Drugs, Health Technologies, Health Systems
Reimbursement Review
Leuprolide Mesylate (Camcevi)
Sponsor: Accord Healthcare Inc.
Therapeutic area: Prostate cancer
Abbreviations
ACE
angiotensin-converting enzyme
ADT
androgen-deprivation therapy
AE
adverse event
AUA
American Urological Association
AUC0-4wk
area under the concentration-time curve, calculated using the linear up, log down trapezoidal method from 0 weeks to 4 weeks after dosing
AUC0-6mon
area under the concentration-time curve, calculated using the linear up, log down trapezoidal method from 0 months to 6 months after dosing
BIA
budget impact analysis
Cavg(0-6mon)
mean serum concentration in the 6 months after dosing
CDA-AMC
Canada's Drug Agency
CI
confidence interval
Cmax
maximum observed serum concentration
Cmon6
observed serum concentration 6 months after dosing
Cwk4
observed serum concentration 4 weeks after dosing
EAU
European Association of Urology
ECG
electrocardiogram
ECOG PS
Eastern Cooperative Oncology Group Performance Status
EOS
end of study
GnRH
gonadotropin-releasing hormone
GU DAC
Genitourinary Cancers Drug Advisory Committee
ITC
indirect treatment comparison
ITT
intention to treat
LH
luteinizing hormone
LHRH
luteinizing hormone–releasing hormone
LMIS
leuprolide mesylate injectable suspension
MID
minimal important difference
OS
overall survival
PD
pharmacodynamics
PFS
progression-free survival
PK
pharmacokinetic
PP
per protocol
PSA
prostate-specific antigen
QoL
quality of life
SAE
serious adverse event
SC
subcutaneous
SD
standard deviation
TEAE
treatment-emergent adverse event
Tmax
time to maximum serum concentration
TNM
tumour, node, metastasis
VAS
visual analogue scale
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Leuprolide mesylate (Camcevi), extended-release emulsion, 42 mg leuprolide, subcutaneous injection |
Sponsor | Accord Healthcare Inc. |
Indication | For the treatment of adult patients with advanced prostate cancer |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | November 16, 2021 |
Recommended dose | 42 mg administered every 6 months as a single subcutaneous injection |
NOC = Notice of Compliance.
Sources: Drug Reimbursement Review Sponsor Submission,1 product monograph for leuprolide injectable emulsion (Camcevi).2
Introduction
Prostate cancer is one of the most commonly diagnosed cancers in males in Canada, with an estimated 27,900 patients diagnosed in 2024, accounting for 22% of all new cancer cases in males.3 Prostate cancer is also one of the most common causes of cancer deaths in Canada, with an estimated 5,000 deaths in 2024.3 The median age at diagnosis is 66 years, but 20% of patients diagnosed are older than 75 years.4,5 Most patients with prostate cancer do not have initial or early symptoms.6 As the tumour grows locally, or as it metastasizes, symptoms can intensify and start to interfere with the physiological functions of the body. Diagnostic tests for prostate cancer include blood screening for prostate-specific antigen (PSA), physical digital rectal examination, biopsy of the prostate, and imaging with CT or MRI scans.6-9 The tumour, node, metastasis (TNM) classification system, commonly used to determine tumour stage in patients with prostate cancer, has 4 stages (stage I to stage IV), and the higher the stage, the more the tumour has spread.7,8,10 Terms such as localized prostate cancer (limited to the prostate), locally advanced prostate cancer (spread outside of the prostate but not metastatic), and metastatic prostate cancer (spread beyond the tissue surrounding the prostate to lymph nodes or other parts of the body, such as the lungs, liver, or bones) are also used to describe the growth and spread of prostate cancer.11,12 For patients with localized prostate cancer at the time of diagnosis, the 5-year overall survival (OS) rate is reported to be nearly 100%, whereas for patients with distant metastases, the 5-year OS rate is only 29%.6
Both the sponsor and the clinical experts consulted by Canada's Drug Agency (CDA-AMC) review team noted that androgen-deprivation therapy (ADT) is the backbone of treatment when systemic anticancer therapies are considered for the treatment of metastatic prostate cancer. According to the clinical experts consulted by the review team, ADT is continued throughout a patient’s life after diagnosis, and other medicines are used on top of ADT. The clinical experts consulted by the review team noted that ADT is typically continued even when a patient’s disease progresses or when a patient transitions to best supportive care near the end of life. The clinical experts also noted that some treatment protocols use ADT for a finite period (e.g., as neoadjuvant treatment before definitive surgery or radiotherapy, or as adjuvant therapy with radiotherapy). Gonadotropin-releasing hormone (GnRH) analogues, a type of ADT, are available as antagonists, such as degarelix and relugolix,13,14 or as GnRH agonists, such as leuprolide products, which include leuprolide acetate (Eligard, Lupron Depot, Zeulide Depot) and goserelin acetate (Zoladex).15-18 The main goals of treatment for advanced prostate cancer are to delay disease progression with the achievement of castrate levels of testosterone, which is defined as less than 50 ng/dL by the American Urological Association (AUA) and European Association of Urology (EAU).19,20 The EAU also indicates that testosterone levels of less than 20 ng/dL might be associated better outcomes than levels that range from 20 ng/dL to 50 ng/dL.19
This is a tailored review submission of a new salt formulation of leuprolide, leuprolide mesylate (Camcevi). The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of leuprolide mesylate, extended-release emulsion, 42 mg of leuprolide, administered by subcutaneous (SC) injection for the treatment of adult patients with advanced prostate cancer.
Perspectives of Patient, Clinicians, and Drug Programs
The information in this section is a summary of input provided by the patient and clinician groups that responded to our call for input and from clinical experts consulted by for the purpose of this review.
Patient Input
No patient group input was received by the review team for this review.
Clinician Input
Input From Clinical Experts Consulted for This Review
According to the clinical experts consulted by the CDA-AMC review team, there is no universal definition for advanced prostate cancer, and the phrase “patients with advanced prostate cancer,” which is used in the Health Canada–approved indication, is subject to interpretation. The clinical experts consulted by the CDA-AMC review team noted that ADT can be used in patients with intermediate-risk or high-risk localized prostate cancer, as well as in those with biochemically recurrent or metastatic disease, which is the more common definition of advanced prostate cancer. According to the clinical experts consulted by the CDAAMC review team, for patients with biochemically recurrent or metastatic disease, the treatment goal is to achieve a castrate testosterone level, which controls the disease by decreasing PSA levels, delaying the development of metastatic disease (in the context of biochemical recurrence), and reducing disease burden (in metastatic prostate cancer), which often results in improved symptoms, OS, and progression-free survival (PFS). According to the clinical experts consulted by the review team, the long-term use of ADT with currently available GnRH analogues typically results in the development of refractory disease or castrate resistance, which was considered one of the main unmet needs. Leuprolide mesylate, a GnRH analogue, does not address this major unmet need in the clinical experts’ opinions. However, according to the clinical experts consulted by the CDA-AMC review team, the availability of leuprolide mesylate, which does not require reconstitution like Eligard does, provides an additional formulation option for patients and clinicians.
According to the clinical experts consulted by the review team, there are currently several approved preparations of leuprolide products in Canada, including Lupron Depot (3-month, 4-month, and 6-month depots administered as intramuscular injections), Eligard (1-month, 3-month, 4-month, and 6-month depots administered as SC injections), and goserelin acetate (3-month depot administered as SC pellet implants). Despite prescribing heterogeneity in Canada, most patients do not receive the 6-month preparations. The clinical experts consulted by the review team noted that leuprolide mesylate is not expected to shift the current treatment paradigm; rather, leuprolide mesylate would share the same place in therapy as other GnRH analogues and provide an additional option to the existing GnRH analogues.
According to the clinical experts consulted by the review team, patients with advanced prostate cancer are most likely to respond to treatment with leuprolide mesylate. The clinical experts consulted by the review team noted that the indications for GnRH analogue injections for patients with advanced prostate cancer have been well established for several decades, and that there are several other drugs with very similar chemical formulations and pharmaceutical actions already on the market. According to the clinical experts consulted by the review team, there are no particular disease characteristics that would warrant or contraindicate the use of leuprolide mesylate relative to other GnRH analogues, and there are no identifiable patient or disease features (clinical, histological, or biochemical) that render patients more suited to leuprolide mesylate than to other GnRH analogues.
According to the clinical experts consulted by the review team, the outcome measures for advanced prostate cancer treatment used in clinical practice are aligned with those used in clinical trials. The clinical experts consulted by the review team noted that for advanced prostate cancer, efficacy is determined by evaluating a number of metrics, including the clinical status of the patient, radiologic response and/or progression, and PSA dynamics. The clinical experts consulted by the review team noted that the definition of a clinically meaningful response is well established in current clinical guidelines and should not vary among physicians, and the parameters applied to leuprolide mesylate should be identical to those already accepted for other GnRH analogue drugs. The clinical experts consulted by the CDA-AMC review team noted that a serum testosterone level of 50 ng/dL or less is an accepted surrogate end point for efficacy, and that recent evidence suggests that the suppression of serum testosterone levels to less than 20 ng/dL might be associated with better biochemical relapse-free survival.
According to the clinical experts consulted by the review team, ADT with GnRH analogues is typically intended to be continued throughout a patient’s lifetime; however, in some instances, GnRH analogues are prescribed for a finite period as an adjunct to other definitive therapies (e.g., as neoadjuvant treatment before surgery or as neoadjuvant and adjuvant therapy to radiotherapy). The clinical experts consulted by the review team noted that GnRH analogue therapy is typically not discontinued, even when there is disease progression or when a patient transitions to best supportive care near the end of life; instead, other medicines are added to ADT. According to the clinical experts consulted by the review team, ADT with GnRH analogues may be occasionally discontinued in cases of significant or intolerable side effects or at patient or clinician discretion.
According to the clinical experts consulted by the review team, appropriate treatment settings for leuprolide mesylate include outpatient or ambulatory care clinics, physicians’ offices, and injection clinics in the community setting. The clinical experts consulted by the review team also noted that treatment may be administered through a home injection program by nursing personnel. According to the clinical experts consulted by the review team, a specialist (most commonly a urologist) usually establishes the diagnosis; treatment may be administered by a urologist, a radiation oncologist, or a medical oncologist, depending on the stage of disease; and monitoring is provided by a specialist or by a primary care physician, depending on treatment response and stability of disease.
Clinician Group Input
The clinician group input on leuprolide mesylate was received from 1 clinician group: the Ontario Health (Cancer Care Ontario) Genitourinary Cancers Drug Advisory Committee (GU DAC). A total of 2 clinicians provided the input.
According to the GU DAC, the treatment goals for patients with hormone-dependent advanced prostate cancer are to improve survival, delay disease progression, reduce cancer-related complications (e.g., skeletal-related events with reduced need for palliative radiotherapy, spinal cord compression, urinary obstruction, and the need for palliative procedures, such as transurethral resection of the prostate), improve patients’ quality of life (QoL), maintain the independence of patients, and reduce the burden on caregivers. The GU DAC noted that there are unmet needs for effective methods of suppressing testosterone with reduced side effects in patients with advanced prostate cancer, as well as an unmet need for reduced drug costs for these patients.
According to the GU DAC, leuprolide mesylate is a 6-month depot injection, and the Eligard 6-month depot injection is currently available and reimbursed in Canada. According to the GU DAC, leuprolide mesylate may improve convenience for patients who prefer a 6-month dosing schedule because of their lifestyle, the location of their residence, as well as their ability to travel to get treatment, to have vacations, or to work. According to the GU DAC, leuprolide mesylate would not change the current treatment paradigm, but it would provide an alternative to the 6-month Eligard product. According to the GU DAC, any patients who require GnRH agonist therapy for prostate cancer would be candidates for leuprolide mesylate. The GU DAC noted that the selection of leuprolide mesylate versus other available drugs would be based mostly on the preferences of the prescriber and patient for a longer, 6-month injection interval.
According to the GU DAC, testosterone levels, PSA response, and radiographic response are used to determine whether a patient is responding to treatment in clinical practice and in clinical trials. The GU DAC noted that a common standard follow-up would be the measurement of a PSA level and testosterone level every 3 months, and interval imaging would depend on the scenario. The GU DAC also noted that for patients with symptoms related to either locally advanced disease or metastatic disease, a clinically meaningful response is a reduction or resolution of urinary tract obstruction or pain from bone metastases. The magnitude of response is standard among prescribers and should not depend on the drug chosen if the testosterone level is suppressed adequately.
Echoing the clinical experts consulted by the review team, the GU DAC noted that for patients with metastatic prostate cancer, current evidence supports the continuation of GnRH agonist therapy indefinitely through the next lines of therapy, even with disease progression. The GU DAC noted that GnRH agonist therapy can be discontinued because of severe intolerance or side effects that significantly interfere with a patient’s QoL. The GU DAC noted that community, academic, outpatient, and hospital settings are all appropriate for treatment with leuprolide mesylate.
Drug Program Input
Input was obtained from the drug programs that participate in the reimbursement review process. The following were identified as key factors that could potentially affect the implementation of a recommendation for leuprolide mesylate:
relevant comparators
considerations for continuation or renewal of therapy
considerations for discontinuation of therapy
considerations for prescribing of therapy
generalizability
funding algorithm
system and economic issues.
The responses to the issues raised are presented in Table 3.
Clinical Evidence
The CDA-AMC Clinical Review was based on a summary of clinical evidence provided by the sponsor, in accordance with the CDA-AMC tailored review process.
Description of Studies
A phase III, multicentre, single-arm, open-label pivotal study (FP01C-13-001) and a phase III, single-arm, open-label, safety extension study (FP01C-13-001-EX) were submitted by the sponsor and assessed by the CDA-AMC review team.
The FP01C-13-001 study evaluated the efficacy, safety, and pharmacokinetics of leuprolide mesylate in 137 adult patients with histologically confirmed prostate carcinoma and a baseline morning serum testosterone level of more than 150 ng/dL. Patients were enrolled in the FP01C-13-001 study at 26 sites (no sites in Canada) and scheduled to receive a total of 2 doses of leuprolide mesylate with a 6-month interval. There were 3 primary objectives of the FP01C-13-001 study: to establish the efficacy of leuprolide mesylate for up to 1 year, measured as the percentage of patients with a serum testosterone concentration suppressed to castrate levels (≤ 50 ng/dL) by day 28 with or without 1 day after the first injection of leuprolide mesylate and the percentage of patients with serum testosterone suppression (≤ 50 ng/dL) from day 28 through day 336 (remaining duration of the study); to determine the safety and tolerability of leuprolide mesylate for up to 1 year of exposure; and to evaluate the pharmacokinetic behaviour of serum leuprolide. The median age of the patients in the FP01C-13-001 study was 71.0 years (range, 51 to 88 years). The race composition of the study population was 89.8% white, 5.8% Black or African American, 3.6% Asian, and 0.7% unknown. Approximately 2.9%, 22.6%, 27.0%, and 23.4% had, respectively, stage I, stage II, stage III, or stage IV prostate cancer; the disease stage in the remaining 24.1% of the study population was unknown.
The FP01C-13-001-EX extension study was conducted at 7 sites in the US and enrolled 30 patients who participated in the FP01C-13-001 pivotal study and assessed the safety and tolerability of leuprolide mesylate in these patients for up to 1 year.
Efficacy Results
Serum Testosterone Levels of 50 ng/dL or Less (Primary End Point)
In the FP01C-13-001 study, serum testosterone level suppression to castrate levels (≤ 50 ng/dL) was reached by 98.5% (95% confidence interval [CI], 94.8% to 99.8%) of the intention-to-treat (ITT) population by day 28 with or without 1 day after the first dose of leuprolide mesylate. By day 336, 97.0% (95% CI, 92.2% to 98.9%) of the ITT population achieved castrate testosterone levels (≤ 50 ng/dL).
The sponsor provided post hoc subgroup analyses, including subgroup results by disease stage and by disease characteristics at enrolment in response to the CDA-AMC information request21 seeking the sponsor’s input on how to define advanced prostate cancer. The summary of the subgroup analyses on the proportion of patients who achieved a castrate testosterone level (≤ 50 ng/dL) is presented in Appendix 1. In general, the proportion of patients who achieved a castrate testosterone level (≤ 50 ng/dL) was similar across subgroups.
Pharmacokinetic Parameters (Primary End Point)
In the FP01C-13-001 study, after the first and second doses of leuprolide mesylate, mean serum leuprolide concentrations reached the maximum observed serum concentration (Cmax) of 94.5 ng/mL (standard deviation [SD] = 53.7) and 99.0 ng/mL (SD = 73.0), respectively, at 3.23 hours and 2.08 hours (median time to the maximum serum concentration [Tmax]). After the first dose of leuprolide mesylate, the mean values of observed serum concentration 4 weeks after dosing (Cwk4); observed serum concentration 6 months after dosing (Cmon6); area under the concentration-time curve calculated using the linear up, log down trapezoidal method from 0 weeks to 4 weeks after dosing (AUC0-4wk); area under the concentration-time curve calculated using the linear up, log down trapezoidal method from 0 months to 6 months after dosing (AUC0-6mon); and mean serum concentration in the 6 months after dosing (Cavg[0-6mon)] were, respectively, 1.04 ng/mL, 0.497 ng/mL, 91.6 day ng/mL, 224 day ng/mL, and 1.34 ng/mL. After the second dose of leuprolide mesylate, the mean values of Cwk4, Cmon6, AUC0-4wk, AUC0-6mon, and Cavg(0-6mon) were, respectively, 1.64 ng/mL, 0.511 ng/mL, 125 day ng/mL, 268 day ng/mL, and 1.59 ng/mL.
Postsuppression Excursion of Serum Testosterone to More Than 50 ng/dL (Secondary End Point)
The FP01C-13-001 study examined the proportion of patients who exhibited postsuppression excursions of serum testosterone (> 50 ng/dL), either through breakthrough (i.e., episodes unrelated to leuprolide mesylate dosing) or through the acute-on-chronic surge (i.e., related to the second dose of leuprolide mesylate). Overall, no postsuppression breakthrough effect was observed, other than the acute-on-chronic surge, during the study period. Two of 137 patients did not reach the castrate testosterone level (≤ 50 ng/dL) by day 28. Two patients exhibited postsuppression excursions of serum testosterone (> 50 ng/dL) after the second dose of leuprolide mesylate, between day 28 and day 336, but their serum testosterone returned to the castration levels on day 336.
Serum Testosterone Suppression of Less Than 20 ng/dL (Secondary End Point)
In the ITT population of the FP01C-13-001 study, 69.3% and 95.9% of patients achieved a testosterone suppression level of less than 20 ng/dL by day 28 and by day 336, respectively.
PSA Levels (Secondary End Point)
In the ITT population (N = 137) of the FP01C-13-001 study, the mean baseline PSA level was ██████ █████ ███ █ █████████, and it decreased to ██████ █████ ███ █ ████████ on day 28. The mean PSA level was █████ █████ ███ █ ███████ on day 168 and ██████ █████ ███ █ ████████ on day 336.
Luteinizing Hormone Levels (Secondary End Point)
In the ITT population of the FP01C-13-001 study, the mean luteinizing hormone (LH) level at baseline was █████ ████ ███ █ ████████ The mean serum LH level was ███████ ████ ███ █ █████████ on day 28, ███████ ████ ███ █ █████████ on day 168, and ███████ ████ ███ █ █████████ on day 336.
QoL (Primary End Point)
In the ITT population of the FP01C-13-001 study, approximately 68.6% (94 of 137) of patients felt satisfied (rated QoL as 0, indicating delighted; as 1, indicating pleased; or as 2, indicating mostly satisfied) with their condition at baseline. Of the patients who reported QoL on day 168, approximately 69.7% (90 of 129 patients) felt satisfied. Of the patients who reported QoL on day 336, approximately 65.9% (87 of 132 patients) felt satisfied.
Harms Results
The FP01C-13-001 Study
In the pivotal FP01C-13-001 study, the median duration of follow-up was ███ ████ ███████ ██ ██ ████ and approximately █████ ████ ██ ████ of the study population received both doses of leuprolide mesylate. The most common treatment-emergent adverse event (TEAE) in the safety analysis set of the FP01C-13-001 study (N = 137) was hot flush (48.9%), followed by hypertension (14.6%), pain in extremity (9.5%), injection-site pain (7.3%), arthralgia (6.6%), fatigue (6.6%), nocturia (5.8%), back pain (5.1%), and nasopharyngitis (5.1%). Serious adverse events (SAEs) occurred in █████ of the safety analysis set, with injury, poisoning, and procedural complications ██████ being the most frequent SAEs. There were ██████ deaths: ███ due to stroke, ███ due to metastatic prostate cancer and acute renal failure, and ███ ██ for unknown reasons. Five patients (3.6%) discontinued the study because of TEAEs, which included acute kidney injury, atrial fibrillation, cerebrovascular accident, death, hormone-refractory prostate cancer, and metastatic prostate cancer.
The FP01C-13-001-EX Extension Study
In the FP01C-13-001-EX extension study (██), the median duration of follow-up was ███ ████ ███████ ██ ██ █████ The most common TEAEs that occurred during the extension study were acute kidney injury ████████ increased blood triglycerides (6.67%), dehydration ███████, dizziness ███████, fall ███████, fatigue ███████, and hypertension ███████. Four patients ████████ reported SAEs, and no discontinuations due to adverse events (AEs) or death were reported during the extension study.
Critical Appraisal
Internal Validity
The evidence for leuprolide mesylate included in the sponsor’s summary was based on 1 phase III, single-arm, open-label pivotal study (FP01C-13-001) and 1 phase III, single-arm, open-label, safety extension study (FP01C-13-001-EX). From a methodological perspective, the absence of an internal comparison group in the FP01C-13-001 pivotal study is a key limitation; because of the intrinsic nature of the single-arm design, this absence leads to low confidence in how well the findings reflect the truth. Consequently, it is a challenge to make inferences about the efficacy and safety of leuprolide mesylate. However, from a regulatory perspective, it was considered acceptable for the pivotal FP01C-13-001 study to adopt the single-arm design, per current FDA guidance for the establishment of the efficacy and safety of GnRH analogues for the treatment of advanced prostate cancer.22 Health Canada was in agreement with the FDA guidance, finding that the single-arm study design was appropriate for the assessment of leuprolide mesylate.23 The design of the FP01C-13-001 study was, overall, well aligned with the FDA guidance because it met the major criteria outlined in the guidance.22
The FP01C-13-001-EX safety extension study provided 2 additional doses of leuprolide mesylate, approximately 6 months apart, to patients from the pivotal FP01C-13-001 study and assessed the safety of leuprolide mesylate for up to 2 years (i.e., 1 year in the FP01C-13-001 study plus 1 year in the extension study). Although all ██ ████████ received the first dose of leuprolide mesylate, it was noted that approximately ███ ███ ██ ███ of the patients in the FP01C-13-001-EX extension study did not receive the second dose, including ████████ who discontinued due to early termination unrelated to AEs and ████████ who discontinued due to drug supply expiration. The large proportion of patients missing the second dose of treatment might result in a potential risk of underestimation in the incidence of harms, although the magnitude of the impact remains unknown.
A gap remains in the sponsor-submitted evidence because of the absence of direct or indirect evidence (i.e., the pivotal FP01C-13-001 study used a single-arm design), which limited the ability of the CDA-AMC review team to draw any evidence-based conclusions about the efficacy of leuprolide mesylate relative to other ADTs (including leuprolide products) currently available in Canada.
External Validity
The pivotal FP01C-13-001 study does not completely align with the population in the Health Canada–approved indication in terms of advanced prostate cancer. The Health Canada–approved indication targets patients with advanced prostate cancer, whereas the pivotal FP01C-13-001 study did not limit enrolment to patients with advanced prostate cancer; it included patients with stage I to stage IV disease. Of note, according to the clinical experts consulted by the CDA-AMC review team, there is no universal definition for advanced prostate cancer; the phrase “patients with advanced prostate cancer” used in the Health Canada–approved indication is subject to interpretation. The clinical experts consulted by the CDA-AMC review team noted that advanced prostate cancer commonly refers to biochemically recurrent or metastatic disease, yet patients with intermediate-risk or high-risk localized prostate cancer can also be eligible for ADT. The sponsor, in its response to a CDA-AMC information request,21 stated that: “In clinical practice, the standard definition [of advanced prostate cancer] is disease that has progressed beyond localized treatment options, including locally advanced disease (e.g., T3 or T4 classification) that is not amenable to curative treatment, metastatic disease, and castration-resistant prostate cancer.” Treatment recommendations and guidelines for prostate cancer recognize that advanced prostate cancer encompasses both localized disease with high-risk features and metastatic disease, and that all patients with advanced prostate cancer would typically require ADT.8,24,25 Nonetheless, both clinical experts consulted by the CDA-AMC review team agreed that the results generated from the FP01C-13-001 study population would still be generalizable to the Health Canada–indicated population.
The clinical experts consulted by the CDA-AMC review team noted that the pivotal FP01C-13-001 study excluded patients who had an Eastern Cooperative Oncology Group Performance Status (ECOG PS) of greater than 2; who received combination therapy with chemotherapy, immunotherapy, cryotherapy, radiotherapy, concomitant ADT, or LH–releasing hormone (LHRH) therapy during the study; and who had a baseline morning serum testosterone level of 150 ng/dL or less. In contrast, the Health Canada indication did not restrict the use of leuprolide mesylate in these patients. According to the clinical experts consulted by the CDA-AMC review team, the patients who were excluded from the FP01C-13-001 study would benefit from leuprolide mesylate treatment and account for a large proportion of patients with advanced prostate cancer. Nonetheless, the clinical experts felt that the results from the pivotal FP01C-13-001 study would still be generalizable to the patients who were excluded.
Cost Information
The sponsor submitted a cost comparison that evaluated the annual drug and health care resource-use costs associated with leuprolide mesylate and other available ADTs.
At the submitted price of $1,499.00 per 42 mg extended-release emulsion for injection, the annual drug acquisition cost of leuprolide mesylate is estimated to be $2,998 per patient. Based on publicly available list prices, the annual cost of leuprolide mesylate is expected to be lower than the annual cost of all other ADTs (i.e., buserelin acetate, degarelix, goserelin acetate, leuprolide acetate [Eligard], leuprolide acetate [Lupron Depot], relugolix, and triptorelin). Leuprolide mesylate is to be administrated once every 6 months; compared with the 6-month ADT comparator formulations (i.e., 45 mg of leuprolide acetate [Eligard] and 22.5 mg of triptorelin), leuprolide mesylate is estimated to be associated with cost savings of $322 annually. The incremental savings associated with leuprolide mesylate are based on publicly available list prices and may not reflect actual prices paid by drug plans.
The sponsor estimated health care resource-use costs based on treatment-specific administration times and the cost of nonsteroidal antiandrogen therapy for GnRH agonists during the first year. Differences in administration times among ADTs were deemed to be uncertain, as clinical expert feedback received by CDA-AMC indicated that some patients receiving ADT treatments are enrolled in patient support programs, which cover administration costs, and the time differences associated with administration may not result in clinically meaningful differences in administration times; therefore, it is uncertain whether these differences will translate to cost savings to the health care system.
The cost comparison assumes clinical similarity between leuprolide mesylate, and the other ADTs included in the analysis. The CDA-AMC Clinical Review was unable to draw a conclusion on the relative efficacy and safety of leuprolide mesylate and other ADTs currently available in Canada, given the absence of direct and indirect comparative evidence. However, according to the CDA-AMC Clinical Review, Health Canada and the clinical experts consulted by the CDA-AMC review team did not express any major concerns with the lack of such evidence in their assessment of the relative efficacy and safety of leuprolide mesylate. As well, any conclusions regarding incremental savings associated with the reimbursement of leuprolide mesylate are uncertain, given the uncertainty in the comparative clinical effectiveness.
Conclusions
Leuprolide mesylate is a new salt formulation of leuprolide that has been in use broadly for more than 30 years for the treatment of prostate cancer. A phase III, single-arm, open-label pivotal study submitted by the sponsor (FP01C-13-001) assessed the efficacy and safety of leuprolide mesylate in 137 adult patients with histologically confirmed prostate carcinoma and a baseline serum testosterone level greater than 150 ng/dL. The absence of a comparator group in the pivotal FP01C-13-001 study is a key limitation that, from the methodological perspective, leads to low certainty in the evidence of leuprolide’s efficacy in treating prostate cancer. However, the single-arm study design of FP01C-13-001 is acceptable, from the regulatory perspective, as it met the requirements of current guidance for establishing the efficacy and safety of GnRH analogues for advanced prostate cancer.
The results of the pivotal FP01C-13-001 study showed that leuprolide mesylate could effectively suppress serum testosterone to the castrate level (≤ 50 ng/dL) in 98.5% (95% CI, 94.8% to 99.8%) of the ITT population by day 28 after the first dose of leuprolide mesylate and help maintain the castrate testosterone level in 97.0% (95% CI, 92.2% to 98.9%) of the ITT population by day 336. The ability of leuprolide mesylate to suppress testosterone levels was further supported by findings that showed a low incidence of testosterone excursions ██ █████████ of more than 50 ng/dL during the study, as well as the high proportion of the ITT population that achieved (69.3%) and maintained (95.9%) testosterone suppression at 20 ng/dL or less. Based on the harms data from the pivotal FP01C-13-001 study and the phase III, single-arm, FP01C-13-001-EX extension study, which enrolled 30 patients who participated in the FP01C-13-001 pivotal study, the clinical experts consulted by the CDA-AMC review team considered leuprolide mesylate to be, overall, safe and tolerable.
The CDA-AMC review team was unable to draw any conclusions about the relative efficacy and safety of leuprolide mesylate and other ADTs (including leuprolide products) currently available in Canada in the absence of direct and indirect comparative evidence, although Health Canada and the clinical experts consulted by the CDA-AMC review team did not express any major concerns about the lack of such evidence in their assessment of the relative efficacy and safety of leuprolide mesylate.
At the sponsor’s submitted price, drug-acquisition costs for leuprolide mesylate are estimated to be $2,998 annually, which, at public list prices, is less costly than the annual costs associated with other available ADTs. The sponsor’s cost comparison assumes clinical similarity between leuprolide mesylate and other ADTs; however, no direct or indirect evidence was submitted to support this assumption. According to the CDA-AMC Clinical Review, Health Canada and the clinical experts consulted by the CDA-AMC review team did not express any major concerns about the lack of direct or indirect evidence in their assessment of the relative efficacy and safety of leuprolide mesylate. Incremental savings are based on publicly available list prices and may not reflect the actual prices paid by public drug plans in Canada. Overall, given the lack of direct or indirect comparative efficacy data for leuprolide mesylate versus other ADTs, there is insufficient evidence for leuprolide mesylate to have a price premium over currently reimbursed ADTs.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of leuprolide mesylate (extended-release emulsion, 42 mg of leuprolide, and SC injection) in the treatment of advanced prostate cancer in adults.
Disease Background
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the review team.
Prostate cancer is one of the most commonly diagnosed cancers in males in Canada, with an estimated 27,900 patients diagnosed in 2024, accounting for 22% of all new cancer cases in males.3 Prostate cancer is also one of the most common causes of cancer deaths in Canada, with an estimated 5,000 deaths in 2024.3 The median age at diagnosis is 66 years, but 20% of patients are older than 75 years.4,5 Most patients with prostate cancer do not have initial or early symptoms.6 As the tumour grows locally, or as it metastasizes, symptoms can intensify and start to interfere with the physiological functions of the body.
Diagnostic tests for prostate cancer include blood screening for PSA, physical digital rectal examination, biopsy of the prostate, and imaging with CT or MRI scans.6-9 According to the clinical experts consulted by the review team, the Gleason grading system is often used to give a histological grade for prostate cancer; however, a newer Grade Group system is increasingly used around the world for this purpose. The TNM classification system, commonly used to determine tumour stage in patients with prostate cancer, has 4 stages (stage I to stage IV), and the higher the stage, the more the tumour has spread.7,8,10
Terms such as localized prostate cancer (limited to the prostate), locally advanced prostate cancer (spread outside of the prostate but not metastatic), and metastatic prostate cancer (spread beyond the tissue surrounding the prostate to lymph nodes or other parts of the body, such as the lungs, liver, or bones), are also used to describe the growth and spread of prostate cancer.11,12 For patients with localized prostate cancer at the time of diagnosis, the 5-year OS rate is reported to be nearly 100%, whereas for patients with distant metastases, the 5-year OS rate is only 29%.6
Standards of Therapy
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the review team.
According to the clinical experts consulted by the CDA-AMC review team, metastatic prostate cancer is incurable in nature, but there are a wide variety of clinical therapeutic options, and survival times range from weeks to many years.
Both the sponsor and the clinical experts consulted by the review team noted that ADT is the backbone of treatment when systemic anticancer therapies are considered for the treatment of metastatic prostate cancer. According to the clinical experts consulted by the review team, ADT is continued throughout a patient’s life after diagnosis, and other medicines are used in addition to ADT. The clinical experts consulted by the review team noted that ADT is typically continued even when a patient’s disease progresses or when a patient transitions to best supportive care near the end of life. The clinical experts also noted that some treatment protocols use ADT for a finite period (e.g., as neoadjuvant treatment before definitive surgery or radiotherapy, or as adjuvant therapy with radiotherapy).
GnRH analogues, a type of ADT, are available as antagonists, such as degarelix and relugolix,13,14 or as GnRH agonists, such as leuprolide products, which include leuprolide acetate (Eligard, Lupron Depot, and Zeulide Depot) and goserelin acetate.15-18 The main goals of treatment for advanced prostate cancer are to delay disease progression with the achievement of castrate levels of testosterone, which is defined as less than 50 ng/dL by the AUA and the EAU.19,20 The EAU also indicates that testosterone levels of less than 20 ng/dL might be associated with better outcomes than levels that range between 20 ng/dL and 50 ng/dL.19
Drug Under Review
Leuprolide is a GnRH receptor agonist, and the continuous use of leuprolide in therapeutic doses decreases testosterone in males to levels associated with castration (≤ 50 ng/dL in serum).2,26-28
Leuprolide mesylate is one of the various formulations of leuprolide but is the first available formulation that does not require reconstitution before administration. Leuprolide mesylate received a Notice of Compliance from Health Canada on November 16, 2021, for the treatment of adult patients with advanced prostate cancer.2 The sponsor’s reimbursement request is in line with the Health Canada indication. Leuprolide mesylate has not been previously reviewed by CDA-AMC.
The recommended dose of leuprolide mesylate is 42 mg, administered every 6 months as a single SC injection.2
The key characteristics of leuprolide mesylate and other relevant treatments available for adult patients with advanced prostate cancer are summarized in Table 2.
Table 2: Key Characteristics of Leuprolide Mesylate and Its Main Comparators
Drug | Mechanism of action | Indicationa | Route of administration and recommended dose | Serious adverse effects or safety issues |
|---|---|---|---|---|
Leuprolide mesylate (Camcevi) | Nonapeptide analogue of naturally occurring GnRH or LHRH; inhibits pituitary gonadotropin secretion and suppresses testicular testosterone production | For the treatment of adult patients with advanced prostate cancer | SC injection 42 mg administered every 6 months as a single SC injection |
|
Leuprolide acetate (Eligard) | Nonapeptide analogue of naturally occurring GnRH or LHRH; inhibits pituitary gonadotropin secretion and suppresses testicular testosterone production | For the treatment of patients with advanced prostate cancer | SC injection After mixing with a special polymer formulation:
|
|
Leuprolide acetate for depot suspension (Lupron Depot) | Nonapeptide analogue of naturally occurring GnRH or LHRH; inhibits pituitary gonadotropin secretion and suppresses testicular testosterone production | For the palliative treatment of sex hormone-responsive advanced (stage D2) carcinoma of the prostate | Intramuscular injection After reconstitution with the special diluent:
|
|
Leuprolide acetate for depot suspension (Zeulide Depot) | Nonapeptide analogue of naturally occurring GnRH or LHRH; inhibits pituitary gonadotropin secretion and suppresses testicular testosterone production | For the palliative treatment of patients with advanced and/or metastatic prostate cancer | SC injection After reconstitution with the ready-to-use diluent:
|
|
Relugolix (Orgovyx) | GnRH receptor antagonist at pituitary gland; reduces release of LH and FSH, thereby suppressing testosterone release by the testes | For the treatment of adult patients with advanced prostate cancer | Oral tablets Loading dose of 360 mg (3 tablets) on the first day and continue treatment with 120 mg (1 tablet) once daily at approximately the same time each day |
|
Buserelin acetate (Suprefact Depot) | Peptide analogue of the natural GnRH or LHRH; inhibits gonadotropin release and subsequently reduces serum testosterone | For the palliative treatment of patients with hormone-dependent advanced carcinoma of the prostate gland (stage D) | SC implant:
|
|
Degarelix (Firmagon) | Competitively and reversibly binds to GnRH receptors at the pituitary gland, reducing the release of LH and FSH, which leads to reduced secretion of testosterone by the testes | For testosterone suppression in patients with advanced hormone-dependent prostate cancer for whom androgen deprivation is warranted | SC injection A starting dose of 240 mg once, followed by a maintenance dose of 80 mg once monthly |
|
Goserelin acetate (Zoladex) | Decapeptide analogue of GnRH or LHRH; inhibits gonadotropin (LH) production, resulting in gonadal and, consequently, accessory sex-organ regression (suppressing testosterone) |
| SC injection When given in combination with a nonsteroidal antiandrogen and radiotherapy for patients with stage T2b to stage T4 prostatic carcinoma, treatment should be started 8 weeks before the initiation of radiotherapy and continue until the completion of radiation therapy; a treatment regimen using 3.6 mg of goserelin acetate 8 weeks before radiotherapy, followed in 28 days by 10.8 mg, can be administered by SC injection into the anterior abdominal wall Alternatively, 4 SC injections of goserelin acetate, 3.6 mg, into the anterior abdominal wall can be administered in 28-day intervals, with 2 depots preceding and 2 during radiation therapy until the completion of radiation therapy |
|
Triptorelin (Trelstar) | Decapeptide analogue of naturally occurring GnRH or LHRH; inhibits gonadotropin secretion and suppresses testosterone production at the testes | For the palliative treatment of patients with hormone-dependent advanced carcinoma of the prostate gland (stage D2) | Intramuscular injection:
|
|
AUA = American Urological Association; FSH = follicle-stimulating hormone; GnRH = gonadotropin-releasing hormone; LH = luteinizing hormone; LHRH = luteinizing hormone–releasing hormone; SC = subcutaneous; TNM = tumour, node, metastasis.
aHealth Canada–approved indication.
Sources: Product monographs for leuprolide mesylate (Camcevi),2 Eligard,17 Lupron Depot,15 Zeulide Depot,16 relugolix (Orgovyx),13 buserelin acetate (Suprefact Depot),29 degarelix (Firmagon),14 goserelin acetate (Zoladex),18 triptorelin (Trelstar).30
Perspectives of Patients, Clinicians, and Drug Programs
The full patient and clinician group submissions received are available in the consolidated patient and clinician group input document for this review on the project website.
Patient Group Input
No patient group input was received by the CDA-AMC review team for this review.
Clinician Input
Input From Clinical Experts Consulted for This Review
All CDA-AMC review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of advanced prostate cancer.
Unmet Needs
According to the clinical experts consulted by the CDA-AMC review team, there is no universal definition for advanced prostate cancer, and the phrase “patients with advanced prostate cancer” used in the Health Canada–approved indication is subject to interpretation. The clinical experts consulted by the CDA-AMC review team noted that ADT can be used in intermediate-risk and high-risk localized prostate cancer, as well as in the context of biochemically recurrent and metastatic disease, which would be the more common definition of advanced prostate cancer. According to the clinical experts consulted by the CDAAMC review team, for patients with biochemically recurrent or metastatic disease, the treatment goal is to achieve a castrate testosterone level, which controls the disease by decreasing PSA levels, delaying the development of metastatic disease (in the context of biochemical recurrence), and reducing disease burden (in metastatic prostate cancer), which often result in improved symptoms, OS, and PFS.
According to the clinical experts consulted by the review team, the long-term use of ADT with currently available GnRH analogues typically results in the development of refractory disease or castrate resistance, which was considered one of the main unmet needs. Leuprolide mesylate, as a GnRH analogue, does not address this major unmet need in the clinical experts’ opinions.
According to the clinical experts consulted by the CDA-AMC review team, the availability of leuprolide mesylate, which does not require reconstitution like Eligard does, provides an additional formulation option for patients and clinicians.
Place in Therapy
According to the clinical experts consulted by the review team, there are currently several approved preparations of leuprolide products in Canada, including Lupron Depot (3-month, 4-month, and 6-month depots given as intramuscular injections), Eligard (1-month, 3-month, 4-month, and 6-month depots given as SC injections), and goserelin acetate (3-month depot given as SC pellet implants). Despite prescribing heterogeneity in Canada, most patients do not receive the 6-month preparations. The clinical experts consulted by the review team noted that leuprolide mesylate is not expected to shift the current treatment paradigm; rather, leuprolide mesylate would share the same place in therapy as other GnRH analogues, providing an additional option to the existing GnRH analogues.
Patient Population
According to the clinical experts consulted by the review team, patients with advanced prostate cancer are most likely to respond to treatment with leuprolide mesylate. The clinical experts consulted by the review team noted that the indications for GnRH analogue injections for patients with advanced prostate cancer have been well established for several decades, and that there are several other drugs with very similar chemical formulations and pharmaceutical actions already on the market. According to the clinical experts consulted by the review team, there are no particular disease characteristics that would warrant or contraindicate the use of leuprolide mesylate relative to other GnRH analogues, and there are no identifiable patient or disease features (clinical, histological, or biochemical), that render patients more suited for leuprolide mesylate than to other GnRH analogues.
The clinical experts consulted by the review team noted that although patients with very high-grade prostate cancer and patients with unusual histology (e.g., sarcomatoid or neuroendocrine) are likely to become refractory to treatment with GnRH analogues sooner than those with lower-grade prostate cancer, it is not possible to identify patients who are more likely to respond to leuprolide mesylate than to other GnRH analogues already on the market.
Assessing the Response Treatment
According to the clinical experts consulted by the review team, the outcome measures for advanced prostate cancer treatment used in clinical practice are aligned with those used in clinical trials. The clinical experts consulted by the review team noted that for advanced prostate cancer, efficacy was determined by evaluating a number of metrics, including the clinical status of the patient, radiologic response and/or progression, and PSA dynamics. The clinical experts consulted by the review team noted that the definition of a clinically meaningful response is well established in current clinical guidelines, and the parameters applied to leuprolide mesylate should be identical to those already accepted for other GnRH analogue drugs. The clinical experts consulted by the CDA-AMC review team noted that a serum testosterone level of 50 ng/dL or less is an accepted surrogate end point for efficacy, and that recent evidence suggests that suppression of serum testosterone levels to less than 20 ng/dL might be associated with better biochemical relapse-free survival.31
Discontinuing Treatment
According to the clinical experts consulted by the review team, ADT with GnRH analogues is typically intended to be continued for a patient’s lifetime; however, in some instances, GnRH analogues are prescribed for a finite period as an adjunct to other definitive therapies (e.g., as neoadjuvant treatment before surgery or as neoadjuvant and adjuvant therapy to radiotherapy). The clinical experts consulted by the review team noted that GnRH analogue therapy is typically not discontinued, even when there is disease progression or when a patient transitions to best supportive care near the end of life; instead, other medicines are added to ADT. According to the clinical experts consulted by the review team, ADT with GnRH analogues may be occasionally discontinued in cases of significant or intolerable side effects or at patient or clinician discretion.
Prescribing Considerations
According to the clinical experts consulted by the review team, appropriate treatment settings for leuprolide mesylate include outpatient or ambulatory care clinics, physicians’ offices, and injection clinics in the community setting. The clinical experts consulted by the review team also noted that treatment may be administered through a home injection program by nursing personnel.
According to the clinical experts consulted by the review team, a specialist (most commonly a urologist) usually establishes the diagnosis, and treatment may be administered by a urologist, a radiation oncologist, or a medical oncologist, depending on the stage of disease. The clinical experts consulted by the review team noted that monitoring is provided by either a specialist or by a primary care physician, depending on treatment response and stability of disease.
Clinician Group Input
This section was prepared by the review team based on the input provided by clinician groups.
The clinician group input on the review of leuprolide mesylate was received from 1 clinician group: the Ontario Health (Cancer Care Ontario) GU DAC. A total of 2 clinicians provided the input.
According to the GU DAC, the treatment goals for patients with hormone-dependent advanced prostate cancer are to improve survival, delay disease progression, reduce cancer-related complications (e.g., skeletal-related events with reduced need for palliative radiotherapy, spinal cord compression, urinary obstruction, and the need for palliative procedures, such as transurethral resection of the prostate), improve patients’ QoL, maintain the independence of patients, and reduce the burden on caregivers.
According to the GU DAC, there are unmet needs for effective methods of suppressing testosterone with reduced side effects in patients with advanced prostate cancer. In addition, there is also a need for reduced drug costs for these patients.
According to the GU DAC, leuprolide is a GnRH agonist and is the active ingredient not only in leuprolide mesylate, but also in Eligard, Lupron Depot, and Zeulide Depot, which have been approved by Health Canada for patients with prostate cancer. The GU DAC noted that what distinguishes these drugs are the mode of injection and the delivery system, which create long-acting release and suppress testosterone for 1 to 6 months. Leuprolide mesylate is a 6-month depot injection; the Eligard 6-month depot injection is currently available and reimbursed in Canada. According to the GU DAC, leuprolide mesylate may improve convenience for patients who prefer a 6-month dosing schedule because of their lifestyle, the location of their residence, as well as their ability to travel to get treatment, to have vacations, or to work.
According to the GU DAC, leuprolide mesylate would not change the current treatment paradigm, but it would provide an alternative to the 6-month Eligard product. The GU DAC noted that leuprolide mesylate would fit into first-line therapy.
According to GU DAC, any patients who require GnRH agonist therapy for prostate cancer would be candidates for leuprolide mesylate. The GU DAC noted that the selection of leuprolide mesylate versus other available drugs would be based mostly on the preferences of the prescriber and patients for a longer, 6-month injection interval. According to the GU DAC, patients who need customization of their GnRH agonist treatment interval may not be candidates (e.g., intermittent therapy, which most commonly involves 9-month courses treatment rather than a 6-month depot), but this is a very small population.
According to the GU DAC, testosterone levels, PSA response, and radiographic response are used to determine whether a patient is responding to treatment in clinical practice and in clinical trials. The GU DAC noted that a common standard follow-up would be the measurement of a PSA level and testosterone level every 3 months, and interval imaging would depend on the scenario. The GU DAC also noted that for patients with symptoms related to either locally advanced disease or metastatic disease, a clinically meaningful response is a reduction or resolution of urinary tract obstruction or pain from bone metastases. The magnitude of response is standard across prescribers and should not depend on the drug chosen if the testosterone level is suppressed adequately.
The GU DAC noted that for patients with metastatic prostate cancer, current evidence supports the continuation of GnRH agonist therapy indefinitely through the next lines of therapy, even with disease progression. The GU DAC noted that GnRH agonist therapy can be discontinued because of severe intolerance or side effects that significantly interfere with a patient’s QoL.
The GU DAC noted that community, academic, outpatient, and hospital settings are all appropriate for treatment with leuprolide mesylate.
Drug Program Input
The drug programs provide input on each drug being reviewed through the CDA-AMC reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CDA-AMC are summarized in Table 3.
Table 3: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
There was no comparator used in the FP01C-13-001 or the FR01C-13-001-EX single-arm trials. Relevant comparators include leuprolide, goserelin acetate, buserelin acetate, degarelix, and relugolix. | This is a comment from the drug plans to inform pERC deliberations. |
Considerations for the continuation or renewal of therapy | |
The primary efficacy end points were the percentage of patients with a serum testosterone level ≤ 50 ng/dL by day 28 and the percentage with a testosterone level ≤ 50 ng/dL from day 28 through day 336. Question: Is this appropriate for monitoring therapeutic response? | According to the clinical experts consulted by the review team, in clinical practice, serum testosterone levels are monitored as biochemical responses (not as clinical responses) to ensure that patients have achieved castrate testosterone levels after receiving ADT. The clinical experts consulted by the review team noted that defining the castrate testosterone level as the suppression of the serum testosterone level to ≤ 50 ng/dL was appropriate. The clinical experts consulted by the review team also noted that recent evidence suggests that the suppression of serum testosterone levels to less than 20 ng/dL might be associated with better biochemical relapse-free survival.31 The clinical experts consulted by the review team noted that monitoring serum testosterone levels at day 28 was appropriate. Given that leuprolide mesylate is a 6-month preparation, the clinical experts consulted by the review team noted that monitoring serum testosterone levels close to 6 months after injection would help determine whether leuprolide mesylate could maintain castrate testosterone levels. |
Considerations for the discontinuation of therapy | |
In the FR01C-13-001-EX extension study, 30 patients went on to receive an additional 2 doses, for a total treatment duration of 2 years. There are no safety or efficacy data beyond 2 years. Question: What are the discontinuation criteria? | According to the clinical experts consulted by the review team, ADT with GnRH analogues is intended to be continued for a patient’s lifetime. The clinical experts consulted by the review team noted that GnRH analogue therapy is typically not discontinued, even when there is disease progression or when a patient transitions to best supportive care near the end of life; instead, other medicines are added to ADT. According to the clinical experts consulted by the review team, ADT with GnRH analogues may be occasionally discontinued because of significant or intolerable side effects at the patient’s or clinician’s discretion. |
Considerations for the prescribing of therapy | |
No concomitant anticancer therapies were permitted in the trial. Concomitant radiation was not permitted. Question: Can leuprolide mesylate be used in combination with other treatments for prostate cancer? | The clinical experts consulted by the review team noted that although it is acceptable in the clinical trial setting to not allow concomitant anticancer therapies, in clinical practice, leuprolide mesylate would usually be used in combination with other prostate cancer treatments. |
Generalizability | |
Patients with an ECOG PS > 2 were excluded from the trial. Question: Can patients with an ECOG PS > 2 be considered eligible? | The clinical experts consulted by the review team noted that patients with an ECOG PS > 2 may be considered eligible for leuprolide mesylate. |
Funding algorithm | |
Request the initiation of a rapid provisional funding algorithm. | This is a comment from the drug plans to inform pERC deliberations. |
Leuprolide mesylate may change the place in therapy of comparator drugs. | This is a comment from the drug plans to inform pERC deliberations. |
Consider aligning the reimbursement criteria of leuprolide mesylate with criteria for other leuprolide treatments currently available in Canada. | This is a comment from the drug plans to inform pERC deliberations. |
Question: In what situations would leuprolide mesylate be preferred over other ADT options? | Neither clinical expert consulted by the review team identified a situation in which the selection of leuprolide mesylate over other ADT options would be absolutely necessary. According to the clinical experts consulted by the review team, there are no particular disease characteristics that would warrant or contraindicate the use of leuprolide mesylate relative to other GnRH analogues, and there are no identifiable patient or disease features (e.g., clinical, histological, or biochemical) that would render a patient more suited to leuprolide mesylate than to other GnRH analogues. |
System and economic issues | |
If the CDA-AMC recommendation is positive, jurisdictions would not be willing to pay more than the lowest price currently negotiated for an LHRH analogue. | This is a comment from the drug plans to inform pERC deliberations. |
ADT = androgen-deprivation therapy; CDA-AMC = Canada's Drug Agency; ECOG PS = Eastern Cooperative Oncology Group Performance Status; GnRH = gonadotropin-releasing hormone; LHRH = luteinizing hormone–releasing hormone; pERC = pan-Canadian Oncology Drug Review Expert Review Committee.
Clinical Evidence
Note that the clinical evidence summarized in this section was prepared by the sponsor in accordance with the CDA-AMC tailored review process and has not been modified by CDA-AMC except for some editorial changes.
Sponsor Submitted Pivotal Study and Long Extension Study
Effective testosterone suppression through medical castration is the standard of care for patients diagnosed with advanced prostate cancer. There is considerable clinical and real-world evidence demonstrating the effectiveness of leuprolide in achieving castrate levels.32-41
CAMCEVI® (leuprolide mesylate injectable suspension - LMIS 50 mg) differs from existing leuprolide products as the salt used is mesylate instead of acetate. LMIS 50 mg is a pre-mixed product containing leuprorelin mesylate equivalent to 42 mg of leuprorelin and is equivalent to existing 45 mg leuprorelin therapies. Accordingly, based on the same active moiety and therapeutic use, similar clinical pharmacodynamics (PD) and pharmacokinetic (PK) characteristics, LMIS 50 mg can be compared to therapies such as Eligard® (leuprolide acetate). Similar to the clinical development of Eligard, which was studied in a 12-month, open-label, multicentre trial, a 12-month, phase III, open-label, multicentre clinical trial was conducted to study the efficacy, safety and PK/PD of LMIS 50 mg. A 12-month extension was conducted to assess the safety of LMIS 50 mg over a prolonged period.
Table 4: Details of the Included Study
Characteristics | FP01C-13-001 | FP01C-13-001-EX |
|---|---|---|
Design Population | ||
Study design | Phase III, single arm, open label, multicentre | Phase III, single arm, open label, multicentre, safety extension study |
Locations | 26 sites in eight countries: Austria, Czech Republic, Germany, Lithuania, Poland, Slovakia, Taiwan, and the United States | 7 sites in the United States |
Patient enrolment dates | Start: 08/12/2014 (First patient enrolled) End: 09/02/2016 (Last patient completed) | Start: 02/18/2016 (First patient enrolled) End: 06/12/2017 (Last patient completed) |
Randomized (N) | N= 137 Part I: n = 33 Part II: n = 104 | N = 30 |
Inclusion criteria |
|
|
Exclusion criteria |
|
|
Drug | ||
Intervention | Leuprolide mesylate (LMIS): Two doses of 50 mg (42 mg leuprolide free base) as SC injection every 24 weeks (approximately every 6 months) | Leuprolide mesylate (LMIS): Two doses of 50 mg (42 mg leuprolide free base) as SC injection every 24 weeks (approximately every 6 months) |
Comparator(s) | NA | NA |
Duration | ||
Phase | ||
Screening phase | Up to 28 days prior to D0 of treatment | NA |
Treatment phase | 6 months (Days 1-168) | NA |
Follow-up phase | 6 months (Days 169-336) | NA |
Outcomes | ||
Primary end point |
| To determine the safety and tolerability by:
|
Secondary and exploratory end points | Secondary end points:
Exploratory end points: N/A | NA |
Notes | ||
Publications | Shore et al. 202042 NCT0223411543 | NCT0271232044 |
AE = adverse event; ALP = alkaline phosphatase; ALT = alanine transaminase; AST = aspartate aminotransferase; BP = blood pressure; BUN = blood urea nitrogen; Ca = calcium; Cr = creatinine; dL = decilitre; ECG = electrocardiogram; ECOG = Eastern Cooperative Oncology Group; HDL = high density lipoprotein; HR = heart rate; LDL = low density lipoprotein; HbA1c = hemoglobin A1c; K = potassium; LH = luteinizing hormone; LHRH = luteinizing hormone–releasing hormone; LMIS = leuprolide mesylate injectable suspension; mg = milligram; Mg = magnesium; Na = sodium; ng = nanogram; OTC = over the counter; P = phosphorus; PSA = prostate-specific antigen; SC = subcutaneous
Source: FPC01C-13 to 001 Clinical Study Report (Sections 9 and 10);45 FP01C-13-001-EX Clinical Study Report (Section 9)46
Description of Study
FP01C-13-001 was a phase III, single-arm, open-label, multicentre trial that enrolled a total of 137 patients with histologically confirmed prostate cancer eligible for androgen ablation therapy. Patients were enrolled between August 12, 2014 and September 2, 2016 from 26 sites in 8 countries in Europe, Taiwan, and the United States. There were no sites in Canada.
The trial was conducted in two parts. Part I was designed to assess the safety of LMIS 50 mg after receiving the first dose. Thirty-three patients were enrolled in Part I and assessed at Week 4 (Day 28). Of these 33 patients, the first 10 to enrol served as a ‘sentinel’ group for safety. For these subjects, additional safety reviews were conducted at the end of Week 2, Month 1, Month 3, and Month 6. At the interim review of safety, serum leuprolide concentrations and serum testosterone suppression assessments were completed. Enrollment was temporarily suspended until the interim safety review was completed. Since3 90% of Part I patients achieved serum testosterone suppression to castrate levels (≤ 50 ng/dL), enrollment was re-opened for Part II and the remaining patients were enrolled.
Figure 1: Study Design of FP01C-13-001
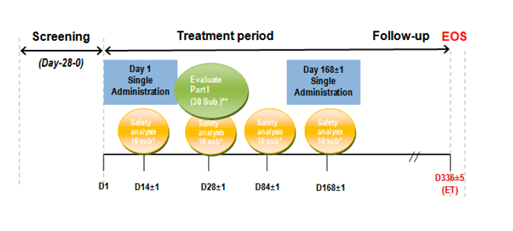
D = day; EOS = end of study; ET = extension trial; sub = subjects
The primary efficacy end points in the FP01C-13-001 trial were the percentage of patients with serum testosterone ≤ 50 ng/dL by Day 28 (i.e., within 28 days following the first dose of LMIS 50 mg) and the percentage of patients with testosterone suppression ≤ 50 ng/dL from Day 28 through Day 336. The secondary end points were the proportion of patients exhibiting post-suppression excursion of serum testosterone > 50 ng/dL, either through ‘breakthrough’ (i.e., episodes unrelated to LMIS 50 mg) or through the ‘acute-on-chronic’ effect (i.e., related to the second dose of LMIS 50 mg), commonly referred to as ‘surge’. Other secondary end points included the effect of LMIS 50 mg on serum PSA and serum LH levels. The primary safety endpoint was measured through laboratory parameters, adverse events, local skin tolerability, bone pain, and urinary symptom exacerbations. The PK behavior of leuprolide was evaluated by full PK profiles from serum leuprolide concentrations in Part I subjects. The safety objective was to assess the tolerability and safety of LMIS 50 mg.
The extension study evaluated the safety of LMIS 50 mg of subjects who participated in FP01C-13-001. Subjects were administered 2 doses of LMIS 50 mg approximately 6 months apart over 48 weeks. As this was a safety study, no efficacy analysis was performed.
Populations
Inclusion and Exclusion Criteria
In FP01C-13-001, males3 18 years of age with confirmed advanced prostate cancer and ECOG performance status ≤ 2 with a life expectancy of at least 18 months were included in the trial. Baseline morning testosterone serum needed to be < 150 ng/dL at the Screening Visit.
If patients had received chemotherapy, immunotherapy, cryotherapy, radiotherapy, or concomitant ADT within 8 weeks of the Screening Visit, they were excluded from the study. Patients should not have received any luteinizing hormone–releasing hormone (LHRH) therapy within 6 months of the Screening Visit. If patients had a clinically significant abnormal ECG or cardiovascular disease, as judged by the investigator, they were not included in the trial. Another exclusion criterion was the presence of type 1 diabetes, however, patients with type 2 diabetes were allowed if they were well-controlled on oral hypoglycemics.
The extension study enrolled patients who participated in FP01C-13-001. If participants enrolled in the study > 28 days after completion in FP01C-13-001, their baseline serum testosterone was measured to confirm castrate-level testosterone was confirmed. Additionally, examination of ECOG, PE, ECG, lab and PSA tests were repeated. These were not repeated if enrollment was < 28 days after completion in FP01C-13-001.
Baseline Characteristics
The baseline characteristics of patients enrolled in the FP01C-13-001 study are provided in Table 5. A total of 137 patients were enrolled and received LMIS 50 mg. Thirty-three patients were enrolled in Part I and additional 104 patients were enrolled in Part II.
For all subjects in Part I and Part II of the study (N = 137), the median (range) age was 74.0 (54-86) years, and most subjects were White (75.8%). For the total study population (N = 137), median (min, max) LH was 4.320 (1.54, 16.20) IU/L and median (min, max) PSA was 8.310 (0.06, 2,748.44) ng/mL.
Median (min, max) days since diagnosis was 633.0 (0, 13,290) for the full study population and most patients were Stage III (27%). The ECOG status of most patients was 0 (83.2%). These were similar to the baseline characteristics in the extension study with the key difference being that most patients were Stage II (30%).
Table 5: Summary of Baseline Characteristics – ITT Set For FP01C-13-001 and Safety Set for FP01C-13-001-EX
Characteristics | FP01C-13-001 | FP01C-13-001-EX | ||
|---|---|---|---|---|
Part I n = 33 | Part II n = 104 | Total N = 137 | Total N = 30 | |
Age, yearsa | ||||
Median (range) | 74.0 (54–86) | 70.0 (51–88) | 71.0 (51–88) | 76 (52–88) |
Race, n (%) | ||||
White | 25 (75.8) | 98 (94.2) | 123 (89.8) | 25 (83.3) |
Black or African American | 4 (12.1) | 4 (3.8) | 8 (5.8) | 3 (10.0) |
Asian | 4 (12.1) | 1 (1.0) | 5 (3.6) | 1 (3.3) |
Unknown | 0 (0.0) | 1 (1.0) | 1 (0.7) | 1 (3.3) |
LH, IU/L | ||||
Median (min, max) | NR | NR | ████ | NAd |
PSA, ng/mL | ||||
Median (min, max) | NR | NR | ████ | NAd |
Diagnosis, Daysb | ||||
median (min, max) | 2,254.0 (12, 9066) | 158.5 (0, 13290) | 633.0 (0, 13290) | NA |
Prostate cancer stage, n (%)c | ||||
I | 1 (3.0) | 3 (2.9) | 4 (2.9) | 4 (13.3) |
II | 8 (24.2) | 23 (22.1) | 31 (22.6) | 9 (30.0) |
III | 5 (15.2) | 32 (30.8) | 37 (27.0) | 5 (16.7) |
IV | 9 (27.3) | 23 (22.1) | 32 (23.4) | 7 (23.3) |
Unknown | 10 (30.3%) | 23 (22.1) | 33 (24.1) | 5 (16.7) |
ECOG performance status | ||||
0 | 30 (90.9) | 84 (80.8) | 114 (83.2) | 24 (80.0) |
1 | 3 (9.1) | 19 (18.3) | 22 (16.1) | 2 (6.7) |
2 | 0 (0.0) | 1 (1.0) | 1 (0.7) | 1 (3.3) |
aAge was calculated as (Date of informed consent – Date of birth)/365.25 and rounded down to integer. bdiagnosis of prostate carcinoma history (days) was calculated as
(Date of informed consent - Date of diagnosis). Subject TW12-001's date of diagnosis was the same as date of informed consent. cStaging of prostate carcinoma history.dEfficacy analysis was not conducted for the extension study
CI = confidence interval; ECOG = Eastern Cooperative Oncology Group; IU = international units; L = litre; LH = luteinizing hormone; n/N = number; NA = not available; NR = not reported; PSA = prostate-specific antigen; SD = standard deviation
Source: FP01C-13-001 Clinical Study Report (Section 11.2);45 FP01C-13-001-EXClinical Study Report (Table 11 to 6)46
Interventions
In the FP01C-13-001 and FP01C-13-001-EX trials, LMIS 50 mg was administered via SC injection by study personnel and no reconstitution was required. Patients received a single SC injection of LMIS 50 mg every 24 weeks (approximately every 6 months) with the first dose administered on Day 1 and the second dose on Day 168.
The dosage form of the intervention was leuprolide mesylate injection suspension, a pre-mixed drug product containing 50 mg leuprolide mesylate equivalent to 42 mg of leuprolide free base, formulated in a solution of N-methyl-2-pyrrolidone (NMP) and poly (D, L-lactide) (PLA) to control and sustain the release over a 6-month period after SC administration.
Concomitant Medications and Co-Interventions
Therapies permitted in the pivotal and extension trials included bisphosphonates and denosumab. Patients were also allowed to take vitamin D and calcium supplementation, if needed, as well as plain, over-the-counter multi-vitamins. Non-prescription and prescription pain medication, if prescribed by a physician, were allowed, in addition to radiation for pain. Patients were also permitted glucocorticosteroids for replacement therapy. For patients with Type 2 diabetes, oral hypoglycemic drugs were allowed.
Outcomes
A listing of trial outcomes, including the end point type, as well as their respective assessment timepoints, is provided in Table 6.
Outcome measure | FP01C-13-001 | FP01C-13-001-EX | ||
|---|---|---|---|---|
Timepoint | End point | Timepoint | End point | |
Efficacy outcomes | ||||
Percentage of subjects with a serum testosterone concentration suppressed to castrate levels (≤ 50 ng/dL) by Day 28 ± 1 day following the first injection of LMIS 50 mg (Part I) | Baseline to 28 days | Primary | NA | NA |
Percentage of subjects with serum testosterone suppression (≤ 50 ng/dL) from Day 28 through Day 336 (remaining duration of the study) (Part II) | 28 days to 336 days | Primary | NA | NA |
Suppression of serum testosterone level (≤ 20 ng/dL) | On Day 28 On Day 336 | Secondary | NA | NA |
The proportion of subjects exhibiting post-suppression excursions of serum testosterone to > 50 ng/dL, either through ‘breakthrough’ (i.e., episodes unrelated to LMIS 50 mg dosing), or through the “acute-on-chronic” phenomenon (i.e., related to the second dose of LMIS 50 mg) | NA | Secondary | NA | NA |
PSA response rate | From baseline to Day 336 | Secondary | NA | NA |
LH level | From baseline to Day 336 | Secondary | NA | NA |
Pharmacokinetics | ||||
Full pharmacokinetic profiles from serum leuprolide concentrations in Part I subjects | Up to 336 days | Primary | NA | NA |
Additional serum leuprolide concentrations during Part II | Up to 336 days | Primary | NA | NA |
Safety and tolerability | ||||
Change in bone pain measurement (by VAS scale) | Baseline to 28 days, 28 days to 336 days | Primary | NA | NA |
Change in urinary pain measurement (by VAS scale) | Baseline to 28 days, 28 days to 336 days | Primary | NA | NA |
Change in urinary signs and symptoms (by AUA Symptom Score sheet) | Baseline to 28 days, 28 days to 336 days | Primary | NA | NA |
Change in vital signs (BP, HR, RR) | Baseline to 28 days, 28 days to 336 days | Primary | Up to 48 weeks | Primary |
Change in physical examinations (including weight) | Baseline to 28 days, 28 days to 336 days | Primary | Up to 48 weeks | Reported but not included as a primary endpoint |
Assessment for local skin tolerability | Baseline to 28 days, 28 days to 336 days | Primary | NA | NA |
Change in lab data, including liver function (AST, ALT, ALP), renal function (BUN, SCr), complete blood count with platelets, clinical chemistries (K, Na, Mg, Ca, and P), urinalysis, serum glucose, lipid profile (LDL, HDL, triglycerides), and HgbA1c | Baseline to 28 days, 28 days to 336 days | Primary | Up to 48 weeks | Primary |
AE reporting | Baseline to 28 days, 28 days to 336 days | Primary | Up to 48 weeks | Primary |
Clinically significant changes in 12-lead resting ECGs per the investigator’s judgment | Baseline to 28 days, 28 days to 336 days | Primary | Up to 48 weeks | Primary |
QoL | Baseline to 28 days, 28 days to 336 days | Primary | NA | NA |
AE = adverse event; ALP = alkaline phosphatase; ALT = alanine transaminase;; AST = aspartate aminotransferase; AUA = American Urological Association; BP = blood pressure; BUN = blood urea nitrogen; Ca = calcium; dL = decilitre; ECG = electrocardiogram; HgbA1c = hemoglobin A1c; HDL = high density lipoprotein; HR = heart rate; LDL = low density lipoprotein; LH = luteinizing hormone; LMIS = leuprolide mesylate injectable suspension; K = potassium; mg = milligram; Mg = magnesium; Na = sodium; NA = not available; ng = nanogram; P = phosphorus; PSA = prostate-specific antigen; QoL = quality of life; RR = length of ventricular cardiac cycle measured between two successive R waves; SCr = serum creatine; VAS = visual analogue scale
Source: FP01C-13-001 Clinical Study Report (Section 9.5)45
Testosterone Suppression ≤ 50 ng/dL
Testosterone suppression ≤ 50 ng/dL was a primary outcome in the FP01C-13-001. Proliferation and survival of prostate cancer cells are dependent upon androgen stimulation via testosterone and dihydrotestosterone (DHT).47 Thus it is critical to achieve serum testosterone levels to castrate levels to minimize stimulation of cancer cells.48 Accordingly, the threshold of ≤ 50 ng/dL has been well established to achieve chemical castration and is a recognized outcome in clinical trials.48
Testosterone Suppression ≤ 20 ng/dL
Testosterone suppression to ≤ 20 ng/dL was a secondary outcome in the FP01C-13-001. Orchiectomy or surgical castration achieves castrate levels of serum testosterone ≤ 20 ng/dL and has been suggested to be a clinically meaningful outcome for patients undergoing androgen deprivation therapy.49
Prostate-Specific Antigen (PSA) Response Rate
PSA response rate was a secondary outcome measure outcome in the FP01C-13-001 trial. PSA is a well-established biomarker to assess treatment response.50 PSA increases with age and age-based reference ranges are suggested: men < 50 years, 0–2.5 ng/mL; 50–59 years, 0–3.5 ng/mL; 60–69 years, 0–4.5 ng/mL; and 70+ years, 0–6.5 ng/mL.50 In the FP01C-13-001 study, PSA > 4 ng/mL was considered elevated.42
Luteinizing Hormone (LH)
LH was a secondary outcome measure outcome in the FP01C-13-001. LH acts on the Leydig cells to increase testosterone production.51 A reduction in LH manifests as a reduction in testosterone.52 No minimally important difference (MID) was identified in the literature for LH.
Serum Leuprolide Levels
Evaluating the pharmacokinetic properties of LMIS 50 mg was conducted through measurement of serum leuprolide levels, which was a primary outcome. A concentration > 90 ng/mL was characterized as a high serum leuprolide concentration in FP01C-13-001.42 There is no MID level identified in the literature.
Adverse Events (AEs)
In the trial, safety was assessed through the incidence of adverse events (AEs), serious adverse events (SAEs), and treatment emergent adverse events (TEAEs). All AE terms in FP01C-13-001 were coded using the MedDRA dictionary version 19.1 and classified by system organ class (SOC) and preferred terms (PTs) whereas those in FP01C-13-001-EX were coded as 20.1 using the MedDRA dictionary.
Visual Analog Scale (Bone and Urinary Pain)
The visual analog scale (VAS) is a validated pain rating scale that includes a 10-point continuum. One end notes ‘no pain’ (0) and the other end notes ‘worst pain’ (10). The values can be used to track pain progression and to compare between baseline and different points in the study.53 In FPC01-13-001, the VAS was used to track bone and urinary pain progression.
Bone and urinary pain were used to evaluate the safety of LMIS 50 mg as part of the primary safety end point outcome in the FP01C-13-001 trial. Leuprolide acetate causes an increase in serum testosterone in the first week of treatment and this may cause some patients to experience symptoms such as bone pain. No MID was identified in literature for bone or urinary pain.
AUA Symptom Score Sheet (Urinary Symptoms)
The American Urological Association (AUA) Symptom Score sheet includes six questions on urinary signs and one question on urinary symptoms. The first six questions are scaled from 0 to 5 with 0 indicating no symptom at all, 1 suggesting 1 out of 5 times, 2 indicating 2 out of 5 times, 3 indicating 3 out of 5 times, 4 indicating 4 out of 5 times, and 5 indicating constant presence of symptoms. The last question is also scaled from 0 to 5, however 1 indicates one time, 2 indicates two times, 3 indicates three times, 4 indicates 4 times, and 5 indicates 5 or more times. The questions were:
Over the past month, how often have you had the sensation of not emptying your bladder completely after you finished urinating?
During the past month, how often have you had to urinate again less than 2 hours after you finished urinating?
During the past month, how often have you found you stopped and started again several times when you urinate?
During the past month, how often have you found it difficult to postpone urination?
During the past month, how often have you had a weak urinary stream?
During the past month, how often have you had to push or strain to begin urination?
Over the past month, how many times did you most typically get up to urinate during the night?
Vital Signs, Weight, and Physical Examination
Vital signs, weight, and physical examination were included as part of the primary safety end point.
Hematological, Biochemical Assessments, and Urinalysis
As part of the primary safety end point in both the FP01C-13-001 and FP01C-13-001-EX trials, blood samples were collected, and laboratory tests run to obtain complete blood counts, as well as alanine transaminase (ALT), aspartate aminotransferase (AST), alkaline phosphatase (ALP), blood urea nitrogen (BUN) total bilirubin, serum creatinine (Cr), electrolyte (potassium (K), magnesium (Mg), calcium (Ca), and phosphorus (P)), blood glucose, and lipid profile levels. Reference ranges are dependent on the assays, equipment, and technique used.
Skin Tolerability
Skin tolerability assessment was completed to assess tolerability to the SC injection in the F901C-13 to 001 trial. The skin tolerability assessment included itchiness, erythema, burning, and stinging sensation. Grading of local tolerability symptoms were from 0 (none) to 3 (severe).
Electrocardiogram
ECG in test subjects was performed by standard 12-lead at baseline and by end of study to assess the cardiac effects of leuprolide and examine the QT interval (QTc) durations as a primary safety outcome in both the FP01C-13-001 and FP01C-13-001-EX trials. Normal values for QTc range from 350 to 450 ms for adult men.54
Quality of Life Assessment
Quality of life as a result of urinary symptoms was also assessed through the use of a validated questionnaire in FP01C-13-001. Subjects were asked ‘How would you feel if you had to live with your current condition for the rest of your life?’ Respondents could answer on a scale from 0 to 6, with 0 being delighted, 1 being pleased, 2 being most pleased, 3 being mixed, 4 being mostly dissatisfied, 5 being unhappy, and 6 being terrible.
Statistical Analysis
Statistical analysis was conducted using SAS® (version not specified). Continuous variables were described using descriptive statistics such as number of observations, mean, median, standard deviation, minimum, maximum, Hodges−Lehmann estimator, and 95% confidence intervals. The paired t test or Wilcoxon signed-rank test were used to test the change from baseline for continuous variables.
Frequencies and percentages were used for summarizing discrete variables. The denominator (unless specified otherwise) was the number of subjects in the specified population.
Additional details on the statistical model used in the pivotal and extension study are provided in Table 7.
Table 7: Statistical Analysis of Efficacy and Safety End Points
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
FP01C-13-001 | ||||
Percentage of subjects with a serum testosterone concentration suppressed to castrate levels (≤ 50 ng/dL) by Day 28 ± 1 day following first injection of LMIS 50 mg | Standard large sample normal approximation to a binomial distribution | NR | Not performed | NR |
Percentage of subjects with serum testosterone suppression (≤ 50 ng/dL) from Day 28 through Day 336 (remaining duration of study) | Kaplan−Meier | NR | Subject with an event, it was analyzed as an event on day of first testosterone escape. Subject without more than one missing testosterone value was censored on day of last measurement prior to discontinuation. Any subject having more than one missing testosterone value was censored on day of the last measurement before the first missing testosterone measurement. If subject does not have suppression by Day 28, the event date was Day 28 for the analysis of percentage of subjects with testosterone suppression (≤ 50 ng/dL) from Day 28 through Day 336. | Duration of serum testosterone levels ≤ 50 ng/dL:
|
PSA response rate | Descriptive statistics and paired t test or Wilcoxon signed-rank test at a significance level of 0.05 | NR | Not performed | NR |
Change in bone pain measurement (by VAS scale) | Descriptive statistics and paired t-test or Wilcoxon signed-rank test at a significance level of 0.05 | NR | Not performed | NR |
Change in urinary pain measurement (by VAS scale) | Descriptive statistics and paired t-test or Wilcoxon signed-rank test at a significance level of 0.05 | NR | Not performed | NR |
Change in urinary signs and symptoms (by AUA Symptom Score Sheet) | Descriptive statistics and paired t-test or Wilcoxon signed-rank test at a significance level of 0.05 | NR | Not performed | NR |
Change in vital signs (BP, HR, RR)a | Descriptive statistics and paired t-test or Wilcoxon signed-rank test at a significance level of 0.05 | NR | Not performed | NR |
Change in physical examinations (including weight)a | Descriptive statistics and paired t-test or Wilcoxon signed-rank test at a significance level of 0.05 | NR | Not performed | NR |
Assessment for local skin tolerability | Descriptive statistics and paired t-test or Wilcoxon signed-rank test at a significance level of 0.05 | NR | Not performed | NR |
Change in lab data, including liver function (AST, ALT, ALP), renal function (BUN, SCr), complete blood count with platelets, clinical chemistries (K, Na, Mg, Ca, and P), urinalysis, serum glucose, lipid profile (LDL, HDL, triglycerides), and HbA1ca | Descriptive statistics and paired t-test or Wilcoxon signed-rank test at a significance level of 0.05 | NR | Not performed | NR |
AE reporta | Descriptive statistics | NR | Not performed | NR |
Clinically significant changes in 12-lead resting ECGs per the investigator’s judgementa | Descriptive statistics | NR | Not performed | NR |
Full pharmacokinetic profiles from serum leuprolide concentrations in Part I subjects | Non-compartmental analysis model | NR | Not performed | NR |
Additional leuprolide concentrations during Part II | Non-compartmental analysis model | NR | Not performed | NR |
aThese were analyses conducted for both the FP01C-13-001 and FP01C-13-001-EX studies
AE = adverse event; ALT = alanine transaminase; ALP = alkaline phosphatase; AST = aspartate aminotransferase; AUA = American Urological Association; BP = blood pressure; BUN = blood urea nitrogen; Ca = calcium; dL = decilitre; ECG = electrocardiogram; HbA1c = hemoglobin A1c; HDL = high density lipoprotein; HR = heart rate; LDL = low density lipoprotein; LH = luteinizing hormone; LMIS = leuprolide mesylate injectable suspension; K = potassium; mg = milligram; Mg = magnesium; Na = sodium; ng = nanogram; NR = not reported; P = phosphorus; PSA = prostate-specific antigen; QoL = quality of life; RR = length of ventricular cardiac cycle measured between two successive R waves; SCr = serum creatinine; VAS = visual analogue scale
Primary Outcomes
Power Calculation
██████ ████ ██ ███ ███ █████████ ██ ███████ ███ ███ █████ ██ ██████ █ ██████████ ███ █ ███ ██ ██████ █████ █ █████████ ████████ █████ ███ ██████ ████████████ █████ ███ ███████ ████ ███████ ████ ███ ██████████ ██████████ █████ ███ ████ ██████████ ███ ██████ ███ █████ ███ ███████████ ███ █████ ███ ████████ ████ ███ ████████ ██ ██ ███ ██████████████ ██ █████████ ██████ ███ ████ █ █████ ██ ███ ████████ ████ ████████ ██ ███ ██████. A total of 137 subjects were enrolled in the study.
No sample size calculations were performed for FP01C-13-001-EX as it was a safety study and no efficacy analysis was conducted.
Statistical Test or Model
The baseline values that were included in the statistical model for each outcome are provided in Table 7. FP01C-13-001 had three primary end points and FP01C-13-001-EX only had the safety end point. The results presented in this report are the final analyses.
Data Imputation Methods
Missing data related to an efficacy and safety end point did not have a method of imputation applied. Details on how missing data was handled is reported in Table 7. No data imputation was applied for missing data in FP01C-13-001-EX.
Subgroup Analyses
No subgroup analyses were conducted in FP01C-13-001 or FP01C-13-001-EX.
Sensitivity Analyses
The sensitivity analysis was performed for the primary efficacy end point in FP01C-13-001 to determine whether censoring for any missing data would affect the analysis of duration time of subject with a serum testosterone of ≤ 50 ng/dL from Day 28 through Day 336. In this analysis, the subject was censored at the last testosterone measurement before the first missing visit if no prior escape occurred. The subject was considered having event at the escape visit otherwise. If a subject maintained suppression from Day 28 through Day 336 and completed the study without missing any testosterone tests, the subject was censored at the end of study (EOS) visit. Also, given that the missing testosterone value may not have been missing at random, a sensitivity analysis of the duration time of a subject with serum £ 50 ng/dL from Day 28 through Day 336 was performed for the subset of subjects who did complete the study and did not have any testosterone data missing. This analysis was done for the ITT and PP set.
Sensitivity analyses for the duration of serum testosterone levels ≤ 50 ng/dL was conducted, in which patients missing testosterone level at any visit were censored for the analysis of duration time of subject with a serum testosterone of ≤ 50 ng/dL from Day 28 through Day 336. Subjects were censored at the last testosterone measurement before the first missing visit if no prior escape occurred; otherwise, subjects were considered to have an event at the escape visit. If a subject maintained suppression from Day 28 through Day 336 and completed the study without missing any testosterone tests, the subject was censored at the EOS visit. The analysis with censoring for any missing data was performed for patients in the ITT as well as the PP population.
No sensitivity analyses were conducted in FP01C-13-001-EX.
Secondary Outcomes
Statistical analyses for secondary outcomes for FP01C-13-001 are described in Table 7. There were no secondary outcomes in the extension study.
Analysis Populations
All efficacy analyses were conducted for the ITT set and the PP set in FP01C-13-001. Safety analyses were conducted for the Safety set. The ITT set included patients who received at least one dose of LMIS 50 mg, and the PP set included those who received both doses of LMIS 50 mg with no major protocol deviations. The Safety set included patients who received a dose of LMIS 50 mg in both FP01C-13-001 and FP01C-13-001-EX. Details are provided in Table 8.
Table 8: Analysis Populations of FP01C-13-001 and FP01C-13-001-EX
Study | Population | Definition | Application |
|---|---|---|---|
FP01C-13-001 | Intention-to-Treat (ITT) Set | Any subject who received at least one dose of LMIS 50 mg | All efficacy analyses |
Per Protocol (PP) Set | Any subject who received two doses of LMIS 50 mg, followed inclusion/exclusion requirements, and had no major protocol violations | All efficacy analyses | |
Safety Set | Any subject who received a dose of LMIS 50 mg | Safety analyses | |
FP01C-13-001-EX | Safety Set | Any subject who received a dose of LMIS 50 mg | Safety analyses |
ITT = intention-to-treat; LMIS = leuprolide mesylate injectable suspension; mg = milligram; PP = per protocol
Source: FP01C-13-001 Clinical Study Report (Section 11);45 FP01C-13-001-EX Clinical Study Report (Section 11.1)46
Sponsor’s Summary of the Results
Patient Disposition
In Part I of FP01C-13-001, 4 patients (12.1%) discontinued, out of whom 1 discontinued as a result of AEs (25% of total discontinuations). In Part II, 11 patients (10.6%) discontinued, out of whom 4 discontinued because of AEs. Overall, a total of 15 patients (10.9%) discontinued, 5 (33.3%) as a result of AEs. These events were investigated to be unrelated to LMIS 50 mg. One patient in Part I discontinued as a result of a protocol violation. There were no protocol violations in Part II.
In the extension study, 5 subjects discontinued, but none were due to AEs.
Characteristics | FP01C-13-001 | FP01C-13-001-EX | ||
|---|---|---|---|---|
Part I n = 33 | Part II n = 104 | Total N = 137 | Total N = 30 | |
Screened, n | 33 | 104 | 137 | 30 |
Discontinued, n (%) | ██████ | ██ ███ | 15 (10.9) | 5 (16.7) |
Reason for discontinuation, n (%) | ||||
AEs | █████ | █████ | 5 (33.3) | █████ |
Lack of efficacy | ██████ | ██████ | █████ | ██ |
Lost to follow-up | █████ | █████ | ██████ | 1 (20.0) |
Subject withdrew consent | ██████ | ██████ | █████ | 1 (20.0) |
Treated with prohibited medications | █████ | █████ | ██████ | 2 (40.0) |
Protocol violation: subject taking Humalog 18 units TID (3x day) | ██████ | ██████ | █████ | ██ |
Other: disease progression, PSA rise | █████ | █████ | ██████ | ██ |
Other: Subject transferred to skilled nursing facility | ██ | ██ | █████ | ██████ |
ITT, n | ██ ██ | ███ ██ | 137 (100.0) | NA |
PP, n | ██ ██ | ██ ███ | 124 (90.5) | NA |
Safety, n | ██ ██ | ███ ██ | 137 (100.0) | 30 (100.0) |
AE = adverse event; ITT = intention-to-treat; N = number; NA = not applicable; NR = not reported; PP = per protocol; PSA = prostate-specific antigen; TID = three times a day
Source: FP01C-13-001 Clinical Study Report (Tables 11 to 6, 14.1.2);45 FP01C-13-001-EX Clinical Study Report (Table 10 to 2)46
Exposure to Study Treatments
Study Treatments
The summary of study drug exposure is provided in Table 10. All patients who received one dose were included in the safety analysis in both the pivotal and extension trials. In Part I (n = 33) of FP01C-13-001, all patients (100%) received the first dose and most (87.9%) went on to receive the second dose. The mean (SD) duration was 308.6 (80.89) days. In Part II (n = 104), all patients (100%) received the first administration and 95.2% received the second dose. The mean (SD) duration was 325.7 (42.44) days. In the total population (N = 137), 93.4% of patients received both doses, with a mean (SD) study duration of 321.6 (54.39) days. Compliance to treatment was 93.4% in FP01C-13-001. In the extension study, compliance was 56.7%.
In the extension trial however, only 56.7% of subjects received both doses. 9 subjects did not receive their second dose as a result of expiration of drug supply in November 2016 and 4 subjects terminated the study early.
Table 10: Patient Exposure (Safety Population)
Patient disposition | FP01C-13-001 | FP01C-13-001-EX | ||
|---|---|---|---|---|
Part I (N = 33) | Part II (N = 104) | Total (N = 137) | Total (N = 30) | |
Received first administration | 33 (100.0) | 104 (100.0) | 137 (100.0) | 30 (100.0) |
Received second administration | ██ ██████ | ██ ██████ | ███ ██ | ██ ██████ |
Study duration, daysa | ||||
n | 33 | 104 | 137 | 30 |
Mean (SD) | █████ ██ | █████ | █████ █ | █████ ███ |
Median (min, max) | █████ ██ | █████ █ | █████ █ | ███ ████ █ |
Hodges–Lehmann estimator | ██████ | ██████ | ██████ | ██ |
95% CI | ███████ | ███████ | ███████ | ██ |
Compliance, n (%) | ██ | ██ | ███ ███ | ██ ██████ |
CI = confidence interval; N = number; NR = not reported; SD = standard deviation
Source: FP01C-13-001 Clinical Study Report (Table 12-30);45 FP01C-13-001-EX Clinical Study Report (Table 12-7)46
Concomitant Medications
In Part I (n = 33), ███ ████████ ██████ took concomitant medication, with the most common therapies being anti-thrombotic agents ████████ lipid-modifying agents ████████ ACE inhibitors ████████ and systemic antibacterials ████████ In Part II (n = 104), most subjects ███████ took concomitant medications, the most common being ACE inhibitors ███████ and systemic antibacterials ████████ Overall, in the total population (N = 137), most patients ███████ took concomitant medications. Table 11 provides the most common (³ 10% of subjects) concomitant medications used in FP01C-13-001.
Concomitant medications such as oral antihyperglycemics, statins, pain, antiplatelets were taken by subjects in FP01C-13-001-EX. Table 11 provides details on concomitant medications used in FP01C-13-001. While individual therapies taken by subjects was available, a summary of concomitant drug classes was not available for FP01C-13-EX.
Table 11: Concomitant Medications in FP01C-13-001
Exposure | FP01C-13-001 | ||
|---|---|---|---|
Part I n = 33 | Part II N = 104 | Total N = 137 | |
Concomitant medications and exposure, n (%) | ██ ██████ | ██ ██████ | ███ ██████ |
Agents acting on the renin-angiotensin system / ACE inhibitors | ██ ██████ | ██ ██████ | ██ ██████ |
Analgesics | ██ ██████ | ██ ██████ | ██ ██████ |
Antibacterials, systemic | ██ ██████ | ██ ██████ | ██ ██████ |
Antihypertensives | ██ ██████ | ██ ██████ | ██ ██████ |
Anti-inflammatory and antirheumatic products | ██ ██████ | ██ ██████ | ██ ██████ |
Antithrombotic agents | ██ ██████ | ██ ██████ | ██ ██████ |
Beta blocking agents | ██ ██████ | ██ ██████ | ██ ██████ |
Calcium channel blockers | ██ ██████ | ██ ██████ | ██ ██████ |
Diuretics | ██ ██████ | ██ ██████ | ██ ██████ |
Drugs for acid-related disorders | ██ ██████ | ██ ██████ | ██ ██████ |
Drugs for obstructive airway diseases | ██ ██████ | ██ █████ | ██ ██████ |
Drugs used in diabetes | ██ ██████ | ██ ██████ | ██ ██████ |
Lipid-modifying agents | ██ ██████ | ██ ██████ | ██ ██████ |
Mineral supplements | ██ ██████ | ██ ██████ | ██ ██████ |
Not available | ██ ██████ | ██ ██████ | ██ ██████ |
Ophthalmologic | ██ ██████ | ██ ██████ | ██ ██████ |
Psychoanaleptics | ██ ██████ | ██ ██████ | ██ ██████ |
Urologic | ██ ██████ | ██ ██████ | ██ ██████ |
Vitamins | ██ ██████ | ██ ██████ | ██ ██████ |
ACE = angiotensin converting enzyme; N = number
Source: FP01C-13-001 Clinical Study Report (Table 14.3.10)45
Efficacy (FP01C-13-001 Only)
Summary of Efficacy Outcomes
Serum Testosterone Levels ≤ 50 ng/dL (Primary End Point)
The primary efficacy end point of serum testosterone level suppressed to castrate levels ≤ 50 ng/dL was reached by 98.5% of patients in the ITT population and 99.2% of subjects in the PP population by Day 28 ± 1 day following the first dose of LMIS 50 mg. The administration of LMIS 50 mg successfully suppressed serum testosterone levels at castrate levels (≤ 50 ng/dL) from Day 28 until Day 336 in 97% and 97.6% of subjects in the ITT and PP populations, respectively (Table 12).
Figure 2: Serum Testosterone Over Time (ITT Population)
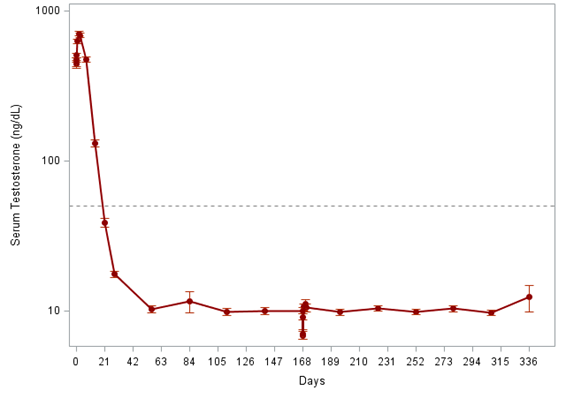
dL = decilitre; ITT = intent-to-treat; ng = nanogram
Source: FP01C-13-001 Clinical Study Report (Figure 11-3)45
Figure 3: Serum Testosterone Over Time (PP Population) [Redacted]

dL = decilitre; ITT = intent-to-treat; ng = nanogram
Source: FP01C-13-001 Clinical Study Report (Figure 11-4)45
Table 12: Serum Testosterone Levels (≤ 50 ng/dL) – Primary End Point
Characteristics | Part I | Part II | Total | |||
|---|---|---|---|---|---|---|
ITT n = 33 | PP n = 27 | ITT n = 104 | PP n = 97 | ITT N = 137 | PP N = 124 | |
Serum testosterone levels (≤ 50 ng/dL) | ||||||
Baseline | ||||||
N | 33 | ██ | 104 | ██ | 137 | 124 |
Serum testosterone ≤ 50 ng/dL, n (%) | █████ | █████ | █████ | █████ | █████ | █████ |
95% CI | █████ | █████ | █████ | █████ | █████ | █████ |
Day 28 | ||||||
n | 33 | ██ | 104 | ██ | 137 | ███ |
Serum testosterone ≤ 50 ng/dL, n (%) | ██ ██████ | █████ | █████ | █████ | 135 (98.5) | 123 (99.2) |
95% CI | █████ | █████ | █████ | █████ | 94.8, 99.8 | 95.6, 100.0 |
Days 28 to 336a | ||||||
n | ██ | ██ | ███ | ██ | ███ | ███ |
Event number (%) | █████ | █████ | █████ | █████ | █████ | █████ |
Mean time (days) | ███ | ███ | █████ | █████ | █████ | █████ |
Median time (days) | ██ | ██ | ██ | ██ | ██ | ██ |
95% CI of median (days) | ██ | ██ | ██ | ██ | ██ | ██ |
Suppression rateb by Day 336 (95% CI) | █████ | █████ | █████ | █████ | 97.0 (92.2, 98.9) | 97.6 (92.7, 99.2) |
95% RCI by Day 336c | ██ | ██ | ██ | ██ | █████ | ██ |
aThe Kaplan–Meier method was used to analyze the duration of subjects with testosterone suppression from Day 28 through Day 336. Event was defined as subjects had testosterone to > 50 ng/dL. bThe suppression rate of subjects with testosterone suppression (≤ 50 ng/dL) by Day 336 was provided. cThe 95% RCI by Day 336 was provided by ADDPLAN software and manually scribed to the SAS generated table.
CI = confidence interval; dL = decilitre; ITT = intention-to-treat; n/N = number; ng = nanogram; NR = not reported; PP = per protocol; RCI = repeated-confidence interval
Source: FP01C-13-001 Clinical Study Report (Tables 11-0, Table 11-9, 14.2.5, 14.2.6)45
Pharmacokinetic Parameters of LMIS 50 mg (Primary End Point)
Part I
After the first and second doses of LMIS 50 mg, mean serum leuprolide concentrations rose to reach the Cmax of 94.5 ± 53.7 and 99.0 ± 73.0 ng/mL at 3.23 and 2.08 hours (median Tmax), respectively. The mean values of Cwk4, Cmon6, AUC0-4wk, AUC0-6mon, and Cavg(0-6mon) were 1.04 ng/mL, 0.497 ng/mL, 91.6 day ng/mL, 224 day ng/mL, and 1.34 ng/mL, respectively, post the first dose of LMIS 50 mg SC injection. The mean values of Cwk4, Cmon6, AUC0-4wk, AUC0-6mon, and Cavg(0-6mon) were 1.64 ng/mL, 0.511 ng/mL, 125 day ng/mL, 268 day ng/mL, and 1.59 ng/mL, respectively, post the second dose of LMIS 50 mg SC injection (Table 13).
Figure 4: Arithmetic Mean Leuprolide and Testosterone Serum Concentration−Time After LMIS 50 mg SC in Part I [Redacted]

dL = decilitre; LMIS = leuprolide mesylate injectable suspension; ng = nanogram; SC = subcutaneous
Source: FP01C-13-001 Clinical Study Report (Figure 11-11)45
Table 13: Summary of Serum Pharmacokinetic Parameters of Leuprolide After LMIS 50 mg SC in Part I
Characteristic | First dose | Second dose | ||||
|---|---|---|---|---|---|---|
PK parameter | n | Mean | SD | n | Mean | SD |
Cmax (ng/mL) | 31 | 94.5 | 53.7 | 29 | 99 | 73 |
Tmax (min, max)(h)ab | 31 | 3.23 | NR | 29 | 2.08 | NR |
Cwk4 (ng/mL) | 31 | 1.04 | 0.863 | 29 | 1.64 | 0.983 |
Cmon6 (ng/mL) | 29a | 0.497 | 0.610 | 29 | 0.511 | 0.488 |
AUC0-4wks (day ng/mL) | 31 | 91.6 | 47.9 | 29 | 125 | 57.3 |
AUC0-6mon (day ng/mL) | 29a | 224 | 87.3 | 29 | 268 | 88.1 |
Cavg(0-6mon) (ng/mL) | 29a | 1.34 | 0.519 | 29 | 1.59 | 0.525 |
aNot reportable for two subjects; bTmax is reported as median (min, max)
AUC0-4wk = area under the concentration curve from time 0 to 4 weeks; AUC0-6mon = area under the concentration time curve from time 0 to 24 weeks; Cavg(0-6mon) = mean serum concentration within 24 weeks post dosing; Cmax = maximum concentration; h = hour; mL = millilitre; n = number; ng = nanogram; NR = not reported; SD = standard deviation; Tmax = time to maximum serum concentration
Source: FP01C-13-001 Clinical Study Report (Table 11-20)45
Part II
After the first and second doses of LMIS 50 mg SC injection, mean serum leuprolide concentrations rose rapidly to reach Cmax of 99.7 ± 65.6 and 93.7 ± 60.8 ng/mL at 3.67 and 3.78 hours (median Tmax), respectively. The mean values of Cwk4, Cmon6, AUC0-4wk, AUC0-6mon, and Cavg(0-6mon) were 1.47 ng/mL, 0.370 ng/mL, 103 day ng/mL, 219 day ng/mL, and 1.31 ng/mL, respectively, post the first dose of LMIS 50 mg SC injection. The mean values of Cwk4, Cmon6, AUC0-4wks, AUC0-6mon, and Cavg(0-6mon) were 2.40 ng/mL, 0.410 ng/mL, 131 day ng/mL, 250 day ng/mL, and 1.49 ng/mL, respectively, post the second dose of LMIS 50 mg SC injection (Table 14).
Figure 5: Arithmetic Mean Leuprolide and Testosterone Serum Concentration−Time After LMIS 50 mg SC in Part II [Redacted]

dL = decilitre; LMIS = leuprolide mesylate injectable suspension; ng = nanogram; SC = subcutaneous
Source: FP01C-13-001 Clinical Study Report (Figure 11-12)45
In general, the LMIS 50-mg formulation resulted in multi-phasic leuprolide concentration versus time profiles characterized by a distinctive burst phase and a plateau phase. The initial acute increase of leuprolide mesylate concentration, followed by the rapid decline to a steady-state level, was similar to the release pattern seen with the other leuprolide depot formulations. After an initial burst phase characterized by mean high serum leuprolide concentrations (> 90 ng/mL), mean serum leuprolide levels maintained relatively constant over the majority of each 6-month dosing interval.
Table 14: Summary of Serum Pharmacokinetic Parameters of Leuprolide After LMIS 50 mg SC in Part II
Characteristic | First dose | Second dose | ||||
|---|---|---|---|---|---|---|
PK parameter | N | Mean | SD | N | Mean | SD |
Cmax (ng/mL) | 94 | 99.7 | 65.6 | 97 | 93.7 | 60.8 |
Tmax (h) (min, max)ad | 94 | 3.67 | NR | 97 | 3.78 | NR |
Cwk4 (ng/mL) | 94 | 1.47 | 2.57 | 96b | 2.40 | 4.05 |
Cmon6 (ng/mL) | 92a | 0.370 | 0.313 | 94b | 0.410 | 0.538 |
AUC0-4wks (day ng/mL) | 94 | 103 | 62.4 | 96b | 131 | 91.4 |
AUC0-6mon (day ng/mL) | 92a | 219 | 108 | 94c | 250 | 160 |
Cavg(0-6mon) (ng/mL) | 92a | 1.31 | 0.643 | 94c | 1.49 | 0.950 |
aNot reportable for two subjects; bNot reportable for one subject; cNot reportable for three subjects; d bTmax is reported as median (min, max)
AUC0−4wk = area under the concentration curve from time 0−4 weeks; AUC0−6mon = area under the concentration time curve from time 0 to 24 weeks; Cavg(0−6mon) = mean serum concentration within 24 weeks post dosing; Cmax = maximum concentration; h = hour; mL = millilitre; N = number; ng = nanogram; NR = not reported; SD = standard deviation; Tmax = time to maximum serum concentration
Source: FP01C-13-001 Clinical Study Report (Table 11-25)45
Post-Suppression Excursion of Serum Testosterone to > 50 ng/dL (Secondary End Point)
Most subjects did not exhibit post-suppression testosterone > 50 ng/dL after achieving castration levels of testosterone on Day 28. Two patients did exhibit post-suppression excursions of serum testosterone > 50 ng/dL during the study period. However, the percentage of subjects exhibiting post-suppression excursion of serum testosterone > 50 ng/dL was 0% in both the ITT and PP sets at the end of study (Day 336). Results are summarized in Table 15.
Table 15: Post-Suppression Excursions of Testosterone to > 50 ng/dL
Characteristic | Part I | Part II | Total | |||
|---|---|---|---|---|---|---|
ITT N = 33 | PP N = 27 | ITT N = 104 | PP N = 97 | ITT N = 137 | PP N = 124 | |
Day 168 - Prior Dosing | ||||||
n | ██ | ██ | ███ | ██ | ███ | ███ |
Serum testosterone > 50 ng/dL, n (%) | ████ | ████ | ████ | ████ | ████ | ███ |
95% CI | ████ █ | ████ █ | ████ █ | ████ █ | ████ █ | ████ █ |
Day 336 | ||||||
n | ██ | ██ | ███ | ██ | ███ | ███ |
Serum testosterone > 50 ng/dL, n (%) | ████ | ████ | ████ | ████ | ████ | ████ |
95% CI | ████ █ | ████ █ | ████ █ | ████ █ | ████ █ | ████ █ |
CI = confidence interval; dL = decilitre; ITT = intention-to-treat; n/N = number; ng = nanogram; PP = per protocol
Source: FP01C-13-001 Clinical Study Report (Tables 11-11)45
Serum Testosterone Suppression < 20 ng/dL (Secondary End Point)
Results are only reported for the total ITT (N = 137) population. By Day 28, 69.3% of patients achieved testosterone suppression < 20 ng/dL and by Day 336, most patients (95.9%) were able to achieve testosterone suppression < 20 ng/dL (Table 16).
Table 16: Summary of Patients With Testosterone Suppression
Characteristic | ITT (N = 137) |
|---|---|
Testosterone suppression on Day 28 | |
n | 137 |
Subject with testosterone suppression, n (%) | 135 (98.5) |
Testosterone < 20 ng/dL, n (%) | 95 (69.3) |
Testosterone between 20–50 ng/dL, n (%) | ██ ██████ |
Testosterone suppression on Day 336 | |
n | ███ |
Testosterone < 20 ng/dL, n (%) | 117 (95.9) |
Testosterone between 20–50 ng/dL, n (%) | █████ |
dL = decilitre; ITT = intention-to-treat; n/N = number; ng = nanogram
Source: FP01C-13-001 Clinical Study Report (Tables 11-11)45
PSA Levels (Secondary End Point)
The administration of LMIS 50 mg significantly reduced serum PSA levels after its first injection and this effect remained until end of study. In the total ITT population (N = 137), mean baseline of PSA level was ██████ █ ████████ █████ and it decreased to ██████ █ ███████ █████ on Day 28. PSA levels appeared to approach normal on Day 168 with mean PSA level of ████████████ █████ (Table 17) The decline from baseline to Day 28 was significant (p < 0.001) and from baseline to Day 336 (p < 0.001) (Table 18). These findings were similar for Part I and Part II. In Part I, patients achieved a significant change from baseline to end of study (p < 0.001). Similarly, in Part II, patients experienced a significant reduction in PSA levels from baseline to Day 336 (p < 0.001). Table 19 additionally reports that most patients in ITT group were able to achieve PSA decreases between 50% and < 90% by Day 28. By Day 336, █████ of subjects achieved a reduction of ³ 95%. For subjects with elevated PSA at baseline, █████ were able to achieve PSA ≤ 4 ng/mL by Day 336 (Table 20).
Figure 6: PSA Levels Over Time (ITT Population) [Redacted]

ITT = intent-to-treat; mL = millilitre; ng = nanogram; PSA = prostate-specific antigen
Source: FP01C-13-001 Clinical Study Report (Figure 11-5)45
Figure 7: PSA Levels Over time (PP Population) [Redacted]

mL = millilitre; ng = nanogram; PP = per protocol; PSA = prostate-specific antigen
Source: FP01C-13-001 Clinical Study Report (Figure 11-6)45
Table 17: PSA Levels in the ITT and PP Populations (Part I, Part II, and Total)
Characteristic | Part I | Part II | Total | |||
|---|---|---|---|---|---|---|
ITT n = 33 | PP n = 27 | ITT N = 104 | PP N = 97 | ITT N = 137 | PP N = 124 | |
Baseline | ||||||
n | ██ | ██ | ███ | ██ | ███ | ███ |
Mean (SD) | █████ | █████ | █████ | █████ | █████ | █████ |
Median (min, max) | █████ | █████ | █████ | █████ | █████ | █████ |
Hodges–Lehmann estimator | █████ | █████ | █████ | █████ | █████ | █████ |
95% CI | █████ | █████ | █████ | █████ | █████ | █████ |
Day 28 | ||||||
n | █████ | █████ | █████ | █████ | █████ | █████ |
Mean (SD) | █████ | █████ | █████ | █████ | █████ | █████ |
Median (min, max) | █████ | █████ | █████ | █████ | █████ | █████ |
Hodges–Lehmann estimator | █████ | █████ | █████ | █████ | █████ | █████ |
95% CI | █████ | █████ | █████ | █████ | █████ | █████ |
Day 168 | ||||||
n | █████ | █████ | █████ | █████ | █████ | █████ |
Mean (SD) | █████ | █████ | █████ | █████ | █████ | █████ |
Median (min, max) | █████ | █████ | █████ | █████ | █████ | █████ |
Hodges–Lehmann estimator | █████ | █████ | █████ | █████ | █████ | █████ |
95% CI | █████ | █████ | █████ | █████ | █████ | █████ |
Day 336 | ||||||
n | █████ | █████ | █████ | █████ | █████ | █████ |
Mean (SD) | █████ | █████ | █████ | █████ | █████ | █████ |
Median (min, max) | █████ | █████ | █████ | █████ | █████ | █████ |
Hodges–Lehmann estimator | █████ | █████ | █████ | █████ | █████ | █████ |
95% CI | █████ | █████ | █████ | █████ | █████ | █████ |
CI = confidence interval; dL = decilitre; ITT = intention-to-treat; n/N = number; ng = nanogram; PP = per protocol; SD = standard deviation
Source: FP01C-13-001 Clinical Study Report (Table 14.2.13)45
Table 18: PSA Level (ng/mL) Change From Baseline
Characteristic | Part I | Part II | Total | |||
|---|---|---|---|---|---|---|
ITT n = 33 | PP n = 27 | ITT n = 104 | PP n = 97 | ITT N = 137 | PP N = 124 | |
Change from baseline: Day 28 | ||||||
n | █████ | █████ | █████ | █████ | █████ | █████ |
Mean (SD) | █████ | █████ | █████ | █████ | █████ | █████ |
Median (min, max) | █████ | █████ | █████ | █████ | █████ | █████ |
Hodges–Lehmann estimator | █████ | █████ | █████ | █████ | █████ | █████ |
95% CI | █████ | █████ | █████ | █████ | █████ | █████ |
P-valuea | █████ | █████ | █████ | █████ | █████ | █████ |
Change from baseline: Day 168 | ||||||
n | █████ | █████ | █████ | █████ | █████ | █████ |
Mean (SD) | █████ | █████ | █████ | █████ | █████ | █████ |
Median (min, max) | █████ | █████ | █████ | █████ | █████ | █████ |
Hodges–Lehmann estimator | █████ | █████ | █████ | █████ | █████ | █████ |
95% CI | █████ | █████ | █████ | █████ | █████ | █████ |
P-valuea | █████ | █████ | █████ | █████ | █████ | █████ |
Change from baseline: Day 336 | ||||||
N | █████ | █████ | █████ | █████ | █████ | █████ |
Mean (SD) | █████ | █████ | █████ | █████ | █████ | █████ |
Median (min, max) | █████ | █████ | █████ | █████ | █████ | █████ |
Hodges–Lehmann estimator | █████ | █████ | █████ | █████ | █████ | █████ |
95% CI | █████ | █████ | █████ | █████ | █████ | █████ |
P-valuea | █████ | █████ | █████ | █████ | █████ | █████ |
aPaired t test or Wilcoxon signed-rank test used to test the change from baseline for continuous variables
CI = confidence interval; dL = decilitre; ITT = intention-to-treat; n/N = number; ng = nanogram; PP = per protocol; SD = standard deviation
Source: FP01C-13-001 Clinical Study Report (Table 14.2.13)45
Table 19: Summary of PSA Level Percent Change From Baseline (ITT Population), n (%)
Day | n | < 50% decrease | 50 to < 90% decrease | 90 to < 95% decrease | ≥ 95% decrease | Increase |
|---|---|---|---|---|---|---|
Day 28 | 137 | █████ | ██ ███ | ██ ███ | ██ ███ | █████ |
Day 84 | 136 | █████ | ██ ███ | ██ ███ | ██ ███ | █████ |
Day 168 | 129 | █████ | ██ ███ | ██ ███ | ██ ███ | █████ |
Day 252 | 124 | █████ | ██ ███ | ██ ███ | ██ ████ | █████ |
Day 336 | 122 | █████ | ██ ███ | ██ ███ | ██ ████ | █████ |
ITT = intention-to-treat; n = number; PP = per protocol
Source: FP01C-13-001 Clinical Study Report (Table 14.2.24)45
Table 20: Summary of PSA Level Percent Change From Baseline for Subjects With Elevated PSA at Baseline (ITT Population), n (%)
Day | n | < 50% decrease | 50 to < 90% decrease | 90 to < 95% decrease | ≥ 95% decrease | Increase | ≤ 4 ng/mL | ≥ 4 ng/mL |
|---|---|---|---|---|---|---|---|---|
Day 28 | 99 | 26 (26.3) | 65 (65.7) | 4 (4.0) | 1 (1.0) | 3 (3.0) | 47 (47.5) | 52 (52.5) |
Day 84 | 98 | 4 (4.1) | 31 (31.6) | 22 (22.4) | 41 (41.8) | 0 (0.0) | 75 (76.5) | 23 (23.5) |
Day 168 | 94 | 2 (2.1) | 24 (25.5) | 11 (11.7) | 57 (60.6) | 0 (0.0) | 80 (85.1) | 14 (14.9) |
Day 252 | 90 | 2 (2.2) | 16 (17.8) | 13 (14.4) | 58 (64.4) | 1 (1.1) | 76 (84.4) | 14 (15.6) |
Day 336 | 88 | 4 (4.5) | 17 (19.3) | 11 (12.5) | 55 (62.5) | 1 (1.1) | 76 (86.4) | 12 (13.6) |
ITT = intention-to-treat; mL = millilitre; n = number; ng = nanogram
Source: FP01C-13-001 Clinical Study Report (Table 14.2.30 and 14.2.31)45
LH Levels (Secondary End Point) (Results Measured by BA Lab)
The data demonstrated that the administration of LMIS 50 mg significantly reduced serum LH levels after the first dose. In the ITT (N = 137) population, the mean LH level at baseline was 5.125 ± 3.0421 IU/L. On Day 168 the mean serum LH level was 0.10699 ± 0.251897 IU/L, and that this effect remained until the end of study (Day 336).
A significant decrease in mean serum LH level was observed by end of study on Day 336 (0.10650 ± 0.256015 IU/L) in the PP population. This was also observed in the PP population with mean level at baseline being 5.016 ± 2.9666 IU/L and 0.08545 ± 0.186483 IU/L by Day 336. Summary of results are presented in Table 21 and Table 22.
Figure 8: LH Levels Over Time (ITT Population)
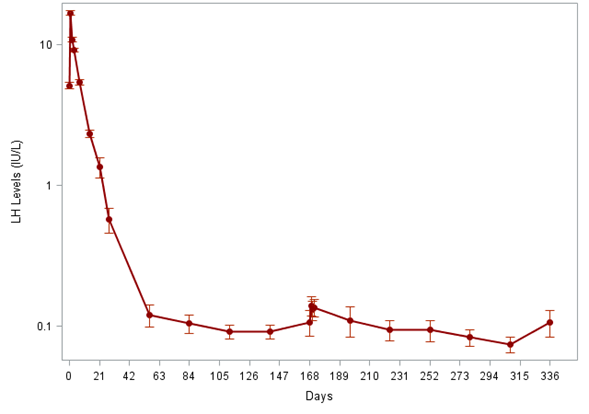
Note: Analysis was run at two labs, and were similar. The results presented are those from the BA lab.
ITT = intent-to-treat; IU = international units; L = litre; LH = luteinizing hormone
Source: FP01C-13-001 Clinical Study Report (Figure 11-7)45
Figure 9: LH Levels Over Time (PP Population) [Redacted]

Note: Analysis was run at two labs, and were similar. The results presented are those from the BA lab.
IU = international units; L = litre; LH = luteinizing hormone; PP = per protocol
Source: FP01C-13-001 Clinical Study Report (Figure 11-8)45
Table 21: Luteinizing Hormone Levels
Characteristic | Part I | Part II | Total | |||
|---|---|---|---|---|---|---|
ITT n = 33 | PP n = 27 | ITT n = 104 | PP n = 97 | ITT N = 137 | PP N = 124 | |
Baseline | ||||||
n | ████ | ████ | ████ | ████ | ████ | ████ |
Mean (SD) | ████ | ████ | ████ | ████ | ████ | ████ |
Median (min, max) | ████ | ████ | ████ | ████ | ████ | ████ |
Hodges–Lehmann estimator | ████ | ████ | ████ | ████ | ████ | ████ |
95% CI | ████ | ████ | ████ | ████ | ████ | ████ |
Day 28 | ||||||
n | ████ | ████ | ████ | ████ | ████ | ████ |
Mean (SD) | ████ | ████ | ████ | ████ | ████ | ████ |
Median (min, max) | ████ | ████ | ████ | ████ | ████ | ████ |
Hodges–Lehmann estimator | ████ | ████ | ████ | ████ | ████ | ████ |
95% CI | ████ | ████ | ████ | ████ | ████ | ████ |
Day 168 | ||||||
n | ████ | ████ | ████ | ████ | ████ | ████ |
Mean (SD) | ████ | ████ | ████ | ████ | ████ | ████ |
Median (min, max) | ████ | ████ | ████ | ████ | ████ | ████ |
Hodges–Lehmann estimator | ████ | ████ | ████ | ████ | ████ | ████ |
95% CI | ████ | ████ | ████ | ████ | ████ | ████ |
Day 336 | ||||||
n | ████ | ████ | ████ | ████ | ████ | ████ |
Mean (SD) | ████ | ████ | ████ | ████ | ████ | ████ |
Median (min, max) | ████ | ████ | ████ | ████ | ████ | ████ |
Hodges–Lehmann estimator | ████ | ████ | ████ | ████ | ████ | ████ |
95% CI | ████ | ████ | ████ | ████ | ████ | ████ |
CI = confidence interval; dL = decilitre; ITT = intention-to-treat; n/N = number; PP = per protocol; SD = standard deviation
Source: FP01C-13-001 Clinical Study Report (Table 14.2.19)45
Table 22: Luteinizing Hormone Level Change From Baseline (IU/L)
Characteristic | Part I | Part II | Total | |||
|---|---|---|---|---|---|---|
ITT n = 33 | PP n = 27 | ITT n = 104 | PP n = 97 | ITT N = 137 | PP N = 124 | |
Change from baseline: Day 28 | ||||||
n | ████ | ████ | ████ | ████ | ████ | ████ |
Mean (SD) | ████ | ████ | ████ | ████ | ████ | ████ |
Median (min, max) | ████ | ████ | ████ | ████ | ████ | ████ |
Hodges–Lehmann estimator | ████ | ████ | ████ | ████ | ████ | ████ |
95% CI | ████ | ████ | ████ | ████ | ████ | ████ |
p-valuea | ████ | ████ | ████ | ████ | ████ | ████ |
Change from baseline: Day 168 | ||||||
n | ████ | ████ | ████ | ████ | ████ | ████ |
Mean (SD) | ████ | ████ | ████ | ████ | ████ | ████ |
Median (min, max) | ████ | ████ | ████ | ████ | ████ | ████ |
Hodges–Lehmann estimator | ████ | ████ | ████ | ████ | ████ | ████ |
95% CI | ████ | ████ | ████ | ████ | ████ | ████ |
p-valuea | ████ | ████ | ████ | ████ | ████ | ████ |
Change from baseline: Day 336 | ||||||
n | ████ | ████ | ████ | ████ | ████ | ████ |
Mean (SD) | ████ | ████ | ████ | ████ | ████ | ████ |
Median (min, max) | ████ | ████ | ████ | ████ | ████ | ████ |
Hodges–Lehmann estimator | ████ | ████ | ████ | ████ | ████ | ████ |
95% CI | ████ | ████ | ████ | ████ | ████ | ████ |
p-valuea | ████ | ████ | ████ | ████ | ████ | ████ |
aPaired t test or Wilcoxon signed-rank test used to test the change from baseline for continuous variables
CI = confidence interval; dL = decilitre; ITT = intention-to-treat; n/N = number; ng = nanogram; PP = per protocol; SD = standard deviation
Source: FP01C-13-001 Clinical Study Report (Tables 14.2.16 and 14.2.17)45
Harms
The evaluation of safety was based on the following parameters: adverse events, change in bone and urinary pain, change in urinary symptoms, change in vital signs, physical examination, local skin tolerability, lab data including liver function, complete blood counts, clinical chemistries, lipid levels, HbA1c, and significant changes in 12-lead resting ECG. Safety assessment was done on the Safety set for both FP01C-13-001 and FP01C-13-001-EX.
Safety Evaluation Plan
AEs were recorded throughout the study, with five interim analyses. The first four interim analyses were conducted in the first 10 patients at the end of Week 2, Month 1, Month 3 and Month 6. Safety interim analysis was conducted for the first 30 patients after Day 28. Similarly, in FP01C-13-001-EX, safety parameters were collected throughout the study.
An AE was defined as any untoward medical occurrence in a patient administered a pharmaceutical product and does not necessarily have a causal relationship to the study treatment. It can therefore be any unfavourable and unintended sign, symptom, or disease, temporally associated with the use of study medication, irrespective of its relationship. However, abnormal laboratory values or changes were not automatically reported as AEs if they were not clinically significant. They were only recorded as AEs if the investigator judged them to be clinically significant or if therapeutic action was required.
Overview of Safety
Key harms data including treatment emergent adverse events (TEAEs) and serious adverse events (SAEs) are presented in Table 23 below.
Table 23: Key Harms Data in FP01C-13-001 and FP01C-13-001-EX
Safety population | FP01C-13-001 | FP01C-13-001-EX | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
Part I (N = 33) | Part II (N = 104) | Total (N = 137) | Total (N = 30)a | |||||||||
Event | Subject | % | Event | Subject | % | Event | Subject | % | Event | Subject | % | |
Summary of TEAEs | ||||||||||||
Total | 160 | 31 | 93.9 | 393 | 83 | 79.8 | 553 | 114 | 83.2 | ██ | ██ | ██ |
TEAEs by severity | ||||||||||||
Mild | 116 | 29 | 87.9 | 279 | 76 | 73.1 | 395 | 105 | 76.6 | ██ | ██ | ██ |
Moderate | 36 | 19 | 57.6 | 94 | 40 | 38.5 | 130 | 59 | 43.1 | ██ | ██ | ██ |
Severe | 6 | 5 | 15.2 | 18 | 13 | 12.5 | 24 | 18 | 13.1 | ██ | ██ | ██ |
Drug-related AEsb | ||||||||||||
Mild | 35 | 19 | 57.6 | 81 | 56 | 53.8 | 116 | 75 | 54.7 | ██ | ██ | ██ |
Moderate | 7 | 5 | 15.2 | 18 | 13 | 12.5 | 25 | 18 | 13.1 | ██ | ██ | ██ |
Severe | 2 | 2 | 6.1 | 1 | 1 | 1.0 | 3 | 3 | 2.2 | ██ | ██ | ██ |
Total | 44 | 21 | 63.6 | 100 | 64 | 61.5 | 144 | 85 | 62.0 | ██ | ██ | ██ |
Most common drug-related AEs | ||||||||||||
Hot flush | 18 | 18 | 54.5 | 50 | 48 | 46.2 | 68 | 66 | 48.2 | ██ | ██ | ██ |
Injection site pain | 6 | 5 | 15.2 | 7 | 5 | 4.8 | 13 | 10 | 7.3 | ██ | ██ | ██ |
Fatigue | 2 | 2 | 6.1 | 6 | 6 | 5.8 | 8 | 8 | 5.8 | ██ | ██ | ██ |
████ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ |
Summary of incidence of TEAEs in ≥ 5% of overall subjects by System Organ Classc | ||||||||||||
Vascular disorders | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ |
Infections and infestations | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ |
Musculoskeletal and connective tissue disorders | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ |
General disorders and administration site conditions | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ |
Renal and urinary disorders | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ |
Investigations | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ |
Gastrointestinal disorders | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ |
Injury, poisoning and procedural complications | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ |
Metabolism and nutrition disorders | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ |
Respiratory, thoracic, and mediastinal disorders | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ |
Nervous system disorders | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ |
Skin and SC tissue disorders | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ |
Cardiac disorders | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ |
Psychiatric disorders | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ |
Neoplasms benign, malignant, and unspecified (incl. cysts and polyps) | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ |
Summary of incidence of TEAEs leading to discontinuation | ||||||||||||
Overall | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ |
Atrial fibrillation | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ |
Death | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ |
Due to cerebrovascular accident | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ |
Due to acute kidney injury | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ |
Hormone refractory prostate cancer | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ |
Prostate cancer, metastatic | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ |
Cerebrovascular accident | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ |
Acute kidney injury | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ |
Summary of incidence of death | ||||||||||||
Overall | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ |
Due to general disorders and administration site conditions | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ |
Due to metastatic prostate cancer | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ |
Due to cerebrovascular accident | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ |
Due to acute kidney injury | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ |
Summary of incidence of SAEs | ||||||||||||
Cardiac disorders | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ |
Ear and labyrinth disorders | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ |
Eye disorders | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ |
Gastrointestinal disorders | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ |
General disorders and administration site conditions | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ |
Infections and infestations | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ |
Injury, poisoning and procedural complications | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ |
Metabolism and nutrition disorders | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ |
Musculoskeletal and connective tissue disorders | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ |
Neoplasms benign, malignant, and unspecified (incl. cysts and polyps) | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ |
Nervous system disorders | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ |
Respiratory, thoracic, and mediastinal disorders | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ |
Skin and SC tissue disorders | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ |
Vascular disorders | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ | ██ |
aAE terms in the extension study were coded using MedDRA version 20.1, otherwise were coded using MedDRA version 19.1 in the pivotal study; bCausal relationship to study drug: AEs related to study drug include AEs classified as Possible, Probably, or Definite. AEs not related to study drug include AEs that were None or Improbable; cTEAEs/SAEs most frequently occurring as per the pivotal study are captured in this table. Summary of Incidence of new TEAEs≥5% in overall subjects also included Eye Disorders (6.7%) and summary of incidence of SAEs included Surgical and Medical Procedures (3.3%) in the extension study
AE = adverse event; incl. = including; n/N = number; NA = not applicable; SAE = serious adverse event; SC = suibcutaneous; TEAE = treatment emergent adverse event
Source: FP01C-13-001 Clinical Study Report (Table 12-31),45 FP01C-13-001-EX Clinical Study Report (Tables 12-8, 12-9)46
Adverse Events
In FP01C-13-001, out of 137 total subjects, there were a total 553 TEAEs in 114 subjects (83.2%). The most common TEAEs (≥ 5%) in the total population (N = 137) were vascular disorder ███████, followed by infections and infestations ████████ musculoskeletal and connective tissue disorders ████████ general disorders and administration site conditions ████████ and renal and urinary disorders ████████ These were similar to the frequency of TEAEs experienced by subjects in Part I and Part II of the study.
Serious Adverse Events
A summary of SAEs reported in FP01C-13-001 is reported in Table 23. Overall, there were a total of ██ ████ ██ ██ ████████ ████████ The most frequently occurring SAEs were cardiac disorders ███████ injury, poisoning, and procedural complications ███████ neoplasms ███████ general disorders and administration site concerns ███████ and infections and infestations ███████ Out of ██ █████ ████ █ ████ determined to be related to LMIS 50 mg. Drug-related SAE reports included a participant with blurred vision, a participant with left hip fracture, and a participant with myocardial infarction.
There were █████ ██████ ████████ ██████ with cause of death being cerebrovascular accident, pulmonary embolism, prostate cancer, and acute lung injury (one subject presented with the latter 2 events). These were determined to be unrelated to LMIS 50 mg by the study investigator.
Withdrawals Due to Adverse Events
As demonstrated in Table 23, a total of 5 subjects (3.6%) withdrew from the study due to AEs: acute kidney injury, atrial fibrillation, cerebrovascular accident, death, hormone refractory prostate cancer, and metastatic prostate cancer. It was determined that these were not related to the administration of LMIS 50 mg by the investigator.
Brief Summary of Adverse Events in the Extension Study
In the extension study FPC01 to 13 to 001-EX, ██ ████████ ████ ██ ███ █████ had at least 1 TEAE, with most being mild or moderate. There were ██████ ███ ██ █ ████████ and ████████████████ ███ ██ █ ████████ which were determined to be unrelated to LMIS 50 mg. There ████ ██ ██████ in reported. There were ██ ███ that led to discontinuation. There were a total of ████ ██ █ ████████.
Bone and Urinary Pain
The safety of LMIS 50 mg was also assessed in FP01C-13-001 by determining change in bone pain and urinary pain from baseline using the VAS scale, with 0 indicating no pain and 10 indicating worst pain ever. Bone and urinary pain were assessed throughout the study.
Table 24 provides a summary of the change in bone and urinary pain from baseline at Day 336 (end of study).
Table 24: Change in Bone Pain From Baseline
Parameter | Part I N = 33 | Part II N = 104 | Total N = 137 |
|---|---|---|---|
Baseline | |||
n | ██ | ███ | ███ |
Mean (SD) | ████ █████ | ████ █████ | ████ ██████ |
Median (min, max) | ████ █████ | ████ █████ | ████ █████ █ |
Hodges−Lehmann estimator | ████ | ████ | ████ |
95% CI | █████ ████ | █████ ████ | █████ ████ |
Change on Day 336 | |||
n | ██ | ███ | ██████ |
Mean (SD) | ████ ███████ | ████ ██████ | ████ ██████ |
Median (min, max) | ████ ██████ | ████ ██████ | ████ ██████ |
Hodges−Lehmann estimator | ████ | ████ | ████ |
95% CI | ██████ ████ | ██████ ████ | ██████ ████ |
P-valuea | ██████ | ██████ | ██████ |
aPaired t-test or Wilcoxon signed-rank test used to test the change from baseline for continuous variables
CI = confidence interval; max = maximum; min = minimum; SD = standard deviation
Source: FP01C-13-001 Clinical Study Report (Table 12-55)45
By the end of the trial (Day 336), for the total population, bone pain was ████ ██████). Mean change measured by the paired t test or Wilcoxon signed-rank test from baseline to end of trial was not clinically significant.
For the total population, the mean (SD) urinary pain was ████ ███████ at baseline and ████ ███████ by Day 336. Mean change in urinary pain from baseline to end of trial did not demonstrate a statistically significant difference.
Table 25: Change in Urinary Pain From Baseline
Needs heading | Part I n = 33 | Part II n = 104 | Total N = 137 |
|---|---|---|---|
Baseline | |||
n | ██ | ███ | ███ |
Mean (SD) | ████ █████ | ████ █████ | ████ █████ |
Median (min, max) | ████ █████ | ████ █████ | ████ █████ |
Hodges−Lehmann estimator | ████ | ████ | ████ |
95% CI | ██████ ████ | █████ ████ | █████ ████ |
Change on Day 366 | |||
N | ██ | ███ | ██████ |
Mean (SD) | ████ ██████ | ████ ██████ | ████ ██████ |
Median (min, max) | ████ ██████ | ████ ██████ | ████ ██████ |
Hodges−Lehmann estimator | ████ | ████ | ████ |
95% CI | ██████ ████ | ██████ ████ | ██████ ████ |
p-valuea | ██████ | ██████ | ██████ |
aPaired t-test or Wilcoxon signed-rank test used to test the change from baseline for continuous variables
CI = confidence interval; max = maximum; min = minimum; SD = standard deviation
Source: FP01C-13-001 Clinical Study Report (Table 12-55)45
Vital Signs, Physical Examination, Hematologic and Biochemistry Parameters
██ █████████████ ███████████ ██████ was observed for vital signs. Physical examination was also normal in most subjects by Day 336, with ███ ███████ reported to have abnormal findings in the head, heart, and neurological exams. ███ ████████ had abnormal findings in the lungs, ████ ██ ███ chest and musculoskeletal exams, and ██ ████████ had abnormal findings in extremities in the pivotal study. There were ██ ██████████ ███████████ abnormalities noted for the overall interpretation of the 12-lead ECG assessment in the pivotal or extension study.
There were ██ ██████████ ███████████ ███████ observed in hematological parameters. Similarly, while █████████████ ███████████ ███████ ████ ████████ in biochemical parameters, they were ███ █████ ██ ██ ██████████ ███████████ and were possibly related to pre-existing conditions. This was also similar for the extension study.
Quality of Life (FP01C-13-001 Study Only)
Most subjects felt satisfied by Day 168 and Day 336 (scales 0, 1, and 2). At Day 168, █████ of Part I subjects and ███ of Part II subjects were satisfied. By Day 336, █████ of Part I subjects and ███ of Part II subjects were satisfied. By trial end, most subjects ███████ in the total population were satisfied.
Bioequivalence
Cross-Study PK/PD Comparison of LMIS 50 mg to Marketed Leuprolide Acetate Depot Products
A cross-study compared PK and PD after exposure to LMIS 50 mg in the FP01C-13-001 trial with literature data on different formulations of leuprolide acetate products. As part of the analysis, LMIS 50 mg was compared to Eligard® 45mg (6-month formulation).55
Eligard data were obtained from public sources by targeted desktop searches using the following search terms, singularly or in combination: Eligard 45 mg +PK/pharmacokinetics +profile +exposure +depot/depo + 6 month + 6M +review +approval +AUC +Cmax +leuprolide +leuprolide acetate. Only results containing PK data of leuprolide were retained. In order to derive exposure parameters of mean concentration–time profiles, applicable figures were digitized and area under the curve (AUC) and concentration measures were obtained via non-compartmental analysis. Where individual profiles were available, individual PK parameters were obtained first. Thereafter, bioequivalence was assessed.
The FDA Approval Packages were found to provide the most complete summary of publicly available Eligard 45 mg data as it contained tabulated mean concentration data over 336 days (n = 27 patients). A non-compartmental PK analysis of the mean concentration data presented was performed to obtain Cmax and AUC data. Concentrations at week 4 and month 6 (Cwk4 and Cmon6, respectively) were read directly from the profile. Finally, Cavg(0 to 6mo) and Cwk4:Cmon6 ratios were calculated.
The results of the analysis showed that leuprolide exposure (Cmax, AUC4wks, AUC6mon, and Cavg(0 to 6mo)) appeared similar between LMIS 50 mg and Eligard 45 mg (see Table 26 below). The time to castration level was very similar for both formulations (20.2 versus 19.1 days for LMIS and Eligard, respectively).
Table 26: Summary of Exposure Parameters in Humans After Administration of LMIS 50 mg and Eligard 45 mg
Product | Cmax (ng/mL) | AUC4wks (day*ng/mL) | AUC6mon (day*ng/mL) | Cavg(0-6mo) (ng/mL) | Cwk4 (ng/mL) | Cmon6 (ng/mL) | Cwk4:Cmon6 ratio |
|---|---|---|---|---|---|---|---|
Eligard 45 mg first dose | ████ | ████ | ███ | ████ | ████ | ████ | ████ |
Eligard 45 mg second dose | ████ | ████ | ███ | ████ | ████ | ████ | ████ |
LMIS 50 mg first dose | ████ | ████ | ███ | ████ | ████ | ████ | ████ |
LMIS 50 mg second dose | ████ | ████ | ███ | ████ | ████ | ████ | ████ |
AUC4wks = area under the concentration time curve at 4 weeks; AUC0-6mon = area under the concentration time curve from time 0 to 24 weeks; Cavg(0-6mon) = mean serum concentration within 24 weeks post dosing; Cmax = maximum concentration; Cwk4 = concentration at 4 weeks; LMIS = leuprolide mesylate injectable suspension
Source: Cross-Study (PK/PD) Comparison of LMIS 50 mg (6 month) and LMIS 25 mg to Marketed Leuprolide Acetate Depot Products56
Indirect Treatment Comparison
An indirect treatment comparison (ITC) was determined by the sponsor to be not feasible. According to the sponsor’s feasibility assessment, an indirect comparison is difficult to apply in this context, for multiple reasons. First, regardless of the selected ITC method, only unanchored approaches are feasible because FP01C-13-001, the only trial investigating the SC leuprolide mesylate injectable suspension 50 mg, is a single-arm trial. As mentioned previously, unanchored ITC methods rely on strong assumptions that are rarely met in reality, and existing guidelines warn that their use is problematic.57 Second, the ITC feasibility assessment concluded that only 2 of the considered studies reported the baseline concentration of testosterone. Because this is likely the most important confounding factor, only 2 studies can be compared to the FP01C-13-001 trial with adjustment for potential confounding factors, heavily limiting the number of comparators that can be included in the analysis. Moreover, the limited available data prevents any further statistical combination of the analysis results (such as a meta-analysis of the single matching-adjusted indirect comparison, which would be feasible if there were multiple estimates of comparative efficacy from different matching-adjusted indirect comparisons for each relevant comparator).
CDA-AMC Critical Appraisal of the Clinical Evidence
CDA-AMC conducted a critical appraisal of the clinical study for leuprolide mesylate, based on the summary of the evidence provided by the sponsor.
Internal Validity
The evidence on leuprolide mesylate included in the sponsor’s summary was based on 1 phase III, single-arm, open-label pivotal study (FP01C-13-001) and 1 phase III, single-arm, open-label, safety extension study (FP01C-13-001-EX). The absence of an internal comparison group in the FP01C-13-001 pivotal study is a key methodological limitation; with the single-arm design, efficacy can be interpreted only with low confidence, given the absence of comparative data from alternative treatment options. Consequently, it is a challenge to make inferences about the efficacy and safety of leuprolide mesylate. From a regulatory perspective, it was considered acceptable for the pivotal FP01C-13-001 study to adopt the single-arm design, per current FDA guidance for establishing the efficacy and safety of GnRH analogues for the treatment of advanced prostate cancer.22 Health Canada is in agreement with the FDA guidance that the single-arm study design was appropriate for the assessment of leuprolide mesylate.23
The design of the FP01C-13-001 study was, overall, well aligned with FDA guidance and met several major criteria.22 First, the FP01C-13-001 study determined that leuprolide mesylate could be used to attain castrate testosterone levels, defined as a testosterone level of 50 ng/dL or less, in patients with advanced prostate cancer. Although the FDA guidance suggests measuring plasma testosterone levels, the FP01C-13-001 study measured serum testosterone levels. Nonetheless, the clinical experts consulted by the CDA-AMC review team did not consider this difference to be a serious concern and noted that a serum testosterone level of 50 ng/dL or less is a widely accepted surrogate end point for efficacy. Second, the FP01C-13-001 study evaluated whether leuprolide mesylate could maintain castrate testosterone levels during a dosing period and whether leuprolide mesylate (a 6-month long-acting formulation) could maintain castrate testosterone levels immediately after a subsequent dosing period by extending the treatment period by 2 dosing intervals. Third, to address a potential drawback — mean testosterone levels do not reveal individual treatment failures — the FP01C-13-001 study adopted the percentage of patients who achieved and maintained castrate testosterone levels to demonstrate the efficacy of leuprolide mesylate.
In the FP01C-13-001 trial, the percentage of patients with a serum castrate testosterone level (≤ 50 ng/dL) was presented at day 28 with or without 1 day after first dose of leuprolide mesylate, at day 336 (i.e., the end of the second dose of leuprolide mesylate), and between day 28 and day 336 (analyzed using the Kaplan-Meier method). The selection of these time points was appropriate, according to the FDA guidance.22
The FP01C-13-001 study reported on QoL and subjective harm outcomes, such as pain on VAS. Despite favouring leuprolide mesylate, findings on these outcomes were prone to performance and detection bias because of the open-label study design, in which QoL and subjective harm outcomes had no blinded assessment. Furthermore, efficacy of leuprolide mesylate in the FP01C-13-001 study was assessed up to 336 days after treatment (less than 1 year), and as such, the longer-term efficacy of leuprolide mesylate is uncertain. Despite the uncertainty, the clinical experts consulted by the CDA-AMC review team noted that there are no serious concerns about missing longer-term results, based on their understanding of the efficacy of leuprolide.
The sponsor confirmed that information on the grade of prostate cancer at baseline was not available in the FP01C-13-001 study.21 According to the clinical experts consulted by the CDA-AMC review team, the PSA levels in the FP01C-13-001 study were not interpretable without information on the grade of prostate cancer.
In the FP01C-13-001 study, the sponsor conducted post hoc analyses on subgroups by disease stage (i.e., I or II, III, IV, or unknown) and by disease characteristics at enrolment (metastatic castration-sensitive prostate cancer, nonmetastatic castration-sensitive prostate cancer with locally advanced disease, or nonmetastatic castration-sensitive prostate cancer with high-risk localized disease). Both subgroup analyses involved a large proportion of patients in whom disease stage was unknown. Specifically, 24.1% of the ITT population (33 of 137) had an unknown disease stage, and 59 of the 137 patients could not be classified by disease characteristics at enrolment. Given the post hoc nature of these subgroup analyses, the CDA-AMC review team considered the findings exploratory and was unable to draw any conclusions about response to treatment by subgroup.
The FP01C-13-001-EX extension study provided 2 additional doses of leuprolide mesylate, approximately 6 months apart, to patients from the pivotal FP01C-13-001 study and assessed the safety of leuprolide mesylate for up to 2 years (i.e., 1 year in the FP01C-13-001 study plus 1 year in the extension study). Although all ██ ████████ received the first dose of leuprolide mesylate, it was noted that approximately ███ ███ ███ ██ ███ of the patients in the FP01C-13-001-EX extension study did not receive the second dose, including ████████ discontinued early for reasons unrelated to AEs and ████████ because of drug supply expiration. The proportion of patients missing the second dose of treatment might result in a potential risk of an underestimation of the incidence of harms, although the magnitude of the impact remains unknown.
A gap remains in the sponsor-submitted evidence because of the absence of direct or indirect comparative evidence (FP01C-13-001 was a single-arm study), which limited the ability of the CDA-AMC review team to draw any evidence-based conclusions about the efficacy of leuprolide mesylate relative to other ADTs (including leuprolide products) currently available in Canada.
External Validity
The type of disease investigated in the pivotal FP01C-13-001 study was carcinoma of the prostate, whereas the Health Canada–approved indication for leuprolide mesylate is for prostate cancer. The clinical experts consulted by the CDA-AMC review team noted that carcinoma of the prostate and prostate cancer in general can be used interchangeably.
The pivotal FP01C-13-001 study does not completely align with the population in the Health Canada–approved indication in terms of advanced prostate cancer. The Health Canada–approved indication targets patients with advanced prostate cancer, whereas the pivotal FP01C-13-001 study did not limit enrolment to patients with advanced prostate cancer; it also included patients with stage I to stage IV disease. Of note, according to the clinical experts consulted by the CDA-AMC review team, there is no universal definition of advanced prostate cancer; the phrase ”patients with advanced prostate cancer” used in the Health Canada–approved indication is subject to interpretation. The clinical experts consulted by the CDA-AMC review team noted that advanced prostate cancer commonly refers to biochemically recurrent or metastatic disease, yet patients with intermediate-risk or high-risk localized prostate cancer can also be eligible for ADT. The sponsor, in its response to a CDA-AMC information request,21 stated that: “In clinical practice, the standard definition [of advanced prostate cancer] is disease that has progressed beyond localized treatment options, including locally advanced disease (e.g., T3 or T4 classification) that is not amenable to curative treatment, metastatic disease, and castration-resistant prostate cancer.” Treatment recommendations and guidelines for prostate cancer recognize that advanced prostate cancer encompasses both localized disease with high-risk features and metastatic disease, and that all patients with advanced prostate cancer typically require ADT.8,24,25 Nonetheless, both clinical experts consulted by the CDA-AMC review team agreed that the results generated from the FP01C-13-001 study population would still be generalizable to the Health Canada–indicated population.
The clinical experts consulted by the CDA-AMC review team noted that the pivotal FP01C-13-001 study excluded patients who had an ECOG PS of greater than 2; those who received combination therapy with chemotherapy, immunotherapy, cryotherapy, radiotherapy, concomitant ADT, or LHRH therapy during study; and those who had a baseline morning serum testosterone level of 150 ng/dL or less, whereas the Health Canada indication did not restrict the use of leuprolide mesylate in these patients. According to the clinical experts consulted by the CDA-AMC review team, the patients who were excluded from the FP01C-13-001 study would benefit from leuprolide mesylate treatment and account for a large proportion of patients with advanced prostate cancer. Nonetheless, the clinical experts said that the results from the pivotal FP01C-13-001 study would still be generalizable to these patients.
The composition of race in the FP01C-13-001 study population was 89.8% white, 5.8% Black or African American, 3.6% Asian, and 0.7% unknown. There was an overrepresentation of patients who were white compared to the patient population in Canada, per clinical expert input. However, the clinical experts consulted by the CDA-AMC review team did not note any serious concerns about the generalizability of findings from the pivotal FP01C-13-001 study to the population in Canada.
Economic Review
Sponsor-Submitted Cost Comparison
Leuprolide mesylate is a new salt formulation of leuprolide.58 The sponsor submitted a cost comparison for leuprolide mesylate compared with leuprolide acetate products (i.e., Eligard and Lupron Depot), relugolix, buserelin acetate, degarelix, goserelin acetate, and triptorelin for the treatment of adult patients with advanced prostate cancer.59 The cost comparison included a comparison of drug and health care resource-use costs.
Drug costs were calculated based on the sponsor’s submitted price for leuprolide mesylate, the price listed on the Ontario Drug Benefit Formulary for comparators, and recommended dosages from the respective product monographs (Table 27).60,61
Health care costs consisted of administration costs (calculated as a nursing wage multiplied by a treatment-specific duration of administration) and the cost of nonsteroidal antiandrogen therapy.58 Differences in administration time were related to differences in modes of administration (i.e., intramuscular, SC, or oral) and differences in reconstitution times for treatments requiring injections (Table 31 and Table 32 in Appendix 2).58,62 Administration times were sourced from literature or assumed (Table 31 in Appendix 2).62,63 Nursing wage was sourced from Statistics Canada (Table 30 in Appendix 2).64 The costs of nonsteroidal antiandrogen therapy (i.e., bicalutamide) were included for GnRH agonists to prevent testosterone flares (Table 31 and Table 32 in Appendix 2).58,65-67
The sponsor’s analysis reported an annual drug cost for leuprolide mesylate of $2,998 per year, based on a unit price of $1,499.00 per 42 mg extended-release emulsion for injection (Table 27).59,60 Annual drug costs for comparators ranged from $3,287 to $5,575, based on list prices from the Ontario Drug Benefit Formulary (Table 27).61,68 Incremental cost savings associated with leuprolide mesylate ranged from $289 to $2,557 annually (Table 27, Table 28).
The sponsor reported an annual health care cost of $31.76 for leuprolide mesylate in the first year (Table 33 in Appendix 2), and $3.57 in subsequent years (Table 34 in Appendix 2).58 Annual health care costs for comparators ranged from $0.00 to $103.11 in the first year, and from $0.00 to $74.91 in subsequent years (Table 33 and Table 34 in Appendix 2).58 Therefore, the sponsor estimated annual health care cost savings of $71.35 and an incremental cost of $31.76 annually in the first year, and a cost saving of $71.34 and an incremental cost of $3.57 in subsequent years, per patient (Table 28).58 Cost-savings estimates took into account the higher frequency and longer duration of administration of triptorelin compared to leuprolide mesylate, and incremental cost estimates took into account the oral mode of administration and the lack of nonsteroidal antiandrogen therapy for relugolix compared to leuprolide mesylate.
Table 27: Sponsor’s Drug-Acquisition Cost Comparison
Generic name (brand name) | Strength | Dosage form | Pricea ($) | Recommended dosage regimenc | Annual drug cost ($) | Difference in annual cost ($) |
|---|---|---|---|---|---|---|
Leuprolide mesylate (CAMCEVI) 50 mg | 42 mg | SC | 1499.00 | 42 mg 2x | 2998.00 | — |
Comparators | ||||||
Leuprolide acetate (Eligard) 7.5 mg | 7.5 mg | SC | 310.72 | 7.5 mg 12x | 3,728.64 | 730.64 |
Leuprolide acetate (Eligard) 22.5 mg | 22.5 mg | SC | 891.00 | 22.5 mg 4x | 3,564.00 | 566.00 |
Leuprolide acetate (Eligard) 30 mg | 30 mg | SC | 1,285.20 | 30 mg 3x | 3,855.60 | 857.60 |
Leuprolide acetate (Eligard) 45 mg | 45 mg | SC | 1,659.90 | 45 mg 2x | 3,319.80 | 321.80 |
Leuprolide acetate (Lupron Depot) 7.5 mg | 7.5 mg | IM | 387.97 | 7.5 mg 12x | 4,655.64 | 1,657.64 |
Leuprolide acetate (Lupron Depot) 22.5 mg | 22.5 mg | IM | 1,071.00 | 22.5 mg 4x | 4,284.00 | 1,286.00 |
Leuprolide acetate (Lupron Depot) 30 mg | 30 mg | IM | 1,428.00 | 30 mg 3x | 4,284.00 | 1,286.00 |
Relugolix (Orgovyx) 360/120mg, induction + maintenance – first year of treatment | 120 mg | Oral | 9.00b | Induction: 120 mg x3 on day 1 Maintenance: 120 mg x 364.25 days | 3,305.25 | 307.25 |
Relugolix (Orgovyx) 120 mg, maintenance – subsequent years of treatment | 120 mg | Oral | 9.00b | Maintenance: 120 mg x 365.25 days/year | 3,287.25 | 289.25 |
Buserelin acetate depot (Suprefact) 6.3 mg | 6.3 mg | SC | 929.23 | 6.3 mg 6x | 5,575.41 | 2,577.41 |
Buserelin acetate depot (Suprefact) 9.45 mg | 9.45 mg | SC | 1,376.99 | 9.45 mg 4x | 5,507.98 | 2,509.98 |
Degarelix (Firmagon) 120/80 mg, induction + maintenance – first year of treatment | 120/ 80 mg | SC | 370.94 (120 mg)/ 274.18 (80 mg) | Induction: 120 mg 2x on day 1 Maintenance: 80 mg once per month (11x) | 3,757.82 | 759.82 |
Degarelix (Firmagon) 80 mg, maintenance – subsequent years of treatment | 80 mg | SC | 274.18 | Maintenance: 80 mg once per month (12x year) | 3,290.11 | 292.11 |
Goserelin acetate (Zoladex) 10.8 mg | 10.8 mg | SC | 1,204.73 | 10.8 mg 4x | 4,818.93 | 1,820.93 |
Triptorelin (Trelstar) 3.75 mg | 3.75 mg | IM | 346.31 | 3.75 mg 12x | 4,155.72 | 1,157.72 |
Triptorelin (Trelstar) 11.25 mg | 11.25 mg | IM | 1,038.97 | 11.25 mg 4x | 4,155.88 | 1,157.88 |
Triptorelin (Trelstar) 22.5 mg | 22.5 mg | IM | 1,659.90 | 22.5 mg 2x | 3,319.80 | 321.80 |
IM = intramuscular; SC = subcutaneous.
aOntario Drug Benefit Formulary.61
bCDA-AMC recommendation for relugolix.69
cProduct monographs.61,68,70-74
Table 28: Sponsor’s Summary of Comparative Treatment Costs
Generic name (brand name) | Difference in drug-acquisition costs per year ($) | Difference in total health care costs per year ($) | Difference in total costs per year ($) |
|---|---|---|---|
Leuprolide mesylate (Camcevi) 50 mg | Reference | Reference | Reference |
Comparators | |||
Leuprolide acetate (Eligard) 7.5 mg | 730.64 | 48.66 | 779.30 |
Leuprolide acetate (Eligard) 22.5 mg | 566.00 | 13.84 | 579.84 |
Leuprolide acetate (Eligard) 30 mg | 857.60 | 9.49 | 867.09 |
Leuprolide acetate (Eligard) 45 mg | 321.80 | 5.14 | 326.94 |
Leuprolide acetate (Lupron Depot) 7.5 mg | 1,657.64 | 17.84 | 1,675.48 |
Leuprolide acetate (Lupron Depot) 22.5 mg | 1,286.00 | 3.57 | 1,289.57 |
Leuprolide acetate (Lupron Depot) 30 mg | 1,286.00 | 1.78 | 1,287.78 |
Relugolix (Orgovyx) 360 mg/120 mg for the first year | 307.25 | −31.76 | 275.49 |
Relugolix (Orgovyx) 120 mg for subsequent years | 289.25 | −3.57 | 285.68 |
Buserelin acetate depot (Suprefact Depot) 6.3 mg | 2,577.41 | 7.13 | 2,584.54 |
Buserelin acetate depot (Suprefact Depot) 9.45 mg | 2,509.98 | 3.57 | 2,513.54 |
Degarelix (Firmagon) 120 mg + 80 mg, induction + maintenance for the first year | 759.82 | 49.39 | 809.22 |
Degarelix (Firmagon) 80 mg, maintenance for subsequent years | 292.11 | 71.35 | 363.46 |
Goserelin acetate (Zoladex) 10.8 mg | 1,820.93 | 3.57 | 1,824.50 |
Triptorelin (Trelstar) 3.75 mg | 1,157.72 | 71.35 | 1,229.07 |
Triptorelin (Trelstar) 11.25 mg | 1,157.88 | 21.40 | 1,179.28 |
Triptorelin (Trelstar) 22.5 mg | 321.80 | 8.92 | 330.72 |
Critical Appraisal of Cost Information
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the cost comparison:
Assumption of clinical similarity is uncertain. The sponsor’s cost comparison assumes that the clinical efficacy of leuprolide mesylate is similar to that of other ADTs. The clinical trial that investigated the efficacy and safety of leuprolide mesylate was a single-arm trial.45,46 In the absence of direct and indirect comparative evidence, the CDA-AMC Clinical Review was unable to draw any conclusions about the efficacy and safety of leuprolide mesylate relative to other ADT products (including leuprolide products) currently available in Canada. However, the Clinical Review noted that Health Canada and the clinical experts consulted by CDA-AMC for this review considered the efficacy and safety of leuprolide mesylate observed in the sponsor-submitted evidence to be consistent with the efficacy and safety of leuprolide products currently used in Canada, and did not express any major concerns about the lack of direct or indirect comparative evidence in their assessments of the efficacy and safety of leuprolide mesylate relative to other leuprolide products.
CDA-AMC was unable to address this limitation in its reanalysis. However, according to the CDA-AMC Clinical Review, Health Canada and the clinical experts consulted by CDA-AMC for this review did not express any major concerns about the lack of direct or indirect comparative evidence in their assessments of the relative efficacy and safety of leuprolide mesylate. If the efficacy of leuprolide mesylate is expected to be similar to existing ADTs, a cost comparison may be appropriate.
Administration costs are uncertain and are potentially overestimated. Health care costs associated with each treatment were based on administration costs and the cost of nonsteroidal antiandrogen therapy in the first year.58 The administration cost of each treatment was calculated using the time required for administration associated with each treatment, multiplied by nursing wages.58,64 The sponsor cited a survey conducted by Goldfischer et al.62 to derive the estimated time required to administer both leuprolide acetate and its comparators. Clinical expert feedback elicited by CDA-AMC indicated that some patients receiving ADTs are enrolled in patient support programs, in which the manufacturer covers the administration costs associated with ADT treatment.75,76 Additionally, clinical expert feedback received by CDA-AMC indicated that the cited time differences associated with administration may not result in clinically meaningful differences in administration times, and it is uncertain whether these differences will translate to cost savings to the health care system.
CDA-AMC was unable to address this limitation in reanalyses. Differences in health care resource use associated with treatment administration are considered highly uncertain.
Confidential pricing agreements. Although the submitted price for leuprolide mesylate results in treatment-acquisition costs that are similar to or lower than costs for other reimbursed ADTs, based on publicly available list prices, list prices may be higher than the price paid by jurisdictional drug plans because of confidential pricing agreements. Therefore, the submitted price of leuprolide mesylate may require a price reduction to avoid incurring additional costs relative to its comparators.
CDA-AMC was unable to address this limitation in its reanalyses, as the negotiated prices of the comparators are unknown.
Comparator dosage form missing from the sponsor’s analysis: The 3.6 mg goserelin acetate dosage form was not included in the sponsor’s cost comparison table.77
CDA-AMC reanalyses included the 3.6 mg goserelin acetate product.
CDA-AMC Reanalyses
CDA-AMC conducted a reanalysis to present drug-acquisition costs on daily and 28-day cycles and dosage forms that aligned with the dosage form in comparator’s respective product monographs (Table 29). CDA-AMC reanalyses also included the dosing regimen for goserelin acetate of 3.6 mg every 4 weeks. In CDA-AMC reanalyses, leuprolide mesylate remained less costly than other ADTs, based on public list prices. CDA-AMC was unable to address several key limitations of the sponsor’s submission, including the inability to draw a conclusion regarding the efficacy of leuprolide mesylate relative to other ADTs currently available in Canada, the uncertainty of health care resource use, and the uncertainty in comparator drug prices because of confidential pricing agreements. Based on the publicly available list prices of all comparators and the sponsor’s submitted price of leuprolide mesylate, reimbursement of leuprolide mesylate is expected to be associated with cost savings in terms of drug costs.
Table 29: CDA-AMC Cost Comparison
Drug | Strength | Dosage form | Price ($) | Recommended dosea | Average annual drug cost ($) | Daily cost ($) | Cost per 28 days ($) |
|---|---|---|---|---|---|---|---|
Leuprolide mesylate (Camcevi) | 42 mg | Extended-release emulsion for injection | 1,499.0000b | 42 mg every 6 months | 2,998 | 8.21 | 230 |
GnRH agonist | |||||||
Buserelin acetate (Suprefact Depot) | 6.3 mg 9.45 mg | Implant | 929.2348 1,376.9938 | 6.3 mg every 2 months 9.45 mg every 3 months | 5,508 to 5,575 | 15.08 to 15.26 | 422 to 427 |
Goserelin acetate (Zoladex, Zoladex LA) | 3.6 mg 10.8 mg | Depot for SC injection | 422.6778 1,204.7322 | 3.6 mg every 4 weeks 10.8 mg every 3 months | 4,819 to 5,072 | 13.19 to 13.89 | 369 to 389 |
Leuprolide acetate (Eligard) | 7.5 mg 22.5 mg 30 mg 45 mg | Prefilled, dual-chambered syringe with powder for reconstitution | 310.7200 891.0000 1,285.2000 1,659.9000 | 7.5 mg every month 22.5 mg every 3 months 30 mg every 4 months 45 mg every 6 months | 3,320 to 3,856 | 9.09 to 10.56 | 254 to 296 |
Leuprolide acetate (Lupron Depot) | 7.5 mg 22.5 mg 30 mg | Prefilled, dual-chambered syringe with powder for reconstitution | 387.9700 1,071.0000 1,428.0000 | 7.5 mg every month 22.5 mg every 3 months 30 mg every 4 months | 4,284 to 4,656 | 11.73 to 12.75 | 328 to 357 |
Triptorelin (Trelstar, Trelstar LA) | 3.75 mg 11.25 mg 22.5 mg | Powder for injectable suspension | 346.3100 1,038.9700 1,659.9000 | 3.75 mg every month 11.25 mg every 3 months 22.5 mg every 6 months | 3,320 to 4,156 | 9.09 to 11.38 | 254 to 319 |
GnRH antagonist | |||||||
Degarelix (Firmagon) | 80 mg 120 mg | Powder for injection | 274.1760 370.9440 | Initiation: 240 mg on day 1 Maintenance: 80 mg every month | Year 1: 3,758 Year 2+: 3,290 | First month: 24.39 Year 2+: 9.01 | First cycle: 683 Year 2+: 252 |
Relugolix (Orgovyx) | 120 mg | Tablet | 9.0000c | Initiation: 360 mg on day 1 Maintenance: 120 mg daily | Year 1: 3,305 Year 2+: 3,287 | First cycle: 9.64 Year 2+: 9.00 | First cycle: 270 Year 2+: 252 |
CDA-AMC = Canada's Drug Agency; GnRH = gonadotropin-releasing hormone; SC = subcutaneous.
Note: All prices are from the Ontario Drug Benefit Formulary (accessed March 2025)61 unless otherwise indicated and do not include dispensing fees. A year was assumed to last 365.25 days, and a month was assumed to be 365/12 (30.42) days.
aAll dose regimens are from the respective product monographs.63,65,66,78-86
bSponsor’s submitted price of leuprolide mesylate.58,60
cRelugolix price is based on the CDA-AMC relugolix reimbursement review.68
Price Reduction Analyses
At the submitted price for leuprolide mesylate and public list prices for comparators, leuprolide mesylate is less costly than other reimbursed ADTs. Therefore, at public list prices, no price reduction is required for leuprolide mesylate to result in cost savings compared with other ADTs. Given the sponsor’s claim of clinical similarity between leuprolide mesylate and other ADTs and given the lack of direct or indirect comparative efficacy data for leuprolide mesylate compared to other ADTs, there is insufficient evidence to support a price premium for leuprolide mesylate over currently reimbursed ADTs.
Issues for Consideration
Relugolix received a recommendation to reimburse with clinical criteria and/or conditions from CDAAMC68 and is currently undergoing negotiations with the pan-Canadian Pharmaceutical Alliance.87 As such, it is uncertain whether it will be listed on public drug plan formularies.
Analyses by the sponsor and CDA-AMC are based on publicly available list prices for all comparators. Actual costs paid by public drug plans are unknown.
The sponsor-submitted cost comparison used treatment acquisition costs from the Ontario Drug Benefit Formulary for comparator treatments, with the exception of relugolix.58 As the price of each comparator may be different across the public drug plans, the sponsor-submitted cost comparison may not be reflective of the costs incurred by every public drug plan.
Coverage of ADTs varies across drug plans, and not all drug plans may cover all ADTs considered in the sponsor’s analysis.
Discussion
Summary of Available Evidence
A phase III, multicentre, single-arm, open-label pivotal study of 137 patients (FP01C-13-001) was submitted by the sponsor and assessed by the CDA-AMC review team. The pivotal FP01C-13-001 study evaluated the efficacy, safety, and pharmacokinetic behaviour of leuprolide mesylate in adult patients with histologically confirmed prostate carcinoma and a baseline serum testosterone level of greater than 150 ng/dL. Patients enrolled in the FP01C-13-001 study were from 26 sites in 8 countries (no sites in Canada). Patients were scheduled to receive a total of 2 doses of leuprolide mesylate, with a 6-month interval. The primary objectives of the FP01C-13-001 study were to establish the efficacy of leuprolide mesylate for up to 1 year, measured by the percentage of patients with a serum testosterone concentration suppressed to castrate levels (≤ 50 ng/dL) by day 28 with or without 1 day after the first injection of leuprolide mesylate and the percentage of patients with serum testosterone suppression (≤ 50 ng/dL) from day 28 through day 336 (the remaining duration of the study); to determine the safety and tolerability of leuprolide mesylate after up to 1 year of exposure; and to evaluate the pharmacokinetic behaviour of serum leuprolide. The median age of patients in the FP01C-13-001 study was 71.0 years (range, 52 to 88 years). The race composition of the study population was 89.8% white, 5.8% Black or African American, 3.6% Asian, and 0.7% unknown. Approximately 2.9%, 22.6%, 27.0%, and 23.4% of patients, respectively, had stage I, stage II, stage III, or stage IV prostate cancer; the disease stage in the remaining 24.1% of the study population was unknown.
A phase III, single-arm, open-label, safety extension study (FP01C-13-001-EX) was also submitted by the sponsor. The FP01C-13-001-EX extension study, conducted in 7 sites in the US, enrolled 30 patients who participated in the FP01C-13-001 pivotal study and assessed the safety and tolerability of leuprolide mesylate in these patients for up to 1 year.
Interpretation of Results
Efficacy
In the pivotal FP01C-13-001 study, the proportion of patients in the ITT population who achieved the castrate testosterone level (≤ 50 ng/dL) was 98.5% (95% CI, 94.8% to 99.8%) by day 28 after the first dose of leuprolide mesylate. Patients treated with leuprolide mesylate also maintained the castrate testosterone level (≤ 50 ng/dL) between day 28 and day 336: The proportion of patients in the ITT population who achieved the castrate testosterone level (≤ 50 ng/dL) was 97.0% (95% CI, 92.2% to 98.9%) by day 336. These proportions were clinically meaningful, according to the clinical experts consulted by the CDA-AMC review team. Furthermore, the lower bounds of the 95% CIs for the proportions of patients in the ITT population who achieved the castrate testosterone level (≤ 50 ng/dL) were greater than 90%, which meets the requirement to demonstrate the efficacy of GnRH analogues proposed in the FDA guidance.22 The ability of leuprolide mesylate to inhibit the testosterone level in patients with prostate cancer was further supported by the low incidence of testosterone excursions of more than 50 ng/dL during the study, including 2 patients who did not achieve castrate testosterone levels (≤ 50 ng/dL) by day 28 after the first dose of leuprolide mesylate, as well as 2 patients who had testosterone excursions of more than 50 ng/dL after having achieved castrate testosterone levels (≤ 50 ng/dL) on day 28.
In addition to the castrate testosterone level (≤ 50 ng/dL), the percentage of patients who achieved a testosterone level of less than 20 ng/dL on day 28 and day 336 was examined to determine the efficacy of leuprolide mesylate. Approximately 69.3% (95 of 137) of patients the ITT patient population reached testosterone suppression of less than 20 ng/dL by day 28, and the suppressed testosterone level (< 20 ng/dL) was maintained in approximately 95.9% (117 of 122) of the ITT population out to day 336. According to the clinical experts consulted by the CDA-AMC review team, the assessment of patients who attained a castrate testosterone level of less than 20 ng/dL could be used to conduct an in-depth examination of the ability of leuprolide mesylate to reduce testosterone levels, as existing evidence suggests that suppression of the serum testosterone level to less than 20 ng/dL might be associated with better biochemical relapse-free survival.31
Of note, survival outcomes such as OS and PFS were not reported in the pivotal FP01C-13-001 study, which was not a concern, according to the clinical experts consulted by the CDA-AMC review team. The clinical experts noted that survival outcomes are not used to assess GnRH analogues when prescribed as monotherapy, as this class of drug is known to be noncurative for prostate cancer when used in isolation.
QoL was assessed in the FP01C-13-001 study on a scale of 0 (being delighted) to 6 (being terrible). The proportion of patients feeling most satisfied or better, including those who reported being delighted, being pleased, and being mostly satisfied (scores of 0, 1, and 2, respectively), did not show drastic changes at several time points; rates were 68.6% on day 0, 69.7% on day 168, and 65.9% on day 336. However, the CDA-AMC review team determined that these findings were not adequate to draw any conclusions on the impact of leuprolide mesylate on QoL, mainly because performance and detection bias could not be ruled out because of the single-arm, open-label study design.
The single-arm design of the pivotal FP01C-13-001 study and the lack of ITC evidence prevented the CDAAMC review team from making comparisons between leuprolide mesylate and other leuprolide products or other ADTs available in Canada, based on the sponsor-submitted evidence. Health Canada commented that: “Although this is a limitation of the study, in light of the fact that ADME (absorption, distribution, metabolism, and excretion] data from Eligard PM [product monograph]) is being utilized in the current PM for CAMCEVI, a comparator in a controlled setting would help further confirm the adoption of the ADME data into the PM. Nevertheless, such a trial would need more patients and delay the authorization of this leuprolide-based product; a product with which the clinicians have over 30 years of experience with and one that has the potential to reduce medication errors. Moreover, testosterone level suppression is an accepted surrogate for clinical efficacy of leuprolide based products and based on the new GnRH guidance released by the FDA a comparator trial is not required.”23 According to the clinical experts consulted by the CDAAMC review team, the efficacy of leuprolide mesylate observed in the pivotal FP01C-13-001 study is similar to the efficacy of leuprolide products currently used in Canada (e.g., Eligard or Lupron Depot).
Harms
In the pivotal FP01C-13-001 study, the median duration of follow-up was ███ ████ ███████ ██ ██ ████ and approximately █████ ████ ██ ████ of the study population received both doses of leuprolide mesylate. The most common TEAE in the safety analysis set of the FP01C-13-001 study (N = 137) was hot flush (48.9%), followed by hypertension (14.6%), pain in extremity (9.5%), injection-site pain (7.3%), arthralgia (6.6%), fatigue (6.6%), nocturia (5.8%), back pain (5.1%), and nasopharyngitis (5.1%). SAEs occurred in █████ of the safety analysis set, with injury, poisoning, and procedural complications ██████ being the most frequent SAEs. There were ██████ deaths: ███ ███ to stroke, ███ ██ to metastatic prostate cancer and acute renal failure, and ███ to unknown reasons. Five patients (3.6%) discontinued the study because of TEAEs, including acute kidney injury, atrial fibrillation, cerebrovascular accident, death, hormone-refractory prostate cancer, and metastatic prostate cancer. Treatment with leuprolide products can cause the stimulation of testosterone production, especially during the initial weeks of treatment, which in turn may result in symptom flare, such as bone pain and exacerbation of obstructive symptoms.88 In the pivotal FP01C-13-001 study, the tolerability of leuprolide mesylate was also examined by assessing changes in bone pain and urinary pain (on a VAS scale, with 0 indicating no pain and 10 indicating the worst pain). The results suggest that the administration of leuprolide mesylate did not cause additional bone or urinary pain in patients, as there was no statistically significant change in VAS bone pain or VAS urinary pain scores from day 0 to day 168 or to day 336.
In the FP01C-13-001-EX extension study (N = 30), the median duration of follow-up ███ ███ ████ ███████ ██ ██ █████ All 30 patients received the first dose of leuprolide mesylate, whereas only ██ ████████ ███████ received the second dose. The most common TEAEs that occurred during the extension period were acute kidney injury ████████ increased blood triglycerides ████████ dehydration ████████ dizziness ████████ fall ████████ fatigue ████████ and hypertension ████████ ████ ████████ ████████ reported SAEs, and ██ discontinuation due to AEs or deaths were reported during the extension period.
Overall, the clinical experts consulted by the CDA-AMC review team did not have serious concerns about the harms outcomes, they noted that the safety profile of leuprolide mesylate is consistent with the profiles reported for the currently available leuprolide products, and they considered leuprolide mesylate to be safe and tolerable.
Cost
At the submitted price of $1,499.00 per 42 mg extended-release emulsion for injection, the annual drug-acquisition cost of leuprolide mesylate is estimated to be $2,998 per patient. Based on publicly available list prices, the annual cost of leuprolide mesylate is expected to be lower than the associated annual cost of all ADTs (i.e., buserelin acetate, degarelix, goserelin acetate, leuprolide acetate [Eligard], leuprolide acetate [Lupron depot], relugolix, and triptorelin). Compared with the 6-month ADT formulations (i.e., leuprolide acetate [Eligard] and triptorelin), leuprolide mesylate is estimated to be associated with cost savings of $322 annually when compared to 45 mg of leuprolide acetate (Eligard) and 22.5 mg of triptorelin. Incremental savings associated with leuprolide mesylate are based on publicly available list prices and may not reflect actual prices paid by drug plans.
The sponsor estimated health care resource-use costs based on treatment-specific administration times and the cost of nonsteroidal antiandrogen therapy for GnRH agonists during the first year. Differences in administration times among ADTs were deemed to be uncertain, as clinical expert feedback received by CDA-AMC indicated that patients receiving ADT treatments are often enrolled in patient support programs, which cover administration costs, and the time differences associated with administration may not result in clinically meaningful differences in administration times; therefore, its uncertain whether these differences will translate to cost savings to the health care system.
The cost comparison assumes clinical similarity between leuprolide mesylate, and the other ADTs included in the analysis. The CDA-AMC Clinical Review was unable to draw a conclusion on the relative efficacy and safety of leuprolide mesylate and other ADTs currently available in Canada in the absence of direct and indirect comparative evidence. Therefore, given the lack of evidence regarding clinical similarity, the appropriateness of conducting a cost comparison is unknown. As well, any conclusions regarding incremental savings associated with the reimbursement of leuprolide mesylate are uncertain.
Conclusion
Leuprolide mesylate is a new salt formulation of leuprolide that has been in use broadly for more than 30 years for the treatment of prostate cancer. A phase III, single-arm, open-label pivotal study submitted by the sponsor (FP01C-13-001) assessed the efficacy and safety of leuprolide mesylate in 137 adult patients with histologically confirmed prostate carcinoma and a baseline serum testosterone level of greater than 150 ng/dL. The absence of a comparator group in the pivotal FP01C-13-001 study is a key limitation that, from the methodological perspective, leads to low certainty about the evidence of leuprolide’s efficacy in treating prostate cancer. However, the single-arm design of the FP01C-13-001 study is acceptable from the regulatory perspective, as it met the requirements of current guidance for establishing the efficacy and safety of GnRH analogues for advanced prostate cancer.
The results of the pivotal FP01C-13-001 study showed that leuprolide mesylate effectively suppressed serum testosterone to the castrate level (≤ 50 ng/dL) in 98.5% (95% CI, 94.8% to 99.8%) of the ITT population by day 28 after the first dose of leuprolide mesylate and helped maintain the castrate testosterone level in 97.0% (95% CI, 92.2% to 98.9%) of the ITT population by day 336. The ability of leuprolide mesylate to suppress testosterone levels was further supported by findings that showed a low incidence of testosterone excursions ██ █████████ of more than 50 ng/dL during the study, as well as the high proportion of the ITT population who achieved (69.3%) and maintained (95.9%) testosterone suppression at 20 ng/dL or less. Based on the harms data from the pivotal FP01C13001 study and from the phase III, single-arm FP01C-13-001-EX extension study, which enrolled 30 patients who had participated in the FP01C-13-001 pivotal study, the clinical experts consulted by the CDA-AMC review team considered leuprolide mesylate to be, overall, safe and tolerable.
The CDA-AMC review team was unable to draw a conclusion about the relative efficacy and safety of leuprolide mesylate relative to other ADTs (including leuprolide products) currently available in Canada in the absence of direct and indirect comparative evidence, although Health Canada and the clinical experts consulted by the CDA-AMC review team did not express any major concerns about the lack of such evidence in their assessment of the relative efficacy and safety of leuprolide mesylate.
At the sponsor’s submitted price, drug-acquisition costs for leuprolide mesylate are estimated to be $2,998 annually, which, at public list prices, is less costly than the annual costs associated with other available ADTs. The sponsor’s cost comparison assumes clinical similarity between leuprolide mesylate and other ADTs; however, no direct or indirect evidence was submitted to support this assumption. According to the CDA-AMC Clinical Review, Health Canada and the clinical experts consulted by the CDA-AMC review team did not express any major concerns about the lack of direct or indirect evidence in their assessment of the relative efficacy and safety of leuprolide mesylate. Incremental savings are based on publicly available list prices and may not reflect actual prices paid by public drug plans in Canada. Overall, given the lack of direct or indirect comparative efficacy data for leuprolide mesylate versus other ADTs, there is insufficient evidence for leuprolide mesylate to have a price premium over currently reimbursed ADTs.
References
1.Accord Healthcare Inc. Drug Reimbursement Review sponsor submission: Leuprolide injectable emulsion (Camcevi), Extended release emulsion, 42 mg leuprolide (as leuprolide mesylate), Subcutaneous injection [internal sponsor's package]. 2024.
2.Accord Healthcare Inc. Leuprolide injectable emulsion (Camcevi), Extended release emulsion, 42 mg leuprolide (as leuprolide mesylate), Subcutaneous injection [product monograph]. 2021.
3.Brenner DR, Gillis J, Demers AA, et al. Projected estimates of cancer in Canada in 2024. CMAJ. 2024;196(18):E615-E623. doi:10.1503/cmaj.240095 PubMed
4.Siegel DA, O'Neil ME, Richards TB, Dowling NF, Weir HK. Prostate Cancer Incidence and Survival, by Stage and Race/Ethnicity - United States, 2001-2017. MMWR Morb Mortal Wkly Rep. 2020;69(41):1473-1480. doi:10.15585/mmwr.mm6941a1 PubMed
5.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7-34. doi:10.3322/caac.21551 PubMed
6.Leslie SW, Soon-Sutton TL, Skelton WP. Prostate Cancer. StatPearls. 2024.
7.Canadian Cancer Society. Prostate cancer. Accessed December 11, 2024, https://cancer.ca/en/cancer-information/cancer-types/prostate
8.Schaeffer EM, Srinivas S, Adra N, et al. Prostate Cancer, Version 4.2023, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2023;21(10):1067-1096. doi:10.6004/jnccn.2023.0050 PubMed
9.Gleason DF, Mellinger GT. Prediction of prognosis for prostatic adenocarcinoma by combined histological grading and clinical staging. J Urol. 1974;111(1):58-64. doi:10.1016/s0022-5347(17)59889-4 PubMed
10.Rosen R, Sapra A. TNM Classification. [Updated 2023 Feb 13]. 2024. Accessed December 12, 2024. https://www.ncbi.nlm.nih.gov/books/NBK553187/
11.Canadian Cancer Society. Grading prostate cancer. Accessed December 11, 2024, https://cancer.ca/en/cancer-information/cancertypes/prostate/grading
12.Canadian Cancer Society. Stages of prostate cancer. Accessed December 11, 2024, https://cancer.ca/en/cancer-information/cancertypes/prostate/staging
13.Sumitomo Pharma Switzerland GmbH. Orgovyx (relugolix): 120 mg oral tablets [product monograph]. 2023.
14.Ferring Pharmaceuticals. Firmagon (Degarelix): 120 mg and 80 mg degarelix injection [product monograph]. 2016.
15.AbbVie Corporation. Lupron (leuprolide acetate): 3.75. 7.5, 11.25, 22.5, 30 mg pre-filled syringe for intramuscular injection [product monograph]. 2024.
16.Verity Pharmaceuticals Inc. Zeulide depot (leuprolide acetate): 3.75 mg and 22.5 mg powder for intramuscular injection [product monograph]. 2024.
17.Tolmar International Ltd. Eligard (leuprolide acetate): 7.5 mg, 22.5 mg, 30 mg and 45 mg powder for subcutaneous use [product monograph]. 2024.
18.TerSera Therapeutics LLC. Zoladex (goserelin): 13.6 mg depot injection [product monograph]. 2017.
19.Cornford P, Bellmunt J, Bolla M, et al. EAU-ESTRO-SIOG Guidelines on Prostate Cancer. Part II: Treatment of Relapsing, Metastatic, and Castration-Resistant Prostate Cancer. Eur Urol. 2017;71(4):630-642. doi:10.1016/j.eururo.2016.08.002 PubMed
20.Lowrance WT, Breau RH, Chou R, et al. Advanced Prostate Cancer: AUA/ASTRO/SUO Guideline PART II. J Urol. 2021;205(1):22-29. doi:10.1097/JU.0000000000001376 PubMed
21.Accord Healthcare, Inc response to Jan 23, 2025 CDA-AMC request for additional information regarding Leuprolide mesylate (Camcevi) CDA-AMC review [internal additional sponsor's information]. Jan 30, 2025.
22.Food and Drug Administration. Advanced Prostate Cancer: Developing Gonadotropin Releasing Hormone Analogues Guidance for Industry. 2022. https://www.fda.gov/media/129027/download
23.Accord Healthcare Inc. Health Canada reviewer's report: Leuprolide injectable emulsion (Camcevi) [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Leuprolide injectable emulsion (Camcevi), Extended release emulsion, 42 mg leuprolide (as leuprolide mesylate), Subcutaneous injection. 2021.
24.Moul JW. The evolving definition of advanced prostate cancer. Rev Urol. 2004;6 Suppl 8(Suppl 8):S10-7.
25.Canada's Drug Agency. Provisional Funding Algorithm: Prostate cancer. Accessed December 12, 2024, https://www.cda-amc.ca/prostate-cancer-0
26.Swayzer DV, Gerriets V. Leuprolide. StatPearls. 2024.
27.Plosker GL, Brogden RN. Leuprorelin. A review of its pharmacology and therapeutic use in prostatic cancer, endometriosis and other sex hormone-related disorders. Drugs. 1994;48(6):930-67. doi:10.2165/00003495-199448060-00008 PubMed
28.Wilson AC, Meethal SV, Bowen RL, Atwood CS. Leuprolide acetate: a drug of diverse clinical applications. Expert Opin Investig Drugs. 2007;16(11):1851-63. doi:10.1517/13543784.16.11.1851 PubMed
29.Cheplapharm Arzneimittel GmbH. Suprefact (buserelin): 1 mg/mL injection and 1 mg/mL Nasal Solution [product monograph]. 2020.
30.Knight Therapeutics Inc. Trelstar (triptorelin): 3.75 mg, 11.25 mg and 22.5 mg powder for injectable suspension [product monograph]. 2024.
31.Ozyigit G, Hurmuz P, Yuce D, Akyol F. Prognostic significance of castrate testosterone levels for patients with intermediate and high risk prostate cancer. World J Clin Oncol. 2019;10(8):283-292. doi:10.5306/wjco.v10.i8.283 PubMed
32.Crawford ED, Sartor O, Chu F, Perez R, Karlin G, Garrett JS. A 12-month clinical study of LA-2585 (45.0 mg): a new 6-month subcutaneous delivery system for leuprolide acetate for the treatment of prostate cancer. J Urol. 2006;175(2):533-6. doi:10.1016/S0022-5347(05)00161-8 PubMed
33.Prezioso D, Lotti T, Montironi R, Polito M. Leuprorelin 1-month depot as neoadjuvant therapy for prostate cancer. Takeda NHT Italian Group. Urologia internationalis. 1998;60 Suppl 2.
34.Sethi R, Sanfilippo N. Six-month depot formulation of leuprorelin acetate in the treatment of prostate cancer [sponsor provided reference]. Clinical interventions in aging2009. PubMed
35.Sharifi R, Bruskewitz RC, Gittleman MC, Graham SD, Jr., Hudson PB, Stein B. Leuprolide acetate 22.5 mg 12-week depot formulation in the treatment of patients with advanced prostate cancer. Clin Ther. 1996;18(4):647-57. doi:10.1016/s0149-2918(96)80215-3 PubMed
36.Sharifi R, Knoll LD, Smith J, Kramolowsky E. Leuprolide acetate (30-mg depot every four months) in the treatment of advanced prostate cancer. Urology. 1998;51(2):271-6. doi:10.1016/s0090-4295(97)00500-1 PubMed
37.Bourdin S, Karam G, Clemain P, et al. Clinical experience with leuprorelin acetate before radiotherapy for prostatic cancer. Journal of International Medical Research. 1990;18(SUPPL. 1). doi:10.1177/03000605900180s111 PubMed
38.David Crawford E, Phillips JM. Six-month gonadotropin releasing hormone (GnRH) agonist depots provide efficacy, safety, convenience, and comfort Cancer Management and Research. 2011;3. doi:10.2147/cmr.s12700 PubMed
39.de Freitas CSM, Soares AN. Efficacy of Leuprorelide acetate (Eligard(R)) in daily practice in Brazil: a retrospective study with depot formulations in patients with prostate cancer. Int Braz J Urol. 2020;46(3):383-389. doi:10.1590/S1677-5538.IBJU.2019.0212 PubMed
40.Sergio Lages P, Zullo GA, Bergerot PG, et al. Real world data of leuprorelin castration efficacy in patients with prostate cancer: A comprehensive assessment of 1,744 tests [sponsor provided reference]. Journal of Clinical Oncology; 2024:
41.Tunn UW. A 6-month depot formulation of leuprolide acetate is safe and effective in daily clinical practice: a non-interventional prospective study in 1273 patients. BMC Urol. 2011;11:15. doi:10.1186/1471-2490-11-15 PubMed
42.Shore N, Mincik I, DeGuenther M, et al. A phase 3, open-label, multicenter study of a 6-month pre-mixed depot formulation of leuprolide mesylate in advanced prostate cancer patients. World J Urol. 2020;38(1):111-119. doi:10.1007/s00345-019-02741-7 PubMed
43.Foresee Pharmaceuticals Inc. NCT02234115 Safety, Efficacy, and Pharmacokinetic Behavior of Leuprolide Mesylate in Subjects With Advanced Prostate Carcinoma [sponsor provided reference]. Accessed 25 August, 2024. https://clinicaltrials.gov/study/NCT02234115?intr=leuprolide%20mesylate&rank=2
44.Foresee Pharmaceuticals Inc. NCT02712320 Open-Label Safety Extension Study in Patients Who Have Previously Participated and Have Benefited From LMIS 50 mg [sponsor provided reference]. Accessed 26 October, 2024. https://clinicaltrials.gov/study/NCT02712320?intr=leuprolide%20mesylate&rank=5&tab=results
45.Foresee Pharmaceuticals Ltd. FP01C-13-001Clinical Study Report [sponsor provided reference]. 2018.
46.Foresee Pharmaceuticals Inc. FP01C-13-001-EX Clinical Study Report [sponsor provided reference]. 2018.
47.Murray TBJ. The Pathogenesis of Prostate Cancer. In: Bott SRJ, Ng KL, eds. Prostate Cancer. 2021.
48.Sharifi N, Gulley JL, Dahut WL. Androgen deprivation therapy for prostate cancer. JAMA. 2005;294(2):238-44. doi:10.1001/jama.294.2.238 PubMed
49.Klotz L, Shayegan B, Guillemette C, et al. Testosterone suppression in the treatment of recurrent or metastatic prostate cancer - A Canadian consensus statement. Can Urol Assoc J. 2018;12(2):30-37. doi:10.5489/cuaj.5116 PubMed
50.Lacher DA, Hughes JP. Total, free, and complexed prostate-specific antigen levels among US men, 2007-2010. Clin Chim Acta. 2015;448:220-7. doi:10.1016/j.cca.2015.06.009 PubMed
51.Nassar GN, Leslie SW. Physiology, Testosterone [sponsor provided reference]. StatPearls2023.
52.Schriefer J. Luteinizing Hormone. In: Enna SJ, Bylund DB, eds. xPharm: The Comprehensive Pharmacology Reference. Elsevier; 2007:1-4.
53.Alexander I. Electronic medical records for the orthopaedic practice. Clinical Orthopaedics and Related Research. 2007;457. doi:10.1097/BLO.0b013e3180342802 PubMed
54.Rezus C, Moga VD, Ouatu A, Floria M. QT interval variations and mortality risk: is there any relationship? Anatol J Cardiol. 2015;15(3):255-8. doi:10.5152/akd.2015.5875 PubMed
55.Certara. Data on File [sponsor supplied reference]. 2020.
56.Foresee Pharmaceuticals Inc. Cross-Study (PK/PD) Comparison of LMIS 50 mg (6 month) and LMIS 25 mg to Marketed Leuprolide acetate Depot Products [sponsor provided reference]. 2020.
57.Phillippo D, Ades T, Dias S, Palmer S, Abrams KR, Welton N. Methods for population-adjusted indirect comparisons in submissions to NICE [sponsor provided reference]. vol Document 18. NICE DSU Technical Support Documents. 2016.
58.Accord Healthcare Inc. Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Leuprolide mesylate (PrCAMCEVI®), 42 mg leuprolide [internal sponsor's package]. 2024.
59.Accord Healthcare Inc. PrCAMCEVI® (leuprolide injectable emulsion): Extended Release Emulsion, 42 mg leuprolide (as leuprolide mesylate), Subcutaneous Injection (6-Month) [product monograph]. 2021.
60.Accord Healthcare Inc. Drug Reimbursement Review sponsor submission: Leuprolide mesylate (PrCAMCEVI®), 42 mg leuprolide [internal sponsor's package]. 2024.
61.Ontario Ministry of Health and Long-Term Care. Ontario drug benefit formulary/comparative drug index. Accessed January 2025, https://www.formulary.health.gov.on.ca/formulary/
62.Goldfischer ER, Grubb E, Nisbet P. Preparation and administration of androgen deprivation therapy products: Retrospective survey results. International Journal of Urological Nursing. 2023;17(2):97-102. doi:10.1111/ijun.12342
63.Ferring Pharmaceuticals. Firmagon - Product monograph [sponsor provided reference]. 2016. Accessed October 2024. https://pdf.hres.ca/dpd_pm/00034229.PDF
64.Statistics Canada. Employee wages by occupation, annual, 1997 to 2022 [sponsor provided reference]. Accessed 28 October, 2024. https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1410034001
65.Cheplapharm Arzneimittel Gmb H. Suprefact Depot - Product monograph [sponsor supplied reference]. 2020. Accessed October 2024. https://dhpp.hpfb-dgpsa.ca/dhpp/resource/43499
66.TerSera Therapeutics LLC. Zoledex Product Monograph. 2024. Accessed October 2024. https://documents.tersera.com/zoladex-ca/10.8mg_ProductMonograph.pdf
67.Cancer Care Alberta. Advanced/Metastatic Prostate Cancer - Clinical Practice Guideline [sponsor supplied reference]. Accessed October, 2024. https://www.albertahealthservices.ca/assets/info/hp/cancer/if-hp-cancer-guide-gu010-met-prostate.pdf
68.Canadian Agency for Drugs and Technologies in Health. Relugolix (Orgovyx) - Reimbursement recommendations [sponsor supplied reference]. Canadian Journal of Health Technologies. 2024;4(10)
69.Canada's Drug Agency. Relugolix Reimbursement Recommendation [sponsor provided reference]. 2024. 8 August. https://www.cda-amc.ca/sites/default/files/DRR/2024/PC0342_Final_Recommendation.pdf
70.Manitoba Health. Drug Formulary Lookup [sponsor provided reference]. Accessed October 28, 2024, https://web22.gov.mb.ca/eFormulary/searchResults.aspx?query=buserelin&type=basic
71.Newfoundland Labrador. NLPDP Drug Product Database [sponsor provided reference]. Accessed 28 October, 2024. https://www.health.gov.nl.ca/health/prescription/newformulary.asp
72.Nova Scotia Department of Health. Formulary [sponsor provided reference]. Accessed 28 October, 2024. https://novascotia.ca/dhw/pharmacare/documents/formulary.pdf
73.BC PharmaCare. BC PharmaCare Formulary Search [sponsor provided reference]. Accessed 28 October, 2024. https://pharmacareformularysearch.gov.bc.ca/Search.xhtml
74.Saskatchewan Drug Plan. Saskatchewan Online Formulary Database [sponsor provided reference]. Accessed 28 October, 2024. https://formulary.drugplan.ehealthsask.ca/SearchFormulary/BG/964071
75.AbbVie Corporation. AbbVie Care Program. Accessed March 2025, https://luprondepotmen.abbviecare.ca/en
76.Medicum. ELIGARD Home Injection Program. Accessed March 2025, https://medicum.ca/en/eligardmd-home-injection-program/
77.TerSera Therapeutics LLC. PrZOLADEX® Goserelin Depot, 3.6 mg Goserelin/depot (as goserelin acetate), Luteinizing Hormone - Releasing Hormone Analog (LHRH Analog) [product monograph]. 2017.
78.AbbVie Corporation. Lupron Depot - Product monograph [sponsor provided reference]. 2013. Accessed October 2024. https://dhpp.hpfb-dgpsa.ca/dhpp/resource/10318
79.Accord Healthcare Inc. Camcevi - Product monograph [sponsor provided reference]. 2021. Accessed October 2024.
80.AstraZeneca Canada Inc. Zoladex LA - Product monograph [sponsor provided reference]. 2024. https://health-products.canada.ca/dpd-bdpp/info?lang=eng&code=43433
81.Health Canada. Zoladex - Product monograph [sponsor provided reference]. https://pdf.hres.ca/dpd_pm/00042728.PDF
82.Cheplapharm Arzneimittel Gmb H. Suprefact - Product monograph [sponsor supplied reference]. 2020. Accessed October 2024. https://pdf.hres.ca/dpd_pm/00056627.PDF
83.Knight Therapeutics. Trelstar - Product monograph [sponsor provided reference]. 2024. Accessed October 2024. https://dhpp.hpfb-dgpsa.ca/dhpp/resource/67797
84.Sumitomo Pharma Canada Inc. Orgovyx - Product monograph [sponsor provided reference]. 2023. Accessed October 2024. https://dhpp.hpfb-dgpsa.ca/dhpp/resource/103073
85.TerSera Therapeutics LLC. Zoledex LA Product Monograph. 2024. Accessed October 2024. https://documents.tersera.com/zoladex-ca/10.8mg_ProductMonograph.pdf
86.Tolmar International L. Eligard - Product monograph. 2022. Accessed October 2024. https://dhpp.hpfb-dgpsa.ca/dhpp/resource/73384
87.Pan-Canadian Pharmaceutical Alliance. Orgovyx (relugolix) [sponsor provided reference]. Accessed 7 November, 2024. https://www.pcpacanada.ca/negotiation/22728
88.Chrisp P, Sorkin EM. Leuprorelin. A review of its pharmacology and therapeutic use in prostatic disorders. Drugs Aging. 1991;1(6):487-509. doi:10.2165/00002512-199101060-00008 PubMed
89.Pan-Canadian Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Leuprolide mesylate (PrCAMCEVI®), 42 mg leuprolide [internal sponsor's package].
90.So AI, Chi K, Danielson B, et al. 2022 UPDATE: Canadian Urological Association-Canadian Urologic Oncology Group guideline: Metastatic castration-naive and castration-sensitive prostate cancer Full-text. Can Urol Assoc J. 2022;16(12):E581-E589. doi:10.5489/cuaj.8148 PubMed
91.Kokorovic A, So AI, Serag H, et al. Canadian Urological Association guideline on androgen deprivation therapy: Adverse events and management strategies. Can Urol Assoc J. 2021;15(6):E307-E322. doi:10.5489/cuaj.7355 PubMed
92.Statistics Canada. Table 17-10-0005-01 Population estimates on July 1, by age and gender [sponsor provided reference]. Accessed 27 October, 2024. https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1710000501
93.Statistics Canada. Table 17-10-0009-01 Population estimates, quarterly [sponsor provided reference]. Accessed 3 November, 2024. https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1710000901
94.Government of Canada. Non-Insured Health Benefits program: First Nations and Inuit Health Branch: Annual report 2022 to 2023 [sponsor supplied reference] Accessed 27 October, 2024. https://www.sac-isc.gc.ca/eng/1713194236054/1713194280612#sec2
95.Patented Medicine Prices Review Board. Alignment Among Public Formularies in Canada, Part 2: Oncology Medicines [sponsor provided reference]. Accessed 27 October, 2024. https://www.canada.ca/en/patented-medicine-prices-review/services/npduis/analytical-studies/formularies-part2-oncology-medicines.html#oncology-medicine
96.Canadian Cancer Society. Canadian Cancer Statistics - A 2018 special report on cancer incidence by stage [sponsor provided reference]. Accessed 25 October, 2024. https://cdn.cancer.ca/-/media/files/research/cancer-statistics/2018-statistics/canadian-cancer-statistics-2018-en.pdf
97.Canadian Cancer Society. Stages of prostate cancer [sponsor provided reference]. Accessed 27 October, 2024. https://cancer.ca/en/cancer-information/cancer-types/prostate/staging#:~:text=For%20prostate%20cancer%20there%20are,into%20A%2C%20B%20or%20C.
98.Sydes MR, Spears MR, Mason MD, et al. Adding abiraterone or docetaxel to long-term hormone therapy for prostate cancer: directly randomised data from the STAMPEDE multi-arm, multi-stage platform protocol. Ann Oncol. 2018;29(5):1235-1248. doi:10.1093/annonc/mdy072 PubMed
99.Warde P, Mason M, Ding K, et al. Combined androgen deprivation therapy and radiation therapy for locally advanced prostate cancer: a randomised, phase 3 trial. Lancet. 2011;378(9809):2104-11. doi:10.1016/S0140-6736(11)61095-7 PubMed
100.Canadian Cancer Society. Survival statistics for prostate cancer [sponsor provided reference]. Accessed 25 October, 2024. https://cancer.ca/en/cancer-information/cancer-types/prostate/prognosis-and-survival/survival-statistics#:~:text=Net%20survival&text=It%20is%20used%20to%20give,for%20at%20least%205%20years.
101.Sutherland G, Dihn T. Understanding the gap: a pan-Canadian analysis of prescription drug insurance coverage. The Conference Board of Canada; 2017. Accessed February 2025. https://www.conferenceboard.ca/e-library/abstract.aspx?did=9326
102.IQVIA. PharmaStat. 2024. Accessed January 21, 2025. https://www.iqvia.com/
103.ACCORD Healthcare Inc. CAMCEVI Market Share Projection - Technical Report [sponsor supplied reference]. 2024.
Appendix 1: Findings From Subgroup Analyses in the FP01C-13-001 Study
Please note that this appendix has not been copy-edited.
As discussed in the external validity section, the pivotal FP01C-13-001 study does not completely align with the population indicated in the Health Canada–approved indication. Specifically, the Health Canada–approved indication approved the use of leuprolide mesylate in patients with advanced prostate cancer, whereas the FP01C-13-001 study did not limit the enrolment to patients with advanced prostate cancer but included patients with stage I to stage IV disease. Although the study protocol of FP01C-13-001 did not plan any subgroup analyses, the sponsor provided post hoc subgroup analyses in the response to the CDA-AMC information request21 seeking the sponsor’s input on how to define advanced prostate cancer. A summary of the subgroup analyses on the proportion of patients who achieved castrate testosterone level (≤ 50 ng/dL) is subsequently presented.
Subgroup Results by Disease Stage (Stage I, II, III, IV, Unknown)
███ ██████████ ██ ████████ ███ ████████ ████████ ████████████ █████ ██ ██ ██████ ██ ███ ██ ███ ██████ ███ ██ ███ ███ ███ ████ ██ ██████ ██ █████ █ ██ ██ █████████ ██████ ███ ██ ███ ███ ███ ████ ██ ██████ ██ █████ ███ █████████ █████ ███ ██ ███ ███ ███ ████ ██ █████ ██ █████ ██ █████████ ███ █████ ███ ██ ███ ███ ███ ████ ██ █████ ██ ████████ ████ ███████ ██████ ██ ███ ████ ███ ██████████ ██ ████████ ███ ████████ ████████ ████████████ █████ ██ ██ ██████ ██ ███ ██ ███ ██████ ███ ██ ███ ███ ███ █████ ██ ██████ ██ █████ █ ██ ██ █████████ █████ ███ ██ ███ ███.
Subgroup Results by Disease Characteristics at Enrolment
In patients with metastatic castration-sensitive prostate cancer (N1 or M1), the proportion of patients who achieved castrate testosterone level (≤ 50 ng/dL) was █████ ███ ██ ███ ███ ███ ████ ██ █████ ██ ███ ██ ███ █████ ███ ██ ███ ███ ███ ████ ██ █████ by day 336.
In patients with nonmetastatic castration-sensitive prostate cancer with locally advanced disease (T3-N0 or T4-N0), the proportion of patients who achieved castrate testosterone level (≤ 50 ng/dL) was ██████ ███ ██ ███ ███ ███ ████ ██ ██████ ██ ███ ██ ███ █████ ███ ██ ███ ███ ███ ████ ██ █████ ██ ███ ████
In patients with nonmetastatic castration-sensitive prostate cancer with high-risk localized disease (i.e., patients in previous subgroup with PSA > 20 ng/mL at screening), the proportion of patients who achieved castrate testosterone level (≤ 50 ng/dL) was ██████ ███ ██ ███ ███ ███ ████ ██ ██████ ██ ███ ██ ███ ██████ ███ ██ ███ ███ ███ █████ ██ ██████ ██ ███ ████.
Appendix 2: Additional Economic Information
Please note that this appendix has not been copy-edited.
Additional Details on the Sponsor’s Submission
Table 30: Hourly Wage for Professional Occupation in Nursing
Year | 2018 | 2019 | 2020 | 2021 | 2022 | 2023 | 2024 |
|---|---|---|---|---|---|---|---|
Reported wage ($) | 36.56 | 38.04 | 39.34 | 39.59 | 40.73 | — | — |
Linearly extrapolated wage ($) | — | — | — | — | — | 41.82 | 42.81 |
Source: Statistics Canada.64
Table 31: Sponsor’s Assumptions Made for Health Care Resource Use
Assumption | Rationalea |
|---|---|
All SC or IM GnRH agonists/antagonists are assumed to be administered in an outpatient setting. | Based on the interview with the Canadian prostate cancer specialist consulted to inform the cost economic analysis. According to the specialist, most patients receive ADTs in an outpatient setting, with only a small minority receiving them in-hospital. |
The time to prepare and inject Eligard is set to 6.1 minutes. | Based on the study from Goldfischer et al.62 |
The time to prepare and inject Lupron Depot is set to 2.5 minutes. | Based on the study from Goldfischer et al.62 |
The time to inject LMIS 50 mg, buserelin acetate, and goserelin acetate is set to 2.5 minutes. | These products do not require reconstitution. The conservative assumption is made that the time to inject them equals the time to prepare and inject Lupron Depot. |
The time to prepare and inject degarelix is set to 8.8 minutes. | According to the product monograph, reconstitution usually lasts a few minutes but may take up to 15 minutes.63 Based on this, the average between 2.5 minutes (preparation and administration time of Lupron Depot) and 15 minutes (max time reported in the product monograph) was assumed. |
The time to prepare and inject triptorelin is set to 8.8 minutes. | The time to prepare and inject triptorelin is assumed to be equal to the time to prepare and inject degarelix.63 This is based on the fact the 2 products have a similar delivery system and need to be pre-mixed by “gentle swirling”. |
Bicalutamide 50 mg/day is assumed to be administered for an average of 22.2 days after initiation of GnRH agonists to block the potential initial testosterone flare. | Non-steroidal antiandrogens are recommended to be administered together with GnRH agonists to block the initial testosterone flare associated with these drugs. Alberta guidelines recommend the administration of non-steroidal antiandrogens (such as bicalutamide 50 mg daily) for a minimum of 14 days to a maximum of 1 month. The average between 14 and 30.4 days is used in the analysis). |
ADT = androgen-deprivation therapy; GnRH = gonadotropin-releasing hormone; IM = intramuscular; LMIS = leuprolide mesylate injectable suspension; SC = subcutaneous.
aTime for administration based on survey by Goldfischer et al. or product monographs.61,68,70-74
Table 32: Sponsor’s Health Care Resource Use
Health care resource | Frequency and duration per yeara | Treatmenta |
|---|---|---|
Drug administration by health care professional | 2.5 minute 2x year | LMIS 50 mg, Lupron Depot 45 mg |
6.1 minute 12x year | Leuprolide acetate (Eligard) 7.5 mg | |
6.1 minute 4x year | Leuprolide acetate (Eligard) 22.5 mg | |
6.1 minute 3x year | Leuprolide acetate (Eligard) 30 mg | |
6.1 minute 2x year | Leuprolide acetate (Eligard) 45 mg | |
2.5 minute 3x year | Leuprolide acetate (Lupron Depot) 30 mg | |
2.5 minute 4x year | Leuprolide acetate (Lupron Depot) 22.5 mg, goserelin acetate (Zoladex) 10.8 mg, buserelin acetate depot (Suprefact) 9.45 mg | |
2.5 minute 6x year | Buserelin acetate depot (Suprefact) 6.3 mg | |
2.5 minute 12x year | Leuprolide acetate (Lupron Depot) 7.5 mg | |
8.75 minute 13x year | Degarelix (Firmagon) first year: Induction (2x 120 mg) followed by maintenance (11x 80 mg) | |
8.75 minute 12x year | Degarelix (Firmagon) subsequent years: maintenance (12x 80 mg) | |
8.75 minute 12x year | Triptorelin (Trelstar) 3.75 mg | |
8.75 minute 4x year | Triptorelin (Trelstar) 11.25 mg | |
8.75 minute 2x year | Triptorelin (Trelstar) 22.5 mg | |
Non-steroidal anti-androgen therapy (bicalutamide 50 mg) | 50 mg/day for an average of 22.2 days (only considered for the first year of treatment) | LMIS 50 mg, Eligard (all formulations), Lupron Depot (all formulations), buserelin acetate (Suprefact; all formulations), goserelin acetate (Zoladex; all formulations), triptorelin (Trelstar; all formulations) |
ADT = androgen deprivation therapy; LMIS = leuprolide mesylate injectable suspension.
aADT formulations based on product monograph.61,68,70-74
Table 33: Sponsor’s Associated Health Care Costs — First Year
Generic name (brand name) | Administration cost ($)a | Cost of non-steroidal antiandrogen therapy ($) | Aggregated health care cost per year ($) | Difference in health care costs per year ($) |
|---|---|---|---|---|
LMIS (Camcevi) 50 mg | 3.57 | 28.20 | 31.76 | — |
Comparators | ||||
Leuprolide acetate (Eligard) 7.5 mg | 52.23 | 28.20 | 80.42 | 48.66 |
Leuprolide acetate (Eligard) 22.5 mg | 17.41 | 28.20 | 45.60 | 13.84 |
Leuprolide acetate (Eligard) 30 mg | 13.06 | 28.20 | 41.25 | 9.49 |
Leuprolide acetate (Eligard) 45 mg | 8.70 | 28.20 | 36.90 | 5.14 |
Leuprolide acetate (Lupron Depot) 7.5 mg | 21.40 | 28.20 | 49.60 | 17.84 |
Leuprolide acetate (Lupron Depot) 22.5 mg | 7.13 | 28.20 | 35.33 | 3.57 |
Leuprolide acetate (Lupron Depot) 30 mg | 5.35 | 28.20 | 33.55 | 1.78 |
Relugolix (Orgovyx) 120 mg | 0.00 | 0.00 | 0.00 | −31.76 |
Buserelin acetate depot (Suprefact) 6.3 mg | 10.70 | 28.20 | 38.90 | 7.13 |
Buserelin acetate depot (Suprefact) 9.45 mg | 7.13 | 28.20 | 35.33 | 3.57 |
Degarelix (Firmagon) 120/80 mg, induction + maintenance | 81.16 | 0.00 | 81.16 | 49.39 |
Goserelin acetate (Zoladex) 10.8 mg | 7.13 | 28.20 | 35.33 | 3.57 |
Triptorelin (Trelstar) 3.75 mg | 74.91 | 28.20 | 103.11 | 71.35 |
Triptorelin (Trelstar) 11.25 mg | 24.97 | 28.2 | 53.17 | 21.40 |
Triptorelin (Trelstar) 22.5 mg | 12.49 | 28.2 | 40.68 | 8.92 |
LMIS = leuprolide mesylate injectable suspension.
aThe total administration cost was calculated by multiplying the hourly wage of nursing professionals by the time for drug administration (in hours) and the number of administrations during the first year.61,68,70-74
Table 34: Sponsor’s Associated Health Care Costs — Subsequent Years
Generic name (brand name) | Administration cost ($)a | Cost of nonsteroidal antiandrogen therapy ($)b | Aggregated health care cost per year ($) | Difference in health care costs per year ($) |
|---|---|---|---|---|
LMIS (Camcevi) 50 mg | 3.57 | 0.00 | 3.57 | — |
Comparators | ||||
Leuprolide acetate (Eligard) 7.5 mg | 52.23 | 0.00 | 52.23 | 48.66 |
Leuprolide acetate (Eligard) 22.5 mg | 17.41 | 0.00 | 17.41 | 13.84 |
Leuprolide acetate (Eligard) 30 mg | 13.06 | 0.00 | 13.06 | 9.49 |
Leuprolide acetate (Eligard) 45 mg | 8.70 | 0.00 | 8.70 | 5.14 |
Leuprolide acetate (Lupron Depot) 7.5 mg | 21.40 | 0.00 | 21.40 | 17.84 |
Leuprolide acetate (Lupron Depot) 22.5 mg | 7.13 | 0.00 | 7.13 | 3.57 |
Leuprolide acetate (Lupron Depot) 30 mg | 5.35 | 0.00 | 5.35 | 1.78 |
Relugolix (Orgovyx) 120 mg | 0.00 | 0.00 | 0.00 | −3.57 |
Buserelin depot (Suprefact) 6.3 mg | 10.70 | 0.00 | 10.70 | 7.13 |
Buserelin depot (Suprefact) 9.45 mg | 7.13 | 0.00 | 7.13 | 3.57 |
Degarelix (Firmagon) 80 mg, maintenance | 74.91 | 0.00 | 74.91 | 71.35 |
Goserelin (Zoladex) 10.8 mg | 7.13 | 0.00 | 7.13 | 3.57 |
Triptorelin (Trelstar) 3.75 mg | 74.91 | 0.00 | 74.91 | 71.35 |
Triptorelin (Trelstar) 11.25 mg | 24.97 | 0.00 | 24.97 | 21.40 |
Triptorelin (Trelstar) 22.5 mg | 12.49 | 0.00 | 12.49 | 8.92 |
GnRH = gonadotropin-releasing hormone; LMIS = leuprolide mesylate injectable suspension.
aThe total administration cost was calculated by multiplying the hourly wage of nursing professionals by the time for drug administration (in hours) and the number of administrations during the first year.61,68,70-74
bNonsteroidal antiandrogen therapy is assumed to be administered only at the beginning of the treatment with GnRH agonists to prevent the testosterone flare associated with these products. Therefore, they are not associated with any cost in the subsequent years of treatment.
Appendix 3: Submitted Budget Impact Analysis and CDA-AMC Appraisal
Please note that this appendix has not been copy-edited.
Table 35: Summary of Key Take-Aways
Key take-aways of the BIA |
|---|
CDA-AMC identified several key limitations in the sponsor’s analysis:
CDA-AMC conducted reanalyses including adding stage I and II intermediate-risk and high-risk prostate cancer, adjusting public coverage rates in some jurisdictions, and revising capture rate assumptions for leuprolide mesylate. CDA-AMC reanalyses suggest that reimbursing leuprolide mesylate for advanced prostate cancer would be associated with cost savings of $433,416 over 3 years (year 1: -$47,846; year 2: -$134,555; and year 3: -$251,014). |
ADT = androgen deprivation therapy; BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency; GnRH = gonadotropin-releasing hormone; LMIS = leuprolide mesylate injectable suspension.
Summary of Sponsor’s BIA
In the sponsor-submitted budget impact analysis (BIA), the sponsor assessed the budget impact of reimbursing leuprolide mesylate for the treatment of adult patients with advanced prostate cancer.89 The BIA was conducted from the perspective of a public drug plan in Canada over a 3-year time horizon (January 2026 to December 2028).89
An epidemiological approach was taken to determine the number of patients eligible for leuprolide mesylate using data from literature.90-99 The sponsor compared a reference scenario where patients are treated with leuprolide acetate (Eligard and Lupron Depot), relugolix, buserelin acetate, degarelix, goserelin acetate, and triptorelin, to a new drug scenario in which leuprolide mesylate would be reimbursed.89 In the new drug scenario, leuprolide mesylate was not assumed to be the dominant therapy.68,89 Reference scenario market shares were estimated based on the CDA-AMC relugolix reimbursement review, and new drug scenario market shares were based on the sponsor’s internal projection of leuprolide mesylate and an assumption that it will result in equal displacement of comparators.68,89 Drug costs were derived from drug formularies with available drug prices, or the average cost of said available drug prices.61,70-74 The sponsor included mark-ups, dispensing fees, allowable upcharges, and copayments for all provincial jurisdictions. Key inputs to the BIA are documented in Table 36.
The key assumptions included:
Market size was estimated by incidence.100 The incidence of locally advanced and metastatic prostate cancer was assumed to be equivalent to the incidence of stage III and stage IV prostate cancer.89
Market uptake of leuprolide mesylate was assumed to capture market share equally from all comparators, except relugolix.89
Patients receiving stable ADT treatment are unlikely to switch to a different ADT treatment, meaning that all uptakes came from new diagnoses (and existing patients were excluded from the analysis).89
ADT treatment is continued until death.89
OS for all ADT treatments was calculated using a log-normal function based on the docetaxel arm of the STAMPEDE clinical trial.98
Nonsteroidal antiandrogens are coadministered with GnRH agonists.58,65-67,89
Table 36: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as Year 1 / Year 2 / Year 3 if appropriate) |
|---|---|
Target population | |
Eligible population covered by public drug plans Incidence of stage III and IV prostate cancer (ASIR per 100,000) Proportion of patients receiving ADT | 67%a95 30.6a96 100% |
Number of patients eligible for drug under review | 3,209 / 3,280 / 3,352 |
Market uptake (3 years) | |
Uptake (reference scenario) leuprolide mesylate leuprolide acetate (Eligard) leuprolide acetate (Lupron Depot) Relugolix Buserelin acetate Degarelix Goserelin Triptorelin | 0.0% / 0.0% / 0.0% 36.3% / 36.1% / 35.9% 23.0% / 22.8% / 22.7% 2.9% / 3.4% / 3.9% 0.05% / 0.05% / 0.05% 3.6% / 3.6% / 3.5% 32.0% / 31.8% / 31.7% 2.2% / 2.2% / 2.2% |
Uptake (new drug scenario) leuprolide mesylate leuprolide acetate (Eligard) leuprolide acetate (Lupron Depot) Relugolix Buserelin acetate Degarelix Goserelin Triptorelin | 3.7% / 6.8% / 9.4% 35.0% / 33.6% / 32.4% 22.1% / 21.2% / 20.5% 2.9% / 3.4% / 3.9% 0.1% / 0.0% / 0.0% 3.4% / 3.3% / 3.2% 30.8% / 29.6% / 28.6% 2.1% / 2.1% / 2.0% |
Cost of treatment (per patient, per annum)b | |
leuprolide mesylate leuprolide acetate (Eligard) leuprolide acetate (Lupron Depot) Relugolix (induction year) Relugolix (maintenance year) Buserelin acetate Degarelix (induction year) Degarelix (maintenance year) Goserelin Triptorelin | $2,998 $3,433 $4,284 $3,305 $3,287 $5,389 $3,758 $3,290 $4,883 $3,996 |
ADT = androgen-deprivation therapy; ASIR = age-standardized incidence ratio.
aBased on a pan-Canadian average.
bFor comparators with multiple dosing options, sponsor selected dosages based on clinical input. Costs are sourced from available drug formularies, except relugolix, which was sourced from the CDA-AMC recommendation.61,68,70-74
Summary of the Sponsor’s BIA Results
The sponsor estimated that funding leuprolide mesylate for the treatment of adult patients with advanced prostate cancer would be associated with cumulative cost savings of $1,230,815 over a 3-year time horizon (year 1: -$134,787; year 2: -$381,664; and year 3: -$714,364).
CDA-AMC Appraisal of the Sponsor’s BIA
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
The market size is likely underestimated. The Health Canada indication for leuprolide mesylate is for the treatment of adult patients with advanced prostate cancer.59 According to the CDA-AMC Clinical Review, there is no universal definition for advanced prostate cancer, and “patients with advanced prostate cancer” noted in the Health Canada–approved indication is subject to interpretation. The sponsor’s base-case BIA assumed that advanced prostate cancer comprised stage IIIb, IIIc, and IV prostate cancer.89 Therefore, the eligible patient population was estimated based on stage III and IV incidence rates. As noted in the sponsor’s pharmacoeconomic evaluation and confirmed by clinical expert feedback received by CDA-AMC, some patients with unfavourable intermediate-risk and high-risk localized prostate cancer in stages II or earlier would also be treated with ADT.58 These patients were not included in the sponsor’s base case but were accounted for in a scenario analysis conducted by the sponsor.89 Additionally, the sponsor’s submitted base case was conducted using an incidence based approach of patients with de novo stage III and IV prostate cancer, which excludes the patients with stage I or II prostate cancer who progress to stage III or IV. The exclusion of patients who are treated with ADTs in stage II or earlier and patients who are diagnosed as stage I or II but progressed to stage III or IV likely results in an underestimate of the market size.
In reanalysis, CDA-AMC included patients with intermediate-risk and high-risk localized prostate cancer in the market size.
The provincial coverage rates are likely underestimated. The sponsor’s submitted BIA assumed coverage rates per jurisdiction based on the proportion of drug expenses associated with take-home oncology medicines covered by public payers.95 This approach assumes that the proportion paid by public drug plans is independent of whether a drug is listed in a given jurisdiction. To be considered a relevant comparator included in the BIA, the drug should either be reimbursed by at least 1 participating plan or should have received a recommendation in favour of reimbursement; as such, through its inclusion in the BIA, it is assumed that it is publicly reimbursed, and take-home cancer drugs are fully covered in Manitoba, Saskatchewan, Alberta, and British Columbia.95 As noted in the Patented Medicine Prices Review Board report, British Columbia, Alberta, Saskatchewan, and Manitoba provide coverage for all oncology medications regardless of the setting of treatment.95 As well, many jurisdictions have oncology drug coverage for patients that are aged 65 years or older.101
Coverage rates for patients were adjusted to account for 100% oncology drug coverage in the western provinces (i.e., British Columbia, Alberta, Saskatchewan, and Manitoba).
Reference scenario market shares and patient distribution across ADT formulations is uncertain. Reference scenario market shares in the sponsor’s submitted BIA were estimated from the CDA-AMC relugolix pharmacoeconomic report, where uncertainty in the uptake of relugolix was a key limitation.69 The reference scenario market shares are uncertain due to the variability of ADT coverage in different provinces. As well based on clinical expert feedback received by CDA-AMC, market shares may not align with clinical practice as experts expected degarelix to have a higher market share than assumed in the analysis.
As well, for each comparator with multiple dosage forms (i.e., 3-month preparations and 6-month preparations), the sponsor attributed a distribution of patients across the available dosage forms which was informed by sponsor-elicited clinical input.89 For some comparators (i.e., leuprolide acetate [Lupron Depot], and buserelin acetate), patients were only receiving 1 dose regimen. According to clinical expert feedback consulted by CDA-AMC, all patients may not be suitable for only 1 dose regimen due to clinician visit frequency, and patient preference. Additionally, the 3.6 mg dose of goserelin was not considered in the sponsor’s BIA model.
To address the uncertainty in the distribution of patients across dosage forms, CDA-AMC conducted a scenario analysis using the treatment distribution of leuprolide acetate (Eligard and Lupron Depot), and triptorelin based on public claims data from the IQVIA Pharmastat database, accessed January 2025102 (i.e., Leuprolide acetate [Eligard]: 7.5 mg-0%, 22.5 mg-36%, 30 mg-20%, and 45 mg-44%; leuprolide acetate [Lupron Depot]: 7.5 mg-4%, 22.5 mg-56%, and 30 mg-39%; triptorelin 3.75 mg-1%, 11.25 mg-63%, and 22.5 mg-36%).102
CDA-AMC was unable to address the uncertainty of missing goserelin (3.6 mg) due to inflexibility in the sponsor’s model.
Market uptake of leuprolide mesylate and displacement of comparators are uncertain and potentially overestimated. The sponsor estimated the market uptake of leuprolide mesylate by using US market data and determining volume of sales for leuprolide mesylate relative to leuprolide acetate 45 mg.103 Clinical experts noted that uptake of leuprolide mesylate is uncertain and may be lower than estimated by the sponsor. Additionally, the sponsor assumed that leuprolide mesylate would proportionally displace all comparators except for relugolix.89 Clinician expert feedback indicated that primarily 6-month ADT formulations would be displaced by leuprolide mesylate.
In reanalysis, CDA-AMC revised the market capture of leuprolide mesylate to only assume to displace 6-month ADT formulations. CDA-AMC was unable to revise market uptake in the absence of more appropriate estimates.
The price of drugs paid by public drug plans is uncertain. The analyses by both the sponsor and CDA-AMC are based on publicly available list prices for all comparators. Actual costs paid by public drug plans are unknown. Confidential negotiated prices for ADT comparators may lead to budgetary savings associated with the reimbursement of leuprolide mesylate being limited or eliminated.
CDA-AMC could not address this limitation in reanalysis.
CDA-AMC Reanalyses of the BIA
CDA-AMC base-case reanalyses revised the market uptake displacement and displacement of leuprolide mesylate; revised the patient population; and adjusted the reference scenario market shares and patient distribution of each comparator (Table 37).
Table 37: CDA-AMC Revisions to the Submitted BIA
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Correctionsa to sponsor’s base case | ||
1. Drug mark-ups, fees, and copayments | Included | Excluded |
Changes to derive the CDA-AMC base case | ||
1. Eligible patient population | Patients with stage III and IV prostate cancer were included | Stage III and IV along with stage I and II intermediate and high-risk prostate cancer were included |
2. Public coverage rates | British Columbia: 83% Alberta: 88% Saskatchewan: 88% Manitoba: 87% | British Columbia: 100% Alberta: 100% Saskatchewan: 100% Manitoba: 100% |
3. Market capture | Leuprolide mesylate displaces all comparators | Leuprolide mesylate displaces 6-formulation of leuprolide acetate [Eligard] (95%) and triptorelin (5%). |
CDA-AMC base case | 1 + 2 + 3 | |
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency.
aCorrections are minor errors (e.g., transcription errors between report and model, misapplication of distributions or SEs in probabilistic analyses) that are not identified as limitations.
The results of the CDA-AMC step-wise reanalysis are presented in summary format in Table 38, and a more detailed breakdown is presented in Table 39. Based on the CDA-AMC base-case reanalyses, the cost savings for funding leuprolide mesylate for the treatment of advanced prostate cancer is estimated to be $433,416 over a 3-year time horizon (year 1: -$47,846; year 2: -$134,555; and year 3: -$251,014). The CDA-AMC reanalyses are based on publicly available list prices for all comparators.
Table 38: Summary of the CDA-AMC Reanalyses of the BIA
Stepped analysis | Three-year total |
|---|---|
Submitted base case | -$1,230,815 |
Sponsor’s corrected base case | -$1,236,485 |
CDA-AMC reanalysis 1- stage I and II | -$1,867,335 |
CDA-AMC reanalysis 2 – public drug coverage | -$1,342,662 |
CDA-AMC reanalysis 3 – market capture | -$268,544 |
CDA-AMC base case | -$433,416 |
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency.
Table 39: Detailed Breakdown of the CDA-AMC Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
Submitted base case | Reference | $13,055,518 | $13,266,725 | $25,950,542 | $37,685,655 | $76,902,922 |
New drug | $13,055,518 | $13,131,938 | $25,568,878 | $36,971,291 | $75,672,106 | |
Budget impact | $0 | -$134,787 | -$381,664 | -$714,364 | -$1,230,815 | |
CDA-AMC base case | Reference | $21,775,824 | $22,061,049 | $42,582,285 | $61,514,007 | $126,157,341 |
New drug | $21,775,824 | $22,013,203 | $42,447,729 | $61,262,993 | $125,723,926 | |
Budget impact | $0 | -$47,846 | -$134,555 | -$251,014 | -$433,416 | |
CDA-AMC scenario analysis 1: treatment distribution | Reference | $21,824,967 | $22,109,813 | $42,676,407 | $61,649,890 | $126,436,110 |
New drug | $21,824,967 | $22,064,549 | $42,549,113 | $61,412,422 | $126,026,085 | |
Budget impact | $0 | -$45,264 | -$127,294 | -$237,468 | -$410,026 |
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.