Drugs, Health Technologies, Health Systems
Reimbursement Review
Brentuximab Vedotin (Adcetris)
Sponsor: BC Cancer Agency and Pediatric Oncology Group of Ontario
Therapeutic area: Hodgkin lymphoma
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
ABVD
doxorubicin-bleomycin-vinblastine-dacarbazine
ABVE-PC
doxorubicin-bleomycin-vincristine-etoposide–prednisone-cyclophosphamide
AE
adverse event
ASCT
autologous stem cell transplant
AVD
doxorubicin-vinblastine-dacarbazine
AVEPC
doxorubicin-vincristine-etoposide-prednisone-cyclophosphamide
BEACOPP
bleomycin-etoposide-doxorubicin-cyclophosphamide-vincristine-procarbazine-prednisone
BV
brentuximab vedotin
CI
confidence interval
CR
complete response
ECOG PS
Eastern Cooperative Oncology Group Performance Status
EFS
event-free survival
EMA
European Medicines Agency
EORTC QLQ-C30
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30
G-CSF
granulocyte colony-stimulating factor
HL
Hodgkin lymphoma
HR
hazard ratio
HRQoL
health-related quality of life
IPFP
International Prognostic Factor Project
IPS
International Prognostic Score
IRF
independent review facility
ITC
indirect treatment comparison
ITT
intention to treat
MedDRA
Medical Dictionary for Regulatory Activities
mPFS
modified progression-free survival
OH-CCO
Ontario Health (Cancer Care Ontario)
OS
overall survival
PET2
PET response after 2 cycles of chemotherapy
PFS
progression-free survival
POGO
Pediatric Oncology Group of Ontario
PPS
post-progression survival
RCT
randomized controlled trial
SAE
serious adverse event
TEAE
treatment-emergent adverse event
TESAE
treatment-emergent serious adverse event
VAS
visual analogue scale
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Information on drug submitted for review | |
Drug product | Brentuximab vedotin for injection (Adcetris), 50 mg per vial, lyophilized powder for reconstitution, IV infusion |
Sponsor | BC Cancer Agency Pediatric Oncology Group of Ontario |
Approved indication | For the treatment of previously untreated patients with stage IV HL, in combination with doxorubicin, vinblastine, and dacarbazine |
Reimbursement request | Brentuximab vedotin in combination with doxorubicin, vinblastine, and dacarbazine for the treatment of previously untreated patients with advanced-stage HL Brentuximab vedotin in combination with doxorubicin, vincristine, etoposide, prednisone, and cyclophosphamide in previously untreated high-risk HL in the pediatric population |
Health Canada approval status | Unlabelled indication |
Health Canada review pathway | NA |
NOC date | NA |
Recommended dose | 1.2 mg/kg up to a maximum of 120 mg in combination with doxorubicin, vinblastine, and dacarbazine administered every 2 weeks for a maximum of 12 doses or until disease progression or unacceptable toxicity occurs |
HL = Hodgkin lymphoma; NA = not applicable; NOC = Notice of Compliance.
Source: Manufacturer’s Summary of Clinical Evidence1 and brentuximab vedotin product monograph.2
Introduction
Hodgkin lymphoma (HL) is a B-cell malignancy that originates in the lymphocytes.3 Classical HL accounts for 95% of all HL cases. The estimated incidence in Canada in 2022 was 2.6 cases per 100,000.4 Based on 2018 Canadian Cancer Statistics, which report cancer incidence by stage, approximately 23.3% of patients in Canada presenting with HL have stage III disease and 22.7% have stage IV disease.5 Childhood HL represents 6% of all cancers and has an incidence rate of 12 cases per million (1.2 cases per 100,000) per year in patients aged 0 to 14 years.6 In 2019, a total of 25 children in Canada in this age group were diagnosed with HL.7 The clinical experts consulted by the review team noted that advanced-stage HL in adult patients refers to Ann Arbor stage III and IV HL. The clinical experts consulted for this review noted that, in clinical practice, pediatric patients with HL are usually classified into low-, intermediate-, and high-risk groups, and the high-risk group is generally considered equivalent to those with advanced-stage classical HL in adults. The clinical experts reported that pediatric patients with high-risk or advanced-stage HL are generally defined as having stage II with bulk tumour, stage III with B symptoms (stage IIIB), and stage IV with or without B symptoms (stage IVA and stage IVB). These patients are treated the same in clinical practice: as having advanced-stage HL. However, the clinical experts consulted by the review team also indicated that the definition of advanced-stage HL in pediatric patients is evolving and may vary by centre, with some centres defining any stage III or IV case of HL in pediatric patients as advanced-stage disease.
The goal of therapy in patients with advanced HL is curative.8,9 The clinical experts consulted by the review team noted that patients with advanced HL are treated the same, regardless of stage. Current first-line treatment regimens for adult patients with advanced-stage HL rely on chemotherapy. For patients with stage IV HL, the preferred regimen uses brentuximab vedotin (BV) in combination with doxorubicin-vinblastine-dacarbazine (AVD). For patients with advanced HL, treatment approaches also include doxorubicin-bleomycin-vinblastine-dacarbazine (ABVD) for up to 6 cycles with PET response after 2 cycles of chemotherapy (PET2)-directed treatment adaptation, and based on upfront PET2-driven treatment adaptation with bleomycin-etoposide-doxorubicin-cyclophosphamide-vincristine-procarbazine-prednisone (BEACOPP).
For pediatric patients, the clinical experts consulted by the review team also noted that most clinical centres in Canada use doxorubicin-bleomycin-vincristine–etoposide-prednisone-cyclophosphamide (ABVE-PC) for 5 cycles with radiation therapy determined by PET2, while relatively fewer centres use vincristine-etoposide-prednisone-doxorubicin–cyclophosphamide-vincristine-prednisone-dacarbazine.
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of brentuximab (50 mg per vial, IV infusion) for the treatment of previously untreated patients with advanced-stage HL, in combination with AVD. BV has been previously reviewed and recommended for reimbursement by the CDA-AMC pan-Canadian Oncology Review Expert Review Committee for the treatment of previously untreated patients with stage IV HL in combination with AVD.10
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient and clinician groups who responded to the review team’s call for input and from clinical experts consulted by the review team for the purpose of this review.
Patient Input
One patient group, Lymphoma Canada, provided input on the use of BV to treat previously untreated patients with advanced-stage HL, in combination with AVD. Patient input was gathered from an online anonymous patient survey from March 14 to May 2, 2023. A total of 26 responses were gathered, and 3 respondents reported receiving BV + AVD treatment.
Patients were asked questions regarding the physical and psychosocial symptoms experienced at the time of diagnosis, current quality of life, and how these symptoms affected their daily activities. At the time of their lymphoma diagnosis, most of the patients described fatigue (79%) as the most consequential symptom (5 out of 5), followed by enlarged lymph nodes (58%), shortness of breath (63%), and weight loss (47%). In addition, 74% of patients reported experiencing anxiety or worry, 68% stressing about their diagnosis, 63% difficulty in sleeping, and 58% fearing progression of their lymphoma. Regarding physical symptoms that currently affect their quality of life, out of 7 responses, fatigue (29%) and headaches (14%) were identified as the most significant factors having a negative impact on quality of life. The most psychosocial factors with the greatest impact were the stress of having cancer (71%), fear of progression (71%), anxiety or worry (71%), difficulty sleeping (43%), problems concentrating (43%), and inability to attend work or school (43%).
Among the surveyed patients, 3 reported receiving ABVD in the front-line setting, 2 were treated with other forms of chemotherapy, and 1 was treated with cyclophosphamide-doxorubicin-prednisone-rituximab-vincristine. While evaluating the importance of the outcomes of new treatments, patients from the survey prioritized the need for a novel lymphoma therapy to control disease symptoms, prolong disease remission, extend life spans, and improve quality of life.
While describing the experience with the treatment under review, 1 of the 3 patients who had received BV + AVD indicated they have been in remission for between 6 months and a year, another was in remission for longer than a year, and the other patient was in post-treatment (and unsure about their remission status). Side effects from the BV + AVD treatment reported by patients were fatigue (n = 3), neutropenia (n = 2), constipation (n = 2), joint or muscle pain (n = 2), low platelet count (n = 1), low blood pressure (n = 1), and decreased appetite (n = 1). Two patients reported experiencing financial setbacks — 1 due to absence from work and 1 due to the cost of other medications. One of these patients mentioned having a poor experience with BV, and the other 2 rated their experience as very good.
Clinician Input
Input From Clinical Experts Consulted by the Review Team
The clinical experts consulted by the review team noted that improving the proportion of patients cured with first-line treatment is an important unmet need for patients with advanced-stage HL. The clinical experts also emphasized the need to reduce treatment failure, prevent disease progression or relapse, and avoid late side effects (e.g., secondary malignancies and cardiac and pulmonary late effects) and therapies that are toxic (e.g., autologous stem cell transplant [ASCT]), particularly for younger patients diagnosed with advanced-stage HL and older patients who have poor tolerance to treatment.
The clinical experts noted that BV + AVD is considered a front-line therapy for advanced HL. The clinical experts noted that, at the time of the review, BV + AVD had been approved only for the treatment of previously untreated patients with stage IV HL, and pointed out that the use of BV + AVD in patients with stage III classical HL could shift the current treatment paradigm for those patients. The clinical experts indicated that, in pediatric patients, BV would be used in combination with a different chemotherapy backbone, namely the therapy investigated in a phase III randomized controlled trial (AHOD1331)11 in pediatric patients: BV in combination with doxorubicin-vincristine-etoposide-prednisone-cyclophosphamide (AVEPC). Trials of BV + AVD in pediatric patients had not been completed at the time of this review.12
The clinical experts noted that any previously untreated adult patients with stage III or IV classical HL who meet the eligibility criteria of the ECHELON-1 trial are best suited for the use of BV + AVD. The clinical experts indicated that pediatric patients with advanced-stage classical HL could also be eligible for BV in combination with chemotherapy, and eligibility for therapy should be determined by the treating physician or based on the eligibility criteria of the AHOD1331 trial.
The clinical experts noted that PET scans, typically PET2 and a PET scan after all 6 cycles (end of treatment), are used to assess responses. In patients who have a complete response (CR), follow-up visits take place every 3 to 4 months for 2 years, then typically every 6 months for 3 more years. Patients with a partial response may undergo radiation therapy, and patients with refractory disease undergo further investigations (e.g., a biopsy) and treatment with a second-line regimen. One of the clinical experts, who specializes in pediatrics, noted that outcomes used in clinical practice to assess treatment response are generally aligned with outcomes typically used in adult therapeutics. The clinical experts noted that overall survival (OS) is the most clinically meaningful outcome to assess the efficacy of BV in combination with chemotherapy in patients with advanced-stage HL, and progression-free survival (PFS) is an important outcome. In pediatric patients, event-free survival (EFS) is also important.
The clinical experts agreed that discontinuation of BV + AVD is uncommon overall because unacceptable toxicity or refractory HL is not common. The clinical experts identified several situations in which BV + AVD can be discontinued, including completion of treatment, clear evidence of progression of disease, and an unacceptable adverse event (AE).
The clinical experts noted that diagnosis of the disease in adult patients must be made by an experienced pathologist. Selection of patients for BV + AVD should be made by a hemato-oncologist experienced with treating HL. Treatment can be delivered in the specialty clinics of nonacademic centres. The clinical expert specializing in pediatrics noted that all pediatric oncology patients are cared for by pediatric oncology teams at tertiary care centres. Some aspects of care may be provided at satellite centres after diagnostic and management decisions are made.
Clinician Group Input
Clinician group input on the review of BV was received from the Ontario Health (Cancer Care Ontario) (OH-CCO) Hematology Cancer Drug Advisory Committee and the Pediatric Oncology Group of Ontario (POGO). Six clinicians provided input on behalf of OH-CCO. POGO is a collaboration of Ontario’s 5 specialized childhood cancer centres. The input collected from POGO was prepared in a consultative manner, with 1 clinician discussing the indication with other members of the submission panel and seeking input from POGO’s Therapeutic and Technology Advisory Committee. POGO’s final submission was based on this process.
Input from OH-CCO emphasized the need to improve outcomes with first-line therapy to avoid the need for second-line therapy. The group noted that patients with stage III and IV disease would be best suited for current treatment. The group indicated that typical lymphoma response measures, including PET scans, are used in clinical practice to assess patients’ response to treatment. They noted they would discontinue treatment with BV + AVD in the event of significant toxicity or disease progression.
Input from POGO noted that, while a variety of chemotherapy and radiation approaches are available for use as standard of care, these vary by region and between pediatric- and adult-focused practitioners in Canada. POGO reported that, historically, the ABVD chemotherapy backbone used with BV in adult patients has not been used by pediatric oncologists to treat pediatric patients due to concerns regarding higher anthracycline (doxorubicin) and bleomycin exposure, as well as known dose-dependent cardiac and pulmonary toxicities. POGO noted that BV has been studied and used in combination with another chemotherapy regimen (AVEPC) in patients aged 2 to 21 years with previously untreated high-risk HL.11 POGO indicated that this alternative chemotherapy backbone is more commonly used in the pediatric setting, and BV + AVEPC has become standard care for high-risk pediatric patients in Ontario. Regarding treatment goals in pediatric patients with HL, POGO emphasized the need to avoid disease recurrence to minimize potential late effects from subsequent chemotherapies and ASCT received at relapse, and the associated impact on health-related quality of life (HRQoL). While describing the outcomes used to determine whether a pediatric patient is responding to treatment for HL, POGO emphasized the importance of OS and EFS, considering the higher chance of experiencing late effects of therapy after treatment among the younger patient population. Like OH-COO, POGO suggested treatment be discontinued at disease progression.
Drug Program Input
Input was obtained from the drug programs that participate in our reimbursement review process. The following were identified as key factors that could potentially affect the implementation of a recommendation for BV:
relevant comparators
consideration for initiation of therapy
consideration of discontinuation of therapy
consideration for prescribing of therapy
generalizability
funding algorithm (oncology only)
care provision issues.
The clinical experts consulted by the review team provided advice on the potential implementation issues raised by the drug programs (Table 4).
Clinical Evidence
Pivotal Studies and Randomized Controlled Trial Evidence
Description of Studies
One phase III, open-label, randomized, active-controlled, superiority trial (ECHELON-1, N = 1,334) was identified from a systematic literature review conducted by the sponsor.13,14 The primary objective of the ECHELON-1 trial was to determine the efficacy of BV + AVD relative to ABVD as measured by modified progression-free survival (mPFS). The key secondary objective was to compare OS between BV + AVD and ABVD. The ECHELON-1 trial is ongoing. Data gathered at the cut-off dates of April 20, 2017, and June 1, 2021, were assessed for this review. New data from a descriptive analysis of OS conducted in response to a request for supplementary information from the European Medicines Agency (EMA) with a data cut-off date of March 11, 2023, was also included in this report.15
Participants eligible to be included in the ECHELON-1 trial were previously untreated adult patients with histologically confirmed advanced-stage classical HL, consisting of stage III and stage IV patients as determined by the Ann Arbor classification system. Patients with nodular lymphocyte-predominant HL and those with sensory or motor peripheral neuropathy were excluded. The median age of enrolled patients was 36 years (range = 18 to 83); most (66%) were younger than 45 years, and 14% were aged 60 years or older. Of the total number of patients enrolled, 58% were male and 84% were white. Notably, most patients had stage IV disease (64%), 2 or 3 (53%) had International Prognostic Factor Project (IPFP) risk factors, an Eastern Cooperative Oncology Group Performance Status (ECOG PS) of 0 (57%), extranodal involvement at diagnosis (62%), and B symptoms (59%) at baseline.
Efficacy Results
The efficacy end points that were noted to be important to patients and clinicians in stakeholder input are summarized in Table 2. Outcomes of OS, PFS (as determined by an investigator), percentage of patients alive without HL, HRQoL as measured by the European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 (EORTC QLQ-C30), and EQ-5D-3L questionnaire were obtained from data with a cut-off date of June 1, 2021, while mPFS as determined by an independent review facility (IRF) was based on data with a cut-off date of April 20, 2017.
Overall Survival
As of the data cut-off date of June 1, 2021, the median follow-up was 73.3 months (95% confidence interval [CI], 72.61 to 74.05) in the BV + AVD group and 72.4 months (95% CI, 71.10 to 73.63) in the ABVD group. In the intention-to-treat (ITT) population, the hazard ratio (HR) for OS was 0.59 (95% CI, 0.396 to 0.879; P = 0.009), favouring BV + AVD treatment. The absolute difference in the number of OS events between the BV + AVD (6%) and ABVD (10%) arms was 4%. The median OS was not reached for patients with advanced-stage classical HL in either the BV + AVD group or the ABVD group. In a subgroup analyses by disease stage, the HR for OS was 0.863 (95% CI, 0.452 to 1.648; P = 0.654) for patients with stage III classical HL and 0.478 (95% CI, 0.286 to 0.799; P = 0.004) patients with stage IV classical HL.
As of the data cut-off date of March 11, 2023, the descriptive analysis for OS had a median follow-up of approximately 88 months for the ITT population: The median follow-up duration was 89.7 months (95% CI, 86.57 to 90.55) for the BV + AVD group and 86.3 months (95% CI, 84.53 to 89.33) in the ABVD group. This analysis included 111 OS events (deaths): 44 deaths (7%) were reported in the BV + AVD group and 67 deaths (10%) in the ABVD group. Median OS was not reached for either group. The HR for OS was 0.61 (95% CI, 0.414 to 0.892, descriptive P = 0.010). In the stage III subgroup, the median OS was not reached for either treatment arm, and the HR for OS was 1.004 (95% CI, 0.540 to1.866) for BV + AVD patients, compared with ABVD patients. In the stage IV subgroup, the median OS was not reached for either treatment arm, and the HR for OS was 0.48 (95% CI, 0.291 to 0.784; descriptive P = 0.003) for BV + AVD patients, compared with that for ABVD patients.
Alive Without HL
In the ITT population, the 3-year rates of being alive without HL were 96% (546 of 567) in the BV + AVD group and 93% (503 of 540) in the ABVD group. The 5-year rates of being alive without HL were about 94% (450 of 480) in the BV + AVD group and 92% (408 of 443) in the ABVD group. No subgroup analyses by disease stage were reported for this efficacy end point.
PFS According to Investigator
As of the data cut-off date of June 1, 2021, the median follow-up was 73.2 months (95% CI, 72.48 to 74.05) in the BV + AVD group and 71.6 months (95% CI, 70.37 to 72.87) in the ABVD group. In the ITT population, the HR of PFS according to investigator was 0.678 (95% CI, 0.532 to 0.863; P = 0.002), favouring treatment with BV + AVD. There was a 7% absolute difference in the number of PFS events between the BV + AVD group (17%) and the ABVD group (24%). The median PFS according to investigator was not reached for patients with advanced-stage classical HL for either the BV + AVD or ABVD group. In subgroup analyses by disease stage, the HRs for PFS according to investigator were 0.603 (95% CI, 0.391 to 0.930; P = 0.021) for patients with stage III classical HL and 0.715 (95% CI, 0.534 to 0.959; P = 0.024) patients with stage IV classical HL.
Modified PFS According to IRF
As of the data cut-off date of April 20, 2017, the median mPFS was not reached in either the BV + AVD group or the ABVD group. In the ITT population, the HR for mPFS according to IRF was 0.770 (95% CI, 0.603 to 0.982; P = 0.035). There was a 4% absolute difference in number of mPFS events between the BV + AVD arm and the ABVD arm, favouring BV + AVD (18% versus 22%). In subgroup analyses by disease stage, the HRs for mPFS according to IRF were 0.923 (95% CI, 0.600 to 1.420; P = 0.716) for patients with stage III classical HL and 0.712 (95% CI, 0.530 to 0.957; P = 0.024) for patients with stage IV classical HL.
Harms Results
Harms results for the safety population are summarized in Table 2. Deaths and secondary malignancies were from the data cut-off date of June 1, 2021, while the remaining data were from the data cut-off of April 20, 2017.
In the safety population, the proportions of patients experiencing treatment-emergent adverse events (TEAEs) up to 30 days after the last front-line dose were similar between patients treated with BV + AVD (99%) and those treated with ABVD (98%). Higher percentages of patients in the BV + AVD group experienced treatment-emergent serious adverse events (TESAEs) up to 30 days after the last front-line dose, compared to the percentages of patients in the ABVD group (43% versus 27%, respectively). Deaths were reported in 6% of the patients in the BV + AVD arm and 10% of the patients in the ABVD arm. Treatment discontinuation due to AEs occurred in 13% of the patients in the BV + AVD arm and 16% of those in the ABVD arm. In terms of notable harms, 67% of the patients in the BV + AVD group and 43% in the ABVD group experienced at least 1 peripheral neuropathy event. About 3% of the patients in the BV + ABVD group and 5% of the patients in the ABVD group developed secondary malignancies. The proportion of patients who experienced neutropenia as TEAEs of grade 3 or higher was higher in the BV + ABVD group than in the ABVD group (54% versus 39%, respectively). Similarly, the proportions of patients who experienced febrile neutropenia as TEAEs of grade 3 or higher were also higher in the BV + ABVD group than in the ABVD group (19% versus 8%). Fewer patients in the BV + AVD arm experienced AEs of pulmonary-related toxicity than in the ABVD arm (13% versus 25%). The most common AE of pulmonary-related toxicity for either group was dyspnea (12% versus 24%).
Table 2: Summary of Key Results From Pivotal Studies and RCT Evidence
Outcome | ECHELON-1 | |
|---|---|---|
BV + AVD | ABVD | |
Efficacy end points (ITT population) | N = 664 | N = 670 |
Overall survival (data cut-off date: June 1, 2021) | ||
Number of patients contributing to the analysis, n (%) | 664 (100) | 670 (100) |
Number with events (%) | 39 (6) | 64 (10) |
Number censored (%) | 625 (94) | 606 (90) |
Median, months (95% CI) | NE (NE to NE) | NE (NE to NE) |
Hazard ratio (95% CI)a | 0.59 (0.396 to 0.879) | |
P value between treatment groups | 0.009 | |
Median follow-up time, months (95% CI)b | 73.3 (72.61 to 74.05) | 72.4 (71.10 to 73.63) |
Overall survival (data cut-off date: March 11, 2023) | ||
Number of patients contributing to the analysis, n (%) | 664 (100) | 670 (100) |
Number with events (%) | 44 (7) | 67 (10) |
Number censored (%) | 620 (93) | 603 (90) |
Median, months (95% CI) | NE (115.1 to NE) | NE (NE to NE) |
Hazard ratio (95% CI)a | 0.607 (0.414 to 0.892) | |
P value between treatment groups | 0.010 | |
Median follow-up time, months (95% CI)b | 89.7 (86.57 to 90.55) | 86.3 (84.53 to 89.33) |
Alive without HL, n (%) | ||
Number of patients alive at 3 years after randomization | 567 (85) | 540 (81) |
Patients who were alive without HL | 546 (96) | 503 (93) |
P value | 0.016 | |
Number of patients alive at 5 years after randomization | 480 (72) | 443 (66) |
Patients who were alive without HL | 452 (94) | 408 (92) |
P value | 0.194 | |
Progression-free survival according to investigator | ||
Number of patients contributing to the analysis, n (%) | 664 (100) | 670 (100) |
Number with events (%) | 112 (17) | 159 (24) |
Number censored (%) | 552 (83) | 511 (76) |
Median (95% CI) | NE (NE to NE) | NE (NE to NE) |
Hazard ratio (95% CI)a | 0.678 (0.532 to 0.863) | |
P value between treatment groups | 0.002 | |
Median follow-up time, months (95% CI)c | 73.2 (72.48 to 74.05) | 71.6 (70.37 to 72.87) |
Modified progression-free survival according to IRF | ||
Number of patients contributing to the analysis, n (%) | 664 (100) | 670 (100) |
Number with events (%) | 117 (18) | 146 (22) |
Number censored (%) | 547 (82) | 524 (78) |
Median, months (95% CI) | NE (48.2 to NE) | NE (NE to NE) |
Hazard ratio (95% CI)a | 0.770 (0.603 to 0.982) | |
P value between treatment groups | 0.035 | |
Median follow-up time, months (95% CI)d | 24.9 (24.64 to 25.03) | 24.9 (24.61 to 25.07) |
Harms end points (safety population) | N = 662 | N = 659 |
Patients with ≥ 1 TEAE up to 30 days after last front-line dose, n (%) | 653 (99) | 646 (98) |
Patients with ≥ 1 SAE up to 30 days after last front-line dose, n (%) | 284 (43) | 178 (27) |
Deaths, n (%) | 39 (6) | 64 (10) |
Patients who discontinued treatment due to AEs, n (%) | 88 (13) | 105 (16) |
Notable harms, n (%) | ||
Treatment-emergent peripheral neuropathy (grade 3 or higher) | 70 (10) | 11 (2) |
Secondary malignancy | 23 (3) | 32 (5) |
Treatment-emergent neutropeniae (grade 3 or higher) | 430 (65) | 317 (48) |
Treatment-emergent febrile neutropenia (grade 3 or higher) | 128 (19) | 52 (8) |
Pulmonary-related toxicityf | 89 (13) | 165 (25) |
Dyspnea | 82 (12) | 155 (24) |
Lung infiltration | 6 (< 1) | 0 |
Pneumonitis | 6 (< 1) | 18 (3) |
Hypoxia | 4 (< 1) | 10 (2) |
Interstitial lung disease | 1 (< 1) | 6 (< 1) |
Pulmonary toxicity | 0 | 16 (2) |
ABVD = doxorubicin-bleomycin-vinblastine-dacarbazine; AVD = doxorubicin-vinblastine-dacarbazine; BV = brentuximab vedotin; CI = confidence interval; HL = Hodgkin lymphoma; IRF = intendent review facility; ITT = intention to treat; OS = overall survival; mPFS = modified progression-free survival; NE = not estimable; PFS = progression-free survival; RCT = randomized controlled trial.
Note: Multiplicities were only adjusted for OS and mPFS, and P values for other efficacy end points were provided for descriptive purposes only.
aThe HR and 95% CI were based on a stratified Cox’s proportional hazard regression model with stratification factors region and number of IPFP risk factors at baseline with treatment as the explanatory variable in the model. A HR of less than 1 favours BV + AVD group.
bThe median OS follow-up was calculated from the Kaplan-Meier method switching the OS event and censored status, i.e., OS event as censored and censored as OS event.
cThe median PFS follow-up was calculated from the Kaplan-Meier method switching the PFS according to investigator event and censored status, i.e., PFS according to investigator event as censored and censored as PFS according to investigator event.
dThe median mPFS follow-up was calculated from the Kaplan-Meier method switching the mPFS event and censored status, i.e., mPFS event as censored and censored as mPFS event.
ePreferred terms of neutropenia and decreased neutrophil count are counted as neutropenia.
fPulmonary-related toxicity include preferred terms of “dyspnea” and “hypoxia,” and all preferred terms in an interstitial lung disease standardized Medical Dictionary for Regulatory Activities query, and preferred terms with the high-level term “respiratory and pulmonary function diagnostic procedures.”
Sources: ECHELON-1 original Clinical Study Report (data cut-off: April 20, 2017),13 ECHELON-1 Clinical Study Report Addendum 1 (data cut-off: June 01, 2021),14 Drug Reimbursement Review sponsor submission,16, and the sponsor’s Summary of Clinical Evidence.1
Critical Appraisal
Generally, no serious concerns were identified in the conduct of the ECHELON-1 trial. However, the validity of the primary outcome (mPFS) is a key consideration in evaluating the evidence for BV + AVD. The mPFS was adopted in the ECHELON-1 trial to capture all events that reflect a failure of front-line chemotherapy by counting a response that was less than complete at the end of the front-line therapy as an event. The ECHELON-1 trial defined a response of less than complete as “receipt of anticancer therapy or radiotherapy for HL after completion of front-line therapy for patients who were confirmed non-complete responders.” However, the clinical experts consulted by the review team noted that this definition is not consistent with practice in defining disease progression or first-line treatment failure in advanced HL, and receipt of radiotherapy does not necessarily indicate disease progression in clinical practice. Despite the end-of-treatment PET scans conducted by the IRF, there is a concern that the results for mPFS could be biased given that the administration of new anticancer therapy was at the discretion of the treating physician. The clinical experts consulted by the review team noted that OS and PFS are more clinically relevant to assessing patient benefits from treatment, and no evidence was included in the submission to the review team that empirically validated mPFS as an outcome measure or established a correlation with OS. High percentages in loss to follow-up and withdrawal by patients were noted in both OS and PFS analyses. Although the percentages of loss to follow-up and withdrawal by patients were balanced between treatment arms, reasons for loss to follow-up and withdrawal could be differential between groups, which could lead to biased estimates of treatment effects. Moreover, no sensitivity analyses assessing the potential impact of the loss to follow-up and withdrawal on OS and PFS results were available. Subgroup analyses by HL stage signal that there might be a difference in treatment effects between patients with stage III and those with stage IV classical HL for mPFS and OS. However, the review team’s ability to make a definitive conclusion as to whether the difference between the disease stage subgroups is true was limited by several concerns, such as the possibility that the balance of known and unknown factors between treatment groups achieved by randomization was not preserved in stage III or stage IV subgroups. In addition, the trial was not specifically designed to test statistical inferences between BV + AVD and ABVD in stage III and stage IV subgroups.
All participants in the ECHELON-1 trial were required to be aged 18 years or older and diagnosed with classical HL. The ECHELON-1 trial therefore did not reflect results for pediatric patients or patients with nodular lymphocyte-predominant HL. The eligibility criteria of the ECHELON-1 trial in general were aligned with selection criteria in the Canadian settings when identifying suitable candidates for BV + AVD, according to the clinical experts consulted by the review team. However, the clinical experts noted that, in clinical practice, a small percentage of patients who were excluded from the ECHELON-1 trial, such as patients with HIV, might be eligible to receive BV + AVD, if the disease is well managed, and to patients with a borderline left ventricle ejection fraction after consultation with a cardiologist. The clinical experts also noted that patients with ECOG PS scores higher than 2 could be considered for treatment with BV + AVD on a case-by-case basis. The clinical experts noted that the doses of BV + AVD and ABVD used in the ECHELON-1 trial generally reflected the standard dose schedules used for adults in Canada. The clinical experts also confirmed that the direct comparator, ABVD up to 6 cycles (not adapted based on PET response), is a relevant therapy used in current standard of care, although, because it is not the only standard-of-care front-line therapy used in Canada, other relevant comparators are available. In addition, the clinical experts noted that the percentages of patients who received a transplant as subsequent treatment were lower in either group than they would have expected to see in clinical practice. According to the clinical experts consulted by the review team, the characteristics of the study population were generally reflective of patients who would be eligible for BV + AVD in Canadian practice. However, the experts noted that the percentage of patients with stage IV HL in the trial population and the percentage of participants who are white were higher than what would be seen in clinical practice.
Long-Term Extension Studies
No long-term extension studies were submitted by the sponsor.
Indirect Comparisons
No indirect treatment comparisons (ITCs) were submitted by the sponsor. The sponsor provided a feasibility assessment that determined it would be infeasible to conduct ITCs of BV + AVD versus other front-line therapies examined in clinical studies for advanced HL.
Studies Addressing Gaps in the Pivotal and RCT Evidence
To address gaps in the pivotal randomized controlled trial (RCT) evidence related to the use of BV in pediatric patients with advanced HL, the review team reviewed evidence from an additional phase III RCT.
Description of Studies
The AHOD1331 trial (N = 587),11 published in The New England Journal of Medicine, is a phase III, multicentre, open-label, randomized active-controlled trial comparing BV + AVEPC with doxorubicin-bleomycin-vincristine-etoposide-prednisone- cyclophosphamide (ABVE-PC) in previously untreated patients aged 2 to 21 years with high-risk classical HL, defined as Ann Arbor stage IIB with bulk tumour, stage IIIB, stage IVA and stage IVB patients. The primary objective of the AHOD1331 trial was to determine the efficacy of BV + AVEPC relative to ABVE-PC as measured by EFS. Harms and OS were also reported. The AHOD1331 trial is ongoing. The final analysis of EFS was based on the database lock date of December 31, 2021.
In the AHOD1331 trial, the median age of participants was 15.6 years (range = 3.4 to 21.99); most (84.7%, 497 of 587) were aged between 12 and 22 years. Of the 587 patients enrolled, 47% (276) were female and 57.6% (338) were non-Hispanic white. The proportions of patients by disease stage were 20.6% (121) for stage IIB with bulk tumour, 19.3% (113) for stage IIIB, 28.4% (167) for stage IVA, and 31.7% (186) for stage IVB. Most of the patients had nodular-sclerosis classical HL (76.5%, 449 of 587).
Efficacy Results
The median follow-up time was 42.1 months (range = 0.1 to 80.9). In terms of the 3-year OS in the ITT population, the proportions of patients who were censored were 99.3% (95% CI, 97.3 to 99.8) in the BV + AVEPC group and 98.5% (95% CI, 96.0 to 99.4) in the ABVE-PC group. The HR for 3-year OS was not provided.
In terms of the 3-year EFS in the ITT population, the proportions of patients who were censored were 92.1% (95% CI, 88.4 to 94.7) in the BV + AVEPC group and 82.5% (95% CI, 77.4 to 86.5) in the ABVE-PC group. The HR for 3-year EFS was 0.41 (95% CI, 0.25 to 0.67; P < 0.001), favouring the BV + AVEPC arm.
Harms Results
The incidence of any AEs of grade 3 or higher was 73.5% in patients treated with BV plus AVEPC and 68.2% in patients treated with ABVE-PC. Peripheral neuropathy of grade 3 or higher occurred in 6.7% of the patients in the BV plus AVEPC arm and 5.5% % of the patients in the ABVE-PC arm. Febrile neutropenia occurred in 30.9% and 32.5% of patients in the BV plus AVEPC and ABVE-PC arms, respectively. None of the patients in the BV plus AVEPC group experienced pneumonitis compared to 1 patient in the ABVE-PC group.
Critical Appraisal
Although details about the randomization process and allocation concealment were not reported in the research protocol or the main article, the risk of bias in the AHOD1331 trial is anticipated to be low given that baseline characteristics between the treatment arms were generally similar for clinically important factors. The AHOD1331 trial was open-label but had blinded outcome assessors, the definition of EFS was aligned with accepted definitions from regulators, and treatment response was assessed via centralized review, helping reduce the risk of detection bias related to the open-label design. Although patients were aware of the treatment allocation, which may result in performance bias, this risk is considered low as the 3-year PFS in the ABVE-PC group (82.5%) and the types of AEs were generally in line with what the consulted clinical expert who specializes in pediatrics expected. Those patients who remained PET2-positive received response-adapted involved-site radiation therapy guided by blinded central assessment of PET scans. This could bias the EFS results if the radiation therapy could improve response, reduce the likelihood of relapse, and/or increase the risk of secondary malignancy. However, the risk of this potential bias was mitigated by the requirement that radiation therapy could not be administered until directed by the blinded assessment. Also, the percentages of patients who received involved-site radiation therapy were similar between the BV + AVEPC and ABVE-PC groups (53.4% versus 56.8%, respectively). Concomitant anticancer medications were not allowed. Antibiotics and supportive medications (e.g., antiemetics) were permitted as needed. Patients also received granulocyte colony-stimulating factor (G-CSF) support. None of the permitted medications would likely influence the results for either treatment group. However, after a progression event, the treating physician could treat the patient at their discretion, which may affect the longer-term OS results.
The AHOD1331 trial was appraised in this section to address an important gap with respect to the unmet needs of using BV + chemotherapy in pediatric patients with classical HL. However, several notable issues need to be considered when generalizing results from the AHOD1331 trial. First, although the chemotherapy backbone used in the AHOD1331 trial (AVEPC) is a preferred backbone for pediatric patients, according to POGO and the clinical experts consulted by the review team, it is different from the backbone used in adults (ABVD). Regarding the regulatory status of the pediatric regimen, the review team confirmed that BV is not approved for use in combination with the pediatric regimen, and the sponsor confirmed it is not planning to request Health Canada approval for BV + AVEPC. Second, the clinical experts consulted by the review team noted the definition of high-risk or advanced-stage HL in pediatric patients varies. While the AHOD1331 trial adopted the definition of advanced-stage HL in pediatric patients as stage II with bulk tumour, stage IIIB, stage IVA, and stage IVB, some medical centres may define any stage III or IV as advanced-stage disease in pediatric patients. Finally, the AHOD1331 trial involved both nonadults and young adults (up to 22 years old), while the pivotal ECHELON-1 trial enrolled patients aged 18 years and older. This created an overlap in patient age between the pivotal ECHELON-1 trial and the AHOD1331 trial. The clinical experts consulted by the review team noted that the chemotherapy backbone used in the AHOD1331 trial (AVEPC) would not typically be used in patients aged 18 years or older in Canada, and the chemotherapy backbone ABVD investigated in the pivotal ECHELON-1 trial may be used in adolescents with HL aged close to 18 years.
Conclusions
Overall, evidence from the phase III, open-label, randomized ECHELON-1 trial suggests that BV + AVD is an effective front-line treatment for previously untreated adult patients diagnosed with advanced-stage classical HL. The clinically relevant efficacy end points examined in the report (OS, PFS, and mPFS) were consistently in favour of BV + AVD compared to ABVD in the ITT population. However, the clinical significance of the magnitude of the treatment differences is uncertain, and concerns remain about the validity of the primary outcome (mPFS) as well as the high percentages in loss to follow-up and withdrawal by patients in both OS and PFS analyses. In addition, although subgroup analyses of the OS and mPFS results signalled that BV + AVD may be more effective in patients with stage IV classical HL than those with stage III HL, conclusions regarding subgroup differences between stage III versus stage IV patients are uncertain because the study was not designed to detect differences between these subgroups and patients were not stratified according to disease stage at randomization. The safety profile of BV + AVD is consistent with the known AEs for the individual components of the regimen, but more patients treated with BV + AVD experienced serious adverse events (SAEs) compared with those in the ABVD group. An evidence gap remains with respect to the clinical efficacy and safety of BV + AVD in the pediatric population as all participants in the pivotal ECHELON-1 trial were required to be aged 18 years or older. To address unmet needs in the pediatric patient population, the AHOD1331 trial was examined. Efficacy results from the AHOD1331 trial indicated that BV + AVEPC provides a clinically meaningful benefit in EFS compared to ABVE-PC in patients aged 2 to 21 years with high-risk classical HL defined as Ann Arbor stage IIB with bulk tumour, stage IIIB, stage IVA, and stage IVB HL.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of BV (50 mg per vial, lyophilized powder for reconstitution, IV infusion) for the treatment of previously untreated patients with advanced-stage HL, in combination with AVD.
This is a submission from a tumour group. However, the review is based on a previous sponsor-initiated submission (Project Number PC0311 to 000) that was withdrawn before CADTH adopted the Grading of Recommendations Assessment, Development and Evaluation framework for each reimbursement review. As such, the framework was not used for the current review.
Disease Background
Contents within this section were informed by materials submitted by the sponsor and clinical expert input. The following summary was validated by the review team.
Hodgkin lymphoma is a B-cell lymphoid malignancy that originates in the lymphocytes.3 Classical HL accounts for 95% of all HL cases and is characterized by the presence of Hodgkin Reed-Sternberg cells, which express the surface antigen CD30.17,18 While the median age of diagnosis for HL is 39 years, the incidence of HL is age-related and bimodal, predominantly affecting people in their 20s, early 30s, and those older than 55 years.19,20 A common early sign of HL is painless enlargement of 1 or more lymph nodes.9 Other signs and symptoms of HL include fatigue, shortness of breath, itchiness on the trunk of the body, unusual back or abdominal pain, and abdominal swelling.21 A diagnosis of HL is done by a lymph node biopsy.8,9,22
In 2022, an estimated 1,050 new cases of HL occurred in Canada overall, for an incidence rate of 2.6 cases per 100,000.4 Based on the 2018 Canadian Cancer Statistics, approximately 23.3% of Canadian patients presenting with HL have stage III disease and 22.7% have stage IV disease.5 Childhood HL represents 6% of all cancers and has an incidence rate of 12 cases per million (1.2 cases per 100,000) per year among children aged 0 to 14 years.6 In 2019, a total of 25 children in Canada in this age group were diagnosed with HL.7
The clinical experts consulted by the review team noted that advanced-stage HL in adult patients refers to Ann Arbor stage III and IV HL (Appendix 1). The clinical experts noted that pediatric patients with HL are usually classified into low-, intermediate-, and high-risk groups in clinical practice, and the high-risk group is generally considered equivalent to the advanced-stage classical HL group in adults. The clinical experts reported that pediatric patients with high-risk or advanced-stage HL are generally defined as stage II with bulk tumour, stage III with B symptoms (stage IIIB), and stage IV with or without B symptoms (stage IVA and stage IVB). However, the clinical experts consulted by the review team also indicated that the definition of advanced-stage HL in pediatric patients is evolving and may vary by centre, with some centres defining any stage III or IV as advanced-stage disease in pediatric patients.
HL is considered a curable disease.23 In 2021, the age-standardized mortality rate was 0.2 per 100,000 with 5- and 10-year net survival rates of 85% and 81%, respectfully.24
Standards of Therapy
Contents within this section were informed by materials submitted by the sponsor and clinical expert input. The following summary was validated by the review team.
The goal of therapy in patients with advanced-stage (stage III and IV) HL is curative.8,9 The clinical experts consulted by the review team noted that the treatments for patients in the advanced-stage HL group (stage III and IV) are similar. The clinical experts also emphasized that treatments may differ between classical HL and nodular lymphocyte-predominant HL, the latter of which does not have classical Reed-Sternberg cells and may behave more like indolent non-HL.
The clinical experts consulted by the review team noted that current treatment regimens for adult patients in Canada with advanced-stage HL mainly include: an approach based on ABVD for 6 cycles with PET2-directed de-escalation for PET2-negative patients to AVD and, in some jurisdictions, PET2-directed escalation to escalated BEACOPP in PET2-positive patients; an approach based on upfront escalated BEACOPP with PET2-driven treatment adaptation; and BV + AVD for patients with stage IV HL. For pediatric patients, the clinical experts consulted by the review team noted that most clinical centres in Canada have adopted ABVE-PC for 5 cycles with radiation therapy determined by PET2 response, while relatively few centres use vincristine-etoposide-prednisone-doxorubicin–cyclophosphamide-vincristine-prednisone-dacarbazine.
The clinical experts consulted by the review team noted that current drugs for HL that are essentially cytotoxic and/or DNA-damaging do not target the underlying pathogenetic mechanisms, except for programmed cell-death protein 1 inhibitors, which target the underlying immune escape essential for classical HL proliferation. The clinical experts noted that no drugs are currently accessed through special programs to treat advanced-stage HL in the front-line setting, and radiotherapy is usually reserved for earlier-stage (I and II) disease or may be given at the end of therapy for patients with advanced-stage HL.
Drug Under Review
Brentuximab vedotin is an antibody-drug conjugate directed against CD30.2 Currently, BV has an indication approved by Health Canada2 for the treatment of previously untreated patients with stage IV HL, in combination with AVD; BV was reviewed and recommended for reimbursement for that indication in 2020.25
The recommended dose of BV for previously untreated advanced-stage HL is 1.2 mg/kg up to a maximum of 120 mg in combination with AVD administered every 2 weeks for a maximum of 12 doses or until disease progression or unacceptable toxicity occurs. BV is administered as an IV infusion over 30 minutes.
Key characteristics of BV are summarized in Table 3, along with other treatments available for advanced-stage HL.
Table 3: Key Characteristics of BV and Bleomycin
Characteristic | BV | Bleomycin |
|---|---|---|
Mechanism of action | The biological activity of BV results from a multistep process. Binding of the ADC to CD30 on cell surfaces initiates internalization of the ADC-CD30 complex, which then traffics to the lysosomal compartments. Within cells, MMAE is released via proteolytic cleavage. Binding of MMAE to tubulin disrupts the microtubule network within cells, induces cell-cycle arrest, and results in apoptotic death of CD30-expressing tumour cells. | Although the exact mechanism of action of bleomycin is unknown, available evidence indicates that the main mode of action is inhibition of DNA synthesis, with some evidence of inhibition of RNA and protein synthesis. |
Approved indicationa | For the treatment of previously untreated patients with stage IV HL, in combination with AVD. | Bleomycin for injection is indicated in Hodgkin and non-Hodgkin lymphoma. |
Route of administration | IV infusion | IV infusion |
Recommended dose | 1.2 mg/kg via IV infusion up to a maximum of 120 mg in combination with AVD | 10 units/m2 IV infusion on days 1 and 15 |
Serious adverse effects or safety issues | Serious warnings for JC virus infection resulting in PML and death, Stevens-Johnson syndrome and toxic epidermal necrolysis, serious and opportunistic infections, acute pancreatitis, gastrointestinal complications, and pulmonary toxicity. Serious warnings in combination therapy with AVD for febrile neutropenia. BV is contraindicated for patients who are hypersensitive to this drug or any ingredient in the formulation, for patients receiving concomitant bleomycin due to pulmonary toxicity, and for patients who have or have had PML. | Extreme caution in patients with significant impairment of renal function or compromised pulmonary function. Bleomycin for injection is contraindicated in patients who have demonstrated hypersensitivity to the drug. |
ADC = antibody-drug conjugate; AVD = doxorubicin-vinblastine-dacarbazine; BV = brentuximab vedotin; MMAE = monomethyl auristatin E; NA = not applicable; PML = progressive multifocal leukoencephalopathy.
aHealth Canada–approved indication.
Sources: Sponsor’s Clinical Summary Evidence,1 BV product monograph,2, and bleomycin for injection product monograph.26
Stakeholder Perspectives
Patient-Group Input
This section was prepared by the review team based on the input provided by patient groups. The full original patient input(s) received by the review team are included in the stakeholder section at the end of this report.
Patient Input
One patient group, Lymphoma Canada, provided input for BV for the treatment of previously untreated patients with advanced-stage HL, in combination with AVD. Patient input was gathered from an online anonymous patient survey from March 14 to May 2, 2023. A total of 26 responses were gathered, and 3 of these patients reported receiving BV + AVD treatment.
Patients were asked questions regarding the physical and psychosocial symptoms experience at the time of diagnosis, current quality of life, and how these symptoms affected their daily activities. At the time of their lymphoma diagnosis, most of the patients reported fatigue (79%) as the symptom with the greatest impact (5 out of 5), followed by enlarged lymph nodes (58%), shortness of breath (63%), and weight loss (47%). In addition, 74% of patients reported experiencing anxiety or worry, 68% stressing about their diagnosis, 63% difficulty in sleeping, and 58% fearing progression of their lymphoma. Regarding physical symptoms that currently affect their quality of life, out of 7 responses, fatigue (29%) and headaches (14%) were identified as the most significant factors imposing a negative impact on quality of life. The most consequential psychosocial factors affected patients’ current quality of life were the stress of having cancer (71%), fear of progression (71%), anxiety or worry (71%), difficulty sleeping (43%), problems concentrating (43%), and inability to attend work or school (43%).
Among the surveyed patients, 3 reported receiving ABVD in the front-line setting, 2 were treated with other forms of chemotherapy, and 1 was treated with cyclophosphamide-doxorubicin-prednisone-rituximab-vincristine. While evaluating the importance of outcomes of new treatments, patients from the survey emphasized the need for a novel lymphoma therapy to control disease symptoms, lengthen disease remission, extend life spans, and improve quality of life.
While describing the experience with the treatment under review, 1 of the 3 patients who had received BV + AVD indicated they had been in remission for between 6 months and a year, another had been in remission for longer than a year, and the third patient was in post-treatment (and unsure about their remission status). Side effects from the BV + AVD treatment reported by patients were fatigue (n = 3), neutropenia (n = 2), constipation (n = 2), joint or muscle pain (n = 2), low platelet count (n = 1), low blood pressure (n = 1), and decreased appetite (n = 1). Two patients reported experiencing financial setbacks — 1 due to absence from work and 1 due to cost of other medications. One of these patients mentioned having a poor experience with BV, and the other 2 rated their experience as very good.
Clinician Input
Input From Clinical Experts Consulted by the Review Ream
All our review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 3 clinical specialists with expertise in the diagnosis and management of previously untreated patients with advanced-stage HL.
Unmet Needs
The clinical experts consulted by the review team noted that advanced-stage HL in adult patients refers to Ann Arbor stage III and IV HL and agreed that the treatment goals as well as unmet needs are the same between adult patients with stage III HL and those with stage IV HL. The clinical experts noted that, in clinical practice, pediatric patients with HL are usually classified into low-, intermediate-, and high-risk groups, and the high-risk group is generally considered equivalent to advanced-stage classical HL in adults. The clinical experts reported that pediatric patients with high-risk or advanced-stage HL are generally defined as having stage II with bulk tumour, stage III with B symptoms (stage IIIB), and stage IV with or without B symptoms (stage IVA and stage IVB), and these patients are treated the same in clinical practice — as advanced-stage HL. However, the clinical experts consulted by the review team indicated that the definition of advanced-stage HL in pediatric patients is evolving and may vary by centre, with some centres defining any stage III or IV as advanced-stage disease in pediatric patients.
The clinical experts noted that cure of the disease is the main goal of treatment. Moreover, the cure should be achieved with an acceptable level of short- and long-term toxicity. In younger patients with advanced-stage HL, the goal of therapy is also to restore normal life expectancy and productivity while minimizing toxicity by increasing the cure rate, lowering the risk of relapse, and reducing the need for more cytotoxic and/or DNA-damaging therapies.
According to the clinical experts, an improved cure is an important unmet need for patients with advanced-stage HL to reduce treatment failure, prevent disease progression or relapse, and avoid late side effects (e.g., secondary malignancies, cardiac and pulmonary late effects) and further therapies that are toxic (e.g., ASCT), particularly for younger patients diagnosed with advanced-stage HL and older patients who have poor tolerance to treatment. In addition, 1 clinical expert noted that a combination therapy without bleomycin (i.e., part of the standard AVD) is needed for older patients with advanced-stage HL due the potentially fatal lung toxicity of bleomycin.
Place in Therapy
The clinical experts noted that BV + AVD is considered a front-line therapy and should not be used for patients who are intolerant to other therapies. The purpose of using BV + AVD is for curing the disease, not for symptomatic control of disease. The clinical experts noted that, at the time of the review, BV + AVD had been approved only for the treatment of previously untreated patients with stage IV HL, and the use of BV + AVD in patients with stage III classical HL would cause a shift in the current treatment paradigm for those patients. The clinical experts noted that BV + AVD would not be added to other treatments, except that radiotherapy may be given at the end of therapy. The clinical experts indicated that, in pediatric patients, BV would be used in combination with a different chemotherapy backbone that has been investigated in a phase III randomized controlled trial11 (i.e., BV + AVEPC) instead of AVD, as trials of BV + AVD in pediatric patients had not been completed at the time of this review.12
Patient Population
The clinical experts noted that any previously untreated adult patients with stage III or IV classical HL who meet the eligibility criteria of the ECHELON-1 trial are best suited for the use of BV + AVD. The clinical experts indicated that patients with nodular lymphocyte-predominant HL are not suitable for BV + AVD. The clinical experts emphasized that BV + AVD should be used with caution in older patients (i.e., those aged ≥ 60 years) due to potential toxicity. The clinical experts also noted that factors such as older age, stage IV, International Prognostic Score (IPS), and ECOG PS are accepted risk factors and may be associated with adverse outcomes. The clinical experts indicated that liver function tests, ECOG PS, and blood counts are important when considering giving BV + AVD. The clinical experts also noted that any pediatric patient with advanced-stage classical HL could also be eligible for BV in combination with chemotherapy, and eligibility for therapy should be determined by the treating physician.
Assessing the Response Treatment
The clinical experts noted that PET scans, typically PET2 and a PET scan after all 6 cycles (end of treatment), is used for response assessment. The desired outcome is a complete metabolic response on the end-of-treatment PET scan. In follow-ups of patients who are found to have achieved a CR, visits take place every 3 to 4 months for 2 years, then typically every 6 months for 3 more years, and there is no serial imaging to monitor disease. In the absence of a CR, patients with a partial response may undergo radiation therapy, and patients with refractory disease would undergo further investigations (e.g., biopsy) and treatment with a second-line regimen.
Whether patients who receive BV + AVD and remain PET-positive on PET2 should be escalated to a different treatment remained an unresolved question for 1 clinical expert, while a second expert suggested continuing BV + AVD. The clinical experts pointed out that, although PET2 positivity is important for most regimens, its role in the BV + AVD regimen is not yet clearly established.
The clinical expert specializing in pediatrics noted that outcomes used in clinical practice to assess treatment response are generally aligned with outcomes typically used in adult therapeutics. All 3 clinical experts noted that OS is the most clinically meaningful outcome to assess the efficacy of BV in combination with chemotherapy in patients with advanced-stage HL, and PFS is an important outcome. In pediatric patients, EFS is also important. However, the importance of mPFS remains controversial. The clinical experts noted that end points such as duration of response and duration of CR are not typically used in a disease such as HL.
Discontinuing Treatment
The clinical experts agreed that, overall, discontinuation of BV + AVD is uncommon because toxicity or refractory HL is not common. The clinical experts identified several situations in which BV + AVD can be discontinued: if treatment is complete; there is clear progression of disease (although a Deauville 4 or 5 PET2 without new disease would not warrant discontinuing therapy); or there is an AE such as an allergic reaction to BV, severe neuropathy, pneumonitis, elevated liver enzymes, or neutropenia. For elevated liver enzymes and neutropenia, dose delays or dose adjustments are required before discontinuation.
Prescribing Considerations
The clinical experts noted that diagnosis of the disease in adult patients must be made by an experienced pathologist. Selection of patients for BV + AVD should be made by a hemato-oncologist experienced in treating HL. However, the treatment can be delivered in specialty clinics of nonacademic centres.
The clinical expert specializing in pediatrics noted that all pediatric oncology patients are cared for by pediatric oncology teams at tertiary care centres. Some aspects of care may be provided at satellite centres after diagnostic and management decisions are made.
Clinician Group Input
This section was prepared by the review team based on the input provided by clinician groups. The full original clinician group inputs received by the review team are included in the stakeholder section at the end of this report.
Clinician group input on the review of BV was received from the OH-CCO Hematology Cancer Drug Advisory Committee and POGO. Six clinicians provided input on behalf of OH-CCO. POGO is a collaboration of Ontario’s 5 specialized childhood cancer centres. The input collected from POGO was prepared in a consultative manner, with 1 clinician discussing the indication with other members of the submission panel and seeking input from POGO’s Therapeutic and Technology Advisory Committee, and the final submission was based on the results of this process.
The OH-CCO input highlighted the need to improve outcomes with first-line therapy to avoid the need for second-line therapy. The group noted that patients with stage III and IV disease would be best suited for current treatment. The group indicated that typical lymphoma response measures, including PET scans, are used in clinical practice to assess patients’ response to treatment. They indicated they would discontinue treatment with BV + AVD in cases of significant toxicities or disease progression.
The POGO input noted that, while a variety of chemotherapy and radiation approaches are available as options for the standard of care, these vary by region and between pediatric- and adult-focused practitioners in Canada. POGO reported that, historically, the ABVD chemotherapy backbone used with BV in adult patients has not been used by pediatric oncologists to treat pediatric patients due to concerns about higher anthracycline (doxorubicin) and bleomycin exposure, and known dose-dependent cardiac and pulmonary toxicities. POGO noted that BV has been studied and used in combination with another chemotherapy regimen (AVEPC) in patients aged 2 to 21 years with previously untreated high-risk HL.11 POGO indicated that this alternative chemotherapy backbone is more commonly used in the pediatric setting, and BV + AVEPC has become standard care for high-risk pediatric patients in Ontario. Regarding treatment goals for HL in pediatric patients with, POGO emphasized the need to avoid disease recurrence to minimize potential late effects from subsequent chemotherapies and ASCT received at relapse, and the associated impact on HRQoL. While describing the outcomes used to determine whether a pediatric patient is responding to treatment for HL, the POGO group emphasized the importance of OS and EFS, considering the higher chance of experiencing late effects of therapy after treatment among the younger patient population. Like OH-COO input, POGO suggested treatment be discontinued at disease progression.
Drug Program Input
The drug programs provide input on each drug being reviewed through the reimbursement review processes by identifying issues that may affect their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by the review team are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
The ECHELON-1 trial included ABVD, which is the current standard front-line treatment for HL, as comparator. An alternative regimen for young and healthy patients for whom the infertility implications are acceptable is BEACOPP, which can be given in fixed or escalated dosing. PET-CT scans guide treatment (i.e., number of cycles, change in therapy between ABVD and BEACOPP). In some provinces, CVPP can be given to patients with contraindications to anthracyclines and/or bleomycin. The ECHELON-1 trial compared BV + AVD to ABVD. The PAG is also seeking comparison of BV with PET-adapted BEACOPP and ABVD. | For adults patients in Canada, escalated BEACOPP and PET-adapted BEACOPP are only used in a few centres, and CVPP is not commonly used. For pediatric patients, relevant comparators to BV in combination with chemotherapy include ABVD, ABVE-PC, and OEPA-COPDAC. |
Considerations for initiation of therapy | |
Are there disease-specific features or prognostic features that would influence choice of therapy between PET-guided BEACOPP or ABVD and BV + AVD? | The toxicity of BEACOPP and the improvement in OS with BV + AVD make BV + AVD a preferred treatment regimen in adult patients. Currently in clinical practice, the clinical experts favour BV + AVD over PET-guided ABVD or BEACOPP in adult patients with stage IV HL. The clinical experts also noted that, if BV + AVD is approved for adult patients with stage III HL, BV + AVD will be preferred in this patient population as well. |
The indication requested for review is advanced-stage HL. Is the advanced-stage definition limited to stage III and IV? What staging system should be used (Lugano or Ann Arbor)? | Advanced-stage HL in adults refers to stage III and IV HL according to the Ann Arbor staging system. The clinical expert specialized in pediatric oncology noted that, in current clinical practice, pediatric patients with HL are usually classified into low, intermediate, and high-risk groups, and the high-risk group is generally considered equivalent to the advanced-stage classical HL in adults. The Children’s Oncology Group trial (AHOD133111) defined pediatric patients with high-risk or advanced-stage HL as stage II with bulk tumour, stage III with B symptoms (stage IIIB), and stage IV with or without B symptoms (stage IVA and stage IVB) as determined by Ann Arbor staging. However, the definition of advanced-stage HL in pediatric patients is evolving and may vary across centres, with some centres defining any stage III or IV as advanced-stage disease in pediatric patients.12 Lugano criteria allow an investigator not only to stage the lymphoma but also to assess response. In the Lugano criteria, staging is performed using the Ann Arbor system. |
According to the current provisional funding algorithm, patients who relapse would be eligible for BV re-treatment if relapse occurs more than 12 months after completion of prior BV therapy, with at least 6 months of response. Is pERC in agreement with this guidance, which was informed by the pERC recommendation for BV + AVD in stage IV HL? | In Ontario and Quebec, BV cannot be given unless a patient who relapsed has had a transplant, in which case BV can be used as maintenance or if the patient has relapsed after the transplant. |
Should BV + AVD be available to patients:
| BV + AVD should be available to patients:
CNS involvement is rare in patients with advanced HL. PML is a rare condition in these patients and fatal. The experts indicated they would not treat patients who had PML and HL for their lymphoma with BV + AVD. |
Should BV + AVD be available to patients aged less than 18 years? | BV in combination with chemotherapy should be available to patients aged less than 18 years. However, the ABVD chemotherapy backbone used with BV in adult patients is currently not used for pediatric patients because the ECHELON-1 trial excluded pediatric patients and there are toxicity concerns. In current practice, BV would be given in combination with a pediatric chemotherapy backbone such as AVPEC, as used in the AHOD1331 trial. One expert suggested that clinicians could consider pediatric patients who will become eligible for transfer to adult care during therapy (for example, turning 18 within 6 months of diagnosis) as having the option to receive BV + AVD instead of BV + AVEPC to address the transition issue. However, as a pediatric oncology practitioner, the expert would prefer to use BV + AVEPC over BV + AVD based on the evidence available at the time of this review. |
Considerations for discontinuation of therapy | |
PAG requested that pERC consider consistency with discontinuation criteria associated with other drugs reviewed by the review team in the same therapeutic space. Treatment with BV + AVD should continue until disease progression, unacceptable toxicity or until a maximum of 6 cycles, whichever comes first. | The clinical experts agreed with the current discontinuation criteria. |
Considerations for prescribing of therapy | |
For patients unable to receive doxorubicin, can etoposide be substituted (i.e., BV with etoposide, bleomycin, vinblastine, or dacarbazine)? | The clinical experts consulted by the review team did not agree with the substitution. |
Consider alignment with prescribing criteria for BV + AVD in stage IV disease. | To inform pERC deliberations. |
Generalizability | |
Should patients who recently initiated ABVD or BEACOPP be eligible to switch to BV + AVD? The PAG noted that the previous review for BV + AVD for stage IV disease the CGP indicated it would be reasonable to switch patients initiated on ABVD to BV + AVD on a time-limited basis. The CPG note that patients initiated on BEACOPP should not be offered BV + AVD on a time-limited basis. | The clinical experts who treat adult patients indicated it is good to have the option to switch on a time-limited basis. However, the clinical expert who treats pediatric patients noted that such situation would not come up when treating pediatric patients as the pediatric setting is different from the adult setting. |
Funding algorithm | |
Existing algorithm to be updated to include use of BV + AVD for stage III disease if recommended for reimbursement | To inform pERC deliberations. |
Care provision | |
Primary prophylaxis with G-CSF is typically prescribed with BV + AVD and is associated with additional cost. | To inform pERC deliberations. |
Is it appropriate for patients initiated on ABVD who experience treatment-related adverse effects to be switched to BV + AVD to complete a maximum of 6 cycles? | The clinical experts consulted by the review team did not consider the switch appropriate. |
Can BV be combined with any other regimens other than AVD (i.e., substituting etoposide in patients unable to receive doxorubicin)? | The clinical expert who specializes in pediatric oncology noted that BV can be combined with other regimens other than AVD in the pediatric setting (i.e., pediatric chemotherapy backbones). |
ABVD = doxorubicin-bleomycin-vinblastine-dacarbazine; ABVE-PC = adriamycin-bleomycin-vincristine-etoposide-prednisone-cyclophosphamide; ADV = doxorubicin-vinblastine-dacarbazine; AVPEC = doxorubicin-vincristine-etoposide-prednisone-cyclophosphamide; BEACOPP = bleomycin-etoposide-doxorubicin-cyclophosphamide, vincristine, procarbazine, and prednisone; BV = brentuximab vedotin; CNS = central nervous system; CPG = clinical practice guidelines; CVPP = cyclophosphamide-vinblastine-procarbazine-prednisone; ECOG PS = Eastern Cooperative Oncology Group Performance Status; G-CSF = granulocyte colony-stimulating factor; HL = Hodgkin lymphoma; OPEA-COPDAC = vincristine-etoposide-prednisone-doxorubicin–cyclophosphamide-vincristine-prednisone-dacarbazine; OS = overall survival; PAG = Provincial Advisory Group; pERC = pan-Canadian Oncology Review Expert Review Committee; PML = progressive multifocal leukoencephalopathy.
Clinical Evidence
The objective of the Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of BV (50 mg per vial, lyophilized powder for reconstitution, IV infusion), in combination with AVD, in the treatment of previously untreated patients with advanced-stage HL. The focus will be placed on comparing BV + AVD to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of BV + AVD is presented in 2 sections, and a critical appraisal of the evidence is included after this section. The first section, the systematic review, includes pivotal studies and RCTs selected according to the sponsor’s systematic review protocol. No long-term extension studies or indirect evidence was submitted by the sponsor. The next section includes an additional study that was considered to address important gaps in the pivotal RCT evidence.
Included Studies
Clinical evidence from 1 pivotal phase III, multicentre, open-label RCT (ECHELON-1) and 1 additional phase III, multicentre, open-label, RCT addressing gaps in the pivotal RCT evidence (AHOD1331) is included in the review and appraised in this document:
Pivotal Study and RCT Evidence
Contents within this section were informed by materials submitted by the sponsor. The following summary was validated by the review team.
Description of Studies
One study (ECHELON-1),13,14 which was conducted by the sponsor, met the inclusion criteria for the sponsor-submitted systematic review. Characteristics of the included studies are summarized in Table 5.
The ECHELON-1 trial is a phase III, open-label, randomized, active-controlled, superiority trial comparing BV + AVD with ABVD in previously untreated adult patients with advanced-stage classical HL, consisting of stage III and stage IV patients as determined by the Ann Arbor classification system.9,27,28 This study is being conducted in 1,334 participants from 21 countries, and 60 patients in Canada have been enrolled. Participants were randomized 1:1 and stratified by geographical region (i.e., Americas versus Asia versus Europe) and number of IPFP risk factors (i.e., 0 to1 versus 2 to 3 versus 4 to 7), with 664 patients randomized to the BV + AVD arm and 670 patients to the ABVD arm. The primary objective of the ECHELON-1 trial was to determine the efficacy of BV + AVD relative to ABVD as measured by mPFS. The key secondary objective was to compare OS between BV + AVD and ABVD.
The ECHELON-1 trial is ongoing. Data gathered at the cut-off dates of April 20, 2017, and June 1, 2021, were assessed for this review. New data from a descriptive analysis of OS conducted in response to a request for supplementary information from the EMA with a data cut-off date of March 11, 2023 was also included in this report.15
Table 5: Details of Pivotal Studies and RCT Evidence Identified by the Sponsor
Detail | ECHELON-1 |
|---|---|
Designs and populations | |
Study design | Open-label, randomized, 2-arm, global, multicentre, phase III trial |
Locations |
|
Key dates |
|
Randomized (N) | 1,334 patients were randomized to receive BV + AVD (n = 664) or ABVD (n = 670) |
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Intervention | BV + AVD (brentuximab vedotin 1.2 mg/kg IV, doxorubicin 25 mg/m2 IV, vinblastine 6 mg/m2 IV, dacarbazine 375 mg/m2 IV) administered as infusions on days 1 and 15 of each 28-day cycle up to a maximum of 6 cycles. |
Comparator | ABVD (doxorubicin 25 mg/m2 IV, bleomycin 10 units/m2 IV, vinblastine 6 mg/m2 IV, dacarbazine 375 mg/m2 IV) administered as infusions on days 1 and 15 of each 28-day cycle up to a maximum of 6 cycles. |
Study duration | |
Screening phase | Up to 28 days before the first dose of study drug |
Treatment phase | Up to 6 cycles of study treatment |
Follow-up phase | Starting 30 days after the last dose of study drug and lasting until the first instance of consent withdrawal, lost to follow-up, study closure, or after being followed for 10 years (initially defined as 5 years in the published protocol) |
Outcomes | |
Primary end point | Modified progression-free survival |
Secondary and exploratory end points | Key secondary
Secondary
Exploratory
|
Publication status | |
Publications | |
ABVD = doxorubicin-bleomycin-vinblastine-dacarbazine; ADV = doxorubicin-vinblastine-dacarbazine; ALT = alanine transaminase; AST = aspartate transaminase; BV = brentuximab vedotin; ECOG PS = Eastern Cooperative Oncology Group Performance Status; EMA = European Medicines Agency; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; FACT/GOG-Ntx Abbreviated = abbreviated Functional Assessment of Cancer Therapy/Gynecologic Oncology Group – Neurotoxicity; FACIT-Dyspnea 10 = Functional Assessment of Chronic Illness Therapy – Dyspnea 10-item short-form questionnaire; HBV = hepatitis B virus; HCV = hepatitis C virus; HL = Hodgkin lymphoma; HRQoL = health-related quality of life; NYHA = New York Heart Association; SAE = serious adverse event; TEAE = treatment-emergent adverse event; ULN = upper limit of normal.
aWHO classification includes nodular-sclerosis, mixed-cellularity, lymphocyte-rich, lymphocyte-depleted, or classical HL not otherwise specified.
bSpecified clinical laboratory values include: absolute neutrophil count of 1,500/μL or higher and platelet count of 75,000/μL or greater unless known HL marrow involvement, total bilirubin less than 1.5 times the ULN unless due to Gilbert syndrome, ALT or AST less than 3 times the ULN range (AST and ALT may be elevated up to 5 times the ULN if reasonably ascribed to presence of HL in liver), serum creatinine less than 2.0 mg/dL and/or creatinine clearance or calculated creatinine clearance 40 mL/minute or greater, hemoglobin 8 g/dL or greater.
cNonmelanoma skin cancer or carcinoma in situ of any type are not excluded if they had been completely resected.
Sources: ECHELON-1 original Clinical Study Report (data cut-off: April 20, 2017)13 and Addendum 1 (data cut-off: June 01, 2021)14 and the sponsor’s Summary of Clinical Evidence.1
Populations
Inclusion and Exclusion Criteria
The ECHELON-1 trial included previously untreated adult patients with histologically confirmed stage III or stage IV classical HL and with an ECOG PS of 2 or lower (Table 5). Patients with nodular lymphocyte-predominant HL and those with sensory or motor peripheral neuropathy were excluded.
Interventions
In the ECHELON-1 trial, patients in both groups received combination therapy (i.e., intervention group: BV + AVD; control group: ABVD), with each drug administered as an IV infusion on days 1 and 15 of a 28-day cycle. Treatment was continued up to a maximum of 6 cycles, which were completed before the final mPFS analysis (data cut-off: April 20, 2017). No new study treatments were administered during the post-treatment follow-up period leading up to Addendum 1 (data cut-off date: June 1, 2021).
The BV + AVD regimen consisted of the following drugs administered in sequential order: doxorubicin 25 mg/m2 IV infusion, vinblastine 6 mg/m2 IV infusion, and dacarbazine 375 mg/m2 IV infusion. Then, approximately 1 hour after the end of the dacarbazine infusion, BV (i.e., 1.2 mg/kg) was administered via IV infusion over approximately 30 minutes. The dose of BV was calculated based on actual weight, except for patients weighing more than 100 kg. In such cases, the dose was calculated based on a weight of 100 kg. During the trial, 9% of patients (n = 58) randomized to BV + AVD received the 100 mg capped dose of BV. Dose adjustments were performed in patients who experienced a change in weight of 10% or more from baseline.
The ABVD regimen consisted of the following agents administered in sequential order: doxorubicin 25 mg/m2 IV infusion, bleomycin 10 units/m2 IV infusion, vinblastine 6 mg/m2 IV infusion, and dacarbazine 375 mg/m2 IV infusion.
Dose modification was permitted, based on treatment-associated toxicity. Administration of growth factors (i.e., G-CSF or granulocyte-macrophage colony-stimulating factor) were permitted to treat neutropenia, as were platelets and/or red blood cell–supportive growth factors or transfusions when needed. Due to the higher incidence of neutropenia in the BV + AVD group, the independent data and safety monitoring committee of the ECHELON-1 trial recommended primary prophylaxis with G-CSF for patients newly randomized to BV + AVD treatment. This change was made after 75% of enrolment was complete; primary prophylaxis was defined as use of G-CSF by day 5 of the treatment cycle.
Prophylactic antiemetics and antidiarrheals were not included in the study protocol but were permitted at the physician’s discretion. Corticosteroids were permitted as part of a chemotherapy premedication regimen or for treatment of HL according to an institution’s standards. If a PET scan performed on day 25 resulted in a Deauville score of 5, physicians had the option of switching the patient to an alternative treatment regimen. Any switch in therapy made before completion of front-line treatment was not considered an mPFS event. At the end of front-line treatment, patients in partial remission with persistent PET-positive disease were permitted to receive radiation therapy at the discretion of the investigator. However, such patients were counted has having an mPFS event only if they were deemed to have a non-CR (i.e., a Deauville score ≥ 3) confirmed by the IRF.
Outcomes
A list of end points assessed in this clinical review report is provided in Table 6 and further summarized in the following section. Summarized end points are based on those included in the sponsor’s Summary of Clinical Evidence as well as any identified as important to this review according to stakeholders (the clinical experts, clinician groups, or patient groups). In addition to the end points listed in the table, the review team noted that post-progression survival (PPS), which was defined as time from progression to death and calculated for patients who experienced disease progression by subtracting PFS from OS obtained from the ECHELON-1 trial, was submitted by the sponsor to support pharmacoeconomic analyses (Appendix 2). Of note, PPS was not prespecified in the ECHELON-1 trial protocol.
Table 6: Outcomes Summarized From the ECHELON-1 Trial
Outcome measure | Time point | End point |
|---|---|---|
Overall survival | Until death due to any cause or patient withdrawal (monitored for up to 10 years) | Key secondary (data cut-off dates: June 1, 2021, and March 11, 2023) |
Percentage of patients alive without HL | After 3 years and after 5 years | Exploratory (data cut-off date: June 1, 2021) |
PFS according to investigatora | At the first instance of documented disease progression, death due to any cause, modified progression, or patient withdrawal (monitored for up to 10 years) | Exploratory (data cut-off date: June 1, 2021) |
mPFS according to IRF | At the first instance of documented disease progression, death due to any cause, or modified progression (monitored for up to 10 years) | Primary (data cut-off date: April 20, 2017) |
Complete response rate according to IRF | At the end of the assigned treatment regimen | Secondary (data cut-off date: April 20, 2017) |
EOQTC QLQ-C30 | Until the final visit, death, or patient withdrawal | Secondary (data cut-off date: June 1, 2021) |
EQ-5D-3L | Until the first instance of disease progression or 3 years after the last dose of front-line therapy | Exploratory (data cut-off date: June 1, 2021) |
Harms outcomes | Up to 30 days after the last dose of front-line therapy and post-treatment follow-up period | Secondary (data cut-off dates: April 20, 2017, and June 1, 2021) |
EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; IRF = independent review facility; mPFS = modified progression-free survival; PFS = progression-free survival.
aPFS according to IRF (data cut-off date: April 20, 2017) was also reported by the ECHELON-1 trial. Results can be found in Appendix 4.
Sources: ECHELON-1 original Clinical Study Report (data cut-off: April 20, 2017)13 and Addendum 1 (data cut-off: June 01, 2021),14 and the sponsor’s Summary of Clinical Evidence.1
Efficacy Outcomes
Several definitions of key efficacy outcomes were provided in the ECHELON-1 trial.37,38
Overall Survival
In the ECHELON-1 trial, OS was defined as the time from the date of randomization to the date of death.
Percentage of Patients Alive Without HL
The percentage of patients alive without HL was defined as the proportion of patients who were alive without classical HL at 3 years or 5 years after the patient’s randomization date.
Progression-Free Survival According to Investigator
Progression-free survival was defined as the time from the date of randomization to the date of the first of documentation of progressive disease or death due to any cause.
Modified Progression-Free Survival According to IRF
The mPFS37,38 was assessed by the IRF according to the Revised Response Criteria for Malignant Lymphoma and defined as the time from the date of randomization to the date of the first of documentation of progressive disease, or death due to any cause, or modified progression, which was defined as receipt of anticancer therapy or radiotherapy for HL after completion of front-line therapy for patients who were confirmed noncomplete responders according to IRF. Completion of front-line therapy was defined upon receipt of the planned study drug regimen with no more than 2 missed doses of BV + AVD or ABVD in patients who did not switch therapy, or upon conclusion of 1 alternative anticancer regiment for HL after BV + AVD or ABVD discontinuation in patients who switched therapy before completion of BV + AVD or ABVD. The mPFS event date of these patients was the date of the first PET scan after completion of front-line therapy demonstrating the absence of a complete remission, defined as a Deauville score of 3 or higher.
CR Rate According to IRF
A CR according to IRF was defined as the proportion of patients who achieved a CR at the end of front-line therapy as determined by an IRF.
Health-Related Quality of Life
The summary of EORTC QLQ-C30 and EQ-5D-3L is shown in Table 7.
Table 7: Summary of HRQoL Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
EORTC QLQ-C30 | A widely used, cancer-specific HRQoL instrument consisting of 30 items measuring 5 functional dimensions (physical, role, cognitive, emotional, and social), 3 symptom dimensions (fatigue, nausea and/or vomiting, and pain), 6 additional items (dyspnea, sleep disturbance, appetite loss, constipation, diarrhea, and financial impact), and a global HRQoL measure. The responses are converted into a scale score from 0 to 100; higher scores indicate better quality of life for functional and Global Health Status/quality-of-life scales, whereas lower scores indicated better quality of life for symptom scales.39-42 | Validity, reliability, and responsiveness: |
No MID specific to relapsed or refractory HL |
EQ-5D-3L | A 2-part standardized instrument that measures health status and consists of the EQ-5D descriptive system and EQ VAS. The descriptive system consists of 5 dimensions (i.e., mobility, self-care, usual activities, pain and/or discomfort, and anxiety and/or depression). Patients are asked to describe their health related to each dimension according to 3 response levels of severity, ranging from no problems, some problems, to extreme problems. A summary index score is derived using value sets that provide weights for each health-state description based on preferences of the general population in a specific region or country.48,49 The EQ VAS is a subjective quantitative measure of health outcomes and measures a patient’s self-rated health on a vertical scale, between best to worst health that the patient can imagine. The EQ VAS has end points of 0 and 100, with respective anchors of “worst imaginable health state” and “best imaginable health state.” Respondents are asked to rate their health by drawing a line from an anchor box to the point on the EQ VAS that best represents their health on that day.50 The lowest possible overall score for the EQ-5D-3L (corresponding to severe problems on all 5 attributes) varies depending on the utility function that is applied to the descriptive system (e.g., −0.59 for the UK algorithm and −0.109 for the US algorithm). Scores of less than 0 represent health states that are valued by society as being worse than dead, while scores of 0 and 1.00 are assigned to the health states “dead” and “perfect health,” respectively.51 | Validity: Moderate to poor ability to distinguish between cancer severity by 3 scales (self-reported health status, ECOG PS, stage of cancer)52 Reliability: No literature identified that assessed responsiveness in patients with relapsed or refractory HL Responsiveness: No literature identified that assessed responsiveness in patients with relapsed or refractory HL | MID 0.033 to 0.074 estimated for the general population51 MID 0.07 to 0.11 for UK index scores and 0.05 to 0.08 for US index scores for patients with cancer53 VAS MID of 6 to 10 for patients with cancer53 No MID identified in patients with relapsed or refractory classical HL |
ECOG PS = Eastern Cooperative Oncology Group Performance Status; EORTC QLQ-C30 = European Organisation for the Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EQ VAS = visual analogue scale; HL = Hodgkin lymphoma; HRQoL = health-related quality of life; MID = minimal important difference.
Harms Outcomes
The harms outcomes assessed in the ECHELON-1 trial included TEAEs, TESAEs, deaths, withdrawals due to AEs, and notable harms (treatment-emergent peripheral neuropathy, secondary malignancy, neutropenia, febrile neutropenia, and pulmonary-related toxicity). AEs were coded using the Medical Dictionary for Regulatory Activities (MedDRA). TEAEs were defined as any AE that occurs after administration of the first dose of study drug and through 30 days after the last dose of front-line therapy. Pulmonary-related toxicity included preferred terms of “dyspnea” and “hypoxia,” and all preferred terms in interstitial lung disease standardized MedDRA query, and preferred terms with the high-level term “respiratory and pulmonary function diagnostic procedures.”
Statistical Analysis
Sample Size and Power Calculation
Sample size was calculated based on the primary end point of mPFS according to IRF, with a planned enrolment of approximately 1,240 patients. To detect an HR of 0.67 in mPFS (assuming an emergent plateau in the mPFS rate after 2 years), which would indicate a 73% improvement in 2-year mPFS in the ABVD arm compared to the 81% in the BV + AVD arm, approximately 260 mPFS events were needed to achieve 90% power at a 1-sided significance level of 0.025 using a log-rank test. With 1,240 participants, there was a 95% likelihood of obtaining 260 mPFS over 60 months (assuming 36-month accrual, 24 months mPFS follow-up after the last patient recruitment, and a 5% annual dropout rate).
Statistical Testing
Details of the statistical analysis of selected efficacy end points are summarized in Table 8.
Both OS and mPFS were compared between treatment arms using the stratified log-rank test, with the stratification factors being the number of IPFP risk factors at baseline and region. A stratified Cox regression model was used to estimate HRs and 95% CIs. The Kaplan-Meier approach was used to estimate distribution of OS or mPFS end points for each treatment group. The model included covariates such as age, race, baseline ECOG PS score, baseline cancer stage, baseline B symptoms, and PET2 results, along with treatment.
Two formal analyses for OS, including an OS interim analysis at the time of the final mPFS analysis (data cut-off date of April 20, 2017) and a final analysis when 112 deaths had occurred were planned in the original protocol. An additional interim analysis of OS, which was submitted by the sponsor for the present reimbursement request, was added to protocol Amendment 9 and performed after the accrual of 103 deaths. The final OS analysis will be carried out after accrual of 112 deaths or 10 years from the randomization date of the last patient, whichever occurs first. When there was no documented death at the time of analysis, patients were censored at the date last known to be alive.
Modified PFS was tested at a 1-sided significance level of 0.025. OS was tested at a 1-sided significance level of 0.025 given that the test of mPFS reached statistical significance in the final mPFS analysis, which was prespecified in the protocol. The overall type I error was controlled using the O’Brien-Fleming boundary with a Lan-DeMets alpha spending function. The alpha level of OS at the final analysis will be adjusted for the final observed death count to maintain the overall type I error. Statistical testing adjusted for multiplicities were only conducted for OS and mPFS; P values were used for descriptive purposes only for other secondary and exploratory end points.
Sensitivity and subgroup analyses are shown in Table 8. Two additional exploratory analyses were performed, including 1 for mPFS restricted to patients who did not switch therapy and 1 for PFS according to investigator. The statistical methods of PFS according to investigator were like those of mPFS. PFS was censored at the last known alive date for those who did not have events. Patients with PFS events according to investigator after more than 1 missed visit were censored at the date of the last adequate assessment.
The percentage of patients alive without HL and the CR rate according to IRF between the 2 treatment arms were compared using a stratified Cochran-Mantel-Haenszel test. Descriptive summaries of observed EORTC QLQ-C30 and EQ-5D-3L scores were generated at each scheduled assessment time point by treatment.
All patients who received at least 1 dose of the study drug were included in the safety analysis set and were analyzed according to the actual treatment received. AEs were categorized according to the Medical Dictionary for Regulatory Activities and intensity was measured using National Cancer Institute Common Terminology Criteria for Adverse Events version 4.03. TEAEs, TESAEs, mortality, and withdrawals due to AEs are presented in this report as harms outcomes. Peripheral neuropathy, secondary malignancy, neutropenia, febrile neutropenia, pneumonitis, and pulmonary toxicity were reported as notable harms.
Table 8: Statistical Analysis of Efficacy End Points in the ECHELON-1 Trial
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity or subgroup analyses |
|---|---|---|---|---|
OS |
|
| Censoring
| Sensitivity analyses
Subgroup analyses
|
Percentage of patients alive without HL |
| None | Censoring | None |
PFS according to investigator |
| None | Censoring
| Subgroup analyses
|
mPFS by IRF |
|
| Censoring
| Sensitivity analyses
Subgroup analyses
|
CR rate according to IRF |
| None | Nonevaluable patients were treated as if they did not achieve CR | Sensitivity analyses
|
EORTC QLQ-C30 |
| None | Imputed according to the published tool manual | Sensitivity analyses
|
EQ-5D-3L |
| None | Imputed according to the published tool manual | None |
CI = confidence interval; CR = complete response; ECOG PS = Eastern Cooperative Oncology Group Performance Status; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; HL = Hodgkin lymphoma; IPFP = International Prognostic Factor Project; IRF = intendent review facility; ITT = intention to treat; mPFS = modified progression-free survival; OS = overall survival; PET2 = PET response after 2 cycles of chemotherapy; PFS = progression-free survival; SD = standard deviation; VAS = visual analogue scale; vs. = versus.
Sources: Drug Reimbursement Review sponsor submission16 and the sponsor’s Summary of Clinical Evidence.1
Analysis Populations
Analysis populations of the ECHELON-1 trial are summarized in Table 9.
Table 9: Analysis Populations of the ECHELON-1 Trial
Population | Definition | Application |
|---|---|---|
ITT population | All patients randomized to treatment | All patients analyzed according to the treatment arm to which they were randomized; the ITT population was used for all efficacy end points |
Per-protocol population | All randomized patients who met the eligibility criteria and did not have major protocol violations as determined by the project clinician | This population was used to supplement the analysis of the primary efficacy end point (i.e., mPFS) using the ITT population; all patients were analyzed according to the actual treatment received |
Safety population | All enrolled patients who received at least 1 dose of any study drug | All patients analyzed according to the actual treatment received; all safety analyses were performed using this set |
ITT = intention-to-treat; mPFS = modified progression-free survival.
Sources: ECHELON-1 original Clinical Study Report (data cut-off: April 20, 2017)13 and the sponsor’s Summary of Clinical Evidence.1
Protocol Amendments and Deviations
Nine amendments were made to the protocol. The original protocol and the protocol with the ninth amendment were issued on March 29, 2012, and September 24, 2021, respectively. A summary of key protocol amendments implemented in the ECHELON-1 trial is presented in Appendix 3. Patients were not enrolled into the study until Amendment 4. The sample size was increased in Amendment 7. In Amendment 9, an additional interim analysis of OS, which was submitted by the sponsor for the present reimbursement request, was performed.
Major protocol deviations occurred in 16 patients who were enrolled despite not satisfying eligibility criteria, and in 10 patients who received an incorrect treatment or dose of the study drug. No deviations were identified in patients who received excluded medications or were not discontinued from the study despite study withdrawal criteria not being met.
Results
Patient Disposition
A summary of patient disposition in the ECHELON-1 trial is presented in Table 10. Overall, patient dispositions were balanced between the BV + AVD and the ABVD arms. For example, similar proportions of patients completed front-line therapy between the BV + AVD and ABVD groups (92% [608 of 664] versus 93% [622 of 670]). Other than the completed maximum number of cycles per protocol (89% [593 of 664] in the BV + AVD group versus 91% [607 of 670] in the ABVD group), the second-most common reason for treatment discontinuation was AEs (4% [28 of 664] in the BV + AVD group versus 3% [22 of 670] in the ABVD group). About 12% (12 of 664) and 14% (95 of 670) were lost to follow-up.
Table 10: Summary of Patient Disposition From Pivotal Studies and RCT Evidence Submitted by the Sponsor
Patient disposition | ECHELON-1 | |
|---|---|---|
BV + AVD (N = 664) | ABVD (N = 670) | |
Screened, N | 1,585 | |
Reason for screening failure, N (%) | 251 (15.8) | |
Fail to meet at least 1 inclusion criterion | 187 | |
Excluded by an exclusion criterion | 64 | |
Randomized, N | 1,334 | |
ITT populationa | 664 | 670 |
Safety populationb | 662 | 659 |
Patients completing treatment per protocol, N (%)c | 628 (95) | 634 (95) |
Completed front-line therapyd | 608 (92) | 622 (93) |
Randomized regimen only | 594 (89) | 613 (91) |
Randomized regimen with alternative front-line medication | 14 (2) | 9 (1) |
Experienced progressive disease or died before completion of front-line therapy | 20 (3) | 12 (2) |
Primary reason off study treatment, N (%) | 664 (100) | 670 (100) |
Adverse event | 28 (4) | 22 (3) |
Completed maximum number of cycles per protocol | 593 (89) | 607 (91) |
Lost to follow-up | 2 (< 1) | 2 (< 1) |
Progressive disease | 17 (3) | 10 (1) |
Protocol violation | 1 (< 1) | 0 |
Unsatisfactory therapeutic response | 1 (< 1) | 2 (< 1) |
Withdrawal by patient | 7 (1) | 15 (2) |
Other | 15 (2) | 12 (2) |
Patients who have participated in PFS follow-up, N (%) | 572 (86) | 544 (81) |
Patients who have participated in OS follow-up, N (%) | 155 (23) | 189 (28) |
Patients currently in PFS follow-up, N (%) | 335 (50) | 277 (41) |
Patients currently in OS follow-up, N (%) | 59 (9) | 73 (11) |
Death, N (%) | 39 (6) | 64 (10) |
On-study deathe | 9 (1) | 13 (2) |
Death during post-treatment follow-upf | 30 (5) | 51 (8) |
Reason for discontinuing study, N (%) | 270 (41) | 320 (48) |
Lost to follow-up | 81 (12) | 95 (14) |
Withdrawal by patient | 131 (20) | 143 (21) |
Death | 39 (6) | 64 (10) |
Other | 19 (3) | 18 (3) |
ABVD = doxorubicin-bleomycin-vinblastine-dacarbazine; AVD = doxorubicin-vinblastine-dacarbazine; BV = brentuximab vedotin; HL = Hodgkin lymphoma; ITT = intention-to-treat; OS = overall survival; PFS = progression-free survival.
aThe ITT population was defined as patients who were randomized to treatment.
bThe safety population was defined as all enrolled patients who received at least 1 dose of study medication.
cPatients were considered to have completed study treatment per protocol if they completed front-line treatment or experienced progressive disease according to investigator or died before completion of front-line treatment.
dCompletion of front-line treatment was defined as: upon receipt of planned study drug regimen with no more than 2 missed doses of BV + AVD or ABVD, or upon conclusion of 1 alternative anticancer regiment for HL after discontinuation of BV + AVD or ABVD.
eOn-study deaths were defined as deaths that occurred within 30 days of the last dose of front-line therapy.
fFollow-up deaths were defined as deaths that occurred after 30 days of the last dose of front-line therapy.
Sources: ECHELON-1 Clinical Study Report Addendum 1 (data cut-off: June 1, 2021)14 and the sponsor’s Summary of Clinical Evidence.1
Baseline Characteristics
A summary of baseline patient demographics and disease characteristics in the ECHELON-1 trial is shown in Table 11.
A total of 1,334 patients were randomly assigned to receive BV + AVD (n = 664) or ABVD (n = 670). Baseline demographics and disease characteristics were generally well balanced between the 2 treatment groups. Overall, the median age of enrolled patients was 36 years (range = 18 to 83); most (66%, 874 of 1,334) were younger than 45 years, and 14% (186 of 1,334) were aged 60 years or older. Of the 1,334 patients enrolled, 58% (776) were male and 84% (1,114) were white. A majority (64%) of patients had stage IV disease (846), 2 or 3 (53%, or 705) IPFP risk factors, an ECOG PS of 0 (57%, or 754), extranodal involvement at diagnosis (62%, or 827), and B symptoms (59%, or 781) at baseline.
Table 11: Summary of Baseline Characteristics of Pivotal Studies and RCT Evidence Submitted by the Sponsor
Characteristic | ECHELON-1 (ITT population) | |
|---|---|---|
BV + AVD (N = 664) | ABVD (N = 670) | |
Age in yearsa | ||
n | 664 | 670 |
Mean (SD) | 38.8 (15.83) | 40.2 (16.05) |
Median (minimum to maximum) | 35.0 (18 to 82) | 37.0 (18 to 83) |
Age categories in years, n (%)a | ||
< 45 | 451 (68) | 423 (63) |
45 to 59 | 129 (19) | 145 (22) |
60 to 64 | 24 (4) | 40 (6) |
≥ 65 | 60 (9) | 62 (9) |
Sex, n (%) | ||
Male | 378 (57) | 398 (59) |
Female | 286 (43) | 272 (41) |
Regions, n (%) | ||
Americas | 261 (39) | 262 (39) |
Europe | 333 (50) | 336 (50) |
Asia Pacific | 70 (11) | 72 (11) |
Race, n (%) | ||
White | 560 (84) | 554 (83) |
Asian | 56 (8) | 57 (9) |
Black or African American | 20 (3) | 25 (4) |
Other | 18 (3) | 17 (3) |
Not reported | 10 (2) | 17 (3) |
Time since initial diagnosis, monthsb | ||
n | 662 | 659 |
Mean (SD) | 1.09 (1.12) | 1.18 (3.34) |
Median (minimum to maximum) | 0.92 (0.1 to 21.4) | 0.89 (0.0 to 81.4) |
Missing | 2 | 11 |
Disease type | ||
HL | 661 (100) | 664 (99) |
Nodular lymphocyte-predominant HL | 0 | 0 |
Classical HLc | 144 (22) | 140 (21) |
Nodular-sclerosis classical HL | 425 (64) | 386 (58) |
Lymphocyte-rich classical HL | 12 (2) | 20 (3) |
Mixed-cellularity classical HL | 78 (12) | 111 (17) |
Lymphocyte-depleted classical HL | 2 (< 1) | 7 (1) |
Otherd | 3 (< 1) | 6 (< 1) |
Ann Arbor stage at initial diagnosis, n (%) | ||
Stage I | 0 | 0 |
Stage IIe | 1 (< 1) | 0 |
Stage III | 237 (36) | 246 (37) |
Stage IV | 425 (64) | 421 (63) |
Not applicable | 1 (< 1) | 1 (< 1) |
Missing | 0 | 2 |
Number of IPFP risk factors | ||
0 to 1 | 141 (21) | 141 (21) |
2 to 3 | 354 (53) | 351 (52) |
4 to 7 | 169 (25) | 178 (27) |
ECOG PS, n (%) | ||
0 | 376 (57) | 378 (57) |
1 | 260 (39) | 263 (39) |
2 | 28 (4) | 27 (4) |
Missing | 0 | 2 |
Bone marrow involvement at initial diagnosis, study entry, n (%) | ||
Yes | 147 (22) | 151 (23) |
No | 502 (76) | 509 (76) |
Unknown or missing | 15 (2) | 10 (1) |
Evidence of extranodal involvement at initial diagnosis, n (%) | ||
Yes | 411 (62) | 416 (62) |
1 extranodal site | 217 (33) | 223 (33) |
> 1 extranodal sites | 194 (29) | 193 (29) |
No | 217 (33) | 228 (34) |
Unknown or missing | 36 (5) | 25 (4) |
B symptomsf | ||
Patients with any B symptoms, n (%) | 400 (60) | 381 (57) |
ABVD = doxorubicin-bleomycin-vinblastine- dacarbazine; AVD = doxorubicin-vinblastine-dacarbazine; BV = brentuximab vedotin; ECOG PS = Eastern Cooperative Oncology Group Performance Status; HL = Hodgkin lymphoma; IPFP = International Prognostic Factor Project; ITT = intention-to-treat; SD = standard deviation.
Note: Percentages are based on nonmissing values in the ITT population in each column.
aAge on date of informed consent.
bTime since initial diagnosis = (first dose date of study drug – date of initial diagnosis) / 30.4375.
cClassical HL included patients who were diagnosed with classical HL not otherwise specified.
dPatients whose initial diagnosis was HL, then subsequently found to have been misdiagnosed.
eStage II disease was captured as a protocol violation.
fPatients who presented with a B symptom for ≥ 1 visit before the start of study drug administration.
Sources: ECHELON-1 Clinical Study Report Addendum 1 (data cut-off: June 1, 2021)14 and the sponsor’s Summary of Clinical Evidence.1
Exposure to Study Treatments
Overall, patients in the BV + AVD group and those in the ABVD group received a similar median number of treatment cycles, which were administered over a similar median duration of treatment at similar median relative dose intensities. Details on the extent of exposure to study treatments in the ECHELON-1 trial are summarized in Table 12. The assigned treatments had been completed by the data cut-off date of April 20, 2017, and no new study treatments were administered as of the data cut-off on June 1, 2021.
Before the completion of the front-line therapy, of the physician’s choice, 15 patients (2%) in the BV + AVD arm and 9 (1%) in the ABVD arm switched to an alternative front-line treatment; all were treated with another form of chemotherapy only. ABVD was the most frequently reported alternative front-line treatment for the BV + AVD group and BEACOPP was the most frequently reported alternative front-line treatment for the ABVD group. In addition, in the BV + AVD arm, the switch was made mainly due to AEs (12 patients), followed by other reasons (2 patients), and the Deauville score after the PET2 assessment (1 patient). In the ABVD arm, 4 patients switched to another form of chemotherapy due to a Deauville score and other reasons, while only 1 patient switched due to AEs.
Concomitant Medications
As of April 20, 2017, at least 1 concomitant medication was reported for 659 patients (100%) treated with BV + AVD and for 653 patients (99%) treated with ABVD. Details about commonly reported concomitant medications (used in ≥ 20% of patients in either group) are shown in Table 13.
Table 12: Summary of Patient Exposure From Pivotal Studies and RCT Evidence Submitted by the Sponsor
Exposure | ECHELON-1 (safety population) | |||||||
|---|---|---|---|---|---|---|---|---|
BV + AVD (N = 662) | ABVD (N = 659) | |||||||
BV (mg/kg) | Doxorubicin (mg/m2) | Vinblastine (mg/m2) | Dacarbazine (mg/m2) | Bleomycin (IU/m2) | Doxorubicin (mg/m2) | Vinblastine (mg/m2) | Dacarbazine (mg/m2) | |
Duration of treatment, weeksa | ||||||||
n | 662 | 656 | 661 | 661 | 659 | 649 | 659 | 659 |
Mean (SD) | 23.19 (5.646) | 23.88 (5.362) | 23.60 (5.600) | 23.89 (5.335) | 22.38 (5.694) | 23.88 (4.669) | 23.65 (4.880) | 23.86 (4.658) |
Median | 24.21 | 24.57 | 24.43 | 24.57 | 24.00 | 24.00 | 24.00 | 24.00 |
Range | 2.0 to 35.0 | 2.0 to 48.9 | 2.0 to 48.9 | 2.0 to 48.9 | 2.0 to 39.1 | 2.0 to 45.4 | 2.0 to 45.4 | 2.0 to 45.4 |
Total number of doses received | ||||||||
n | 662 | 656 | 661 | 661 | 659 | 649 | 659 | 659 |
Mean (SD) | 10.8 (2.60) | 11.2 (2.38) | 11.0 (2.50) | 11.2 (2.37) | 10.7 (2.64) | 11.4 (2.00) | 11.3 (2.13) | 11.4 (2.02) |
Median | 12.0 | 12.0 | 12.0 | 12.0 | 12.0 | 12.0 | 12.0 | 12.0 |
Range | 1 to 12 | 1 to 12 | 1 to 12 | 1 to 12 | 1 to 12 | 1 to 12 | 1 to 12 | 1 to 12 |
Number of treated cyclesb | ||||||||
n | 662 | 656 | 661 | 661 | 659 | 649 | 659 | 659 |
Mean (SD) | 5.5 (1.21) | 5.6 (1.13) | 5.6 (1.18) | 5.6 (1.12) | 5.4 (1.24) | 5.7 (0.95) | 5.7 (1.01) | 5.7 (0.96) |
Median | 6.0 | 6.0 | 6.0 | 6.0 | 6.0 | 6.0 | 6.0 | 6.0 |
Range | 1 to 6 | 1 to 6 | 1 to 6 | 1 to 6 | 1 to 6 | 1 to 6 | 1 to 6 | 1 to 6 |
Relative dose intensity (%)c | ||||||||
n | 661 | 656 | 661 | 661 | 659 | 649 | 659 | 659 |
Mean (SD) | 94.01 (12.003) | 99.11 (5.358) | 96.56 (10.305) | 99.12 (4.214) | 93.51 (16.395) | 99.54 (4.112) | 96.91 (9.715) | 98.93 (6.104) |
Median | 99.46 | 99.99 | 99.10 | 99.99 | 99.85 | 100.00 | 99.36 | 100.00 |
Range | 16.7 to 114.3 | 4.1 to 109.2 | 15.4 to 115.2 | 66.0 to 111.9 | 8.1 to 119.4 | 59.6 to 111.1 | 9.3 to 116.2 | 13.9 to 114.0 |
ABVD = doxorubicin-bleomycin-vinblastine-dacarbazine; AVD = doxorubicin-vinblastine-dacarbazine; BV = brentuximab vedotin; RCT = randomized controlled trial; SD = standard deviation.
aDuration of treatment was defined as (last dose date – first dose date + 14) / 7.
bA treated cycle was defined as a 28-day period, during which the patient received any amount of individual drugs within the BV + AVD regimen or the ABVD regimen.
cRelative dose intensity (%) was defined as: 100 × (total dose received) / (total dose intended). Total dose intended was the summation of the intended doses in all treatment cycles before patients switch to alternative front-line medication. The intended dose in each cycle was determined by the dose at the time the patient was randomized. For BV, total dose intended = sum of intended dose at each dosing visit over treated dosing visit, where intended dose = prescribed dose at cycle 1, day 1 × actual weight; For ABVD, total dose intended = sum of intended dose at each cycle over treated cycle, where intended dose = (prescribed dose level at cycle 1, day 1 × body surface area (m2). The unit for bleomycin is IU/m2.
Sources: ECHELON-1 original Clinical Study Report (data cut-off: April 20, 2017)13 and the sponsor’s Summary of Clinical Evidence.1
Table 13: Concomitant Medications for 20% or Greater of Patients in the ECHELON-1 Trial
Exposure | ECHELON-1 (safety population) | |
|---|---|---|
BV + AVD (N = 662) | ABVD (N = 659) | |
Patients with at least 1 concomitant medication, n (%) | 659 (100) | 653 (99) |
Ondansetron | 486 (73) | 493 (75) |
Filgrastim | 405 (61) | 286 (43) |
Dexamethasone | 381 (58) | 388 (59) |
Paracetamol | 333 (50) | 331 (50) |
Allopurinol | 280 (42) | 277 (42) |
Metoclopramide | 190 (29) | 170 (26) |
Lorazepam | 183 (28) | 181 (27) |
Sodium chloride | 173 (26) | 133 (20) |
Bactrim (sulfamethoxazole and trimethoprim) | 156 (24) | 164 (25) |
Omeprazole | 162 (24) | 142 (22) |
Palonosetron | 154 (23) | 172 (26) |
Acyclovir | 148 (22) | 101 (15) |
Prochlorperazine | 135 (20) | 161 (24) |
Levofloxacin | 131 (20) | 106 (16) |
Pegfilgrastim | 131 (20) | 70 (11) |
Aprepitant | 126 (19) | 145 (22) |
ABVD = doxorubicin-bleomycin-vinblastine-dacarbazine; AVD = doxorubicin-vinblastine-dacarbazine; BV = brentuximab vedotin.
Sources: ECHELON-1 original Clinical Study Report (data cut-off: April 20, 2017)13 and the sponsor’s Summary of Clinical Evidence.1
Subsequent Treatment
As of the data cut-off date of June 1, 2021, 20% (135 of 662) of the patients in the BV + AVD group and 24% (157 of 659) of the patients in the ABVD group received at least 1 subsequent anticancer therapy after completion of the front-line therapy (Table 14).
The most common subsequent therapy was chemotherapy (12% [79 of 662] in the BV + AVD group; 17% [111 of 659] in the ABVD group). The most common chemotherapy for the BV + AVD group was cisplatin plus cytarabine plus dexamethasone (3% [17 of 662]). In the BV + AVD arm, 3 patients (< 1%) received ABVD as subsequent treatment. In the ABVD arm, 48 (7%) patients received BV as subsequent anticancer treatment.
Other subsequent treatments included radiation therapy (8% [54 of 662] in the BV + AVD group; 8% [54 of 659] in the ABVD group), high-dose chemotherapy in combination with transplant (7% [45 of 662] in the BV + AVD group; 9% [62 of 659] in the ABVD group), immunotherapy (3% [18 of 662] in the BV + AVD group; 4% [28 of 659] in the ABVD group), hormonal therapy (0 in the BV + AVD group; < 1% [2 of 659] in the ABVD group), and other investigational drug (0 in the BV + AVD group; < 1% [2 of 659] in the ABVD group).
Table 14: Summary of Subsequent Treatment From Pivotal Studies and RCT Evidence Submitted by the Sponsor
Exposure | ECHELON-1 (Safety population) | |
|---|---|---|
BV + AVD (N = 662) | ABVD (N = 659) | |
Patients with at least 1 subsequent anticancer therapy, n (%) | 135 (20) | 157 (24) |
Chemotherapy | 79 (12) | 111 (17) |
Cisplatin + cytarabine + dexamethasone | 17 (3) | 12 (2) |
BV | 8 (1) | 48 (7) |
ABVD | 3 (< 1) | 3 (< 1) |
Radiation | 54 (8) | 54 (8) |
High-dose chemotherapy plus transplant | 45 (7) | 62 (9) |
Immunotherapy | 18 (3) | 28 (4) |
Hormonal | 0 | 2 (< 1) |
Other investigational drug | 0 | 1 (< 1) |
ABVD = doxorubicin-bleomycin-vinblastine-dacarbazine; AVD = doxorubicin-vinblastine-dacarbazine; BV = brentuximab vedotin; RCT = randomized controlled trial.
Sources: ECHELON-1 Clinical Study Report Addendum 1 (data cut-off: June 01, 2021),14 Drug Reimbursement Review sponsor submission,16, and the sponsor’s Summary of Clinical Evidence.1
Efficacy
Key efficacy results in the ITT population of the ECHELON-1 trial are presented in Table 15. The data cut-off dates for OS were June 1, 2021 (i.e., the cut-off date in the sponsor’s Clinical Study Report) and March 11, 2023 (i.e., the cut-off date in the sponsor’s response to a request for supplementary information from the EMA). Data for PFS according to investigator, percentage of patients alive without HL, EOQTC QLQ-C30, and EQ-5D-3L were obtained from the data with a cut-off date of June 1, 2021, while data for mPFS according to IRF and CR rate according to IRF were from the data with a cut-off date of April 20, 2017. Results of subgroup analyses by disease stage (stage III only versus stage IV only) are shown in Table 16. Subgroup analyses by other factors are presented in Appendix 4.
Table 15: Summary of Key Efficacy Results From the ECHELON-1 Trial (ITT Population)
Key efficacy outcome | BV + AVD (N = 664) | ABVD (N = 670) |
|---|---|---|
OS (data cut-off date: June 1, 2021) | ||
Number of patients contributing to the analysis, n (%) | 664 (100) | 670 (100) |
Number with events (%) | 39 (6) | 64 (10) |
Number censored (%) | 625 (94) | 606 (90) |
Median, months (95% CI) | NE (NE to NE) | NE (NE to NE) |
Minimum, months | 0.0 | 0.0 |
Maximum, months | 100.6 | 97.9 |
Hazard ratio (95% CI)a | 0.59 (0.396 to 0.879) | |
P value between treatment groups | 0.009 | |
Median follow-up time, months (95% CI)b | 73.3 (72.61 to 74.05) | 72.4 (71.10 to 73.63) |
Reason leading to OS event, n (%) | ||
Death due to any cause | 39 (6) | 64 (10) |
Reason for censoring, n (%) | ||
End of study | 210 (32) | 234 (35) |
Lost to follow-up | 65 (10) | 76 (11) |
Withdrawal by patient | 126 (19) | 141 (21) |
Other | 19 (3) | 17 (3) |
Alive on date of last contact | 415 (63) | 372 (56) |
Alive without HL, n (%) | ||
Number of patients at 3 years after randomization | 567 (85) | 540 (81) |
Patients who were alive without HL | 546 (96) | 503 (93) |
P value | 0.016 | |
Number of patients at 5 years after randomization | 480 (72) | 443 (66) |
Patients who were alive without HL | 452 (94) | 408 (92) |
P value | 0.194 | |
PFS according to investigator | ||
Number of patients contributing to the analysis, n (%) | 664 (100) | 670 (100) |
Number with events (%) | 112 (17) | 159 (24) |
Number censored (%) | 552 (83) | 511 (76) |
Median (95% CI) | NE (NE to NE) | NE (NE to NE) |
Minimum, months | 0.0 | 0.0 |
Maximum, months | 100.6 | 97.9 |
Hazard ratio (95% CI)a | 0.678 (0.532 to 0.863) | |
P value between treatment groups | 0.002 | |
Median follow-up time, months (95% CI)c | 73.2 (72.48 to 74.05) | 71.6 (70.37 to 72.87) |
Reason leading to PFS event, n (%) | ||
Progressive disease | 96 (14) | 129 (19) |
Death due to any cause | 16 (2) | 30 (4) |
Reason for censoring, n (%) | ||
No baseline and/or no postbaseline assessment | 9 (1) | 22 (3) |
PFS event after more than 1 missed visit | 10 (2) | 8 (1) |
Treatment discontinuation for undocumented disease progression | 0 | 0 |
Loss to follow-up | 73 (11) | 72 (11) |
Withdrawal by patient | 98 (15) | 101 (15) |
No documented PFS event | 362 (55) | 308 (46) |
mPFS by IRF | ||
Number of patients contributing to the analysis, n (%) | 664 (100) | 670 (100) |
Number with events (%) | 117 (18) | 146 (22) |
Number censored (%) | 547 (82) | 524 (78) |
Median, months (95% CI) | NE (48.2 to NE) | NE (NE to NE) |
Minimum, months | 0.0 | 0.0 |
Maximum, months | 48.8 | 49.3 |
Hazard ratio (95% CI)a | 0.770 (0.603 to 0.982) | |
P value between treatment groups | 0.035 | |
Median follow-up time, months (95% CI)d | 24.9 (24.64 to 25.03) | 24.9 (24.61 to 25.07) |
Reason leading to mPFS event, n (%) | ||
Progressive disease | 90 (14) | 102 (15) |
Death due to any cause | 18 (3) | 22 (3) |
Receipt of additional therapy after non-CR | 9 (1) | 22 (3) |
Reason for censoring, n (%) | ||
No baseline and/or no postbaseline assessment | 11 (2) | 24 (4) |
mPFS event after more than 1 missed visit | 1 (< 1) | 3 (< 1) |
Treatment discontinuation for undocumented disease progression | 4 (< 1) | 4 (< 1) |
Loss to follow-up | 14 (2) | 20 (3) |
Withdrawal by patient | 22 (3) | 20 (3) |
No documented mPFS event | 495 (75) | 453 (68) |
CR rate according to IRF | ||
CR rate at the end of front-line therapy, n (%) | 488 (73) | 474 (71) |
Relative risk (95% CI) | 1.038 (0.97 to 1.11) | |
EORTC QLQ-C30e | ||
Summary score | ||
Baseline | ||
n | 648 | 651 |
LS mean (SE) | 79.294 (0.7486) | 78.955 (0.7456) |
LS mean difference (SE) [95% CI] | 0.339 (0.9084) [−1.443 to 2.121] | |
P value | 0.709 | |
End of treatment | ||
n | 566 | 564 |
LS mean (SE) | 2.540 (0.6141) | 5.861 (0.5625) |
LS mean difference (SE) [95% CI] | −3.321 (0.7811) [−4.853 to −1.788] | |
P value | < 0.001 | |
36 months after end of treatment | ||
n | 332 | 311 |
LS mean (SE) | 9.336 (0.5632) | 9.697 (0.5957) |
LS mean difference (SE) [95% CI] | −0.361 (0.7666) [−1.865 to 1.143] | |
P value | 0.638 | |
Global Health Status/QoL subscore | ||
Baseline | ||
n | 649 | 654 |
LS mean (SE) | 63.765 (0.9974) | 62.282 (0.9927) |
LS mean difference (SE) [95% CI] | 1.483 (1.2089) [−0.888 to 3.855] | |
P value | 0.220 | |
End of treatment | ||
n | 570 | 566 |
LS mean (SE) | 3.873 (0.8301) | 8.254 (0.7622) |
LS mean difference (SE) [95% CI] | −4.381 (1.0383) [−6.418 to −2.343] | |
P value | < 0.001 | |
36 months after end of treatment | ||
n | 334 | 313 |
LS mean (SE) | 13.487 (0.8269) | 12.791 (0.8714) |
LS mean difference (SE) [95% CI] | 0.696 (1.1244) [−1.510 to 2.902] | |
P value | 0.536 | |
EQ-5D-3L visual analogue scale scoref | ||
Baseline | ||
n | 652 | 653 |
Mean (SD) [95% CI] | 66.44 (23.778) [64.61 to 68.27] | 67.04 (23.542) [65.23 to 68.85] |
End of treatment | ||
n | 578 | 577 |
Mean (SD) [95% CI] | 74.11 (20.408) [72.44 to 75.78] | 76.89 (19.158) [75.33 to 78.46] |
36 months after end of treatment | ||
n | 281 | 266 |
Mean (SD) [95% CI] | 83.98 (16.553) [82.04 to 85.93] | 84.20 (16.317) [82.23 to 86.17] |
ABVD = doxorubicin-bleomycin-vinblastine-dacarbazine; AVD = doxorubicin-vinblastine-dacarbazine; BV = brentuximab vedotin; CI = confidence interval; CR = complete response; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; HL = Hodgkin lymphoma; IPFP = International Prognostic Factor Project; IRF = intendent review facility; ITT = intention-to-treat; LS = least squares; mPFS = modified progression-free survival; NE = not estimable; OS = overall survival; PFS = progression-free survival; QoL = quality of life; SD = standard deviation; SE = standard error.
aHR and 95% CI were based on a stratified Cox’s proportional hazard regression model with stratification factors region and number of IPFP risk factors at baseline with treatment as the explanatory variable in the model. A HR of less than 1 favours the BV + AVD group.
bMedian OS follow-up was calculated from the Kaplan-Meier method switching the OS event and censored status, i.e., OS event as censored and censored as OS event.
cMedian PFS follow-up was calculated from the Kaplan-Meier method switching the PFS according to investigator event and censored status, i.e., PFS according to investigator event as censored and censored as PFS according to investigator event.
dMedian mPFS follow-up was calculated from the Kaplan-Meier method switching the mPFS event and censored status, i.e., mPFS event as censored and censored as mPFS event.
eThe EORTC QLQ-C30 score range is 0 to 100. A high summary score represents a high quality of life.
fThe range of EQ-5D VAS scores is 0 to 100. A higher score indicates a more preferred health status.
Sources: ECHELON-1 original Clinical Study Report (data cut-off: April 20, 2017),13 ECHELON-1 Clinical Study Report Addendum 1 (data cut-off: June 01, 2021),14 Drug Reimbursement Review sponsor submission,16, and the sponsor’s Summary of Clinical Evidence.1
Table 16: Summary of Key Efficacy Results From the ECHELON-1 Trial (Stage III Only Versus Stage IV Only)
Outcome | Stage III only | Stage IV only | ||
|---|---|---|---|---|
BV + AVD (N = 237) | ABVD (N = 246) | BV + AVD (N = 425) | ABVD (N = 421) | |
OS (data cut-off date: June 1, 2021) | ||||
Number of patients contributing to analysis, n (%) | 237 (100) | 246 (100) | 425 (100) | 421 (100) |
Number with events (%) | 17 (7) | 20 (8) | 22 (5) | 43 (10) |
Number censored (%) | 220 (93) | 226 (92) | 403 (95) | 378 (90) |
Median, months (95% CI) | NE (NE to NE) | NE (NE to NE) | NE (NE to NE) | NE (NE to NE) |
Minimum, months | 0.0 | 0.1 | 0.3 | 0.0 |
Maximum, months | 97.5 | 97.9 | 100.6 | 94.6 |
Hazard ratio (95% CI)a | 0.863 (0.452 to 1.648) | 0.478 (0.286 to 0.799) | ||
P value between treatment groups | 0.654 | 0.004 | ||
P value between stage III only vs. stage IV only | NR | |||
Median follow-up time, months (95% CI)b | 72.8 (71.49 to 74.02) | 72.6 (69.75 to 74.71) | 73.6 (72.67 to 74.68) | 72.3 (70.87 to 73.63) |
Reason leading to OS event, n (%) | ||||
Death due to any cause | 17 (7) | 20 (8) | 22 (5) | 43 (10) |
Reason for censoring, n (%) | ||||
End of study | 78 (33) | 92 (37) | 130 (31) | 140 (33) |
Lost to follow-up | 26 (11) | 39 (16) | 39 (9) | 37 (9) |
Withdrawal by patient | 46 (19) | 48 (20) | 79 (19) | 93 (22) |
Other | 6 (3) | 5 (2) | 12 (3) | 10 (2) |
Alive on date of last contact | 142 (60) | 134 (54) | 273 (64) | 238 (57) |
PFS according to investigator | ||||
Number of patients contributing to analysis, n (%) | 237 (100) | 246 (100) | 425 (100) | 421 (100) |
Number with events (%) | 33 (14) | 54 (22) | 79 (19) | 103 (24) |
Number censored (%) | 204 (86) | 192 (78) | 346 (81) | 318 (76) |
Median (95% CI) | NE (NE to NE) | NE (NE to NE) | NE (NE to NE) | NE (NE to NE) |
Minimum, months | 0.0 | 0.0 | 0.0 | 0.0 |
Maximum, months | 97.5 | 97.9 | 100.6 | 94.6 |
Hazard ratio (95% CI)a | 0.603 (0.391 to 0.930) | 0.715 (0.534 to 0.959) | ||
P value between treatment groups | 0.021 | 0.024 | ||
P value between stage III only vs. stage IV only | NR | |||
Median follow-up time, months (95% CI)c | 72.8 (71.49 to 74.05) | 71.3 (68.57 to 73.95) | 73.4 (72.48 to 74.51) | 71.8 (70.47 to 73.40) |
Reason leading to PFS event, n (%) | ||||
Progressive disease | 25 (11) | 44 (18) | 71 (17) | 83 (20) |
Death due to any cause | 8 (3) | 10 (4) | 8 (2) | 20 (5) |
Reason for censoring, n (%) | ||||
No baseline and/or no postbaseline assessment | 5 (2) | 7 (3) | 3 (< 1) | 14 (3) |
PFS event after more than 1 missed visit | 3 (1) | 4 (2) | 7 (2) | 4 (< 1) |
Treatment discontinuation for undocumented disease progression | 0 | 0 | 0 | 0 |
Loss to follow-up | 30 (13) | 42 (17) | 43 (10) | 30 (7) |
Withdrawal by patient | 39 (16) | 34 (14) | 59 (14) | 67 (16) |
No documented PFS event | 127 (54) | 105 (43) | 234 (55) | 203 (48) |
mPFS by IRF | ||||
Number of patients contributing to analysis, n (%) | 237 (100) | 246 (100) | 425 (100) | 421 (100) |
Number with events (%) | 40 (17) | 43 (17) | 77 (18) | 102 (24) |
Number censored (%) | 197 (83) | 203 (83) | 348 (82) | 319 (76) |
Median, months (95% CI) | 48.2 (48.2 to NE) | NE (NE to NE) | NE (NE to NE) | NE (NE to NE) |
Minimum, months | 0.0 | 0.0 | 0.0 | 0.0 |
Maximum, months | 48.5 | 49.3 | 48.8 | 43.9 |
Hazard ratio (95% CI)a | 0.923 (0.600 to 1.420) | 0.712 (0.530 to 0.957) | ||
P value between treatment groups | 0.716 | 0.024 | ||
P value between stage III only vs. stage IV only | NR | |||
Median follow-up time, months (95% CI)d | 24.7 (22.34 to 25.10) | 24.6 (21.13 to 24.97) | 24.9 (24.71 to 25.10) | 25.0 (24.64 to 25.17) |
Reason leading to mPFS event, n (%) | ||||
Progressive disease | 29 (12) | 33 (13) | 61 (14) | 69 (16) |
Death due to any cause | 9 (4) | 8 (3) | 9 (2) | 14 (3) |
Receipt of additional therapy after non-CR | 2 (< 1) | 2 (< 1) | 7 (2) | 19 (5) |
Reason for censoring, n (%) | ||||
No baseline and/or no postbaseline assessment | 5 (2) | 7 (3) | 5 (1) | 15 (4) |
mPFS event after more than 1 missed visit | 1 (< 1) | 2 (< 1) | 0 | 1 (< 1) |
Treatment discontinuation for undocumented disease progression | 2 (< 1) | 2 (< 1) | 2 (< 1) | 2 (< 1) |
Loss to follow-up | 5 (2) | 12 (5) | 9 (2) | 8 (2) |
Withdrawal by patient | 9 (4) | 9 (4) | 13 (3) | 11 (3) |
No documented mPFS event | 175 (74) | 171 (70) | 319 (75) | 282 (67) |
ABVD = doxorubicin-bleomycin-vinblastine-dacarbazine; AVD = doxorubicin-vinblastine-dacarbazine; BV = brentuximab vedotin; CI = confidence interval; IRF = intendent review facility; ITT = intention-to-treat; mPFS = modified progression-free survival; NE = not estimable; OS = overall survival; PFS = progression-free survival; vs. = versus.
Note: OS, PFS according to investigator, the percentage of patients alive without HL, European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30, and EQ-5D-3L were obtained from the data with a cut-off date of June 1, 2021, while mPFS according to IRF, CR rate according to IRF were from the data with a cut-off date of April 20, 2017. Subgroup analyses for OS and PFS according to investigator were obtained from the data with a cut-off date of June 1, 2021, while subgroup analyses for mPFS according to IRF was from the data with a cut-off date of April 20, 2017.
aThe HR and 95% CI were based on an unstratified Cox’s proportional hazard regression model. A hazard ratio of less than 1 favours the BV + AVD group.
bMedian OS follow-up was calculated from the Kaplan-Meier method switching the OS event and censored status, i.e., OS event as censored and censored as OS event.
cMedian PFS follow-up was calculated from the Kaplan-Meier method switching the PFS according to investigator event and censored status, i.e., PFS according to investigator event as censored and censored as PFS according to investigator event.
dThe median mPFS follow-up was calculated from the Kaplan-Meier method switching the mPFS event and censored status, i.e., mPFS event as censored and censored as mPFS event.
Source: ECHELON-1 original Clinical Study Report (data cut-off: April 20, 2017),13 ECHELON-1 Clinical Study Report Addendum 1 (data cut-off: June 01, 2021),14 Drug Reimbursement Review sponsor submission,16, and the sponsor’s Summary of Clinical Evidence.1
Overall Survival
As of the data cut-off date of June 1, 2021, the median follow-up times were 73.3 months (95% CI, 72.61 to 74.05) in the BV + AVD group and 72.4 months (95% CI, 71.10 to 73.63) in the ABVD group (Table 15). In the ITT population, the HR for OS was 0.59 (95% CI, 0.396 to 0.879; P = 0.009), indicating that patients treated with BV + AVD had a 41% lower risk of death than those treated with BV + AVD. The median OS was not reached for patients with advanced-stage classical HL for either the BV + AVD group or the ABVD group. The absolute difference in number of events between the BV + AVD and ABVD arms was 4%, favouring BV + AVD (6% versus 10%). The proportion of patients who were alive on the date of last contact was 63% (415 of 664) in the BV + AVD group and 56% (372 of 670%) in the ABVD group, respectively. Of note, 10% (65 of 664) and 19% (126 of 664) of patients in the BV + AVD group were censored due to loss to follow-up and withdrawal by participants, respectively, while these proportions were 11% (76 of 670) and 21% (141 of 670), respectively, for patients receiving ABVD.
The HRs for OS were 0.863 (95% CI, 0.452 to 1.648; P = 0.654) for patients with stage III classical HL and 0.478 (95% CI, 0.286 to 0.799; P = 0.004) for patients with stage IV classical HL (Table 16). Kaplan-Meier plots of OS for the ITT population, the subpopulation with stage III classical HL, and the subpopulation with stage IV classical HL are presented in Figure 1, Figure 2, and Figure 3, respectively.
Figure 1: Kaplan-Meier Plot of OS for Patients With Advanced-Stage Classical HL (ITT Population, Data Cut-Off Date: June 1, 2021)
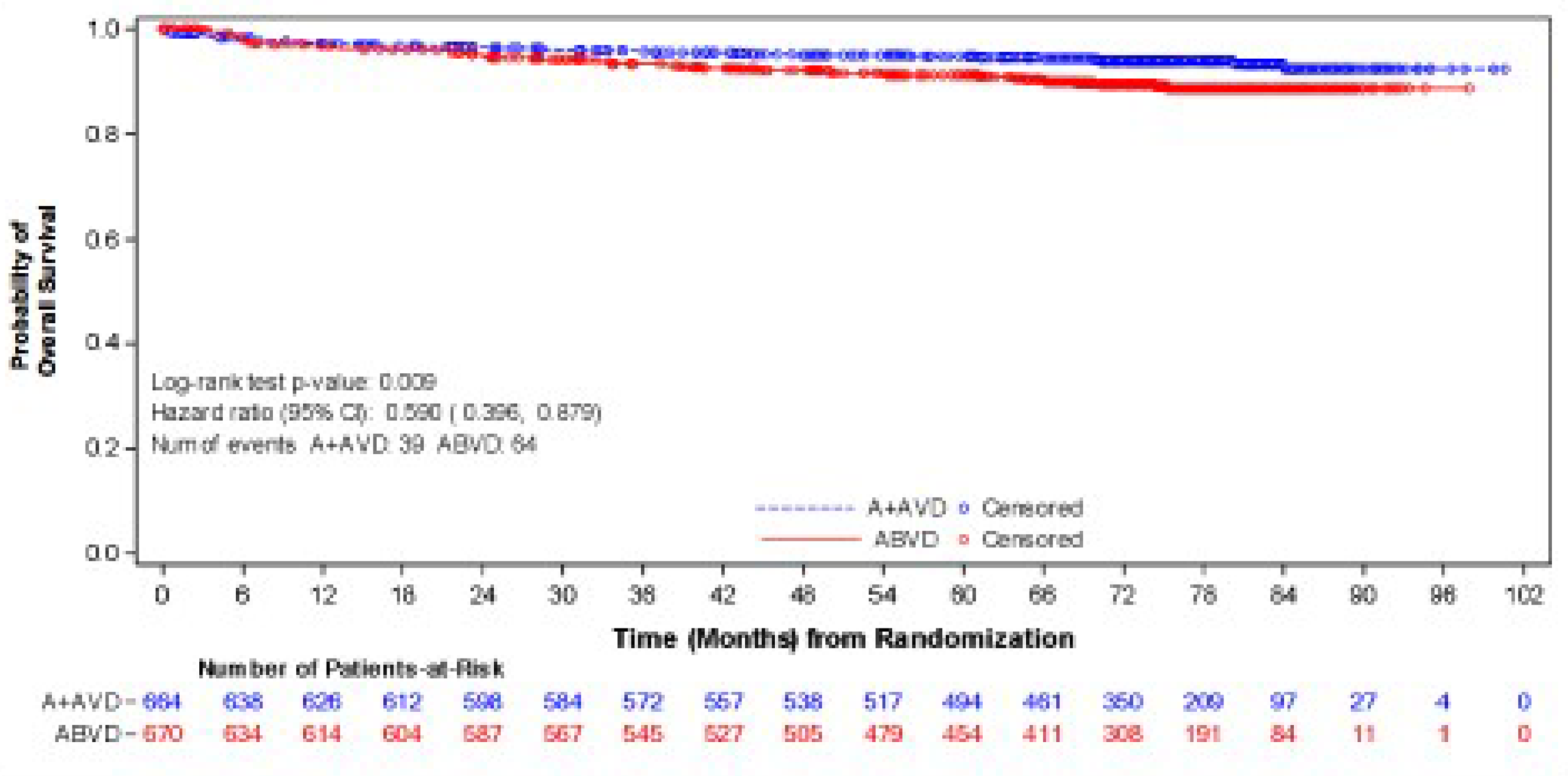
ABVD (denoted as A + AVD in the sponsor’s Clinical Study Report) = doxorubicin-bleomycin-vinblastine-dacarbazine; AVD = doxorubicin-vinblastine-dacarbazine; BV = brentuximab vedotin; CI = confidence interval; HL = Hodgkin lymphoma; HR = hazard ratio; IPFP = International Prognostic Factor Project; ITT = intention-to-treat; OS = overall survival.
Note: HR ([BV + AVD] / ABVD) and 95% CI were based on a stratified Cox’s proportional hazard regression model with the stratification factors, region, and number of IPFP risk factors at baseline with treatment as the explanatory variable in the model. An HR of less than 1 favours the BV + ABVD arm.
Sources: ECHELON-1 Clinical Study Report Addendum 1 (data cut-off: June 01, 2021)14 and the sponsor’s Summary of Clinical Evidence.1
Figure 2: Kaplan-Meier Plot of OS for Patients With Stage III Classical HL (Data Cut-Off Date: June 1, 2021)
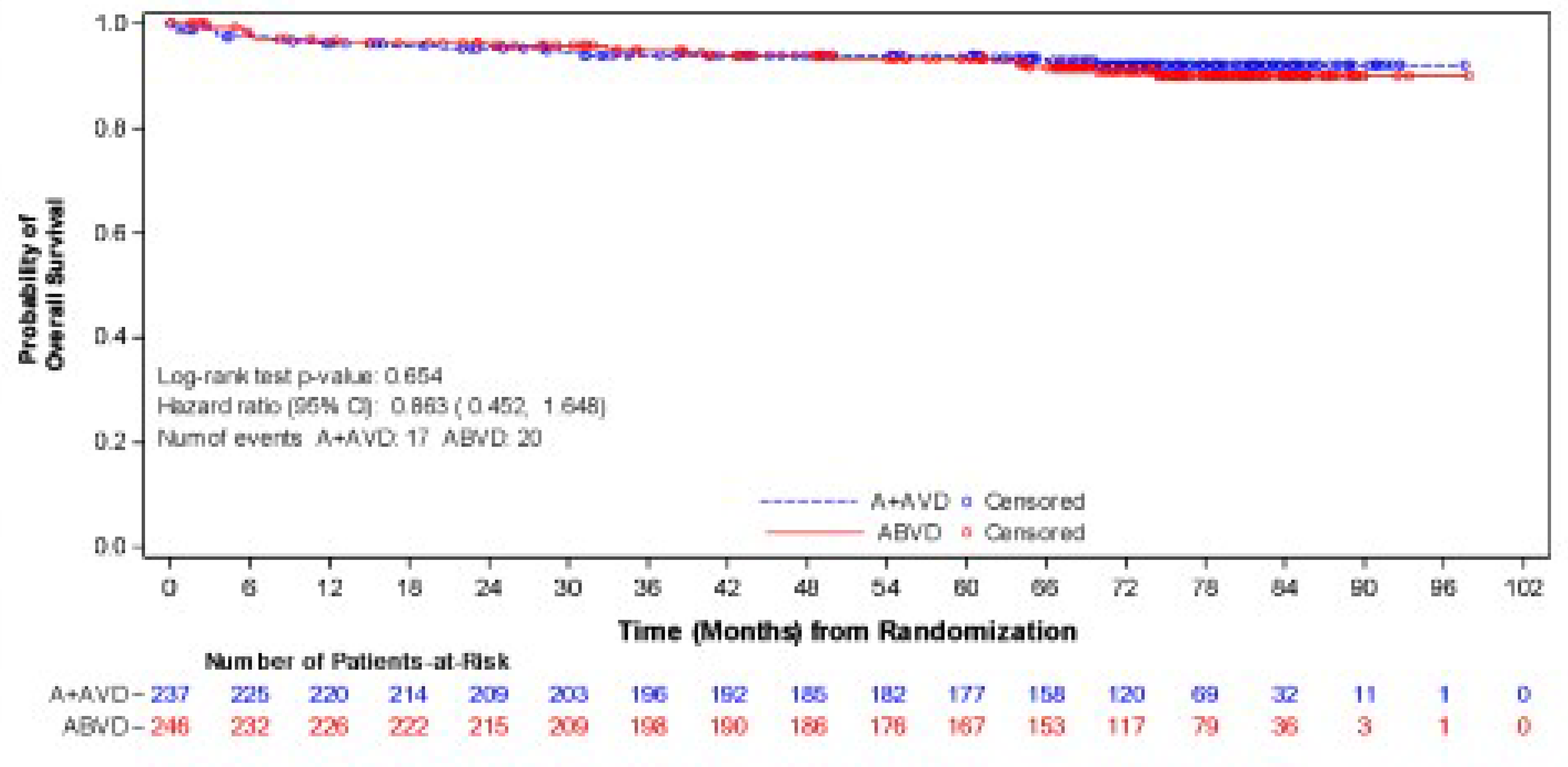
ABVD (denoted as A + AVD in the sponsor’s Clinical Study Report) = doxorubicin-bleomycin-vinblastine-dacarbazine; AVD = doxorubicin-vinblastine-dacarbazine; BV = brentuximab vedotin; CI = confidence interval; HL = Hodgkin lymphoma; HR = hazard ratio; ITT = intention-to-treat; OS = overall survival.
Note: HR ([BV + AVD] / ABVD) and 95% CI were based on unstratified Cox’s proportional hazard regression model. An HR of less than 1 favours the BV + ABVD arm.
Sources: ECHELON-1 Clinical Study Report Addendum 1 (data cut-off: June 01, 2021)14 and the sponsor’s Summary of Clinical Evidence.1
Figure 3: Kaplan-Meier Plot of OS for Patients With Stage IV Classical HL (Data Cut-Off Date: June 1, 2021)
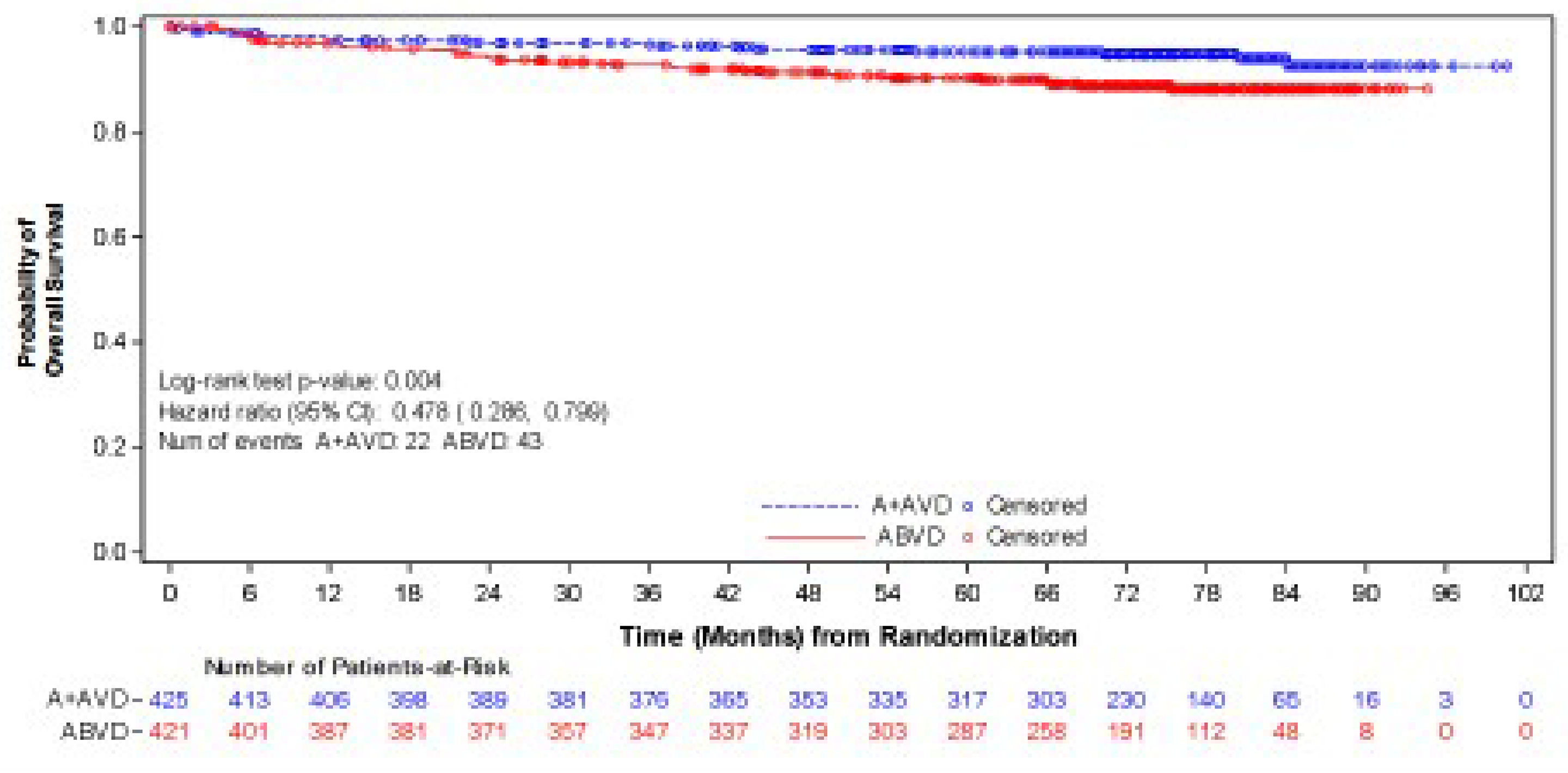
ABVD (denoted as A + AVD in the sponsor’s Clinical Study Report) = doxorubicin-bleomycin-vinblastine-dacarbazine; AVD = doxorubicin-vinblastine-dacarbazine; BV = brentuximab vedotin; CI = confidence interval; HL = Hodgkin lymphoma; HR = hazard ratio; ITT = intention-to-treat; OS = overall survival.
Note: HR ([BV + AVD] / ABVD) and 95% CI were based on unstratified Cox’s proportional hazard regression model. An HR of less than 1 favours the BV + ABVD arm.
Sources: ECHELON-1 Clinical Study Report Addendum 1 (data cut-off: June 01, 2021)14 and the sponsor’s Summary of Clinical Evidence.1
New OS Data — 2023 Descriptive Analysis15
As of the data cut-off date of March 11, 2023, the descriptive analysis had a median follow-up of approximately 88 months for the ITT population (Table 17). The median follow-up durations were 89.7 months (95% CI, 86.57 to 90.55) for the BV + AVD group and 86.3 months (95% CI, 84.53 to 89.33) for the ABVD group. This analysis included 111 OS events (deaths): 44 deaths (7%) occurred in the BV + AVD group and 67 (10%) in the ABVD group. The median OS was not reached for either group. The HR for OS was 0.61 (95% CI, 0.414 to 0.892).
In the stage III subgroup (Table 18), the median OS was not reached for either treatment arm, and the HR for OS was 1.004 (95% CI, 0.540 to1.866) for BV + AVD patients compared with ABVD patients. In the stage IV subgroup, the median OS was not reached for either treatment arm, and the HR for OS was 0.48 (95% CI, 0.291 to 0.784) for BV + AVD patients compared with ABVD patients. Kaplan-Meier plots of OS for the ITT population, the subpopulation with stage III classical HL, and the subpopulation with stage IV classical HL are presented in Figure 4, Figure 5, and Figure 6, respectively.
Table 17: Summary of OS Results From the ECHELON-1 Trial (ITT Population) — 2023 Descriptive Analysis (Data Cut-Off Date: March 11, 2023)
OS | BV + AVD (N = 664) | ABVD (N = 670) |
|---|---|---|
Number with events (%) | 44 (7) | 67 (10) |
Number censored (%) | 620 (93) | 603 (90) |
Median (95% CI), months | NE (115.1 to NE) | NE (NE to NE) |
Minimum, months | 0.0 | 0.0 |
Maximum, months | 118.0 | 118.7 |
Hazard ratio (95% CI)a | 0.607 (0.414 to 0.892) | |
Descriptive P value between treatment groups | 0.010 | |
Median follow-up time, months (95% CI)b | 89.7 (86.57 to 90.55) | 86.3 (84.53 to 89.33) |
Reason leading to OS event, n (%) | ||
Death due to any cause | 44 (7) | 67 (10) |
Reason for censoring, n (%) | ||
End of study | 247 (37) | 255 (38) |
Lost to follow-up | 86 (13) | 86 (13) |
Withdrawal by patient | 142 (21) | 149 (22) |
Other | 19 (3) | 20 (3) |
Alive on date of last contact | 373 (56) | 348 (52) |
ABVD = doxorubicin-bleomycin-vinblastine-dacarbazine; AVD = doxorubicin-vinblastine-dacarbazine; BV = brentuximab vedotin; CI = confidence interval; ITT = intention-to-treat; NE = not estimable; OS = overall survival.
aThe HR ([BV + AVD] / ABVD) and 95% CI were based on a stratified Cox’s proportional hazard regression model. A hazard ratio of less than 1 favours the BV + AVD group.
bThe median OS follow-up was calculated from the Kaplan-Meier method switching the OS event and censored status (i.e., OS event as censored and censored as OS event).
Source: EMA Assessment Report: Adcetris.15
Table 18: Summary of OS Results From the ECHELON-1 Trial in Stage III Versus Stage IV Disease Subgroups — 2023 Descriptive Analysis (Data Cut-Off Date: March 11, 2023)
OS | Stage III only | Stage IV only | ||
|---|---|---|---|---|
BV + AVD (N = 237) | ABVD (N = 246) | BV + AVD (N = 425) | ABVD (N = 421) | |
Number with events (%) | 20 (8) | 20 (8) | 22 (5) | 43 (10) |
Number censored (%) | 217 (92) | 226 (92) | 403 (95) | 378 (90) |
Median, months (95% CI) | NE (NE to NE) | NE (NE to NE) | NE (NE to NE) | NE (NE to NE) |
Minimum, months | 0.0 | 0.1 | 0.3 | 0.0 |
Maximum, months | 116.1 | 118.7 | 100.6 | 94.6 |
Hazard ratio (95% CI)a | 1.004 (0.540 to 1.866) | 0.478 (0.286 to 0.799) | ||
Descriptive P value between treatment groups | 0.990 | 0.004 | ||
Median follow-up time, months (95% CI)b | 89.6 (83.75 to 90.87) | 85.0 (81.35 to 89.53) | 73.6 (72.67 to 74.68) | 72.3 (70.87 to 73.63) |
Reason leading to OS event, n (%) | — | — | — | — |
Death due to any cause | 17 (7) | 20 (8) | 22 (5) | 43 (10) |
Reason for censoring, n (%) | — | — | — | — |
End of study | 78 (33) | 92 (37) | 130 (31) | 140 (33) |
Lost to follow-up | 26 (11) | 39 (16) | 39 (9) | 37 (9) |
Withdrawal by patient | 46 (19) | 48 (20) | 79 (19) | 93 (22) |
Other | 6 (3) | 5 (2) | 12 (3) | 10 (2) |
Alive on date of last contact | 142 (60) | 134 (54) | 273 (64) | 238 (57) |
ABVD = doxorubicin-bleomycin-vinblastine-dacarbazine; AVD = doxorubicin-vinblastine-dacarbazine; BV = brentuximab vedotin; CI = confidence interval; NE = not estimable; OS = overall survival.
aThe HR ([BV + AVD] / ABVD) and 95% CI were based on an unstratified Cox’s proportional hazard regression model. A HR of less than 1 favours the BV + AVD group.
bThe median OS follow-up was calculated from the Kaplan-Meier method switching the OS event and censored status (i.e., OS event as censored and censored as OS event).
Source: EMA Assessment Report: Adcetris.15
Figure 4: Kaplan-Meier Plot of OS for Patients With Advanced-Stage Classical HL (ITT Population) — 2023 Descriptive Analysis (Data Cut-Off Date: March 11, 2023)
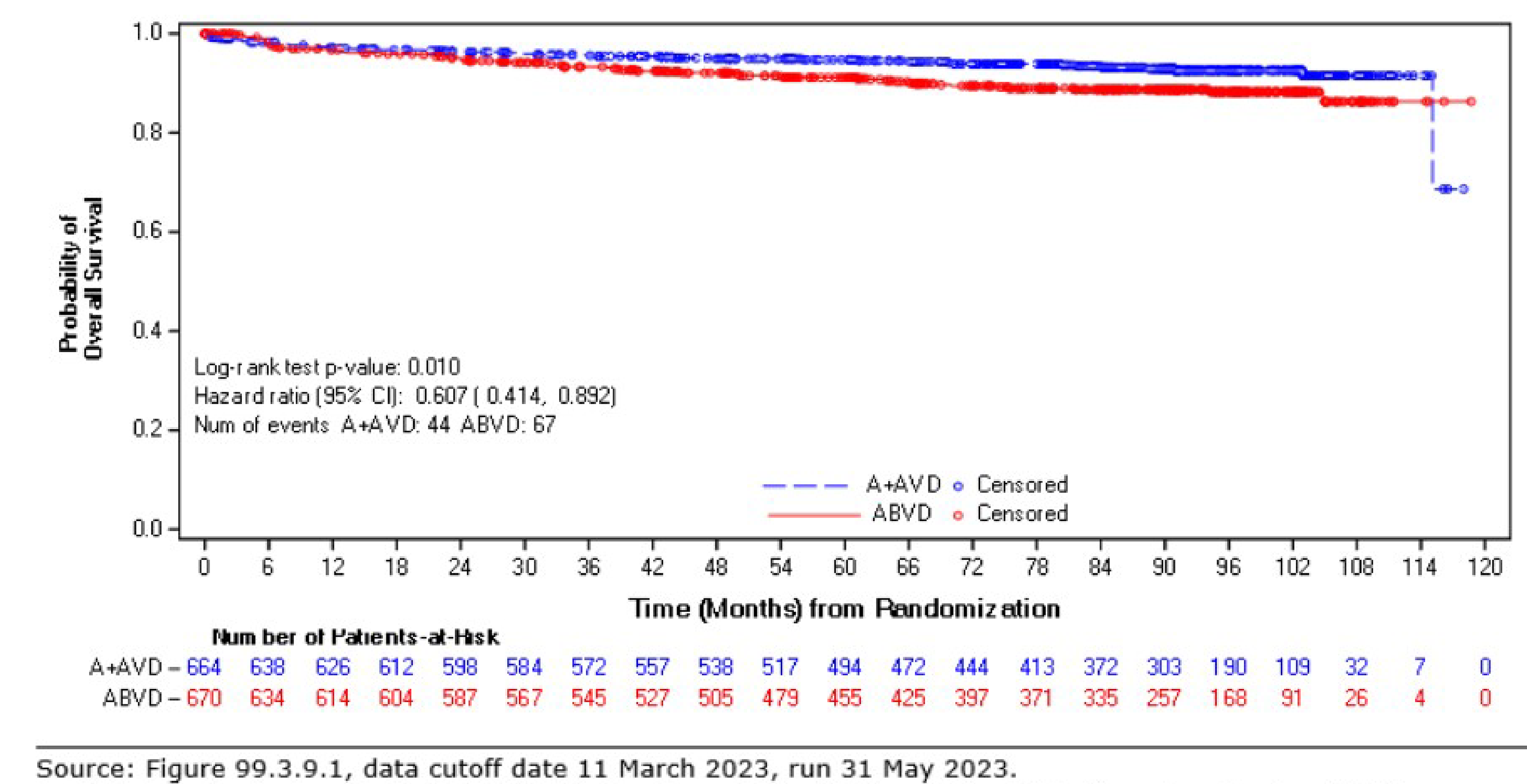
ABVD (denoted as A + AVD) = doxorubicin-bleomycin-vinblastine-dacarbazine; AVD = doxorubicin-vinblastine-dacarbazine; BV = brentuximab vedotin; CI = confidence interval; HL = Hodgkin lymphoma; ITT = intention-to-treat; OS = overall survival.
Note: The hazard ratio ([BV + AVD] / ABVD) and 95% CI were based on a stratified Cox’s proportional hazard regression model. A HR of less than 1 favours the BV + ABVD arm.
Source: EMA Assessment Report: Adcetris.15
Figure 5: Kaplan-Meier Plot of OS for Patients With Stage III Classical HL — 2023 Descriptive Analysis (Data Cut-Off Date: March 11, 2023)
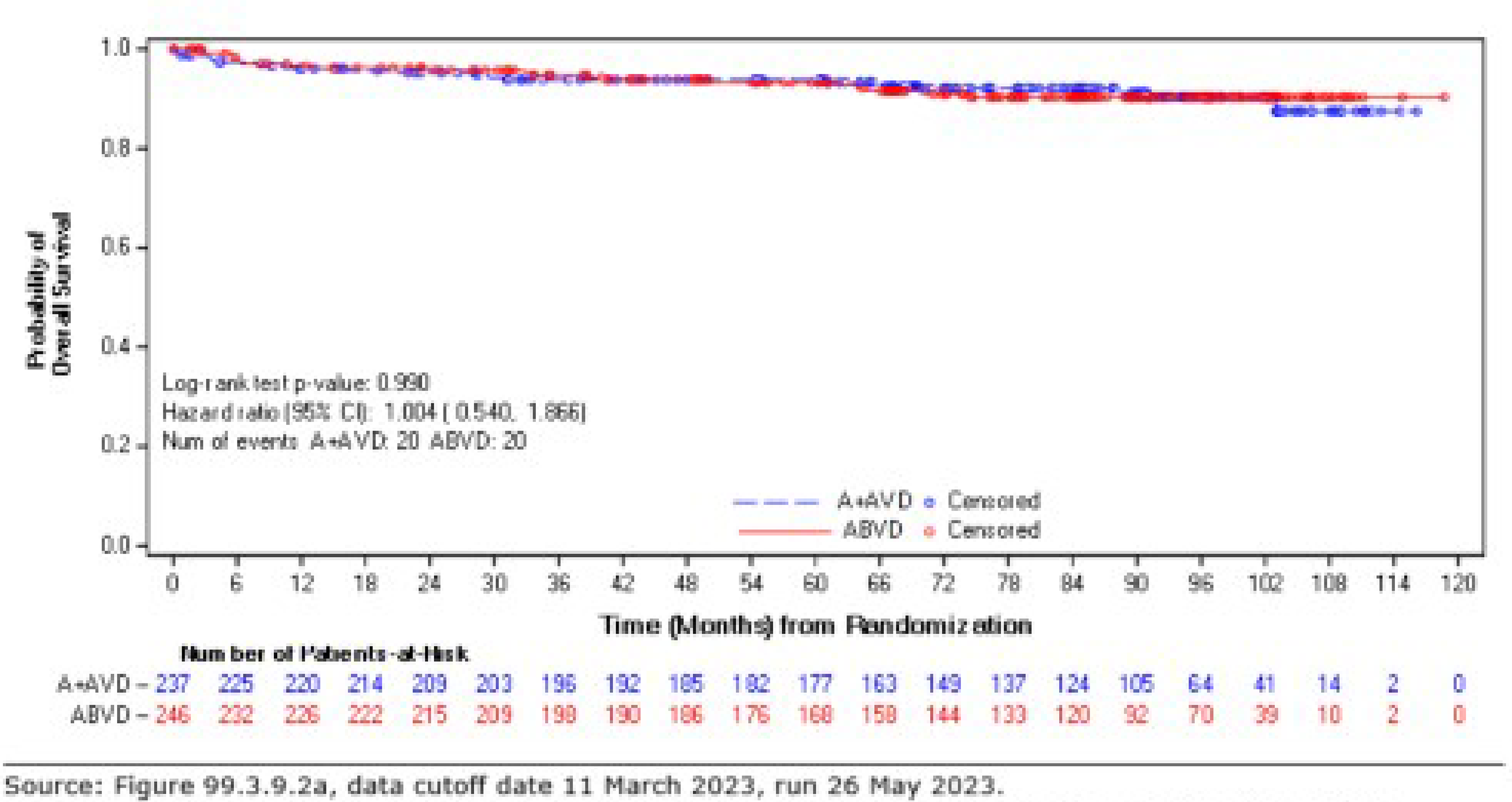
ABVD (denoted as A + AVD) = doxorubicin-bleomycin-vinblastine-dacarbazine; AVD = doxorubicin-vinblastine-dacarbazine; BV = brentuximab vedotin; CI = confidence interval; HL = Hodgkin lymphoma; OS = overall survival.
Note: The HR ([BV + AVD] / ABVD) and 95% CI were based on a stratified Cox’s proportional hazard regression model. A HR of less than 1 favours the BV + ABVD arm.
Source: EMA Assessment Report: Adcetris.15
Figure 6: Kaplan-Meier Plot of OS for Patients With Stage IV Classical HL — 2023 Descriptive Analysis (Data Cut-Off Date: March 11, 2023)
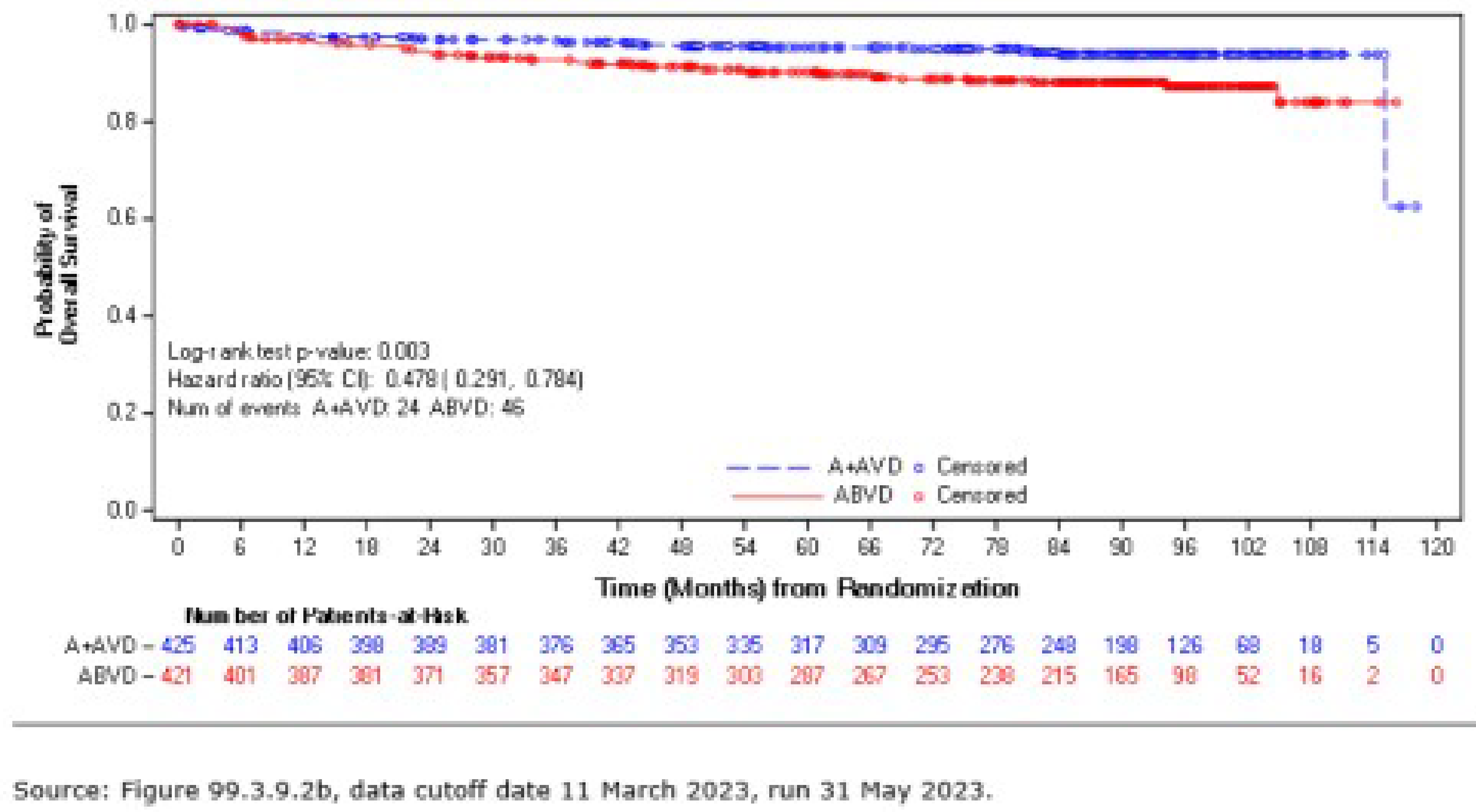
ABVD (denoted as A + AVD) = doxorubicin-bleomycin-vinblastine-dacarbazine; AVD = doxorubicin-vinblastine-dacarbazine; BV = brentuximab vedotin; CI = confidence interval; HL = Hodgkin lymphoma; OS = overall survival.
Note: The HR ([BV + AVD] / ABVD) and 95% CI were based on a stratified Cox’s proportional hazard regression model. A HR of less than 1 favours the BV + ABVD arm.
Source: EMA Assessment Report: Adcetris.15
Alive Without HL
In the ITT population as of the data cut-off date June 1, 2021, the 3-year rates of being alive without HL were about 96% (546 of 567) in the BV + AVD group and 93% (503 of 540) in the ABVD group (Table 15). The 5-year rates of being alive without HL were about 94% (450 of 480) in the BV + AVD group and 92% (408 of 443) in the ABVD group. No subgroup analyses by disease stage were reported for this efficacy end point.
PFS According to Investigator
As of the data cut-off date June 1, 2021, the median follow-ups were 73.2 months (95% CI, 72.48 to 74.05) in the BV + AVD group and 71.6 months (95% CI, 70.37 to 72.87) in the ABVD group (Table 15). In the ITT population, the HR of PFS according to investigator was 0.678 (95% CI, 0.532 to 0.863; P = 0.002), indicating that patients treated with BV + AVD had a 33.2% lower risk of experiencing PFS events compared with those treated with BV +AVD. There was a 7% absolute difference in number of PFS events between the BV + AVD group and the ABVD group, favouring BV + AVD (17% versus 24%, respectively). The median PFS according to investigator was not reached for patients with advanced-stage classical HL for either the BV + AVD group or the ABVD group. The most common reason for PFS event was progressive disease, and the most common reason for being censored in both treatment groups was the absence of a documented PFS event. Of note, 11% (73 of 664) and 15% (98 of 664) of patients in the BV + AVD group were censored due to loss to follow-up and withdrawal by participants, respectively, while these proportions were 11% (72 of 670) and 15% (101 of 670), respectively, for patients receiving ABVD.
The HRs for PFS according to investigator were 0.603 (95% CI, 0.391 to 0.930; P = 0.021) for patients with stage III classical HL and 0.715 (95% CI, 0.534 to 0.959; P = 0.024) for patients with stage IV classical HL (Table 16). Kaplan-Meier plots of PFS according to investigator for the ITT population, the subpopulation with stage III classical HL, and the subpopulation with stage IV classical HL are presented in Figure 7, Figure 8, and Figure 9, respectively.
Figure 7: Kaplan-Meier Plot of PFS According to Investigator for Patients With Advanced-Stage Classical HL (ITT Population)
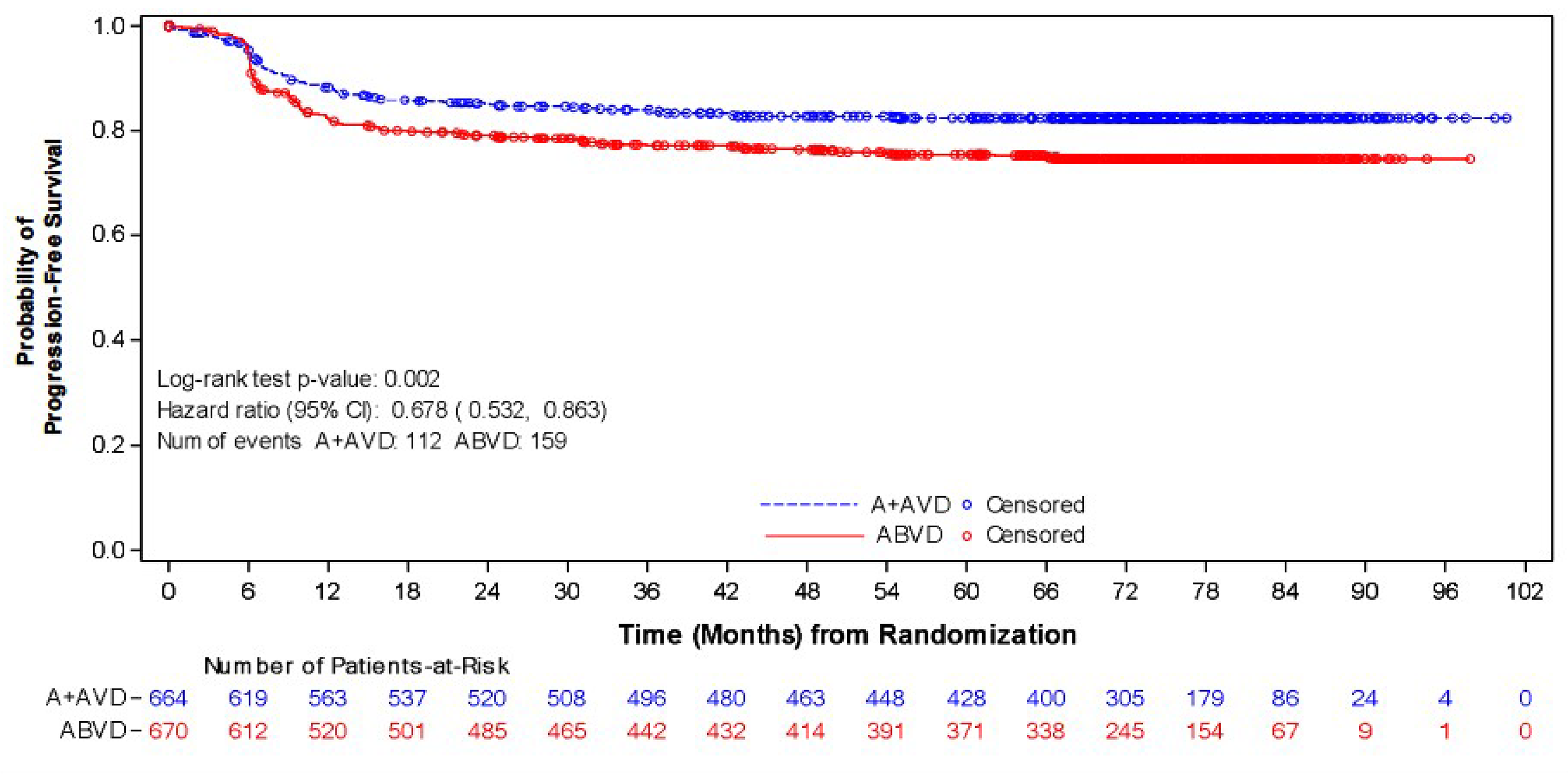
ABVD (denoted as A + AVD in the sponsor’s Clinical Study Report) = doxorubicin-bleomycin-vinblastine-dacarbazine; AVD = doxorubicin-vinblastine-dacarbazine; BV = brentuximab vedotin; CI = confidence interval; HL = Hodgkin lymphoma; HR = hazard ratio; ITT = intention-to-treat; PFS = progression-free survival.
Note: The HR ([BV + AVD] / ABVD) and 95% CI were based on a stratified Cox’s proportional hazard regression model with the stratification factors, region, and number of IPFP risk factors at baseline with treatment as the explanatory variable in the model. An HR of less than 1 favours the BV + ABVD arm.
Sources: ECHELON-1 Clinical Study Report Addendum 1 (data cut-off: June 01, 2021)14 and the sponsor’s Summary of Clinical Evidence.1
Figure 8: Kaplan-Meier Plot of PFS According to Investigator for Patients With Stage III Classical HL
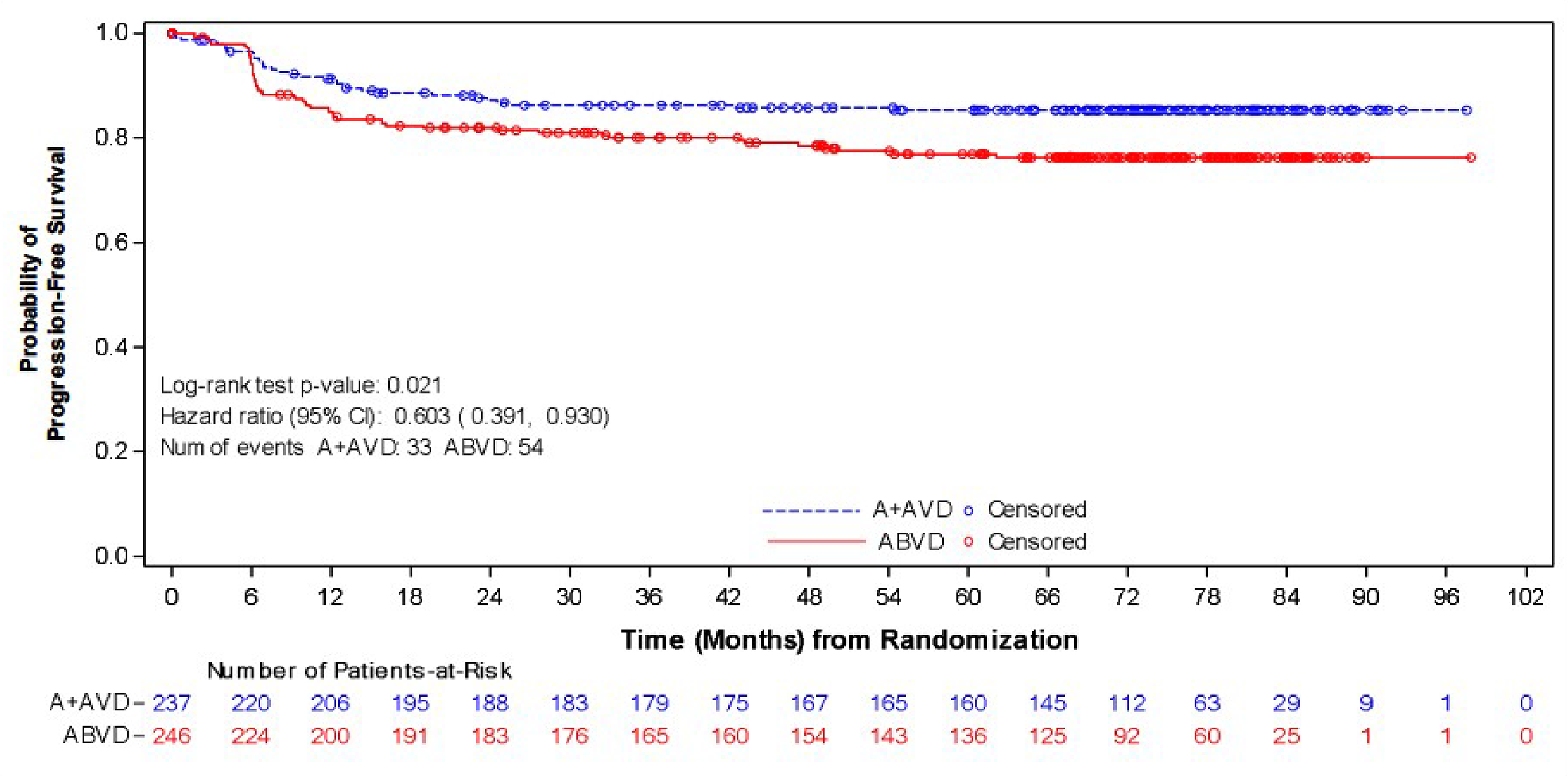
ABVD (denoted as A + AVD in the sponsor’s Clinical Study Report) = doxorubicin-bleomycin-vinblastine-dacarbazine; BV + AVD = brentuximab vedotin plus doxorubicin, vinblastine, dacarbazine; HL = Hodgkin lymphoma; HR = hazard ratio; PFS = progression-free survival.
Note: The HR ([BV + AVD] / ABVD) and 95% CI were based on unstratified Cox’s proportional hazard regression model. A HR of less than 1 favours the BV + ABVD arm.
Sources: ECHELON-1 Clinical Study Report Addendum 1 (data cut-off: June 01, 2021)14 and the sponsor’s Summary of Clinical Evidence.1
Figure 9: Kaplan-Meier Plot of PFS According to Investigator for Patients With Stage IV Classical HL
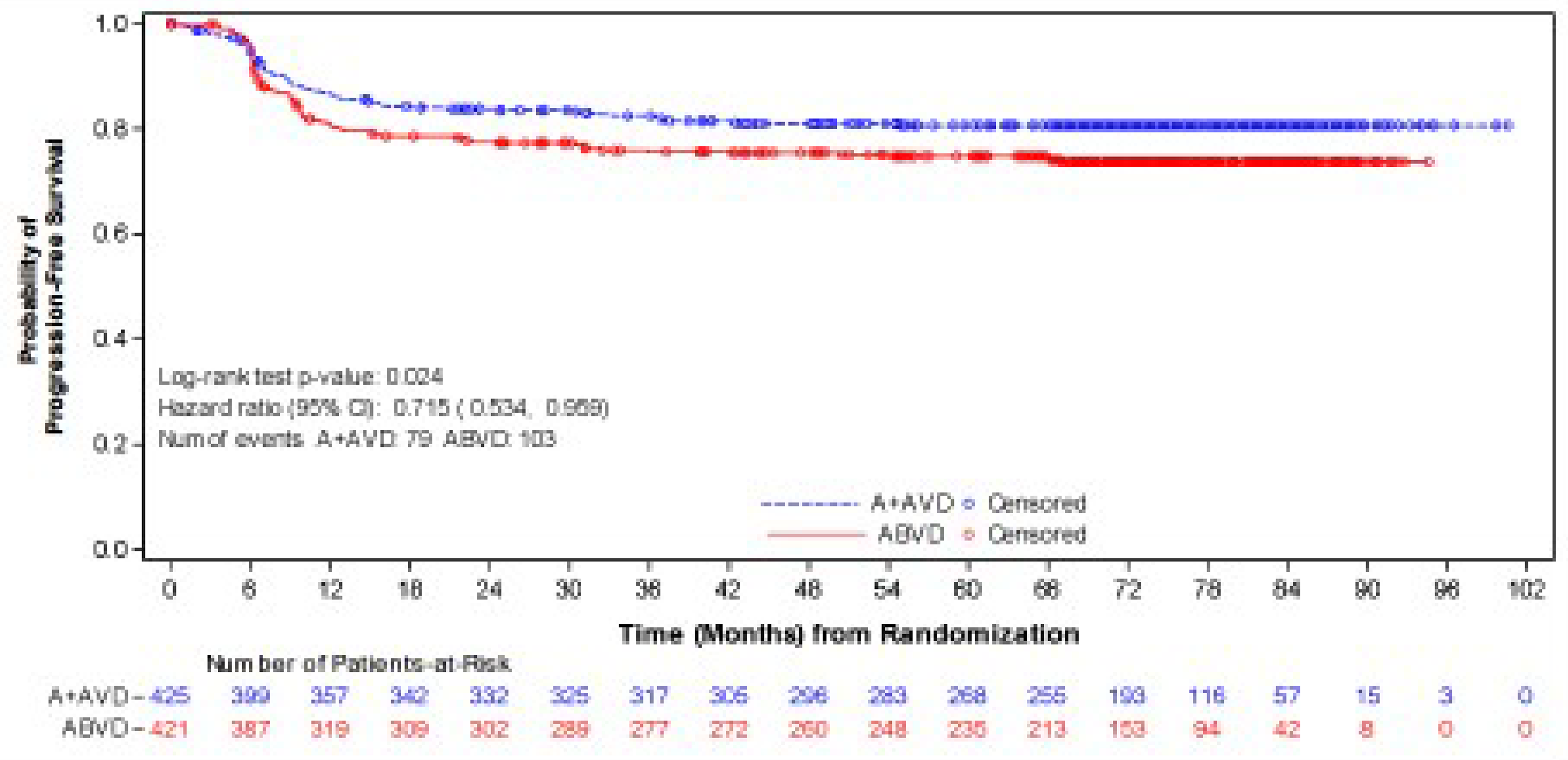
ABVD (denoted as A + AVD in the sponsor’s Clinical Study Report) = doxorubicin-bleomycin-vinblastine-dacarbazine; AVD = doxorubicin-vinblastine-dacarbazine; BV = brentuximab vedotin; CI = confidence interval; HL = Hodgkin lymphoma; HR = hazard ratio; PFS = progression-free survival.
Note: The HR ([BV + AVD] / ABVD) and 95% CI were based on unstratified Cox’s proportional hazard regression model. An HR of less than 1 favours the BV + ABVD arm.
Sources: ECHELON-1 Clinical Study Report Addendum 1 (data cut-off: June 01, 2021)14 and the sponsor’s Summary of Clinical Evidence.1
Modified PFS According to IRF
As of the data cut-off date April 20, 2017, the median mPFS was not reached in either the BV + AVD group or the ABVD group (Table 15). In the ITT population, the HR for mPFS according to IRF was 0.770 (95% CI, 0.603 to 0.982; P = 0.035). The absolute difference in number of mPFS events between the BV + AVD arm and the ABVD arm was 4%, favouring BV + AVD (18% versus 22%). The most common reason for being censored in both treatment groups was no documented mPFS event.
The HR for mPFS according to IRF was 0.923 (95% CI, 0.600 to 1.420; P = 0.716) for patients with stage III classical HL and 0.712 (95% CI, 0.530 to 0.957; P = 0.024) for patients with stage IV classical HL (Table 16). Only the Kaplan-Meier plot of mPFS according to IRF for the ITT population was available (Figure 10).
Figure 10: Kaplan-Meier Plot of mPFS According to IRF for Patients With Advanced-Stage Classical HL (ITT Population)
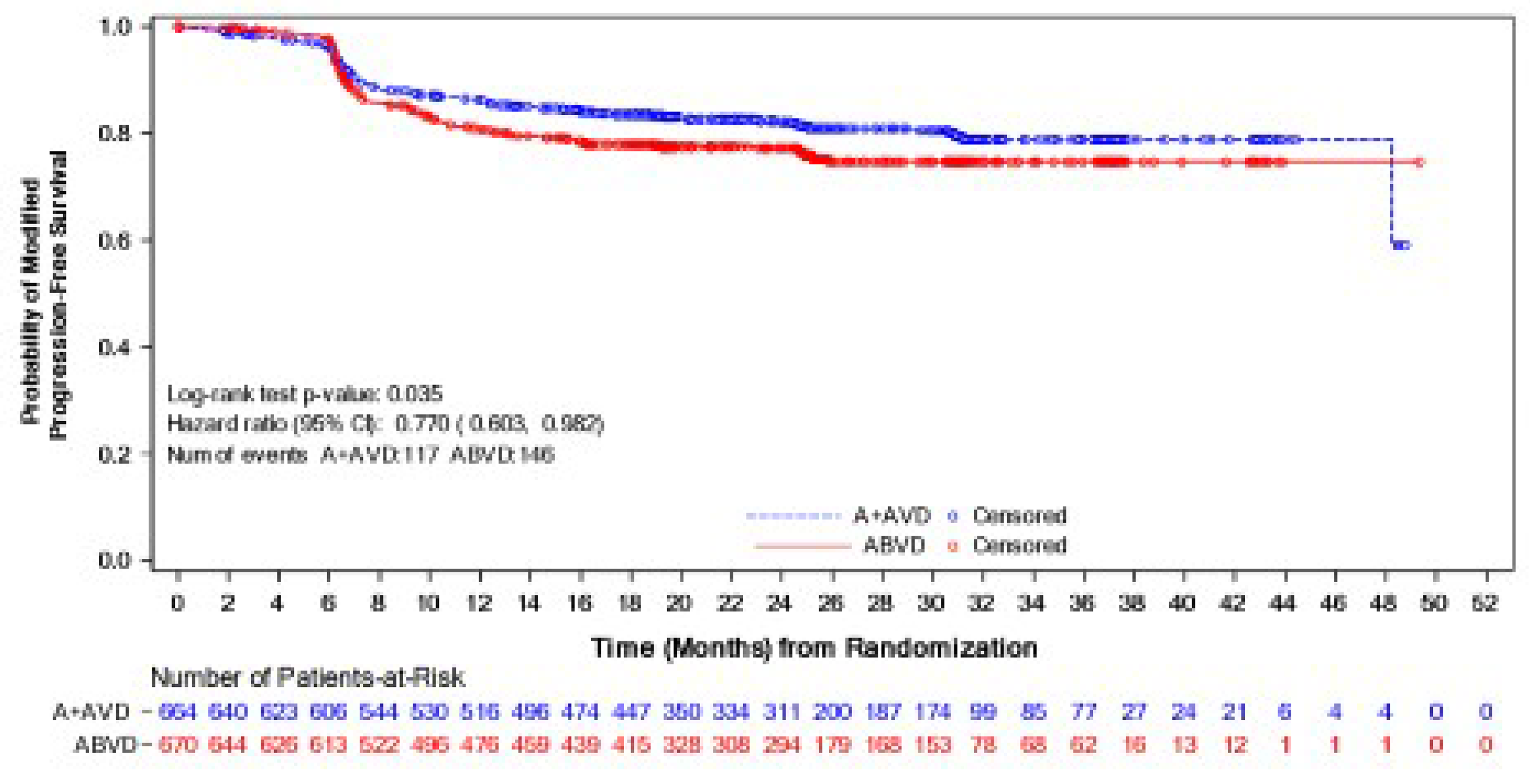
ABVD (denoted as A + AVD in the sponsor’s Clinical Study Report) = doxorubicin-bleomycin-vinblastine-dacarbazine; AVD = doxorubicin-vinblastine-dacarbazine; BV = brentuximab vedotin; CI = confidence interval; HL = Hodgkin lymphoma; HR = hazard ratio; IRF = independent review facility; ITT = intention-to-treat; mPFS = modified progression-free survival.
Note: The HR ([BV + AVD] / ABVD) and 95% CI were based on a stratified Cox’s proportional hazard regression model with the stratification factors, region, and number of IPFP risk factors at baseline with treatment as the explanatory variable in the model. An HR of less than 1 favours the BV + ABVD arm.
Sources: ECHELON-1 original Clinical Study Report (data cut-off: April 20, 2017)13 and the sponsor’s Summary of Clinical Evidence.1
CR Rate According to IRF
As of April 20, 2017, in the ITT population, the CR rates after the end of front-line therapy were 73% in the BV + AVD group and 71% in the ABVD group, with a corresponding relative risk of 1.038 (95% CI, 0.97 to 1.11) (Table 15). No subgroup analyses by disease stage were reported for CR rate according to IRF.
EORTC QLQ-C30
Summary and subscale scores for the EORTC QLQ-C30 were reported in the ECHELON-1 trial. Table 15 summarizes the summary scores as well as the EORTC QLQ-C30 Global Health Status/quality-of-life subscale scores at baseline, end of treatment, and 36 months after end of treatment. The mean EORTC QLQ-C30 summary scores and the Global Health Status/quality-of-life subscale scores were similar between the BV + AVD group and the ABVD group between 3 months and 36 months after end of treatment. No subgroup analyses by disease stage were reported for this efficacy end point.
EQ-5D-3L
Visual analogue scale (VAS) scores for the EQ-5D-3L were reported in the ECHELON-1 trial. Table 15 summarizes the VAS scores at baseline, end of treatment, and 36 months after end of treatment. No subgroup analyses by disease stage were reported for this efficacy end point. The scores were similar between the BV + AVD group and the ABVD group between 3 months and 36 months after end of treatment. No subgroup analyses by disease stage were reported for this efficacy end point.
Harms
A summary of harms in the ECHELON-1 trial is provided in Table 19. Deaths during the post-treatment follow-up period, resolution or improvement in peripheral neuropathy events, and secondary malignancies were from the data cut-off date of June 1, 2021, while the remaining data were from the data cut-off of April 20, 2017.
Adverse Events
The proportions of patients experiencing TEAEs up to 30 days after the last front-line dose were similar between patients treated with BV + AVD (99%) and those treated with ABVD (98%) (Table 19). However, compared to patients treated with ABVD, higher proportions of patients treated with BV + AVD were found in several TEAEs, such as neutropenia (58% versus 45%, respectively) and peripheral sensory neuropathy (29% versus 17%, respectively).
Serious Adverse Events
Higher percentages of patients in the BV + AVD group experienced TESAEs up to 30 days after the last front-line dose, compared to the percentages of patients in the ABVD group (43% versus 27%, respectively) (Table 19). Notably, 17% of patients treated with BV + AVD and 7% of patients treated with ABVD experienced febrile neutropenia.
Mortality
Deaths were reported in 6% of the patients in the BV + AVD arm and 10% of the patients in the ABVD arm (Table 19). Among them, deaths occurred in 1% of the patients treated with BV + AVD and 2% of the patients treated with ABVD within 30 days of the last dose of front-line therapy, as well as in 5% of the patients in the BV + AVD group and 8% of the patients in the ABVD group during the post-treatment follow-up period (> 30 days after the last dose of front-line therapy).
Withdrawal Due to AEs
Treatment discontinuation due to AEs occurred in 13% of the patients in the BV + AVD arm and 16% of the patients in the ABVD arm (Table 19). The most common reason for withdrawal in the BV + AVD arm was peripheral sensory neuropathy (3%), followed by neuropathy peripheral (2%) and peripheral motor neuropathy (2%).
Notable Harms
Details on notable harms are shown in Table 19. In the safety population, 67% of the patients in the BV + AVD group and 43% in the ABVD group experienced at least 1 peripheral neuropathy event. The percentages of patients who experienced treatment-emergent peripheral neuropathy of grade 3 or higher were 10% in the BV + AVD group and 2% in the ABVD group. Resolution or improvement was reported for 379 patients (86%) treated with BV + AVD and 249 patients (87%) treated with ABVD at the last follow-up.
Approximately 3% of the patients in the BV + ABVD group and 5% of the patients in the ABVD group developed secondary malignancies. The proportions of patients who experienced neutropenia as TEAEs or TEAEs of grade 3 or higher were higher in the BV + ABVD group compared with those in the ABVD group (TEAEs: 58% versus 45%; TEAEs grade 3 or higher: 54% versus 39%). Similarly, the proportions of patients who experienced febrile neutropenia as TEAEs or TEAEs of grade 3 or higher were also higher in the BV + ABVD group than in the ABVD group (TEAEs: 19% versus 8%; TEAEs grade 3 or higher: 19% versus 8%). Primary prophylaxis with G-CSF was used in 83 patients in the BV + AVD arm and 43 patients in the ABVD arm to prevent neutropenia or febrile neutropenia (Table 20). Implementation of G-CSF prophylaxis in patients receiving BV + AVD resulted in relatively lower rates of neutropenia (any events and grade 3 or higher events) and febrile neutropenia compared to those who had not received primary prophylaxis, although these rates remained higher than the rates in patients in the ABVD group who received G-CSF prophylaxis.
The ECHELON-1 trial defined pulmonary-related toxicity to include the preferred terms of “dyspnea” and “hypoxia,” all preferred terms in an interstitial lung disease standardized MedDRA query, and preferred terms with the high-level term “respiratory and pulmonary function diagnostic procedures.” Overall, fewer patients in the BV + AVD arm experienced AEs of pulmonary-related toxicity compared with those in the ABVD arm (13% versus 25%). The most common AE of pulmonary-related toxicity for either group was dyspnea (12% versus 24%).
Table 19: Summary of Harms — Pivotal and RCT Evidence
Adverse events | ECHELON-1 (safety population) | |
|---|---|---|
BV + AVD (N = 662) | ABVD (N = 659) | |
Treatment-emergent adverse event up to 30 days after last front-line dose, n (%) | ||
Patients with ≥ 1 treatment-emergent adverse event | 653 (99) | 646 (98) |
Neutropeniaa | 454 (69) | 361 (55) |
Nausea | 348 (53) | 371 (56) |
Constipation | 279 (42) | 241 (37) |
Vomiting | 216 (33) | 183 (28) |
Fatigue | 211 (32) | 211 (32) |
Treatment-emergent serious adverse event up to 30 days after last front-line dose, n (%) | ||
Patients with ≥ 1 serious adverse event | 284 (43) | 178 (27) |
Febrile neutropenia | 114 (17) | 43 (7) |
Pyrexia | 44 (7) | 28 (4) |
Death, n (%) | ||
Patients who died | 39 (6) | 64 (10) |
Deaths within 30 days of last dose of front-line therapy | 9 (1) | 13 (2) |
Deaths > 30 days of last dose of front-line therapy (post-treatment follow-up deaths) | 30 (5) | 51 (8) |
Withdrawals due to adverse events, n (%) | ||
Patients who discontinued treatment due to adverse events | 88 (13) | 105 (16) |
Notable harms | ||
Peripheral neuropathy, n (%) | ||
Treatment-emergent adverse event | 443 (67) | 286 (43) |
Treatment-emergent adverse event (grade 3 or higher) | 70 (10) | 11 (2) |
At end of treatment,b n (%) | ||
Patients with resolution or improvementc,d | 236 (53) | 180 (63) |
Patients with no resolution or improvement | 207 (47) | 106 (37) |
Patients with ongoing peripheral neuropathy eventse | 310 (70) | 140 (49) |
At last follow-up, n (%) | ||
Patients with resolution or improvementc,d | 379 (86) | 249 (87) |
Patients with no resolution or improvement | 64 (14) | 37 (13) |
Patients with ongoing peripheral neuropathy eventsf | 125 (28) | 59 (21) |
Secondary malignancy, n (%) | 23 (3) | 32 (5) |
Neutropenia,a n (%) | ||
Treatment-emergent adverse event | 454 (69) | 361 (55) |
Treatment-emergent adverse event (grade 3 or higher) | 430 (65) | 317 (48) |
Febrile neutropenia, n (%) | ||
Treatment-emergent adverse event | 128 (19) | 52 (8) |
Treatment-emergent adverse event (grade 3 or higher) | 128 (19) | 52 (8) |
Pulmonary-related toxicity,g n (%) | 89 (13) | 165 (25) |
Dyspnea | 82 (12) | 155 (24) |
Lung infiltration | 6 (< 1) | 0 |
Pneumonitis | 6 (< 1) | 18 (3) |
Hypoxia | 4 (< 1) | 10 (2) |
Interstitial lung disease | 1 (< 1) | 6 (< 1) |
Pulmonary toxicity | 0 | 16 (2) |
ABVD = doxorubicin-bleomycin-vinblastine-dacarbazine; AVD = doxorubicin-vinblastine-dacarbazine; BV = brentuximab vedotin.
Note: Deaths during post-treatment follow-up period, resolution or improvement in peripheral neuropathy events, and secondary malignancies were from data cut-off date June 1, 2021, while the remaining were from data as of April 20, 2017.
aPreferred terms of neutropenia and neutrophil count decreased are counted as neutropenia.
bFor patients without end-of-treatment date, the date was imputed as the last dose of front-line therapy plus 30 days.
cResolution was defined as event outcome of “resolved” or “resolved with sequelae.” Patients with resolution of all peripheral neuropathy events at end of treatment (or last follow-up) were patients with all peripheral neuropathy resolved and all the resolution dates were on or before end of treatment or last follow-up date.
dResolution implies improvement. In addition, for events that were not resolved, improvement was defined as decrease by at least 1 grade from the worst grade with no higher grade thereafter. Patients with improvement in any event at end of treatment or at last follow-up were those with at least 1 improved event and with a date of improvement before the end-of-treatment date or last follow-up date. Patients with all events resolved were excluded.
eAn ongoing event at end of treatment was defined as event end date after the end-of-treatment date if the event end date was not missing or the last follow-up date was on or after the end-of-treatment date if the event end date is missing. Maximum severity is the maximum among grades that were occurring at end of treatment.
fAn ongoing event at last follow-up was defined as event outcome of “unresolved” or “unknown” or events without an end date. Maximum severity is the maximum among grades that were occurring at last follow-up.
gPulmonary-related toxicity include preferred terms of “dyspnea” and “hypoxia,” and all preferred terms in an interstitial lung disease standardized Medical Dictionary for Regulatory Activities query, and preferred terms with the high-level term “respiratory and pulmonary function diagnostic procedures.”
Sources: ECHELON-1 original Clinical Study Report (data cut-off: April 20, 2017),13 ECHELON-1 Clinical Study Report Addendum 1 (data cut-off: June 01, 2021),14 Drug Reimbursement Review sponsor submission,16, and the sponsor’s Summary of Clinical Evidence.1
Table 20: Treatment-Emergent Neutropenia and Febrile Neutropenia by G-CSF Primary Prophylaxis (ECHELON-1 Safety Population)
Adverse events | BV + AVD (N = 662) | ABVD (N = 659) | ||
|---|---|---|---|---|
Primary prophylaxis with G-CSF (N = 83) | No primary prophylaxis with G-CSF (N = 579) | Primary prophylaxis with G-CSF (N = 43) | No primary prophylaxis with G-CSF (N = 61) | |
Treatment-emergent neutropeniaa | 29 (35) | 425 (73) | 9 (21) | 352 (57) |
Grade 3 or higher neutropenia | 24 (29) | 406 (70) | 8 (19) | 309 (50) |
Febrile neutropenia | 9 (11) | 119 (21) | 3 (7) | 49 (8) |
ABVD = doxorubicin-bleomycin-vinblastine-dacarbazine; AVD = doxorubicin-vinblastine-dacarbazine; BV = brentuximab vedotin; G-CSF = granulocyte colony-stimulating factor.
aPreferred term of neutropenia and decreased neutrophil count are counted as neutropenia.
Sources: ECHELON-1 original Clinical Study Report (data cut-off: April 20, 2017),13 Drug Reimbursement Review sponsor submission,16, and the sponsor’s Summary of Clinical Evidence.1
Critical Appraisal
Internal Validity
The ECHELON-1 trial was an international, multicentre, phase III, open-label, randomized, active-controlled superiority trial. A stratified randomization procedure based on region (i.e., Americas versus Europe versus Asia) and number of IPFP risk factors (i.e., IPS of 0 to 1 versus 2 to 3 versus 4 to 7) was used to minimize potential imbalances between the study groups that might bias results. Baseline characteristics were similar overall between the treatment groups, although patients in the ABVD group were aged about 2 years older on average, and a larger percentage of those randomized to BV + ABVD had nodular-sclerosis classical HL (64% versus 58%, respectively); however, these differences were determined not to be clinically important. The sampled population had a generally good performance status at baseline, and approximately two-thirds of patients had stage IV disease. Neither of these factors were likely to have influenced the results in a meaningful way. The ECHELON-1 trial therefore did not appear to have major concerns related to selection bias.
As an open-label trial, the investigators, patients, and sponsor in the ECHELON-1 trial were aware of the patients’ treatment allocation. The primary outcome of mPFS and secondary outcome of CR were assessed by an IRF that was blinded, which would help limit detection bias related to the open-label design.54 The level of concordance between mPFS according to IRF and mPFS according to investigators was reported as 91%, suggesting good agreement between the methods of assessing the primary outcome (the Internal Validity section of this review provides further discussion of mPFS). However, other important outcomes, such as PFS and HRQoL, had no blinded assessment and are therefore more prone to detection bias and performance bias. The sponsor indicated that, once the primary analysis of mPFS according to IRF was met, the trial protocol no longer required scans and disease-progression information to be sent to the IRF. As a result, PFS according to IRF was discontinued. PFS was assessed by the investigators instead in the long-term follow-up period. There was no clear evidence that the knowledge of treatment assignment among investigators influenced the assessment of PFS and the results were consistent with those from the mPFS analyses. The sponsor confirmed that no sensitivity analyses were conducted to explore the robustness of the PFS findings, including the potential impact of investigator assessment.16 Similarly, there were no obvious differences between the treatment groups in the HRQoL measurements, indicating that, if performance bias was present, it likely affected both groups.
Generally, no serious concerns were identified in the conduct of the ECHELON-1 trial, including protocol amendments and protocol deviations. The use of concomitant medications was considered by the clinical experts consulted by the review team to reflect practice settings, and the types of medications used were not expected to modify treatment effects. It was noted that, before the completion of the front-line treatment to which patients were randomized, patients were allowed to switch to an alternative front-line therapy of the physician’s choice. Overall, 15 patients (2%) in the BV + AVD arm and 9 (1%) in the ABVD arm switched to another form of chemotherapy. ABVD was the most frequently reported alternative front-line treatment for the patients originally randomized to the BV + AVD group, and BEACOPP was the most common alternative for patients randomized to the ABVD group. One concern regarding the switching was that study outcomes in those who switched might not adequately reflect the effects of the randomized study therapies. However, given that the switching rates were small (1% to 2%) and ITT analyses were applied, the risk of bias was considered low.
After completion of the front-line treatment to which patients were randomized, patients were permitted to receive subsequent treatment for HL. Receipt of subsequent therapy was considered an mPFS event. As of June 1, 2021, overall, 20% (135 of 662) of patients in the BV + AVD group and 24% (157 of 659) in the ABVD group received at least 1 subsequent anticancer therapy. The difference between the groups was primarily related to subsequent use of chemotherapy in the ABVD group (17% versus 12% in the BV + AVD group). The frequencies of use of other anticancer therapies were similar between the groups. Among the subsequent chemotherapies, 7% (48 of 659) of the patients randomized to the ABVD group received BV and 3% of patients received BV as maintenance therapy after transplant. Numerous other subsequent chemotherapy regimens were reported, with no single regimen standing out as a preferred alternative (generally used in < 2% of patients). Receiving subsequent treatment might confound the assessment of PFS and OS by delaying progression and prolonging survival beyond what would have occurred with front-line treatment alone. The overall difference between the groups in subsequent therapy use was 5%, which may or may not be important. However, as described, the only apparent difference between the groups was in the subsequent use of BV in the ABVD group. Any bias would therefore likely be in the direction of the null. The sponsor confirmed that no sensitivity analyses were conducted on PFS or OS outcomes to assess the impact of subsequent therapies. The review team concluded that the limited potential bias from the use of subsequent therapies was unlikely to have a large influence on the PFS and OS results.
The validity of the primary outcome of mPFS is a key consideration in evaluating the evidence for BV + AVD. The outcome of mPFS was adopted in the ECHELON-1 trial to capture all events that reflect a failure of front-line chemotherapy by counting a response that was less than complete at the end of the front-line therapy as an event. The ECHELON-1 trial defined a response that was less than complete as “receipt of anticancer therapy or radiotherapy for HL after completion of front-line therapy for patients who were confirmed noncomplete responders.” However, the clinical experts consulted by the review team noted that this definition is not consistent with practice in defining disease progression or first-line treatment failure in advanced HL. Furthermore, receipt of radiotherapy does not necessarily indicate disease progression in clinical practice because certain patients (e.g., those with bulky disease) may receive radiotherapy as part of their initial treatment. Despite the end-of-treatment PET scans conducted by IRF, there is a concern that the results would be biased given the administration of new anticancer therapy was at the discretion of the treating physician. Health Canada also raised this concern in its original review of BV.55 While sensitivity analyses, including additional analyses requested by the EMA, showed the mPFS results were robust to various censoring rules. However, 2 sensitivity analyses related to how subsequent therapies were treated in the primary analysis suggested sensitivity to the definitions. In sensitivity analysis 6 of mPFS according to IRF, “subsequent therapy after completion of front-line therapy without confirmed non-CR was considered as event.”13 The HR for the analysis was reported to be 0.837 (95% CI, 0.679 to 1.033). In sensitivity analysis 12a of mPFS according to IRF, “for the purpose of determining a modified event, the definition of subsequent therapy was restricted to anticancer chemotherapy,” thereby excluding radiotherapy from the definition. The HR for the analysis was reported to be 0.791 (95% CI 0.618 to 1.013). In both sensitivity analyses the upper confidence limit crosses 1 and in analysis 6 the HR increases. The treatment of the “modified” component of mPFS in the analysis therefore appears to affect the results. The clinical experts noted that OS and PFS are more clinically relevant to assessing patient benefits from treatment in adults with advanced HL, while no evidence was included in the submission to the review team empirically validating mPFS as an outcome measure or that established the correlation with OS.
The proportional hazards assumption for mPFS, as well as PFS and OS, was evaluated and visual inspection of the curves indicated that the assumption was met. Multiplicities were only adjusted for the mPFS and OS, and P values for other end points, including patient-reported outcomes, were provided for descriptive purposes only. Data for OS (as of March 11, 2023) and PFS (as of June 1, 2022) were not mature (i.e., the median was not reached). A median follow-up time of approximately 88 months for OS or 70 months for PFS was considered by the clinical experts consulted by the review team to be sufficient to capture early events because most events are expected to occur within about 24 months of initial treatment in clinical practice. The clinical experts consulted by the review team also noted that a longer follow-up (≥ 10 years) for patients with HL may be needed to show a survival signal. High percentages in loss to follow-up and withdrawal by patients were noted in both OS and PFS analyses. Although the percentages of loss to follow-up and withdrawal by patients appeared to be balanced between treatment arms, reasons for loss to follow-up and withdrawal could be different between groups, which may lead to biased estimates of treatment effects. Because each patient’s outcome status is unknown and any ITT assumptions for such patients are unverifiable, ITT analysis alone is not appropriate for minimizing bias introduced by patients who are lost to follow-up. Moreover, relevant sensitivity analyses were not available for OS or PFS, and the potential impact of high loss to follow-up and withdrawal could not be determined further. Subgroup analyses by HL stage signalled that there might be a difference in treatment effects between patients with stage III classical HL and patients with stage IV classical HL for mPFS and OS. The review team’s ability to make a definitive conclusion as to whether the difference is true was limited by several concerns. First, sample sizes differed between the stages, with approximately two-thirds of patients classified with stage IV disease. Patient randomization was not stratified according to disease stage; the IPS was a stratification factor, and it includes disease stage as a component. However, the balance of known and unknown factors between treatment groups achieved by randomization may not have been preserved in stage III or stage IV subgroups. Additionally, although subgroup analyses by stage were prespecified, the study was not specifically designed to test statistical inferences between BV + AVD and ABVD in these subgroups. It is therefore impossible to rule out chance as a factor in subgroup effects between treatment arms given that no formal statistical tests for interaction between subgroups were done. It is biologically plausible that people with stage IV disease would show more benefit from treatment with BV + AVD than those with stage III disease because they may have a poorer prognosis. Because stage III disease is a lower risk, and progression or death occurs less frequently compared with stage IV HL, the treatment effect may not be observed in a trial.
External Validity
All participants in the pivotal ECHELON-1 trial were required to be aged 18 years or older. An evidence gap therefore remains with respect to the clinical efficacy and safety of BV + AVD in the pediatric population. In addition, the ECHELON-1 trial only included patients with classical HL. As a result, the pivotal trial did not reflect results for patients with nodular lymphocyte-predominant HL.
The inclusion and exclusion criteria of the ECHELON-1 trial in general were aligned with selection criteria in the Canadian settings when identifying suitable candidates for BV + AVD, according to the clinical experts consulted by the review team. However, the clinical experts noted that, in clinical practice, a small percentage of patients who were excluded from the ECHELON-1 trial may be eligible to receive BV + AVD. For example, BV + AVD can be given to patients with HIV if the disease is well managed, and to patients with a borderline left ventricle ejection fraction, after consultation with a cardiologist. The clinical experts also noted that patients with a higher ECOG PS (> 2) could be considered for treatment with BV + AVD on a case-by-case basis.
The clinical experts consulted by the review team noted that the doses of BV + AVD and ABVD used in the ECHELON-1 trial generally reflected the standard dose schedules used for adults in Canada. The clinical experts also confirmed that ABVD up to 6 cycles that is not adapted based on PET response is a relevant therapy used in the current standard of care and therefore a reasonable comparator for the adult population. However, the clinical experts also noted that ABVD up to 6 cycles not adapted based on PET response is not the only standard of care in Canada. In addition, the percentages of patients who received a transplant as subsequent treatment were lower in both groups than the experts would expect to see in clinical practice.
According to the clinical experts consulted by the review team, the study population characteristics generally reflected those of patients who would be eligible for BV + AVD in Canadian practice, and are representative of HL in Canada. However, the clinical experts noted that the percentage of patients with stage IV HL in the trial population and the percentage of white participants were higher than what would be seen in clinical practice.
Long-Term Extension Studies
No long-term extension studies were submitted by the sponsor.
Indirect Evidence
Contents within this section were informed by materials submitted by the sponsor. The following summary was validated by the review team.
No ITCs were submitted by the sponsor. The sponsor provided a feasibility assessment that determined it would be infeasible to conduct ITCs of BV + AVD versus other front-line therapies examined in clinical studies for advanced HL.
Studies Addressing Gaps in the Pivotal and RCT Evidence
In consultation with stakeholders, the review team identified a gap in the pivotal RCT evidence regarding the use of BV in pediatric patients (those aged < 18 years). The review team received input from POGO that front-line BV in combination with a chemotherapy backbone would be considered a treatment option in pediatric patients with HL based on the ECHELON-1 trial results and evidence from a published RCT that studied the addition of BV to a classic pediatric chemotherapy backbone in pediatric patients (the AHOD1331 trial11). Furthermore, the sponsor identified the AHOD1331 trial as a study that could address a gap in the evidence regarding the use of BV in combination with chemotherapy in pediatric patients with high-risk HL, as the ECHELON-1 trial only enrolled adult patients (those aged ≥ 18 years). The review team confirmed with Health Canada and the sponsor that sponsor-supported data from pediatric patients with HL was not submitted to Health Canada for the supplement to a new drug submission. The review team, in consultation with the drug programs and a clinical expert specialized in treating pediatric patients with HL, identified children and adolescents with advanced or high-risk HL as having a need for new treatments, and that their exclusion resulted in an important gap in the evidence from the ECHELON-1 trial. With agreement from the sponsor, the review team therefore summarized and critically appraised the AHOD1331 trial.
Description of the AHOD1331 Trial
The AHOD1331 trial,11 published in The New England Journal of Medicine, is a phase III, multicentre, open-label, randomized active-controlled trial comparing BV + AVEPC to ABVE-PC in previously untreated patients aged 2 to 21 years with high-risk classical HL, defined as patients with Ann Arbor stage IIB with bulk, stage IIIB, stage IVA and stage IVB HL. This trial was conducted in 587 participants across 153 Children’s Oncology Group institutions in North America. The number of trial sites in Canada was not reported. Patients were randomly assigned 1:1 to the BV + AVEPC or ABVE-PC group, stratified by the Ann Arbor stage (i.e., stage IIB with bulk tumour versus stage IIIB versus stage IVA versus stage IVB). The primary objective of the AHOD1331 trial was to determine the efficacy of BV + AVEPC relative to ABVE-PC as measured by EFS. Characteristics of the AHOD1331 trial are summarized in Table 21.
The AHOD1331 trial is ongoing. The final analysis of EFS was based on the database lock date of December 31, 2021.
Table 21: Details of the AHOD1331 Trial
Detail | AHOD1331 |
|---|---|
Designs and populations | |
Study design | Phase III, multicentre, open-label, randomized active-controlled trial |
Locations | 153 Children’s Oncology Group institutions in North America |
Patient enrolment dates: | Start date: March 16, 2015 End date: August 2, 2019 |
Randomized and enrolled (N) | 587 |
Key inclusion criteria |
|
Key exclusion criteria |
|
Drugs | |
Intervention | BV + AVEPC (brentuximab vedotin 1.8 mg/kg/dose IV; doxorubicin: 25 mg/m2/dose IV or intermittent infusion; vincristine: 1.4 mg/m2/dose IV or infusion; etoposide: 125 mg/m2/dose IV; prednisone: 20 mg/m2/dose twice a day oral; cyclophosphamide: 600 mg/m2/dose IV), 5 cycles (21 days per cycle) |
Comparator(s) | ABVE-PC (doxorubicin: 25 mg/m2/dose IV or intermittent infusion; bleomycin: the dose is different on each day of administration, IV or subcutaneous; etoposide: 125 mg/m2/dose IV; prednisone: 20 mg/m2/dose twice a day oral; cyclophosphamide: 600 mg/m2/dose IV; vincristine: 1.4 mg/m2/dose IV or infusion), 5 cycles (21 days per cycle) |
Study duration | |
Treatment period | 5 cycles of study treatment |
Follow-up phase | 48 ± 6 months after end of therapy |
Outcomesb | |
Primary end point | Event-free survival |
Secondary and exploratory end points | Secondary
Exploratory
|
Publication status | |
Publication | Castellino et al. (2022)11 |
ABVE-PC = doxorubicin-bleomycin-etoposide-prednisone-cyclophosphamide-vincristine; ALT = alanine transaminase; AST = aspartate aminotransferase; AVEPC = doxorubicin-vincristine-etoposide-prednisone-cyclophosphamide; BV = brentuximab vedotin; FEV1 = forced expiratory volume in 1 second; FVC = forced vital capacity; HL = Hodgkin lymphoma; HRQoL = health-related quality of life; SRL = slow-responding lesion or nodal site; ULN = upper limit of normal.
aBulk was defined as a contiguous extramediastinal nodal aggregate that measures greater than 6 cm across the longest transverse diameter (transaxial measurement) or craniocaudal dimension (measured on reformatted CT), or a mediastinal mass where the tumour diameter is greater than one-third the maximal thoracic diameter on an upright posteroanterior chest radiograph. If study eligibility by staging is uncertain, consultation with Imaging and Radiation Oncology Core Group Rhode Island may be obtained before study enrolment.
bA formal list of end points was not available. End points were mainly identified from the objective section in the trial protocol.
cOverall survival was not explicitly listed an exploratory end point.
Source: Castellino et al. (2022).11
Populations
Inclusion and Exclusion Criteria
Previously untreated patients aged 2 to 21 years with classic HL of Ann Arbor stage IIB with bulk tumour, stage IIIB, stage IVA, or stage IVB were eligible (Table 21). Patients with nodular lymphocyte-predominant HL were excluded.
Interventions
In the AHOD1331 trial, patients in both groups received combination therapy (i.e., intervention group: BV + AVEPC, 5 cycles with 21 days for each cycle; control group: ABVE-PC, 5 cycles with 21 days for each cycle).
The BV + AVEPC regimen consisted of BV (1.8 mg/kg/dose IV) administered on day 1 before other chemotherapy, doxorubicin 25 mg/m2/dose IV or intermittent infusion on days 1 and 2, etoposide 125 mg/m2/dose IV from day 1 to day 3, prednisone 20 mg/m2/dose twice a day oral from day 1 to day 7, cyclophosphamide 600 mg/m2/dose IV on days 1 and 2, vincristine 1.4 mg/m2/dose IV or infusion (maximum dose: 2.8 mg) on day 8 only.
The ABVE-PC regimen (1 cycle) consisted of doxorubicin 25 mg/m2/dose IV or intermittent infusion on days 1 and 2, bleomycin with the dose varying on each day of administration (IV or subcutaneous), vincristine 1.4 mg/m2/dose IV or infusion (maximum dose: 2.8 mg) on days 1 and 8, etoposide 125 mg/m2/dose IV from day 1 to day 3, prednisone 20 mg/m2/dose twice a day oral from day 1 to day 7, and cyclophosphamide 600 mg/m2/dose IV on days 1 and 2.
Dose modification was permitted based on treatment-associated toxicity. G-CSF (i.e., filgrastim or pegfilgrastim) was given to all participants in the AHOD1331 trial.
Following the completion of cycle 2, patients were assessed for response to treatment. All participants received 3 additional cycles of chemotherapy unless they met criteria for removal from protocol therapy, such as progressive disease, physician’s determination, or development of a second malignancy. Patients with progressive or recurrent disease could receive subsequent anticancer therapy at the physician's discretion.
Patients without initial large mediastinal adenopathy and without any slow-responding lesion or nodal site of disease completed therapy after receiving 3 additional cycles of chemotherapy. Patients with initial large mediastinal adenopathy or any slow-responding lesion or nodal site continued to receive radiation therapy after chemotherapy. The percentages of patients who received radiation therapy were 53.4% (95% CI, 47.7 to 59.0) after receipt of BV + AVEPC and 56.8% (95% CI, 51.0 to 62.5) after the receipt of ABVE-PC.
Outcomes
A list of end points assessed in this clinical review report are provided in Table 22 and summarized further in the following section. Summarized end points are based on those included in the sponsor’s Summary of Clinical Evidence as well as any identified as important to this review according to stakeholders, for example the clinical expert, clinician groups, or patient groups. OS was not a protocol-specified end point but was reported in the published results of the AHOD1331 trial.
Table 22: Outcomes Summarized From the AHOD1331 Trial
Outcome measure | Time point | ECHELON-1 |
|---|---|---|
3-year overall survival | Randomization until death due to any cause | Other |
3-year event-free survival | Randomization until disease progression or relapse, second malignancy, or death | Primary |
Harms outcomesa | After the first 2 cycles and after all 5 cycles of chemotherapy (Grade 3 or higher peripheral neuropathy) | Secondary (grade 3 or higher peripheral neuropathy) |
aOnly grade 3 or higher peripheral neuropathy was explicitly protocol prespecified. All other harms outcomes reported were not explicitly prespecified in the protocol.
Source: Castellino et al. (2022).11
Overall Survival
Overall survival was defined as the time from randomization to death from any cause.
Event-Free Survival
EFS was defined as the time from randomization until disease progression or relapse, second malignancy, or death.
Harms Outcomes
Adverse events, which were considered clinically significant by the AHOD1331 trial investigators, were defined as AEs of grade 3 or higher graded with the National Cancer Institute Common Toxicity Criteria versions 4.0 and 5.0. The grade of peripheral neuropathy was determined using the Balis Pediatric Scale.57
Statistical Analysis
Sample Size and Power Calculation
Limited information regarding the calculation of sample size was reported. The trial protocol stated that 580 eligible patients would be accrued to a maximum of 600 patients. The accrual rate for eligible patients was estimated to be about 146 patients per year, and the accrual duration was estimated to be about 4 years. An estimate of 77 observed events was considered necessary to have an 86% power to detect a difference of 8% in EFS in the BV + AVEPC group compared with the predicted EFS of 82% in the ABVE-PC group.
Statistical Testing
Details of statistical analysis of selected efficacy end points are summarized in Table 23.
For the primary end point of EFS in the ITT population, patients without an EFS event were censored at last contact. In the statistical analysis plan of the AHOD1331 trial, EFS analysis was claimed to be based on a 2-sided log-rank test with a significance level of 0.10 between the 2 randomized arms per ITT principle, and 90% CIs were planned. In the published results, EFS results were reported using a 2-sided log-rank test at a significance level of 0.05, and 95% CIs were used.
Interim and final EFS analyses were conducted. The planned final EFS analysis was scheduled to be performed after 77 events or 3 years after the last patient enrolled, whichever comes first. However, the actual final analysis of EFS was based on 74 EFS events due to the slow pace of events. The overall type I error of the EFS analyses was controlled using the Lan-DeMets alpha method.
Statistical analysis details about OS were not provided. No subgroup factors or related hypotheses were prespecified in the AHOD1331 trial protocol; post hoc subgroup analyses were conducted. No information about sensitivity analyses were identified from the AHOD1331 trial.
Table 23: Statistical Analysis of Efficacy End Points in the AHOD1331 Trial
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity and subgroup analyses |
|---|---|---|---|---|
3-year OS | NR | |||
3-year EFS |
| None | NR | Sensitivity analysis: NR Subgroup analysis:
|
CI = confidence interval; EFS = event-free survival; NR = not reported; OS = overall survival; vs. = versus.
Source: Castellino et al. (2022).11
Analysis Populations
In the AHOD1331 trial, the ITT population, including all the patients who had undergone randomization, was used for both efficacy and harms outcomes summarized in the report.
Protocol Amendments and Deviations
The original and final protocols were issued on December 9, 2014, and June 22, 2020, respectively. The AHOD1331 trial initially enrolled patients younger than 18 years of age, and was later expanded to patients aged less than 22 years. Details on protocol deviations were not reported.
Results
Patient Disposition
Out of 600 participants screened, 13 were excluded due to a stage not consistent with eligibility at central review (n = 9), tests for eligibility out of window or out of range (n = 3), or incorrect consent process (n = 1).
Baseline Characteristics
A summary of baseline patient demographics and disease characteristics in the AHOD1331trial is shown in Figure 11.
A total of 587 patients were randomly assigned to receive BV plus chemotherapy (i.e., BV + AVEPC; n = 298) or standard care (ABVE-PC; n = 289). Baseline demographics and disease characteristics were generally well balanced between the 2 treatment groups. The median age of participants was 15.6 years (range = 3.4 to 21.99); most (84.7%) were between 12 and 22 years. Of the total number of patients enrolled, 47% were female and 57.6% were non-Hispanic white. The proportions of patients by disease stage were 20.6% for stage IIB with bulk tumour, 19.3% for stage IIIB, 28.4% for stage IVA, and 31.7% for stage IVB. Most of the patients had nodular-sclerosis classical HL (76.5%).
Figure 11: Summary of Baseline Characteristics of the AHOD1331 Trial
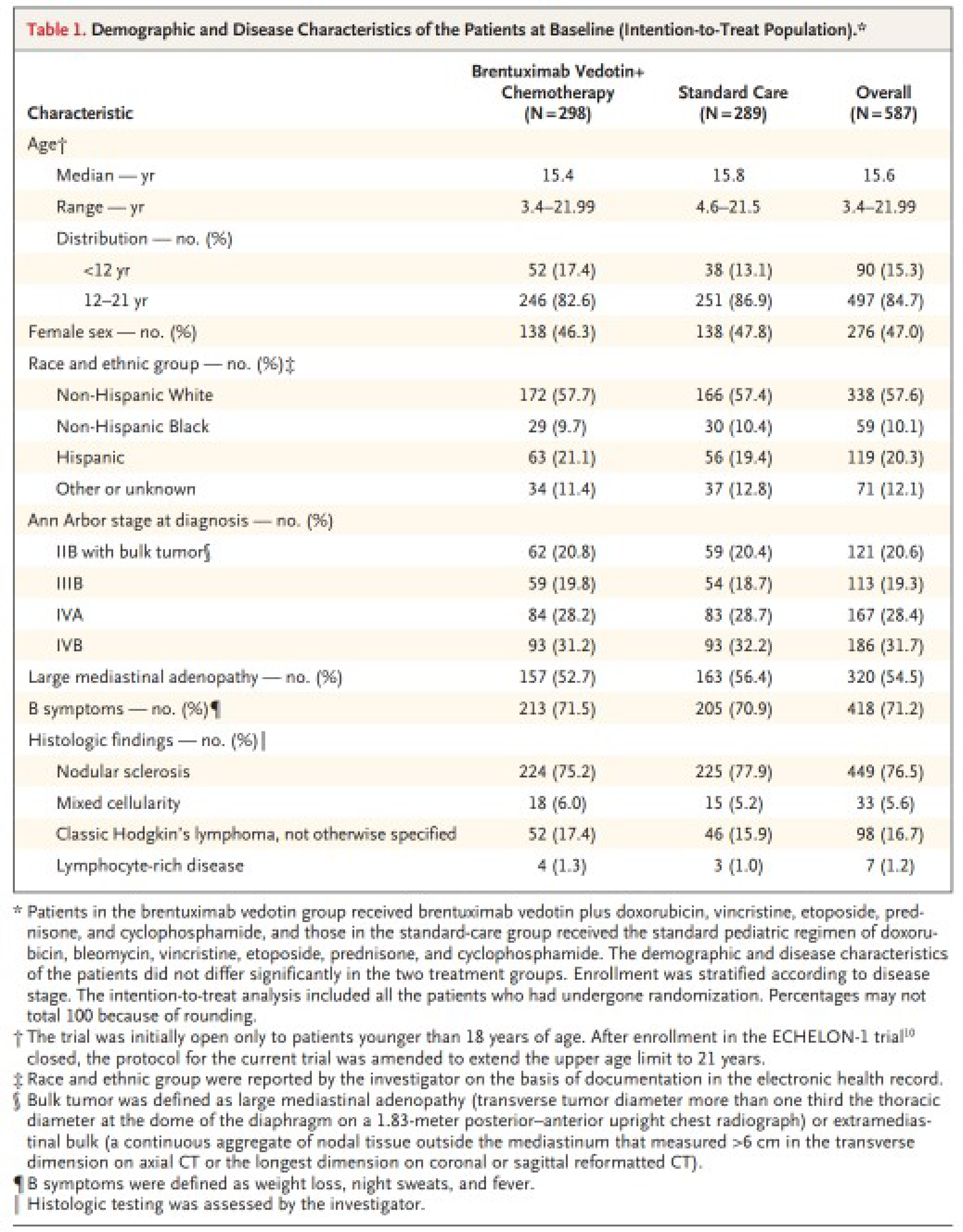
From: NEJM, Castellino et al,11 Brentuximab vedotin with chemotherapy in pediatric high-risk Hodgkin’s lymphoma, Vol 387, p 1649 to 1660. Copyright © 2022 Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society.
Exposure to Study Treatments, Concomitant Medications, Subsequent Treatment
Dose modification on any drug occurred in 28.2% of patients in the BV + AVEPC arm and 26.0% in the ABVE-PC arm. Dose modification on BV occurred in 8.1% of patients in the BV + AVEPC group. No concomitant cancer chemotherapy or immunomodulating agents were allowed. Corticosteroid therapy was only allowed for anaphylactic reactions, adrenal insufficiency, and severe asthma or as a breakthrough antiemetic. Antibiotics, blood products, antiemetics, fluids, electrolytes, and general supportive care were used.
Efficacy
The median follow-up time was 42.1 months (range = 0.1 to 80.9).
Three-Year Overall Survival
In terms of the 3-year OS in the ITT population, 99.3% (95% CI, 97.3 to 99.8) of patients were censored in the BV + AVEPC group and 98.5% (95% CI, 96.0 to 99.4) were censored in the ABVE-PC group. The HR for 3-year OS was not provided.
Three-Year EFS
In terms of the 3-year EFS in the ITT population, 92.1% (95% CI, 88.4 to 94.7) were censored in the BV + AVEPC group and 82.5% (95% CI, 77.4 to 86.5) were censored in the ABVE-PC group. The HR for 3-year EFS was 0.41 (95% CI, 0.25 to 0.67; P < 0.001) favouring the BV + AVEPC arm. First events occurred in 23 out of 298 patients and 51 out of 289 patients in the BV + AVEPC and ABVE-PC groups, respectively. Each group included 1 patient who developed secondary cancer (BV + AVEPC: acute myeloid leukemia; ABVE-PC: papillary thyroid carcinoma). One patient in the ABVE-PC group died due to a motor vehicle accident.
Harms
As shown in Figure 12, the occurrences of any AEs of grade 3 or higher were 73.5% in patients treated with BV + AVEPC and 68.2% in patients treated with ABVE-PC. Peripheral neuropathies of grade 3 or higher occurred in 6.7% of the patients in the BV + AVEPC arm and 5.5% % of the patients in the ABVE-PC arm. Febrile neutropenia occurred in 30.9% and 32.5% of patients in the BV + AVEPC and ABVE-PC arms, respectively. None of the patients in the BV + AVEPC group experienced pneumonitis, while only 1 patient in the ABVE-PC group did.
Figure 12: Summary of Harms — AHOD1331 Trial
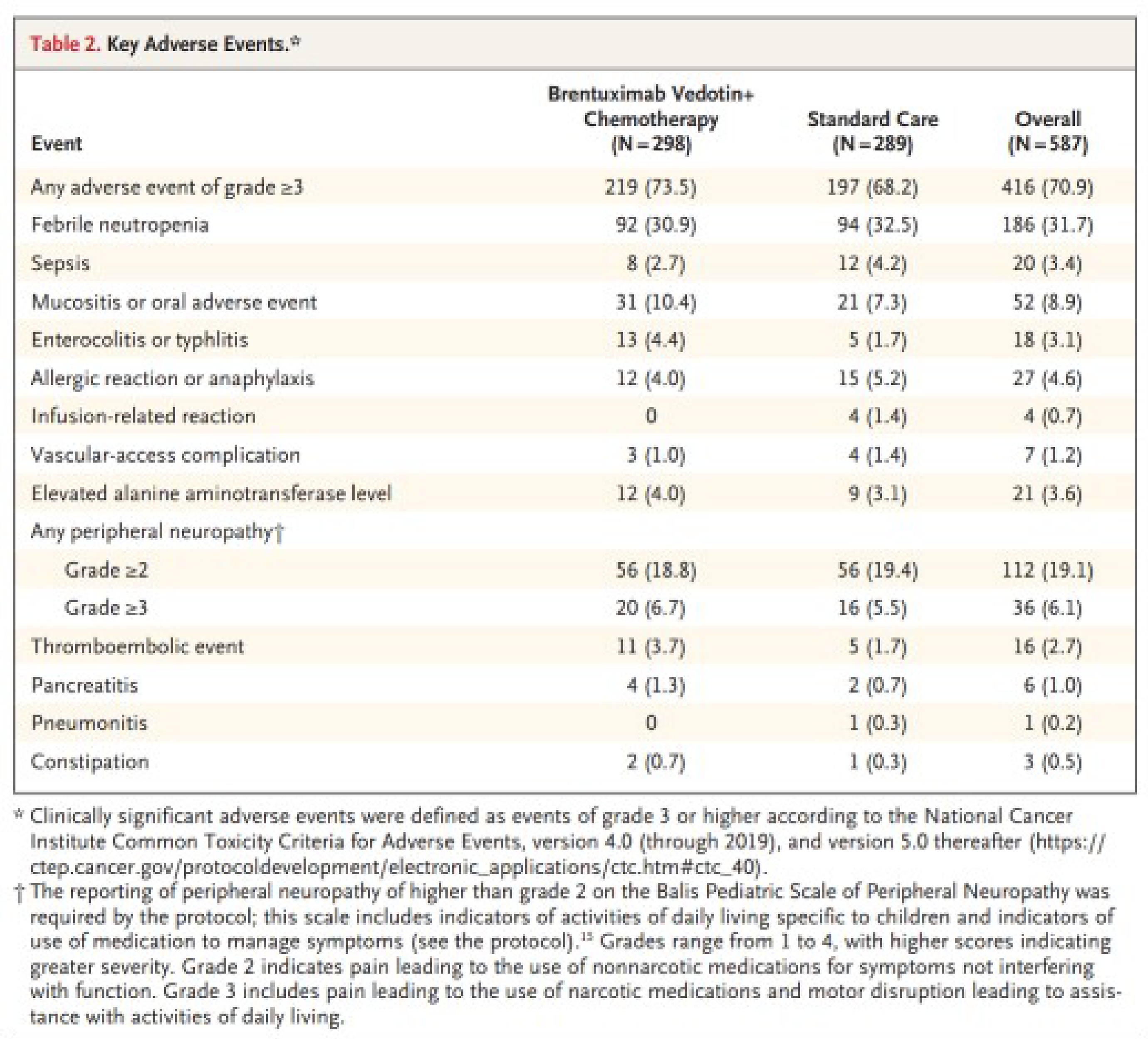
From: NEJM, Castellino et al,11 Brentuximab vedotin with chemotherapy in pediatric high-risk Hodgkin’s lymphoma, Vol 387, p 1649 to 1660. Copyright © 2022 Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society.
Critical Appraisal
Internal Validity
Although details about the randomization process and allocation concealment were not reported in the research protocol or the main article, the risk of bias is anticipated to be low given that baseline characteristics between the treatment arms were generally similar for clinically important factors. The AHOD1331 trial was open-label but had blinded outcome assessors, the definition of EFS was aligned with accepted definitions from regulators, and treatment response was assessed via a centralized review, helping reduce the risk of detection bias related to the open-label design. Even though patients were aware of the treatment allocation, which could result in performance bias, the risk is considered low because the 3-year PFS in the ABVE-PC group (82.5%) and the types of AEs were generally in line with what the clinical experts consulted by the review team expected.
Those patients who remained PET-positive after 2 cycles of chemotherapy received response-adapted involved-site radiation therapy guided by blinded central assessment of PET scans. This could bias EFS results if the radiation therapy improved responses, reduced the likelihood of relapse, and/or increased the risk of secondary malignancy. However, the risk of this potential bias was mitigated by the requirement that radiation therapy could not be administered until directed to do so by the blinded assessment. Also, the percentages of patients who received involved-site radiation therapy were similar between the BV + AVEPC group and the ABVE-PC group (53.4% versus 56.8%, respectively).
Concomitant anticancer medications were not allowed. Antibiotics and supportive medications (e.g., antiemetics) were permitted as needed. Patients also received G-CSF support. None of the permitted medications would likely influence the results for either treatment group. However, after progression events, the treating physician could treat the patient at their discretion, which may affect the longer-term OS results.
External Validity
The AHOD1331 trial was appraised in this section to address an important gap with respect the unmet needs of using BV + chemotherapy in pediatric patients with high-risk HL. However, several notable issues need to be considered when generalizing results from the AHOD1331 trial. First, although the chemotherapy backbone used in the AHOD1331 trial (AVEPC) is a preferred backbone for pediatric patients according to POGO and the clinical expert consulted by the review team, it is different from the backbone used in adults (ABVD). Regarding the regulatory status of the pediatric regimen, the review team confirmed that BV is not approved for use in combination with the pediatric regimen and the sponsor confirmed that it is not planning to file for Health Canada approval of BV + AVEPC. Second, the clinical experts consulted by the review team noted the definition of high-risk or advanced-stage HL in pediatric patients varies. While the AHOD1331 trial adopted the definition of advanced-stage HL in pediatric patients as stage II with bulk tumour, stage IIIB, stage IVA, and stage IVB, some medical centres define any stage III or IV disease as advanced-stage disease in pediatric patients. Finally, the AHOD1331 trial involved both nonadults and young adults (up to the age of 22 years), while the pivotal ECHELON-1 trial enrolled patients aged 18 years and older. This created an overlap in patient ages between the pivotal ECHELON-1 trial and the AHOD1331 trial. The clinical experts consulted by the review team noted that the chemotherapy backbone AVEPC used in the AHOD1331 trial would not typically be used in patients aged 18 years or older in Canada and the chemotherapy backbone ABVD investigated in the pivotal ECHELON-1 trial may be used in adolescents aged close to 18 years with HL.
Discussion
Summary of Available Evidence
One ongoing phase III, open-label, randomized, active-controlled, superiority trial (ECHELON-1, N = 1,334, with 60 patients in Canada enrolled) was included. The primary objective of the ECHELON-1 trial was to determine the efficacy of BV + AVD relative to ABVD as measured by mPFS, while the key secondary objective was to compare OS. Other efficacy and safety end points were also examined, including PFS, percent alive without HL, CR rate, EORTC QLQ-30, EQ-5D-3L, TEAEs, TESAEs, deaths, withdrawals due to AEs, and notable harms (i.e., treatment-emergent peripheral neuropathy, secondary malignancy, neutropenia, febrile neutropenia, and pulmonary-related toxicity). The ECHELON-1 trial enrolled previously untreated adult patients with histologically confirmed stage III or stage IV classical HL with an ECOG PS of 2 or lower. The median age of enrolled participants was 36 years (range = 18 to 83), with 66% (874 of 1,334) aged younger than 45 years and 14% (186 of 1,334) aged 60 years or older. Of the total number of participants, 58% (n = 776) were male and 84% (n = 1,114) were white. Most patients had stage IV disease (64%, n = 846), 2 or 3 (53%, n = 705) IPFP risk factors, an ECOG PS of 0 (57%, n = 754), extranodal involvement at diagnosis (62%, n = 827), and B symptoms (59%, n = 781) at baseline.
No ITCs or long-term extension studies were submitted by the sponsor. The sponsor provided a feasibility assessment that determined it would be infeasible to conduct ITCs of BV + AVD versus other front-line therapies examined in clinical studies for advanced HL.
A phase III, multicentre, open-label, randomized active-controlled trial (AHOD1331, N = 587), published in The New England Journal of Medicine, was included to address the evidence gap regarding the use of BV + chemotherapy in pediatric patients with advanced-stage classical HL. The primary objective of the AHOD1331 trial was to assess the efficacy of BV + AVEPC versus ABVE-PC as measured by EFS. OS and safety outcomes were also reported in the published results. The AHOD1331 trial involved patients aged 2 to 21 years who were previously untreated for high-risk classical HL (i.e., Ann Arbor stage IIB with bulk tumour, stage IIIB, stage IVA, stage IVB). The median age of enrolled patients was 15.6 years (range = 3.4 to 21.99); most (84.7%, 497 of 587) were between 12 and 21 years. Of the 587 patients enrolled, 47% (276) were female and 57.6% (338) were non-Hispanic white. The proportions of patients by disease stage were 20.6% (121 of 587) for stage IIB with bulk tumour, 19.3% (113 of 587) for stage IIIB, 28.4% (167 of 587) for stage IVA, and 31.7% (186 of 587) for stage IVB. Most of the patients (76.5%, 449 of 587) had nodular-sclerosis classical HL.
Interpretation of Results
Efficacy
Overall, efficacy evidence from the ECHELON-1 trial indicated that BV + AVD is more efficacious than ABVD in patients with advanced-stage HL. This conclusion was made after considering the efficacy outcomes (i.e., OS, mPFS according to IRF, PFS according to investigator, percentage of patients who were alive without HL, and CR rate according to IRF) examined in the report and selected with input from the consulted clinical experts, and by input to the review team from patient and clinician groups. These efficacy outcomes aligned with patients’ expectation of important outcomes, including controlling disease symptoms, achieving longer disease remission, and prolonging life.
In general, the OS results (in the ITT population as well as the stage III and IV subgroups) were consistent between the updated descriptive analysis as of March 11, 2023, and the analysis as of June 1, 2021. OS was identified as the most clinically important outcome in our previous25 and current reviews of BV + AVD for the treatment of advanced classical HL. An important consideration in our previous review was the relatively short duration of the follow-up in the OS analyses (median < 30 months), which was determined to be immature at that time. Given the natural history of HL and use of standard-of-care chemotherapy as a comparator (which includes ABVD), a much longer follow-up period would be required to assess the effects of BV + AVD on OS. The updated OS data (cut-off date of March 11, 2023) had a median follow-up of approximately 88 months for the ITT population. The HR for OS (data cut-off date of March 11, 2023) was 0.607 (95% CI, 0.414 to 0.892; P = 0.010). The absolute reduction in number of OS events between the BV + AVD group and the ABVD group was 3%, favouring the BV + AVD arm over the follow-up period, which the clinical experts considered to be meaningful. The 4-year (extent of separation: 2.8 months), 5-year (3.6 months), 6-year (4.4 months), and 7-year (4.7 months) Kaplan-Meier survival estimates suggest an increasing separation of the survival curves with longer follow-up; however, the extent of separation was slightly decreased at year 8 (4.3 months). Given that later estimates are obviously based on decreasing sample sizes and associated with less precision, as well as the fact that the median OS has not yet been reached in either group, the longer-term differences in OS remain unclear. As classical HL is generally considered a curable disease, the clinical experts consulted by the review team noted that the overall 3% absolute difference was, in their opinions, clinically meaningful. However, no empirically validated minimal important difference exists for OS in patients with advanced classical HL, making it difficult to interpret the size of the between-group difference in OS and to gauge the added clinical value of BV + AVD versus ABVD beyond statistical significance. Because patient-group input to the review team stated that treatments that allow for longer life are important to them, the observed OS difference with BV + AVD may be meaningful to patients. Patient-group input also highlighted the importance of improved HRQoL. The HRQoL outcome measures, the EORTC QLQ-C30, and EQ-5D-3L VAS, showed improvements in both treatment groups and similar scores between the BV + AVD and ABVD groups. However, interpretation of these results is complicated by the analyses’ exploratory nature and the absence of formal between-group comparisons for the EQ-5D-3L VAS. Combining the OS and HRQoL results indicates a favourable effect on OS with BV + AVD versus ABVD and no apparent meaningful difference in HRQoL. However, the percentage of patients who experienced SAEs and notable harms with BV + AVD versus ABVD was nearly double (as discussed in the Harms section).
Although uncertainty remains as to the magnitude of the OS benefit, the results are supported by evidence that BV + AVD reduces disease progression to a greater degree than does ABVD. Benefit with BV + AVD was also found in mPFS according to IRF (data cut-off date of April 20, 2017; HR for mPFS: 0.770; 95% CI, 0.603 to 0.982; absolute difference in number of mPFS events: 4%, favouring BV + AVD) and PFS according to investigator (data cut-off date of June 1, 2021; HR for PFS: 0.678; 95% CI, 0.532 to 0.863; absolute difference in number of PFS events: 7%, favouring BV + AVD). Despite the failure to reach the median OS, median mPFS according to IRF, and median PFS according to investigator, the follow-up time (i.e., median of about 88 months for OS, > 70 months for PFS, and 24.9 months for mPFS) was considered sufficient to capture early events, as in clinical practice most events are expected to occur within 24 months after treatment, according to the clinical experts consulted by the review team. Again, the clinical experts considered the results of the PFS assessments to be clinically meaningful, although there is no empirically derived threshold for clinical significance to determine this objectively. Further uncertainty comes from the validity of mPFS as an outcome in classical HL (as mentioned in the Critical Appraisal section). However, patient-group input identified delayed disease progression as an important outcome.
Other sources of uncertainty include the relatively high percentages in loss to follow-up and withdrawal by patients in both analyses for OS and PFS according to investigator. For example, in the interim OS data analysis (data cut-off date of March 11, 2023), 13% of the patients in the BV + AVD group and 13% of patients in the ABVD group were lost to follow-up; overall withdrawals occurred in 21% of the patients in the BV + AVD group and 22% in the ABVD group. The percentages were balanced between the 2 treatment arms. However, the reasons for the loss to follow-up and withdrawal were not reported and could differ between groups, which could lead to biased estimates. ITT analysis alone cannot minimize bias introduced by patients who are lost to follow-up because each patient’s outcome status is unknown and any ITT assumptions for such patients is unverifiable. Moreover, with no relevant sensitivity analyses available, as confirmed by the sponsor,16 the impact of higher loss to follow-up and withdrawal could not be further determined. It is acknowledged that larger losses to follow-up occur in longer-term studies such as the ECHELON-1 trial.
Another key source of uncertainty is the subgroup effect regarding the stage of advanced classical HL (stage III versus IV) at baseline. Potentially small treatment effects (and uncertain benefit-to-risk ratios) were identified in 2019 by Health Canada, which restricted the indication to stage IV disease.55 Although PFS improved at both stage III (HR = 0.603; 95% CI, 0.391 to 0.930; data cut-off of June 1, 2021) and stage IV (HR = 0.715; 95% CI, 0.534 to 0.959; data cut-off date of June 1, 2021) subgroups, the hazards for OS and mPFS were lower in patients with stage IV classical HL (the HR for OS as of March 11, 2023 was 0.478; 95% CI, 0.286 to 0.799 and the HR for mPFS as of April 20, 2017 was 0.712; 95% CI, 0.530 to 0.957), compared to patients with stage III disease (HR for OS as of March 11, 2023 = 1.004, 95% CI, 0.540 to 1.866; HR for mPFS as of April 20, 2017 = 0.923; 95% CI, 0.600 to 1.420). However, as described in the Critical Appraisal section, there are numerous reasons the review team was not able to make definitive conclusions about whether there is a true difference in efficacy between patients with stage IV HL and those with stage III HL. This includes, among others, the fact that the majority (> 60%) of enrolled patients had stage IV disease, which caused an imbalance in sample size between the stage subgroups. This complicates interpretation of the subgroup analyses by stage, but it also means that the results of ECHELON-1 are easier to generalize to those with stage IV disease. The review team acknowledged that the inclusion of more patients with stage IV disease is practical for the trial, as it would allow for detection of a difference between treatments over a shorter duration of follow-up given the poorer prognosis in stage IV than stage III. However, determining whether the magnitude of benefit with BV + AVD is greater in those with stage IV disease than in those with stage III disease is challenging. While there is a signal of a subgroup difference, the design and analysis of the study precludes the review team from drawing any certain conclusion. The clinical experts consulted by the review team emphasized that the approach to treatment, particularly with for those with experience using ABVD, is to treat stage III and stage IV classical HL the same. It was therefore their opinion that BV + AVD would be used in the same manner as ABVD for advanced classical HL, regardless of stage.
An important gap in the evidence from the ECHELON-1 trial is the lack of data in people with advanced HL who are aged younger than 18 years. The review team received input from a clinical expert who specializes in treating pediatric patients with HL and a clinician group (POGO) indicating that BV in combination with chemotherapy would be used as front-line treatment for pediatric patients with advanced or high-risk HL. To address this gap, the review team reviewed evidence regarding the use of BV in combination with a pediatric chemotherapy backbone in patients aged 2 to 21 years with high-risk or advanced-stage classical HL from the AHOD1331 trial. Regarding the regulatory status of the pediatric regimen, the review team confirmed that BV is not approved for use in combination with the pediatric chemotherapy regimen and the sponsor confirmed that it is not planning to file for Health Canada approval for BV + AVEPC. Evidence from the AHOD1331 trial indicated that BV + AVEPC is more effective than ABVE-PC for EFS in patients aged 2 to 21 years with high-risk classical HL defined as Ann Arbor stage IIB with bulk tumour, stage IIIB, stage IVA, and stage IVB HL. According to the clinical experts consulted by the review team, the EFS in the ABVE-PC group (82.5% of patients were censored) reflected the effectiveness of ABVE-PC seen in clinical practice. The 6.6% absolute difference in the number of EFS events between the BV + AVEPC group and the ABVE-PC group favouring the former was clinically meaningful in the opinion of the clinical experts. The median follow-up time for EFS was considered adequate given that most events in clinical practice are expected to occur within 24 months after treatment. However, in the AHOD1331 trial, no difference between treatment groups was found in OS. The clinical expert consulted by the review team stated this would be expected, as 42 months of follow-up would be too short for OS events to occur in pediatric and young adult patients because HL is curable in these patients. The experts therefore indicated that EFS was a clinically relevant outcome.
Harms
In the pivotal ECHELON-1 trial, the observed AEs were consistent with the safety profiles of BV + AVD and ABVD according to the clinical experts consulted by the review team. Fewer patients treated with BV + AVD in the ECHELON-1 trial discontinued treatment due to AEs or experienced secondary malignancy and pulmonary-related toxicity compared with those receiving ABVD. More patients in the BV + AVD group experienced TEAEs and TESAEs, as well as notable harms such as grade 3 or higher treatment-emergent peripheral neuropathy, neutropenia, and febrile neutropenia, compared to those in the ABVD group. The clinical experts noted that the AEs were consistent with what they typically manage in clinical practice and were encouraged that key AEs with BV, such as peripheral neuropathy, resolved or improved in most patients. In the AHOD1331 trial, no difference was identified overall across harms outcomes between the BV + AVEPC group and the ABVE-PC group.
Conclusion
Overall, evidence from the phase III, open-label, randomized, ECHELON-1 trial suggested that BV + AVD is an effective front-line treatment for previously untreated adult patients diagnosed with advanced-stage classical HL. The clinically relevant efficacy end points examined in the report (OS, PFS, and mPFS) were consistently in favour of BV + AVD compared to ABVD in the ITT population. However, the clinical significance of the magnitude of the treatment differences is uncertain and concerns remain about the validity of the primary outcome (mPFS), as well as the high percentages in loss to follow-up and withdrawal by patients in both OS and PFS analyses. In addition, although subgroup analyses of the OS and mPFS results signalled that BV + AVD may be more effective in patients with stage IV classical HL than in those with stage III HL, conclusions regarding subgroup differences between stage III versus stage IV patients are uncertain because the study was not designed to detect differences between these subgroups and patients were not stratified at randomization according to disease stage. The safety profile of BV + AVD is consistent with the known AEs for the individual components of the regimen, but more patients treated with BV + AVD experienced SAEs compared with those in the ABVD group. An evidence gap remains with respect to the clinical efficacy and safety of BV + AVD in the pediatric population as all participants in the pivotal ECHELON-1 trial were required to be aged 18 years or older. To address the unmet needs in the pediatric patient population, the AHOD1331 trial was examined. Efficacy results from the AHOD1331 trial indicated that BV + AVEPC provides a clinically meaningful benefit in EFS compared to ABVE-PC in patients aged 2 to 21 years with high-risk classical HL defined as Ann Arbor stage IIB with bulk tumour, stage IIIB, stage IVA, and stage IVB HL.
References
1.Manufacturer Summary of Clinical Evidence [internal manufacturer's report]. In: Drug Reimbursement Review manufacturer submission: Brentuximab vedotin for injection (Adcetris), 50 mg/vial, lyophilized powder for reconstitution, intravenous infusion. Mississauga (ON): Seagen Canada Inc.; 2023 Feb.
2.Adcetris (brentuximab vedotin): Lyophilized powder for reconstitution with 10.5 mL of sterile water for injection, USP 50 mg [product monograph]. Bothell (WA): Seagen Inc.; 2021 Jun 11: https://pdf.hres.ca/dpd_pm/00061748.PDF. Accessed 2024 May 27.
3.Canadian Cancer Society. What is Hodgkin Lymphoma? [manufacturer-provided reference]. [2023]; https://cancer.ca/en/cancer-information/cancer-types/hodgkin-lymphoma/what-is-hodgkin-lymphoma. Accessed 2023 Jan 13.
4.Brenner DR, Poirier A, Woods RR, et al. Projected estimates of cancer in Canada in 2022. CMAJ. 2022;194(17):E601-E607. PubMed
5.Canadian Cancer Statistics: A 2018 special report on cancer incidence by stage [manufacturer-provided reference]. Toronto (ON): Canadian Cancer Society; 2018: https://cdn.cancer.ca/-/media/files/research/cancer-statistics/2018-statistics/canadian-cancer-statistics-2018-en.pdf.
6.Nagpal P, Akl MR, Ayoub NM, et al. Pediatric Hodgkin lymphoma: biomarkers, drugs, and clinical trials for translational science and medicine. Oncotarget. 2016;7(41):67551-67573. PubMed
7.Canadian Cancer Society. Childhood Hodgkin lymphoma statistics. 2024; https://cancer.ca/en/cancer-information/cancer-types/hodgkin-lymphoma-childhood/statistics. Accessed 2024 Mar 19.
8.Ansell SM. Hodgkin lymphoma: A 2020 update on diagnosis, risk-stratification, and management. Am J Hematol. 2020;95(8):978-989. PubMed
9.NCCN Clinical Practice Guidelines in Oncology: Hodgkin Lymphoma Version 1.2023 [manufacturer-provided reference]. Plymouth Meeting (PA): National Comprehensive Cancer Network (NCCN); 2022: https://www.nccn.org/.
10.CADTH Drug Reimbursement Expert Review Committee final recommendation: Brentuximab Vedotin (Adcetris - Seattle Genetics, Inc.). Ottawa (ON): CADTH; 2020 Dec 3: https://www.cda-amc.ca/sites/default/files/pcodr/Reviews2020/10214BrentuximabVedotinHLAVD_FnRec_approvedbyChair_REDACT_Post03Dec2020_final.pdf. Accessed 2023 Apr 26.
11.Castellino SM, Pei Q, Parsons SK, et al. Brentuximab Vedotin with Chemotherapy in Pediatric High-Risk Hodgkin’s Lymphoma. N Engl J Med. 2022;387(18):1649-1660. PubMed
12.Herrera AF, LeBlanc ML, Castellino SM, et al. SWOG S1826, a randomized study of nivolumab(N)-AVD versus brentuximab vedotin(BV)-AVD in advanced stage (AS) classic Hodgkin lymphoma (HL). J Clin Oncol. 2023;41(17_suppl):LBA4-LBA4.
13.Clinical Study Report: C25003. A randomized, open-label, phase 3 trial of A+AVD versus ABVD as front-line therapy in patients with advanced classical Hodgkin lymphoma [internal manufacturer's report]. Cambridge (MA): Millennium Pharmaceuticals, Inc.; 2017 Aug 28.
14.Clinical Study Report: C25003. A randomized, open-label, phase 3 trial of A+AVD versus ABVD as front-line therapy in patients with advanced classical Hodgkin lymphoma addendum 1 [internal manufacturer's report]. Lexington (MA): Takeda Development Center Americas, Inc.; 2022 Jun 16.
15.Committee for Medicinal Products for Human Use. Assessment Report: Adcetris. (European public assessment report). Amsterdam (NL): European Medicines Agency; 2023: https://www.ema.europa.eu/en/documents/variation-report/adcetris-h-c-002455-ii-0107-epar-assessment-report-variation_en.pdf. Accessed 2024 Mar 14.
16.Drug Reimbursement Review manufacturer submission: Brentuximab vedotin for injection (Adcetris), 50 mg/vial, lyophilized powder for reconstitution, intravenous infusion [internal manufacturer's package]. Mississauga (ON): Seagen Canada Inc.; 2023 Apr 12.
17.Kuppers R. The biology of Hodgkin's lymphoma. Nat Rev Cancer. 2009;9(1):15-27. PubMed
18.Ansell SM. Hodgkin lymphoma: 2018 update on diagnosis, risk-stratification, and management. Am J Hematol. 2018;93(5):704-715. PubMed
19.Leukemia and Lymphoma Society of Canada. Facts and Statistics About Blood Cancers [manufacturer-provided reference]. 2015; https://www.llscanada.org/disease-information/facts-and-statistics.
20.Shanbhag S, Ambinder RF. Hodgkin lymphoma: A review and update on recent progress. CA Cancer J Clin. 2018;68(2):116-132. PubMed
21.BC Cancer. Cancer Mangement Manual: Hodgkins Lymphoma [manufacturer-provided reference]. 2013; http://www.bccancer.bc.ca/health-professionals/clinical-resources/cancer-management-manual/lymphoma-chronic-leukemia-myeloma/hodgkin-lymphoma.
22.Lymphoma Clinical Practice Guidelines [manufacturer-provided reference]. Edmonton (AB): Cancer Care Alberta; 2019: https://www.albertahealthservices.ca/assets/info/hp/cancer/if-hp-cancer-guide-lyhe002-lymphoma.pdf.
23.Brice P, de Kerviler E, Friedberg JW. Classical Hodgkin lymphoma. Lancet. 2021;398(10310):1518-1527. PubMed
24.Canadian Cancer Statistics 2021 [manufacturer-provided reference]. Toronto (ON): Canadian Cancer Society; 2021: https://cdn.cancer.ca/-/media/files/research/cancer-statistics/2021-statistics/2021-pdf-en-final.pdf.
25.CADTH PCODR Final Clinical Guidance Report: Brentuximab Vedotin (Adcetris) [manufacturer-provided reference]. Ottawa (ON): CADTH; 2020: https://www.cadth.ca/sites/default/files/pcodr/Reviews2020/10214BrentuximabVedotinHLAVD_fnCGR_REDACT_Post03Dec2020_final.pdf.
26.Bleomycin for Injection [product monograph; manufacturer-provided reference]. Fresenius Kabi Canada, Ltd; 2016.
27.Cheson BD, Fisher RI, Barrington SF, et al. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: the Lugano classification. J Clin Oncol. 2014;32(27):3059-3068. PubMed
28.Kaseb H, Babiker HM. Hodgkin Lymphoma [manufacturer-provided reference]. StatPearls. Treasure Island (FL): StatPearls; 2022.
29.Ansell SM, Radford J, Connors JM, et al. Overall Survival with Brentuximab Vedotin in Stage III or IV Hodgkin's Lymphoma. N Engl J Med. 2022;387(4):310-320. PubMed
30.Connors JM, Jurczak W, Straus DJ, et al. Brentuximab Vedotin with Chemotherapy for Stage III or IV Hodgkin's Lymphoma. N Engl J Med. 2018;378(4):331-344. PubMed
31.Evens AM, Connors JM, Younes A, et al. Older patients (aged ≥60 years) with previously untreated advanced-stage classical Hodgkin lymphoma: a detailed analysis from the phase III ECHELON-1 study. Haematologica. 2022;107(5):1086-1094. PubMed
32.Hutchings M, Radford J, Ansell SM, et al. Brentuximab vedotin plus doxorubicin, vinblastine, and dacarbazine in patients with advanced-stage, classical Hodgkin lymphoma: A prespecified subgroup analysis of high-risk patients from the ECHELON-1 study. Hematol Oncol. 2021;39(2):185-195. PubMed
33.Ramchandren R, Advani RH, Ansell SM, et al. Brentuximab Vedotin plus Chemotherapy in North American Subjects with Newly Diagnosed Stage III or IV Hodgkin Lymphoma. Clin Cancer Res. 2019;25(6):1718-1726p. PubMed
34.Straus DJ, Dlugosz-Danecka M, Connors JM, et al. Brentuximab vedotin with chemotherapy for stage III or IV classical Hodgkin lymphoma (ECHELON-1): 5-year update of an international, open-label, randomised, phase 3 trial. Lancet Haematol. 2021;8(6):e410-e421. PubMed
35.Straus DJ, lstrokugosz-Danecka M, Alekseev S, et al. Brentuximab vedotin with chemotherapy for stage III/IV classical Hodgkin lymphoma: 3-year update of the ECHELON-1 study. Blood. 2020;135(10):735-742. PubMed
36.Takeda. NCT01712490: A Randomized, Open-label, Phase 3 Trial of A+AVD Versus ABVD as Frontline Therapy in Patients With Advanced Classical Hodgkin Lymphoma [manufacturer-provided reference]. ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine; 2018: https://clinicaltrials.gov/ct2/show/NCT01712490.
37.C25003: Statistical Analysis Plan Amendment 1 [internal manufacturer's report]. Cambridge (MA): Millennium Pharmaceuticals, Inc.; 2021 Dec 7.
38.C25003: Clinical Study Protocol Incorporating Amendment 9 [internal manufacturer's report]. Lexington (MA): Takeda Development Center Americas, Inc.; 2021 Sept 24.
39.Giesinger JM, Kieffer JM, Fayers PM, et al. Replication and validation of higher order models demonstrated that a summary score for the EORTC QLQ-C30 is robust. J Clin Epidemiol. 2016;69:79-88. PubMed
40.EORTC Quality of Life. EORTC Quality of life: FAQs. [2023]; https://qol.eortc.org/faq/. Accessed 2023 Jun 19.
41.Aaronson NK, Ahmedzai S, Bergman B, et al. The European Organisation for Research and Treatment of Cancer QLQ-C30: a quality-of-life instrument for use in international clinical trials in oncology. J Natl Cancer Inst. 1993;85(5):365-376. PubMed
42.EORTC QLC-C30 scoring manual. Brussels (BE): EORTC Data Center; 2001: https://www.eortc.org/app/uploads/sites/2/2018/02/SCmanual.pdf. Accessed 2023 Apr 26.
43.Davda J, Kibet H, Achieng E, Atundo L, Komen T. Assessing the acceptability, reliability, and validity of the EORTC Quality of Life Questionnaire (QLQ-C30) in Kenyan cancer patients: a cross-sectional study. J Patient Rep Outcomes. 2021;5(1):4. PubMed
44.Luo N, Fones CS, Lim SE, Xie F, Thumboo J, Li SC. The European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire (EORTC QLQ-c30): validation of English version in Singapore. Qual Life Res. 2005;14(4):1181-1186. PubMed
45.Osoba D, Rodrigues G, Myles J, Zee B, Pater J. Interpreting the significance of changes in health-related quality-of-life scores. J Clin Oncol. 1998;16(1):139-144. PubMed
46.Snyder CF, Blackford AL, Sussman J, et al. Identifying changes in scores on the EORTC-QLQ-C30 representing a change in patients' supportive care needs. Qual Life Res. 2015;24(5):1207-1216. PubMed
47.Bedard G, Zeng L, Zhang L, et al. Minimal important differences in the EORTC QLQ-C30 in patients with advanced cancer. Asia Pac J Clin Oncol. 2014;10(2):109-117. PubMed
48.Brooks R. EuroQol: the current state of play. Health Policy. 1996;37(1):53-72. PubMed
49.EuroQol--a new facility for the measurement of health-related quality of life. Health Policy. 1990;16(3):199-208. PubMed
50.EuroQol Research Foundation. EQ-5D-3L | About [manufacturer-provided reference]. 2022; https://euroqol.org/eq-5d-instruments/eq-5d-3l-about/.
51.Sinnott PL, Joyce VR, Barnett PG. Guidebook: preference measurement in economic analysis. Menlo Park (CA): Health Economics Research Center; 2007.
52.Teckle P, Peacock S, McTaggart-Cowan H, et al. The ability of cancer-specific and generic preference-based instruments to discriminate across clinical and self-reported measures of cancer severities. Health Qual Life Outcomes. 2011;9:106. PubMed
53.Pickard AS, Neary MP, Cella D. Estimation of minimally important differences in EQ-5D utility and VAS scores in cancer. Health Qual Life Outcomes. 2007;5(1):70. PubMed
54.Clinical trial end points for the approval of cancer drugs and biologics - guidance for industry. Silver Spring (MD): U.S. Food and Drug Administration; 2018: https://www.fda.gov/media/71195/download. Accessed 2023 Apr 26.
55.Health Canada reviewer's report: Adcetris (brentuximab vedotin) [internal manufacturer' report]. In: Drug Reimbursement Review manufacturer submission: Brentuximab vedotin for injection (Adcetris), 50 mg/vial, lyophilized powder for reconstitution, intravenous infusion. Bothell (WA): Seattle Genetics, Inc.; 2019.
56.Schwartz GJ, Gauthier B. A simple estimate of glomerular filtration rate in adolescent boys. J Pediatr. 1985;106(3):522-526. PubMed
57.Lavoie Smith EM, Li L, Hutchinson RJ, et al. Measuring vincristine-induced peripheral neuropathy in children with acute lymphoblastic leukemia. Cancer Nurs. 2013;36(5):E49-60. PubMed
58.Parker C, Woods B, Eaton J, et al. Brentuximab vedotin in relapsed/refractory Hodgkin lymphoma post-autologous stem cell transplant: a cost-effectiveness analysis in Scotland. J Med Econ. 2017;20(1):8-18. PubMed
59.Large S, Hettle R, Balakumaran A, Wu E, Borse RH. Cost-effectiveness of pembrolizumab versus brentuximab vedotin for patients with relapsed or refractory classical Hodgkin's lymphoma: a United States payer perspective. J Med Econ. 2018:1-10. PubMed
60.Petrelli F, Barni S. Correlation of progression-free and post-progression survival with overall survival in advanced colorectal cancer. Ann Oncol. 2013;24(1):186-192. PubMed
61.Hotta K, Kiura K, Fujiwara Y, et al. Role of survival post-progression in phase III trials of systemic chemotherapy in advanced non-small-cell lung cancer: a systematic review. PLoS One. 2011;6(11):e26646. PubMed
62.Sundar S, Wu J, Hillaby K, Yap J, Lilford R. A systematic review evaluating the relationship between progression free survival and post progression survival in advanced ovarian cancer. Gynecol Oncol. 2012;125(2):493-499. PubMed
Appendix 1: Ann Arbor Staging Classification for HL
Please note that this appendix has not been copy-edited.
Following the initial diagnosis the disease is then staged using imaging and laboratory blood tests and categorized according to the Ann Arbor stage I through IV of disease severity where higher stages indicate more widespread disease (Table 24).9,27,28
Table 24: Ann Arbor Definitions for HL
Stages | Definition |
|---|---|
Stage I | Single lymph node region (I) or localized involvement of a single extralymphatic organ or site (IE) |
Stage II | Two or more lymph node regions on the same side of the diaphragm (II) or localized involvement of a single associated extralymphatic organ or site and its regional lymph node(s), with or without involvement of other lymph node regions on the same side of the diaphragm (IIE) |
Stage III | Lymph node regions on both sides of the diaphragm (III), which may also be accompanied by localized involvement of an associated extralymphatic organ or site (IIIE), involvement of the spleen (IIIS), or both (IIIE+S) |
Stage IV | Disseminated (multifocal) involvement of 1 or more extralymphatic organs, with or without associated lymph node involvement, or isolated extralymphatic organ involvement with distant (nonregional) nodal involvement |
E = extranodal contiguous extension; HL = Hodgkin lymphoma; S = involvement of the spleen.
Source: NCCN (2022),9 Cheson et al. (2014),27 A Kaseb et al. (2022).9,27,28
Each Ann Arbor stage is further subdivided into A and B categories. Category A denotes that no systemic symptoms are present, and category B is assigned to patients who have unexplained fevers, drenching night sweats or unexplained weight loss. In addition, patients with classical HL are usually divided in 3 groups:
early-stage favourable (stage I to II with no unfavourable factors)
early-stage unfavourable (stage I to II with unfavourable factors such as large mediastinal adenopathy; > 3 nodal sites of disease; B symptoms; extranodal involvement; or significantly elevated erythrocyte sedimentation rate)
advanced-stage disease (stage III to IV).9
Appendix 2: Appraisal of Post-Progression Survival
Please note this appendix has not been copy-edited.
PPS, which was defined as time from progression to death and calculated for participants who experienced disease progression by subtracting PFS from OS, was used by the sponsor in the pharmacoeconomic analysis of BV + AVD as front-line therapy for advance stage classical HL submitted to the review team.16 PPS was not a prespecified end point in the ECHELON-1 trial, and data for PPS analyses were obtained via post hoc analysis based on the ECHELON-1 trial data. PPS was estimated based on parametric survival distributions fit to individual participant data on PPS from the ECHELON-1 trial. The PPS analyses were conducted for cohorts of patients from the ECHELON-1 trial who had evidence of receipt of ASCT (N = 99) and those who did not (N = 126). Within these cohorts, analyses were stratified by front-line treatment arm (i.e., BV + AVD versus ABVD). Receipt of ASCT was determined using data on subsequent therapies received from the ECHELON-1 trial data.
PPS has been adopted in several cost-effectiveness analyses in oncology, including HL.58,59 However, no studies with respect to the relationship between PPS and OS or PFS in the patient population with hematological malignancy (e.g., HL) were identified by the review team from the body of literature, while a few studies have been identified in patients with solid tumours. For instance, between PPS and OS, a systematic review of 34 RCTs indicated that PPS might be an appropriate surrogate for OS in patients with advanced colorectal cancer: Improvements in PPS were found strongly associated with improvements in OS.60 Similarly, in patients with advanced non–small cell lung cancer,61 a systematic review of 70 phase III trials published between 1988 and 2007 found that PPS had become increasingly associated with OS over years. Between PPS and PFS, findings from an examination of 37 RCTs published between 1990 and 2010 suggested that percentage gains in PFS among patients with advanced ovarian cancer were related to no percentage gains or slight percentage gains or losses in PPS.62 The referenced systematic reviews pooled data from heterogeneous published studies that had wide ranging follow-up durations, differences in progression definitions, and limited information regarding post-progression disease management to mention some of the limitations. The authors of some of the reviews identify their analyses as not definitive of a correlation between PPS and OS. Given these limitations, lacking relevant evidence in hematological cancers, and the apparent differences between hematological and solid tumours, the association between PPS and OS/PFS in patients with classical HL remains unclear.
Appendix 3: Key Protocol Amendments in the ECHELON-1 Trial
Please note this appendix has not been copy-edited.
Table 25: Summary of Key Protocol Amendments in the ECHELON-1 Trial
Amendment number (date) | Summary of key changes |
|---|---|
Amendment 4 (August 3, 2012) |
|
Amendment 5 (February 6, 2014) |
|
Amendment 6 (May 27, 2014) |
|
Amendment 7 (March 2, 2015) |
|
Amendment 8 (July 16, 2018) |
|
Amendment 9 (September 24, 2021) |
|
ABVD = doxorubicin, bleomycin, vinblastine, dacarbazine; BSA = body surface area; BV + AVD = brentuximab vedotin plus doxorubicin, vinblastine, dacarbazine; CR = complete remission; EOT = end of treatment; HL = Hodgkin lymphoma; IRF = independent review facility; mPFS = modified progression-free survival; OS = overall survival; PRO = patient-reported outcome; RT = radiation therapy; SAE = serious adverse event.
Source: Drug Reimbursement Review manufacturer submission.16
Appendix 4: Additional Data From the ECHELON-1 Trial
Please note this appendix has not been copy-edited.
Figure 13: Forest Plot of HR in OS for Subgroup Analyses (ITT Population) in the ECHELON-1 Trial
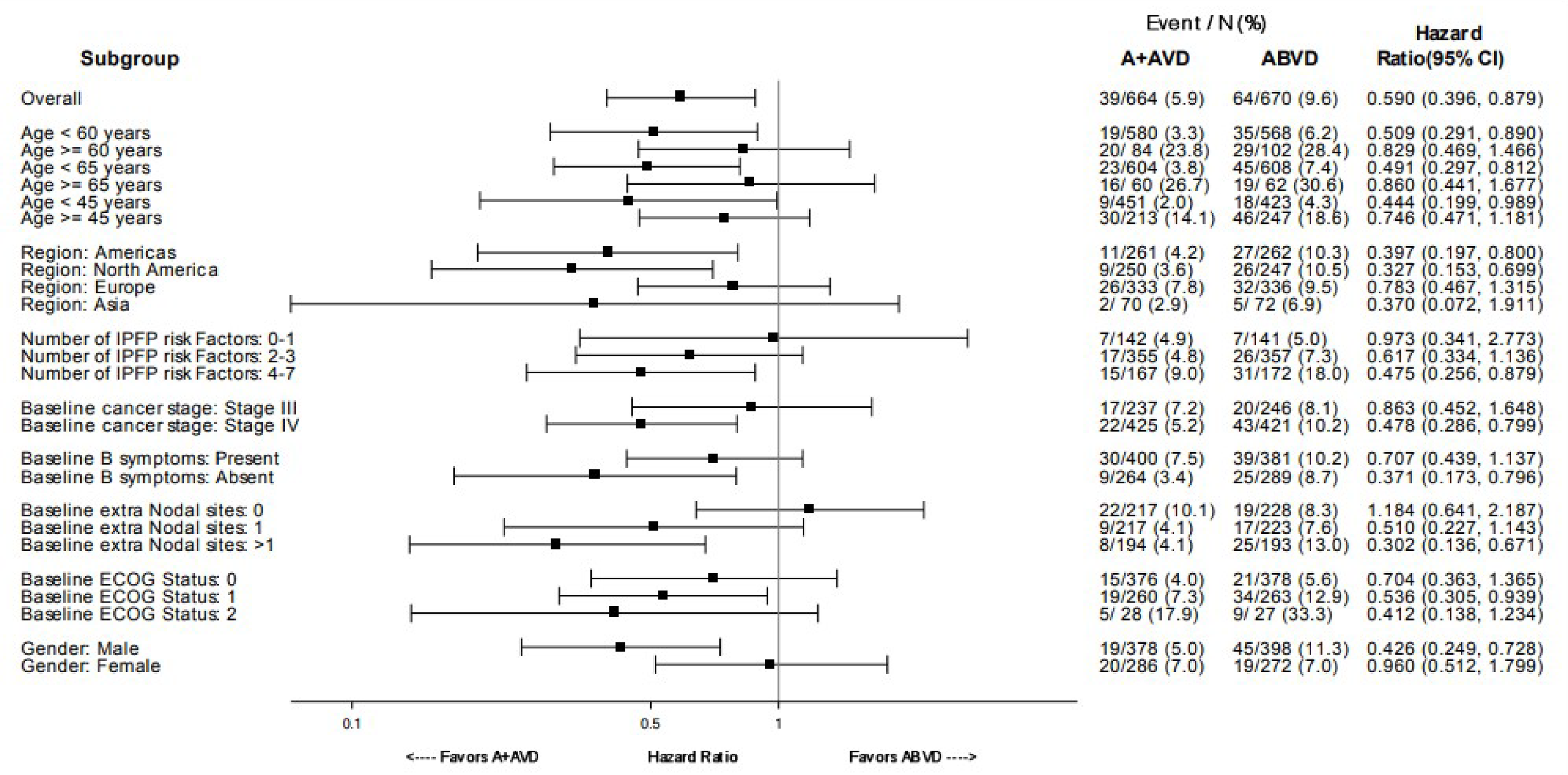
ABVD (denoted as A + AVD in the manufacturer’s Clinical Study Report) = doxorubicin-bleomycin-vinblastine-dacarbazine; BV + AVD = brentuximab vedotin plus doxorubicin, vinblastine, dacarbazine; ECOG = Eastern Cooperative Oncology Group; IPFP = International Prognostic Factor Project; ITT = intention to treat; OS = overall survival.
Sources: ECHELON-1 Clinical Study Report Addendum 1 (data cut-off: June 01, 2021)14 and the sponsor’s Summary of Clinical Evidence.1
Figure 14: Forest Plot of HR in mPFS According to IRF for Subgroup Analyses (ITT Population) in the ECHELON-1 Trial
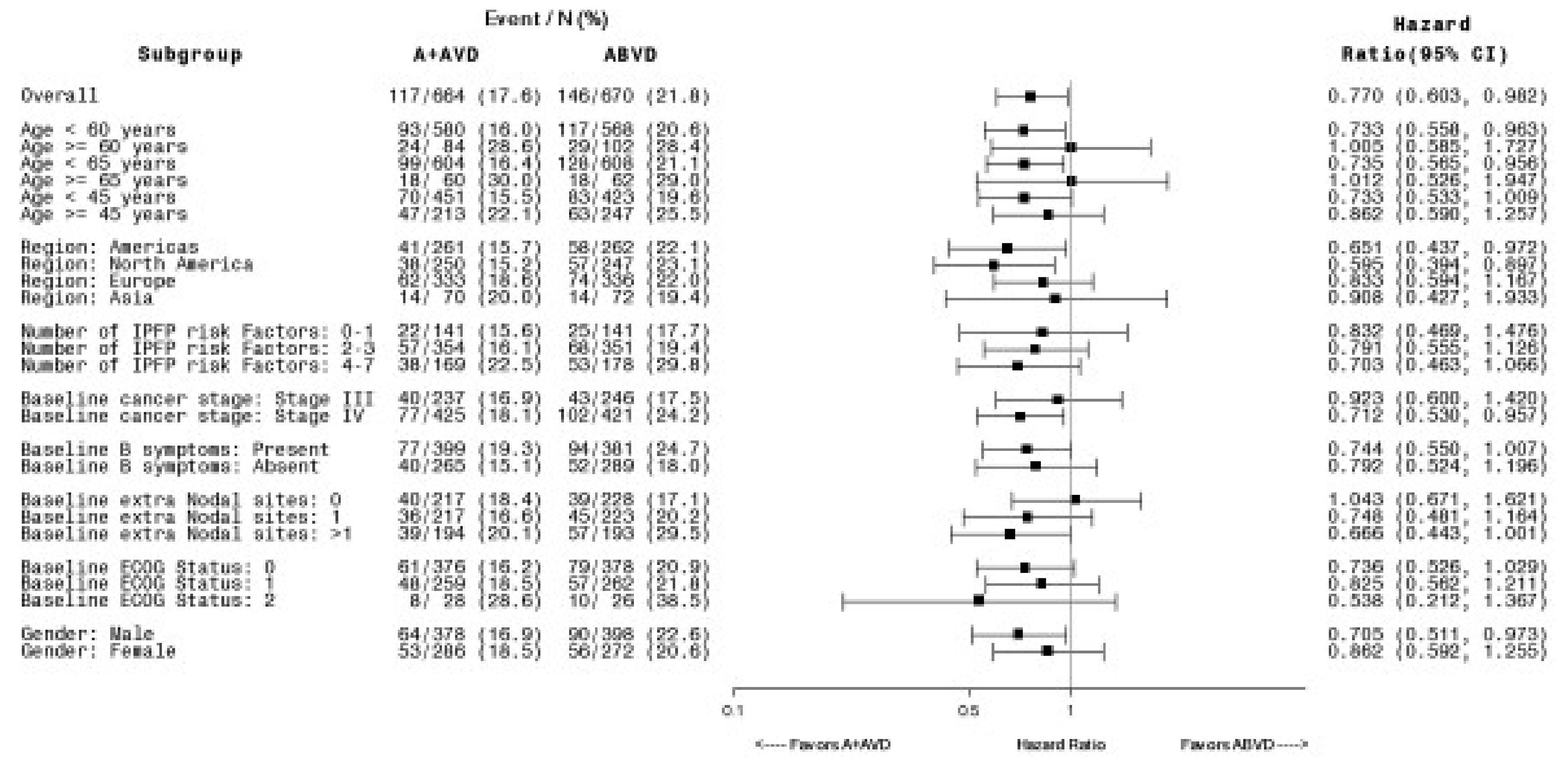
ABVD (denoted as A + AVD in the manufacturer’s Clinical Study Report) = doxorubicin-bleomycin-vinblastine-dacarbazine; BV + AVD = brentuximab vedotin plus doxorubicin, vinblastine, dacarbazine; ECOG = Eastern Cooperative Oncology Group; IPFP = International Prognostic Factor Project; ITT = intention to treat; mPFS = modified progression-free survival.
Source: ECHELON-1 original Clinical Study Report (data cut-off: April 20, 2017)13 and the sponsor’s Summary of Clinical Evidence1.
Figure 15: Forest Plot of HR in PFS According to Investigator for Subgroup Analyses (ITT Population) in the ECHELON-1 Trial
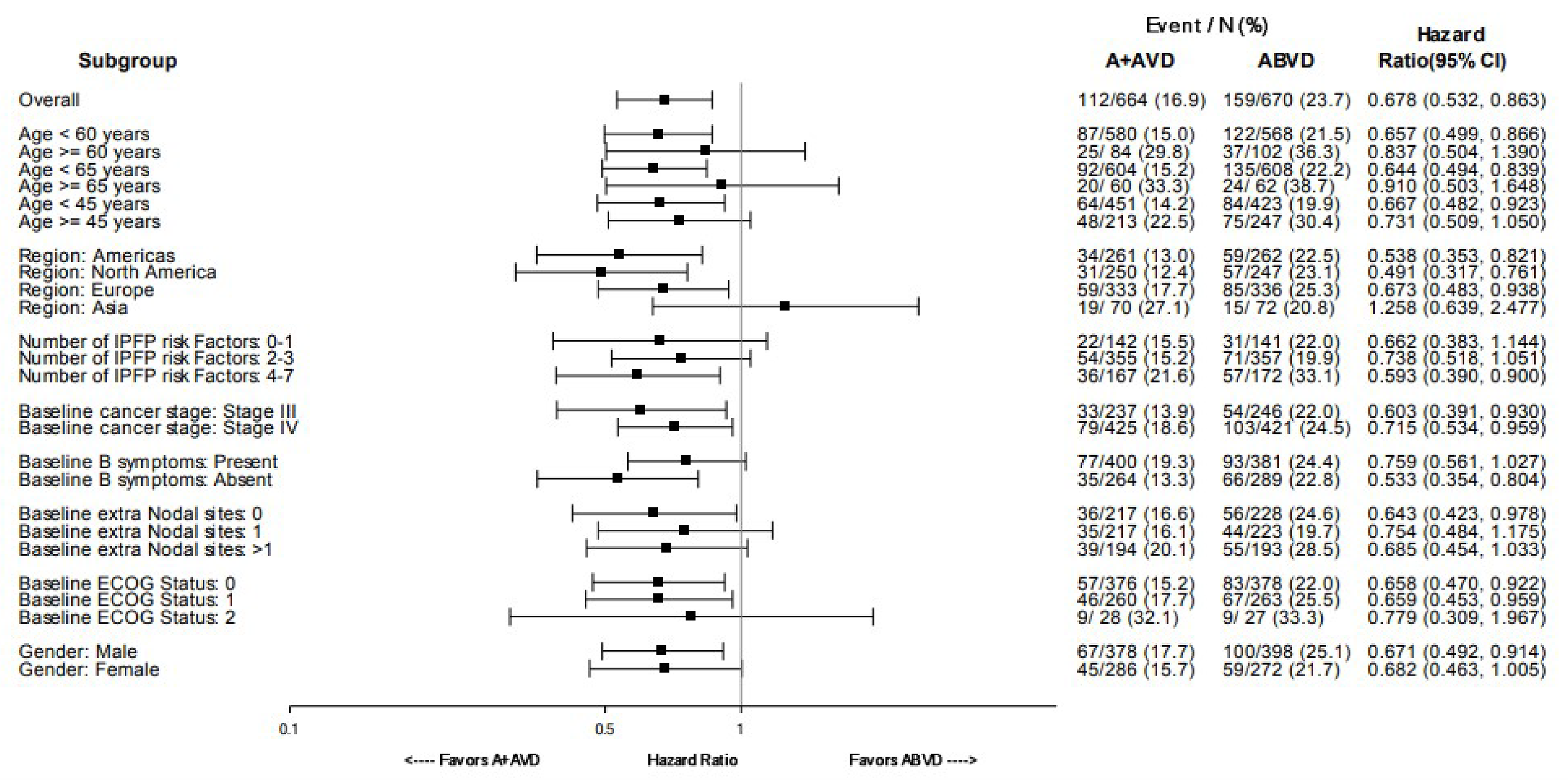
ABVD (denoted as A + AVD in the manufacturer’s Clinical Study Report) = doxorubicin-bleomycin-vinblastine-dacarbazine; BV + AVD = brentuximab vedotin plus doxorubicin, vinblastine, dacarbazine; ECOG = Eastern Cooperative Oncology Group; IPFP = International Prognostic Factor Project; ITT = intention to treat; PFS = progression-free survival.
Source: ECHELON-1 Clinical Study Report Addendum 1 (data cut-off: June 01, 2021)14 and the sponsor’s Summary of Clinical Evidence1.
Table 26: PFS According to IRF (ITT Population) in the ECHELON-1 Trial
Outcome | BV +AVD (N = 664) | ABVD (N = 670) |
|---|---|---|
PFS according to IRF | ||
Number of patients contributing to the analysis, n (%) | 664 (100) | 670 (100) |
Number with events (%) | 110 (17) | 128 (19) |
Number censored (%) | 554 (83) | 542 (81) |
Median (95% CI) | NE (48.2 to NE) | NE (NE to NE) |
Min, months | 0.0 | 0.0 |
Max, months | 48.8 | 49.3 |
Hazard ratio (95% CI)a | 0.829 (0.642 to 1.071) | |
P value between treatment groups | 0.150 | |
Median follow-up time, months (95% CI)b | 24.9 (24.61 to 25.03) | 24.8 (24.51 to 25.03) |
Reason leading to PFS event, n (%) | ||
Progressive disease | 96 (14) | 106 (16) |
Death due to any cause | 18 (3) | 22 (3) |
Reason for censoring, n (%) | ||
No baseline and/or no postbaseline assessment | 11 (2) | 24 (4) |
PFS event after more than 1 missed visit | 1 (< 1) | 4 (< 1) |
Treatment discontinuation for undocumented disease progression | 5 (< 1) | 4 (< 1) |
Loss to follow-up | 14 (2) | 20 (3) |
Withdrawal by patient | 22 (3) | 20 (3) |
No documented PFS event | 501 (75) | 470 (70) |
ABVD = doxorubicin-bleomycin-vinblastine-dacarbazine; BV + AVD = brentuximab vedotin in combination with doxorubicin, vinblastine, dacarbazine; 95% CI = 95% confidence interval; HL = Hodgkin lymphoma; IPFP = International Prognostic Factor Project; IRF = intendent review facility; ITT = intention to treat; NE = not estimable; PFS = progression-free survival.
Note; Data cut-off date for PFS according to IRF was April 20, 2017.
aHR and 95% CI were based on a stratified Cox’s proportional hazard regression model with stratification factors region and number of IPFP risk factors at baseline with treatment as the explanatory variable in the model. A HR of less than 1 favours the BV + AVD group.
bMedian PFS follow-up was calculated from the Kaplan-Meier method switching the PFS according to IRF event/censored status, i.e., PFS event as censored and censored as PFS event.
Source: ECHELON-1 original Clinical Study Report (data cut-off: April 20, 2017).13
Pharmacoeconomic Review
Abbreviations
ABVD
doxorubicin-bleomycin-vinblastine-dacarbazine
ABVE-PC
doxorubicin-bleomycin-vincristine-etoposide–prednisone-cyclophosphamide
AE
adverse event
ASCT
autologous stem-cell transplant
AVD
doxorubicin-vinblastine-dacarbazine
AVEPC
doxorubicin-vincristine-etoposide-prednisone-cyclophosphamide
BEACOPP
bleomycin-etoposide-doxorubicin-cyclophosphamide-vincristine-procarbazine
BIA
budget impact analysis
BV
brentuximab vedotin
CI
confidence interval
EFS
event-free survival
G-CSF
granulocyte colony-stimulating factor
HL
Hodgkin lymphoma
HR
hazard ratio
ICER
incremental cost-effectiveness ratio
ITT
intention-to-treat
LY
life-years
mPFS
modified progression-free survival
OEPA-COPDAC
vincristine-etoposide-prednisone-doxorubicin–cyclophosphamide-vincristine-prednisone-dacarbazine
OS
overall survival
PD-1
programmed cell death protein 1
PFS
progression-free survival
PPS
post-progression survival
RDI
relative dose intensity
QALY
quality-adjusted life-year
WTP
willingness to pay
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Brentuximab vedotin (Adcetris), 50 mg lyophilized powder, for IV infusion following reconstitution |
Submitted price | Brentuximab vedotin, 50 mg, IV infusion: $4,840 per vial |
Approved indication | For the treatment of previously untreated patients with stage IV HL, in combination with doxorubicin, vinblastine, and dacarbazine |
Reimbursement request | Brentuximab vedotin in combination with doxorubicin, vinblastine, and dacarbazine for the treatment of previously untreated patients with advanced-stage HL Brentuximab vedotin in combination with doxorubicin, vincristine, etoposide, prednisone, and cyclophosphamide in previously untreated high-risk HL in the pediatric population |
Health Canada approval status | Unlabelled indication |
Health Canada review pathway | NA |
NOC date | NA |
Sponsor | BC Cancer Agency Pediatric Oncology Group of Ontario |
Submission history | Previously reviewed: Yes Indication: For the treatment of previously untreated patients with advanced-stage HL, in combination with doxorubicin, vinblastine, and dacarbazine Recommendation: NA; withdrawn by sponsor Indication: HL at high risk of relapse or progression post-ASCT Recommendation date: February 21, 2018 Recommendation: Recommended with clinical criteria and/or conditions Indication: Stage IV HL in combination with AVD Recommendation date: December 3, 2020 Recommendation: Recommended with clinical criteria and/or conditions Indication: HL after failure of ASCT or after failure of at least 2 prior therapies in patients who are not ASCT candidates Recommendation date: August 29, 2013 Recommendation: Recommended with clinical criteria and/or conditions |
AVD = doxorubicin-vinblastine-dacarbazine; HL = Hodgkin lymphoma; NA = not applicable; NOC = Notice of Compliance; ASCT = autologous stem-cell transplant.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Semi-Markov model |
Target population | Previously untreated patients with advanced-stage HL |
Treatment | BV + AVD |
Comparator | ABVD |
Perspective | Canadian publicly funded health care payer |
Outcome | QALYs, LYs |
Time horizon | Lifetime (60 years) |
Key data source | ECHELON-1, an open-label, multicentre, randomized phase III trial |
Submitted results | ICER = $118,104 per QALY gained (incremental costs: $93,282; incremental QALYs: 0.79) |
Key limitations |
|
CDA-AMC reanalysis results |
|
ABVD = doxorubicin-bleomycin-vinblastine-dacarbazine; AE = adverse event; ASCT = autologous stem-cell transplant; AVD = doxorubicin- vinblastine-dacarbazine; AVEPC = doxorubicin-vincristine-etoposide–prednisone-cyclophosphamide; BEACOPP = bleomycin-etoposide-doxorubicin-cyclophosphamide-vincristine-procarbazine; BV = brentuximab vedotin; ICER = incremental cost-effectiveness ratio; HL = Hodgkin lymphoma; LY = life-year; PFS = progression-free survival; QALY = quality-adjusted life-year; RDI = relative dose intensity.
Conclusions
Based on the CDA-AMC Clinical Review of the ECHELON-1 trial, a phase III, open-label, randomized controlled trial, overall survival (OS), progression-free survival (PFS), and modified progression-free survival (mPFS) were consistently in favour of brentuximab vedotin (BV) in combination with doxorubicin-vinblastine-dacarbazine (AVD) compared to doxorubicin-bleomycin-vinblastine-dacarbazine (ABVD) in the intention-to-treat (ITT) population, which comprised previously untreated adult patients with advanced-stage classical Hodgkin lymphoma (HL). However, the clinical significance of the magnitude of the treatment differences is uncertain and concerns remain about the high percentages in loss to follow-up and withdrawal of patients in both OS and PFS analyses. In addition, although subgroup analyses of the OS and mPFS results signalled that BV plus AVD may be more effective in patients with stage IV classical HL than in those with stage III HL, conclusions regarding subgroup differences between stage III versus stage IV patients are uncertain because the study was not designed to detect differences between these subgroups, and patients were not stratified according to disease stage at randomization. More patients treated with BV plus AVD experienced severe adverse events (AEs) compared with those in the ABVD group. Given that all patients in the pivotal ECHELON-1 trial were required to be aged 18 years or older, the ECHELON-1 trial did not reflect results for pediatric patients. The AHOD1331 trial indicated that BV plus doxorubicin-vincristine-etoposide-prednisone-cyclophosphamide (AVEPC) provides a clinically meaningful benefit in event-free survival (EFS) compared to doxorubicin-bleomycin-vincristine-etoposide–prednisone-cyclophosphamide (ABVE-PC) in patients aged 2 years to 21 years with high-risk classical HL.
CDA-AMC undertook reanalyses to address limitations in the economic evaluation submitted by the manufacturer by removing treatment-specific utility values, adding AE disutilities, and eliminating relative dose intensity (RDI). CDA-AMC could not resolve all outstanding limitations in the analysis and relied on scenario analyses to explore the impact of some of these limitations.
In the CDA-AMC reanalysis, the incremental cost-effectiveness ratio (ICER) for BV plus AVD compared to ABVD was $115,865 per quality-adjusted life-year (QALY) gained (incremental costs: $105,110; incremental QALYs: 0.91). As the manufacturer’s model used ECHELON-1 trial data, which excluded pediatric patients, results represent the cost-effectiveness of BV plus AVD for the treatment of previously untreated adult patients with advanced-stage HL. CDA-AMC’s base case was in line with the manufacturer’s results, which estimated an ICER of $118,104 per QALY gained when comparing BV plus AVD with ABVD. The incremental benefit of BV plus AVD was driven mainly by an improvement in PFS (incremental life-years [LYs] in front-line progression-free: 1.94). A reduction in the price of BV of at least 55% would be necessary for BV plus AVD to be considered cost-effective at a willingness-to-pay (WTP) threshold of $50,000 per QALY gained. A 55% price reduction would reduce the unit price of a 50 mg vial of BV from $4,840 to $2,178, which would reduce 28-day cycle costs for BV from $19,360 to $8,712. No information was provided for the population aged less than 18 years.
A number of limitations could not be addressed given the availability of clinical information and the nature of the manufacturer’s economic model. CDA-AMC was unable to address the lack of comparative clinical information for BV plus AVD versus other relevant comparators, including bleomycin-etoposide-doxorubicin-cyclophosphamide-vincristine-procarbazine (BEACOPP) and fluorodeoxyglucose PET-adapted BEACOPP and PET-adapted ABVD; as such, the cost-effectiveness of BV plus AVD versus these comparators is unknown. CDA-AMC was also unable to estimate the cost-effectiveness of BV plus AVEPC compared with ABVE-PC. The manufacturer’s submitted model structure did not allow for the exploration of the treatment effect of BV plus AVD by disease stage. As such, the cost-effectiveness of BV plus AVD by disease stage is unknown. Finally, as patients aged less than 18 years were excluded from the ECHELON-1 trial, which was used to populate the economic model, the clinical effectiveness, and therefore the cost-effectiveness, of BV plus AVD compared with most relevant comparators for those aged less than 18 years is unknown.
Additional limitations that were explored in scenario analyses included the potential bias around investigator-assessed PFS given the open-label nature of the ECHELON-1 trial, which could have biased results in favour of BV plus AVD. If the actual treatment effect of BV plus AVD on PFS was aligned more closely with the mPFS outcome (which was assessed by an independent review facility), the ICER for BV plus AVD is expected to be higher than estimated in the CDA-AMC base case. Additionally, increasing the proportion of patients in the model undergoing an autologous stem-cell transplant (ASCT), which was deemed to be lower than what the clinical experts consulted by CDA-AMC for this review expected to see clinical practice in Canada, resulted in a slightly higher ICER. These scenario analyses indicate that the CDA-AMC base case may overestimate the benefit associated with BV plus AVD and therefore likely represents a lower limit on what the ICER may be in actual practice.
Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CDA-AMC review process.
One patient group, Lymphoma Canada, provided input for this review. Patient input was received from an online survey focused on patients with HL. Twenty-six responses were received, with 5 responders being from Canada. Among the patients who answered the survey, 3 indicated they were treated with ABVD, 2 with other forms of chemotherapy, and 1 with R-CHOP regimen, which consists of cyclophosphamide, doxorubicin hydrochloride (hydroxydaunomycin), and vincristine sulphate. These patients reported being satisfied with the treatment provided to them. Patient feedback emphasized that controlling disease symptoms, maintaining disease remission, increasing length of life, and improving quality of life were the most important factors to be considered in a novel treatment. Three patients had experience with BV plus AVD. Two of these patients reported to be still in remission, and 1 did not report remission status. Patients reported the most common AEs during their experience with BV to be fatigue, neutropenia, constipation, and joint or muscle pain, and 2 patients referred to the AEs as manageable. Two patients rated their experience with BV plus AVD as very good, while 1 patient reported having had a poor experience.
Two clinician groups provided input for this review: the Ontario Health (Cancer Care Ontario) Hematology Cancer Drug Advisory Committee and Pediatric Oncology Group of Ontario. Clinician input included ABVD or BEACOPP as current first-line treatments for patients with stage III and IV HL. In the pediatric setting, ABVE-PC and vincristine-etoposide-prednisone-doxorubicin, followed by cyclophosphamide-vincristine-prednisone-dacarbazine (OEPA-COPDAC) regimens are the 2 treatments used in practice. In the pediatric setting, the chemotherapy regimen AVEPC is the current backbone used in practice with BV. Clinician input emphasized that treatment goals are to improve outcomes with first-line therapy to avoid the need for second-line therapy. For the pediatric setting, clinician input indicated that the main treatment goal is to avoid recurrence and consequently to minimize potential late effects which impact quality of life, add to health care utilization costs in survivors. Additionally, patients experiencing treatment failure or relapse may require additional cytotoxic therapy and ASCT.
Drug plan input indicated concerns with missing relevant comparators such as the BEACOPP regimen and PET-adapted regimens; according to the current provisional funding algorithm, patients who relapse would be eligible for BV re-treatment if a relapse occurs more than 12 months after completion of prior BV therapy with at least 6 months of response. The drug plans asked whether BV would be funded for patients who are aged less than 18 years, and those with an Eastern Cooperative Oncology Group Performance Status greater than 2, with nodular lymphocyte-predominant HL (no CD30 expression), with stage IIB disease at high risk, or with central nervous system involvement and symptoms of progressive multifocal leukoencephalopathy. Drug plan input indicated that primary prophylaxis with granulocyte colony-stimulating factor (G-CSF) is typically prescribed with BV plus AVD, and is associated with an additional cost.
Several of these concerns were addressed in the manufacturer’s model:
The manufacturer’s submitted model accounted for quality of life and length of life by using QALYs as the primary outcome.
The cost of G-CSF prophylaxis was included.
CDA-AMC was unable to address the following concerns raised from stakeholder input:
BEACOPP was not included as a relevant comparator in the analysis.
The pediatric population was not included in the ECHELON-1 study, and relevant comparators were not included in the analysis.
Economic Review
The current review is for BV (Adcetris) for previously untreated patients with advanced-stage HL and considers 2 reimbursement requests: BV in combination with ABVD for the treatment of previously untreated patients with advanced-stage HL, and BV in combination with AVEPC in previously untreated high-risk HL in the pediatric population.1 This is a tumour group submission sponsored by the BC Cancer Agency and Pediatric Oncology Group of Ontario. However, the review is based on a previous manufacturer-initiated submission (Project Number PC0311 to 000) that was withdrawn. As such, the cost-effectiveness evidence is based on the evidence submitted by the manufacturer for Project Number PC0311 to 000.
Economic Evaluation
Summary of Manufacturer’s Economic Evaluation
Overview
The manufacturer submitted a cost-utility analysis assessing BV plus AVD compared with ABVD in patients with previously untreated patients with advanced-stage HL.1 The modelled population was aligned with the patient population in the ECHELON-1 trial, which included adults with untreated advanced-stage HL.2
The recommended dose of BV is 1.2 mg/kg of body weight, given intravenously, on days 1 and 15 of each 28-day cycle for up to 6 cycles, according to the product monograph.3 BV is indicated in combination with 25 mg/m2 of doxorubicin, 6 mg/m2 of vinblastine, and 375 mg/m2 of dacarbazine. The manufacturer-submitted price for BV is $4,840 per 50 mg vial. Assuming wastage, as well as a mean body weight (75 kg) and body surface area (1.88 m2) based on the ECHELON-1 trial, the cost of BV in the adult population is estimated to be $19,360 over a 28-day cycle and $116,160 over 6 cycles in the manufacturer’s model. The BV plus AVD regimen is estimated to cost $21,584 over a 28-day cycle and $129,506 over 6 cycles. The comparator regimen, ABVD, consisted of 25 mg/m2 of doxorubicin, 10 units/m2 of bleomycin, 6 mg/m2 of vinblastine, and 375 mg/m2 of dacarbazine on days 1 and 15 of each 28-day cycle for up to 6 cycles. The ABVD regimen is estimated to cost $3,902 over a 28-day cycle and $23,412 over 6 cycles.
The clinical outcome of interest was QALYs and LYs. The economic analysis was undertaken using a lifetime time horizon (60 years) from the perspective of a public health care payer. Discounting of 1.5% per year was applied to both costs and outcomes.
Model Structure
A semi-Markov model was developed in Microsoft Excel. The model consisted of 4 health states defined as front-line progression-free, post-progression and not receiving an ASCT, post-progression and receipt of an ASCT, and death (Figure 1). All patients entered the model in the front-line (i.e., receipt of their first-ever treatment) progression-free health state and received either BV plus AVD or ABVD. Patients in the front-line progression-free health state were at risk of disease progression or death. Upon progression, patients could go on to receive ASCT or no ASCT. Patients who failed front-line therapy and did not receive ASCT remained in a post-progression, no-ASCT state until death and incurred costs of subsequent therapies, including programmed cell death protein 1 (PD-1) inhibitors, such as nivolumab and pembrolizumab. Patients receiving ASCT after front-line failure were at risk of ASCT failure. Patients in whom ASCT failed less than 9 months after completion of front-line treatment were eligible to receive post-ASCT consolidation with BV, and incurred BV treatment costs. The proportion of patients who received BV consolidation was dependent on front-line treatment. In addition, a proportion of patients in whom ASCT failed incurred costs of subsequent therapies, which included nivolumab and pembrolizumab, and/or BV salvage therapy. However, patients who had received BV consolidation were assumed to be ineligible for BV salvage after ASCT failure. A proportion of patients progressing after ASCT were also assumed to undergo repeat ASCT, or an allogenic stem-cell transplant.
Model Inputs
Baseline characteristics (proportion of males: 58%; mean age: 39.5 years) and the probability of progression following front-line treatment were derived from the ECHELON-1 trial, an open-label, multicentre, randomized phase III trial.2 Investigator-assessed PFS and post-progression survival (PPS) of the ITT population obtained from the ECHELON-1 trial were used to inform the base-case analysis. Patient progression from the front-line progression-free health state to the post-progression states or death was based on extrapolation curves fit to PFS data from the ECHELON-1 trial. Due to an observation that PFS curves in the ECHELON-1 trial plateaued, the manufacturer used a mixture cure model. The probability that the progression event, defined according to PFS, was death, was based on the ECHELON-1 trial and was treatment-specific. Based on the best-fitting mixture cure model, different front-line therapy cure proportions were observed for BV plus AVD and ABVD (proportion cured from front-line therapy at 8 years: 82.2% versus 74.7%, respectively). All extrapolated survival curves used in the model were selected based on the best fit according to the Akaike information criterion, Bayesian information criterion, visual inspection, and clinical plausibility. After 8 years, patients who remained in the front-line progression-free health state were assumed to be cured and no longer transitioned to post-progression health states.1 At this time, patients remaining in the front-line progression-free health state could progress to death based on age- and gender-specific general population mortality, weighted to the gender distribution of patients in ECHELON-1 trial,4 and adjusted for non-HL–related excess mortality obtained from the literature.5 Transitions to death from the post-progression health states were based on PPS estimates derived from the ITT population of the ECHELON-1 trial who experienced disease progression and stratified by patients who had or did not have evidence of receipt of ASCT post-progression, as well as the general population mortality from Canadian life tables,4 adjusted for non-HL–related excess mortality.5 Survival distributions were fit separately for PPS with or without ASCT, independent of front-line therapy received. For the PPS of patients who had evidence of receipt of ASCT, the Gompertz distribution was chosen as it implied a cure fraction of approximately 83%. For the PPS of patients without evidence of receipt of ASCT, the log-normal distribution was chosen based on statistical fit and clinical plausibility.
The proportion of patients eligible for ASCT (44%) was based on data from the ECHELON-1 trial.2 Among patients who failed therapy with ABVD, 68% were assumed to received BV consolidation therapy based on a previous CDA-AMC submission.6 The proportion of patients receiving BV consolidation after ASCT for who failed front-line therapy with BV plus AVD (33%) was based on clinician input. The manufacturer assumed that all patients who did not receive BV consolidation therapy and who progressed after ASCT would receive BV post–transplant failure. In addition, 75% of these patients were assumed to receive a PD-1 inhibitor (pembrolizumab or nivolumab) after BV post–transplant failure, based on a pivotal phase II trial.7 It was assumed that all patients who progress after receiving ASCT with BV consolidation would receive a PD-1 inhibitor as salvage therapy. The proportion of patients receiving G-CSF prophylaxis was obtained from clinician input and a previous CDA-AMC submission.6 Several other probabilities related to the likelihood of receiving an allogenic stem-cell transplant after ASCT failure, and receiving a repeat ASCT after initial ASCT failure were identified from the published literature and clinician input. Among patients who were not eligible for ASCT, the manufacturer assumed 50% would receive nivolumab and 50% pembrolizumab as subsequent therapies.
Probabilities of AEs were based on frequencies observed in the ECHELON-1 trial.2 AEs considered in the model included serious neutropenic events, pulmonary toxicity, peripheral neuropathy, and anemia.
Health-state utility values for patients in the front-line PFS health state were derived from the ECHELON-1 trial using the EQ-5D-3L instrument and values sets from the UK.1 These values were specific to the treatment regimen and stratified as to whether the patient was on or off treatment. The manufacturer also accounted for declining utilities with age, by setting utilities to the minimum of the state–specific or age- and sex-matched general population norm utility values obtained from literature using the Health Utilities Index Mark 3 questionnaire.8 Treatment-related AE disutilities were assumed to be included in these measures and were not explicitly modelled in the base case. Health-state utility values related to the post-progression ASCT state, which included separate values for the first 3 months and subsequent months following ASCT, were identified from the literature.9,10 The utility for post-progression and not receiving ASCT was obtained from literature.10
Costs in the model included those for drug acquisition, drug administration, supportive care (e.g., clinician assessments, laboratory tests), AE management, subsequent therapy, and terminal care. Costs associated with drug acquisition were acquired from the manufacturer for the submitted drug, and a combination of the Ontario Drug Benefit Formulary, l’Association Québécoise des pharmaciens propriétaires, and a previous CDA-AMC submission.6,11,12 The number of cycles for each drug was based on the mean number of cycles from various trials.7,13,14 Costs associated with drug administration and subsequent treatment were obtained from publicly available Canadian sources and literature, including Statistics Canada, Job Bank Canada, and Cancer Care Ontario.15-17 The cost of AEs was identified using the Ontario Case Costing Analysis Tool.18 Costs related to stem-cell transplants were based on the interprovincial billing rates for designated high-cost transplants and induction therapy from Raymakers et al.19,20 Monitoring costs were considered only for post-progression states, and the frequency of resource use was based on monitoring guidance from National Comprehensive Cancer Network guidelines.21 Monitoring costs were obtained from the Ontario Schedule of Benefits.22 The model incorporated terminal-care costs for patients based on a value identified in the literature.23 All costs were presented in 2022 Canadian dollars. Costs obtained from other years were inflated to 2022 values using the consumer price index from Statistics Canada.24
Summary of Manufacturer’s Economic Evaluation Results
All analyses were run probabilistically (5,000 iterations for the base-case and scenario analyses). The deterministic and probabilistic results were similar. The probabilistic findings are presented in the following section.
Base-Case Results
In the manufacturer’s base-case analysis, treatment with BV plus AVD was associated with incremental costs of $93,282 and a gain of 0.79 QALYs compared with ABVD over the lifetime (60 years) time horizon, resulting in an ICER of $118,104 per QALY gained (Table 3). The probability of BV plus AVD being cost-effective at a WTP threshold of $50,000 per QALY gained compared to ABVD was 0%. Additional results from the manufacturer’s submitted economic evaluation base case are available in Appendix 3.
Results were driven by the drug acquisition costs of BV plus AVD (incremental costs of $93,282), the predicted gain in LYs (incremental: 0.98), and the predicted reduction in costs associated with the post-progression ASCT state (incremental savings of $10,349) and post-progression no-ASCT state (incremental savings of $2,539). All the incremental LYs gained for BV plus AVD were accrued in the front-line progression-free health state. CDA-AMC noted that approximately 12% of the incremental QALYs in the manufacturer’s base case were accrued during the first 8.3 years (100 months) of the model (the maximum follow-up of the ECHELON-1 trial), indicating that 88% of the incremental benefit was accrued during the post-trial period. The submitted analysis is based on Association Québécoise des pharmaciens propriétaires and wholesale list prices for all treatments, other than the BV, including subsequent therapies.
Table 3: Summary of the Manufacturer’s Economic Evaluation Results
Drug | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER vs. ABVD ($ per QALY) |
|---|---|---|---|---|---|
ABVD | 72,827 | Reference | 19.55 | Reference | Reference |
BV + AVD | 166,108 | 93,282 | 20.34 | 0.79 | 118,104 |
ABVD = doxorubicin-bleomycin-vinblastine-dacarbazine; AVD = doxorubicin-vinblastine-dacarbazine; BV = brentuximab vedotin; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Source: Manufacturer’s pharmacoeconomic submission.1
Sensitivity and Scenario Analysis Results
The manufacturer undertook scenario analyses which varied several parameters, including the time horizon, discount rate, alternative percentage of patients receiving ASCT after front-line failure, and the application of disutilities for AEs. Shortening the time horizon had the greatest impact, with the ICER ranging from $558,449 per QALY gained with a 10-year time horizon to $148,840 per QALY gained with a 30-year time horizon.
CDA-AMC Appraisal of the Manufacturer’s Economic Evaluation
CDA-AMC identified several key limitations to the manufacturer’s analysis that have notable implications on the economic analysis:
Investigator-assessed PFS is uncertain: Extrapolations of investigator-assessed PFS from the ECHELON-1 trial were used to determine the duration patients spent in the front-line progression-free health state. As of the June 1, 2021, data cut-off date, the hazard ratio (HR) for investigator-assessed PFS was 0.678 (95% confidence interval, 0.532 to 0.863) and the absolute difference in the number of PFS events was 7%, favouring BV plus AVD.2 The difference in PFS between BV plus AVD and ABVD was the main driver of clinical benefit for BV plus AVD in the manufacturer’s submitted model. According to the CDA-AMC Clinical Review, as ECHELON-1 was an open-label trial, the investigators, patients, and the manufacturer were aware of the patients’ treatment allocation. Important outcomes such as PFS therefore had no blinded assessment and are prone to detection bias. The manufacturer confirmed that no sensitivity analyses were conducted to explore the robustness of the PFS findings, including the potential impact of investigator assessment. Results of the PFS assessed by an independent review facility were reported for the April 20, 2017, data cut-off date, but were not reported for the June 1, 2021, data cut-off date.
In addition, according to the CDA-AMC Clinical Review, high percentages of loss to follow-up and withdrawal by patients were noted in the investigator-assessed PFS analyses. Although the percentages of loss to follow-up and withdrawal by patients appeared to be balanced between treatment arms, the reasons for loss to follow-up and withdrawal could differ between groups, which could lead to biased estimates of treatment effects. Moreover, as the manufacturer confirmed, no relevant sensitivity analyses were available for investigator-assessed PFS, due to which the potential impact of high loss to follow-up and withdrawal could not be further determined.
CDA-AMC was not able to address this limitation. As the time spent progression-free is driving the benefit of BV plus AVD, bias in PFS estimates could result in the cost-effectiveness results favouring BV plus AVD. The primary outcome of the ECHELON-1 trial, mPFS, was assessed by an independent review facility, which would help limit detection bias related to the open-label trial design, had a HR of 0.770 (95% confidence interval, 0.603 to 0.982). However, the clinical experts consulted by CDA-AMC noted that the definition of mPFS (counting a response that was less than complete at the end of the front-line therapy as an event) is not consistent with clinical practice in defining disease progression or first-line treatment failure in advanced HL, and there is a concern that the results for mPFS would be biased given that the administration of new anticancer therapy was at the discretion of the treating physician. To explore the influence of potential bias in investigator-assessed PFS, CDA-AMC conducted a scenario analysis using the mPFS HR.
The manufacturer’s model does not assess the cost-effectiveness of BV in pediatrics: The manufacturer’s submitted analysis reflected the cost-effectiveness of BV plus AVD among untreated adult patients with advanced-stage HL, with effectiveness informed by the ECHELON-1 trial.2 The ECHELON-1 trial did not include patients aged younger than 18 years.2 As such, the clinical- and cost-effectiveness of BV plus AVD in those under 18 is unknown. The manufacturer’s analysis therefore reflects the cost-effectiveness of BV plus AVD only in those aged 18 and older.
Clinical expert feedback and clinician group input indicated that BV would be considered for use in the advanced HL pediatric population; however, it would be used with a different chemotherapy backbone, such as AVEPC. To address the significant unmet needs in the pediatric patient population, the AHOD1331 trial was examined in the Clinical Review.25 However, as the chemotherapy backbone used in the AHOD1331 trial (AVEPC) is different from the backbone studied in the ECHELON-1 trial used to populate the economic model, the clinical efficacy and cost-effectiveness of BV plus AVD in the pediatric population remains unknown. Efficacy results from the AHOD1331 trial indicated that BV plus AVEPC provides a clinically meaningful benefit in EFS compared to ABVE-PC in patients aged 2 to 21 years with high-risk classical HL, defined as Ann Arbor stage IIB with bulk tumour, stage IIIB, stage IVA, and stage IVB HL.
CDA-AMC was not able to address this limitation due to a lack of clinical data for BV plus AVD in the pediatric population. As noted in its Clinical Review, CDA-AMC was unable to draw conclusions related to the efficacy of BV plus AVD in the pediatric population as the pivotal ECHELON-1 trial did not include patients aged younger than 18 years. As such, the clinical- and cost-effectiveness of BV plus AVD in the pediatric population is unknown.
CDA-AMC was also unable to address the lack of cost-effectiveness evidence for BV plus AVEPC in the pediatric population as it is beyond the scope of our reviews to incorporate clinical data that were not provided as part of the manufacturer’s submission. Additionally, the manufacturer’s submitted model relied on PFS and PPS outcomes, neither of which were included in the AHOD1331 trial. As such, the cost-effectiveness of BV plus AVEPC is unknown.
Relevant comparators were excluded: The relevant comparator considered in the manufacturer’s base case was restricted to the ABVD regimen. According to clinical expert feedback received for this review, as well as registered clinician input, while most adult patients with advanced HL will be treated with ABVD, between 10% and 20% of adult patients with advanced HL will be treated with BEACOPP, making BEACOPP a relevant comparator. Additionally, clinical expert feedback received for this review indicated that some jurisdictions and centres across Canada use PET-adapted ABVD or BEACOPP, which allows for front-line therapy dose escalation or de-escalation or a change in treatment regimen after the second treatment cycle. PET-adapted regimens were not included in the manufacturer’s economic evaluation. CDA-AMC Economic Evaluation Guidelines state that all interventions currently used for treatment and potentially displaced by a new technology should be considered in an economic evaluation. Excluding BEACOPP and PET-adapted treatments as relevant comparators was therefore not appropriate. As ABVD in the ECHELON-1 trial was not PET-guided, the efficacy of PET-adapted ABVD versus BV plus AVD is unknown. In addition, according to clinical experts consulted by the review team, the current treatment options for pediatric patients with advanced HL include ABVE-PC and OEPA-COPDAC. Neither of these regimens were included in the economic evaluation.
CDA-AMC could not address this limitation. According to the CDA-AMC Clinical Review report, the manufacturer provided a feasibility assessment that determined it would be infeasible to conduct indirect treatment comparisons of BV plus AVD versus other front-line therapies examined in clinical studies for advanced HL. The manufacturer therefore did not submit direct or indirect evidence for BV plus AVD versus other relevant comparators, and no conclusions regarding the comparative effectiveness of BV plus AVD versus BEACOPP or PET-adapted regimens could be made.
The model structure lacked face validity: The manufacturer’s submitted model structure included 4 health states: front-line progression-free, post-progression and receipt of ASCT, post-progression and no receipt of ASCT, and death.1 Patients who experienced progression after front-line therapy and received ASCT remained in this health state until death. However, this health state did not differentiate between patients who were cured by ASCT and those who were not. Although costs associated with relapse after undergoing ASCT were considered in the model, all patients in the health state were expected to have the same survival, regardless of whether they were or were not cured by ASCT. Additionally, all patients in the health state were assigned the same utility value of 0.879. According to clinical experts consulted by CDA-AMC for this review, patients who are not cured by ASCT are expected to have a poorer life expectancy and utility value compared to those who are cured by ASCT. From a methodological perspective, a health state in an economic model should represent a homogenous group of patients who have similar expected costs and quality-of-life considerations; this is not captured by the modelled post-progression and receipt-of-ASCT health states. The implications of heterogeneity in health states are well documented in the literature.26
Second, the manufacturer-submitted model did not stratify patients by disease stage (i.e., stage III and IV patients).1 According to the Clinical Review, stage IV HL patients have a poorer prognosis compared with stage III patients. Additionally, according to the Clinical Review, subgroup analyses by HL stage suggest there may be a difference in treatment effects between patients with stage III and IV classical HL for mPFS, PFS, and OS. However, CDA-AMC was not able to make definitive conclusions about whether there is a true difference in efficacy between patients with stage IV classical HL and those with stage III HL due to several limitations, including the failure to stratify patients based on disease stage at randomization. CDA-AMC Economic Evaluation Guidelines indicate that potential sources of heterogeneity that may lead to differences in parameter-input values across distinct subgroups, including those due to differences in natural history, should be examined in the submitted model.27 However, it was not possible to address heterogeneity among the different stages of the target population through stratified analysis in the manufacturer’s submitted model. As such, CDA-AMC could not assess the cost-effectiveness of BV plus AVD stratified by disease stage. Additionally, the distribution of disease stage was not reflective of the distribution of advanced HL patients in Canadian clinical practice. According to the clinical expert input received by the review team, the percentage of patients with stage IV HL in the ECHELON-1 trial population (64%) is higher than the proportion in Canadian clinical practice (approximately 50%).28 The impact of this could also not be explored due to the limitations of the manufacturer’s model structure.
CDA-AMC also noted that the model structure restricted patients to either receive or not receive ASCT if they relapsed after receiving front-line therapy. In the ECHELON-1 trial, 22% of the safety population (n = 1,321) received at least 1 subsequent treatment after front-line therapy. This simplification of the HL treatment path did not reflect clinical practice, where, based on the provisional funding algorithm, patients can opt for subsequent treatments before deciding to undergo ASCT.
CDA-AMC could not address these model structure limitations. The direction and magnitude of the impact of these structural limitations could not be evaluated and are therefore unknown.
Use of treatment-specific health-utility values is inappropriate: The manufacturer’s submitted base case utilized treatment-specific health-utility values in both the progression-free and post-progression health states.1 According to the CDA-AMC Guidelines for the Economic Evaluation of Health Technologies, utility values should not be treatment-specific but differentiated by health state and/or events, with treatment efficacy captured via health-state occupancy and event occurrence driving the difference in observed QALYs.27 Given the use of treatment-specific utility values, disutilities related to occurrence of AEs were not directly included in the base-case analysis. This is not aligned with the CDA-AMC Guidelines for the Economic Evaluation of Health Technologies, which recommends the adjustment of the utility for a specific health state by applying a disutility for an AE to allow the utility for the health state with an AE to be estimated.27
In addition, the manufacturer used the UK time trade-off value set instead of a Canadian value set, which is available for both the EQ-5D-3L and EQ-5D-5L questionnaires. It would be more appropriate to adopt utility values reflective of the preferences of the general population in Canada.27
CDA-AMC addressed this limitation by using the same utility values by health state for both BV plus AVD and ABVD. CDA-AMC applied the values obtained for the standard-of-care arm in the ECHELON-1 trial, as well as incorporating AE-related disutilities based on AE event rates. CDA-AMC noted that the difference between BV plus AVD and ABVD treatment-specific utilities (0.059) was higher than the disutility modelled by including AEs (0.005). Based on clinical expert feedback obtained for this review, it is expected that most of the differences in utilities between treatments can be explained by differences in AE frequencies. This indicates that it is possible that not all AE effects are captured by the manufacturer’s incorporation of AE disutilities. Any additional disutility due to AEs not captured by the analysis will favour BV plus AVD, as AE frequencies were higher for BV plus AVD patients compared to ABVD.
CDA-AMC could not address the use of UK instead of Canadian values sets. The impact of using a UK value set instead of Canadian value sets when valuing patient health-state utilities in the model is unknown, adding uncertainty to the results.
The proportion of patients undergoing ASCT is uncertain: The manufacturer indicated that the proportion of patients undergoing ASCT after failure of front-line therapy (44%) was derived from the ECHELON-1 trial.2 When validating this estimate, CDA-AMC noted that, among patients whose disease progressed (n = 225), 107, or 47%, underwent ASCT.2 However, this simple calculation did not account for patient censoring during trial follow-up. By the June 1, 2021, data cut-off date, 105 patients (11% of the total sample size) were censored due to loss of follow-up and 199 (15% of total sample size) were censored due to withdrawal by patient. As such, the calculated proportion of patients undergoing ASCT is uncertain. In addition, clinical expert feedback indicated that the proportion of patients undergoing ASCT is expected to be approximately 70% in Canadian clinical practice, given that advanced HL patients are generally young and eligible for subsequent therapy with ASCT.
CDA-AMC addressed the uncertainty in the proportion of patients undergoing ASCT by increasing it to 70% in a scenario analysis.
Patients’ monitoring costs were not considered in front-line progression-free health state: The model did not include any disease monitoring or health care resource costs, such as physicians’ visits, or laboratory or imaging assessments, in the progression-free health state. This is inappropriate according to clinical expert input received by CDA-AMC, as patients would have follow-up visits every 3 months during the first 2 years of diagnosis, and every 6 months for the subsequent 3 years, even while remaining progression-free. In addition, patients would be expected to undergo blood work at those visits.
CDA-AMC could not address this limitation. As patients receiving BV plus AVD spend more time in the progression-free state than those receiving ABVD, not considering monitoring costs in the progression-free health states favours BV plus AVD.
The number of cycles of treatment received is likely underestimated: The manufacturer used the mean dosage per course and mean number of cycles obtained from the ECHELON-1 trial to calculate drugs dosage.1 As such, the manufacturer’s base case incorporated reduced dose intensities for all therapies (expected versus observed doses). This assumes the RDI correlates perfectly with expected cost. Given the inability to link distinct dose intensity with outcomes, the CDA-AMC base case does not incorporate RDI. A reduced or enhanced RDI can be derived from many factors, including clinical discretion, delayed dose, missed dose, or reduction in dose. When considering wastage and discontinuation, each component can have a different influence on drug costs. Likewise, it is unclear how treatment discontinuation influences RDI.
RDI calculations were excluded from the CDA-AMC base case.
Additionally, the following key assumptions were made by the manufacturer and have been appraised by CDA-AMC (Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Manufacturer’s key assumption | CDA-AMC comment |
|---|---|
Patients who did not experience disease progression within 8 years were considered cured. | Appropriate, according to clinical experts consulted by CDA-AMC for this review. |
Association Québécoise des Pharmaciens Propriétaires costs were used for drug acquisition pricing. | Inappropriate. CDA-AMC corrected the drug acquisition costs using costs from the Delta PA database. |
Patients undergoing ASCT who had progressed for less than 9 months after completion of front-line treatment were eligible to receive BV consolidation following ASCT. | Uncertain. Clinical experts consulted by CDA-AMC for this review indicated that there is heterogeneity in practice when deciding if patients will be treated with consolidation BV therapy after quick progression after BV + AVD front-line therapy due to a lack of evidence of benefit in this scenario. However, alternative assumptions had minimal impact on the cost-effectiveness results. |
PPS curves were used to populate death from the post-progression health states in the economic model. | Uncertain. PPS, which was defined as time from progression to death and calculated for patients who experienced disease progression by subtracting PFS from OS obtained the ECHELON-1 trial and stratified by ASCT status, was submitted by the manufacturer to support pharmacoeconomic analyses. However, PPS was not prespecified in the ECHELON-1 trial protocol and data were obtained via post hoc analysis. Its estimates are therefore considered uncertain by CDA-AMC clinical team. |
The proportion of PFS events that are death were 14.3% and 18.9% for BV + AVD and ABVD, respectively, obtained from ECHELON-1 trial. | Inappropriate. First, the values used by the manufacturer assumed that the proportion of events that are death will remain constant over time, which is not a suitable assumption for a semi-Markov model structure. Second, these values were calculated using the total number of events and total number of deaths at 60 months, which does not account for censoring or time dependencies. The impact of these assumptions is unknown. |
ASCT = autologous stem-cell transplant; ABVD = doxorubicin-bleomycin-vinblastine-dacarbazine; AVD = doxorubicin-vinblastine-dacarbazine; BV = brentuximab vedotin; HL = Hodgkin lymphoma; PPS = post-progression survival.
CDA-AMC Reanalyses of the Economic Evaluation
Base-Case Results
The CDA-AMC base case was derived by making changes in model parameter values and assumptions, in consultation with clinical experts. Table 5 details each change made to derive the CDA-AMC revised base case, which was conducted in a stepwise approach to highlight the impact of each change.
CDA-AMC was unable to address other key limitations of the model (described previously), including potential bias around investigator-assessed PFS given the open-label nature of the ECHELON-1 trial, missing relevant comparators, lack of generalizability of findings to the pediatric population, uncertainty on the treatment effect for the distinct disease stages, and lack of face validity of the model structure. Due to these key limitations, it is uncertain that costs and health outcomes have been appropriately captured.
CDA-AMC undertook a stepped analysis, incorporating each change in Table 5 to the manufacturer’s base case to highlight the impact of each change (Table 6). Results from the CDA-AMC base-case reanalysis suggest that, compared to ABVD, BV plus AVD was associated with higher costs ($105,110) and yielded more QALYs (0.91 QALYs), resulting in an ICER of $115,865 per QALY gained. Like the manufacturer's base case, BV plus AVD had a 0% probability of being cost-effective at a WTP threshold of $50,000 per QALY gained. Of the 0.91 QALYs gained for BV plus AVD, 0.15 (16%) were gained during the trial period, meaning 84% of the incremental QALYs for BV plus AVD were extrapolated. All incremental benefit for BV plus AVD accrued in the front-line progression-free health state (Table 11). The additional drug acquisition costs were the main driver for the incremental cost difference between the 2 treatments. The disaggregated results for CDA-AMC reanalysis are available in Table 11.
Table 5: CDA-AMC Revisions to the Submitted Economic Evaluation
Stepped analysis | Manufacturer’s value or assumption | CDA-AMC value or assumption | |
|---|---|---|---|
Corrections to manufacturer’s base case | |||
Acquisition cost of doxorubicin | $285.00 | $255.00a | |
Acquisition cost of vinblastine | $13.49 | $176.79a | |
Acquisition cost of dacarbazine | $242.80 | $251.82a | |
Changes to derive the CDA-AMC base case | |||
1. Health-state utilities | Treatment-specific health-state utility values obtained from the ECHELON-1 trial (BV + AVD: 0.747 for initial PFS, and 0.864 for long-term PFS; ABVD: 0.806 for initial PFS and 0.876 for long-term PFS) AE disutilities were not included in the analysis | Health state–specific utility values based on the standard-of-care values derived from the ECHELON-1 trial. (0.747 for initial PFS, and 0.864 for long-term PFS for both BV + AVD and ABVD) AEs disutilities were included | |
2. RDI | Included | Excluded | |
CDA-AMC base case | — | 1 + 2 | |
AE = adverse events; RDI = relative dose intensity.
aSource: Delta PA database.29
CDA-AMC undertook a stepped analysis, incorporating each change in Table 5 to the manufacturer’s base case to highlight the impact of each change (Table 6). Results from the CDA-AMC base-case reanalysis suggest that, compared to ABVD, BV plus AVD was associated with higher costs ($105,110) and yielded more QALYs (0.91 QALYs), resulting in an ICER of $115,865 per QALY gained. Like the manufacturer's base case, BV plus AVD had a 0% probability of being cost-effective at a WTP threshold of $50,000 per QALY gained. Of the 0.91 QALYs gained for BV plus AVD, 0.15 (16%) were gained during the trial period, meaning 84% of the incremental QALYs for BV plus AVD were extrapolated. All incremental benefit for BV plus AVD accrued in the front-line progression-free health state (Table 11). The additional drug acquisition costs were the main driver for the incremental cost difference between the 2 treatments. The disaggregated results for CDA-AMC reanalysis are available in Table 11.
Table 6: Summary of the Stepped Analysis of the CDA-AMC Reanalysis Results
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($ per QALY) |
|---|---|---|---|---|
Manufacturer’s base case (probabilistic) | ABVD | 72,827 | 19.55 | Reference |
BV + AVD | 166,108 | 20.34 | 118,104 | |
Manufacturer’s base case (deterministic) | ABVD | 77,872 | 19.66 | Reference |
BV + AVD | 169,970 | 20.41 | 123,371 | |
Manufacturer’s corrected base case (deterministic) | ABVD | 81,448 | 19.66 | Reference |
BV + AVD | 173,428 | 20.41 | 123,215 | |
CDA-AMC reanalysis 1 | ABVD | 81,448 | 19.66 | Reference |
BV + AVD | 173,428 | 20.54 | 105,635 | |
CDA-AMC reanalysis 2 | ABVD | 83,035 | 19.66 | Reference |
BV + AVD | 186,960 | 20.41 | 139,215 | |
CDA-AMC base case (deterministic) | ABVD | 83,035 | 19.66 | Reference |
BV + AVD | 186,960 | 20.54 | 119,353 | |
CDA-AMC base case (probabilistic) | ABVD | 77,989 | 19.55 | Reference |
BV + AVD | 183,099 | 20.46 | 115,865 |
ABVD = doxorubicin-bleomycin-vinblastine-dacarbazine; AVD = doxorubicin-vinblastine-dacarbazine; BV = brentuximab vedotin; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Scenario Analysis Results
Price-reduction analyses were conducted on the CDA-AMC base case (Table 7). The analyses demonstrated that a price reduction of 55% in BV would be necessary for BV plus AVD to be considered cost-effective at a WTP threshold of $50,000 per QALY gained.
Table 7: CDA-AMC Price-Reduction Analyses
Analysis | ICERs for BV + AVD vs. ABVD ($ per QALY) | |
|---|---|---|
Price reduction | Manufacturer base case | CDA-AMC reanalysis |
No price reduction | 118,104 | 115,865 |
10% | 105,656 | 103,752 |
20% | 93,209 | 91,640 |
30% | 80,761 | 79,527 |
40% | 68,314 | 67,414 |
50% | 55,866 | 55,302 |
60% | 43,418 | 43,189 |
ABVD = doxorubicin-bleomycin-vinblastine-dacarbazine; AVD = doxorubicin-vinblastine-dacarbazine; BV = brentuximab vedotin; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Scenario analyses were conducted to explore the impact of alternative assumptions on the cost-effectiveness of BV plus AVD versus ABVD, including:
Adopting a time horizon of 70 months (the maximum observation time in ECHELON-1 trial) to account for uncertainty in long-term OS.
Applying an mPFS HR to BV plus AVD PFS extrapolations, to account for uncertainty in investigator PFS.
Assuming 70% of patients will undergo ASCT based on feedback from clinical experts.
The results of these scenario analyses are presented in Table 12. When the time horizon of the analysis was reduced to 70 months the ICER increased substantially to $729,491 per QALY gained. This indicates that the cost-effectiveness of BV plus AVD is contingent on long-term health gains being realized. A second scenario analysis using the mPFS HR to model BV plus AVD PFS extrapolations resulted in an increased ICER of $186,120 per QALY gained, due to the reduction of the incremental QALY benefit from 0.91 to 0.60 in comparison with the CDA-AMC base-care analysis. Finally, when it was assumed that 70% of patients will undergo ASCT, the ICER increased to $125,025 per QALY gained.
Issues for Consideration
This review of BV plus AVD for the treatment of previously untreated patients with advanced-stage HL is based on a previous sponsor-initiated submission (Project Number PC0311 to 000) that was withdrawn.30
BV plus AVD was previously reviewed by CDA-AMC for stage IV HL and received a recommendation to reimburse with clinical criteria and/or conditions.6 This review concluded that BV plus AVD was not cost-effective at the submitted price. The submitted price for BV in the previous review was the same as the current review ($4,840 per 50 mg vial).
Overall Conclusions
Based on the CDA-AMC Clinical Review of ECHELON-1, a phase III, open-label, randomized controlled trial, OS, PFS, mPFS were consistently in favour of BV plus AVD compared to ABVD in the ITT population of previously untreated adult patients with advanced-stage classical HL. However, the clinical significance of the magnitude of the treatment differences is uncertain and concerns remain about the high percentages in loss to follow-up and withdrawal of patients in both OS and PFS analyses. In addition, although subgroup analyses of the OS and mPFS results signalled that BV plus AVD may be more effective in patients with stage IV classical HL than those with stage III HL, conclusions regarding subgroup differences between stage III versus stage IV patients are uncertain because the study was not designed to detect differences between these subgroups and patients were not stratified according to disease stage at randomization. More patients treated with BV plus AVD experienced severe AEs than in the ABVD group. As all patients in the pivotal ECHELON-1 trial were required to be aged 18 years or older, the ECHELON-1 trial did not reflect results for pediatric patients. The AHOD1331 trial indicated that BV plus AVEPC provides a clinically meaningful benefit in EFS compared to ABVE-PC in patients aged 2 to 21 years with high-risk classical HL.
CDA-AMC undertook reanalyses to address limitations in the economic evaluation submitted by the manufacturer by removing treatment-specific utility values, adding AE disutilities, and eliminating the RDI. CDA-AMC could not resolve all outstanding limitations in the analysis and relied on scenario analyses to explore the impact of some of these limitations.
In the CDA-AMC reanalysis, the ICER for BV plus AVD compared to ABVD was $115,865 per QALY gained (incremental costs: $105,110; incremental QALYs: 0.91). Note that, as the manufacturer’s model used ECHELON-1 trial data, which excluded pediatric patients, results represent the cost-effectiveness of BV plus AVD for the treatment of previously untreated adult patients with advanced-stage HL. CDA-AMC’s base case was in line with the manufacturer’s results, which estimated an ICER of $118,104 per QALY gained when comparing BV plus AVD with ABVD. The incremental benefit of BV plus AVD was driven mainly by an improvement in PFS (incremental LYs in front-line progression-free: 1.94). A reduction in the price of BV of at least 55% would be necessary for BV plus AVD to be considered cost-effective at a WTP threshold of $50,000 per QALY gained. This would reduce the unit price of a 50 mg vial of BV from $4,840 to $2,178, which would reduce 28-day cycle costs for BV from $19,360 to $8,712. No information was provided for the population aged less than 18 years.
A number of limitations could not be addressed given the availability of clinical information and the nature of the manufacturer’s economic model. CDA-AMC was unable to address the lack of comparative clinical information for BV plus AVD versus other relevant comparators, including BEACOPP and PET-adapted BEACOPP and PET-adapted ABVD; as such, the cost-effectiveness of BV plus AVD versus these comparators is unknown. Additionally, CDA-AMC was unable to estimate the cost-effectiveness of BV plus AVEPC compared with ABVE-PC. Because the manufacturer’s submitted model structure did not allow for the exploration of the treatment effect of BV plus AVD by disease stage, the cost-effectiveness of BV plus AVD by disease stage is unknown. Finally, as patients aged less than 18 years were excluded from the ECHELON-1 trial, which was used to populate the economic model, the clinical effectiveness, and as such the cost-effectiveness, of BV plus AVD compared with most relevant comparators for those aged under 18 years is unknown.
Additional limitations that were explored in scenario analysis included the potential bias around investigator-assessed PFS given the open-label nature of the ECHELON-1 trial, which could have biased results in favour of BV plus AVD. If the actual treatment effect of BV plus AVD on PFS was aligned more closely with the mPFS outcome (which was assessed by an independent review facility), the ICER for BV plus AVD would be expected to be higher than estimated in the CDA-AMC base case. Additionally, increasing the proportion of patients undergoing ASCT in the model, which was deemed to be lower than expected in Canadian clinical practice by the clinical experts consulted by CDA-AMC for this review, resulted in a slightly higher ICER. These scenario analyses indicate that the CDA-AMC base case may overestimate the benefit associated with BV plus AVD and therefore likely represents a lower limit on what the ICER may be in actual practice.
References
1.Pharmacoeconomic evaluation [internal manufacturer's report]. In: Drug Reimbursement Review manufacturer submission: Brentuzimab vedotin, 50 mg lyophilized powder for reconstitution. Mississauga (ON): Seagen Canada Inc.; 2023 March 8.
2.Clinical Study Report: C25003. A Randomized, Open-label, Phase III Trial of A+AVD Versus ABVD as Front-line Therapy in Patients With Advanced Classical Hodgkin Lymphoma [internal manufacturer's report]. Cambridge (MA): Millennium Pharmaceuticals, Inc.; 2017 Aug 28.
3.Adcetris (brentuximab vedotin): Lyophilized powder for reconstitution with 10.5 mL of sterile water for injection, USP 50 mg [product monograph]. Bothell (WA): Seagen Inc.; 2022 Jul 14.
4.Statistics Canada. Table: 13-10-0114-01. Life expectancy and other elements of the life table, Canada, all provinces except Prince Edward Island. 2022; https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310011401. Accessed 2023 Mar 8.
5.Aleman BM, van den Belt-Dusebout AW, Klokman WJ, Van't Veer MB, Bartelink H, van Leeuwen FE. Long-term cause-specific mortality of patients treated for Hodgkin's disease. J Clin Oncol. 2003;21(18):3431-3439. PubMed
6.Drug Reimbursement Review pharmacoeconomic report: Brentuximab Vedotin (Adcetris) for the treatment of previously untreated patients with Stage IV Hodgkin lymphoma, in combination with doxorubicin, vinblastine, and dacarbazine [manufacturer-provided reference]. Ottawa (ON): CADTH; 2020 Dec 3: https://www.cadth.ca/sites/default/files/pcodr/Reviews2020/10214BrentuximabVedotinHLAVD_fnEGR_NOREDACT-ABBREV_03Dec2020_final.pdf.
7.Chen R, Gopal AK, Smith SE, et al. Five-year survival and durability results of brentuximab vedotin in patients with relapsed or refractory Hodgkin lymphoma. Blood. 2016;128(12):1562-1566. PubMed
8.Guertin JR, Feeny D, Tarride JE. Age- and sex-specific Canadian utility norms, based on the 2013-2014 Canadian Community Health Survey. CMAJ. 2018;190(6):E155-E161. PubMed
9.Swinburn P, Shingler S, Acaster S, Lloyd A, Bonthapally V. Health utilities in relation to treatment response and adverse events in relapsed/refractory Hodgkin lymphoma and systemic anaplastic large cell lymphoma. Leuk Lymphoma. 2015;56(6):1839-1845. PubMed
10.Ramsey SD, Nademanee A, Masszi T, et al. Quality of life results from a phase III study of brentuximab vedotin consolidation following autologous haematopoietic stem cell transplant for persons with Hodgkin lymphoma. Br J Haematol. 2016;175(5):860-867. PubMed
11.Association québécoise des pharmaciens propriétaires. AQPP Price List [manufacturer-provided reference]. 2022.
12.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario drug benefit formulary/comparative drug index. 2022; https://www.formulary.health.gov.on.ca/formulary/. Accessed 2023 May 10.
13.Moskowitz CH, Nademanee A, Masszi T, et al. Brentuximab vedotin as consolidation therapy after autologous stem-cell transplantation in patients with Hodgkin's lymphoma at risk of relapse or progression (AETHERA): a randomised, double-blind, placebo-controlled, phase III trial. Lancet. 2015;385(9980):1853-1862. PubMed
14.Younes A, Gopal AK, Smith SE, et al. Results of a pivotal phase II study of brentuximab vedotin for patients with relapsed or refractory Hodgkin's lymphoma. J Clin Oncol. 2012;30(18):2183-2189. PubMed
15.Cancer Care Ontario. Drug Formulary: funded evidence-informed regimens. 2022; https://www.cancercareontario.ca/en/drugformulary/regimens. Accessed 2023 Mar 8.
16.Government of Canada. Job Bank - Wage Report [manufacturer-provided reference]. 2019: https://www.jobbank.gc.ca/wagereport/location/geo9219.
17.Statistics Canada. Average usual and actual hours worked in a reference week by type of work (full- and part-time), annual (Table: 14-10-0043-01) [manufacturer-provided reference]. 2019; https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1410004301. Accessed 2019 Oct 30.
18.Ontario Case Costing Initiative (OCCI). Toronto (ON): Ontario Health and Long-Term Care; 2017: https://data.ontario.ca/dataset/ontario-case-costing-initiative-occi. Accessed 2023 Mar 8.
19.Raymakers AJN, Costa S, Cameron D, Regier DA. Cost-effectiveness of brentuximab vedotin in advanced-stage Hodgkin's lymphoma: a probabilistic analysis. BMC Cancer. 2020;20(1):992. PubMed
20.Interprovincial Health Insurance Agreements Coordinating Committee (IHIACC). Interprovincial Billing Rates for Designated High Cost Transplants Effective for Discharges on or after April 1, 2019 [manufacturer-provided reference]. 2019; http://www.health.gov.on.ca/en/pro/programs/ohip/bulletins/na_85/high_cost.pdf. Accessed 2019 Nov 1.
21.NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines): Hodgkin Lymphoma. Version 2.2023 - November 8, 2022 [manufacturer-provided reference]. Plymouth Meeting (PA): National Comprehensive Cancer Network; 2022: https://www.nccn.org/professionals/physician_gls/pdf/hodgkins.pdf.
22.Ministry of Health and Long-Term Care of Ontario. Schedule of Benefits: Physician Services Under the Health Insurance Act (Effective October 1st, 2019) [manufacturer-provided reference]. 2019.
23.de Oliveira C, Pataky R, Bremner KE, et al. Phase-specific and lifetime costs of cancer care in Ontario, Canada. BMC Cancer. 2016;16(1):809. PubMed
24.Statistics Canada. Table: 18-10-0004-13 Consumer Price Index by product group, monthly, percentage change, not seasonally adjusted, Canada, provinces, Whitehorse, Yellowknife and Iqaluit. 2022; https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1810000413. Accessed 2022 Oct 20.
25.Castellino SM, Pei Q, Parsons SK, et al. Brentuximab Vedotin with Chemotherapy in Pediatric High-Risk Hodgkin's Lymphoma. N Engl J Med. 2022;387(18):1649-1660. PubMed
26.Zaric GS. The impact of ignoring population heterogeneity when Markov models are used in cost-effectiveness analysis. Med Decis Making. 2003;23(5):379-396. PubMed
27.Guidelines for the economic evaluation of health technologies: Canada. 4th ed. Ottawa (ON): CADTH; 2017: https://www.cadth.ca/guidelines-economic-evaluation-health-technologies-canada-4th-edition. Accessed 2023 Jun 15.
28.Canadian Cancer Statistics: A 2018 special report on cancer incidence by stage [manufacturer-provided reference]. Toronto (ON): Canadian Cancer Society; 2018: https://cdn.cancer.ca/-/media/files/research/cancer-statistics/2018-statistics/canadian-cancer-statistics-2018-en.pdf.
29.DeltaPA. [Ottawa (ON)]: IQVIA; 2022: https://www.iqvia.com/. Accessed 2023 May 10.
30.CADTH. Drug Reimbursement Review: brentuximab vedotin (Adcetris) for the treatment of previously untreated patients with advanced-stage Hodgkin lymphoma (HL), in combination with doxorubicin, vinblastine, and dacarbazine [withdrawn]. 2023; https://www.cda-amc.ca/brentuximab-vedotin. Accessed 2024 May 6.
31.CADTH Reimbursement Review: Pembrolizumab (Keytruda) for classical Hodgkin lymphoma. Can J Health Technol. 2021;1(12). https://www.cda-amc.ca/sites/default/files/DRR/2021/PC0236%20Keytruda-combined-report.pdf. Accessed 2024 May 6.
32.Budget Impact Analysis [internal manufacturer's report]. In: Drug Reimbursement Review manufacturer submission: Brentuzimab vedotin, 50 mg lyophilized powder for reconstitution. Mississauga (ON): Seagen Canada Inc.; 2023 Mar 8.
33.Brenner DR, Poirier A, Woods RR, et al. Projected estimates of cancer in Canada in 2022. CMAJ. 2022;194(17):E601-E607. PubMed
34.Institut national d'excellence en santé et en services sociaux (INESSS). ADCETRIS: Avis transmis au ministre en octobre 2020 [manufacturer-provided reference]. 2020.
35.Patented Medicine Prices Review Board. Alignment Among Public Formularies in Canada, Part 2: Oncology Medicines [manufacturer-provided reference]. 2021.
Appendix 1: Cost Comparison
Please note this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical expert(s) and drug plan. Comparators may be recommended (appropriate) practice or actual practice. Existing product listing agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 8: CDA-AMC Cost-Comparison Table for Front-Line Regimens for the Treatment of Advanced-Stage HL
Treatment | Strength / concentration | Form | Price | Recommended dosage | Daily cost | 28-day cycle cost |
|---|---|---|---|---|---|---|
BV (Adcetris) | 50 mg / vial | IV | 4,840.0000 | 1.2 mg/kg on days 1 and 15 for each 28-day cycle of up to 6 cycles | 691.43 | 19,360 |
BV + AVD | ||||||
Dacarbazine (generic) | 600 mg / 100 mL vial | IV | 251.8200 | 375 mg/m2 on days 1 and 15 of each 28-day cycle for up to 6 cycles | 35.97 | 1,007 |
Doxorubicin (generic) | 10 mg / 5 mL 50 mg / 25 mL vial 200 mg / 100 mL | IV | 50.0000 255.0000 770.0000 | 25 mg/m2 on days 1 and 15 of each 28-day cycle for up to 6 cycles | 18.21 | 510 |
Vinblastine (generic) | 10 mg / 10 mL vial | IV | 176.7900 | 6 mg/m2 on days 1 and 15 of each 28-day cycle for up to 6 cycles | 25.26 | 707 |
BV-AVD | 770.87 | 21,584 | ||||
ABVD | ||||||
Bleomycin (generic) | 15 units / 10mL vial | IV | 419.4000 | 10 units/m2 on days 1 and 15 of each 28-day cycle for up to 6 cycles | 59.91 | 1,678 |
Dacarbazine (generic) | 600 mg / 100 mL vial | IV | 251.8200 | 375 mg/m2 on days 1 and 15 of each 28-day cycle for up to 6 cycles | 35.97 | 1,007 |
Doxorubicin (generic) | 10 mg / 5 mL 50 mg / 25 mL vial 200 mg / 100 mL | IV | 50.0000 255.0000 770.0000 | 25 mg/m2 on days 1 and 15 of each 28-day cycle for up to 6 cycles | 18.21 | 510 |
Vinblastine (generic) | 10 mg / 10 mL vial | IV | 176.7900 | 6 mg/m2 on days 1 and 15 of each 28-day cycle for up to 6 cycles | 25.26 | 707 |
ABVD | 139.35 | 3,902 | ||||
BEACOPP | ||||||
Cyclophosphamide (generic) | 500 mg vial 1,000 mg vial 2000 mg vial | IV | 101.7100 184.3600 339.2000 | 1,250 mg/m2 on day 1 of each 21-day cycle for up to 8 cycles | 21.00 | 588 |
Doxorubicin (generic) | 10 mg / 5 mL 50 mg / 25 mL vial 200 mg / 100 mL | IV | 50.0000 255.0000 770.0000 | 35 mg/m2 on day 1 of each 21-day cycle for up to 8 cycles | 16.90 | 473 |
Etoposide (generic) | 100 mg / 5mL 200 mg / 10 mL 500 mg / 25 mL 1,000 mg / 50 mL | IV | 75.0000 150.0000 375.0000 750.0000 | 200 mg/m2 on days 1 to 3 of each 21-day cycle for up to 8 cycles | 42.86 | 1,200 |
Procarbazine (generic) | 50 mg | PO | 77.5199a | 100 mg/m2 daily on days 1 to 7 of each 21-day cycle for up to 8 cycles | 103.36 | 2,894 |
Vincristine (generic) | 1 mg/mL | IV | 30.6000a | 1.4 mg/m2 on day 8 (max of 2mg) of each 21-day cycle for up to 8 cycles | 2.91 | 82 |
Bleomycin (generic) | 15 units / 10mL vial | IV | 419.4000 | 10 units/m2 on day 8 of each 21-day cycle for up to 8 cycles | 79.89 | 2,237 |
BEACOPP | 266.92 | 7,474 | ||||
ABVD = doxorubicin-bleomycin-vinblastine-dacarbazine; AVD = doxorubicin-vinblastine-dacarbazine; BEACOPP = bleomycin-etoposide-doxorubicin-cyclophosphamide-vincristine-procarbazine; BV = brentuximab vedotin.
Note: Costs assume a mean patient weight of 75.06 kg and BSA = 1.88 m2 consistent with the ECHELON-1 trial.1 All prices are from the Delta PA database (accessed May 2023),29 unless otherwise indicated, and do not include dispensing fees.
aPrice obtained from Ontario Drug Benefit Formulary (accessed May 2023).12
Table 9: CDA-AMC Cost-Comparison Table for Front-line Regimens for the Treatment of Previously Untreated High-Risk HL Patients in the Pediatric Population
Treatment | Strength / concentration | Form | Price | Recommended dosage | Daily cost | 28-day cycle cost |
|---|---|---|---|---|---|---|
BV (Adcetris) | 50 mg / vial | IV | 4,840.0000 | 1.8 mg/kg for each 21-day cycle of up to 5 cycles | 691.43a | 19,360a |
BV + AVEPC | ||||||
Doxorubicin (generic) | 10 mg / 5 mL 50 mg / 25 mL vial 200 mg / 100 mL | IV | 50.0000 255.0000 770.0000 | 25 mg/m2 on days 1 and 2 of each 21-day cycle for up to 5 cycles | 19.05 | 533 |
Vincristine (generic) | 1 mg / mL | IV | 30.6000a | 1.4 mg/m2 on day 1 of each 21-day cycle for up to 5 cycles | 4.37 | 122 |
Etoposide (generic) | 100 mg / 5mL 200 mg / 10 mL 500 mg / 25 mL 1,000 mg / 50 mL | IV | 75.0000 150.0000 375.0000 750.0000 | 125 mg/m2 on days 1 to 3 of each 21-day cycle for up to 5 cycles | 21.43 | 600 |
Prednisone (generic) | 5 mg 50 mg | Tablet | 0.0220b 0.1735b | 20 mg/m2 twice daily on days 1 to 7 of each 21-day cycle for up to 5 cycles | 0.10 | 3 |
Cyclophosphamide (generic) | 500 mg vial 1,000 mg vial 2,000 mg vial | IV | 101.7100 184.3600 339.2000 | 600 mg/m2 on day 1 and 2 of each 21-day cycle for up to 5 cycles | 17.56 | 492 |
BV + AVEPC | 753.94 | 21,110 | ||||
ABVE-PC | ||||||
Doxorubicin (generic) | 10 mg / 5 mL 50 mg / 25 mL vial 200 mg / 100 mL | IV | 50.0000 255.0000 770.0000 | 25 mg/m2 on days 1 and 2 of each 21-day cycle for up to 5 cycles | 19.05 | 533 |
Bleomycin (Generic) | 15 units / 10 mL vial | IV | 419.4000 | 5 mg/m2 on day 1 and 10 mg/m2 on day 8 of each 21-day cycle for up to 5 cycles | 39.94 | 1,118 |
Etoposide (generic) | 100 mg / 5mL 200 mg / 10 mL 500 mg / 25 mL 1,000 mg / 50 mL | IV | 75.0000 150.0000 375.0000 750.0000 | 125 mg/m2 on days 1 to 3 of each 21-day cycle for up to 5 cycles | 21.43 | 600 |
Prednisone (generic) | 5 mg 50 mg | Tablet | 0.0220b 0.1735b | 20 mg/m2 twice daily on days 1 to 7 of each 21-day cycle for up to 5 cycles | 0.10 | 3 |
Cyclophosphamide (generic) | 500 mg vial 1,000 mg vial 2,000 mg vial | IV | 101.7100 184.3600 339.2000 | 600 mg/m2 on day 1 and 2 of each 21-day cycle for up to 5 cycles | 17.56 | 492 |
Vincristine (generic) | 1 mg / mL vial | IV | 30.6000a | 1.4 mg/m2 on days 1 and 8 of each 21-day cycle for up to 5 cycles | 8.74 | 245 |
ABVE-PC | 106.82 | 2,991 | ||||
ABVE-PC = doxorubicin-bleomycin-etoposide, prednisone-vincristine-cyclophosphamide; AVPE = brentuximab-doxorubicin-vincristine-etoposide-prednisone-cyclophosphamide; BV = brentuximab vedotin.
Note: Costs assume a mean patient weight of 58.5 kg and BSA = 1.6 m2 consistent with a previous HL review.31 All prices are from the Delta PA database (accessed June 2024),29 unless otherwise indicated, and do not include dispensing fees.
aDaily and 28-day cycle costs represent costs for patients with weights ranging from 55.7 kg to 83 kg. For patients weighing 0 to 55.6 kg, daily and 28-day cycle costs would be $460.95 and $12,910, respectively.
bPrice obtained from Ontario Drug Benefit Formulary (accessed June 2024).12
Appendix 2: Submission Quality
Please note that this appendix has not been copy-edited.
Description | Yes/No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | Concerns with generalizability of findings from ECHELON-1 trial population to pediatric population and lack of relevant comparator; refer to CDA-AMC appraisal |
Model has been adequately programmed and has sufficient face validity | No | Limitations identified with the face validity of the model; refer to CDA-AMC appraisal |
Model structure is adequate for decision problem | No | Limitations identified with the model structure; refer to CDA-AMC appraisal |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | Yes | No comment |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | Yes | No comment |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | Yes | No comment |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Please note that this appendix has not been copy-edited.
Appendix 4: Additional Details on the CDA-AMC Reanalyses and Sensitivity Analyses of the Economic Evaluation
Please note that this appendix has not been copy-edited.
Table 11: Disaggregated Summary of CDA-AMC’s Economic Evaluation Results
Parameter | BV + AVD | ABVD | Incremental |
|---|---|---|---|
Discounted LYs | |||
Total | 24.58 | 23.60 | 0.98 |
Front-line PF | 21.88 | 19.93 | 1.94 |
Post-progression, ASCT | 1.41 | 1.90 | −0.50 |
Post-progression, no ASCT | 1.30 | 1.76 | −0.46 |
Discounted QALYs | |||
Total | 20.46 | 19.55 | 0.91 |
Front-line PF | 18.41 | 16.77 | 1.63 |
Post-progression, ASCT | 1.17 | 1.59 | −0.42 |
Post-progression, no ASCT | 0.87 | 1.18 | −0.31 |
Discounted costs ($) | |||
Total | 183,099 | 77,989 | 105,110 |
PF health state | |||
Acquisition | 129,507 | 23,412 | 106,094 |
Administration | 3,347 | 3,120 | 227 |
Immunostimulant therapies | 14,645 | 937 | 13,707 |
AEs | 2,459 | 1,732 | 727 |
PP health state | |||
Post-progression, ASCT | 20,193 | 30,543 | −10,349 |
Post-progression, no ASCT | 7,151 | 9,691 | −2,539 |
End of life | 5,798 | 8,555 | −2,757 |
ICER ($ per QALY) | 115,865 | ||
ASCT = autologous stem-cell transplant; ICER = incremental cost-effectiveness ratio; LY = life-year; PF = progression-free; PP = post-progression; QALY = quality-adjusted life-year.
Scenario Analyses
Stepped analysis | Comparator | Total costs ($) | Total QALYs | ICER ($ per QALY) |
|---|---|---|---|---|
CDA-AMC’s base case | ABVD | 77,989 | 19.55 | Reference |
BV + AVD | 183,099 | 20.46 | 115,865 | |
CDA-AMC scenario 1: Time horizon set to maximum follow-up of trial (70 months) | ABVD | 78,612 | 4.61 | Reference |
BV + AVD | 179,836 | 4.76 | 729,491 | |
CDA-AMC scenario 2: application of modified PFS hazard ratio to BV + AVD PFS extrapolations | ABVD | 72,340 | 19.86 | Reference |
BV + AVD | 183,099 | 20.46 | 186,120 | |
CDA-AMC scenario 3: Proportion of patients receiving ASCT increased to 70% | ABVD | 89,525 | 19.94 | Reference |
BV + AVD | 190.224 | 20.74 | 125,025 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Appendix 5: Submitted Budget Impact Assessment and CDA-AMC Appraisal
Please note that this appendix has not been copy-edited.
Table 13: Summary of Key Take Aways
Key take aways of the Budget Impact Analysis |
|---|
|
Summary of Manufacturer’s Budget Impact Analysis
The manufacturer submitted a budget impact analysis (BIA) estimating the budget impact of expanding the use of BV plus AVD as a treatment of previously untreated patients with advanced-stage Hodgkin lymphoma (HL).32 The analytic framework, which used a top-down epidemiology-based approach, leveraged data from the Canadian Cancer Society and literature to determine the estimated population size.28,33 The relevant comparator was assumed to be ABVD regimen (i.e., doxorubicin, bleomycin, vinblastine, and dacarbazine). The manufacturer compared a reference scenario where BV was only reimbursed for patients with stage IV HL, with an expanded indication scenario, where BV was funded for patients with advanced HL, as per the Health Canada indication.
The BIA base case was undertaken from a publicly funded drug plan perspective considering only drug costs over a 3-year time horizon. Drug costs considered included that of front-line treatment only. BV costs were calculated by multiplying the recommended dosage (1.2 mg/kg) by an average patient weight of 75.06 kg, resulting in the need for 4 vials of 50 mg per patient. The manufacturer assumed vials would not be shared among patients. The manufacturer assumed that all patients would complete 6 cycles of treatment. Based on literature, the proportion of patients presenting in each stage of the disease was approximately 50%.28 The market shares for the reference scenario were obtained from the Institut National D’excellence en Santé et en Services Sociaux (INESSS) review of BV.34 Market share for the new drug scenario were based on manufacturer’s assumptions. Key inputs to the BIA are documented in Table 14.
Table 14: Summary of Key Model Parameters
Parameter | Manufacturer’s estimate (reported as year 1 / year 2 / year 3 if appropriate) |
|---|---|
Target population | |
Public coverage rate | Jurisdiction specific35 |
Incidence of HL | 2.6 per 100,00033 |
Proportion with stage III and IV | 46%28 |
Proportion of patients treated | 90%a |
Number of patients eligible for drug under review | 320 / 320 / 328 |
Market Uptake (3 years) | |
Uptake (reference scenario) BV + AVD ABVD | 28% / 32% / 32% 72% / 68% / 68% |
Uptake (new drug scenario) BV + AVD ABVD | 48% / 60% / 60% 52% / 40% / 40% |
Cost of treatment (per patient) | |
Cost of treatment over 6-cycles BV + AVD ABVD | $126,902 $18,291 |
ABVD = doxorubicin, bleomycin, vinblastine, and dacarbazine; BV plus AVD = brentuximab vedotin, doxorubicin, vinblastine, and dacarbazine; HL = Hodgkin lymphoma.
aManufacturer’s assumption,
Summary of the Manufacturer’s BIA Results
Results of the manufacturer’s BIA base case indicated that the budget impact associated with the reimbursement of BV plus AVD as a front-line treatment regimen for previously untreated patients with advanced-stage HL is expected to be $6,955,759 in year 1, $9,564,11,568 in year 2, and $11,568,301 in year 3 for a total budget impact of $28,088,229 over 3 years.
CDA-AMC Appraisal of the Manufacturer’s BIA
CDA-AMC identified several key limitations to the manufacturer’s analysis that have notable implications on the results of the BIA:
Relevant comparators were excluded: The relevant comparator considered in the manufacturer’s base case were restricted to the ABVD regimen. According to clinical expert feedback received by CDA-AMC for this review, BEACOPP is a relevant comparator, with between 10% and 20% of advanced HL patients being treated with BEACOPP as front-line treatment. Additionally, PET-guided ABVD or BEACOPP are also used in some jurisdictions. Therefore, the exclusion of BEACOPP and PET-guided treatments as relevant comparators was not appropriate.
In addition, according to clinical experts consulted by CDA-AMC, the current treatment options for pediatric patients with advanced HL include ABVE-PC and OEPA-COPDAC. Neither of these regimens were included as comparators in the BIA.
CDA-AMC could not address this limitation owing to the structure of the BIA.
Public coverage was inappropriate: The manufacturer informed the oncology drug coverage rates using a PMPRB report, where public coverage varied by jurisdiction due to the fact that there may be medicines that the hospital formularies do not list.35 However, in the context of this specific BIA, all medicines are funded and therefore, the coverage rates do not apply. Per the provisions of the Canada Health Act, oncology medicines administered in Canadian hospitals are fully covered by hospital budgets and provided at no cost to the patient.35
CDA-AMC assumed all IV products had the public coverage of 100% in the base-case reanalysis.
The market uptake for BV plus AVD in the stage III and IV HL was underestimated: In the reference scenario, the manufacturer’s BIA estimated that 55% of patients with stage IV HL would be already receiving BV plus AVD in the first year and that this would increase to 65% in the second and third year.34 According to the clinical expert consulted by CDA-AMC for this review, the manufacturer’s market shares for stage IV patients are likely underestimated, since BV plus AVD is currently funded and used to treat stage IV HL. The clinical expert consulted indicated that most (at least 80%) stage IV patients are currently using BV plus AVD, with only those with advanced age/comorbidities, pre-existing neuropathy, or preference for other regimens would be considered for treatment with other options. In addition, clinical expert feedback indicated that the expected market uptake for stage III HL will be at least 60% to 80% since clinicians have experience with this therapy by using it to treat patients with stage IV HL.
CDA-AMC increased the market shares in the reference and new drug scenarios for stage IV HL to 80%, 85%, and 90% for years 1, 2 and 3, respectively. In addition, based on clinical expert feedback CDA-AMC increased the market shares for stage III HL to 60%, 65%, and 70% for years 1, 2 and 3, respectively. These changes resulted in a total market share of 70%, 75%, and 80% for the expanded indication scenario for years 1, 2 and 3, respectively.
The clinical and cost-effectiveness of BV plus AVD in those under 18 is unknown: The manufacturer’s submitted BIA included both adult and pediatric populations of previously untreated patients with advanced HL. As the ECHELON-1 trial did not include patients younger than 18 years old, the clinical and cost-effectiveness of BV plus AVD in those under 18 is unknown. In addition, the chemotherapy backbone used in pediatric population is different than in adults.
CDA-AMC conducted a scenario analysis considering if BV plus AVD was only reimbursed in adults with untreated advanced HL (i.e., considering a population of adults only).
Note that in the manufacturer’s and CDA-AMC’s base cases, pediatric patients were included, and it is assumed that the chemotherapy backbone and comparator treatment they will receive is identical to adults, which is contradictory to feedback provided by clinical experts for this review. CDA-AMC did not address this in reanalyses as it is beyond the scope of a CDA-AMC review.
Additional limitations were identified but were not considered to be key limitations. These limitations include:
The manufacturer did not consider the drug acquisition costs of subsequent therapies: The manufacturer only considered the acquisition costs of first-line therapy in the BIA. The exclusion of drug acquisition costs of subsequent therapies may be conservative, as if fewer people experience disease progression on BV plus AVD, fewer people may receive subsequent therapies. In this case, the budget impact could be overestimated.
The calculated number of vials per cycle in the BIA were not aligned with the approach used in the pharmacoeconomic analysis: In the BIA, the manufacturer calculated the number of vials considering only the total drug dose needed for that cycle. However, because some drug therapies, such as vinblastine, dacarbazine, and bleomycin, should be administered in 2 distinct days of the cycle (e.g., day 1 and 15), the method used by the manufacturer resulted in less vials than the amount necessary to administer the dose necessary each day. For instance, for dacarbazine, the dose necessary per day is 709.5 mg requiring 2 vials on day 1 and 2 vials on day 15, while using the manufacturer approach, the dose necessary per cycle would be 1,419mg, requiring 3 vials in total. The method used to calculate number of vials in the BIA was not aligned with the approach used in the manufacturer’s pharmacoeconomic analysis, or in CDA-AMC’s cost table.
CDA-AMC addressed this limitation by changing the number of vials per cycle from 3 to 4 for vinblastine, dacarbazine, and bleomycin.
CDA-AMC Reanalyses of the BIA
Table 15: CDA-AMC Revisions to the Submitted BIA
Stepped analysis | Manufacturer’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Correctionsa to manufacturer’s base case | ||
1. Number of patients with stage III and IV at year 2 | 356 | 360 |
2. Number of patients treated at year 2 | 320 | 324 |
3. Doxorubicin’s acquisition cost | $225.25 | $255.00 |
Changes to derive the CDA-AMC base case | ||
1. Public coverage rate | Jurisdiction specific | 100% |
2. Market shares | Stage IV HL – reference and new drug scenario Year 1: 55% Year 2: 65% Year 3: 65% Stage III HL – new drug scenario Year 1: 40% Year 2: 55% Year 3: 65% | Stage IV HL– reference and new drug scenario Year 1: 80% Year 2: 85% Year 3: 90% Stage III HL – new drug scenario Year 1: 60% Year 2: 65% Year 3: 70% |
3. Number of vials per cycle for vinblastine, dacarbazine, and bleomycin | 3 | 4 |
CDA-AMC base case | 1 + 2 + 3 | |
aCorrections are minor errors (e.g., transcription errors between report and model, misapplication of distributions, or SEs in probabilistic analyses) that are not identified as limitations.
BIA = budget impact analysis.
The results of the CDA-AMC stepwise reanalysis are presented in summary format in Table 16 and a more detailed breakdown is presented in Table 17.
Based on CDA-AMC’s base-case, the expected budget impact for funding BV plus AVD is expected to be $10,658,052in year 1, $11,681,771 in year 2, and $12,726,344 in year 3, for a three-year budget impact of $35,066,167. The number of patients in the CDA-AMC base case in year 1, year 2 and year 3 were 335, 339, and 343, respectively.
Table 16: Summary of the CDA-AMC Reanalyses of the BIA
Stepped analysis | Three-year total ($) |
|---|---|
Submitted base case | 28,088,229 |
Submitted base case – corrected | 28,200,426 |
CDA-AMC reanalysis 1 | 29,490,528 |
CDA-AMC reanalysis 2 | 34,327,514 |
CDA-AMC reanalysis 3 | 27,547,051 |
CDA-AMC base case | 35,066,167 |
BIA = budget impact analysis.
CDA-AMC also conducted an additional scenario analysis to address remaining uncertainty, using the CDA-AMC base-case. The scenario conducted was:
Inclusion of administration fees.
Inclusion of only adult population with advanced HL (assuming 6.4 cases per 1,000,000 are pediatric).
Price reduction of BV was 55% (aligned with the price reduction from the economic evaluation appraisal).
In the scenario analysis that considered reimbursement in the adult only population, the expected three-year budget impact was estimated to be $33,766,360. In this scenario, the total adult patient population was 322, 326 and 330 in years 1, 2 and 3, respectively. Therefore, in the CDA-AMC base case, there are 13 pediatric patient per years (39 pediatric patients over 3 years). Based on the cost-comparison table, the 28-day cycle costs of treating pediatric patients with BV plus AVEPC is $21,110, or $105,551 for 5 cycles of treatment. As such, the cost of treating pediatric HL patients with BV plus AVEPC would be approximately $1,372,165 per year (assuming 13 patients are treated per year).
Table 17: Detailed Breakdown of the CDA-AMC Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
Submitted base case | Reference | $12,662,560 | $15,421,277 | $17,160,217 | $17,562,828 | $50,144,322 |
New drug | $12,662,560 | $22,377,036 | $26,724,385 | $29,131,129 | $78,232,550 | |
Budget impact | $0 | $6,955,759 | $9,564,169 | $11,568,301 | $28,088,229 | |
CDA-AMC base case | Reference | $14,758,834 | $22,039,523 | $23,196,857 | $24,375,044 | $69,611,424 |
New drug | $14,758,834 | $32,697,575 | $34,878,628 | $37,101,389 | $104,677,591 | |
Budget impact | $0 | $10,658,052 | $11,681,771 | $12,726,344 | $35,066,167 | |
CDA-AMC scenario analysis 1: include administration costs | Reference | $16,217,640 | $23,437,967 | $24,601,893 | $25,786,444 | $73,826,304 |
New drug | $16,217,640 | $33,979,482 | $36,155,934 | $38,373,636 | $108,509,052 | |
Budget impact | $0 | $10,541,515 | $11,554,041 | $12,587,192 | $34,682,748 | |
CDA-AMC scenario analysis 2: include only adult population | Reference | $14,212,819 | $21,226,065 | $22,347,415 | $23,489,394 | $67,062,874 |
New drug | $14,212,819 | $31,485,594 | $33,595,992 | $35,747,648 | $100,829,233 | |
Budget impact | $0 | $10,259,529 | $11,248,577 | $12,258,254 | $33,766,360 | |
CDA-AMC scenario analysis 3: 55% price reduction | Reference | $10,530,349 | $13,482,092 | $13,997,847 | $14,521,897 | $42,001,836 |
New drug | $10,530,349 | $17,722,071 | $18,645,081 | $19,584,683 | $55,951,834 | |
Budget impact | $0 | $4,239,979 | $4,647,234 | $5,062,785 | $13,949,998 |
BIA = budget impact analysis.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.