Drugs, Health Technologies, Health Systems
Reimbursement Review
Durvalumab (Imfinzi), Carboplatin, Paclitaxel
Sponsor: AstraZeneca Canada Inc.
Therapeutic area: Endometrial cancer that is mismatch repair deficient (dMMR)
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
AE
adverse event
AESI
adverse event of special interest
BICR
blinded independent central review
CCRAN
Colorectal Cancer Resource & Action Network
CI
confidence interval
CR
complete response
dMMR
mismatch repair deficient
DOR
duration of response
ECOG
Eastern Cooperative Oncology Group
EORTC QLQ-C30
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30
EORTC QLQ-EN24
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire – Endometrial Cancer Module
ESS
effective sample size
FIGO
International Federation of Gynecology and Obstetrics
GOC
Gynecologic Oncology Society of Canada
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HR
hazard ratio
HRQoL
health-related quality of life
IA
interim analysis
ICI
immune checkpoint inhibitor
IHC
immunohistochemistry
ITT
intention to treat
IVRS
interactive voice response system
MAIC
matching-adjusted indirect comparison
MID
minimal important difference
MMR
mismatch repair
MSI
microsatellite instability
MSI-H
microsatellite instability-high
MSS
microsatellite stable
OH (CCO)
Ontario Health (Cancer Care Ontario)
OR
odds ratio
ORR
objective response rate
OS
overall survival
PFS
progression-free survival
pMMR
mismatch repair proficient
PR
partial response
RCT
randomized controlled trial
RECIST
Response Evaluation Criteria in Solid Tumours
SAE
serious adverse event
SD
standard deviation
SLR
systematic literature review
SOC
standard of care
TDT
time to study treatment discontinuation or death
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Durvalumab (Imfinzi) 50 mg/mL, concentrate for IV infusion |
Sponsor | AstraZeneca Canada Inc. |
Indication | Durvalumab in combination with carboplatin and paclitaxel for the first-line treatment of adults with primary advanced or recurrent mismatch repair deficient (dMMR) endometrial cancer who are candidates for systemic therapy, followed by durvalumab as monotherapy. |
Reimbursement request | As per indication |
Health Canada approval status | Pre-NOC |
Health Canada review pathway | Standard |
NOC date | January 17, 2025 |
Recommended dose | Durvalumab 1,120 mg in combination with platinum-based chemotherapy every 3 weeks (21 days) for a minimum of 4 and up to 6 cycles, followed by maintenance with 1,500 mg every 4 weeks as monotherapy until disease progression or unacceptable toxicity. |
NOC = Notice of Compliance.
Introduction
Endometrial cancer is a type of uterine cancer originating in the lining of the uterus and is the most common gynecological malignancy,1 accounting for approximately 95% of uterine cancers.2 In Ontario, 2,909 new endometrial cancer cases were diagnosed in 2018, with an age-standardized incidence rate of 36.2 per 100,000.3 In 2024, the Canadian Cancer Society estimated that 8,600 females would be diagnosed with uterine cancer that year, and 1,600 would die from it.4 Endometrial cancer primarily affects females who are postmenopausal, with an average age at diagnosis of 60 years.5
Endometrial cancer is staged using the International Federation of Gynecology and Obstetrics (FIGO) system, ranging from stage I (tumour confined to the uterus), to stage IV (cancer has spread to bladder, bowel, or distant organs).6 In Canada, the overall 5-year net survival for uterine cancer is 82%, with stage-specific survival rates of 70% for stage I, 45% for stage II, 30% for stage III, and 15% for stage IV.7
The symptoms of advanced (stage III or IV) and recurrent (return following primary treatment) disease are variable, but include abnormal vaginal bleeding,8,9 pelvic or back pain, the presence of a palpable mass in the lower abdomen, and unintentional weight loss.8 Patients often experience additional issues like sexual dysfunction, anxiety, depression, and the long-term effects of chemotherapy, all of which further reduce health-related quality of life (HRQoL).1,10,11
The diagnosis of endometrial cancer involves clinical assessments, radiological imaging, and histopathological analysis of biopsy samples. For recurrent endometrial cancer cases, biopsies may help identify molecular subtypes of the cancer.12,13 Molecular testing during endometrial biopsy is crucial for treatment decisions and risk assessment.14 Endometrial cancer is characterized as microsatellite instability (MSI)-high (MSI-H), MSI-low, or microsatellite stable (MSS) based on MSI testing,15 and mismatch repair (MMR) proficient (pMMR) or MMR deficient (dMMR) based on MMR testing.16 According to the clinical experts consulted by the review team, MMR testing, assessed by immunohistochemistry (IHC), is currently performed as the standard of care (SOC) for patients with endometrial cancer in Canada.
Durvalumab is a fully human, high-affinity, immunoglobulin G1 kappa monoclonal antibody that selectively blocks the interaction of PD-L1, a protein expressed on tumour cells, with PD-1 and CD80 on T cells. PD-L1 binds to PD-1 and CD80 on T cells, inhibiting their activation and reducing their ability to detect and destroy cancer cells. The selective blockade of PD-L1 and PD-1 and PD-L1 and CD80 interactions releases the inhibition of immune responses and enhances antitumour immune responses.17,18 It is administered at 1,120 mg every 3 weeks in combination with platinum-based chemotherapy for 4 to 6 cycles. This is followed by maintenance therapy at 1,500 mg every 4 weeks, as monotherapy, until disease progression or unacceptable toxicity.19
Durvalumab was reviewed by Health Canada (Notice of Compliance issued January 17, 2025) as a first-line treatment for adult patients with primary advanced or recurrent endometrial cancer who are eligible for systemic therapy. The sponsor’s indication and reimbursement request is for durvalumab in combination with carboplatin and paclitaxel for the first-line treatment of adults with primary advanced or recurrent dMMR endometrial cancer who are candidates for systemic therapy, followed by durvalumab as monotherapy. Durvalumab was previously reviewed by Canada’s Drug Agency (CDA-AMC) for extensive-stage small-cell lung cancer; locally advanced, unresectable non–small cell lung cancer; biliary tract cancer; and unresectable hepatocellular carcinoma, all of which received conditional reimbursement recommendations.
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of durvalumab (concentrate for solution for IV infusion, 50 mg/mL) in combination with platinum-based chemotherapy, followed by maintenance with durvalumab as monotherapy for the first-line treatment of patients with advanced or recurrent endometrial cancer that is dMMR.
Perspectives of Patients, Clinicians, and Drug Programs
The information in this section is a summary of input provided by the patient and clinician groups that responded to the call by CDA-AMC for input and by the clinical experts consulted for the purpose of this review.
Patient Input
One patient group, the Colorectal Cancer Resource & Action Network (CCRAN), in collaboration with the Canadian Cancer Survivor Network and HPV Global Action, submitted input on durvalumab for endometrial cancer. Information for this submission was gathered through outreach by CCRAN to 12 clinicians in Canada in September 2024, 17 US-based clinician investigators involved in the DUO-E clinical trial in October 2024, and the Society of Gynecologic Oncology and the Gynecologic Cancer Initiative. Additionally, HPV Global Action collaborated with several medical advisors to identify patients with experience using the therapy under review. These efforts led to 2 telephone interviews with patients with MSS endometrial cancer who participated in the DUO-E trial. Also, data from a 2023 online survey, which generated 6 responses and gathered information on the experiences of 4 patients, were used for this input.
The patient group input highlighted that patients with endometrial cancer face significant inequities, including research underfunding; rising incidence rates, particularly among females who are postmenopausal; and increasing mortality rates, despite advancements in oncology. According to the patient group input, a diagnosis of endometrial cancer is profoundly distressing, placing significant emotional strain on both patients and their caregivers and triggering fears related to personal health and family welfare.
Respondents to the survey reported using various treatment options, including radiation, surgical resection, targeted therapy, hormonal therapy, immunotherapy, chemotherapy, and complementary medicines. Common symptoms included neuropathy, fatigue, vaginal dryness, itching, burning sensations, changes in sexual function, fluid retention, nausea, constipation, and cognitive impairment known as “chemo brain.” Many patients described chemotherapy as “tough,” with significant nausea and fatigue impacting daily life. The effects of treatment often extend to sexual health, which is frequently overlooked in clinical care and research.
The input highlighted that early-stage endometrial cancer is typically treated with surgery, often combined with chemotherapy, radiation, or hormone therapy. However, treatment options for patients with recurrent or metastatic endometrial cancer are limited, and the prognosis remains poor due to stagnant access to new therapeutics over the past decades. Patient groups highlighted an urgent unmet need for additional precision therapeutics in advanced or recurrent endometrial cancer in Canada, with 60% of patients with endometrial cancer ranking “prolong life” as the most important issue they hope new treatments will address.
The 2 patients who participated in telephone interviews shared their experiences with durvalumab in combination with olaparib during the DUO-E trial, reporting complete responses (CRs) and minimal side effects, while a third patient receiving durvalumab treatment in Australia highlighted the positive impact of targeted therapies on their quality of life.
Clinician Input
Input From the Clinical Experts Consulted for This Review
The clinical experts consulted for this review noted that the goals of treatment for advanced or metastatic endometrial cancer are to improve quality of life by reducing pain and suffering, improve disease-related symptoms, control the cancer proliferation, and improve survival, if possible. They noted that achieving a longer duration of response would be strongly linked to improvement in relevant patient outcomes. In dMMR endometrial cancer, 1 expert noted that between 20% and 40% of patients do not experience a response to the current first-line treatment paradigm; for context, they noted that, in general, approximately 40% of patients are not expected to experience a response to chemotherapy, and the duration of response (DOR) to chemotherapy alone is often very short. While the addition of an immune checkpoint inhibitor (ICI) improves the overall treatment response rate, it has the most significant impact on the improvement in DOR. The unmet needs in patients with dMMR endometrial cancer include better identification of potential nonresponders, determining the therapy their cancer might respond to, and determining the ideal duration of maintenance treatment among the patients with cancer that does respond to therapy.
The experts indicated that durvalumab would be an additional first-line ICI option, alongside dostarlimab, that could be added to chemotherapy and used as maintenance monotherapy in patients with dMMR endometrial cancer. In general, the clinical experts noted that the choice of ICI is often based on availability but, when multiple approved options are available, any of them could be offered. If a patient did not experience a response to ICI therapy, the clinical experts noted it was unlikely they would trial the drug class again; however, if a patient had a recurrence more than 12 months after therapy and had previously experienced a response, then re-treatment might be considered.
The clinical experts noted that the DUO-E trial results did not identify specific molecular subtypes of cancer (beyond dMMR) that could respond to the new treatments; therefore, all patients who meet the other clinical trial criteria would likely be candidates for treatment with durvalumab.
According to the experts, the outcomes from the DUO-E trial corresponded to standard clinical assessments, with the exception of HRQoL measures, which are not routinely used in clinical practice. In general, symptomatic benefit would be assessed every cycle through conversations with the patient, and radiologic treatment response would be assessed every 2 to 3 cycles. They noted that the ideal outcome to assess response would be survival but, in the face of measurable disease, achieving stability (i.e., prolongation of progression-free survival [PFS]) while being tolerated well would be a minimum response to continue therapy. They indicated that the definition of a treatment benefit may vary across physicians and patients.
Disease progression and serious adverse events (SAEs) would be grounds to discontinue therapy, according to the experts. Examples of SAEs could include grade 3 or 4 toxicities such as hepatitis, colitis, or pneumonitis, as well as any other significant immune-related toxicity. Both clinical experts agreed that an oncologist or specialist in administering chemotherapy or biologic therapy would be essential to manage the complexities of treatment toxicity.
Clinician Group Input
Two inputs, 1 from the Ontario Health (Cancer Care Ontario) (OH [CCO]) Gynecologic Cancer Drug Advisory Committee (7 clinicians contributed to the input) and 1 from the Gynecologic Oncology Society of Canada (GOC) (1 clinician contributed to the input) were provided for this review. The OH (CCO) input was gathered through conference calls and emails, while the GOC input was based on data from completed clinical trials and expert opinions from board members on treating advanced or recurrent endometrial cancer.
According to the clinician groups, treatment for dMMR endometrial cancer involves platinum-based chemotherapy (carboplatin and paclitaxel) and radiation, with pembrolizumab funded for recurrent dMMR disease after failure of chemotherapy. There are no publicly funded first-line immunotherapies for patients with dMMR endometrial cancer, although compassionate access to dostarlimab in combination with chemotherapy is available.
The clinician groups agreed with the experts consulted for this review that the primary treatment goals are to prolong survival, delay disease progression, control symptoms, improve HRQoL and, when possible, cure the disease. Both the clinician input and the clinical experts agreed that chemotherapy alone fails to provide a durable response in patients with dMMR endometrial cancer. The input from both the clinician groups and the clinical experts also agreed that durvalumab would be suitable as a first-line option for patients with advanced (stage III or IV) or recurrent endometrial cancer. They also noted that patients with dMMR endometrial cancer can receive durvalumab treatment without prior chemotherapy.
The clinician groups agreed that treatment response is assessed through imaging (CT or MRI) and clinical evaluations. The GOC added that tolerability is evaluated before each treatment cycle (i.e., every 3 weeks) with radiologic assessments every 6 to 9 weeks. A clinically meaningful response includes tolerable toxicity and improved PFS.
The clinician groups indicated that treatment may be withheld due to disease progression and intolerable toxicity or adverse events (AEs). The GOC further mentioned that such decisions could also be related to patient preference. In line with the clinical experts consulted for this review, the clinician groups stated that durvalumab plus carboplatin and paclitaxel should be administered in an outpatient setting and is best prescribed by specialist physicians experienced in systemic therapy.
Drug Program Input
Input was obtained from the drug programs that participate in the CDA-AMC Reimbursement Review process. The following were identified as key factors that could potentially impact the implementation of a recommendation for durvalumab combined with carboplatin and paclitaxel followed by durvalumab monotherapy:
relevant comparators
considerations for initiation of therapy
considerations for continuation or renewal of therapy
considerations for prescribing of therapy
generalizability
care provision issues.
The clinical experts provided advice on the potential implementation issues raised by the drug program input. Refer to Table 5 for more details.
Clinical Evidence
Systematic Review
Description of Studies
The DUO-E study is an ongoing phase III, multicentre, double-blind, placebo-controlled randomized controlled trial (RCT) of durvalumab in combination with platinum-based chemotherapy (carboplatin and paclitaxel) followed by maintenance with durvalumab monotherapy or with durvalumab in combination with olaparib in patients with recurrent or newly diagnosed advanced endometrial cancer compared with SOC alone (carboplatin and paclitaxel). Enrolment for the DUO-E study ended in March 2022; however, treatment and follow-up are still ongoing. In total, 718 patients were randomized 1:1:1 to SOC (arm A; N = 241), durvalumab plus SOC (arm B; N = 239), or durvalumab plus olaparib plus SOC (arm C; N = 238). In the chemotherapy phase, arm A received a regimen of platinum-based chemotherapy consisting of carboplatin and paclitaxel plus durvalumab placebo for 6 cycles; after the chemotherapy phase, patients who had no evidence of progressive disease then received maintenance-phase therapy consisting of durvalumab placebo IV every 4 weeks plus olaparib placebo tablets twice daily. Patients in arms B and C received the same platinum-based chemotherapy as those in the SOC arm plus durvalumab for 6 cycles, then arm B received maintenance-phase therapy consisting of durvalumab plus olaparib placebo tablets twice daily, while arm C received both durvalumab and olaparib. The primary end point of the DUO-E study was PFS, with secondary end points consisting of overall survival (OS), objective response rate (ORR), DOR, time to study treatment discontinuation or death (TDT), and HRQoL.
Of note, the DUO-E trial randomized patients with pMMR and dMMR status to all 3 study arms. To align with the Health Canada indication and reimbursement request (durvalumab in combination with carboplatin and paclitaxel in patients with dMMR endometrial cancer, followed by maintenance durvalumab), this Clinical Review Report focuses only on the population of patients from the DUO-E trial with dMMR endometrial cancer who received durvalumab in combination with carboplatin and paclitaxel (i.e., only a subset of the patients included in arm B of the DUO-E study).
Efficacy Results
Progression-Free Survival
In the dMMR subgroup, there were a total of 25 PFS-related events in the SOC arm (51.0%; N = 49 patients) and 15 PFS-related events in the SOC plus durvalumab arm (32.6%; N = 46 patients). There were ██████ censored deaths in the SOC arm and ██████ in the SOC plus durvalumab arm. The median PFS in the SOC arm was 7.0 months (95% confidence interval [CI], 6.7 to 14.8), while the median PFS was not calculable in the SOC plus durvalumab arm (hazard ratio [HR], 0.42; 95% CI, 0.22 to 0.80 in favour of durvalumab). The risk difference between study arms for the proportion of patients with progression-free disease was █████ (95% CI, ████ ██ ██████) at 6 months; █████ (95% CI, ████ ██ ██████) at 12 months; and █████ (95% CI, █████ ██ ██████) at 18 months.
Overall Survival
In the dMMR subgroup, there were a total of ██ ███████ deaths in the SOC arm and ███████ deaths in the SOC plus durvalumab arm. The median OS in the SOC arm was ████ months (95% CI ███ ██████████), while the median OS was ███ ██████████ in the SOC plus durvalumab arm (HR, ████; 95% CI, ████ ██ ████ in favour of durvalumab). The risk difference between study arms for the proportion of patients who were alive was █████ (95% CI, █████ ██ ██████) at 6 months, ████% (95% CI, ████ ██ █████) at 12 months, and █████ (95% CI, ████ ██ ██████) at 18 months.
Objective Response Rate
In the dMMR subgroup, a total of ██ patients (████%) had a response out of ██ patients with measurable disease at baseline in the SOC arm, and ██ patients (████%) had a response out of ██ with measurable disease at baseline in the SOC plus durvalumab arm. The odds ratio (OR) for response was ████ (95% CI, ████ ██ ████) in favour of durvalumab.
Duration of Response
In the dMMR subgroup, █ patients (████%) out of the ██ patients in the SOC arm who experienced a response subsequently died or experienced disease progression. The median DOR (measured from the onset of response) was 10.5 months. Out of the ██ patients in the SOC plus durvalumab arm who experienced a response, █ patients (████%) subsequently died or experienced disease progression; the median DOR was not calculable. The difference between study arms for the proportion of patients who continued to experience a response was ████% (95% CI, ████% to ████%) at 6 months, ████% (95% CI, █████ ██ ████%) at 12 months, and ████% (95% CI, █████ ██ ████%) at 18 months.
Time to Treatment Discontinuation or Death
In the dMMR subgroup, ██ ███████ patients in the SOC arm and ██ ███████ patients in the SOC plus durvalumab arm had an event of treatment discontinuation or death (HR, ████; 95% CI, ████ ██ ████ in favour of durvalumab).
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30
The European Organisation for Research and Treatment of Cancer (EORTC) Quality of Life Questionnaire Core 30 (EORTC QLQ-C30) functional scales range from 0 to 100, with higher scores indicating better functioning. In the dMMR subgroup, the mean baseline score was █████ points (standard deviation [SD] = █████) in the SOC arm and █████ points (SD = █████) in the SOC plus durvalumab arm. At week 18 (corresponding to the sixth cycle of the chemotherapy phase), the mean change in score from baseline was ██████ points (SD = █████) in the SOC arm (N = ██) and █████ points (SD = █████) in the SOC plus durvalumab arm (N = ██). The absolute mean difference between study arms was █████ points (95% CI, –█████ ██ ████). At week 42 (corresponding to the sixth month of the maintenance phase), the mean change in scores from baseline was ██████ points (SD = █████) in the SOC arm (N = ██) and ████ points (SD = █████) in the SOC plus durvalumab arm (N = ██). The absolute mean difference between study arms was █████ points (95% CI, ████ ██ █████).
EORTC Quality of Life Questionnaire – Endometrial Cancer Module Pain in Back and Pelvis Score
The EORTC Quality of Life Questionnaire – Endometrial Cancer Module (EORTC QLQ-EN24) functional scales range from 0 to 100, with higher scores indicating better functioning, and the 10 symptom scales range from 0 to 100, with higher scores indicating more severe symptoms. In the dMMR subgroup, the mean baseline score was █████ points (SD = █████) in the SOC arm and █████ points (SD = █████) in the SOC plus durvalumab arm. At week 18, the mean change in score from baseline was ████ points (SD = █████) in the SOC arm (N = ██) and ████ points (SD = █████) in the SOC plus durvalumab arm (N = ██). The absolute mean difference between study arms was ████ points (95% CI, █████ ██ █████). At week 42, the mean change in scores from baseline was █████ points (SD = █████) in the SOC arm (N = ██) and ████ points (SD = █████) in the SOC plus durvalumab arm (N = ██). The absolute mean difference between study arms was ████ points (95% CI, █████ ██████).
EORTC QLQ-EN24 Urological Symptoms Score
In the dMMR subgroup, the mean baseline score was █████ points (SD = █████) in the SOC arm and █████ points (SD = █████) in the SOC plus durvalumab arm. At week 18, the mean change in score from baseline was ████ points (SD = █████) in the SOC arm (N = ██) and █████ points (SD = █████) in the SOC plus durvalumab arm (N = ██). The absolute mean difference between study arms was ████ points (95% CI, █████ ██████). At week 42, the mean change in scores from baseline was ████ points (SD = █████) in the SOC arm (N = ██) and ████ points (SD = █████) in the SOC plus durvalumab arm (N = ██). The absolute mean difference between study arms was ████ points (95% CI, █████ ██ ████).
Harms Results
Adverse Events
In the dMMR subgroup, all patients in each study arm experienced an AE during the DUO-E trial. A total of ████% of patients in the SOC arm and ████% of patients in the SOC plus durvalumab arm experienced an AE with a maximum grade of 3 or 4. The most common AEs were nausea (████% of patients in the SOC plus durvalumab arm, ████% of patients in the SOC arm), alopecia (█████ in the SOC plus durvalumab arm, █████ in the SOC arm), arthralgia (█████ in the SOC plus durvalumab arm, █████ in the SOC arm), and anemia (█████ in the SOC plus durvalumab arm, █████ in the SOC arm).
There were differences between study arms in the proportion of patients with several AEs. Of note, a numerically higher proportion of patients in the SOC plus durvalumab arm reported nausea (█████ versus █████ in the SOC arm), alopecia (█████ versus █████ in the SOC arm), arthralgia (█████ versus █████ in the SOC arm), hypomagnesemia (█████ versus █████ in the SOC arm), cough (█████ versus █████ in the SOC arm), peripheral neuropathy (█████ versus █████ in the SOC arm), dyspnea (█████ versus ████ in the SOC arm), and injury, poisoning, or procedural complications (█████ versus █████ in the SOC arm).
Serious Adverse Events
In the dMMR subgroup, a total of █████ patients in the SOC arm and █████ patients in the SOC plus durvalumab arm experienced an SAE during the DUO-E trial. The most common SAEs were as follows: gastrointestinal disorders (█████ in the SOC plus durvalumab arm consisting of █ reports of fecaloma and █ report each of abdominal hernia, colitis, intestinal obstruction, and nausea, and ███% in the SOC arm consisting of █ report each of diarrhea, nausea, and vomiting), blood and lymphatic system disorders (████% in the SOC arm consisting of █ reports of anemia, █ reports of febrile neutropenia, and █ reports of neutropenia, and ███% in the SOC plus durvalumab arm consisting of █ report of autoimmune hemolytic anemia), infections and infestations (███% in the SOC arm consisting of █ report each of COVID-19, neutropenic sepsis, urinary tract infection and urosepsis, and ███% in the SOC plus durvalumab arm consisting of █ report each of appendicitis, gastroenteritis, and sepsis), and renal and urinary disorders (███% in the SOC arm consisting of █ report each of acute kidney injury, renal failure, ureteric obstruction and urinary bladder hemorrhage, and █% in the SOC plus durvalumab arm).
Withdrawals Due to Adverse Events
In the dMMR subgroup. a total of ████% of patients experienced AEs leading to the withdrawal of durvalumab in the SOC plus durvalumab arm, and ████% reported this in the SOC arm (durvalumab placebo). In the SOC plus durvalumab arm, the reasons for discontinuation were anemia, interstitial lung disease, maculopapular rash, symmetric drug-related intertriginous and flexural exanthema, fatigue, and procedural pain (each reported in █ patient). In the SOC arm, the reasons for discontinuation of durvalumab placebo were anemia (█ patients), cerebrovascular accident, tinnitus, and asthenia (each reported in █ patient). A total of ████% of patients in the SOC plus durvalumab arm and ███% of patients in the SOC arm experienced an AE leading to discontinuation of SOC.
Mortality
In the dMMR subgroup, a total of ██ ███████ patients died in the SOC arm and ███████ patients died in the SOC plus durvalumab arm. The submission did not provide details on the specific causes of death in patients in the dMMR subgroup.
Notable Harms
Immune-mediated AEs (exact conditions not specified) and infusion reactions were identified by the clinical experts as adverse events of special interest (AESIs). In the dMMR subgroup, ████% of patients experienced an immune-mediated AE in the SOC plus durvalumab arm and ████% experienced this in the SOC arm. A total of ███% of patients experienced infusion reactions in the SOC plus durvalumab arm compared with ████% of patients in the SOC arm.
A total of ████% of patients in the SOC plus durvalumab arm and ████% of patients in the SOC arm experienced an AESI for durvalumab (or its placebo). The most common AEs in both arms were diarrhea (████% in the SOC plus durvalumab arm, ████% in the SOC arm) and rash (████% in the SOC plus durvalumab arm, ████% in the SOC arm). Hypothyroidism (████% of patients) was the next most common AE in the SOC plus durvalumab arm, and hyperthyroidism (███% of patients) was the next most common AE in the SOC arm.
Critical Appraisal
The DUO-E study is an ongoing phase III trial assessing the efficacy and safety of durvalumab plus SOC compared with SOC alone in the treatment of primary advanced or recurrent endometrial cancer. Despite the adequate randomization, concealment, and blinding, there were numerically higher numbers of patients in the SOC arm who discontinued treatment relative to the number in the SOC plus durvalumab arm, which suggests the possibility that patients may have become unblinded to their treatment assignment and discontinued treatment more readily than those in the SOC plus durvalumab arm. As part of the study design, patients with no evidence of progressive disease during the 6-cycle chemotherapy phase were eligible to proceed to the maintenance phase. Results separating the efficacy outcomes in the chemotherapy and maintenance phases were not provided; thus, the impact of the chemotherapy phase versus the maintenance phase individually on response to treatment remains unknown. The design of the DUO-E trial included patients with either dMMR and pMMR endometrial cancer. However, given the Health Canada indication and reimbursement request, the focus of this review is on the subgroup of patients with dMMR endometrial cancer. Therefore, all results focusing on the dMMR subgroup can only be considered exploratory and supportive of the overall effect of durvalumab. Furthermore, the subgroup was not controlled for multiple comparisons and there is an increased risk of type I error, which is particularly important, given that the sample sizes in each study arm were small. The results from the DUO-E trial were from an interim analysis (IA), and the P value cut-off for the IA of the subgroups themselves is not known; this carries an increased risk of overestimating the true effect. While the primary end point was reached for the intention-to-treat (ITT) population, the median PFS and OS were not estimable for the dMMR subgroup in the respective analyses. In the ITT population, this analysis was based on an information fraction of 61.0% while, in the dMMR subgroup, only ████% of PFS events had occurred, suggesting the data were immature, particularly for OS. As a result, there is increased uncertainty in the PFS and OS results, and it is therefore unclear how confidently the long-term results associated with durvalumab can be predicted. For the HRQoL end points, the baseline estimates and the estimates at weeks 18 and 42 have a substantial amount of missing data. The end points were analyzed using a mixed model for repeated measures, which assumes the missing data are missing at random; however, given the nature of the disease and the design of the trial, the missing-at-random assumption is not appropriate. Because patients are censored at disease progression or death, among other reasons, it is likely that these patients are systematically different from the patients who are included in the analysis; thus, there is a likelihood of bias in the results.
Per the clinical experts, the inclusion and exclusion criteria were broadly representative of patients who would be candidates for durvalumab. However, the study is subject to some limitations impacting external validity. The results of patient screening were not available for the dMMR subgroup; therefore, the reasons for screening failure and the distribution of screening failures between study arms is not known, so it is unclear whether there are systematic differences between the patients who failed screening and those who did not. Similarly, the exact causes of death in participants who died during the study were not reported, and it is therefore not known whether there are systematic differences between the patients who died and those who did not. Patients with an Eastern Cooperative Oncology Group (ECOG) status greater than 1 were not included in the trial; however, according to the clinical experts consulted for this review, these patients would likely be considered for treatment with durvalumab in clinical practice. Apart from this, the study did not assess whether other molecular subtypes (e.g., POLE mutation, HER expression, p53 mutation) would have an impact on the results observed. The clinical experts noted that additional molecular classifications are usually undertaken in clinical practice and that there is a proportion of patients with dMMR cancer that does not respond to current ICI therapy, the reason for which is not always clear. Therefore, it is possible the same nonresponder population may also exist for patients treated with durvalumab.
GRADE Summary of Findings and Certainty of the Evidence
For the pivotal studies and RCTs identified in the sponsor’s systematic review, the Grading of Recommendations Assessment, Development and Evaluation (GRADE) was used to assess the certainty of the evidence for outcomes considered most relevant to inform expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group.20,21
Following the GRADE approach, the evidence from the RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty-of-evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null. For the PFS and OS outcomes, the target of the assessment was the presence or absence of an important effect based on thresholds provided by the clinical experts. For the HRQoL outcomes, the target of the assessment was the presence or absence of any effect based on a null threshold.
The selection of outcomes for GRADE assessment was based on the sponsor’s summary of clinical evidence, consultation with clinical experts, and the input received from patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members:
PFS (difference in the proportion of patients with progression-free disease at 12 months and 18 months)
OS (difference in the proportion of patients alive at 12 months and 18 months)
EORTC QLQ-C30 (change from baseline to 18 weeks and change from baseline to 42 weeks)
EORTC QLQ-EN24 (change from baseline to 18 weeks and change from baseline to 42 weeks)
Notable harms: infusion-mediated AEs and infusion-related reactions.
Table 2: Summary of Findings for Durvalumab in Combination With Carboplatin-Paclitaxel Versus Placebo in Combination With Carboplatin-Paclitaxel for Patients With Primary Advanced or Recurrent dMMR Endometrial Cancer
Outcome and follow-up | Patients (studies), N | Absolute effects (95% CI) | Certainty | What happens | ||
|---|---|---|---|---|---|---|
SOC | SOC + durvalumab | Difference | ||||
Survival outcomes | ||||||
Progression-free survivala | ||||||
Proportion of patients whose cancer is progression-free at 12 months Median follow-up:b ██ ███ ███ | 95 (1 RCT) | ███ ██ | ███ ███ ████ █████ ██ ███ | ███ ███ ███ ████ ██ ███ | Lowd,e | Durvalumab in combination with carboplatin and paclitaxel, followed by durvalumab monotherapy, may result in an increase in the proportion of patients whose cancer is progression-free at 12 months when compared with carboplatin and paclitaxel alone. |
Proportion of patients whose cancer is progression-free at 18 months Median follow-up:b ██ ███ ███ ██ | 95 (1 RCT) | ███ ███ | ███ ███ ████ ██ | ███ ███ ██ | Lowd,e | Durvalumab in combination with carboplatin and paclitaxel, followed by durvalumab monotherapy, may result in an increase in the proportion of patients whose cancer is progression-free at 18 months when compared with carboplatin and paclitaxel alone. |
Overall survivala | ||||||
Proportion of patients who are alive at 12 months Median follow-up:b ██ ███ ████ | 95 (1 RCT) | ███ ███ | ███ ███ ████ ██ | ███ ███ ██ | Lowf,g | Durvalumab in combination with carboplatin and paclitaxel, followed by durvalumab monotherapy, may result in an increase in the proportion of patients who are alive at 12 months when compared with carboplatin and paclitaxel alone. |
Proportion of patients who are alive at 18 months Median follow-up: b ██ ███ ████ | 95 (1 RCT) | ███ ██ | ███ ███ ████ ██ | ███ ███ ██ | Lowf,g | Durvalumab in combination with carboplatin and paclitaxel, followed by durvalumab monotherapy, may result in an increase in the proportion of patients who are alive at 18 months when compared with carboplatin and paclitaxel alone. |
Health-related quality of lifea | ||||||
EORTC QLQ-C30 global health status score (100 = best; 0 = worst)c | ||||||
Change from baseline, points Follow-up: ██ █████ | 95 (1 RCT) | ███ ███ | ███ ███ ████ ██ | ███ ███ █ | Very lowh,i | The evidence is very uncertain about the effect of durvalumab in combination with carboplatin and paclitaxel followed by durvalumab monotherapy on the change in EORTC QLQ-C30 global health status score from baseline to 18 weeks when compared with carboplatin and paclitaxel alone. |
Change from baseline, points Follow-up: ██ █████ | 95 (1 RCT) | ███ ███ | ███ ███ ████ ██ | ███ ███ █ | Lowi | Durvalumab in combination with carboplatin and paclitaxel, followed by durvalumab monotherapy, may result in an increase in EORTC QLQ-C30 global health status score from baseline to 42 weeks when compared with carboplatin and paclitaxel alone. |
EORTC QLQ-EN24 pain in back and pelvis score (100 = best; 0 = worst)c | ||||||
Change from baseline, points Follow-up: ██ █████ | 95 (1 RCT) | █████ | ████████████ | █████████ | Very lowh,i | The evidence is very uncertain about the effect of durvalumab in combination with carboplatin and paclitaxel, followed by durvalumab monotherapy, on the change in EORTC QLQ-C30 global health status score from baseline to 18 weeks when compared with carboplatin and paclitaxel alone. |
Change from baseline, points Follow-up: ██ █████ | 95 (1 RCT) | ██████ | ██████████ | █████████ | Very lowh,i | The evidence is very uncertain about the effect of durvalumab in combination with carboplatin and paclitaxel, followed by durvalumab monotherapy, on the change in EORTC QLQ-C30 global health status score from baseline to 42 weeks when compared with carboplatin and paclitaxel alone. |
EORTC QLQ-EN24 urological symptoms score (100 = best; 0 = worst)c | ||||||
Change from baseline, points Follow-up: ██ █████ | 95 (1 RCT) | ██████ | ███████████ | █████████ | Very lowh,i | The evidence is very uncertain about the effect of durvalumab in combination with carboplatin and paclitaxel, followed by durvalumab monotherapy, on the change in EORTC QLQ-C30 global health status score from baseline to 18 weeks when compared with carboplatin and paclitaxel alone. |
Change from baseline, points Follow-up: ██ █████ | 95 (1 RCT) | ██████ | ███████████ | ████████ | Very lowh,i | The evidence is very uncertain about the effect of durvalumab in combination with carboplatin and paclitaxel, followed by durvalumab monotherapy, on the change in EORTC QLQ-C30 global health status score from baseline to 42 weeks, when compared with carboplatin and paclitaxel alone. |
Harms | ||||||
Proportion of patients with immune-mediated adverse events Follow-up: ███ █████ | 95 (1 RCT) | ███ ███ | ███ ███ ████ | ██ █████ | Lowj | Durvalumab in combination with carboplatin and paclitaxel, followed by durvalumab monotherapy, may result in an increase in the proportion of patients with immune-mediated adverse events when compared with carboplatin and paclitaxel alone. |
Proportion of patients with infusion reactions Follow-up: ███ █████ | 95 (1 RCT) | ███ ███ | ██ ███ ████ ██ | ██ █████ | Very lowj | The evidence is very uncertain about the effect of durvalumab in combination with carboplatin and paclitaxel, followed by durvalumab monotherapy, on the proportion of patients with infusion reactions when compared with carboplatin and paclitaxel alone. |
CI = confidence interval; dMMR = mismatch repair deficient; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-EN24 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire – Endometrial Cancer Module; HRQoL = health-related quality of life; NR = not reported; OS = overall survival; PFS = progression-free survival; RCT = randomized controlled trial; SOC = standard of care.
Note: Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aIn the trial, testing for this outcome in the dMMR subgroup was not adjusted for multiplicity. The results are considered supportive evidence.
bFollow-up presented as durvalumab + carboplatin and paclitaxel vs. placebo + carboplatin and paclitaxel.
cAdditional information was requested from the sponsor to obtain absolute differences and 95% CIs. This information was not necessarily part of the sponsor’s statistical plan and is considered exploratory evidence.
dRated down 1 level for serious indirectness. At the time of data cut-off, median PFS was not reached and the study did not meet the primary end point in the dMMR subgroup. This implies that the PFS data are immature and there is high uncertainty in the trends observed to date; therefore, the confidence with which the results predict the outcome in the long-term is not clear.
eRated down 1 level for serious imprecision. Based on a nontrivial target certainty assessment with a minimal important difference threshold of 150 per 1,000 at 12 months and 100 per 1,000 at 18 months provided by the clinical experts, the point estimate of the effect is larger than the threshold but the 95% CI includes the possibility of a trivial effect as well as a nontrivial effect at 12 months, the number of PFS events in each arm were not provided by the sponsor, and the sample size is very small, raising concern for prognostic imbalance and potential overestimation of the true effect.
fRated down 1 level for serious indirectness. At the time of data cut-off, median OS was not reached and the study did not meet the secondary end point in the dMMR subgroup. This implies that the OS data are immature and there is high uncertainty in the trends observed to date; therefore, the confidence with which the results predict the outcome in the long-term is not clear.
gRated down 1 level for serious imprecision. Based on a nontrivial target certainty assessment with a minimal important difference threshold of 100 per 1,000 provided by the clinical experts, the point estimate of the effect is larger than the threshold but the 95% CI includes the possibility of a trivial effect as well as a nontrivial effect, the number of OS events at 12 months and 18 months in each arm were not provided by the sponsor, and the sample size is very small, raising concern for prognostic imbalance and potential overestimation of the true effect.
hRated down 2 levels for serious study limitations. A considerable amount of patient data was missing at the time points and, based on the study design, it is likely that the missingness is informative.
IRated down 1 level for serious imprecision. Based on a non-null target certainty assessment, the CI for the estimate contains the possibility of reduced HRQoL as well as improved HRQoL.
jRated down 2 levels for very serious imprecision due to the small number of events in a small patient population and the lack of specified follow-up duration.
Sources: Details included in the table are from the sponsor’s summary of clinical evidence22 and additional information provided by the sponsor.23,24
Long-Term Extension Studies
The submission did not contain any long-term extension studies.
Indirect Comparisons
The DUO-E trial included SOC chemotherapy (carboplatin and paclitaxel) but did not include a comparison with dostarlimab, a relevant first-line comparator for patients with dMMR endometrial cancer, and information from indirect comparisons was included in the pharmacoeconomic model. Therefore, a review of the indirect evidence was undertaken. The body of indirect evidence consisted of 2 sponsor-submitted multiple-adjusted indirect comparisons (MAICs).
Description of Studies
A systematic literature review (SLR) was conducted with a focus on systemic anticancer treatments and which excluded all other therapies. The patient population of interest comprised those newly diagnosed with stage III or IV disease or recurrent disease who were either naive to first-line therapy or had been treated with 1 prior line of chemotherapy. The submission did not provide details on the date when the SLR was conducted, the range of publication dates that were used in the search terms, or the search terms used.
An anchored MAIC was performed using a frequentist approach and Bucher methodology. The treatment effects for the 2 efficacy outcomes, PFS and OS, were reported as HRs and associated 95% CIs for SOC plus durvalumab versus SOC plus dostarlimab, with an HR of less than 1 favouring SOC plus durvalumab. For the safety end points, the ORs were calculated for SOC plus durvalumab versus SOC plus dostarlimab, with ORs of less than 1 favouring SOC plus durvalumab. ORs were estimated from AE frequency data and the number of patients included in the safety analysis sets.
Efficacy Results
The submission did not provide a Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) diagram of the results from the SLR but reported that the SLR identified 2 phase III, double-blind, placebo-controlled RCTs relevant to the indirect treatment comparison: the DUO-E study and the RUBY Part 1 study. Differences were noted in the median duration of follow-up as well as the molecular characteristics of the patients’ cancer; the primary analysis for the MAICs used data from the post hoc subgroup of the DUO-E trial, which contained only patients with dMMR and/or MSI-H cancer (and therefore could contain patients with MSI-H pMMR endometrial cancer), and the main analysis in the RUBY Part 1 study enrolled patients who had either dMMR or MSI-H tumours. The submission did not provide a breakdown of other baseline characteristics before and after matching. Of note, after the matching procedure, minor differences remained in the proportion of patients who were Black or African American (███% in the DUO-E study after weighting, and 8.5% in the RUBY Part 1 study), and the proportion of patients who were aged 65 years and older (████% in the DUO-E trial after weighting, and 49.2% in the RUBY Part 1 study).
The results for PFS from the anchored, weighted MAIC suggested there was insufficient evidence to detect a difference between durvalumab plus SOC and dostarlimab plus SOC (adjusted HR, ████ ████ ███ ████ ██ ██████; effective sample size [ESS] = ████). Similarly, the results of OS using the anchored, unweighted MAIC suggested there was insufficient evidence to detect a difference between durvalumab plus SOC and dostarlimab plus SOC (adjusted HR, ████ ████ ███ ████ ██ ██████).
Harms Results
The harms results from the anchored, unweighted MAIC suggested there was insufficient evidence to detect a difference between durvalumab plus SOC and dostarlimab plus SOC for AEs leading to discontinuation (adjusted OR, ████ ████ ███ ████ ██ ████) and any SAEs (adjusted OR, ████ ████ ███ ████ ██ █████); however, for grade 3 or greater AEs, durvalumab plus SOC was favoured over dostarlimab plus SOC (adjusted OR, ████ ████ ███ ████ ██ ███████).
Critical Appraisal
The procedures as described for screening, data extraction, and the quality assessment steps used were considered generally accepted methods. However, the date on which the SLR was undertaken is not provided, and it is not known whether the most recent publications on any relevant comparators would have been captured in the search. The submission did not provide the results of the quality assessment; therefore, the risk of bias in the studies is not known. However, because the DUO-E and RUBY Part 1 trials were phase III trials, the risk of bias may be lower. For both MAIC analyses, data from a subgroup of the RUBY Part 1 study and a subgroup from the DUO-E trial were used (patients with dMMR and/or MSI-H cancer). In the case of the DUO-E trial, this resulted in the inclusion of some patients with pMMR MSI-H cancer in the subgroup used for the analysis, which is likely to somewhat bias the results because MMR status is a known treatment-effect modifier. The 2 studies also differ considerably in the median follow-up time because the follow-up in the RUBY Part 1 study was longer than in the DUO-E trial (twice as long follow-up time for PFS). This increases the likelihood of bias to possibly favour durvalumab because there was a shorter follow-up period over which events could accrue. Lastly, in both studies, the subgroup arms had small sample sizes and a low number of events, which impacts the power of the analysis. In addition, other baseline characteristics not included in the matching were not reported and, therefore, it is not known whether there are other potential sources of confounding in these characteristics. There were also no details reported on model fit, convergence, or model selection, which is another potential source of bias or uncertainty.
In addition to the limitations in the comparability of the studies and matching, there are additional limitations specific to each efficacy outcome. The MAIC for PFS was a weighted, anchored MAIC, and the weighting procedure was largely able to balance the baseline characteristics reported. However, the ESS was approximately ███ smaller (N = ████) than the number of patients prematching (N = ███), suggesting that data from a smaller number of patients may be driving the results, which increases the imprecision. These results are subject to high uncertainty due to the limitations in the comparability of the studies as well as the limitations around the size of the ESS. The analyses undertaken for OS and harms were unweighted, anchored MAICs, which did not employ the propensity score measurement process to reweight the results and was considered a naive comparison of the DUO-E and RUBY Part 1 trials. For OS, this was due to the small number of events observed (8 events for dMMR or MSI-H in SOC plus durvalumab and 7 events for SOC plus dostarlimab), which would generate potentially unstable results if reweighted and compared using MAIC methodology. There were still notable differences between the 2 studies at baseline, and it is not known how the sample size in the 2 studies compares with the sample in the MAIC for OS. It is highly likely that not all possible effect modifiers were controlled for in the analysis for these outcomes. The limitations in the comparability of the studies and analysis method significantly undermines the validity of the results for OS and harms.
Studies Addressing Gaps in the Evidence From the Systematic Review
The submission did not contain any studies addressing gaps in the evidence from the systematic review.
Conclusions
One phase III ongoing multicentre, double-blind, placebo-controlled RCT (the DUO-E trial) provided evidence for the efficacy and safety of durvalumab plus SOC for 6 cycles followed by durvalumab maintenance therapy as first-line treatment in the subgroup of 95 adult patients with primary advanced or recurrent dMMR endometrial cancer.
Patients and clinicians identified a need for new treatments for primary advanced or recurrent endometrial cancer — which is the most common gynecological malignancy — and prolonged survival was considered the most important outcome. In the subgroup of patients with dMMR endometrial cancer, the subgroup results of the DUO-E trial suggested improved PFS and OS in patients treated with durvalumab. The clinical experts consulted for this review considered these results compelling, and the results of the GRADE assessment suggested that the minimal important differences (MIDs) for the proportion of patients with progression-free disease or alive at 12 and 18 months were surpassed. However, because these results were based on a subgroup of the ITT population, the statistical analyses were not powered to detect differences in this subgroup, and no adjustments were made for multiple testing. Furthermore, although the median results for the time-to-event end points were not reached, the certainty in the magnitude of benefit was reduced because they were based on a small sample size and an IA with largely immature data and based on an information fraction of 42.1% for the subgroup and treatment arms presented. Furthermore, despite results for HRQoL that suggested no detriment in patients receiving durvalumab, there was a high rate of attrition. As such, the results of these important efficacy outcomes can be considered only as supportive of the overall effect of durvalumab, and causal conclusions cannot be drawn. Indirect evidence comparing durvalumab with dostarlimab, a relevant comparator in the Canadian clinical setting, suggested no difference between the 2 treatment regimens for PFS; however, the considerable limitations in the indirect evidence preclude conclusions on efficacy (as measured by OS) and harms. While the reporting of harms in the DUO-E study is also immature and lacks important details, in particular with regard to AESIs and the specific causes of death, the clinical experts consulted for this review considered the harms to be typical for patients undergoing chemotherapy with an ICI.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of durvalumab 1,120 mg administered intravenously every 3 weeks in combination with carboplatin and paclitaxel, followed by durvalumab 1,500 mg IV every 4 weeks until disease progression or unacceptable toxicity, in the treatment of primary advanced or recurrent endometrial cancer in adult patients who are candidates for systemic therapy.
Disease Background
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following was summarized and validated by the review team.
Endometrial cancer is a type of uterine cancer originating in the lining of the uterus and is the most common gynecological malignancy,1 accounting for approximately 95% of uterine cancers.2 In Ontario, 2,909 new endometrial cancer cases were diagnosed in 2018, with an age-standardized incidence rate of 36.2 per 100,000.3 In 2024, the Canadian Cancer Society estimated that 8,600 females would be diagnosed with uterine cancer that year, and 1,600 would die from it.4 Endometrial cancer primarily affects females who are postmenopausal, with an average age of diagnosis of 60 years.5
Endometrial cancer is staged using the FIGO system: stage I (tumour confined to the uterus), stage II (cervical stroma invasion), stage III (regional spread), and stage IV (spread to bladder, bowel, or distant organs).6 In Canada, the overall 5-year net survival for uterine cancer is 82%, with stage-specific survival rates of 70% for stage I, 45% for stage II, 30% for stage III, and 15% for stage IV, highlighting significant variability based on disease progression.7
Both primary advanced (stage III or IV) and recurrent (return following primary treatment) endometrial cancer are associated with numerous debilitating symptoms that significantly impair physical functioning and HRQoL. Key symptoms include abnormal vaginal bleeding, which may be periodic or continuous.8,9 Additional manifestations encompass pelvic or lower back pain, the presence of a palpable mass in the lower abdomen, and unintentional weight loss.8 Patients often experience abdominal distension, early satiety, alterations in bowel or bladder habits, and dyspareunia.10 The debilitating nature of the disease has a profound impact on daily activities, confidence, and self-esteem. Symptoms such as menopausal-like effects, sexual dysfunction, anxiety, depression, and long-term side effects of chemotherapy further diminish HRQoL.1,10,11
A diagnosis of endometrial cancer relies on clinical assessments to evaluate tumour location, size, and spread, supported by radiological imaging of the uterus and histopathological analysis of biopsy samples to determine histological type and grade. In cases of recurrent endometrial cancer, biopsies may also be used to identify molecular subtypes of the cancer.12,13
Molecular testing during endometrial biopsy is crucial for treatment decisions and risk assessment.14 MMR protein expression and MSI are key biomarkers for evaluating DNA repair functionality.25 Endometrial cancer is categorized as MSI-H, microsatellite instability-low, or MSS based on MSI testing,15 and as either pMMR or dMMR based on MMR status.16 Additional molecular tests are also recommended to assess for other molecular subtypes such as mutations in p53, DNA POLE, and estrogen receptor and HER2 expression.
MMR dysfunction results in MSI-H, marked by defective DNA repair and elevated mutation rates. IHC is a standard technique for detecting the expression of MMR proteins (hMLH1, hPMS2, hMSH2, and hMSH6). Loss of any protein indicates dMMR, while intact expression signifies pMMR.16 Clinically, dMMR endometrial cancer, which accounts for 20% to 30% of endometrial cancer cases,27 is linked to higher tumour mutation burdens and greater sensitivity to immunotherapies, such as PD-1 inhibitors.
Standards of Therapy
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following was summarized and validated by the review team.
The goals of treatment for advanced or metastatic endometrial cancer, according to the clinical experts consulted for this review, are to improve quality of life by reducing pain and suffering, reduce disease-related symptoms, control cancer proliferation, and improve survival, if possible.
The National Comprehensive Cancer Network (NCCN) and European Society for Medical Oncology (ESMO) guidelines recommend surgery, if possible, and adjuvant chemotherapy treatment for advanced endometrial cancer (advanced being stage III or IV) upon initial diagnosis of metastatic disease.28,29 The clinical experts consulted for this review noted that targeted radiation therapy is also an option in some cases (e.g., cancer confined to the pelvis); the NCCN does not recommend whole-abdomen radiotherapy due to toxicity. Surgery is recommended to be accompanied by adjuvant chemotherapy rather than observation alone.30
Patients who are not surgical candidates, or patients with recurrent or metastatic endometrial cancer (e.g., after treatment of localized disease) typically undergo systemic therapy. ESMO guidelines recommend platinum-based chemotherapy, doxorubicin or paclitaxel, dostarlimab, or pembrolizumab plus lenvatinib as treatment options for dMMR endometrial cancer that is recurrent or metastatic. According to the clinical experts, the current treatment paradigm in Canada for patients with dMMR endometrial cancer is to treat all eligible patients with platinum-taxane chemotherapy (i.e., carboplatin and paclitaxel or, in rare cases, with a substitution of 1 of the 2 agents) plus an ICI for up to 6 cycles followed by maintenance ICI alone. Dostarlimab is the only ICI therapy currently recommended for reimbursement for this indication. It is used alongside chemotherapy and then as maintenance monotherapy for up to 3 years if well tolerated and there is no evidence of disease progression. If patients with dMMR endometrial cancer do not receive an ICI in the first line, they are eligible to receive it in the second line. If there is disease progression, patients who did not receive an ICI in the first-line setting would receive a single-drug ICI in the second-line setting.
Other potential first-line treatments for advanced recurrent endometrial cancer include hormone therapies, which may be considered for specific patients with indolent or low-volume disease and with low-grade carcinomas.26 The clinical experts noted that patients with low-grade endometrioid histologies may receive hormone therapies such as letrozole or medroxyprogesterone acetate, either on their own or in combination with tamoxifen. For patients with HER2-overexpressing serous tumours, trastuzumab and chemotherapy are suggested over other regimens.30
Re-treatment with platinum-based chemotherapy is considered an acceptable approach for those experiencing relapse more than 6 months after initiating their last platinum-based chemotherapy because their disease is considered platinum-sensitive.29 The clinical experts consulted for this review noted that if a patient’s cancer does not respond to therapy, they are unlikely to be re-treated with the same class of treatment but, if the cancer recurs more than 12 months after treatment in a patient who initially experienced a response, then re-treatment with the same therapy may be considered. If a patient’s cancer progresses after an interval of less than 6 months, then treatment with second-line therapies such as pembrolizumab are options.31
Drug Under Review
The Health Canada indication for durvalumab (Notice of Compliance date: January 17, 2025), in combination with carboplatin and paclitaxel, is as a first-line treatment for adult patients with primary advanced or recurrent dMMR endometrial cancer who are eligible for systemic therapy. Durvalumab is administered at 1,120 mg every 3 weeks in combination with platinum-based chemotherapy for 4 to 6 cycles. This is followed by maintenance therapy at 1,500 mg every 4 weeks as monotherapy until disease progression or unacceptable toxicity.19
The sponsor’s reimbursement request is the same as the Health Canada indication. Durvalumab was previously reviewed by CDA-AMC for extensive-stage small-cell lung cancer; locally advanced, unresectable non–small cell lung cancer; biliary tract cancer; and unresectable hepatocellular carcinoma, all of which received conditional reimbursement recommendations.
Durvalumab is a fully human, high-affinity, immunoglobulin G1 kappa monoclonal antibody that selectively blocks the interaction of PD-L1 (a protein expressed on tumour cells) with PD-1 and CD80 on T cells. PD-L1 binds to PD-1 and CD80 on T cells, inhibiting their activation and reducing their ability to detect and destroy cancer cells. Selective blockade of PD-L1 and PD-1 interactions and PD-L1 and CD80 interactions releases the inhibition of immune responses and enhances antitumour immune responses.17,18
The key characteristics of durvalumab and other treatments available for primary advanced or recurrent endometrial cancer are summarized in Table 3.
Table 3: Key Characteristics of Durvalumab, Dostarlimab, Carboplatin, and Paclitaxel
Characteristic | Durvalumab | Dostarlimab | Carboplatin | Paclitaxel |
|---|---|---|---|---|
Mechanism of action | Humanized monoclonal antibody that selectively blocks the interaction of PD-L1 with PD-1 and CD80. | Monoclonal antibody that blocks PD-1 receptor, allowing cytotoxic T cells to kill tumour cells. | Synthetic analogue of cisplatin that causes DNA crosslinks in cancer cells, preventing proper DNA replication and transcription, triggering apoptosis (cell death) and inhibiting tumour growth. | Antimicrotubule agent that promotes abnormal microtubule assembly, disrupting the cell's ability to divide by blocking the G2 and M (mitotic) phases of the cell cycle. |
Indicationa | Durvalumab in combination with carboplatin and paclitaxel is indicated for the first-line treatment of adult patients with primary advanced or recurrent dMMR endometrial cancer who are candidates for systemic therapy, followed by maintenance treatment with durvalumab as monotherapy. | In combination with carboplatin and paclitaxel for the treatment of adult patients with primary advanced or recurrent dMMR or MSI-H endometrial cancer who are candidates for systemic therapy. | Although this regimen is considered appropriate as part of the first-line standard of care for patients,32 the indication for endometrial cancer is not specified in the product monograph. | Although this regimen is considered appropriate as part of the standard of care for patients,33 the indication for endometrial cancer is not specified in the product monograph. |
Route of administration | IV infusion | IV infusion | IV infusion | IV infusion |
Recommended dose | The recommended dose of durvalumab is 1,120 mg in combination with platinum-based chemotherapy (carboplatin and paclitaxel) every 3 weeks (21 days) for a minimum of 4 and up to 6 cycles, followed by maintenance with 1,500 mg every 4 weeks as monotherapy until disease progression or unacceptable toxicity. Patients with a body weight of 30 kg or less during the maintenance phase must receive weight-based dosing of durvalumab at 20 mg/kg until weight increases to greater than 30 kg. | 500 mg every 3 weeks for 6 doses followed by 1,000 mg every 6 weeks for all cycles thereafter. | 6 mg/mL/min administered every 3 weeks for a maximum of 6 cycles. | 175 mg/m2 IV infusion over 3 hours administered every 3 weeks for a maximum of 6 cycles. |
Serious adverse effects or safety issues |
|
| Hypersensitivity reactions, bone marrow suppression, and renal and hepatic toxicity. | Bone marrow suppression, cardiac abnormalities, hepatotoxicity. |
dMMR = deficient mismatch repair; MSI-H = microsatellite instability-high.
aHealth Canada–approved indication.
Sources: Product monographs for durvalumab,19 dostarlimab,34 carboplatin,35 and paclitaxel.36
Testing Procedure Considerations
In patients with endometrial cancer, MMR status is considered an important prognostic factor, and its determination can help in clinical decision-making.29,37,38 Confirmation of the molecular subtype based on MMR status (i.e., dMMR or pMMR) can inform risk stratification, eligibility for ICI therapy, and further germline testing for inherited conditions such as Lynch syndrome.39 Current international guidelines recommend testing all endometrial cancer tumours for MMR status along with other molecular or genetic tests, such as those for p53 status and, when indicated, POLE mutations.29,38
MMR status can be determined using IHC. This technique uses antibodies to target and stain specific MMR proteins such as MLH1, MSH2, MSH6, and PMS2, which are the most clinically relevant in cancer.39,40 MMR is considered deficient (i.e., dMMR) if there is an absence of staining in any of the 4 MMR proteins.39 Each of the MMR proteins pair with another to form heterodimers (i.e., PMS2 with MLH1 and MSH6 with MSH2); using a 2-antibody approach targeting PMS2 and MSH6 first, instead of targeting all 4 proteins, is a more cost-effective and quicker method for determining MMR status that is already being implemented in some jurisdictions in Canada.41,42 To offset a potential decrease in accuracy with the 2-antibody method, dMMR samples are further tested using IHC for MLH1 and MSH2 expression if PMS2 or MSH6 expression appears abnormal.42 IHC for MMR testing is conducted using tissue samples obtained through biopsies or surgical resections. IHC is widely available and relatively inexpensive. When considering ICI therapy, using IHC to determine MMR status is recommended over other methods, such as next-generation sequencing or polymerase chain reaction, due to factors such as lower costs, shorter turnaround times, and being less resource-intensive.42-44
The clinical experts consulted for this review noted that testing for MMR status with IHC using the diagnostic biopsy sample is routine in most jurisdictions in Canada and is the SOC for endometrial cancer. Jurisdictions that are not currently using IHC testing for MMR in patients with endometrial cancer should already have the capability (e.g., availability of antibodies) and the technical expertise in place. The clinical experts stated that the results of MMR testing are usually reported simultaneously with the diagnostic pathology report and that MMR testing can also be ordered by the treating oncologist.
The potential impacts of MMR testing for establishing treatment eligibility for durvalumab in adult patients with primary advanced or recurrent endometrial cancer were considered for this review, including impacts to health systems, patients (including families and caregivers), and costs. No new impacts are anticipated because MMR testing is currently performed as the SOC for endometrial cancer in Canada. Key considerations and relevant information available from materials submitted by the sponsor, input from the clinical experts consulted by the review team, and sources from the literature were validated by the review team, when possible, and are summarized in Table 4.
Table 4: Considerations for MMR Testing for Establishing Treatment Eligibility With Durvalumab in Primary Advanced or Recurrent Endometrial Cancer
Consideration | Criterion | Available information |
|---|---|---|
Health system–related | Number of individuals in Canada expected to require the test (e.g., per year). | The sponsor estimated that around 5,500 patients with incident EC will be tested for MMR status in 2024.22 According to the clinical experts consulted for this review, this is an accurate estimate of the number of individuals with EC expected to require MMR testing per year in Canada. The clinical experts noted that most, if not all, individuals with recurrent EC would have been tested for MMR status at the time of their initial diagnosis. Because MMR testing by IHC is already part of the standard of care for all patients with EC, the testing volume is not expected to increase as part of establishing treatment eligibility with durvalumab. |
Availability and reimbursement status of the testing procedure in jurisdictions across Canada. | According to the clinical experts, MMR testing by IHC is broadly available and funded across all jurisdictions in Canada. | |
Testing procedure as part of routine care. | According to the clinical experts, MMR testing by IHC is currently performed as part of the standard of care at diagnosis for all patients with EC. | |
Repeat testing requirements. | According to the clinical experts, MMR testing needs to be performed only once and does not need to be repeated. They also confirmed that individuals with recurrent EC would have been tested for MMR status at the time of their initial diagnosis and would not need to be retested. | |
Impact on human and other health care resources by provision of the testing procedure. | MMR testing by IHC is currently the standard of care for all patients with EC; therefore, no additional impact on human health and other resources is anticipated from the testing as part of establishing treatment eligibility for durvalumab. | |
Patient-related | Accessibility of the testing procedure in jurisdictions across Canada. | Based on the input from the clinical experts, MMR testing is accessible to most, if not all, patients across the country. |
Expected turnaround time for the testing procedure. | According to the clinical experts, turnaround times for IHC testing of MMR status are minimal. Testing is done on the diagnostic biopsy sample, with results usually being received simultaneously with the pathology report, or up to 1 or 2 days later. They anticipate no or minimal additional impact for patients or caregivers due to wait times if durvalumab is made available. | |
Burden associated with the testing procedure for patients, families, and/or caregivers. | In patients with EC, IHC testing for MMR status is routinely conducted on biopsy samples collected as part of the diagnostic work-up. According to the clinical experts, in rare instances, a new biopsy sample may be required to test for MMR status (e.g., when an individual had EC resection surgery abroad). Individuals whose results show a dMMR status would likely need genetic counselling (and potentially their families, as well) and additional testing to determine the presence of a hereditary condition (e.g., Lynch syndrome).45 However, because MMR testing by IHC is already part of the standard of care for all patients with EC, there is no additional burden to patients, families, and/or caregivers anticipated from the testing as part of establishing treatment eligibility with durvalumab. | |
Clinical | Clinical utility and validity of the testing procedure. | There is evidence that demonstrates the diagnostic accuracy and clinical utility of IHC testing for MMR status.a,39,42 |
Risks of harm associated with the testing procedure. | According to the clinical experts, because testing for MMR status by IHC is part of the standard of care at diagnosis for all patients with EC, there is no additional risk of harm anticipated from the testing as part of establishing treatment eligibility with durvalumab. | |
Cost | Projected cost of the testing procedure. | Materials provided by the sponsor show an estimated cost of less than $200 per IHC test performed to determine MMR status.46,47 Because testing for MMR status is currently part of the standard of care at diagnosis for all patients with EC, there is no additional cost anticipated from the testing as part of establishing treatment eligibility with durvalumab. |
dMMR = mismatch repair deficient; EC = endometrial cancer; IHC = immunohistochemistry; MMR = mismatch repair.
aCanada’s Drug Agency has not evaluated or critically appraised this evidence to determine its validity or reliability.
Perspectives of Patients, Clinicians, and Drug Programs
The full patient and clinician group submissions received are available in the consolidated patient and clinician group input document for this review on the project website for durvalumab (Imfinzi) in combination with carboplatin and paclitaxel.
Patient Group Input
This section was prepared by the review team based on the input provided by patient groups.
One patient group, CCRAN, in collaboration with the Canadian Cancer Survivor Network and HPV Global Action, submitted input on durvalumab for endometrial cancer. CCRAN gathered information for this submission in September 2024 through outreach to 12 clinical trial investigators and/or clinicians in Canada to identify patients with experience using durvalumab in combination with carboplatin and paclitaxel for dMMR and/or MSI-H cancer, followed by olaparib in patients with pMMR or MSS cancer. CCRAN also reached out to 17 US-based clinician investigators involved in the DUO-E clinical trial via email in October 2024, as well as to the Society of Gynecologic Oncology and the Gynecologic Cancer Initiative. Additionally, HPV Global Action collaborated with several of its medical advisors to identify patients with experience using the therapy under review. These efforts led to 2 telephone interviews with patients with MSS endometrial cancer who participated in the DUO-E trial. Also, data from a 2023 online survey, which generated 6 responses and gathered information on the experiences of 4 patients, were used for this input.
The patient group input highlighted that patients with endometrial cancer face significant inequities, including research underfunding; rising incidence rates, particularly among females who are postmenopausal; and increasing mortality rates, despite advancements in oncology. According to the patient group input, a diagnosis of endometrial cancer is profoundly distressing, placing significant emotional strain on both patients and their caregivers, and triggering fears related to personal health and family welfare.
Respondents to the survey reported using various treatment options, including radiation, surgical resection, targeted therapy, hormonal therapy, immunotherapy, chemotherapy, and complementary medicines. Common symptoms included neuropathy, fatigue, vaginal dryness, itching, burning sensations, changes in sexual function, fluid retention, nausea, constipation, and cognitive impairment known as “chemo brain.” Many patients described chemotherapy as “tough,” with significant nausea and fatigue impacting daily life. The effects of treatment often extend to sexual health, which is frequently overlooked in clinical care and research.
The input highlighted that early-stage endometrial cancer is typically treated with surgery, often combined with chemotherapy, hormone therapy, or radiation. However, treatment options for patients with recurrent or metastatic endometrial cancer are limited, and the prognosis remains poor due to stagnant access to new therapeutics over the past decades. Thus, the patient groups highlighted an urgent unmet need for additional precision therapeutics in advanced or recurrent endometrial cancer in Canada. When asked about desired improvements in cancer treatment, the 2 patients who participated in telephone interviews emphasized the necessity of timely access to new therapies for patients with similar genomic markers in different tumour types. Among the survey respondents, 60% of patients with endometrial cancer ranked “prolong life” as the most important issue they hope new treatments will address.
The 2 patients who were interviewed shared their experiences with durvalumab in combination with olaparib during the DUO-E trial, reporting CRs and minimal side effects, while a third patient who was receiving durvalumab treatment in Australia highlighted, in their survey response, the positive impact of targeted therapies on their quality of life.
Clinician Input
Input From the Clinical Experts Consulted for This Review
All CDA-AMC review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of endometrial cancer.
Unmet Needs
The goals of treatment for advanced or metastatic endometrial cancer, according to the clinical experts, are to improve quality of life by reducing pain and suffering, improve disease-related symptoms, control the cancer proliferation, and improve survival, if possible. They noted that achieving a longer DOR would be strongly linked to improvement in relevant patient outcomes.
In dMMR endometrial cancer, 1 expert noted that between 20% and 40% of patients do not experience a response to the current first-line treatment paradigm; for context, they noted that, in general, approximately 40% of patients are not expected to experience a response to chemotherapy, and the DOR to chemotherapy alone is often very short. While the addition of an ICI improves the overall treatment response rate, it has the most significant impact on the improvement in DOR. The unmet needs in patients with dMMR endometrial cancer include better identification of potential nonresponders, determining the therapy their cancer might respond to, and determining the ideal duration of maintenance treatment among the patients with cancer that does respond to therapy.
Place in Therapy
The experts indicated that durvalumab would be an additional first-line ICI option, alongside dostarlimab, that could be added to chemotherapy and used as maintenance monotherapy in patients with dMMR endometrial cancer. In general, the clinical experts noted that the choice of ICI is often based on availability but, in a situation when multiple approved options are available, any of them could be offered. If a patient did not experience a response to ICI therapy, the clinical experts noted it was unlikely they would trial the same drug class again; however, if a patient had a recurrence more than 12 months after therapy and had previously experienced a response, then re-treatment might be considered.
Patient Population
Endometrial cancer is not considered a rare disease, and the indication places durvalumab as first-line therapy. The clinical experts noted that the DUO-E trial results did not identify specific molecular subtypes of cancers (beyond dMMR) that could respond to the new treatments; therefore, all patients who meet the other clinical trial criteria would likely be candidates for treatment with durvalumab.
Assessing the Response to Treatment
The clinical experts consulted for this review noted that the outcomes from the DUO-E trial corresponded to standard clinical assessments, with the exception of HRQoL measures, which are not routinely used in clinical practice. In general, symptomatic benefit would be assessed every treatment cycle through conversations with the patient, and radiologic treatment response would be assessed every 2 to 3 cycles. They noted that the ideal outcome to assess response would be survival but, in the face of measurable disease, achieving stability (i.e., prolongation of PFS) while being tolerated well would be a minimum response to continue therapy. They indicated that the definition of a treatment benefit may vary across physicians and patients.
Discontinuing Treatment
The clinical experts noted that disease progression and SAEs would be grounds to discontinue therapy. Examples of SAEs could include grade 3 or 4 toxicities such as hepatitis, colitis, or pneumonitis as well as any other significant immune-related toxicity.
Prescribing Considerations
Both clinical experts agreed that an oncologist or specialist in administering chemotherapy or biologic therapy would be essential to manage the complexities of treatment toxicity.
Clinician Group Input
This section was prepared by the review team based on the input provided by clinician groups.
Two inputs, 1 from the OH (CCO) Gynecologic Cancer Drug Advisory Committee (7 clinicians contributed to the input) and 1 from the GOC (1 clinician contributed to the input), were provided for this review. The OH (CCO) input was gathered through conference calls and emails, while the GOC input was based on data from completed clinical trials and expert opinions from its board members on treating advanced or recurrent endometrial cancer.
According to the clinician groups, treatment for dMMR endometrial cancer involves platinum-based chemotherapy (carboplatin and paclitaxel) and radiation. The clinician groups underscored the lack of publicly funded first-line immunotherapy options, though both groups acknowledged that pembrolizumab is funded for recurrent dMMR disease after failure of chemotherapy. The OH (CCO) noted that compassionate access to dostarlimab in combination with chemotherapy is available.
According to both clinician groups, the primary goals of treatment are to prolong survival, delay disease progression, control symptoms, improve HRQoL and, when possible, cure the disease. The OH (CCO) emphasized that chemotherapy fails to provide a durable response in patients with dMMR endometrial cancer.
Both clinician groups recommended durvalumab as a first-line option for patients with advanced (stage III or IV) or recurrent endometrial cancer. The OH (CCO) noted its suitability for patients with dMMR endometrial cancer, specifically highlighting that patients with dMMR endometrial cancer can receive durvalumab treatment without prior chemotherapy. The GOC emphasized that treatment suitability is determined by pathologic and radiologic staging, underscoring that it is best suited for patients with stage III or IV disease.
The clinician groups noted that treatment response is assessed through imaging (CT or MRI) and clinical evaluations. The GOC added that tolerability is evaluated before each treatment cycle (i.e., every 3 weeks) and that radiologic assessments occur every 2 to 3 cycles (i.e., every 6 to 9 weeks). The GOC highlighted that a clinically meaningful response includes tolerable toxicity and improved PFS. However, the clinician groups indicated that treatment may be withheld due to disease progression and intolerable toxicity or AEs. The GOC further mentioned that such decisions could also be related to patient preference.
In line with the clinical experts consulted for this review, the clinician groups stated that durvalumab plus carboplatin and paclitaxel should be administered in an outpatient setting and is best prescribed by specialist physicians experienced in systemic therapy.
Drug Program Input
The drug programs provide input on each drug being reviewed through the Reimbursement Review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted for this review are summarized in Table 5.
Table 5: Summary of Drug Plan Input and Clinical Expert Responses
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
The DUO-E study compared treatment arms (durvalumab with carboplatin and paclitaxel then maintenance durvalumab vs. durvalumab with carboplatin and paclitaxel then maintenance with durvalumab and olaparib) with 6 cycles of carboplatin and paclitaxel chemotherapy. In Canada, carboplatin and paclitaxel treatment is the current standard. At the time of this review, dostarlimab in combination with carboplatin and paclitaxel (for patients with dMMR tumours) has completed pCPA negotiations and is currently at the jurisdictional level to approve funding. Dostarlimab is under review to expand use beyond dMMR to all patients. Pembrolizumab is also under review for the first-line treatment of primary advanced or recurrent endometrial cancer regardless of the MMR status. | This is a comment from the drug plans to inform pERC deliberations. |
In the DUO-E study, durvalumab maintenance continued until progression or toxicity. Dostarlimab continues for a maximum of 3 years in this indication. Pembrolizumab continues for a maximum of 2 years. | This is a comment from the drug plans to inform pERC deliberations. |
Considerations for initiation of therapy | |
MMR testing is not reflexive in all jurisdictions for endometrial carcinoma. Is there a standard definition of dMMR to help define treatment for eligible patients? | The clinical experts noted that MMR testing is reflexive in a majority of care settings, and the standard definition most use would be based on immunohistochemistry. |
Can durvalumab be administered with alternate chemotherapy if a patient cannot receive or tolerate carboplatin and/or paclitaxel? | The experts indicated that patients could receive alternate chemotherapy such as single-agent carboplatin, paclitaxel, or another single-platinum agent. |
Patients who received previous systemic therapy were eligible only if previous treatment was in the adjuvant setting and there was ≥ 12 months between the last dose and subsequent relapse. Should patients be eligible for treatment if there was less than 12 months between the last dose of adjuvant therapy and subsequent relapse? | The experts indicated there are no data in this setting, but speculated that patients with dMMR endometrial cancer who have a rapid recurrence after platinum-based therapy would probably be treated with immunotherapy. |
In the DUO-E trial, patients had to complete a minimum of 4 cycles before being eligible for the maintenance phase. If patients must discontinue carboplatin and paclitaxel before the fourth cycle, should they be considered for maintenance treatment? | The clinical experts indicated that the reason for the discontinuation would be a factor in decision-making, and that it would be a clinical decision. However, patients with dMMR cancer would likely proceed with maintenance treatment in this scenario. |
In the DUO-E trial, treatment was continued until disease progression or unacceptable toxicity. If a patient stopped treatment for a reason other than progression or unacceptable toxicity and then wanted to resume at time of progression, would they be eligible? If yes, for which components of the regimen would they be eligible? | The clinical experts indicated that the reason for stopping treatment initially would be a factor in the decision-making in this scenario; however, they noted that disease progression or toxicity are the main reasons they do not rechallenge with the same therapy. As such, they likely would resume treatment at time of progression, although the components of the regimen would depend on when in treatment the stoppage occurred. For example, the experts noted that if a patient stopped treatment after 1 week, they would likely be considered naive and resume treatment at the beginning. However, if they stopped treatment after 5 cycles, then they could be considered to have received the whole chemotherapy regimen and could be evaluated for maintenance therapy. |
If recommended for reimbursement, would it be appropriate to consider aligning the initiation criteria of the drug under review with the initiation criteria for dostarlimab and pembrolizumab in endometrial cancer? | The experts noted that in the absence of direct comparative evidence, they consider these treatments to be somewhat equivalent; therefore, the initiation criteria could be aligned with dostarlimab and pembrolizumab in the same care setting. |
Considerations for continuation or renewal of therapy | |
If there is progression during a “drug holiday,” can treatment be resumed? According to what time frame? | The experts indicated that the reason for the drug holiday would be an important factor in the decision-making for this situation, and this would likely be a patient-specific clinical decision. Examples of factors they would take into consideration included the reason for the holiday, how much treatment the patient had received before the holiday, and after which interval their disease recurred. For example, if a longer interval had elapsed, then the experts would be more likely to consider re-treatment, while a shorter interval may imply a lack of benefit. |
Considerations for prescribing of therapy | |
Most jurisdictions use weight-based dosing up to a cap for durvalumab: 15 mg/kg to 20 mg/kg (up to a maximum of 1,500 mg) every 3 weeks in combination with chemotherapy, then 20 mg/kg (up to a maximum of 1,500 mg) every 4 weeks. | This is a comment from the drug plans to inform pERC deliberations. |
Generalizability | |
The exclusion criteria for the DUO-E study excluded patients with an ECOG PS of > 1 and patients with sarcomas. Should either of these groups of patients be considered for treatment? | The clinical experts indicated they would likely treat patients with an ECOG PS of 2, but there are no data on pure sarcomas and they would not consider them for treatment. |
Should patients who are currently receiving first-line chemotherapy be eligible to add durvalumab? | The experts indicated that if the treatment becomes available and the patients otherwise meet the eligibility criteria, then they would add durvalumab to their chemotherapy regimen. |
Should patients currently receiving dostarlimab in combination with chemotherapy be eligible to switch to durvalumab? Should patients currently receiving dostarlimab maintenance be eligible to switch to durvalumab? | The clinical experts indicated they would not generally switch 1 ICI for another unless data were to become available to support a switch or if there were a contract change in their care centre. In general, if a patient’s cancer failed to respond to ICI treatment, they also would not rechallenge with another drug from that treatment class. A change from durvalumab to a drug with a more convenient treatment schedule (every 6 weeks in the maintenance setting for dostarlimab or pembrolizumab) might be considered, which could improve patient QoL and reduce resource use at the treating centre. |
Funding algorithm (oncology only) | |
Durvalumab may change the place in therapy of comparator drugs and may change the place in therapy of drugs reimbursed in subsequent lines. Under what circumstances would durvalumab be chosen over dostarlimab or pembrolizumab if reimbursed in the same line of therapy? | The experts indicated that if all 3 treatments were available, they did not see a compelling reason to use 1 ICI over another, other than the potential convenience of 6 weekly infusions of drugs such as dostarlimab or pembrolizumab. |
System and economic issues | |
Funding of oral drugs varies by province. | These are comments from the drug plans to inform pERC deliberations. |
Confidential prices exist for other treatment options. | |
PAG would like to inform pERC that there is concern about the budget impact of durvalumab in the first-line treatment of all patients with endometrial cancer compared with chemotherapy alone and other ICIs. Dostarlimab continues for up to 3 years, pembrolizumab continues for up to 2 years, and durvalumab would continue until progression or unacceptable toxicity with no maximum cut-off. There is no comparative data to determine whether durvalumab provides a significant advantage over dostarlimab or pembrolizumab. | |
dMMR = mismatch repair deficient; ECOG PS = Eastern Cooperative Oncology Group Performance Status; ICI = immune checkpoint inhibitor; MMR = mismatch repair; PAG = Provincial Advisory Group; pCPA = pan-Canadian Pharmaceutical Alliance; pERC = pan-Canadian Oncology Drug Review Expert Review Committee; QoL = quality of life; vs. = versus.
Clinical Evidence
The objective of this Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of durvalumab plus carboplatin and paclitaxel, and durvalumab monotherapy in the treatment of advanced or recurrent endometrial cancer in adult patients aged 18 years or older. The focus will be placed on comparing durvalumab with relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of durvalumab plus carboplatin and paclitaxel is presented in 2 sections, with the CDA-AMC critical appraisal of the evidence included at the end of each section. The first section, the systematic review, includes pivotal studies and RCTs that were selected according to the sponsor’s systematic review protocol. The review team’s assessment of the certainty of the evidence in this first section using the GRADE approach follows the critical appraisal of the evidence. The second section includes indirect evidence from the sponsor. No long-term extension studies or studies addressing gaps in the evidence were included.
Included Studies
Clinical evidence from the following is included in the review and appraised in this document:
1 pivotal study or RCT identified in systematic review
1 indirect treatment comparison consisting of 2 MAICs.
Systematic Review
Contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
Description of Studies
One study (the DUO-E study) was included in this review. Characteristics of the DUO-E study are summarized in Table 6.
The DUO-E study is an ongoing phase III, multicentre, double-blind, placebo-controlled RCT of durvalumab in combination with platinum-based chemotherapy (carboplatin and paclitaxel), followed by maintenance durvalumab or maintenance durvalumab in combination with olaparib, in patients with newly diagnosed advanced or recurrent endometrial cancer compared with SOC alone (carboplatin and paclitaxel). The primary end point of the DUO-E study was PFS, with secondary end points consisting of OS, ORR, DOR, TTD, and HRQoL. In total, 718 patients were randomized 1:1:1 to SOC (arm A; N = 241), durvalumab plus SOC (arm B; N = 239), or durvalumab plus olaparib plus SOC (arm C; N = 238). Randomization was stratified according to MMR status (dMMR versus pMMR), disease status (recurrent versus newly diagnosed), and geographic region (Asia versus the rest of the world), and was undertaken using an interactive voice response system (IVRS) or interactive web response system. The study took place in 22 countries worldwide and included 7 centres in Canada. Enrolment for the DUO-E study ended in March 2022; however, treatment and follow-up are still ongoing. Full details of the DUO-E study design are included in Figure 1.
Of note, the DUO-E trial randomized patients with either pMMR or dMMR status to all 3 study arms. To align with the Health Canada indication and reimbursement request (durvalumab in combination with carboplatin and paclitaxel in patients with dMMR endometrial cancer, followed by maintenance durvalumab), this Clinical Review Report focuses only on the population of patients from the DUO-E study with dMMR status who received durvalumab in combination with carboplatin and paclitaxel (i.e., only a subset of patients included in arm B of the DUO-E study).
Other trial results, such as the use of concomitant medications and efficacy and safety results for the full ITT population (i.e., all patients with either dMMR or pMMR endometrial cancer) are presented in Appendix 1.
Table 6: Details of Studies Included in the Systematic Review
Detail | DUO-E study |
|---|---|
Designs and populations | |
Study design | Ongoing phase III, randomized, multicentre, double-blind, placebo-controlled RCT |
Locations | 202 sites in 22 countries: Australia (5 centres), Belgium (9 centres), Brazil (11 centres), Canada (7 centres), China (25 centres), Columbia (9 centres), Estonia (2 centres), Germany (4 centres), Greece (4 centres), Hong Kong (1 centre), Hungary (6 centres), India (2 centres), Israel (5 centres), Japan (18 centres), Lithuania (3 centres), Mexico (7 centres), Poland (5 centres), Republic of Korea (9 centres), Russia (8 centres), Singapore (3 centres), Spain (7 centres), and US (52 centres). |
Patient enrolment dates | Start date: May 5, 2020 End date: March 30, 2022 |
Randomized (N) | Randomized (N = 718):
|
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Intervention | Induction: Platinum-based chemotherapy (carboplatin: AUC 5 mg/mL/min or 6 mg/mL/min q.3.w. for 6 cycles; paclitaxel: 175 mg/m2 q.3.w. for 6 cycles) plus durvalumab 1,120 mg intravenously once q.3.w. for 6 cycles. Platinum-based chemotherapy was to have continued for a maximum of 6 cycles. A minimum of 4 cycles of platinum-based chemotherapy was acceptable if treatment was stopped due to toxicity. Maintenance:
|
Comparator | SOC, which consisted of both of the following:
|
Study duration | |
Screening phase | Up to 28 days |
On-treatment phase | Chemotherapy phase: Cycles 1 to 6 Maintenance phase: Maintenance treatments were continued until objective disease progression (per RECIST 1.1), unless there was unacceptable toxicity, withdrawal of consent, or another discontinuation criterion was met. To start maintenance therapy, patients had to meet the eligibility criteria for organ function, bone marrow function, and CrCl after the last cycle of chemotherapy. |
Follow-up phase | If a patient’s cancer progressed, the patient was followed as per local clinical practice. In the case of a second progression, assessments were planned for every 12 weeks and survival status was assessed every 2 months until the final analysis of the study. |
Outcomes | |
Primary end point | PFS per RECIST 1.1, as assessed by an investigator, was defined as the time from randomization until the date of objective disease progression or death (from any cause in the absence of progression). This was assessed by determining the efficacy of durvalumab in combination with platinum-based chemotherapy (carboplatin and paclitaxel) and continued as maintenance vs. upfront SOC platinum-based chemotherapy (carboplatin and paclitaxel) alone. |
Secondary and exploratory end points | Secondary:
Exploratory:
|
Publication status | |
Publications | Westin et al. (2024) Clinicaltrials.gov entry NCT04269200 |
ALT = alanine aminotransferase; AML = acute myeloid anemia; ANC = absolute neutrophil count; anti-HBc = hepatitis B core antibody; AST = aspartate transferase; AUC = area under the curve; CrCl = creatinine clearance; DOR = duration of response; dMMR = mismatch repair deficient; ECG = electrocardiogram; ECOG = Eastern Cooperative Oncology Group; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-EN24 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire – Endometrial Cancer Module; FFPE = formalin-fixed paraffin-embedded; HbsAg = hepatitis B virus surface antigen; HBV = hepatitis B virus; HCV = hepatitis C virus; HRD = homologous recombination deficiency; IP = investigational product; MDS = myelodysplastic syndrome; MMR = mismatch repair; MSI = microsatellite instability; NCI CTCAE = National Cancer Institute Common Terminology Criteria for Adverse Events; ORR = objective response rate; q.3.w. = every 3 weeks; q.4.w. = every 4 weeks; QT = electrocardiogram interval measured from the beginning of the QRS complex to the end of the T wave; PFS = progression-free survival; PGI-BR = Patient Global Impression of Benefit–Risk; PGI-S = Patient Global Impression–Severity; PGI-TT = Patient Global Impression of Treatment Tolerability; PGIC = Patient Global Impression of Change; PR = partial response; PRO-CTCAE = Patient-Reported Outcomes Version of the Common Terminology Criteria for Adverse Events; RCT = randomized controlled trial; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1; SOC = standard of care; TDT = time to study treatment discontinuation or death; TFST = time to first subsequent therapy or death; TMB = tumour mutational burden; TSST = time to second subsequent therapy or death; ULN = upper limit of normal; vs. = versus.
aThese inclusion and exclusion criteria were checked during prescreening; the remainder were verified during screening.
Sources: Details included in the table are from the sponsor’s summary of clinical evidence22 and the DUO-E Clinical Study Report.48
The data provided in the submission were from a global population data cut-off of April 12, 2023, when clinical study protocol version 5.0 was in effect. The study design for the DUO-E trial consisted of a prescreening phase, screening phase, on-treatment phase (split into chemotherapy and maintenance phases) as well as a postdiscontinuation follow-up phase. During prescreening, tissue for MMR screening at a central laboratory was collected and the main inclusion and exclusion criteria (indicated as footnote “a” in Table 6) were checked. Patients underwent the screening phase while the MMR status of their sample was being tested and, following this, were randomized to begin treatment on day 1 of the study. Investigators and patients were to remain blinded to treatment assignment throughout the course of the study, except in specific circumstances. Washout periods or prespecified lengths of time were required for patients taking concomitant moderate or strong CYP3A4 inhibitors or inducers.
Figure 1: DUO-E Study Design — ITT
![The image shows the study design for the ITT population in the DUO-E study. Patients were screened for the key eligibility criteria (newly diagnosed stage III or IV, or recurrent endometrial cancer; naive to first-line systemic anticancer treatment; prior adjuvant chemotherapy allowed if 12 months or more from the last treatment to relapse; known MMR status; prior radiotherapy allowed; all histologies except sarcomas) then randomized 1:1:1 to arm A (platinum chemotherapy plus placebo for durvalumab for 6 cycles, then placebo for olaparib plus placebo for durvalumab in the maintenance phase [cycles 7 and beyond]), arm B (platinum chemotherapy plus durvalumab for 6 cycles, then placebo for olaparib plus durvalumab in the maintenance phase [cycles 7 and beyond]), or arm C (platinum chemotherapy plus durvalumab for 6 cycles, then olaparib plus durvalumab in the maintenance phase [cycles 7 and beyond]). Randomization was stratified by MMR status, recurrent disease status, and geographic region.](https://canjhealthtechnol.ca/index.php/cjht/article/download/PC0366r/version/1255/2765/10769/PC0366-Clinical_Review-fig01.png)
bd = twice a day; CTX = platinum-based chemotherapy; MMR = mismatch repair; Q3W = every 3 weeks; Q4W = every 4 weeks.
aChemotherapy was to have been administered for a maximum of 6 cycles.
bMaintenance treatment was to continue until progression of disease or other discontinuation criteria were met.
cMMR status was to be known before randomization.
Source: details included in the figure are from the sponsor’s summary of clinical evidence.22
Notable changes to the protocol between versions 1.0 and 5.0 included: the definitions for end of treatment and when a patient was considered to have completed the study (April 2021) were revised, the PFS futility boundary and analysis time point were updated (June 2022), and the statistical analysis was updated so that 2 PFS comparisons (durvalumab plus SOC versus SOC alone, and durvalumab plus olaparib plus SOC versus SOC alone) could be tested independently. Other updates to the statistical analysis included updates to the statistical analysis method, sample size assumptions, timing of the primary analysis of PFS, multiplicity testing strategy, and an assumption of a 3-month lag for durvalumab plus SOC (June 2022). Version 6.0 of the study protocol was implemented in January 2023.
Populations
Inclusion and Exclusion Criteria
The DUO-E study enrolled adult (aged 18 years or older) female patients with a histologically confirmed diagnosis of epithelial endometrial carcinoma (excluding sarcomas). The cancer status was measurable per Response Evaluation Criteria in Solid Tumours Version 1 (RECIST 1.1) and could be in any of the following categories: newly diagnosed stage III following surgery or diagnostic biopsy, newly diagnosed stage IV with or without disease following surgery or diagnostic biopsy, or recurrent disease when the potential for cure by surgery alone or in combination was poor. Patients had to be naive to first-line systemic anticancer therapy, except when prior adjuvant chemotherapy had been used (e.g., in the context of prior surgery or radiotherapy) and at least 12 months or more had elapsed from the last treatment to the recurrence of disease. Key exclusion criteria included unresolved toxicities from prior anticancer therapy (with exceptions), prior radiotherapy administered to more than 30% of the bone marrow or with a wide field of radiation within 4 weeks of beginning the study, any previous immune-mediated therapy, concurrent hormonal therapy for noncancer conditions, history of another primary malignancy (with exceptions) or major surgeries within 2 weeks of starting the study treatment. Full detailed inclusion and exclusion criteria are in Table 6.
Tumour markers were not used for determining patient eligibility, but were used to allow for stratification by MMR status. Other putative DNA damage response markers (BRCA gene mutation status, presence of HRR gene mutation, and so forth) and putative immune-oncology markers (for example, PD-L1 expression on tumour and immune cells) were tested retrospectively for subgroup analyses.
Interventions
Patients began treatment on the day of randomization or, in the event of logistical challenges, no later than 3 calendar days following randomization. Treatment in each group was divided into an initial chemotherapy phase for up to 6 cycles, followed by a maintenance phase for cycles 7 and beyond in patients who experienced a response. Patients received the following interventions based on their assigned treatment arm:
Arm A (SOC): A chemotherapy phase regimen of platinum-based chemotherapy consisting of carboplatin (area under the curve [AUC] 5 mg/mL/min or 6 mg/mL/min once every 3 weeks for 6 cycles) and paclitaxel (175 mg/m2 once every 3 weeks for 6 cycles) plus durvalumab placebo for 6 cycles. After the chemotherapy phase, patients who had no evidence of progressive disease (per RECIST 1.1) then received maintenance-phase therapy consisting of durvalumab placebo IV every 4 weeks plus olaparib placebo tablets twice daily.
Arm B (SOC plus durvalumab): A chemotherapy phase regimen of the same platinum-based chemotherapy as the SOC arm plus durvalumab 1,120 mg IV every 3 weeks for 6 cycles. After the chemotherapy phase, patients who had no evidence of progressive disease (per RECIST 1.1) then received maintenance-phase therapy consisting of durvalumab 1,500 mg IV every 4 weeks plus olaparib placebo tablets twice daily.
Arm C (SOC plus durvalumab plus olaparib): The same chemotherapy phase regimen as arm B. After the chemotherapy phase, patients who had no evidence of progressive disease (per RECIST 1.1) then received maintenance-phase therapy consisting of durvalumab 1,500 mg IV every 4 weeks plus olaparib 300 mg twice daily. This study arm was not appraised in this review.
The carboplatin and paclitaxel regimens were to be continued for a maximum of 6 cycles but could be reduced to a minimum of 4 cycles if required due to toxicity.
Durvalumab or its placebo were available in a 500 mg vial containing 50 mg/mL solution for IV infusion after dilution. The dose was administered using an IV bag with 0.9% (w/v) saline or 5% (w/v) dextrose. If a patient’s weight fell to 30 kg or less using treatment, weight-based dosing of 20 mg/kg was administered. Dose delays of up to 12 weeks were permitted for durvalumab under prespecified circumstances, but dose reductions were not permitted. A cycle was counted if treatment was started, even if the full dose was not delivered.
The order of infusions for the chemotherapy phase was durvalumab or placebo followed by paclitaxel and then carboplatin. Dosing intervals in the maintenance phase were permitted to be shortened as clinically feasible to align treatment cycles with the schedule for the assessments of tumour efficacy (RECIST 1.1) and HRQoL. The subsequent time between 2 consecutive doses was not permitted to be less than 21 days, based on the half-life of durvalumab. Maintenance treatments were continued until objective disease progression (per RECIST 1.1), unless there was unacceptable toxicity, withdrawal of consent, or another discontinuation criterion was met. In cases of doubt regarding radiological disease progression, patients could continue study treatment(s) for the purpose of confirming disease progression, with a follow-up scan in 4 weeks or less if they were not experiencing any significant, unacceptable, or irreversible toxicities, in the opinion of the investigator. The discontinuation criteria were any of the following occurrences:
Objective disease progression according to RECIST 1.1 criteria.
Clinical deterioration was used as a reason for discontinuation only when objective disease progression had not yet been or could not be determined. Patients with symptomatic or clinical deterioration requiring discontinuation of treatment without objective radiologic evidence of disease progression at that time were to continue undergoing tumour assessments.
Withdrawal of consent from further treatment. Per the submission, patients could discontinue treatment at any time; patients who discontinued would be expected to continue to participate in the study (e.g., for safety and survival follow-up) unless they specifically withdrew consent to all further participation in any study procedures and assessments.
An AE that, in the opinion of the investigator or the sponsor, contraindicated further dosing.
Any AE that met criteria for discontinuation as defined in the study’s dosing modification and toxicity management guidelines or as defined in the local prescribing information for the chemotherapy agents (carboplatin and paclitaxel).
Pregnancy or intent to become pregnant.
Nonadherence with the study protocol that, in the opinion of the investigator or the sponsor, warranted withdrawal from treatment.
Bone marrow findings consistent with myelodysplastic syndrome or acute myeloid leukemia.
Initiation of an alternative anticancer therapy, including another investigational drug.
All patients continued to be assessed for radiological tumours, according to the study schedule, until objective radiological disease progression, irrespective of any treatment delays or discontinuation of study treatment. Once a patient progressed, they were followed as per local clinical practice; in the case of a second progression, assessments were planned for every 12 weeks and survival status was to be assessed every 2 months until the final analysis of the study.
Restrictions, Concomitant Medications, and Rescue Medications During the DUO-E Trial
Patients who were enrolled in the study and had been on moderate or strong cytochrome P450 3A (CYP3A) inducers or inhibitors were required to discontinue them during a 2-week washout period before beginning the study. The consumption of grapefruit juice was prohibited during the maintenance phase of the study (i.e., during the administration of olaparib or its placebo). Strong and moderate CYP3A inhibitors or inducers were prespecified not to be taken with olaparib, but if no suitable alternative was found, then the dose of olaparib was reduced for the period of concomitant administration. Medications that are substrates for CYP3A4, including CYP2B6, OATP1B1, OACT3, OCT1, OCT2, MATE1, and MATEK1, were to be used with caution. Warfarin was permitted with a recommendation to monitor the international normalized ratio at prespecified intervals. Palliative radiotherapy was permitted for the treatment of pain at the site of bony metastases that were present at baseline, provided the investigator did not feel they were indicative of clinical disease progression, and the study treatment was to be discontinued and resumed at prespecified time points.
Prohibited medications included other anticancer drugs, immunosuppressive medications (e.g., systemic corticosteroids at doses exceeding 10 mg/kg/day of prednisone or equivalent, methotrexate, tumour necrosis factor alpha blockers), EGFR tyrosine kinase inhibitors, live virus vaccines, live bacterial vaccines, and herbal or natural remedies that may have immunomodulating effects. Patients were permitted to continue the use of denosumab or bisphosphonate for bone disease provided these were started at least 4 weeks before beginning study treatment. For each prohibited medication class, prespecified guidance was provided in the study protocol. Full details of the restricted concomitant therapies are in Table 7.
Two rescue treatments, infliximab (e.g., for colitis) and mycophenolate mofetil (e.g., for hepatitis), were permitted for any immune-mediated AEs experienced by patients. Dose modifications for the management of AEs (including renal impairment) for durvalumab were prespecified in the study protocol.
Table 7: Relevant Prohibited and Restricted Medications for the DUO-E Study — dMMR Subgroup
Treatments prohibited or restricted for concomitant use during study | Medication or class of drug |
|---|---|
Prohibited medications | |
Patients receiving durvalumab or placebo | Immunosuppressive medications, including but not limited to, systemic corticosteroids at doses exceeding 10 mg/day of prednisone or equivalent, methotrexate, azathioprine, and blockers of tumour necrosis factor alpha |
Herbal and natural remedies that may have immune-modulating effects | |
Other anticancer therapy:
| |
| |
EGFR tyrosine kinase inhibitors | |
Restricted medications | |
Patients receiving durvalumab or placebo | Palliative radiotherapy |
Administration of other anticancer drugs | |
dMMR = mismatch repair deficient; mAb = monoclonal antibody.
Note: Hormone replacement therapy was acceptable.
Source: Details included in the table are from the DUO-E study protocol.49
Outcomes
A list of efficacy end points assessed in this Clinical Review Report is provided in Table 8, followed by descriptions of the outcome measures. Summarized end points are based on outcomes included in the sponsor’s summary of clinical evidence as well as any outcomes identified as important to this review according to the clinical experts consulted for this review and input from patient and clinician groups and public drug plans. Using the same considerations, the review team selected end points that were considered to be most relevant to inform expert committee deliberations and finalized this list of end points in consultation with members of the expert committee.
The included time-to-event end point outcomes for PFS and OS have been summarized in this report and assessed at 12- and 18-month time points using GRADE; these measures and time points were chosen because they are standard measures of response to treatment in oncology and provide the longest follow-up available from the DUO-E trial. To provide additional context for efficacy, ORR, DOR, and TDT have been summarized in this report but were not included in GRADE. The EORTC QLQ-C30 and EORTC QLQ-EN24 scores were summarized in this report and included in GRADE; these measures were chosen as they measure quality of life in patients with cancer and with endometrial cancer, respectively. The HRQoL outcomes at 18 weeks, which corresponds to the last infusion of the chemotherapy phase, have been summarized, and summarized again at 42 weeks, which corresponds to the sixth month of maintenance therapy. Select notable harms outcomes considered important for informing expert committee deliberations were also assessed using GRADE; these included immune-mediated AEs and infusion-related reactions.
Of note, results for arm B relative to arm A, and arm C relative to arm A, were reported separately in the ITT population. Results for arm B relative to arm A are reported only for the dMMR subgroup in this review. The submission also included a post hoc analysis of arm B versus arm C, but this was not appraised in the review because the comparison was not relevant to the reimbursement request.
Table 8: Outcomes Summarized From the DUO-E Study
Outcome measure | Time point | DUO-E study |
|---|---|---|
PFS | Time from randomization until the date of objective disease progression or death from any cause in the absence of progression. | Primarya |
OS | Time from the date of randomization until death due to any cause. | Secondarya |
ORR | A response of CR or PR recorded at 1 visit and confirmed by repeat imaging no fewer than 4 weeks after the visit at which the response was first observed, with no evidence of progression between the initial visit and the visit to confirm the CR or PR. | Secondary |
DOR | Time from the date of first documented response until date of documented progression or death in the absence of disease progression. | Secondary |
TDT | Time from randomization to the earlier of the date of study treatment discontinuation or date of death. | Secondary |
EORTC QLQ-C30 global health status (QoL) score change from baseline | Assessed at baseline (before dosing on day 1 of cycle 1), then q.3.w. (± 3 days) for the first 18 weeks, and then q.4.w. (± 3 days) until a second disease progression or death. Patients were also assessed at the study treatment discontinuation visit (+ 2 days) unless completed within 3 days before the visit and, for those who discontinued for reasons other than PD, also at the PD assessment visit (+ 2 days) unless an assessment had been completed within 3 days before the visit. Patients had an assessment at the visit marking the start of maintenance therapy unless an assessment had been completed within 3 days before the visit. | Secondary |
EORTC QLQ-EN24:
| Assessed at baseline (before dosing on day 1 of cycle 1), then q.3.w. (± 3 days) for the first 18 weeks, and then q.4.w. (± 3 days) until a second disease progression or death. Patients were also assessed at the study treatment discontinuation visit (+ 2 days) unless an assessment had been completed within 3 days before the visit and, for those who discontinued for reasons other than PD, also at the PD visit (+ 2 days) unless an assessment had been completed within 3 days before the visit. Patients had an assessment at the start of maintenance therapy visit unless an assessment had been completed within 3 days before the visit. | Secondary |
CR = complete response; dMMR = mismatch repair deficient; DOR = duration of response; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-EN24 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire – Endometrial Cancer Module; ITT = intention to treat; ORR = objective response rate; OS = overall survival; PD = progressive disease; PR = partial response; q.3.w. = every 3 weeks; q.4.w. = every 4 weeks; QoL = quality of life; PFS = progression-free survival; TDT = time to study treatment discontinuation or death.
aStatistical testing for these end points was adjusted for multiple comparisons (e.g., hierarchal testing) in the ITT analysis. Statistical testing was not controlled for multiplicity in the dMMR subgroup analysis.
Sources: Details included in the table are from the sponsor’s summary of clinical evidence22 and additional information provided by the sponsor.50
Survival Outcomes
All patients were given radiological tumour assessments according to the study schedule (every 9 weeks for the first 18 weeks relative to randomization, then every 12 weeks, plus or minus 1 week, until RECIST 1.1–defined radiological progression).
Progression-Free Survival
PFS was defined as the time from randomization to the date of objective disease progression or death from any cause in the absence of progression, regardless of whether the patient withdrew from therapy or received another anticancer therapy before progression. PFS determined by the investigator was the primary outcome in the DUO-E trial; PFS assessment by blinded independent central review (BICR) was used as a prespecified sensitivity analysis. The PFS time was always derived based on scan/assessment dates and not visit dates. RECIST 1.1 assessments or scans contributing toward a particular visit were performed on different dates. The following rules were applied:
For investigator assessments, the date of progression was determined based on the earliest of the scan dates of the component that triggered progression.
For BICR assessments, the date of progression was determined based on the earliest of the scan dates of the component that triggered the determination of progression by the adjudicated reviewer who assessed progressive disease, or by the reviewer who read the baseline data first if there was no adjudication for BICR data.
For both BICR and investigator assessments, when censoring a patient for PFS, the patient was censored at the latest of the scan dates contributing to a particular overall visit assessment.
Overall Survival
OS was defined as the time from the date of randomization until death due to any cause, regardless of whether the patient withdrew from randomized therapy or received another anticancer therapy (i.e., date of death or censoring minus date of randomization plus 1). OS determined by the investigator was a secondary outcome.
Objective Response Rate
ORR was defined as the proportion of patients with measurable disease at baseline who had a CR or partial response (PR), as determined by the investigator. A confirmed response of CR or PR meant that the response had been recorded at 1 visit and confirmed by repeat imaging no sooner than 4 weeks after the visit when the response was first observed, with no evidence of progression between the initial and confirmation visit. Patients who discontinued treatment without progression, or who received a subsequent anticancer therapy and then experienced a response, were not included as responders in the ORR assessment. If PRs were recorded for a patient at 2 nonconsecutive visits, the patient was defined as a responder if the time between the 2 visits was greater than 4 weeks and there was no progressive disease between the 2 visits. Similarly, if a patient had a response of “CR not evaluable” recorded at 1 visit, and then a CR was recorded at another visit, then a best response of CR was assigned if the time between the 2 visits was greater than 4 weeks.
Duration of Response
DOR was defined as the time from the date of first documented response until the date of documented progression or death in the absence of disease progression (i.e., date of PFS event or censoring minus the date of first response plus 1). DOR was defined only for patients classified as responders in the ORR analysis. The end of response had to have coincided with the date of progression or death from any cause that was used for the PFS outcome. The time of the initial response was defined as the latest of the visit dates at which the first CR or PR was recorded and subsequently confirmed. If a patient’s cancer did not progress following a response, then their DOR used the PFS censoring time.
Time to Study Treatment Discontinuation or Death
TDT was defined as the time from randomization to the earlier of either the date of study treatment discontinuation or death. Treatment discontinuation was defined as all investigational products being discontinued. For example, if a patient had received both durvalumab and olaparib and they were discontinued on different dates, the date of discontinuation was the later date; if a patient had received only 1 of the study treatments, then the date of discontinuation was the date that drug was discontinued; if a patient had not received either study treatment, they were censored at day 1.
HRQoL Outcomes
HRQoL measures, including the EORTC QLQ-C30 and EORTC QLQ-EN24, were assessed at baseline (before dosing on day 1 of cycle 1), then every 3 weeks (± 3 days) for the first 18 weeks, and then every 4 weeks (± 3 days) until a second disease progression or death. Patients were also assessed at the study treatment discontinuation visit (+ 2 days) unless an assessment had been completed within 3 days before the visit; for those who discontinued for reasons other than progressive disease, HRQoL was assessed for patients at the progressive disease assessment visit (+ 2 days) unless an assessment had been completed within 3 days before the visit. Patients also had an HRQoL assessment at the visit marking the start of maintenance therapy, unless an assessment had been completed within 3 days before the visit. Full details of the HRQoL measures, including validation and MIDs, are presented in Table 9.
EORTC Quality of Life Questionnaire Core 30
The EORTC QLQ-C30 is a 30-item, cancer-specific, patient-reported HRQoL questionnaire designed to assess the physical, psychological, and social functioning of patients with cancer.51 The functional scales range from 0 to 100, with higher scores indicating better functioning. An MID of 10 points, based on the recommendation to use 10% of the scale as a cut-off of meaningful change, has been identified in patients with breast and small-cell lung cancer.52 No MID was available in the literature for patients with endometrial cancer.
EORTC Quality of Life Questionnaire – Endometrial Cancer Module
The EORTC QLQ-EN24 is a 24-item, cancer-specific patient-reported outcome measure designed to assess the HRQoL of patients with endometrial cancer. It is intended to be used alongside the EORTC QLQ-C30 to capture both disease and treatment-specific aspects of quality of life in patients with endometrial cancer. All questions are rated on a 4-point Likert scale.53 The functional scales range from 0 to 100, with higher scores indicating better functioning. The 10 symptom scales range from 0 to 100, with higher scores indicating more severe symptoms. The pain in back and pelvis score and the urological symptoms score are included in this review. No MID was identified for patients with endometrial cancer.
Table 9: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
EORTC QLQ-C30 | The EORTC QLQ-C30 is a 30-item, cancer-specific, patient-reported HRQoL questionnaire designed to assess the physical, psychological, and social functioning of patients with cancer.51 It uses 4- and 7-point Likert scales and covers 15 domains, which include both functional and symptom scales. The functional scales (ranging from 0 to 100, with higher scores indicating better functioning) assess global health quality of life and physical, role, emotional, cognitive, and social functioning. The symptom scales (ranging from 0 to 100, with higher scores indicating more severe symptoms) assess fatigue, pain, nausea and vomiting, dyspnea (shortness of breath), insomnia, appetite loss, constipation, diarrhea, and financial difficulty.51 | Validity: Construct, criterion, and discriminate validity were demonstrated in patients with ovarian, gestational trophoblastic disease, and other types of cancers.54-56 The EORTC QLQ-30 was found to adequately assess the effect of expected toxicities on patients’ HRQoL during and following treatment.57 No reported validation studies were found for patients with endometrial cancer. Reliability: Minimum reliability with Cronbach alpha > 0.70 was met in 7 of 9 subscales in patients with gestational trophoblastic disease, ovarian cancer, and other types of gynecological cancers.55 Responsiveness: There were no data available for responsiveness. | A literature search was conducted to identify MIDs for the EORTC- QLQ-EN30 in patients with endometrial cancer and none were identified. In a study analyzing data from 13,015 patients across 9 cancer types, anchor-based MIDs for EORTC QLQ-C30 scores mostly ranged from 5 to 10 points, with differences between improvement and deterioration typically within a 2-point range.58 Osoba et al. (1998)52 identified 5 to 10 points for a small change, 10 to 20 points for a moderate change, and more than 20 points for a large change, aligning closely with findings from King et al. (2014).59 This led to the recommendation by Osoba et al. to use 10% of the scale as a cut-off to define a clinically meaningful change.60 |
EORTC QLQ-EN24 | The EORTC QLQ-EN24 is a 24-item, cancer-specific patient-reported outcome measure designed to assess the HRQoL of patients with endometrial cancer. It is intended to be used alongside the EORTC QLQ-C30 to capture both disease- and treatment-specific aspects of quality of life in patients with endometrial cancer. All questions are rated on a 4-point Likert scale.53 The questionnaire consists of 24 items distributed across 10 symptom scales and 3 functional scales. The functional scales (ranging from 0 to 100, with higher scores indicating better functioning) include assessments of sexual interest, sexual activity, and sexual enjoyment. The 10 symptom scales (ranging from 0 to 100, with higher scores indicating more severe symptoms) assess lymphedema, urological symptoms, gastrointestinal symptoms, body image problems, and sexual or vaginal problems, along with single items addressing back or pelvic pain, tingling or numbness, muscular or joint pain, hair loss, and taste changes.53 | Validity: Among patients with endometrial cancer, the EORTC QLQ-EN24 demonstrated good convergent and discriminant validity compared with the EORTC QLQ-C30.53,61,62 Reliability: Test–retest reliability was observed for all multi-item scales (range, 0.81 to 0.92) and the single-item questions (range, 0.66 to 0.97).53 For the urological symptoms scale, good test–retest reliability was shown (Cronbach alpha = 0.92).53 Good internal consistency was demonstrated for 5 multi-item scales (Cronbach alpha range, 0.74 to 0.86). For the urological symptoms scale, internal consistency ranged from 0.75 to 0.86.53,61,62 Responsiveness: There were no data available for responsiveness. | A literature search was conducted to identify the MID for the EORTC- QLQ-EN24 In patients with endometrial cancer and none were identified. |
EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-EN24 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire – Endometrial Cancer Module; HRQoL = health-related quality of life; ICC = interclass correlation; MID = minimal important difference.
Statistical Analysis
Sample Size and Power Calculations
Sample size calculations were performed for the overall study following the prespecified statistical analysis plan and included patients with either pMMR or dMMR endometrial cancer. No prespecified sample size calculations were performed for patients with a specific MMR status; thus, none of the comparisons for the dMMR cohort were sufficiently powered to detect statistically significant differences between durvalumab plus carboplatin and paclitaxel versus SOC.
Globally, the aim was to randomize approximately 699 eligible patients with endometrial cancer at a 1:1:1 ratio to the study treatments. The sample size calculations were based on the following assumptions:
A median PFS of 12 months was assumed for the control arm, which is in line with the data reported for carboplatin plus paclitaxel in first-line endometrial cancer from the GOG-209 study,63 as reported in the GOG-86P manuscript.64
The sample size was derived on the assumption of a 3-month delay in the separation of the PFS curves between arm B versus arm A and between arm C versus arm A. The true average HR for arm B versus arm A was assumed to be 0.70 (corresponding to an improvement in median PFS of 5.5 months over the assumed median PFS of 12 months in the control arm), and 0.55 for arm C (corresponding to an improvement in median PFS of 11.2 months).
If the average true PFS HR was 0.70 for arm B versus arm A, the study would provide 80% power to demonstrate a statistically significant difference for PFS with an overall 2-sided significance level of 2.5%, which would translate to a 5.5-month benefit in median PFS over 12 months relative to arm A. The smallest treatment difference that would be statistically significant would be an HR of 0.77. If the average true PFS HR was 0.55 for arm C versus arm A, the study would provide more than 99% power to demonstrate a statistically significant difference for PFS with an overall 2-sided significance level of 2.5%; this would translate to a 11.2-month benefit in median PFS over 12 months relative to the control arm. The smallest treatment difference that would be statistically significant would be an HR of 0.76.
In addition, the sample size was derived on the following assumptions:
27-month period of recruitment
approximately 10% uniform dropout rate over the study period.
The power calculations for OS were based on the following assumptions:
The median OS for the control arm was assumed to be 22.7 months.
The sample size was derived on the assumption of a 3-month delay in separation of the OS curves between arm B versus arm A, and arm C versus arm A, hence the use of an average HR for OS. The assumed true average OS HR was 0.75 for both comparisons, corresponding to an improvement in median OS of approximately 7.9 months over the assumed median OS of 22.7 months in arm A.
The first IA of OS was performed at the time of the primary PFS analysis. For the comparison of arm B versus arm A and arm C versus arm A, the submission anticipated that 74% of the target number of OS events would have occurred at this time (i.e., approximately 208 of 280 OS events per comparison). A further analysis of OS was performed at the same calendar time when approximately 244 OS events (87% of the target number of OS events) occurred for the comparisons of interest, approximately 51 months after the first patient was randomized. A final analysis of OS was performed at the same calendar time when approximately 280 OS events occurred (60% maturity) for the comparisons of interest, approximately 63 months after the first patient was randomized.
If the average true OS HR was 0.75 for the comparison of the experimental arm versus control, the study would provide 55% power to demonstrate a statistically significant difference for OS with an overall 2-sided significance level of 2.5%, or a 7.9-month benefit in median OS over 22.7 months relative to arm A. The smallest treatment difference that would be statistically significant is an HR of 0.76. These estimates were based on the assumption that no confounding would occur.
Statistical and Analytical Plans
Details of the statistical analyses for both the primary analysis (i.e., the ITT population) and the dMMR subgroup for the included outcomes are summarized in Table 10. Briefly, the primary analyses separately compared arm B (durvalumab plus SOC) with arm A (SOC), and arm C (durvalumab plus olaparib plus SOC) with arm A (SOC). As previously noted, the results for the analysis comparing arm C with arm A are not included in this report. The study protocol was modified in June 2022, updating the ITT population statistical analysis so that 2 PFS comparisons could be tested independently; the changes included updates to the statistical analysis method, sample size assumptions, timing of the primary analysis PFS, multiplicity testing strategy, and an assumption of a 3-month lag for arm B. The primary analysis was prespecified to be undertaken when approximately 299 PFS events had occurred (64% maturity) for the arm B versus arm A comparison, and when approximately 281 PFS events had occurred (60% maturity) for the comparison of arm C versus arm A. No information was provided on any prespecified statistical analyses of the dMMR subgroup indicated in the reimbursement request, and no outcomes were adjusted for multiple testing in this cohort of patients. The data cut-off for the primary analysis of PFS was April 12, 2023; the first IA for OS was also performed using this data.
Statistical Tests and Models
Some general principles were prespecified in the submission and followed for the statistical analysis. Among the key principles were the following: for safety end points, the last observation before the first dose of study treatment was considered the baseline measurement, unless otherwise specified; for the efficacy and HRQoL end points, baseline was defined as the last assessment before randomization. However, if an evaluable assessment was not available before randomization but was available before the first dose of study treatment, then this assessment was to be used as baseline. Change from baseline was calculated as the posttreatment value of the variable minus the value at baseline. The results of all statistical analyses were presented using corresponding CIs and 2-sided P values, unless otherwise stated.
Time-to-Event Analyses: All time-to-event end points, including the primary end point of PFS, were analyzed in the ITT population using a log-rank test stratified by MMR status (proficient versus deficient), disease status (recurrent versus newly diagnosed), and geographic region (Asia versus rest of world) for each pairwise comparison independently. Of note, stratification factors were excluded from the model (region first, followed by MMR status and then disease status) until there were at least 5 events in each stratum of interest across the 3 treatment arms. The HR and its CI were estimated from a stratified Cox proportional hazards model that included the same stratification factors as strata. The CIs were calculated using a profile likelihood approach. In the dMMR subgroup, the HR and corresponding 95% CI were estimated from an unstratified Cox proportional hazards model. Details on patient censoring are described in Table 10 under Handling of missing data.
HRQoL and Descriptive Analyses: Descriptive statistics were used for all variables, as appropriate, and were presented by treatment group. Continuous variables were summarized by the number of observations, mean, SD, median, minimum, and maximum. For log-transformed data, the geometric mean, coefficient of variation, and the median, minimum, and maximum were reported. Categorical variables were summarized by frequency counts and percentages for each category. Unless otherwise stated, percentages were calculated out of the total population for the corresponding treatment arm.
Analysis models were stratified by the randomization stratification factors: MMR status (MMR-proficient tumours versus dMMR tumours), disease status (recurrent endometrial cancer versus newly diagnosed endometrial cancer), and geographic region (Asia versus rest of world). The strata obtained from the randomization code were used, as opposed to the values recorded in the electronic case report form. The submission provided rationales for the choice of stratification factors. Specifically, it was noted that MMR status and disease recurrence status impact treatment response, and Asian countries may have different approaches to managing advanced disease (e.g., greater emphasis on surgery versus systemic therapy). The submission did not contain details on whether other factors were considered for stratification and what the results of those deliberations might be.
Table 10: Statistical Analysis of Efficacy End Points for the DUO-E Study
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
PFS Primary analysis of arm B vs. arm A using investigator assessments (RECIST 1.1) | The PFS analysis was performed using a log-rank test stratified in accordance with the predefined pooling strategy.a The HR and its CI were estimated for the ITT population from a stratified Cox proportional hazards model (with ties = Efron and the stratification factors as strata) and the CI calculated using a profile likelihood approach. The HR and CI for the dMMR subgroup were estimated from an unstratified Cox proportional hazard model. Summaries of the number and percentage of patients experiencing a PFS event, and the type of event (RECIST 1.1 progression or death) were provided along with median PFS for each treatment. The proportion of patients alive with progression-free disease at 6 monthly intervals from randomization were summarized (using a KM analysis) and presented by treatment group. | The stratification variables were based on the values entered into the IVRS at randomization:
No details were provided on adjustment factors in the dMMR subgroup model. | If the patient had no evaluable visits or did not have baseline data, they were censored at day 1 unless they died within 2 visits (19 weeks) of baseline (2 × 9 = 18 weeks plus 1 week to allow for a late assessment within the visit window). Patients with cancer that had not progressed, who had died, or who had not died and whose cancer had not progressed after 2 missed assessments were censored at the latest study visit or the latest visit before the 2 missed assessments, or at day 1. | Sensitivity analyses (durvalumab vs. control) using investigator assessments (RECIST 1.1):
Sensitivity analysis using a stratified log-rank test using BICR assessments (RECIST 1.1) to assess ascertainment bias:
Additional analysis using Cox proportional hazards models to determine the consistency of treatment effect between subgroups using the approach of Gail and Simon (1985).65 |
OS | Stratified log-rank test for arm B vs. arm A. The number and proportion of patients alive at 6-month intervals from randomization (month 6, 12, 18, 24, and so forth) were summarized using a KM analysis and presented by treatment group. The HR and its CI were estimated for the ITT population using a stratified Cox proportional hazards model (with ties = Efron and the stratification factors as strata) and the CI calculated using a profile likelihood approach. The HR and CI for the dMMR subgroup was estimated from an unstratified Cox proportional hazard model. | The stratification variables were based on the values entered into the IVRS at randomization:
No details were provided on adjustment factors in the dMMR subgroup model. | If a patient was known to have died but only a partial death date was available, then the date of death was imputed as the last date (from the database) at which they were known to be alive plus 1. The available death date information was then used, with any missing data added as follows:
If there was evidence of death but the date was missing entirely, it was treated as missing, i.e., OS was censored at the last known alive date. | Sensitivity analysis using a KM plot of time to censoring, where the censoring indicator of the primary analysis is reversed to assess attrition bias:
|
TDT | The TDT analysis was performed using a log-rank test stratified in accordance with the predefined pooling strategy. The HR and its CI were estimated for the ITT population from a stratified Cox proportional hazards model (with ties = Efron and the stratification factors as strata) and the CI calculated using a profile likelihood approach. The HR and CI for the dMMR subgroup were estimated from an unstratified Cox proportional hazard model. | The stratification variables were based on the values entered into the IVRS at randomization:
No details were provided on adjustment factors in the dMMR subgroup model. | Any patient not known to have died at the time of analysis and not known to have discontinued study treatment was censored based on the last recorded date on which the patient was known to be alive. | None. |
ORR | Logistic regression models (durvalumab vs. control), adjusting for the same stratification factors as the primary end point as covariates in the model. The results of the analyses were presented in terms of an OR (OR > 1 favours investigational treatment arm) together with its associated profile likelihood 95% CI and P value (based on twice the change in log-likelihood resulting from the addition of a treatment factor to the model). If there were not enough responses for a meaningful analysis using logistic regression, then a CMH test was presented. A descriptive summary of the number and percentage of patients with a confirmed tumour response (CR or PR) according to the investigator based on the number of patients with measurable disease at baseline. | The stratification variables were based on the values entered into the IVRS at randomization:
No details were provided on adjustment factors in the dMMR subgroup model. | Patients who discontinued treatment without progression and received a subsequent anticancer therapy and then experienced a response were not included as responders in the ORR (i.e., both visits contributing to a response had to be before subsequent therapy for the patient to be considered a responder). | None. |
DOR | Descriptive data were provided in patients whose disease responded, including the associated KM curves (without any formal comparison or P value attached). Median DOR was calculated from the KM curve. Only patients with a confirmed response were included in this summary table. | NA | If a patient’s cancer did not progress following a response, then their DOR used the PFS censoring time. A DOR was defined only for patients classified as responders in the ORR analysis. | None. |
Change from baseline (EORTC QLQ-C30 and EORTC QLQ-EN24 scores) | MMRM analysis: Arm B vs. arm A. | NA | The following protocol was used for missing items: If at least half of the items from the multi-item scale were answered, then all items that were completed were used, and any items with missing values were ignored when making the calculations. If fewer than half of the items were answered, the scale score was set to missing. For unanswered single-item measures, the score was set to missing. | None. |
BICR = blinded independent central review; CI = confidence interval; CMH = Cochran-Mantel-Haenszel; CR = complete response; dMMR = mismatch repair deficient; DOR = duration of response; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-EN24 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire – Endometrial Cancer Module; HR = hazard ratio; ITT = intention to treat; IVRS = interactive voice response system; KM = Kaplan-Meier; MMR = mismatch repair; MMRM = mixed model for repeated measures; NA = not applicable; OR = odds ratio; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; PR = partial response; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1; SOC = standard of care; TDT = time to study treatment discontinuation or death; vs. = versus.
aFor the analysis of the PFS primary end point, if there were fewer than 5 events per stratum, the following pooling strategy was applied across all 3 arms: Strata were calculated as stratum 1 × stratum 2 × …stratum X × treatment; so, with 3 stratification factors for every 2 levels and 3 treatments, the total was 24 strata (2 × 2 × 2 × 3). Stratification factors were removed in the following order until there were at least 5 events in each stratum: Region (Asia vs. rest of world), MMR status (proficient vs. deficient), and disease status (recurrent vs. newly diagnosed). The same pooling was applied for both primary comparisons (arm B vs. arm A and arm C vs. arm A). All analyses were then conducted with the same stratification factors that were used for the primary analysis of PFS in the ITT population. If there were secondary end points or sensitivity analyses that still did not conform to the 5-event rule per stratum, an unstratified analysis was conducted.
Source: Details included in the table are from the sponsor’s summary of clinical evidence.22
Sensitivity and Subgroup Analyses
Prespecified subgroup analyses of the ITT population were conducted for several end points. The subgroup analyses for the stratification factors were based on the values entered into the IVRS; all other factors were based on values recorded on the electronic case report form, as indicated previously, or third-party vendor data. No adjustment to the significance level for testing was made. Because the subgroup analyses were undertaken for the ITT population, the results were not included in the report.
Multiple-Testing Procedure
A multiple-testing procedure with a gatekeeping strategy was used for PFS and OS and for the treatment comparisons of interest for the ITT population. Details of the multiple-comparisons strategy are in Figure 2. An alpha-exhaustive recycling strategy was employed in which the test mass that became available after each rejected null hypothesis was recycled to a secondary hypothesis not yet rejected. If 1 test in the family could be rejected, the family was rejected and the total alpha assigned to the family was recycled to the next multiple-testing level. The testing procedure stopped when the entire test mass was allocated to nonrejected null hypotheses.
Timing of Efficacy Analyses
The timing of the efficacy analyses for PFS and OS in the ITT population is summarized in Table 11. Briefly, the analyses were prespecified to occur after specific thresholds for PFS events were reached. Once a PFS threshold was reached, the OS IA was undertaken. Two IAs and a final analysis for OS were planned.
In the ITT population analysis, the OS tests for the same comparison were to be considered as 1 test family. The alpha level allocated to OS was controlled at the interim and primary time points by using the Lan DeMets66 spending function that approximated an O’Brien-Fleming approach; with this method, the alpha level applied at the interim time point depends on the proportion of information available. If an IA or primary analysis was found to be statistically significant, the overall 2-sided alpha was to be allocated to the next level. For a given hypothesis, if the IA did not meet the criterion for stopping for superiority, the follow-up would continue until the final target number of OS events for that comparison had been observed, following which the hypothesis would be retested. If the null hypothesis was rejected, subsequent testing would continue hierarchically.
Included in the sponsor’s current submission were the results from the primary PFS analysis and first IA for the OS analysis in the ITT population. The dMMR subgroup was derived from the same data cut-off; however, no information was provided on whether similar testing methods (i.e., the Lan DeMets and O’Brien-Fleming approach) were undertaken for the analysis of this subgroup.
Figure 2: Multiple-Comparisons Strategy — DUO-E Study ITT Population
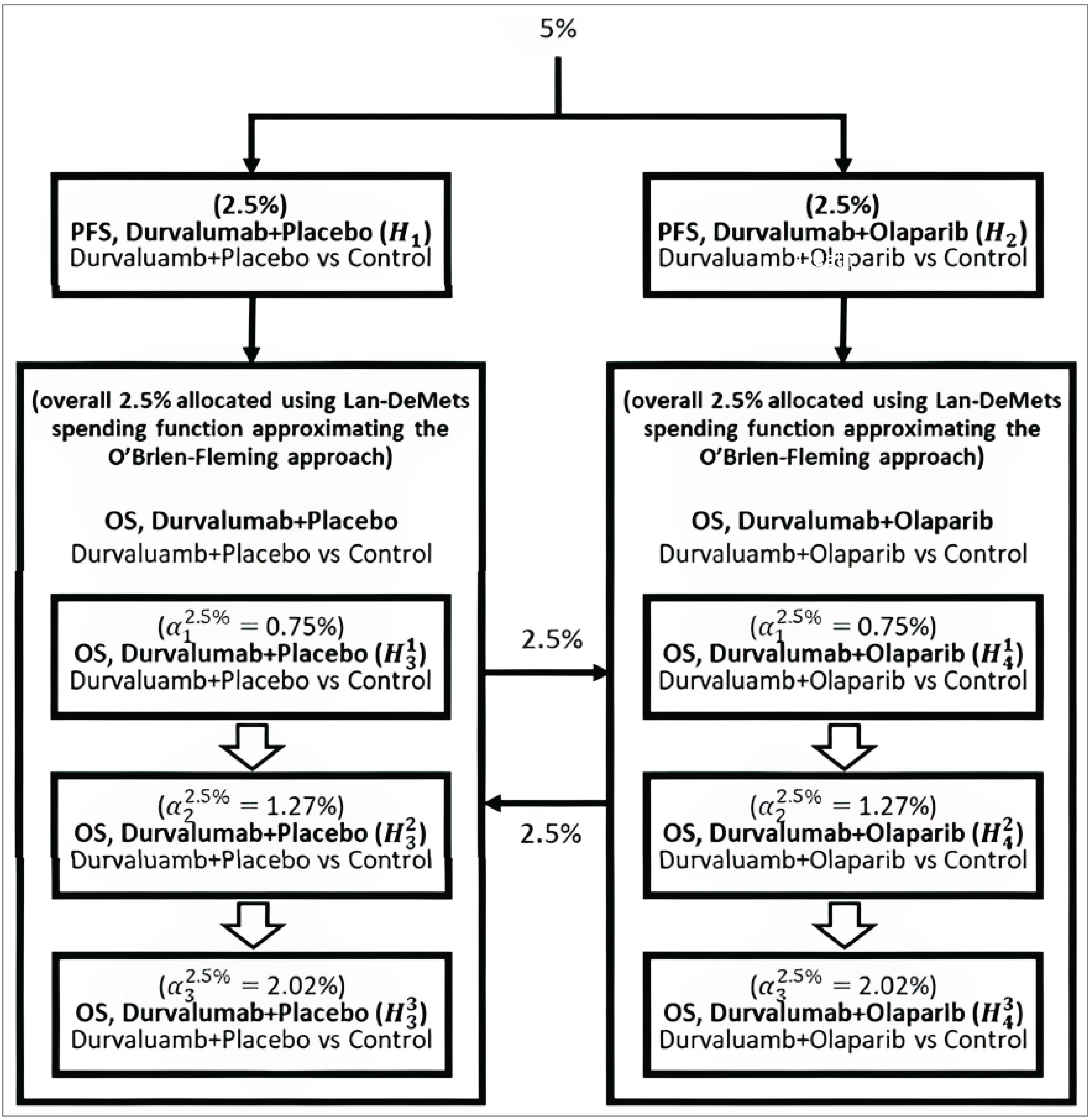
H = hypothesis; ITT = intention to treat; OS = overall survival; PFS = progression-free survival; vs = versus.
Note: The alpha levels presented in the figure are 2-sided.
Source: Details included in the figure are from the sponsor’s summary of clinical evidence.22
The submission noted that the IA and primary analysis boundaries for the OS analysis would be derived from the number of events observed in the study, and that the numbers in this figure are for reference only. As noted, no multiple testing was conducted for the dMMR subgroup.
Table 11: Timing of Efficacy Analyses for PFS and OS in the DUO-E Study — ITT Population
Analysis | Time point |
|---|---|
Primary PFS and first IA for OS | The same calendar time when there were approximately 299 PFS events across arm B and arm A, and when there were approximately 281 events across arm C and arm A |
Second IA for OS | The same calendar time when there were approximately 244 deaths across arm B and arm A, and approximately 244 deaths across arm C and arm A (not included in submission) |
Final OS analysis | The same calendar time when there were approximately 280 deaths across arm B and arm A, and approximately 280 deaths across arm C and arm A (not included in submission) |
IA = interim analysis; ITT = intention to treat; OS = overall survival; PFS = progression-free survival.
Sources: Details included in the table are from the sponsor’s summary of clinical evidence22 and the DUO-E Clinical Study Report.48
Analysis Populations
Details of the analysis sets are available in Table 12. Briefly, the full analysis set (FAS) included all randomized patients within treatment arms assigned in accordance with the randomization, regardless of the treatment actually received. Patients who were randomized but did not subsequently receive treatment were included in the FAS. The analysis of data using the FAS comprised an ITT population. Study population, demographic data, and efficacy and HRQoL results were summarized and analyzed based on the FAS, and patients were summarized based on the treatment arm they were assigned to, regardless of the treatment they received.
The safety analysis set consisted of all randomized patients who received any amount of study treatment (durvalumab or its placebo, or olaparib or its placebo). Patients who initially received a dose of durvalumab or durvalumab placebo were summarized according to the arm to which they had been randomized. Safety and treatment exposure were summarized based on the safety analysis set. For the maintenance phase, the number of patients in the safety analysis set who entered the maintenance phase (defined as having received at least 1 dose of olaparib or olaparib placebo) were also summarized.
Table 12: DUO-E Study Analysis Populations
Population | Definition | Application |
|---|---|---|
FAS | The FAS included all randomized patients within treatment arms assigned in accordance with the randomization, regardless of the treatment actually received. Patients who were randomized but did not subsequently receive treatment were included in the FAS. The analysis of data using the FAS followed intention-to-treat principles. | The FAS was used to analyze efficacy data (including HRQoL), baseline characteristics data, and biomarker data. Patients were summarized based on the treatment arm to which they were randomized, regardless of the treatment they actually received. |
SAS | The SAS consisted of all randomized patients who received any amount of study treatment (durvalumab or placebo, olaparib or placebo). Patients who initially received a dose of durvalumab or placebo were summarized according to the arm to which they were randomized. Maintenance phase: Number of patients in the SAS who entered the maintenance phase (defined as having received at least 1 dose of olaparib or olaparib placebo) were summarized and additional summaries of safety by treatment phase were also generated. | Exposure, adverse events, laboratory measurements, vital signs, ECGs. Safety data were not formally analyzed but were summarized using the SAS. |
PK analysis set | All patients who received at least 1 dose of durvalumab per the protocol for whom any postdose data were available and who did not violate or deviate from the protocol in ways that would have significantly affected the PK analyses were included in the PK analysis set. The population was defined by the study physician, pharmacokineticist, and statistician before any analyses were performed. | PK data. |
ADA analysis set | The ADA-evaluable patients are those in the SAS who received at least 1 dose of durvalumab and had nonmissing baseline ADA data and at least 1 postbaseline ADA result. | Immunogenicity data. |
dMMR subgroup | Subset to isolate the patients with dMMR cancer within arm A (SOC) and arm B (SOC plus durvalumab). | Aligned with the Health Canada indication and appraised in this review. |
ADA = antidrug antibody; dMMR = mismatch repair deficient; ECG = electrocardiogram; FAS = full analysis set; HRQoL = health-related quality of life; PK = pharmacokinetic; SAS = safety analysis set; SOC = standard of care.
Source: Details included in the table are from the sponsor’s summary of clinical evidence.22
Results
Patient Disposition
Results of the patient disposition for the ITT population, including the number of screening failures and other reasons for nonrandomization, are shown in Table 13. Specific reasons for screen failure were not provided. Briefly, out of 875 patients enrolled, a total of 718 were randomized into the study and comprised the ITT population (containing patients with either pMMR or dMMR cancers). A summary of the efficacy and harms results for the ITT population is included in Appendix 1.
Table 13: Summary of Patient Disposition From the DUO-E Study — ITT Population
Patient disposition | DUO-E study |
|---|---|
Enrolled, N | 875 |
Not randomized, N | 157 |
Reasons, n (%) | |
Screen failure | 145 (92.4) |
Withdrawal by patient | 7 (4.5) |
Death | 5 (3.2) |
COVID-19 pandemic | 0 |
ITT, N | 718 |
Results of randomization, N | Arm A (receiving SOC): 241 Arm B (receiving SOC + durvalumab): 238 Arm C (receiving SOC + durvalumab + olaparib): 239 |
ITT = intention to treat; SOC = standard of care.
Source: Details included in the table are from the sponsor’s DUO-E Clinical Study Report.48
A subset of the enrolled population (i.e., patients with dMMR status) within arm A (SOC) and arm B (SOC plus durvalumab) was isolated to align the trial population with the Health Canada indication. Details of the patient disposition for this subgroup are summarized in Table 14. Of the 718 patients included in the ITT population, 143 had dMMR status: 49 were randomized to the SOC arm, 46 were randomized to the durvalumab plus SOC arm, and 48 were randomized to the durvalumab plus olaparib and SOC arm. Data related to the durvalumab plus olaparib and SOC arm in the dMMR population, and all data from all patients in the pMMR population, were omitted from this review, given that these data are not relevant to the reimbursement request or the Health Canada indication.
At the time of data cut-off, all patients had discontinued carboplatin and paclitaxel, per the protocol. The most common reason for discontinuing carboplatin and paclitaxel was reaching the maximum number of chemotherapy cycles. In the dMMR subgroup, the proportion of patients discontinuing durvalumab was numerically greater in the SOC arm relative to the SOC arm. The most common reason for the discontinuation of durvalumab (or its placebo) was objective disease progression (█████ ██ █████). A numerically higher proportion of patients in the dMMR subgroup receiving SOC (██████) discontinued the study relative to the durvalumab plus SOC arm (█████). A numerically higher proportion of patients in the dMMR subgroup receiving durvalumab (█████) reported starting the maintenance phase relative to patients receiving SOC (█████).
Table 14: Summary of Patient Disposition From the DUO-E Study — dMMR Subgroup
Patient disposition | Patients with dMMR cancer | |
|---|---|---|
SOC (N = 49) | SOC + durvalumab (N = 46) | |
Patients who received any treatment, n (%)a | 46 (93.9) | 44 (95.7) |
SOC | 46 (93.9) | 44 (95.7) |
Durvalumab or its placebo | 46 (93.9) | 44 (95.7) |
Olaparib or its placebo | ██ ███████ | ██ ███████ |
Patients who did not receive any treatmenta | ██ ███████ | ██ ███████ |
Patients receiving ongoing treatment with durvalumab or its placebo at data cut-offb | ██ ███████ | ██ ███████ |
Patients who discontinued treatment with durvalumab or its placebo, n (%)b | ██ ███████ | ██ ███████ |
Objective disease progression | ██ ███████ | ██ ███████ |
Adverse event | ██ ███████ | ██ ███████ |
Patient decision | ██ ███████ | ██ ███████ |
Other | ██ ███████ | ██ ███████ |
Clinical deterioration | ██ ███████ | ██ ███████ |
Development of study-specific discontinuation criteria | ██ ███████ | ██ ███████ |
Patient lost to follow-up | ██ ███████ | ██ ███████ |
Severe noncompliance to protocol | ██ ███████ | ██ ███████ |
Due to COVID-19 pandemic | ██ ███████ | ██ ███████ |
Patients receiving ongoing treatment with olaparib or its placebo at data cut-offb | ██ ███████ | ██ ███████ |
Patients who discontinued treatment with olaparib or its placebo, n (%)b | ██ ███████ | ██ ███████ |
Objective disease progression | ██ ███████ | ██ ███████ |
Adverse event | ██ ███████ | ██ ███████ |
Patient decision | ██ ███████ | ██ ███████ |
Clinical deterioration | ██ ███████ | ██ ███████ |
Other | ██ ███████ | ██ ███████ |
Development of study-specific discontinuation criteria | ██ ███████ | ██ ███████ |
Patient lost to follow-up | ██ ███████ | ██ ███████ |
Due to COVID-19 pandemic | ██ ███████ | ██ ███████ |
Severe noncompliance to protocol | ██ ███████ | ██ ███████ |
Patients receiving ongoing treatment with carboplatin or substitute at data cut-offb | ██ ███████ | ██ ███████ |
Patients who discontinued treatment with carboplatin or substitute, n (%)b | ██ ███████ | ██ ███████ |
Maximum cycle of chemotherapy reached | ██ ███████ | ██ ███████ |
Adverse event | ██ ███████ | ██ ███████ |
Objective disease progression | ██ ███████ | ██ ███████ |
Other | ██ ███████ | ██ ███████ |
Patient decision | ██ ███████ | ██ ███████ |
Clinical deterioration | ██ ███████ | ██ ███████ |
Severe noncompliance to protocol | ██ ███████ | ██ ███████ |
Due to COVID-19 pandemic | ██ ███████ | ██ ███████ |
Development of study-specific discontinuation criteria | ██ ███████ | ██ ███████ |
Patient lost to follow-up | ██ ███████ | ██ ███████ |
Patients receiving ongoing treatment with paclitaxel or substitute at data cut-offb | ██ ███████ | ██ ███████ |
Patients who discontinued treatment with paclitaxel or substitute, n (%)b | ██ ███████ | ██ ███████ |
Maximum cycle of chemotherapy reached | ██ ███████ | ██ ███████ |
Adverse event | ██ ███████ | ██ ███████ |
Objective disease progression | ██ ███████ | ██ ███████ |
Other | ██ ███████ | ██ ███████ |
Patient decision | ██ ███████ | ██ ███████ |
Clinical deterioration | ██ ███████ | ██ ███████ |
Severe noncompliance to protocol | ██ ███████ | ██ ███████ |
Due to COVID-19 pandemic | ██ ███████ | ██ ███████ |
Development of study-specific discontinuation criteria | ██ ███████ | ██ ███████ |
Patient lost to follow-up | ██ ███████ | ██ ███████ |
Patients receiving any ongoing treatment at data cut-offb | ██ ███████ | ██ ███████ |
Patients who discontinued all initiated treatments (carboplatin, paclitaxel, durvalumab or its placebo, and olaparib or its placebo)b | ██ ███████ | ██ ███████ |
Patients ongoing study at data cut-off | ██ ███████ | ██ ███████ |
Patients who terminated study, n (%)c | ██ ███████ | ██ ███████ |
Death | ██ ███████ | ██ ███████ |
Withdrawal by patient | ██ ███████ | ██ ███████ |
Lost to follow-up | ██ ███████ | ██ ███████ |
Due to COVID-19 pandemic | ██ ███████ | ██ ███████ |
Screen failure | ██ ███████ | ██ ███████ |
Other | ██ ███████ | ██ ███████ |
Patients who started maintenance phase, n (%) | ██ ███████ | ██ ███████ |
Patients who received durvalumab or its placebo in maintenance phase | ██ ███████ | ██ ███████ |
Patients who discontinued durvalumab or its placebo in maintenance phase | ██ ███████ | ██ ███████ |
Patients who did not start the maintenance phase but started durvalumab or its placebo at the maintenance dosed | ██ ███████ | ██ ███████ |
FAS, Ne | 49 | 46 |
SAS, Ne | ███ | ███ |
dMMR = mismatch repair deficient; FAS = full analysis set; NR = not reported; OS = overall survival; SAS = safety analysis set; SOC = standard of care.
aAny of the following treatments: durvalumab or its placebo, olaparib or its placebo, carboplatin or substitute, or paclitaxel or substitute. The submission did not specify substitutes.
bPercentages were calculated from the number of patients who received the treatment.
cRepresents patients’ disposition status at time of discontinuation from study. Some patients subsequently had vital status information collected from publicly available resources (when it was possible to do so under applicable local laws) for the purpose of the OS analysis, as detailed in the DUO-E protocol.67
dComprises the patients who continued treatment with durvalumab or its placebo after the end of chemotherapy treatment; however, because they did not initiate treatment with olaparib or its placebo, they were not included in the maintenance phase.
eAll randomized patients who received any amount of study treatment (durvalumab or its placebo or olaparib or its placebo).
Source: Details included in the table are from the sponsor’s summary of clinical evidence.22
Baseline Characteristics
The baseline characteristics of the dMMR population outlined in Table 15 are limited to those that are most relevant to this review or that were felt to affect the outcomes or interpretation of the study results. The demographic characteristics were broadly balanced between study arms; the mean age of patients was roughly ██ years and approximately half of the patients in each study arm were older than 65 years. The study arms were approximately evenly split between patients with newly diagnosed versus recurrent cancer (████% in the SOC arm and ████% in the SOC plus durvalumab arm had a recurrence of earlier cancer). There were no details of additional molecular classifications within the dMMR subgroup (e.g., p53 mutations).
The majority of patients had metastatic disease (████% in the SOC arm, ████% in the SOC plus durvalumab arm) and the majority of tumours were endometroid (83.7% in the SOC arm and 71.7% in the SOC plus durvalumab arm). Patients with recurrent disease in the SOC plus durvalumab arm had a numerically longer mean time since initial diagnosis (█████ weeks; SD = ██████) relative to the SOC arm (█████ weeks; SD = ██████). There was a numeric difference in the proportion of patients who had locally advanced disease between study arms (███% in the SOC arm, ████% in the SOC plus durvalumab arm). There were also notable differences in the proportion of patients with a history of debulking surgery (████% in the SOC arm, ████% in the SOC plus durvalumab arm), the proportion of patients with a grade G2 tumour (████% in the SOC arm, ████% in the SOC plus durvalumab arm), and in the proportion of patients with an ECOG status of 0 (59.2% in the SOC arm, 50.0% in the SOC plus durvalumab arm) or 1 (40.8% in the SOC arm, 50.0% in the SOC plus durvalumab arm).
The baseline characteristics of the ITT population can be found in Appendix 1.
Table 15: Summary of Baseline Characteristics From the DUO-E Study — dMMR Subgroup
Characteristic | Patients with dMMR endometrial cancer | |
|---|---|---|
SOC (N = 49) | SOC + durvalumab (N = 46) | |
Demographic characteristics | ||
Age (years), mean (SD) | ████ █████ | ████ ███████ |
Age group (years), n (%) | ||
< 65 | 25 (51.0) | 25 (54.3) |
≥ 65 | 24 (49.0) | 21 (45.7) |
Race, n (%) | ||
White | 30 (61.2) | 29 (63.0) |
Asian | 15 (30.6) | 14 (30.4) |
Black or African American | 2 (4.1) | 0 |
Other | ██ ███████ | ██ ███████ |
American Indian or Alaska Native [wording from original source] | ██ ███████ | ██ ███████ |
Native Hawaiian or Other Pacific Islander | ██ ███████ | ██ ███████ |
Not reported | ██ ███████ | ██ ███████ |
Medical characteristics | ||
Time since initial diagnosis (weeks), mean (SD) | ||
Patients with newly diagnosed cancer | ██ ███████ | ██ ███████ |
Patients with recurrent cancer | ██ ███████ | ██ ███████ |
Time from recent progression to randomization (weeks), mean (SD)a | ██ ███████ | ██ ███████ |
ECOG performance status, n (%) | ||
0: Normal activity | 29 (59.2) | 23 (50.0) |
1: Restricted activity | 20 (40.8) | 23 (50.0) |
2: In bed ≤ 50% of the time | ██ | ██ |
Histology type, n (%)b | ||
Endometroid | 41 (83.7) | 33 (71.7) |
Serous | 2 (4.1) | 2 (4.3) |
Carcinosarcoma | ██ ███████ | ██ ███████ |
Mixed, epithelial | ██ ███████ | ██ ███████ |
Other | ██ ███████ | ██ ███████ |
Clear cell | ██ ███████ | ██ ███████ |
Undifferentiated | ██ ███████ | ██ ███████ |
Mucinous | ██ ███████ | ██ ███████ |
Tumour gradeb | ||
G1: Well differentiated | ██ ███████ | ██ ███████ |
G2: Moderately differentiated | ██ ███████ | ██ ███████ |
G3: Poorly differentiated | ██ ███████ | ██ ███████ |
GX: Unassessable | ██ ███████ | ██ ███████ |
Missing | ██ ███████ | ██ ███████ |
FIGO stageb | ||
IA | ██ ███████ | ██ ███████ |
IB | ██ ███████ | ██ ███████ |
II | ██ ███████ | ██ ███████ |
IIIA | ██ ███████ | ██ ███████ |
IIIB | ██ ███████ | ██ ███████ |
IIIC | ██ ███████ | ██ ███████ |
IIIC1 | ██ ███████ | ██ ███████ |
IIIC2 | ██ ███████ | ██ ███████ |
IVA | ██ ███████ | ██ ███████ |
IVB | ██ ███████ | ██ ███████ |
Missing | ██ ███████ | ██ ███████ |
Recurrence of earlier cancerc | ||
Yes | ██ ███████ | ██ ███████ |
No | ██ ███████ | ██ ███████ |
Baseline overall disease classification | ||
Metastaticd | ██ ███████ | ██ ███████ |
Locally advancede | ██ ███████ | ██ ███████ |
Missing | ██ ███████ | ██ ███████ |
MMR statusc | ||
Proficient | ██ ███████ | ██ ███████ |
Deficient | ██ ███████ | ██ ███████ |
Unknown | ██ ███████ | ██ ███████ |
Debulking surgery history | ||
Yes | ██ ███████ | ██ ███████ |
No | ██ ███████ | ██ ███████ |
Unknown | ██ ███████ | ██ ███████ |
Prior chemotherapy | ||
Yes | ██ ███████ | ██ ███████ |
No | ██ ███████ | ██ ███████ |
dMMR = mismatch repair deficient; ECOG = Eastern Cooperative Oncology Group; FFPE = formalin-fixed paraffin-embedded; FIGO = International Federation of Gynecology and Obstetrics; G = grade; IVRS = interactive voice response system; MMR = mismatch repair; pMMR = mismatch repair proficient; SD = standard deviation; SOC = standard of care (carboplatin + paclitaxel); vs. = versus.
aPatients with recurrent disease only.
bPathology-related disease characteristics were collected at the time of primary diagnosis of disease under investigation.
cMMR status (proficient vs. deficient) was per central laboratory result using the FDA-cleared Class II Ventana MMR immunohistochemistry panel (based on evaluation of tumour cells from an FFPE tumour tissue sample) and disease status (recurrent vs. newly diagnosed) was as collected on the electronic case report form. Note: 2 patients with “unknown” MMR status (per central laboratory) were randomized as having dMMR cancer per the IVRS based on local testing. Two additional patients were mis-stratified in the IVRS: 1 patient with dMMR cancer (per central laboratory) was randomized in the IVRS as having pMMR, and 1 patient with pMMR cancer (per central laboratory) was randomized in the IVRS as having dMMR.
dAny metastatic site of disease.
ePatient had only locally advanced sites of disease.
Source: Details included in the table are from the sponsor’s summary of clinical evidence.22
Exposure to Study Treatments
The exposure to study treatments for the study arms and subgroups relevant to the reimbursement request are presented in Table 16. Of note, patients in the SOC plus durvalumab arm had a numerically longer actual exposure to treatment in the study (mean = █████ weeks; SD = █████) relative to the SOC arm (mean = █████ weeks; SD = █████). A total of ██ patients in the SOC arm and ██ patients in the SOC plus durvalumab arm received maintenance therapy. The majority of infusions in both treatment arms were reported as taking place in the maintenance phase. In the SOC arm, the mean number of infusions was █████ infusions (SD = ████; range, ██ ██) in the overall study and ████ infusions (SD = ████; range, █ to ██) in the maintenance phase. In the SOC plus durvalumab arm, the mean number of infusions was █████ infusions (SD = ████; range, █ to ██) in the overall study and █████ infusions (SD = ████; range, █ to ██) in the maintenance phase.
Table 16: Summary of Patient Exposure From the DUO-E Study — dMMR Subgroup
Exposure | Overall | Maintenance | ||
|---|---|---|---|---|
SOC (N = 46) | SOC + durvalumab (N = 44) | SOC (N = 25) | SOC + durvalumab (N = 33) | |
Number of infusions | ||||
Mean (SD) | ██ ████ | ██ ███████ | ██ █ | ██ ███████ |
Median (range) | ██ ████ | ██ ███████ | ██ █ | ██ ███████ |
Number of infusions, n (%) | ██ ████ | ██ ███████ | ██ █ | ██ ███████ |
0 | ██ ████ | ██ ███████ | ██ █ | ██ ███████ |
1 to 3 | ██ ████ | ██ ███████ | ██ █ | ██ ███████ |
4 to 6 | ██ ████ | ██ ███████ | ██ █ | ██ ███████ |
7 to 10 | ██ ████ | ██ ███████ | ██ █ | ██ ███████ |
11 to 15 | ██ ████ | ██ ███████ | ██ █ | ██ ███████ |
16 to 20 | ██ ████ | ██ ███████ | ██ █ | ██ ███████ |
More than 20 | ██ ████ | ██ ███████ | ██ █ | ██ ███████ |
Intended exposure (weeks), mean (SD)a | ██ ████ | ██ ███████ | ██ █ | ██ ███████ |
Actual exposure (weeks), mean (SD)b | ██ ████ | ██ ███████ | ██ █ | ██ ███████ |
dMMR = mismatch repair deficient; SD = standard deviation; SOC = standard of care (carboplatin + paclitaxel).
aIntended exposure (weeks) = ([earliest of: last dose date when dose was > 0 mg plus C days, date of death, or date of data cut-off] minus [the first dose date plus 1 day]) divided by 7, where C is equal to the scheduled number of days between doses minus 1. Note: C = 20 if the last dose date falls in the chemotherapy phase; C = 27 if the last dose date falls in the maintenance phase.
bActual exposure (weeks) = intended exposure minus total duration of dose delays in days. Total duration of dose delays in days = (date of the dose minus date of the previous dose minus D days) divided by 7. Note: D = 21 if it happens during the chemotherapy phase; D = 28 if it happens during the maintenance phase.
Source: Details included in the table are from the sponsor’s summary of clinical evidence22 and additional information provided by the sponsor.68
Results on the proportion of patients receiving the prespecified rescue medications were not available. Results for the most common permitted concomitant medications and the number of patients reporting the use of prohibited medications for the ITT population are presented in Appendix 1; concomitant medications specific to the dMMR subgroup were not provided in the submission.
For patients who discontinued the study drug, details on the therapy they received after discontinuation is presented in Table 17. Of note, patients who discontinued the study drug received immunotherapy, chemotherapy, hormonal therapy, or targeted therapy.
Table 17: Postdiscontinuation Disease-Related Anticancer Therapy — dMMR Subgroup
Variable | Patients, n (%) | |
|---|---|---|
SOC N = 49 | SOC + durvalumab N = 46 | |
Immunotherapy | ██ █ | ██ █ |
BgB-A445 monoclonal antibody | ██ █ | ██ █ |
Dostarlimab | ██ █ | ██ █ |
Investigational antineoplastic drugs | ██ █ | ██ █ |
Pembrolizumab | ██ █ | ██ █ |
Hormonal therapy | ██ █ | ██ █ |
Letrozole | ██ █ | ██ █ |
Megestrol | ██ █ | ██ █ |
Megestrol acetate | ██ █ | ██ █ |
Tamoxifen | ██ █ | ██ █ |
Cytotoxic chemotherapy | ██ █ | ██ █ |
Carboplatin | ██ █ | ██ █ |
Cisplatin | ██ █ | ██ █ |
Doxorubicin | ██ █ | ██ █ |
Doxorubicin hydrochloride | ██ █ | ██ █ |
Gemcitabine | ██ █ | ██ █ |
Liposomal doxorubicin hydrochloride | ██ █ | ██ █ |
Nedaplatin | ██ █ | ██ █ |
Paclitaxel | ██ █ | ██ █ |
Topotecan | ██ █ | ██ █ |
Targeted therapy | ██ █ | ██ █ |
Capivasertib | ██ █ | ██ █ |
Cediranib | ██ █ | ██ █ |
Everolimus | ██ █ | ██ █ |
Lenvatinib | ██ █ | ██ █ |
Lenvatinib mesilate | ██ █ | ██ █ |
Olaparib | ██ █ | ██ █ |
Radiotherapy | ██ █ | ██ █ |
Missing | ██ █ | ██ █ |
dMMR = mismatch repair deficient; SOC = standard of care (carboplatin + paclitaxel).
Source: Details included in the table are from additional information provided by the sponsor.23
Efficacy
Survival Outcomes
Detailed results of the survival analysis to date (final analysis for PFS, IA for OS) at the data cut-off (April 12, 2023) are presented in Table 18.
Progression-Free Survival
In the dMMR subgroup, there were a total of 25 PFS-related events (51.0%; N = 49 patients) in the SOC arm and 15 events (32.6%; N = 46 patients) in the SOC plus durvalumab arm. There were ██████ censored deaths in the SOC arm and ██████ in the SOC plus durvalumab arm. The median PFS in the SOC arm was 7.0 months (95% CI, 6.7 to 14.8), while the median PFS was not calculable in the SOC plus durvalumab arm (HR, 0.42; 95% CI, 0.22 to 0.80 in favour of durvalumab). The risk difference between study arms for the proportion of patients whose cancer had not progressed was ████% (95% CI, ████ ██ ████) at 6 months; ████% (95% CI, ████ █████) at 12 months; and ████% (95% CI, █████ ██ ████) at 18 months.
Overall Survival
In the dMMR subgroup, there were a total of ██ ███████ deaths in the SOC arm and ███████ deaths in the SOC plus durvalumab arm. The median OS in the SOC arm was ████ months (95% CI, ███ ██████████), while the median OS was ███ ██████████ in the SOC plus durvalumab arm (HR, ████; 95% CI, ████ ██ ████, in favour of durvalumab). The risk difference between study arms for the proportion of patients who were alive was ████% (95% CI, █████ ██ █████) at 6 months; ████% (95% CI, ████ ██ ████) at 12 months; and ████% (95% CI, ████ ██ █████) at 18 months.
Objective Response Rate
In the dMMR subgroup, a total of ██ ███████ patients had a response out of ██ patients with measurable disease at baseline in the SOC arm, and ██ ███████ patients had a response out of ██ patients with measurable disease at baseline in the SOC plus durvalumab arm. The OR for response was ████ ████ ███ ████ ██ ████ in favour of durvalumab.
Duration of Response
In the dMMR subgroup in the SOC arm, ███████ patients out of the ██ patients who experienced a response subsequently died or experienced disease progression. The median DOR (measured from the onset of response) was ████ months. Out of the ██ patients in the SOC plus durvalumab arm who experienced a response, ████████ patients subsequently died or experienced disease progression; the median DOR was not calculable. The difference between study arms for the proportion of patients who continued to experience a response was █████ (95% CI, █████ ██ █████) at 6 months, █████ (95% CI, █████ ██ █████) at 12 months, and █████ (95% CI, ████% to ████%) at 18 months.
Time to Treatment Discontinuation or Death
In the dMMR subgroup, 37 patients (75.5%) in the SOC arm and 22 patients (47.8%) in the SOC plus durvalumab arm had an event of treatment discontinuation or death (HR = 0.47; 95% CI, 0.27 to 0.79 in favour of durvalumab).
Table 18: Summary of Survival Analysis Results From the DUO-E Study — dMMR Subgroup
Variable | Patients with dMMR cancer | |
|---|---|---|
SOC N = 49 | SOC + durvalumab N = 46 | |
PFS: Primary outcomea | ||
Total events, n (%) | 25 (51.0) | 15 (32.6) |
Censored patients, n (%) | ████ █████ | ████ █████ |
Progression-free at time of analysis | ████ █████ | ████ █████ |
Censored death | ████ █████ | ████ █████ |
Withdrawn consent | ████ █████ | ████ █████ |
No postbaseline evaluable tumour assessment | ████ █████ | ████ █████ |
Censored RECIST progression | ████ █████ | ████ █████ |
Lost to follow-up | ████ █████ | ████ █████ |
Duration of follow-up in censored patients (months), median (range) | ████ █████ | ████ █████ |
PFS (months), median (95% CI) | 7.0 (6.7 to 14.8) | NR (NR to NR) |
HR (95% CI) vs. SOCb | 0.42 (0.22 to 0.80) | |
P value | NR | |
Proportion (95% CI) of patients with progression-free disease | ||
At 6 months | 73.1 (56.6 to 84.2) | 90.6 (76.9 to 96.4) |
Survival difference (95% CI) vs. SOC | ████ █████ █████ | |
At 12 months | 43.3 (27.3 to 58.3) | 67.9 (51.1 to 80.0) |
Survival difference (95% CI) vs. SOC | ████ █████ █████ | |
At 18 months | 31.7 (16.7 to 47.9) | 67.9 (51.1 to 80.0) |
Survival difference (95% CI) vs. SOC | ████ ██████ █████ | |
OS: Secondary outcomea | ||
Deaths, n (%) | ████ █████ | ████ █████ |
Censored patients, n (%) | ████ █████ | ████ █████ |
Still in survival follow-up | ████ █████ | ████ █████ |
Terminated before deathc | ████ █████ | ████ █████ |
Withdrawn consent | ████ █████ | ████ █████ |
Lost to follow-up | ████ █████ | ████ █████ |
Duration of follow-up in censored patients (months), median (range) | ████ █████ | ████ █████ |
OS (months), median (95% CI) | ████ █████ | ████ █████ |
HR (95% CI) vs. SOCb | ████ █████ | |
P value | ████ █████ | |
Proportion (95% CI) of patients who were alive | ||
At 6 months | ████ █████ | ████ █████ |
Survival difference (95% CI) vs. SOC | ████ █████ | |
At 12 months | ████ █████ | ████ █████ |
Survival difference (95% CI) vs. SOC | ████ █████ | |
At 18 months | ████ █████ | ████ █████ |
Survival difference (95% CI) vs. SOC | ████ █████ | |
ORR: secondary outcome | ||
Number of patients with response out of the number of patients with measurable disease at baseline, n/N (%) | ████ █████ | ████ █████ |
OR (95% CI) vs. SOC | ████ █████ | |
CR (%) | ████ █████ | ████ █████ |
PR (%) | ████ █████ | ████ █████ |
DOR | ||
Number of responders who died or whose cancer subsequently progressed out of the number of patients who experienced a response, n/N (%) | ████ █████ | ████ █████ |
DOR from onset of response (months), mediand | ████ █████ | ████ █████ |
Proportion of patients continuing to experience a response (%) | ||
6 months | ████ █████ | ████ █████ |
Difference in DOR (%) vs. SOC | ████ █████ | |
12 months | ████ █████ | ████ █████ |
Difference in DOR (%) vs. SOC | ████ █████ | |
18 months | ████ █████ | ████ █████ |
Difference in DOR (%) vs. SOC | ████ █████ | |
TDT | ||
Total events, n (%) | ████ █████ | ████ █████ |
Treatment discontinuation | ████ █████ | ████ █████ |
Death | ████ █████ | ████ █████ |
Censored patients, n (%)e | ████ █████ | ████ █████ |
Median TDT (95% CI), (months) | ████ █████ | ████ █████ |
HR (95% CI) vs. SOCb | ████ █████ | |
P value | ████ █████ | |
Event-free rate (95% CI) | ||
At 6 months | ████ █████ | ████ █████ |
Difference in event vs. SOC, % (95% CI) | ████ █████ | |
At 12 months | ████ █████ | ████ █████ |
Difference in event vs. SOC, % (95% CI) | ████ █████ | |
At 18 months | ████ █████ | ████ █████ |
Difference in event vs. SOC, % (95% CI) | ████ █████ | |
CI = confidence interval; CR = complete response; dMMR = mismatch repair deficient; DOR = duration of response; HR = hazard ratio; ITT = intention to treat; NR = not reported; OR = odds ratio; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; PR = partial response; RECIST = Response Evaluation Criteria in Solid Tumours; SOC = standard of care (carboplatin + paclitaxel); TDT = time to study treatment discontinuation or death; vs. = versus.
aA multiple-testing procedure was applied to the analysis of PFS and OS for the ITT population; however, this procedure was not applied to the analysis of the data from the dMMR subgroup.
bThe HR and CI were estimated from an unstratified Cox proportional hazards model. An HR of less than 1 favoured the treatment arm of interest over the reference arm.
cIncludes patients with unknown survival status and patients who were lost to follow-up.
dCalculated as the time from the first confirmed CR or PR until either the date of progression or the date of censoring for PFS.
eAny patient not known to have died and not known to have discontinued all investigational treatments was censored at the last recorded date on which the patient was known to be alive.
Source: Details included in the table are from the sponsor’s summary of clinical evidence and additional information provided by the sponsor.23
HRQoL Outcomes
Details of the HRQoL results are presented in Table 19.
EORTC QLQ-C30 Global Health Score
In the dMMR subgroup, the mean baseline score was █████ points (SD = █████) in the SOC arm and █████ points (SD = █████) in the SOC plus durvalumab arm. At week 18 (corresponding to the sixth cycle of the chemotherapy phase), the mean change in score from baseline was ██████ points (SD = █████) in the SOC arm (N = ██) and █████ points (SD = █████) in the SOC plus durvalumab arm (N = ██). The absolute mean difference between study arms was █████ points (95% CI, ██████ to ████). At week 42 (corresponding to the sixth month of the maintenance phase), the mean change in scores from baseline was ██████ points (SD = █████) in the SOC arm (N = ██) and ████ points (SD = █████) in the SOC plus durvalumab arm (N = ██). The absolute mean difference between study arms was █████ points (95% CI, ████ to █████).
EORTC QLQ-EN24 Pain in Back and Pelvis Score
In the dMMR subgroup, the mean baseline score was █████ points (SD = █████) in the SOC arm and █████ points (SD = █████) in the SOC plus durvalumab arm. At week 18, the mean change in score from baseline was ████ points (SD = █████) in the SOC arm (N = ██), and ████ points (SD = █████) in the SOC plus durvalumab arm (N = ██). The absolute mean difference between study arms was ████ points (95% CI, █████ to █████). At week 42, the mean change in scores from baseline was █████ points (SD = █████) in the SOC arm (N = ██), and ████ points (SD = █████) in the SOC plus durvalumab arm (N = ██). The absolute mean difference between study arms was ████ points (95% CI, █████ to █████).
EORTC QLQ-EN24 Urological Symptoms Score
In the dMMR subgroup, the mean baseline score was █████ points (SD = █████) in the SOC arm and █████ points (SD = █████) in the SOC plus durvalumab arm. At week 18, the mean change in score from baseline was ████ points (SD = █████) in the SOC arm (N = ██) and −████ points (SD = █████) in the SOC plus durvalumab arm (N = ██). The absolute mean difference between study arms was ████ points (95% CI, █████ to █████). At week 42, the mean change in scores from baseline was ████ points (SD = █████) in the SOC arm (N = ██) and ████ points (SD = █████) in the SOC plus durvalumab arm (N = ██). The absolute mean difference between study arms was ████ points (95% CI, █████ to ████).
Table 19: Summary of HRQoL Results From the DUO-E Study — dMMR Subgroup
Variable | Patients with dMMR cancer | |
|---|---|---|
SOC N = 49 | SOC + durvalumab N = 46 | |
Change from baseline in EORTC QLQ-C30 global health status score | ||
Number of patients contributing at baseline, n | █████ ███████ | █████ ███████ |
Baseline score, mean (SD) | █████ ███████ | █████ ███████ |
Number of patients contributing at week 18, na | █████ ███████ | █████ ███████ |
Week 18 score, mean (SD) | █████ ███████ | █████ ███████ |
Change from baseline at week 18, mean (95% CI)a | █████ ███████ | █████ ███████ |
Absolute difference, mean (95% CI) | █████ ████████ █████ | |
Number of patients contributing at week 42, nb | █████ ███████ | █████ ███████ |
Week 42 score, mean (SD) | █████ ███████ | █████ ███████ |
Change from baseline at week 42, mean (95% CI)b | ██████ ██████ | ████ ███████ |
Absolute difference, mean (95% CI) | █████ ██████ ██████ | |
Change from baseline in EORTC QLQ-EN24 pain in back and pelvis score | ||
Number of patients contributing at baseline, n | █████ ███████ | █████ ███████ |
Baseline score, mean (SD) | █████ ███████ | █████ ███████ |
Number of patients contributing at week 18, na | █████ ███████ | █████ ███████ |
Week 18 score, mean (SD) | █████ ███████ | █████ ███████ |
Change from baseline at week 18, mean (95% CI)a | █████ ███████ | █████ ███████ |
Absolute difference, mean (95% CI) | █████ ███████ | |
Number of patients contributing at week 42, nb | █████ ███████ | █████ ███████ |
Week 42 score, mean (SD) | █████ ███████ | █████ ███████ |
Change from baseline at week 42, mean (95% CI)b | █████ ███████ | █████ ███████ |
Absolute difference, mean (95% CI) | █████ ███████ | |
Change from baseline in EORTC QLQ-EN24 urological symptoms score | ||
Number of patients contributing at baseline, n | █████ ███████ | █████ ███████ |
Baseline score, mean (SD) | █████ ███████ | █████ ███████ |
Number of patients contributing at week 18, na | █████ ███████ | █████ ███████ |
Week 18 score, mean (SD) | █████ ███████ | █████ ███████ |
Change from baseline at week 18, mean (95% CI)a | █████ ███████ | █████ ███████ |
Absolute difference, mean (95% CI) | █████ ███████ | |
Number of patients contributing at week 42, nb | █████ ███████ | █████ ███████ |
Week 42 score, mean (SD) | █████ ███████ | █████ ███████ |
Change from baseline at week 42, mean (95% CI)b | █████ ███████ | █████ ███████ |
Absolute difference, mean (95% CI) | █████ ███████ | |
CI = confidence interval; dMMR = mismatch repair deficient; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-EN24 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire – Endometrial Cancer Module; HRQoL = health-related quality of life; SD = standard deviation; SOC = standard of care (carboplatin + paclitaxel or substitutes).
aCorresponds to cycle 6 of the chemotherapy phase.
bAfter the chemotherapy phase, the assessment of outcomes shifted from every 3 weeks to every 4 weeks; week 42 corresponds to the sixth month of the maintenance phase.
Sources: Details contained in the table are from the sponsor’s summary of clinical evidence22 and additional information provided by the sponsor.69
Harms
Results for harms from both the chemotherapy and maintenance phases (overall phase) and maintenance phase alone of the DUO-E trial, specifically in the dMMR subgroup, are summarized subsequently and in Table 20. Harms data for the entire ITT population of the DUO-E trial are presented in the Appendix.
Adverse Events
In the dMMR subgroup, all patients in each study arm experienced an AE during the DUO-E trial. A total of ████% of patients in the SOC arm and ████% of patients in the SOC plus durvalumab arm experienced an AE with a maximum grade of 3 or 4. The most common AEs were nausea (████% of patients in the SOC plus durvalumab arm, ████% of patients in the SOC arm), alopecia (████% in the SOC plus durvalumab arm, ████% in the SOC arm), arthralgia (████% in the SOC plus durvalumab arm, ████% in the SOC arm), and anemia (████% in the SOC plus durvalumab arm, ████% in the SOC arm).
There were differences between study arms in the proportion of patients with several AEs. Of note, a numerically higher proportion of patients in the SOC plus durvalumab arm reported nausea (████% versus ████% in the SOC arm), alopecia (████% versus ████% in the SOC arm), alopecia (████% versus ████% in the SOC arm), arthralgia (████% versus ████% in the SOC arm), hypomagnesemia (████% versus ████% in the SOC arm), cough (████% versus 15.2% in the SOC arm), peripheral neuropathy (████% versus ████% in the SOC arm), dyspnea (████% versus ███% in the SOC arm), and injury, poisoning or procedural complications (████% versus ████%). A numerically higher proportion of patients in the SOC arm reported anemia (████% versus ████% in the SOC plus durvalumab arm), fatigue (████% versus ████% in the SOC plus durvalumab arm), peripheral sensory neuropathy (████% versus ████% in the SOC plus durvalumab arm), decreased appetite (████% versus ████% in the SOC plus durvalumab arm), neutrophil count decreased (████% versus ████% in the SOC plus durvalumab arm), and neutropenia (████% versus ████% in the SOC plus durvalumab arm).
Serious Adverse Events
In the dMMR subgroup, a total of ████% of patients in the SOC arm and ████% in the SOC plus durvalumab arm experienced an SAE during the DUO-E trial. The most common SAEs were as follows: gastrointestinal disorders (████% in the SOC plus durvalumab arm consisting of ███████ reports of fecaloma, ███████ report each of abdominal hernia, colitis, intestinal obstruction and nausea, and ███% in the SOC arm consisting of ███████ report each of diarrhea, nausea, and vomiting), blood and lymphatic system disorders (████% in the SOC arm consisting of ███████ reports of anemia, ███████ reports of febrile neutropenia, and ███████ reports of neutropenia, and ███% in the SOC plus durvalumab arm consisting of 1 report of autoimmune hemolytic anemia), infections and infestations (███% in the SOC arm consisting of ███████ report each of COVID-19, neutropenic sepsis, urinary tract infection and urosepsis, and ███% in the SOC plus durvalumab arm consisting of ███████ report each of appendicitis, gastroenteritis, and sepsis), and renal and urinary disorders (███% in the SOC arm consisting of ███████ report each of acute kidney injury, renal failure, ureteric obstruction and urinary bladder hemorrhage, and ███████% in the SOC plus durvalumab arm).
Withdrawals Due to Adverse Events
In the dMMR subgroup. a total of ████% of patients experienced AEs leading to the withdrawal of durvalumab in the SOC plus durvalumab arm, and ████% reported this in the SOC arm. In the SOC plus durvalumab arm, the reasons for discontinuation were anemia, interstitial lung disease, maculopapular rash, symmetric drug-related intertriginous and flexural exanthema, fatigue, and procedural pain (each reported in ██ patient) In the SOC arm, the reasons for discontinuation of durvalumab placebo were anemia (2 patients), cerebrovascular accident, tinnitus, and asthenia (each reported in ██ patient). A total of ██% of patients in the SOC plus durvalumab arm and ██% of patients in the SOC arm experienced an AE leading to discontinuation of SOC.
Mortality
In the dMMR subgroup, a total of ██ patients (██%) died in the SOC arm and ███ patients (██%) died in the SOC plus durvalumab arm. The submission did not provide details on the specific causes of death in patients in the dMMR subgroup.
Notable Harms
Immune-mediated AEs (exact conditions not specified) and infusion reactions were identified by the clinical experts consulted for this review as AESIs. In the dMMR subgroup, ██% of patients experienced an immune-mediated AE in the SOC plus durvalumab arm and ██% experienced this in the SOC arm. A total of ███% of patients experienced infusion reactions in the SOC plus durvalumab arm versus ██% of patients in the SOC arm.
A total of ██% of patients in the SOC plus durvalumab arm and ███% of patients in the SOC arm experienced 1 of the AESIs that had been identified by the clinical experts for this review. The most common AEs in both arms were diarrhea (██% in the SOC plus durvalumab arm, ██% in the SOC arm) and rash (███% in the SOC plus durvalumab arm, ██% in the SOC arm). Hypothyroidism (██% of patients) was the next most common reason in the SOC plus durvalumab arm, and hyperthyroidism (██% of patients) the next most common reason in the SOC arm.
Table 20: Summary of Harms Results From the DUO-E Study — dMMR Subgroup
AEs | Overall | Maintenance phase only | ||
|---|---|---|---|---|
SOC N = 46 | SOC + durvalumab N = 44 | SOC N = 25 | SOC + durvalumab N = 33 | |
AEs, n (%) | ||||
≥ 1 AE | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
≥ 1 grade 3 or 4 AE | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Most common (≥ 20%) AEs, n (%)a | ||||
Anemia | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Neutropenia | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Decreased appetite | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Hypomagnesemia | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Headache | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Neuropathy peripheral | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Peripheral sensory neuropathy | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Cough | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Dyspnea | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Diarrhea | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Nausea | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Vomiting | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Alopecia | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Rash | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Arthralgia | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Fatigue | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Pain in extremity | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Neutrophil count decreased | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Injury, poisoning, and procedural complications | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
SAEs, n (%) | ||||
Patients with any SAE, n (%) | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Most common (≥ 2 patients) SAEs | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Gastrointestinal disorders | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Fecaloma | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Infections and infestations | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Respiratory, thoracic, and mediastinal disorders | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Skin and subcutaneous tissue disorders | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
General disorders and administration site conditions | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Injury, poisoning, procedural complications | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Nervous system disorders | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Blood and lymphatic system disorders | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Anemia | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Febrile neutropenia | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Neutropenia | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Renal and urinary disorders | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Metabolism and nutrition disorders | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Hypokalemia | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Hyponatremia | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Patients who stopped treatment due to AEs, n (%) | ||||
Any AE leading to discontinuation of any study treatment | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
AE leading to discontinuation of durvalumab or its placebo | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Anemia | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Cerebrovascular accident | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Tinnitus | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Interstitial lung disease | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Rash maculopapular | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Symmetric drug-related intertriginous and flexural exanthema | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Asthenia | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Fatigue | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Procedural pain | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Any AE leading to discontinuation of SOC | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Anemia | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Neutropenia | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Neuropathy periphera | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Neurotoxicity | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Peripheral sensory neuropathy | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Tinnitus | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Alopecia | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Asthenia | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Fatigue | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
ECOG Performance Status decreased | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Platelet count decreased | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Procedural pain | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Deaths, n (%) | ||||
Patients who died | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
AEs of special interest, n (%) | ||||
Any durvalumab-related AE of special interest | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Diarrhea | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Rash | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Hypothyroidism | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Eczema | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Hyperthyroidism | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Pneumonitis | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Rash, maculopapular | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Autoimmune hemolytic anemia | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Colitis | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Dermatitis | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Immune-mediated thyroiditis | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Infusion-related reaction | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Interstitial lung disease | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Myositis | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Rash, erythematous | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Rash, popular | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Rash, pustular | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Thyroiditis | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Urticaria | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Psoriasis | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Rash, macular | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Drug hypersensitivity | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Immune-mediated AEs | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
Infusion reactions | ██ ███ | ██ ███ | ██ ███ | ██ ███ |
AE = adverse event; dMMR = mismatch repair deficient; ECOG = Eastern Cooperative Oncology Group; NR = not reported; SAE = serious adverse event; SOC = standard of care.
aPatients with multiple AEs in the same category were counted only once for that preferred term. Patients with AEs in more than 1 category were counted once in each of these categories.
bPatients with multiple AEs leading to discontinuation of any study treatment or SOC were counted once for each system organ class.
cDeath not due to disease progression.
Sources: Details included in the table are from the sponsor’s summary of clinical evidence22 and additional information provided by the sponsor.23
Critical Appraisal
Internal Validity
The DUO-E trial is an ongoing phase III trial assessing the efficacy and safety of durvalumab plus SOC compared with SOC alone in the treatment of primary advanced or recurrent endometrial cancer. Included in the submission was an IA containing results up to the data cut-off on April 13, 2023. The randomization, allocation concealment, and laboratory assessments were at low risk of bias because randomization was stratified and conducted centrally using an IVRS or interactive web response system, and placebo descriptions were provided. Likewise, the outcome ascertainment methods were at low risk of bias because the time-to-event outcomes were assessed by investigator; although BICR results were not available for the subgroups to compare, the impact is likely minimal. Blinding was likewise continued for the duration of the study; therefore, the impact on subjective outcomes (i.e., the HRQoL measures) was likely minimal. However, there were numerically higher numbers of patients in the SOC arm who discontinued treatment relative to the SOC plus durvalumab arm, which suggests the possibility that patients may have become unblinded to their treatment arm and discontinued treatment more readily than those in the SOC plus durvalumab arm. As part of the study design, patients with no evidence of progressive disease during the 6-cycle chemotherapy phase were eligible to proceed to the maintenance phase. Results separating the efficacy outcomes in the chemotherapy and maintenance phases were not provided; thus, the impact of the chemotherapy phase versus the maintenance phase individually on response to treatment remains unknown.
There are several limitations to note regarding the study design and the statistical model. First, the design of the DUO-E study comprised 3 study arms that contained patients with either dMMR and pMMR endometrial cancer. However, given the Health Canada indication and reimbursement request, the focus of this review is based on a subgroup of patients with dMMR endometrial cancer. Therefore, all results focusing on the dMMR subgroup can only be considered exploratory and supportive of the overall effect of durvalumab. Furthermore, the subgroup was not controlled for multiple comparisons and there is an increased risk of type I error, which is particularly important, given that the sample sizes in each study arm were small. Second, the study protocol was amended numerous times after patient enrolment started and, although the level of potential impact of these protocol changes was considered low based on the feedback from the clinical experts, the potential for bias due to these changes is unknown.
Results from the DUO-E trial were from an IA, and the P value cut-off for the IA of the subgroups themselves is not known; this carries an increased risk of overestimating the true effect. While the primary end point was reached for the ITT population, the median PFS and OS were not estimable for the dMMR subgroup in the respective analyses. In the ITT population, this analysis was based on an information fraction of 61.0% while, in the dMMR subgroup, only ████% of PFS events had occurred, suggesting the data were immature, particularly for OS. Lastly, the analysis for PFS, OS, and TDT in the subgroup was undertaken using an unstratified Cox proportional hazards model; an unstratified logistic regression model was used for ORR. While randomization was stratified on MMR status, disease status, and region, which are known effect modifiers described in the submission, these were not accounted for in the time-to-event analysis and might have impacted PFS, OS, ORR, and TDT outcome estimates. As such, there is increased uncertainty in the PFS and OS results, and it is therefore unclear how confidently the long-term results associated with durvalumab can be predicted.
There are additional limitations specific to the HRQoL measures reported in the DUO-E study. HRQoL end points were secondary in the DUO-E trial and the results were not adjusted for multiplicity; thus, there is an increased risk of type I error. In addition, the baseline estimates and the estimates at weeks 18 and 42 have a substantial amount of missing data (N = ██ patients in the SOC arm and ██ patients in the SOC plus durvalumab arm contributed data at 18 weeks; ██ patients in the SOC arm and ██ patients in the SOC plus durvalumab arm contributed data at 42 weeks). The end points were analyzed using a mixed model for repeated measures, which assumes that the missing data are missing at random; however, given the nature of the disease and design of the trial, the missing patient data are likely missing due to reasons related to the interventions; therefore, the missing-at-random assumption is not appropriate. The nature of the disease also means that, because patients are censored at disease progression or death, among other reasons, it is likely that these patients are systematically different from the patients included in the analysis; thus, there is also a likelihood of bias in the results. Furthermore, missing answers in the surveys were ignored and this imparts an additional risk of bias because the completeness of the survey responses was not reported. Lastly, there were no validation studies or MIDs identified specific to endometrial cancer; therefore, a direct assessment of the clinical meaningfulness of the HRQoL results was not possible.
External Validity
Per the clinical experts, the inclusion and exclusion criteria were broadly representative of patients who would be candidates for durvalumab. However, the results of patient screening were not available for the dMMR subgroup; therefore, the reasons for study failure and the distribution of screening failures between study arms is not known, so it is unclear whether there are systematic differences between the patients who passed screening and those who did not. Similarly, the exact causes of death in participants who died during the study were not reported, and it is therefore not known whether there are systematic differences between the patients who died and those who did not.
The clinical experts noted that, for the dMMR subgroup, the numeric differences in baseline patient characteristics that occurred would not be of great clinical importance, apart from the fact that the definition of “locally advanced disease” versus “metastatic disease” was not provided in the submission, and further noted that classifications within these disease categories may vary between clinicians. They also noted that patients with an ECOG status greater than 1 were not included in the trial; however, these patients would likely be considered for treatment with durvalumab in clinical practice. Apart from this, the study did not assess whether other molecular subtypes (e.g., POLE mutated, HER expression, p53 mutated) would have an impact on the results observed. The clinical experts noted that additional molecular classifications are usually undertaken in clinical practice and that a proportion of patients with dMMR cancer do not experience a response to current ICI therapy, the reason for which is not always clear. Therefore, it is possible the same nonresponder population may also exist for patients treated with durvalumab.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For the pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered most relevant to inform expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group:20,21
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate. The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited. The true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate. The true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as “very uncertain.”
Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty-of-evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null. For the PFS and OS outcomes, the target of the assessment was the presence or absence of an important effect based on thresholds provided by the clinical experts. For the HRQoL outcomes, the target of the assessment was the presence or absence of any effect based on a null threshold.
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for durvalumab plus carboplatin and paclitaxel followed by durvalumab maintenance therapy versus carboplatin and paclitaxel alone in patients with recurrent endometrial cancer that is dMMR.
Long-Term Extension Studies
The submission did not contain any long-term extension studies.
Indirect Evidence
Contents within this section have been informed by materials submitted by the sponsor. The following was summarized and validated by the review team.
Objectives for the Summary of Indirect Evidence
The DUO-E trial included SOC chemotherapy (carboplatin and paclitaxel) but did not include a comparison with dostarlimab, a relevant first-line comparator for patients with dMMR endometrial cancer, and information from indirect comparisons were included in the pharmacoeconomic model. Therefore, a review of the indirect evidence was undertaken.
Description of Indirect Comparisons
The body of indirect evidence consisted of 2 sponsor-submitted MAICs. Details of the study selection criteria and methods are presented in Table 21.
Indirect Treatment Comparison Design
Objectives
The objective of the MAIC was to assess the comparative efficacy and safety of durvalumab plus chemotherapy followed by maintenance durvalumab monotherapy, relative to dostarlimab plus chemotherapy, followed by maintenance dostarlimab in patients with dMMR and/or MSI-H advanced endometrial cancer.
Table 21: Study Selection Criteria and Methods for the SLR Informing the MAIC
Characteristics | MAIC |
|---|---|
Population |
|
Interventions and comparators | Systemic anticancer treatment from the following classes:
Durvalumab 1,120 mg given in combination with platinum-based chemotherapy (carboplatin AUC 4 mg/mL/min to 5 mg/mL/min, paclitaxel 175 mg/m2), followed by maintenance durvalumab 1,120 mg monotherapy, referred to as “SOC plus durvalumab.” |
Outcomes | Efficacy outcomes:
Safety and tolerability outcomes:
HRQoL outcomes, from generic or disease-specific measures:
The outcomes of interest specified in the submission for the ITC were PFS, OS, and safety. Results contained in the submission were for PFS and OS. |
Study designs |
|
Publication characteristics | Published and unpublished |
Exclusion criteria | Patient population:
Intervention:
Outcomes:
Study design:
Language:
Other:
|
Databases searched |
Grey literature was also searched, including clinical trials registries and conference proceedings. The submission did not provide a list of specific registries and trial proceedings. |
Selection process | Every title and abstract was reviewed against the eligibility criteria by 2 independent reviewers. When the applicability of the inclusion criteria was unclear, the article was included at this stage to ensure that all potentially relevant studies were captured. The independent reviewers then compared their results, and any disagreements were resolved by discussion until a consensus was reached. If necessary, a third independent reviewer made the final decision. Each full-text article was then reviewed against the eligibility criteria by 2 independent reviewers. If an article did not give enough information to confirm it met the inclusion criteria, the article was excluded at this stage, ensuring that only relevant articles were ultimately included in the systematic review. The independent reviewers compared their results, and any disagreements were resolved through discussion until a consensus was reached. If necessary, a third reviewer made the final decision. |
Data extraction process | Data extraction was performed by a single individual for each included study. When the initial extraction was complete, a second individual independently verified the extracted information and checked that no relevant information had been missed. Any discrepancies or missing information identified by the second individual were discussed by both individuals until a consensus was reached on the information that should be presented in the extraction grid. When necessary, a third individual was enlisted to arbitrate the final decision. For each included study, key information was extracted into a prespecified data extraction grid in Microsoft Excel. Extracted data were stratified in the grid by subgroups, when appropriate, including MMR status (dMMR and pMMR) and PD-L1 expression (positive and negative). |
Quality assessment | The quality of all included RCTs was assessed using the quality assessment tool developed by the University of York Centre for Reviews and Dissemination. The quality of included nonrandomized interventional studies and observational studies was assessed using the ROBINS-I tool. The quality assessment was completed by 1 individual and verified by a second independent reviewer. |
AUC = area under the curve; dMMR = mismatch repair deficient; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire C30; EORTC QLQ-EN24 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire – Endometrial Cancer Module; FACT-G = Functional Assessment of Cancer Therapy–General; HRQoL = health-related quality of life; ITC = indirect treatment comparison; MAIC = matching-adjusted indirect comparison; MMR = mismatch repair; mTOR = mammalian target of rapamycin; OS = overall survival; PARP = poly(adenosine diphosphate ribose) polymerase; PFS = progression-free survival; PFS2 = progression-free survival from time from randomization to the earliest progression event; pMMR = mismatch repair proficient; RCT = randomized controlled trial; ROBINS-I = Risk of Bias in Non-randomised Studies of Interventions; SF-36 = Short Form (36) Health Survey; SF-6D = Short Form 6 Dimensions; SLR = systematic literature review; SOC = standard of care; VAS = visual analogue scale.
Source: Details included in the table are from the sponsor’s summary of clinical evidence.22
Study Selection Methods
Details of the methods used for study selection are in Table 21. In summary, an SLR was conducted focusing on systemic anticancer treatments and excluded all other therapies. The patient population of interest comprised those newly diagnosed with stage III or IV disease or recurrent disease who were either naive to first-line therapy or had been treated with 1 prior line of chemotherapy. The submission did not provide details on the date when the SLR was conducted, the range of publication dates that was used in the search terms, or the search terms used.
MAIC Analysis Methods
Details of the analysis methods for the MAIC are presented in Table 22. Briefly, an anchored MAIC was performed using a frequentist approach and Bucher methodology. The treatment effects for the 2 efficacy outcomes, PFS and OS, were reported as an HR and associated 95% CI for SOC plus durvalumab versus SOC plus dostarlimab, with an HR of less than 1 favouring SOC plus durvalumab. For safety end points, ORs were calculated for SOC plus durvalumab versus SOC plus dostarlimab, with ORs of less than 1 favouring SOC plus durvalumab. ORs were estimated from AE frequency data and the number of patients included in the safety analysis sets.
Table 22: MAIC Analysis Methods
Methods | Description |
|---|---|
Analysis methods | Anchored MAIC using a frequentist approach and the Bucher methodology. |
Priors | None. |
Assessment of model fit | None reported. |
Assessment of consistency | None reported. |
Assessment of convergence | None reported. |
Outcomes | The same outcome measures were assessed between the 2 studies; the submission did not provide any details on how censoring may have been similar or different between the 2 studies. |
Follow-up time points | The median follow-up times for patients censored for PFS and OS were reported, as well as the data cut-off for the studies. |
Construction of nodes | The network was constructed based on common comparators; the submission did not provide details on how the SOC arm treatment regimens were compared. |
Sensitivity analyses | None reported. |
Subgroup analysis | None reported. |
Methods for pairwise meta-analysis | None reported. |
Identifying matching variables | Variables were identified from the literature and in consultation with medical and clinical experts from the sponsor. The results of prespecified subgroup analyses for platinum-based chemotherapy plus immunotherapy were reviewed from the following trials: DUO-E, RUBY, NRG-GY018 (NCT03914612), and AtTend (NCT03603184). |
Matching procedure | Matching was performed using the method of moments approach described in NICE DSU guidance70 using a propensity score derived from logistic regression. Histograms of weights were inspected for the presence of extreme weights; the submission did not provide a threshold for what was considered extreme. |
Comparison of baseline characteristics | Baseline characteristics before and after matching were compared. The submission did not provide details on whether statistical testing was undertaken in the matching. |
MAIC = matching-adjusted indirect comparison; NICE DSU = National Institute for Health and Care Excellence Decision Support Unit; OS = overall survival; PFS = progression-free survival; SOC = standard of care.
Source: Details included in the table are from the sponsor’s summary of clinical evidence22 and the MAIC technical report.71
Results of MAIC
Summary of Included Studies
The submission did not provide a PRISMA diagram of results from the SLR but reported that the SLR identified 2 phase III double-blind, placebo-controlled RCTs relevant to the indirect treatment comparison: the DUO-E and RUBY Part 1 studies. A summary of key study characteristics is in Table 23. Briefly, differences were noted in the median duration of follow-up as well as the molecular characteristics of patients; the primary analysis for the MAICs used data from the post hoc subgroup of the DUO-E study, which contained only patients with dMMR and/or MSI-H cancer (and therefore could contain patients with MSI-H pMMR endometrial cancer), and the main analysis in the RUBY Part 1 study enrolled patients who had either dMMR or MSI-H tumours.
Table 23: Summary of Key Study Characteristics for the DUO-E and RUBY Part 1 Trials
Characteristics | DUO-E study | RUBY Part 1 study |
|---|---|---|
Study design | Ongoing phase III, randomized, multicentre, double-blind, placebo-controlled trial | Phase III, double-blind, randomized, placebo-controlled trial |
Trial eligibility criteria |
|
|
Randomization stratification |
|
|
Chemotherapy phase | Durvalumab 1,120 mg (or placebo) IV in combination with carboplatin (AUC 5 mg/mL/min) and paclitaxel (175 mg per m2) q.3.w. for 4 to 6 cycles | Dostarlimab 500 mg (or placebo) in combination with carboplatin (AUC 5 mg/mL/min) and paclitaxel (175 mg per m2) q.3.w. for 6 cycles |
Maintenance phase | Durvalumab 1,500 mg (or placebo) IV every 4 weeks until disease progression | Dostarlimab 1,000 mg every 6 weeks for up to 36 months (approximately 26 cycles) |
Placebo response | Not reported | Not reported |
Definitions of key end points |
|
|
Timing of end point evaluation |
|
|
Data maturity (PFS) | 61% (438 events in 718 patients) | 61.9% (312 events in 504 patients) |
Data maturity (OS) | 27.7% (199 events in 718 patients) | 32.7% (165 events in 504 patients) |
Withdrawal frequency | Time-to-event analysis | Time-to-event analysis |
Clinical trial setting | Not reported | Not reported |
AUC = area under the curve; BICR = blinded independent central review; dMMR = mismatch repair deficient; ECOG = Eastern Cooperative Oncology Group; FIGO = International Federation of Gynecology and Obstetrics; MAIC = matching-adjusted indirect comparison; MMR = mismatch repair; MSI-H = microsatellite instability-high; OS = overall survival; PFS = progression-free survival; q.3.w. = every 3 weeks; vs. = versus.
Source: Details included in the table are from the sponsor’s summary of clinical evidence22 and MAIC technical report.71
Results
The base case of the MAIC for PFS matched on the following variables: region (Asia versus rest of world) and disease status. The summary of select baseline characteristics before and after matching is in Table 24. Of note, after the matching procedure, minor differences remained in the proportion of patients who were Black or African American (███% in the DUO-E trial after weighting and 8.5% in the RUBY Part 1 study), and the proportion of patients who were aged 65 years or older (████% in the DUO-E trial after weighting and 49.2% in the RUBY Part 1 study).
Table 24: Summary of Baseline Characteristics Before and After Matching — DUO-E and RUBY Part 1 Studies
Variable | Subgroups | DUO-E study | RUBY Part 1 study | |
|---|---|---|---|---|
Before weighting | After weighting | |||
Age group (years) | < 65 | 55.1% | ██ ███ | ██ ███ |
≥ 65 | 44.9% | ██ ███ | ██ ███ | |
Disease status | Primary stage III | 9.3% | ██ ███ | ██ ███ |
Primary stage IV | 40.2% | ██ ███ | ██ ███ | |
Recurrent | 50.5% | ██ ███ | ██ ███ | |
ECOG Performance Status | 0: Normal activity | 57.0% | ██ ███ | ██ ███ |
1: Restricted activity | 43.0% | ██ ███ | ██ ███ | |
Histologic type | Endometrioid | 78.5% | ██ ███ | ██ ███ |
Serous | 3.7% | ██ ███ | ██ ███ | |
Other | 17.8% | ██ ███ | ██ ███ | |
Race | American Indian or Alaska Native [wording from original source] | ██ ███ | ██ ███ | ██ ███ |
Asian | ██ ███ | ██ ███ | ██ ███ | |
Black or African American | ██ ███ | ██ ███ | ██ ███ | |
Not reported | ██ ███ | ██ ███ | ██ ███ | |
Other | ██ ███ | ██ ███ | ██ ███ | |
White | ██ ███ | ██ ███ | ██ ███ | |
ECOG = Eastern Cooperative Oncology Group; MAIC = matching-adjusted indirect comparison.
Sources: Details included in the table are from the sponsor’s summary of clinical evidence22 and MAIC technical report.71
Detailed MAIC results for the efficacy end points are presented in Table 25. Briefly, the results for PFS from the anchored, weighted MAIC and the results of OS using the anchored, unweighted MAIC suggested there was insufficient evidence to detect a difference between durvalumab plus SOC and dostarlimab plus SOC. The harms results from the anchored, unweighted MAIC also suggested there was insufficient evidence to detect a difference between durvalumab plus SOC and dostarlimab plus SOC for AEs leading to discontinuation and any SAEs. However, durvalumab plus SOC was favoured over dostarlimab plus SOC for grade 3 or greater AEs.
Critical Appraisal of MAIC
The body of indirect evidence consisted of 2 MAIC analyses informed by an SLR. The procedures as described for screening, data extraction, and the quality assessment steps used were considered generally accepted methods. However, the date on which the SLR was undertaken was not provided, and it is not known whether the most recent publications on any relevant comparators would have been captured in the search. In addition, the search terms were not provided in the submission and there was no description of the numbers of papers retrieved, screened, or extracted. As such, the results from the SLR, including the steps leading to the selection of the comparator study, are not known. Lastly, while the number of reviewers and steps for the assessment of study quality were conducted using generally accepted methods, the submission did not provide the results of the quality assessment; therefore, the risk of bias in the studies is not known. Because both the DUO-E and RUBY Part 1 studies were phase III trials, the risk of bias may be lower; however, the results of the MAIC analyses was based on subgroups from these trials, so the potential for bias remains.
Study | Population | Treatment | Events, n (%) | Unadjusted results | MAIC-adjusted results | ||
|---|---|---|---|---|---|---|---|
Sample size | Unadjusted HR (95% CI) | ESS | Adjusted HR (95% CI) | ||||
Progression-free survival: Anchored, weighted MAIC | |||||||
DUO-E | dMMR | SOC | ██ ███ | ██ ███ | ██ ███ | ██ | ██ ██████ |
SOC + durvalumab | ██ ███ | ██ ███ | |||||
DUO-E | dMMR or MSI-H | SOC | ██ ███ | ██ ███ | ██ ███ | ██ | ██ ██████ |
SOC + durvalumab | ██ ███ | ██ ███ | |||||
RUBY Part 1 | dMMR or MSI-H | SOC | ██ ███ | ██ ███ | ██ ███ | ||
SOC + dostarlimab | ██ ███ | ██ ███ | |||||
Overall survival: Anchored, unweighted MAIC | |||||||
DUO-E | dMMR | SOC | ██ ███ | ██ ███ | ██ ███ | ██ | ██ ██████ |
SOC + durvalumab | ██ ███ | ██ ███ | |||||
DUO-E | dMMR or MSI-H | SOC | ██ ███ | ██ ███ | ██ ███ | ██ | ██ ██████ |
SOC + durvalumab | ██ ███ | ██ ███ | |||||
RUBY Part 1 | dMMR or MSI-H | SOC | ██ ███ | ██ ███ | ██ ███ | ||
SOC + dostarlimab | ██ ███ | ██ ███ | |||||
Any AEs grade 3 or more: Anchored, unweighted MAIC | |||||||
DUO-E | Safety dataset | SOC | ██ ███ | ██ ███ | ██ ███ | ██ | ██ ██████ |
SOC + durvalumab | ██ ███ | ██ ███ | |||||
RUBY Part 1 | Safety dataset | SOC | ██ ███ | ██ ███ | ██ ███ | ██ | |
SOC + dostarlimab | ██ ███ | ██ ███ | |||||
AEs leading to discontinuation of drug or placebo: Anchored, unweighted MAIC | |||||||
DUO-E | Safety dataset | SOC | ██ ███ | ██ ███ | ██ ███ | ██ | ██ ██████ |
SOC + durvalumab | ██ ███ | ██ ███ | |||||
RUBY Part 1 | Safety dataset | SOC | ██ ███ | ██ ███ | ██ ███ | ██ | |
SOC + dostarlimab | ██ ███ | ██ ███ | |||||
Any SAEs: Anchored, unweighted MAIC | |||||||
DUO-E | Safety dataset | SOC | ██ ███ | ██ ███ | ██ ███ | ██ | ██ ██████ |
SOC + durvalumab | ██ ███ | ██ ███ | |||||
RUBY Part 1 | Safety dataset | SOC | ██ ███ | ██ ███ | ██ ███ | ██ | |
SOC + dostarlimab | ██ ███ | ██ ███ | |||||
AE = adverse event; CI = confidence interval; dMMR = mismatch repair deficient; ESS = effective sample size; HR = hazard ratio; MAIC = matching-adjusted indirect comparison; MSI-H = microsatellite instability-high; NA = not applicable; NR = not reported; SAE = serious adverse event; SOC = standard of care.
aNot included in the MAIC analysis.
Sources: Details included in the table are from the sponsor’s summary of clinical evidence22 and the MAIC technical document.71
There are also limitations in the body of evidence that add uncertainty to both MAIC analyses. For both MAIC efficacy analyses, data from a subgroup of the RUBY Part 1 study and a subgroup from the DUO-E trial were used (patients with dMMR and/or MSI-H cancers). In the case of the DUO-E trial, this resulted in the inclusion of some patients with pMMR cancer with MSI-H status in the subgroup used for the analysis, which is likely to somewhat bias the results because MMR status is a known treatment-effect modifier. Likewise, a subgroup from the RUBY Part 1 study was used in the analysis, and it is not known whether the trial itself was adequately powered to detect an effect in the subgroup or whether the subgroup was prespecified. In addition to this, the subgroup from the DUO-E trial is subject to the same limitations that were described in the critical appraisal of the pivotal trial i.e., the dMMR subgroup had inestimable PFS or OS, likely due to immature data. In addition to this, the 2 studies differ considerably in the median follow-up time because the follow-up period in the RUBY Part 1 study was longer than in the DUO-E trial (twice as long follow-up time for PFS). This impacts the interpretation of the results because there is a longer time frame over which events could have occurred, which increases the likelihood of bias to possibly favour durvalumab because there was a shorter follow-up period over which events could accrue. Lastly, in both studies, the subgroup arms had small sample sizes and a low number of events, which impacts the power of the analysis. For both MAICs, the procedure for identifying the characteristics to match on was provided in the submission and the list of characteristics was comprehensive, per the clinical experts. However, only select baseline characteristics were reported after matching; therefore, it is not known whether there are other potential sources of confounding in other baseline clinical and demographic characteristics across studies. Furthermore, there were no details reported on model fit, convergence, or model selection, which is another potential source of bias or uncertainty.
In addition to the limitations in the comparability of the studies and matching, there are additional limitations specific to each efficacy outcome. The MAIC for PFS was a weighted anchored MAIC, and the weighting procedure was largely able to balance the baseline characteristics reported. However, the ESS was approximately ██% smaller (N = ████) than the number of patients prematching (N = 107), suggesting that data from a smaller number of patients may be driving the results, which increases the imprecision. Taken as a whole, these results are subject to high uncertainty due to the limitations in the comparability of the studies as well as the limitations around the size of the ESS. With regard to the MAICs undertaken for the OS and harms outcomes, the results provided in the submission were for unweighted, anchored MAICs that did not employ the propensity score measurement process to reweight the results; thus, this is a largely a naive comparison of the DUO-E and RUBY Part 1 studies. For OS, this was due to the small number of events observed (8 events for dMMR or MSI-H in SOC plus durvalumab and 7 events for SOC plus dostarlimab), which would generate potentially unstable results if reweighted and compared using MAIC methodology. There were still notable differences between the 2 studies at baseline. These included disease status (the proportion of patients with primary stage III disease in the DUO-E study at baseline was 9.3% and in the RUBY Part 1 study, it was 20.3%; the proportion of patients with primary stage IV cancer in the DUO-E study at baseline was 40.2% and in the RUBY Part 1 study it was 29.7%). Furthermore, an ESS was not reported for the OS or harms MAICs, and it is not known how the sample size in the 2 studies compares with the sample in the MAIC. It is highly likely that not all possible effect modifiers were controlled for in the analysis for these outcomes. Taken as a whole, this limitation along with the limitations in the comparability of the studies significantly undermine the results for OS and harms.
Studies Addressing Gaps in the Systematic Review Evidence
The submission did not contain any additional studies addressing gaps in the systematic review evidence.
Discussion
Summary of Available Evidence
This report summarizes the evidence for durvalumab in combination with carboplatin and paclitaxel, followed by treatment with durvalumab monotherapy, for the first-line treatment of adult patients with primary advanced or recurrent dMMR endometrial cancer who are candidates for systemic therapy. The evidence appraisal was based on results from the first IA of an ongoing phase III multisite, double-blind, placebo-controlled RCT (the DUO-E trial), as well as indirect treatment comparisons consisting of 2 MAIC analyses comparing durvalumab plus carboplatin and paclitaxel followed by durvalumab monotherapy, versus dostarlimab, carboplatin, and paclitaxel followed by dostarlimab monotherapy for up to 36 months. No long-term extension studies or studies addressing gaps were included in the review. The primary outcome of the DUO-E trial was PFS, and secondary end points included OS, ORR, DOR, TDT, and HRQoL measures (change from baseline at 18 and 42 weeks in EORTC QLQ-C30 global health status, EORTC QLQ-EN24 pain in back and pelvis score, and EORTC QLQ-EN24 urological symptoms score).
The DUO-E trial randomized patients with either pMMR or dMMR endometrial cancer into all 3 study arms; however, the evidence appraised from the DUO-E trial was based on a subgroup of patients with dMMR status who received SOC (defined as carboplatin and paclitaxel) plus placebo, or SOC plus durvalumab, to align with the Health Canada indication and reimbursement request. In total, 49 patients with dMMR status were enrolled to receive SOC for 6 cycles followed by placebo maintenance therapy, and 46 patients with dMMR status were enrolled to receive SOC plus durvalumab for 6 cycles followed by durvalumab maintenance therapy.
The patients in the dMMR subgroup of the DUO-E trial were approximately evenly split between those with newly diagnosed versus recurrent disease status, and most patients had metastatic disease. Patients with recurrent disease in the SOC plus durvalumab arm had a numerically longer mean time since initial diagnosis, and there were numeric differences between study arms in the proportion of patients who had locally advanced disease, the proportion of patients with debulking surgery history, the proportion of patients with a grade G2 tumour, and in the proportion of patients with an ECOG status of 0 or 1. The clinical experts also noted that the study inclusion and exclusion criteria, as well as the baseline characteristics, broadly characterized patients in the Canadian clinical setting who would be candidates for durvalumab, with the exception of patients with an ECOG status greater than 1, because those patients would likely be offered treatment.
Interpretation of Results
Efficacy
The clinical experts consulted for this review indicated that OS is the clinical outcome of most interest, and the clinician group input indicated that PFS is also an important outcome. Per FDA guidance, PFS — which was the primary end point of the DUO-E study — can be used for regulatory approval depending on other factors such as effect size, duration, and the benefits of other available therapies.72 The clinical experts consulted for this review noted there is a current unmet need for treatment options in patients with dMMR endometrial cancer because some patients do not experience a response to current therapies and, in general, patients on chemotherapy alone tend to experience disease progression despite initial treatment response. With the addition of ICI therapy, the experts noted that many patients do not experience disease progression; however, dostarlimab — which received a recommendation to reimburse in May 2024 — remains the only ICI available in Canada for these patients. Both the patient and clinician groups also noted that toxicities related to current treatments impact HRQoL. The outcomes in the DUO-E trial included survival outcomes and HRQoL measures; therefore, the study evaluated several outcomes of importance to patients and clinicians.
PFS and OS were the primary and secondary outcomes of the DUO-E trial. The clinical experts consulted for this review agreed that a PFS benefit of 15% to 20% at 12 months, and a 10% to 15% benefit at 18 months, would be clinically meaningful. Based on this threshold, the results of the DUO-E study suggest that durvalumab plus SOC may increase the proportion of patients who do not experience any disease progression at 12 months and 18 months relative to SOC alone. However, the PFS analysis was only prespecified to be undertaken once a specific number of events was reached in the ITT population and not in the dMMR subgroup; thus, these results can only be considered supportive of the overall effect of durvalumab. In the SOC plus durvalumab arm, the median PFS was not reached (51.0% of patients in the SOC arm and 32.6% of patients in the SOC plus durvalumab arm had reported a PFS event at the data cut-off). The clinical experts consulted for this review noted that the point estimate for the HR was compelling; however, it had a wide CI, suggesting imprecision in the results. The wide CI and inestimable median PFS suggests that the results as of the data cut-off were immature for patients with dMMR endometrial cancer based on an information fraction of ████% (██ events per ████ patients across both study arms), although the information fraction for the ITT population was 61.0%. This implies there is high uncertainty in the results observed, and the confidence with which the results predict PFS in the long-term is not clear.
Similarly, for OS, the clinical experts consulted for this review agreed that an OS benefit of 10% at 12 months and 10% at 18 months would be clinically meaningful. Based on this threshold, the results of the DUO-E study suggest that durvalumab plus SOC may increase the proportion of patients alive at 12 months and at 18 months relative to SOC alone. The first IA for OS was prespecified to occur when the criteria for the PFS analysis were met; however, at this data cut-off, the median OS was not reached for either the SOC arm or the SOC plus durvalumab arm in the dMMR subgroup, and only ███████ OS events had occurred, suggesting immaturity in the results. Similarly, the point estimates of the HR were considered compelling, according to the clinical experts. However, the estimate also had a wide CI, suggesting imprecision. Furthermore, the analysis for all survival outcomes in the subgroup was conducted using an unstratified model and was not controlled for multiple comparison. Overall, this suggests that the survival data at the data cut-off are immature for patients with dMMR cancer and, combined with the study limitations, this implies there is high uncertainty in the results; therefore, the confidence with which the results predict OS in the long-term is not clear.
HRQoL was assessed in the DUO-E study using 2 cancer-related measures, the EORTC QLQ-C30 and EORTC QLQ-EN24, which were assessed at 18 and 42 weeks. The results for HRQoL were impacted by high levels of attrition, with only 55% to 65% of patients completing HRQoL measures at week 18, and 54% to 61% completing HRQoL measures at week 42. The HRQoL measures were modelled using a mixed model for repeated measures with no imputation for missing data, and the model assumes that data are missing at random. This assumption is likely inappropriate because the study design is based on time-to-event principles; therefore, patients who are lost to follow-up are most likely lost because they had an event. Given the high attrition rate and missing data, the results for HRQoL are uncertain and may be biased. Lastly, there are no MIDs for these measures in patients with endometrial cancer, so the clinical impact of the results is unclear. Despite the limitations in data availability, the choice of model, and the lack of MIDs, the results do not suggest any explicit detriment in HRQoL; however, firm conclusions regarding the impact of durvalumab plus SOC followed by durvalumab maintenance on HRQoL cannot be drawn.
The clinical experts noted that in this setting, dostarlimab would be the main comparator for durvalumab. As such, 2 sponsor-submitted MAIC analyses assessing the comparative PFS and OS of durvalumab plus SOC from the DUO-E study and dostarlimab plus SOC from the RUBY Part 1 trial were included in this review. The heterogeneity in the patient populations with respect to mutation status, the immaturity of results for each trial, and the substantial reduction in ESS increase the risk of bias in the results as well as the uncertainty in the effect estimates. The results from the PFS analysis suggested there was no difference between the 2 treatments, but the results are highly uncertain due to the described limitations. The OS analysis was an anchored unweighted MAIC and although the results suggested there was no difference between durvalumab and dostarlimab, the limitations in the analysis and the body of evidence did not allow conclusions to be drawn regarding the comparative efficacy of the treatments for this outcome. As noted, results for both outcomes were associated with wide 95% CIs, reducing the precision of the results.
The indication is for first-line therapy for patients with dMMR cancer who are candidates for systemic therapy. The results from the DUO-E trial were from a relatively small sample size and there were no sensitivity analyses provided to determine the robustness of the results across subgroups (for example, patients with recurrent versus newly diagnosed cancer). In addition, in the dMMR subgroup, the total treatment exposure was ████ weeks in the SOC plus durvalumab arm; therefore, longer-term efficacy and safety results are unavailable, which is important in the context of a treatment that is intended to be administered for potentially long periods of time. The clinical experts also referenced pembrolizumab as a potential future comparator in the first-line setting. This drug is currently under review by CDA-AMC in combination with chemotherapy for the treatment of adult patients with primary advanced or recurrent endometrial cancer, followed by monotherapy.73 The experts noted that they would consider all 3 therapies to be interchangeable, although if a patient’s cancer failed to respond to a therapy, they would not re-trial with a treatment in that same drug class. Therefore, the list of relevant comparators may change for durvalumab, and the approval of durvalumab may impact therapeutic options in later lines of therapy.
Harms
Harms were reported for both the “overall phase” of the DUO-E study (i.e., including both the chemotherapy and maintenance phases) and the maintenance phase alone. Although the overall frequency of AEs was similar between the maintenance phase and overall phase (100% in the SOC and SOC plus durvalumab groups in the overall phase versus 88.0% and 93.9% in the SOC and SOC plus durvalumab groups in the maintenance phase), the frequency of individual AEs was higher in the overall phase of the study. Though harms results for the chemotherapy phase alone were not provided, it is expected that the increased harms in the overall phase may be due to chemotherapy; however, this is unclear.
As noted, all patients experienced AEs during the study, and the majority of patients (60.9% of patients in the SOC arm and 52.3% of patients in the SOC plus durvalumab arm) experienced an AE with a maximum grade of 3 or 4. The most commonly reported AEs (of any grade) included nausea, alopecia, arthralgia, and anemia. One expert noted that rates of alopecia (41.3% of patients in the SOC arm and 52.3% of patients in the SOC plus durvalumab arm) were lower in the DUO-E study than what they would expect to see in practice with the chemotherapy regimen of carboplatin and paclitaxel because this regimen causes grade 2 alopecia in nearly all patients, which raises questions about the accuracy of harms reporting in this study. SAEs were reported in 32.6% of patients in the SOC arm and 29.5% in the SOC plus durvalumab arm. The most commonly reported SAEs were gastrointestinal disorders (11.4% in the SOC plus durvalumab arm and 4.3% in the SOC arm), blood and lymphatic system disorders (10.9% in the SOC arm and 2.3% in the SOC plus durvalumab arm), infections and infestations (6.5% in the SOC arm and 6.8% in the SOC plus durvalumab arm), and renal and urinary disorders (8.7% in the SOC arm and 0% in the SOC plus durvalumab arm).
The majority of patients in both study arms also experienced an AESI identified by the clinical experts for this review. The causes of death in the study were not reported, which is an important limitation because it is not known whether disease progression, immune suppression, or other factors caused the deaths reported. Overall, the clinical experts consulted for this review noted that the degree and types of harms reported to date were similar to what is typically expected for patients undergoing chemotherapy with an ICI. The comparative safety of durvalumab was analyzed in the indirect evidence using anchored, unweighted MAICs. As noted, the comparative safety remains uncertain, given the limitations in the sponsor-submitted indirect evidence.
The DUO-E study is ongoing; therefore, the harms reporting is not yet complete and some additional harms may not have been captured in the data. In addition, the lack of a long-term extension study means that long-term harms data are not available. However, durvalumab has been available for the treatment of other malignancies since 2017, so the AE profile is generally well understood.
Conclusion
One phase III, ongoing, multicentre, double-blind, placebo-controlled RCT (the DUO-E trial) provided evidence for the efficacy and safety of durvalumab plus SOC for 6 cycles followed by durvalumab maintenance therapy as first-line treatment in the subgroup of 95 adult patients with primary advanced or recurrent dMMR endometrial cancer.
Patients and clinicians identified a need for new treatments for primary advanced or recurrent endometrial cancer — which is the most common gynecological malignancy — and prolonged survival was considered the most important outcome. In the subgroup of patients with dMMR endometrial cancer, the subgroup results of the DUO-E trial suggested improved PFS and OS in patients treated with durvalumab. The clinical experts consulted for this review considered these results compelling, and the results of the GRADE assessment suggested that the MIDs for the proportion of patients who do not experience disease progression or who are alive at 12 and 18 months were surpassed. However, because these results were based on a subgroup of the ITT population, statistical analyses were not powered to detect differences in this subgroup, and no adjustments were made for multiple testing. Furthermore, although the median results for time-to-event end points were not reached, the certainty in the magnitude of benefit was reduced because the results were based on a small sample size and an IA with largely immature data based on an information fraction of ████% for the subgroup and treatment arms presented. Furthermore, despite results for HRQoL that suggested no detriment in patients receiving durvalumab, there was a high rate of attrition. As such, the results of these important efficacy outcomes can only be considered supportive of the overall effect of durvalumab and causal conclusions cannot be drawn. Indirect evidence comparing durvalumab with dostarlimab, a relevant comparator in the Canadian clinical setting, suggested no difference between the 2 treatment regimens for PFS; however, the considerable limitations in the indirect evidence preclude conclusions on efficacy (as measured by OS) and harms. While the reporting of harms in the DUO-E study is also immature and lacks important details, in particular with regard to AESIs and the specific causes of death, the clinical experts consulted for this review considered the harms to be typical for patients undergoing chemotherapy with an ICI.
References
1.Makker V, MacKay H, Ray-Coquard I, et al. Endometrial cancer. Nat Rev Dis Primers. 2021;7(1):88. doi: 10.1038/s41572-021-00324-8 PubMed
2.Canadian Cancer Society. Cancerous tumours of the uterus. 2024. https://cancer.ca/en/cancer-information/cancer-types/uterine/what-is-uterine-cancer/cancerous-tumours [sponsor submitted reference]
3.Ontario Health (Cancer Care Ontario). Ontario Cancer Statistics 2022. 2022. Accessed November 26, 2024. https://www.cancercareontario.ca/en/data-research/view-data/statistical-reports/ontario-cancer-statistics-2022/ch-4-cancers-incidence-2022
4.Canadian Cancer Society. Uterine Cancer Statistics. 2024. Accessed November 25, 2024. https://cancer.ca/en/cancer-information/cancer-types/uterine/statistics
5.National Cancer Institute. PDQ® Screening and Prevention Editorial Board. PDQ Endometrial Cancer Prevention. 2024. Accessed November 25, 2024. https://www.cancer.gov/types/uterine/hp/endometrial-prevention-pdq
6.American Cancer Society. Endometrial cancer stages. 2019. Accessed November 25, 2024. https://www.cancer.org/cancer/types/endometrial-cancer/detection-diagnosis-staging/staging.html
7.Canadian Cancer Society. Survival Statistics for Uterine Cancer. 2024. Accessed November 25, 2024. https://cancer.ca/en/cancer-information/cancer-types/uterine/prognosis-and-survival/survival-statistics
8.American Cancer Society. Signs and Symptoms of Endometrial Cancer. Accessed October 5, 2023. https://www.cancer.org/cancer/types/endometrial-cancer/detection-diagnosis-staging/signs-and-symptoms.html [sponsor submitted reference]
9.Braun MM, Overbeek-Wager EA, Grumbo RJ. Diagnosis and management of endometrial cancer. Am Fam Physician. 2016;93(6):468-474. PubMed
10.Passarello K, Kurian S, Villanueva V. Endometrial Cancer: An Overview of Pathophysiology, Management, and Care. Semin Oncol Nurs. 2019;35(2):157-165. doi: https://doi.org/10.1016/j.soncn.2019.02.002 PubMed
11.Ferrandina G, Petrillo M, Mantegna G, et al. Evaluation of quality of life and emotional distress in endometrial cancer patients: A 2-year prospective, longitudinal study. Gynecol Oncol. 2014;133(3):518-525. doi: https://doi.org/10.1016/j.ygyno.2014.03.015 PubMed
12.Mahdy H, Casey MJ, Vadakekut ES, Crotzer D. Endometrial Cancer. StatPearls. StatPearls Publishing; 2024.
13.Canadian Cancer Society. Diagnosis of Uterine Cancer. 2024. Accessed November 25, 2024. https://cancer.ca/en/cancer-information/cancer-types/uterine/diagnosis
14.Yang Y, Wu SF, Bao W. Molecular subtypes of endometrial cancer: Implications for adjuvant treatment strategies. Int J Gynaecol Obstet. 2024;164(2):436-459. doi: 10.1002/ijgo.14969 PubMed
15.Boland CR, Thibodeau SN, Hamilton SR, et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998;58(22):5248-57. PubMed
16.Mendiola M, Heredia-Soto V, Ruz-Caracuel I, et al. Comparison of Methods for Testing Mismatch Repair Status in Endometrial Cancer. Int J Mol Sci. 2023;24(19). doi: 10.3390/ijms241914468 PubMed
17.European Medicines Agency. IMFINZI (durvalumab) SmPC. Accessed November 2023. https://www.ema.europa.eu/en/documents/product-information/imfinzi-epar-product-information_en.pdf [sponsor submitted reference]
18.AstraZeneca. Data on File. DUO-E Mechanism of Action. 2024 [sponsor submitted reference].
19.IMFINZI (durvalumab). Product Monograph [draft]. AstraZeneca Canada Inc. [sponsor submitted reference].
20.Balshem H, Helfand M, Schünemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-6. doi: 10.1016/j.jclinepi.2010.07.015 PubMed
21.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. doi: 10.1016/j.jclinepi.2019.10.014 PubMed
22.Astrazeneca Inc. Sponsor's Clinical Report Template: Durvalumab plus Olaparib (IMFINZI® plus LYNPARZA®) [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: durvalumab, carboplatin, paclitaxel. October 16, 2024.
23.Astrazeneca Inc. Astrazeneca Inc. response to Canada's Drug Agency request for additional information regarding durvalumab, carboplatin, paclitaxel CADTH review [internal additional sponsor's information]. November 21, 2024.
24.Astrazeneca Inc. Astrazeneca Inc. response to Canada's Drug Agency request for additional information regarding durvalumab, carboplatin, paclitaxel CADTH review [internal additional sponsor's information]. December 4, 2024.
25.Kurnit KC, Westin SN, Coleman RL. Microsatellite instability in endometrial cancer: New purpose for an old test. Cancer. 2019;125(13):2154-2163. doi: 10.1002/cncr.32058 PubMed
26.Abu-Rustum N, Yashar C, Arend R, et al. Uterine Neoplasms, Version 1.2023, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2023;21(2):181-209. doi: 10.6004/jnccn.2023.0006 PubMed
27.Deshpande M, Romanski PA, Rosenwaks Z, Gerhardt J. Gynecological Cancers Caused by Deficient Mismatch Repair and Microsatellite Instability. Cancers (Basel). 2020;12(11). doi: 10.3390/cancers12113319 PubMed
28.Abu-Rustum N, Yashar C, Arend R, et al. Uterine Neoplasms, Version 2.2024, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2024.
29.Oaknin A, Bosse TJ, Creutzberg CL, et al. Endometrial cancer: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann Oncol. 2022;33(9):860-877. doi: 10.1016/j.annonc.2022.05.009 PubMed
30.Campos SM, Cohn DE. Initial treatment of metastatic endometrial cancer. UptoDate; 2025. Accessed January 8, 2025. https://www.uptodate.com/
31.Campos SM, Cohn DE. Subsequent line systemic therapy for metastatic endometrial cancer. UpToDate; 2025. Accessed January 8, 2025. https://www.uptodate.com/
32.CDA provisional funding algorithm for Endometrial Cancer 2024. Accessed December 5, 2024. https://www.cda-amc.ca/endometrial-cancer [sponsor submitted reference]
33.Cancer Care Ontario. Drug Formulary: paclitaxel. Accessed December 5, 2024. https://www.cancercareontario.ca/en/drugformulary/drugs/monograph/44151
34.JEMPERLI (dostarlimab). GlaxoSmithKline Inc. Product Monograph. Apr 2024. [sponsor submitted reference].
35.CARBOPLATIN INJECTION BP. Pfizer Canada ULC. Product Monograph. Aug 2023. [sponsor submitted reference].
36.ABRAXANE® for Injectable Suspension (paclitaxel). Bristol-Myers Squibb Canada. Product Monograph. Mar 2024. [sponsor submitted reference].
37.Berek JS, Matias-Guiu X, Creutzberg C, et al. FIGO staging of endometrial cancer: 2023. Int J Gynaecol Obstet. 2023;162(2):383-394. doi: 10.1002/ijgo.14923 PubMed
38.Concin N M-GX, Vergote I, et al. ESGO/ESTRO/ESP guidelines for the management of patients with endometrial carcinoma. Int J Gynecol Cancer. 2021;31(1):12-39. doi: 10.1136/ijgc-2020-002230 PubMed
39.Yoshida H, Takigawa W, Kobayashi-Kato M, Nishikawa T, Shiraishi K, Ishikawa M. Mismatch Repair Protein Expression in Endometrial Cancer: Assessing Concordance and Unveiling Pitfalls in Two Different Immunohistochemistry Assays. J Pers Med. 2023;13(8). doi: 10.3390/jpm13081260 PubMed
40.Bateman AC. DNA mismatch repair proteins: scientific update and practical guide. J Clin Pathol. 2021;74(4):264-268. doi: 10.1136/jclinpath-2020-207281 PubMed
41.BC Cancer. Endometrium. 2023. Accessed January 8, 2025. http://www.bccancer.bc.ca/health-professionals/clinical-resources/cancer-management-manual/gynecology/endometrium
42.Aiyer KTS, Doeleman T, Ryan NA, et al. Validity of a two-antibody testing algorithm for mismatch repair deficiency testing in cancer; a systematic literature review and meta-analysis. Mod Pathol. 2022;35(12):1775-1783. doi: 10.1038/s41379-022-01149-w PubMed
43.Vikas P MH, Compton C, et al. Mismatch Repair and Microsatellite Instability Testing for Immune Checkpoint Inhibitor Therapy: ASCO Endorsement of College of American Pathologists Guideline. J Clin Oncol 2023;41(10):1943-1948. doi: 10.1200/JCO.22.02462 PubMed
44.Bartley AN MA, Konnick E, et al. Mismatch Repair and Microsatellite Instability Testing for Immune Checkpoint Inhibitor Therapy: Guideline From the College of American Pathologists in Collaboration With the Association for Molecular Pathology and Fight Colorectal Cancer. Arch Pathol Lab Med. 2022;146(10):1194-1210. doi: 10.5858/arpa.2021-0632-CP PubMed
45.Lawrence J RL, Arseneau J, Zeng X, Chong G, Weber E, Foulkes W, Palma L. Mismatch Repair Universal Screening of Endometrial Cancers (MUSE) in a Canadian Cohort. Current Oncology. 2021;28(1):509-522. doi: https://doi.org/10.3390/curroncol28010052 PubMed
46.AstraZeneca Inc. Sponsor BIA dMMR [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: durvalumab, carboplatin, paclitaxel. October 16, 2024.
47.AstraZeneca Inc. Sponsor BIA pMMR [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: durvalumab, carboplatin, paclitaxel. October 16, 2024.
48.AstraZeneca. DUO-E Clinical Study Report: A Randomised, Multicentre, Double-blind, Placebo-controlled, Phase III Study of First-line Carboplatin and Paclitaxel in Combination with Durvalumab, Followed by Maintenance Durvalumab with or without Olaparib in Patients with Newly Diagnosed Advanced or Recurrent Endometrial Cancer. 2023 [sponsor submitted reference].
49.AstraZeneca Inc. DUO-E. Appendix 16.1.1 Protocol and Protocol Amendments (Version 6). 24 Jan 2023. [sponsor submitted reference].
50.Astrazeneca Inc. Astrazeneca Inc. response to Canada's Drug Agency request for additional information regarding durvalumab, carboplatin, paclitaxel CADTH review [internal additional sponsor's information]. November 15, 2024.
51.Aaronson NK, Ahmedzai S, Bergman B, et al. The European Organization for Research and Treatment of Cancer QLQ-C30: a quality-of-life instrument for use in international clinical trials in oncology. JNCI: Journal of the National Cancer Institute. 1993;85(5):365-376. PubMed
52.Osoba D, Rodrigues G, Myles J, Zee B, Pater J. Interpreting the significance of changes in health-related quality-of-life scores. J Clin Oncol. 1998;16(1):139-144. doi: 10.1200/JCO.1998.16.1.139 PubMed
53.Greimel E, Nordin A, Lanceley A, et al. Psychometric validation of the European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire-Endometrial Cancer Module (EORTC QLQ-EN24). Eur J Cancer. 2011;47(2):183-90. doi: 10.1016/j.ejca.2010.08.014 PubMed
54.Osoba D, Zee B, Pater J, Warr D, Kaizer L, Latreille J. Psychometric properties and responsiveness of the EORTC quality of Life Questionnaire (QLQ-C30) in patients with breast, ovarian and lung cancer. Qual Life Res. 1994;3(5):353-64. doi: 10.1007/bf00451727 PubMed
55.Zhao H, Kanda K. Translation and validation of the standard Chinese version of the EORTC QLQ-C30. Qual Life Res. 2000;9(2):129-37. doi: 10.1023/a:1008981520920 PubMed
56.Davda J, Kibet H, Achieng E, Atundo L, Komen T. Assessing the acceptability, reliability, and validity of the EORTC Quality of Life Questionnaire (QLQ-C30) in Kenyan cancer patients: a cross-sectional study. Journal of Patient-Reported Outcomes. 2021;5(1):4. doi: 10.1186/s41687-020-00275-w PubMed
57.Butler L, Bacon M, Carey M, Zee B, Tu D, Bezjak A. Determining the relationship between toxicity and quality of life in an ovarian cancer chemotherapy clinical trial. J Clin Oncol. 2004;22(12):2461-8. doi: 10.1200/jco.2004.01.106 PubMed
58.Musoro JZ, Coens C, Sprangers MAG, et al. Minimally important differences for interpreting EORTC QLQ-C30 change scores over time: A synthesis across 21 clinical trials involving nine different cancer types. Eur J Cancer. 2023;188:171-182. doi: 10.1016/j.ejca.2023.04.027 PubMed
59.King MT, Bell ML, Costa D, Butow P, Oh B. The Quality of Life Questionnaire Core 30 (QLQ-C30) and Functional Assessment of Cancer-General (FACT-G) differ in responsiveness, relative efficiency, and therefore required sample size. J Clin Epidemiol. 2014;67(1):100-7. doi: 10.1016/j.jclinepi.2013.02.019 PubMed
60.Cocks K, King MT, Velikova G, Fayers PM, Brown JM. Quality, interpretation and presentation of European Organisation for Research and Treatment of Cancer quality of life questionnaire core 30 data in randomised controlled trials. Eur J Cancer. 2008;44(13):1793-8. doi: 10.1016/j.ejca.2008.05.008 PubMed
61.Gallardo-Rincón D, Toledo-Leyva A, Bahena-González A, et al. Validation of the QLQ-EN24 instrument for the assessment of health-related quality of life for women with endometrial cancer in México. Arch Gynecol Obstet. 2021;304(3):773-782. doi: 10.1007/s00404-021-05990-3 PubMed
62.Stukan M, Zalewski K, Mardas M, et al. Independent psychometric validation of European Organization for Research and Treatment of Cancer Quality of Life Questionnaire-Endometrial Cancer Module (EORTC QLQ-EN24). Eur J Cancer Care (Engl). 2018;27(1). doi: 10.1111/ecc.12639 PubMed
63.Miller D, Filiaci V, Fleming G, et al. Late-Breaking Abstract 1: Randomized phase III noninferiority trial of first line chemotherapy for metastatic or recurrent endometrial carcinoma: A Gynecologic Oncology Group study. Gynecol Oncol. 2012;125(3):771.
64.Aghajanian C, Filiaci V, Dizon DS, et al. A phase II study of frontline paclitaxel/carboplatin/bevacizumab, paclitaxel/carboplatin/temsirolimus, or ixabepilone/carboplatin/bevacizumab in advanced/recurrent endometrial cancer. Gynecol Oncol. 2018;150(2):274-281. doi: 10.1016/j.ygyno.2018.05.018 PubMed
65.Gail M, Simon R. Testing for qualitative interactions between treatment effects and patient subsets. Biometrics. 1985;41(2):361-72. PubMed
66.Gordon Lan KK, DeMets DL. Discrete sequential boundaries for clinical trials. Biometrika. 1983;70(3):659-663. doi: 10.2307/2336502
67.AstraZeneca. DUO-E Clinical Study Protocol: A Randomised, Multicentre, Double-blind, Placebo-controlled, Phase III Study of First-line Carboplatin and Paclitaxel in Combination with Durvalumab, Followed by Maintenance Durvalumab with or without Olaparib in Patients with Newly Diagnosed Advanced or Recurrent Endometrial Cancer (DUO-E). 2023 [sponsor submitted reference].
68.Astrazeneca Inc. Astrazeneca Inc. response to Canada's Drug Agency request for additional information regarding durvalumab, carboplatin, paclitaxel CADTH review: [internal additional sponsor's information]. October 28, 2024.
69.Astrazeneca Inc. Astrazeneca Inc. response to Canada's Drug Agency request for additional information regarding durvalumab, carboplatin, paclitaxel CADTH review [internal additional sponsor's information]. November 7, 2024.
70.Phillippo DM, Ades AE, Dias S, Palmer S, Abrams KR, Welton NJ. NICE DSU technical support document 18: methods for population-adjusted indirect comparisons in submissions to NICE. 2016 [sponsor submitted reference].
71.AstraZeneca Canada Inc. Indirect treatment comparison of durvalumab plus chemotherapy versus dostarlimab plus chemotherapy in MMR-deficient and/or MSI-H primary advanced or recurrent endometrial cancer [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: durvalumab (Imfinzi) 50 mg/mL, concentrate for IV infusion. October 16, 2024.
72.U.S. Food & Drug Administration. Clinical Trial Endpoints for the Approval of Cancer Drugs and Biologics Guidance for Industry 2018. Accessed December 18, 2024. https://www.fda.gov/media/71195/download
73.Canada’s Drug Agency. Pembrolizumab for advanced or recurrent endometrial carcinoma. 2025. Accessed December 19 2024. https://www.cda-amc.ca/pembrolizumab-18
Appendix 1: Detailed Outcome Data
Please note that this appendix has not been copy-edited.
Table 26: Most Common (20% of Patients or More) Permitted Concomitant Medications — ITT
Exposure | DUO-E study ITT population | ||
|---|---|---|---|
SOC (N = 241) | SOC + durvalumab (N = 238) | SOC + durvalumab + olaparib (N = 239) | |
Number of patients with allowed concomitant medication, n (%) | ███ ██████ | ███ ██████ | ███ ██████ |
Serotonin (5HT3) antagonists | ███ ██████ | ███ ██████ | ███ ██████ |
Ondansetron | ███ ██████ | ███ ██████ | ███ ██████ |
Glucocorticoids | ███ ██████ | ███ ██████ | ███ ██████ |
Dexamethasonea | ███ ██████ | ███ ██████ | ███ ██████ |
H2 receptor antagonists | ███ ██████ | ███ ██████ | ███ ██████ |
Famotidine | ███ ██████ | ███ ██████ | ███ ██████ |
Anilides | ███ ██████ | ███ ██████ | ███ ██████ |
Acetaminophen | ███ ██████ | ███ ██████ | ███ ██████ |
Aminoalkyl ethers | ███ ██████ | ███ ██████ | ███ ██████ |
Diphenhydramine | ███ ██████ | ███ ██████ | ███ ██████ |
5HT3 = serotonin; CSP = clinical study protocol; FAS = full analysis set; H2 = histamine type 2; ITT = intention to treat; SOC = standard of care (carboplatin + paclitaxel).
Note: Included medication started on or after the date of first dose of durvalumab, olaparib, or placebo up until the initiation of the first subsequent anticancer therapy following discontinuation of study treatment or until the end of the safety follow-up period (latest of either 30 days following discontinuation of olaparib/placebo or 90 days following discontinuation of durvalumab/placebo), whichever occurred first.
aThere was a restriction change for dexamethasone (from restricted to allowed) that was implemented in version 3.0 of the CSP (April 15, 2021).
Source: Details included in the table are from the sponsor’s summary of clinical evidence.22
Table 27: Most Common (2 Patients or More) Disallowed Concomitant Medications — ITT
Exposure | DUO-E study ITT population | ||
|---|---|---|---|
SOC (N = 241) | SOC + durvalumab (N = 238) | SOC + durvalumab + olaparib (N = 239) | |
Number of patients with disallowed concomitant medication | ███ ██████ | ███ ██████ | ███ ██████ |
Glucocorticoids | ███ ██████ | ███ ██████ | ███ ██████ |
Dexamethasonea | ███ ██████ | ███ ██████ | ███ ██████ |
Prednisone | ███ ██████ | ███ ██████ | ███ ██████ |
Prednisolone | ███ ██████ | ███ ██████ | ███ ██████ |
Herbal immunomodulators | ███ ██████ | ███ ██████ | ███ ██████ |
Diyushengbai tablet | ███ ██████ | ███ ██████ | ███ ██████ |
Appetite stimulants | ███ ██████ | ███ ██████ | ███ ██████ |
Megestrol acetate (Megace F suspension) | ███ ██████ | ███ ██████ | ███ ██████ |
Herbal anticancer remedies | ███ ██████ | ███ ██████ | ███ ██████ |
Kanglaite injection | ███ ██████ | ███ ██████ | ███ ██████ |
ITT = intention to treat; SOC = standard of care (carboplatin and paclitaxel).
Note: Included medication started on or after the date of first dose of durvalumab, olaparib, or placebo up until the initiation of the first subsequent anticancer therapy following discontinuation of study treatment or until the end of the safety follow-up period (latest of either 30 days following discontinuation of olaparib or placebo or 90 days following discontinuation of durvalumab or placebo), whichever occurred first.
aThere was a restriction change for dexamethasone (from restricted to allowed) that was implemented in clinical study protocol version 3.0 (April 15, 2021).
Source: Details included in the table are from the sponsor’s summary of clinical evidence.22
Table 28: Results of the Survival Analysis of the ITT Population From the DUO-E Study
Variable | ITT population | ||
|---|---|---|---|
SOC N = 241 | SOC + durvalumab N = 238 | SOC + durvalumab + olaparib N = 239 | |
PFS: Primary outcomea | |||
Total events, n (%)b | 173 (71.8) | 139 (58.4) | 126 (52.7) |
Censored patients, n (%) | ███ ██████ | ███ ██████ | ███ ██████ |
Progression-free disease at time of analysis | ███ ██████ | ███ ██████ | ███ ██████ |
Censored deathc | ███ ██████ | ███ ██████ | ███ ██████ |
Withdrawn consent | ███ ██████ | ███ ██████ | ███ ██████ |
No postbaseline evaluable tumour assessment | ███ ██████ | ███ ██████ | ███ ██████ |
Censored RECIST progressiond | ███ ██████ | ███ ██████ | ███ ██████ |
Lost to follow-up | ███ ██████ | ███ ██████ | ███ ██████ |
Duration of follow-up in censored patients (months), median (range) | ███ ██████ | ███ ██████ | ███ ██████ |
PFS (months), median (95% CI) | 9.6 (9.0 to 9.9) | 10.2 (9.7 to 14.7) | 15.1 (12.6 to 20.7) |
HR (95% CI) vs. SOCe | NA | 0.71 (0.57 to 0.89) | 0.55 (0.43 to 0.69) |
P value vs. SOC | NA | 0.003 | < 0.0001 |
Proportion of patients whose cancer was progression-free, % (95% CI) | |||
At 6 months | 82.5 (76.9 to 86.8) | 83.8 (78.4 to 88.0) | 83.9 (78.6 to 88.0) |
Survival difference (95% CI) vs. SOC | NA | NR | NR |
At 12 months | 41.1 (34.6 to 47.5) | 48.5 (41.8 to 54.9) | 61.5 (54.9 to 67.4) |
Survival difference (95% CI) vs. SOC | NA | NR | NR |
At 18 months | 21.7 (16.0 to 27.9) | 37.8 (31.0 to 44.5) | 46.3 (39.2 to 52.0) |
Survival difference (95% CI) vs. SOC | NA | NR | NR |
OS: Secondary outcomea | |||
Deaths, n (%) | ███ ██████ | ███ ██████ | ███ ██████ |
Censored patients, n (%) | ███ ██████ | ███ ██████ | ███ ██████ |
Still in survival follow-up | ███ ██████ | ███ ██████ | ███ ██████ |
Terminated before deathf | ███ ██████ | ███ ██████ | ███ ██████ |
Withdrawn consent | ███ ██████ | ███ ██████ | ███ ██████ |
Lost to follow-up | ███ ██████ | ███ ██████ | ███ ██████ |
Duration of follow-up in censored patients (months), median (range) | 18.6 (0.5 to 32.9) | 18.4 (2.1 to 33.0) | 18.7 (1.1 to 33.4) |
OS (months), median (95% CI) | 25.9 (23.9 to NR) | NR | NR |
HR (95% CI) vs. SOC | NA | 0.77 (0.56 to 1.07) | 0.59 (0.42 to 0.83) |
P value | NA | 0.120 | 0.003 |
Proportion (95% CI) of patients who were alive | |||
At 6 months | ███ ██████ | ███ ██████ | ███ ██████ |
Survival difference (95% CI) vs. SOC | ███ ██████ | ███ ██████ | ███ ██████ |
At 12 months | ███ ██████ | ███ ██████ | ███ ██████ |
Survival difference (95% CI) vs. SOC | ███ ██████ | ███ ██████ | ███ ██████ |
At 18 months | ███ ██████ | ███ ██████ | ███ ██████ |
Survival difference (95% CI) vs. SOC | ███ ██████ | ███ ██████ | ███ ██████ |
ORR | |||
Number of patients with response out of number of patients with measurable disease at baseline, n/N (%) | ███ ██████ | ███ ██████ | ███ ██████ |
OR (95% CI) vs. SOC | ███ ██████ | ███ ██████ | ███ ██████ |
CR (%) | ███ ██████ | ███ ██████ | ███ ██████ |
PR (%) | ███ ██████ | ███ ██████ | ███ ██████ |
DOR | |||
Number of responders who died or whose cancer subsequently progressed out of number of patients whose cancer responded, n/N (%) | ███ ██████ | ███ ██████ | ███ ██████ |
DOR from onset of response (months), median | ███ ██████ | ███ ██████ | ███ ██████ |
Proportion of patients remaining in response (%) | |||
6 months | ███ ██████ | ███ ██████ | ███ ██████ |
12 months | ███ ██████ | ███ ██████ | ███ ██████ |
18 months | ███ ██████ | ███ ██████ | ███ ██████ |
24 months | ███ ██████ | ███ ██████ | ███ ██████ |
TDT | |||
Total events, n (%) | ███ ██████ | ███ ██████ | ███ ██████ |
Treatment discontinuation | ███ ██████ | ███ ██████ | ███ ██████ |
Death | ███ ██████ | ███ ██████ | ███ ██████ |
Censored patients, n (%) | ███ ██████ | ███ ██████ | ███ ██████ |
Median TDT (95% CI), (months) | ███ ██████ | ███ ██████ | ███ ██████ |
HR (95% CI) vs. SOC | ███ ██████ | ███ ██████ | ███ ██████ |
P value | ███ ██████ | ███ ██████ | ███ ██████ |
Event-free rate (95% CI) | |||
At 6 months | ███ ██████ | ███ ██████ | ███ ██████ |
At 12 months | ███ ██████ | ███ ██████ | ███ ██████ |
At 18 months | ███ ██████ | ███ ██████ | ███ ██████ |
CI = confidence interval; CR = complete response; dMMR = mismatch repair deficient; DOR = duration of response; HR = hazard ratio; ITT = intention to treat; MMR = mismatch repair; OR = odds ratio; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; PR = partial response; NA = not applicable; NR = not reported; RECIST = Response Evaluation Criteria in Solid Tumours; SOC = standard of care (carboplatin + paclitaxel); TDT = time to study treatment discontinuation or death; vs. = versus.
aControlled for multiple comparisons.
bPatients who have not died or whose cancer has not progressed (or who have died or whose cancer has progressed after 2 missed assessments) are censored at the latest visit (or the latest visit before the 2 missed assessments), or at day 1.
cDeath that occurred after 2 or more missed RECIST assessments in the absence of progression.
dProgression event occurred after 2 or more missed assessments or within 2 assessments of day 1 if the patient had no evaluable assessments or did not have a baseline assessment.
eA pooling strategy was applied to the primary comparisons of PFS whereby stratification factors were removed until there were at least 5 events in each stratum of interest across the 3 treatment arms. The HR and CI were estimated from a Cox proportional hazard model stratified by MMR and disease status. An HR of less than 1 favoured the treatment arm of interest over the reference arm.
fIncludes patients with unknown survival status or patients who were lost to follow-up.
gEstimated using an unstratified Cox proportional hazards model, with the CI estimated using a profile likelihood approach.
Source: Details included in the table are from the sponsor’s summary of clinical evidence.22
Table 29: HRQoL Results From ITT Population of the DUO-E Study
Variable | ITT population | ||
|---|---|---|---|
SOC N = 241 | SOC + durvalumab N = 238 | SOC + durvalumab + olaparib N = 239 | |
Change from baseline in EORTC QLQ-C30 global health status | |||
Number of patients contributing at baseline, n | ███ ███ | ███ ██████ | ███ ██████ |
Baseline score, mean (SD) | ███ ███ | ███ ██████ | ███ ██████ |
Number of patients contributing at week 18, na | ███ ███ | ███ ██████ | ███ ██████ |
Change from baseline at week 18, mean (SD)a | ███ ███ | ███ ██████ | ███ ██████ |
Number of patients contributing at week 42, nb | ███ ███ | ███ ██████ | ███ ██████ |
Change from baseline at week 30, mean (SD)b | ███ ███ | ███ ██████ | ███ ██████ |
Change from baseline in EORTC QLQ-EN24 pain in back and pelvis | |||
Number of patients contributing at baseline, n | ███ ███ | ███ ███ | ███ ███ |
Baseline score, mean (SD) | ███ ███ | ███ ███ | ███ ███ |
Number of patients contributing at week 18, na | ███ ███ | ███ ███ | ███ ███ |
Change from baseline at week 18, mean (SD)a | ███ ███ | ███ ███ | ███ ███ |
Number of patients contributing at week 42, nb | ███ ███ | ███ ███ | ███ ███ |
Change from baseline at week 42, mean (SD)b | ███ ███ | ███ ███ | ███ ███ |
Change from baseline in EORTC QLQ-EN24 urological symptoms | |||
Number of patients contributing at baseline, n | ███ ███ | ███ ███ | ███ ███ |
Baseline score, mean (SD) | ███ ███ | ███ ███ | ███ ███ |
Number of patients contributing at week 18, na | ███ ███ | ███ ███ | ███ ███ |
Change from baseline at week 18, mean (SD)a | ███ ███ | ███ ███ | ███ ███ |
Number of patients contributing at week 42, nb | ███ ███ | ███ ███ | ███ ███ |
Change from baseline at week 42, mean (SD)b | ███ ███ | ███ ███ | ███ ███ |
dMMR = mismatch repair deficient; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-EN24 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire – Endometrial Cancer Module; HRQoL = health-related quality of life; ITT = intention to treat; SD = standard deviation; SOC = standard of care (carboplatin + paclitaxel or substitutes).
aCorresponds to cycle 6 of the chemotherapy phase.
bAfter the chemotherapy phase, assessment of outcomes shifted from every 3 weeks to every 4 weeks; week 42 corresponds to the sixth month of the maintenance phase.
Sources: Details contained in the table are from the sponsor’s summary of clinical evidence22 and the DUO-E Clinical Study Report.48
Table 30: Summary of Harms Data From the ITT Population of the DUO-E Study
AEs | Overall | Maintenance phase only | ||||
|---|---|---|---|---|---|---|
SOC N = 236 | SOC + durvalumab N = 235 | SOC + durvalumab + olaparib N = 238 | SOC N = 169 | SOC + durvalumab N = 183 | SOC + durvalumab + olaparib N = 192 | |
AEs, n (%) | ||||||
≥ 1 AE | 236 (100.0) | 232 (98.7) | 237 (99.6) | 143 (84.6) | 158 (86.3) | 184 (95.8) |
Most common AEs, n (%)a | ||||||
Nausea | 105 (44.5) | 96 (40.9) | 130 (54.6) | 25 (14.8) | 22 (12.0) | 79 (41.1) |
Constipation | 81 (34.3) | 64 (27.2) | 78 (32.8) | 9 (5.3) | 13 (7.1) | 13 (6.8) |
Diarrhea | 66 (28.0) | 74 (31.5) | 67 (28.2) | 20 (11.8) | 28 (15.3) | 34 (17.7) |
Vomiting | 43 (18.2) | 49 (20.9) | 61 (25.6) | 16 (9.5) | 13 (7.1) | 39 (20.3) |
Neuropathy peripheral | 66 (28.0) | 61 (26.0) | 60 (25.2) | 5 (3.0) | 5 (2.7) | 12 (6.3) |
Peripheral sensory neuropathy | 66 (28.0) | 60 (25.5) | 60 (25.2) | 2 (1.2) | 6 (3.3) | 3 (1.6) |
Alopecia | 118 (50.0) | 118 (50.2) | 121 (50.8) | 1 (0.6) | 2 (1.1) | 5 (2.6) |
Fatigue | 87 (36.9) | 82 (34.9) | 93 (39.1) | 19 (11.2) | 13 (7.1) | 43 (22.4) |
Anemia | 128 (54.2) | 111 (47.2) | 147 (61.8) | 17 (10.1) | 16 (8.7) | 70 (36.5) |
Neutropenia | 31 (13.1) | 36 (15.3) | 49 (20.6) | 1 (0.6) | 6 (3.3) | 21 (10.9) |
Neutrophil count decreased | 63 (26.7) | 44 (18.7) | 50 (21.0) | 6 (3.6) | 8 (4.4) | 16 (8.3) |
Arthralgia | 58 (24.6) | 71 (30.2) | 58 (24.4) | 16 (9.5) | 34 (18.6) | 22 (11.5) |
COVID-19 | 32 (13.6) | 36 (15.3) | 48 (20.2) | 20 (11.8) | 21 (11.5) | 34 (17.7) |
Urinary tract infection | 50 (21.2) | 33 (14.0) | 48 (20.2) | 23 (13.6) | 14 (7.7) | 25 (13.0) |
Decreased appetite | 46 (19.5) | 42 (17.9) | 55 (23.1) | 6 (3.6) | 9 (4.9) | 28 (14.6) |
SAEs, n (%) | ||||||
Patients with any SAE, n (%) | 73 (30.9) | 73 (31.1) | 85 (35.7) | 19 (11.2) | 22 (12.0) | 42 (21.9) |
Most common (≥ 1%) SAEs, n (%) | ||||||
Anemia | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Febrile neutropenia | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Neutropenia | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Aplasia pure red cell | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Urinary tract infection | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Sepsis | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
COVID-19 | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
COVID-19 pneumonia | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Pneumonia | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Urosepsis | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Diarrhea | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Vomiting | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Nausea | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Constipation | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Pneumonitis | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Pulmonary embolism | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Pyrexia | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Deep vein thrombosis | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Hyponatremia | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Fall | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Infusion-related reaction | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Hydronephrosis | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Neutrophil count decreased | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Patients who stopped treatment due to AEs, n (%) | ||||||
Patients with any AE leading to discontinuation of any study treatment or SOC | 44 (18.6) | 49 (20.9) | 58 (24.4) | 7 (4.1) | 11 (6.0) | 27 (14.1) |
AEs (≥ 1%) leading to discontinuation of any study treatment or SOC, n (%)b | ||||||
Anemia | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Neutropenia | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Neuropathy peripheral | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Peripheral sensory neuropathy | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Pneumonitis | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Interstitial lung disease | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Infusion-related reaction | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Rash maculopapular | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Drug hypersensitivity | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Hypersensitivity | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Fatigue | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Deaths, n (%) | ||||||
Patients who died | 82 (34.0) | 65 (27.3) | 52 (21.8) | NR | NR | NR |
Death with unknown reason | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
AEs with the outcome of death, n (%) | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Patients with any AE with outcome of death, n (%) | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Cardiac arrest | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Myocardial infarction | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Multiple organ dysfunction syndrome | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Death | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
General physical health deterioration | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Sepsis | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
COVID-19 | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Pneumonia aspiration | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Septic shock | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Urosepsis | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Acute respiratory failure | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Pulmonary embolism | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Respiratory failure | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Renal failure | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
AEs of special interest, n (%) | ||||||
Olaparib: New primary malignancies, n (%) | ||||||
Number of patients | 3 (1.3) | 2 (0.9) | 2 (0.8) | 2 (1.2) | 1 (0.5) | 1 (0.5) |
Adenocarcinoma of colon | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Neuroendocrine tumour | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Invasive breast carcinoma | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Malignant melanoma | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Papillary thyroid cancer | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Renal neoplasm | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Olaparib: Pneumonitis, n (%) | ||||||
Number of patients with pneumonitis | 1 (0.4) | 4 (1.7) | 12 (5.0) | 0 | 3 (1.6) | 8 (4.2) |
Pneumonitis | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Interstitial lung disease | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Bronchiolitis | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Durvalumab, n | ||||||
Adrenal insufficiency | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Dermatitis | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Dermatitis bullous | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Eczema | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Erythema multiforme | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Immune-mediated dermatitis | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Psoriasis | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Seborrheic dermatitis | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Rash | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Rash, erythematous | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Rash, macular | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Rash, maculopapular | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Rash, papular | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Rash, pruritic | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Rash, pustular | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Colitis | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Enterocolitis | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Diarrhea | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Drug-induced liver injury | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Hepatitis | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Immune-mediated hepatitis | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Hyperthyroidism | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Central hypothyroidism | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Hypothyroidism | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Anaphylactic reaction | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Anaphylactic shock | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Drug hypersensitivity | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Hypersensitivity | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Infusion-related reaction | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Urticaria | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Myasthenia gravis | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Dermatomyositis | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Myositis | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Autoimmune hemolytic anemia | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Cutaneous vasculitis | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Hemolytic anemia | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Immune thrombocytopenia | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Sarcoidosis | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Uveitis | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Vasculitis | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Interstitial lung disease | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Pneumonitis | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Immune-mediated nephritis | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Autoimmune thyroiditis | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Immune-mediated thyroiditis | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Thyroiditis | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Type 1 diabetes mellitus | ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
AE = adverse event; ITT = intention to treat; SAE = serious adverse event; SOC = standard of care.
Source: Details included in the table are from the sponsor’s summary of clinical evidence.22
Pharmacoeconomic Review
Abbreviations
AE
adverse event
BIA
budget impact analysis
CAR-PAC
carboplatin and paclitaxel
CDA-AMC
Canada’s Drug Agency
dMMR
mismatch repair deficient
HR
hazard ratio
ICER
incremental cost-effectiveness ratio
ITC
indirect treatment comparison
OS
overall survival
PD
progressed disease
PF
progression-free
PFS
progression-free survival
PSM
partitioned survival model
QALY
quality-adjusted life-year
RDI
relative dose intensity
TTD
time-to-treatment discontinuation
WTP
willingness to pay
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Durvalumab (Imfinzi), 50 mg/mL solution for infusion |
Indication | For the first-line treatment of adult patients with primary advanced or recurrent mismatch repair deficient (dMMR) endometrial cancer who are candidates for systemic therapy, followed by maintenance treatment with Imfinzi as monotherapy. |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | January 17, 2025 |
Reimbursement request | As per indication |
Sponsor | AstraZeneca Canada Inc. |
Submission history | Previously reviewed: Yes. CDA-AMC has previously reviewed durvalumab as monotherapy or in combination with other products. Indication: Tremelimumab (Imjudo) for injection in combination with durvalumab for the first-line treatment of adult patients with unresectable hepatocellular carcinoma who require systemic therapy. Recommendation date: November 3, 2023. Recommendation: Reimburse with clinical criteria and/or conditions. Indication: Durvalumab in combination with gemcitabine-based chemotherapy for the treatment of patients with locally advanced or metastatic biliary tract cancer. Recommendation date: February 3, 2023. Recommendation: Reimburse with clinical criteria and/or conditions. Indication: Durvalumab in combination with etoposide and either carboplatin or cisplatin is indicated for the first-line treatment of adult patients with extensive-stage small-cell lung cancer. Recommendation date: July 27, 2021. Recommendation: Reimburse with clinical criteria and/or conditions. |
CDA-AMC = Canada’s Drug Agency; NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis PSM |
Target populations | Adult patients with primary advanced or recurrent endometrial cancer that is mismatch repair deficient. |
Treatments | CAR-PAC plus durvalumab. |
Dose regimen | 1,120 mg durvalumab in combination with platinum-based chemotherapy every 21 days for 4 to 6 cycles, followed by maintenance with 1,500 mg every 4 weeks as monotherapy. |
Submitted price | Durvalumab (50 mg/mL): $938.67 per 2.4 mL vial; $3,911.11 per 10 mL vial. |
Submitted treatment cost | Chemotherapy phase: $13,996 every 21 days (durvalumab = $8,671; carboplatin = $1,195; paclitaxel = $4,040) Maintenance phase: $11,733 every 28 days. |
Comparators | CAR-PAC alone CAR-PAC plus dostarlimab |
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | Lifetime (37.4 years) |
Key data sources | DUO-E trial, sponsor-submitted ITC |
Submitted results | CAR-PAC plus durvalumab was dominated by CAR-PAC plus dostarlimab: CAR-PAC plus durvalumab is more costly by $231,661 and results in a 0.61 loss of incremental QALYs. |
Key limitations |
|
CDA-AMC reanalysis results |
|
CAR-PAC = carboplatin and paclitaxel; CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; ITC = indirect treatment comparison; LY = life-year; OS = overall survival; PFS = progression-free survival; pMMR = mismatch repair proficient; PSM = partitioned survival model; QALY = quality-adjusted life-year; RDI = relative dose intensity.
Conclusions
The Clinical Review by Canada’s Drug Agency (CDA-AMC) of the DUO-E trial concluded with low certainty that carboplatin and paclitaxel (CAR-PAC) plus durvalumab is more effective than CAR-PAC alone in terms of overall survival (OS) and progression-free survival (PFS) for the treatment of adult patients with primary advanced or recurrent endometrial cancer that is mismatch repair deficient (dMMR). A definitive conclusion could not be reached for either outcome in the indirect treatment comparison (ITC) that was used to inform the relative effectiveness of CAR-PAC plus durvalumab and CAR-PAC plus dostarlimab in the submitted model. This was attributed to the imprecision of the relative effect estimates as well as concerns relating to the sample size and the power of the analysis. These limitations in the comparative clinical evidence translate to uncertainty surrounding the estimated cost-effectiveness of CAR-PAC plus durvalumab relative to other comparators.
The results from the CDA-AMC base case are similar to the sponsor’s base case: CAR-PAC plus durvalumab is dominated by CAR-PAC plus dostarlimab; CAR-PAC plus durvalumab results in greater total costs (additional $234,585) and 0.68 fewer quality-adjusted life-years (QALYs). As a result, CAR-PAC plus durvalumab would not be cost-effective at a willingness-to-pay (WTP) threshold of $50,000 per QALY gained. At a 47% reduction in the price of durvalumab, CAR-PAC plus durvalumab was equally costly and less effective compared with CAR-PAC plus dostarlimab. At this price reduction, CAR-PAC plus durvalumab was associated with an incremental cost-effectiveness ratio (ICER) of $33,074 compared with CAR-PAC alone. The vast majority of the predicted incremental LYs and QALYs for CAR-PAC plus durvalumab compared with CAR-PAC was estimated through extrapolation beyond the period of observed evidence. The uncertainty in the evidence for long-term OS and PFS adds uncertainty to the estimates of relative costs and QALYs compared with CAR-PAC, and a greater price reduction may be required to achieve cost-effectiveness.
Input Relevant to the Economic Review
This section is a summary of the feedback relevant to this economic review received from the patient groups, clinician groups, and drug plans that participated in the CDA-AMC review process.
Patient input was received as a joint submission from the Colorectal Cancer Resource & Action Network, the Canadian Cancer Survivor Network, and HPV Global Action. Information was collected using an online survey of patients with endometrial cancer conducted from October to November 2023. All 6 respondents to the survey were self-reported residents of Canada. In addition, telephone interviews were conducted with 2 patients in Canada with endometrial cancer who had enrolled in the DUO-E trial. Survey respondents reported treatment histories that included radiation therapy, surgical resection, targeted therapy, hormonal therapy, immunotherapy, and complementary medicines. Before enrolling in the DUO-E trial, both interview participants had undergone a hysterectomy followed by chemotherapy treatment. Survey respondents ranked prolonging life as the most important issue they hoped a new treatment would address. In addition, the interview participants expressed a desire for improved and timely access to new therapies. Both interview participants reported they had been allocated to 1 of the intervention arms of the DUO-E trial. However, the blinded protocol meant this could not be confirmed. Both patients reported that the therapy they received was effective and offered manageable, minimal side effects. They also appreciated the shorter IV administration time required for their treatment relative to prior chemotherapy regimens.
Clinician input was received from 2 groups: the Gynecologic Oncology Society of Canada and the Ontario Health Gynecologic Cancer Drug Advisory Committee. Both submissions stated that the objectives of therapy include prolonging life, delaying disease progression, reducing symptoms, improving health-related quality of life, and potentially curing the disease. The current pathway of care for patients with endometrial cancer includes surgery with or without adjuvant radiation therapy, although patients with advanced disease (stage III or IV) will require further intervention. Current treatment options at this stage include hormone therapy, cytotoxic chemotherapy (usually CAR-PAC), and radiation. Both submissions cited a desire for more treatments with tolerable adverse effects that are effective at delaying progression and improving OS relative to the current standard of care (defined as cytotoxic chemotherapy with CAR-PAC). The input stated that the regimen under review would be positioned for use as a first-line treatment option for patients with advanced or recurrent endometrial cancer. This change in treatment practice is expected to lead to improved patient outcomes.
Drug plan input identified concerns relating to the estimated budget impact and the presence of confidential negotiated prices for comparator treatments. Several of these concerns were addressed in the sponsor’s model:
The comparators in the economic evaluation included the current standard of care, defined as cytotoxic chemotherapy with CAR-PAC.
In addition, CDA-AMC addressed some of these concerns as follows:
A scenario analysis was conducted for the budget impact analysis (BIA) that considered a 30% reduction in price for durvalumab.
CDA-AMC was unable to address the following concerns raised by the input received:
The estimated budget impact is subject to uncertainty.
Economic Review
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted an economic evaluation of durvalumab compared with existing alternatives for the treatment of adults with primary advanced or recurrent endometrial cancer. Treatment with durvalumab was considered as monotherapy for patients with dMMR cancer.1 This population of patients is aligned with the Health Canada indication and the reimbursement request.
Durvalumab is available as a solution for injection at a concentration of 50 mg/mL. It is packaged as a 2.4 mL or 10 mL single-use vial with a unit cost of $938.67 and $3,911.11 respectively. Treatment is provided in 2 phases. In the chemotherapy phrase, the recommended dose is 1,120 mg durvalumab in combination with CAR-PAC. Carboplatin is available as a single-use 10 mg/mL vial at volumes of 5 mL ($70), 15 mL ($210), 45 mL ($600), and 60 mL ($775). Paclitaxel is available as a single-use 6 mg/mL vial at volumes of 5 mL ($300), 16.7 mL ($1,197), and 50 mL ($971). Regimens which included CAR-PAC, with a recommended dose for carboplatin of area under the curve (AUC) 4 mg/mL/min to 6 mg/mL/min and 175 mg/m2 of paclitaxel every 21 days for 4 to 6 cycles.2,3 At the submitted price, the total regimen cost in the chemotherapy phase for a patient with a body surface area of 1.77 m2 and a glomerular filtration rate of 125 mL/min/1.73 m2 would be $13,996 every 21 days (durvalumab = $8,761; carboplatin = $1,195; paclitaxel = $4,040). In the maintenance phase, the recommended monotherapy dose is 1,500 mg every 4 weeks, resulting in a 28-day cost of $11,733.
Two comparators were considered in the economic evaluation: CAR-PAC and CAR-PAC plus dostarlimab. At publicly available list prices, the total cost of the CAR-PAC regimen for a patient with a body surface area of 1.77 m2 and glomerular filtration rate of 125 mL/min/1.73 m2 would be $5,235. Meanwhile, dostarlimab is available as a single-use 50 mg/mL vial at a volume of 10 mL ($10,031). In the chemotherapy phase, the recommended dose is 500 mg dostarlimab every 3 weeks in combination with CAR-PAC. In the maintenance phase, the recommended dose is 1,000 mg dostarlimab monotherapy every 6 weeks. This would cost $15,266 per 21 days (dostarlimab = $10,031; carboplatin = $1,195; paclitaxel = $4,040) in the chemotherapy phase and $10,031 per 21 days in the maintenance phase.
Modelled outcomes included life-years and QALYs. Costs were estimated from the perspective of the Canadian public health care payer. Model outputs were generated over a lifetime horizon of 37.4 years, with a cycle length of 1 month. Costs and outcomes were discounted at 1.5% annually.
Model Structure
The costs and effects for each alternative were estimated using a partitioned survival model (PSM). As illustrated in Figure 1, the model comprised 3 mutually exclusive health states: progression-free (PF), progressed disease (PD), and death. At model entry, patients were assumed to occupy the PF state and initiate therapy. In each cycle, patients could remain in their current state or transition to a different state until they entered the death state.1
Model Inputs
Data summarizing baseline characteristics for both subpopulations were obtained from the DUO-E trial.4 This was a randomized phase III trial that involved the direct comparison of durvalumab plus CAR-PAC with CAR-PAC alone. Patients were randomized to 1 of 3 arms: CAR-PAC, durvalumab plus CAR-PAC (dMMR subpopulation), and durvalumab plus olaparib plus CAR-PAC (mismatch repair proficient subpopulation).4 Baseline characteristics were assumed to be consistent with all randomized patients from the trial and included age (mean = 62.6 years; SD = 10.03), body surface area (mean = ████ m2), and weight (mean = ████ kg; SD = █████).1,4
Estimates of the relative efficacy of the comparators excluded from the DUO-E trial were obtained from the sponsor-submitted systematic review and ITC.5 This was necessary to incorporate CAR-PAC plus dostarlimab as a comparator in the economic evaluation. The ITC was conducted using 2 trials of treatments for patients in the dMMR subpopulation.4-6 These formed a connected network between CAR-PAC plus durvalumab, CAR-PAC, and CAR-PAC plus dostarlimab. An anchored matching-adjusted indirect comparison was used to estimate hazard ratios (HRs) for 2 outcomes, OS and PFS. For PFS, the sponsor estimated an HR of ████ (95% CI, ████ ██ ████) for CAR-PAC plus durvalumab relative to CAR-PAC plus dostarlimab. For OS, the sponsor estimated an HR of ████ (95% CI, ████ ██ ████) for CAR-PAC plus durvalumab relative to CAR-PAC plus dostarlimab.1,5
Direct and indirect approaches were used to estimate the treatment-specific survival probabilities needed to estimate state membership. In both subpopulations, parametric survival models were fit for time-to-treatment discontinuation (TTD), PFS, and OS. Treatment-dependent and -independent models were fit using the exponential, log-logistic, log-normal, Weibull, Gompertz, gamma, and generalized gamma distributions. In addition, the sponsor explored treatment-dependent and -independent flexible-spline models for OS and PFS. Selection of the base-case configuration was informed by visual inspection of the Kaplan-Meier curve, model fit statistics, and clinical plausibility.1
In the sponsor’s base case, PFS was predicted using the treatment-independent 2- and 1-knot flexible-spline models for CAR-PAC plus durvalumab and CAR-PAC, respectively. Predicted values for CAR-PAC plus dostarlimab were estimated indirectly by adjusting the PFS for CAR-PAC plus durvalumab using the HR estimated from the submitted ITC.1,5 Meanwhile, the base-case OS values were predicted using the treatment-independent parametric models, which followed the log-normal distribution. It was assumed there was no difference in OS between CAR-PAC plus durvalumab and CAR-PAC plus dostarlimab.1 Predicted values for TTD, which were used to inform the initial and acquisition costs for durvalumab for maintenance treatment, were assumed to follow a gamma distribution.1
To ensure that the risk of death would not fall below that of the general population, the sponsor assumed that the predicted values for OS could not fall below the corresponding mortality risk for the general population.1 Age-matched and sex-specific mortality risks were obtained from life tables published by Statistics Canada.7 In addition, the sponsor assumed that the predicted values for PFS could not exceed the corresponding OS curve.1
In addition to tracking the proportion of the cohort in each health state, the model also tracked the occurrence of adverse events (AEs). This was restricted to grade 3 and 4 AEs that occurred in at least 5% of patients from any arm of the DUO-E trial and the CAR-PAC plus dostarlimab arm of the RUBY part 1 trial.1,4,6 The identified AEs included anemia; neutropenia; decreased neutrophil, lymphocyte, or white cell counts; hypertension; pulmonary embolism; and hypokalemia.1
Health-related quality of life was captured in the model using utility values specific to every state but death. In the submitted base case, the sponsor carried forward health state utility values from a CDA-AMC reanalysis in a prior submission involving pembrolizumab for the treatment of advanced endometrial cancer.1,8 It was assumed that the PF state would have a utility value of 0.782 (95% CI, 0.769 to 0.796) and the PD state would have a utility value of 0.731 (95% CI, 0.709 to 0.754). These values were selected for the submitted base case to avoid criticism that trial-estimated utility values would underestimate the disutility associated with progressive disease.1,8 A separate scenario analysis was conducted in which health state utility values were calculated using patient preference data collected from the DUO-E trial indirectly using the EQ-5D questionnaire.1 To reflect the preferences of the general population in Canada, EQ-5D responses were converted to utilities using Canada-specific tariffs.1,9 In this scenario analysis, the health state utility values were █████ (95% CI, █████ ██ █████) and █████ (95% CI, █████ ██ █████) for the PF and PD states, respectively.1
The submission also included the option (which was applied in the base case) to adjust the utility values for age. In the absence of Canada-specific utility values, the adjustment was made using estimates reflecting the general population of the UK.1,10 The base case also included changes in utility as a result of AEs as a 1-time event in the first model cycle. The change in utility was assumed to be −0.037 for hypertension, −0.051 for pulmonary embolism, and −0.020 for all other AEs.1,11,12
Costs in the model included treatment acquisition and administration, monitoring, the management of AEs, and end-of-life care. Treatment acquisition costs were calculated from the price per unit consumed based on the recommended dosage for each alternative. The price for durvalumab represented the sponsor’s submitted price, while all other drug prices were obtained from the Ontario Drug Benefit Formulary or wholesale list prices.1,13,14 For durvalumab, a relative dose intensity (RDI) of 98.54% was assumed. Following entry to the PD state, it was assumed that patients could initiate 1 of several anticancer systemic therapies indicated for a later line of treatment. These costs were calculated as a weighted average reflecting the proportion of receiving each subsequent regimen, the time on treatment, and the corresponding acquisition costs. Treatment administration costs were applied for any therapy that required IV administration. It was assumed that the unit cost of treatment administration reflected the average IV administration cost in Canada ($149.76) estimated by Sohi et al.1,15 Monitoring costs included outpatient visits (physician and nurse), laboratory tests (liver function, complete blood count, renal function, thyroid function), and imaging (chest X-ray, CT scan).1 Unit costs for these services were obtained from the Ontario Schedule of Benefits for Physician and Laboratory Services, while the frequency of service utilization was informed by expert opinion.1,16-18 Costs associated with the management of AEs were applied as a lump sum at model entry using Canada-specific unit costs.19 End-of-life care ($11,573) was incorporated as a 1-time cost upon entry to the death state. It reflected adult palliative care estimates from 2021 to 2022, inflated to a 2024 price year.1,19
Summary of Sponsor’s Economic Evaluation Results
The base-case costs and QALYs for each alternative were generated in a Monte Carlo simulation of 1,000 iterations. All scenario analyses were simulated deterministically. Results from the deterministic and probabilistic base-case simulations were similar.
Base-Case Results
The submitted analyses were based on publicly available prices of the comparator treatments. Results from the base case of the submitted economic evaluation for each subpopulation are presented in Table 3.
CAR-PAC and CAR-PAC plus dostarlimab were on the efficiency frontier. CAR-PAC plus durvalumab was more costly and less effective (incremental costs: $231,661; incremental QALYs: −0.61) than CAR-PAC plus dostarlimab. CAR-PAC plus durvalumab had a 2.7% probability of being cost-effective at a WTP threshold of $50,000 per QALY gained. In the deterministic base case, 95.8% and 100% of the incremental QALYs for CAR-PAC plus durvalumab compared with CAR-PAC and CAR-PAC plus dostarlimab, respectively, were gained beyond the duration of observation from the DUO-E trial.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|
CAR-PAC | 140,918 | 3.59 | Reference |
CAR-PAC plus dostarlimab | 419,350 | 10.77 | $38,784 vs. CAR-PAC |
CAR-PAC plus durvalumab | 651,011 | 10.16 | Dominated by CAR-PAC plus dostarlimab |
CAR-PAC = carboplatin and paclitaxel; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Source: Sponsor’s pharmacoeconomic submission.1
Additional results from the sponsor’s submitted base case are presented in Appendix 3.
Sensitivity and Scenario Analysis Results
In addition to the submitted base case, the sponsor considered 21 distinct scenario analyses. These scenarios explored alternate inputs or assumptions relating to the discount rates, drug wastage, inclusion of the diagnostic testing cost, source of the utility values, and selection of the parametric survival functions for OS, PFS, and TTD.1 While each scenario had a slight impact on the expected costs and benefits, none had a meaningful effect on the conclusion of the economic evaluation.
The submission included the ability to conduct a scenario analysis from the societal perspective. This enabled the model to consider the additional costs associated with productivity loss and caregiver costs. However, the sponsor did not conduct any scenario analyses from a perspective other than the health care payer.1
CDA-AMC Appraisal of the Sponsor’s Economic Evaluation
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis:
The relative efficacy of durvalumab with relevant comparators in uncertain: The submitted economic evaluation compared durvalumab (in combination with CAR-PAC) with relevant alternatives that could be used to treat primary advanced or recurrent dMMR endometrial cancer in adults. Estimates of relative efficacy were obtained from the DUO-E trial as well as the sponsor-submitted systematic review and ITC. The CDA-AMC Clinical Review concluded with low certainty that durvalumab may be more effective than CAR-PAC alone in terms of OS and PFS. The uncertainty in the conclusion was attributed to imprecision in the estimated HRs, immature data, and an increased risk of type I error due to multiplicity in subgroups. Meanwhile, findings from the sponsor’s ITC indicated no difference in efficacy in terms of PFS or OS. However, the imprecision of the effect estimate and concerns related to the sample size and power of the analysis prevented any definitive conclusions from being reached.
The limitations in the underlying clinical evidence could not be addressed through reanalysis.
Improper methods were used to calculate membership in a health state: When estimating state membership with a PSM, the estimates for each mutually exclusive health state (PF, PD, death) must be calculated using a series of non–mutually exclusive survival functions.20 In the submitted model, this requirement was not satisfied due to the decision to cap the predicted OS curves by the general population mortality risk. This is problematic because the 2 types of probabilities are not interchangeable. The general population mortality risk refers to the probability of death at a specific age. In contrast, survival probability refers to the probability that death will not occur after a specific point in time. Given that the general population mortality risk is not a survival probability, it cannot be used to estimate state membership in a PSM.
CDA-AMC modified the economic evaluation to remove the assumption capping the OS curve by the general population mortality risk.
Influence of RDI on drug acquisition costs: The sponsor adjusted the unit price of durvalumab to reflect an assumption that the RDI was equal to ██████%. This parameter was calculated as the actual dose received divided by the standard calculated dose during the DUO-E trial. While the purpose of this adjustment was to ensure that drug exposure was consistent with the trial data, it did not consider other factors that may affect dosage adjustments. The clinical experts consulted by CDA-AMC indicated that dosage may vary as a result of attempts to manage toxicity like dose reductions, or delays and subsequent escalation. It is unclear why the sponsor assumed that patients would be subject to toxicity-related dose modifications for durvalumab but no other therapy. The inconsistent nature of the RDI assumption may have led to the mischaracterization of the treatment acquisition costs in the sponsor’s base case.
CDA-AMC modified the economic evaluation to consider an RDI of 100% for durvalumab.
The methods for the age adjustment of health state utility values were inappropriate: Following the calculation of health state utilities, the sponsor implemented a procedure to adjust the values by age.1,10 The intent of this method was to reflect the fact that quality of life for the general population declines with age. In the absence of Canada-specific data, the sponsor applied the age-adjustment utility values reflecting the general population of the UK. The correct implementation of the age adjustment required a comparison using the EQ-5D utility values of the general population in Canada, matched by age to the simulated cohort.
CDA-AMC modified the economic evaluation to remove the age adjustment.
Additionally, the following key assumptions were made by the sponsor and appraised by CDA-AMC (refer to Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation
Sponsor’s key assumption | CDA-AMC comment |
|---|---|
Predicted PFS values were obtained using the treatment-independent 2- and 1-knot flexible-spline models for CAR-PAC plus durvalumab and CAR-PAC alone, respectively. In addition, predicted OS values were obtained using the treatment-independent models that followed the log-normal distribution. | Appropriate. Alternate configurations of the parametric survival models for PFS and OS did not impact the results of the economic evaluation. Each alternate configuration did influence the expected (mean) costs and the QALYs returned from the model. However, no configuration influenced the order of the alternatives in terms of QALYs, the order of alternatives in terms of cost, the composition of the efficiency frontier, or which alternatives are subject to dominance or the reason(s) they are subject to dominance. |
Consideration of treatment switching following entry to the PD state. | It is inappropriate to consider the costs, but not the effects of subsequent lines of therapy. This choice introduced an assumption that the risk of death is unaffected by a patient’s disease progression status. For the conceptualized model structure, the proper consideration of treatment switching should have been the use of a Markov model to estimate state occupancy. |
CAR-PAC = carboplatin and paclitaxel; CDA-AMC = Canada’s Drug Agency; OS = overall survival; PD = progressed disease; PFS = progression-free survival; QALY = quality-adjusted life-year.
CDA-AMC Reanalyses of the Economic Evaluation
Base-Case Results
CDA-AMC undertook reanalyses that addressed the key limitations within the submitted economic models, as summarized in Table 5. The CDA-AMC base case was derived by making changes in the model parameter values and assumptions, in consultation with the clinical experts. CDA-AMC was unable to address limitations related to the uncertainty in the clinical evidence identified in the CDA-AMC Clinical Review and the characterization of parameter uncertainty in the economic evaluation. Consistent with the sponsor’s base case, the costs and effects for each alternative were generated from a Monte Carlo simulation of 1,000 iterations.
Table 5: CDA-AMC Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Changes to derive the CDA-AMC base case | ||
1. Predicted values for OS | Parametric OS curve capped by general population mortality risk. | Removed. Membership in a health state was estimated using unmodified survival probabilities. |
2. Age-adjusted utility values | General population utilities from the UK are interchangeable with Canada. | Age adjustment removed. Canada-specific utility values are required for the sponsor’s age adjustment. |
3. RDI | Durvalumab RDI assumed to equal █████%. | RDI assumed to equal 100%. |
CDA-AMC base case | ― | 1 + 2 + 3 |
CDA-AMC = Canada’s Drug Agency; OS = overall survival; RDI = relative dose intensity.
Results from the CDA-AMC base case are presented in Table 6. As with the sponsor’s base case, the results were generated using publicly available prices for the comparator treatments. The impact of each individual modification on the deterministic results is reported in Table 11, and the disaggregated summary of the probabilistic base case is reported in Table 12.
In the CDA-AMC base case, the efficiency frontier comprised CAR-PAC and CAR-PAC plus dostarlimab. CAR-PAC plus durvalumab did not lie on the efficiency frontier because it was dominated by CAR-PAC plus dostarlimab. This means that CAR-PAC plus durvalumab is not cost-effective because it is more costly (incremental costs: $234,585) and less effective (incremental QALYs: −0.68) than CAR-PAC plus dostarlimab. CAR-PAC plus durvalumab had a 1.8% probability of being cost-effective at a WTP threshold of $50,000 per QALY gained. In the deterministic base case, 96% and 99% of the incremental QALYs for CAR-PAC plus durvalumab compared with CAR-PAC and CAR-PAC plus dostarlimab were gained beyond the duration of observation from the DUO-E trial.
Table 6: Summary of the CDA-AMC Reanalysis Results
Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|
Sponsor base case (probabilistic) | |||
CAR-PAC | 140,918 | 3.59 | Reference |
CAR-PAC plus dostarlimab | 419,350 | 10.77 | $38,784 vs. CAR-PAC |
CAR-PAC plus durvalumab | 651,011 | 10.16 | Dominated by CAR-PAC plus dostarlimab |
CDA-AMC base case (probabilistic) | |||
CAR-PAC | 140,935 | 3.57 | Reference |
CAR-PAC plus dostarlimab | 423,917 | 12.07 | 33,301 vs. CAR-PAC |
CAR-PAC plus durvalumab | 658,502 | 11.39 | Dominated by CAR-PAC plus dostarlimab |
CAR-PAC = carboplatin and paclitaxel; CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Scenario Analysis Results
CDA-AMC undertook price reduction analyses based on the sponsor’s results and the CDA-AMC base case. A price reduction of 47% (durvalumab price of $488.11 per 2.4 mL vial and $2,033.78 per 10 mL vial; $4,556 per 21-day initiation cycle and $6,101 per 28-day maintenance cycle) would be needed for CAR-PAC plus durvalumab to lie on the cost-effectiveness frontier. The ICER at that price reduction would be $33,074 per QALY gained compared with CAR-PAC.
Table 7: CDA-AMC Price Reduction Analyses
Analysis | Unit drug cost ($) | ICERs for CAR-PAC plus durvalumab ($/QALY) | |
|---|---|---|---|
Price reduction | 2.4 mL vial; 10 mL vial | Sponsor base case | CDA-AMC base case |
No price reduction | 938.67; 3,911.11 | Dominated by CAR-PAC dostarlimab | Dominated by CAR-PAC plus dostarlimab |
10% | 844.80; 3,519.99 | Dominated | Dominated |
20% | 750.94; 3,128.89 | Dominated | Dominated |
30% | 657.07; 2,737.78 | Dominated | Dominated |
40% | 563.20; 2,346.67 | Dominated | Dominated |
47% | 497.50; 2,072.89 | Dominated | 33,074 vs. CAR-PAC |
48% | 488.11; 2,033.78 | 37,977 vs. CAR-PAC | 32,369 vs. CAR-PAC |
50% | 469.34; 1,955.56 | 36,324 vs. CAR-PAC | 30,960 vs. CAR-PAC |
60% | 375.47; 1,564.44 | 28,059 vs. CAR-PAC | 23,913 vs. CAR-PAC |
70% | 281.60; 1,173.33 | 19,795 vs. CAR-PAC | 16,866 vs. CAR-PAC |
80% | 187.73; 782.22 | 11,531 vs. CAR-PAC | 9,819 vs. CAR-PAC |
90% | 93.87; 391.11 | 3,266 vs. CAR-PAC | 2,772 vs. CAR-PAC |
CAR-PAC = carboplatin and paclitaxel; CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Issues for Consideration
The pan-Canadian Pharmaceutical Alliance (pCPA) concluded negotiations with a letter of intent for dostarlimab for the treatment of adults with primary advanced or recurrent dMMR or microsatellite instability-high endometrial cancer who are candidates for systemic therapy. As a result, dostarlimab has a confidential negotiated price and is currently funded by jurisdictional cancer formularies. CDA-AMC also notes that the sponsor’s analysis relies on public list prices for carboplatin and paclitaxel that may not reflect existing confidential prices for those therapies. The pCPA also concluded negotiations with a letter of intent for durvalumab in the treatment of metastatic biliary tract cancer and unresectable hepatocellular carcinoma in February 2024. It is likely that the current unit costs for durvalumab, dostarlimab, carboplatin, and paclitaxel are lower than the unit prices used within the economic evaluation. Therefore, the actual unit prices may influence the results of cost- effectiveness and the BIAs.
CDA-AMC is reviewing durvalumab in combination with olaparib for the treatment of adult patients with primary advanced or recurrent endometrial cancer that is mismatch repair proficient. The comparative efficacy and cost-effectiveness of durvalumab in this context is unknown and may further affect price negotiations with drug plans. CDA-AMC is also reviewing pembrolizumab plus chemotherapy for the treatment of adult patients with advanced or recurrent endometrial cancer irrespective of mismatch repair status.
The pan-Canadian Oncology Drug Review Expert Review Committee has reviewed CAR-PAC plus dostarlimab for the treatment of adult patients with primary advanced or recurrent endometrial cancer. This review considered all adult patients, irrespective of mismatch repair status. The committee’s final recommendation (published in May 2025) is to reimburse with conditions.
The recommended dosage of CAR-PAC plus durvalumab from the product monograph does not include an explicit stopping rule. Instead, the product monograph states that treatment should continue until disease progression or unacceptable toxicity. The pharmacoeconomic evaluation did not consider a stopping rule, although it did consider treatment discontinuation and assumed that no patient would receive CAR-PAC plus durvalumab after 40 cycles.
Overall Conclusions
The CDA-AMC Clinical Review of the DUO-E trial concluded with low certainty that CAR-PAC plus durvalumab is more effective than CAR-PAC alone in terms of OS and PFS for the treatment of adult patients with primary advanced or recurrent dMMR endometrial cancer. A definitive conclusion could not be reached for either outcome in the ITC that was used to inform the relative effectiveness of CAR-PAC plus durvalumab and CAR-PAC plus dostarlimab in the submitted model. This was attributed to the imprecision of the relative effect estimates as well as concerns relating to the sample size and power of the analysis. These limitations in the comparative clinical evidence translate to uncertainty surrounding the estimated cost-effectiveness of CAR-PAC plus durvalumab relative to other comparators.
CDA-AMC identified several additional limitations with the sponsor’s economic evaluation. These included improper methods to calculate membership in a health state, the influence of RDI on treatment acquisition costs, the omission of a decline in effectiveness over time, the improper characterization of uncertainty, and inappropriate methods to calculate age-adjusted utility values. CDA-AMC attempted to address some of these limitations through reanalysis. In the CDA-AMC base case, this involved removing the cap on OS, removing the age-adjusted utility values, and setting the RDI for durvalumab to 100%.
In the CDA-AMC base case, CAR-PAC plus durvalumab was more costly (incremental costs: $517,567) and more effective (incremental QALYs: 7.82) than CAR-PAC. However, CAR-PAC plus durvalumab was dominated (incremental costs: $234,585; incremental QALYs: −0.68) by CAR-PAC plus dostarlimab. CAR-PAC plus durvalumab was not cost-effective at any possible WTP threshold. There was a 1.8% probability of CAR-PAC plus durvalumab being cost-effective at a WTP threshold of $50,000 per QALY gained.
In the CDA-AMC base case, durvalumab was no longer dominated by CAR-PAC plus dostarlimab at a 47% reduction in the price of durvalumab. At this price reduction, CAR-PAC plus durvalumab was associated with an ICER of $33,074 per QALY gained. The cost-effectiveness of CAR-PAC plus durvalumab and the price reduction required to bring CAR-PAC plus durvalumab onto the cost-effectiveness frontier are sensitive to the acquisition cost of durvalumab, dostarlimab, carboplatin, and paclitaxel. All of these drugs have negotiated prices that are likely lower than the publicly available list prices used in the CDA-AMC base case.
References
1.AstraZeneca Canada Inc. Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Durvalumab for endometrial cancer - dMMR, 120mg/2.4mL and 500 mg/10mL, intravenous injection. October 16, 2024.
2.Cancer Care Ontario. CRBPPACL Monograph. https://www.cancercareontario.ca/en/drugformulary/regimens/monograph/45981 [sponsor submitted reference]
3.AstraZeneca Inc. Imfinzi (durvalumab): concentrate for solution for infusion, 50 mg / mL, Intravenous [product monograph]. 2024.
4.AstraZeneca Inc. Clinical Study Report: NCT04269200. A Randomized, Multicentre, Double-blind, Placebo-controlled, Phase III Study of First-line Carboplatin and Paclitaxel in Combination with Durvalumab, Followed by Maintenance Durvalumab with or without Olaparib in Patients with Newly Diagnosed Advanced or Recurrent Endometrial Cancer [internal sponsor's report]. 2023.
5.AstraZeneca Inc. Indirect treatment comparison of durvalumab plus chemotherapy versus dostarlimab plus chemotherapy in MMR-deficient and/or MSI-H primary advanced or recurrent endometrial cancer [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Duravalumab for endometrial cancer - dMMR, 120mg/2.4mL and 500 mg/10mL, intravenous injection. October 1, 2024.
6.Mirza MR, Sharma S, Herrstedt J, et al. 740MO Dostarlimab+ chemotherapy for the treatment of primary advanced or recurrent endometrial cancer (pA/rEC): Analysis of progression free survival (PFS) and overall survival (OS) outcomes by molecular classification in the ENGOT-EN6-NSGO/GOG-3031/RUBY trial. Ann Oncol. 2023;34:S507. doi: 10.1016/j.annonc.2023.09.1919
7.Statistics Canada. Table: 13-10-0114-01. Life expectancy and other elements of the life table, Canada, all provinces except Prince Edward Island. 2023. Accessed November 6, 2024. https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310011401
8.Canada's Drug Agency. Pembrolizumab for advanced endometrial cancer. 2023. https://www.cda-amc.ca/pembrolizumab-8 [sponsor submitted reference]
9.Xie F, Pullenayegum E, Gaebel K, et al. A time trade-off-derived value set of the EQ-5D-5L for Canada. Med Care. 2016;54(1):98-105. doi: 10.1097/MLR.0000000000000447 PubMed
10.Alava MH, Pudney S, Wailoo A. Estimating EQ-5D by age and sex for the UK. 2022. NICE DSU Report. www.nicedsu.org.uk. [sponsor submitted reference]
11.Sullivan PW, Slejko JF, Sculpher MJ, Ghushchyan V. Catalogue of EQ-5D scores for the United Kingdom. Med Decis Making. 2011;31(6):800-804. doi: 10.1177/0272989X11401031 PubMed
12.National Institute for Health Clinical Excellence. Necitumumab for untreated advanced or metastatic squamous non-small-cell lung cancer [TA411]. 2016. Accessed March 2024. https://www.nice.org.uk/guidance/ta411 [sponsor submitted reference]
13.Ontario Ministry of Health. Ontario drug benefit formulary/comparative drug index. 2024. Accessed November 6, 2024. https://www.formulary.health.gov.on.ca/formulary/
14.IQVIA. DeltaPA. 2024. Accessed November 6, 2024. https://www.iqvia.com/
15.Sohi GK, Levy J, Delibasic V, et al. The cost of chemotherapy administration: A systematic review and meta-analysis. American Society of Clinical Oncology; 2020 [sponsor submitted reference].
16.Government of Ontario. Schedule of Physician Benefits. 2024. https://www.ontario.ca/files/2024-08/moh-schedule-benefit-2024-08-30.pdf [sponsor submitted reference]
17.Government of Ontario. Schedule of Benefits for Laboratory Services. 2024. https://www.ontario.ca/files/2024-01/moh-ohip-schedule-of-benefits-laboratory-services-2024-01-24.pdf [sponsor submitted reference]
18.AstraZeneca. Canadian KOL Survey [data on file]. 2024 [sponsor submitted reference].
19.Canadian Institute for Health Information. Patient Cost Estimator. 2024. https://www.cihi.ca/en/patient-cost-estimator [sponsor submitted reference]
20.Woods B, Sideris E, Palmer S, Latimer N, Soares M. Partitioned survival analysis for decision modelling in health care: a critical review. Decision Support Unit, ScHARR, University of Sheffield; 2017. Accessed October 17, 2024. http://nicedsu.org.uk/wp-content/uploads/2017/06/Partitioned-Survival-Analysis-final-report.pdf
21.Calvert AH, Newell DR, Gumbrell LA, et al. Carboplatin dosage: prospective evaluation of a simple formula based on renal function. J Clin Oncol. 1989;7(11):1748-1756. PubMed
22.AstraZeneca Canada Inc. Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Durvalumab, 120mg/2.4mL and 500 mg/10mL and intravenous. October 16, 2024.
23.Canada's Drug Agency. Dostarlimab Submission for dMMR EC. 2024. https://www.cda-amc.ca/sites/default/files/DRR/2024/PC0325_Combined_Report.pdf [sponsor submitted reference]
24.Di Dio C, Bogani G, Di Donato V, et al. The role of immunotherapy in advanced and recurrent MMR deficient and proficient endometrial carcinoma. Gynecol Oncol. 2023;169:27-33. doi: 10.1016/j.ygyno.2022.11.031 PubMed
25.Cortes-Ciriano I, Lee S, Park WY, Kim TM, Park PJ. A molecular portrait of microsatellite instability across multiple cancers. Nat Commun. 2017;8:15180. doi: 10.1038/ncomms15180 PubMed
26.Evrard C, Alexandre J. Predictive and Prognostic Value of Microsatellite Instability in Gynecologic Cancer (Endometrial and Ovarian). Cancers (Basel). 2021;13(10). doi: 10.3390/cancers13102434 PubMed
27.Statistics Canada. Table 17-10-0005-01 Population estimates on July 1, by age and gender. 2024. https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1710000501 [sponsor submitted reference]
28.Brenner DR, Gillis J, Demers AA, et al. Projected estimates of cancer in Canada in 2024. CMAJ. 2024;196(18):E615-E623. doi: 10.1503/cmaj.240095 PubMed
29.Cancer Care Ontario. Ontario Cancer Facts. 2019. https://www.cancercareontario.ca/en/cancer-facts/endometrial-cancer-starting-rise-younger-women-ontario [sponsor submitted reference]
30.Government of Canada. Canadian Cancer Data Tool. 2021. https://health-infobase.canada.ca/ccdt/age-distribution?index=8429 [sponsor submitted reference]
31.Canadian Cancer Society. Cancerous tumours of the uterus. 2024. https://cancer.ca/en/cancer-information/cancer-types/uterine/what-is-uterine-cancer/cancerous-tumours [sponsor submitted reference]
32.Martins D, O’Sullivan DE, Boyne DJ, et al. Understanding characteristics, treatment patterns, and clinical outcomes for individuals with advanced or recurrent endometrial cancer in Alberta, Canada: a retrospective, population-based cohort study. Current Oncology. 2023;30(2):2277-2289. doi: 10.1200/JCO.2022.40.16_suppl.e17624 PubMed
33.Huijgens ANJ, Mertens HJMM. Factors predicting recurrent endometrial cancer. Facts Views Vis Obgyn. 2013;5(3):179-186. PubMed
34.Rutten H, Verhoef C, van Weelden WJ, et al. Recurrent Endometrial Cancer: Local and Systemic Treatment Options. Cancers (Basel). 2021;13(24). doi: 10.3390/cancers13246275 PubMed
35.Cancer Care Ontario. Systemic Therapy for Advanced or Recurrent Endometrial Cancer and Advanced or Recurrent Uterine Papillary Serous Carcinoma. 2019. https://www.cancercareontario.ca/en/guidelines-advice/types-of-cancer/501 [sponsor submitted reference]
36.IQVIA. DeltaPA Canadian Drug Pricing. 2024. https://www.iqvia.com/locations/canada/library/fact-sheets/iqvia-deltapa [sponsor submitted reference]
37.Peterse EFP, Naber SK, Daly C, et al. Cost-effectiveness of Active Identification and Subsequent Colonoscopy Surveillance of Lynch Syndrome Cases. Clin Gastroenterol Hepatol. 2020;18(12):2760-2767 e12. doi: 10.1016/j.cgh.2019.10.021 PubMed
Appendix 1: Cost Comparison Table
Please note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical expert(s). Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 8: CDA-AMC Cost Comparison Table for Advanced or Recurrent dMMR Endometrial Cancer
Treatment | Strength or concentration | Form | Price | Recommended dosage | Daily cost ($) | 21-day cost ($) |
|---|---|---|---|---|---|---|
Carboplatin | 10 mg/mL | 5 mL vial 15 mL vial 45 mL vial 60 mL vial | 70.0000 210.0000 600.0000 775.0020 | AUC 5 to 6 on day 1 for 3, 21-day cycles | $46.90 to $56.90 | $985 to $1,195 |
Paclitaxel | 6 mg/mL | 5 mL vial 16.7 mL vial 25 mL vial 50 mL vial | 300.0000 1,196.8000 $1,870.0000 $3,740.0000 | 175 to 200 mg/m2 on day 1 for 3, 21-day cycles | $174.61 to $206.67 | $3,667 to $4,340 |
Durvalumab (Imfinzi) | 50 mg/mL | 2.4 mL vial 10 mL vial | 938.6700 3,911.1100 | 1,120 mg every 21 days for 4 to 6 cycles Maintenance: 1,500 mg every 28-days as monotherapy | $417.19 $419.05 | $8,761 $8,800 |
dMMR subpopulation: Carboplatin-paclitaxel plus durvalumab | $638.70 to $680.76 | $13,413 to $14,296 | ||||
Durvalumab (maintenance)a | $419.05 | $8,800 | ||||
Carboplatin-paclitaxel | ||||||
Carboplatin | 10 mg/mL | 5 mL vial 15 mL vial 45 mL vial 60 mL vial | 70.0000 210.0000 599.9850 775.0020 | AUC 5 to 6 on day 1 for 3, 21-day cyclesa | $46.90 to $56.90 | $985 to $1,195 |
Paclitaxel | 6 mg/mL | 5 mL vial 16.7 mL vial 25 mL vial 50 mL vial | 300.0000 1,196.8000 $1,870.0000 $3,740.0000 | 175 to 200 mg/m2 on day 1 for 3, 21-day cycles | $174.61 to $206.67 | $3,667 to $4,340 |
Carboplatin-paclitaxel | $221.51 to $263.57 | $4,652 to $5,535 | ||||
Carboplatin-paclitaxel plus dostarlimab | ||||||
Carboplatin | 10 mg/mL | 5 mL vial 15 mL vial 45 mL vial 60 mL vial | 70.0000 210.0000 599.9850 775.0020 | AUC 5 to 6 on day 1 for 3, 21-day cyclesb | $46.90 to $56.90 | $985 to |
Paclitaxel | 6 mg/mL | 5 mL vial 16.7 mL vial 25 mL vial 50 mL vial | 300.0000 1,196.8000 $1,870.0000 $3,740.0000 | 175 to 200 mg/m2 on day 1 for 3 21-day cycles | $174.61 to $206.67 | $3,667 to $4,340 |
Dostarlimab (Jemperli) | 50 mg/mL | 10 mL vial | 10,031.0800 | 500 mg on day 1 for 6, 21-day cycles Maintenance: 1,000 mg every 6 weeks thereafter. | $477.67 $477.67 | $10,031 $10,031 |
Carboplatin-Paclitaxel plus dostarlimab | $699.18 to $741.24 | $14,683 to $15,566 | ||||
Dostarlimab (maintenance) | $477.67 | $10,031 | ||||
AUC = area under the curve; CDA-AMC = Canada’s Drug Agency; dMMR = mismatch repair deficient; pMMR = mismatch repair proficient.
Note: All prices are IQVIA DeltaPA wholesale list prices (accessed November 2024), unless otherwise indicated, and do not include dispensing fees.14 Costs were calculated assuming a hypothetical patient weighing 73.8 kg with a body surface area of 1.77 m2 and a glomerular filtration rate of 125.
Follow wastage guidance in the CDA-AMC process document.
aThe maintenance doses in the table have been standardized to a 21-day cycle. The 28-day cost of durvalumab is $11,733.
bThe recommended dose is calculated using a Calvert formula: Dose (mg) = Target AUC × (glomerular filtration rate + 25).21
Appendix 2: Submission Quality
Please note that this appendix has not been copy-edited.
Description | Yes or no | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | Yes | No comment. |
Model has been adequately programmed and has sufficient face validity | Yes | No comment. |
Model structure is adequate for decision problem | Yes | No comment. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | No | Refer to limitations relating to the: improper calculation of state membership, influence of RDI on drug acquisition costs, and methods for the age adjustment of health state utility values. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | No | Refer to limitation relating to the improper characterization of parameter uncertainty. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | Yes | No comment. |
RDI = relative dose intensity.
Appendix 3: Additional Information on the Submitted Economic Evaluation
Please note that this appendix has not been copy-edited.
Detailed Results of the Sponsor’s Base Case
Table 10: Disaggregated Summary of the Sponsor’s Economic Evaluation Results
Parameter | CAR-PAC plus durvalumab | CAR-PAC plus dostarlimab | CAR-PAC |
|---|---|---|---|
Discounted LYs | |||
Total | 14.13 | 14.99 | 4.83 |
By health state or data source | |||
Progression-free | 8.41 | 9.14 | 3.03 |
Progressed disease | 5.72 | 5.84 | 1.80 |
Discounted QALYs | |||
Total | 10.16 | 10.77 | 3.59 |
By health state or data source | |||
Progression-free | 6.37 | 6.87 | 2.34 |
Progressed disease | 3.80 | 3.90 | 1.25 |
AE disutility | −0.0034 | −0.0048 | −0.0029 |
Discounted costs ($) | |||
Total | 651,011 | 419,350 | 140,918 |
Acquisition | 578,355 | 347,410 | 27,290 |
Administration | 8,999 | 4,407 | 1,624 |
Disease management (progression-free) | 29,297 | 31,721 | 10,207 |
Disease management (progressed disease) | 19,211 | 19,684 | 6,041 |
Subsequent therapy | 1,919 | 1,886 | 80,921 |
AEs | 4,168 | 5,338 | 4,053 |
Terminal care | 9,062 | 8,905 | 10,781 |
AE = adverse event; CAR = carboplatin; dMMR = mismatch repair deficient; PAC = paclitaxel.
Source: Sponsor’s pharmacoeconomic submission.1
Appendix 4: Additional Details on the CDA-AMC Reanalyses and Sensitivity Analyses of the Economic Evaluation
Please note that this appendix has not been copy-edited.
Detailed Results of the CDA-AMC Base Case
Table 11: Summary of the Stepped Analysis of the CDA-AMC Base-Case Results
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
Sponsor base case | CAR-PAC | 139,204 | 3.04 | Reference |
CAR-PAC plus dostarlimab | 426,787 | 10.23 | 39,991 | |
Dominated treatments | ||||
CAR-PAC plus durvalumab | 642,246 | 10.21 | Dominated by CAR-PAC plus dostarlimab | |
CDA-AMC reanalysis 1 | CAR-PAC | 139,226 | 3.06 | Reference |
CAR-PAC plus dostarlimab | 430,858 | 11.40 | 34,962 | |
Dominated treatments | ||||
CAR-PAC plus durvalumab | 646,317 | 11.37 | Dominated by CAR-PAC plus dostarlimab | |
CDA-AMC reanalysis 2 | CAR-PAC | 139,204 | 3.14 | Reference |
CAR-PAC plus dostarlimab | 426,787 | 11.03 | 36,443 | |
Dominated treatments | ||||
CAR-PAC plus durvalumab | 642,246 | 11.00 | Dominated by CAR-PAC plus dostarlimab | |
CDA-AMC reanalysis 3 | CAR-PAC | 139,204 | 3.04 | Reference |
CAR-PAC plus dostarlimab | 426,787 | 10.23 | 39,991 | |
Dominated treatments | ||||
CAR-PAC plus durvalumab | 650,307 | 10.21 | Dominated by CAR-PAC plus dostarlimab | |
CDA-AMC base case (deterministic) 1 + 2 + 3 | CAR-PAC | 139,226 | 3.06 | Reference |
CAR-PAC plus dostarlimab | 430,858 | 11.40 | 34,962 | |
Dominated treatments | ||||
CAR-PAC plus durvalumab | 654,378 | 11.37 | Dominated by CAR-PAC plus dostarlimab | |
CDA-AMC base case (probabilistic) 1 + 2 + 3 | CAR-PAC | 140,935 | 3.57 | Reference |
CAR-PAC plus dostarlimab | 423,917 | 12.07 | 33,301 | |
Dominated treatments | ||||
CAR-PAC plus durvalumab | 658,502 | 11.39 | Dominated by CAR-PAC plus dostarlimab | |
CAR = carboplatin; CDA-AMC = Canada’s Drug Agency; dMMR = mismatch repair proficient; ICER = incremental cost-effectiveness ratio; PAC = paclitaxel; QALY = quality-adjusted life-year.
Note: The CDA-AMC reanalysis is based on publicly available prices of the comparator treatments. The results of all steps are presented deterministically unless otherwise indicated, while the cumulative CDA-AMC base case is always presented both deterministically and probabilistically.
Table 12: Disaggregated Summary of CDA-AMC Economic Evaluation Results
Parameter | CAR-PAC plus durvalumab | CAR-PAC plus dostarlimab | CAR-PAC |
|---|---|---|---|
Discounted LYs | |||
Total | 16.23 | 17.16 | 4.80 |
By health state or data source | |||
Progression-free | 8.49 | 9.22 | 2.96 |
Progressed disease | 7.74 | 7.94 | 1.84 |
Discounted QALYs | |||
Total | 12.33 | 13.04 | 3.78 |
By health state or data source | |||
Progression-free | 6.43 | 6.96 | 2.30 |
Progressed disease | 4.97 | 5.11 | 1.18 |
AE Disutility | −0.0030 | −0.0048 | −0.0029 |
Discounted costs ($) | |||
Total | 658,502 | 423,917 | 140,935 |
Acquisition | 581,634 | 347,203 | 27,289 |
Administration | 8,904 | 4,394 | 1,621 |
Disease management (progression-free) | 29,735 | 32,252 | 10,010 |
Disease management (progressed disease) | 25,971 | 26,701 | 6,185 |
Subsequent therapy | 1,894 | 1,874 | 81,042 |
AEs | 4,031 | 5,338 | 4,034 |
Terminal care | 6,333 | 6,155 | 10,754 |
AE = adverse event; CAR-PAC = carboplatin and paclitaxel; CDA-AMC = Canada’s Drug Agency; dMMR = mismatch repair deficient; LY = life-year; QALY = quality-adjusted life-year.
Appendix 5: Submitted Budget Impact Analysis and CDA-AMC Appraisal
Please note that this appendix has not been copy-edited.
Table 14: Summary of Key Take-Aways
Key take-aways of the budget impact analysis |
|---|
|
CAR-PAC = carboplatin and paclitaxel; CDA-AMC = Canada’s Drug Agency; dMMR = mismatch repair deficient.
Summary of Sponsor’s Budget Impact Analysis
The sponsor submitted a BIA estimating the incremental budget impact of reimbursing durvalumab (Imfinzi) for patients with primary advanced or recurrent endometrial cancer who are eligible for systemic therapy.3 The analysis evaluated the introduction of durvalumab in combination with CAR-PAC, followed by durvalumab monotherapy for the dMMR population.3 The BIA was conducted from the perspective of Canadian public drug plans (excluding Quebec) over a 3-year time horizon.22 Key inputs to the BIA are documented in Table 17.
To estimate the eligible population, the sponsor used an epidemiological approach incorporating Canadian data, clinical trial evidence, and expert input.22 The incidence of uterine cancer was derived from national sources, with annual growth rates applied over the time horizon. Among the uterine endometrial cancer cases, the sponsor estimated that 27.5% were dMMR.23-26 The total eligible population was calculated after accounting for diagnostic testing and the proportion of patients initiating systemic therapy. The BIA incorporated the costs of primary and subsequent treatments but excluded diagnostic testing and administration costs from the base case. The sponsor projected durvalumab uptake to reach ███ by year 3.22 The model incorporated TTD and PFS estimates primarily derived from clinical trial data, specifically the DUO-E study, with median values calculated for each treatment arm. Among patients with dMMR cancer, the median TTD was 21.85 months for durvalumab in combination with CAR-PAC, followed by durvalumab maintenance, compared with 4.14 months for CAR-PAC alone. The corresponding PFS was 40.0 months for the durvalumab arm versus 9.0 months for the patients on carboplatin and paclitaxel, patients in the dostarlimab with CAR-PAC had the longest PFS of 45 months.1 The budget impact model assumed that costs were spread annually to reflect expenditure from the public health payer perspective.
Key assumptions:
The sponsor assumed that 100% of patients in the CAR-PAC arm would receive pembrolizumab as subsequent therapy, compared with 0% in the comparator arms.18
The sponsor assumed 100% RDI across all treatments in the BIA.22
Table 15: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3, if appropriate) |
|---|---|
Target population | |
Canadian adult female population | 16,134,760 / 16,690,168 / 17,265,18127 |
Annual % growth in the female population | 3.4%27 |
Incidence rate of uterine cancer | 36.7 / 100,00028 |
Uterine cancer growth rate | |
Proportion with endometrial cancer | 95%31 |
Proportion with advanced EC | |
Proportion of advanced dMMR | |
Proportion of advanced dMMR receiving 1L therapy | 75.2%32 |
Proportion with recurrent stage I or II EC | |
Proportion of recurrent stage I or II that is dMMR | |
Proportion of dMMR recurrent stage I or II receiving 1L therapy | 75.2%32 |
Number of patients eligible for drug under review | 288 / 304 / 321 |
Market uptake (3 years) | |
Uptake (reference scenario) Carboplatin + paclitaxel Dostarlimab + carboplatin + paclitaxel | ███ / ███ / ███22 ███ / ███ / ███22 |
Uptake (new drug scenario) Durvalumab + carboplatin + paclitaxel Carboplatin + paclitaxel Dostarlimab + carboplatin + paclitaxel | ███ / ███ / ███22 ███ / ███ / ███22 ███ / ███ / ███22 |
Cost of treatment (per patient, per cycle) | |
Durvalumab + carboplatin + paclitaxel Carboplatin + paclitaxel Dostarlimab + carboplatin + paclitaxel | $12,850.8936 $4,090.0036 $14,016.0836 |
Cost of diagnostic testing (per patient) | |
Immunohistochemistry test unit cost | $177.2437 |
Proportion of patients in each arm advancing to 2L therapy | |
Durvalumab + CP arm CP arm Dostarlimab + CP arm | 39.5%18 49.4%18 39.5%18 |
Proportion of subsequent treatment (market share) | |
Carboplatin Cisplatin Doxorubicin Paclitaxel | Durvalumab + CP arm ███18 ███18 ███18 ███18 |
Pembrolizumab | CP arm ████18 |
Carboplatin Cisplatin Doxorubicin Paclitaxel | Dostarlimab + CP arm ███18 ███18 ███18 ███18 |
1L = first line; CP = carboplatin + paclitaxel; dMMR = deficient mismatch repair; EC = endometrial cancer.
Summary of the Sponsor’s BIA Results
The cumulative net budget impact of funding durvalumab plus CAR-PAC for patients with primary advanced or recurrent dMMR endometrial cancer, accounting for subsequent therapy costs was $5,782,905 in year 1, $10,330,546 in year 2, and $11,916,217 in year 3, for a 3-year net budget impact of $28,029,667.
CDA-AMC Appraisal of the Sponsor’s BIA
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Uncertainty in the proportion initiating subsequent therapy: The sponsor assumed that █████ of patients in the durvalumab and dostarlimab arms would not initiate second-line therapy, with only █████ progressing to second-line treatment. This assumption was applied to the dostarlimab arm, based on clinical trial evidence demonstrating comparable efficacy and the expectation of similar outcomes between the 2 treatments. For patients initiating second-line therapy, the sponsor assumed the following distribution: carboplatin (███), cisplatin (███), doxorubicin hydrochloride (███), and paclitaxel (███). In contrast, the sponsor assumed that █████ of patients in the CAR-PAC arm would initiate second-line therapy, with 100% of these patients receiving pembrolizumab. This assumption increases the cost of subsequent therapy for the CAR-PAC arm, introducing a bias in favour of durvalumab arm.
CDA-AMC conducted a scenario analysis assuming that 70% of patients receiving durvalumab or dostarlimab and 80% of patients receiving CAR-PAC initiated second-line therapy based on expert opinion. Additionally, clinical experts consulted by CDA-AMC indicated that at least 30% of patients would receive doxorubicin as a second-line therapy, with the proportion reallocated equally from the comparators.
CDA-AMC Reanalyses of the BIA
In the absence of more reliable estimates to inform the parameters of the BIA, the sponsor’s base case was maintained. CDA-AMC anticipates that the budget impact of durvalumab will be sensitive to inputs and assumptions related to the proportion of patients initiating subsequent therapy and the market share of doxorubicin. This was reflected in a scenario analysis conducted by CDA-AMC, which examined the impact of increasing both the proportion of patients initiating subsequent treatment and doxorubicin’s market share. In this scenario, it was assumed that 70% of patients in the durvalumab and dostarlimab arms and 80% of patients in the CAR-PAC arm would initiate subsequent treatment, with doxorubicin capturing 30% of the subsequent treatment market share.
Table 16: CDA-AMC Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
1. Proportion initiating subsequent therapy | ████% (durvalumab + CP arm) ████% (CP arm) | 70% (durvalumab + CP arm) 80% (CP arm) |
2. The market share of doxorubicin | 15% | 30% |
3. Durvalumab unit price | $938 $3,911 | 47% price reduction ($497) 47% price reduction ($2,073) |
CDA-AMC = Canada’s Drug Agency; CP = carboplatin + paclitaxel.
The results of the CDA-AMC stepwise reanalysis are presented in summary format in Table 17 and a more detailed breakdown is presented in Table 18.
Increasing the proportion of patients initiating subsequent therapy to 70% in the durvalumab arm and 80% in the CAR-PAC arm reduced the net budget impact to $21,065,803, demonstrating the sensitivity of the budget impact to changes in subsequent therapy assumptions. When doxorubicin was assumed to capture 30% of the market share, the net budget impact remained the same at $28,029,667.
Additionally, a scenario analysis was conducted to assess the effect of a price reduction for durvalumab identified in the economic evaluation. Assuming a 47% price reduction, the net budget impact of reimbursing durvalumab with CAR-PAC was estimated at $6,077,097.
Table 17: Summary of the CDA-AMC Reanalyses of the Budget Impact Analysis
Stepped analysis | Three-year total ($) |
|---|---|
Submitted base case | $28,029,667 |
CDA-AMC scenario 1: Proportion initiating subsequent therapy | $21,065,803 |
CDA-AMC scenario 2: Doxorubicin market share | $28,029,667 |
CDA-AMC scenario 3: 47% price reduction | $6,077,097 |
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency.
Note: All reanalyses are based on IQVIA DeltaPA wholesale list prices (accessed November 2024), unless otherwise indicated.
CDA-AMC conducted the following scenario analyses to address remaining uncertainty, using the CDA-AMC base case (results are provided in Table 18:
The proportion initiating subsequent treatment was assumed to be 70% for both durvalumab and dostarlimab and 80% among the CAR-PAC arm.
Increased the proportion of patients receiving doxorubicin hydrochloride to 30% by reallocating 15% equally from the other comparators.
Assuming that the price of durvalumab is reduced by 47% (CDA-AMC estimated price reduction from the cost-utility analysis).
Table 18: Detailed Breakdown of the CDA-AMC Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|---|
Submitted base case | Reference | 20,368,090 | 47,831,448 | 67,390,760 | 83,395,764 | 218,986,062 |
New drug | 20,368,090 | 53,614,353 | 77,721,306 | 95,311,981 | 247,015,729 | |
Budget impact | 0 | 5,782,905 | 10,330,546 | 11,916,217 | 28,029,667 | |
CDA-AMC scenario analysis: 47% price reduction | Reference | 20,368,090 | 47,831,448 | 67,390,760 | 83,395,764 | 218,986,062 |
New drug | 20,368,090 | 50,508,250 | 69,916,088 | 84,270,730 | 225,063,159 | |
Budget impact | 0 | 2,676,802 | 2,525,328 | 874,966 | 6,077,097 | |
CDA-AMC scenario analysis: 70% and 80% initiating subsequent therapy for durvalumab and CAR-PAC, respectively | Reference | 22,952,368 | 57,772,314 | 81,557,554 | 100,011,173 | 262,293,408 |
New drug | 22,952,368 | 63,043,676 | 89,639,073 | 107,724,094 | 283,359,211 | |
Budget impact | 0 | 5,271,362 | 8,081,520 | 7,712,922 | 21,065,803 | |
CDA-AMC scenario analysis: 30% uptake for doxorubicin | Reference | 20,368,090 | 47,831,448 | 67,390,760 | 83,395,764 | 218,986,062 |
New drug | 20,368,090 | 53,614,353 | 77,721,306 | 95,311,981 | 247,015,729 | |
Budget impact | 0 | 5,782,905 | 10,330,546 | 11,916,217 | 28,029,667 |
BIA = budget impact analysis; CAR-PAC = carboplatin and paclitaxel; CDA-AMC = Canada’s Drug Agency.
Note: All reanalyses are based on IQVIA DeltaPA wholesale list prices (accessed November 2024), unless otherwise indicated.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@cda-amc.ca.