Drugs, Health Technologies, Health Systems
Reimbursement Review
Momelotinib (Ojjaara)
Sponsor: GlaxoSmithKline Inc.
Therapeutic area: Myelofibrosis
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
AE
adverse event
AESI
adverse event of special interest
AML
acute myeloid leukemia
BAT
best available therapy
CDA-AMC
Canada’s Drug Agency
CI
confidence interval
CMH
Cochran–Mantel–Haenszel
CMPNN
Canadian MPN Network
CTCAE
Common Terminology Criteria for Adverse Events
DIPSS
Dynamic International Prognostic Scoring System
ECOG PS
Eastern Cooperative Oncology Group Performance Status
ESA
erythropoiesis-stimulating agent
ET
essential thrombocythemia
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HRQoL
health-related quality of life
HSCT
hematopoietic stem cell transplant
IPSS
International Prognostic Scoring System
ITT
intention to treat
IWG-MRT
International Working Group for Myelofibrosis Research and Treatment
JAK
Janus kinase
LLSC
Leukemia & Lymphoma Society of Canada
LOCF
last observation carried forward
MF
myelofibrosis
MFSAF
Myelofibrosis Symptom Assessment Form
MPN
myeloproliferative neoplasm
MPN-SAF
Myeloproliferative Neoplasm Symptom Assessment Form
NR
not reported
OH-CCO
Ontario Health – Cancer Care Ontario
OS
overall survival
PMF
primary myelofibrosis
PV
polycythemia vera
RBC
red blood cell
RCT
randomized controlled trial
SAE
serious adverse event
SD
standard deviation
SRR
splenic response rate
TEAE
treatment-emergent adverse event
TSS
total symptom score
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Momelotinib (Ojjaara), 200 mg, 150 mg, and 100 mg tablets |
Sponsor | GlaxoSmithKline Inc. |
Indication | For the treatment of splenomegaly and/or disease-related symptoms, in adult patients with intermediate or high-risk primary myelofibrosis (MF), post polycythemia vera MF or post essential thrombocythemia MF who have moderate to severe anemia |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | November 8, 2024 |
Recommended dose | 200 mg taken orally once daily |
NOC = Notice of Compliance.
Introduction
Myelofibrosis (MF) is a rare, chronic, and progressive bone marrow disorder categorized as a Philadelphia chromosome-negative myeloproliferative neoplasm (MPN).1,2 Characterized by the excessive production of reticulin and collagen fibres, MF leads to bone marrow fibrosis, bone marrow failure, systemic inflammation, and splenomegaly.3 MF can develop as primary myelofibrosis (PMF) or as secondary forms following essential thrombocythemia (ET) or polycythemia vera (PV). PMF is the most aggressive type and has the potential to progress into acute myeloid leukemia (AML).3 The incidence of PMF in Canada is estimated at 0.80 per 100,000 person-years,4 with approximately 200 new cases diagnosed annually, accounting for 1% of all hematological malignancies.5 Key clinical manifestations of MF include severe anemia, thrombocytopenia, marked hepatosplenomegaly, and constitutional symptoms such as fatigue, night sweats, and unintentional weight loss. Current treatment options primarily include Janus kinase (JAK) inhibitors like ruxolitinib, which are aimed at reducing splenomegaly and managing symptoms.6-9 However, unmet needs remain, especially for patients who experience disease progression after JAK inhibitor therapy.
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of momelotinib, administered orally at a dosage of 200 mg once daily, for the treatment of splenomegaly and/or disease-related symptoms in adult patients with intermediate-risk or high-risk PMF, post-PV MF, or post-ET MF who have moderate to severe anemia. Momelotinib, which also inhibits ACVR1, may provide additional benefits, particularly in managing anemia, by restoring iron homeostasis and reducing the need for RBC transfusions. This drug has not been previously reviewed by Canada’s Drug Agency (CDA-AMC).
Perspectives of Patients, Clinicians, and Drug Programs
The information in this section is a summary of the input provided by the patient and clinician groups who responded to the CDA-AMC call for input and from the clinical experts consulted by CDA-AMC for the purpose of this review.
Patient Input
The Leukemia & Lymphoma Society of Canada (LLSC) and the Canadian MPN Network (CMPNN) jointly provided patient input for this review, sourcing information from 3 online surveys conducted between March 2024 and May 2024, with a total of 73 respondents. Heal Canada also provided input for this review, sourcing information mainly from surveys and interviews. The surveys included in both inputs gathered insights from patients with MF and their caregivers, focusing on their lived experiences and specific interactions with the drug under review, momelotinib. MF profoundly impacts patients and their families, affecting physical, emotional, and financial aspects of daily life. Many patients reported relying heavily on caregiver support, which placed significant burdens on both parties. Key outcomes important to patients include the management of fatigue, anemia, and spleen size, with a particular emphasis on reducing symptom burden, improving quality of life, and decreasing the need for blood transfusions. Notably, 73% of respondents with experience using momelotinib felt it improved their quality of life.
Clinician Input
Input From Clinical Experts
Clinical experts consulted by CDA-AMC identified significant unmet needs in the current treatment landscape for MF. While existing JAK inhibitors like ruxolitinib and fedratinib effectively address symptoms such as splenomegaly and constitutional symptoms, they do not modify the underlying disease or delay its progression. Additionally, hematopoietic stem cell transplant (HSCT), the only potentially curative treatment, is viable for less than 10% of patients due to its high associated morbidity and mortality. Experts emphasized the need for therapies that provide more durable responses, better management of anemia, and potential modification of disease progression.
Experts suggested that momelotinib could be an important option for patients with MF who require JAK inhibitor therapy and have clinically significant anemia. Momelotinib would be particularly beneficial for patients not previously treated with JAK inhibitors or who have developed anemia or intolerance on existing JAK inhibitor therapy. The experts noted that momelotinib could be used in first-line settings and as a second-line or third-line treatment for patients with clinically relevant anemia and MPN symptoms. However, the experts noted that momelotinib might be less suitable for patients whose primary issue is symptomatic splenomegaly in the context of ruxolitinib resistance or intolerance.
Based on the input provided by the clinical experts, the patient population most likely to benefit from momelotinib includes those with MF who have not been previously treated with JAK inhibitors and have splenomegaly or MPN symptoms and clinically relevant anemia, as well as those experiencing anemia or intolerance on other JAK inhibitor therapies. Patients whose main issue is splenomegaly without accompanying anemia or MPN symptoms may be less likely to benefit from momelotinib.
The experts recommended assessing the response to momelotinib through patient-reported outcomes, physical examinations (including spleen size), and anemia parameters such as hemoglobin levels and transfusion frequency. The experts suggested that responses should be evaluated approximately every 3 months, with a clinically meaningful response being indicated by subjective improvements, reduced spleen size, and improved anemia metrics. Treatment discontinuation should be considered if there is no response after about 6 months, if there is a loss of a prior response, or if the patient experiences grade 3 adverse events (AEs) that do not resolve with dose modification.
The experts advised that momelotinib should be prescribed and monitored by hematologists or oncologists with expertise in MF, ideally in hospital outpatient clinics or specialty settings where appropriate expertise is available. Regional access to such specialists should be considered when prescribing momelotinib.
Clinician Group Input
Clinician group input on the review of momelotinib was provided by 15 clinicians from LLSC and the CMPNN Clinician Group, as well as the Ontario Health – Cancer Care Ontario (OH-CCO) Hematology Disease Site Drug Advisory Committee. Both clinician groups emphasized the significant unmet need for effective treatments to manage anemia in MF, aligning with the clinical experts consulted by CDA-AMC, who also identified anemia management as a critical challenge. The clinician groups and the clinical experts all recognized the potential of momelotinib to benefit patients with MF-associated anemia; the clinician groups noted, however, a lack of evidence on the reduction in the risk of progression to acute leukemia associated with momelotinib. The clinician groups highlighted that assessment of response to momelotinib in clinical practice should include improvements in hemoglobin, reductions in transfusions, and stable disease or improvement in symptom burden, which are also consistent with the views of the clinical experts consulted by CDA-AMC. These clinician groups believe that momelotinib could be relevant to clinical practice, especially for patients who experience anemia and transfusion dependence, although the clinician groups also caution that it does not address all aspects of disease progression.
Drug Program Input
The drug plans submitted questions relating to the initiation and generalizability of momelotinib and the associated funding algorithm.
Clinical Evidence
Systematic Review
Description of Studies
Three pivotal randomized controlled trials (RCTs) were included in the sponsor’s submission to assess the efficacy and safety of momelotinib for MF in adults. The SIMPLIFY-1 trial (N = 432) was a phase III, double-blind, multicentre study that compared momelotinib with ruxolitinib in patients with PMF, post-PV MF, or post-ET MF who had not previously been treated with JAK inhibitors. The primary end point was the splenic response rate (SRR) at week 24, defined as a reduction of 35% or more in spleen volume from baseline. The secondary outcomes included the total symptom score (TSS) response rate, defined as the proportion of patients experiencing a reduction of 50% or more from baseline in symptom burden, and transfusion independence, defined as the proportion of patients who do not require any red blood cell (RBC) transfusions for a period of 12 weeks while maintaining hemoglobin levels at or more than 8 g/dL. The SIMPLIFY-2 trial (N = 156) was a phase III, open-label, multicentre study that evaluated the efficacy of momelotinib versus the best available therapy (BAT) — 88.5% of patients received ruxolitinib as the BAT of choice — in patients with MF who had been previously treated with ruxolitinib but experienced either an inadequate response or intolerance. The primary end point was the SRR at week 24, with the secondary outcomes including the TSS response rate and overall survival (OS). The MOMENTUM trial (N = 195) was a phase III, double-blind, multicentre study that focused on patients with symptomatic and anemic MF who had received prior JAK inhibitor therapy. The trial compared momelotinib with danazol, with the primary end point being the TSS response rate at week 24. The secondary outcomes included SRR, transfusion independence, and OS.
Baseline characteristics across the studies showed a population predominantly composed of patients with intermediate-2–risk or high-risk MF. Across the 3 trials, more than one-half of patients were male, the majority were white, and the mean age was mid-to-late 60s. In the SIMPLIFY-1 trial, 56.5% of patients were male and 43.5% were female, with 9.2% identifying as Asian, 0.9% as Black, 82.6% as white, and 7.9% as “other” or “not reported.” In the SIMPLIFY-2 trial, 59.6% of patients were male and 40.4% were female, with 3.8% identifying as Black, 81.4% as white, and 14.7% as “other” or “not reported.” In the MOMENTUM trial, 63.1% of patients were male and 36.9% were female, with 9.2% identifying as Asian, 2.1% as Black, 80.5% as white, and 6.2% as “other.” With the exception of anemia-related characteristics in the MOMENTUM trial, where only patients with a hemoglobin level of less than 10 g/dL were included, the rest of the baseline characteristics were relatively consistent across the 3 trials, with relatively balanced demographic and clinical characteristics between treatment arms.
Efficacy Results
In the SIMPLIFY-1 trial, 66.5% of patients treated with momelotinib experienced transfusion independence at week 24, compared to 49.3% in the ruxolitinib group, with a proportion difference of 0.18 (95% confidence interval [CI], 0.09 to 0.26). In the SIMPLIFY-2 trial, 43.3% of patients in the momelotinib group experienced transfusion independence at week 24, compared with 21.2% in the BAT group, with a proportion difference of 0.23 (95% CI, 0.09 to 0.37). In the MOMENTUM study, 30.8% of patients treated with momelotinib experienced transfusion independence at week 24, compared to 20.0% in the danazol group (proportion difference = 10.99%; 95% CI, –0.80% to 22.77%), with an adjusted proportion difference noninferiority test, which targeted 80% retention of the effect of danazol, at 14.77% (95% CI, 3.13% to 26.41%; P = 0.0064).
The mean rate of RBC transfusions at week 24 in the SIMPLIFY-1 trial was 0.5 units per patient-month in the momelotinib group, versus 1.0 unit in the ruxolitinib group, with a transfusion rate ratio of 0.28 (95% CI, 0.19 to 0.43). In the SIMPLIFY-2 trial, the mean transfusion rate was 1.6 units in the momelotinib group, compared to 1.8 units in the BAT group (transfusion rate ratio = 0.80; 95% CI, 0.49 to 1.31). In the MOMENTUM trial, patients in the momelotinib group received a mean 6.6 units, compared with a mean 10.9 units in the danazol group, with a treatment difference of –5.66 units (95% CI, –10.65 to –0.68 units).
In the SIMPLIFY-1 trial, 26.5% of patients in the momelotinib group experienced a splenic response at week 24, compared to 29.5% in the ruxolitinib group. Momelotinib met the noninferiority criterion, with an adjusted proportion difference (targeting 60% retention of the effect of ruxolitinib) of 0.09 (95% CI, 0.02 to 0.16; P = 0.014), but it did not demonstrate superiority (proportion difference = –0.03; 95% CI, –0.12 to 0.05; P = 0.45). In the SIMPLIFY-2 trial, the SRR was 6.7% in the momelotinib group and 5.8% in the BAT group (proportion difference = 0.01; 95% CI, –0.09 to 0.10; P = 0.90). In the MOMENTUM trial, the SRR was 23.1% in the momelotinib group versus 3.1% in the danazol group (proportion difference = 19.37%; 95% CI, 10.96% to 27.77%; P = 0.001).
In the SIMPLIFY-1 trial, 28.4% of patients in the momelotinib group experienced a TSS response at week 24, compared to 42.2% in the ruxolitinib group (proportion difference = –14.0%; 95% CI, –23.0% to –5.0%; P = 0.9985). A noninferiority test that targeted 67% retention of ruxolitinib failed to the predefined noninferiority margin, where the lower bound of the 2-sided 95% CI should be greater than 0. Specifically, the adjusted proportion difference noninferiority testing was 0.00 (95% CI, –0.08 to 0.08; P = 0.98). In the SIMPLIFY-2 trial, 26.2% of patients in the momelotinib group experienced a TSS response, compared to 5.9% in the BAT group, with a proportion difference of 0.20 (95% CI, 0.09 to 0.32). In the MOMENTUM study, 24.6% of patients in the momelotinib group experienced a TSS response, compared to 9.2% in the danazol group, with a proportion difference of 15.67% (95% CI, 5.54% to 25.81%; P = 0.0095).
Harms Results
Across the trials, most patients treated with momelotinib experienced at least 1 AE. In the SIMPLIFY-1 trial, 92.5% of patients in the momelotinib group experienced at least 1 AE, compared to 95.4% in the ruxolitinib group. In the SIMPLIFY-2 trial, the rates were 97.1% in the momelotinib group and 88.5% in the BAT group. For the MOMENTUM study, 93.8% of patients in the momelotinib group reported at least 1 AE, compared to 95.4% in the danazol group. Thrombocytopenia and anemia were commonly reported AEs across these trials. In the SIMPLIFY-1 trial, thrombocytopenia occurred in 18.7% of patients in the momelotinib group and 29.2% of patients in the ruxolitinib group, while anemia was reported in 14.5% of patients in the momelotinib group and 37.5% of patients in the ruxolitinib group. In the SIMPLIFY-2 trial, thrombocytopenia was observed in 10.6% of patients in the momelotinib group and 5.8% of patients in the BAT group, and anemia was reported in 13.5% of patients in the momelotinib group, compared to 17.3% in the BAT group. In the MOMENTUM study, thrombocytopenia was seen in 22.3% of patients in the momelotinib group versus 10.8% of patients in the danazol group, while anemia was observed in 7.7% of patients in the momelotinib group and 6.2% of patients in the danazol group.
Grade 3 or 4 AEs were observed in a proportion of patients across all studies. In the SIMPLIFY-1 trial, 34.6% of patients in the momelotinib group experienced grade 3 or 4 AEs, compared to 43.5% in the ruxolitinib group. In the SIMPLIFY-2 trial, 54.8% of patients in the momelotinib group experienced grade 3 or 4 AEs versus 42.3% in the BAT group. In the MOMENTUM trial, 48.5% of patients in the momelotinib group reported grade 3 or 4 AEs, compared to 63.1% in the danazol group. Thrombocytopenia and anemia were the most common grade 3 or 4 AEs. In the SIMPLIFY-1 trial, grade 3 or 4 thrombocytopenia was reported in 7.0% of patients in the momelotinib group and 4.6% of patients in the ruxolitinib group, while grade 3 or 4 anemia was reported in 6.1% of patients in the momelotinib group and 22.7% of patients in the ruxolitinib group. In the SIMPLIFY-2 trial, grade 3 or 4 thrombocytopenia was observed in 10.6% of patients in the momelotinib group versus 5.8% of patients in the BAT group, and grade 3 or 4 anemia was reported in 13.5% of patients in the momelotinib group compared to 17.3% in the BAT group. In the MOMENTUM trial, grade 3 or 4 thrombocytopenia was seen in 16.9% of patients in the momelotinib group and 7.7% of patients in the danazol group, while grade 3 or 4 anemia was reported in 7.7% of patients in the momelotinib group and 6.2% of patients in the danazol group.
Serious AEs (SAEs) were frequent across the trials. In the SIMPLIFY-1 trial, 22.9% of patients in the momelotinib group experienced at least 1 SAE, compared to 18.1% in the ruxolitinib group. In the SIMPLIFY-2 trial, 35.6% of patients in the momelotinib group experienced at least 1 SAE versus 23.1% in the BAT group. In the MOMENTUM trial, 34.6% of patients in the momelotinib group reported at least 1 SAE, compared to 40.0% in the danazol group. Common SAEs included anemia, pneumonia, and sepsis. In the SIMPLIFY-1 trial, anemia was observed in 1.9% of patients in the momelotinib group and 3.7% of patients in the ruxolitinib group, and pneumonia was reported in 1.9% of patients in the momelotinib group and 1.4% of patients in the ruxolitinib group. In the SIMPLIFY-2 trial, sepsis was observed in 2.9% of patients in the momelotinib group; no cases were reported in the BAT group. In the MOMENTUM trial, anemia was seen in 3.8% of patients in the momelotinib group versus 4.6% in the danazol group, and pneumonia was reported in 2.3% of patients in the momelotinib group and 9.2% of patients in the danazol group.
Discontinuations due to AEs were relatively common. In the SIMPLIFY-1 trial, 12.6% of patients in the momelotinib group discontinued treatment due to AEs, compared to 5.6% in the ruxolitinib group. In the SIMPLIFY-2 trial, discontinuation rates were 21.2% in the momelotinib group versus 1.9% in the BAT group. In the MOMENTUM trial, 17.7% of patients in the momelotinib group discontinued treatment, compared to 23.1% in the danazol group. Thrombocytopenia was a key reason for discontinuation, especially in the SIMPLIFY-2 trial, where it led to treatment cessation in 4.8% of patients in the momelotinib group; there were no reported cases in the BAT group. In the MOMENTUM trial, thrombocytopenia caused discontinuation in 0.8% of patients in the momelotinib group versus 3.1% of patients in the danazol group.
Mortality rates varied across the studies. In the SIMPLIFY-1 trial, 3.7% of patients in the momelotinib group died, compared to 2.8% of patients in the ruxolitinib group. In the SIMPLIFY-2 trial, mortality was 7.7% in the momelotinib group and 9.6% in the BAT group. In the MOMENTUM trial, 29.2% of patients in the momelotinib group died, compared to 30.8% in the danazol group. In the SIMPLIFY-1 trial, most deaths in the momelotinib group were due to treatment-emergent AEs (TEAEs), while in the MOMENTUM trial, a notable number of deaths were linked to TEAEs in both the momelotinib and danazol groups.
Notable harms included peripheral neuropathy, reported in 10.3% of patients in the momelotinib group in the SIMPLIFY-1 trial and 11.5% in the SIMPLIFY-2 trial, with fewer cases in the comparator groups (5.6% in the ruxolitinib group in the SIMPLIFY-1 trial and none reported in the BAT group in the SIMPLIFY-2 trial). In the MOMENTUM trial, infections were prevalent, affecting 33.8% of patients in the momelotinib group and 35.4% of patients in the danazol group. Other significant AEs in the MOMENTUM trial included hemorrhage (21.5% in the momelotinib group versus 18.5% in the danazol group), malignancies (5.4% in the momelotinib group versus 9.2% in the danazol group), thromboembolism (3.8% in the momelotinib group versus 9.2% in the danazol group), and transformation to AML (3.1% in the momelotinib group versus 4.6% in the danazol group).
Critical Appraisal
The studies included in this review are generally well designed, with RCTs and active comparator arms, which strengthen their internal validity. The SIMPLIFY-1 and MOMENTUM trials were double-blind studies, while the SIMPLIFY-2 trial was open label, increasing the potential for bias, particularly in subjective outcomes like the TSS. The studies used robust randomization and allocation concealment methods, with noninferiority to be met if the lower 95% CI did not go below the null; this margin was established based on prior evidence, which was supported by clinical experts. However, limited clinical rationale was provided for the threshold used to determine the comparator efficacy preservation. The open-label design of the SIMPLIFY-2 trial introduces a risk of bias in favour of momelotinib, especially for patient-reported outcomes. A significant limitation across all studies is the high rate of treatment discontinuation, which was particularly imbalanced in the MOMENTUM trial, where more patients discontinued treatment in the danazol group than in the momelotinib group. Additionally, the lack of adjustment for type I error (multiple testing) in several efficacy outcomes further complicates the interpretation of these results, particularly in the SIMPLIFY-2 trial, where the primary objectives were not met, rendering subsequent analyses nominal and unadjusted.
The external validity of the studies is supported by their attempt to capture a representative population of patients with MF, including those who have not previously been treated with a JAK inhibitor, those who have previously been treated with a JAK inhibitor, and those who have anemia. The baseline characteristics of the study populations were consistent with those seen in clinical practice in Canada, according to the clinical experts. However, the studies have limitations in generalizability due to the lack of comparisons against certain relevant treatments, such as fedratinib or hydroxyurea, particularly in the Canadian context. The use of danazol in the MOMENTUM trial, which is uncommon in Canadian practice, further limits the applicability of the results. Additionally, the short 24-week duration of the studies is insufficient to assess long-term outcomes such as survival and disease progression, which are critical in MF management. The high rates of treatment discontinuation also limit the generalizability of the findings to patients who are likely to remain on therapy, potentially skewing results toward those who respond well to treatment. Lastly, the absence of established minimal important differences for key outcomes diminishes the ability to interpret the clinical significance of the differences observed between momelotinib and comparators.
Table 2: Summary of Findings for Momelotinib vs. Ruxolitinib for Patients With Myelofibrosis Not Previously Treated With JAK Inhibitors
Outcome and follow-up | Patients (studies), N | Relative effect (95% CI) | Absolute effects | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
Ruxolitinib | Momelotinib | Difference | |||||
Blood transfusion | |||||||
Transfusion independence response rate Follow-up: week 24 | 432 (1 RCT) | NR | 49.3 per 100 | 66.5 per 100 (95% CI, 59.8 to 72.8 per 100) | 18.0 more per 100 (95% CI, 9.0 to 26.0 more) | Higha,b | Momelotinib results in an increase in the number of patients who are transfusion independent compared to ruxolitinib. The clinical relevance of the increase is uncertain. |
Rate of RBC transfusion Follow-up: week 24 | 432 (1 RCT) | Rate ratio = 0.28 (0.19 to 0.43) | 1.0 mean units per month | 0.5 mean units per month (SD = 1.27) | NR | Highb,c | Momelotinib results in a decrease in amount of blood transfusion units per month when compared to ruxolitinib. The clinical relevance of the decrease is uncertain. |
Splenic response (spleen volume reduction of ≥ 35% from baseline at the week 24 assessment as measured by MRI or CT scans) | |||||||
Splenic response rate Follow-up: week 24 | 432 (1 RCT) | NR | 29.5 per 100 | 26.5 per 100 (95% CI, 20.74 to 32.94 per 100) | 3 less per 100 (95% CI, 12.0 less to 5.0 more) | Moderateb,d | Momelotinib likely results in little-to-no difference in splenic response rate when compared to ruxolitinib. |
TSS response (≥ 50% reduction in TSS from baseline at the week 24 assessment as measured by the modified MPN-SAF) | |||||||
TSS response rate Follow-up: week 24 | 432 (1 RCT) | NR | 42.2 per 100 | 28.4 per 100 (95% CI, 22.45 to 35.03 per 100) | 14.0 less per 100 (95% CI, 23.0 to 5.0 less) | Higha,b | Momelotinib results in a decrease in the number of patients who experience response based on total symptom score compared to ruxolitinib. The clinical relevance of the decrease is uncertain. |
Harms | |||||||
Serious adverse events Follow-up: week 24 | 432 (1 RCT) | NR | 18.2 per 100 | 22.9 per 100 (NR) | 5 more per 100 (95% CI, 3 less to 12 more) | Lowb,e | Momelotinib may result in an increase in the proportion of patients who experience ≥ 1 serious adverse event compared with ruxolitinib. The clinical importance of the increase is uncertain. |
CDA-AMC = Canada’s Drug Agency; CI = confidence interval; JAK = Janus kinase; MID = minimal important difference; MPN-SAF = Myeloproliferative Neoplasm Symptom Assessment Form; NR = not reported; RBC = red blood cell; RCT = randomized controlled trial; SD = standard deviation; TSS = total symptom score; vs. = versus.
Notes: Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aNo published between-group MID was identified, and the clinical experts consulted by CDA-AMC were unable to estimate a threshold for clinically important effects; therefore, the null was used. Did not rate down for imprecision; a between-group difference of larger than the null and a CI that excludes the null suggest benefit as judged by the CDA-AMC review team.
bEnd point not adjusted for multiple testing, thus should be used as supportive evidence.
cResults for absolute between-group difference with 95% CI for the full study population were not available. Furthermore, no MID was identified, and the clinical experts consulted by CDA-AMC were unable to estimate a threshold for clinically important effects. Therefore, the null was used in relation to the relative treatment effect. Did not rate down for imprecision; a relative treatment effect larger than the null and a CI that excludes the null suggest benefit as judged by the CDA-AMC review team.
dNo published between-group MID was identified, and the clinical experts consulted by CDA-AMC were unable to estimate a threshold for clinically important effects; therefore, the null was used. Rated down 1 level for serious imprecision as the lower bound of the CI suggests harm and the upper bound of the 95% CI suggests benefit and/or little-to-no difference.
eNo published between-group MID was identified, and the clinical experts consulted by CDA-AMC were unable to estimate a threshold for clinically important effects. Rated down 2 levels for very serious imprecision as the lower bound of the CI suggests benefit and the upper bound of the 95% CI suggests harm.
Sources: GSK Data on File, 2021 (SIMPLIFY-1 Clinical Study Report);10 GSK Data on File, 2021 (SIMPLIFY-2 Clinical Study Report);11 GSK Data on File, 2023 (MOMENTUM Clinical Study Report).12 Details included in the table are from the sponsor’s summary of clinical evidence.
Table 3: Summary of Findings for Momelotinib vs. Best Available Therapy for Patients With Myelofibrosis Previously Treated With JAK Inhibitors
Outcome and follow-up | Patients (studies), N | Relative effect (95% CI) | Absolute effects | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
Best available therapy | Momelotinib | Difference | |||||
Blood transfusion | |||||||
Transfusion independence response rate Follow-up: week 24 | 156 (1 RCT) | NR | 21.2 per 100 | 43.3 per 100 (95% CI, 33.59 to 53.35 per 100) | 23.0 more per 100 (95% CI, 9.0 to 37.0 more) | Moderatea,f | Momelotinib likely results in an increase in the number of patients who are transfusion independent compared to best available therapy. The clinical relevance of the increase is uncertain. |
Rate of RBC transfusion Follow-up: week 24 | 156 (1 RCT) | Rate ratio = 0.80 (0.49 to 1.31) | 1.8 mean units per month | 1.6 mean units per month (SD = 2.09) | NR | Lowb,c | Momelotinib may result in a decrease in amount of blood transfusion units per month when compared to best available therapy. The clinical relevance of the increase is uncertain. |
Splenic response (spleen volume reduction of ≥ 35% from baseline at the week 24 assessment as measured by MRI or CT scans) | |||||||
Splenic response rate Follow-up: week 24 | 156 (1 RCT) | NR | 5.8 per 100 | 6.7 per 100 (95% CI, 2.75 to 13.38 per 100) | 1 more per 100 (95% CI, 9 less to 10.0 more) | Very lowd | The evidence is very uncertain about the effect of momelotinib on splenic response rate when compared to best available therapy. |
TSS response (≥ 50% reduction in TSS from baseline at week 24 assessment as measured by the modified MPN-SAF) | |||||||
TSS response rate Follow-up: week 24 | 156 (1 RCT) | NR | 5.9 per 100 | 26.2 per 100 (95% CI, 18.04 to 35.80 per 100) | 20.0 more per 100 (95% CI, 9 to 32 more) | Lowc,e | Momelotinib may result in an increase in the number of patients who experience response based on total symptom score compared to best available therapy. The clinical relevance of the increase is uncertain. |
Harms | |||||||
Serious adverse events Follow-up: week 24 | 156 (1 RCT) | NR | 23.1 per 100 | 35.6 per 100 (NR) | 13 more per 100 (95% CI, 2 less to 27 more) | Lowc,f | Momelotinib may result in an increase in the proportion of patients who experience ≥ 1 serious adverse event compared with best available therapy. The clinical importance of the increase is uncertain. |
CDA-AMC = Canada’s Drug Agency; CI = confidence interval; JAK = Janus kinase; MID = minimal important difference; MPN-SAF = Myeloproliferative Neoplasm Symptom Assessment Form; NR = not reported; RBC = red blood cell; RCT = randomized controlled trial; SD = standard deviation; TSS = total symptom score; vs. = versus.
aNo published between-group MID was identified, and the clinical experts consulted by CDA-AMC were unable to estimate a threshold for clinically important effects; therefore, the null was used. Did not rate down for imprecision; a between-group difference of larger than the null and a CI that excludes the null suggest benefit as judged by the CDA-AMC review team. Rated down 1 level for serious risk of bias due to missing data and the lack of a washout period.
bResults for absolute between-group difference with 95% CI for the full study population were not available. Furthermore, no MID was identified, and the clinical experts consulted by CDA-AMC were unable to estimate a threshold for clinically important effects Rated down 2 levels for very serious imprecision as the lower bound of the CI suggests comparative harm and the upper bound of the 95% CI suggests comparative benefit.
cEnd point not adjusted for multiple testing, thus should be used as supportive evidence.
dNo published between-group MID was identified, and the clinical experts consulted by CDA-AMC were unable to estimate a threshold for clinically important effects; therefore, the null was used. Rated down 2 levels for very serious imprecision as the lower bound of the 95% CI suggests serious harm and the upper bound of the 95% CI suggests serious benefit. Rated down 1 level for serious risk of bias due to missing data and lack of a washout period.
eNo published between-group MID was identified, and the clinical experts consulted by CDA-AMC were unable to estimate a threshold for clinically important effects; therefore, the null was used. Rated down 2 levels for very serious risk of bias due to open-label design in a subjective outcome, missing data, and lack of a washout period.
fNo published between-group MID was identified, and the clinical experts consulted by CDA-AMC were unable to estimate a threshold for clinically important effects. Rated down 2 levels for very serious imprecision as the lower bound of the CI suggests benefit and the upper bound of the 95% CI suggests harm.
Sources: GSK Data on File, 2021 (SIMPLIFY-1 Clinical Study Report);10 GSK Data on File, 2021 (SIMPLIFY-2 Clinical Study Report);11 GSK Data on File, 2023 (MOMENTUM Clinical Study Report).12 Details included in the table are from the sponsor’s summary of clinical evidence.
Table 4: Summary of Findings for Momelotinib vs. Danazol for Patients With Myelofibrosis Previously Treated With JAK Inhibitors and Who Have Anemia
Outcome and follow-up | Patients (studies), N | Relative effect (95% CI) | Absolute effects | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
Danazol | Momelotinib | Difference | |||||
Blood transfusion | |||||||
Transfusion independence response rate Follow-up: week 24 | 195 (1 RCT) | NR | 20.0 per 100 | 30.8 per 100 (95% CI, 22.98 to 39.46) | 10.99 more per 100 (95% CI, 0.8 less to 22.77 more per 100) | Lowa,b | Momelotinib may result in an increase in the number of patients who are transfusion independent compared to danazol. The clinical relevance of the increase is uncertain. |
Number of RBCs or whole blood units transferred Follow-up: week 24 | 195 (1 RCT) | NR | Mean = 10.9 | Mean = 6.6 (SD = 8.41) | –5.66 (95% CI, –10.65 to –0.68) | Moderateb,c | Momelotinib likely results in a decrease in amount of blood transfusion units when compared to danazol. The clinical relevance of the decrease is uncertain. |
Splenic response (spleen volume reduction of ≥ 35% from baseline at the week 24 assessment as measured by MRI or CT scans) | |||||||
Splenic response rate Follow-up: week 24 | 195 (1 RCT) | NR | 3.1 per 100 | 23.1 per 100 (95% CI, 16.14 to 31.28 per 100) | 19.37 more per 100 (95% CI, 10.96 to 27.77 more) | Moderated | Momelotinib likely results in an increase in splenic response rate when compared to danazol. The clinical relevance of the increase is uncertain. |
TSS response (≥ 50% reduction in TSS from baseline at week 24 assessment as measured by MFSAF) | |||||||
TSS response rate Follow-up: week 24 | 195 (1 RCT) | NR | 9.2 per 100 | 24.6 per 100 (95% CI, 17.49 to 32.94 per 100) | 15.67 more per 100 (95% CI, 5.54 to 25.81 more) | Moderated | Momelotinib likely results in an increase in the number of patients who experience response based on total symptom score compared to danazol. The clinical relevance of the decrease is uncertain. |
Harms | |||||||
Serious adverse events Follow-up: week 24 | 195 (1 RCT) | NR | 40 per 100 | 34.6 per 100 (NR) | 5 less per 100 (95% CI, 20 less to 9 more) | Lowe,f | Momelotinib may result in a decrease in the proportion of patients who experience ≥ 1 serious adverse events compared with danazol. The clinical importance of the increase is uncertain. |
CDA-AMC = Canada’s Drug Agency; CI = confidence interval; JAK = Janus kinase; MFSAF = Myelofibrosis Symptom Assessment Form; MID = minimal important difference; NR = not reported; RBC = red blood cell; RCT = randomized controlled trial; SD = standard deviation; TSS = total symptom score; vs. = versus.
aNo published between-group MID was identified, and the clinical experts consulted by CDA-AMC were unable to estimate a threshold for clinically important effects; therefore, the null was used. Rated down 1 level for serious imprecision as the lower bound of the 95% CI suggests minimal harm and/or no difference and the upper bound of the 95% CI suggests benefit. Rated down 1 level for serious risk of bias due to missing data.
bEnd point not adjusted for multiple testing, thus should be used as supportive evidence.
cNo published between-group MID was identified, and the clinical experts consulted by CDA-AMC were unable to estimate a threshold for clinically important effects; therefore, the null was used. Rated down 1 level for serious risk of bias due to the large and imbalanced number of treatment discontinuations and the lack of data imputation methods for this outcome.
dNo published between-group MID was identified, and the clinical experts consulted by CDA-AMC were unable to estimate a threshold for clinically important effects; therefore, the null was used. Did not rate down due to imprecision. Rated down 1 level for serious risk of bias due to the large and imbalanced number of treatment discontinuations.
eNo published between-group MID was identified, and the clinical experts consulted by CDA-AMC were unable to estimate a threshold for clinically important effects. Rated down 2 levels for very serious imprecision as the lower bound of the CI suggests benefit and the upper bound of the 95% CI suggests harm.
Sources: GSK Data on File, 2021 (SIMPLIFY-1 Clinical Study Report);10 GSK Data on File, 2021 (SIMPLIFY-2 Clinical Study Report);11 GSK Data on File, 2023 (MOMENTUM Clinical Study Report).12 Details included in the table are from the sponsor’s summary of clinical evidence.
GRADE Summary of Findings and Certainty of the Evidence
The selection of outcomes for the Grading of Recommendations Assessment, Development and Evaluation (GRADE) was based on the sponsor’s summary of clinical evidence, consultation with clinical experts, and input received from patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members:
Transfusion independence response rate — follow-up: week 24
Rate of RBC transfusion — follow-up: week 24
SRR — follow-up: week 24
TSS response rate — follow-up: week 24
SAEs — follow-up: week 24.
Long-Term Extension Studies
This section summarizes the open-label extension studies of the SIMPLIFY-1, SIMPLIFY-2, and MOMENTUM trials.
Description of Studies
The open-label long-term extension of the SIMPLIFY-1 trial evaluated the open-label treatment with momelotinib for up to 216 weeks (i.e., through week 240) after the randomized, double-blind phase. The open-label extension of the SIMPLIFY-2 trial evaluated the open-label treatment with momelotinib for up to 204 weeks (i.e., through week 228) after the randomized treatment phase. All patients who completed the 24-week randomized treatment phase in the SIMPLIFY-1 and SIMPLIFY-2 trials were eligible to participate in the extended treatment phases.
The open-label extension of the MOMENTUM trial evaluated the open-label treatment with momelotinib for up to 180 weeks (i.e., through week 204) or with danazol for up to 24 weeks after the randomized, double-blind phase. Patients who completed the 24-week randomized treatment phase in the MOMENTUM trial, discontinued treatment early due to splenic progression, or discontinued treatment early for other reasons but completed scheduled assessments through week 24 had the option to continue on momelotinib.
The median total duration of follow-up (combined randomized and open-label extension phases) was 35.3 months (range, 0.4 to 59.3 months) in the SIMPLIFY-1 trial and 28.2 months (range, 0.3 to 50.4 months) in the SIMPLIFY-2 trial. In the open-label extension phase of the SIMPLIFY-1 trial, the majority of patients in the continuing momelotinib and switch to momelotinib treatment groups (40.4% and 48.7%, respectively) had high-risk MF per the International Prognostic Scoring System (IPSS) criteria and a positive JAK2V617F mutation status (58.5% and 64.0% in the continuing momelotinib and switch to momelotinib treatment groups, respectively) at baseline. The proportion of patients with a hemoglobin level less than 10 g/dL was higher in the switch to momelotinib treatment group (56.3%) than in the continuing momelotinib group (37.4%).
In the SIMPLIFY-2 trial, the majority of patients in the continuing momelotinib group (64.1%) and the switch to momelotinib treatment group (55.0%) had intermediate-2–risk MF per the Dynamic International Prognostic Scoring System (DIPSS) criteria, and more than 60% of patients in both treatment groups had a positive JAK2V617F mutation status (60.9% versus 72.5% in the continuing momelotinib group and switch to momelotinib treatment group, respectively). A numerically larger proportion of patients in the continuing momelotinib group (57.8%) were transfusion dependent than in the switch to momelotinib treatment group (50.0%). The proportion of patients with an Eastern Cooperative Oncology Group Performance Status (ECOG PS) of 1 was higher in the continuing momelotinib group (64.1%) than in the switch to momelotinib treatment group (47.5%). A numerically smaller proportion of patients in the continuing momelotinib group (4.7%) had an ECOG PS of 2 than in the switch to momelotinib treatment group (15.0%). There were no patients with an ECOG PS of 3 in the continuing momelotinib group; 5% of patients in the switch to momelotinib treatment group had an ECOG PS of 3. The proportion of patients with a hemoglobin level less than 8 g/dL was higher in the continuing momelotinib group (28.1%) than in the switch to momelotinib treatment group (7.5%).
Efficacy Results
██ ███████████ ██ ███████ ██ ███ ███ ████████ ██ ███ ███████████ █████ ███ █ ███████ ████████ ██ ███ ████ ██████ ███ ████████████ ██ ██████████ █████████ ██████ ██ ██████████ ██ ███████ █████████ ████ ████████ ██ ███ ██████████████████████ █████ █████████ █████████ ██ ███████████ ██ ███ ██████████ █████████ ██████ ██ ███ █████ ███████████ ████████ ███████ ███ ███████ ██ ███ ████████ ███ █ ███████ ████████ ██ ███ █████ ██ ███████████ ██ ███████ ██ ███ ███ ████████ ██ ███ ███████████ █████ ███ █ ███████ ██ ██ ████████ ██ ███ ███ █████ ██████████ ████████ ███ ████████ ████ ███ ██ ████████████ ███ █ ███████ ████████ ██ ███ ████ ██ ███ ██████████ ██ ██████████ █████████ ██████ █████ ███████ ██ ██ ████████ ██████████ ██ ███ ███ ████████ ████ ███ ██ ███████████ ██████ ███ ██████████ █████████ █████ ████ ██████████ ██ ███ ██████████ In the MOMENTUM trial, most patients (n = 19 of 24; 79.2%) in the continuing momelotinib group and 50.0% of patients (n = 1 of 2) in the switch to momelotinib treatment group who experienced splenic response at week 24 were also classified as experiencing splenic response at week 48. Of those not experiencing splenic response at week 24 in the continuing to momelotinib group (n = 43) and the switch to momelotinib treatment group (n = 28), 23.3% and 10.7%, respectively, were classified as experiencing a splenic response at week 48.
In the MOMENTUM trial, the majority of patients were transfusion independent at week 24 and at week 48, including 88.2% of patients (n = 30 of 34) in the continuing momelotinib group and 80.0% of patients (n = 8 of 10) in the switch to momelotinib treatment group. A majority of patients with at least a 50% reduction from baseline Myelofibrosis Symptom Assessment Form (MFSAF) TSS at week 24 were also experiencing a TSS response at week 48, including 72.0% (n = 18 of 25) in the continuing momelotinib group and all patients (n = 5 of 5; 100%) in the switch to momelotinib treatment group.
Harms Results
In the SIMPLIFY-1 trial, the overall frequencies of TEAEs were numerically higher in patients who switched from ruxolitinib to momelotinib than those who continued on momelotinib (89.8% versus 78.4%, respectively) after 24 weeks of treatment with momelotinib in the open-label extension phase. Similar trends were observed for the most common grade 3 or 4 AEs (37.6% versus 27.5%), for SAEs (23.4% versus 15.8%), and for TEAEs leading to treatment discontinuation (14.7% versus 8.8%), with numerically higher proportions for patients who switched from ruxolitinib to momelotinib than for those who continued on momelotinib. The most reported AEs in both groups, occurring in at least 10% of patients, were diarrhea, thrombocytopenia, anemia, fatigue, nausea, and cough. The most common AEs leading to treatment discontinuation were thrombocytopenia, fatigue, and peripheral sensory neuropathy (no events in the continuing momelotinib group and relatively few [2.0% to 2.5% of patients] in the switch to momelotinib treatment group). Among the continuing momelotinib group and the switch to momelotinib treatment group, the following AEs of special interest (AESIs) were reported: peripheral neuropathy (5.3% versus 7.6% of patients), nonhematological AEs (77.2% versus 87.3% of patients), cataract (4.7% versus 3.6% of patients), and first dose effect (not reported [NR] versus 2.0% of patients). In the continuing momelotinib group, 10.5% of patients died from TEAEs not related to disease progression; in the switch to momelotinib treatment group, this percentage was 8.6%.
In the SIMPLIFY-2 trial, the overall frequencies of the following were numerically higher in patients who switched from BAT to momelotinib than in those who continued on momelotinib: TEAEs (100% versus 93.8%), grade 3 or 4 AEs (55.0% versus 28.1%), SAEs (27.5% versus 20.3%), TEAEs leading to treatment discontinuation (37.5% versus 7.8%), and AEs leading to treatment interruption and/or dose reduction (19.2% versus 16.3%). The most commonly reported AEs occurring in at least 15% of patients were cough and diarrhea in patients who continued momelotinib in the extended treatment phase, and asthenia, pyrexia, diarrhea, thrombocytopenia, cough, and anemia in patients who switched from BAT to momelotinib. The most reported SAEs occurring in at least 5% of patients were anemia, pyrexia, and confusional state in patients who switched from BAT to momelotinib. No patient in the continuing momelotinib group experienced any of these SAEs. The most common AEs leading to treatment discontinuation were thrombocytopenia, diarrhea, and headache (no events in the continuing momelotinib group and events affecting 5.0% to 7.5% of patients in the switch to momelotinib treatment group). Among the continuing momelotinib group and the switch to momelotinib treatment group, the following AESIs were reported: peripheral neuropathy (10.9% versus 20.0%), nonhematological AEs (98.4% versus 100%), cataract (1.6% versus 0%), and first-dose effect (NR versus 7.5%). Deaths due to TEAEs not related to disease progression were reported in 21.9% of patients who continued treatment with momelotinib and 7.5% of patients who switched from BAT to momelotinib.
In the MOMENTUM trial, after 24 weeks of treatment with momelotinib in the open-label treatment phase, the overall frequencies of TEAEs (89.2% versus 85.4%), grade 3 or higher TEAEs (51.6% versus 48.8%), and serious TEAEs (32.3% versus 29.3%) were slightly higher in patients who continued on momelotinib than in those who switched from danazol to momelotinib. The most reported AEs occurring in at least 10% of patients were diarrhea, thrombocytopenia, pyrexia, asthenia, and anemia in patients who continued momelotinib, and thrombocytopenia and diarrhea in those who switched from danazol to momelotinib. The most commonly reported SAEs occurring in at least 2% of patients were urinary tract infection, acute kidney injury, febrile neutropenia, and squamous cell carcinoma of the skin in patients who continued momelotinib, and acute kidney injury and urinary tract infection in those who switched from danazol to momelotinib. The most common AEs leading to treatment discontinuation were anemia, AML, and transformation to AML (no events in the continuing momelotinib group for AML or transformation to AML, and no events in the switch from danazol to momelotinib group for anemia). No deaths due to TEAE not related to disease progression were reported in either of the treatment groups.
Critical Appraisal
Internal Validity
The open-label extension phase design of the SIMPLIFY-1, SIMPLIFY-2, and MOMENTUM trials may have biased the reporting of some end points because awareness of the study treatment received may have influenced the perception of improvement and/or harms by patients and clinicians, particularly for outcomes that are subjective in measurement and interpretation (e.g., TSS response rate and subjective AEs). In the open-label extension phases, all patients were taking momelotinib. As such, there was no relevant randomized comparison group (i.e., for any active comparator of interest), which precludes causal conclusions. In terms of protocol deviations, for the SIMPLIFY-2 trial, the proportion of patients with at least 1 important protocol violation was higher in the continuing momelotinib group (20.3%) than in the switch to momelotinib treatment group (10.0%) in the extended treatment phase. No information on protocol deviation for the open-label extension phase of the MOMENTUM study was reported separately; as such, the risk of bias due to deviations from the intended interventions is uncertain. The results are reflective of patients who were able to tolerate and stay on momelotinib (in the continuing momelotinib group). No information on missing data imputations was reported for the open-label extension phase in the SIMPLIFY-1, SIMPLIFY-2, or MOMENTUM Clinical Study Reports provided by the sponsor. In the SIMPLIFY-1 and SIMPLIFY-2 trials, the number of patients who discontinued treatment before week 24 of the open-label treatment phase was higher among those who switched from ruxolitinib to momelotinib or from BAT to momelotinib than among those who continued on momelotinib. The main reason behind this imbalance in both groups was AEs. This imbalance may potentially bias the safety results as patients who were still continuing the open-label extension phase had better tolerability of momelotinib than those who had discontinued.
External Validity
Because the patients who took part in the open-label extension phases were originally from the pivotal trials and the eligibility criteria remained the same, it is reasonable to expect that the same limitations to generalizability are relevant to the open-label extension phases for all 3 studies as were identified for the randomized phases. The trials included patients who were transfusion dependent and patients who were transfusion independent, making the results generalizable to more patients.
Indirect Comparisons
None submitted.
Studies Addressing Gaps in the Evidence From the Systematic Review
The sponsor submitted 2 retrospective analyses and the interim results of 1 ongoing extended access study to address gaps related to long-term survival by baseline transfusion independence status.1,13,14 These studies were not included in the clinical review report as they provided supplementary evidence rather than addressing specific gaps in the evidence.
Conclusions
Three pivotal RCTs informed the efficacy and safety of momelotinib for the treatment of MF in adults compared to the following: ruxolitinib in patients not previously treated with a JAK inhibitor (SIMPLIFY-1 trial), BAT (which was mostly ruxolitinib) in patients who had been previously treated with a JAK inhibitor (SIMPLIFY-2 trial), and danazol in patients who had been previously treated with a JAK inhibitor and who had anemia (MOMENTUM trial). In patients not previously treated with a JAK inhibitor, momelotinib, as compared to ruxolitinib, resulted in an increased number of patients who were transfusion independent, likely led to little-to-no difference in SRR, but resulted in a decrease in the number of patients with a TSS response. In patients who had been previously treated with a JAK inhibitor, the evidence on the effect of momelotinib compared to BAT on the outcome of SRR was uncertain but showed that momelotinib may increase TSS response and may be likely to result in more patients who are transfusion independent. In patients who had been previously treated with a JAK inhibitor and who had anemia, compared to danazol, momelotinib may result in an increased number of patients with transfusion independence and is likely to increase the SRR and TSS response. As such, it is unclear if momelotinib offers an advantage over existing therapies in SRR or offers better symptom resolution compared to ruxolitinib in patients not previously treated with a JAK inhibitor. However, momelotinib is likely to result in an improvement in blood transfusion outcomes when compared to ruxolitinib.
The safety of momelotinib is in line with that of other JAK inhibitors. There is low-certainty evidence that momelotinib will lead to an increase or decrease in patients with SAEs compared to ruxolitinib, BAT, or danazol.
There is no evidence to inform the comparative efficacy and safety of momelotinib versus fedratinib, and there remains no long-term comparative data on important clinical outcomes such as survival and leukemia progression.
The results available from the long-term extension studies suggest that more than two-thirds of patients experienced sustained efficacy with momelotinib over extended periods. In the open-label extension phases of the SIMPLIFY-1, SIMPLIFY-2, and MOMENTUM trials, momelotinib appeared to provide ongoing benefits in terms of transfusion independence, splenic response, and symptom relief. Splenic responses were observed in a proportion of patients, particularly those continuing momelotinib treatment, and the majority of patients who were transfusion independent at week 24 maintained this status through extended follow-up.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of momelotinib, 200 mg, administered orally once daily, with 150 mg and 100 mg alternative strengths, for the treatment of splenomegaly and/or disease-related symptoms in adult patients with intermediate-risk or high-risk PMF, post-PV MF, or post-ET MF who have moderate to severe anemia.
Disease Background
The contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the CDA-AMC review team.
MF is a rare, chronic, and progressive bone marrow disorder classified as a Philadelphia chromosome-negative MPN.1,2 It is characterized by excessive production of reticulin and collagen fibres in the bone marrow.3 MF can present de novo as PMF or secondary to other conditions, such as ET and PV, termed post-ET MF and post-PV MF, respectively.1,2 PMF is the most aggressive form of MF and can progress into AML.3 The 10-year risk of transformation to AML for patients with PMF ranges from 10% to 20%.15
MF originates from acquired mutations targeting hematopoietic stem cells, leading to dysregulation of kinase signalling, clonal myeloproliferation, and abnormal cytokine expression. A mutation in the JAK2 gene is found in nearly all patients (95%) with post-PV MF and approximately 50% to 60% of those with post-ET MF or PMF.16-18
Diagnosis of MF is based on the 2017 WHO criteria and involves a combination of physical examination, blood tests, imaging, bone marrow biopsy, and molecular testing.4 The median age at diagnosis for patients with MF is 67 years.19 Approximately 30% of patients are asymptomatic at the time of diagnosis, but as the disease progresses, all patients eventually develop symptoms due to bone marrow fibrosis, bone marrow failure, systemic inflammation, and splenomegaly.19 Key clinical manifestations include severe anemia, thrombocytopenia, marked hepatosplenomegaly, and constitutional symptoms such as fatigue, night sweats, low-grade fevers, itching, bone pain, unintentional weight loss, and cachexia.16-18 Splenomegaly, a common feature in MF, can result in several complications, including pressure and pain in the left upper quadrant of the abdomen, pain in the left shoulder, early satiety, portal hypertension, splanchnic vein thrombosis, hepatic extramedullary hematopoiesis, and obliterative portal venopathy.20 Massive splenomegaly can lead to severe sequelae, like lower extremity edema and painful splenic infarctions, significantly impacting patients’ quality of life.21
MF prognosis can be estimated using scoring systems such as the IPSS and the DIPSS.17,18,22 These systems evaluate risk factors including age, constitutional symptoms, hemoglobin levels, white blood cell count, and blood blasts. The median OS varies significantly: patients considered to be at low risk have a median OS of 11.3 years, patients considered to be at intermediate-1 risk have a median OS of 7.9 years, patients considered to be at intermediate-2 risk have a median OS of 4 years, and patients considered to be at high risk have a median OS of 1.5 to 2.3 years. Approximately 70% of patients with MF fall into the intermediate-2–risk or high-risk categories.17,18,22
Anemia and transfusion dependence are negative prognostic factors in MF.23 At diagnosis, about 40% of patients have moderate anemia (hemoglobin level < 10 g/dL), and nearly all patients develop anemia over time.20 Anemia’s multifactorial pathophysiology in MF can be driven by the disease itself or by myelosuppressive treatments such as JAK inhibitors.16-18 The median OS for patients with even mild anemia is significantly lower (4.9 years) than for patients without anemia (6 years).20 Transfusion dependence further worsens survival outcomes, with a median survival of 2.6 years for patients who are transfusion dependent versus 8 years for those not requiring transfusions at diagnosis.23
There is limited evidence on the incidence of MF in Canada. In 2016, approximately 200 individuals were diagnosed with MF in Canada, accounting for 1% of all hematological malignancies.5 A Canadian study estimated that between 2011 and 2015, the age-standardized incidence rate of PMF in Canada was 0.80 per 100,000 person-years.4 Data from the Ontario Cancer Registry shows that between 2004 and 2016, 1,031 patients were diagnosed with PMF in Ontario, which increased to 1,385 between 2004 and 2019.24,25 The incidence rates are slightly higher among males than among females.24 As with incidence, the prevalence estimates of PMF in Canada are limited. The prevalence of MF in Canada is estimated to be 6.2 per 100,000 person-years.5 In 2016, an estimated 1,800 people in Canada were living with MF, with estimates ranging from 1,400 to 2,177.26 The proportion of patients with intermediate-risk and high-risk MF is estimated to be 15% to 37% and 33% to 37%, respectively.27,28
Standards of Therapy
The contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the CDA-AMC review team.
The only curative treatment option for MF is allogenic HSCT; however, patient eligibility for this treatment is limited, and the procedure is associated with morbidity and mortality.21 JAK inhibitors are the current standard of care for MF, with demonstrated efficacy in improving splenomegaly and constitutional symptoms.6-9 International and national guidelines recommend that patients with HSCT-ineligible intermediate-risk to high-risk MF receive a JAK inhibitor as initial therapy, and ruxolitinib has become standard of care for patients not previously treated with JAK inhibitors who have sufficient platelet and RBC levels.20 In Canada, the treatment recommendation after ruxolitinib is to offer patients HSCT (if eligible), administer a second-line JAK inhibitor (i.e., fedratinib), or enrol patients in a clinical trial.6-9 However, although fedratinib is approved for patients not previously treated with JAK inhibitors and those previously treated with ruxolitinib, reimbursement is limited to patients who are intolerant of ruxolitinib or for whom ruxolitinib is contraindicated and whose MF did not progress while receiving ruxolitinib.29 Patients who do not have access to fedratinib or another JAK inhibitor after ruxolitinib are treated with suboptimal doses of ruxolitinib or other therapies (i.e., hydroxyurea, steroids, palliative splenectomy, or radiation).
Several supportive therapies are used to address symptomatic anemia in MF, such as androgens (danazol), erythropoiesis-stimulating agents (ESAs), immunomodulating drugs (thalidomide or lenalidomide), corticosteroids (prednisone or prednisolone), and chronic RBC transfusions.20 However, these supportive therapies have demonstrated limited clinical activity in MF-related anemia, and in Canada these supportive therapies are used off-label for managing anemia in MF.
Relief of symptoms and improved health-related quality of life (HRQoL) are important goals for all patients with PMF.21 Before the introduction of JAK inhibitors, conventional cytoreductive therapies for patients with symptomatic splenomegaly included hydroxyurea and interferon therapy.6-9 Hydroxyurea is an oral ribonucleotide reductase inhibitor and is often employed as first-line cytoreductive therapy in post-PV MF and post-ET MF, while for patients with PMF, hydroxyurea is typically reserved for those who have hyperproliferative features including splenomegaly, leukocytosis, and constitutional symptoms who are not eligible for JAK2 inhibitors.30 Adverse effects related to the use of hydroxyurea include dose-limiting cytopenias and oral and lower extremity skin ulcers.30
Currently, the mainstay of pharmacologic-induced spleen reduction in MF is the use of JAK1 and JAK2 inhibitors, such as ruxolitinib. Ruxolitinib is a JAK1- and JAK2-selective inhibitor and is indicated for the treatment of splenomegaly and/or its associated symptoms in adult patients with PMF, post-PV MF, or post-ET MF. The effects of ruxolitinib in reducing spleen volume and improving disease-related symptoms have been demonstrated in clinical trials involving patients with intermediate-risk and high-risk MF, as well as patients who did not experience response with or were intolerant of hydroxyurea.8 Although the initial response rate to ruxolitinib therapy is high, patients eventually develop intolerance due to dose-dependent drug-related cytopenias or resistance to ruxolitinib after 2 to 3 years of therapy.2,8 After ruxolitinib failure, strategies to overcome ruxolitinib resistance or intolerance are limited and mainly involve different approaches to continued ruxolitinib therapy, including dosing modifications and ruxolitinib rechallenge. The goals of treatment are to reduce the symptoms of MF, reduce splenomegaly, and improve OS. Before ruxolitinib becoming the standard of care, patients were treated with hydroxyurea or interferons. These treatments may still be used in select groups of patients (e.g., interferon for patients with the potential to become pregnant, hydroxyurea for patients with severe thrombocytopenia).2,8
Despite effective pharmacologic options, there are patients who have experienced disease progression but have limited treatment options; more invasive treatment modalities may then be given. Splenectomy, splenic irradiation, and partial splenic artery embolization remain valid therapeutic tools to consider in the management of MF-related splenomegaly.31-33 Another treatment option for patients with intermediate-risk or high-risk MF is HSCT, but this requires an appropriate donor, and the associated rates of morbidity and mortality limit eligibility.14,15,33 AEs related to HSCT include treatment-related mortality, graft failure, graft versus host disease, and infection.34
Drug Under Review
Key characteristics of momelotinib are summarized in Table 5, along with other treatments available for MF.
Momelotinib is an orally bioavailable, small-molecule inhibitor of JAK1 and JAK2, and the first JAK1 and JAK2 inhibitor with a differentiated mechanism of action that also inhibits ACVR1.35-37 Details included in the table are from the sponsor’s summary of clinical evidence. Momelotinib’s mechanism of action involves inhibition of JAK1 and JAK2 signalling, which reduces constitutional symptoms and splenomegaly through decreased inflammatory cytokine signalling, hematopoietic stem cell proliferation, and RBC sequestration. Inhibition of ACVR1 decreases hepcidin expression, restores iron homeostasis, increases serum iron availability for erythropoiesis, facilitates improvements in hemoglobin levels, and decreases the need for RBC transfusion, thus improving anemia.35-37
Momelotinib is indicated for the treatment of disease-related splenomegaly or symptoms and anemia in adult patients with PMF, post-PV MF, or post-ET MF who have or have not been previously treated with a JAK inhibitor.35-37 The recommended starting dosage for momelotinib is 200 mg administered once daily, with 150 mg and 100 mg strengths available for dose modification. Momelotinib can be taken with or without food. The sponsor’s reimbursement request is aligned with this proposed indication. Momelotinib received a Notice of Compliance from Health Canada on November 8, 2024.
Table 5: Key Characteristics of Momelotinib, Ruxolitinib, and Fedratinib
Characteristic | Momelotinib | Ruxolitinib | Fedratinib |
|---|---|---|---|
Mechanism of action | JAK1/2 inhibitor | JAK1/2 inhibitor | JAK2 inhibitor |
Indicationa | The treatment of splenomegaly and/or disease-related symptoms, in adult patients with intermediate or high-risk primary myelofibrosis (MF), post polycythemia vera myelofibrosis or post essential thrombocythemia MF who have moderate to severe anemia. | For the treatment of splenomegaly and/or its associated symptoms in adult patients with PMF (also known as chronic idiopathic MF), post-PV MF or post-ET MF; For the control of hematocrit in adult patients with PV resistant to or intolerant of a cytoreductive agent. | For the treatment of splenomegaly and/or disease-related symptoms. |
Route of administration | Oral | Oral | Oral |
Recommended dosing | 200 mg taken orally q.d. | Starting dosage for patients with MF:
Starting dose for patients with PV:
| 400 mg orally q.d. for patients with a baseline platelet count of ≥ 50 × 109/L |
Serious adverse effects or safety issues | New onset of severe (grade ≥ 3) thrombocytopenia. A complete blood count, including platelet count, should be obtained before initiating treatment, periodically during treatment, and as clinically indicated. Dose interruption or reduction may be required. Infections, including serious and sometimes fatal bacterial and viral infections; should not be initiated in patients with active infections. | Serious bacterial, mycobacterial, fungal, and viral infections; some cases were life-threatening or led to death. Decrease in blood cell count; a complete blood count must be performed before initiating therapy with ruxolitinib and during therapy. | Serious and fatal encephalopathy, including Wernicke encephalopathy; assess thiamine levels in all patients before starting fedratinib, periodically during treatment, and as clinically indicated. |
b.i.d. = twice daily; ET = essential thrombocythemia; JAK = Janus kinase; JAKi = Janus kinase inhibitor; MF = myelofibrosis; PMF = primary myelofibrosis; PV = polycythemia vera; q.d. = once daily.
aHealth Canada–approved indication.
Sources: Novartis Pharmaceuticals Canada Inc., 2023 (Health Canada product monograph for ruxolitinib);38 Celgene Inc., 2020 (Health Canada product monograph for fedratinib);39 GSK Data on File draft product monograph momelotinib.40
Perspectives of Patients, Clinicians, and Drug Programs
Patient Group Input
This section was prepared by the CDA-AMC review team based on the input provided by patient groups. The full original patient input(s) have been included in the Perspectives of Patients, Clinicians, and Drug Programs section of this report.
Two patient groups, LLSC and CMPNN, provided a joint input for this review. Another patient group, Heal Canada, also provided input. Information from the joint input was sourced from 3 online surveys conducted between March 2024 and May 2024. Two surveys (survey 1 and survey 2) were developed by CMPNN and were distributed by both CMPNN and LLSC. Another survey (survey 3) was developed and distributed by both CMPNN and LLSC. Survey 1 (33 respondents) and survey 3 (29 respondents) gathered input from patients and caregivers who had lived experience with MF; survey 2 (11 respondents) was intended to gather information on patient experience with the drug under review. LLSC also conducted 1-on-1 interviews with 2 patients currently living with MF and being treated with momelotinib. Most respondents (70.37%) in survey 3 indicated that they were a patient (past or present), and 25.93% of respondents indicated that they were a caregiver. All respondents from survey 1 and survey 3 were living in Canada; survey 2 received responses from patients living in Canada as well as patients living in the US and Germany. Heal Canada gathered information from Cheryl Petruk, who was a former caregiver of a patient with post-ET MF and the founder of CMPNN. She was also involved in the international MPN landmark survey conducted in 2016, she also supported the recruitment of 64 patients living in Canada, including 28 patients with MF. A total of 174 patients participated in this survey. Other information was gathered from interviews with patients living in Canada being treated for MF (10 respondents), from an online survey conducted in 2024 to assess the impact of frequent transfusion on patients living in Canada (24 respondents), and from literature review.
The joint input from LLSC and CMPNN highlighted that patients often rely heavily on caregiver support to navigate daily life and manage their symptoms, which puts a significant burden on both patients and their caregivers. Caregivers for this input stated that witnessing the decline in their loved one’s health and daily functioning caused substantial stress and anxiety. Patients with MF in this joint input noted that fatigue, enlarged spleen, anemia, night sweats, bone or joint pain, weight loss, early satiety, brain fog, itching or pruritus, gout, and frequent infections were some of the side effects of MF that affected them. Fifty-five percent of respondents mentioned losing income due to MF treatment. When asked about the impact of MF, 85% respondents replied that it had a negative to extremely negative impact on their personal or home life and social life and 65% replied that it had a negative to extremely negative impact on their mental health. Respondents highlighted that low energy, symptom burden, depression and anxiety, fear of infections, and frequent hospital visits were some of the factors that contributed to these negative impacts. Respondents in the Heal Canada landmark survey expressed similar concerns regarding symptom frequency and severity, with 83% of respondents stating that MPN symptoms impacted their quality of life and 58% particularly mentioned bone pain.
Many respondents in the joint input indicated that expenses to attend treatments (e.g., parking, travel) had an impact on them (20 of 33, 60.6%), as did the need to take time off work for treatments (13 of 33; 39.4%); difficulties taking treatments, such as the transfusion process, swallowing pills, and injections (11 of 33; 33.3%); and transfusion dependency (6 of 33; 18.2%). Respondents in the Heal Canada landmark survey also reported a high impact of MF on ability to work, with only 14% of patients being able to work full-time and 11% being able to work part-time. In the 2024 online survey conducted by Heal Canada, 75% of respondents mentioned experiencing or caring for adverse reactions during and after blood transfusion. All respondents from this survey added that the benefits of blood transfusion wear off before the next transfusion. One-half of the respondents from this survey also indicated that receiving regular transfusion had impacted their and their caregivers’ working status.
When respondents in the joint input were asked to choose the top 3 factors that are most important to them when considering new MF treatment options, most respondents (94.74%) selected the amount and severity of side effects as the most important factor, followed by quality of life during treatment (84.21%), the length of time in potential remission (63.16%), financial costs (31.58%), the length and frequency of hospital visits or stays (26.32%), outpatient treatment options (21.05%), distance from home (15.79%), number of treatments (10.53%), and physician recommendation (10.53%).
The joint input highlighted that MPN specialists are not universally available at every community cancer centre, which means some patients and caregivers may need to travel outside their local area to access health care services for MF. The patient groups noted that despite the availability of the current treatment options for MF, not all patients may respond adequately, and even those who do may experience only temporary benefits. Additionally, many patients may not qualify for a potentially curative stem cell transplant. Considering patient responses and health status, along with patient preference, the joint input highlighted that the range of currently available treatment options may be quite limited.
The joint input noted that managing anemia was a significant challenge for patients living with MF, with 63% of respondents receiving blood or platelet transfusions due to MF. When asked about the desired improvements they would like to experience from new treatments, the respondents highlighted reducing symptom burden, enhancing quality of life, and reducing the need for transfusions as some of the desired factors. When respondents were asked about the aspects of the disease they felt were important to control, 58% noted fatigue, 54% noted anemia or need for transfusions, and 37% noted spleen size. The majority of respondents (73%) from survey 2 who had experience with the drug under review stated that they felt momelotinib treatment improved their quality of life.
Clinician Input
Input From Clinical Experts
All CDA-AMC review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of MF.
Unmet Needs
According to the clinical experts, current treatments for MF, including JAK inhibitors like ruxolitinib and fedratinib, address symptoms such as splenomegaly and constitutional symptoms but do not modify the underlying disease or delay its progression. It was noted that the only potentially curative therapy, HSCT, is applicable to less than 10% of patients due to significant associated morbidity and mortality. Not all patients experience a response to available therapies, with only approximately 30% experiencing a splenic response to JAK inhibitors and up to 50% experiencing a response to ESAs for anemia. The experts noted that even when responses are experienced, they are often not durable, with a median response duration of 3 years for ruxolitinib and 2 years for ESAs. Therefore, there is an unmet need for therapies that can provide durable responses, better manage anemia, and possibly modify disease progression.
Place in Therapy
According to the clinical experts, momelotinib would fit into the current treatment paradigm for MF as an option for patients who require JAK inhibitor therapy and have clinically significant anemia. It would be particularly beneficial for patients who have not previously been treated with JAK inhibitors and those who have experienced intolerance or ongoing symptomatic or transfusion-dependent anemia on existing JAK inhibitor therapy. The experts noted that momelotinib could be used both in the first-line setting and as a second-line or third-line treatment, especially in patients with clinically relevant anemia and MPN symptoms. However, it may not be the best option for patients whose primary clinical issue is symptomatic splenomegaly in the context of ruxolitinib resistance or intolerance.
Patient Population
According to the clinical experts, the patients most suited for treatment with momelotinib include those with MF who have not previously been treated with JAK inhibitors and have splenomegaly or MPN symptoms and clinically relevant anemia, those who have experienced intolerance or anemia on JAK inhibitor therapy, and those in whom JAK inhibitor therapy has failed, particularly if anemia or MPN symptoms are present. The experts noted that the patients best suited for treatment can be identified through clinical examination, spleen palpation or measurement, and assessment of anemia parameters such as hemoglobin levels and frequency of RBC transfusions. Patients whose main clinical issue is splenomegaly without anemia or MPN symptoms may be less suitable for momelotinib.
Assessing the Response Treatment
In clinical practice, according to the clinical experts consulted by the review team, response to momelotinib is assessed through a combination of patient-reported outcomes (e.g., improvement in anemia or spleen symptoms), physical examination (including spleen size), and anemia parameters (e.g., hemoglobin levels, transfusion frequency). Response assessments should be conducted approximately every 3 months. A clinically meaningful response includes subjective improvements reported by the patient, objective reduction in spleen size, and improved anemia metrics. The experts noted that the magnitude of the response considered necessary to continue treatment is likely to vary among physicians but that consistency in monitoring is key.
Discontinuing Treatment
According to the clinical experts, treatment with momelotinib should be discontinued if there is no evidence of response after approximately 6 months, if there is a loss of previous response (e.g., worsening of symptoms, spleen size, or anemia), or if grade 3 AEs occur that do not resolve with dose interruption or reduction.
Prescribing Considerations
Momelotinib, as noted by the clinical experts, should be prescribed and monitored by hematologists or oncologists specializing in the treatment of MF. The administration and continued monitoring of patients receiving momelotinib should occur in a hospital outpatient clinic or a specialty clinic setting where appropriate expertise is available. Consideration should also be given to potential regional variations in access to such specialists.
Clinician Group Input
This section was prepared by the CDA-AMC review team based on the input provided by clinician groups. The full original clinician group inputs have been included in the Perspectives of Patients, Clinicians, and Drug Programs section of this report.
Clinician group input on the review of momelotinib was received from 2 clinician groups: a joint input from LLSC and the CMPNN Clinician Group, and another input from the OH-CCO Hematology Cancer Disease Site Drug Advisory Committee. A total of 15 clinicians (9 from LLSC and the CMPNN Clinician Group, and 6 from OH-CCO’s drug advisory committees) provided input for this submission.
LLSC and the CMPNN Clinician Group emphasized that the management of anemia remains a critical challenge in MF care. While discussing the unmet needs of patients, the groups highlighted that while the currently available JAK inhibitors can be effective in addressing some symptoms and splenomegaly, they may fail to improve anemia and may exacerbate anemia as an on-target side effect. The groups also noted the lack of approved treatments for MF-associated anemia. They pointed out that anemia, as well as the need for life-saving transfusions, significantly impact patients’ quality of life. The groups further stated that treatments that can decrease the need for transfusions are highly desired in this patient population. The groups noted that for patients considered to be at high risk who are eligible for allogeneic stem cell transplant, fewer transfusions may lead to better outcomes, whereas for patients who are not transplant eligible or who have low-risk MF, the focus is on improving quality of life. In addition, the OH-CCODrug Advisory Committee highlighted that current treatments for this patient population are designed to reduce symptoms, improve blood counts or splenomegaly, and reduce transfusion requirements. Both groups agreed that some treatments are not well tolerated and may not be very effective.
Both clinician groups agreed that patients with MF-associated anemia may benefit from momelotinib in comparison to other available therapies. However, LLSC and the CMPNN Clinician Group added that momelotinib has not yet demonstrated a reduction in the risk of transformation to acute leukemia or mitigated the need for curative allogeneic stem cell transplant in eligible patients. The OH-CCO Drug Advisory Committee noted that treatment responses with momelotinib are based on symptom burden, blood counts, and splenomegaly, whereas LLSC and the CMPNN Clinician Group noted stable disease or improvement in symptom burden, decrease in spleen volume, improvement in hemoglobin, and/or reduction in transfusions as determining factors for outcome assessment. LLSC and the CMPNN Clinician Group elaborated that at least 6 months may be required for assessment of the efficacy of the drug under review. They also added some factors to consider in determining lack of efficacy or deciding on discontinuation of treatment; these factors were increase in transfusion requirement, symptom burden, spleen volume, or severe therapy-related thrombocytopenia or neutropenia. Significant intolerance and clear worsening or lack of response were some factors noted by the OH-CCO Drug Advisory Committee to be considered in deciding on discontinuation of treatment.
Drug Program Input
The drug programs provide input on each drug being reviewed through the reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CDA-AMC are summarized in Table 6.
Table 6: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Considerations for initiation of therapy | |
Would use of momelotinib be limited to patients with anemia due to myelofibrosis? | The experts indicated that the use of momelotinib would likely be prioritized for patients with myelofibrosis who have anemia or are borderline for anemia. The clinical experts highlighted the importance of carefully considering the threshold for anemia, particularly for patients with mild anemia (hemoglobin levels between 100 g/L and 120 g/L). Momelotinib could be particularly beneficial in cases where treatment with ruxolitinib has led to anemia. |
Generalizability | |
At the time of funding, should patients receiving alternative therapies (e.g., ruxolitinib, fedratinib, hydroxyurea) be eligible to switch to momelotinib? | The experts indicated that momelotinib should be available as an upfront treatment, including as a second-line option after initial treatment with other therapies. The experts noted that patients currently receiving alternative therapies, such as ruxolitinib or fedratinib, could be eligible to switch to momelotinib, especially if they develop anemia. However, the switch might be more appropriate in cases where splenomegaly is not the primary concern and anemia is the predominant issue. |
Funding algorithm | |
Is there evidence for downstream treatment options following progression on momelotinib? | The experts indicated that while there is no direct evidence for downstream treatment options following progression on momelotinib, other JAK inhibitors like fedratinib may be considered as subsequent lines of therapy. Momelotinib could be used as a first-line treatment, with fedratinib as a potential second-line option. In cases where the primary concern is anemia rather than splenomegaly, momelotinib might be more suitable in third-line settings. However, the evidence is limited, and treatment decisions should be individualized based on patient response and specific clinical scenarios. |
Clinical Evidence
The objective of the CDA-AMC clinical review is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of oral momelotinib 200 mg once daily for the treatment of splenomegaly and/or disease-related symptoms in adult patients with intermediate-risk or high-risk PMF, post-PV MF, or post-ET MF who have moderate to severe anemia. The focus will be placed on comparing momelotinib to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of momelotinib is presented in 4 sections, with critical appraisal of the evidence by CDA-AMC included at the end of each section. The first section, the systematic review, includes the pivotal studies and RCTs selected according to the sponsor’s systematic review protocol. The CDA-AMC assessment of the certainty of the evidence in this first section using the GRADE approach follows the critical appraisal of the evidence. The second section includes sponsor-submitted long-term extension studies. The third section would include indirect evidence from the sponsor (none submitted). The fourth section would include additional studies considered by the sponsor to address important gaps in the systematic review evidence. The sponsor submitted 2 retrospective analyses and 1 set of interim results of an ongoing extended access study to address gaps related to long-term survival by baseline transfusion independence status; however, these were not included in the clinical review report.
Included Studies
Clinical evidence from the following are included in the CDA-AMC review and appraised in this document:
3 pivotal studies or RCTs identified in the systematic review
3 long-term extension studies.
Systematic Review
The contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the CDA-AMC review team.
Description of Studies
Characteristics of the included studies are summarized in Table 7.
The clinical efficacy and safety of momelotinib were assessed in 3 international phase III trials — the SIMPLIFY-1 trial (patients not previously treated with JAK inhibitors) and the SIMPLIFY-2 and MOMENTUM trials (patients previously treated with JAK inhibitors) — in patients with symptomatic PMF, post-ET MF, or post-PV MF and anemia.10-12,43,44,49
SIMPLIFY-1 Trial
The SIMPLIFY-1 trial was a randomized, double-blind study of momelotinib versus ruxolitinib in patients with symptomatic PMF, post-ET MF, or post-PV MF who had not previously received a JAK inhibitor therapy.10,43 The study was conducted in 112 sites across 22 countries, including Edmonton, Vancouver, Hamilton, and Toronto in Canada (Table 7). Thirteen of the enrolled patients were living in Canada.10,51 The study design of the SIMPLIFY-1 trial is presented in Figure 1.
Patients were randomly assigned in a 1:1 ratio to receive oral momelotinib plus ruxolitinib placebo or ruxolitinib plus momelotinib placebo for 24 weeks, following 5 weeks of screening.10 Randomization was stratified by transfusion dependence (presence or absence; defined as ≥ 4 units of RBC transfusions or hemoglobin levels < 8 g/dL in the 8 weeks before randomization, excluding patients with clinically overt bleeding) and by platelet count (< 100 × 109/L, ≥ 100 × 109/L and ≤ 200 × 109/L, or > 200 × 109/L). At the time of the SIMPLIFY-1 study initiation, ruxolitinib was the only approved JAK inhibitor indicated for the treatment of intermediate-risk or high-risk MF in adults and therefore was chosen as the active control for this trial.10,38,56
Following the completion of the double-blind treatment phase, patients treated with momelotinib or ruxolitinib were eligible to receive momelotinib as an open-label treatment for up to 216 weeks (i.e., through week 240).10 The planned overall study duration, including screening, double-blind randomized treatment, open-label treatment, and survival follow-up periods, was approximately 5 years.10
Figure 1: Study Design of the SIMPLIFY-1 Trial
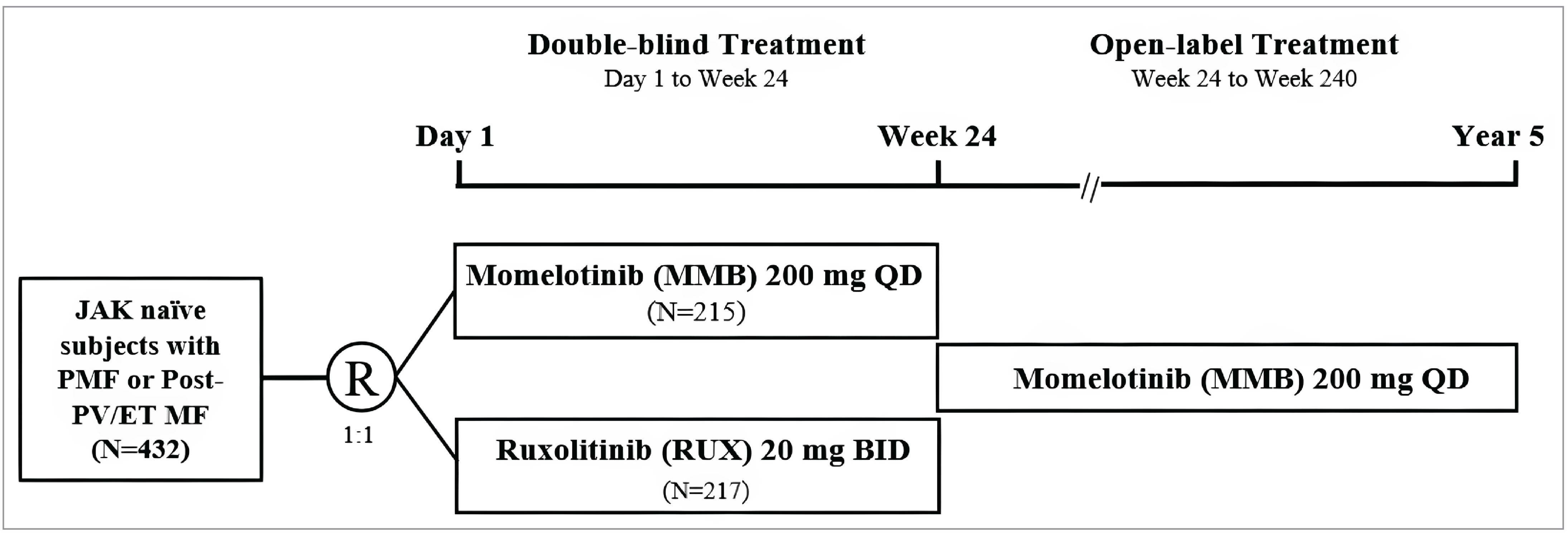
BID = twice daily; ET = essential thrombocythemia; JAK = Janus kinase; MF = myelofibrosis; MMB = momelotinib; QD = once daily; PMF = primary myelofibrosis; PV = polycythemia vera; R = randomization; RUX = ruxolitinib.
Source: GSK Data on File 2021 (SIMPLIFY-2 Clinical Study Report).10
Table 7: Details of Studies Included in the Systematic Review
Detail | SIMPLIFY-1 (JAK inhibitor naive, noninferiority) | SIMPLIFY-2 (JAK inhibitor experienced, superiority) | MOMENTUM (JAK inhibitor experienced with symptomatic anemia, superiority) |
|---|---|---|---|
Designs and populations | |||
Study design | Phase III, double-blind RCT | Phase III, open-label RCT | Phase III, double-blind, active-controlled RCT |
Locations | 23 sites in 22 countries, including Canada | 52 sites in 8 countries, including Canada | 107 sites in 21 countries, including Canada |
Patient enrolment dates | Start date: December 6, 2013 End date: May 2, 2019 | Start date: June 19, 2014 End date: April 25, 2019 | Start date: February 7, 2020 End date: December 29, 2022 |
Randomized (N) | 432 (MMB: 215; RUX: 217) | 156 (MMB: 104; BAT: 52) | 195 (MMB: 130; DAN: 65) |
Inclusion criteria |
|
|
|
Exclusion criteria |
|
|
|
Drugs | |||
Intervention | MMB 200 mg q.d. + RUX PBO b.i.d. | MMB 200 mg q.d. | MMB 200 mg q.d.+ DAN PBO b.i.d. |
Comparator | RUX 20 mg b.i.d. + MMB PBO q.d. | BAT | DAN 300 mg b.i.d. + MMB PBO q.d. |
Study duration | |||
Screening phase | 5 weeks | 30 days | ≤ 6 weeks before randomization |
Treatment phase | Blinded treatment: 24 weeks Open-label extension: up to 216 weeks | Open label: 24 weeks Extended: up to 204 weeks | Blinded treatment: 24 weeks Extended DAN: up to 48 weeks Patients who completed MMB treatment at 24 weeks could transition to the extension study |
Follow-up phase | 12 weeks posttreatment Long-term survival follow-up every 6 months up to 5 years | 12 weeks posttreatment Long-term survival follow-up every 6 months up to 5 years | 30 days posttreatment Long-term survival follow-up for 7 years |
Outcomes | |||
Primary end point | SRR ≥ 35% from baseline at week 24 | SRR ≥ 35% from baseline at week 24 | MFSAF TSS response at week 24 |
Secondary and exploratory end points | Secondary:
Exploratory:
| Secondary:
Exploratory:
| Secondary:
Exploratory:
|
Publication status | |||
Publications | Gupta et al. (2024)41 Oh et al. (2024)42 Mesa et al. (2022)1 Mesa et al. (2017)43 ClinicalTrials.gov, NCT01969838 | Mesa et al. (2022)1 Harrison et al. (2018)44 ClinicalTrials.gov, NCT02101268 | Verstovsek et al. (2023a)45
Gerds et al. (2023)46 Verstovsek et al. (2023b)47 Mesa et al. (2023)48 Verstovsek et al. (2021)49 ClinicalTrials.gov, NCT04173494 |
AE = adverse event; ALT = alanine transaminase; ANC = absolute neutrophil count; ASCT = allogenic stem cell transplant; AST = aspartate transaminase; BAT = best available therapy; b.i.d. = twice daily; CR = complete remission; CrCl = creatinine clearance; DAN = danazol; DIPSS = Dynamic International Prognostic Scoring System; ECOG PS = Eastern Cooperative Oncology Group Performance Status; ELN = European Leukemia Net; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EMH = extramedullary hematopoiesis; ESA = erythropoiesis-stimulating agent; ET = essential thrombocythemia; HAV = hepatitis A virus; HBV = hepatitis B virus; HCV = hepatitis C virus; HRQoL = health-related quality of life; ICT = iron chelation therapy; IWG-MRT = International Working Group for Myelofibrosis Research and Treatment; JAK = Janus kinase; LCM = Left Coastal Margin; LFS = leukemia-free survival; MF = myelofibrosis; MF-8D = Myelofibrosis 8 dimensions; MFSAF = Myelofibrosis Symptom Assessment Form; MMB = momelotinib; MPN-SAF = Myeloproliferative Neoplasm Symptom Assessment Form; ORR = overall response rate; OS = overall survival; PBC = peripheral blast count; PBO = placebo; PGI-C = Patient Global Impressions — Change; PMF = primary myelofibrosis; PR = partial remission; PRO = patient-reported outcome; PROMIS = Patient-Reported Outcomes Measurement Information System; PV = polycythemia vera; q.d. = once daily; RBC = red blood cell; RCT = randomized controlled trial; RUX = ruxolitinib; SAE = serious adverse event; SF-36 = Short Form (36) Health Survey; SRR = splenic response rate; TD = transfusion dependence; TEAE = treatment-emergent adverse event; TI = transfusion independence; TSS = total symptom score; ULN = upper limit of normal; VAS = visual analogue scale.
Sources: ClinicalTrials.gov 2023 [MOMENTUM];50 ClinicalTrials.gov 2023 [SIMPLIFY-1];51 ClinicalTrials.gov 2023 [SIMPLIFY-2];52 GSK Data on File 2016 [SIMPLIFY-1 statistical analysis plan];35 GSK Data on File 2016 [SIMPLIFY-2 statistical analysis plan];36 GSK Data on File 2017 [SIMPLIFY-1 clinical study protocol];53 GSK Data on File 2017 [SIMPLIFY-2 clinical study protocol];54 GSK Data on File 2020 [MOMENTUM clinical study protocol];55 GSK Data on File 2021 [SIMPLIFY-1 Clinical Study Report];10 GSK Data on File 2021 [SIMPLIFY-2 Clinical Study Report];11 GSK Data on File 2021 [MOMENTUM statistical analysis plan];37 GSK Data on File 2023 [MOMENTUM Clinical Study Report];12 Harrison et al. (2018); Mesa et al. (2017); Verstovsek et al. (2023).45 Details included in the table are from the sponsor’s summary of clinical evidence.
SIMPLIFY-2 Trial
The SIMPLIFY-2 trial was a randomized, open-label study of momelotinib versus BAT in patients with PMF, post-ET MF, or post-PV MF who experienced suboptimal responses or hematological toxic effects after treatment with ruxolitinib and who experienced National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE) grade 3 or 4 anemia, thrombocytopenia, or hematoma during ruxolitinib treatment.11,44 The study was conducted in 55 sites across 8 countries, including Edmonton, Toronto, and Montreal in Canada (Table 7). Ten of the enrolled patients were living in Canada.11,52 The study design of the SIMPLIFY-2 trial is presented in Figure 2.
Patients were randomly assigned in a 2:1 ratio to receive oral momelotinib or BAT for 24 weeks, following 30 days of screening.11 Randomization was stratified based on transfusion dependence (presence or absence; defined as ≥ 4 units of RBC transfusions or hemoglobin levels < 8 g/dL in the 8 weeks before randomization) and by baseline TSS (< 18 or ≥ 18).11
Following the completion of the open-label treatment phase, patients treated with momelotinib or BAT were eligible to receive momelotinib as an open-label treatment for up to 204 weeks (i.e., through week 228) in the open-label extension phase.11 The planned overall study duration, including screening, randomized treatment, posttreatment follow-up, and survival follow-up periods, was approximately 5 years.11
Figure 2: Study Design of the SIMPLIFY-2 Trial
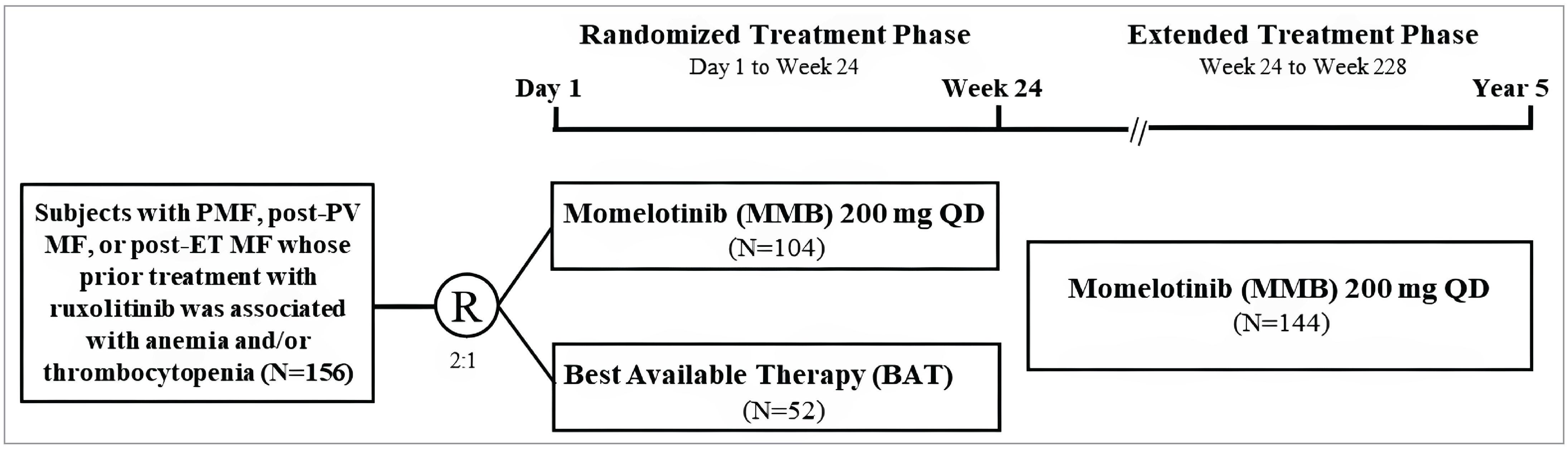
BAT = best available therapy; ET = essential thrombocythemia; MF = myelofibrosis; MMB = momelotinib; QD = once daily; PMF = primary myelofibrosis; PV = polycythemia vera; R = randomization.
Source: GSK Data on File 2021 (SIMPLIFY-2 Clinical Study Report).11
MOMENTUM Trial
The MOMENTUM trial was a randomized, double-blind study of momelotinib versus danazol in patients with symptomatic PMF, post-ET MF, or post-PV MF who had anemia (hemoglobin levels < 10 g/dL) and had previously received JAK inhibitor therapy.45 The study was conducted in 107 sites across 21 countries, including Vancouver, Halifax, Hamilton, Toronto, Montreal, and Quebec City in Canada (Table 7). Five of the patients were living in Canada.12,45,50 The study design of the MOMENTUM trial is presented in Figure 3.12
Patients were randomly assigned in a 2:1 ratio to receive oral momelotinib plus danazol placebo or danazol plus momelotinib placebo, following a baseline period of 7 days.45 Danazol was selected as an active comparator as per the National Comprehensive Cancer Network and European Society for Medical Oncology clinical treatment recommendation for the use of danazol in the treatment of patients with anemia and MF.7,45,57 Patients receiving JAK inhibitor therapy at screening tapered therapy over at least 1 week, with a lesser (or no) tapering period for those receiving a low-dose JAK inhibitor therapy. Patients then completed a nontreatment period of at least 2 weeks beginning at least 7 days before the first day of the baseline period. Patients were stratified based on MFSAF TSS (< 22 or ≥ 22), baseline palpable spleen length below the left costal margin (< 12 cm or ≥ 12 cm), baseline RBC or whole blood units transfused in the 8 weeks before randomization (0 units, 1 to 4 units, or ≥ 5 units), and study site.12,45
Patients treated with momelotinib or danazol were eligible to receive momelotinib as an open-label treatment for up to 180 weeks (i.e., through week 204) in the open-label extension phase.12,45 The planned overall study duration, including screening, baseline, double-blind randomized treatment, open-label treatment, safety follow-up, and long-term follow-up periods, was approximately 7 years.12
Populations
Inclusion and Exclusion Criteria
The SIMPLIFY-1, SIMPLIFY-2, and MOMENTUM trials included adult patients (aged ≥ 18 years) with a confirmed diagnosis of PMF per the WHO criteria or of post-PV MF or post-ET MF per the International Working Group for Myelofibrosis Research and Treatment (IWG-MRT) criteria.10-12,43-45 Patients with high-risk, intermediate-2–risk, or intermediate-1–risk MF, as defined by the IPSS (the SIMPLIFY-1 trial), the DIPSS (the SIMPLIFY-2 trial), or the DIPSS-plus (the MOMENTUM trial), were included in the studies. Patients had baseline palpable splenomegaly (≥ 5 cm below the left costal margin), an ECOG PS of less than or equal to 2, and a life expectancy of more than 24 weeks. The SIMPLIFY-1 trial comprised patients who had not previously received a JAK inhibitor, and the SIMPLIFY-2 and MOMENTUM trials comprised patients who had previously received treatment with a JAK inhibitor.10-12,43-45
Patients in the SIMPLIFY-2 study had experienced complications with prior ruxolitinib treatment, such as hematologic toxicity, as characterized by a requirement for RBC transfusion while on ruxolitinib, or a dosage adjustment of ruxolitinib to less than 20 mg twice daily at the start of or during treatment and the occurrence of CTCAE grade 3 or 4 thrombocytopenia, anemia, or hematoma while receiving ruxolitinib.11,44 While the SIMPLIFY-1 trial included patients with a baseline platelet count of at least 50 × 109/L and the MOMENTUM trial included patients with a baseline platelet count of at least 25 × 109/L, there was no minimum platelet count eligibility criterion for the SIMPLIFY-2 trial.10-12 The MOMENTUM study also required patients to have a baseline hemoglobin level of less than 10 g/dL.12,49
Patients were excluded from all the trials if they had prior splenectomy, were unwilling or unable to undergo MRI or CT imaging, had splenic irradiation 3 months or less before the first dose of the study drug, had received prior treatment with momelotinib, and (for the SIMPLIFY-1 trial only) prior treatment with a JAK1 inhibitor or JAK2 inhibitor.10-12,43-45
Figure 3: Study Design of the MOMENTUM Trial
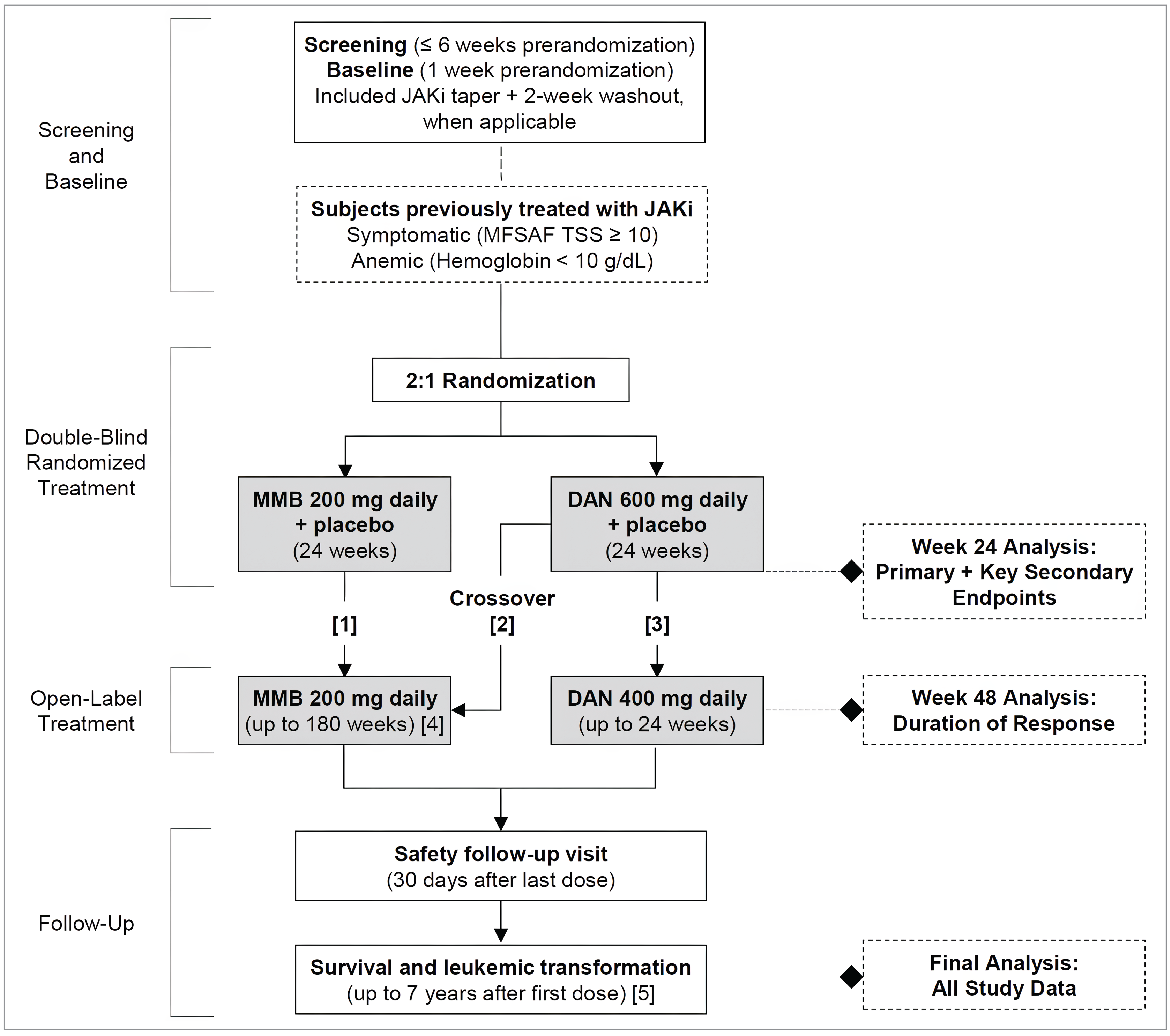
DAN = danazol; JAKi = Janus kinase inhibitor; MFSAF = Myelofibrosis Symptom Assessment Form; MMB = momelotinib; TSS = treatment symptom score.
[1] Patients treated with MMB who completed at least 24 weeks of randomized treatment were eligible for open-label MMB (patient remained blinded). Patients treated with MMB who discontinued treatment early and continued assessments through week 24 or experienced splenic progression were ineligible for open-label MMB (patient was unblinded).
[2] Crossover from randomized DAN to open-label MMB was allowed, as follows: at end of week 24 if the patient completed the randomized treatment period (patient remained blinded); at end of week 24 if the patient discontinued DAN early but continued study assessments and did not receive prohibited medications unless approved by the sponsor (patient was unblinded); or before the end of week 24 if the patient met criteria for confirmed splenic progression (patient was unblinded).
[3] Patients treated with DAN receiving clinical benefits after 24 weeks of randomized treatment were eligible for open-label DAN (patient was unblinded).
[4] Transition to an MMB extension study (SRA-MMB-4365, XAP) was allowed for patients who completed at least 24 weeks of open-label treatment with MMB.
[5] Patients who completed the safety follow-up visit in the MOMENTUM trial were transitioned to extension study for long-term survival follow-up.
Source: GSK Data on File 2023 (MOMENTUM Clinical Study Report).12
Interventions
In the SIMPLIFY-1, SIMPLIFY-2, and MOMENTUM trials, patients randomized to the momelotinib arm self-administered momelotinib at a dosage of 200 mg once daily, orally, followed by postdose observation period of at least 4 hours.10-12,53,54 Treatment with momelotinib was continued until disease progression, confirmed splenic progression, leukemic transformation, pregnancy, or toxicity.10-12 In the SIMPLIFY-1 and MOMENTUM trials, the momelotinib placebo tablets were visually identical to the momelotinib tablets but without any active ingredients.10-12
In the SIMPLIFY-1 trial, ruxolitinib was self-administered orally (1 to 2 tablets), with the starting dosage ranging from 5 mg to 20 mg twice daily based on platelet count, creatinine clearance, and aspartate aminotransferase or alanine aminotransferase levels.10 In the SIMPLIFY-2 trial, patients receiving BAT received treatment at doses and schedules as determined by the investigator per standard of care.11 Regimens for BAT included chemotherapy (e.g., hydroxyurea), anagrelide, corticosteroid, hematopoietic growth factor, immunomodulating agent, androgen, and interferon. No active treatment was also an option for patients randomized to the BAT arm of the SIMPLIFY-2 trial.11 Treatment switching, interruptions, or holds and treatment with ruxolitinib were also permitted in the BAT arm.11,54 In the MOMENTUM trial, the active comparator, danazol, was self-administered by patients orally, with a starting dosage of 300 mg twice daily (for a total dose of 600 mg per day).12
In the SIMPLIFY-1, SIMPLIFY-2, and MOMENTUM trials, treatment with momelotinib or comparators (ruxolitinib, BAT, or danazol, respectively) was interrupted if any of the following occurred: thrombocytopenia, an absolute neutrophil count of less than 0.5 × 109/L, grade 3 or 4 nonhematologic toxicity, or a grade 2 or higher bleeding event.10-12 Dose re-escalation was allowed following the resolution of toxicity or return to baseline grade, at the investigator’s discretion.10-12
In the event of insufficient splenic response (a < 50% reduction from the pretreatment baseline in palpable spleen length or a < 35% reduction in spleen volume, as measured by CT or MRI) in the SIMPLIFY-1 and SIMPLIFY-2 trials, for patients receiving less than 200 mg per day of momelotinib, the dose was increased in 50 mg increments up to 200 mg per day, provided there were no new or recurrent instances of the following: momelotinib-related grade 3 or 4 nonhematologic toxicities, grade 2 or higher bleeding, a platelet count less than 50 × 109/L, and/or an absolute neutrophil count of less than 0.75 × 109/L.10,11 In the SIMPLIFY-1 and SIMPLIFY-2 trials, momelotinib was permanently discontinued if grade 3 or 4 treatment-related nonhematologic toxicity led to treatment interruption at the 100 mg once daily dosage and recurred after restarting momelotinib at the same dosage.10,11
In the MOMENTUM trial, dose reduction with re-escalation following resolution of symptoms was recommended for patients who experienced mild dizziness or light headedness, hypotension, flushing, nausea, or headache that persisted or recurred beyond the first day of dosing.12 During randomized treatment, doses of the study treatment (i.e., momelotinib plus danazol placebo or danazol plus momelotinib placebo) were reduced by sequential decrements as presented in Table 8.
In the SIMPLIFY-1, SIMPLIFY-2, and MOMENTUM trials, anti-hypertensive therapy was withheld for at least 4 hours after study drug administration on the first day of dosing.10-12 Patients requiring anti-hypertensive therapy were observed, and anti-hypertensive medication could be administered if considered clinically necessary. Potent CYP3A4 inducers in the MOMENTUM trial and dual CYP3A4 and CYP2C9 inducers were permitted with prior sponsor approval.10-12
In the MOMENTUM trial, alternatives to breast cancer resistance protein transporter or modified breast cancer resistance protein transporter substrates were permitted, as clinically indicated during the study treatment. Steroids were permitted for the treatment non-MF conditions as supportive care if initiated before screening and continuing at a stable dosage equivalent to no more than 10 mg prednisone daily. If clinically indicated, the steroids could be temporarily increased for up to 10 days (cumulative) in any 28-day period or, if clinically indicated, could be initiated for up to 10 days (cumulative) in any 28-day period. Temporary use of restricted anti-MF medications was permitted for severe splenic progression that required immediate action for patients continuing in the study, until confirmed splenic progression.12
Concomitant use of the following agents was prohibited:10-12
SIMPLIFY-1 trial: Experimental therapy other than momelotinib, anti-MF therapy, chemotherapy, immunomodulators, systemic corticosteroids, androgens for the treatment of MF, ESAs, interferons, JAK inhibitors other than ruxolitinib, and granulocyte colony-stimulating factor (unless administered for the treatment of neutropenic fever).
SIMPLIFY-2 trial: Experimental therapy other than momelotinib and anti-MF therapy, including hematopoietic growth factor.
MOMENTUM trial: Concomitant use of anti-MF therapy, including JAK inhibitors, alkylating agents, hypomethylating agents, interferons, ESAs, immunomodulating agents, corticosteroids, androgens, growth factors, splenic irradiation, and splenectomy, as well as the use of investigational agents within 3 months before randomization. Steroids for the treatment of MF were prohibited within 1 week before the day 1 baseline until study drug discontinuation.
Table 8: Treatment Dose Reduction in the MOMENTUM Trial
Dosing stage | Total daily dose (mg) | |
|---|---|---|
Momelotinib | Danazol | |
Starting dose | 200 | 600 |
Dose decrement 1 | 150 | 400 |
Dose decrement 2 | 100 | 300 |
Dose decrement 3 | 50 | 200 |
Source: GSK Data on File, 2023 (MOMENTUM Clinical Study Report).12
In the double-blind treatment phase of the SIMPLIFY-1 and MOMENTUM trials, the identity of the study drug was concealed by central blinding of study drug assignments and randomization was achieved via the Interactive Response System. Treatment assignment remained blinded unless unblinding was required for emergency medical care.10,12 In the SIMPLIFY-1 trial, treatment assignments in the randomized treatment phase remained blinded until all patients had completed week 24 and the study was unblinded for the final efficacy analysis.10 In the MOMENTUM study, patients in the momelotinib plus danazol placebo arm who received momelotinib in the open-label treatment period remained blinded to their randomized treatment, whereas patients who received danazol in the open-label treatment period as well as those with confirmed symptomatic splenic progression before week 24 were unblinded to their treatment assignment after completing week 24 to confirm their treatment in the randomized treatment period.12
Outcomes
A list of efficacy end points assessed in this clinical review report is provided in Table 9, followed by descriptions of the outcome measures. The summarized end points are based on the outcomes included in the sponsor’s summary of clinical evidence as well as any outcomes identified as important to this review according to the clinical experts consulted by CDA-AMC and input from patient and clinician groups and public drug plans. Using the same considerations, the review team selected the end points that were considered to be most relevant to inform the expert committee deliberations and finalized this list of end points in consultation with members of the expert committee. All summarized efficacy end points were assessed using GRADE. Select notable harms outcomes considered important for informing the expert committee deliberations were also assessed using GRADE. End points are associated with time points that would allow the information to be clinically relevant. Given the design of the trials (with crossovers to momelotinib after week 24), end points reported after the double-blind period would have significant limitations in interpretations and generalizability. Thus, any clinically relevant end points with a clinically relevant time point beyond 24 weeks were not included as part of the main outcomes of this report.
Table 9: Outcomes Summarized From the Studies Included in the Systematic Review
Outcome measure | Time point | SIMPLIFY-1 (JAK inhibitor naive, noninferiority) | SIMPLIFY-2 (JAK inhibitor experienced, superiority) | MOMENTUM (JAK inhibitor experienced with symptomatic anemia, superiority) |
|---|---|---|---|---|
Transfusion independence | Week 24 | Secondarya | Secondarya | Key secondarya |
Rate of RBC transfusion | Week 24 | Secondarya | Secondarya | Exploratory (total number of units) |
Splenic response rate | Week 24 | Primarya | Primarya | Key secondarya |
Duration of splenic response | Up to 1 year | NR (only up to week 24 as an exploratory outcome) | NR (only up to week 24 as an exploratory outcome) | NR |
MFSAF total symptom score response rate | Week 24 | NR | NR | Primarya |
MPN-SAF total symptom score response rate | Week 24 | Secondarya | Secondarya | NR |
Overall survival | Up to 2 years | NR (only up to week 24; subsequently, patients were eligible to receive momelotinib) | NR (only up to week 24; subsequently, patients were eligible to receive momelotinib) | NR (only up to week 24; subsequently, patients were eligible to receive momelotinib) |
Leukemia-free survival | Up to 2 years | NR (only up to week 24; subsequently, patients were eligible to receive momelotinib) | NR (only up to week 24; subsequently, patients were eligible to receive momelotinib) | NR (only up to week 24; subsequently, patients were eligible to receive momelotinib) |
Serious adverse events | Week 24 | Safety | Safety | Safety |
JAK = Janus kinase; MFSAF = Myelofibrosis Symptom Assessment Form; MPN-SAF = Myeloproliferative Neoplasm Symptom Assessment Form; NR = not reported; RBC = red blood cell.
aStatistical testing for these end points was adjusted for multiple comparisons through hierarchal testing.
Source: Details included in the table are from the sponsor’s summary of clinical evidence.
Transfusion Independence Response Rate at Week 24
Across all 3 trials, transfusion independence was defined as the absence of RBC transfusions and no hemoglobin levels less than 8 g/dL in the prior 12 weeks, except in the case of clinically overt bleeding.53-55
SRR at Week 24
Across all 3 trials, SRR was defined as the proportion of patients with at least a 35% reduction from baseline in spleen volume as measured by MRI or CT at week 24.53-55 The clinical experts consulted by CDA-AMC identified this threshold as clinically meaningful.
Rate of RBC Transfusion at Week 24
In the SIMPLIFY-1 and SIMPLIFY-2 trials, the rate of RBC transfusion was defined as the average number of RBC units per patient-month during the randomized treatment phase (through week 24).35,36 The rate of RBC transfusion was not reported in the MOMENTUM trial; instead, the total number of transferred units was used to inform this outcome.
MFSAF TSS Response Rate at Week 24
The MFSAF is a validated MF-specific quality-of-life and symptom assessment form that evaluates the severity of 7 key symptoms experienced by patients with MF: fatigue, night sweats, pruritus, abdominal discomfort, pain under the left ribs, early satiety, and bone pain.58 The 7 symptom domains are assessed on an 11-point numeric rating scale, ranging from 0 to 10, with the TSS representing the sum of the scores across these 7 domains, ranging from 0 to 70. Higher symptom scores correspond to more severe symptoms.37 In the MOMENTUM trial, MFSAF version 4.0 was used.12 Validation was conducted in the MOMENTUM trial as outline in Table 10.
The MFSAF TSS response rate was the primary end point in the MOMENTUM trial.12 It was defined as the proportion of patients with at least a 50% reduction from baseline in mean MFSAF TSS over the 28 days immediately before the end of week 24. No clear rationale was given for this threshold being chosen. However, the clinical experts consulted by CDA-AMC suggested that this threshold is clinically meaningful. The baseline MFSAF TSS was calculated as the mean of the daily TSS values collected over the baseline period of the 7 consecutive days before randomization.12,37
MPN-SAF TSS Response Rate at Week 24
The Myeloproliferative Neoplasm Symptom Assessment Form (MPN-SAF) is a validated HRQoL measure that evaluates symptom burden and symptom status during the treatment of patients with MPN.10,11 The modified MPN-SAF version 2.0 is an 8-item questionnaire that consists of 9 symptom domains assessed on an 11-point numeric rating scale, ranging from 0 to 10, with the TSS representing the sum of the scores across these 9 domains, with a range from 0 to 80. The items assess the worst daily occurrences of tiredness, early satiety, abdominal discomfort, night sweats, itching, bone pain, pain under the left ribs, and inactivity. Higher symptom scores correspond to more severe symptoms.10,11 No information was available on the validity of the questionnaire, as indicated in Table 10.
In the SIMPLIFY-1 and SIMPLIFY-2 trials, the MPN-SAF TSS response rate was a secondary efficacy end point. It was defined as the proportion of patients who have experienced at least a 50% reduction from baseline in mean MFSAF TSS over the 28 days immediately before the end of week 24. No clear rationale was given for this threshold being chosen. However, the clinical experts consulted by CDA-AMC suggested that this threshold is clinically meaningful. The baseline MPN-SAF TSS was calculated as the mean of the daily TSS values collected over the baseline period of the 7 consecutive days before randomization.53,54
Overall Survival
OS was defined as the interval from the first study drug dosing date in the randomized treatment phase (or randomization in patients who did not receive treatment) to death from any cause.35-37
Comparative data are only available up to week 24 and are not considered to be clinically relevant according to the feedback received from the clinical experts consulted by CDA-AMC. In the 3 included studies, patients switched to momelotinib after the randomized period (week 24). Results from the extension phase are outlined in Appendix 1.
Leukemia-Free Survival
Across all 3 trials, leukemia-free survival was defined as the interval from the first study drug dosing date (or randomization date for patients who did not receive treatment) to the date of any evidence of leukemic transformation or death from any cause. Leukemic transformation was defined according to IWG-MRT and European Leukemia Net criteria as a bone marrow blast count of at least 20% or a peripheral blood blast content of at least 20% associated with an absolute blast count of at least 1 × 109/L that lasts for 2 weeks or longer.10-12
Comparative data are only available up to week 24 and are not considered to be clinically relevant according to the feedback received from the clinical experts consulted by CDA-AMC. In the 3 included studies, patients switched to momelotinib after the randomized period (week 24). Results from the extension phase are outlined in Appendix 1.
Safety
The safety of momelotinib in patients with untreated PMF, post-ET MF, or post-PV MF was assessed in the safety analysis set of the SIMPLIFY-1 and SIMPLIFY-2 trials and the in safety population of the MOMENTUM trial using descriptive analyses.10-12 The Medical Dictionary for Regulatory Activities version 22.0 was used to classify AEs by system organ class and preferred term in the SIMPLIFY-1 and SIMPLIFY-2 trials, and the Medical Dictionary for Regulatory Activities version 24.0 was used in the MOMENTUM trial. AEs were graded using CTCAE version 4.03 in the SIMPLIFY-1 and SIMPLIFY-2 trials and using CTCAE version 5.0 in the MOMENTUM trial.10-12
Safety data reported in this submission are based on the respective Clinical Study Reports for the most recent database locks in each trial — July 1, 2019, for the SIMPLIFY-1 trial; June 25, 2019, for the SIMPLIFY-2 trial; and December 3, 2021, for the MOMENTUM trial — unless otherwise specified.10-12
Table 10: Summary of Outcome Measures and Their Measurement Properties
Change outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
MFSAF TSS | A validated MF-specific quality-of-life and symptom assessment form that evaluates the severity of 7 key symptoms experienced by patients with MF. The 7 symptom domains are assessed on an 11-point numeric rating scale, ranging from 0 to 10, with the TSS representing the sum of the scores across these 7 domains, ranging from 0 to 70. Higher symptom scores correspond to more severe symptoms.58 | Validity: MFSAF was validated in a prospective trial of patients with MF involving patient and provider feedback. The MSAS and the BPI instruments were used to establish correlation for agreement between instruments (the MFSAF and BPI or MFSAF and MSAS).58 The MFSAF demonstrated good performance in terms of the assessment of specific symptoms addressed in the validated MSAS instrument. Specifically, corresponding questions from the MSAS were all highly correlated with MFSAF counterparts (P < 0.01), including lack of energy (fatigue), cough, pain (both abdominal pain and bone pain), sweats (night sweats), itching, and weight loss. Further validation of pain measurements in the MFSAF was established in comparison to the BPI scale, where the presence and intensity of pain (both abdominal and bone) were highly correlated (P < 0.01).58 Reliability: No information was found on the reliability of the MFSAF for patients with MF in a literature search. Responsiveness: No information was found on the responsiveness of the MFSAF for patients with MF in a literature search. | In the MOMENTUM trial, an MID analysis was conducted to derive the MID for MFSAF TSS using PGI-S as an anchor (PGI-C symptom and fatigue anchors demonstrated low correlation with TSS [r < 0.30] and were excluded from the MID analyses).12,58,59 The MID analysis was conducted on the modified ITT population, which included all randomized patients from both the momelotinib and danazol arms; patients with a missing baseline MFSAF TSS were excluded. The primary MID was determined by weighing correlations between the PGI-S MF symptom and fatigue, and the mean percent change from baseline in TSS to weeks 12 and 24.59 Using the anchor-based MID analysis, a 50% reduction in TSS was identified as a suitable MID for evaluating symptom response using the MFSAF version 4.0. Individual items in the MFSAF, which have 11 levels (0 to 10), were considered to have a response if the change from baseline was ≥ 3.12,58,59 |
MPN-SAF TSS | A validated HRQoL measure that evaluates symptom burden and symptom status during treatment in patients with MPN. The modified MPN-SAF version 2.0 is an 8-item questionnaire that consists of 9 symptom domains assessed on an 11-point numeric rating scale, ranging from 0 to 10, with the TSS representing the sum of the scores across these 9 domains. Higher symptom scores correspond to more severe symptoms.10,11 | No information was found on the validity, reliability, and responsiveness of the MPN-SAF for patients with MF. | No MID was established for patients with MF. |
BPI = Brief Pain Inventory; HRQoL = health-related quality of life; ITT = intention to treat; MID = minimal important difference; MF = myelofibrosis; MFSAF = Myelofibrosis Symptom Assessment Form; MPN = myeloproliferative neoplasm; MPN-SAF = Myeloproliferative Neoplasm Symptom Assessment Form; MSAS = Memorial Symptom Assessment Scale; PGI-C = Patient Global Impressions — Change; PGI-S = Patient Global Impressions — Severity; TSS = total symptom score.
Statistical Analysis
Clinical Trial End Points
Transfusion Independence Status at Week 24
In the SIMPLIFY-1 and SIMPLIFY-2 trials, transfusion-independent status at week 24 was a secondary end point; it was a key secondary end point in the MOMENTUM trial.35-37
In the SIMPLIFY-1 and SIMPLIFY-2 trials, the 95% exact CI was calculated using the Clopper-Pearson method without stratification.35,36 The 12-week period at week 24 for the absence of transfusion was the consecutive 84-day period with the last day as the earliest date of day 176, or the last randomized treatment phase participation date if the last date is after or on day 162. For the primary analysis of transfusion-independent status at week 24, a patient whose last randomized treatment phase participation was before day 162 was considered not transfusion independent. The noninferiority of momelotinib over ruxolitinib or BAT was evaluated in a similar way as for SRR at week 24 using the Cochran-Mantel-Haenszel (CMH) approach (refer to the SRR at Week 24 section). Three sensitivity analyses were conducted (Table 11).35,36
In the MOMENTUM trial, a synthesis approach was used to determine the noninferiority of momelotinib versus danazol, wherein the treatment effect of the active control (danazol) was not prespecified, but the proportion of the active control effect to be preserved was specified (i.e., 80% of the response rate in the danazol arm should be preserved in the momelotinib arm). A stratum-adjusted 2-sided 95% CI was calculated to determine the noninferiority treatment difference (lower bound of CI > 0 demonstrates noninferiority of momelotinib versus danazol).37
Rate of RBC Transfusion
In the SIMPLIFY-1 and SIMPLIFY-2 trials, the rate of RBC transfusion was a secondary end point. A negative binomial regression method with an offset parameter was used to analyze the rate of RBC transfusion.35,36 A time-to-event analysis employing the proportional means model was used as a supportive analysis. To accommodate the presence of multiple units in a transfusion, the recurrence times were perturbed, ensuring they did not appear identical. When calculating RBC transfusion-related end points, transfusions due to clinically overt bleeding were excluded from the calculation. Missing transfusion units were imputed by 2 units.35,36
In the MOMENTUM trial, a proportional rate or mean model was performed as an exploratory analysis, treating each transfusion unit as a recurrent event, including follow-up time after the last transfusion, and censoring patients at their last follow-up date. A weighting based on the number of units transfused was applied to the transfusion event to calculate the number of units used in a transfusion.37
TSS Response Rate at Week 24
The TSS response rate at week 24, as measured using the modified MPN-SAF, was the first key secondary end point in the SIMPLIFY-1 and SIMPLIFY-2 trials.35,36 The TSS at week 24 was calculated as the average of daily TSS from a 28-day consecutive period before week 24. The response rate in TSS at week 24 was analyzed for patients who had a baseline TSS greater than or equal to 0 but a nonzero or missing TSS at week 24. Patients with a missing week 24 TSS but a nonmissing baseline TSS were considered not to experience a response. The noninferiority of momelotinib over ruxolitinib or BAT was evaluated in a similar way as for SRR at week 24 using the CMH approach (refer to the SRR at Week 24 section). In the SIMPLIFY-1 trial, the noninferiority of momelotinib was assessed by comparing the difference in TSS response rates at week 24, aiming for a 67% preservation of the ruxolitinib effect, using the CMH approach to adjust for stratification factors at a 2-sided 0.05 level. If noninferiority was established, the 2-sided 95% CI for the difference was then used to evaluate superiority. No clinical rationale was given for the decision to preserve 67% of the treatment effect. The percentage change in TSS from baseline to week 24 was calculated as:35,36
percentage change = 100 × (week 24 TSS – baseline TSS)/baseline TSS
To evaluate the robustness of the primary results, 5 sensitivity analyses were performed in the SIMPLIFY-1 trial and 4 in the SIMPLIFY-2 trial (Table 11).35,36
The TSS response rate at week 24, as measured by the MFSAF, was the primary end point in the MOMENTUM trial.37 The TSS at week 24 was calculated as the average of the daily TSS from a 28-day consecutive period before week 24. Patients with a missing week 24 TSS, as well as those with daily TSS measurements for fewer than 20 days during the consecutive 28-day period, were classified as not experiencing a response. The primary analysis of MFSAF TSS response was performed on the intention-to-treat (ITT) analysis set using a stratified CMH test. Primary inference was based on the asymptotic P value, in turn based on the Wald statistic from this CMH test. To evaluate the robustness of the primary results, 3 sensitivity analyses were performed. Further, a sensitivity analysis was conducted on data collected before the treatment switch (danazol to momelotinib), in case more than 10% of patients crossed over from danazol to momelotinib treatment before the end of week 24.37
SRR at Week 24
In the statistical hierarchy of primary and secondary end points in the SIMPLIFY-1 and SIMPLIFY-2 trials, the primary analysis was for SRR at week 24.35,36 In the MOMENTUM trial, SRR at week 24 was a key secondary end point.37
In the SIMPLIFY-1 and SIMPLIFY-2 trials, the noninferiority of momelotinib over ruxolitinib or BAT was determined using a conventional 2-sided 95% CI greater than 0, based on a stratum-adjusted CMH proportion.35,36 Additionally, a 2-sided 95% exact CI was provided for each treatment arm based on the Clopper-Pearson method. The noninferiority test aimed to preserve 60% of the treatment effect size of ruxolitinib in the SIMPLIFY-1 trial. No clinical rationale was provided to support the preservation threshold. To evaluate the robustness of the primary results, 4 sensitivity analyses were performed.35,36
In the MOMENTUM trial, the primary analysis of splenic response was performed on the ITT analysis set, as well as on the per-protocol analysis set for sensitivity purposes, using a CMH test after adjusting for stratification factors.37
Across the 3 trials, patients with a missing baseline spleen volume, a missing scan, or unavailable week 24 spleen volume due to early discontinuation of the randomized treatment phase were considered not to experience a response.35-37 In addition, patients with splenic assessments (by MRI or CT) taken more than 10 days after the beginning of the open-label treatment phase and patients receiving other anti-MF therapy during the randomized treatment phase were considered not to experience a response in the MOMENTUM trial.37
Safety Outcomes
Safety outcomes were reported by treatment arm and phase using descriptive statistics and graded on the National Cancer Institute CTCAE version 4.03 in the SIMPLIFY-1 and SIMPLIFY-2 trials and CTCAE version 5.0 in the MOMENTUM trial. Safety assessments included AEs, SAEs, AEs leading to premature discontinuation of the study drug, AEs leading to a dose reduction of the study drug, AEs leading to deaths, AESI, and laboratory evaluations.35-37
Handling of Missing Data
The conventions used for inputting missing data are presented in Table 11. In the SIMPLIFY-1 and SIMPLIFY-2 studies, missing data for the transfusion independence outcome were addressed in the primary analysis by classifying patients with incomplete transfusion independence data as nonresponders; additional imputation methods were applied in the sensitivity analyses to assess the robustness of the results to different assumptions. The MOMENTUM study used a similar approach, categorizing patients with missing transfusion independence data as nonresponders. For the rate of RBC transfusion, both SIMPLIFY studies imputed missing transfusion units as 2 units and applied standard rules for partial date imputation. Assessments up to 10 days after the first momelotinib dose in the extended treatment phase and between days 141 and 197 from the date of randomization were used for week 24 spleen volume if no assessment was available in the randomized phase before or on the date of the first momelotinib dose in the extended treatment phase and between days 141 and 197.11,36 Patients who discontinued early in the randomized treatment phase before an efficacy assessment visit or with missing assessments at the corresponding visit for MRI spleen response, TSS response, and transfusion independence response were considered to be patients with missing transfusion independence data.36
The MOMENTUM study handled missing data by using a recurrent event model, treating each transfusion as a separate event and censoring patients at their last follow-up. For the SRR and TSS response at week 24 in the primary analysis, all studies classified patients with missing TSS data as nonresponders. Sensitivity analyses, including the last observation carried forward (LOCF) method, were used to assess the impact of the approach for handling missing data.
For safety data, missing data were generally not imputed across the studies. Analyses focused on the observed data, with safety outcomes like AEs reported descriptively.
Sample Size and Power Calculation
SIMPLIFY-1 Trial
The study sample size for the SIMPLIFY-1 trial was based on the primary end point of SRR at week 24.35 The sample size was calculated to compare the SRR at week 24 among participants randomized to receive momelotinib plus ruxolitinib placebo versus ruxolitinib plus momelotinib placebo.35
A sample size of 420 was estimated for randomization to the 2 treatment arms in a 1:1 ratio, based on the assumption of the common treatment effect of momelotinib and ruxolitinib on SRR being 34% (based on the lower bound of the 95% CI of the ruxolitinib effect on SRR observed in Study 351 COMFORT-I).35 This sample size provided more than 90% power for testing the noninferiority hypothesis of SRR at week 24. The sample size calculation also considered the comparison of the TSS response rate at week 24 among participants randomized to receive momelotinib plus ruxolitinib placebo versus ruxolitinib plus momelotinib placebo.35
A sample size of 420 was estimated to provide more than 90% power for testing the noninferiority hypothesis of TSS response rate at week 24, based on the assumption of the common treatment effect of momelotinib and ruxolitinib on TSS response rate at week 24 being 38% (based on the lower bound of the 95% CI of the ruxolitinib effect on TSS response rate observed in Study 351 COMFORT-I).35
With a total sample size of 420, the study was estimated to yield 85% power to detect for testing at least a 15% difference between the 2 treatment arms in the transfusion independence and transfusion dependence rates using a chi-square test.35
SIMPLIFY-2 Trial
The study sample size for the SIMPLIFY-2 trial was based on the primary end point of SRR at week 24.36 The sample size was calculated to compare the SRR at week 24 among participants randomized to receive momelotinib plus BAT-placebo versus BAT plus momelotinib placebo.36
Based on historical data from momelotinib studies and BAT trials, a sample size of 150 was estimated for randomization to the 2 treatment arms in a 2:1 ratio.36 This sample size was based on the common treatment effect on SRR at week 24 in patients previously treated with ruxolitinib being 20% for momelotinib (SRR at week 24 being 23.5% and 33.3% from the CCL09101 and YM387-II-02 studies, respectively) and 1% for BAT (0 out of 73 patients experienced splenic response at week 24 in Study 352 COMFORT-II). With a total sample size of 150, the study was estimated to yield more than 95% power at a 2-sided level of 0.05 using the Fisher exact test.36
MOMENTUM Trial
The study sample size for the MOMENTUM trial was based on the first primary end point of TSS response rate at week 24, the second primary end point of transfusion independence response rate at week 24, and the first key secondary end point of SRR at week 24.37 The sample size was calculated to compare the TSS response rate, the transfusion independence response rate, and SRR at week 24 among participants randomized to receive momelotinib plus danazol placebo versus danazol plus momelotinib placebo.37
A sample size of 180 was estimated for randomization to the 2 treatment arms in a 2:1 ratio.37 With a total sample size of 180, the study was estimated to yield 98.8% power to detect a true difference of 21% (23% with momelotinib versus 2% with danazol) or 90% power to detect a true difference of 15% (17% with momelotinib versus 2% with danazol) in TSS response rate at week 24 using a 2-sided significance of 0.05. Further, with a total sample size of 180, the study was estimated to yield 90% power to detect a true difference of 24% in the proportion of patients with transfusion-independent status (45% with momelotinib versus 21% with danazol) and a true difference of 14% in SRR at week 24 (15% with momelotinib versus 1% with danazol).37
Statistical Testing
SIMPLIFY-1 Trial
A hierarchical testing procedure controlled at 5% (2-sided) was used to control for type I errors for the primary end point and key secondary end points.35
The primary analysis was for SRR at week 24.35 Sequential testing of key secondary end points was conducted only if the primary efficacy analysis was noninferior at the applicable alpha level (0.05). If the SRR at week 24 was statistically significant, then the key secondary end points — TSS response rate at week 24, transfusion independence response rate at week 24, transfusion dependence rate at week 24, and rate of RBC transfusion in the randomized treatment phase — were to be tested (Figure 4). Other end points were not formally tested in a hierarchal testing procedure. The primary analysis of the primary efficacy end point and the secondary end points was conducted when each patient completed the randomized treatment period (July 1, 2019).35
Figure 4: SIMPLIFY-1 Trial Gatekeeping Process for Multiple Hypothesis Tests
RBC = red blood cell; RUX = ruxolitinib; SRR = splenic response rate; TD = transfusion dependence; TI = transfusion independence; TSS = total symptom score.
Note: Statistical significance at P ≤ 0.05.
Source: GSK Data on File, 2016 (SIMPLIFY-1 statistical analysis plan).35
SIMPLIFY-2 Trial
The hierarchical testing procedure in the SIMPLIFY-2 trial was similar to that in the SIMPLIFY-1 trial. A hierarchical testing procedure controlled at 5% (2-sided) was used to control for type I errors for the primary end point and key secondary end points.36
The primary analysis was for SRR at week 24.36 Sequential testing of key secondary end points was conducted only if the primary efficacy analysis was significantly superior at the applicable alpha level (0.05). If the SRR at week 24 was statistically significant, then the key secondary end points — TSS response rate at week 24, rate of RBC transfusion in the randomized treatment phase, transfusion independence response rate at week 24, and transfusion dependence rate at week 24 — were to be tested (Figure 5). Other end points were not formally tested in a hierarchal testing procedure.36 The primary analysis of the primary efficacy end point and the secondary end points was conducted when each patient completed the randomized treatment period (June 25, 2019).11,36
Figure 5: SIMPLIFY-2 Trial Gatekeeping Process for Multiple Hypothesis Tests
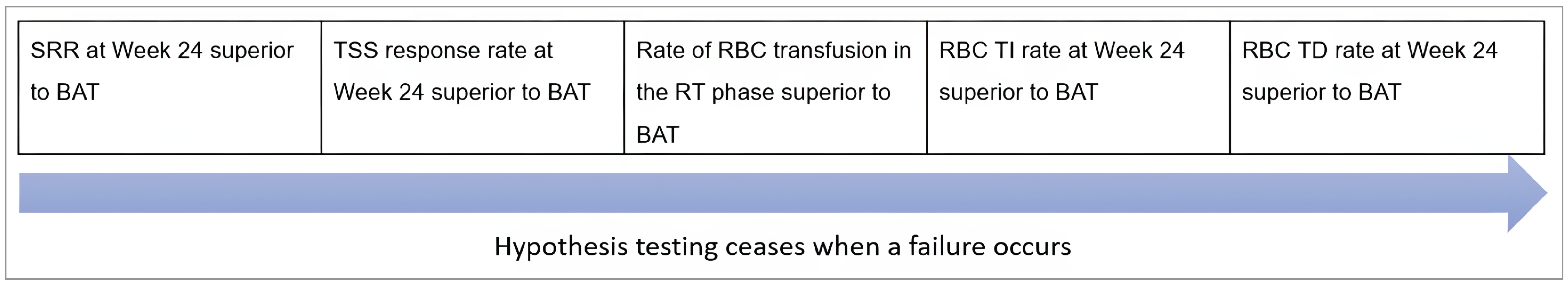
BAT = best available therapy; RBC = red blood cell; RT = randomized treatment; SRR = splenic response rate; TD = transfusion dependence; TI = transfusion independence; TSS = total symptom score.
Note: Statistical significance at P ≤ 0.05.
Source: GSK Data on File, 2016 (SIMPLIFY-2 statistical analysis plan).36
MOMENTUM Trial
A hierarchical testing procedure controlled at 5% (2-sided) was used to control for type I errors for the primary end point and key secondary end points.37
The primary analysis was for MFSAF TSS response rate at week 24.37 Sequential testing of key secondary end points was conducted only if the primary efficacy analysis was significantly superior at the applicable alpha level (0.05). If the MFSAF TSS response rate at week 24 was statistically significant, then the key secondary end points — transfusion independence status at week 24, SRR at week 24 (based on the 25% reduction criterion), MFSAF TSS change from baseline at week 24, SRR at week 24 (based on the 35% reduction criterion), and the rate of no transfusion at week 24 — were to be tested (Figure 6). Other end points were not formally tested in a hierarchal testing procedure.37
The primary analysis of the primary efficacy end point was conducted when each patient completed the randomized treatment period, crossed over early, or dropped out from the randomized treatment (December 3, 2021).12,37 The study was unblinded at the data cut-off date of December 3, 2021, and all other study end points were analyzed, except for transfusion independence status and SRR at week 24, which were analyzed at the database lock of January 17, 2023.12 All the data before the data cut-off, including the data from the open-label extended treatment period, were included in the primary analysis.
Figure 6: MOMENTUM Trial Gatekeeping Process for Multiple Hypothesis Tests

DAN = danazol; MFSAF = Myelofibrosis Symptom Assessment Form; SRR = splenic response rate; TI = transfusion independence; TSS = total symptom score.
Notes: If noninferiority is established for TI status, then the P value associated with the test of superiority was estimated. Statistical significance at P ≤ 0.05.
Source: GSK Data on File, 2021 (MOMENTUM statistical analysis plan).37
Subgroup Analyses
SIMPLIFY-1 Trial
Prespecified subgroup analyses were performed by age, sex, race, geographical region, baseline transfusion dependence, baseline spleen volume, TSS at baseline, baseline hemoglobin level (< 8 g/dL or ≥ 8 g/dL), baseline platelet count (< 100, ≥ 100 and ≤ 200, and > 200 × 109/L), IPSS risk category (intermediate risk or high risk), baseline JAK2V617F mutation status (positive or negative), baseline MF disease status (PMF, post-PV MF, or post-ET MF).35
Subgroup analyses were presented as a forest plot of proportion differences (SRR, TSS response rate, rate of RBC transfusion, rate of transfusion independence and transfusion dependence) or treatment arm least squares mean differences by subgroup.35
In addition to the prespecified subgroup analyses, post hoc subgroup analyses were performed for splenic response, TSS, transfusion independence, and transfusion dependence at week 24 as well as for transfusion independence by week 24 and transfusion rate across the following subgroups:10
transfusion independence at baseline
non–transfusion independence at baseline
baseline TSS (≥ 10)
baseline hemoglobin (< 10 g/dL, < 12 g/dL, and ≥ 12 g/dL)
baseline TSS ≥ 10 and hemoglobin < 10 g/dL
baseline platelet count (≤ 150, > 150 and ≤ 300, > 300 × 109/L).
SIMPLIFY-2 Trial
Prespecified subgroup analyses were performed by age, sex, race, baseline transfusion dependence, baseline spleen volume, TSS at baseline, baseline hemoglobin level (< 8 g/dL or ≥ 8 g/dL), DIPSS risk category (intermediate risk or high risk), baseline JAK2V617F mutation status (positive or negative), baseline MF disease status (PMF, post-PV MF, or post-ET MF), duration of ruxolitinib received before randomization (< 12 weeks or ≥ 12 weeks), and highest dosage of ruxolitinib received since randomization (≥ 20 mg twice daily or < 20 mg twice daily; for BAT treatment arm only).36
Subgroup analyses were presented as a forest plot of proportion differences (SRR, TSS response rate, rate of RBC transfusion, rate of transfusion independence and transfusion dependence) or treatment arm least squares mean differences by subgroup.36
In addition to the prespecified subgroup analyses, post hoc subgroup analyses were performed for splenic response, TSS, transfusion independence, and transfusion dependence at week 24 as well as for transfusion independence by week 24 and transfusion rate across the following subgroups:11
transfusion independence at baseline
non–transfusion independence at baseline
baseline TSS (< 10 or ≥ 10)
baseline hemoglobin (< 10 g/dL or ≥ 10 g/dL)
baseline TSS ≥ 10 and hemoglobin < 10 g/dL
baseline platelet count (< 100, < 150, ≥ 100 and ≤ 200, > 200 × 109/L).
MOMENTUM Trial
Prespecified subgroup analyses were performed by age, sex, race, geographical region, baseline platelet count (< 50, 50 to 150, 150 to 300, or > 300 × 109/L; ≤ 150 or > 150 × 109/L; ≤ 200 or > 200 × 109/L), baseline transfusion dependence and transfusion independence, baseline MFSAF TSS (< 22 or ≥ 22), baseline spleen volume, baseline hemoglobin level (< 8 g/dL or ≥ 8 g/dL), RBC or whole blood units transfused in the 8-week period before randomization (0, 1 to 4, or ≥ 5 units), baseline DIPSS risk category (low risk, intermediate-1 risk, intermediate-2 risk, or high risk), baseline JAK2V617F mutation status (positive, negative, or unknown), baseline MF disease status (PMF, post-PV MF, or post-ET MF), JAK1 inhibitor treatment received before randomization (< 12 weeks or ≥ 12 weeks), ongoing JAK1 inhibitor at screening, prior JAK1 inhibitor total daily dosage received before enrolment (none, < 20 mg twice daily, or ≥ 20 mg twice daily of ruxolitinib or ≤ 200 mg or > 200 mg of fedratinib), and baseline glomerular filtration rate (30 mL/min/1.73 m² to 60 mL/min/1.73 m², or ≥ 60 mL/min/1.73 m²).37
Patients with missing subgroup-determining variables were included in a subgroup of their own.37 Subgroups with too few patients were combined as necessary. Subgroup analyses were presented as a forest plot of proportion differences (TSS response, transfusion independence status, splenic response) or treatment arm least squares mean differences (TSS change from baseline) by subgroup. Analyses of the duration of response or improvement were restricted to those patients whose data qualified for the response or improvement of interest. Unstratified exact 95% CIs were produced for differences in treatment proportions across subgroups.37
Analysis Populations
The main analysis populations in the SIMPLIFY-1, SIMPLIFY-2, and MOMENTUM studies are summarized in Table 12. In all 3 studies, the primary end point was analyzed using the ITT analysis set, which included all randomized patients. Safety analysis populations were defined as all patients who received at least 1 dose of the study drug.
Results
Patient Disposition
A total of 432, 156, and 195 patients were included in the randomized treatment phase of the SIMPLIFY-1, SIMPLIFY-2, and MOMENTUM trials, respectively.30,50,51 This included 180 patients in the SIMPLIFY-1 trial (momelotinib: 86; ruxolitinib: 94) and 105 patients in the SIMPLIFY-2 trial (momelotinib: 66; BAT: 39) who had anemia, with hemoglobin levels less than 10 g/dL.52,72
In the SIMPLIFY-1 and SIMPLIFY-2 trials, a numerically greater proportion of patients discontinued momelotinib than ruxolitinib or BAT in the randomized treatment phase. In the MOMENTUM trial, a greater proportion of patients in the danazol arm discontinued treatment in the randomized treatment phase than in the momelotinib arm. The most common reasons for treatment discontinuation were AEs in the SIMPLIFY-1 and MOMENTUM trials and patient decision in the SIMPLIFY-2 trial. In the SIMPLIFY-1 trial, more patients in the momelotinib group than the ruxolitinib group discontinued due to AEs (4.7% versus 1.8%) and death (2.3% versus 0.9%). In the SIMPLIFY-2 trial, the most noticeable imbalances were AEs (5.8% versus 0%). In the MOMENTUM trial, more discontinuations due to AEs occurred in the danazol group than in the momelotinib group (16.9% versus 12.3%), with additional imbalances seen in discontinuations due to insufficient efficacy, which were slightly higher in the danazol group.30,50,51
Table 11: Statistical Analysis of Efficacy End Points
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
SIMPLIFY-1 | ||||
Rate of TI at week 24 | 95% exact CI was calculated using the Clopper-Pearson method without stratification | NR | None |
|
Rate of RBC transfusion in the double-blind phase |
| NR | Negative binomial regression method handles missing data under missing at random assumption | Analysis based on per-protocol analysis set |
SRR at week 24 |
| Adjustment for stratification factors: TD (yes or no) and platelet count (< 100 × 109/L, ≥ 100 × 109/L and ≤ 200 × 109/L, or > 200 × 109/L) | Patients with missing baseline spleen volume or with unavailable week 24 spleen volume due to early discontinuation from the double-blind phase or a missing scan were considered nonresponders |
|
MPN-SAF TSS response |
| Adjustment for stratification factors: TD (yes or no) and platelet count (< 100 × 109/L, ≥ 100 × 109/L and ≤ 200 × 109/L, or > 200 × 109/L) | Patients with missing data classified as nonresponder |
|
Safety |
| NR | None | NR |
SIMPLIFY-2 | ||||
Rate of TI at week 24 | 95% exact CI was calculated using the Clopper-Pearson method without stratification | NR | None |
|
Rate of RBC transfusion |
| NR | Missing transfusion units were imputed by 2 units | Analysis based on per-protocol analysis set |
SRR at week 24 |
| Adjustments made for stratification factors: TD (yes or no) and baseline TSS (< 18 or ≥ 18) | Missing spleen volume at week 24 was imputed using the last postbaseline value |
|
MPN-SAF TSS response rate at week 24 | 2-sided 95% CI based on stratum-adjusted CMH proportion | NR | Patients missing a week 24 TSS but not missing a baseline TSS were considered nonresponders |
|
Safety |
| None | None | None |
MOMENTUM | ||||
Rate of TI at week 24 |
| Adjustments made for stratification factors: MFSAF TSS baseline score (< 22 or ≥ 22), baseline palpable spleen length below LCM (< 12 cm or ≥ 12 cm), baseline RBC or whole blood transfusion in the 8-week period before randomization (0, 1 to 4, and ≥ 5 units) | Patients with missing data for TI at week 24 were categorized as nonresponders |
|
Total number of RBC transfusion units |
| NR | None | NR |
SRR at week 24 | 2-sided 95% CI based on stratum-adjusted CMH proportion | Adjustments made for stratification factors: MFSAF TSS baseline score (< 22 or ≥ 22), baseline palpable spleen length below LCM (< 12 cm or ≥ 12 cm), baseline RBC or whole blood transfusion in the 8-week period before randomization (0, 1 to 4, and ≥ 5 units) | Patients with missing evaluation at baseline or week 24 and patients with differing spleen scanning modalities at baseline and week 24 were set to nonresponder for SRR at week 24 | Analysis based on per-protocol analysis set |
MFSAF TSS response rate at week 24 | Asymptomatic P value for primary inference was based on the Wald statistics from the CMH test | Adjustments made for stratification factors: MFSAF TSS baseline score (< 22 or ≥ 22), baseline palpable spleen length below LCM (< 12 cm or ≥ 12 cm), baseline RBC or whole blood transfusion in the 8-week period before randomization (0, 1 to 4, and ≥ 5 units) | Patients with missing data classified as nonresponder |
|
Safety | Descriptive statistics, graded for severity using the NCI CTCAE version 5.0 | None | None | NR |
AESI = adverse event of special interest; CI = confidence interval; CMH = Cochran-Mantel-Haenszel; DAN = danazol; KM = Kaplan-Meier; LCM = left costal margin; LOCF = last observation carried forward; MFSAF = Myelofibrosis Symptom Assessment Form; MMB = momelotinib; MPN-SAF = Myeloproliferative Neoplasm Symptom Assessment Form; NCI CTCAE = National Cancer Institute Common Terminology Criteria for Adverse Events; NR = not reported; RBC = red blood cell; SRR = splenic response rate; TD = transfusion dependence; TI = transfusion independence; TSS = total symptom score; vs. = versus.
Sources: GSK Data on File, 2016 (SIMPLIFY-1 statistical analysis plan);35 GSK Data on File, 2016 (SIMPLIFY-2 statistical analysis plan);36 GSK Data on File, 2021 (MOMENTUM statistical analysis plan).37 Details included in the table are from the sponsor’s summary of clinical evidence.
Table 12: Analysis Populations of the SIMPLIFY-1, SIMPLIFY-2, and MOMENTUM Trials
Study | Population | Definition | Application |
|---|---|---|---|
SIMPLIFY-1 | ITTa | All patients who were randomized in the study |
|
ITT anemia analysis seta | Patients in the ITT population with baseline hemoglobin < 10 g/dL |
| |
Per-protocol analysis setb | Patients in the ITT analysis set who:
|
| |
Safety analysis setb |
| Safety analyses | |
PRO analysis seta |
| PRO analyses | |
PK analysis setb | Patients in the SAS who have ≥ 1 nonmissing postdose concentration value for the corresponding analyte in plasma | PK parameters | |
PK substudy analysis setb | Patients in the SAS who participated in the PK substudy and have intensive concentration data to provide interpretable results for the specific parameters of interest (i.e., ≥ 1 nonmissing PK parameter) for the corresponding analyte | PK parameters (not included in this submission) | |
SAS anemia analysisb | Patients in the safety population with baseline hemoglobin < 10 g/dL | Safety analyses | |
SIMPLIFY-2 | ITTa | All patients who were randomized in the study |
|
ITT anemia analysis seta | Patients in the ITT population with baseline hemoglobin < 10 g/dL | Efficacy analyses, PRO analyses, demographic and baseline characteristics | |
Per-protocol analysis setb | Patients in the ITT analysis set who:
|
| |
Safety analysis setb |
| Safety analyses | |
PRO analysis setb |
| PRO analyses | |
PK analysis setb | Patients in the SAS who have ≥ 1 nonmissing postdose concentration value for the corresponding analyte in plasma | PK parameters (not included in this submission) | |
PK substudy analysis setb | Patients in the SAS who participated in the PK substudy and have intensive concentration data to provide interpretable results for the specific parameters of interest (i.e., ≥ 1 nonmissing PK parameter) for the corresponding analyte | PK parameters (not included in this submission) | |
SAS anemia analysisb | Patients in the safety population with baseline hemoglobin < 10 g/dL | Safety analyses | |
MOMENTUM | ITTa | All randomized patients | Efficacy analyses except for TI status at week 24, PRO analyses |
Per-protocol analysis setb | Randomized patients who received ≥ 1 dose of the study drug and had no important protocol deviation | Sensitivity analyses of efficacy | |
Safety analysis setb | All patients in the ITT population who received ≥ 1 dose of the study drug | Safety analyses |
ITT = intention to treat; MF = myelofibrosis; PK = pharmacokinetic; PP = per protocol; PRO = patient-reported outcome; SAS = safety analysis set; TI = transfusion independence; TSS = total symptom score.
aPatients were grouped according to randomized treatment.
bPatients were grouped according to the actual treatment received.
Sources: GSK Data on File, 2016 (SIMPLIFY-1 statistical analysis plan);35 GSK Data on File, 2016 (SIMPLIFY-2 statistical analysis plan);36 GSK Data on File, 2021 (SIMPLIFY-1 Clinical Study Report);10 GSK Data on File, 2021 (SIMPLIFY-2 Clinical Study Report);11 GSK Data on File, 2021 (MOMENTUM statistical analysis plan).37 Details included in the table are from the sponsor’s summary of clinical evidence.
Patient disposition in the randomized phase of the SIMPLIFY-1, SIMPLIFY-2, and MOMENTUM trials is summarized in Table 13.
Table 13: Summary of Patient Disposition From Studies Included in the Systematic Review
Patient disposition | SIMPLIFY-1 | SIMPLIFY-2 | MOMENTUM | |||
|---|---|---|---|---|---|---|
MMB (N = 215) | RUX (N = 217) | MMB (N = 104) | BAT (N = 52) | MMB (N = 130) | DAN (N = 65) | |
Screened, n | 602 | 244 | 307 | |||
Reason for not proceeding past screening, n | 21 | 22 | NR | |||
AEs | 2 | 2 | NR | |||
Investigator discretion | 2 | 1 | NR | |||
Withdrew consent | 6 | 3 | NR | |||
Lost to follow-up | 1 | 0 | NR | |||
Outside of visit window | 6 | 10 | NR | |||
Other | 4 | 6 | NR | |||
Randomized, N | 215 | 217 | 104 | 52 | 130 | 65 |
Completed randomized treatment phase, n (%) | 188 (87.4) | 208 (95.9) | 77 (74.0) | 41 (78.8) | 94 (72.3) | 38 (58.5) |
Discontinued randomized treatment phase, n (%) | 27 (12.6) | 9 (4.1) | 27 (26.0) | 11 (21.2) | 36 (27.7) | 27 (41.5) |
Reason for discontinuation, n (%) | ||||||
AEs | 10 (4.7) | 4 (1.8) | 6 (5.8) | 0 | 16 (12.3) | 11 (16.9) |
Death | 5 (2.3) | 2 (0.9) | 5 (4.8) | 4 (7.7) | 4 (3.1) | 3 (4.6) |
Patient decision | 4 (1.9) | 2 (0.9) | 8 (7.7) | 4 (7.7) | 6 (4.6) | 5 (7.7) |
Disease progression | 2 (0.9) | 0 | 4 (3.8) | 1 (1.9) | 1 (0.8) | 2 (3.1) |
Investigator discretion | 5 (2.3) | 1 (0.5) | 4 (3.8) | 1 (1.9) | 0 | 1 (1.5) |
Symptomatic spleen growth | 1 (0.5) | 0 | 0 | 1 (1.9) | 0 | 0 |
Insufficient efficacy | 0 | 0 | 0 | 0 | 6 (4.6) | 3 (4.6) |
Leukemic transformation | 0 | 0 | 0 | 0 | 2 (1.5) | 2 (3.1) |
Lost to follow-up | 0 | 0 | 0 | 0 | 1 (0.8) | 0 |
AE = adverse event; BAT = best available therapy; DAN = danazol; MMB = momelotinib; NR = not reported; RUX = ruxolitinib.
Sources: GSK Data on File, 2021 (SIMPLIFY-1 Clinical Study Report);10 GSK Data on File, 2021 (SIMPLIFY-2 Clinical Study Report);11 GSK Data on File, 2023 (MOMENTUM Clinical Study Report).12 Details included in the table are from the sponsor’s summary of clinical evidence.
Baseline Characteristics
In the SIMPLIFY-1 trial, the baseline characteristics between the momelotinib and ruxolitinib treatment groups were generally well balanced. Both groups had similar mean ages (65.0 years for momelotinib and 64.4 years for ruxolitinib) and a comparable proportion of male participants (57.7% for momelotinib versus 55.3% for ruxolitinib). The distribution of MF subtypes was also similar, with PMF being the most common subtype in both groups (59.5% for momelotinib and 53.5% for ruxolitinib). The risk categories, based on the IPSS, showed a slightly higher proportion of patients considered to be at high risk in the ruxolitinib group (49.3%, versus 43.3% for momelotinib). Other characteristics, such as mean TSS, ECOG PS, and JAK2V617F mutation status, were also comparable between the groups, indicating minimal baseline imbalances.
In the SIMPLIFY-2 trial, the momelotinib and BAT treatment groups were generally similar in baseline characteristics, though some differences were observed. The mean age was slightly higher in the BAT group (69.4 years) than in the momelotinib group (66.4 years). The momelotinib group had a higher proportion of males (66.3%, versus 46.2% for BAT). The distribution of MF subtypes and DIPSS risk categories was similar, though there was a higher proportion of patients considered to be at intermediate-2 risk in the momelotinib group (59.6%, versus 53.8% for BAT). The ECOG PS showed a slightly higher percentage of patients having an ECOG PS of 1 in the momelotinib group (58.7%, versus 50% for ruxolitinib) and a similar proportion of patients with an ECOG PS of 0. However, more patients in the BAT group had an ECOG PS of 2 (13.5%, versus 6.7% for momelotinib). JAK2V617F mutation status was similar across the groups, with the majority of patients being positive. However, patients in the BAT group had slightly lower mean hemoglobin levels and platelet counts.
In the MOMENTUM study, differences in baseline characteristics were noted between the momelotinib and danazol groups. The danazol group had a slightly older population on average (71.46 years, versus 69.85 years for momelotinib). The proportion of males was higher in the danazol group (67.7%, versus 60.8% in momelotinib). The distribution of DIPSS risk categories showed more patients considered to be at high risk in the momelotinib group (38.5%, versus 29.2% for danazol). Both groups had a majority of patients with an ECOG PS of 1 (63.8% for momelotinib versus 52.3% for danazol). The danazol group had a slightly higher proportion of patients with an ECOG PS of 2 (24.6%, versus 23.8% for momelotinib). A smaller proportion of patients in both groups had an ECOG PS of 0: 12.3% in the momelotinib group and 23.1% in the danazol group. Both groups had very low mean hemoglobin levels (8.06 g/dL for momelotinib and 7.86 g/dL for danazol), reflective of the patient population targeted in the MOMENTUM trial, with all participants having hemoglobin levels less than 10 g/dL (contrasted with mean hemoglobin levels of 10.6 g/dL and 10.7 g/dL [momelotinib versus ruxolitinib] in the SIMPLIFY-1 trial and 9.4 g/dL and 9.5 g/dL [momelotinib versus BAT] in the SIMPLIFY-2 trial). There were also some differences in prior treatment exposure, with a higher proportion of patients in the momelotinib group having received JAK inhibitor treatment for 12 weeks or more before randomization.
The baseline characteristics outlined in Table 14 are limited to those that are most relevant to this review or were felt to affect the outcomes or interpretation of the study results by the CDA-AMC team in consultation with the clinical experts.
Exposure to Study Treatments
A summary of patient exposure to study treatments is provided in Table 15.
Across all 3 trials, the duration of exposure to the study drug was similar, irrespective of the treatment received.10-12 In the ITT population, the mean momelotinib treatment adherence rate was approximately 99% in the SIMPLIFY-1 trial and 80% in the SIMPLIFY-2 trial.10,11
In the ITT population, patients randomized to the BAT arm of the SIMPLIFY-2 trial, received ruxolitinib (88.5%), hydroxyurea (23.1%), prednisone or prednisolone (11.5%), danazol (5.8%), no therapy (3.8%), ESA (3.8%), anagrelide (1.9%), darbepoetin alfa (1.9%), acetylsalicylic acid (1.9%), and thalidomide (1.9%). The mean treatment duration of ruxolitinib in the BAT arm of the SIMPLIFY-2 trial among this patient population was 19.6 weeks (standard deviation [SD] = 7.69).11
In patients with hemoglobin levels less than 10 g/dL in the SIMPLIFY-2 trial, those randomized to the BAT arm received ruxolitinib (89.7%), hydroxyurea (15.4%), prednisone or prednisolone (10.3%), danazol (7.7%), no therapy (5.1%), anagrelide (2.6%), darbepoetin alfa (2.6%), ESA (2.6%), and thalidomide (2.6%). The mean treatment duration of ruxolitinib in the BAT arm of the SIMPLIFY-2 trial among this patient population was 20.2 weeks (SD = 7.37).11
Concomitant medication use was reported by more than 95% of patients across the 3 trials.12,60,61 No information on subsequent therapy was provided.
Efficacy
Transfusion Independence Response Rate at Week 24
In the SIMPLIFY-1 trial, 66.5% of patients in the momelotinib group experienced transfusion independence at week 24 compared to 49.3% in the ruxolitinib group. The proportion difference was 0.18 (95% CI, 0.09 to 0.26). In the subpopulation with hemoglobin levels less than 10 g/dL, the response rates were 46.5% for momelotinib and 26.6% for ruxolitinib, with a treatment difference of 0.20 (95% CI, 0.05 to 0.34). For patients with hemoglobin levels less than 12 g/dL, response rates were 62.3% for momelotinib and 37.2% for ruxolitinib. In the SIMPLIFY-2 trial, 43.3% of patients in the momelotinib group experienced transfusion independence, compared to 21.2% in the BAT group, with a proportion difference of 0.23 (95% CI, 0.09 to 0.37). In the subpopulation with hemoglobin levels less than 10 g/dL, the rates were 33.3% for momelotinib and 12.8% for BAT, with a treatment difference of 0.21 (95% CI, 0.01 to 0.39). For patients with hemoglobin levels less than 12 g/dL, response rates were 40.9% for momelotinib and 15.2% for BAT. In the MOMENTUM study, 30.8% of patients in the momelotinib group experienced transfusion independence at week 24, compared to 20.0% in the danazol group, with a proportion difference of 10.99% (95% CI, –0.80% to 22.77%), with an adjusted proportion difference noninferiority test, which targeted 80% retention of the effect of danazol, at 14.77% (95% CI, 3.13% to 26.41%; P = 0.0064). The results of the sensitivity analyses in the SIMPLIFY-1 and SIMPLIFY-2 trials using the per-protocol analysis set and the LOCF method were in line with those of the ITT analysis. The results of the sensitivity analyses in the MOMENTUM trial were not available.
Table 14: Summary of Baseline Characteristics From Studies Included in the Systematic Review (ITT Analysis Set)
Characteristic | SIMPLIFY-1 | SIMPLIFY-2 | MOMENTUM | |||
|---|---|---|---|---|---|---|
MMB (N = 215) | RUX (N = 217) | MMB (N = 104) | BAT (N = 52) | MMB (N = 130) | DAN (N = 65) | |
Age (years), mean (SD) | 65.0 (10.67) | 64.4 (10.59) | 66.4 (8.13) | 69.4 (7.42) | 69.85 (8.24) | 71.46 (6.99) |
Male, n (%) | 124 (57.7) | 120 (55.3) | 69 (66.3) | 24 (46.2) | 79 (60.8) | 44 (67.7) |
Female, n (%) | 91 (42.3) | 97 (44.7) | 35 (33.7) | 28 (53.8) | 51 (39.2) | 21 (32.3) |
Race, n (%) | ||||||
Asian | 17 (7.9) | 20 (9.2) | NR | NR | 12 (9.2) | 6 (9.2) |
Black | 2 (0.9) | 2 (0.9) | 6 (5.8) | 0 | 2 (1.5) | 2 (3.1) |
White | 179 (83.3) | 178 (82.0) | 83 (79.8) | 44 (84.6) | 107 (82.3) | 50 (76.9) |
Not permitted (patient did not consent to share race information) | 15 (7.0) | 16 (7.4) | 15 (14.4) | 8 (15.4) | NR | NR |
Other | 2 (0.9) | 1 (0.5) | NR | NR | 7 (5.4) | 5 (7.7) |
BMI (kg/m2), mean (SD) | 24.9 (4.02) | 25.3 (3.99) | 26.7 (4.81) | 26.2 (3.82) | 25.20 (3.69) | 25.66 (6.02) |
MF subtype, n (%) | ||||||
Primary | 128 (59.5) | 116 (53.5) | 64 (61.5) | 30 (57.7) | 78 (60.0) | 46 (70.8) |
Post PV | 48 (22.3) | 50 (23.0) | 18 (17.3) | 12 (23.1) | 27 (20.8) | 11 (16.9) |
Post ET | 39 (18.1) | 51 (23.5) | 22 (21.2) | 10 (19.2) | 25 (19.2) | 8 (12.3) |
IPSS (SIMPLIFY-1) or DIPPS (SIMPLIFY-2 and MOMENTUM) risk category, n (%) | ||||||
Intermediate-1 | 46 (21.4) | 43 (19.8) | 23 (22.1) | 16 (30.8) | 7 (5.4) | 3 (4.6) |
Intermediate-2 | 76 (35.3) | 67 (30.9) | 62 (59.6) | 28 (53.8) | 72 (55.4) | 40 (61.5) |
High | 93 (43.3) | 107 (49.3) | 19 (18.3) | 8 (15.4) | 50 (38.5) | 19 (29.2) |
Missing | NA | NA | 0 | 0 | 1 (0.8) | 3 (4.6) |
TSS, mean (SD) | 19.4 (13.18) | 17.9 (11.47) | 18.5 (12.97) | 20.5 (16.03) | 27.96 (13.84) | 25.70 (12.79) |
ECOG PS, n (%) | ||||||
0 | 76 (35.3) | 72 (33.2) | 36 (34.6) | 19 (36.5) | 16 (12.3) | 15 (23.1) |
1 | 122 (56.7) | 120 (55.3) | 61 (58.7) | 26 (50.0) | 83 (63.8) | 34 (52.3) |
2 | 17 (7.9) | 25 (11.5) | 7 (6.7) | 7 (13.5) | 31 (23.8) | 16 (24.6) |
JAK2V617F mutation, n (%) | ||||||
Positive | 125 (58.1) | 141 (65.0) | 70 (67.3) | 37 (71.2) | 97 (74.6) | 51 (78.5) |
Negative | 61 (28.4) | 53 (24.4) | 31 (29.8) | 12 (23.1) | 28 (21.5) | 12 (18.5) |
Not previously tested or unknown | 29 (13.5) | 23 (10.6) | 3 (2.9) | 3 (5.8) | 3 (2.3) | 0 |
Duration of JAK inhibitor treatment, n (%) | ||||||
Missing | NA | NA | 13 (12.5) | 9 (17.3) | NR | NR |
< 12 weeks | NA | NA | 16 (15.4) | 10 (19.2) | 3 (2.3) | 2 (3.1) |
≥ 12 weeks | NA | NA | 75 (72.1) | 33 (63.5) | 127 (97.7) | 63 (96.9) |
Hemoglobin (g/dL), mean (SD) | 10.6 (2.09) | 10.7 (2.37) | 9.4 (1.92) | 9.5 (1.59) | 8.06 (1.14) | 7.86 (0.83) |
Hemoglobin level, n (%) | ||||||
< 8 g/dL | 28 (13.0) | 21 (9.7) | 27 (26.0) | 6 (11.5) | 62 (47.7) | 32 (49.2) |
≥ 8 g/dL | 187 (87.0) | 195 (89.9) | 77 (74.0) | 46 (88.5) | 67 (51.5) | 33 (50.8) |
< 10 g/dL | 86 (40.0) | 94 (43.3) | 66 (63.5) | 39 (75.0) | 130 (100) | 65 (100) |
≥ 10 g/dL | 129 (60.0) | 122 (56.2) | 38 (36.5) | 13 (25.0) | NR | NR |
< 12 g/dL | NR | NR | 93 (89.4) | 46 (88.5) | NR | NR |
Transfusion independent, n (%) | 147 (68.4) | 152 (70.0) | 32 (30.8) | 19 (36.5) | 17 (13.1) | 10 (15.4) |
Transfusion dependent, n (%) | 53 (24.7) | 52 (24.0) | 58 (55.8) | 27 (51.9) | 63 (48.5) | 34 (52.3) |
Platelet count (× 109/L), mean (SD) | 300.9 (206.86) | 301.5 (255.85) | 170.8 (148.01) | 126.5 (95.92) | 151.68 (130.90) | 130.69 (101·0) |
ANC (× 109/L), mean (SD) | 12.0 (13.39) | 11.3 (11.04) | 10.2 (13.47) | 8.0 (9.89) | 8.55 (11.26) | 6.93 (8.25) |
ANC = absolute neutrophil count; BAT = best available therapy; BMI = body mass index; DAN = danazol; DIPSS = Dynamic International Prognostic Scoring System; ECOG PS = Eastern Cooperative Oncology Group Performance Status; ET = essential thrombocythemia; IPSS = International Prognostic Scoring System; ITT = intention to treat; JAK = Janus kinase; MF = myelofibrosis; MMB = momelotinib; NA = not applicable; NR = not reported; PV = polycythemia vera; RUX = ruxolitinib; SD = standard deviation; TSS = total symptom score.
Sources: GSK Data on File, 2021 (SIMPLIFY-1 Clinical Study Report);10 GSK Data on File, 2021 (SIMPLIFY-2 Clinical Study Report);11 GSK Data on File, 2023 (MOMENTUM Clinical Study Report).12 Details included in the table are from the sponsor’s summary of clinical evidence.
Table 15: Summary of Patient Exposure From Studies Included in the Systematic Review (Safety Analysis Set)
Exposure | SIMPLIFY-1 | SIMPLIFY-2 | MOMENTUM | |||
|---|---|---|---|---|---|---|
MMB (N = 214) | RUX (N = 216) | MMB (N = 104) | BAT (N = 52) | MMB (N = 130) | DAN (N = 65) | |
Daily dose intensity (mg/day)a | ||||||
Mean (SD) | 188.4 (24.31) | 28.0 (11.06) | 187.1 (26.14)b | NR | 188.84 (18.41) | 576.47 (65.28) |
Median (Q1, Q3) | 200.0 (187.6, 200.0) | 30.0 (18.8, 38.9) | 197.6 (184.6, 200.0)b | NR | 200.00 (181.82, 200.00) | 600.00 (600.00, 600.00) |
Relative dose intensity (%), mean (SD)c | NR | NR | NR | NR | 94.42 (9.20) | 96.08 (10.88) |
On-treatment adherence rate (%)b,d | ||||||
Mean (SD) | 98.5 (5.24) | 97.8 (10.66) | 77.9 (31.53)b | NR | NR | NR |
Median (range) | 100.0 (67 to 111) | 99.2 (41 to 182) | 97.0 (5 to 113)b | NR | NR | NR |
Total duration of exposure to study drug (weeks)e | ||||||
Mean (SD) | 21.3 (6.37) | 23.3 (3.13) | 19.5 (7.71) | 21.0 (6.91) | 20.61 (6.21) | 17.29 (7.99) |
Median (range) | 23.9 (0.3 to 26.1) | 24.0 (1.3 to 26.9) | 23.9 (1 to 26) | 24.1 (2 to 25) | 24.00 (0.3 to 26.7) | 23.71 (0.7 to 26.9) |
BAT = best available therapy; DAN = danazol; MMB = momelotinib; NR = not reported; Q1 = quarter 1; Q3 = quarter 3; RUX = ruxolitinib; SD = standard deviation.
Notes: The safety analysis set includes all patients in the intention-to-treat population who received at least 1 dose of the study drug. Patients were evaluated based on actual treatment received. In the pooled data from all trials, the relative dose intensity was 100 (range, 0 to 114) for patients randomized to MMB during the treatment phase and was 97.2 (range, 0 to 387) for those who received MMB during the open-label phase.47
aDose intensity = actual cumulative dose / actual duration of the treatment (days).
bn = 102.
cRelative dose intensity = actual dose intensity / planned dose intensity.
dAdherence rate (%) = [total study drug administered / total study drug prescribed per protocol] × 100. Only patients who returned at least 1 bottle of study drug were included in the analysis.
eDuration of exposure to study drug = ([treatment end date – treatment start date] + 1) / 7.
Sources: GSK Data on File, 2021 (SIMPLIFY-1 Clinical Study Report);10 GSK Data on File, 2021 (SIMPLIFY-2 Clinical Study Report);11 GSK Data on File, 2023 (MOMENTUM Clinical Study Report).12 Details included in the table are from the sponsor’s summary of clinical evidence.
Rate of RBC Transfusion at Week 24
In the SIMPLIFY-1 trial, the mean rate of RBC transfusion at week 24 was 0.5 units per patient-month (SD = 1.27) in the momelotinib group, compared to 1.0 unit (SD = 1.62) in the ruxolitinib group, with a transfusion rate ratio of 0.28 (95% CI, 0.19 to 0.43). Among the subpopulation with hemoglobin levels less than 10 g/dL, the mean rates were 1.2 units (SD = 1.75) for momelotinib and 1.8 units (SD = 2.01) for ruxolitinib, with a rate ratio of 0.46 (95% CI, 0.30 to 0.70). For patients with hemoglobin levels less than 12 g/dL, the rates were 0.7 units for momelotinib and 1.3 units for ruxolitinib. In the SIMPLIFY-2 trial, the mean transfusion rate was 1.6 units (SD = 2.09) in the momelotinib group compared to 1.8 units (SD = 1.91) in the BAT group, with a transfusion rate ratio of 0.80 (95% CI, 0.49 to 1.31). Among the subpopulation with hemoglobin levels less than 10 g/dL, the mean rates were 2.1 units (SD = 2.12) for momelotinib and 2.0 units (SD = 1.97) for BAT, with a rate ratio of 1.10 (95% CI, 0.71 to 1.70). In the MOMENTUM trial, the total transfusions were 6.6 units (SD = 8.41) in the momelotinib group compared to 10.9 units (SD = 13.20) in the danazol group, with a treatment difference of –5.66 units (95% CI, –10.65 to –0.68 units). The results of the sensitivity analyses in the SIMPLIFY-1 and SIMPLIFY-2 trials using the per-protocol analysis set were in line with those of the ITT analysis. The results of the sensitivity analyses in the MOMENTUM trial were not available.
SRR at Week 24
In the SIMPLIFY-1 trial, 26.5% of patients in the momelotinib group experienced a splenic response at week 24, compared to 29.5% in the ruxolitinib group. Momelotinib achieved the definition of noninferiority, with a proportion difference for noninferiority of 0.09 (95% CI, 0.02 to 0.16; P = 0.014), while the proportion difference for superiority was –0.03 (95% CI, –0.12 to 0.05; P = 0.45). In the subpopulation with hemoglobin levels less than 10 g/dL, the response rates were 31.4% for momelotinib and 33.0% for ruxolitinib, with a treatment difference of –0.02 (95% CI, –0.16 to 0.13). For patients with hemoglobin levels less than 12 g/dL, the response rates were 28.9% for momelotinib and 29.3% for ruxolitinib. In the SIMPLIFY-2 trial, a splenic response was observed in 6.7% of patients in the momelotinib group versus 5.8% in the BAT group, with a proportion difference of 0.01 (95% CI, –0.09 to 0.10; P = 0.90). In the subpopulation with hemoglobin levels less than 10 g/dL, the response rates were 9.1% for momelotinib and 5.1% for BAT. The MOMENTUM study reported an SRR of 23.1% in the momelotinib group compared to 3.1% in the danazol group, with a proportion difference of 19.37% (95% CI, 10.96% to 27.77%; P = 0.001). The sensitivity analyses in the SIMPLIFY-1 and SIMPLIFY-2 trials using the per-protocol analysis set and the LOCF method were in line with those of the ITT analysis. The results of the sensitivity analyses in the MOMENTUM trial were not available.
TSS Response Rate at Week 24
In the SIMPLIFY-1 trial, 28.4% of patients in the momelotinib group experienced a TSS response at week 24, compared to 42.2% in the ruxolitinib group (using the MPN-SAF score). The proportion difference was –14.0% (95% CI, –23.0% to –5.0%; P = 0.9985). A noninferiority test that targeted 67% retention of ruxolitinib failed to the predefined noninferiority margin, where the lower bound of the 2-sided 95% CI should be greater than 0. Specifically, the adjusted proportion difference noninferiority testing was 0.00 (95% CI, –0.08 to 0.08; P = 0.98). Among the subpopulation with hemoglobin levels less than 10 g/dL, the response rates were 25% for momelotinib and 35.5% for ruxolitinib, with a treatment difference of –0.10 (95% CI, –0.25 to 0.04). For patients with hemoglobin levels less than 12 g/dL, response rates were 29.7% for momelotinib and 39.9% for ruxolitinib. In the SIMPLIFY-2 trial, TSS response was observed in 26.2% of patients in the momelotinib group versus 5.9% in the BAT group (using the MPN-SAF score), with a proportion difference of 0.20 (95% CI, 0.09 to 0.32; P < 0.001). In the subpopulation with hemoglobin levels less than 10 g/dL, the response rates were 32.3% for momelotinib and 2.6% for BAT. The MOMENTUM study reported a TSS response rate of 24.6% in the momelotinib group compared to 9.2% in the danazol group (using the MFSAF score), with a proportion difference of 15.67% (95% CI, 5.54% to 25.81%; P = 0.0095). The results of the sensitivity analyses in all 3 trials were in line with ITT analysis.
Table 16: Summary of Key Efficacy Results From Studies Included in the Systematic Review (ITT Analysis Set)
Variable | SIMPLIFY-1 | SIMPLIFY-2 | MOMENTUM | |||
|---|---|---|---|---|---|---|
MMB (N = 215) | RUX (N = 217) | MMB (N = 104) | BAT (N = 52) | MMB (N = 130) | DAN (N = 65) | |
Transfusion independence response rate at week 24 | ||||||
Patients experience response, n (%) | 143 (66.5) | 107 (49.3) | 45 (43.3) | 11 (21.2) | 40 (30.8) | 13 (20.0) |
Noninferiority test, adjusted proportion difference (95% CI)a | NR | NR | 14.77% (3.13% to 26.41%) | |||
P value, noninferiority | NR | NR | 0.0064 | |||
Proportion difference, superiority (95% CI) | 18.0% (9.0% to 26.0%) | 23.0% (9.0% to 37.0%) | 10.99% (–0.80% to 22.77%) | |||
P value | < 0.001b | 0.001b | 0.0861b | |||
Rate of RBC transfusion (units per month) at week 24 | ||||||
Mean (SD) | 0.5 (1.27) | 1.0 (1.62) | 1.6 (2.09) | 1.8 (1.91) | NR | NR |
Transfusion rate ratio (95% CI) | 0.28 (0.19 to 0.43) | 0.80 (0.49 to 1.31) | NR | |||
Treatment difference (95% CI) | NR | NR | NR | |||
P value | < 0.001b | 0.38b | NR | |||
Number of RBC or whole blood units | ||||||
Mean (SD) | NR | NR | 6.6 (8.41) | 10.9 (13.20) | ||
Treatment difference (95% CI) | NR | NR | –5.66 (–10.65 to –0.68) | |||
P value | NR | NR | 0.0260b | |||
Splenic response rate ≥ 35% at week 24 | ||||||
Patients experiencing response, n (%) | 57 (26.5) | 64 (29.5) | 7 (6.7) | 3 (5.8) | 30 (23.1) | 2 (3.1) |
Adjusted proportion difference, noninferiority (95% CI)c | 9.0% (2.0% to 16.0%) | NR | NR | NR | NR | |
P value, noninferiority | 0.014 | NR | NR | NR | NR | |
Proportion difference, superiority (95% CI) | –3.0% (–12.0% to 5.0%) | 1.0% (–9.0% to 10.0%) | 19.37% (10.96% to 27.77%) | |||
P value, superiority | 0.45b | 0.90 | 0.001 | |||
Total symptom score response rate at week 24 | ||||||
Patients experiencing response, n (%) | 60 (28.4) | 89 (42.2) | 27 (26.2) | 3 (5.9) | 32 (24.6) | 6 (9.2) |
Adjusted proportion difference, noninferiority (95% CI)d | 0.00 (–8.0% to 8.0%) | NR | NR | NR | NR | |
P value | 0.98 | NR | NR | NR | NR | |
Proportion difference, superiority (95% CI) | –14.0% (–23.0% to –5.0%) | 0.20 (0.09 to 0.32) | 15.67% (5.54% to 25.81%) | |||
P value | 0.9985b | < 0.001b | 0.0095 | |||
BAT = best available therapy; CI = confidence interval; DAN = danazol; ITT = intent to treat; MMB = momelotinib; NR = not reported; RBC = red blood cell; RUX = ruxolitinib; SD = standard deviation.
aDelta for noninferiority (defined as p[MMB] – 0.8 × p[DAN]) of 14.77% (95% CI, 3.13 to 26.41) ruling out 0% (i.e., noninferiority was to be declared if 80% of the response rate in the DAN group was preserved in the MMB group).
bP values lie outside of a statistical testing hierarchy and are thus not adjusted for multiple testing.
cDelta for noninferiority (defined as p[MMB] – 0.6 × p[RUX]) of 9.0% (95% CI, 2.0% to 16.0%) ruling out 0% (i.e., noninferiority was to be declared if 60% of the response rate in the RUX group was preserved in the MMB group).
dDelta for noninferiority (defined as p[MMB] – 0.67 × p[RUX]) of 0.0% (95% CI, –8.0% to 8.0%) including 0% (i.e., noninferiority was not achieved).
Sources: GSK Data on File, 2021 (SIMPLIFY-1 Clinical Study Report);10 GSK Data on File, 2021 (SIMPLIFY-2 Clinical Study Report);11 GSK Data on File, 2023 (MOMENTUM Clinical Study Report).12 Details included in the table are from the sponsor’s summary of clinical evidence.
Table 17: Summary of Key Efficacy Results in the SIMPLIFY-1 Trial in ITT Analysis Subsets — Hemoglobin < 10 g/dL and Hemoglobin < 12 g/dL
Variable | Hemoglobin < 10 g/dL | Hemoglobin < 12 g/dL | ||
|---|---|---|---|---|
MMB (n = 86) | RUX (n = 94) | MMB (n = 159) | RUX (n = 164) | |
Transfusion independence response rate at week 24 | ||||
Patients experiencing response, n (%) | 40 (46.5) | 25 (26.6) | 99 (62.3) | 61 (37.2) |
Treatment difference, superior (95% CI) | 0.20 (0.05 to 0.34) | 0.26 (0.16 to 0.36) | ||
Rate of RBC transfusion at week 24 | ||||
Mean (SD) | 1.2 (1.75) | 1.8 (2.01) | 0.7 (1.44) | 1.3 (1.75)a |
Rate ratio (95% CI) | 0.46 (0.30 to 0.70) | 0.31 (0.21 to 0.47) | ||
Splenic response rate of ≥ 35% at week 24 | ||||
Patients experiencing response, n (%) | 27 (31.4) | 31 (33.0) | 46 (28.9) | 48 (29.3) |
Treatment difference, noninferior (95% CI) | –0.02 (–0.16 to 0.13) | 0.11 (0.03 to 0.20) | ||
Total symptom score at week 24 | ||||
Patients experiencing response, n (%) | 21 (25) | 33 (35.5) | 46 (29.7) | 63 (39.9) |
Treatment difference, noninferior (95% CI) | –0.10 (–0.25 to 0.04) | 0.03 (–0.06 to 0.12) | ||
CI = confidence interval; ITT = intention to treat; MMB = momelotinib; RBC = red blood cell; RUX = ruxolitinib; SD = standard deviation.
aN = 163.
Sources: GSK Data on File, 2021 (SIMPLIFY-1 Clinical Study Report).10 Details included in the table are from the sponsor’s summary of clinical evidence.
Table 18: Summary of Key Efficacy Results in the SIMPLIFY-2 Trial in ITT Analysis Subsets — Hemoglobin < 10 g/dL and Hemoglobin < 12 g/dL
Variable | Hemoglobin < 10 g/dL | Hemoglobin < 12 g/dL | ||
|---|---|---|---|---|
MMB (n = 66) | BAT (n = 39) | MMB (n = 66) | BAT (n = 39) | |
Transfusion independence response rate at week 24 | ||||
Patients experiencing response, n (%) | 22 (33.3) | 5 (12.8) | 38 (40.9) | 7 (15.2) |
Treatment difference, superior (95% CI) | 0.21 (0.01 to 0.39) | 0.26 (0.08 to 0.42) | ||
Rate of RBC transfusion at week 24 | ||||
Mean (SD) | 2.1 (2.12) | 2.0 (1.97) | 1.7 (2.13) | 1.9 (1.93) |
Rate ratio (95% CI) | 1.10 (0.71 to 1.70) | 0.84 (0.52 to 1.35) | ||
Splenic response rate of ≥ 35% at week 24 | ||||
Patients experiencing response, n (%) | 6 (9.1) | 2 (5.1) | 7 (7.5) | 2 (4.3) |
Treatment difference, noninferior (95% CI) | 0.04 (–0.16 to 0.23) | 0.03 (–0.14 to 0.21) | ||
Total symptom score at week 24 | ||||
Patients experiencing response, n (%) | 21 (32.3) | 1 (2.6) | 26 (28.3) | 2 (4.4) |
Treatment difference, noninferior (95% CI) | 0.30 (0.10 to 0.48) | 0.24 (0.06 to 0.41) | ||
BAT = best available therapy; CI = confidence interval; ITT = intention to treat; MMB = momelotinib; RBC = red blood cell; SD = standard deviation.
Sources: GSK Data on File, 2021 (SIMPLIFY-2 Clinical Study Report).11 Details included in the table are from the sponsor’s summary of clinical evidence.
Harms
Refer to Table 19 for harms data.10-12
Adverse Events
The majority of patients experienced at least 1 AE across all studies. Thrombocytopenia was a commonly reported AE across all studies, with higher incidence in the ruxolitinib group of the SIMPLIFY-1 trial (29.2%) and the momelotinib group of the MOMENTUM trial (22.3%) than in the momelotinib groups in the SIMPLIFY-1 trial (18.7%) and the SIMPLIFY-2 trial (10.6%). Anemia was frequently observed, particularly in the ruxolitinib group of the SIMPLIFY-1 trial, where it occurred in 37.5% of patients. In comparison, anemia was reported in 14.5% of patients in the momelotinib group of the SIMPLIFY-1 trial and in 13.5% of the momelotinib group in the SIMPLIFY-2 trial (compared with 17.3% in the BAT group). Diarrhea was notably common in the momelotinib groups, with 32.7% in the SIMPLIFY-2 trial and 22.3% in the MOMENTUM trial; it was less frequent in the comparator groups (15.4% in the BAT group in the SIMPLIFY- 2 trial and 9.2% in the danazol group in the MOMENTUM trial). Asthenia (weakness) was observed in the SIMPLIFY-2 trial, occurring in 19.2% of patients in the momelotinib group patients and 21.2% of patients in the BAT group.
Serious AEs
In the SIMPLIFY-1 trial, 74 patients (34.6%) in the momelotinib group and 94 patients (43.5%) in the ruxolitinib group experienced at least 1 grade 3 or 4 AE. In the SIMPLIFY-2 trial, 57 patients (54.8%) in the momelotinib group and 22 patients (42.3%) in the BAT group experienced such AEs. In the MOMENTUM study, 63 patients (48.5%) in the momelotinib group and 41 patients (63.1%) in the danazol group experienced at least 1 grade 3 or 4 AE.
The most common grade 3 or 4 AEs across the studies were thrombocytopenia and anemia. Thrombocytopenia was reported in 15 patients (7.0%) in the momelotinib group of the SIMPLIFY-1 trial, 11 patients (10.6%) in the momelotinib group of the SIMPLIFY-2 trial, and 22 patients (16.9%) in the momelotinib group of the MOMENTUM trial. It was less frequent in the comparator groups but still notable, affecting 10 patients (4.6%) in the ruxolitinib group of the SIMPLIFY-1 trial, 3 patients (5.8%) in the BAT group of the SIMPLIFY-2 trial, and 5 patients (7.7%) in the danazol group of the MOMENTUM trial. Anemia was another frequent grade 3 or 4 AE, occurring in 13 patients (6.1%) in the momelotinib group of the SIMPLIFY-1 trial, 14 patients (13.5%) in the momelotinib group of the SIMPLIFY-2 trial, and 10 patients (7.7%) in the momelotinib group of the MOMENTUM trial, with the highest incidence observed in the ruxolitinib group of the SIMPLIFY-1 trial: 49 patients (22.7%). Grade 3 to 4 anemia occurred among 17.3% of patients in the BAT group of the SIMPLIFY-2 trial and 6.2% of patients in the danazol group of the MOMENTUM trial.
At least 1 SAE was reported in 49 patients (22.9%) in the momelotinib group and 39 patients (18.1%) in the ruxolitinib group in the SIMPLIFY-1 trial. In the SIMPLIFY-2 trial, 37 patients (35.6%) in the momelotinib group and 12 patients (23.1%) in the BAT group experienced at least 1 SAE. The MOMENTUM study reported that 45 patients (34.6%) in the momelotinib group and 26 patients (40.0%) in the danazol group experienced at least 1 SAE.
Common SAEs included anemia, which was reported in 4 patients (1.9%) in the momelotinib group of the SIMPLIFY-1 trial, 4 patients (3.8%) in the momelotinib group of the SIMPLIFY-2 trial, and 5 patients (3.8%) in the momelotinib group of the MOMENTUM trial. In comparison, 8 patients (3.7%) in the ruxolitinib group of the SIMPLIFY-1 trial and 3 patients (4.6%) in the danazol group of the MOMENTUM trial experienced anemia. No cases of anemia were reported as SAEs in the BAT group in the SIMPLIFY-2 trial. Pneumonia was another significant SAE, occurring in 4 patients (1.9%) in the momelotinib group of the SIMPLIFY-1 trial and 6 patients (9.2%) in the danazol group of the MOMENTUM trial. In contrast, it was reported in 3 patients (1.4%) in the ruxolitinib group of the SIMPLIFY-1 trial, while no cases were reported in the SIMPLIFY-2 trial. Additionally, sepsis was reported as an SAE in 3 patients (2.9%) in the momelotinib group of the SIMPLIFY-2 trial, 2 (1.5%) in the momelotinib group of MOMENTUM, and 3 patients (4.6%) in the danazol group of the MOMENTUM trial. There were no reported cases of sepsis as a SAE in the momelotinib group of the SIMPLIFY-1 trial, the ruxolitinib group of the SIMPLIFY-1 trial, or the BAT group of the SIMPLIFY-2 trial.
Withdrawals due to AEs
Across the 3 studies, a number of patients discontinued treatment due to AEs. In the SIMPLIFY-1 trial, 27 patients (12.6%) in the momelotinib group and 12 patients (5.6%) in the ruxolitinib group discontinued treatment due to AEs. In the SIMPLIFY-2 trial, 22 patients (21.2%) in the momelotinib group and 1 patient (1.9%) in the BAT group discontinued treatment due to AEs. In the MOMENTUM study, 23 patients (17.7%) in the momelotinib group and 15 patients (23.1%) in the danazol group discontinued treatment due to AEs.
Among the specific AEs leading to treatment discontinuation, thrombocytopenia was a common cause, reported in 3 patients (1.4%) in the momelotinib group of the SIMPLIFY-1 trial, 5 patients (4.8%) in the momelotinib group of the SIMPLIFY-2 trial, and 1 patient (0.8%) in the momelotinib group of the MOMENTUM trial. In the comparator groups, thrombocytopenia led to discontinuation in 4 patients (1.9%) in the ruxolitinib group of the SIMPLIFY-1 trial, with no cases reported in the BAT group of the SIMPLIFY-2 trial, and 2 cases (3.1%) in the danazol group of the MOMENTUM trial.
Thrombocytopenia was the most common AE leading to treatment interruption or dose reduction, affecting 13 patients (6.1%) in the momelotinib group of the SIMPLIFY-1 trial, 5 patients (4.8%) in the momelotinib group of the SIMPLIFY-2 trial, and 13 patients (10.0%) in the momelotinib group of the MOMENTUM trial. In the comparator groups, thrombocytopenia led to these events in 53 patients (24.5%) in the ruxolitinib group of the SIMPLIFY-1 trial, 3 patients (5.8%) in the BAT group of the SIMPLIFY-2 trial, and 2 patients (3.1%) in the danazol group of the MOMENTUM trial. Other notable AEs leading to treatment interruption or dose reduction included anemia, experienced by 3 patients (1.4%) in the momelotinib group and 12 patients (5.6%) in the ruxolitinib group of the SIMPLIFY-1 trial, as well as diarrhea, experienced by 3 patients (1.4%) in the momelotinib group and 4 patients (1.9%) in the ruxolitinib group of the SIMPLIFY-1 trial, and 3 patients (2.3%) in the momelotinib group of the MOMENTUM trial; no cases were reported in the danazol group of the MOMENTUM trial.
Mortality
In the SIMPLIFY-1 trial, 8 patients (3.7%) in the momelotinib group and 6 patients (2.8%) in the ruxolitinib group died. In the SIMPLIFY-2 trial, there were 8 deaths (7.7% of patients) in the momelotinib group and 5 deaths (9.6% of patients) in the BAT group. The MOMENTUM study reported a higher number of deaths: 38 patients (29.2%) in the momelotinib group and 20 patients (30.8%) in the danazol group.
The causes of death varied across the studies. In the SIMPLIFY-1 trial, 7 deaths (3.3% of patients) in the momelotinib group and 6 deaths (2.8% of patients) in the ruxolitinib group were attributed to TEAEs not related to disease progression, while 1 death (0.5% of patients) in the momelotinib group was due to disease progression. In the SIMPLIFY-2 trial, 5 deaths (4.8% of patients) in the momelotinib group and 3 deaths (5.8% of patients) in the BAT group were due to TEAEs not related to disease progression. Additionally, 2 deaths in both the momelotinib group (1.9% of patients) and BAT group (3.8% of patients) were related to disease progression.
In the MOMENTUM study, 16 deaths (12.3% of patients) in the momelotinib group and 11 deaths (16.9% of patients) in the danazol group were due to TEAEs not related to disease progression. Notably, 1 patient (0.8%) in the momelotinib group died due to progression to AML.
Notable Harms
Peripheral Neuropathy
In the SIMPLIFY-1 trial, 22 patients (10.3%) in the momelotinib group and 12 patients (5.6%) in the ruxolitinib group experienced peripheral neuropathy. In the SIMPLIFY-2 trial, this event was reported in 12 patients (11.5%) in the momelotinib group, with no cases reported in the BAT group. In the MOMENTUM study, peripheral neuropathy occurred in 5 patients (3.8%) in the momelotinib group and 1 patient (1.5%) in the danazol group.
Infections
In the MOMENTUM study, 44 patients (33.8%) in the momelotinib group and 23 patients (35.4%) in the danazol group experienced infections. A matching category was not included in the SIMPLIFY-1 trial or the SIMPLIFY-2 trial.
Nonhematological AEs
In the SIMPLIFY-1 trial, nonhematological AEs occurred in 193 patients (90.2%) in the momelotinib group and 200 patients (92.6%) in the ruxolitinib group. In the SIMPLIFY-2 trial, they occurred in 100 patients (96.2%) in the momelotinib group and 45 patients (86.5%) in the BAT group. A matching category was not included in the MOMENTUM trial.
Hemorrhage
In the MOMENTUM study, 28 patients (21.5%) in the momelotinib group and 12 patients (18.5%) in the danazol group experienced hemorrhage. A matching category was not included in the SIMPLIFY-1 trial or the SIMPLIFY-2 trial.
Malignancies
In the MOMENTUM study, 7 patients (5.4%) in the momelotinib group and 6 patients (9.2%) in the danazol group developed malignancies. A matching category was not included in the SIMPLIFY-1 trial or the SIMPLIFY-2 trial.
Thromboembolism
In the MOMENTUM study, 5 patients (3.8%) in the momelotinib group and 6 patients (9.2%) in the danazol group experienced thromboembolism. A matching category was not included in the SIMPLIFY-1 trial or the SIMPLIFY-2 trial.
AML or Transformation to AML
In the MOMENTUM trial, AML or transformation to AML occurred in 4 patients (3.1%) in the momelotinib group and 3 patients (4.6%) in the danazol group. A matching category was not included in the SIMPLIFY-1 trial or the SIMPLIFY-2 trial.
Table 19: Summary of Harms Results From Studies Included in the Systematic Review
Adverse events | SIMPLIFY-1 | SIMPLIFY-2 | MOMENTUM | |||
|---|---|---|---|---|---|---|
MMB (N = 214) | RUX (N = 216) | MMB (N = 104) | BAT (N = 52) | MMB (N = 130) | DAN (N = 65) | |
Most common AEs (≥ 20%), n (%) (any grade) | ||||||
≥ 1 AE | 198 (92.5) | 206 (95.4) | 101 (97.1) | 46 (88.5) | 122 (93.8) | 62 (95.4) |
Thrombocytopenia | 40 (18.7) | 63 (29.2) | 11 (10.6) | 3 (5.8) | 29 (22.3) | 7 (10.8) |
Anemia | 31 (14.5) | 81 (37.5) | 14 (13.5) | 9 (17.3) | 10 (7.7) | 4 (6.2) |
Diarrhea | NR | NR | 34 (32.7) | 8 (15.4) | 29 (22.3) | 6 (9.2) |
Asthenia | NR | NR | 20 (19.2) | 11 (21.2) | NR | NR |
Most common grade 3 or 4 AEs (≥ 5%), n (%) | ||||||
Any grade 3 or 4 AE | 74 (34.6) | 94 (43.5) | 57 (54.8) | 22 (42.3) | 63 (48.5) | 41 (63.1) |
Thrombocytopenia | 15 (7.0) | 10 (4.6) | 11 (10.6) | 3 (5.8) | 22 (16.9) | 5 (7.7) |
Anemia | 13 (6.1) | 49 (22.7) | 14 (13.5) | 9 (17.3) | 10 (7.7) | 4 (6.2) |
Neutropenia | 1 (0.5) | 3 (1.4) | 5 (4.8) | 1 (1.9) | 3 (2.3) | 1 (1.5) |
Platelet count decreased | NR | NR | NR | NR | 7 (5.4) | 3 (4.6) |
Acute kidney injury | NR | NR | NR | NR | 4 (3.1) | 6 (9.2) |
Pneumonia | NR | NR | NR | NR | 2 (1.5) | 6 (9.2) |
Serious AEs, n (%) | ||||||
Patients with ≥ 1 serious AE | 49 (22.9) | 39 (18.1) | 37 (35.6) | 12 (23.1) | 45 (34.6) | 26 (40.0) |
Anemia | 4 (1.9) | 8 (3.7) | 4 (3.8) | 0 | 5 (3.8) | 3 (4.6) |
Pneumonia | 4 (1.9) | 3 (1.4) | NR | NR | 3 (2.3) | 6 (9.2) |
Atrial fibrillation | 4 (1.9) | 1 (0.5) | NR | NR | NR | NR |
Sepsis | NR | NR | 3 (2.9) | 0 | 2 (1.5) | 3 (4.6) |
Diarrhea | 4 (1.9) | 1 (0.5) | NR | NR | NR | NR |
Pyrexia | 2 (0.9) | 3 (1.4) | NR | NR | 3 (2.3) | 0 |
Thrombocytopenia | NR | 3 (1.4) | 5 (4.8) | 0 | 3 (2.3) | 0 |
COVID-19 pneumonia | NR | NR | NR | NR | 3 (2.3) | 0 |
Transformation to AML | NR | NR | NR | NR | 2 (1.5) | 2 (3.1) |
General physical health deterioration | NR | NR | 2 (1.9) | 2 (3.8) | 2 (1.5) | 2 (3.1) |
Splenic infarction | NR | NR | NR | NR | 1 (0.8) | 2 (3.1) |
Patients (≥ 1%) who discontinued treatment due to AEs, n (%) | ||||||
Patients who discontinued treatment due to AEs | 27 (12.6) | 12 (5.6) | 22 (21.2) | 1 (1.9) | 23 (17.7) | 15 (23.1) |
Thrombocytopenia | 3 (1.4) | 4 (1.9) | 5 (4.8) | 0 | 1 (0.8) | 2 (3.1) |
Anemia | NR | NR | NR | NR | 1 (0.8) | 2 (3.1) |
Transformation to AML | NR | NR | NR | NR | 1 (0.8) | 2 (3.1) |
Patients (≥ 2%) with AEs leading to treatment interruption and/or dose reduction, n (%) | ||||||
Total all-cause AEs leading to treatment interruption and/or dose reduction | 39 (18.2) | 79 (36.6) | 17 (16.3) | 10 (19.2) | 44 (33.8) | 19 (29.2) |
Thrombocytopenia | 13 (6.1) | 53 (24.5) | 5 (4.8) | 3 (5.8) | 13 (10.0) | 2 (3.1) |
Alanine aminotransferase increased | 4 (1.9) | 0 | NR | NR | NR | NR |
Anemia | 3 (1.4) | 12 (5.6) | NR | NR | NR | NR |
Hypotension | 3 (1.4) | 0 | NR | NR | NR | NR |
Abdominal pain | 3 (1.4) | 0 | NR | NR | NR | NR |
Diarrhea | 3 (1.4) | 4 (1.9) | NR | NR | 3 (2.3) | 0 |
Neutropenia | 1 (0.5) | 3 (1.4) | NR | NR | 3 (2.3) | 1 (1.5) |
Platelet count decreased | NR | NR | NR | NR | 5 (3.8) | 0 |
Blood creatinine increased | NR | NR | NR | NR | 1 (0.8) | 2 (3.1) |
Acute kidney injury | NR | NR | NR | NR | 0 | 3 (4.6) |
Deaths, n (%) | ||||||
Patients who died | 8 (3.7) | 6 (2.8) | 8 (7.7) | 5 (9.6) | 38 (29.2) | 20 (30.8) |
Due to TEAE not related to disease progressiona | 7 (3.3) | 6 (2.8) | 5 (4.8) | 3 (5.8) | 16 (12.3) | 11 (16.9) |
Due to disease progression | 1 (0.5) | 0 | 2 (1.9) | 2 (3.8) | 1 (0.8) | NR |
Other reasonb | NR | NR | 1 (1.0) | 0 | NR | NR |
AEs of special interest, n (%) | ||||||
Peripheral neuropathy | 22 (10.3) | 12 (5.6) | 12 (11.5) | 0 | 5 (3.8) | 1 (1.5) |
Nonhematological AEs | 193 (90.2) | 200 (92.6) | 100 (96.2) | 45 (86.5) | NR | NR |
Cataract | 7 (3.3) | 5 (2.3) | 4 (3.8) | 0 | NR | NR |
First dose effect | 14 (6.5) | 2 (0.9) | 4 (3.8) | 0 | NR | NR |
Infections | NR | NR | NR | NR | 44 (33.8) | 23 (35.4) |
Hemorrhage | NR | NR | NR | NR | 28 (21.5) | 12 (18.5) |
Malignancies | NR | NR | NR | NR | 7 (5.4) | 6 (9.2) |
Thromboembolism | NR | NR | NR | NR | 5 (3.8) | 6 (9.2) |
AML or transformation to AML | NR | NR | NR | NR | 4 (3.1) | 3 (4.6) |
AE = adverse event; AML = acute myeloid leukemia; BAT = best available therapy; DAN = danazol; MMB = momelotinib; NR = not reported; RUX = ruxolitinib; TEAE = treatment-emergent adverse event.
Notes: AEs mapped according to Medical Dictionary for Regulatory Activities version 22.0. Severity grades were defined by Common Terminology Criteria for Adverse Events version 4.03. Multiple AEs were counted only once per patient.
aThe term disease progression included the following: disease progression, myelofibrosis, and AML.
bIncluded disease progression more than 30 days after the last dose of the study drug, unknown, and other reported causes of death.
Sources: GSK Data on File, 2021 (SIMPLIFY-1 Clinical Study Report);10 GSK Data on File, 2021 (SIMPLIFY-2 Clinical Study Report);11 GSK Data on File, 2023 (MOMENTUM Clinical Study Report).12 Details included in the table are from the sponsor’s summary of clinical evidence.
Critical Appraisal
Internal Validity
All studies are randomized with an active control arm. Two of the studies — the SIMPLIFY-1 and MOMENTUM trials — are double masked (or double blind); the SIMPLIFY-2 trial was an open-label study. The randomization and allocation concealment procedures (where applicable) were described in detail and likely to have been effective in balancing out baseline characteristics (with the exception of some imbalance in ECOG PS across trials and anemia in the MOMENTUM trial). Two of the studies aimed to assess noninferiority as a primary outcome (SIMPLIFY-1 and MOMENTUM trials). The lower margin to achieve noninferiority (0 at the lower end of the 95% CI) was considered acceptable by the clinical experts consulted by CDA-AMC. However, despite using previous evidence to gauge the power needed to achieve noninferiority, the clinical rationale of preserving 60% of the ruxolitinib effect is not explicitly clear. The studies provided a comprehensive and appropriate approach to assessing various efficacy outcomes with a predefined statistical hierarchy to control type I error. Data imputation methods were implemented, with an overall approach of imputing missing data as a ‘nonresponse’ and several sensitivity analyses were implemented to assess the robustness of data imputation. All results from the subgroup analyses have considerable limitations due to the potential lack of power and type I error control.
Primary limitations to the internal validity of the included studies lie in the open-label design of the SIMPLIFY-2 trial, large and imbalanced treatment discontinuation, and lack of control for type I error (i.e., no adjustment for multiple testing). Furthermore, the lack of a washout period may prove a potential confounder.
In the SIMPLIFY-1 trial, more patients discontinued momelotinib (27; 12.6%) than ruxolitinib (9; 4.1%). Within the SIMPLIFY-1 trial, the primary analysis of the rate of transfusion independence at week 24 does not include an approach to handle missing data. The sponsor conducted a sensitivity analysis where missing data were classified as nonresponders with a result similar to the ITT analysis. The outcomes of splenic response and TSS response used a conservative approach of imputing missing data as nonresponders. However, additional sensitivity analysis using other methods (e.g., LOCF) suggest similar results to the ITT analysis, despite the assumption of LOCF being unlikely true. As such, the effect of the disproportional treatment discontinuation on the internal validity of the SIMPLIFY-1 trial remains unclear but is unlikely to be a major limitation. However, a larger limitation affecting the reported outcomes is the lack of adjustment for type I error (multiple testing) for the outcomes of transfusion independence, SRR, and TSS. This is due to the failure of the testing hierarchy at the point of the secondary outcome of noninferiority of momelotinib versus ruxolitinib in TSS response rate. As such, there is an increased risk that statistically significant results among these end points (i.e., transfusion independence response rate) are false-positives. Thus, in the context of causal inference, results for these end points should only be considered as supportive evidence.
The SIMPLIFY-2 trial was of an open-label design which would increase the risk of bias, potentially in favour of momelotinib for subjective outcomes. This risk of bias is likely to apply mostly to the TSS response rate as it is a patient-reported outcome covering symptoms related to the disease. There was a large, but similar, proportion of treatment discontinuation in both treatment groups. This may affect both internal and external validity, where the comparative end point could have looked different had the missing data existed. Finally, the SIMPLIFY-2 trial failed to achieve its primary objective (i.e., the superiority of momelotinib versus BAT for SRR), rendering all comparative testing nominal in nature and not adjusted for type I error. As such, there is an increased risk that statistically significant results for subsequently tested end points (i.e., transfusion independence response rate and TSS response rate) are false-positives. Thus, in the context of causal inference, results for these end points should only be considered as supportive evidence. Adding a limitation to the SIMPLIFY-2 trial is the lack of a washout period at screening combined with a relatively large proportion of patients who were on ruxolitinib or fedratinib within 12 weeks of randomization. Specifically, 15.4% of patients in the momelotinib treatment group were on a JAK inhibitor within 12 weeks of randomization as opposed to 19.2% in the BAT treatment group. The implications are that the observed results may not have been solely driven by the intervention or control and may have been influenced, to an unknown extent, by the prior JAK inhibitor therapy.
The MOMENTUM trial had a large proportion of treatment discontinuation in both treatment groups as well as a large imbalance of treatment discontinuation between the 2 groups (27.7% in momelotinib and 41.5% in danazol). For the primary efficacy end point of TSS, missing data were imputed as patients not experiencing a response. This could possibly bias the result in favour of momelotinib. The sponsor conducted a number of sensitivity analyses (no imputation, no stratification, LOCF, and controlled-based multiple imputation). The control-based multiple imputation delivered a P value of 0.062, while all other sensitivity analyses supported the primary ITT analysis with a similar direction of effect. While these analyses may increase the overall robustness of the primary analysis results, the large number of missing data and the imbalance between treatment groups is still a major limitation with an unknown directional effect on the results. This limitation applies to the rest of the efficacy end points presented here, including transfusion independence response rate, rate of RBC transfusion, and SRR. Within the reported efficacy end points of the MOMENTUM trial, the transfusion independence response rate superiority testing and the rate of RBC transfusion are outside of the statistical hierarchy and are thus not adjusted for type I error (multiple testing). As such, there is an increased risk that the result for the rate of RBC transfusion is a false-positive; in the context of causal inference, this end point should only be interpreted as supportive evidence. The superiority test for transfusion independence response rate yielded a result that was not statistically significant.
External Validity
The 3 studies reviewed in this submission attempted to capture various representations of the patient population with MF. The SIMPLIFY-1 trial enrolled patients who had not previously been treated with a JAK inhibitor, regardless of anemia status; the SIMPLIFY-2 trial enrolled patients who had previously been treated with a JAK inhibitor, regardless of anemia status; and the MOMENTUM trial enrolled patients who had previously been treated with a JAK inhibitor and who had anemia. All the studies included patients who were considered at high risk or intermediate risk. According to the clinical experts consulted by CDA-AMC, more than one-half the patients seen in their clinics would be eligible to participate in 1 of these trials. This is further reinforced by the clinical expert opinion that the baseline characteristics of the patients in these studies are generally representative of the majority of the patient population in Canada.
Overall, limitations to external validity in the studies include lack of comparison against relevant comparators, a large number of treatment discontinuations, the short duration of assessment for survival and progression outcomes, and the lack of established minimal important differences for the outcomes of interest.
The momelotinib intervention is in accordance with the Health Canada indication. The comparator in the SIMPLIFY-1 trial (ruxolitinib) is an appropriate representation of clinical practice in Canada. According to the clinical experts, currently patients who experience intolerance or anemia can be given the option of switching to fedratinib. However, clinical practice in Canada tends to attempt to maintain patients on ruxolitinib for as long as possible, even in cases where there is disease progression. In the SIMPLIFY-2 trial, the comparison was BAT, where the majority of patients (89.7%) ended up receiving ruxolitinib but none received fedratinib. Due to the large proportion of patients receiving ruxolitinib in the SIMPLIFY-2 trial, it is likely that the results observed would mostly apply to the comparative efficacy and safety of momelotinib versus ruxolitinib but not versus other modalities of treatment. The use of danazol in the MOMENTUM trial does not reflect Canadian clinical practice. According to the clinical experts consulted by CDA-AMC, patients who experience anemia are usually treated with hydroxyurea with the possible add-on of ESAs, and danazol is uncommonly used. Both hydroxyurea and ESAs were prohibited in the MOMENTUM trial.
All the included studies exhibited either a notable imbalance in treatment discontinuation between the treatment groups, a high proportion of treatment discontinuation, or both. This reduces the overall generalizability of the results to only those who are likely to finish the trial (commonly the ones who are responding well to treatment and can tolerate it). This is especially true in the SIMPLIFY-2 and MOMENTUM trials.
A limitation of the 24-week study design is the lack of information on clinical outcomes related to survival and disease progression, as 24 weeks is considered too short to be informative and patients cross over to momelotinib after 24 weeks. According to the clinical experts, a time point of 2 years for a comparative clinical end point would provide clinically meaningful results.
The lack of established minimal important difference of the outcomes of interests reduces the ability to better understand the clinical meaningfulness of the difference between the intervention and the comparators. The clinical experts consulted on this review suggested that the definition of “patients experiencing a response” across the outcomes of interest is clinically meaningful. However, they were unable to determine a magnitude of difference in the rate of patients experiencing a response that would be clinically meaningful in determining which treatment is better. Moreover, there was no clear clinical justification behind the thresholds of the definitions of “patients experiencing a response.”
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For the pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for the outcomes considered most relevant to inform the expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group:62,63
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate — The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited — The true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate — The true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as “very uncertain.”
Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null. In the case of the outcomes presented in this report, the target of certainty was assessed based on the location of the point estimate relative to the null.
For the GRADE assessments, the SIMPLIFY-1, SIMPLIFY-2, and MOMENTUM trials were assessed individually because of the differences in the patient population and comparators included in these trials.
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for momelotinib versus ruxolitinib. Table 3 presents the GRADE summary of findings for momelotinib versus BAT. Table 4 presents the GRADE summary of findings for momelotinib versus danazol.
Long-Term Extension Studies
The contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the CDA-AMC review team.
Description of Studies
SIMPLIFY-1 Trial
The open-label extension of the SIMPLIFY-1 trial evaluated open-label treatment with momelotinib for up to 216 weeks (i.e., through week 240) after the randomized, double-blind phase.10
SIMPLIFY-2 Trial
The open-label extension of the SIMPLIFY-2 trial evaluated open-label treatment with momelotinib for up to 204 weeks (i.e., through week 228) after the randomized treatment phase.11
For the open-label extension of both the SIMPLIFY-1 and SIMPLIFY-2 trials, a posttreatment follow-up visit was scheduled 12 weeks after the discontinuation of the study treatment and patients were followed up with approximately every 6 months for 5 years or until study termination for the long-term assessment for survival. The 12-week and survival follow-up visits were not required for patients who continued treatment with momelotinib following the end of the study.10,11
MOMENTUM Trial
The open-label extension of the MOMENTUM trial evaluated open-label treatment with momelotinib for up to 180 weeks (i.e., through week 204) and danazol for up to 24 weeks after the randomized, double-blind phase of the MOMENTUM trial.12
A safety follow-up visit was scheduled for 30 days after the last dose of the study drug, and long-term follow-up assessments were made for survival and leukemic transformation 7 years after the first dose.12 Patients who completed at least 24 weeks of open-label treatment with momelotinib had the option to continue treatment in the momelotinib extension study, and those who completed the safety follow-up visit in the MOMENTUM trial were transitioned to the open-label extension study.12
Populations
Patients who completed the 24-week randomized treatment phase in the SIMPLIFY-1 and SIMPLIFY-2 trials had the option to participate in the open-label extension phases.10,11
Patients who completed the 24-week randomized treatment phase in the MOMENTUM trial but discontinued treatment early due to splenic progression or discontinued treatment early for other reasons but completed scheduled assessments through week 24 had the option to continue momelotinib in the open-label extension phase. Patients treated with momelotinib who completed treatment through the end of week 24 had the option to continue momelotinib as open-label treatment for up to 180 weeks (i.e., through week 204) for a total of approximately 4 years.12 Patients treated with danazol had the option to cross over to open-label momelotinib treatment in the following situations: if they had completed the 24-week randomized treatment period; if they had discontinued treatment with danazol but continued study assessments through week 24 without receiving any prohibited medications; or if they had met the criteria for confirmed splenic progression before the end of week 24.12 Patients treated with danazol who continued to receive clinical benefit at the end of week 24 of the double-blind randomized treatment phase had the option to continue danazol as open-label treatment through week 48.12
Interventions
In the open-label extension phase in the SIMPLIFY-1 and SIMPLIFY-2 trials, all patients received momelotinib 200 mg once daily.10,11
In the open-label extension phase of the MOMENTUM trial, patients received momelotinib at a dosage of 200 mg orally once daily, unless the dose of momelotinib was reduced in the randomized treatment phase due to an AE.12 The dosage of momelotinib was eventually increased to 200 mg once daily after the resolution of events leading to dose reduction at the investigator’s discretion. Patients were followed up with for at least a 4-hour postdose observation period after the first dose.12
For those patients who received open-label danazol treatment, danazol was administered orally at a total daily dose of 400 mg.12 Patients who received a reduced dose of danazol during the randomized treatment phase continued to receive the same dose, which was then progressively reduced to the minimum dose to maintain a response.12
Outcomes
For the open-label extension phases, only outcomes relevant to the review were reported. This included splenic response at any time for the SIMPLIFY-1 and SIMPLIFY-2 trials and SRR (≥ 35% reduction from baseline spleen volume at week 24) and transfusion independence response rate at week 48 for the MOMENTUM trial. Rate of RBC transfusion was not reported for the open-label extension phases of the SIMPLIFY-1, SIMPLIFY-2, or MOMENTUM trials.10-12 For the MOMENTUM trial, the TSS response rate was reported during the open-label extension phase at week 48.12 The TSS response rate was not reported for the open-label extension phases of the SIMPLIFY-1 and SIMPLIFY-2 trials.10,11 For the SIMPLIFY-1, SIMPLIFY-2, and MOMENTUM trials, the safety outcomes — including AEs, SAEs, treatment-related discontinuations, deaths, and AESIs — were reported for the safety population for the open-label extension phases.10-12
Statistical Analysis
In the SIMPLIFY-1 and SIMPLIFY-2 trials, baseline assessments were carried out as part of the randomized phase of the pivotal trials. The baseline assessment for patients continuing momelotinib (momelotinib to momelotinib) was done before or on the date of randomization in both the SIMPLIFY-1 and SIMPLIFY-2 trials. For those patients who switched to momelotinib (ruxolitinib to momelotinib in the SIMPLIFY-1 trial or BAT to momelotinib in the SIMPLIFY-2 trial), the baseline assessment was done before or on the date of the first dose of momelotinib in the open-label extended treatment phase.35
In the SIMPLIFY-1 and SIMPLIFY-2 trials, the open-label analysis set included patients who had received at least 1 dose of momelotinib in the open-label extension phase. Patients were grouped based on their treatment group assignment in the pivotal treatment phase.35
For both the SIMPLIFY-1 and SIMPLIFY-2 trials, data pertaining to the open-label extension phases were collected after the date of the first open-label dose of momelotinib, with the exception that MRI or CT spleen volume or IWG-MRT or European Leukemia Net overall response rate assessments collected within 10 days of the first extended open-label dose of momelotinib were included in the analysis of the randomized phase due to the challenge of scheduling MRI and CT scans. It was anticipated that an additional 10 days of treatment with momelotinib would not have a significant impact on the spleen volume.35 Data on concomitant medications in the open-label extension phase were collected with a start date of the date of the first extended open-label dose of momelotinib.35
In the SIMPLIFY-1 and SIMPLIFY-2 trials, exposure to momelotinib was presented for the duration of the extended treatment phase for patients who switched to momelotinib from ruxolitinib or BAT, and the combined duration of the randomized and extended treatment phases was presented for patients who were continuing momelotinib treatment.35
In the SIMPLIFY-1 and SIMPLIFY-2 trials, AEs with an onset date of the day of the first extended open-label dose of momelotinib and reported as occurring in the extended open-label phase or with no information on which phase the event occurred in were classified as occurring in the open-label extension phase.35
In the open-label extension phase of the MOMENTUM trial, all patients who received momelotinib were grouped based on their treatment group assignment in the double-blind randomized treatment phase into continuing (momelotinib to momelotinib), continuing (danazol to danazol), and switch (danazol to momelotinib).35 Data pertaining to the open-label, long-term extension phase were collected following the completion of the randomized week 24 treatment or following the crossover visit for early crossover in cases of confirmed splenic progression.35
During the open-label extension phase of the MOMENTUM study, averages for any TSS data available were calculated. For the calculation of mean TSS during the open-label phase, TSS was only considered missing if all the 7 daily TSS results were missing.35 All statistical tests were 2-sided and performed at the 5% significance level. No control for multiple testing was reported by the sponsors for the open-label extension phase.35 AEs were described using descriptive statistics by the treatment group from the randomized period; data for patients continuing with danazol treatment up to week 48 and for patients switching from danazol to momelotinib were reported separately.35
Results
Patient Disposition
In the SIMPLIFY-1, SIMPLIFY-2 and MOMENTUM trials, the proportions of patients who either reached open-label extension phase week 24 or discontinued the study drug before week 24 in the open-label phase were 83.6% (n = 361), 64.7% (n = 101), and 68.7% (n = 134), respectively.10-12 Among patients randomized in the SIMPLIFY-1 trial, 78.6% in the momelotinib group and 88.5% in the ruxolitinib group reached the open-label extension phase week 24 or discontinued the study drug before week 24.10 Among the patients randomized in the SIMPLIFY-2 trial, 60.6% in the momelotinib group and 73.1% in the BAT group reached the open-label extension phase week 24 or discontinued the study drug before week 24.11 Among the patients randomized in the MOMENTUM trial, 71.5% in the momelotinib group and 63.1% in the danazol group reached the open-label extension phase week 24 or discontinued the study drug before week 24.12
In the SIMPLIFY-1 trial, the proportion of patients who discontinued treatment before week 24 of open-label treatment was higher among those who switched from ruxolitinib to momelotinib (20.3%) than among those who continued with momelotinib (10.2%). Across both populations, AEs followed by disease progression were the most common reasons for treatment discontinuation, with a greater proportion of patients who had received ruxolitinib in the double-blind phase than those treated with momelotinib discontinuing treatment due to AEs (21 [9.7%] versus 10 [4.7%]) and disease progression (9 [4.1%] versus 2 [1.0%]).10
In the SIMPLIFY-2 trial, the proportion of patients discontinuing treatment in the open-label extended treatment phase was numerically higher among those in the switch to momelotinib treatment group (34.6%; n = 18) than among those in the continuing momelotinib group (13.5%; n = 14). AEs were the most common reason for treatment discontinuation in both treatment groups, with a numerically larger proportion of patients who had received BAT in the double-blind phase discontinuing treatment due to AEs (25.0%; n = 13) than those treated with momelotinib (5.8%; n = 6).11
In the MOMENTUM trial, the proportion of patients discontinuing treatment was similar between the 2 treatment groups. The most common reasons for treatment discontinuation in both treatment groups were AEs (11 [8.5%] versus 1 [1.5%] in the continuing momelotinib group and switch to momelotinib treatment group, respectively) and death (6 [4.6%] versus 3 [4.6%] in the continuing momelotinib group and switch to momelotinib treatment group, respectively).12
The disposition of patients in the open-label extension phase of the SIMPLIFY-1, SIMPLIFY-2, and MOMENTUM trials in the ITT population is presented in Table 20.
Pagebreak
Table 20: Summary of Patient Disposition in the Open-Label Extension Phase of the SIMPLIFY-1, SIMPLIFY-2, and MOMENTUM Trials
Patient disposition | SIMPLIFY-1 | SIMPLIFY-2 | MOMENTUM | |||
|---|---|---|---|---|---|---|
MMB (N = 215) | RUX (N = 217) | MMB (N = 104) | BAT (N = 52) | MMB (N = 130) | DAN (N = 65) | |
Either reached open-label phase week 24 or discontinued study drug before week 24 in the open-label extension phase, n (%) | 169 (78.6) | 192 (88.5) | 63 (60.6) | 38 (73.1) | 93 (71.5) | 41 (63.1) |
Reached week 24 of open-label MMB treatment,a n (%) | 147 (68.4) | 148 (68.2) | 49 (47.1) | 20 (38.5) | 67 (51.5) | 32 (49.2) |
Discontinued study drug before week 24 of open-label treatment,b n (%) | 22 (10.2) | 44 (20.3) | 14 (13.5) | 18 (34.6) | NA | NA |
Reason for premature treatment discontinuation, n (%) | ||||||
Continuing in MMB open-label extension study (only for MOMENTUM) | NA | NA | NA | NA | 61 (46.9) | 27 (41.5) |
Adverse events | 10 (4.7) | 21 (9.7) | 6 (5.8) | 13 (25.0) | 11 (8.5) | 1 (1.5) |
Disease progression | 2 (1.0) | 9 (4.1) | 2 (1.9) | 2 (3.8) | 1 (0.8) | 1 (1.5) |
Patient decision | 6 (2.8) | 2 (1.0) | 1 (1.0) | 1 (1.9) | 4 (3.1) | 3 (4.6) |
Insufficient efficacy | 2 (1.0) | 5 (2.3) | 2 (1.9) | 1 (1.9) | 6 (4.6) | 2 (3.1) |
Investigator discretion | 2 (1.0) | 2 (1.0) | 2 (1.9) | 1 (1.9) | 4 (3.1) | 3 (4.6) |
Death | 0 | 5 (2.3) | 1 (1.0) | 0 | 6 (4.6) | 3 (4.6) |
Leukemic transformation | NR | NR | NR | NR | 0 | 1 (1.5) |
BAT = best available therapy; DAN = danazol; MMB = momelotinib; NA = not applicable; NR = not reported; RUX = ruxolitinib.
Note: Results are from the final analysis at the database lock of July 1, 2019, for the SIMPLIFY-1 trial; June 25, 2019, for the SIMPLIFY-2 trial; and January 17, 2023, for the MOMENTUM trial.
a“Reached week 24 of open-label treatment” refers to patients whose last extended open-label treatment phase participation date was more than 176 days after the first extended open-label treatment dose of MMB for the SIMPLIFY-1 and SIMPLIFY-2 trials.
b“Discontinued study drug before week 24 of open-label treatment” refers to patients who discontinued the extended open-label treatment phase study drug and had a last extended open-label treatment phase participation date less than 162 days after the first open-label treatment dose of MMB for the SIMPLIFY-1 and SIMPLIFY-2 trials.
Sources: GSK Data on File, 2021 (SIMPLIFY-1 Clinical Study Report);10 GSK Data on File, 2021 (SIMPLIFY-2 Clinical Study Report);11 GSK Data on File, 2023 (MOMENTUM Clinical Study Report).12 Details included in the table are from the sponsor’s summary of clinical evidence.
Baseline Characteristics
The baseline characteristics of the patients enrolled in the open-label extension phase of the SIMPLIFY-1 and SIMPLIFY-2 trials in the ITT population are presented in Table 21.
In the open-label extension phase of the SIMPLIFY-1 trial, the mean age of the patients in the ITT population was 64.2 years. There were slightly more males in the continuing momelotinib group (59.1%) than in the switch to momelotinib treatment group (54.8%). The majority of patients in the continuing momelotinib group (40.4%) and the switch to momelotinib treatment group (48.7%) had high-risk MF per the IPSS criteria and a positive JAK2V617F mutation status (58.5% and 64.0% in the continuing momelotinib and switch to momelotinib groups, respectively) at baseline. The proportion of patients who were transfusion dependent was similar in both the continuing momelotinib group and the switch to momelotinib treatment group (22.8% versus 21.8%, respectively). Other characteristics, such as mean TSS, ECOG PS, and mean hemoglobin, were also similar in both groups. There were some imbalances between the 2 groups in hemoglobin level at baseline: The proportion of patients with a hemoglobin level less than 10 g/dL was higher in the switch to momelotinib treatment group (56.3%) than in the continuing momelotinib group (37.4%).10
In the open-label extension phase of the SIMPLIFY-2 trial, the mean age of the patients in the ITT population was 68.1 years. There were more males in the continuing momelotinib group (64.1%) than in the switch to momelotinib treatment group (40.0%). The majority of patients in the continuing momelotinib group (64.1%) and the switch to momelotinib treatment group (55.0%) had intermediate-2–risk MF per the DIPSS criteria, and more than 60% of patients in both treatment groups had a positive JAK2V617F mutation status (60.9% versus 72.5% in the continuing momelotinib group and switch to momelotinib treatment group, respectively). A numerically larger proportion of patients were transfusion dependent in the continuing momelotinib group (57.8%) than in the switch to momelotinib treatment group (50.0%).11 There were some imbalances between the 2 groups in ECOG PS and hemoglobin level at baseline. The proportion of patients with an ECOG PS of 1 was higher in the continuing momelotinib group (64.1%) than in the switch to momelotinib treatment group (47.5%). A numerically smaller proportion of patients had an ECOG PS of 2 in the continuing momelotinib group (4.7%) than in the switch to momelotinib treatment group (15.0%). There were no patients with an ECOG PS of 3 in the continuing momelotinib group, but 5% of patients in the switch to momelotinib treatment group had an ECOG PS of 3. The proportion of patients with a hemoglobin level less than 8 g/dL was higher in the continuing momelotinib group (28.1%) than in the switch to momelotinib treatment group (7.5%).11
The population demographics or baseline disease characteristics were not presented separately for patients who participated in the open-label extension phase of the MOMENTUM trial.12
Table 21: Summary of Baseline Characteristics in the Open-Label Extension Phase of the SIMPLIFY-1 and SIMPLIFY-2 Trials (ITT Analysis Set)
Characteristic | SIMPLIFY-1 | SIMPLIFY-2 | ||
|---|---|---|---|---|
Continuing (MMB to MMB) (n = 171) | Switch (RUX to MMB) (n = 197) | Continuing (MMB to MMB) (n = 64) | Switch (BAT to MMB) (n = 40) | |
Age (years), mean (SD)a | 64.2 (10.76) | 64.2 (10.62) | 66.9 (8.28) | 70.0 (6.89) |
Sex, n (%) | ||||
Male | 101 (59.1) | 108 (54.8) | 41 (64.1) | 16 (40.0) |
Female | 70 (40.9) | 89 (45.2) | 23 (35.9) | 24 (60.0) |
BMI (kg/m2), mean (SD) | 24.7 (3.88) | 26.7 (4.15) | 28.2 (5.21) | 26.0 (3.92) |
MF subtype, n (%) | ||||
Primary | 104 (60.8) | 104 (52.8) | 37 (57.8) | 23 (57.5) |
Post-PV | 37 (21.6) | 45 (22.8) | 12 (18.8) | 10 (25.0) |
Post-ET | 30 (17.5) | 48 (24.4) | 15 (23.4) | 7 (17.5) |
IPSS (SIMPLIFY-1) or DIPSS (SIMPLIFY-2) risk category, n (%) | ||||
Intermediate-1 | 41 (24.0) | 40 (20.3) | 13 (20.3) | 12 (30.0) |
Intermediate-2 | 61 (35.7) | 61 (31.0) | 41 (64.1) | 22 (55.0) |
High | 69 (40.4) | 96 (48.7) | 10 (15.6) | 6 (15.0) |
TSS, mean (SD) | 18.5 (12.52) | 17.6 (11.46) | 17.5 (13.40) | 21.4 (16.68) |
ECOG PS, n (%) | ||||
0 | 63 (36.8) | 76 (38.6) | 20 (31.3) | 13 (32.5) |
1 | 95 (55.6) | 111 (56.3) | 41 (64.1) | 19 (47.5) |
2 | 13 (7.6) | 10 (5.1) | 3 (4.7) | 6 (15.0) |
3 | 0 | 0 | 0 | 2 (5.0) |
JAK2V617F mutation, n (%) | ||||
Positive | 100 (58.5) | 126 (64.0) | 39 (60.9) | 29 (72.5) |
Negative | 49 (28.7) | 48 (24.4) | 23 (35.9) | 9 (22.5) |
Not previously tested | 22 (12.9) | 23 (11.7) | 2 (3.1) | 2 (5.0) |
Hemoglobin (g/dL), mean (SD) | 10.7 (2.05) | 9.9 (1.67) | 9.2 (1.69) | 9.5 (1.54) |
Hemoglobin level, n (%) | ||||
< 8 g/dL | 20 (11.7) | 20 (10.2) | 18 (28.1) | 3 (7.5) |
≥ 8 g/dL | 151 (88.3) | 177 (89.8) | 46 (71.9) | 37 (92.5) |
< 10 g/dL | 64 (37.4) | 111 (56.3) | 43 (67.2) | 27 (67.5) |
≥ 10 g/dL | 107 (62.6) | 86 (43.7) | 21 (32.8) | 13 (32.5) |
Transfusion independent, n (%) | 121 (70.8) | 142 (72.1) | 20 (31.3) | 14 (35.0) |
Transfusion dependent, n (%) | 39 (22.8) | 43 (21.8) | 37 (57.8) | 20 (50.0) |
Platelet count (× 103/µL), mean (SD) | 293.8 (195.16) | 199.5 (154.79) | 180.3 (148.49) | 126.6 (91.14) |
ANC (× 103/µL), mean (SD) | 11.3 (13.05) | 8.3 (10.26) | 6.7 (6.54) | 7.7 (7.72) |
ANC = absolute neutrophil count; BAT = best available therapy; BMI = body mass index; DIPSS = Dynamic International Prognostic Scoring System; ECOG PS = Eastern Cooperative Oncology Group Performance Status; ET = essential thrombocythemia; IPSS = International Prognostic Scoring System; ITT = intention to treat; MF = myelofibrosis; MMB = momelotinib; PV = polycythemia vera; RUX = ruxolitinib; SD = standard deviation; TSS = total symptom score.
Notes: For the continuing (MMB to MMB) treatment group, the baseline is the last assessment before or on the date of randomization. For the switch (RUX to MMB in the SIMPLIFY-1 trial or BAT to MMB in the SIMPLIFY-2 trial) treatment group, the baseline is the last assessment before or on the date of the first dose of MMB in the extended treatment phase.
aAge was calculated in years at the date of randomization.
Sources: GSK Data on File, 2021 (SIMPLIFY-1 Clinical Study Report);10 GSK Data on File, 2021 (SIMPLIFY-2 Clinical Study Report).11 Details included in the table are from the sponsor’s summary of clinical evidence.
Exposure to Study Treatments
A summary of patient exposure in the open-label extension phases of the SIMPLIFY-1, SIMPLIFY-2, and MOMENTUM trials is presented in Table 22. In the overall safety population of the SIMPLIFY-1 trial, the mean treatment adherence rates were more than 90% and were approximately similar across the continuing momelotinib group and switch to momelotinib treatment group (94.2% versus 94.8%, respectively). The mean daily dose of momelotinib was slightly higher in the continuing momelotinib treatment group than in the switch to momelotinib treatment group (180.3 mg/day versus 174.6 mg/day, respectively).10
In the overall safety population in the extended treatment phase of the SIMPLIFY-2 trial, the mean treatment adherence rates were numerically higher in the continuing momelotinib group than in the switch to momelotinib treatment group (39.2% versus 31.6%, respectively). The mean daily dose of momelotinib was numerically higher in the switch to momelotinib treatment group than in the continuing momelotinib group (176.3 mg/day versus 163.9 mg/day, respectively).11
The mean duration of momelotinib treatment in the open-label extension phase of the MOMENTUM trial was similar for patients who continued momelotinib treatment and those who switched to momelotinib after randomized treatment with danazol. The mean relative dose intensity was similar in both groups.12
Concomitant Medications and Co-Interventions
Concomitant medication use was reported by approximately 99% of patients in the overall safety population who transitioned to the open-label extension phase of the SIMPLIFY-1 trial.10 Concomitant medication use was reported by all patients (100%) in the overall safety population who transitioned to the open-label treatment phase of the SIMPLIFY-2 trial.11 Concomitant medications and co-interventions were not reported separately for patients who participated in the open-label extension phase of the MOMENTUM trial.12
Details on subsequent treatments were not available for the open-label phase.
Efficacy
The overall median duration of follow-up in the SIMPLIFY-1 trial safety analysis set (combined randomized and open-label extension phases) was 35.3 months (range, 0.4 to 59.3 months; momelotinib: 35.4 months and ruxolitinib: 35.2 months).10 In the SIMPLIFY-2 trial, the overall median duration of follow-up in the safety analysis set (combined randomized and open-label extension phases, and long-term follow-up) was 28.2 months (range, 0.3 to 50.4 months; momelotinib: 28.2 months and BAT: 27.2 months).11
Table 22: Summary of Patient Exposure in the Open-Label Extension Phase of the SIMPLIFY-1, SIMPLIFY-2, and MOMENTUM Trials (Safety Analysis Set)
Exposure | SIMPLIFY-1 (weeks 24 to 48) | SIMPLIFY-2 (weeks 24 to 48) | MOMENTUM (weeks 24 to 48) | |||
|---|---|---|---|---|---|---|
Continuing (MMB to MMB) (n = 171) | Switch (RUX to MMB) (n = 197) | Continuing (MMB to MMB) (n = 64) | Switch (BAT to MMB) (n = 40) | Continuing (MMB to MMB) (n = 93) | Switch (DAN to MMB) (n = 41) | |
Daily dose intensity (mg/day)a | ||||||
Mean (SD) | 180.3 (57.64) | 174.6 (35.98) | 163.9 (45.29) | 176.3 (34.15) | 181.78 (34.79) | 177.95 (34.60) |
Median (Q1, Q3) | 193.2 (164.3, 199.0) | 191.8 (155.4, 199.2) | 181.8 (145.1, 196.3) | 188.2 (166.2, 197.7) | 200.00 (181.40, 200.00) | 200.00 (164.44, 200.00) |
Relative dose intensity (%), mean (SD)b | NR | NR | NR | NR | 90.89 (17.40) | 88.98 (17.30) |
On-treatment adherence rate (%)c | ||||||
Mean (SD) | 94.2 (13.67) | 94.8 (12.41) | 39.2 (31.26) | 31.6 (28.45) | NR | NR |
Median (range) | 98.5 (0 to 124) | 99.0 (0 to 119) | 31.9 (0 to 103) | 32.9 (0 to 92) | NR | NR |
Total duration of exposure to study drug (weeks)c,d | ||||||
Mean (SD) | 88.7 (56.49) | 79.5 (57.28) | 72.0 (54.03) | 56.3 (52.32) | 26.35 (10.86) | 25.20 (10.57) |
Median (range) | 81.6 (1.9 to 218.3) | 74.3 (2.1 to 209.4) | 60.6 (0 to 176) | 38.1 (1 to 161) | 24.14 (2.0 to 53.1) | 24.00 (3.6 to 48.3) |
BAT = best available therapy; DAN = danazol; MMB = momelotinib; NR = not reported; Q1 = quarter 1; Q3 = quarter 3; RUX = ruxolitinib; SD = standard deviation.
Note: The Safety population comprises all patients in the intention-to-treat population who received at least 1 dose of the study drug. Patients were evaluated based on actual treatment received.
aDose intensity = actual cumulative dose / actual duration of the treatment (days).
bRelative dose intensity = actual dose intensity / planned dose intensity. In the pooled data from all trials, the relative dose intensity was 100 (range, 0 to 114) for patients randomized to MMB during the treatment phase and was 97.2 (range, 0 to 387) for those who received MMB during the open-label phase.47
cAdherence rate (%) = [total study drug administered / total study drug prescribed per protocol] × 100. Only patients who returned at least 1 bottle of the study drug were included in the analysis.
dDuration of exposure to study drug = ([treatment end date – treatment start date] + 1) / 7.
Sources: GSK Data on File, 2021 (SIMPLIFY-1 Clinical Study Report);10 GSK Data on File, 2021 (SIMPLIFY-2 Clinical Study Report);11 GSK Data on File, 2023 (MOMENTUM Clinical Study Report).12 Details included in the table are from the sponsor’s summary of clinical evidence.
Transfusion Independence Response Rate, Week 48
The transfusion independence response rate was not reported for the open-label extension phases of the SIMPLIFY-1 and SIMPLIFY-2 trials.10,11 In the open-label extension phase of the MOMENTUM trial, transfusion independence response was reported as the transfusion independence response rate at week 48 and the transfusion independence response rate by week 48, as described in Table 23. Transfusion independence at week 48 was defined as the absence of RBC transfusion and no hemoglobin level less than 8 g/dL during the 12 weeks immediately before the week 48 visit. Transfusion independence by week 48 was defined as the absence of RBC transfusion and no hemoglobin level less than 8 g/dL in any rolling period of 12 weeks (i.e., 84 consecutive days) before the week 48 visit.12
The transfusion independence response rate during the first 24 weeks of the open-label extension phase for the MOMENTUM trial is presented in Table 23. In the MOMENTUM trial, the majority of patients were transfusion independent at week 24 and week 48, including 88.2% of patients (n = 30 of 34) in the continuing momelotinib group and 80.0% of patients (n = 8 of 10) in the switch to momelotinib treatment group. Further, 24.2% of patients (n = 8 of 33) in the continuing momelotinib group and 50.0% of patients (n = 10 of 20) in the switch to momelotinib treatment group without transfusion-independence status at week 24 and met the criteria for transfusion independence response at week 48.12
Table 23: Summary of Transfusion Independence Response During the Open-Label Extension Phase in the MOMENTUM Trial (ITT Analysis Set)
Characteristic | Continuing (MMB to MMB) | Switch (DAN to MMB) | ||
|---|---|---|---|---|
Week 24 responder (N = 39) | Week 24 nonresponder (N = 54) | Week 24 responder (N = 13) | Week 24 nonresponder (N = 28) | |
Transfusion independence response rate at week 48 | ||||
Evaluable at week 48, n | 34 | 33 | 10 | 20 |
Responders,a n (%) | 30 (88.2) | 8 (24.2) | 8 (80.0) | 10 (50.0) |
Nonresponders,b n (%) | 4 (11.8) | 25 (75.8) | 2 (20.0) | 10 (50.0) |
Transfusion independence response rate by week 48 | ||||
Evaluable by week 48, n | 36 | 48 | 12 | 24 |
Responders,c n (%) | 34 (94.4) | 10 (20.8) | 10 (83.3) | 10 (41.7) |
Nonresponders,d n (%) | 2 (5.6) | 38 (79.2) | 2 (16.7) | 14 (58.3) |
DAN = danazol; ITT = intention to treat; MMB = momelotinib; RBC = red blood cell.
aCompleted the week 48 visit and had no RBC or whole blood transfusion, had no hemoglobin level less than 8 g/dL, and provided hemoglobin assessments during the 12 weeks immediately before the week 48 visit.
bCompleted the week 48 visit and did not meet the transfusion independence criteria.
cHad no RBC or whole blood transfusion, had no hemoglobin level less than 8 g/dL, and provided hemoglobin assessments during any rolling 12-week period from the first day of the open-label treatment period up to the week 48 visit.
dCompleted the week 48 visit and did not meet the transfusion independence criteria.
Source: GSK Data on File, 2023 (MOMENTUM Clinical Study Report).12 Details included in the table are from the sponsor’s summary of clinical evidence.
Rate of RBC Transfusion at Week 48
The rate of RBC transfusion was not reported for the SIMPLIFY-1 and SIMPLIFY-2 trials and was not reported for the open-label extension phase of the MOMENTUM trial.10-12
Splenic Response at Any Time
███████ ████████ ██ ███ ████ ███ ███ ██████████ █████████ █████ ██ ██████████ ███ ██████████ ███ ████ █████████ ██ █████ ███ ███████ ████████ ████ ████ ████ ███ ███████ ██ ███ ██████████ ██ ████████ ███ ████████ █ ██████ ██████ █████████ ██ █ ███ ████ ████████ ██ ████████ ██ ███ ██ ██ ██████ ██ ███████████ ██ ███████ ██ ███ ███ ████████ ██ ███ ███████████ █████ ███ █ ███████ ████████ ██ ███ ████ ██████ ███ ████████████ ██ ██████████ █████████ ██████ ██ ██████████ ██ ███████ █████████ ████ ████████ ██ ███ ██████████████████████ █████ █████████ █████████ ██ ███████████ ██ ███ ██████████ █████████ ██████ ████████████ ████████ ██ ███ ████████ ██████████ ██ ███████████ ███ ████████ ██ ███████████ ██████ ███ ██████████ █████████ █████ ████ ██████████ ██ ███ ██████████ ██ ███ ███████ ██████ ██ ███████████ █████████ ███ ███████ ██ ███ ████████ ███ █ ███████ ████████ ██ ███ ██████ ███████████ ██ ███████ ██ ███ ███ ████████ ██ ███ ███████████ █████ ███ █ ███████ ██ ██ ████████ ██ ███ ███ █████ ██████████ ████████ ███ ████████ ████ ███ ██ ████████████ ███ █ ███████ ████████ ██ ███ ████ ██ ███ ██████████ ██ ██████████ █████████ ██████ ██ ███ ███████ ██████ ██ ███████████ █████████ ██ ███████ ████████ ███ █ ███████ ████████ ██ ███ █████ ██ ███ ██████████ █████████ ██████ ██ ███████ ██ ██ ████████ ██████████ ██ ███████████ ███ ████████ ██ ███████████ ████ ██████████ ██ ███ ██████████ █████ ███████ ██ ██ ████████ ██████████ ██ ███ ███ ████████ ████ ███ ██ ███████████ ██████ ███ ██████████ █████████ █████ ████ ██████████ ██ ███ █████████
The proportion of patients with and without a splenic response at week 24 in the MOMENTUM trial who were classified as having a splenic response at week 48 is shown in Table 25. Most patients (n = 19 of 24; 79.2%) in the continuing momelotinib group and 50.0% of patients (n = 1 of 2) in the switch to momelotinib treatment group who experienced splenic response at week 24 were also classified as experiencing splenic response at week 48. Of those not experiencing splenic response at week 24 in the continuing momelotinib group (n = 43) and the switch to momelotinib treatment group (n = 28), 23.3% and 10.7%, respectively, were classified as experiencing a splenic response at week 48.12
Table 24: Splenic Response at Any Time in Combined Double-Blind and Open-Label Extension Phases of the SIMPLIFY-1 and SIMPLIFY-2 Trials (ITT Analysis Set)
Splenic response at any time | SIMPLIFY-1 | SIMPLIFY-2 | ||
|---|---|---|---|---|
MMB (N = 215) | RUX (N = 217) | MMB (N = 104) | BAT (N = 52) | |
████████ ████ ██ ███ ██████ ██████ █████ | ██ ██████ | ███ ██████ | ██ ██████ | ██████ |
████████ ████ ██ ███ ██████ ██████ ████ | ███ ██████ | ███ ██████ | ██ ██████ | ██ ██████ |
████████ ████ ██ ███ ██████ ██████ ████ | ███ █████ | ███ █████ | ██ █████ | ██ █████ |
████████ ████ ██ ███ ██████ ██████ █████ | ██████ █ | ██████ ██ | █████ █ ███ | ████ █ ███ |
BAT = best available therapy; ITT = intention to treat; MMB = momelotinib; RUX = ruxolitinib.
Note: The baseline spleen volume used is from the last assessment before or on the date of randomization.
Sources: GSK Data on File, 2021 (SIMPLIFY-1 Clinical Study Report);10 GSK Data on File, 2021 (SIMPLIFY-2 Clinical Study Report).11 Details included in the table are from the sponsor’s summary of clinical evidence.
Table 25: Splenic Response at Week 48 in the Open-Label Extension Phase of the MOMENTUM Trial
Splenic response at week 48 | Continuing (MMB to MMB) | Switch (DAN to MMB) | ||
|---|---|---|---|---|
Experiencing splenic response at week 24a (n = 29) | Not experiencing splenic response at week 24 (n = 64) | Experiencing splenic response at week 24a (n = 2) | Not experiencing response at week 24 (n = 39) | |
Evaluable at week 48, n | 24 | 43 | 2 | 28 |
Experiencing splenic response, n (%) | 19 (79.2) | 10 (23.3) | 1 (50.0) | 3 (10.7) |
Not experiencing splenic response, n (%) | 5 (20.8) | 33 (76.7) | 1 (50.0) | 25 (89.3) |
DAN = danazol; ITT = intent to treat; MMB = momelotinib.
Notes: Splenic response is defined as having at least a 35% reduction from baseline spleen volume. Splenic response at week 48 was assessed in patients who crossed over to open-label treatment with MMB and had a week 48 spleen scan. The imaging modality was required to match at baseline and week 48. Results are from the final analysis at the database lock on January 17, 2023.
aPatients with at least a 35% reduction from baseline spleen volume at week 48.
Source: GSK Data on File, 2023 (MOMENTUM Clinical Study Report).12 Details included in the table are from the sponsor’s summary of clinical evidence.
TSS Response Rate, Week 48
TSS response rates were not reported for the SIMPLIFY-1 and SIMPLIFY-2 open-label extension studies.10,11
Table 26 presents the results for MFSAF TSS response during the first 24 weeks of the open-label extension phase for the MOMENTUM trial, expressed as “at week 48” and “by week 48.” Patients experiencing a TSS response at week 48 were defined as patients with at least a 50% reduction from baseline in mean MFSAF TSS during the 28-day period immediately before the end of week 48. Patients experiencing a TSS response by week 48 were defined as patients with at least a 50% reduction from baseline in mean MFSAF TSS during any rolling 28-day evaluation period from the first day of the open-label extension period up to week 48.12 A majority of patients with at least a 50% reduction from baseline MFSAF TSS at week 24 were also experiencing a TSS response at week 48, including 72.0% of patients (n = 18 of 25) in the continuing momelotinib group and all patients (n = 5 of 5; 100%) in the switch to momelotinib treatment group. Further, 27.9% (n = 12 of 43) and 40.0% (n = 10 of 25) of nonresponders at week 24 in the continuing momelotinib group and the switch to momelotinib treatment group had a TSS response at week 48.12 A majority of patients with at least a 50% reduction from baseline MFSAF TSS at week 24 were experiencing a TSS response by week 48, including 96.7% of patients (n = 29 of 30) in the continuing momelotinib group and all patients (n = 5 of 5; 100%) in the switch to momelotinib treatment group. Further, 37.0% (n = 17 of 46) and 51.9% (n = 14 of 27) of nonresponders at week 24 in the continuing momelotinib group and the switch to momelotinib treatment group had a TSS response by week 48.12
Table 26: MFSAF TSS Response Rate at Week 48 or by Week 48 in the Open-Label Extension Phase of the MOMENTUM Trial (Weeks 24 to 48)
MFSAF TSS response | Continuing (MMB to MMB) | Switch (DAN to MMB) | ||
|---|---|---|---|---|
Experiencing TSS response at week 24 (n = 32) | Not experiencing TSS response at week 24 (n = 61) | Experiencing TSS response at week 24 (n = 6) | Not experiencing TSS response at week 24 (n = 25) | |
MFSAF TSS response rate at week 48 | ||||
Evaluable at week 48, n | 25 | 43 | 5 | 25 |
Experiencing TSS response,a n (%) | 18 (72.0) | 12 (27.9) | 5 (100) | 10 (40.0) |
Nonresponders, n (%)b | 7 (28.0) | 31 (72.1) | 0 | 15 (60.0) |
MFSAF TSS response rate by week 48 | ||||
Evaluable by week 48, n | 30 | 46 | 5 | 27 |
Experiencing TSS response,c n (%) | 29 (96.7) | 17 (37.0) | 5 (100) | 14 (51.9) |
Nonresponders, n (%)d | 1 (3.3) | 29 (63.0) | 0 | 13 (48.1) |
DAN = danazol; DBL = database lock; MFSAF = Myelofibrosis Symptom Assessment Form; MMB = momelotinib; TSS = total symptom score.
Note: Results are from the final analysis at the database lock on January 17, 2023.
aHad at least a 50% reduction from baseline in mean MFSAF TSS during the 28-day period immediately before the end of week 48.
bCompleted the week 48 visit and did not meet response criteria.
cHad a at least a 50% reduction from baseline in mean MFSAF TSS during any rolling 28-day evaluation period from the first day of the open-label treatment period up to week 48.
dCompleted the week 48 visit and had MFSAF TSS data but did not meet response criteria.
Source: GSK Data on File, 2023 (MOMENTUM Clinical Study Report).12 Details included in the table are from the sponsor’s summary of clinical evidence.
Harms
A summary of safety outcomes in the overall safety population of the SIMPLIFY-1, SIMPLIFY-2, and MOMENTUM trials in the open-label extension phase is provided in Table 27.
Across the overall safety population in the SIMPLIFY-1 trial, the frequencies of TEAEs were numerically higher in patients who switched from ruxolitinib to momelotinib than those who continued on momelotinib (89.8% versus 78.4%, respectively) after 24 weeks of treatment with momelotinib in the open-label extension phase. Similar trends were observed for the most common grade 3 or 4 AEs (37.6% versus 27.5%), for SAEs (23.4% versus 15.8%), and for TEAEs leading to treatment discontinuation (14.7% versus 8.8%), with numerically higher proportions for patients who switched from ruxolitinib to momelotinib than for those who continued on momelotinib after 24 weeks of treatment with momelotinib in the open-label extension phase.10 The most commonly reported AEs in both groups, occurring in at least 10% of patients, were diarrhea, thrombocytopenia, anemia, fatigue, nausea, and cough. No SAE occurred in more than 2% of patients in either group.10 In the overall safety population, the most common AEs leading to treatment discontinuation were thrombocytopenia, fatigue, and peripheral sensory neuropathy (no events in the continuing momelotinib group and relatively few, 2.0% to 2.5%, in the switch to momelotinib group). In the continuing momelotinib and the switch to momelotinib groups, the following AESIs were reported: peripheral neuropathy (5.3% versus 7.6% of patients), nonhematological AEs (77.2% versus 87.3% of patients), cataract (4.7% versus 3.6% of patients), and first dose effect (NR versus 2.0% of patients). In the continuing momelotinib group, 10.5% of patients died from TEAEs not related to disease progression; in the switch to momelotinib group, this percentage was 8.6%.10
Across the overall safety population in the SIMPLIFY-2 trial, the frequencies of the following were numerically higher in patients who switched from BAT to momelotinib than in those who continued on momelotinib after 24 weeks of treatment with momelotinib in the open-label extension phase: TEAEs (100% versus 93.8%), grade 3 or 4 AEs (55.0% versus 28.1%), SAEs (27.5% versus 20.3%), TEAEs leading to treatment discontinuation (37.5% versus 7.8%), and AEs leading to treatment interruption and/or dose reduction (19.2% versus 16.3%).11 The most commonly reported AEs occurring in at least 15% of patients were, in the continuing momelotinib group, cough and diarrhea and, in the switch to momelotinib treatment group, asthenia, pyrexia, diarrhea, thrombocytopenia, cough, and anemia. The most commonly reported SAEs in patients who switched from BAT to momelotinib, occurring in at least 5% of patients, were anemia, pyrexia, and confusional state.11 The most common AEs leading to treatment discontinuation were thrombocytopenia, diarrhea, and headache (no events in the continuing momelotinib group, and 5.0% to 7.5% of patients experiencing events in the switch to momelotinib group). Among the continuing momelotinib group and the switch to momelotinib treatment group, the following AESIs were reported: peripheral neuropathy (10.9% versus 20.0%), nonhematological AEs (98.4% versus 100%), cataract (1.6% versus 0%), and first dose effect (NR versus 7.5%). Deaths due to TEAEs not related to disease progression in the overall safety population were reported in 21.9% of patients who continued treatment with momelotinib and 7.5% of patients who switched from BAT to momelotinib.11
In the MOMENTUM trial, after 24 weeks of treatment with momelotinib in the open-label treatment phase, the overall frequencies of TEAEs (89.2% versus 85.4%), grade 3 or higher TEAEs (51.6% versus 48.8%), and serious TEAEs (32.3% versus 29.3%) were numerically slightly higher in patients who continued on momelotinib than in those who switched from danazol to momelotinib.12 The most commonly reported AEs occurring in at least 10% of patients were, among patients who continued on momelotinib, diarrhea, thrombocytopenia, pyrexia, asthenia, and anemia and, among those who switched from danazol to momelotinib, thrombocytopenia and diarrhea. The most commonly reported SAEs occurring in at least 2% of patients were, among patients who continued on momelotinib, urinary tract infection, acute kidney injury, febrile neutropenia, and squamous cell carcinoma of the skin and, among those who switched from danazol to momelotinib, acute kidney injury and urinary tract infection. In the overall safety population, the most common AEs leading to treatment discontinuation were anemia, AML, and transformation to AML (no events in the continuing momelotinib group for AML or transformation to AML and no events in the switch to momelotinib treatment group for anemia). No deaths due to TEAEs not related to disease progression were reported in either of the treatment groups.12
Table 27: Summary of Harms in the Open-Label Extension Phase of the SIMPLIFY-1, SIMPLIFY-2, and MOMENTUM Trials (Safety Analysis Set)
AEs | SIMPLIFY-1 | SIMPLIFY-2 | MOMENTUM | |||
|---|---|---|---|---|---|---|
Continuing (MMB to MMB) (n = 171) | Switch (RUX to MMB) (n = 197) | Continuing (MMB to MMB) (n = 64) | Switch (BAT to MMB) (n = 40) | Continuing (MMB to MMB) (n = 93) | Switch (DAN to MMB) (n = 41) | |
Most common AEs (any grade; ≥ 10% of patients), n (%) | ||||||
≥ 1 AE | 134 (78.4) | 177 (89.8) | 60 (93.8) | 40 (100) | 83 (89.2) | 35 (85.4) |
Thrombocytopenia | 16 (9.4) | 28 (14.2) | 3 (4.7) | 8 (20.0) | 13 (14.0) | 7 (17.1) |
Anemia | 14 (8.2) | 21 (10.7) | 7 (10.9) | 6 (15.0) | 10 (10.8) | 4 (9.8) |
Diarrhea | 18 (10.5) | 28 (14.2) | 13 (20.3) | 9 (22.5) | 16 (17.2) | 5 (12.2) |
Cough | 12 (7.0) | 22 (11.2) | 12 (18.8) | 8 (20.0) | 8 (8.6) | 2 (4.9) |
Fatigue | 11 (6.4) | 26 (13.2) | 4 (6.3) | 4 (10.0) | 6 (6.5) | 3 (7.3) |
Nausea | 6 (3.5) | 28 (14.2) | 7 (10.9) | 3 (7.5) | 8 (8.6) | 0 |
Pyrexia | 7 (4.1) | 5 (2.5) | 9 (14.1) | 9 (22.5) | 13 (14.0) | 4 (9.8) |
Asthenia | 6 (3.5) | 6 (3.0) | 4 (6.3) | 9 (22.5) | 12 (12.9) | 0 |
Edema peripheral | 8 (4.7) | 3 (1.5) | 6 (9.4) | 5 (12.5) | 4 (4.3) | 1 (2.4) |
Dizziness | 11 (6.4) | 13 (6.6) | 5 (7.8) | 5 (12.5) | 1 (1.1) | 4 (9.8) |
Headache | 4 (2.3) | 13 (6.6) | 2 (3.1) | 5 (12.5) | 3 (3.2) | 1 (2.4) |
Urinary tract infection | 8 (4.7) | 11 (5.6) | 6 (9.4) | 4 (10.0) | 5 (5.4) | 1 (2.4) |
Weight decreased | NR | NR | 1 (1.6) | 4 (10.0) | 9 (9.7) | 7 (17.1) |
Peripheral sensory neuropathy | 7 (4.1) | 15 (7.6) | 2 (3.1) | 5 (12.5) | 2 (2.2) | 1 (2.4) |
Bronchitis | 3 (1.8) | 11 (5.6) | 3 (4.7) | 4 (10.0) | 1 (1.1) | 0 |
Decreased appetite | 7 (4.1) | 11 (5.6) | 3 (4.7) | 4 (10.0) | 4 (4.3) | 1 (2.4) |
Vomiting | 6 (3.5) | 8 (4.1) | 4 (6.3) | 4 (10.0) | 3 (3.2) | 1 (2.4) |
Hypotension | 3 (1.8) | 7 (3.6) | 0 | 4 (10.0) | 3 (3.2) | 1 (2.4) |
Most common grade 3 or 4 AEs (≥ 5% of patients), n (%) | ||||||
Any grade 3 or 4 AE | 47 (27.5) | 74 (37.6) | 18 (28.1) | 22 (55.0) | 48 (51.6) | 20 (48.8) |
Thrombocytopenia | 6 (3.5) | 17 (8.6) | 1 (1.6) | 5 (12.5) | 8 (8.6) | 6 (14.6) |
Anemia | 8 (4.7) | 10 (5.1) | 6 (9.4) | 6 (15.0) | 8 (8.6) | 1 (2.4) |
Diarrhea | 0 | 5 (2.5) | 1 (1.6) | 2 (5.0) | 1 (1.1) | 0 |
Neutropenia | 2 (1.2) | 4 (2.0) | 1 (1.6) | 3 (7.5) | 5 (5.4) | 0 |
Asthenia | 0 | 1 (0.5) | 1 (1.6) | 4 (10.0) | 4 (4.3) | 0 |
Cardiac failure | 0 | 0 | 1 (1.6) | 2 (5.0) | 1 (1.1) | 0 |
Acute kidney injury | 1 (0.6) | 1 (0.5) | 1 (1.6) | 1 (2.5) | 2 (2.2) | 3 (7.3) |
Serious AEs (any grade; n ≥ 2 patients), n (%) | ||||||
Patients with ≥ 1 serious AE | 27 (15.8) | 46 (23.4) | 13 (20.3) | 11 (27.5) | 30 (32.3) | 12 (29.3) |
Pneumonia | 3 (1.8) | 4 (2.0) | 3 (4.7) | 0 | 1 (1.1) | 1 (2.4) |
Acute kidney injury | 2 (1.2) | 2 (1.0) | 1 (1.6) | 0 | 2 (2.2) | 2 (4.9) |
Anemia | 0 | 3 (1.5) | 0 | 3 (7.5) | 1 (1.1) | 1 (2.4) |
Urinary tract infection | 1 (0.6) | 2 (1.0) | NR | NR | 2 (2.2) | 1 (2.4) |
Sepsis | 2 (1.2) | 0 | 2 (3.1) | 1 (2.5) | NR | NR |
Febrile neutropenia | 2 (1.2) | 0 | NR | NR | 2 (2.2) | 0 |
GI hemorrhage | NR | NR | 2 (3.1) | 0 | NR | NR |
Pyrexia | 0 | 0 | 2 (3.1) | 3 (7.5) | 1 (1.1) | 1 (2.4) |
Confusional state | NR | NR | 0 | 2 (5.0) | NR | NR |
Squamous cell carcinoma of skin | NR | NR | NR | NR | 2 (2.2) | 0 |
COVID-19 pneumonia | NR | NR | NR | NR | 4 (4.3) | 0 |
Patients (n ≥ 2) who discontinued treatment due to AEs, n (%) | ||||||
Patients who discontinued treatment | 15 (8.8) | 29 (14.7) | 5 (7.8) | 15 (37.5) | 17 (18.3) | 5 (12.2) |
Thrombocytopenia | 1 (0.6) | 5 (2.5) | 0 | 3 (7.5) | 1 (1.1) | 0 |
Fatigue | 0 | 5 (2.5) | NR | NR | 1 (1.1) | 0 |
Diarrhea | 0 | 3 (1.5) | 0 | 3 (7.5) | 1 (1.1) | 0 |
Peripheral sensory neuropathy | 0 | 4 (2.0) | 0 | 1 (2.5) | NR | NR |
Headache | NR | NR | 0 | 2 (5.0) | NR | NR |
Anemia | NR | NR | NR | NR | 2 (2.2) | 0 |
Deaths, n (%) | ||||||
Patients who died | 44 (25.7) | 54 (27.4) | 29 (45.3) | 18 (45.0) | 10 (10.8) | 10 (10.8) |
Due to TEAE not related to disease progression | 18 (10.5) | 17 (8.6) | 14 (21.9) | 3 (7.5) | NR | NR |
Due to infections and infestations | NR | NR | NR | NR | 5 (5.4) | 1 (2.4) |
AEs of special interest, n (%) | ||||||
Peripheral neuropathy | 9 (5.3) | 15 (7.6) | 7 (10.9) | 8 (20.0) | 2 (2.2) | 0 |
Nonhematological AEs | 132 (77.2) | 172 (87.3) | 63 (98.4) | 40 (100) | NR | NR |
Cataract | 8 (4.7) | 7 (3.6) | 1 (1.6) | 0 | NR | NR |
First-dose effect | 0 | 4 (2.0) | 0 | 3 (7.5) | NR | NR |
Infections | NR | NR | NR | NR | NR | NR |
Hemorrhage | NR | NR | NR | NR | NR | NR |
Malignancies | NR | NR | NR | NR | NR | NR |
Thromboembolism | NR | NR | NR | NR | NR | NR |
AML or transformation to AML | NR | NR | NR | NR | NR | NR |
AE = adverse event; AML = acute myeloid leukemia; BAT = best available therapy; DAN = danazol; GI = gastrointestinal; MMB = momelotinib; NR = not reported; RUX = ruxolitinib; TEAE = treatment-emergent adverse event.
Notes: All patients in the intention-to-treat population who received at least 1 dose of the study drug. Patients were evaluated based on actual treatment received.
Sources: GSK Data on File, 2021 (SIMPLIFY-1 Clinical Study Report);10 GSK Data on File, 2021 (SIMPLIFY-2 Clinical Study Report);11 GSK Data on File, 2023 (MOMENTUM Clinical Study Report).12 Details included in the table are from the sponsor’s summary of clinical evidence.
Critical Appraisal
Internal Validity
The open-label extension phase design of the SIMPLIFY-1, SIMPLIFY-2, and MOMENTUM trials may have biased the reporting of some end points because awareness of the study treatment received may have influenced the perception of improvement and/or harms by patients and clinicians, particularly for outcomes that are subjective in measurement and interpretation (e.g., TSS response rate and subjective AEs). Although there will be a longer-term follow-up period for all 3 studies once the ongoing open-label extension phase is completed, at the time of this assessment data were available up to 24 weeks in the extension phase of the 3 studies, which may not be long enough to observe important safety and efficacy outcomes. Moreover, in the open-label extension phases, all patients were taking momelotinib. As such, there was no relevant randomized comparison group (i.e., for any active comparator of interest), which precludes causal conclusions.
In terms of protocol deviations, for the SIMPLIFY-2 trial, the proportion of patients with at least 1 important protocol violation was higher in the continuing momelotinib treatment group (20.3%) than in the switch to momelotinib treatment group (10.0%) in the extended treatment phase. The reasons behind the protocol violations were missing data related to key end points (10.9% versus 5.0%, respectively), good clinical practice violation (7.8% versus 5.0%, respectively), and other treatment compliance (3.1% versus 0, respectively). This difference may lead to some biases and affect the efficacy outcomes. No information on protocol deviation for the open-label extension phase of the MOMENTUM study was reported separately; as such, any risk of bias due to deviations from the intended interventions is uncertain. The results are reflective of patients who were able to tolerate and stay on momelotinib (in the continuing momelotinib group). No information on missing data imputations were reported for the open-label extension phase in the SIMPLIFY-1, SIMPLIFY-2, or MOMENTUM trials.
The proportion of patients who were transfusion dependent was similar across the groups of patients continuing on momelotinib and switching to momelotinib in the open-label extension phases of the SIMPLIFY-1 trial. In the SIMPLIFY-2 trial, a higher proportion of patients in the group continuing on momelotinib was transfusion dependent than among those who switched to momelotinib in the open-label extension phase. Among patients continuing on momelotinib, the results are representative of those who were able to continue in the trial (commonly the ones who are responding well to treatment and can tolerate it). This is especially true for the open-label extension phases of the SIMPLIFY-1 and SIMPLIFY-2 studies. In the SIMPLIFY-1 and SIMPLIFY-2 trials, the number of patients who discontinued treatment before week 24 of the open-label treatment phase were higher among those who switched from ruxolitinib to momelotinib or BAT to momelotinib than among those who continued on momelotinib. The main reason for this imbalance in both groups was due to AEs. This imbalance may potentially bias the safety results, as patients who were still in the open-label extension phase had better tolerability of momelotinib than those who had discontinued. Another reason for discontinuation was disease progression, which was higher among those who switched from ruxolitinib to momelotinib or BAT to momelotinib than among those who continued on momelotinib in both the SIMPLIFY-1 and SIMPLIFY-2 trials. This difference may potentially bias the results, as patients who discontinued are likely to have had poorer outcomes if they were on momelotinib. In contrast, in the MOMENTUM trial, a greater proportion of patients who had received momelotinib in the double-blind phase discontinued treatment due to toxicity during the open-label extension phase than those treated with danazol in the double-blind phase.
External Validity
Because the patients who took part in the open-label extension phases were originally from the pivotal trials and the eligibility criteria remained the same, similar issues related to the generalizability of the enrolled patient population are expected for the open-label extension phases for all 3 studies as were identified for the randomized phases. The trials included patients who were transfusion dependent and patients who were transfusion independent, making the results generalizable to more patients.
Indirect Evidence
None.
Studies Addressing Gaps in the Systematic Review Evidence
The sponsor submitted 2 retrospective analyses and 1 set of interim results of an ongoing extended access study to address gaps related to long-term survival by baseline transfusion independence status.1,13,14 These studies are not included in the clinical review report as they provided supplementary evidence rather than addressing specific gaps in the evidence.
Discussion
Summary of Available Evidence
The clinical efficacy and safety of momelotinib were evaluated across 3 pivotal phase III RCTs: the SIMPLIFY-1, SIMPLIFY-2, and MOMENTUM trials. These studies enrolled adult patients with PMF, post-PV MF, or post-ET MF. The patient populations varied between not having been previous treated with JAK inhibitors (SIMPLIFY-1 trial [N = 432]) and having previously received JAK inhibitor treatment (SIMPLIFY-2 trial [N = 156] and MOMENTUM trial [N = 195]), with the MOMENTUM trial specifically targeting patients with symptomatic anemia. The trials compared momelotinib to relevant comparators: ruxolitinib in the SIMPLIFY-1 trial, BAT in the SIMPLIFY-2 trial, and danazol in the MOMENTUM trial. The primary outcomes were SRR in the SIMPLIFY-1 and SIMPLIFY-2 trials, with secondary outcomes related to transfusion and quality-of-life measures. The primary outcome in the MOMENTUM trial was TSS response, with secondary and exploratory outcomes covering SRR and transfusion measures.
The baseline characteristics were generally balanced across the treatment arms within each study, although there were some differences. The average age of the participants was approximately 65 to 71 years, with a higher proportion of males in most groups. Patients in the SIMPLIFY-2 and MOMENTUM trials had previously received JAK inhibitor treatment and had more severe anemia at baseline (mean hemoglobin level < 10 g/dL). Risk stratification showed a predominance of intermediate-2–risk MF in the SIMPLIFY-2 and MOMENTUM trials and high-risk MF in the SIMPLIFY-1 trial, with most patients having a confirmed JAK2V617F mutation across the trials. The MOMENTUM study uniquely included patients with significantly lower baseline hemoglobin levels, reflecting its focus on symptomatic anemia. Treatment discontinuation was considered high, imbalanced, or both in all the included studies.
Interpretation of Results
Efficacy
Across the 3 reported clinical trials — the MOMENTUM, SIMPLIFY-1, and SIMPLIFY-2 trials — there are consistent treatment effects demonstrating that momelotinib positively influences patients’ blood transfusion needs when compared with ruxolitinib among patients not previously treated with JAK inhibitors and when compared with BAT and danazol among patients who have previously received JAK inhibitor treatment. The positive trends in transfusion independence were also consistently observed in the subgroup analysis of patients with anemia (defined as having a hemoglobin level < 10 g/dL at baseline). The clinical experts consulted by CDA-AMC suggested that, due to the positive effect on blood transfusion needs, the use of momelotinib should be considered in patients who are at risk of anemia. However, the efficacy of momelotinib compared with ruxolitinib, BAT, and danazol on other outcomes, specifically SRR and HRQoL, as measured by the TSS response rate, is less consistent across the trials and must be interpreted within the context of the each of the studies’ eligibility criteria, populations, comparators, and limitations.
In the SIMPLIFY-1 trial, momelotinib achieved the primary outcome of showing noninferiority to ruxolitinib in SRR (26.5% versus 29.5% of patients who met this radiographic threshold for spleen shrinkage at the 24‑week landmark) but did not demonstrate superiority. In the SIMPLIFY-2 trial, momelotinib failed to meet the primary end point, and no significant difference in SRR was shown between momelotinib and BAT. These results indicate that, in patients not previously treated with JAK inhibitors, momelotinib likely provides similar SRR outcomes to ruxolitinib, as seen in the SIMPLIFY-1 trial. This suggests that momelotinib might be a reasonable alternative to ruxolitinib for reducing spleen size in this population. However, the SIMPLIFY-2 trial, which enrolled patients previously treated with JAK inhibitors and in which most of the patients in the BAT arm received ruxolitinib, demonstrated only a low certainty that momelotinib may result in little-to-no difference in SRR when compared to BAT. This outcome is confounded by the lack of a washout period at the beginning of the trial, and it is not clear how much of an effect is driven only by momelotinib in this patient population. In the MOMENTUM trial, momelotinib was shown to provide a likely improvement over danazol in SRR. However, this improvement is to be expected as danazol does not affect spleen size and is not used in practice to reduce spleen size.
The TSS response rate for momelotinib shows varying degrees of efficacy compared with ruxolitinib, BAT, and danazol across the trials. In the SIMPLIFY-1 trial, momelotinib was shown to demonstrate a poorer TSS response rate than ruxolitinib, indicating that while momelotinib may control spleen size effectively and reduce the need for blood transfusion, it may be less beneficial for symptom relief in patients not previously treated with JAK inhibitors. Conversely, the results of the SIMPLIFY-2 trial suggest that momelotinib may result in a better TSS response rate than BAT, suggesting that for patients who have been previously treated with JAK inhibitors, momelotinib may offer improved symptom management. However, with the confounding effects of an open-label study design, the lack of a washout period, and the large number of treatment discontinuations in the SIMPLIFY-2 trial, the certainty that momelotinib would improve HRQoL in this patient population is reduced. The MOMENTUM trial shows that there is likely a benefit of treatment with momelotinib in comparison with danazol in terms of TSS.
When considered within the outlined limitations on external validity, the results from the MOMENTUM trial become less applicable in the Canadian setting due to the nature of the comparator (danazol is not commonly used in Canadian practice) as well as the fact that hydroxy urea and ESA were forbidden, when these are commonly used in patients with anemia in Canadian practice. In contrast, that the majority of patients in the SIMPLIFY-2 trial received ruxolitinib is in line with Canadian practice.
However, there is no evidence to support the comparative efficacy or safety of momelotinib versus fedratinib. Moreover, there is insufficient information on the clinically important comparative outcomes of survival and leukemia progression. Specifically, the lack of long-term comparative results reduces the ability to assess and generalize important clinical outcomes that require a longer time horizon to be clinically meaningful. This is especially true considering there is a strong unmet need for an intervention that would increase OS.
Patient group input emphasizes the importance of HRQoL outcomes, particularly symptom management and the reduction of transfusion dependence, as critical measures of treatment success. While momelotinib likely reduces transfusion needs compared with ruxolitinib, BAT, and danazol, the TSS response varying in different studies may suggest that not all patient-prioritized outcomes are consistently improved with momelotinib.
Harms
Across the 3 clinical trials — the MOMENTUM, SIMPLIFY-1, and SIMPLIFY-2 trials — the safety profile of momelotinib was consistent with known AEs associated with JAK inhibitors; anemia, thrombocytopenia, and gastrointestinal events were among the AEs most frequently reported. The SIMPLIFY-1 and SIMPLIFY-2 trials showed a numerically higher proportion of patients treated with momelotinib experiencing SAEs than patients treated with ruxolitinib or BAT. Specifically, 22.9% of patients in the momelotinib group in the SIMPLIFY-1 trial experienced at least 1 SAE, compared to 18.1% of patients in the ruxolitinib group. In the SIMPLIFY-2 trial, the difference was more pronounced, with 35.6% of patients in the momelotinib group reporting SAEs versus 23.1% in the BAT group. However, this trend was not observed in the MOMENTUM trial, where the proportion of patients with SAEs in the momelotinib group was 34.6% and in the danazol group was 40.0%. There was no clear single SAE that accounted for the imbalance in SAEs between momelotinib and the comparators.
Across the 3 trials, the incidence of death varied, with the highest rates observed in the MOMENTUM trial, where 29.2% of patients in the momelotinib group and 30.8% in the danazol group died. In all trials, a significant proportion of deaths were attributed to TEAEs not related to disease progression. The mortality rates for momelotinib were, numerically, comparable to those for the comparators.
In the clinical trials for momelotinib, several AESIs were noted. Peripheral neuropathy was more common in patients treated with momelotinib, particularly in the SIMPLIFY-2 trial, where 11.5% of patients experienced this AE contrasted with 0 in the BAT group. In the SIMPLIFY-1 trial, peripheral neuropathy occurred in 10.3% of patients in the momelotinib group compared to 5.6% of patients in the ruxolitinib group. In the MOMENTUM trial, peripheral neuropathy was less frequent, reported in 3.8% of patients in the momelotinib group contrasted with 1.5% in the danazol group. Cataracts were observed in small percentages across the studies: 3.3% of patients in the momelotinib group in the SIMPLIFY-1 trial compared to 2.3% in the ruxolitinib group, and 3.8% of patients in the momelotinib group in the SIMPLIFY-2 trial. Infections were a notable AESI in the MOMENTUM trial, affecting 33.8% of patients in the momelotinib group, with a slightly higher incidence in the danazol group at 35.4%. However, infections were not reported for the SIMPLIFY-1 and SIMPLIFY-2 trials for either momelotinib or its respective comparators. Thromboembolism and transformation to AML were also reported, though these were relatively uncommon.
Conclusion
Three pivotal RCTs informed the efficacy and safety of momelotinib for the treatment of MF in adults compared to the following: ruxolitinib in patients not previously treated with a JAK inhibitor (SIMPLIFY-1 trial), BAT (which was mostly ruxolitinib) in patients who had been previously treated with a JAK inhibitor (SIMPLIFY-2 trial), and danazol in patients who had been previously treated with a JAK inhibitor and who had anemia (MOMENTUM trial). In patients not previously treated with a JAK inhibitor, momelotinib, as compared to ruxolitinib, resulted in an increased number of patients who were transfusion independent, likely led to little-to-no difference in SRR, but resulted in a decrease in the number of patients with a TSS response. In patients who had been previously treated with a JAK inhibitor, the evidence on the effect of momelotinib compared to BAT on the outcome of SRR was uncertain but showed that momelotinib may increase TSS response and may be likely to result in more patients who are transfusion independent. In patients who had been previously treated with a JAK inhibitor and who had anemia, compared to danazol, momelotinib may result in an increased number of patients with transfusion independence and is likely to increase the SRR and TSS response. As such, it is unclear if momelotinib offers an advantage over existing therapies in SRR or offers better symptom resolution compared to ruxolitinib in patients not previously treated with a JAK inhibitor. However, momelotinib is likely to result in an improvement in blood transfusion outcomes when compared to ruxolitinib.
The safety of momelotinib is in line with that of other JAK inhibitors. There is low-certainty evidence that momelotinib will lead to an increase or decrease in patients with SAEs compared to ruxolitinib, BAT, or danazol.
There is no evidence to inform the comparative efficacy and safety of momelotinib versus fedratinib, and there remains no long-term comparative data on important clinical outcomes such as survival and leukemia progression.
The results available from the long-term extension studies suggest that more than two-thirds of patients experienced sustained efficacy with momelotinib over extended periods. In the open-label extension phases of the SIMPLIFY-1, SIMPLIFY-2, and MOMENTUM trials, momelotinib appeared to provide ongoing benefits in terms of transfusion independence, splenic response, and symptom relief. Splenic responses were observed in a proportion of patients, particularly those continuing momelotinib treatment, and the majority of patients who were transfusion independent at week 24 maintained this status through extended follow-up.
References
1.Mesa R, Harrison C, Oh ST, et al. Overall survival in the SIMPLIFY-1 and SIMPLIFY-2 phase 3 trials of momelotinib in patients with myelofibrosis. Leukemia. 2022;36(9):2261-2268. doi:10.1038/s41375-022-01637-7 PubMed
2.Bose P, Verstovsek S. Management of Myelofibrosis-Related Cytopenias. Curr Hematol Malig Rep. 2018;13(3):164-172. doi:10.1007/s11899-018-0447-9 PubMed
3.Chifotides HT, Bose P, Verstovsek S. Momelotinib: an emerging treatment for myelofibrosis patients with anemia. J Hematol Oncol. 2022;15(1):7. doi:10.1186/s13045-021-01157-4 PubMed
4.Heppner J, Nguyen LT, Guo M, Naugler C, Rashid-Kolvear F. Incidence of myeloproliferative neoplasms in Calgary, Alberta, Canada. BMC Res Notes. 2019;12(1):286. doi:10.1186/s13104-019-4321-1 PubMed
5.CADTH. Reimbursement Review: fedratinib (Inrebic). Can J Health Technol. 2021;1(7):1-159. doi:10.51731/cjht.2021.124
6.National Comprehensive Cancer Network (NCCN). Myeloproliferative Neoplasms, version 3.2022. Updated September, 2022. Accessed by sponsor, no date provided. https://www.nccn.org/
7.Vannucchi AM, Barbui T, Cervantes F, et al. Philadelphia chromosome-negative chronic myeloproliferative neoplasms: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2015;26 Suppl 5:v85-99. doi:10.1093/annonc/mdv203 PubMed
8.Gupta V, Cerquozzi S, Foltz L, et al. Patterns of Ruxolitinib Therapy Failure and Its Management in Myelofibrosis: Perspectives of the Canadian Myeloproliferative Neoplasm Group. JCO Oncol Pract. 2020;16(7):351-359. doi:10.1200/JOP.19.00506 PubMed
9.Guideline Resource Unit, Cancer Care Alberta. Myelofibrosis: clinical practice guideline, version 2. Updated March, 2021. Accessed by sponsor, no date provided. https://www.albertahealthservices.ca/assets/info/hp/cancer/if-hp-cancer-guide-lyhe011-mf.pdf
10.Clinical Study Report: GS-US-352-0101. A Phase 3, Randomized, Double-blind Active-controlled Study Evaluating Momelotinib versus Ruxolitinib in Subjects with Primary Myelofibrosis (PMF) or Post-polycythemia Vera or Post-essential Thrombocythemia Myelofibrosis (Post-PV/ET MF). Sierra Oncology Inc.; March 30, 2021.
11.Clinical Study Report: GS-US-352-1214. A Phase 3, Randomized Study to Evaluate the Efficacy of Momelotinib Versus Best Available Therapy in Anemic or Thrombocytopenic Subjects with Primary Myelofibrosis, Post-polycythemia Vera Myelofibrosis, or Post-essential Thrombocythemia Myelofibrosis who were Treated with Ruxolitinib. Sierra Oncology, Inc.; January 13, 2021.
12.GSK Data on File. End of Study Clinical Report - A Randomized, Double-Blind, Phase 3 Study to Evaluate the Activity of Momelotinib (MMB) Versus Danazol (DAN) in Symptomatic, Anemic Subjects with Primary Myelofibrosis (PMF), Post-Polycythemia Vera (PV) Myelofibrosis, or Post-Essential Thrombocythemia (ET) Myelofibrosis who were Previously Treated with JAK Inhibitor Therapy. 2023 [sponsor provided reference].
13.Gangat N, Begna KH, Al-Kali A, et al. Predictors of anemia response to momelotinib therapy in myelofibrosis and impact on survival. Am J Hematol. 2023;98(2):282-289. doi:10.1002/ajh.26778 PubMed
14.Bartoszko J, Panzarella T, Lau A, et al. Effect of Red Blood Cell Transfusion Dependence on the Natural History of Myeloproliferative Neoplasm-Associated Myelofibrosis. Clin Lymphoma Myeloma Leuk. 2015;15(11):e151-e156. doi:10.1016/j.clml.2015.09.001 PubMed
15.Dunbar AJ, Rampal RK, Levine R. Leukemia secondary to myeloproliferative neoplasms. Blood. 2020;136(1):61-70. doi:10.1182/blood.2019000943 PubMed
16.Gangat N, Caramazza D, Vaidya R, et al. DIPSS plus: a refined Dynamic International Prognostic Scoring System for primary myelofibrosis that incorporates prognostic information from karyotype, platelet count, and transfusion status. J Clin Oncol. 2011;29(4):392-397. doi:10.1200/JCO.2010.32.2446 PubMed
17.Passamonti F, Cervantes F, Vannucchi AM, et al. Dynamic International Prognostic Scoring System (DIPSS) predicts progression to acute myeloid leukemia in primary myelofibrosis. Blood. 2010;116(15):2857-2858. doi:10.1182/blood-2010-06-293415 PubMed
18.Cervantes F, Dupriez B, Pereira A, et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood. 2009;113(13):2895-2901. doi:10.1182/blood-2008-07-170449 PubMed
19.Gerds AT, Bose P, Hobbs GS, et al. Treating Anemic Patients With Myelofibrosis in the New Janus Kinase Inhibitor Era: Current Evidence and Real-world Implications. Hemasphere. 2022;6(10):e778. doi:10.1097/HS9.0000000000000778 PubMed
20.Naymagon L, Mascarenhas J. Myelofibrosis-Related Anemia: Current and Emerging Therapeutic Strategies. Hemasphere. 2017;1(1):e1. doi:10.1097/HS9.0000000000000001 PubMed
21.Tefferi A. Primary myelofibrosis: 2023 update on diagnosis, risk-stratification, and management. Am J Hematol. 2023;98(5):801-821. doi:10.1002/ajh.26857 PubMed
22.Zahr AA, Salama ME, Carreau N, et al. Bone marrow fibrosis in myelofibrosis: pathogenesis, prognosis and targeted strategies. Haematologica. 2016;101(6):660-671. doi:10.3324/haematol.2015.141283 PubMed
23.Elena C, Passamonti F, Rumi E, et al. Red blood cell transfusion-dependency implies a poor survival in primary myelofibrosis irrespective of IPSS and DIPSS. Haematologica. 2011;96(1):167-170. doi:10.3324/haematol.2010.031831 PubMed
24.Bankar A, Zhao H, Iqbal J, et al. Healthcare resource utilization in myeloproliferative neoplasms: a population-based study from Ontario, Canada. Leuk Lymphoma. 2020;61(8):1908-1919. doi:10.1080/10428194.2020.1749607 PubMed
25.Bankar A, Chan WC, Liu N, Cheung M, Alibhai S, Gupta V. Prevalence of frailty and its association with clinical outcomes in myeloproliferative neoplasms: a population-based study. Blood Adv. 2023;7(17):5014-5026. doi:10.1182/bloodadvances.2023009825 PubMed
26.Leukemia & Lymphoma Society of Canada. Blood Cancer in Canada - Facts & Stats [sponsor provided reference].
27.Tefferi A, Lasho TL, Jimma T, et al. One thousand patients with primary myelofibrosis: the mayo clinic experience. Mayo Clin Proc. 2012;87(1):25-33. doi:10.1016/j.mayocp.2011.11.001 PubMed
28.Barraco D, Elala YC, Lasho TL, et al. Molecular correlates of anemia in primary myelofibrosis: a significant and independent association with U2AF1 mutations. Blood Cancer J. 2016;6(4):e415. doi:10.1038/bcj.2016.22 PubMed
29.CADTH. Reimbursement Recommendation final: fedratinib (Inrebic). Can J Health Technol. 2021;1(6):1-12. doi:10.51731/cjht.2021.86
30.Musiałek MW, Rybaczek D. Hydroxyurea-The Good, the Bad and the Ugly. Genes (Basel). 2021;12(7):1096. doi:10.3390/genes12071096 PubMed
31.Wang Q, Xiong B, Zheng C, Liang M, Han P. Splenic Arterial Embolization in the Treatment of Severe Portal Hypertension Due to Pancreatic Diseases: The Primary Experience in 14 Patients. Cardiovasc Intervent Radiol. 2016;39(3):353-358. doi:10.1007/s00270-015-1199-8 PubMed
32.He XH, Gu JJ, Li WT, et al. Comparison of total splenic artery embolization and partial splenic embolization for hypersplenism. World J Gastroenterol. 2012;18(24):3138-3144. doi:10.3748/wjg.v18.i24.3138 PubMed
33.Palandri F, Palumbo GA, Bonifacio M, et al. Baseline factors associated with response to ruxolitinib: an independent study on 408 patients with myelofibrosis. Oncotarget. 2017;8(45):79073-79086. doi:10.18632/oncotarget.18674 PubMed
34.Styczyński J, Tridello G, Koster L, et al. Death after hematopoietic stem cell transplantation: changes over calendar year time, infections and associated factors. Bone Marrow Transplant. 2020;55(1):126-136. doi:10.1038/s41409-019-0624-z PubMed
35.GSK Data on File. Statistical Analysis Plan Version 1.0 - A Phase 3, Randomized, Double-Blind Active-Controlled Study Evaluating Momelotinib vs. Ruxolitinib in Subjects with Primary Myelofibrosis (PMF) or Post-Polycythemia Vera or Post-Essential Thrombocythemia Myelofibrosis (Post-PV/ET MF). 2016 [sponsor provided reference].
36.GSK Data on File. Statistical Analysis Plan Version 1.0 - A Phase 3, Randomized Study To Evaluate the Efficacy of Momelotinib Versus Best Available Therapy in Anemic or Thrombocytopenic Subjects with Primary Myelofibrosis, Post-polycythemia Vera Myelofibrosis, or Post-essential Thrombocythemia Myelofibrosis who were Treated with Ruxolitinib. 2016 [sponsor provided reference].
37.GSK Data on File. Statistical Analysis Plan Version 2.0 - A Randomized, Double-Blind, Phase 3 Study to Evaluate the Activity of Momelotinib (MMB) versus Danazol (DAN) in Symptomatic, Anemic Subjects with Primary Myelofibrosis (PMF), Post-Polycythemia Vera (PV) Myelofibrosis, or Post Essential Thrombocythemia (ET) Myelofibrosis who were Previously Treated with JAK Inhibitor Therapy. 2021 [sponsor provided reference].
38.Novartis Pharmaceuticals Canada Inc. Jakavi (ruxolitinib): 5 mg, 10 mg, 15 mg and 20 mg, oral tablets [product monographs]. Updated 2023. Accessed by sponsor, no date provided.
39.Celgene Inc. Inrebic (fedratinib): 100 mg oral capsules [product monograph]. Updated July 23, 2020.
40.GlaxoSmithKline Inc. Ojjaara (momelotinib): 100 mg, 150 mg, and 200 mg, oral tablets [product monograph]. Updated November 8, 2024.
41.Gupta V, Oh S, Devos T, et al. Momelotinib vs. ruxolitinib in myelofibrosis patient subgroups by baseline hemoglobin levels in the SIMPLIFY-1 trial. Leuk Lymphoma. 2024;65(7):965-977. doi:10.1080/10428194.2024.2328800 PubMed
42.Oh ST, Verstovsek S, Gupta V, et al. Changes in bone marrow fibrosis during momelotinib or ruxolitinib therapy do not correlate with efficacy outcomes in patients with myelofibrosis. EJHaem. 2024;5(1):105-116. doi:10.1002/jha2.854 PubMed
43.Mesa RA, Kiladjian JJ, Catalano JV, et al. SIMPLIFY-1: A Phase III Randomized Trial of Momelotinib Versus Ruxolitinib in Janus Kinase Inhibitor-Naïve Patients With Myelofibrosis. J Clin Oncol. 2017;35(34):3844-3850. doi:10.1200/jco.2017.73.4418 PubMed
44.Harrison CN, Vannucchi AM, Platzbecker U, et al. Momelotinib versus best available therapy in patients with myelofibrosis previously treated with ruxolitinib (SIMPLIFY 2): a randomised, open-label, phase 3 trial. Lancet Haematol. 2018;5(2):e73-e81. doi:10.1016/s2352-3026(17)30237-5 PubMed
45.Verstovsek S, Gerds AT, Vannucchi AM, et al. Momelotinib versus danazol in symptomatic patients with anaemia and myelofibrosis (MOMENTUM): results from an international, double-blind, randomised, controlled, phase 3 study. Lancet. 2023;401(10373):269-280. doi:10.1016/s0140-6736(22)02036-0 PubMed
46.Gerds AT, Verstovsek S, Vannucchi AM, et al. Momelotinib versus danazol in symptomatic patients with anaemia and myelofibrosis previously treated with a JAK inhibitor (MOMENTUM): an updated analysis of an international, double-blind, randomised phase 3 study. Lancet Haematol. 2023;10(9):e735-e746. doi:10.1016/s2352-3026(23)00174-6 PubMed
47.Verstovsek S, Mesa R, Gupta V, et al. Momelotinib long-term safety and survival in myelofibrosis: integrated analysis of phase 3 randomized controlled trials. Blood Adv. 2023;7(14):3582-3591. doi:10.1182/bloodadvances.2022009311 PubMed
48.Mesa RA, Harrison C, Palmer JM, et al. Patient-reported Outcomes and Quality of Life in Anemic and Symptomatic Patients With Myelofibrosis: Results From the MOMENTUM Study. Hemasphere. 2023;7(11):e966. doi:10.1097/hs9.0000000000000966 PubMed
49.Verstovsek S, Chen CC, Egyed M, et al. MOMENTUM: momelotinib vs danazol in patients with myelofibrosis previously treated with JAKi who are symptomatic and anemic. Future Oncol. 2021;17(12):1449-1458. doi:10.2217/fon-2020-1048 PubMed
50.Sierra Oncology LLC - a GSK company. NCT04173494: A Study of Momelotinib Versus Danazol in Symptomatic and Anemic Myelofibrosis Participants (MOMENTUM). ClinicalTrials.gov. Updated November 1, 2023. Accessed February 16, 2024. https://clinicaltrials.gov/study/NCT04173494
51.Sierra Oncology LLC - a GSK company. NCT01969838: Momelotinib Versus Ruxolitinib in Subjects With Myelofibrosis (SIMPLIFY 1). ClinicalTrials.gov. Updated May 12, 2023. Accessed by sponsor, no date provided. https://clinicaltrials.gov/study/NCT01969838
52.Sierra Oncology LLC - a GSK company. NCT02101268: Efficacy of Momelotinib Versus Best Available Therapy in Anemic or Thrombocytopenic Subjects With Primary Myelofibrosis (MF), Post-polycythemia Vera MF, or Post-essential Thrombocythemia MF (SIMPLIFY 2). ClinicalTrials.gov. Updated May 23, 2023. Accessed Febrary 16, 2024. https://clinicaltrials.gov/study/NCT02101268
53.GSK Data on File. Final Clinical Study Protocol Amendment 3 - A Phase 3, Randomized, Double-blind Active-controlled Study Evaluating Momelotinib vs. Ruxolitinib in Subjects with Primary Myelofibrosis (PMF) or Post-polycythemia Vera or Post-essential Thrombocythemia Myelofibrosis (Post-PV/ET MF). 2017 [sponsor provided reference].
54.GSK Data on File. Final Clinical Study Protocol Amendment 2 - A Phase 3, Randomized Study To Evaluate the Efficacy of Momelotinib Versus Best Available Therapy in Anemic or Thrombocytopenic Subjects with Primary Myelofibrosis, Post-polycythemia Vera Myelofibrosis, or Post-essential Thrombocythemia Myelofibrosis who were Treated with Ruxolitinib. 2017 [sponsor provided reference].
55.GSK Data on File. Clinical Study Protocol Version 2.0 - A Randomized, Double-blind, Phase 3 Study to Evaluate the Activity of Momelotinib (MMB) versus Danazol (DAN) in Symptomatic, Anemic Subjects with Primary Myelofibrosis (PMF), Post-polycythemia Vera (PV) Myelofibrosis, or Post-essential Thrombocythemia (ET) Myelofibrosis who were Previously Treated with JAK Inhibitor Therapy. 2020 [sponsor provided reference].
56.CADTH. Drug Reimbursement Review clinical guidance report: ruxolitinib (Jakavi) for myelofibrosis. Updated January 14, 2013. Accessed by sponsor, no date provided. https://www.cda-amc.ca/sites/default/files/pcodr/pcodr-jakavi-myelofibro-fn-cgr.pdf
57.National Comprehensive Cancer Network (NCCN). Myeloproliferative Neoplasms, version 2.2023. Updated 2023. Accessed by sponsor, no date provided. https://www.nccn.org/
58.Mesa RA, Schwager S, Radia D, et al. The Myelofibrosis Symptom Assessment Form (MFSAF): an evidence-based brief inventory to measure quality of life and symptomatic response to treatment in myelofibrosis. Leuk Res. 2009;33(9):1199-1203. doi:10.1016/j.leukres.2009.01.035 PubMed
59.GSK Data on File. Meaningful Change Threshold Analysis Plan - A Randomized, Double-blind, Phase 3 Study to Evaluate the Activity of Momelotinib (MMB) versus Danazol (DAN) in Symptomatic, Anemic Subjects with Primary Myelofibrosis (PMF), Post-polycythemia Vera (PV) Myelofibrosis, or Post-essential Thrombocythemia (ET) Myelofibrosis who were Previously Treated with JAK Inhibitor Therapy. 2021 [sponsor provided reference].
60.GSK Data on File. A Phase 3, Randomized, Double-blind Active-controlled Study Evaluating Momelotinib versus Ruxolitinib in Subjects with Primary Myelofibrosis (PMF) or Post-polycythemia Vera or Post-essential Thrombocythemia Myelofibrosis (Post-PV/ET MF) - Anemic analysis set results. 2023 [sponsor provided reference].
61.GSK Data on File. A Phase 3, Randomized Study to Evaluate the Efficacy of Momelotinib Versus Best Available Therapy in Anemic or Thrombocytopenic Subjects with Primary Myelofibrosis, Post-polycythemia Vera Myelofibrosis, or Post-essential Thrombocythemia Myelofibrosis who were Treated with Ruxolitinib - Anemic analysis set results 2023 [sponsor provided reference].
62.Balshem H, Helfand M, Schünemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-406. doi:10.1016/j.jclinepi.2010.07.015 PubMed
63.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. doi:10.1016/j.jclinepi.2019.10.014 PubMed
Appendix 1: Overall Survival and Leukemia-Free Survival Long-Term Extension Study Data
Please note that this appendix has not been copy-edited.
Overall Survival
Table 28: OS in the Overall Treatment Perioda in the SIMPLIFY-1 Trial (Safety Analysis Set)
Overall survival (OS) | MMB (N = 214) | RUX (N = 216) |
|---|---|---|
Events (deaths), n (%) | 61 (28.5) | 66 (30.6) |
Deaths in double-blind phaseb | 8 (13.1) | 4 (6.1) |
Deaths in open-label phasec | 18 (29.5) | 17 (25.8) |
Deaths during follow-upd | 35 (57.4) | 45 (68.2) |
KM estimate of median OS (95% CI), months | NR (46.19 to NR) | 53.06 (48.72 to NR) |
HR (95% CI) | 0.99 (0.70 to 1.41) | |
Stratified log-rank test P value | 0.97 | |
CI = confidence interval; CSR = Clinical Study Report; DBL = database lock; HR = hazard ratio; KM = Kaplan-Meier; MMB = momelotinib; NR = not reached; OS = overall survival; RUX = ruxolitinib.
Note: Results of final analyses at the DBL of July 1, 2019.
aIncluded randomized and extended treatment phase.
bDeath in the double-blind phase is death that occurred on or after the first double-blind treatment dose up to the earliest of last dose in the double-blind phase + 30 days, or first dose in the open-label phase –1 day.
cDeath in the open-label phase is death that occurred on or after the first open-label phase dose up to the last open-label phase MMB dose + 30 days.
dDeath in the follow-up phase is the death that occurred after 30 days of the last dose in the double-blind or open-label phase, whichever the latest.
Source: GSK Data on File, 2021 (SIMPLIFY-1 Clinical Study Report).10
Table 29: OS in the Overall Treatment Perioda in the SIMPLIFY-2 Trial (Safety Analysis Set)
Overall survival (OS) | MMB (N = 104) | BAT (N = 52) |
|---|---|---|
Events (deaths), n (%) | 46 (44.2) | 23 (44.2) |
KM estimate of median OS (95% CI), months | 34.33 (27.33 to NR) | 37.52 (21.29 to NR) |
HR (95% CI)b | 0.96 (0.58 to 1.59) | |
Stratified log-rank test P valuec | 0.86 | |
BAT = best available therapy; CI = confidence interval; CSR = Clinical Study Report; DBL = database lock; HR = hazard ratio; ITT = intent to treat; KM = Kaplan-Meier; MMB = momelotinib; NE = not estimable; OS = overall survival; TD = transfusion dependence; TSS = total symptom score.
Note: Results from the final analysis at the DBL on June 25, 2019.
aIncluded randomized and extended treatment phase.
bAdjusted HR and 95% CI from Cox proportional hazard model with covariates of treatment, baseline TD (yes versus no), and baseline TSS (< 18 versus ≥ 18).
cStratified two-sided P value is from a log-rank test comparing treatment stratified by baseline TD (yes versus no) and baseline TSS (< 18 versus ≥ 18).
Source: GSK Data on File, 2021 (SIMPLIFY-2 Clinical Study Report).11
Table 30: OS in the Overall Treatment Perioda in the MOMENTUM Trial (ITT Analysis Set)
Overall survival (OS) | MMB (N = 130) | DAN (N = 65) |
|---|---|---|
Events (deaths), n (%) | 32 (29.2) | 20 (30.8) |
KM estimate of median OS (95% CI), days | 624.0 (582.0 to NE) | NE (471.0 to NE) |
HR (95% CI)b | 0.890 (0.504 to 1.572) | |
Stratified log-rank test P valuec | 0.6879 | |
CI = confidence interval; CSR = Clinical Study Report; DAN = danazol; DBL = database lock; HR = hazard ratio; ITT = intent to treat; KM = Kaplan-Meier; LCM = left costal margin; MFSAF = Myelofibrosis Symptom Assessment Form; MMB = momelotinib; NE = not estimable; OS = overall survival; RBC = red blood cell; TSS = total symptom score.
Note: Results from the final analysis at the DBL on January 17, 2023.
aIncluded randomized and open-label treatment phase.
bAdjusted HR and 95% CI from Cox proportional hazard model with a single factor of treatment group by MFSAF TSS baseline score (< 22 versus ≥ 22), baseline palpable spleen length below LCM (≥ 12 cm versus < 12 cm), and baseline RBC units transfused in the 8-week period before randomization (0, 1 to 4, and ≥ 5).
cStratified two-sided p value is from a log-rank test comparing treatment stratified by MFSAF TSS baseline score (< 22 versus ≥ 22), baseline palpable spleen length below LCM (≥ 12 cm versus < 12 cm), and baseline RBC units transfused in the 8-week period before randomization (0, 1 to 4, and ≥ 5).
Source: GSK Data on File, 2023 (MOMENTUM Clinical Study Report).12
Leukemia-Free Survival
Table 31: LFS in the Overall Treatment Perioda in the SIMPLIFY-1 Trial (ITT Analysis Set)b
Leukemia-free survival | MMB (N = 215) | RUX (N = 217) |
|---|---|---|
Events, n (%) | 67 (31.2) | 68 (31.3) |
Leukemic transformation | 12 (5.6) | 9 (4.1) |
Death | 55 (25.6) | 59 (27.2) |
KM estimate of median OS (95% CI), months | NR (44.09 to NR) | 53.06 (48.72 to NR) |
HR (95% CI)c | 1.07 (0.76 to 1.50) | |
Stratified log-rank test P valued | 0.70 | |
CI = confidence interval; CSR = Clinical Study Report; DBL = database lock; HR = hazard ratio; ITT = intent to treat; KM = Kaplan-Meier; MMB = momelotinib; NR = not reached; OS = overall survival; RUX = ruxolitinib; TD = transfusion dependence.
Note: Results from the final analysis at the DBL on July 1, 2019.
aIncluded randomized and extended treatment phase.
bInsert text
cAdjusted HR and 95% CI are from a Cox proportional hazards model with covariates of treatment, baseline TD (yes/no), and baseline platelet count (< 100, 100 to 200, > 200 × 109/L).
dStratified two-sided p value is from a log-rank test comparing treatment stratified by baseline TD (yes/no) and baseline platelet count (< 100, 100 to 200, > 200 × 109/L).
Source: GSK Data on File, 2021 (SIMPLIFY-1 Clinical Study Report).10
Table 32: LFS in the Overall Treatment Perioda in the SIMPLIFY-2 Trial (Safety Analysis Set)b
Leukemia-free survival (LFS) | MMB (N = 104) | BAT (N = 52) |
|---|---|---|
Events, n (%) | 48 (46.2) | 24 (46.2) |
Leukemic transformation | 7 (6.7) | 1 (1.9) |
Death | 41 (39.4) | 23 (44.2) |
KM estimate of median LFS (95% CI), months | 33.97 (27.33 to NR) | 37.52 (20.27 to NR) |
HR (95% CI)c | 0.95 (0.58 to 1.57) | |
Stratified log-rank test P valued | 0.85 | |
BAT = best available therapy; CI = confidence interval; CSR = Clinical Study Report; DBL = database lock; HR = hazard ratio; ITT = intent to treat; KM = Kaplan-Meier; LFS = leukemia-free survival; MMB = momelotinib; NE = not estimable; TD = transfusion dependence; TSS = total symptom score.
Note: Results from the final analysis at the DBL on 25 June 2019.
aIncluded randomized and extended treatment phase.
bInsert text
cAdjusted HR and 95% CI from Cox proportional hazard model with covariates of treatment, baseline TD (yes versus no), and baseline TSS (< 18 versus ≥ 18).
dStratified two-sided p value is from a log-rank test comparing treatment stratified by baseline TD (yes versus no) and baseline TSS (< 18 versus ≥ 18).
Source: GSK Data on File, 2021 (SIMPLIFY-2 Clinical Study Report).11
Table 33: LFS in the Overall Treatment Perioda in the MOMENTUM Trial (ITT Analysis Set)
Leukemia-free survival (LFS) | MMB (N = 130) | DAN (N = 65) |
|---|---|---|
Events, n (%) | 40 (30.8) | 22 (33.8) |
KM estimate of median LFS (95% CI), days | 624.0 (582.0 to NE) | NE (471.0 to NE) |
HR (95% CI)b | 0.804 (0.466 to 1.386) | |
Stratified log-rank test P valuec | 0.4320 | |
CI = confidence interval; CSR = Clinical Study Report; DAN = danazol; DBL = database lock; HR = hazard ratio; ITT = intent to treat; KM = Kaplan-Meier; LCM = left costal margin; LFS = leukemia-free survival; MFSAF = Myelofibrosis Symptom Assessment Form; MMB = momelotinib; NE = not estimable; RBC = red blood cell; TSS = total symptom score.
Note: Results from the final analysis at the DBL on January 17, 2023.
aIncluded randomized and open-label treatment phase.
bAdjusted HR and 95% CI from Cox proportional hazard model with a single factor of treatment group by MFSAF TSS baseline score (< 22 versus ≥ 22), baseline palpable spleen length below LCM (≥ 12 cm versus < 12 cm), and baseline RBC units transfused in the 8-week period before randomization (0, 1 to 4, and ≥ 5).
cStratified two-sided p value is from a log-rank test comparing treatment stratified by MFSAF TSS baseline score (< 22 versus ≥ 22), baseline palpable spleen length below LCM (≥ 12 cm versus < 12 cm), and baseline RBC units transfused in the 8-week period before randomization (0, 1 to 4, and ≥ 5).
Source: GSK Data on File, 2023 (MOMENTUM Clinical Study Report).12
Pharmacoeconomic Review
Abbreviations
AE
adverse event
BAT
best available therapy
BIA
budget impact analysis
CDA-AMC
Canada’s Drug Agency
ICER
incremental cost-effectiveness ratio
JAK
Janus kinase
MF
myelofibrosis
OS
overall survival
QALY
quality-adjusted life-year
RBC
red blood cell
TD
transfusion dependent
TI
transfusion independent
TR
transfusion requiring
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Momelotinib (Ojjaara), 200 mg, 150 mg, and 100 mg tablets |
Indication | For the treatment of splenomegaly and/or disease-related symptoms, in adult patients with intermediate or high-risk primary myelofibrosis (MF), post polycythemia vera MF or post essential thrombocythemia MF who have moderate to severe anemia |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | November 8, 2024 |
Reimbursement request | As per indication |
Sponsor | GlaxoSmithKline Inc. |
Submission history | Previously reviewed: No |
NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Markov model |
Target population | Adult patients with intermediate-risk or high-risk primary myelofibrosis, post–polycythemia vera myelofibrosis, or post–essential thrombocythemia myelofibrosis who have moderate to severe anemia |
Treatment | Momelotinib |
Dose regimen | 200 mg daily |
Submitted price | $230.86 per tablet |
Submitted treatment cost | $6,464.11 per 28-day cycle |
Comparators | Patients not previously treated with JAK inhibitors: ruxolitinib Patients previously treated with JAK inhibitors: BAT |
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, life-years |
Time horizon | Lifetime (33 years) |
Key data sources | The SIMPLIFY-1 and SIMPLIFY-2 trials were used to inform efficacy and safety data for the patient populations who, respectively, had not and had previously been treated with JAK inhibitors. The MOMENTUM trial was used to identify relevant adverse events. |
Submitted results | In the blended population (JAK inhibitor naive [15%] and JAK inhibitor experienced [85%]), momelotinib was dominant relative to ruxolitinib or BAT (cost savings: $138,451; incremental QALYs: 0.092). |
Key limitations |
|
CDA-AMC reanalysis results |
|
BAT = best available therapy; CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; JAK = Janus kinase; QALY = quality-adjusted life-year.
Conclusions
The Clinical Review of the SIMPLIFY-1 and SIMPLIFY-2 trials by Canada’s Drug Agency (CDA-AMC) concluded that there is moderate to high certainty that momelotinib will provide benefits to patients in terms of transfusion independence and reduction of transfusion needs in adults with myelofibrosis (MF) and that there is moderate evidence showing a benefit in disease-related symptoms compared to best available therapy (BAT). The Clinical Review further concluded that momelotinib was noninferior to ruxolitinib in terms of splenic response and that ruxolitinib improved disease-related symptoms compared to momelotinib. There was no long-term comparative evidence for momelotinib compared to BAT, ruxolitinib, or any other Janus kinase (JAK) inhibitor treatment. Given that 98% of the incremental quality-adjusted life-years (QALYs) estimated for momelotinib versus BAT were accrued through extrapolation beyond the period of the trial evidence, the lack of long-term comparative evidence contributes much uncertainty to the cost-effectiveness of momelotinib.
While incremental QALYs were similar in the sponsor’s base case and the CDA-AMC base case, momelotinib was no longer cost-saving in the CDA-AMC base case due to the inclusion of ruxolitinib as subsequent therapy. In the CDA-AMC reanalysis for patients not previously treated with JAK inhibitors, momelotinib was more costly and more effective than ruxolitinib (incremental costs: $23,841; incremental QALYs: 0.097), with an incremental cost-effectiveness ratio (ICER) of $245,628 per QALY gained. In the CDA-AMC reanalysis for patients previously treated with JAK inhibitors, momelotinib was more costly and more effective than BAT (incremental costs: $30,087; incremental QALYs: 0.092). Using publicly available list prices for momelotinib and all comparators, a price reduction of approximately 27% was needed for momelotinib to be cost-effective compared to ruxolitinib and BAT at a willingness-to-pay threshold of $50,000 per QALY gained.
Treatment with momelotinib is more costly than treatment with ruxolitinib in patients with MF who have not previously been treated with JAK inhibitors. The clinical evidence suggests that in such patients, momelotinib and ruxolitinib result in similar splenic response but that ruxolitinib has a more favourable disease-related symptom profile. In the sponsor and the CDA-AMC base cases, estimates of incremental QALYs were small and highly uncertain due to a lack of long-term comparative evidence and a model that did not include splenic response (i.e., the noninferiority outcome that was measured in the SIMPLIFY-2 trial). As such, there is no robust clinical or economic evidence to support a price premium for momelotinib compared with ruxolitinib.
Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, clinician groups, and drug plans that participated in the CDA-AMC review process.
Patient group input was received jointly from the Leukemia & Lymphoma Society of Canada and the Canadian MPN Network and additionally from Heal Canada. Information was gathered from online surveys and interviews. Overall, patient group input noted that MF is a disease associated with high disease burden, reduced quality of life, and shorted survival. Patient groups indicated that JAK inhibitors improved the treatment landscape of MF; however, they are not curative. As such, patients expressed a desire for additional treatments that could improve clinical outcomes with minimal adverse events (AEs). Of the respondents, a small number of patients (21 in total) had experience with momelotinib. The majority felt that momelotinib improved their quality of life but commented that patients’ experiences can vary and expressed the importance of having continued alternative treatment options.
Clinician group input was received by the Leukemia & Lymphoma Society of Canada Clinical Network and the Ontario Health – Cancer Care Ontario Hematology Cancer Disease Site Drug Advisory Committee. Both groups noted that MF is a complex and debilitating disease that impacts a patient’s quality of life. It was noted that current treatments include ruxolitinib, fedratinib, and supportive care (such as transfusion and erythropoietin) with the goal of treatment being to reduce symptoms, improve blood counts or splenomegaly, and reduce the number of transfusions required. Clinician group input noted that many patients still experience a lack of effective therapy for their disease and that clinical trial data for momelotinib shows that it has promise in being a novel drug that can target some of the unmet needs of patients.
Drug plan input noted that there is a lack of comparative data between momelotinib and fedratinib, which would be a relevant comparator in this patient population. The plans further asked for clarification on which patients should receive momelotinib and if patients currently receiving alternative therapies for MF would also be eligible for treatment with momelotinib. The drug plans asked about the anticipated place in therapy for momelotinib and what alternative treatments options are available following disease progression on momelotinib. The drug plans commented that confidential pricing exists for ruxolitinib and fedratinib.
The following concern was addressed in the sponsor’s model:
Health-related quality of life was incorporated in the sponsor’s model by use of EQ-5D-5L data captured in the SIMPLIFY-1 and SIMPLIFY-2 trials for patients who, respectively, have not and have previously been treated with JAK inhibitors.
CDA-AMC was unable to address the following concerns raised from input relevant to the economic review:
The uncertainty associated with the long-term efficacy of momelotinib could not be addressed due to a lack of long-term data.
The lack of comparative evidence between momelotinib and fedratinib could not be addressed.
Economic Review
This review is for momelotinib (Ojjaara) for the treatment of splenomegaly and/or disease-related symptoms in adult patients with intermediate-risk or high-risk primary MF, post–polycythemia vera MF, or post–essential thrombocythemia MF who have moderate to severe anemia.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis to assess the cost-effectiveness of momelotinib versus ruxolitinib (among patients not previously treated with JAK inhibitors) and versus BAT (among patients previously treated with JAK inhibitors) for the treatment of splenomegaly and/or disease-related symptoms in adult patients with intermediate-risk or high-risk primary MF, post–polycythemia vera MF, or post–essential thrombocythemia MF who have moderate to severe anemia. The model population aligns with the Health Canada indication.1
Momelotinib is available as 100 mg, 150 mg, and 200 mg oral tablets.2 The recommended dosage is 200 mg orally once daily; complete blood cell count and liver function tests must be performed before initiation and periodically during treatment.2 The submitted price for momelotinib is $230.8610 per tablet or $6,464.11 per 28-day cycle.1 The BAT used in the SIMPLIFY-2 trial was used to inform the BAT in the model. It was assumed that patients could receive more than 1 therapy as BAT.
The outcomes of the model included QALYs and life-years over a time horizon of 33 years. Discounting (1.5% per annum) was applied for both costs and outcomes, and a cycle length of 4 weeks was used with a half-cycle correction applied.1
Model Structure
The sponsor submitted a Markov model to track a cohort of patients with MF.1 As noted in Figure 1, the model consists of 2 submodels (1 for patients who have not previously received JAK inhibitor treatment and 1 for patients who have), each consisting of 4 health states: transfusion independent (TI), transfusion requiring (TR), transfusion dependent (TD), and death.1 TI status was defined as an absence of red blood cell (RBC) transfusion and no hemoglobin level below 8 g/dL in the prior 3 model cycles. TD status was defined as at least 4 units of RBC transfusion or a hemoglobin level below 8 g/dL in the prior 2 model cycles. TR status was defined as not meeting the TI or TD criteria. Movement between each health state was determined by transition probabilities informed by the SIMPLIFY-1 and SIMPLIFY-2 trials.1
Patients were distributed among each health state based on baseline observations in the SIMPLIFY-1 and SIMPLIFY-2 trials. From each health state, patients could remain or transition to any of the other transfusion states (e.g., if a patient started in the TI state, they could remain in that state or transition to the TD or TR states). At any point in the model, patients could transition to death.
Model Inputs
The baseline population characteristics used to inform the model were based on the SIMPLIFY-1 trial for the JAK inhibitor–naive population (mean age: 64.7 years; male: 56.5%) and on the SIMPLIFY-2 trial for the JAK inhibitor–experienced population (mean age: 67.4 years; male: 59.6%).
Efficacy in the model was represented by achieving and maintaining TI status. The initial distribution of patients into the model health states was informed by observations at baseline in the SIMPLIFY-1 and SIMPLIFY-2 trials. Generally, transition data for the first 6 cycles (i.e., 24 weeks) were generated from patient-level data from the SIMPLIFY-1 and SIMPLIFY-2 trials. However, in the absence of data for the first 12 weeks due to the trial definition of TI status, the sponsor assumed that patients would experience no change in baseline during this time.
In the absence of data beyond 24 weeks, the sponsor used extrapolation methods to inform transition probabilities for momelotinib and its comparators. Specifically, transition count data for momelotinib and for BAT or ruxolitinib were pooled for cycles 5 and 6. The sponsor noted that doing so increased the sample size. Health state transitions were assumed to be the same regardless of treatment from cycle 6 (i.e., 24 weeks) onward.
The sponsor fitted parametric survival curves to Kaplan-Meier estimates derived from overall survival (OS) time-to-event data from the SIMPLFY trials. The transfusion status of patients assessed at 24 weeks was used to stratify OS data. The sponsor assumed that patients in the same health state regardless of treatment had equal mortality. The parametric survival distributions used in the base case were selected based on fit statistics, visual inspection, and clinical and external validity. The curve selections used in the sponsor’s base-case analysis for the JAK inhibitor–naive and JAK inhibitor–experienced populations can be found in Table 10.
Treatment discontinuation for momelotinib and ruxolitinib was applied in the sponsor’s model based on extrapolated time to treatment discontinuation data from the SIMPLIFY-1 and SIMPLIFY-2 trials. Refer to Table 11 for the curve selections used to inform the sponsor’s base-case analysis for the JAK inhibitor–naive and JAK inhibitor–experienced populations. It was assumed that patients treated with BAT did not experience treatment discontinuation. Treatment discontinuation was assumed to have no impact on a patient’s health state.
The model included grade 3 or 4 AEs occurring in 5% of patients in any treatment arm in the SIMPLIFY-1, SIMPLFY-2, and MOMENTUM trials, including anemia, thrombocytopenia, asthenia, and neutropenia. AE rates were informed by the SIMPLIFY-1 and SIMPLIFY-2 trials, depending on the subpopulation’s JAK inhibitor experience. The sponsor used a hemoglobin level of less than 12 g/dL to inform treatment-specific AE rates in the base-case analysis.
Health state utility values in the model were informed by EQ-5D-5L health index scores from the SIMPLIFY trials and adjusted using Canadian tariffs from Xie et al.3 For the JAK inhibitor–naive subgroup, patients in the TI, TR, and TD health states had utility values of ████, ████, and ████, respectively. For the JAK inhibitor–experienced subgroup, patients in the TI, TR, and TD health states had a utility value of ████, ████, and ████, respectively.1 Additional disutilities associated with AEs such as anemia, thrombocytopenia, asthenia, and neutropenia were included and informed by published literature.4-6
The costs included in the model consisted of treatment acquisition costs, administration costs, RBC transfusion costs, subsequent therapy costs, monitoring and disease management costs, and AEs. Acquisition costs were based on the sponsor’s submitted price for momelotinib. Acquisition costs for ruxolitinib and BAT were informed by either the Ontario Drug Benefit Formulary or the Ontario Exceptional Access Program pricing list.7,8 Oral treatments were assumed to have no administration cost, with the exception of thalidomide, which was assumed to receive an oral outpatient chemotherapy administration cost.1 Subcutaneous injections were assumed to use prefilled syringes, and patients receiving such injections were assumed to incur a one-off administration fee for initial education and support. The cost was assumed to be 20 minutes of a registered nurse’s time.1 The model considered subsequent therapy after patients discontinued their initial therapy. Patients receiving first-line momelotinib received BAT as subsequent therapy. Patients receiving first-line ruxolitinib also received BAT as subsequent therapy, and some of these patients received subsequent ruxolitinib as part of BAT. Patients previously treated with JAK inhibitors who received momelotinib in the second line were assumed to receive subsequent BAT. Patients previously treated with JAK inhibitors who received BAT were assumed to remain on BAT. Resource use and costs associated with RBC transfusions were applied to patients for each cycle of the model. RBC unit costs and transfusion fees were informed by a previous CADTH review. The frequency of RBC transfusion per health state was informed by the SIMPLIFY-1 and SIMPLIFY-2 trials, where patients with TR status were assumed to require ██ RBC unit every 3 months and patients with TD status were assumed to require approximately ███ RBC units every month.1
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically with 5,000 iterations. The deterministic results were aligned with submitted probabilistic results. The probabilistic findings are presented in the following sections.
Base-Case Results
The sponsor considered a “blended” population that comprised 15% of patients not previously treated with JAK inhibitors and 85% of patients who had previously been treated with JAK inhibitors. In the sponsor’s probabilistic base-case analysis, for the blended population, momelotinib when compared to ruxolitinib and BAT was associated with an additional 0.092 QALYs at lower costs (incremental costs: –$138,451). Therefore, momelotinib was dominant relative to ruxolitinib and BAT. Nearly all (approximately 98%) of the incremental QALYs for momelotinib were estimated through extrapolation beyond the trial period.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER vs. BAT and RUX ($/QALY) |
|---|---|---|---|---|---|
BAT and RUX blended | 320,184 | Reference | 2.934 | Reference | Reference |
MMB blended | 181,734 | −138,451 | 3.026 | 0.092 | Dominant |
BAT = best available therapy; ICER = incremental cost-effectiveness ratio; MMB = momelotinib; QALY = quality-adjusted life-year; RUX = ruxolitinib; vs. = versus.
Source: Sponsor’s pharmacoeconomic submission.1
Sensitivity and Scenario Analysis Results
In addition to the base-case analysis, the sponsor conducted several scenario analyses. The analyses conducted included those that examined the impact of alternative discount rates, the impact of JAK inhibitor status, alternative efficacy sources, alternative mortality assumptions, and alternative utility assumptions. No scenario had a significant impact on the relative cost-effectiveness of momelotinib versus ruxolitinib and BAT.
The sponsor additionally conducted a scenario analysis from a societal perspective, which included additional costs associated with productivity loss and caregivers. In that analysis, momelotinib was associated with an additional 0.11 QALYs for a cost reduction of $141,479 relative to ruxolitinib and BAT. The results were similar to the sponsor’s base-case analysis using a health care payer perspective where momelotinib was dominant versus ruxolitinib and BAT.
CDA-AMC Appraisal of the Sponsor’s Economic Evaluation
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications for the economic analysis:
The long-term efficacy and survival benefit of momelotinib is uncertain. The sponsor’s submitted economic analysis models efficacy through achieving and maintaining TI status. Data from the SIMPLIFY-1 and SIMPLIFY-2 trials were used to inform baseline distribution and transitions between the transfusion health states for the first 24 weeks in the JAK inhibitor–naive and JAK inhibitor–experienced groups, respectively. In the absence of data beyond 24 weeks, the sponsor applied pooled transition data from momelotinib and BAT or ruxolitinib by subpopulation at cycles 5 to 6 to both treatment arms for the remainder of the time horizon, thus assuming no additional benefit for momelotinib after cycle 6. In the absence of long-term efficacy data informing how momelotinib impacts patients’ long-term transfusion status, the long-term efficacy of momelotinib is uncertain. The modelled outcomes are highly sensitive to the proportion of patients achieving and maintaining transfusion independence over time, and 98% of incremental QALYs were estimated during the extrapolated period beyond cycle 6.
Additionally, the sponsor assumed that patients would not move to a different health state when they discontinued treatment (informed by extrapolation of time to discontinuation data from the SIMPLIFY trials). This assumption means the sponsor’s submitted economic analysis does not account for treatment waning. Clinical expert feedback noted that the efficacy of JAK inhibitor treatments is not expected to persist indefinitely, and therefore the sponsor’s model may overestimate the long-term benefit of momelotinib over a lifetime horizon.
Mortality in the model was derived by data from the SIMPLIFY trials. Specifically, the sponsor estimated OS in terms of health state (i.e., TI or non-TI status [TR status and TD status assumed to be the same]) by JAK inhibitor status. Because crossover was allowed in the SIMPLIFY trials, OS data for patients treated with ruxolitinib or BAT beyond week 24 was unavailable. In the absence of long-term survival data, the sponsor used parametric distributions fitted to OS Kaplan-Meier data from the trials (Table 10). Clinical expert feedback received by CDA-AMC noted that while the proposed OS extrapolations by the sponsor appear reasonable, due to the lack of long-term data from the SIMPLIFY trials relating to OS on momelotinib, there remains uncertainty in the long-term effect of momelotinib on mortality.
CDA-AMC was unable to address this limitation due to the absence of long-term data about the duration of momelotinib treatment response.
Use of transfusion dependence alone does not accurately reflect the clinical management of MF. Clinical effectiveness in the sponsor’s model was modelled through a patient’s ability to achieve and maintain TI status, which was defined as the absence of RBC transfusions and no hemoglobin levels less than 8 g/dL in the prior 12 weeks (i.e., 3 model cycles). Despite the primary outcome of the SIMPLIFY trials being splenic response, the sponsor noted that splenic response was not used to model efficacy for the following reasons: momelotinib is expected to have similar efficacy to ruxolitinib on splenic effect; clinical expert feedback received by the sponsor noted that treating anemia and its associated symptoms would be considered as clinically relevant as spleen volume reduction; and feedback from advisory boards noted that the TI end point of the SIMPLIFY trials would be considered the most clinically relevant end point. Clinical expert feedback received by CDA-AMC indicated that transfusion status is an important element to consider for the clinical management of MF; however, treatment decisions are generally based on many outcomes, including patient-reported symptoms and spleen volume. This is consistent with the National Comprehensive Cancer Network’s 2024 guidelines, which recommend that response assessment should be based on the improvement of disease-related symptoms.9 While the sponsor captured symptoms and spleen response indirectly in the model through health-related quality-of-life assessments, the model did not reflect how treatment decisions are made in clinical practice, which introduces uncertainty around health state occupancy and, consequently, the incremental costs and QALYs associated with treatment.
CDA-AMC was unable to address this limitation.
Subsequent therapy for patients treated with momelotinib may not reflect clinical practice. In the sponsor’s submitted base-case analysis, patients were assumed to receive subsequent treatment upon discontinuation of initial therapy. Patients not previously treated with JAK inhibitors who were treated with first-line momelotinib were assumed to receive BAT, excluding an alternative JAK inhibitor. The exclusion of an alternative JAK inhibitor as a subsequent therapy option was due to the following assumptions made by the sponsor: patients treated with momelotinib would not be suitable for JAK inhibitor re-treatment as disease progression due to worsening of splenomegaly or anemia would not be alleviated by an alternate JAK inhibitor therapy; there are reimbursement restrictions on ruxolitinib and fedratinib in some jurisdictions for patients experiencing disease progression on a prior JAK inhibitor. In contrast, patients not previously treated with JAK inhibitors who received first-line ruxolitinib were modelled to transition to subsequent BAT including a JAK inhibitor. Approximately 88% of subsequent therapy was assumed to be ruxolitinib. Clinical expert feedback received by CDA-AMC noted that in current practice in Canada, patients who experience disease progression may continue to receive suboptimal ruxolitinib or transition to an alternative JAK inhibitor (e.g., fedratinib). As such, in the absence of data suggesting otherwise, the clinical experts did not agree with the assumption that patients who received momelotinib in the first line would not receive subsequent JAK inhibitor treatment.
In the JAK inhibitor–experienced population, a similar subsequent therapy assumption was made for patients on momelotinib, where subsequent therapy consisted of BAT without a JAK inhibitor. Patients previously treated with a JAK inhibitor who received BAT (with JAK inhibitor) were assumed to maintain BAT (with JAK inhibitor). Clinical expert feedback stated that while not ideal, in the absence of alternative therapies, patients may continue to receive BAT consisting of a JAK inhibitor. Therefore, the sponsor’s assumption that patients with or without JAK inhibitor experience who are on momelotinib cannot go on to receive BAT with JAK inhibitor may underestimate the subsequent therapy cost associated with momelotinib. This assumption would introduce a bias that favours the cost-effectiveness of momelotinib. In the absence of robust evidence to inform the use of JAK inhibitor therapy following disease progression on momelotinib, the true impact of subsequent therapy in the submitted analysis remains uncertain.
In reanalysis, CDA-AMC included the cost of ruxolitinib as part of BAT subsequent therapy for patients both with and without JAK inhibitor experience who were receiving momelotinib using a built-in option in the sponsor’s model.
CDA-AMC considered a scenario analysis where patients continued to receive suboptimal momelotinib after disease progression, using the cost of momelotinib to inform the JAK inhibitor portion of BAT subsequent therapy for patients previously treated with JAK inhibitors.
Relevant comparators were excluded. The sponsor’s pharmacoeconomic submission assessed the cost-effectiveness analysis of momelotinib versus ruxolitinib (JAK inhibitor–naive population) or BAT (JAK inhibitor–experienced population). Fedratinib was excluded based on lack of clinical differentiation from ruxolitinib, absence of comparative data to momelotinib in patients with MF and anemia, and increased drug costs. Although CDA-AMC approved the sponsor’s request to exclude fedratinib from its analysis in a deviation request, clinical expert feedback received by CDA-AMC noted that fedratinib remains an option for this patient population and therefore is a relevant comparator for momelotinib despite funding criteria being limited.
CDA-AMC was unable to address this limitation. The cost-effectiveness of momelotinib versus fedratinib is unknown.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CDA-AMC (refer to Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CDA-AMC comment |
|---|---|
A “blended” population was applied with 15% patients who had not previously treated with JAK inhibitors and 85% of patients who had been previously treated with JAK inhibitors. | Reasonable. |
Grade ≥ 3 AEs reported in > 5% of patients were modelled and assumed to incur a utility decrement per cycle. | Reasonable. Clinical expert feedback received by CDA-AMC noted that AEs may occur at any point. |
AE = adverse event; CDA-AMC = Canada’s Drug Agency; JAK = Janus kinase.
CDA-AMC Reanalyses of the Economic Evaluation
Base-Case Results
The CDA-AMC base case was derived by making changes in model parameter values and assumptions, in consultation with clinical experts (Table 5). Several limitations with the sponsor’s submission could not be adequately addressed (i.e., exclusion of relevant comparators, uncertainty regarding long-term efficacy of momelotinib, and uncertainty in the model structure). CDA-AMC conducted stepped analysis that separated the JAK inhibitor–naive and JAK inhibitor–experienced populations and assumed alternative subsequent therapies for patients receiving momelotinib.
Details for each stepwise change to derive the CDA-AMC analysis and summary results of the CDA-AMC reanalysis are presented in Table 6.
Table 5: CDA-AMC Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Changes to derive the CDA-AMC base case | ||
1a. Subpopulation | Blended | JAK inhibitor naive |
1b. Subpopulation | JAK inhibitor experienced | |
2. Subsequent therapies | Exclude ruxolitinib as a subsequent therapy for patients after momelotinib | Include ruxolitinib as a subsequent therapy for patients after momelotinib |
CDA-AMC base case | ― | Reanalysis 1 + 2 |
CDA-AMC = Canada’s Drug Agency; JAK = Janus kinase.
For the JAK inhibitor–naive population, momelotinib was associated with an ICER of $245,628 per QALY gained compared to ruxolitinib (incremental costs: $23,841; incremental QALYs: 0.097). For the JAK inhibitor–experienced population, momelotinib was associated with an ICER of $327,295 per QALY gained compared to ruxolitinib (incremental costs: $30,087; incremental QALYs: 0.092). Incremental costs in both analyses were primarily due to the drug acquisition cost of momelotinib and subsequent treatment. Incremental QALYs in both analyses were primarily due to more patients achieving transfusion independence. While the incremental QALY results were similar to the sponsor’s submitted base case, the inclusion of subsequent ruxolitinib costs in the momelotinib arm changed the incremental cost results for momelotinib from being cost-saving to more costly than ruxolitinib or BAT in the JAK inhibitor–naive and JAK inhibitor–experienced populations, respectively.
Table 6: Summary of the Stepped Analysis of the CDA-AMC Reanalysis Results
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
Sponsor’s base case (blended analysis; probabilistic) | BAT and ruxolitinib | 320,184 | 2.934 | Reference |
Momelotinib | 181,734 | 3.026 | Dominant | |
CDA-AMC reanalysis 1a (JAK inhibitor naive) | Ruxolitinib | 404,150 | 4.013 | Reference |
Momelotinib | 227,025 | 4.099 | Dominant | |
CDA-AMC reanalysis 1b (JAK inhibitor experienced) | BAT | 307,717 | 2.814 | Reference |
Momelotinib | 172,750 | 2.928 | Dominant | |
CDA-AMC reanalysis 2 | Ruxolitinib | 322,346 | 2.974 | Reference |
Momelotinib | 352,630 | 3.083 | 277,771 | |
CDA-AMC base case, JAK inhibitor naive (reanalysis 1a + 2) | Ruxolitinib | 404,150 | 4.013 | Reference |
Momelotinib | 426,276 | 4.099 | 259,380 | |
CDA-AMC base case, JAK inhibitor naive (reanalysis 1a + 2; probabilistic) | Ruxolitinib | 419,050 | 4.187 | Reference |
Momelotinib | 442,890 | 4.284 | 245,628 | |
CDA-AMC base case, JAK inhibitor experienced (reanalysis 1b + 2) | BAT | 307,717 | 2.814 | Reference |
Momelotinib | 339,459 | 2.928 | 278,761 | |
CDA-AMC base case, JAK inhibitor experienced (reanalysis 1b + 2; probabilistic) | BAT | 303,606 | 2.748 | Reference |
Momelotinib | 333,694 | 2.840 | 327,295 |
BAT = best available therapy; CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; JAK = Janus kinase; QALY = quality-adjusted life-year.
Notes: The CDA-AMC reanalysis is based on publicly available prices of the comparator treatments. The results of all steps are presented deterministically unless otherwise indicated; the cumulative CDA-AMC base case is always presented both deterministically and probabilistically.
Scenario Analysis Results
CDA-AMC conducted a scenario analysis to explore the impact of assuming that patients previously treated with JAK inhibitors who were on momelotinib would continue to receive momelotinib as subsequent therapy. The results of this scenario analysis were similar to the base case in that momelotinib was associated with higher costs and QALYs compared to BAT (incremental cost: $18,582; incremental QALYs: 0.092; ICER: $201,446).
CDA-AMC undertook price reduction analysis based on the 2 CDA-AMC reanalyses. The results from the CDA-AMC reanalysis suggest that a price reduction of approximately 15% is required for momelotinib to achieve cost-effectiveness versus ruxolitinib for the JAK inhibitor–naive population. Comparatively, a price reduction of approximately 29% is required for momelotinib to achieve cost-effectiveness versus BAT for the JAK inhibitor–experienced population. Assuming a distribution of 15% patients who have not previously been treated with JAK inhibitors and 85% patients who have previously been treated with JAK inhibitors, a price reduction of approximately 26.9% would be required for momelotinib to achieve cost-effectiveness versus ruxolitinib or BAT in the blended population.
Additional scenario analyses were performed to examine the cost-effectiveness of momelotinib among patients with hemoglobin levels less than 10 g/dL. Further details of this analysis, including related price reductions, are available in Appendix 4.
Table 7: CDA-AMC Price Reduction Analyses
Price reduction | Unit drug cost ($) | ICERs for momelotinib ($/QALY) | |
|---|---|---|---|
JAK inhibitor naive (vs. ruxolitinib) | JAK inhibitor experienced (vs. BAT) | ||
No price reduction | 231 | 245,628 | 327,295 |
10% | 208 | 117,369 | 231,407 |
20% | 185 | Dominant | 135,520 |
30% | 162 | Dominant | 39,633 |
40% | 139 | Dominant | Dominant |
50% | 115 | Dominant | Dominant |
60% | 92 | Dominant | Dominant |
70% | 69 | Dominant | Dominant |
80% | 46 | Dominant | Dominant |
90% | 23 | Dominant | Dominant |
BAT = best available therapy; CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; JAK = Janus kinase; QALY = quality-adjusted life-year; vs. = versus.
Overall Conclusions
The CDA-AMC Clinical Review of the SIMPLIFY-1 and SIMPLIFY-2 trials concluded that there is moderate to high certainty that momelotinib will provide benefits to patients in terms of transfusion independence and reduction of transfusion needs in adults with MF and that there is moderate evidence showing a benefit in disease-related symptoms compared to BAT. The Clinical Review further concluded that momelotinib was noninferior to ruxolitinib in terms of splenic response and that ruxolitinib improved disease-related symptoms compared to momelotinib. There was no long-term comparative evidence for momelotinib compared to BAT, ruxolitinib, or any other JAK inhibitor treatment. Given that 98% of incremental QALYs estimated for momelotinib versus BAT were accrued through extrapolation beyond the period of the trial evidence, the lack of long-term comparative evidence contributes much uncertainty to the cost-effectiveness of momelotinib.
The economic review found additional limitations, including an inappropriate removal of subsequent treatment costs for patients treated with momelotinib, a model structure that did not fully reflect clinical decision-making in MF treatment, and the exclusion of a potentially relevant comparator. In reanalysis, CDA-AMC included ruxolitinib as subsequent therapy for patients who discontinue momelotinib. CDA-AMC was not able to address the other limitations identified in the sponsor’s submission.
In the CDA-AMC reanalysis for patients not previously treated with JAK inhibitors, momelotinib was more costly and more effective than ruxolitinib (incremental costs: $23,841; incremental QALYs: 0.097), with an ICER of $245,628 per QALY gained. In the CDA-AMC reanalysis for patients who had been previously treated with JAK inhibitors, momelotinib was more costly and more effective than BAT (incremental costs: $30,087; incremental QALYs: 0.092). While incremental QALYs were similar in the sponsor’s base case and the CDA-AMC base case, momelotinib was no longer cost-saving in the CDA-AMC base case due to the inclusion of ruxolitinib as subsequent therapy. CDA-AMC also conducted a scenario analysis that considered the possibility that patients previously treated with JAK inhibitors would continue to receive momelotinib as subsequent therapy following disease progression on momelotinib. In this scenario, treatment costs for patients receiving momelotinib increased by $36,701. Using publicly available list prices for momelotinib and all comparators, a price reduction of approximately 27% was needed for momelotinib to be cost-effective compared to ruxolitinib and BAT at a willingness-to-pay threshold of $50,000 per QALY gained.
For patients with MF, treatment with momelotinib is more costly than treatment with ruxolitinib in those not previously treated with JAK inhibitors and more costly than treatment with BAT in those who have been previously treated with JAK inhibitors. The clinical evidence suggested that among patients not previously treated with JAK inhibitors, momelotinib and ruxolitinib resulted in similar splenic response but that ruxolitinib had a more favourable disease-related symptom profile. In the CDA-AMC base case, estimates of incremental QALYs were marginal and highly uncertain due to a lack of long-term comparative evidence and a model that did not include splenic response (i.e., the noninferiority outcome that was measured in the SIMPLIFY-2 trial). Overall, there is no robust clinical or economic evidence supporting a price premium for momelotinib over ruxolitinib.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Ojjaara (momelotinib), 100 mg, 150 mg, and 200 mg oral tablets. Mississauga (ON): GlaxoSmithKline Inc.; June 19, 2024.
2.GlaxoSmithKline Inc. Ojjaara (momelotinib): 100 mg, 150 mg, and 200 mg oral tablets [product monograph]. Updated November 8, 2024.
3.Xie F, Pullenayegum E, Gaebel K, et al. A time trade-off-derived value set of the EQ-5D-5L for Canada. Med Care. 2016;54(1):98-105. doi:10.1097/MLR.0000000000000447 PubMed
4.Beusterien KM, Davies J, Leach M, et al. Population preference values for treatment outcomes in chronic lymphocytic leukaemia: a cross-sectional utility study. Health Qual Life Outcomes. 2010;8:50. doi:10.1186/1477-7525-8-50 PubMed
5.Tolley K, Goad C, Yi Y, Maroudas P, Haiderali A, Thompson G. Utility elicitation study in the UK general public for late-stage chronic lymphocytic leukaemia. Eur J Health Econ. 2013;14(5):749-59. doi:10.1007/s10198-012-0419-2 PubMed
6.Shao T, Zhao M, Tang W. Cost-effectiveness analysis of sintilimab vs. placebo in combination with chemotherapy as first-line therapy for local advanced or metastatic oesophageal squamous cell carcinoma. Front Oncol. 2022;12:953671. doi:10.3389/fonc.2022.953671 PubMed
7.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Exceptional Access Program (EAP). Accessed July 30, 2024. http://www.health.gov.on.ca/en/pro/programs/drugs/odbf/odbf_except_access.aspx
8.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario drug benefit formulary/comparative drug index. Accessed 2024 Jul 30, https://www.formulary.health.gov.on.ca/formulary/
9.National Comprehensive Cancer Network (NCCN). Myeloproliferative neoplasms, version 1.2024. Updated December 21, 2023. Accessed May 28, 2024. https://www.nccn.org/
Appendix 1: Cost Comparison Table
Please note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical expert(s). Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 8: CDA-AMC Cost Comparison Table for Treatments Indicated for Myelofibrosis
Treatment | Strength / concentration | Form | Price | Recommended dosage | Daily cost ($) | Cost per 28-day cycle ($) |
|---|---|---|---|---|---|---|
Momelotinib | 100 mg 150 mg 200 mg | Tablet | 230.8610a | 200 mg once daily | 230.8610 | 6,464 |
Antineoplastic agent | ||||||
Fedratinib (Inrebic) | 100 mg | Tablet | 84.3930 | 400 mg once daily | 337.57 | 9,452 |
Ruxolitinib (Jakavi) | 5 mg 10 mg 15 mg 20 mg | Tablet | 86.6275 97.1600 97.1600 97.1600 | 5 mg to 20 mg twice daily | 173.26 to 194.32 | 4,851 to 5,441 |
CDA-AMC = Canada’s Drug Agency.
Note: All prices are from the Ontario Exceptional Access Program (accessed July 2024), unless otherwise indicated, and do not include dispensing fees.7
aSponsor-submitted price.
Appendix 2: Submission Quality
Please note that this appendix has not been copy-edited.
Description | Yes or no | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing and no relevant outcome missing | No | Refer to CDA-AMC critical appraisal. |
Model has been adequately programmed and has sufficient face validity | Yes | No comment. |
Model structure is adequate for decision problem | No | Refer to CDA-AMC critical appraisal. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | Yes | No comment. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | Yes | No comment. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | Yes | No comment. |
CDA-AMC = Canada’s Drug Agency.
Appendix 3: Additional Information on the Submitted Economic Evaluation
Please note that this appendix has not been copy-edited.
Figure 1: Model Structure – JAKi Naive and JAKi Experienced
TD = transfusion dependent; TI = transfusion independent; TR = transfusion requiring.
Source: Sponsor’s pharmacoeconomic submission.1
Figure 2: Markov Model Structure
TD = transfusion dependent; TI = transfusion independent; TR = transfusion requiring.
Source: Sponsor’s pharmacoeconomic submission.1
Details of the Sponsor’s Base Case
Table 10: Sponsor’s Base-Case Overall Survival Curve Selections
Treatment | Transfusion independent | Transfusion dependent |
|---|---|---|
JAKi naive | Log-logistic | Generalized gamma |
JAKi experienced | Log-logistic | Exponential |
JAKi = Janus kinase inhibitor.
Table 11: Sponsor’s Base-Case Time to Treatment Discontinuation Curve Selections
Treatment | Momelotinib | Ruxolitinib |
|---|---|---|
JAKi naive | Generalized gamma | Generalized gamma |
JAKi experienced | Gompertz | NA |
JAKi = Janus kinase inhibitor; NA = not applicable.
Table 12: Disaggregated Summary of the Sponsor’s Economic Evaluation (Blended Population)
Parameter | Momelotinib | Ruxolitinib/BAT |
|---|---|---|
Discounted LYs | ||
Total LYs | 4.089 | 3.967 |
TI | 2.220 | 1.903 |
TR | 0.770 | 0.816 |
TD | 1.099 | 1.247 |
Discounted QALYs | ||
Total QALYs | 3.026 | 2.934 |
TI | 1.675 | 1.440 |
TR | 0.559 | 0.594 |
TD | 0.792 | 0.900 |
Discounted costs ($) | ||
Total cost | 181,734 | 320,184 |
Drug acquisition | 93,483 | 223,896 |
Drug administration | 0 | 316 |
Health state cost | 29,745 | 32,917 |
Adverse event cost | 10,302 | 5,915 |
Subsequent treatment cost | 25,617 | 34,509 |
Terminal care cost | 22,587 | 22,631 |
Additional costs | 0 | 0 |
Societal costs | 0 | 0 |
BAT = best available therapy; LY = life-year; QALY = quality-adjusted life-year; TD = transfusion dependent; TI = transfusion independent; TR = transfusion requiring.
Source: Sponsor’s pharmacoeconomic submission.1
Appendix 4: Additional Details on the CDA-AMC Reanalyses and Sensitivity Analyses of the Economic Evaluation
Please note that this appendix has not been copy-edited.
Detailed Results of CDA-AMC Base Case
Table 13: Disaggregated Summary of CDA-AMC Economic Evaluation Results – JAKi Naive
Parameter | Momelotinib | Ruxolitinib |
|---|---|---|
Discounted LYs | ||
Total | 5.245 | 5.134 |
TI | 2.903 | 2.604 |
TR | 0.396 | 0.422 |
TD | 1.945 | 2.107 |
Discounted QALYs | ||
Total | 4.284 | 4.187 |
TI | 2.412 | 2.164 |
TR | 0.322 | 0.344 |
TD | 1.550 | 1.679 |
Discounted costs ($) | ||
Total | 442,890 | 419,050 |
Drug acquisition | 126,636 | 112,384 |
Drug administration | 0 | 0 |
Health state cost | 41,433 | 44,491 |
Adverse event cost | 10,193 | 9,977 |
Subsequent treatment cost | 242,496 | 230,025 |
Terminal care cost | 22,132 | 22,173 |
Additional costs | 0 | 0 |
Societal costs | 0 | 0 |
CDA-AMC = Canada’s Drug Agency; JAKi = Janus Kinase inhibitor; LY = life-year; QALY = quality-adjusted life-year; TD = transfusion dependent; TI = transfusion independent; TR = transfusion requiring.
Table 14: Disaggregated Summary of CDA-AMC Economic Evaluation Results – JAKi Experienced
Parameter | Momelotinib | BAT |
|---|---|---|
Discounted LYs | ||
Total | 3.898 | 3.773 |
TI | 2.109 | 1.788 |
TR | 0.836 | 0.885 |
TD | 0.954 | 1.100 |
Discounted QALYs | ||
Total | 2.840 | 2.748 |
TI | 1.567 | 1.332 |
TR | 0.602 | 0.640 |
TD | 0.671 | 0.776 |
Discounted costs ($) | ||
Total | 333,694 | 303,606 |
Drug acquisition | 87,657 | 244,398 |
Drug administration | 0 | 374 |
Health state cost | 27,729 | 30,917 |
Adverse event cost | 10,345 | 5,213 |
Subsequent treatment cost | 185,303 | 0 |
Terminal care cost | 22,660 | 22,705 |
Additional costs | 0 | 0 |
Societal costs | 0 | 0 |
CDA-AMC = Canada’s Drug Agency; JAKi = Janus Kinase inhibitor; LY = life-year; QALY = quality-adjusted life-year; TD = transfusion dependent; TI = transfusion independent; TR = transfusion requiring.
Scenario Analyses
Table 15: Summary of the CDA-AMC Scenario Analysis Results
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
Sponsor’s base case (blended analysis; probabilistic) | BAT and ruxolitinib | 320,184 | 2.934 | Reference |
Momelotinib | 181,734 | 3.026 | Dominant | |
CDA-AMC base case JAKi naive (reanalysis 1a + 2; probabilistic) | Ruxolitinib | 419,050 | 4.187 | Reference |
Momelotinib | 442,890 | 4.284 | 245,628 | |
CDA-AMC base case JAKi experienced (reanalysis 1b + 2; probabilistic) | BAT | 303,606 | 2.748 | Reference |
Momelotinib | 333,694 | 2.840 | 327,295 | |
CDA-AMC Scenario Analysis JAKi experienced (momelotinib subsequent therapy) | BAT | 351,813 | 2.745 | Reference |
Momelotinib | 370,395 | 2.837 | 201,446 | |
CDA-AMC Scenario Analysis JAKi naive (hemoglobin levels < 10 g/dL) | Ruxolitinib | 356,101 | 3.419 | Reference |
Momelotinib | 376,487 | 3.485 | 309,768 | |
CDA-AMC Scenario Analysis JAKi experienced (hemoglobin levels < 10 g/dL) | BAT | 227,818 | 1.960 | Reference |
Momelotinib | 253,419 | 1.999 | 666,547 |
BAT = best available therapy; CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; JAKi = Janus kinase inhibitor; QALY = quality-adjusted life-year.
Note: The CDA-AMC reanalysis is based on publicly available prices of the comparator treatments. The results of all steps are presented deterministically unless otherwise indicated, while the cumulative CDA-AMC base case is always presented both deterministically and probabilistically.
A scenario analysis was performed to estimate the cost-effectiveness in patients with JAKi-naive and JAKi-experienced patients with hemoglobin levels less than 10 g/dL. Among JAKi-naive patients, momelotinib was associated with an ICER of $309,768 per QALY gained compared to ruxolitinib, and a 14.6% price reduction was needed to be considered cost-effective at a willingness-to-pay threshold of $50,000 per QALY gained. Among JAKi-experienced patients, momelotinib was associated with an ICER of $666,547 per QALY gained, and a 27.3% price reduction was needed to be considered cost-effective at this threshold. Based on an assumption that 15% of eligible patients are JAKi naive and the remaining 85% are JAKi experienced, a price reduction of 25.4% would be required for momelotinib to be considered cost-effective in patients with hemoglobin levels less than 10 g/dL.
Appendix 5: Submitted Budget Impact Analysis and CDA-AMC Appraisal
Please note that this appendix has not been copy-edited.
Table 16: Summary of Key Take-Aways
Key take-aways of the budget impact analysis |
|---|
|
CDA-AMC = Canada’s Drug Agency.
Summary of Sponsor’s Budget Impact Analysis
The sponsor submitted a budget impact analysis (BIA) to estimate the three-year budget impact of reimbursing momelotinib for the treatment of splenomegaly and/or disease-related symptoms, in adult patients with intermediate or high-risk primary MF, post polycythemia vera MF or post essential thrombocythemia MF who have moderate to severe anemia. The analysis was taken from the perspective of the Canadian public drug plan. A three-year time horizon was used from 2025 to 2027, with 2024 as the base year.
The target population size was derived with an epidemiological approach. For the JAKi-naive population, the sponsor estimated the total eligible population by applying the MF incidence to the total population in Canada (excluding Quebec) and accounted for the proportion who have intermediate/high-risk at the time of diagnosis and confirmed anemia. For the JAKi-experienced population, the sponsor estimated the proportion of eligible patients by applying the MF prevalence rate to total adult population of Canada (excluding Quebec) and then accounting for the proportion who had intermediate or high-risk disease at the time of diagnosis, the proportion of patients who had confirmed anemia, and the proportion of patients who have previously received a JAK inhibitor. Key inputs to the BIA are documented in Table 16.
The BIA compared 2 scenarios to determine the incremental budget impact of reimbursing momelotinib. The reference case scenario assumed that momelotinib was not available and therefore patients received either ruxolitinib or fedratinib. The new drug scenario included momelotinib as an option. In the sponsor’s base case, costs related to drug acquisition were considered.
Table 17: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3 if appropriate) |
|---|---|
Target population | |
JAKi-naive population | |
Total patient population (Canada excluding Quebec) in 2024 | 31,015,000 |
Myelofibrosis incidence | 0.0008% |
% of patients with intermediate/high-risk disease at the time of diagnosis | 84.00% |
% of patients with confirmed anemia | █████% |
% under 65 years | █████% |
% under 65 years with public coverage | 66.85% |
% of patients 65 years and older | █████% |
% of patients 65 years and older with public coverage | 98.43% |
Transfusion status distributions | █████████████████████████ |
Mortality rates | █████████████████████████ |
JAKi-experienced population | |
Total patient population in Canada excluding Quebec | 31,015,000 |
Proportion of adults | 81.5% |
Myelofibrosis prevalence | 0.0062% |
% of patients with intermediate/high-risk at time of diagnosis | 89.00% |
% of patients with confirmed anemia | █████% |
% of patients that had received a JAK inhibitor | 70.00% |
% of patients under 65 | █████% |
% of patients under 65 with public coverage | 66.85% |
% of patients 65 years old and over | █████% |
% of patients 65 years and older with public coverage | 98.43% |
Transfusion status distributions | ███████████████████████████ |
Mortality rates | Assumed constant |
Number of patients eligible for drug under review | 893 / 1,006 / 1,086 |
Market uptake (3 years) | |
Uptake (reference scenario) | |
JAKi naive Ruxolitinib Fedratinib | ██% / ██% / ██% ██% / ██% / ██% |
JAKi experienced BAT | 100% / 100% / 100% |
Uptake (new drug scenario) | |
JAKi naive Momelotinib Ruxolitinib Fedratinib | ████% / ████% / ████% ████% / ████% / ███% ███% / ███% / ███% |
JAKi experienced Momelotinib BAT | ███% / ████% / ████% ████% / ████% / ████% |
Cost of treatment (per patient, per year) | |
Momelotinib Ruxolitinib Fedratinib BAT | $84,322 $70,975 $123,298 $64,731 |
BAT = best available therapy; JAKi = Janus kinase inhibitor.
Summary of the Sponsor’s BIA Results
In the sponsor’s base-case analysis, the estimated incremental budget impact of funding momelotinib for the treatment of splenomegaly and/or disease-related symptoms, in adult patients with intermediate or high-risk primary MF, post-polycythemia vera MF or post-essential thrombocythemia MF who have moderate to severe anemia was $1,488,224 in year 1, $4,613,504 in year 2, and $6,305,271 in year 3. Therefore, the 3-year incremental budget impact was $12,407,000.
CDA-AMC Appraisal of the Sponsor’s BIA
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
The number of eligible patients is uncertain. In the sponsor’s base-case analysis, the JAKi-naive population was derived by applying an age-standardized incidence rate for primary MF obtained from Heppner et al., (2019) to the pan-Canadian population from Statistics Canada. However, given the Health Canada indication is for adult patients, the sponsor’s use of the total pan-Canadian population overestimates the patient population as it includes individuals less than 18 years old.
In the reanalysis, CDA-AMC used the sponsor’s provided “% adult” (i.e., 81.5%) to calculate the pan-Canadian adult population for the JAKi-naive eligible population.
Due to the limitations associated with using an age-standardized incidence rate versus an age-specific incidence rate (i.e., ≥ 18 years of age), CDA-AMC conducted a scenario analysis where the MF incidence rate was increased by 10%.
Estimated market uptake of momelotinib is uncertain. In the sponsor’s base-case analysis, momelotinib market uptake was informed by internal market assumptions made by the sponsor. Upon reimbursement, in the JAKi-naive population, the sponsor assumed that the market share for momelotinib would be 30.7% in year 1, 70.0% in year 2, and 95.6% in year 3. Comparatively, in the JAKi-experienced population the sponsor assumed that market share would be 6.6% in year 1, 19.1% in year 2, and 19.2% in year 3. Clinical expert feedback received by CDA-AMC noted that the anticipated market share estimates for momelotinib in the JAKi-naive and JAKi-experienced populations were higher and lower than expected, respectively. It was noted that for JAKi-naive patients, momelotinib is not expected to nearly completely displace ruxolitinib by year 3 whereas for the JAKi-experienced population given the lack of available alternatives, momelotinib use is expected to be higher than the proposed 19.2% in year 3.
In the CDA-AMC base case, market share estimates for momelotinib for JAKi-naive patients was set to 30.7%, 45%, and 60% in years 1, 2, and 3 respectively. For the JAKi-experienced population, market share estimates for momelotinib were set to 6.6%, 19.1%, and 25% in years 1, 2, and 3 respectively. CDA-AMC additionally explored a scenario where the market uptake in year 3 for the JAKi-experienced population was increased to 35%.
CDA-AMC Reanalyses of the BIA
Table 18: CDA-AMC Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Changes to derive the CDA-AMC base case | ||
1. Population | JAKi naive: pan-Canadian population | JAKi naive: adult pan-Canadian population |
2. Momelotinib market share | JAKi naive: 30.7% / 70.0% / 95.6% JAKi experienced: 6.6% / 19.1% / 19.2% | JAKi naive: 30.7% / 45% / 60% JAKi experienced: 6.6% / 19.1% / 25% |
CDA-AMC base case | reanalysis 1 + 2 | |
CDA-AMC = Canada’s Drug Agency; JAKi = Janus kinase inhibitor.
The results of the CDA-AMC stepwise reanalysis are presented in summary format in Table 18 and a more detailed breakdown is presented in Table 19. All CDA-AMC reanalysis were based on publicly available prices of the comparator treatments.
In the CDA-AMC base case, the estimated incremental budget impact of reimbursing momelotinib for the treatment of disease-related splenomegaly or symptoms, and anemia in adult patients with primary MF, post-polycythemia vera MF or post-essential thrombocythemia MF who are JAKi naive or have been treated with a JAKi is expected to be $10,966,008 (year 1: $1,394,787; year 2: $3,946,755; year 3: $5,624,465).
Table 19: Summary of the CDA-AMC Reanalyses of the Budget Impact Analysis
Stepped analysis | Three-year total ($) |
|---|---|
Submitted base case | 12,407,000 |
CDA-AMC reanalysis 1 | 11,349,162 |
CDA-AMC reanalysis 2 | 11,740,519 |
CDA-AMC base case | 10,966,008 |
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency.
CDA-AMC conducted the following scenario analyses to address remaining uncertainty, using the CDA-AMC base case (results are provided in Table 19):
assuming the MF incidence is increased by 10%
assuming market uptake for momelotinib in year 3 for the JAKi-experienced population was increased to 35%
assuming that the price of momelotinib is reduced by 15% and 29% (CDA-AMC estimated price reductions from the cost-utility analysis).
Table 20: Detailed Breakdown of the CDA-AMC Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|---|
Submitted base case | Reference | 58,669,824 | 58,812,491 | 66,940,345 | 72,692,022 | 198,444,858 |
New drug | 58,669,824 | 60,300,715 | 71,553,849 | 78,997,294 | 210,851,857 | |
Budget impact | 0 | 1,488,224 | 4,613,504 | 6,305,271 | 12,407,000 | |
CDA-AMC base case | Reference | 56,916,309 | 57,032,612 | 63,658,504 | 68,347,318 | 189,038,433 |
New drug | 56,916,309 | 58,427,399 | 67,605,259 | 73,971,783 | 200,004,441 | |
Budget impact | 0 | 1,394,787 | 3,946,755 | 5,624,465 | 10,966,008 | |
CDA-AMC scenario analysis 1: increased incidence rate | Reference | 57,689,867 | 57,817,800 | 65,106,281 | 70,263,976 | 193,188,057 |
New drug | 57,689,867 | 59,253,807 | 69,163,075 | 76,078,860 | 204,495,742 | |
Budget impact | 0 | 1,436,007 | 4,056,794 | 5,814,884 | 11,307,684 | |
CDA-AMC scenario analysis 2: increased market share | Reference | 56,916,309 | 57,032,612 | 63,658,504 | 68,347,318 | 189,038,433 |
New drug | 56,916,309 | 58,427,399 | 67,605,259 | 75,460,134 | 201,492,792 | |
Budget impact | 0 | 1,394,787 | 3,946,755 | 7,112,817 | 12,454,359 | |
CDA-AMC scenario analysis 3: hemoglobin levels < 10 g/dL | Reference | 41,407,758 | 41,472,824 | 45,123,765 | 47,657,808 | 134,254,397 |
New drug | 41,407,758 | 42,451,768 | 47,806,203 | 51,525,378 | 141,783,350 | |
Budget impact | $0 | 978,944 | 2,682,439 | 3,867,570 | 7,528,952 | |
CDA-AMC scenario analysis 3: 15% price reduction | Reference | 56,916,309 | 57,032,612 | 63,658,504 | 68,347,318 | 189,038,433 |
New drug | 56,916,309 | 57,369,203 | 64,778,759 | 69,932,119 | 192,080,081 | |
Budget impact | 0 | 336,591 | 1,120,255 | 1,584,802 | 3,041,648 | |
CDA-AMC scenario analysis 4: 29% price reduction | Reference | 56,916,309 | 57,032,612 | 63,658,504 | 68,347,318 | 189,038,433 |
New drug | 56,916,309 | 56,381,552 | 62,140,693 | 66,161,766 | 184,684,011 | |
Budget impact | 0 | −651,059 | −1,517,811 | −2,185,551 | −4,354,422 |
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.