Drugs, Health Technologies, Health Systems
Reimbursement Recommendation
Mirvetuximab Soravtansine (Elahere)
Indication: As monotherapy indicated for the treatment of adult patients with folate receptor-alpha positive, platinum-resistant epithelial ovarian, fallopian tube, or primary peritoneal cancer, who have received one to three prior systemic treatment regimens
Sponsor: AbbVie Corporation
Final recommendation: Reimburse with conditions
Summary
What Is the Reimbursement Recommendation for Elahere?
Canada’s Drug Agency (CDA-AMC) recommends that Elahere be reimbursed by public drug plans for the treatment of adults with FR-alpha–positive, platinum-resistant epithelial ovarian, fallopian tube, or primary peritoneal cancer who have received 1 to 3 prior systemic treatment regimens if certain conditions are met.
Which Patients Are Eligible for Coverage?
Elahere should only be covered to treat patients with a confirmed diagnosis of epithelial ovarian, primary peritoneal, or fallopian tube cancer who have platinum-resistant disease, a tumour that is positive for FR-alpha expression confirmed by a validated test, disease that progressed on or after their most recent line of therapy, and received at least 1 prior systemic line of anticancer therapy.
What Are the Conditions for Reimbursement?
Elahere should only be reimbursed if the patient is under the care of a physician with expertise in managing gynecological malignancies, if the treatment is administered in an oncology health facility with access to ophthalmic services and FR-alpha testing, and if the cost of Elahere is reduced.
Why Did CDA-AMC Make This Recommendation?
Evidence from 1 clinical trial demonstrated that Elahere led to treatment response, delayed cancer progression, and may help patients live longer compared to chemotherapy. Elahere did not lead to a meaningful difference in the number of patients who reported an improvement in their health-related quality of life (HRQoL) compared to chemotherapy.
Elahere potentially met several patient-identified needs, including controlling disease progression, prolonging survival, improving response rate, and addressing the underlying disease while maintaining HRQoL.
Based on the CDA-AMC assessment of the health economic evidence, Elahere does not represent good value to the health care system at the public list price. A price reduction is therefore required.
Based on public list prices, Elahere is estimated to cost the public drug plans approximately $120 million over the next 3 years.
Additional Information
What Is Epithelial Ovarian, Fallopian Tube, or Primary Peritoneal Cancer?
Epithelial ovarian cancer is a serious and aggressive type of ovarian cancer that starts in the surface cells of the ovaries, fallopian tubes, or lining of the abdomen, and causes symptoms such as pelvic pain and changes in appetite or urination. It is usually diagnosed at a late stage, making it difficult to treat. The lifetime risk of developing epithelial ovarian cancer is about 1.3%.
Unmet Needs in Epithelial Ovarian, Fallopian Tube, or Primary Peritoneal Cancer
Patients with epithelial ovarian, fallopian tube, or primary peritoneal cancer do not often have disease that responds to available nonplatinum chemotherapy treatments. Treatment options that use biomarkers such as FR-alpha expression levels and can help improve response rate, prolong survival, and address the underlying disease while maintaining HRQoL are needed.
How Much Does Elahere Cost?
Treatment with Elahere is expected to cost approximately $29,307 per patient per 28-day cycle.
Recommendation
The pan-Canadian Oncology Drug Review Expert Review Committee (pERC) recommends that mirvetuximab soravtansine be reimbursed for the treatment of adults with FR-alpha–positive, platinum-resistant epithelial ovarian, fallopian tube, or primary peritoneal cancer who have received 1 to 3 prior systemic treatment regimens only if the conditions listed in Table 1 are met.
Rationale for the Recommendation
Evidence from 1 randomized, open-label, phase III study (MIRASOL; N = 453) demonstrated that treatment with mirvetuximab soravtansine resulted in added clinical benefit for adults with FR-alpha–positive (≥ 75% of viable tumour cells with ≥ 2+ staining intensity), platinum-resistant epithelial ovarian cancer who had received 1 to 3 prior systemic treatment regimens compared with investigator’s choice (IC) of chemotherapy. In the MIRASOL study, treatment with mirvetuximab soravtansine (6 mg/kg of adjusted ideal body weight every 3 weeks) resulted in an improved progression-free survival (PFS) and overall survival (OS), and a higher objective response rate (ORR) compared to chemotherapy but resulted in little to no clinically important difference in the proportion of patients who reported an improvement in HRQoL. Regarding safety, pERC noted that a greater proportion of patients in the mirvetuximab soravtansine treatment group compared to the IC chemotherapy group reported notable harms, including keratopathy, pneumonitis, and blurred vision. While the harms present potential safety concerns, they were seen as manageable with appropriate care and monitoring. An indirect treatment comparison (ITC) estimated the comparative effectiveness of mirvetuximab soravtansine versus bevacizumab plus IC chemotherapy. The results were not statistically conclusive and generally suggest a similar treatment efficacy between the groups.
Input from the clinical experts indicated that platinum-resistant epithelial ovarian cancer often becomes refractory to current nonplatinum chemotherapies with or without bevacizumab. The patient and clinician groups noted that the treatment outcomes valued by patients include controlling disease progression, prolonging survival, improving response rate, and addressing the underlying disease while maintaining HRQoL. pERC concluded that mirvetuximab soravtansine potentially meets all of the needs identified by patients relative to IC chemotherapy alone.
Using the sponsor-submitted price for mirvetuximab soravtansine and publicly listed prices for all other drug costs, the incremental cost-effectiveness ratio (ICER) for mirvetuximab soravtansine was $642,249 per quality-adjusted life-year (QALY) gained compared with pooled chemotherapy. At this ICER, mirvetuximab soravtansine is not cost-effective at a $50,000 per QALY gained willingness-to-pay threshold for the submitted indication. A price reduction is required for mirvetuximab soravtansine to be considered cost-effective at a $50,000 per QALY threshold.
Table 1: Reimbursement Conditions and Reasons
Reimbursement condition | Reason | Implementation guidance |
|---|---|---|
Initiation | ||
1. Treatment should only be reimbursed when initiated in patients with a confirmed diagnosis of epithelial ovarian cancer, primary peritoneal cancer, or fallopian tube cancer who have all the following: 1.1. platinum-resistant disease 1.2. a tumour that is positive for FR-alpha expression confirmed by a validated test 1.3. disease that has progressed on or after their most recent line of therapy 1.4. received at least 1 prior systemic line of anticancer therapy. | Evidence from the MIRASOL study suggested that treatment with mirvetuximab soravtansine demonstrated a clinically meaningful benefit in patients with these characteristics for this condition. | pERC noted that in the MIRASOL study, positive FR-alpha expression was defined as at least 75% of viable tumour cells having exhibited level 2 and/or 3 membrane staining intensity according to the VENTANA FOLR1 (FOLR1-2.1) RxDx Assay. pERC acknowledged that the indication is for patients who have received 1 to 3 systemic treatment regimens; however, patients who have received more than 3 prior lines of therapy before the availability of mirvetuximab soravtansine may be considered eligible for treatment on a time-limited basis. |
2. Treatment should not be reimbursed for patients with: 2.1. chronic corneal disorders, history of corneal transplant, or ocular conditions requiring treatment and/or monitoring 2.2. previous clinical diagnosis of noninfectious ILD, including noninfectious pneumonitis. | Patients with any of these characteristics were excluded from the MIRASOL study. As such, the potential benefit of mirvetuximab soravtansine in these patients has not been demonstrated. | — |
Renewal | ||
3. Patients must meet the following criteria for renewal: 3.1. tumour assessments per institution policy 3.2. toxicity monitoring requirement per institutional policy. | This criterion is per the MIRASOL study, clinical expert opinion, and standard clinical practice. | — |
Discontinuation | ||
4. Reimbursement of mirvetuximab soravtansine should be discontinued on occurrence of any of the following: 4.1. significant disease progression 4.2. intolerable toxicity. | This criterion is per the MIRASOL study, clinical expert opinion, and standard clinical practice. | — |
Prescribing | ||
5. Mirvetuximab soravtansine should be prescribed by physicians with expertise in managing gynecological malignancies and should be administered in an oncology health facility with access to ophthalmic services. | This is to ensure that mirvetuximab soravtansine is prescribed only for appropriate patients and adverse effects are managed in an optimized and timely manner. | pERC noted that, due to the ocular toxicity associated with mirvetuximab soravtansine, it should only be used in a setting with access to facilities and clinician specialists (i.e., ophthalmologists or optometrists) to monitor and manage ocular adverse events. |
6. Testing for FR-alpha expression should be available and accessible. | Evidence from the MIRASOL study suggested that treatment with mirvetuximab soravtansine demonstrated a clinically meaningful benefit in patients whose tumour was positive for FR-alpha expression. | FR-alpha expression is determined using an immunohistochemistry-based assay on tissue samples. Implementation of FR-alpha expression testing would likely impact human and other health care resources. Cost of testing, as well as that for infrastructure and training of laboratory personnel, should be considered. |
Pricing | ||
7. A reduction in price | The ICER for mirvetuximab soravtansine is $642,249 per QALY gained compared with pooled chemotherapy. A price reduction greater than 80% would be required for mirvetuximab soravtansine to achieve an ICER of $50,000 per QALY gained compared to pooled chemotherapy. Price reductions for different thresholds are available in the Pharmacoeconomic Review report. | — |
Feasibility of adoption | ||
8. The economic feasibility of adoption of mirvetuximab soravtansine must be addressed. | At the submitted price, the incremental budget impact of mirvetuximab soravtansine is expected to be greater than $40 million after year 2. | The CDA-AMC estimate of the budget impact is based on the 92% of eligible patients with high-grade serous epithelial ovarian cancer. The true budget impact is therefore higher than the CDA-AMC estimate. |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; ILD = interstitial lung disease; pERC = pan-Canadian Oncology Drug Review Expert Review Committee; QALY = quality-adjusted life-year.
Discussion Points
Available treatment options and unmet patient needs: pERC discussed that patients with platinum-resistant epithelial ovarian cancer often have disease that becomes refractory or resistant to current nonplatinum chemotherapy, including paclitaxel, pegylated liposomal doxorubicin (PLD), topotecan, and gemcitabine, with or without bevacizumab, and that the condition is associated with a poor prognosis. pERC also noted input from clinical experts that emphasized that there is a significant unmet need for therapeutic options that leverage the use of biomarkers such as FR-alpha expression levels. Based on this input, pERC recognized the need for an additional treatment option for patients with platinum-resistant epithelial ovarian cancer.
Certainty of evidence for PFS and OS: pERC discussed the Grading of Recommendations Assessment, Development and Evaluation (GRADE) assessment of PFS and OS and noted that the certainty of evidence improved at later time points. Though the evidence for PFS at 6 months was assessed to be of moderate certainty because the between-group difference included both a meaningful benefit and the possibility of no effect; at 12 months, there is a high certainty in the evidence that mirvetuximab soravtansine results in an increase in PFS compared to IC chemotherapy. Similarly, there is a low certainty of evidence for OS at 6 months, which included both a clinically meaningful benefit and the possibility of no survival benefit; however, at 18 months, there is moderate certainty in the evidence that mirvetuximab soravtansine likely results in an increase in OS compared to IC chemotherapy.
HRQoL: While pERC acknowledged that improvement in HRQoL is an important outcome for patients, mirvetuximab soravtansine did not demonstrate a clinically meaningful improvement in HRQoL as measured by the proportion of patients with a 15-point symptom subscale decrease (i.e., improvement) on the European Organisation for Research and Treatment of Cancer (EORTC) Quality of Life Questionnaire ovarian cancer module (QLQ-OV28) abdominal/gastrointestinal symptom subscale (which is a 28-item scale that assesses the quality of life [QoL] of patients with ovarian cancer). The certainty of the evidence was assessed to be low because the observed effect included the possibility of no improvement, suggesting that mirvetuximab soravtansine may result in little to no improvement in abdominal and gastrointestinal symptoms compared to IC chemotherapy. However, pERC also acknowledged that treatment with mirvetuximab soravtansine was not associated with a deterioration in HRQoL based on the EORTC QLQ-OV28 abdominal/gastrointestinal symptom subscale. pERC additionally noted that the EORTC QLQ-OV28 abdominal/gastrointestinal symptom subscale does not measure QoL related to ocular effects and lung symptoms, which may be an important consideration for QoL of patients treated with mirvetuximab soravtansine.
Indirect evidence: pERC discussed the sponsor-submitted ITC that evaluated mirvetuximab soravtansine versus bevacizumab plus chemotherapy. The ITC results suggested inconsistent findings: mirvetuximab soravtansine was associated with longer median OS (hazard ratio [HR] = ████; 95% confidence interval [CI], ████ ██ ████), suggesting a potential benefit, and higher ORR rate, but potentially shorter median PFS (HR = ████; 95% CI, ████ ██ ████) indicating a possibility of no benefit. Further, pERC concluded that the comparative results were not statistically conclusive and, overall, may suggest similar treatment efficacy between mirvetuximab soravtansine and bevacizumab plus chemotherapy, though several methodological and clinical limitations affecting the validity of the indirect comparison were identified. These included the temporal gap of more than 10 years between the MIRASOL and AURELIA trials, differences in the clinical management of treatment-emergent adverse events (TEAEs), heterogeneity in patient characteristics (e.g., tumour histology, FR-alpha expression status, prior PARP inhibitor or bevacizumab use), and variations in chemotherapy regimens across the trials. pERC noted that these factors introduce potential biases and concluded that the clinical benefit of mirvetuximab soravtansine compared to bevacizumab plus chemotherapy in patients with platinum-resistant epithelial ovarian cancer is uncertain.
Toxicity and safety monitoring: pERC discussed the safety profile of mirvetuximab soravtansine, with particular attention to ocular toxicities (i.e., blurred vision, keratopathy, and photophobia) and pneumonitis, which require additional monitoring and supportive care. The clinical experts emphasized that while these events are generally reversible and manageable with prophylactic eye drops, they require early recognition and coordinated ophthalmologic monitoring. Pneumonitis was also more frequently reported, highlighting the need for timely intervention. pERC concluded that these toxicities, while clinically significant, can be addressed with appropriate safety assessments and multidisciplinary management strategies. As such, pERC agreed with clinical experts that mirvetuximab soravtansine should be administered in specialized oncology settings with access to ophthalmic services.
FR-alpha expression testing: FR-alpha expression testing is not performed as part of routine care for patients with epithelial ovarian cancer. The immunohistochemistry (IHC)-based testing platform is under review by Health Canada and is not currently available or publicly funded in Canada. The clinical experts noted that implementation of FR-alpha expression testing would likely lead to an increase in human resources and laboratory costs. The committee also noted that there would be costs associated with testing when accounting for reagents and human resources, but this is not expected to be high. Additional costs would be incurred to ensure tests are available and accessible to patients across the country. Furthermore, pERC noted that while the sponsor recommendation was to test for FR-alpha expression reflexively at the time of epithelial ovarian cancer diagnosis, the optimal time for testing is unclear.
Background
Epithelial ovarian cancer is the most common type of ovarian cancer and the deadliest gynecologic malignancy. Epithelial ovarian cancer arises from the epithelium covering the fimbria of the fallopian tubes, the ovaries, or the peritoneal cavity. About 85% to 95% of ovarian cancers are epithelial in origin. Most females with advanced ovarian cancer will have disease that initially responds to platinum-based chemotherapy; however, up to 70% experience disease recurrence. Females with disease that shows a response to platinum-based chemotherapy and subsequently experience relapse 6 months or more after completion of platinum-based therapy are classified as “platinum sensitive,” while those who experience relapse within 6 months of completing platinum-based therapy are known to have disease that is “platinum resistant.” Up to 70% of patients with recurrent disease eventually experience platinum resistance. Compared to other late-stage cancers, platinum-resistant ovarian cancer (PROC) is difficult to treat; patients experience severe symptoms that impair their QoL and face a very poor prognosis with limited treatment options. PROC is characterized by low expected ORR (10% to 15%), short PFS (3 to 6 months), and limited survival (median OS = 9 to 12 months). In addition to poor survival, patients experience severe impairments in HRQoL, including frequent bowel obstruction, abdominal pain, residual neuropathy from prior therapies, and anxiety and/or fear, all of which have a direct, severe impact on their daily lives.
There is no cure for PROC; therefore, the conventional treatment goals are to maximize or maintain HRQoL while improving OS by controlling disease or delaying further progression. Current therapies in PROC consist primarily of nonplatinum-based chemotherapy (e.g., weekly paclitaxel, PLD, or topotecan), administered either as a single drug or in combination with bevacizumab. Single-drug, nonplatinum-based chemotherapy treatments are associated with many toxicities, including sensory and motor neurotoxicity. Chemotherapy is also associated with many significant severe adverse events (AEs) often experienced without any improvement in disease-related symptoms. Nonplatinum-based chemotherapy has low ORRs of 4% to 13%, median PFS of 3 to 4 months, and median OS of approximately 9 to 12 months, as well as significant adverse effects that negatively impact HRQoL. Although bevacizumab in combination with chemotherapy shows modest improvements in PFS (median PFS = 6.7 months) and ORR (30.9%), it can be associated with higher rates of gastrointestinal perforation, hypertension, thrombotic complications, and proteinuria in patients with ovarian cancer. Additional challenges in the treatment of PROC include the lack of meaningful, predictive biomarkers, which impedes targeted treatment. There is a significant unmet need for a novel therapeutic option for PROC that takes advantage of molecular targets such as FR-alpha, which have yet to be leveraged.
Mirvetuximab soravtansine is a first-in-class antibody-drug conjugate targeting FR-alpha, a protein that is commonly overexpressed on ovarian carcinomas and minimally expressed on normal tissues. Mirvetuximab soravtansine is approved by Health Canada as monotherapy for the treatment of adults with FR-alpha–positive, platinum-resistant epithelial ovarian, fallopian tube, or primary peritoneal cancer who have received 1 to 3 prior systemic treatment regimens. The sponsor is requesting reimbursement per the approved Health Canada indication. The recommended dose of mirvetuximab soravtansine is 6 mg/kg of adjusted ideal body weight administered once every 3 weeks (21-day cycle) as an IV infusion until disease progression or unacceptable toxicity according to the draft product monograph.
Sources of Information Used by the Committee
To make its recommendation, the committee considered the following information:
a review of 1 phase III open-label trial (MIRASOL) designed to compare the efficacy and safety of mirvetuximab soravtansine against select IC chemotherapies for patients with platinum-resistant high-grade epithelial ovarian, primary peritoneal, or fallopian tube cancer whose tumours express a high level of FR-alpha and 1 indirect treatment comparisons
patients’ perspectives gathered by 1 patient group, Ovarian Cancer Canada (OCC)
input from public drug plans that participate in the reimbursement review process
2 clinical specialists with expertise diagnosing and treating patients with platinum-resistant high-grade epithelial ovarian cancer
input from 3 clinician groups, the BC Cancer Gynecologic Oncology Provincial Tumor Group, the Ontario Health (Cancer Care Ontario) (OH [CCO]) Gynecologic Cancer Drug Advisory Committee, and the Society of Gynecologic Oncology of Canada (GOC)
a review of testing procedure considerations for FR-alpha expression in patients with platinum-resistant high-grade epithelial ovarian, primary peritoneal, or fallopian tube cancer
a review of the pharmacoeconomic model and report submitted by the sponsor.
Perspectives of Patients, Clinicians, and Drug Programs
The information in this section is a summary of input provided by the patient and clinician groups that responded to our call for input and from the clinical experts consulted by for the purpose of this review.
Patient Input
Input for this review was submitted by 1 patient group, OCC. Information for this input was gathered through 10 patient and/or caregiver interviews between December 16 and February 28, 2025, and an online survey in Canada available from February 12 to March 3, 2025. The survey received 41 respondents, 34 of whom were patients living with PROC and 7 of whom were caregivers. Most survey respondents had epithelial ovarian cancer (59%), were diagnosed between 2022 to 2024 (52%) at stage III or IV (95%), and had experienced a recurrence (91%). A total of 3 interviewees (1 patient and 2 caregivers) had experience with mirvetuximab soravtansine.
Patients highlighted that ovarian cancer led to a severe impact on their self-esteem, ability to participate in physical activity, sleep, and ability to care for themselves and their family. Patients noted that their ovarian cancer has resulted in an inability to plan their future, particularly due to the known poor prognosis with recurrence or platinum resistance. Patients also noted a profound fear of leaving their families and children. Regarding prior treatment, 24% strongly disagreed and 43% neither agreed nor disagreed that prior treatments were able to manage their cancer. Survey respondents ranked prolonged survival (70%), improved QoL (65%), and lengthening time to recurrence (70%) as the most important outcomes for a new treatment. Eighty percent of patients indicated that they have considered taking mirvetuximab soravtansine. According to patients, they are willing to tolerate hair loss (100%), fatigue (95%), aching joints (79%), neuropathy (70%), and eye problems (55%) if mirvetuximab soravtansine prolongs their life and improves daily functioning. Most respondents (71%) felt that the benefits of mirvetuximab soravtansine outweigh the risks.
OCC interviewed 1 patient (69 years of age) diagnosed with stage III ovarian cancer in 2021 who was treated with mirvetuximab soravtansine in the US between 2023 and 2024, and a caregiver whose mother was diagnosed in 2022 at age 77 with PROC who also received treatment in the US between June and August 2024. Both patients paid out of pocket for the treatment, with 1 patient requiring a reverse mortgage on their home to pay the approximate $108,000 in treatment costs, excluding travel, to receive only 3 treatments.
A total of 2 patients and 2 caregivers had experience with FR-alpha expression testing to confirm treatment with mirvetuximab soravtansine. The patient group highlighted that to access FR-alpha expression testing in the US from Canada, patients need time, funds, ability to travel, and understanding of how to access tests in the US, often having to obtain this information through self-advocacy and self-education. Patients reported that the FR-alpha expression testing process is costly, lengthy, confusing, and stressful, though they reported no side effects from the testing. FR-alpha expression testing in the US was an additional out-of-pocket cost (US$4,500) and there were delays associated with receiving testing results and starting treatment.
Clinician Input
Input From Clinical Experts Consulted for This Review
Two clinical experts with experience in the diagnosis and medical management of patients with platinum-resistant epithelial ovarian, fallopian tube, or primary peritoneal cancer were consulted by CDA-AMC for the review of mirvetuximab soravtansine.
These clinical experts noted that most patients have disease that becomes resistant to platinum-based therapy during their disease course and that PROC is characterized by high recurrence rates. The clinical experts noted that the treatment goals for PROC are to control disease, delay progression, manage symptoms, and maintain HRQoL. Current standard nonplatinum chemotherapies, including paclitaxel, PLD, topotecan, and gemcitabine, with or without bevacizumab, offer limited clinical benefit, with low response rates (approximately 10% to 15%) and short expected duration of response (less than 6 months). As a result, new therapies that can meaningfully improve response rates, prolong survival, and address the underlying disease process while maintaining HRQoL are urgently needed.
The clinical experts noted that compared to current available therapies, which primarily offer symptom control, mirvetuximab soravtansine selectively targets tumour cells with high FR-alpha expression and delivers a cytotoxic payload, offering a more targeted and disease-specific treatment. According to the clinical experts, mirvetuximab soravtansine should be considered as a first-line therapy on confirmation of platinum resistance in eligible patients, given its response rate and survival benefit over existing available therapies. Both clinical experts noted that mirvetuximab soravtansine should be used as a single drug in patients with recurrent progressive PROC with high FR-alpha expression, which should be assessed using the VENTANA FOLR1 (FOLR1 to 2.1) RxDx Assay (an IHC-based assay that identifies the signal intensity and percentage of stained tumour cells, which corresponds to a FR-alpha expression signal). Patients with low or absent FR-alpha expression or those unable to tolerate antibody-drug conjugate-related toxicities are less suitable candidates for treatment. According to the clinical experts, the diagnostic infrastructure for FR-alpha expression is not yet standardized across Canada.
The clinical experts indicated that response to treatment should be assessed through a combination of symptom improvement, clinical and radiologic assessments, and standard outcome measures, including PFS, OS, and HRQoL. Treatment should be discontinued on evidence of disease progression or the development of significant unresolved AEs, particularly interstitial lung disease. The clinical experts emphasized that mirvetuximab soravtansine should be administered in specialized oncology settings with multidisciplinary support, including optometry, ophthalmology, and pulmonology specialists, to monitor and manage drug-specific toxicities and ensure appropriate patient counselling and follow-up.
Clinician Group Input
Input for this review was submitted by 3 clinician groups, the BC Cancer Gynecologic Oncology Provincial Tumor Group, the OH (CCO) Gynecologic Cancer Drug Advisory Committee, and the GOC. Information was gathered through published literature and clinical experience of physicians who treat patients with platinum-resistant cancer.
The groups noted that while initial ovarian cancer treatment is typically effective with high response rates, most patients will subsequently experience relapse and develop recurrent disease, which is associated with poor prognosis (survival is between 6 to 12 months). The clinician groups highlighted that patient diagnosed with PROC represents a group of patients with a high unmet need. According to the clinician groups, treatment goals consist of improving PFS, delaying disease progression, and improving QoL by reducing symptom severity. The clinician groups noted the main treatment option is bevacizumab in combination with a nonplatinum-based chemotherapy, which has shown minimal impact on OS historically, has response rates lower than 30%, is associated with significant toxicity, and disease rapidly becomes refractory to subsequent lines of treatment, which is in agreement with the input from the clinical experts consulted for the review.
The clinician group expressed differing opinions on the place in therapy for mirvetuximab soravtansine. The OH (CCO) Gynecologic Cancer Drug Advisory Committee indicated it would provide an additional treatment option for patients whose disease has not responded to existing platinum-resistant options; GOC indicated that mirvetuximab soravtansine would fit into the current treatment paradigm as an additional treatment option for platinum-resistant recurrent high-grade serous ovarian cancer, and BC Cancer noted it would be used as the first treatment option in the setting of platinum resistance with high FR-alpha expression, replacing nonplatinum-based chemotherapy with or without bevacizumab. GOC noted that mirvetuximab soravtansine would be used as monotherapy and not combined with other treatments.
The clinician groups agreed that the patients best suited for mirvetuximab soravtansine are those with platinum-resistant, high-grade serous epithelial ovarian cancer with a high FR-alpha expression (≥ 75% of cells with ≥ 2 staining intensity). In clinical practice, the clinician groups noted that PFS and OS are the standard measures for assessing treatment response. The clinician groups agreed that a clinically meaningful treatment response would be defined as radiographic disease control (i.e., tumour response or stabilization on CT and/or MRI) with improvement in cancer-related symptom burden and tolerable toxicity. Treatment discontinuation should be considered in the event of disease progression, intolerable AEs, or patient choice. Patients should receive treatment in an outpatient setting under the supervision (or guidance, in remote areas) of an oncologist. The clinician groups noted that access to optometry or ophthalmology will be necessary to ensure ocular toxicities are appropriately assessed and managed.
Drug Program Input
Input was obtained from the drug programs that participate in our reimbursement review process. Please refer to Table 2 for further information. The following were identified as key factors that could potentially impact the implementation of mirvetuximab soravtansine:
relevant comparators
considerations for initiation of therapy
considerations for continuation or renewal of therapy
considerations for prescribing of therapy
generalizability
funding algorithm
care provision issues
system and economic issues.
The clinical experts consulted for the review provided advice on the potential implementation issues raised by the drug programs.
Table 2: Responses to Questions From the Drug Programs
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
Currently funded treatment options for PROC include single-drug nonplatinum chemotherapy (i.e., paclitaxel, pegylated liposomal doxorubicin, topotecan, gemcitabine, docetaxel) with or without bevacizumab. Is there evidence comparing mirvetuximab soravtansine to chemotherapy plus bevacizumab? | The 2 clinical experts consulted for this review indicated that they are not aware of any head-to-head trials comparing mirvetuximab soravtansine and chemotherapy plus bevacizumab. pERC agreed with the clinical experts. |
Considerations for initiation of therapy | |
The trial defined platinum resistance as patients who had received 1 line of platinum-based therapy, at least 4 cycles of an initial platinum-containing regimen, had disease response, and had disease progression between 3 and 6 months after their last dose. Is this appropriate for clinical practice in Canada? | The 2 clinical experts indicated that the definition of patients with platinum-resistant disease in the MIRASOL trial is consistent with the definition used in clinical practice in Canada. They further noted that fewer cycles (3) of an initial platinum-containing regimen avert toxicities and facilitates transition to second-line treatment. The experts also indicated that the concept of platinum resistance is evolving in current clinical practice. Although patients with platinum-refractory disease are often excluded from clinical trials due their poor prognosis, the experts indicated that a patient receiving first-line therapy for advanced ovarian cancer that is platinum refractory would be treated with a nonplatinum-based therapy, such as mirvetuximab soravtansine. pERC agreed with the clinical experts. |
Should patients who have received more than 3 prior lines of therapy be eligible? | The clinical experts agreed that patients who have received more than 3 prior lines of therapy should be eligible; however, there are very few patients who have received more than 3 prior lines of nonplatinum-based therapy. The clinical experts also noted that patients currently receiving fourth-line treatment who are considering switching to mirvetuximab soravtansine should have high FR-alpha expression and no other significant comorbidities. pERC agreed with the clinical experts, noting that it is anticipated that this scenario will only apply to a few patients when mirvetuximab soravtansine is available. As such, patients who have received more than 3 prior lines of therapy may be considered eligible for treatment on a time-limited basis. |
Should patients who cycle through multiple platinum-sensitive regimens (exceeds 3 prior lines) and then have disease that becomes platinum resistant be considered? | The 2 clinical experts noted that patients who become platinum resistant after more than 3 prior lines of therapy should be eligible if platinum rechallenging is no longer available. pERC agreed with the clinical experts. |
Considerations for continuation or renewal of therapy | |
Mirvetuximab soravtansine is dosed at 6 mg/kg of adjusted ideal body weight and requires a titration for the first infusion. There is potential for drug wastage given the vial size. | This is a comment from the drug plans to inform the expert committee deliberations. |
Considerations for prescribing of therapy | |
Consider alignment with the prescribing criteria for bevacizumab in PROC. | This is a comment from the drug plans to inform the expert committee deliberations. |
Should ophthalmic exams be necessary for consideration and, if so, when should ophthalmic exams take place and should they be conducted by an ophthalmologist or would an optometrist suffice? | The 2 clinical experts noted that frequent assessments by either an optometrist or ophthalmologist may be necessary. The clinical experts indicated that the protocol should include an ophthalmic examination at screening, and regular monitoring is required during the treatment period and at follow-up. pERC agreed that regular monitoring for ocular toxicities should be required. pERC also noted the potential for challenges associated with managing the significant ocular toxicities outside of the clinical trial setting, particularly in smaller or remote or rural care settings. |
Generalizability | |
Should patients with an ECOG PS > 1 be considered eligible? | The 2 clinical experts noted that patients with an ECOG PS of 1 and 2 should be eligible for mirvetuximab soravtansine treatment. pERC agreed with the clinical experts. |
Should patients with more than grade 1 peripheral neuropathy per CTCAE be eligible for mirvetuximab soravtansine? | The clinical experts indicated that patients with more than grade 1 peripheral neuropathy should be eligible. They further noted that this will require close monitoring for worsening symptoms and should be at the discretion of the treating physician. pERC agreed with the clinical experts. |
Should patients with endometrioid, clear cell, mucinous, or sarcomatous histology; mixed tumours containing any of the previously noted histologies; or a low-grade or borderline ovarian tumour be eligible for mirvetuximab soravtansine? | The clinical experts noted that a patient’s eligibility for mirvetuximab soravtansine treatment should be based on their FR-alpha expression status. The experts also noted that PROC with serous histology is most common, resulting in a lack of data for other histology types; however, patients with other histology types should still be considered for treatment. pERC agreed with the clinical experts and noted concerns with the status of testing and nationwide access to FR-alpha expression testing. |
Funding algorithm | |
The sponsor assumes that mirvetuximab soravtansine will displace another treatment option in the algorithm. What available evidence supports sequencing between mirvetuximab soravtansine and other drugs (e.g., bevacizumab-based regimens)? | The 2 clinical experts suggested that treatment with mirvetuximab soravtansine should be prioritized before other bevacizumab-based regimens once platinum resistance is diagnosed. pERC agreed with the clinical experts and noted that earlier treatment should be recommended in patients with eligible FR-alpha expression. |
Care provision issues | |
Prophylactic glucocorticoid eye drops are needed for 9 days each cycle. Preservative-free lubricating eye drops are recommended daily. Ocular testing and therapies may represent an out-of-pocket expense for some patients. | This is a comment from the drug plans to inform the expert committee deliberations. |
FR-alpha testing is not currently standard in Canada, representing additional costs to the health care system. AbbVie recommends reflex testing to reduce burden on laboratories. | This is a comment from the drug plans to inform the expert committee deliberations. |
System and economic issues | |
Confidential pricing exists for bevacizumab and there are several marketed biosimilars. | This is a comment from the drug plans to inform the expert committee deliberations. |
CTCAE = common terminology criteria for adverse events; ECOG PS = Eastern Cooperative Oncology Group Performance Status; pERC = pan-Canadian Oncology Drug Review Expert Review Committee; PROC = platinum-resistant ovarian cancer.
Clinical Evidence
Systematic Review
Description of Studies
One randomized controlled trial (RCT), MIRASOL, was included in the sponsor’s systematic review. Between February 2020 and July 25, 2022, 453 patients (including patients from 8 sites in Canada) were enrolled in the MIRASOL trial. Patients were randomized at a ratio of 1:1 to receive single-drug mirvetuximab soravtansine (3-week regimen) (N = 227) or IC chemotherapy (n = 226) (i.e., paclitaxel, PLD, topotecan for both 3- and 4-week regimens). Randomization was stratified by the number of previous lines of therapy (1, 2, or 3) and chemotherapy drug. Female patients aged 18 years or older with platinum-resistant disease and a confirmed diagnosis of high-grade serous epithelial ovarian, primary peritoneal, or fallopian tube cancer were included. Key inclusion criteria were patients whose disease had progressed radiographically on or after their most recent line of therapy, were positive for FR-alpha expression as defined by the VENTANA FOLR1 (FOLR1 to 2.1) RxDx Assay, had received at least 1 but no more than 3 prior systemic lines of anticancer therapy, and for whom single-drug therapy was appropriate as the next line of treatment. All patients randomized to mirvetuximab soravtansine received it at 6 mg/kg of adjusted ideal body weight administered by IV every 3 weeks. For patients randomized to IC chemotherapy, the choice of chemotherapy (i.e., paclitaxel, PLD, or topotecan) was made before randomization and body weight from baseline was used to calculate body surface area to determine the required dose. No dose modifications were anticipated unless the patient’s body weight changed by more or less than 10% from baseline. Patients continued to receive mirvetuximab soravtansine or the matching IC chemotherapy until disease progression, unacceptable toxicity, withdrawal of consent, death, or the sponsor terminated the study (whichever came first).
The primary objective of the MIRASOL trial was to compare PFS of patients randomized to mirvetuximab soravtansine versus IC chemotherapy. Key secondary objectives were to compare ORR, OS, and HRQoL assessed with EORTC QLQ-OV28 between patients randomized to mirvetuximab soravtansine versus IC chemotherapy. Safety outcomes were AEs, serious AEs, AEs of special interest, and deaths. Notable harms included peripheral neuropathy, fatigue, keratopathy, vision blurred, and pneumonitis.
Demographic and baseline characteristics were similar between the mirvetuximab soravtansine and IC chemotherapy groups. The median age was 64.0 years in the mirvetuximab soravtansine group and 62.0 years in the IC group. In the mirvetuximab soravtansine group, 80% had epithelial ovarian cancer, 12% had fallopian tube cancer, and 7% had primary peritoneal cancer, and 13% had BRCA-positive disease(87% had negative or unknown BRCA status); in the IC group, these proportions were 81% for epithelial ovarian cancer, 10% for fallopian tube cancer, 9% for primary peritoneal cancer, and 16% had BRCA-positive disease. High-grade serous histology that was poorly differentiated (██%) or unknown (██% versus ██%) was common in both groups, and the most frequent stages at diagnosis were stage IIIC (53%) and stage IV (29%), with a median time since diagnosis of █████ months.
The primary data cut-off (DCO) was March 6, 2023, and the latest DCO was September 26, 2024.
Efficacy Results
The efficacy outcomes in the MIRASOL trial were PFS, OS, ORR, duration of response (DOR), and EORTC QLQ-OV28 score. Results are reported at the latest DCO unless otherwise stated.
Progression-Free Survival
The primary end point was PFS per investigator assessment. PFS was assessed based on radiological imaging and determined by Response Evaluation Criteria in Solid Tumor version 1.1 (RECIST 1.1) CT or MRI scans and tested using a stratified log-rank test. The PFS curve was estimated using the Kaplan-Meier estimator. The DCO for primary PFS analysis was March 6, 2023, by which time 330 events had occurred. The median PFS was 5.62 months (95% CI, 4.34 to 5.95) in the mirvetuximab soravtansine treatment group compared to 3.98 (95% CI, 2.86 to 4.47) in the IC chemotherapy. The HR for PFS was 0.65 (95% CI, 0.521 to 0.808; P < 0.0001). The Kaplan-Meier estimated differences in PFS probabilities between treatment groups at 6 months was ████ (95% CI, ████ ██ ████) in favour of mirvetuximab soravtansine. At 12 months, the estimated difference in PFS probability was ████ (95% CI, ████ ██ ████) in favour of mirvetuximab soravtansine. The results of the subgroup analyses of PFS by blinded independent central review (BICR) and sensitivity analysis were consistent with the overall population analyses and investigator assessment, respectively. PFS results at the latest DCO were consistent with those at the primary DCO.
Overall Survival
OS was a key secondary outcome, and its curve was estimated using the Kaplan-Meier estimator. The comparison of OS between treatment groups was performed using a Cox proportional hazard regression and log-rank test. The DCO for OS analysis was September 26, 2024, by which time 300 events had occurred. The median OS was 16.85 months (95% CI, 14.36 to 19.78) in patients randomized to mirvetuximab soravtansine and 13.34 months (95% CI, 11.37 to 15.15) in the IC chemotherapy group. The HR for OS was 0.68 (95% CI, 0.543 to 0.840; P = 0.0004). The Kaplan-Mier estimated differences in OS probabilities between groups at 6 months was ████ (95% CI, █████ ██ ████) in favour of mirvetuximab soravtansine. At 18 months, the estimated difference in OS probability was ████ (95% CI, ████ ██ ████) in favour of mirvetuximab soravtansine. The subgroup analyses showed a trend for benefit of mirvetuximab soravtansine in OS over IC chemotherapy at both DCOs and were consistent with the primary analyses.
Objective Response Rate
ORR refers to the proportion of patients who experienced a pre-established reduction in tumour volume as complete response or partial response. ORR was a key secondary outcome and was assessed by RECIST 1.1. At the latest DCO, a higher percentage of patients treated with mirvetuximab soravtansine achieved an ORR compared to those treated with IC chemotherapy (████% versus ████%) with a between-group difference of ████% (95% CI, ████ ██ ████) in favour of mirvetuximab soravtansine. The results of the subgroup analyses were consistent with the overall primary analysis at both DCOs. ORR per BICR was evaluated as a sensitivity analysis and the results were consistent with the investigator assessment.
Duration of Response
DOR was a secondary outcome and was only defined for patients who had a confirmed best overall response of complete response or partial response. The comparison of DOR between treatment group was conducted using a Cox proportional hazard regression and log-rank test. At the latest DCO, among the ██ patients with DOR events in the mirvetuximab soravtansine group, the median DOR was ████ months (95% CI, ████ ██ ████) compared to ████ months (95% CI, ████ ██ ████) in the ██ patients with DOR events in the IC chemotherapy group, with a between-group difference of ███ months (95% CI, ████ ██ ████) in favour of mirvetuximab soravtansine. In a sensitivity analysis, the median DOR by BICR was ████ months (95% CI, ████ ██ ████ months) in the mirvetuximab soravtansine group compared to ████ (95% CI, ████ ██ █████) in the IC chemotherapy group. No subgroup analyses were performed.
Health-Related Quality of Life
HRQoL was assessed using the EORTC QLQ-OV28 abdominal/gastrointestinal subscale at week 8 to 9, which was a key secondary end point. EORTC QLQ-OV28 abdominal/gastrointestinal subscale items are scored 1 (“not at all”) to 4 (“very much”). Scores are derived via linearly transforming scale and/or standalone item raw scores to a 0 to 100 scale. Higher scores indicate worse symptoms. At the primary DCO, 21% of patients in the mirvetuximab soravtansine group compared to 15% in the IC chemotherapy group achieved a 15-point symptom subscale decrease on the QLQ-OV28 abdominal/gastrointestinal subscale at the week 8 to 9 assessment, with a between-group difference of 5.65 (95% CI, –2.86 to 14.17) in favour of mirvetuximab soravtansine. In the change from baseline analysis, the difference in change from baseline in EORTC QLQ-OV28 abdominal/ gastrointestinal subscale at week 8 to 9 was –5.0 in favour of mirvetuximab soravtansine. These results were consistent with those at the primary DCO.
Harms Results
A total of 210 patients (96%) treated with mirvetuximab soravtansine reported TEAEs of any grade compared to 94% of patients in the IC chemotherapy group. Patients treated with mirvetuximab soravtansine reported fewer serious AEs compared to those treated with IC chemotherapy (24% versus 33%). Compared to 16% of patients in the IC chemotherapy, 9% of patients treated with mirvetuximab soravtansine reported TEAEs leading to treatment discontinuation. Among patients treated with mirvetuximab soravtansine, ██ patients (████%) died compared to ███ patients (████%) in the IC chemotherapy group. In both treatment groups, the primary cause of death was disease progression: ███% in the mirvetuximab soravtansine group versus ███% in the IC chemotherapy group.
Notable harms included peripheral neuropathy, fatigue, keratopathy, vision blurred, and pneumonitis. The proportion of patients with grade 3 peripheral neuropathy was similar in both treatment groups. Five patients in the mirvetuximab soravtansine group reported grade 3 fatigue compared to 11 patients in the IC chemotherapy group. Compared to IC chemotherapy, a greater proportion of patients treated with mirvetuximab soravtansine reported ocular TEAEs, including keratopathy (32% versus 0%) and blurred vision (41% versus 2%). The percentage of patients with pneumonitis was higher in the mirvetuximab soravtansine group than in the IC chemotherapy group (██% versus ██%).
Critical Appraisal
Internal Validity
The MIRASOL trial used stratified randomization of the treatment based on the following prognostic factors: prior number of previous lines of therapy (1, 2, or 3) and IC choice of chemotherapy (paclitaxel, PLD, or topotecan); however, details on allocation concealment were not provided by the sponsor. Baseline characteristics were balanced between treatment groups and all analysis were conducted in the intention-to-treat population. Missing data were low for efficacy outcomes, including PFS, OS and ORR, while ██% (n = ██) and ██% (n = ██) of patients randomized to mirvetuximab soravtansine and IC chemotherapy had missing data on HRQoL assessed via EORTC QLQ-OV28 score. The sponsor did not fully describe their imputation methods for missing data. The open-label design of the trial introduces the possibility of reporting, performance, and detection biases in efficacy estimations of subjective harms and benefits. Among tumour efficacy outcomes (i.e., PFS, ORR), bias was minimized through the use of BICR, with high concordance between investigator- and BICR-assessed end points, while OS is considered to be less susceptible to bias due to its objective nature. The trial reported that ██% of patients randomized to mirvetuximab soravtansine and ██% to IC chemotherapy received subsequent therapies. Notably, subsequent anticancer therapies were not formally balanced by protocol and were administered at the investigator’s discretion. There was no formal sensitivity analysis adjusting OS estimates for postprogression therapies in the clinical study reports submitted by the sponsor. CDA-AMC notes subsequent anticancer therapies could be a risk of bias for OS estimates if differential access or effectiveness of subsequent therapies existed between treatment groups. For the primary end point of PFS, the widths of the 95% CIs were not adjusted for multiplicity. For the key secondary end points of ORR, OS, and the abdominal/gastrointestinal symptom scale of EORTC QLQ-OV28, a hierarchical testing procedure was applied to control the family-wise type I error only if the null hypothesis for the primary end point was rejected at a 2-sided alpha level of 0.05.
External Validity
The population enrolled in the MIRASOL trial was females with platinum-resistant epithelial ovarian cancer who showed high FR-alpha expression, defined as 75% or more of tumour cells staining at 2+ or higher intensity by the VENTANA FOLR1 (FOLR1 to 2.1) RxDx Assay. According to the clinical experts consulted for this review, only 35% to 40% of patients with platinum-resistant epithelial ovarian cancer would meet this biomarker threshold, suggesting that mirvetuximab soravtansine is specific for this subgroup of females with epithelial ovarian cancer. In addition, only patients with an Eastern Cooperative Oncology Group Performance Status (ECOG PS) of 0 or 1 were eligible, whereas, in practice, a considerable proportion of patients with platinum-resistant disease have an ECOG PS of 2 or higher, particularly after multiple lines of therapy. The exclusion of patients with primary refractory disease, active ocular conditions, significant neuropathy, or serious comorbidities from the trial limits the generalizability of the efficacy results to patients with a worse prognostic status who may still be considered eligible for treatment.
The clinical experts consulted for this review indicated that the comparator arm of IC chemotherapy, consisting of weekly paclitaxel, PLD, or topotecan, reflects not only the standard of care treatment options for platinum-resistant epithelial ovarian cancer in Canada, but their dosing schedules were consistent with the Health Canada–approved labelling. Inputs from the clinical experts and the patient and clinician groups suggested that the trial measured meaningful outcomes relevant to patients, including PFS, OS, and HRQoL assessed via EORTC QLQ-OV28 score. However, a key limitation was the incomplete measurement of HRQoL — the primary HRQoL analysis was assessed at week 8 to 9 only, with limited longer-term data reported. Given that HRQoL was identified as a key outcome by the patient and clinician input, the lack of long-term assessment of these data is a notable limitation.
GRADE Summary of Findings and Certainty of the Evidence
For pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered most relevant to inform expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group.
Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null.
The reference points for the certainty of evidence assessment for PFS, OS, ORR, HRQoL assessed using EORTC QLQ-OV28 score, and notable harms, including peripheral neuropathy, fatigue, keratopathy, blurred vision, and pneumonitis, were set according to the presence or absence of an important effect based on thresholds informed by the clinical experts consulted for this review. The reference point for the certainty of the evidence assessment for EORTC QLQ-OV28 abdominal/gastrointestinal symptom subscale score was set according to the 15-point symptom subscale decrease threshold that was informed by the literature.
Results of GRADE Assessments
Table 3 presents the GRADE summary of findings for mirvetuximab soravtansine from the MIRASOL trial, for the treatment of adults with FR-alpha–positive, platinum-resistant epithelial ovarian, fallopian tube, or primary peritoneal cancer who have received 1 to 3 prior systemic treatment regimens.
The selection of outcomes for GRADE assessment was based on the sponsor’s Summary of Clinical Evidence, consultation with the clinical experts, and the input received from patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with the expert committee members:
PFS
OS
ORR
HRQoL measured using the EORTC QLQ-OV28
notable harms (peripheral neuropathy, fatigue, keratopathy, vision blurred, and pneumonitis).
Table 3: Summary of Findings for Mirvetuximab Soravtansine vs. IC Chemotherapy for Patients With FR-Alpha–Positive, Platinum-Resistant Epithelial Ovarian, Fallopian Tube, or Primary Peritoneal Cancer (DCO: September 26, 2024)
Outcome and follow-up | Patients (studies), N | Relative effect (95% CI) | Absolute effects (95% CI) | Certaintya | What happens | ||
|---|---|---|---|---|---|---|---|
IC chemotherapy | Mirvetuximab soravtansine | Difference | |||||
PFS by investigator | |||||||
Probability of PFS at 6 months Median follow-up: █████ months | 453 (1 RCT) | NR | ██████ | ██████ | ██████ | Moderateb | Mirvetuximab soravtansine likely results in an increase in PFS compared to IC chemotherapy at 6 months. |
Probability of PFS at 12 months Median follow-up: █████ months | 453 (1 RCT) | NR | ██████ | ██████ | ██████ | Highc | Mirvetuximab soravtansine results in an increase in PFS compared to IC chemotherapy at 12 months. |
OS | |||||||
Probability of OS at 6 months Median (range) follow-up: █████ months | 453 (1 RCT) | NR | ██████ | ██████ | ██████ | Lowd | Mirvetuximab soravtansine may result in little to no difference in OS compared to IC chemotherapy at 6 months. |
Probability of OS at 18 months Median follow-up: █████ months | 453 (1 RCT) | NR | ██████ | ██████ | ██████ | Moderatee | Mirvetuximab soravtansine likely results in an increase in OS compared to IC chemotherapy at 18 months. |
ORR per investigator | |||||||
ORR (by investigator) Median follow-up: █████ months | 453 (1 RCT) | NR | ██████ | ██████ | ██████ | Highf | Mirvetuximab soravtansine results in a higher proportion of patients experiencing overall response compared to IC chemotherapy. |
HRQoL | |||||||
N (%) of patients achieving at least a 15% (≥ 15 points) absolute improvement on the EORTC QLQ-OV28 abdominal/gastrointestinal symptom subscale score Time point: 8 to 9 weeks | 312 (1 RCT) | NR | 23 (15.3) | 34 (21.0) | 5.65 (–2.86 to 14.17) | Lowg | Mirvetuximab soravtansine may result in little to no difference in HRQoL compared to IC chemotherapy. |
Harms | |||||||
Patients with peripheral neuropathy Follow-up: NR Time point: end of trial | 425 (1 RCT) | ██████ | ██████ | ██████ | ██████ | Lowh | Mirvetuximab soravtansine may result in little to no difference in peripheral neuropathy compared to IC chemotherapy. |
Patients with fatigue Follow-up: NR Time point: end of trial | 425 (1 RCT) | ██████ | ██████ | ██████ | ██████ | Lowi | Mirvetuximab soravtansine may result in little to no difference in fatigue compared to IC chemotherapy. |
Patients with keratopathy Follow-up: NR Time point: end of trial | 425 (1 RCT) | ██████ | ██████ | ██████ | ██████ | Moderatej | Mirvetuximab soravtansine likely results in an increase in keratopathy compared to IC chemotherapy. |
Patients with blurred vision Follow-up: NR Time point: end of trial | 425 (1 RCT) | ██████ | ██████ | ██████ | ██████ | Highj | Mirvetuximab soravtansine results in an increase in blurred vision compared to IC chemotherapy. |
Patients with pneumonitis Follow-up: NR Time point: end of trial | 425 (1 RCT) | ██████ | ██████ | ██████ | ██████ | Moderatek | Mirvetuximab soravtansine likely results in an increase in pneumonitis compared to IC chemotherapy. |
CI = confidence interval; DCO = data cut-off; EORTC QLQ-OV28 = European Organisation for Research and Treatment of Cancer Core Quality of Life Questionnaire ovarian cancer module; HRQoL = health-related quality of life; IC = investigator’s choice; MID = minimal important difference; NR = not reported; OS = overall survival; ORR = overall response rate; PFS = progression-free survival; RCT = randomized controlled trial; vs. = versus.
aStudy limitation (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the following footnotes.
bRated down 1 level for imprecision. In the absence of a validated MID, the threshold was informed by the clinical experts consulted for this review. A between-group absolute risk difference of 5% (50 fewer or more events per 1,000 patients) at 6 and 12 months was considered clinically significant by the clinical experts. The 95% CI included both a meaningful benefit and the possibility of no effect.
cIn the absence of a validated MID, the threshold was informed by the clinical experts consulted for this review. A between-group absolute risk difference of 5% (50 fewer or more events per 1,000 patients) at 6 and 12 months was considered clinically significant by the clinical experts. The point estimate and entire CI exceeded the threshold.
dRated down 2 levels for serious imprecision. In the absence of a validated MID, the threshold was informed by the clinical experts consulted for this review. A between-group absolute risk difference of 5% (50 fewer or more events per 1,000 patients) at 6 months was considered clinically significant by the clinical experts. The 95% CI included both a meaningful benefit and the possibility of no effect.
eRated down 1 level for imprecision. In the absence of a validated MID, the threshold was informed by the clinical experts consulted for this review. A between-group absolute risk difference of 5% (50 fewer or more events per 1,000 patients) at 18 months was considered clinically significant by the clinical experts
fIn the absence of a validated MID, the threshold was informed by the clinical experts consulted for this review. A between-group absolute risk difference of 10% (100 fewer or more events per 1,000 patients) was considered clinically significant by the clinical experts. The point estimate and entire CI exceeded the threshold.
gRated down 2 levels for serious imprecision. In the absence of a validated MID, the threshold was informed by the clinical experts consulted for this review. A between-group absolute risk difference of 10% (100 fewer or more events per 1,000 patients) was considered clinically significant by the clinical experts. The 95% CI included both a meaningful benefit and the possibility of no effect.
hRated down 2 levels for serious imprecision. In the absence of a validated MID, the threshold was informed by the clinical experts consulted for this review. A between-group absolute risk difference of 10% (100 fewer or more events per 1,000 patients) was considered clinically significant by the clinical experts. The point estimate did not exceed this threshold and the CI included the possibility of no effect, introducing uncertainty.
iRated down 2 levels for serious imprecision. In the absence of a validated MID, the threshold was informed by the clinical experts consulted for this review. A between-group absolute risk difference of 10% (100 fewer or more events per 1,000 patients) was considered clinically significant by the clinical experts. The point estimate was below this threshold, and the CI included both a meaningful increase and no effect, introducing uncertainty.
jIn the absence of a validated MID, the threshold was informed by the clinical experts consulted for this review. The point estimate and CI exceeded the 10% (100 per 1,000 patients) threshold for clinical significance.
kRated down 1 level for imprecision. In the absence of a validated MID, the threshold was informed by the clinical experts consulted for this review. A between-group absolute risk difference of 10% (100 fewer or more events per 1,000 patients) was considered clinically significant by the clinical experts. Although the point estimate and upper CI exceed this threshold, the lower CI is slightly below this threshold, introducing uncertainty.
Sources: MIRASOL Clinical Study Report. Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Long-Term Extension Studies
There were no relevant long-term extension studies submitted for this review.
Indirect Comparisons
In the absence of direct head-to-head trials evaluating the efficacy of mirvetuximab soravtansine compared to bevacizumab and chemotherapy in adults with PROC who have received 1 to 3 prior systemic treatment regimens, the sponsor conducted and submitted an ITC. The objective of this section is to summarize and critically appraise the sponsor-submitted ITC, and to inform the pharmacoeconomic model.
Description of Studies
The sponsor conducted a matching-adjusted indirect comparison (MAIC) using individual-level patient data from the MIRASOL study and published aggregate data from the AURELIA study (bevacizumab plus IC chemotherapy). The MAIC adjusted for the following baseline characteristics: IC chemotherapy, age, number of prior lines of therapy, platinum-free interval, ECOG PS, and CA-125 levels. It was not feasible to adjust additional baseline characteristics due to lack of testing or reporting in the AURELIA study, more restrictive inclusion criteria in the MIRASOL study, or impact on sample size.
Efficacy Results
Risk of progression or death in the mirvetuximab soravtansine arm was comparable to that in the bevacizumab and chemotherapy arm both before matching (HR = ████; 95% CI, ████ ██████) and after (HR = ████; 95% CI, ████ ██ ████). Risk of death in the mirvetuximab soravtansine arm was lower than in the bevacizumab and chemotherapy arm, with an HR of ████ (95% CI, ████ ██ ████) before matching and an HR of ████ (95% CI, ████ ██ ████) after matching for mirvetuximab soravtansine versus bevacizumab and chemotherapy. For ORR, the odds ratio (OR) of mirvetuximab soravtansine versus bevacizumab and chemotherapy was ████ (95% CI, ████ ██ ████) before matching and increased to ████ (95% CI, ████ ██ ████) after matching, indicating numerically higher odds of overall response in the mirvetuximab soravtansine arm than in the bevacizumab and chemotherapy arm.
Harms Results
After matching, the OR of a grade 3 or higher TEAE in the mirvetuximab soravtansine arm versus the bevacizumab and chemotherapy arm was ████ (95% CI, ████ ██ ████), indicating numerically lower odds of a grade 3 or higher TEAE in the mirvetuximab soravtansine arm than the bevacizumab and chemotherapy arm. The OR of discontinuation due to any TEAE for mirvetuximab soravtansine versus bevacizumab and chemotherapy was ████ (95% CI, ████ ██ ████) before matching and ████ (95% CI, ████ ██ ████) after matching, indicating lower odds of discontinuation due to any TEAE in the mirvetuximab soravtansine arm than in the bevacizumab and chemotherapy arm.
Critical Appraisal
The sponsor-submitted ITC was informed by a systematic literature review. The 2 included studies, MIRASOL and AURELIA, are phase III, global, multicentre, open-label RCTs among adults with epithelial ovarian, fallopian tube, or primary peritoneal cancer, focusing on adults with platinum-resistant disease, and with similar median follow-up durations, conducted 10 years apart. The sponsor conducted a methodologically sound MAIC and reported on key aspects, including patient population matching, outcome definitions, statistical methods, and assessment of heterogeneity. Some key patient characteristics could feasibly be adjusted in the MAIC, including the number of prior lines of therapy, platinum-free interval, choice of preassigned chemotherapy, and ECOG PS. Other key characteristics identified by the clinical experts consulted for this review, such as tumour histology, FR-alpha expression status, prior PARP use, and prior use of bevacizumab, could not be adjusted due to lack of testing or reporting in the AURELIA study, more restrictive inclusion criteria in the MIRASOL study, or impact on sample size.
The sponsor and the clinical experts have noted that the treatment landscape of ovarian cancer has evolved in the time between both trials, so all patients were naive to PARP inhibitors and had prior antiangiogenic therapy at baseline in the AURELIA trial. The clinical experts noted that the serous histology subtype, which was a required subtype for inclusion in the MIRASOL trial but not the AURELIA trial, generally has a better prognosis than other subtypes. The sponsor and clinical expert noted that FR-alpha expression has been shown to be an effect modifier for both mirvetuximab soravtansine and pooled chemotherapy; FR-alpha expression status was not reported in the AURELIA study. As a result of this heterogeneity between patient populations in both trials, the clinical experts concluded that no meaningful cross-trial comparison could be made. In addition, due to poor overlap in baseline characteristics between trials, the small effective sample size after matching (██ for the mirvetuximab soravtansine arm and ██ for the chemotherapy arm in the MIRASOL trial) limited the power to detect statistically significant differences in treatment effects and constrained the ability to adjust for additional baseline characteristics.
ITC results were presented for OS, PFS, and ORR, as well as harms outcomes; other outcomes of relevance to patients (e.g., HRQoL) were not reported.
Studies Addressing Gaps in the Evidence From the Systematic Review
No relevant studies addressing gaps in the evidence from the systematic review were submitted by the sponsor.
Testing Procedure Assessment
FR-alpha expression is determined using the VENTANA FLOR1 (FOLR1 to 2.1) RxDx Assay. To facilitate the implementation of FR-alpha testing, the sponsor and the manufacturer of the VENTANA FOLR1 (FOLR1 to 2.1) RxDx Assay have identified key laboratory sites throughout Canada where the assay could be performed to enable equitable access to patients. However, it is yet to be determined if this approach would ensure equitable access to testing for patients living in other jurisdictions regardless of the intended implementation strategy.
Key considerations and relevant information available from materials submitted by the sponsor, input from the clinical experts and patient groups, and sources from the literature were validated by the review team when possible and are summarized in Table 4.
Table 4: Considerations for FR-Alpha Expression Testing for Establishing Treatment Eligibility for Mirvetuximab Soravtansine
Consideration | Criterion | Available Information |
|---|---|---|
Health system–related | Number of individuals in Canada expected to require the test (e.g., per year) | The clinical experts noted that ideally all patients with epithelial ovarian cancer would require testing. Accordingly, the number of individuals in Canada expected to require FR-alpha expression testing would be best represented by the total number of patients with epithelial ovarian cancer, which is estimated to be between 2,137 and 2,146 patients per year over the next 3 years based on the sponsor-submitted information. |
Availability and reimbursement status of the testing procedure in jurisdictions across Canada | FR-alpha expression testing is not currently available or funded in Canada. | |
Testing procedure as part of routine care | FR-alpha expression testing is not performed as part of routine care for patients with epithelial ovarian cancer in Canada. | |
Repeat testing requirements | FR-alpha is a stable biomarker, and as such, repeat testing is not necessary at the time of platinum resistance development. | |
Impacts on human and other health care resources by provision of the testing procedure | The clinical experts noted that implementation of FR-alpha expression testing would likely lead to an expected increase in human resource and laboratory costs due to upscaling the capacity associated with additional IHC-based testing and appropriate laboratory equipment needs. | |
Patient-related | Accessibility of the testing procedure in jurisdictions across Canada | There is limited access to FR-alpha expression testing for patients with epithelial ovarian cancer. According to patient group input, patients can access FR-alpha expression testing by travelling to the US, but is associated with major logistical and accessibility concerns. |
Expected turnaround times for the testing procedure | The estimated turnaround time for testing using the VENTANA FOLR1 (FOLR1 to 2.1) RxDx Assay is approximately 48 to 72 hours based on US commercial laboratory information; however, the clinical experts indicated that for most testing procedures, turnaround time in Canada may be closer to 7 days once a sample is made available. | |
Burden associated with the testing procedure for patients, families, and/or care partners | According to the patient group input, patients who experienced FR-alpha expression testing noted that waiting on testing results caused anxiety. Only patients who are considered to have high FR-alpha expression would be eligible for mirvetuximab soravtansine; thus, patients with lower FR-alpha expression levels may not have the same access to treatment options. No additional burdens associated with FR-alpha expression testing are anticipated for patients, families, or care partners. | |
Clinical | Diagnostic test accuracy and clinical validity of the testing procedure | No specific measurements for test sensitivity and specificity are available because the VENTANA FOLR1 (FOLR1 to 2.1) RxDx Assay is the evidentiary standard FR-alpha expression test, and no relevant comparators have been approved. The results of the precision analysis studies showed high overall percent agreement for each measurement of intermediate precision, reader precision, and interlaboratory and interreader precision. |
Risks of harm associated with the testing procedure | FR-alpha expression testing is done using tumour tissue samples, which involves a procedure that is often invasive. However, FR-alpha expression testing can be performed on tissue samples already collected during diagnosis and any additional risk of harms associated with tissue sample collection is likely low. | |
Cost | Projected cost of the testing procedure | According to the sponsor’s budget impact analysis, the estimated cost of FR-alpha expression testing is $2,600 for 50 tests or $52 per test; however, the sponsor also reported that the total estimated cost would likely vary between $150 and $200 per test when accounting for reagents, laboratory technologists, and pathologist time. The clinical experts noted that no significant impact is anticipated in terms of cost on the health care system. |
IHC = immunohistochemistry.
Economic Evidence
Cost and Cost-Effectiveness
Mirvetuximab soravtansine is available as a 5 mg/mL single use vial (100 mg in 20 mL).1 At the submitted price of $5,495.00 per vial, the 28-day cycle cost of mirvetuximab soravtansine is expected to be $29,307 per patient, based on the Health Canada–recommended dosage. FR-alpha expression testing is required to determine FR-alpha expression status and patient eligibility for mirvetuximab soravtansine.
Clinical efficacy in the economic analysis for mirvetuximab soravtansine versus pooled chemotherapy (i.e., paclitaxel and PLD) was derived from the MIRASOL trial. Evidence submitted by the sponsor indicates that mirvetuximab soravtansine is likely to improve PFS with moderate certainty when compared with pooled chemotherapy among patients with FR-alpha–positive, platinum-resistant epithelial ovarian, fallopian tube, or primary peritoneal cancer who have received 1 to 3 prior systemic treatment regimens. Moreover, the CDA-AMC clinical review concluded that mirvetuximab soravtansine may result in little to no difference in OS compared to pooled chemotherapy at 6 months with low certainty, and likely improve OS compared to pooled chemotherapy at 18 months with moderate certainty.
Mirvetuximab soravtansine was also compared to bevacizumab plus chemotherapy, informed by the sponsor-submitted MAIC. The MAIC displayed inconsistent results (e.g., mirvetuximab soravtansine may have a shorter PFS, but higher OS, than bevacizumab plus chemotherapy). Clinical heterogeneity in the patient populations of both trials, including in key variables that could not feasibly be adjusted (i.e., tumour histology, FR-alpha expression status, prior PARP inhibitor use, and prior use of bevacizumab), were identified by CDA-AMC, resulting in the magnitude of comparative effectiveness of mirvetuximab soravtansine versus bevacizumab plus chemotherapy to be too uncertain to determine.
The results of the CDA-AMC base case suggest:
Mirvetuximab soravtansine is predicted to be associated with higher costs to the health care system than pooled chemotherapy (incremental costs = $195,857), primarily driven by increased drug acquisition costs associated with mirvetuximab soravtansine.
Mirvetuximab soravtansine is predicted to be associated with a gain of 0.43 life-years (LYs) and may result in a gain of 0.30 QALYs compared to pooled chemotherapy.
The ICER for mirvetuximab soravtansine compared to pooled chemotherapy is $642,249 per QALY gained in the CDA-AMC base case. The estimated ICER was highly sensitive to assumptions about long-term OS.
Although the CDA-AMC base case estimated a gain in LYs with mirvetuximab soravtansine compared to pooled chemotherapy (incremental LYs = 0.43), the magnitude of the survival benefit is highly uncertain and the incremental LYs for mirvetuximab soravtansine predicted in the CDA-AMC base case may be overestimated. Additional price reductions may therefore be required.
CDA-AMC estimates that the budget impact of reimbursing mirvetuximab soravtansine in the indicated population will be approximately $120 million over the first 3 years of reimbursement compared to the amount currently spent on pooled chemotherapy, with an estimated expenditure of $140 million on mirvetuximab soravtansine over this period. The actual budget impact of reimbursing mirvetuximab soravtansine will depend on the placement of FR-alpha expression test in the current practice (i.e., patients could be tested at diagnosis, after first recurrence, or after platinum resistance) and potential costs associated with additional resources needed to implement FR-alpha expression testing. With the budget impact of mirvetuximab soravtansine exceeding $40 million in year 3 of the analysis, the budget impact must be addressed to ensure the feasibility of adoption.
Figure 1: Summary of the CDA-AMC Economic Analysis and Price Reduction
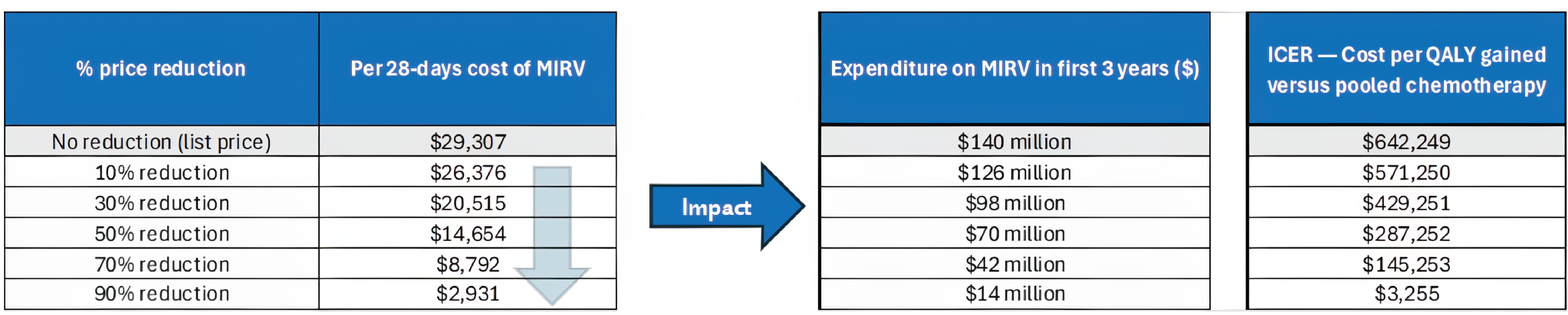
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; MIRV = mirvetuximab soravtansine; QALY = quality-adjusted life-year.
Note: Expenditure includes only the mirvetuximab soravtansine drug cost.
pERC Information
Members of the Committee
Dr. Catherine Moltzan (Chair), Dr. Kelvin Chan (Vice Chair), Dr. Phillip Blanchette, Dr. Matthew Cheung, Dr. Michael Crump, Annette Cyr, Dr. Jennifer Fishman, Dr. Jason Hart, Terry Hawrysh, Dr. Yoo-Joung Ko, Dr. Aly-Khan Lalani, Amy Peasgood, Dr. Anca Prica, Dr. Adam Raymakers, Dr. Patricia Tang, Dr. Pierre Villneuve, and Danica Wasney
Meeting date: August 13, 2025
Regrets: Four expert committee members did not attend.
Conflicts of interest: None
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@cda-amc.ca.