Drugs, Health Technologies, Health Systems
Reimbursement Review
Durvalumab (Imfinzi)
Sponsor: AstraZeneca Canada Inc.
Therapeutic area: Limited-stage small cell lung cancer
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
AE
adverse event
BICR
blinded independent central review
CCSN
Canadian Cancer Survivor Network
CDA-AMC
Canada’s Drug Agency
CI
confidence interval
CRT
chemoradiotherapy
DOR
duration of response
ECOG
Eastern Cooperative Oncology Group
EORTC QLQ-C30
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30
EORTC QLQ-LC13
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Lung Cancer 13
ES-SCLC
extensive-stage small cell lung cancer
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HR
hazard ratio
HRQoL
health-related quality of life
LCC
Lung Cancer Canada
LCC MAC
Lung Cancer Canada Medical Advisory Committee
LHF
Lung Health Foundation
LS-SCLC
limited-stage small cell lung cancer
NR
not reported
NSCLC
non–small cell lung cancer
OH (CCO)
Ontario Health (Cancer Care Ontario)
OLA
Ontario Lung Association
OS
overall survival
PCI
prophylactic cranial irradiation
PFS
progression-free survival
PRO
patient-reported outcome
RCT
randomized controlled trial
RECIST 1.1
Response Evaluation Criteria in Solid Tumours version 1.1
SAE
serious adverse event
SCLC
small cell lung cancer
TEAE
treatment-emergent adverse event
TTDM
time to death or distant metastasis
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information on Application Submitted for Review
Item | Description |
|---|---|
Drug product | Durvalumab (Imfinzi), 50 mg/mL, concentrate for IV infusion |
Sponsor | AstraZeneca Canada Inc. |
Indication | Imfinzi (durvalumab), as monotherapy, indicated for the treatment of adult patients with limited-stage small cell lung cancer (LS-SCLC) whose disease has not progressed following platinum-based chemoradiation therapy (CRT). |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Priority review and Project Orbis |
NOC date | April 8, 2025 |
Recommended dose | 1,500 mg every 4 weeks. Therapy should continue for 24 months or until disease progression or unacceptable toxicity. |
NOC = Notice of Compliance.
Introduction
Lung cancer is the most common and deadliest cancer in Canada. It was estimated that in 2024 there would be 32,100 new cases of lung cancer in the country and that it would account for 23% of all cancer-related deaths.1 Small cell lung cancer (SCLC), the most aggressive form of lung cancer, accounts for 12% of all diagnoses.2-4 Approximately one-third of SCLC cases are classified as limited-stage SCLC (LS-SCLC), in which the disease is confined to the thorax and regional lymph nodes.5 Without treatment, patients with LS-SCLC have a life expectancy of only 10 to 12 weeks, and even with the current standard of care — platinum-based chemoradiotherapy (CRT) involving cisplatin or carboplatin combined with etoposide — most patients survive only 12 to 16 months.6 For patients who respond well to CRT, prophylactic cranial irradiation (PCI) may be administered to reduce the risk of brain metastases. However, nearly 90% of patients relapse after treatment, and long-term survival remains poor, with only up to 25% of patients surviving 5 years.7-12 Despite significant efforts over the past 3 decades, treatment advances have been unsuccessful, underscoring the urgent need for novel therapeutic approaches to improve outcomes and reduce progression to metastatic disease.
Based on provincial population estimates, lung cancer has a 10-year prevalence of 151.1 cases per 100,000 people, with SCLC representing 12% of these cases.12,13 Approximately 33% of SCLC cases are diagnosed as limited-stage disease, and in approximately 90% of patients with LS-SCLC, the disease is considered medically inoperable.14 Among patients with inoperable LS-SCLC, it is estimated that 80% receive CRT8 These estimates highlight the attrition that occurs throughout the treatment pathway and the limited number of patients with LS-SCLC who experience meaningful survival outcomes. Together, these statistics emphasize the aggressive nature of LS-SCLC and the critical importance of developing new strategies to improve survival and quality of life for these patients.
The objective of this report is to review and critically appraise the clinical evidence submitted by the sponsor on the efficacy and safety of durvalumab (1,500 mg administered intravenously every 4 weeks) for the treatment of LS-SCLC in patients who do not experience disease progression after platinum-based CRT. Durvalumab has previously been reviewed and has received conditional reimbursement recommendations for the following indications: extensive-stage SCLC (ES-SCLC), unresectable hepatocellular carcinoma, advanced or metastatic biliary tract cancer, and for the treatment of locally advanced non–small cell lung cancer (NSCLC) after CRT.
Perspectives of Patient, Clinicians, and Drug Programs
The information in this section is a summary of the input provided by the patient and clinician groups who responded to the call from Canada’s Drug Agency (CDA-AMC) for input and from the clinical experts consulted by CDA-AMC for the purpose of this review.
Patient Input
CDA-AMC received a joint submission from the Canadian Cancer Survivor Network (CCSN), Lung Cancer Canada (LCC), and the Lung Health Foundation (LHF). The information was gathered through an online survey conducted from August to November 2024. There was 1 respondent to this survey, who was a patient with NSCLC who had experience with durvalumab. In November 2024, LCC also conducted 3 interviews with patients with SCLC who had direct experience with durvalumab. The patient group submitting input believed that, in the absence of input from patients with LS-SCLC, the information obtained from the survey respondent and the interviewees would still be valuable to include in the submission. In the submitted input, the survey respondent explained their experience with the disease as coughing, difficulty fighting infection, fatigue, reduced appetite, weight loss, nausea, waking up in the night or early morning because of breathing problems, feeling cold, negative impacts on emotional well-being, and excessive time spent attending medical appointments. The important outcomes identified by the survey respondent for a new treatment included reduced cost, improved quality of life, and improved energy levels. The 3 interviewees with SCLC reported their experience with the disease as coughing and noted some of the side effects they had experienced with the currently available treatments as difficulties swallowing and eating, stomach pain, voice loss, hair loss, nausea, problems completing day-to-day activities, tiredness, and hearing problems. One of the interviewees had experienced no side effects after receiving 2 treatments with durvalumab. The second interviewee, who had received 2 treatments of durvalumab through a compassionate access program, reported that he had felt more nauseous after the treatments but that his energy levels had since recovered significantly. The third interviewee only had 2 treatments of durvalumab in 2021 before he had to stop it because he had no appetite, was vomiting constantly, had diarrhea, and had lost around 22 kg of his body weight. One of the patients noted that she was relying on her pension and that if she had had to pay for durvalumab, she would not have been able to afford it.
Clinician Input
Input From Clinical Experts Consulted by CDA-AMC
The clinical experts emphasized that LS-SCLC remains an area of high unmet medical need due to the limited survival benefits of current standard treatments. After completing concurrent CRT, patients are left with surveillance as the only option, which frequently leads to disease recurrence with poor survival outcomes (median overall survival [OS] of 25 to 30 months and a 5-year survival rate of 29% to 34%). The experts highlighted the need for therapies that reduce the risk of recurrence or disease progression, particularly given the rapid progression associated with relapses.
The clinical experts indicated that durvalumab would be used as a consolidation therapy for patients who have completed CRT and whose disease has not progressed. They noted that durvalumab would be added as a consolidation therapy rather than replacing CRT, and they agreed that this would represent a significant addition to the treatment paradigm for LS-SCLC, potentially shifting standard practice.
The experts identified patients with LS-SCLC who experience complete or partial response or stable disease after CRT as the most suitable candidates for durvalumab. They noted that patients with good Eastern Cooperative Oncology Group (ECOG) Performance Status (0 or 1), minimal comorbidities, and a positive response to CRT would derive the most benefit. The inclusion of patients with medically operable stage I/II disease as candidates for durvalumab was considered reasonable based on clinical practice in Canada.
The clinical experts indicated that response to durvalumab should be assessed using imaging and clinical evaluation every 2 to 3 months. Important outcomes include progression-free survival (PFS), OS, and symptom management. A clinically meaningful response was defined as measurable improvements in survival (e.g., at least 2 additional months of PFS or OS) and symptom stabilization or improvement. The experts emphasized the importance of long-term survival data, such as 5-year OS rates, to understand the drug’s long-term impact.
The clinical experts outlined factors for discontinuing durvalumab, including evidence of disease progression; development of intolerable or potentially life-threatening immune-mediated toxicities, such as pneumonitis, colitis, hepatitis, myocarditis, or nephritis; and significant deterioration in patient quality of life. One clinical expert suggested that treatment could continue when radiologic progression is observed early after chemoradiation or within a time frame compatible with durvalumab-mediated pseudoprogression. This observation was based on the clinician’s clinical experience that posttreatment imaging may show apparent tumour enlargement due to treatment effects, such as radiation-induced inflammation or transient mediastinal mass enlargement. In such cases, a follow-up CT scan after 2 months may help determine true progression before a decision is made to discontinue treatment, provided the patient’s overall condition remains stable and their symptoms do not worsen.
The clinical experts noted that durvalumab should be prescribed by oncologists experienced in managing systemic cancer therapies and checkpoint inhibitor–related toxicities. They highlighted that initial treatments should be administered in centres equipped to manage severe immune-mediated adverse events (AEs), with subsequent cycles transitioning to outpatient settings under the supervision of trained oncology practitioners.
Clinician Group Input
CDA-AMC received 2 clinician group input submissions: 1 from the LCC Medical Advisory Committee (LCC MAC) comprising contributions from 27 clinicians and 1 from the Ontario Health (Cancer Care Ontario) (OH [CCO]) Lung Cancer Drug Advisory Committee comprising contributions from 5 clinicians. Both clinician groups agreed that the current standard treatment for LS-SCLC is 4 cycles of cytotoxic platinum-based chemotherapy (cisplatin or carboplatin) and etoposide combined with concurrent or sequential radiation and that the treatment goal is to prevent or delay disease recurrence and improve OS. The clinician input from the OH (CCO) Lung Cancer Drug Advisory Committee anticipated that durvalumab would be used after standard systemic therapy with platinum-based chemotherapy and etoposide, as well as radiation treatments. If the cancer recurs while the patient is on durvalumab, the use of more durvalumab in the metastatic setting would not occur. It was noted that the mechanism of action of durvalumab is different than that of chemotherapy or radiation therapy; therefore, durvalumab would not replace either of those therapies. The LCC MAC added that platinum-etoposide combined with either durvalumab or atezolizumab, followed by maintenance immunotherapy as monotherapy, is the standard of care in Canada for patients with ES-SCLC with good disease-related performance status and no contraindications to therapy. The OH (CCO) Lung Cancer Drug Advisory Committee believed that patients with LS-SCLC who have completed chemotherapy and radiation therapy and who have not experienced significant pneumonitis, disease progression, or autoimmune disease would be most suitable for treatment with durvalumab. Patients with poor disease-related performance status, and those who have radiation pneumonitis, would not be candidates for treatment with durvalumab. The LCC MAC added that patients who have shown disease stabilization or shrinkage after standard concurrent treatment with cytotoxic platinum–etoposide chemotherapy and thoracic radiation and patients with an ECOG Performance Status of 0 to 1 (or an ECOG Performance Status of 2 in the real-world setting) after chemotherapy and radiation would be suitable candidates. According to the OH (CCO) Lung Cancer Drug Advisory Committee input, the outcomes to determine whether a patient is responding to treatment in clinical practice include OS and disease progression based on signs, symptoms, radiology, and laboratory tests. Chest imaging (CT or X-ray) should be done every 3 to 6 months, and imaging of the abdomen, bones, brain, and pelvis should be done on a symptom-derived basis. The OH (CCO) Lung Cancer Drug Advisory Committee added that improved survival is clinically meaningful if the absolute number is greater than 5% or if the median is greater than 6 months. The LCC MAC noted that quality of life is another important outcome. The LCC MAC added that in addition to the 3-month to 4-month assessments, patients who are on durvalumab would be assessed clinically every 4 weeks before each new treatment cycle. Both clinician groups noted that disease progression and intolerable treatment-related adverse effects are the main reasons for discontinuation of durvalumab. Based on the clinician groups’ input, durvalumab after chemoradiation can be administered in an outpatient setting at a systemic therapy treatment unit and can be performed in the community oncology setting. Treatment most often would be given in a specialized cancer hospital with chemotherapy and immunotherapy experience. Treatment should be under the supervision of the appropriate oncology care team.
Drug Program Input
Input was obtained from the drug programs that participate in the CDA-AMC reimbursement review process. The following were identified as key factors that could potentially impact the implementation of a CDA-AMC recommendation for durvalumab:
relevant comparators
considerations for initiation of therapy
considerations for discontinuation of therapy
considerations for prescribing of therapy
generalizability
care provision issues
system and economic issues.
The clinical experts consulted by CDA-AMC provided advice on the potential implementation issues raised by the drug programs. Refer to Table 4.
Clinical Evidence
Systematic Review
Description of Studies
One trial, the ADRIATIC study (N = 730), was included in the sponsor’s submission. The objective of the ADRIATIC trial was to evaluate the efficacy and safety of durvalumab consolidation therapy compared with placebo in patients with LS-SCLC following concurrent CRT. This was a randomized, double-blind, placebo-controlled, phase III trial. The participants were adults who had completed CRT without experiencing disease progression and who had an ECOG Performance Status of 0 or 1. Patients were excluded if they had received prior immune checkpoint inhibitor therapy or if they had active autoimmune diseases or uncontrolled comorbidities.
The Health Canada indication and reimbursement request aligned with the trial population. Outcomes relevant to the CDA-AMC review included the dual primary end points of OS and PFS. Secondary outcomes included health-related quality of life (HRQoL) and safety. Additional efficacy end points included duration of response (DOR) and time to death or distant metastasis (TTDM). Efficacy and safety data were evaluated at multiple prespecified interim analyses.
Baseline characteristics were generally balanced between the treatment groups. The median age was 62 years, and 100% of patients had an ECOG Performance Status of 0 or 1. The trial population consisted of 50% white, 48% Asian, and 2% other racial groups. A total of 91.3% of patients had a history of smoking, and common comorbidities included hypertension (37.9% of patients) and chronic obstructive pulmonary disease (16.2%). Prior treatments included platinum-based chemotherapy with concurrent radiotherapy. Approximately 54% of patients had received PCI.
Efficacy Results
At the data cut-off date (January 15, 2024), the hazard ratio (HR) for OS was 0.73 (95% confidence interval [CI], 0.569 to 0.928; P = 0.01042), favouring durvalumab, representing a 27% reduction in the risk of death. The median OS was 55.9 months (95% CI, 37.3 months to not estimable]) in the durvalumab group compared to 33.4 months (95% CI, 25.5 to 39.9 months) in the placebo group. Survival probabilities at 24 and 36 months were higher in the durvalumab group (68.0% and 56.5%, respectively) than in the placebo group (58.5% and 47.6%, respectively).
Durvalumab also significantly improved PFS, with an HR of 0.76 (95% CI, 0.606 to 0.950; P = 0.01608), translating to a 24% reduction in the risk of progression or death. The median PFS was 16.6 months (95% CI, 10.2 to 28.2 months) in the durvalumab group versus 9.2 months (95% CI, 7.4 to 12.9 months) in the placebo group. At the 24-month landmark analysis, 46.2% of patients in the durvalumab group were progression-free, compared to 34.2% in the placebo group.
There was no difference in TTDM between treatment with durvalumab and placebo (███ █████ ███ ███ ██████ █████) at this interim analysis.
Patient-reported outcomes (PROs) assessed using the European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 (EORTC QLQ-C30) revealed no clinically meaningful differences between treatment groups in global health status/quality of life scores or functional scales. Chest pain was the only symptom that showed improvement with durvalumab treatment compared to placebo (odds ratio = 2.28; P = 0.0308).
Harms Results
Treatment-emergent AEs (TEAEs) were reported for 94.3% of patients in the durvalumab group and 88.3% of patients in the placebo group. Serious AEs (SAEs) were reported for 29.8% and 24.2% of patients in the durvalumab and placebo groups, respectively. The most commonly reported SAEs in the durvalumab group were radiation pneumonitis (5.0% of patients), pneumonia (4.6%), and pneumonitis (3.1%).
Immune-mediated AEs occurred more frequently in the durvalumab group (32.1% of patients, versus 10.2% in the placebo group). Moreover, the following AEs accrued more frequently in durvalumab group than in the placebo group: hypothyroidism (16.0% versus 3.8%), hyperthyroidism (10.3% versus 1.5%), and dermatitis/rash (28.2% versus 17.4%). Discontinuation due to AEs was also higher in the durvalumab group than in the placebo group (16.4% of patients versus 10.6%), with the primary reasons in the durvalumab group being radiation pneumonitis (3.8%) and pneumonitis (3.1%).
AEs resulting in death occurred in 2.7% of patients in the durvalumab group and 1.9% in the placebo group. Deaths in the durvalumab group were primarily attributed to pneumonia (0.8% of patients), bacterial pneumonia (0.8%), cardiac failure (0.4%), encephalopathy (0.4%), and pneumonitis (0.4%).
Critical Appraisal
Internal Validity
In the phase III ADRIATIC trial, randomization and allocation concealment procedures were appropriately conducted using clinically relevant stratification factors (disease stage and receipt of PCI), with allocation managed through an interactive response system. Blinding was maintained with placebo infusions, though some unblinding likely occurred due to imbalances in immune-mediated AEs in the durvalumab group. This potential unblinding could introduce bias in subjective outcomes like HRQoL, but not in objective end points like OS.
A total of ██ patients (█████) had at least 1 protocol deviation: ██ patients (█████) in the durvalumab group and ██ patients ██ (█████) in the placebo group. The most frequently reported protocol deviations included deviation from key eligibility criteria (██ patients in the durvalumab group [█████ versus ██ patients in the placebo group [██████, primarily due to █████████ ███ ██████ █████████ ██████ ████ ███ ██████████ ██ █████████████████ ██ █████████████. Another common deviation was █████████ █████████ ██████████████ (██ patients ██████ in the durvalumab group versus ███ patients ██████ in the placebo group). These protocol deviations were not considered to be major by the review team and, therefore, were not expected to have a major impact on the interpretability of the trial data.
The trial’s hierarchical testing strategy for OS and PFS controlled for multiplicity. The primary outcomes were measured using Response Evaluation Criteria in Solid Tumours version 1.1 (RECIST 1.1) criteria and assessed by blinded independent central review (BICR), reducing the potential for information (or measurement) bias. Sensitivity analyses were conducted to test the robustness of OS and PFS results, addressing potential biases from missing data, censoring rules, and assessment methods. These included alternative censoring rules (e.g., censoring patients with missed tumour assessments at their last evaluable visit) and comparing investigator-assessed PFS with BICR results, both of which yielded consistent HR estimates. A Cox model adjusting for stratification factors also confirmed the robustness of OS results. While these analyses reinforced the reliability of findings, moderate imprecision was noted due to variations in censoring assumptions.
External Validity
The ADRIATIC trial population and interventions are largely generalizable to practice in Canada with some limitations. The trial excluded patients with medically operable stage I/II disease, which does not reflect routine practice in Canada, in which surgery may be considered in select cases. The trial population was approximately 50% white and 48% Asian, with Black and other racial groups underrepresented, potentially limiting generalizability to the more racially diverse population living in Canada. The median age of 62 years also reflects a younger-than-expected population compared to real-world Canadian cases, according to the clinical experts consulted by CDA-AMC. In addition, patients with an ECOG Performance Status of 0 or 1 represented a relatively healthy subset of patients with LS-SCLC; as such, generalizability to patients with an ECOG Performance Status of 2 may be limited. The clinical experts noted that the dosing schedule of durvalumab used in the ADRIATIC trial is consistent with what would be used in clinical practice in Canada; however, the requirement for close monitoring during early cycles may pose challenges to the implementation of the drug for the condition under review in community settings. The review team considered placebo to be an appropriate comparator in this treatment space, given the current lack of a standard of care for LS-SCLC. While survival benefits were clinically meaningful, long-term follow-up beyond 36 months may be necessary to fully evaluate the generalizability of OS results from the ADRIATIC trial.
GRADE Summary of Findings and Certainty of the Evidence
For the pivotal studies and randomized controlled trials (RCTs) identified in the sponsor’s systematic review, Grading of Recommendations Assessment, Development and Evaluation (GRADE) was used to assess the certainty of the evidence for the outcomes considered most relevant to inform the expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group.
Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null.
The reference points for the certainty of evidence assessment for OS, PFS, any immune-mediated TEAEs, and any infusion-related reactions were set according to the presence or absence of an important effect based on thresholds informed by the clinical experts consulted for this review. The reference point for the certainty of evidence assessment for EORTC QLQ-C30 and European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Lung Cancer 13 (EORTC QLQ-LC13) global health status scores were set according to the presence or absence of an important effect based on a threshold suggested by the sponsor that was informed by the literature.
The selection of outcomes for the GRADE assessment was based on the sponsor’s summary of clinical evidence, consultation with clinical experts, and input received from patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members:
survival outcomes (OS and PFS)
HRQoL outcome (EORTC QLQ-C30 and EORTC QLQ-LC13 global health status/quality of life)
notable harms (SAEs and pneumonitis).
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for durvalumab versus placebo.
Table 2: Summary of Findings for Durvalumab vs. Placebo for Patients With Limited-Stage Small Cell Lung Cancer, ADRIATIC Trial
Outcome and follow-up | Patients (studies), N | Relative effect (95% CI) | Absolute effects (95% CI) | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
Placebo | Durvalumab | Difference | |||||
OS, full analysis set | |||||||
Probability of survival at 24 months Median follow-up for all patients: 37.2 months | 530 (1 RCT) | NA | ███ per 1,000 | ███ per 1,000 (███ to ███) | ██ ████ per 1,000 (██ ████ to ███ ████) | Moderatea | Durvalumab likely results in a clinically important increase in the probability of survival at 24 months compared to placebo. |
Probability of survival at 36 months Median follow-up for all patients: 37.2 months | 530 (1 RCT) | NA | ███ per 1,000 | ███ per 1,000 (███ to ███) | ██ ████ per 1,000 (| █████ to ███ █████) | Moderateb | Durvalumab likely results in a clinically important increase in the probability of survival at 36 months compared to placebo. |
PFS, full analysis set | |||||||
Probability of PFS at 18 months Median follow-up: 27.4 months (durvalumab) and 27.7 months (placebo) | 530 (1 RCT) | NA | ███ per 1,000 | ███ per 1,000 (███ to ███) | ███ ████ per 1,000 (| ████ to ███ █████) | Moderatec | Durvalumab likely results in a clinically important improvement in PFS at 18 months compared to placebo. |
Probability of PFS at 24 months Median follow-up: 27.4 months (durvalumab) and 27.7 months (placebo) | 530 (1 RCT) | NA | ███ per 1,000 | ███ per 1,000 (███ to ███) | ███ ████ per 1,000 (██ █████ to ███ █████) | Moderatec | Durvalumab likely results in a clinically important improvement in PFS at 24 months compared to placebo. |
HRQoL, full analysis set | |||||||
Global health status/quality of life: Average over 24 months | 418 (1 RCT) | NA | ████ | ████ █████ to █████ | ████ █████ to ████ | Lowd | Due to the limited certainty of evidence, the effect of durvalumab on HRQoL remains uncertain. |
Harms, safety analysis set | |||||||
SAEs Median follow-up: 27.4 months (durvalumab) and 27.7 months (placebo) | 527 (1 RCT) | NA | ███ per 1,000 | ███ per 1,000 (███ to ███) | ██ ████ per 1,000 (██ █████ to ███ ████) | Moderatee | Durvalumab likely increases the risk of SAEs (notably radiation pneumonitis and pneumonia) compared to placebo. |
Pneumonitis Median follow-up: 27.4 months (durvalumab) and 27.7 months (placebo) | 530 (1 RCT) | NA | ██ per 1,000 | ███ per 1,000 (██ to ███) | ██ ████ per 1,000 (| █████ to ██ ████) | Moderatee | Durvalumab likely increases the risk of pneumonitis compared to placebo. |
CI = confidence interval; HRQoL = health-related quality of life; NA = not applicable; OS = overall survival; PFS = progression-free survival; RCT = randomized controlled trial; SAE = serious adverse event; vs. = versus.
Notes: First interim analysis data cut-off date: January 15, 2024. Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aA between-group absolute risk difference of 5% (at least 30 fewer events per 1,000 patients) at 24 and 36 months was clinically important according to the clinical experts. The point estimate exceeded the threshold. Certainty was rated down 1 level for imprecision due to wide CIs, which include large effect estimates.
bA between-group absolute risk difference of 5% (at least 30 fewer events per 1,000 patients) at 24 and 36 months was clinically important according to the clinical experts. The point estimate exceeded the threshold. Certainty was rated down 1 level for imprecision due to wide CIs, which include null value.
cA between-group absolute risk difference of 5% (at least 50 fewer events per 1,000 patients) at 18 and 24 months was clinically important according to the clinical experts. The point estimate exceeded the threshold. Certainty was rated down 1 level for imprecision due to wide CIs.
dThere was no meaningful change in HRQoL; the clinical experts emphasized that this was acceptable because the comparator was placebo and maintenance of HRQoL was viewed positively. However, certainty was rated down 2 levels for imprecision due to wide CIs, which include null the value, and there is uncertainty based on the loss to follow-up at later times.
eRated down 1 level for imprecision due to wide CIs, which include large effect estimates.
Sources: Details included in the table are from the ADRIATIC Clinical Study Report, Section 12, and additional information provided in the sponsor's submission (data cut-off date: January 15, 2024).
Long-Term Extension Studies
No materials on long-term extension studies were submitted by the sponsor.
Indirect Comparisons
No indirect treatment comparisons were submitted by the sponsor.
Studies Addressing Gaps in the Evidence From the Systematic Review
No additional studies were submitted by the sponsor.
Conclusions
The results of the phase III, randomized, double-blind, placebo-controlled ADRIATIC trial suggest that durvalumab as consolidation therapy likely results in improved OS and PFS compared to placebo in adult patients with LS-SCLC after concurrent CRT based on moderate certainty evidence. Placebo was considered a relevant comparator in this setting because active surveillance is currently used after platinum-based CRT in the patient population under review.
A higher incidence of SAEs and immune-mediated AEs was observed in the durvalumab group than in the placebo group; however, these AEs were deemed by the clinical experts to be manageable with appropriate monitoring and intervention. The clinical experts noted that, overall, the safety profile of durvalumab was consistent with the expected AE profile for anti–PD-L1 therapies. However, missing data that were not accounted for in the ADRIATIC trial, such as data from patients who exited the study early, may result in some level of uncertainty in the estimates of harms effects.
The PRO results assessed using the EORTC QLQ-C30 in the ADRIATIC trial showed that durvalumab treatment as consolidation therapy after concurrent CRT may result in improvement in chest pain symptoms compared to placebo. No statistically meaningful differences were reported in global health status/quality of life scores or functional scales between the durvalumab and placebo groups; however, the PRO results were uncertain due to a notable amount of missing data and the exploratory nature of some PRO analyses.
Introduction
Disease Background
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the review team.
Lung cancer has the highest incident rate of cancers in Canada and is the leading cause of cancer death in the country.1 It was estimated that by the end of 2024, lung cancer would account for 32,100 new cancer diagnoses in Canada and approximately 23% of all cancer-related deaths.1 SCLC is an aggressive form of lung cancer with high mortality and a high relapse rate and represents around 12% of all lung cancers in Canada. SCLC can be classified as limited-stage disease or extensive-stage disease.2-4 Almost 30% of patients with SCLC are diagnosed with limited-stage disease.5 Based on the information published by the Canadian Cancer Society in 2020, patients with LS-SCLC have a median survival of 12 to 16 months with treatment.6 According to the information published by BC Cancer in 2014, the prognosis of people with LS-SCLC with no therapy is poor, with a median survival of 10 to 12 weeks, and the median survival of patients with LS-SCLC treated with radiotherapy alone is reported as 5 to 6 months.15 The 5-year OS rate of patients with LS-SCLC treated with concurrent CRT has been reported as 34%.16 The physical, emotional, and social toll of an SCLC diagnosis negatively impacts patient HRQoL.17 Patient health is further compromised by toxicities during chemotherapy and the side effects of current therapies (e.g., anemia, neutropenia, infection, anorexia, weight loss, nausea, vomiting, nephrotoxicity, neurotoxicity, and fatigue).18
LS-SСԼС is defined by the presence of tumours that are limited to the ipsilateral hemithorax and can be encompassed in a safe rаԁiοtheraру field.12 SCLC often presents with a large hilar mass with bulky mediastinal adenopathy and is characterized by a rapid doubling time, high growth fraction, propensity to metastasize, and transient responses to conventional chemotherapy and radiotherapy.19 Important prognostic factors in SCLC include disease stage at diagnosis, performance status, age, sex, and markers of excessive bulk disease.20 A diagnosis of lung cancer starts with a complete medical and family history and clinical examination, along with standard laboratory tests, imaging, and biopsy.21 Some of the signs and symptoms of lung cancer include a cough that gets worse or does not go away, shortness of breath, chest pain that gets worse with deep breathing or coughing, blood in mucus coughed up from the lungs, wheezing, weight loss, fatigue, hoarseness, difficulty swallowing, swollen lymph nodes in the neck or above the collarbone, and headache.21 Some patients may also experience recurrent lung infections such as bronchitis or pneumonia.22
Standards of Therapy
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the review team.
Treatment priorities for patients with LS-SCLC whose disease has not progressed after CRT are prolonging survival, delaying disease progression, and improving HRQoL.16,23 According to the clinical experts consulted for this review, the current standard of care in Canada after definitive concurrent CRT for LS-SCLC is surveillance, with no active consolidation treatment. Surveillance typically involves repeat imaging every 3 months, along with laboratory assessments and physical examinations.
At present, there are no approved systemic consolidation therapies available in Canada for patients with LS-SCLC after concurrent CRT. There is an unmet need for effective interventions to extend PFS and OS beyond what is achievable with current practices.24,25 According to the existing literature, treatment options are limited to palliative care or systemic therapy upon disease recurrence; these treatment options include rechallenging with platinum-based chemotherapy, introducing second-line chemotherapeutic agents, or considering immunotherapies approved for recurrent disease.3,16
Drug Under Review
Durvalumab is a fully human, high-affinity, immunoglobulin G1 kappa monoclonal antibody that selectively blocks the interaction of PD-L1 with PD-1 and CD80 (B7.1) while leaving PD-1/PD-L2 interaction intact. Durvalumab does not induce antibody-dependent, cell-mediated cytotoxicity. The selective blockade of PD-L1/PD-1 and PD-L1/CD80 interactions releases the inhibition of immune responses and enhances antitumour immune responses. The key characteristics of durvalumab are summarized in Table 3.
The recommended dosage of durvalumab for the indication under review is 1,500 mg every 4 weeks, administered as an IV infusion. Therapy should continue for 24 months or until disease progression or unacceptable toxicity. Patients with a body weight of 30 kg or less must receive weight-based dosing, equivalent to durvalumab 20 mg/kg every 4 weeks as monotherapy until weight increases to greater than 30 kg.
Durvalumab is also indicated for the following:26
Treatment of patients with locally advanced or metastatic urothelial carcinoma who:
experience disease progression during or after platinum-containing chemotherapy
experience disease progression within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy.
Treatment of patients with locally advanced, unresectable, stage III NSCLC whose disease has not progressed after platinum-based CRT.
First-line treatment of adult patients with ES-SCLC in combination with etoposide and either carboplatin or cisplatin.
Treatment of patients with locally advanced or metastatic biliary tract cancer in combination with gemcitabine-based chemotherapy.
Durvalumab has been previously reviewed by CDA-AMC and CADTH for other indications. On July 27, 2021, the pan-Canadian Oncology Drug Review Expert Committee recommended that durvalumab in combination with etoposide and platinum (cisplatin or carboplatin) chemotherapy should be reimbursed for the treatment of adult patients with ES-SCLC.
Durvalumab received a Notice of Compliance from Health Canada on April 8, 2025, for use as monotherapy in adult patients with LS-SCLC whose disease has not progressed after platinum-based CRT. Durvalumab is under review by the FDA and the European Medicines Agency for the treatment of patients with LS-SCLC whose disease has not progressed after platinum-based CRT.
Table 3: Key Characteristics of Durvalumab
Characteristic | Durvalumab |
|---|---|
Mechanism of action | A human monoclonal antibody that enhances antitumour immune responses. |
Indication | Durvalumab, as monotherapy, indicated for the treatment of adult patients with limited-stage small cell lung cancer (LS-SCLC) whose disease has not progressed following platinum-based chemoradiation therapy (CRT). |
Route of administration | IV |
Recommended dosage | 1,500 mg every 4 weeks. Patients with a body weight of 30 kg or less must receive weight-based dosing, equivalent to durvalumab 20 mg/kg every 4 weeks, as monotherapy until weight increases to greater than 30 kg. Treatment should continue until disease progression, unacceptable toxicity, or a maximum of 24 months. |
Serious adverse effects or safety issues |
|
Other | Should be administered under the supervision of health care practitioners experienced in the treatment of cancer. |
Source: Product monograph for durvalumab.26
Perspectives of Patients, Clinicians, and Drug Programs
The full patient and clinician group submissions received are available in the consolidated patient and clinician group input document for this review on the project website.
Patient Group Input
This section was prepared by the review team based on the input provided by patient groups.
CDA-AMC received a joint submission from CCSN, LCC, and LHF. CCSN is a national network of patients, families, survivors, community partners, funders, and sponsors with the aim of promoting the standards of care and providing support for patients living with cancer and for issues related to survivorship or quality of end-of-life care. LCC is a registered national charitable organization that supports patients through education, research, and advocacy. LHF is a registered charity that assists and empowers people living with or caring for others with lung disease.
The information was gathered through an online survey conducted from August to November 2024. One patient from Canada with NSCLC, who had experience with durvalumab, responded to the survey. No patients with LS-SCLC who had not experienced disease progression after definitive platinum-based concurrent CRT participated in the survey. However, in November 2024, LCC conducted 3 interviews with patients with SCLC (1 with ES-SCLC, 1 with stage III SCLC, and 1 with unknown stage SCLC) who had direct experience with durvalumab. The patient group submitting input believed that, in the absence of input from patients with LS-SCLC, the information obtained from the survey respondent and the interviewees would still be valuable to include in the submission.
The survey respondent described their experience with the disease as coughing, difficulty fighting infection, fatigue, reduced appetite, weight loss, nausea, waking up in the night or early morning because of breathing problems, feeling cold, negative impacts on emotional well-being, and excessive time spent attending medical appointments. Fatigue was identified as an aspect of the disease that is more important to control than others. The important outcomes identified by the survey respondent for a new treatment included reduced cost, improved quality of life, and improved energy levels. In terms of experience with durvalumab, the survey participant indicated that fatigue and constipation were the 2 main adverse effects and that the ease of administration was the main benefit.
The 3 interviewees with SCLC reported their experience with the disease as coughing, and they noted chemotherapy and radiation as the currently available treatments. Some of the side effects of the currently available treatments experienced by those patients included difficulty swallowing and eating, stomach pain, voice loss, hair loss, nausea, problems with completing day-to-day activities, tiredness, hearing problems, and sensitivity to noise.
One of the patients would be receiving durvalumab once a month for 12 months, and she had experienced no side effects after receiving 2 treatments. She had experienced no issues going about her life as usual and felt that she had experienced no change in independence or functionality. When asked to rate her experience with durvalumab versus other treatments, the respondent rated chemotherapy and durvalumab “about the same,” but she would much prefer durvalumab to radiation. The second interviewee, who had received 2 durvalumab treatments through a compassionate access program, reported feeling more nauseous after the treatments, but his energy had since recovered significantly. His quality of life on durvalumab had been better than on chemotherapy or radiation, but he was unable to comment if durvalumab had a better or worse treatment effect compared with other treatment options because he did not know if the drug was working on his tumours yet. The third interviewee had only received 2 treatments of durvalumab in 2021 before he had to stop it because he had no appetite, was vomiting constantly, had diarrhea, and had lost around 22 kg of his body weight.
One of the patients noted that she was relying on her pension and that if she had had to pay for durvalumab, she would not have been able to afford it.
Given the limited number of patients with SCLC in the current submission, CDA-AMC provides the following additional information from the Patient Group Input sections of previous Clinical Review Reports of patients with SCLC.
Atezolizumab in combination with platinum-based chemotherapy and etoposide for the first line treatment of patients with ES-SCLC (PC0156 review, recommended not to reimbursed in 2020): Two patient advocacy groups, LCC and the Ontario Lung Association (OLA), provided input for atezolizumab with etoposide and a platinum-based chemotherapy for the first-line treatment of patients with SCLC. LCC and OLA noted that, from a patient’s perspective, SCLC is an aggressive condition with limited treatment options available for patients. The fear and stress experienced by patients and caregivers related to receiving a diagnosis of SCLC was mentioned by both LCC and OLA, as SCLC is associated with poor survival. Symptoms of lung cancer were stated to impact a patient’s ability to engage with family and friends and to take part in daily activities or work. Immunotherapy and chemotherapy were treatments patients had received to treat SCLC. Both chemotherapy and immunotherapy were stated to be effective; however, the side effects of immunotherapy were much more tolerable, with some patients being able to resume their daily tasks. In terms of expectations for alternative treatment options, OLA and LCC highlighted the following patient values: extension of life, improvement of quality of life, manageable side effects, and additional and affordable treatment choices. In addition, the following expectations for a better coordinated health system were expressed: incorporating more respiratory and lung cancer specialists and administering more treatments at home rather than in the hospital to remove the need for patients and caregivers to take time off work.
Durvalumab (Imfinzi) in combination with etoposide and either carboplatin or cisplatin for the first-line treatment of patients with ES-SCLC (PC0234 review, recommended to reimburse in 2021): Two patient groups responded to the call from CADTH to provide input about patients’ experience, values, and preferences related to SCLC and its treatment. LCC provided information collected through environmental scans as well as interviews with 7 patients (1 male and 6 females, aged between 40 and 70 years) and their families and/or caregivers to address their thoughts and experiences with SCLC. The information was accessed from November to December 2020. LHF, previously known as the OLA, provided information from online surveys completed by people living with lung cancer and phone interviews with 14 patients living in Ontario. According to the patient input received, a diagnosis of lung cancer and the subsequent treatment have a major impact on the life of the patient and their loved ones. Many caregivers are involved in the care, well-being, and management of their loved ones. More than half of patient respondents from the LHF surveys reported current issues with work, day-to-day chores, and socialization. LCC reported that with certain treatments, such as chemotherapy and immunotherapy, caregivers need to take their loved ones to the hospital or specialized clinics for their treatments and care for them afterward for variable periods of time. In some cases, caregivers may need to take time off work to provide this care, which affects work productivity and finances and can cause mental stress. The emotional and physical toll during and after treatment may affect the caregivers’ ability to fulfill their role in the family and at work and affect their ability to participate in activities they enjoy. The current therapy for ES-SCLC is chemotherapy or immunotherapy. According to the patient input received, chemotherapy continues to be a viable form of treatment for this type of cancer. This treatment has been shown to work well in this group of patients, resulting in improved symptoms and increasing patients’ quality of life. However, chemotherapy can lower patients’ immunity and, in some cases, may result in an inability to return to normal activities, have visitors, or spend quality time with family and loved ones. Patients reported that immunotherapy is a form of treatment that has allowed many patients to hope for improved outcomes and has been shown to improve quality of life with more manageable side effects. Patients report feeling better within days of their first treatment with immunotherapy. Because patients with lung cancer, and patients with SCLC in particular, have a high symptom burden, no deterioration in quality of life and better tolerability are important aspects of this form of treatment. Patients on immunotherapy reported side effects that were mild and easily managed. In a few cases, there were stronger side effects that had to be managed by either over-the-counter or prescription drugs. Most found that the treatment was tolerable and did not interfere with day-to-day life. LCC highlighted that no form of immunotherapy has currently been approved for patients with SCLC in Canada. The important patient outcomes included control of the cancer, improved symptoms and delayed deterioration, manageable side effects, effectiveness on the central nervous system, delayed progression, extended survival with a good quality of life, and longer lasting and durable treatment. Both LCC and LHF were unable to include patients’ experiences with durvalumab because the treatment was not currently accessible to patients with SCLC in Canada, and there were no Canadian trial sites for the requested treatment. LCC was able to look at the outcomes of patients with SCLC treated with atezolizumab in combination with chemotherapy to highlight patients’ experiences with an immunotherapy similar to durvalumab. This combination was shown to work well for patients in terms of tolerability. The patient input emphasized that SCLC is a devastating disease, with very few treatment options. Treatment for SCLC has not changed in the past 30 years, and this puts these patients at a huge disadvantage. Patient groups identified that there is an unmet need for patients with ES-SCLC, unlike for patients with NSCLC, who have a larger range of treatment options available to them. The LCC group emphasized that there is a need for treatment options and that there is no reason to delay patients’ access to this treatment.
Atezolizumab (Tecentriq) in combination with carboplatin and etoposide for the first-line treatment of adult patients with ES-SCLC (PC0277 review, recommended to reimburse in 2022): The patient and caregiver input received for this review was collected by LCC from interviews with patients with SCLC and their caregivers, gathered from December 2021 to February 2022, as well as information from previous LCC submissions. Six respondents with SCLC had experience with atezolizumab (in combination with chemotherapy or as a single treatment), 4 of whom had extensive-stage disease. Five patients had access to atezolizumab through a clinical trial, and 1 through a compassionate access program. Four of these respondents resided in Ontario, 1 resided in British Columbia, and 1 resided in Quebec. Respondents indicated that a diagnosis of SCLC and the subsequent treatment had a major impact on the lives of patients and their family members. They reported that they expect the following key outcomes to be improved from any new drug or treatment: relief of disease symptoms, manageable side effects, improved quality of life, maintenance of independence and functionality, greater access across jurisdictions, disease stability, longer periods of remission, and prolonged survival. Patients with SCLC have a very high unmet need, as there have been no new treatment options for SCLC in the past 30 years until the last 12 months, when durvalumab was approved for treatment of ES-SCLC. Six respondents who received or continue to receive atezolizumab indicated that this drug has had promising and durable treatment results with tolerable side effects. They also mentioned that atezolizumab helped them regain their independence, functionality, and livelihood, which reduced the burden on their caregivers and loved ones.
Lurbinectedin (Zepzelca), for treatment of adult patients with stage III or metastatic SCLC who have experienced disease progression on or after platinum-containing therapy (PC0281 review, recommended not to reimburse in 2022): Two patient groups provided input for this review: LCC and LHF (previously known as OLA). LCC conducted phone interviews with 2 patients from Canada with SCLC (1 localized and 1 metastatic) and environmental scans with 1 patient and 2 caregivers of patients with metastatic SCLC from the US in March 2022; all had experience with lurbinectedin. LHF conducted an online survey (2 respondents; no demographic or disease information collected) and phone interviews (3 patients from Canada with lung cancer; type and stage not reported) from September 2021 to December 2021 and collected input from 2 additional individuals (1 registered nurse and 1 certified respiratory educator); none had experience with lurbinectedin. Patients highlighted the nonspecific early symptoms of SCLC and the resulting delays in diagnosis, as well as the physical (e.g., shortness of breath, cough, fatigue, pain), emotional, and social toll of an SCLC diagnosis. Patients acknowledged that while existing treatments for SCLC (e.g., surgery, radiation, chemotherapy, targeted therapy, immunotherapy) prolonged survival and delayed disease progression, the side effects of currently available second-line and third-line chemotherapies for metastatic SCLC (e.g., nausea, fatigue, weight, and hair loss) were sometimes severe and negatively impacted HRQoL, employment, and ability to perform activities of daily living. Patients identified an unmet need for additional second-line treatment options for metastatic SCLC that can prolong survival, delay disease progression, manage cancer symptoms, and maintain HRQoL while having minimal side effects. Patients emphasized that stopping or delaying disease progression was the most important factor in choosing treatments and that they were more receptive to the potential side effects of efficacious therapies. Patients who had experience with lurbinectedin felt that the drug had reduced or stabilized tumour size, delayed disease progression, helped them continue or resume activities of daily living, including employment, and had more manageable side effects and a shorter recovery time compared with other SCLC therapies they had received.
For the review of tarlatamab, for the treatment of adult patients with ES-SCLC who had experienced disease progression on or after at least 2 prior lines of therapy including platinum-based chemotherapy (CDA-AMC project number PC0351 to 000): CDA-AMC received a joint patient group submission from LCC, LHF, and CCSN in 2024. Information provided for this submission consisted of the thoughts and experiences of 3 patients with ES-SCLC and their caregivers. Data were collected by LCC through virtual interviews conducted with patients. All interviews were conducted between July and August 2024. All patients (2 males, 1 female) with ES-SCLC had experience with tarlatamab; 2 patients resided in Canada (1 in Ontario and the other in New Brunswick) and 1 in the United States. The patient groups emphasized that SCLC is an aggressive type of lung cancer, with a high symptom burden, rapid disease progression, and poorer health outcomes. Given the lack of developments in new treatment options for patients with SCLC beyond first-line therapy, the patient groups highlighted an urgent need for a new treatment beyond the first-line setting for patients with ES-SCLC. The patient groups noted that new treatments should be effective in controlling the disease and symptoms, minimizing side effects of the treatments, allowing patients to maintain a meaningful quality of life, minimizing caregiver burden, delaying disease progression, and offering patients an additional treatment option upon disease progression or when other treatments are exhausted. Regarding currently available treatment options for ES-SCLC, patients noted that chemotherapy is associated with limited DOR, harsh side effects, increased dependence on caregivers in daily activities, and an impact on the patients’ functionality. Two patients also had experience with immunotherapy either in combination with chemotherapy or after successful completion of chemotherapy. However, 1 patient stopped treatment with immunotherapy due to neutropenia and the other due to disease progression. As noted, all patients had experience with tarlatamab as a third-line or beyond therapy. Two patients accessed tarlatamab through a clinical trial and 1 through a special access program. All patients indicated that they had significant side effects when receiving the first dose of tarlatamab; however, these side effects improved over time. Two patients experienced CRS during their first infusion, which improved over time. One patient continued to work during treatment with tarlatamab. Overall, patients indicated that tarlatamab was effective in treating their disease and delaying disease progression. They also indicated that tarlatamab significantly improved their quality of life, similar to before their diagnosis and that they had a better experience with tarlatamab than with previous therapies.
Clinician Input
Input From Clinical Experts Consulted for This Review
All CDA-AMC review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of lung cancer.
Unmet Needs
The clinical experts emphasized that LS-SCLC remains an area of high unmet medical need due to the current standard treatments offering limited survival benefits. The clinical experts noted that, after completing concurrent CRT, most patients are left with surveillance as the only option. This approach often results in disease recurrence, with an OS of approximately 25 to 30 months and a 5-year survival rate of 29% to 34%.7,27
In the absence of active and effective consolidation treatments to extend PFS or OS in this population, the clinical experts highlighted the need for therapies that reduce the risk of recurrence (relapse) or disease progression, particularly given the rapid progression observed when relapses occur.
Place in Therapy
The clinical experts noted that durvalumab would be used as a consolidation therapy for patients who have completed CRT and whose disease has not progressed; it would be the first approved agent for this purpose. While durvalumab would not replace CRT, it would be added as a consolidation therapy. Both experts agreed that this would represent a new therapeutic option in the treatment paradigm.
Patient Population
The clinical experts agreed that patients with LS-SCLC who experienced complete or partial response or stable disease after CRT would be the most suitable candidates for durvalumab. They noted that predictive biomarkers for treatment response are not yet available and that patient selection would rely on clinical judgment. Patients with a good ECOG Performance Status (0 or 1) and minimal comorbidities and patients whose disease responded well to CRT might derive the most benefit from treatment with durvalumab. The experts expressed that considering medically operable stage I/II cases eligible to receive durvalumab may be a reasonable extrapolation based on clinical practice in Canada.
Assessing the Response Treatment
The clinical experts noted that response to durvalumab should be assessed using imaging and clinical evaluation every 2 to 3 months. Important outcomes include PFS, OS, and symptom management. Both experts agreed that the treatment outcomes in the ADRIATIC trial align with those used in their clinical practice.
A clinically meaningful response was defined as a measurable improvement in survival (e.g., at least 2 months of additional PFS or OS) and stabilization or improvement of symptoms. The experts emphasized the importance of long-term survival data, with 1 noting that 5-year OS rates would provide valuable insights into the long-term impact of durvalumab.
Discontinuing Treatment
The clinical experts outlined the following factors for discontinuing durvalumab: evidence of disease progression based on imaging or clinical evaluation; development of immune-mediated toxicities, such as pneumonitis, colitis, hepatitis, myocarditis, or nephritis, that are intolerable or unmanageable; and significant deterioration in patient quality of life. One clinical expert suggested that treatment could continue in cases of radiologic progression being observed early after chemoradiation or within a time frame compatible with durvalumab-mediated pseudoprogression. This observation was based on clinical experience that posttreatment imaging may show apparent tumour enlargement due to treatment effects, such as radiation-induced inflammation or transient mediastinal mass enlargement. In such cases, a follow-up CT scan after 2 months may help determine true progression before a decision is made to discontinue treatment, provided the patient’s overall condition remains stable and their symptoms do not worsen.
Prescribing Considerations
The clinical experts noted that durvalumab should be prescribed by oncologists experienced in managing systemic cancer therapies and checkpoint inhibitor–related toxicities. The experts highlighted the need for initial treatments to be administered in centres equipped to manage severe immune-mediated AEs. After the initial cycles, transitioning to outpatient administration under the supervision of trained oncology practitioners would be feasible.
Clinician Group Input
This section was prepared by the review team based on the input provided by clinician groups.
CDA-AMC received 2 clinician group input submissions: 1 from the LCC MAC and 1 from the OH (CCO) Lung Cancer Drug Advisory Committee. LCC is a national charity that aims to increase awareness about lung cancer, provide support and education to lung cancer patients and their families, support research, and advocate for access to the best care for all lung cancer patients in all provinces and territories. The LCC MAC consists of clinicians in the field of lung cancer across the country. The OH (CCO) Cancer Drug Advisory Committees provide timely evidence-based clinical and health system guidance on drug-related issues in support of CCO’s mandate. The LCC MAC gathered information from publicly available sources, primarily published manuscripts and conference presentations, together with the clinical experience of members of the committee, comprising 27 clinicians. The OH (CCO) Lung Cancer Drug Advisory Committee gathered information by email from 5 clinicians.
Both clinician groups agreed that the current standard treatment for LS-SCLC is 4 cycles of cytotoxic platinum-based (cisplatin or carboplatin) and etoposide chemotherapy combined with concurrent or sequential radiation. The clinician groups noted that the treatment goal is to prevent or delay disease recurrence and improve OS. The input received from the LCC MAC noted that patients with confirmed LS-SCLC are treated for cure with aggressive chemotherapy and radiation. Failure to cure the disease can result in disease progression, particularly in sites such as the brain, liver, and bone, leading to significant symptomatic burden, impacting patients’ quality of life and survival. The OH (CCO) Lung Cancer Drug Advisory Committee added that the majority of patients with LS-SCLC experience disease recurrence and die from their disease.
The clinician input from the OH (CCO) Lung Cancer Drug Advisory Committee anticipated that durvalumab would be used after standard systemic therapy with platinum-based chemotherapy and etoposide, as well as radiation treatments. If the cancer recurs while the patient is on durvalumab, the use of more durvalumab in the metastatic setting would not occur. It was noted that the mechanism of action of durvalumab is different than that of chemotherapy or radiation therapy; therefore, durvalumab would not replace either of those therapies. The LCC MAC added that platinum-etoposide combined with either durvalumab or atezolizumab, followed by maintenance immunotherapy as monotherapy, is the standard of care in Canada for patients with ES-SCLC with good disease-related performance status and no contraindications to therapy.
The OH (CCO) Lung Cancer Drug Advisory Committee believed that patients with LS-SCLC who have completed CRT and who have not had significant pneumonitis, disease progression, or autoimmune disease would be most suitable for treatment with durvalumab. Patients with poor disease-related performance status, and those who have radiation pneumonitis, would not be candidates for treatment with durvalumab. Patients who have disease with mixed histology (small cell and non–small cell) would be suitable candidates and would be considered for treatment with durvalumab. Patients with LS-SCLC who complete 2 years of adjuvant durvalumab treatment and then experience disease relapse would be considered for treatment with durvalumab plus chemotherapy if there is a durvalumab-free interval of 6 months or greater. The LCC MAC added that patients who have experienced disease stabilization or shrinkage after standard concurrent treatment with cytotoxic platinum-etoposide chemotherapy and thoracic radiation, and those with an ECOG Performance Status of 0 to 1 (or an ECOG Performance Status of 2 in the real-world setting) after chemotherapy and radiation, would be suitable candidates for durvalumab. Patients with severe or symptomatic autoimmune disorders are generally not suitable for treatment with durvalumab.
According to the OH (CCO) Lung Cancer Drug Advisory Committee input, the outcomes to determine whether a patient is responding to treatment in clinical practice included OS and disease progression based on signs, symptoms, radiology, and laboratory tests. Chest imaging (CT or X-ray) should be done every 3 to 6 months, and imaging of the abdomen, bones, brain, and pelvis should be done on a symptom-derived basis. Improved survival is clinically meaningful if the absolute number is greater than 5% or if the median is greater than 6 months. The LCC MAC noted that quality of life is another important outcome. The LCC MAC added that in addition to every 3-month to 4-month assessment, patients who are on durvalumab will be assessed clinically every 4 weeks before each new cycle of treatment. Patients who are disease-free after the completion of 5 years’ post-chemoradiation surveillance are considered cured from their LS-SCLC.
Both clinician groups noted that disease progression and intolerable treatment-related adverse effects are the main reasons for discontinuation of durvalumab. The OH (CCO) Lung Cancer Drug Advisory Committee highlighted the following discontinuation criteria: completion of 2 years of therapy with no evidence of disease progression, occurrence of life-threatening or life-limiting conditions, and significant immune-mediated toxicity. The LCC MAC added patient choice as another reason for the discontinuation of durvalumab.
According to the clinician groups, durvalumab after chemoradiation can be received as an outpatient in a systemic therapy treatment unit and can be performed in a community oncology setting. Treatment most often would be given in a specialized cancer hospital with chemotherapy and immunotherapy experience. Treatment should be under the supervision of the appropriate oncology care team.
Drug Program Input
The drug programs provide input on each drug being reviewed through the reimbursement review process by identifying issues that may impact the drug programs’ ability to implement a recommendation. The implementation questions and the corresponding responses from the clinical experts consulted for this review are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Responses
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
Issues with the choice of comparator in the submitted trial(s): In the ADRIATIC trial, the comparator to durvalumab was placebo. The current standard of care in Canada is active surveillance, so the choice of placebo was an appropriate comparator. Patients in both groups were treated to a maximum of 24 months. | Comment from the drug plans to inform pERC deliberations. |
Considerations for initiation of therapy | |
Eligibility for re-treatment:
| The clinical experts noted that re-treatment eligibility depends on the timing of disease progression. More specifically:
|
Considerations for discontinuation of therapy | |
Treatment interruptions: For patients who stop for reasons other than disease progression, can durvalumab be restarted if the disease progresses while off therapy? | The clinical experts noted that, for patients who stop durvalumab treatment for reasons unrelated to disease progression (e.g., adverse events or unrelated medical interventions), treatment can be resumed following interruption, or after toxicity resolves to acceptable levels, to complete the planned 2 years if no disease progression occurs during the interruption. It was also noted that, if disease progression occurs during the interruption, re-treatment with durvalumab alone would not be appropriate. In such cases, treatment should follow the extensive-stage paradigm, which currently involves combination chemotherapy. |
Considerations for prescribing of therapy | |
Dosing, schedule/frequency, dose intensity: If therapy is funded or implemented, most jurisdictions are likely to implement a weight-based durvalumab dose, as is used for other funded indications (e.g., 20 mg/kg up to a maximum of 1,500 mg per dose). | Comment from the drug plans to inform pERC deliberations. |
Generalizability | |
Populations of interest matching the indication but with insufficient data:
| ECOG Performance Status: The clinical experts suggested that patients with an ECOG Performance Status of 2 should be considered eligible for treatment, as there is supporting data from similar settings, including NSCLC (e.g., the PACIFIC trial). Eligibility for those with an ECOG Performance Status of 3 is uncertain and warrants further expert input. Mixed SCLC and NSCLC: Patients with mixed SCLC and NSCLC were excluded from the ADRIATIC trial. The clinical experts suggested that these patients should be considered eligible to receive durvalumab, as the SCLC component of their condition is more aggressive. They also noted that results from the PACIFIC trial suggested a benefit for consolidation therapy with durvalumab after chemoradiation in patients with NSCLC. Brain metastases: The clinical experts noted that patients with brain metastases may be eligible to receive durvalumab if the metastases are stable, treated, and not causing clinical problems. They indicated that modern approaches, such as stereotactic body radiation therapy, often allow for treatment with curative intent in this context. However, patients with progressing or uncontrolled brain metastases are not considered eligible. |
Patients on active treatment with a time-limited opportunity to switch to the drug(s) under review: Can patients who have recently finished concurrent chemoradiotherapy be allowed to switch over to durvalumab? | The clinical experts suggested that patients who have recently finished concurrent chemoradiotherapy may switch to durvalumab. However, the timing is important:
|
Care provision issues | |
Drug preparation, storage, administration, or dispensing: Preparation of durvalumab is familiar to many jurisdictions due to its use in other indications. | Comment from the drug plans to inform pERC deliberations. |
System and economic issues | |
Concerns regarding the anticipated budget impact and sustainability: Feasibility of adoption (budget impact), as this will become the new standard of care. | Comment from the drug plans to inform pERC deliberations. |
ECOG = Eastern Cooperative Oncology Group; ES-SCLC = extensive-stage small cell lung cancer; LS-SCLC = limited-stage small cell lung cancer; NSCLC = non–small cell lung cancer; pERC = pan-Canadian Oncology Drug Review Expert Review Committee; SCLC = small cell lung cancer.
Clinical Evidence
The objective of this Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of durvalumab (1,500 mg, IV) in the treatment of LS-SCLC in patients whose disease has not progressed after platinum-based CRT. The focus will be placed on comparing durvalumab to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of durvalumab is presented in 4 sections, with the CDA-AMC critical appraisal of the evidence included at the end of each section. The first section, the systematic review, includes the pivotal study selected according to the sponsor’s systematic review protocol. The CDA-AMC assessment of the certainty of the evidence in this first section using the GRADE approach follows the critical appraisal of the evidence. The second, third, and fourth sections would include long-term extension studies, indirect evidence, and additional studies to address important gaps in the systematic review evidence, respectively; however, none were submitted by the sponsor.
Included Studies
Clinical evidence from the following is included in the review and appraised in this document:
1 phase III, randomized, pivotal trial included in the systematic review (ADRIATIC trial).
Systematic Review
Contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
Description of Studies
The ADRIATIC trial (NCT03703297) is a completed phase III, randomized, double-blind, placebo-controlled, multicentre study that aimed to evaluate the efficacy and safety of durvalumab with or without tremelimumab as consolidation therapy in patients with LS-SCLC who did not experience disease progression after concurrent CRT (Figure 1). The trial was conducted across 164 sites in 19 countries, including 5 Canadian sites, and enrolled a total of 730 patients. Patients were randomized in a 1:1:1 ratio to receive either durvalumab monotherapy, durvalumab in combination with tremelimumab, or placebo. The data cut-off date for the results presented is January 15, 2024. Additional characteristics of the trial are summarized in Table 5.
The study included a screening phase conducted 1 to 42 days before randomization, during which patients who completed definitive platinum-based concurrent CRT were assessed for eligibility. Eligible patients had histologically or cytologically confirmed LS-SCLC, experienced disease control (complete response, partial response, or stable disease) after concurrent CRT, and demonstrated an ECOG Performance Status of 0 or 1. Patients with mixed histology or ES-SCLC were excluded. Treatment randomization was stratified by disease stage (I/II versus III) and receipt of PCI (yes versus no). The primary objectives of the trial were to assess PFS and OS in the durvalumab versus placebo groups.
The treatment phase involved administration of the study drugs every 4 weeks for up to 24 months or until disease progression, intolerable toxicity, or patient withdrawal. Durvalumab was administered at 1,500 mg intravenously, while tremelimumab (if applicable) was given at 75 mg intravenously for the first 4 cycles.
The trial also included a follow-up phase to monitor safety and survival outcomes. Safety assessments occurred up to 90 days after the last dose of the study drug, and survival was monitored at predefined intervals (8, 12, 16, 24, 32, 40, and 48 weeks, and every 8 weeks thereafter).
Although the trial included a treatment group for durvalumab in combination with tremelimumab, the sponsor did not seek approval from Health Canada for this combination. Details relating to this group are summarized in this section for completeness.
Table 5: Details of Study Included in the Systematic Review
Study characteristic | ADRIATIC trial |
|---|---|
Designs and populations | |
Study design | Phase III, randomized, double-blind, placebo-controlled, multicentre, international study |
Locations | 164 sites across 19 countries randomized ≥ 1 patient into the global cohort: Argentina (5 sites), Belgium (4 sites), Canada (6 sites screened patients; 16 patients were randomized from 5 sites), China (24 sites), Czech Republic (4 sites), Germany (11 sites), India (2 sites), Italy (4 sites), Japan (16 sites), Netherlands (4 sites), Poland (5 sites), Russia (10 sites), South Korea (10 sites), Spain (9 sites), Taiwan (9 sites), Turkey (9 sites), UK (1 site), US (26 sites), and Vietnam (6 sites). |
Patient enrolment dates | Start date: October 17, 2018 (first patient randomized) End date: September 27, 2021 (last patient randomized) |
Randomized | Randomized: N = 730
|
Key inclusion criteria |
|
Key exclusion criteria |
|
Drugs | |
Intervention | Durvalumab monotherapy: Durvalumab (1,500 mg IV) q.4.w. in combination with tremelimumab-matching placebo (IV) q.4.w. for 4 doses or cycles each, followed by durvalumab 1,500 mg q.4.w. starting 4 weeks after the final dose of durvalumab in combination with tremelimumab-matching placebo. Patients received treatment until clinical or RECIST 1.1 radiologic progression, until intolerable toxicity, or for a maximum of 24 months, whichever occurred first. After closure of randomization to the durvalumab + tremelimumab group, patients newly randomized to the durvalumab group were to receive only 1 infusion of durvalumab from cycle 1 onward for the duration of treatment (i.e., a maximum of 24 months). Patients no longer received the placebo infusion that was intended to mask the tremelimumab infusion because the actively enrolling experimental group did not include tremelimumab infusion. Therefore, there was no need to maintain blinding with a second placebo infusion. |
Comparators | Placebo: Durvalumab-matching placebo (IV) q.4.w. in combination with tremelimumab-matching placebo (IV) q.4.w. for 4 doses or cycles each, followed by durvalumab-matching placebo q.4.w. starting 4 weeks after the final dose of the 2 placebos in combination. Durvalumab + tremelimumab: Durvalumab (1,500 mg IV) q.4.w. in combination with tremelimumab (75 mg IV) q.4.w. for 4 doses or cycles each, followed by durvalumab 1,500 mg q.4.w. starting 4 weeks after the final dose of durvalumab in combination with tremelimumab. Patients received the following treatments until clinical or RECIST 1.1 radiologic progression, until intolerable toxicity, or for a maximum of 24 months, whichever occurred first. The durvalumab + tremelimumab group is not being considered in the review. |
Study duration | |
Screening phase | The screening period was from the last day of the final cycle of chemotherapy or the last day of radiotherapy (whichever occurred later) and lasted 1 to 42 days before randomization and the first dose of the study drug. |
Treatment phase | From the first dose of the study drug until investigator-determined progression or intolerable toxicity, or for a maximum of 24 months, whichever occurred first. |
Follow-up phase | After radiological progression, there was a follow-up scan no earlier than 4 weeks later and no later than the next regularly scheduled imaging visit. Survival status was assessed at 8, 12, 16, 24, 32, 40, and 48 weeks after the last dose of the study treatment and every 8 weeks thereafter until study termination or death. The safety follow-up period was 90 days after the last dose of the study drug. |
Outcomes | |
Primary end point | Dual primary end points
|
Secondary and exploratory end points | Key secondary:
Other secondary:
Supportive:
Exploratory:
Safety:
|
Publication status | |
Publications | Cheng et al. (2024)24 NCT03703297 |
AE = adverse event; AESI = adverse event of special interest; BICR = blinded independent central review; BOR = best objective response; CRT = chemoradiotherapy; DOR = duration of response; ECOG = Eastern Cooperative Oncology Group; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-LC13 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Lung Cancer 13; GHS = global health status; HRQoL = health-related quality of life; LS-SCLC = limited-stage small cell lung cancer; NSCLC = non–small cell lung cancer; ORR = objective response rate; OS = overall survival; PCI = prophylactic cranial irradiation; PFS = progression-free survival; PGIS = Patient Global Impression of Severity; PRO-CTCAE = Patient-Reported Outcomes version of the Common Terminology Criteria for Adverse Events; q.4.w. = every 4 weeks; QoL = quality of life; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours version 1.1; SCLC = small cell lung cancer; TFST = time to first subsequent therapy or death; TTDM = time to death or distant metastasis; vs. = versus; WDAE = withdrawal due to adverse event.
Sources: ADRIATIC Clinical Study Report and clinical study protocol; Cheng et al. (2024).24 Details included in the table are from the sponsor’s summary of clinical evidence.
Figure 1: ADRIATIC Clinical Trial Design
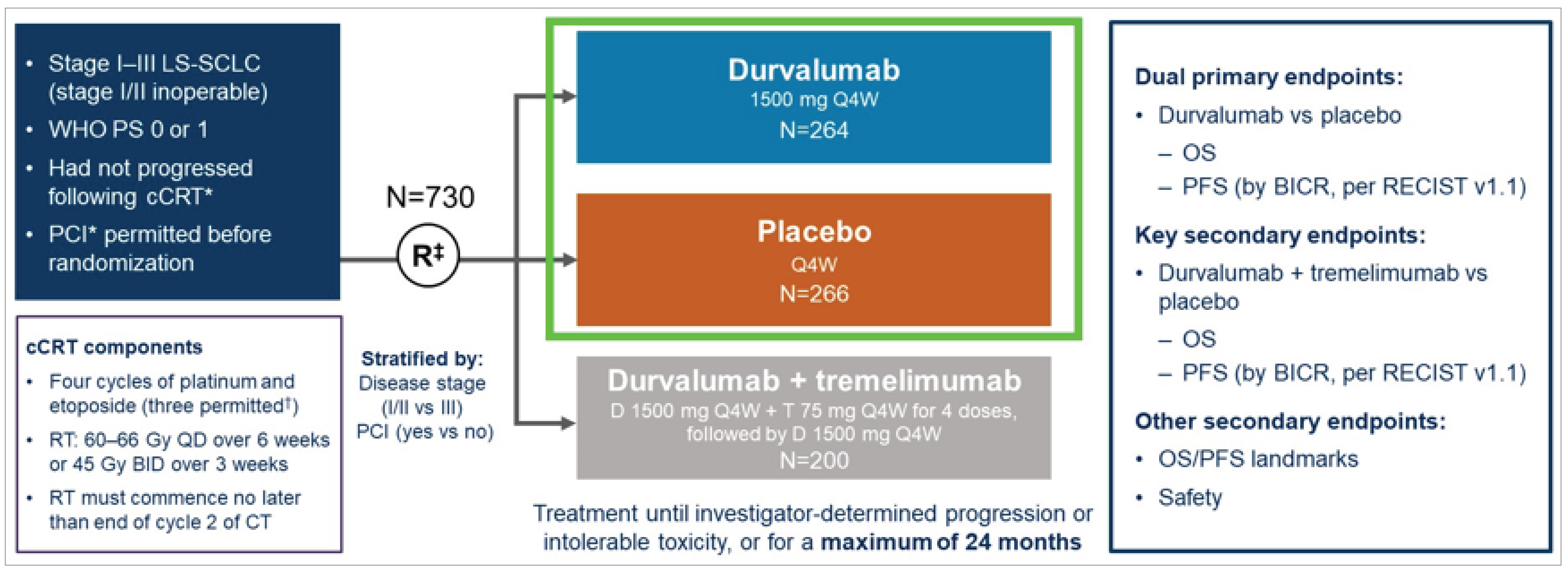
BICR = blinded independent central review; BID = twice daily; cCRT = concurrent chemoradiotherapy; CT = chemotherapy; D = durvalumab; LS-SCLC = limited-stage small cell lung cancer; OS = overall survival; PCI = prophylactic cranial irradiation; PFS = progression-free survival; PS = Performance Status; Q4W = every 4 weeks; QD = once daily; RECIST = Response Evaluation Criteria in Solid Tumours; RT = radiotherapy; T = tremelimumab; vs = versus.
*cCRT and PCI treatment. If received per local standard of care, must have been completed 1 to 42 days before randomization.
†If disease control was achieved and no additional benefit was expected with an additional cycle of chemotherapy, in the opinion of the investigator.
‡The first 600 patients were randomized in a 1:1:1 ratio to the 3 treatment groups; subsequent patients were randomized 1:1 to either durvalumab or placebo.
Sources: ADRIATIC Clinical Study Report and clinical study protocol; Cheng et al. (2024).24 Details included in the figure are from the sponsor’s summary of clinical evidence.
Populations
Inclusion and Exclusion Criteria
Eligible patients in the ADRIATIC trial were aged 18 years or older with histologically or cytologically confirmed LS-SCLC and had not experienced disease progression after concurrent CRT. Patients were required to have a ECOG Performance Status of 0 or 1, to have experienced disease control (complete response, partial response, or stable disease) after completing concurrent CRT, and to have received their last concurrent CRT treatment 1 to 42 days before randomization. Patients with mixed histology of SCLC and NSCLC or ES-SCLC were excluded.
Interventions
In the ADRIATIC trial, patients were randomized to receive 1 of 3 treatments: durvalumab monotherapy, durvalumab in combination with tremelimumab (not considered in this review), or placebo. Durvalumab was administered at a dose of 1,500 mg IV every 4 weeks. In the placebo group, matched placebos were administered on the same schedule. The assigned study treatment was continued for up to 24 months unless disease progression, unacceptable toxicity, withdrawal of consent, or investigator decision required treatment cessation.
Patients were closely monitored throughout the study for AEs, disease progression, and treatment responses. All study treatments were administered in clinical settings under supervision by trained health care professionals. After disease progression or completion of the study treatment, patients could transition to subsequent anticancer therapies at investigator discretion.
PCI was permitted before randomization if completed 1 to 42 days before the first dose of the study treatment. Patients were pretreated with appropriate medications for infusion-related reactions as per the protocol, and rescue medications for AEs were made available at all study sites. Criteria for dose interruptions or treatment discontinuation due to toxicities were prespecified. For immune-mediated AEs or other toxicities linked to study treatments, modifications were not permitted, but treatment interruptions and discontinuations were allowed as needed.
Outcomes
A list of efficacy end points assessed in this Clinical Review Report is provided in Table 6, followed by descriptions of the outcome measures. Summarized end points are based on outcomes included in the sponsor’s submission (summary of clinical evidence) as well as any outcomes identified as important to this review according to the clinical expert(s) consulted for this review and input from patient and clinician groups and public drug plans. The selected end points were those considered most relevant to inform expert committee deliberations, and a list was finalized in consultation with members of the expert committee. All summarized efficacy end points were assessed using GRADE. Select notable harms outcomes considered important for informing expert committee deliberations were also assessed using GRADE.
Table 6: Outcomes Summarized From the Study Included in the Systematic Review
Outcome measure | Time point | End point type |
|---|---|---|
OSa | 24 months, 36 months (data cut-off date: January 15, 2024) | Primary |
PFSa | 18 months, 24 months (data cut-off date: January 15, 2024) | Primary |
TTDM | Median (data cut-off date: January 15, 2024) | Secondary |
DOR | Median (data cut-off date: January 15, 2024) | Secondary |
SAE | Any reported event from the date of informed consent until 90 days after the last dose of the study treatment (data cut-off date: January 15, 2024) | Secondary |
Pneumonitis (safety outcome) | Any reported event from the date of informed consent until 90 days after the last dose of the study treatment (data cut-off date: January 15, 2024) | Secondary |
EORTC QLQ-C30 | Change from baseline | Secondary (items 29 and 30), tertiary/exploratory (items 1 to 8) |
EORTC QLQ-LC13 | Change from baseline | Tertiary/exploratory |
DOR = duration of response; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-LC13 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Lung Cancer 13; OS = overall survival; PFS = progression-free survival; SAE = serious adverse event; TTDM = time to death or distant metastasis.
aStatistical testing for these outcomes was adjusted for multiple comparisons (e.g., hierarchal testing).
Sources: ADRIATIC Clinical Study Report and clinical study protocol; Cheng et al. (2024).24 Details included in the table are from the sponsor’s summary of clinical evidence.
Dual Primary End Points
The dual primary end points were OS and PFS, specifically for the comparison of durvalumab monotherapy versus placebo.
OS was defined as the time from the date of randomization until death due to any cause. Patients who were alive at the time of the data cut-off or who were lost to follow-up were censored at the last known date they were confirmed alive.28-30
PFS (per RECIST 1.1, as assessed by BICR) was defined as the time from the date of randomization until the date of disease progression or death, regardless of whether the patient withdrew from therapy or received another anticancer therapy before progression.28,29 PFS has been evaluated as a potential surrogate end point for OS in LS-SCLC.31
Secondary End Points
TTDM (per RECIST 1.1, as assessed by BICR) was defined as the time from the date of randomization until the first occurrence of distant metastasis or death in the absence of distant metastasis. Distant metastasis was characterized by the presence of new lesions outside the primary tumour site, as confirmed by BICR. Patients who did not experience distant metastasis or death by the data cut-off date were censored at their last evaluable assessment. Patients who had distant metastasis or who died after 2 or more missed visits were censored at the latest RECIST assessment before the missed visits. Patients with no evaluable visits or no baseline data were censored at day 1, unless they died within 2 visits of the baseline.
DOR (per RECIST 1.1, as assessed by BICR) was defined as the time from the date of the first documented response (complete response or partial response) to the first occurrence of documented progression or to death in the absence of disease progression. For patients who did not experience disease progression or death, the duration was censored at the date of their last evaluable assessment.
Health-Related Quality of Life
HRQoL in the ADRIATIC trial was assessed using 2 PRO measures: the EORTC QLQ-C30 and its lung cancer–specific module, the EORTC QLQ-LC13.
The EORTC QLQ-C30 consists of 30 items grouped into 5 functional scales (physical, role, cognitive, emotional, and social functioning); 3 symptom scales (fatigue, pain, and nausea/vomiting); 1 global health status/quality of life scale; and 6 single-item measures assessing additional symptoms frequently reported by cancer patients (dyspnea, appetite loss, insomnia, constipation, diarrhea, and financial difficulties). Responses were transformed into scores ranging from 0 to 100, for which higher scores on the functional and global health status/quality of life scales indicate better functioning and health, and higher scores on symptom scales or item scales represent greater symptom severity. Patients complete the questionnaire based on a 1-week recall period by rating most items on a 4-point Likert-type scale (1 = not at all; 2 = a little; 3 = quite a bit; 4 = very much). For the 2 items in the global health status/quality of life scale, the response format is a 7-point Likert-type scale (1 = very poor; 7 = excellent).32
The EORTC QLQ-LC13 is designed to assess lung cancer–specific symptoms and treatment-related side effects. It includes items measuring symptoms such as cough, hemoptysis, and dyspnea, as well as treatment-related effects like sore mouth, dysphagia, and neuropathy. Most items are scored on a 1 to 4 scale (not at all to very much), except for the multi-item dyspnea scale, which combines 3 items if all are scored.
Harms End Points
AEs and SAEs were collected throughout the study, from the date of informed consent until 90 days after the last dose of the study treatment. The Medical Dictionary for Regulatory Activities version 26.1 was used to code the AEs. AEs were also graded for severity according to Common Terminology Criteria for Adverse Events version 4.03. A TEAE was defined as an AE that begins, or a pre-existing AE that worsens (taking the last grade before dosing as the reference), on or after the first dose of the study treatment through to 90 days after the last dose of the study treatment or the initiation of the first subsequent therapy (whichever occurred first). All AEs are listed in this report; however, only TEAEs are summarized.
AEs of special interest and AEs of potential interest were reported in the ADRIATIC trial. AEs of special interest were defined as AEs with a likely inflammatory or immune-mediated pathophysiological basis resulting from the mechanism of action of durvalumab and requiring more frequent monitoring and/or interventions such as corticosteroids, immunosuppressants, and/or endocrine therapy. AEs of potential interest were defined as AEs that could potentially have an inflammatory or immune-mediated pathophysiological basis resulting from the mechanism of action of durvalumab but are more likely to have occurred due to other pathophysiological mechanisms; thus, the likelihood of the event being inflammatory or immune-mediated in nature is not high and/or is most often or usually explained by other causes. Immune-mediated AEs were identified from both AEs of special interest and AEs of potential interest based on programmatic rules that considered interventions involving systemic steroid therapy, immunosuppressant use, and/or endocrine therapy.
Table 7: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
EORTC QLQ-C30 | A multidimensional, cancer-specific, patient-reported measure used to assess HRQoL in response to treatment in clinical trials.33 The core questionnaire consists of 30 items across 5 multi-item functional scales (physical [5 items], role [2 items], emotional [4 items], cognitive [2 items], and social [2 items] functioning), 3 multi-item symptom scales (fatigue [3 items], nausea/vomiting [2 items], and pain [2 items]), 6 single-item symptom scales (dyspnea, insomnia, appetite loss, constipation, diarrhea and financial impact), and a 2-item global health status/QoL scale. Raw scores for each scale are computed as the average of the items that contribute to a particular scale. Each raw scale score is converted to a standardized score that ranges from 0 to 100 using a linear transformation. A decline in the symptom scale score reflects an improvement, whereas an increase in the function and QoL scale scores reflects an improvement.32 | In studies of patients with lung cancer Validity: Based on completed questionnaires from 184 patients with various cancer types (breast [n = 66], lung [n = 61], and colorectal [n = 57]),34 moderate correlations were observed between the 5 EORTC QLQ-C30 functioning scales (r = 0.41 to 0.77), and FACT-G and EORTC QLQ-C30 scales (r = 0.64 to 0.76).34 In another study of 111 patients with lung cancer (n = 101) or pleural mesothelioma (n = 11), a correlation was observed between EORTC QLQ-C30 emotional functioning and the HADS anxiety scale (r = 0.75).35 The HADS depression scale correlated substantially with all functioning scales (r = −0.40 to −0.55), fatigue (r = 0.52), and appetite loss (r = 0.48).35 Correlations between clinical measures of severity and self-reported health status (r > 0.50) were reported for key domains — such as physical functioning and fatigue, EORTC QLQ-C30 functioning scales (r = 0.41 to 0.77),34 HADS with all EORTC QLQ-C30 scales (r = 0.28 to 0.75) excluding nausea/vomiting,35 BPI scales with all EORTC QLQ-C30 scales (r = 0.20 to 0.72), except nausea/vomiting and financial difficulties35 — supporting convergent validity. Reliability: Cronbach alpha coefficients were used to measure internal consistency. For functional scales, the Cronbach alpha values ranged from 0.70 to 0.85 and supported internal consistency. For symptom scales, the Cronbach alpha values ranged from 0.70 to 0.90.36 Additionally, a cluster-based analysis of several cancer types showed correlations of 0.91 for physical function–related measures (role, physical and social functions and fatigue, pain and global health status); 0.68 for psychological function (emotional functioning, cognitive functioning, and insomnia); and 0.63 for gastrointestinal symptoms (nausea, appetite loss, and vomiting).37 Responsiveness: The EORTC QLQ-C30 was administered before treatment and once during treatment to 305 patients with nonresectable lung cancer from centres in 13 countries.36 Over a 28-day period, there was a statistically significant difference in the global QoL scores (P < 0.01) between patients whose condition improved or worsened based on ECOG Performance Status during pretreatment and on-treatment periods. No significant difference was observed among patients whose ECOG Performance Status remained stable or unchanged.36 | Between-group MIDs for improvement (5 points) and deterioration (−5 points) in global health status were identified for patients with lung cancer.38 The MID estimates in patients with SCLC and breast cancer who reported “a little” change in the SSQ had corresponding changes in the EORTC QLQ-C30 of 5 to 10 points, those who reported a “moderate” change had corresponding changes of about 10 to 20 points, and those who reported “very much” change had corresponding changes of > 20 points.39 In a study of patients with advanced cancer, MID estimates of the EORTC QLQ-C30 included a meaningful change for improvement ranging from 9.1 units (cognitive functioning) to 23.5 units (pain) and a meaningful change for deterioration ranging from 7.2 units (physical functioning) to 13.5 units (role functioning). Distribution-based estimates were closest to 0.5 SD.40 |
EORTC QLQ-LC13 | The EORTC QLQ-LC13 is a lung cancer–specific questionnaire used to supplement the EORTC QLQ-C30 and contains 13 items related to lung cancer symptoms and treatment side effects, including a 3-item scale assessing dyspnea and 9 single items: pain in chest, pain in arm or shoulder, pain in other parts, coughing, hemoptysis, sore mouth or tongue, dysphagia, peripheral neuropathy, and alopecia.36 All the scales range in score from 0 to 100. Higher scores on the symptom scales reflect worse symptoms.36 | Validity: Construct validity was supported by the evidence of a correlation between pain score and disease type (P < 0.001). Conditional on ECOG Performance Status, correlation was observed in dyspnea, coughing, and pain scores (P < 0.001).41 Correlation between spirometry result and dyspnea score was found to be weak (r = 0.24). Moderate correlation was observed between BPI intensity score and EORTC QLQ-LC13 pain score (r > 0.4).35 Reliability: Internal consistency measured by Cronbach alpha values ranging from 0.52 to 0.89 in functioning and symptom scales.36,39,41 Internal consistency was found to be unacceptable for pain scores (alpha = 0.53 to 0.54) when EORTC QLQ-LC13 was used alone without the EORTC QLQ-C30 pain items.41 In another study, the Cronbach alpha for dyspnea was considered to be acceptable (alpha = 0.76).35 Responsiveness: Dyspnea, coughing, and pain scores improved over time between pretreatment and during treatment (statistically significant; P < 0.001).41 | No relevant studies on MID in patients with SCLC were identified. No studies with an MID were available in the overall population of patients with lung cancer or with NSCLC. |
BPI = Brief Pain Inventory; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-LC13 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Lung Cancer 13; FACT-G = Functional Assessment of Cancer Therapy–General; HADS = Hospital Anxiety and Depression Scale; HRQoL = health-related quality of life; MID = minimal important difference; NSCLC = non–small cell lung cancer; QoL = quality of life; SCLC = small cell lung cancer; SD = standard deviation; SSQ = subjective significance questionnaire.
Statistical Analysis
Sample Size and Power Calculation
The sample size was calculated based on a dual primary hypothesis for PFS and OS. Power calculations considered an alternative hypothesis with the following assumptions:
PFS has an HR of 0.65 when comparing durvalumab versus placebo; this corresponds to a 5.4-month median PFS benefit assumed under an exponential model for survival data (10 months for placebo versus 15.4 months for durvalumab).
OS has an HR of 0.73 when comparing durvalumab versus placebo; this corresponds to a 8.9-month difference in median OS under a treatment-specific exponential distribution (24 months for placebo versus 32.9 months for durvalumab).
Recruitment was planned for 38 months, with a total of 965 patients globally, to randomize approximately 724 patients across 3 groups: durvalumab (262), durvalumab plus tremelimumab (200), and placebo (262). A total of 370 PFS events (70.6% maturity) was required to achieve 90% power at a desired type I error rate of 5% (2 sided), controlled using the Lan-DeMets alpha spending function with O’Brien-Fleming boundaries.
Statistical Testing
The type I error was controlled for multiple hypotheses and interim analyses using a multiple testing hierarchical procedure. This testing procedure differed from the proposed strategy in the statistical analysis plan. An alpha of 0.5% was allocated for PFS, and a 4.5% alpha was allocated to OS. Should 1 of these tests be significant, the alpha could be recycled to the other primary analysis. If significance was achieved for both primary outcomes, testing proceeded to the secondary end points of OS and then PFS (durvalumab plus tremelimumab versus placebo).
The interim analyses based on the number of observed events were as follows. For the first interim analysis, the OS null hypothesis was rejected based on a significance level of 0.01679. The allocated 4.5% type I error was recycled to the PFS analysis, which rejected the first interim analysis hypothesis at the 0.02805 significance level.
A description of the statistical analysis of efficacy outcomes is included in Table 8. The dual primary outcomes of PFS and OS were tested for group differences using a stratified log-rank test. Treatment effect was summarized using HRs and associated 2-sided CIs based on a stratified Cox proportional hazards model. A Kaplan-Meier estimator was used to estimate PFS and OS distributions, and median values for each group were constructed along with 95% CIs using the Brookmeyer-Crowley method.
Subgroup Analyses
Prespecified subgroup analyses examined treatment effect difference in both PFS and OS using Cox proportional hazards models for various baseline characteristics, including:
tumour stage (stage I or II versus III)
timing from radiotherapy or chemotherapy to randomization
best response to prior treatment
age at randomization (< 65 versus ≥ 65 years).
These exploratory subgroup analyses included Kaplan-Meier curves as estimates of survival probabilities and forest plots of HRs. According to the clinical experts consulted for this review, tumour stage (I/II versus III) and ECOG Performance Status (0/1 versus 2) are key prognostic factors for OS and PFS in LS-SCLC. While ECOG Performance Status was not a subgroup analysis factor in the ADRIATIC trial, it is considered by the clinical experts to be an important determinant of treatment effect in clinical practice.
Sensitivity Analyses
The following analyses were conducted to evaluate the robustness of the PFS results based on the potential for bias due to attrition bias, informative censoring, misclassification, or stratification factors. Sensitivity analyses were conducted to evaluate the robustness of the PFS and OS results, addressing potential biases due to attrition, informative censoring, and stratification misclassification:
Alternative censoring rules were applied to assess attrition bias; patients with 2 or more missed RECIST 1.1 assessments before progression or death were censored at their last evaluable visit.
Informative censoring was explored by comparing stratification factors recorded at randomization with those derived from the electronic case report form data. Additionally, discrepancies between investigator-assessed and BICR-assessed progression events were analyzed to determine their impact on PFS estimates.
Stratification factor adjustments were made using electronic case report form data, ensuring standardized patient characteristics at randomization.
For OS, sensitivity analyses were adjusted for attrition, informative censoring, and misclassification of stratification factors to confirm the robustness of survival estimates under different assumptions.
Overall, these sensitivity analyses reinforced the reliability of OS and PFS findings, though moderate imprecision was noted due to variations in censoring assumptions.
Statistical Analysis of Secondary Outcomes
Two secondary end points — the proportions of patients alive and progression-free at 18 months and 24 months after randomization and the number of patients alive at 24 months and 36 months after randomization — were also included, as these are clinically meaningful landmark time points. These end points were estimated using the Kaplan-Meier method.
For secondary end points, statistical methods varied depending on the outcome. A mixed-effects model for repeated measures was used for longitudinal analyses of quality of life outcomes where appropriate, while other secondary end points were analyzed using descriptive statistics (e.g., DOR) or survival analysis methods (e.g., TTDM). Adjustment factors included baseline values and visit interactions for HRQoL analyses. However, these analyses were not controlled for multiplicity.
Table 8 summarizes the statistical approaches used, adjustment factors, handling of missing data, and sensitivity analyses performed for primary and secondary end points.
Table 8: Statistical Analysis of Efficacy End Points in the ADRIATIC Trial
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
OS | Stratified log-rank test and stratified Cox proportional hazards model. The treatment effect was estimated using the HR and corresponding CI. Landmark OS was estimated using the Kaplan-Meier method. | Adjusted for receipt of PCI (yes vs. no). | Patients not known to have died were censored at the last date they were known to be alive. | Sensitivity analyses included investigating potential attrition bias and misclassification of stratification factors. |
PFS | Stratified log-rank test and stratified Cox proportional hazards model. The treatment effect was estimated using the HR and corresponding CI. Landmark PFS was estimated using the Kaplan-Meier method. | Adjusted for TNM stage (I/II vs. III) and receipt of PCI (yes vs. no). | Patients who progressed or died after ≥ 2 missed visits were censored at the last evaluable assessment before the missed visits; patients without baseline data were censored at day 1 unless they died within 2 visits of baseline. | Sensitivity analyses included investigating evaluation time bias, attrition bias, ascertainment bias (using investigator-assessed RECIST 1.1 criteria), and alternative censoring rules for progression or death. |
TTDM | Stratified log-rank test and stratified Cox proportional hazards model. Kaplan-Meier curves were also estimated. | Adjusted for the stratification factors used in the analysis of PFS (TNM stage and PCI). | Patients were censored at the last evaluable RECIST 1.1 assessment if distant metastasis or death occurred after ≥ 2 missed visits. Patients with no baseline data or evaluable visits were censored at day 1 unless they died within 2 visits of baseline. | Investigator-assessed TTDM was analyzed as part of the sensitivity analyses. |
DOR | Kaplan-Meier estimator used to summarize DOR. Descriptive statistics were reported. | None. | Patients without documented disease progression following response were censored at the PFS censoring time. | None. |
EORTC QLQ-C30 and EORTC QLQ-LC13 time to deterioration | Stratified log-rank test. | Stratified by PCI status and stage. | Patients censored at the last evaluable assessment if ≥ 2 consecutive visits were missed or no baseline data are available. | None specified. |
EORTC QLQ-C30 and EORTC QLQ-LC13 symptom improvement rate | Logistic regression. | Adjusted for the same set of covariates as the primary PFS analysis. | Missing data imputed using the last observation carried forward method. | None specified. |
EORTC QLQ-C30 and EORTC QLQ-LC13 HRQoL or function improvement rate | Descriptive statistics. | NA. | Aligned with the analysis of change from baseline. | None specified. |
CI = confidence interval; DOR = duration of response; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-LC13 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Lung Cancer 13; HR = hazard ratio; HRQoL = health-related quality of life; NA = not applicable; OS = overall survival; PCI = polycranial irradiation; PFS = progression-free survival; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours version 1.1; TNM = tumour, node, metastasis; TTDM = time to death or distant metastasis; vs. = versus.
Sources: ADRIATIC Clinical Study Report and clinical study protocol; Cheng et al. (2024).24 Details included in the table are from the sponsor’s summary of clinical evidence.
Analysis Populations
The analysis populations of the ADRIATIC trial are summarized in Table 9.
Table 9: Analysis Populations of ADRIATIC Trial
Population | Definition | Application |
|---|---|---|
Full analysis set | The full analysis set includes all randomized patients. The treatment groups were compared on the basis of randomized study treatment, regardless of the treatment actually received. Patients who were randomized but did not subsequently go on to receive study treatment are included in the analysis in the treatment group to which they were randomized. | The full analysis set was used for all efficacy analyses (including patient-reported outcomes). |
Safety analysis set | The safety analysis set consists of all patients who received at least 1 dose of the study treatment. | Safety data are summarized according to the treatment received. |
Sources: ADRIATIC Clinical Study Report and clinical study protocol; Cheng et al. (2024).24
Results
In accordance with the interim analysis Clinical Study Report submitted by the sponsor, this Clinical Review Report presents data for the durvalumab and placebo groups as per data cut-off point of January 15, 2024. The OS analysis did not reach the prespecified O’Brien-Fleming boundary for statistical significance in the durvalumab plus tremelimumab versus placebo comparison. As a result, the durvalumab plus tremelimumab group remains blinded until the next scheduled analysis. Additionally, the sponsor did not pursue Health Canada approval or public reimbursement for durvalumab plus tremelimumab for this indication.
Patient Disposition
Patient disposition in the ADRIATIC study is summarized in Table 10. Of the 939 patients screened, 209 (22.3%) were enrolled but not randomized, with the main reasons being not meeting the screening criteria (19.6%), patient withdrawal (2.4%), or death (0.2%). A total of 530 patients were randomized across the 2 treatment groups considered in this review: 264 patients to durvalumab monotherapy and 266 patients to placebo. Most patients (99.6%) in both treatment groups received at least 1 dose of the study treatment.
Table 10: Summary of Patient Disposition in ADRIATIC Trial
Patient disposition | Durvalumab (N = 264) | Placebo (N = 266) |
|---|---|---|
Screened, N | 939 | |
Patients enrolled but not randomized, N (%) | 209 (22.3) | |
Patients randomized to durvalumab + tremelimumab group,a N (%) | 200 (21.3) | |
Reason not randomized, n (%) | ||
Did not meet screening criteria | 184 (19.6) | |
Withdrawal by patient | 23 (2.4) | |
Death | 2 (0.2) | |
Randomized, N | 264 | 266 |
Patients who received at least 1 dose of study treatment, n (%) | 263b (99.6) | 265c (99.6) |
Completed study treatmentd | 88 | 70 |
Ongoing on any study treatment, n (%) | 0 (0) | 0 (0) |
Discontinued from study treatment, n (%) | 175 (66.3) | 195 (73.3) |
Reason for discontinuation, n (%) | ||
Patient decision | 10 (3.8) | 11 (4.1) |
Adverse event | 43 (16.3) | 29 (10.9) |
Severe noncompliance to protocol | 0 (0) | 1 (0.4) |
Condition under investigation worsened | 121 (45.8) | 154 (57.9) |
Other | 1 (0.4) | 0 (0) |
Ongoing on study, n (%) | 140 (53.0) | 111 (41.7) |
Terminated study, n (%) | 124 (47.0) | 155 (58.3) |
Reason for termination of study, n (%) | ||
Death | 115 (43.6) | 144 (54.1) |
Withdrawal by patient | 9 (3.4) | 11 (4.1) |
FAS, N | 264 (100.0) | 266 (100.0) |
SAS, Ne | 262 (99.2) | 265 (99.6) |
FAS = full analysis set; SAS = safety analysis set.
Note: Data cut-off date of January 15, 2024.
aDurvalumab plus tremelimumab group remains blinded.
bOne patient from 264 patients missed the 3-day dosing window and subsequently withdrew.
cOne patient from 266 patients missed the 3-day dosing window and subsequently died.
dPatients who completed durvalumab have “maximum cycle of immunotherapy reached” on electronic case report form.
eOne patient randomized to the durvalumab group received 2 cycles of tremelimumab. This patient was not included in the durvalumab or placebo SAS.
Sources: ADRIATIC Clinical Study Report and clinical study protocol; Cheng et al. (2024).24 Details included in the table are from the sponsor’s summary of clinical evidence.
Treatment discontinuation was reported in 66.3% of patients in the durvalumab group and 73.3% in the placebo group. The primary reasons for discontinuation were disease progression (45.8% for durvalumab versus 57.9% for placebo), AEs (16.3% versus 10.9%), and patient decision (3.8% versus 4.1%). At the January 15, 2024, data cut-off date, 115 patients (43.6%) in the durvalumab group and 144 patients (54.1%) in the placebo group had died.
Baseline Characteristics
The baseline characteristics for the intention-to-treat population are outlined in Table 11 and include key factors relevant to this review because they are expected to affect the outcomes or interpretation of the trial results. The key characteristics were generally balanced across treatment groups. The trial population had a median age of 62 years, with 60.6% of patients aged younger than 65 years. The majority of participants were male (approximately 69%), and the majority were white (approximately 50%) or Asian (approximately 48%). All patients had a baseline WHO or ECOG Performance Status of 0 (approximately 49%) or 1 (approximately 51%), indicating relatively good functional status. Most patients (87%) had stage III disease based on American Joint Committee on Cancer classification, while smaller proportions had stage I disease (3%) or II disease (9%). Approximately 91% of patients had a history of smoking.
In terms of prior treatment, all participants had received concurrent CRT as per protocol requirements. The majority (88%) had completed 4 cycles of platinum-based chemotherapy, with cisplatin used in approximately 66% of cases and carboplatin in 34%. Around 5% of patients switched their platinum agent during treatment, with similar proportions in both treatment groups. Radiotherapy was administered once daily (total dose: 60 Gy to 66 Gy) in approximately 73.9% of patients in the durvalumab group and 70.3% of patients in the placebo group, and twice daily (total dose: 45 Gy) in 26.1% of patients and 29.7% patients in the placebo group. Approximately 54% of patients received PCI.
Table 11: Summary of Baseline Characteristics in ADRIATIC Trial
Characteristic | Durvalumab (N = 264) | Placebo (N = 266) |
|---|---|---|
Age (years) | ||
Mean (SD) | 61.8 (8.93) | 61.2 (9.40) |
Median (range) | 62.0 (28 to 84) | 62.0 (28 to 79) |
Age group (years), n (%) | ||
< 50 | 21 (8.0) | 26 (9.8) |
≥ 50 to < 65 | 139 (52.7) | 136 (51.1) |
≥ 65 to < 75 | 89 (33.7) | 86 (32.3) |
≥ 75 | 15 (5.7) | 18 (6.8) |
Sex, n (%) | ||
Male | 178 (67.4) | 188 (70.7) |
Female | 86 (32.6) | 78 (29.3) |
Race, n (%) | ||
White | 130 (49.2) | 137 (51.5) |
Black or African American | 1 (0.4) | 3 (1.1) |
Asian | 131 (49.6) | 121 (45.5) |
Other | 2 (0.8) | 5 (1.9) |
Ethnic group, n (%) | ||
Hispanic or Latino | 8 (3.0) | 14 (5.3) |
Not Hispanic or Latino | 255 (96.6) | 249 (93.6) |
Missing | 1 (0.4) | 3 (1.1) |
Smoking history, n (%) | ||
Never | 23 (8.7) | 26 (9.8) |
Smoker | 241 (91.3) | 240 (90.2) |
Ex-smoker | 178 (67.4) | 185 (69.5) |
Current smoker | 63 (23.9) | 55 (20.7) |
ECOG Performance Status, n (%) | ||
(0) Normal activity | 132 (50.0) | 126 (47.4) |
(1) Restricted activity | 132 (50.0) | 140 (52.6) |
AJCC overall stage,a n (%) | ||
I | 8 (3.0) | 11 (4.1) |
II | 25 (9.5) | 23 (8.6) |
III | 231 (87.5) | 232 (87.2) |
PD-L1 status,b n (%) | ||
Tumour cell AND immune cell < 1% | 78 (29.5) | 73 (27.4) |
Tumour cell OR immune cell ≥ 1% | 84 (31.8) | 98 (36.8) |
Missing | 102 (38.6) | 95 (35.7) |
Extent of disease at baseline,c n (%) | ||
No evidence of disease | 32 (12.1) | 34 (12.8) |
Locally advanced (total) | 232 (87.9) | 232 (87.2) |
Respiratory | 199 (75.4) | 209 (78.6) |
Lymph nodes | 167 (63.3) | 148 (55.6) |
Number of chemotherapy cycles, n (%) | ||
2 | 0 | 1 (0.4) |
3 | 29 (11.0) | 31 (11.7) |
4 | 234 (88.6) | 234 (88.0) |
6 | 1 (0.4) | 0 |
Chemotherapy regimen,d n (%) | ||
Cisplatin + etoposide | 173 (65.5) | 178 (66.9) |
Carboplatin + etoposide | 91 (34.5) | 88 (33.1) |
Radiotherapy regimen,e n (%) | ||
Once daily | 195 (73.9) | 187 (70.3) |
Twice daily | 69 (26.1) | 79 (29.7) |
Best response to concurrent CRT, n (%) | ||
Complete response | 31 (11.7) | 34 (12.8) |
Partial response | 191 (72.3) | 200 (75.2) |
Stable disease | 42 (15.9) | 32 (12.0) |
PCI regimen, n (%) | ||
Yes | 142 (53.8) | 143 (53.8) |
No | 122 (46.2) | 123 (46.2) |
AJCC = American Joint Committee on Cancer; CRT = chemoradiotherapy; ECOG = Eastern Cooperative Oncology Group; PCI = prophylactic cranial irradiation; SD = standard deviation.
Note: Data cut-off date of January 15, 2024.
aAJCC stage is derived from the tumour, node, and metastasis stage; AJCC overall stage is from the time of diagnosis as reported by the investigator on the electronic case report form. The AJCC 8th Edition is used.
bTesting was retrospective and not required for randomization.
cA patient could have 1 or more sites of disease reported.
dChemotherapy regimen was based on the first cycle of chemotherapy.
eChest irradiation.
Sources: ADRIATIC Clinical Study Report and clinical study protocol; Cheng et al. (2024).24 Details included in the table are from the sponsor’s summary of clinical evidence.
Exposure to Study Treatments
By the data cut-off date of January 15, 2024, in the ADRIATIC trial, the median duration of treatment was 40.0 weeks (mean = 54.5 weeks; range, 4 to 108 weeks) in the durvalumab group and 35.9 weeks (mean = 49.0 weeks; range, 4 to 108 weeks) in the placebo group. The on-treatment time, excluding interruptions and delays, was similar between the groups, with a median actual on-treatment time of 36.6 weeks in the durvalumab group and 35.9 weeks in the placebo group. The median number of treatment cycles was 9.0 in both groups, with 76.0% of patients in the durvalumab group and 75.5% of patients in the placebo group receiving at least 4 cycles. Additionally, 29.4% of patients in the durvalumab group and 23.4% in the placebo group received at least 24 cycles.
Concomitant Medications and Co-Interventions
Most patients (93.6%) in the ADRIATIC trial received at least 1 concomitant medication during the study, with slightly higher use in the durvalumab group (95.8%) than in the placebo group (91.4%). Commonly used medication classes included glucocorticoids (47.9%), proton pump inhibitors (43.6%), anilides (23.8%), and HMG-CoA reductase inhibitors (23.0%). Prohibited corticosteroid use exceeding 10 mg/day of prednisone or equivalent occurred in 2.1% of patients (4 patients in the durvalumab group and 7 in the placebo group).
Subsequent Treatment
Treatments administered after study discontinuation are summarized in Table 12. After treatment discontinuation, 95 patients (36.0%) in the durvalumab group and 127 patients (47.7%) in the placebo group received post-discontinuation anticancer therapy. Cytotoxic chemotherapy was the most common posttreatment intervention, received by 47 patients (17.8%) in the durvalumab group and 57 patients (21.4%) in the placebo group as single-agent therapy. Platinum-based doublet chemotherapy was the most frequently administered regimen, used by 54 patients (20.5%) in the durvalumab group and 56 patients (21.1%) in the placebo group. Immunotherapy was administered to 7 patients (2.7%) in the durvalumab group and 7 patients (2.6%) in the placebo group as single-agent therapy, while combination chemotherapy and immunotherapy was used in 13 patients (4.9%) and 28 patients (10.5%) in the durvalumab and placebo groups, respectively. The number of patients receiving second-line therapy was 92 (34.8%) for durvalumab and 124 (46.6%) for placebo, with third-line or later treatment administered in 22 patients (8.3%) in both groups.
Table 12: Post-Discontinuation Anticancer Therapy in ADRIATIC Trial, FAS
Treatment | Durvalumab (N = 264) | Placebo (N = 266) |
|---|---|---|
Discontinued study treatment, n (%) | 175 (66.5) | 195 (73.6) |
Received post-discontinuation disease-related anticancer therapy, n (%) | 95 (36.0) | 127 (47.7) |
Types of posttreatment anticancer therapy received, n (% of FAS) [% of patients who discontinued treatment]a | ||
Cytotoxic chemotherapy, single agent | 47 (17.8) [49.5] | 57 (21.4) [44.9] |
Cytotoxic chemotherapy, platinum doublet | 54 (20.5) [56.8] | 56 (21.1) [44.1] |
Chemotherapy + immunotherapyb | 13 (4.9) [13.7] | 28 (10.5) [22.0] |
Chemotherapy + targeted therapy | 6 (2.3) [6.3] | 7 (2.6) [5.5] |
Chemotherapy + immunotherapy + targeted therapyb | 1 (0.4) [1.1] | 4 (1.5) [3.1] |
Other chemotherapy combination | 16 (6.1) [16.8] | 18 (6.8) [14.2] |
Hormonal therapy | 0 | 0 |
Immunotherapy, single agentb | 7 (2.7) [7.4] | 7 (2.6) [5.5] |
Immunotherapy + immunotherapyb | 2 (0.8) [2.1] | 1 (0.4) [0.8] |
Immunotherapy + targeted therapyb | 1 (0.4) [1.1] | 3 (1.1) [2.4] |
Immunotherapy + antibody drug conjugateb | 0 | 0 |
Radiopharmaceuticals | 0 | 0 |
Targeted therapy, single agent | 5 (1.9) [5.3] | 5 (1.9) [3.9] |
Targeted therapy + targeted therapy | 1 (0.4) [1.1] | 0 |
Targeted therapy + antibody drug conjugate therapy | 0 | 0 |
Antibody drug conjugate, single agent | 1 (0.4) [1.1] | 1 (0.4) [0.8] |
Investigational agent | 0 | 1 (0.4) [0.8] |
Investigational antineoplastic drugs | 0 | 1 (0.4) [0.8] |
Other | 0 | 0 |
Line of treatment, n (% of FAS) [% of patients who discontinued treatment]a,c | ||
Second line | 92 (34.8) [96.8] | 124 (46.6) [97.6] |
Third line | 47 (17.8) [49.5] | 56 (21.1) [44.1] |
> Third line | 22 (8.3) [23.2] | 22 (8.3) [17.3] |
Not applicable | 4 (1.5) [4.2] | 1 (0.4) [0.8] |
FAS = full analysis set.
Note: Data cut-off date of January 15, 2024.
aThe number of patients is shown with the first percentages (%) calculated as the proportion of patients in the FAS and the second percentages [%] as the proportion of patients who discontinued randomized study treatment.
bImmunotherapy includes any therapy in which at least 1 mechanism of action involves modulation of the immune system.
cA patient can be counted in 1 or more of the categories.
Sources: ADRIATIC Clinical Study Report and clinical study protocol; Cheng et al. (2024).24 Details included in the table are from the sponsor’s summary of clinical evidence.
Efficacy
The primary efficacy outcomes were OS and PFS. The primary results include estimated HRs. The secondary results include the probability of OS at 24 and 36 months and the probability of PFS at 12 and 24 months. Summary statistics for objective response rate and TTDM are also reported. The data cut-off date for this analysis was January 15, 2024.
Overall Survival
A summary of the results for OS is provided in Table 13. At the time of the interim analysis for OS, 261 deaths had been reported across the treatment groups. The median duration of OS follow-up was 30.75 months in the durvalumab group and 28.63 months in the placebo group.
Treatment with durvalumab resulted in a statistically significant improvement in OS compared to placebo. The estimated HR was 0.73 (95% CI, 0.569 to 0.928; P = 0.01042), which corresponds to a 27% reduced hazard of death on treatment with durvalumab compared to placebo over the study period. The median OS was 55.9 months (95% CI, 37.3 months to not estimable) in the durvalumab group compared to 33.4 months (95% CI, 25.5 to 39.9 months) in the placebo group, indicating an estimated improvement in median OS of 22.5 months (95% CI, 1.1 to 28.6 months).
At the 24-month and 36-month landmark analyses, the survival probabilities were higher in the durvalumab group (68.0% and 56.5%, respectively) than in the placebo group (58.5% and 47.6%, respectively), with differences of 9.5% (95% CI, 1.0% to 17.8%) and 8.9% (95% CI, −0.1% to 17.8%), respectively. Figure 2 illustrates the Kaplan-Meier plot of OS and demonstrates a sustained separation in favour of durvalumab beginning around 8 months after randomization.
Sensitivity Analyses: OS
The prespecified sensitivity analyses supported the robustness of the primary OS analysis (Appendix 1, Table 19 and Figure 5) by addressing potential biases related to censoring, attrition, and baseline covariates.
Cox proportional hazards model: The Cox proportional hazards model, stratified by relevant factors and adjusted for baseline covariates (e.g., age, sex, smoking status, region, and prior treatments), produced an HR of 0.75 (95% CI, 0.585 to 0.966), consistent with the primary analysis (Appendix 1, Table 20).
Censoring patterns analysis: A Kaplan-Meier analysis of time to censoring was conducted to explore attrition bias. The censoring indicator for OS was reversed to evaluate whether differences in censoring patterns between treatment arms influenced results. No major imbalances were detected, supporting the validity of the OS analysis (Appendix 1, Figure 5).
Effect of covariates on OS HR: A multivariable Cox proportional hazards model was used to examine the effect of key baseline covariates on OS. This analysis confirmed that the OS findings were consistent across patient subgroups, reinforcing the robustness of the primary OS results (Appendix 1, Table 20).
OS Subgroup Analyses
The subgroup analysis for OS included the following main covariates: tumour stage (stage I/II versus III), timing from radiotherapy or chemotherapy to randomization, best response to prior treatment, and age at randomization (< 65 years versus ≥ 65 years).
The results for these subgroups are presented in Figure 3 with HRs and CIs, indicating the observed OS estimates across these predefined categories. The OS HRs across the selected subgroups indicate comparable OS outcomes. Some subgroups, such as patients with stage I/II disease and patients experiencing stable disease, have wider CIs, likely due to smaller sample sizes. No significant differences in OS were observed among these subgroups. However, due to small sample sizes and wide CIs, no firm conclusions can be made.
Table 13: OS (Using BICR Assessments According to RECIST 1.1) (FAS, OS-IA1), ADRIATIC Trial
OS | Durvalumab (N = 264) | Placebo (N = 266) |
|---|---|---|
Deaths, n (%) | 115 (43.6) | 146 (54.9) |
Censored patients, n (%) | 149 (56.4) | 120 (45.1) |
OS (months),a median (95% CI) | 55.9 (37.3 to not estimable) | 33.4 (25.5 to 39.9) |
Difference in medians, months (95% CI) | 22.5 (1.1 to 28.6) | |
HR, durvalumab vs. placebob | 0.73 | |
95% CI for HRb | 0.569 to 0.928 | |
2-sided P valuec | 0.01042 | |
Survival probability at 24 months,a % (95% CI) | 68.0 (61.9 to 73.3) | 58.5 (52.3 to 64.3) |
Difference in survival probability, % (95% CI) | 9.5 (1.0 to 17.8) | |
Survival probability at 36 months,a % (95% CI) | 56.5 (50.0 to 62.5) | 47.6 (41.3 to 53.7) |
Difference in survival probability, % (95% CI) | 8.9 (−0.1 to 17.8) | |
BICR = blinded independent central review; CI = confidence interval; FAS = full analysis set; HR = hazard ratio; NR = not reported; OS = overall survival; OS-IA1 = first interim analysis of overall survival; PCI = prophylactic cranial irradiation; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours version 1.1; vs. = versus.
Notes: Data cut-off date of January 15, 2024.
One month is calculated as 30.4375 days.
The stratification factor is based on the values entered the interactive voice response system.
aCalculated using the Kaplan-Meier technique. The CI for the median OS is derived based on the Brookmeyer-Crowley method with log-log transformation, and the Kaplan-Meier method is based on log-log transformation.
bThe HR and CI were calculated using a stratified Cox proportional hazards model, adjusting for receipt of PCI (yes vs. no), with treatment as the only covariate and ties handled by the Efron approach. CIs were calculated using the profile likelihood approach.
cThe analysis was performed using the stratified log-rank test, adjusting for receipt of PCI (yes vs. no).
Sources: ADRIATIC Clinical Study Report and clinical study protocol; Cheng et al. (2024).24 Details included in the table are from the sponsor’s summary of clinical evidence.
Figure 2: Kaplan-Meier Plot of Overall Survival in ADRIATIC Trial (FAS, OS-IA1)
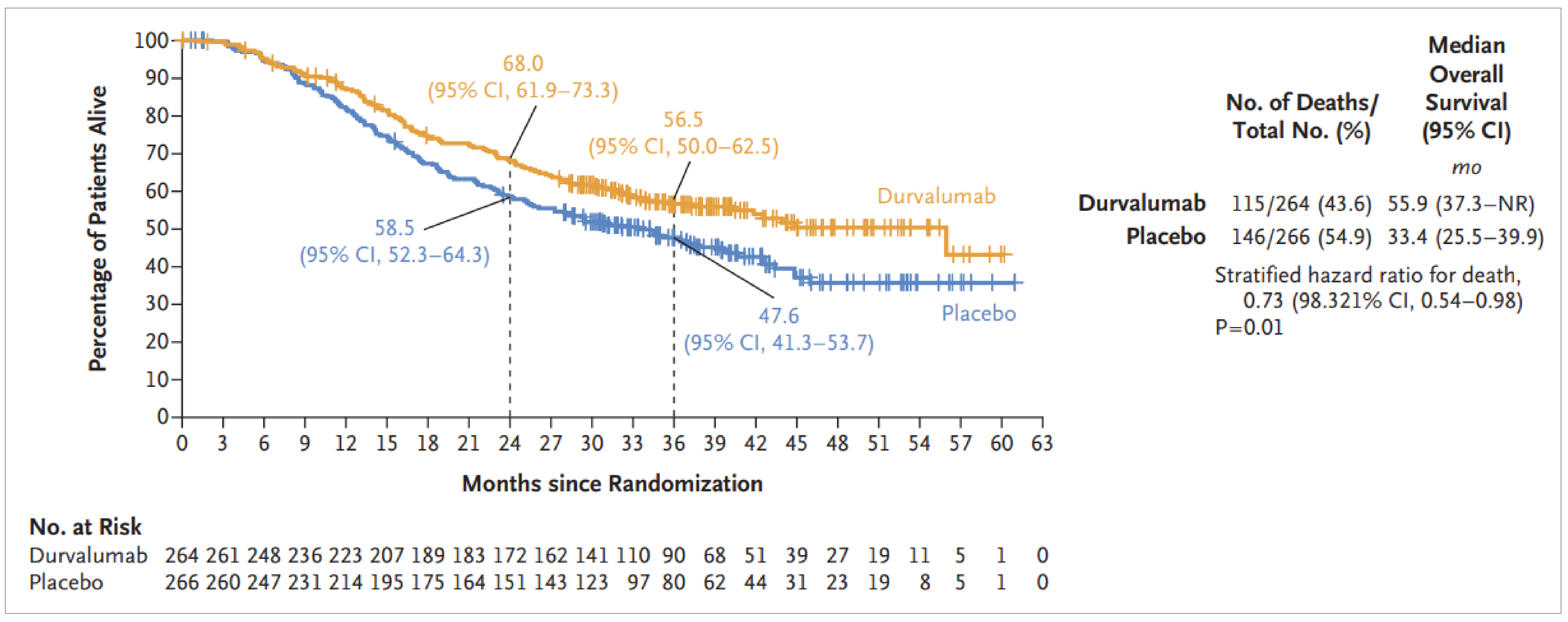
CI = confidence interval; FAS = full analysis set; mo = months; NR = not reached; OS = overall survival; OS-IA1 = first interim analysis of overall survival; PCI = prophylactic cranial irradiation.
Notes: Data cut-off: January 15, 2024. OS was analyzed using a stratified log-rank test adjusted for receipt of PCI (yes versus no). The significance level for testing OS in this interim analysis was 0.01679 (2 sided) at the overall 4.5% level, allowing for strong alpha control across interim and final analysis time points.
Sources: ADRIATIC Clinical Study Report and clinical study protocol; Cheng et al. (2024).24
Figure 3: Overall Survival in ADRIATIC Trial: Forest Plot by Subgroup (FAS, OS-IA1)
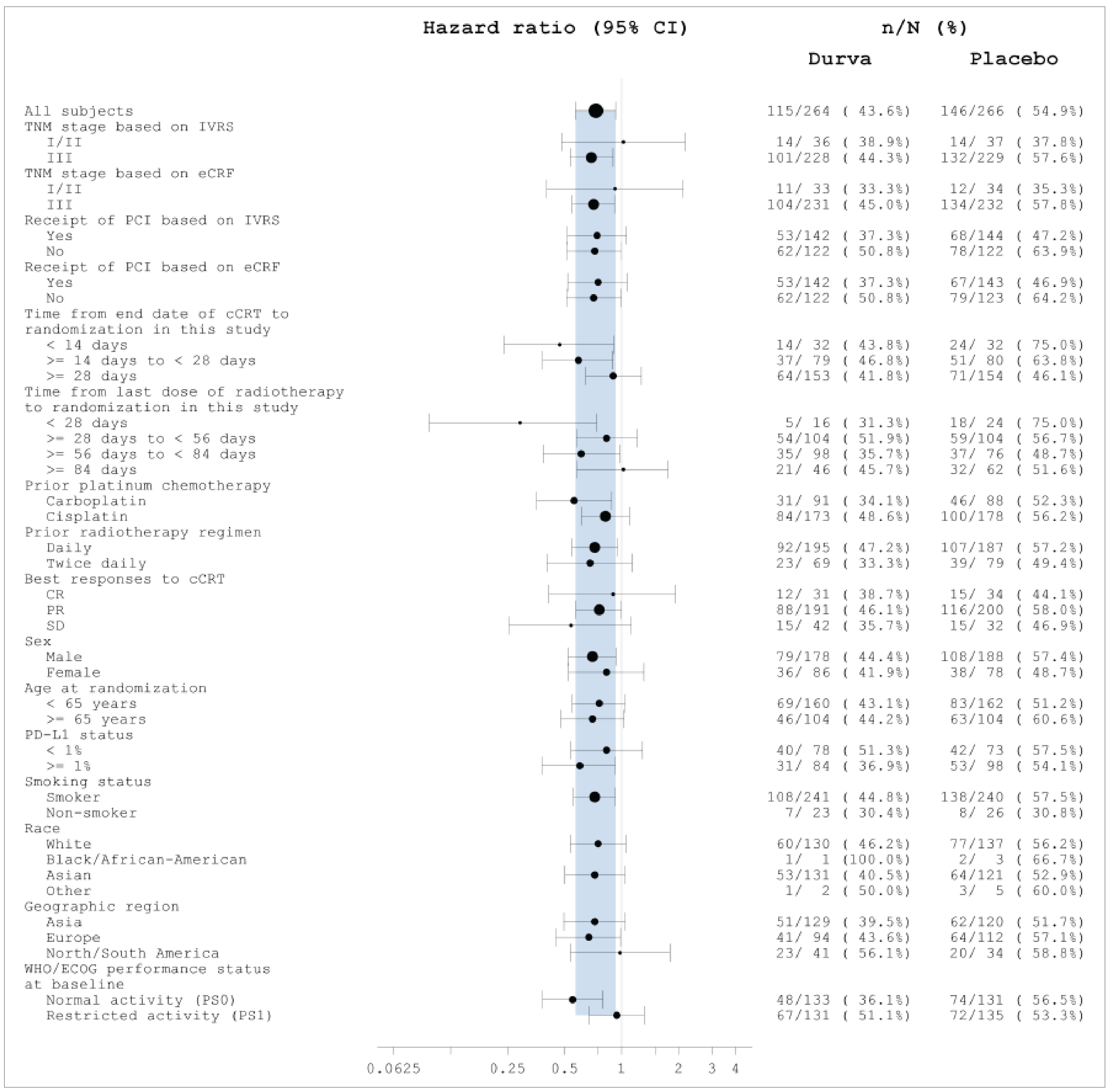
cCRT = concurrent chemoradiotherapy; CI = confidence interval; CR = complete response; Durva = durvalumab; ECOG = Eastern Cooperative Oncology Group; eCRF = electronic case report form; FAS = full analysis set; IVRS = interactive voice response system; OS-IA1 = first interim analysis of overall survival; PCI = prophylactic cranial irradiation; PR = partial response; SD = stable disease; TNM = tumour, node, metastasis.
Notes: Data cut-off: January 15, 2024. Hazard ratios (durvalumab versus placebo) and 95% CIs are displayed on a logarithmic scale. The size of the circle is proportional to the number of events. The band represents the 95% CI for the main overall survival hazard ratio.
Sources: ADRIATIC Clinical Study Report and clinical study protocol; Cheng et al. (2024).24
Progression-Free Survival
At the planned interim analysis of PFS (data cut-off: January 15, 2024), the median follow-up duration for PFS was 9.07 months in the durvalumab group and 7.39 months in the placebo group, with a total of 308 PFS events observed across both groups.
The HR for PFS was 0.76 (95% CI, 0.606 to 0.950; P = 0.01608) in favour of durvalumab, which corresponds to a 24% reduced hazard of progression or death on treatment compared to placebo over the study period. The median PFS was 16.6 months (95% CI, 10.2 to 28.2 months) in the durvalumab group, compared to 9.2 months (95% CI, 7.4 to 12.9 months) in the placebo group, translating to a difference of 7.4 months (95% CI, −0.1 to 20.5 months) in median PFS. Table 14 summarizes the key PFS findings.
The Kaplan-Meier plot of PFS illustrated separation between the curves for durvalumab and placebo starting at around 6 months, which persisted throughout the analysis period (Figure 4). At the 18-month and 24-month landmark analyses, more patients in the durvalumab group than in the placebo group remained progression-free: 48.8% (95% CI, 42.2% to 55.0%) versus 36.1% (95% CI, 29.9% to 42.2%) at 18 months, and 46.2% (95% CI, 39.6% to 52.5%) versus 34.2% (95% CI, 28.2% to 40.3%) at 24 months, with differences of 12.7% (95% CI, −0.2% to 17.5%) and 12.0% (95% CI, 3.2% to 21.1%), respectively.
Sensitivity Analysis
Sensitivity analyses were performed to evaluate the consistency of PFS results under different conditions:
The HR for PFS based on BICR was 0.76 (95% CI, 0.606 to 0.950), with nearly the same number of patients experiencing PFS in each group: 139 of 264 (52.7%) in the durvalumab group and 169 of 266 (63.5%) in the placebo group.
To address potential evaluation time bias, an interval-censored analysis produced an HR of 0.75 (95% CI, 0.599 to 0.940).
An analysis using alternative censoring rules for PFS resulted in an HR of 0.77 (95% CI, 0.616 to 0.964). The number of PFS events was slightly higher: 142 of 264 (53.8%) events in the durvalumab group and 169 of 266 (63.5%) events in the placebo group.
An analysis of PFS based on investigator assessments yielded an HR of 0.76 (95% CI, 0.615 to 0.948). The number of events further increased: 153 of 264 (58.0%) in the durvalumab group and 179 of 266 (67.3%) in the placebo group (Figure 6 in Appendix1).
Table 14: PFS (Using BICR Assessments According to RECIST 1.1) (FAS, PFS-IA), ADRIATIC Trial
PFS | Durvalumab (N = 264) | Placebo (N = 266) |
|---|---|---|
Total PFS events,a n (%) | 139 (52.7) | 169 (63.5) |
RECIST 1.1 progression | 126 (47.7) | 158 (59.4) |
Death in absence of progression | 13 (4.9) | 11 (4.1) |
Censored patients, n (%) | 125 (47.3) | 97 (36.5) |
Median PFS (months),b (95% CI) | 16.6 (10.2 to 28.2) | 9.2 (7.4 to 12.9) |
Difference in medians, months (95% CI) | 7.4 (−0.1 to 20.5) | |
HR, durvalumab vs. placeboc,d | 0.76 | |
95% CI for HRc | 0.606 to 0.950 | |
2-sided P valuee | 0.01608 | |
PFS at 18 months,b % (95% CI) | 48.8 (42.2 to 55.0) | 36.1 (29.9 to 42.2) |
Difference in PFS rate, % (95% CI) | 12.7 (−0.2 to 17.5) | |
PFS at 24 months,b % (95% CI) | 46.2 (39.6 to 52.5) | 34.2 (28.2 to 40.3) |
Difference in PFS rate, % (95% CI) | 12.0 (3.2 to 21.1) | |
BICR = blinded independent central review; CI = confidence interval; FAS = full analysis set; HR = hazard ratio; PCI = prophylactic cranial irradiation; PFS = progression-free survival; PFS-IA = interim analysis of progression-free survival; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours version 1.1; TNM = tumour, node, metastasis; vs. = versus.
Note: Data cut-off: January 15, 2024.
aPatients who had not experienced disease progression or who died, or who experienced disease progression or died after 2 or more missed visits, were censored at the latest evaluable RECIST 1.1 assessment or on day 1 if there were no evaluable visits. Patients who experienced RECIST 1.1 progression within 2 visits of baseline who did not have any evaluable visits or did not have a baseline assessment were censored at day 1.
bCalculated using the Kaplan-Meier technique. The CI for median PFS is derived based on the Brookmeyer-Crowley method with log-log transformation. The CIs for PFS at 18 and 24 months are derived based on a log(-log(.)) transformation.
cThe HR and CI were calculated using a stratified Cox proportional hazards model, adjusting for TNM stage (stage I/II vs. III) and receipt of PCI (yes vs. no), with treatment as the only covariate and ties handled by the Efron approach. CIs were calculated using the profile likelihood approach.
dAn HR less than 1 favours durvalumab being associated with longer event-free survival than placebo.
eThe analysis was performed using the stratified log-rank test, adjusting for TNM stage (stage I/II vs. III) and receipt of PCI (yes vs. no).
Sources: ADRIATIC Clinical Study Report and clinical study protocol; Cheng et al. (2024).24 Details included in the table are from the sponsor’s summary of clinical evidence.
Figure 4: Kaplan-Meier Plot of PFS (Using BICR per RECIST 1.1) (FAS, PFS-IA), ADRIATIC Trial
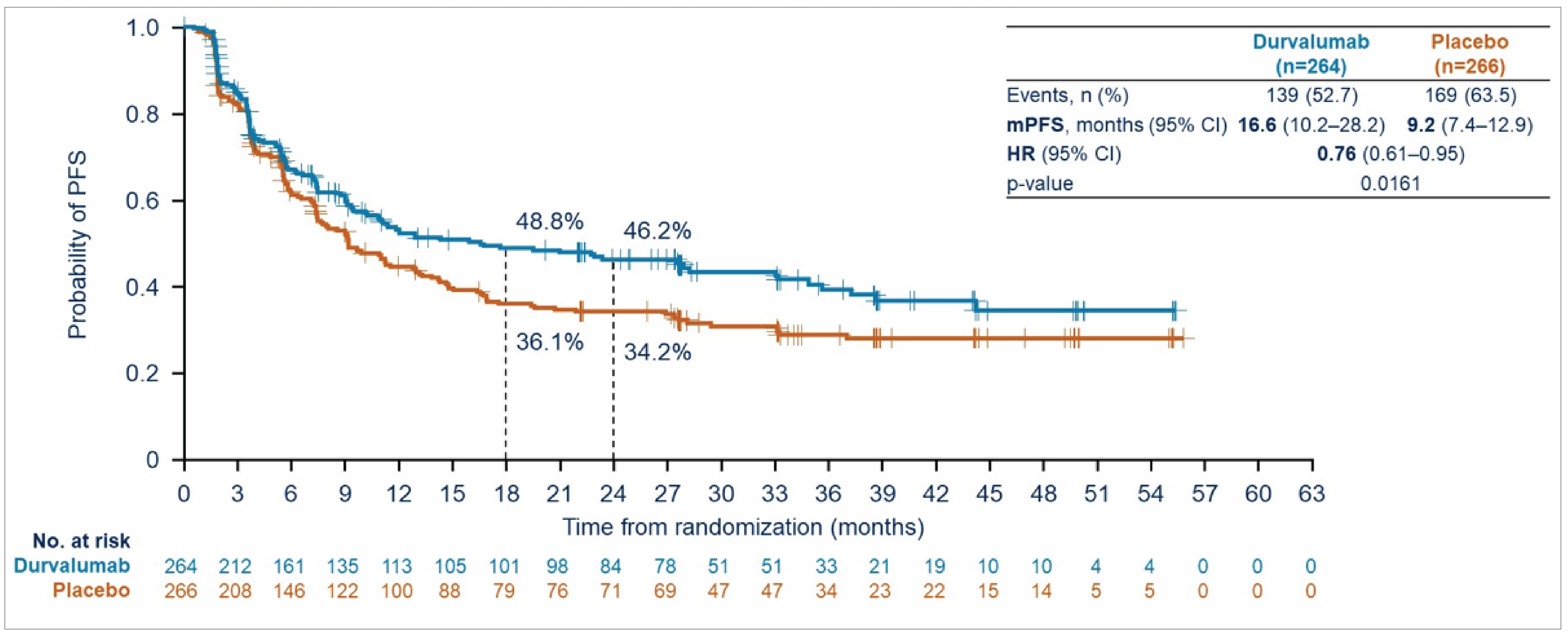
BICR = blinded independent central review; CI = confidence interval; FAS = full analysis set; HR = hazard ratio; mPFS = median progression-free survival; PCI = prophylactic cranial irradiation; PFS = progression-free survival; PFS-IA = interim analysis of progression-free survival; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours version 1.1.
Notes: Data cut-off: January 15, 2024. PFS was analyzed using a stratified log-rank test adjusted for disease stage (I/II versus III) and receipt of PCI (yes versus no). The significance level for testing PFS at this interim analysis was 0.00184 (2 sided) at the 0.5% level and 0.02805 (2 sided) at the overall 5% level. Statistical significance for PFS was achieved through the recycling multiple testing procedure framework and testing at the 5% (2-sided) alpha level (adjusted for an interim and final analysis).
Sources: ADRIATIC Clinical Study Report and clinical study protocol; Cheng et al. (2024).24
Duration of Response
A summary of DOR results is presented in Table 15 at the time of the interim analysis (data cut-off date: January 15, 2024).
For unconfirmed responses, the median DOR was 33.0 months (95% CI, 22.4 months to not estimable) in the durvalumab group and 27.7 months (95% CI, 9.6 months to not estimable) in the placebo group. For confirmed responses, the median DOR was 38.8 months (95% CI, 25.9 months to not estimable) in the durvalumab group compared to 27.8 months (95% CI, 9.9 months to not estimable) in the placebo group.
The Kaplan-Meier estimated probability of maintaining response at 6, 12, and 18 months was higher in the durvalumab group than in the placebo group for both confirmed and unconfirmed responses. After 18 months, the Kaplan-Meier–estimated probability of remaining in response for unconfirmed responses was 71.5% (95% CI, not estimable) versus 55.2% (95% CI, not estimable) in the durvalumab and placebo groups, respectively. For confirmed responses, █████ (95% CI, ██) versus █████ (95% CI, ██) of patients had disease that remained in response at 18 months in the durvalumab and placebo groups, respectively.
Table 15: DOR (Using BICR per RECIST 1.1) (PFS-IA), ADRIATIC Trial
DOR | Durvalumab (N = 264) | Placebo (N = 266) |
|---|---|---|
Unconfirmed response | ||
Patients who experienced response, N | 53 | 54 |
Patients who experienced response and subsequently experienced disease progression or died, n (%) | 22 (41.5) | 23 (42.6) |
DOR from onset of response (months)a,b | ||
Median (95% CI) | 33.0 | 27.7 |
Difference in medians (95% CI) | 5.3 (−1.9 to 28.8) | |
Hazard ratio (95% CI) | NR | |
P value | NR | |
Share of patients whose disease is remaining in response,b % | ||
At 18 months | 71.5 | 55.2 |
Difference (95% CI) | 16.3 (NR) | |
Confirmed response | ||
Patients who experienced response, N | 45 | 44 |
Patients who experienced response and subsequently | 16 (35.5) | 19 (43.2) |
DOR from onset of response (months)a,b | ||
Median (95% CI) | 38.8 | 27.8 |
Difference in medians (95% CI) | 11.0 (−1.8 to 24.0) | |
Hazard ratio (95% CI) | NR (NR) | |
P value | NR | |
Share of patients whose disease is remaining in response,b % (95% CI) | ||
At 18 months | ████ ████ | ████ ████ |
Difference (95% CI) | ████ ████ | |
BICR = blinded independent central review; CI = confidence interval; DOR = duration of response; NR = not reported; PFS-IA = interim analysis of progression-free survival; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours version 1.1.
Note: Data cut-off: January 15, 2024.
aDOR is the time from the first documented response (complete response or partial response) until the date of first documented progression, or death in the absence of disease progression. Patients who had not experienced disease progression or who had died, or who experienced disease progression or died after 2 or more missed visits, were censored at the latest evaluable RECIST 1.1 assessment, or day 1 if there were no evaluable visits.
bCalculated using the Kaplan-Meier technique. CI for median DOR is derived based on the Brookmeyer-Crowley method with log-log transformation.
Sources: ADRIATIC Clinical Study Report and clinical study protocol; Cheng et al. (2024).24 Details included in the table are from the sponsor’s summary of clinical evidence.
Time to Death or Distant Metastasis
The results are summarized in Table 16. During the study, it was observed that some new lesions retrospectively identified by BICR had incomplete or inaccurate date and location descriptions. While this may have affected TTDM assessment by BICR, it did not impact the identification or timing of progression events for the primary end point of PFS. TTDM as assessed by the investigators remained unaffected.
At the time of analysis (data cut-off date: January 15, 2024), TTDM events had occurred in ██ ███████ patients in the durvalumab group and ███ ███████ patients in the placebo group. Among these, distant metastasis was reported in ██ ███████ and ██ ███████ patients, respectively, while death in the absence of distant metastasis occurred in ██ ██████ and ██ ██████ patients, respectively.
The median TTDM was ███ ███████ in the durvalumab group (95% CI, █████ ███ ███████) and was ████ ██████ (95% CI, █████ ███ ████████ in the placebo group. The estimated HR for TTDM was ████ (95% CI, ██████ ██████, with a 2-sided P value of ██████.
Table 16: Time to Death or Distant Metastasis (Using BICR per RECIST 1.1) (FAS, PFS-IA), ADRIATIC Trial
TTDM | Durvalumab (N = 264) | Placebo (N = 266) |
|---|---|---|
Total events, n (%) | ██ ██████ | ███ ██████ |
Distant metastasis | ██ ██████ | ██ ██████ |
Death in the absence of distant metastasis | ██ █████ | ██ █████ |
Censored patients, n (%) | ███ ██████ | ███ ██████ |
TTDM (months),a median (95% CI) | ███ ███████ █ | ████ ██████ █ |
HR, durvalumab vs. placebob (95% CI) | ████ ███████ ██████ | |
2-sided P value | ██████ | |
TTDM at landmark time points | NR | NR |
BICR = blinded independent central review; CI = confidence interval; FAS = full analysis set; HR = hazard ratio; NR = not reported; PCI = prophylactic cranial irradiation; PFS-IA = interim analysis for progression-free survival; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours version 1.1; TNM = tumour, node, metastasis; TTDM = time to death or distant metastasis; vs. = versus.
Note: Data cut-off: January 15, 2024.
aEstimated using the Kaplan-Meier technique. CI for median time to event is derived based on the Brookmeyer-Crowley method with log-log transformation.
bThe HR and CI were calculated using a stratified Cox proportional hazards model, adjusting for TNM stage (stage I/II vs. III) and receipt of PCI (yes vs. no), with treatment as the only covariate and ties handled by the Efron approach. CIs were calculated using the profile likelihood approach. The P value was calculated using the stratified log-rank test, adjusting for TNM stage (stage I/II vs. III) and receipt of PCI (yes vs. no).
Sources: ADRIATIC Clinical Study Report and clinical study protocol; Cheng et al. (2024).24 Details included in the table are from the sponsor’s summary of clinical evidence.
Patient-Reported Outcomes
PRO Completion and Compliance
At baseline, completion rates for the EORTC QLQ-C30 were 81.8% in the durvalumab group and 79.7% in the placebo group. Compliance remained at least 73% through week 16, more than 60% through week 36, and more than 50% through week 84 in both groups. The overall compliance rate for EORTC QLQ-C30 was 57.5% in the durvalumab group and 59.6% in the placebo group.
For EORTC QLQ-LC13, baseline compliance rates were 82.2% for the durvalumab group and 80.8% for the placebo group. Compliance remained more than 70% through week 16, more than 60% through week 36, and more than 50% through week 84 in both groups. The overall compliance rate for EORTC QLQ-LC13 was 61.2% in the durvalumab group and 63.3% in the placebo group.
Baseline PRO Scores
Baseline global health status/quality of life scores were comparable between groups, with mean scores above 70, indicating good functional health at study entry. Functional scores exceeded 80, surpassing general LS-SCLC reference values. Among symptom scales, fatigue had the highest baseline burden (mean score for durvalumab: 23.66; mean score for placebo: 25.21). The primary symptoms assessed from the EORTC QLQ-LC13 (dyspnea, cough, and chest pain) were comparable between groups, with mean scores below 27 at baseline.
Change From Baseline in PRO Scores
Both groups experienced declines in functioning over time, with no statistically significant differences between treatment groups for global health status, physical functioning, role functioning, fatigue, dyspnea, cough, or chest pain (all P > 0.05). The only statistically significant difference was observed for appetite loss, for which the placebo group showed greater improvement than the durvalumab group: the adjusted mean change from baseline for durvalumab was −6.1 (95% CI, −8.345 to −3.869) and for placebo was −8.8 (95% CI, −11.092 to −6.603). The least squares mean difference was 2.7 (95% CI, 0.151 to 5.331; P = 0.0381). Table 17 shows details for the change from baseline in PROs.
Improvement Rates
There was no statistically significant difference between durvalumab and placebo for most symptom end points assessed in the ADRIATIC trial. The only statistically significant difference was observed in chest pain, for which durvalumab was associated with greater improvement than placebo (odds ratio = 2.28; 95% CI, 1.078 to 4.951; P = 0.0308). There was no significant difference between treatment groups for fatigue (odds ratio = 1.30; 95% CI, 0.845 to 2.006; P = 0.2328), appetite loss (odds ratio = 1.04; 95% CI, 0.551 to 1.951; P = 0.9092), dyspnea (odds ratio = 1.28; 95% CI, 0.822 to 1.986; P = 0.2780), or cough (odds ratio = 1.36; 95% CI, 0.831 to 2.221; P = 0.2235). Table 21 in Appendix 2 shows the details for improvement rates in PROs.
Table 17: Adjusted Mean Change From Baseline in Key EORTC QLQ-C30 and QLQ-LC13 End Points (FAS, PFS-IA), ADRIATIC Trial
PRO scales | Treatment group | N | Adjusted mean change (95% CI) from baseline (average over 24 months) | Average difference in change from baseline in meansa (durvalumab – placebo) (95% CI) | 2-sided P value |
|---|---|---|---|---|---|
EORTC QLQ-C30 | |||||
GHS/QoL | Durvalumab | 214 | −3.6 (−5.893 to −1.297) | −1.1 ████████ █ | ████ |
Placebo | 204 | −2.5 (−4.781 to −0.179) | |||
Physical functioning | Durvalumab | 214 | −1.5 (−3.450 to 0.376) | −0.3 ████████ █ | ████ |
Placebo | 204 | −1.3 (−3.200 to 0.660) | |||
Role functioning | Durvalumab | 214 | −2.1 (−4.596 to 0.391) | 1.3 ████████ █ | ████ |
Placebo | 204 | −3.4 (−5.945 to −0.948) | |||
Fatigue | Durvalumab | 214 | −1.4 (−3.955 to 1.164) | −0.9 ████████ █ | ████ |
Placebo | 204 | −0.5 (−3.064 to 2.078) | |||
Appetite loss | Durvalumab | 214 | −6.1 (−8.345 to −3.869) | 2.7 ███████ █ | ████ |
Placebo | 204 | −8.8 (−11.092 to −6.603) | |||
EORTC QLQ-LC13 | |||||
Dyspnea | Durvalumab | 216 | 2.4 (0.278 to 4.492) | 0.3 ████████ █ | ████ |
Placebo | 211 | 2.1 (0.003 to 4.230) | |||
Coughing | Durvalumab | 216 | 1.2 (−1.288 to 3.645) | 0.3 ████████ █ | ████ |
Placebo | 211 | 0.9 (−1.600 to 3.353) | |||
Chest pain | Durvalumab | 216 | 1.4 (−0.474 to 3.198) | −1.2 ████████ █ | ████ |
Placebo | 211 | 2.6 (0.743 to 4.419) | |||
CI = confidence interval; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-L13 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Lung Cancer 13; FAS = full analysis set; GHS = global health status; MMRM = mixed model for repeated measures; PFS-IA = interim analysis for progression-free survival; PRO = patient-reported outcome; QoL = quality of life.
Notes: Data cut-off: January 15, 2024. Negative change from baseline indicates deterioration in GHS/QoL and functioning scales. A positive change from baseline indicates deterioration in symptom scales.
aFor symptom scales, an estimated difference < 0 favours durvalumab over placebo. For function scales and GHS/QoL, an estimated difference > 0 favours durvalumab over placebo.
Sources: ADRIATIC Clinical Study Report and clinical study protocol; Cheng et al. (2024).24 Details included in the table are from the sponsor’s summary of clinical evidence.
Harms
The harms data presented are based on the data cut-off date of January 15, 2024, and include all randomized patients who received at least 1 dose of the study treatment, which corresponds to the safety analysis set. The safety analysis set includes 262 patients in the durvalumab group and 265 in the placebo group (Table 18).
Adverse Events
The majority of patients in both the durvalumab group (94.3%) and the placebo group (88.3%) reported at least 1 TEAE. Common TEAEs (experienced by ≥ 5% of patients) included radiation pneumonitis (22.9% in the durvalumab group versus 23.4% in the placebo group), decreased appetite (16.8% versus 12.8%), hypothyroidism (16.0% versus 3.8%), cough (15.3% versus 12.1%), pruritus (13.0% versus 7.2%), and fatigue (12.2% versus 12.8%). A higher proportion of hypothyroidism (16.0% versus 3.8%) and hyperthyroidism (10.3% versus 1.5%) was observed in the durvalumab group than in the placebo group.
Serious AEs
SAEs were more frequent in the durvalumab group (29.8% of patients) than in the placebo group (24.2% of patients). The most common SAEs in the durvalumab group were radiation pneumonitis (5.0% of patients), pneumonia (4.6%), and pneumonitis (3.1%). Some SAEs, such as interstitial lung disease (2.3% of patients) and immune-mediated lung disease (1.5%), were exclusively observed in the durvalumab group.
Withdrawals Due to AEs
A higher proportion of patients discontinued treatment due to AEs in the durvalumab group (16.4%) than in the placebo group (10.6%). Key reasons for discontinuation in the durvalumab group included radiation pneumonitis (3.8% of patients, versus 1.9% in the placebo group) and pneumonitis (3.1%, versus 1.1% in the placebo group).
Mortality
AEs leading to death were reported in 2.7% of patients in the durvalumab group and 1.9% of patients in the placebo group. Deaths in the durvalumab group were attributed to pneumonia (0.8% of patients), bacterial pneumonia (0.8%), cardiac failure (0.4%), encephalopathy (0.4%), and pneumonitis (0.4%).
Notable Harms
Patients in the durvalumab group experienced higher rates of the following notable harms than patients in the placebo group: hypothyroid events (17.2% versus 4.9%) and pneumonitis categorized as an AE of special interest: 16.4% versus 6.4%. Other notable events included adrenal insufficiency (1.1% of patients) and thyroiditis (1.1% of patients) in the durvalumab group, which were not observed in the placebo group. Dermatitis/rash was also more frequent in the durvalumab group than the placebo group (28.2% versus 17.4% of patients).
Table 18: Key Harms Data (SAS, PFS-IA), ADRIATIC Trial
AEs | Durvalumab (N = 262) | Placebo (N = 265) |
|---|---|---|
Most common AEs, n (%) [reported in ≥ 10% of patients in either treatment group] | ||
Patients with any AE | 247 (94.3) | 234 (88.3) |
Radiation pneumonitis | 60 (22.9) | 62 (23.4) |
Decreased appetite | 44 (16.8) | 34 (12.8) |
Hypothyroidism | 42 (16.0) | 10 (3.8) |
Cough | 40 (15.3) | 32 (12.1) |
Pruritus | 34 (13.0) | 19 (7.2) |
Nausea | 33 (12.6) | 29 (10.9) |
Dizziness | 32 (12.2) | 20 (7.5) |
Fatigue | 32 (12.2) | 34 (12.8) |
Diarrhea | 29 (11.1) | 22 (8.3) |
Pneumonia | 29 (11.1) | 20 (7.5) |
Pneumonitis | 28 (10.7) | 16 (6.0) |
Rash | 28 (10.7) | 16 (6.0) |
Constipation | 27 (10.3) | 26 (9.8) |
Hyperthyroidism | 27 (10.3) | 4 (1.5) |
SAEs, n (%) [reported in ≥ 1% of patients in either treatment group] | ||
Patients with any SAE | 78 (29.8) | 64 (24.2) |
Radiation pneumonitis | 13 (5.0) | 7 (2.6) |
Pneumonia | 12 (4.6) | 10 (3.8) |
Pneumonitis | 8 (3.1) | 6 (2.3) |
Interstitial lung disease | 6 (2.3) | 0 |
Immune-mediated lung disease | 4 (1.5) | 0 |
Pneumonia bacterial | 3 (1.1) | 0 |
Chronic obstructive pulmonary disease | 1 (0.4) | 4 (1.5) |
Asthenia | 0 | 3 (1.1) |
Patients who stopped treatment due to AEs, n (%) [reported in ≥ 1% of patients in either treatment group] | ||
Patients with any AE leading to discontinuation | 43 (16.4) | 28 (10.6) |
Radiation pneumonitis | 10 (3.8) | 5 (1.9) |
Pneumonitis | 8 (3.1) | 3 (1.1) |
Immune-mediated lung disease | 4 (1.5) | 0 |
Pneumonia | 3 (1.1) | 2 (0.8) |
Deaths, n (%) [reported in ≥ 1 patient in either treatment group] | ||
Patients with any AE with outcome of death | 7 (2.7) | 5 (1.9) |
Pneumonia | 2 (0.8) | 2 (0.8) |
Pneumonia bacterial | 2 (0.8) | 0 |
Cardiac failure | 1 (0.4) | 0 |
Encephalopathy | 1 (0.4) | 0 |
Pneumonitis | 1 (0.4) | 0 |
COVID-19 | 0 | 1 (0.4) |
Sepsis | 0 | 1 (0.4) |
Squamous cell carcinoma of the hypopharynx | 0 | 1 (0.4) |
AESIs and AEPIs (by category), n (%) | ||
Adrenal insufficiency | 3 (1.1) | 0 |
Dermatitis/rash | 74 (28.2) | 46 (17.4) |
Diarrhea/colitis | 30 (11.5) | 24 (9.1) |
Guillain-Barre syndrome | 1 (0.4) | 0 |
Hepatic events | 27 (10.3) | 24 (9.1) |
Hyperthyroid events | 32 (12.2) | 5 (1.9) |
Hypophysitis | 2 (0.8) | 0 |
Hypothyroid events | 45 (17.2) | 13 (4.9) |
Infusion/hypersensitivity reactions | 6 (2.3) | 3 (1.1) |
Myocarditis | 2 (0.8) | 1 (0.4) |
Myositis | 2 (0.8) | 3 (1.1) |
Other rare or miscellaneous AESI/AEPI | 24 (9.2) | 33 (12.5) |
Pancreatic events | 12 (4.6) | 9 (3.4) |
Pneumonitis | 43 (16.4) | 17 (6.4) |
Renal events | 10 (3.8) | 5 (1.9) |
Thyroiditis | 3 (1.1) | 0 |
Type 1 diabetes mellitus | 1 (0.4) | 0 |
AE = adverse event; AEPI = adverse event of possible interest; AESI = adverse event of special interest; PFS-IA = interim analysis for progression-free survival; PT = preferred term; SAE = serious adverse event; SAS = safety analysis set.
Notes: Data cut-off date: January 15, 2024. Patients with multiple events in the same PT are counted only once in that PT. Patients with more than 1 PT are counted once in each of those PTs.
Sources: ADRIATIC Clinical Study Report and clinical study protocol; Cheng et al. (2024).24 Details included in the table are from the sponsor’s summary of clinical evidence.
Critical Appraisal
Internal Validity
In the ADRIATIC trial, randomization and allocation concealment were appropriately implemented to prevent selection and allocation bias. An interactive response system was used to conceal allocation, preventing investigators and patients from predicting or influencing treatment assignments. This minimized selection bias and ensured that baseline patient characteristics were evenly distributed across treatment groups, reducing the potential for confounding. Stratification factors included disease stage (I/II versus III) and receipt of PCI (yes versus no), both clinically relevant for LS-SCLC.
The trial was blinded to both patients and investigators, with placebo infusions used to maintain blinding. However, potential unblinding may have occurred due to imbalances in TEAEs, such as immune-mediated AEs in the durvalumab group. Unblinding could have introduced performance and detection bias, particularly in subjectively measured outcomes like HRQoL. However, objective end points such as OS are less susceptible to bias from unblinding.
Patient characteristics were well balanced at baseline, with no major imbalances in key factors such as age, sex, ECOG Performance Status, and disease stage. The use of concomitant medications was similar across groups and aligned with the trial protocol. Seventy-nine patients (14.9%) had at least 1 important protocol deviation, including deviations from key eligibility criteria, incorrect treatment administration, and use of prohibited medications (i.e., medications restricted per protocol that should not have been used concomitantly with study treatment). However, these deviations were balanced between treatment groups and were unlikely to have introduced significant bias.
The study report does not indicate major missing data issues for key outcomes. However, different censoring rules were applied depending on the end point. OS data were censored at the last known alive date for patients lost to follow-up or alive at data cut-off. PFS and TTDM applied alternative censoring rules for patients with missed assessments, while DOR used PFS censoring for patients without documented progression. Safety data were generally not imputed, except for laboratory values at quantification limits. Some missing data, such as those arising from early dropout, could not be accounted for solely by censoring and may contribute to uncertainty in the estimates. These limitations should be considered when interpreting the trial results.
The placebo comparator was appropriate, as there is no established consolidation therapy following concurrent CRT in LS-SCLC. From an ethical standpoint, placebo use was justified given the lack of an approved standard of care in this setting, ensuring that all patients received the best available treatment during the induction phase.
A hierarchical testing strategy was used for the dual primary outcomes (OS and PFS) to control for multiplicity. The power calculations and sample size were sufficient for the primary outcomes but were not designed to detect significant differences for secondary outcomes. Importantly, there was no multiplicity control for DOR or TTDM, which should be considered when interpreting these end points. Prespecified subgroup analyses may have been underpowered due to small sample sizes, limiting confidence in the subgroup findings.
A total of 175 patients (66.5%) in the durvalumab group and 195 patients (73.6%) in the placebo group discontinued study treatment. The primary reasons were disease progression (46.0% versus 58.1%) and AEs (16.3% versus 10.9%). The lower rate of treatment discontinuation due to disease progression in the durvalumab group suggests a potential treatment benefit, while the higher rate of discontinuation due to AEs in the durvalumab group indicates a greater toxicity burden. Differential discontinuation rates between groups can introduce attrition bias if follow-up differs based on treatment assignment. To minimize attrition bias, patients who permanently discontinued treatment were followed for survival, and those who discontinued for reasons other than RECIST-defined radiological progression continued to undergo scheduled RECIST 1.1 assessments. This approach ensured that survival and progression data remained systematically collected, reducing the risk of informative censoring.
PFS has been evaluated as a potential surrogate end point for OS in LS-SCLC. Regulatory agencies such as the FDA and the European Medicines Agency acknowledge PFS as a meaningful end point when the observed benefit is substantial. In the ADRIATIC trial, PFS was assessed by BICR using RECIST 1.1, minimizing assessment bias. Sensitivity analyses confirmed the robustness of the PFS findings, addressing potential biases such as evaluation time bias, attrition bias, and ascertainment bias, reinforcing the credibility of PFS as a surrogate end point for OS. However, strong surrogacy between PFS and OS in LS-SCLC remains uncertain compared to first-line ES-SCLC studies, in which chemotherapy has demonstrated a more established correlation between these end points.
External Validity
The ADRIATIC trial population and interventions are largely generalizable to Canadian clinical practice, but some limitations exist.
Eligible patients included those with ECOG Performance Status 0 or 1, representing a relatively healthier subset of patients with LS-SCLC. Patients with extensive-stage disease, mixed histology, or significant comorbidities were excluded, which may limit the generalizability of the results to less fit patients, commonly encountered in Canadian practice. While the study population was globally representative, it had notable demographic differences from the broader population of patients with LS-SCLC seen in Canada. The majority of patients were white (50.4%) or Asian (47.5%), with Black or African American (0.8%) and other ethnicities (1.3%) underrepresented. This differs from the racial diversity seen in Canadian clinical practice, which includes a larger proportion of Black, Indigenous, and other racial groups. More than 90% of patients were current or former smokers, consistent with LS-SCLC epidemiology. The median age of the patients was 62 years, with 39.2% of patients aged older than 65 years. This represents a younger-than-expected population compared to real-world practice in Canada, where a higher proportion of such patients are older. The demographic differences and the exclusion of patients with significant comorbidities should be considered when interpreting the generalizability of the findings.
The study allowed for prior treatment with either cisplatin-etoposide or carboplatin-etoposide, which reflect the current treatment options in Canada. PCI use was 53.8%, which aligns with Canadian practice but varies depending on clinician preference and patient characteristics. According to the clinical experts consulted for the review, the specialized hospital admission required during initial treatment cycles (to monitor for immune-mediated AEs) may pose challenges in community settings. Most trial sites were based in hospitals or specialized oncology centres, ensuring appropriate management of these events. Clinical experts noted that while initial treatments should be administered in well-equipped centres, later cycles could transition to outpatient settings where feasible.
The clinical experts noted that the dosing schedule of durvalumab used in the ADRIATIC trial is consistent with what would be used in clinical practice in Canada; however, the requirement for close monitoring during early cycles may pose challenges to the implementation of the drug for the condition under review in community settings. The review team considered placebo to be an appropriate comparator in this treatment space, given the current lack of a standard of care for LS-SCLC.
While survival benefits were clinically meaningful, long-term follow-up beyond 36 months may be necessary to fully evaluate the generalizability of the OS results from the ADRIATIC trial.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For the pivotal study identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for the outcomes considered most relevant to inform expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group.42,43
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate — The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited — The true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate — The true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as “very uncertain.”
Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null.
The selection of outcomes for GRADE assessment was based on the sponsor’s summary of clinical evidence, consultation with clinical experts, and input received from patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members:
OS
PFS
HRQoL (EORTC QLQ-C30 and EORTC QLQ-LC13)
SAEs
pneumonitis.
The reference points for the certainty of evidence assessment for OS, PFS, DOR, and TTDM were set according to the presence or absence of an important effect based on thresholds informed by the clinical experts consulted for this review. The reference points for the certainty of evidence assessment for the EORTC QLQ-C30 and EORTC QLQ-LC13 global health status/quality of life scores were determined based on a threshold suggested by the sponsor, informed by the literature, which identified a 10-point change from baseline as clinically meaningful. The reference points for the certainty of evidence assessment for SAEs and pneumonitis were set according to the presence or absence of a clinically important increase in the frequency of these harms, as informed by clinical expert input.
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for durvalumab versus placebo for patients with LS-SCLC.
Long-Term Extension Studies
No long-term extension studies were submitted by the sponsor.
Indirect Evidence
No indirect treatment comparisons were submitted by the sponsor.
Studies Addressing Gaps in the Systematic Review Evidence
No additional studies were submitted by the sponsor.
Discussion
Summary of Available Evidence
The Clinical Review summarized evidence from the ADRIATIC trial, a pivotal phase III, randomized, double-blind study comparing durvalumab monotherapy with placebo, when used as consolidation treatment in patients with LS-SCLC who had not progressed after definitive platinum-based concurrent CRT. The trial enrolled and randomly assigned 539 patients to durvalumab (N = 269) or placebo (N = 270). Eligible participants included those aged 18 years or older with histologically or cytologically confirmed LS-SCLC, a WHO Performance Status of 0 or 1, and no evidence of metastatic disease. Patients who had received prior systemic anticancer therapy, PCI, or definitive thoracic radiation therapy were excluded. The dual primary end points of the trial were OS and PFS. Secondary end points included TTDM, DOR, HRQoL, and safety. The outcomes of interest were evaluated using stratified analyses, including subgroups by patient baseline characteristics. The data cut-off date for the most recent analysis was January 15, 2024. Overall, the trial population included predominantly white (approximately 50%) and Asian (48%) patients, with a median age of 62 years. Most patients presented with stage III disease (87%) and received platinum-etoposide as part of their concurrent CRT regimen. Baseline characteristics were generally balanced between treatment groups.
Interpretation of Results
Efficacy
The ADRIATIC trial demonstrated a statistically significant improvement in OS with durvalumab compared to placebo when used as consolidation treatment in patients with LS-SCLC who had not progressed after definitive platinum-based concurrent CRT. The median OS was 55.9 months (95% CI, 37.3 months to not estimable) in the durvalumab group compared to 33.4 months (95% CI, 25.5 to 39.9 months) in the placebo group, indicating an estimated improvement in median OS of 22.5 months. At key time points, survival rates were consistently higher with durvalumab. At 24 months, OS was 68.0% with durvalumab compared to 58.5% with placebo, reflecting an absolute difference of 9.5% (95% CI, 1.0% to 17.8%). At 36 months, OS was 56.5% in the durvalumab group versus 47.6% in the placebo group, with an absolute difference of 8.9% (95% CI, −0.1% to 17.8%). According to the GRADE assessment, the certainty of evidence for OS was rated moderate, with some imprecision in the CIs, but the observed OS benefit was considered clinically meaningful. The clinical experts suggested that an absolute survival improvement of 5% to 10% represents a clinically meaningful improvement in OS, and the observed differences align with these expectations. While subgroup analyses generally supported the benefit of durvalumab, many subgroups had only a few observed events and limited statistical power.
A statistically significant improvement in PFS was also observed in the durvalumab group compared to the placebo group. The median PFS was 16.6 months (95% CI, 10.2 to 28.2 months) in the durvalumab group versus 9.2 months (95% CI, 7.4 to 12.9 months) in the placebo group, reflecting a 7.4-month extension in progression-free time. At 24 months, 46.2% of patients in the durvalumab group remained progression-free compared to 34.2% in the placebo group, with an absolute difference of 12.0% (95% CI, 3.2% to 21.1%). The Kaplan-Meier curves indicated that PFS separation between durvalumab and placebo occurred early, around 6 months after randomization, and remained consistent throughout the study period. According to the GRADE assessment, the certainty of evidence for PFS was rated moderate, primarily due to imprecision in the CIs. Despite this, the observed PFS improvement was considered clinically meaningful, as the absolute difference exceeded the 5% to 10% threshold identified by the clinical experts.
For patients who experienced a confirmed response, the median DOR was 38.8 months (95% CI, 25.9 months to not estimable) in the durvalumab group, compared to 27.8 months (95% CI, 9.9 months to not estimable) in the placebo group. This result indicates a longer sustained response with durvalumab.
TTDM also favoured durvalumab, with an ██ ██ ████ ████ ███ ██████ ███████. However, the result did not achieve statistical significance (██████), likely due to limited events and incomplete follow-up data.
Throughout the trial, there were no statistically significant differences in most PRO end points, except for appetite loss, for which the placebo group showed greater improvement than the durvalumab group. Chest pain was the only symptom to demonstrate a statistically significant improvement favouring durvalumab, with an odds ratio of 2.28 (P = 0.0308). This suggests that while durvalumab may not significantly impact overall quality of life, it could provide symptomatic relief for specific issues such as chest pain.
Overall, according to the clinical experts, the trial results align with the existing evidence on the use of immune checkpoint inhibitors in the treatment of SCLC. Patient group input emphasized the importance of improving survival and maintaining quality of life, both of which are supported by the trial findings. Based on the results of the ADRIATIC trial, which demonstrated statistically significant results in prolonging OS and PFS, the clinical experts commented that durvalumab may fulfill the unmet need for the patient population under review, for whom treatment options for consolidation therapy after concurrent CRT are limited. However, some key gaps remain in the evidence, including limited data on the long-term safety of durvalumab in the patient population under review and on the effects of durvalumab on certain subgroups such as patients with poor performance status.
From an implementation perspective, the need for close monitoring of immune-mediated toxicities was identified by the clinical experts as an important consideration. The clinical experts indicated that the feasibility of integrating durvalumab into existing treatment protocols will depend on health care system capacity, particularly in community settings where early monitoring may be less accessible.
Harms
In the ADRIATIC trial, durvalumab treatment was associated with a higher frequency of immune-mediated events and treatment-related discontinuations. Notably, SAEs, including radiation pneumonitis and pneumonitis, occurred more frequently in the durvalumab group, contributing to a higher discontinuation rate than for placebo. The increase in immune-mediated AEs, such as thyroid dysfunction and dermatitis, aligns with expectations for this class of therapy.
Based on the input from the clinical experts consulted by the review team, the safety profile of durvalumab was consistent with its mechanism of action as an anti–PD-L1 therapy, and the observed safety signals are in line with known toxicities of immune checkpoint inhibitors. The clinical experts indicated that these risks are expected and that they can be managed with appropriate monitoring and supportive care. However, the potential for serious immune-mediated toxicities underscores the need for careful patient selection and access to specialized care, particularly in community settings where close monitoring may be challenging.
Although mortality due to AEs was slightly higher in the durvalumab group, the clinical experts believed that the overall benefit-risk profile should be considered in the context of improved survival outcomes. The GRADE assessment suggests that the increase in SAEs is likely clinically important, which should be weighed against the survival benefits when considering treatment decisions.
Conclusion
The results of the phase III, randomized, double-blind, placebo-controlled ADRIATIC trial suggest that durvalumab as consolidation therapy likely results in improved OS and PFS compared to placebo in adult patients with LS-SCLC after concurrent CRT based on moderate certainty evidence. Placebo was considered a relevant comparator in this setting because active surveillance is currently used after platinum-based CRT in the patient population under review.
A higher incidence of SAEs and immune-mediated AEs was observed in the durvalumab group than in the placebo group; however, these AEs were deemed by the clinical experts to be manageable with appropriate monitoring and intervention. The clinical experts noted that, overall, the safety profile of durvalumab was consistent with the expected AE profile for anti–PD-L1 therapies. However, missing data that were not accounted for in the ADRIATIC trial, such as data arising from early dropouts, may result in some level of uncertainty in the estimates of harms effects.
The PRO results assessed using the EORTC QLQ-C30 in the ADRIATIC trial showed that durvalumab treatment as consolidation therapy after concurrent CRT may result in improvement in chest pain symptoms when compared to placebo. No statistically meaningful differences were reported in global health status/quality of life scores or functional scales between the durvalumab and placebo groups; however, the PRO results were uncertain due to a notable amount of missing data and the exploratory nature of some PRO analyses.
References
1.Brenner DR, Gillis J, Demers AA, et al. Projected estimates of cancer in Canada in 2024. CMAJ. 2024;196(18):E615-E623. doi: 10.1503/cmaj.240095 PubMed
2.Micke P, Faldum A, Metz T, et al. Staging small cell lung cancer: Veterans Administration Lung Study Group versus International Association for the Study of Lung Cancer--what limits limited disease? Lung Cancer. 2002;37(3):271-6. doi: 10.1016/s0169-5002(02)00072-7 PubMed
3.Melosky BL, Leighl NB, Dawe D, et al. Canadian Consensus Recommendations on the Management of Extensive-Stage Small-Cell Lung Cancer. Curr Oncol. 2023;30(7):6289-6315. doi: 10.3390/curroncol30070465 PubMed
4.Doherty J, Dawe DE, Pond GR, Ellis PM. The effect of age on referral to an oncologist and receipt of chemotherapy among small cell lung cancer patients in Ontario, Canada. J Geriatr Oncol. 2019;10(3):449-458. doi: 10.1016/j.jgo.2018.10.001 PubMed
5.Zhong L, Suo J, Wang Y, et al. Prognosis of limited-stage small cell lung cancer with comprehensive treatment including radical resection. World J Surg Oncol. 2020;18(1):27. doi: 10.1186/s12957-020-1807-1 PubMed
6.Canadian Cancer Society. Survival Statistics for small cell lung cancer. 2020. Accessed January 7, 2025. https://cancer.ca/en/cancer-information/cancer-types/lung/prognosis-and-survival/small-cell-lung-cancer-survival-statistics
7.Faivre-Finn C, Snee M, Ashcroft L, et al. Concurrent once-daily versus twice-daily chemoradiotherapy in patients with limited-stage small-cell lung cancer (CONVERT): an open-label, phase 3, randomised, superiority trial. Lancet Oncol. 2017;18(8):1116-1125. doi: 10.1016/S1470-2045(17)30318-2 PubMed
8.Bogart JA, Waqar SN, Mix MD. Radiation and Systemic Therapy for Limited-Stage Small-Cell Lung Cancer. J Clin Oncol. 2022;40(6):661-670. doi: 10.1200/JCO.21.01639 PubMed
9.Garcia-Campelo R, Sullivan I, Arriola E, et al. SEOM-GECP Clinical guidelines for diagnosis, treatment and follow-up of small-cell lung cancer (SCLC) (2022). Clin Transl Oncol. 2023;25(9):2679-2691. doi: 10.1007/s12094-023-03216-3 PubMed
10.Moore S, Zhan LJ, Chowdhury D, et al. 1793P Treatment and outcomes of limited disease small cell lung cancer (LD-SCLC) in the Canadian small cell lung cancer database (CASCADE). Ann Oncol. 2024;35(suppl_2):S1062-S1076. doi: 10.1016/j.annonc.2024.08.1884
11.Rudin CM, Brambilla E, Faivre-Finn C, Sage J. Small-cell lung cancer. Nat Rev Dis Primers. 2021;7(1):3. doi: 10.1038/s41572-020-00235-0 PubMed
12.Dawe DE, Rittberg R, Syed I, et al. Real-world predictors of survival in patients with limited-stage small-cell lung cancer in Manitoba, Canada. Front Oncol. 2023;13:1191920. doi: 10.3389/fonc.2023.1191920 PubMed
13.Statistics Canada. Table 13-10-0111-01 Number and rates of new cases of primary cancer, by cancer type, age group and sex [sponsor supplied reference]. 2024. Accessed April 2, 2024. https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310011101
14.Canadian Cancer Statistics Advisory Committee. Canadian Cancer Statistics: A 2020 special report on lung cancer [sponsor supplied reference]. Canadian Cancer Society; 2020. https://cancer.ca/Canadian-Cancer-Statistics-2020-EN
15.BC Cancer Agency. Small cell lung cancer [accessed by sponsor]. 2014. http://www.bccancer.bc.ca/books/lung/management/small-cell-lung-cancer
16.Khurshid H, Ismaila N, Bian J, et al. Systemic Therapy for Small-Cell Lung Cancer: ASCO-Ontario Health (Cancer Care Ontario) Guideline. J Clin Oncol. 2023;41(35):5448-5472. doi: 10.1200/JCO.23.01435 PubMed
17.Bennett BM, Wells JR, Panter C, Yuan Y, Penrod JR. The Humanistic Burden of Small Cell Lung Cancer (SCLC): A Systematic Review of Health-Related Quality of Life (HRQoL) Literature. Front Pharmacol. 2017;8:339. doi: 10.3389/fphar.2017.00339 PubMed
18.Midthun DE. Post TW, ed. Overview of the initial treatment and prognosis of lung cancer. UpToDate; 2024. Accessed December 11, 2024. http://www.uptodate.com
19.Byers LA, Gay CM. Post TW, ed. Pathobiology and staging of small cell carcinoma of the lung. UpToDate; 2023. Accessed August 16, 2024. http://www.uptodate.com
20.National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: small cell lung cancer (Version 3. 2023), 21 December [sponsor supplied reference]. 2022.
21.Canadian Cancer Society. Lung cancer [accessed by sponsor]. https://www.cancer.ca/en/cancer-information/cancer-type/lung/lung-cancer/?region=on
22.Qin A. Small Cell Lung Cancer. National Organization for Rare Disorders; 2023. Accessed October 31, 2023. https://rarediseases.org/rare-diseases/small-cell-lung-cancer/
23.Sun A, Durocher-Allen LD, Ellis PM, et al. Guideline for the Initial Management of Small Cell Lung Cancer (Limited and Extensive Stage) and the Role of Thoracic Radiotherapy and First-line Chemotherapy. Clin Oncol (R Coll Radiol). 2018;30(10):658-666. doi: 10.1016/j.clon.2018.06.008 PubMed
24.Cheng Y, Spigel DR, Cho BC, et al. Durvalumab after Chemoradiotherapy in Limited-Stage Small-Cell Lung Cancer. N Engl J Med. 2024;391(14):1313-1327. doi: 10.1056/NEJMoa2404873 PubMed
25.Spigel DR, Cheng Y, Cho BC, et al. ADRIATIC: Durvalumab (D) as consolidation treatment (tx) for patients (pts) with limited-stage small-cell lung cancer (LS-SCLC). J Clin Oncol. 2024;42(17_suppl):LBA5-LBA5. doi: 10.1200/JCO.2024.42.17_suppl.LBA5
26.AstraZeneca Canada Inc. Imfinzi (durvalumab): solution, 50 mg / mL, intravenous infusion [product monograph] [sponsor supplied reference]. September 21, 2020.
27.Bogart J, Wang X, Masters G, et al. High-Dose Once-Daily Thoracic Radiotherapy in Limited-Stage Small-Cell Lung Cancer: CALGB 30610 (Alliance)/RTOG 0538. J Clin Oncol. 2023;41(13):2394-2402. doi: 10.1200/JCO.22.01359 PubMed
28.European Medicines Agency. Guidelines on the evaluation of anticancer medicinal products. November 2023. EMA/CHMP/205/95 Rev.6. Accessed February 6, 2025. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-clinical-evaluation-anticancer-medicinal-products-revision-6_en.pdf
29.Food and Drug Administration. Clinical trial endpoints for the approval of cancer drugs and biologics. Guidance for industry. December 2018 [sponsor supplied reference]. https://www.fda.gov/regulatory-information/search-fda-guidance documents/clinical-trialendpoints-approval-cancer-drugs-and-biologics.
30.Food and Drug Administration. Clinical trial endpoints for the approval of non-small cell lung cancer drugs and biologics. Guidance for industry [sponsor supplied reference]. April 2015. https://www.fda.gov/regulatory-information/search-fda-guidancedocuments/clinical-trial-endpoints-approval-non-small-cell-lung-cancer-drugs-and-biologics
31.Yang Y, Wang J, Wang W, et al. Progression-Free Survival and Time to Progression as Potential Surrogate Endpoints for Overall Survival in Chemoradiotherapy Trials in Limited-Stage Small-Cell Lung Cancer: A Systematic Review and Meta-Analysis. Front Oncol. 2022;12:810580. doi: 10.3389/fonc.2022.810580 PubMed
32.Fayers PM AN, Bjordal K. The EORTC QLC-C30 scoring manual (3rd Edition). 2001. Accessed May 25, 2024. https://www.eortc.org/app/uploads/sites/2/2018/02/SCmanual.pdf
33.Giesinger JM, Kieffer JM, Fayers PM, et al. Replication and validation of higher order models demonstrated that a summary score for the EORTC QLQ-C30 is robust. J Clin Epidemiol. 2016;69:79-88. doi: 10.1016/j.jclinepi.2015.08.007 PubMed
34.Teckle P, Peacock S, McTaggart-Cowan H, et al. The ability of cancer-specific and generic preference-based instruments to discriminate across clinical and self-reported measures of cancer severities. Health Qual Life Outcomes. 2011;9(106):106. doi: 10.1186/1477-7525-9-106 PubMed
35.Nicklasson M, Bergman B. Validity, reliability and clinical relevance of EORTC QLQ-C30 and LC13 in patients with chest malignancies in a palliative setting. Qual Life Res. 2007;16(6):1019-28. doi: 10.1007/s11136-007-9210-8 PubMed
36.Aaronson NK, Ahmedzai S, Bergman B, et al. The European Organization for Research and Treatment of Cancer QLQ-C30: a quality-of-life instrument for use in international clinical trials in oncology. J Natl Cancer Inst. 1993;85(5):365-76. doi: 10.1093/jnci/85.5.365 PubMed
37.Machingura A, Taye M, Musoro J, et al. Clustering of EORTC QLQ-C30 health-related quality of life scales across several cancer types: Validation study. Eur J Cancer. 2022;170:1-9. doi: 10.1016/j.ejca.2022.03.039 PubMed
38.Musoro JZ, Coens C, Sprangers MAG, et al. Minimally important differences for interpreting EORTC QLQ-C30 change scores over time: A synthesis across 21 clinical trials involving nine different cancer types. Eur J Cancer. 2023;188:171-182. doi: 10.1016/j.ejca.2023.04.027 PubMed
39.Osoba D, Zee B, Pater J, Warr D, Kaizer L, Latreille J. Psychometric properties and responsiveness of the EORTC quality of Life Questionnaire (QLQ-C30) in patients with breast, ovarian and lung cancer. Qual Life Res. 1994;3(5):353-64. doi: 10.1007/BF00451727 PubMed
40.Bedard G, Zeng L, Zhang L, et al. Minimal important differences in the EORTC QLQ-C30 in patients with advanced cancer. Asia Pac J Clin Oncol. 2014;10(2):109-17. doi: 10.1111/ajco.12070 PubMed
41.Bergman B, Aaronson NK, Ahmedzai S, Kaasa S, Sullivan M. The EORTC QLQ-LC13: a modular supplement to the EORTC Core Quality of Life Questionnaire (QLQ-C30) for use in lung cancer clinical trials. EORTC Study Group on Quality of Life. Eur J Cancer. 1994;30A(5):635-42. doi: 10.1016/0959-8049(94)90535-5 PubMed
42.Balshem H, Helfand M, Schunemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-6. doi: 10.1016/j.jclinepi.2010.07.015 PubMed
43.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. doi: 10.1016/j.jclinepi.2019.10.014 PubMed
Appendix 1: Sensitivity Analysis
Please note that this appendix has not been copy-edited.
Overall Survival
Figure 5: Kaplan-Meier Plot of Overall Survival (With Censoring Indicators Reversed), Time to Censoring (FAS, OS-IA1), ADRIATIC Trial
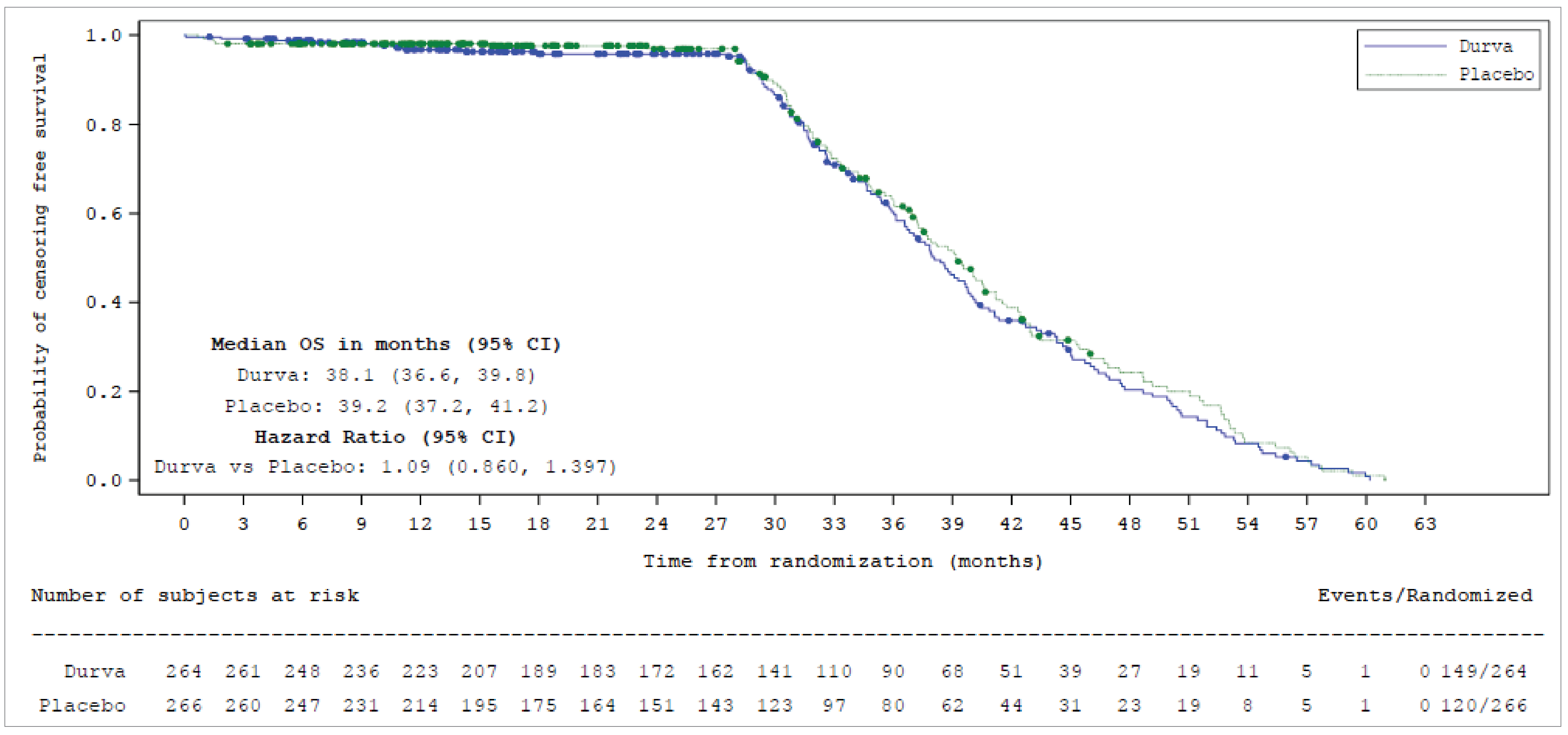
CI = confidence interval; Durva = durvalumab; FAS = full analysis set; OS = overall survival; OS-IA1 = first interim analysis of overall survival.
Note: Data cut-off date of January 15, 2024.
Sources: ADRIATIC Clinical Study Report and clinical study protocol; Cheng et al. (2024).24
Table 19: Overall Survival, Summary of Duration of Follow-Up (FAS, OS-IA1), ADRIATIC Trial
Overall survival | Durvalumab (N = 264) | Placebo (N = 266) |
|---|---|---|
Duration of follow-up in all patients (months) | ||
n | 264 | 266 |
Median | 30.75 | 28.63 |
Min, max | 0.1 to 60.2 | 0.7 to 60.9 |
Q1, Q3 | 16.31 to 39.46 | 14.13 to 37.65 |
Duration of follow-up in censored patients (months) | ||
na | 149 | 120 |
Median | 37.19 | 37.24 |
Min, max | 0.1 to 60.2 | 0.7 to 60.9 |
Q1, Q3 | ██████ █████ | ██████ █████ |
Patients prematurely censoredb, n (%) | ██ █████ | ██ █████ |
Censored patients, n (%) | 149 (56.4) | 120 (45.1) |
Censored ≤ 8 weeks before DCO | ███ ██████ | ███ ██████ |
Censored > 8 weeks to ≤ 16 weeks before DCO | ███ ██████ | ███ ██████ |
Censored > 16 weeks to ≤ 24 weeks before DCO | █████ | █████ |
Censored > 24 weeks before DCO | ██ █████ | █████ |
DCO = data cut-off; FAS = full analysis set; min = minimum; OS-IA1 = first interim analysis of overall survival; Q1 = first quartile; Q3 = third quartile; max = maximum.
Note: Data cut-off date of January 15, 2024.
an is the number of censored patients.
bA patient would be defined as prematurely censored if their survival status was not defined at the DCO.
Sources: ADRIATIC Clinical Study Report and clinical study protocol; Cheng et al. (2024).24 Details included in the table are from the sponsor’s summary of clinical evidence.
Table 20: Overall Survival, Effect of Covariates on Primary Analysis (FAS, OS-IA1), ADRIATIC Trial
Model | Group | N | Number (%) of patients with events | Comparison between groups | |
|---|---|---|---|---|---|
Hazard ratio | 95% CI | ||||
Analysis with stratification factors – excluding covariatesa | Durvalumab | 264 | 115 (43.6) | 0.73 | 0.569 to 0.928 |
Placebo | 266 | 146 (54.9) | |||
Analysis with stratification factors – including covariatesb | Durvalumab | 264 | 115 (43.6) | 0.75 | 0.585 to 0.966 |
Placebo | 266 | 146 (54.9) | |||
cCRT = concurrent chemoradiotherapy; CI = confidence interval; CR = complete response; FAS = full analysis set; IVRS = interactive voice response system; OS = overall survival; OS-IA1 = first interim analysis of overall survival; PR = partial response; SD = standard deviation.
Notes: Data cut-off date of January 15, 2024. A hazard ratio < 1 favours Durva to be associated with a longer survival than placebo. Stratification factor is based on the values entered into the IVRS.
aCorresponds to the primary OS.
bThe hazard ratio and CI were calculated using a stratified Cox proportional hazards model, adjusting for receipt of PCI (yes vs. no), with ties handled by Efron approach and with treatment, sex (male vs. female), age at randomization (< 65 vs. > = 65 years), smoking status (smoker vs. nonsmoker), WHO/ECOG performance at baseline (normal activity vs. restricted activity), region (Asia vs. Europe vs. North America), and race (white vs. Black/African American vs. Asian vs. Other), time from last dose of concurrent CRT to randomization (< 14 days vs. > = 14 days to < 28 days vs. > = 28 days), prior platinum chemotherapy (cisplatin vs. carboplatin), prior radiotherapy regimen (once daily vs. twice daily), best response to concurrent CRT (CR vs. PR vs. stable disease) as covariates. Patients in the Argentina category are grouped with those in North America and Smoker category includes current and former smokers.
Sources: ADRIATIC Clinical Study Report and clinical study protocol; Cheng et al. (2024).24 Details included in the table are from the sponsor’s summary of clinical evidence.
Progression-Free Survival
Figure 6: Progression-Free Survival (Using BICR per RECIST 1.1) — Forest Plot by Primary and Sensitivity Analyses (FAS, PFS-IA), ADRIATIC Trial
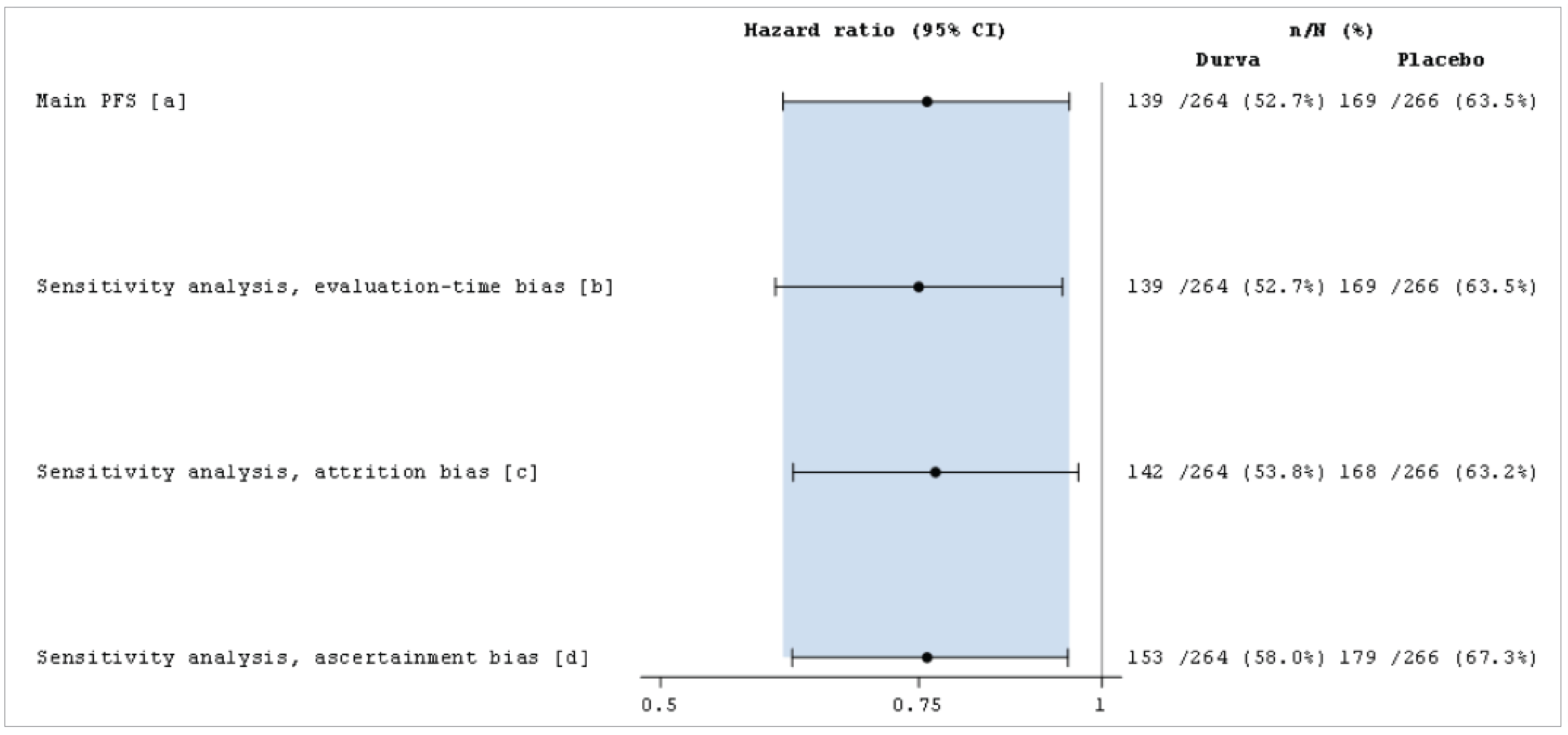
BICR = blinded independent central review; CI = confidence interval; durva = durvalumab; FAS = full analysis set; PFS = progression-free survival; PFS-IA = PFS interim analysis; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours version 1.1.
Data cut-off: January 15, 2024.
Source: ADRIATIC Clinical Study Report and clinical study protocol; Cheng et al. (2024).24
Appendix 2: HRQoL Improvement Rate
Please note that this appendix has not been copy-edited.
Table 21: Improvement Rate of EORTC QLQ-C30 and QLQ-LC13 Subscales/Items (FAS, PFS-IA), ADRIATIC Trial
EORTC subscale/item | Summary statistics | Durvalumab (N = 264) | Placebo (N = 266) |
|---|---|---|---|
QLQ-C30 GHS/QoL | Patients with baseline score ≤ 90, n | ███ | ███ |
Patients with improvement, n (%) | ██ ██████ | ██ ██████ | |
Improvement rate, n (%) | ██ (30.4) | ██ (33.5) | |
Odds ratio, durvalumab vs placebo | 0.85 | ||
95% CI | 0.545 to 1.338 | ||
2-sided P value | ██████ | ||
QLQ-C30 physical functioning | Patients with baseline score ≤ 90, n | ███ | ███ |
Patients with improvement, n (%) | ██ ██████ | ██ ██████ | |
Improvement rate, n (%) | ██ (35.4) | ██ (38.1) | |
Odds ratio, durvalumab vs placebo | 0.87 | ||
95% CI | 0.513 to 1.483 | ||
2-sided P value | ██████ | ||
QLQ-C30 role functioning | Patients with baseline score ≤ 90, n | ██ | ██ |
Patients with improvement, n (%) | ██ ██████ | ██ ██████ | |
Improvement rate, n (%) | ██ (52.1) | ██ (54.1) | |
Odds ratio, durvalumab vs placebo | 0.92 | ||
95% CI | 0.479 to 1.760 | ||
2-sided P value | ██████ | ||
QLQ-C30 fatigue symptom | Patients with baseline score ≥ 10, n | ███ | ███ |
Patients with improvement, n (%) | ███ ██████ | ███ ██████ | |
Improvement rate, n (%) | ██ (51.2) | ██ (44.9) | |
Odds ratio, durvalumab vs placebo | 1.30 | ||
95% CI | 0.845 to 2.006 | ||
2-sided P value | ██████ | ||
QLQ-C30 appetite loss symptom | Patients with baseline score ≥ 10, n | ██ | ██ |
Patients with improvement, n (%) | ██ ██████ | ██ ██████ | |
Improvement rate, n (%) | ██ (66.3) | ██ (65.1) | |
Odds ratio, durvalumab vs placebo | 1.04 | ||
95% CI | 0.551 to 1.951 | ||
2-sided P value | ██████ | ||
QLQ-LC13 dyspnea symptom | Patients with baseline score ≥ 10, n | ███ | ███ |
Patients with improvement, n (%) | ███ ██████ | ███ ██████ | |
Improvement rate, n (%) | ██ (55.6) | ██ (49.4) | |
Odds ratio, durvalumab vs placebo | 1.28 | ||
95% CI | 0.822 to 1.986 | ||
2-sided P value | ██████ | ||
QLQ-LC13 cough symptom | Patients with baseline score ≥ 10, n | ███ | ███ |
Patients with improvement, n (%) | ██ ██████ | ██ ██████ | |
Improvement rate, n (%) | ██ (46.9) | ██ (40.1) | |
Odds ratio, durvalumab vs placebo | 1.36 | ||
95% CI | 0.831 to 2.221 | ||
2-sided P value | ██████ | ||
QLQ-LC13 chest pain symptom | Patients with baseline score ≥ 10, n | ██ | ██ |
Patients with improvement, n (%) | ██ ██████ | ██ ██████ | |
Improvement rate, n (%) | ██ (67.3) | ██ (47.8) | |
Odds ratio, durvalumab vs placebo | 2.28 | ||
95% CI | 1.078 to 4.951 | ||
2-sided P value | ██████ | ||
CI = confidence interval; EORTC = European Organisation for Research and Treatment of Cancer; QLQ-C30 = EORTC 30-item core quality of life self-administered questionnaire; QLQ-LC13 = EORTC 13-item lung cancer module self-administered questionnaire; PCI = prophylactic cranial irradiation; QoL = quality of life; TNM = tumour, node, metastasis.
Note: Data cut-off date of January 15, 2024.
Sources: ADRIATIC Clinical Study Report and clinical study protocol; Cheng et al. (2024).24
Pharmacoeconomic Review
Abbreviations
AE
adverse event
BIA
budget impact analysis
CDA-AMC
Canada’s Drug Agency
CRT
chemoradiotherapy
CUA
cost-utility analysis
ES-SCLC
extensive-stage small cell lung cancer
ICER
incremental cost-effectiveness ratio
LS-SCLC
limited-stage small cell lung cancer
OS
overall survival
PFS
progression-free survival
QALY
quality-adjusted life-year
SCLC
small cell lung cancer
WTP
willingness to pay
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Durvalumab (Imfinzi), 50 mg/mL, concentrate for IV infusion |
Indication | Treatment, as monotherapy, of adult patients with limited-stage small cell lung cancer (LS-SCLC) whose disease has not progressed following platinum-based chemoradiation therapy (CRT). |
Health Canada approval status | NOC |
Health Canada review pathway | Priority review and Project Orbis |
NOC date | April 8, 2025 |
Reimbursement request | As per indication |
Sponsor | AstraZeneca Canada Inc. |
Submission history | Previously reviewed: Yes Indication: For the treatment of patients with locally advanced, unresectable non–small cell lung cancer following curative intent platinum-based chemoradiation therapy, for up to a maximum of 12 months Recommendation date: May 3, 2019 Recommendation: Reimburse with clinical criteria and/or conditions Indication: In combination with etoposide and either carboplatin or cisplatin, for the first-line treatment of adult patients with extensive-stage small cell lung cancer Recommendation date: July 27, 2021 Recommendation: Reimburse with clinical criteria and/or conditions Indication: In combination with Imjudo (tremelimumab for injection), for the first-line treatment of adult patients with unresectable hepatocellular carcinoma who require systemic therapy Recommendation date: November 3, 2023 Recommendation: Reimburse with clinical criteria and/or conditions Indication: In combination with gemcitabine-based chemotherapy, for the treatment of patients with locally advanced or metastatic biliary tract cancer Recommendation date: February 3, 2023 Recommendation: Reimburse with clinical criteria and/or conditions |
NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis PSM |
Target population | Adult patients with LS-SCLC who did not experience disease progression after platinum-based chemoradiotherapy |
Treatment | Durvalumab |
Dose regimen | 1,500 mg every 4 weeks until disease progression, unacceptable toxicity, or a maximum of 24 months |
Submitted price | Durvalumab: $938.67 per 120 mg/2.4 mL single-use vial for IV infusion Durvalumab: $3,911.11 per 500 mg/10 mL single-use vial for IV infusion |
Submitted treatment cost | $11,733 per 28-day cycle |
Comparator | Active surveillance |
Perspective | Publicly funded health care payer in Canada |
Outcomes | QALYs, LYs |
Time horizon | Lifetime (38 years) |
Key data source | ADRIATIC trial informed PFS, OS, TTD, and health state utility values |
Submitted results | ICER vs. active surveillance = $69,481 per QALY gained (incremental costs = $123,021; incremental QALYs = 1.77) |
Key limitations |
|
CDA-AMC reanalysis results |
|
AE = adverse event; CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; LS-SCLC = limited-stage small cell lung cancer; LY = life-year; OS = overall survival; PFS = progression-free survival; PSM = partitioned survival model; QALY = quality-adjusted life-year; TTD = time to treatment discontinuation; vs. = versus; WTP = willingness to pay.
Conclusions
The Canada’s Drug Agency (CDA-AMC) Clinical Review of the ADRIATIC trial found that durvalumab likely resulted in a clinically meaningful reduction in the risk of death and disease progression, with moderate certainty evidence, at 24 and 36 months. The CDA-AMC Clinical Review also noted concerns with the generalizability of the trial findings to patients typically encountered in clinical practice in Canada because the trial reflected a relatively healthy subset of patients with limited-stage small cell lung cancer (LS-SCLC). The CDA-AMC Clinical Review also noted that a higher incidence of serious adverse events (AEs) and immune-mediated AEs was associated with durvalumab than with placebo.
CDA-AMC undertook a reanalysis that addressed some of the limitations with the sponsor’s economic submission: overestimation of overall survival (OS) for patients under active surveillance, uncertainty regarding the impact of durvalumab on long-term OS, and modelling of AEs that lacked face validity. The results of the CDA-AMC reanalyses align with those of the sponsor’s submitted analysis. In the CDA-AMC base case, durvalumab is associated with an incremental cost-effectiveness ratio (ICER) of $79,547 per quality-adjusted life-year (QALY) gained compared to active surveillance in the patient population aligned with the ADRIATIC trial (incremental costs: $121,169; incremental QALYs: 1.52). A price reduction of approximately 34% would be required for durvalumab to be considered cost-effective at a willingness-to-pay (WTP) threshold of $50,000 per QALY gained. With this price reduction, the 28-day cycle drug acquisition costs for durvalumab would be approximately $7,744 per patient. The majority of the total costs for durvalumab were related to treatment acquisition costs, which were offset by reduced costs of subsequent therapy. As such, the economic results are sensitive to parameters that impact treatment acquisition costs and assumptions about subsequent therapy.
The CDA-AMC estimates of cost-effectiveness may be biased in favour of durvalumab because the CDA-AMC reanalysis was unable to fully address concerns with the modelled impact of AEs and the limited generalizability of the ADRIATIC trial. When it was assumed that patients receiving active surveillance have no treatment-emergent AEs, the ICER for durvalumab increased to $90,744 per QALY gained compared to active surveillance. The cost-effectiveness of durvalumab is also sensitive to the uncertainty in the long-term effectiveness of durvalumab treatment, and the results of the CDA-AMC reanalysis require that patients realize 1.83 additional life-years compared to those on active surveillance. Approximately 77% of the long-term survival benefit associated with durvalumab was predicted beyond the trial period through extrapolation.
Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, clinician groups, and drug plans that participated in the CDA-AMC review process.
Patient input was received from a joint submission by the Canadian Cancer Survivor Network, Lung Cancer Canada, and the Lung Health Foundation, which collected, through interviews, the perspectives of patients with small cell lung cancer (SCLC) who had experience with durvalumab through the ADRIATIC clinical trial or a compassionate access program. All 3 interviewees were older adults and former smokers. No symptoms were consistently reported by patients; however, coughing was a common theme that was often attributed to lifestyle factors rather than identified as a potential indicator of disease. Patients described their experience with chemotherapy and radiation under the current standard of care. The treatment goals described by patients included improved quality of life, improved energy, and reduced cost burden. It was unclear, though, if durvalumab had an impact in shrinking or stabilizing their cancers because all 3 patients had received only 2 cycles of durvalumab. One patient on durvalumab reported side effects such as appetite loss, vomiting, diarrhea, weight loss, and fatigue.
Clinician input was received from Ontario Health–Cancer Care Ontario and the Lung Cancer Canada Medical Advisory Committee, gathered through interviews with clinical experts and from published literature. The current standard of treatment for LS-SCLC is 4 cycles of cytotoxic platinum (cisplatin or carboplatin) and etoposide chemotherapy combined with concurrent or sequential thoracic radiation, followed by prophylactic cranial irradiation, if needed. The treatment goal was identified as curative intent, which would be achieved by delaying recurrences and improving OS. Improved quality of life was also important. The clinician input noted that in clinical practice, patients on durvalumab would be clinically assessed every 4 weeks for treatment toxicity, particularly immune-related AEs. The clinicians anticipated that the use of durvalumab after chemoradiation may result in fewer cases progressing to extensive-stage SCLC (ES-SCLC). However, it was also noted that if patients relapse after at least 6 months following 2 years of treatment, re-treatment with durvalumab plus chemotherapy may be considered for ES-SCLC. The clinician input noted that patients with an Eastern Cooperative Oncology Group Performance Status of 0 to 2 may be treated with durvalumab in clinical practice, although they were not included in the ADRIATIC trial. It was noted that treatment should be discontinued if there is unequivocal disease progression or intolerable treatment-related adverse effects or if the patient choses to discontinue. Durvalumab may be administered in community oncology clinics.
CDA-AMC–participating drug plans indicated that most jurisdictions will likely implement weight-based dosing of 20 mg/kg up to a maximum of 1,500 mg per dose and noted a maximum treatment duration of 24 months in the ADRIATIC trial. The drug plan input also anticipated that durvalumab may become the new standard of care for the indicated population. The plans also noted concerns about the eligibility criteria for downstream immunotherapy for ES-SCLC if patients are treated with durvalumab during earlier disease stages, and about use of durvalumab in patients with an Eastern Cooperative Oncology Group Performance Status of 2 or more.
Several of these concerns were addressed in the sponsor’s model:
Clinical outcomes valued by patients and clinicians (i.e., improved OS and quality of life) were considered.
In addition, CDA-AMC addressed some of these concerns as follows:
CDA-AMC explored the impact of adopting weight-based dosing for durvalumab in the pharmacoeconomic and budget impact analysis (BIA) based on feedback received from the drug plans.
CDA-AMC was unable to address the following concerns raised by the input relevant to the economic review:
Concerns regarding the generalizability of the cost-effectiveness results to patients with an Eastern Cooperative Oncology Group Performance Status of 2 or greater could not be addressed.
Economic Review
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis (CUA) of durvalumab compared with active surveillance (informed by the placebo arm of the ADRIATIC trial).1,2 The model population comprised adult patients with LS-SCLC who did not experience disease progression after platinum-based chemoradiotherapy (CRT). This was consistent with the intention-to-treat population in the ADRIATIC trial.2 The sponsor’s modelled population was aligned with the Health Canada indication and the reimbursement request.
Durvalumab is available as a solution for IV infusion (120 mg/2.4 mL and 500 mg/10 mL).3 The recommended dosage of durvalumab is 1,500 mg every 4 weeks for 24 months or until disease progression or unacceptable toxicity.3 Patients with a body weight of 30 kg or less must receive weight-based dosing of durvalumab at 20 mg/kg every 4 weeks as monotherapy until weight increases to greater than 30 kg. The sponsor submitted a price of $3,911.11 per 500 mg vial and $938.67 per 120 mg vial. The sponsor estimated the cost of treatment under the assumption that the 500 mg vial would be used exclusively, with no consideration of the 120 mg vial in the calculation. At the submitted price, the cost of durvalumab per 28-day cycle was estimated to be $11,733 per patient. The sponsor included the cost of drug wastage in the calculation of drug costs (i.e., no vial sharing was assumed). The comparator in this analysis was active surveillance, which was assumed by the sponsor to incur no treatment costs.
The clinical outcomes modelled were OS, progression-free survival (PFS), and time to treatment discontinuation. The model simulated life-years, QALYs, and costs for each treatment over a lifetime time horizon (38 years), discounted at an annual rate of 1.5%. The analysis was undertaken from the perspective of the Canadian public health care payer.
Model Structure
The sponsor submitted a partitioned survival model with 3 mutually exclusive health states: progression-free, progressed disease, and death (Appendix 3, Figure 1). The model cycle length was 4 weeks. All patients began in the progression-free health state and were assumed to be stable or responding to treatment, as defined by the PFS measure assessed in the ADRIATIC trial.2 The proportion of patients who were progression-free and the proportion of patients alive at each time point were estimated independently, using PFS and OS curves, respectively. The proportion of patients in the progressed disease health state was calculated as the difference between the proportion of living patients (estimated from the OS curve) and the proportion of patients who were progression-free (estimated from the PFS curve). In the progression-free health state, time on treatment was informed by the time to treatment discontinuation curve from the ADRIATIC trial.2 Patients were subject to a probability of death in each cycle.
Model Inputs
Baseline patient characteristics were aligned with the intention-to-treat population of the ADRIATIC trial,2 a phase III, randomized, placebo-controlled trial designed to evaluate the efficacy and safety of durvalumab in patients with LS-SCLC whose disease had not progressed after platinum-based CRT. The average patient in the modelled cohort was aged 62 years; had a mean body surface area of ████ m2, weight of █████ kg, height of ██████ cm, and creatinine clearance of ████ mL/min; and was more likely to be male (69.1%).2
Key inputs used to inform the clinical efficacy of durvalumab and active surveillance (i.e., OS and PFS) were derived from the ADRIATIC clinical trial,2 which had a maximum follow-up of approximately 5 years (data cut-off date: January 15, 2024). The sponsor adopted independent parametric models for each arm because the log odds diagnostic plots demonstrated violations of the proportional hazard assumption. All clinical outcomes (i.e., OS and PFS) were extrapolated beyond the trial duration by fitting the 1-spline odds distribution for OS and generalized gamma distribution for PFS. Model selection was based on visual inspection, statistical fit (Akaike information criterion, Bayesian information criterion), review of hazard plots and external validation against sponsor-sought expert opinion, and data from the CONVERT trial and the Canadian Small Cell Lung Cancer Database.4,5 The Kaplan-Meier curve for time to treatment discontinuation was adopted, and a maximum treatment duration was set at 24 months for durvalumab. The model was adjusted for the crossing over of PFS and OS curves (i.e., PFS could not be greater than OS), and OS was capped using the general population mortality rates. The sponsor assumed no treatment effectiveness waning in its base-case analysis.
The progression-free (█████) and progressed disease (█████) health utility for patients with LS-SCLC was derived from the ADRIATIC trial.2 In the trial, patient-reported data on health-related quality of life was measured using EQ-5D-5L questionnaires, which were mapped to EQ-5D-5L utility values using Canadian tariffs.2,6 Utilities were adjusted for age-related changes in quality of life.
The sponsor included grade 3 or 4 AEs that occurred in any treatment arm of the ADRIATIC trial.2 Disutilities and costs associated with AEs were incorporated in the first cycle of the model for all patients. The model incorporated a 1-time utility decrement for durvalumab (−0.3931) and for active surveillance (−0.2889), which were estimated using incidence data from the ADRIATIC trial; disutilities were obtained from published literature.7-13 Hospitalization costs for proxy conditions, which were assumed to reflect the modelled AEs, were obtained from the Canadian Institute for Health Information Patient Cost Estimator for the age group of 60 to 79 years and weighted by the incidence rates of AEs for durvalumab and active surveillance.14 This weighted average cost was applied as a 1-time cost associated with treating AEs.
The economic model included costs associated with drug acquisition, administration, routine medical care, and end-of-life care. Prices for durvalumab and subsequent therapy were obtained from the IQVIA DeltaPA database.15 A fixed dosage (i.e., 1,500 mg every 4 weeks) was adopted for durvalumab in alignment with the product monograph and trial protocol.2,3 Dosing for subsequent therapy was sourced from published literature,16,17 the Cancer Drug Formulary,18 and the CASPIAN trial.19 Subsequent therapy was modelled as a basket of treatments used for ES-SCLC (such as single-drug chemotherapy, platinum-based chemotherapy, immunotherapy plus chemotherapy, and combination chemotherapy). The cost of subsequent therapy was calculated as a weighted average of these treatments, with their relative use based on the ADRIATIC trial2 and sponsor-sought expert opinion. The weighted average cost of subsequent therapy for durvalumab and active surveillance was applied to 85% of patients who experienced disease progression based on sponsor-sought expert opinion. An administration cost for IV infusions and injections included the costs of physician fees and chair time, which were obtained from the Ontario Health Insurance Plan Schedule of Benefits and Fees and published literature, respectively.20,21 Costs associated with health care resource use for routine medical care were incurred by patients in the progression-free health state on and off treatment with durvalumab, patients in the progression-free health state under active surveillance considered “off treatment,” and patients in the progressed disease health state. The frequency of health care use was sourced from published literature.22,23 Radiotherapy costs were applied as a 1-time cost at the time of progression. Terminal care cost was also applied as a 1-time cost to patients who survived for 180 days or more after diagnosis; the cost was obtained from published literature.24
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically (1,000 iterations for the base-case and scenario analyses). The deterministic and probabilistic results were similar. The probabilistic findings are presented in the following subsections.
Base-Case Results
The results of the sponsor’s probabilistic base-case analysis for the reimbursement request population suggested that durvalumab was associated with an additional 1.77 QALYs at an additional cost of $123,021, relative to active surveillance. This resulted in an ICER of $69,481 per QALY gained (Table 3). The probability that durvalumab would be cost-effective compared to active surveillance was 23% at a WTP threshold of $50,000 per QALY gained.
The sponsor’s analysis predicted that durvalumab would be associated with a longer duration of life than active surveillance (incremental life-years: 1.98). At the end of 10 years, approximately 29% of the modelled patients in the durvalumab arm and 19% in the active surveillance arm were alive in the model. Given the duration of follow-up in the ADRIATIC trial (median follow-up for durvalumab: 30.75 months; maximum follow-up: 60.2 months), in contrast to the model’s lifetime horizon of 38 years, approximately 79% of the incremental QALYs gained by patients receiving durvalumab were accrued beyond the trial follow-up and were based on the sponsor’s extrapolations of the trial data. Durvalumab was associated with lower disutilities due to AEs than active surveillance. The key cost driver in the analysis was the drug acquisition cost of durvalumab. The drug acquisition cost accounted for the majority of the incremental costs for durvalumab, which were partially offset by the reduced costs of subsequent treatment (Table 10, Appendix 3). The submitted analysis is based on the publicly available prices for all drug treatments. Additional results from the sponsor’s submitted economic evaluation base case are presented in Appendix 3.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Treatment | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER vs. active surveillance ($/QALY) |
|---|---|---|---|---|---|
Active surveillance | 72,701 | Reference | 4.50 | Reference | Reference |
Durvalumab | 195,722 | 123,021 | 6.27 | 1.77 | 69,481 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Source: Sponsor’s pharmacoeconomic submission.1
Sensitivity and Scenario Analysis Results
The sponsor conducted scenario analyses, which included exploring alternative discount rates, time horizon (25 years), survival distributions for OS and PFS, utility values, and treatment waning effect applied at 10 years. No scenario analyses presented by the sponsor were key drivers of the cost-effectiveness results. There are no scenario analyses that drive the analysis as reported by the sponsor.
The sponsor’s model allowed exploration of the results from a societal perspective and included costs associated with informal care, travel to appointments, and lost working days. In this analysis, relative to active surveillance, the ICER was $75,013 per QALY gained. The results were similar to the sponsor’s base-case analysis using a health care payer perspective.
CDA-AMC Appraisal of the Sponsor’s Economic Evaluation
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications for the economic analysis:
Long-term OS of patients on active surveillance is overestimated: The sponsor used parametric modelling, adopting a 1-spline odds model, to extrapolate long-term OS beyond the observable time points in the ADRIATIC trial (median follow-up for durvalumab: 30.75 months; maximum follow-up: 60.2 months) to a lifetime horizon of 38 years.2 While the median OS is consistent with the trial data, the proportion of patients alive beyond the trial follow-up period does not align with feedback from clinical experts on the proportion of patients alive in their clinical practice under current standard of care (i.e., active surveillance). This discrepancy suggests that long-term survival beyond the trial follow-up, for which there is no direct evidence available to validate the extrapolated OS, has been overestimated. Although the modelled distribution performed well on statistical measures of fit for the period for which data are available (i.e., ranking first on Akaike information criterion and second on Bayesian information criterion), these metrics do not account for clinical plausibility, particularly in the absence of long-term survival data for LS-SCLC. In the published literature, the median OS ranges from 12 to 20 months and approximately 10% to 22% of patients survive past 5 years.25,26 In contrast, in the pharmacoeconomic model, the median OS is 31.2 months and approximately 33% of patients survive past 5 years. The modelled OS for patients on active surveillance is not aligned with clinical expert feedback that LS-SCLC is a progressive disease with poor prognosis. As such, the clinical plausibility of the modelled OS for active surveillance lacks face validity.
CDA-AMC evaluated parametric models based on the clinical plausibility of the long-term OS estimates derived through extrapolation. The Weibull distribution stood out as a more clinically plausible option for modelling OS in patients with LS-SCLC. The Weibull distribution estimated a median OS that aligned with the ADRIATIC trial (both 2.8 years) and estimated that 24% of patients would be alive at 5 years. Overall, the Weibull distribution was more aligned with the estimates from the published literature and the expectations of the clinical experts consulted by CDA-AMC regarding the long-term survival of patients with LS-SCLC and reflected the progressive nature of the disease.
In its reanalysis, CDA-AMC selected the Weibull distribution to model OS for patients on active surveillance. To implement this change, CDA-AMC corrected a formula used for probabilistic sampling.
Impact of durvalumab on long-term OS is uncertain: To extrapolate the long-term survival of patients treated with durvalumab, the sponsor also adopted a 1-spline odd distribution to extrapolate long-term OS, which estimated a median OS of 46.9 months (3.9 years). The sponsor’s model predicted that patients on durvalumab would have an average life expectancy of 70.66 years. Similar to the OS projections for patients under active surveillance, the OS for patients receiving durvalumab appeared to have been overestimated. According to the CDA-AMC Clinical Review,2 there are concerns regarding the generalizability of the treatment effect observed in the ADRIATIC trial because the trial population was deemed to be healthier than the patients who would be treated in clinical practice based on the expert feedback received for the review. As such, it is uncertain if the treatment efficacy of durvalumab in the trial would be observed in real-world conditions. CDA-AMC notes that approximately 57% of life-years derived from durvalumab treatment in the sponsor’s model were accrued after the trial duration (maximum follow-up: 60.2 months) and, as a result, represent an outcome of modelling instead of observed data. The results of this cost-effectiveness analysis are sensitive to the sponsor’s extrapolation assumptions, which were based on a limited sample size near the end of the trial and a high rate of censoring close to the median follow-up. This adds uncertainty in the long-term clinical efficacy of durvalumab treatment.
CDA-AMC evaluated parametric models based on the clinical plausibility of the long-term OS estimates for durvalumab. The clinical expert input expected to observe a plateauing effect in OS after 10 years for patients who would receive durvalumab. Among the alternatives, the exponential model was the most reflective of expert feedback. While it does not explicitly model a plateauing effect, the OS declines at a slower rate than OS for patients under active surveillance. As such, the predicted OS benefit is maintained over time, with a 7% survival benefit for patients treated with durvalumab compared to active surveillance at 15 years. The exponential model also produced a similar median OS estimate to the sponsor’s selected extrapolation (3.8 years). CDA-AMC notes that the CDA-AMC–selected distribution amplifies the estimated treatment effect of durvalumab (evident through an increased gap between the OS curves of durvalumab and active surveillance) compared to the sponsor’s selected distributions up to 7 years. However, the exponential model was deemed to be the best compromise between aligning the OS curve with the expected treatment effect of durvalumab observed in the trial and ensuring consistency with the anticipated long-term survival for patients with LS-SCLC.
In its reanalysis, CDA-AMC selected the exponential distribution to model OS for patients on durvalumab. To implement this change, CDA-AMC corrected a formula used for probabilistic sampling.
Modelled PFS does not meet face validity: The sponsor’s base case adopted a generalized gamma distribution to model PFS for patients on durvalumab and active surveillance. The modelled PFS and OS curves for both durvalumab and active surveillance merge, resulting in all modelled events after the merging time point being deaths (i.e., no patients experience disease progression). As a result, patients accrue only the costs of the progression-free health state and no subsequent therapy costs. This also likely results in the overestimation of PFS because patients do not transition to the progressed disease health state before death, and there is resulting uncertainty in the estimated PFS for both durvalumab and active surveillance. As such, the modelled PFS lacks face validity and contributes to the uncertainty in the modelled outcomes. Expert feedback obtained for this review noted that the PFS curve should be conceptually shifted to the left of the OS curve, reflecting that in clinical practice, patients who progress survive approximately 1 year. CDA-AMC evaluated parametric models based on the clinical plausibility of the modelled relationship between OS and PFS. Although some of the alternative distributions, such as log-logistic distribution, improved clinical plausibility by pushing the merging point between OS and PFS further into the future, they also resulted in a postprogression survival benefit, which was considered implausible. As a result, CDA-AMC was unable to address the lack of face validity in the modelled PFS.
CDA-AMC was unable to address this limitation in reanalysis.
Modelled impact of AEs lacks face validity: In the sponsor’s base-case analysis, the disutility associated with the AE profile experienced by patients under active surveillance (−0.39) was greater than that of patients being treated with durvalumab (−0.29), suggesting that patients receiving active surveillance experience greater quality-of-life impairments from AEs than patients on active durvalumab. However, this finding does not meet face validity because it is implausible that patients under active surveillance experience treatment-emergent AEs, and it has not been demonstrated that durvalumab has a protective effect against some of these grade 3 or 4 AEs. In the ADRIATIC trial,2 16.4% of patients discontinued treatment due to AEs compared to 10.6% of patients receiving placebo, suggesting that there may be important treatment-related AEs associated with durvalumab. Additionally, according to the clinical expert feedback received for this review, there is insufficient evidence of clinical plausibility linking some of the modelled AEs to immunotherapy. For example, the clinical expert input flagged that chronic obstructive pulmonary disease is likely unrelated to treatment and, rather, likely associated with smoking. The AEs that are not related to treatment are expected to be implicitly accounted for in health state utilities, and modelling them separately risks double counting their impact on quality-of-life estimates.
In reanalysis, CDA-AMC excluded AEs identified to be likely unrelated to immunotherapy based on clinical expert feedback. These AEs included pneumonia, pulmonary embolism, radiation pneumonitis, hypertension, and chronic obstructive pulmonary disease. CDA-AMC additionally explored the impact of assuming patients under active surveillance have no AEs in a scenario analysis.
Subsequent therapy is uncertain: The sponsor assumed that patients with LS-SCLC who had experienced disease progression would receive treatment for ES-SCLC. According to sponsor-sought expert opinion, the majority of patients who experience disease progression under active surveillance receive the more costly immunotherapy in combination with etoposide and platinum-based chemotherapy as subsequent therapy. In contrast, the majority of patients on durvalumab received the less costly etoposide and platinum-based chemotherapy as subsequent therapy. Although this met face validity for patients who experienced disease progression while on treatment, according to expert feedback received by CDA-AMC for this review, patients may be re-treated with immunotherapy if their disease progresses after 6 months of completing durvalumab treatment. The sponsor’s modelling approach did not account for the timing of disease progression, which would influence the choice and cost of subsequent therapy. Additionally, the CDA-AMC Clinical Review reported that ████ of patients who were treated with durvalumab in the ADRIATIC trial received immunotherapy as subsequent therapy, compared to █████ of patients on active surveillance receiving immunotherapy as subsequent therapy. In the sponsor’s economic model, 10% of patients treated with durvalumab and 65% of patients on active surveillance were assumed to receive immunotherapy. Because cost offsets associated with durvalumab are derived from avoiding subsequent therapy costs, the results of this analysis are sensitive to the sponsor’s assumptions about subsequent therapy, particularly changes in the proportion of patients receiving immunotherapies, which are more costly than chemotherapies.
Given the uncertainty in the distribution of subsequent therapies patients will receive, CDA-AMC explored the impact of excluding subsequent therapy in a scenario analysis. While most patients in clinical practice in Canada would receive a subsequent therapy at the time of progression, this scenario analysis aims to explore the impact of the uncertainty in the sponsor’s subsequent therapy assumptions.
Dosing of durvalumab is uncertain: In the ADRIATIC trial,2 durvalumab was administered at a fixed dosage of 1,500 mg every 4 weeks. Similarly, in the economic model, the sponsor assumed that all patients would receive 1,500 mg every 4 weeks. Input from the participating public drug plans received by CDA-AMC for this review indicated that weight-based dosing is likely to be implemented for durvalumab. CDA-AMC notes that if a weight-based dosing approach would reduce drug acquisition costs, it would lower the ICER for durvalumab compared to active surveillance. However, it remains uncertain whether a weight-based regimen would result in the same clinical outcomes as the fixed-dose regimen studied in the trial.
In CDA-AMC scenario analysis, weight-based dosing (i.e., 20 mg/kg every 4 weeks) and vial sharing were adopted for durvalumab.
Poor modelling practices were employed: The sponsor’s submitted model included numerous IFERROR statements, which lead to situations in which the parameter value may be overwritten with an alternative value without alerting the user to the automatized overwriting. The systematic use of IFERROR statements makes thorough auditing of the sponsor’s model impractical. It remains unclear whether the model is running inappropriately by overriding errors.
CDA-AMC also identified an inaccuracy with the sponsor’s approach to modelling costs related to health care resource use for patients who have not experienced disease progression under active surveillance. In the sponsor-submitted model, there are no costs related to routine medical care for the proportion patients who have not experienced disease progression “on treatment,” which underestimates the costs associated with active surveillance.
CDA-AMC made a correction that all patients who have not experienced disease progression under active surveillance incurred costs related to health care resource use. CDA-AMC was unable to address the use of IFERROR statements in the model and notes that a thorough validation of the sponsor’s model was not possible.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CDA-AMC (refer to Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CDA-AMC comment |
|---|---|
The sponsor assumed that background mortality for the modelled population is reflective of the general population. | Inappropriate. It is not reasonable to assume no excess smoking-related mortality in a patient population consisting predominantly of current and former smokers (91%), as seen in the ADRIATIC trial population. The sponsor’s approach leads to an underestimation of overall mortality for the modelled population. |
The sponsor used general population utility norms from a study that used the Health Utilities Index. | Inappropriate. Using utilities measured using different measurement instruments contradicts best practice. General population utility norms for the population living in Canada measured using EQ-5D are available and would have been more appropriate to use. |
CDA-AMC = Canada’s Drug Agency.
CDA-AMC Reanalyses of the Economic Evaluation
Base-Case Results
The CDA-AMC base case was derived by making changes in model parameter values and assumptions in consultation with clinical experts (Table 5). CDA-AMC corrected the sponsor’s base case by assuming that all patients who do not experience disease progression under active surveillance incur costs related to health care resource use. CDA-AMC undertook a stepped analysis, sequentially incorporating each adjustment outlined in Table 6 into the sponsor’s model, to demonstrate the impact of each change. These adjustments included adopting the Weibull distribution to extrapolate OS for patients under active surveillance, adopting an exponential distribution to model OS for patients on durvalumab, and excluding AEs not related to durvalumab treatment.
Table 5: CDA-AMC Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
1. Health care resource use costs for patients in the progression-free health state under active surveillance | Not incurred by patients “on treatment” | Incurred by all patients in the progression-free health state |
Changes to derive the CDA-AMC base case | ||
1. OS extrapolation for patients under active surveillance | 1-spline odds | Weibull |
2. OS extrapolation for patients on durvalumab | 1-spline odds | Exponential |
3. AEs such as pneumonia, pulmonary embolism, radiation pneumonitis, hypertension, and chronic obstructive pulmonary disease | Included | Excluded |
CDA-AMC base case | ― | Reanalysis 1 + 2 + 3 |
AE = adverse event; CDA-AMC = Canada’s Drug Agency; OS = overall survival.
In the CDA-AMC base case, durvalumab treatment was associated with an ICER of $79,547 per QALY gained compared with active surveillance (incremental costs: $121,169; incremental QALYs: 1.52) (Table 6). There was a 3.26% probability that durvalumab was cost-effective at a WTP threshold of $50,000 per QALY gained. The CDA-AMC base case is based on publicly available prices for all drug treatments. A detailed breakdown of the disaggregated results is available in Appendix 4, Table 11.
The CDA-AMC base-case results align with those of the sponsor’s submitted analysis, indicating that durvalumab is not cost-effective at a WTP threshold of $50,000 per QALY gained relative to active surveillance. Consistent with the sponsor’s analysis, the CDA-AMC reanalysis estimates that the majority (77%) of incremental QALYs gained by patients receiving durvalumab relative to active surveillance were derived in the model on the basis of extrapolation.
Table 6: Summary of the Stepped Analysis of the CDA-AMC Reanalysis Results
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
Sponsor’s base case | Active surveillance | 67,158 | 4.43 | Reference |
Durvalumab | 194,154 | 6.20 | 71,835 | |
Sponsor’s corrected base case | Active surveillance | 68,096 | 4.43 | Reference |
Durvalumab | 194,154 | 6.20 | 71,304 | |
CDA-AMC reanalysis 1: OS extrapolation for active surveillance | Active surveillance | 67,515 | 2.58 | Reference |
Durvalumab | 194,154 | 6.20 | 34,952 | |
CDA-AMC reanalysis 2: OS extrapolation for durvalumab | Active surveillance | 68,096 | 4.43 | Reference |
Durvalumab | 192,798 | 4.26 | Dominated by active surveillance | |
CDA-AMC reanalysis 3: excluding adverse events unrelated to treatment | Active surveillance | 67,345 | 4.70 | Reference |
Durvalumab | 193,551 | 6.29 | 79,091 | |
CDA-AMC base case (reanalyses 1 + 2 + 3) | Active surveillance | 66,764 | 2.84 | Reference |
Durvalumab | 192,195 | 4.36 | 82,824 | |
CDA-AMC base case (reanalyses 1 + 2 + 3) (probabilistic) | Active surveillance | 71,192 | 2.85 | Reference |
Durvalumab | 192,361 | 4.37 | 79,547 |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; OS = overall survival; QALY = quality-adjusted life-year.
Notes: The CDA-AMC reanalysis is based on publicly available prices of the comparator treatments. The results of all steps are presented deterministically unless otherwise indicated; the cumulative CDA-AMC base case is always presented both deterministically and probabilistically.
Scenario Analysis Results
CDA-AMC undertook price reduction analyses deterministically based on the sponsor’s and the CDA-AMC base case (Table 7). The CDA-AMC base case suggested that a 34% price reduction is required for durvalumab to be considered cost-effective relative to active surveillance at a WTP threshold of $50,000 per QALY gained.
Table 7: CDA-AMC Price Reduction Analyses
Price reduction | Unit drug cost ($) | ICERs for durvalumab vs. active surveillance ($/QALY) | |
|---|---|---|---|
Sponsor base case | CDA-AMC reanalysis | ||
No price reduction | 3,911 | 68,091 | 79,547 |
10% | 3,520 | 60,719 | 70,818 |
20% | 3,129 | 53,348 | 62,089 |
30% | 2,738 | 45,976 | 53,359 |
40% | 2,347 | 38,605 | 44,630 |
50% | 1,956 | 31,234 | 35,901 |
60% | 1,564 | 23,862 | 27,172 |
70% | 1,173 | 16,491 | 18,442 |
80% | 782 | 9,119 | 9,713 |
90% | 391 | 1,748 | 984 |
100% | 0 | Dominant | Dominant |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Note: The price reduction for the sponsor’s base case was conducted using the probabilistic results of the corrected sponsor’s base case.
CDA-AMC conducted scenario analyses to explore the impact of alternative assumptions on the cost-effectiveness of durvalumab:
assuming patients under active surveillance have no treatment-emergent AEs
excluding subsequent therapy costs
adopting weight-based dosage (20 mg/kg every 4 weeks) and vial sharing for durvalumab based on the drug plan input received for this review.
The results of the scenario analyses are presented in Appendix 4, Table 12.
The cost-effectiveness of durvalumab was sensitive to the impact of AEs and subsequent therapy. When assuming that patients under active surveillance have no treatment-emergent AEs, the ICER for durvalumab increased to $90,744 per QALY gained compared to active surveillance. When excluding subsequent therapy from the analysis, the ICER for durvalumab increased to $105,319 per QALY gained compared to active surveillance.
Issues for Consideration
CADTH previously reviewed and recommended durvalumab for several conditions, including for the first-line treatment of adult patients with ES-SCLC, in combination with etoposide and either carboplatin or cisplatin.27 The cost-effectiveness results of these evaluations may not be directly comparable to those in the current review because of differences in target population, model structure, clinical effectiveness parameters, health state utility values, and cost inputs. CDA-AMC further notes that the pan-Canadian Pharmaceutical Alliance concluded negotiations with a letter of intent for durvalumab for the aforementioned indication.28 As such, durvalumab has a confidential negotiated price and is currently funded by jurisdictional cancer formularies.29-31 The CDA-AMC reanalyses are based on the publicly available price of durvalumab, which may be different than the confidential price and may influence the results of the cost-effectiveness analyses and BIAs.
Durvalumab is available as 120 mg/2.4 mL and 500 mg/10 mL vials.32 The sponsor’s analysis used the 500 mg vial in estimating treatment costs. Given that both vials have the same price per milligram, this approach does not affect the overall cost estimates.
Overall Conclusions
The CDA-AMC Clinical Review of the ADRIATIC trial found that durvalumab likely resulted in a clinically meaningful reduction in the risk of death and disease progression, with moderate certainty evidence, at 24 and 36 months. The CDA-AMC Clinical Review also noted concerns with the generalizability of the trial findings to patients typically encountered in clinical practice in Canada because the trial reflected a relatively healthy subset of patients with LS-SCLC. The CDA-AMC Clinical Review also noted a higher incidence of serious AEs and immune-mediated AEs associated with durvalumab than with placebo.
CDA-AMC undertook a reanalysis that addressed some of the limitations with the sponsor’s economic submission: overestimation of OS for patients under active surveillance, uncertainty regarding the impact of durvalumab on long-term OS, and modelling of AEs that lacked face validity. The results of the CDA-AMC reanalyses align with those of the sponsor’s submitted analysis. In the CDA-AMC base case, durvalumab is associated with an ICER of $79,547 per QALY gained compared to active surveillance in the patient population aligned with the ADRIATIC trial (incremental costs: $121,169; incremental QALYs: 1.52). A price reduction of approximately 34% would be required for durvalumab to be considered cost-effective at a WTP threshold of $50,000 per QALY gained. With this price reduction, the 28-day cycle drug acquisition costs for durvalumab would be approximately $7,744 per patient. The majority of the total costs for durvalumab were related to treatment acquisition costs, which were offset by reduced costs of subsequent therapy. As such, the economic results are sensitive to parameters that impact treatment acquisition costs and assumptions about subsequent therapy.
The CDA-AMC estimates of cost-effectiveness may be biased in favour of durvalumab because the CDA-AMC reanalysis was unable to fully address concerns with the modelled impact of AEs and the limited generalizability of the ADRIATIC trial. When it was assumed that patients receiving active surveillance have no treatment-emergent AEs, the ICER for durvalumab increased to $90,744 per QALY gained compared to active surveillance. The cost-effectiveness of durvalumab is also sensitive to the uncertainty in the long-term effectiveness of durvalumab treatment, and the results of the CDA-AMC reanalysis require that patients realize 1.83 additional life-years compared to those on active surveillance. Approximately 77% of the long-term survival benefit associated with durvalumab was predicted beyond the trial period through extrapolation.
References
1.AstraZeneca Canada Inc. Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Imfinzi (durvalumab), concentrate for solution for infusion, 50 mg / mL, intravenous. November 14, 2024.
2.AstraZeneca. ADRIATIC CSR [sponsor supplied reference].
3.AstraZeneca Canada. IMFINZI (durvalumab), solution, 50 mg / mL, intravenous infusion [product monograph]. September 21, 2020.
4.Faivre-Finn C, Snee M, Ashcroft L, et al. Concurrent once-daily versus twice-daily chemoradiotherapy in patients with limited-stage small-cell lung cancer (CONVERT): an open-label, phase 3, randomised, superiority trial. Lancet Oncol. 2017;18(8):1116-1125. doi: 10.1016/S1470-2045(17)30318-2 PubMed
5.Moore S, Zhan LJ, Chowdhury D, et al. 1793P Treatment and outcomes of limited disease small cell lung cancer (LD-SCLC) in the Canadian small cell lung cancer database (CASCADE). Ann Oncol. 2024;35:S1066. doi: 10.1016/j.annonc.2024.08.1884
6.Xie F, Pullenayegum E, Gaebel K, et al. A Time Trade-off-derived Value Set of the EQ-5D-5L for Canada. Med Care. 2016;54(1):98-105. doi: 10.1097/MLR.0000000000000447 PubMed
7.Goeree R, Villeneuve J, Goeree J, Penrod JR, Orsini L, Tahami Monfared AA. Economic evaluation of nivolumab for the treatment of second-line advanced squamous NSCLC in Canada: a comparison of modeling approaches to estimate and extrapolate survival outcomes. J Med Econ. 2016;19(6):630-44. doi: 10.3111/13696998.2016.1151432 PubMed
8.Li LY, Wang H, Chen X, Li WQ, Cui JW. First-line atezolizumab plus chemotherapy in treatment of extensive small cell lung cancer: a cost-effectiveness analysis from China. Chin Med J (Engl). 2019;132(23):2790-2794. doi: 10.1097/CM9.0000000000000536 PubMed
9.Morris S, Gurusamy KS, Patel N, Davidson BR. Cost-effectiveness of early laparoscopic cholecystectomy for mild acute gallstone pancreatitis. Br J Surg. 2014;101(7):828-35. doi: 10.1002/bjs.9501 PubMed
10.Monahan M, Ensor J, Moore D, Fitzmaurice D, Jowett S. Economic evaluation of strategies for restarting anticoagulation therapy after a first event of unprovoked venous thromboembolism. J Thromb Haemost. 2017;15(8):1591-1600. doi: 10.1111/jth.13739 PubMed
11.Tachkov K, Kamusheva M, Pencheva V, Mitov K. Evaluation of the economic and social burden of chronic obstructive pulmonary disease (COPD). Biotechnology & Biotechnological Equipment. 2017;31:1-7. doi: 10.1080/13102818.2017.1335616
12.Sullivan PW, Ghushchyan V. Preference-Based EQ-5D index scores for chronic conditions in the United States. Med Decis Making. 2006;26(4):410-20. doi: 10.1177/0272989X06290495 PubMed
13.Smith-Palmer J, Bae JP, Boye KS, Norrbacka K, Hunt B, Valentine WJ. Evaluating health-related quality of life in type 1 diabetes: a systematic literature review of utilities for adults with type 1 diabetes. Clinicoecon Outcomes Res. 2016;8:559-571. doi: 10.2147/CEOR.S114699 PubMed
14.Canadian Institute for Health Information. Patient cost estimator [accessed by sponsor]. 2024. https://www.cihi.ca/en/patient-cost-estimator
15.IQVIA. DeltaPA. Accessed September 25, 2024. https://www.iqvia.com/
16.Horn L, Mansfield AS, Szczesna A, et al. First-Line Atezolizumab plus Chemotherapy in Extensive-Stage Small-Cell Lung Cancer. N Engl J Med. 2018;379(23):2220-2229. doi: 10.1056/NEJMoa1809064 PubMed
17.Sehouli J, Chekerov R, Reinthaller A, et al. Topotecan plus carboplatin versus standard therapy with paclitaxel plus carboplatin (PC) or gemcitabine plus carboplatin (GC) or pegylated liposomal doxorubicin plus carboplatin (PLDC): a randomized phase III trial of the NOGGO-AGO-Study Group-AGO Austria and GEICO-ENGOT-GCIG intergroup study (HECTOR). Ann Oncol. 2016;27(12):2236-2241. doi: 10.1093/annonc/mdw418 PubMed
18.Cancer Care Ontario. Drug Formulary [accessed by sponsor].
19.Paz-Ares L, Dvorkin M, Chen Y, et al. Durvalumab plus platinum-etoposide versus platinum-etoposide in first-line treatment of extensive-stage small-cell lung cancer (CASPIAN): a randomised, controlled, open-label, phase 3 trial. Lancet. 2019;394(10212):1929-1939. doi: 10.1016/S0140-6736(19)32222-6 PubMed
20.OHIP. Schedule of benefits and fees. Accessed September 25, 2024. https://www.ontario.ca/page/ohip-schedule-benefits-and-fees
21.Tam VC, Ko YJ, Mittmann N, et al. Cost-effectiveness of systemic therapies for metastatic pancreatic cancer. Curr Oncol. 2013;20(2):e90-e106. doi: 10.3747/co.20.1223 PubMed
22.National Institute for Health and Care Excellence. Durvalumab for maintenance treatment of unresectable non-small-cell lung cancer after platinum-based chemoradiation (NICE technology appraisal TA798) [accessed by sponsor]. https://www.nice.org.uk/guidance/ta798/history
23.O'Sullivan DE, Cheung WY, Syed IA, et al. Real-World Treatment Patterns, Clinical Outcomes, and Health Care Resource Utilization in Extensive-Stage Small Cell Lung Cancer in Canada. Curr Oncol. 2021;28(4):3091-3103. doi: 10.3390/curroncol28040270 PubMed
24.Bremner KE, Krahn MD, Warren JL, et al. An international comparison of costs of end-of-life care for advanced lung cancer patients using health administrative data. Palliat Med. 2015;29(10):918-28. doi: 10.1177/0269216315596505 PubMed
25.Dawe DE, Rittberg R, Syed I, et al. Real-world predictors of survival in patients with limited-stage small-cell lung cancer in Manitoba, Canada. Front Oncol. 2023;13:1191920. doi: 10.3389/fonc.2023.1191920 PubMed
26.Cancer Canadian Society. Survival statistics for small cell lung cancer. Accessed February 4, 2025. https://cancer.ca/en/cancer-information/cancer-types/lung/prognosis-and-survival/small-cell-lung-cancer-survival-statistics
27.CADTH Canadian Drug Expert Committee (CDEC) final recommendation: Durvalumab for ES-SCLC. Can J Health Technol. 2021;1(7). doi:10.51731/cjht.2021.109
28.pCPA negotiations for Imfinzi (durvalumab). pan-Canadian Pharmaceutical Alliance; 2021. Accessed February 4, 2025. https://www.pcpacanada.ca/negotiation/21550
29.Cancer Care Ontario. CRBPETOP+DURV [product monograph] [sponsor supplied reference]. Accessed February 4, 2025. https://www.cancercareontario.ca/en/drugformulary/regimens/68436
30.Cancer Care Ontario. CISPETOP+DURV [product monograph] [sponsor supplied reference]. Accessed February 4, 2025. https://www.cancercareontario.ca/en/drugformulary/regimens/68431
31.Cancer Care Ontario. DURV(MNT) [product monograph]. Accessed February 4, 2025. https://www.cancercareontario.ca/en/drugformulary/regimens/68426
32.AstraZeneca Canada Inc. Imfinzi (durvalumab): concentrate for solution for infusion, 50 mg / mL, intravenous [product monograph]. September 9, 2024.
33.AstraZeneca Canada Inc. Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Imfinzi (durvalumab), concentrate for solution for infusion, 50 mg / mL, intravenous. November 14, 2024.
34.Canadian Cancer Statistics Advisory Committee in collaboration with the Canadian Cancer Society, Statistics Canada, and the Public Health Agency of Canada. Canadian Cancer Statistics (Table 1, Appendix Data Table 2). Canadian Cancer Society. Accessed October 3, 2024. https://cancer.ca/en/research/cancer-statistics/canadian-cancer-statistics
35.Canadian Cancer Statistics Advisory Committee. Canadian Cancer Statistics. A 2020 special report on lung cancer. Canadian Cancer Society. Accessed September 24, 2024. https://cdn.cancer.ca/-/media/files/cancer-information/resources/publications/2020-canadian-cancer-statistics-special-report/2020-canadian-cancer-statistics-special-report-en.pdf
36.Sun A, Durocher-Allen LD, Ellis PM, et al. Guideline for the Initial Management of Small Cell Lung Cancer (Limited and Extensive Stage) and the Role of Thoracic Radiotherapy and First-line Chemotherapy. Clin Oncol (R Coll Radiol). 2018;30(10):658-666. doi: 10.1016/j.clon.2018.06.008 PubMed
37.Gronberg BH, Halvorsen TO, Flotten O, et al. Randomized phase II trial comparing twice daily hyperfractionated with once daily hypofractionated thoracic radiotherapy in limited disease small cell lung cancer. Acta Oncol. 2016;55(5):591-7. doi: 10.3109/0284186X.2015.1092584 PubMed
38.Bogart J, Wang X, Masters G, et al. High-Dose Once-Daily Thoracic Radiotherapy in Limited-Stage Small-Cell Lung Cancer: CALGB 30610 (Alliance)/RTOG 0538. J Clin Oncol. 2023;41(13):2394-2402. doi: 10.1200/JCO.22.01359 PubMed
39.Medina DA, Cotté FE, Gaudin AF, Lévy P. An exploration of the methodology for considering treatment duration in budget impact analyses in oncology: A case study in 2L advanced renal cell carcinoma [accessed by sponsor]. 2018. https://www.ispor.org/
40.Royston P, Parmar MK. Restricted mean survival time: an alternative to the hazard ratio for the design and analysis of randomized trials with a time-to-event outcome. BMC Med Res Methodol. 2013;13(1):152. doi: 10.1186/1471-2288-13-152 PubMed
41.Cancer Care Ontario. Etoposide [product monograph] [sponsor supplied reference]. Accessed October 23, 2024. https://www.cancercareontario.ca/en/drugformulary/drugs/monograph/43711
42.Cancer Care Ontario. CRBPETOP [product monograph] [sponsor supplied reference]. Accessed October 23, 2024. https://www.cancercareontario.ca/en/drugformulary/regimens/monograph/52371
43.Cancer Care Ontario. CRBPETOP+DURV [product monograph] [sponsor supplied reference]. https://www.cancercareontario.ca/en/drugformulary/regimens/monograph/68436
44.Cancer Care Ontario. CISPTOPO [product monograph] [sponsor supplied reference]. Accessed October 23, 2024. https://www.cancercareontario.ca/en/drugformulary/regimens/monograph/45526files/16149/45526.html
45.Cancer Care Ontario. CISPIRIN [product monograph] [sponsor supplied reference]. Accessed October 23, 2024. https://www.cancercareontario.ca/en/drugformulary/regimens/monograph/51316files/16152/51316.html
46.Cancer Care Ontario. CISPETOP+DURV [product monograph] [sponsor supplied reference]. https://www.cancercareontario.ca/en/drugformulary/regimens/monograph/68431
47.Cancer Care Ontario. CRBPETOP(PO)+ATEZ [product monograph] [sponsor supplied reference]. Accessed October 23, 2024. https://www.cancercareontario.ca/en/drugformulary/regimens/monograph/62096files/16146/62096.html
48.Cancer Care Ontario. CISPETOP(3D) [product monograph] [sponsor supplied reference]. Accessed October 23, 2024. https://www.cancercareontario.ca/en/drugformulary/regimens/monograph/52341files/16151/52341.html
49.Cancer Care Ontario. CAV [product monograph] [sponsor supplied reference]. Accessed October 23, 2024. https://www.cancercareontario.ca/en/drugformulary/regimens/monograph/52361files/16145/52361.html
50.Irinotecan [product monograph]. Cancer Care Ontario. Accessed October 23, 2024. https://www.cancercareontario.ca/en/drugformulary/drugs/monograph/44181files/16154/44181.html
51.Topotecan [product monograph]. Cancer Care Ontario. Accessed October 23, 2024. https://www.cancercareontario.ca/en/drugformulary/drugs/monograph/44216
Appendix 1: Cost Comparison Table
Please note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical expert(s) and drug plan. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 8: CDA-AMC Cost Comparison Table for LS-SCLC
Treatment | Strength / concentration | Form | Price | Recommended dosage | Average daily cost ($) | Average 28-day cost ($) |
|---|---|---|---|---|---|---|
Durvalumab | 120 mg/2.4 mL 500 mg/10 mL | Vial for IV infusion | $938.6700a $3,911.1100a | 1,500 mg every 4 weeks for 24 months or until disease progression or unacceptable toxicityb | 419.05 | 11,733 |
Weight-based dosage: 20 mg/kg every 4 weeksc | 402.29 | 11,264 |
CDA-AMC = Canada’s Drug Agency; LS-SCLC = limited-stage small cell lung cancer.
Note: Costs do not include dispensing fees. Vial sharing was not assumed.
aSponsor’s submitted price.1
bDosing was obtained from product monograph.32
cInput from clinical experts and participating drug plans indicated that weight-based dosage may be used for durvalumab in patients weighing more than 30 kg. Weight-based dosage assumes a mean weight of 72 kg and vial sharing.
Appendix 2: Submission Quality
Please note that this appendix has not been copy-edited.
Description | Yes or no | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | Yes | No comment. |
Model has been adequately programmed and has sufficient face validity | No | Refer to key limitation “Poor modelling practices were employed.” Additionally, the sponsor’s probabilistic analysis failed to produce plausible results when selecting several parametric models for both overall survival and progression-free survival. CDA-AMC identified an error in their probabilistic sampling formula in which values were log-transformed and not converted back to the appropriate scale for use in the model. |
Model structure is adequate for decision problem | Yes | No comment. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | Yes | No comment. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | Yes | No comment. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | Yes | No comment. |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Please note that this appendix has not been copy-edited.
OS = overall survival; PFS = progression-free survival.
Source: Sponsor’s pharmacoeconomic submission.1
Detailed Results of the Sponsor’s Base Case
Table 10: Disaggregated Summary of the Sponsor’s Economic Evaluation Results
Parameter | Durvalumab | Active surveillance |
|---|---|---|
Discounted LYs | ||
Total | 7.69 | 5.71 |
Discounted QALYs | ||
Total | 6.27 | 4.50 |
By health state | ||
Progression-free | 5.21 | 3.61 |
Progressed disease | 1.35 | 1.28 |
Death | 0.00 | 0.00 |
Adverse events | −0.29 | −0.39 |
Discounted costs ($) | ||
Total | 195,722 | 72,701 |
Primary treatment | ||
Acquisition | 154,714 | 0 |
Administration | 3,862 | 0 |
Adverse event management | 1,231 | 1,158 |
Subsequent treatment cost | 15,917 | 53,360 |
Health care resource use (i.e., routine medical care cost) | ||
Progression-free on treatment | 946 | 0 |
Progression-free off treatment | 2,037 | 1,328 |
Progressed disease | 8,381 | 7,931 |
Radiotherapy | 669 | 667 |
End of life | 8,021 | 8,295 |
LY = life-year; QALY = quality-adjusted life-year.
Source: Sponsor’s pharmacoeconomic submission.1
Appendix 4: Additional Details on the CDA-AMC Reanalyses and Sensitivity Analyses of the Economic Evaluation
Please note that this appendix has not been copy-edited.
Detailed Results of CDA-AMC Base Case
Table 11: Disaggregated Summary of CDA-AMC Economic Evaluation Results
Parameter | Durvalumab | Active surveillance |
|---|---|---|
Discounted LYs | ||
Total | 5.21 | 3.38 |
Discounted QALYs | ||
Total | 4.37 | 2.85 |
By health state | ||
Progression-free | 3.67 | 2.12 |
Progressed disease | 0.90 | 0.85 |
Death | 0.00 | 0.00 |
Adverse events | −0.20 | −0.13 |
Discounted costs ($) | ||
Total | 192,361 | 71,192 |
Primary treatment | ||
Acquisition | 154,833 | 0 |
Administration | 3,833 | 0 |
Adverse event management | 628 | 407 |
Subsequent treatment cost | 16,307 | 54,706 |
Health care resource use (i.e., routine medical care cost) | ||
Progression-free on treatment | 946 | 0 |
Progression-free off treatment | 1,306 | 1,582 |
Progressed disease | 5,488 | 5,226 |
Radiotherapy | 666 | 667 |
End of life | 8,391 | 8,624 |
CDA-AMC = Canada’s Drug Agency; LY = life-year; QALY = quality-adjusted life-year.
Scenario Analyses
Table 12: Scenario Analyses Conducted on the CDA-AMC Base Case
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
Sponsor’s base case (probabilistic) | Active surveillance | 72,701 | 4.50 | Reference |
Durvalumab | 195,722 | 6.27 | 69,481 | |
CDA-AMC base case (probabilistic) | Active surveillance | 71,192 | 2.85 | Reference |
Durvalumab | 192,361 | 4.37 | 79,547 | |
CDA-AMC scenario 1: No AEs for patients under active surveillance | Active surveillance | 66,356 | 2.97 | Reference |
Durvalumab | 192,195 | 4.36 | 90,744 | |
CDA-AMC scenario 2: Exclude subsequent therapy costs | Active surveillance | 16,410 | 2.84 | Reference |
Durvalumab | 175,909 | 4.36 | 105,319 | |
CDA-AMC scenario 3: Assuming weight-based dosing and vial sharinga | Active surveillance | 65,435 | 2.84 | Reference |
Durvalumab | 185,724 | 4.36 | 79,428 |
AE = adverse event; CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
aAssuming weight-based dosing only does not impact total costs and ICER because the same number of vials are used to administer weight-based dose if the cost of drug wastage is included.
Appendix 5: Submitted BIA and CDA-AMC Appraisal
Please note that this appendix has not been copy-edited.
Table 13: Summary of Key Take-Aways
Key Take-Aways of the Budget Impact Analysis |
|---|
|
Summary of Sponsor’s BIA
The submitted BIA assessed the expected budgetary impact of reimbursing durvalumab for the treatment of adult patients with LS-SCLC who did not progress following platinum-based CRT.33 The BIA was undertaken from the perspective of the Canadian public drug plans at base year (2024) and over a 3-year time horizon (2025 to 2027). Key inputs to the BIA are documented in Table 14.
The sponsor estimated the number of eligible patients for durvalumab using an epidemiologic approach with data obtained from various sources including published literature, the ADRIATIC clinical trial, sponsor’s internal market research and sponsor’s assumptions. 2, 25,34,35,36-38 The sponsor estimated the number of new patients using annual incident rates of lung cancer for each jurisdiction and narrowed the population to LS-SCLC patients deemed medically inoperable who have not progressed within 42 days of receiving CRT. The time period used to define nonprogression (i.e., 42 days) was selected to align with ADRIATIC trial.2 The sponsor used a technique called restricted mean survival time to estimate the mean number of months patients spent in progression-free and progressed states using PFS curve from the accompanying pharmacoeconomic analysis.39,40 This was used to estimate the number of patients who remain progression-free and progress based on the year they start treatment. The proportion of patients who progressed discontinued treatment with durvalumab and 85% of progressed patients incurred the cost of subsequent therapy. Time on treatment was estimated using time to treatment discontinuation curve from the accompanying pharmacoeconomic analysis. The sponsor estimated that patients initiating durvalumab treatment in year 1 remain on treatment, on average, for 7.85 months in year 1 and 4.34 months in year 2.
The sponsor included active surveillance as the only comparator, which was assumed to have no cost to the drug plans. Drug acquisition costs for durvalumab and subsequent therapy were included. The cost of subsequent therapy was calculated as a weighted average of treatments for extensive-stage SCLC, with their relative use based on the ADRIATIC trial and sponsor-sought expert opinion.2 Prices for durvalumab and subsequent therapy were obtained from IQVIA DeltaPA database.15 Dosing was obtained from product monograph for durvalumab and sourced from Cancer Care Ontario drug formulary and published literature for subsequent therapy.16,17,19,29,30,41-51
Table 14: Summary of Key Model Parameters
Parameter | Sponsor’s estimate |
|---|---|
Target population | |
Annual lung cancer incidence | 0.055% to 0.078% |
Proportion of patients with SCLC | 12% |
Proportion of LS-SCLC patients in stages I to III | 33% |
Proportion of patients deemed medically inoperable | 90% |
Proportion of patients receiving chemoradiotherapy | 80% |
Proportion of patients that have not progressed after receiving chemoradiotherapy | 97.5% |
Proportion of patients that are eligible for systemic therapy | 100% |
Number of patients eligible for drug under review | 432 / 441 / 449 |
Market uptake (3 years) | |
Uptake (reference scenario) Active surveillance | 100% / 100% / 100% |
Uptake (new drug scenario) Durvalumab Active surveillance | ███ █ ███ █ ███ ███ █ ███ █ ███ |
Cost of treatment (per patient, per 28 days) | |
Durvalumab Active surveillance | $11,733 $0 |
Cost of subsequent treatment (per patient, 1-time cost)a | |
Durvalumab Active surveillance | $9,313 $45,194 |
LS = limited stage; SCLC = small cell lung cancer.
Note: Patient characteristics used to calculate treatment costs were obtained from the ADRIATIC trial (mean age: 60.3 years, proportion of males: 34.1%, mean weight: 65.30 kg, mean height: 1.66 m, body surface area: 1.74 m2, creatin clearance of 89.4 mL/min). The cost of drug wastage was included (i.e., vial sharing was not assumed). Mark-ups and dispensing fees were not included.
aThe cost of subsequent therapy is the weighted sum of acquisition cost, treatment duration, and proportion of patients on each treatment used in the second line (i.e., single-drug chemotherapy, platinum-based chemotherapy, immunotherapy plus chemotherapy and combination chemotherapy).
Summary of the Sponsor’s BIA Results
The sponsor estimated the 3-year budget impact of reimbursing durvalumab for the treatment of adult patients with LS-SCLC who did not progress following platinum-based CRT to be $76,242,169 (year 1: $13,560,945; year 2: $27,229,367; year 3: $35,451,856).
CDA-AMC Appraisal of the Sponsor’s BIA
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Restricting eligibility to medically inoperable patients does not reflect anticipated clinical practice: The sponsor restricted the eligible population to medically inoperable patients and assumed that these patients would be the only ones receiving durvalumab in clinical practice. However, clinical expert feedback received for this review highlighted that even those deemed medically operable would receive durvalumab in clinical practice due to delays in scheduling surgeries and the associated costs and burden of recovery. The exclusion of patients who are medically operable does not reflect the anticipated clinical practice and underestimates the number of eligible patients.
In reanalysis, 100% of patients were assumed to be medically inoperable. Doing this removed the restriction on eligibility by operable status.
Market share of durvalumab is underestimated: The sponsors assumed that durvalumab would have a market share of ███ in year 1, ███ in year 2, and ███ by year 3, if it is reimbursed. The clinical expert feedback received by CDA-AMC for this review anticipated a higher uptake of durvalumab in clinical practice. The expert input noted that the proportion of patients who would be actively surveilled if durvalumab is reimbursed would be very low. The current standard of care is active surveillance, which offers no therapeutic benefit. As such, if durvalumab is reimbursed, its uptake is expected to be rapid in clinical practice.
In reanalysis, CDA-AMC assumed that the market uptake of durvalumab would be 60%, 70% and 80% in years 1, 2 and 3, respectively, based on clinical expert feedback.
Treatment duration is uncertain: The sponsor used restricted mean survival time to estimate the mean number of months patients using the time to treatment discontinuation curve from the CUA. In this approach, the sponsor assumed that patients who initiate treatment accrue the costs of 7.85 months in the first year of treatment, and 4.34 in the second year of treatment. While this amounts to 12.19 months total time on treatment for patients who initiate Imfinzi in years 1 and 2 in the BIA and aligns with the average number of cycles of durvalumab received in the ADRIATIC trial (12.9 infusions; average treatment duration of 54.5 weeks), patients who initiate in year 3 only accrue the cost of 7.85 months of treatment. As such, the sponsor’s approach may have underestimated the treatment duration of patients who initiate durvalumab treatment in year 3 of the BIA.
In reanalysis, CDA-AMC assumed that patients receive 12 months of treatment continuously in the year they initiate, and accrue no treatment costs in years 2 and 3 following initiation. This aligns with the average number of infusions received in the ADRIATIC trial. CDA-AMC explored the impact of adopting sponsor’s estimates of treatment duration in scenario analysis.
Subsequent therapy is uncertain: As noted in the “CDA-AMC Appraisal of the Sponsor’s Economic Evaluation,” the choice and cost of subsequent therapy would be based on the timing of disease progression, which was not captured in the sponsor’s analysis. As such, the cost of subsequent therapy is uncertain.
CDA-AMC explored the impact of excluding subsequent therapy in scenario analysis.
Dosing of durvalumab is uncertain: In the ADRIATIC trial, durvalumab was administered at a fixed dosage of 1,500 mg every 4 weeks. Similarly, in the economic model, the sponsor assumed that all patients would receive 1,500 mg every 4 weeks. Participating public drug plan input received by CDA-AMC for this review indicated that a weight-based dosage is likely to be adopted for durvalumab (20 mg/kg [up to 1,500 mg] every 4 weeks).
CDA-AMC explored the impact of adopting weight-based dosage with vial sharing in scenario analysis.
Patient weight is not aligned with the CUA or the ADRIATIC trial: In the BIA, the sponsor adopted an average patient weight of 65.30 kg, however, the patient weight in the CUA is █████ kg and is aligned with the patient population in the ADRIATIC trial.
CDA-AMC corrected the average patient weight by aligning it with the CUA.
CDA-AMC Reanalyses of the BIA
CDA-AMC corrected the sponsor’s base case by adopting the average weight of the patient population in the ADRIATIC trial. CDA-AMC revised the sponsor’s base case by increasing the proportion of patients deemed medically inoperable, increasing the market share of durvalumab, and assuming that 12 months of treatment costs would be incurred the year of treatment initiation (Table 15).
Table 15: CDA-AMC Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
Weight (kg) | 65.30 | █████ |
Changes to derive the CDA-AMC base case | ||
Proportion of patients deemed medically inoperable | 90% | 100% |
Market share of durvalumab (year 1 / year 2 / year 3) | █████ | 60% / 70% / 80% |
Treatment duration (months) | Year 1: 7.85 Year 2: 4.34 | Year 1: 12 Year 2: 0 |
CDA-AMC base case | Reanalysis 1 + 2 | |
CDA-AMC = Canada’s Drug Agency.
The results of the CDA-AMC step-wise reanalysis are presented in summary format in Table 16 and a more detailed breakdown is presented in Table 17.
In the CDA-AMC reanalysis, the three-year budget impact of reimbursing durvalumab for the treatment of adult patients with LS-SCLC who did not experience disease progression following CRT increased to $133,319,319 (year 1: $39,053,199; year 2: $44,425,786; year 3: $49,840,333). The estimated budget impact is primarily driven by market share of durvalumab. The submitted analysis is based on the publicly available prices of the durvalumab and comparator treatments.
Table 16: Summary of the CDA-AMC Reanalyses of the Budget Impact Analysis
Stepped analysis | Three-year total ($) |
|---|---|
Submitted base case | 76,242,169 |
Submitted base case, corrected | 76,242,018 |
CDA-AMC reanalysis 1 | 84,713,353 |
CDA-AMC reanalysis 2 | 102,344,898 |
CDA-AMC reanalysis 3 | 91,443,533 |
CDA-AMC base case | 133,319,319 |
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency.
CDA-AMC conducted the following scenario analyses to address remaining uncertainty, using the CDA-AMC base case (results are provided in Table 17):
Adopting sponsor’s estimates of treatment duration (year 1: 7.85 months; year 2: 4.34 months).
Assuming weight-based dosing of durvalumab.
Excluding subsequent therapy costs.
Table 17: Detailed Breakdown of the CDA-AMC Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|---|
Submitted base case | Reference | 9,044,980 | 9,216,835 | 13,077,086 | 17,036,441 | 39,330,361 |
New drug | 9,044,980 | 22,777,780 | 40,306,452 | 52,488,297 | 115,572,530 | |
Budget impact | 0 | 13,560,945 | 27,229,367 | 35,451,856 | 76,242,169 | |
Submitted base case, corrected | Reference | 9,045,581 | 9,217,447 | 13,077,954 | 17,037,572 | 39,332,972 |
New drug | 9,045,581 | 22,778,357 | 40,307,261 | 52,489,371 | 115,574,990 | |
Budget impact | 0 | 13,560,911 | 27,229,308 | 35,451,800 | 76,242,018 | |
CDA-AMC base case | Reference | 10,050,645 | 10,241,607 | 14,531,060 | 18,930,635 | 53,753,947 |
New drug | 10,050,645 | 49,294,807 | 58,956,846 | 68,770,969 | 187,073,267 | |
Budget impact | 0 | 39,053,199 | 44,425,786 | 49,840,333 | 133,319,319 | |
CDA-AMC scenario 1: Sponsor’s estimates of treatment duration | Reference | 10,050,645 | 10,241,607 | 14,531,060 | 18,930,635 | 53,753,947 |
New drug | 10,050,645 | 34,032,679 | 56,772,646 | 66,614,530 | 167,470,501 | |
Budget impact | 0 | 23,791,071 | 42,241,587 | 47,683,895 | 113,716,553 | |
CDA-AMC scenario analysis 2: Assuming weight-based dosing and vial sharing | Reference | 10,158,631 | 10,351,645 | 14,687,184 | 19,134,029 | 44,172,858 |
New drug | 10,158,631 | 48,044,523 | 57,638,331 | 67,466,388 | 173,149,242 | |
Budget impact | 0 | 37,692,878 | 42,951,148 | 48,332,359 | 128,976,384 | |
CDA-AMC scenario analysis 3: Exclude subsequent therapy | Reference | 0 | 0 | 0 | 0 | 0 |
New drug | 0 | 44,121,175 | 52,452,723 | 61,084,943 | 157,658,840 | |
Budget impact | 0 | 44,121,175 | 52,452,723 | 61,084,943 | 157,658,840 |
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.