Drugs, Health Technologies, Health Systems
Reimbursement Review
Pembrolizumab (Keytruda)
Sponsor: Merck Canada Inc.
Therapeutic area: Non-small cell lung cancer
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
ACT
adjuvant chemotherapy
AE
adverse event
AEOSI
adverse event of special interest
AJCC
American Joint Committee on Cancer
ALK
anaplastic lymphoma kinase
APaT
all participants as treated
CDA-AMC
Canada’s Drug Agency
DFS
disease-free survival
ECOG PS
Eastern Cooperative Oncology Group Performance Status
EGFR
epidermal growth factor receptor
EORTC QLQ-C30
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core-30
EORTC QLQ-LC13
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Lung Cancer Module
FAS
full analysis set
GHS
Global Health Status
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HR
hazard ratio
HRQoL
health-related quality of life
IA3
third interim analysis
ITC
indirect treatment comparison
ITT
intention-to-treat
LCC
Lung Cancer Canada
MID
minimal important difference
NSCLC
non–small cell lung cancer
OS
overall survival
PRO
patient-reported outcome
QoL
quality of life
RCT
randomized controlled trial
RECIST v1.1
Response Evaluation Criteria in Solid Tumors Version 1.1
SAE
serious adverse event
T2a
tumour stage 2a
TPS
tumour proportion score
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Pembrolizumab (Keytruda), 100 mg per 4 mL vial, solution for infusion |
Sponsor | Merck Canada Inc. |
Indication | For the adjuvant treatment of adult patients with stage IB (T2a ≥ 4 cm), II, or IIIA NSCLC who have undergone complete resection and platinum-based chemotherapy |
Reimbursement request | For the adjuvant treatment of adult patients with stage IB (T2a ≥ 4 cm), II, or IIIA NSCLC who have undergone complete resection and platinum-based chemotherapy and whose tumors have a programmed cell-death ligand tumour proportion score < 50%, as determined by a validated test |
Health Canada approval status | NOC |
Health Canada review pathway | Standard review |
NOC date | April 19, 2023 |
Recommended dose | Either 200 mg every 3 weeks or 400 mg every 6 weeks given through IV over 30 minutes, for up to 1 year or until disease recurrence or unacceptable toxicity |
NOC = Notice of Compliance; NSCLC = non–small cell lung cancer; T2a = tumour stage 2a.
Introduction
Lung cancer is the most diagnosed cancer and leading cause of cancer deaths in Canada.1,2 Lung cancer survival in all stages and histologies are poor, with an overall 5-year net survival of 22%,2,3 and only 3% for those diagnosed with stage IV disease.2 In 2024, it was estimated that there would be 32,100 new cases of lung cancer diagnosed and 20,700 deaths from lung cancer.2 Non–small cell lung cancer (NSCLC) accounts for approximately 88% of all cases of lung cancer in Canada.1 It is estimated that 30% to 35% of NSCLCs are diagnosed at an early stage (I to IIIA)4-6 and approximately 20% to 25% of patients with NSCLC have surgically resectable disease.7 After surgery, 45% of patients with stage IB disease and 76% of patients with stage III disease will experience disease recurrence and subsequently die over a median follow-up of 5 years, regardless of the use of adjuvant chemotherapy (ACT) or immunotherapy.8
Standard treatment for patients with stage IB to IIIA NSCLC is surgical resection.9 Perioperative treatments (neoadjuvant or adjuvant) are used depending on stage. According to the clinical experts consulted by the review team, the goal of any adjuvant therapy following complete resection in early-stage NSCLC is to improve cure rates by reducing the risk of relapse, after which no curative therapies are currently available. In the perioperative setting, the current treatment standard for patients with resectable NSCLC without actionable oncogenic alterations is neoadjuvant platinum-doublet chemotherapy in combination with immunotherapy (nivolumab), or adjuvant platinum-doublet chemotherapy followed by immunotherapy (atezolizumab) for patients with a PD-L1 tumour proportion score (TPS) of 50% or greater.10-13 However, the clinical experts consulted for this review indicated that not all patients receive immunotherapy or platinum-based chemotherapy in the perioperative setting as they may decline, not be offered a referral to medical oncology, or are ineligible. The clinical experts noted that there is a need to reduce the recurrence rates in patients with early-stage NSCLC, and provide options for those who have not had neoadjuvant immunochemotherapy and are ineligible for adjuvant atezolizumab (e.g., patients with a PD-L1 TPS of less than 50%) to cure disease, delay disease recurrence, improve survival, and maintain quality of life (QoL).10,14,15 According to the clinical experts consulted by the review team, PD-L1 TPS testing is currently performed as standard of care for patients with NSCLC in Canada.
This report reviews and critically appraises the evidence submitted by the sponsor on the beneficial and harmful effects of pembrolizumab (Keytruda) administered by IV infusion as either 200 mg every 3 weeks or 400 mg every 6 weeks for the adjuvant treatment of adult patients with stage IB (tumour stage 2a [T2a] ≥ 4 cm), II, or IIIA NSCLC who have undergone complete resection and platinum-based chemotherapy and whose tumours have a PD-L1 TPS of less than 50%, as determined by a validated test.
Perspectives of Patients, Clinicians, and Drug Programs
The information in this section is a summary of input provided by the patient and clinician groups who responded to a call for input from Canada’s Drug Agency (CDA-AMC) and from the clinical experts consulted for the purpose of this review.
Patient Input
This review received a joint submission by 3 patient groups: the Lung Health Foundation (LHF), Lung Cancer Canada (LCC), and the Canadian Cancer Survivor Network. The input was based on information collected by the LHF from individuals living with lung cancer, 3 interviews with patients from Ottawa, Vancouver, and Toronto who had experience with pembrolizumab, and 33 responses to an online survey available between June 2023 to June 2024.
Survey respondents reported similar symptoms and challenges due to their lung cancer, some of which included fatigue (53%), shortness of breath (50%), coughing (23%), and pain (20%). Most respondents indicated that living with lung cancer negatively affects their emotional well-being through feelings of isolation, challenges with symptom management, and perceived burdens on caregivers and family. Disease aspects that were most important to responders to control included symptoms and pain and side effects from therapy. Patients who previously received surgery reported experiencing deconditioning and chronic fatigue, and medication side effects that included extreme itching affecting sleep, brain fog, fatigue, nausea, vomiting, mood changes, diminished appetite, weight loss, hair loss, anemia, and neuropathy. These patients also experienced challenges accessing some therapies due to high treatment costs, as well as difficulty navigating the health care system and locating disease information and support. The input also noted concerns from patients on targeted therapy as to their ability to access the next line of treatment if or when their current therapy stops working.
Respondents indicated that key treatment outcomes to consider when evaluating new therapies include stopping or slowing disease progression with minimal side effects, and effectiveness in advanced disease. Three LHF interviewees had experience with pembrolizumab, although these patients were not part of the eligible population of the current indication under review, which is limited to those with stage IB (T2a ≥ 4 cm), II, or IIIA NSCLC. One interviewee with epidermal growth factor receptor (EGFR)-positive stage 4 lung cancer was taking pembrolizumab in combination with chemotherapy, although it was unclear if the interviewees with NSCLC were taking pembrolizumab similarly or as monotherapy. One patient discontinued pembrolizumab after 19 months due to progression, while another experienced sufficient tumour shrinkage and inactivity to discontinue pembrolizumab after 3 years, before re-initiating shortly after upon tumour reactivation. After re-initiation of pembrolizumab, the patient’s tumours once again decreased in size. Side effects reported by these patients included nausea, fatigue, muscle soreness, constipation, diarrhea, and worsening of their diabetes, eczema, and liver-enzyme levels. Patients did not report that these side effects impeded their ability to participate in daily activities or exercise, and overall they reported experiencing improved QoL while on therapy.
Clinician Input
Input From Clinical Experts Consulted by CDA-AMC
According to the clinical experts consulted by the review team, the key treatment goals for patients with early-stage NSCLC who receive adjuvant therapy following complete surgical resection is to improve cure rates by reducing the risk of relapse. The clinical experts noted that there are currently no adjuvant immunotherapy options for the patients in Canada with stage IB to IIIA NSCLC who have a PD-L1 TPS of less than 50%, creating an unmet need for this group. The clinical experts noted that adjuvant pembrolizumab would fill a treatment gap, as currently patients can only access this treatment after relapse. The clinical experts pointed out that overall survival (OS) is the outcome most important to patients. The clinical experts indicated that pembrolizumab treatment should be discontinued if 1 of the following has been met: a total of 18 cycles (1 year) of adjuvant immunotherapy has been completed, disease progression has been detected, or unacceptable toxicity develops. The clinical experts noted that pembrolizumab should be administered in a specialty setting that has surgical and medical oncology multidisciplinary staff with the expertise to provide systemic therapy, monitor the patient, and manage treatment-related toxicities.
Clinician Group Input
This review received input from the Ontario Health (Cancer Care Ontario) Lung Cancer Drug Advisory Committee and the LCC Clinician Group. Six clinicians from the Drug Advisory Committee and 35 clinicians from LCC provided input for this review. The clinician groups supplied input aligning with input from the clinical experts consulted for this review with respect to the unmet needs, patient population, key outcomes, discontinuing treatment, and prescribing considerations. The LCC group emphasized that, in patients with early-stage resected IB to IIIA NSCLC, current therapies do not adequately achieve high cure rates or prevent recurrences. The LCC group noted that this is particularly important for patients with NSCLC, as the risk of relapse increases substantially with each subsequent disease stage. The LCC group also pointed out that patient relapse and metastatic disease impose substantial costs on patient health, QoL, utilization of health care resources, and economic loss of productivity, and overall costs to society. The LCC group expected that pembrolizumab would shift the current treatment paradigm, as it represents the first adjuvant immunotherapy option for this patient population. The LCC group also anticipated that treatment with pembrolizumab for eligible patients with a sensitizing EGFR mutation would require case-by-case consideration by treating clinicians, weighing the risks and benefits of adjuvant sequential chemotherapy and immunotherapy versus those of adjuvant osimertinib. The clinician groups agreed that treatment benefits in the adjuvant setting are primarily determined by disease recurrence, which the LCC group noted typically occurs within 2 to 3 years for patients with stage IB to IIIA NSCLC. The LCC group indicated that cure rates, as measured by 5-year OS, can also determine response, but typically requires additional years of follow-up. The LCC group suggested implementing clinical and laboratory follow-ups every 3 weeks to evaluate toxicity and disease recurrence, as well as imaging scans at 3- to 4-month intervals, given pembrolizumab is administered over 1 year. The LCC group noted that, overall, immunotherapies are well tolerated by patients, and autoimmune side effects can often be readily managed by oncologists.
Drug Program Input
Input was obtained from the drug programs that participate in the reimbursement review process. The following were identified as key factors that could potentially affect the implementation of a recommendation for pembrolizumab:
consideration for initiation of therapy
consideration for discontinuation of therapy
generalizability.
The clinical experts consulted for this review provided advice on the potential implementation issues raised by the drug programs. Table 5 provides more details.
Clinical Evidence
Systematic Review
Description of Studies
One ongoing, multicentre, triple-blind, phase III randomized controlled trial (RCT), KEYNOTE-091 (N = 1,177) submitted by the sponsor was included for this review. The study compared pembrolizumab (200 mg every 3 weeks or 400 mg every 6 weeks, by IV infusion, for up to 1 year or until disease recurrence or unacceptable toxicity) with placebo as adjuvant therapy for completely resected stage IB to IIIA NSCLC. Eligible patients were adults with pathologically confirmed NSCLC (any histology) of stage IB (T2a ≥ 4 cm), II or IIIA as defined by the seventh edition of the American Joint Committee on Cancer (AJCC) staging system16 after complete surgical resection (lobectomy, sleeve lobectomy, bi-lobectomy, or pneumonectomy) and negative margins (R0). Eligible patients had an available tumour sample obtained during resection for PD-L1 assessment, a known PD-L1 expression status, no evidence of disease on clinical examination and radiographic assessment according to the Response Evaluation Criteria in Solid Tumors Version 1.1 (RECIST 1.1)17 as determined by local investigator review after surgery but within 12 weeks before randomization, an Eastern Cooperative Oncology Group Performance Status (ECOG PS) of 0 or 1, and adequate organ function within 10 days of treatment initiation. Previous neoadjuvant or adjuvant radiotherapy for the current malignancy was not permitted. ACT was not mandatory but was to be considered for patients with stage IB disease and strongly recommended for those with stage II or IIIA disease according to local practice and national guidelines. Patients with prior treatment with an anti–PD-1, anti–PD-L1 or anti–PD-L2, anti-CD137, CTLA-4 modulators, or any other immune-modulating drugs were excluded from the KEYNOTE-091 study. The subpopulation of interest (the reimbursement request population [n = 363 in the pembrolizumab group, and n = 363 in the placebo group]), were patients receiving ACT and a PD-L1 TPS of less than 50%. The outcomes relevant to this review included OS, disease-free survival (DFS), health-related quality of life (HRQoL), and safety.
The reimbursement request population of the KEYNOTE-091 study had a median age of 64 to 65 years. The proportion of male patients (65% to 68%) was higher than that of female patients (32% to 35%). Most patients (77% to 78%) were white, followed by Asians (17% to 18%), among others. A relatively small proportion of patients (11% to 12%) had stage IB disease and more than half (55% to 59%) had stage II disease. Most patients (68% to 73%) were former smokers, followed by those who had never smoked (13% to 19%), and current smokers (13% to 14%). More patients had an ECOG PS of 0 (56% to 62%) compared to those with an ECOG PS of 1 (38% to 44%). Most patients (41%) had a lymph-node stage of N0 (no regional lymph-node involvement) and 38% had a lymph-node stage of N1 (nearby lymph-node involvement). More patients had a nonsquamous histology (62.5% to 72.2%) compared with those with squamous histology (27.8% to 37.5%). The proportion of patients who had an EGFR mutation (5.8% to 8.5%) or anaplastic lymphoma kinase (ALK) translocation (0.8% to 1.7%) was low. More than half of the patients had an unknown status for the EGFR mutation (51.8% to 57.4%) or ALK translocation (56.7% to 66.4%).
Efficacy Results
The key efficacy results from the KEYNOTE-091 trial are summarized in Table 2 in order from the most important to the less important outcomes as suggested by the clinical experts consulted for this review. The efficacy and harms outcomes of the KEYNOTE-091 study reported in this review were based on the protocol-prespecified third interim analysis (IA3), for which the data cut-off date was January 24, 2023.
Overall Survival
As of the data cut-off date (January 24, 2023) and among the reimbursement request population (N = 726), the median duration of follow-up was 46.6 months (range = 0.6 to 84.2). The median OS was not reached in either group ███████ █████ █ █████ ███ ██████████ ████████ █████ ████ ██ █████ | | ██████. The Kaplan-Meier estimates of the probability of OS in the pembrolizumab and placebo groups were █████ ████ ███ ████ ██ █████ versus █████ ████ ███ ████ ██ █████ at 36 months; and █████ ████ ███ ████ ██ █████ versus █████ ████ ███ ████ ██ █████ at 48 months, respectively.
Disease-Free Survival
As of the data cut-off date among the reimbursement request population, the median DFS was 51.7 months (95% confidence interval [CI], 39.0 to 70.4) for patients treated with pembrolizumab and 34.5 months (95% CI, 23.3 to 46.4) for patients who received placebo (hazard ratio [HR] = 0.72; 95% CI, 0.58 to 0.89; P < 0.001). The Kaplan-Meier estimates of the probability of DFS in the pembrolizumab and placebo groups were 67.2% (95% CI, 61.9 to 71.9) versus 55.0% (95% CI, 49.7 to 60.0) at 24 months; and 51.2% (95% CI, 45.2 to 56.9) versus 42.4% (95% CI, 36.7 to 47.9) at 48 months, respectively.
Health-Related Quality of Life
The HRQoL outcomes assessed with the European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 (EORTC QLQ-C30) Global Health Status (GHS)/QoL measure and the European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Lung Cancer Module (EORTC QLQ-LC13) symptom scales at 48 weeks were available for the patient-reported outcome (PRO) full analysis set (FAS) population (N = 1,161). The compliance rates for the EORTC QLQ-C30 and EORTC QLQ-LC13 were 98.6% at baseline and 85.8% at week 48 in the pembrolizumab group, and 99.8% at baseline and 90.0% at week 48 in the placebo group, respectively. At week 48, the questionnaire completion rates were 77.9% and 84.9% in the pembrolizumab and placebo groups, respectively.
The proportion of patients with a deteriorated score in EORTC QLQ-C30 GHS/QoL (a 10-point or greater deterioration from baseline at any time during the trial when the criteria for improved or stable is not met) in the pembrolizumab group was higher than in the placebo group (18.1% and 12.9%, respectively; difference = 5.2%; 95% CI, 1.0 to 9.4; P = 0.015).
At week 48, the proportion of patients with a deteriorated score in EORTC QLQ-LC13 symptom scales was similar between the 2 groups for chest pain (difference = −1.7%, 95% CI, −4.9 to 1.5; P = 0.295), coughing (difference = −0.1%, 95% CI, −3.9 to 3.6; P = 0.945), and dyspnea (difference = 3.2%, 95% CI, −1.5 to 7.8; P = 0.181).
Harms Results
The harms outcomes were available among the all participants as treated (APaT) population in the KEYNOTE-091 study (N = 1,161). Adverse events (AEs) were reported in 96% and 91% of patients in the pembrolizumab and placebo groups, respectively. The most common AEs were increased weight (23% in the pembrolizumab group and 29% in the placebo group, respectively), pruritus (22% and 13%), and hypothyroidism (21% and 5%). The incidence was higher in the pembrolizumab group than in the placebo group for grade 3 to 5 AEs (34% and 26%), serious adverse events (SAEs) (25% and 16%), AEs resulting in treatment discontinuation (20% and 6%), AEs resulting in death (2% and 1%), and adverse events of special interest (AEOSIs) such as immune-mediated events and infusion-related reactions (39% and 13%). The most frequently reported AEOSIs in the pembrolizumab and placebo groups were hypothyroidism (21% and 5%, respectively), hyperthyroidism (11% and 3%), and pneumonitis (7% and 3%).
Critical Appraisal
In the KEYNOTE-091 study, the review team and the clinical experts consulted for this review did not identify major issues that would affect the validity of the study results as a consequence of presenting the DFS and OS outcomes through ad hoc analyses in the reimbursement request population, based on the fact that the PD-L1 TPS category was a stratification variable. The patient demographic and disease characteristics appeared to be generally balanced between the treatment groups in both overall population and reimbursement request population, suggesting that the benefits of the randomization were reasonably maintained in the subpopulation for reimbursement request. The review team noted that histologic status was unbalanced between the 2 groups (27.8% squamous in the pembrolizumab group versus 37.5% in the placebo group). To what extent this imbalance could bias the results is unknown. In the reimbursement request population, a higher proportion of patients (48%) in the pembrolizumab group discontinued from the study medication compared with the placebo group (37%), mainly due to AEs. The clinical experts commented that the between-group imbalance and the reasons for the study medication discontinuation were reasonable and in line with the safety outcomes, in that larger proportions of patients in the pembrolizumab group experienced AEOSIs compared with those in the placebo group. In the reimbursement request population, nearly all of the study patients (95%) received at least 1 concomitant medication and the proportions of patients with the use of most medications were similar between treatment groups. However, there was a larger proportion of some concomitant medications (e.g., antihistamines, corticosteroids, and thyroid replacement therapy) in the pembrolizumab group compared to the placebo group, which may have affected the assessment of HRQoL and biased the results in favour of pembrolizumab, as these concomitant drug uses were most likely for the control or treatment of drug-related side effects associated with pembrolizumab. The proportion of patients receiving subsequent anticancer treatment during the trial was smaller in the pembrolizumab group than in the placebo group, for both antineoplastic therapy and immunotherapies. Although these uneven uses of anticancer therapies may have biased the efficacy results against pembrolizumab as compared to the placebo group for OS, the extent of any important impact on interpretation of the observed effect could not be determined. The triple-blind design of the trial likely mitigated the risks associated with knowledge of group assignment for these outcomes. The risk of bias due to missing outcome data for OS, DFS, and safety outcomes appeared to be low as losses to follow-up for reasons other than death were low, and sensitivity analysis with the different censoring rule for DFS in the reimbursement request population was consistent. The risk of bias due to missing outcome data for the HRQoL outcome is low as only a small proportion of patients (1% to 6%) had “no assessment” for the select measures in the PRO FAS population (N = 1,161). OS and DFS were tested by applying a multiplicity hierarchical testing procedure to account for the potential inflated type I error rates across multiple end points and interim analyses. However, OS and DFS results were based on interim analyses, which may have overestimated the treatment-effect estimates.18,19 The presence and extent of any overestimate that may have been introduced could not be determined.
Patients in the KEYNOTE-091 study were recruited from multiple countries, including Canada. The clinical experts described the eligibility criteria of patients in the KEYNOTE-091 study as appropriate and noted that the demographic diversity of the patients in the study was mostly in line with what is seen in clinical practice in Canada. The clinical experts noted that pembrolizumab is often offered to patients with an ECOG PS of up to 2 in clinical practice in Canada, and these patients might benefit from pembrolizumab, even though only patients with an ECOG PS of 0 to 1 were enrolled in the KEYNOTE-091 study as specified by the study inclusion criteria. The clinical experts noted that presenting the survival outcomes among the subgroup of patients in the KEYNOTE-091 study who had a PD-L1 TPS of less than 50% and had received ACT was appropriate for this review, aligns with the reimbursement request, and addresses the unmet therapeutic needs.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
The Grading of Recommendations Assessment, Development and Evaluation (GRADE) framework was used to assess the certainty of the evidence in the pivotal studies and RCTs identified in the sponsor’s systematic review regarding outcomes considered most relevant to deliberations of the CDA-AMC expert committee, and a final certainty rating was determined as outlined by the GRADE Working Group.20,21 Following the GRADE approach, evidence from RCTs started as high certainty and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
The selection of outcomes for the GRADE assessment was based on the sponsor’s Summary of Clinical Evidence, consultation with clinical experts, and input received from patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members:
probability of OS at months 36 and 48
probability of DFS at months 24 and 48
HRQoL as measured by the EORTC QLQ-C30 (GHS/QoL) and EORTC QLQ-LC13 symptomatic scales (chest pain, coughing, and dyspnea) at week 48
grade 3 to 5 AEs.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty-of-evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null.
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for pembrolizumab versus placebo in adult patients with stage IB (T2a ≥ 4 cm), II, or IIIA NSCLC who have undergone complete resection and platinum-based chemotherapy and whose tumours have a PD-L1 TPS of less than 50%.
Table 2: Summary of Findings for Pembrolizumab Versus Placebo for Adult Patients With Stage IB, II, or IIIA NSCLC With a PD-L1 TPS of Less Than 50%
Outcome and follow-up | Patients, N (studies) | Relative effect (95% CI) | Absolute effects (95% CI) | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
Placebo | Pembrolizumab | Difference | |||||
OS | |||||||
Probability of survival at 36 months Median follow-up: 46.6 (range = 0.6 to 84.2) months | 726 (1 RCT) | NR | ███ ███ █████ ████ ██ ███ ███ ██████ | ███ ███ █████ ████ ██ ███ ███ ██████ | ██ ████ ███ █████ █████ █ █████ ██ ███ ████ ███ ██████ | Lowa | Pembrolizumab may result in little to no difference in OS compared to placebo at 36 months |
Probability of survival at 48 months Median follow-up: 46.6 (range = 0.6 to 84.2) months | 726 (1 RCT) | NR | 699 per 1,000 (646 to 745 per 1,000) | 778 per 1,000 | ██ ███ █████ █████ ██ ████ ██ ███ ████ ███ ██████ | Moderateb | Pembrolizumab likely results in an increase in OS compared to placebo at 48 months |
DFS | |||||||
Probability of DFS at 24 months Median follow-up: 46.6 (range = 0.6 to 84.2) months | 726 (1 RCT) | NR | 550 per 1,000 (497 to 600 per 1,000) | 672 per 1,000 (619 to 719 per 1,000) | ███ ███ █████ █████ ██ ████ ██ ███ ████ ███ ██████ | Moderatec | Pembrolizumab likely results in a clinically important increase in DFS compared to placebo at 24 months |
Probability of DFS at 48 months Median follow-up: 46.6 (range = 0.6 to 84.2) months | 726 (1 RCT) | NR | 424 per 1,000 (367 to 479 per 1,000) | 512 per 1,000 (452 to 569 per 1,000) | ██ ███ █████ █████ █ ████ ██ ███ ████ ███ ██████ | Moderatec | Pembrolizumab likely results in a clinically important increase in DFS compared to placebo at 48 months |
HRQoL (measured with EORTC QLQ-C30 and EORTC QLQ-LC13) | |||||||
Proportion of patients with a ≥ 10-point deterioration in EORTC QLQ-C30 GHS/QoL score from baseline (0 [worst] to 100 [best]) Follow-up: 48 weeks | 1,161 (1 RCT) | NR | 129 per 1,000 (103 to 159 per 1,000) | 181 per 1,000 (151 to 215 per 1,000) | ██ ███ █████ ████ █████ ██ ████ ██ ██ ████ ███ ██████ | Highd,e | Pembrolizumab results in little to no clinically important difference in EORCT QLQ-C30 GHS/QoL compared to placebo |
Proportion of patients with a ≥ 10-point deterioration in EORTC QLQ-LC13 chest pain score from baseline (0 [best] to 100 [worst]) Follow-up: 48 weeks | 1,161 (1 RCT) | NR | 91 per 1,000 (69 to 118 per 1,000) | 74 per 1,000 (54 to 99 per 1,000) | ██ ███ █████ █████ █████ ██ █████ ██ ██ ████ ███ ██████ | Lowd,f | Pembrolizumab may result in little to no clinically important difference in EORTC QLQ-LC13 chest pain compared to placebo |
Proportion of patients with a ≥ 10-point deterioration in EORTC QLQ-LC13 coughing score from baseline (0 [best] to 100 [worst]) Follow-up: 48 weeks | 1,161 (1 RCT) | NR | 119 per 1,000 (94 to 148 per 1,000) | 117 per 1,000 (92 to 147 per 1,000) | ██ █████ █████ █████ ██ █████ ██ ██ ████ ███ ██████ | Lowd,f | Pembrolizumab may result in little to no clinically important difference in EORTC QLQ-LC13 coughing compared to placebo |
Proportion of patients with a ≥ 10-point deterioration in EORTC QLQ-LC13 dyspnea score from baseline (0 [best] to 100 [worst]) Follow-up: 48 weeks | 1,161 (1 RCT) | NR | 189 per 1,000 (158 to 224 per 1,000) | 221 per 1,000 (188 to 257 per 1,000) | ██ ███ █████ ████ █████ ██ █████ ██ ██ ████ ███ ██████ | Lowd,f | Pembrolizumab may result in little to no clinically important difference in EORTC QLQ-LC13 dyspnea compared to placebo |
Harms | |||||||
Grade 3 to 5 AEs Median follow-up: 46.7 (range = 0.6 to 84.2) months | 1,161 (1 RCT) | NR | 258 per 1,000 (NR) | 341 per 1,000 (NR) | ██ ███ █████ ████ █████ ██ ████ ██ ███ ████ ███ ██████ | Highd,e | Pembrolizumab likely results in an increase in grade 3 to 5 AEs compared to placebo. |
AE = adverse event; CDA-AMC = Canada’s Drug Agency; CI = confidence interval; DFS = disease-free survival; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-LC13 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Lung Cancer Module; GHS = Global Health Status; HRQoL = health-related quality of life; MID = minimal important difference; NR = not reported; NSCLC = non–small cell lung cancer; OS = overall survival; QoL = quality of life; RCT = randomized controlled trial; TPS = tumour proportion score.
Notes: Data presented in this table were based on analyses at clinical cut-off date of January 24, 2023. The OS data were not mature as of January 24, 2023. The between-group differences for all the outcomes in this table were requested from the sponsor to aid in interpretation and were not part of the sponsor’s analysis plan. Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aRated down 2 levels for very serious imprecision. At the data cut-off date, the OS results data were immature. An empirically derived and validated between-group MID for OS was not identified. Based on a threshold that is usually used by the CDA-AMC for effect assessment of an adjuvant treatment in patients with NSCLC of similar severity or stage, a between-group difference in the probability of OS of 5% may be clinically meaningful. The 95% CI of the absolute effect included the “no effect” threshold of 0 as well as the clinical importance threshold of 5%.
bRated down 1 level for serious imprecision. At the data cut-off date, the OS results data were immature. An empirically derived and validated between-group MID for OS was not identified. Based on a threshold that is usually used by the CDA-AMC for effect assessment of an adjuvant treatment in patients with NSCLC of similar severity or stage, a between-group difference in the probability of OS of 5% may be clinically meaningful. At 48 months, the 95% CIs of the absolute effect excluded the “no effect” threshold of 0, and the point estimate and the upper bound of the 95% CI suggest a clinical important increase in OS based on the threshold of 5%.
cRated down 1 level for serious imprecision. An empirically derived and validated between-group MID for DFS was not identified. Based on the threshold usually used by the CDA-AMC for effect assessment of an adjuvant treatment in patients with NSCLC of similar severity or stage, a between-group difference in the probability of DFS of 10% may be clinically meaningful. At both 24 months and 48 months, the 95% CIs of the absolute effect excluded the “no effect” threshold of 0, and the upper bound of the 95% CIs suggests a clinical important increase in DFS based on the threshold of 10%.
dIndirectness was not rated down. The outcome data for HRQoL and harms were based on the total population in the KEYNOTE-091 study. Although there may be uncertainties regarding the presence and magnitude of any potential differences in these outcomes between the total population and the reimbursement request population, the review team and the clinical experts consulted for this review did not identify major issues that would affect the study results as the PD-L1 TPS category was a stratification variable, and the patient characteristics appeared balanced between the treatment groups in both overall population and reimbursement request population, suggesting that the benefits of the randomization were reasonably maintained in the subpopulation for reimbursement request.
eImprecision did not result in the level of certainty being rated down, as the 95% CI of the absolute effect excluded the null threshold of 0. The clinical experts consulted for this review could not provide a threshold of important difference; however, the review team judged that the point estimate and 95% CI of the absolute effect were unlikely to include any important difference.
fRated down 2 levels for very serious imprecision. The review team was unable to identify the MID to assess a between-group difference from literature or the clinical experts consulted for this review; the null was therefore used to assess certainty. The 95% CI of the absolute effect included the “no effect” threshold of 0.
Sources: BARDS Health Technology Assessment Statistical Report: Baseline Characteristics and Efficacy (April 9, 2024),22 Clinical Study Report: P091V02MK3475,23 and sponsor’s submission.24
Long-Term Extension Studies
The KEYNOTE-091 study is ongoing for OS follow-up. No other long-term extension studies are currently ongoing or completed.
Indirect Comparisons
In the absence of direct head-to-head trials evaluating the comparative efficacy of pembrolizumab and atezolizumab for the adjuvant treatment of adult patients with early-stage NSCLC who have undergone complete resection and platinum-based chemotherapy, the sponsor conducted 1 indirect treatment comparison (ITC) including only the subpopulation of participants with a TPS of 50% or greater. The findings from this ITC were used to support the sponsor’s reimbursement request and a request for a deviation from pharmacoeconomic requirements that excludes this subpopulation.
Description of Studies
The sponsor included 2 studies, KEYNOTE-091 and IMpower010, in its ITC. For the KEYNOTE-091 study, the sponsor included only the ongoing trial patients after excluding patients who discontinued treatment. The sponsor did not report the median follow-up duration for this subpopulation; however, the median follow-up time for the intention-to-treat (ITT) population of patients with a PD-L1 TPS of 50% or greater who were treated with pembrolizumab was 46.8 months (range = 3.4 to 83.5). For the IMpower010 study, from the published data, the median follow-up time for patients with a PD-L1 TPS of 50% or greater who were treated with atezolizumab was 35.98 months (range = 0.2 to 54.2). The sponsor did not report any assessment of homogeneity or any handling of potential effect modifiers.
Efficacy Results
The ITC of pembrolizumab versus atezolizumab in patients with a TPS of 50% or greater and stage II to IIIA cancer and who had received prior adjuvant chemotherapy showed that pembrolizumab appears to be less effective than atezolizumab in this subpopulation, with an HR of ████ ████ ███ ████ ██ ██████.
Harms Results
Harms outcomes were not reported.
Critical Appraisal
The sponsor-submitted ITC was used to support its reimbursement request and request for deviation from pharmacoeconomic requirements that excludes this subpopulation of adult patients with early-stage NSCLC who have undergone complete resection and platinum-based chemotherapy with a TPS of 50% or greater. The sponsor did not conduct an additional ITC for the reimbursement request population (patients with a TPS of less than 50%). It did not conduct a systematic literature review for this ITC. The apparent 10-month difference in median follow-up time between both trials may have an impact on the time to event outcomes. The clinical experts consulted for this review commented that the baseline patient characteristics from both trials appeared to be well matched. However, the sponsor did not report or appear to assess homogeneity between the 2 studies, and could only include published aggregate level data from the Impower010 study. It is therefore unclear if sources of clinical or methodological heterogeneity biased effect estimates of ITC.
Harms outcomes and other outcomes of relevance to patients (e.g., HRQoL) were not reported.
The clinical experts noted that, while no therapy options are currently available after adjuvant chemotherapy for patients with resected stage IB to IIIA NSCLC who have a PD-L1 TPS of less than 50%, atezolizumab is currently the treatment of choice for patients with a TPS of 50% or greater. They added that the results from this ITC support the current therapy guidelines and the sponsor’s reimbursement request.
Studies Addressing Gaps in the Evidence From the Systematic Review
No studies addressing gaps in the pivotal and RCT evidence were identified for this review.
Conclusions
One triple-blind, phase III RCT comparing the efficacy and safety of adjuvant pembrolizumab and placebo in adult patients with stage IB to IIIA NSCLC who have undergone complete resection and platinum-based chemotherapy and whose tumours have a PD-L1 TPS of less than 50% showed a clinically meaningful benefit of adjuvant pembrolizumab in the probability of DFS at 24 and 48 months. Uncertainty remains in the OS results because the data were immature (the median OS was not reached in either group, with OS events observed in 23% and 30% of patients in the pembrolizumab and placebo groups, respectively), even though there is a trend toward improved OS in favour of pembrolizumab. The HRQoL of the patients in both the pembrolizumab and placebo groups was relatively stable over 48 weeks. According to the clinical experts consulted for this review, the safety profile of pembrolizumab was consistent with their expectations for this drug.
The sponsor submitted an ITC comparing pembrolizumab and atezolizumab in patients with a TPS of 50% or greater to support its reimbursement request for patients with a TPS of less than 50%. The indirect comparative evidence suggests that atezolizumab is superior in patients with a TPS of 50% or greater; however, it is unclear if sources of clinical or methodological heterogeneity biased the effect estimates in the ITC as no assessment of homogeneity was conducted. The sponsor did not submit an ITC comparing pembrolizumab and atezolizumab in patients with a TPS of less than 50% because, according to the clinician input, atezolizumab is not indicated for this subpopulation of patients.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of pembrolizumab administered by IV infusion as either 200 mg every 3 weeks or 400 mg every 6 weeks for the adjuvant treatment of adult patients with stage IB (T2a ≥ 4 cm), II, or IIIA NSCLC who have undergone complete resection and platinum-based chemotherapy and whose tumours have a PD-L1 TPS of less than 50%, as determined by a validated test.
Disease Background
Contents within this section were informed by materials submitted by the sponsor and clinical expert input. The following summary was validated by the CDA-AMC review team.
Lung cancer is the most diagnosed cancer and the leading cause of cancer deaths in Canada.1,2 Lung cancer survival at all stages and with all histologies is poor, with an overall 5-year net survival of 22%,2,3 and only 3% for those diagnosed with stage IV disease.2 In 2024, it was estimated that there would be 32,100 new cases of lung cancer diagnosed and 20,700 deaths from lung cancer.2 It is estimated that 1 in 21 Canadians (4.8%) will die from lung cancer.2
Lung cancer is classified into NSCLC or small-cell lung cancer, with NSCLC accounting for approximately 88% of all lung cancer cases in Canada.1 NSCLC is further classified by histology into squamous and nonsquamous (including adenocarcinoma, large-cell carcinoma, and other less common histologies).1 To determine a patient’s prognosis and treatment, NSCLC is staged using the AJCC staging criteria, which involves tumour-node-metastasis classification of the disease based on the size and spread of the primary tumour, lymph-node involvement, and occurrence of metastasis.25 The clinical experts consulted for this review pointed out that such changes in staging were made relatively recently as there has been some movement between stage I and II. As a consequence, comparing a more recent study’s results with previous trials may be problematic.
It is estimated that 30% to 35% of patients with NSCLC are diagnosed at an early stage (I to IIIA)4-6 and approximately 20% to 25% of patients with NSCLC have surgically resectable disease.7 After surgery, 45% of patients with stage IB disease and 76% of patients with stage III disease will experience disease recurrence and subsequently die over a median follow-up of 5 years, regardless of the use of ACT or immunotherapy.8 As NSCLC may be asymptomatic or minimally symptomatic, patients may have a late presentation.26 The most common symptoms include coughing, chest and shoulder pain, hemoptysis, weight loss, dyspnea, hoarseness, bone pain, fever, and recurring infections with bronchitis and pneumonia.26,27 Diagnostic procedures include lung imaging and tissue biopsy.28
Standards of Therapy
Contents within this section were informed by materials submitted by the sponsor and clinical expert input. The following summary was validated by the CDA-AMC review team.
Standard treatment for patients with stage IB to IIIA NSCLC, as staged by the AJCC,16 is surgical resection.9 Perioperative treatments (neoadjuvant or adjuvant) are used depending on the stage. According to the clinical experts consulted by the review team, the goal of any adjuvant therapy following complete resection for early-stage NSCLC is to improve cure rates by reducing the risk of relapse, after which no curative therapies are currently available.
In the perioperative setting, the current treatment standard for patients with resectable NSCLC without actionable oncogenic alterations is neoadjuvant platinum-doublet chemotherapy in combination with immunotherapy (nivolumab), or adjuvant platinum-doublet chemotherapy followed by immunotherapy (atezolizumab for patients with a PD-L1 TPS of 50% or greater).11,13 However, the clinical experts consulted for this review indicated that there is currently no consensus on whether neoadjuvant or adjuvant therapy is best, or if a combination of the 2 is beneficial. The clinical experts noted that the recommended neoadjuvant regimen for patients with resectable stage IB to IIIA NSCLC is 3 cycles of platinum-doublet chemotherapy in combination with nivolumab immunotherapy, and the adjuvant regimen is platinum-doublet chemotherapy, followed by atezolizumab immunotherapy if eligible. Neoadjuvant nivolumab is indicated for patients with tumours measuring 4 cm or greater or those that are node-positive, while adjuvant atezolizumab is indicated for patients with a PD-L1 TPS of 50% or greater.29,30 However, the clinical experts indicated that not all patients receive immunotherapy or platinum-based chemotherapy in the perioperative setting as they may decline, not be offered a referral to medical oncology, or are ineligible. Additionally, the experts noted that patients with a PD-L1 TPS of less than 50% are not eligible for adjuvant atezolizumab, and these patients currently have no adjuvant immunotherapy options. There is a need to improve the rate of recurrences in early-stage NSCLC, as well as provide options for those who have not had neoadjuvant immunochemotherapy and are ineligible for adjuvant atezolizumab (as their tumours have a PD-L1 TPS of less than 50%) to cure disease, delay disease recurrence, improve survival, and maintain QoL.10,14,15
Drug Under Review
The key characteristics of pembrolizumab are summarized in Table 3, with other treatments available as monotherapy for the adjuvant treatment of adult patients with stage IB (T2a ≥ 4 cm), II, or IIIA NSCLC who have undergone complete resection and platinum-based chemotherapy. The recommended dose of pembrolizumab in adults is 200 mg every 3 weeks or 400 mg every 6 weeks, administered as an IV infusion over 30 minutes.31
Pembrolizumab is a high-affinity antibody against PD-1, an immune checkpoint receptor that limits T-lymphocyte activity in peripheral tissues.31 Pembrolizumab reactivates tumour-specific cytotoxic T lymphocytes in the tumour microenvironment by exerting a dual-ligand blockade of the PD-1 pathway, including both PD-L1 and PD-L2, on antigen-presenting or tumour cells.31
Pembrolizumab has been approved by Health Canada as monotherapy for the adjuvant treatment of adult patients with stage IB (T2a ≥ 4 cm), II, or IIIA NSCLC who have undergone complete resection and platinum-based chemotherapy.31 The sponsor has requested reimbursement for pembrolizumab as monotherapy for the adjuvant treatment of adult patients with stage IB (T2a ≥ 4 cm), II, or IIIA NSCLC, and with a PD-L1 TPS of less than 50% who have undergone complete resection and platinum-based chemotherapy.
Table 3: Key Characteristics of Pembrolizumab and Atezolizumab
Characteristic | Pembrolizumab | Atezolizumab |
|---|---|---|
Mechanism of action | High-affinity antibody that inhibits the PD-1 receptor on antigen-presenting or tumour cells, reactivating tumour-specific cytotoxic T lymphocytes in the tumour microenvironment | Monoclonal antibody that binds directly to PD-L1 to block interaction with PD-1 and B7.1 receptors, reactivating the antitumour immune response while leaving the PD-L1/PD-1 interaction intact |
Indicationa | As monotherapy for adjuvant treatment of adult patients with stage IB (T2a ≥ 4 cm), II, or IIIA NSCLC who have undergone complete resection and platinum-based chemotherapy | As monotherapy for adjuvant treatment following complete resection and no progression after platinum-based adjuvant chemotherapy for adult patients with stage II to IIIA NSCLC whose tumours have PD-L1 expression of ≥ 50% of tumour cells |
Route of administration | IV infusion over 30 minutes | IV infusion |
Recommended dose | 200 mg every 3 weeks or 400 mg every 6 weeks | 840 mg every 2 weeks, 1,200 mg every 3 weeks, or 1,680 mg every 4 weeks |
Serious adverse effects or safety issues | Warnings and precautions Immune-mediated adverse reactions:
Infusion-related reactions:
Complications of allogeneic HSCT:
Embryo-fetal toxicity:
Adverse reactions
| Warnings and precautions Immune-mediated adverse reactions:
Infusion-related reactions:
Complications of allogeneic HSCT:
Ophthalmologic:
Embryo-fetal toxicity:
Adverse reactions
|
HSCT = hematopoietic stem-cell transplant; NSCLC = non–small cell lung cancer.
aHealth Canada–approved indication.
Sources: Product monographs.29,31
Testing Procedure Considerations
Tumour PD-L1 expression is determined using immunohistochemistry, which uses antibodies to detect the presence of specific biomarkers. PD-L1 testing results are commonly reported as the percentage of tumour cells that stain in the presence of an antibody, known as the TPS.32,33 Approximately 72% to 85% of patients with early-stage NSCLC have a PD-L1 TPS of 49% or greater.34 Multiple immunohistochemistry testing platforms are available for detecting and measuring PD-L1 expression in NSCLC, each co-developed as a companion or complementary diagnostic for a specific immune checkpoint inhibitor.32,33,35 According to the clinical experts consulted by the review team, the 22C3 clone, used to determine PD-L1 expression status in the KEYNOTE-091 study, is a standard assay used in Canada.9
Testing for PD-L1 TPS at diagnosis is the recommended standard of care for all patients with NSCLC.13,36 The clinical experts consulted by the review team verified that, in Canada, PD-L1 testing is routinely conducted from the biopsy sample. If no biopsy is performed, PD-L1 expression can be assessed using tissue from the surgically resected tumour. The clinical experts estimated that 95% of patients in Canada with NSCLC are tested for PD-L1 expression, with this proportion approaching 100% for patients seen in cancer centres.
We considered the potential impacts of PD-L1 TPS testing to determine eligibility for adjuvant treatment with pembrolizumab for patients with stage IB to IIIA NSCLC following complete tumour resection and platinum-based chemotherapy. We considered impacts on health systems and patients (including families and caregivers), and costs; these impacts are not anticipated to be substantial. Key considerations and relevant information available from materials submitted by the sponsor, input from the clinical experts consulted by the review team, and sources from the literature were validated by the review team when possible and are summarized in Table 4.
Table 4: Considerations for PD-L1 Testing for Establishing Treatment Eligibility for Pembrolizumab in Stage IB to IIIA NSCLC with Complete Resection and Platinum-Based Chemotherapy
Consideration | Criterion | Available information |
|---|---|---|
Health system–related | Number of individuals in Canada expected to require the test (e.g., per year) | The clinical experts consulted by the review team estimated that 20,000 incident patients with NSCLC will be reflexively tested for PD-L1 TPS annually in Canada (excluding Québec).37 Because PD-L1 TPS testing is already part of routine care for NSCLC regardless of anticipated treatment plan, no additional impact on health systems is expected as part of establishing treatment eligibility for pembrolizumab. |
Availability of the testing procedure in jurisdictions across Canada | According to the clinical experts, PD-L1 TPS testing is broadly available across Canada as part of the current standard of care for NSCLC. | |
Testing procedure as part of routine care | According to the clinical experts, PD-L1 TPS testing is currently performed as part of the diagnostic standard of care for all stages and histologic subtypes of NSCLC. | |
Repeat testing requirements | According to the clinical experts, testing for PD-L1 TPS in patients with NSCLC is performed once and would likely not need to be repeated. | |
Impact on human and other health care resources by provision of the testing procedure | Testing for PD-L1 TPS is currently part of the standard of care for NSCLC and is publicly funded across jurisdictions. No additional substantial impact on human and other health care resources is anticipated by provision of the testing procedure. | |
Patient-related | Accessibility of the testing procedure in jurisdictions across Canada | Because PD-L1 TPS testing is part of the current diagnostic standard of care for NSCLC in Canada, no additional access implications are anticipated from the testing as part of establishing treatment eligibility for pembrolizumab. |
Expected turnaround time for the testing procedure | The turnaround time for PD-L1 TPS testing results can be up to 8 weeks.34 According to the clinical experts, PD-L1 TPS testing is generally done early in the diagnostic pathway, well before adjuvant therapy is initiated. They anticipated no or minimal additional impact for patients or caregivers due to wait times. | |
Burden associated with the testing procedure for patients, families, and/or caregivers | Because testing for PD-L1 TPS is currently part of the standard of care for NSCLC, no additional burden to patients, families, and/or caregivers is anticipated from the testing as part of establishing treatment eligibility for pembrolizumab. | |
Clinical | Clinical utility and validity of the testing procedure | Some evidencea demonstrates the diagnostic accuracy and clinical utility of immunohistochemistry testing for PD-L1 TPS using a validated test.35,38 PD-L1 assays can be interchangeable if the therapeutic drug–specific cutoff is used when interpreting the results.32,33 |
Risks of harm associated with the testing procedure | Because testing for PD-L1 TPS is currently part of the standard of care for NSCLC, there is no additional risk of harm associated from the testing as part of establishing treatment eligibility for pembrolizumab. | |
Cost | Projected cost of the testing procedure | The cost to assess PD-L1 TPS is about $105 per test.34 Because testing for PD-L1 TPS is currently part of the standard of care for NSCLC, no additional cost implications are anticipated from the testing as part of establishing treatment eligibility for pembrolizumab. |
NSCLC = non–small cell lung cancer; TPS = tumour proportion score.
aCanada’s Drug Agency has not evaluated or critically appraised this evidence to determine its validity or reliability.
Perspectives of Patients, Clinicians, and Drug Programs
Patient Group Input
This section was prepared by the CDA-AMC review team based on the input provided by patient groups. The full original patient inputs received by CDA-AMC have been included in the Perspectives of Patients, Clinicians, and Drug Programs section of this report.
This review received a joint submission by 3 patient groups, the LHF, LCC, and the Canadian Cancer Survivor Network. The input was based on information collected by the LHF from individuals living with lung cancer, conducting 3 interviews with patients from Ottawa, Vancouver and Toronto who had experience with pembrolizumab, and gathering 33 responses to an online survey available between June 2023 to June 2024.
When asked about their disease experience, interviewees reported substantial challenges obtaining an accurate and timely diagnosis, which resulted in substantial declines in quality of life until diagnosis. Most had difficulty verbally communicating during this period due to interruptions by coughing fits, which resolved for some upon treatment initiation. Survey respondents reported similar symptoms and challenges due to their lung cancer, some of which included: fatigue (53%), shortness of breath (50%), cough (23%), and pain (20%). Respondents also noted chest tightness, wheezing, weight loss, diminished appetite, low mood and/or depressive periods, and challenges with physical and emotional intimacy. When asked how lung cancer negatively affects their day-to-day life, respondents highlighted their inability to work (48%), inability to participate in physical activities (33%), do housework (21%), use stairs (22%), or engage in hobbies (21%). Most respondents indicated that living with lung cancer negatively affects their emotional well-being through feelings of isolation, challenges with symptom management, and perceived burden on caregivers and family. Disease aspects that were most important to responders to control included improved management of disease symptoms, as well as pain and side effects from therapy.
Respondents reported some benefit from previous treatment with alectinib, lorlatinib, metoclopramide, gefitinib, entrectinib, osimertinib, and brigatinib, such as reduced cough and shortness of breath, increased participation in daily activities, ability to exercise, prolonged life, delayed disease progression and a reduction in the severity of other disease-related symptoms. The input also noted that patients on oral therapies value the flexibility such therapies provide in allowing them to work and travel without restrictions. However, respondents reported struggling with lingering side effects. Patients who previously received surgery reported experiencing deconditioning and chronic fatigue, and medication side effects included extreme itching that affects sleep, brain fog, fatigue, nausea, vomiting, mood changes, diminished appetite, weight loss, hair loss, anemia, and neuropathy. These patients also experienced challenges accessing some therapies due to high treatment costs, as well as difficulty navigating the health care system and locating disease information and support. The input also noted concerns from patients on targeted therapy as to their ability to access the next line of treatment if or when their current therapy stops working.
Respondents indicated that key treatment outcomes to consider when evaluating new therapies include stopping or slowing disease progression with minimal side effects, and effectiveness in advanced disease. Respondents also highlighted efficacy as an outcome of interest, with 1 respondent noting they would be more receptive to side effects if there was robust evidence that the medication would stop or slow their lung cancer progression.
Three LHF interviewees had experience with the drug under review. These patients (2 of whom had NSCLC with PD-L1 expression) were diagnosed with stage IV lung cancer and were initiated on pembrolizumab shortly after surgical resection. However, these patients were not part of the eligible population of the current indication under review, which is limited to stage IB (T2a ≥ 4 cm), II, or IIIA NSCLC. One patient was diagnosed with stage I lung cancer, received surgery, and was initiated on pembrolizumab after progression to stage IV following a 3-year disease-free period. One interviewee with EGFR-positive, stage IV lung cancer was taking pembrolizumab in combination with chemotherapy, although it was unclear if the interviewees with NSCLC were taking pembrolizumab similarly or as monotherapy. One patient discontinued pembrolizumab after 19 months due to progression, while another experienced sufficient tumour shrinkage and inactivity to discontinue pembrolizumab after 3 years, before re-initiating shortly after tumour reactivation. After re-initiation of pembrolizumab, the patient’s tumours once again decreased in size. Side effects reported by these patients included nausea, fatigue, muscle soreness, constipation, diarrhea, and worsening of their diabetes, eczema, and liver enzymes. Patients did not report that these side effects impeded their ability to participate in activities of daily living or exercise and, overall, described an improved QoL while on therapy.
Clinician Input
Input From Clinical Experts Consulted by CDA-AMC
All CDA-AMC review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of lung cancer.
Unmet Needs
According to the clinical experts consulted by the review team, the key treatment goals for patients with early-stage NSCLC who receive adjuvant therapy following complete surgical resection is to improve cure rates through reducing risk of relapse.
The clinical experts noted that a risk of recurrence remains even after complete resection. The experts highlighted 2 approaches to curative-intent treatment: neoadjuvant therapy and adjuvant therapy. For patients with resectable NSCLC without actionable mutations, the current patient management in Canada is neoadjuvant platinum-based chemotherapy in combination with nivolumab, or adjuvant platinum-based chemotherapy, followed by atezolizumab if they have a PD-L1 TPS of 50% or greater. The clinical experts added that no adjuvant immunotherapy options are available for patients with stage IB to IIIA NSCLC who have a PD-L1 TPS of less than 50% in Canada, defining an unmet need for this group.
Place in Therapy
According to the clinical experts consulted by the review team, pembrolizumab would represent a new adjuvant immunotherapy option for patients with resected stage IB to IIIA NSCLC who have a PD-L1 TPS of less than 50% and who have not had access to neoadjuvant immunochemotherapy. The clinical experts agreed that adjuvant pembrolizumab would fill a treatment gap, as currently patients can only access this treatment after relapse.
Patient Population
The clinical experts consulted for this review indicated that the patient population most suited for treatment with adjuvant pembrolizumab are those with resectable NSCLC and a PD-L1 TPS of less than 50% using a validated PD-L1 assay.
Assessing the Response Treatment
Both clinical experts consulted by the review team agreed that the gold-standard outcome to use when determining a patient’s response to treatment in clinical practice will be overall survival.
Discontinuing Treatment
Clinical experts consulted for this review indicated that pembrolizumab treatment should be discontinued if 1 of the following has been met: a total of 18 cycles (1 year) of adjuvant immunotherapy has been completed, disease progression has been detected, or unacceptable toxicity develops. One expert noted that toxicity related to gastrointestinal, skin, lung, heart, central nervous system, or endocrine functions is particularly important when determining whether to discontinue therapy.
Prescribing Considerations
The clinical experts noted that pembrolizumab should be administered in a specialty setting by multidisciplinary surgical and medical oncology staff with the expertise to administer systemic therapy, monitor the patient, and manage treatment-related toxicities.
Clinician Group Input
This section was prepared by the CDA-AMC review team based on the input provided by clinician groups. The full original clinician group inputs received by the CDA-AMC review team have been included in the Perspective of Patients, Clinicians, and Drug Programs section of this report.
Clinician group input on this review was received from 2 clinician groups: The Ontario Health (Cancer Care Ontario Drug Advisory Committee and LCC. Six clinicians from the Drug Advisory Committee and 35 clinicians from LCC provided input for this review.
For the treatment of resected stage IB to IIIA NSCLC, the LCC group indicated that chemotherapy, immunotherapy, radiation, and targeted drugs play a key role in improving outcomes and cure rates. Aligning with clinical expert input, the Drug Advisory Committee emphasized that, in the subset of patients with resected stage IB to IIIA NSCLC who have a PD-L1 of less than 50%, there are currently no other therapy options available after adjuvant chemotherapy, creating an urgent unmet need for this patient population. The LCC group also highlighted that, in patients with early-stage resected IB to IIIA NSCLC, current therapies do not achieve sufficiently high cure rates or prevent recurrences. The LCC group described this as particularly important for patients with NSCLC, as the risk of relapse substantially increases with each subsequent disease stage. The LCC group also emphasized that patient relapse and metastatic disease also come with substantial costs to patient health, QoL, utilization of health care resources, economic loss of productivity, and overall costs to society.
Aligning with clinical expert input, the clinicians agreed that pembrolizumab is an option for patients with stage IB to IIIA NSCLC with a PD-L1 TPS of less than 50% and proposed using it as a second adjuvant therapy following adjuvant chemotherapy. The LCC group expected that pembrolizumab will shift the current treatment paradigm, as it represents the first adjuvant immunotherapy option for this patient population. Clinician groups agreed that all patients with stage IB to IIIA NSCLC who have undergone complete resection and who have a PD-L1 TPS of less than 50% would be suitable for pembrolizumab in the absence of contraindications to immunotherapy. The LCC group expected that clinicians will continue to treat patients with a PD-L1 TPS of 50% or greater or ALK translocations with other available drugs that have demonstrated superior efficacy in these subgroups. The LCC group also pointed out that treatment with pembrolizumab for eligible patients with a sensitizing EGFR mutation should be considered on case-by-case basis by treating clinicians, weighing the risks and benefits of adjuvant sequential chemotherapy and immunotherapy versus adjuvant osimertinib. The Ontario Health Drug Advisory Committee and clinical experts agreed that pembrolizumab should be considered for patients who are ineligible for chemotherapy, as study patients were permitted to receive it even without chemotherapy.
The groups agreed that treatment benefit in the adjuvant setting is primarily determined by disease recurrence, which the LCC group noted typically occurs within 2 to 3 years for patients with stage IB to IIIA NSCLC. The LCC group indicated that cure rates, as measured by 5-year OS, can also determine response, but typically require even more years of follow up. The LCC group suggested implementing clinical and laboratory follow up every 3 weeks to evaluate toxicity and disease recurrence, as well as imaging scans at 3- to 4-month intervals, as pembrolizumab is administered over 1 year. The LCC group noted that, overall, immunotherapies are well tolerated by patients, and autoimmune side effects can often be readily managed by oncologists. Both clinician groups agreed that treatment could be delivered in an outpatient setting under the supervision of a medical oncologist; however, the Drug Advisory Committee suggested that a pulmonologist experienced in managing thoracic malignancies could also diagnose, treat, and monitor patients on pembrolizumab. Both clinician groups, as well as the consulted clinical experts, agreed that treatment would be discontinued in the event of disease recurrence or progression, SAEs, or completion of therapy after 18 cycles.
Drug Program Input
The drug programs provide input on each drug being reviewed through the CDA-AMC reimbursement review process by identifying issues that may affect their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by the CDA-AMC review team are summarized in Table 5.
Table 5: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
Pembrolizumab is an add-on to ACT and other currently available adjuvant therapies and is not expected to replace atezolizumab or osimertinib in the adjuvant setting There was an indirect treatment comparison study vs. atezolizumab. There is no direct comparator for the adjuvant treatment of NSCLC with stage IB (T2a ≥ 4cm), II, or IIIA NSCLC who have undergone complete resections and platinum-based chemotherapy with PD-L1 < 50%. | This is a comment from the drug plans to inform pERC deliberations. |
Considerations for initiation of therapy | |
Exclusion criteria in the KEYNOTE-091 study:
Would patients who had planned to receive neoadjuvant or adjuvant radiotherapy or chemotherapy be eligible for treatment? | Based on the inclusion and exclusion criteria in the KEYNOTE-091 study as well as the clinical practice in Canada, the following patients would be eligible to receive treatment with pembrolizumab:
ACT was not mandatory in the pivotal trial but considered for patients with stage IB (T2a ≥ 4 cm) and strongly recommended for those with stage II and IIIA NSCLC and was administered according to national and local guidelines. The following patients would not be eligible to receive treatment with pembrolizumab:
In addition, adjuvant radiotherapy is not currently the standard of care for the patients in Canada. As such, the impact on application of not recommending patients receiving adjuvant radiotherapy would be minimal. |
In the KEYNOTE-091 study, patients must undergo complete resection of their NSCLC (lobectomy, sleeve lobectomy, bi-lobectomy, or pneumonectomy) Resection margins proved microscopically free (R0). Can patients with a partial resection be eligible for treatment? If a patient’s complete resection failed, would they be eligible to receive this treatment? In the KEYNOTE-091 study, patients should also complete a maximum of 4 cycles of ACT with a platinum-based regimen. The first dose of pembrolizumab should be administered at least 3 weeks but no more than 12 weeks after the last dose of ACT. If there are delays in treatment or complications with surgery, would patients be eligible for treatment > 12 weeks after the last dose of ACT? What conditions or time frame would be advisable? | If a patient does not have a complete resection, they would not be eligible to receive pembrolizumab. It is usually recommended ACT start within 8 weeks surgery; and an immunotherapy (e.g., pembrolizumab) can be started within 12 weeks of ACT completion. For patients who have not had 4 full cycles of platinum-based chemotherapy ACT, conditions may preclude them from having an immunotherapy. Some examples of these conditions include patients’ refusal, treatment toxicity (i.e., neurologic or auditory AEs), and eligibility of receiving an immunotherapy (e.g., renal issues). Patients receiving adjuvant chemotherapy should receive no more than 4 cycles of adjuvant chemotherapy and have their immunotherapy start within 12 weeks of completion of adjuvant chemotherapy, based on the patients included in the KEYNOTE-091 study (i.e., patients who are not receiving adjuvant chemotherapy should receive their immunotherapy within 12 weeks of surgery). |
Should patients who complete 1 year of treatment and experience disease progression/recurrence off of pembrolizumab treatment be eligible for up to 1 year (18 cycles) of pembrolizumab re-treatment? | This question deals with the management of relapsed disease, and the evidence from the KEYNOTE-091 study is not applicable. In clinical practice in Canada, patients in the incurable setting can receive up to 2 years of pembrolizumab, when a patient relapses 6 months or later after completion of their adjuvant immunotherapy. |
Considerations for discontinuation of therapy | |
The study treated patients for 18 doses (for approximately 1 year). Should therapy end after 18 doses or 1 year, whichever comes first? | Pembrolizumab should end after 18 doses or 1 year, whichever comes first. |
Considerations for prescribing of therapy | |
Jurisdictions utilize weight-based dosing to a cap: 2 mg/kg to a maximum of 200 mg every 3 weeks or 4 mg/kg to a maximum of 400 mg every 6 weeks, as is outlined in other indications. | This is a comment from the drug plans to inform pERC deliberations. |
Consider alignment with atezolizumab criteria: For patients who have received a full course of treatment with nivolumab (i.e., 3 cycles) in combination with platinum-doublet chemotherapy in the NAT setting, the expert panel acknowledges that further immunotherapy in the adjuvant setting is not supported by the available evidence and most jurisdictions restrict this use for atezolizumab. | This is a comment from the drug plans to inform pERC deliberations. |
Generalizability | |
The study was in patients with an ECOG PS of 0 to 1. Can patients with an ECOG status > 1 be eligible to receive treatment? | Although there is a lack in data in patients with an ECOG PS of 2 based on the KEYNOTE-091 study, patients with an ECOG PS of 0 to 2 would be eligible for treatment with pembrolizumab, because in clinical practice in Canada, treatment with pembrolizumab is often offered to patients with an ECOG PS of up to 2. Clinicians need to evaluate patients with an ECOG PS of 2 thoroughly, and following clinicians’ assessment, consider the patients’ individual status when considering treating those with an ECOG PS of 2 with pembrolizumab and ensure close follow-up and compliance. |
Funding algorithm | |
Complex therapeutic space with multiple lines of therapy, subpopulations, or competing products. | This is a comment from the drug plans to inform pERC deliberations. |
Care provision issues | |
PD-L1 testing is widely available at many institutions. | This is a comment from the drug plans to inform pERC deliberations. |
ACT = adjuvant chemotherapy; AE = adverse event; ECOG = Eastern Cooperative Oncology Group Performance Status; NAT = neoadjuvant treatment; NSCLC = non–small cell lung cancer; pERC = pan-Canadian Oncology Drug Review Expert Review Committee; vs. = versus.
Clinical Evidence
The objective of this CDA-AMC Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of pembrolizumab (administered as an IV infusion of either 200 mg every 3 weeks or 400 mg every 6 weeks) for the adjuvant treatment of adult patients with stage IB (T2a ≥ 4 cm), II, or IIIA NSCLC who have undergone complete resection and platinum-based chemotherapy and whose tumours have a PD-L1 TPS of less than 50%, as determined by a validated test. The focus will be placed on comparing pembrolizumab to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of pembrolizumab is presented in 4 sections, with the CDA-AMC critical appraisal of the evidence included at the end of each section. The first section, the systematic review, includes pivotal studies and RCTs that were selected according to the sponsor’s systematic review protocol. The CDA-AMC assessment of the certainty of the evidence in this first section using the GRADE approach follows the critical appraisal of the evidence. The second section includes sponsor-submitted long-term extension studies. The third section includes indirect evidence from the sponsor. The fourth section includes additional studies that were considered by the sponsor to address important gaps in the systematic review evidence. No long-term extension studies (Section 2) or additional studies to address important gaps in the systematic review evidence (Section 4) were submitted by the sponsor.
Included Studies
Clinical evidence from the following is included in the CDA-AMC review and appraised in this document:
1 pivotal placebo-controlled RCT
1 indirect treatment comparison.
Systematic Review
Contents within this section have been informed by materials submitted by the sponsor. The following have been summarized and validated by the CDA-AMC review team.
Description of Studies
Characteristics of the included studies are summarized in Table 6.
Table 6: Details of Studies Included in the Systematic Review
Detail | KEYNOTE-091 |
|---|---|
Designs and populations | |
Study design | Phase III, triple-blind, placebo-controlled RCT |
Locations | 206 sites in 29 countries from continents of Australia, the Americas (Canada, 5 sites with 10 patients), Asia, and Europe |
Patient enrolment dates | Start: January 20, 2016 End: May 6, 2020 |
Data cut-off date | For this review: January 24, 2023 |
Randomized (N) | Total N = 1,177 Pembrolizumab = 590 Placebo = 587 |
Key inclusion criteria | Male and female patients aged at least 18 years were eligible to enrol in the study if they had:
|
Key exclusion criteria |
|
Drugs | |
Intervention | Pembrolizumab: IV infusion, 200 mg every 3 weeks for approximately 1 year (18 doses) |
Comparator | Placebo: IV infusion, 200 mg every 3 weeks for approximately 1 year (18 doses) |
Study duration | |
Screening phase | Maximum of 8 weeks from consent from signature to randomization |
Treatment phase | 1 year |
Follow-up phase | Safety and survival follow-up
|
Outcomes | |
Primary end point | DFS in PD-L1 TPS ≥ 50% or overall population |
Secondary and exploratory end points |
Tertiary and/or exploratory
|
Publication status | |
Publications | O’Brien et al. (2022)9 Clinicaltrials.gov Identifier: NCT02504372 |
DFS = disease-free survival; ECOG PS = Eastern Cooperative Oncology Group Performance Status; HRQoL = health-related quality of life; IHC = immunohistochemistry; NSCLC = non–small cell lung cancer; OS = overall survival; RCT = randomized controlled trial; TPS = tumour proportion score; UICC v7 = Union for International Cancer Control version 7.
Note: One additional report was included: O’Brien et al. (2022).9
Source: Clinical Study Report for P091V02MK3475.23
One pivotal, multicentre, triple-blind, placebo-controlled randomized phase III trial (KEYNOTE-091) met the inclusion criteria for the sponsor’s systemic review, in which a total of 206 sites randomized 1,177 patients (Table 6). The KEYNOTE-091 trial investigated the efficacy and safety of pembrolizumab compared with placebo as adjuvant therapy for completely resected stage IB to IIIA NSCLC.23,39 Eligible patients were randomized 1:1 to receive pembrolizumab or placebo using a central interactive voice-response system (Figure 1). Randomization was stratified by disease stage (IB versus II versus IIIA), receipt of ACT (no versus yes), PD-L1 TPS (< 1% [negative] versus 1% to 49% versus ≥ 50%) and geographic region (Asia versus Eastern Europe versus Western Europe versus the rest of the world). Patients, investigators, and individuals who collected or analyzed the data, including sponsor representatives, were masked to treatment assignment.39,40
Scans of the chest and upper abdomen were performed by CT within 12 weeks before randomization, 12 weeks after the first dose of study treatment, and every 12 weeks thereafter during year 1, every 6 months during years 2 and 3, annually during years 4 and 5, and according to local standard of care thereafter until disease recurrence or withdrawal of consent. CT or MRI scans of the brain were performed within 12 weeks before randomization, and only if clinically indicated thereafter. After treatment discontinuation, survival was assessed every 12 weeks through year 5 and every 6 months thereafter.39,40
The KEYNOTE-091 study is ongoing, and the results reported in this review are based on the protocol-prespecified IA3, for which the data cut-off date was January 24, 2023.23,39
Figure 1: Study Design for the KEYNOTE-091 Study
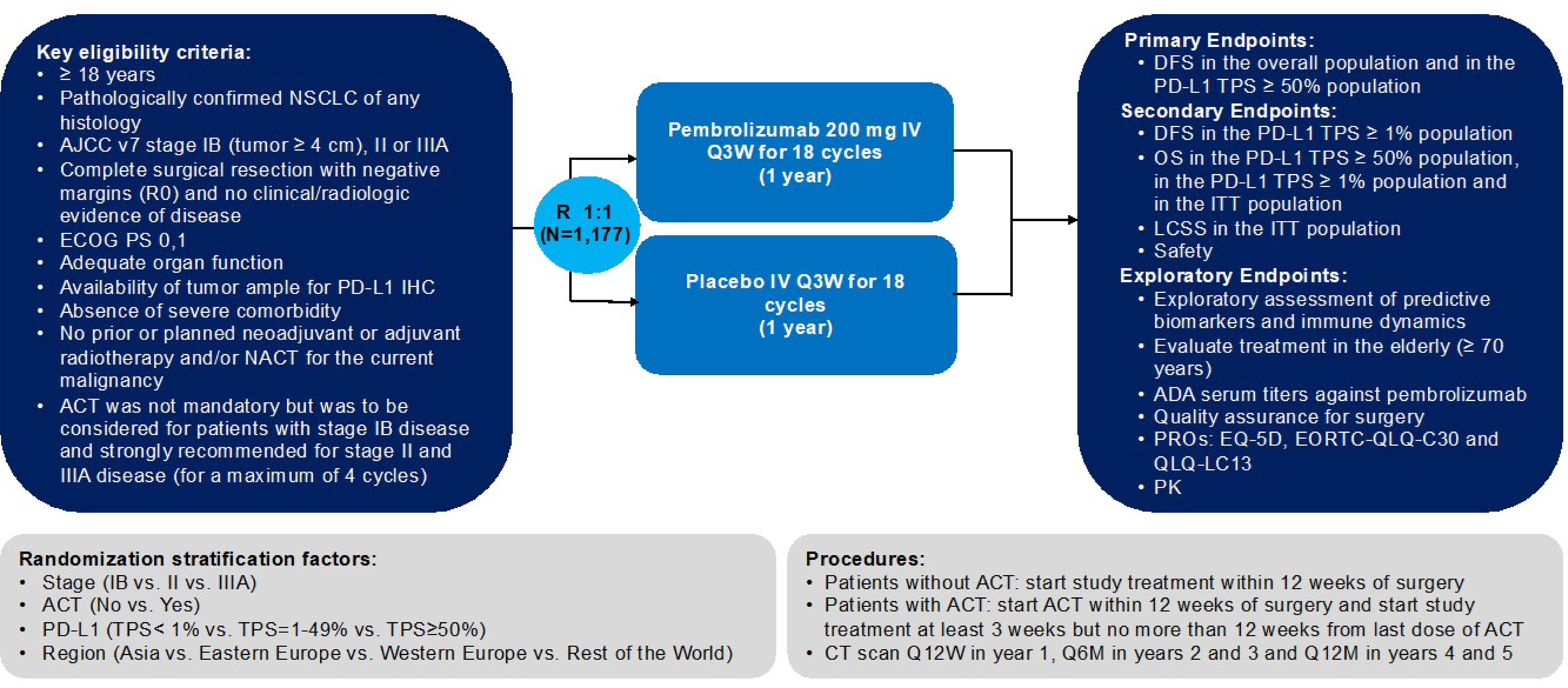
ACT = adjuvant chemotherapy; ADA = antidrug antibody; AJCC v7 = American Joint Committee on Cancer version 7; DFS = disease-free survival; ECOG PS = Eastern Cooperative Oncology Group Performance Status; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; IHC = immunochemistry; LCSS = lung cancer–specific survival; NACT = neoadjuvant chemotherapy; NSCLC = non–small cell lung cancer; OS = overall survival; PK = pharmacokinetics; PRO = patient-reported outcome; Q3W = every 3 weeks; Q6M = every 6 months; Q12M = every 12 months; Q12W = every 12 weeks; QLQ-LC13 Quality of Life Questionnaire Lung Cancer Module; TPS = tumour proportion score.
Source: Sponsor’s submission.39
Populations
Inclusion and Exclusion Criteria
In the KEYNOTE-091 study, eligible patients were aged 18 years or older, had pathologically confirmed NSCLC (any histology) of stage IB (T2a ≥ 4 cm), II, or IIIA according to the AJCC staging system16 after complete surgical resection (lobectomy, sleeve lobectomy, bi-lobectomy, or pneumonectomy) and negative margins (R0). Eligible patients had an available tumour sample obtained during resection for PD-L1 assessment, known PD-L1 expression status, no evidence of disease on clinical examination and radiographic assessment according to RECIST 1.1)17 as determined by local investigator review after surgery but within 12 weeks before randomization, an ECOG PS of 0 or 1, and adequate organ function within 10 days of treatment initiation. Previous neoadjuvant or adjuvant radiotherapy for the current malignancy was not permitted. ACT was not mandatory but was to be considered for patients with stage IB disease and strongly recommended for those with stage II or IIIA disease according to local practice and national guidelines. Patients without ACT had to receive their first study treatment dose within 12 weeks of surgery. Patients who received ACT had to initiate it within 12 weeks of surgery and receive a maximum of 4 chemotherapy cycles; the first dose of study treatment was administered at least 3 weeks but no more than 12 weeks after the last dose of chemotherapy. Patients with prior treatment with an anti–PD-1, anti–PD-L1 or anti–PD-L2, anti-CD137, CTLA-4 modulators, or any other immune-modulating drugs were excluded from the study.39,40
Interventions
Pembrolizumab 200 mg or placebo was administered by IV infusion on day 1 of every 3-week cycle until recurrence (as determined by the investigator according to RECIST 1.1),17 new malignancy, unacceptable toxicity, withdrawal of consent, completion of 18 cycles (approximately 1 year) of treatment or other reason (intercurrent illness, failure to comply with study treatment or procedure requirements, pregnancy, investigator’s decision, or administrative reasons).39,40
Outcomes
A list of efficacy end points assessed in this Clinical Review is provided in Table 7. Summarized end points are based on outcomes included in the sponsor’s Summary of Clinical Evidence as well as any outcomes identified as important to this review by the clinical experts consulted for this review and input from patient and clinician groups and public drug plans. Using the same considerations, the review team selected end points that were considered to be most relevant to inform the expert committee deliberations and finalized this list of end points in consultation with members of the expert committee. Select efficacy and harms outcomes that were considered important for informing the expert committee deliberations were assessed using GRADE.
Table 7: Outcomes Summarized From the KEYNOTE-091 Study
Outcome measure | Time point | KEYNOTE-091 |
|---|---|---|
DFS | DFS was defined as the time from randomization to either the date of disease recurrence or death (whatever the cause) as assessed by the investigator by database cut-off of January 24, 2023. The study is ongoing. Recurrence of disease was defined as local regional recurrence, a distant (metastatic) recurrence, or a second primary cancer. Occurrence of a second extra-pulmonary malignancy was considered to be an event. | Primarya |
OS | OS was defined as the time from randomization to the date of death (whatever the cause) by database cut-off of January 24, 2023. The study is ongoing. | Secondarya |
EORTC QLQ-C30 v3 | Baseline assessed during screening period (within 8 weeks to randomization) and every 12 weeks after treatment initiation during the first year; every 6 months during the second year, and then yearly until year 5. | Tertiary or exploratory |
EORTC QLQ-LC13 | Same as above | Tertiary or exploratory |
Grade 3 to 5 AEsb | Same as above | Secondary |
AE = adverse event; DFS = disease-free survival; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-LC13 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Quality of Life Questionnaire Lung Cancer Module; OS = overall survival.
aStatistical testing for these end points was adjusted for multiple comparisons.
bAEs were recorded according to the Common Terminology Criteria for Adverse Events version 4.03.
Source: Clinical Study Report for P091V02MK3475.23
In the KEYNOTE-091 study, the dual primary end points were DFS in the PD-L1 strong positive (TPS ≥ 50%) population or in the overall population. Secondary and exploratory end points included DFS in the population with a PD-L1 TPS of 1% or greater, OS in different populations, HRQoL, and safety, among others.39,40
Overall survival was defined as the time from randomization to death due to any cause.
Disease-free survival was defined as the time from randomization to locoregional or metastatic recurrence assessed under RECIST 1.117 by investigator review, appearance of a second primary NSCLC or other malignancy, or death from any cause.
A summary of EORTC QLQ-C30 and EORTC QLQ-LC13 results is provided in Table 8. All HRQoL questionnaires were filled out at the hospital during scheduled visits. Pre-treatment questionnaires were completed within 12 weeks before randomization. Subsequent questionnaires were filled in every 12 weeks during the first year after randomization (starting on day 1 cycle 1), every 6 months during the second year and then yearly until the fifth year. HRQoL data were collected regardless of the patient’s progression status, before the pembrolizumab infusion.
Any AEs, irrespective of causality, were reported from the time of treatment randomization through 30 days after the last dose of study treatment (intensity was assessed by the investigator according to the National Cancer Institute Common Terminology Criteria for Adverse Events, version 4.03).
Immune-mediated events and infusion-related reactions associated with pembrolizumab were considered AEOSIs. A predefined list of preferred terms was developed by the sponsor to consistently characterize the nature and frequency of each AEOSI regardless of causality as reported by investigators. These preferred terms are considered medically equivalent to the immune-mediated events and infusion-related reactions. AEOSIs were reported from the time of randomization through 90 days after the last dose of study treatment or 30 days following discontinuation of study treatment if the patient initiated a new anticancer treatment, whichever occurred first.
Considerations that informed the selection of efficacy outcomes to be summarized and assessed using GRADE include the following:
Survival outcomes were identified by the patient and clinician group input, and specified by the clinical experts consulted for this review to include OS and DFS. Of the Kaplan-Meier estimates of probability of the survival outcomes at multiple time points, those at 36 months and 48 months for OS and 24 months and 48 months for DFS were specified by the clinical experts.
HRQoL outcomes were identified by the patient and clinician group input, and specified by the clinical experts to include the EORTC QLQ-C30 GHS/QoL and the select EORTC QLQ-LC13 symptom scales (chest pain, coughing, and dyspnea).
Table 8: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
EORTC QLQ-C30 v3 | Cancer-specific self-reported measure of HRQoL 30-item questionnaire, consisting of 5 functional scales (physical, role, emotional, social, and cognitive), 9 symptom scales (fatigue, nausea/vomiting, pain, dyspnea, insomnia, appetite loss, constipation, diarrhea, and financial difficulties), and a GHS scale All scales range in score from 0 to 100; a higher score for functional scales and for GHS represents better functioning ability or HRQoL; a higher score for symptom scales represents a worsening of symptoms41 | In studies with lung cancer patients Validity: Moderate to strong correlations between the 5 QLQ-C30 functioning scales (r = 0.41 to 0.77); FACT-G and QLQ-C30 scales (r = 0.64 to 0.76);42 HADS with all QLQ-C30 functioning scales (r = 0.28 to 0.75); BPI scales with all QLQ-C30 scales except for nausea/vomiting (r = 0.20 to 0.72),43 supporting convergent validity Reliability: Cronbach alpha ranging from 0.56 to 0.93 with 7 scales having acceptable internal consistency (alpha > 0.70)44 Responsiveness: Group differences (improved vs. deteriorated based on ECOG PS) over 28 days between pre- and on-treatment periods showed a statistically difference in global quality of life (P < 0.01) scale; no such difference was identified in patients whose ECOG PS remained unchanged41 | In a study with NSCLC patients:45 MID estimates in score for improvement (deterioration) using the ECOG PS and weight change as anchors:
In a study with lung cancer patients: an anchor-based approach in which patients who reported “a little” change on the SSQ had subsequent changes on a scale of the EORTC QLQ-C30 of 5 to 10 points46 |
EORTC QLQ-LC13 | The QLQ-LC13 is a tumour-specific questionnaire used to supplement the EORTC QLQ-C30 and contains 13 items related to lung cancer symptoms and treatment side effects including: a 3-item scale assessing dyspnea and 9 single items: pain in chest, pain in arm or shoulder, pain in other parts, coughing, hemoptysis, sore mouth or tongue, dysphagia, peripheral neuropathy, and alopecia41 All scales range in score from 0 to 100; higher scores on the symptom scales indicate worse symptoms41 | Validity: Construct validity has been established between pain score and disease type (P < 0.001); based on ECOG PS, construct validity was confirmed in dyspnea, coughing, and pain (P < 0.001) scores;47 correlation between spirometry result and dyspnea score was found to be weak (r = 0.24); BPI intensity score and QLQ-LC13 pain score were found to be modestly correlated (r > 0.4)43 Reliability: Good internal consistency reliability for the dyspnea multi-item scale (alpha = 0.81);47 however, internal consistency was found to be unacceptable for pain scores (alpha = 0.53 to 0.54) when QLQ-LC13 was used alone without QLQ-C30 questionnaire pain items;47 reliability estimate for dyspnea scale has been confirmed to be acceptable, i.e., alpha = 0.76 in another study43 Responsiveness: Dyspnea, coughing, and pain scores improved significantly over time between pre-treatment and on-treatment period (P < 0.001 for all except for extra thoracic pain which showed P < 0.05); responsiveness of chest pain (P < 0.01), dyspnea (P < 0.001) and coughing (P < 0.001) to change in ECOG PS was also noted47 | No relevant studies on MID in patients with NSCLC were identified For the sponsor-submitted study, a 10-point or more change in score in categorical definitions of improved, stable, and deteriorated status was used for subscales from the QLQ-LC1323,39 |
BPI = Brief Pain Inventory; ECOG PS = Eastern Cooperative Oncology Group Performance Status; EORTC = European Organisation for Research and Treatment of Cancer; FACT-G = Functional Assessment of Cancer Therapy–General; GHS = Global Health Status; HADS = Hospital Anxiety and Depression Scale; HRQoL = health-related quality of life; MID = minimal important difference; NSCLC = non–small cell lung cancer; QLQ-C30 = Quality of Life Questionnaire Core 30; QLQ-LC13 = Quality of Life Questionnaire Lung Cancer Module; SSQ = subjective significance questionnaire; vs. = versus.
Statistical Analysis
Clinical Trial End Points
Estimates of the median OS, and DFS were obtained by the Kaplan-Meier technique. The 95% CIs for the medians were calculated using the reflected CI. Estimates of the event-free rate at a fixed time point were obtained using the Kaplan-Meier technique and 95% CIs were calculated by the Greenwood formula for standard deviation. Estimates of HRs and their 95% CI were obtained by Cox regression.39,40 For patient-reported HRQoL outcomes, no PRO analyses were planned in any subpopulation.39 Details about the statistical analyses for each of the selected end points are presented in Table 9.
Table 9: Statistical Analysis of Efficacy End Points for the KEYNOTE-091 Study (All Patients)
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
OS | Wald test with multivariate Cox regression model with the Efron tie-handling method | Stage, PD-L1 expression, adjuvant chemotherapy, regions, and additional factors, including histology and smoking status | Censored at the date patient last known to be alive |
|
DFS | Primary approach: permutation test with multivariate Cox regression model with the Efron tie handling method Secondary approach:
| Primary approach: Adjusted by stage (IB vs. II vs. IIIA), PD-L1 IHC expression (0 vs. 1% to 49% vs. ≥ 50%), adjuvant chemo (no chemotherapy vs. adjuvant platinum-based chemotherapy), histology (squamous vs. nonsquamous), smoking status (smokers vs. nonsmokers), and regions (Western Europe vs. Eastern Europe vs. rest of the world vs. Asia) | Event at earlier date of documented recurrence and death censored at last disease assessment for patients with no recurrence and no death |
|
HRQoL (EORTC QLQ-C30 v3, EORTC QLQ-LC13) | Unstratified Miettinen and Nurminen test, least squares mean change in scores | NR | NR | NR |
DFS = disease-free survival; EORTC QLQ-C30 v3 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 version 3; EORTC QLQ-LC13 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Lung Cancer Module; HRQoL = health-related quality of life; NR = not reported; OS = overall survival; vs. = versus.
Sources: Clinical Study Report: P091V02MK347523 and sponsor’s submission.24
Sample Size and Power Calculation
The KEYNOTE-091 study was designed to enroll approximately 1,180 patients randomized 1:1 into the experimental and control arms. For DFS, the study has approximately 86% power at an alpha of 1.25% (1-sided) and approximately 92% power at an alpha of 2.5% (1-sided) in the overall study population based on a target number of approximately 551 events at final analysis. It was expected that there would be approximately 334 patients with a PD-L1 TPS of 50% or greater in the study to achieve about 90% power for DFS at an alpha of 1.25% (1-sided) and approximately 94% power at an alpha of 2.5% (1-sided) in the final DFS analysis in this subgroup.
Statistical Testing
The KEYNOTE-091 study was designed with 2 primary end points: DFS in the whole population and DFS in the subgroup with a PD-L1 TPS of 50% or greater. The 1-sided family-wise error rate for DFS and OS hypotheses was strongly controlled at 2.5%.23 The study used the graphic method of Maurer and Bretz to provide strong multiplicity control for multiple hypotheses as well as interim efficacy analyses. The alpha was equally split to test DFS in the overall population and in patients in the subgroup with a TPS of 50% or greater (Figure 2). If the null hypothesis of DFS was rejected in that subgroup, its alpha would be fully reallocated to the DFS hypothesis in the subgroup with a TPS of 1% or greater. The OS hypotheses testing in the overall population and the subgroup with a TPS of 50% or greater would not occur until the DFS hypothesis was rejected in the overall populations. The actual boundaries were updated based on the number of events observed at the time of analysis using the Hwang-Shih-DeCani spending function with a gamma of −4.40
The protocol specified 2 event-driven interim analyses for DFS. Analyses of OS were planned in all analyses conducted for DFS. Additional OS analyses were also planned after the final DFS analysis. The final OS analysis was scheduled to be conducted no later than 120 months after the first randomized patient if the accumulation of OS events was much slower than expected and the final target OS events could not be reached by 120 months.40 In this situation, all remaining alpha would be used in the final OS analysis. An alpha reallocation strategy was used to address multiplicity issues across multiple end points and interim analyses (Figure 2).40
Analyses of GHS/QoL scores of the EORTC QLQ-C30 were prespecified key exploratory PRO end points. Analyses of the following disease-related symptoms of the EORTC QLQ-C30 and EORTC QLQ-LC13 were prespecified supportive exploratory PRO end points: pain, fatigue, appetite loss, shortness of breath, cough, chest pain, and dyspnea.39 In the statistical analysis plan, week 48 was selected as the primary time point for the mean change from baseline analysis to ensure that completion rates reached or exceeded 60% and compliance rates reached or exceeded 80% across the treatment groups.23,39
The results reported in this review are based on IA3, which is the final analysis for DFS and an interim analysis for OS. The data cut-off date for IA3 was January 24, 2023 (first patient first visit, November 10, 2015; database lock date, March 15, 2023).39
Figure 2: Multiplicity Graph for Alpha Reallocation Strategy in the KEYNOTE-091 Study
DFS = disease-free survival; OS = overall survival; PD-L1+ = PD-L1 TPS of 1% or greater; PD-L1++ = PD-L1 TPS of 50% or greater; TPS = tumour proportion score.
Source: KEYNOTE-091 study protocol.40
Subpopulation of Interest
Efficacy analyses for OS and DFS were carried out on an ad hoc basis in the subpopulation of patients with ACT and a PD-L1 TPS of less than 50% (the reimbursement request population). The models used in these analyses are the same as those described in Table 9.
Analysis Populations
Analyses of OS and DFS were conducted on all randomized patients according to the ITT principle, as well as in the subpopulation of interest in this review, i.e., all randomized patients who received prior ACT and had a PD-L1 TPS of less than 50% (the reimbursement request population). Safety analyses were based on all patients who received at least 1 dose of study treatment (APaT).40 Key analysis populations are presented in Table 10.
Table 10: Analysis Populations in the KEYNOTE-091 Study
Population | Definition | Application |
|---|---|---|
ITT | All randomized patients included in the treatment group to which they were randomized | All efficacy analyses |
APaT | All randomized patients who received at least 1 dose of study medication | All safety analyses |
PRO FAS | All randomized patients who completed at least 1 PRO (HRQoL) assessment available and who received at least 1 dose of study drug | All PRO analyses |
Reimbursement request population (prior ACT and PD-L1 TPS < 50%) | All randomized patients who had prior ACT with PD-L1 TPS < 50%, whether or not treatment was administered | All efficacy analyses in the CDA-AMC reimbursement request population |
ACT = adjuvant chemotherapy; APaT = all participants as treated; FAS = full analysis set; HRQoL = health-related quality of life; ITT = intention-to-treat; PRO = patient-reported outcome; TPS = tumour proportion score.
Sources: Clinical Study Report for P091V02MK347523 and BARDS Health Technology Assessment Statistical Report: Baseline Characteristics and Efficacy (April 9, 2024).22
Results
Patient Disposition
Patient screening in all patients and patient disposition as of the IA3 data cut-off date (January 24, 2023) in the reimbursement request population is summarized in Table 11. In the pembrolizumab (N = 363) and placebo (N = 363) groups, 171 (48%) and 133 (37%) patients from these 2 groups discontinued treatment before completing 18 cycles, respectively. The most common reasons for treatment discontinuation were recurrence, relapse, or death due to NSCLC (13% in the pembrolizumab group and 23% in the placebo group, respectively) and toxicity related to study medication (18% and 4%, respectively). The median numbers of treatment cycles administered were 17 in the pembrolizumab group and 18 in the placebo group.
Table 11: Summary of Patient Disposition in the KEYNOTE-091 Study
Patient disposition | Pembrolizumab | Placebo |
|---|---|---|
KEYNOTE-091: All patients | ||
Screened, N | 1,955 | |
Primary reason for screening failure, n (%) | ||
Central confirmation of PD-L1 expression was non-eligible | 7 (0.9) | |
Patient could not be randomized within the protocol timelines | 74 (9.5) | |
Patient does not meet criteria for — no evidence of disease | 137 (17.6) | |
Patient does not meet ECOG PS criteria | 11 (1.4) | |
Patient does not meet the protocol-defined surgical criteria | 97 (12.5) | |
Patient was ineligible for another reason | 131 (16.9) | |
Patient's refusal | 320 (41.2) | |
Randomized (all patients), N | 590 | 587 |
KEYNOTE-091: Prior ACT and PD-L1 TPS < 50% (reimbursement request population) | ||
Randomized, N | 363 | 363 |
Randomized with ITT, N | 363 | 363 |
Randomized and treated, N | 357 | 359 |
Discontinued from trial, n (%) | 101 (27.8) | 121 (33.3) |
Reason for discontinuation from trial, n (%) | ||
Death | 84 (23.1) | 110 (30.3) |
Lost to follow-up | 0 | 2 (0.6) |
Withdrawal of consent | 17 (4.7) | 9 (2.5) |
Discontinued from study medication, N (%) | 171 (47.9) | 133 (37.0) |
Reason for discontinuation from study medication, n (%) | ||
Administrative reasons | 1 (0.3) | 0 |
Adverse event not related to study medication | 8 (2.2) | 3 (0.8) |
Ineligible | 1 (0.3) | 0 |
Investigator's decision | 13 (3.6) | 9 (2.5) |
Lost to follow-up | 2 (0.6) | 0 |
Other malignancy | 5 (1.4) | 6 (1.7) |
Patient's decision not related to toxicity | 31 (8.7) | 16 (4.5) |
Recurrence, relapse, or death due to progressive disease | 45 (12.6) | 83 (23.1) |
Toxicity due to study medication | 63 (17.6) | 14 (3.9) |
Other | 2 (0.6) | 2 (0.6) |
PRO FAS, N | NAa | NAa |
Safety population (all participants as treated), N | NAb | NAb |
ACT = adjuvant chemotherapy; ECOG PS = Eastern Cooperative Oncology Group Performance Status; FAS = full analysis set; ITT = intention to treat; NA = not available; PRO = patient-reported outcome; TPS = tumour proportion score.
aThe PRO results were only analyzed in randomized patients who received at least 1 dose of study treatment and who completed at least 1 PRO assessment. These analyses were not conducted in the reimbursement request population (i.e., the subpopulation of patients who received prior ACT and had a PD-L1 TPS of less than 50%).
bSafety data were reported in the overall APaT population (i.e., in all patients randomized who received at least 1 dose of study treatment) to provide an exhaustive picture of pembrolizumab safety in the adjuvant setting (i.e., larger population exposed to treatment than the subgroup constituting the reimbursement request population).
Sources: BARDS Health Technology Assessment Statistical Report: Baseline Characteristics and Efficacy (April 9, 2024)22 and sponsor’s submission.24
Baseline Characteristics
The baseline characteristics outlined in Table 12 are limited to those that are most relevant to this review or were deemed to affect the outcomes or interpretation of the study results.
In the KEYNOTE-091 study, the median age across the groups in the overall ITT population and the reimbursement request population was 64 to 65 years. The proportion of male patients (64.5% to 68.7%) was higher than that of female patients (31.3% to 35.5%). Most enrolled patients were white (76.3% to 77.7%), followed by Asian (17.4% to 18.2%), of multiple ethnicities (0 to 1.1%), other (0.3% to 1.0%), or Black or African American (0 to 0.8%). Most patients in the overall ITT population (86%) had received 3 or 4 cycles of prior ACT with a cisplatin- or carboplatin-based regimen or both. All patients in the reimbursement request population (100%) had received ACT. A relatively small proportion of patients had stage IB disease (11.0% to 14.8% across the groups in both populations of the study) and more than half (55.4% to 59.0%) had stage II disease. Most patients (68.0% to 73.4%) were former smokers, followed by those who had never smoked (11.2% to 19.0%), and current smokers (12.7% to 15.3%). More patients (56.2% to 64.4%) had an ECOG PS of 0 compared to those with an ECOG PS of 1 (35.6% to 43.8%). The proportions of patients who had the EGFR mutation (5.8% to 8.5%) or ALK translocation (0.8% to 1.7%) were low. More than half of the patients (51.8% to 57.4%) had an unknown status for EGGR mutations or ALK translocations (56.7% to 66.4%). A total of 333 patients (28%) in the overall ITT population had a PD-L1 TPS of 50% or greater and 844 (72%) had a PD-L1 TPS of less than 50%. Most patients in the reimbursement request population had a lymph-node stage of N0 (no regional lymph-node involvement, 41.3%) or N1 (nearby lymph-node involvement, 37.5%), followed by N2 (involvement of lymph nodes in the mediastinum, 21.2%).48 Overall, the baseline characteristics were well balanced between both treatment groups in the overall 2 ITT populations. Despite that, there was a slightly higher variation in histology in the reimbursement request population: 27.8% in the pembrolizumab group versus 37.5% in the placebo group.48
Table 12: Summary of Baseline Characteristics in the KEYNOTE-091 Study
Characteristic | All patients (overall ITT population) | Prior ACT and PD-L1 TPS < 50% (reimbursement request population) | ||
|---|---|---|---|---|
Pembrolizumab (N = 590) | Placebo (N = 587) | Pembrolizumab (N = 363) | Placebo (N = 363) | |
Age, years | ||||
Mean (SD) | 64.1 (8.5) | 64.5 (8.4) | 63.3 (8.2) | 63.7 (7.9) |
Median (range) | 65.0 (31 to 87) | 65.0 (37 to 85) | 64.0 (35 to 80) | 65.0 (42 to 84) |
< 65 years, n (%) | 285 (48.3) | 273 (46.5) | 189 (52.1) | 178 (49.0) |
≥ 65 years, n (%) | 305 (51.7) | 314 (53.5) | 174 (47.9) | 185 (51.0) |
Sex, n (%) | ||||
Male | 401 (68.0) | 403 (68.7) | 234 (64.5) | 247 (68.0) |
Female | 189 (32.0) | 184 (31.3) | 129 (35.5) | 116 (32.0) |
Race, n (%) | ||||
Asian | 107 (18.1) | 107 (18.2) | 63 (17.4) | 65 (17.9) |
Black or African American | 0 | 3 (0.5) | 0 | 3 (0.8) |
White | 450 (76.3) | 455 (77.5) | 278 (76.6) | 282 (77.7) |
Multiple | 4 (0.7) | 1 (0.2) | 4 (1.1) | 0 |
Other | 6 (1.0) | 2 (0.3) | 3 (0.8) | 1 (0.3) |
Missing | 22 (3.7) | 19 (3.2) | 15 (4.1) | 12 (3.3) |
Geographic region, n (%) | ||||
Western Europe | 303 (51.4) | 301 (51.3) | 186 (51.2) | 188 (51.8) |
Eastern Europe | 116 (19.7) | 113 (19.3) | 76 (20.9) | 71 (19.6) |
Rest of world | 65 (11.0) | 68 (11.6) | 39 (10.7) | 41 (11.3) |
Asia | 106 (18.0) | 105 (17.9) | 62 (17.1) | 63 (17.4) |
Stage at baseline per AJCC, n (%) | ||||
IB | 85 (14.4) | 87 (14.8) | 45 (12.4) | 40 (11.0) |
II | 330 (55.9) | 338 (57.6) | 201 (55.4) | 214 (59.0) |
IIIA | 175 (29.7) | 160 (27.3) | 117 (32.2) | 107 (29.5) |
IV | 0 | 2 (0.3) | 0 | 2 (0.6) |
Adjuvant chemotherapy, n (%) | ||||
No | 84 (14.2) | 83 (14.1) | 0 (0.0) | 0 (0.0) |
Yes | 506 (85.8) | 504 (85.9) | 363 (100.0) | 363 (100.0) |
PD-L1 TPS, n (%) | ||||
< 1% | 233 (39.5) | 232 (39.5) | 198 (54.5) | 198 (54.5) |
1% to 49% | 189 (32.0) | 190 (32.4) | 165 (45.5) | 165 (45.5) |
≥ 50% | 168 (28.5) | 165 (28.1) | 0 (0.0) | 0 (0.0) |
Smoking status, n (%) | ||||
Never smoker | 87 (14.7) | 66 (11.2) | 69 (19.0) | 47 (12.9) |
Former smoker | 428 (72.5) | 431 (73.4) | 247 (68.0) | 266 (73.3) |
Current smoker | 75 (12.7) | 90 (15.3) | 47 (12.9) | 50 (13.8) |
Baseline ECOG PS, n (%) | ||||
0 | 380 (64.4) | 343 (58.4) | 226 (62.3) | 204 (56.2) |
1 | 210 (35.6) | 244 (41.6) | 137 (37.7) | 159 (43.8) |
Histology, n (%) | ||||
Squamous | 192 (32.5) | 224 (38.2) | 101 (27.8) | 136 (37.5) |
Non-squamous | 398 (67.5) | 363 (61.8) | 262 (72.2) | 227 (62.5) |
EGFR mutation status, n (%) | ||||
No | 218 (36.9) | 216 (36.8) | 144 (39.7) | 135 (37.2) |
Yes | 39 (6.6) | 34 (5.8) | 31 (8.5) | 25 (6.9) |
Unknown | 333 (56.4) | 337 (57.4) | 188 (51.8) | 203 (55.9) |
ALK mutation status, n (%) | ||||
Negative | 226 (38.3) | 190 (32.4) | 154 (42.4) | 118 (32.5) |
Positive | 7 (1.2) | 7 (1.2) | 3 (0.8) | 6 (1.7) |
Unknown | 357 (60.5) | 390 (66.4) | 206 (56.7) | 239 (65.8) |
Lymph-node stage,a n (%) | ||||
N0 | NR | NR | 146 (40.2) | 154 (42.4) |
N1 | NR | NR | 134 (36.9) | 138 (38.0) |
N2 | NR | NR | 83 (22.9) | 71 (19.6) |
ACT = adjuvant chemotherapy; AJCC = American Joint Committee on Cancer version 7; ALK = anaplastic lymphoma kinase; ECOG PS = Eastern Cooperative Oncology Group Performance Status; EGFR = epidermal growth factor receptor; ITT = intention-to-treat; NR = not reported; SD = standard deviation; TPS = tumour proportion score.
aThe lymph nodes stages in the KEYNOTE-091 study included N0 (no regional lymph-node involvement), N1 (nearby lymph-node involvement), and N2 (involvement of lymph nodes located in the mediastinum).
Sources: Clinical Study Report for P091V02MK3475,23 BARDS Health Technology Assessment Statistical Report: Baseline Characteristics and efficacy (April 9, 2024),22 and sponsor’s submission.48
Exposure to Study Treatments
In the reimbursement request population, the mean durations of treatment exposure were 266 days in the pembrolizumab group and 302 days in the placebo group; and the median duration of treatment was similar in the 2 treatment arms (Table 13). Among patients who received at least 1 dose of their assigned study treatment, 244 (68%) and 292 (81%) in the pembrolizumab and placebo groups, respectively, remained on study treatment for 6 months or longer (Table 13).
Table 13: Summary of Patient Exposure in the KEYNOTE-091 Study (Reimbursement Request Population)
Treatment exposure | Prior ACT and PD-L1 TPS < 50% (reimbursement request population) | |
|---|---|---|
Pembrolizumab (N = 357) | Placebo (N = 359) | |
Duration on therapy, days | ||
Mean (SD) | 266.1 (135.4) | 301.6 (109.5) |
Median (range) | 357 (1.0 to 470.0) | 358 (1.0 to 551.0) |
Number of cycles, n | ||
Mean (SD) | 12.9 (6.2) | 14.8 (5.0) |
Median (range) | 17 (1.0 to 18.0) | 18 (1.0 to 19.0) |
Duration of exposure, n (%),a person-years | ||
> 0 monthb | 357 (100.0), 260.1 | 359 (100.0), 296.5 |
≥ 1 monthb | 333 (93.3), 259.3 | 351 (97.8), 296.2 |
≥ 3 monthsb | 286 (80.1), 250.9 | 325 (90.5), 291.0 |
≥ 6 monthsb | 244 (68.3), 235.1 | 292 (81.3), 278.2 |
≥ 12 monthsb | 77 (21.6), 82.1 | 84 (23.4), 88.8 |
ACT = adjuvant chemotherapy; SD = standard deviation; TPS = tumour proportion score.
aEach patient is counted once on each applicable duration category row. Duration of exposure is the time from the first dose date to the last dose date.
bOne month = 30.4367 days.
Source: BARDS Health Technology Assessment Statistical Report: Baseline Characteristics and efficacy (April 9, 2024).22
Concomitant Medications and Cointerventions
In the reimbursement request population, 346 patients (95.3%) in the pembrolizumab group and 342 patients (94.2%) in the placebo group received 1 or more concomitant medications during the study.49 The concomitant medications were generally balanced between the treatment groups for most reported medications.49 The use of the following concomitant medications were higher in the pembrolizumab group compared with the placebo group: “antidiarrheals, intestinal anti-inflammatory/anti-infective agents” (42.7% versus 26.4%, respectively), systemic antihistamines (28.1% versus 17.6%), systemic corticosteroids (43.3% versus 27.3%), and thyroid replacement therapy (23.7% versus 12.1%).49
Subsequent Treatment
In the reimbursement request population, a smaller proportion of patients initiated subsequent antineoplastic therapy in the pembrolizumab group (19.3%) compared with the placebo group (28.1%). Similarly, a smaller proportion of patients initiated subsequent immunotherapies in the pembrolizumab group (4.1%) compared with the placebo group (14.6%) (Table 14). In the reimbursement request population, the most prescribed subsequent systemic anticancer therapies in the pembrolizumab group were carboplatin or pemetrexed (4.7%), docetaxel (3.0%), carboplatin or paclitaxel (2.8%), and pemetrexed (2.5%). In the placebo group, the most prescribed subsequent therapies were pembrolizumab (6.6%), carboplatin or pemetrexed (5.2%), atezolizumab (5.0%), pemetrexed (3.9%), docetaxel (3.3%), carboplatin (3.0%), and nivolumab (2.2%) (Table 14).
Table 14: Summary of Subsequent Treatment in the KEYNOTE-091 Study (Reimbursement Request Population)
Exposure | Prior ACT and PD-L1 TPS < 50% (reimbursement request population) | |
|---|---|---|
Pembrolizumab (N = 363) | Placebo (N = 363) | |
Antineoplastic drugs | ||
≥ 1 antineoplastic drugs, n (%) | 70 (19.3) | 102 (28.1) |
Carboplatin; pemetrexed | 17 (4.7) | 19 (5.2) |
Docetaxel | 11 (3.0) | 12 (3.3) |
Carboplatin; paclitaxel | 10 (2.8) | 5 (1.4) |
Pemetrexed | 9 (2.5) | 14 (3.9) |
Carboplatin | 6 (1.7) | 11 (3.0) |
Cisplatin; pemetrexed | 6 (1.7) | 4 (1.1) |
Pembrolizumab | 5 (1.4) | 24 (6.6) |
Paclitaxel | 5 (1.4) | 3 (0.8) |
Atezolizumab | 4 (1.1) | 18 (5.0) |
Bevacizumab | 4 (1.1) | 5 (1.4) |
Nivolumab | 3 (0.8) | 8 (2.2) |
Osimertinib | 3 (0.8) | 5 (1.4) |
Durvalumab | 3 (0.8) | 4 (1.1) |
Gefitinib | 3 (0.8) | 4 (1.1) |
Carboplatin; vinorelbine | 0 | 4 (1.1) |
Vinorelbine | 0 | 4 (1.1) |
Immunomodulating drugs | ||
≥ 1 immunomodulating drugs, n (%) | 15 (4.1) | 53 (14.6) |
Antineoplastic drugs | 15 (4.1) | 53 (14.6) |
Pembrolizumab | 5 (1.4) | 24 (6.6) |
Atezolizumab | 4 (1.1) | 18 (5.0) |
Nivolumab | 3 (0.8) | 8 (2.2) |
Durvalumab | 3 (0.8) | 4 (1.1) |
Carboplatin; pembrolizumab; pemetrexed | 1 (0.3) | 1 (0.3) |
Carboplatin; paclitaxel; pembrolizumab | 1 (0.3) | 0 |
Ipilimumab | 1 (0.3) | 0 |
Ipilimumab; nivolumab | 0 | 1 (0.3) |
Immunostimulants | 0 | 2 (0.6) |
Eftilagimod alfa | 0 | 1 (0.3) |
Interleukin-2 | 0 | 1 (0.3) |
ACT = adjuvant chemotherapy; TPS = tumour proportion score.
Source: BARDS Health Technology Assessment Statistical Report: Baseline Characteristics and Efficacy (April 9, 2024).22
Efficacy
Survival Outcomes
The OS and DFS outcomes for the reimbursement request population are presented in Table 15, Figure 3, and Figure 4. As of the data cut-off date of January 24, 2023, the median duration of follow-up was 46.6 months (range = 0.6 to 84.2) for the reimbursement request population.24
Overall Survival
As of the data cut-off date of January 24, 2023, the median OS was not reached in either group ███████ █████ █ █████ █████████████ ████████ █████ ████ ██ █████ | | ██████. The Kaplan-Meier estimates of the probability of OS in the pembrolizumab and placebo groups were █████ ████ ███ ████ ██ █████ versus █████ ████ ███ ████ ██ █████ at 36 months; and █████ ████ ███ ████ ██ █████ versus █████ ████ ███ ████ ██ █████ at 48 months, respectively.
The results from the prespecified sensitivity analyses of OS in the overall ITT population were generally consistent with those of the primary analysis. No results are available for the reimbursement request population.23
In the reimbursement request population, the OS results were consistent for point estimates of the HR across subgroups; however, the 95% CIs of HRs of nearly all categories (except for age) in the subgroups included the null value (P values for interaction tests for all the subgroups > 0.05) (Table 21).
Disease-Free Survival
As of the data cut-off date of January 24, 2023, the median DFS values were 51.7 months (95% CI, 39.0 to 70.4) for patients treated with pembrolizumab and 34.5 months (95% CI, 23.3 to 46.4) for patients who received placebo (HR = 0.72; 95% CI, 0.58 to 0.89; P < 0.001). The Kaplan-Meier estimates of the probability of DFS in the pembrolizumab and placebo groups were 67.2% (95% CI, 61.9 to 71.9) versus 55.0% (95% CI, 49.7 to 60.0) at 24 months; and 51.2% (95% CI, 45.2 to 56.9) versus 42.4% (95% CI, 36.7 to 47.9) at 48 months, respectively.
The results from the prespecified sensitivity analysis of DFS using actual stages according to the AJCC were consistent with the primary analysis results for the reimbursement request population.24
In the reimbursement request population, the DFS results were generally consistent across subgroups, including those for age, sex, race, region, disease stage, smoking status (never smoker, former smoker, or current smoker), histology (squamous or nonsquamous), ECOG PS (0 or 1), EGFR mutation status (no, yes, or unknown), and PD-L1 status (< 1% or 1% to 49%), with the 95% CIs of HRs of a few categories in subgroups marginally crossing 1 (P values for interaction test for all the subgroups > 0.05). The subgroup of patients with disease stage IB showed a wide 95% CI crossing the null value (HR = 0.66; 95% CI, 0.32 to 1.35) compared with patients with stage II to IIIA NSCLC (HR = 0.79; 95% CI, 0.63 to 0.98) (Table 22).
Table 15: Summary of Key Efficacy Results in the KEYNOTE-091 Study (Reimbursement Request Population)
Efficacy outcomes | Prior ACT and PD-L1 TPS < 50% (reimbursement request population) | ||
|---|---|---|---|
Pembrolizumab (N = 363) | Placebo (N = 363) | ||
OS | |||
OS events, n (%) | ██ ██████ | 110 (30.3) | |
OS, months, median (95% CI) | Not reached (NR) | Not reached (NR) | |
HR (95% CI)a | ████ █████ ██ █████ | ||
P value | ███████| | ||
Kaplan-Meier estimate of OS rate, % (95% CI) | |||
At 12 months | ████ █████ ██ █████ | 94.7 (91.9 to 96.6) | |
Difference in survival probability | ███ █████ ██ ████ | ||
At 18 months | ████ █████ ██ █████ | 90.0 (86.4 to 92.7) | |
Difference in survival probability | ███ █████ ██ ████ | ||
At 24 months | ████ █████ ██ █████ | 86.4 (82.4 to 89.5) | |
Difference in survival probability | ███ █████ ██ ████ | ||
At 36 months | ████ █████ ██ █████ | 76.1 (71.3 to 80.2) | |
Difference in survival probability | ███ █████ ██ █████ | ||
At 48 months | ████ █████ ██ █████ | 69.9 (64.6 to 74.5) | |
Difference in survival probability | ███ ████ ██ █████ | ||
At 60 months | ████ █████ ██ █████ | 66.1 (60.2 to 71.4) | |
Difference in survival probability | ███ █████ ██ █████ | ||
DFS | |||
DFS events, n (%) | 168 (46.3) | 199 (54.8) | |
Type of first event in DFS analysis, n (%)c | |||
Not disease-free at baseline | 3 (0.8) | 4 (1.1) | |
Recurrence | 118 (32.5) | 158 (43.5) | |
New malignancyd | 25 (6.9) | 20 (5.5) | |
Death | 22 (6.1) | 17 (4.7) | |
DFS in months, median (95% CI) | 51.7 (39.0 to 70.4) | 34.5 (23.3 to 46.4) | |
HR (95% CI)a | 0.72 (0.58 to 0.89) | ||
P value | 0.00096b | ||
Kaplan-Meier estimate of DFS rate, % (95% CI) | |||
At 12 months | 78.3 (73.5 to 82.3) | 69.3 (64.3 to 73.8) | |
Difference in survival probability | 9.0 (2.5 to 15.5) | ||
At 18 months | 74.1 (69.1 to 78.4) | 60.5 (55.2 to 65.4) | |
Difference in survival probability | 13.6 (6.7 to 20.5) | ||
At 24 months | 67.2 (61.9 to 71.9) | 55.0 (49.7 to 60.0) | |
Difference in survival probability | 12.2 (5.0 to 19.4) | ||
At 36 months | 57.4 (51.9 to 62.6) | 47.5 (42.1 to 52.6) | |
Difference in survival probability | 9.9 (2.4 to 17.4) | ||
At 48 months | 51.2 (45.2 to 56.9) | 42.4 (36.7 to 47.9) | |
Difference in survival probability | 8.8 (0.7 to 16.9) | ||
At 60 months | 42.9 (35.4 to 50.1) | 39.2 (33.0 to 45.4) | |
Difference in survival probability | 3.7 (−5.9 to 13.3) | ||
ACT = adjuvant chemotherapy; CI = confidence interval; DFS = disease-free survival; HR = hazard ratio; NR = not reported; NSCLC = non–small cell lung cancer; OS = overall survival; TPS = tumour proportion score; vs. = versus.
aBased on the multivariate Cox regression model with treatment adjusted by the following covariates: stage (IB vs. II vs. IIIA), region (Western Europe vs. Eastern Europe vs. rest of world vs. Asia), histology (squamous vs. nonsquamous), and smoking status (never vs. former or current).
bOne-sided P value based on the Wald test with a multivariate Cox regression model.
cA summary of disease status, showing number and percentage of participants by treatment arm per type of first event in DFS analysis (no event vs. event [not disease-free at baseline/local and/or regional recurrence/distant metastasis/both/new malignancy/death]). DFS was defined as the time from randomization to the first documented loco-regional recurrence, occurrence of distant metastasis(es), a second primary NSCLC or second malignancy, based on investigator assessment or death due to any cause, whichever occurred first, expressed in months.
dNew malignancy included the second primary and second malignancies.
Sources: BARDS Health Technology Assessment Statistical Report: Baseline Characteristics and Efficacy (April 9, 2024)22 and sponsor’s submission.49
Figure 3: Kaplan-Meier Estimates of Overall Survival in Patients With Prior ACT and PD-L1 TPS Less Than 50% in the KEYNOTE-091 Study (Reimbursement Request Population) [Redacted]

Figure 4: Kaplan-Meier Estimates of Disease-Free Survival in Patients With Prior ACT and PD-L1 TPS Less Than 50% in the KEYNOTE-091 Study (Reimbursement Request Population)
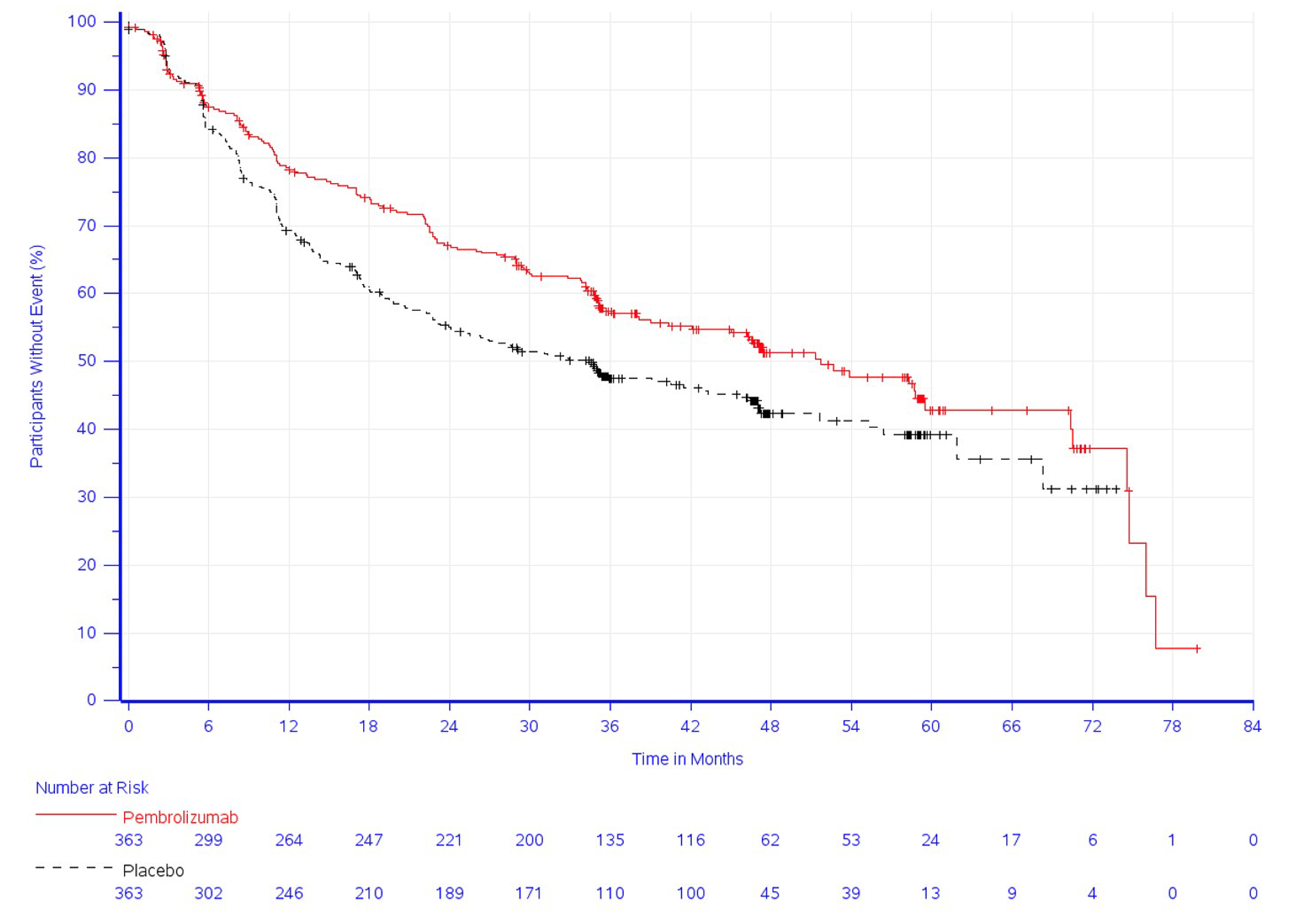
ACT = adjuvant chemotherapy; TPS = tumour proportion score.
Source: BARDS Health Technology Assessment Statistical Report: Baseline Characteristics and Efficacy (April 9, 2024).22
Health-Related Quality of Life
According to the sponsor, HRQoL outcome data for the reimbursement request population were not available. The key HRQoL outcomes among the PRO FAS population in the KEYNOTE-091 study (data cut-off date: January 24, 2023) at 48 weeks are summarized in Table 16. Additional HRQoL outcomes (select EORTC QLQ-C30 subscales) are summarized in Table 23.
In the PRO FAS population of the KEYNOTE-91 study, compliance rates for the EORTC QLQ-C30 and EORTC QLQ-LC13 at baseline through week 48 were high and similar between the treatment groups (98.6% at baseline and 85.8% at week 48 in the pembrolizumab group, 99.8% at baseline and 90.0% at week 48 in the placebo group for EORTC QLQ-C30 and EORTC QLQ-LC13, respectively). At week 48, the overall questionnaire completion rates were 77.9% and 84.9%, in the pembrolizumab and placebo groups, respectively.23,39
EORTC QLQ-C30 GHS/QoL
At week 48, the majority of participants achieved an “improved” or “stable” GHS/QoL score over time in the pembrolizumab (72.4%; 95% CI, 68.6 to 76.0) and placebo (82.4%; 95% CI, 79.1 to 85.5) groups. The proportion of patients with a deteriorated score (greater than or equal to a 10-point deterioration from baseline at any time during the trial when the criteria for improved or stable is not met) in the pembrolizumab group was higher than in the placebo group (18.1% and 12.9%, respectively; difference = 5.2%; 95% CI, 1.0 to 9.4; P = 0.015) (Table 16).
The empirical mean change from baseline in GHS/QoL scores was stable over time (Figure 5). According to the sponsor, there were no clinically meaningful differences across the treatment groups at different time points, including week 48.23,39
EORTC QLQ-LC13 Symptom Scales
At week 48, the proportion of patients with a deteriorated score in EORTC QLQ-LC13 symptom scales was similar between the 2 groups for chest pain (difference = −1.7%, 95% CI, −4.9 to 1.5; P = 0.295), coughing (difference = −0.1%, 95% CI, −3.9 to 3.6; P = 0.945), and dyspnea (difference = 3.2%, 95% CI, −1.5 to 7.8; P = 0.181) (Table 16).
Table 16: Key HRQoL Outcomes in the KEYNOTE-091 Study (PRO FAS Population at 48 Weeks)
HRQoL outcome | Measure | Pembrolizumab (N = 580) | Placebo (N = 581) |
|---|---|---|---|
EORTC QLQ-C30 Global Health Status/Quality of Life | |||
Deteriorateda, n (%; 95 CIb) | 105 (18.1; 15.1 to 21.5) | 75 (12.9; 10.3 to 15.9) | |
Difference in % deteriorateda | Estimate (95% CI)c | 5.2 (1.0 to 9.4) | Reference |
P valued | 0.0145 | ||
EORTC QLQ-LC13 Symptom Scales — Chest Pain | |||
Deteriorateda, n (%; 95 CIa) | 43 (7.4; 5.4 to 9.9) | 53 (9.1; 6.9 to 11.8) | |
Difference in % deteriorateda | Estimate (95% CI)c | −1.7 (−4.9 to 1.5) | Reference |
P valued | 0.2948 | ||
EORTC QLQ-LC13 Symptom Scales — Coughing | |||
Deteriorateda, n (%; 95 CIa) | 68 (11.7; 9.2 to 14.7) | 69 (11.9; 9.4 to 14.8) | |
Difference in % deteriorateda | Estimate (95% CI)c | −0.1 (−3.9 to 3.6) | Reference |
P valued | 0.9446 | ||
EORTC QLQ-LC13 Symptom Scales — Dyspnea | |||
Deteriorateda, n (%; 95 CIb) | 128 (22.1; 18.8 to 25.7) | 110 (18.9; 15.8 to 22.4) | |
Difference in % deteriorateda | Estimate (95% CI)c | 3.2 (−1.5 to 7.8) | Reference |
P valued | 0.1809 | ||
CI = confidence interval; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-LC13 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Lung Cancer Module; FAS = full analysis set; HRQoL = health-related quality of life; PRO = patient-reported outcome; vs. = versus.
aDeteriorated is defined as a deterioration of 10 points or greater in the score from baseline at any time during the trial when the criteria for improved or stable is not met.
bBased on binomial exact confidence interval method.
cBased on unstratified Miettinen and Nurminen method.
dTwo-sided P value for testing. H0: difference in % = 0 vs. H1: difference in % ≠ 0.
Source: Clinical Study Report: P091V02MK3475.23
Figure 5: Empirical Mean Change From Baseline and 95% CI for the EORTC QLQ-C30 GHS/QoL Over Time by Treatment (PRO FAS Population)
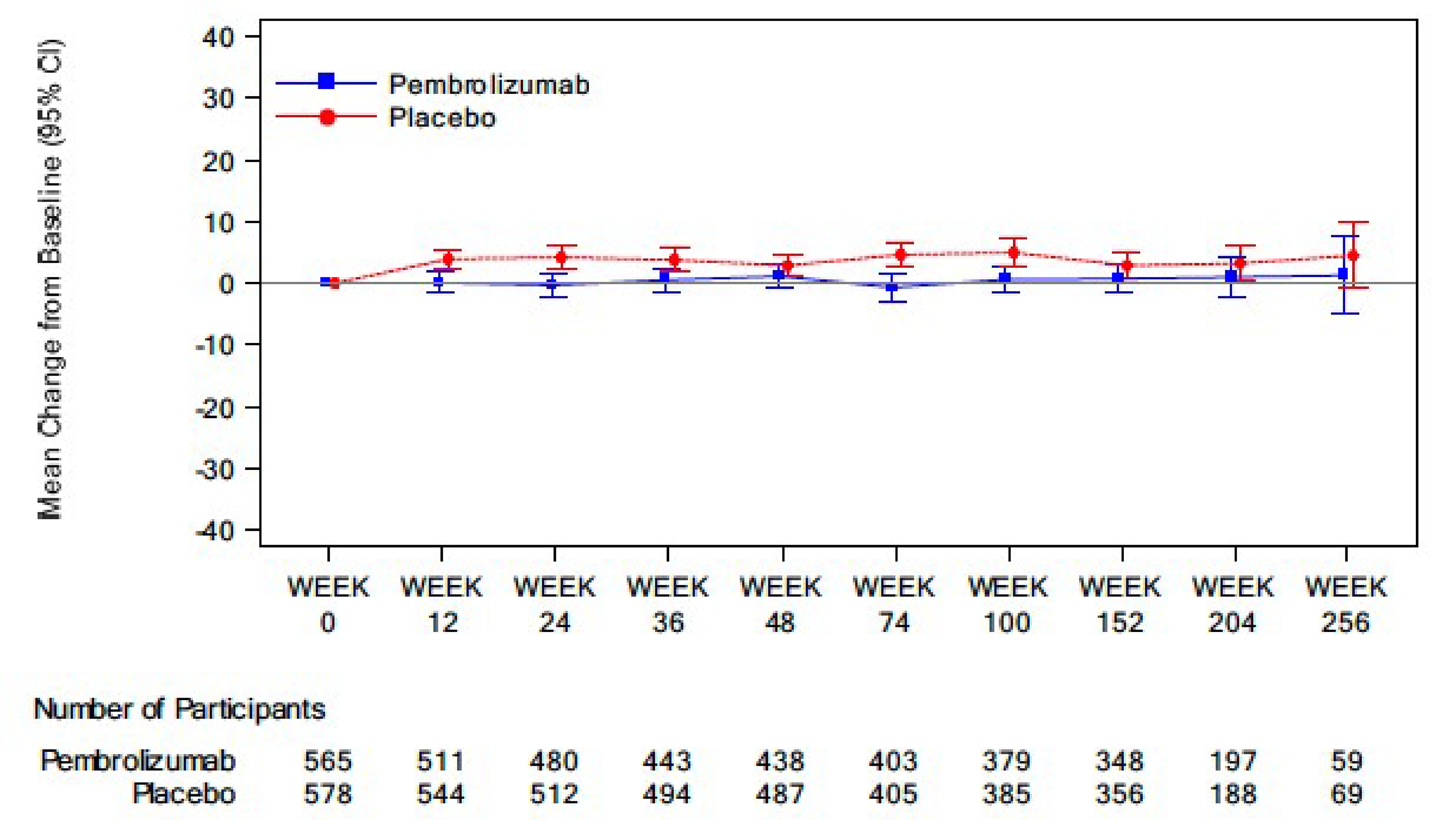
CI = confidence interval; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; FAS = full analysis set; GHS Global Health Status; PRO = patient-reported outcome.
Source: Clinical Study Report for P091V02MK3475.23
Harms
According to the sponsor, the harms outcome data among the reimbursement request population were not available. The harms observed among all patient population as treated in the KEYNOTE-091 study (data cut-off date of January 24, 2023) are summarized in Table 17.
Adverse Events
Adverse events were reported in 95.9% and 91.0% of patients in the pembrolizumab and placebo groups, respectively. The most common AEs in the pembrolizumab and placebo groups were increased weight (22.8% and 28.9%, respectively), pruritus (21.6% and 12.7%), and hypothyroidism (20.7% and 4.6%).
Grade 3 to 5 Adverse Events
The incidence of grade 3 to 5 AEs was higher in the pembrolizumab group (34.1%) than in the placebo group (25.8%). The most common grade 3 to 5 AEs in the pembrolizumab and placebo groups were hypertension (6.0% and 5.5%, respectively), and pneumonia (2.1% and 1.2%).
Serious Adverse Events
The incidence of SAEs was higher in the pembrolizumab group (24.5%) than in the placebo group (15.5%). The most common SAEs in the pembrolizumab and placebo groups were pneumonia (2.2% and 1.5%, respectively), and pneumonitis (2.1% and 0.7%).
Withdrawals due to Adverse Events
The incidence of AEs resulting in treatment discontinuation was higher in the pembrolizumab group (20.0%) than that in the placebo group (5.9%). The most common AEs leading to discontinuation were pneumonitis (3.6%) and diarrhea (1.2%) in the pembrolizumab group, and no AE led to treatment discontinuation in at least 1% of patients in the placebo group.
Mortality
Eleven patients (1.9%) in the pembrolizumab group and 6 patients (1.0%) in the placebo group had AEs resulting in death. Based on the study investigators’ assessment, 4 patients (0.7%) in the pembrolizumab group and 0 patients in the placebo group died due to a drug-related AEs. The reasons for these 4 deaths were cardiogenic shock and myocarditis, septic shock and myocarditis, pneumonia, and sudden death.
Notable Harms
The incidence of AEOSIs, which are considered to be medically equivalent to the immune-mediated events and infusion-related reactions, was higher in the pembrolizumab group (39.1%) compared with the placebo group (13.1%). Most AEOSIs were grade 1 or 2 in severity, nonserious and generally consistent with the known safety profile of pembrolizumab. Grade 3 to 5 AEOSIs occurred in 7.9% of pembrolizumab-treated patients compared to 1.9% in the placebo group.23,39 The most frequently reported AEOSIs in the pembrolizumab and placebo groups were hypothyroidism (20.7% and 4.6%, respectively), hyperthyroidism (10.7% and 2.9%), and pneumonitis (6.9% and 2.9%).23,39
Table 17: Summary of Harms Results From the KEYNOTE-091 Study (Overall APaT Population; Data Cut-Off: January 24, 2023)
Adverse events | Pembrolizumab (N = 580) | Placebo (N = 581) |
|---|---|---|
Most common AEs (≥ 10% in either treatment group),a,b,c,d n (%) | ||
Patients with ≥ 1 AE | 556 (95.9) | 529 (91.0) |
Weight increased | 132 (22.8) | 168 (28.9) |
Pruritus | 125 (21.6) | 74 (12.7) |
Hypothyroidism | 120 (20.7) | 27 (4.6) |
Arthralgia | 107 (18.4) | 72 (12.4) |
Diarrhea | 106 (18.3) | 83 (14.3) |
Fatigue | 96 (16.6) | 89 (15.3) |
Cough | 87 (15.0) | 98 (16.9) |
Hypertension | 67 (11.6) | 74 (12.7) |
Dyspnea | 66 (11.4) | 72 (12.4) |
Hyperthyroidism | 62 (10.7) | 17 (2.9) |
Grade 3 to 5 AEs (≥ 1% in 1 or more treatment groups),a,b,c,d n (%) | ||
Patients with ≥ 1 grade 3 to 5 AE | 198 (34.1) | 150 (25.8) |
RD, % (95% CI) | 8.3 (3.1 to 13.6) | |
Hypertension | 35 (6.0) | 32 (5.5) |
Pneumonia | 12 (2.1) | 7 (1.2) |
Diarrhea | 8 (1.4) | 2 (0.3) |
Dyspnea | 8 (1.4) | 7 (1.2) |
Hyponatremia | 8 (1.4) | 6 (1.0) |
Pneumonitis | 7 (1.2) | 4 (0.7) |
Increased weight | 6 (1.0) | 9 (1.5) |
SAEs (≥ 1% in either treatment group),a,c,d,e n (%) | ||
Patients with ≥ 1 SAE | 142 (24.5) | 90 (15.5) |
Pneumonia | 13 (2.2) | 9 (1.5) |
Pneumonitis | 12 (2.1) | 4 (0.7) |
Diarrhea | 7 (1.2) | 1 (0.2) |
Patients with AEs resulting in treatment discontinuation (≥ 0.5% in either treatment group),a,d n (%) | ||
Patients who discontinued | 116 (20.0) | 34 (5.9) |
Pneumonitis | 21 (3.6) | 4 (0.7) |
Diarrhea | 7 (1.2) | 3 (0.3) |
Colitis | 5 (0.9) | 1 (0.2) |
Hypothyroidism | 5 (0.9) | 0 |
Hypophysitis | 4 (0.7) | 0 |
Interstitial lung disease | 4 (0.7) | 1 (0.2) |
Hepatitis | 3 (0.5) | 0 |
Immune-mediated hepatitis | 3 (0.5) | 0 |
Myocarditis | 3 (0.5) | 1 (0.2) |
Psoriasis | 3 (0.5) | 0 |
Increased alanine transaminase | 2 (0.3) | 4 (0.7) |
Increased aspartate transaminase | 2 (0.3) | 4 (0.7) |
Patients who discontinued drug due to a serious AE | 48 (8.4) | 15 (2.6) |
Deaths,a,b,c n (%) | ||
Patients with any AE resulting in death | 11 (1.9)f | 6 (1.0) |
Myocarditis | 2 (0.3) | 0 |
Cardiac arrest | 1 (0.2) | 0 |
Cardiac death | 1 (0.2) | 0 |
Cardiogenic shock | 1 (0.2) | 0 |
Completed suicide | 1 (0.2) | 0 |
Myocardial infarction | 1 (0.2) | 1 (0.2) |
Myocardial ischemia | 1 (0.2) | 0 |
Pneumonia | 1 (0.2) | 1 (0.2) |
Respiratory tract infection | 1 (0.2) | 0 |
Sepsis | 1 (0.2) | 0 |
Septic shock | 1 (0.2) | 0 |
Sudden death | 1 (0.2) | 0 |
Aortic aneurysm rupture | 0 | 1 (0.2) |
Death | 0 | 1 (0.2) |
Pneumonia bacterial | 0 | 1 (0.2) |
Postprocedural pneumonia | 0 | 1 (0.2) |
AEs of special interest,h n (%) | ||
Patients with ≥ 1 AEOSI | 227 (39.1) | 76 (13.1) |
Patients with grade 3 to 5 AEOSI | 46 (7.9) | 11 (1.9) |
Hypothyroidism | 120 (20.7) | 27 (4.6) |
Hyperthyroidism | 62 (10.7) | 17 (2.9) |
Pneumonitis | 40 (6.9) | 17 (2.9) |
Severe skin reactions | 16 (2.8) | 4 (0.7) |
Colitis | 14 (2.4) | 5 (0.9) |
Adrenal insufficiency | 10 (1.7) | 0 |
Hepatitis | 9 (1.6) | 4 (0.7) |
Hypophysitis | 7 (1.2) | 0 |
Thyroiditis | 6 (1.0) | 1 (0.2) |
Infusion reactions | 5 (0.9) | 4 (0.7) |
Myocarditis | 5 (0.9) | 1 (0.2) |
Nephritis | 4 (0.7) | 0 |
Pancreatitis | 2 (0.3) | 2 (0.3) |
Arthritis | 1 (0.2) | 0 |
Hypoparathyroidism | 1 (0.2) | 0 |
Myositis | 1 (0.2) | 0 |
Optic neuritis | 0 | 1 (0.2) |
Sarcoidosis | 1 (0.2) | 0 |
Type 1 diabetes mellitus | 1 (0.2) | 0 |
Vasculitis | 1 (0.2) | 0 |
APaT = all participants as treated; AE = adverse event; AEOSI = adverse event of special interest; CI = confidence interval; MedDRA = Medical Dictionary for Regulatory Activities; NCI CTCAE = National Cancer Institute Common Terminology Criteria for Adverse Events; RD = risk difference.
Note: AEs were monitored throughout the study and graded in severity according to the guideline outlined in the NCI CTCAE, Version 4.03.
aEvery patient is counted once for each applicable specific AE.
bNonserious AEs up to 30 days of last dose, SAEs up to 90 days of last dose and AEOSIs up to 90 days of last dose are included.
cThe MedDRA-preferred terms “neoplasm progression,” “malignant neoplasm progression,” and “disease progression” not related to the drug are excluded.
dA specific AE appears in this report only if its incidence in 1 or more groups meets the incidence criterion in the table section title, after rounding.
eSAEs up to 90 days after the last dose are included.
fFour deaths were deemed related to pembrolizumab by the investigator: 1 death was due to both cardiogenic shock and myocarditis, 1 death was due to both septic shock and myocarditis, 1 death was due to pneumonia, and 1 death was due to sudden death.
gTreatment-emergent AEs were not reported in KEYNOTE-091. The reported drug-related AEs were determined by the investigator to be related to the drug.
hAEOSIs are potentially immune-mediated AEs and infusion reactions based on a list of terms prepared by the sponsor and were considered regardless of attribution to trial treatment by the investigator. In addition to the specific preferred terms listed, related terms were included.
Source: Clinical Study Report: P091V02MK3475.23
Critical Appraisal
Internal Validity
The KEYNOTE-091 study is a randomized, multicentre, parallel-group, placebo-controlled, triple-blinded, phase III trial. Randomization was performed using an appropriate methodology with adequate allocation concealment (a central interactive voice-response system). Randomization stratification was prespecified and was based on relevant prognostic factors of patients as well as factors related to practice and access to health care resources (i.e., disease stage, receipt of ACT, PD-L1 TPS, and geography).
In the sponsor’s reimbursement request for pembrolizumab as adjuvant treatment for patients with a PD-L1 of less than 50%, the survival outcomes (OS and DFS) submitted for this review were based on a subpopulation of the original study (ITT population, N = 1,177) among those with a PD-L1 of less than 50% and had received an ACT (N = 726). The review team and the clinical experts consulted for this review did not identify major issues that would affect the validity of study results with presenting DFS and OS outcomes through ad hoc analyses in the reimbursement request population, based on the fact that the PD-L1 TPS category was a stratification variable. The patient demographic and disease characteristics appeared to be generally balanced between the treatment groups in both the overall population and reimbursement request population, suggesting that the benefits of the randomization were maintained reasonably well in the subpopulation for reimbursement request. However, the review team noted that histologic status was slightly unbalanced between the 2 groups (27.8% squamous in the pembrolizumab group versus 37.5% in the placebo group). To what extent that this imbalance could bias the results is unknown.
In the reimbursement request population, a higher proportion of patients discontinued from the trial in the placebo group (33%) compared with those in the pembrolizumab group (28%), mainly due to death. A higher proportion of patients in the pembrolizumab group (48%) discontinued from the study medication compared with those in the placebo group (37%), mainly because of toxicity associated with the study medication (18% in the pembrolizumab group versus 4% in the placebo group). The clinical experts noted that the between-group imbalance and the reasons for the discontinuation from the study medication were reasonable and in line with the safety outcomes, specifically that higher proportions of patients in the pembrolizumab group experienced AEOSIs compared with those in the placebo group. However, these intercurrent events, particularly the high and uneven proportion of early discontinuation from study medication, would add challenges in an appropriate interpretation of efficacy results.50 As such, the effect observed in this subpopulation was primarily from those patients who could have continued to use the study drug throughout the entire treatment period, introducing uncertainty in the study results.
In the reimbursement request population, nearly all of the study patients (95%) received at least 1 concomitant medication and the proportion of patients using the most medications was similar between treatment groups. The review team noted that there was a higher proportion of patients using some concomitant medications in the pembrolizumab group compared to the placebo group, which might have affected the assessment of HRQoL and biased the results in favour of pembrolizumab, as these concomitant drug uses could control or treat adverse effects associated with pembrolizumab, including “antidiarrheals, intestinal anti-inflammatory/anti-infective agents” (43% versus 26%), systemic antihistamines (28% versus 18%), systemic corticosteroids (43% versus 27%), and thyroid replacement therapy (24% versus 12%).
In the reimbursement request population, the proportion of patients receiving subsequent anticancer treatment during the trial was lower in the pembrolizumab group than in the placebo group, for both antineoplastic therapy (19% versus 28%, respectively) and immunotherapies (4% versus 15%). Although these uneven uses of anticancer therapies may have biased the efficacy results against pembrolizumab compared to placebo for OS, the extent of any important impact on interpretation of the observed effect could not be determined.
In the KEYNOTE-091 study, the triple-blind approach, which involved masking patients, investigators, and individuals who collected or analyzed the data regarding treatment allocation as prescribed in the study protocol, were appropriate. OS is considered an objective outcome and is not prone to bias due to knowledge of group assignment. DFS and intensity of AEs were assessed by the investigators blinded to treatment assignment. The HRQoL outcome assessed with EORTC QLQ-C30 and EORTC QLQ-LC13 was a patient-reported measure. Although these subjective outcomes may be influenced by knowledge of treatment assignment, the triple-blind design of the trial likely mitigated this risk. The sponsor’s use of a prespecified cut-off threshold of a 10-point or greater deterioration in score from baseline to define the proportion of patients with a deteriorated outcome for EORTC QLQ-C30 scales and EORTC QLQ-LC13 symptom scales was regarded as appropriate by the clinical experts. The risk of bias due to missing outcome data for OS, DFS, and safety outcomes appeared to be low as losses to follow-up for reasons other than death were low and sensitivity analysis with the different censoring rule for DFS in the reimbursement request population was consistent. The risk of bias from missing outcome data for the HRQoL outcome was low as only a small proportion of patients had a category of “no assessment” for the select measures (1% to 6%) in the PRO FAS population (N = 1,161) (Table 23).
Analysis of efficacy results for the reimbursement request population followed the same defined statistical plan and employed appropriate censoring criteria as in the overall population. The efficacy end points of OS and DFS were tested by applying a multiplicity hierarchical testing procedure to account for the potential inflated type I error rates across multiple end points and interim analyses. Both OS and DFS were modelled using a proportional hazards assumption (the multivariate Cox regression model with treatment adjusted by several covariates). Although the hazards assumption underlying the HRs for OS and DFS was not evaluated, in a visual inspection, the curves appeared to be relatively parallel after approximately 12 months for OS and 8 months for DFS. Of note, OS and DFS results were based on interim analyses, which may have overestimated the treatment-effect estimates.18,19 The presence and extent of any overestimate that may have been introduced could not be determined.
External Validity
Patients in the KEYNOTE-091 study were recruited from multiple countries, including Canada. More than half of the overall or reimbursement request population were from Western Europe. In the reimbursement request population, 2 patients in the placebo group (0.6%) with stage IV disease were enrolled and included in the analyses, despite the inclusion criteria specifying that only patients with IB to IIIA stages were eligible. Problems with diagnoses led to screening failure for between 1% and 18% of patients. The clinical experts did not anticipate that this would influence the generalizability of study’s results. The clinical experts considered the eligibility criteria of patients in the KEYNOTE-091 study to be appropriate, and the demographic characteristics of the patients from the diversity aspect in the study were mostly in line with those of patients seen in clinical practice in Canada. Moreover, the clinical experts noted that, even though only patients with an ECOG PS of 0 to 1 were enrolled in the KEYNOTE-091 study as specified by the inclusion criteria, patients with an ECOG PS of 2 may benefit from pembrolizumab. The clinical experts pointed out that pembrolizumab is often offered to patients with an ECOG PS of up to 2 in clinical practice in Canada.
The clinical experts noted that presenting the survival outcomes among the subgroup of patients in the KEYNOTE-091 study who had a PD-L1 TPS of less than 50% and had received ACT was appropriate for this review, aligns with the reimbursement request, and addresses unmet therapeutic needs.
The clinical experts consulted for this review noted that the dosing and schedule of pembrolizumab in the KEYNOTE-091 study (200 mg every 3 weeks) as well as that specified in the drug’s product monograph (either 200 mg every 3 weeks or 400 mg every 6 weeks for up to 1 year or until disease recurrence or unacceptable toxicity) is in line with clinical practice in Canada.
The KEYNOTE-091 study included outcomes that were important to patients. OS, DFS, HRQoL, and safety were considered appropriate outcomes by the clinical experts and the clinician group. A study showed that, from the perspective of lung cancer survivors, DFS is a meaningful end point and addresses patients’ expectation for rapid approval of treatments that have been shown to improve DFS.51 The use of DFS as a surrogate end point for OS is accepted by the FDA for both accelerated and regular approval.52 The clinician groups noted that DFS is generally considered the most common surrogate survival end point for adjuvant treatments and described it as an appropriate end point for evidence in the current review.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered most relevant to inform CDA-AMC expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group:20,21
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate — The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited — The true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate — The true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as “very uncertain.”
Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null. An empirically derived and validated between-group minimal important difference (MID) for OS or DFS was not identified. Based on the thresholds typically used by the CDA-AMC to assess the effects of an adjuvant treatment in patients with NSCLC of similar severity or stage, between-group differences of 5% for OS and 10% for DFS were considered clinically meaningful. Due to the lack of a formal MID estimate, the target of the certainty of evidence assessment was the presence or absence of any (non-null) effect for HRQoL and grade 3 to 5 AEs.
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for pembrolizumab versus placebo in adult patients with stage IB (T2a ≥ 4 cm), II, or IIIA NSCLC who have undergone complete resection and platinum-based chemotherapy and whose tumours have a PD-L1 TPS of less than 50%.
Long-Term Extension Studies
Contents within this section have been informed by materials submitted by the sponsor. The following have been summarized and validated by the CDA-AMC review team.
Description of Studies
The KEYNOTE-091 study is still ongoing for OS follow-up. No other long-term extension studies are ongoing or completed.39
Indirect Evidence
Contents within this section have been informed by materials submitted by the sponsor. The following have been summarized and validated by the CDA-AMC review team.
Objectives for the Summary of Indirect Evidence
In the absence of direct head-to-head trials evaluating the comparative efficacy of pembrolizumab and atezolizumab in the adjuvant treatment of adult patients with early-stage NSCLC who have undergone complete resection and platinum-based chemotherapy, the sponsor conducted 1 ITC including only the subpopulation of participants with a TPS of 50% or greater. The findings from this ITC are used to support the sponsor’s reimbursement request and its request for a deviation from pharmacoeconomic requirements that excludes this subpopulation.
Indirect Treatment Comparison Design
Objectives
The objective of the sponsor-submitted ITC was to evaluate the comparative efficacy of pembrolizumab versus atezolizumab for the adjuvant treatment of stage II to IIIA NSCLC in adult patients with a TPS of 50% or greater who had received prior adjuvant chemotherapy.
Study Selection Methods
The sponsor did not conduct a systematic literature review, and included data from 2 phase III RCTs in its ITC:
The KEYNOTE-091 study, which is a 2-arm, multicentre, international, triple-blind, placebo-controlled, randomized, phase III trial with anti–PD-1 monoclonal antibody pembrolizumab (MK-3475) versus placebo for participants with early-stage NSCLC after resection and completion of standard adjuvant therapy (PEARLS). The database cut-off date was January 24, 2023.
The IMpower010 study, which is a randomized, multicentre, open-label, phase III trial of atezolizumab versus best supportive care after adjuvant cisplatin-based chemotherapy in participants with completely resected stage IB to IIIA NSCLC. The database cut-off date was January 21, 2021.
Indirect Treatment Comparison Analysis Methods
Indirect comparison of pembrolizumab and atezolizumab was conducted using the Bucher method.53 The sponsor calculated the HR and its 95% CI as an exponent of the treatment effect and its corresponding 95% CI, and the standard error was calculated using the regular variance formula for 2 additive normal distributions.
For the KEYNOTE-091 study, the sponsor used individual participant data, and DFS survival curves were estimated using the Kaplan-Meier method. A Cox proportional hazards model was used with treatment as a covariate and the Efron method of tie handling was used to assess the magnitude of the treatment effect.
For the IMpower010 study, the sponsor used the treatment effect and the hazard ratio and its corresponding standard error (derived from the 95% CI) as extracted from the published literature.
Results of Indirect Treatment Comparison
Summary of Included Studies
A summary of the KEYNOTE-091 and IMpower010 studies is presented in Table 18. A summary of the baseline characteristics of patients in both studies with a PD-L1 TPS of 50% or greater is presented in Table 19.
For the KEYNOTE-091 study, the sponsor included only the ongoing trial patients after excluding patients who discontinued treatment. The sponsor did not report the median follow-up duration for this subpopulation; however, the median follow-up time for the ITT population of patients with a PD-L1 TPS of 50% or greater who were treated with pembrolizumab was ████ months (range = ███ ██ █████. For the IMpower010 study, from the published data, the median follow-up time for patients with a PD-L1 TPS of 50% or greater who were treated with atezolizumab was █████ months ███████ ███ ██ █████.
The sponsor did not report any assessment of homogeneity or any handling of potential effect modifiers.
Table 18: Summary of Included Randomized Controlled Trials
Characteristic | KEYNOTE-091 (pembrolizumab) | IMpower010 (atezolizumab) |
|---|---|---|
Population | Participants with early-stage NSCLC after resection and completion of standard adjuvant therapy (PEARLS) 1,180 eligible participants were planned to be randomized (either without receiving adjuvant chemotherapy or after receiving adjuvant chemotherapy) in a 1:1 ratio | Participants with completely resected stage IB–IIIA NSCLC 1,280 eligible participants were planned to be randomized in a 1:1 ratio |
Intervention | Anti–PD-1 monoclonal antibody pembrolizumab (MK-3475) | Atezolizumab |
Control | Placebo | Best supportive care after adjuvant cisplatin-based chemotherapy |
Outcome | DFS | DFS |
Study design | 2-arm, multicentre, international, triple-blind, placebo-controlled, randomized phase III trial | Randomized, multicentre, open-label, phase III study |
DFS = disease-free survival; NSCLC = non–small cell lung cancer.
Source: Sponsor’s network meta-analysis study report.54
Table 19: Summary of Baseline Characteristics of Patients in the KEYNOTE-091 and IMpower010 Studies With a PD-L1 TPS of 50% or Greater
Characteristic, n (%) | KEYNOTE-091 | IMpower010 | ||
|---|---|---|---|---|
Pembrolizumab (Na = 128) | Placebo (Na = 124) | Atezolizumab (N = 115) | BSC (N = 114) | |
Mean age, years (SD) | 63.1 (8.0) | 63.0 (8.1) | 61.1 (8.5) | 61.3 (9.2) |
Age group | ||||
< 65 | 67 (52.3) | 66 (53.2) | 70 (60.9) | 68 (59.6) |
≥ 65 | 61 (47.7) | 58 (46.8) | 45 (39.1) | 46 (40.4) |
Sex | ||||
Male | 93 (72.7) | 85 (68.5) | 89 (77.4) | 78 (68.4) |
Female | 35 (27.3) | 39 (31.5) | 26 (22.6) | 36 (31.6) |
Race | ||||
Asian | 24 (18.8) | 23 (18.5) | 36 (31.3) | 26 (22.8) |
White | 95 (74.2) | 95 (76.6) | 75 (65.2) | 86 (75.4) |
Other | 3 (2.3) | 1 (0.8) | 2 (1.7) | 0 |
Missing | 6 (4.7) | 5 (4.0) | 2 (1.7) | 2 (1.8) |
Smoking status | ||||
Never | 11 (8.6) | 10 (8.1) | 16 (13.9) | 15 (13.2) |
Current | 14 (10.9) | 20 (16.1) | 16 (13.9) | 22 (19.3) |
Previous | 103 (80.5) | 94 (75.8) | 83 (72.2) | 77 (67.5) |
ECOG PS | ||||
0 | 88 (68.8) | 75 (60.5) | 71 (61.7) | 60 (52.6) |
1 | 40 (31.3) | 49 (39.5) | 44 (38.3) | 53 (46.5) |
2 | 0 | 0 | 0 | 1 (0.9) |
Histology | ||||
Squamous | 45 (35.2) | 43 (34.7) | 47 (40.9) | 45 (39.5) |
Nonsquamous | 83 (64.8) | 81 (65.3) | 68 (59.1) | 69 (60.5) |
Stage of initial diagnosis | ||||
Stage II | 82 (64.1) | 81 (65.3) | 62 (53.9) | 57 (50) |
Stage IIIA | 46 (35.9) | 43 (34.7) | 53 (46.1) | 57 (50) |
EGFR mutation status | ||||
Detected | 4 (3.1) | 5 (4.0) | 6 (5.2) | 8 (7.0) |
Not detected | 43 (33.6) | 53 (42.7) | 60 (52.2) | 64 (56.1) |
Unknown | 81 (63.3) | 66 (53.2) | 49 (42.6) | 42 (36.8) |
ALK mutation status | ||||
Yes | 3 (2.3) | 0 | 3 (2.6) | 3 (2.6) |
No | 39 (30.5) | 44 (35.5) | 62 (53.9) | 62 (54.4) |
Unknown | 86 (67.2) | 80 (64.5) | 50 (43.5) | 49 (43.0) |
ALK = anaplastic lymphoma kinase; BSC = best supportive care; ECOG PS = Eastern Cooperative Oncology Group Performance Status; EGFR = epidermal growth factor receptor; SD = standard deviation; TPS = tumour proportion score.
aOngoing trial patients after excluding patients who discontinued treatment.
Sources: Sponsor’s network meta-analysis study report54 and CADTH Reimbursement Review Report for atezolizumab (Tecentriq).55
Results
The ITC of pembrolizumab versus atezolizumab in patients with a TPS of 50% or greater and stage II to IIIA and prior adjuvant chemotherapy is presented in Table 20. Pembrolizumab appears to be less effective (██ █ █████ ███ ███ ████ ██ ████ when compared to placebo) than atezolizumab (██ █ █████ ███ ███ ████ ██ ████ when compared to best supportive care) on DFS in this subpopulation with indirect comparison by applying the Bucher method, which resulted in ██ █ █████ ███ ███ ████ ██ ████.
Table 20: Analysis of DFS Based on an Investigator-Assessment ITC of Pembrolizumab vs. Atezolizumab With a TPS of 50% or Greater, Stage II to IIIA NSCLC, and Prior Adjuvant Chemotherapy (ITT Population)
ITC | Na | Events, n (%) | Hazard ratiob (95% CI) | ITC hazard ratioc (95% CI) |
|---|---|---|---|---|
Pembrolizumab | 128 | ██ ██████ | ██████████ ██ █████ | ██████████ ██ █████ |
Atezolizumab | 115 | ██ ██████ | ██████████ ██ █████ |
CI = confidence interval; DFS = disease-free survival; ITC = indirect treatment comparison; ITT = intention to treat; TPS = tumour proportion score; vs. = versus.
aNumber of participants: ITT, participants with a TPS of 50% or greater and stage II to IIIa and prior adjuvant chemotherapy. For pembrolizumab this includes only ongoing trial patients after excluding patients who discontinued treatment.
bBased on a Cox regression with treatment as a covariate.
cBucher methodology using separate study results (estimate and its standard error) with a common control arm to perform an indirect comparison of the effect of pembrolizumab (KEYNOTE-091) vs. atezolizumab (IMpower010).
Source: Sponsor’s Network Meta-Analysis Study Report.54
Critical Appraisal of Indirect Treatment Comparison
The sponsor-submitted ITC was used to support its reimbursement request and request for deviation from pharmacoeconomic requirements that excludes this subpopulation of adult patients with early-stage NSCLC who have undergone complete resection and platinum-based chemotherapy with a TPS of 50% or greater. The sponsor did not conduct an additional ITC for the reimbursement request population (those with a TPS of less than 50%); therefore, this ITC does not provide evidence regarding the comparative efficacy of pembrolizumab versus atezolizumab in the population that is the subject of this review. The sponsor did not conduct a systematic literature review for this ITC and included only the 2 pivotal trials that included this subpopulation. There appears to be a 10-month difference in median follow-up times between both trials, which may have an impact on the time to event outcomes. The clinical experts consulted for this review pointed out that the baseline patient characteristics from both trials appeared to be well matched. However, the sponsor did not report or appear to assess homogeneity between the 2 studies, and could only include published aggregate level data from the IMpower010 study. It is therefore unclear if sources of clinical or methodological heterogeneity biased the effect estimates of ITC, and the results should be interpreted with caution.
Harms outcomes and other outcomes of relevance to patients (e.g., HRQoL) were not reported.
The clinical experts noted that, while no therapy options are currently available after adjuvant chemotherapy for patients with resected stage IB to IIIA NSCLC who have a PD-L1 TPS of less than 50%, atezolizumab is now the treatment of choice for patients with a TPS of 50% or greater. They noted that the results from this ITC support both the current therapy guidelines and the sponsor’s reimbursement request.
Studies Addressing Gaps in the Systematic Review Evidence
No studies addressing gaps in the pivotal and RCT evidence were identified for this review.
Discussion
Summary of Available Evidence
This review included 1 phase III RCT and 1 ITC, both conducted by the sponsor.
The KEYNOTE-091 study is an ongoing, multicentre, triple-blind, phase III RCT investigating the efficacy and safety of pembrolizumab versus placebo as adjuvant therapy for completely resected, ECOG PS of 0 or 1, stage IB to IIIA NSCLC (any histology). Patients were randomized in a 1:1 ratio to receive 200 mg of pembrolizumab by IV infusion every 3 weeks or 400 mg every 6 weeks for up to 1 year or until disease recurrence or unacceptable toxicity, or placebo. This review focused on the population of patients enrolled in the KEYNOTE-09 trial that aligned with the sponsor’s reimbursement request (N = 726), which is for the adjuvant treatment of adult patients with stage IB (T2a ≥ 4 cm), II, or IIIA NSCLC who have undergone complete resection and received platinum-based chemotherapy and whose tumours have a PD-L1 TPS of less than 50%, as determined by a validated test. The outcomes assessed in the reimbursement review included OS, DFS, HRQoL with EORTC QLQ-C30 and EORTC QLQ-LC13, and safety.
The reimbursement request population had a median age of 64 to 65 years. The proportion of male patients (65% to 68%) was higher than that of female patients (32% to 35%). A relatively small proportion of patients had stage IB disease (11% to 12%) and more than half had stage II disease (55% to 59%). Most patients were former smokers (68% to 73%), followed by those who had never smoked (13% to 19%) and current smokers (13% to 14%). More patients had an ECOG PS of 0 (56% to 62%) compared to those with an ECOG PS of 1 (38% to 44%). Most patients had the lymph node stage of N0 (41%) or N1 (38%). More patients had a nonsquamous histology (62.5% to 72.2%) compared with a squamous histology (27.8% to 37.5%). The proportion of patients who had the EGFR mutation (5.8% to 8.5%) or ALK translocation (0.8% to 1.7%) was low.
No direct comparative evidence between pembrolizumab and atezolizumab were identified. The sponsor submitted an ITC comparing pembrolizumab and atezolizumab only in the subpopulation of patients with a TPS of 50% or greater to support its reimbursement request for patients with a TPS of less than 50%. The sponsor did not submit an ITC comparing pembrolizumab and atezolizumab in patients with a TPS of less than 50%, as atezolizumab is not indicated for this group of patients and not a comparator for the reimbursement request population for pembrolizumab.
Interpretation of Results
Efficacy
The patient groups indicated that stopping or slowing disease progression is the most important outcome. The clinical experts consulted for this review and the clinician groups noted that the key treatment goals in early-stage NSCLC following complete resection is to improve cure rates. In Canada, pembrolizumab was approved on April 19, 2023, for the adjuvant treatment of adult patients with stage IB (T2a ≥ 4 cm), II, or IIIA NSCLC who have undergone complete resection and platinum-based chemotherapy.31
The sponsor identified that the analysis in subgroups of the overall population of KEYNOTE-091 showed a larger effect of pembrolizumab in patients with a PD-L1 TPS of less than 50% than in those with a TPS of 50% or greater with respect to DFS and OS. According to the sponsor, the difference in treatment effect for DFS and OS between the populations is best explained by the overperformance of placebo in patients with a PD-L1 TPS of 50% or greater, who were excluded from the total ITT population in the survival analyses for reimbursement request population.39 Also, a sponsor-submitted ITC study54 reported results that the DFS benefit of pembrolizumab in patients with a PD-L1 TPS of 50% or greater is not as significant as the benefit of atezolizumab, the current standard of care for these patients.39 In addition, the clinical experts and clinician groups consulted for this review noted that the current practice in Canada is neoadjuvant platinum-based chemotherapy in combination with nivolumab, or adjuvant platinum-based chemotherapy, followed by atezolizumab if patients have a PD-L1 TPS of 50% or greater; that is, no adjuvant immunotherapy options area available for patients in Canada with stage IB to IIIA NSCLC who have a PD-L1 TPS of less than 50%, creating an unmet need for this group.
Given these data and the unmet treatment need, the sponsor’s reimbursement request for this submission is for the adjuvant treatment of adult patients with stage IB (T2a ≥ 4 cm), II, or IIIA NSCLC who have undergone complete resection and platinum-based chemotherapy and whose tumours have a PD-L1 TPS of less than 50%, as determined by a validated test. The review team and the clinical experts consulted for this review did not identify any major issues with presenting survival outcomes through an ad hoc analyses in the reimbursement request population (N = 726, which accounted for approximately 62% patients of the total KEYNOTE-091 ITT population39). The review team also did not identify major issues with the using placebo as the comparator group in the KEYNOTE-091 study, as it is not mandatory in clinical practice in most countries to use adjuvant chemotherapy in patients similar to those in the reimbursement request population.9
The clinical experts consulted for this review were unable to suggest MID thresholds for between-group differences in the proportions of patients with OS or DFS, as an MID likely depends on patients’ individual values and preferences, and therefore may vary among different patients and at different follow-up time points. The assessment of precision in the GRADE certainty of evidence was based on a threshold usually used by CDA-AMC to assess the effects of a treatment in patients with NSCLC of similar condition (i.e., 5% for OS and 10% for DFS). In the reimbursement request population, median OS was not reached in any treatment group at interim phase. The risk of death was reduced by 27% with pembrolizumab compared to placebo ████ █████ ███ ███ ████ ██ █████ ███████ | | ██████. There were uncertainties in the GRADE assessment for OS as its data were immature, based on the prespecified analysis plan by the sponsor for the KEYNOTE-091 trial.23,40 Specifically, the median OS was not reached in either group (the observed Kaplan-Meier curves did not meet their medians), with OS events observed in 23.1% of patients in the pembrolizumab group and in 30.3% of patients in the placebo group at IA3. The median DFS at IA3 was 17 months longer in the pembrolizumab group than in the placebo group (51.7 months versus 34.5 months; HR = 0.72; 95% CI, 0.58 to 0.89; nominal P < 0.001), with a 28% lower risk of disease recurrence, other malignancy, or death. Evidence with a moderate level of certainty showed that pembrolizumab likely results in a clinically important increase in the probability of DFS at both 24 months and 48 months.
Two subgroup analyses in the reimbursement request population — patient smoking status and EGFR mutation at baseline — are relevant to the pharmacoeconomic analysis of this review. For OS, interpretation of subgroup results (current smoker versus former smoker versus nonsmoker; and EGFR mutation status, yes versus no) is limited, because of immature data for OS. For DFS, the review team noted a numerically larger effect of pembrolizumab among the patients who were current smokers (HR = 0.51; 95% CI, 0.27 to 0.97) compared to former smokers (HR = 0.80; 95% CI, 0.62 to 1.02) as well as nonsmokers (HR = 0.71; 95% CI, 0.44 to 1.16), for the point estimate of the HR, seemingly suggesting the effect of pembrolizumab in DFS increases with the presence and recency of smoking (current smokers > former smokers > nonsmokers). However, no conclusion could be drawn based on this subgroup results from the KEYNOTE-091 study data as the 95% CIs around the HR values were relatively wide because of small subgroup sample sizes. In addition, the interaction P value was 0.549, indicating no statistically significant difference among the categories for different smoking status (Table 22). Pembrolizumab showed a larger effect in DFS among patients with an EGFR mutation (event rates of 55% versus 84% in the pembrolizumab and placebo groups, respectively; HR = 0.44; 95% CI, 0.23 to 0.84) than in those without an EGFR mutation (event rates of 51% versus 62%; HR = 0.76; 95% CI, 0.56 to 1.04) in terms of the differences in the point estimate and 95% CI of HRs. However, the interaction P value was 0.334, indicating no statistically significant difference among the 3 categories of EGFR mutation status (Table 22). The review team noted that a possible limitation of the KEYNOTE-091 study could be that EGFR testing was not required.40 As a result, more than half of patients had “unknown” information for EGFR mutation status at baseline (Table 12). Although an EGFR mutation could be an important prognostic factor in the reimbursement request population, our ability to interpret the consequence of this assumption is limited because of the small sample size. Moreover, all the subgroup results in the reimbursement request population were based on ad hoc analyses, making interpretation of any subgroup effects findings uncertain.
The patient and clinician groups also regarded HRQoL as an outcome of importance. Both the EORTC QLQ-C30 and EORTC QLQ-LC13 are validated tools commonly used to measure HRQoL among patients with NSCLC. The clinical experts consulted for this review acknowledged that the sponsor’s use of a 10-point cutoff threshold in change from baseline for these 0-to-100 measurements in defining categorical outcomes (improved, stable, deteriorated, and unconfirmed) was appropriate. Of these categories, the clinical experts indicated the proportion of patients who had a deteriorated outcome in HRQoL was important. The missing data for HRQoL outcomes important to this review was low as only a small proportion of patients (1% to 6%) had no assessment in the PRO FAS population at week 48. The between-group difference in the EORTC QLQ-C30 GHS/QoL change was not regarded as clinically meaningful. The 95% CIs of risk differences in the proportion of patients with a deteriorated outcome in EORTC QLQ-LC13 symptoms of chest pain, coughing, and dyspnea all included the null threshold of 0. These outcome data, together with the empirical mean change from baseline data of the EORTC QLQ-C30 GHS/QoL (Figure 5), suggest that both the patients who received pembrolizumab and those who received placebo had comparable and relatively stable HRQoL over 48 weeks.
In the ITC of pembrolizumab versus atezolizumab in patients with a TPS of 50% or greater and stage II to IIIA and prior adjuvant chemotherapy, pembrolizumab appears to be less effective (██ █ ████ ████ ███ ████ ██ █████ when compared to placebo) than atezolizumab (HR = ████ ████ ███ ████ ██ █████ when compared to best supportive care) with respect to DFS in this subpopulation, with an indirect comparison by the Bucher method resulting in a HR of ████ ████ ███ ████ ██ ████). However, the sponsor did not report or appear to assess homogeneity between the 2 included studies and could only include published aggregate level data from study on atezolizumab. It is therefore unclear if the sources of clinical or methodological heterogeneity biased the effect estimates of the ITC. The sponsor did not submit an ITC comparing pembrolizumab and atezolizumab in patients with a TPS of less than 50%, which is the population of patients included in the sponsor’s reimbursement request.
Harms
In the overall APaT population of the KEYNOTE-091 study, the incidence of harms was higher in the pembrolizumab group than in the placebo group for grade 3 to 5 AEs (34% versus 26%, respectively), SAEs (25% versus 16%), and AEs resulting in treatment discontinuation (20% versus 6%). The clinical experts consulted for this review noted that, in a placebo-controlled study, it is expected that patients in the pembrolizumab group would experience AEs more frequently than would patients in the placebo group. The most common AEOSIs in the pembrolizumab group, including hypothyroidism (21%) and hyperthyroidism (11%), were potentially immune-mediated AEs that may be associated with immune checkpoint inhibitors such as pembrolizumab.56 The clinical experts noted that, overall, the safety outcomes in the KEYNOTE-091 study were consistent with their expectations for pembrolizumab. They added that the difference in incidence of grade 3 to 5 AEOSIs between the pembrolizumab group (7.9%) and the placebo group (1.9%) was small, indicating that the tolerability of pembrolizumab is acceptable. The clinical experts also noted that the incidence of AEs important to patients or AEs associated with immune checkpoint inhibitor therapy,57 including thyroiditis (1.0% versus 0.2%), myocarditis (0.9% versus 0.2%), and type 1 diabetes mellitus (0.2% versus 0), was low.
Harms outcomes were not reported in the sponsor-submitted ITC.
Conclusion
One triple-blind, phase III RCT comparing the efficacy and safety of adjuvant pembrolizumab and placebo in adult patients with stage IB to IIIA NSCLC who have undergone complete resection and platinum-based chemotherapy and whose tumours have a PD-L1 TPS of less than 50% showed a clinically meaningful benefit of adjuvant pembrolizumab in the probability of DFS at 24 and 48 months. The OS results are uncertain due to the immaturity of the data (the median OS was not reached in either group, with OS events observed in 23% and 30% of patients in the pembrolizumab and placebo groups, respectively), although there is a trend toward improved OS in favour of pembrolizumab. Patients who received either pembrolizumab or placebo had comparable and relatively stable HRQoL over 48 weeks. According to the clinical experts consulted for this review, the safety profile of pembrolizumab was consistent with their expectations for this drug.
The sponsor submitted an ITC comparing pembrolizumab and atezolizumab in patients with a TPS of 50% or greater to support its reimbursement request for patients with a TPS of less than 50%. Indirect comparative evidence suggests that atezolizumab is superior in patients with a TPS of 50% or greater; however, it is unclear if sources of clinical or methodological heterogeneity biased the effect estimates of the ITC, because no assessment of homogeneity was conducted. The sponsor did not submit an ITC comparing pembrolizumab and atezolizumab in patients with a TPS of less than 50% because, according to clinician input, atezolizumab is not indicated for this subpopulation of patients.
References
1.Canadian Cancer Statistics Advisory Committee. Canadian Cancer Statistics: A 2020 special report on lung cancer. Toronto (ON): Canadian Cancer Society; 2020: https://cdn.cancer.ca/-/media/files/cancer-information/resources/publications/2020-canadian-cancer-statistics-special-report/2020-canadian-cancer-statistics-special-report-en.pdf. Accessed 2024 Aug 15.
2.Canadian Cancer Statistics Advisory in collaboration with the Canadian Cancer Society, Statistics Canada, and the Public Health Agency of Canada. Canadian Cancer Statistics 2023. Toronto (ON): Canadian Cancer Society; 2023: https://cdn.cancer.ca/-/media/files/research/cancer-statistics/2023-statistics/2023_PDF_EN.pdf. Accessed 2024 Aug 15.
3.Cancer-Specific Stats 2022: Snapshot of incidence, mortality and survival estimates by cancer type. Toronto (ON): Canadian Cancer Society; 2022: https://cdn.cancer.ca/-/media/files/research/cancer-statistics/2022-statistics/2022_cancer-specific-stats.pdf. Accessed 2024 Aug 15.
4.Ehrenstein V, Eriksen K, Taylor A, Servidio L, Jakobsen E. Characteristics and overall survival of patients with early-stage non–small cell lung cancer: A cohort study in Denmark. Cancer Med. 2023;12(1):30-37. PubMed
5.Molina JR, Yang P, Cassivi SD, Schild SE, Adjei AA. Non-small cell lung cancer: Epidemiology, risk factors, treatment, and survivorship. Mayo Clin Proc. 2008;83(5):584-594. PubMed
6.Muthusamy B, Patil PD, Pennell NA. Perioperative systemic therapy for resectable non–small cell lung cancer. J Natl Compr Canc Netw. 2022;20(8):953-961. PubMed
7.Forde PM, Spicer J, Lu S, et al. Neoadjuvant nivolumab plus chemotherapy in resectable lung cancer. N Engl J Med. 2022;386(21):1973-1985. PubMed
8.Pignon JP, Tribodet H, Scagliotti GV, et al. Lung adjuvant cisplatin evaluation: A pooled analysis by the LACE Collaborative Group. J Clin Oncol. 2008;26(21):3552-3559. PubMed
9.O'Brien M, Paz-Ares L, Marreaud S, et al. Pembrolizumab versus placebo as adjuvant therapy for completely resected stage IB-IIIA non–small-cell lung cancer (PEARLS/KEYNOTE-091): An interim analysis of a randomised, triple-blind, phase III trial. Lancet Oncol. 2022;23(10):1274-1286. PubMed
10.CADTH Reimbursement Recommendation: Atezolizumab (Tecentriq) [accessed by sponsor]. Can J Health Technol. 2022;2(9). https://www.cda-amc.ca/sites/default/files/DRR/2022/PC0269%20Tecentriq%20for%20NSCLC%20-%20CADTH%20Final%20Recommendation-Final-meta.pdf.
11.CADTH Reimbursement Review Provisional Funding Algorithm: Non-small cell lung cancer without actionable oncogenic alterations. Ottawa (ON): CADTH; 2023: https://www.cadth.ca/sites/default/files/DRR/2023/PH0029-Algorithm_secured.pdf. Accessed 2024 Mar 12.
12.Felip E, Altorki N, Zhou C, et al. Adjuvant atezolizumab after adjuvant chemotherapy in resected stage IB-IIIA non–small-cell lung cancer (IMpower010): A randomised, multicentre, open-label, phase III trial. Lancet. 2021;398(10308):1344-1357. PubMed
13.Tankel J, Spicer J, Chu Q, et al. Canadian consensus recommendations for the management of operable stage II/III non–small-cell lung cancer: Results of a modified Delphi process. Current Oncology. 2023;30(12):10363-10384. PubMed
14.CADTH Reimbursement Recommendation: Osimertinib (Tagrisso) [accessed by sponsor]. Can J Health Technol. 2022;2(1). https://www.cadth.ca/osimertinib.
15.CADTH Reimbursement Recommendation: Nivolumab (Opdivo). Can J Health Technol. 2023;3(4). https://www.cadth.ca/nivolumab-2. Accessed 2024 Mar 12.
16.Edge SB, Compton CC. The American Joint Committee on Cancer: The 7th edition of the AJCC cancer staging manual and the future of TNM. Ann Surg Oncol. 2010;17(6):1471-1474.
17.Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45(2):228-247. PubMed
18.Bassler D, Briel M, Montori VM, et al. Stopping randomized trials early for benefit and estimation of treatment effects: systematic review and meta-regression analysis. JAMA. 2010;303(12):1180-1187. PubMed
19.Guyatt GH, Briel M, Glasziou P, Bassler D, Montori VM. Problems of stopping trials early. BMJ. 2012;344:e3863. PubMed
20.Balshem H, Helfand M, Schunemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-406. PubMed
21.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: Informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. PubMed
22.BARDS Health Technology Assessment Statistical Report. Pembrolizumab for Adjuvant Treatment of Early Stage Non-Small Cell Lung Cancer After Resection and Completion of Standard Adjuvant Therapy (PEARLS). Protocol 091 IA3 Canada. Baseline Characteristics and Efficacy [internal sponsor's report]. In: Keytruda (pembrolizumab), solution for infusion 100 mg/4 mL vial. Rahway (NJ): BARDS, Merck & Co., Inc.; 2024.
23.Clinical Study Report: P091V02MK3475. A randomized, phase III trial with anti PD-1 monoclonal antibody pembrolizumab (MK 3475) versus placebo for patients with early-stage NSCLC after resection and completion of standard adjuvant therapy (PEARLS) KEYNOTE 091 [internal sponsor's report]. Rahway (US): Merck Sharp & Dohme LLC; 2023 Jul 3.
24.Merck Canada Inc. response to August 21, 2024 CADTH request for additional information regarding Keytruda (pembrolizumab) CADTH review [internal additional sponsor's information]. Kirkland (QC): Merck Canada Inc.; 2024 Aug 23.
25.Brenner DR, Gillis J, Demers AA, et al. Projected estimates of cancer in Canada in 2024. CMAJ. 2024;196(18):E615-E623. PubMed
26.Canadian Cancer Society. What is lung cancer? https://cancer.ca/en/cancer-information/cancer-types/lung/what-is-lung-cancer. Accessed 2024 Aug 15.
27.Birring SS, Peake MD. Symptoms and the early diagnosis of lung cancer. Thorax. 2005;60(4):268-269. PubMed
28.BC Cancer. Lung. 2013; http://www.bccancer.bc.ca/health-professionals/clinical-resources/cancer-management-manual/lung/lung. Accessed 2024 Aug 15.
29.Tecentriq (atezolizumab for injection), concentrate for solution for infusion, 60 mg/mL, 1200 mg/20 mL and 840 mg/14 mL single use vials [product monograph]. Mississauga (ON): Hoffmann-La Roche Limited; 2024 Mar 13: https://pdf.hres.ca/dpd_pm/00074930.PDF. Accessed 2024 Aug 15.
30.Opdivo (nivolumab for injection), intravenous infusion, 10 mg/mL, 40 mg and 100 mg single use vials [product monograph]. . Saint-Laurent (QC): Bristol-Myers Squibb Canada; 2024 Jun 28: https://pdf.hres.ca/dpd_pm/00076152.PDF. Accessed 2024 Aug 22.
31.Keytruda (pembrolizumab), solution for infusion 100 mg/4 mL vial [product monograph]. Kirkland (QC): Merck Canada Inc.; 2024 Jun 20.
32.Hirsch FR, McElhinny A, Stanforth D, et al. PD-L1 immunohistochemistry assays for lung cancer: Results from Phase 1 of the Blueprint PD-L1 IHC Assay Comparison Project. J Thorac Oncol. 2017;12(2):208-222. PubMed
33.Tsao MS, Kerr KM, Kockx M, et al. PD-L1 immunohistochemistry comparability study in real-life clinical samples: Results of Blueprint Phase 2 project. J Thorac Oncol. 2018;13(9):1302-1311. PubMed
34.Institut national d'excellence en santé et en services sociaux. Test compagnon de Tecentriq: Cancer du poumom non à petites cellules. Avis transmis au ministre en janvier 2023. Montreal (QC): INESSS; 2023: https://www.inesss.qc.ca/fileadmin/doc/INESSS/Inscription_medicaments/Avis_au_ministre/Mars_2023/TC_Tecentriq_CPNPC_2023_01.pdf. Accessed 2024 Aug 6.
35.Lantuejoul S, Sound-Tsao M, Cooper WA, et al. PD-L1 testing for lung cancer in 2019: Perspective from the IASLC Pathology Committee. J Thorac Oncol. 2020;15(4):499-519. PubMed
36.National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology: Non-small cell lung cancer. v6.2024. 2024; https://www.nccn.org/professionals/physician_gls/pdf/nscl.pdf. Accessed 2024 Jun 25.
37.Canadian Cancer Statistics Advisory Committee. Canadian Cancer Statistics. A 2020 special report on lung cancer. Toronto (ON): Canadian Cancer Society; 2020: https://cdn.cancer.ca/-/media/files/research/cancer-statistics/2020-statistics/canadian-cancer-statistics/2020-special-report/res-cancerstatistics-canadiancancerstatistics-2020_special-report_en.pdf. Accessed 2024 Jun 12.
38.Ionescu DN, Downes MR, Christofides A, Tsao MS. Harmonization of PD-L1 testing in oncology: A Canadian pathology perspective. Curr Oncol. 2018;25(3):e209-e216. PubMed
39.Keytruda (pembrolizumab) for patients with early-stage NSCLC after resection and completion of standard adjuvant therapy: Clinical summary [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Keytruda (pembrolizumab), solution for infusion 100 mg/4 mL vial. Kirkland (QC): Merck Canada Inc.; 2024.
40.Intergroup Study (EORTC-1416-LCG). A randomized, phase III trial with anti-PD-1 monoclonal antibody pembrolizumab (MK-3475) versus placebo for patinets with early stage NSCLC after resection and completion of standard adjuvant therapy (PEARLS) KEYNOTE-091 - Study protocol v5.0 [internal sponsor's report]. Brussels (BE): EORTC Lung Cancer Group, Merck & Co., Inc.; 2022 Sep 23.
41.Aaronson NK, Ahmedzai S, Bergman B, et al. The European Organisation for Research and Treatment of Cancer QLQ-C30: A quality-of-life instrument for use in international clinical trials in oncology. J Natl Cancer Inst. 1993;85(5):365-376. PubMed
42.Teckle P, Peacock S, McTaggart-Cowan H, et al. The ability of cancer-specific and generic preference-based instruments to discriminate across clinical and self-reported measures of cancer severities. Health Qual Life Outcomes. 2011;9:106. PubMed
43.Nicklasson M, Bergman B. Validity, reliability and clinical relevance of EORTC QLQ-C30 and LC13 in patients with chest malignancies in a palliative setting. Qual Life Res. 2007;16(6):1019-1028. PubMed
44.Ozturk A, Sarihan S, Ercan I, Karadag M. Evaluating quality of life and pulmonary function of long-term survivors of non–small cell lung cancer treated with radical or postoperative radiotherapy. Am J Clin Oncol. 2009;32(1):65-72. PubMed
45.Maringwa JT, Quinten C, King M, et al. Minimal important differences for interpreting health-related quality of life scores from the EORTC QLQ-C30 in lung cancer patients participating in randomized controlled trials. Support Care Cancer. 2011;19(11):1753-1760. PubMed
46.Osoba D, Rodrigues G, Myles J, Zee B, Pater J. Interpreting the significance of changes in health-related quality-of-life scores. J Clin Oncol. 1998;16(1):139-144. PubMed
47.Bergman B, Aaronson NK, Ahmedzai S, Kaasa S, Sullivan M. The EORTC QLQ-LC13: A modular supplement to the EORTC Core Quality of Life Questionnaire (QLQ-C30) for use in lung cancer clinical trials. EORTC Study Group on Quality of Life. Eur J Cancer. 1994;30A(5):635-642. PubMed
48.Merck Canada Inc. response to September 10, 2024 CADTH request for additional information regarding Keytruda (pembrolizumab) CADTH review [internal additional sponsor's information]. Kirkland (QC): Merck Canada Inc.; 2024 Sept 12.
49.Merck Canada Inc. response to August 30, 2024 CADTH request for additional information regarding Keytruda (pembrolizumab) CADTH review [internal additional sponsor's information]. Kirkland (QC): Merck Canada Inc.; 2024 Sept 5.
50.ICH E9 (R1) addendum on estimands and sensitivity analysis in clinical trials to the guideline on statistical principles for clinical trials. EMA/CHMP/ICH/436221/2017. Amsterdam (NL): European Medicines Agency (EMA); 2020 Feb 17: https://www.ema.europa.eu/en/documents/scientific-guideline/ich-e9-r1-addendum-estimands-and-sensitivity-analysis-clinical-trials-guideline-statistical-principles-clinical-trials-step-5_en.pdf. Accessed 2024 Sep 20.
51.Bever A, Manthorne J, Rahim T, Moumin L, Johnston K, Szabo S. The importance of the disease-free survival (DFS) endpoint to survivors of lung cancer. Eur Respir J. 2021;58(suppl 65):PA2190.
52.Clinical trial endpoints for the approval of cancer drugs and biologics: Guidance for industry. Silver Spring (MD): U.S. Food and Drug Administration; 2018: https://www.fda.gov/media/71195/download. Accessed 2024 Aug 23.
53.Bucher HC, Guyatt GH, Griffith LE, Walter SD. The results of direct and indirect treatment comparisons in meta-analysis of randomized controlled trials. J Clin Epidemiol. 1997;50(6):683-691. PubMed
54.BARDS Health Technology Assessment Statistical Report. Compound: Pembrolizumab (MK-3475). Indication: Adjuvant Treatment of Early Stage Non-Small Cell Lung Cancer after Resection and Completion of Standard Adjuvant Therapy (PEARLS). Protocol 091 IA3. Indirect Treatment Comparison: DFS [internal sponsor's report]. In: Keytruda (pembrolizumab), solution for infusion 100 mg/4 mL vial. Rahway (NJ): BARDS, Merck & Co., Inc.; 2024.
55.CADTH Reimbursement Review: Atezolizumab (Tecentriq). Can J Health Technol. 2022;2(12). https://www.cda-amc.ca/sites/default/files/DRR/2022/PC0269-Tecentriq%20NSCLC_combined.pdf. Accessed 2024 Sep 20.
56.Postow MA, Sidlow R, Hellmann MD. Immune-related adverse events associated with immune checkpoint blockade. N Engl J Med. 2018;378(2):158-168. PubMed
57.Rubio-Infante N, Ramirez-Flores YA, Castillo EC, Lozano O, Garcia-Rivas G, Torre-Amione G. Cardiotoxicity associated with immune checkpoint inhibitor therapy: A meta-analysis. Eur J Heart Fail. 2021;23(10):1739-1747. PubMed
Appendix 1: Detailed Outcomes Data
Please note that this appendix has not been copy-edited.
Additional data of the KEYNOTE-091 study are presented in the following.
Table 21: Subgroup Analysis Results of Overall Survival in the KEYNOTE-091 Study (Reimbursement Request Population) [Redacted]
██████ | █████████████ | ███████ | ███████ | ██████ | ||||
|---|---|---|---|---|---|---|---|---|
██ | █████ | ██████ | ██ | ██████ | ███████ | |||
████ █████ | ||||||||
██ | ███ | ████ | ██████ | ███ | █████ | ███████ | ███████ | █████ |
██ | ███ | ████ | ██████ | ███ | █████ | ███████ | ███████ | |
███ | ||||||||
████ | ███ | ████ | ██████ | ███ | █████ | ███████ | ███████ | █████ |
█████ | ███ | ████ | ██████ | ███ | █████ | ███████ | ███████ | |
████ | ||||||||
█████ | ███ | ████ | ██████ | ███ | █████ | ███████ | ███████ | █████ |
█████ | ██ | ████ | ██████ | ██ | █████ | ███████ | ███████ | |
██████████ ██████ | ||||||||
████ | ██ | ████ | ██████ | ██ | █████ | ███████ | ███████ | █████ |
█████ | ██ | ████ | ██████ | ██ | █████ | ███████ | ███████ | |
█████ | ██ | ████ | ██████ | ██ | █████ | ███████ | ███████ | |
█████ | ███ | ████ | ██████ | ███ | █████ | ███████ | ███████ | |
█████ ██ ████████ ███ ████ ██ | ||||||||
██ | ██ | ████ | ██████ | ██ | █████ | ███████ | ███████ | █████ |
██ | ███ | ████ | ██████ | ███ | █████ | ███████ | ███████ | |
████ | ███ | ████ | ██████ | ███ | █████ | ███████ | ███████ | |
█████ ███ | ||||||||
██ | ███ | ████ | ██████ | ███ | █████ | ███████ | ███████ | █████ |
█████ | ███ | ████ | ██████ | ███ | █████ | ███████ | ███████ | |
███████ ██████ | ||||||||
█████ | ██ | ████ | █████ | ██ | █████ | ███████ | ███████ | █████ |
█████ | ███ | ████ | ██████ | ███ | █████ | ███████ | ███████ | |
█████ | ██ | ████ | ██████ | ██ | █████ | ███████ | ███████ | |
████████ ████ ███████████ ██████ | ||||||||
██ | ███ | ████ | █████ | ███ | █████ | ███████ | ███████ | █████ |
██ | ███ | ████ | █████ | ███ | █████ | ███████ | ███████ | |
█████████ | ||||||||
█████ | ███ | ████ | ██████ | ███ | █████ | ███████ | ███████ | █████ |
█████ | ███ | ████ | ██████ | ███ | █████ | ███████ | ███████ | |
████ ████████ ██████ | ||||||||
██ | ███ | ████ | █████ | ███ | █████ | ███████ | ███████ | █████ |
███ | ██ | ████ | ██████ | ██ | █████ | ███████ | ███████ | |
█████ | ███ | ████ | █████ | ███ | █████ | ███████ | ███████ | |
███ █ ████████ █████████████ ████ █ ████████ █████ █████████ ██ ███████ ███ █ ██████████ ████████ ███████ ████ █ ███████ ███████████ ████████ ██████ ████ █ █████████ ██████ ██████ █████████ ███ █ ███████████████████ ██ █ ███ █████████ █████ █ █████████ ████ ████ ███████ ████ █ ██████████ ████ █████ ███████ ██ ██████ ██████████ ████ ████████████ ██ ██ █ ████████ ██████████ ███ █ █████ ██████████ ████████████ ████ █████████ ██ ████ █████ ████ █████ ██ ████████ ██ ████████ ███████ ████ ██ ███████ ███ █████ ██ ███ ███ ██████████ ██ ████████ ████ █████ ███ █ ███ ███ █████ █████|████ █████████████ ██████████████ ██████ ███ ████████ ██████|███ █████ █████████ ████████ ███ █████ ██ ████████████ ███ ██████████ █████ ████ ██████████ ████████ ██ ███ █████████ ███████████ █████ ███ ███ ██ ███ ██████ █████ ██████ ██ ███ ███ █████ ███ █ ████ ██████ ████████ ██████ ███ ███████ ██████ ███ ████ ██ █████ ███ ██████ █████████ █████████ ███ ██████████████ ███ ███████ ██████ ██████ ███ ████████████████ █████ ████ ██████████ █████████ ███ █████ ██████████ ████████ ███ █████ ██ ███ ██████████ █████ ████ █████████ ██ █ █████████ █████ ████ ██████████ ██████████|███ █████ █████████ ████████ ███ █████ ██ ████████████ ███ ██████████ █████ ████ █████████ ████████ ██ ███ █████████ ███████████ █████ ███ ███ ██ ███ ██████ █████ ██████ ██ ███ ███ █████ ███ █ ████ ██████ ████████ ██████ ███ ███████ ██████ ███ ████ ██ █████ ███ ██████ █████████ █████████ ███ ██████████████ ███████ ██████ ██████ ███ ████████████████ ███ █████████████████████ ███████████ ██ █████ ██ ██████████ █████ ████ ███ ███████████ ██████ ███ █████ ██████████ ████████ ███ █████ ██ ███ ██████████ █████ ████ █████████ ███ ████████ ██ ███████████ ███ █████████████████████ ███████████ ██ █████ ██ ██████████ █████ ████ ███ ███████████ ██████████████ █████████ █████████████
Table 22: Subgroup Analysis Results of Disease-Free Survival in the KEYNOTE-091 Study (Reimbursement Request Population)
Subgroups | Pembrolizumab | Placebo | Pembrolizumab vs. placebo HR (95% CI)b | P value for interaction testc | ||||
|---|---|---|---|---|---|---|---|---|
N | DFS event, n (%) | DFS, months, median (95% CI)a | N | DFS event, n (%) | DFS, months, median (95% CI)a | |||
Age, years | ||||||||
< 65 | 189 | 86 (45.5) | 58.7 (35.6; 74.7) | 178 | 94 (52.8) | 45.4 (22.5; 61.9) | 0.77 (0.57; 1.04) | 0.745 |
≥ 65 | 174 | 82 (47.1) | 51.7 (34.4; 58.7) | 185 | 105 (56.8) | 28.6 (20.5; 41.5) | 0.74 (0.55; 0.99) | |
Sex | ||||||||
Male | 234 | 108 (46.2) | 51.7 (35.2; 58.7) | 247 | 131 (53.0) | 35.1 (24.1; 47.0) | 0.83 (0.64; 1.07) | 0.281 |
Female | 129 | 60 (46.5) | 59.5 (36.2; 74.7) | 116 | 68 (58.6) | 25.4 (16.9; 51.6) | 0.63 (0.44; 0.90) | |
Race | ||||||||
White | 278 | 124 (44.6) | 58.3 (42.1; 74.5) | 282 | 145 (51.4) | 39.1 (24.1; NR) | 0.79 (0.62; 1.01) | 0.635 |
All others | 70 | 37 (52.9) | 45.0 (22.8; NR) | 69 | 47 (68.1) | 24.0 (16.8; 35.0) | 0.67 (0.43; 1.04) | |
Geographic region | ||||||||
Asia | 62 | 32 (51.6) | 46.2 (22.8; NR) | 63 | 42 (66.7) | 24.1 (16.9; 35.0) | 0.68 (0.43; 1.09) | 0.949 |
Eastern Europe | 76 | 35 (46.1) | 47.4 (29.5; NR) | 71 | 34 (47.9) | 35.9 (17.5; NR) | 0.85 (0.52; 1.36) | |
Rest of world | 39 | 12 (30.8) | 70.4 (35.6; NR) | 41 | 17 (41.5) | Not reached (35.1; NR) | 0.72 (0.34; 1.55) | |
Western Europe | 186 | 89 (47.8) | 51.3 (35.1; 74.7) | 188 | 106 (56.4) | 29.1 (18.9; 47.0) | 0.75 (0.56; 1.00) | |
Stage at baseline per AJCC v7 | ||||||||
IB | 45 | 15 (33.3) | 74.7 (46.2; NR) | 40 | 16 (40.0) | Not reached (25.5; NR) | 0.66 (0.32; 1.35) | 0.768 |
II | 201 | 82 (40.8) | 70.5 (47.1; 76.0) | 214 | 106 (49.5) | 47.0 (29.4; NR) | 0.74 (0.56; 1.00) | |
IIIA | 117 | 71 (60.7) | 29.8 (22.1; 45.0) | 107 | 75 (70.1) | 18.0 (13.6; 25.4) | 0.81 (0.59; 1.12) | |
PD-L1 TPS | ||||||||
< 1% | 198 | 92 (46.5) | 51.7 (42.1; 74.5) | 198 | 108 (54.5) | 34.8 (20.5; 51.6) | 0.74 (0.56; 0.98) | 0.739 |
1% to 49% | 165 | 76 (46.1) | 52.6 (34.2; 76.7) | 165 | 91 (55.2) | 32.9 (22.3; 47.2) | 0.70 (0.51; 0.96) | |
Smoking status | ||||||||
Never smoker | 69 | 37 (53.6) | 34.8 (22.5; NR) | 47 | 30 (63.8) | 22.8 (13.6; 55.4) | 0.71 (0.44; 1.16) | 0.549 |
Former smoker | 247 | 114 (46.2) | 51.3 (39.0; 70.4) | 266 | 142 (53.4) | 35.1 (25.4; 47.0) | 0.80 (0.62; 1.02) | |
Current smoker | 47 | 17 (36.2) | 59.5 (38.1; NR) | 50 | 27 (54.0) | 31.3 (15.9; NR) | 0.51 (0.27; 0.97) | |
Baseline ECOG performance status | ||||||||
0 | 226 | 107 (47.3) | 53.8 (39.0; 70.5) | 204 | 108 (52.9) | 35.9 (24.0; 51.6) | 0.78 (0.59; 1.02) | 0.607 |
1 | 137 | 61 (44.5) | 47.4 (34.8; NR) | 159 | 91 (57.2) | 27.6 (18.0; 47.2) | 0.73 (0.53; 1.02) | |
Histology | ||||||||
Squamous | 101 | 33 (32.7) | 76.0 (51.3; NR) | 136 | 60 (44.1) | Not reached (35.9; NR) | 0.68 (0.44; 1.05) | 0.801 |
Non-squamous | 262 | 135 (51.5) | 39.0 (34.2; 58.7) | 227 | 139 (61.2) | 24.0 (18.0; 34.8) | 0.73 (0.57; 0.92) | |
EGFR mutation status | ||||||||
No | 144 | 74 (51.4) | 46.7 (29.4; 59.5) | 135 | 83 (61.5) | 24.1 (18.0; 45.4) | 0.76 (0.56; 1.04) | 0.334 |
Yes | 31 | 17 (54.8) | 35.2 (22.8; NR) | 25 | 21 (84.0) | 14.9 (9.2; 29.4) | 0.44 (0.23; 0.84) | |
Unknown | 188 | 77 (41.0) | 58.7 (46.2; 76.0) | 203 | 95 (46.8) | 47.0 (28.4; NR) | 0.79 (0.58; 1.07) | |
ACT = adjuvant chemotherapy; AJCC = American Joint Committee on Cancer; CI = confidence interval; DFS = disease-free survival; ECOG = Eastern Cooperative Oncology Group; EGFR = epidermal growth factor receptor; ITT = intention-to-treat; NR = not reported; NSCLC = non–small cell lung cancer; TPS = tumour proportion score.
Note: Data presented in this table were based on analyses at clinical cut-off date of January 24, 2023, in the ITT population of patients with a PD-L1 TPS of less than 50% and prior ACT.
aFrom product-limit (Kaplan-Meier) method for censored data.
bFor PD-L1 subgroup, analysis was based on multivariate Cox regression model with treatment, adjusted by the following covariates: stage (IB vs. II vs. IIIA), PD-L1 status (≥ 50% vs. 1% to 49% vs. < 1%), ACT (yes vs. no), region (Western Europe vs. Eastern Europe vs. Rest of World vs. Asia), histology (squamous vs. non-squamous), and smoking status (never vs. former/current), using Wald confidence interval. For other subgroups, analysis was based on Cox regression model with treatment as a covariate using Wald confidence interval.
cFor PD-L1 subgroup, analysis was based on multivariate Cox regression model with treatment adjusted by the following covariates: stage (IB vs. II vs. IIIA), PD-L1 status (≥ 50% vs. 1% to 49% vs. < 1%), ACT (yes vs. no), region (Western Europe vs. Eastern Europe vs. Rest of World vs. Asia), histology (squamous vs. non-squamous), smoking status (never vs. former/current), and treatment-by-subgroup interaction (P value of likelihood ratio test for interaction term). For other subgroups, analysis was based on Cox regression model with treatment and subgroup as covariates, and treatment-by-subgroup interaction (P value of likelihood ratio test for interaction term).
Source: Sponsor’s submission.24
Table 23: Additional HRQoL Outcomes in the KEYNOTE-091 Study (PRO FAS Population)
HRQoL outcomes | Pembrolizumab (N = 580) | Placebo (N = 581) |
|---|---|---|
EORTC QLQ-C30 Global Health Status/QoL | ||
N | 580 | 581 |
Improved, n (%; 95 CIa) | 156 (26.9; 23.3 to 30.7) | 205 (35.3; 31.4 to 39.3) |
Stable, n (%; 95 CIa) | 264 (45.5; 41.4 to 49.7) | 274 (47.2; 43.0 to 51.3) |
Improved + Stable, n (%; 95 CIa) | 420 (72.4; 68.6 to 76.0) | 479 (82.4; 79.1 to 85.5) |
Deteriorated, n (%; 95 CIa) | 105 (18.1; 15.1 to 21.5) | 75 (12.9; 10.3 to 15.9) |
Unconfirmed, n (%; 95 CIa) | 21 (3.6; 2.3 to 5.5) | 20 (3.4; 2.1 to 5.3) |
No assessment, n (%; 95 CIa) | 34 (5.9; 4.1 to 8.1) | 7 (1.2; 0.5 to 2.5) |
Difference in % improved | ||
Estimate (95% CI)b | −8.4 (−13.7 to −3.1) | Reference |
P valuec | 0.0020 | |
Difference in % Improved + Stable | ||
Estimate (95% CI)b | −10.0 (−14.8 to −5.2) | Reference |
P valuec | < 0.0001 | |
EORTC QLQ-C30 Symptom Scales — Appetite Loss | ||
N | 580 | 581 |
Improved, n (%; 95 CIa) | 104 (17.9; 14.9 to 21.3) | 141 (24.3; 20.8 to 28.0) |
Stable, n (%; 95 CIa) | 348 (60.0; 55.9 to 64.0) | 362 (62.3; 58.2 to 66.3) |
Improved + Stable, n (%; 95 CIa) | 452 (77.9; 74.3 to 81.2) | 503 (86.6; 83.5 to 89.2) |
Deteriorated, n (%; 95 CIa) | 67 (11.6; 9.1 to 14.4) | 48 (8.3; 6.2 to 10.8) |
Unconfirmed, n (%; 95 CIa) | 29 (5.0; 3.4 to 7.1) | 20 (3.4; 2.1 to 5.3) |
No assessment, n (%; 95 CIa) | 32 (5.5; 3.8 to 7.7) | 10 (1.7; 0.8 to 3.1) |
Difference in % improved | ||
Estimate (95% CI)b | −6.3 (−11.0 to −1.6) | Reference |
P valuec | 0.0082 | |
Difference in % Improved + Stable | ||
Estimate (95% CI)b | −8.6 (−13.0 to −4.3) | Reference |
P valuec | 0.0001 | |
EORTC QLQ-C30 Symptom Scales — Dyspnea | ||
N | 580 | 581 |
Improved, n (%; 95 CIa) | 149 (25.7; 22.2 to 29.4) | 184 (31.7; 27.9 to 35.6) |
Stable, n (%; 95 CIa) | 282 (48.6; 44.5 to 52.8) | 292 (50.3; 46.1 to 54.4) |
Improved + Stable, n (%; 95 CIa) | 431 (74.3; 70.6 to 77.8) | 476 (81.9; 78.6 to 85.0) |
Deteriorated, n (%; 95 CIa) | 87 (15.0; 12.2 to 18.2) | 76 (13.1; 10.4 to 16.1) |
Unconfirmed, n (%; 95 CIa) | 27 (4.7; 3.1 to 6.7) | 20 (3.4; 2.1 to 5.3) |
No assessment, n (%; 95 CIa) | 35 (6.0; 4.2 to 8.3) | 9 (1.5; 0.7 to 2.9) |
Difference in % improved | ||
Estimate (95% CI)b | −6.0 (−11.2 to −0.8) | Reference |
P valuec | 0.0243 | |
Difference in % Improved + Stable | ||
Estimate (95% CI)b | −7.6 (−12.4 to −2.9) | Reference |
P valuec | 0.0017 | |
EORTC QLQ-C30 Symptom Scales — Fatigue | ||
N | 580 | 581 |
Improved, n (%; 95 CIa) | 240 (41.4; 37.3 to 45.5) | 287 (49.4; 45.3 to 53.5) |
Stable, n (%; 95 CIa) | 160 (27.6; 24.0 to 31.4) | 148 (25.5; 22.0 to 29.2) |
Improved + Stable, n (%; 95 CIa) | 400 (69.0; 65.0 to 72.7) | 435 (74.9; 71.1 to 78.3) |
Deteriorated, n (%; 95 CIa) | 132 (22.8; 19.4 to 26.4) | 126 (21.7; 18.4 to 25.3) |
Unconfirmed, n (%; 95 CIa) | 20 (3.4; 2.1 to 5.3) | 13 (2.2; 1.2 to 3.8) |
No assessment, n (%; 95 CIa) | 28 (4.8; 3.2 to 6.9) | 7 (1.2; 0.5 to 2.5) |
Difference in % improved | ||
Estimate (95% CI)b | −8.0 (−13.7 to −2.3) | Reference |
P valuec | 0.0061 | |
Difference in % Improved + stable | ||
Estimate (95% CI)b | −5.9 (−11.1 to −0.7) | Reference |
P valuec | 0.0252 | |
EORTC QLQ-C30 Symptom Scales — Pain | ||
N | 580 | 581 |
Improved, n (%; 95 CIa) | 161 (27.8; 24.1 to 31.6) | 178 (30.6; 26.9 to 34.6) |
Stable, n (%; 95 CIa) | 256 (44.1; 40.0 to 48.3) | 262 (45.1; 41.0 to 49.2) |
Improved + Stable, n (%; 95 CIa) | 417 (71.9; 68.0 to 75.5) | 440 (75.7; 72.0 to 79.2) |
Deteriorated, n (%; 95 CIa) | 115 (19.8; 16.7 to 23.3) | 126 (21.7; 18.4 to 25.3) |
Unconfirmed, n (%; 95 CIa) | 21 (3.6; 2.3 to 5.5) | 9 (1.5; 0.7 to 2.9) |
No assessment, n (%; 95 CIa) | 27 (4.7; 3.1 to 6.7) | 6 (1.0; 0.4 to 2.2) |
Difference in % improved | ||
Estimate (95% CI)b | −2.9 (−8.1 to 2.4) | Reference |
P valuec | 0.2810 | |
Difference in % Improved + Stable | ||
Estimate (95% CI)b | −3.8 (−8.9 to 1.2) | Reference |
P valuec | 0.1374 | |
EORTC QLQ-LC13 Symptom Scales — Chest Pain | ||
N | 580 | 581 |
Improved, n (%; 95 CIa) | 123 (21.2; 18.0 to 24.8) | 117 (20.1; 16.9 to 23.6) |
Stable, n (%; 95 CIa) | 350 (60.4; 56.3 to 64.5) | 372 (64.0; 60.0 to 67.9) |
Improved + Stable, n (%; 95 CIa) | 473 (81.7; 78.3 to 84.8) | 489 (84.2; 80.9 to 87.0) |
Deteriorated, n (%; 95 CIa) | 43 (7.4; 5.4 to 9.9) | 53 (9.1; 6.9 to 11.8) |
Unconfirmed, n (%; 95 CIa) | 31 (5.4; 3.7 to 7.5) | 25 (4.3; 2.8 to 6.3) |
No assessment, n (%; 95 CIa) | 32 (5.5; 3.8 to 7.7) | 14 (2.4; 1.3 to 4.0) |
Difference in % improved | ||
Estimate (95% CI)b | 1.1 (−3.6 to 5.8) | Reference |
P valuec | 0.6422 | |
Difference in % Improved + Stable | ||
Estimate (95% CI)b | −2.5 (−6.8 to 1.9) | Reference |
P valuec | 0.2633 | |
EORTC QLQ-LC13 Symptom Scales — Coughing | ||
N | 580 | 581 |
Improved, n (%; 95 CIa) | 143 (24.7; 21.2 to 28.4) | 163 (28.1; 24.4 to 31.9) |
Stable, n (%; 95 CIa) | 309 (53.4; 49.2 to 57.5) | 314 (54.0; 49.9 to 58.2) |
Improved + Stable, n (%; 95 CIa) | 452 (78.1; 74.5 to 81.4) | 477 (82.1; 78.7 to 85.1) |
Deteriorated, n (%; 95 CIa) | 68 (11.7; 9.2 to 14.7) | 69 (11.9; 9.4 to 14.8) |
Unconfirmed, n (%; 95 CIa) | 31 (5.4; 3.7 to 7.5) | 24 (4.1; 2.7 to 6.1) |
No assessment, n (%; 95 CIa) | 28 (4.8; 3.2 to 6.9) | 11 (1.9; 0.9 to 3.4) |
Difference in % improved | ||
Estimate (95% CI)b | −3.4 (−8.4 to 1.7) | Reference |
P valuec | 0.1947 | |
Difference in % Improved + Stable | ||
Estimate (95% CI)b | −4.0 (−8.6 to 0.6) | Reference |
P valuec | 0.0855 | |
EORTC QLQ-LC13 Symptom Scales — Dyspnea | ||
N | 580 | 581 |
Improved, n (%; 95 CIa) | 191 (33.0; 29.2 to 37.0) | 218 (37.5; 33.6 to 41.6) |
Stable, n (%; 95 CIa) | 195 (33.7; 29.8 to 37.7) | 216 (37.2; 33.2 to 41.3) |
Improved + Stable, n (%; 95 CIa) | 386 (66.7; 62.7 to 70.5) | 434 (74.7; 71.0 to 78.2) |
Deteriorated, n (%; 95 CIa) | 128 (22.1; 18.8 to 25.7) | 110 (18.9; 15.8 to 22.4) |
Unconfirmed, n (%; 95 CIa) | 30 (5.2; 3.5 to 7.3) | 21 (3.6; 2.3 to 5.5) |
No assessment, n (%; 95 CIa) | 35 (6.0; 4.2 to 8.3) | 16 (2.8; 1.6 to 4.4) |
Difference in % improved | ||
Estimate (95% CI)b | −4.5 (−10.0 to 1.0) | Reference |
P valuec | 0.1063 | |
Difference in % Improved + Stable | ||
Estimate (95% CI)b | −8.0 (−13.2 to −2.8) | Reference |
P valuec | 0.0027 | |
CI = confidence interval; EORTC = European Organisation for Research and Treatment of Cancer; FAS = full analysis set; PRO = patient-reported outcomes; QLQ-C30 = Quality of Life Questionnaire-Core 30; QLQ-LC13 = Quality of Life Questionnaire Lung Cancer module; QoL = quality of life; vs. = versus.
Note: Categories of the outcome definitions are as follows:
Improved is defined as a 10-point or more increase in score (in the positive direction) at any time during the trial and confirmed by a 10-point or more increase in score at the next consecutive visit.
Stable is defined as a 10-point or more increase (in the positive direction) or less than 10-point change in score (in the positive or negative direction) from baseline and confirmed by a less than 10-point change in score at the next consecutive visit; OR a less than 10-point change in score and a 10-point or more increase in score at the next consecutive visit.
Improved + stable is defined as the composite of the improved and stable.
Deteriorated is defined as a ≥ 10-point deterioration in score from baseline at any time during the trial when the criteria for improved or stable is not met.
Unconfirmed is defined as when the criteria for improved or stable with confirmation or deterioration is not met.
No assessment is defined as participants who do not have baseline or post-baseline assessments available.
aBased on binomial exact confidence interval method.
bBased on unstratified Miettinen and Nurminen method
c2-sided P value for testing. H0: difference in % = 0 vs. H1: difference in % ≠ 0.
Source: Clinical Study Report for P091V02MK3475.23
EORTC QLQ-C30
The least squares (LS) mean change from baseline in global health status/QOL score did not show a 10-point change in either treatment group with little to no separation between the treatment groups at Week 48, indicating stable scores with no clinically meaningful differences across the treatment groups. The LS mean change from baseline in functioning scores were generally stable from baseline to Week 48 in both treatment groups (Figure 6).23,39
Figure 6: Summary LS Mean Change From Baseline to Week 48 and 95% Confidence Interval in EORTC QLQ-C30 Functional Scales/Global Health Status/QoL (PRO FAS Population)
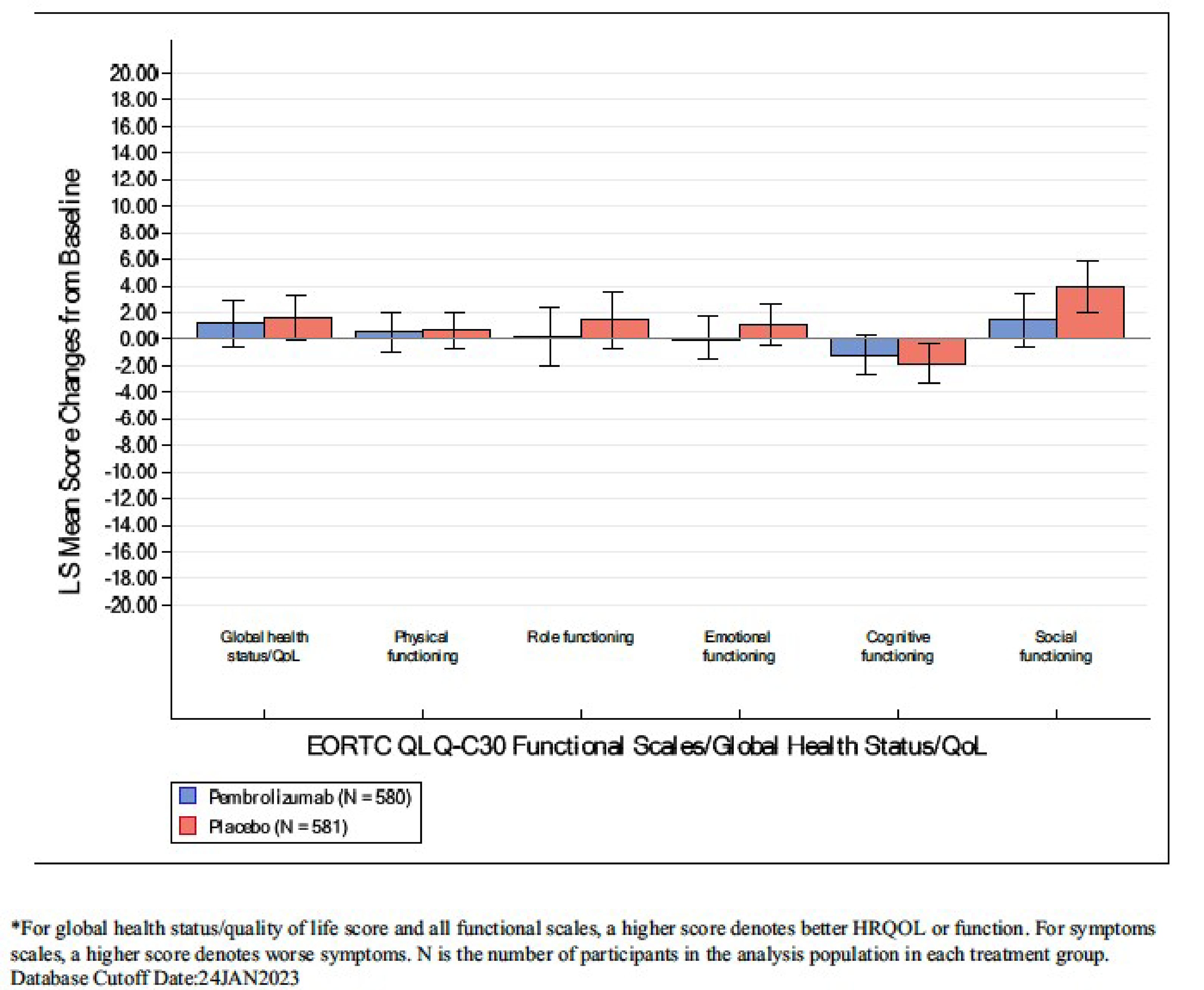
EORTC = European Organisation for Research and Treatment of Cancer; FAS = full analysis set; LS = least squares; PRO = patient-reported outcomes; QLQ-C30 = Quality of Life Questionnaire Core 30; QoL = quality of life.
Source: Clinical Study Report: P091V02MK3475.23
Pharmacoeconomic Review
Abbreviations
AE
adverse event
ALK
anaplastic lymphoma kinase
BIA
budget impact analysis
CDA-AMC
Canada’s Drug Agency
CUA
cost-utility analysis
DF
disease free
DFS
disease-free survival
DM
distant metastasis
EGFR
epidermal growth factor receptor
HRQoL
health-related quality of life
I-ELCAP
International Early Lung Cancer Action Program
ICER
incremental cost-effectiveness ratio
IPD
individual patient-level data
LR
local-regional recurrence
LY
life-year
NSCLC
non–small cell lung cancer
OS
overall survival
PFS
progression-free survival
QALY
quality-adjusted life-year
SEER
Surveillance, Epidemiology, and End Results Program
T2a
tumour stage 2a
ToT
time on treatment
TPS
tumour proportion score
WTP
willingness to pay
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Pembrolizumab (Keytruda), IV solution |
Indication | For the adjuvant treatment of adult patients with stage IB (T2a ≥ 4 cm), II, or IIIA NSCLC who have undergone complete resection and platinum-based chemotherapy |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | April 19, 2023 |
Reimbursement request | For the adjuvant treatment of adult patients with stage IB (T2a ≥ 4 cm), II, or IIIA NSCLC who have undergone complete resection and platinum-based chemotherapy and whose tumours have a PD-L1 TPS < 50%, as determined by a validated test |
Sponsor | Merck Canada Inc. |
Submission history | Pembrolizumab (Keytruda) has been reviewed for numerous indications by CDA-AMC. Indication: For the treatment of patients with metastatic squamous NSCLC in combination with carboplatin and either paclitaxel or nab-paclitaxel, in adults with no prior systemic chemotherapy treatment for metastatic NSCLC. Recommendation: Reimburse with clinical criteria and/or conditions Recommendation date: January 3, 2020 Indication: In combination with pemetrexed and platinum chemotherapy, for the treatment of metastatic non-squamous NSCLC, in adults with no EGFR or ALK genomic tumour aberrations, and no prior systemic chemotherapy treatment for metastatic NSCLC. Recommendation: Reimburse with clinical criteria and/or conditions Recommendation date: May 31, 2019 Indication: For previously untreated patients with metastatic NSCLC whose tumours express PD-L1 and who do not harbor a sensitizing EGFR mutation or ALK translocation. Funding requested for patients with a TPS of PD-L1 ≥ 50%. Recommendation: Reimburse with clinical criteria and/or conditions Recommendation date: August 23, 2017 Indication: For the treatment of patients with metastatic NSCLC whose tumours express PD-L1 (as determined by a validated test) and who have disease progression on or after platinum-containing chemotherapy. Funding requested for patients with a TPS of PD-L1 ≥ 1%. Recommendation: Reimburse with clinical criteria and/or conditions Recommendation date: November 3, 2016 |
ALK = anaplastic lymphoma kinase; CDA-AMC = Canada’s Drug Agency; EGFR = epidermal growth factor receptor; NOC = Notice of Compliance; NSCLC = non–small cell lung cancer; T2a = tumour stage 2a; TPS = tumour proportion score.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Markov model |
Target population | Adult patients with stage IB (T2a ≥ 4 cm), II, or IIIA NSCLC who have undergone complete resection and received platinum-based chemotherapy and whose tumours have a PD-L1 TPS < 50% as determined by a validated test |
Treatment | Pembrolizumab |
Dose regimen | 200 mg every 3 weeks or 400 mg every 6 weeks for up to 1 year or until disease recurrence or unacceptable toxicity |
Submitted price | Pembrolizumab: $4,400 per 100 mg per 4mL vial |
Submitted treatment cost | $8,800 per 21-day cycle ($158,400 for 18 cycles)a |
Comparator | Active surveillance |
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | Lifetime (36 years) |
Key data sources | KEYNOTE-091 provided DFS data to estimate transitions from DF to LR, DM, and death for pembrolizumab and active surveillance. A SEER-Medicare real-world evidence study (calibrated with OS data from KEYNOTE-091) estimated transitions from LR to DM. KEYNOTE-189 and KEYNOTE-407 informed OS and PFS data used to estimate transitions from DM to death. |
Submitted results | ICER vs. active surveillance = $68,241 per QALY gained ($84,667 incremental costs and 1.24 incremental QALYs) |
Key limitations |
|
CDA-AMC reanalysis results |
|
CDA-AMC = Canada’s Drug Agency; DF = disease free; DM = distant metastasis; DFS = disease-free survival; ICER = incremental cost-effectiveness ratio; LR = local-regional recurrence; LY = life-year; NSCLC = non–small cell lung cancer; OS = overall survival; PFS = progression-free survival; QALY = quality-adjusted life-year; SEER = Surveillance, Epidemiology, and End Results Program; T2a = tumour stage 2a; TPS = tumour proportion score; WTP = willingness to pay; vs. = versus.
aThe sponsor assumed patients received a fixed dose of pembrolizumab of 200 mg every 3 weeks for up to 18 cycles, which incorporated vial sharing.
Conclusions
Evidence from the phase III, randomized, placebo-controlled KEYNOTE-091 trial comparing the efficacy and safety of adjuvant pembrolizumab and placebo in adult patients with stage IB to IIIA non–small cell lung cancer (NSCLC) who have undergone complete resection and received platinum-based chemotherapy and whose tumours have a PD-L1 tumour proportion score (TPS) of less than 50% showed adjuvant pembrolizumab provided a clinically meaningful benefit in disease-free survival (DFS). The Clinical Review concluded that pembrolizumab likely results in an increase in the probability of DFS at 24 and 48 months compared to placebo, with a moderate certainty of evidence. Although there is a trend toward improved overall survival (OS) in favour of pembrolizumab, the OS data are uncertain due to immaturity (the data cut-off date was January 24, 2023, with a median follow-up period of 47 months). The clinical experts consulted for this review emphasized that a longer follow-up is needed to determine the OS benefit of pembrolizumab relative to placebo.
The CDA-AMC base-case results align with those of the sponsor’s submitted analysis, indicating that adjuvant pembrolizumab is not cost-effective at a willingness-to-pay (WTP) threshold of $50,000 per quality-adjusted life-year (QALY) gained. In the CDA-AMC base case, adjuvant treatment with pembrolizumab was associated with an incremental cost-effectiveness ratio (ICER) of $103,900 per QALY gained compared with active surveillance. When compared to the sponsor’s analysis, the CDA-AMC base case estimated a reduced benefit with pembrolizumab (i.e., 0.73 incremental QALYs compared with 1.24 QALYs in the sponsor’s analysis) at a similar cost ($75,957 incremental costs in the CDA-AMC base case versus $84,667 in the sponsor’s analysis). The estimated ICER was driven by the selection of alternative distributions for extrapolating DFS, the cure assumption applied to patients who achieve long-term DFS, and the dosage of pembrolizumab. In the CDA-AMC base case, a price reduction of 54% is required for pembrolizumab to be considered cost-effective relative to active surveillance at a WTP threshold of $50,000 per QALY gained. This would reduce the price of pembrolizumab from $4,400 to $2,044 per 4 mL vial. With this price reduction, the per-patient 28-day drug acquisition costs for pembrolizumab would be $4,088, assuming weight-based dosing and vial sharing (i.e., no wastage).
The CDA-AMC base-case results assume that patients treated with pembrolizumab gain an additional life-year (LY) compared to patients on active surveillance. In the absence of robust, long-term clinical evidence, the extent of this survival benefit is highly uncertain. Should the OS benefit of pembrolizumab be less than 0.9 years, the ICER would be higher than that in the CDA-AMC base case, requiring larger price reductions to achieve cost-effectiveness. Moreover, when comparing the median follow-up in the KEYNOTE-091 trial to the model’s time horizon (47 months versus 36 years, respectively), it is important to note that most of the QALY and LY benefits realized by patients treated with pembrolizumab in the CDA-AMC base case accrue in the posttrial period of the model based on extrapolation.
Finally, the sponsor did not consider adjuvant osimertinib as a relevant comparator in its economic analysis. The cost-effectiveness of pembrolizumab relative to osimertinib in the adjuvant setting is therefore unknown.
Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, clinician groups, and drug plans that participated in the CDA-AMC review process.
Patient input was received in the form of a joint submission from the Lung Health Foundation (also known as the Ontario Lung Association), Lung Cancer Canada, and the Canadian Cancer Survivor Network, which collected perspectives of caregivers and patients with lung cancer through surveys and interviews. Patients with lung cancer reported fatigue, shortness of breath, coughing, pain, chest tightness, wheezing, weight loss, diminished appetite and a negative impact on their mental health and ability to carry out daily activities. Respondents described experience with a variety of treatments, which included surgery, radiation, chemotherapy, targeted therapy, immunotherapy and medications such as Alecensaro, lorlatinib, gefitinib, entrectinib, osimertinib, and brigatinib. Patients reported that these treatments reduced some symptoms of disease but also had lingering side effects. Patients also reported difficulties obtaining a diagnosis, accessing information about available treatments, and receiving timely access to treatment options, including next-line therapies, if their current targeted therapy becomes ineffective. Treatment goals identified by patients included stopping or slowing the progression of the disease with minimal side effects. Patient input indicated that 3 patients had experience with pembrolizumab after resection. One patient received pembrolizumab in combination with chemotherapy, the second patient received pembrolizumab after progression for 3 years, while the third was rechallenged with a double dose of pembrolizumab 2 months after stopping treatment. All patients receiving pembrolizumab after resection reported that treatment was associated with adverse events (AEs), such as constipation, nausea, diarrhea, eczema, worsened diabetes, sore muscles, fatigue, and worsened liver enzymes.
Clinician input was received from the Ontario Health (Cancer Care Ontario) Lung Cancer Drug Advisory Committee and Lung Cancer Canada, based on interviews with clinical experts and published literature. Current treatment for patients with stage IB (tumour stage 2a [T2a] ≥ 4 cm), II, or IIIA NSCLC who have undergone complete resection and received platinum-based chemotherapy and whose tumours have a PD-L1 TPS of less than 50% is limited to active surveillance. Clinician input noted that adjuvant atezolizumab is a therapeutic option among patients whose tumours have a PD-L1 TPS of 50% or greater. The clinician input also highlighted that, in the case of patients with resected NSCLC and a sensitizing epidermal growth factor receptor (EGFR) mutation along with an identified PD-L1 TPS of less than 50%, clinicians would need to choose between adjuvant pembrolizumab and adjuvant osimertinib. The identified treatment goal was a cure, which would be achieved by delaying recurrences and improving OS. The clinician input described DFS and OS as acceptable outcomes for assessing treatment response. Clinicians anticipated that the reimbursement of pembrolizumab as adjuvant therapy following platinum-based chemotherapy would fill in a treatment gap among patients with a PD-L1 TPS of less than 50%. Clinicians also noted that they would consider rechallenging with pembrolizumab following its use in the adjuvant setting, particularly if cancer recurred at least 6 months after completing adjuvant therapy, in line with historically accepted clinical practices.
The drug plans participating in this review noted that pembrolizumab is an add-on therapy to adjuvant chemotherapy. The drug plans also anticipated that pembrolizumab would not replace atezolizumab in the adjuvant setting. The drug plans expressed concerns with the eligibility criteria, such as having to complete a maximum of 4 cycles of adjuvant chemotherapy, and emphasized that weight-based dosing would be implemented for pembrolizumab.
Two of these concerns were addressed in the sponsor’s model:
DFS and OS, outcomes that are valued by patients and clinicians, were included in the model.
The impact of disease and treatment on patient health-related quality of life (HRQoL) was captured with utility values.
In addition, CDA-AMC addressed some of these concerns:
The time to consider patients to be rechallenged with pembrolizumab was aligned with clinical input.
Weight-based dosing approach was applied for pembrolizumab in the base case, in line with input received from the drug plans.
CDA-AMC was unable to address the concern raised that the cost-effectiveness of pembrolizumab relative to osimertinib in the adjuvant setting is unknown.
Economic Review
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis (CUA) of pembrolizumab as adjuvant monotherapy compared with active surveillance. The model population comprised adult patients with stage IB (T2a ≥ 4 cm), II, or IIIA NSCLC who have undergone complete resection and received platinum-based chemotherapy.1 The sponsor requested a deviation to focus the base-case analysis exclusively on patients with a PD-L1 TPS of less than 50%, as determined by a validated test, in alignment with the reimbursement criteria. CDA-AMC approved the deviation, recognizing that pembrolizumab is less effective than the standard treatment for patients with a PD-L1 TPS of 50% or greater. As a result, the modelled cohort is narrower than the full Health Canada–indicated population but aligns with the reimbursement request population.
Pembrolizumab is available as a solution for IV infusion (100 mg per 4 mL vial).2 The recommended dosage of pembrolizumab is either 200 mg every 3 weeks or 400 mg every 6 weeks for up to 1 year or until disease recurrence or unacceptable toxicity.2 At the submitted price of $4,400 per 4 mL vial, the cost of pembrolizumab per 21-day cycle was estimated by the sponsor to be $8,800 per patient.1 The sponsor incorporated vial sharing in its calculation of drug costs. The comparator in this analysis was active surveillance.
The clinical outcomes modelled were OS, DFS, and time on treatment (ToT).1 The model simulated LYs, QALYs, and costs for each treatment over a lifetime time horizon of 36 years, discounted at an annual rate of 1.5%. The analysis was undertaken from the perspective of the Canadian public health care payer.
Model Structure
The sponsor submitted a Markov model consisting of 4 mutually exclusive health states: disease free (DF), local-regional recurrence (LR), distant metastases (DM), and death, with transitions occurring on a weekly cycle (Appendix 3, Figure 1).1 All patients entered the model in the DF health state, receiving either adjuvant pembrolizumab or active surveillance following surgical resection and adjuvant platinum-based chemotherapy. Patients in the DF health state may experience 1 of 2 types of progression: LR or DM. Patients in the LR health state faced the risk of further progression to the DM health state. Patients in all health states were subject to a probability of death in each cycle.
Model Inputs
Baseline patient characteristics were derived from the KEYNOTE-091 study, a phase III, randomized, placebo-controlled trial designed to evaluate the efficacy and safety of pembrolizumab as adjuvant therapy in patients with completely resected stage IB to IIIA NSCLC and known PD-L1 expression.3 The modelled population is aligned with the analytical sample restricted to patients who received adjuvant chemotherapy with a PD-L1 TPS of less than 50% (N = 726), henceforth referred to as the reimbursement request population. The average patient in the modelled cohort, whom the sponsor assumed reflected the patient population in Canada, was 64 years old, weighed 75 kg, had a mean body surface area of 1.9 m2, and was more likely to be male (68%).1 These characteristics were used to inform the drug dosage regimens, as well as the age- and sex-specific distribution of the general population mortality risk, which the sponsor used to cap the lower bound for all-cause mortality in the model.
Clinical efficacy parameters used to characterize pembrolizumab and active surveillance (including OS, DFS, and ToT) were derived from various data sources. Transition probabilities starting from the DF health state were estimated based on individual patient-level data (IPD) from the reimbursement request population of the KEYNOTE-091 trial using the third interim analysis (data cut-off date of January 24, 2023; median follow-up of 46.6 months).3 The sponsor used a parametric multistate modelling approach to estimate transition probabilities between health states,4,5 specifically from DF to LR, DM, and death. Parametric functions were fitted by a treatment strategy to each of the 3 individual transitions starting from DF, accounting for competing risks. The selection of parametric survival models used in the base case was based on the clinical plausibility of long-term survival projections, visual inspection of model fit, and mean-squared error of statistical fit.1 Transition probabilities from LR to DM and LR to death were estimated using IPD from the Surveillance, Epidemiology, and End Results (SEER)-Medicare database (SEER data for 2007 to 2017; associated Medicare claims data for 2007 to 2019), which included a real-world cohort study of patients (N = 392) with completely resected stage IB to IIIA NSCLC, with or without receipt of adjuvant chemotherapy.6 The sponsor used an exponential model to estimate these transition probabilities and calibrated values that optimized the statistical fit between the predicted and observed OS in each arm of KEYNOTE-091. Transition probabilities from DM to death were estimated for each intervention using OS and progression-free survival (PFS) data from the KEYNOTE-189,7 KEYNOTE-407,8 and other trials of treatments for metastatic NSCLC.9-15 These transition probabilities from DM depended on market shares of first-line treatments for metastatic NSCLC and the efficacy of those treatments with respect to OS. The sponsor used Canadian market shares of first-line treatments for metastatic NSCLC16,17 coupled with evidence from network meta-analyses to compare treatments for metastatic NSCLC in terms of OS and PFS.18 The sponsor used exponential OS and PFS distributions for each first-line treatment based on trials involving metastatic NSCLC. The expected OS in the DM health state was calculated for each intervention as a market share–weighted average of expected OS under different treatments for metastatic NSCLC.
The 3 health-state transition rates (LR to DM, LR to death, and DM to death) were simultaneously calibrated to OS data in each arm of the KEYNOTE-091 trial by rescaling the 3 rates by the same multiplicative factor.1 Calibration effects and differential transition rates between adjuvant pembrolizumab and active surveillance from the LR and DM health states were gradually reduced starting at year 7, and fully attenuated (using the same transition rates for both interventions) by the start of year 10. Additionally, the per-cycle transition probability from each health state (DF, LR, and DM) to death was set to the higher of 2 values: the estimated probability from parametric modelling or the background mortality rate. As a result, the risk of death in any given cycle, regardless of health state, was at least as high as the age- and sex-adjusted all-cause mortality rate of the general population in Canada.
The sponsor did not incorporate treatment-effectiveness waning in the submitted model.1 Instead, the model assumes that treatment with adjuvant pembrolizumab remains equally effective indefinitely, with sustained survival and HRQoL improvements over the lifetime horizon. In addition, the sponsor applied a cure assumption to patients who achieved long-term DFS. Beginning in year 7, the per-cycle risk of progression from the DF health state gradually decreased, culminating in a 95% reduction by year 10, achieving the cure landmark.1 The sponsor argued that the cure assumption is supported by evidence, citing a 20-year follow-up data from the International Early Lung Cancer Action Program (I-ELCAP) cohort study.19
Health-state utility values for the DF (0.806), LR (0.776), and preprogression DM (0.743) were informed by the responses to the EQ-5D-3L questionnaire collected in the reimbursement request population of the KEYNOTE-091 trial and estimated through a UK-specific algorithm.20 The utility value for postprogression DM (0.667) was derived using EQ-5D-3L data collected from the KEYNOTE-189 and KEYNOTE-407 trials in the metastatic NSCLC setting, and was also estimated with a UK-specific algorithm. The utility for the DM health state was adjusted for the time spent in pre- versus postprogression. The sponsor did not apply an age-related disutility in the submitted base case. Disutilities related to AEs were calculated based on the mean duration and frequency of episodes, along with the estimated disutility associated with an active grade 3 or worse AE based on analyses of EQ-5D-3L data from the KEYNOTE-091 trial.3
The economic model included costs associated with drug acquisition and administration for adjuvant therapy and subsequent therapy, as well as costs associated with radiotherapy, salvage surgery, disease management, terminal care, and PD-L1 testing. Drug acquisition costs for pembrolizumab were calculated as a function of the sponsor-submitted unit price, dosing schedule (consistent with that described in the overview section), and the proportion of patients on treatment based on ToT curves reported in the KEYNOTE-091 trial.3 Costs incurred in the LR health state included one-time costs of chemotherapy, radiotherapy, and salvage surgery. The distribution of patients on these therapies were based on a study by Agulnik et al. (2020).21 The dosing schedules were based on prescribing information and treatment protocols from clinical trials, while the mean ToT was derived from data from patients who experienced LR in the KEYNOTE-091 study, along with data from the KEYNOTE-671 study and relevant published literature.22 The sponsor assumed that nearly half of the patients receiving chemotherapy with radiotherapy would incur the cost of durvalumab treatment. The dosing schedule of durvalumab was sourced from Cancer Care Ontario.23 The one-time cost for patients entering the LR health state was calculated as a weighted average, combining treatment frequencies and associated costs. Patients entering the DM health state were assumed to incur a one-time cost of first- and second-line treatments for metastatic NSCLC.1 The distribution of patients across these treatments was sourced from sponsor’s internal analysis of the Oncology Continuous Audit of Patients and Prescriptions Syndicated database.24 Dosing schedules were based on prescribing information, treatment protocols in pivotal clinical trials, and clinical guidelines.7,8,11,13-15,22,25-32 The duration of treatments was determined using the exponential rates of PFS failure from relevant trials, empirical data from the Flatiron database of adult patients with metastatic NSCLC who initiated second-line treatments, and findings from published literature.13,14 Patients who transitioned to the death state incurred a one-time cost associated with terminal care.33
Disease management costs included weekly costs of medical resource use, which were estimated from the Canadian Institute for Health Information Patient Cost Estimator,34 the Schedule of Benefits,35 and a previous reimbursement review.35,36 Finally, for PD-L1–positive patients, the model applied a cost of $105 for PD-L1 biomarker testing, sourced from Quebec’s medical biology procedures directory.37 In the KEYNOTE-091 study, 72% of patients received an PD-L1 test.3 The total average cost to identify 1 patient eligible for adjuvant pembrolizumab was therefore $146.1
Summary of Sponsor’s Economic Evaluation Results
The sponsor conducted all analyses probabilistically over 5,000 iterations. The deterministic and probabilistic results were similar. The probabilistic findings are presented in the following section.
Base-Case Results
The results of the sponsor’s probabilistic base-case analysis for the reimbursement request population suggested that adjuvant pembrolizumab was associated with an additional 1.24 QALYs at an increased cost of $84,667, relative to active surveillance. This resulted in an ICER of $68,241 per QALY gained (Table 3).
The sponsor’s analysis predicted that pembrolizumab was associated with a longer duration of life (1.53 incremental LYs) compared with active surveillance. Given the duration of follow-up in the KEYNOTE-091 trial (a median follow-up of 46.6 months and a maximum follow-up of 84.2 months) in contrast with the model’s lifetime horizon of 36 years, it is important to highlight that the near entirety (92%) of the incremental QALYs gained by patients receiving pembrolizumab was derived from a period for which observed trial data are not available (i.e., the extrapolated period).
The probability that pembrolizumab would be cost-effective compared to active surveillance, at a WTP threshold of $50,000 per QALY gained, was 25%. The submitted analysis is based on the publicly available prices for all drug treatments. Additional results from the sponsor’s submitted economic evaluation base case are presented in Appendix 3.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER vs. active surveillance ($ per QALY) |
|---|---|---|---|---|---|
Active surveillance | 229,461 | Reference | 6.96 | Reference | Reference |
Pembrolizumab | 314,128 | 84,667 | 8.20 | 1.24 | 68,241 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Source: Sponsor’s pharmacoeconomic submission.1
Sensitivity and Scenario Analysis Results
The sponsor conducted several scenario analyses, which included exploring alternative discount rates and alternative distributions and calibration factors to estimate transition probabilities between health states and assumed equal distributions of first- and second-line treatments across interventions. Additional scenarios involved using alternative utility estimates derived from the KEYNOTE-091, KEYNOTE-189, and KEYNOTE-407 trials, incorporating age-adjusted disutility, excluding AE-related disutility, and assuming no vial sharing. Across all scenarios, adjuvant pembrolizumab resulted in ICERs ranging from $53,064 to $80,949 per QALY gained compared with active surveillance. The ICER was most sensitive to applying no calibration for transitions from the LR and DM health states, resulting in an ICER of $80,949 per QALY gained.
The sponsor’s model allowed for exploration of the results based on a societal perspective by providing an option to include additional costs associated with productivity losses for both patients and caregivers. In this analysis, the ICER of pembrolizumab relative to active surveillance was $65,829 per QALY gained. The results were similar to the sponsor’s base-case analysis using a health care payer perspective.
Appraisal of the Sponsor’s Economic Evaluation
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications for the economic analysis:
The impact of pembrolizumab on long-term OS is highly uncertain. The sponsor’s base case predicts a survival advantage with adjuvant pembrolizumab compared with active surveillance (1.53 incremental LYs). However, as of the data cut-off date of January 24, 2023, the median OS had not been reached in either intervention group, indicating OS data immaturity. According to the Clinical Review of the KEYNOTE-091 trial, the certainty of the OS evidence is low due to serious imprecision, with few OS events reported during the median follow-up of 46.6 months. While pembrolizumab may provide an OS benefit compared to placebo at 48 months, the clinical significance of this increase is uncertain, as an empirically derived and validated minimal important difference in OS between the intervention groups was not identified. Clinical expert feedback suggested that delayed disease recurrence, as reflected by DFS in the KEYNOTE-091 trial, may translate into OS benefits, although the extent remains uncertain because of limited OS data.
Additional uncertainty is associated with the predicted OS for pembrolizumab because of the modelling approach adopted by the sponsor. Data informing disease progression once patients experienced an LR or DM health state were not routinely collected in the KEYNOTE-091 trial. Instead, this information was derived from the SEER-Medicare database and relevant published literature. The sponsor used an exponential distribution to model transition probabilities from the LR and DM health states for pembrolizumab and active surveillance, making the hazard rates time-invariant. However, the use of an exponential distribution, which assumes a constant transition risk over time, may lead to oversimplification, particularly when the risk of disease progression fluctuates. For example, if the risk of developing DM among patients who experience LR is assumed to decrease over time, the exponential model may overestimate the number of transitions in the long term, potentially biasing survival estimates. As acknowledged by the sponsor, the submitted model requires calibration due to an underestimation of OS in both the pembrolizumab and active surveillance groups in the KEYNOTE-091 trial. This indicates that postrecurrence transitions should have been less frequent, suggesting the exponential distribution may have oversimplified postrecurrence dynamics and overestimated long-term transition probabilities for both interventions. The predicted incremental gain in LYs associated with pembrolizumab is therefore highly uncertain, given the limitations of the immature OS data observed in the KEYNOTE-091 trial, the lack of available long-term efficacy data for pembrolizumab and active surveillance, and the sponsor’s modelling approach when extrapolating OS.
CDA-AMC could not address this limitation because of the immaturity of the OS data and the structure of the submitted model. Approximately 75% of incremental LYs gained by patients treated with pembrolizumab accrued beyond the trial’s maximum follow-up of 84 months, representing model-generated outcomes rather than trial-based evidence.
The long-term DFS of active surveillance is uncertain. The sponsor used parametric modelling to extrapolate long-term DFS beyond the observable time points in the KEYNOTE-091 trial (a median follow-up period of 46.6 months and a maximum follow-up period of 84 months) to a lifetime horizon of 36 years. Parametric functions were fitted by treatment strategy to each of the 3 transitions originating from the DF health state — transitioning to LR, DM, or death — while accounting for competing risks. As a result, the projected long-term DFS curve in each treatment strategy was influenced by the combination of distributions applied to these transitions. The parametric models selected by the sponsor for transitions originating from the DF health state, along with other modelling assumptions, such as the inclusion of a cure assumption, resulted in an incremental gain of 1.18 QALYs in the DF health state for patients treated with pembrolizumab. As a result, 95% of the total incremental QALYs accrued through extrapolation.
The sponsor evaluated 67 potential combinations of parametric functions for transitions from the DF health state and selected the following distributions for both interventions: log-normal for the transitions from DF to LR and from DF to DM, and exponential for the transition from DF to death. When comparing the different parametric functions used to model DFS in the active surveillance arm, the combination selected by the sponsor was ranked 16th among 67. Additionally, the sponsor externally validated the long-term extrapolation of DFS for patients on active surveillance using IPD data obtained from the SEER-Medicare database. However, a direct comparison between these 2 patient populations is inappropriate because not all patients in the SEER-Medicare database received adjuvant chemotherapy, whereas all patients in the KEYNOTE-091 trial did. Consequently, the DFS curve from this real-world cohort is expected to differ from — and likely underestimates — the predicted DFS for patients on active surveillance.
The review team evaluated parametric models based on both their statistical fit to the observed DFS data during the trial period and the clinical plausibility of the long-term DFS estimates derived through extrapolation. The generalized gamma distribution was selected to extrapolate the transition from DF to LR as it provided an improved visual fit between the predicted and observed cumulative incidence of LR in the KEYNOTE-091 trial; an improved statistical fit between the predicted and observed DFS in KEYNOTE-091, ranking second out of 67 functional combinations; and improved the clinical plausibility when validated against the Patient360 NSCLC dataset, which the clinical experts consulted for this review considered a more relevant real-world cohort for external validation because all patients receive adjuvant chemotherapy.
In reanalysis, CDA-AMC selected the generalized gamma distribution to extrapolate the transition from DF to LR for patients on active surveillance in the long term.
The cure assumption is uncertain. The model assumed that the proportion of patients who are deemed cured increases linearly from the start of year 7 and reaches a maximum of 95% at year 10, citing 20-year follow-up data from the I-ELCAP cohort study.19 The sponsor noted that, because lung cancer–related deaths are typically a consequence of disease recurrence, the absence of such deaths beyond 10 years in the I-ELCAP study suggests that recurrence is unlikely beyond this period, supporting the assumption that long-term survivors are effectively cured. However, the study referenced by the sponsor involved a population in which 81% of the patients had clinical stage I disease at pretreatment CT (indicating less-advanced disease and potentially better prognosis) while only 12% of patients in the reimbursement request population for the KEYNOTE-091 trial had stage I disease at baseline. In addition, the cure assumption implemented by the sponsor equated the long-term survival outcomes of current or former tobacco users with those of the average person in Canada, an assumption that lacks face validity. The model presumed that cured patients would experience the same mortality outcomes as a general age- and sex-matched population in Canada, suggesting no excess cancer-related mortality. However, feedback from the clinical experts indicated that it is unlikely that cured patients experience the same long-term health outcomes as the general population in Canada. While the assumption of no excess cancer-related mortality may be plausible, it likely overestimates survival given the predominance of early-stage lung cancer in the I-ELCAP study. More importantly, it is not reasonable to assume no excess smoking-related mortality in a patient population consisting predominantly of current and former smokers (84%), as seen in the KEYNOTE-091 reimbursement group. The clinical experts noted that cancer is not the only prognostic indicator of higher mortality in this patient group, as current and former tobacco users often have significant comorbidities, including chronic obstructive pulmonary disease, making them more prone to increased mortality from cardiovascular and respiratory diseases.
CDA-AMC acknowledges the uncertainty regarding the risk of late recurrence that patients with NSCLC may experience beyond the 10-year landmark for DFS. Evidence points to the magnitude of a late recurrence risk in patients who remain recurrence-free 10 years after resection, which demonstrates that the recurrence-free probability may be 89%, 84%, and 65% for patients with lymph-node stage N0, N1, and N2 cancers, respectively.38
CDA-AMC conducted a base-case reanalysis that applied an instantaneous cure assumption at 10 years, with an 82% cure fraction for patients who remained DF. This was based on recurrence-free probabilities from the literature, weighted according to the distribution of the N status in the reimbursement request population. This reanalysis is optimistic as it does not account for excess smoking-related mortality in a population predominantly comprising current and former smokers.
CDA-AMC conducted an additional scenario analysis assuming no cure assumption.
The time points used to attenuate the differential transition rates between interventions are uncertain. The sponsor fitted exponential models for the transitions from LR to DM and LR to death using real-world evidence from the SEER-Medicare database. The sponsor estimated the cause-specific hazards of LR to DM and LR to death by following a subset of patients (identified as having LR at least 30 days before any DM occurrence) from the time of LR to the earliest of the competing event, loss to follow-up, or end of the data-capture period. Using this approach, the sponsor estimated the weekly probability of experiencing DM (0.005) and death (0.002) among patients with LR, for both pembrolizumab and active surveillance. The predicted OS (based on the transition probabilities derived from real-world evidence) underestimated the observed OS in both the pembrolizumab and active surveillance arms of the KEYNOTE-091 trial, indicating that at least 1 of the postrecurrence transition probabilities (LR to DM, LR to death, or DM to death) should occur less frequently for both interventions. To address this, the sponsor conducted a calibration approach that relied on several key assumptions. First, the sponsor assumed that, in the first 7 years from adjuvant treatment initiation, the probability of experiencing DM or death among patients with LR differs based on the intervention received. Specifically, patients on active surveillance are expected to progress faster than patients treated with pembrolizumab. Second, the sponsor assumed that transition probabilities starting from the LR state in patients treated with pembrolizumab converge with those of patients on active surveillance between 7 and 10 years from adjuvant treatment initiation. Third, the sponsor assumed that, from 10 years onward, the probability of experiencing DM or death among patients with LR is equal between patients who received adjuvant pembrolizumab and active surveillance.
Due to the substantial underestimate of OS produced by the sponsor’s modelling approach (the limitation on long-term OS uncertainty), calibration is necessary to better align the predicted OS with the observed OS from the KEYNOTE-091 trial. However, because there are no time-to-event trial data for subsequent transitions to support the sponsor’s assumptions regarding the extent and duration of differential progression rates between interventions, assumptions regarding the time points used to initiate and complete the capping of the calibration effect are highly uncertain. The sponsor’s choice of calibration time points implicitly assumes that patients on active surveillance progress faster than patients treated with pembrolizumab well beyond the KEYNOTE-091 trial’s maximum follow-up. To address this, CDA-AMC initiated the capping of the calibration effect at the trial’s median follow-up of 46.6 months (3.9 years) and capped it at the trial’s maximum follow-up of 84.2 months (7 years). The median follow-up time represents the point at which outcomes are robust for half of the patients in the trial. However, beyond the median follow-up, fewer participants were observed, leading to greater uncertainty in long-term outcomes. It is therefore reasonable to start reducing the weight (or calibration effect) of trial data as the level of certainty about survival outcomes diminishes over time. The maximum follow-up time represents the point beyond which the trial no longer provides data. Beyond this point, assumptions about treatment effects become highly speculative. Attenuating the effect from the median follow-up ensures a gradual transition, while phasing it out completely by the maximum follow-up helps avoid over-extrapolation.
CDA-AMC conducted a base-case reanalysis that initiated capping of the calibration effect at the trial’s median follow-up of 46.6 months (3.9 years) and capped it at the trial’s maximum follow-up of 84.2 months (7 years).
Age-related disutility was not incorporated in the submitted base case. The sponsor’s submitted base case did not incorporate age-related disutilities, which is a critical omission in a cohort model with a lifetime horizon that follows an aging population. Age-related disutilities account for the natural decline in HRQoL as patients age, reflecting the cumulative impact of comorbidities and decreased physical and cognitive functioning. Ignoring these factors can result in overestimating the QALYs gained with treatment, particularly for older patients, as the baseline utility values remain artificially high over time. This could lead to an inflated perception of pembrolizumab’s effectiveness over time, as it does not account for the natural decline in quality of life unrelated to the treatment itself. Consequently, this omission biases the incremental QALYs in favour of adjuvant pembrolizumab, skewing the cost-effectiveness results to appear more favourable. Incorporating age-related disutilities provides a more accurate assessment of patient health outcomes and the cost-effectiveness of adjuvant pembrolizumab within a population that is progressively aging.
CDA-AMC conducted a base-case reanalysis that included age-related disutilities.
Pembrolizumab dosage does not align with public drug plans’ implementation strategy. In the KEYNOTE-091 trial, pembrolizumab was administered at a fixed dosage of 200 mg every 3 weeks. Similarly, in the economic model, the sponsor assumed that all patients would receive 200 mg every 3 weeks. Participating public drug plan input and clinical expert feedback received by the review team indicates that a weight-based dosage is likely to be adopted for pembrolizumab (2 mg/kg [up to 200 mg] every 3 weeks or 4 mg/kg [up to 400 mg] every 6 weeks). Using weight-based dosing would reduce drug acquisition costs and lower the ICER for pembrolizumab compared to active surveillance, while offering improved dosing flexibility. However, whether a weight-based regimen would result in the same clinical outcomes as a fixed-dose regimen remains uncertain. Lower doses may affect the safety profile and discontinuation rates, which could, in turn, influence overall treatment efficacy.
CDA-AMC conducted a base-case reanalysis that implemented weight-based dosing for pembrolizumab. In a scenario analysis, a fixed dosage was adopted for pembrolizumab based on the product monograph.2 CDA-AMC was unable to fully address this limitation, given the uncertainty around the impact of different dosage regimens on treatment efficacy.
The selection of relevant comparators does not align with current clinical practice. The sponsor’s analysis excluded adjuvant osimertinib. Adjuvant osimertinib is indicated for patients with stage IB to IIIA NSCLC whose tumours have EGFR exon 19 deletions or exon 21 substitution mutations. Given that the Health Canada indication and reimbursement request for adjuvant pembrolizumab do not specify EGFR or anaplastic lymphoma kinase (ALK) expression status, osimertinib should have been considered a comparator. In the KEYNOTE-091 trial population, 8% of patients had EGFR mutations and 1% had ALK translocations. Clinician input suggests that, for patients with resected NSCLC who have both a sensitizing EGFR mutation and known PD-L1 expression, clinicians would need to choose between adjuvant pembrolizumab and adjuvant osimertinib. There is therefore a subgroup within the reimbursement request for whom adjuvant pembrolizumab may replace adjuvant osimertinib. The sponsor justified the exclusion of adjuvant osimertinib by stating that the KEYNOTE-091 trial was not designed to evaluate outcomes based on EGFR status. As a result, indirect comparisons between the KEYNOTE-091 and the ADAURA trials were deemed neither feasible nor appropriate. The cost-effectiveness of adjuvant pembrolizumab compared with adjuvant osimertinib remains unknown.
CDA-AMC could not address this limitation due to the lack of comparative effectiveness data.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CDA-AMC (refer to Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation
Sponsor’s key assumption | CDA-AMC comment |
|---|---|
100% of patients who experience LR or DM receive subsequent therapy | Uncertain. In the reimbursement request population of the KEYNOTE-091 trial, 24% and 9% of patients received subsequent antineoplastic agents and subsequent immunotherapies, respectively. Although the sponsor’s pharmacoeconomic model was calibrated to align with the OS observed in the KEYNOTE-091 trial, a greater number of patients incurred the cost of subsequent therapy in the model compared to the proportion of patients treated in the trial. |
Drug wastage was assumed | Uncertain. The sponsor assumed that vial sharing would occur, resulting in 5% of vial contents being wasted. While vial sharing is common practice in large centres, no data are available to quantify the percentage of excess drug wasted under such circumstances. The sponsor’s assumption of 5% drug wastage when vial sharing is applied remains uncertain. Assuming alternative proportions of drug wastage under vial sharing has a minimal impact on the results. |
CDA-AMC = Canada’s Drug Agency; DM = distant metastasis; LR = local-regional recurrence; OS = overall survival.
Reanalyses of the Economic Evaluation
Base-Case Results
The CDA-AMC base case was derived by making changes in model parameter values and assumptions, in consultation with clinical experts. CDA-AMC undertook a stepped analysis, sequentially incorporating each adjustment outlined in Table 5 into the sponsor’s model, to demonstrate the impact of each change. These included: adopting an alternative parametric distribution to extrapolate the transition from DF to LR in patients receiving active surveillance; assuming that 82% of patients who are disease free 10 years after treatment initiation would be considered cured; aligning the time points used for calibration of health-state transition rates between adjuvant pembrolizumab and active surveillance with the median and maximum follow-up observed in the KEYNOTE-091 trial; including age-related disutility; and adopting weight-based dosing for pembrolizumab.
The summary results of the CDA-AMC reanalyses for the weighted population are presented in Table 6.
Table 5: CDA-AMC Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Changes to derive the CDA-AMC base case | ||
1. Parametric distribution from DF to LR (active surveillance) | Log-normal | Generalized gamma |
2. Cure assumption |
|
|
3. Time points for calibration |
|
|
4. Age-related disutility | Excluded | Included |
5. Dosage for pembrolizumab | Flat dose: 200 mg every 3 weeks | Weight-based: 2 mg/kg every 3 weeks |
CDA-AMC base case | ― | CDA-AMC reanalysis 1 + 2 + 3 + 4 + 5 |
CDA-AMC = Canada’s Drug Agency; DF = disease free; LR = local-regional recurrence.
In the CDA-AMC base case, which is based on publicly available prices for all drug treatments, adjuvant treatment with pembrolizumab was associated with an ICER of $103,900 per QALY gained compared with active surveillance ($75,957 in incremental costs and 0.73 incremental QALYs) (Table 6). There was a 6% probability that pembrolizumab was cost-effective at a WTP threshold of $50,000 per QALY gained. A detailed breakdown of the disaggregated results is available in Appendix 4.
The CDA-AMC base-case results align with those of the sponsor’s submitted analysis, indicating that pembrolizumab is not cost-effective at a WTP threshold of $50,000 per QALY gained, relative to active surveillance. Consistent with the sponsor’s analysis, the CDA-AMC reanalysis estimates that the majority (85%) of incremental QALYs gained by patients receiving pembrolizumab relative to active surveillance were derived in the model on the basis of extrapolation.
Table 6: Summary of the Stepped Analysis of the CDA-AMC Reanalysis Results
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($ per QALY) |
|---|---|---|---|---|
Sponsor’s base case (Deterministic) | Active surveillance | 228,053 | 6.95 | Reference |
Pembrolizumab | 312,900 | 8.21 | 67,568 | |
CDA-AMC reanalysis 1 — Parametric distribution from DF to LR (active surveillance) | Active surveillance | 215,228 | 7.26 | Reference |
Pembrolizumab | 312,900 | 8.21 | 102,348 | |
CDA-AMC reanalysis 2 — Cure assumption | Active surveillance | 237,016 | 6.72 | Reference |
Pembrolizumab | 323,500 | 7.93 | 70,972 | |
CDA-AMC reanalysis 3 — Time points for calibration | Active surveillance | 226,453 | 6.88 | Reference |
Pembrolizumab | 309,004 | 8.02 | 71,893 | |
CDA-AMC reanalysis 4 — Age-related disutility | Active surveillance | 228,053 | 6.74 | Reference |
Pembrolizumab | 312,900 | 7.93 | 71,396 | |
CDA-AMC reanalysis 5 — Dosage for pembrolizumab | Active surveillance | 214,137 | 6.95 | Reference |
Pembrolizumab | 276,863 | 8.21 | 49,952 | |
CDA-AMC base case (Reanalyses 1 + 2 + 3 + 4 + 5) (Deterministic) | Active surveillance | 206,471 | 6.77 | Reference |
Pembrolizumab | 282,670 | 7.50 | 103,679 | |
CDA-AMC base case (Reanalyses 1 + 2 + 3 + 4 + 5) (Probabilistic) | Active surveillance | 207,887 | 6.76 | Reference |
Pembrolizumab | 283,844 | 7.49 | 103,900 |
CDA-AMC = Canada’s Drug Agency; DF = disease free; ICER = incremental cost-effectiveness ratio; LR = local-regional recurrence; QALY = quality-adjusted life-year.
Note: The CDA-AMC reanalysis is based on publicly available prices of the comparator treatments. The results of all steps are presented deterministically unless otherwise indicated, while the cumulative CDA-AMC base case is always presented both deterministically and probabilistically.
Scenario Analysis Results
CDA-AMC undertook price reduction analyses deterministically based on the sponsor and CDA-AMC base case. The CDA-AMC base case suggested that a 53.54% price reduction is required for pembrolizumab to be considered cost-effective relative to active surveillance at a WTP threshold of $50,000 per QALY gained (Table 7).
Table 7: CDA-AMC Price Reduction Analyses
Analysis: price reduction | Unit drug cost ($) | ICERs for pembrolizumab vs. active surveillance ($ per QALY) | |
|---|---|---|---|
Sponsor’s base case | CDA-AMC reanalysis | ||
No price reduction | 4,400 | 67,568 | 103,679 |
10% | 3,960 | 60,210 | 93,653 |
20% | 3,520 | 52,851 | 83,627 |
30% | 3,080 | 45,493 | 73,601 |
40% | 2,640 | 38,135 | 63,575 |
50% | 2,200 | 30,777 | 53,549 |
60% | 1,760 | 23,418 | 43,523 |
70% | 1,320 | 16,060 | 33,497 |
80% | 880 | 8,702 | 23,471 |
90% | 440 | 1,344 | 13,445 |
100% | 0 | Dominant | 3,419 |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
CDA-AMC conducted 2 scenario analyses to explore the impact of alternative assumptions on the cost-effectiveness of adjuvant pembrolizumab. One assumed no cure and the other adopted fixed dosage (200 mg every 3 weeks) for pembrolizumab based on the product monograph.2
Results of the scenario analyses are presented in Appendix 4, Table 12.
The cost-effectiveness of pembrolizumab was sensitive to assumptions concerning a cure among patients who achieve long-term DFS. When assuming that a patient population consisting predominantly of current and former smokers (84%) would not have the same age- and sex-adjusted general population mortality risk as the average person in Canada (i.e., no cure assumption), the ICER for pembrolizumab increased to $122,164 per QALY gained compared to active surveillance. When adopting fixed dosage for pembrolizumab based on the product monograph, the ICER for pembrolizumab increased to $135,566 per QALY gained compared to active surveillance.
Issues for Consideration
Pembrolizumab is currently under review for the treatment of adult patients with resectable stage II, IIIA, or IIIB (T3 to 4N2) NSCLC, in combination with platinum-based chemotherapy as a neoadjuvant treatment, followed by adjuvant monotherapy (perioperative pembrolizumab). The ongoing review of perioperative pembrolizumab is PD-L1 TPS–agnostic. As such, it encompasses both the Health Canada–indicated population of the current review, including patients with a PD-L1 TPS greater than or equal to 50%, and the reimbursement request population of patients with a PD-L1 TPS of less than 50%. According to clinical expert feedback, clinical practices are likely to change in both the Health Canada–indicated population and the reimbursement request population if pembrolizumab were reimbursed in the perioperative setting.
Pembrolizumab was previously reviewed for several conditions: for the treatment of patients with metastatic NSCLC whose tumours express PD-L1 greater than or equal to 1% and who have disease progression on or after platinum-containing chemotherapy;39 for the treatment of patients with untreated metastatic, squamous NSCLC (in combination with carboplatin and paclitaxel or nab-paclitaxel chemotherapy);40 for the treatment of patients with previously untreated metastatic NSCLC whose tumours express PD-L1 greater than or equal to 50% and who do not harbour a sensitizing EGFR mutation or ALK translocation;41 and for the treatment of patients with metastatic nonsquamous NSCLC with no EGFR or ALK genomic tumour aberrations and no prior systemic chemotherapy treatment for metastatic NSCLC (in combination with pemetrexed and platinum chemotherapy).42 The cost-effectiveness results of these evaluations may not be directly comparable to those in the current review because of differences in the target populations, model structures, clinical effectiveness parameters, health-state utility values, and cost inputs. The pan-Canadian Pharmaceutical Alliance concluded negotiations with a letter of intent for pembrolizumab for the aforementioned indications.43-45 As such, pembrolizumab has a confidential negotiated price, and is currently funded by jurisdictional cancer formularies.46-48 The CDA-AMC reanalyses are based on the publicly available price of pembrolizumab, which may differ from the confidential price and may influence the results of the cost-effectiveness and budget impact analyses (BIAs).
Overall Conclusions
Evidence from the phase III, randomized, placebo-controlled KEYNOTE-091 trial comparing the efficacy and safety of adjuvant pembrolizumab and placebo in adult patients with stage IB to IIIA NSCLC who have undergone complete resection and platinum-based chemotherapy and whose tumours have a PD-L1 TPS of less than 50% showed a clinically meaningful benefit of adjuvant pembrolizumab in DFS. The Clinical Review concluded that pembrolizumab likely results in an increase in the probability of being DF at 24 and 48 months compared to placebo, with a moderate certainty of evidence. Although there is a trend toward improved OS in favour of pembrolizumab, the OS data remain uncertain because of immaturity (the data cut-off was January 24, 2023, and the median follow-up period was 47 months). The clinical experts consulted for this review emphasized that a longer follow-up is needed to determine the OS benefit of pembrolizumab relative to placebo.
In addition to the limitations with the clinical evidence, CDA-AMC identified several limitations with the sponsor’s economic submission: uncertainty regarding the impact of pembrolizumab on long-term OS, additional uncertainty surrounding predicted OS because of the use of time-invariant extrapolations across all postprogression health states, uncertainty regarding the long-term DFS of active surveillance, uncertainty regarding the assumption of cure among patients who remain disease free, uncertainty regarding the duration of differential progression rates between adjuvant pembrolizumab and active surveillance used in calibration, inappropriate exclusion of age-related disutility in the submitted base case, misalignment of the adopted pembrolizumab dosage in the submitted base case and that observed in clinical practice, and omission of adjuvant osimertinib from the economic analysis.
The CDA-AMC base case was derived by making changes in model parameter values and assumptions, in consultation with clinical experts. These included: adopting an alternative parametric distribution to extrapolate the transition from DF to LR in patients receiving active surveillance, assuming that 82% of patients who are disease free 10 years after treatment initiation would be considered cured, aligning the time points used for calibration of health state transition rates between adjuvant pembrolizumab and active surveillance with the median and maximum follow-up observed in the KEYNOTE-091 trial, including age-related disutility, and adopting weight-based dosage for pembrolizumab.
The CDA-AMC base-case results align with those of the sponsor’s submitted analysis, indicating that adjuvant pembrolizumab is not cost-effective at a WTP threshold of $50,000 per QALY gained. In the CDA-AMC base case, adjuvant treatment with pembrolizumab was associated with an ICER of $103,900 per QALY gained compared with active surveillance ($75,957 incremental costs and 0.73 incremental QALYs). When compared to the sponsor’s analysis, the CDA-AMC base case estimated a reduced QALY benefit with pembrolizumab (i.e., 0.73 incremental QALYs versus 1.24 QALYs in the sponsor’s analysis), at a similar cost (i.e., $75,957 incremental costs in the CDA-AMC base case versus $84,667 in the sponsor’s analysis). The estimated ICER was driven by the selection of alternative distributions for extrapolating DFS, the cure assumption among patients who achieve long-term DFS, and the dosage adopted for pembrolizumab. In the CDA-AMC base case, a price reduction of 54% is required for pembrolizumab to be considered cost-effective relative to active surveillance at a WTP threshold of $50,000 per QALY gained. This would reduce the price of pembrolizumab from $4,400 to $2,044 per 4 mL vial. With this price reduction, the per-patient 28-day drug acquisition costs for pembrolizumab would be $4,088, assuming weight-based dosage and vial sharing (i.e., no wastage).
In addition, CDA-AMC conducted scenario analyses to evaluate the impact of the cure assumption and the dosage adopted for pembrolizumab. When assuming that a patient population consisting predominantly of current and former smokers would not have the same age- and sex-adjusted general population mortality risk as the average person in Canada (i.e., no cure assumption), the ICER for pembrolizumab increased to $122,164 per QALY gained compared to active surveillance. When adopting a fixed dosage for pembrolizumab based on the product monograph, the ICER for pembrolizumab increased to $135,566 per QALY gained compared to active surveillance.
The CDA-AMC base-case results assume that patients treated with pembrolizumab gain an additional LY compared to patients on active surveillance. In the absence of robust, long-term clinical evidence, the extent of this survival benefit is highly uncertain. Should the OS benefit of pembrolizumab be less than 0.9 years, the ICER would be higher than that in our base case, requiring larger price reductions to achieve cost-effectiveness. Moreover, when comparing the median follow-up in the KEYNOTE-091 trial to the model’s time horizon (47 months versus 36 years), it is important to note that most of the QALY and LY benefits realized by patients treated with pembrolizumab in the CDA-AMC base case accrue during the posttrial period of the model based on extrapolation.
Finally, because the sponsor did not consider adjuvant osimertinib as a relevant comparator in the economic analysis, the cost-effectiveness of pembrolizumab relative to osimertinib in the adjuvant setting is unknown.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Keytruda (pembrolizumab), 100 mg/4 mL vial, solution for infusion. Kirkland (QC): Merck Canada Inc.; 2024 Jul 15.
2.Keytruda (pembrolizumab): 100 mg/4 mL vial, solution for infusion [product monograph]. Kirkland (QC): Merck Canada Inc.; 2023 Apr 19.
3.O'Brien M, Paz-Ares L, Marreaud S, et al. Pembrolizumab versus placebo as adjuvant therapy for completely resected stage IB-IIIA non-small-cell lung cancer (PEARLS/KEYNOTE-091): an interim analysis of a randomised, triple-blind, phase 3 trial. Lancet Oncol. 2022;23(10):1274-1286. PubMed
4.Williams C, Lewsey JD, Briggs AH, Mackay DF. Cost-effectiveness Analysis in R Using a Multi-state Modeling Survival Analysis Framework: A Tutorial. Med Decis Making. 2017;37(4):340-352. PubMed
5.Williams C, Lewsey JD, Mackay DF, Briggs AH. Estimation of Survival Probabilities for Use in Cost-effectiveness Analyses: A Comparison of a Multi-state Modeling Survival Analysis Approach with Partitioned Survival and Markov Decision-Analytic Modeling. Med Decis Making. 2017;37(4):427-439. PubMed
6.Merck, Co. Analysis of SEER-Medicare database (2022) - Cohort I: Patients with completely resected stage IB-IIIA NSCLC (AJCC 7th edition). https://healthcaredelivery.cancer.gov/seermedicare/. [Data on file].
7.Gadgeel S, Rodriguez-Abreu D, Speranza G, et al. Updated Analysis From KEYNOTE-189: Pembrolizumab or Placebo Plus Pemetrexed and Platinum for Previously Untreated Metastatic Nonsquamous Non-Small-Cell Lung Cancer. J Clin Oncol. 2020;38(14):1505-1517. PubMed
8.Paz-Ares L, Luft A, Vicente D, et al. Pembrolizumab plus Chemotherapy for Squamous Non-Small-Cell Lung Cancer. N Engl J Med. 2018;379(21):2040-2051. PubMed
9.Reckamp KL, Redman MW, Dragnev KH, et al. Phase II Randomized Study of Ramucirumab and Pembrolizumab Versus Standard of Care in Advanced Non-Small-Cell Lung Cancer Previously Treated With Immunotherapy-Lung-MAP S1800A. J Clin Oncol. 2022;40(21):2295-2306. PubMed
10.Ramalingam SS, Vansteenkiste J, Planchard D, et al. Overall Survival with Osimertinib in Untreated, EGFR-Mutated Advanced NSCLC. N Engl J Med. 2020;382(1):41-50. PubMed
11.Paz-Ares LG, Ramalingam SS, Ciuleanu TE, et al. First-Line Nivolumab Plus Ipilimumab in Advanced NSCLC: 4-Year Outcomes From the Randomized, Open-Label, Phase 3 CheckMate 227 Part 1 Trial. J Thorac Oncol. 2022;17(2):289-308. PubMed
12.de Castro G, Jr., Kudaba I, Wu YL, et al. Five-Year Outcomes With Pembrolizumab Versus Chemotherapy as First-Line Therapy in Patients With Non-Small-Cell Lung Cancer and Programmed Death Ligand-1 Tumor Proportion Score >/= 1% in the KEYNOTE-042 Study. J Clin Oncol. 2023;41(11):1986-1991. PubMed
13.Herbst RS, Giaccone G, de Marinis F, et al. Atezolizumab for First-Line Treatment of PD-L1-Selected Patients with NSCLC. N Engl J Med. 2020;383(14):1328-1339. PubMed
14.Ozguroglu M, Kilickap S, Sezer A, et al. First-line cemiplimab monotherapy and continued cemiplimab beyond progression plus chemotherapy for advanced non-small-cell lung cancer with PD-L1 50% or more (EMPOWER-Lung 1): 35-month follow-up from a mutlicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2023;24(9):989-1001. PubMed
15.Gogishvili M, Melkadze T, Makharadze T, et al. Cemiplimab plus chemotherapy versus chemotherapy alone in non-small cell lung cancer: a randomized, controlled, double-blind phase 3 trial. Nat Med. 2022;28(11):2374-2380. PubMed
16.Onco CAPPs. Patient Treatment Dynamics for Pembrolizumab Advanced 1L Non-Small Cell Lung Cancer (NSCLC),2023 Q3 update (3-months new starts) [sponsor provided reference]. 2023.
17.Onco CAPPs. NSCLC Market Sizing & Treatment Rate Audit, 2023 Q3 (Stade IV) [sponsor provided reference]. 2023.
18.Merck, Co. Network meta-analysis of interventions for the first-line treatment of metastatic NSCLC patients: technical report. 19 July 2023. [Data on file].
19.Henschke CI, Yip R, Shaham D, et al. A 20-year Follow-up of the International Early Lung Cancer Action Program (I-ELCAP). Radiology. 2023;309(2):e231988. PubMed
20.Dolan P. Modeling valuations for EuroQol health states. Medical Care [sponsor provided reference]. 1997. PubMed
21.Agulnik J, Kasymjanova G, Pepe C, et al. Understanding clinical practice and survival outcomes in patients with unresectable stage III non-small-cell lung cancer in a single centre in Quebec. Curr Oncol. 2020;27(5):e459-e466. PubMed
22.Danson S, Middleton MR, O'Byrne KJ, et al. Phase III trial of gemcitabine and carboplatin versus mitomycin, ifosfamide, and cisplatin or mitomycin, vinblastine, and cisplatin in patients with advanced nonsmall cell lung carcinoma. Cancer. 2003;98(3):542-553. PubMed
23.Cancer Care Ontario. Monograph 54276. Drug Formulary [accessed by sponsor]. https://www.cancercareontario.ca/en/drugformulary/regimens/monograph/54276.
24.Onco CAPPs. Advanced 2L+ Non-Small Cell Lung Cancer (NSCLC), Patient treatment dynamics for pembrolizumab [sponsor provided reference]. 2019.
25.Gandhi L, Rodriguez-Abreu D, Gadgeel S, et al. Pembrolizumab plus Chemotherapy in Metastatic Non-Small-Cell Lung Cancer. N Engl J Med. 2018;378(22):2078-2092. PubMed
26.Socinski MA, Nishio M, Jotte RM, et al. IMpower150 Final Overall Survival Analyses for Atezolizumab Plus Bevacizumab and Chemotherapy in First-Line Metastatic Nonsquamous NSCLC. J Thorac Oncol. 2021;16(11):1909-1924. PubMed
27.Paz-Ares L, Ciuleanu TE, Cobo M, et al. First-line nivolumab plus ipilimumab combined with two cycles of chemotherapy in patients with non-small-cell lung cancer (CheckMate 9LA): an international, randomised, open-label, phase 3 trial. Lancet Oncol. 2021;22(2):198-211. PubMed
28.Patel JD, Socinski MA, Garon EB, et al. PointBreak: a randomized phase III study of pemetrexed plus carboplatin and bevacizumab followed by maintenance pemetrexed and bevacizumab versus paclitaxel plus carboplatin and bevacizumab followed by maintenance bevacizumab in patients with stage IIIB or IV nonsquamous non-small-cell lung cancer. J Clin Oncol. 2013;31(34):4349-4357. PubMed
29.Fossella FV, DeVore R, Kerr RN, et al. Randomized phase III trial of docetaxel versus vinorelbine or ifosfamide in patients with advanced non-small-cell lung cancer previously treated with platinum-containing chemotherapy regimens. The TAX 320 Non-Small Cell Lung Cancer Study Group. J Clin Oncol. 2000;18(12):2354-2362. PubMed
30.Stinchcombe TE, Harper HD, Hensing TA, et al. The feasibility of adjuvant carboplatin and docetaxel in patients with curatively resected non-small cell lung cancer. J Thorac Oncol. 2008;3(2):145-151. PubMed
31.National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology: Non-Small Cell Lung Cancer. v3.2023 [accessed by sponsor]. 2023.
32.Cancer Care Ontario. Monograph 69631. Drug Formulary [accessed by sponsor]. https://www.cancercareontario.ca/en/drugformulary/regimens/monograph/69631.
33.Oliveira CD, Pataky R, Bremner KE, et al. Estimating the Cost of Cancer Care in British Columbia and Ontario: A Canadian Inter-Provincial Comparison. Healthcare Policy [accessed by sponsor]. 2017. PubMed
34.Canadian Institute for Health Information. Patient Cost Estimator [accessed by sponsor]. 2020; https://www.cihi.ca/en/patient-cost-estimator.
35.Physician Services under the Health Insurance Act. Schedule of Benefits (June 29, 2023 (effective July 24, 2023)). Toronto (ON): Ministry of Health; 2023: https://www.ontario.ca/files/2024-01/moh-ohip-schedule-of-benefits-2024-01-24.pdf. Accessed 2024 Jun 29.
36.CADTH Reimbursement Review: Osimertinib (Tagrisso) [accessed by sponsor]. Can J Health Technol. 2022;2(3). https://www.cadth.ca/sites/default/files/DRR/2022/PC0246-Tagrisso.pdf.
37.Appendix B, code 60046. Quebec directory and system for measuring medical biology procedures [accessed by sponsor]. Quebec (QC): Ministère de la Santé et des Services sociaux; 2022: https://publications.msss.gouv.qc.ca/msss/fichiers/2022/22-922-02W.pdf.
38.Maeda R, Yoshida J, Hishida T, et al. Late recurrence of non-small cell lung cancer more than 5 years after complete resection: incidence and clinical implications in patient follow-up. Chest. 2010;138(1):145-150. PubMed
39.Drug Reimbursement Review pharmacoeconomic report: Pembrolizumab (Keytruda) for Non-small cell lung cancer Ottawa (ON): CADTH; 2016: https://www.cda-amc.ca/keytruda-non-small-cell-lung-cancer-second-line-or-beyond-details. Accessed 2016 Nov 18.
40.Drug Reimbursement Review pharmacoeconomic report: Pembrolizumab (Keytruda) for Squamous NSCLC. Ottawa (ON): CADTH; 2020: https://www.cda-amc.ca/keytruda-squamous-nsclc-details. Accessed 2020 Jan 20.
41.Drug Reimbursement Review pharmacoeconomic report: Pembrolizumab (Keytruda) for Advanced Non-Small Cell Lung Carcinoma (First Line). Ottawa (ON): CADTH; 2017: https://www.cda-amc.ca/keytruda-advanced-non-small-cell-lung-carcinoma-first-line-details. Accessed 2017 Sep 8.
42.Drug Reimbursement Review pharmacoeconomic report: Pembrolizumab (Keytruda) for Non-Squamous NSCL. Ottawa (ON): CADTH; 2019: https://www.cda-amc.ca/keytruda-non-squamous-nsclc-details. Accessed 2019 Jun 17.
43.Pan-Canadian Pharmaceutical Alliance. Keytruda (pembrolizumab) for Non-small Cell Lung Cancer. 2017; https://www.pcpacanada.ca/negotiation/20927. Accessed 2024 Oct 2.
44.Pan-Canadian Pharmaceutical Alliance. Keytruda (pembrolizumab) for Squamous Non-Small Cell Lung Cancer. 2020; https://www.pcpacanada.ca/negotiation/21168. Accessed 2024 Oct 2.
45.Pan-Canadian Pharmaceutical Alliance. Keytruda (pembrolizumab) for Non-Squamous Non-Small Cell Lung Cancer. 2020; https://www.pcpacanada.ca/negotiation/21137. Accessed 2024 Oct 2.
46.Cancer Care Ontario. CISPPEME+PEMB: Drug formulary [accessed by sponsor]. https://www.cancercareontario.ca/en/drugformulary/regimens/53611.
47.Cancer Care Ontario. CRBPPACL+PEMB: Drug formulary [accessed by sponsor]. https://www.cancercareontario.ca/en/drugformulary/regimens/60431.
48.Drug formulary. Regina (SK): Saskatchewan Cancer Agency: https://saskcancer.ca/sites/default/files/2024-10/SCA%20Drug%20Formulary%20-%202024-10-01.pdf. Accessed 2024 Oct 2.
49.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Keytruda (pembrolizumab), 100 mg/4 mL vial, solution for infusion. Kirkland (QC): Merck Canada Inc.; 2024 Jul 15.
50.CADTH Reimbursement Recommendation: Osimertinib (Tagrisso) [accessed by sponsor]. Can J Health Technol. 2022;2(1). https://www.cadth.ca/osimertinib.
51.Wu YL, Tsuboi M, He J, et al. Osimertinib in Resected EGFR-Mutated Non-Small-Cell Lung Cancer. N Engl J Med. 2020;383(18):1711-1723. PubMed
52.Iqvia. DeltaPA Drug Formulary. 2024. Accessed 2024 Jan 15.
53.Drug Intelligence. ONCO-CAPPS: 1L NSCLC New Start Treatment Dynamics Q4 2023 [sponsor provided reference]. 2024. Accessed 2024 Jun 25.
Appendix 1: Cost Comparison Table
Please note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical experts and drug plans. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 8: CDA-AMC Cost Comparison Table for Adjuvant Treatment of NSCLC
Treatment | Strength / concentration | Form | Price ($) | Recommended dosage | Average daily cost ($) | Average cost per 28-days ($) |
|---|---|---|---|---|---|---|
Pembrolizumab | 100 mg vial | 100 mg/4 mL IV solution | 4,400.0000a | Weight-based dosage: 2 mg/kg (up to 200 mg) every 3 weeks or 4 mg/kg (up to 400 mg) every 6 weeksb | 314.29 | 8,800 |
Fixed dosage: 200 mg every 3 weeks or 400 mg every 6 weeks for up to 1 year or until disease recurrence or unacceptable toxicity | 419.05 | 11,733 |
NSCLC = non–small cell lung cancer.
Note: Costs do not include dispensing fees.
aSponsor’s submitted price.1
bInput from clinical experts and participating drug plans indicated that weight-based dosage may be used for pembrolizumab. Weight-based dosage assumes a mean weight of 75 kg (aligned with the baseline patient characteristics from the KEYNOTE-091 trial) and vial sharing.
Appendix 2: Submission Quality
Please note that this appendix has not been copy-edited.
Description | Yes or No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | Refer to key limitation “Selection of relevant comparators does not align with current clinical practice.” |
Model has been adequately programmed and has sufficient face validity | Yes | No comment. |
Model structure is adequate for decision problem | Yes | No comment. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | Yes | No comment. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | Yes | No comment. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | Yes | No comment. |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Please note that this appendix has not been copy-edited.
Detailed Results of the Sponsor’s Base Case
Table 10: Disaggregated Summary of the Sponsor’s Economic Evaluation Results
Parameter | Pembrolizumab | Active surveillance |
|---|---|---|
Discounted LYs | ||
Total | 10.42 | 8.88 |
By health state | ||
DF | 7.81 | 6.35 |
LR | 1.25 | 1.09 |
DM | 1.35 | 1.44 |
Discounted QALYs | ||
Total | 8.20 | 6.96 |
By health state or data source | ||
DF | 6.30 | 5.12 |
LR | 0.97 | 0.85 |
DM | 0.94 | 1.00 |
AE-related disutility | −0.0094 | −0.0096 |
Age-related disutility | 0 | 0 |
Discounted costs ($) | ||
Total | 314,128 | 229,461 |
By cost category | ||
Adjuvant treatment costs | 115,623 | 0 |
Drug acquisition costs | 115,359 | 0 |
Drug administration costs | 264 | 0 |
Subsequent treatment costs (LR state) | 23,468 | 25,077 |
Drug acquisition costs | 18,368 | 19,626 |
Drug administration costs | 205 | 219 |
Radiotherapy costs | 4,393 | 4,695 |
Salvage surgery costs | 502 | 537 |
Subsequent treatment costs (DM state) | 56,852 | 86,874 |
Drug acquisition costs | 56,576 | 86,541 |
Drug administration costs | 276 | 333 |
AE costs | 484 | 276 |
Disease management costs | 49,094 | 46,742 |
DF | 3,294 | 2,807 |
LR | 25,343 | 22,086 |
DM | 20,458 | 21,849 |
Terminal care costs | 68,460 | 70,492 |
Testing cost | 146 | 0 |
By health state | ||
DF | 119,547 | 3,084 |
LR | 48,812 | 47,163 |
DM | 77,309 | 108,723 |
Death | 68,460 | 70,492 |
AE = adverse event; DF = disease free; DM = distant metastases; LR = local-regional recurrence; LY = life-year; QALY = quality-adjusted life-year.
Source: Sponsor’s pharmacoeconomic submission.1
Appendix 4: Additional Details on the CDA-AMC Reanalyses and Sensitivity Analyses of the Economic Evaluation
Please note that this appendix has not been copy-edited.
Detailed Results of CDA-AMC Base Case
Table 11: Disaggregated Summary of the CDA-AMC Economic Evaluation Results
Parameter | Pembrolizumab | Active surveillance |
|---|---|---|
Discounted LYs | ||
Total | 9.84 | 8.90 |
By health state | ||
DF | 7.33 | 6.60 |
LR | 1.25 | 0.96 |
DM | 1.26 | 1.35 |
Discounted QALYs | ||
Total | 7.49 | 6.76 |
By health state or data source | ||
DF | 5.91 | 5.31 |
LR | 0.97 | 0.74 |
DM | 0.88 | 0.94 |
AE-related disutility | −0.0094 | −0.0096 |
Age-related disutility | −0.25 | −0.22 |
Discounted costs ($) | ||
Total | 283,844 | 207,887 |
By cost category | ||
Adjuvant treatment costs | 88,007 | 0 |
Drug acquisition costs | 87,743 | 0 |
Drug administration costs | 264 | 0 |
Subsequent treatment costs (LR state) | 25,487 | 22,010 |
Drug acquisition costs | 19,948 | 17,228 |
Drug administration costs | 223 | 193 |
Radiotherapy costs | 4,771 | 4,119 |
Salvage surgery costs | 545 | 471 |
Subsequent treatment costs (DM state) | 52,905 | 72,504 |
Drug acquisition costs | 52,609 | 72,173 |
Drug administration costs | 296 | 331 |
AE costs | 484 | 276 |
Disease management costs | 47,514 | 42,627 |
DF | 3,166 | 2,857 |
LR | 25,303 | 19,387 |
DM | 19,045 | 20,384 |
Terminal care costs | 69,301 | 70,469 |
Testing cost | 146 | 0 |
By health state | ||
DF | 91,803 | 3,133 |
LR | 50,790 | 41,397 |
DM | 71,950 | 92,888 |
Death | 69,301 | 70,469 |
AE = adverse event; CDA-AMC = Canada’s Drug Agency; DF = disease free; DM = distant metastases; LR = local-regional recurrence; LY = life-year; QALY = quality-adjusted life-year.
Scenario Analyses
Table 12: Summary of CDA-AMC Scenario Analyses
Scenario analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
CDA-AMC base case (Probabilistic) | Active surveillance | 207,887 | 6.76 | Reference |
Pembrolizumab | 283,844 | 7.49 | 103,900 | |
Scenario 1: No cure assumption | Active surveillance | 218,313 | 6.51 | Reference |
Pembrolizumab | 300,919 | 7.18 | 122,164 | |
Scenario 2: Fixed dosage for pembrolizumab | Active surveillance | 221,767 | 6.76 | Reference |
Pembrolizumab | 320,875 | 7.49 | 135,566 |
CDA-AMC = Canada’s Drug Agency.
Note: The CDA-AMC base case is based on publicly available prices of the comparator treatments. Scenario analyses are conducted probabilistically.
Appendix 5: Submitted BIA and CDA-AMC Appraisal
Please note that this appendix has not been copy-edited.
Table 13: Summary of Key Take-Aways
Key take-aways of the budget impact analysis |
|---|
|
Summary of Sponsor’s BIA
The sponsor submitted a BIA49 assessing the expected budgetary impact associated with the introduction of pembrolizumab for the adjuvant treatment of adult patients with stage IB (T2a ≥ 4 cm), II, or IIIA NSCLC who have undergone complete resection and platinum-based chemotherapy and whose tumours have a PD-L1 TPS less than 50%, as determined by a validated test. The BIA was undertaken from the perspective of the pan-Canadian participating public drug plans over a 3-year time horizon (2025 to 2027).
The sponsor estimated the size of the eligible population using an epidemiologic approach, with data obtained from publications, previous CDA-AMC submissions and sponsor’s assumptions.3 The sponsor assumed a patient weight of 75 kg and a mean body surface area of 1.9 m2 in the calculation of drug costs, as reported in the KEYNOTE-091 trial.3 Pembrolizumab’s price, dosage and ToT was also sourced from KEYNOTE-091.3 Comparators in the adjuvant setting included osimertinib; with price and dosage obtained from CADTH’s reimbursement review of osimertinib for NSCLC and ToT derived from the ADAURA trial.50,51 The sponsor also assumed that a proportion of eligible patients would be on active surveillance. Subsequent therapy costs for patients who progressed to LR and DM were also included. The impact of adjuvant pembrolizumab on treatments received in LR and DM settings was captured using transition probabilities from the sponsor’s submitted base case.3 Drug acquisition costs for subsequent therapies were sourced from CDA-AMC reimbursement reviews and IQVIA DeltaPA.52 The distribution of patients across subsequent treatments was derived from published literature and the sponsor’s internal analysis of the ONCO-CAPPS database.21,53
Table 14: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3 if appropriate) |
|---|---|
Target population | |
Incident cases of lung cancer | 23,165 |
Patients with NSCLC | 88.00% |
Distribution of NSCLC from stage I to III at diagnosis | |
Stage I | 23.10% |
Stage II | 9.10% |
Stage III | 19.00% |
Proportion of patient from stage IB to IIIA at diagnosis | |
Stage IB | 39.60% |
Stage II | 100.00% |
Stage IIIA | 69.30% |
Proportion of resected patient (stage IB to IIIA) | 60.00% |
Patients with no prior neoadjuvant treatment | 60.00% |
Patients who receive adjuvant chemotherapy following surgery | 26.00% |
Patients eligible for adjuvant immunotherapy | 95.00% |
Patients tested for PD-L1 expression | 90.00% |
Patients with PD-L1 < 50% | 72.00% |
Number of patients eligible for drug under review | 369 / 381 / 393 |
Market Uptake (3 years) | |
Uptake (reference scenario) | |
Active surveillance | 85.0% / 85.0% / 85.0% |
Osimertinib | 15.0% / 15.0% / 15.0% |
Clinical Trials | 0.0% / 0.0% / 0.0% |
Uptake (new drug scenario) | |
Pembrolizumab | 17.1% / 46.0% / 50.0% |
Active surveillance | 67.9% / 39.0% / 35.0% |
Osimertinib | 15.0% / 15.0% / 15.0% |
Clinical Trials | 0.0% / 0.0% / 0.0% |
Cost of treatment (per patient, per cycle) | |
Adjuvant setting | |
Pembrolizumaba | $8,800 |
Osimertinib | $1,889 |
Active surveillance | $0 |
NSCLC = non–small cell lung cancer.
aPembrolizumab’s cost of treatment is based on fixed dosage of 200 mg every 3 weeks and vial sharing (i.e., drug wastage is not included).
Summary of the Sponsor’s BIA Results
The sponsor estimated the 3-year budget impact of reimbursing pembrolizumab for the adjuvant treatment of adult patients with stage IB (T2a ≥ 4 cm), II, or IIIA NSCLC who have undergone complete resection and platinum-based chemotherapy and whose tumours have a PD-L1 TPS less than 50% to be $34,756,346 (Year 1: $2,823,220; Year 2: $13,625,780; Year 3: $18,307,346).
CDA-AMC Appraisal of the Sponsor’s BIA
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Proportion of patients tested for PD-L1 expression is underestimated. The sponsor assumed that 90% of patients would be tested for PD-L1 expression in the NSCLC adjuvant setting. However, clinical expert feedback received for this review indicated that PD-L1 testing is part of routine clinical practice in NSCLC and the sponsor’s adopted testing rate is underestimated.
CDA-AMC performed a reanalysis by adopting a PD-L1 testing rate of 95% baseline on clinical expert opinion.
Market uptake of pembrolizumab is underestimated. The sponsor assumed a linear uptake of adjuvant pembrolizumab, resulting in a market share of 17% by Year 1, 46% by Year 2 and a maximum of 50% by Year 3. However, clinical expert feedback indicated that the market uptake of pembrolizumab is highly uncertain in the face of emerging perioperative treatments. Clinical experts further indicated that a rapid uptake of adjuvant pembrolizumab is anticipated for patients who have already undergone surgery and have a PD-L1 expression in less than 50% of tumour cells.
CDA-AMC performed a reanalysis by adopting a rapid uptake (using a logarithmic curve), which resulted in a market share of 35%, 49% and 50% in Year 1, 2, and 3, respectively.
Pembrolizumab dosage does not align with public drug plans’ implementation strategy. In the sponsor’s base case, pembrolizumab was administered at a fixed dosage of 200 mg every 3 weeks. Input received from participating public drug plans and clinical experts indicates that a weight-based dosage is likely to be adopted for pembrolizumab (2 mg/kg [up 200 mg] every 3 weeks or 4 mg/kg [up to 400 mg] every 6 weeks).
CDA-AMC conducted a base case reanalysis that implemented weight-based dosing for pembrolizumab. CDA-AMC explored the impact of fixed dosage for pembrolizumab (200 mg every 3 weeks) in scenario analysis.
Costs associated with subsequent therapy are uncertain. The costs associated with subsequent therapy are uncertain. The sponsor included the impact of adjuvant pembrolizumab on subsequent treatment costs in the LR and DM settings. To model these costs in the BIA, the sponsor used the proportion of DF patients who progressed to LR and DM at each weekly cycle based on the submitted base case of the CUA. However, the CDA-AMC Pharmacoeconomic Review of the sponsor's CUA identified limitations regarding DFS extrapolation, which were addressed in reanalyses. CDA-AMC used the DF transition probabilities from the CDA-AMC base case of the CUA to estimate subsequent therapy costs in the BIA.
CDA-AMC conducted a base case reanalysis that aligned the proportion of DF patients who progressed to LR and DM at each weekly cycle with the CDA-AMC base case of the CUA. Additionally, CDA-AMC conducted a scenario analysis that excluded costs associated with subsequent therapy.
CDA-AMC Reanalyses of the BIA
CDA-AMC revised the sponsor’s submitted analyses by assuming 95% of patients are tested for PD-L1 expression, adopting a rapid uptake of pembrolizumab in the adjuvant setting, and adopting weight-based dosing for pembrolizumab.
Table 15: CDA-AMC Revisions to the Submitted BIA
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
None | — | — |
Changes to derive the CDA-AMC base case | ||
1. Patients tested for PD-L1 expression | 90% | 95% |
2. Market uptake of pembrolizumab | Linear (slow uptake) | Logarithmic (rapid uptake) |
3. Pembrolizumab dosage | Fixed: 200 mg every 3 weeks | Weight-based: 2 mg/kg every 3 weeks |
4. Subsequent therapy | Aligned the DF to LR and DF to DM transitions for pembrolizumab and active surveillance with the sponsor’s submitted base case of the CUA | Aligned the DF to LR and DF to DM transitions for pembrolizumab and active surveillance with the CDA-AMC base case of the CUA |
CDA-AMC base case | CDA-AMC base case (Reanalyses 1 + 2 + 3 + 4) | |
BIA = budget impact analysis; CUA = cost-utility analysis; DF = disease free; LR = local-regional recurrence.
The results of the CDA-AMC step-wise reanalysis are presented in summary format in Table 16 and a more detailed breakdown is presented in Table 17.
Based on the CDA-AMC base case, the budget impact associated with the reimbursement of pembrolizumab for the adjuvant treatment of adult patients with stage IB (T2a ≥ 4 cm), II, or IIIA NSCLC who have undergone complete resection and platinum-based chemotherapy and whose tumours have a PD-L1 TPS less than 50% is expected to be $5,297,747 in Year 1, $12,988,648 in Year 2, $13,650,607 in Year 3, for a 3-year total budgetary impact of $31,937,001. The 3-year total budgetary impact increased to $43,183,321 when fixed dosing for pembrolizumab was adopted.
Table 16: Summary of the CDA-AMC Reanalyses of the BIA
Stepped analysis | Three-year total ($) |
|---|---|
Submitted base case | 34,756,346 |
CDA-AMC reanalysis 1 | 36,687,254 |
CDA-AMC reanalysis 2 | 41,512,738 |
CDA-AMC reanalysis 3 | 25,842,559 |
CDA-AMC reanalysis 4 | 34,364,720 |
CDA-AMC base case | 31,937,001 |
BIA = budget impact analysis.
CDA-AMC conducted the following scenario analyses to address remaining uncertainty, using the CDA-AMC base case (results are provided in Table 17:
Assuming fixed dosing of pembrolizumab
Excluding subsequent therapy costs
Table 17: Detailed Breakdown of the CDA-AMC Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|---|
Submitted base case | Reference | 4,965,079 | 15,719,328 | 24,971,462 | 32,147,282 | 72,838,072 |
New drug | 4,965,079 | 18,542,548 | 38,597,242 | 50,454,628 | 107,594,418 | |
Budget impact | 0 | 2,823,220 | 13,625,780 | 18,307,346 | 34,756,346 | |
CDA-AMC base case | Reference | 4,892,297 | 15,589,981 | 24,082,367 | 30,163,861 | 69,836,209 |
New drug | 4,892,297 | 20,887,728 | 37,071,014 | 43,814,468 | 101,773,210 | |
Budget impact | 0 | 5,297,747 | 12,988,648 | 13,650,607 | 31,937,001 | |
CDA-AMC scenario analysis 1: Fixed dosage of pembrolizumab | Reference | 5,216,569 | 17,009,338 | 26,694,062 | 33,879,950 | 77,583,349 |
New drug | 5,216,569 | 24,143,651 | 44,273,900 | 52,349,119 | 120,766,670 | |
Budget impact | 0 | 7,134,313 | 17,579,838 | 18,469,170 | 43,183,321 | |
CDA-AMC scenario analysis 2: Subsequent therapy costs excluded | Reference | 2,389,704 | 5,812,308 | 7,959,468 | 8,880,321 | 22,652,097 |
New drug | 2,389,704 | 11,567,171 | 23,316,975 | 26,481,187 | 61,365,334 | |
Budget impact | 0 | 5,754,863 | 15,357,507 | 17,600,866 | 38,713,237 |
BIA = budget impact analysis.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.