Drugs, Health Technologies, Health Systems
Reimbursement Review
Durvalumab (Imfinzi) and Tremelimumab (Imjudo)
Sponsor: AstraZeneca Canada Inc.
Therapeutic area: Metastatic non–small cell lung cancer (NSCLC)
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
AE
adverse event
AEPI
adverse event of potential interest
AESI
adverse event of special interest
AUC
area under the plasma drug concentration-time curve
BICR
blinded independent central review
BoR
best objective response
bTMB12
blood tumour mutational burden of 12 mutations per megabase or greater
bTMB16
blood tumour mutational burden of 16 mutations per megabase or greater
bTMB20
blood tumour mutational burden of 20 mutations per megabase or greater
CCSN
Canadian Cancer Survivor Network
CI
confidence interval
CNS
central nervous system
CR
complete response
DAC
Drug Advisory Committee
DCO
data cut-off
DSU
Decision Support Unit
DoR
duration of response
ECOG PS
Eastern Cooperative Oncology Group Performance Status
EORTC
European Organisation for Research and Treatment of Cancer
EORTC QLQ-C30
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30
EORTC QLQ-LC13
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Lung Cancer 13
ESS
effective sample size
FAS
full analysis set
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HR
hazard ratio
HRQoL
health-related quality of life
HTA
health technology assessment
ICI
immune checkpoint inhibitor
imAE
immune-mediated adverse event
ITC
indirect treatment comparison
ITT
intention to treat
IVRS
interactive voice response system
IWRS
interactive voice web system
LCC
Lung Cancer Canada
LCC-MAC
Lung Cancer Canada Medical Advisory Committee
LHF
Lung Health Foundation
MAIC
matching adjusted indirect comparison
MID
minimal important difference
MTP
multiple testing procedure
NICE
National Institute for Health and Care Excellence
NSCLC
non–small cell lung cancer
OR
odds ratio
ORR
objective response rate
OS
overall survival
PFS
progression-free survival
PR
partial response
PRO
patient-reported outcome
QoL
quality of life
RECIST 1.1
Response Evaluation Criteria in Solid Tumours Version 1.1
RCT
randomized controlled trial
SAE
serious adverse event
SAF
safety analysis set
SLR
systematic literature review
SOC
standard of care
SD
standard deviation
TC
tumour cell
TEM
treatment-effect modifier
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Durvalumab (Imfinzi), 50 mg/mL, concentrate for IV infusion (in combination with tremelimumaba (Imjudo), 20 mg/mL, concentrate for IV infusion) |
Sponsor | AstraZeneca Canada Inc. |
Indication | Durvalumab in combination with tremelimumab and platinum-based chemotherapy is indicated for the first-line treatment of adult patients with metastatic NSCLC with no sensitizing epidermal growth factor receptor mutations or anaplastic lymphoma kinase genomic tumour aberrations |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Standard review |
NOC date | April 10, 2025 |
Recommended dosage | Patients ≥ 30 kg During chemotherapy: durvalumab 1,500 mg in combination with tremelimumab 75 mg and platinum-based chemotherapy q.3.w. (21 days) for 4 cycles Post−platinum chemotherapy:
Patients < 30 kg During chemotherapy: durvalumab 20 mg/kg in combination with tremelimumab 1 mg/kg and platinum-based chemotherapy q.3.w. (21 days) for 4 cycles Post−platinum chemotherapy:
Durvalumab and tremelimumab are administered by IV infusion. |
NOC = Notice of Compliance; NSCLC = non–small cell lung cancer; q.3.w. = every 3 weeks; q.4.w. = every 4 weeks.
aTremelimumab to be used in combination with durvalumab. Tremelimumab is not intended for monotherapy use.
Sources: Sponsor’s Summary of Clinical Evidence1 and Imfinzi product monograph.2
Table 2: Recommended Regimen and Dosing for Durvalumab in Combination With Tremelimumab and Platinum-Based Chemotherapy for Treatment of Metastatic NSCLC
Tumour histology | Patient weighta | Durvalumab dose | Tremelimumab dose | Chemotherapy regimen |
|---|---|---|---|---|
Nonsquamous | ≥ 30kg | 1,500 mg | 75 mg |
|
< 30kg | 20 mg/kg | 1 mg/kg | ||
Squamous | ≥ 30kg | 1,500 mg | 75 mg |
|
< 30kg | 20 mg/kg | 1 mg/kg |
NSCLC = non–small cell lung cancer.
aPatients with a body weight of 30 kg or less must receive weight-based dosing, until the weight increases to greater than 30 kg.
Source: Imfinzi product monograph.2
Table 3: Recommended Dosing Schedule for Durvalumab in Combination With Tremelimumab and Platinum-Based Chemotherapy for the Treatment of Metastatic NSCLC
Regimen details | During chemotherapy (combination) stage 1 cycle = 3 weeks (21 days) | Postchemotherapy (maintenance) stage 1 cycle = 4 weeks (28 days) | ||||||
|---|---|---|---|---|---|---|---|---|
Weeka | ||||||||
0 | 3 | 6 | 9 | 12 | 16 | 20 | 24 | |
Cycle | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
Durvalumabb, c | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
Tremelimumabc | Yes | Yes | Yes | Yes | No | Yes | No | No |
Chemotherapyd | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
NSCLC = non–small cell lung cancer.
aThe dosing interval changes from every 3 weeks to every 4 weeks starting at cycle 5.
bContinue until disease progression or unacceptable toxicity.
cIV infusion over 60 minutes.
dFor patients with nonsquamous NSCLC treated with pemetrexed and carboplatin or cisplatin: pemetrexed maintenance therapy from week 12 until disease progression or unacceptable toxicity is based on physician discretion.
Sources: Sponsor’s Summary of Clinical Evidence1 and Imfinzi product monograph.2
Introduction
Lung cancer is the leading cause of cancer-related death in Canada; it is heterogeneous in nature, comprising several different disease subtypes categorized by histology, staging, and mutation driver status.3 Non–small cell lung cancer (NSCLC) is the most common type, representing approximately 89% of all lung cancer diagnoses in Canada.4 NSCLC can be categorized into histologic subtypes: squamous-cell carcinoma (17% to 27% of NSCLC cases) and non–squamous cell carcinoma (73% to 83% of NSCLC cases).3,5 At early stages, NSCLC may be asymptomatic,3 with symptoms developing only when the cancer has become more advanced and is no longer amenable to curative-intent therapy. Presenting symptoms can sometimes be nonspecific (common manifestations include coughing, chest pain, hemoptysis, fatigue, weight loss, dyspnea, hoarseness, and recurring infections with bronchitis and pneumonia), contributing to delays in diagnosis.3,5,6 Screening programs for NSCLC are being established throughout Canada, but many patients who would qualify for NSCLC screening live in jurisdictions where it is not yet available. Although EGFR mutations and ALK aberrations have been identified as oncogenic driver mutations,7 which collectively occur in approximately 25% of patients,8 an estimated 74.5% of NSCLC cases do not have EGFR mutations or ALK aberrations.
Nearly half (48.4%) of NSCLC cases are metastatic (stage IV).3,8 For the majority of patients with metastatic NSCLC whose tumours lack actionable genomic alterations, treatment is selected based on tumour histology (squamous or nonsquamous); PD-L1 expression, which is predictive of response to immune checkpoint inhibitors (ICIs); and patient symptom burden (as measured by Eastern Cooperative Oncology Group Performance Status [ECOG PS]), comorbidities, and preferences.9-11 In recent years, new treatment options include ICI-based therapies (either as monotherapy or in combination with chemotherapy), which are now the standard first-line treatment for metastatic NSCLC without targetable genomic alterations.10,11 For patients with any or unknown tumoural PD-L1 expression, first-line options include pembrolizumab plus platinum-based chemotherapy; nivolumab and ipilimumab plus platinum-based chemotherapy; or cemiplimab plus platinum-based chemotherapy.10,11 Monotherapy with pembrolizumab or cemiplimab may be selected as first-line treatment for patients with high PD-L1 expression (PD-L1 expression in ≥ 50% of tumour cells [TCs]).10,11 Cemiplimab (with or without platinum-based chemotherapy) is not currently funded in Canada.12 Platinum-based chemotherapy alone is also a potential first-line treatment option; however, the clinical experts consulted by Canada’s Drug Agency (CDA-AMC) noted that this would be reserved for patients with significant contraindications to immunotherapy for whom the perceived risks of ICI-based treatment outweigh any potential benefits. The clinical experts consulted for this review indicated that immuno-oncology treatment is considered the backbone of therapy for the first-line treatment of patients with metastatic NSCLC without targetable oncogenic aberrations. The goals of therapy for patients with metastatic NSCLC, as identified by the clinical experts, include prolonging survival, extending the time before disease progression, decreasing cancer-related symptoms, and maintaining or improving quality of life (QoL).
Tremelimumab in combination with durvalumab was previously reviewed by CDA-AMC for the first-line treatment of adult patients with unresectable hepatocellular carcinoma who require systemic therapy. The final recommendation issued in November 2023 was “reimburse with clinical criteria and/or conditions.”
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of durvalumab (50 mg/mL concentrate for IV infusion) in combination with tremelimumab (20 mg/mL concentrate for IV infusion) and platinum-based chemotherapy in the first-line treatment of metastatic NSCLC with no sensitizing EGFR mutations or ALK genomic tumour aberrations.
Perspectives of Patients, Clinicians, and Drug Programs
The information in this section is a summary of input provided by the patient and clinician groups who responded to our call for input and from clinical experts consulted by for the purpose of this review.
Patient Input
A joint patient group submission was received from the Canadian Cancer Survivor Network (CCSN), Lung Cancer Canada (LCC), and the Lung Health Foundation (LHF). CCSN is a national network of patients, families, survivors, friends, community partners, funders, and sponsors who have come together to promote the best standard of care (SOC), including early diagnosis, timely treatment and follow-up care, and support for patients with cancer, and regarding issues related to survivorship or quality of end-of-life care. LCC is a registered national charitable organization that serves as Canada’s leading resource for lung cancer education, patient support, research and advocacy. The LHF is a registered charity that assists and empowers people living with or caring for others with lung disease. It is a recognized leader, voice, and primary resource in the prevention and control of respiratory illness, tobacco cessation and prevention, and the effects on lung health.
CCSN, LCC, and LHF collectively produced a survey and circulated it among their networks. The survey was disseminated through the 3 organizations’ social media platforms, as well as CCSN’s monthly newsletter to gather responses from August 1, 2024, to the date of writing of the patient group submission. LCC also conducted 1 interview on October 8, 2024, with a patient who was a part of the POSEIDON trial.
Respondents from a previous survey and submission on durvalumab in combination with chemotherapy as neoadjuvant treatment, followed by durvalumab as monotherapy after surgery indicated for the treatment of patients with NSCLC with resectable tumours (≥ 4 cm and/or node-positive) and no known EGFR or ALK rearrangements shared their experiences with lung cancer and durvalumab. The following symptoms associated with lung cancer and affecting QoL were reported: fatigue; pain in chest, shoulder, back or arms; shortness of breath; infections; pneumonia and/or bronchitis; and loss of weight, appetite, hair, and teeth. Chemotherapy, immunotherapy, targeted therapy, radiation, surgical therapy, and participation in clinical trials were identified as current treatment options by respondents. The respondents emphasized that these regimens were generally well tolerated; however, they described several side effects (e.g., joint and muscle pain, fatigue, diarrhea, neuropathy, weight loss, anemia, nausea, vomiting, constipation, migraines, change in vision and hearing, and forgetfulness). One respondent reported doing well until a CT scan showed disease progression.
Fatigue was identified by the respondents as the main side effect of durvalumab. In comparison to other therapies, at least 2 respondents indicated that symptom management was much better with durvalumab; however, there was little to no difference in side effects, ease of use, and disease progression.
Respondents noted several unmet needs, such as better mental health support, availability of immunotherapy for a longer duration, access to counselling, and help with travel costs associated with therapy. The respondents highlighted the following outcomes as important: disease management, prolonged life, a cure, QoL, delayed onset of symptoms, easy-to-use medication, reduction in side effects, and access to new options for treatment.
The patient from the POSEIDON trial reported that it took a while after diagnosis before they could begin receiving treatment. The physician who treated the patient confirmed that the patient received durvalumab and tremelimumab in combination with platinum-based chemotherapy. The patient started with first-line chemotherapy and received durvalumab simultaneously. While on chemotherapy, the patient received appropriate care and dealt with minimal side effects, including mild diarrhea, bone pain, itching, and tiredness, particularly when receiving chemotherapy. While on durvalumab, the patient noted temporary side effects such as occasional diarrhea, itching, and hot flashes. The patient emphasized that they were still able to carry out regular activities during treatment.
Clinician Input
Input From Clinical Experts Consulted for This Review
The clinical experts consulted by CDA-AMC identified several goals of first-line treatment of metastatic NSCLC: improving survival, extending the time before disease progression, decreasing cancer-related symptoms, and improving QoL. The experts acknowledged that currently available first-line treatment options for metastatic NSCLC have been associated with a survival benefit compared to chemotherapy alone but noted that there remains an unmet need among patients with low or negative PD-L1 expression, including options that have lower rates of immune-related adverse events (AEs). Clinical experts recognized that there is a significant unmet need among subgroups of patients with mutations associated with a lower chance of responding to immuno-oncology therapy or with a generally poor prognosis, such as KRAS, STK11, and KEAP mutations. The clinical experts noted that durvalumab and tremelimumab plus platinum-based chemotherapy would be used as a standalone treatment for patients with previously untreated metastatic NSCLC not harbouring a targetable oncogenic aberration with an associated SOC first-line targeted therapy option (e.g., an EGFR mutation or ALK rearrangement) and that this regimen would not be expected to cause a shift in the current treatment paradigm but would be an additional first-line option combining immuno-oncology treatment and chemotherapy. According to the clinical experts, patients ideally suited for this treatment would have previously untreated metastatic NSCLC, a good ECOG PS (0 or 1), no actionable oncogenic mutations or translocations (including EGFR mutations and ALK rearrangements), any tumoural PD-L1 expression, no significant comorbidities, and the ability to report adverse effects as soon as possible. The clinical experts pointed out that, in practice, durvalumab and tremelimumab plus platinum-based chemotherapy is anticipated to be used most often in patients with PD-L1 levels of less than 50%, but they added that, because patients with any PD-L1 status were included in the POSEIDON trial, approval and funding should be in accordance with trial outcomes. The clinical experts also noted that durvalumab and tremelimumab plus platinum-based chemotherapy may be an option for patients with tumours that have biomarkers that may predict a poor or no response to currently available ICI-based therapies (e.g., those that do not express PD-L1 or have KRAS, STK11, or KEAP mutations). The clinical experts stated that the treatment strategy would be based on an informed decision by the patients in consultation with a medical oncologist, and that response to treatment would be determined through clinical and radiological assessments. Treatment and monitoring would be administered locally for patients who live close to regional cancer clinics and at community oncology networks for patients living in remote settings, where specially trained general practitioners or internists would oversee day-to-day treatment. The clinical experts acknowledged that it would be appropriate for any systemic therapy unit and lung cancer treatment team currently administering other combinations of immuno-oncology and chemotherapy to deliver treatment with durvalumab and tremelimumab plus platinum-based chemotherapy. According to the clinical experts, discontinuation of first-line treatment for metastatic NSCLC would occur when there is disease progression, unacceptable toxicity, or the patient chooses to stop treatment. One clinical expert also noted that discontinuation of treatment may occur when there is suspicion of progression and a risk of rapid clinical deterioration if treatment is continued. According to the clinical experts, early identification and management of immune-related AEs is paramount to reduce the risk of treatment-related morbidity or mortality, as is the case with any immunotherapy-containing regimen. Subspecialist consultations may be required to effectively manage immune toxicity.
Clinician Group Input
Two clinician groups, the Lung Cancer Canada Medical Advisory Committee (LCC-MAC) and the Ontario Health (Cancer Care Ontario) Lung Cancer Drug Advisory Committee (DAC) provided input for this submission. The LCC-MAC is a national charity that has been offering support and education to patients with lung cancer and their families and supporting research, and has been providing clinician input for submissions of new lung cancer drugs to the health technology assessment (HTA) process for many years. The DAC provides evidence-based clinical and health system guidance on drug-related issues to support the mandate of Cancer Care Ontario, including the Provincial Drug Reimbursement Programs and the Systemic Treatment Program.
A total of 19 clinicians from the LCC-MAC and 5 clinicians from the DAC provided input for this submission. The submission from the LCC-MAC was informed by data and information from publicly available sources, primarily published manuscripts and conference presentations, together with clinical experience of the LCC-MAC members. Information from Cancer Care Ontario was gathered via teleconference meeting and emails.
The DAC highlighted that the treatment combination of durvalumab and tremelimumab plus platinum-based chemotherapy would serve as a first-line treatment. The LCC-MAC noted that most thoracic oncologists in Canada had experience with using the treatment combination through clinical trials. It also emphasized that the treatment combination would not replace other regimens that were already approved or funded but would serve as an alternative. Chemotherapy and immunotherapy options were noted as appropriate comparators to the treatment combination by the LCC-MAC.
In terms of unmet needs, both clinician groups agreed on the need for treatments with better tolerability. The DAC noted the following additional unmet needs: not all patients respond to available treatments, patients become refractory to current treatment options, and the lack of treatments and formulations that improve compliance and that convenience.
The clinician groups highlighted treatment goals similar to those indicated by the clinical experts, in addition to tumour shrinkage. The LCC-MAC clinician group further noted that an important outcome for immunotherapy combinations is survival with the goal of improving the number of patients with durable benefits of treatment, as manifested by increases in progression-free survival (PFS) and overall survival (OS) at 5 years.
The clinician groups noted that those best suited for the treatment combination included patients without actionable driver mutations with any PD-L1 status and those with KRAS, STK11 and KEAP mutations not currently actionable in the first-line setting. The DAC also noted that patients with stage IV or incurable NSCLC considering first-line therapy would be suitable candidates for treatment.
Both groups highlighted the need for clinical assessment of symptoms and imaging (e.g., CT scans and chest X-rays) to monitor response to treatment. The DAC advised treatment response assessment every 6 weeks initially, and then less often. However, the LCC-MAC suggested that scans be made every 3 months, with symptom assessments every 3 to 4 weeks, depending on if a patient receives chemotherapy or single-drug immunotherapy during the maintenance phase.
The clinician groups agreed that disease progression is a factor when deciding whether to discontinue treatment. The LCC-MAC indicated that immunotherapy may be interrupted or discontinued because of immune-related AEs, and most low-grade toxicities were manageable with symptom management, topical or oral steroids, and potential treatment interruption. The DAC also identified intolerable side effects and patient withdrawal as factors when considering treatment discontinuation. The LCC-MAC indicated the initial patient assessment should be carried out by a medical oncologist. Both clinician groups stated that the treatment combination should be administered in facilities with expertise in managing cytotoxic anticancer therapies.
Drug Program Input
Input was obtained from the drug programs that participate in the reimbursement review process. The following were identified as key factors that could potentially affect the implementation of a recommendation for durvalumab and tremelimumab: relevant comparators; considerations for prescribing, initiation, continuation or renewal, and discontinuation of therapy; generalizability; funding algorithm; care provision; and system and economic issues. The details of the drug program input along with advice from the clinical experts consulted for this review are provided in Table 6.
Clinical Evidence
Systematic Review
Description of Studies
One phase III, randomized, open-label, global study, POSEIDON, assessed the efficacy and safety of durvalumab with or without tremelimumab for the first-line treatment of patients (total N = 1,013) with metastatic NSCLC with tumours without activating EGFR mutations and ALK fusions. Patients were randomized 1:1:1 to 1 of 3 study arms: durvalumab and tremelimumab plus SOC chemotherapy; durvalumab plus SOC chemotherapy; or SOC chemotherapy alone. Randomization was stratified according to PD-L1 expression status (TC level ≥ 50% versus < 50%), disease stage (IVA versus IVB), and histology (nonsquamous versus squamous). The primary objective of the POSEIDON trial was to assess the efficacy of durvalumab plus SOC chemotherapy compared to SOC chemotherapy alone in terms of dual primary end points of PFS and OS. The key secondary objectives were to assess the efficacy of durvalumab and tremelimumab plus SOC chemotherapy compared to SOC chemotherapy alone in terms of PFS as assessed by blinded independent central review (BICR) using Response Evaluation Criteria in Solid Tumours Version 1.1 (RECIST 1.1) and OS. Other secondary end points included objective response rate (ORR) and health-related quality of life (HRQoL), as measured by the European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 (EORTC QLQ-C30) and European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Lung Cancer 13 (EORTC QLQ-LC13), to compare durvalumab and tremelimumab plus SOC chemotherapy versus SOC chemotherapy alone. Results from this comparison (durvalumab and tremelimumab plus SOC chemotherapy versus SOC chemotherapy alone) are presented in this report, and include results from the prespecified main analyses of the POSEIDON trial, referred to as the “final analysis” (data cut-off [DCO]: July 24, 2019, for PFS and other RECIST-related end points; March 12, 2021, for OS and safety data), and a 5-year OS analysis update (DCO: August 24, 2023). The primary end points are based on the evaluation of durvalumab plus SOC chemotherapy versus SOC chemotherapy alone, which is outside the scope of the indication approved by Health Canada and the sponsor’s reimbursement request; the primary end points are therefore not the focus of this review.
Patients eligible for participation in the POSEIDON trial were aged 18 years or older with stage IV NSCLC not amenable to curative surgery or radiation with tumours lacking activating EGFR mutations and ALK fusions. Across all study arms, the median age of patients was 64 years, and the majority were male (76.0%), white (55.9%), and either current or former smokers (78%), with an ECOG PS of 1 (66.5%). The included patients had not received prior chemotherapy or any other systemic therapy for metastatic NSCLC, including prior exposure to immune-mediated therapy. Participants were required to have a WHO performance status or ECOG PS of 0 or 1. Patients with active or prior documented autoimmune or inflammatory disorders (with some exceptions) were not eligible for the trial.
Efficacy Results
As of March 12, 2021, the median OS in the durvalumab and tremelimumab plus SOC chemotherapy group was 14.0 months (95% confidence interval [CI], 11.7 to 16.1 months) compared with 11.7 months (95% CI, 10.5 to 13.1 months) in the SOC chemotherapy alone group, with a hazard ratio (HR) of 0.77 (95% CI, 0.650 to 0.916; P = 0.00304). For the OS final analysis results, the median follow-ups were 13.63 months in the durvalumab and tremelimumab plus SOC chemotherapy group and 11.17 months in the SOC chemotherapy alone group. At the 5-year OS analysis DCO, the median OS in the durvalumab and tremelimumab plus SOC chemotherapy group was 14.0 months (95% CI, 11.7 to 16.1 months) versus 11.6 months (95% CI, 10.5 to 13.1 months) in the SOC chemotherapy alone group, with an HR of 0.76 (95% CI, 0.642 to 0.893). For the 5-year OS analysis results, the median follow-up periods were 13.63 months in the durvalumab and tremelimumab plus SOC chemotherapy group and 11.10 months in the SOC chemotherapy alone group. The difference in median OS was statistically significant in favour of durvalumab and tremelimumab plus SOC chemotherapy at the final analysis time point (statistical significance could not be determined for the 5-year time point results because these estimates were not alpha-controlled). Results for longer-term survival probability also demonstrated improvement for durvalumab and tremelimumab plus SOC chemotherapy versus SOC chemotherapy alone at 36 months (25.3% versus 13.3%, respectively), 48 months (20.9% versus 8.5%), and 60 months (15.7% versus 6.8%). The clinical experts consulted by CDA-AMC considered the differences in median OS at both time points and the differences in survival probabilities at 36 months, 48 months, and 60 months to be clinically meaningful.
As of July 24, 2019, the median PFS assessed by BICR in the durvalumab and tremelimumab plus SOC chemotherapy arm was 6.2 months (95% CI, 5.0 to 6.5 months) compared with 4.8 months (95% CI, 4.6 to 5.8 months) in the SOC chemotherapy alone arm, with an HR of 0.72 (95% CI, 0.600 to 0.860; 2-sided P = 0.00031). The difference was statistically significant in favour of durvalumab and tremelimumab plus SOC chemotherapy. The PFS rate at 12 months for patients in the durvalumab and tremelimumab plus SOC chemotherapy arm was 26.6% and for patients in the SOC chemotherapy alone arm it was 13.1%. For the PFS results, the median follow-ups for censored patients were 11.9 months in the durvalumab and tremelimumab plus SOC chemotherapy group and 5.5 months in the SOC chemotherapy alone group. The clinical experts considered the differences in median PFS and in PFS rate at 12 months to be clinically meaningful.
In terms of other secondary end points, the ORR for durvalumab and tremelimumab plus SOC chemotherapy was 46.3% compared with 33.4% for SOC chemotherapy alone, with an odds ratio (OR) of 1.72 (95% CI, 1.260 to 2.367; nominal P < 0.001). The statistical significance of this result could not be determined because the ORR end point was not included in the hierarchical statistical analysis plan. However, the clinical experts considered the difference between groups to be clinically meaningful. The median follow-up times for censored patients were 11.9 months in the durvalumab and tremelimumab plus SOC chemotherapy group and 5.5 months in the SOC chemotherapy alone group. In the assessment of HRQoL, time to deterioration in the EORTC QLQ-C30 Global Health Status (GHS)/QoL was 8.3 months (95% CI, 6.4 to 10.2) in the durvalumab and tremelimumab plus SOC chemotherapy arm compared with 5.6 months (95% CI, 4.4 to 7.5) in the SOC chemotherapy alone arm. The statistical significance of this result could not be determined; however, the clinical experts described the difference between groups as clinically meaningful. The median durations of follow-up were 13.63 months in the durvalumab and tremelimumab plus SOC chemotherapy group and 11.17 months in the SOC chemotherapy alone group.
No published between-group minimal important difference (MID) values were provided by the sponsor for OS, PFS, ORR, and time to deterioration in the EROTC QLQ-C30 GHS/QoL in the first-line treatment of metastatic NSCLC; as such, the thresholds used to judge the target of certainty in the Grading of Recommendations Assessment, Development and Evaluation (GRADE) assessment (Table 4) are based on input from clinical experts consulted by CDA-AMC.
Harms Results
Similar proportions of patients experience at least 1 AE in the durvalumab and tremelimumab plus SOC chemotherapy arm (97.3%) and the SOC chemotherapy alone arm (96.1%), with the most commonly reported AEs being anemia (49.7% versus 48.9%, respectively), nausea (41.5% versus 36.6%), neutropenia (30.0% versus 23.4%), decreased appetite (28.2% versus 24.6%), fatigue (24.5% versus 22.2%), diarrhea (21.5% versus 15.3%), rash (19.4% versus 6.6%), and constipation (19.1% versus 23.7%). Serious adverse events (SAEs) were reported in a higher percentage of patients in the durvalumab and tremelimumab plus SOC chemotherapy arm (44.2%) than in the SOC chemotherapy alone arm (35.1%), as were AEs leading to discontinuation of any study treatment (22.1% versus 15.3%). AEs leading to death were reported in 12.4% of patients in the durvalumab and tremelimumab plus SOC chemotherapy arm and 9.0% of patients in the SOC chemotherapy alone arm.
Adverse events of special interest (AESIs) or adverse events of potential interest (AEPIs) were reported in a higher percentage of patients in the durvalumab and tremelimumab plus SOC chemotherapy arm than in the SOC chemotherapy alone arm (█████ ██████ █████). These included grade 3 or 4 AESIs or AEPIs (█████ ██████ ████) and AESIs or AEPIs that led to study treatment discontinuation █████ ██████ ████). AESIs or AEPIs with an outcome of death were reported in | ██████ patients in the durvalumab and tremelimumab plus SOC chemotherapy arm and | ██████ patient in the SOC chemotherapy alone arms.
The percentages of patients experiencing select AEs and AESIs (identified by clinical experts as AEs of greatest clinical importance) in the durvalumab and tremelimumab plus SOC chemotherapy and SOC chemotherapy alone groups are as follows: neutropenia (█████ ██████ ███████ pneumonitis (████ ██████ ████), hepatitis (████ ██████ ██ ████████), colitis (████ ██████ ████), type 1 diabetes mellitus (0.3% versus no patients), and myocarditis (0.3% versus no patients).
For the safety analysis, the median follow-up times were 13.63 months in the durvalumab and tremelimumab plus SOC chemotherapy arm and 11.17 months in the SOC chemotherapy alone arm.
Critical Appraisal
The POSEIDON trial was a phase III, randomized, open-label, comparative study. Methods of randomization and treatment allocation were adequate. Reported baseline characteristics were generally balanced across the study arms and the clinical experts consulted by CDA-AMC did not identify any considerable differences between the durvalumab and tremelimumab plus SOC chemotherapy and SOC chemotherapy alone treatment arms that would be expected to affect the interpretation of results. The prespecified sample size was achieved and actual screening failures did not exceed the expected number. The POSEIDON trial was powered for the dual primary end points and power calculations were provided for all end points included in the multiple testing procedure (MTP), which included the key secondary end points of OS and PFS for the comparison of durvalumab and tremelimumab plus SOC chemotherapy against SOC chemotherapy alone. The study used an open-label design because of differences in administration schedule and duration of study treatments. Of the efficacy end points of interest for this review, OS is an objective end point, and PFS, ORR, best objective response (BoR), and duration of response (DoR) were assessed by BICR according to RECIST 1.1 criteria, which helped to mitigate potential bias. Patient-reported efficacy assessments (i.e., HRQoL end points) and reporting of AEs can be influenced by knowledge of the treatment assignment by patients and clinicians. Other limitations related to internal validity include the potential impact of disproportionate treatment discontinuation between study arms, higher treatment exposure to SOC chemotherapy, overall higher exposure to study treatment in the durvalumab and tremelimumab plus SOC chemotherapy arm, and the possible confounding effect of subsequently received anticancer treatments on survival. Greater study treatment exposure (to SOC chemotherapy in particular) in the durvalumab and tremelimumab plus SOC chemotherapy group may have biased efficacy results in favour of this treatment regimen, whereas the higher proportion of patients in the SOC arm who received subsequent systemic anticancer therapy may have diluted the survival benefit observed with the combination treatment compared to SOC chemotherapy alone. Additionally, non-key secondary end points were not controlled for multiplicity. Overall adherence rates to the patient-reported outcome (PRO) scales generally decreased over time and were lower in the SOC chemotherapy arm. The missing data present a challenge in evaluating effects on HRQoL.
The clinical experts consulted for this review commented that the POSEIDON trial used standard inclusion and exclusion criteria that would be expected in a study of first-line treatment of patients with metastatic NSCLC. However, the experts identified differences between the POSEIDON trial participants and patients who would receive first-line immuno-oncology plus SOC chemotherapy for metastatic NSCLC in clinical practice in Canada. For example, patients in practice would typically be older, have relative contraindications to immunotherapy (i.e., quiescent inflammatory or autoimmune conditions that would not result in significant morbidity if reactivated), would include patients with an ECOG PS of 2, and those who had received immunotherapy for earlier-stage cancer. Also, the POSEIDON trial did not include any sites in Canada. However, the clinical experts noted that, overall, they did not have concerns regarding the generalizability of the study findings to patients seen in clinical practice. The main efficacy and harms outcomes assessed in the POSEIDON trial align with outcomes of importance identified by patients and clinicians.
An important limitation of the POSEIDON trial is that durvalumab and tremelimumab plus SOC chemotherapy was compared to SOC chemotherapy alone. When the trial was conducted, platinum-based chemotherapy was the standard therapy for the indicated population; however, current first-line treatment of metastatic NSCLC without targetable genomic alterations consists of immuno-oncology treatment (on its own or in combination with chemotherapy). Concerning subsequent anticancer treatments received in the POSEIDON trial, the clinical experts noted that rechallenging patients with immunotherapy and the low rate of patients who received immunotherapy following first-line chemotherapy in the POSEIDON trial are not reflective of practice in Canada.
GRADE Summary of Findings and Certainty of the Evidence
The selection of outcomes for GRADE (Table 4) was based on the sponsor’s Summary of Clinical Evidence, consultation with the clinical experts, and input received from patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with the expert committee members:
OS (median and probability of survival at 36, 48, and 60 months)
PFS (median and PFS rate at 12 months)
ORR
HRQoL (time to deterioration in EORTC QLQ-C30 GHS/QoL)
AEs (AESIs or AEPIs).
Table 4: Summary of Findings for Durvalumab and Tremelimumab Plus SOC Chemotherapy vs. SOC Chemotherapy for Patients With Metastatic NSCLC With No Sensitizing EGFR Mutations or ALK Genomic Tumour Aberrations
Outcome and follow-up | Patients (studies), N | Relative effect (95% CI) | Absolute effects (95% CI) | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
Chemotherapy | D + T + Chemotherapy | Difference | |||||
Overall survival | |||||||
OS, median (final analysisa) Median follow-up (months): D + T + chemotherapy: 13.63 Chemotherapy: 11.17 | N = 675 (1 RCT) | HR = 0.77 (0.650 to 0.916) | 11.7 months (10.5 to 13.1) | 14.0 months (11.7 to 16.1) | 2.3 more months ████ █████ ██ ███ █████ | Moderateb | D + T + chemotherapy likely results in a clinically important increase in OS compared with chemotherapy alone |
OS, median (5-year OS updatec,d) Median follow-up (months): D + T + chemotherapy: 13.63 Chemotherapy: 11.10 | N = 675 (1 RCT) | HR = 0.76 (0.642 to 0.893) | 11.6 months (10.5 to 13.1) | 14.0 months (11.7 to16.1) | 2.4 more months ████ █████ ██ ███ █████ | Moderatee | D + T + chemotherapy likely results in a clinically important increase in OS compared with chemotherapy alone |
Probability of survival at 36 months (final analysisa) Median follow-up (months): D + T + chemotherapy: 13.63 Chemotherapy: 11.17 | N = 675 (1 RCT) | NA | 13.3 per 100 people (9.8 to 17.4 per 100) | 25.3 per 100 people (20.8 to 30.2 per 100) | 12 more per 100 people ████ ██ ████ ████ ███ ████ | Highf | D + T + chemotherapy results in a clinically important increase in the probability of survival at 36 months compared with chemotherapy alone |
Probability of survival at 48 months (5-year OS updatec,d) Median follow-up (months): D + T + chemotherapy: 13.63 Chemotherapy: 11.10 | N = 675 (1 RCT) | NA | 8.5 per 100 people (5.8 to 11.9 per 100) | 20.9 per 100 people (16.7 to 25.4 per 100) | 12.4 more per 100 people ████ ██ ████ ████ ███ ████ | Highg | D + T + chemotherapy results in a clinically important increase in the probability of survival at 48 months compared with chemotherapy alone |
Probability of survival at 60 months (5-year OS updatec,d) Median follow-up (months): D + T + chemotherapy: 13.63 Chemotherapy: 11.10 | N = 675 (1 RCT) | NA | 6.8 per 100 people (4.4 to 10.0 per 100) | 15.7 per 100 people (12.0 to 19.9 per 100 people) | 8.9 more per 100 people ████ ██ ████ ████ ███ ████ | Highg | D + T + chemotherapy results in a clinically important increase in the probability of survival at 60 months compared with chemotherapy alone |
PFS according to RECIST 1.1 by BICR | |||||||
PFS,h median Median follow-up for censored patients (months): D + T + chemotherapy: 11.9 Chemotherapy: 5.5 | N = 675 (1 RCT) | HR = 0.72 (0.600 to 0.860) | 4.8 months (4.6 to 5.8) | 6.2 months (5.0 to 6.5) | 1.4 more months ████ █████ ██ ███ █████ | Moderatei | D + T + chemotherapy likely results in a clinically important increase in PFS compared with chemotherapy alone |
PFS rate at 12 monthsh,j Median follow-up for censored patients (months): D + T + chemotherapy: 11.9 Chemotherapy: 5.5 | N = 675 (1 RCT) | NA | 13.1 per 100 people (9.3 to 17.6 per 100) | 26.6 per 100 people (21.7 to 31.7 per 100) | 13.5 more per 100 people ████ ██ ████ ████ ███ ████ | Highf | D + T + chemotherapy results in a clinically important increase in the PFS rate at 12 months compared with chemotherapy alone |
ORR | |||||||
ORRj (unconfirmed responses) Median follow-up for censored patients (months): D + T + chemotherapy: 11.9 Chemotherapy: 5.5 | N = 667 (1 RCT) | OR = 1.72 (1.260 to 2.367) | 33.4 per 100 people █████ ██ ████ ███ ████ | 46.3 per 100 people █████ ██ ████ ███ ████ | 12.8 more per 100 people ████ ██ ████ ████ ███ ████ | Moderatek | D + T + chemotherapy likely results in a clinically important increase in ORR compared with chemotherapy alone |
HRQoL | |||||||
Time to deterioration (median) in EORTC QLQ-C30 GHS/QoLa,j Median follow-up (months): D + T + chemotherapy: 13.63 Chemotherapy: 11.17 | N = 637 (1 RCT) | HR = 0.78 (0.631 to 0.961) | 5.6 months (4.4 to 7.5) | 8.3 months (6.4 to 10.2) | 2.7 more months ████ █████ ██ ███ ████ ███████ | Very lowl | The evidence is very uncertain about the effect of D + T + chemotherapy on time to deterioration in EORTC QLQ-C30 GHS/QoL compared with chemotherapy alone |
Harms | |||||||
AESIs or AEPIs Median follow-up (months): D + T + chemotherapy: 13.63 Chemotherapy: 11.17 | N = 663 (1 RCT) | NA | AEs of special or potential interest were reported in ███ ███████ patients in the D + T + chemotherapy arm and ███ ███████ patients in the chemotherapy arm. Grade 3 or 4 AESIs or AEPIs were reported in ██ ███████ patients in the D + T + chemotherapy arm and ██ ██████ patients in the chemotherapy arm. | Highm | D + T + chemotherapy results in a higher incidence of AEs of special or potential interest (including those of grade 3 or 4) compared with chemotherapy alone | ||
AE = adverse event; AEPI = adverse event of potential interest; AESI = adverse event of special interest; BICR = blinded independent central review; CI = confidence interval; D = durvalumab; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; GHS = Global Status of Health; HR = hazard ratio; MID = minimal important difference; NA = not applicable; NSCLC = non–small cell lung cancer; OR = odds ratio; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; QoL = quality of life; RCT = randomized controlled trial; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1; SOC = standard of care; t = tremelimumab; vs. = versus.
Note: Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aFinal analysis data cut-off March 12, 2021.
bRated down 1 level for serious imprecision. Because no MID from the literature was provided, the target of certainty appraisal was the MID of 2 to 3 months based on clinical expert input; CI for difference between groups includes the possibility of trivial effects and no difference.
cFive-year OS update data cut-off August 24, 2023.
dFive-year estimates and CIs are not alpha-controlled.
eRated down 1 level for serious imprecision. Because no MID from the literature was provided, the target of certainty appraisal was the MID of 2.5 months based on clinical expert input; CI for difference between groups includes the possibility of trivial effects and no difference.
fBecause no MID from the literature was provided, the target of certainty appraisal was the MID of 5 to 10 per 100 people based on clinical expert input; CI for difference between groups included potential for important benefit.
gBecause no MID from the literature was provided, the target of certainty appraisal was the MID of 5 per 100 people based on clinical expert input; CI for difference between groups included potential for important benefit.
hFinal analysis data cut-off July 24, 2019.
iRated down 1 level for serious imprecision. Because no MID from the literature was provided, the target of certainty appraisal was the MID of 1 to 2 months based on clinical expert input; CI for difference between groups includes the possibility of trivial effects and no difference.
jSecondary efficacy end point not adjusted for multiplicity (considered supportive evidence).
kRated down 1 level for serious imprecision. Because no MID from the literature was provided, the target of certainty appraisal was the MID of 10 per 100 people based on clinical expert input; CI for difference between groups includes the possibility of trivial effects.
lRated down 2 levels for serious risk of bias because of knowledge of treatment assignment having the potential to affect reporting or recording of HRQoL outcomes and because of missing data (adherence rates fell below 60% after 88 weeks in the durvalumab and tremelimumab plus chemotherapy arm and after 24 weeks in the chemotherapy alone arm). Rated down 1 level for serious imprecision. Because no MID from the literature was provided, the target of certainty appraisal was the MID of 1.5 months based on clinical expert input; CI for difference between groups includes the possibility of trivial effects and no difference.
mNo statistical tests were performed. The difference in effects between groups was considered certain based on input from clinical experts.
Sources: Sponsor’s Summary of Clinical Evidence1 and sponsor’s data on file.13
Long-Term Extension Studies
No long-term extension studies were submitted by the sponsor.
Indirect Comparisons
Description of Studies
The sponsor conducted indirect treatment comparisons (ITCs) to estimate the relative efficacy of durvalumab and tremelimumab plus platinum-based chemotherapy versus other approved ICI plus platinum-based chemotherapy combinations among patients with metastatic NSCLC who lack EGFR mutations and ALK aberrations.
Efficacy Results
Based on the sponsor-submitted matching adjusted indirect comparisons (MAICs), overall, the efficacy, in terms of OS and PFS, appeared comparable when durvalumab and tremelimumab plus platinum-based chemotherapy was compared with relevant comparators for the first-line treatment of patients with metastatic NSCLC and no sensitizing EGFR mutations or ALK genomic tumour aberrations.
In the comparison of durvalumab and tremelimumab plus platinum-based chemotherapy versus pembrolizumab plus chemotherapy, the OS HR (95% CI) was ████ ██████ █████. The PFS HR (95% CI) was ████ ██████ █████ in the nonsquamous population, the OS HR (95% CI) was ████ ██████ █████, and PFS HR (95% CI) was ████ ██████ █████ in the squamous population.
In the comparison of durvalumab and tremelimumab plus platinum-based chemotherapy versus nivolumab plus ipilimumab plus chemotherapy, the OS HR was ████ ██████ ████). The PFS was ████ ██████ █████ in the squamous plus nonsquamous (intention to treat [ITT]) population and the OS HR was ████ ██████ █████. The PFS HR was ████ ██████ █████ in the nonsquamous population, the OS HR was ████ ██████ ██████ and the PFS HR was ████ ██████ █████ in the squamous population.
In the comparison of durvalumab and tremelimumab plus platinum-based chemotherapy versus cemiplimab plus chemotherapy, the OS HR was ████ ██████ █████. The PFS HR was ████ ██████ █████ in the squamous plus nonsquamous (ITT) population; the OS HR was ████ ██████ ██████ The PFS HR was ████ ██████ █████ in the nonsquamous population, the OS HR was ████ ██████ ██████, and the PFS HR was ████ ██████ █████ in squamous population.
Harms Results
The sponsor-submitted ITCs did not report harms outcomes.
Critical Appraisal
Overall, the MAICs were conducted according to accepted methodological guidance. The potential key limitations of the MAICs were that the analysis did not adjust for all the important effect modifiers (such as PD-L1 status, sex, age, ECOG PS, presence of brain metastases, disease stage, and type of chemotherapy), likely leading to residual heterogeneity between studies. In addition, the MAIC method reduced the effective sample size (by up to 40%). Imprecision observed in the effect point estimates, as indicated by the wide 95% CI, precluded making conclusions about the comparative effectiveness of durvalumab and tremelimumab plus chemotherapy versus other comparators in terms of OS and PFS.
Studies Addressing Gaps in the Evidence From the Systematic Review
No studies addressing gaps in the systematic review evidence were submitted by the sponsor.
Conclusions
Based on evidence from the POSEIDON study, durvalumab and tremelimumab plus SOC chemotherapy demonstrated a benefit compared to SOC chemotherapy alone in OS, PFS, and ORR for the first-line treatment of adult patients with metastatic NSCLC with tumours that lack activating EGFR mutations and ALK fusions. The GRADE assessment suggested that durvalumab and tremelimumab plus SOC chemotherapy likely results in a clinically important increase in median OS, median PFS, and ORR compared to SOC chemotherapy, and a high certainty that durvalumab and tremelimumab plus SOC chemotherapy results in a clinically important increase in the survival rate at 36 months, 48 months, and 60 months, as well as in the PFS rate at 12 months. However, the evidence is very uncertain about the effect of durvalumab and tremelimumab plus SOC chemotherapy on HRQoL, as measured by time to deterioration in EORTC QLQ-C30 GHS/QoL when compared with SOC chemotherapy alone.
In the POSEIDON study, AEPIs and AESIs (including those that were grade 3 or 4) were experienced by a higher percentage of patients receiving durvalumab and tremelimumab plus SOC chemotherapy than those receiving SOC chemotherapy alone, with the GRADE assessment suggesting with high certainty that durvalumab and tremelimumab plus SOC chemotherapy results in a higher incidence of AEPIs and AESIs when compared to SOC chemotherapy alone. The clinical experts consulted for this review stated that the types and rates of AEs in the POSEIDON trial were not unexpected. They indicated that immune-mediated adverse events (imAEs) caused by ICI treatment are typically of low grade and reversible and that health care providers treating lung cancer have a decade of experience using ICIs in routine clinical practice. The experts also noted that some imAEs can be very serious or life-threatening and that interventions required for the treatment of imAEs can negatively impact patient QoL.
Given that SOC chemotherapy was the comparator in the POSEIDON study, the lack of direct comparative data for the efficacy and safety between durvalumab and tremelimumab plus platinum-based chemotherapy and currently used first-line treatments (i.e., immuno-oncology plus chemotherapy options) represents a key gap in the evidence.
The findings of the MAICs demonstrated a largely comparable benefit in terms of OS and PFS when comparing combinations of durvalumab and tremelimumab plus platinum-based chemotherapy with relevant comparators for first-line treatment in patients with metastatic NSCLC without EGFR mutations and ALK aberrations. Harms were not investigated in the MAICs.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of durvalumab (50 mg/mL concentrate for IV infusion) in combination with tremelimumab (20 mg/mL concentrate for IV infusion) and platinum-based chemotherapy in the first-line treatment of metastatic NSCLC with no sensitizing EGFR mutations or ALK genomic tumour aberrations.
Disease Background
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the review team.
Lung cancer is among the most commonly diagnosed cancers14 and is the leading cause of cancer-related death in Canada.3 In Canada, the 5-year net survival rate for lung cancer (all stages and histological subtypes) is 22%.14 Lung cancer is heterogenous in nature, comprising several different disease subtypes and categorized by histology, staging, and mutation driver status.3 NSCLC is the most common type, representing approximately 89% of all lung cancer diagnoses in Canada.4 NSCLC can be categorized into histologic subtypes: squamous-cell carcinoma (17% to 27% of NSCLC cases), and non–squamous cell carcinoma (73% to 83% of NSCLC cases).3,5 At early stages, NSCLC may be asymptomatic,3 with symptoms developing only when the cancer has become more advanced and is no longer amenable to curative-intent therapy. Presenting symptoms can sometimes be nonspecific (common manifestations include coughing, chest pain, hemoptysis, fatigue, weight loss, dyspnea, hoarseness, and recurring infections with bronchitis and pneumonia), contributing to delays in diagnosis.3,5,6 Screening programs for NSCLC are being established throughout Canada, but many patients who would qualify for NSCLC screening live in jurisdictions where it is not yet available.
To determine a patient’s prognosis and treatment, a tissue biopsy is performed for histologic confirmation and the tumour is staged using the most recent American Joint Committee on Cancer staging criteria, which involve tumour-node-metastasis classification of the disease, based on assessment of the size and spread of the primary tumour, lymph node involvement, and occurrence of distance metastasis.15 Recent advances in NSCLC, including molecular profiling, allow for the identification of genomic alterations or mutations (i.e., oncogenic drivers).16 Several oncogenic driver mutations have been identified, where frequently occurring mutations include EGFR mutations and ALK aberrations,7 which collectively occur in approximately 25% of patients.8 An estimated 74.5% of NSCLC cases do not have EGFR or ALK aberrations.
A diagnosis starts with a complete medical history and physical exam, assessment of risk factor exposure (e.g., history of tobacco use or exposure to radon gas), along with standard tests, including lung function, routine hematology, renal and hepatic function, and bone biochemistry tests.17-19 Patients also undergo radiographic staging, which may include CT scans or PET-CT scans of the chest, abdomen, and pelvis, and MRI scans of the central nervous system (CNS).17-19 Upon receiving a confirmed diagnosis of NSCLC that includes histopathological subtyping, patients undergo testing for oncogenic drivers and PD-L1 levels via biomarker testing at an accredited laboratory. Tumours with a nonsquamous histology are tested for a wide panel of oncogenic drivers including EGFR mutations (common and uncommon), ALK rearrangements, ROS1 rearrangements, and BRAF mutations, in addition to PD-L1 immunohistochemical testing.20 Tumours with squamous histology are typically tested only for PD-L1, with most jurisdictions only performing molecular testing in squamous tumours on request in specific cases (e.g., a tumour from a patient with little or no tobacco use history). Biomarker testing is performed by molecular tissue testing on a tissue sample most commonly obtained via biopsy.20 Molecular tissue testing in NSCLC is well-established and a standard diagnostic workup is performed at the time of diagnoses based on current clinical practices in Canada.20
Standards of Therapy
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the review team.
Although currently available ICI-based therapies can produce deep and durable responses in a small subset of patients, there is no cure for NSCLC in the metastatic stage.21 The goals of therapy for patients with metastatic NSCLC, as identified by the clinical experts consulted by CDA-AMC, include prolonging survival, extending the time before disease progression, decreasing cancer-related symptoms, and maintaining or improving QoL. In current practice, the treatment of metastatic NSCLC is guided by the presence or absence of oncogenic drivers, including EGFR mutations and ALK rearrangements (among others).11,17 Nearly half (48.4%) of NSCLC cases are metastatic (stage IV).3,8 For the majority of patients with metastatic NSCLC whose tumours lack actionable genomic alterations, treatment is selected based on tumour histology (squamous or nonsquamous); tumoural PD-L1 expression (which is predictive of response to ICIs); and patient symptom burden (as measured by ECOG PS), comorbidities, and preferences.9-11
In the past, SOC first-line systemic therapy for patients with advanced NSCLC was treatment with a platinum doublet chemotherapy regimen.22 Advances in recent years have led to ICI-based therapies, either as monotherapy or in combination with chemotherapy, being the current standard first-line treatment for metastatic NSCLC without targetable genomic alterations.10,11 The sponsor has described the current treatment algorithm for metastatic NSCLC without targetable genomic alterations, as presented in Figure 1. For patients with any or unknown PD-L1 expression levels, first-line options include pembrolizumab plus platinum-based chemotherapy, nivolumab and ipilimumab plus platinum-based chemotherapy, or cemiplimab plus platinum-based chemotherapy.10,11 Monotherapy with pembrolizumab or cemiplimab may be selected as first-line treatment for patients with high PD-L1 expression (≥ 50% TCs).10,11 Cemiplimab (with or without platinum-based chemotherapy) is not currently funded in Canada.12 Platinum-based chemotherapy alone is also a potential first-line treatment option; however, the clinical experts noted that this would be reserved for patients with significant contraindications to immunotherapy and that few patients would receive this option. The clinical experts indicated that immuno-oncology treatment is considered the backbone of therapy for the first-line treatment of metastatic NSCLC without EGFR mutations or ALK aberrations.
Figure 1: Current Treatment Algorithm for Metastatic NSCLC Without Targetable Genomic Alterations
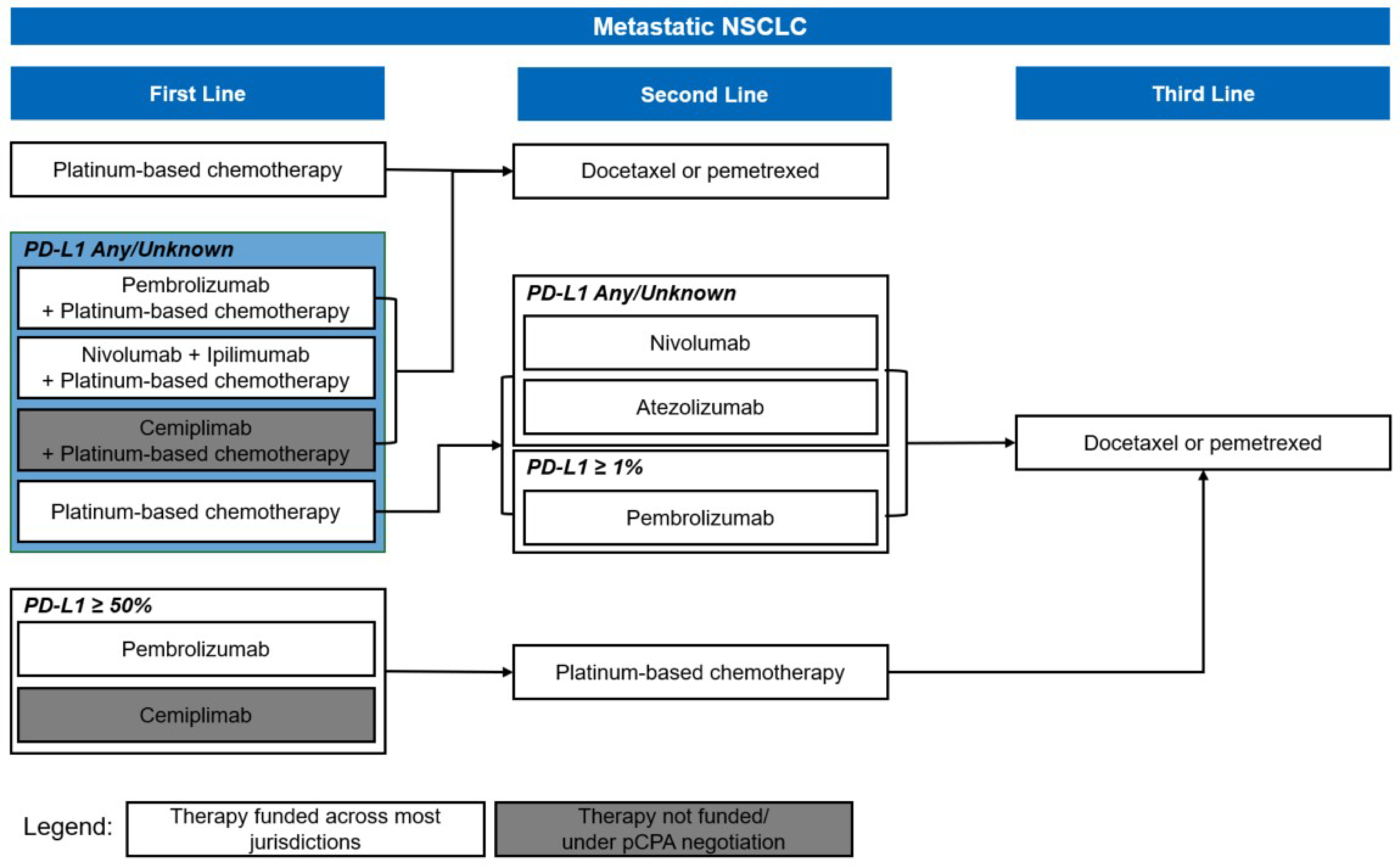
CDA-AMC = Canada’s Drug Agency; ICI = immune checkpoint inhibitor; NSCLC = non–small cell lung cancer; pCPA = pan-Canadian Pharmaceutical Alliance.
Note: The sponsor noted that the indication of interest for this review is for ICI-based regimens in combination with platinum-based chemotherapy, as indicated by the blue shading in the PD-L1 Any/Unknown box. A deviation request was approved by CDA-AMC allowing for the exclusion of monotherapy with immuno-oncology drugs (i.e., those given without platinum-based chemotherapy) as comparators. Cemiplimab has a “reimburse with clinical criteria and/or conditions” CDA-AMC recommendation for patients who have a PD-L1 status of 50% or greater and in combination with chemotherapy for patients with an “Any/Unknown” PD-L1 status, but it is not currently listed on any provincial drug plan or cancer agency.12,23
Sources: Developed by the sponsor based on the CDA-AMC provisional funding algorithm12 and Clinical Practice Guidelines.10,11
Drug Under Review
Key characteristics of durvalumab and tremelimumab are summarized in Table 5 with other treatments available for the treatment of metastatic NSCLC.
According to the product monograph, the recommended dosing for durvalumab in combination with tremelimumab and platinum-based chemotherapy differs based on the patients’ body weight.2 The recommended dosage during chemotherapy is either durvalumab 1,500 mg in combination with tremelimumab 75 mg and platinum-based chemotherapy (body weight ≥ 30 kg) or durvalumab 20 mg/kg in combination with tremelimumab 1 mg/kg and platinum-based chemotherapy (body weight < 30 kg) every 3 weeks (21 days) for 4 cycles. After platinum-based chemotherapy, the recommended dosage of durvalumab is 1,500 mg (body weight ≥ 30 kg) or 20 mg/kg (body weight < 30kg) every 4 weeks and histology-based pemetrexed maintenance therapy every 4 weeks. A fifth dose of tremelimumab is to be given at week 16 alongside durvalumab dose 6. During cycle 1, tremelimumab is followed by durvalumab starting approximately 1 hour (maximum 2 hours) after the end of the tremelimumab infusion. Platinum-based chemotherapy infusion should start approximately 1 hour (maximum 2 hours) after the end of the durvalumab infusion. If there are no clinically significant concerns during cycle 1, then subsequent cycles of durvalumab can be given at the physician’s discretion immediately after tremelimumab, and the time between the end of the durvalumab infusion and the start of chemotherapy can be reduced to 30 minutes.2
Durvalumab is an engineered monoclonal antibody that blocks the interaction of PD-L1 with its receptors PD-1 and CD80.2 Selective blockade of interactions between PD-L1 and PD-1 and between PD-L1 and CD80 lead to inhibition of immune responses and enhanced antitumour activity.2 Tremelimumab is a selective, fully human immunoglobulin G2 antibody that blocks CTLA-4 interactions with CD80 and CD86, enhancing T-cell activation and proliferation, which may result in increased T-cell diversity and enhanced antitumour immune activity.24
The reimbursement request is for durvalumab in combination with tremelimumab and platinum-based chemotherapy for the first-line treatment of adult patients with metastatic NSCLC with no sensitizing EGFR mutations or ALK genomic tumour aberrations, which is aligned with the Health Canada–approved indication. The HTA agency in France (Haute Autorité de Santé)25 and the FDA26 approved the use of the durvalumab in combination with tremelimumab and platinum-based chemotherapy for the same indication approved by Health Canada. The European Medicines Agency initially provided a positive recommendation for tremelimumab for the same indication (December 15, 2022)27 and granted market authorization on February 20, 2023.28 However, it withdrew the market authorization at the request of the marketing authorization holder, AstraZeneca AB, on November 6, 2023.29 The HTA agency in England, the National Institute for Health and Care Excellence (NICE)30 was asked to carry out a single technology appraisal of durvalumab with tremelimumab for untreated EGFR-negative and ALK-negative locally advanced and metastatic NSCLC by the Department for Health and Social Care. In August 2022, the appraisal was suspended as the company advised that it is was no longer pursuing a marketing authorization from the Medicines and Health care Products Regulatory Agency for the indication. In August 2023, appraisal was discontinued as no further information was received from the company.
Tremelimumab in combination with durvalumab has been previously reviewed by CDA-AMC for the first-line treatment of adult patients with unresectable hepatocellular carcinoma who require systemic therapy, with a recommendation to “reimburse with clinical criteria and/or conditions” (issued in November 2023).31 Durvalumab has been previously reviewed and received conditional positive recommendations for various therapeutic areas (unresectable NSCLC, extensive-stage small-cell lung cancer, and biliary tract cancer).32-34
Table 5: Key Characteristics of Durvalumab in Combination With Tremelimumab; Nivolumab in Combination With Ipilimumab; Cemiplimab; and Pembrolizumab
Characteristic | Durvalumab + tremelimumab | Nivolumab + ipilimumab | Cemiplimab | Pembrolizumab |
|---|---|---|---|---|
Mechanism of action | The combination of durvalumab, a PD-L1 inhibitor, and tremelimumab, a CTLA-4 inhibitor, enhances antitumour T-cell activation and function at multiple stages of the immune response, maximizing antitumour immunity | Inhibition mediated by a combination of nivolumab (anti–PD-1) and ipilimumab (anti–CTLA-4) results in enhanced T-cell function | Inhibits T-cell proliferation and cytokine production | Inhibits the PD-1 receptor from binding to its ligands, which reactivates tumour-specific cytotoxic T lymphocytes in the tumour microenvironment |
Indicationa | First-line treatment of adult patients with metastatic NSCLC with no sensitizing EGFR mutations or ALK genomic tumour aberrations in combination with tremelimumab and platinum-based chemotherapy | Metastatic NSCLC with no EGFR or ALK genomic tumour aberrations and no prior systemic therapy for metastatic NSCLC, in combination with ipilimumab and 2 cycles of platinum doublet chemotherapy | In combination with platinum-based chemotherapy for first‐line treatment of adult patients with NSCLC whose tumours have no EGFR, ALK, or ROS1 aberrations and whose disease is locally advanced and who are not candidates for surgical resection or definitive chemoradiation, or for patients with metastatic NSCLC | For the first-line treatment of adult patients with metastatic NSCLC or stage III disease who are not candidates for surgical resection or definitive chemoradiation, and who express PD-L1 (Tumour Proportion Score ≥ 1%), determined by a validated test, but who have no EGFR or ALK genomic tumour aberrations In combination with pemetrexed and platinum-based chemotherapy, for treatment of adult patients with metastatic nonsquamous NSCLC with no EGFR or ALK genomic tumour aberrations and who have received no prior systemic chemotherapy for metastatic NSCLC In combination with carboplatin and either paclitaxel or nab-paclitaxel, for treatment of adult patients with metastatic squamous NSCLC who have received no prior systemic chemotherapy for metastatic NSCLC |
Route of administration | IV | IV | IV | IV |
Recommended dose | During chemotherapy: Durvalumab 1,500 mg in combination with tremelimumab 75 mg and platinum-based chemotherapy (BW ≥ 30kg) or durvalumab 20 mg/kg in combination with tremelimumab 1 mg/kg and platinum-based chemotherapy every 3 weeks for 4 cycles (BW < 30 kg) After chemotherapy: durvalumab 1,500 mg (BW ≥ 30 kg) or 20 mg/kg (BW < 30kg) and histology-based pemetrexed maintenance therapy every 4 weeks | Nivolumab 3 mg/kg every 2 weeks or 360 mg every 3 weeks (30-minute IV) with ipilimumab 1 mg/kg every 6 weeks (30-minute IV) | 350 mg every 3 weeks, administered by IV infusion over 30 minutes | 200 mg every 3 weeks or 400 mg every 6 weeks, administered by IV infusion over 30 minutes |
Serious adverse effects or safety issues | Can cause immune-mediated adverse reactions (pneumonitis, hepatitis, colitis, endocrinopathies, adrenal insufficiency, hypophysitis or hypopituitarism, type 1 diabetes, nephritis, rash, myocarditis) infections, infusion-related reactions | Nivolumab as monotherapy or in combination with ipilimumab can cause severe and fatal immune-mediated adverse reactions, including pneumonitis, interstitial lung disease, encephalitis, myocarditis, Stevens-Johnson syndrome, toxic epidermal necrolysis and autoimmune hemolytic anemia | Contraindicated in the case of hypersensitivity; should be administered under the supervision of health care practitioners experienced in the treatment of cancer Can cause immune-mediated adverse reactions, such as pneumonitis, colitis, hepatitis, endocrinopathies, thyroid disorders, hypophysitis, nephritis, skin reactions, meningitis, myositis, encephalomyelitis, and myocarditis, adrenal insufficiency, and type 1 diabetes | Contraindicated in cases of hypersensitivity; should be administered under the supervision of physicians experienced in the treatment of cancer Can cause severe and fatal immune-mediated adverse reactions, such as pneumonitis, colitis, hepatitis, endocrinopathies, thyroid disorders, hypophysitis, nephritis, skin reactions, uveitis, arthritis, myositis, encephalitis, sarcoidosis, myasthenic syndrome and/or myasthenia gravis, vasculitis, Guillain-Barré syndrome, hemolytic anemia, pancreatitis, myelitis, and hypoparathyroidism, adrenal insufficiency, and type 1 diabetes |
BW = body weight; NSCLC = non–small cell lung cancer.
aHealth Canada–approved indication.
Sources: Product monographs of durvalumab,2 tremelimumab,24 nivolumab,35 cemiplimab,36 and pembrolizumab.37
Perspectives of Patients, Clinicians, and Drug Programs
The full patient and clinician group submissions received are available in the consolidated patient and clinician group input document for this review on the project website.
Patient Group Input
This section was prepared by the review team based on input provided by patient groups.
A joint patient group submission was received from CCSN, LCC, and LHF. CCSN is a national network of patients, families, survivors, friends, community partners, funders, and sponsors who have come together to take action to promote the best SOC, including early diagnosis, timely treatment and follow-up care, support for patients with cancer, and regarding issues related to survivorship or quality of end-of-life care. LCC is a registered national charitable organization that serves as Canada’s leading resource for lung cancer education, patient support, research and advocacy. LHF (previously the Ontario Lung Association) is a registered charity that assists and empowers people living with or caring for others with lung disease. It is a recognized leader, voice, and primary resource in the prevention and control of respiratory illness, tobacco cessation and prevention, and its effects on lung health. The Foundation provides programs and services to patients and health care providers, invests in lung research, and advocates for improved policies in lung health.
CCSN, LCC, and LHF collectively produced a survey that was disseminated through the 3 organizations’ social media platforms, as well as CCSN’s monthly newsletter to gather responses from August 1, 2024, to the date of writing of the patient group submission. LCC also interviewed 1 patient who was a part of the POSEIDON trial, on October 8, 2024.
The experience of the patient with lung cancer who participated in the POSEIDON trial has been summarized. The patient’s diagnosis took a while before they could get started on treatments. The patient started with first-line chemotherapy and received durvalumab simultaneously. The treatment regimen of this patient was unclear to the interviewer; however, upon clarification with the physician who treated the patient, confirmation was received that the patient was treated with the drug under review (durvalumab and tremelimumab in combination with platinum-based chemotherapy). While on chemotherapy, the patient received appropriate care and dealt with minimal side effects, including mild diarrhea, bone pain, itching and tiredness, especially on the days of chemotherapy. While on durvalumab, the patient reported experiencing temporary side effects, such as occasional diarrhea, itching, and hot flashes. The patient emphasized that they were still able to carry out regular activities during treatment.
Respondents from a previous survey and submission on durvalumab in combination with chemotherapy as neoadjuvant treatment, followed by durvalumab as monotherapy after surgery indicated for the treatment of patients with NSCLC and resectable tumours (≥ 4 cm and/or node-positive) and no known EGFR or ALK rearrangements, shared their experiences with lung cancer and durvalumab. There were 5 respondents, including 2 with stage IVA lung cancer, 1 with stage IVB, and 2 others (1 with stage IV that had spread only to the brain and 1 with stage IV metastatic NSCLC). The following symptoms affecting QoL were reported: fatigue; pain in chest, shoulder, back or arms; shortness of breath; infections; pneumonia and/or bronchitis; and loss of weight, appetite, hair, and teeth. Respondents identified chemotherapy, immunotherapy, targeted therapy, radiation, surgical therapy, and participation in clinical trials as current treatment options. Respondents noted several unmet needs, including better mental health support and greater availability of immunotherapy for longer durations. In terms of accessing any therapies, 1 respondent reported not being able to acquire counselling and another mentioned burdensome travel costs associated with treatment.
On current treatment regimens, the respondents mentioned several side effects, with 4 in 6 patients reporting joint and muscle pain. Other side effects included fatigue, diarrhea, neuropathy, weight loss, anemia, nausea, vomiting, constipation, migraines, change in vision and hearing, and forgetfulness. Of the 5 respondents, 2 described their AEs as well tolerated. The respondents indicated they were managing their current treatments generally well.
The respondents rated several outcomes that they hoped to see in newer treatments to manage their disease (from most to least important based on average ranking): prolonged life, provision of a cure, QoL, delay in onset of symptoms, ease of use and reduction in side effects from medications or treatments, and access to new options for treatment.
Fatigue was noted as the main side effect for treatment with durvalumab. One of the respondents also mentioned the development of hives. For symptom management, 2 of the 5 respondents noted it was much better, and 1 of the 5 respondents noted little or no difference. For side effects, 3 of the 5 respondents noted little or no difference. For ease of use, 2 of the 5 respondents noted little or no difference. For disease progression, 1 of the 5 respondents noted it was much better, and 2 of the 5 respondents noted little or no difference.
In comparison to other therapies, at least 2 respondents indicated that symptom management was much better with durvalumab; however, little to no difference was reported for side effects, ease of use, and disease progression.
Overall, the patient group highlighted the need for treatments that improve survival (6 months or greater) regardless of the level of side effects. They also noted that patients experienced fewer AEs while receiving durvalumab and felt better versus their experience with previous lines of care.
Clinician Input
Input From Clinical Experts Consulted for This Review
All CDA-AMC review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of NSCLC.
Unmet Needs
The goals of first-line treatment of metastatic NSCLC identified by the clinical experts consulted by CDA-AMC were to improve survival, delay progression of cancer, decrease cancer-related symptoms, and improve QoL. The clinical experts acknowledged that currently available first-line treatment options for metastatic NSCLC have been associated with a survival benefit compared to chemotherapy alone but noted that there remains an unmet need with overall poor outcomes for the great majority of patients with advanced NSCLC, particularly among those whose tumours have low or negative tumoural PD-L1 expression, who are less likely to have a durable benefit from single-drug immuno-oncology treatment plus chemotherapy. The clinical experts also expressed the need for treatment options in patients with low or negative tumoural PD-L1 expression who have lower rates of immune-related AEs. The experts recognized that there is significant unmet need among subgroups of patients with mutations associated with a lower chance of responding to immuno-oncology therapy or with a generally poor prognosis, including KRAS, STK11, and KEAP mutations.
Place in Therapy
The clinical experts noted that durvalumab and tremelimumab plus platinum-based chemotherapy would be used as a standalone treatment for patients with previously untreated metastatic NSCLC not harbouring a targetable oncogenic aberration with an associated SOC first-line targeted therapy option (e.g., an EGFR mutation or ALK rearrangement) and without any absolute contraindications. The experts did not anticipate that durvalumab and tremelimumab plus platinum-based chemotherapy would shift the current treatment paradigm; instead, this regimen would fit into the treatment algorithm as an additional first-line option combining immuno-oncology treatment and chemotherapy.
Patient Population
In describing patients who would be best and least suited for treatment with durvalumab and tremelimumab plus chemotherapy, the clinical experts stated that there is no reliable biomarker that can be used to predict which patients will respond to this treatment. The experts noted that patients ideally suited for this treatment would have previously untreated metastatic NSCLC, a good ECOG PS (0 or 1), no actionable oncogenic mutations or translocations (including EGFR mutations and ALK rearrangements), any tumoural PD-L1 expression, no significant comorbidities (i.e., uncontrolled autoimmune or inflammatory diseases), and the ability to recognize and report potential adverse effects to their oncology care team in a timely manner. The experts stated that the eligibility criteria for the POSEIDON trial were aligned with those used in clinical trials in this patient population, but added that certain patients excluded from the trial would be considered for treatment with immuno-oncology therapy in clinical practice, including those with an ECOG PS of 2, those with autoimmune or inflammatory disorders or with infections such as HIV or hepatitis C, a subpopulation with prior exposure to immunotherapy for earlier-stage cancer, and a broader population of patients with brain metastases. The clinical experts pointed out that, in practice, durvalumab and tremelimumab plus platinum-based chemotherapy is anticipated to be used most often in patients with PD-L1 levels below 50%, but they noted that, because patients with any PD-L1 status were included in the POSEIDON trial, approval and funding should be in accordance with the trial outcomes. The experts stated that durvalumab and tremelimumab plus platinum-based chemotherapy may be an option for patients whose tumours do not express PD-L1 (based on a post hoc analysis showing that these patients may derive the most benefit from the addition of an anti–CTLA-4 antibody to an anti–PD-1 antibody), noting that reliable biomarkers to determine which patients will derive the most benefit are lacking. The experts identified durvalumab and tremelimumab plus platinum-based chemotherapy as an option for patients with KRAS, STK11, or KEAP mutations, while acknowledging that the current evidence is post hoc and hypothesis-generating. The treatment strategy for the indicated population would be based on an informed decision by the patients in consultation with a medical oncologist. According to the clinical experts, misdiagnosis of advanced NSCLC or errors in biomarker testing does not occur often because of rigorous quality control measures in place at pathology labs in Canada that perform cancer diagnoses and biomarker testing. The experts expected that all centres would have access to routine testing for KRAS, EGFR, and ALK mutations and PD-L1 status but that testing for STK11 and KEAP mutations may vary by centre.
Assessing the Response to Treatment
The clinical experts commented that patients with metastatic NSCLC receiving systemic therapy would typically be clinically assessed before each treatment (every 3 weeks) by a medical oncologist, general practitioner in oncology, or nurse practitioner with special training in medical oncology. Radiological assessment for response to treatment would typically be performed every 2 to 3 months by CT scan, and less commonly with MRI or PET-CT scan, unless symptoms suggesting disease progression prompt earlier investigations. The clinical experts noted that surveillance imaging in clinical trials is typically performed every 6 to 9 weeks initially. The experts did not expect that results would vary among physicians. Clinically meaningful end points identified by the clinical experts are improvement in survival, decrease in cancer-related symptoms, and improvement in QoL. Tumour evaluation in the POSEIDON trial was performed by CT scan or MRI at weeks 6 and 12 after randomization and every 8 weeks thereafter.38
Discontinuing Treatment
The clinical experts stated that discontinuation of first-line treatment for metastatic NSCLC would be considered when there is significant disease progression or unacceptable toxicity, or if the patient chooses to stop treatment. One clinical expert added that discontinuation of treatment may be considered when there is suspicion of progression and a risk of rapid clinical deterioration if treatment is continued. Clinicians treating NSCLC already have experience diagnosing and managing immune-related adverse effects of ICIs in clinical practice, and the clinical experts stated that immuno-oncology treatment may be held temporarily but can often be safety resumed (even for high-grade immune-related adverse effects) and that permanent discontinuation may not be needed. However, they also pointed out that rechallenge with immunotherapy would typically not be undertaken if the AE was life-threatening. The clinical experts noted that, upon development of an AE, the risks and benefits of resuming treatment would be considered through consultation between the clinician and patient.
Prescribing Considerations
The clinical experts noted that first-line treatments for metastatic NSCLC would typically be initiated following consultation with a medical oncologist at a regional cancer clinic. Treatment would be administered and monitoring conducted locally for patients who live close to the regional clinic, and often at community oncology networks for patients living in remote settings, where specially trained general practitioners or internists would oversee day-to-day management of the treatment protocol. The clinical experts agreed that it would be appropriate for any systemic therapy unit and lung cancer treatment team currently administering other combinations of immuno-oncology and chemotherapy to deliver treatment with durvalumab and tremelimumab plus platinum-based chemotherapy. Clinical experts added that all patients must be closely monitored to detect early evidence of toxicity or progression, and that consultation with a subspecialist (e.g., a gastroenterologist or hepatologist in the event of liver toxicity) may be required if there is evidence of toxicity.
Additional Considerations
CDA-AMC approved a deviation request from the sponsor allowing for the exclusion of monotherapy with immuno-oncology drugs (i.e., those given without platinum-based chemotherapy) as comparators in both the pharmacoeconomic model and budget impact analysis. The sponsor’s rationale was that the proposed place in therapy for durvalumab and tremelimumab plus platinum-based chemotherapy is the same as other immuno-oncology drugs that are indicated for use in combination with platinum-based chemotherapy, and as such, the sponsor proposed that the use of durvalumab and tremelimumab plus platinum-based chemotherapy would only displace the use of other regimens that include platinum-based chemotherapy. The clinical experts described immuno-oncology monotherapy as a potential comparator for the combination of durvalumab and tremelimumab plus platinum-based chemotherapy. They also noted that, although durvalumab and tremelimumab plus platinum-based chemotherapy is anticipated to be used most often in patients with tumoural PD-L1 expression below 50% in clinical practice, approval and funding should be based on the outcomes of the POSEIDON trial, adding that patients with any PD-L1 status were included in the study. The experts stated that platinum-based chemotherapy alone does not represent a reasonable comparator to durvalumab and tremelimumab plus platinum-based chemotherapy because immuno-oncology treatment is the backbone of therapy for first-line treatment of metastatic NSCLC. Platinum-based chemotherapy alone would typically only be given to patients who are not able to or choose not to receive immuno-oncology treatment, and these patients would not be eligible for treatment with the regimen used in the POSEIDON trial. The clinical experts pointed out that platinum-based chemotherapy has been shown to be inferior to immuno-oncology treatment plus platinum-based chemotherapy or immuno-oncology monotherapy (depending on the clinical trial).
Clinician Group Input
This section was prepared by the review team based on input provided by clinician groups.
Two clinician groups, the LLC-MAC and the Cancer Care Ontario DAC, provided input for this submission. The LCC-MAC is a national charity providing support and education, supporting research, and providing clinician input for submissions to reviews of new lung cancer drugs by the HTA process for many years. The DAC provides evidence-based clinical and health system guidance on drug-related issues to support the mandate of Cancer Care Ontario, including the Provincial Drug Reimbursement Programs and the Systemic Treatment Program.
A total of 19 clinicians from the LCC-MAC and 5 clinicians from the DAC provided input for this submission. The submission from LCC-MAC was informed by data and information from publicly available sources, primarily published manuscripts and conference presentations, together with clinical experience of the LCC-MAC members. Information from the DAC was gathered via teleconference meeting and emails.
The LCC-MAC indicated that multiple treatment options are available in Canada for patients without driver mutations (e.g., EGFR mutations or ALK translocations). In addition, they noted that, for those with tumours with PD-L1 expression of any level or unknown, current treatment options include platinum doublet (cisplatin or carboplatin with pemetrexed) plus pembrolizumab (nonsquamous histology), platinum doublet (carboplatin and paclitaxel) plus pembrolizumab (squamous histology), or chemotherapy-sparing combinations of 2 cycles of platinum doublet with doublet immunotherapy in the form of nivolumab and ipilimumab (most often used in PD-L1–negative or squamous histology). Both clinician groups acknowledged the use of single-drug pembrolizumab in patients with a PD-L1 expression level of 50% or greater. The DAC noted current treatment options included chemotherapy with pembrolizumab, and chemotherapy with nivolumab and ipilimumab.
The DAC emphasized that the treatment combination of durvalumab with tremelimumab and platinum-based chemotherapy would serve as a first-line treatment. The LCC-MAC noted that most thoracic oncologists in Canada have experience with using the treatment combination through clinical trials. They also pointed out that the treatment combination would not replace other regimens that were already approved or funded but would serve as an alternative. Chemotherapy and immunotherapy options were described by the LCC-MAC as appropriate comparators to the treatment combination.
In terms of unmet needs, both clinician groups agreed that treatments with better tolerability are needed. The DAC also noted that not all patients respond to available treatments, patients become refractory to current treatment options, and treatments and formulations that improve compliance and convenience are needed. Important treatment goals identified by both clinician groups were tumour shrinkage, improvement of symptoms and QoL, and improved OS, with the LCC-MAC adding that improvement in PFS is an important goal. The clinician groups further noted that an important aspect to be looked at with immunotherapy combinations was landmark survival with the goal of improving the number of patients with durable benefits of treatment as manifested in increases in PFS and OS at 5 years.
The clinicians indicated that patients without actionable driver mutations (i.e., no EGFR or ALK alterations, no ROS and RET fusions) would be best suited for treatment. The DAC noted that patients with stage IV or incurable NSCLC considering first-line therapy would be suitable candidates for treatment. In addition, the LCC-MAC noted that patients in the POSEIDON trial with KRAS, STK11, and KEAP mutations, which are not currently actionable in the first-line setting, seem to benefit from the combination. Both groups highlighted the need for clinical assessment of symptoms and imaging (e.g., CT scans and chest X-rays) to monitor response to treatment. The DAC advised assessing treatment response every 6 weeks initially, and then less often. However, the LCC-MAC suggested that scans be performed every 3 months, with symptom assessments every 3 to 4 weeks, depending on whether a patient receives chemotherapy or single-drug immunotherapy during the maintenance phase.
The LCC-MAC also noted that this treatment regimen would be an appropriate choice for patients without an actionable driver mutation with any PD-L1 status. The groups agreed that disease progression is a factor when deciding whether to discontinue treatment. The LCC-MAC indicated that immunotherapy may be interrupted or discontinued because of imAEs. They added that most low-grade toxicities were manageable with symptom management, topical or oral steroids, and potential treatment interruption. The DAC also identified intolerable side effects and patient withdrawal as factors when considering treatment discontinuation.
The LCC-MAC indicated that initial patient assessments should be conducted by a medical oncologist. They noted that, with high rates of lung cancer, Canada has an established network of experts, including family physicians and general practitioners in oncology who can assist in the assessment of these patients on treatment, particularly in remote areas. Both clinician groups agreed that the treatment combination should be administered in facilities with expertise in managing cytotoxic anticancer therapies.
Drug Program Input
The drug programs provide input on each drug being reviewed through the Reimbursement Review processes by identifying issues that may affect their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by for this review are summarized in Table 6.
Table 6: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
Issues with the choice of comparator in the submitted trial(s) In addition to platinum chemotherapy, relevant comparators include pembrolizumab-platinum chemotherapy, single-agent pembrolizumab (if PD-L1 > 50%), ipilimumab-nivolumab-platinum chemotherapy, and cemiplimab-platinum chemotherapy. How does durvalumab-tremelimumab compare to other immunotherapy with or without platinum chemotherapy regimens? | The clinical experts noted that patients with any PD-L1 status were included in the POSEIDON trial. Addition of a CTLA-4 inhibitor (i.e., tremelimumab) may be of greatest benefit to patients with a low or negative PD-L1 status. One clinical expert also noted that this may also be of benefit to patients with very aggressive, high-burden disease. According to the clinical experts, the regimen that is most similar to durvalumab and tremelimumab plus chemotherapy is nivolumab and ipilimumab plus chemotherapy. The rates and severity of immune-mediated AEs between durvalumab and tremelimumab plus chemotherapy and nivolumab and ipilimumab plus chemotherapy are likely very similar. However, there may be indirect evidence for less-severe AEs with durvalumab and tremelimumab than with nivolumab and ipilimumab based on lower rates of discontinuation because of AEs observed in the POSEIDON trial than in the CheckMate 9LA trial of nivolumab and ipilimumab. However, there is no published, direct evidence to inform this comparison. Conversely, the clinical experts noted that the durvalumab and tremelimumab regimen uses 4 cycles of chemotherapy whereas the nivolumab and ipilimumab regimen uses 2 cycles; and nivolumab and ipilimumab may be more suitable in patients for whom fewer cycles of platinum doublet chemotherapy is preferred. |
Considerations for initiation of therapy | |
Prior therapies required for eligibility The trial allowed patients who received prior adjuvant or neoadjuvant platinum-chemo or platinum-chemo with definitive chemoradiation provided disease progression occurred ≥ 12 months from the last dose of treatment. What if the disease-free interval was ≥ 6 months? What is the minimum number of chemotherapy cycles that the patient must have to be eligible for continued durvalumab-tremelimumab? | According to the clinical experts, patients should receive a minimum of 1 chemotherapy cycle to be eligible for continued treatment with durvalumab and tremelimumab because the 2 drugs in the pivotal trial were administered in combination with chemotherapy. Discontinuation of chemotherapy in the event of a chemotherapy-related AE and continuation with durvalumab and tremelimumab would align with current clinical practice. |
Eligibility for re-treatment In the trial, patients who completed 5 cycles of durvalumab-tremelimumab combination therapy with clinical benefit but who had subsequent radiological progression during durvalumab maintenance could be re-treated with an additional 4 cycles of tremelimumab alongside durvalumab. What are the appropriate re-treatment criteria for durvalumab plus tremelimumab? If patients discontinue therapy for reasons other than toxicity or progressive disease and/or loss of clinical benefit, should patients be eligible for re-treatment? If yes, what re-treatment protocol and duration would be appropriate? | There is no precedent for re-treatment with a CTLA-4 inhibitor following progression; in the CheckMate 9LA regimen of nivolumab and ipilimumab, ipilimumab was not discontinued after a set number of cycles, but continued for the duration of therapy. As such, it was suggested by the clinical experts that appropriate re-treatment criteria for durvalumab and tremelimumab would align with the POSEIDON clinical trial at the clinician’s discretion. Re-treatment with tremelimumab following progression would be expected to be uncommon in clinical practice. Reasons for discontinuation of therapy other than toxicity or progressive disease and/or loss of clinical benefit would comprise patient choice and unforeseeable circumstances (e.g., a pandemic). In these cases, eligibility for re-treatment and the re-treatment protocol and duration would need to be determined and individualized on a case-by-case basis. |
Consistency with initiation criteria associated with other drugs reviewed by CDA-AMC in the same therapeutic space There are several other therapies in this treatment space previously reviewed by CDA-AMC, including pembrolizumab + PBC; ipilimumab and nivolumab + PBC; and cemiplimab + PBC. | Comment from the drug programs to inform pERC deliberations. |
Considerations for continuation or renewal of therapy | |
Consistency with renewal criteria associated with other drugs reviewed by CDA-AMC in the same therapeutic space | Comment from the drug programs to inform pERC deliberations. |
Considerations for discontinuation of therapy | |
Definition of loss of response, absence of clinical benefit, or disease progression In the trial, patients who experienced radiological disease progression during durvalumab maintenance could continue with treatment if they continued to derive benefit and met the trial criteria. What are the appropriate discontinuation criteria for durvalumab plus tremelimumab? | Clinical experts stated that appropriate discontinuation criteria for durvalumab and tremelimumab would comprise clear widespread progression that is not pseudoprogression and is not oligoprogression amenable to local therapies delivered with the intent of control of all areas of significant progression. |
Consistency with discontinuation criteria associated with other drugs reviewed by CDA-AMC in the same therapeutic space | Comment from the drug programs to inform pERC deliberations. |
Considerations for prescribing of therapy | |
Dosing, schedule/frequency, dose intensity In the POSEIDON trial, patients in the study arm received tremelimumab 75 mg and durvalumab 1,500 mg flat dosing. If therapy is funded and implemented, jurisdictions are likely to implement a weight-based durvalumab dose used for other funded indications (e.g., 20 mg/kg to a maximum of 1,500 mg per dose). | Comment from the drug programs to inform pERC deliberations. |
Consistency with prescribing criteria associated with other drugs reviewed by CDA-AMC in the same therapeutic space | Comment from the drug programs to inform pERC deliberations. |
Generalizability | |
Populations of interest matching the indication but with insufficient data Should the following patients be considered:
| According to the clinical experts, patients with locally advanced unresectable NSCLC of any stage should be considered if the disease is not amenable to any curative-intent option. The clinical experts stated that, based on the PAPILLON study, the preferred first-line treatment option for patients with metastatic NSCLC whose tumours have an exon 20 insertion mutation is chemotherapy and amivantamab. Immunotherapy-based treatments in this subgroup would not be recommended as immunotherapy is known to have little to no activity in patients whose tumours harbour an EGFR driver mutation. According to the clinical experts, for patients judged to be at high risk for AEs from platinum-based chemotherapy, there is insufficient evidence to consider the POSEIDON regimen of durvalumab and tremelimumab plus chemotherapy. Clinical experts stated that patients with an ECOG PS of 2 could be considered for treatment with durvalumab and tremelimumab plus chemotherapy (but not those with an ECOG PS of 3 or 4) if the perceived benefits exceed predicted risks. However, the POSEIDON trial included only patients with an ECOG PS of 0 or 1 at treatment initiation. |
Patients on active treatment with a time-limited opportunity to switch to the drug(s) under review Should patients currently on other chemo ± immuno-oncology treatments be allowed to switch over to durvalumab-tremelimumab? | One clinical expert stated that switching from another immuno-oncology treatment to durvalumab and tremelimumab plus chemotherapy would only make sense if there was toxicity to the former. The other noted that patients should only be allowed to switch to durvalumab and tremelimumab if there was a very serious infusion reaction. |
Funding algorithm | |
Request an initiation of a rapid provisional funding algorithm Note that if the final reimbursement recommendation for this drug under review is “Do not Reimburse,” the project will be suspended indefinitely. | Comment from the drug programs to inform pERC deliberations. |
Complex therapeutic space with multiple lines of therapy, subpopulations, or competing products | Comment from the drug programs to inform pERC deliberations. |
Other aspects Under what clinical circumstances will durvalumab-tremelimumab be preferred over other immuno-oncology ± platinum chemotherapy regimens? | Durvalumab and tremelimumab would typically be used in patients with tumoural PD-L1 < 50%. (This statement is only intended as information regarding how durvalumab and tremelimumab may be used in clinical practice; it does not mean that durvalumab and tremelimumab would necessarily be preferred over other immuno-oncology ± platinum chemotherapy regimens in this population). As the POSEIDON trial included patients with all PD-L1 expression levels, durvalumab and tremelimumab plus chemotherapy should not be restricted to those with tumoural PD-L1 < 50%. One clinical expert commented that durvalumab and tremelimumab plus chemotherapy may also be used in patients with rapidly progressive disease and a high tumour burden. The clinical experts stated that for patients with certain mutations (STK11, KRAS, KEAP) post hoc analyses suggest that durvalumab and tremelimumab may be active; this regimen may be preferred in patients with these mutations. |
Care provision issues | |
Drug preparation, storage, administration or dispensing Preparation of both durvalumab and tremelimumab is familiar to many jurisdictions because of use in other indication(s). | Comment from the drug programs to inform pERC deliberations. |
System and economic issues | |
Presence of confidential negotiated prices for comparators Confidential pricing exists for pembrolizumab, ipilimumab, and nivolumab. | Comment from the drug programs to inform pERC deliberations. |
AE = adverse event; CDA-AMC = Canada’s Drug Agency; ECOG PS = Eastern Cooperative Oncology Group Performance Status; NSCLC = non–small cell lung cancer; PBC = platinum-based chemotherapy; pERC = pan-Canadian Oncology Drug Review Expert Review Committee.
Clinical Evidence
The objective of this Clinical Review report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of durvalumab (50 mg/mL, concentrate for IV infusion) in combination with tremelimumab (20 mg/mL, concentrate for IV infusion) and platinum-based chemotherapy in the first-line treatment of adult patients with metastatic NSCLC but no sensitizing EGFR mutations or ALK genomic tumour aberrations. The focus is on comparing durvalumab in combination with tremelimumab and platinum-based chemotherapy to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of durvalumab in combination with tremelimumab and platinum-based chemotherapy is presented in 4 sections with the CDA-AMC critical appraisal of the evidence included at the end of each section. The first section, the systematic review, includes pivotal studies and randomized controlled trials (RCTs) that were selected according to the sponsor’s systematic review protocol. The CDA-AMC assessment of the certainty of the evidence in this first section using the GRADE approach follows the critical appraisal of the evidence. The third section includes indirect evidence from the sponsor. The sponsor did not include long-term extension studies (second section) or additional studies addressing important gaps in the pivotal and RCT evidence (fourth section).
Included Studies
Clinical evidence from 1 pivotal study identified in systematic review and 1 ITC is included in the review and appraised in this document.
Systematic Review
Contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
Description of Studies
Characteristics of the included study are summarized in Table 7.
Table 7: Details of Studies Included in the Systematic Review
Detail | POSEIDON study |
|---|---|
Designs and populations | |
Study design | Phase III, randomized, open-label, comparative, multicentre, global study |
Locations | The study was conducted at study centres in North and Latin America, Europe, Asia Pacific, and Africa. There were no Canadian study sites. Patients were recruited from 142 centres overall in Brazil (13 centres), Bulgaria (6 centres), Germany (10 centres), Hong Kong (1 centre), Hungary (5 centres), Japan (18 centres), South Korea (9 centres), Mexico (9 centres), Peru (5 centres), Poland (4 centres), Russia (9 centres), South Africa (7 centres), Taiwan (10 centres), Thailand (6 centres), Ukraine (10 centres), UK (5 centres), US (12 centres), and Vietnam (3 centres). |
Patient enrolment dates | First patient enrolled: June 1, 2017 First patient randomized: June 27, 2017 Last patient randomized: September 19, 2018 |
Randomized (N) | Total N = 1,013 n = 338 for durvalumab and tremelimumab + chemotherapy n = 338 for durvalumab + chemotherapya n = 337 for chemotherapy |
Inclusion criteria |
|
Exclusion criteria |
(Brain metastases were not recorded as RECIST 1.1 target lesions at baseline.)
|
Drugs | |
Intervention | Durvalumab and tremelimumab + chemotherapyc Combination stage:
Maintenance stage:
Durvalumab + chemotherapya,f Combination stage:
Maintenance stage:
|
Comparator(s) | Chemotherapy:
Patients could receive 2 additional doses of chemotherapy at weeks 12 and 15 (a total of 6 doses postrandomization), as clinically indicated. Patients with nonsquamous tumour histology who received carboplatin or cisplatin + pemetrexed and who had not progressed after 4 to 6 cycles of carboplatin or cisplatin + pemetrexed could receive pemetrexed maintenance therapy q.3.w. or q.4.w. |
Study duration | |
Screening phase | 4 weeks (28 days) |
Treatment phase | During chemotherapy (combination stage): 12 weeks Following chemotherapy (maintenance stage): until disease progression |
Follow-up phase | Following discontinuation or completion of treatment, patients were followed for survival at 8, 12, and 16 weeks, and then every 8 weeks until the end of the study |
Outcomes | |
Primary end point | Dual primary end pointsa
|
Secondary end points | Key alpha-controlled secondary
Secondary
Safety
|
Publication status | |
Publications | Johnson et al. (2023)39 Peters et al. (2024)40 Garon et al. (2023)41 Garon et al. (2024)42 NCT03164616 trial |
AE = adverse event; AJCC = American Joint Committee on Cancer; ALT = alanine transaminase; AST = aspartate transaminase; AUC = area under the curve; BICR = blinded independent central review; bTMB = blood tumour mutational burden; CrCL = creatinine clearance; ECOG PS = Eastern Cooperative Oncology Group Performance Status; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-LC13 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Lung Cancer 13; HBsAg = hepatitis B surface antigen; HBV = hepatitis B virus; HCV = hepatitis C virus; IASLC = International Association for the Study of Lung Cancer; ICF = informed consent form; ILD = interstitial lung disease; NSCLC = non–small cell lung cancer; OS = overall survival; PFS = progression-free survival; PFS2: time from randomization to second progression; q.3.w. = every 3 weeks; q.4.w = every 4 weeks; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1; ULN = upper limit normal; SAE = serious adverse event; vs. = versus.
aEfficacy data for the durvalumab plus chemotherapy arm of the POSEIDON trial will not be presented in the systematic review because this combination is not seeking approval in Canada and therefore is not aligned with the Health Canada product monograph.
bThe IASLC is the group responsible for conducting research to inform future versions of the AJCC. Version 8 of the IASLC Staging Manual in Thoracic Oncology was endorsed and published by the AJCC as the AJCC 8th edition.15,43,44 The 2 are considered interchangeable.45,46
cPatients whose weight fell to 30 kg or below received weight-based dosing of 20 mg/kg of durvalumab and 1 mg/kg of tremelimumab q.3.w. (in the combination stage) or q.4.w. (in the maintenance stage) until their weight improved to greater than 30 kg, at which point the patient started receiving the fixed dosing of 1,500 mg durvalumab at 75 mg tremelimumab.
dPatients were treated until clinical or radiological progression unless there was unacceptable toxicity, consent withdrawal, or another discontinuation criterion was met.
ePatients with nonsquamous tumour histology who received carboplatin or cisplatin plus pemetrexed and who had not progressed after 4 cycles of carboplatin or cisplatin plus pemetrexed could receive pemetrexed maintenance therapy, unless contraindicated as determined by the investigator.
fPatients whose weight fell to 30 kg or below received weight-based dosing of 20 mg/kg of durvalumab q.3.w. (in the combination stage) or q.4.w. (in the maintenance stage) until the weight improved to greater than 30 kg, at which point the patient started receiving the fixed dosing of durvalumab at 1,500 mg.
Sources: Sponsor’s Summary of Clinical Evidence,1 POSEIDON Clinical Study Report,38 POSEIDON Clinical Study Protocol,47 and POSEIDON Statistical Analysis Plan.48
The POSEIDON trial was a phase III, randomized, open-label, multicentre, global study investigating the efficacy and safety of durvalumab and tremelimumab plus SOC chemotherapy versus SOC chemotherapy alone or durvalumab monotherapy plus SOC chemotherapy versus SOC chemotherapy alone for the first-line treatment in patients (total N = 1,013) with metastatic NSCLC with tumours without activating EGFR mutations and ALK fusions.1,38,47 There were no study sites in Canada.1,38
Patients were randomized 1:1:1 by interactive voice response system (IVRS) or interactive web response system (IWRS) over a 15-month period to 1 of 3 study arms: durvalumab and tremelimumab plus SOC chemotherapy (arm 1; n = 338); durvalumab plus SOC chemotherapy (arm 2; n = 338); or SOC chemotherapy alone (arm 3; n = 337). SOC chemotherapy consisted of the investigator’s choice of 1 of the following regimens: nab-paclitaxel plus carboplatin (patients with squamous and nonsquamous tumour histology); pemetrexed plus cisplatin or carboplatin (patients with nonsquamous tumour histology only); or gemcitabine plus cisplatin or carboplatin (patients with squamous tumour histology only). Randomization was stratified according to PD-L1 tumour-expression status (TCs ≥ 50% versus < 50%), disease stage (IVA versus IVB), and histology (nonsquamous versus squamous).1,38 The study design for the POSEIDON trial is presented in Figure 2.
The primary objective of the POSEIDON trial was to assess the efficacy of durvalumab plus SOC chemotherapy compared to SOC chemotherapy alone in terms of dual primary end points of PFS and OS in all patients. The key secondary objectives were to assess the efficacy of durvalumab and tremelimumab plus SOC chemotherapy compared to SOC chemotherapy alone in terms of PFS and OS in all patients.1,38
The study arm that evaluated durvalumab plus SOC chemotherapy is not aligned with the indication approved by Health Canada and therefore results for this arm are not reported in this CDA-AMC review. Only results from the comparison of durvalumab and tremelimumab plus SOC chemotherapy versus SOC chemotherapy alone are presented in this review and include results from the final analysis (DCO: July 24, 2019, for PFS and other RECIST 1.1–related end points; DCO: March 12, 2021, for OS, safety, and all other data). Results from an updated 5-year OS analysis (DCO: August 24, 2023) have also been reported for this comparison.1,38 The POSEIDON trial was supported by AstraZeneca.39
The POSEIDON trial included a 4-week screening period to complete eligibility assessments, during which informed consent was obtained, patient data and medical history were obtained, and various laboratory and baseline assessments were performed.47 The choice of SOC chemotherapy was patient-specific based on the treatment most appropriate for the patient and was selected before receiving a randomization number in the IVRS or IWRS. The trial comprised 2 treatment periods following randomization, referred to as the combination (during chemotherapy) stage and the maintenance (post−chemotherapy) stage. In the combination stage, patients in arm 1 received treatment with 4 cycles of durvalumab and tremelimumab plus SOC chemotherapy and patients in arm 3 received 4 cycles of SOC chemotherapy. In the maintenance stage, patients in arm 1 received treatment with durvalumab until disease progression as well a single additional dose of tremelimumab; patients with nonsquamous histology may have also received pemetrexed maintenance therapy until disease progression. In the maintenance stage, patients in arm 3 could receive an additional 2 cycles of SOC chemotherapy, if clinically indicated, before entering follow-up; patients with nonsquamous histology could also receive pemetrexed maintenance therapy. Dose reductions of durvalumab and tremelimumab were not permitted.1,38 Crossover was not permitted as part of the study.38 Continuation of durvalumab monotherapy beyond disease progression was permitted for patients who continued to receive a benefit and met the criteria to remain on treatment. Patients who had received 5 cycles of durvalumab plus tremelimumab and then experienced disease progression during durvalumab monotherapy were permitted to receive re-treatment once with up to 4 additional cycles of tremelimumab alongside durvalumab.1,39,47 Tumour evaluation for patients in all treatment arms was by CT scan or MRI, with scans performed at screening (as baseline), week 6 (± 1 week) and week 12 (± 1 week) from the date of randomization, and then every 8 weeks (± 1 week) until radiological progression.1,38,47
Figure 2: Study Design for the POSEIDON Trial
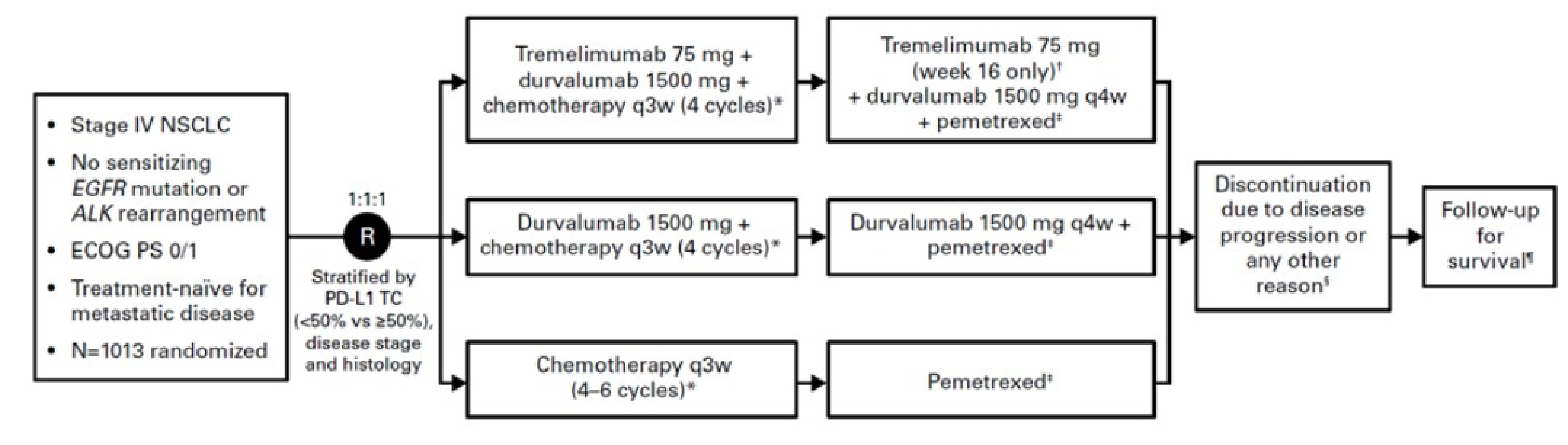
ECOG PS = Eastern Cooperative Oncology Group Performance Status; NSCLC = non–small cell lung cancer; q3w = every 3 weeks; q4w = every 4 weeks; TC = tumour cell.
* Chemotherapy options comprised nab-paclitaxel plus carboplatin (patients with either squamous or nonsquamous histology); gemcitabine plus cisplatin or carboplatin (patients with squamous histology); and pemetrexed plus cisplatin or carboplatin (patients with nonsquamous histology). The choice of chemotherapy was at the discretion of the investigator.
† Patients received an additional dose of tremelimumab postchemotherapy (total of 5 durvalumab plus tremelimumab combination doses).
‡ Patients with nonsquamous histology who received pemetrexed-platinum doublet and who had not progressed after 4 to 6 cycles received pemetrexed maintenance therapy unless contraindicated by the investigator.
§ Patients continued treatment until disease progression, unacceptable toxicity, or withdrawal of consent. In the durvalumab plus tremelimumab and chemotherapy arm and the durvalumab plus chemotherapy arm, continuation of durvalumab monotherapy after objective radiological progression was permitted at the investigator’s discretion if there was evidence of clinical benefit and the patient met the criteria for treatment in the setting of progressive disease. Patients allocated to durvalumab plus tremelimumab and chemotherapy who had completed 5 dosing cycles of durvalumab plus tremelimumab combination therapy with clinical benefit per investigator judgment, but subsequently had radiological progression during durvalumab monotherapy, could receive re-treatment with up to 4 additional cycles of tremelimumab alongside durvalumab.
¶ Anticancer therapy utilized after discontinuation of study treatment was recorded as part of the follow-up schedule.
Source: Sponsor’s Summary of Clinical Evidence.1
Populations
Inclusion and Exclusion Criteria
Detailed key inclusion and exclusion criteria are provided in Table 7. Patients eligible for participation in the POSEIDON trial were aged 18 years or older with histologically or cytologically documented stage IV NSCLC (according to Version 8 of the IASLC Staging Manual in Thoracic Oncology [2016]44) not amenable to curative surgery or radiation with tumours that lacked activating EGFR mutations and ALK fusions. Patients had not received prior chemotherapy or any other systemic therapy for metastatic NSCLC. Patients who had received prior platinum-containing adjuvant, neoadjuvant, or definitive chemoradiation for advanced disease were eligible if progression had occurred more than 12 months after completing therapy. Patients had no prior exposure to immune-mediated therapy (including other anti–CTLA-4, anti–PD-1, anti–PD-L1, and anti–PD-L2 antibodies). Participants were required to have a WHO performance status or ECOG PS of 0 or 1. Patients who had suspected brain metastases at screening were required to have an IV contrast-enhanced MRI or CT scan of the brain before study entry. Those with brain metastases were required to be treated before randomization and were eligible if a stable condition was confirmed after the intervention, there was a return neurologically to baseline, and associated steroid treatment had been completed at least 5 days before randomization. Patients with active or prior documented autoimmune or inflammatory disorders (e.g., colitis, Crohn disease, systemic lupus erythematosus, rheumatoid arthritis), with some exceptions (e.g., vitiligo, alopecia, and chronic skin conditions not requiring systemic treatment), were not eligible for the trial.1,38
Interventions
In the POSEIDON trial, patients were randomized 1:1:1, to 1 of 3 treatment arms:1,38
arm 1: Durvalumab and tremelimumab plus SOC chemotherapy (n = 338)
arm 2: Durvalumab plus SOC chemotherapy (n = 338)
arm 3: SOC chemotherapy alone (n = 337).
Arm 1: Durvalumab and Tremelimumab Plus SOC Chemotherapy
Combination (During Chemotherapy) Stage1,38
Durvalumab, 1,500 mg by 60-minute IV infusion, every 3 weeks at weeks 0, 3, 6, and 9
Tremelimumab, 75 mg by 60-minute IV infusion, every 3 weeks at weeks 0, 3, 6, and 9
SOC chemotherapy for 4 cycles:
nab-paclitaxel (patients with squamous and nonsquamous tumour histology) (100 mg/m2) IV infusion on days 1, 8, and 15 of each 21-day cycle plus carboplatin (area under the plasma drug concentration-time curve [AUC] 5 or 6) IV infusion on day 1 of each 21-day cycle, for 4 cycles; or
gemcitabine (patients with squamous tumour histology only) (1,000 mg/m2 or 1,250 mg/m2) IV infusion on days 1 and 8 of each 21-day cycle plus cisplatin (75 mg/m2) or carboplatin (AUC 5 or 6) IV infusion on day 1 of each 21-day cycle, for 4 cycles; or
pemetrexed (patients with nonsquamous tumour histology only) (500 mg/m2) IV infusion plus cisplatin (75 mg/m2) or carboplatin (AUC 5 or 6) IV infusion on day 1 of each 21-day cycle, for 4 cycles.
If a dosing delay occurred during chemotherapy while on the every 3 weeks schedule, all future dosing days were delayed to ensure that the intervals between study treatment doses were always a minimum of 21 days.1,38
Maintenance (Postchemotherapy) Stage1,38
Durvalumab, 1,500 mg by 60-minute IV infusion, every 4 weeks from week 12 to disease progression
Patients were treated until clinical or radiological progression unless there was unacceptable toxicity, consent withdrawal, or another criterion for discontinuation was met.
Tremelimumab, 75 mg by 60-minute IV infusion, 1 dose at week 16
In the case of dose delay(s), more than 1 tremelimumab plus durvalumab combination dose could be administered at and after week 16 postchemotherapy to ensure that up to 5 combination doses were given. If patients received fewer than 4 cycles of platinum doublet chemotherapy, the remaining cycles of combined durvalumab and tremelimumab (up to a total of 5) were to be given after a combination of platinum doublet chemotherapy (with maintenance pemetrexed, if applicable).
Pemetrexed (patients with nonsquamous tumour histology only), 500 mg/m2 by IV infusion, every 4 weeks from week 12 to disease progression
Patients with nonsquamous tumour histology who received pemetrexed plus carboplatin or cisplatin and who had not progressed after 4 cycles of pemetrexed plus carboplatin or cisplatin could receive maintenance therapy with pemetrexed, unless contraindicated as determined by the investigator.
Patients were treated until clinical progression or RECIST 1.1–defined radiological progression unless there was unacceptable toxicity or consent withdrawal or another criterion for discontinuation was met.
In both stages, weight-based doses were received by patients whose weight fell to 30 kg or less, equivalent to 20 mg/kg of durvalumab and 1 mg/kg of tremelimumab (every 3 weeks in the combination stage, every 4 weeks in the maintenance stage) until the weight improved to greater than 30 kg, at which point the fixed dosing of durvalumab at 1,500 mg and tremelimumab at 75 mg was started.38 Dose reductions of durvalumab and tremelimumab were not permitted.1,38
Arm 2: Durvalumab Plus SOC Chemotherapy
Combination (During Chemotherapy) Stage1,38
Durvalumab, 1,500 mg by 60-minute IV infusion, every 3 weeks at weeks 0, 3, 6, and 9
SOC chemotherapy for 4 cycles
nab-paclitaxel (patients with squamous and nonsquamous tumour histology) (100 mg/m2) IV infusion on days 1, 8, and 15 of each 21-day cycle plus carboplatin (AUC 5 or 6) IV infusion on day 1 of each 21-day cycle, for 4 cycles; or
gemcitabine (patients with squamous tumour histology only) (1,000 mg/m2 or 1,250 mg/m2) IV infusion on days 1 and 8 of each 21-day cycle plus cisplatin (75 mg/m2) or carboplatin (AUC 5 or 6) IV infusion on day 1 of each 21-day cycle, for 4 cycles; or
pemetrexed (patients with nonsquamous tumour histology only) (500 mg/m2) IV infusion plus cisplatin (75 mg/m2) or carboplatin (AUC 5 or 6) IV infusion on day 1 of each 21-day cycle, for 4 cycles.
If a dosing delay occurred during chemotherapy while on the every 3 weeks schedule, all future dosing days were delayed to ensure that the intervals between dosing study treatment were always a minimum of 21 days.1,47
Maintenance (Postchemotherapy) Stage1,38
Durvalumab, 1,500 mg by 60-minute IV infusion, every 4 weeks from week 12 to disease progression
Patients were treated until clinical or radiological progression unless there was unacceptable toxicity, consent withdrawal, or another criterion for discontinuation was met.
Pemetrexed (patients with nonsquamous tumour histology only), 500 mg/m2 by IV infusion, every 4 weeks from week 12 to disease progression
Patients with nonsquamous tumour histology who received pemetrexed plus carboplatin or cisplatin and who had not progressed after 4 cycles of pemetrexed plus carboplatin or cisplatin could receive maintenance therapy with pemetrexed, unless contraindicated per the investigator.
Patients were treated until clinical progression or RECIST 1.1–defined radiological progression unless there was unacceptable toxicity or consent withdrawal or another criterion for discontinuation was met.
In both stages, weight-based dosing was received by patients whose weight fell to 30 kg or less, equivalent to 20 mg/kg of durvalumab every 3 weeks in the combination stage and every 4 weeks in the maintenance stage, until the weight improved to greater than 30 kg, at which point the fixed dosing of durvalumab at 1,500 mg was started.38 Dose reductions of durvalumab were not permitted.1,38
Results from the durvalumab plus SOC chemotherapy arm are not included in the systematic review because this regimen is not part of the Health Canada–approved indication and reimbursement request under review by CDA-AMC.
Arm 3: SOC Chemotherapy1,38
Nab-paclitaxel (patients with squamous and nonsquamous tumour histology) (100 mg/m2) IV infusion on days 1, 8, and 15 of each 21-day cycle plus carboplatin (AUC 5 or 6) IV infusion on day 1 of each 21-day cycle, for 4 cycles
Gemcitabine (patients with squamous tumour histology only) (1,000 mg/m2 or 1,250 mg/m2) IV infusion on days 1 and 8 of each 21-day cycle plus cisplatin (75 mg/m2) or carboplatin (AUC 5 or 6) IV infusion on day 1 of each 21-day cycle, for 4 cycles
Pemetrexed (patients with nonsquamous tumour histology only) (500 mg/m2) IV infusion plus cisplatin (75 mg/m2) or carboplatin (AUC 5 or 6) IV infusion on day 1 of each 21-day cycle, for 4 cycles
An additional 2 doses of SOC chemotherapy (weeks 12 and 15) could have been given at the investigator’s discretion, if clinically indicated.
Patients with nonsquamous tumour histology who received pemetrexed plus carboplatin or cisplatin and who had not progressed after 4 to 6 cycles of pemetrexed plus carboplatin or cisplatin could receive maintenance therapy with pemetrexed given every 3 weeks or every 4 weeks.
If a dosing delay occurred during chemotherapy while on the every 3 weeks schedule, all future dosing days were delayed to ensure that the intervals between dosing study treatment were always a minimum of 21 days.38
A summary of the overall dosing scheme for POSEIDON trial is provided in Table 8.
Table 8: POSEIDON Trial Dosing Scheme
Treatment arm | Combination (during chemotherapy) stage 1 cycle = 3 weeks (21 days) | Maintenance (postchemotherapy) stage 1 cycle = 4 weeks (28 days) | |||||
|---|---|---|---|---|---|---|---|
Cycle 1 week 0 | Cycle 2 week 3 | Cycle 3 week 6 | Cycle 4 week 9 | Week 12 | Week 16 | Week 20 to progressive disease | |
D + T + chemotherapy (arm 1) | D + T + chemotherapy | D + T + chemotherapy | D + T + chemotherapy | D + T + chemotherapy | D + pemea | D + Tb + pemea | D + pemea |
D + chemotherapyc (arm 2) | D + chemotherapy | D + chemotherapy | D + chemotherapy | D + chemotherapy | D + pemea | D + pemea | D + pemea |
Chemotherapy (arm 3) | Chemotherapy | Chemotherapy | Chemotherapy | Chemotherapyd | pemea | pemea | pemea |
D = durvalumab; peme = pemetrexed; q.3.w. = every 3 weeks; q.4.w. = every 4 weeks; t = tremelimumab.
Note: Response Evaluation Criteria in Solid Tumours Version 1.1 assessment was performed at week 12 ± 1 week from the date of randomization, and every 8 weeks ± 1 week thereafter until radiological progression (regardless of whether q.3.w. or q.4.w. regimen was chosen). Weight-based dosing was received by patients whose weight fell to 30 kg or less, equivalent to durvalumab 20 mg/kg and tremelimumab 1 mg/kg until the weight improved to greater than 30 kg, at which point the fixed dosing of durvalumab 1,500 mg and tremelimumab 75 mg was started. The durvalumab dose was 1,500 mg during chemotherapy and 1,500 mg postchemotherapy; the tremelimumab dose was 75 mg.
aPemetrexed maintenance therapy was for patients with nonsquamous NSCLC who received treatment with pemetrexed and carboplatin or cisplatin during chemotherapy and did not progress after 4 to 6 cycles, unless contraindicated as determined by the investigator. Pemetrexed maintenance therapy could have been given q.3.w. or q.4.w. (i.e., q.4.w. for arms 1 and 2; for arm 3, pemetrexed maintenance therapy could have been given either q.3.w. or q.4.w. depending on investigator decision and local standards, unless contraindicated per the investigator).
bFor patients in arm 1, an additional dose of durvalumab and tremelimumab was given at week 16 postchemotherapy. In the case of dose delay(s), more than 1 durvalumab and tremelimumab combination dose could have been given at and after week 16 postchemotherapy to ensure that up to 5 combination doses were administered in arm 1. If patients received fewer than 4 cycles of platinum doublet chemotherapy, the remaining cycles of combined durvalumab and tremelimumab (up to a total of 5) were to be given after combination of platinum doublet chemotherapy (with maintenance pemetrexed, if applicable).
cThe durvalumab plus chemotherapy arm is not included in the systematic review because this regimen is not approved by Health Canada.
dIn arm 3, chemotherapy could have been given for an additional 2 cycles q.3.w. on weeks 12 and 15 (i.e., a total of 6 cycles postrandomization) if clinically indicated, at the investigator’s discretion before patients entered follow-up. This did not alter the planned scan schedule of every 8 weeks starting at week 12 for patients in arm 3.
Sources: Sponsor’s Summary of Clinical Evidence1 and POSEIDON Clinical Study Report.38
Duration of Treatment
Treatment with chemotherapy in arms 1 and 2 was limited to 4 cycles administered at 3-week intervals subsequent to randomization. Patients in arm 3 (SOC chemotherapy alone) could receive an additional 2 doses of SOC chemotherapy at weeks 12 and 15 (a total of 6 doses postrandomization), as clinically indicated, at the investigator’s discretion. Treatment with immunotherapy plus SOC chemotherapy in arms 1 and 2, as well as treatment with SOC chemotherapy alone in arm 3, was administered beginning on cycle 1 day 1.1,38,47
For patients randomized to arms 1 and 2, treatment with durvalumab monotherapy continued until clinical or radiological progression, unless there was unacceptable toxicity or consent withdrawal, or another criterion for discontinuation was met. Patients in arm 1 received an additional dose of durvalumab and tremelimumab at week 16 postchemotherapy. If dose delay(s) occurred, more than 1 dose of the durvalumab and tremelimumab combination could have been given at and after week 16 postchemotherapy to ensure that up to 5 combination doses were administered in arm 1. If patients received fewer than 4 cycles of platinum doublet chemotherapy, the remaining cycles of combined durvalumab and tremelimumab (up to a total of 5) were to be given after a combination of platinum doublet chemotherapy (with maintenance pemetrexed if applicable). All patients with nonsquamous tumour histology who received a pemetrexed doublet in the initial part of the study were to receive maintenance therapy with pemetrexed in the postchemotherapy phase of the study, unless contraindicated as determined by the investigator.1,38,47
In arms 1 and 2, when SOC chemotherapy was discontinued because of treatment-related toxicity, patients could continue to receive durvalumab monotherapy or durvalumab plus tremelimumab at the investigator’s discretion when toxicity resolved to at least grade 2 or less.1,38,47
Treatment Through Progression (Arms 1 and 2)
In arms 1 and 2, patients with objective radiological progression who continued to receive a benefit from their assigned treatment (in the investigator’s opinion) and who met the criteria for treatment in the setting of disease progression could continue to receive durvalumab monotherapy for as long as they were gaining clinical benefit.1,38,47
Patients with rapid tumour progression or with symptomatic progression requiring urgent medical intervention (e.g., CNS metastasis, respiratory failure because of tumour compression, spinal cord compression) were not eligible to continue any study treatment.1,38,47
In arms 1 and 2, the investigator ensured that all patients who were treated through progression did not have any significant, unacceptable, or irreversible toxicities that indicated continuing treatment would not further benefit the patient. Patients could continue receiving their assigned treatment in the setting of unconfirmed disease progression, at the investigator’s discretion, until disease progression was confirmed with a subsequent scan.1,38,47
Re-Treatment (Arm 1)
Patients in arm 1 (durvalumab and tremelimumab plus SOC chemotherapy) with radiological progression who, in the investigator’s opinion, continued to receive a benefit from their assigned treatment and who met the criteria for re-treatment in the setting of disease progression, could have re-treatment with durvalumab and tremelimumab combination therapy.1,38 Patients continued durvalumab dosing at 1,500 mg every 4 weeks with tremelimumab at 75 mg every 4 weeks for 4 doses per cycle each and then continued with durvalumab monotherapy at 1,500 mg every 4 weeks (with weight-based dosing used for patients whose weight fell to 30 kg or below).47
Patients with rapid tumour progression or with symptomatic progression that required urgent medical intervention (e.g., CNS metastasis, respiratory failure because of tumour compression, or spinal cord compression) were not eligible to begin re-treatment with durvalumab and tremelimumab combination therapy.1,38
In arm 1, the investigator ensured that all patients who began re-treatment did not have any significant, unacceptable, or irreversible toxicities that indicated restarting treatment would not further benefit the patient. Patients who met the re-treatment criteria followed the same treatment guidelines followed during the original, every 4 weeks, postchemotherapy-maintenance treatment period. Those who met the re-treatment criteria were permitted to receive re-treatment only once.1,38
Treatment Through Progression and Re-Treatment (Arm 3)
In arm 3, treatment through progression and re-treatment was not permitted. Crossing over to durvalumab and tremelimumab plus SOC chemotherapy or durvalumab plus SOC chemotherapy was not permitted following objective disease progression. The investigator could offer other therapies following objective disease progression, and patients were still followed for survival if another treatment for NSCLC was initiated.1,38,47
Discontinuation of Study Treatment
Reasons for discontinuation of any further study treatment (i.e., durvalumab and tremelimumab combination therapy, durvalumab monotherapy, or SOC chemotherapy) included clinical progression (investigator determination that the patient is no longer benefiting from treatment in arms 1, 2, and 3), radiological progressive disease (based on RECIST 1.1 criteria and investigator determination that the patient is no longer benefiting from treatment in arms 1 and 2), RECIST 1.1-defined radiological progression (arm 3), AE meeting discontinuation criteria, consent withdrawal, and other reasons (e.g., nonadherence to study protocol or initiation of an alternative anticancer therapy).38,47 Patients who discontinued study treatment for reasons other than objective disease progression continued to receive RECIST 1.1 scans until RECIST 1.1–defined radiological progression plus an additional subsequent scan, or death (whichever came first). Survival status was assessed for all patients until the end of the study, including those who initiated alternative treatment for their NSCLC.38
Concomitant Medications
Concomitant medications that were permitted included:47
medications necessary for prophylactic or supportive care (e.g., acetaminophen, diphenhydramine), except those identified as prohibited
best supportive care (e.g., antibiotics, optimal symptom control, pain management [including palliative radiotherapy to nontarget lesions])
inactivated viruses (e.g., influenza vaccine).
Concomitant medications that were prohibited for all treatment arms included:47
other investigational anticancer therapies
other monoclonal antibodies against CTLA4, PD-1, or PD-L1
other concurrent chemotherapy, radiotherapy, immunotherapy, or biologic or hormonal therapy for cancer treatment (local treatment of isolated lesions excluding target lesions for palliative intent was acceptable by radiotherapy or surgically)
live attenuated vaccines (not to be given until 30 days following the last dose of study treatment).
Concomitant medications that were prohibited for arms 1 and 2 included:
immunosuppressive medications, with some exceptions (e.g., management of study treatment-related AEs; short-term premedication as required by prescribing information for those receiving combination agents SOC; inhaled, topical, or intranasal corticosteroids; temporary management of non–immunotherapy-related events)
medications with laxative properties, herbal or natural remedies for constipation (use with caution up until 90 days following the last dose of tremelimumab)
sunitinib (not to be used concomitantly or up until 90 days following the last dose of tremelimumab)
epidermal growth factor receptor tyrosine kinase inhibitors (not to be used concomitantly and to be used with caution in the 90 days following the last dose of durvalumab)
herbal or natural remedies with the potential for immune-modulating effects.
Outcomes
A list of efficacy and safety end points included in this Clinical Review report is provided in Table 9, followed by descriptions of the outcome measures. Summarized end points are based on outcomes included in the sponsor’s Summary of Clinical Evidence as well as any outcomes identified as important by the clinical experts consulted for this review and input from patient and clinician groups and public drug plans. Using the same considerations, we selected end points considered to be most relevant to inform expert committee deliberations and finalized this list of end points in consultation with members of the expert committee. Select efficacy end points and notable harms outcomes considered important for informing expert committee deliberations were assessed using GRADE.
Table 9: Outcomes Summarized From the Study Included in the Systematic Review
Outcome measure | Time point | POSEIDON studya |
|---|---|---|
OS (D + T + chemotherapy vs. chemotherapy) | From randomization until death because of any cause | Key secondaryb |
OS, Kaplan-Meier survival probability | 12 months | OS was a key secondary end point |
18 months | ||
24 months | ||
36 months | ||
48 months | ||
60 months | ||
PFS (D + T + chemotherapy vs. chemotherapy) | From randomization until the date of objective disease progression or death (by any cause in the absence of progression) regardless of whether the patient withdraws from randomized therapy or receives another anticancer therapy before progression | Key secondaryb |
PFS at 12 months, Kaplan-Meier estimate | At 12 months | PFS was a key secondary end point |
ORR (using unconfirmed responsesc) | Data obtained up until progression, or last evaluable assessment in the absence of progression, were included in the assessment of ORR; patients who discontinued randomized treatment without progression, received a subsequent anticancer therapy (radiotherapy was not considered a subsequent anticancer therapy for this analysis), and then responded were not included as responders in the ORR | Secondary |
DoR | Time from the date of first documented response until the first date of documented progression or death in the absence of disease progression | Secondary |
BoR | BoR was calculated based on the overall visit responses from each RECIST 1.1 assessment and was the best response a patient had during their time in the study up until RECIST progression or the last evaluable assessment in the absence of RECIST 1.1 progression, as determined by BICR | Secondary |
Time to deterioration (EORTC QLQ-C30 and EORTC QLQ-LC13 end points) | From the date of randomization until the date of the first clinically meaningful deterioration that is confirmed at a subsequent visit or death (by any cause) in the absence of a clinically meaningful deterioration, regardless of whether the patient withdraws from study treatment or receives another anticancer therapy before HRQoL or function deterioration | Secondary |
Function improvement rate (EORTC QLQ-C30 end points) | Function improvement rate was defined as the number (percentage) of patients with 2 consecutive assessments at least 14 days apart that showed a clinically meaningful improvement in that function from baseline | Secondary |
Symptom improvement Rate (EORTC QLQ-C30 and EORTC QLQ-LC13 end points) | Symptom improvement rate was defined as the number (percentage) of patients with 2 consecutive assessments at least 14 days apart that showed a clinically meaningful improvement in that symptom from baseline | Secondary |
Incidence of AEs, SAEs, and AEs of special interest | Collected throughout the study | Harms |
AE = adverse event; BICR = blinded independent central review; BoR = best objective response; D = durvalumab; DoR = duration of response; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-LC13 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Lung Cancer 13; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1; SAE = serious adverse event; SOC = standard of care; t = tremelimumab; vs. = versus.
aThe primary end point was a dual end point of OS and PFS for the durvalumab plus SOC chemotherapy vs. SOC chemotherapy comparison. Because this combination is not aligned with the Health Canada product monograph indication for durvalumab, results for the combination of durvalumab plus SOC chemotherapy are not presented in this review.
bStatistical testing for these end points was adjusted for multiple comparisons (hierarchal testing).
cThe prespecified secondary outcome of ORR (based on BICR) using unconfirmed responses was defined as the number (percentage) of patients with at least 1 visit response of complete response or partial response. Post hoc sensitivity analysis of ORR (based on BICR) was conducted using confirmed responses; confirmed ORR was defined as the number (percentage) of patients with at least 1 visit response of complete response or partial response and a confirmatory scan no sooner than 4 weeks after the initial complete response or partial response.
Sources: Sponsor’s Summary of Clinical Evidence,1 POSEIDON Clinical Study Report,38 POSEIDON Study Protocol,47 and the sponsor’s Summary of Clinical Evidence.
Survival Assessments
In the POSEIDON trial, survival assessments were made for patients who completed or discontinued treatment at months 2, 3, and 4, and then every 2 months (8 weeks ± 2 weeks) following the last dose of study treatment. Patients on treatment or in survival follow-up were also contacted to provide survival data following the DCO for the primary and all subsequent survival analyses.47
Tumour Assessments
Tumour evaluation was performed at screening (as baseline), at week 6 (± 1 week) and week 12 (± 1 week) from the date of randomization, and then every 8 weeks (± 1 week) until radiological disease progression. RECIST 1.1 assessments were performed on images from CT scans (preferred) or MRI scans.1,38,47 Patients who discontinued study treatment because of toxicity without objective disease progression continued with scheduled tumour assessments until disease progression or death.47,48 End points of PFS, ORR, DoR, and BoR were derived using a BICR of all radiologic scans, according to RECIST 1.1.47,48
Primary End Point
The primary objective of the POSEIDON trial was to assess the efficacy of durvalumab plus SOC chemotherapy compared to SOC chemotherapy alone in terms of dual primary end points of PFS and OS.1,38,48 The primary end points are not the focus of this review because this objective is outside the scope of the approved Health Canada indication and the sponsor’s reimbursement request.
Key Secondary End Points
Key secondary end points of PFS and OS were assessed for the comparison of durvalumab and tremelimumab plus SOC chemotherapy versus SOC chemotherapy alone.38 These end points were alpha-controlled and included in the hierarchical statistical analysis plan.1
Overall Survival
OS was defined as the time from randomization to death because of any cause.1,47 Any patients not known to have died at the time of analysis were censored at the date last known to be alive. If confirmation of a patient being alive or having died occurred after the DCO date, patients were censored at the DCO date.47 The sponsor stated that there is no reported MID for OS in the first-line treatment of metastatic NSCLC.1
Progression-Free Survival
PFS was defined as the time from the date of randomization until the date of objective disease progression or death (because of any cause in the absence of disease progression), regardless of whether the patient withdraws from study treatment or receives another anticancer treatment before progression. PFS was evaluated by BICR according to RECIST 1.1,1,47 which defines progressive disease as a 20% or greater increase in the sum of diameters of target lesions in addition to an absolute increase of 5 mm or more (with reference to the smallest sum of diameters since initiation of treatment, including the baseline sum if this is the smallest sum on study). Progressive disease is also characterized by fulfillment of progression criteria for nontarget lesions or by the appearance of 1 or more new lesions.47,48 Patients who did not have disease progression or death at the time of analysis were censored at the time of the latest date of assessment from their last evaluable RECIST 1.1 assessment. Patients who experienced disease progression or death following 2 or more missed visits were censored at the time of the latest evaluable RECIST 1.1 assessment.47,48 The sponsor stated that there is no reported MID for PFS in the first-line treatment of metastatic NSCLC.1
Other Secondary End Points
Objective Response Rate
ORR was defined as the number (percentage) of patients with at least 1 visit response of complete response (CR) or partial response (PR). Data obtained up until progression, or the last evaluable assessment in the absence of progression, were included in the assessment of ORR.1,47 Patients who discontinued study treatment without disease progression, received subsequent (nonradiotherapy) anticancer treatment, and then responded were not included as responders in the ORR.47,48 The prespecified secondary outcome of ORR (based on BICR) using unconfirmed responses was defined as the number (percentage) of patients with at least 1 visit response of CR or PR.1,38,47 Post hoc sensitivity analysis of ORR (based on BICR) was conducted using confirmed responses; confirmed ORR was defined as the number (percentage) of patients with at least 1 visit response of CR or PR and a confirmatory scan no sooner than 4 weeks after the initial CR or PR.1,38 The sponsor stated that there is no reported MID for ORR in the first-line treatment of metastatic NSCLC.1
Duration of Response
DoR was defined as the time from the date of first documented response until the first date of documented progression or death in the absence of disease progression.1,47 For patients who did not progress following a response, PFS censoring time was used for their DoR. For patients who did not have a documented response, DoR was not defined.47 The sponsor stated that there is no reported MID for DoR in the first-line treatment of metastatic NSCLC.1
Best Objective Response
BoR was defined as the best response a patient had during the study but before starting any subsequent cancer treatment, and up until RECIST 1.1 progression or the last evaluable assessment in the absence of RECIST 1.1 progression, as determined by BICR. BoR categorization was based on RECIST 1.1 using CR, PR, stable disease, progressive disease, and not-evaluable response categories.1,48 The sponsor stated that there is no reported MID for BoR in the first-line treatment of metastatic NSCLC.1
HRQoL End Points
HRQoL was assessed using the EORTC QLQ-C30 and EORTC QLQ-LC13 questionnaires.1,49,50 PRO assessments were completed by patients during clinic visits using an electronic tablet.47
Version 3 of the EORTC QLQ-C30 is a self-administered measure used to assess HRQoL in response to treatment in clinical trials.1,47,48,51 It consists of 30 questions that can be combined to produce:1,47,48
5 functional scales (physical, role, cognitive, emotional, social)
3 symptom scales (fatigue, pain, nausea and vomiting)
6 individual items, including 5 symptoms (dyspnea, insomnia, appetite loss, constipation, diarrhea) and 1 item assessing financial difficulties
2-item global measure of health status.
For each of the 15 domains, final scores are converted to a standardized score that ranges from 0 to 100 using a linear transformation, with a higher score indicating better functioning on the functional scales, higher (i.e., worse) symptoms on the symptom scales, and better HRQoL on the GHS scale.1,47-49
The EORTC QLQ-LC13 is a 13-item, lung cancer–specific, self-administered questionnaire. The module consists of both multi-item and single-item measures of lung cancer–associated symptoms (i.e., coughing, hemoptysis, dyspnea, and pain) and side effects from conventional chemotherapy and radiotherapy (i.e., hair loss, neuropathy, sore mouth, and dysphagia).47,48,50 The scoring approach is similar to that used for the symptom scales and items of the EORTC QLQ-C30.47,48
In the POSEIDON trial, a clinically meaningful change for the EORTC QLQ-C30 and for the EORTC QLQ-LC13 was defined as an absolute change in the score from baseline of 10 or more points. For each postbaseline assessment, the change from baseline for the scales and items were categorized as improvement, no change, or deterioration.47,48 This threshold for clinically meaningful change was based on a study by Osoba et al. (1998)52 evaluating the significance to patients of changes in EORTC QLQ-C30 scores. Patients who received chemotherapy for either breast cancer or small-cell lung cancer completed a subjective significance questionnaire and the EORTC QLQ-C30, with “a little” change in the subjective significance questionnaire corresponding to changes in the EORTC QLQ-C30 of 5 to 10 points, “moderate” change corresponding with changes of about 10 to 20 points, and “very much” change corresponding with changes of more than 20 points.1,52
The POSEIDON protocol predefined the following primary PRO symptoms (key symptoms): fatigue, appetite loss (from the EORTC QLQ-C30), coughing, dyspnea, and chest pain (from the EORTC QLQ-LC13). The GHS/QoL and physical functioning domains of the EORTC QLQ-C30 were also prespecified PRO domains of interest.1,47 PROs were not included in the MTP; however, the overall type 1 (5% [2-sided]) error was controlled across the 5 key symptoms.1,47,48
Time to deterioration: Time to deterioration was derived for all EORTC QLQ-C30 items except financial difficulties and for all lung cancer symptoms scale items in the EORTC QLQ-LC13. Time to deterioration is defined as the time from the date of randomization until the date of the first clinically meaningful deterioration that is confirmed at a subsequent visit or death (by any cause) in the absence of a clinically meaningful deterioration, regardless of whether the patient withdraws from study treatment or receives another anticancer therapy before HRQoL or symptom deterioration.1,48
HRQoL/function improvement rate: The function improvement rate was calculated for the EORTC QLQ-C30 and is defined as the number (percentage) of patients with 2 consecutive assessments at least 14 days apart that show a clinically meaningful improvement in that function scale from baseline.1,48
Symptom improvement rate: Symptom improvement rate was calculated for the EORTC QLQ-LC13 and is defined as the number (percentage) of patients with 2 consecutive assessments at least 14 days apart that show a clinically meaningful improvement in that symptom from baseline.1,47
Table 10: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
EORTC QLQ-C30 | A multidimensional, cancer-specific, patient-reported measure used to assess HRQoL in response to treatment in clinical trials.51 The core questionnaire consists of 30 items that make 5 multi-item functional scales (physical [5 items], role [2 items], emotional [4 items], cognitive [2 items], and social [2 items] functioning); 3 multi-item symptom scales (fatigue [3 items], nausea and vomiting [2 items], and pain [2 items]); 6 single-item symptom scales (dyspnea, insomnia, appetite loss, constipation, diarrhea and financial impact); and a 2-item global QoL scale. Patients complete the questionnaire based on a 1-week recall period by rating most items on a 4-point Likert scale (1 = not at all; 2 = a little; 3 = quite a bit; 4 = very much). For the 2 items in the global QoL scale, the response format is a 7-point Likert scale (1 = very poor; 7 = excellent).53 Raw scores for each scale are computed as the average of the items that contribute to a particular scale. Each raw scale score is converted to a standardized score that ranges from 0 to 100 using a linear transformation. A decline in the symptom scale score reflects an improvement, whereas an increase in the function and quality of life scale scores reflects an improvement.53 According to the QLQ-C30 scoring algorithm, if a scale item is missing, the score for that scale can still be computed if there are responses for at least one-half of the items. In calculating the scale score, missing items are ignored.53 | In studies including lung cancer patients: Validity The EORTC QLQ-C30 was administered before treatment and once during treatment to 305 patients with nonresectable lung cancer from centres in 13 countries.49 The authors noted the strongest correlations before and during treatment between the physical functioning, role functioning and fatigue scales (ranging from 0.54 to 0.63). In another study of 112 patients with lung cancer (n = 101), pleural mesothelioma (n = 11), and colon cancer (n = 1), the authors observed a strong correlation between EORTC QLQ-C30 emotional functioning and the HADS anxiety scale (r = −0.75).54 Additionally, the authors noted a substantial correlation between the HADS depression scale with all functioning scales (ranging from r = −0.40 to −0.55), fatigue (r = 0.52) and appetite loss (r = 0.48).54 Reliability The EORTC QLQ-C30 was administered before treatment and once during treatment to 305 patients with nonresectable lung cancer from centres in 13 countries.49 The authors of the study noted reliability coefficients (Cronbach alphas) for the multi-item scales ranging from 0.54 to 0.86 (before treatment) and from 0.52 to 0.89 (during treatment). For functional scales, alpha values ranged from 0.54 to 0.86 (before treatment) and 0.52 to 0.89 (during treatment). For symptom scales, the alpha ranged from 0.65 to 0.82 (before treatment) and 0.73 to 0.85 (during treatment).49 In another study of 112 patients with advanced lung cancer (n = 101), pleural mesothelioma (n = 11), and colon cancer (n = 1) the authors noted an internal consistency of > 0.70 for all functional scales, except cognitive functioning (alpha = 0.57). They also noted alpha > 0.80 for items fatigue and nausea or vomiting of the symptom scale.54 Responsiveness The EORTC QLQ-C30 was administered before treatment and once during treatment to 305 patients with nonresectable lung cancer from centres in 13 countries.49 Over a 28-day period, there was a statistically significant difference in the global QoL scores (P < 0.01) between patients whose condition improved or worsened based on ECOG PS during pretreatment and on-treatment periods. No significant difference was observed among patients whose ECOG PS remained stable/unchanged.49 | Between-group MIDs for improvement (5 points) and deterioration (−5 points) on Global Health Status were identified for patients with lung cancer.55 The MID estimates in patients with small-cell lung cancer and breast cancer who reported “a little” change in the SSQ had corresponding changes in the EORTC QLQ-C30 of 5 to 10 points, those who reported a “moderate” change had corresponding changes of about 10 to 20 points, and those who reported “very much” change had corresponding changes of more than 20 points.52 In a study of patients with advanced cancer, MID estimates of the EORTC QLQ-C30 ranged from a meaningful change for improvement of 9.1 units (cognitive functioning) to 23.5 units (pain), and a meaningful change for deterioration ranging from 7.2 units (physical functioning) to 13.5 units (role functioning). Distribution-based estimates were closest to 0.5 SD.56 In another study of patients with multiple myeloma (n = 239), MID estimates for the EORTC QLQ-C30, mean scores ranged from 7.6 (improved global QoL) to –12.1 (deteriorated global QoL).57 |
EORTC QLQ LC13 | The EORTC QLQ-LC13 is a lung cancer–specific questionnaire used to supplement the EORTC QLQ-C30 and contains 13 items related to lung cancer symptoms and treatment side effects including: a 3-item scale assessing dyspnea and 9 single items: pain in chest, pain in arm or shoulder, pain in other parts, coughing, hemoptysis, sore mouth or tongue, dysphagia, peripheral neuropathy, and alopecia.49 All the scales range in score from 0 to 100. Higher scores on the symptom scales reflect worse symptoms.49 | Validity The EORTC QLQ-LC13 was administered to 735 patients with nonresectable lung cancer from 17 countries. The authors confirmed construct validity between pain score and disease type (metastatic vs. local disease) before treatment (P < 0.001). Also, based on ECOG PS, the authors confirmed construct validity in dyspnea, coughing, and pain (P < 0.001) scores.50 Changes in symptom measures over time were significantly associated with either chemotherapy (tingling in arms and legs and hair loss) or radiotherapy (difficulty swallowing) (P < 0.001).50 Reliability The EORTC QLQ-LC13 was administered (once before and during treatment) to 735 patients with nonresectable lung cancer from 17 countries. The authors noted pretreatment reliability estimates (alphas) of 0.81 and 0.83 for overall dyspnea scale (3-item scale).50 However, for overall pain items (3-item scale) they noted an alpha of 0.53 and 0.54.50 In another study of 112 patients with advanced lung cancer (n = 101), pleural mesothelioma (n = 11), and colon cancer (n = 1) the authors noted reliability estimates for dyspnea item of the symptom scale as acceptable (alpha = 0.76).54 Responsiveness The EORTC QLQ-LC13 was administered (before and during treatment) to 735 patients with nonresectable lung cancer from 17 countries. The authors noted a statistically significant improvement (P < 0.001) for various symptoms over time between pretreatment and during treatment, including dyspnea, coughing, pain (shoulder and chest).50 | No studies with an MID were available in the overall lung cancer population or for NSCLC. |
ECOG PS = Eastern Cooperative Oncology Group Performance Status; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-LC13 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Lung Cancer 13; HRQoL = health-related quality of life; HADS = Hospital Anxiety and Depression Scale; MID = minimal important difference; NSCLC = non–small cell lung cancer; QoL = quality of life; SD = standard deviation; SSQ = subjective significance questionnaire; vs. = versus.
Harms
AEs included any AE with a date of onset on or after the date of the first dose or pretreatment AEs that increased in severity on or after the date of first dose and up to and including the earlier of 90 days following the date of the last dose of study treatment or the date of initiation of the first subsequent anticancer treatment (including radiotherapy, with the exception of palliative radiotherapy) following discontinuation of study treatment (whichever occurred first).1,48
Coding of AEs was based on the Medical Dictionary for Regulatory Activities dictionary (version 23.1) and grading for severity was according to Common Terminology Criteria for Adverse Events (version 4.03).1,38,47 AE categories included: all AEs (including those causally related to study treatment), AEs of Common Terminology Criteria for Adverse Events grade 3 or 4 (including those causally related to study treatment), AEs leading to dose modification, AEs leading to hospitalization, AEs with an outcome of death (including those causally related to study treatment), SAEs (including those causally related to study treatment), AEs leading to discontinuation (including those causality related to study treatment), and infusion-related AEs.1,38
Per the POSEIDON study protocol, for durvalumab with or without tremelimumab, AESIs included events with a potential immune-mediated or inflammatory mechanism and that may require more frequent monitoring and/or interventions (e.g., steroids, immunosuppressants, and/or hormone replacement therapy).1,47,48
Per the POSEIDON statistical analysis plan, the following were considered AESIs:1,48
diarrhea or colitis
pneumonitis or interstitial lung disease
increases in alanine transaminase or aspartate transaminase, hepatitis, and hepatotoxicity
neuropathy or neuromuscular toxicity (e.g., Guillain-Barré, myasthenia gravis)
endocrinopathies (i.e., events of hypophysitis, hypopituitarism, adrenal insufficiency, diabetes insipidus, hyper- and hypothyroidism, type I diabetes mellitus)
rash or dermatitis
nephritis or blood creatinine increases
pancreatitis (or laboratory results suggestive of pancreatitis)
other inflammatory responses that are rare with a potential immune-mediated etiology include, but are not limited to, myocarditis, pericarditis, uveitis, vasculitis, noninfectious meningitis, and noninfectious encephalitis
infusion-related reactions and hypersensitivity or anaphylactic reactions with a different underlying pharmacological etiology.
While the POSEIDON study was being conducted, the process for identifying imAEs was redefined, which resulted in revision to the criteria for defining AESIs and AEPIs. The definitions for AESIs, AEPIs, and imAEs in the study were:38
AESIs: AEs with a likely inflammatory or immune-mediated pathophysiological basis resulting from the mechanism of action of durvalumab and requiring more frequent monitoring and/or interventions (e.g., corticosteroids, immunosuppressants, endocrine therapy)
AEPIs: AEs that could have a potential inflammatory or immune-mediated pathophysiological basis resulting from the mechanism of action of durvalumab but are more likely to have occurred because of other pathophysiological mechanisms; the likelihood of the event being inflammatory or immune-mediated in nature is not high and/or is most often or usually explained by the other causes.
Statistical Analysis
A summary of the statistical analyses relevant to the efficacy end points of interest from the POSEIDON trial is presented in the following section and in Table 11.
Sample Size and Power Calculation
The POSEIDON trial planned to enrol approximately 2,000 patients and randomize approximately 1,000 patients in a 1:1:1 ratio (approximately 333 patients in each treatment arm) to durvalumab and tremelimumab plus SOC chemotherapy, durvalumab plus SOC chemotherapy, or SOC chemotherapy alone, including at least 250 patients in each treatment arm with PD-L1 TC level of less than 50%. Participants were randomized in a stratified manner according to PD-L1 tumour-expression status (PD-L1 TC ≥ 50% versus < 50%), disease stage (IVA versus IVB), and histology (nonsquamous versus squamous). The study was powered for dual primary end points to characterize PFS and OS effects for the comparison of durvalumab plus SOC chemotherapy versus SOC chemotherapy alone in the ITT population.1,38,48 A statistically significant benefit in either PFS or OS for this comparison had to be demonstrated for the study to be declared positive.1
One interim analysis and 1 final analysis of the primary and key secondary PFS end points were performed and 3 interim analyses and 1 final analysis were planned to be performed for the primary and key secondary OS end points.1,48
The final PFS (primary end point) analysis for superiority was prespecified to be performed when approximately 497 BICR PFS events from the global cohort had occurred across the durvalumab plus SOC chemotherapy and SOC chemotherapy alone arms (75% maturity). The final OS (primary end point) analysis for superiority was prespecified to be performed when approximately 532 OS events had occurred across the durvalumab plus SOC chemotherapy and SOC chemotherapy alone arms (80% maturity).1,38,48
Key Secondary End Points
Durvalumab and Tremelimumab Plus SOC Chemotherapy Versus SOC Chemotherapy (PFS in ITT Population)
Assuming a true PFS HR of 0.51 and median PFS of 6 months in the SOC chemotherapy arm, 465 PFS events from the global cohort (70% maturity) would provide greater than 90% power to demonstrate statistical significance at the 2-sided alpha level of 0.9% (with an overall alpha for PFS of 1%), allowing for 1 interim analysis conducted at approximately 80% of the target events (information fraction). The smallest statistically significant treatment difference would be an HR of 0.78. This analysis was anticipated to be 25 months from first patient in (assuming a recruitment period of 16 months).1,38,48
Durvalumab and Tremelimumab Plus SOC Chemotherapy Versus SOC Chemotherapy (OS in ITT Population)
Assuming a true OS HR of 0.7 and median OS of 12.9 months in the SOC chemotherapy arm, 532 OS events (80% maturity) would provide greater than 90% power to demonstrate statistical significance at the 2-sided alpha level of 3.3% (with an overall alpha for OS of 4%), allowing for 3 interim analyses conducted at approximately 45%, 61%, and 84% of the target events (information fraction). The smallest statistically significant treatment difference would be an HR of 0.83. This analysis was anticipated to be 46 months from first patient in (assuming a recruitment period of 16 months).1,38,48
Statistical Test or Model
Statistical analysis of efficacy end points for the POSEIDON trial are presented in Table 11. Efficacy analyses were based on the ITT population (full analysis set [FAS]), and safety analyses were based on the safety analysis set (SAF) (Table 12).
Multiple Testing Procedure
To control the type I error at 5% (2-sided), an MTP with a gatekeeping strategy was used across the following end points included in the MTP:1,38
Dual primary end points: PFS (using BICR assessments according to RECIST 1.1) and OS for the durvalumab plus SOC chemotherapy versus SOC chemotherapy comparison in the ITT population
Key secondary end points: PFS (using BICR assessments according to RECIST 1.1) and OS for the durvalumab and tremelimumab plus SOC chemotherapy versus SOC chemotherapy comparison in the ITT population
Secondary end point: OS for the durvalumab and tremelimumab plus SOC chemotherapy versus SOC chemotherapy comparison in the populations of patients of with a blood tumour mutational burden of 20 mutations per megabase or greater (bTMB20 high), 16 mutations per megabase or greater (bTMB16 high), and 12 mutations per megabase or greater (bTMB12 high).
Hypotheses were tested using an MTP with an alpha-exhaustive recycling strategy and were tested in a predefined order (Figure 3).48 According to alpha (test mass) splitting and alpha recycling, if the higher-level hypothesis in the MTP was rejected for superiority, the next lower-level hypothesis was then tested. The test mass that became available after each rejected hypothesis was recycled to the lower-level hypotheses not yet rejected. This testing procedure stopped when the entire test mass was allocated to nonrejected hypotheses.1,38,48
The alpha level allocated to the interim or final analyses was determined by the Lan-DeMets spending function that approximates an O’Brien-Fleming approach. The alpha level applied at the interims was set to the proportion of information available; that is the actual observed number of PFS or OS events and percent of maturity at the time of interim analyses. A separate Lan-DeMets (O’Brien-Fleming) spending function accounting for interim and final analysis was applied for OS and PFS end points.1,38,48
In the MTP, the overall 5% type I error was first split between the dual primary end points of PFS and OS; an alpha level of 1% was allocated to the primary PFS analysis and an alpha of 4% was allocated to the primary OS analysis (durvalumab plus SOC chemotherapy versus SOC chemotherapy) (Figure 3). The MTP involved the following strategy:1,38,48
If the primary PFS analysis was significant, the 1% alpha level could be recycled to the key secondary PFS comparison (durvalumab and tremelimumab plus SOC chemotherapy versus SOC chemotherapy).
If the primary OS analysis was significant, the 4% alpha level could be recycled to the key secondary OS comparison (durvalumab and tremelimumab plus SOC chemotherapy versus SOC chemotherapy).
If the key secondary PFS comparison between durvalumab and tremelimumab plus SOC chemotherapy versus SOC chemotherapy was significant, the 1% alpha level could be recycled to the OS comparison between durvalumab and tremelimumab plus SOC chemotherapy versus SOC chemotherapy.
If the key secondary OS comparison between durvalumab and tremelimumab plus SOC chemotherapy versus SOC chemotherapy was significant, the 4% alpha level could be recycled to the key secondary PFS comparison between durvalumab and tremelimumab plus SOC chemotherapy versus SOC chemotherapy.
If both key secondary PFS and OS comparisons of durvalumab and tremelimumab plus SOC chemotherapy versus SOC chemotherapy were significant, the available alpha level could be recycled to the OS comparison between durvalumab and tremelimumab plus SOC chemotherapy versus SOC chemotherapy alone in the bTMB20 high population. If the comparison was significant, the alpha level could be recycled to the OS comparison between durvalumab and tremelimumab plus SOC chemotherapy versus SOC chemotherapy in the bTMB16 high population. If the comparison was significant, the alpha level could be recycled to the OS comparison between durvalumab and tremelimumab plus SOC chemotherapy versus SOC chemotherapy in the bTMB12 high population.
Figure 3: Multiple Testing Procedures for Controlling the Type I Error Rate
bTMB12 = blood tumour mutational burden of 12 mutations per megabase or greater; bTMB16 = blood tumour mutational burden of 16 mutations per megabase or greater; bTMB20 = blood tumour mutational burden of 20 mutations per megabase or greater; combo = durvalumab and tremelimumab plus chemotherapy; ITT = intention to treat; mono = durvalumab plus chemotherapy; OS = overall survival; PFS = progression-free survival; SoC = standard of care chemotherapy.
Sources: Sponsor’s Summary of Clinical Evidence1 and POSEIDON Clinical Study Report.38
One interim analysis of PFS was planned for when approximately 80% of the target PFS events (approximately 497) had occurred across the durvalumab plus SOC chemotherapy and SOC chemotherapy arms (397 out of 497 events).1,38,48
Three interim analyses of OS were planned:1,38,48
the first at the time of the interim PFS analysis (approximately 45% of the target OS events [approximately 532] in the durvalumab plus SOC chemotherapy and SOC chemotherapy arms [243 out of 532 events])
the second at the time of the final PFS analysis (approximately 61% of the target OS events in the durvalumab plus SOC chemotherapy and SOC chemotherapy arms [328 out of 532 events])
the third when approximately 84% of the target OS events had occurred in the durvalumab plus SOC chemotherapy and SOC chemotherapy arms (449 out of 532 events).
The interim analyses were performed for the analyses specified in MTP. Global recruitment (excluding China) was completed before the results of the interim analyses becoming available (data presented in this review report are for the global cohort excluding China).1,38
The DCO for the final analysis of the POSEIDON trial occurred on July 24, 2019, for PFS and all RECIST 1.1–related end points, and on March 12, 2021, for OS, safety, and all other data.1,38
A prespecified long-term survival follow-up analysis of the POSEIDON study was scheduled to occur at least 3 years after the last patient was randomized and has been completed. This is referred to as the 4-year OS analysis and the DCO date of this analysis was March 11, 2022.1
A subsequent long-term survival follow-up analysis of the POSEIDON trial was scheduled to occur at least 4 years after the last patient was randomized and has been completed. This is referred to as the 5-year OS analysis and the DCO date of this analysis was August 24, 2023.1
Data from the final analysis and 5-year OS analysis (but not the 4-year OS analysis) are presented in this Clinical Review.
The 5-year OS analysis included all 3 treatment arms and was limited to an updated summary of OS (including sensitivity and subgroup analyses) and a summary of key safety data occurring since the final analysis DCO date (as well as other updated data). All safety and efficacy end points were evaluated using the same statistical methodology as previously defined for the final analysis.58 The updated OS estimates and CIs were not alpha-controlled.1
PRO symptoms and HRQoL end points were not part of the MTP. However, the overall type I error (5% [2-sided]) was controlled across the 5 predefined primary PRO measures (key symptoms) of fatigue and appetite loss (from EORTC QLQ-C30) and coughing, dyspnea, and pain in chest (from EORTC QLQ-LC13) using the Bonferroni-Holm procedure. The EORTC QLQ-C30 overall health status and physical functioning domains were prespecified end points of interest.1,48
Data Imputation Methods
Missing efficacy and safety data were generally not imputed.48
Subgroup Analyses
The following prespecified subgroups analyses were performed comparing OS and PFS between treatments of the primary and key secondary end points to evaluate the consistency of treatment effect across expected prognostic and/or predictive factors:1,48
sex (male versus female)
age at randomization (< 65 years versus ≥ 65 years)
PD-L1 status (PD-L1 TC ≥ 50% versus TC < 50%)
histology (squamous versus nonsquamous)
chemotherapy (nab-paclitaxel doublet versus pemetrexed doublet versus gemcitabine doublet)
smoking (current smoker, former smoker, or never smoker)
race (Asian versus non-Asian)
PD-L1 using cut points of 1% and 25% tumour expression (< 1% versus ≥ 1%, and < 25% versus ≥ 25%)
performance status (normal activity [0] versus restricted activity [1])
brain metastases (yes versus no)
disease stage (IVA versus IVB).
The study was not powered for any of the individual subgroup evaluations and no adjustments were made for multiple testing subgroup analyses.1,38,48
The HR and 95% CI was calculated from an unstratified Cox proportional hazards model with treatment as the only covariate for each subgroup level of a factor.48
Interactions between treatment and stratification factors were to be tested to rule out any qualitative interaction using the Gail and Simon approach.48,59 This test was to be performed separately for the treatment comparisons of the dual primary and key secondary end points based on the FAS.48
Sensitivity Analyses
The POSEIDON study conducted sensitivity analyses for the key secondary end points of OS and PFS in the comparison of durvalumab and tremelimumab plus SOC chemotherapy versus SOC chemotherapy alone, adjusting for further prespecified covariates of age at randomization, sex, smoking history, and race (Table 11). Additional sensitivity analyses were conducted for OS to rule out attrition bias, for PFS to assess the robustness of the PFS effect to potential sources of bias, and for OS (post hoc) using confirmed responses (Table 11).
Table 11: Statistical Analysis of Efficacy End Points for POSEIDON Study
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
OS (D + T + chemotherapy vs. chemotherapy) | Analyzed using a stratified log-rank test for the ITT population. HRs and CIs were estimated from a stratified Cox Proportional hazards model. Subgroup analyses used an unstratified Cox proportional hazards model. | Adjusted for stratification variables: PD-L1 (TC ≥ 50% vs. < 50%), disease stage (IVA and IVB), and histology (nonsquamous vs. squamous). | Any patient not known to have died at the time of analysis will be censored based on the last recorded date on which the patient was known to be alive. | Sensitivity analysis adjusted for further prespecified covariates (age at random assignment, sex, smoking history, race). A sensitivity analysis examined the censoring patterns to rule out attrition bias, using a Kaplan-Meier plot of time to censoring where the censoring indicator of OS is reversed. |
PFS (D + T + chemotherapy vs. chemotherapy) | Analyzed using a stratified log-rank test for the ITT population. HRs and CIs were estimated from a stratified Cox proportional hazards model. Subgroup analyses used an unstratified Cox proportional hazards model. | Adjusted for stratification variables: PD-L1 (TC ≥ 50% vs. < 50%), disease stage (IVA and IVB), and histology (squamous vs. nonsquamous). | Patients who have not progressed or died at the time of analysis will be censored at the time of the latest date of assessment from their last evaluable RECIST 1.1 assessment. However, if the patient progresses or dies after 2 or more missed visits, the patient will be censored at the time of the latest evaluable RECIST 1.1 assessment. If the patient has no evaluable visits or does not have baseline data, they will be censored at 0 days unless they die within 2 visits of baseline. | Sensitivity analysis adjusted for further prespecified covariates (age at random assignment, sex, smoking history, and race). Sensitivity analyses were conducted to assess robustness of the PFS effect to the potential source of bias in PFS measurement, including the possibility of evaluation-time bias, attrition bias, ascertainment bias, and using variables derived from electronic case report forms. |
Objective response rate | Logistic regression model. Odds ratios and 95% CIs were calculated. | Adjusted for stratification variables: PD-L1 (TC ≥ 50% vs. < 50%), disease stage (IVA and IVB), and histology (nonsquamous vs. squamous). | Missing or nonevaluable were categorized as nonresponse. | Post hoc sensitivity analysis requiring confirmation of response no sooner than 4 weeks after the initial complete or partial response was conducted. |
Duration of response | Determined using Kaplan-Meier estimates. | NA | If a patient does not progress following a response, then their duration of response will be censored at the PFS censoring time. | Not performed |
Best objective response | Determined programmatically based on RECIST 1.1 from the overall visit response using all BICR data up until the first progression event. | NA | Missing or nonevaluable were categorized as nonresponse. | Not performed |
Time to deterioration (EORTC QLQ-C30 and EORTC QLQ-LC13) | Stratified log-rank test | Adjusted for stratification variables: PD-L1 (TC ≥ 50% vs. < 50%), disease stage (IVA and IVB), and histology (nonsquamous vs. squamous). | Patients will be censored at the time of their last PRO assessment where the HRQoL or symptom could be evaluated. | Not performed |
HRQoL/function improvement rate (EORTC QLQ-C30) | Logistic regression | Adjusted for stratification variables: PD-L1 (TC ≥ 50% vs. < 50%), disease stage (IVA and IVB), and histology (nonsquamous vs. squamous). | Patients will be censored at the time of their last PRO assessment where the HRQoL or symptom could be evaluated. | Not performed |
Symptom improvement rate (EORTC QLQ-C30 and QLQ-LC13) | Logistic regression | Adjusted for stratification variables: PD-L1 (TC ≥ 50% vs. < 50%), disease stage (IVA and IVB), and histology (nonsquamous vs. squamous). | Patients will be censored at the time of their last PRO assessment where the HRQoL or symptom could be evaluated. | Not performed |
BICR = blinded independent central review; CI = confidence interval; D = durvalumab; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-LC13 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Lung Cancer 13; HR = hazard ratio; HRQoL = health-related quality of life; ITT = intention to treat; NA = not available; OR = odds ratio; OS = overall survival; PFS = progression-free survival; PRO = patient-reported outcomes; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1; t = tremelimumab; TC = tumour cell; vs. = versus.
Sources: Sponsor’s Summary of Clinical Evidence,1 POSEIDON Clinical Study Report,38 and POSEIDON Statistical Analysis Plan.48
Analysis Populations
Analysis populations relevant to the outcomes reported in this Clinical Review are presented in Table 12.
Table 12: Analysis Populations of the POSEIDON Trial
Population | Definition | Application |
|---|---|---|
Full analysis set (FAS) (ITT) N (total) = 1,013 n (D + T + chemotherapy) = 338 n (chemotherapy) = 337 | Includes all randomized patients. Treatment arms were compared on the basis of randomized study treatment, regardless of the treatment actually received. Patients who were randomized but did not subsequently go on to receive study treatment were included in the analysis in the treatment arm to which they were randomized. | Efficacy analyses:
|
Safety analysis set (SAF) N (total) = 997 n (D + T + chemotherapy) = 330 n (chemotherapy) = 333 | Includes all patients who received at least 1 dose of study treatment. Safety data were not formally analyzed but summarized according to the treatment received, that is, erroneously treated patients (e.g., those randomized to treatment A but actually given treatment B) are summarized according to the treatment they actually received. Patients in the D + T + chemotherapy arm or D + CT arm who received only chemotherapy and no doses of D or T are summarized in the chemotherapy-alone arm. | Safety analyses:
|
AE = adverse event; BoR = best objective response; D = durvalumab; DoR = duration of response; FAS = full analysis set; GHS = Global Health Status; HRQoL = health-related quality of life; ITT = intention to treat; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; PRO = patient-reported outcome; SAF = safety analysis set; t = tremelimumab.
aPatients who are evaluable for the analysis of ORR are those with measurable disease at baseline.
bPatients who are evaluable for the analysis of DoR are those who responded in the ORR analysis.
cThe population for analysis of time to GHS/HRQoL or function deterioration will include a subset of the FAS population who have baseline scores of 10 or higher; the population for analysis of time to symptom deterioration will include a subset of the FAS population who have baseline scores of 90 or lower. For symptom improvement rate, the denominator will consist of a subset of the FAS who have a baseline symptom score of 10 or higher. For HRQoL/function improvement rate, the denominator will consist of a subset of the FAS who have a baseline HRQoL/function score of 90 or lower.
Sources: Sponsor’s Summary of Clinical Evidence,1 POSEIDON Clinical Study Report,38 and POSEIDON Statistical Analysis Plan.48
Outcomes of the MTP
The outcomes of the MTP at the final analysis (DCO: July 24, 2019, for PFS; March 12, 2021, for OS) are presented Figure 4. At level 1, the dual primary end point of PFS and OS for the comparison of durvalumab plus SOC chemotherapy versus SOC chemotherapy alone was tested and at level 2, the key secondary end points of PFS and OS for the comparison of durvalumab and tremelimumab plus SOC chemotherapy versus SOC chemotherapy alone were tested. At level 1, the HR for PFS favoured durvalumab plus SOC chemotherapy and was statistically significant at the 1% alpha level; the 1% alpha was therefore recycled to test PFS for the durvalumab and tremelimumab plus SOC chemotherapy versus SOC chemotherapy alone comparison at level 2. The HR for PFS favoured durvalumab and tremelimumab plus SOC chemotherapy and was statistically significant at the 1% alpha level. At level 1, the HR for OS numerically favoured durvalumab plus chemotherapy but did not cross the prespecified statistical significance threshold at the 4% alpha level. For this reason, at level 2, the 1% alpha level from the PFS for durvalumab and tremelimumab plus SOC chemotherapy versus SOC chemotherapy alone comparison was recycled to test OS for this same comparison. The HR for OS favoured durvalumab and tremelimumab plus SOC chemotherapy and was statistically significant at the 1% alpha level. At level 3, OS in the bTMB20 population for the comparison between durvalumab and tremelimumab plus SOC chemotherapy versus SOC chemotherapy alone was tested at the recycled 1% alpha level. The HR for OS favoured durvalumab and tremelimumab plus SOC chemotherapy but did not cross the prespecified statistical significance threshold; bTMB16 and bTMB12 populations were therefore not tested.1,38
Efficacy results for the comparison between durvalumab and tremelimumab plus SOC chemotherapy versus SOC chemotherapy alone are presented in this Clinical Review, because this treatment arm aligns with the indication approved by Health Canada. Results for the dual primary end points (OS and PFS) for the comparison between durvalumab and chemotherapy versus SOC chemotherapy alone are presented in Appendix 1.
Figure 4: Actual Multiple Testing Procedure at Final Analysis in the POSEIDON Trial
bTMB = blood tumour mutational burden; ITT = intention to treat; mut/Mb = mutations per megabase; OS = overall survival; PFS = progression-free survival.
Source: Sponsor’s Summary of Clinical Evidence.1
Results
Patient Disposition
Patient disposition in the POSEIDON trial is summarized in Table 13.
A total of 1,807 patients were enrolled into the study, of whom 1,013 were randomized to 1 of the 3 study arms (arm 1: N = 338; arm 2: N = 338; arm 3: N = 337).
In the FAS for treatment arm 1 and arm 3, 7 patients (2.1%) and 6 patients (1.8%), respectively, were randomized but did not receive treatment.38 Of the patients who received treatment in arm 1 (n = 331), 294 (88.8%) had discontinued durvalumab treatment at the final analysis DCO (March 12, 2021), and 311 (94.0%) had discontinued durvalumab treatment at the 5-year OS analysis DCO (August 24, 2023).
The most common reason for discontinuation of durvalumab treatment was the patient’s “condition under investigation worsened” (216 [65.3%] and 219 [66.2%] at the final analysis and 5-year OS analysis DCO dates, respectively). Of the patients who received treatment in arm 1, at both DCO dates, 117 patients (35.3%) had discontinued tremelimumab treatment and 213 patients (64.4%) had completed tremelimumab treatment. The most common reason for discontinuation of tremelimumab was that the patient’s “condition under investigation worsened” (69 [20.8%] for both DCO dates).
Of the patients who received treatment in arm 1, 311 (94.0%) had discontinued chemotherapy at the final analysis DCO and 320 (96.7%) had discontinued chemotherapy at the 5-year OS analysis DCO. The most common reason for discontinuation of chemotherapy in arm 1 was that the patient’s “condition under investigation worsened” (138 [41.7%] at both DCO dates); 95 patients (28.7%) at both DCO dates discontinued because the maximum number of chemotherapy cycles had been reached.
Of the patients who received treatment in arm 3 (n = 331), 326 (98.5%) had discontinued chemotherapy at the final analysis DCO and 330 (99.7%) had discontinued chemotherapy at the 5-year OS analysis DCO. The most common reason for discontinuation of chemotherapy was also that the patient’s “condition under investigation worsened” (183 [55.3%] and 185 [55.9%] at the final analysis and 5-year OS analysis DCO dates, respectively); 73 patients (22.1%) at the final analysis and 74 patients (22.4%) at the 5-year analysis discontinued because the maximum number of chemotherapy cycles had been reached. In the full analysis set (FAS), a higher proportion of patients in arm 3 had terminated the study at the final analysis and 5-year OS analysis DCOs (88.1% and 94.4%, respectively) than in arm 1 (76.3% and 85.2%, respectively), with death the most common reason for study termination in both groups.
Table 13: Patient Disposition in the POSEIDON Trial
Patient disposition | D + T + chemotherapy (N = 338) | D + chemotherapy (N = 338) | Chemotherapy (N = 337) | |||
|---|---|---|---|---|---|---|
Final analysis | 5-year OS analysisa | Final analysis | 5-year OS analysisa | Final analysis | 5-year OS analysisa | |
Screened, N | 1,807 | |||||
Reason for screening failure, N (%) | 794 | |||||
Screening failure | 760 | |||||
Withdrawal by patient | 27 | |||||
Death | 6 | |||||
Other | 1 | |||||
Randomized,b N (%) | 338 | 338 | 337 | |||
Full analysis set | 338 (100) | 338 (100) | 338 (100.0) | 338 (100) | 337 (100.0) | 337 (100) |
Patients who received treatmentc | 331 (97.9) | 331 (97.9) | 335 (99.1) | 335 (99.1) | 331 (98.2) | 331 (98.2) |
Patients who did not receive treatmentc | 7 (2.1) | 7 (2.1) | 3 (0.9) | 3 (0.9) | 6 (1.8) | 6 (1.8) |
Patients ongoing D treatment at DCOd | 36 (10.9) | 19 (5.7) | 31 (9.3) | 13 (3.9) | NA | NA |
Patients who discontinued D treatmentd | 294 (88.8) | 311 (94.0) | 303 (90.4) | 321 (95.8) | NA | NA |
Patient decision | 14 (4.2) | 17 (5.1) | 16 (4.8) | 18 (5.4) | NA | NA |
Adverse event | 61 (18.4) | 70 (21.1) | 56 (16.7) | 61 (18.2) | NA | NA |
Condition under investigation worsened | 216 (65.3) | 219 (66.2) | 226 (67.5) | 236 (70.4) | NA | NA |
Patient lost to follow-up | 0 | 0 | 1 (0.3) | 1 (0.3) | NA | NA |
Other | 3 (0.9) | 5 (1.5) | 4 (1.2) | 5 (1.5) | NA | NA |
Patients ongoing T treatment at DCOd,e | 0 | 0 | NA | NA | NA | NA |
Patients who completed T treatmentd,e,f | 213 (64.4) | 213 (64.4) | NA | NA | NA | NA |
Patients who discontinued T treatmentd | 117 (35.3) | 117 (35.3) | NA | NA | NA | NA |
Patient decision | 9 (2.7) | 9 (2.7) | NA | NA | NA | NA |
Adverse event | 35 (10.6) | 35 (10.6) | NA | NA | NA | NA |
Condition under investigation worsened | 69 (20.8) | 69 (20.8) | NA | NA | NA | NA |
Other | 4 (1.2) | 4 (1.2) | NA | NA | NA | NA |
Patients ongoing SOC chemotherapy at DCOd | 19 (5.7) | 10 (3.0) | 21 (6.3) | 8 (2.4) | 5 (1.5) | 1 (0.3) |
Patients who discontinued SOC chemotherapyd,g | 311 (94.0) | 320 (96.7) | 314 (93.7) | 327 (97.6) | 326 (98.5) | 330 (99.7) |
Patient decision | 13 (3.9) | 15 (4.5) | 17 (5.1) | 18 (5.4) | 20 (6.0) | 20 (6.0) |
Adverse event | 62 (18.7) | 67 (20.2) | 63 (18.8) | 66 (19.7) | 44 (13.3) | 44 (13.3) |
Severe noncompliance to protocol | 0 | 0 | 0 | 0 | 2 (0.6) | 2 (0.6) |
Condition under investigation worsened | 138 (41.7) | 138 (41.7) | 132 (39.4) | 140 (41.8) | 183 (55.3) | 185 (55.9) |
Patient lost to follow-up | 0 | 0 | 1 (0.3) | 1 (0.3) | 1 (0.3) | 1 (0.3) |
Maximum cycle of chemotherapy reached | 95 (28.7) | 95 (28.7) | 95 (28.4) | 95 (28.4) | 73 (22.1) | 74 (22.4) |
Other | 3 (0.9) | 5 (1.5) | 6 (1.8) | 7 (2.1) | 3 (0.9) | 4 (1.2) |
Patients ongoing treatment at DCOd,h | 37 (11.2) | 20 (6.0) | 31 (9.3) | 13 (3.9) | 5 (1.5) | 1 (0.3) |
Patients who discontinued treatmentd,i | 294 (88.8) | 311 (94.0) | 304 (90.7) | 322 (96.1) | 326 (98.5) | 330 (99.7) |
Patients ongoing T re-treatment at DCOd | 1 (0.3) | 0 | NA | NA | NA | NA |
Patients completed T re-treatmentd | 2 (0.6) | 3 (0.9) | NA | NA | NA | NA |
Patients discontinued T re-treatmentd | 7 (2.1) | 7 (2.1) | NA | NA | NA | NA |
Patients alive and ongoing study at FA DCOc,j and completed study at 5Y DCOc,k | 80 (23.7) | 50 (14.8) | 65 (19.2) | 38 (11.2) | 40 (11.9) | 19 (5.6) |
Patients who terminated studyc | 258 (76.3) | 288 (85.2) | 273 (80.8) | 300 (88.8) | 297 (88.1) | 318 (94.4) |
Death | 250 (74.0) | 278 (82.2) | 264 (78.1) | 290 (85.8) | 279 (82.8) | 298 (88.4) |
Lost to follow-up | 2 (0.6) | 3 (0.9) | 2 (0.6) | 2 (0.6) | 2 (0.6) | 3 (0.9) |
Withdrawal by patient | 6 (1.8) | 7 (2.1) | 7 (2.1) | 8 (2.4) | 16 (4.7) | 17 (5.0) |
5Y = 5-year; CDA-AMC = Canada’s Drug Agency’ D = durvalumab; DCO = data cut-off; eCRF = electronic case report form; FA = final analysis; NA = not available; OS = overall survival; SOC = standard of care; t = tremelimumab.
Notes: Results from the durvalumab plus SOC chemotherapy arm are not relevant to the systematic review because this regimen is not part of the Health Canada–approved indication and reimbursement request under review by CDA-AMC.
For patients who underwent re-treatment with T, definitions were as follows:60 If a patient discontinues T re-treatment for the reason “maximum cycle of immunotherapy reached,” they are classed as “completed.” If a patient discontinues T re-treatment for any reason other than “maximum cycle of immunotherapy reached,” they are classed as “discontinued.” If a patient started T re-treatment, but has either not discontinued T re-treatment or has not filled out the DOSDISC8 form, they are classed as “ongoing.” DCO dates: March 12, 2021 (final analysis), and August 24, 2023 (5-year OS analysis).
aThe median duration of follow-up in censored patients was 63.44 months (range, 0 to 73.9 months) at the 5-year DCO date.
bPercentages are calculated from number of patients enrolled.
cPercentages are calculated from number of patients in the FAS.
dPercentages are calculated from number of patients who received treatment.
ePatients who received tremelimumab re-treatment were not included.
fPatients who completed tremelimumab have “maximum cycle of immunotherapy reached” reported for tremelimumab on the eCRF. Patients who completed SOC chemotherapy have “maximum cycle of chemotherapy reached” reported for any SOC chemotherapy molecule on the eCRF.
gA patient is considered as having discontinued SOC chemotherapy when all molecules are discontinued. If different reasons for discontinuation are collected, the last discontinuation reason by date is selected.
hA patient is considered to be ongoing treatment when receiving at least 1 study treatment at DCO. Treatment includes D, T, CT, and T re-treatment where applicable.
iA patient is considered as having discontinued treatment when the patient has discontinued all study treatments. Treatment includes D, T, CT, and T re-treatment where applicable.
jRepresents patients who were alive and continuing in the study at the DCO date of the FA.
kPatients who completed the study includes patients who are alive and remain in the study at 5-year DCO. One patient was marked as completed (alive) at OS final analysis (March 12, 2021), and updated information is not available. One patient was marked as completed (alive) at the 4-year OS analysis (March 11, 2022).
Sources: Sponsor’s Summary of Clinical Evidence,1 POSEIDON Clinical Study Report,38 and POSEIDON Clinical Study Report 5-year OS Addendum.58
Baseline Characteristics
Demographic and baseline tumour characteristics of patients in the POSEIDON trial (Table 14) were largely similar between arm 1 and arm 3 of the POSEIDON trial. Across all 3 treatment arms (FAS), the median age of patients was 64 years (range, 27 to 87), with approximately 47% of patients aged 65 years or older and the majority of patients being male (76.0%) and white (55.9%). The durvalumab and tremelimumab plus SOC chemotherapy group (arm 1) had a lower percentage of female patients compared to the SOC chemotherapy group (arm 3) (20.4% versus 26.4%) as well as a lower percentage of Asian patients compared to the chemotherapy group (29.3% versus 38.0%). Overall, most patients were either current or former smokers (78%) and 21.9% of patients reported having never smoked, with a lower proportion of patients in the durvalumab and tremelimumab plus SOC chemotherapy arm being never smokers compared with the SOC chemotherapy arm (17.5% versus 23.4%). Overall, the majority of patients had an ECOG PS of 1 (66.5%), nonsquamous histology (62.9%), a PD-L1 TC level of less than 50% (71.1%), and 10.5% of patients had brain and/or CNS metastases at study entry. Between arm 1 and arm 3, histology type, PD-L1 status, and receipt of prior anticancer therapy (cytotoxic chemotherapy or radiotherapy) were generally well balanced.1,38
Table 14: Summary of Baseline Characteristics of the POSEIDON Trial (Full Analysis Set)
Characteristic | D + T + chemotherapy (N = 338) | D + chemotherapy (N = 338) | Chemotherapy (N = 337) |
|---|---|---|---|
Age (years)a | |||
Mean (SD) | 62.6 (9.43) | 63.5 (9.10) | 63.1 (9.87) |
Median (range) | 63.0 (27 to 87) | 64.5 (32 to 87) | 64.0 (32 to 84) |
Age group (years), n (%)a | |||
≥ 18 to < 50 | 29 (8.6) | 27 (8.0) | 30 (8.9) |
≥ 50 to < 65 | 162 (47.9) | 142 (42.0) | 146 (43.3) |
≥ 65 to < 75 | 112 (33.1) | 130 (38.5) | 121 (35.9) |
≥ 75 | 35 (10.4) | 39 (11.5) | 40 (11.9) |
Sex, n (%) | |||
Male | 269 (79.6) | 253 (74.9) | 248 (73.6) |
Female | 69 (20.4) | 85 (25.1) | 89 (26.4) |
Race, n (%) | |||
American Indian [wording from original source] or Alaska Native | 12 (3.6) | 17 (5.0) | 9 (2.7) |
Asian | 99 (29.3) | 123 (36.4) | 128 (38.0) |
Black or African American | 8 (2.4) | 4 (1.2) | 8 (2.4) |
Native Hawaiian or other Pacific Islander | 2 (0.6) | 0 | 0 |
White | 205 (60.7) | 182 (53.8) | 179 (53.1) |
Other | 12 (3.6) | 12 (3.6) | 13 (3.9) |
Geographic region, n (%) | |||
Eastern Europe | 122 (36.1) | 103 (30.5) | 95 (28.2) |
Asia | 96 (28.4) | 120 (35.5) | 124 (36.8) |
North America | 44 (13.0) | 46 (13.6) | 40 (11.9) |
South America | 34 (10.1) | 32 (9.5) | 41 (12.2) |
Western Europe | 29 (8.6) | 26 (7.7) | 28 (8.3) |
Africa | 13 (3.8) | 11 (3.3) | 9 (2.7) |
ECOG Performance Status,b n (%) | |||
0 | 110 (32.5) | 109 (32.2) | 119 (35.3) |
1 | 228 (67.5) | 229 (67.8) | 217 (64.4) |
Missing | 0 | 0 | 1 (0.3) |
AJCC disease stagec | |||
IIIA | 1 (0.3) | 0 | 0 |
IIIB | 1 (0.3) | 1 (0.3) | 0 |
IVA | 171 (50.6) | 170 (50.3) | 166 (49.3) |
IVB | 165 (48.8) | 167 (49.4) | 170 (50.4) |
Missing | 0 | 0 | 1 (0.3) |
BMI group (kg/m2), n (%) | |||
n | 335 | 338 | 335 |
Underweight (< 18.5) | 21 (6.3) | 23 (6.8) | 29 (8.7) |
Normal (18.5 to 25) | 184 (54.9) | 187 (55.3) | 181 (54.0) |
Overweight (25 to 30) | 93 (27.8) | 96 (28.4) | 91 (27.2) |
Obese (> 30) | 37 (11.0) | 32 (9.5) | 34 (10.1) |
Missing | 3 | 0 | 2 |
Smoking status, n (%) | |||
Never | 59 (17.5) | 84 (24.9) | 79 (23.4) |
Current | 84 (24.9) | 64 (18.9) | 66 (19.6) |
Former | 195 (57.7) | 190 (56.2) | 191 (56.7) |
Missing | 0 | 0 | 1 (0.3) |
Histology type, n (%) | |||
Squamous | 124 (36.7) | 128 (37.9) | 122 (36.2) |
Squamous-cell carcinoma | 124 (36.7) | 127 (37.6) | 122 (36.2) |
Other | 0 | 1 (0.3) | 0 |
Nonsquamous | 214 (63.3) | 209 (61.8) | 214 (63.5) |
Adenocarcinoma | 208 (61.5) | 203 (60.1) | 211 (62.6) |
Large-cell carcinoma | 2 (0.6) | 5 (1.5) | 3 (0.9) |
Other | 4 (1.2) | 1 (0.3) | 0 |
Other | 0 | 1 (0.3) | 0 |
Missing | 0 | 0 | 1 (0.3) |
Overall disease classification, n (%) | |||
Metastaticd | 337 (99.7) | 336 (99.4) | 336 (99.7) |
Locally advancede | 0 | 2 (0.6) | 0 |
Missing | 1 (0.3) | 0 | 1 (0.3) |
PD-L1 status,f n (%) | |||
TC < 50% | 237 (70.1) | 243 (71.9) | 240 (71.2) |
TC ≥ 50% | 101 (29.9) | 94 (27.8) | 97 (28.8) |
Missing | 0 | 1 (0.3) | 0 |
Central nervous system metastases, n (%) | 33 (9.8) | 28 (8.3) | 45 (13.4) |
Liver metastases, n (%) | 69 (20.4) | 62 (18.3) | 80 (23.7) |
Prior anticancer therapy, n (%) | |||
Cytotoxic chemotherapy | 13 (3.8) | 11 (3.3) | 14 (4.2) |
Adjuvant | 10 (3.0) | 7 (2.1) | 8 (2.4) |
Neoadjuvant | 2 (0.6) | 1 (0.3) | 0 |
Definitive | 1 (0.3) | 2 (0.6) | 7 (2.1) |
Missing | 1 (0.3) | 1 (0.3) | 0 |
Radiotherapy | 50 (14.8) | 43 (12.7) | 52 (15.4) |
Adjuvant | 8 (2.4) | 6 (1.8) | 2 (0.6) |
Neoadjuvant | 1 (0.3) | 2 (0.6) | 2 (0.6) |
Palliative | 34 (10.1) | 32 (9.5) | 42 (12.5) |
Definitive | 9 (2.7) | 2 (0.6) | 7 (2.1) |
Not applicable | 0 | 1 (0.3) | 0 |
AJCC = American Joint Committee on Cancer; BMI = body mass index; CDA-AMC = Canada’s Drug Agency; D = durvalumab; ECOG = Eastern Cooperative Oncology Group; eCRF = electronic case report form; IASLC = The International Association for the Study of Lung Cancer; SD = standard deviation; t = tremelimumab; TC = tumour cell.
Notes: Results from the durvalumab plus SOC chemotherapy arm are not relevant to the systematic review because this regimen is not part of the Health Canada–approved indication and reimbursement request under review by CDA-AMC.
Percentages are calculated from number of patients in the full analysis set in that treatment group.
aAge at randomization.
bECOG performance status at baseline, where baseline is defined as the last evaluable assessment before randomization.
cThe IASLC is the group responsible for conducting research to inform future versions of the AJCC. Version 8 of the IASLC Staging Manual in Thoracic Oncology was endorsed and published by the AJCC as the AJCC 8th edition.15,43 The 2 are considered interchangeable.45,46
dMetastatic disease: patient has any metastatic site of disease.
eLocally advanced: patient has only locally advanced sites of disease. Two patients in the D + T + CT arm and 1 in the durvalumab plus chemotherapy arm were incorrectly randomly assigned with stage III disease; these were reported as protocol deviations.
fStratification factor recorded on an eCRF. PD-L1 tumour-expression status is summarized based on laboratory data outside of the eCRF.
Sources: Sponsor’s Summary of Clinical Evidence1 and POSEIDON Clinical Study Report.38
Exposure to Study Treatments
Patient exposure to study treatments in arm 1 and arm 3 of the POSEIDON trial is summarized in Table 15. In the SAF, the mean and median duration of exposure to durvalumab and tremelimumab plus SOC chemotherapy was longer than that to SOC chemotherapy for both DCO dates. For the final analysis DCO (March 12, 2021), the median extents of exposure were 29.93 weeks (range, 1.10 to 189.6) and 18.00 weeks (range, 0.7 to 184.4) in arms 1 and 3, respectively. For the same DCO, the mean extents of exposure were 49.62 weeks (standard deviation [SD] = 48.151) and 25.83 weeks (SD = 29.004) in arms 1 and 3, respectively. For the 5-year OS analysis DCO (August 24, 2023), the median extents of exposure were 29.93 weeks (range, 1.1 to 317.4) and 18.00 weeks (range, 0.7 to 284.1) in arms 1 and 3, respectively. For the 5-year DCO, the mean extent of exposure was 60.37 weeks (SD = 75.397) and 27.26 weeks (SD = 36.968) in arms 1 and 3, respectively. At the final analysis, the total durations of treatment across all patients were 313.8 years in the durvalumab and tremelimumab plus SOC chemotherapy arm and 164.9 years in the SOC chemotherapy alone arm. At the 5-year OS update analysis the total duration of treatment across all patients had increased to 381.8 years in the durvalumab and tremelimumab plus SOC chemotherapy arm and 174.0 years in the SOC chemotherapy alone arm.
Patient exposure for durvalumab, tremelimumab, and SOC chemotherapy treatments in the durvalumab and tremelimumab plus SOC chemotherapy arm (arm 1, SAF) is summarized in Table 16. The median number of infusions and median duration of exposure to durvalumab and to SOC chemotherapy was the same for both the final analysis and 5-year OS DCOs. With the longer duration of follow-up, the maximum number of infusions and the maximum duration of exposure (in weeks) increased from 49 to 80, and 189.6 to 317.4, respectively for both durvalumab and SOC chemotherapy between the 2 DCOs. In arm 1 at the 5-year OS DCO, there are an additional 65.5 patient-years of exposure to durvalumab and an additional 35 patient-years for chemotherapy (with longer exposure to chemotherapy because of patients who received pemetrexed maintenance therapy). There was no change in the total number of infusions of tremelimumab or duration of exposure in arm 1 between the 2 DCOs.
Patient exposure to chemotherapy in the SOC chemotherapy arm (arm 3, SAF) is summarized in Table 17. The median numbers of infusions and median duration of exposure to chemotherapy were the same for both the final analysis and 5-year OS DCOs. The maximum number of chemotherapy infusions and the maximum duration of exposure (in weeks) increased from 57 to 94, and 184.4 to 284.1, respectively between the 2 DCOs. In arm 3 at the 5-year OS DCO, there are 9.1 additional patient-years of exposure to SOC chemotherapy because of patients who received pemetrexed maintenance therapy.
Among patients who received SOC chemotherapy, patients with nonsquamous histology most commonly received the pemetrexed-platinum regimen (598 of 626 [95.5%]) and patients with squamous histology most commonly received the gemcitabine platinum regimen (326 of 369 [88.3%]).39
Table 15: Summary of Patient Exposure From the POSEIDON Trial (Safety Analysis Set)
Total treatment duration (weeks)a | D + T + chemotherapy (N = 330) | Chemotherapy (N = 333) | ||
|---|---|---|---|---|
Final analysis | 5-year OS analysis | Final analysis | 5-year OS analysis | |
n | 330 | 330 | 333 | 333 |
Mean | 49.62 | 60.37 | 25.83 | 27.26 |
SD | 48.151 | 75.397 | 29.004 | 36.968 |
Median | 29.93 | 29.93 | 18.00 | 18.00 |
Minimum | 1.10 | 1.1 | 0.7 | 0.7 |
Maximum | 189.6 | 317.4 | 184.4 | 284.1 |
Total treatment years | 313.8 | 381.8 | 164.9 | 174.0 |
D = durvalumab; DCO = data cut-off; OS = overall survival; SD = standard deviation; t = tremelimumab.
Note: Percentages are calculated from number of patients in the safety analysis set in that treatment group. Chemotherapy for a patient who received it during re-treatment is also included in this table. DCO dates: March 12, 2021 (final analysis), and August 24, 2023 (5-year OS analysis).
aTotal treatment duration = minimum of (last infusion or dose date of the last cycle + 20 days [if last infusion or dose date was during chemotherapy] or last infusion or dose date of the last cycle + 27 days [if last infusion or dose date was postchemotherapy], date of death, date of DCO) – first infusion or dose date of first cycle + 1.
Sources: Sponsor’s Summary of Clinical Evidence,1 POSEIDON Clinical Study Report,38 and POSEIDON Clinical Study Report 5-year OS Addendum.58
Table 16: Patient Exposure in the Durvalumab and Tremelimumab Plus Chemotherapy Arm (N = 330) of the POSEIDON Trial (Safety Analysis Set)
Exposure | Durvalumab | Tremelimumab | Chemotherapy | |||
|---|---|---|---|---|---|---|
Final analysis | 5-year OS analysisa | Final analysis | 5-year OS analysisa | Final analysis | 5-year OS analysisa | |
Total number of infusions | ||||||
Mean (SD) | 12.5 (11.74) | 14.9 (17.93) | 4.3 (1.43) | 4.3 (1.43) | 10.7 (9.77) | 11.9 (14.22) |
Median | 8.0 | 8.0 | 5.0 | 5.0 | 8.0 | 8.0 |
Minimum to maximum | 1 to 49 | 1 to 80 | 1 to 9 | 1 to 9 | 1 to 49 | 1 to 80 |
Total exposure duration (weeks)b | ||||||
Duration, mean (SD) | 48.83 (47.979) | 59.19 (74.569) | 17.78 (7.361) | 17.78 (7.361) | 35.35 (41.733) | 40.91 (60.542) |
Duration, median | 29.79 | 29.79 | 20.0 | 20.0 | 15.0 | 15.0 |
Minimum to maximum | 1.1 to 189.6 | 1.1 to 317.4 | 1.1 to 38.3 | 1.1 to 38.3 | 1.1 to 189.6 | 1.1 to 317.4 |
Total treatment (patient-years) | 308.8 | 374.3 | 112.4 | 112.4 | 222.9 | 257.9 |
DCO = data cut-off; OS = overall survival; SD = standard deviation.
Note: DCO dates: March 12, 2021 (final analysis), and August 24, 2023 (5-year OS analysis).
aThe median duration of follow-up in censored patients was 63.44 months (range, 0 to 73.9 months) at the 5-year data cut-off date.
bTotal treatment duration = minimum of (last infusion or dose date of the last cycle + 20 days [if last infusion or dose date was during combination] or last infusion or dose date of the last cycle + 27 days [if last infusion or dose date was in maintenance or tremelimumab re-treatment], date of death, date of DCO) – first infusion or dose date of first cycle + 1.
Sources: Sponsor’s Summary of Clinical Evidence,1 POSEIDON Clinical Study Report,38 and POSEIDON Clinical Study Report 5-year OS Addendum.58
Table 17: Patient Exposure in the Chemotherapy Arm (N = 333) of the POSEIDON Trial (SAF)
Exposure | Chemotherapy | |
|---|---|---|
Final analysis | 5-year OS analysisa | |
Total number of infusions | ||
Mean (SD) | 9.1 (8.33) | 9.5 (10.74) |
Median | 8.0 | 8.0 |
Minimum to maximum | 1 to 57 | 1 to 94 |
Total exposure duration (weeks)b | ||
Duration, mean (SD) | 25.83 (29.004) | 27.26 (36.968) |
Duration, median | 18.0 | 18.0 |
Minimum to maximum | 0.7 to 184.4 | 0.7 to 284.1 |
Total treatment (patient-years) | 164.9 | 174.0 |
DCO = data cut-off OS = overall survival; SAF = safety analysis set; SD = standard deviation.
Note: DCO dates: March 12, 2021 (final analysis), and August 24, 2023 (5-year OS analysis).
aThe median duration of follow-up in censored patients was 63.44 months (range, 0 to 73.9 months) at the 5-year data cut-off date.
bTotal treatment duration = minimum of (last infusion or dose date of the last cycle + 20 days [if last infusion or dose date was during combination] or last infusion or dose date of the last cycle + 27 days [if last infusion or dose date was in maintenance], date of death, date of DCO) – first infusion or dose date of first cycle + 1.
Sources: Sponsor’s Summary of Clinical Evidence,1 POSEIDON Clinical Study Report,38 and POSEIDON Clinical Study Report 5-Year OS Addendum.58
Durvalumab Treatment Through Progression
Patients in arm 1 (durvalumab and tremelimumab plus SOC chemotherapy) who were clinically stable at the initial RECIST 1.1–defined progressive disease assessment continued to receive durvalumab monotherapy, at the discretion of the investigator and patient, until confirmation of progression at the next subsequent scan (no sooner than 4 weeks and preferably at the next scheduled imaging visit). Patients who were considered by the investigator to be still benefiting from durvalumab monotherapy following confirmed progression could continue receiving durvalumab monotherapy as long as criteria for treatment in the setting of objective radiological progression were fulfilled.
The durvalumab and tremelimumab plus SOC chemotherapy arm included 132 patients (40.0%) with a RECIST 1.1–defined disease progression followed by a subsequent scan confirming RECIST 1.1–defined disease progression. In the durvalumab and tremelimumab plus SOC chemotherapy arm, 36 patients (10.9%) received treatment with durvalumab following the date of disease progression confirmation and 18 patients (5.5%) continued to receive durvalumab 8 weeks (56 days) after progression confirmation, with a median duration of treatment postprogression confirmation of 8.4 weeks. Data for patients receiving durvalumab after the date of disease progression confirmation in the durvalumab and tremelimumab plus SOC chemotherapy arm are presented in Table 18.60
Tremelimumab Re-Treatment
Patients in the durvalumab and tremelimumab plus SOC chemotherapy arm with radiological progression who, in the investigator’s opinion, continued to receive benefit from their assigned treatment and who met the criteria for re-treatment in the setting of disease progression, could be re-treated with 4 doses per cycle of tremelimumab alongside durvalumab followed by continued durvalumab monotherapy.1,38,47
At the final analysis, 11 patients (3.3%) in the durvalumab and tremelimumab plus SOC chemotherapy arm received 6 or more cycles of tremelimumab (i.e., patients receiving tremelimumab re-treatment).38 Table 13 reports a total of 10 patients as having received tremelimumab re-treatment (presented in the rows of ongoing, completed, and discontinued tremelimumab re-treatment) as the data in this table align with patient disposition data from the 5-year follow-up Clinical Study Report. The difference in the 1 additional patient being reported was because of incorrect data entry.60
Table 18: Summary of Patients Who Received Durvalumab Through Confirmed Progression (Based on Site-Investigator Assessments According to RECIST 1.1) (Safety Analysis Set)
Category | Durvalumab + tremelimumab + chemotherapy (N = 330) |
|---|---|
Durvalumab-treated patients with 2 consecutive RECIST 1.1 progressionsa | ███ ██████ |
Date of last durvalumab dose > date of progression confirmation | ██ ██████ |
Date of last durvalumab dose > 56 days postprogression confirmation | ██ █████ |
Duration of treatment postprogression (weeks) | |
Minimum | ███ |
First quartile | ███ |
Mean | ████ |
Median | ███ |
Third quartile | ████ |
Maximum | █████ |
RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1; SOC = standard of care.
Note: The date of progression confirmation is defined as the date of the second progression assessment of the first case of 2 consecutive assessments of progression. Duration of treatment postprogression is defined as the time (weeks) from progression confirmation until the date of last durvalumab dose + 1 day. There were 6 patients in the durvalumab and tremelimumab plus SOC chemotherapy arm (N = 330) in whom the durvalumab component was not the last to be discontinued. Data cut-off: March 12, 2021.
aProgression based on site-investigator assessments according to RECIST 1.1.
Source: Sponsor’s data on file.60
Concomitant Medications and Cointerventions
A summary of allowed concomitant medications that were taken during study treatment (by ≥ 20% of patients in any treatment arm) in the POSEIDON trial is presented in Table 19. At the final analysis DCO (March 12, 2021), almost all patients in the durvalumab and tremelimumab plus SOC chemotherapy and SOC chemotherapy alone arms ██████ ███ █████, respectively) received allowed concomitant medications during study treatment. The most commonly received concomitant medications were serotonin (5HT3) antagonists and glucocorticoids. Concomitant medications with a 5% or greater difference between the durvalumab and tremelimumab plus SOC chemotherapy and SOC chemotherapy alone arms, respectively, were serotonin antagonists (█████ ██ █████); anilides ██████ ██ ██████; combinations of penicillin, including beta-lactamase inhibitors ██████ ██ ██████; and colony-stimulating factors ██████ ██ ██████.38
Table 19: Allowed Concomitant Medications During Study Treatment Received by 20% or More of Patients in Any Treatment Arm (Full Analysis Set)
Concomitant medications (by ATC classification) | POSEIDON | ||
|---|---|---|---|
D + T + chemotherapy (N = 338) | D + chemotherapy (N = 338) | Chemotherapy (N = 337) | |
Number of patients with allowed concomitant medications | ███ ██████ | ███ █████ | ███ █████ |
Serotonin (5HT3) antagonists | ███ ██████ | ███ █████ | ███ █████ |
Glucocorticoids | ███ ██████ | ███ █████ | ███ █████ |
Folic acid and derivatives | ███ ██████ | ███ █████ | ███ █████ |
Vitamin B12 (cyanocobalamin and analogues) | ███ ██████ | ███ █████ | ███ █████ |
Proton pump inhibitors | ███ ██████ | ███ █████ | ███ █████ |
Propulsives | ███ ██████ | ███ █████ | ███ █████ |
Anilides | ███ ██████ | ███ █████ | ███ █████ |
Electrolyte solutions | ███ ██████ | ███ █████ | ██ ██████ |
H2-receptor antagonists | ██ ██████ | ███ █████ | ██ ██████ |
Natural opium alkaloids | ██ ██████ | ███ █████ | ██ ██████ |
Osmotically acting laxatives | ██ ██████ | ██ ██████ | ██ ██████ |
Other antiemetics | ██ ██████ | ██ ██████ | ██ ██████ |
Fluoroquinolones | ██ ██████ | ██ ██████ | ██ ██████ |
Contact laxatives | ██ ██████ | ██ ██████ | ██ ██████ |
Combinations of penicillins, including beta-lactamase inhibitors | ██ ██████ | ██ ██████ | ██ ██████ |
Colony-stimulating factors | ██ ██████ | ██ ██████ | ██ ██████ |
Propionic acid derivatives | ██ ██████ | ██ ██████ | ██ ██████ |
Other opioids | ██ ██████ | ██ ██████ | ██ ██████ |
Other blood products | ██ ██████ | ██ ██████ | ██ ██████ |
ATC = Anatomic Therapeutic Chemical; D = durvalumab; H2 = histamine H2; t = tremelimumab.
Note: A patient can have 1 or more generic terms reported under a given ATC classification. Includes medications that began before randomization and were ongoing after randomization, or medications that started on or after date of randomization and before the date of last dose of study drug plus 90 days. WHO Drug Dictionary version 202009. Percentages are calculated from number of patients in the full analysis set in that treatment arm. Data cut-off dates: March 12, 2021 (final analysis).
Sources: Sponsor’s Summary of Clinical Evidence1 and POSEIDON Clinical Study Report.38
Subsequent Treatment
A summary of subsequent anticancer treatments received by patients in the POSEIDON trial following discontinuation of study treatment is presented in Table 20. At both the final analysis and 5-year OS analysis DCO dates, a smaller proportion of patients in the durvalumab and tremelimumab plus SOC chemotherapy group (arm 1) received subsequent anticancer therapy compared to patients in the SOC chemotherapy group (arm 3). At the final analysis DCO date (March 12, 2021), 40.8% and 60.2% of patients in arms 1 and 3, respectively, had received subsequent anticancer treatment; at the 5-year OS analysis (August 24, 2023), these percentages were 42.9% and 60.8%, respectively. Systemic therapy was used more commonly than radiotherapy in both treatment arms. At the 5-year OS DCO (August 24, 2023), 37.9% of patients in the durvalumab and tremelimumab plus SOC chemotherapy arm and 57.9% of patients in the SOC chemotherapy alone arm had received subsequent systemic therapy. At the same DCO, generally similar proportions of patients in arms 1 and 3 (33.4% and 36.2%, respectively) had received cytotoxic chemotherapy; however, a smaller proportion of patients in the durvalumab and tremelimumab plus SOC chemotherapy arm had received subsequent immunotherapy compared to those in the SOC chemotherapy alone arm (7.4% versus 33.2%).
Table 20: Subsequent Anticancer Treatments in the POSEIDON Trial (Full Analysis Set)
Subsequent treatmentsa | POSEIDON | |||||
|---|---|---|---|---|---|---|
D + T + chemotherapy (N = 338) | D + chemotherapy (N = 338) | Chemotherapy (N = 337) | ||||
Final analysis | 5-year OS analysisb | Final analysis | 5-year OS analysisb | Final analysis | 5-year OS analysisb | |
Patients with postdiscontinuation anticancer therapy, n (%) | 138 (40.8) | 145 (42.9) | 150 (44.4) | 159 (47.0) | 203 (60.2) | 205 (60.8) |
Systemic therapy | 123 (36.4) | 128 (37.9) | 139 (41.1) | 145 (42.9) | 194 (57.6) | 195 (57.9) |
Cytotoxic chemotherapy | 107 (31.7) | 113 (33.4) | 128 (37.9) | 131 (38.8) | 122 (36.2) | 122 (36.2) |
Single drug | 76 (22.5) | 80 (23.7) | 95 (28.1) | 96 (28.4) | 87 (25.8) | 85 (25.2) |
Platinum doublet | 37 (10.9) | 41 (12.1) | 31 (9.2) | 33 (9.8) | 24 (7.1) | 24 (7.1) |
Other combination | 16 (4.7) | 18 (5.3) | 28 (8.3) | 31 (9.2) | 28 (8.3) | 31 (9.2) |
Immunotherapy | 22 (6.5) | 25 (7.4) | 22 (6.5) | 24 (7.1) | 112 (33.2) | 112 (33.2) |
IO only | 17 (5.0) | 18 (5.3) | 20 (5.9) | 20 (5.9) | 97 (28.8) | 97 (28.8) |
IO + chemotherapy | 1 (0.3) | 1 (0.3) | 0 | 0 | 9 (2.7) | 10 (3.0) |
IO + IOc | — | 1 (0.3) | — | 2 (0.6) | — | 1 (0.3) |
IO + other | 4 (1.2) | 5 (1.5) | 3 (0.9) | 4 (1.2) | 6 (1.8) | 5 (1.5) |
Targeted therapy | 14 (4.1) | 14 (4.1) | 13 (3.8) | 17 (5.0) | 19 (5.6) | 22 (6.5) |
Other | 4 (1.2) | 5 (1.5) | 2 (0.6) | 2 (0.6) | 6 (1.8) | 5 (1.5) |
Radiotherapy | 48 (14.2) | 50 (14.8) | 57 (16.9) | 63 (18.6) | 65 (19.3) | 66 (19.6) |
CDA-AMC = Canada’s Drug Agency; D = durvalumab; DCO = data cut-off; IO = immuno-oncology; OS = overall survival; SOC = standard of care; t = tremelimumab.
Note: Results from the durvalumab plus SOC chemotherapy arm are not relevant to the systematic review because this regimen is not part of the Health Canada–approved indication and reimbursement request under review by CDA-AMC.
Patients with therapies in more than 1 category are counted once in each of those categories. Percentages are calculated from the number of patients in the FAS in that treatment arm. DCO dates: March 12, 2021 (final analysis), and August 24, 2023 (5-year OS analysis).
aTherapies after discontinuation of study treatment.
bThe duration of follow-up in censored patients was 63.44 months (range, 0 to 73.9 months) at the 5-year DCO date.
cOne patient had their subsequent therapy updated from “IO + other” at the FA DCO to “IO + IO” at the 5-year DCO.
Sources: Sponsor’s Summary of Clinical Evidence,1 POSEIDON Clinical Study Report,38 and POSEIDON Clinical Study Report 5-year OS Addendum.58
Efficacy
Summary of Efficacy Outcomes
Efficacy results presented are for the final analysis based on DCO dates of July 24, 2019, for all RECIST 1.1–related end points and March 12, 2021, for all other data. Results from a long-term survival follow-up analysis are also presented for OS (5-year OS DCO: August 24, 2023).1,38,58
A summary of results for key efficacy outcomes for the durvalumab and tremelimumab plus SOC chemotherapy arm (arm 1) and SOC chemotherapy alone arm (arm 3) of the POSEIDON trial is presented in Table 21.
Table 21: Summary of Key Efficacy Results From the POSEIDON Trial
Result | D + T + chemotherapy | Chemotherapy |
|---|---|---|
Overall survival (final analysis; DCO: March 12, 2021) | N = 338 | N = 337 |
Death events, n (%) | 251 (74.3) | 285 (84.6) |
Overall survival (months), median (95% CI) | 14.0 (11.7 to 16.1) | 11.7 (10.5 to 13.1) |
Difference, months (95% CI) | ███ ██████ ████ | |
HR (95% CI)a,b | 0.77 (0.650 to 0.916) | |
P valuec | 0.00304c | |
Survival probability (%) at 12 months (95% CI)d | 54.8 (49.3 to 60.0) | 49.1 (43.6 to 54.4) |
Difference in survival probability, % (95% CI) | ███ ██████ █████ | |
Survival probability (%) at 18 months (95% CI)d | 41.3 (36.0 to 46.5) | 34.1 (29.0 to 39.2) |
Difference in survival probability, % (95% CI) | ███ ██████ █████ | |
Survival probability (%) at 24 months (95% CI)d | 32.9 (27.9 to 37.9) | 22.1 (17.8 to 26.8) |
Difference in survival probability, % (95% CI) | ████ █████ █████ | |
Survival probability (%) at 36 months (95% CI)d | 25.3 (20.8 to 30.2) | 13.3 (9.8 to 17.4) |
Difference in survival probability, % (95% CI) | ██ █████ █████ | |
Overall survival (5-year OS update; DCO: August 24, 2023)e | N = 338 | N = 337 |
Death events, n (%) | 279 (82.5) | 304 (90.2) |
Overall survival (months), median (95% CI) | 14.0 (11.7 to 16.1) | 11.6 (10.5 to 13.1) |
Difference, months (95% CI) | ███ ██████ ████ | |
HR (95% CI)a,b | 0.76 (0.642 to 0.893) | |
P value | NAf | |
Survival probability (%) at 48 months (95% CI)d | 20.9 (16.7 to 25.4) | 8.5 (5.8 to 11.9) |
Difference in survival probability, % (95% CI) | ████ █████ █████ | |
Survival probability (%) at 60 months (95% CI)d | 15.7 (12.0 to 19.9) | 6.8 (4.4 to 10.0) |
Difference in survival probability, % (95% CI) | ███ █████ █████ | |
Progression-free survival (final analysis; DCO: July 24, 2019) | N = 338 | N = 337 |
Events, n (%) | 238 (70.4) | 258 (76.6) |
Progression-free survival (months), median (95% CI) | 6.2 (5.0 to 6.5) | 4.8 (4.6 to 5.8) |
Difference, months (95% CI) | ███ ██████ ████ | |
HR | 0.72 | |
99.265% CIg | ██████ █████ | |
95% CI | 0.600 to 0.860 | |
2-sided P valueh | 0.00031 | |
Progression-free survival rate (%) at 12 months (95% CI) | 26.6 (21.7 to 31.7) | 13.1 (9.3 to 17.6) |
Difference in PFS rate, % (95% CI) | ████ █████ █████ | |
Objective response rate (unconfirmed responses) | N = 335 | N = 332 |
Objective response rate, n (% [95%CI]) | 155 (46.3 [40.9 to 51.8]) | 111 (33.4 [28.4 to 38.8]) |
Difference, % (95% CI) | ████ █████ █████ | |
Odds ratio, T + D + chemotherapy vs. chemotherapy alonei | 1.72 | |
95% CI for odds ratio | 1.260 to 2.367 | |
2-sided P value | < 0.001 | |
Best objective response, n (%) | N = 335 | N = 332 |
Response | 155 (46.3) | 111 (33.4) |
Complete response | 2 (0.6) | 0 |
Partial response | 153 (45.7) | 111 (33.4) |
Nonresponse | 180 (53.7) | 221 (66.6) |
Stable disease | 120 (35.8) | 150 (45.2) |
Progression | 48 (14.3) | 61 (18.4) |
Not evaluable | 12 (3.6) | 10 (3.0) |
Duration of response in patients with objective response | N = 335 | N = 332 |
Patients with confirmed objective response, n of N (%) | 130 of 335 (38.8) | 81 of 332 (24.4) |
Median duration of response, months (95% CI) | 9.5 (7.2 to NE) | 5.1 (4.4 to 6.0) |
In response at 12 months (95% CI) | 49.7 (40.4 to 58.4) | 21.4 (12.7 to 31.6) |
Difference, % (95% CI) | ████ ██████ █████ | |
HRQoL — time to deterioration | ||
EORTC QLQ-C30 Global Health Status/QoL | ||
n | 319 | 318 |
Total events, n (%)j | 175 (54.9) | 186 (58.5) |
Median time to deterioration, months (95% CI)d | 8.3 (6.4 to 10.2) | 5.6 (4.4 to 7.5) |
Difference, months (95% CI) | ███ ██████ ████ | |
HR (95% CI)a | 0.78 (0.631 to 0.961) | |
2-sided P valueh | █████ | |
EORTC QLQ-C30 physical functioning | ||
n | 323 | 320 |
Total events, n (%)j | 189 (58.5) | 193 (60.3) |
Median time to deterioration, months (95% CI)d | 7.7 (5.9 to 9.4) | 5.3 (4.2 to 6.5) |
HR (95% CI)a | 0.75 (0.610 to 0.920) | |
2-sided P valueh | █████ | |
EORTC QLQ-C30 fatigue | ||
n | 317 | 314 |
Total events, n (%)j | 232 (73.2) | 220 (70.1) |
Median time to deterioration, months (95% CI)d | 3.7 (2.8 to 5.0) | 2.8 (2.1 to 3.7) |
HR (95% CI)a | 0.90 (0.746 to 1.084) | |
2-sided P valueh | █████ | |
EORTC QLQ-C30 appetite loss | ||
n | 308 | 305 |
Total events, n (%)j | 179 (58.1) | 158 (51.3) |
Median time to deterioration, months (95% CI)d | 7.2 (5.7 to 9.4) | 7.0 (5.6 to 9.6) |
HR (95% CI)a | 0.94 (0.754 to 1.169) | |
2-sided P valueh | █████ | |
EORTC QLQ-LC13 coughing | ||
n | 302 | 295 |
Total events, n (%)j | 162 (53.6) | 142 (48.1) |
Median time to deterioration, months (95% CI)d | 9.7 (7.3 to 13.3) | 8.8 (6.8 to12.3) |
HR (95% CI)a | 0.91 (0.722 to 1.146) | |
2-sided P valueh | █████ | |
EORTC QLQ-LC13 dyspnea | ||
n | 325 | 316 |
Total events, n (%)j | 220 (67.7) | 211 (66.8) |
Median time to deterioration, months (95% CI)d | 5.4 (4.2 to 6.4) | 3.6 (2.6 to 4.5) |
HR (95% CI)a | 0.77 (0.635 to 0.936) | |
2-sided P valueh | █████ | |
EORTC QLQ-LC13 pain in chest | ||
n | 319 | 309 |
Total events, n (%)j | 172 (53.9) | 151 (48.9) |
Median time to deterioration, months (95% CI)d | 10.0 (7.7 to 13.3) | 8.6 (6.8 to 11.4) |
HR (95% CI)a | 0.85 (0.681 to 1.066) | |
2-sided P valueh | ██████ | |
CI = confidence interval; D = durvalumab; DCO = data cut-off; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-LC13 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Lung Cancer 13; HR = hazard ratio; HRQoL; health-related quality of life; NA = not available; OS = overall survival; QoL = quality of life; t = tremelimumab; vs. = versus.
Note: DCO dates: July 24, 2019, and March 12, 2021 (final analysis), and August 24, 2023 (5-year OS analysis).
aThe HR and CI were estimated from a stratified Cox proportional hazards model with the Efron method to control for ties, the stratification factors PD-L1 tumour expression (≥ 50% vs. < 50%), histology (squamous vs. nonsquamous), and disease stage (IVA vs. IVB) in the strata statement, and the CI calculated using a profile likelihood approach.
bAn HR of less than 1 favours D + T + CT to be associated with a longer OS than CT alone.
cP value has been adjusted for multiple testing.
dCalculated using the Kaplan-Meier technique.
eThe median duration of follow-up in censored patients was 63.44 months (range, 0 to 73.9 months) at the 5-year DCO date.
fThe 5-year estimates and CIs are not alpha-controlled.
gBased on a Lan-DeMets alpha spending function with an O’Brien-Fleming boundary with the actual number of events observed, the boundaries for declaring statistical significance are 0.00735 for a 1% overall alpha. Corresponding CIs are shown.
hP values were generated using the stratified log-rank test adjusting for PD-L1 (≥ 50% vs. < 50%), histology (squamous vs. nonsquamous), and disease stage (IVA vs. IVB) and using the Breslow approach for handling ties.
iAn odds ratio greater than 1 favours D + T + CT compared to CT alone.
jPatients who have not shown a clinically meaningful deterioration, or who showed clinically meaningful deterioration or died after 2 or more missed visits, will be censored at the time of the latest evaluable PRO assessment (or at the latest evaluable PRO assessment before the 2 missed visits), or at the date of randomization if there are no evaluable visits or no baseline data.
Sources: Sponsor’s Summary of Clinical Evidence,1 POSEIDON Clinical Study Report,38 POSEIDON Clinical Study Report 5-year OS update,58 and sponsor’s data on file.13
Overall Survival
As of the March 12, 2021, final analysis DCO, there were 251 deaths in the durvalumab and tremelimumab plus SOC chemotherapy arm and 285 deaths in the SOC chemotherapy alone arm for a total of 536 deaths across the 2 groups (79.4% maturity for OS overall).1,38 The final analysis of OS met the prespecified boundary for declaring statistical significance between the durvalumab and tremelimumab plus SOC chemotherapy versus SOC chemotherapy alone arms (2-sided P value boundary of 0.00797 for a 1% overall alpha recycled from the significant PFS test of durvalumab and tremelimumab plus SOC chemotherapy versus SOC chemotherapy alone in the FAS). The median OS was greater in the durvalumab and tremelimumab plus SOC chemotherapy group (14.0 months; 95% CI, 11.7 to 16.1 months) than in the SOC chemotherapy alone group (11.7 months; 95% CI, 10.5 to 13.1 months), with an HR of 0.77 (95% CI, 0.650 to 0.916; P = 0.00304), as presented in Table 21.
As of the August 24, 2023, DCO for 5-year OS, there were 279 deaths in the durvalumab and tremelimumab plus SOC chemotherapy arm and 304 deaths in the SOC chemotherapy alone arm for a total of 583 deaths across the 2 groups (86.4% maturity for OS overall, representing an approximate 7% increase in data maturity from the final analysis DCO).1,38 The median OS was greater in the durvalumab and tremelimumab plus SOC chemotherapy group (14.0 months; 95% CI, 11.7 to 16.1 months) than in the SOC chemotherapy alone group (11.6 months; 95% CI, 10.5 to 13.1 months), with an HR of 0.76 (95% CI, 0.642 to 0.893), as presented in Table 21. The 5-year estimates and CIs are not alpha-controlled.
The median duration of follow-up for the final analysis DCO of March 12, 2021, in censored patients in the durvalumab and tremelimumab plus SOC chemotherapy arm was 35.09 months (range, 1.5 to 43.9) and in the SOC chemotherapy alone arm was 34.18 months (range, 0.0 to 43.9). Overall, the median duration of follow-up in censored patients across the 3 treatment arms was 34.89 months (range, 0.0 to 44.5). In all patients, the median duration of follow-up in the durvalumab and tremelimumab plus SOC chemotherapy arm was 13.63 months (range 0.3 to 43.9) and in the SOC chemotherapy alone arm it was 11.17 months (range, 0.0 to 43.9); the median duration of follow-up in all patients across the 3 treatment arms was 12.52 months (range, 0.0 to 44.5).38
The median duration of follow-up for the 5-year OS DCO of August 24, 2023, in censored patients was 63.44 months (range, 0 to 73.9). In the durvalumab and tremelimumab plus SOC chemotherapy arm the median duration of follow-up in censored patients was 64.30 months (range, 1.5 to 73.3) and in the SOC chemotherapy alone arm it was 61.04 months (range, 0 to 73.3). In all patients, the median duration of follow-up in the durvalumab and tremelimumab plus SOC chemotherapy arm was 13.63 months (range, 0.3 to 73.3) and in the SOC chemotherapy alone arm it was 11.10 months (range, 0 to 73.3).58
Kaplan-Meier plots of OS for the comparison of durvalumab and tremelimumab plus SOC chemotherapy versus SOC chemotherapy alone at the final analysis and the 5-year OS analysis are presented in Figure 5 and Figure 6, respectively.
Figure 5: Kaplan-Meier Plot of OS — Durvalumab and Tremelimumab Plus Chemotherapy vs. Chemotherapy Alone, Final Analysis (FAS)
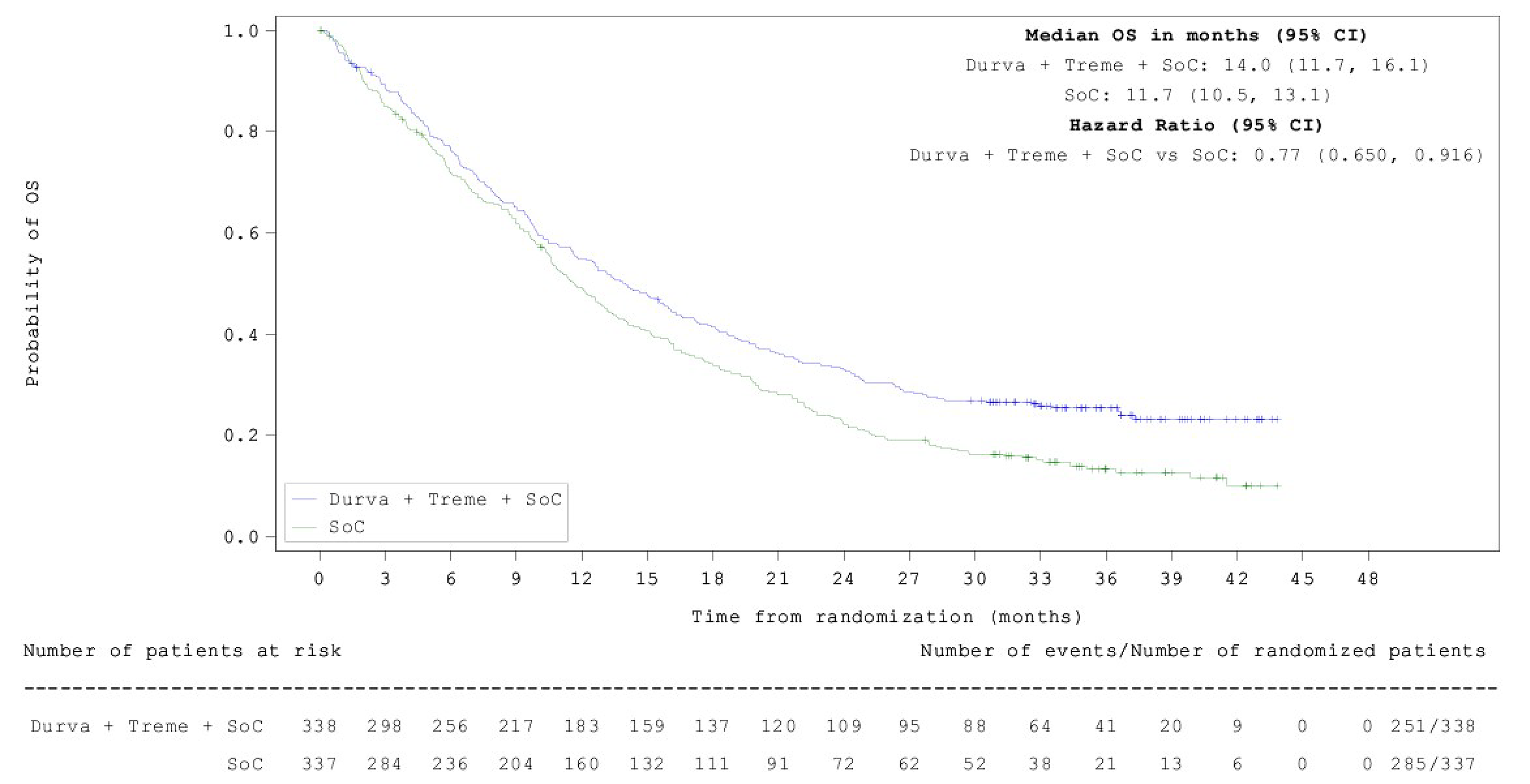
CI = confidence interval; Durva = durvalumab; FAS = full analysis set; OS = overall survival; SoC = standard of care chemotherapy; Treme = tremelimumab; vs. = versus.
Notes: One patient died 1 day before randomization and was censored at day 1. Data cut-off date: March 12, 2021.
The + symbol on the curves represents censored observations.
Source: POSEIDON Clinical Study Report.61
Figure 6: Kaplan-Meier Plot of OS — Durvalumab and Tremelimumab Plus Chemotherapy vs. Chemotherapy Alone, 5-Year OS Update (FAS)
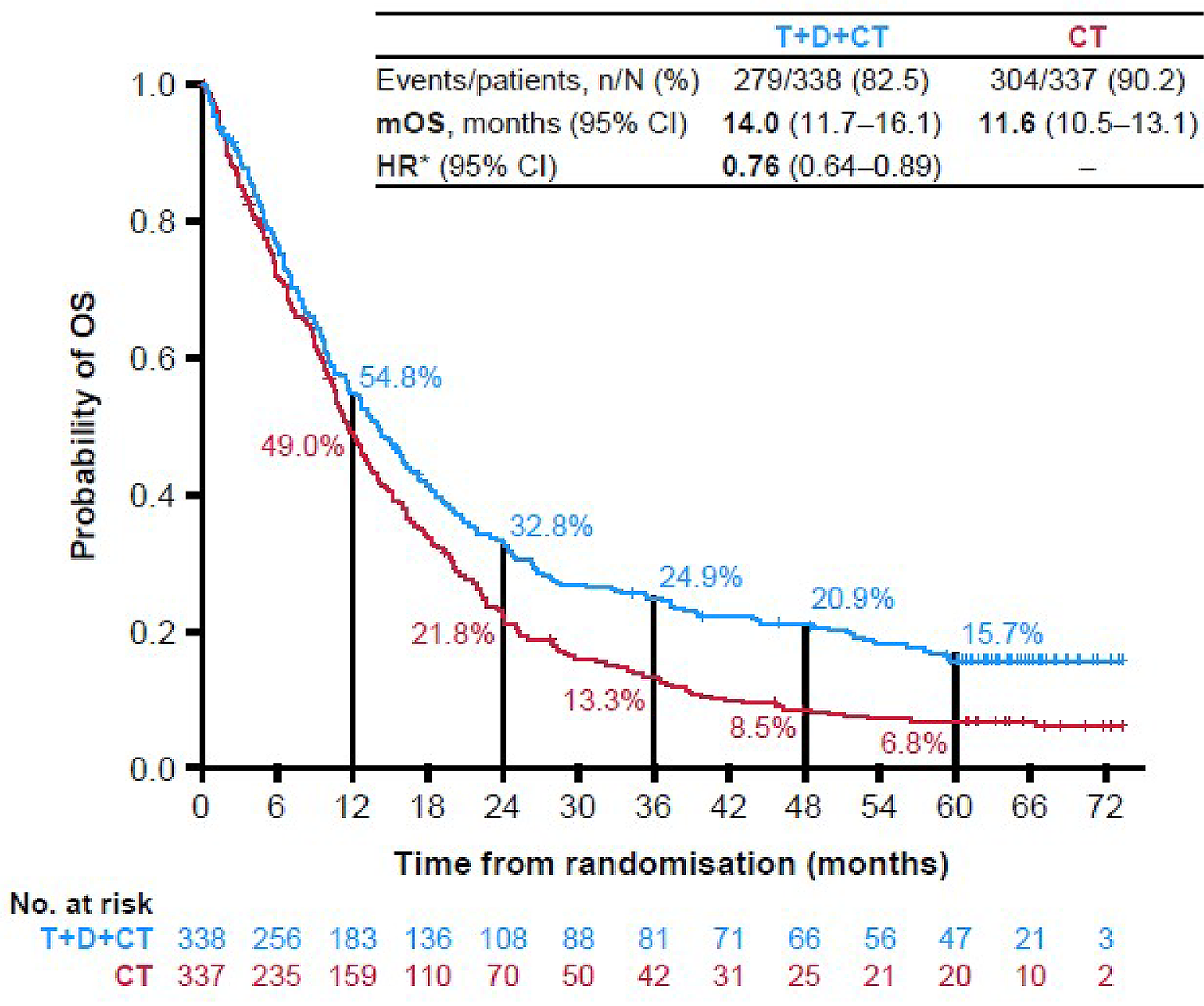
CI = confidence interval; CT = standard-of-care chemotherapy; D = durvalumab; FAS = full analysis set; HR = hazard ratio; mOS = median overall survival; OS = overall survival; t = tremelimumab; vs. = versus.
Source: Sponsor’s Summary of Clinical Evidence.1
Sensitivity Analyses of OS
Sensitivity analysis for OS adjusting for electronic case report form–derived stratification variables of PD-L1 status (≥ 50% versus < 50%), histology (squamous versus nonsquamous), and disease stage (IVA versus IVB) resulted in HRs of 0.78 (95% CI, 0.663 to 0.922) at the 5-year OS analysis and 0.79 (95% CI, 0.667 to 0.940, 2-sided P value = 0.008) at the previous final analysis.1,58,61
Two multivariate Cox models were used: model 1 adjusted for treatment and the stratification factors only and model 2 adjusted for treatment, stratification factors, and additional prespecified covariates. The HR results were 0.78 (95% CI, 0.658 to 0.925) from model 1 and 0.76 (95% CI, 0.635 to 0.903) from model 2.1,38
The impact of nonproportional hazards was assessed in a sensitivity analysis using a 3-component, stratified max-combo test with the same stratification factors as the primary analysis; a restricted mean survival time of an AUC approach was also performed. These analyses were generally consistent with the primary analyses.1
Subgroup Analyses of OS
At the final analysis DCO (March 12, 2021), the survival benefit of durvalumab and tremelimumab plus SOC chemotherapy compared to SOC chemotherapy alone was consistent across all subgroups, except for the subgroup of never smokers (HR = 1.15; 95% CI, 0.792 to 1.665). An OS benefit was observed in the subgroup of patients with nonsquamous histology for durvalumab and tremelimumab plus SOC chemotherapy compared to SOC chemotherapy alone (HR = 0.70; 95% CI, 0.558 to 0.870).1,38,61
At the 5-year OS analysis DCO (August 24, 2023), the survival benefit of durvalumab and tremelimumab plus SOC chemotherapy compared to SOC chemotherapy alone was also consistent across subgroups (estimated HRs were < 1), except for the subgroup of never smokers (HR = 1.17; 95% CI, 0.820 to 1.661), as shown in Figure 7. An OS benefit of durvalumab and tremelimumab plus SOC chemotherapy compared to SOC chemotherapy alone was also observed in patients with nonsquamous histology (HR = 0.69; 95% CI, 0.56 to 0.85) at the 5-year analysis, with OS rates of 20.5% in the durvalumab and tremelimumab plus SOC chemotherapy arm and 9.1% in the SOC chemotherapy alone arm, as presented in Figure 8.1,58
The POSEIDON study was not powered for any individual subgroup evaluations, adjustments were not made for multiple testing, and there were low numbers of patients and events across individual subgroups.1,38
Post Hoc Exploratory Subgroup Analysis of OS in KRAS and STK11 Mutation Subgroups
Post hoc exploratory subgroup analyses were performed in patients with nonsquamous histology for subgroups with KRAS mutations and those with STK11 mutations. At the 5-year OS analysis DCO (August 24, 2023), an OS benefit of durvalumab and tremelimumab plus SOC chemotherapy compared to SOC chemotherapy alone was observed in both subgroups.1 For the KRAS mutation subgroup, the median OS in the durvalumab and tremelimumab plus SOC chemotherapy group (n = 60) was 25.7 months (95% CI, 9.9 to 36.7) and in the SOC chemotherapy alone group (n = 53) it was 10.4 months (95% CI, 7.3 to 12.6) with an HR of 0.55 (95% CI, 0.36 to 0.83). For the STK11 mutation subgroup, the median OS in the durvalumab and tremelimumab plus SOC chemotherapy group (n = 31) was 15.0 months (95% CI, 8.2 to 23.8) and in the SOC chemotherapy alone group (n = 22) it was 10.7 months (95% CI, 6.0 to 14.9), with an HR of 0.57 (95% CI, 0.32 to 1.04).1,62
The POSEIDON study was not powered for these post hoc subgroup evaluations and there were low numbers of patients and events across the subgroups.1
Figure 7: Overall Survival in Patient Subgroups With Durvalumab and Tremelimumab Plus Chemotherapy vs. Chemotherapy Alone, 5-Year OS Analysis
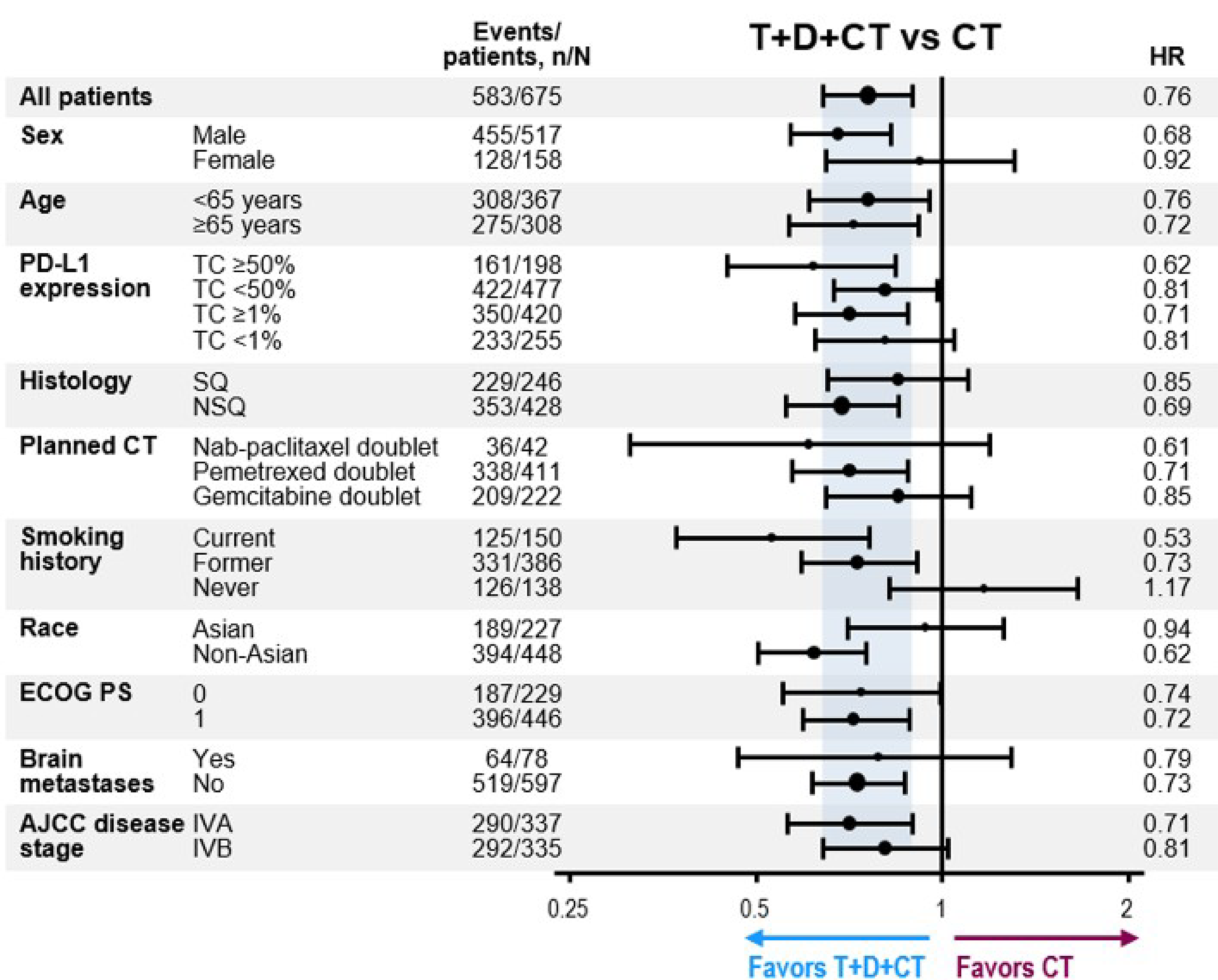
AJCC = American Joint Committee on Cancer; CT = standard-of-care chemotherapy; D = durvalumab; DCO = data cut-off; ECOG = Eastern Cooperative Oncology Group Performance Status; HR = hazard ratio; NSQ = nonsquamous; OS = overall survival; SQ = squamous; t = tremelimumab; TC = tumour cell; vs. = versus.
Note: HRs and 95% confidence intervals in subgroups were estimated using an unstratified Cox proportional hazards model. DCO: August 24, 2023 (5-year OS analysis).
Source: Sponsor’s Summary of Clinical Evidence.1
Figure 8: Kaplan-Meier Plot of Overall Survival — Durvalumab and Tremelimumab Plus Chemotherapy vs. Chemotherapy Alone (Nonsquamous Histology Subgroup), 5-Year OS Analysis
CI = confidence interval; CT = standard-of-care chemotherapy; D = durvalumab; DCO = data cut-off; HR = hazard ratio; mOS = median overall survival; OS = overall survival; t = tremelimumab; vs. = versus.
Note: DCO: 24 August 2023 (5-year OS analysis).
a HRs and 95% CIs in subgroups were estimated using an unstratified Cox proportional hazards model.
Source: Sponsor’s Summary of Clinical Evidence.1
Progression-Free Survival
At the July 24, 2019, final analysis DCO, 238 PFS events had occurred in the durvalumab and tremelimumab plus SOC chemotherapy arm and 258 PFS events had occurred in the SOC chemotherapy alone arm, for a total of 496 PFS events across the 2 groups (73.5% maturity for PFS overall). The final analysis of PFS met the prespecified boundary for declaring statistical significance between the durvalumab and tremelimumab plus chemotherapy versus chemotherapy alone arms (2-sided P value boundary of 0.00735 for a 1% overall alpha recycled from the significant PFS test of durvalumab plus chemotherapy versus chemotherapy alone in the FAS).1,38
The median PFS assessed by BICR in the durvalumab and tremelimumab plus SOC chemotherapy arm (6.2 months; 95% CI, 5.0 to 6.5 months) was greater than that in the SOC chemotherapy alone arm (4.8 months; 95% CI, 4.6 to 5.8 months), with an HR of 0.72 (95% CI 0.600 to 0.860; 2-sided P = 0.00031), as presented in Table 21.1,38 The PFS rate at 12 months for patients in the durvalumab and tremelimumab plus SOC chemotherapy arm was 26.6% (95% CI, 21.7% to 31.7%) and for patients in the SOC chemotherapy alone arm it was 13.1% (95% CI, 9.3% to 17.6%).
At the final analysis DCO, 24.6% and 12.8% of randomized patients were still progression-free at the time of analysis in the durvalumab and tremelimumab plus SOC chemotherapy and SOC chemotherapy alone arms, respectively. In total, 18.9% and 16.6% of patients had a progression event because of death in the absence of RECIST progression in the durvalumab and tremelimumab plus SOC chemotherapy and SOC chemotherapy alone arms, respectively.
The median durations of follow-up in censored patients for the final analysis DCO of July 24, 2019, were 11.99 months (range, 0.0 to 23.1) in the durvalumab and tremelimumab plus SOC chemotherapy arm and 5.5 months (range, 0.0 to 22.8) in the SOC chemotherapy alone arm. Overall, the median duration of follow-up in censored patients across the 3 treatment arms was 10.33 months (range, 0.0 to 23.1).38
A summary of PFS in the durvalumab and tremelimumab plus SOC chemotherapy arm versus SOC chemotherapy alone arm is presented in Table 22. A Kaplan-Meier plot of PFS for the comparison of durvalumab and tremelimumab plus chemotherapy versus chemotherapy alone is presented in Figure 9.
Table 22: Progression-Free Survival for Durvalumab and Tremelimumab Plus SOC Chemotherapy vs. SOC Chemotherapy Alone Based on BICR According to RECIST 1.1 (FAS)
Progression-free survival | D + T + chemotherapy (N = 338) | Chemotherapy (N = 337) |
|---|---|---|
Events, n (%)a | 238 (70.4) | 258 (76.6) |
RECIST 1.1 progression | 174 (51.5) | 202 (59.9) |
Death in the absence of progression | 64 (18.9) | 56 (16.6) |
Censored patients, n (%) | 100 (29.6) | 79 (23.4) |
Censored RECIST 1.1 progressionb | 0 | 2 (0.6) |
Censored deathc | 11 (3.3) | 24 (7.1) |
Progression-free at time of analysis | 83 (24.6) | 43 (12.8) |
Lost to follow-up | 0 | 0 |
Withdrawn consent | 4 (1.2) | 9 (2.7) |
Discontinued study | 2 (0.6) | 1 (0.3) |
Progression-free survival (months), median (95% CI)d | 6.2 (5.0 to 6.5) | 4.8 (4.6 to 5.8) |
HRe,f | 0.72 | |
99.265% CIg | ██████ █████ | |
95% CI | 0.600 to 0.860 | |
2-sided P valueh | 0.00031 | |
PFS rate (%) at 12 months (95% CI) | 26.6 (21.7 to 31.7) | 13.1 (9.3 to 17.6) |
Difference in PFS rate (%) (95% CI) | ████ ████ | |
BICR = blinded independent central review; CI = confidence interval; D = durvalumab; FAS = full analysis set; HR = hazard ratio; PFS = progression-free survival; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1; SOC = standard of care; t = tremelimumab; vs. = versus.
Note: One patient died 1 day before randomization and was censored on day 1. Data cut-off: July 24, 2019.
aPatients who did not progress or die, or who progressed or died after 2 or more missed visits, were censored at the latest evaluable RECIST assessment or at day 1 if there were no evaluable visits or no baseline data and patient did not die within 2 visits of baseline.
bRECIST 1.1 progression event occurred after 2 or more missed visits or within 2 visits of baseline without any evaluable visits or baseline data.
cDeath occurred after 2 or more missed visits in the absence of progression.
dCalculated using the Kaplan-Meier technique.
eThe HR and CI were estimated from a stratified Cox proportional hazards model with the Efron method to control for ties, the stratification factors PD-L1 (≥ 50% vs. < 50%), histology (squamous vs. nonsquamous), and disease stage (IVA vs. IVB) in the strata statement, and the CI was calculated using a profile likelihood approach.
fAn HR of less than 1 favours T + D + chemotherapy to be associated with a longer PFS than chemotherapy alone.
gBased on a Lan-DeMets alpha spending function with O’Brien-Fleming type boundary with the actual number of events observed, the boundaries for declaring statistical significance are 0.00735 for a 1% overall alpha. Corresponding CIs are shown.
hP values were generated using the stratified log-rank test adjusting for PD-L1 (≥ 50% vs. < 50%), histology (squamous vs. nonsquamous), and disease stage (IVA vs. IVB) and using the Breslow approach for handling ties.
Sources: Sponsor’s Summary of Clinical Evidence,1 POSEIDON Clinical Study Report,38 and the sponsor’s Summary of Clinical Evidence.
Figure 9: Kaplan-Meier Plot of PFS for Durvalumab and Tremelimumab Plus Chemotherapy vs. Chemotherapy Alone (FAS)
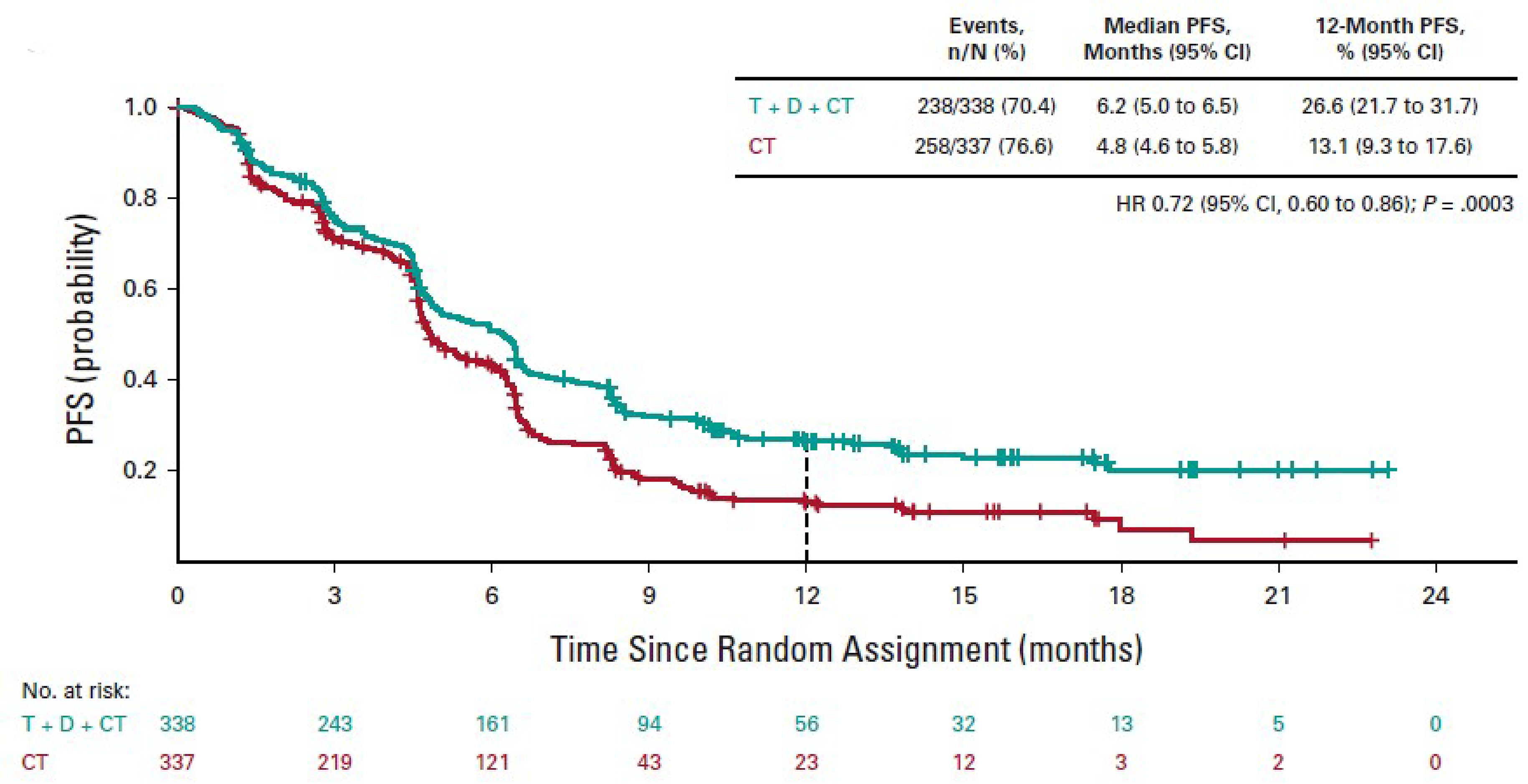
CI = confidence interval; CT = standard-of-care chemotherapy; D = durvalumab; FAS = full analysis set; HR = hazard ratio; PFS = progression-free survival; t = tremelimumab; vs. = versus.
Note: Data cut-off: July 24, 2019.
Source: Sponsor’s Summary of Clinical Evidence.1
Sensitivity Analyses of PFS
Robustness of the PFS effect to possible sources of bias in the measurement of PFS were assessed via sensitivity analyses. HRs for sensitivity analyses ranged from 0.66 to 0.74.38,61
Censoring for PFS occurred for 100 patients (29.6%) in the durvalumab and tremelimumab plus SOC chemotherapy arm and 79 patients (23.4%) in the SOC chemotherapy alone arm. A Kaplan-Meier plot of time to censoring that was used to further assess any potential attrition bias showed that some censoring occurred earlier in the SOC chemotherapy alone arm in comparison to the durvalumab and tremelimumab plus SOC chemotherapy arm.38
Two multivariate Cox models were used: model 1 adjusted for treatment and the stratification factors only, and model 2 adjusted for treatment, stratification factors, and additional prespecified covariates. The HRs were 0.71 (95% CI, 0.598 to 0.854) from model 1 and 0.70 (95% CI, 0.585 to 0.845) from model 2.38
Subgroup Analyses of PFS
Prespecified subgroup analyses to investigate the consistency of treatment effect across prespecified stratification factors, demographics, planned chemotherapy administered, and baseline disease characteristics showed that the PFS benefit of durvalumab and tremelimumab plus SOC chemotherapy versus SOC chemotherapy alone was observed across all prespecified subgroups (Figure 10). Estimated HRs for all subgroups were in favour of durvalumab and tremelimumab plus SOC chemotherapy (HR < 1) and consistent with the overall HR for PFS. A PFS benefit of durvalumab and tremelimumab plus SOC chemotherapy compared to SOC chemotherapy alone was observed in patients with nonsquamous histology (HR = 0.66; 95% CI, 0.52 to 0.84).1
No significant qualitative interaction was found when the stratification factors of PD-L1 status (TC ≥ 50% versus < 50%), histology (squamous versus nonsquamous) and disease stage (IVA versus IVB) were tested for any interaction with treatment (P = 0.500 for all factors).1,38
The POSEIDON study was not powered for any individual subgroup evaluations, adjustments were not made for multiple testing, and there were low numbers of patients and events across individual subgroups.1,38
Figure 10: PFS in Patient Subgroups With Durvalumab and Tremelimumab Plus SOC Chemotherapy vs. SOC Chemotherapy Alone
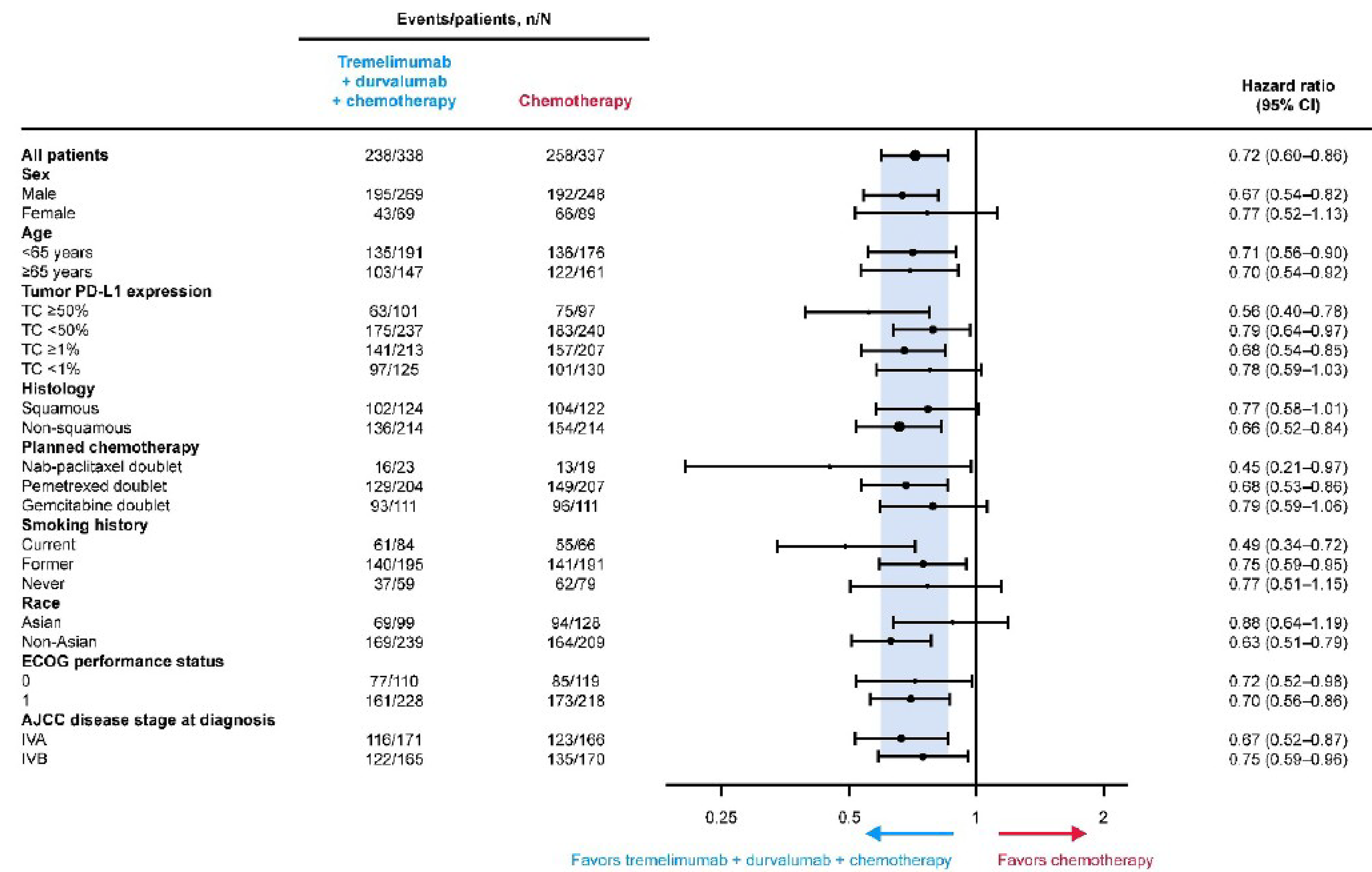
AJCC = American Joint Committee on Cancer; CI = confidence interval; ECOG = Eastern Cooperative Oncology Group; HR = hazard ratio; SOC = standard of care; TC = tumour cell; vs. = versus.
Note: Data cut-off: July 24, 2019. HRs and 95% CIs in the intention-to-treat population were estimated using a Cox proportional hazards model stratified by PD-L1 expression status, histology, and disease stage. HRs and 95% CIs in subgroups were estimated using an unstratified Cox proportional hazards model.
Source: Sponsor’s Summary of Clinical Evidence.1
Objective Response Rate
At the July 24, 2019, final analysis DCO, the prespecified end point of ORR using unconfirmed responses based on BICR was greater in the durvalumab and tremelimumab plus SOC chemotherapy arm than in the SOC chemotherapy alone arm (46.3% versus 33.4%, respectively), with an OR for durvalumab and tremelimumab plus SOC chemotherapy versus SOC chemotherapy alone of 1.72 (95% CI, 1.260 to 2.367; nominal P < 0.001), as presented in Table 21.1,38
Post hoc analysis of ORR using confirmed responses based on BICR showed that the ORR was greater in the durvalumab and tremelimumab plus SOC chemotherapy arm than in the SOC chemotherapy alone arm (38.8% versus 24.4%), with an OR for durvalumab and tremelimumab plus SOC chemotherapy versus SOC chemotherapy alone of 2.00 (95% CI, 1.428 to 2.807; nominal P < 0.001), as presented in Table 23.1,38
Table 23: Objective Response Rate (Confirmed and Unconfirmed) Based on BICR Assessments According to RECIST 1.1 (FAS, Patients With Measurable Disease at Baseline) for Durvalumab and Tremelimumab Plus Chemotherapy vs. Chemotherapy Alone
Results | Unconfirmed responses | Confirmed responses only | ||
|---|---|---|---|---|
D + T + chemotherapy (N = 335) | Chemotherapy (N = 332) | D + T + chemotherapy (N = 335) | Chemotherapy (N = 332) | |
ORR, n (%) | 155 (46.3) | 111 (33.4) | 130 (38.8) | 81 (24.4) |
Odds ratio, T + D + chemotherapy vs. chemotherapy alonea | 1.72 | 2.00 | ||
95% CI for odds ratio | 1.260 to 2.367 | 1.428 to 2.807 | ||
2-sided P value | < 0.001 | < 0.001 | ||
BICR = blinded independent central review; CI = confidence interval; D = durvalumab; FAS = full analysis set; ORR = objective response rate; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1; t = tremelimumab; vs. = versus.
Note: The prespecified secondary outcome of ORR (based on BICR) using unconfirmed responses was the number (percentage) of patients with at least 1 visit response of complete response or partial response.1,38,47 Post hoc sensitivity analysis of ORR (based on BICR) was conducted using confirmed responses, which were defined as the number (percentage) of patients with at least 1 visit response of complete response or partial response and a confirmatory scan no sooner than 4 weeks after the initial complete response or partial response.1,38 The analysis was performed using logistic regression adjusting for PD-L1 (≥ 50% vs. < 50%), histology (squamous vs. nonsquamous), and disease stage (IVA vs. IVB), with the CI calculated using a profile likelihood approach and the P value calculated based on twice the change in log-likelihood resulting from the addition of a treatment factor to the model. One patient died 1 day before randomization and was censored at day 1. Data cut-off date: July 24, 2019.
aAn odds ratio greater than 1 favours T + D + chemotherapy compared to chemotherapy alone.
Sources: Sponsor’s Summary of Clinical Evidence1 and POSEIDON Clinical Study Report.38
Best Objective Response
BoR results in the durvalumab and tremelimumab plus SOC chemotherapy and SOC chemotherapy alone arms at the July 24, 2019, DCO are presented in Table 24. The BoR was a CR for 2 patients (0.6%) in the durvalumab and tremelimumab plus SOC chemotherapy arm whereas no patients in the SOC chemotherapy alone arm had a BoR of a CR. BoR was a PR for 153 patients (45.7%) in the durvalumab and tremelimumab plus SOC chemotherapy arm and 111 patients (33.4%) in the SOC chemotherapy alone arm. In the durvalumab and tremelimumab plus SOC chemotherapy and SOC chemotherapy alone arms, 120 (35.8%) and 150 (45.2%) of the patients, respectively, had a BoR of stable disease of 6 weeks or longer; 26 patients (7.8%) and 36 patients (10.8%), respectively, had a BoR of RECIST 1.1–based progression, and 22 (6.6%) and 25 (7.5%), respectively, died.1,38
Table 24: Best Objective Response Based on BICR Assessments According to RECIST 1.1 (Unconfirmed and Confirmed Responses) (FAS, Patients With Measurable Disease at Baseline) for Durvalumab and Tremelimumab plus SOC Chemotherapy vs. SOC Chemotherapy Alone
Method | Best objective response | D + T + chemotherapy (N = 335) | Chemotherapy (N = 332) |
|---|---|---|---|
Response | Total | 155 (46.3) | 111 (33.4) |
Complete responsea | 2 (0.6) | 0 | |
Partial responsea | 153 (45.7) | 111 (33.4) | |
Nonresponse | Total | 180 (53.7) | 221 (66.6) |
Stable disease ≥ 6 weeksb | 120 (35.8) | 150 (45.2) | |
Progression | 48 (14.3) | 61 (18.4) | |
RECIST 1.1 progression | ██ █████ | ██ ██████ | |
Death | ██ █████ | ██ █████ | |
Not evaluable | 12 (3.6) | 10 (3.0) | |
Stable disease < 6 weeksb | 0 | 0 | |
Incomplete postbaseline assessments | 12 (3.6) | 10 (3.0) |
BICR = blinded independent central review; D = durvalumab; FAS = full analysis set; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1; SOC = standard of care; t = tremelimumab; vs. = versus.
Note: Data cut-off date: July 24, 2019.
aResponse did not require confirmation.
bIn practice, 5 weeks was considered the threshold to allow for the 1-week permitted time window.
Sources: Sponsor’s Summary of Clinical Evidence1 and POSEIDON Clinical Study Report.38
Duration of Response
At the July 24, 2019, final analysis DCO, in the prespecified analysis of DoR using unconfirmed responses based on BICR, the durvalumab and tremelimumab plus SOC chemotherapy and SOC chemotherapy alone arms both had a median time to response of 1.5 months (Table 25). Median DoR was longer in the durvalumab and tremelimumab plus SOC chemotherapy arm (7.4 months) than in the SOC chemotherapy alone arm (4.2 months), as presented in Table 25. The proportion of responders with an estimated DoR of 12 months or longer was greater in the durvalumab and tremelimumab plus SOC chemotherapy arm (42.5%) than in the SOC chemotherapy alone arm (16.4%) (Figure 11).1,38
Post hoc analysis of DoR using confirmed ORR assessed by BICR also demonstrated a similar median time to response in the durvalumab and tremelimumab plus SOC chemotherapy and SOC chemotherapy alone arms (1.5 months and 1.4 months, respectively), as presented in Table 25. In this analysis, the median DoR was also longer in the durvalumab and tremelimumab plus SOC chemotherapy arm (9.5 months) than in the SOC chemotherapy alone arm (5.1 months), as presented in Table 25. The proportion of responders with an estimated DoR of 12 months or longer was greater in the durvalumab and tremelimumab plus SOC chemotherapy arm (49.7%) than in the SOC chemotherapy alone arm (21.4%).1,38 Curves for the proportion of patients remaining in response over time using confirmed responses39 follow a similar pattern as those presented using unconfirmed responses.
Table 25: Duration and Onset of Unconfirmed and Confirmed Objective Response Based on BICR Assessments According to RECIST 1.1 (FAS, Patients With Measurable Disease at Baseline) for Durvalumab and Tremelimumab Plus SOC Chemotherapy vs. SOC Chemotherapy Alone
Results | Unconfirmed responses | Confirmed responses only | ||
|---|---|---|---|---|
D + T + chemotherapy (N = 155) | Chemotherapy (N = 111) | D + T + chemotherapy (N = 130) | Chemotherapy (N = 81) | |
Number of responders who subsequently progressed or died | 87 | 84 | 65 | 60 |
Time to onset of response from randomization (months)a | ||||
25th percentile | ███ | ███ | ███ | ███ |
Median | ███ | ███ | ███ | ███ |
75th percentile | ███ | ███ | ███ | ███ |
DoR from onset of response (months)b,c | ||||
25th percentile | 3.5 | 3.0 | 5.0 | 3.7 |
Median | 7.4 | 4.2 | 9.5 | 5.1 |
75th percentile | NR | 6.9 | NR | 7.5 |
Percentage remaining in responsec | ||||
6 months | 57.2 | 31.0 | 67.0 | 40.4 |
12 months | 42.5 | 16.4 | 49.7 | 21.4 |
18 months | 34.7 | NR | 40.7 | NR |
BICR = blinded independent central review; D = durvalumab; DoR = duration of response; FAS = full analysis set; NR = not reached; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1; SOC = standard of care; t = tremelimumab; vs. = versus.
Note: Data cut-off date: July 24, 2019.
aCalculated using summary descriptive statistics, given that only patients with measurable disease at baseline that have an objective response were considered.
bDoR is the time from the first documentation of complete response or partial response until the date of progression, death in absence of progression, or the last evaluable RECIST 1.1 assessment for patients who progressed or died after 2 or more missed visits.
cCalculated using the Kaplan-Meier technique.
Sources: Sponsor’s Summary of Clinical Evidence1 and POSEIDON Clinical Study Report.38
Figure 11: Kaplan-Meier Plot of Duration of Response (Unconfirmed) Based on BICR Assessments per RECIST 1.1 for Patients With Measurable Disease at Baseline (FAS), All Study Treatments
BICR = blinded independent central review; Durva = durvalumab; FAS = full analysis set; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1; SoC = standard of care chemotherapy; Treme = tremelimumab.
Notes: Data cut-off date: July 24, 2019.
The + symbol on the curves represents censored observations.
Source: POSEIDON Clinical Study Report.38
Health-Related Quality of Life
For the EORTC QLQ-C30 and EORTC QLQ-L13, overall compliance rates were 73.0% and 72.8%, respectively, in the durvalumab and tremelimumab plus SOC chemotherapy arm and 65.0% and 64.8%, respectively, in the SOC chemotherapy alone arm. In both treatment arms, compliance rates generally decreased over time and were lower in the SOC chemotherapy alone arm. For both scales, compliance rates greater than 60% were observed continuously in the durvalumab and tremelimumab plus SOC chemotherapy arm for up to 88 weeks and were observed continuously in the SOC chemotherapy alone arm for up to 24 weeks.1,38 Scores for EORTC QLQ-C30 and EORTC QLQ-LC13 were comparable between durvalumab and tremelimumab plus SOC chemotherapy and SOC chemotherapy alone arms at baseline.38
Time to Deterioration (EORTC QLQ-C30 and EORTC QLQ-LC13)
Time to deterioration results are presented in Table 26. At the March 12, 2021, final analysis DCO, the median time to deterioration in EORTC QLQ-C30 GHS/QoL in the durvalumab and tremelimumab plus SOC chemotherapy arm (8.3 months; 95% CI, 6.4 to 10.2) was numerically greater than that in the SOC chemotherapy alone arm (5.6 months; 95% CI, 4.4 to 7.5) (HR = 0.78; 95% CI, 0.631 to 0.961; ███████ ███████). For the EORTC QLQ-C30 physical functioning domain, the median time to deterioration was also numerically greater in the durvalumab and tremelimumab plus SOC chemotherapy arm (7.7 months; 95% CI, 5.9 to 9.4) than in the SOC chemotherapy alone arm (5.3 months; 95% CI, 4.2 to 6.5) (HR = 0.75; 95% CI, 0.610 to 0.920 |███████ ███████). Regarding results for the prespecified key symptoms of fatigue and appetite loss (from the EORTC QLQ-C30) and coughing, dyspnea, and chest pain (from the QLQ-LC13), the median time to deterioration was numerically longer but not statistically significantly different in the durvalumab and tremelimumab plus SOC chemotherapy arm compared to the SOC chemotherapy alone arm for fatigue (HR = 0.90; 95% CI, 0.746 to 1.084; ███████ ███████), appetite loss (HR = 0.94; 95% CI, 0.754 to 1.169; ███████ ███████), coughing (HR = 0.91; 95% CI, 0.722 to 1.146; ███████ ███████), dyspnea (HR = 0.77; 95% CI, 0.635 to 0.936 | ███████ ███████), and chest pain (HR = 0.85; 95% CI, 0.681 to 1.066; ███████ ███████).1
Figure 12: Forest Plot of Time to Deterioration in EORTC QLQ-C30 and EORTC QLQ-L13 Scales and Items, Durvalumab and Tremelimumab Plus SOC Chemotherapy vs. SOC Chemotherapy Alone (FAS)
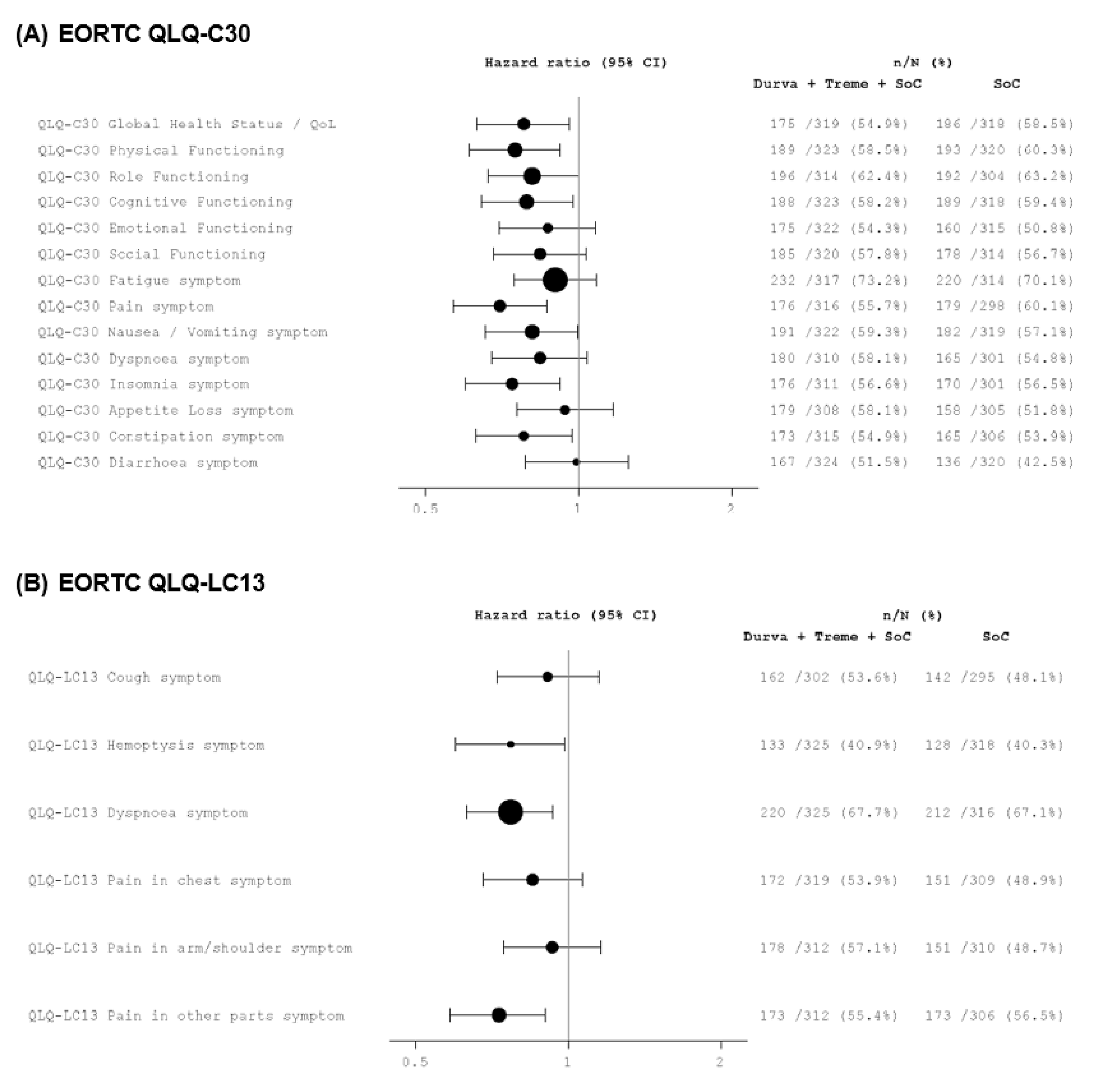
CI = confidence interval; Durva = durvalumab; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-LC13 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Lung Cancer 13; FAS = full analysis set; SoC = standard of care chemotherapy; Treme = tremelimumab; vs. = versus.
Note: A hazard ratio of less than 1 favours durvalumab and tremelimumab plus chemotherapy to be associated with a longer TTD than chemotherapy alone. The size of the circles is proportional to the number of events. Percentages are calculated from the number of patients in the full analysis set with baseline symptom scores of 90 or lower or baseline HRQoL/function scores of 10 or higher in that treatment arm. Data cut-off: March 12, 2021.
Sources: Sponsor’s Summary of Clinical Evidence1 and POSEIDON Clinical Study Report.38
Table 26: Time to Deterioration in EORTC QLQ-C30 and EORTC QLQ-LC13 Scales and Items — Durvalumab and Tremelimumab Plus SOC Chemotherapy vs. SOC Chemotherapy Alone (FAS)
Scale or item | D + T + chemotherapy (N = 338) | Chemotherapy (N = 337) |
|---|---|---|
EORTC QLQ-C30 Global Health Status/QoL | ||
n | 319 | 318 |
Total events, n (%)a | 175 (54.9) | 186 (58.5) |
Median time to deterioration, months (95% CI)b | 8.3 (6.4 to 10.2) | 5.6 (4.4 to 7.5) |
HR (95% CI)c | 0.78 (0.631 to 0.961) | |
2-sided P valued | █████ | |
EORTC QLQ-C30 physical functioning | ||
n | 323 | 320 |
Total events, n (%)a | 189 (58.5) | 193 (60.3) |
Median time to deterioration, months (95% CI)b | 7.7 (5.9 to 9.4) | 5.3 (4.2 to 6.5) |
HR (95% CI)c | 0.75 (0.610 to 0.920) | |
2-sided P valued | █████ | |
EORTC QLQ-C30 role functioning | ||
n | 314 | 304 |
Total events, n (%)a | 196 (62.4) | 192 (63.2) |
Median time to deterioration, months (95% CI)b | 6.6 (5.4 to 8.3) | 4.8 (3.6 to 6.2) |
HR (95% CI)c | 0.81 (0.664 to 0.999) | |
2-sided P valued | █████ | |
EORTC QLQ-C30 cognitive functioning | ||
n | 323 | 318 |
Total events, n (%)a | 188 (58.2) | 189 (59.4) |
Median time to deterioration, months (95% CI)b | 7.6 (6.1 to 9.7) | 5.8 (4.6 to 7.5) |
HR (95% CI)c | 0.79 (0.644 to 0.975) | |
2-sided P valued | █████ | |
EORTC QLQ-C30 emotional functioning | ||
n | 322 | 315 |
Total events, n (%)a | 175 (54.3) | 160 (50.8) |
Median time to deterioration, months (95% CI)b | 8.5 (6.6 to 11.3) | 7.5 (5.8 to 11.0) |
HR (95% CI)c | 0.87 (0.697 to 1.082) | |
2-sided P valued | █████ | |
EORTC QLQ-C30 social functioning | ||
n | 320 | 314 |
Total events, n (%)a | 185 (57.8) | 177 (56.4) |
Median time to deterioration, months (95% CI)b | 6.4 (5.6 to 9.4) | 5.7 (4.6 to 7.4) |
HR (95% CI)c | 0.85 (0.687 to 1.045) | |
2-sided P valued | █████ | |
EORTC QLQ-C30 fatigue | ||
n | 317 | 314 |
Total events, n (%)a | 232 (73.2) | 220 (70.1) |
Median time to deterioration, months (95% CI)b | 3.7 (2.8 to 5.0) | 2.8 (2.1 to 3.7) |
HR (95% CI)c | 0.90 (0.746 to 1.084) | |
2-sided P valued | █████ | |
EORTC QLQ-C30 pain | ||
n | 316 | 298 |
Total events, n (%)a | 176 (55.7) | 180 (60.4) |
Median time to deterioration, months (95% CI) b | 8.9 (6.2 to 10.9) | 5.7 (4.6 to 7.2) |
HR (95% CI)c | 0.70 (0.563 to 0.862) | |
2-sided P valued | ██████ | |
EORTC QLQ-C30 nausea and vomiting | ||
n | 322 | 319 |
Total events, n (%)a | 191 (59.3) | 182 (57.1) |
Median time to deterioration, months (95% CI)b | 7.8 (5.6 to 10.0) | 5.6 (4.2 to 7.5) |
HR (95% CI)c | 0.81 (0.655 to 0.994) | |
2-sided P valued | █████ | |
EORTC QLQ-C30 dyspnea | ||
n | 310 | 301 |
Total events, n (%)a | 180 (5,801) | 164 (54.5) |
Median time to deterioration, months (95% CI)b | 7.9 (5.8 to 9.8) | 6.7 (5.5 to 8.8) |
HR (95% CI)c | 0.84 (0.678 to 1.047) | |
2-sided P valued | █████ | |
EORTC QLQ-C30 insomnia | ||
n | 311 | 301 |
Total events, n (%)a | 176 (56.6) | 170 (56.5) |
Median time to deterioration, months (95% CI)b | 8.3 (6.4 to 10.4) | 5.8 (4.2 to 7.2) |
HR (95% CI)c | 0.74 (0.598 to 0.921) | |
2-sided P valued | █████ | |
EORTC QLQ-C30 appetite loss | ||
n | 308 | 305 |
Total events, n (%)a | 179 (58.1) | 158 (51.3) |
Median time to deterioration, months (95% CI)b | 7.2 (5.7 to 9.4) | 7.0 (5.6 to 9.6) |
HR (95% CI)c | 0.94 (0.754 to 1.169) | |
2-sided P valued | █████ | |
EORTC QLQ-C30 constipation | ||
n | 315 | 306 |
Total events, n (%)a | 173 (54.9) | 165 (53.9) |
Median time to deterioration, months (95% CI)b | 9.2 (6.4 to 12.2) | 6.1 (4.8 to 7.5) |
HR (95% CI)c | 0.78 (0.627 to 0.972) | |
2-sided P valued | █████ | |
EORTC QLQ-C30 diarrhea | ||
n | 324 | 320 |
Total events, n (%)a | 167 (51.5) | 135 (42.2) |
Median time to deterioration, months (95% CI)b | 11.0 (9.3 to 13.6) | 10.8 (9.2 to 15.1) |
HR (95% CI)c | 1.00 (0.792 to 1.260) | |
2-sided P valued | █████ | |
EORTC QLQ-LC13 cough | ||
n | 302 | 295 |
Total events, n (%)a | 162 (53.6) | 142 (48.1) |
Median time to deterioration, months (95% CI)b | 9.7 (7.3, 13.3) | 8.8 (6.8, 12.3) |
HR (95% CI)c | 0.91 (0.722, 1.146) | |
2-sided P valued | █████ | |
EORTC QLQ-LC13 hemoptysis | ||
n | 325 | 318 |
Total events, n (%)a | 133 (40.9) | 128 (40.3) |
Median time to deterioration, months (95% CI)b | 17.8 (12.5 to 31.3) | 11.4 (9.3 to 19.3) |
HR (95% CI)c | 0.77 (0.598 to 0.984) | |
2-sided P valued | █████ | |
EORTC QLQ-LC13 dyspnea | ||
n | 325 | 316 |
Total events, n (%)a | 220 (67.7) | 211 (66.8) |
Median time to deterioration, months (95% CI)b | 5.4 (4.2 to 6.4) | 3.6 (2.6 to 4.5) |
HR (95% CI)c | 0.77 (0.635 to 0.936) | |
2-sided P valued | █████ | |
EORTC QLQ-LC13 pain in chest | ||
n | 319 | 309 |
Total events, n (%)a | 172 (53.9) | 151 (48.9) |
Median time to deterioration, months (95% CI)b | 10.0 (7.7 to 13.3) | 8.6 (6.8 to 11.4) |
HR (95% CI)c | 0.85 (0.681 to 1.066) | |
2-sided P valued | █████ | |
EORTC QLQ-LC13 pain in arm and/or shoulder | ||
n | 312 | 310 |
Total events, n (%)a | 178 (57.1) | 151 (48.7) |
Median time to deterioration, months (95% CI)b | 8.9 (6.6 to 10.9) | 8.8 (6.2 to 12.0) |
HR (95% CI)c | 0.98 (0.745 to 1.161) | |
2-sided P valued | █████ | |
EORTC QLQ-LC13 pain in other parts | ||
n | 312 | 306 |
Total events, n (%)a | 174 (55.8) | 172 (56.2) |
Median time to deterioration, months (95% CI)b | 9.7 (6.8 to 12.1) | 5.8 (4.9 to 9.3) |
HR (95% CI)c | 0.74 (0.597 to 0.921) | |
2-sided P valued | █████ | |
CI = confidence interval; D = durvalumab; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-LC13 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Lung Cancer 13; FAS = full analysis set; HR = hazard ratio; HRQoL = health-related quality of life; PRO = patient-reported outcome; SOC = standard of care; t = tremelimumab; vs. = versus.
Note: A hazard ratio of less than 1 favours D + T + chemotherapy to be associated with a longer time to deterioration than chemotherapy alone. Percentages are calculated from the number of patients in the full analysis set with baseline symptom scores of 90 or lower or baseline HRQoL/function scores of 10 or higher in that treatment group. Data cut-off: March 12, 2021.
aPatients who have not shown a clinically meaningful deterioration, or who showed clinically meaningful deterioration or died after 2 or more missed visits, will be censored at the time of the latest evaluable PRO assessment (or at the latest evaluable PRO assessment before the 2 missed visits), or at the date of randomization if there are no evaluable visits or no baseline data.
bCalculated using the Kaplan-Meier technique.
cThe hazard ratio and CI are estimated from a stratified Cox proportional hazards model with the Efron method to control for ties, the stratification factors PD-L1 (≥ 50% vs. < 50%), histology (squamous vs. nonsquamous), and disease stage (IVA vs. IVB) in the strata statement, and the CI was calculated using a profile likelihood approach.
dP values were generated using the stratified log-rank test adjusting for PD-L1 (≥ 50% vs. < 50%), histology (squamous vs. nonsquamous), and disease stage (IVA vs. IVB) and using the Breslow approach for handling ties.
Sources: Sponsor’s Summary of Clinical Evidence1 and POSEIDON Clinical Study Report (Tables and Figures).61
Improvement Rate (EORTC QLQ-C30 and EORTC QLQ-LC13)
Improvement rate results are presented in Table 27. At the March 12, 2021, final analysis DCO, the improvement rate in EORTC QLQ-C30 GHS/QoL in the durvalumab and tremelimumab plus SOC chemotherapy arm (38.7%) was numerically greater but not statistically significantly different compared to the SOC chemotherapy alone arm (31.4%) (OR = 1.38; 95% CI 0.980 to 1.933; ███████ ███████). For the EORTC QLQ-C30 physical functioning domain, the improvement rate was numerically greater in the durvalumab and tremelimumab plus SOC chemotherapy group (41.7%) compared to the SOC chemotherapy alone group (31.9%) (OR = 1.56; 95% CI, 1.061 to 2.317; ███████ ███████). Regarding results for the prespecified key symptoms of fatigue and appetite loss (from the EORTC QLQ-C30) and cough, dyspnea, and chest pain (from the EORTC QLQ-LC13), the improvement rate was numerically greater but not statistically significantly different in the durvalumab and tremelimumab plus SOC chemotherapy arm compared to the SOC chemotherapy alone arm for symptoms of fatigue (OR = 1.26; 95% CI, 0.896 to 1.770; ███████ ███████), appetite loss (OR = 1.43; 95% CI, 0.905 to 2.253; ███████ ███████), cough (OR = 1.08; 95% CI, 0.759 to 1.541; ███████ ███████), dyspnea (OR = 1.38; 95% CI, 0.972 to 1.966 | ███████ ███████), and chest pain (OR = 1.35; 95% CI, 0.860 to 2.137; ███████ ███████).
Figure 13: Forest Plot of Improvement Rate in EORTC QLQ-C30 and EORTC QLQ-L13 Scales and Items, Durvalumab and Tremelimumab Plus SOC Chemotherapy vs. SOC Chemotherapy Alone (FAS)
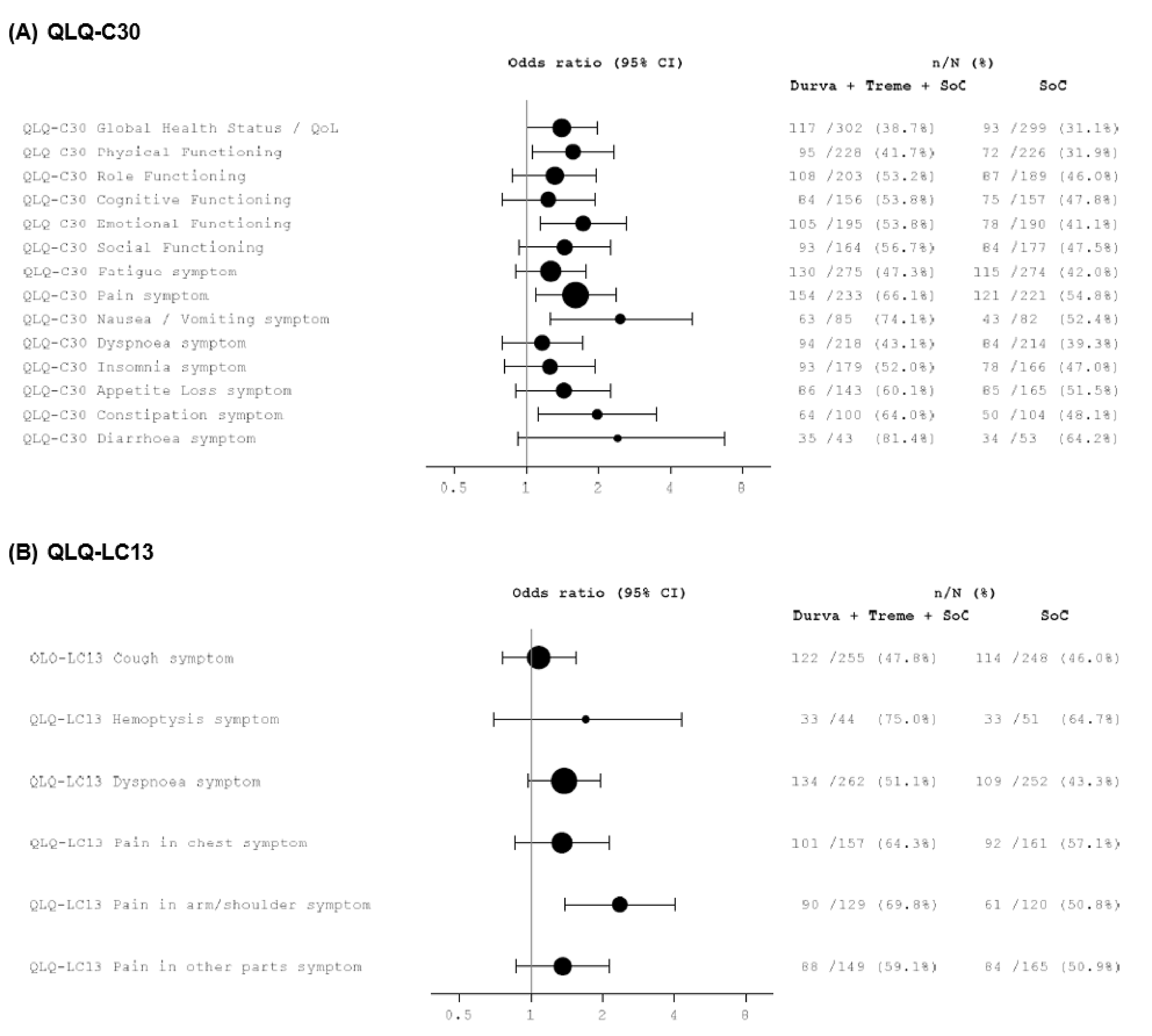
CI = confidence interval; Durva = durvalumab; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-LC13 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Lung Cancer 13; QoL = quality of life SoC = standard of care; Treme = tremelimumab; vs. = versus.
Note: An odds ratio greater than 1 favours durvalumab and tremelimumab plus SOC chemotherapy to be associated with a higher probability of improvement than SOC chemotherapy alone. Size of the circles is proportional to the number of patients with improvement. Percentages are calculated from the number of patients in the full analysis set with baseline symptom scores of 10 or higher or baseline HRQoL/function scores of 90 or lower in that treatment arm. Data cut-off date: March 12, 2021.
Sources: Sponsor’s Summary of Clinical Evidence1 and POSEIDON Clinical Study Report.38
Table 27: Improvement Rate of EORTC QLQ-C30 and EORTC QLQ-LC13 Scales and Items — Logistic Regression (FAS)
Scale or item | D + T + chemotherapy (N = 338) | Chemotherapy (N = 337) |
|---|---|---|
EORTC QLQ-C30 Global Health Status/QoL | ||
n | 302 | 299 |
Patients with improvement, n (%) | 173 (57.3) | 150 (50.2) |
Improvement rate (% [95%CI])a | 117 (38.7 [33.3 to 44.5]) | 94 (31.4 [26.3 to 37.1]) |
Difference, % (95% CI) | 7.3 (−0.6 to 15.2) | |
Odds ratio (95% CI) | 1.38 (0.980 to 1.933) | |
2-sided P value | █████ | |
EORTC QLQ-C30 physical functioning | ||
n | 228 | 226 |
Patients with improvement, n (%) | 138 (60.5) | 113 (50.0) |
Improvement rate (%)a | 95 (41.7) | 72 (31.9) |
Odds ratio (95% CI) | 1.56 (1.061 to 2.317) | |
2-sided P value | █████ | |
EORTC QLQ-C30 role functioning | ||
n | 203 | 189 |
Patients with improvement, n (%) | 150 (73.9) | 139 (73.5) |
Improvement rate (%)a | 108 (53.2) | 87 (46.0) |
Odds ratio (95% CI) | 1.31 (0.874 to 1.955) | |
2-sided P value | █████ | |
EORTC QLQ-C30 cognitive functioning | ||
n | 156 | 157 |
Patients with improvement, n (%) | 127 (81.4) | 115 (73.2) |
Improvement rate (%)a | 84 (53.8) | 75 (47.8) |
Odds ratio (95% CI) | 1.23 (0.788 to 1.931) | |
2-sided P value | █████ | |
QLQ-C30 emotional functioning | ||
n | 195 | 190 |
Patients with improvement, n (%) | 140 (71.9) | 123 (64.7) |
Improvement rate (%) a | 105 (53.8) | 78 (41.1) |
Odds ratio (95% CI) | 1.72 (1.142 to 2.595) | |
2-sided P value | █████ | |
EORTC QLQ-C30 social functioning | ||
n | 164 | 177 |
Patients with improvement, n (%) | 132 (80.5) | 130 (73.4) |
Improvement rate (%)a | 93 (56.7) | 84 (47.5) |
Odds ratio (95% CI) | 1.44 (0.935 to 2.231) | |
2-sided P value | █████ | |
EORTC QLQ-C30 fatigue | ||
n | 275 | 274 |
Patients with improvement, n (%) | 204 (74.2) | 174 (63.5) |
Improvement rate (%)a | 130 (47.3) | 115 (42.0) |
Odds ratio (95% CI) | 1.26 (0.896 to 1.770) | |
2-sided P value | █████ | |
EORTC QLQ-C30 pain | ||
n | 233 | 221 |
Patients with improvement, n (%) | 196 (84.1) | 160 (72.4) |
Improvement rate (%)a | 154 (66.1) | 121 (54.8) |
Odds ratio (95% CI) | 1.60 (1.094 to 2.362) | |
2-sided P value | █████ | |
EORTC QLQ-C30 nausea and vomiting | ||
n | 85 | 82 |
Patients with improvement, n (%) | 74 (87.1) | 70 (85.4) |
Improvement rate (%)a | 63 (74.1) | 43 (52.4) |
Odds ratio (95% CI) | 2.46 (1.254 to 4.909) | |
2-sided P value | █████ | |
EORTC QLQ-C30 dyspnea | ||
n | 218 | 214 |
Patients with improvement, n (%) | 144 (66.1) | 129 (60.3) |
Improvement rate (%)a | 94 (43.1) | 84 (39.3) |
Odds ratio (95% CI) | 1.16 (0.790 to 1.712) | |
2-sided P value | █████ | |
EORTC QLQ-C30 insomnia | ||
n | 179 | 166 |
Patients with improvement, n (%) | 144 (80.4) | 114 (68.7) |
Improvement rate (%)a | 93 (52.0) | 78 (47.0) |
Odds ratio (95% CI) | 1.25 (0.808 to 1.923) | |
2-sided P value | █████ | |
QLQ-C30 appetite loss | ||
n | 143 | 165 |
Patients with improvement, n (%) | 111 (77.6) | 121 (73.3) |
Improvement rate (%)a | 86 (60.1) | 85 (51.5) |
Odds ratio (95% CI) | 1.43 (0.905 to 2.253) | |
2-sided P value | █████ | |
EORTC QLQ-C30 constipation | ||
n | 100 | 104 |
Patients with improvement, n (%) | 88 (88.0) | 78 (75.0) |
Improvement rate (%)a | 64 (64.0) | 50 (48.1) |
Odds ratio (95% CI) | 1.97 (1.119 to 3.494) | |
2-sided P value | █████ | |
EORTC QLQ-C30 diarrhea | ||
n | 43 | 53 |
Patients with improvement, n (%) | 39 (90.7) | 41 (77.4) |
Improvement rate (%)a | 35 (81.4) | 34 (64.2) |
Odds ratio (95% CI) | 2.40 (0.924 to 6.707) | |
2-sided P value | █████ | |
EORTC QLQ-LC13 cough | ||
n | 255 | 248 |
Patients with improvement, n (%) | 168 (65.9) | 162 (65.3) |
Improvement rate (%)a | 122 (47.8) | 114 (46.0) |
Odds ratio (95% CI) | 1.08 (0.759 to 1.541) | |
2-sided P value | █████ | |
EORTC QLQ-LC13 hemoptysis | ||
n | 44 | 51 |
Patients with improvement, n (%) | 39 (88.6) | 41 (80.4) |
Improvement rate (%)a | 33 (75.0) | 33 (64.7) |
Odds ratio (95% CI) | 1.70 (0.697 to 4.301) | |
2-sided P value | █████ | |
EORTC QLQ-LC13 dyspnea | ||
n | 262 | 252 |
Patients with improvement, n (%) | 189 (72.1) | 159 (63.1) |
Improvement rate (%)a | 134 (51.1) | 109 (43.3) |
Odds ratio (95% CI) | 1.38 (0.972 to 1.966) | |
2-sided P value | █████ | |
EORTC QLQ-LC13 pain in chest | ||
n | 157 | 161 |
Patients with improvement, n (%) | 127 (80.9) | 129 (80.1) |
Improvement rate (%)a | 101 (64.3) | 92 (57.1) |
Odds ratio (95% CI) | 1.35 (0.860 to 2.137) | |
2-sided P value | █████ | |
EORTC QLQ-LC13 pain in arm and/or shoulder | ||
n | 129 | 120 |
Patients with improvement, n (%) | 109 (84.5) | 95 (79.2) |
Improvement rate (%)a | 50 (69.8) | 61 (50.8) |
Odds ratio (95% CI) | 2.36 (1.396 to 4.049) | |
2-sided P value | █████ | |
EORTC QLQ-LC13 pain in other parts | ||
n | 148 | 165 |
Patients with improvement, n (%) | 121 (81.8) | 121 (73.3) |
Improvement rate (%)a | 87 (58.8) | 84 (50.9) |
Odds ratio (95% CI) | 1.35 (0.857 to 2.131) | |
2-sided P value | █████ | |
CI = confidence interval; D = durvalumab; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-LC13 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Lung Cancer 13; FAS = full analysis set; HRQoL = health-related quality of life; QoL = quality of life; t = tremelimumab; vs. = versus.
Note: The analysis was performed using logistic regression adjusting for PD-L1 (≥ 50% vs. < 50%), histology (squamous vs. nonsquamous), and disease stage (IVA vs. IVB), with the CI calculated using a profile likelihood approach and the P value calculated based on twice the change in log-likelihood resulting from the addition of a treatment factor to the model. An odds ratio of greater than 1 favours durvalumab and tremelimumab plus standard-of-care chemotherapy compared to standard-of-care chemotherapy alone. Percentages are calculated from the number of patients in the full analysis set with baseline symptom scores of 10 or higher or a baseline HRQoL/function scores of 90 or lower in that treatment group. Data cut-off: March 12, 2021.
aImprovement rate is defined as the number (%) of patients with 2 consecutive assessments at least 14 days apart that show a clinically meaningful improvement (decrease from baseline score of 10 or higher for symptom scales or increase from baseline score of 10 or higher for functional scales and Global Health Status) in that symptom or function from baseline.
Source: Sponsor’s Summary of Clinical Evidence,1 POSEIDON Clinical Study Report,38 and sponsor’s data on file.13
Harms
A summary of key harms in arms 1 and 3 of the POSEIDON trial from the final analysis (DCO: March 12, 2021) for the SAF is presented in Table 28. The SAF consisted of all patients who received at least 1 dose of study treatment. Safety data were summarized according to the treatment actually received.1,38
Table 28: Summary of Harms Results From the POSEIDON Trial (Safety Analysis Set)
Adverse events | D + T + chemotherapy (N = 330) | Chemotherapy (N = 333) |
|---|---|---|
Most common adverse events (occurring in ≥ 10% of patients), n (%) | ||
At least 1 adverse event | 321 (97.3) | 320 (96.1) |
Anemia | 164 (49.7) | 163 (48.9) |
Nausea | 137 (41.5) | 122 (36.6) |
Neutropenia | 99 (30.0) | 78 (23.4) |
Decreased appetite | 93 (28.2) | 82 (24.6) |
Fatigue | 81 (24.5) | 74 (22.2) |
Diarrhea | 71 (21.5) | 51 (15.3) |
Rash | 64 (19.4) | 22 (6.6) |
Constipation | 63 (19.1) | 79 (23.7) |
Thrombocytopenia | 60 (18.2) | 57 (17.1) |
Vomiting | 60 (18.2) | 45 (13.5) |
Asthenia | 56 (17.0) | 41 (12.3) |
Pyrexia | 53 (16.1) | 23 (6.9) |
Pneumonia | 47 (14.2) | 32 (9.6) |
Increased alanine aminotransferase | 46 (13.9) | 44 (13.2) |
Increased aspartate aminotransferase | 42 (12.7) | 38 (11.4) |
Leukopenia | 42 (12.7) | 39 (11.7) |
Arthralgia | 41 (12.4) | 21 (6.3) |
Decreased neutrophil count | 39 (11.8) | 59 (17.7) |
Hypothyroidism | 39 (11.8) | 4 (1.2) |
Headache | 37 (11.2) | 25 (7.5) |
Pruritus | 36 (10.9) | 15 (4.5) |
Cough | 33 (10.0) | 22 (6.6) |
Alopecia | 33 (10.0) | 20 (6.0) |
Serious adverse eventsa (occurring in ≥ 1% of patients), n (%) | ||
Patients with ≥ 1 serious adverse event | 146 (44.2) | 117 (35.1) |
Pneumonia | 36 (10.9) | 16 (4.8) |
Anemia | 18 (5.5) | 21 (6.3) |
Thrombocytopenia | 8 (2.4) | 3 (0.9) |
Diarrhea | 8 (2.4) | 2 (0.6) |
Pyrexia | 8 (2.4) | 1 (0.3) |
Febrile neutropenia | 7 (2.1) | 4 (1.2) |
Pneumonitis | 6 (1.8) | 1 (0.3) |
Acute kidney injury | 6 (1.8) | 1 (0.3) |
Pulmonary embolism | 5 (1.5) | 9 (2.7) |
Colitis | 5 (1.5) | 0 |
Sepsis | 5 (1.5) | 2 (0.6) |
Neutropenia | 4 (1.2) | 3 (0.9) |
Cerebrovascular accident | 4 (1.2) | 1 (0.3) |
Death | 3 (0.9) | 1 (0.3) |
Hemoptysis | 2 (0.6) | 3 (0.9) |
Pancytopenia | 2 (0.6) | 3 (0.9) |
Chronic obstructive pulmonary disease | 2 (0.6) | 5 (1.5) |
Patients who stopped treatment because of adverse eventsb (reported for ≥ 2 patients in the D + T + chemotherapy arm), n (%) | ||
Any adverse event leading to discontinuation of any study treatment | 73 (22.1) | 51 (15.3) |
Pneumonia | 8 (2.4) | 7 (2.1) |
Anemia | 5 (1.5) | 4 (1.2) |
Acute kidney injury | 4 (1.2) | 1 (0.3) |
Increased blood creatinine | 4 (1.2) | 0 |
Pneumonitis | 3 (0.9) | 1 (0.3) |
Sepsis | 3 (0.9) | 0 |
Pulmonary embolism | 2 (0.6) | 4 (1.2) |
Colitis | 2 (0.6) | 0 |
Diarrhea | 2 (0.6) | 0 |
Nausea | 2 (0.6) | 1 (0.3) |
Drug-induced liver injury | 2 (0.6) | 0 |
Autoimmune nephritis | 2 (0.6) | 0 |
Fatigue | 2 (0.6) | 1 (0.3) |
Decreased neutrophil count | 2 (0.6) | 1 (0.3) |
Patients who had dose delay or interruptionc because of adverse events | ||
Patients with an adverse event leading to dose delay/interruption | 189 (57.3) | 143 (42.9) |
Deaths (occurring in ≥ 2 patients in any treatment group), n (%) | ||
Patients with any adverse event with an outcome of death | 41 (12.4) | 30 (9.0) |
Pneumonia | 7 (2.1) | 7 (2.1) |
Sepsis | 3 (0.9) | 1 (0.3) |
Febrile neutropenia | 1 (0.3) | 2 (0.6) |
Embolism | 0 | 1 (0.3) |
Cerebrovascular accident | 2 (0.6) | 1 (0.3) |
Cardiac failure | 2 (0.6) | 1 (0.3) |
Cardiopulmonary failure | 2 (0.6) | 1 (0.3) |
Acute myocardial infarction | 1 (0.3) | 1 (0.3) |
Pulmonary embolism | 1 (0.3) | 5 (1.5) |
Pneumonia aspiration | 0 | 0 |
Pulmonary hemorrhage | 0 | 2 (0.6) |
Acute kidney injury | 2 (0.6) | 0 |
Death | 3 (0.9) | 1 (0.3) |
Sudden death | 1 (0.3) | 0 |
Adverse events of special or potential interest (by category), n (%) | ||
Pneumonitis | 16 (4.8) | 2 (0.6) |
Hepatic events | 77 (23.3) | 56 (16.8) |
Diarrhea or colitis | 81 (24.5) | 51 (15.3) |
Adrenal insufficiency | 8 (2.4) | 0 |
Type 1 diabetes mellitus | 1 (0.3) | 0 |
Hyperthyroid events | 22 (6.7) | 3 (0.9) |
Hypophysitis | 6 (1.8) | 0 |
Hypothyroid events | 44 (13.3) | 7 (2.1) |
Thyroiditis | 4 (1.2) | 1 (0.3) |
Renal events | 24 (7.3) | 17 (5.1) |
Dermatitis/Rash | 116 (35.2) | 45 (13.5) |
Pancreatic events | 45 (13.6) | 20 (6.0) |
Myocarditis | 2 (0.6) | 0 |
Myositis | 2 (0.6) | 1 (0.3) |
Infusion or hypersensitivity reactions | 15 (4.5) | 8 (2.4) |
Other rare or miscellaneous | 47 (14.2) | 23 (6.9) |
Adverse events of special interest (by category), n (%) | ||
Pneumonitis | 16 (4.8) | 2 (0.6) |
Hepatic events | 12 (3.6) | 0 |
Diarrhea or colitis | 81 (24.5) | 51 (15.3) |
Adrenal insufficiency | 7 (2.1) | 0 |
Type 1 diabetes mellitus | 1 (0.3) | 0 |
Hyperthyroid events | 19 (5.8) | 2 (0.6) |
Hypophysitis | 6 (1.8) | 0 |
Hypothyroid events | 39 (11.8) | 4 (1.2) |
Thyroiditis | 4 (1.2) | 1 (0.3) |
Renal events | 2 (0.6) | 0 |
Dermatitis or rash | 89 (27.0) | 31 (9.3) |
Pancreatic events | 7 (2.1) | 2 (0.6) |
Myocarditis | 1 (0.3) | 0 |
Myositis | 2 (0.6) | 1 (0.3) |
Infusion-related reaction | 13 (3.9) | 5 (1.5)d |
Hypersensitivity or anaphylactic reactions | 3 (NR) | 3 (NR) |
D = durvalumab; NR = not reported; t = tremelimumab.
Note: Patients with multiple serious adverse events are counted once for each preferred term. Includes adverse events with an onset date on or after the date of first dose or pretreatment adverse events that increase in severity on or after the date of first dose up to and including 90 days following the date of last dose of study medication or up to and including the date of initiation of the first subsequent therapy (whichever occurs first). Data cut-off: March 12, 2021.
aSeriousness as assessed by the investigator. An adverse event with missing seriousness is considered serious.
bAction taken, study treatment permanently discontinued.
cDose interruptions were defined as same day interruptions of infusion during administration of D, T or chemotherapy. Dose delays were defined as administration of treatment outside of the scheduled visit window.
dN = 337 for these data.
Sources: Sponsor’s Summary of Clinical Evidence,1 POSEIDON Clinical Study Report,38 and POSEIDON Clinical Study Report (Tables and Figures).61
Adverse Events
In the SAF, the proportions of patients who experienced at least 1 AE were 97.3% in the durvalumab and tremelimumab plus SOC chemotherapy arm and 96.1% in the SOC chemotherapy alone arm. In the durvalumab and tremelimumab plus SOC chemotherapy arm compared with the SOC chemotherapy alone arm, the most commonly reported AEs were anemia (49.7% versus 48.9%, respectively), nausea (41.5% versus 36.6%), neutropenia (30.0% versus 23.4%), decreased appetite (28.2% versus 24.6%), fatigue (24.5% versus 22.2%), diarrhea (21.5% versus 15.3%), rash (19.4% versus 6.6%), and constipation (19.1% versus 23.7%). AEs were reported in a higher proportion of patients (≥ 5% difference between groups) in the durvalumab and tremelimumab plus SOC chemotherapy arm than in the SOC chemotherapy alone arm were neutropenia (30.0% versus 23.4%), diarrhea (21.5% versus 15.3%), rash (19.4% versus 6.6%), pyrexia (16.1% versus 6.9%), arthralgia (12.4% versus 6.3%), hypothyroidism (11.8% versus 1.2%), pruritus (10.9% versus 4.5%), and hyperthyroidism (5.8% versus 0.6%). A lower proportion of patients in the durvalumab and tremelimumab plus SOC chemotherapy arm had reported neutrophil count decreased than in the SOC chemotherapy alone arm (11.8% versus 17.7%).1
Serious Adverse Events
SAEs were reported in 44.2% of patients in the durvalumab and tremelimumab plus SOC chemotherapy arm and 35.1% of patients in the SOC chemotherapy alone arm. SAEs with a 2% or greater difference between groups were reported in a higher proportion of patients in the durvalumab and tremelimumab plus SOC chemotherapy group than in the SOC chemotherapy alone group: pneumonia (10.9 versus 4.8%, respectively) and pyrexia (2.4% versus 0.3%).1
Withdrawals and Dose Delays or Interruptions of Study Treatment Because of AEs
AEs leading to discontinuation of any study treatment were reported in 22.1% of patients in the durvalumab and tremelimumab plus SOC chemotherapy arm, and 15.3% of patients in the SOC chemotherapy alone arm. In the durvalumab and tremelimumab plus SOC chemotherapy arm compared to the SOC chemotherapy alone arm, there was a 2% or greater difference between total AEs leading to discontinuation of study treatment reported in the system organ classes of infections and infestations (4.5% versus 2.4%, respectively) and gastrointestinal disorders (3% versus 0.9%). AEs leading to at least 1 dose delay or interruption of any study treatment occurred in 57.3% of patients in the durvalumab and tremelimumab plus SOC chemotherapy arm and 42.9% of patients in the SOC chemotherapy alone arm.38
Mortality
AEs leading to death were reported in 12.4% of patients in the durvalumab and tremelimumab plus SOC chemotherapy arm and 9.0% of patient in the SOC chemotherapy alone arm.
Notable Harms
AESIs or AEPIs were reported ███ ██████ patients in the durvalumab and tremelimumab plus SOC chemotherapy arm and 165 ███████ patients in the SOC chemotherapy alone arm. Grade 3 or 4 AESIs or AEPIs were experienced by ██ ███████ patients in the durvalumab and tremelimumab plus SOC chemotherapy arm and ██ ██████ patients in the SOC chemotherapy alone arm. AESIs or AEPIs with an outcome of death were reported in | ██████ patients and | ██████ patient in the durvalumab and tremelimumab plus SOC chemotherapy and SOC chemotherapy alone arms, respectively. In the durvalumab and tremelimumab plus SOC chemotherapy arm, AESIs or AEPIs that led to any study treatment discontinuation were reported in ██ ██████ patients; these were reported in | ██████ patients in the SOC chemotherapy alone arm.38 Separate data for overall rates of AESIs and overall rates of AEPIs were not provided in the evidence submitted by the sponsor.
A total of 111 patients (33.6%) patients in the durvalumab and tremelimumab plus SOC chemotherapy arm reported imAEs compared with 17 (5.1%) patients in the SOC chemotherapy alone arm. Grade 3 or 4 imAEs were reported in 33 patients (10.0%) and 5 patients (1.5%), respectively. Two patients (0.6%) in the durvalumab and tremelimumab plus SOC chemotherapy arm and no patients in the SOC chemotherapy alone arm experienced imAEs with an outcome of death. AEs leading to discontinuation of any study treatment occurred in 19 patients (5.8%) in the durvalumab and tremelimumab plus SOC chemotherapy arm and 2 patients (0.6%) in the SOC chemotherapy alone arm. Up to █████ of patients in the durvalumab and tremelimumab plus SOC chemotherapy arm experienced imAEs requiring treatment with systemic corticosteroids, high-dose steroids, and endocrine therapy; this was reported for up to ████ of patients in the SOC chemotherapy alone arm. ████ ██████ patients in the durvalumab and tremelimumab plus SOC chemotherapy arm received other immunosuppressants whereas no patients in the SOC chemotherapy alone arm received this treatment intervention.38
Summaries of select AEs and AESIs, described by clinical experts as AEs of greatest clinical importance, are presented in the following section.
Neutropenia
Neutropenia was reported in 30.0% of patients in the durvalumab and tremelimumab plus SOC chemotherapy arm and 23.4% of patients in the SOC chemotherapy alone arm, with grade 3 or 4 neutropenia occurring in 17.0% and 12.3% of patients, respectively.1,61
Pneumonitis
The proportions of patients who experienced AESIs of pneumonitis (grouped term) were 4.8% in the durvalumab and tremelimumab plus SOC chemotherapy arm and 0.6% in the SOC chemotherapy alone arm. Grade 3 or 4 events were reported in 1.2% of patients in the durvalumab and tremelimumab plus SOC chemotherapy arm and 0.6% of patients in the SOC chemotherapy alone arm and serious AESIs of pneumonitis were reported in ████ ███ ████ of patients, respectively. AESIs of pneumonitis that led to discontinuation of any study treatment occurred in ████ of patients in the durvalumab and tremelimumab plus SOC chemotherapy arm and ████ of patients in the SOC chemotherapy alone arm.1
Hepatic Events
The proportion of patients who experienced AESIs of hepatic events (grouped term) was 3.6% in the durvalumab and tremelimumab plus SOC chemotherapy arm. In this group, grade 3 or 4 AESIs of hepatic events were reported in 1.5% of patients and serious AESIs of hepatic events were reported in 1.8% of patients. No AESIs of hepatic events (including grade 3 or 4 and serious events) were reported in the SOC chemotherapy alone arm. AESIs of hepatic events that led to discontinuation of any study treatment occurred in 1.2% of patients in the durvalumab and tremelimumab plus SOC chemotherapy arm and no patients in the SOC chemotherapy alone arm.1 AESIs of hepatitis were reported in 2.1% of patients in the durvalumab and tremelimumab plus SOC chemotherapy arm, with grade 3 or 4 hepatitis occurring in 0.3% of patients; no such events were reported in patients in the SOC chemotherapy alone arm.61
Diarrhea or Colitis
The proportion of patients who experienced AESIs of diarrhea or colitis (grouped term) was 24.5% in the durvalumab and tremelimumab plus SOC chemotherapy arm and 15.3% in the SOC chemotherapy alone arm. Grade 3 or 4 AESIs of diarrhea or colitis were reported in 3.6% of patients in the durvalumab and tremelimumab plus SOC chemotherapy arm and 1.8% of patients in the SOC chemotherapy alone arm, and serious AESIs of diarrhea or colitis were reported in 4.2% and 0.6% of patients, respectively. AESIs of diarrhea or colitis that led to discontinuation of any study treatment occurred in 1.5% of patients in the durvalumab and tremelimumab plus SOC chemotherapy arm and no patients in the SOC chemotherapy alone arm. Diarrhea was reported in 21.5% of patients in the durvalumab and tremelimumab plus SOC chemotherapy arm and 15.3% of patients in the SOC chemotherapy alone arm, and colitis was reported in ████ ███ ████ of patients, respectively.1
Type 1 Diabetes Mellitus
AESIs of type 1 diabetes mellitus (grouped term) was reported in 1 patient (0.3%) in the durvalumab and tremelimumab plus SOC chemotherapy arm, which was reported as serious and grade 3 or 4 in severity. No reports of AESIs of type 1 diabetes mellitus were made in the SOC chemotherapy alone arm.1
Myocarditis
AESIs of myocarditis events (grouped term) was reported in 1 patient (0.3%) in the durvalumab and tremelimumab plus SOC chemotherapy arm, which was serious (grade 5), and resulted in death. No AESIs of myocarditis events were reported in the SOC chemotherapy alone arm.1
Critical Appraisal
Internal Validity
The POSEIDON trial was a phase III, randomized, open-label, comparative, multicentre, global study. Methods of randomization and treatment allocation (by IVRS or IWRS) were adequate. Reported baseline characteristics were generally balanced across the study arms, suggesting that randomization was effective. The durvalumab and tremelimumab plus SOC chemotherapy arm had a lower percentage of female patients, Asian patients, and never smokers than in the SOC chemotherapy arm. Clinical experts consulted by CDA-AMC did not identify any considerable differences between these treatment arms that would be expected to affect the interpretation of results; they acknowledged that, generally, an imbalance in the proportion of never smokers between groups could affect treatment effect but were uncertain if the difference between durvalumab and tremelimumab plus SOC chemotherapy and SOC chemotherapy alone arms in the POSEIDON trial was substantial enough to have an effect. The prespecified sample size was achieved and actual screening failures did not exceed the expected number. The POSEIDON trial was powered for the dual primary end points of PFS and OS for the comparison of durvalumab plus SOC chemotherapy versus SOC chemotherapy. Power calculations were provided for all end points included in the MTP, which included key secondary end points of OS and PFS for the comparison between durvalumab and tremelimumab plus SOC chemotherapy and SOC chemotherapy alone.
The study used an open-label design because of differences in administration schedule and duration of study treatments. Of the efficacy end points of interest for this review, OS is an objective end point, and PFS, ORR, BoR, and DoR were assessed by BICR according to RECIST 1.1 criteria, which helped to mitigate potential bias. However, the patient-reported efficacy assessments (i.e., HRQoL end points) and reporting of AEs can be influenced by knowledge of the treatment assignment by patients and clinicians.
Allowed concomitant medications received by patients during the trial were mostly balanced between the durvalumab and tremelimumab plus SOC chemotherapy and SOC chemotherapy alone arms; the clinical experts consulted for this review noted that medication categories that were less balanced would not be expected to affect treatment effect. The experts noted that proton pump inhibitors may affect the efficacy of immunotherapy, and administration to these patients may be discouraged, but they added that oncology patients receiving immunotherapy will still be given this class of medication in practice when it is indicated.
Continuation of durvalumab monotherapy or re-treatment with tremelimumab upon disease progression was permitted in the POSEIDON trial at the investigator’s discretion. In the durvalumab and tremelimumab plus SOC chemotherapy arm, 36 patients (10.9%) received treatment with durvalumab following the date of disease progression confirmation and 18 patients (5.5%) continued to receive durvalumab 8 weeks after progression confirmation. The clinical experts did not expect the percentage of patients in the study who received continuation of durvalumab treatment to have an effect on the efficacy observed in the trial. At the 5-year DCO, a total of 10 patients had received tremelimumab re-treatment. The clinical experts also stated that, for patients who had an initial response to durvalumab and tremelimumab plus SOC chemotherapy and then subsequently progressed, the benefit of re-treatment with tremelimumab is unknown, and they did not expect the proportion of patients who were re-treated with tremelimumab in the POSEIDON trial to have an impact on observed efficacy.
Loss to follow-up was low and occurred at the same rate in both arms at the final analysis (0.6%) and 5-year analysis (0.9%). At both DCOs, a higher proportion of patients had discontinued treatment in the SOC chemotherapy alone arm (final analysis DCO: 98.5%; 5-year DCO: 99.7%) than in the durvalumab and tremelimumab plus SOC chemotherapy arm (88.8% and 94.0%, respectively). Most treatment-discontinuation events were because of reasons anticipated in the protocol (i.e., disease progression or death, AEs, or maximum cycles of therapy achieved). The clinical experts stated that, overall, the patient rates of discontinuation in the POSEIDON trial were not unreasonable. However, the treatment exposure to SOC chemotherapy alone was significantly lower in the SOC chemotherapy arm (164.9 patient-years with a mean duration of 25.83 weeks) than in the durvalumab and tremelimumab plus SOC chemotherapy arm (222.9 patient-years with mean duration of 35.35 weeks) at the final analysis. This is most likely attributable to a larger proportion of patients discontinuing SOC chemotherapy because their “condition under investigation worsened” in the SOC chemotherapy arm (55.3%) compared with the durvalumab and tremelimumab plus SOC chemotherapy arm (41.7%). Greater exposure to SOC chemotherapy in the durvalumab and tremelimumab plus SOC chemotherapy arm would have biased the results in favour of this treatment regimen compared to SOC chemotherapy alone.
Overall, there was also higher treatment exposure in the durvalumab and tremelimumab plus SOC chemotherapy arm (313.8 patient-years with a mean duration of 49.62 weeks) than in the SOC chemotherapy arm (164.9 patient-years with a mean duration of 25.83 weeks) at the final analysis. Although higher overall treatment exposure in the durvalumab and tremelimumab plus SOC chemotherapy group may be anticipated because of the treatment protocol, some of the difference may be attributable to treatment through progression with durvalumab and re-treatment with tremelimumab. Continued treatment with durvalumab monotherapy following disease progression may not affect the estimation of PFS, but it could have affected the exact assessment and interpretation of overall treatment benefit as continuation of treatment was subjectively based on investigator judgment. The ICH E9 (R1) addendum of the International Council for Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use63 defines an estimand as a precise description of the treatment effect reflecting the clinical question posed by a given clinical trial objective. Continuation of treatment through progression as well as subsequent anticancer treatments could constitute intercurrent events after initiation of the study treatment.
Subsequent therapies in the POSEIDON trial may introduce a confounding effect on survival. At both DCO dates, subsequent systemic anticancer treatment was received by a larger proportion of patients in the SOC chemotherapy alone arm (final analysis DCO: 60.2%; 5-year DCO: 60.8%) than in the durvalumab and tremelimumab plus SOC chemotherapy arm (40.8% and 42.9%, respectively). For example, subsequent immunotherapy treatment was received by 33.2% and 6.5% of patients in these treatment arms, respectively, at the final analysis. This may have diluted the survival benefit of durvalumab and tremelimumab plus SOC chemotherapy compared to SOC chemotherapy alone.
Primary and key secondary efficacy end points in the POSEIDON trial were analyzed according to the prespecified analysis plan that controlled for multiple comparisons and was finalized before the interim PFS analysis. Other secondary end points were not controlled for multiplicity, and the 5-year survival estimates and CIs were not alpha-controlled.
Sensitivity analyses were performed for the key secondary end points of OS and PFS. For OS, sensitivity analyses adjusting for electronic case report form–derived stratification variables showed consistency between the 5-year OS analysis and the previous final analysis; the results of other conducted sensitivity analyses were also generally consistent with the primary analysis. For PFS, sensitivity analyses for robustness of the PFS effect to possible sources of bias in the measurement of PFS and sensitivity analyses adjusting for treatment, stratification factors, and/or additional prespecified covariates were generally consistent with those of the primary PFS analysis. Post hoc analysis of ORR using confirmed responses by BICR also demonstrated a greater ORR for durvalumab and tremelimumab plus SOC chemotherapy compared to SOC chemotherapy alone.
Subgroup analyses for OS and PFS end points were conducted according to prespecified subgroups, except for the KRAS and STK11 mutation subgroup analyses (conducted for OS), which were post hoc and exploratory. However, the POSEIDON trial was not powered for any individual subgroup evaluations, adjustments were not made for multiple testing, and there were low numbers of patients and events across individual subgroups, which leads to uncertainty in their estimates. No survival benefit of durvalumab and tremelimumab plus SOC chemotherapy compared to SOC chemotherapy alone was observed in the subgroup of never smokers (HR = 1.17; 95% CI, 0.820 to 1.661 at the 5-year DCO analysis). Moreover, the overall OS benefit in the trial was driven primarily by patients with nonsquamous histology (n = 428 in the durvalumab and tremelimumab plus SOC chemotherapy and SOC chemotherapy alone groups) (HR = 0.69; 95% CI, 0.56 to 0.85 at the 5-year DCO analysis). In patients with squamous histology (n = 246 in the durvalumab and tremelimumab plus SOC chemotherapy and SOC chemotherapy alone groups), the difference in OS numerically favoured durvalumab and tremelimumab plus SOC chemotherapy (HR = 0.85) but its 95% CI crossed the null value.
The psychometric properties of the EORTC QLQ-C30 and EORTC QLQ-LC13 scales, including supportive evidence for their validity, reliability, and responsiveness in patients with lung cancer, are described in the literature. References from the literature report a range of MID values for EORTC QLQ-C30 in various types of cancers (including lung cancer), with evidence to support a clinically meaningful change for the EORTC QLQ-C30 as an absolute change in the score from baseline of 10 or more points, as was used in the POSEIDON trial. The POSEIDON trial also used this threshold to define a clinically meaningful change in the EORTC QLQ-LC13; however, no evidence was provided for MID values for this scale in the overall lung cancer population or for patients with NSCLC. Similarly, between-group MID values were not submitted for time to deterioration or improvement-rate measures for either scale. Overall adherence rates to the PRO scales generally decreased over time and were lower in the SOC chemotherapy arm (the adherence rate fell below 60% after 24 weeks in the SOC chemotherapy arm and after 88 weeks in the durvalumab and tremelimumab plus SOC chemotherapy arm). The missing data present a challenge to evaluating the effects on HRQoL.
External Validity
The clinical experts consulted by CDA-AMC commented that the POSEIDON trial used standard inclusion and exclusion criteria that would be expected in a study of first-line treatment of patients with metastatic NSCLC, but noted certain eligibility criteria that would not necessarily apply in clinical practice. For example, exclusion criteria for brain metastases in the trial were stricter than what would be used in practice; patients with infections such as HIV or hepatitis C would routinely be offered therapy, a trial of immunotherapy may be recommended for most patients with autoimmune or inflammatory disorders, and exclusion of patients with a history of cancer in the previous 5 years may not be applicable to practice. The experts identified differences between the POSEIDON trial participants and patients who would receive first-line immuno-oncology plus SOC chemotherapy for metastatic NSCLC in clinical practice in Canada. Specifically, patients in practice would typically be older than 70 years of age; have relative contraindications to immunotherapy (i.e., quiescent inflammatory or autoimmune conditions that would not result in significant morbidity if reactivated); include those who have an ECOG PS score of 2; and include those who had received immunotherapy for earlier-stage cancer. Female patients were underrepresented in the POSEIDON trial and a higher proportion of patients with liver and CNS metastases would be expected in practice. The clinical experts commented that the ratio of patients with squamous-cell to non–squamous cell carcinomas in the POSEIDON trial may be representative of areas of Canada with more prevalent tobacco use and therefore more tobacco-related cancers but less representative of areas with less tobacco use, where a lower proportion of squamous-cell NSCLC may be observed. The proportions of patients with a PD-L1 status below 50% or 50% or greater in the trial is reflective of the population of patients with metastatic NSCLC seen in Canada. The clinical experts noted that, overall, they did not have concerns regarding the generalizability of the study findings to patients seen in clinical practice. The POSEIDON trial did not include any sites in Canada. The experts also did not have any major concerns regarding the generalizability of the trial findings based on the study locations.
The clinical experts expected that regional variability would exist in the use of fixed versus weight-based dosing of durvalumab and tremelimumab based on the typical patient population (e.g., some areas of Canada may implement weight-based dosing for a generally lower-weight patient population, who may receive too much medication with the fixed-dose regimen).
In the POSEIDON trial, patients who discontinued SOC chemotherapy because of AEs were permitted to continue with durvalumab monotherapy or durvalumab and tremelimumab combination therapy; the clinical experts acknowledged that this is reflective of clinical practice and would be expected to occur frequently. Regarding treatment and re-treatment through progression, the experts agreed that they would not expect treatment beyond progression to occur in clinical practice (noting that pseudo- and oligoprogression may be exceptions), nor would they expect re-treatment with tremelimumab to occur commonly; patients who experience disease progression would be expected to subsequently receive a different type of systemic therapy.
An important limitation of the POSEIDON trial is that durvalumab and tremelimumab plus SOC chemotherapy was compared to SOC chemotherapy alone. At the time the trial was conducted, platinum-based chemotherapy was the standard of therapy for the indicated population; however, current first-line treatment of metastatic NSCLC without targetable genomic alterations consists of immuno-oncology treatment (on its own or in combination with chemotherapy). The clinical experts commented that SOC chemotherapy regimens used in the POSEIDON trial represent those used in clinical practice in Canada with some caveats. Nab-paclitaxel would not be a standard regimen (it is not funded in Canada); paclitaxel plus carboplatin would be used in its place and its efficacy is considered to be comparable to that of nab-paclitaxel plus carboplatin. Also, the use of gemcitabine in a platinum doublet with immunotherapy varies in clinical practice across Canada. One expert commented that, in combination with immuno-oncology, patients with nonsquamous NSCLC would typically receive the pemetrexed regimen and patients with squamous NSCLC would typically receive paclitaxel plus carboplatin. The other clinical expert reported using gemcitabine in clinical practice, with approximately half of patients with squamous NSCLC receiving the gemcitabine regimen and approximately half of those patients receiving the paclitaxel regimen. In the POSEIDON trial, most patients with nonsquamous histology received the pemetrexed regimen and most patients with squamous histology received the gemcitabine regimen. Patients in the SOC chemotherapy alone arm of the POSEIDON trial were able to receive 2 additional doses of chemotherapy, if clinically indicated, at the investigator’s discretion. The clinical experts noted that this may or may not be done in practice and that there may be regional variability in this practice depending on the underlying health status of the typical patient population.
Concerning subsequent anticancer treatments received in the POSEIDON trial, the clinical experts consulted for this review pointed out that patients who received first-line immunotherapy cannot be rechallenged with immunotherapy outside of a clinical trial setting, and this is not reflective of clinical practice. In addition, it would be expected that, in Canada, patients who received chemotherapy alone as first-line treatment would have a much higher rate of subsequent treatment with immunotherapy because of current access to this treatment.
According to the clinical experts, the goals of therapy include helping patients feel better (which can be determined from trials through ORR, QoL, and other PROs) and improvement in survival (PFS and OS). Outcomes of importance identified by clinician groups and patient groups include prolonging survival, maintenance or improvement in QoL, improvement in symptoms, and reduced adverse effects. As such, the main efficacy and harms outcomes assessed in the POSEIDON trial align with outcomes of importance to patients and clinicians.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered most relevant to inform expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group:64,65
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate — The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited — The true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate — The true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as “very uncertain.”
Following the GRADE approach, evidence from the RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null.
Results of GRADE Assessments
Table 4 presents the GRADE summary of findings for durvalumab and tremelimumab plus SOC chemotherapy versus SOC chemotherapy.
Long-Term Extension Studies
No long-term extension studies were submitted by the sponsor.
Indirect Evidence
Contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
Objectives for the Summary of Indirect Evidence
The objective of this section is to summarize and critically appraise the sponsor-submitted ITC.
Description of Indirect Comparison
Objectives
In the absence of head-to-head trial data comparing the combination of durvalumab and tremelimumab plus chemotherapy versus other ICIs plus chemotherapy for the first-line treatment of patients with metastatic NSCLC with no sensitizing EGFR mutations or ALK genomic tumour aberrations, the sponsor submitted an ITC of the relative efficacy of durvalumab and tremelimumab in combination with platinum-based chemotherapy versus other approved ICIs plus platinum-based chemotherapy combination regimens in this patient population. The ITC was carried out using a MAIC approach. CDA-AMC accepted the sponsor’s deviation request to include only regimens of other ICI plus chemotherapy in the economic analyses to align with the anticipated Health Canada indication for durvalumab and tremelimumab plus chemotherapy. As such, the submitted ITC did not include comparisons to ICI monotherapies.
Study Selection Methods
An initial systematic literature review (SLR) was conducted to identify clinical evidence from RCTs that addressed the efficacy and safety of first-line treatments of adults with metastatic NSCLC without EGFR mutations or ALK aberrations. This initial SLR included relevant literature up to March 2022. The SLR was updated in March 2024, giving the literature search a range of January 2022 to March 2024. Selected conference papers from 2023 were also screened for relevant material. In addition, hand searches were performed to identify post hoc, follow-up, and subgroup analysis of trials identified both in the original and updated SLRs.
The original systematic review identified 15 studies (90 publications) reporting on the efficacy, safety, and HRQoL of first-line treatments recommended by treatment guidelines in patients with metastatic NSCLC without EGFR mutations or ALK aberrations. Out of 90 publications, 55 reports were not extracted as these publications reported on results from intermediate DCOs rather than the DCOs of interest (i.e., primary, and latest). Four new trials were identified in the search for the updated SLR. Collectively, the updated SLR included 19 trials, with results reported across 127 publications.
The MAICs focused on ICI plus chemotherapy regimens used in first-line treatment of metastatic NSCLC (without oncogenic driver mutations) in Canada, and included only the following comparators:
pembrolizumab in combination with platinum-based chemotherapy: considered to represent the SOC ICI plus chemotherapy regimen, and the preferred combination regimen in National Comprehensive Cancer Network guidelines66 for first-line metastatic NSCLC without actionable mutations (across all PD-L1 expression levels)
nivolumab in combination with ipilimumab and platinum-based chemotherapy: the only currently approved dual ICI plus chemotherapy regimen, which also includes the combination of an anti–PD-1 and anti–CTLA-4 drugs
cemiplimab in combination with platinum-based chemotherapy: since the initial MAIC analyses were conducted, cemiplimab in combination with platinum-based chemotherapy has been approved to treat patients diagnosed with NSCLC; as such, an additional MAIC was conducted to compare durvalumab and tremelimumab plus chemotherapy versus cemiplimab in combination with platinum-based chemotherapy.
The comparator data used in the MAICs are from the registrational phase III RCTs for each of these regimens. The relevant studies for each of these comparators identified in the trials are:
the KEYNOTE-189 study (pembrolizumab plus chemotherapy in nonsquamous NSCLC)
the KEYNOTE-407 study (pembrolizumab plus chemotherapy in squamous NSCLC)
the CheckMate 9LA study (nivolumab plus ipilimumab plus chemotherapy in NSCLC)
the EMPOWER-Lung 3 study (cemiplimab plus chemotherapy in NSCLC).
Since the searches of the SLR were conducted, more recent OS data from the POSEIDON trial have become available (DCO: August 24, 2023 [5-year OS analysis]), and survival data from a longer duration of follow-up have been presented at clinical congresses for the KEYNOTE-189 trial (5-year follow-up), the KEYNOTE-407 trial (5-year follow-up), and the CheckMate 9LA trial (4-year follow-up).62,67-69 Results are therefore also presented for OS MAICs using the more recent data from the POSEIDON and comparator trials. PFS was not analyzed at the 5-year OS analysis (or 4-year OS analysis) in POSEIDON and no updates for PFS MAICs were conducted.58,70
Study selection criteria and methods for the MAICs are summarized in Table 29.
Table 29: Study Selection Criteria and Methods for Indirect Comparisons
Characteristics | Indirect comparison |
|---|---|
Population | Adults with untreated metastatic NSCLC without sensitizing EGFR mutations or ALK fusions (including both squamous and nonsquamous NSCLC) No restrictions based on PD-L1 status |
Intervention | Durvalumab and tremelimumab in combination with platinum-based chemotherapy |
Comparator |
|
Outcome |
|
Study designs | Randomized controlled trials |
Publication characteristics |
|
Exclusion criteria |
|
Databases searched | The following databases were searched (until March 2024)
Conference proceedings from the following congresses were hand-searched to identify any relevant abstracts for inclusion: ASCO, ESMO, ELCC, WCLC, ACCR, ESMO-ASIA, ESMO IO |
Selection process | Trials were identified through an SLR. Titles and abstracts of studies identified from the search strategy were reviewed independently by 2 researchers in accordance with the prespecified inclusion and exclusion criteria. Potentially relevant articles were then further reviewed for eligibility. A full-text review of these articles was also conducted by 2 researchers independently and in parallel, using the prespecified inclusion and exclusion criteria. Any discrepancy at the title, abstract, or full-text review stage was resolved by discussion with a third reviewer. After the full-text review was completed, a list of the excluded studies was created with the reason for exclusion. |
Data extraction process | For each trial, the following data components were extracted: trial acronym, year of publication, primary publication, study phase, blinding, names of intervention arms, treatment details (dose, cycles, and frequency of administration), arm sample size, total sample size, key baseline characteristics (sex, age, region, ethnicity, histology, PD-L1 tumour proportion score distribution, ECOG PS, smoking status, histology, EGFR and ALK mutation status, stage, and metastasis), key efficacy, safety and HRQoL outcomes, as well as efficacy and safety results for histology, PD-L1 tumour proportion score (cut-offs), mutations, and combinations thereof. Where eligible trials were represented by multiple references (e.g., post hoc analysis and conference presentations). All study characteristics, patient baseline characteristics, and patient disease characteristics were extracted from the primary publication only. All data extracted as part of the original SLR were used as is, from the original SLR (MS Word) document and accompanying appendices. |
Quality assessment | To assess the quality of the included clinical trials, the list of assessment questions provided in the Revised Cochrane risk-of-bias tool for randomized trials was used. |
ALK = anaplastic lymphoma kinase; ASCO = American Society of Clinical Oncology; ECOG PS = Eastern Cooperative Oncology Group Performance Status; EGFR = epidermal growth factor receptor; ELCC = European Lung Cancer Congress; ESMO = European Society of Medical Oncology; IO = immuno-oncology; NSCLC = non–small cell lung cancer; OS = overall survival; PFS = progression-free survival; SLR = systematic literature review; WCLC = World Conference on Lung Cancer.
Sources: Global POSEIDON MAIC Report.71,1
Indirect Comparison Analysis Methods
Matching Adjusted Indirect Comparison
According to the sponsor, the MAIC approach was chosen for these analyses, as it is more commonly used than simulated treatment comparisons, and has been used in similar analyses of NSCLC.72,73 The use of the MAIC approach may be limited in cases involving an uneven distribution of weights, which can occur when there is low overlap between the different study populations. This has been inspected for each of the analyses conducted in the sponsor-submitted MAIC, and in each case the distribution of weights and effective sample size has been considered acceptable by the sponsor.
Identification of Treatment-Effect Modifiers
The identification of treatment-effect modifiers (TEMs) was based on exploratory analyses of the POSEIDON trial, for which individual patient-level data were available. A global interaction test was conducted to first assess the consistency of treatment effects for durvalumab and tremelimumab plus chemotherapy versus chemotherapy alone for OS across subgroups (based on POSEIDON OS final analysis; DCO March 12, 2021). A model that included all the following baseline characteristics as covariates, plus all interaction terms for covariate and treatment, was compared against a model that excluded all interaction terms. The baseline characteristics were as follows:
histology (squamous versus nonsquamous)
disease stage (IVA versus IVB)
PD-L1 expression (< 50% versus ≥ 50%)
smoking status (current or former versus never)
ECOG PS (0 versus 1)
age (< 65 years versus ≥ 65 years)
planned chemotherapy (Abraxane, gemcitabine versus pemetrexed)
sex (female versus male)
race (Asian versus non-Asian)
brain metastases (yes versus no)
bTMB (< 20, ≥ 20 versus unknown).
The list of characteristics included in the model was based on the characteristics that were planned for the prespecified subgroup analyses of the POSEIDON trial.
Evidence from a global interaction test (P = 0.00714) suggests that significant interactions between treatment and baseline characteristics exist, when all subgroups are considered. Interactions between treatment and baseline characteristics for durvalumab and tremelimumab plus chemotherapy versus chemotherapy alone for OS were further explored using a stepwise backward-selection model. In this approach, the least significant interaction terms are removed from the model 1 at a time until a final model is reached in which all interactions are significant and all excluded interactions are nonsignificant, with a significance threshold of 0.1. Of the baseline characteristics explored, race, smoking status, and histology were identified as having a significant interaction with treatment (P < 0.05) in the stepwise backward-selection model. Race (non-Asian versus Asian), smoking status (current and former versus never) and histology (nonsquamous versus squamous) were therefore considered potential TEMs for the MAIC.
Anchored MAIC Methodology
Anchored MAICs were conducted following the methodology described in the NICE Decision Support Unit (DSU) Technical Supporting Document 18.72 The steps followed for conducting the MAICs are described in the following section. Individual patient-level data were available from the POSEIDON trial, with only aggregate data available from the comparator trials.
Step 1: Patients in the relevant patient population for the comparisons (ITT, nonsquamous and squamous) were identified from the POSEIDON trial to match the eligibility criteria of the comparator trials. In all other respects, the main eligibility criteria were considered similar between studies.
Step 2: To adjust for the imbalances in TEM between studies, patients from the durvalumab and tremelimumab plus chemotherapy and chemotherapy alone arms in the relevant POSEIDON subset (ITT, nonsquamous or squamous) were weighted according to the comparator trial’s baseline characteristics (race, smoking status, and histology [where applicable]) using propensity-score weighting, such that the weighted mean baseline characteristics for these TEMs in the pooled durvalumab and tremelimumab plus chemotherapy and chemotherapy alone POSEIDON subset matched those in the comparator trial.
Patient-level weights were derived from a logistic regression model that included only the relevant TEMs for each analysis. These weights represent the odds of an individual being enrolled in the POSEIDON subset versus the comparator trial given their race, smoking status, and histology (where applicable). With only aggregate data available from the comparator trial, the method of moments was used to estimate the patient-level weights such that the mean baseline characteristics after weighting matched those in the comparator trial.74,75
Step 3: Kaplan-Meier plots for survival outcomes were generated from the weighted durvalumab and tremelimumab plus chemotherapy and chemotherapy alone arms and HRs for durvalumab and tremelimumab plus chemotherapy versus chemotherapy alone were derived from weighted Cox proportional hazards models. Standard errors for weighted survival outcomes in the MAIC were derived using robust sandwich estimators.
Step 4: The weighted HR for durvalumab and tremelimumab plus chemotherapy versus chemotherapy alone was included in a Bucher ITC with the HR from the comparator trial (e.g., pembrolizumab plus chemotherapy versus chemotherapy alone, or nivolumab and ipilimumab plus chemotherapy versus chemotherapy alone, or cemiplimab plus chemotherapy versus chemotherapy alone) to estimate a HR for durvalumab and tremelimumab plus chemotherapy versus the MAIC comparator (e.g., durvalumab and tremelimumab plus chemotherapy versus pembrolizumab plus chemotherapy, or versus nivolumab and ipilimumab plus chemotherapy, or versus cemiplimab plus chemotherapy).
Following the NICE DSU Technical Supporting Document 18 guidance for anchored MAICs, only TEMs and not prognostic factors were included in this propensity-score weighting.72 After weighting, baseline characteristics were also compared across trials to ensure that imbalances in TEMs had been addressed.
The distribution of weights in POSEIDON following propensity-score weighting was inspected to identify whether the analyses was highly sensitive to extreme weights. The weights were also used to estimate the effective sample size (ESS) of the postweighting POSEIDON population. A low ESS is reflective of an uneven distribution of weights and limited overlap between study populations with respect to TEMs, with the outcomes in the postweighting POSEIDON population driven by a small number of individual patients.
The analyses were conducted using R version 4.1 statistical software. The MAIC analysis was performed using the MAIC version 0.1.4 package, per Signorovitch et al. (2012) as recommended in the NICE DSU guidelines.72,75
Since the searches of the SLR were conducted, survival data from longer durations of follow-up have been presented at clinical congresses for the KEYNOTE-189 trial (5-year follow-up), the KEYNOTE-407 trial (5-year follow-up), and the CheckMate 9LA trial (3-year follow-up).68,76,77 The MAICs were run again based on the most recent data from the POSEIDON trial and comparator trials.
Table 30: Indirect Comparison Analysis Methods
Methods | Description |
|---|---|
Analysis methods | Anchored MAICs conducted following the methodology described in NICE DSU TSD |
Priors | Not reported |
Assessment of model fit | Not reported |
Assessment of consistency | Not reported |
Assessment of convergence | Not reported |
Outcomes | Progression-free survival and overall survival |
Follow-up time points | Not reported |
Construction of nodes | Not reported |
Sensitivity analyses | Not reported |
Subgroup analysis | POSEIDON intention-to-treat subsets:
|
Methods for pairwise meta-analysis | Not reported |
MAIC = matching adjusted indirect comparison; NICE DSU TSD = National Institute for Health and Care Excellence Decision Support Unit Technical Supporting Document.
Source: Global POSEIDON MAIC Report.71,1
Results
Summary of Included Studies
An overview of POSEIDON and comparator studies for the MAICs is provided in Table 31. As confirmed in the submitted deviation request, only ICI plus chemotherapy regimens have been included.
Table 31: Overview of Studies Included in the Matching Adjusted Treatment Comparisons
Study or trial design | Intervention and comparator | Population | Analysis set and patient population | Reported outcomes |
|---|---|---|---|---|
POSEIDON Clinical Study Report Randomized, controlled, double-blind, phase III trial | Intervention: Durvalumab + tremelimumab + platinum-based chemotherapy Durvalumab 1,500 mg q.3.w. (4 cycles) followed by 1,500 mg q.4.w. (until progression) + tremelimumab 75 mg q.3.w. (4 cycles) followed by 1 dose of 75 mg at week 16 + SOC platinum-based chemotherapy (refer to comparator) (4 cycles only) Comparator: Platinum-based chemotherapy Investigator’s choice of: Gemcitabine 1,000 or 1,250 mg/m2 + carboplatin AUC 5 or 6 or cisplatin 75 mg/m2 q.3.w. (4 to 6 cycles) (squamous histology only) or Pemetrexed 500 mg/m2 q.3.w. (4 to 6 cycles) followed by pemetrexed 500 mg/m2 q.4.w. or q.3.w.a (until progression) + carboplatin AUC 5 or 6 or cisplatin 75 mg/m2 q.3.w. (4 to 6 cycles) (nonsquamous histology only) or Nab-paclitaxel 100 mg/m2 on days 1, 8, and 16 of each 21-day cycle + carboplatin AUC 5 or 6 q.3.w. (4 to 6 cycles) (nonsquamous or squamous histology) | Adult patients with metastatic (stage IV) NSCLC without activating EGFR mutations or ALK fusions; no prior chemotherapy or any other systemic therapy for metastatic disease; WHO or ECOG performance status of 0 or 1 | ITT (nonsquamous and squamous) | OS (dual primary end point): the time from the date of randomization until death from any cause regardless of whether the subject withdraws from randomized therapy or receives another anticancer therapy PFS (dual primary end point): the time from randomization until the date of objective disease progression or death (by any cause in the absence of progression) regardless of whether the subject withdraws from randomized therapy or receives another anticancer therapy before progression. (based on BICR assessment; RECIST 1.1) |
KEYNOTE-189 Gandhi et al. (2018)78 Randomized, controlled, double-blind, phase III trial | Intervention: Pembrolizumab + platinum-based chemotherapy Pembrolizumab 200 mg q.3.w. (up to 35 cycles) + investigator’s choice of cisplatin 75 mg/m2 q.3.w. or carboplatin AUC 5 mg/mL/min q.3.w. + pemetrexed 500 mg/m2 q.3.w. (4 cycles) followed by pemetrexed 500 mg/m2 q.3.w. (until progression) Comparator: Platinum-based chemotherapy Placebo q.3.w. (up to 35 cycles) + investigator’s choice of cisplatin 75 mg/m2 q.3.w. or carboplatin 5 mg/mL/min q.3.w. + pemetrexed 500 mg q.3.w. (4 cycles) followed by pemetrexed q.3.w. | Adult patients with metastatic nonsquamous NSCLC without sensitizing EGFR or ALK mutations; received no previous systemic therapy for metastatic disease; ECOG performance status of 0 or 1 | Nonsquamous only | OS (primary end point): the time from randomization to death from any cause PFS (primary end point): the time from randomization to disease progression, as assessed using BICR, or death from any cause, whichever occurred first |
KEYNOTE-407 Paz-Ares et al. (2018)79 Randomized, controlled, double-blind, phase III trial | Intervention: Pembrolizumab + platinum-based chemotherapy Pembrolizumab 200 mg q.3.w. (up to 35 cycles) + carboplatin AUC 6 mg/mL/min q.3.w. + either paclitaxel 200 mg/m2 on day 1 or nab-paclitaxel 100 mg/m2 on days 1, 8, or 15 of each 21-day cycle (4 cycles) Comparator: Platinum-based chemotherapy Placebo q.3.w. (up to 35 cycles) + carboplatin AUC 6 mg/mL/min q.3.w. + either paclitaxel 200 mg/m2 on day 1 or nab-paclitaxel 100 mg/m2 on days 1, 8, or 15 of each 21-day cycle (4 cycles) | Adult patients with metastatic (stage IV) squamous NSCLC; received no previous systemic therapy for metastatic disease; ECOG performance status 0 or 1 | Squamous only | OS (dual primary end point): the time from randomization to death from any cause PFS (dual primary end point): the time from randomization to the first documented disease progression per RECIST 1.1 based on BICR or death from any cause, whichever occurs first |
CheckMate 9LA Paz-Ares et al. (2021)80 Randomized, controlled, open-label, phase III trial | Intervention: Nivolumab + ipilimumab + platinum-based chemotherapy Nivolumab 360 mg q.3.w. + ipilimumab 1 mg/kg q.6.w. (until progression and maximum 2 years) + carboplatin AUC 6 mg/mL/min q.3.w. + paclitaxel 200 mg/m2 q.3.w. (2 cycles) (squamous histology) or + cisplatin 75 mg/m2 q.3.w. or carboplatin AUC 5 or 6 q.3.w. + pemetrexed 500 mg/m2 q.3.w. (2 cycles) (nonsquamous histology) Comparator: Platinum-based chemotherapy Carboplatin AUC 6 mg/mL/min q.3.w. + paclitaxel 200 mg/m2 q.3.w. (4 cycles) (squamous histology) or + cisplatin 75 mg/m2 q.3.w. or carboplatin AUC 5 or 6 q.3.w. + pemetrexed 500 mg/m2 q.3.w. (4 cycles) followed by optional pemetrexed 500 mg/m2 q.3.w. (until progression) (nonsquamous histology) | Adult patients with metastatic (stage IV) or recurrent squamous or nonsquamous NSCLC; received no previous systemic therapy for advanced or metastatic disease; ECOG performance status 0 or 1 | ITT (nonsquamous + squamous) | OS (primary end point): the time from randomization to death from any cause. PFS (secondary end point): the time from randomization to the date of the first documented tumour progression, or death from any cause, whichever occurred first. (based on BICR assessment; RECIST 1.1) |
EMPOWER-Lung 3 Gogishvili et al. (2022)81 Two-part, randomized, controlled, double-blind, phase III trial | Intervention: Cemiplimab + platinum-based chemotherapy Cemiplimab 350 mg q.3.w. (until progression) + SOC platinum-based chemotherapy (refer to comparator) (4 cycles only) Comparator: Platinum-based Investigator’s choice of: Paclitaxel 200 mg/m2 + carboplatin AUC 5 or 6 q.3.w. (4 cycles) or Paclitaxel 200 mg/m2 + cisplatin 75 mg/m2 q.3.w. (4 cycles) or Pemetrexed 500 mg/m2 + carboplatin AUC 5 or 6 q.3.w. (4 cycles) followed by pemetrexed 500 mg/m2 maintenance or Pemetrexed 500 mg/m2 + cisplatin 75mg/m2 q.3.w. (4 cycles) followed by pemetrexed 500 mg/m2 maintenance | Adult patients with locally advanced or metastatic NSCLC without activating EGFR mutations or ALK fusions | ITT (nonsquamous + squamous) | OS (primary end point): the time from randomization to the date of death PFS (secondary end point): defined as the time from randomization to the date of the first documented tumour progression or death from any cause |
AUC = area under the curve; BICR = blinded independent central review; ECOG = Eastern Cooperative Oncology Group; ITT = intention to treat; NSCLC = non–small cell lung cancer; OS = overall survival; PFS = progression-free survival; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1; q.3.w. = every 3 weeks; q.4.w. = every 4 weeks; q.6.w. = every 6 weeks; SOC = standard of care.
Source: Global POSEIDON MAIC Report.71,1
Comparison of Study Characteristics and Selection of Treatment-Effect Modifiers
Based on the identification of possible TEMs in the POSEIDON trial (race, smoking status and histology [where applicable]), and imbalances in these TEMs between the POSEIDON and comparator trials (CheckMate 9LA, KEYNOTE-407, KEYNOTE-189, and EMPOWER-Lung 3), the use of MAICs was considered justified and appropriate.
ITCs that do not adjust for the differences between trials in these TEMs would result in biased estimates of the relative efficacy of durvalumab and tremelimumab plus chemotherapy versus pemetrexed plus chemotherapy; durvalumab and tremelimumab plus chemotherapy versus nivolumab and ipilimumab plus chemotherapy, and durvalumab and tremelimumab plus chemotherapy versus cemiplimab plus chemotherapy. Given the impact of these TEMs on the treatment effect in POSEIDON (e.g., greater treatment effect in the non-Asian versus Asian subgroup) and the imbalances between trials in these TEMs (e.g., considerably lower proportion of patients from Asia enrolled in KEYNOTE-189 versus POSEIDON), this bias would appear to be in favour of the ICI comparator (i.e., pemetrexed plus chemotherapy, nivolumab and ipilimumab plus chemotherapy, or cemiplimab plus chemotherapy).
Table 32: Assessment of Homogeneity and Description of TEMs Considered
Characteristics | Description and handling of potential effect modifiers |
|---|---|
Trial eligibility | The main trial eligibility criteria differ between the POSEIDON (mixed histology) and KEYNOTE-189 (nonsquamous only) and KEYNOTE-407 (squamous only) trials with respect to tumour histology, but are otherwise consistent across studies. The POSEIDON trial only allowed patients with metastatic cancer, while the EMPOWER-Lung 3 trial included patients with locally advanced or metastatic NSCLC. |
End points | The definitions for OS and PFS are consistent across all trials. For PFS, BICR was used for the assessment of radiological progression (according to RECIST 1.1) across all trials |
Platinum-based chemotherapy | For patients with nonsquamous histology, pemetrexed in combination with platinum-based chemotherapy was used across all studies, with pemetrexed maintenance therapy permitted in the control arms of all trials. In the control arm of POSEIDON, 214 patients had nonsquamous histology and 204 patients received pemetrexed plus cisplatin or carboplatin.1,38 In the CheckMate 9LA and KEYNOTE-189 trials, pemetrexed in combination with platinum-based chemotherapy was the only available therapy for patients with nonsquamous histology.78,80 However, for patients with squamous histology, gemcitabine plus carboplatin or cisplatin was permitted in the POSEIDON trial but was not included in any of the comparator trials. Paclitaxel or nab-paclitaxel plus carboplatin was instead included as the only chemotherapy regimens for patients with squamous histology in the comparator trials. In the control arm of POSEIDON, 122 patients had squamous histology and 112 patients received gemcitabine plus cisplatin or carboplatin, whereas only 17 patients received nab-paclitaxel plus carboplatin.38 For the purpose of conducting anchored comparisons that include patients with squamous histology, it was assumed that the platinum-based chemotherapy regimens included in the different studies can be treated as common comparators. There is evidence from clinical trials to suggest that OS may be similar between regimens such as gemcitabine plus cisplatin or carboplatin and nab-paclitaxel or paclitaxel plus carboplatin; however the equivalence of these chemotherapy regimens was not assessed here via a systematic literature review or meta-analysis.22,82 It is important to note, however, that these trials did not include these chemotherapy regimens in combination with immuno-oncology, but as platinum-based chemotherapy alone. Planned chemotherapy was not identified as a TEM based on the results of the interaction tests. Given that the chemotherapy options used in the POSEIDON trial were dependent on histology to a large extent (e.g., pemetrexed doublet only for patients with nonsquamous histology and gemcitabine doublet only for patients with squamous histology), weighting based on histology in the POSEIDON ITT MAICs will also affect the distribution of chemotherapy drugs received. However, the MAICs do not account for potential differences in the chemotherapy drugs received within each histology subgroup. Gemcitabine plus cisplatin or carboplatin was highly utilized in the POSEIDON trial as an option for patients with squamous histology, but was not an option in the KEYNOTE-407, CheckMate 9LA, and EMPOWER-Lung 3 trials (nab-paclitaxel or paclitaxel plus carboplatin only). In POSEIDON, patients receiving gemcitabine plus cisplatin or carboplatin appeared to have lower treatment benefit with durvalumab and tremelimumab plus chemotherapy vs. chemotherapy alone compared with those receiving nab-paclitaxel plus carboplatin, but the results of these subgroup analyses should be interpreted with caution given the small sample sizes. Restricting the ITC to only those patients with squamous histology treated with nab-paclitaxel plus carboplatin (to align with the chemotherapy regimens available in the MAIC comparator trials) was not considered possible because of the prohibitively small number of patients receiving nab-paclitaxel plus carboplatin in the POSEIDON trial (24 patients in the durvalumab and tremelimumab plus chemotherapy arm and 17 patients in the chemotherapy alone arm).70 The MAICs (including patients with squamous histology) should also be interpreted with caution because the possible impact that the use of different chemotherapy regimens (for squamous histology) between trials might have on treatment effects is not accounted for in the ITCs. |
Race (Asian vs. non-Asian) | ITT population: The proportion of Asian patients in the POSEIDON trial (33.6%) was considerably higher than the proportion of patients enrolled as Asian in the CheckMate 9LA trial (8.1%) and the EMPOWER-Lung 3 trial (12.4%). Nonsquamous population: The proportion of Asian patients in the POSEIDON trial (36.7%) was considerably higher than the proportion of Asian patients in the KEYNOTE-189 trial (1.6%), the CheckMate 9LA trial (8.1%), and the EMPOWER-Lung 3 trial (12.4%). Squamous population: The proportion of Asian patients in the POSEIDON trial (28.0%) was higher than the proportion of Asian patients in the KEYNOTE-407 trial (19.0%), the CheckMate 9LA trial (8.1%), and the EMPOWER-Lung 3 trial (12.4%). However, the difference vs. the KEYNOTE-407 trial was less pronounced when compared to that seen vs. the KEYNOTE-189 trial nonsquamous subgroup. The proportion of patients in the POSEIDON trial described as Asian (28.0%) was higher than the proportion of Asian patients in the EMPOWER-Lung 3 trial (12.4%). |
Smoking status (current vs. former smoker) | ITT population: The proportion of patients in the POSEIDON trial who were “current/former smokers” (79.4%) was lower than in the CheckMate 9LA trial (86.4%) and the EMPOWER-Lung 3 trial (85.6%). Nonsquamous population: The proportion of patients in the POSEIDON trial who were “current/former smokers” (74.8%) was lower than in the KEYNOTE-189 trial (88.1%), the CheckMate 9LA trial (86.4%), and the EMPOWER-Lung 3 trial (85.6%). Squamous population: The proportion of patients in the POSEIDON trial who were “current/former smokers” (87.8%) was lower than in the KEYNOTE-407 trial (92.7%) but was similar to those in the CheckMate 9LA trial (86.4%) and the EMPOWER-Lung 3 trial (85.6%). |
Histology (nonsquamous vs. squamous) | ITT population: The proportion of patients in the POSEIDON trial with nonsquamous histology (63.4%) was lower than in the CheckMate 9LA trial (68.8%) and higher than in the EMPOWER-Lung 3 trial (57.1%) For the comparisons vs. pembrolizumab plus chemotherapy (and vs. nivolumab and ipilimumab plus chemotherapy, and vs. cemiplimab plus chemotherapy) in the nonsquamous and squamous histology subgroups, it was assumed that the TEMs identified in the POSEIDON ITT population would also be applicable in each of the histology subgroups. Given the preponderance of patients with squamous histology known to be current or former smokers (87.8% in POSEIDON squamous subgroup and 92.7% in the KEYNOTE-407 trial), the impact of smoking status as a TEM in the squamous subgroup MAICs may be limited, regardless of the magnitude of effect modification in this subgroup. |
Other baseline characteristics | Other baseline characteristics were generally well balanced across trials.
Sex, CNS or brain metastases, and ECOG PS at baseline were not identified as significant TEMs based on these analyses for the POSEIDON trial. |
BICR = blinded independent central review; CNS = central nervous system; ECOG PS = Eastern Cooperative Oncology Group Performance Status; ITC = indirect treatment comparison; ITT = intention to treat; MAIC = matching adjusted indirect comparison; NSCLC = non–small cell lung cancer; OS = overall survival; PFS = progression-free survival; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1; TEM = treatment-effect modifier; vs. = versus.
Source: Global POSEIDON MAIC Report.71,1
Efficacy
Durvalumab and Tremelimumab Plus Platinum-Based Chemotherapy Versus Pembrolizumab Plus Platinum-Based Chemotherapy
Squamous Plus Nonsquamous Population
There was no ITC for this mixed squamous plus nonsquamous population in this comparison.
Nonsquamous Population
Data from the POSEIDON trial versus the KEYNOTE-189 trial was used for this MAIC. The HRs for durvalumab and tremelimumab plus chemotherapy versus chemotherapy alone before and after weighting to the KEYNOTE-189 trial were used in Bucher indirect comparisons versus pembrolizumab plus chemotherapy, with chemotherapy as a common comparator in both trials. The HRs for durvalumab and tremelimumab plus chemotherapy versus pembrolizumab plus chemotherapy before and after weighting are presented in Table 33, using the data identified in the SLR from the KEYNOTE-189 trial (DCO of August 28, 2020).83
After weighting the Bucher ITC, the OS HR for durvalumab and tremelimumab plus chemotherapy versus pembrolizumab plus chemotherapy appeared numerically in favour of durvalumab and tremelimumab plus chemotherapy (██ ███ ██ ████ ████ ████ ██████ ████) (Table 33). But for PFS, the HR appeared numerically in favour of pembrolizumab plus chemotherapy (██ ███ ██ ████ ████ ████ ██████ ████) (Table 33). Similar results were also seen in the updated MAIC using the more recent OS data for the POSEIDON trial (5-year follow-up; August 24, 2023) and the KEYNOTE-189 trial (5-year follow-up; March 8, 2022).62,68
Because the 95% Cls for the estimated HRs include 1, the differences in outcomes were not statistically significant.
Squamous Population
Data from the POSEIDON trial versus the KEYNOTE-407 trial was used for this MAIC. The HRs for durvalumab and tremelimumab plus chemotherapy versus chemotherapy alone before and after weighting to the KEYNOTE-407 trial were used in Bucher indirect comparisons versus pembrolizumab plus chemotherapy, with chemotherapy as a common comparator in both trials. The HRs for durvalumab and tremelimumab plus chemotherapy versus pembrolizumab plus chemotherapy before and after weighting are presented in Table 33, using the data identified in the SLR from the KEYNOTE-407 trial (DCO of September 30, 2020).84
After weighting, the Bucher indirect comparison of durvalumab and tremelimumab plus chemotherapy versus pembrolizumab plus chemotherapy for both OS and PFS HRs appeared to numerically favour pembrolizumab plus chemotherapy (██ ██), with an OS HR of ████ ██████ ██████ and a PFS HR of ████ ██████ █████ (Table 33). Similar results were also seen in the updated MAIC using the more recent OS data for the POSEIDON trial (5-year follow-up; August 24, 2023) and the KEYNOTE-407 trial (5-year follow-up; February 23, 2022).62,67
Because the 95% Cl for the estimated HRs include 1, the differences in outcomes were not statistically significant.
Table 33: Hazard Ratios for Durvalumab and Tremelimumab Plus Chemotherapy vs. Pembrolizumab Plus Chemotherapy From Bucher ITCs Before and After Weighting
Hazard ratio (95% CI) | Before weighting D + T + chemotherapy vs. P + chemotherapy (POSEIDON nonsquamous subgroup, N = 428) | After weighting D + T + chemotherapy vs. P + chemotherapy (POSEIDON ESS = 256.71) |
|---|---|---|
Nonsquamous population | ||
OS (original analysis) | ████ ██████ █████ | ████ ██████ █████ |
OS (more recent OS data) | ████ ██████ █████ | ████ ██████ █████ |
PFS | ████ ██████ █████ | ████ ██████ █████ |
Squamous population | ||
OS (original analysis) | ████ ██████ █████ | ████ ██████ █████ |
OS (more recent OS data) | ████ ██████ █████ | ████ ██████ █████ |
PFS | ████ ██████ █████ | ████ ██████ █████ |
CI = confidence interval; D = durvalumab; ESS = effective sample size; ITC = indirect treatment comparison; OS = overall survival; P = pembrolizumab; PFS = progression-free survival; t = tremelimumab; vs. = versus.
Source: Global POSEIDON MAIC Report.71,1
Durvalumab and Tremelimumab Plus Platinum-Based Chemotherapy Versus Nivolumab and Ipilimumab Plus Platinum-Based Chemotherapy
Squamous and Nonsquamous Population (ITT Population)
Data from the POSEIDON trial versus the CheckMate 9LA trial was used for this MAIC. The HRs for durvalumab and tremelimumab plus chemotherapy versus chemotherapy alone in the POSEIDON trial both before and after weighting to the CheckMate 9LA trial were used in Bucher indirect comparisons versus nivolumab and ipilimumab plus chemotherapy, with chemotherapy as a common comparator in both trials (POSEIDON and CheckMate 9LA). The HRs for durvalumab and tremelimumab plus chemotherapy versus nivolumab and ipilimumab plus chemotherapy before and after weighting are presented in Table 34, using data from the CheckMate 9LA trial identified in the SLR (DCO of February 18, 2021).85 Similar results were also seen in the updated MAIC using the more recent OS data for the POSEIDON trial (5-year follow-up; August 24, 2023) and the CheckMate 9LA trial (4-year follow-up; February 13, 2023).62,69
After weighting (i.e., based on the TEMs of race, smoking status, and histology), the OS and PFS HRs for durvalumab and tremelimumab plus chemotherapy versus nivolumab and ipilimumab plus chemotherapy appeared to numerically favour durvalumab and tremelimumab plus chemotherapy (███ ██), with an OS HR ████ ██████ █████ and a PFS HR ████ ██████ █████ (Table 34).
For both OS and PFS, the 95% Cl for the estimated HRs include 1, so the differences in outcomes were not statistically significant.
Table 34: Hazard Ratios for Durvalumab and Tremelimumab Plus Chemotherapy vs. Nivolumab and Ipilimumab Plus Chemotherapy From Bucher ITCs Before and After Weighting
Hazard ratio (95% CI) | Before weighting D + T + chemotherapy vs. N + I + chemotherapy (POSEIDON ITT, N = 675) | After weighting D + T + chemotherapy vs. N + I + chemotherapy (POSEIDON ITT, ESS = 491.1) |
|---|---|---|
Squamous and nonsquamous population (ITT population) | ||
OS (original analysis) | ████ ██████ █████ | ████ ██████ █████ |
OS (more recent OS data) | ████ ██████ █████ | ████ ██████ █████ |
PFS | ████ ██████ █████ | ████ ██████ █████ |
Nonsquamous population | ||
OS (original analysis) | ████ ██████ █████ | ████ ██████ █████ |
OS (more recent OS data) | ████ ██████ █████ | ████ ██████ █████ |
PFS | ████ ██████ █████ | ████ ██████ █████ |
Squamous population | ||
OS (original analysis) | ████ ██████ █████ | ████ ██████ █████ |
OS (more recent OS data) | ████ ██████ █████ | ████ ██████ █████ |
PFS | ████ ██████ █████ | ███████████████ |
CI = confidence interval; D = durvalumab; ESS = effective sample size; I = ipilimumab; ITC = indirect treatment comparison; ITT = intentional to treat; N = nivolumab; OS = overall survival; PFS = progression-free survival; t = tremelimumab; vs. = versus.
Source: Global POSEIDON MAIC Report.71,1
Nonsquamous Population
Data from the POSEIDON trial versus the CheckMate 9LA trial was used for this MAIC. The HRs for durvalumab and tremelimumab plus chemotherapy versus chemotherapy alone before and after weighting to the CheckMate 9LA trial were used in Bucher indirect comparisons versus nivolumab and ipilimumab plus chemotherapy, with chemotherapy as a common comparator in both trials. The HRs for durvalumab and tremelimumab plus chemotherapy versus nivolumab and ipilimumab plus chemotherapy before and after weighting are presented in Table 34, using data identified in the SLR from the CheckMate 9LA trial (DCO of February 18, 2021).85
After weighting, the HR was ████ ██████ █████ for OS, and ████ ██████ █████ for PFS, and both were numerically in favour of durvalumab and tremelimumab plus chemotherapy versus nivolumab and ipilimumab plus chemotherapy (Table 34). Similar results were also seen in the updated MAIC using the more recent OS data for the POSEIDON trial (5-year follow-up; August 24, 2023) and the CheckMate 9LA trial (4-year follow-up; February 13, 2023).62,69
Because the 95% Cl for the estimated HRs include 1 for both OS and PFS, the differences were not statistically significant.
Squamous Population
Data from the POSEIDON trial versus the CheckMate 9LA trial were used for this MAIC. The HRs for durvalumab and tremelimumab plus chemotherapy versus chemotherapy alone before and after weighting to the CheckMate 9LA trial were used in Bucher indirect comparisons versus nivolumab and ipilimumab plus chemotherapy, with chemotherapy as a common comparator in both trials. The HRs for durvalumab and tremelimumab plus chemotherapy versus nivolumab and ipilimumab plus chemotherapy before and after weighting are presented in Table 34, using data identified in the SLR from the CheckMate 9LA trial (DCO: February 18, 2021).85
After weighting, the HR was ████ ██████ █████ for OS and ████ ██████ █████ for PFS, and both were numerically in favour of nivolumab and ipilimumab plus chemotherapy (██ ██) (Table 34). Similar results were also seen in the updated MAIC using the more recent OS data for the POSEIDON trial (5-year follow-up; August 24, 2023) and the CheckMate 9LA trial (4-year follow-up; February 13, 2023).62,69
Because the 95% Cl for the estimated HRs include 1, the differences in outcomes were not statistically significant.
Durvalumab and Tremelimumab Plus Platinum-Based Chemotherapy Versus Cemiplimab Plus Platinum-Based Chemotherapy
Squamous and Nonsquamous Population (ITT Population)
Data from the POSEIDON trial versus the EMPOWER-Lung 3 trial were used for this MAIC. The HRs for durvalumab and tremelimumab plus chemotherapy versus chemotherapy alone in the POSEIDON trial both before and after weighting to EMPOWER-Lung 3 were used in Bucher indirect comparisons versus cemiplimab plus chemotherapy, with chemotherapy as a common comparator in both trials. The HRs for durvalumab and tremelimumab plus chemotherapy versus cemiplimab plus chemotherapy before and after weighting are listed in Table 35, using data from the EMPOWER-Lung 3 trial identified in the SLR.86
After weighting, the OS HR was ████ ██████ ██████ which demonstrated similar survival between durvalumab and tremelimumab plus chemotherapy and cemiplimab plus chemotherapy. The PFS HR was ████ ██████ █████, which appeared to numerically favour cemiplimab plus chemotherapy (Table 35).
Because the 95% Cl for the estimated HRs for OS and PFS include 1, the differences in outcomes were not statistically significant.
Table 35: Hazard Ratios for Durvalumab and Tremelimumab Plus Chemotherapy vs. Cemiplimab Plus Chemotherapy From Bucher ITCs Before and After Weighting
Hazard ratios (95% CI) | Before weighting D + T + chemotherapy vs. C + chemotherapy (POSEIDON ITT, N = 675) | After weighting D + T + chemotherapy vs. C + chemotherapy (POSEIDON ITT, ESS = 546.5) |
|---|---|---|
Squamous and nonsquamous population (ITT population) | ||
OS | ████ ██████ █████ | ████ ██████ █████ |
PFS | ████ ██████ █████ | ████ ██████ █████ |
Nonsquamous population | ||
OS | ████ ██████ █████ | ████ ██████ █████ |
PFS | ████ ██████ █████ | ████ ██████ █████ |
Squamous population | ||
OS | ████ ██████ █████ | ████ ██████ █████ |
PFS | ████ ██████ █████ | ████ ██████ █████ |
C = cemiplimab; CI = confidence interval; D = durvalumab; ESS = effective sample size; ITC = indirect treatment comparison; ITT = intention to treat; N = nivolumab; OS = overall survival; PFS = progression-free survival; t = tremelimumab; vs. = versus.
Source: Global POSEIDON MAIC Report.71,1
Nonsquamous Population
Data from the POSEIDON trial versus the EMPOWER-Lung 3 trial were used for this MAIC. The HRs for durvalumab and tremelimumab plus chemotherapy versus cemiplimab plus chemotherapy before and after weighting are presented in Table 35.
After weighting, the OS HR for durvalumab and tremelimumab plus chemotherapy versus cemiplimab plus chemotherapy appeared to numerically favour durvalumab and tremelimumab plus chemotherapy (█████ ██ ████ ████ ████ ██████ ███████ For PFS, the HR appeared to numerically favour of cemiplimab plus chemotherapy (█████ ██ ████ ████ ████ ██████ ██████ (Table 35).
Because the 95% Cl for the estimated HRs include 1, the differences in outcomes were nominally not statistically significant.
Squamous Population
Data from the POSEIDON trial versus the EMPOWER-Lung 3 trial were used for this MAIC. The HRs for durvalumab and tremelimumab plus chemotherapy versus cemiplimab plus chemotherapy before and after weighting are presented in Table 35.
After weighting, both the OS and PFS HRs for durvalumab and tremelimumab plus chemotherapy versus cemiplimab plus chemotherapy appeared to numerically favour of cemiplimab plus chemotherapy (████), with an OS HR of ████ ██████ █████, and PFS HR of ████ ██████ █████ (Table 35).
Because the 95% Cl for the estimated HRs include 1, the differences in outcomes were nominally not statistically significant.
Harms
Harms outcomes were not assessed in the sponsor-submitted ITCs.
In summary, the results of the MAICs demonstrated a largely similar efficacy for durvalumab and tremelimumab plus chemotherapy versus other ICI combination regimens in the treatment of the patients with metastatic NSCLC whose tumours are lacking sensitizing EGFR mutations or ALK rearrangements.
Critical Appraisal of the Indirect Treatment Comparison
Overall, the MAICs were conducted according to accepted methodological guidance. The search for potentially eligible studies was reasonably comprehensive and the methods used to select relevant studies, extract data, and appraise risk of bias of included studies were adequate.
One of the key limitations associated with the ITC was the heterogeneity across the included studies. The sponsor identified potential effect modifiers, including race (non-Asian versus Asian), smoking status (current and former versus never) and histology (nonsquamous versus squamous) based on exploratory analyses of the POSEIDON trial. To account for imbalances at baseline in the 3 effect modifiers, MAICs were conducted in which the POSEIDON population was weighted to more closely match the relevant comparator trials. According to the clinical experts consulted for this review, the important effect modifiers would also include PD-L1 status, sex, age, ECOG PS, presence of brain metastases, and disease stage. The presence of KRAS, STK11, and KEAP mutations and use of gemcitabine-containing chemotherapy regimens (for patients with squamous NSCLC) may also be predictive effect modifiers. However, the sponsor-submitted ITC did not fully report on these potential effect modifiers. Further, after weighting, it is unknown if more imbalances were introduced in other potential effect modifiers, such as ECOG PS, disease stage, and sex.
Given the limited or lack of reporting on the potential EMs a comprehensive assessment of their impact on the reported findings was not possible. While this limitation is not unique to the current MAIC, it remains critical.
Anchor-based approaches assumed all the chemotherapies have identical treatment effects. Although previous studies reported that it was likely this assumption may hold, it is uncertain to what extent it would be applicable in this ITC, which included diverse populations.
Furthermore, reductions in ESS after adjustment (from 6% to 40% reduction) were observed in various MAIC analyses. The reduced ESS contributed to imprecision and suggested a greater influence of subsets of patients in the comparator trial. The 95% CIs for the comparative effect estimates (HRs) were wide, further indicating a lack of precision in the comparative efficacy results.
HRQoL was identified by patient groups as an important outcome. However, no indirect comparison was conducted for HRQoL and safety outcomes in the sponsor-submitted ITC.
Summary
As a first-line treatment for adult patients with metastatic NSCLC without EGFR mutations or ALK aberrations, the findings of the MAICs showed that, when compared with pembrolizumab plus chemotherapy, the combination of durvalumab and tremelimumab plus chemotherapy demonstrated similar effects in terms of OS and PFS in the nonsquamous and squamous NSCLC populations. When compared with nivolumab and ipilimumab plus chemotherapy, the combination of durvalumab and tremelimumab plus chemotherapy demonstrated similar effects in terms of OS and PFS in the squamous plus nonsquamous (ITT) population, nonsquamous NSCLC population, and squamous NSCLC population. When compared with cemiplimab plus chemotherapy, the combination of durvalumab and tremelimumab plus chemotherapy demonstrated a similar effect in OS and PFS in the nonsquamous plus squamous population, nonsquamous population, and squamous NSCLC population.
The clinical experts consulted for this review indicated that the underperformance of durvalumab and tremelimumab plus chemotherapy in the squamous population in all MAICs could be related at least in part to the use of gemcitabine platinum doublet therapy in the durvalumab and tremelimumab plus chemotherapy regimens in the POSEIDON trial. One clinical expert indicated that, based on a theoretical difference in efficacy that has not been consistently demonstrated in clinical trials, some clinicians prefer carboplatin-paclitaxel over platinum-gemcitabine in conjunction with immunotherapy for squamous NSCLC.
In all MAICs, the 95% Cl of the estimated HRs include 1 for both OS and PFS. The efficacy, in terms of OS and PFS, therefore appeared comparable when comparing durvalumab and tremelimumab plus chemotherapy with pembrolizumab plus chemotherapy, nivolumab and ipilimumab plus chemotherapy, and cemiplimab plus chemotherapy for the first-line treatment of patients with metastatic NSCLC with no sensitizing EGFR mutations or ALK genomic tumour aberrations.
Interpretation of these findings should consider the potential limitations associated with MAICs, such as the failure to use all potential effect modifiers in the adjustment and the impact of the reductions in ESS after adjustment. However, the clinical experts CDA-AMC consulted for this review indicated that the findings of the MAICs are largely aligned with what is expected in clinical practice in Canadian settings.
Studies Addressing Gaps in the Systematic Review Evidence
No studies addressing gaps in the systematic review evidence were submitted by the sponsor.
Discussion
Summary of Available Evidence
The POSEIDON trial was a phase III, randomized, open-label, global study investigating the efficacy and safety of durvalumab with or without tremelimumab for the first-line treatment in patients (N = 1,013) with metastatic NSCLC and tumours that lack activating EGFR mutations and ALK fusions. There were no study sites in Canada. Patients were randomized 1:1:1 to 1 of 3 study arms: durvalumab and tremelimumab plus SOC chemotherapy; durvalumab plus SOC chemotherapy; or SOC chemotherapy alone. The primary objective of the POSEIDON trial was to assess the efficacy of durvalumab plus SOC chemotherapy compared to SOC chemotherapy alone in terms of dual primary end points of PFS and OS. The key secondary objectives were to assess the efficacy of durvalumab and tremelimumab plus SOC chemotherapy compared to SOC chemotherapy alone in terms of PFS and OS. Other secondary end points included ORR, BoR, DoR, and HRQoL for the comparison of durvalumab and tremelimumab plus SOC chemotherapy versus SOC chemotherapy alone. Harms outcomes were also assessed. Results from the comparison of durvalumab and tremelimumab plus SOC chemotherapy versus SOC chemotherapy alone are presented in this review and include results from the final analysis and from an updated 5-year OS analysis.
Patients eligible for participation in the POSEIDON trial were 18 years of age or older with stage IV NSCLC not amenable to curative surgery or radiation with tumours that lacked activating EGFR mutations and ALK fusions. Across all study arms, the median age of patients was 64 years and the majority of patients were male (76.0%), white (55.9%), either current or former smokers (78%), and had an ECOG PS of 1 (66.5%). The included patients had received no prior chemotherapy or other systemic therapy for metastatic NSCLC, including prior exposure to immune-mediated therapy. Participants were required to have a WHO or ECOG PS of 0 or 1. Patients with active or prior documented autoimmune or inflammatory disorders (with some exceptions) were not eligible for the trial.
In the absence of direct evidence from randomized trials between durvalumab and tremelimumab plus SOC chemotherapy and relevant comparators, the sponsor conducted MAICs to assess the comparative OS and PFS efficacy of durvalumab and tremelimumab plus SOC chemotherapy versus other approved ICIs plus platinum-based chemotherapy combination regimens in the patient population for the indication.
Interpretation of Results
Efficacy
In the comparison of durvalumab and tremelimumab plus SOC chemotherapy versus SOC chemotherapy alone, the POSEIDON trial demonstrated improvement in median OS at both the final analysis DCO (March 12, 2021) and 5-year OS analysis DCO (August 24, 2023) and improvement in median PFS at the final analysis DCO (July 24, 2019). The PFS rate at 12 months also demonstrated improvement for durvalumab and tremelimumab plus SOC chemotherapy compared to SOC chemotherapy alone. The clinical experts consulted for this review described these results as clinically meaningful. The experts also noted that the end points of median OS, median PFS, and PFS at 12 months and their HRs do not fully capture clinical meaningfulness. They emphasized that OS and PFS results at key landmark time points (e.g., 2, 3, 4, and 5 years) are important indicators of long-term benefits in clinical trials of immuno-oncology treatments. The GRADE assessment suggested moderate certainty in the median OS and median PFS results (both rated down because of imprecision). The differences in the median OS (at the final analysis) and the median PFS between groups were statistically significant (statistical significance could not be determined for other end points as these were not part of the hierarchical statistical analysis plan). Separation of the Kaplan-Meier OS curves for durvalumab and tremelimumab plus SOC chemotherapy versus SOC chemotherapy alone occurred at approximately 3 months, with separation becoming more pronounced between 9 and 12 months and maintained through 60 months. Results for survival probability at 36 months, 48 months, and 60 months demonstrated improvement for durvalumab and tremelimumab plus SOC chemotherapy versus SOC chemotherapy alone, which the clinical experts identified as clinically meaningful and for which the GRADE assessment suggested high certainty in the results. Separation of Kaplan-Meier PFS curves for durvalumab and tremelimumab plus SOC chemotherapy and SOC chemotherapy alone occurred between 3 and 6 months, with separation maintained through 12 months. The GRADE assessment suggested high certainty in the results for PFS at 12 months. Results for the probability of PFS beyond this time point were not provided in the sponsor’s submission. Results for the secondary end point of ORR (using unconfirmed responses) also demonstrated improvement for durvalumab and tremelimumab plus SOC chemotherapy versus SOC chemotherapy alone. This difference was considered clinically significant by the clinical experts and the GRADE assessment suggested moderate certainty in these results. The sponsor indicated that there are no reported MID values for OS, PFS, and ORR in the first-line treatment of metastatic NSCLC; as such, the thresholds used to judge the target of certainty in the GRADE assessment is based on input from clinical experts consulted by CDA-AMC.
Results of sensitivity analyses conducted for OS and PFS were generally consistent with the primary analyses. Analysis of PFS to assess potential attrition bias showed that some censoring occurred earlier in the SOC chemotherapy alone arm in comparison to the durvalumab and tremelimumab plus SOC chemotherapy arm.
Differential treatment exposure to SOC chemotherapy between groups may have biased the overall efficacy results in favour of durvalumab and tremelimumab plus SOC chemotherapy. Specifically, the treatment exposure to SOC chemotherapy alone was significantly lower in the SOC chemotherapy arm (164.9 patient-years, with a mean duration of 25.83 weeks) than in the durvalumab and tremelimumab plus SOC chemotherapy arm (222.9 patient-years, with a mean duration of 35.35 weeks) at the final analysis. This can most likely be attributed to a higher proportion of patients discontinuing SOC chemotherapy because their “condition under investigation worsened” in the SOC chemotherapy arm (55.3%) than in the durvalumab and tremelimumab plus SOC chemotherapy arm (41.7%). Greater exposure to SOC chemotherapy in the durvalumab and tremelimumab plus SOC chemotherapy arm would have biased the results in favour of this treatment regimen compared to SOC chemotherapy alone. Another consideration is that subsequent systemic anticancer treatment was received by a larger proportion of patients in the SOC chemotherapy alone arm (final analysis DCO: 60.2%; 5-year DCO: 60.8%) than in the durvalumab and tremelimumab plus SOC chemotherapy arm (40.8% and 42.9%). For example, subsequent immunotherapy treatment was received by 33.2% and 6.5% of patients in these treatment arms, respectively, at the final analysis. This would have biased OS against the durvalumab and tremelimumab plus SOC chemotherapy arm. Other intercurrent events also complicated the interpretation of the efficacy findings, including continuation of durvalumab treatment through progression and disproportionate treatment discontinuation because of AEs between study arms. Overall, at the final analysis DCO, 88.8% of patients in the durvalumab and tremelimumab plus SOC chemotherapy arm and 98.5% of patients in the SOC chemotherapy alone arm had discontinued treatment, with AEs leading to discontinuation of any study treatment occurring in 22.1% and 15.3% of patients, respectively, and AEs leading to dose delay or interruption of study treatment occurring in 57.3% and 42.9% of patients in the durvalumab and tremelimumab plus SOC chemotherapy and SOC chemotherapy arms, respectively (as described in the following Harms section). Overall, the magnitude of these effects on OS was difficult to determine given the various potential biases, and interpretation of the estimated between-group differences in OS should consider that the effects of biases may be counterbalanced, and in the context of substantially incomplete per-protocol treatments.
Factors related to the generalizability of the results of the POSEIDON trial were identified by the clinical experts consulted for this review. The POSEIDON trial did not include certain patients who would be considered for treatment with immuno-oncology therapy in clinical practice, including those with brain metastases (criteria for brain metastases in the trial were stricter than what would be used in practice), infections such as HIV or hepatitis C, autoimmune or inflammatory disorders, and potentially those with a history of cancer in the previous 5 years. The clinical experts also identified differences between the POSEIDON trial participants and the target population in clinical practice in Canada: patients in practice would typically be older and would include patients with an ECOG PS of 2 and those who had received immunotherapy for earlier-stage cancer. Female patients were underrepresented in the trial and a higher proportion of patients with liver and CNS metastases would be expected in practice. Clinical experts also pointed out that the ratio of patients with squamous-cell carcinomas to those with non–squamous cell carcinomas in the POSEIDON trial may be representative of areas of Canada with more prevalent tobacco use than those with less tobacco use. The POSEIDON trial did not include any sites in Canada. The clinical experts noted that, overall, they had no concerns about the generalizability of the study findings to patients seen in clinical practice based on study patient characteristics or locations. The experts acknowledged some differences between the chemotherapy regimens used in combination with durvalumab and tremelimumab in the POSEIDON trial and those used in clinical practice in Canada; paclitaxel plus carboplatin would be used in practice instead of the nab-paclitaxel regimen (the efficacy of these regimens is considered comparable), and the paclitaxel regimen would be expected to be administered to a greater proportion of patients with squamous histology in practice than was reported in the trial (most of these patients in the study received gemcitabine). The clinical experts also noted that subsequent anticancer therapies received by patients in the POSEIDON trial are not fully representative of clinical practice because patients who received first-line immunotherapy cannot be rechallenged with immunotherapy and a higher proportion of patients in practice would be expected to receive subsequent immunotherapy following first-line treatment with chemotherapy alone.
Prespecified subgroup analyses for OS at the final analysis and 5-year OS analysis showed that the survival benefit was consistent across subgroups, with the exception of the subgroup of never smokers, for whom durvalumab and tremelimumab plus SOC chemotherapy was not favoured. In addition, at both analysis DCO dates, a more pronounced OS benefit of durvalumab and tremelimumab plus SOC chemotherapy compared to SOC chemotherapy alone was observed in patients with nonsquamous histology. Post hoc exploratory subgroup analyses of OS in the KRAS and STK11 mutation subgroups performed in patients with nonsquamous histology showed that an OS benefit of durvalumab and tremelimumab plus SOC chemotherapy compared to SOC chemotherapy was observed in both subgroups. Prespecified subgroup analyses showed the PFS benefit of durvalumab and tremelimumab plus SOC chemotherapy versus SOC chemotherapy alone was observed across all prespecified subgroups, with a more pronounced improvement in PFS of durvalumab and tremelimumab plus SOC chemotherapy compared to SOC chemotherapy alone in patients with nonsquamous histology. Prespecified subgroup analyses for key secondary OS and PFS end points were conducted in a small number of patients, and no adjustments were made for multiple testing, precluding the ability to produce conclusive results. Similarly, conclusions cannot be drawn from analyses of the KRAS and STK11 subgroups because the trial was not powered for these evaluations, there were low numbers of patients and events, and because of the post hoc and exploratory nature of these analyses. The clinical experts emphasized that these subgroups would not be relevant to informing reimbursement criteria for durvalumab and tremelimumab; results for these subgroups are hypothesis-generating and may perhaps inform treatment choice. The experts noted that smoking status is a strong predictor of efficacy with immunotherapy-based regimens. One expert pointed out that underperformance in the squamous subgroup may be related in part to the use of gemcitabine chemotherapy in that population, noting that preclinical data suggest that gemcitabine attenuates the efficacy of immunotherapy. The potential differential effect in the aforementioned subgroups also requires further research.
An important limitation of the POSEIDON trial is that durvalumab and tremelimumab plus SOC chemotherapy was compared to SOC chemotherapy alone. At the time the trial was conducted, platinum-based chemotherapy was the standard of therapy for the indicated population. However, current first-line treatment of metastatic NSCLC without targetable genomic alterations consists of immuno-oncology treatment (on its own or in combination with chemotherapy). The sponsor submitted an ITC comparing durvalumab and tremelimumab plus chemotherapy to other available regimens of immuno-oncology plus chemotherapy for the indication under review. A deviation request was approved by CDA-AMC allowing for the exclusion of monotherapy with immuno-oncology drugs (i.e., those given without platinum-based chemotherapy) as comparators in both the pharmacoeconomic model and budget impact analysis. The clinical experts consulted for this review stated that immuno-oncology monotherapy could be considered a potential comparator for the combination of durvalumab and tremelimumab plus SOC chemotherapy. They also stated that platinum-based chemotherapy alone does not represent a reasonable comparator to durvalumab and tremelimumab plus platinum-based chemotherapy; immuno-oncology treatment is considered the backbone of therapy for first-line treatment of metastatic NSCLC, and platinum-based chemotherapy alone would typically only be used for patients who are not able to, or choose not to, receive immuno-oncology treatment. Because no data were submitted by the sponsor comparing durvalumab and tremelimumab plus platinum-based chemotherapy to immuno-oncology monotherapy, this represents a gap in the available evidence for this review.
The clinical experts consulted for this review indicated that the underperformance of durvalumab and tremelimumab plus SOC chemotherapy in the squamous population in all MAICs could be related at least in part to the use of gemcitabine platinum doublet therapy in the durvalumab and tremelimumab plus SOC chemotherapy regimen in the POSEIDON trial. In clinical practice, some clinicians prefer an immuno-oncology plus chemotherapy combination with carboplatin and paclitaxel as opposed to platinum and gemcitabine for those patients with squamous NSCLC.
The ITC evidence indicated no statistically significant differences between durvalumab and tremelimumab plus chemotherapy and their comparators. The efficacy, in terms of OS and PFS, therefore appeared likely comparable when comparing durvalumab and tremelimumab plus chemotherapy with pembrolizumab plus chemotherapy, nivolumab and ipilimumab plus chemotherapy, and cemiplimab plus chemotherapy for the first-line treatment of patients with metastatic NSCLC with no EGFR mutations or ALK genomic tumour aberrations. Because of the potential limitations associated to these MAICs, including the failure to use all potential effect modifiers in the adjustment, the impact of the reductions in ESS after adjustment (ranging from a 6% to 40% reduction), there is no clear evidence as to whether durvalumab and tremelimumab plus chemotherapy has favourable or unfavourable efficacy compared with all other comparators. However, the clinical experts consulted for this review indicated that the findings of the MAICs largely align with what is expected in clinical practice in Canadian settings.
The clinical experts reported that efficacy outcomes between durvalumab and tremelimumab plus chemotherapy and the available immuno-oncology plus chemotherapy treatment options appear comparable and not different from each other. They noted that the most relevant comparator to durvalumab and tremelimumab is nivolumab and ipilimumab, for which long-term outcomes appear comparable.
In terms of HRQoL, the time to deterioration in the EORTC QLQ-C30 GHS/QoL was greater for durvalumab and tremelimumab plus SOC chemotherapy compared to SOC chemotherapy alone, with the difference between groups considered clinically meaningful by the clinical experts; however, the GRADE assessment suggested very low certainty in this result. Interpretation of the HRQoL results is limited by potential attrition bias and limitations associated with the subjectivity of PROs. In addition, lower response rates to EORTC QLQ-C30 and EORTC QLQ-L13 questionnaires were seen in the SOC chemotherapy alone arm (65.0% and 64.8%, respectively) compared to the durvalumab and tremelimumab plus SOC chemotherapy arm (73.0% and 72.8%). Missing data may not have occurred at random because of patient-selective factors (e.g., patients who were sicker may have been less likely to complete PRO assessments), which has the potential to introduce bias in the effect estimate. The open-label study design and subjectivity in the PROs added to the difficulty in attributing differences between treatment arms in HRQoL measures to the addition of durvalumab and tremelimumab. Prespecified key domains and symptoms of interest in the POSEIDON trial included those identified by clinical experts as most relevant to patients with metastatic lung cancer. The sponsor-submitted ITC did not evaluate HRQoL for durvalumab and tremelimumab plus SOC chemotherapy compared to other available immuno-oncology plus chemotherapy options.
The clinical experts consulted for this review stated that durvalumab and tremelimumab plus platinum-based chemotherapy would be used for treatment of patients with previously untreated metastatic NSCLC not harbouring a targetable oncogenic aberration with an associated SOC first-line targeted therapy option (e.g., EGFR mutation or ALK rearrangement) and would serve as an additional first-line option combining immuno-oncology treatment and chemotherapy. They noted that, in practice, the durvalumab and tremelimumab plus platinum-based chemotherapy regimen is anticipated to be used most often in patients with a PD-L1 expression level of less than 50%. The experts also stated that patients with a negative or low tumoural PD-L1 status may benefit more from regimens using a CTLA-4 inhibitor. As such, this may be a population in which durvalumab and tremelimumab plus platinum-based chemotherapy may be used (although reliable biomarkers to determine which patients will derive the most benefit are lacking). At the primary analysis DCOs, post hoc exploratory analyses of POSEIDON subgroups of patients with a PD-L1 level of 1% or higher and those with a PD-L1 level of less than 1% suggested improvement in both OS and PFS for durvalumab and tremelimumab plus SOC chemotherapy compared to SOC chemotherapy alone in both subgroups.42 The clinical experts noted that patients with any tumoural PD-L1 status were included in the POSEIDON trial and commented that approval and funding should be based on the trial outcomes. Durvalumab and tremelimumab plus platinum-based chemotherapy was also identified by the experts as a potential treatment option for patients with KRAS, STK11, or KEAP mutations, but the experts acknowledged that the current evidence is post hoc and hypothesis-generating and that testing for all of these mutations is not routinely performed or may not be accessible in practice.
The main outcomes of importance identified by patient groups and clinicians were assessed in the POSEIDON trial. The clinical experts consulted for this review identified additional outcomes of interest that were not reported in the evidence submitted by the sponsor, including treatment-free interval and PFS at key landmarks beyond 12 months. They indicated that the treatment-free interval is of interest because it is a measure of the amount of time patients are living without active disease or progression of disease and not having to go for systemic treatment, which correlates with the best possible QoL. The experts noted that key landmark analyses for PFS add information regarding efficacy in terms of deep and durable response.
Harms
In both the durvalumab and tremelimumab plus SOC chemotherapy and SOC chemotherapy alone groups, similar proportions experienced any AE (97.3% versus 96.1%). The most frequent AEs in the durvalumab and tremelimumab plus SOC chemotherapy group were anemia, nausea, neutropenia, decreased appetite, fatigue, diarrhea, and rash. However, the proportions of patients who experienced SAEs, AEs leading to treatment discontinuation, AEs leading dose delays or interruptions, and AEs leading to death were numerically higher with durvalumab and tremelimumab plus SOC chemotherapy than with SOC chemotherapy alone.
The proportions of patients who experienced AESIs or AEPIs, including grade 3 or 4 AEs and those that led to study treatment discontinuation, were numerically higher with durvalumab and tremelimumab plus SOC chemotherapy than with SOC chemotherapy alone. AESIs or AEPIs with an outcome of death were reported in 2 patients (0.6%) in the durvalumab and tremelimumab plus SOC chemotherapy arm and 1 patient (0.3%) in the SOC chemotherapy alone arms. The proportions of patients who experienced imAEs, including grade 3 or 4 imAEs and those leading to discontinuation of any study treatment, were also numerically higher with durvalumab and tremelimumab plus SOC chemotherapy than with SOC chemotherapy alone. Two patients (0.6%) in the durvalumab and tremelimumab plus SOC chemotherapy arm and no patients in the SOC chemotherapy alone arm experienced imAEs with an outcome of death.
Because data were not provided for overall rates for the AESI category on its own, the GRADE assessment was based on data for the category of AESIs or AEPIs. The GRADE assessment suggested with high certainty that the combination of durvalumab and tremelimumab plus SOC chemotherapy results in a higher incidence of AESIs or AEPIs (including those that are grade 3 or 4) when compared with SOC chemotherapy alone. Input from the clinical experts informed this assessment, as they attributed the difference in these AEs between the 2 arms to the addition of durvalumab and tremelimumab. The experts added that the types and rates of AEs in the POSEIDON trial were not unexpected, and that grade 3 and 4 immune-related AEs did not occur at very high rates in the durvalumab and tremelimumab plus SOC chemotherapy arm. The clinical experts stated that imAEs caused by ICI treatment are typically of a low grade and reversible, and that health care providers treating lung cancer now have a decade of experience using ICIs in routine clinical practice. The experts also pointed out that some of these imAEs can be very serious or life-threatening, early identification and involvement of subspecialists are important, and that patients who live outside of major cancer centres are at higher risk of delays in diagnosis and treatment of imAEs, which can cause considerable morbidity or even mortality. The clinical experts noted that interventions required for the treatment of imAEs can negatively impact patient QoL.
The patient groups reported that maintaining QoL and reducing side effects from current medications and/or treatments are important outcomes and that patients were willing to endure a considerable level of side effects should they gain a significant amount of time in return (6 months or greater).
There was no evidence directly comparing the safety of durvalumab and tremelimumab plus SOC chemotherapy against other immuno-oncology plus chemotherapy combinations, and the ITC submitted by the sponsor did not evaluate harms outcomes. The clinical experts commented that imAEs between durvalumab and tremelimumab plus SOC chemotherapy and nivolumab and ipilimumab plus chemotherapy are likely very similar, and that AE profiles of these 2 regimens would not be expected to differ.
Other Considerations
The sponsor stated that the fixed dosing and short course duration of tremelimumab used in the POSEIDON regimen offers a convenient option that alleviates the need for vial sharing. The clinical experts consulted by CDA-AMC expected to see regional variability in the use of fixed versus weight-based dosing of durvalumab and tremelimumab based on the typical patient population (e.g., some areas of Canada may implement weight-based dosing for a generally lower-weight patient population who may receive too much medication with the fixed-dose regimen). The clinical experts noted that the choice of dosing regimen is an important factor for access to therapy as it may determine whether patients can be treated close to home or if they have to travel farther to a major systemic therapy unit to avoid drug wastage.
Conclusion
Based on evidence from the POSEIDON study, durvalumab and tremelimumab plus SOC chemotherapy demonstrated a benefit compared to SOC chemotherapy alone in OS, PFS, and ORR for the first-line treatment of adult patients with metastatic NSCLC with tumours that lack activating EGFR mutations and ALK fusions. The GRADE assessment suggested that durvalumab and tremelimumab plus SOC chemotherapy likely results in a clinically important increase in median OS, median PFS, and ORR compared to SOC chemotherapy, and a high certainty that durvalumab and tremelimumab plus SOC chemotherapy results in a clinically important increase in the survival rate at 36 months, 48 months, and 60 months, as well as in the PFS rate at 12 months. However, the evidence is very uncertain about the effect of durvalumab and tremelimumab plus SOC chemotherapy on HRQoL, as measured by time to deterioration in QLQ-C30 GHS/QoL, when compared with SOC chemotherapy alone.
In the POSEIDON study, AESI and AEPIs (including those that were grade 3 or 4) were experienced by a higher percentage of patients receiving durvalumab and tremelimumab plus SOC chemotherapy than those receiving SOC chemotherapy alone, with the GRADE assessment suggesting with high certainty that durvalumab and tremelimumab plus SOC chemotherapy results in a higher incidence of AESI or AEPIs when compared to SOC chemotherapy alone. The clinical experts consulted for this review stated that the types and rates of AEs in the POSEIDON trial were not unexpected. They indicated that imAEs caused by ICI treatment are typically of low grade and reversible, and that health care providers treating lung cancer now have a decade of experience using ICIs in routine clinical practice. The experts also noted that some imAEs can be very serious or life-threatening and that interventions required for the treatment of imAEs can negatively impact patient QoL.
Given that SOC chemotherapy was the comparator in the POSEIDON study, the lack of direct comparative data for the efficacy and safety between durvalumab and tremelimumab plus platinum-based chemotherapy and currently used first-line treatments (i.e., immuno-oncology plus chemotherapy options) represents a key gap in the evidence.
The findings of the MAICs demonstrated a largely comparable benefit in terms of OS and PFS when comparing combinations of durvalumab and tremelimumab plus platinum-based chemotherapy with relevant comparators for first-line treatment in patients with metastatic NSCLC without EGFR mutations and ALK aberrations. Harms were not investigated in the MAICs.
References
1.AstraZeneca Canada Inc. Sponsor's clinical summary report title [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Infinzi (durvalumab) 50 mg/mL concentrate for intravenous infusion and Imjudo (tremelimumab) 20 mg/mL concentrate for intravenous infusion. October 2024.
2.AstraZeneca Canada Inc. IMFINZI (durvalumab): 50 mg/mL, intravenous [product monograph]. 2025. Updated April 10, 2025.
3.Canadian Cancer Statistics Advisory Committee. Canadian Cancer Statistics: A 2020 special report on lung cancer [accessed by sponsor]. Canadian Cancer Society. https://cancer.ca/Canadian-Cancer-Statistics-2020-EN
4.Canadian Cancer Statistics Advisory in collaboration with the Canadian Cancer Society, Statistics Canada, and the Public Health Agency of Canada. Canadian Cancer Statistics: A 2022 special report on cancer prevalence [accessed by sponsor]. Canadian Cancer Society. https://cancer.ca/Canadian-Cancer-Statistics-2022-EN
5.Canadian Cancer Society. Lung cancer [accessed by sponsor]. http://www.cancer.ca/en/cancer-information/cancer-type/lung/lung-cancer/?region = on.
6.Birring SS, Peake MD. Symptoms and the early diagnosis of lung cancer. Thorax. 2005;60(4):268-9. doi: 10.1136/thx.2004.032698 PubMed
7.Hirsch FR, Suda K, Wiens J, Bunn PA, Jr. New and emerging targeted treatments in advanced non–small-cell lung cancer. Lancet. 2016;388(10048):1012-24. doi: 10.1016/S0140-6736(16)31473-8 PubMed
8.AACR Project GENIE Consortium. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017;7(8):818-831. doi: 10.1158/2159-8290.CD-17-0151 PubMed
9.Neal J. Post TW, ed. Initial systemic therapy for advanced non-small cell lung cancer lacking a driver mutation. UpToDate; 2024. Accessed October 15, 2024. http://www.uptodate.com
10.Hendriks LE, Kerr KM, Menis J, et al. Non-oncogene-addicted metastatic non–small-cell lung cancer: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann Oncol. 2023;34(4):358-376. doi: 10.1016/j.annonc.2022.12.013 PubMed
11.National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®). Non–small Cell Lung Cancer Version 9.2024 — September 9, 2024 [accessed by sponsor].
12.CADTH Provisional Funding Algorithm: Non-small lung cancer without actionable oncogenic alterations [accessed by sponsor]. 2023.
13.AstraZeneca. AstraZeneca response to December 3, 2024 CDA request for additional information regarding Durvalumab and Tremelimumab CDA review: Absolute effects for GRADE [internal additional sponsor's information]. December 10, 2024.
14.Canadian Cancer Statistics Advisory in collaboration with the Canadian Cancer Society, Statistics Canada, and the Public Health Agency of Canada. Canadian Cancer Statistics 2023 [accessed by sponsor]. Canadian Cancer Society. https://cancer.ca/Canadian-Cancer-Statistics-2023-EN
15.Detterbeck FC, Boffa DJ, Kim AW, Tanoue LT. The Eighth Edition Lung Cancer Stage Classification. Chest. 2017;151(1):193-203. doi: 10.1016/j.chest.2016.10.010
16.Kris MG, Johnson BE, Berry LD, et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA. 2014;311(19):1998-2006. doi: 10.1001/jama.2014.3741 PubMed
17.Hendriks LE, Kerr KM, Menis J, et al. Oncogene-addicted metastatic non–small-cell lung cancer: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann Oncol. 2023;34(4):339-357. doi: 10.1016/j.annonc.2022.12.009 PubMed
18.B.C. Cancer Agency. Cancer Management Manual -- Lung [accessed by sponsor] http://www.bccancer.bc.ca/health-professionals/clinical-resources/cancer-management-manual/lung/lung
19.Cancer Care Ontario. Lung Cancer Diagnosis Pathway Map, Version 2023.04 [accessed by sponsor]. https://www.cancercareontario.ca/sites/ccocancercare/files/assets/LungCancerDiagnosisPathwayMap.pdf
20.Melosky B, Blais N, Cheema P, et al. Standardizing biomarker testing for Canadian patients with advanced lung cancer. Curr Oncol. 2018;25(1):73-82. doi: 10.3747/co.25.3867 PubMed
21.Singh N, Jaiyesimi IA, Ismaila N, et al. Therapy for Stage IV Non-small-Cell Lung Cancer Without Driver Alterations: ASCO Living Guideline, Version 2023.1. J Clin Oncol. 2023;41(15):e51-e62. doi: 10.1200/JCO.23.00282 PubMed
22.Schiller JH, Harrington D, Belani CP, et al. Comparison of four chemotherapy regimens for advanced non–small-cell lung cancer. N Engl J Med. 2002;346(2):92-8. doi: 10.1056/NEJMoa011954 PubMed
23.CDA-AMC. CADTH Drug Reimbursement Review: Libtayo (cemiplimab) for non–small cell lung cancer. Accessed December 21, 2024. https://www.cda-amc.ca/cemiplimab-0
24.AstraZeneca Canada Inc. IMJUDO (tremelimumab): 20 mg / mL, intravenous infusion [product monograph]. 2024.
25.IMFINZI/TREMELIMUMAB ASTRAZENECA (durvalumab/tremelimumab) - Metastatic non–small cell lung cancer (NSCLC). Haute Autorité de Santé; 2023. Accessed November 27, 2024. https://www.has-sante.fr/jcms/p_3459834/en/imfinzi−/−tremelimumab-astrazeneca-durvalumab−/−tremelimumab-metastatic-non–small-cell-lung-cancer-nsclc
26.U.S. Food and Drug Administation. FDA approves tremelimumab in combination with durvalumab and platinum-based chemotherapy for metastatic non–small cell lung cancer. 2022. Accessed December 23, 2024. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-tremelimumab-combination-durvalumab-and-platinum-based-chemotherapy-metastatic-non
27.European Medicines Agency. Summary of Opinion: Initial Authorization: Tremelimumab AstraZeneca. 2022. Accessed November 27, 2024. https://www.ema.europa.eu/en/documents/smop-initial/chmp-summary-positive-opinion-tremelimumab-astrazeneca_en.pdf
28.European Medicines Agency. Tremelimumab AstraZeneca. 2024. Accessed November 27, 2024. https://www.ema.europa.eu/en/medicines/human/EPAR/tremelimumab-astrazeneca#assessment-history
29.European Medicines Agency. Public Statement: Tremelimumab AstraZeneca. 2024. Accessed November 27, 2024. https://www.ema.europa.eu/en/documents/public-statement/public-statement-tremelimumab-astrazeneca-withdrawal-marketing-authorisation-european-union_en.pdf
30.National Institute for Health and Care Excellence. Durvalumab with tremelimumab for untreated EGFR- and ALK-negative locally advanced and metastatic non–small-cell lung cancer. (NICE technology appraisal TA1538). 2023. Accessed November 27, 2024. https://www.nice.org.uk/guidance/discontinued/gid-ta10422
31.CADTH Reimbursement Recommendation: Tremelimumab (Imjudo) in Combination With Durvalumab (Imfinzi). Can J Health Technol. 2023;3(11). doi:10.51731/cjht.2023.788
32.pCODR expert review committee (PERC) final recommendation: Durvalumab (Imfinzi). 2019. Accessed December 23, 2024. https://www.cda-amc.ca/sites/default/files/pcodr/Reviews2019/10131DurvalumabNSCLC_fnRec_02May2019_approvedbyChair_Post_03May2019_final.pdf
33.CADTH Reimbursement Recommendation: Durvalumab (Imfinzi). Can J Health Technol. 2021;1(7). doi:10.51731/cjht.2021.109
34.CADTH Reimbursement Recommendation: Durvalumab (Imfinzi). Can J Health Technol. 2023;3(2). doi:10.51731/cjht.2023.580
35.Bristol-Myers Squibb Canada. Opdivo (nivolumab for injection): intravenous infusion, 10 mg nivolumab /mL 40 mg and 100 mg single-use vials [product monograph]. September 25, 2015. Updated June 28, 2024. https://pdf.hres.ca/dpd_pm/00076152.PDF
36.Regeneron Pharmaceuticals Inc. Libtayo(cemiplimab for injection): solution for intravenous infusion, 50mg/mL [product monograph]. April 10, 2019. Updated September 25, 2024. https://pdf.hres.ca/dpd_pm/00077201.PDF
37.Merck Canada Inc. Keytruda (pembrolizumab): solution for infusion, 100mg/4mL vial [product monograph]. May 19, 2015. Updated December 2, 2024. https://pdf.hres.ca/dpd_pm/00077917.PDF
38.AstraZeneca. Clinical Study Report: D419MC00004. A Phase III, Randomized, Multi-Center, Open-Label, Comparative Global Study to Determine the Efficacy of Durvalumab or Durvalumab and Tremelimumab in Combination With Platinum-Based Chemotherapy for First-Line Treatment in Patients With Metastatic Non–small-Cell Lung Cancer (NSCLC) (POSEIDON) [internal sponsor's report]. September 6, 2021.
39.Johnson ML, Cho BC, Luft A, et al. Durvalumab With or Without Tremelimumab in Combination With Chemotherapy as First-Line Therapy for Metastatic Non–small-Cell Lung Cancer: The Phase III POSEIDON Study. J Clin Oncol. 2023;41(6):1213-1227. doi: 10.1200/JCO.22.00975 PubMed
40.Peters S, Cho BC, Luft AV, et al. Durvalumab With or Without Tremelimumab in Combination With Chemotherapy in First-Line Metastatic NSCLC: Five-Year Overall Survival Outcomes From the Phase 3 POSEIDON Trial. J Thorac Oncol. 2025;20(1):76-93. doi: 10.1016/j.jtho.2024.09.1381 PubMed
41.Garon EB, Cho BC, Luft A, et al. Patient-reported outcomes with durvalumab, with or without tremelimumab, plus chemotherapy as first-line treatment for metastatic non–small-cell lung cancer (POSEIDON). Lung Cancer. 2023;186:107422. doi: 10.1016/j.lungcan.2023.107422 PubMed
42.Garon EB, Cho BC, Luft A, et al. A Brief Report of Durvalumab With or Without Tremelimumab in Combination With Chemotherapy as First-Line Therapy for Metastatic Non–small-Cell Lung Cancer: Outcomes by Tumor PD-L1 Expression in the Phase 3 POSEIDON Study. Clin Lung Cancer. 2024;25(3):266-273 e5. doi: 10.1016/j.cllc.2024.03.003 PubMed
43.Goldstraw P, Chansky K, Crowley J, et al. The IASLC Lung Cancer Staging Project: Proposals for Revision of the TNM Stage Groupings in the Forthcoming (Eighth) Edition of the TNM Classification for Lung Cancer. J Thorac Oncol. 2016;11(1):39-51. doi: 10.1016/j.jtho.2015.09.009 PubMed
44.Rami-Porta R, International Association for the Study of Lung Cancer. IASLC Staging Manual in Thoracic Oncology. Editorial Rx Press; 2016.
45.Highlights of the 8th Edition of the TNM Staging System: Practicalities and Tools. IASLC Lung Cancer News. Accessed December 23, 2024. https://old.lungcancernews.org/2018/06/01/highlights-of-the-8th-edition-of-the-tnm-staging-system-practicalities-and-tools/
46.Summary of the 8th Edition of the TNM Classification for Thoracic Cancers. IASLC Lung Cancer News. Accessed December 23, 2024. https://www.iaslc.org/research-education/publications-resources-guidelines/summary-8th-edition-tnm-classification
47.AstraZeneca. Clinical Study Protocol: D419MC00004. A Phase III, Randomized, Multi-Center, Open-Label, Comparative Global Study to Determine the Efficacy of Durvalumab or Durvalumab and Tremelimumab in Combination With Platinum-Based Chemotherapy for First-Line Treatment in Patients With Metastatic Non–small-Cell Lung Cancer (NSCLC) (POSEIDON) [internal sponsor's report]. April 20, 2020.
48.AstraZeneca. Statistical Analysis Plan D419MC00004. A Phase III, Randomized, Multi-Center, Open-Label, Comparative Global Study to Determine the Efficacy of Durvalumab or Durvalumab and Tremelimumab in Combination With Platinum-Based Chemotherapy for First-Line Treatment in Subjects With Metastatic Non–small-Cell Lung Cancer (NSCLC) (POSEIDON) [internal sponsor's report]. February 25, 2021.
49.Aaronson NK, Ahmedzai S, Bergman B, et al. The European Organization for Research and Treatment of Cancer QLQ-C30: a quality-of-life instrument for use in international clinical trials in oncology. J Natl Cancer Inst. 1993;85(5):365-76. doi: 10.1093/jnci/85.5.365 PubMed
50.Bergman B, Aaronson NK, Ahmedzai S, Kaasa S, Sullivan M. The EORTC QLQ-LC13: a modular supplement to the EORTC Core Quality of Life Questionnaire (QLQ-C30) for use in lung cancer clinical trials. EORTC Study Group on Quality of Life. Eur J Cancer. 1994;30A(5):635-42. doi: 10.1016/0959-8049(94)90535-5 PubMed
51.Giesinger JM, Kieffer JM, Fayers PM, et al. Replication and validation of higher order models demonstrated that a summary score for the EORTC QLQ-C30 is robust. J Clin Epidemiol. 2016;69:79-88. doi: 10.1016/j.jclinepi.2015.08.007 PubMed
52.Osoba D, Rodrigues G, Myles J, Zee B, Pater J. Interpreting the significance of changes in health-related quality-of-life scores. J Clin Oncol. 1998;16(1):139-44. doi: 10.1200/JCO.1998.16.1.139 PubMed
53.Fayers PM, Aaronson NK, Bjordal K, Groenvold M, Curran D, Bottomley A. The EORTC QLQ-C30 Scoring Manual (3rd Edition). EORTC; 2001. Accessed November 28, 2024. https://www.eortc.org/app/uploads/sites/2/2018/02/SCmanual.pdf
54.Nicklasson M, Bergman B. Validity, reliability and clinical relevance of EORTC QLQ-C30 and LC13 in patients with chest malignancies in a palliative setting. Qual Life Res. 2007;16(6):1019-28. doi: 10.1007/s11136-007-9210-8 PubMed
55.Musoro JZ, Coens C, Sprangers MAG, et al. Minimally important differences for interpreting EORTC QLQ-C30 change scores over time: A synthesis across 21 clinical trials involving nine different cancer types. Eur J Cancer. 2023;188:171-182. doi: 10.1016/j.ejca.2023.04.027 PubMed
56.Bedard G, Zeng L, Zhang L, et al. Minimal important differences in the EORTC QLQ-C30 in patients with advanced cancer. Asia Pac J Clin Oncol. 2014;10(2):109-17. doi: 10.1111/ajco.12070 PubMed
57.Kvam AK, Fayers PM, Wisloff F. Responsiveness and minimal important score differences in quality-of-life questionnaires: a comparison of the EORTC QLQ-C30 cancer-specific questionnaire to the generic utility questionnaires EQ-5D and 15D in patients with multiple myeloma. Eur J Haematol. 2011;87(4):330-7. doi: 10.1111/j.1600-0609.2011.01665.x PubMed
58.AstraZeneca. Clinical Study Report Addendum 2: D419MC00004. A Phase III, Randomized, Multi-Center, Open-Label, Comparative Global Study to Determine the Efficacy of Durvalumab or Durvalumab and Tremelimumab in Combination With Platinum-Based Chemotherapy for First-Line Treatment in Patients With Metastatic Non–small-Cell Lung Cancer (NSCLC) (POSEIDON) [internal sponsor's report]. January 16, 2024.
59.Gail M, Simon R. Testing for qualitative interactions between treatment effects and patient subsets. Biometrics. 1985;41(2):361-72. PubMed
60.AstraZeneca. AstraZeneca response to December 2, 2024 CDA request for additional information regarding Durvalumab and Tremelimumab CDA review: [internal additional sponsor's information]. December 9, 2024.
61.AstraZeneca. Clinical Study Report: D419MC00004 (Tables and Figures). A Phase III, Randomized, Multi-Center, Open-Label, Comparative Global Study to Determine the Efficacy of Durvalumab or Durvalumab and Tremelimumab in Combination With Platinum-Based Chemotherapy for First-Line Treatment in Patients With Metastatic Non–small-Cell Lung Cancer (NSCLC) (POSEIDON) [internal sponsor's report]. September 6, 2021.
62.Peters S, Cho BC, Luft A, et al. LBA3 Durvalumab (D) ± tremelimumab (T) + chemotherapy (CT) in first-line (1L) metastatic NSCLC (mNSCLC): 5-year (y) overall survival (OS) update from the POSEIDON study. Immuno-Oncology and Technology. 2023;20. doi: 10.1016/j.iotech.2023.100693
63.ICH E9(R1) Estimands and Sensitivity Analysis in Clinical Trials. International Council for Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. Accessed Jan 4, 2025. https://admin.ich.org/sites/default/files/inline-files/ICH_E9%28R1%29_TrainingMaterial_COMPILATION_2021_1214.pdf
64.Balshem H, Helfand M, Schunemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-6. doi: 10.1016/j.jclinepi.2010.07.015 PubMed
65.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. doi: 10.1016/j.jclinepi.2019.10.014 PubMed
66.National Comprehensive Cancer Network (NCCN) Guidelines: Non-small Cell Lung Cancer. Version 5.2022 [sponsor supplied reference].
67.Novello S, Kowalski DM, Luft A, et al. Pembrolizumab Plus Chemotherapy in Squamous Non–small-Cell Lung Cancer: 5-Year Update of the Phase III KEYNOTE-407 Study. J Clin Oncol. 2023;41(11):1999-2006. doi: 10.1200/JCO.22.01990 PubMed
68.Garassino MC, Gadgeel S, Speranza G, et al. Pembrolizumab Plus Pemetrexed and Platinum in Nonsquamous Non–small-Cell Lung Cancer: 5-Year Outcomes From the Phase 3 KEYNOTE-189 Study. J Clin Oncol. 2023;41(11):1992-1998. doi: 10.1200/JCO.22.01989 PubMed
69.Carbone DP, Ciuleanu T-E, Schenker M, et al. First-line (1L) nivolumab (N) + ipilimumab (I) + chemotherapy (C) vs. C alone in patients (pts) with metastatic NSCLC (mNSCLC) from CheckMate 9LA: 4-y clinical update and outcomes by tumor histologic subtype (THS). J Clin Oncol. 2023;41(17_suppl):LBA9023-LBA9023. doi: 10.1200/JCO.2023.41.17_suppl.LBA9023
70.AstraZeneca. Clinical Study Report Addendum: A Phase III, Randomized, Multi-Center, Open-Label, Comparative Global Study to Determine the Efficacy of Durvalumab or Durvalumab and Tremelimumab in Combination With Platinum-Based Chemotherapy for First-Line Treatment in Patients With Metastatic Non–small-Cell Lung Cancer (NSCLC) (POSEIDON) [internal sponsor's report]. May 25, 2022.
71.AstraZeneca Canada Inc. Matching-Adjusted Indirect Comparison (MAIC) Report [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: IMFINZI (durvalumab), 50 mg / mL, intravenous. February 2024.
72.Phillippo DM, Ades AE, Dias S, Palmer S, Abrams KR, Welton NJ. NICE DSU Technical Support Document 18: Methods for population-adjusted indirect comparisons in submission to NICE [accessed by sponsor]. National Institute for Health and Care Excellence; 2016. http://www.nicedsu.org.uk
73.Halmos B, Burke T, Kalyvas C, et al. A Matching-Adjusted Indirect Comparison of Pembrolizumab + Chemotherapy vs. Nivolumab + Ipilimumab as First-Line Therapies in Patients with PD-L1 TPS >/=1% Metastatic NSCLC. Cancers (Basel). 2020;12(12). doi: 10.3390/cancers12123648 PubMed
74.Signorovitch JE, Wu EQ, Yu AP, et al. Comparative effectiveness without head-to-head trials: a method for matching-adjusted indirect comparisons applied to psoriasis treatment with adalimumab or etanercept. Pharmacoeconomics. 2010;28(10):935-45. doi: 10.2165/11538370-000000000-00000 PubMed
75.Signorovitch JE, Sikirica V, Erder MH, et al. Matching-adjusted indirect comparisons: a new tool for timely comparative effectiveness research. Value Health. 2012;15(6):940-7. doi: 10.1016/j.jval.2012.05.004 PubMed
76.Novello S, Kowalski DM, Luft A, et al. 974MO 5-year update from KEYNOTE-407: Pembrolizumab plus chemotherapy in squamous non–small cell lung cancer (NSCLC). Ann Oncol. 2022;33:S993-S994. doi: 10.1016/j.annonc.2022.07.1102
77.Paz-Ares LG, Ciuleanu T-E, Cobo-Dols M, et al. First-line (1L) nivolumab (NIVO) + ipilimumab (IPI) + 2 cycles of chemotherapy (chemo) versus chemo alone (4 cycles) in patients (pts) with metastatic non–small cell lung cancer (NSCLC): 3-year update from CheckMate 9LA. J Clin Oncol. 2022;40(17_suppl):LBA9026-LBA9026. doi: 10.1200/JCO.2022.40.17_suppl.LBA9026
78.Gandhi L, Rodriguez-Abreu D, Gadgeel S, et al. Pembrolizumab plus Chemotherapy in Metastatic Non–small-Cell Lung Cancer. N Engl J Med. 2018;378(22):2078-2092. doi: 10.1056/NEJMoa1801005 PubMed
79.Paz-Ares L, Luft A, Vicente D, et al. Pembrolizumab plus Chemotherapy for Squamous Non–small-Cell Lung Cancer. N Engl J Med. 2018;379(21):2040-2051. doi: 10.1056/NEJMoa1810865 PubMed
80.Paz-Ares L, Ciuleanu TE, Cobo M, et al. First-line nivolumab plus ipilimumab combined with two cycles of chemotherapy in patients with non–small-cell lung cancer (CheckMate 9LA): an international, randomised, open-label, phase 3 trial. Lancet Oncol. 2021;22(2):198-211. doi: 10.1016/S1470-2045(20)30641-0 PubMed
81.Gogishvili M, Melkadze T, Makharadze T, et al. Cemiplimab plus chemotherapy versus chemotherapy alone in non–small cell lung cancer: a randomized, controlled, double-blind phase 3 trial. Nat Med. 2022;28(11):2374-2380. doi: 10.1038/s41591-022-01977-y PubMed
82.Wang Z, Huang C, Yang JJ, et al. A randomised phase II clinical trial of nab-paclitaxel and carboplatin compared with gemcitabine and carboplatin as first-line therapy in advanced squamous cell lung carcinoma (C-TONG1002). Eur J Cancer. 2019;109:183-191. doi: 10.1016/j.ejca.2019.01.007 PubMed
83.Gray J, Rodríguez-Abreu D, Powell SF, et al. FP13.02 Pembrolizumab + Pemetrexed-Platinum vs. Pemetrexed-Platinum for Metastatic NSCLC: 4-Year Follow-up From KEYNOTE-189. J Thorac Oncol. 2021;16(3):S224. doi: 10.1016/j.jtho.2021.01.141
84.Robinson AG, Vicente D, Tafreshi A, et al. 97O First-line pembrolizumab plus chemotherapy for patients with advanced squamous NSCLC: 3-year follow-up from KEYNOTE-407. J Thorac Oncol. 2021;16(4):S748-S749. doi: 10.1016/s1556-0864(21)01939-0
85.Reck M, Ciuleanu TE, Cobo M, et al. First-line nivolumab plus ipilimumab with two cycles of chemotherapy versus chemotherapy alone (four cycles) in advanced non–small-cell lung cancer: CheckMate 9LA 2-year update. ESMO open. 2021;6(5):100273. doi: 10.1016/j.esmoop.2021.100273 PubMed
86.Makharadze T, Gogishvili M, Melkadze T, et al. Cemiplimab Plus Chemotherapy Versus Chemotherapy Alone in Advanced NSCLC: 2-Year Follow-Up From the Phase 3 EMPOWER-Lung 3 Part 2 Trial. J Thorac Oncol. 2023;18(6):755-768. doi: 10.1016/j.jtho.2023.03.008 PubMed
Appendix 1: Detailed Outcome Data
Please note that this appendix has not been copy-edited.
Table 36: Overall Survival for Durvalumab Plus Chemotherapy vs. Chemotherapy Alone (Primary End Point, FAS)
Results | D + chemotherapy (N = 338) | Chemotherapy (N = 337) |
|---|---|---|
HR,a D + chemotherapy vs. chemotherapy aloneb | 0.86 | |
97.121% CI for HRc | 0.710, 1.036 | |
95% CI for HRa | 0.724, 1.016 | |
2-sided P valued | 0.07581 | |
Death, n (%) | 264 (78.1) | 285 (84.6) |
Censored patients, n (%) | 74 (21.9) | 52 (15.4) |
Still in survival follow-upe | 65 (19.2) | 40 (11.9) |
Terminated before deathf | 9 (2.7) | 12 (3.6) |
Lost to follow-up | 2 (0.6) | 2 (0.6) |
Withdrawn consent | 6 (1.8) | 10 (3.0) |
Other | 1 (0.3) | 0 |
Median OS (95%), monthsg | 13.3 | 11.7 |
95% CI for median OSg | 11.4, 14.7 | 10.5, 13.1 |
OS rate at 12 months (%)g | 53.2 | 49.1 |
95% CI for OS rate at 12 monthsg | 47.7, 58.4 | 43.6, 54.4 |
OS rate at 18 months (%)g | 38.1 | 34.1 |
95% CI for OS rate at 18 monthsg | 32.9, 43.3 | 29.0, 39.2 |
OS rate at 24 months (%)g | 29.6 | 22.1 |
95% CI for OS rate at 24 monthsg | 24.8, 34.6 | 17.8, 26.8 |
OS rate at 36 months (%)g | 20.3 | 13.3 |
95% CI for OS rate at 36 monthsg | 16.1, 25.0 | 9.8, 17.4 |
CI = confidence interval; D = durvalumab; FAS = full analysis set; HR = hazard ratio; OS = overall survival; vs. = versus.
aThe HR and CI were estimated from a stratified Cox proportional hazards model with the Efron method to control for ties, the stratification factors PD-L1 tumour expression (PD-L1 ≥ 50% vs. PD-L1 < 50%), histology (squamous vs. nonsquamous), and disease stage (Stage IVA vs. Stage IVB) in the strata statement, and the CI calculated using a profile likelihood approach.
bAn HR < 1 favours durvalumab plus standard of care chemotherapy to be associated with a longer OS than standard-of-care chemotherapy alone.
cBased on a Lan-DeMets alpha spending function with O'Brien Fleming type boundary with the actual number of events observed, the boundary for declaring statistical significance is 0.02879 for a 4% overall alpha. Corresponding CI is shown.
dP values were generated using the stratified log-rank test adjusting for PD-L1 tumour expression (PD-L1 ≥ 50% vs. PD-L1 < 50%), histology (squamous vs. nonsquamous), and disease stage (Stage IVA vs. Stage IVB) and using the Breslow approach for handling ties.
eIncludes patients known to be alive at data cut-off.
fIncludes patients with unknown survival status or patients who were lost to follow-up.
gCalculated using the Kaplan-Meier technique.
Data cut-off date: March 12, 2021.
Source: POSEIDON Clinical Study Report.38
Table 37: Progression-Free Survival Based on BICR Assessments per RECIST 1.1 for Durvalumab Plus Chemotherapy vs. Chemotherapy Alone (Primary End Point, FAS)
Results | D + chemotherapy (N = 338) | Chemotherapy (N = 337) |
|---|---|---|
HR, D + chemotherapy vs. chemotherapy alonea,b | 0.74 | |
99.181% CI for HRc | 0.583, 0.942 | |
95% CI for HRa | 0.620, 0.885 | |
2-sided P valued | 0.00093 | |
Total number of events,e n (%) | 253 (74.9) | 258 (76.6) |
RECIST 1.1 progression | 193 (57.1) | 202 (59.9) |
Death in the absence of progression | 60 (17.8) | 56 (16.6) |
Censored patients, n (%) | 85 (25.1) | 79 (23.4) |
Censored RECIST progressionf | 0 | 2 (0.6) |
Censored deathg | 8 (2.4) | 24 (7.1) |
Progression-free at time of analysis | 72 (21.3) | 43 (12.8) |
Lost to follow-up | 0 | 0 |
Withdrawn consent | 3 (0.9) | 9 (2.7) |
Discontinued study | 2 (0.6) | 1 (0.3) |
Median PFS (months)h | 5.5 | 4.8 |
95% CI for median PFSh | 4.7, 6.5 | 4.6, 5.8 |
PFS rate at 12 months (%)h | 24.4 | 13.1 |
95% CI for PFS rate at 12 monthsh | 19.7, 29.5 | 9.3, 17.6 |
BICR = blinded independent central review; CI = confidence interval; D = durvalumab; FAS = full analysis set; HR = hazard ratio; PFS = progression-free survival; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours, version 1.1; vs. = versus.
aThe HR and CI were estimated from a stratified Cox proportional hazards model with the Efron method to control for ties, the stratification factors PD-L1 (PD-L1 ≥ 50% vs. PD-L1 < 50%), histology (squamous vs. nonsquamous), and disease stage (Stage IVA vs. Stage IVB) in the strata statement, and the CI calculated using a profile likelihood approach.
bA hazard ratio < 1 favours durvalumab plus standard-of-care chemotherapy to be associated with a longer PFS than standard-of-care chemotherapy alone.
cBased on a Lan-DeMets alpha spending function with O'Brien Fleming type boundary with the actual number of events observed, the boundary for declaring statistical significance is 0.00819 for a 1% overall alpha. Corresponding CI is shown.
dP values were generated using the stratified log-rank test adjusting for PD-L1 (PD-L1 ≥ 50% vs. PD-L1 < 50%), histology (squamous vs. nonsquamous), and disease stage (Stage IVA vs. Stage IVB) and using the Breslow approach for handling ties.
ePatients who did not progress or die, or who progressed or died after 2 or more missed visits, were censored at the latest evaluable RECIST assessment or at Day 1 if there were no evaluable visits or no baseline data and the patient did not die within 2 visits of baseline.
fRECIST progression event occurred after 2 or more missed visits or within 2 visits of baseline without any evaluable visits or baseline data.
gDeath occurred after 2 or more missed visits in the absence of progression.
hCalculated using the Kaplan-Meier technique.
There was 1 patient who died 1 day before randomization and was censored on day 1.
Data cut-off: July 24, 2019.
Source: POSEIDON Clinical Study Report.38
Pharmacoeconomic Review
Abbreviations
AE
adverse event
BIA
budget impact analysis
CDA-AMC
Canada’s Drug Agency
ECOG PS
Eastern Cooperative Oncology Group Performance Status
HR
hazard ratio
ICER
incremental cost-effectiveness ratio
ITT
intention-to-treat
LCC
Lung Cancer Canada
MAIC
matching adjusted indirect comparison
NSCC
non–squamous cell carcinoma
NSCLC
non–small cell lung cancer
OS
overall survival
PFS
progression-free survival
QALY
quality-adjusted life-year
SCC
squamous-cell carcinoma
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Durvalumab (Imfinzi), 50 mg/mL, concentrate for IV infusion (in combination with tremelimumaba (Imjudo), 20 mg/mL, concentrate for IV infusion) |
Indication | Durvalumab in combination with tremelimumab and platinum-based chemotherapy is indicated for the first-line treatment of adult patients with metastatic NSCLC with no sensitizing epidermal growth factor receptor (EGFR) mutations or anaplastic lymphoma kinase (ALK) genomic tumour aberrations |
Health Canada approval status | NOC |
Health Canada review pathway | Standard review |
NOC date | April 10, 2025 |
Reimbursement request | As per indication |
Sponsor | AstraZeneca Canada Inc. |
Submission history | Previously reviewed: Yes Indication: First-line treatment of adult patients with unresectable hepatocellular carcinoma who require systemic therapy. Recommendation date: November 3, 2023 Recommendation: Reimburse with clinical criteria and/or conditions |
NOC = Notice of Compliance; NSCLC = non–small cell lung cancer.
aTremelimumab is to be used in combination with durvalumab. Tremelimumab is not intended for monotherapy use.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Partitioned survival model |
Target population | Adult patients with metastatic NSCLC who lack activating EGFR mutations and ALK fusions who have not received prior therapy for metastatic disease |
Treatment | Durvalumab and tremelimumab in combination with PBC |
Dose regimena |
|
Submitted price | Durvalumab (120 mg/2.4 mL vial): $938.67 Durvalumab (500 mg/10 mL vial): $3,911.11 Tremelimumab (25 mg/1.25 mL vial): $2,859.97 Tremelimumab (300 mg/15 mL vial): $34,319.58 |
Submitted treatment costb | $22,010, per 21-day cycle in the initial treatment stage (up to week 12), $15,387 per 21-day cycle in the initial maintenance phase (weeks 13 to 16), and $8,952 per 21-day cycle in the subsequent maintenance phase (beyond week 16) |
Comparators |
|
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | Lifetime (20 years) |
Key data sources | Efficacy inputs for durvalumab and tremelimumab + PBC and PBC alone were informed by the POSEIDON trial (NCT03164616). Efficacy inputs for nivolumab and ipilimumab + PBC, pembrolizumab + PBC, and cemiplimab + PBC were derived from sponsor-submitted anchored MAICs. Efficacy for nivolumab and ipilimumab + PBC was informed by the CHECKMATE-9LA trial; efficacy for pembrolizumab + PBC was informed by the KEYNOTE-407 (SCC population) and KEYNOTE-189 (NSCC population) trials, and efficacy for cemiplimab + PBC was informed by the EMPOWER-Lung3 trial |
Submitted resultsc | Pairwise comparisons vs:
|
Key limitations |
|
CDA-AMC reanalysis results |
|
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; IO = immuno-oncology; LY = life-year; MAIC = matching adjusted indirect comparison; NSCLC = non–small cell lung cancer; OS = overall survival; PBC = platinum-based chemotherapy; PFS = progression-free survival; q.3.w. = every 3 weeks; q.4.w. = every 4 weeks; QALY = quality-adjusted life-year; vs. = versus.
aFor patients weighing less than 30 kg the initial dosing is as follows. During chemotherapy, patients receive durvalumab 20 mg/kg in combination with tremelimumab 1 mg/kg and platinum-based chemotherapy q.3.w. (21 days) for 4 cycles. Following platinum chemotherapy, patients receive durvalumab 20 mg/kg q.4.w. and histology-based pemetrexed maintenance therapy q.4.w.
bSubmitted treatment cost included the combined cost of durvalumab with tremelimumab, in combination with PBC.
cSponsor results are presented pairwise as the sponsor noted that conducting a sequential analysis is not appropriate when using a MAIC.
Conclusions
The sponsor submitted evidence from 1 pivotal randomized, placebo-controlled study (the POSEIDON study) that demonstrated durvalumab and tremelimumab plus chemotherapy was associated with a benefit compared with chemotherapy alone in terms of overall survival (OS), progression-free survival (PFS), and objective response rate for the first-line treatment of patients with metastatic NSCLC with tumours that lack activating EGFR mutations and ALK fusions. However, the most relevant comparator for durvalumab and tremelimumab plus platinum-based chemotherapy in practice in Canada was determined to be other immuno-oncology therapies in combination with platinum-based chemotherapies. In the absence of head-to-head evidence for durvalumab and tremelimumab plus platinum-based chemotherapy compared with other immuno-oncology therapies plus chemotherapy (i.e., nivolumab and ipilimumab plus platinum-based chemotherapy, pembrolizumab plus platinum-based chemotherapy and cemiplimab plus platinum-based chemotherapy), the sponsor submitted matching adjusted indirect comparisons (MAICs) to inform the comparative efficacy used within the economic evaluation. The MAIC results indicated that the credible intervals for OS and PFS crossed the null and durvalumab and tremelimumab plus platinum-based chemotherapy could therefore be considered comparable to other immuno-oncology therapies plus platinum-based chemotherapy. However, the Clinical Review by Canada’s Drug Agency (CDA-AMC) noted that the analysis did not adjust for important effect modifiers, such as PD-L1 status, sex, age, Eastern Cooperative Oncology Group Performance Status (ECOG PS), disease stage, type of chemotherapy, and presence of brain metastases, likely leading to residual heterogeneity between the studies. As a result, the Clinical Review reported that no conclusions could be drawn as to whether durvalumab and tremelimumab plus platinum-based chemotherapy was favoured over other comparators, or vice versa. The clinical experts consulted by CDA-AMC for this review indicated that durvalumab and tremelimumab plus platinum-based chemotherapy is expected to have a similar treatment benefit compared to other immuno-oncology plus platinum-based chemotherapy regimens.
CDA-AMC identified several limitations with the sponsor’s pharmacoeconomic analysis, which mostly resulted from limitations with the submitted comparative evidence. The sponsor suggests that durvalumab and tremelimumab plus platinum-based chemotherapy is associated with an incremental cost-effectiveness ratio (ICER) of at least $174,337 per quality-adjusted life-year (QALY) gained compared with other immuno-oncology therapies plus platinum-based chemotherapy. The incremental benefit is largely based on assumed long-term benefits from capping the treatment effect of comparator immuno-oncology therapies to a maximum of 5 years, while the incremental costs are driven by differences in the cost of treatment, and using durvalumab and tremelimumab plus platinum-based chemotherapy until disease progression or unacceptable tolerability (i.e., no maximum duration) compared with other immune-oncology treatments being used for a maximum of 2 years. When testing the sponsor’s model (e.g., attempting to remove the sponsor’s treatment effect cap of 5 years that was applied to the comparator immuno-oncology plus platinum-based chemotherapy), the results of the model did not meet face validity. For example, while pembrolizumab plus platinum-based chemotherapy showed improved PFS relative to durvalumab and tremelimumab plus platinum-based chemotherapy based on the point estimates from the MAIC, the modelled results suggested durvalumab and tremelimumab plus platinum-based chemotherapy was associated with more QALYs in the PFS health state. As such, any changes made to the model could not be interpreted with accuracy, and the results of the submitted cost-utility analysis could not be effectively validated. Overall, there is insufficient clinical and economic evidence to suggest that durvalumab and tremelimumab plus platinum-based chemotherapy should cost more than currently reimbursed immuno-oncology plus platinum-based chemotherapy regimens for adult patients with metastatic non–small cell lung cancer (NSCLC) that lack activating genomic EGFR mutations and ALK fusions and who have not received prior therapy for metastatic disease.
Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, clinician groups, and drug plans that participated in the CDA-AMC review process.
Patient input was received from a joint submission by the Canadian Cancer Survivor Network, Lung Cancer Canada (LCC) and the Lung Health Foundation. A survey was conducted from August 1, 2024, to October 28, 2024, via the 3 organizations’ social media platforms. Five patients completed the survey. One interview was also conducted with a patient who was part of the POSEIDON trial. Respondents noted that managing pain and side effects, preventing tumour and nodule growth, and reducing shortness of breath were important aspects of their disease to control. The respondents indicated differing tolerances for side effects to extend survival, generally accepting higher levels for longer life extensions. The interviewed patient from the POSEIDON trial reported significant benefits from durvalumab, including minimal side effects, stabilized health, and a sense of normalcy. Participants valued durvalumab for symptom management and slowing disease progression, with most recommending its availability for eligible patients. Many respondents described quality of life and family time as key factors in treatment decisions.
CDA-AMC received 2 registered clinician group inputs for this review from the Ontario Health (Cancer Care Ontario) Drug Advisory Committee and LCC. The Drug Advisory Committee input noted that current treatments for NSCLC include chemotherapy with pembrolizumab, ipilimumab with nivolumab, or platinum-based chemotherapy, with goals focused on tumour shrinkage, symptom relief, quality of life improvement, and survival prolongation. Gaps in treatment include limited patient responsiveness, treatment resistance, and issues with tolerance, compliance, and convenience. Clinician input suggested durvalumab and tremelimumab is best suited as a first-line treatment for people with stage IV or incurable NSCLC who do not have sensitizing EGFR mutations or ALK fusions, offering an additional option without significantly altering the treatment paradigm. Treatment response is typically monitored through symptom assessments and imaging every 6 weeks initially, with discontinuation considered upon disease progression, intolerable side effects, or patient withdrawal. LCC input noted that current treatments for metastatic NSCLC without actionable driver mutations include combinations of chemotherapy and immunotherapy, aiming to improve symptoms, quality of life, and survival. The POSEIDON regimen, combining durvalumab, tremelimumab, and platinum doublet chemotherapy, shows promising survival benefits, particularly in poor-risk populations like those with KRAS mutations, while maintaining a manageable toxicity profile. Regular assessments, including imaging and symptom evaluation, help monitor treatment response, while discontinuation decisions consider disease progression and treatment tolerability.
Drug plan input for this review noted that tremelimumab and durvalumab were prescribed in the POSEIDON trial with flat dosing. However, the drug plans noted that, if the therapy is funded, jurisdictions are likely to implement a weight-based durvalumab dose. The drug plans questioned if patients on other chemotherapy or immuno-oncology treatments should be permitted to switch to tremelimumab and durvalumab plus platinum-based chemotherapy and what the minimum number of chemotherapy cycles would be for eligible patients. The drug plans also asked what the appropriate discontinuation criteria would be for the treatment under review, and when patients could be eligible for re-treatment.
CDA-AMC was unable to address the following concerns raised from patient, clinician, and drug plan input:
histology-based subgroups could not be evaluated separately in the CDA-AMC reanalysis
weight-based dosing for durvalumab or other IO therapies.
Economic Review
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis comparing the costs and outcomes for durvalumab and tremelimumab in combination with platinum-based chemotherapy with relevant comparators for the treatment of adult patients with metastatic NSCLC that lack activating EGFR mutations and ALK fusions who have not received prior therapy for metastatic disease. The modelled population was aligned with the POSEIDON trial intention-to-treat (ITT) population.1 The target population aligned with the Health Canada–indicated population.
The relevant comparators the sponsor included in the economic analysis were: nab-paclitaxel, gemcitabine, pemetrexed, or paclitaxel plus cisplatin or carboplatin (platinum-based chemotherapy), nivolumab in combination with ipilimumab, paclitaxel, and carboplatin or cisplatin (nivolumab and ipilimumab plus platinum-based chemotherapy), pembrolizumab in combination with pemetrexed, cisplatin or carboplatin for non–squamous cell carcinoma (NSCC) or pembrolizumab in combination with carboplatin, paclitaxel or nab-paclitaxel for squamous-cell carcinoma (SCC) (pembrolizumab plus platinum-based chemotherapy), and cemiplimab, pemetrexed, or paclitaxel, and carboplatin or cisplatin (cemiplimab plus platinum-based chemotherapy).
Durvalumab is available as a concentrate for solution for IV infusion in 2.4 mL or 10 mL vials (containing 120 mg or 500 mg of durvalumab, respectively). Tremelimumab is available as a concentrate for solution for IV infusion in 1.25 mL or 15 mL vials (containing 25 mg and 300 mg of tremelimumab, respectively). According to the product monograph, the recommended dosage for patients with a body weight of 30 kg or more during chemotherapy is 1,500 mg of durvalumab in combination with 75 mg of tremelimumab with platinum-based chemotherapy every 3 weeks (21 days) for 4 cycles.2,3 Following platinum chemotherapy, the recommended dosage for patients with a body weight of 30 kg or more is 1,500 mg of durvalumab every 4 weeks in combination with histology-based pemetrexed maintenance therapy every 4 weeks. Alternatively, patients with a body weight of less than 30 kg undergoing chemotherapy are recommended to receive durvalumab at 20 mg/kg with tremelimumab at 1 mg/kg plus platinum-based chemotherapy every 3 weeks (21 days) for 4 cycles. Following platinum chemotherapy, patients with a body weight of less than 30 kg are recommended to receive durvalumab at 20 mg/kg every 4 weeks and histology-based pemetrexed maintenance therapy every 4 weeks. Durvalumab should be taken until disease progression and tremelimumab should be taken for up to 5 doses or until disease progression, whichever occurs first. A fifth dose of 75 mg of tremelimumab should be given at week 16 alongside durvalumab dose 6 for all patients. Platinum-based chemotherapy was defined as 4 combination regimens of either nab-paclitaxel plus cisplatin or carboplatin, gemcitabine plus cisplatin or carboplatin, pemetrexed plus cisplatin or carboplatin, and paclitaxel plus cisplatin or carboplatin. Dosing and costing for each regimen are provided in Appendix 1. Using patient characteristics derived from the POSEIDON trial ITT population (68.5 kg, 1.79 m2 of body surface area), the cost of durvalumab ($11,733.33 per 1,500 mg dose) and tremelimumab ($8,579.90 per 75 mg dose), and assuming vial sharing, the weekly cost of durvalumab and tremelimumab plus platinum-based chemotherapy was $7,337 up to week 12, $5,129 in the initial maintenance phase (weeks 13 to week 16), and $2,984 in the subsequent maintenance phase (beyond week 16). The cost of platinum-based chemotherapy alone was estimated to be $525 per week up week 12, and $60 per week beyond 12 weeks. Due to the changing frequency of treatment, a cost per 21 days or 28 days was not provided by the sponsor.
The clinical outcomes modelled were PFS and OS. The economic outcomes of interest were QALYs and life-years. Patient age at the model entry was 63.1 years and the economic analysis was undertaken over a lifetime time horizon (assumed to be 20 years) from the perspective of the Canadian publicly funded health care system. Costs and QALYs were discontinued at a rate of 1.5% per year.
Model Structure
The sponsor submitted a partitioned survival model with 3 health states: preprogression, postprogression, and death. The estimated proportion of patients in each health state over time was based on the OS and PFS curves for each intervention. All patients entered the model in the preprogression health state and received 1 of the treatment options, with state occupancy defined by PFS. The cycle length was 1 week to account for different treatment schedules. As a result, the sponsor converted all nonweekly cost inputs to weekly costs. A half-cycle correction was applied in the model.
Model Inputs
The modelled population generally reflected the baseline characteristics of the enrolled population in the POSEIDON trial, a phase III, randomized, open-label, multicentre, global study that was designed to evaluate the efficacy and safety of durvalumab and tremelimumab plus platinum-based chemotherapy. The sponsor’s model used a mean age of 63.1 years, a mean weight of 68.5 kg, a creatine clearance rate of 90.8 mL/min, and a mean height of 167.6 cm (to determine body surface area).4 The ITT population included patients irrespective of PD-L1 expression levels, with either NSCC or SCC histology, and excluded patients with specific EGFR mutations or ALK fusions.
Key clinical efficacy inputs (OS and PFS) for durvalumab and tremelimumab plus platinum-based chemotherapy and platinum-based chemotherapy alone were derived from the POSEIDON trial. Efficacy inputs for remaining comparators were estimated based on results from MAICs. In particular, 2 independent trials were identified for pembrolizumab plus platinum-based chemotherapy based on histology (KEYNOTE-189 for the NSCC population and KEYNOTE-407 for the SCC population).5,6 The remaining trials informing the MAICs for the other comparators were CheckMate-9LA for nivolumab and ipilimumab plus platinum-based chemotherapy and EMPOWER-Lung 3 for cemiplimab plus platinum-based chemotherapy.7,8
The sponsor extrapolated survival and time to treatment discontinuation using the POSEIDION trial’s Kaplan-Meier curves for durvalumab and tremelimumab plus platinum-based chemotherapy versus platinum-based chemotherapy alone and plotted curves alongside the Kaplan-Meier data. Based on visual inspections of the logarithmic risk curves for OS, PFS, and time to treatment discontinuation, the proportional hazards assumption for durvalumab and tremelimumab plus platinum-based chemotherapy versus platinum-based chemotherapy alone could either not be confirmed or there was evidence of nonproportionality. Schoenfeld tests also suggested that the proportional hazards were either inconclusive or nonlinear. While the sponsor indicated that dependent curves could be used for OS, given that the different mechanisms of action between the treatment groups may result in different hazards of death over time (durvalumab and tremelimumab plus platinum-based chemotherapy and platinum-based chemotherapy alone), independent curves were used in the base case. Several parametric functions were fitted to the OS and PFS data to select candidate distributions based on Akaike and Bayesian information criteria and visual inspections of the curves. For OS, the sponsor selected nonparametric spline models, suggesting these captured the tail of the Kaplan-Meier curve well; the spline-hazard (2 knots) and spline-normal (3 knots) curves were selected to model durvalumab and tremelimumab plus platinum-based chemotherapy and platinum-based chemotherapy, respectively. For PFS, the sponsor selected nonparametric spline models: the spline-normal (3 knots) and spline-odds (3 knots) curves for modelling durvalumab and tremelimumab plus platinum-based chemotherapy, respectively.
Time to treatment discontinuation was defined in the POSEIDON trial as the time from randomization to discontinuation of study treatment or death in the absence of discontinuation. As a result, the sponsor modelled time to treatment discontinuation independently of PFS. Based on the Akaike or Bayesian information criteria and visual inspection of the curves, the sponsor selected nonparametric spline models; spline-hazard (2 knots) curves were selected for modelling durvalumab and tremelimumab plus platinum-based chemotherapy and platinum-based chemotherapy.
The sponsor obtained relative effects for durvalumab and tremelimumab plus platinum-based chemotherapy versus nivolumab and ipilimumab plus platinum-based chemotherapy, pembrolizumab plus platinum-based chemotherapy, and cemiplimab plus platinum-based chemotherapy from MAICs using a propensity-score weighting approach. Hazard ratios (HRs) derived from the MAIC were used to estimate the OS and PFS of nivolumab and ipilimumab plus platinum-based chemotherapy, pembrolizumab plus platinum-based chemotherapy, and cemiplimab plus platinum-based chemotherapy relative to durvalumab and tremelimumab plus platinum-based chemotherapy. A weighted-average approach using data from squamous trials (37.1%) and nonsquamous trials (62.9%) was used to derive costs and outcomes associated with pembrolizumab plus platinum-based chemotherapy to represent the ITT population. As the relative time to treatment discontinuation was not assessed in the MAICs, the sponsor applied the HRs from the PFS analyses to the durvalumab and tremelimumab plus platinum-based chemotherapy time-to-treatment-discontinuation curve.
In its base case, based on the maturity of the durvalumab and tremelimumab plus platinum-based chemotherapy data from the POSEIDON trial (86.4% OS maturity and 70.4% PFS maturity) showing sustained treatment benefit observed in 5-year data, the sponsor assumed the predicted benefit associated with durvalumab and tremelimumab plus platinum-based chemotherapy would be maintained throughout the lifetime horizon of the model. Alternatively, stopping rules are recommended for nivolumab and ipilimumab plus platinum-based chemotherapy, pembrolizumab plus platinum-based chemotherapy, and cemiplimab plus platinum-based chemotherapy, and a cap was placed on OS and PFS benefits for these treatments at 5 years from treatment initiation.
Overall QALYs were calculated as the utility-weighted time in each health state, with a half-cycle correction and discounting applied. Overall health states were calculated from the utility of the health state and disutilities due to adverse events (AEs). Utility values used in the base-case analysis were based on progression status (PFS and progressive disease) regardless of the treatment arm and were obtained from the EQ-5D-5L data collected in the POSEIDON trial. Mixed models for repeated measures were used to estimate utility values from the EQ-5D-5L data and were adjusted to Canadian value sets developed by Xie et al.9 Utility values for the preprogression and postprogression health states were █████ and █████, respectively. AEs considered in the analysis were those that were treatment-related, grade 3 or 4, and occurred in at least 2% of any treatment arm. The most reported AEs in any treatment arm were anemia, neutropenia, and thrombocytopenia. AE disutilities were used to capture the negative effects of AEs on health-related quality of life. The total decrement from AE-related disutilities was calculated using the published disutility values and the estimated duration for each AE. Mean durations of each event were derived from the POSEIDON trial and supplemented by values from the literature and assumptions.10,11 An age adjustment to health-state utilities using an age-based utility multiplier12 was implemented in the base case and was based on the Canadian EQ-5D-5L valuation study.13
Costs included drug acquisition and administration for both first-line and subsequent therapies, resource use, the management of AEs, and terminal care. Drug acquisition costs for tremelimumab were derived from sponsor data on file, and remaining drug costs were derived from the IQVIA Delta PA database.14 Relative dose intensity for all treatments was assumed to be 100%. Chemotherapy was characterized as a grouped comparator, based on the POSEIDON trial,4 the CheckMate-9LA trial,7 the KEYNOTE-189 trial,15 the KEYNOTE-407 trial,5 and the EMPOWER-Lung 3 trial.8 To estimate costs and outcomes for pembrolizumab plus PCC, a weighted-average approach was used, based on the NSCC versus SCC distribution in the POSEIDON trial. Immuno-oncology therapy costs were capped after 2 years (104 weeks) in the sponsor-submitted model. The costs of subsequent therapy were calculated per line, using the distribution of treatments utilized, and the duration of those treatments in each line. Administration costs were obtained from the Ontario Schedule of Benefits for Physician Services,16 and costs of managing AEs were sourced from OCCI 2015 and 2017, and from Meyers et al.16-18 End-of-life (terminal care) costs were included as a one-time cost, applied at the time of death, sourced from Chaudhary et al. (2021).19
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically with 1,000 iterations. Deterministic and probabilistic results were consistent. The probabilistic findings are presented in the following section.
Base-Case Results
The submitted analyses were based on publicly available prices of the comparator treatments. Results from the base case of the submitted economic evaluation are presented in Table 3. Additional results from the sponsor’s submitted economic evaluation base case are presented in Appendix 3.
In the sponsor’s base-case analysis, over a 20-year time horizon, the expected total costs and QALYs for durvalumab and tremelimumab plus platinum-based chemotherapy were $343,237 and 2.37, respectively. Compared to platinum-based chemotherapy, durvalumab and tremelimumab plus platinum-based chemotherapy resulted in 0.95 QALYs gained at an incremental cost of $266,150, for an estimated ICER of $279,849 per QALY gained. Given the sponsor’s comparison to other immuno-oncology plus platinum-based chemotherapy regimens via MAICs, all results were presented as pairwise analyses. When compared to pembrolizumab plus platinum-based chemotherapy, the incremental QALYs from durvalumab and tremelimumab plus platinum-based chemotherapy were 0.41 at an increased cost of $126,805, resulting in an ICER of $307,101 per QALY gained. When compared to nivolumab and ipilimumab plus platinum-based chemotherapy, durvalumab and tremelimumab plus platinum-based chemotherapy generated 0.63 more QALYs at an increased cost of $110,188, resulting in an ICER of $174,337 per QALY gained. When compared to cemiplimab plus platinum-based chemotherapy, durvalumab and tremelimumab plus platinum-based chemotherapy generated 0.21 more QALYs at an increased cost of $142,086, resulting in an ICER of $680,318 per QALY gained. The majority of the incremental benefit (between 63% and 104%) gained by durvalumab and tremelimumab plus platinum-based chemotherapy was incurred in the extrapolated period (after the 5-year data cut in the POSEIDON trial).
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Total QALYs | ICER ($ per QALY) |
|---|---|---|---|
Durvalumab and tremelimumab + PBC vs. PBC | |||
PBC | 77,088 | 1.42 | Reference |
Durvalumab and tremelimumab + PBC | 343,237 | 2.37 | 279,849 |
Durvalumab and tremelimumab + PBC vs. cemiplimab + PBC | |||
Cemiplimab + PBC | 201,151 | 2.16 | Reference |
Durvalumab and tremelimumab + PBC | 343,237 | 2.37 | 680,318 |
Durvalumab and tremelimumab + PBC vs. pembrolizumab plus PBC | |||
Pembrolizumab plus PBC | 216,432 | 1.96 | Reference |
Durvalumab and tremelimumab + PBC | 343,237 | 2.37 | 307,101 |
Durvalumab and tremelimumab + PBC vs. nivolumab and ipilimumab + PBC | |||
Nivolumab and ipilimumab + PBC | 233,050 | 1.74 | Reference |
Durvalumab and tremelimumab + PBC | 343,237 | 2.37 | 174,337 |
ICER = incremental cost-effectiveness ratio; PBC = platinum-based chemotherapy; QALY = quality-adjusted life-year; vs. = versus.
Source: Sponsor’s pharmacoeconomic submission.1
Additional results from the sponsor’s submitted economic evaluation base case are presented in Appendix 3.
Sensitivity and Scenario Analysis Results
The sponsor performed scenario analyses to assess the impact of the model’s structural and methodological assumptions, using a spline-normal OS curve, assuming a steeper decline in OS (e.g., with a gamma curve), and changing assumptions regarding utility decrements related to AEs. Key drivers of the cost-effectiveness of durvalumab and tremelimumab plus platinum-based chemotherapy were related to utilities and disease-management costs in the progression-free health state, the distribution of males in the baseline characteristics, and the pemetrexed maintenance for durvalumab and tremelimumab plus platinum-based chemotherapy. No scenario analysis was conducted using a perspective other than that of the health care payer.
CDA-AMC Appraisal of the Sponsor’s Economic Evaluation
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis:
The comparative efficacy of relevant comparators versus durvalumab and tremelimumab plus platinum-based chemotherapy is uncertain: In the absence of head-to-head evidence for durvalumab and tremelimumab plus platinum-based chemotherapy compared with other immuno-oncology therapies in combination with chemotherapy (nivolumab and ipilimumab plus platinum-based chemotherapy, pembrolizumab plus platinum-based chemotherapy and cemiplimab plus platinum-based chemotherapy), the sponsor used MAICs to inform the relative efficacy of these comparators in the submitted model. Based on the sponsor-submitted MAICs, there were no statistically significant differences in terms of OS and PFS, although the results ███████████ ████████ cemiplimab plus platinum-based chemotherapy (PFS) and pembrolizumab plus platinum-based chemotherapy (OS and PFS for SCC, and PFS for NSCC) when compared to durvalumab and tremelimumab plus platinum-based chemotherapy (Table 7). However, the Clinical Review noted that the analysis did not adjust for important effect modifiers, such as PD-L1 status, sex, age, ECOG PS, disease stage, type of chemotherapy, and presence of brain metastases, likely leading to residual heterogeneity between studies. Additionally, the MAIC method reduced the effective sample size, resulting in wide confidence intervals. As a result, the Clinical Review reported that no conclusions could be drawn as to whether durvalumab and tremelimumab plus platinum-based chemotherapy was favoured over other comparators, or vice versa. The uncertainty in the sponsor’s MAICs directly translates into uncertainty in the modelled OS and PFS curves for nivolumab and ipilimumab plus platinum-based chemotherapy, pembrolizumab plus 6platinum-based chemotherapy, and cemiplimab plus platinum-based chemotherapy because they are directly modelled using the HRs derived from the MAICs. The clinical experts consulted for this review indicated that durvalumab and tremelimumab plus platinum-based chemotherapy is expected to have a treatment benefit similar to that of other immuno-oncology plus platinum-based chemotherapy regimens.
Given the inability to draw conclusions from the MAIC, and uncertainty in the long term effects of all comparator regimens, and the lack of more robust data, CDA-AMC was unable to address this limitation in reanalyses.
The OS and PFS benefit cap for immuno-oncology plus platinum-based chemotherapy comparator regimens is inappropriate: In the sponsor-submitted model, given that the duration of treatment was limited to 2 years for immuno-oncology therapy in line with prior recommendations from CDA-AMC,20-23 the sponsor assumed that the duration of OS and PFS benefits for immuno-oncology comparator regimens (nivolumab and ipilimumab plus platinum-based chemotherapy, cemiplimab plus platinum-based chemotherapy, and pembrolizumab plus platinum-based chemotherapy) would be capped after 5 years after initiation of treatment. However, there is published evidence of persistence of benefit with immuno-oncology therapies plus chemotherapy over at least 5 years,24-26 which is the duration of benefit observed for durvalumab and tremelimumab plus platinum-based chemotherapy within this review referenced by the sponsor in its submission. As a result, there is no robust evidence to suggest that any treatment effect for durvalumab and tremelimumab plus platinum-based chemotherapy will be maintained for longer than it would with currently available immuno-oncology therapies. This interpretation was validated by the clinical experts consulted for this review, who confirmed that the long-term duration of benefit is expected to be similar as it is unreasonable to assume that 1 regimen would have a higher risk of late recurrences (i.e., after 5 years) than another based on the available evidence. Furthermore, published literature suggests that any waning of the treatment effect for immuno-oncology therapies would be applied over time and in a transparent manner.27
CDA-AMC explored revising the sponsor’s model to assume no difference between the long-term efficacy assumptions (e.g., removing capping for the comparator immuno-oncology therapies), but the results from this analysis did not meet face validity, indicating an issue with the programming of the sponsor-submitted model. The following limitation explores this programming issue further.
Model programming results in output that lacks face validity: During the model validation process, CDA-AMC noted issues with the sponsor’s approach to modelling the comparative effectiveness of durvalumab and tremelimumab plus platinum-based chemotherapy compared with the comparators, particularly pembrolizumab. In the data trace for durvalumab and tremelimumab plus platinum-based chemotherapy derived from the ITT population in the POSEIDON trial, OS was 99.7% in the first cycle. However, when reviewing the NSCC and SCC subgroup populations (indicated to be informed from those specific subgroups within the POSEIDON trial) within the same trace, OS was 99.5% and 99.2%, respectively. This indicates that there may be patients missing between the ITT and NSCC and SCC subgroup populations. To compare pembrolizumab plus platinum-based chemotherapy and durvalumab and tremelimumab plus platinum-based chemotherapy, OS was derived from the NSCC and SCC subgroup populations and a weighted analysis was undertaken in which the HRs derived from the MAIC were applied to the subgroup estimates. This resulted in patients in the pembrolizumab plus platinum-based chemotherapy treatment arm starting with a lower OS, to which the HR was applied, resulting in OS of 99.4% and 99.2% in the first cycle according to fitted curves, which was not reflective of the durvalumab and tremelimumab plus platinum-based chemotherapy treatment arm ITT population (in which OS was 99.7% in the first cycle according to the fitted curve). The same observations were apparent for PFS. To accurately investigate the relationship, the sponsor should have conducted a separate analysis that weighted the durvalumab and tremelimumab plus platinum-based chemotherapy treatment arm to align with the approach taken in the pembrolizumab plus platinum-based chemotherapy treatment arm. In other words, the sponsor should have compared pembrolizumab plus platinum-based chemotherapy for the NSCC population to durvalumab and tremelimumab plus platinum-based chemotherapy for the NSCC population. It should also have compared pembrolizumab plus platinum-based chemotherapy for the SCC population to durvalumab and tremelimumab plus platinum-based chemotherapy to the SCC population from the POSEIDON trial, and weighted durvalumab and tremelimumab plus platinum-based chemotherapy in the same manner as pembrolizumab plus platinum-based chemotherapy, as opposed to using the ITT population results for durvalumab and tremelimumab plus platinum-based chemotherapy. As a result, the sponsor’s approach is inappropriate and produces results that do not meet face validity.
When removing the 5-year cap on the treatment effect for immuno-oncology plus platinum-based chemotherapy comparator regimens (Table 8), given the output of the MAICs and the HRs favouring cemiplimab plus platinum-based chemotherapy and pembrolizumab plus platinum-based chemotherapy compared to durvalumab and tremelimumab plus platinum-based chemotherapy in most circumstances relevant to the model, these treatments should result in more QALYs than durvalumab and tremelimumab plus platinum-based chemotherapy (particularly in the preprogression state within the model). Table 11, highlights the disaggregated outputs of durvalumab and tremelimumab plus platinum-based chemotherapy and pembrolizumab plus platinum-based chemotherapy (NSCC and SCC) within the model. When viewing these sets of data in combination, pembrolizumab plus platinum-based chemotherapy should result in PFS and OS outcomes superior to those of durvalumab and tremelimumab plus platinum-based chemotherapy.
Additionally, to derive the results for pembrolizumab plus platinum-based chemotherapy, model users were required to run a macro after any change was made to any parameter, resulting in a lack of model transparency. If a user does not run the macro after a single change, errors within the calculation of the pembrolizumab plus platinum-based chemotherapy treatment arm are realized. Furthermore, the sponsor’s model reports multiple different probabilistic QALY results for the analysis (1 on the “incremental” tab and 1 on the “PSA” tab) and the probabilistic costs are consistent throughout.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CDA-AMC (Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CDA-AMC comment |
|---|---|
Utilities for PF and PD were derived from the POSEIDON trial. | Uncertain. Based on data obtained from the POSEIDON trial, the PF health state was calculated to have a utility value of █████, and the PD health state was calculated to have a utility value of █████. Results of the KEYNOTE-189 and KEYNOTE-407 trials for pembrolizumab plus platinum-based chemotherapy noted that utilities for PF and PD for first-line metastatic NSCC were 0.770 and 0.700, respectively, and 0.770 and 0.680 for SCC, respectively. Clinical expert feedback obtained by CDA-AMC emphasized that the utility values are likely closer to the published values for pembrolizumab plus platinum-based chemotherapy. |
Use of spline models for extrapolated OS, PFS and time-to-treatment-discontinuation outcomes. | Uncertain. In the sponsor’s submitted model, spline-hazard curves were selected for modelling durvalumab and tremelimumab plus platinum-based chemotherapy, and platinum-based chemotherapy alone curves for OS, PFS and time to treatment discontinuation in the base case. While spline models may be attractive in recognizing the nonstandard shape of both hazards and hazard ratios for the period covered by the trial duration, there is the potential that the modelling of long-term outcomes is based on extrapolation from the tail of the Kaplan-Meier curve and therefore derived from a lesser volume of data.28 |
The use of a MAIC to derive comparative efficacy for immuno-oncology plus platinum-based chemotherapy treatments meant that results should be presented as pairwise analysis instead of a sequential analysis within the base case. | Inappropriate. While in general, use of multiple MAICs should mean analyses are presented as pairwise analyses, this is a case when the values for the drug under review also differ based on the population used in the MAIC analysis. In the sponsor’s analysis, the same baseline values for durvalumab and tremelimumab plus platinum-based chemotherapy inform output for all comparisons, although the inputs informing the comparison with pembrolizumab plus platinum-based chemotherapy were derived from subgroup estimates and differed from the other immuno-oncology plus platinum-based chemotherapy comparisons. As noted in the discussion of study limitations, the sponsor should not have taken this approach when determining the efficacy of durvalumab and tremelimumab plus platinum-based chemotherapy vs. pembrolizumab plus platinum-based chemotherapy (i.e., it should have used data from the durvalumab and tremelimumab plus platinum-based chemotherapy SCC and NSCC populations to inform durvalumab and tremelimumab plus platinum-based chemotherapy for the comparison with pembrolizumab plus platinum-based chemotherapy). |
As time to treatment discontinuation was not an outcome that was considered for the MAICs, HRs obtained for PFS were then applied to the time to discontinuation curve of durvalumab and tremelimumab plus platinum-based chemotherapy (derived from the POSEIDON trial), to estimate time to treatment discontinuation for the immuno-oncology plus platinum-based chemotherapy comparators. | Uncertain. The application of HRs to the PFS derived from the POSEIDON trial for durvalumab and tremelimumab plus platinum-based chemotherapy suggests that treatment was discontinued at the time of disease progression. The clinical expert feedback obtained by CADTH noted that, for all therapies, most patients would switch to a second-line therapy at the time of disease progression, and this was not captured in the sponsor’s model. |
The target population aligned with the POSEIDON trial ITT population, which included mixed histology types and all patients irrespective of their PD-L1 levels. | Uncertain. The CDA-AMC Clinical Review noted that prespecified subgroup analyses for OS showed that the survival benefit was consistent across subgroups, except for the subgroup of never smokers, for whom durvalumab and tremelimumab plus platinum-based chemotherapy was not favoured. Additionally, a more pronounced OS benefit was observed in patients with nonsquamous histology for durvalumab and tremelimumab plus platinum-based chemotherapy vs. platinum-based chemotherapy. Clinical expert feedback emphasized that, because smoking is a strong predictor of efficacy to immunotherapy, the underperformance of the squamous subgroup may be related to the use of gemcitabine chemotherapy in the population, as gemcitabine may reduce the efficacy of immunotherapy. As a result, the assumption of equivalent benefit assumed for patients regardless of histology in the economic model may be inappropriate. |
According to the submitted deviation request, monotherapy with immuno-oncology treatments was not included as a comparator. | Uncertain. Clinical expert feedback noted that immuno-oncology monotherapy could be considered a potential comparator for the durvalumab and tremelimumab plus platinum-based chemotherapy. Additionally, at the time that the POSEIDON trial was conducted, platinum-based chemotherapy was the standard of therapy for the indication population. However, the clinical experts noted that platinum-based chemotherapy alone does not represent a reasonable comparator to durvalumab and tremelimumab plus platinum-based chemotherapy as immuno-oncology treatment is considered the backbone of therapy for first-line treatment of metastatic NSCLC in the current landscape. The clinical experts noted that patients would only receive platinum-based chemotherapy alone if they are unable to, or choose not to, receive immuno-oncology treatment. |
Durvalumab (120 mg per 2.4mL vial) and tremelimumab (25 mg per 1.25 vial) were noted in the sponsor’s submission as formulations that were not relevant for the target population; as such the formulations were not considered in the sponsor’s submission. | Uncertain. The price per mg is the same regardless of dose, and the sponsor’s assumption of vial sharing for all immuno-oncology therapies and lack of wastage means that alternative vial sizes do not affect the results. However, the model did not allow for these formulations to be used in calculations. If there is no vial sharing, the costs associated with all immuno-oncology therapies are underestimated. |
CDA-AMC = Canada’s Drug Agency; HR = hazard ratio; MAIC = matching adjusted indirect comparison; NSCC = non–squamous cell carcinoma; OS = overall survival; PD = progressed disease; PF = progression-free; PFS = progression-free survival; SCC = squamous-cell carcinoma.
CDA-AMC Reanalyses of the Economic Evaluation
Base-Case Results
Given the limitations identified with the sponsor’s economic submission, CDA-AMC was unable to derive a reliable estimate of the cost-effectiveness of durvalumab and tremelimumab plus platinum-based chemotherapy compared with other immuno-oncology therapies plus platinum-based chemotherapy, which were deemed the most relevant comparators. The estimates of relative effect obtained from the sponsor’s MAICs and assumptions regarding long-term comparative effectiveness represent the largest sources of uncertainty.
Under the assumption of equivalent clinical efficacy and safety, treatment with durvalumab and tremelimumab plus platinum-based chemotherapy results in incremental costs when compared to immuno-oncology plus platinum-based chemotherapy regimens based on the publicly available prices at the time of this submission.
Issues for Consideration
CDA-AMC reanalyses are based on a sponsor-submitted price of durvalumab and tremelimumab and the public list prices of other comparator treatments which may differ from any confidential prices paid by the participating drug plans and may influence the results of the cost-effectiveness and budget impact analysis.
Overall Conclusions
The sponsor submitted evidence from 1 pivotal randomized, placebo-controlled study (the POSEIDON study) that demonstrated that durvalumab and tremelimumab plus chemotherapy was associated with a benefit compared with chemotherapy alone in terms of OS, PFS, and objective response rate for the first-line treatment of patients with metastatic NSCLC with tumours that lack activating EGFR mutations and ALK fusions. However, the most relevant comparator for durvalumab and tremelimumab plus chemotherapy in practice in Canada was determined to be other immuno-oncology therapies in combination with platinum-based chemotherapies. In the absence of head-to-head evidence for durvalumab and tremelimumab plus platinum-based chemotherapy compared with other immuno-oncology therapies plus chemotherapy (i.e., nivolumab and ipilimumab plus platinum-based chemotherapy, pembrolizumab plus platinum-based chemotherapy, and cemiplimab plus platinum-based chemotherapy), the sponsor submitted MAICs to inform the comparative efficacy used within the economic evaluation. The MAIC results indicated that, because the credible intervals for OS and PFS crossed the null, durvalumab and tremelimumab plus platinum-based chemotherapy could be considered comparable to other immuno-oncology therapies plus platinum-based chemotherapy. However, the Clinical Review by CDA-AMC noted that the analysis did not adjust for important effect modifiers, such as PD-L1 status, sex, age, ECOG PS, disease stage, type of chemotherapy, and presence of brain metastases, likely leading to residual heterogeneity between the studies. As a result, the Clinical Review reported that no conclusions could be drawn as to whether durvalumab and tremelimumab plus platinum-based chemotherapy was favoured over other comparators, or vice versa. The clinical experts consulted for this review indicated that durvalumab and tremelimumab plus platinum-based chemotherapy is expected to have treatment benefit similar to that of other immuno-oncology plus platinum-based chemotherapy regimens.
CDA-AMC identified several limitations with the sponsor’s pharmacoeconomic analysis, which mostly resulted from limitations with the submitted comparative evidence. The sponsor suggests that durvalumab and tremelimumab plus platinum-based chemotherapy is associated with an ICER of at least $174,337 per QALY gained compared with other immuno-oncology therapies plus platinum-based chemotherapy. The incremental benefit is largely based on assumed long-term benefits from capping the treatment effect of comparator immuno-oncology therapies at a maximum of 5 years, while the incremental costs are driven by differences in the cost of treatment, and durvalumab and tremelimumab plus platinum-based chemotherapy used until disease progression or unacceptable tolerability (i.e., no maximum duration) compared with other immuno-oncology treatments being used for a maximum of 2 years. When testing the sponsor’s model (e.g., attempting to remove the sponsor’s treatment effect cap of 5 years that was applied to the comparator immuno-oncology therapies plus platinum-based chemotherapy), the results of the model did not meet face validity. For example, while pembrolizumab plus platinum-based chemotherapy showed improved PFS relative to durvalumab and tremelimumab plus platinum-based chemotherapy based on the point estimates from the MAIC, the modelled results suggested durvalumab and tremelimumab plus platinum-based chemotherapy was associated with greater QALYs in the PFS health state. As such, any changes made to the model could not be interpreted with accuracy, and the results of the submitted cost-utility analysis could not be effectively validated. Overall, there is insufficient clinical and economic evidence to suggest that durvalumab and tremelimumab plus platinum-based chemotherapy should cost more than currently reimbursed immuno-oncology plus platinum-based chemotherapy regimens for adult patients with metastatic NSCLC that lack activating EGFR mutations and ALK fusions and who have not received prior therapy for metastatic disease.
References
1.AstraZeneca Canada Inc. Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Imfinzi (durvalumab), 50 mg/mL, concentrate for solution for intravenous infusion and Imjudo (tremelimumab), 20 mg/mL, concentrate for solution for intravenous infusion. October 17, 2024.
2.AstraZeneca Canada, Inc. Durvalumab (Imfinzi) Product Monograph. Submitted to Health Canada [accessed by sponsor]. 2023. https://pdf.hres.ca/dpd_pm/00073952.PDF
3.AstraZeneca Canada Inc. Imjudo (tremelimumab), 20 mg/mL, concentrate for solution for intravenous infusion [product monograph]. August 31, 2023.
4.AstraZeneca. Clinical Study Report: A Phase III, Randomized, Multi-Center, Open-Label, Comparative Global Study to Determine the Efficacy of Durvalumab or Durvalumab and Tremelimumab in Combination With Platinum-Based Chemotherapy for First-Line Treatment in Patients With Metastatic Non-Small-Cell Lung Cancer (NSCLC) (POSEIDON) [internal sponsor's report]. 2021.
5.Novello S, Kowalski DM, Luft A, et al. Pembrolizumab Plus Chemotherapy in Squamous Non-Small-Cell Lung Cancer: 5-Year Update of the Phase III KEYNOTE-407 Study. J Clin Oncol. 2023;41(11):1999-2006. doi: 10.1200/JCO.22.01990 PubMed
6.Garassino MC, Gadgeel S, Speranza G, et al. Pembrolizumab Plus Pemetrexed and Platinum in Nonsquamous Non-Small-Cell Lung Cancer: 5-Year Outcomes From the Phase 3 KEYNOTE-189 Study. J Clin Oncol. 2023;41(11):1992-1998. doi: 10.1200/JCO.22.01989 PubMed
7.Paz-Ares L, Ciuleanu TE, Cobo M, et al. First-line nivolumab plus ipilimumab combined with two cycles of chemotherapy in patients with non-small-cell lung cancer (CheckMate 9LA): an international, randomised, open-label, phase 3 trial. Lancet Oncol. 2021;22(2):198-211. doi: 10.1016/S1470-2045(20)30641-0 PubMed
8.Makharadze T, Gogishvili M, Melkadze T, et al. Cemiplimab Plus Chemotherapy Versus Chemotherapy Alone in Advanced NSCLC: 2-Year Follow-Up From the Phase 3 EMPOWER-Lung 3 Part 2 Trial. J Thorac Oncol. 2023;18(6):755-768. doi: 10.1016/j.jtho.2023.03.008 PubMed
9.Xie F, Pullenayegum E, Gaebel K, et al. A Time Trade-off-derived Value Set of the EQ-5D-5L for Canada. Med Care. 2016;54(1):98-105. doi: 10.1097/MLR.0000000000000447 PubMed
10.Lee MY, Kang HJ, Park SY, Kim HL, Han E, Lee EK. Cost-effectiveness of tolvaptan for euvolemic or hypervolemic hyponatremia. Clin Ther. 2014;36(9):1183-94. doi: 10.1016/j.clinthera.2014.07.010 PubMed
11.Misra UK, Kalita J, Nair PP. Diagnostic approach to peripheral neuropathy. Ann Indian Acad Neurol. 2008;11(2):89-97. doi: 10.4103/0972-2327.41875 PubMed
12.Guidelines for the economic evaluation of health technologies: Canada [accessed by sponsor]. 4th ed. CADTH; 2017. https://www.cda-amc.ca/sites/default/files/pdf/guidelines_for_the_economic_evaluation_of_health_technologies_canada_4th_ed.pdf
13.Yan J, Xie S, Johnson JA, et al. Canada population norms for the EQ-5D-5L. Eur J Health Econ. 2024;25(1):147-155. doi: 10.1007/s10198-023-01570-1 PubMed
14.DeltaPA [accessed by sponsor]. IQVIA; 2022. https://www.iqvia.com/
15.Gandhi L, Rodriguez-Abreu D, Gadgeel S, et al. Pembrolizumab plus Chemotherapy in Metastatic Non-Small-Cell Lung Cancer. N Engl J Med. 2018;378(22):2078-2092. doi: 10.1056/NEJMoa1801005 PubMed
16.Government of Ontario. OHIP Schedule of Benefits and Fees [accessed by sponsor]. 2024. https://www.ontario.ca/page/ohip-schedule-benefits-and-fees
17.Government of Ontario. Ontario Case Costing Initiative (OCCI) [accessed by sponsor]. 2017. https://data.ontario.ca/en/dataset/ontario-case-costing-initiative-occi
18.Meyers BM, Vogel A, Marotta P, et al. The Cost-Effectiveness of Lenvatinib in the Treatment of Advanced or Unresectable Hepatocellular Carcinoma from a Canadian Perspective. Can J Gastroenterol Hepatol. 2021;2021:8811018. doi: 10.1155/2021/8811018 PubMed
19.Chaudhary MA, Holmberg C, Lakhdari K, et al. Cost-effectiveness of nivolumab in squamous and non-squamous non-small cell lung cancer in Canada and Sweden: an update with 5-year data. J Med Econ. 2021;24(1):607-619. doi: 10.1080/13696998.2021.1917139 PubMed
20.CADTH Reimbursement Recommendation: Cemiplimab (Libtayo). Can J Health Technol. 2024;4(5). doi:10.51731/cjht.2024.896
21.pCODR Expert Review Committee (pERC) Final Recommendation: Pembrolizumab (Keytruda) for Squamous Non-Small Cell Lung Cancer [accessed by sponsor]. 2019. https://www.cadth.ca/sites/default/files/pcodr/Reviews2019/10176PembrolizumabSQ-NSCLC_fnRec_NOREDACT_ApprovedbyChair_Post_03Jan2020_final.pdf
22.pCODR Expert Review Committee (pERC) Final Recommendation: Pembrolizumab (Keytruda) Non-squamous cell lung cancer [accessed by sponsor]. CADTH; 2019. https://www.cadth.ca/sites/default/files/pcodr/Reviews2019/10153PembroNSQ-NSCLC_FnRec_approvedbyChair_Post_NOREDACT_31May2019_final.pdf
23.pCODR Expert Review Committee (pERC) Final Recommendation: Nivolumab (Opdivo) Plus Ipililumab (Yervoy) for Non-Small Cell Lung Cancer [accessed by sponsor]. CADTH; 2021. https://www.cadth.ca/sites/default/files/pcodr/Reviews2021/10218Nivolumab-IpilimumabNSCLC_FnRec_pERC%20Chair%20Approved_redact%20Post04Mar2021_final.pdf
24.de Castro G, Jr., Kudaba I, Wu YL, et al. Five-Year Outcomes With Pembrolizumab Versus Chemotherapy as First-Line Therapy in Patients With Non-Small-Cell Lung Cancer and Programmed Death Ligand-1 Tumor Proportion Score >/= 1% in the KEYNOTE-042 Study. J Clin Oncol. 2023;41(11):1986-1991. doi: 10.1200/JCO.21.02885 PubMed
25.Cali Daylan AE, Halmos B. Long-term benefit of immunotherapy in metastatic non-small cell lung cancer: the tale of the tail. Transl Lung Cancer Res. 2023;12(7):1636-1642. doi: 10.21037/tlcr-23-245 PubMed
26.Brahmer JR, Lee JS, Ciuleanu TE, et al. Five-Year Survival Outcomes With Nivolumab Plus Ipilimumab Versus Chemotherapy as First-Line Treatment for Metastatic Non-Small-Cell Lung Cancer in CheckMate 227. J Clin Oncol. 2023;41(6):1200-1212. doi: 10.1200/JCO.22.01503 PubMed
27.Taylor K, Latimer NR, Douglas T, et al. Treatment Effect Waning in Immuno-oncology Health Technology Assessments: A Review of Assumptions and Supporting Evidence with Proposals to Guide Modelling. Pharmacoeconomics. 2024;42(11):1181-1196. doi: 10.1007/s40273-024-01423-6 PubMed
28.Coyle D, Haines A, Lee K. Extrapolating Clinical Evidence Within Economic Evaluations: CADTH Methods and Guidelines. Can J Health Technol. 2023;3(5). doi: 10.51731/cjht.2023.649 PubMed
29.Cancer Care Ontario. Funded evidence-informed regimens. 2024. Accessed January 2, 2025. https://www.cancercareontario.ca/en/drugformulary/regimens
30.AstraZeneca Canada Inc. Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Imfinzi (durvalumab), 50 mg/mL, concentrate for solution for intravenous infusion and Imjudo (tremelimumab), 20 mg/mL, concentrate for solution for intravenous infusion. October 17, 2024.
31.Statistics Canada. Population estimates on July 1st, by age and sex. Accessed June 20, 2023. https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1710000501
32.Statistics Canada. Number of new cases and age-standardized rates of primary cancer, by cancer type and sex [accessed by sponsor]. https://www150.statcan.gc.ca/t1/tbl1/en/cv.action?pid=1310074701
33.Canadian Cancer Statistics Advisory Committee. Canadian Cancer Statistics: A 2020 special report on lung cancer [accessed by sponsor]. Canadian Cancer Society; 2020. https://cancer.ca/Canadian-Cancer-Statistics-2020-EN
34.AACR Project GENIE Consortium. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017;7(8):818-831. doi: 10.1158/2159-8290.CD-17-0151 PubMed
35.INESSS. Cancer du poumon non à petites cellules. Avis transmis au ministre en octobre 2022 [accessed by sponsor]. https://www.inesss.qc.ca/fileadmin/doc/INESSS/Inscription_medicaments/Avis_au_ministre/Novembre_2022/Libtayo_CPNPC_2022_10.pdf
36.Bigras G, Mairs S, Swanson PE, Morel D, Lai R, Izevbaye I. Small Biopsies Misclassify up to 35% of PD-L1 Assessments in Advanced Lung Non-Small Cell Lung Carcinomas. Appl Immunohistochem Mol Morphol. 2018;26(10):701-708. doi: 10.1097/PAI.0000000000000698 PubMed
37.Brenner DR, Gillis J, Demers AA, et al. Projected estimates of cancer in Canada in 2024. CMAJ. 2024;196(18):E615-E623. doi: 10.1503/cmaj.240095 PubMed
Appendix 1: Cost-Comparison Table
Please note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical experts and drug plan. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 5: CDA-AMC Cost-Comparison Table for Metastatic NSCLC
Treatment | Strength | Form | Price ($) | Recommended dosage | Average daily cost ($) | Average 21-day course ($) |
|---|---|---|---|---|---|---|
Durvalumab + tremelimumab + PBC | ||||||
Durvalumab (Imfinzi) | 50 mg/mL | 10 mL (500 mg) vial for IV infusion | 3,911.1100 | 1,500 mg on day 1 every 3 weeks for 4 cycles, then day 1 of each 28-day cycle, until loss of clinical benefit | 558.73 | 11,733 |
Tremelimumab (Imjudo) | 20 mg/mL | 15 mL (300 mg) vial for IV infusion | 2,859.9700 | 75 mg on day 1 every 3 weeks for 4 cycles, then 1 dose at week 16 (5 total cycles) | 408.57 | 8,580 |
Cisplatin + pemetrexed + durvalumab + tremelimumab (PBC for a total of 24 cycles) | 1,008.01 | 21,168 | ||||
Carboplatin + pemetrexed + durvalumab + tremelimumab (carboplatin dose: AUC 5; PBC for a total of 4 cycles) | 1,035.63 | 21,748 | ||||
Cisplatin + paclitaxel + durvalumab + tremelimumab (paclitaxel dose: 200 mg/m2; PBC for a total of 4 cycles) | 1,193.25 | 25,058 | ||||
Carboplatin + paclitaxel + durvalumab + tremelimumab (paclitaxel dose: 200 mg/m2; carboplatin dose: AUC 6; PBC for a total of 4 cycles) | 1,230.87 | 25,848 | ||||
Carboplatin + nab-paclitaxel + durvalumab + tremelimumab (PBC for a total of 4 cycles) | 1,035.92 | 21,754 | ||||
Cisplatin + gemcitabine+ durvalumab + tremelimumab (PBC for a total of 4 cycles) | 1,065.63 | 22,373 | ||||
Carboplatin + gemcitabine+ durvalumab + tremelimumab (PBC for a total of 4 cycles) | 1,038.01 | 21,798 | ||||
Pemetrexed (MNT) (until disease progression) | 21.43 | 450 | ||||
Cemi + PBC | ||||||
Cemiplimab (Cemi) (Libtayo) | 50 mg/mL IV infusion | 7 mL Vial | 8,200.0000 | 350 mg on Day 1 every 3 weeks for a total 24 months | 390.48 | 8,200 |
Cisplatin + pemetrexed + Cemi (PBC for a total of 4 cycles) | 431.19 | 9,055 | ||||
Carboplatin + pemetrexed + Cemi (carboplatin dose: AUC 5; PBC for a total of 4 cycles) | 458.81 | 9,635 | ||||
Cisplatin + paclitaxel + Cemi (paclitaxel dose: 200 mg/m2; PBC for a total of 4 cycles) | 616.43 | 12,945 | ||||
Carboplatin + paclitaxel + Cemi (paclitaxel dose: 200 mg/m2; carboplatin dose: 750 mg; PBC for a total of 4 cycles) | 654.05 | 13,735 | ||||
Pemetrexed (MNT) (until disease progression) | 21.43 | 450 | ||||
Platinum-based chemotherapy (PBC alone) | ||||||
Gemcitabine (generic) | 1,000 mg vial 2,000 mg vial | 40 mg/mL lyophilized powder | 270.0000 540.0000 | 1,000 mg/ m2 on Day 1 and 8 every 3 weeks | 51.43 | 1,080 |
Pemetrexed (generic) | 100 mg vial 500 mg vial | 10 mg/mL IV solution | 50.0000 250.0000 | 500 mg /m2 on Day 1 every 3 weeks | 21.43 | 450 |
Nab-paclitaxel (Abraxane) | 50 mL vial | 100 mg/mL IV solution | 450.0000 | 100 mg/m2 on Day 1, 8 and 15 every 3 weeks | 21.71 | 456 |
Paclitaxel (generic) | 30 mg vial 96 mg vial 150 mg vial 300 mg vial | 6 mg/mL IV solution | 300.0000 1,196.8000 1,870.0000 3,740.0000 | 175 to 200 mg /m2 on Day 1 every 3 weeks | 192.38 to 206.67 | 4,040 to 4,340 |
Cisplatin (generic) | 50 mg vial 100 mg vial | 1 mg/mL IV solution | 135.0000 270.0000 | 75 mg /m2 on Day 1 every 3 weeks | 19.29 | 405 |
Carboplatin (generic) | 50 mg vial 150 mg vial 450 mg vial 600 mg vial | 10 mg/mL IV solution | 70.0000 210.0000 600.0000 775.0000 | AUC 5 to 6 on Day 1 every 3 weeks | 46.90 to 56.90 | 985 to 1,195 |
Cisplatin + pemetrexed (for a total of 3 cycles) | 40.71 | 855 | ||||
Carboplatin + pemetrexed (carboplatin dose: AUC 5; for a total of 3 cycles) | 68.33 | 1,435 | ||||
Cisplatin + paclitaxel (paclitaxel dose: 200 mg/m2; for a total of 3 cycles) | 225.95 | 4,745 | ||||
Carboplatin + paclitaxel (paclitaxel dose: 200 mg/m2; carboplatin dose: AUC 6; for a total of 4 to 6 cycles) | 263.57 | 5,535 | ||||
Carboplatin + nab-paclitaxel (carboplatin dose: AUC 6; for a total of 4 to 6 cycles) | 68.62 | 1,441 | ||||
Gemcitabine + carboplatin (for a total of 4 to 6 cycles) | 98.33 | 2,065 | ||||
Gemcitabine + cisplatin (for a total of 4 to 6 cycles) | 70.71 | 1,485 | ||||
Pemetrexed (MNT) (until disease progression) | 21.43 | 450 | ||||
Pembro + PBC | ||||||
Pembrolizumab (Pembro) (Keytruda) | 100 mg vial | 100 mg / 4 mL IV solution | 4,400.0000a | 2 mg/kg (max 200 mg) on Day 1 every 3 weeksb | 287.05 | 6,028 |
Pembro (MNT) (for a total of 24 months) | 287.05 | 6,028 | ||||
Cisplatin + pemetrexed + Pembro (PBC for a total of 4 to 6 cycles) | 327.76 | 6,883 | ||||
Carboplatin + pemetrexed + Pembro (carboplatin dose: AUC 5; for a total of 4 to 6 cycles) | 355.38 | 7,463 | ||||
Carboplatin + nab-paclitaxel + pemetrexed (carboplatin dose: AUC 6; for a total of 4 to 6 cycles) | 355.67 | 7,469 | ||||
Carboplatin + paclitaxel + Pembro (paclitaxel dose: 200 mg/m2; carboplatin dose: AUC 6; PBC for a total of 4 to 6 cycles) | 550.62 | 11,563 | ||||
Pemetrexed (MNT) (until disease progression) | 21.43 | 450 | ||||
Nivo + Ipi + PBC | ||||||
Nivolumab (Nivo) | 40 mg vial 100 mg vial | 10 mg/mL IV solution | 782.2200c 1,955.5600c | 4.5 mg/kg (max 360 mg) on Day 1 every 3 weeks | 287.05 | 6,028 |
Ipilimumab (Ipi) | 50 mg vial | 5 mg/mL IV solution | 5,800.0000c | Cycle 1: 1 mg/kg on Day 1 every 3 weeks Cycle 2: No dose Maintenance cycles: 1 mg/kg on Day 1 every 6 weeks | Cycle 1: 552.38 Cycle 2: 0 Maintenance cycles: 276.19 | Cycle 1: 11,600 Cycle 2: 0 Maintenance cycles: 5,800 |
Cisplatin + pemetrexed + Nivo + Ipi (PBC for a total of 2 cycles) | Cycle 1: 880.14 Cycle 2: 327.76 | Cycle 1: 18,483 Cycle 2: 6,883 | ||||
Carboplatin + pemetrexed + Nivo + Ipi (carboplatin dose: AUC 5; PBC for a total of 2 cycles) | Cycle 1: 907.76 Cycle 2: 355.38 | Cycle 1: 19,063 Cycle 2: 7,463 | ||||
Carboplatin + paclitaxel + Nivo + Ipi (paclitaxel dose: 200 mg/m2; carboplatin dose: AUC 6; PBC for a total of 2 cycles) | Cycle 1: 1,103.00 Cycle 2: 550.62 | Cycle 1: 23,163 Cycle 2: 11,563 | ||||
Nivo + Ipi (MNT) (ipilimumab maintenance dose; for a total of 22 months followed by Cycle 1 and Cycle 2) | Maintenance cycles: 563.24 | Maintenance cycles: 11,828 | ||||
AUC = area under the curve; CDA-AMC = Canada’s Drug Agency; Cemi = cemiplimab; Ipi = ipilimumab; MNT = monotherapy; Nivo = nivolumab; PBC = platinum-based chemotherapy; Pembro = pembrolizumab.
Note: All prices are from IQVIA drug price database (accessed January 2025),14 unless otherwise indicated, and do not include dispensing fees. Dosing is based on Cancer Care Ontario Drug Formulary,29 unless otherwise indicated. Drug wastage is included, expect for pembrolizumab and nivolumab.
aCADTH review of pembrolizumab.21
bDespite a fixed-dosing in the product monograph, Cancer Care Ontario Drug Formulary references a weight-based dosing (confirmed by clinical experts and drug plans feedback as the current practice across jurisdictions) and does not include wastage.
cCADTH review of nivolumab.23
Appendix 2: Submission Quality
Please note that this appendix has not been copy-edited.
Description | Yes or no | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | Sponsor submission did not include relevant comparators in model, refer to limitation. |
Model has been adequately programmed and has sufficient face validity | No | Refer to CDA-AMC Appraisal. |
Model structure is adequate for decision problem | Yes | No comment. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | No | Refer to CDA-AMC Appraisal. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | No | Refer to CDA-AMC Appraisal. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | No | Refer to CDA-AMC Appraisal. Additionally, the pharmacoeconomic report reported information in a manner that lacked clarity. |
CDA-AMC = Canada’s Drug Agency.
Appendix 3: Additional Information on the Submitted Economic Evaluation
Please note that this appendix has not been copy-edited.
Table 7: Relative Treatment Effects of Durvalumab and Tremelimumab Plus PBC vs. Comparators
Comparator | Durvalumab + tremelimumab + PBC vs. comparator hazard ratio | |
|---|---|---|
OS | PFS | |
Base case (ITT) | ||
NIVO + IPI + PBC | ████ ██████ █████ | ████ ███████████ |
CEMI + PBC | ████ ██████ █████ | ████ ██████ █████ |
NSCC | ||
pembrolizumab plus PBC | ████ ██████ █████ | ████ ██████ █████ |
SCC | ||
pembrolizumab plus PBC | ████ ██████ █████ | ████ ██████ █████ |
CEMI = cemiplimab; IPI = ipilimumab; ITT = intention-to-treat; NSCC = non–squamous cell carcinoma; OS = overall survival; PBC = platinum—based chemotherapy; PEMBRO = pembrolizumab; PFS = progression-free survival; SCC = squamous cell carcinoma; vs. = versus.
Source: Sponsor’s pharmacoeconomic submission.1
Table 8: Capping Rules Implemented in Model
Comparator | Base-case approach | Time of OS and PFS benefit cap |
|---|---|---|
PEMBRO + PBC | Cap applied to all patients at specified time since immuno-oncology plus PBC initiation | 5 years |
NIVO + IPI + PBC | Cap applied to all patients at specified time since immuno-oncology plus PBC initiation | 5 years |
CEMI + PBC | Cap applied to all patients at specified time since immuno-oncology plus PBC initiation | 5 years |
CEMI = cemiplimab; IPI = ipilimumab; OS = overall survival; PBC = platinum-based chemotherapy; PEMBRO = pembrolizumab; PFS = progression-free survival.
Source: Sponsor’s pharmacoeconomic submission.1
Detailed Results of the Sponsor’s Base Case
Table 9: Disaggregated Summary of the Sponsor’s Economic Evaluation Results
Parameter | Durvalumab + Tremelimumab + PBC | PBC | PEMBRO + PBC | NIVO + IPI + PBC | CEMI + PBC |
|---|---|---|---|---|---|
Discounted LYs | |||||
Total | 2.87 | 1.75 | 2.37 | 2.11 | 2.61 |
Preprogression | 1.89 | 0.84 | 1.69 | 1.49 | 2.03 |
Progressed disease | 0.99 | 0.91 | 0.68 | 0.62 | 0.58 |
Discounted QALYs | |||||
Total | 2.37 | 1.42 | 1.96 | 1.74 | 2.16 |
Preprogression | 1.58 | 0.70 | 1.41 | 1.24 | 1.70 |
Progressed disease | 0.79 | 0.73 | 0.55 | 0.50 | 0.47 |
AE disutilities | −0.004 | −0.004 | −0.003 | −0.002 | −0.001 |
Discounted costs ($) | |||||
Total | 343,237 | 77,088 | 216,432 | 233,050 | 201,151 |
Acquisition (first-line) | 272,289 | 7,772 | 152,563 | 172,995 | 137,358 |
Acquisition (subsequent) | 5,388 | 16,806 | 5,682 | 5,419 | 5,680 |
Administration (first-line) | 1,462 | 388 | 1,148 | 955 | 1,142 |
Administration (subsequent) | 142 | 254 | 167 | 143 | 168 |
Radiotherapy | 53 | 71 | 66 | 53 | 66 |
Disease management | 33,814 | 21,145 | 27,900 | 24,751 | 30,243 |
AEs | 7,183 | 6,844 | 5,449 | 5,112 | 3,169 |
Terminal care | 22,906 | 23,856 | 23,457 | 23,621 | 23,324 |
AE = adverse event; CEMI = cemiplimab; IPI = ipilimumab; LY = life-year; PBC = platinum-based chemotherapy; PEMBRO = pembrolizumab; QALY = quality-adjusted life-year.
Source: Sponsor’s pharmacoeconomic submission.1
Appendix 4: Additional Details on the CDA-AMC Reanalyses and Sensitivity Analyses of the Economic Evaluation
Please note that this appendix has not been copy-edited.
Results of CDA-AMC Exploratory Results
Table 10: Summary of the Exploratory Analysis Removing the Treatment Benefit Cap
Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|
Durvalumab + tremelimumab + PBC vs. PBC | |||
PBC | 77,364 | 1.43 | Reference |
Durvalumab + tremelimumab + PBC | 344,703 | 2.39 | 279,565 |
Durvalumab + tremelimumab + PBC vs. CEMI + PBC | |||
CEMI + PBC | 207,777 | 2.43 | Reference |
Durvalumab + tremelimumab + PBC | 344,703 | 2.39 | Dominated by CEMI + PBC |
Durvalumab + tremelimumab + PBC vs. PEMBRO + PBC | |||
PEMBRO + PBC | 219,681 | 2.15 | Reference |
Durvalumab + tremelimumab + PBC | 344,703 | 2.39 | 527,542 |
Durvalumab + tremelimumab + PBC vs. NIVO + IPI + PBC | |||
NIVO + IPI + PBC | 237,681 | 1.86 | Reference |
Durvalumab + tremelimumab + PBC | 344,703 | 2.39 | 205,096 |
CDA-AMC = Canada’s Drug Agency; CEMI = cemiplimab; ICER = incremental cost-effectiveness ratio; IPI = ipilimumab; NIVO = nivolumab; PBC = platinum-based chemotherapy; PEMBRO = pembrolizumab; QALY = quality-adjusted life-year; vs. = versus.
To highlight the concerns related to the model output, CDA-AMC reported disaggregated results from the reanalysis of removing the 5-year treatment benefit cap, focusing on the key comparators of concern (Table 11).
Table 11: Disaggregated Summary of CDA-AMC Exploratory Analysis Results
Parameter | Durvalumab + tremelimumab + PBC | CEMI + PBC | PEMBRO + PBC |
|---|---|---|---|
Discounted LYs | |||
Total | 2.89 | 2.93 | 2.60 |
Preprogression | 1.91 | 2.27 | 1.86 |
Progressed disease | 0.98 | 0.67 | 0.74 |
Discounted QALYs | |||
Total | 2.39 | 2.43 | 2.15 |
Progression-free | 1.60 | 1.90 | 1.55 |
Progressed disease | 0.79 | 0.54 | 0.60 |
AE disutility | –0.0037 | –0.0014 | –0.0029 |
Discounted costs ($) | |||
Total | 344,703 | 207,777 | 219,681 |
Acquisition – first line | 273,371 | 140,621 | 153,524 |
Acquisition – subsequent lines | 5,377 | 5,544 | 5,675 |
Administration – first line | 1,464 | 1,153 | 1,144 |
Administration – subsequent lines | 141 | 163 | 167 |
Radiotherapy | 53 | 65 | 66 |
Disease management | 34,279 | 34,296 | 30,553 |
AEs | 7,186 | 3,172 | 5,449 |
Terminal care | 22,831 | 22,762 | 23,103 |
AE = adverse event; CDA-AMC = Canada’s Drug Agency; CEMI = cemiplimab; ICER = incremental cost-effectiveness ratio; LY = life-year; PBC = platinum-based chemotherapy; PEMBRO = pembrolizumab; QALY = quality-adjusted life-year.
Appendix 5: Submitted Budget Impact Analysis and CDA-AMC Appraisal
Please note that this appendix has not been copy-edited.
Table 12: Summary of Key Take-Aways
Key take-aways of the budget impact analysis |
|---|
|
Summary of Sponsor’s Budget Impact Analysis
In the submitted budget impact analysis (BIA), the sponsor assessed the expected budgetary impact resulting from reimbursing durvalumab and tremelimumab plus platinum-based chemotherapy for the first-line treatment of patients with metastatic NSCLC, with no sensitizing EGFR mutation or ALK genomic tumour aberrations.30 The BIA was conducted from the perspective of the Canadian public drug plans over a 3-year time horizon (2025 to 2027) using an epidemiologic approach The sponsor compared a reference scenario in which patients received either platinum-based chemotherapy, pembrolizumab + platinum-based chemotherapy, NIVO + IPI + platinum-based chemotherapy or cemiplimab + platinum-based chemotherapy, to a new drug scenario in which durvalumab and tremelimumab plus platinum-based chemotherapy was reimbursed. The sponsor’s submission only considered annual drug acquisition costs in which the costs were obtained from IQVIA.14 The sponsor’s pan-Canadian estimates reflect the aggregated results from provincial budgets (excluding Quebec). As durvalumab and tremelimumab are infused oncology products that will be administered in a hospital or infusion clinic setting, the Non-Insured Health Benefits (NIHB) program was not included. The sponsor also presented the health care payer perspective, including first and second-line therapy costs, administration, disease management and AE management costs. Key inputs to the BIA are documented in Table 13.
The following key assumptions were made by the sponsor:
Durvalumab (120 mg/2.4mL vial) and tremelimumab (25 mg/1.25 vial) were not relevant for this indication and were not considered in the BIA, aligning with the cost-utility analysis.
NIHB-covered patients would access IV NSCLC therapies through their respective provincial cancer programs.
In the sponsor’s health care payer perspective, disease management and AE management included as one-time costs and aligned with the disease-management costs in the cost-utility analysis.
In the sponsor’s health care payer perspective, to calculate the administration costs, a $75 IV administration cost, sourced from the Ontario Schedule of Benefits, was multiplied by the number of active days per cycle for each treatment.
The reimbursement of durvalumab and tremelimumab plus platinum-based chemotherapy would not capture any market shares from platinum-based chemotherapy.
Table 13: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3 if appropriate) |
|---|---|
Target population | |
Population across Canada | 31,637,83031 |
Lung cancer incidence cases | 15,02832 |
Percentage with NSCLC | 89%33 |
Percentage without EGFR mutation and ALK aberration | 75.5%34 |
Percentage stage IV | 48.4%33 |
Percentage treated | |
Percentage treated with immune checkpoint inhibitor and platinum doublet chemotherapy | 65%36 |
Previous year incidence | 76 |
Number of patients eligible for drug under review | 1,407 / 1,373 / 1,340 |
Market uptake (3 years) | |
Uptake (reference scenario) PEMBRO + PBC CEMI + PBC NIVO + IPI + PBC PBC | ██ | | ███ █ ███ ██ █ ███ █ ███ ███ █ ███ █ ███ ██ | | | | ██ |
Uptake (new drug scenario) Durvalumab + Tremelimumab + PBC PEMBRO + PBC CEMI + PBC NIVO + IPI + PBC PBC | ██ █ ██ █ ██ █████ █ █████ █ █████ ████ █ ████ █ █████ ███ █ ██ █ ██ ██ █ ██ █ ██ |
Cost of treatment (per patient, per 21-day cycle)a | |
Durvalumab + Tremelimumab + PBC PEMBRO + PBC CEMI + PBC NIVO + IPI + PBC PBC | $21,664 $10,151 $9,551 $14,973 $1,351 |
ALK = anaplastic lymphoma kinase; CEMI = cemiplimab; EGFR = epidermal growth factor receptor; IPI = ipilimumab; NSCLC = non–small cell lung cancer; NIVO = nivolumab; PBC = platinum-based chemotherapy; PEMBRO = pembrolizumab.
aCost of treatments (per patient, per 21-day cycle) were derived from the sponsor’s submitted budget impact model.30
Summary of the Sponsor’s BIA Results
The sponsor’s base case reported that the reimbursement of durvalumab and tremelimumab plus platinum-based chemotherapy for the first-line treatment of patients with metastatic NSCLC, with no sensitizing EGFR mutation or ALK genomic tumour aberrations would lead to an incremental budget impact of $1,509,715 in year 1, $1,835,978 in year 2, and $2,128,008 in year 3. The total 3-year incremental cost was $5,473,701. Scenario analyses were completed to increase the proportion of patients with advanced metastatic NSCLC treated in the first line, explore the impact of increasing and decreasing market capture of durvalumab and tremelimumab plus platinum-based chemotherapy, exclude wastage, include subsequent treatment costs, and include weight-based dosage for pembrolizumab and nivolumab. Results of the scenario analyses resulted in fluctuations of −20% to 46%. A health care payer perspective was conducted and resulted in an incremental budget impact of $6,089,743 over 3 years.
CDA-AMC Appraisal of the Sponsor’s BIA
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Derivation of eligible population does not align with literature: In the sponsor’s submitted model, lung cancer incidence cases were assumed to be 15,028 based on data from Statistics Canada.32 However, recent publications from the Canadian Cancer Society suggests that estimated incident cases for 2024 were approximately 32,100 (including Quebec), when excluding Quebec, the pan-Canadian incidence was 24,075.37 Canadian incidence cases were approximately 31,000 in 2023 (including Quebec), when excluding Quebec, the pan-Canadian incidence was 23,250. This suggests that the values assumed by the sponsor to derive the eligible population are underestimated. Additionally, the sponsor assumes that incidence cases are decreasing over time, as the rate of NSCLC is decreasing. While incidence rates may be stable or decrease over time, the absolute number of cases is impacted by changes in the size of the population. Feedback from clinical experts consulted by CDA-AMC suggested that the absolute number of cases is currently increasing in Canada.
Clinical expert feedback obtained by CDA-AMC also highlighted that the addition of recurrent cases from the previous year underestimates the total number of eligible patients, as lung cancer does not only recur after curative intent therapy for stages I-III in the year after. The experts noted that most patients that do recur, will do so sometime during the 5 years following treatment.
To address this limitation, CDA-AMC conducted a reanalysis to update lung cancer incidence to the Canadian Cancer Society values by diving the total incidence cases by the population living in Canada, removed the decline in cases over the time horizon (suggesting no change from the base year [2021] of the data), and included recurrent cases from the previous 5 years.
Market shares in reference scenario do not align with clinical expectations: In the sponsor-submitted model, the market shares for cemiplimab + platinum-based chemotherapy are estimated to be ██%, ██%, and ██% over the 3-year time horizon. Clinical expert feedback obtained by CDA-AMC suggests that a ██% increase over a 3-year time horizon, captured from PEMBRO + platinum-based chemotherapy is optimistic and does not align with clinical expectations. The experts noted that, assuming pan-Canadian Pharmaceutical Alliance negotiations are successful for cemiplimab, it is more reasonable to expect a shift of up to 10% over a 3-year time horizon.
To address this limitation, CDA-AMC conducted a scenario analysis where cemiplimab + platinum-based chemotherapy captures 2%, 5%, and 10% of the market in years 1, 2, and 3, respectively. In turn, pembrolizumab + platinum-based chemotherapy captures the remaining market, increasing to 78%, 75% and 70% in Year 1, 2, and 3, respectively.
Market uptake of durvalumab and tremelimumab plus platinum-based chemotherapy is uncertain: In the sponsor’s submitted BIA, should durvalumab and tremelimumab plus platinum-based chemotherapy be reimbursed, the sponsor assumed the regimen would capture ██%, ██% and ██% of the market in years 1, 2 and 3, respectively. Clinical expert feedback obtained by CDA-AMC noted that, as the market is already currently crowded and based on the available evidence, it is unreasonable to suggest the regimen would capture this proportion of the market. The experts noted that it is likely that the regimen would capture approximately half of what is suggested by the sponsor, over a 3-year time horizon.
To address this limitation, CDA-AMC conducted a scenario analysis where durvalumab and tremelimumab plus platinum-based chemotherapy captures 2.5%, 3.5% and 4.5% of the market in years 1, 2 and 3, respectively.
The price of drugs paid by public drug plans is uncertain: both the sponsor’s and the CDA-AMC analyses are based on publicly available list prices for all comparators and may not reflect confidential, negotiated prices.
CDA-AMC was unable to address this limitation.
CDA-AMC Reanalyses of the BIA
The CDA-AMC base case was derived by updating lung cancer incidence rates to align with the Canadian Cancer Society values. To derive the rates, the total number of patients with lung cancer across Canada (excluding Quebec) was divided by the total population in Canada. In the sponsor submission, a 3.7% decline in the pan-Canadian population was included to reduce lung cancer incidence rates over the time horizon. To align with clinical expert feedback and the values published by the Canadian Cancer Society, CDA-AMC removed the percent decline in cases. Lastly, as the clinical experts consulted by CDA-AMC highlighted that only including recurrent cases from the previous year underestimates the total number of eligible patients, CDA-AMC included recurrent cases from the previous 5 years.
Table 14: CDA-AMC Revisions to the Submitted BIA
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
No corrections. | — | — |
Changes to derive the CDA-AMC base case | ||
1. Lung cancer incidence | 2023: 0.0517% (population n = 15,405) 2024: 0.0495% (population n = 15,028) | 2023: 0.0748% (population n = 23,250) 2024: 0.0761% (population n = 24,075) |
2. Removal of decline in cases | Pan-Canadian: –3.7% | Pan-Canadian: 0% |
3. Recurrent cases | Only including for previous year | Including for past 5 years |
CDA-AMC base-case | Reanalysis 1 + 2 + 3 | |
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency.
The results of the CDA-AMC step-wise reanalysis are presented in summary format in Table 15 and a more detailed breakdown is presented in Table 16. The CDA-AMC reanalysis results in an increase in the budget impact, with an estimated 3-year total impact of $11,508,110, which stems from the increase in total eligible patients. In the sponsor’s base case, the total numbers of eligible patients were 1,407; 1,373; and 1,340 in years 1, 2, and 3, respectively. In the CDA-AMC reanalysis, the total numbers of eligible patients were 2,822; 2,871; and 2,922 in years 1, 2, and 3, respectively.
Table 15: Summary of the CDA-AMC Reanalyses of the BIA
Stepped analysis | Three-year total ($) |
|---|---|
Submitted base case | 5,473,701 |
CDA-AMC reanalysis 1 | 8,758,321 |
CDA-AMC reanalysis 2 | 6,801,563 |
CDA-AMC reanalysis 3 | 6,629,319 |
CDA-AMC base case | 11,508,110 |
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency.
CADTH conducted the following scenario analyses to address remaining uncertainty, using the CDA-AMC base case (results are provided in Table 16):
Assuming a decrease in the market shares captured by cemiplimab + platinum-based chemotherapy in the reference and new drug scenario, an increase in the market shares captured by pembrolizumab + platinum-based chemotherapy in the reference and new drug scenario, and a decrease in the market capture of durvalumab and tremelimumab plus platinum-based chemotherapy in the new drug scenario to align with clinical expert feedback.
Table 16: Detailed Breakdown of the CDA-AMC Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|---|
Submitted base case | Reference | 171,831,315 | 165,461,716 | 159,321,080 | 153,400,634 | 478,183,430 |
New drug | 171,831,315 | 166,971,431 | 161,157,058 | 155,528,642 | 483,657,131 | |
Budget impact | 0 | 1,509,715 | 1,835,978 | 2,128,008 | 5,473,701 | |
CDA-AMC base case | Reference | 329,207,596 | 331,834,758 | 333,205,158 | 334,523,758 | 999,563,673 |
New drug | 329,207,596 | 334,862,503 | 337,044,934 | 339,164,347 | 1,011,071,784 | |
Budget impact | 0 | 3,027,745 | 3,839,776 | 4,640,589 | 11,508,110 | |
CDA-AMC scenario analysis 1: Market shares | Reference | 329,207,596 | 334,461,254 | 337,659,491 | 339,056,310 | 1,011,177,054 |
New drug | 329,207,596 | 335,756,252 | 341,637,175 | 342,037,074 | 1,019,430,501 | |
Budget impact | 0 | 1,294,998 | 3,977,684 | 2,980,765 | 8,253,447 |
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.