that the differences in OS and PFS were clinically meaningful; however, the magnitude is uncertain given the identified concerns related to censoring within the trial (refer to Clinical Review report).
In the absence of head-to-head trials of Glofit-GemOx and pola-BR or glofitamab monotherapy, an indirect treatment comparison was submitted by the sponsor. Based on the submitted indirect evidence, there is insufficient evidence of superiority for Glofit-GemOx compared with pola-BR or glofitamab monotherapy among the indicated population. The CDA-AMC Clinical Review report noted that the submitted ITCs were associated with uncertainty and generalizability concerns due to the small sample size after adjustment and wide confidence intervals. HRQoL and AEs were not included in the ITCs. As such, there remains significant uncertainty associated with the comparative evidence between Glofit-GemOx compared with pola-BR or glofitamab monotherapy for the indicated population.
Key Issues of the Submitted Economic Evaluation
CDA-AMC identified the following key issues with the sponsor’s analysis:
The comparative efficacy of Glofit-GemOx is uncertain: There is a lack of direct head-to-head evidence comparing Glofit-GemOx to comparators other than R-GemOx. To inform the economic model, the sponsor submitted ITCs in the form of propensity score analyses to provide estimates of the relative treatment effects for Glofit-GemOx versus pola-BR and Glofit-GemOx versus glofitamab monotherapy. Data were obtained from the STARGLO trial for Glofit-GemOx, the phase Ib/2 GO29365 study for pola-BR, and the phase I/II NP30179 study for glofitamab monotherapy. Results of these analyses do not support a difference in PFS or OS between Glofit-GemOx and pola-BR or glofitamab monotherapy. However, as noted in the CDA-AMC Clinical Review, there is substantial uncertainty in the comparative efficacy, owing to, for example, small sample sizes after adjustment and wide confidence intervals that crossed the null value (refer to the Clinical Review report). The clinical evidence for both Glofit-GemOx and pola-BR that informed these comparisons were from small, short-term, nonrandomized, early-phase studies, which introduces additional uncertainty into the comparative effect estimates.
CDA-AMC was unable to address uncertainty in the comparative data. CDA-AMC conducted a scenario analysis assuming equal efficacy for Glofit-GemOx and pola-BR.
The sponsor’s pharmacoeconomic analysis does not adequately reflect the clinical pathway or cost-effectiveness by line of therapy. The draft Health Canada indication for Glofit-GemOx is for the treatment of relapsed or refractory DLBCL NOS in patients who are not candidates for ASCT (i.e., for use in the second or later line). As noted in the prior key issue, the sponsor utilized observations from the STARGLO trial and the results of ITCs to provide comparative efficacy data for the economic model for the comparisons to pola-BR (GO29365) and glofitamab monotherapy (NP30179). The STARGLO and GO29365 trials enrolled patients who had received 1 to 4 prior lines of treatment for DLBCL, while NP30179 enrolled patients with 2 to 4 prior lines. Thus, the comparative efficacy estimates, and hence the cost-effectiveness estimates, reflect the comparison of Glofit-GemOx versus R-GemOx and pola-BR in the second or later line and versus glofitamab monotherapy in the third or later line.
The care pathway for patients with relapsed or refractory DLBCL depends on prior treatment received as well as ASCT and CAR T eligibility.11 Therefore, relevant comparators in each line of therapy vary by prior treatment exposure and eligibility. In the second line, relevant comparators are pola-BR and chemotherapy (with or without rituximab), while relevant comparators in the third line include glofitamab monotherapy, pola-BR, chemotherapy with or without rituximab, epcoritamab, and CAR Ts (axicabtagene ciloleucel, tisagenlecleucel, lisocabtagene maraleucel) depending on prior treatment exposure and CAR T eligibility. The sponsor’s choice of a partitioned survival model is not suitable for capturing complex relationships in the care pathway. Although the sponsor attempted to incorporate subsequent treatment in a manner that was dependent on previous treatment received, there were numerous issues with the approaches taken to modelling subsequent therapy (refer to subsequent key issue).
CDA-AMC was unable to address the structural limitations of the sponsor’s modelling approach. The CDA-AMC base case includes comparison of Glofit-GemOx to R-GemOx and pola-BR in the second or later line, which is aligned with the draft Health Canada indication. The cost-effectiveness of Glofit-GemOx versus glofitamab monotherapy, which reflects usage in the third or later line, was explored in scenario analysis.
The impact of Glofit-GemOx on long-term survival is uncertain: There are several sources of uncertainty related to the impact of Glofit-GemOx on survival. First, the sponsor’s analysis predicts an incremental gain of 3.51 LYs for Glofit-GemOx compared to R-GemOx, 0.32 LYs compared to pola-BR, and 0.70 LYs compared to glofitamab monotherapy. While a predicted gain in LYs versus R-GemOx is supported by OS data from the STARGLO trial, the magnitude of gain is uncertain due to uncertainty in the OS data from STARGLO (e.g., due to censoring; refer to the CDA-AMC Clinical Review report), generalizability of the STARGLO patient population to clinical practice in Canada, and the extrapolation of OS data from the trial over the 35 year model horizon. Further, data from the sponsor-submitted ITCs does not support a difference in OS between Glofit-GemOx and pola-BR or glofitamab monotherapy.
Second, the sponsor assumed that patients who remain progression-free for 3 or more years after treatment initiation are functionally cured (i.e., no longer at risk of disease progression). While clinical expert feedback received by CDA-AMC indicated that patients who remain progression-free for 3 or more years have a reduced risk of subsequent progression, the risk of progression is not eliminated. Therefore, the incorporation of a cure assumption likely overestimates long-term survival in the economic model, which disproportionately benefits Glofit-GemOx. Additionally, the sponsor assumed that the risk of death for cured patients would be equal to the age-specific general population mortality, adjusted by a standardized mortality ratio of 1.09 to account for increased mortality associated with previously having cancer.6 CDA-AMC notes that this estimate was derived from patients with newly diagnosed DLBCL, and the use of data from earlier lines of therapy may underestimate the risk of mortality for patients with relapsed or refractory DLBCL as newly diagnosed patients may represent a heterogeneous group of patients including those who are eligible for ASCT.
Finally, because OS in the sponsor’s model is derived from the STARGLO trial (and thus implicitly includes efficacy of subsequent therapies), if the subsequent therapies or the distribution of subsequent therapies differs in clinical practice in Canada from that in the STARGLO trial (which had no trial sites in Canada), there may be differences in OS from what was observed in the STARGLO trial.
CDA-AMC was unable to fully address uncertainty in the impact of Glofit-GemOx on long-term survival. CDA-AMC removed the sponsor’s cure assumption from the base case and explored uncertainty in this via scenario analysis.
Costs associated with subsequent therapy are highly uncertain: There are several sources of uncertainty related to subsequent therapy costs in the model. First, the sponsor incorporated the cost of subsequent therapy as a one-time cost for all patients who transition out of the PFS health state (i.e., the difference between the percentage of patients progression-free in the previous cycle and the percentage progression-free in the current cycle). This approach does not account for the proportion of PFS events that are deaths. Instead, the cost of subsequent therapy should be applied only to patients who enter the progressed disease health state (i.e., after accounting for patients who have died); however, the sponsor’s choice of a PSM does not allow for the inclusion of a one-time cost at the time of progression. Applying the cost of subsequent therapy to all patients who transition out of the PFS health state overestimates the costs associated with subsequent therapy for comparators.
Second, applying a one-time cost for subsequent therapy does not adequately capture variability in the duration and timing of treatment. Although the sponsor utilized the mean duration of treatment and the proportion of patients who received each treatment from the STARGLO trial for Glofit-GemOx and R-GemOx and from NP30179 for glofitamab monotherapy, use of a one-time cost assumes that the duration and intensity of treatment is the same for all patients and therefore does not account for variability in treatment regimens. Additionally, the use of a one-time cost means the cost of subsequent treatment is not dependent on time in the progressed health state in the model.
Third, the STARGLO trial was used to inform the distribution of subsequent therapy regimens and the duration of each subsequent treatment for Glofit-GemOx, R-GemOx, and pola-BR (the sponsor assumed that patients who received pola-BR in 2L would receive the same subsequent treatments as those who received Glofit-GemOx in 2L after adjustment to not receive pola-BR again). Although STARGLO was a multinational trial, there were no centres in Canada. The generalizability of the distribution of subsequent treatments to clinical practice in Canada is uncertain, as clinical expert input obtained by CDA-AMC for this review indicated that the distribution of subsequent therapies in the model is not aligned with practice in Canada. For example, patients would have previously received R-CHOP and therefore would not receive it as subsequent therapy. Most notably, patients treated with glofitamab would not typically receive another bi-specific antibody or repeat glofitamab. However, in the sponsor’s submitted analysis, subsequent bi-specific antibody treatment accounted for 35% of Glofit-GemOx subsequent therapy. Experts also indicated that the expected uptake of a bi-specific antibody after R-GemOx or pola-BR 2L treatment would be higher than proposed in the model.
CDA-AMC was unable to address this issue.
Treatment costs are uncertain. In the economic model, the cost of Glofit-GemOx and comparators was based on a mean number of treatment cycles sourced from the STARGLO or other comparator studies (GO29365 and NP30179). Thus, all patients in the model received the same number of treatment cycles (e.g., patients on Glofit-GemOx received 8.5 cycles of glofitamab, 6.1 cycles of gemcitabine, and 6 cycles of oxaliplatin). This approach fails to incorporate variability in treatment exposure, as observed in the STARGLO trial, in which patients in the Glofit-GemOx group received between 1 and 13 cycles of glofitamab (median: 11 cycles), between 1 and 9 cycles of gemcitabine (median: 8 cycles), and between 1 and 9 cycles of oxaliplatin (median 4) (February 16, 2024 analysis). Similarly, this approach does not capture variability in exposure to the components of R-GemOx (refer to the Clinical Review report). Further, it is highly unlikely that all patients in clinical practice will receive the same number of treatments cycles, given that this may be influenced by factors such as tolerability and disease status. Therefore, the use of a defined treatment duration for all patients does not adequately capture variability in expected treatment costs.
In scenario analysis, CDA-AMC assumed that patients would receive treatment until disease progression (up to the max number of cycles as defined by respective dosing regimens), using the sponsor-provided option.
Treatment effectiveness waning was not considered. Treatment effectiveness waning was not considered in the sponsor’s base-case analysis. This is expected to have a minimal impact on the sponsor’s results, owing to the sponsor’s cure assumption (refer to previous key issue). However, if patients are not assumed to be cured after 3 years without disease progression (i.e., as in the CDA-AMC base case), the exclusion of treatment effectiveness waning is not appropriate as this implicitly assumes that treatment effect will be maintained indefinitely, which may overestimate treatment. Clinical expert feedback received by CDA-AMC was in alignment, noting that once off treatment, patients may experience waning of effect. In the absence of long-term efficacy data, the impact of effectiveness waning for patients who discontinue or complete a course of treatment is unknown. The impact of this on the ICER is uncertain.
CDA-AMC explored the impact of treatment effectiveness waning in scenario analysis.
The impact of AEs is uncertain. The sponsor indicated that costs related to grade 3 or higher treatment-related AEs that occurred in at least 2% of patients in the STARGLO and comparator trials were included in their economic evaluation. However, CDA-AMC notes that costs associated with grade 3 or higher CRS, which was defined as a notable harm in clinical expert input, was not included in the model, despite a reported incidence of 2.2% in the Glofit-GemOx group of the STARGLO trial.
Further, the sponsor assumed that the HRQoL impact of AEs would be captured by the use of utility values of separate utility values for patients on or off treatment in the PFS health state. This approach lacks granularity as to the impact of AE on the estimated cost-effectiveness of Glofit-GemOx.
CDA-AMC was unable to address this issue.
Additional issues were identified but were not considered to be key issues:
The model lacked transparency due to poor modelling practices. When the sponsor’s submitted model is run probabilistically, a message box indicates the number of simulations that have been discarded from the analysis “due to numeric instability in the generalized gamma probabilistic distribution.” Upon questioning by CDA-AMC, the sponsor indicated that this issue pertains only to glofitamab monotherapy because it utilizes a generalized gamma distribution for OS and that a number of parameters in the bootstrap sample are sufficiently skewed from the deterministic mean such that there are errors in calculating the distribution. This issue is not expected to impact the CDA-AMC base case, as glofitamab monotherapy was included in scenario analyses only.
CDA-AMC Reanalysis of the Economic Evaluation
The CDA-AMC base case was derived by making changes in model parameter values and assumptions, in consultation with clinical experts (refer to Table 6). The impact of these changes, individually and collectively, is presented in Table 7. The results of the CDA-AMC base case are presented as pairwise comparisons, owing to the use of different sources of efficacy data.
Table 6: Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
1. Cure assumption | Included | Excluded |
CDA-AMC base case | ― | Reanalysis 1 |
CDA-AMC = Canada’s Drug Agency.
Note: CDA-AMC was unable to resolve the issues with comparative efficacy, the failure to capture casual relationships, subsequent therapies, treatment waning, and the lack of model transparency.
Table 7: Summary of the Stepped Analysis
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($ per QALY) |
|---|---|---|---|---|
Glofit-GemOx vs. R-GemOx | ||||
Sponsor’s base case | R-GemOx | 260,446 | 2.87 | Reference |
Glofit-GemOx | 310,079 | 5.30 | 20,376 | |
CDA-AMC reanalysis 1 | R-GemOx | 228,117 | 1.52 | Reference |
Glofit-GemOx | 394,759 | 3.61 | 79,902 | |
CDA-AMC base case: Reanalysis 1 (deterministic) | R-GemOx | 228,117 | 1.52 | Reference |
Glofit-GemOx | 394,759 | 3.61 | 79,902 | |
CDA-AMC base case Reanalysis 1 (probabilistic) | R-GemOx | 228,624 | 1.54 | Reference |
Glofit-GemOx | 394,308 | 3.62 | 79,881 | |
Glofit-GemOx vs. pola-BR | ||||
Sponsor’s base case | pola-BR | 308,225 | 4.54 | Reference |
Glofit-GemOx | 313,570 | 4.80 | 20,387 | |
CDA-AMC reanalysis 1 | pola-BR | 228,117 | 2.79 | Reference |
Glofit-GemOx | 383,468 | 3.14 | 140,607 | |
CDA-AMC base case: Reanalysis 1 (deterministic) | pola-BR | 228,117 | 2.79 | Reference |
Glofit-GemOx | 383,468 | 3.14 | 140,607 | |
CDA-AMC base case Reanalysis 1 (probabilistic) | pola-BR | 334,305 | 2.81 | Reference |
Glofit-GemOx | 383,557 | 3.16 | 141,006 | |
CDA-AMC = Canada’s Drug Agency; Glofit-GemOx = glofitamab in combination with gemcitabine and oxaliplatin; ICER = incremental cost-effectiveness ratio; pola-BR = polatuzumab vedotin plus bendamustine and rituximab; QALY = quality-adjusted life-year; R-GemOx = rituximab in combination with gemcitabine and oxaliplatin; vs. = versus.
Note: The CDA-AMC reanalysis is based on the publicly available prices of the comparator treatments. Deterministic results are presented, unless otherwise indicated.
Table 8: Disaggregated Results of the CDA-AMC Base Case — Glofit-GemOx vs. R-GemOx
Parameter | Glofit-GemOx | R-GemOx |
|---|---|---|
Discounted LYs | ||
Total | 5.17 | 2.22 |
PFS | 3.52 | 0.85 |
Progressed Disease | 1.65 | 1.36 |
Discounted QALYs | ||
Total | 3.62 | 1.54 |
PFS | 2.55 | 0.64 |
Progressed Disease | 1.07 | 0.91 |
Discounted costs ($) | ||
Total | 394,308 | 228,624 |
Treatment Costs | 112,169 | 12,714 |
Drug Administration | 2,473 | 1,187 |
Adverse Events | 10,542 | 4,400 |
PFS Supportive Care | 65,164 | 18,318 |
PD Supportive Care | 62,817 | 51,880 |
Postdiscontinuation Therapy Costs | 141,143 | 140,124 |
CDA-AMC = Canada’s Drug Agency; Glofit-GemOx = glofitamab in combination with gemcitabine and oxaliplatin; LY = life-year; PD = progressed disease; PFS = progression-free survival; QALY = quality-adjusted life-year; R-GemOx = rituximab in combination with gemcitabine and oxaliplatin; vs. = versus.
Table 9: Disaggregated Results of the CDA-AMC Base Case — Glofit-GemOx vs. pola-BR
Parameter | Glofit-GemOx | pola-BR |
|---|---|---|
Discounted LYs | ||
Total | 4.49 | 4.06 |
PFS | 3.17 | 2.02 |
Progressed Disease | 1.32 | 2.03 |
Discounted QALYs | ||
Total | 3.16 | 2.81 |
PFS | 2.30 | 1.49 |
Progressed Disease | 0.86 | 1.32 |
Discounted costs ($) | ||
Total | 383,557 | 334,305 |
Treatment Costs | 118,527 | 74,862 |
Drug Administration | 2,482 | 868 |
Adverse Events | 10,542 | 11,010 |
PFS Supportive Care | 59,757 | 36,628 |
PD Supportive Care | 50,285 | 77,414 |
Postdiscontinuation Therapy Costs | 141,964 | 133,523 |
CDA-AMC = Canada’s Drug Agency; Glofit-GemOx = glofitamab in combination with gemcitabine and oxaliplatin; LY = life-year; PD = progressed disease; PFS = progression-free survival; pola-BR = polatuzumab vedotin plus bendamustine and rituximab; QALY = quality-adjusted life-year; vs. = versus.
Price Reduction Analysis
CDA-AMC conducted price reduction analyses using the sponsor’s base case and the CDA-AMC base case (refer to Table 10).
Table 10: Results of the Price Reduction Analysis — Glofit-GemOx vs. R-GemOx
Price reduction | Unit drug costa ($) | Cost per 21 days of glofitamab ($)b | ICERs for Glofit-GemOx vs. R-GemOx ($ per QALY) | |
|---|---|---|---|---|
Sponsor base case | CDA-AMC base case | |||
No price reduction | 4,160 | 12,480 | 20,376 | 79,881 |
10% | 3,744 | 11,232 | 16,520 | 75,334 |
20% | 3,328 | 9,984 | 12,664 | 70,786 |
30% | 2,912 | 8,736 | 8,808 | 66,239 |
40% | 2,496 | 7,488 | 4,952 | 61,692 |
50% | 2,080 | 6,240 | 1,097 | 57,144 |
60% | 1,664 | 4,992 | Dominant | 52,597 |
70% | 1,248 | 3,744 | Dominant | Dominant |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; Glofit-GemOx = glofitamab in combination with gemcitabine and oxaliplatin; QALY = quality-adjusted life-year; R-GemOx = rituximab in combination with gemcitabine and oxaliplatin; vs. = versus.
aSponsor’s submitted price for glofitamab based on the 10 mL vial (1 mg/mL).3 Glofitamab is also available in 2.5 mL vials (1 mg/mL) at a unit cost of $1,040. For both vial sizes, the cost is $416 per mg glofitamab.
bReflects the costs for cycles 2 to 12.
Table 11: Results of the Price Reduction Analysis — Glofit-GemOx vs. pola-BR
Price reduction | Unit drug cost ($)a | Cost per 21 days of glofitamab ($)b | ICERs for Glofit-GemOx vs. pola-BR ($ per QALY) | |
|---|---|---|---|---|
Sponsor base case | CDA-AMC base case | |||
No price reduction | 4,160 | 12,480 | 20,387 | 141,006 |
10% | 3,744 | 11,232 | Dominant | 114,001 |
20% | 3,328 | 9,984 | Dominant | 86,995 |
30% | 2,912 | 8,736 | Dominant | 59,989 |
40% | 2,496 | 7,488 | Dominant | Dominant |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; Glofit-GemOx = glofitamab in combination with gemcitabine and oxaliplatin; QALY = quality-adjusted life-year; pola-BR = polatuzumab vedotin plus bendamustine and rituximab; vs. = versus.
aSponsor’s submitted price for glofitamab based on the 10 mL vial (1 mg/mL).3 Glofitamab is also available in 2.5 mL vials (1 mg/mL) at a unit cost of $1,040. For both vial sizes, the cost is $416 per mg glofitamab.
bReflects the costs for cycles 2 to 12.
Assessment of Uncertainty
CDA-AMC used the CDA-AMC base case to conduct scenario analyses to address uncertainty within the economic evaluation. The results are provided in Table 12.
Scenario 1: Assuming that patients who remain in the PFS health state for at least 5 years are cured, using the sponsor-provided option.
Scenario 2: Assuming that treatment will be received until disease progression (up to the maximum number of treatment cycles), using the sponsor-provided option.
Scenario 3: Inclusion of treatment effectiveness waning (PFS and OS), using the sponsor-provided option.
Scenario 4: Assuming equal efficacy between Glofit-GemOx and pola-BR, based on the results of the submitted ITC suggesting there may be no difference in OS or PFS between treatments.
Scenario 5: Glofit-GemOx versus glofitamab monotherapy in the 3L+ setting.
Table 12: Results of CDA-AMC Scenario Analyses — Glofit-GemOx vs. R-GemOx
Analysisa | Drug | Total costs ($) | Total QALYs | ICER ($ per QALYs) |
|---|---|---|---|---|
CDA-AMC base case | R-GemOx | 228,624 | 1.54 | Reference |
Glofit-GemOx | 394,308 | 3.62 | 79,881 | |
CDA-AMC scenario 1: 5-year cure assumption | R-GemOx | 285,837 | 2.81 | Reference |
Glofit-GemOx | 372,948 | 5.25 | 35,779 | |
CDA-AMC scenario 2: Treatment until disease progression | R-GemOx | 243,104 | 1.55 | Reference |
Glofit-GemOx | 416,465 | 3.63 | 83,470 | |
CDA-AMC scenario 3: Treatment effectiveness waning | R-GemOx | 227,685 | 1.53 | Reference |
Glofit-GemOx | 392,958 | 3.61 | 79,329 |
CDA-AMC = Canada’s Drug Agency; Glofit-GemOx = glofitamab in combination with gemcitabine and oxaliplatin; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
aProbabilistic analyses.
Table 13: Results of CDA-AMC Scenario Analyses — Glofit-GemOx vs. pola-BR
Analysisa | Drug | Total costs ($) | Total QALYs | ICER ($ per QALYs) |
|---|---|---|---|---|
CDA-AMC base case | pola-BR | 334,305 | 2.81 | Reference |
Glofit-GemOx | 383,557 | 3.16 | 141,006 | |
CDA-AMC scenario 1: 5-year cure assumption | pola-BR | 365,204 | 4.51 | Reference |
Glofit-GemOx | 370,463 | 4.78 | 19,219 | |
CDA-AMC scenario 2: Treatment until disease progression | pola-BR | 356,060 | 2.82 | Reference |
Glofit-GemOx | 413,710 | 3.19 | 154,824 | |
CDA-AMC scenario 3: Treatment effectiveness waning | pola-BR | 331,873 | 2.78 | Reference |
Glofit-GemOx | 381,541 | 2.94 | 313,150 | |
CDA-AMC scenario 4: Equal efficacy | pola-BR | 323,811 | 3.20 | Reference |
Glofit-GemOx | 384,458 | 3.20 | Dominated |
CDA-AMC = Canada’s Drug Agency; Glofit-GemOx = glofitamab in combination with gemcitabine and oxaliplatin; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; pola-BR = polatuzumab vedotin plus bendamustine and rituximab; vs. = versus.
aProbabilistic analyses.
Table 14: Results of CDA-AMC Scenario Analyses — Glofit-GemOx vs. Glofitamab Monotherapy
Analysisa | Drug | Total costs ($) | Total QALYs | ICER ($ per QALYs) |
|---|---|---|---|---|
CDA-AMC scenario 5: Glofit-GemOx vs. glofitamab monotherapy | Glofitamab monotherapy | 299,888 | 2.57 | Reference |
Glofit-GemOx | 388,203 | 3.07 | 177,652 |
CDA-AMC = Canada’s Drug Agency; Glofit-GemOx = glofitamab in combination with gemcitabine and oxaliplatin; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
aProbabilistic analyses.
Issues for Consideration
Negotiations with pan-Canadian Pharmaceutical Alliance concluded with a letter of intent for glofitamab for the treatment of relapsed or refractory diffuse DLBCL NOS, DLBCL arising from follicular lymphoma, or primary mediastinal B-cell lymphoma, who have received 2 or more lines of systemic therapy and are ineligible to receive or cannot receive CAR T-cell therapy or have previously received CAR T-cell therapy, following obinutuzumab pre-treatment. As such, a confidential negotiated price likely exists for glofitamab, while the sponsor’s base case and all CDA-AMC reanalyses rely on the price submitted by the sponsor. Similarly, confidential prices that are lower than the publicly available list prices for likely exist for polatuzumab vedotin, bendamustine, and rituximab but could not be factored into the CDA-AMC analyses.
Appendix 5: Budget Impact Analysis
Please note that this appendix has not been copy-edited.
Summary of the Submitted BIA
The sponsor submitted a BIA that estimated the expected incremental budgetary impact of reimbursing glofitamab for use in combination with GemOx for the treatment of adult patients with relapsed or refractory DLBCL NOS who are ineligible for ASCT. The BIA was conducted from the perspective of public drug plan payers over a 3-year time horizon (2026 to 2028), with 2025 as the base year. The sponsor’s estimate reflects the aggregated results from the jurisdictional provincial budgets (excluding Quebec). The sponsor estimated the eligible population using an epidemiological approach. The sponsor’s base case included drug acquisition costs. The market uptake for Glofit-GemOx was estimated using market research undertaken by the sponsor and validated by clinical experts consulted by the sponsor. The key inputs to the BIA are documented in Table 15.
The sponsor estimated the 3-year incremental budget impact associated with reimbursing glofitamab for use in combination with GemOx for the Health Canada–indicated population to be $43,323,364 (year 1 = $9,563,254; year 2 = $15,611,797; year 3 = $18,148,313).
Table 15: Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3 if appropriate) | ||
|---|---|---|---|
Reference | New drug | ||
Target population | |||
Number of incident NHL cases | 9,320 / 9,412 / 9,50812 | ||
Percentage of NHL that is DLBCL | 35%13 | ||
Receive 1L treatment, % | |||
Relapse or refractory after 1L treatment, % | 40%16 | ||
Eligible for 2L ASCT, % | 50%17 | ||
Eligible for 2L ASCT who intend to receive ASCT, % | 28% / 23% / 23%a | ||
Eligible for 2L ASCT but receive 2L CAR T, % | 72% / 77% / 77%a | ||
Require subsequent treatment after 2L CAR T, % | 55%18 | ||
Intend to receive ASCT and receive ASCT, % | 50%17 | ||
Intend to receive ASCT but refractory to 2L salvage chemotherapy, % | 50%17 | ||
Receive transplant after 2L CAR T, % | 11%19 | ||
Ineligible for transplant after 2L CAR T, % | 89%19 | ||
Relapse and refractory after 2L ASCT, % | 50%17 | ||
Receive 3L+ if not eligible for 2L ASCT or 2L CAR T (reference scenario),%b | 49% / 49% / 49%a | ||
Receive 3L+ if not eligible for 2L ASCT or 2L CAR T (new drug scenario), %c | 44% / 41% / 39%a | ||
Eligible Patients in 2L transplant ineligible | 587 / 593 / 599 | ||
Eligible Patients in 3L post 2L ASCT eligibility | 124 / 103 / 104 | ||
Eligible Patients in 3L after 2L CAR T | 206 / 223 / 225 | ||
Eligible Patients in 3L post 2L ASCT ineligibility (Reference) | 290 / 293 / 296 | ||
Eligible Patients in 3L post 2L ASCT ineligibility (New Drug Scenario) | 258 / 242 / 233 | ||
Market shares (Eligible Patients in 2L Transplant Ineligible) | |||
Treatment | |||
Glofit-GemOx | 0% / 0% / 0% | 25% / 40% / 50% | |
Pola + BR | 45% / 45% / 45% | 30% / 20% / 12.5% | |
Salvage Chemotherapy | 55% / 55% / 55% | 45% / 40% / 37.5% | |
Market shares (Eligible Patients in 3L Post 2L ASCT Eligibility) | |||
Treatment | |||
Glofit-GemOx | 0% / 0% / 0% | 3% / 5% / 5% | |
Glofitamab monotherapy | 17% / 18% / 22% | 14% / 13% / 17% | |
Epcoritamab | 8% / 9% / 11% | 8% / 9% / 11% | |
Pola + BR | 5% / 3% / 2% | 5% / 3% / 2% | |
Salvage Chemotherapy | 20% / 15% / 10% | 20% / 15% / 10% | |
Axicabtagene ciloleucel | 17% / 18% / 18% | 17% / 18% / 18% | |
Tisagenlecleucel | 17% / 18% / 18% | 17% / 18% / 18% | |
Lisocabtagene maraleucel | 17% / 18% / 18% | 17% / 18% / 18% | |
Market Shares (Eligible Patients in 3L after 2L CAR T) | |||
Treatment | |||
Glofit-GemOx | 0% / 0% / 0% | 10% / 20% / 25% | |
Glofitamab monotherapy | 27% / 33% / 40% | 24% / 27% / 29% | |
Epcoritamab | 13% / 17% / 20% | 12% / 13% / 14% | |
Pola + BR | 20% / 15% / 10% | 20% / 15% / 10% | |
Salvage Chemotherapy | 40% / 35% / 30% | 34% / 25% / 22% | |
Market Shares (Eligible Patients in 3L Post 2L ASCT Ineligibility) | |||
Treatment | |||
Glofit-GemOx | 0% / 0% / 0% | 1% / 2% / 3% | |
Glofitamab monotherapy | 27% / 32% / 35% | 15% / 12% / 10% | |
Epcoritamab | 13% / 16% / 18% | 7% / 6% / 5% | |
pola-BR | 5% / 3% / 2% | 22% / 31% / 37% | |
Salvage Chemotherapy | 30% / 24% / 20% | 30% / 24% / 20% | |
Axicabtagene ciloleucel | 10% / 10% / 10% | 10% / 10% / 10% | |
Tisagenlecleucel | 10% / 10% / 10% | 10% / 10% / 10% | |
Lisocabtagene maraleucel | 5% / 5% / 5% | 5% / 5% / 5% | |
Cost of treatment (per patient per 21-day cycle)d | |||
Glofit-GemOxe | Cycle 1: $7,829 Cycle 2+: $138,536 Premedication: $5,482 | ||
Glofitamab monotherapy | Cycle 1: $5,200 Cycle 2+: $68,640 Premedication: $5,481 | ||
Epcoritamabe | Cycle 1: $14,319.50 Cycle 2 to 3: $52,872.00 Cycle 4 to 9: $79,308.00 Cycle 10+: $19,827.00 Premedication: $43.24 | ||
pola-BR | $89,524 | ||
Salvage chemotherapyf | $16,009 | ||
Axicabtagene ciloleucel | $485,021 (one-time cost) | ||
Tisagenlecleucel | $450,000 (one-time cost) | ||
Lisocabtagene maraleucel | $501,900 (one time cost) | ||
ASCT = autologous stem cell transplant; CAR T = chimeric antigen receptor; DLBCL = diffuse large B-cell lymphoma; Glofit-GemOx = glofitamab in combination with gemcitabine and oxaliplatin; NHL = non-Hodgkin Lymphoma; pola-BR = polatuzumab vedotin plus bendamustine and rituximab; R-GemOx = rituximab in combination with gemcitabine and oxaliplatin; R-GDP = rituximab-gemcitabine-dexamethasone-cisplatin); 1L = first line, 2L = second line, 3L+ = third line and later.
aCalculated by the sponsor.
bThe sponsor assumed that the number of 2L transplant ineligible patients who will require subsequent therapy is based on the therapy they received in 2L. This was calculated as the weighted average from Time to Next Treatment and New Antilymphoma Therapy events from 2L treatments multiplied by respective market shares in Reference Scenario
cThe sponsor assumed that the number of 2L transplant ineligible patients who will require subsequent therapy is based on the therapy they received in 2L. This was calculated as the weighted average from Time to Next Treatment and New Antilymphoma Therapy events from 2L treatments multiplied by respective market shares in New Drug Scenario.
dComparators listed may not align with those presented in the cost-utility analysis as comparators differ by line of therapy in the BIA.
eIncludes cost of premedication
fSponsor assumed that salvage chemotherapy was 50% R-GDP and 50% R-GemOx.
Key Issues of the Submitted BIA
CDA-AMC identified several key issues to the sponsor’s analysis that have notable implications on the results of the BIA:
The number of patients eligible for Glofit-GemOx treatment is uncertain: The sponsor estimated the number of eligible patients by based on a patient’s treatment pathway (refer to Table 15). This approach is considered appropriate given that treatment experience and transplant/CAR T eligibility impacts what treatments may be received in later lines of therapy. However, CDA-AMC was unable to fully validate the implementation of this approach, given the nuances involved in identifying patients eligible for Glofit-GemOx treatment across all lines of therapy.
In the BIA, the sponsor noted that “The number of patients in 3L post 2L ASCT ineligibility is lower in New Drug Scenario. This is due to the use of COLUMVI plus GemOx is expected to lower treatment progression from 2L to 3L.” Based on the results of the STARGLO trial, in which PFS was improved with Glofit-GemOx versus R-GemOx, this is a reasonable assumption, however the magnitude of difference is uncertain owing to uncertainty in the PFS estimates (Refer to the Clinical Review report). If the rate of progression from second to third line treatment with Glofit-GemOx treatment is lesser than estimated by the sponsor, the budget impact of reimbursing Glofit-GemOx would be higher than predicted in the sponsor’s analysis.
CDA-AMC identified an error in the sponsor submitted BIA where the “% of patients that receive new antilymphoma therapy after 2L treatment” for pola-BR was pointing to the value for Tafa-Len.
In the CDA-AMC base case, CDA-AMC corrected the reference error in the Excel model, where the “% of patients that receive new antilymphoma therapy after 2L treatment” for pola-BR was using the value for Tafa-Len.
To explore the uncertainty in the rate of progression from second line to third line treatment for patients who receive Glofit-GemOx, CDA-AMC conducted a scenario in which the proportion of patients ineligible for ASCT who require third or later line treatment after receiving Glofit-GemOx in the second line was set equal to pola-BR (i.e., 41%).
The market uptake of Glofit-GemOx is uncertain: The sponsor’s submitted BIA indicated that, by the third year of reimbursement, Glofit-GemOx would acquire up to 50% market share for patients who are transplant ineligible in 2L, up to 5% for patients in 3L post 2L ASCT eligibility, up to 25% for patients in 3L after 2L CAR T, and up to 3% for patients in 3L post 2L ineligible for ASCT. Clinical expert feedback received by CDA-AMC noted that the general trends of the proposed market shares of Glofit-GemOx appear reasonable but that there is considerable uncertainty as to the uptake of Glofit-GemOx in later lines of therapy given that patients who receive Glofit-GemOx in earlier lines would not receive repeat treatment with glofitamab (as Glofit-GemOx or glofitamab monotherapy) in later lines. Additionally, it is uncertain as to which therapies will be displaced by earlier use of Glofit-GemOx.
CDA-AMC was unable to address this limitation.
Treatment duration may not reflect clinical practice. Treatment duration in the sponsor submitted BIA was informed by mean treatment duration from clinical trials, public formularies, or previous CDA-AMC reports. CDA-AMC noted that in many cases, treatment duration was a set number of cycles based on a treatment’s product monograph; however, some drugs are given until disease progression (e.g., epcoritamab) and therefore the reported duration may not be reflective of clinical practice. Should treatment duration in clinical practice differ from that obtained from these sources, the budget impact of reimbursing Glofit-GemOx is likely to differ from that estimated by the sponsor. However, the direction of impact of this issue on the estimated budget impact is unknown.
CDA-AMC was unable to address this key issue.
The price of drugs paid by public drug plans is uncertain: Both the sponsor’s and the CDA-AMC analyses are based on publicly available list prices for all comparators, as well as the sponsor’s submitted cost for glofitamab. Actual costs paid by public drug plans are unknown.
CDA-AMC was unable to address this issue.
CDA-AMC Reanalyses of the BIA
CDA-AMC revised the sponsor’s submitted analyses by making the following corrections outlined in Table 15.
Table 16: Revisions to the Submitted BIA
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
1. Percentage of patients that receive new antilymphoma therapy after 2L pola-BR (transplant ineligible) | 40% (referencing the value for Tafa-Len) | 41% |
CDA-AMC base case | ― | Reanalysis 1 |
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency; pola-BR = polatuzumab vedotin plus bendamustine and rituximab; Tafa-Len = tafasitamab plus lenalidomide; 2L = second line.
Note: CDA-AMC was unable to resolve the issues with market uptake, treatment duration, or drug prices.
The results of the CDA-AMC step-wise reanalysis are presented in summary format in Table 17 and a more detailed breakdown is presented in Table 18. In the CDA-AMC base case, the 3-year budget impact of reimbursing Glofit-GemOx for treatment of adult patients with relapsed or refractory DLBCL NOS who are ineligible for ASCT was $42,543,307 (year 1 = $9,407,842; year 2 = $15,344,009; year 3 = $17,791,456).
Table 17: Summary of the Stepped Analysis of the CDA-AMC Base Case
Stepped analysis | Three-year total ($) |
|---|---|
Submitted base case | $43,323,364 |
CDA-AMC reanalysis 1 | $42,543,307 |
CDA-AMC base case: Reanalysis 1 | $42,543,307 |
CDA-AMC = Canada’s Drug Agency.
Note: The CDA-AMC reanalysis is based on publicly available prices of the comparator treatments
CDA-AMC used the CDA-AMC base case to explore uncertainty in the estimated budget impact of reimbursing Glofit-GemOx, by adopting an alternative percentage of patients in the transplant ineligible subgroup who receive new antilymphoma therapy after 2L Glofit-GemOx treatment. The results are provided in Table 18.
Table 18: Disaggregated Summary of the BIA
Stepped analysis | Scenario | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|---|
Submitted base case | Reference total | 126,121,845 | 126,404,772 | 127,910,260 | 132,104,090 | 386,419,121 |
Glofitamab + GemOx | 0 | 0 | 0 | 0 | 0 | |
All other comparators | 126,121,845 | 126,404,772 | 127,910,260 | 132,104,090 | 386,419,121 | |
New drug total | 126,121,845 | 135,968,026 | 143,522,057 | 150,252,402 | 429,742,485 | |
Glofitamab + GemOx | 0 | 26,288,851 | 44,311,179 | 55,850,002 | 126,450,032 | |
All other comparators | 126,121,845 | 109,679,175 | 99,210,878 | 94,402,401 | 303,292,453 | |
Budget Impact | 0 | 9,563,254 | 15,611,797 | 18,148,313 | 43,323,364 | |
CDA-AMC base case | Reference total | 126,544,129 | 126,856,405 | 128,382,890 | 132,592,430 | 387,831,725 |
Glofitamab + GemOx | 0 | 0 | 0 | 0 | 0 | |
All other comparators | 126,544,129 | 126,856,405 | 128,382,890 | 132,592,430 | 387,831,725 | |
New drug total | 126,544,129 | 136,264,247 | 143,726,900 | 150,383,885 | 430,375,032 | |
Glofitamab + GemOx | 0 | 26,291,526 | 44,314,781 | 55,853,410 | 126,459,717 | |
All other comparators | 126,544,129 | 109,972,721 | 99,412,119 | 94,530,475 | 303,915,315 | |
Budget Impact | 0 | 9,407,842 | 15,344,009 | 17,791,456 | 42,543,307 | |
CDA-AMC scenario analysis | ||||||
Scenario 1: Higher % of patients who receive NALT after 2L Glofit-GemOxa | Reference total | 126,544,129 | 126,856,405 | 128,382,890 | 132,592,430 | 387,831,725 |
New drug total | 126,544,129 | 140,189,183 | 150,240,903 | 158,746,186 | 449,176,272 | |
Budget Impact | 0 | 13,332,778 | 21,858,013 | 26,153,756 | 61,344,547 | |
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency; Glofit-GemOx = glofitamab in combination with gemcitabine and oxaliplatin; NALT = new antilymphoma treatment; pola-BR = polatuzumab vedotin plus bendamustine and rituximab; 2L = second line.
Note: The CDA-AMC reanalysis is based on the publicly available prices of the comparator treatments.
aThe percentage of patients who receive a new antilymphoma treatment after receiving glofitamab in the second line was assumed to be 25.1% in the sponsors’ base case. In this scenario, this value was assumed to be equivalent to that for pola-BR (41%).
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@cda-amc.ca.