Drugs, Health Technologies, Health Systems
Reimbursement Review
Ribociclib (Kisqali)
Sponsor: Novartis Pharmaceuticals Canada Inc.
Therapeutic area: Adjuvant treatment of hormone receptor–positive, HER2-negative early breast cancer
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
AE
adverse event
AI
aromatase inhibitor
AJCC
American Joint Committee on Cancer
ALN
axillary lymph node
BCC
Breast Cancer Canada
CBCN
Canadian Breast Cancer Network
CDK4/6
cyclin-dependent kinase 4 and kinase 6
CI
confidence interval
DDFS
distant disease–free survival
DRFS
distant relapse–free survival
EBC
early breast cancer
ECOG
Eastern Cooperative Oncology Group
EORTC QLQ-C30
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30
ET
endocrine therapy
FAS
full analysis set
GRADE
Grading of Recommendations, Assessment, Development and Evaluation
HR
hormone receptor
HRQoL
health-related quality of life
IA3
interim analysis 3
iDFS
invasive disease–free survival
IPD
individual patient data
ITC
indirect treatment comparison
LHRH
luteinizing hormone–releasing hormone
MAIC
matching-adjusted indirect comparison
OH (CCO)
Ontario Health (Cancer Care Ontario)
OR
odds ratio
OS
overall survival
RCT
randomized controlled trial
RFS
recurrence-free survival
SAE
serious adverse event
SD
standard deviation
SE
standard error
SLR
systematic literature review
TEAE
treatment-emergent adverse event
TNM
tumour, node, and metastasis
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Ribociclib (Kisqali), 200 mg, tablets, oral |
Sponsor | Novartis Pharmaceuticals Canada Inc. |
Indication | In combination with an aromatase inhibitor for the adjuvant treatment of adult patients with HR-positive, HER2-negative stage II-III early breast cancer at high risk of recurrence. In pre- or perimenopausal women, or men, the aromatase inhibitor should be combined with a luteinizing hormone–releasing hormone agonist. |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | June 12, 2025 |
Recommended dose | 400 mg taken orally, once daily for 21 consecutive days followed by 7 days off treatment resulting in a complete cycle of 28 days |
HR = hormone receptor; NOC = Notice of Compliance.
Note: The sponsor’s application was filed on a pre-NOC basis. This review was conducted before the confirmation of the Health Canada indication.
Introduction
Breast cancer is the most commonly diagnosed cancer in females and the second leading cause of cancer-related death among females in Canada.1 In 2022, the 5-year prevalence of breast cancer in females and males was 138,072 in Canada.2 Although breast cancer primarily affects women, it can also occur in men.3 In 2024, an estimated 30,500 new cases were projected in females and 290 in males in Canada.1 The age-standardized mortality rate for breast cancer is 11.8 per 100,000 for both females and males, and 21.8 per 100,000 in females.1
The most common subtype of breast cancer is hormone receptor (HR)-positive, HER2-negative disease, which accounts for 73% of all breast cancer cases.4 Early breast cancer (EBC) refers to cancer that has not spread outside the breast to other parts of the body.5
The majority of breast cancer cases are diagnosed in the early stage; stages I to III (nonmetastatic) account for 94% of all new breast cancer diagnoses in Canada.6 Generally, breast cancer does not induce symptoms in the early stage and regular mammograms are crucial for the early diagnosis of breast cancer.7-10 Among symptomatic cases, a painless mass in the breast is the most common sign.7-10 Other early signs and symptoms of breast cancer may include changes in nipple shape, breast or nipple pain, new breast lumps, nipple retraction, skin changes (such as redness, dryness, flaking, or thickening), nipple discharge, or swelling around the collarbone or under the arm.7-10 In contrast to advanced or metastatic breast cancer, EBC is potentially curable.11 However, recurrence can occur after the initial treatment, including in cases diagnosed at an early stage.12
Curative-intent therapy in EBC aims to prevent recurrence and improve long-term survival. According to the clinical experts consulted for this review, curative-intent treatment options for patients diagnosed with HR-positive, HER2-negative EBC include surgery, which may be preceded or followed by systemic therapy, such as chemotherapy and/or endocrine therapy (ET), depending on other pathological features and genomic testing results. Based on the type of surgery and pathology, radiation therapy may also be offered, typically in the adjuvant setting.
The clinical experts consulted for this review noted that for patients with HR-positive, HER2-negative stage III EBC, systemic therapy includes chemotherapy, ET, and abemaciclib (patients eligible are those whose disease is node-positive and at high risk of recurrence based on clinicopathological features). For patients with stage II EBC, systemic therapy may or may not include chemotherapy, depending on other pathological features and genomic testing results (e.g., Oncotype DX). However, ET is routinely offered in treating patients with stage II EBC. Additionally, the adjuvant use of olaparib is reserved for patients with a (suspected) deleterious germline breast cancer gene mutation.13
On June 12, 2025, ribociclib was granted a Notice of Compliance by Health Canada. Ribociclib is indicated in combination with an aromatase inhibitor (AI) for the adjuvant treatment of adult patients with HR-positive, HER2-negative stage II to III EBC at high risk of recurrence. In premenopausal or perimenopausal women, or men, the AI should be combined with a luteinizing hormone–releasing hormone (LHRH) agonist.14
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of ribociclib, 200 mg, oral tablets in the adjuvant treatment of HR-positive, HER2-negative stage II and III EBC in adults.
Ribociclib has previously been reviewed by Canada’s Drug Agency (CDA-AMC) for breast cancer in the advanced or metastatic settings.
Perspectives of Patients, Clinicians, and Drug Programs
The information in this section is a summary of input provided by the patient and clinician groups who responded to our call for input and from the clinical experts consulted by CDA-AMC for the purpose of this review. The full patient and clinician group submissions received are available in the consolidated patient and clinician group input document for this review on the project website.
Patient Input
CDA-AMC received 4 patient group submissions, 1 each from the Canadian Breast Cancer Network (CBCN), Rethink Breast Cancer, and Quebec Breast Cancer Foundation (QCBF), and a joint submission from Breast Cancer Canada (BCC) and the McPeak-Sirois Group for Clinical Research in Breast Cancer. CBCN is a national health charity dedicated to ensuring the best quality of care for individuals with breast cancer in Canada through information, education, and advocacy. Rethink educates, empowers, and advocates for system changes to improve outcomes for patients with breast cancer, focusing on historically underserved groups. QCBF is committed to defending the interests and well-being of people affected by breast cancer and their loved ones. BCC focuses on saving lives through precision oncology breast cancer research and education, fostering diversity, accelerating research, innovating with technology, promoting patient leadership, and expanding research networks. The McPeak-Sirois Group unites key players in Quebec’s public health sector to make cutting-edge breast cancer research and treatments accessible, supported by breast cancer survivor Susan McPeak and entrepreneur Charles Sirois.
CBCN collected input through online surveys in 2017 (N = 216) and 2022 (N = 981), including 17 patients in 2017 and 111 patients in 2022 with stage II or III HR-positive, HER2-negative breast cancer and through key informant interviews with 2 patients with metastatic breast cancer, and from a review of current studies and grey literature. Rethink gathered input from 24 key patient advisors, 50 blog contributors, 500 virtual support group participants, 2,200 members of private peer support networks , and 43,000 Instagram followers, along with in-depth telephone interviews with 4 patients experienced with ribociclib for high-risk HR-positive, HER2-negative EBC. The QCBF gathered input from members of its private Facebook group “Club stade 4” (French name) and a peer support helpline member, who has been taking the drug for almost 5 years, while also conducting literature research and social media reviews. From November 14 to 18, 2024, BCC and McPeak-Sirois Group surveyed 188 breast cancer survivors from their communities using validated patient-reported outcome measures to capture the lived experiences of individuals with EBC.
Patients across all groups indicated that HR-positive, HER2-negative EBC has significantly impacted their lives, causing emotional, physical, and financial strain. Rethink highlights that younger patients face additional challenges, such as fertility issues and childcare and career disruptions. Despite these difficulties, patients across all groups are motivated to undergo therapy to reduce recurrence, and strive to maintain normal activities and manage disease-related symptoms.
Patients across all groups prioritize effective treatments with manageable side effects, emphasizing quality of life and personal choice. Financial burdens and long-term impacts of treatment are significant concerns, with unique challenges such as the need for personalized treatment decisions (Rethink), managing daily symptoms and potential insurance loss (QCBF), and the necessity for effective adverse event (AE) management support (BCC and McPeak-Sirois Group).
Overall, patients prioritize treatment efficacy, reducing the risk of recurrence, manageable side effects, and maintaining a good quality of life, with many willing to endure additional side effects for better outcomes. According to Rethink, younger patients emphasize the importance of long-term remission to continue their personal and professional lives. QCBF highlighted concerns about long-term medication effects and the significant impact of side effects on daily life.
In the inputs from BCC and McPeak-Sirois Group and Rethink, there were 8 and 4 patients with EBC, respectively, who had experience with ribociclib. In the inputs from CBCN and QBCF, 2 patients and 1 patient, respectively, had experience with ribociclib in the metastatic setting. Across the 4 groups, patients consistently reported positive experiences with ribociclib, emphasizing its efficacy in controlling cancer and the manageable nature of its side effects (e.g., neutropenia, nausea, liver issues, fatigue, dry eyes). They valued the treatment for providing reassurance, hope, and a sense of control over their cancer.
Clinician Input
Input From the Clinical Experts Consulted for This Review
The clinical experts consulted by CDA-AMC emphasized the significant unmet needs in treating stage II and III HR-positive, HER2-negative breast cancer, noting the high rates of recurrence despite aggressive treatments, and indicated that the goal of treatment is to eliminate the disease and reduce the risk of recurrence, minimize side effects and impact on quality of life, maintain employment and independence, and reduce caregiver burden and health care system strain.
For patients with HR-positive, HER2-negative EBC, the clinical experts indicated that ribociclib would be added to existing ET when it is deemed appropriate. Both clinical experts indicated that patients with high risk of recurrence (based on genomic testing, such as Oncotype DX and MammaPrint) are those most in need of new treatments. Currently, there is no evidence or specific biomarkers to predict response, but 1 clinical expert mentioned that patients with poor prognostic features, such as positive lymph nodes, large tumours, and high recurrence scores, are most likely to respond and benefit from ribociclib when added to ET. However, ribociclib is not suitable for patients with severe liver dysfunction, individuals who are pregnant or breastfeeding, or those with a history of QT prolongation or severe neutropenia.
Both clinical experts indicated that for patients with stage II and III HR-positive, HER2-negative EBC, preventing recurrence (measured by recurrence-free survival [RFS]) is considered a clinically meaningful outcome to measure the response to treatment. Other meaningful outcomes to measure responses include improved overall survival (OS), quality of life, and toxicity. The clinical experts indicated that routine surveillance visits are typically conducted every 3 to 6 months during the first 2 to 3 years after treatment, depending on the initial extent of disease, with clinical examinations and/or imaging conducted regularly to check for recurrence, especially for disease that is at high risk of recurrence (e.g., node-positive, large tumours). The clinical experts noted that with ribociclib treatment, frequent toxicity assessments are also required in addition to routine surveillance with frequent bloodwork and electrocardiograms (i.e., every 2 weeks), and increased physician appointments, compared to ET alone. The clinical experts indicated that disease recurrence during ribociclib therapy and severe, unmanageable side effects (e.g., myelosuppression, liver toxicity, QT prolongation, and pulmonary toxicity) necessitate stopping treatment with ribociclib. Additionally, 1 clinical expert mentioned that factors influencing the continuation of therapy include significant impact on quality of life, overall health, and patient preferences. The clinical experts indicated that ribociclib should be prescribed and monitored in a hospital outpatient or community oncology practice, with a medical oncologist or breast oncologist involved in patient care.
Clinician Group Input
Clinician group input for this review was received from 2 clinician groups: the Research Excellence, Active Leadership (REAL) Canadian Breast Cancer Alliance and the Ontario Health (Cancer Care Ontario) (OH [CCO]) Breast Cancer Drug Advisory Committee. A total of 19 clinicians provided input for this submission: 11 clinicians from REAL Canadian Breast Cancer Alliance plus 3 clinicians who agree with the REAL Canadian Breast Cancer Alliance clinician input and are in support of the submission, and 5 clinicians from the OH (CCO) Breast Cancer Drug Advisory Committee.
The input from the clinician groups aligned with that of the clinical experts consulted for this review with regard to treatment goals, the unmet needs of this patient population, assessing treatment response, the drug’s place in therapy, deciding when to discontinue treatment, which specialists should manage these patients, and where patients should be treated with ribociclib. Clinicians from the OH (CCO) Breast Cancer Drug Advisory Committee noted that the duration of treatment with ribociclib is 3 years, which may increase the need for clinical monitoring for patients (e.g., blood work and clinic visits) compared to abemaciclib, which is administered for 2 years.
Drug Program Input
Input was obtained from the drug programs that participate in the reimbursement review process. The following were identified as key factors that could potentially affect the implementation of a recommendation for ribociclib: consideration for initiation of therapy, considerations for discontinuation of therapy, and generalizability. The clinical experts consulted for the purpose of this review provided advice on the potential implementation issues raised by the drug programs. Refer to Table 4 for more details.
Clinical Evidence
Systematic Review
Description of Studies
One phase III, open-label, active-controlled randomized controlled trial (RCT) (NATALEE; N = 5,101) was included that evaluated the efficacy and safety of ribociclib plus AI compared to AI alone as an adjuvant treatment in premenopausal and postmenopausal women, and men, with HR-positive, HER2-negative stage II and III EBC. The primary end point was invasive disease–free survival (iDFS) assessed by the investigator using the Standardized Definitions for Efficacy End Points criteria. Secondary end points included RFS, distant disease–free survival (DDFS), OS, health-related quality of life (HRQoL), and safety. Patients were randomized 1:1 to receive either ribociclib plus AI (n = 2,549) or AI alone (n = 2,552). Randomization was stratified by menopausal status (premenopausal versus postmenopausal, with men grouped in the premenopausal category), anatomical stage (II versus III), prior adjuvant or neoadjuvant chemotherapy (yes versus no), and geographical region (North America, Western Europe, and Oceania versus rest of the world). Eligible patients were recruited in 393 centres in 20 countries. The study enrolled 171 patients across 23 sites in Canada.
Demographic and baseline characteristics were well balanced between treatment groups. The median age of all patients was 52.0 years, with ages ranging from 24 to 90 years. Most patients were postmenopausal (55.8%) and female (99.6%). The majority of the patients were white (73.4%), but the trial also included patients who self-reported as American Indian or Alaska Native, Asian, Black or African American, and Native Hawaiian or other Pacific Islander. Most patients had an Eastern Cooperative Oncology Group (ECOG) Performance Status score of 0 (83.1). More than half (59.6%) of patients had disease that was American Joint Committee on Cancer (AJCC) stage III, nearly half of patients (41.2%) had 1 to 3 positive axillary lymph nodes (ALNs), and most patients (88.1%) had received prior adjuvant or neoadjuvant chemotherapy. There were 676 patients (13.3%) who had genomic tests; Oncotype DX was the most reported test (249; 4.9% of all patients).
Efficacy Results
Three analysis time points are described in this report. The NATALEE trial met its primary end point (iDFS) at interim analysis 3 (IA3) (data cut-off date: January 11, 2023). At IA3, the median duration of follow-up was 27.7 months (range, 0 to 45 months). At the time of end of ribociclib analysis (data cut-off date: April 29, 2024), the median duration of follow-up was 44.2 months (range, 0 to 63 months). At the time of the 5-year follow-up analysis (data cut-off date: May 28, 2025), the median duration of follow-up was 55.4 months. Results for iDFS were reported at IA3, the end of ribociclib analysis, and the 5-year follow-up analysis. No formal statistical analysis of secondary outcomes was planned at any time point; only the results for the end of ribociclib analysis and the 5-year follow-up analysis are summarized in detail for RFS, DDFS, and OS. Because no results were available at the end of ribociclib analysis, only the results for HRQoL at IA3 are presented for this outcome.
Invasive Disease–Free Survival
At IA3, 189 patients (7.4%) in the ribociclib plus AI group and 237 patients (9.3%) in the AI alone group experienced an iDFS event. Distant recurrence (4.7% versus 6.7%) was the most commonly reported iDFS event for both groups. Though the median iDFS was not estimable for either treatment group, there was a statistically significant improvement in iDFS in the ribociclib plus AI group compared with the AI alone group (hazard ratio = 0.75; 95% confidence interval [CI], 0.62 to 0.91; P = 0.0014). The Kaplan-Meier probability estimate of iDFS at 36 months was 90.4% (95% CI, 88.6% to 91.9%) for the ribociclib plus AI group, and 87.1% (95% CI, 85.3% to 88.8%) for the AI alone group. The between-group difference was 3.3% (95% CI, 0.9% to 5.7%). The iDFS results were consistent across all prespecified and additional sensitivity analyses and subgroups.
At the end of ribociclib analysis, a total of 263 patients (10.3%) in the ribociclib plus AI group and 340 patients (13.3%) in the AI alone group experienced an iDFS event. Distant recurrence (6.9% versus 9.6%) was the most reported iDFS event for both groups. The median iDFS was not estimable at the end of ribociclib analysis for either treatment group. The hazard ratio for the ribociclib plus AI group versus the AI alone group was 0.72 (95% CI, 0.61 to 0.84; nominal P < 0.0001), in favour of ribociclib plus AI. The Kaplan-Meier probability estimate of iDFS at 36 months for both groups was consistent with IA3. At 48 months, the Kaplan-Meier probability estimate of iDFS was 88.5% (95% CI, 87.1% to 89.8%) for the ribociclib plus AI group, and 83.6% (95% CI, 81.8% to 85.2%) for the AI alone group, resulting in a between-group difference of 4.9% (95% CI, 2.7% to 7.1%).
At the 5-year follow-up analysis, the hazard ratio for the ribociclib plus AI group versus the AI alone group was 0.72 (95% CI, 0.62 to 0.83). At 60 months, the Kaplan-Meier probability estimate of iDFS was 85.5% (95% CI, 83.9% to 87.0%) for the ribociclib plus AI group, and 81.0% (95% CI, 79.2% to 82.7%) for the AI alone group, resulting in a between-group difference of 4.5% (95% CI, 2.1% to 6.9%).
The subgroup and sensitivity analyses of iDFS at IA3 and the end of ribociclib analysis time points were generally consistent with the primary analysis across all prespecified subgroups.
Recurrence-Free Survival
At the end of ribociclib analysis, a total of 224 patients (8.8%) in the ribociclib plus AI group and 298 patients (11.7%) in the AI alone group experienced an RFS event. The median RFS was not estimable for either treatment group, although the treatment benefit in RFS favoured ribociclib plus AI (hazard ratio = 0.70; 95% CI, 0.58 to 0.83; nominal P < 0.0001). ███ ████████████ ███████████ ████████ ██ ███ ██ ██ ██████ ███ █████ ████ ███ █████ ██ ██████ ███ ███ ██████████ ████ ██ ██████ ███ ████ █ ████ ███ █████ ██ ██████ ███ ███ ██ ██████ ███ ████ █████ ████████ ██ ████ ███ ███████ ███████ ████ ████████ ██ ███ ██████ █████████ █████████.
Distant Disease–Free Survival
At the end of ribociclib analysis, a total of 240 patients (9.4%) in the ribociclib plus AI group and 311 patients (12.2%) in the AI alone group experienced a DDFS event. The median DDFS was not estimable for either treatment group, though the treatment benefit in DDFS favoured ribociclib plus AI compared with AI alone (hazard ratio = 0.72; 95% CI, 0.60 to 0.85; nominal P < 0.0001). ███ ████████████ ███████████ ████████ ██ ████ ██ ██ ██████ ███ █████ ████ ███ █████ ██ ██████ The between-group difference was 4.5% ████ ███ █. These results were consistent with those observed at IA3 and similar results were observed at the 5-year follow-up analysis.
Overall Survival
At the end of ribociclib analysis, a total of 105 patients (4.1%) in the ribociclib plus AI group and 121 patients (4.7%) in the AI alone group had died. The median OS was not estimable for either treatment group. There was no difference in the risk of death between ribociclib plus AI compared with AI alone (hazard ratio = 0.83; 95% CI, 0.64 to 1.07; nominal P = 0.0766). ███ ████████████ ███████████ ████████ ██ ██ ██ ██ ██████ ███ █████ ████ ███ █████ ██ ██████ ███ ███ ██████████ ███████ █████ ██ ████. These results were consistent with those observed at IA3 and similar results were observed at the 5-year follow-up analysis.
Health-Related Quality of Life
At IA3, the mean global health status and quality of life subscale score of the European Organisation for Research and Treatment of Cancer (EORTC) Quality of Life Questionnaire Core 30 (QLQ-C30) was 73.7 points (standard deviation [SD] = 17.7 points) in the ribociclib plus AI group (N = 462; 18.1%), and 73.8 points (SD = 17.8 points) in the AI alone group (N = 497; 19.5%). At end of treatment as of IA3, patients in the ribociclib plus AI group reported a least squares mean decrease (deterioration) from baseline in the global health status and quality of life subscale score of the EORTC QLQ-C30 of –10.40 points (standard error [SE] = 1.36 points) compared to –10.04 points (SE = 1.29 points) in patients in the AI alone group. The between-group difference was –0.36 points (95% CI, –3.12 to 2.39 points; nominal P = 0.7957).
Harms Results
At the time of the end of ribociclib analysis, 2,478 patients (98.1%) in the ribociclib plus AI group and 2,155 patients (88.3%) in the AI alone group experienced at least 1 treatment-emergent adverse event (TEAE). The most common TEAEs, occurring in more than 25% of patients, were neutropenia (41.7% versus 2.9% for ribociclib plus AI versus AI alone), and arthralgia (38.8% versus 44.4%). Grade 3 or higher AEs were reported more frequently in the ribociclib plus AI group than in the AI alone group (64.2% versus 19.7%), with grade 3 or higher neutropenia (27.0% versus 0.5%) and decreased neutrophil count (17.7% versus 0.3%) being the most frequently reported.
Serious adverse events (SAEs) were reported among 14.8% of patients in the ribociclib plus AI group and 10.9% of patients in the AI alone group, with COVID-19 (0.8% versus 0.5%) being the most reported SAE.
A total of 21.1% of patients in the ribociclib plus AI group and 5.3% of patients in the AI alone group had TEAEs leading to discontinuation of study treatment. The most common TEAEs that caused treatment discontinuation were increased ALT (7.2% versus 0.1%) and increased AST (2.9% versus 0).
There were 104 patients (4.1%) in the ribociclib plus AI group and 122 patients (5.0%) in the AI alone group who died before the end of follow-up at the end of ribociclib analysis. Overall, the leading reported cause of death was disease recurrence or progression (3.0% versus 4.1%). AEs were reported as the primary cause of death for 16 patients (0.6%) in the ribociclib plus AI group and 6 patients (0.2%) in the AI alone group. At the 5-year follow-up analysis, 137 patients (5.4%) in the ribociclib plus AI group and 163 patients (6.7%) in the AI alone group had died, primarily due to disease recurrence (101 patients; 4.0%) or progression (133 patients; 5.4%). There were 19 patients (0.8%) and 8 patients (0.3%) in the NATALEE trial who had fatal AEs in the ribociclib plus AI group and AI group, respectively.
The clinical experts consulted by CDA-AMC identified the following notable harms that were reported more frequently in the ribociclib plus AI group than in the AI alone group: hepatobiliary toxicity (26.7% versus 11.4%), QT interval prolongation (5.4% versus 1.6%), and pulmonary toxicity (interstitial lung disease or pneumonitis) (1.6% versus 0.9%).
Critical Appraisal
The Health Canada–approved indication for ribociclib is for patients with HR-positive, HER2-negative EBC at high risk of recurrence. However, the eligibility criteria for the NATALEE trial population were based on disease stage and included a heterogenous population of patients with varying degrees of risk. There was no definition of high risk provided in the trial, nor were there any prespecified subgroup analyses specifically for high-risk patients in the NATALEE trial. As such, it was unclear what proportion of patients in the NATALEE trial were at high risk of recurrence.
The choice of AI as the comparator in the NATALEE trial was clinically relevant because it is reflective of clinical practice in Canada for the treatment of patients with HR-positive and HER2-negative EBC, according to the clinical experts. The NATALEE trial was open label, which may introduce performance and detection bias, particularly for subjective, self-reported outcomes. Additionally, fewer patients discontinued the study in the ribociclib plus AI group compared to the AI alone group at both the end of ribociclib analysis (17.7% versus 22.1%) and the 5-year follow-up (21.5% versus 26.0%).
The median follow-up was 44.2 months at the end of ribociclib analysis, which was considered reasonable by the clinical experts, but longer follow-up is needed for the accurate assessment of treatment effect because recurrence can occur beyond 10 years. The analysis of iDFS was predominately influenced by the initial treatment response, which is represented by an event rate of 10.3% and 13.3% in the ribociclib plus AI group compared to the AI alone group, respectively, at the end of ribociclib analysis, and 12.4% and 16.0%, respectively, at the 5-year follow-up analysis. Though more events had occurred at the 5-year follow-up analysis and the proportion of patients who were censored was generally balanced between treatment groups at previous data cut-off dates, there was still a substantial amount of censoring, with rapid drops in the number of patients at risk, which may introduce uncertainties in the estimates of the treatment effects of ribociclib plus an AI on iDFS, RFS, DDFS, and OS at 36, 48, and 60 months. Additionally, the treatment effect observed over 5 years does not permit definitive conclusions about the long-term effectiveness of ribociclib plus an AI.
Relevant outcomes of RFS, DDFS, OS, and HRQoL were not adjusted for multiple testing, limiting the ability to draw definitive conclusions. There was a high attrition rate in the analysis of HRQoL, which may be due to the limited number of patients who had reached the end-of-treatment stage; there were only █████ patients contributing to the analysis, potentially introducing bias due to differing characteristics between those who continued the trial and those who withdrew. No sensitivity analyses were conducted to evaluate missing data or attrition.
According to the clinical experts consulted for this review, the NATALEE trial’s eligibility criteria were standard, but stricter than clinical practice, excluding patients with significant comorbidities or poorer performance status. However, the clinical experts noted that they would consider patients with an ECOG Performance Status of 2 to be potential candidates for ribociclib based on the extensive experience with ribociclib in the metastatic setting. Most patients in the NATALEE trial were white (73.4%), and 55.9% were postmenopausal. The clinical expert indicated there is a more diversified patient population in their clinical practice compared to the patient population in the NATALEE trial, and that there would be a higher proportion of postmenopausal patients aged 60 to 70 years in their clinical practice who are candidates for ribociclib than were enrolled in the NATALEE trial. Within the NATALEE trial, patients with high genomic risk for the purposes of staging were identified by genomic testing using Oncotype DX. The clinical experts noted that genomic risk testing (e.g., Oncotype DX) is used to determine eligibility for chemotherapy rather than ribociclib, and thus the use of genomic testing in the NATALEE trial may not reflect current clinical practice. More than 88% of patients in the NATALEE trial received prior chemotherapy. The clinical experts noted that most patients with no nodal involvement would have been tested for genomic risk; thus, fewer patients in clinical practice would receive chemotherapy compared to the NATALEE trial. This suggests that the study results may not be generalizable due to differences in pretreatment. More than half of the patients in the NATALEE trial (62.8%) completed the 3-year treatment duration of ribociclib at the time of the end of ribociclib analysis, and no patients were receiving ribociclib treatment at the end of the 5-year follow-up. The clinical experts commented that adherence to treatment is a challenge in the adjuvant setting due to the longer disease-free intervals. The clinical experts noted that patients in the NATALEE trial were predominantly younger women with more aggressive disease, who may be less willing to endure the side effects (e.g., nausea and fatigue) of ribociclib for the full 3 years.
GRADE Summary of Findings and Certainty of the Evidence
For the pivotal NATALEE trial identified in the sponsor’s systematic review, Grading of Recommendations, Assessment, Development and Evaluation (GRADE) was used to assess the certainty of the evidence for outcomes considered most relevant to inform expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group.15,16 Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null. The reference points for the certainty of evidence assessment for iDFS, RFS, DDFS, OS, HRQoL, and harms were set according to the presence of an important effect based on thresholds agreed upon by the clinical experts consulted for this review.
The selection of outcomes for GRADE assessment was based on the sponsor’s Summary of Clinical Evidence, consultation with clinical experts, and input received from patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members:
clinical outcomes (iDFS, RFS, DDFS, and OS)
HRQoL
safety.
Table 2: Summary of Findings for Ribociclib Plus AI vs. AI Alone for Patients With Early Breast Cancer
Outcome and follow-up | Patients (studies), N | Relative effect (95% CI) | Absolute effects (95% CI) | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
AI alone | Ribociclib + AI | Difference | |||||
iDFS | |||||||
Probability of being alive and invasive disease–free at 36 months Follow-up (median): 27.7 months | 5,101 (1 RCT) | Hazard ratio = 0.75 (0.62 to 0.91) | 871 per 1,000 | 904 per 1,000 (886 to 919 per 1,000) | 33 per 1,000 (9 to 57 per 1,000) | Moderatea,b | Ribociclib plus AI likely results in a little to small increase in the probability of being alive and invasive disease–free at 36 months when compared with AI alone. The clinical importance of the increase is unclear. |
Probability of being alive and invasive disease–free at 48 months Follow-up (median): 44.2 months | 5,101 (1 RCT) | Hazard ratio = 0.72 (0.61 to 0.84) | 836 per 1,000 | 885 per 1,000 (871 to 898 per 1,000) | 49 per 1,000 (27 to 71 per 1,000) | Moderatea,b | Ribociclib plus AI likely results in a little to small increase in the probability of being alive and invasive disease–free at 48 months when compared with AI alone. The clinical importance of the increase is unclear. |
Probability of being alive and invasive disease–free at 60 months Follow-up (median): 55.4 months | 5,101 (1 RCT) | Hazard ratio = 0.716 (0.618 to 0.829) | 810 per 1,000 | 855 per 1,000 (839 to 870 per 1,000) | 45 per 1,000 (21 to 69 per 1,000) | Moderatea,b | Ribociclib plus AI likely results in a little to small increase in the probability of being alive and invasive disease–free at 60 months when compared with AI alone. The clinical importance of the increase is unclear. |
RFS | |||||||
Probability of being alive and recurrence-free at 48 months Follow-up (median): 44.2 months | 5,101 (1 RCT) | Hazard ratio = 0.70 (0.58 to 0.83) | ███ ███ █████ | ███ ███ █████ ████ ██ | ██ ███ █████ ███ ██ ██ ███ | Moderatea | Ribociclib plus AI likely results in a little to small increase in the probability of being alive and recurrence-free at 48 months when compared with AI alone. The clinical importance of the increase is unclear. |
Probability of being alive and recurrence-free at 60 months Follow-up (median): 55.4 months | 5,101 (1 RCT) | Hazard ratio = 0.70 (0.598 to 0.82) | 834 per 1,000 | 876 per 1,000 (861 to 890 per 1,000) | 43 per 1,000 (22 to 64 per 1,000) | Moderatea | Ribociclib plus AI likely results in a little to small increase in the probability of being alive and recurrence-free at 60 months when compared with AI alone. The clinical importance of the increase is unclear. |
DDFS | |||||||
Probability of being alive and distant disease–free at 48 months Follow-up (median): 44.2 months | 5,101 (1 RCT) | Hazard ratio = 0.72 (0.60 to 0.85) | ███ ███ █████ | ███ ███ █████ ████ ██ | 45 per 1,000 ███ ██ ██ ███ | Moderatea | Ribociclib plus AI likely results in a little to small increase in the probability of being alive and distant disease–free at 48 months when compared with AI alone. The clinical importance of the increase is unclear. |
Probability of being alive and distant disease–free at 60 months Follow-up (median): 55.4 months | 5,101 (1 RCT) | Hazard ratio = 0.709 (0.608 to 0.827) | 825 per 1,000 | 868 per 1,000 (852 to 882 per 1,000) | 43 per 1,000 (22 to 64 per 1,000) | Moderatea | Ribociclib plus AI likely results in a little to small increase in the probability of being alive and distant disease–free at 60 months when compared with AI alone. The clinical importance of the increase is unclear. |
OS | |||||||
Probability of being alive at 48 months Follow-up (median): 44.2 months | 5,101 (1 RCT) | Hazard ratio = 0.83 (0.64 to 1.07) | ███ ███ █████ | ███ ███ █████ ████ ██ | ███ █████ ██ █████ ██ | Moderatec | Ribociclib plus AI likely results in little to no clinically important difference in the probability of being alive at 48 months when compared with AI alone. |
Probability of being alive at 60 months Follow-up (median): 55.4 months | 5,101 (1 RCT) | Hazard ratio = 0.8 (0.637 to 1.003) | 925 per 1,000 | 941 per 1,000 (930 to 950 per 1,000) | 16 per 1,000 (1 to 31 per 1,000) | Moderatec | Ribociclib plus AI likely results in little to no clinically important difference in the probability of being alive at 60 months when compared with AI alone. |
Health-related quality of life | |||||||
Change from baseline in GHS and QoL subscale score of the EORTC QLQ-C30 at the end of treatment Follow-up (median): NR | 5,101 (1 RCT) | NA | –10.04 (NR) | –10.40 (NR) | –0.36 (–3.12 to 2.39) | Lowd | Ribociclib plus AI may result in little to no clinically important difference in the change from baseline in GHS and QoL subscale score of the EORTC QLQ-C30 at the end of treatment when compared with AI alone. |
Harms | |||||||
Hepatobiliary toxicity Follow-up: NR | 4,967 (1 RCT) | NR | 114 per 1,000 | 267 per 1,000 (NR) | 153 per 1,000 (132 to 174 per 1,000) | Moderatee | Ribociclib plus AI likely results in a clinically important increase in the incidence of hepatobiliary toxicity when compared with AI alone. |
QT interval prolongation Follow-up: NR | 4,967 (1 RCT) | NR | 16 per 1,000 | 54 per 1,000 | 38 per 1,000 (28 to 48 per 1,000) | Moderatee | Ribociclib plus AI likely results in a clinically important increase in the incidence of QT interval prolongation when compared with AI alone. |
Interstitial lung disease or pneumonitis Follow-up: NR | 4,967 (1 RCT) | NR | 9 per 1,000 | 16 per 1,000 | 7 per 1,000 (2 to 14 per 1,000) | Moderatee | Ribociclib plus AI likely results in a clinically important increase in the incidence of interstitial lung disease or pneumonitis when compared with AI alone. |
AI = aromatase inhibitor; CI = confidence interval; DDFS = distant disease–free survival; EBC = early breast cancer; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; GHS = global health status; iDFS = invasive disease–free survival; MID = minimal important difference; NA = not applicable; NR = not reported; OS = overall survival; QoL = quality of life; RCT = randomized controlled trial; RFS = recurrence-free survival; vs. versus.
Note: Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
The statistical testing for probabilities of iDFS, RFS, DDFS, and OS at 48 months and the change from baseline in GHS and QoL subscale score of the EORTC QLQ-C30 at the end of treatment were not adjusted for multiplicity in the NATALEE trial and should be considered as supportive evidence.
aRated down 1 level for serious imprecision. There is no established between-group MID for iDFS, RFS, and DDFS at 36, 48, or 60 months, but the clinical experts suggested that a 5% difference between groups in the probabilities of iDFS, RFS, and DDFS could be considered a threshold of minimal clinical importance. In the review of abemaciclib for a similar population, a between-group difference in the probabilities of iDFS used a threshold range of 3% to 5%. As such, this review adopts the threshold range of 3% to 5% for consistency. Supposing the threshold was 5%, the point estimate and the lower bound of the 95% CI for the between-group difference suggested no minimal clinically important difference between the 2 groups, while the upper bound of the 95% CI suggested a clinically important difference for ribociclib plus AI vs. AI alone based on this threshold. Supposing the threshold was 3%, the point estimate would exceed the threshold of clinical meaningfulness at 36, 48, and 60 months; however, because the 95% CI would continue to suggest both the possibility of no benefit and the possibility of benefit and would be rated down 1 level for serious imprecision, the certainty of evidence would remain “moderate.” However, at 3%, the majority of the effect lies higher than the threshold, which would result in greater confidence that the result is higher than the MID.
bThe absolute difference in Kaplan-Meier probability estimates of iDFS between study groups at 36 months varied notably across different data cut-offs (3.3% at the data cut-off of January 11, 2023, vs. 2.7% at the data cut-off of April 29, 2024). Additionally, the shapes of the iDFS curves differed notably at different data cut-offs.
cRated down 1 level for serious risk of bias due to the low number of patients at risk. There were significant uncertainties at 48 and 60 months due to the limited number of patients at risk and the rapid depletion of the risk set. Imprecision was not rated down. There is no established between-group MID for OS at 48 and 60 months, but the clinical experts suggested that a 3% difference between groups in the probabilities of being alive could be considered a threshold of clinical importance. At 48 and 60 months, the point estimate and the 95% CI for the between-group difference suggested no clinically important difference between the 2 groups for ribociclib plus AI vs. AI alone based on a 3% threshold.
dRated down 2 levels for very serious risk of bias due to the large amount of missing data and for performance and detection biases due to the trial’s open-label design. Imprecision was not rated down. There is no established between-group MID for change from baseline in GHS and QoL subscale score of the EORTC QLQ-C30 in patients with EBC, but the clinical experts considered that a 5-point difference between groups in the change from baseline in GHS and QoL subscale score of the EORTC QLQ-C30 could be considered a threshold of clinical importance in patients with EBC. The point estimate and the upper and lower bounds of the 95% CI for the between-group difference suggested no clinically important difference for ribociclib plus AI vs. AI alone based on a 5-point threshold.
eThe analyses of incidences of hepatobiliary toxicity, QT interval prolongation, and interstitial lung disease or pneumonitis did not account for the participants lost to follow-up or those who dropped out. This omission may introduce a risk of bias, although the direction of this bias is unknown due to the limited data available. Imprecision was not rated down. There is no established between-group MID for the incidence of hepatobiliary toxicity and the incidence of QT interval prolongation, but the clinical experts suggested that a 5% difference in the incidence of hepatobiliary toxicity, a 2% difference in the incidence of QT interval prolongation, and any difference for the incidence of interstitial lung disease or pneumonitis between groups could be considered a threshold of clinical importance. The point estimate and the 95% CI for the between-group difference suggested a clinically important difference between the 2 groups for ribociclib plus AI vs. AI alone based on a 5% threshold for the incidence of hepatobiliary toxicity, 2% threshold for the incidence of QT interval prolongation, and non-null threshold for incidence of interstitial lung disease or pneumonitis.
Sources: NATALEE Clinical Study Reports for the end of ribociclib analysis (2024)17 interim analysis 3 (2023),18 and sponsor-provided additional data.19,20 Details included in the table are from the sponsor’s Summary of Clinical Evidence.21
Long-Term Extension Studies
No long-term extension studies were submitted for this review.
Indirect Comparison
Description of Studies
The sponsor conducted an indirect treatment comparison (ITC) using a matching-adjusted indirect comparison (MAIC) to estimate the relative efficacy of ribociclib plus AI compared with abemaciclib plus ET in the adjuvant treatments of patients with HR-positive, HER2-negative EBC whose disease is at high risk of recurrence based on clinicopathological features.
Efficacy Results
After applying the MAIC weights, the estimated iDFS hazard ratio was 0.901 for ribociclib plus AI versus abemaciclib plus ET (95% CI, 0.677 to 1.197; P = 0.4701). ███ █████ ███ █████████ ██████ █████ ███ █████ ████ ███ █████ ██ ████████████████ ███ ███ ███ █████████ ██████ █████ ██ ██████████ ████ ██ ██████. HRQoL was not assessed in the MAIC.
Harms Results
Grade 3 or higher TEAEs (that occurred in ≥ 5% of patients in either group) were assessed in the MAIC. In the weighted comparison, ribociclib plus AI also had significantly increased odds of neutropenia (odds ratio [OR] = 1.56; 95% CI, 1.23 to 1.97) compared with abemaciclib plus ET and increased ALT (OR = 3.94; 95% CI, 2.56 to 6.05). ORs for diarrhea, leukopenia, and lymphopenia were reduced with ribociclib plus AI versus abemaciclib plus ET, with estimated ORs of 0.11 (95% CI, 0.03 to 0.40), 0.27 (95% CI, 0.16 to 0.46), and 0.14 (95% CI, 0.04 to 0.53), respectively.
Critical Appraisal
Overall, the MAICs were conducted according to accepted methodological guidance. A key limitation of the MAICs was heterogeneity across the included studies. In the primary analysis, the MAIC was adjusted for all baseline characteristics identified in published reports of cohort 1 from the monarchE study, but excluded patients from both studies outside of this subcohort (i.e., excluded patients from cohort 2 of the monarchE study, which included patients with 1 to 3 positive ALNs plus a Ki-67 score of 20% or more if tumour size was < 5 cm and disease was not grade 3). The omission of these patients, who are within the Health Canada indication for ribociclib, leads to uncertainty in the generalizability of the efficacy results to the clinical context. Second, a Bucher analysis of iDFS via common comparator was conducted as a second analysis (subgroup analysis) using data reported for a subgroup of patients receiving AI as the ET component of each treatment arm in cohort 1 of the monarchE trial, but excluding patients who received tamoxifen as the endocrine partner. However, this subgroup analysis was not weighted and not adjusted for the prognostic and treatment-effect modifiers. Overall, the results for iDFS, distant relapse–free survival (DRFS), and OS generally suggested no difference between ribociclib plus AI and abemaciclib plus ET in cohort 1 of the monarchE trial, but the generalizability of the results to the Canadian clinical setting is uncertain. A reduced effective sample size (approximately ███ across various analyses), and wide 95% CIs for the comparative effect estimates (hazard ratios) contributed to imprecision and suggested either benefit or harm in efficacy outcomes.
Studies Addressing Gaps in the Evidence From the Systematic Review
No studies addressing gaps in the evidence from the systematic review were submitted for this review.
Conclusions
The patient and clinician input highlighted the need for new effective treatments for EBC that reduce the risk of recurrence with manageable side effects and that maintain quality of life. Evidence from 1 phase III, open-label, active-controlled RCT (NATALEE) comparing the efficacy and safety of ribociclib plus AI with AI alone as adjuvant treatment in patients with HR-positive, HER2-negative stage II or III EBC, demonstrated that when compared to AI alone, the addition of ribociclib to an AI results in statistically significantly prolonged iDFS, although the magnitude of the benefit was not clinically meaningful based on the 5% threshold suggested by the clinical experts. However, using a threshold of 3%, as has been done previously, the results for iDFS at 48 months and 60 months may be considered clinically meaningful because most of the observed effect lies higher than the threshold, though this remained uncertain. Although results for RFS, DDFS, and OS were consistent with the primary outcome, they were secondary and were not controlled for multiplicity; therefore, they were considered only supportive of the overall effect of ribociclib plus AI. Additionally, the differences between treatment groups for RFS, DDFS, and OS are likely not clinically meaningful based on the thresholds suggested by the clinical experts. The results were uncertain for all time-to-event efficacy outcomes because the medians of these end points were not reached in either study group at the time of the end of ribociclib analysis, indicating the data were immature. There was a decrease in overall quality of life for both ribociclib plus AI and AI alone, though there was little to no difference in HRQoL between treatments, and the results were difficult to interpret due to the large amount of missing data, precluding definitive conclusions.
There is extensive experience with ribociclib, having been available to patients in Canada since 2018. In the NATALEE trial, no new safety signals were identified, and the safety profile was considered manageable based on the clinical experts’ experience with the drug in the metastatic setting.
Indirect evidence via 1 sponsor-submitted MAIC comparing ribociclib plus AI to abemaciclib plus ET suggested no difference between the 2 treatment regimens. However, there were limitations in the indirect evidence, specifically, that the population of patients from the NATALEE study was matched to cohort 1 of the monarchE trial and was not representative of the entire population within the scope of this Reimbursement Review. Additionally, considerable reductions in effective sample size resulting in wide 95% CIs undermined the validity and precision of the results, precluding conclusions on the comparative efficacy of ribociclib plus AI.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of ribociclib, 200 mg, oral tablets in the adjuvant treatment of HR-positive, HER2-negative stage II and III EBC in adults.
Disease Background
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following was summarized and validated by the review team.
Breast cancer is the most commonly diagnosed cancer in females and the second leading cause of cancer-related death among females in Canada.1 In 2022, the 5-year prevalence of breast cancer in females and males was 138,072 in Canada.2 Although breast cancer primarily affects women, it can also occur in men.3 In 2024, an estimated 30,500 new cases were projected in females and 290 in males in Canada.1 The age-standardized mortality rate for breast cancer is 11.8 per 100,000 for females and males, and 21.8 per 100,000 in females.1 The most common subtype of breast cancer is HR-positive, HER2-negative disease, which accounts for 73% of all breast cancer cases.4 Given its prevalence and impact, early detection and diagnosis of breast cancer are critical.22
The majority of breast cancer cases are diagnosed in the early stage, with stages I to III (nonmetastatic) accounting for 94% of all new breast cancer cases in Canada.6 Early breast cancer refers to cancer that has not spread outside the breast to other parts of the body.5 Generally, breast cancer does not induce symptoms in the early stage.7-10 Among symptomatic cases, a painless mass in the breast is the most common sign.7-10 Only 5% of patients with a malignant mass experience pain in the breast.7-10 Other early signs and symptoms of breast cancer may include changes in nipple shape, breast or nipple pain, new breast lumps, nipple retraction, skin changes (such as redness, dryness, flaking, thickening), nipple discharge, or swelling around the collarbone or under the arm.7-10
Breast cancer can recur after the initial treatment, even when diagnosed at an early stage.12 Despite efforts to eradicate all cancer cells during the initial treatment, some may remain and be undetected.23 Over time, these residual cells can multiply and lead to a recurrence of the cancer.23 Recurrence can happen months or even years after the initial treatment,23 and can be categorized as local (cancer reappearing at the original site), regional (cancer returning in nearby tissues or lymph nodes), or distant (cancer metastasizing and reappearing in a different part of the body).24 Various risk factors of recurrence have been highlighted in the literature, including, but not limited to, nodal status, primary tumour size, young age at diagnosis, tumour grade, Ki-67 status, or genomic signatures.12,25 Thus, even in patients with the most favourable prognosis, the risk of breast cancer recurrence remains a significant health concern.12
Early cancer detection can prevent the use of more aggressive treatments and improve patient outcomes, including survival rates.22 Regular mammograms are crucial for the early diagnosis of breast cancer. While mammography serves as the primary method for breast cancer screening, other screening methods can be used such as breast exam or ultrasound.22 MRI may be used to screen patients at high risk of breast cancer. Following suspicious findings, a core biopsy, which is sometimes accompanied by clip placement, is conducted to confirm diagnosis.26 Subsequently, biomarker testing, including estrogen receptor, progesterone receptor, and HER2, conducted via immunochemistry or fluorescence in situ hybridization on tumour samples, plays a crucial role in directing personalized treatment pathways.27,28 Clinical staging is determined using imaging and clinical examination findings. Pathological staging postsurgery involves examining the tumour and lymph nodes to further refine treatment strategies and prognostic assessment.29
Standards of Therapy
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following was summarized and validated by the review team.
In contrast to advanced or metastatic breast cancer, EBC is potentially curable.11 Curative-intent therapy in EBC aims to prevent recurrence and improve long-term survival. According to the clinical experts consulted for this review, curative-intent therapy options for patients diagnosed with HR-positive, HER2-negative EBC include surgery, which may be preceded or followed by systemic therapy (i.e., chemotherapy and/or ET, depending on other pathological features and genomic testing results). Based on the type of surgery and pathology, radiation therapy may also be offered, typically in the adjuvant setting. The clinical experts consulted for this review noted that for patients with HR-positive, HER2-negative stage III EBC, systemic therapy includes chemotherapy, ET, and abemaciclib (for eligible patients whose disease is node-positive and at high risk of recurrence based on clinicopathological features). For patients with stage II EBC, systemic therapy may or may not include chemotherapy, depending on other pathological features and genomic testing results (e.g., Oncotype DX). The clinical experts indicated the typical chemotherapy options can include anthracyclines and taxanes (although anthracyclines may be excluded in certain settings), and ET (e.g., AIs such as anastrozole, letrozole, or exemestane, or tamoxifen), which would be offered as the standard of care for all patients with stage II EBC, presuming no contraindications.30-33 ET is typically prescribed for 5 to 10 years, depending on the risk of recurrence, patients’ preferences, or menopausal status. Ovarian function suppression or ablation is offered concurrently in premenopausal or perimenopausal patients.30-33 In men with breast cancer, the treatment regimen is similar to that in postmenopausal patients, and the use of androgen suppression with LHRH agonists is the current standard treatment.33 Additionally, the adjuvant use of olaparib is reserved for patients with (suspected) deleterious germline breast cancer gene mutation.13
Drug Under Review
Key characteristics of ribociclib are summarized in Table 3 with other treatments available for the adjuvant treatment of patients with HR-positive, HER2-negative, stage II and III EBC.
Ribociclib is a cyclin-dependent kinase 4 and kinase 6 (CDK4/6) inhibitor that reduces cell cycle progression and proliferation by decreasing retinoblastoma protein phosphorylation, leading to tumour regression and a senescent phenotype in breast cancer models. In vivo, ribociclib treatment resulted in well-tolerated tumour regressions correlated with retinoblastoma protein phosphorylation inhibition.14 Ribociclib is available in 200 mg oral tablets. The recommended dosage of ribociclib is 400 mg taken orally once daily for 21 consecutive days followed by 7 days off treatment, resulting in a complete cycle of 28 days.14
On June 12, 2025, ribociclib received a Health Canada Notice of Compliance in combination with an AI for the adjuvant treatment of adult patients with HR-positive, HER2-negative stage II to III EBC at high risk of recurrence. In premenopausal or perimenopausal women, or men, the AI should be combined with an LHRH agonist.14 In September 2024, the FDA approved ribociclib in combination with an AI for the adjuvant treatment of adults with HR-positive, HER2-negative, stage II and III EBC at high risk of recurrence, including those with no nodal involvement.34 More recently, in November 2024, the European Commission also approved ribociclib in combination with an AI for the adjuvant treatment of patients with HR-positive, HER2-negative EBC at high risk of recurrence.35 The sponsor is seeking reimbursement for ribociclib as per the proposed Health Canada indication.
Ribociclib was previously reviewed by CDA-AMC for breast cancer in the advanced or metastatic settings:
On June 4, 2020, a recommendation for reimbursement was issued for ribociclib in combination with an AI and an LHRH agonist for the treatment of premenopausal or perimenopausal patients with HR-positive, HER2-negative advanced or metastatic breast cancer as initial endocrine-based therapy.36
On April 22, 2020, a recommendation for reimbursement was issued for ribociclib in combination with fulvestrant for the treatment of postmenopausal patients with HR-positive, HER2-negative advanced or metastatic breast cancer as initial therapy or following disease progression.37
On April 18, 2018, a recommendation for reimbursement was issued for ribociclib in combination with letrozole for the treatment of postmenopausal patients with HR-positive, HER2-negative advanced or metastatic breast cancer as initial ET.38
Table 3: Key Characteristics of Ribociclib, Abemaciclib, Anastrozole, Exemestane, Goserelin Acetate, Letrozole, and Tamoxifen
Treatment | Mechanism of action | Indicationa | Route of administration | Recommended dosage | Serious adverse effects or safety issues | Other |
|---|---|---|---|---|---|---|
Ribociclib14 | Ribociclib is a CDK4/6 inhibitor that reduces cell cycle progression and proliferation by decreasing retinoblastoma protein phosphorylation, leading to tumour regression and a senescent phenotype in breast cancer models. | Proposed: For the adjuvant treatment of patients with HR-positive, HER2-negative, stage II and III EBC, in combination with an AI. In premenopausal or perimenopausal patients, or men, the AI should be combined with an LHRH agonist. | Oral | 400 mg (2 × 200 mg film-coated tablets) once a day for 21 consecutive days followed by 7 days off treatment resulting in a complete cycle of 28 days. For EBC, continue ribociclib until completion of 3 years of treatment or until disease recurrence or unacceptable toxicity occurs. |
| NA |
Abemaciclib39 | Abemaciclib is a CDK4/6 inhibitor. It blocks retinoblastoma protein phosphorylation, suppressing tumour growth, inducing senescence and apoptosis in ER-positive breast cancer, and reducing tumour size in xenograft models alone or with antiestrogens. | In combination with ET for the adjuvant treatment of adult patients with HR-positive, HER2-negative, node-positive, EBC at high risk of disease recurrence based on clinicopathological features. | Oral | 150 mg twice daily. For EBC, continue abemaciclib until completion of either 2 years of treatment or until disease recurrence or unacceptable toxicity. |
| NA |
Anastrozole40 | Anastrozole is a selective nonsteroidal AI that lowers serum estradiol concentrations, helping to reduce estrogen levels and thereby decreasing tumour mass or slowing tumour progression in many breast cancers, which often grow due to estrogen stimulation. | For the adjuvant treatment of postmenopausal patients with HR-positive EBC. | Oral | 1 mg once a day. In the adjuvant setting, it is currently recommended that treatment be given for 5 years. |
|
|
Exemestane41 | Exemestane is a potent aromatase inactivator that significantly lowers circulating estrogen levels in postmenopausal patients by irreversibly binding to the aromatase enzyme, effectively suppressing estrogen production and inhibiting tumour growth in hormone-sensitive breast cancer. | For the sequential adjuvant treatment of postmenopausal patients with estrogen receptor–positive EBC who have received 2 to 3 years of initial adjuvant tamoxifen therapy. | Oral | 25 mg once daily after a meal in early and advanced breast cancer. In postmenopausal patients with EBC, treatment should continue until completion of 5 years of adjuvant ET or until local or distant recurrence or new contralateral breast cancer. |
|
|
Goserelin acetate42 | Goserelin acetate is a synthetic decapeptide analogue of GnRH that, when administered chronically, inhibits gonadotropin production, leading to regression of gonadal and accessory sex organs, and is used to treat hormone-dependent breast cancer, endometriosis, and other conditions by suppressing serum estradiol. | As an alternative to standard adjuvant chemotherapy in premenopausal and perimenopausal patients with EBC who are unsuitable for, intolerant to, or decline chemotherapy, and whose tumour contains estrogen and/or progesterone receptors. | Subcutaneous | 3.6 mg should be administered subcutaneously every 28 days into the anterior abdominal wall. |
|
|
Letrozole43 | Letrozole is an AI that inhibits the aromatase enzyme by competitively binding to the heme of the cytochrome P450 subunit of the enzyme, resulting in a reduction of estrogen biosynthesis in all tissues. | For the adjuvant treatment of postmenopausal patients with HR-positive EBC. For the extended adjuvant treatment of HR-positive EBC in postmenopausal patients who have received approximately 5 years of prior standard adjuvant tamoxifen therapy. | Oral | 2.5 mg once a day. In the adjuvant setting, the intended duration of treatment is 5 years. In the extended adjuvant setting, treatment with letrozole is intended for 5 years. |
|
|
Tamoxifen44 | Tamoxifen is a nonsteroidal drug with potent antiestrogenic properties that competes with estrogen for binding sites in target tissues, inhibiting and causing regression of dimethylbenz(a)anthracene-induced mammary carcinomas by binding to estrogen receptors. | For the adjuvant treatment of EBC in patients with estrogen receptor–positive tumours. | Oral | 20 mg to 40 mg per day in a single dose or 2 divided doses. The clinical experts consulted by CDA-AMC indicated that the typical dose in clinical practice is 20 mg per day. |
|
|
AI = aromatase inhibitor; BMI = body mass index; CDA-AMC = Canada’s Drug Agency; CDK4/6 = cyclin-dependent kinase 4 and kinase 6; EBC = early breast cancer; ET = endocrine therapy; ER = estrogen receptor; GnRH = gonadotropin-releasing hormone; HR = hormone receptor; INR = international normalized ratio; LHRH = luteinizing hormone–releasing hormone; NA = not applicable.
aHealth Canada–approved indication.
Sources: Product monographs for ribociclib,14 abemaciclib,39 anastrozole,40 exemestane,41 goserelin acetate,42 letrozole,43 and tamoxifen.44
Perspectives of Patients, Clinicians, and Drug Programs
The full patient and clinician group submissions received are available in the consolidated patient and clinician group input document for this review on the project website.
Patient Group Input
This section was prepared by the review team based on the input provided by patient groups.
CDA-AMC received 4 patient group submissions from CBCN, Rethink Breast Cancer, QCBF, and a joint submission from BCC and the McPeak-Sirois Group for Clinical Research in Breast Cancer. CBCN is a national health charity dedicated to ensuring the best quality of care for individuals with breast cancer in Canada through information, education, and advocacy. Rethink educates, empowers, and advocates for system changes to improve outcomes for patients with breast cancer, focusing on historically underserved groups. QCBF is committed to defending the interests and well-being of people affected by breast cancer and their loved ones. BCC focuses on saving lives through precision oncology breast cancer research and education, fostering diversity, accelerating research, innovating with technology, promoting patient leadership, and expanding research networks. The McPeak-Sirois Group unites key players in Quebec’s public health sector to make cutting-edge breast cancer research and treatments accessible, supported by breast cancer survivor Susan McPeak and entrepreneur Charles Sirois.
CBCN collected input through online surveys in 2017 (N = 216) and 2022 (N = 981), including 17 patients in 2017 and 111 patients in 2022 with stage II or III HR-positive, HER2-negative breast cancer, respectively, as well as key informant interviews with 2 patients with metastatic breast cancer, and a review of current studies and grey literature. Rethink gathered input from 24 key patient advisors, 50 blog contributors, 500 virtual support group participants, 2,200 private peer support network members, and 43,000 Instagram followers, along with in-depth telephone interviews with 4 patients experienced with ribociclib for high-risk HR-positive, HER2-negative EBC. The QCBF gathered inputs from members of its Facebook private group “Club stade 4” (French name) and a peer support helpline member, who has been taking the drug for almost 5 years, while also conducting literature research and social media reviews. From November 14 to 18, 2024, BCC and McPeak-Sirois Group surveyed 188 breast cancer survivors from their communities using validated patient-reported outcomes measures to capture the lived experiences of individuals with EBC.
Patients across all groups indicated that HR-positive, HER2-negative EBC significantly impacts their lives, causing emotional, physical, and financial strain. Rethink highlights that younger patients face additional challenges such as fertility issues and disruptions to childcare and career. Despite these difficulties, patients across all groups are motivated to undergo therapy to reduce recurrence and strive to maintain normal activities and manage disease-related symptoms.
Patients across all groups prioritize effective treatments with manageable side effects, emphasizing quality of life and personal choice. Financial burdens and long-term impacts of treatment are significant concerns, with unique challenges such as the need for personalized treatment decisions (Rethink), managing daily symptoms and potential insurance loss (QCBF), and the necessity for effective AE management support (BCC and McPeak-Sirois Group).
Overall, patients prioritize treatment efficacy, reducing the risk of recurrence, manageable side effects, and maintaining a good quality of life, with many willing to endure additional side effects for better outcomes. According to Rethink, younger patients emphasize the importance of long-term remission to continue their personal and professional lives. QCBF highlighted concerns about long-term medication effects and the significant impact of side effects on daily life.
In the joint submission from BCC and McPeak-Sirois Group and the input from Rethink, there were 8 and 4 patients with EBC, respectively, who had experience with ribociclib. In the inputs from CBCN and QBCF, 2 patients and 1 patient, respectively, had experience with ribociclib in the metastatic setting. Across the 4 groups, patients consistently reported positive experiences with ribociclib, emphasizing its efficacy in controlling cancer and the manageable nature of its side effects (e.g., neutropenia, nausea, liver issues, fatigue, dry eyes). They valued the treatment for providing reassurance, hope, and a sense of control over their cancer.
The inputs from CBCN focused on patients with metastatic breast cancer, Rethink's detailed account of side effects, QCBF's concern about long-term access and insurance, and BCC and McPeak-Sirois Group's emphasis on maintaining daily functionality and the importance of clinical support. Additionally, inputs from Rethink and BCC and McPeak-Sirois Group highlight the financial burden on younger patients and the importance of public funding and accessible care to alleviate these challenges. While the input from BCC and McPeak-Sirois Group focuses on the financial burden and the need for public funding, the input from Rethink underscored the positive impact of CDK4/6 inhibitors and the necessity of addressing barriers to care for younger patients.
Clinician Input
Input From Clinical Experts Consulted for This Review
All CDA-AMC review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of HR-positive, HER2-negative stage II and III EBC.
Unmet Needs
The clinical experts consulted by CDA-AMC emphasized the significant unmet needs in treating stage II and III HR-positive, HER2-negative breast cancer, noting the high rates of recurrence despite aggressive treatments. According to the clinical experts, the estimated recurrence rates are 10% to 30% for patients with stage II disease and 30% to 50% for patients with stage III disease over 10 years. Over 20 years after diagnosis, the clinical experts estimated the recurrence rates to be 27% to 37% for patients with stage II disease and 46% to 57% for patients with stage III disease. The clinical experts underscored the necessity for improved long-term management strategies to enhance patient outcomes and reduce the chances of recurrence. The clinical experts highlighted that considering that treatment for HR-positive, HER2-negative, EBC is with curative intent, the goal is to eliminate the disease and reduce the risk of recurrence, minimize side effects and impact on quality of life, maintain employment and independence, and reduce caregiver burden and health care system strain.
Place in Therapy
For patients with HR-positive, HER2-negative EBC, the clinical experts indicated that ribociclib would be added to existing ET when it is deemed appropriate. One clinical expert noted that ribociclib works synergistically with ET (e.g., AIs or tamoxifen) by targeting cancer cell proliferation while ET blocks estrogen-driven tumour growth. The clinical experts expect ribociclib to shift the treatment paradigm for high-risk HR-positive, HER2-negative EBC, replacing abemaciclib when appropriate, particularly for patients at high risk of recurrence who benefit from the combination of ET and a CDK4/6 inhibitor. According to the clinical experts, the choice between ribociclib and abemaciclib would depend on differences in trial eligibility, patient comorbidities, patient preferences, and potential toxicities.
Patient Population
Both clinical experts indicated that patients with a high risk of recurrence (based on genomic testing, such as Oncotype DX and MammaPrint) are those most in need of new treatments. Currently, there is no evidence or specific biomarkers to predict response, but 1 clinical expert mentioned that patients with poor prognostic features, such as positive lymph nodes, large tumours, and high recurrence scores, are most likely to respond and benefit from ribociclib when added to ET. One clinical expert mentioned that patients with minimal contraindications to CDK4/6 inhibitors and good performance status are also ideal candidates. However, 1 expert noted that ribociclib is not suitable for patients with severe liver dysfunction, patients who are pregnant or breastfeeding, those with a history of QT prolongation, or those with severe neutropenia. The clinical experts stated that clinical judgment of tumour size, lymph node involvement, and risk of recurrence, as well as genomic testing and laboratory tests are essential for identifying patients at high risk of recurrence. Additionally, patients’ comorbidities and tolerability should be considered to ensure safety before initiating treatment with ribociclib.
Assessing the Response to Treatment
The clinical experts highlighted that given the early stage of the disease, response to treatment, generally measured by recurrence (i.e., RFS), is a clinically meaningful outcome; however, this takes time and may not be evaluable during the ribociclib treatment period. Other meaningful outcomes to measure responses include improved OS, quality of life, and toxicity, which are assessed frequently during follow-up.
The clinical experts indicated that routine surveillance visits are typically conducted every 3 to 6 months during the first 2 to 3 years after treatment, with clinical examinations and/or imaging conducted regularly to check for recurrence, especially for disease that is at high risk of recurrence (e.g., node-positive, large tumours). Additionally, the clinical experts stated that with ribociclib, close monitoring for treatment toxicity is required, with frequent bloodwork and electrocardiograms (i.e., every 2 weeks) and increased physician appointments, compared to ET alone.
Discontinuing Treatment
The clinical experts indicated that disease recurrence during ribociclib therapy and severe, unmanageable side effects (e.g., myelosuppression, liver toxicity, QT prolongation, and pulmonary toxicity) necessitate stopping treatment with ribociclib. Additionally, 1 clinical expert mentioned that factors influencing the continuation of therapy include significant impacts on quality of life, overall health, and patient preferences.
Prescribing Considerations
The clinical experts indicated that ribociclib should be prescribed and monitored in a hospital outpatient or community oncology practice with a medical oncologist or breast oncologist involved in patient care. The clinical experts highlighted the importance of specialized care, side effect management, and timely treatment adjustments, with nurses and pharmacists with oncology expertise playing crucial roles in patient monitoring and support.
Clinician Group Input
This section was prepared by the review team based on the input provided by clinician groups.
Clinician group input for this review was received from 2 clinician groups: the REAL Canadian Breast Cancer Alliance and OH (CCO) Breast Cancer Drug Advisory Committee. A total of 19 clinicians provided input for this submission: 11 clinicians from REAL Canadian Breast Cancer Alliance plus 3 clinicians who agree with the REAL Canadian Breast Cancer Alliance clinician input and are in support of the submission, and 5 clinicians from the OH (CCO) Breast Cancer Drug Advisory Committee.
The input from the clinician groups aligned with that of the clinical experts consulted for this review with regard to treatment goals, the unmet needs of this patient population, assessing treatment response, the drug’s place in therapy, deciding when to discontinue treatment, which specialists should manage these patients, and where patients should be treated with ribociclib. Clinicians from the OH (CCO) Breast Cancer Drug Advisory Committee noted that the duration of treatment with ribociclib is 3 years, which may increase the need for clinical monitoring for patients (e.g., blood work and clinic visits) compared to abemaciclib, which is given for 2 years.
Drug Program Input
The drug programs provide input on each drug being reviewed through the reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by for this review are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
In the NATALEE trial, patients received nonsteroidal aromatase inhibitors (letrozole 2.5 daily or anastrozole 1 mg daily), and goserelin every 28 days in men and premenopausal women. For patients with node-positive HR-positive, HER2-negative breast cancer at high risk of disease recurrence based on clinicopathological features, abemaciclib in combination with ET is funded in Canada as adjuvant therapy and funded in most jurisdictions. | This is a comment from the drug programs to inform pERC deliberations. |
Considerations for initiation of therapy | |
Patients were required to have stage II or III disease based on anatomical stage according to the AJCC Cancer Staging Manual, 8th Edition. All patients with stage IIB or III disease were allowed to participate in the trial, regardless of nodal status. Patients with stage IIA disease were eligible if they had at least 1 lymph node involved. Patients with stage IIA disease with no nodal involvement and grade 3 tumours were also eligible, as well as those with a grade 2 tumour combined with a Ki-67 proliferation index of at least 20% or a high genomic risk (defined as an Oncotype DX score of 26 or higher, or categorized as high risk on PAM50, MammaPrint, or EndoPredict assay) Genomic risk (e.g., Oncotype DX) is not currently used to determine eligibility for abemaciclib. Should the criteria in the NATALEE trial be used in clinical practice to determine eligibility for treatment with ribociclib? | The clinical experts noted that genomic risk testing (e.g., Oncotype DX) is used to determine eligibility for chemotherapy rather than for ribociclib. As part of the standard of care, clinicians would do genomic risk testing (i.e., Oncotype DX) for all patients with stage II (A or B) and grade 2 or grade 3 tumours before initiating chemotherapy. The clinical experts stated that genomic risk is a prognostic factor and predictive of response to chemotherapy but were unsure whether genomic risk is predictive for treatment with ribociclib. The clinical experts indicated that patients with low genomic risk would have a low Oncotype DX score, which means they would not receive chemotherapy nor be eligible for ribociclib based on the NATALEE trial criteria. The clinical experts indicated that generally, they would not give ribociclib to patients who have not previously had adjuvant chemotherapy, considering most patients (88.1%) in the NATALEE trial had previous chemotherapy. According to the clinical experts, the only exception would be for patients with high-risk disease who are eligible for chemotherapy, but for medical reasons cannot receive chemotherapy (e.g., clinicians cannot safely administer the chemotherapy). Additionally, the clinical experts indicated that given the Ki-67 proliferation index criterion has been removed for abemaciclib eligibility, it would not be tested automatically for most patients in the adjuvant setting. Overall, the clinical experts indicated that the same criteria as the NATALEE trial should be used in clinical practice to determine eligibility for ribociclib. |
Can patients be retreated with CDK4/6 inhibitors in the metastatic setting? If yes, what is the minimum disease-free interval required? Similar to abemaciclib, for patients with HR-positive, HER2-negative early-stage breast cancer that recurs, would a 6-month interval between prior CDK4/6 inhibitors be appropriate when considering re-treatment in the advanced and/or metastatic setting? | Although there are no clear data on re-treatment with CDK4/6 inhibitors in the metastatic setting after prior exposure in the adjuvant setting, the clinical experts acknowledged that in practice, clinicians do re-treat with CDK4/6 inhibitors in the metastatic setting, provided there is a treatment-free interval of 6 to 12 months. |
In the monarchE trial with abemaciclib, patients must have been assigned within 16 months of definitive breast cancer surgery. In the NATALEE trial, patients needed to be randomized within 18 months of histologic diagnosis. Is there a maximum allowable time frame since diagnosis or surgery to be eligible for ribociclib? Should the timeline be aligned between abemaciclib and ribociclib and, if so, what is the recommended timeline? | The clinical experts indicated there is no similar time frame listed in the NATALEE trial protocol and highlighted that in the NATALEE trial, patients could have received any adjuvant or neoadjuvant ET for up to 12 months before randomization, and patients were ineligible if they had had major surgery, chemotherapy, or radiotherapy within 14 days before randomization; had received a previous CDK4/6 inhibitor; or had a significant, uncontrolled heart condition. The clinical experts noted they would follow the NATALEE trial protocol to treat patients within 18 months of histologic diagnosis, accounting for potential time lag between biopsy and surgery. The clinical experts indicated that for patients who had a biopsy followed by upfront surgery, the timeline probably aligns with abemaciclib (i.e., 16 months) without any issues. |
Considerations for discontinuation of therapy | |
If a patient has an interruption within 3 years from starting treatment, should a total of 3 years of ribociclib be allowed, or should treatment be stopped at 3 calendar years from the start of treatment? | The clinical experts indicated that the total treatment duration of ribociclib is generally limited to 3 calendar years from the start of treatment. One expert added that this duration depends on the nature of interruptions. If the interruptions are routine, lasting 1 to 2 weeks intermittently for low neutrophils, then a total of 3 years is reasonable. However, if the interruptions are longer, then limiting the treatment to 3 calendar years may be appropriate. |
Should ribociclib be continued until completion of 3 years of treatment or until disease recurrence or unacceptable toxicity occurs? | The clinical experts indicated that ribociclib should be continued until completion of 3 years of treatment or until disease recurrence or unacceptable toxicity occurs. |
Considerations for prescribing of therapy | |
PAG notes that a lower dose of ribociclib is used in early-stage breast cancer compared to the approved regimen for advanced breast cancer (400 mg [2 × 200 mg tablets] once daily; 3 weeks on and 1 week off). | This is a comment from the drug programs to inform pERC deliberations. |
Generalizability | |
Patients with an ECOG Performance Status score > 1 were excluded from the NATALEE trial. Can they be considered eligible for ribociclib? | The clinical experts indicated it is reasonable consider patients with an ECOG Performance Status score of 1 or 2 eligible because they would likely tolerate the treatment with ribociclib. |
In the NATALEE trial, patients could have received any adjuvant or neoadjuvant ET for up to 12 months before randomization. Should existing patients who have been receiving ET for > 12 months be eligible on a time-limited basis? | The clinical experts indicated that existing patients who have been receiving ET for more than 12 months should be eligible on a time-limited basis. |
Funding algorithm (oncology only) | |
Request an initiation of a rapid provisional funding algorithm. | This is a comment from the drug programs to inform pERC deliberations. |
Care provision issues | |
Prolongation of the QTc interval has been observed in patients receiving ribociclib. A significant increase in electrocardiogram tests and monitoring is anticipated due to the volume of patients who may receive ribociclib for early breast cancer which may cause additional health system capacity concerns. | This is a comment from the drug programs to inform pERC deliberations. |
System and economic issues | |
PAG is concerned about the budget impact and affordability, given the large volume of patients with early breast cancer. | This is a comment from the drug programs to inform pERC deliberations. |
Blood work for monitoring neutropenia and liver function will be increased compared to ET alone. | This is a comment from the drug programs to inform pERC deliberations. |
Confidential pricing exists for abemaciclib. | This is a comment from the drug programs to inform pERC deliberations. |
AJCC = American Joint Committee on Cancer; CDK4/6 = cyclin-dependent kinase 4 and kinase 6; ECOG = Eastern Cooperative Oncology Group; ET = endocrine therapy; PAG = Provincial Advisory Group; pERC = pan-Canadian Oncology Drug Review Expert Review Committee; QTc = corrected QT.
Clinical Evidence
The objective of this Clinical Review report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of ribociclib, 200 mg tablets, in the treatment of HR-positive, HER2-negative EBC in adults. The focus will be placed on comparing ribociclib to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of ribociclib is presented in 2 sections, with our critical appraisal of the evidence included at the end of each section. The first section, the systematic review, includes pivotal studies and RCTs that were selected according to the sponsor’s systematic review protocol. Our assessment of the certainty of the evidence in this first section using the GRADE approach follows the critical appraisal of the evidence. The second section includes indirect evidence from the sponsor. No long-term extension studies or studies addressing gaps were included in the review.
Included Studies
Clinical evidence from the following is included in the review and appraised in this document:
one phase III, multicentre, open-label, active-controlled RCT identified in a systematic review (NATALEE)
one ITC using MAICs.
Systematic Review
Contents within this section have been informed by materials submitted by the sponsor. The following was summarized and validated by the review team.
Description of Studies
Characteristics of the included studies are summarized in Table 5.
Table 5: Details of Studies Included in the Systematic Review
Detail | NATALEE trial |
|---|---|
Designs and populations | |
Study design | Phase III, multicentre, open-label, active-controlled RCT |
Locations | 393 sites in 20 countries (Argentina, Australia, Austria, Belgium, Brazil, Canada, China, France, Germany, Hungary, Ireland, Italy, Korea, Poland, Romania, Russia, Spain, Taiwan, UK, and US) |
Patient enrolment dates | Start date: January 10, 2019 End date: April 20, 2021 |
Randomized (N) | A total of 5,101 patients were randomized: 2,549 patients were randomized to the ribociclib plus AI group and 2,552 patients were randomized to the AI alone group |
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Intervention | Patients assigned to the ribociclib plus AI treatment group received ribociclib 400 mg (2 × 200 mg tablets) p.o. once daily on days 1 to 21 of a 28-day cycle for 36 months plus AI continuously for 60 months. Patients also received an AI. The AI could be:
|
Comparator(s) | Patients in the AI treatment group received the same AI received by patients in the ribociclib plus AI group, as previously described |
Study duration | |
Screening phase | 28 days |
Treatment phase | 60 months + 30 days of safety follow-up |
Follow-up phase | Started after the 30-day safety follow-up visit and continued until death, withdrawal of consent, lost to follow-up, or end of trial,a whichever was earliest |
Outcomes | |
Primary end point | iDFS using STEEP criteria, as assessed by the investigator up to the last data cut-off date |
Secondary and exploratory end points | Secondary
Exploratory
|
Publication status | |
Publications | Slamon et al. (2024)68 ClinicalTrials.gov identifier: NCT03701334 |
AE = adverse event; AI = aromatase inhibitor; CDK = cyclin-dependent kinase; CTCAE = Common Terminology Criteria for Adverse Events; ctDNA = circulating tumour DNA; ctRNA = circulating tumour ribonucleic acid; DDFS = distant disease–free survival; ECG = electrocardiogram; ECOG = Eastern Cooperative Oncology Group; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; ER = estrogen receptor; ET = endocrine therapy; iDFS = invasive disease–free survival; LRRFS = locoregional recurrence–free survival; NCI = National Cancer Institute; OS = overall survival; PK = pharmacokinetics; p.o. = orally; PR = progesterone receptor; QoL = quality of life; QTcF = QT corrected for heart rate using the Fridericia formula; RCT = randomized controlled trial; RFS = recurrence-free survival; SC = subcutaneous; STEEP = Standardized Definitions for Efficacy End Points.
aEnd of trial was planned to be declared when 60 months plus 30 days had elapsed from the date the last patient had been randomized.
Source: NATALEE Clinical Study Reports for the end of ribociclib analysis (2024).17 Details included in the table are from the sponsor’s Summary of Clinical Evidence.21
One pivotal trial (NATALEE) was included in the systematic review (Table 5). NATALEE was a phase III, open-label, active-controlled RCT to evaluate the efficacy and safety of ribociclib plus AI compared to AI alone as an adjuvant treatment in premenopausal and postmenopausal women, and men, with HR-positive and HER2-negative EBC. The primary end point was iDFS assessed by the investigator using the Standardized Definitions for Efficacy End Points criteria. Secondary end points included RFS, DDFS, OS, HRQoL, and safety. A total of 5,101 patients were randomized at a 1:1 ratio to receive either ribociclib plus AI (n = 2,549) or AI alone (n = 2,552). Randomization was conducted using an interactive response technology system and stratified by menopausal status (postmenopausal versus premenopausal, with men grouped in the premenopausal category), anatomical stage (II versus III), prior adjuvant or neoadjuvant chemotherapy (yes versus no), and geographical region (North America, Western Europe, and Oceania versus rest of the world). Eligible patients were recruited in 393 centres in 20 countries. The study enrolled 171 patients across 23 sites in Canada.
The NATALEE trial consisted of 3 phases: screening, treatment, and follow-up (Figure 1). The purpose of the screening phase was to assess the inclusion and exclusion criteria and to perform all screening procedures. In the screening phase, patients were screened for eligibility, confirmed by the investigator within 28 days before randomization. The treatment phase consisted of 28-day treatment cycles for up to 60 months plus 30 days of safety follow-up.
Figure 1: NATALEE Study Design
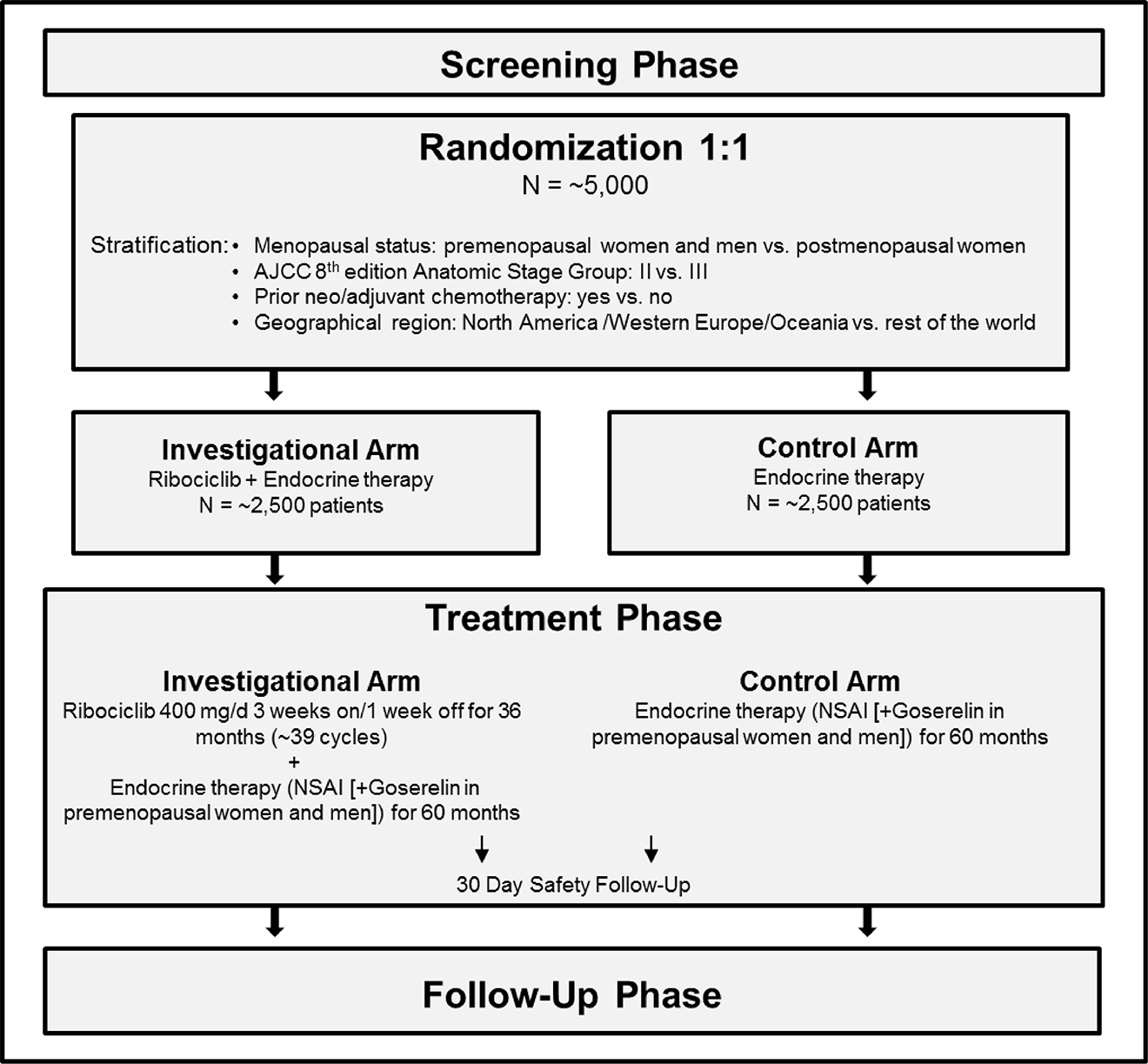
AJCC = American Joint Committee on Cancer; d = day, NSAI = nonsteroidal aromatase inhibitor; vs. = versus.
Source: NATALEE trial statistical analysis plan.45
Three interim analyses were planned after approximately 200, 350, and 425 iDFS events of the approximately 500 targeted iDFS events (i.e., at approximately 40%, 70% and 85% information fractions, respectively) had been documented. The final iDFS analysis was performed when approximately 500 iDFS events had been documented. The end of ribociclib analysis was performed at 603 iDFS events when all patients in the ribociclib plus AI group discontinued ribociclib. Although this CDA-AMC report primarily focuses on the results from the end of ribociclib analysis with a data cut-off date of April 29, 2024, it also reports the results for the primary outcome, iDFS, at IA3 (data cut-off date: January 11, 2023), at which time the NATALEE trial met its primary end point.
Populations
Inclusion and Exclusion Criteria
Eligible patients included men and premenopausal or postmenopausal women who were 18 years of age or older and had histologically confirmed HR-positive, HER2-negative EBC, according to local assessment. Patients were required to have stage II or III disease based on anatomical stage according to the AJCC Cancer Staging Manual, 8th Edition.29 Patients with stage IIA disease were eligible if they had at least 1 lymph node involved. Patients who had no nodal involvement and a grade 2 tumour with a Ki-67 proliferation index of at least 20% or who were classified in a high genomic risk group were also eligible. Patients with stage IIA disease with no nodal involvement and grade 3 tumours were also eligible. All patients with stage IIB or III disease were allowed to participate in the trial regardless of the nodal status. Patients who had received a previous CDK4/6 inhibitor or who had clinically significant uncontrolled heart disease, cardiac repolarization abnormalities, or both, were ineligible.
Interventions
Patients were randomized 1:1 to receive ribociclib (400 mg orally once daily on days 1 to 21 of a 28-day cycle for 36 months from the randomization date plus AI continuously) or AI alone continuously. The AI dosage could either be: letrozole (2.5 mg orally once daily) or anastrozole (1 mg orally once daily) for postmenopausal women or, alternatively, letrozole (2.5 mg orally once daily) or anastrozole (1 mg orally once daily) plus goserelin (3.6 mg subcutaneously once every 4 weeks) for men and premenopausal women. The duration of AI was 60 months starting from the randomization date. Ribociclib and AI were administered daily at the same time with or without food. AI crossovers were not allowed in the study unless there was intolerable toxicity, any important medical event that necessitated a change, or per patient’s request. Patients were discontinued from the ribociclib or ET treatment arms for any of the following reasons:
completion of 36 months of ribociclib treatment (approximately 39 cycles) or 60 months of ET treatment from the randomization date
first recurrence (any of the following or a combination of local, regional, or distant recurrences or contralateral invasive breast cancer, or second primary nonbreast invasive cancer)
adjustments to study treatment due to toxicity that results in treatment discontinuation
interruption of ribociclib dosing for greater than 28 days due to ribociclib-related toxicity
withdrawal of consent by the patient
lost to follow-up or death.
Patients in both groups had safety evaluations performed 30 days after the last dose of treatment.
Dose reduction or interruptions of ribociclib were authorized to allow patients to continue treatment. Patients were permitted to have 1 dose reduction (200 mg) if the recommended dosing schedule was not well tolerated, to allow patients to continue ribociclib. Ribociclib could also be temporarily interrupted until conditions were satisfied, as described in the study protocol. Discontinuation of ribociclib was mandatory when dosing was interrupted for more than 28 days due to ribociclib-related toxicity. For hepatic toxicities, if toxicity recurred after a dose reduction, or if recovery up to a grade 1 AE (or to baseline grade, depending on AE type) took more than 28 days, discontinuation of ribociclib was also required. In the case of a second dose reduction to manage AEs related to ribociclib, the treatment was discontinued.
Medications required to treat AEs or manage cancer symptoms or concurrent diseases, and supportive-care drugs such as pain medications, antiemetics, and antidiarrheals, were allowed during the trial. Prohibited medications during ribociclib included strong inhibitors or inducers of cytochrome P450 3A4/3A5, substrates of cytochrome P450 3A4/3A5 with a narrow therapeutic index, medications with a known risk for QT prolongation and/or torsades de pointes, and concomitant tamoxifen or toremifene use.
Outcomes
A list of efficacy end points assessed in this review is provided in Table 6, followed by descriptions of the outcome measures. The summarized end points are based on outcomes included in the sponsor’s Summary of Clinical Evidence as well as any outcomes identified as important to this review by the clinical experts consulted for this review, and input from patient and clinician groups and public drug plans. Using the same considerations, end points that were considered to be the most relevant to inform expert committee deliberations were selected and finalized in consultation with members of the expert committee. All summarized efficacy end points were assessed using GRADE. Select notable harms outcomes considered important for informing expert committee deliberations were also assessed using GRADE.
Table 6: Outcomes Summarized From the NATALEE Trial
Outcome measure | Time point | NATALEE trial |
|---|---|---|
Efficacy outcomes | ||
Invasive disease–free survival | 36, 48, 60 months | Primarya |
Recurrence-free survival | 48, 60 months | Secondary |
Distant disease–free survival | 48, 60 months | Secondary |
Overall survival | 48, 60 months | Secondary |
Health-related quality of life | ||
Change from baseline in GHS and QoL subscale score of the EORTC QLQ-C30 | 34 months | Secondary |
Safety outcomes | ||
Hepatobiliary toxicity | NR | Secondary |
QT interval prolongation | NR | Secondary |
ILD or pneumonitis | NR | Secondary |
EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; GHS = global health status; ILD = interstitial lung disease; NR = not reported; QoL = quality of life.
aStatistical testing for invasive disease–free survival was adjusted for multiple comparisons across multiple interim analyses.
Sources: NATALEE Clinical Study Reports for the end of ribociclib analysis (2024).17 Details included in the table are from the sponsor’s Summary of Clinical Evidence.21
Efficacy Outcomes
This report primarily focused on the results from the end of ribociclib analysis with a data cut-off date of April 29, 2024 (median duration of follow-up, 44.2 months; range, 0 to 63 months). Results for the primary outcome, iDFS, and for HRQoL were reported at IA3 (data cut-off date: January 11, 2023; median duration of follow-up, 27.7 months; range, 0 to 45 months).
Invasive Disease–Free Survival
The primary efficacy end point of the NATALEE trial was iDFS, which was defined as the time from randomization to the first event of local invasive disease recurrence in the breast, regional invasive recurrence, distant recurrence, death (any cause), contralateral invasive breast cancer, or second primary nonbreast invasive cancer (excluding basal and squamous cell carcinomas of the skin), as defined by the Standardized Definitions for Efficacy End Points criteria:46
Local invasive breast cancer recurrence or ipsilateral invasive breast tumour recurrence. Invasive breast cancer involving the same breast as the original primary tumour. Ductal and lobular carcinoma in situ and tumours in the contralateral breast and/or contralateral lymph nodes are not considered a local invasive recurrence.
Local recurrence has to be confirmed histologically. Histological type of local invasive breast cancer recurrence should be recorded in the case report form.
Regional breast cancer recurrence. Invasive breast cancer in the ipsilateral axilla, regional lymph nodes (all levels), chest wall, or skin of the ipsilateral breast. A tumour in the contralateral breast is not considered a regional recurrence.
Regional recurrence has to be confirmed histologically (preferred) or cytologically. The specific site of regional recurrence should be recorded in the case report form.
Distant recurrence. Distant metastasis of breast cancer (bones, distant lymph nodes, internal organs, central nervous system, bone marrow, and so forth) or any areas of invasive breast cancer recurrence that are not local or regional.
Distant recurrence has to be confirmed histologically (preferred) or cytologically unless there is an unacceptable risk to the patient due to the procedure.
If bone metastases were identified by a bone scan, this must be confirmed histologically (preferred) or radiographically (CT, MRI or fluorodeoxyglucose [FDG]-PET-CT) if biopsy confirmation is not possible.
Metastases in the central nervous system must be confirmed histologically (preferred), cytologically or radiographically (CT or MRI, both with IV contrast material) if biopsy confirmation is not possible.
All other sites of metastases must be confirmed histologically (preferred) or cytologically unless there is an unacceptable risk to the patient due to the procedure.
Location of the first 3 most dominant distant metastases should be reported in the case report form, with preference given to visceral (including brain) metastases, then to bone and then to distant lymph node or skin metastases.
Contralateral invasive breast cancer. Any invasive breast cancer in the contralateral breast with or without contralateral lymph node involvement. Contralateral invasive breast cancer has to be confirmed histologically. In situ or noninvasive contralateral breast cancers are not included in contralateral invasive breast cancers. Histological type and the specific site of contralateral invasive breast cancer should be recorded in the case report form.
Second primary nonbreast invasive cancer. Second primary nonbreast invasive cancer has to be confirmed histologically. In situ or noninvasive cancers and basal or squamous cell carcinomas of the skin are not considered second primary nonbreast invasive cancers. Histological type and the location of the second primary nonbreast invasive cancer should be recorded in the case report form.
Recurrences were detected by scheduled or unscheduled clinical evaluations. Suspected recurrence had to be evaluated by medical imaging and confirmed histologically (or cytologically, when applicable) unless there was an unacceptable risk to the patient or was otherwise specified, according to the study protocol.
Patients who did not have an iDFS event were censored at the last assessment before the data cut-off date (January 11, 2023, for IA3; April 29, 2024, for the end of ribociclib analysis). Any recurrence documented after the initiation of new antineoplastic therapy was considered for the primary analysis, provided that recurrence assessments continued after initiation of new cancer therapy. Discontinuation of treatment due to clinical suspicion of recurrence without histological, cytological, or imaging confirmation was not considered an iDFS event nor a censoring event, which indicated that patients who discontinued treatment were followed up, and treatment discontinuation did not lead to censoring. Detailed measurement properties of iDFS are summarized in Table 7.
Recurrence-Free Survival
RFS was a secondary end point of the NATALEE trial and was defined as the time from randomization to the first event of local invasive disease recurrence in the breast, regional invasive recurrence, distant recurrence, or death (any cause) as described in the previous section. Patients who did not have an RFS event were censored at the last assessment before the data cut-off date for the end of ribociclib analysis.
Distant Disease–Free Survival
Distant disease–free survival was a secondary end point of the NATALEE trial and was defined as the time from randomization to the first event of distant recurrence, death (any cause), or second primary nonbreast invasive cancer (excluding basal and squamous cell carcinomas of the skin), as described in the iDFS section. Patients who did not have a DDFS event were censored at the last assessment before the data cut-off date for the end of ribociclib analysis.
Overall Survival
OS was a secondary end point of the NATALEE trial and was defined as the time from randomization to death due to any cause. If a patient was not known to have died, then OS was censored at the latest date the patient was known to be alive (on or before the data cut-off date for the end of ribociclib analysis).
Health-Related Quality of Life
HRQoL was measured using the EORTC QLQ-C30 and was a secondary end point in the NATALEE trial. The EORTC QLQ-C30 contains 30 items and is composed of both multi-item scales and single-item measures. These include 5 functional scales (physical, role, emotional, cognitive, and social functioning), 3 symptom scales (fatigue, nausea and/or vomiting, and pain), 6 single items (dyspnea, insomnia, appetite loss, constipation, diarrhea, and financial impact) and a global health status and quality of life subscale score.47 All of the scales and single-item measures range in score from 0 to 100. A high functional scale score represents a higher score for a healthy level of functioning; a high score for the global health status and quality of life subscale represents a high quality of life, but a high score for a symptom scale and item represents a high level of symptomatology and problems. All scoring followed the scoring procedures defined by the EORTC QLQ-30 scoring manual. Currently, there is no established minimal important difference in patients with EBC. A change of at least 10 points for improvement and −10 points for deterioration in the global health status and quality of life subscale score was considered meaningful in patients with advanced breast cancer.48 Detailed measurement properties of the EORTC QLQ-C30 are summarized in Table 7.
Safety Outcomes
In the NATALEE trial, AEs were coded using the Medical Dictionary for Regulatory Activities and graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events (Version 4.03). An AE was defined as the appearance of (or worsening of any pre-existing) undesirable sign(s), symptom(s), or medical condition(s). Notable harms from the NATALEE trial included hepatobiliary toxicity, myelosuppression, infections, QT interval prolongation, renal toxicity, pulmonary toxicity (interstitial lung disease or pneumonitis), reproductive toxicity, and second malignancy. Among them, hepatobiliary toxicity, QT interval prolongation, and pulmonary toxicity (interstitial lung disease or pneumonitis) were selected for GRADE assessment because they were considered important by the clinical experts consulted for this review.
Table 7: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
EORTC QLQ-C30 | The EORTC QLQ-C30 is a standardized patient self-administered questionnaire designed to assess HRQoL of adult patients with cancer.47 It consists of 30 items divided into several domains and scales: 5 functional scales (physical, role, cognitive, emotional, and social functioning), 3 symptom scales (fatigue, pain, nausea and vomiting), 6 single-item scales (dyspnea, insomnia, appetite loss, constipation, diarrhea, and financial difficulties), and 1 GHS and QoL scale.47 Each item is scored on a 4-point Likert scale ranging from “not at all” to “very much,” except for the GHS and QoL scale, which uses a 7-point scale. Scores are then converted to a 0 to 100 scale with higher scores indicating better functioning or worse symptoms, or better quality of life, depending on the scale.47,49,50 | Measurement properties of validity, reliability, and responsiveness have not been assessed in patients with EBC. | No MID has been established for patients with EBC. In patients with advanced breast cancer, MIDs for within-group change ranged from 5 to 14 points (improvement) and −14 to −4 points (deterioration). MIDs for between-group change ranged from 4 to 11 points (improvement) and from −18 to −4 points (deterioration). In the GHS and QoL subscale, using the anchor-based method, the estimated MID for within-group change was 12 points for improvement and −8 points for deterioration; the estimated between-group difference in change was 10 points for improvement and −10 points for deterioration.48 |
iDFS | In the NATALEE trial, iDFS was defined as the time from randomization to the first event of local invasive disease recurrence in the breast, regional invasive recurrence, distant recurrence, death (any cause), contralateral invasive breast cancer, or second primary nonbreast invasive cancer (excluding basal and squamous cell carcinomas of the skin).46 | Measurement properties of iDFS in terms of reliability and responsiveness have not been assessed in patients with EBC. Untch et al. (2024) assessed the validity of DFS as a surrogate for OS in patients with HR-positive, HER2-negative EBC. The Spearman coefficient was 0.81 (unweighted) and 0.81 (weighted) between OS and DFS, respectively. It was estimated that 84% of the variability in OS was explained by DFS differences.51 | Not identified for patients with EBC. |
DFS = disease-free survival; EBC = early breast cancer; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; GHS = global health status; HR = hormone receptor; HRQoL = health-related quality of life; iDFS = invasive disease–free survival; MID = minimal important difference; OS = overall survival; QoL = quality of life; STEEP = Standardized Definitions for Efficacy End Points.
Statistical Analysis
A summary of the statistical analysis of the efficacy end points from the NATALEE trial is provided in Table 8.
Sample Size and Power Calculation
The enrolment of patients in the NATALEE trial was based on an expected distribution of 40% of patients having stage II disease and 60% of patients having stage III disease. The 5-year iDFS probability for patients with stage II (excluding low-risk patients) and stage III disease was assumed to be approximately 79% and 72%, respectively, based on data from a retrospective study assessing the prognostic effect of Ki-67 and other disease characteristics in patients with HR-positive, HER2-negative EBC, and data for patients with 4 or more positive lymph nodes treated with AIs in the Early Breast Cancer Trialists’ Collaborative Group meta-analysis.52,53 As a result, the expected overall 5-year iDFS of the control group would be approximately 74.8%. A total of 500 iDFS events would provide approximately 93% and 85% power to reject the null hypothesis of no difference under alternative hypotheses in which the overall hazard ratio is 0.73 and 0.76, respectively. The calculation is based on a 1-sided log-rank test at the overall 2.5% level of significance, patients randomized to the 2 treatment arms in a 1:1 ratio, and a 4-look group sequential design with a Lan-DeMets alpha-spending function and a Lan-DeMets beta-spending function. The final iDFS analysis was planned after approximately 500 documented iDFS events, regardless of the results of the interim analyses. Assuming that enrolment would continue at a rate of approximately 170 patients per month and assuming a 15% dropout rate by the time of the final iDFS analysis, a total of 5,000 patients would need to be randomized to observe the targeted 500 iDFS events at about 44 months after the randomization date of the first patient.
Primary Outcome Analysis
Statistical Test or Model
The primary analysis of iDFS was based on the full analysis set (FAS) and included all data observed up to the cut-off date (January 11, 2023, for IA3; April 29, 2024, for the end of ribociclib analysis). The Kaplan-Meier estimator was used to estimate the distribution of iDFS within each treatment group. A difference in iDFS events across groups was compared using a stratified log-rank test at a 1-sided 2.5% level of significance using the same stratification factors used for randomization. Estimates of hazard ratios were calculated, along with their 95% CIs, using a Cox model stratified by the same stratification factors used for randomization.
Interim and Final Analyses
Three interim analyses were planned after approximately 200, 350, and 425 iDFS events of the approximately 500 targeted iDFS events (i.e., at approximately 40%, 70% and 85% information fractions, respectively) had been documented. These interim analyses were expected to occur around 27, 35, and 39 months from the date the first patient was randomized in the study. An alpha-spending function according to a 3-look group sequential design with a stopping boundary (O’Brien-Fleming type) was used to construct the efficacy stopping boundaries.54 A Lan-DeMets stopping boundary (O’Brien-Fleming type) was used as a beta-spending function to determine the nonbinding futility boundary. The choice of nonbinding nature of the futility-stopping boundary ensures that the efficacy stopping boundaries are not affected. Based on the choice of alpha-spending function described previously, the futility boundary expressed on the P value at the first interim analysis was calculated as P = 0.6752. At the time of the second interim analysis, the efficacy boundary was calculated as P = 0.0074. Similarly, if the third interim analysis was performed exactly at 425 iDFS events, the efficacy boundary was calculated as P = 0.0129. The final iDFS analysis was performed when approximately 500 iDFS events had been documented. If exactly 200, 350, and 425 events were observed at the interim analyses, the trial continued, and if exactly 500 events were obtained at the final analysis, the observed P value had to be less than 0.0202 to conclude statistical significance. The boundary for the final analysis was derived accordingly from the prespecified alpha-spending function such that the overall significance level across all analyses was maintained at 0.025. The end of ribociclib analysis was performed at 603 iDFS events when all patients in the ribociclib plus AI group discontinued ribociclib. Of note, all analyses performed after the final iDFS analysis (i.e., the end of ribociclib analysis) were exploratory.
Multiple-Testing Procedure
The primary end point of iDFS was tested multiple times and stopping boundaries from the group sequential design were calculated to maintain an overall type I error rate of alpha, as described in the previous section. No adjustment for multiple comparisons was planned for secondary and exploratory end points.
The NATALEE trial met its primary end point at IA3 (data cut-off date: January 11, 2023). This report describes the results of the primary end point of iDFS in IA3 because it was inferentially tested, and all other results use the data from the end of ribociclib analysis (data cut-off date: April 29, 2024); because it included more patients and had a longer follow-up time compared to IA3, HRQoL results were available only at IA3. Of note, the 95% CIs and P values were not adjusted for multiplicity for outcomes in the end of ribociclib analysis.
Data Imputation Methods
There were no data imputation methods reported for missing data in the primary analysis of iDFS in the NATALEE trial.
Subgroup Analyses
Subgroup analyses were performed on the primary efficacy end point of iDFS in the FAS population. The subgroups analyses were not controlled for multiplicity. The following subgroup analyses were conducted:
nodal status (N0 versus N1-N3)
menopausal status (postmenopausal versus premenopausal, with men grouped in the premenopausal category)
anatomical stage (II versus III)
prior neoadjuvant or adjuvant chemotherapy (yes versus no)
region (North America, Western Europe, and Oceania versus rest of world).
Among them, the subgroups based on nodal status, menopausal status, anatomical stage, and prior neo- or adjuvant chemotherapy were considered important based on input from the clinical experts consulted by CDA-AMC.
Sensitivity Analyses
In the NATALEE trial, the following 9 sensitivity analyses were performed to evaluate the robustness of the primary end point of iDFS:
Based on per-protocol analysis set.
Stratification per case report form.
Unstratified log-rank test and Cox model.
Stratified Cox model adjusting for baseline covariates.
Using the FAS and excluding an event if it occurred more than 28 weeks after the last assessment (i.e., after at least 2 missing assessments during the first 24 months or after at least 1 missing assessment after the first 24 months).
Using the FAS and backdating events that occurred after 1 or more missing assessments. The date of such an event was changed to the date of the earliest missed scheduled assessment date preceding its detection. This means that, when 1 or more assessments were missed leading up to the event, the event date was backdated to the last assessment date that was missed before the event was captured.
Using the FAS and censoring an iDFS event at the date of the last assessment before the start of new anticancer therapy if no iDFS event was observed before the start of new antineoplastic therapy.
Using the FAS and considering treatment discontinuation due to disease recurrence without a confirmed recurrence event.
Using the FAS and excluding the death due to COVID-19. The iDFS was censored at the last adequate assessment before the COVID-19 death.
Secondary Outcome Analysis
The efficacy analysis of the secondary outcomes was conducted among patients in the FAS population. The following statistics were used to summarize RFS, DDFS, and OS. The hazard ratios were calculated, along with their 95% CIs, using a Cox model stratified by the same stratification factors used for randomization. The survival curve distribution of RFS, DDFS, and OS were compared between the 2 treatment groups using a stratified log-rank test at a 1-sided 2.5% level of significance using the same stratification factors used for randomization.
HRQoL was measured using change from baseline for the global health status and quality of life subscale score of the EORTC QLQ-C30 and presented based on results from IA3 (data cut-off date: January 11, 2023). A repeated-measures model for longitudinal data was used to estimate differences in the global health status and quality of life subscale between treatment groups. The modelling was done on the actual score. This repeated-measure model included terms for treatment, the stratification factors assigned at randomization, time, baseline value as main effects, and an interaction term for treatment by time. This analysis was restricted to patients with an evaluable baseline score and at least 1 evaluable postbaseline score. All data collected until confirmation of first recurrence (including the assessment at confirmation of first recurrence) were included in the analysis. Time was considered a continuous variable expressed in months. This analysis only included assessments up to the time point where there were at least 50 patients on each of the treatments.
Safety Outcome Analysis
All safety analyses were based on the safety analysis set, unless otherwise specified. Descriptive statistics were calculated for continuous safety variables and frequency counts and percentages were tabulated for categorical safety variables.
Table 8: Statistical Analysis of Efficacy End Points
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
iDFS |
|
| iDFS was censored at the date of the last assessment if no relevant event was observed before or on the analysis cut-off date. If no postbaseline recurrence assessments were available and no had death occurred, then the date of randomization was used. |
|
RFS | Same approach as previously described for iDFS | Same factors as previously described for iDFS | Same strategy as previously described for iDFS | NR |
DDFS | Same approach as previously described for iDFS | Same factors as previously described for iDFS | Same strategy as previously described for iDFS | NR |
OS | Same approach as previously described for iDFS | Same factors as previously described for iDFS | Patients were censored at the last contact date. Patients not known to have died were censored for “lost to follow-up” if the time between their last contact date and the analysis cut-off date was longer than 24 weeks + 4 weeks = 182 days. |
|
Change from baseline in global health status and quality of life subscale score of the EORTC QLQ-C30 | Repeated-measures model for longitudinal data | Stratification factors assigned at randomization | Missing items data in a scale were handled based on each instrument manual. No imputation was applied if the total or subscale scores were missing at a visit. For the repeated-measures analysis, patients with a baseline and at least 1 nonmissing postbaseline assessment were included. All available data were used in the repeated-measures models for longitudinal data, which assume the missing scores at any time point were missing at random. | NR |
CRF = case report form; DDFS = distant disease–free survival; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; iDFS = invasive disease–free survival; NR = not reported; OS = overall survival; RFS = recurrence-free survival; vs. = versus.
Source: NATALEE trial statistical analysis plan.45 Details included in the table are from the sponsor’s Summary of Clinical Evidence.21
Analysis Populations
A summary of analysis populations used in the NATALEE trial that are relevant to this review is provided in Table 9.
Table 9: Analysis Populations of the NATALEE Trial
Population | Definition | Application |
|---|---|---|
FAS | All patients to whom study treatment had been assigned by randomization. According to the intention-to-treat principle, patients were analyzed according to the treatment and strata to which they had been assigned during the randomization procedure. | All efficacy analyses |
Per-protocol analysis set | A subset of patients in the FAS who were compliant with the requirements of the protocol. | Sensitivity analyses |
Safety analysis set | Included all randomized patients who received any study treatment (i.e., at least 1 dose of ribociclib or AI). Patients were analyzed according to the study treatment received. | Safety analyses |
AI = aromatase inhibitor; FAS = full analysis set.
Source: NATALEE statistical analysis plan.45 Details included in the table are from the sponsor’s Summary of Clinical Evidence.21
Major Protocol Deviations
A summary of major protocol deviations in the NATALEE trial at IA3 is provided in Table 10. There were 29 patients (1.1%) in the ribociclib plus AI group and 18 patients (0.7%) in the AI alone group who reported major protocol deviations leading to exclusion from analysis sets. The most commonly reported major protocol deviation included inclusion and/or exclusion criteria not being met (1.1% versus 0.7% for ribociclib plus AI versus AI alone), for which 26 patients were excluded because they did not meet the anatomical stage requirement for the study; 18 patients were excluded because their HER2 status was unavailable; and 2 patients were excluded for having metastatic disease at study entry.
Table 10: Summary of Major Protocol Deviation From the NATALEE Trial (FAS; Data Cut-Off Date — January 11, 2023)
Major protocol deviations | NATALEE trial | |
|---|---|---|
Ribociclib + AI (N = 2,549) | AI alone (N = 2,552) | |
Patients with major protocol deviations, n (%) | 29 (1.1) | 18 (0.7) |
Inclusion or exclusion criteria not being met | 29 (1.1) | 17 (0.7) |
Received a wrong treatment | 0 | 1 (< 0.1) |
AI = aromatase inhibitor; FAS = full analysis set.
Source: NATALEE Clinical Study Report for interim analysis 3 (2023).
Results
Although this report primarily focuses on the results from the end of ribociclib analysis, which had a data cut-off date of April 29, 2024, it also reports the results for the primary outcome, iDFS, at IA3 (data cut-off date: January 11, 2023), at which time the NATALEE trial met its primary end point. This report also includes results from the 5-year follow-up analysis with a data cut-off date of May 28, 2025.
Patient Disposition
Patient disposition in the NATALEE trial is summarized in Table 11. At the time of the end of ribociclib analysis, a total of 6,068 patients were screened and 638 patients (10.5%) were excluded, with withdrawal by patient being the most reported reason (203 of 638 patients; 31.8%). A total of 5,101 patients were randomized: 2,549 patients to the ribociclib plus AI group and 2,552 patients to the AI alone group. Generally, a lower proportion of patients in the ribociclib plus AI group were randomized but not treated (0.9% versus 4.3% for ribociclib plus AI versus AI alone) or discontinued from the study (17.1% versus 22.1%) compared with the AI alone group. Fewer patients in the ribociclib plus AI group discontinued from the study compared to the AI alone group: 450 patients (17.7%) versus 565 patients (22.1%); the primary reason for study discontinuation was withdrawal by patient (11.0% versus 15.2%). Completion of the treatment (62.8%) was the most reported reason for ribociclib discontinuation followed by AEs (20.0%). A lower proportion of patients in the ribociclib plus AI group discontinued the AI portion of treatment (28.7% versus 31.9%), with the most reported reason for AI discontinuation being disease relapse (7.7% versus 10.5%). Two patients in the AI alone group received ribociclib but subsequently discontinued its use. Similar patient disposition was observed in the 5-year follow-up analysis (data cut-off date: May 28, 2025), but all patients were no longer receiving treatment with ribociclib. More patients discontinued the AI component of the regimens (72.0% and 71.9%), but 25.4% (1,295 out of 5,101) were still receiving AI treatment.
Based on the final iDFS analysis safety analysis set (data cut-off date: July 21, 2023), 1,091 patients (43.2%) completed the 3-year treatment duration of ribociclib and 1,752 patients (69.4%) completed at least 2 years of ribociclib treatment. At the time of the end of ribociclib analysis, 1,601 patients (62.8%) completed treatment with ribociclib.
Baseline Characteristics
The baseline characteristics outlined in Table 12 are limited to those that are most relevant to this review or were felt by the review team to affect the outcomes or interpretation of the study results. Baseline characteristics in the NATALEE trial were reported in the sponsor’s Clinical Study Report for IA3. Demographic and baseline characteristics were well balanced between treatment groups. The median age of all study patients was 52.0 years, with a range of 24 to 90 years. The NATALEE trial enrolled mostly women (99.6%) who were postmenopausal (55.8%) and had an ECOG Performance Status score of 0 (83.1%). More than half (59.6%) of patients had disease that was AJCC stage III, nearly half of patients (41.2%) had 1 to 3 positive ALNs (i.e., N1 classification), and most patients (88.1%) received prior adjuvant or neoadjuvant chemotherapy. Genomic risk was used to determine the eligibility for patients with stage IIA disease with grade 2 tumours. There were 676 patients (13.3%) who had genomic tests; Oncotype DX was the most reported test (249 [4.9%] of all patients).
Table 11: Summary of Patient Disposition From the NATALEE Trial (FAS)
Patient disposition | NATALEE (April 29, 2024, data cut-off) | NATALEE (May 28, 2025, data cut-off) | ||
|---|---|---|---|---|
Ribociclib + AI (N = 2,549) | AI alone (N = 2,552) | Ribociclib + AI (N = 2,549) | AI alone (N = 2,552) | |
Screened, N | 6,068 | |||
Not eligible after screening, n (%) | 638 (10.5) | |||
Withdrawal by patienta | 203 (31.8) | |||
Adverse eventa | 4 (0.6) | |||
Progressive diseasea | 5 (0.8) | |||
Lost to follow-upa | 2 (0.3) | |||
Othera | 115 (18.0) | |||
Randomized, N (%) | 2,549 (100) | 2,552 (100) | 2,549 (100) | 2,552 (100) |
Randomized but not treated, n (%) | 23 (0.9) | 111 (4.3) | 23 (0.9) | 111 (4.3) |
██████████ ██ ███████ | ██ █████ | ███ ███ | ██ | ██ |
████████ █████████ | █████ | ██████ | ██ | ██ |
█████████ | ██████ | || | ██ | ██ |
█████████ ████████ | █████ | ██████ | ██ | ██ |
Lost to follow-up | 0 | 1 (< 0.1) | NR | NR |
Other | 2 (0.1) | 0 | NR | NR |
Discontinued from study, n (%) | 450 (17.7) | 565 (22.1) | 549 (21.5) | 664 (26.0) |
Withdrawal by patient | 281 (11.0) | 388 (15.2) | 334 (13.1) | 437 (17.1) |
Death | 105 (4.1) | 121 (4.7) | 138 (5.4) | 162 (6.3) |
Lost to follow-up | 30 (1.2) | 36 (1.4) | 43 (1.7) | 45 (1.8) |
█████████ ████████ | ██ █████ | ██ █████ | ██ █████ | ██ █████ |
████████ █████████ | ██████ | ██████ | ██████ | ██████ |
█████████ | ███████ | || | ███████ | || |
█████ | ██████ | ██████ | ██████ | ██████ |
Discontinued all treatment components, n (%) | 732 (28.7) | 813 (31.9) | 1,836 (72.0) | 1,836 (71.9) |
Discontinued ribociclib, n (%) | 2,524 (99.0) | 2 (0.1) | 2,524 (99.0) | 2 (0.1) |
Primary reason for ribociclib discontinuation (≥ 5.0% of patients), n (%) | ||||
Completed | 1,601 (62.8) | NA | 1,600 (62.8) | NA |
Adverse event | 509 (20.0) | NA | 511 (20.0) | NA |
Patient decision to discontinue treatment | 135 (5.3) | NA | 134 (5.3) | NA |
Disease relapse | 127 (5.0) | NA | 127 (5.0) | NA |
Discontinued AI, n (%) | 732 (28.7) | 813 (31.9) | 1,836 (72.0) | 1,836 (71.9) |
Primary reason for AI discontinuation (≥ 5.0% of patients in either group), n (%) | ||||
Disease relapse | 196 (7.7) | 267 (10.5) | 228 (8.9) | 307 (12.0) |
Patient decision to discontinue treatment | 172 (6.7) | 153 (6.0) | 245 (9.6) | 220 (8.6) |
Withdrawal by patient | 139 (5.5) | 177 (6.9) | 159 (6.2) | 184 (7.2) |
Adverse event | 136 (5.3) | 124 (4.9) | 147 (5.8) | 131 (5.1) |
Completed | NR | NR | 930 (36.5) | 877 (34.4) |
Still on treatment, n (%) | 1,794 (70.4) | 1,628 (63.8) | 690 (27.1) | 605 (23.7) |
Entered the follow-up phase, n (%) | 380 (14.9) | 469 (18.4) | NR | NR |
FAS, N | 2,549 | 2,552 | 2,549 | 2,552 |
PP, N | 2,496 | 2,422 | NR | NR |
Safety, N | 2,526 | 2,441 | 2,526 | 2,441 |
AI = aromatase inhibitor; FAS = full analysis set; NA = not applicable; NR = not reported; PP = per protocol.
aPercentage calculated based on the number of patients who were screened but not included in the trial (i.e., 638 patients).
Source: NATALEE Clinical Study Report for the end of ribociclib analysis (2024)17 and sponsor-provided additional information (March 13).55 Details included in the table are from the sponsor’s Summary of Clinical Evidence.21
Table 12: Summary of Baseline Characteristics From NATALEE (FAS; Data Cut-Off Date — January 11, 2023)
Characteristic | NATALEE trial | |
|---|---|---|
Ribociclib + AI (N = 2,549) | AI alone (N = 2,552) | |
Demographic | ||
Age (years) | ||
Mean (SD) | 52.9 (10.75) | 52.7 (10.77) |
Median (range) | 52.0 (24 to 90) | 52.0 (24 to 89) |
Gender, n (%) | ||
Men | 11 (0.4) | 9 (0.4) |
Women | 2,538 (99.6) | 2,543 (99.6) |
Race,a n (%) | ||
American Indian or Alaska Native | 4 (0.2) | 3 (0.1) |
Asian | 341 (13.4) | 334 (13.1) |
Black or African American | 42 (1.6) | 47 (1.8) |
Native Hawaiian or other Pacific Islander | 3 (0.1) | 1 (0.0) |
White | 1,876 (73.6) | 1,868 (73.2) |
Other | 145 (5.7) | 172 (6.7) |
Missing | 138 (5.4) | 127 (5.0) |
ECOG Performance Status score, n (%) | ||
0 | 2,106 (82.6) | 2,132 (83.5) |
1 | 440 (17.3) | 418 (16.4) |
Missing | 3 (0.1) | 2 (0.1) |
Menopausal status, n (%) | ||
Men and premenopausal women | 1,125 (44.1) | 1,128 (44.2) |
Postmenopausal women | 1,424 (55.9) | 1,424 (55.8) |
Region, n (%) | ||
Asia | 281 (11.0) | 290 (11.4) |
Europe | 1,505 (59.0) | 1,506 (59.0) |
North America and Australia | 624 (24.5) | 612 (24.0) |
Latin America | 139 (5.5) | 144 (5.6) |
Disease characteristics | ||
Ki-67 on surgical specimen, n (%) | ||
Number of patients contributing to the analysis, n | 1,269 | 1,332 |
> 20% | 452 (17.7) | 468 (18.3) |
Missing | 1,280 (50.2) | 1,220 (47.8) |
ER and PR combination status, n (%) | ||
ER+ and PR+ | 2,172 (85.2) | 2,132 (83.5) |
ER+ and PR– | 359 (14.1) | 392 (15.4) |
ER– and PR+ | 3 (0.1) | 12 (0.5) |
ER+ and PR unknown | 10 (0.4) | 13 (0.5) |
ER unknown and PR+ | 2 (0.1) | 2 (0.1) |
ER unknown and PR– | 1 (< 0.1) | 1 (< 0.1) |
ER unknown and PR unknown | 2 (0.1) | 0 (0.0) |
T stage on surgical specimen, n (%) | ||
TX | 20 (0.8) | 9 (0.4) |
T0 | 56 (2.2) | 52 (2.0) |
Tis | 16 (0.6) | 19 (0.7) |
T1 | 774 (30.4) | 761 (29.8) |
T2 | 1,162 (45.6) | 1,198 (46.9) |
T3 | 427 (16.8) | 422 (16.5) |
T4 | 92 (3.6) | 91 (3.6) |
Missing | 2 (0.1) | 0 |
N stage on surgical specimen, n (%) | ||
NX | 2 (0.1) | 5 (0.2) |
N0 | 378 (14.8) | 418 (16.4) |
N1 | 1,062 (41.7) | 1,039 (40.7) |
N2 | 733 (28.8) | 690 (27.0) |
N3 | 372 (14.6) | 399 (15.6) |
Missing | 2 (0.1) | 1 (< 0.1) |
AJCC anatomical stage, n (%) | ||
Stage 0 | 0 | 0 |
Stage I | 9 (0.4) | 5 (0.2) |
Stage IIA | 479 (18.8) | 521 (20.4) |
Stage IIB | 532 (20.9) | 513 (20.1) |
Stage IIIA | 939 (36.8) | 894 (35.0) |
Stage IIIB | 168 (6.6) | 149 (5.8) |
Stage IIIC | 421 (16.5) | 469 (18.4) |
Stage IV | 0 | 0 |
Missing | 1 (< 0.1) | 1 (< 0.1) |
Histopathological grade on surgical specimen, n (%) | ||
GX | 32 (1.3) | 30 (1.2) |
G1 | 213 (8.4) | 217 (8.5) |
G2 | 1,460 (57.3) | 1,432 (56.1) |
G3 | 684 (26.8) | 702 (27.5) |
Not done | 159 (6.2) | 168 (6.6) |
Missing | 1 (< 0.1) | 3 (0.1) |
Histologic type, n (%) | ||
Invasive ductal carcinoma not otherwise specified | 1,857 (72.9) | 1,881 (73.7) |
Invasive lobular | 455 (17.9) | 450 (17.6) |
Carcinoma medullary | 1 (< 0.1) | 1 (< 0.1) |
Mucinous | 17 (0.7) | 16 (0.6) |
Papillary | 18 (0.7) | 12 (0.5) |
Tubular | 5 (0.2) | 3 (0.1) |
Ductal carcinoma in situ | 1 (< 0.1) | 0 |
Lobular carcinoma in situ | 0 | 0 |
Other | 194 (7.6) | 189 (7.4) |
Missing | 1 (< 0.1) | 0 |
Genomic test, n (%) | ||
Oncotype DX | 120 (4.7) | 129 (5.1) |
MammaPrint | 46 (1.8) | 51 (2.0) |
PAM50 | 38 (1.5) | 29 (1.1) |
EndoPredict | 23 (0.9) | 28 (1.1) |
Other | 109 (4.3) | 103 (4.0) |
Prior adjuvant or neoadjuvant chemotherapy, n (%) | ||
Yes | 2,249 (88.2%) | 2,245 (88.0%) |
No | 300 (11.8%) | 307 (12.0%) |
AI = aromatase inhibitor; AJCC = American Joint Committee on Cancer; ECOG = Eastern Cooperative Oncology Group; ER = estrogen receptor; FAS = full analysis set; G1 = low-grade tumour; G2 = intermediate-grade tumour; G3 = high-grade tumour; GX = tumour grade cannot be assessed; iDFS = invasive disease–free survival; N0 = no nodal involvement; N1 = 1 to 3 positive axillary lymph nodes; N2 = 4 to 9 positive axillary lymph nodes; N3 = ≥ 10 positive axillary lymph nodes; NX = no nodal information available; PR = progesterone receptor; SD = standard deviation; T0 = main tumour cannot be found; T1 = tumour ≤ 2 cm in size across; T2 = tumour > 2 cm but ≤ 5 cm in size across; T3 = tumour > 5 cm in size across; T4 = tumour growing into chest wall or skin; Tis = carcinoma in situ; TX = main tumour cannot be measured.
Note: Baseline characteristics were reported in the NATALEE Clinical Study Report for interim analysis 3 (data cut-off date: January 11, 2024), considering they were not reported in either the final iDFS analysis report or the end of ribociclib analysis report. The FAS remained the same.
aRacial categories used in the table are as reported in the source and may not align with Canada's Drug Agency inclusive language guidelines.
Sources: NATALEE Clinical Study Report for interim analysis 3 (2023). Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Exposure to Study Treatments
At the end of ribociclib analysis, patients in the ribociclib plus AI group had a median duration of exposure of 35.7 months to ribociclib and 45.01 months to AI. In the AI alone group, patients had a median duration of exposure to the AI of 44.7 months (Table 13). Overall, the relative dose intensity for the AI and goserelin was balanced between treatment groups. The mean relative dose intensity for ribociclib was 83.4%, the mean relative dose intensity for the AI was approximately 99% in both treatment groups, and the mean relative dose intensity for goserelin in men and premenopausal women was 101.37% in the ribociclib plus AI group and 100.19% in the AI alone group. A higher proportion of patients in the ribociclib plus AI group (46.7%) experienced AI dose interruption than in the AI alone group (36.8%), primarily due to included AEs (11.6% versus 5.7%). At the 5-year follow-up analysis (data cut-off date: May 28, 2025), the mean duration of exposure was 46.5 months (SD = 19.46) in the ribociclib plus AI group and 44.6 months (SD = 20.64) in the AI alone group.
Table 13: Summary of Patient Exposure From the NATALEE Study (Safety Analysis Set; Data Cut-Off Date — April 29, 2024)
Exposure | Ribociclib + AI (N = 2,526) | AI alone (N = 2,441) |
|---|---|---|
Duration (months) | ||
Ribociclib | ||
Mean (SD) | 26.5 (13.66) | NA |
Median (range) | 35.7 (0 to 37) | NA |
AI | ||
Mean (SD) | 39.5 (16.09) | 38.2 (17.15) |
Median (range) | 45.0 (0 to 60.8) | 44.7 (0 to 60) |
Relative dose intensity (%), mean (SD) | ||
Ribociclib | 83.4 (19.75) | NA |
AI | 99.0 (3.60) | 99.2 (3.89) |
Goserelin | 101.4 (45.63) | 100.2 (18.49) |
Dose interruption, n (%) | ||
Ribociclib | 2,224 (88.0) | NA |
Adverse event | 1,687 (66.8) | NA |
Logistical issue | 906 (35.9) | NA |
AI | 1,179 (46.7) | 899 (36.8) |
Dosing error | 929 (36.8) | 730 (29.9) |
Adverse event | 293 (11.6) | 140 (5.7) |
AI = aromatase inhibitor; NA = not applicable; SD = standard deviation.
Sources: NATALEE Clinical Study Report for the end of ribociclib analysis (2024). Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Prior Therapies
Prior therapies for patients in the NATALEE trial at IA3 are summarized in (Table 14). Prior therapies were balanced between the treatment groups. There were 2,423 patients (95.1%) in the ribociclib plus AI group and 2,439 patients (95.6%) in the AI alone group who received 1 or more prior antineoplastic medications, the most common being prior antineoplastic radiotherapy (89.9% versus 90.2%).
Table 14: Summary of Prior Therapies From the NATALEE Study (FAS; Data Cut-Off Date — January 11, 2023)
Prior therapies | NATALEE study | |
|---|---|---|
Ribociclib + AI (N = 2,549) | AI alone (N = 2,552) | |
Patients with 1 or more prior antineoplastic medications, n (%) | 2,423 (95.1) | 2,439 (95.6) |
Prior chemotherapy, n (%) | ||
Adjuvant chemotherapy | 1,223 (48.0) | 1,220 (47.8) |
Neoadjuvant chemotherapy | 1,085 (42.6) | 1,095 (42.9) |
Prior antineoplastic radiotherapy, n (%) | 2,292 (89.9) | 2,302 (90.2) |
Prior endocrine therapy, n (%) | 1,824 (71.6) | 1,801 (70.6) |
AI = aromatase inhibitor; FAS = full analysis set.
Sources: NATALEE Clinical Study Report for interim analysis 3 (2023). Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Concomitant Medications
A summary of the concomitant medications used in the safety analysis set at the end of ribociclib analysis and the 5-year follow-up analysis is provided in Table 15. Medications required to treat AEs or manage cancer symptoms or concurrent diseases, and supportive-care drugs such as pain medications, antiemetics, and antidiarrheals, were allowed during the NATALEE trial. Overall, concomitant medications were balanced between the treatment groups. In total, 2,370 patients (93.8%) in the ribociclib plus AI group and 2,220 patients (90.9%) in the AI alone group received 1 or more concomitant medications at the end of ribociclib analysis; similar results were observed at the 5-year follow-up analysis. The most commonly used concomitant medications in both groups were bisphosphonates (end of ribociclib analysis: 17.6% versus 18.3%; 5-year follow-up analysis: 17.7% versus 18.4%).
Table 15: Summary of Concomitant Medications From the NATALEE Study (Safety Analysis Set)
Concomitant medications | NATALEE (April 29, 2024, data cut-off) | NATALEE (May 28, 2025, data cut-off) | ||
|---|---|---|---|---|
Ribociclib + AI (N = 2,526) | AI alone (N = 2,441) | Ribociclib + AI (N = 2,526) | AI alone (N = 2,441) | |
Patients with 1 or more concomitant medications, n (%) | 2,370 (93.8) | 2,220 (90.9) | 2,371 (93.9) | 2,225 (91.2) |
Drug affecting bone structure and mineralization, n (%) | NR | NR | NR | NR |
Bisphosphonates | 444 (17.6) | 446 (18.3) | 448 (17.7) | 449 (18.4) |
Bisphosphonates combination | 17 (0.7) | 11 (0.5) | 16 (0.6) | 11 (0.5) |
Other | 75 (3.0) | 88 (3.6) | 78 (3.1) | 91 (3.7) |
Corticosteroids, n (%) | 85 (3.4) | 58 (2.4) | 85 (3.4) | 60 (2.5) |
Corticosteroids for systemic use | 12 (0.5) | 16 (0.7) | 12 (0.5) | 16 (0.7) |
Corticosteroids acting locally | 3 (0.1) | 5 (0.2) | 3 (0.1) | 5 (0.2) |
Antiemetic medications, n (%) | NR | NR | NR | NR |
Serotonin antagonists | 133 (5.3) | 99 (4.1) | 135 (5.3) | 103 (4.2) |
Other antiemetics | 113 (4.5) | 54 (2.2) | 112 (4.4) | 54 (2.2) |
AI = aromatase inhibitor; NR = not reported.
Sources: NATALEE Clinical Study Report for the end of ribociclib analysis (2024). Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Subsequent Treatment
██ ███ ██████ ████████ ███ ██ ███ ███ ██ ██████████ █████████ ██████████ ███████████████ ██████████ ███ ████████ ██ ███ ███████ ████████ ██ ███ ██████████ ████ ██ █████ ███ ███ ███████ ████████ ██ ███ ██ ██████ ████ ██████ █████████ ██ █████ ███████ ████████ ██ ███ ██████████ ████ ██ █████ ███ █████ ███████ ████████ ██ ███ ██ █████ ██ ███ ██████ █████████ ████████ █████ ███████ █████ ███ ███ █████ ██████ ████ ███ ████ ████████ ██████████ ███████████████ ███████████ █████████ ██ ██ █████ ██ ██ ████████ ██ ██████ ██████ ████████ ██ ████ ██ ██████████ █████████ ████ ████████ ██████ █████████ █████ ███ ███████ ██████████████ ████ ██ ██████████ █████████ ████ █████████ ██████ █████████ █████ ███ ████ ██ ██████████ █████████ ████ ███ █████ ██████ █████████ █████████ ████ ███ █████ ████ ████████ █████████ ███████ █████████ ███████.
Table 16: Summary of Subsequent Treatment From the NATALEE Study (Safety Analysis Set)
Subsequent treatment | NATALEE study (April 29, 2024, data cut-off) | NATALEE study (May 28, 2025, data cut-off) | ||
|---|---|---|---|---|
Ribociclib + AI (N = 2,526) | AI alone (N = 2,441) | Ribociclib + AI (N = 2,526) | AI alone (N = 2,441) | |
Patients with at least 1 subsequent antineoplastic medication, n (%) | ███ ██████ | ███ █████ | ████ ████ | ████ █████ |
Commonly reported subsequent antineoplastic medication (≥ 5% of patients in either group), n (%) | ||||
██ | ███ █████ | ███ █████ | ███ █████ | ███ ██████ |
██████████████ | ███ █████ | ███ █████ | ███ █████ | ███ ██████ |
████████████████ ██████ ██████████ | ██ █████ | ███ █████ | ██ █████ | ███ █████ |
Number of patients with at least 1 antineoplastic radiotherapy, n (%) | ██ █████ | ██ █████ | ██ █████ | ██ █████ |
Number of patients with at least 1 antineoplastic surgery, n (%) | ██ █████ | ██ █████ | ██ █████ | ██ █████ |
AI = aromatase inhibitor.
Source: Sponsor-provided additional data.19
Efficacy
Findings for key efficacy outcomes among the FAS population in the NATALEE trial are summarized in Table 17 and Table 18. The NATALEE trial met its primary end point at IA3 (data cut-off date: January 11, 2023). At IA3, the median duration of follow-up was 27.7 months (range, 0 to 45 months). At the time of end of ribociclib analysis (data cut-off date: April 29, 2024), the median duration of follow-up was 44.2 months (range, 0 to 63 months). At the time of the 5-year follow-up analysis (data cut-off date: May 28, 2025), the median duration of follow-up was 55.4 months. Results for iDFS were reported at IA3, the end of ribociclib analysis, and the 5-year follow-up analysis. No formal statistical analysis of secondary outcomes was planned at any time point; only the results for the end of ribociclib analysis and the 5-year follow-up analysis are summarized in detail for RFS, DDFS, and OS. Because no results were available at other time points, only the results for HRQoL at IA3 are presented for this outcome.
Invasive Disease–Free Survival
At IA3, 189 patients (7.4%) in the ribociclib plus AI group and 237 patients (9.3%) in the AI alone group experienced an iDFS event. Distant recurrence (4.7% versus 6.7%) was the most commonly reported iDFS event for both groups (Table 17). A total of 2,360 patients (92.6%) in the ribociclib plus AI group and 2,315 patients (90.7%) in the AI alone group were censored, primarily due to not experiencing the iDFS event (83.7% versus 77.3%). There was a statistically significant improvement in iDFS in the ribociclib plus AI group compared with the AI alone group (hazard ratio = 0.75; 95% CI, 0.62 to 0.91; P = 0.0014), though the median iDFS was not estimable for either treatment group. The Kaplan-Meier probability estimate of iDFS at 36 months was 90.4% (95% CI, 88.6% to 91.9%) for the ribociclib plus AI group, and 87.1% (95% CI, 85.3% to 88.8%) for the AI alone group (Figure 2a). The between-group difference was 3.3% (95% CI, 0.9% to 5.7%).
Results from the planned sensitivity and supplementary analyses were consistent with the primary analysis at IA3. Subgroup analyses of iDFS were also consistent with the primary analysis, except for the subgroup of patients with lobular breast cancer (hazard ratio = 1.05; 95% CI, 0.70 to 1.59). Refer to Appendix 1 for the detailed subgroup analyses data.
At the end of ribociclib analysis, a total of 263 patients (10.3%) in the ribociclib plus AI group and 340 patients (13.3%) in the AI group experienced an iDFS event, with distant recurrence (6.9% for ribociclib plus AI versus 9.6% for AI alone) being the most reported event for both groups. A total of 2,286 patients (89.7%) in the ribociclib plus AI group and 2,212 patients (86.7%) in the AI alone group were censored. The median iDFS was not estimable at the end of ribociclib analysis for either treatment group. The hazard ratio for the ribociclib plus AI group versus the AI alone group was 0.72 (95% CI, 0.61 to 0.84; nominal P < 0.0001), in favour of ribociclib plus AI. The Kaplan-Meier probability estimate of iDFS at 36 months for both groups was consistent with IA3. At 48 months, the Kaplan-Meier probability estimate of iDFS was 88.5% (95% CI, 87.1% to 89.8%) for the ribociclib plus AI group, and 83.6% (95% CI, 81.8% to 85.2%) for the AI alone group. The between-group difference was 4.9% (95% CI, 2.7% to 7.1%) (Figure 2b).
At the 5-year follow-up analysis, the hazard ratio for the ribociclib plus AI group versus the AI alone group was 0.72 (95% CI, 0.62 to 0.83) (Figure 3). At 60 months, the Kaplan-Meier probability estimate of iDFS was 85.5% (95% CI, 83.9% to 87.0%) for the ribociclib plus AI group, and 81.0% (95% CI, 79.2% to 82.7%) for the AI alone group, resulting in a between-group difference of 4.5% (95% CI, 2.1% to 6.9%).
Results for sensitivity analyses were available only at IA3. Subgroup analyses of iDFS at the end of ribociclib analysis were generally consistent with the primary analysis across all prespecified subgroups. Detailed subgroup analyses are reported in Appendix 1.
At the request of CDA-AMC, the sponsor provided additional data on a subset of patients in the NATALEE trial who would not meet the eligibility criteria for cohort 1 of the monarchE trial (Table 28). The results of a subgroup analysis that was based on patient eligibility for cohort 1 in the monarchE trial were used to inform the pharmacoeconomic model submitted to CDA-AMC to represent patients in the NATALEE trial who would or would not be eligible to receive abemaciclib. Of note, this subgroup was not a predefined subgroup in the NATALEE trial. ███ ████ ███████ ██ ████ ██████ ██ ████████ ████ ██████████ ████ ███ ████ ███████ ███ ███ ███ ███ ██████████ ████ ██ ███ ██ ███████ █████ █ ██████ ███ ███ █████ ██ ████████.
Table 17: Summary of Results for iDFS (Primary Outcome) From the NATALEE Trial (FAS)
End points | Interim analysis 3 (January 11, 2023, DCO) | End of ribociclib analysis (April 29, 2024, DCO) | 5-year follow-up (May 28, 2025, DCO) | ||||
|---|---|---|---|---|---|---|---|
Ribociclib + AI N = 2,549 | AI alone N = 2,552 | Ribociclib + AI N = 2,549 | AI alone N = 2,552 | Ribociclib + AI N = 2,549 | AI alone N = 2,552 | ||
Follow-up time, (months), median (range) | 27.7 (0 to 45) | 44.2 (0 to 63) | 55.4 (NR) | ||||
Patients with event, n (%) | 189 (7.4) | 237 (9.3) | 263 (10.3) | 340 (13.3) | 317 (12.4) | 407 (16.0) | |
Distant recurrence | 120 (4.7) | 170 (6.7) | 176 (6.9) | 246 (9.6) | NR | NR | |
Second primary nonbreast invasive cancer | 30 (1.2) | 28 (1.1) | 39 (1.5) | 40 (1.6) | NR | NR | |
Local and/or regional invasive recurrence | 19 (0.7) | 35 (1.4) | 25 (1.0) | 49 (1.9) | NR | NR | |
Death | 13 (0.5) | 7 (0.3) | 17 (0.7) | 11 (0.4) | NR | NR | |
Invasive contralateral breast cancer | 7 (0.3) | 9 (0.4) | 11 (0.4) | 10 (0.4) | NR | NR | |
Invasive ipsilateral breast tumour recurrence | 8 (0.3) | 7 (0.3) | 8 (0.3) | 9 (0.4) | NR | NR | |
Patients whose data were censored, n (%) | 2,360 (92.6) | 2,315 (90.7) | 2,286 (89.7) | 2,212 (86.7) | NR | NR | |
Ongoing without event | 2,134 (83.7) | 1,972 (77.3) | NR | NR | NR | NR | |
Withdrew consent | 213 (8.4) | 324 (12.7) | NR | NR | NR | NR | |
Lost to follow-up | 13 (0.5) | 19 (0.7) | NR | NR | NR | NR | |
iDFS (months), median (95% CI) | NE (NE to NE) | NE (NE to NE) | NE (NE to NE) | NE (NE to NE) | NR | NR | |
Hazard ratio (95% CI) | 0.75 (0.62 to 0.91) | 0.72 (0.61 to 0.84) | 0.72 (0.62 to 0.83) | ||||
P valuea | 0.0014 | Nominal P value < 0.0001 | Nominal P value < 0.0001 | ||||
iDFS event-free probability at 36 months,b % (95% CI) | 90.4 (88.6 to 91.9) | 87.1 (85.3 to 88.8) | 90.8 (89.5 to 91.9) | 88.1 (86.7 to 89.4) | NR | NR | |
Absolute between-group difference,b % (95% CI) | 3.3 (0.9 to 5.7) | 2.7(0.9 to 4.5) | NR | ||||
iDFS event-free probability at 48 months,b % (95% CI) | NE (NE to NE) | NE (NE to NE) | 88.5 (87.1 to 89.8) | 83.6 (81.8 to 85.2) | NR | NR | |
Absolute between-group difference,b % (95% CI) | NE (NE to NE) | 4.9 (2.7 to 7.1) | NR | ||||
iDFS event-free probability at 60 months,b % (95% CI) | NR | NR | NR | NR | 85.5 (83.9 to 87.0) | 81.0 (79.2 to 82.7) | |
Absolute between-group difference,b % (95% CI) | NR | NR | 4.5 (2.1 to 6.9) | ||||
AI = aromatase inhibitor; CI = confidence interval; DCO = data cut-off; FAS = full analysis set; iDFS = invasive disease–free survival; NE = not estimable; NR = not reported; vs. = versus.
Note: Only the P value in interim analysis 3 (data cut-off date: January 11, 2023) for iDFS was adjusted for multiple comparison. P values for all other outcomes were not adjusted for multiplicity.
aOne-sided P value for log-rank test stratified by postmenopausal women vs. men and premenopausal women, anatomical stage group II vs. anatomical stage group III, prior neoadjuvant or adjuvant chemotherapy (yes vs. no) and North America, Western Europe, and Oceania vs. rest of world.
bEstimated by Kaplan-Meier probabilities.
Sources: NATALEE Clinical Study Reports for the end of ribociclib analysis (2024),17 interim analysis 3 (2023),18 and sponsor-provided additional data.19,20 Details included in the table are from the sponsor’s Summary of Clinical Evidence.21
Recurrence-Free Survival
At the end of ribociclib analysis, a total of 224 patients (8.8%) in the ribociclib plus AI group and 298 patients (11.7%) in the AI alone group experienced an RFS event (i.e., local invasive disease recurrence in the breast, regional invasive recurrence, distant recurrence, or death due to any cause). The median RFS was not estimable at the end of ribociclib analysis for either treatment group, though the risk of an RFS event favoured ribociclib plus AI compared with AI alone (hazard ratio = 0.70; 95% CI, 0.58 to 0.83; nominal P < 0.0001); however, no formal comparison was performed at the time of the end of ribociclib analysis (Table 18). Similar results were observed in IA3 (hazard ratio = 0.72; 95% CI, 0.58 to 0.88; nominal P = 0.0008).
███ ████████████ ███████████ ████████ ██ ███ ██ ██ ██████ ███ █████ ████ ███ █████ ██ ██████ ███ ███ ██████████ ████ ██ ██████ ███ █████ ████ ███ █████ ██ ██████ ███ ███ ██ ██████ ███ █████████████ ██████████ ███ ████ ████ ███ ████ ██ ██████ The results were also consistent with IA3. The Kaplan-Meier curve estimate of RFS distribution among the end of ribociclib analysis population is depicted in Figure 4. Similar results were observed at the 5-year follow-up analysis (Table 18 and Figure 4).
Distant Disease–Free Survival
At the end of ribociclib analysis, a total of 240 patients (9.4%) in the ribociclib plus AI group and 311 patients (12.2%) in the AI alone group experienced a DDFS event (i.e., distant recurrence, death due to any cause, or second primary nonbreast invasive cancer). The median DDFS was not estimable at the time of the end of ribociclib analysis for either treatment group, though the risk of a DDFS event favoured ribociclib plus AI compared with AI alone (hazard ratio = 0.72; 95% CI, 0.60 to 0.85; nominal P < 0.0001); however, no formal comparison was performed at the time of the end of ribociclib analysis. Similar results were observed in IA3 (hazard ratio = 0.74; 95% CI, 0.60 to 0.91; nominal P = 0.0017). ███ ████████████ ███████████ ████████ ██ ████ ██ ██ ██████ ███ █████ ████ ███ █████ ██ ██████ ███ ███ ██████████ ████ ██ ██████ ███ █████ ████ ███ █████ ██ ██████ ███ ███ ██ ██████ the between-group difference was 4.5% ████ ███ ████ ██ █████ (Table 18). The Kaplan-Meier curve estimate of the DDFS event-time distribution is depicted in Figure 5. Similar results were observed at the 5-year follow-up analysis (Table 18 and Figure 5).
Overall Survival
At the end of ribociclib analysis, a total of 105 patients (4.1%) in the ribociclib plus AI group and 121 patients (4.7%) in the AI alone group had died. The median OS was not estimable for either treatment group, and there was no difference in the risk of death between ribociclib plus AI compared with AI alone (hazard ratio = 0.83; 95% CI, 0.64 to 1.07; nominal P = 0.0766). However, no formal comparison was performed at the time of the end of ribociclib analysis. Similar results were observed in IA3 (hazard ratio = 0.76; 95% CI, 0.54 to 1.07; nominal P = 0.0563). ███ ████████████ ███████████ ████████ ██ ██ ██ ██ ██████ ███ █████ ████ ███ █████ ██ ██████ ███ ███ ██████████ ████ ██ ██████ ███ █████ ████ ███ █████ ██ ██████ ███ ███ ██ ██████ ███ █████████████ ██████████ ███ ████ ████ ███ ████ █ ██ ██████. The Kaplan-Meier curve estimate of the OS event-time distribution is depicted in Figure 6. Similar results were observed at the 5-year follow-up analysis (Table 18 and Figure 6).
Health-Related Quality of Life
Change From Baseline in Global Health Status and Quality of Life Subscale Score of the EORTC QLQ-C30
Results for HRQoL were not available at the end of ribociclib analysis. At IA3, the mean global health status and quality of life subscale score of the EORTC QLQ-C30 at baseline was 73.7 points (SD = 17.7 points) in the ribociclib plus AI group and 73.8 points (SD = 17.8 points) in the AI alone group. At the end of treatment as of IA3, patients in the ribociclib plus AI group reported an estimated mean decrease (deterioration) from baseline in the global health status and quality of life subscale score of the EORTC QLQ-C30 of –10.40 points (SE = 1.36 points) compared to –10.04 points (SE = 1.29 points) in patients in the AI alone group, the between-group difference was –0.36 points (95% CI, –3.12 to 2.39 points; nominal P = 0.7957). A plot of change from baseline to end of treatment in the global health status and quality of life subscale score of the EORTC QLQ-C30 is provided in Figure 7.
Table 18: Summary of Key Secondary Efficacy and Health-Related Quality of Life (FAS)
Variable | End of ribociclib analysis (April 29, 2024, DCO) | 5-year follow-up (May 28, 2025, DCO) | ||
|---|---|---|---|---|
Ribociclib + AI (N = 2,549) | AI alone (N = 2,552) | Ribociclib + AI (N = 2,549) | AI alone (N = 2,552) | |
RFS | ||||
Number of RFS events, n (%) | 224 (8.8) | 298 (11.7) | 270 (10.6) | 354 (13.9) |
RFS (months), median (95% CI) | NE (NE to NE) | NE (NE to NE) | NR | NR |
Hazard ratio (95% CI) | 0.70 (0.58 to 0.83) | 0.70 (0.60 to 0.82) | ||
Nominal P valuea | < 0.0001 | < 0.0001 | ||
Kaplan-Meier probability estimate of RFS at 48 months, % (95% CI) | ████ ██████ █████ | ████ ██████ █████ | ██ | ██ |
Absolute between-group difference, % (95% CI) | ███ █████ ████ | ██ | ||
Kaplan-Meier probability estimate of RFS at 60 months, % (95% CI) | ██ | ██ | ████ █████ ██ █████ | ████ █████ ██ █████ |
Absolute between-group difference, % (95% CI) | ██ | ███ ████ ██ ████ | ||
DDFS | ||||
Number of DDFS events, n (%) | 240 (9.4) | 311 (12.2) | 289 (11.3) | 375 (14.7) |
DDFS (months), median (95% CI) | NE (NE to NE) | NE (NE to NE) | NR | NR |
Hazard ratio (95% CI) | 0.72 (0.60 to 0.85) | 0.71 (0.61 to 0.83) | ||
Nominal P valuea | < 0.0001 | < 0.0001 | ||
Kaplan-Meier probability estimate of DDFS event-free at 48 months, % (95% CI) | ████ ██████ █████ | ████ ██████ █████ | ██ | ██ |
Absolute between-group difference, % (95% CI) | 4.5 ████ ██ ████ | ██ | ||
Kaplan-Meier probability estimate of DDFS event-free at 60 months, % (95% CI) | ██ | ██ | ████ █████ ██ █████ | ████ █████ ██ █████ |
Absolute between-group difference, % (95% CI) | ██ | ███ ████ ██ ████ | ||
OS | ||||
Number of OS events, n (%) | 105 (4.1) | 121 (4.7) | 138 (5.2) | 162 (6.3) |
OS (months), median (95% CI) | NE (NE to NE) | NE (NE to NE) | NR | NR |
Hazard ratio (95% CI) | 0.83 (0.64 to 1.07) | 0.80 (0.64 to 1.00) | ||
Nominal P valuea | 0.0766 | 0.026 | ||
Kaplan-Meier probability estimate of OS at 48 months, % (95% CI) | ████ █████ ██ █████ | ████ █████ ██ █████ | ██ | ██ |
Absolute between-group difference, % (95% CI) | ███ █████ ██ ████ | ██ | ||
Kaplan-Meier probability estimate of OS at 60 months, % (95% CI) | ██ | ██ | ████ █████ ██ █████ | ████ █████ ██ █████ |
Absolute between-group difference, % (95% CI) | ██ | ███ ████ ██ ████ | ||
Change from baseline in GHS and QoL subscale score of the EORTC QLQ-C30b | ||||
Number of patients contributing to the analysis at the end of treatment, n | 462 | 497 | NR | NR |
Baseline score, mean (SD) | 73.7 (17.7) | 73.8 (17.8) | NR | NR |
Change from baseline at the end of treatment, LS mean (SE) | –10.40 (1.36) | –10.04 (1.29) | NR | NR |
Absolute between-group difference (95% CI) | –0.36 (–3.12 to 2.39) | NR | ||
Nominal P value | 0.7957 | NR | ||
AI = aromatase inhibitor; CI = confidence interval; DCO = data cut-off; DDFS = distant disease–free survival; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; FAS = full analysis set; GHS = global health status; LS = least squares; NE = not estimable; NR = not reported; OS = overall survival; QoL = quality of life; RFS = recurrence-free survival; SD = standard deviation; SE = standard error; vs. = versus.
aThe P value was not adjusted for multiple testing, 1-sided P value for log-rank test stratified by postmenopausal women vs. men and premenopausal women, anatomical stage group II vs. anatomical stage group III, prior neoadjuvant or adjuvant chemotherapy (yes vs. no), and North America, Western Europe, and Oceania vs. rest of world.
bResults for health-related quality of life are from interim analysis 3, with a DCO of January 11, 2023.
Sources: NATALEE Clinical Study Reports for the end of ribociclib analysis (2024),17 interim analysis 3 (2023),18 and sponsor-provided additional data.19,20 Details included in the table are from the sponsor’s Summary of Clinical Evidence.21
Harms
Refer to Table 19 for harms data in the NATALEE trial at the end of ribociclib analysis for the safety analysis set (data cut-off date: April 29, 2024).
Adverse Events
At the end of ribociclib analysis, 2,478 patients (98.1%) in the ribociclib plus AI group and 2,155 patients (88.3%) in the AI alone group experienced at least 1 TEAE. The most common TEAEs (reported by at least 20% of patients in either group) were neutropenia (41.7% versus 2.9%), which was reported more frequently in the ribociclib plus AI group than in the AI alone group, and arthralgia (38.8% versus 44.4%), which was reported less frequently in the ribociclib plus AI group than in the AI alone group. AEs with differences of 10% or more between groups included decreased neutrophil count (24.2% versus 1.7%), nausea (23.5% versus 7.9%), increased ALT (19.7% versus 5.7%), and increased AST (17.2% versus 5.9%).
Similar to any-grade AEs, grade 3 or higher AEs were reported more frequently in the ribociclib plus AI group than in the AI alone group (64.2% versus 19.7%). Grade 3 or higher neutropenia (27.0% versus 0.5%) and decreased neutrophil count (17.7% versus 0.3%) were the most reported grade 3 AEs and were more frequent in the ribociclib plus AI group than in the AI alone group.
Serious Adverse Events
At the end of ribociclib analysis, 375 patients (14.8%) in the ribociclib plus AI group and 237 patients (10.9%) in the AI alone group reported at least 1 SAE. The most common SAEs were COVID-19 (0.8% versus 0.5%) and pulmonary embolism (0.6% versus 0.2%). All other SAEs were reported at a frequency of less than 0.5%.
Withdrawals Due to Adverse Events
At the end of ribociclib analysis, TEAEs leading to discontinuation of study treatment were reported in 534 patients (21.1%) in the ribociclib plus AI group and 130 patients (5.3%) in the AI alone group. The most common TEAEs leading to treatment discontinuation were increased ALT (7.2% versus 0.1%), increased AST (2.9% versus 0), and arthralgia (1.3% versus 2.1%). All other AEs leading to discontinuation were reported at a frequency of less than 1.0%.
Mortality
There were 104 patients (4.1%) in the ribociclib plus AI group and 122 patients (5.0%) in the AI alone group who had died before the end of follow-up in the end of ribociclib analysis. Overall, the leading cause of death was disease recurrence or progression (3.0% versus 4.1%). AEs were reported as the primary cause of death for 16 patients (0.6%) in the ribociclib plus AI group and 6 patients (0.2%) in the AI alone group. Infections and infestations (0.3% versus 0.1%) and cardiac disorders (0.2% versus 0.1%) were the most reported AEs leading to death. All other AEs that led to death were reported at a frequency of less than 0.1%.
At the 5-year follow-up analysis, 137 patients (5.4%) in the ribociclib plus AI group and 163 patients (6.7%) in the AI alone group had died, primarily due to disease recurrence (101 patients; 4.0%) or progression (133 patients; 5.4%). There were 19 patients (0.8%) and 8 patients (0.3%) in the NATALEE trial who had fatal AEs in the ribociclib plus AI group and AI group, respectively, and the primary reasons were consistent with the end of ribociclib analysis.
Notable Harms
The clinical experts consulted by CDA-AMC identified hepatobiliary toxicity, QT interval prolongation, and pulmonary toxicity (interstitial lung disease or pneumonitis) as notable harms of interest to ribociclib. Hepatobiliary toxicity was observed in 675 patients (26.7%) in the ribociclib plus AI group and 279 patients (11.4%) in the AI alone group. QT interval prolongation was reported in 136 patients (5.4%) in the ribociclib plus AI group and 38 patients (1.6%) in the AI alone group. Interstitial lung disease or pneumonitis was reported in 41 patients (1.6%) in the ribociclib plus AI group and 21 patients (0.9%) in the AI alone group.
Table 19: Summary of Harms Results From the NATALEE Trial (Safety Analysis Set; Data Cut-Off Date — April 29, 2024)
Adverse events | Ribociclib + AI (N = 2,526) | AI alone (N = 2,441) | ||
|---|---|---|---|---|
Any grade | Grade ≥ 3 | Any grade | Grade ≥ 3 | |
Most common adverse events (≥ 20% of patients in either group), n (%) | ||||
≥ 1 adverse event | 2,478 (98.1) | 1,622 (64.2) | 2,155 (88.3) | 481 (19.7) |
Neutropenia | 1,053 (41.7) | 683 (27.0) | 71 (2.9) | 13 (0.5) |
Arthralgia | 979 (38.8) | 25 (1.0) | 1,083 (44.4) | 31 (1.3) |
Decreased neutrophil count | 611 (24.2) | 448 (17.7) | 41 (1.7) | 8 (0.3) |
Nausea | 594 (23.5) | 6 (0.2) | 192 (7.9) | 1 (< 0.1) |
Headache | 579 (22.9) | 11 (0.4) | 420 (17.2) | 4 (0.2) |
Fatigue | 575 (22.8) | 21 (0.8) | 329 (13.5) | 4 (0.2) |
COVID-19 | 569 (22.5) | 20 (0.8) | 358 (14.7) | 12 (0.5) |
Positive SARS-CoV-2 test | 550 (21.8) | 1 (< 0.1) | 340 (13.9) | 0 |
Hot flush | 492 (19.5) | 6 (0.2) | 493 (20.2) | 3 (0.1) |
SAEs (≥ 0.5% of patients in either group), n (%) | ||||
Patients with ≥ 1 SAE | 375 (14.8) | 267 (10.9) | ||
COVID-19 | 20 (0.8) | 13 (0.5) | ||
Pulmonary embolism | 15 (0.6) | 5 (0.2) | ||
Patients who stopped treatment due to adverse events (≥ 1% of patients in either group), n (%) | ||||
Patients who stopped | 534 (21.1) | 130 (5.3) | ||
Increased ALT | 183 (7.2) | 2 (0.1) | ||
Increased AST | 72 (2.9) | 0 | ||
Arthralgia | 34 (1.3) | 51 (2.1) | ||
Deaths (≥ 0.1% of patients in either group), n (%) | ||||
Patients who died | 104 (4.1) | 122 (5.0) | ||
Disease recurrence or progression | 76 (3.0) | 100 (4.1) | ||
Adverse event | 16 (0.6) | 6 (0.2) | ||
Infections and infestations | 7 (0.3) | 3 (0.1) | ||
Cardiac disorders | 5 (0.2) | 2 (0.1) | ||
Other | 12 (0.5) | 16 (0.7) | ||
Adverse events of special interest, n (%) | ||||
Hepatobiliary toxicity | 675 (26.7) | 279 (11.4) | ||
QT interval prolongation | 136 (5.4) | 38 (1.6) | ||
ILD or pneumonitis | 41 (1.6) | 21 (0.9) | ||
AI = aromatase inhibitor; ILD = interstitial lung disease; SAE = serious adverse event; SARS-CoV-2 = severe acute respiratory syndrome coronavirus 2.
Sources: NATALEE Clinical Study Reports for the end of ribociclib analysis (2024)17 and sponsor-provided additional data.19 Details included in the table are from the sponsor’s Summary of Clinical Evidence.21
Critical Appraisal
Internal Validity
The Health Canada–approved indication for ribociclib is for patients with HR-positive, HER2-negative EBC at high risk of recurrence. However, the eligibility criteria for the NATALEE trial population were based on disease stage and included a heterogenous population of patients with varying degrees of risk. There was no definition of high risk provided in the trial, nor were there any prespecified subgroup analyses specifically for high-risk patients in the NATALEE trial. As such, it was unclear what proportion of patients in the NATALEE trial were at high risk of recurrence.
The NATALEE trial was a phase III, multicentre, open-label, active-controlled RCT to evaluate the efficacy and safety of ribociclib plus AI compared to AI alone as an adjuvant treatment in patients with HR-positive, HER2-negative EBC. According to the clinical experts consulted by CDA-AMC, the choice of AI as the comparator was clinically relevant because it is commonly used in clinical practice in Canada for the treatment of patients with HR-positive, HER2-negative EBC.
The NATALEE trial was open label — meaning both the patients and the investigators knew who was receiving ribociclib and who was receiving AI alone — which could introduce performance bias (e.g., differences in how patients are managed or monitored based on their treatment) and detection bias in subjective, self-reported outcomes (e.g., a greater likelihood of reporting AEs or outcomes in the ribociclib plus AI group). Additionally, fewer patients discontinued the study in the ribociclib plus AI group compared to the AI alone group at both the end of ribociclib analysis (17.7% versus 22.1%) and 5-year follow-up (21.5% versus 26.0%). If treatment discontinuations were informed by patients knowing treatment assignment, the observed results may have been influenced by knowledge of and adherence to a treatment regimen rather than solely by the treatment effect.
The clinical experts noted that the median follow-up time at the end of ribociclib analysis (44.2 months) was shorter than ideal, but reasonable considering the NATALEE trial included patients with a high risk of recurrence. However, patients with HR-positive, HER2-negative breast cancer can experience recurrence even after 10 to 15 years. The analysis of iDFS was predominately influenced by the initial treatment response, which represents 10.3% and 13.3% of the probability of an event within each treatment group. In the NATALEE trial, at the end of ribociclib analysis, the survival curves showed a sudden drop beyond 36 months (around 42 months for iDFS, 56 months for RFS and DDFS, and 60 months for OS); this drop is likely artificial and driven by the large amount of censoring observed in the trial. Though more events had occurred at the 5-year follow-up analysis and the proportion of patients who were censored was generally balanced between treatment groups at previous data cut-off dates, there was still a substantial amount of censoring, with rapid drops in the number of patients at risk. The rapid depletion of the risk set may have introduced uncertainties in the estimates of the treatment effects of ribociclib plus an AI on iDFS, RFS, DDFS, and OS at 36, 48, and 60 months. Additionally, the treatment effect observed over 5 years does not permit definitive conclusions about the long-term effectiveness of ribociclib plus an AI.
According to the input provided, patients with EBC and clinicians considered RFS, DDFS, OS, and HRQoL to be relevant outcomes. However, these outcomes were not part of the statistical testing strategy in the NATALEE trial and thus were not controlled for type I error; therefore, the ability to draw causal conclusions from these results may be limited. Additionally, there was a notable attrition rate observed in the analysis of HRQoL measured using the change from baseline in global health status and quality of life subscale score of the EORTC QLQ-C30 at the end of treatment, with only 462 patients (18.8%) contributing to the analysis; considering the median duration of follow-up at the time of the interim analysis was 27.7 months, the attrition was likely due to the insufficient number of patients who had reached the end of treatment, which was defined as completing 5 years of AI in both groups. Given the NATALEE trial was open label and withdrawal by patient was the most common reason for study discontinuation, this would introduce bias because the characteristics of patients who remained in the study may differ from those who did not. Moreover, no sensitivity analyses were conducted for the analysis of HRQoL.
Upon a request from CDA-AMC, the sponsor provided a post hoc subgroup analysis of iDFS for patients who meet the definition of high risk per the monarchE trial (abemaciclib) (Table 28), which was generally consistent with the primary intention-to-treat population; however, this was a post hoc analysis with no statistical testing and was not controlled for overall type I error. As such, the results were purely descriptive and should be considered as supportive data only.
External Validity
The clinical experts highlighted that the NATALEE trial eligibility criteria were standard, but stricter than in clinical practice. For example, patients with significant comorbidities or poorer ECOG Performance Status (e.g., score of 2) were not eligible for the NATALEE trial; however, the clinical experts noted that they would consider patients with an ECOG Performance Status of 2 to be potential candidates for ribociclib based on the extensive experience with ribociclib in the metastatic setting. Additionally, the clinical experts noted there would be a higher proportion of postmenopausal female patients aged 60 to 70 years who are candidates for ribociclib in their clinical practice than the proportion enrolled in the NATALEE trial. Most patients in the NATALEE trial were white (73.4%). The clinical expert indicated there is a more diversified patient population in their clinical practice compared to the patient population in the NATALEE trial. Overall, the patient population in the NATALEE trial may not be fully representative of the patient profile seen in clinical practice.
In the NATALEE trial, genomic test results or Ki-67 levels were used to determine eligibility for patients whose disease was anatomical stage IIA, with no nodal involvement and with an intermediate-grade (grade 2) tumour, but were not used for patients whose disease was stage IIB or III. According to the clinical experts, genomic risk testing (e.g., Oncotype DX) is used to determine eligibility for chemotherapy rather than for ribociclib, and it is unclear whether genomic risk is predictive for treatment with ribociclib. Additionally, the use of genomic testing in the NATALEE trial may not be reflective of current clinical practice because genomic risk testing may be required (i.e., Oncotype DX) for all patients with stage II (A or B) and grade 2 or grade 3 tumour before initiating chemotherapy. Moreover, 88.1% of patients in the NATALEE trial had prior chemotherapy; however, the clinical experts indicated that in clinical practice, most patients with no nodal involvement would have been tested for genomic risk; thus, fewer patients (approximately 70%) in clinical practice would receive chemotherapy than in the NATALEE trial. Therefore, the study results may not be generalizable because patients in the NATALEE trial were more heavily pretreated than patients in clinical practice, which could introduce bias, but the direction of bias is uncertain.
More than half of the patients in the NATALEE trial (62.8%) completed the 3-year treatment duration of ribociclib at the time of the end of ribociclib analysis, and no patients were receiving ribociclib treatment at the end of the 5-year follow-up. The clinical experts commented that adherence to treatment is a challenge in the adjuvant setting due to the longer disease-free intervals. The percentage of patients who completed the 3-year treatment might reflect the clinical effectiveness of treatment in clinical practice, considering patients in the NATALEE trial were predominantly younger women who have more aggressive disease, who may be less willing to endure the side effects (e.g., nausea and fatigue) of ribociclib for the full 3 years.
Corticosteroids and antiemetic medications were allowed to be used concomitantly in the NATALEE trial. According to the clinical experts, these are not commonly used in the adjuvant setting, but patients may receive corticosteroids or antiemetic medications as supportive therapies for prior chemotherapy or to control the side effects of ribociclib, such as nausea and interstitial lung disease. Given the small proportion of patients who used these medications, the impact of the concomitant use of these medications on the study results would likely be minimal.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered most relevant to inform expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group:15,16
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate. The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited. The true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate. The true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as “very uncertain.”
Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null. The reference points for the certainty of evidence assessment for iDFS, RFS, DDFS, OS, HRQoL, and harms were set according to the presence of an important effect based on thresholds agreed upon by clinical experts consulted for this review.
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for ribociclib plus AI versus AI alone.
Long-Term Extension Studies
No long-term extension studies were submitted for this review.
Indirect Evidence
Contents within this section have been informed by materials submitted by the sponsor. The following was summarized and validated by the review team.
Objectives for the Summary of Indirect Evidence
In the absence of head-to-head trial data for ribociclib plus AI compared with abemaciclib plus ET for the adjuvant treatment of patients with HR-positive, HER2-negative EBC, the sponsor submitted an MAIC. The objective of this section is to summarize and critically appraise the sponsor-submitted MAIC comparing ribociclib plus AI versus abemaciclib plus ET in the adjuvant treatment of patients with HR-positive, HER2-negative EBC at high risk of disease recurrence based on clinicopathological features.
Description of Indirect Treatment Comparison
Objectives
The objective of the ITC was to estimate the comparative efficacy and safety of ribociclib plus AI and abemaciclib plus ET to inform the economic model of adjuvant treatments for patients with HR-positive, HER2-negative EBC at high risk of disease recurrence based on clinicopathological features.
Indirect Treatment Comparison Design
Study Selection Methods
A systematic literature review (SLR) was conducted to identify trials for the adjuvant treatment of HR-positive, HER2-negative EBC. The SLR was conducted on March 30, 2023, and updated in October 2023 to identify any supplementary evidence published subsequent to the initial SLR searches. The following electronic databases were searched: MEDLINE All (1946‒), Embase (1974‒), MEDLINE In-Process, and the Cochrane Central Register of Controlled Trials. In addition, conference abstracts from the past 3 years were hand searched to retrieve the latest studies that had not yet been published in journals as full-text articles. Bibliographies of all relevant SLRs and meta-analyses identified during the review were also hand searched to identify any additional relevant studies for inclusion. Searches of the ClinicalTrials.gov registry, health technology assessment websites, and the FDA database were also performed.
The SLR followed the Cochrane Handbook and National Institute for Health and Care Excellence guidelines. Citations were screened by 2 independent reviewers, with discrepancies resolved by a third. Data extraction was performed using a predefined grid and checked by a second reviewer. Subgroup analyses based on the latest NATALEE trial were also extracted, focusing on menopausal status, disease stage, nodal status, and prior chemotherapy. The quality of RCTs was assessed using the York Centre for Reviews and Dissemination checklist.
Table 20: Study Selection Criteria and Methods for Indirect Comparisons
Characteristics | Indirect comparison |
|---|---|
Population |
|
Intervention |
|
Comparator | ET (letrozole, anastrozole, exemestane, or tamoxifen dispensed according to the local clinical guidelines and current local prescribing information) |
Outcome | Efficacy
Safety
|
Study designs | Randomized controlled trials |
Publication characteristics | Studies published in English |
Exclusion criteria | Population
Interventions
Study design
Publication characteristics
|
Databases searched | Biomedical database searching
Conference proceedings searching
Bibliographic searching
|
Selection process | The data collection process followed the Cochrane Handbook and complied with NICE requirements. It involved 2 steps: (1) initial screening based on titles and abstracts, and (2) detailed screening based on full texts. Each citation and full-text publication was reviewed by 2 independent reviewers, with any discrepancies resolved by a third reviewer. |
Data extraction process | Data from the included studies were extracted into a predefined extraction grid. The data extraction process was conducted by a single reviewer and checked by a second reviewer. |
Quality assessment | The quality of randomized controlled trials was assessed using the CRD checklist |
AI = aromatase inhibitor; CENTRAL = Cochrane Central Register of Controlled Trials; CRD = York University Centre for Reviews and Dissemination; DRFS = distant recurrence–free survival; EBC = early breast cancer; ESMO = European Society for Medical Oncology; ET = endocrine therapy; HR = hormone receptor; iDFS = invasive disease–free survival; NICE = National Institute for Health and Care Excellence; OS = overall survival; SLR = systematic literature review; TEAE = treatment-emergent adverse event; TNBC = triple-negative breast cancer.
Source: Ribociclib indirect treatment comparison technical report.56
ITC Analysis Methods
Statistical Model
Based on the feasibility assessment, an unanchored MAIC was used to compare ribociclib plus AI versus abemaciclib plus ET. However, an anchored analysis (Bucher ITC) was conducted for iDFS via a common comparator in a subgroup of patients (i.e., iDFS in ribociclib plus AI in the NATALEE trial and the subgroup of the abemaciclib plus ET group in the monarchE trial that received AIs only).
The unanchored MAICs were conducted using procedures described in the National Institute for Health and Care Excellence Decision Support Unit Technical Support Document 18.57-59 Standardized MAIC weights were generated using the method of moments, with active treatment and control arms matched separately to ensure any residual imbalances between active and control arms in the monarchE trial were replicated in the matched NATALEE sample.59 Although the method of moments used in the MAIC typically ensures covariate balance for the reported characteristics for the 2 trials, standardized mean differences were also calculated for all baseline characteristics to validate the matching and to assess differences between treatments in characteristics not included in the matching algorithm (if any). Distributions of MAIC weights were plotted as histograms to identify outlier observations. The effective sample size for selected patients in the NATALEE trial after weighting was also calculated. A Cox regression model (semiparametric) was chosen to compare ribociclib plus AI with abemaciclib plus ET for time-to-event analyses. The validity of the Cox proportional hazards assumption between ribociclib plus AI and abemaciclib plus ET, before and after weighting, was tested by a visual inspection of the log-cumulative hazard plots and by the Schoenfeld global test of proportionality.
Data Sources
The ITCs used individual patient data (IPD) from the NATALEE trial’s ribociclib plus AI treatment group from the end of ribociclib analysis (April 29, 2024, data cut-off), and aggregate data from the monarchE trial’s abemaciclib plus ET treatment group. The aggregate data from the monarchE trial used in the MAICs come from the prespecified 5-year monarchE trial efficacy analysis.60 However, the AE rates for cohort 1 of the monarchE trial had not been published at the time of these analyses. As such, safety outcomes were based on data reported for the safety set of the monarchE trial.61 The control arms in the NATALEE and monarchE trials consisted of different therapies, preventing a shared comparator necessary to form a connected network.
The NATALEE trial enrolled a broader patient population than the monarchE trial for treatment of EBC. Patients in the monarchE trial were enrolled in 2 cohorts, each with different criteria used to define high-risk characteristics. Cohort 1 of the monarchE trial included patients with 4 or more positive ALNs, or 1 to 3 positive ALNs with grade 3 disease and/or a tumour of 5 cm or greater. Cohort 2 included patients with 1 to 3 positive nodes and a Ki-67 of 20% or greater who did not meet the criteria for cohort 1. Cohort 2 was excluded from the ITC analyses. For the ITC, patients in the NATALEE trial were selected if they met the eligibility criteria for enrolment in cohort 1 of the monarchE trial. Per the NATALEE trial protocol, nodal involvement and tumour size were determined based on the worst of the measurements taken at the time of diagnosis and on a surgical specimen following resection (i.e., anatomical assessment). Information on histologic grade and nodal status were captured at baseline assessment for patients enrolled in the NATALEE study. However, information on explicit tumour size was not captured in the NATALEE study. As such, patients who met the criteria for 1 to 3 positive lymph nodes and a tumour size of 5 cm or greater were identified using information on the tumour, node, and metastasis (TNM) staging as a proxy. Key characteristics of the studies selected for the MAIC are summarized in Table 21.
Table 21: Assessment of Homogeneity
Characteristics | Description and handling of potential effect modifiers |
|---|---|
Study design | NATALEE and monarchE are both phase III, multicentre, global, randomized, open-label clinical trials. Stratification factors for randomization differed slightly between the trials because the NATALEE trial included anatomical stage as a factor and the monarchE trial did not. Tamoxifen use was explicitly disallowed in the NATALEE trial, while no such restriction was specified in the monarchE trial. The extent to which these differences could bias any potential comparisons is unknown. |
Trial eligibility criteria | The trials share most of the key eligibility criteria apart from the nodal involvement requirements. The monarchE trial was focused on a subset of EBC patients, which the trial classified based on whether the patients were at high risk. The population of the NATALEE trial was defined based on disease stage and represents, without being limited to, patients at high risk. Accordingly, the population of the NATALEE trial to include in the ITC was selected in alignment with the eligibility criteria of cohort 1 of the monarchE, as described in the Data Sources section. |
Baseline characteristics | The trials were well balanced for many of the reported characteristics at baseline, and SMDs did not exceed 50% for any characteristic, except region and missing Ki-67 status. MAIC was adjusted for all characteristic variables that were identified from published reports of cohort 1 of the monarchE study to account for the residual differences in baseline characteristics observed between trials. |
Comparators | The NATALEE trial’s control arm was AI, consisting of letrozole and anastrozole, while the monarchE trial control arm was ET, consisting of letrozole, anastrozole, exemestane, and tamoxifen.61,68 The results of previous meta-analyses have shown that tamoxifen has worse efficacy compared with AIs in this population.53,69 An unanchored MAIC was thus conducted because neither comparator arm represented a common comparator through which to form a connected network. |
Definitions of end points | iDFS and OS were predefined end points in both the NATALEE and monarchE trials with the same definition. iDFS was evaluated based on STEEP criteria as the time from randomization until recurrence, second primary nonbreast malignancy, or death. ██ ███ █████████ ██ ███ ████ ████ █████████████ █████ █████ ████ ███ ██████ ██ ████ ███ ███ █ ████ ███████ ████████ ██ ███ ███████ ██████ ██ ███ ██████████ █████ ███ ██████████ ████████ ██ ███ ████████ █████ █████████ ████ ███ ███████ ██ ████ ████ █████████████ ██ ███████ ██████████ ██ █████ ████ ███ ██████ █████████ ██████ ██████ |
Follow-up time | There were differences between the median follow-up times for the latest available data from the NATALEE study. As of the April 29, 2024, data cut-off for the NATALEE trial, the median study follow-up was 49.6 months compared with 54.0 months for the monarchE study. |
Timing of end point evaluation | Patient assessments were more frequent for the monarchE trial than for the NATALEE study during the first 2 years of the trials’ duration. |
AI = aromatase inhibitor; EBC = early breast cancer; ET = endocrine therapy; iDFS = invasive disease–free survival; ITC = indirect treatment comparison; MAIC = matching-adjusted indirect comparison; OS = overall survival; SMD = standardized mean difference; STEEP = Standardized Definitions for Efficacy End Points.
Source: Ribociclib ITC technical report.56
Adjustment Factors Included in the MAIC Analyses
The baseline characteristics reported for cohort 1 of the monarchE trial that were included for adjustment in the MAIC based on a literature review and clinician input are listed in Table 22. The relevance of baseline characteristics as effect modifiers and/or prognostic factors for efficacy was determined through interviews with key opinion leaders and assessed using statistical tests. External clinicians were interviewed to determine which characteristics were known to be prognostic factors for efficacy in EBC, by varying degrees of influence.
Statistical tests were conducted to identify treatment-effect modifiers and prognostic factors, which are summarized in the ITC report.56 Table 22 also reports the results of the statistical tests identifying effect modifiers and prognostic factors. Sensitivity analyses were conducted to assess the range of plausible estimates for treatment effects when adjusting for fewer variables. Two alternative sets of MAIC weights were estimated for characteristics that were of high importance based on consensus among the clinicians, and for characteristics indicated by clinicians as important prognostic factors (high or low), as well as all characteristics with evidence of prognostic influence based on quantitative tests (Table 22). The weights estimated in these sensitivity analyses were then used in the comparison of outcomes, following the same procedures as the primary analysis.
Missing values in the IPD from the NATALEE trial that did not have a corresponding missing category in the summary-level data from the monarchE trial were imputed by randomly assigning values from the nonmissing values. The primary analysis for the unanchored MAIC included all of the available characteristics shown in Table 22. The weighting scheme chosen was adjusted for all available characteristics, provided the effective sample size remained reasonable.
Table 22: Adjustment Factors Included in the MAIC Analyses
Characteristic | Prognostic modifier | Effect modifiera | Included in analysis | ||||
|---|---|---|---|---|---|---|---|
Clinician ranking | Log-rank test | NATALEE trial | monarchE trial | Primaryb | SA 1c | SA 2d | |
Of high importance based on consensus among the clinicians | |||||||
Pathological Dx term | High | — | — | — | Yes | Yes | Yes |
HR status | High | Significant | — | — | Yes | Yes | Yes |
Lymph nodes (n) | High | Significant | — | — | Yes | Yes | Yes |
Histopathology at Dx | High | Significant | — | — | Yes | Yes | Yes |
TNM stage | High | Significant | — | — | Yes | Yes | Yes |
Tumour size (cm) | High | Significant | — | Significant | Yes | Yes | Yes |
Ki-67 index | High | — | — | — | Yes | Yes | Yes |
Assigned some level of importance in EBC prognosis by at least 1 clinician | |||||||
Menopausal status | Low | — | — | — | Yes | NA | Yes |
Age, years | Low | Significant | — | — | Yes | NA | Yes |
Weight, kg | Low | — | — | — | Yes | NA | Yes |
BMI, kg/m2 | Low | — | — | — | Yes | NA | Yes |
Prior chemotherapy | Low | Significant | — | — | Yes | NA | Yes |
Prior radiotherapy | Low | — | — | — | Yes | NA | Yes |
ECOG PS | Low | — | — | — | Yes | NA | Yes |
Tumour side | Low | — | — | — | Yes | NA | Yes |
Not identified as having an important prognostic influence in EBC | |||||||
Female | — | — | — | — | Yes | NA | NA |
Race | — | — | — | — | Yes | NA | NA |
Ethnicity | — | — | — | — | Yes | NA | NA |
Region | — | — | — | — | Yes | NA | NA |
BMI = body mass index; Dx = diagnosis; EBC = early breast cancer; ECOG PS = Eastern Cooperative Oncology Group Performance Status; HR = hormone receptor; NA = not adjusted; SA = sensitivity analysis; TNM = tumour, node, and metastasis.
Note: “—” indicates not important (per clinician interview) or not statistically significant at alpha = 0.05 (if statistical test).
aBased on P value for interaction test.
bAdjusted for all of the available effect or prognostic modifiers.
cAdjusted for high importance of effect or prognostic modifiers based on clinician interviews.
dAdjusted for any importance effect or prognostic modifiers based on clinician interviews and/or statistically significant based on statistical test.
Source: Ribociclib indirect treatment comparison technical report.56
Outcomes
The outcomes assessed in the ITC were iDFS, DRFS, OS, and grade 3 or higher TEAEs with an incidence of at least 5% in either trial. Time-to-event data for iDFS, DRFS, and OS for patients in the monarchE trial were obtained by digitizing published Kaplan-Meier curves.61,62 Reconstructed individual patient time-to-event data for iDFS, DRFS, and OS for the monarchE treatment arms was generated using an adaptation of a published algorithm from Guyot et al. (2012).63 Hazard ratios and 95% CI were calculated with a Cox proportional hazards regression model.59 Safety was assessed based on rates of AEs; ORs and 95% CI were calculated. ORs for TEAEs were calculated using logistic regression. For TEAEs for which there were no events in 1 or more treatment arms, ORs were calculated using a continuity correction by adding 1 patient with the event and 1 without the event to each arm. Results were estimated separately for each of the MAIC analyses: selected unweighted, selected with primary analysis weights applied, and selected with weights applied from the 2 sensitivity analyses.
Secondary Analysis
An anchored analysis of iDFS, via common comparator, was conducted as a second analysis using data reported for a subgroup of patients who received an AI as the ET component of each treatment arm in cohort 1 of the monarchE trial and excluding patients who received tamoxifen as the endocrine partner. This subgroup analysis employed a Bucher ITC.64 It was noted that baseline characteristics for this subgroup of the monarchE trial were not available, which prohibited the calculation of MAIC weights specific to the AI-only subgroup of the monarchE trial. As such, hazard ratios for iDFS in the NATALEE study were based on those estimated for the selected, unweighted sample.
Matching-Adjusted Indirect Comparison Methods
MAIC methods are summarized in Table 23.
Table 23: Matching-Adjusted Indirect Comparison Methods
Methods | Description |
|---|---|
Analysis methods | Unanchored MAIC:
|
Covariates used for weighting | For the primary analysis, the MAICs were adjusted for all baseline characteristics identified in published reports of cohort 1 from the monarchE study (Table 22) |
Outcomes | Efficacy
Safety
|
Follow-up time points | For data used in the primary analysis, median study follow-up was 49.6 months for the NATALEE trial and 54.0 months for the monarchE study |
Construction of nodes | There was no node for the primary analysis because an anchored approach was determined to be infeasible as a primary analysis. Cohort 1 of the monarchE trial is considered the reference population for the analyses |
Population | Both trials studied patients with HR-positive, HER2-negative EBC. Patients were selected from the NATALEE trial who would meet high-risk eligibility criteria for cohort 1 of the monarchE trial:
|
Sensitivity analyses | Two alternative sets of MAIC weights were estimated to explore the impact of adjusting for fewer variables:
|
Secondary analysis | A Bucher analysis of iDFS via common comparator was conducted as a second analysis using data reported for a subgroup of patients receiving AI as the ET component of each treatment arm in cohort 1 of the monarchE trial and excluding patients who received tamoxifen as the endocrine partner |
AI = aromatase inhibitor; CI = confidence interval; DRFS = distant recurrence–free survival; EBC = early breast cancer; ESS = effective sample size; ET = endocrine therapy; HR = hormone receptor; iDFS = invasive disease–free survival; IPD = individual patient data; MAIC = matching-adjusted indirect comparison; OR = odds ratio; OS = overall survival; TEAE = treatment-emergent adverse event.
Source: Ribociclib indirect treatment comparison technical report.56
Results of the ITC
Summary of Included Studies
Search Results
A total of 399 records were initially screened for the SLR. A total of 44 studies reported in 136 publications were included in this SLR. Of the 44 included studies, only 2 studies met the eligibility criteria for the ITC: 1 trial (NATALEE) evaluating ribociclib plus AI and 1 trial (monarchE) evaluating abemaciclib plus ET.
Evidence Networks
Selection of Patients
Patients were selected from the NATALEE trial who would meet high-risk eligibility criteria for cohort 1 of the monarchE trial.
MAIC Weights
The patient baseline characteristics before adjustment are presented in Table 29 in Appendix 1. The patients selected from the NATALEE trial and patients in cohort 1 of the monarchE trial were balanced for many of the reported characteristics at baseline, and standardized mean differences did not exceed 50% for almost all characteristics, except region and missing Ki-67 status. After weighting, the effective sample size for the ribociclib plus AI and AI-only arms was 448 and 453, respectively, constituting a reduction of 73.0% to 72.5% from the selected sample size in each arm, respectively. After weighting, the standardized mean differences between the patient characteristics of the weighted NATALEE trial population and the monarchE study cohort 1 population are relatively small or zero.
The KM curves for iDFS, DRFS, and OS are presented in Figure 7, Figure 8, and Figure 9 in Appendix 1. Kaplan-Meier curves for abemaciclib plus ET treatment and the weighted ribociclib plus AI treatment largely overlapped for all outcomes.
Efficacy
The overall MAIC results are presented in Table 24.
Invasive Disease–Free Survival
Primary Analysis
After applying the MAIC weights, the estimated iDFS hazard ratio was 0.901 for ribociclib plus AI versus abemaciclib plus ET (95% CI, 0.677 to 1.197; P = 0.4701).
Sensitivity Analyses
███████ ███ ███████████ ████████ █ ███ ███████████ ████████ █ ████ ██████████ ████ ███ ███████ ████████ █████ ██████ ██████ █████ ████ ███ █████ ██ ███████ ███ ████ ██████ ██████ █████ ████ ███ █████ ██ ████████.
Table 24: Summary of MAIC Results Comparing Ribociclib Plus AI With Abemaciclib Plus ET
Outcome | MAIC results, hazard ratio (95% CI) | |||
|---|---|---|---|---|
Primary analysis | Sensitivity analysis | |||
Unadjusted | Fully adjusteda | SA1b | SA2c | |
iDFS | 1.055 (0.887 to 1.255) | 0.901 (0.678 to 1.197) | 0.937 (0.743 to 1.182) | 0.933 (0.725 to 1.201) |
████ | ███ █████ ████ | ███ █████ ████ | ███ █████ ████ | ███ █████ ████ |
██ | ███ █████ ████ | ███ █████ ████ | ███ █████ ████ | ███ █████ ████ |
AI = aromatase inhibitor; CI = confidence interval; DRFS = distant recurrence–free survival; ESS = effective sample size; ET = endocrine therapy; iDFS = invasive disease–free survival; MAIC = matching-adjusted indirect comparison; OS = overall survival.
Note: ET in the NATALEE study consisted of letrozole and anastrozole only, while ET in the monarchE trial consisted of letrozole, anastrozole, exemestane, and tamoxifen.
aIn the primary analysis, MAIC weights were adjusted for all available baseline characteristics published for the monarchE trial.
bSensitivity analysis 1 was adjusted for characteristics for which there was a consensus of high prognostic importance among clinicians.
cSensitivity analysis 2 was adjusted when there was evidence of some prognostic importance among clinicians and statistically significant based on statistical test.
Source: Ribociclib indirect treatment comparison technical report.56
Subgroup Analysis (i.e., Secondary Analysis)
The estimated hazard ratio for ribociclib plus AI versus abemaciclib plus AI based on the Bucher ITC was 0.957 (95% CI, 0.726 to 1.261) (Table 25).
Table 25: Anchored Bucher Model ITC of iDFS in the NATALEE Study vs. iDFS in the monarchE Trial AI Subgroup (Unweighted)
Comparison | Source | Hazard ratio (95% CI) |
|---|---|---|
Hazard ratio within trials | ||
Abemaciclib + AI vs. AI | Published cohort 1 AI subgroup | 0.760 (0.618 to 0.935) |
Ribociclib + AI vs. AI | Selected, unweighted NATALEE trial | 0.727 (0.606 to 0.872) |
Hazard ratio across trials | ||
Ribociclib + AI vs. abemaciclib + AI | Selected, unweighted NATALEE trial | 0.957 (0.726 to 1.261) |
AI = aromatase inhibitor; CI = confidence interval; iDFS = invasive disease–free survival; ITC = indirect treatment comparison; vs. = versus.
Source: Ribociclib ITC technical report.56
Distant Relapse–Free Survival
Primary Analysis
█████ ████████ ███ ████ ████████ ███ █████████ ██████ █████ ███ ████ ███ █████ ████ ███ █████ ██ ████████ ██████ ███ ██ ██████████ ███ ██████ ████████ ███ ███ ████ █████████ ███ ██ ████████ █████████ ███ ████████████ █████████ ███████ ████ █████████ ██ █████████ ███ ████ ████ ██████ ████ ████ ████████.
Sensitivity Analyses
███████ ███ ███████████ ████████ ████ ███████████ ████████ █ ████ █████████ ██████████ ████ ███ ███████ ████████ ████ ████████ █████ ██████ ██████ █████ ██████ ██████ █████ ████ ███ █████ ██ ███████ ███ ███ ██████ ██████ █████ ████ ███ █████ ██ ████████.
Overall Survival
Primary Analysis
█████ █████████ ███ █████████ ██ ██████ █████ ██ ██████████ ████ ██ ██████ ███████████ ████ ██ ███ █████ ████ ███ █████ ██ ██████████ ████████.
Sensitivity Analyses
███████ ██ ███████████ ████████ ██ █████ ████████ ███ ███████ ██████ ██ ████ ██████████ █████ ██ █████████ ██████████ ███ ███████████ ████████ █ █████ ████████ ███ ███████ ██ ███ ██████████ █████ ██ █████████ ██████████ ████ ██████████ ████ ███ ███████ ████████ █████ ██████ ██████ █████ ████ ███ █████ ██ ███████ ███ ████ ██████ ██████ █████ ████ ███ █████ ██ ████████.
Harms
Grade 3 or higher TEAEs with an incidence of at least 5% in either trial were compared in the unanchored MAIC. Results of the weighted comparisons are presented in Table 26. In the weighted comparison, ribociclib plus AI also had significantly increased odds of neutropenia (OR = 1.56; 95% CI, 1.23 to 1.97) compared with abemaciclib plus ET, and increased ALT (OR = 3.94; 95% CI, 2.56 to 6.05). Odds ratios for diarrhea, leukopenia, and lymphopenia were reduced with ribociclib plus AI versus abemaciclib plus ET, with estimated ORs of 0.11 (95% CI, 0.03 to 0.40), 0.27 (95% CI, 0.16 to 0.46), and 0.14 (95% CI, 0.04 to 0.53), respectively, based on the weighted analysis. Results from 2 sensitivity analyses were consistent with the primary analyses for TEAEs (Table 26).
Table 26: MAIC Results of Grade 3 or Higher TEAEs
Adverse event | Events rates | MAIC results | ||||
|---|---|---|---|---|---|---|
NATALEE trial | monarchE trial | Ribo + AI vs. abemaciclib + ET | ||||
Ribociclib + AI (%) | AI alone (%) | Abemaciclib + ET (%) | ET (%) | OR (95% CI) | P value | |
Selected, unweighted | ||||||
ALT increased | 7.06 | 0.73 | 2.76 | 0.68 | 2.68 (1.99 to 3.59) | < 0.0001 |
Neutropenia | 28.17 | 0.36 | 19.63 | 0.86 | 1.61 (1.39 to 1.85) | < 0.0001 |
Diarrhea | 0.42 | 0.12 | 7.85 | 0.21 | 0.05 (0.02 to 0.11) | < 0.0001 |
Leukopenia | 3.68 | 0.12 | 11.39 | 0.39 | 0.30 (0.22 to 0.39) | < 0.0001 |
Lymphopenia | 0.42 | 0.00 | 5.41 | 0.50 | 0.07 (0.04 to 0.16) | < 0.0001 |
Selected, weighted | ||||||
ALT increased | 10.05 | 0.75 | 2.76 | 0.68 | 3.94 (2.56 to 6.05) | < 0.0001 |
Neutropenia | 27.54 | 0.12 | 19.63 | 0.86 | 1.56 (1.23 to 1.97) | 0.000 |
Diarrhea | 0.95 | 0.16 | 7.85 | 0.21 | 0.11 (0.03 to 0.40) | 0.001 |
Leukopenia | 3.39 | 0.03 | 11.39 | 0.39 | 0.27 (0.16 to 0.46) | < 0.0001 |
Lymphopenia | 0.82 | 0.00 | 5.41 | 0.50 | 0.14 (0.04 to 0.53) | 0.004 |
AI = aromatase inhibitor; CI = confidence interval; ET = endocrine therapy; OR = odds ratio; TEAE = treatment-emergent adverse event; vs. = versus.
Note: OR < 1 means results were in favour of ribociclib plus ET; OR > 1 means results were in favour of abemaciclib plus ET. Event rates equal to zero were adjusted with continuity correction to allow for the estimation of odds ratios via logistic regression. AI in the NATALEE study consisted of letrozole and anastrozole only, while ET in the monarchE trial consisted of letrozole, anastrozole, exemestane, and tamoxifen.
Source: Ribociclib indirect treatment comparison technical report.56
Critical Appraisal of ITC
The search for potentially eligible studies was reasonably comprehensive, and the methods used to select relevant studies, extract data, and assess the risk of bias of the included studies were adequate. The MAIC estimation procedure was mostly conducted according to best practice. One limitation is the use of matching weights for each treatment arm separately. This approach has been criticized in the past because it may break randomization in the IPD, distorting the balance between treatment arms on covariates that are not accounted for in the weighting, and potentially compromising the internal validity of the within-study estimate.65,66 The unanchored approach makes the interpretation of the efficacy results difficult to translate as an estimate of effectiveness in the Canadian clinical setting. The absence of a comparator that aligns with Canadian clinical practice increases the uncertainty about the magnitude and direction of effect for ribociclib plus AI versus abemaciclib plus ET.
A key limitation of the MAIC was heterogeneity across the included studies. The sponsor identified potential prognostic modifiers and effect modifiers — including pathological diagnosis, HR status, lymph nodes, histopathology at diagnosis, TNM stage, tumour size, and Ki-67 Index — through literature searches and clinical expert opinion. In the primary analysis, the MAIC was adjusted for all of the baseline characteristics identified in published reports of cohort 1 from the monarchE study but excluded patients from both studies outside of this subcohort. The omission of these patients, who are within the Health Canada indication for ribociclib, leads to uncertainty in the generalizability of the efficacy results to the clinical context. It should be noted that CDA-AMC recommended that public drug plans reimburse abemaciclib in combination with ET for the adjuvant treatment of adults with HR-positive, HER2-negative, node-positive, EBC at high risk of recurrence based on the clinicopathological features of the patients included in cohort 1 and cohort 2 of the monarchE trial (4 or more positive ALNs, or 1 to 3 positive ALNs plus histologic grade 3 disease, or 1 to 3 positive ALNs plus a tumour size of 5 cm or more, or 1 to 3 positive ALNs plus a Ki-67 score of 20% or more if tumour size is less than 5 cm and disease is not grade 3). Excluding data from cohort 2 of the monarchE trial leads to uncertainty in the generalizability of the ITC efficacy results to the clinical context.
A Bucher analysis of iDFS, via common comparator (i.e., AI), was conducted as a second analysis (subgroup analysis) using data reported for a subgroup of patients receiving AI as the ET component of each treatment arm in cohort 1 of the monarchE trial and excluding patients who received tamoxifen as the endocrine partner. However, this subgroup analysis was not weighted and adjusted for the prognostic and treatment-effect modifiers; thus, this comparison may not be capturing the real-life treatment regimen used in clinical practice.
Overall, the results for iDFS, DRFS, and OS generally suggested no difference between ribociclib plus AI and abemaciclib plus ET in cohort 1 of the monarchE trial, but the generalizability of the results to the Canadian clinical setting is uncertain. A reduced effective sample size (approximately 70% across various analyses), and wide 95% CIs for the comparative effect estimates (hazard ratios) contributed to imprecision and suggested either benefit or harm in efficacy outcomes. Grade 3 or higher TEAEs (that occurred in ≥ 5% patients) were assessed in the MAIC, but no other safety outcomes such as SAEs and discontinuation due to AEs were assessed. HRQoL was identified by patient groups as an important outcome; however, no indirect comparison was conducted for HRQoL.
Studies Addressing Gaps in the Systematic Review Evidence
No studies addressing gaps in the evidence from the systematic review were submitted for this review.
Discussion
Summary of Available Evidence
The evidence included in this review consisted of 1 pivotal study (NATALEE) and an MAIC. No long-term extension studies or studies addressing gaps in the evidence were included.
The NATALEE trial was a phase III, open-label, active-controlled RCT to evaluate the efficacy and safety of ribociclib plus AI (n = 2,549) compared to AI alone (n = 2,552) in adult patients with HR-positive, HER2-negative, stage II or III EBC, irrespective of nodal status. Randomization was conducted using an interactive response technology system and stratified by menopausal status (postmenopausal versus premenopausal, with men included in the premenopausal category), anatomical stage (II versus III), prior adjuvant or neoadjuvant chemotherapy (yes versus no), and geographical region (North America, Western Europe, and Oceania versus rest of the world). The primary objective of the NATALEE trial was to evaluate the efficacy of ribociclib plus AI compared to AI alone as an adjuvant treatment in premenopausal and postmenopausal women, and men, with HR-positive and HER2-negative EBC in prolonging iDFS, and secondary outcomes included RFS, DDFS, OS, HRQoL, and safety. The baseline demographic and disease characteristics were balanced between the treatment groups. The median age of all patients was 52.0 years, with ages ranging from 24 to 90 years. Most patients were postmenopausal (55.8%) women (99.6%) and most were white (73.4%), but the trial also included patients who self-reported as American Indian or Alaska Native, Asian, Black or African American, and Native Hawaiian or other Pacific Islander. The majority of patients had an ECOG Performance Status score of 0 (83.1%). More than half (59.6%) of patients had disease that was AJCC stage III, nearly half of patients (41.2%) had 1 to 3 positive ALNs, and most patients (88.1%) had received prior adjuvant or neoadjuvant chemotherapy.
In the absence of direct comparative evidence versus relevant comparators, the sponsor conducted an ITC using MAIC methods to estimate the relative efficacy of ribociclib plus AI compared with abemaciclib plus ET in the adjuvant treatment of patients with HR-positive, HER2-negative EBC at high risk of disease recurrence based on clinicopathological features of the monarchE trial. The outcomes of interest in the MAICs included iDFS, DRFS, OS, and safety.
Interpretation of Results
Efficacy
The indication for ribociclib plus AI is for the adjuvant treatment of patients with HR-positive, HER2-negative stage II and III EBC at high risk of recurrence. According to the clinical experts consulted for this review, current systemic therapies for the indicated patient population included chemotherapy, ET (e.g., an AI), and abemaciclib (for patients with a high risk of recurrence). In clinical practice, treatment decisions are often based on tumour pathological features and genomic testing results such as Oncotype DX. In the NATALEE trial, genomic testing results (e.g., Oncotype DX) were used to determine the eligibility for patients with stage IIA disease and a grade 2 tumour. However, the clinical experts commented that genomic risk testing (e.g., Oncotype DX) is used to determine eligibility for chemotherapy rather than for ribociclib because it is unclear whether genomic risk is predictive for treatment with ribociclib. Additionally, genomic risk is not currently used to determine eligibility for abemaciclib. This is a challenge when comparing the drug with other treatments, such as chemotherapy and abemaciclib. Furthermore, patients with an ECOG Performance Status score greater than 1 were excluded from the NATALEE trial; however, the clinical experts consider patients with a poorer ECOG Performance Status (e.g., score of 2) to be potential candidates for ribociclib in clinical practice. Moreover, the NATALEE trial predominantly included patients with a median age of 52 years, which the clinical experts noted is younger than the patients they see in clinical practice. Overall, these limitations may compromise the generalizability of the study results to clinical practice in Canada.
The patient and clinician input highlighted the need for effective treatments for EBC that reduce the risk of recurrence with manageable side effects and that maintain quality of life. Based on the results of the NATALEE trial at IA3 (data cut-off date: January 11, 2023) — when the NATALEE trial met its primary end point — treatment with ribociclib plus AI resulted in an improvement in iDFS compared with AI alone (hazard ratio = 0.75; 95% CI, 0.62 to 0.91; P = 0.0014). The between-group difference in iDFS probability was 3.3% (95% CI, 0.9% to 5.7%) at 36 months. Results from the end of ribociclib analysis with a longer follow-up time (data cut-off date: April 29, 2024) were consistent with the primary analysis in IA3 (hazard ratio = 0.72; 95% CI, 0.61 to 0.84; nominal P < 0.0001).The between-group difference in iDFS probability at 36 months was nearly identical to IA3 and, at 48 months, was 4.9% (95% CI, 2.7% to 7.1%). At the 5-year follow-up analysis, the between-group difference in iDFS probability at 60 months was 4.5% (95% CI, 2.1% to 6.9%). The between-group differences in iDFS event-free probability at 36, 48, and 60 months were considered beneficial but potentially not clinically meaningful, based on the threshold of 5% suggested by the clinical experts. In the review of abemaciclib for a similar population, a threshold range of clinical meaningfulness of 3% to 5% was used. Supposing these results were considered in the context of a 3% threshold, the results for iDFS at 36 months would not be considered clinically meaningful, though there would be moderate certainty that the results for iDFS at 48 and 60 months were clinically meaningful, given that most of the observed effect lies higher than the threshold; however, the 95% CIs also include the possibility of no benefit, though the imprecision may not be considered serious.
Results of the GRADE assessments for RFS, DDFS, and OS also suggested there is moderate certainty that when compared with AI alone, ribociclib plus AI likely results in a little to a small clinically important difference in the probabilities of RFS and DDFS, and little to no clinically important difference in the probability of being alive at 48 and 60 months. The differences between groups were not deemed clinically meaningful at a 3% (for OS) or 5% (for RFS and DDFS) threshold, particularly for OS at 60 months when the majority of the effect lies less than the threshold of 3% suggested by the experts.
The median iDFS, RFS, DDFS, and OS were not reached in either treatment group at IA3, the end of ribociclib analysis, or the 5-year follow-up. The analysis of iDFS is predominately influenced by the initial treatment response, which is represented by the event rate of 10.3% in the ribociclib plus AI group and 13.3% in the AI alone group at the end of ribociclib analysis, and 12.4% and 16.0%, respectively, at the 5-year follow-up analysis. However, the treatment effect observed over 5 years does not permit definitive conclusions about the long-term effectiveness of ribociclib plus AI. There were observed improvements in the iDFS, RFS, and DDFS, though the degree to which the observed benefits in the trial in terms of these outcomes could be translated to an improvement in OS remains uncertain. Additionally, despite the high probabilities of remaining event-free at 48 and 60 months, it remains uncertain how long the treatment response could be sustained. Considering fewer patients in the ribociclib plus AI group received subsequent antineoplastic medications than patients in the AI alone group, there is the potential for bias in the OS results favouring the AI alone group because they received more treatment, which might have led to the dilution of any OS benefit in the ribociclib plus AI group. Moreover, compared to the AI alone group, the ribociclib plus AI group had fewer discontinuations and more patients remaining on treatment. Additionally, patient withdrawal was less frequent in the ribociclib plus AI group. These factors may introduce attrition bias that could lead to better observed efficacy outcomes, not solely due to the treatment effect, but because more patients adhered to ribociclib plus AI. Overall, there were treatment and attrition biases both potentially against and in favour of ribociclib plus AI, which increase the uncertainty in the magnitude of the treatment effect.
As previously noted, results for HRQoL were available only as of IA3 and not at longer follow-ups (i.e., the end of ribociclib analysis or 5-year follow-up). Though the results for HRQoL suggested a decline in quality of life, they were similar between the ribociclib plus AI and AI alone groups. There was a high proportion of missing data, with only 462 of 2,549 patients in the ribociclib plus AI group and 497 of 2,552 patients in the AI alone group contributing to the analysis, which may be due to the limited number of patients who had reached the end-of-treatment stage. This high rate of attrition introduces uncertainty and difficulty in interpreting the effect of ribociclib plus AI on HRQoL.
The indication of ribociclib plus AI is for patients with HR-positive, HER2-negative EBC at high risk of recurrence. However, the eligibility criteria of the NATALEE trial enrolled patients based on disease stage, not exclusively patients at high risk, for which there was no definition. The primary comparator for the high-risk population, abemaciclib, defined high-risk patients as those with either 4 or more positive ALNs, or between 1 and 3 positive ALNs and either grade 3 disease or tumour size of 5 cm or larger.61 Therefore, CDA-AMC asked the sponsor to provide additional data on a subset of patients in the NATALEE trial who would or would not meet the eligibility criteria for cohort 1 of the monarchE trial. Although the subgroup analysis results were consistent with the primary iDFS analysis, this subgroup was not predefined in the NATALEE trial and was not subject to multiplicity controls. As such, these results were considered supportive, and no definitive conclusions were able to be drawn from the subgroup analysis on patients with EBC at high risk of recurrence.
To address the previously mentioned concern about the high-risk population, and given the lack of direct comparative evidence between ribociclib plus AI and relevant comparators, the sponsor conducted an MAIC to estimate the relative efficacy of ribociclib plus AI compared with abemaciclib plus ET in the adjuvant treatment of patients with HR-positive, HER2-negative EBC at high risk of disease recurrence according to the eligibility criteria of cohort 1 of the monarchE trial. Overall, in terms of iDFS, DRFS, and OS, the results of the MAIC suggested there was no difference in efficacy between treatments, but the reduced sample size and wide 95% CIs reduced the precision of the effect. In addition, the MAIC analysis was further limited by the omission of potential effect modifiers from the model, which can result in a biased estimate of comparative efficacy. Importantly, the MAIC was conducted only for patients matching the high-risk characteristics of the monarchE trial, which does not reflect the reimbursement request, and should not be generalized to all patients with HR-positive, HER2-negative EBC. Therefore, the MAIC results should be interpreted with caution, taking into consideration the limitations of the methods to adjust for potential confounding and the key differences in the trial population.
Harms
Ribociclib has been available in Canada for patients with metastatic breast cancer since 2018, providing extensive clinical experience with the drug. Overall, no new safety signals for ribociclib plus AI were identified in the NATALEE trial. Generally, increased incidences of AEs were observed and more patients stopped treatment due to AEs in the ribociclib plus AI group compared to the AI alone group. In addition, grade 3 or higher AEs were reported more frequently in the ribociclib plus AI group than in the AI alone group. These AEs typically require medical intervention and can significantly impact a patient’s daily life. The clinical experts indicated that increased incidences of neutropenia and infections due to the addition of ribociclib would be expected in clinical practice based on their experience with the drug in the metastatic setting. Although more patients died in the AI alone group overall, there was a numerically higher incidence of AEs (e.g., infections and cardiac disorders) leading to death observed in the ribociclib plus AI group. The combination of ribociclib and an AI may offer improved treatment effect; however, the increased risk of death must be weighed against these benefits. In the NATALEE trial, there were increased incidences of hepatobiliary toxicity, QT interval prolongation, and interstitial lung disease or pneumonitis. According to the clinical experts, these AEs are particularly concerning in the adjuvant setting, and there would be added management burden because patients receiving both ribociclib and an AI require more frequent blood work, cardiac monitoring, and clinic visits compared to patients on ET alone. In terms of comparative safety, the MAIC results report increased odds of neutropenia among the ribociclib plus AI group, and decreased odds of TEAEs, such as diarrhea, leukopenia, and lymphopenia, compared to abemaciclib plus ET.
The inputs from patient and clinician groups highlighted that the treatment duration for ribociclib is 3 years and 2 years for abemaciclib, which will increase the need for clinical monitoring for an extra year. Although the median duration of treatment with ribociclib was approximately 3 years (35.7 months) in the NATALEE trial, the mean duration was slightly more than 2 years (26.5 months). The longer median duration may provide more robust data on long-term efficacy and safety effects, but the shorter mean duration suggests that not all patients may benefit from extended treatment with ribociclib. Additionally, the clinical experts and the clinician groups consulted by CDA-AMC noted that adherence is an issue in the adjuvant setting due to longer disease-free intervals, and speculated that the longer treatment duration of ribociclib may cause more adherence issues, based on their experience in the real-world setting.
Conclusion
The input from patients and clinicians highlighted the need for new effective treatments for EBC that reduce the risk of recurrence with manageable side effects and that maintain quality of life. Evidence from 1 phase III, open-label, active-controlled RCT (NATALEE) comparing the efficacy and safety of ribociclib plus AI with AI alone as adjuvant treatment in patients with HR-positive, HER2-negative, stage II or III EBC demonstrated that, when compared to AI alone, the addition of ribociclib to AI results in a statistically significantly prolonged iDFS; however, the magnitude of the benefit was not clinically meaningful based on the 5% threshold suggested by the clinical experts. Using a threshold of 3%, as has been done previously, the results for iDFS at 48 and 60 months may be considered clinically meaningful because most of the observed effect lies higher than the threshold, although this remained uncertain. Results for RFS, DDFS, and OS were consistent with the primary outcome; however, they were secondary and were not controlled for multiplicity. Therefore, they were only considered supportive of the overall effect of ribociclib plus AI. Additionally, the differences between treatment groups for RFS, DDFS, and OS are likely not clinically meaningful based on the thresholds suggested by the clinical experts. The results were uncertain for all time-to-event efficacy outcomes because the medians of these end points were not reached in either study group at the time of the end of ribociclib analysis, indicating the data were immature. There was a decrease in overall quality of life for both ribociclib plus AI and AI alone, although there was little to no difference in HRQoL between treatments and the results were difficult to interpret due to the large amount of missing data, precluding definitive conclusions.
There is extensive experience with ribociclib because it has been available to patients in Canada since 2018. In the NATALEE trial, no new safety signals were identified, and the safety profile was considered manageable based on the clinical experts’ experience with the drug in the metastatic setting.
Indirect evidence via 1 sponsor-submitted MAIC comparing ribociclib plus AI to abemaciclib plus ET suggested no difference between the 2 treatment regimens. However, there were limitations in the indirect evidence, specifically, that the population of patients from the NATALEE study was matched to cohort 1 of the monarchE trial and was not representative of the entire population within the scope of this Reimbursement Review. Additionally, considerable reductions in effective sample size resulting in wide 95% CIs undermined the validity and precision of the results, precluding conclusions on the comparative efficacy of ribociclib plus AI.
References
1.Brenner DR, Gillis J, Demers AA, et al. Projected estimates of cancer in Canada in 2024. Can Med Assoc J. 2024;196(18):E615-E623. doi:10.1503/cmaj.240095 PubMed
2.GLOBOCAN. Breast Cancer: 5-year prevalence in Canada (both sexes) in 2022. 2024. Accessed May 14th. https://gco.iarc.fr/today/en/dataviz/bars-prevalence?mode=cancer&group_populations=1&key=total&sort_by=value1&prev_time=5&sexes=0&populations=124
3.American Cancer Society. What is Breast Cancer? 2023. Accessed 7th August 2024. https://www.cancer.org/cancer/types/breast-cancer/about/what-is-breast-cancer.html
4.Howlader N, Altekruse SF, Li CI, et al. US incidence of breast cancer subtypes defined by joint hormone receptor and HER2 status. J Natl Cancer Inst. 2014;106(5). doi:10.1093/jnci/dju055 PubMed
5.De Polo J. Types of Breast Cancer 2024. Accessed September 5, 2024. https://www.breastcancer.org/types/invasive-ductal-carcinoma
6.Canadian Cancer Statistics Advisory Committee. Canadian Cancer Statistics 2018. 2018. https://cdn.cancer.ca/-/media/files/research/cancer-statistics/2018-statistics/canadian-cancer-statistics-2018-en.pdf
7.Healthline. What are the early signs and symptoms of breast cancer? 2023. Accessed 6th March 2023. https://www.healthline.com/health/breast-cancer/warning-signs#otHER−causes-of-breast-pain.
8.American Cancer Society. Breast cancer signs and symptoms. 2023. Accessed 6th March 2023. https://www.cancer.org/cancer/breast-cancer/screening-tests-and-early-detection/breast-cancer-signs-and-symptoms.html
9.Cancer.Net. Breast cancer: symptoms and signs. 2023. Accessed 6th March 2023. https://www.cancer.net/cancer-types/breast-cancer/symptoms-and-signs
10.Abdolrasoul Talei MA, Maral Mokhtari and Sedigheh Tahmasebi. Surgical and clinical pathology of breast diseases. 2012.
11.Harbeck N, Penault-Llorca F, Cortes J, et al. Breast cancer. Nature Reviews Disease Primers. 2019;5(1):66. doi:10.1038/s41572-019-0111-2 PubMed
12.Pan H, Gray R, Braybrooke J, et al. 20-Year Risks of Breast-Cancer Recurrence after Stopping Endocrine Therapy at 5 Years. N Engl J Med. 2017;377(19):1836-1846. doi:10.1056/NEJMoa1701830 PubMed
13.Canadian Agency for D, Technologies in H. Provisional Funding Algorithm: HR+/HER2- Breast Cancer. 2024. https://www.cadth.ca/sites/default/files/DRR/2024/PH0041-HR%2BHER2-Breast-Cancer.pdf
14.Kisqali (ribociclib): 200mg oral tablets [product monograph]. Novartis Pharmaceuticals Canada, Inc; 2025 June 12. Accessed March 02, 2026. https://pdf.hres.ca/dpd_pm/00081089.PDF
15.Balshem H, Helfand M, Schünemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. Journal of clinical epidemiology. 2011;64(4):401-6. doi:10.1016/j.jclinepi.2010.07.015 PubMed
16.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. Journal of clinical epidemiology. 2020;119:126-135. doi:10.1016/j.jclinepi.2019.10.014 PubMed
17.Clinical Study Report (End of Ribociclib Analysis): CLEE011O12301C (NATALEE). A phase III, multicenter, randomized, open-label trial to evaluate efficacy and safety of ribociclib with endocrine therapy as an adjuvant treatment in patients with hormone receptor-positive, HER2-negative, early breast cancer (New Adjuvant TriAl with Ribociclib [LEE011]: NATALEE) [internal sponsor's report]. Novartis Pharmaceuticals Canada Inc.; 2024 September 12.
18.Clinical Study Report (Interim Analysis 3): CLEE011O12301C (NATALEE). A phase III, multicenter, randomized, open-label trial to evaluate efficacy and safety of ribociclib with endocrine therapy as an adjuvant treatment in patients with hormone receptor-positive, HER2-negative, early breast cancer (New Adjuvant TriAl with Ribociclib [LEE011]: NATALEE) [internal sponsor's report]. Novartis Pharmaceuticals Canada Inc.; 2023.
19.Novartis Pharmaceuticals Canada Inc. response to February 7, 2025 CDA-AMC request for additional information regarding ribociclib (Kisqali) CDA-AMC review: absolute differences [internal additional sponsor's information]. Novartis Pharmaceuticals Canada Inc.; 2025. Accessed February 27, 2025.
20.Novartis Pharmaceuticals Canada Inc. response to August 18, 2025 CDA-AMC request for additional information regarding ribociclib (Kisqali) CDA-AMC review: 95% confidence intervals for between-group difference [internal additional sponsor's information]. Novartis Pharmaceuticals Canada Inc.; 2025. Accessed August 25, 2025.
21.Kisqali (ribociclib) for the treatment of patients with hormone receptor-positive, human epidermal growth factor receptor 2-negative stage II and III early breast cancer: clinical summary [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Kisqali (ribociclib), 200 mg oral tablets Novartis Pharmaceuticals Canada Inc. ; 2025. Accessed March 18, 2025.
22.Mybreastscreening.ca. Your comprehensive guide to breast cancer screening in Canada. 2024. https://mybreastscreening.ca/wp-content/uploads/2024/03/DBC-Screening-Guide-Draft-13_optimized.pdf
23.Mayo Clinic Staff. Recurrent breast cancer. 2024. Accessed August 27, 2024. https://www.mayoclinic.org/diseases-conditions/recurrent-breast-cancer/symptoms-causes/syc-20377135
24.Canadian Cancer Society. Stages of Breast Cancer. 2023.
25.Garutti M, Griguolo G, Botticelli A, et al. Definition of High-Risk Early Hormone-Positive HER2-Negative Breast Cancer: A Consensus Review. Cancers (Basel). 2022;14(8). doi:10.3390/cancers14081898 PubMed
26.Cancer Care Ontario. Breast cancer screening and diagnosis pathway map. Vol. Version 2021.03. 2021. https://www.cancercareontario.ca/sites/ccocancercare/files/assets/BreastCancerScreeningandDiagnosisPathwayMap.pdf
27.Canadian Cancer Society. Hormone receptor status test. 2023. https://cancer.ca/en/treatments/tests-and-procedures/hormone-receptor-status-test
28.Canadian Cancer Society. HER2 Status Test. 2023. Accessed May 14th. https://cancer.ca/en/treatments/tests-and-procedures/her2-status-test#ci_her2_status_test_89_8823_00
29.American Joint Committee on Cancer (AJCC). Cancer Staging Manual, Eighth Edition. 2018. https://cancerstaging.org/references-tools/deskreferences/Documents/AJCC%20Cancer%20Staging%20Form%20Supplement.pdf
30.Canadian Cancer S. Treatments for Breast Cancer. 2024. Accessed February 2024. https://cancer.ca/en/cancer-information/cancer-types/breast/treatment
31.Cancer Care M. Provincial Consensus Recommendations for Adjuvant Systemic Therapy for Breast Cancer. 2017. https://ext-opencms.cancercare.mb.ca/export/sites/default/For-Health-Professionals/.galleries/files/treatment-guidelines-rro-files/practice-guidelines/breast/Adjuvant_Breast_Guideline_July_2017.pdf
32.Cancer Care A. Systemic Therapy for Early Breast Cancer - Quick Reference Guide. 2021. Clinical Practice Guideline BR-014-Version 6. https://www.albertahealthservices.ca/assets/info/hp/cancer/if-hp-cancer-guide-systemic-therapy-early-breast.pdf
33.Drug Reimbursement Review clinical guidance report: abemaciclib (Verzenio) in combination with endocrine therapy for the adjuvant treatment of adult patients with HR-positive, HER2-negative, node-positive early breast cancer at high risk of disease recurrence based on clinicopathologic features and a Ki-67 test score ≥ 20%. CADTH; 2022 Dec 23. Accessed March 20, 2025. https://www.cadth.ca/sites/default/files/DRR/2022/PC0282-Verzenio.pdf
34.FDA approves ribociclib with an aromatase inhibitor and ribociclib and letrozole co-pack for early high-risk breast cancer. September 17, 2024, 2024. Accessed October 17, 2024. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-ribociclib-aromatase-inhibitor-and-ribociclib-and-letrozole-co-pack-early-high-risk-0
35.European C. COMMISSION IMPLEMENTING DECISION amending the marketing authorisation granted by Decision C(2017)5885(final) for “Kisqali - ribociclib”, a medicinal product for human use. 2024. Updated November 25, 2024. https://ec.europa.eu/transparency/documents-register/detail?ref=C(2024)8540&lang=en
36.CADTH Drug Reimbursement Expert Review Committee final recommendation: Ribociclib (Kisqali - Novartis Pharmaceuticals Canada Inc.). CADTH; 2020 Jun 4. Accessed 2025 Jun 4. URL.
37.CADTH Drug Reimbursement Expert Review Committee final recommendation: generic name (Brand name - sponsor's name). CADTH; 1800. Accessed 1800 Jan 1. URL.
38.CADTH Drug Reimbursement Expert Review Committee final recommendation: Ribociclib (Kisqali - Novartis Pharmaceuticals Canada Inc.). CADTH; 2018 Apr 18. Accessed 2025 Mar 3. URL.
39.Verzenio (abemaciclib): 150 mg oral tablets [product monograph]. Eli Lilly Canada, Inc; 2023 Dec 1. Accessed March 03, 2025. https://pi.lilly.com/ca/verzenio-ca-pm.pdf
40.Arimidex (Anastrozole): 1mg oral tablets [product monograph]. AstraZeneca Canada, Inc; 2022. Accessed March 03, 2025.
41.Aromasin (exemestane): 25mg oral tablets [product monograph]. Pfizer Canada Inc.; 2018 Mar 6. Accessed March 03, 2025. https://pdf.hres.ca/dpd_pm/00044156.PDF
42.Zoladex (goserelin depot as goserelin acetate): 3.6 mg subcutaneous injections [product monograph]. AstraZeneca Canada Inc.; 2017 Dec 21. Accessed March 03, 2025. https://pdf.hres.ca/dpd_pm/00042728.PDF
43.Femara (letrozole): 2.5mg oral tablets [product monograph]. Novartis Pharmaceuticals Canada Inc.; 2022 Nov 18. Accessed March 03, 2025. https://pdf.hres.ca/dpd_pm/00071188.PDF
44.AstraZeneca Canada I. Nolvadex (tamoxifen citrate): 10 mg and 20 mg oral tablets [product monograph]. AstraZeneca Canada Inc.; 2003 Aug 25. Accessed August 25, 2003. https://pdf.hres.ca/dpd_pm/00000082.PDF
45.Statistical Analysis Plan: A phase III, multicenter, randomized, open-label trial to evaluate efficacy and safety of ribociclib with endocrine therapy as an adjuvant treatment in patients with hormone receptor-positive, HER2-negative, early breast cancer[internal sponsor's report]. Novartis Pharmaceuticals Canada Inc.; 2023:59. Accessed March 19, 2025.
46.Hudis CA, Barlow WE, Costantino JP, et al. Proposal for standardized definitions for efficacy end points in adjuvant breast cancer trials: the STEEP system. J Clin Oncol. 2007;25(15):2127-32. doi:10.1200/JCO.2006.10.3523 PubMed
47.Aaronson NK, Ahmedzai S, Bergman B, et al. The European Organization for Research and Treatment of Cancer QLQ-C30: a quality-of-life instrument for use in international clinical trials in oncology. J Natl Cancer Inst. 1993;85(5):365-76. doi:10.1093/jnci/85.5.365 PubMed
48.Musoro JZ, Coens C, Fiteni F, et al. Minimally Important Differences for Interpreting EORTC QLQ-C30 Scores in Patients With Advanced Breast Cancer. JNCI Cancer Spectrum. 2019;3(3):pkz037. doi:10.1093/jncics/pkz037 PubMed
49.Abramson JS, Johnston PB, Kamdar M, et al. Health-related quality of life with lisocabtagene maraleucel vs standard of care in relapsed or refractory LBCL. Blood Adv. 2022;6(23):5969-5979. doi:10.1182/bloodadvances.2022008106 PubMed
50.Davda J, Kibet H, Achieng E, Atundo L, Komen T. Assessing the acceptability, reliability, and validity of the EORTC Quality of Life Questionnaire (QLQ-C30) in Kenyan cancer patients: a cross-sectional study. J Patient Rep Outcomes. 2021;5(1):4. doi:10.1186/s41687-020-00275-w PubMed
51.Untch M, Perol D, Mayer EL, et al. Disease-free survival as a surrogate for overall survival in HR+/HER2- early breast cancer: A correlation analysis. Eur J Cancer. 2024;202:113977. doi:10.1016/j.ejca.2024.113977 PubMed
52.Fasching PA, Gass P, Haberle L, et al. Prognostic effect of Ki-67 in common clinical subgroups of patients with HER2-negative, hormone receptor-positive early breast cancer. Breast Cancer Res Treat. 2019;175(3):617-625. doi:10.1007/s10549-019-05198-9 PubMed
53.Early Breast Cancer Trialists' Collaborative G. Aromatase inhibitors versus tamoxifen in early breast cancer: patient-level meta-analysis of the randomised trials. Lancet. 2015;386(10001):1341-1352. doi:10.1016/S0140-6736(15)61074-1 PubMed
54.Lan KKG, DeMets DL. Discrete Sequential Boundaries for Clinical Trials. Biometrika. 1983;70(3):659-663. doi:10.2307/2336502
55.Novartis Pharmaceuticals Canada Inc. response to March 6, 2025 CDA-AMC request for additional information regarding ribociclib (Kisqali) CDA-AMC review: patients who were randomized but not treated [internal additional sponsor's information]. Novartis Pharmaceuticals Canada Inc.; 2025. Accessed March 18, 2025.
56.Health PAIIpoA. Data on file. Indirect Treatment Comparisons of Ribociclib plus Endocrine Therapy vs. Abemaciclib plus Endocrine Therapy in Early Breast Cancer Based on NATALEE and monarchE Trials: April 29, 2024 DCO Analysis Update. 2024.
57.Phillippo DM, Ades AE, Dias S, Palmer S, Abrams KR, Welton NJ. Methods for Population-Adjusted Indirect Comparisons in Health Technology Appraisal. Med Decis Making. 2018;38(2):200-211. doi:10.1177/0272989X17725740 PubMed
58.Signorovitch JE, Sikirica V, Erder MH, et al. Matching-adjusted indirect comparisons: a new tool for timely comparative effectiveness research. Value Health. 2012;15(6):940-7. doi:10.1016/j.jval.2012.05.004 PubMed
59.Phillippo DM, Ades AE, Dias S, Palmer S, Abrams KR, Welton NJ. NICE DSU Technical Support Document 18: Methods for population-adjusted indirect comparisons in submission to NICE. 2016. Available from http://www.nicedsu.org.uk.
60.Rastogi P, O'Shaughnessy J, Martin M, et al. Adjuvant Abemaciclib Plus Endocrine Therapy for Hormone Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative, High-Risk Early Breast Cancer: Results From a Preplanned monarchE Overall Survival Interim Analysis, Including 5-Year Efficacy Outcomes. J Clin Oncol. 2024;42(9):987-993. doi:10.1200/JCO.23.01994 PubMed
61.Johnston SRD, Toi M, O'Shaughnessy J, et al. Abemaciclib plus endocrine therapy for hormone receptor-positive, HER2-negative, node-positive, high-risk early breast cancer (monarchE): results from a preplanned interim analysis of a randomised, open-label, phase 3 trial. The Lancet Oncology. 2023;24(1):77-90. doi:10.1016/S1470-2045(22)00694-5 PubMed
62.Toi M, Boyle F, Im Y-H, et al. Adjuvant Abemaciclib Combined with Endocrine Therapy: Efficacy Results in monarchE Cohort 1. The Oncologist. 2022;28(1):e77-e81. doi:10.1093/oncolo/oyac234 PubMed
63.Guyot P, Ades AE, Ouwens MJ, Welton NJ. Enhanced secondary analysis of survival data: reconstructing the data from published Kaplan-Meier survival curves. BMC Med Res Methodol. 2012;12:9. doi:10.1186/1471-2288-12-9 PubMed
64.Bucher HC, Guyatt GH, Griffith LE, Walter SD. The results of direct and indirect treatment comparisons in meta-analysis of randomized controlled trials. J Clin Epidemiol. 1997;50(6):683-691. doi:10.1016/s0895-4356(97)00049-8 PubMed
65.Remiro-Azócar A, Heath A, Baio G. Marginalization of regression-adjusted treatment effects in indirect comparisons with limited patient-level data. arXiv preprint arXiv:200805951. 2020.
66.Remiro-Azócar A, Heath A, Baio G. Effect modification in anchored indirect treatment comparisons: Comments on” Matching-adjusted indirect comparisons: Application to time-to-event data”. arXiv preprint arXiv:201205127. 2020.
67.Novartis Pharmaceuticals Canada Inc. response to February 24, 2025 CDA-AMC request for additional information regarding ribociclib (Kisqali) CDA-AMC review: results for subgroup analysis [internal additional sponsor's information]. Novartis Pharmaceuticals Canada Inc.; 2025. Accessed March 18, 2025.
68.Slamon D, Lipatov O, Nowecki Z, et al. Ribociclib plus Endocrine Therapy in Early Breast Cancer. N Engl J Med. Mar 21 2024;390(12):1080-1091. doi:10.1056/NEJMoa2305488 PubMed
69.Wolfgang Janni, Michael Untch, Nadia Harbeck, et al. Comparing the efficacy of aromatase inhibitors vs tamoxifen in hormone receptor-positive, human epidermal growth factor receptor 2-negative early breast cancer: a systematic review and trial-level meta-analysis. Cancer Res. 2023;83(5)
Appendix 1: Detailed Outcome Data
Please note that this appendix has not been copy-edited.
Figure 2: Kaplan-Meier Plot for Invasive Disease–Free Survival at IA3 (FAS; Data Cut-Off Date — January 11, 2023) and the End of Ribociclib Analysis (FAS; Data Cut-Off Date — April 29, 2024)
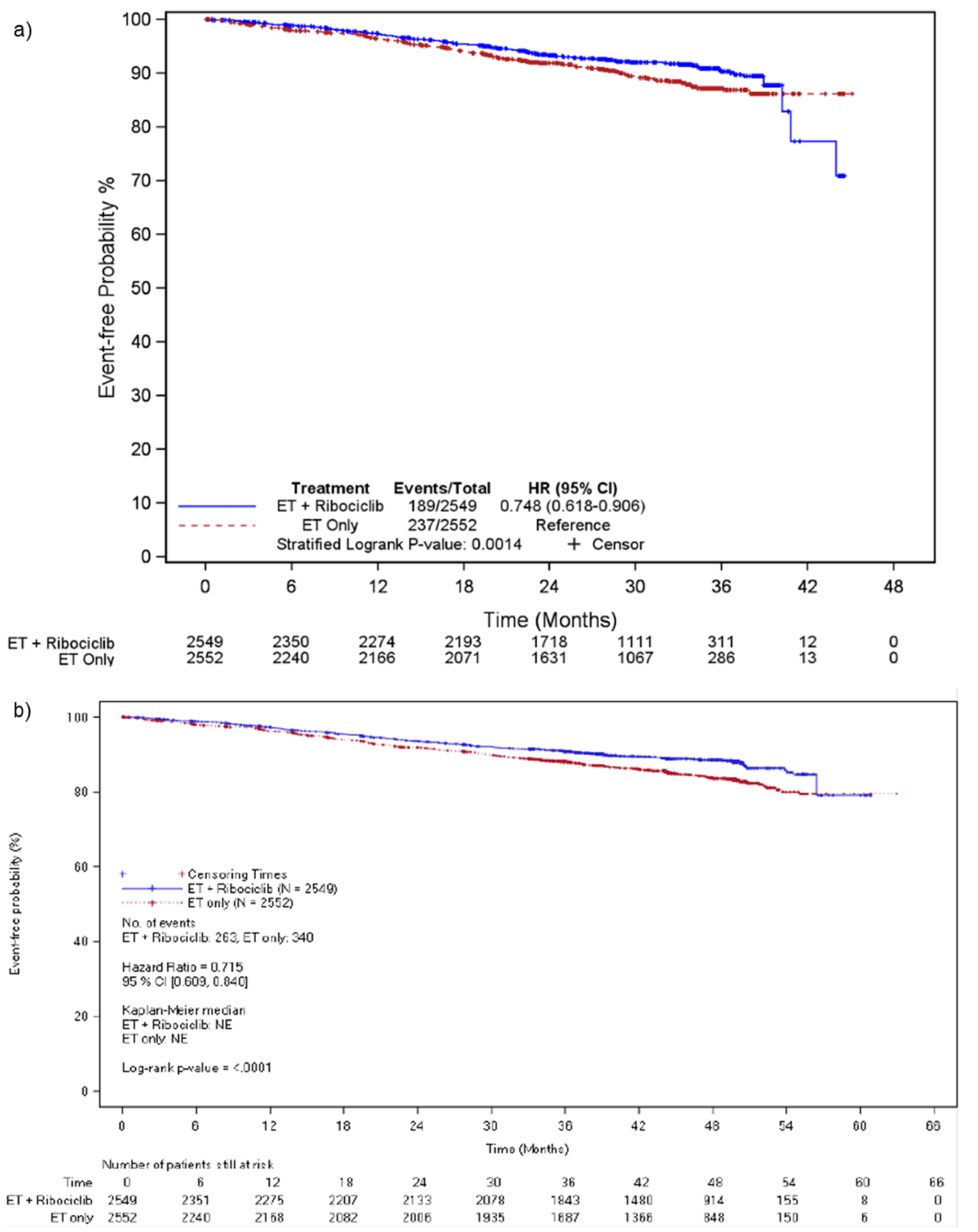
CI = confidence interval; ET = endocrine therapy; NE = not evaluated.
Sources: NATALEE Clinical Study Report for interim analysis 3 (2023)18 and NATALEE Clinical Study Report for the end of ribociclib analysis (2024).17
Table 27: iDFS Results by Subgroups
Subgroup | Interim analysis 3 (Data cut-off date: January 11, 2023) | End of ribociclib analysis (Data cut-off date: April 29, 2024) | ||||
|---|---|---|---|---|---|---|
Events (n/N) | Hazard ratio (95% CI) | Events (n/N) | Hazard ratio (95% CI) | |||
Ribociclib + AI N = 2,549 | AI N = 2,552 | Ribociclib + AI N = 2,549 | AI N = 2,552 | |||
Anatomical stage | ||||||
IIA | 13/479 | 27/521 | 0.500 (0.258 to 0.968) | 18/480 | 43/521 | 0.423 (0.244 to 0.733) |
IIB | 36/532 | 38/513 | 0.930 (0.598 to 1.467) | 44/532 | 53/513 | 0.811(0.544 to 1.209) |
IIIA | 75/939 | 85/894 | 0.792 (0.580 to 1.080) | 94/938 | 119/894 | 0.699 (0.533 to 0.916) |
IIIB | 18/168 | 20/149 | 0.678 (0.355 to 1.295) | 23/168 | 30/149 | 0.622 (0.361 to 1.071) |
IIIC | 47/421 | 67/496 | 0.685 (0.472 to 0.994) | 83/421 | 95/469 | 0.848 (0.632 to 1.139) |
Gender | ||||||
Female | 188/2,538 | 236/2,543 | 0.758 (0.625 to 0.918) | 263/2,539 | 339/2,543 | 0.728 (0.620 to 0.856) |
Prior adjuvant chemotherapy | ||||||
Yes | 63/1,223 | 89/1,220 | 0.671 (0.486 to 0.927) | 96/1,223 | 140/1,220 | 0.644 (0.496 to 0.835) |
No | 126/1,326 | 148/1,332 | 0.814 (0.642 to 1.032) | 167/1,326 | 200/1,332 | 0.786 (0.640 to 0.965) |
Prior neoadjuvant chemotherapy | ||||||
Yes | 111/1,085 | 132/1,095 | 0.785 (0.610 to 1.011) | 150/1085 | 177/1,095 | 0.784 (0.631 to 0.974) |
No | 78/1,464 | 105/1,457 | 0.717 (0.535 to 0.961) | 113/1464 | 163/1,457 | 0.659 (0.519 to 0.838) |
Prior radiation therapy | ||||||
Yes | 170/2,292 | 218/2,302 | 0.739 (0.605 to 0.903) | 238/2,292 | 309/2,302 | 0.723 (0.610 to 0.856) |
No | 19/257 | 19/250 | 0.980 (0.519 to 1.851) | 25/257 | 31/250 | 0.775 (0.457 to 1.312) |
Prior endocrine therapy | ||||||
Yes | 127/1,824 | 157/1,801 | 0.756 (0.598 to 0.955) | 176/1,803 | 227/1,807 | 0.718 (0.589 to 0.874) |
No | 62/725 | 80/751 | 0.774 (0.556 to 1.079) | 87/719 | 113/745 | 0.752 (0.568 to 0.994) |
Prior mastectomy | ||||||
Yes | 147/1,664 | 165/1,691 | 0.853 (0.683 to 1.066) | 198/1,645 | 237/1,678 | 0.799 (0.661 to 0.964) |
No | 42/885 | 72/861 | 0.548 (0.375 to 0.802) | 65/904 | 103/874 | 0.577 (0.423 to 0.787) |
Race | ||||||
Asian | 18/341 | 29/334 | 0.523 (0.290 to 0.942) | 34/341 | 41/334 | 0.707 (0.449 to 1.114) |
Non-Asian | 160/2,070 | 192/2,091 | 0.807 (0.654 to 0.995) | 215/2,070 | 279/2,091 | 0.738 (0.618 to 0.882) |
Region | ||||||
Europe | 122/1,505 | 138/1,506 | 0.875 (0.686 to 1.116) | 159/1,505 | 202/1,506 | 0.769 (0.624 to 0.946) |
North America and Australia | 47/624 | 58/621 | 0.697 (0.475 to 1.025) | 65/624 | 78/612 | 0.709 (0.510 to 0.985) |
Asia | 9/281 | 24/290 | 0.338 (0.157 to 0.728) | 22/281 | 35/290 | 0.563 (0.330 to 0.960) |
Latin America | 11/139 | 17/144 | 0.691 (0.323 to 1.477) | 17/139 | 25/144 | 0.712 (0.383 to 1.322) |
Age category | ||||||
< 45 | 39/611 | 50/591 | 0.683 (0.449 to 1.038) | 58/611 | 72/591 | 0.692 (0.489 to 0.977) |
45 to 54 | 52/849 | 69/895 | 0.750 (0.524 to 1.075) | 71/849 | 98/895 | 0.716 (0.528 to 0.972) |
55 to 64 | 64/682 | 76/700 | 0.839 (0.601 to 1.170) | 88/682 | 102/700 | 0.850 (0.639 to 1.130) |
≥ 65 | 34/407 | 42/366 | 0.723 (0.460 to 1.137) | 46/407 | 68/366 | 0.589 (0.405 to 0.857) |
< Median | 71/1,216 | 95/1,264 | 0.727 (0.535 to 0.989) | 106/1,216 | 140/1,264 | 0.727 (0.565 to 0.935) |
≥ Median | 118/1,333 | 142/1,288 | 0.775 (0.607 to 0.990) | 157/1,333 | 200/1,288 | 0.721 (0.405 to 0.857) |
Type of NSAI | ||||||
Letrozole | 127/1,729 | 154/1,674 | 0.790 (0.624 to 0.999) | 181/1,720 | 224/1,671 | 0.767 (0.630 to 0.933) |
Anastrozole | 62/797 | 83/768 | 0.702 (0.505 to 0.976) | 82/806 | 116/770 | 0.644 (0.486 to 0.855) |
HR status | ||||||
ER+/PR+ | 140/2,172 | 180/2,132 | 0.726 (0.582 to 0.906) | 200/2,172 | 263/2,132 | 0.700 (0.583 to 0.842) |
ER+/PR– | 48/359 | 54/392 | 0.908 (0.615 to 1.339) | 60/359 | 72/392 | 0.837 (0.594 to 1.180) |
Nodal status | ||||||
N0 | 16/285 | 28/328 | 0.630 (0.341 to 1.165) | 23/285 | 38/328 | 0.666 (0.397 to 1.118) |
N1 to N3 | 173/2,261 | 208/2,219 | 0.771 (0.630 to 0.944) | 240/2,261 | 301/2,219 | 0.731 (0.617 to 0.866) |
Tumour category | ||||||
T1 to T3 | 164/2,345 | 212/2,360 | 0.742 (0.605 to 0.910) | 229/2,345 | 301/2,360 | 0.720 (0.606 to 0.855) |
> T3 | 22/189 | 23/181 | 0.827 (0.460 to 1.486) | 29/189 | 36/181 | 0.715 (0.438 to 1.166) |
Histological grade at time of surgery | ||||||
Grade 1 | 9/213 | 12/217 | 0.778 (0.328 to 1.846) | 11/213 | 17/217 | 0.664 (0.311 to 1.419) |
Grade 2 | 102/1,450 | 125/1,432 | 0.749 (0.577 to 0.973) | 136/1,461 | 185/1,432 | 0.663 (0.532 to 0.828) |
Grade 3 | 61/684 | 78/702 | 0.776 (0.555 to 1.065) | 92/684 | 106/702 | 0.852 (0.644 to 1.127) |
Ki-67 status from archival tumour | ||||||
Ki-67 ≤ 20 | 76/1,199 | 95/1,236 | 0.801 (0.593 to 1.083) | 106/1,199 | 142/1,236 | 0.737 (0.573 to 0.948) |
Ki-67 > 20 | 82/920 | 105/938 | 0.746 (0.559 to 0.996) | 113/920 | 149/937 | 0.709 (0.555 to 0.905) |
Histological subtype | ||||||
Ductal | 123/1,858 | 174/1,881 | 0.675 (0.536 to 0.850) | 187/1,916 | 258/1,948 | 0.684 (0.567 to 0.826) |
Lobular | 47/455 | 43/450 | 1.503 (0.696 to 1.592) | 62/467 | 66/459 | 0.897 (0.634 to 1.269) |
Other | 19/235 | 20/221 | 0.887 (0.473 to 1.661) | 14/165 | 16/145 | 0.727 (0.355 to 1.489) |
BMI at screening | ||||||
≥ 25 | 124/1,505 | 142/1,522 | 0.848 (0.667 to 1.079) | 177/1,505 | 206/1,522 | 0.821 (0.672 to 1.004) |
< 25 | 64/1,013 | 93/999 | 0.635 (0.482 to 0.873) | 85/1,014 | 131/999 | 0.596 (0.454 to 0.783) |
AI = aromatase inhibitor; BMI = body mass index; CI = confidence interval; ER = estrogen receptor; PR = progesterone receptor; NSAI = nonsteroidal aromatase inhibitor.
Sources: NATALEE Clinical Study Report for interim analysis 3 (2023)18 and NATALEE Clinical Study Report for the end of ribociclib analysis (2024).17
Table 28: Post Hoc Subgroup Results for the monarchE Trial Cohort 1–Eligible vs. Cohort 1–Ineligible From the NATALEE Study (FAS; Data Cut-Off Date — April 29, 2024)
Subgroup | End of ribociclib analysis (data cut-off date: April 29, 2024) | |
|---|---|---|
Ribociclib + AI N = 2,549 | AI N = 2,552 | |
monarchE cohort 1–eligible | ||
Patients in subgroup, n | █████ | █████ |
Patients with event, n (%) | ███ ████ | ███ ████ |
Nonmetastatic recurrence | ██ ██████ | ██ ██████ |
Distant recurrence | ███ ██████ | ███ ██████ |
Second primary malignancy | ██ ██████ | ██ █████ |
Death | ███████ | ███████ |
Hazard ratio (95% CI) for iDFS | █████ ██████ ██ ██████ | |
monarchE cohort 1–ineligible | ||
Patients in subgroup, n | ███ | ███ |
Patients with event, n (%) | ██ ███ | ██ ███ |
Nonmetastatic recurrence | ██ ██████ | ██ ██████ |
Distant recurrence | ██ ██████ | ██ ██████ |
Second primary malignancy | ██ ██████ | ██ ██████ |
Death | ████████ | ███████ |
Hazard ratio (95% CI) for iDFS | █████ ██████ ██ ██████ | |
CI = confidence interval.
Source: Sponsor-provided additional data.67
Figure 3: Kaplan-Meier Plot for Invasive Disease–Free Survival (FAS; Data Cut-Off Date — May 28, 2025)
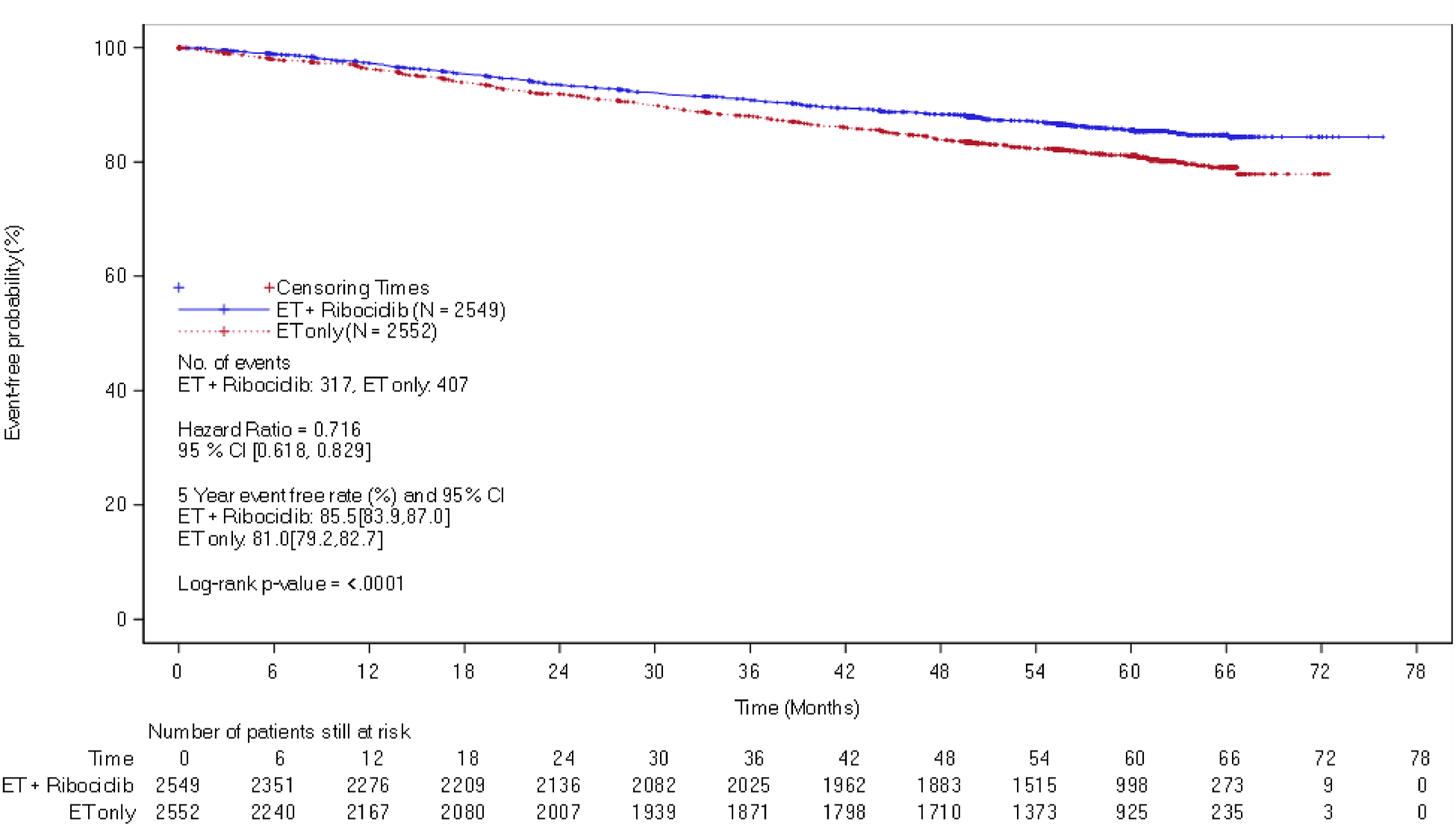
CI = confidence interval; ET = endocrine therapy.
Source: Sponsor-provided additional data.20
Figure 4: Kaplan-Meier Plot for Recurrence-Free Survival at the End of Ribociclib Analysis (FAS; Data Cut-Off Date — April 29, 2024) and 5-Year Follow-Up (FAS; Data Cut-Off Date — May 28, 2025) [Redacted]

CI = confidence interval; ET = endocrine therapy; NE = not evaluated.
Source: NATALEE Clinical Study Report for the end of ribociclib analysis (2024)17 and sponsor-provided additional data.20
Figure 5: Kaplan-Meier Plot for Distant Disease–Free Survival at the End of Ribociclib Analysis (FAS; Data Cut-Off Date — April 29, 2024) and 5-Year Follow-Up (FAS; Data Cut-Off Date — May 28, 2025)
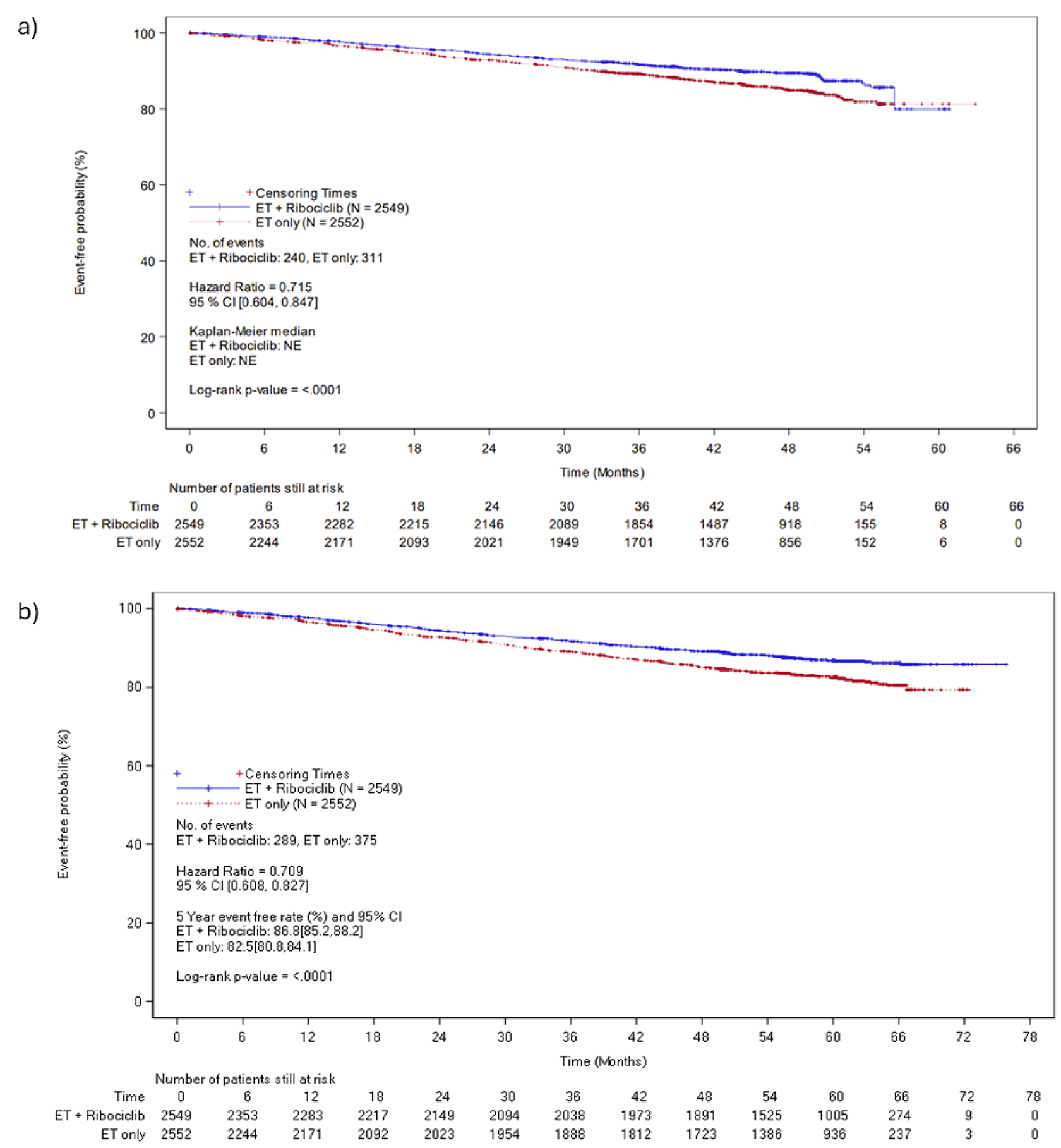
CI = confidence interval; ET = endocrine therapy; NE = not evaluated.
Sources: NATALEE Clinical Study Report for the end of ribociclib analysis (2024)17 and sponsor-provided additional data.20
Figure 6: Kaplan-Meier Plot for Overall Survival at the End of Ribociclib Analysis (FAS; Data Cut-Off Date — April 29, 2024) and 5-Year Follow-Up (FAS; Data Cut-Off Date — May 28, 2025) [Redacted]

CI = confidence interval; ET = endocrine therapy; NE = not evaluated.
Sources: NATALEE Clinical Study Report for the end of ribociclib analysis (2024)17 and sponsor-provided additional data.20
Figure 7: Change From Baseline in the Global Health Status and Quality of Life Subscale Score of the EORTC QLQ-C30 (FAS; Data Cut-Off Date — January 11, 2023)
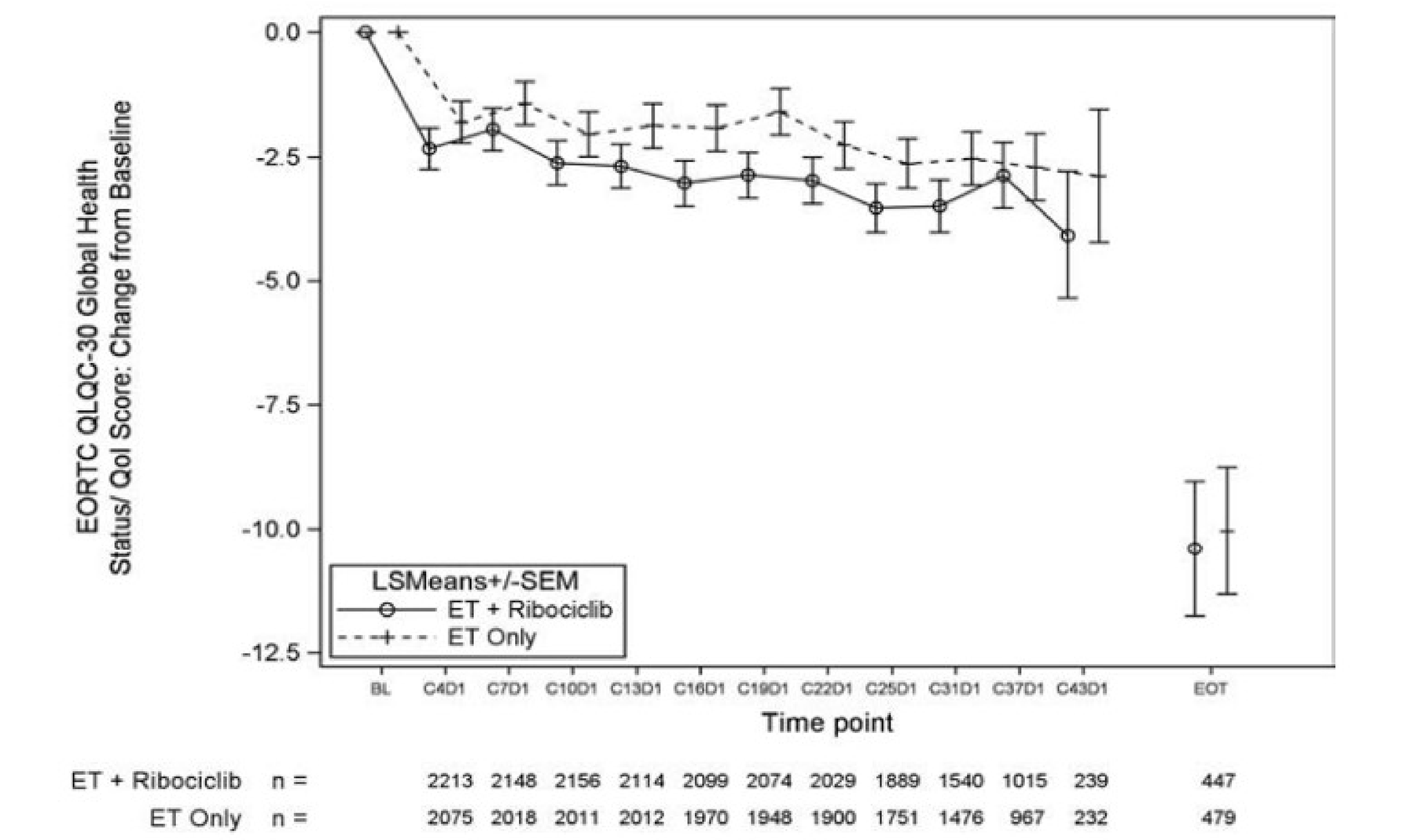
BL = baseline; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EOT = end of treatment; ET = endocrine therapy; FAS = full analysis set; LS = least square; SEM = standard mean error; QoL = quality of life.
Source: NATALEE Clinical Study Report for interim analysis 3 (2023).18
Table 29: Unadjusted Baseline Characteristics in the monarchE and NATALEE Trials (Data Cut-Off Date: April 29, 2024)
Characteristics | monarchE (cohort 1) | NATALEE trial selected, unweighted | SMD | |||
|---|---|---|---|---|---|---|
Abemaciclib + ET (N = 2,555) | ET (N = 2,565) | Ribociclib + ET (N = 1,658) | ET (N = 1,649) | Ribociclib + ET | ET | |
Age, years | ||||||
Mean | 52.2 | 52.2 | 52.3 | 52.5 | 0.0 | 0.0 |
< 65, % | 84.1% | 85.4% | 85.4% | 86.4% | −3.6% | −2.8% |
≥ 65, % | 15.9% | 14.6% | 14.6% | 13.6% | 3.6% | 2.8% |
Female, % | 99.2% | 99.2% | 99.6% | 99.7% | −4.5% | −5.6% |
Race, % | ||||||
Asian | 24.7% | 23.9% | 16.5% | 15.9% | 19.0% | 18.8% |
White | 70.6% | 71.0% | 70.0% | 69.4% | 1.3% | 3.5% |
Other | 4.7% | 5.1% | 13.6% | 14.7% | −42.1% | −43.6% |
Ethnicity, % | ||||||
Hispanic or Latino | 8.0% | 8.9% | 8.0% | 8.3% | 0.0% | 2.1% |
Not Hispanic or Latino | 92.0% | 91.1% | 81.2% | 80.5% | 39.8% | 33.3% |
Missing | 0.0% | 0.0% | 10.7% | 11.2% | 0.0% | 0.0% |
Region, % | ||||||
North America or Europe | 51.8% | 51.9% | 75.9% | 76.2% | −48.2% | −48.6% |
Asia | 20.4% | 20.4% | 13.7% | 14.4% | 16.6% | 14.9% |
Other | 27.8% | 27.7% | 10.4% | 9.5% | 38.8% | 40.7% |
Weight, kg, mean | 71.3 | 71.7 | 71.8 | 71.9 | 0.0 | 0.0 |
BMI, kg/m2, mean | 27.2 | 27.4 | 27.1 | 27.2 | 0.0 | 0.0 |
Pathological diagnosis term, % | ||||||
Ductal carcinoma | 67.3% | 68.7% | 73.2% | 73.4% | −12.6% | −10.1% |
Lobular carcinoma | 13.9% | 13.1% | 20.5% | 20.5% | −19.1% | −22.0% |
Tubular carcinoma | 0.1% | 0.2% | 0.1% | 0.0% | 0.0% | 6.4% |
Other | 18.7% | 18.1% | 6.2% | 6.1% | 32.1% | 31.1% |
HR status, % | ||||||
ER+ | 99.3% | 99.3% | 99.1% | 98.7% | 2.4% | 7.4% |
ER− | 0.5% | 0.6% | 0.1% | 0.4% | 5.6% | 2.5% |
ER missing | 0.2% | 0.0% | 0.8% | 0.8% | −13.7% | −40.3% |
PR+ | 86.4% | 86.8% | 85.3% | 83.5% | 3.4% | 10.2% |
PR− | 10.5% | 10.5% | 13.9% | 15.6% | −11.1% | −16.6% |
PR missing | 3.1% | 2.7% | 0.8% | 0.8% | 18.3% | 16.4% |
Menopausal status, % | ||||||
Premenopause | 43.7% | 43.1% | 45.9% | 44.9% | −4.4% | −3.6% |
Postmenopause | 56.3% | 56.9% | 54.1% | 55.1% | 4.4% | 3.6% |
Positive axillary lymph nodes, % | ||||||
0 | 0.2% | 0.2% | 0.0% | 0.0% | 4.1% | 4.1% |
1 to 3 | 34.2% | 34.6% | 29.3% | 28.4% | 10.3% | 13.0% |
≥ 4 | 65.6% | 65.1% | 70.7% | 71.6% | −10.7% | −13.6% |
Histopathology at Dx, % | ||||||
Grade 1 | 7.3% | 7.4% | 7.0% | 6.7% | 1.2% | 2.7% |
Grade 2 | 46.2% | 46.5% | 51.7% | 51.1% | −11.0% | −9.2% |
Grade 3 | 41.6% | 40.9% | 34.9% | 35.2% | 13.6% | 11.6% |
Not assessed or missing | 4.9% | 5.1% | 6.5% | 7.0% | −7.4% | −8.6% |
ECOG PS | ||||||
0 | 85.4% | 83.7% | 82.6% | 83.2% | 5.6% | 1.0% |
1 | 14.5% | 16.1% | 17.2% | 16.7% | −5.4% | −1.2% |
2 | 0.0% | 0.1% | 0.0% | 0.0% | — | — |
3 | 0.0% | 0.0% | 0.0% | 0.0% | — | — |
Missing | 0.1% | 0.1% | 0.2% | 0.1% | −8.6% | 0.0% |
Tumour side | ||||||
Left | 51.8% | 49.8% | 49.8% | 51.2% | 4.0% | −2.8% |
Right | 46.6% | 49.1% | 50.1% | 48.5% | −7.0% | 1.2% |
Bilateral | 1.6% | 1.1% | 0.1% | 0.4% | 16.6% | 8.5% |
Pathologic tumour size (cm), % | ||||||
< 2 | 26.5% | 25.6% | 12.4% | 11.0% | 32.0% | 33.5% |
2 to 5 | 48.3% | 49.8% | 40.8% | 44.7% | 15.0% | 10.2% |
≥ 5 | 23.5% | 23.6% | 46.5% | 44.3% | −54.3% | −48.7% |
Missing | 1.8% | 1.0% | 0.2% | 0.1% | 12.0% | 9.2% |
Ki-67 index, % | ||||||
< 20 | 37.0% | 37.7% | 27.5% | 28.4% | 19.7% | 19.2% |
≥ 20 | 39.8% | 38.4% | 23.2% | 22.8% | 33.9% | 32.1% |
Missing | 23.2% | 23.9% | 49.3% | 48.8% | −61.8% | −58.4% |
TNM stage, % | ||||||
IA | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | — |
IB | 0.0% | 0.0% | 0.2% | 0.1% | −4.3% | −3.5% |
IIA | 9.0% | 9.7% | 2.6% | 2.6% | 22.4% | 24.0% |
IIB | 11.0% | 11.2% | 6.8% | 8.3% | 13.4% | 9.2% |
IIIA | 40.0% | 39.7% | 56.6% | 54.2% | −33.9% | −29.6% |
IIIB | 3.8% | 3.3% | 8.4% | 6.4% | −24.2% | −17.4% |
IIIC | 35.8% | 36.1% | 25.4% | 28.4% | 21.7% | 16.0% |
Missing | 0.4% | 0.2% | 0.0% | 0.0% | 9.1% | 7.2% |
Prior chemotherapy, % | ||||||
Neoadjuvant | 36.5% | 36.3% | 49.2% | 48.8% | −26.4% | −26.0% |
Adjuvant | 58.7% | 58.6% | 48.9% | 50.3% | 19.9% | 16.9% |
None | 4.8% | 5.1% | 4.3% | 4.7% | 2.2% | 1.6% |
CDK4/6 = cyclin-dependent kinase 4 and kinase 6; Dx = diagnosis; ECOG PS = Eastern Cooperative Oncology Group Performance Status; ER+ = estrogen receptor–positive; ER– = estrogen receptor–negative; ET = endocrine therapy; PR+ = progesterone receptor–positive; PR– = progesterone receptor–negative; TNM = tumour, node, and metastasis; SMD = standardized mean difference.
Source: Indirect treatment comparison report.56
Figure 8: Primary Analysis — iDFS KM Ribociclib Plus ET vs. Abemaciclib Plus ET
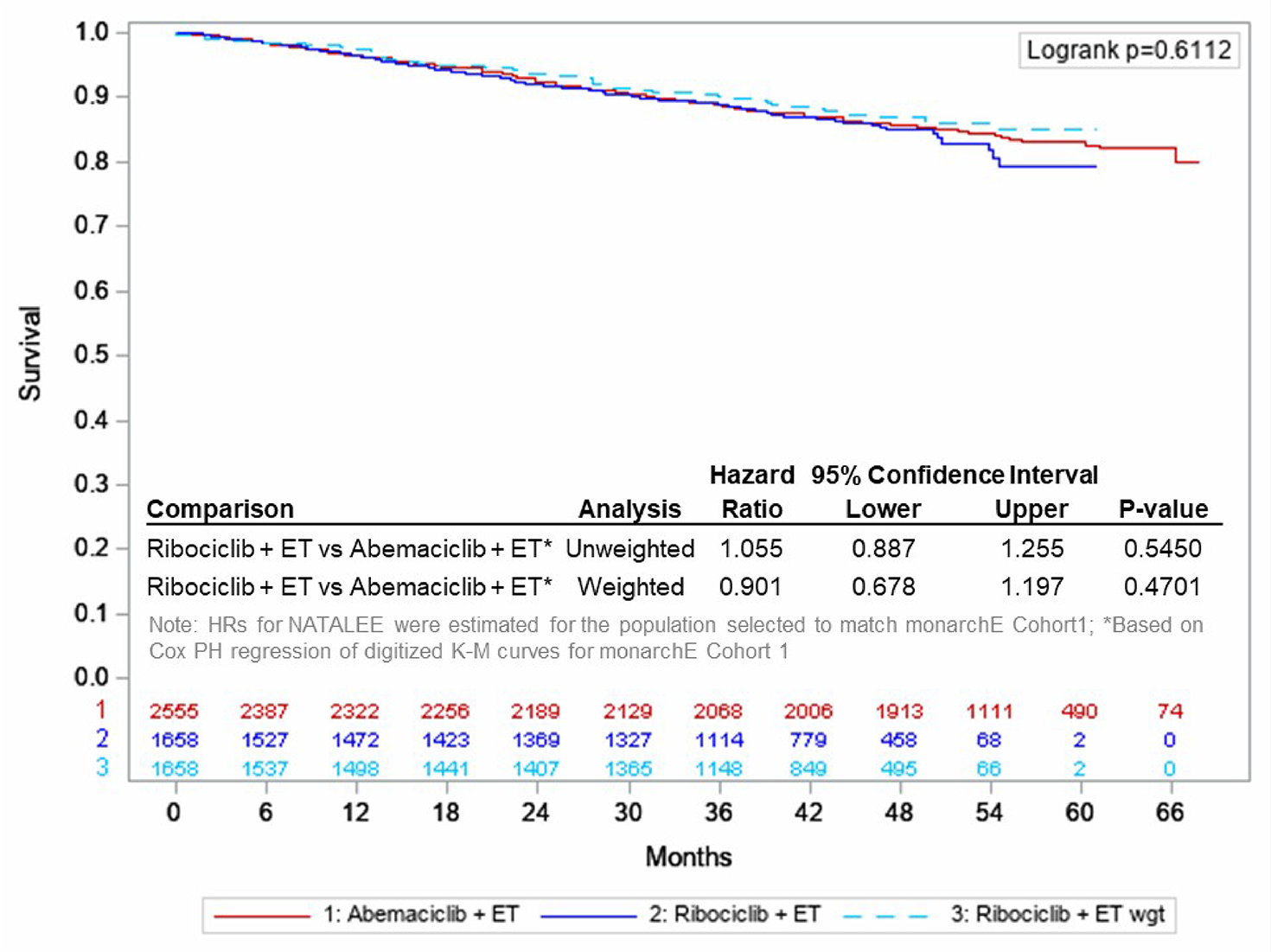
ET = endocrine therapy; iDFS = invasive disease–free survival; K-M = Kaplan-Meier; PH = proportional hazards; vs = versus; wgt = weight.
Source: Indirect treatment comparison report.56
Figure 9: Primary Analysis — DRFS KM Ribociclib Plus ET vs. Abemaciclib Plus ET [Redacted]

DRFS = distant recurrence–free survival; ET = endocrine therapy; KM = Kaplan-Meier; wgt = weight.
Source: Indirect treatment comparison report.56
Figure 10: Primary Analysis — OS KM Ribociclib Plus ET vs. Abemaciclib Plus ET [Redacted]

ET = endocrine therapy; KM = Kaplan-Meier; OS = overall survival; wgt = weight.
Source: Indirect treatment comparison report.56
Pharmacoeconomic Review
Abbreviations
AE
adverse event
AI
aromatase inhibitor
BIA
budget impact analysis
CDA-AMC
Canada’s Drug Agency
ET
endocrine therapy
HR
hormone receptor
HRQoL
health-related quality of life
ICER
incremental cost-effectiveness ratio
iDFS
invasive disease–free survival
ITC
indirect treatment comparison
ITT
intention to treat
LY
life-year
MAIC
matching-adjusted indirect comparison
OS
overall survival
QALY
quality-adjusted life-year
RDI
relative dose intensity
TTD
time to treatment discontinuation
Economic Review
The objective of the economic review undertaken by Canada’s Drug Agency (CDA-AMC) is to review and critically appraise the pharmacoeconomic evidence submitted by the sponsor on the cost-effectiveness and budget impact of ribociclib in combination with an aromatase inhibitor (AI) for the adjuvant treatment of hormone receptor (HR)-positive, HER2-negative early breast cancer. This was conducted by considering 2 subgroups: patients who met the eligibility criteria for cohort 1 of the monarchE trial (4 or more positive axillary lymph nodes, or between 1 and 3 positive nodes and either grade 3 disease or tumour size of 5 cm or larger; abemaciclib-eligible subgroup) and patients who met the ribociclib plus AI criteria but not the criteria for cohort 1 of the monarchE trial (abemaciclib-ineligible subgroup).
Item | Description |
|---|---|
Drug product | Ribociclib (Kisqali), 200 mg, oral tablets |
Indication | Proposed: For the adjuvant treatment of patients with hormone receptor-positive, human epidermal growth factor receptor 2-negative stage II and III early breast cancer, in combination with an aromatase inhibitor. In pre- or perimenopausal women, or men, the aromatase inhibitor should be combined with a luteinizing hormone-releasing hormone agonist. |
Submitted price | Ribociclib: $94.13 per 200 mg tablet |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | June 12, 2025 |
Reimbursement request | Per indication |
Sponsor | Novartis Pharmaceuticals Canada Inc. |
Submission history | Previously reviewed: Yes Indication: Advanced or metastatic breast cancer Recommendation date: April 18, 2018 Recommendation: Recommended with clinical criteria and/or conditions Indication: In combination with fulvestrant for HR-positive, HER2-negative advanced or metastatic breast cancer Recommendation date: April 22, 2020 Recommendation: Recommended with clinical criteria and/or conditions Indication: HR-positive, HER2-negative advanced or metastatic breast cancer Recommendation date: June 4, 2020 Recommendation: Recommended with clinical criteria and/or conditions |
HR = hormone receptor; NOC = Notice of Compliance.
Key Messages
Ribociclib is available as 200 mg tablets. At the submitted price of $94.13 per tablet, the 28-day cycle cost of ribociclib is expected to be $3,953 per patient (ribociclib plus AI: $3,980 to $3,992 per patient), based on the Health Canada–recommended dosage. Ribociclib should be administered for a maximum of 3 years.
Clinical efficacy in the economic analysis for ribociclib plus AI was derived from the NATALEE trial, which suggests that ribociclib plus AI results in prolonged invasive disease–free survival (iDFS) compared to AI alone in the full NATALEE trial population; however, the difference in iDFS between groups may not be clinically meaningful and there may be no difference in overall survival (OS). For the abemaciclib-eligible subgroup, the sponsor submitted an indirect treatment comparison (ITC) (a matching-adjusted indirect comparison [MAIC]) comparing ribociclib plus AI to abemaciclib plus endocrine therapy (ET). The ITC suggests there is no difference in iDFS or OS between treatments but there are increased odds of grade 3 or higher neutropenia and increased ALT compared with abemaciclib plus ET among patients who met the eligibility criteria for cohort 1 of the monarchE trial.
Results of the CDA-AMC base case suggest that:
In the abemaciclib-eligible subgroup, ribociclib plus AI may be associated with lower costs to the health care system compared to abemaciclib plus ET (incremental savings = $9,884), primarily driven by lower predicted drug acquisition costs. There was no difference in incremental quality-adjusted life-years (QALYs) between ribociclib plus AI and abemaciclib plus ET. When compared to AI alone, ribociclib plus AI is predicted to be associated with higher total costs and QALYs compared to AI alone (incremental costs = $86,179; incremental life-years [LYs] = 0.84; incremental QALYs = 0.67), with an incremental cost-effectiveness ratio (ICER) of $127,917 per QALY gained.
In the abemaciclib-ineligible subgroup, ribociclib plus AI is predicted to be associated with higher total costs than AI alone (incremental costs = $97,641), driven by higher drug acquisition costs. Ribociclib plus AI is predicted to be associated with a gain of 1.23 LYs and 0.97 QALYs compared to AI alone, resulting in an ICER of $100,626 per QALY gained.
The estimated ICER for ribociclib plus AI versus AI alone in both subgroups was highly sensitive to assumptions about treatment effectiveness waning. In both subgroups, approximately 99% to 100% of the incremental benefit of ribociclib plus AI versus AI alone was gained in the extrapolated period. In the absence of long-term comparative evidence, the predicted QALYs gained for patients receiving ribociclib plus AI in both subgroups are highly uncertain and may be overestimated. Additional price reductions may therefore be required.
CDA-AMC estimates that the budget impact of reimbursing ribociclib for use in combination with AI for the adjuvant treatment of HR-positive, HER2-negative early breast cancer will be approximately $99 million over the first 3 years of reimbursement compared to the amount currently spent on comparators. The expenditure on ribociclib plus AI over this period is expected to be $212 million. The actual budget impact of reimbursing ribociclib will depend on the total size of the eligible population, the proportion of patients eligible for abemaciclib plus ET, and the uptake of ribociclib plus AI in both the abemaciclib-eligible and abemaciclib-ineligible subgroups. The incremental budget impact of reimbursing ribociclib plus AI for this indication is expected to be greater than $40 million in year 3 of reimbursement, and the economic feasibility of adoption must be addressed.
Summary of the Submitted Economic Evaluation
The sponsor submitted a cost-utility analysis to estimate the cost-effectiveness of ribociclib plus AI from the perspective of the public health care payer in Canada over a lifetime horizon (49 years).1 The modelled population was aligned with the intention-to-treat (ITT) population of the NATALEE trial and comprised adult men and women of known menopausal status with HR-positive, HER2-negative stage II or III breast cancer, which is aligned with the Health Canada indication. In the abemaciclib-eligible subgroup, the comparators were abemaciclib plus ET, AI alone, and tamoxifen, while the comparators in the abemaciclib-ineligible subgroup were AI alone and tamoxifen. The sponsor’s base-case analysis included costs related to drug acquisition (submitted price for ribociclib and public list prices for comparators), administration, follow-up and monitoring, subsequent treatment, adverse events (AEs), and terminal care. Additional information about the sponsor’s submission is provided in Appendix 3.
In the sponsor’s abemaciclib-eligible subgroup analyses, ribociclib plus AI was more effective and less costly than abemaciclib plus ET (incremental QALYs = 0.006, incremental savings = $16,690); compared to AI alone, ribociclib plus AI was associated with higher costs (incremental costs = $29,955) and higher QALYs (incremental QALYs = 0.68), resulting in an ICER of $43,821 per QALY gained. In the abemaciclib-ineligible subgroup, ribociclib plus AI was associated with higher costs (incremental costs = $51,398) and higher QALYs (incremental QALYs = 1.25) relative to AI alone, resulting in an ICER of $41,001 per QALY gained. In both subgroups, tamoxifen was dominated by AI alone.
CDA-AMC identified several key issues with the sponsor’s analysis (refer to Table 2; full details are provided in Appendix 4). A revised base case was therefore developed.
Table 2: Key Issues With the Sponsor’s Economic Submission
Issue | What evidence is there to inform this issue? | How was this issue addressed by CDA-AMC? | Did CDA-AMC explore uncertainty in a scenario analysis? |
|---|---|---|---|
The comparative efficacy and safety of ribociclib plus AI is uncertain. | There is no head-to-head evidence comparing ribociclib plus AI to comparators other than AI alone. The sponsor-submitted MAIC suggests no difference in iDFS and OS between ribociclib plus AI and abemaciclib plus ET. No comparative evidence was submitted for ribociclib plus AI vs. tamoxifen. AEs were included in the model for ribociclib plus AI vs. abemaciclib plus ET via naive comparison. | Tamoxifen was excluded from the CDA-AMC base case because of a lack of comparative evidence. Given the lack of direct evidence, limitations with the sponsor’s MAIC and the use of a naive comparison for AEs, it is uncertain whether ribociclib plus AI provides a clinical benefit compared with abemaciclib plus AI; CDA-AMC could not address this issue. | No scenario analysis was conducted owing to a lack of alternative clinical inputs. It is plausible, based on the results of the sponsor’s submitted ITC, that ribociclib plus AI may be less effective than abemaciclib plus AI. |
The sponsor used a black-box modelling approach to estimate costs and QALYs for the distant recurrence health state. | The sponsor used a fixed payoff, whereby patients who experienced a distant recurrence were assigned a prespecified number of LYs and costs, which were derived from the output of previous economic models (not provided to CDA-AMC as part of this review). | CDA-AMC could not address this issue and was unable to validate the outcomes predicted for the distant recurrence health state. The overall predicted incremental LYs, QALYs, and costs for ribociclib plus AI vs. comparators are thus highly uncertain. | No scenario analysis was conducted because of the model structure. |
The long-term effectiveness of ribociclib plus AI is highly uncertain. | Trial-based efficacy data were extrapolated over a 49-year horizon. Of the incremental QALYs gained for ribociclib plus AI vs. AI alone, 99% to 100% were accrued in the extrapolated period. The sponsor incorporated effectiveness waning in the economic model; however, clinical expert input indicated that there is insufficient evidence to support the sponsor’s assumptions. | In the base case, CDA-AMC adopted alternative assumptions about treatment effectiveness waning (i.e., waning of treatment started at year 8, with duration of 3 years). | Uncertainty in treatment-effectiveness waning was explored in scenario analyses. |
The impact of ribociclib plus AI on survival is highly uncertain. | In the NATALEE trial, there was no statistically significant difference in OS between ribociclib plus AI and AI alone. | CDA-AMC was unable to address this limitation owing to a lack of long-term data. | No scenario analysis was conducted. |
Subsequent treatment is not aligned with clinical practice. | The sponsor assumed that patients who received ribociclib plus AI and subsequently have an ET-sensitive distant recurrence would not receive a CDK4/6 inhibitor in the metastatic setting. Clinical expert input received by CDA-AMC for this review indicated that in Canada, re-treatment with a CDK4/6 inhibitor would be considered if the recurrence were at least 6 months after completion of adjuvant treatment. | CDA-AMC adopted alternative assumptions about the use of CDK4/6 inhibitors among patients with ET-sensitive distant recurrence. | No scenario analysis was conducted. |
Olaparib was omitted as a comparator. | Input received for this review indicated that olaparib is used in Canada for a proportion of patients eligible for ribociclib plus AI (i.e., those with deleterious or suspected deleterious BRCA1- or BRCA2-mutated disease). | CDA-AMC could not address this issue owing to a lack of comparative clinical data. | No scenario analysis was conducted. |
Use of RDI may underestimate actual drug costs. | RDI observations were incorporated from the NATALEE trial for ribociclib plus AI and for AI alone, and from the literature for abemaciclib plus ET. RDI from clinical trials may underestimate the total drug costs in clinical practice. | RDI was assumed to be 100% in the CDA-AMC base case. | RDI values provided by the sponsor were adopted in scenario analyses. |
Treatment duration was modelled inappropriately. | The sponsor utilized trial-based time-on-treatment data in the calculation of the QALYs, which resulted in some patients remaining on adjuvant treatment longer than the maximum specified in the monograph. | In the CDA-AMC base case, maximum treatment durations (3 years for ribociclib, 2 years for abemaciclib, 5 years for AI) were used in the calculation of QALYs.a | No scenario analysis was conducted. |
AE = adverse event; AI = aromatase inhibitor; CDA-AMC = Canada’s Drug Agency; CDK4/6 = cyclin-dependent kinase 4 and kinase 6; ET = endocrine therapy; iDFS = invasive disease–free survival; ITC = indirect treatment comparison; LY = life-year; MAIC = matching-adjusted indirect comparison; OS = overall survival; QALY = quality-adjusted life-year; RDI = relative dose intensity; vs. = versus.
aCosts were modelled separately and were unaffected by this issue in the sponsor’s base case; thus, no changes to cost calculations were made in the CDA-AMC base case.
CDA-AMC Assessment of Cost-Effectiveness
The CDA-AMC base case was derived by making changes to model parameter values and assumptions (refer to Table 9), in consultation with clinical experts. Detailed information about the CDA-AMC base case is provided in Appendix 4.
Impact on Health Care Costs
In the abemaciclib-eligible subgroup, ribociclib plus AI is predicted to be associated with additional health care costs compared to AI alone (incremental costs = $86,179). Compared with abemaciclib plus ET, ribociclib plus AI is predicted to be a cost saving (incremental savings = $10,055). In the abemaciclib-ineligible subgroup, ribociclib plus AI is expected to be associated with additional health care costs compared to AI alone (incremental costs = $97,641). In both subgroups, differences in costs were primarily owing to differences in drug acquisition costs (refer to Figure 1 and to Figure 2).
Figure 1: Impact of Ribociclib Plus AI on Health Care Costs — Abemaciclib-Eligible Subgroup
AE = adverse event; AI = aromatase inhibitor; ET = endocrine therapy.
Note: Other includes costs associated distant recurrence, secondary primary malignancy, and terminal care.
Figure 2: Impact of Ribociclib Plus AI on Health Care Costs — Abemaciclib-Ineligible Subgroup
AE = adverse event; AI = aromatase inhibitor.
Note: Other includes costs associated distant recurrence, secondary primary malignancy, and terminal care.
Impact on Health
In the abemaciclib-eligible subgroup, the CDA-AMC base case predicts there will be no difference in QALYs between ribociclib plus AI and abemaciclib plus ET (refer to Table 10). Relative to AI alone, ribociclib plus AI is expected to increase the amount of time a patient remains in the iDFS health state by approximately 1.29 years, with a total gain of 0.84 LYs (refer to Table 11). Considering the impact of treatment on both quality and length of life, ribociclib plus AI is predicted to result in 0.68 additional QALYs per patient compared to AI alone. Notably, all of the predicted incremental QALYs in the CDA-AMC base case were gained in the extrapolated period. That is, at the end of the NATALEE trial (median follow-up of approximately 44 months), the model predicts higher QALYs for AI alone (QALYs = 3.15) than for ribociclib plus AI (QALYs = 3.13).
In the abemaciclib-ineligible subgroup, ribociclib plus AI is expected to increase the amount of time a patient remains in the iDFS health state by approximately 1.30 years, with a total gain of 1.24 LYs (refer to Table 14). Considering the impact of treatment on both quality and length of life, ribociclib is expected to result in 0.97 additional QALYs per patient compared to AI alone. Approximately 99% of the predicted incremental QALYs were accrued on the basis of extrapolation in the CDA-AMC base case.
Overall Results
In the abemaciclib-eligible subgroup, the CDA-AMC base case suggests that ribociclib plus AI will be associated with comparable QALYs at lower costs to the health care system compared with abemaciclib plus ET. CDA-AMC notes this finding is predicated on the sponsor’s assumption of equivalent efficacy between ribociclib plus AI and abemaciclib plus ET (e.g., iDFS hazard ratio = 1.0); however, based on the results of the sponsor’s ITC, it is plausible that ribociclib could be associated with either improved or worse clinical outcomes compared to abemaciclib plus ET (iDFS hazard ratio = 0.90; 95% CI, 0.68 to 1.20; ██ ██ █ █████ 95% CI, 0.49 to 1.33); uncertainty in these parameters could not be included in the CDA-AMC base case because of the structure of the sponsor’s model. In this subgroup, ribociclib plus AI was associated with an ICER of $127,959 per QALY gained compared to AI alone. In the abemaciclib-ineligible subgroup, ribociclib plus AI was associated with an ICER of $100,626 per QALY gained compared to AI alone.
Table 3: Summary of the CDA-AMC Economic Evaluation Results — Abemaciclib-Eligible Subgroup
Drug | Total costs ($) | Total LYs | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|---|
Ribociclib plus AI vs. abemaciclib plus ET | ||||
Ribociclib plus AI | 196,268 | 15.05 | 11.945 | Reference |
Abemaciclib plus ET | 206,152 | 15.05 | 11.945 | Dominated by ribociclib plus AI |
Ribociclib plus AI vs. AI alone | ||||
AI alone | 110,432 | 14.21 | 11.27 | Reference |
Ribociclib plus AI | 196,611 | 15.05 | 11.95 | 127,959 |
AI = aromatase inhibitor; CDA-AMC = Canada’s Drug Agency; ET = endocrine therapy; ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year; vs. = versus.
Note: Analyses are reported deterministically. The CDA-AMC reanalysis is based on the publicly available prices of the comparator treatments.
Table 4: Summary of the CDA-AMC Economic Evaluation Results — Abemaciclib-Ineligible Subgroup
Drug | Total costs ($) | Total LYs | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|---|
Ribociclib plus AI vs. abemaciclib plus ET | ||||
AI alone | 81,620 | 17.54 | 13.76 | Reference |
Ribociclib plus AI | 179,261 | 18.77 | 14.73 | 100,626 |
AI = aromatase inhibitor; CDA-AMC = Canada’s Drug Agency; ET = endocrine therapy; ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year; vs. = versus.
Note: Analyses are reported deterministically. The CDA-AMC reanalysis is based on the publicly available prices of the comparator treatments.
Uncertainty and Sensitivity
Due to uncertainty in the comparative clinical evidence, including the lack of long-term evidence and lack of data to inform treatment-effectiveness waning over time, as well as the use of a fixed-payoff approach for some health states, the predicted costs and QALYs in the CDA-AMC base case are highly uncertain. The impact of uncertainty in the long-term effectiveness of ribociclib plus AI was explored in scenario analyses and had the greatest impact on estimated QALYs (refer to Table 16 and Table 17).
CDA-AMC notes that the sponsor’s use of the monarchE trial’s cohort 1 criteria to define eligibility for abemaciclib may have resulted in the misclassification of some patients because abemaciclib is reimbursed for patients who meet either the monarchE trial’s cohort 1 criteria (4 or more positive axillary lymph nodes, or between 1 and 3 positive nodes and either grade 3 disease or tumour size of 5 cm or larger) or cohort 2 criteria (between 1 and 3 positive axillary lymph nodes and Ki-67 of at least 20%). CDA-AMC was unable to address this issue; however, owing to comparable iDFS across subgroups, this is not expected to have a large impact on the estimated ICERs.
Summary of the Budget Impact
The sponsor submitted a budget impact analysis (BIA) to estimate the 3-year budget impact of reimbursing ribociclib for use in combination with an AI for the Health Canada–indicated population.2 The sponsor assumed that the payer would be CDA-AMC–participating public drug plans and derived the size of the eligible population using an epidemiologic approach. The price of ribociclib was aligned with the price included in the sponsor’s economic evaluation, while the prices of comparators were based on the publicly available list prices. Additional information pertaining to the sponsor’s submission is provided in Appendix 5.
CDA-AMC identified a number of issues with the sponsor’s estimated budget impact and made changes to model parameters and assumptions in consultation with clinical experts to derive the CDA-AMC base case (Appendix 5). The estimated incremental budget impact of reimbursing ribociclib plus AI is expected to be approximately $99 million over the first 3 years, with an expected expenditure of $212 million on ribociclib plus AI. The actual budget impact will depend on the total size of the eligible population, the proportion of patients eligible for abemaciclib, and the uptake of ribociclib plus AI in both abemaciclib-eligible and abemaciclib-ineligible subgroups. The structure of the sponsor’s model did not permit estimation of the budget impact in subgroups; however, based on an exploratory analysis conducted by CDA-AMC, there is the potential for ribociclib plus AI to be cost saving if reimbursement is restricted to the patients who would otherwise be eligible for abemaciclib; however, the clinical expert input received for this review indicated that clinicians would likely prefer abemaciclib in this patient population. In the abemaciclib-ineligible subgroup, reimbursement of ribociclib will be associated with incremental costs owing to the displacement of less costly treatments.
Conclusions
Results of the CDA-AMC base case suggest that ribociclib plus AI is similarly effective and less costly compared with abemaciclib plus ET in the subgroup of patients who meet the eligibility criteria for abemaciclib (aligned with cohort 1 of the monarchE trial); however, it is plausible, based on the sponsor’s submitted ITC, that ribociclib plus AI may be less effective than abemaciclib plus AI, which would result in overall reduced population health (i.e., lower QALYs). In both the abemaciclib-eligible and abemaciclib-ineligible subgroups, ribociclib plus AI would be considered cost-effective at the submitted price compared to AI alone if the public health care system were willing to pay $100,626 to $127,959 for each additional QALY gained. If the public health care system is not willing to pay that amount, a price reduction should be considered (refer to Figure 3; full details of the impact of price reductions on cost-effectiveness are presented in Table 15). The estimated cost-effectiveness of ribociclib plus AI compared to both abemaciclib plus ET and AI alone is highly uncertain due to uncertainty in the comparative clinical data, including the lack of long-term data. The cost-effectiveness of ribociclib plus AI versus tamoxifen is unknown owing to a lack of comparative clinical data.
The budget impact of reimbursing ribociclib plus AI for the adjuvant treatment of HR-positive, HER2-negative early breast cancer to the public drug plans in the first 3 years is estimated to be approximately $99 million. The 3-year expenditure on ribociclib plus AI (i.e., not accounting for current expenditure on comparators) is estimated to be $212 million. The estimated budget impact is highly uncertain due to uncertainty in the number of patients eligible for abemaciclib and the uptake of ribociclib versus comparators in both subgroups.
Figure 3: Summary of the CDA-AMC Economic Analysis and Price Reduction

AI = aromatase inhibitor; CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: Expenditure includes the drug cost of ribociclib alone. Expenditure on AI is expected to be $10 to $39 per 28-day cycle; refer to Table 3. The term dominant indicates that a drug costs less and provides more QALYs than the comparator.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Kisqali (ribociclib), tablets, 200 mg, oral. Novartis Pharmaceuticals Canada Inc.; 2024 Dec 19.
2.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Kisqali (ribociclib), tablets, 200 mg, oral. Novartis Pharmaceuticals Canada Inc.; 2024 Dec 19.
3.Kisqali (ribociclib): tablets, 200 mg, oral [product monograph]. Novartis Pharmaceuticals Canada Inc.; 2024 Aug 8.
4.Product Monograph: Verzenio (R). Eli Lilly Canada Inc. . 2023 Dec 1. https://pdf.hres.ca/dpd_pm/00073683.PDF
5.Ontario Ministry of Health and Long-Term Care. Ontario drug benefit formulary/comparative drug index. 2024. Accessed 2025 Jan 10. https://www.formulary.health.gov.on.ca/formulary/
6.Exceptional Access Program (EAP). Ontario Ministry of Health; Ontario Ministry of Long-Term Care; 2024. Accessed 2025 Apr 22. http://www.health.gov.on.ca/en/pro/programs/drugs/odbf/odbf_except_access.aspx
7.Canadian Agency for Drugs and Technologies in Health. CADTH Reimbursement Recommendation: Abemaciclib (Verzenio). 2024. https://www.cda-amc.ca/sites/default/files/DRR/2022/PC0282%20Verzenio%20-%20CADTH%20Final%20Recommendation%20(With%20Redactions)_Final.pdf
8.Janni W, Untsch M, Harbeck N, et al. Comparing the efficacy of aromatase inhibitors vs tamoxifen in hormone receptor–positive, human epidermal growth factor receptor 2–negative early breast cancer: a systematic review and trial-level meta-analysis. San Antonio Breast Cancer Symposium Poster P1-02-01. 2022.
9.Rugo HS, O’Shaughnessy J, Boyle F, et al. Adjuvant abemaciclib combined with endocrine therapy for high-risk early breast cancer: safety and patient-reported outcomes from the monarchE study. Ann Oncol. 2022;33(6):616-627. doi:10.1016/j.annonc.2022.03.006 PubMed
10.Table: 13-10-0114-01. Life expectancy and other elements of the life table, Canada, all provinces except Prince Edward Island. Statistics Canada; 2023. Accessed 2024 Dec 19. https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310011401
11.pan-Canadian Oncology Drug R. Final Economic Guidance Report - Ribociclib (Kisqali) plus Fulvestrant for Advanced or Metastatic Breast Cancer. 2020. Accessed [Available at: https://cadth.ca/sites/default/files/pcodr/Reviews2020/10195RibociclibFulvestrantMBC_fnEGR_NOREDACT-ABBREV_EC_22Apr2020_final.pdf; Accessed on: August 20, 2020.
12.Tolaney SM, Garrett-Mayer E, White J, et al. Updated Standardized Definitions for Efficacy End Points (STEEP) in Adjuvant Breast Cancer Clinical Trials: STEEP Version 2.0. J Clin Oncol. 2021;39(24):2720-2731. doi:10.1200/jco.20.03613 PubMed
13.Canadian Agency for Drugs and Technologies in Health. CADTH Reimbursement Review: Abemaciclib (Verzenio). 2024. https://www.cda-amc.ca/sites/default/files/DRR/2022/PC0282-Verzenio.pdf
14.Canadian Cancer Statistics Advisory Committee in collaboration with the Canadian Cancer S, Statistics C, and the Public Health Agency of C. Canadian cancer statistics 2023. Canadian Cancer Society; 2023. Accessed 2024 Dec 19. https://cancer.ca/en/cancer-information/resources/publications/canadian-cancer-statistics-2023
15.Howlader N, Altekruse SF, Li CI, et al. US incidence of breast cancer subtypes defined by joint hormone receptor and HER2 status. JNCI. 2014;106(5). PubMed
16.Sheffield KM, Peachey JR, Method M, et al. A real-world US study of recurrence risks using combined clinicopathological features in HR-positive, HER2-negative early breast cancer. Future Oncol. 2022;18(21):2667-2682. doi:10.2217/fon-2022-0310 PubMed
17.Seung SJ, Traore AN, Pourmirza B, Fathers KE, Coombes M, Jerzak KJ. A population-based analysis of breast cancer incidence and survival by subtype in Ontario women. Curr Oncol. 2020;27(2):e191-e198. doi:10.3747/co.27.5769 PubMed
18.Reeder-Hayes KE, Meyer AM, Dusetzina SB, Liu H, Wheeler SB. Racial disparities in initiation of adjuvant endocrine therapy of early breast cancer. Breast Cancer Res Treat. 2014;145(3):743-51. doi:10.1007/s10549-014-2957-z PubMed
19.Novartis P. A phase III, multicenter, randomized, open-label trial to evaluate efficacy and safety of ribociclib with endocrine therapy as an adjuvant treatment in patients with hormone receptor positive, HER2-negative, early breast cancer: End of Ribociclib Analysis Report. 2024.
20.Sutherland G, Thy D. Understanding the Gap: A Pan-Canadian Analysis, December 2017. 2017.
Appendix 1: Cost Comparison Table
Please note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical experts and CDA-AMC–participating public drug plans. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and, as such, the table may not represent the actual costs to public drug plans.
Table 5: Cost Comparison for HR-Positive, HER2-Negative Early Breast Cancer
Treatment | Strength | Form | Price ($) | Recommended dosage | Daily cost ($) | 28-day cost ($) |
|---|---|---|---|---|---|---|
Ribociclib (Kisqali) | 200 mg | Tablet | 94.12711 | 400 mg, once daily for 3 weeks following by 1 week off, for a maximum of 3 years3 | 141.19 | 3,953 |
Ribociclib + anastrozole | 142.14 | 3,980a | ||||
Ribociclib + exemestane | 142.52 | 3,990a | ||||
Ribociclib + letrozole | 142.57 | 3,992a | ||||
Abemaciclib plus ET | ||||||
Abemaciclibb (Verzenio) | 50 mg 100 mg 150 mg | Tablet | 112.5818 111.5389 111.8550 | 150 mg twice daily, for a maximum of 2 years4 | 223.71 | 6,264 |
Abemaciclib + anastrozole | 224.66 | 6,291a | ||||
Abemaciclib + exemestane | 225.04 | 6,301a | ||||
Abemaciclib + letrozole | 225.09 | 6,302a | ||||
Abemaciclib + tamoxifen | 224.06 | 6,273 | ||||
Aromatase inhibitors | ||||||
Anastrozole | 1 mg | Tablet | 0.9522 | 1 mg, once daily | 0.95 | 27a |
Exemestane | 25 mg | Tablet | 1.3263 | 25 mg, once daily | 1.33 | 37a |
Letrozole | 2.5 mg | Tablet | 1.3780 | 2.5 mg, once daily | 1.38 | 39a |
Tamoxifen | 10 mg 20 mg | Tablet | 0.1750 0.3500 | 20 mg, once daily | 0.35 | 10 |
AI = aromatase inhibitor; ET = endocrine therapy; HR = hormone receptor.
Note: All prices are from the Ontario Drug Benefit Formulary and Exceptional Access Program (accessed April 2025) unless otherwise indicated, and do not include dispensing fees.5,6
Olaparib (Lynparza) has been noted by CDA-AMC as a comparator to abemaciclib for this review for a subset of the patient population (those with deleterious or suspected deleterious germline BRCA1/2-mutated disease). The olaparib unit cost is $69.9482 per 150 mg tablet, the daily cost is $279.79, and the 28-day cost is $7,834 at a recommended dose of 300 mg twice daily (sourced from Exceptional Access Program).6
aWhen used by men and pre- or peri-menopausal women, a luteinizing hormone-releasing hormone agonist should also be used (i.e., goserelin 3.6 mg subcutaneous injection once every 4 weeks), which incurs an additional $422 per patient per 28-day cycle.
bAbemaciclib is reimbursed for patients who meet the eligibility criteria of MonarchE cohort 1 (4 or more positive axillary lymph nodes, or between 1 and 3 positive nodes and either grade 3 disease or tumour size of 5 cm or larger) and cohort 2 (between 1 and 3 positive axillary lymph nodes and Ki-67 of at least 20%).7
Appendix 2: Input Relevant to the Economic Review
Please note that this appendix has not been copy-edited.
This section is a summary of the input received from the patient groups, clinician groups, and drug plans that participated in the CDA-AMC review process.
Patient input was received from Rethink Breast Cancer, Breast Cancer Canada, Canadian Breast Cancer Network, and Quebec Breast Cancer Foundation, collected via online surveys (205 respondents) and interviews (5 interviews) including patients with stage II and III breast cancer in Canada. Patients described the physical symptoms associated with early breast cancer (e.g., fatigue), as well as anxiety associated with fear of recurrence. Patients indicated that the goal of treatment is to cure their breast cancer, reduce the risk of recurrence and metastasis, and maintain quality of life. Of the 12 respondents with ribociclib experience, most described being able to manage AEs with help from their physicians; some noted that dose adjustments had been required to resolve symptoms associated with liver issues and neutropenia, or having to stop treatment due to side effects. However, most indicated that they would be willing to tolerate AEs if the treatment was able to reduce risk of recurrence.
Clinician group input was received from Research Excellence, Active Leadership Canadian Breast Cancer Alliance, and Ontario Health Breast Cancer Drug Advisory Committee. Input indicated that treatments for HR-positive, HER2-negative breast early breast cancer include surgery, radiotherapy, and adjuvant systemic therapy consisting of chemotherapy, ET (tamoxifen or AI), or abemaciclib for eligible patients. Clinician input indicated that the goal of treatment is to prevent recurrence and minimize treatment-related AEs. Clinician input noted that ribociclib is given for up to 3 years (1 year longer than abemaciclib) and that this will increase the need for monitoring for an additional year. The clinician group also noted concerns about ribociclib-associated toxicity and its management.
Input from CDA-AMC–participating drug plans noted that abemaciclib for use in combination with ET is funded in most jurisdictions for patients with node-positive, HR-positive, HER2-negative breast cancer who are at high risk of disease recurrence. Plans questioned whether patients could receive a CDK4/6 inhibitor in the metastatic setting if they received a CDK4/6 inhibitor in the adjuvant setting. Plans indicated that reimbursement of ribociclib will affect health care resource use (e.g., electrocardiography), given the need for monitoring for cardiac AEs. Drug plan input indicated concern about the budget impact and affordability of reimbursing ribociclib because of the large number of patients with early breast cancer.
Several of these concerns were addressed in the sponsor’s model:
iDFS and OS were included in the model.
Health-related quality of life (HRQoL) was included in the model, by use of EQ-5D-5L data collected in the NATALEE trial.
Follow-up and monitoring costs were included in the model.
CDA-AMC addressed some of these concerns as follows:
CDA-AMC adopted alternative assumptions regarding re-treatment with CDK4/6 inhibitors for metastatic disease recurrence.
CDA-AMC was unable to address the following concerns:
The sponsor’s modelling approach precludes full validation of the model findings.
Appendix 3: Summary of the Sponsor’s Submission
Please note that this appendix has not been copy-edited.
Summary of the Sponsor’s Economic Evaluation
For the pharmaceutical reviews program, clinical and economic information is submitted to CDA-AMC by the sponsor. The CDA-AMC health economics team reviews the submitted economic information and appraises the information in collaboration with clinical experts and the clinical review team to evaluate key assumptions, influential parameters, and the overall rigour of the economic submission. Based on what the team learns through this process, adjustments may be made to the sponsor’s model to produce the CDA-AMC base case. The CDA-AMC base case represents the team’s current understanding of the clinical condition, clinical evidence currently available, and best interpretation of the economic evidence based on the information provided.
For the review of ribociclib for use in combination with AI, the sponsor provided a cost-utility analysis and a BIA. The sponsor’s economic submission is summarized in Table 6.
Table 6: Key Components of the Sponsor’s Economic Evaluation
Component | Description |
|---|---|
Treatment information | |
Drug under review | Ribociclib (Kisqali), 200 mg tablets |
Submitted price of drug under review | Ribociclib: $94.13 per 200 mg tablet |
Regimen | Ribociclib 400 mg once daily for 21 consecutive days followed by 7 days off treatment for 36 months, in combination with AI (letrozole 2.5 mg, or anastrozole 1 mg) once daily for 60 monthsa |
28-day cost of drug under review | Ribociclib: $3,297 per patientb,c |
Model information | |
Type of economic evaluation | Cost-utility analysis Semi-Markov model |
Treatment | Ribociclib for use in combination with AI (assumed by the sponsor to be letrozole or anastrozole)a |
Included comparators | Abemaciclib-eligible subgroupd:
Abemaciclib-ineligible subgroup:
|
Perspective | Publicly funded health care payer perspective |
Time horizon | Lifetime (49 years) |
Cycle length | 28 days |
Modelled population |
|
Characteristics of modelled population | Derived from the NATALEE trial (mean age: 52.8 years, 99.6% women, 0.04% men) |
Model health states |
For additional information, refer to Model Structure |
Data sources | |
Comparative efficacy |
|
Natural history and/or clinical pathway |
|
Costs included in the model |
|
Health-related utilities and disutilities |
|
Summary of the submitted results | |
Base-case results |
|
Scenario analysis resultsf |
|
AE = adverse event; AI = aromatase inhibitor; ET = endocrine therapy; HR = hormone receptor;; iDFS = invasive disease-free survival; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-years; RDI = relative dose intensity; TTD = treatment discontinuation.
aFor men and pre- or peri-menopausal women, AI should also be combined with a luteinizing hormone-releasing hormone agonist (i.e., goserelin 3.6 mg subcutaneous injection once every 4 weeks).
bCost of ribociclib alone. Cost of ribociclib plus AI includes an additional $41 per patient per 30-day cycle for patients receiving letrozole, $28 per 30-day cycle for patients receiving anastrozole, and $422 per 28-day cycle for patients receiving goserelin.
cAcquisition cost of ribociclib was adjusted by RDI (83.4%).
dThe abemaciclib-eligible subgroup includes patients who met the eligibility criteria for cohort 1 of the monarchE trial (4 or more positive axillary lymph nodes, or between 1 and 3 positive nodes and either grade 3 disease or tumour size of 5 cm or larger); all other patients who met ribociclib criteria but not the monarchE cohort 1 criteria were included in the abemaciclib-ineligible subgroup.
eET-resistance was defined by the sponsor as disease recurrence 12 or more months after the end of ET. ET-sensitive was defined as disease recurrence at least 12 months after the end of ET.
fAdditional scenarios were submitted that had no meaningful impact on the estimated ICER, including excluding treatment-effectiveness waning and assuming a discount rate of 0%.
Model Structure
The sponsor submitted a semi-Markov model with 6 health states: iDFS, second primary malignancy, nonmetastatic recurrence, remission, distant recurrence, and death (refer to Figure 4). The distant recurrence state is further divided into ET-resistant state (patients with recurrence at least 12 months from the end of ET), and ET-sensitive state (patients with recurrence less than 12 months after the end of ET). All patients entered the model in the iDFS state, and in each cycle could stay in iDFS state, experience a second primary malignancy, nonmetastatic recurrence, distant recurrence, or die. Patients who experience a second primary malignancy were assumed to enter an absorbing state, incur a one-off cost of diagnosis, and leave the model. Patients who experience a nonmetastatic recurrence and remain in the nonmetastatic recurrence state for 1 year were assumed to transition to the remission state, and could remain in that state, experience distant recurrence, or die. The nonmetastatic recurrence and remission states include tunnel states to permit the probabilities of further progression and death to vary by duration of state membership. Patients in the distant recurrence health state were assigned a fixed number of LYs based on external models developed by the sponsor for other submissions (i.e., fixed-payoff approach).11 These LYs were multiplied by costs and utilities to determine total costs and QALYs for patients in the distant recurrence state.
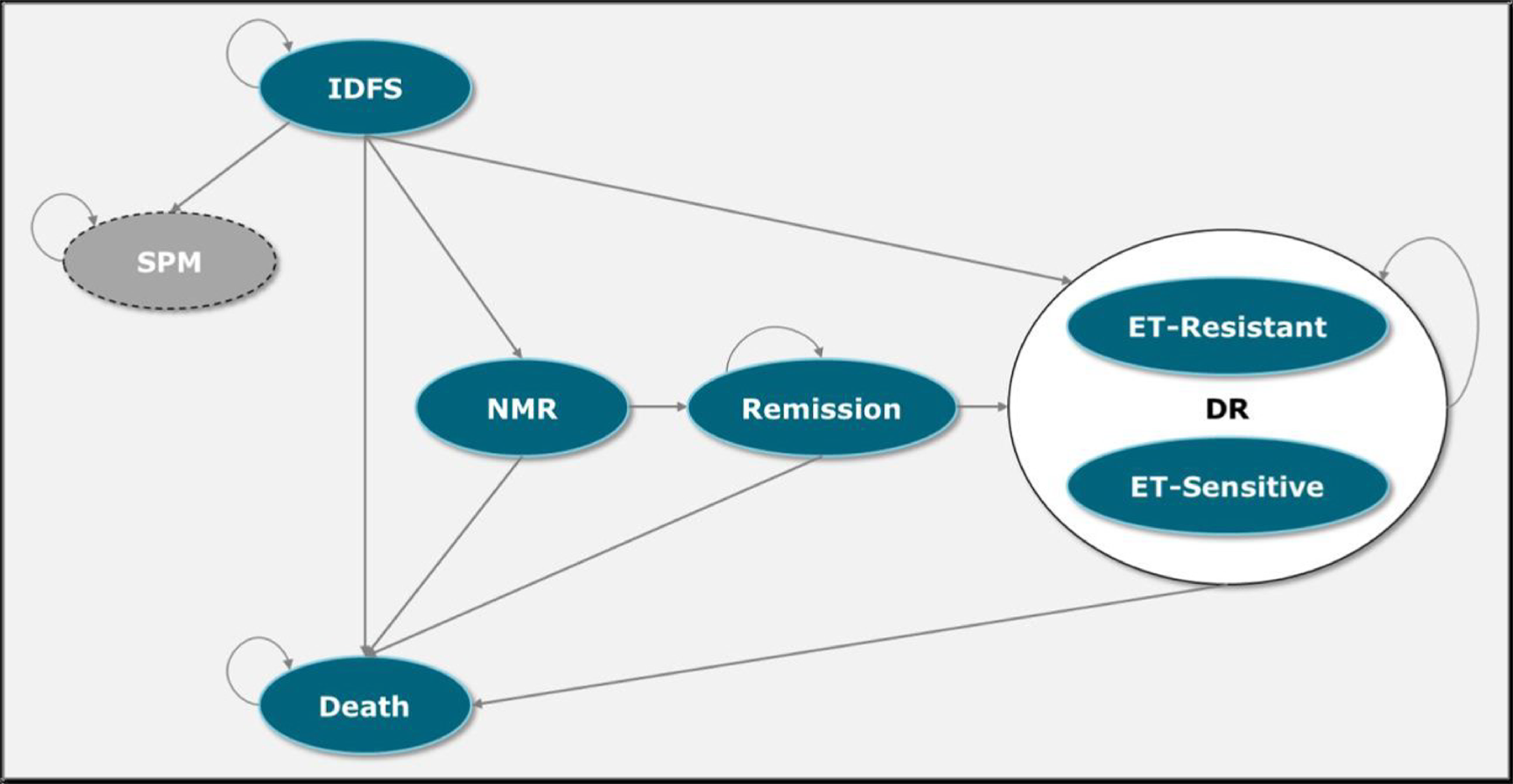
DR = distant recurrence; ET = endocrine therapy; IDFS = invasive disease–free survival; NMR = nonmetastatic recurrence; SPM = secondary primary malignancy.
Source: Sponsor’s pharmacoeconomic submission.1
Table 7: Summary of the Sponsor’s Economic Evaluation Results — Abemaciclib-Eligible Subgroup
Drug | Total costs ($) | Total QALYs | ICER vs. ET alone ($/QALY) |
|---|---|---|---|
AI alone | 125,276 | 12.39 | Reference |
Ribociclib plus AI | 155,231 | 13.08 | 43,821 |
Dominated treatments | |||
Tamoxifen | 133,338 | 10.52 | Dominated by ribociclib plus AI |
Abemaciclib + ET | 171,921 | 13.07 | Dominated by ribociclib plus AI |
AI = aromatase inhibitor; ET = endocrine therapy; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Note: The abemaciclib-eligible subgroup includes patients who met the eligibility criteria for cohort 1 of the monarchE trial (4 or more positive axillary lymph nodes, or between 1 and 3 positive nodes and either grade 3 disease or tumour size of 5 cm or larger); all other patients who met ribociclib criteria but not the monarchE cohort 1 criteria were included in the abemaciclib-ineligible subgroup.
Source: Sponsor’s pharmacoeconomic submission, probabilistic sequential results.1
Table 8: Summary of the Sponsor’s Economic Evaluation Results — Abemaciclib-Ineligible Subgroup
Drug | Total costs ($) | Total QALYs | ICER vs. ET alone ($/QALY) |
|---|---|---|---|
AI | 89,813 | 14.68 | Reference |
Ribociclib + AI | 141,211 | 15.94 | 41,001 |
Dominated treatments | |||
Tamoxifen | 100,489 | 13.09 | Dominated by ribociclib plus AI |
AI = aromatase inhibitor; ET = endocrine therapy; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Note: The abemaciclib-eligible subgroup includes patients who met the eligibility criteria for cohort 1 of the monarchE trial (4 or more positive axillary lymph nodes, or between 1 and 3 positive nodes and either grade 3 disease or tumour size of 5 cm or larger); all other patients who met ribociclib criteria but not the monarchE cohort 1 criteria were included in the abemaciclib-ineligible subgroup.
Source: Sponsor’s pharmacoeconomic submission, probabilistic sequential results.1
Appendix 4: Additional Details of CDA-AMC Reanalyses
Please note that this appendix has not been copy-edited.
Clinical Data in the Economic Model
The key clinical efficacy data (iDFS) used to inform the economic model were derived from the NATALEE trial (data cut-off date: April 29, 2024) for ribociclib plus AI and for AI alone. Based on data for the ITT population of the NATALEE trial, the CDA-AMC clinical review concluded that that ribociclib plus AI likely results little or small clinically important difference in iDFS versus AI alone after 48 months. Similarly, for OS and HRQoL, there may be little or no clinically important benefit associated with ribociclib plus AI versus AI alone. The CDA-AMC clinical review also found that ribociclib plus AI was associated with a clinically important increase in hepatobiliary toxicity, QT interval prolongation, and interstitial lung disease or pneumonitis.
To inform the economic model, the sponsor undertook subgroup analyses to assess the efficacy of ribociclib based on abemaciclib eligibility, with abemaciclib eligibility defined according to the monarchE trial cohort 1 criteria (4 or more positive axillary lymph nodes, or between 1 and 3 positive nodes and either grade 3 disease or tumour size of 5 cm or larger).1 A sponsor-submitted MAIC suggested no difference iDFS or OS between ribociclib plus AI and abemaciclib plus ET in this subgroup of patients; however, wide confidence intervals suggest that ribociclib could be beneficial or harmful compared to abemaciclib. The sponsor’s MAIC suggests that ribociclib plus AI is associated with significantly increased odds of grade 3 or higher neutropenia and increased ALT compared with abemaciclib plus ET, but lower odds of grade 3 or higher diarrhea, leukopenia, and lymphopenia. However, the CDA-AMC clinical review noted that results of the sponsor’s MAIC should be interpreted with consideration of the limitations highlighted in the clinical review (e.g., not all important effect modifiers were included). Further, the population used for the comparison of treatment effects is not representative of the full population for whom abemaciclib is reimbursed (i.e., aligned only with cohort 1 of Monarch, while abemaciclib is additionally reimbursed for patients who meet the monarchE cohort 2 criteria [between 1 and 3 positive axillary lymph nodes and Ki-67 of at least 20%]).7
In the sponsor’s economic model, iDFS was extrapolated beyond the NATALEE trial period over a 49-year lifetime time horizon, assuming waning of treatment effect to begin 96 months after treatment initiation, with total duration of 285 months. Notably, although more incremental QALYs were accrued in the iDFS health state for ribociclib plus AI during the trial period, of the total incremental QALYs gained over the model horizon, more than 100% were accrued in the extrapolated period in both subgroups (that is, at the end of the NATALEE trial period, predicted QALYs were higher for AI than for ribociclib plus AI). Several additional sources of uncertainty related to the clinical efficacy inputs in the economic model were noted (Refer to Key Issues of the Submitted Economic Evaluation). As such, the estimated incremental gain in LYs and QALYs predicted by the sponsor’s model for ribociclib are highly uncertain.
Key Issues of the Submitted Economic Evaluation
CDA-AMC identified the following key issues with the sponsor’s analysis:
The comparative efficacy and safety of ribociclib plus AI is uncertain. There are several sources of uncertainty related to the clinical data in the economic model. First, there is a lack of direct head-to-head evidence comparing ribociclib plus AI to comparators other than AI alone. In the abemaciclib-eligible subgroup, the sponsor assumed equivalent iDFS for ribociclib plus AI and abemaciclib plus ET (i.e., HR 1.0) based on the findings of the submitted MAIC. However, while the results of the sponsor’s MAIC suggest that there may be no difference in iDFS between treatments, the 95% confidence interval for iDFS includes the null value (HR 0.96, 95% CI, 0.73 to 1.26), indicating uncertainty in the comparative efficacy (i.e., the true effect may lie on either side of the null value, thus including the possibility that ribociclib plus AI is associated with lower iDFS versus abemaciclib plus ET; this uncertainty was not captured in the sponsor’s economic analysis. Similarly, for the probability of transition from iDFS to other health states, the sponsor assumed equivalence between ribociclib plus AI and abemaciclib plus ET; these assumptions were not justified by the sponsor, and the uncertainty around these parameters was not included in the sponsor’s economic analysis.
Second, the efficacy of tamoxifen in the model was based on naive comparison; that is, key model parameters were incorporated directly from the literature without adjustment or accounting for differences between trials. Owing to the direct use of data, it is not possible to determine if any observed differences in treatment response between therapies are solely due to the treatment or, rather, due to bias or confounding factors (e.g., differences in study populations, definitions of outcomes, or study designs). CDA-AMC additionally noted that study used to inform the efficacy of tamoxifen reported disease-free survival, which is not interchangeable with iDFS.12 Owing to these limitations, conclusions cannot be drawn as to the relative efficacy of ribociclib versus tamoxifen.
Finally, the sponsor included costs associated with the management of grade 3 or higher AEs in the model, with the incidence of each obtained from the NATALEE trial for ribociclib plus AI and the monarchE trial for abemaciclib plus ET. The impact of each AE on HRQoL (disutility) was assumed to last for 1 month, with disutility values obtained from the literature. In the model, the incidence of AEs was incorporated via naive comparison, without adjustment or accounting for differences in patient characteristics. The sponsor’s submitted MAIC included an analysis of harms, and results suggest that ribociclib plus AI has a significantly increased odds of neutropenia and increased ALT compared with abemaciclib plus ET, but lower odds of diarrhea, leukopenia, and lymphopenia; these data were not utilized in the economic model. Owing to the use of naive safety data in the economic evaluation, value, it is not possible to determine if any observed differences between the therapies (and thus incremental QALYs due to AEs) are solely due to the treatment or, rather, due to bias or confounding factors.
CDA-AMC was unable to incorporate uncertainty around the comparative efficacy of ribociclib plus AI versus abemaciclib plus ET. AEs were excluded from the CDA-AMC base case.
Limitations with the sponsor’s modelling approach. In the sponsor’s model, patients who experienced an iDFS event transitioned to a subsequent model health state. For the nonmetastatic recurrence and secondary primary malignancy health states, the sponsor used a typical Markov cohort approach in which patients accrue costs, LYs, and QALYs based on the duration of time spent in each health state. However, the sponsor adopted a fixed-payoff approach for the distant recurrence state, in which each patient who entered this state was assigned a prespecified number of LYs and costs. To inform this fixed-payoff approach, the sponsor used the output of previous economic models built to assess the cost-effectiveness of ribociclib plus fulvestrant for the treatment of HR-positive, HER2-negative advanced breast cancer.11 Fulvestrant is not an AI, and the assumption that progression-free and postprogression survival of patients who received ribociclib plus fulvestrant in the metastatic setting is equal to that of patients who receive adjuvant ribociclib plus AI was not justified by the sponsor.
Fixed-payoff approaches have previously been described by CDA-AMC and clinical experts as a “black box approach to modelling,” meaning that the method by which results were produced were opaque.13 As such, CDA-AMC was unable to validate the model’s predicted outcomes for patients in the distant recurrence health state because the models used by the sponsor were not provided to CDA-AMC as part of the review process. CDA-AMC additionally notes that these external models were partitioned survival models, which are subject to inherent modelling limitations, and any assumptions made during the development of these models are carried through to the current submission. Therefore, assumptions made by the sponsor regarding a fixed payoff in the distant recurrence health state increase the uncertainty of results.
CDA-AMC was unable to validate the outcomes predicted for patients in the distant recurrence health state. As such, the overall predicted incremental LYs, QALYs, and costs for ribociclib plus AI versus comparators should be considered highly uncertain.
The long-term effectiveness of ribociclib plus AI is highly uncertain. Efficacy data for ribociclib plus AI from the NATALEE trial (median follow-up = 44.2 months) were extrapolated over a lifetime horizon (49 years). The parametric distributions chosen by the sponsor as well as assumptions about treatment-effectiveness waning, resulted in an incremental gain of QALYs for ribociclib plus AI compared to AI alone in both subgroups (incremental QALYs: 0.69 and 1.26 for abemaciclib-eligible and abemaciclib-ineligible subgroups, respectively). Notably, of the total incremental QALYs gained over the model horizon, more than 100% were accrued in the extrapolated period (that is, at the end of the NATALEE trial period, predicted QALYs were higher for AI than for ribociclib plus AI). As noted in the CDA-AMC clinical review, no long-term extension studies were submitted for this review. Given that all of the incremental QALYs predicted by the sponsor’s model for ribociclib and AI versus AI alone were derived through extrapolation, the lack of long-term data introduces considerable uncertainty into the analysis.
CDA-AMC further notes that the relative benefit of ribociclib plus AI versus AI alone in the sponsor’s model continues to increase in the extrapolated period in both subgroups. For example, in the abemaciclib-ineligible subgroup, the incremental benefit in iDFS with ribociclib plus AI versus AI alone is 2.9 percentage points at year 5 and 6.9 percentage points at year 15. The same trend was observed in abemaciclib-eligible group, with the incremental benefit in iDFS for ribociclib plus AI being 6.2 percentage points at year 5 and 10.1 percentage points at year 15. Clinical experts consulted by CDA-AMC for this review indicated that an increase in the relative effectiveness of ribociclib versus AI alone over time is not supported by the available evidence.
Finally, the sponsor assumed that treatment-effectiveness waning for ribociclib plus AI will begin at year 8 (i.e., 5 years after the end of ribociclib treatment) and wane linearly until year 31 (26 years after end of ribociclib treatment), based on a prior submission of abemaciclib to CDA-AMC.13 This implicitly assumes that the treatment effect of ribociclib will be maintained for at least 5 years after the end of ribociclib treatment and that, when waning starts, it will take approximately 23 years for the effects of ribociclib plus AI to fully dissipate. Clinical experts consulted by CDA-AMC for the current review indicated that there is insufficient evidence to support this assumption. Experts additionally noted that it is plausible that effectiveness waning will begin earlier and last for less than 23 years. Similar issues were raised in the prior CDA-AMC review of abemaciclib.13
In the CDA-AMC base case, treatment-effectiveness waning for ribociclib plus AI was assumed to being at year 8 and to end at year 11 (total waning period of 3 years); the same assumptions were made for abemaciclib based on expert input. CDA-AMC explored uncertainty in these assumptions in scenario analyses.
The impact of ribociclib plus AI on survival is highly uncertain. As noted in the prior issue, efficacy data from the NATALEE trial were extrapolated over a 49-year horizon. In both the abemaciclib-eligible and -ineligible subgroups, this extrapolation resulted in an incremental gain of LYs for ribociclib plus AI compared to AI alone (abemaciclib-eligible: incremental LYs = 0.78; abemaciclib-ineligible: incremental LYs = 1.63). As noted in the CDA-AMC clinical report, there was no statistically significant difference in OS between ribociclib plus AI and AI alone (ITT population hazard ratio 0.83, 95% CI 0.64 to 1.07). Additionally, as noted previously, the use of a “fixed-payoff” approach by the sponsor for patients with distant recurrence introduces considerable uncertainty into the sponsor’s findings.
CDA-AMC was unable to address this limitation owing to a lack of long-term data and the structure of the sponsor’s model (i.e., use of a fixed-payoff approach).
Subsequent treatment is not aligned with clinical practice. The sponsor assumed that patients who received with ribociclib plus AI and subsequently have an ET-sensitive distant recurrence (i.e., recurrence more than 12 months after completing adjuvant AI) would not receive a CDK4/6 inhibitor in the metastatic setting. Clinical expert input received by CDA-AMC for this review, as well as input from CDA-AMC–participating drug plans, indicated that in clinical practice in Canada, re-treatment with a CDK4/6 inhibitor patients would be considered if the recurrence was at least 6 months after completion of adjuvant treatment. Given that CDK4/6 inhibitors are more costly than other treatments used as subsequent therapy, as well as the incremental difference in CDK4/6 inhibitor usage assumed by the sponsor between patients who initially received ribociclib plus AI (0%) or AI alone (63%), the incremental costs of subsequent therapy were likely overestimated in the sponsor’s model, which biased the cost-effectiveness results in favour of ribociclib plus AI.
Further, the sponsor assumed that all patients who experience distant recurrence would receive 2 additional lines of treatment (initial treatment for metastatic recurrence, subsequent treatment after metastatic disease progression). As described in a previous issue, the sponsor used a fixed-payoff approach to determine LYs and drug acquisition costs, with the average amount of time spent on each treatment in the basket of subsequent therapy obtained from the literature. After progression of metastatic disease, the sponsor assumed that the distribution of treatments in the basket of subsequent therapies would be the same as for initial treatment of metastatic disease and that patients would receive treatment for the remainder of their lives. Clinical expert input received by CDA-AMC indicated that the treatment received after metastatic disease progression is highly uncertain and is influenced by prior treatments received and ET-sensitivity status, in addition other clinical factors. As such, the costs and LYs predicted by the sponsor’s model, particularly for treatment after metastatic disease progression, are considered highly uncertain.
In the CDA-AMC base case, CDA-AMC adopted alternative assumptions about the use of CDK4/6 inhibitors among patients with ET-sensitive distant recurrence.
Olaparib was omitted as a relevant comparator. The sponsor omitted adjuvant olaparib from the economic analysis. Clinical expert input and clinician group input received for this review indicated that adjuvant olaparib is used in Canada for some patients who would be eligible for ribociclib plus AI (i.e., those with deleterious or suspected deleterious germline BRCA1/2-mutated disease). Hence, the omission of adjuvant olaparib as a relevant comparator for this indication does not reflect current clinical practice in Canada.
CDA-AMC could not address this limitation owing to the absence of comparative clinical data and the structure of the sponsor’s model. The cost-effectiveness of ribociclib plus AI versus olaparib is unknown.
Use of relative dose intensity (RDI) may underestimate actual drug costs. In the sponsor’s analysis, RDI observations were incorporated from the NATALEE trial for ribociclib plus AI and for AI alone, calculated as “dose intensity divided by planned dose intensity, where dose intensity was defined as the ratio of the actual cumulative dose received to the duration of exposure and planned dose intensities were 400 mg/day for ribociclib (for 3 weeks on treatment followed by 1 week off), 2.5 mg/day for letrozole, and 1 mg/day for anastrozole.”1 Owing to the lack of publicly available information on RDI for abemaciclib, the sponsor based RDI for abemaciclib on the RDI for treatment holds only observed for ribociclib in the NATALEE trial. This resulted in a higher RDI for ribociclib (83.4%) than for abemaciclib (91.2%). CDA-AMC notes that the inclusion of RDI may underestimate the total drug costs in clinical practice because changes in RDI can result from numerous factors, including clinical judgment, dose delays, missed doses, or dose reductions, and such adjustments impact drug costs differently.
In the CDA-AMC base case, RDI was assumed to be 100% for all treatments. The impact of this change was explored in a scenario analysis.
Treatment duration was modelled inappropriately. In the pharmacoeconomic model, treatment discontinuation was derived from time to treatment discontinuation (TTD) data from NATALEE and monarchE trials. This resulted in some patients remaining on adjuvant treatment longer than specified in the monograph (i.e., a maximum of 3 years for ribociclib, 2 years for abemaciclib, 5 years for AI). This affected only predicted QALYs (as the sponsor incorporated different utility values for patients who were on or off treatment in the iDFS health state). Drug acquisition costs were correctly capped by the sponsor using the maximum duration specified in the product monographs and were not affected by this issue.
In the CDA-AMC reanalysis, a maximum treatment duration of 3 years for ribociclib, 2 years for abemaciclib, and 5 years for AI was utilized in the calculation of QALYs.
Modelling approach lacks transparency. In addition to the lack of transparency noted in a prior issue (i.e., use of a “black box” fixed-payoff approach), the sponsor’s model included numerous IFERROR statements, which lead to situations in which the parameter value is overwritten with an alternative value without alerting the user to automatic overwriting. The systematic use of IFERROR statements makes thorough auditing of the sponsor’s model impractical, as it remains unclear whether the model is running inappropriately by overriding errors.
CDA-AMC was unable to address this issue and notes that a thorough validation of the sponsor’s model was not possible.
CDA-AMC Reanalysis of the Economic Evaluation
The CDA-AMC base case was derived by making changes in model parameter values and assumptions, in consultation with clinical experts (refer to Table 9). The results of the CDA-AMC base case are presented deterministically, as unexpected results were obtained with the sponsor’s probabilistic model. The impact of these changes, individually and collectively, are presented in Table 10 and Table 13 for the abemaciclib-eligible and -ineligible subgroups, respectively.
For the abemaciclib-eligible subgroup, the results of the CDA-AMC base case are presented as pairwise comparisons, owing to the use of different sources of efficacy data. For both subgroups, tamoxifen was removed from the CDA-AMC base case owing to the lack of comparative efficacy data.
Table 9: Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
1. Treatment effectiveness waning | Treatment waning for ribociclib plus AI and for abemaciclib plus ET was assumed to begin at year 8 and continue until year 31 (total waning period 23 years)a | Treatment waning for ribociclib plus AI and for abemaciclib plus ET was assumed to begin at year 8 and ends at year 11 (total waning period: 3 years)b |
2a. Treatment of ET-sensitive distant recurrenceb |
|
|
2b. Treatment after metastatic disease progression (distant recurrence health state) | The sponsor adopted a fixed-payoff approach to determine costs, LYs, and QALYs for patients | Costs, LYs, and QALYs were excluded for patients with metastatic disease progression |
3. Duration of adjuvant treatment | Modelled using TTD observations from the NATALEE trial extrapolated over the model horizon, with no maximum duration of ribociclib, abemaciclib, or AIc | Maximum duration:
|
4. RDI | Derived from the NATALEE trial and the literature | Assumed to be 100% for all therapies |
5. AEs | Included | Excluded |
CDA-AMC base case (deterministic) | ― | Reanalysis 1 + 2 + 3 + 4 ( + 5d) |
AE = adverse event; AI = aromatase inhibitor; CDA-AMC = Canada’s Drug Agency; ET = endocrine therapy; ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-years; RDI = relative dose intensity.
Note: CDA-AMC was unable to resolve uncertainty in other model parameters.
aThe sponsor assumed that treatment-effectiveness waning would be the same for abemaciclib ET as for ribociclib plus AI. This assumption was maintained in the CDA-AMC base case.
bFor patients who received either ribociclib plus AI or abemaciclib plus AI as adjuvant treatment.
cAffected the calculation of QALYs only. Costs were modelled separately and were not affected by this issue.
dCosts and QALYs related to AEs were excluded only for the pairwise comparison of ribociclib plus AI vs. abemaciclib plus ET.
Table 10: Summary of the Stepped Analysis — Abemaciclib-Eligible Subgroup
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
Ribociclib plus AI vs. abemaciclib plus ET | ||||
Sponsor base case | Ribociclib plus AI | 155,160 | 13.089 | Reference |
Abemaciclib plus ET | 172,506 | 13.083 | Dominated by ribociclib plus AI | |
CDA-AMC reanalysis 1: Effectiveness waning | Ribociclib plus AI | 157,089 | 12.822 | Reference |
Abemaciclib plus ET | 174,434 | 12.819 | Dominated by ribociclib plus AI | |
CDA-AMC reanalysis 2: Treatment of ET-sensitive distant recurrence | Ribociclib plus AI | 169,544 | 12.383 | Reference |
Abemaciclib plus ET | 186,889 | 12.377 | Dominated by ribociclib plus AI | |
CDA-AMC reanalysis 3: Treatment duration | Ribociclib plus AI | 155,160 | 12.949 | Reference |
Abemaciclib plus ET | 172,506 | 12.949 | Dominated by ribociclib plus AI | |
CDA-AMC reanalysis 4: RDI | Ribociclib plus AI | 174,508 | 13.089 | Reference |
Abemaciclib plus ET | 184,563 | 13.083 | Dominated by ribociclib plus AI | |
CDA-AMC reanalysis 5: AEs | Ribociclib plus AI | 154,817 | 13.084 | Reference |
Abemaciclib plus ET | 171,991 | 13.089 | Dominated by ribociclib plus AI | |
CDA-AMC base case Reanalysis 1 + 2 + 3 + 4 + 5 | Ribociclib plus AI | 196,268 | 11.945 | Reference |
Abemaciclib plus ET | 206,152 | 11.945 | Dominated by ribociclib plus AI | |
Ribociclib plus AI vs. AI alone | ||||
Sponsor base case | AI alone | 125,657 | 12.40 | Reference |
Ribociclib plus AI | 155,160 | 13.09 | 42,724 | |
CDA-AMC reanalysis 1: Effectiveness waning | AI alone | 125,657 | 12.40 | Reference |
Ribociclib plus AI | 157,089 | 12.82 | 74,209 | |
CDA-AMC reanalysis 2: Treatment of ET-sensitive distant recurrence | AI alone | 103,526 | 11.38 | Reference |
Ribociclib plus AI | 169,544 | 12.38 | 66,143 | |
CDA-AMC reanalysis 3: Treatment duration | AI alone | 125,657 | 12.29 | Reference |
Ribociclib plus AI | 155,160 | 12.95 | 44,415 | |
CDA-AMC reanalysis 4: RDI | AI alone | 133,180 | 12.40 | Reference |
Ribociclib plus AI | 174,508 | 13.09 | 59,846 | |
CDA-AMC base case Reanalysis 1 + 2 + 3 + 4 | AI alone | 110,432 | 11.27 | Reference |
Ribociclib plus AI | 196,611 | 11.94 | 127,959 | |
AI = aromatase inhibitors; AE = adverse events; CDA-AMC = Canada’s Drug Agency; ET = endocrine therapy; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-years; RDI = relative dose intensity.
Note: All analyses are reported deterministically. The CDA-AMC reanalysis is based on the publicly available prices of the comparator treatments.
Table 11: Disaggregated Results of the CDA-AMC Base Case for the Abemaciclib-Eligible Subgroup — Ribociclib Plus AI vs. Abemaciclib Plus ET
Parameter | Ribociclib plus AI | Abemaciclib plus ET |
|---|---|---|
Discounted LYs | ||
Total | 15.05 | 15.05 |
By health state | ||
iDFS | 12.94 | 12.94 |
Nonmetastatic recurrence | 0.12 | 0.12 |
Remission | 0.85 | 0.85 |
Distant Recurrence, ET-resistant | 0.18 | 0.18 |
Distant Recurrence ET-sensitive | 1.25 | 1.25 |
Discounted QALYs | ||
Total | 11.95 | 11.95 |
By health state | ||
iDFS | 10.38 | 10.38 |
Nonmetastatic recurrence | 0.09 | 0.09 |
Remission | 0.69 | 0.69 |
Distant Recurrence, ET-resistant | 0.12 | 0.12 |
Distant Recurrence, ET-sensitive | 0.67 | 0.67 |
Discounted costs ($) | ||
Total | 196,268 | 206,152 |
Drug acquisition | 123,588 | 135,166 |
Administration and dispensing | 1,609 | 804 |
Follow-up and monitoring | 3,600 | 2,741 |
AEs | 0 | 0 |
Terminal care | 2,964 | 2,954 |
Second primary malignancy | 13 | 13 |
Distant recurrence | 64,495 | 64,495 |
AE = adverse events; CDA-AMC = Canada’s Drug Agency; ET = endocrine therapy; iDFS = invasive disease–free survival; LY = life-year; QALY = quality-adjusted life-year.
Note: All analyses are reported deterministically. The CDA-AMC reanalysis is based on the publicly available prices of the comparator treatments.
Table 12: Disaggregated Results of the CDA-AMC Base Case — Abemaciclib-Eligible Subgroup, Ribociclib Plus AI vs. AI Alone
Parameter | Ribociclib + AI | AI alone |
|---|---|---|
Discounted LYs | ||
Total | 15.05 | 14.21 |
By health state | ||
iDFS | 12.94 | 11.65 |
Nonmetastatic recurrence | 0.12 | 0.15 |
Remission | 0.85 | 1.07 |
Distant recurrence, ET-resistant | 0.18 | 0.38 |
Distant recurrence, ET-sensitive | 1.25 | 1.25 |
Discounted QALYs | ||
Total | 11.95 | 11.27 |
By health state | ||
iDFS | 10.38 | 9.36 |
Nonmetastatic recurrence | 0.09 | 0.11 |
Remission | 0.69 | 0.88 |
Distant recurrence, ET-resistant | 0.12 | 0.25 |
Distant recurrence, ET-sensitive | 0.67 | 0.68 |
Discounted costs ($) | ||
Total | 196,611 | 110,432 |
Drug acquisition | 123,588 | 10,019 |
Administration and dispensing | 1,609 | 1,480 |
Follow-up and monitoring | 3,600 | 2,741 |
AEs | 343 | 11 |
Terminal care | 2,964 | 2,645 |
Second primary malignancy | 13 | 11 |
Distant recurrence | 64,495 | 93,524 |
AE = adverse events; CDA-AMC = Canada’s Drug Agency; ET = endocrine therapy; iDFS = invasive disease–free survival; LY = life-year; QALY = quality-adjusted life-year.
Note: All analyses are reported deterministically. The CDA-AMC reanalysis is based on the publicly available prices of the comparator treatments.
Table 13: Summary of the Stepped Analysis — Abemaciclib-Ineligible Subgroup
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
Sponsor’s base case | AI alone | 91,005 | 14.57 | Reference |
Ribociclib plus AI | 141,378 | 15.91 | 37,656 | |
CDA-AMC reanalysis 1: Effectiveness waning | AI alone | 91,005 | 14.57 | Reference |
Ribociclib plus AI | 143,524 | 15.34 | 68,157 | |
CDA-AMC reanalysis 2: Subsequent treatment | AI alone | 76,928 | 13.90 | Reference |
Ribociclib plus AI | 152,881 | 15.51 | 47,393 | |
CDA-AMC reanalysis 3: Duration of treatment | AI alone | 91,005 | 14.42 | Reference |
Ribociclib plus AI | 141,378 | 15.75 | 38,033 | |
CDA-AMC reanalysis 4: RDI | AI alone | 96,088 | 14.57 | Reference |
Ribociclib plus AI | 160,280 | 15.91 | 47,987 | |
CDA-AMC base case: Reanalysis 1 + 2 + 3 + 4 | AI alone | 81,620 | 13.76 | Reference |
Ribociclib plus AI | 179,261 | 14.73 | 100,626 |
AI = aromatase inhibitors; AE = adverse events; CDA-AMC = Canada’s Drug Agency; ET = endocrine therapy; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-years; RDI = relative dose intensity.
Note: All analyses are reported deterministically. The CDA-AMC reanalysis is based on the publicly available prices of the comparator treatments.
Table 14: Disaggregated Results of the CDA-AMC Base Case for the Abemaciclib-Ineligible Subgroup
Parameter | Ribociclib + AI | AI alone |
|---|---|---|
Discounted LYs | ||
Total | 18.77 | 17.54 |
By health state | ||
iDFS | 17.08 | 15.78 |
Nonmetastatic recurrence | 0.12 | 0.11 |
Remission | 0.81 | 0.75 |
Distant recurrence, ET-resistant | 0.06 | 0.16 |
Distant recurrence, ET-sensitive | 0.70 | 0.75 |
Discounted QALYs | ||
Total | 14.73 | 13.76 |
By health state | ||
iDFS | 13.46 | 12.45 |
Nonmetastatic recurrence | 0.09 | 0.08 |
Remission | 0.65 | 0.60 |
Distant recurrence, ET-resistant | 0.04 | 0.11 |
Distant recurrence, ET-sensitive | 0.49 | 0.52 |
Discounted costs ($) | ||
Total | 179,261 | 81,620 |
Drug acquisition | 123,451 | 10,824 |
Administration and dispensing | 1,704 | 1,602 |
Follow-up and monitoring | 3,931 | 2,668 |
AEs | 296 | 11 |
Terminal care | 5,493 | 4,956 |
Second primary malignancy | 19 | 21 |
Distant recurrence | 44,367 | 61,538 |
AE = adverse events; CDA-AMC = Canada’s Drug Agency; ET = endocrine therapy; iDFS = invasive disease–free survival; LY = life-year; QALY = quality-adjusted life-year.
Note: Deterministic results.
Price Reduction Analysis
CDA-AMC conducted price reduction analyses using the sponsor’s base case and the CDA-AMC base case (refer to Table 15). No price reduction analyses were conducted for ribociclib plus AI versus abemaciclib plus ET because ribociclib plus AI was dominant (more effective, less costly) in both the sponsor’s and the CDA-AMC base case.
Table 15: Results of the Price Reduction Analysis
Price reduction | Unit drug cost ($) | Cost per 28 days ($) | ICERs for ribociclib +AI vs. AI alone ($/QALY) | |||
|---|---|---|---|---|---|---|
Sponsor base case | CDA-AMC base case | |||||
Abemaciclib-eligible | Abemaciclib-ineligible | Abemaciclib-eligible | Abemaciclib-ineligible | |||
No price reduction | 94.1271a | 3,953 | 42,724 | 37,656 | 127,959 | 100,626 |
10% | 84.7144 | 3,558 | 29,095 | 30,668 | 111,203 | 89,075 |
20% | 75.3017 | 3,163 | 15,466 | 23,680 | 94,447 | 77,525 |
30% | 65.8890 | 2,767 | 1,837 | 16,693 | 77,691 | 65,974 |
40% | 56.4763 | 2,372 | Dominant | 9,705 | 60,935 | 54,423 |
50% | 47.0636 | 1,977 | Dominant | 2,717 | 44,179 | 42,873 |
60% | 37.6509 | 1,581 | Dominant | Dominant | 27,423 | 31,322 |
70% | 28.2381 | 1,186 | Dominant | Dominant | 10,667 | 19,771 |
80% | 18.8254 | 791 | Dominant | Dominant | Dominant | 8,220 |
90% | 9.4127 | 395 | Dominant | Dominant | Dominant | Dominant |
AI = aromatase inhibitors; CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; vs. = versus.
aSponsor’s submitted price for ribociclib.1
Assessment of Uncertainty
CDA-AMC conducted scenario analyses to address uncertainty within the economic evaluation, using the CDA-AMC base case for ribociclib plus AI versus AI alone in both the abemaciclib-eligible and -ineligible subgroups. Results are provided in
Assuming no waning of treatment effectiveness over time.
Assuming no further treatment effect beyond the duration of the NATALEE trial.
Including RDI, based on sponsor-provided values.
Table 16: Results of CDA-AMC Scenario Analyses — Abemaciclib-Eligible Subgroup
Stepped analysisa | Drug | Total costs ($) | Total QALYs | ICER ($/QALYs) |
|---|---|---|---|---|
Ribociclib plus AI vs. abemaciclib plus ET | ||||
CDA-AMC base case | Ribociclib plus AI | 196,268 | 11.95 | Reference |
Abemaciclib plus ET | 206,152 | 11.95 | Dominated by ribociclib plus AI | |
CDA-AMC scenario 1: No treatment effectiveness waning | Ribociclib plus AI | 188,631 | 12.44 | Reference |
Abemaciclib plus ET | 198,515 | 12.44 | Dominated by ribociclib plus AI | |
CDA-AMC scenario 2: No effect beyond the within-trial period (i.e., 44 months) | Ribociclib plus AI | 196,846 | 11.50 | Reference |
Abemaciclib plus ET | 206,730 | 11.50 | Dominated by ribociclib plus AI | |
CDA-AMC scenario 3: Including RDI | Ribociclib plus AI | 172,712 | 11.95 | Reference |
Abemaciclib plus ET | 189,885 | 11.95 | Dominated by ribociclib plus AI | |
Ribociclib plus AI vs. AI alone | ||||
CDA-AMC base case | AI alone | 110,432 | 11.27 | Reference |
Ribociclib plus AI | 196,611 | 11.94 | 127,959 | |
CDA-AMC scenario 1: No treatment effectiveness waning | AI alone | 110,432 | 11.27 | Reference |
Ribociclib plus AI | 188,974 | 12.43 | 67,485 | |
CDA-AMC scenario 2: No effect beyond the within-trial period (i.e., 44 months) | AI alone | 110,432 | 11.27 | Reference |
Ribociclib plus AI | 197,189 | 11.50 | 374,443 | |
CDA-AMC scenario 3: Including RDI | AI alone | 103,526 | 11.27 | Reference |
Ribociclib plus AI | 173,055 | 11.95 | 103,235 | |
AI = aromatase inhibitors; CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
aDeterministic analyses.
Table 17: Results of CDA-AMC Scenario Analyses — Abemaciclib-Ineligible Subgroup
Stepped analysisa | Drug | Total costs ($) | Total QALYs | ICER ($/QALYs) |
|---|---|---|---|---|
CDA-AMC base case | AI alone | 81,620 | 13.76 | Reference |
Ribociclib plus AI | 179,261 | 14.73 | 100,626 | |
CDA-AMC scenario 1: No waning effect | AI alone | 81,620 | 13.76 | Reference |
Ribociclib plus AI | 168,598 | 15.75 | 43,747 | |
CDA-AMC scenario 2: Waning effect starts at 50 months | AI alone | 81,620 | 13.76 | Reference |
Ribociclib plus AI | 180,495 | 14.15 | 252,518 | |
CDA-AMC scenario 3: Including RDI | AI alone | 76,928 | 13.76 | Reference |
Ribociclib plus AI | 157,244 | 14.73 | 82,771 |
AI = aromatase inhibitor; CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
aDeterministic analyses.
Issues for Consideration
Abemaciclib has successfully completed negotiations with the pan-Canadian Pharmaceutical Alliance for the treatment of HR-positive, HER2-negative, node-positive, early breast cancer at high risk of recurrence based on clinicopathological features. Ribociclib has previously been negotiated for advanced or metastatic breast cancer. It is therefore likely that both treatments are reimbursed by jurisdictional drug plans at confidential prices that are less than publicly available list prices. Thus, the price of ribociclib and abemaciclib paid by the drug plans may be lower than incorporated in the sponsor’s model, which was based on the submitted price and public list prices, respectively. This would have implications for both the cost-effectiveness analysis and the budget impact assessment; however, the direction of change is unknown.
During the review period, the sponsor provided updated iDFS and OS data to CDA-AMC (data cut-off May 28, 2025). The updated data provides an additional 11.2 months of follow-up versus the data cut from April 2024. Data from the April 2024 data cut inform the sponsor’s and CDA-AMC’s base case; however, the updated data were determined by CDA-AMC to have minimal impact on the results of the CDA-AMC base case.
Appendix 5: Budget Impact Analysis
Please note that this appendix has not been copy-edited.
Summary of the Sponsor’s BIA
The sponsor submitted a BIA that estimated the expected incremental budgetary impact of reimbursing ribociclib for use in the Health Canada–indicated population (adjuvant treatment of HR-positive, HER2-negative stage II and III early breast cancer).2
The BIA was conducted from the perspective of public drug plan payers in Canada over a 3-year time horizon (years not specified by sponsor). The sponsor’s estimate reflects the aggregated results from the jurisdictional provincial budgets (excluding Quebec) as well as the Non-Insured Health Benefits Program. The sponsor estimated the eligible population using an epidemiological approach. The sponsor’s base case included drug acquisition costs, administration costs, dispensing costs, and mark ups. The market uptake for ribociclib plus AI was estimated using sponsor’s assumptions. The sponsor assumed that patients who met the eligibility criteria for cohort 1 of the monarchE trial (stage II with N1 [depending on histopathologic grade and/or tumour size], stage III cases with N1+). In the abemaciclib-eligible subgroup, ribociclib plus AI was assumed to displace only abemaciclib plus ET, while ribociclib plus AI displaced all comparators in the abemaciclib-ineligible subgroup. The key inputs to the BIA are documented in Table 18.
The sponsor estimated that the 3-year incremental budget impact associated with reimbursing ribociclib plus AI for the treatment of HR-positive, HER2-negative stage II and III early breast cancer would be $82,506,937 (year 1 = $10,750,908; year 2 = $23,862,113; year 3 = $47,893,817).
Table 18: Key Model Parameters
Parameter | Sponsor’s estimate (Year 1 / Year 2 / Year 3) | |
|---|---|---|
Abemaciclib-ineligible | Abemaciclib-eligible | |
Target population | ||
Population size | 32,021,468 | |
Age-standardized incidence of breast cancer | Province-specific (range, 62.2 to 91.9 per 100,000)14 | |
Percentage of incident breast cancer that is HR+, HER2-negative | 72.7%15 | |
Percentage of patients with incident breast cancer who undergo surgery | 86.5%16 | |
Stage distribution, percentage of patients | ||
Stage II | 37.0%17 | |
Stage II with N0 | 51.4%a | |
Stage IIB; stage IIA with grade 3 tumours or grade 2 with high-risk genomic profile or Ki67 ≥ 20% | 52.9%a | |
Stage II with N1 | 48.6%a | |
Stage II and N1 with grade 3 or ≥ 5 cm tumour | 25.4%a | |
Stage II with N2+ | 0%a | |
Any stage III | 12.0%17 | |
Stage III with N1 | 34.9%a | |
Stage III and N1 with grade 3 or ≥ 5 cm tumour | 100%a | |
Stage III with N2+ | 61.6%a | |
Percentage of patients who initiate ET within 1 year of diagnosis | 85.9%18 | |
Percentage of early breast cancer that is surgically resected | Province-specific (range, 98.7% to 100%)14 | |
Percentage of patients aged < 65 years | 84.8%19 | |
Percentage of women who are premenopausal | 44.2%19 | |
Percentage of patients covered by public drug plan | Province- and age-specific (range, 7.5% to 100%)20 | |
Number of patients eligible for drug under review | 6,332 / 9,488/ 12,638b | |
Market shares (reference scenario) | ||
Ribociclib plus AI | 0% / 0% / 0% | 0% / 0% / 0% |
Abemaciclib plus ET | NA | 75% / 75% / 75% |
Nonsteroidal aromatase inhibitorsc | 80% / 80% / 80% | 19% / 19% / 19% |
Exemestane | 13% / 13% / 13% | 4% / 4% / 4% |
Tamoxifen | 7% / 7% / 7% | 2% / 2% / 2% |
Market shares (new-drug scenario) | ||
Ribociclib plus AI | 18% / 29% / 40% | 13%/ 25% / 38% |
Abemaciclib plus ET | NA | 62% / 50% / 37% |
Nonsteroidal aromatase inhibitorsc | 64% / 55% / 46% | 19% / 19% / 19% |
Exemestane | 12% / 11% / 10% | 4% / 4% / 4% |
Tamoxifen | 6% / 5% / 4% | 2% / 2% / 2% |
Cost of treatment (per patient per 28-day course)d,e | ||
Ribociclib | $4,143 | |
Abemaciclib | $6,444 | |
Letrozole | $40 | |
Anastrozole | $29 | |
Exemestane | $79 | |
Tamoxifen | $11 | |
AI = aromatase inhibitor; ET = endocrine therapy; NA = not applicable.
Note: N indicates whether the cancer has reached nearby lymph nodes. N0 indicates no nodal involvement, while N1 to N3 indicates involvement of 1 to 3 lymph nodes.
aEstimates derived from analyses of ConcertAI data, a US real-world electronic health record database, based on data from 2015 to 2023.2
bIn the baseline year, the sponsor estimates that the total number of people with and without abemaciclib-eligible characteristics and receiving ET is 2,024 and 3,003 people, respectively.
cNonsteroidal aromatase inhibitors were assumed by the sponsor to be letrozole or anastrozole.
dCosts in the model were adjusted by relative dose intensity by the sponsor.
eTime on treatment in the BIA was based on time to treatment discontinuation observations from the NATALEE trial.
Key Issues of the Submitted BIA
CDA-AMC identified several key issues to the sponsor’s analysis that have notable implications on the results of the BIA:
The number of patients eligible for ribociclib is uncertain. The sponsor used an epidemiologic approach to estimate the number of eligible patients, starting with age-adjusted, province-specific incidence rates estimated using data from Statistics Canada and projected cancer cases by sex and geographic region from the 2023 Canadian Cancer Statistics report. The sponsor then used estimates from the Surveillance, Epidemiology and End Results (SEER) Registries Research Database, an analysis of US electronic health records, and a sponsor-conducted analysis of real-world data to determine the proportion of patients with HR-positive, HER2-negative breast cancer, the proportion of patients who undergo surgical resection, and the distribution of cases by nodal status, grade, and tumour size, respectively. There are several sources of uncertainty associated with these estimates, notably that it is unclear whether the real-world evidence is generalizable to patients in Canada. Real-world evidence from the US may not be relevant to the Canadian setting, for example, because access to health care in the US is dependent, at least in part, on insurance payers and patients’ ability to pay a copayment.
CDA-AMC was unable to address this limitation.
Uncertainty about the budget impact among subgroups. The sponsor assumed that patients who meet the monarchE cohort 1 criteria (4 or more positive axillary lymph nodes, or between 1 and 3 positive nodes and either grade 3 disease or tumour size of 5 cm or larger) would be eligible for abemaciclib and that all other ribociclib-eligible patients would be ineligible for abemaciclib.2 However, the abemaciclib is reimbursed for both cohort 1 and cohort 2 from the monarchE trial (cohort 2: 1 to 3 positive nodes and Ki-67 of at least 20%).7 As such, some patients were misclassified as being abemaciclib-ineligible when they would meet the reimbursement criteria.
The structure of the sponsor’s model did not permit calculation of the expected budget impact among abemaciclib-eligible and -ineligible subgroups. Based on exploratory analyses conducted by CDA-AMC, there is the potential for ribociclib plus AI to be a cost saving if reimbursement were restricted to the patients who would otherwise be eligible for abemaciclib; however, clinical expert input received for this review indicated that clinicians would likely continue to prefer abemaciclib in this patient population. In the abemaciclib-ineligible subgroup, the reimbursement of ribociclib will be associated with incremental costs owing to the displacement of less costly treatments.
CDA-AMC was unable to address this limitation.
Use of RDI to estimate actual drug costs is not appropriate: The sponsor’s base case incorporated RDI for ribociclib, abemaciclib, and endocrine therapies based on data from the NATALEE trial. This consideration of RDI is problematic as this parameter can be influenced by several factors. The dose received by a patient may differ from the full planned dose of the drug due to dose delays, missed doses, dose reductions to manage toxicity, or subsequent dose re-escalation; each of these have differing impacts on drug costs. Furthermore, prescriptions for ribociclib may be filled and reimbursed regardless of treatment adherence.
In the CDA-AMC reanalysis, RDI was set to 100% for all drugs.
The prices paid by public drug plans are uncertain: The prices for ribociclib and comparators were based on publicly available list prices. Both ribociclib and abemaciclib successfully undergone negotiations with pCPA (abemaciclib: for treatment of HR-positive, HER2-negative, node-positive, early breast cancer at high risk of recurrence based on clinicopathological features; ribociclib: advanced or metastatic breast cancer). As such, public list prices for ribociclib and comparators may not reflect the actual prices paid by public drug plans, as any potential confidential rebates are not reflected in this analysis.
CDA-AMC was unable to address this limitation.
The sponsor’s submitted model for the budget impact model is not user-friendly and unnecessarily complicated: Several of the model inputs and assumptions in the sponsor’s submitted budget impact model were difficult to test or modify with alternate inputs or assumptions due to unnecessary complexity (i.e., disconnected worksheets, hard-coded cells, and different values across spreadsheets) and structural constraints.
CDA-AMC was unable to address this key limitation and notes that a thorough validation of the sponsor’s model was not possible.
CDA-AMC Reanalyses of the BIA
CDA-AMC revised the sponsor’s submitted analyses by making changes in model parameter values and assumptions, in consultation with clinical experts, as outlined in Table 19.
Table 19: Revisions to the Submitted BIA
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
1. Remove RDI | RDI obtained from NATALEE trial | Assumed to be 100% for all drugs |
2. Costs associated with administration costs,a dispensing, and mark ups | Included | Excluded |
CDA-AMC base case | ― | Reanalysis 1 + 2 |
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency; RDI = relative dose intensity.
aAdministration costs were removed in the CDA-AMC base case because the sponsor’s model returned improbable results.
The results of the CDA-AMC stepwise reanalysis are presented in summary format in Table 20, and a more detailed breakdown is presented in Table 21. In the CDA-AMC base case, the 3-year budget impact of reimbursing ribociclib plus AI for adjuvant treatment of HR-positive, HER2-negative stage II and III early breast cancer was $103,696,814 (year 1 = $13,845,001; year 2 = $31,247,581; year 3 = $61,604,233).
Table 20: Summary of the Stepped Analysis of the CDA-AMC Base Case
Stepped analysis | Three-year total ($) |
|---|---|
Submitted base case | 82,506,837 |
CDA-AMC reanalysis 1 | 106,696,814 |
CDA-AMC reanalysis 2 | 74,451,096 |
CDA-AMC base case: Reanalysis 1 + 2 | 98,641,073 |
CDA-AMC = Canada’s Drug Agency.
Note: The CDA-AMC reanalysis is based on publicly available prices of the comparator treatments. The sponsor’s base case, as well as the CDA-AMC base case includes mark-up and dispensing fees as submitted by the sponsor.
Table 21: Disaggregated Summary of the BIA
Stepped analysis | Scenario | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|---|
Submitted base case | Reference total | 80,167,736 | 141,299,147 | 148,429,446 | 154,986,675 | 524,883,005 |
Ribociclib plus AI | 0 | 0 | 0 | 0 | 0 | |
All other comparators | 80,167,736 | 141,299,147 | 148,429,446 | 154,986,675 | 524,883,005 | |
New drug total | 80,167,736 | 152,050,055 | 172,291,559 | 202,880,492 | 607,389,842 | |
Ribociclib plus AI | 0 | 24,455,324 | 60,260,192 | 107,890,572 | 192,606,088 | |
All other comparators | 80,167,736 | 127,594,731 | 112,031,367 | 94,989,920 | 414,783,753 | |
Budget Impact | 0 | 10,750,908 | 23,862,113 | 47,893,817 | 82,506,837 | |
CDA-AMC base case | Reference total | 82,656,663 | 145,437,780 | 150,899,323 | 155,882,752 | 534,912,427 |
Ribociclib plus AI | 0 | 0 | 0 | 0 | 0 | |
All other comparators | 82,656,663 | 145,437,780 | 150,899,323 | 155,882,752 | 534,912,427 | |
New drug total | 82,656,663 | 158,281,740 | 179,677,525 | 212,937,572 | 633,553,500 | |
Ribociclib plus AI | 0 | 26,977,889 | 66,476,026 | 119,019,475 | 212,473,390 | |
All other comparators | 82,656,663 | 131,303,851 | 113,201,499 | 93,918,097 | 421,080,110 | |
Budget Impact | 0 | 12,807,961 | 28,778,292 | 57,054,820 | 98,641,073 |
AI = aromatase inhibitor; BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency.
Note: The CDA-AMC reanalysis is based on the publicly available prices of the comparator treatments.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.