Drugs, Health Technologies, Health Systems
Reimbursement Review
Ivosidenib (Tibsovo)
Sponsor: Servir Canada Inc.
Therapeutic area: Acute myeloid leukemia
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Testing Procedure Assessment
Clinical Review
Abbreviations
AE
adverse event
AML
acute myeloid leukemia
CI
confidence interval
CR
complete remission
CRh
complete remission with partial hematologic recovery
Cri
complete remission with incomplete hematologic recovery
Crl
credible interval
DCO
data cut-off
DSU
Decision Support Unit
ECOG
Eastern Cooperative Oncology Group
EFS
event-free survival
EORTC QLQ C-30
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30
ESS
effective sample size
GRADE
Grading of Recommendations, Assessment, Development, and Evaluation
HR
hazard ratio
HRQoL
health-related quality of life
IDMC
Independent Data Monitoring Committee
ITC
indirect treatment comparison
ITT
intention to treat
LDAC
low-dose cytarabine
LLSC
Leukemia & Lymphoma Society of Canada
MAIC
matching-adjusted indirect comparison
MID
minimally important difference
NE
not estimable
NICE
National Institute for Health and Care Excellence
NMA
network meta-analysis
OH-CCO
Ontario Health (Cancer Care Ontario)
OR
odds ratio
ORR
objective response rate
OS
overall survival
QoL
quality of life
RBC
red blood cell
RCT
randomized controlled trial
SAE
serious adverse event
TEAE
treatment-emergent adverse event
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Ivosidenib (Tibsovo), 250 mg, tablet, oral |
Sponsor | Servier Canada Inc. |
Indication | Ivosidenib in combination with azacitidine is indicated for the treatment of adult patients with newly diagnosed AML with an IIDH1 R132 mutation who are not eligible to receive intensive induction chemotherapy |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | July 19, 2024 |
Recommended dose | Ivosidenib 500 mg (2 × 250 mg tablets) taken orally once daily |
AML = acute myeloid leukemia; NOC = Notice of Compliance.
Introduction
Acute myeloid leukemia (AML) is a heterogeneous hematologic malignancy characterized by the clonal expansion of myeloid blasts in the bone marrow, peripheral blood, and/or other tissues.1,2 Typical symptoms of AML include fatigue, pale skin, dyspnea, infection, dizziness, headache, and coldness in hands and feet.3-5 Furthermore, leukopenia and neutropenia increase the risk of infections and fever, while thrombocytopenia increases the likelihood of bruising, bleeding, frequent or severe nosebleeds, bleeding gums, and heavy menstrual bleeding.5 Other symptoms include weight loss, night sweats, and loss of appetite.6,7 AML is 1 of the most aggressive forms of leukemia.5 The Cancer Quality Council of Ontario has reported age-standardized 1-year (2017 to 2018) and 5-year survival rates (2014 to 2018) of 42.1% and 19.9%, respectively.8
The prevalence of AML ranges from 0.6 to 11.0 per 100,000 persons for all age categories, genders, and ethnicities globally.9,10 The national age-standardized incidence rate for AML was reported to be 3.8 per 100,000 persons by Statistics Canada in 2018.11 Approximately 1,600 patients in Canada were diagnosed with AML in 2022.12 It is estimated that 6% to 10% of all people with AML carry an IDH1 mutation, with an estimated incidence ranging from 0.24 to 0.40 per 100,000 persons.13-20 The incidence of IDH1-mutated AML is low, and it is considered to be a rare disease.21 Approximately 40% to 50% of people with newly diagnosed AML are ineligible for standard induction chemotherapy regimens because of older age, insufficient Karnofsky performance status or Eastern Cooperative Oncology Group (ECOG) performance status, and/or comorbid conditions.12,22-25
The treatment goals for patients with AML who are not eligible to receive intensive induction chemotherapy are to prolong life, alleviate symptoms, reduce dependency on blood transfusion, reduce infections, and improve patients’ quality of life (QoL). Treatment options for patients with newly diagnosed AML who carry a mutation in the IDH1 enzyme and are ineligible for standard intensive chemotherapy (because of insufficient performance status, a comorbid medical condition, or age) are limited. In Canada, active treatment options that are currently publicly funded for patients with AML who are ineligible for standard intensive chemotherapy, although not specific to patients carrying an IDH1 mutation, include:1,26-29
venetoclax combined with azacitidine
monotherapy with azacitidine or low-dose cytarabine (LDAC) if the patients are not considered candidates for combination therapy.
Ivosidenib is an inhibitor of the mutant IDH1 enzyme. Mutant IDH1 converts alpha-ketoglutarate to 2-hydroxyglutarate, which blocks cellular differentiation and promotes tumorigenesis in both hematologic and nonhematologic malignancies.30 On July 19, 2024, ivosidenib in combination with azacitidine was approved by Health Canada for the treatment of adult patients with newly diagnosed AML with an IIDH1 R132 mutation who are not eligible to receive intensive induction chemotherapy. The sponsor’s reimbursement request is aligned with the Health Canada–approved indication. IIDH1 R132 mutation must be confirmed before the combination regimen is initiated.30
The recommended dose for ivosidenib is 500 mg (2 × 250 mg tablets) taken orally once daily. Ivosidenib should be started on cycle 1 day 1 and administered once daily during the 28-day cycle. It should be started in combination with azacitidine at 75 mg/m2 of body surface area, intravenously or subcutaneously, once daily on days 1 to 7 of each 28-day cycle. It is recommended that patients be treated for a minimum of 6 cycles. Treatment should be continued until disease progression or until treatment is no longer tolerated by the patient.30
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of ivosidenib (250 mg film-coated tablets) in combination with azacitidine for the treatment of adult patients with newly diagnosed IDH1-mutated AML who are not eligible for intensive induction chemotherapy.
Perspectives of Patients, Clinical Input, and Drug Input
The information in this section is a summary of the input provided by the patient and clinician groups who responded to our call for input and from the clinical experts consulted by the review team for this submission.
Patient Input
Two patient groups, the Leukemia & Lymphoma Society of Canada (LLSC) and Heal Canada, provided input to the review of ivosidenib. The LLSC is a national organization with charitable status dedicated to finding a cure for blood cancers and improving the QoL of people affected by blood cancers and their families by funding life-enhancing research and providing educational resources, services, and support. Heal Canada is a registered not-for-profit organization that aims to empower patients, improve health care outcomes, and advocate for equitable access to quality health care across Canada. Data were gathered through online surveys or emails with people diagnosed with AML and their caregivers. Eighty-three respondents participated in the survey from the LLSC, and 7 of those respondents identified as having the IDH1 mutation. The LLSC also conducted two 1-on-1 interviews with patients currently living with AML. Heal Canada launched an online survey to assess different characteristics of patients living with blood cancer. Of the 22 respondents, 5 had been diagnosed with AML. Information was also gathered from semistructured interviews with 2 patients and 2 caregivers. No patients or caregivers from Heal Canada had experience with ivosidenib; the LLSC interviewed 1 patient with previous experience with ivosidenib.
Most respondents reported that the mental, physical, and financial effects of AML have significant negative impact on the lives of patients and caregivers. The respondents described the challenges linked to the currently available treatments, such as intolerable side effects, lack of treatment response, and the limited options available to patients. Both respondent groups indicated that important patient outcomes included improved health-related QoL (HRQoL) (related to better control of anemia without transfusion or with fewer transfusions, as well as a lower infection rate), improved disease control, and prolonged survival. The patient who had experience with ivosidenib was initially treated with induction chemotherapy after a diagnosis of IDH1-mutated AML. After relapse on chemotherapy, the patient started ivosidenib and reported great response and minimal side effects from the treatment.
Clinician Input
Input From Clinical Experts Consulted by the Review Team for This Submission
The clinical experts identified the following unmet needs associated with the available treatments for patients with AML who are ineligible for intensive induction chemotherapy: first, not all patients respond to available therapies, and the outcomes for patients with AML (with or without IIDH1 R132 mutation) who are not eligible for intensive chemotherapy are poor; second, patients who respond to available therapy eventually relapse and succumb to their disease. Therefore, the clinical experts indicated that for patients in the target population, the most important treatment goals are to prolong remission and survival, reduce transfusion requirement, reduce the risk of infection and bleeding, and improve HRQoL.
The clinical experts indicated that ivosidenib would be reserved as first-line therapy for patients with AML who carry the IIDH1 R132 mutation and who are not eligible for intensive chemotherapy because of their age, comorbidities, or preference. Ivosidenib in combination with azacitidine could potentially replace the currently available combination therapy for these patients.
The clinical experts stated that only patients with a diagnosis of de novo AML with IIDH1 R132 mutation who are not eligible for intensive induction chemotherapy would be eligible to receive treatment with ivosidenib.
According to the experts, important outcomes for patients with AML are survival and improvements in HRQoL, response rates (in particular, complete remission [CR]), transfusion requirements, infection rates, and safety. The experts also noted that in clinical practice, patients’ responses to treatment are typically assessed every 28 days, corresponding to the length of the treatment cycles for azacitidine.
The experts noted that treatment with a combination of ivosidenib and azacitidine will be discontinued if disease progression is detected, if patients experience intolerable adverse events (AEs), and/or based on patient preference.
The clinical experts noted that, in general, patients should be treated by a hematologist and/or a hematologist or oncologist with experience in AML management. Treatment with ivosidenib can be administered in both inpatient and outpatient settings.
Clinician Group Input
Two clinician groups provided input for the review of ivosidenib in combination with azacitidine: the LLSC Clinician Network and the Ontario Health (Cancer Care Ontario) (OH-CCO) Hematology Cancer Drug Advisory Committee.
In general, the clinician group input was consistent with the input provided by the clinical experts consulted by the review team. The treatment goals for this patient population would be to prolong life, improve QoL, reduce transfusion requirement, and experience remission. The clinician groups noted that the current publicly funded treatment options for patients with AML who are not eligible for intensive chemotherapy include venetoclax plus azacitidine, single-drug azacitidine, LDAC, and best supportive care. The OH-CCO Drug Advisory Committee also mentioned venetoclax plus LDAC as an available therapy. However, not all patients respond to these therapies. In addition, both clinician groups suggested that treatment with venetoclax plus azacitidine is associated with increased risk of neutropenic fever and infections compared to azacitidine alone. According to the clinicians, infections may result in hospitalizations, which might last days to weeks depending on severity. The clinicians from the LLSC Clinician Network added that no tumour lysis syndrome monitoring is required with ivosidenib plus azacitidine. The clinician groups noted that specific inhibitors may offer a chance for increased treatment response and suggested ivosidenib plus azacitidine be considered as first-line therapy and become the new standard of care for adult patients with newly diagnosed IDH1-mutated AML who are not eligible for intensive induction chemotherapy or stem cell or bone marrow transplant. Both clinician groups indicated that remission rate, stabilization, and improvement in the frequency and severity of symptoms — such as improvement in blood counts, fewer transfusions, leukemia-free survival, and overall survival (OS), using usual leukemia response timelines — are the outcomes used to determine whether a patient is responding to ivosidenib plus azacitidine. Reasons for treatment discontinuation identified by the clinician groups included disease progression, intolerable side effects, and patient preference. Both clinician groups noted that ivosidenib plus azacitidine can be given in the inpatient and outpatient settings, or even in community centres that have experience treating acute leukemias.
Both the LLSC Clinician Network and the OH-CCO Drug Advisory Committee noted that timely results of testing for IDH1 mutation are required to identify patients who would benefit from and be eligible for this treatment.
Drug Program Input
Input was obtained from the drug programs that participate in our reimbursement review process. Refer to Table 4 for further information. The following were identified as key factors that could potentially impact the implementation of our recommendation for ivosidenib in combination with azacitidine:
considerations for initiation of therapy
considerations for discontinuation of therapy
considerations for prescribing of therapy
generalizability
care provision issues.
Clinical Evidence
Systematic Review
Description of Studies
One international, phase III, multicentre, double-blind randomized controlled trial (RCT), the AGILE trial (N = 146), evaluated the efficacy and safety of ivosidenib plus azacitidine compared to placebo plus azacitidine in adult patients with newly diagnosed AML with an IIDH1 R132 mutation who were not eligible to receive intensive induction chemotherapy. Patients were recruited from 89 study sites across 20 countries. Eligible patients were randomized 1:1 to receive either ivosidenib (500 mg orally once daily) plus azacitidine (75 mg/m2/day, subcutaneous or IV) for 7 days, in 28-day cycles, or placebo in combination with azacitidine. The primary efficacy end point in the AGILE study was event-free survival (EFS). Key secondary end points were CR rates, OS, CR and CR with partial hematologic recovery (CRh), and objective response rate (ORR). Additional secondary end points in this study included HRQoL (measured by the European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 [EORTC QLQ-C30]), transfusion requirement, and harms. The majority of patients (73.3% per investigator [76% per Interactive Web Response System]) had de novo AML at initial diagnosis. There were more male patients in the ivosidenib plus azacitidine group (58.3%) than in the placebo plus azacitidine group (51.4%). According to the WHO classification of AML, fewer patients in the ivosidenib plus azacitidine group (22.2%) had AML with recurrent genetic abnormalities than in the placebo plus azacitidine group (32.4%); more patients in the ivosidenib plus azacitidine group (38.9%) had AML with myelodysplasia-related changes than in the placebo plus azacitidine group (35.1%). IIDH1 R132C was the most common polymorphism (65.8% of patients). In total, 63.9% of patients in the ivosidenib plus azacitidine group and 67.6% of patients in the placebo plus azacitidine group had an ECOG performance status score of 0 to 1. Cytogenetic risk status, as assessed by the investigators based on the 2017 National Comprehensive Cancer Network guidelines, was intermediate (63.0%: 66.7% in the ivosidenib plus azacitidine group versus 59.5% in the placebo plus azacitidine group) or poor (24.7%: 22.2% in the ivosidenib plus azacitidine group versus 27.0% in the placebo plus azacitidine group) for most patients at baseline. The median bone marrow blast at baseline was 52.5% (range, 17% to 100%).
Two data cut-offs (DCOs) were available for the AGILE trial. The first DCO (March 18, 2021) represents an unplanned early interim analysis by the Independent Data Monitoring Committee (IDMC), which occurred before the protocol-specified number of events for the planned analysis. Because of a notable difference in the number of deaths, which favoured ivosidenib, the IDMC recommended that trial recruitment should end early, treatment assignment should be unblinded, and crossover to ivosidenib should be allowed. The stopping boundaries were therefore adjusted, and this interim analysis became the final analysis. A later DCO (June 30, 2022) was available for OS, transfusion requirement, and harms.
Efficacy Results
The AGILE study met its primary end point. As of the DCO of March 18, 2021, the between-group difference in the EFS rate was 19.7% (95% confidence interval [CI], ███ ██ ████) at 6 months and 25.3% (95% CI, ███ ██ ████) at 12 months, favouring ivosidenib. Improvement in EFS was largely driven by the proportion of patients with treatment failure, assigned an event time of the date of randomization: 42 patients (58.3%) in the ivosidenib plus azacitidine group versus 59 patients (79.7%) in the placebo plus azacitidine group had treatment failure. The median EFS in the ivosidenib plus azacitidine group was 0.03 months (95% CI, 0.03 months to 11.01 months) and 0.03 months (95% CI, not estimable [NE] to NE) in the placebo plus azacitidine group. The median did not appear different between groups because the majority of events were treatment failures, which were assigned the date of randomization. The corresponding hazard ratio (HR) was 0.33 (95% CI, 0.16 to 0.69; P = 0.0011). Predefined sensitivity analyses supported the robustness of the primary analysis and suggested an EFS benefit associated with ivosidenib in the short-term.
Treatment with ivosidenib plus azacitidine was associated with prolonged OS and met the prespecified efficacy boundary for a statistically significant OS benefit at the DCO of March 18, 2021. At the updated DCO of June 30, 2022, 37 patients (50.7%) in the ivosidenib plus azacitidine group and 58 (77.3%) in the placebo plus azacitidine group had died. The median OS was 29.3 months (95% CI, 13.2 months to NE) in the ivosidenib plus azacitidine group and 7.9 months (95% CI, 4.1 months to 11.3 months) in the placebo plus azacitidine group (P < 0.0001). The corresponding HR was 0.42 (95% CI, 0.27 to 0.65). The between-group differences in the Kaplan-Meier–estimated OS rate were 24.6% (95% CI, ███ ██ ████) at 12 months and 35.7% (95% CI, ████ ██ ████) at 24 months.
The results of subgroup analyses for OS and EFS (prespecified for EFS) based on various patient baseline characteristics were consistent with those in the overall population.
As of March 18, 2021, the CR rate was 47.2% (95% CI, 35.3% to 59.3%) in the ivosidenib plus azacitidine group and 14.9% (95% CI, 7.7% to 25.0%) in the placebo plus azacitidine group. However, these estimates were affected by high risk of bias due to missing data.
As of the DCO of June 30, 2022, a higher proportion of patients in the ivosidenib plus azacitidine group (██ ████████ ███████) did not require red blood cell (RBC) and/or platelet transfusion than in the placebo plus azacitidine group (██ ████████ ███████). This measurement was from a nonrandomized subset of the population. According to the clinical experts, improved CR rates and a reduced transfusion requirement are considered clinically meaningful changes, and better CR rates and, in their opinion, reduced transfusion can subsequently be translated to improved HRQoL and, potentially, prolonged survival.
Harms Results
Overall, the safety results from the 2 DCOs were consistent.
As of the DCO of March 18, 2021, the proportion of patients who experienced at least 1 AE was 98.6% (70 patients) in the ivosidenib plus azacitidine group and 100% (73 patients) in the placebo plus azacitidine group. Patients treated with ivosidenib plus azacitidine were more likely (5% or more) to report the following AEs than patients treated with placebo plus azacitidine: vomiting (29 patients [40.8%] versus 19 patients [26.0%]), neutropenia (20 [28.2%] versus 12 [16.4%]), thrombocytopenia (20 [28.2%] versus 15 [20.5%]), prolonged electrocardiogram QT interval (14 [19.7%] versus 5 [6.8%]), insomnia (13 [18.3%] versus 9 [12.3%]), differentiation syndrome (10 [14.1%] versus 6 [8.2%]), pain in extremity (10 [14.1%] versus 3 [4.1%]), hematoma (9 [12.7%] versus 1 [1.4%]), arthralgia (8 [11.3%] versus 3 [4.1%]), headache (8 [11.3%] versus 2 [2.7%]), leukocytosis (8 [11.3%] versus 1 [1.4%]), and leukopenia (6 [8.5%] versus 2 [2.7%]).
Grade 3 and higher AEs were reported in 66 patients (93.0%) in the ivosidenib plus azacitidine group and 69 patients (94.5%) in the placebo plus azacitidine group. In both groups, commonly reported grade 3 and higher AEs were anemia (25.4% of patients in the ivosidenib plus azacitidine group versus 26.0% in the placebo plus azacitidine group), febrile neutropenia (28.2% versus 34.2%), neutropenia (26.8% versus 16.4%), thrombocytopenia (23.9% versus 20.5%), and pneumonia (22.5% versus 28.8%).
The proportion of patients who experienced serious adverse events (SAEs) was 69.0% (46 patients) in the ivosidenib plus azacitidine group and 82.2% (60 patients) in the placebo plus azacitidine group. Commonly reported SAEs in the 2 treatment groups were febrile neutropenia (23.9% of patients in the ivosidenib plus azacitidine group versus 27.4% in the placebo plus azacitidine group) and pneumonia (19.7% versus 21.9%).
The overall incidences of treatment-emergent adverse events (TEAEs) that led to combination treatment discontinuation were similar between the treatment groups: 19 patients (26.8%) in the ivosidenib plus azacitidine group and 19 patients (26.0%) in the placebo plus azacitidine group.
Differentiation syndrome and infection were identified by the clinical experts as notable harms for treatment with ivosidenib. As of June 30, 2022, differentiation syndrome was reported in 10 patients (13.9%) in the ivosidenib plus azacitidine group and 6 patients (8.1%) in the placebo plus azacitidine group. Infection was reported in 25 patients (34.7%) in the ivosidenib plus azacitidine group and 38 patients (51.4%) in the placebo plus azacitidine group.
Critical Appraisal
In the AGILE study, there were some imbalances in baseline patient characteristics between the 2 treatment groups, for example sex, WHO classification of AML, and cytogenetic risk status as assessed by the investigator. These imbalances are likely to be the result of the small sample size, within which prognostic balance is not likely to be assured; as such, there is some risk that the observed effects are overestimated or underestimated. In addition, the postbaseline transfusion requirement outcome was measured among approximately half the population who required transfusions at baseline. Randomization is not necessarily upheld in this population. However, the results of transfusion requirement in patients who were dependent on transfusion at baseline did not differ significantly from those in the overall population. Therefore, the potential for bias is unlikely to have an important impact on the study findings specific to this outcome.
The study originally had no planned interim analyses. Observations of a notable difference in the number of deaths (favouring ivosidenib) by the IDMC prompted an unplanned interim analysis before the protocol-defined number of events. To control for multiplicity, new stopping boundaries were calculated based on the observed information fraction that were not outlined in the original statistical analysis plan. Because the results are from an unplanned interim analysis (which became the final analysis), even though the new stopping boundaries are appropriate, there is a risk of overestimation of the true effects of the study drug.
HRQoL was assessed using the EORTC QLQ C-30, although this is not an AML-specific instrument. Even though a minimally important difference (MID) for EORTC QLQ C-30 score for patients with AML was not identified from the literature, a range of potential between-group MIDs (3 to 11 points for improvement and –5 to –13 points for deterioration on the global QoL scale) were established based on clinical trials of 9 cancer types and may provide some guidance when determining the clinical relevance of the findings for HRQoL in the AGILE study. The completion rate of the EORTC QLQ C-30 was low. The completion rates were ██████ █████ ███ ████ at 6 months, 12 months, and 18 months of the study. The evidence for HRQoL was considered to be very uncertain because of large amounts of missing data and imprecision; the CIs included the potential for little-to-no clinically meaningful difference between groups. The missing data imputation approach used may not adequately address the issue. Therefore, there is a high risk of bias because of the large amount of missing HRQoL outcome data in this study; the direction of bias cannot be predicted.
EFS was the primary efficacy outcome in this study. It is a composite end point, and the sample size of the AGILE study was small. In the AGILE study, almost all events occurred at baseline (i.e., 1 component of the end point). As such, there were few patients left at risk postbaseline; as a result, the EFS could not robustly characterize the long-term efficacy of the study drug.31 The correlations between EFS and OS were modest in the published research that provided trial-level information. However, 1 major limitation of these studies was that they were not specific to the population nor the drug class of interest, and therefore the ability to generalize the study findings was not clear.32-34
According to feedback from the clinical experts, the eligibility criteria and baseline characteristics of the patients randomized in the AGILE study generally reflected a patient population in Canadian clinical practice that would receive combination therapy of ivosidenib plus azacitidine. The clinical experts noted that the results from the AGILE study could be generalized to patients with IDH1-mutated AML in Canada who would be treated with ivosidenib plus azacitidine. The clinical experts suggested that some flexibility should be applied in using ivosidenib plus azacitidine in patients with slightly worse ECOG performance status than in the trial. Patients’ IDH1 mutation status should be confirmed before the treatment. The experts indicated that the outcome measures in the AGILE study were generally appropriate and clinically relevant for clinical trials of AML.
In the AGILE study, ivosidenib in combination with azacitidine was compared with azacitidine monotherapy. The clinical experts consulted for this review indicated that azacitidine alone is not the most appropriate comparator for the study drug combination in the study population. Instead, venetoclax plus azacitidine is currently the most commonly used combination therapy in the target patient population. In practice, monotherapy with azacitidine would typically be used for patients who cannot tolerate treatment with the combination of venetoclax and azacitidine. There is a lack of direct evidence within the AGILE study with which to examine the efficacy and safety of the study drug compared with the other combination regimens.
GRADE Summary of Findings and Certainty of the Evidence
For pivotal studies and RCTs identified in the sponsor’s systematic review, the Grading of Recommendations, Assessment, Development, and Evaluations (GRADE) approach was used to assess the certainty of the evidence for the outcomes considered most relevant to inform the expert committee deliberations, and a final certainty rating was determined, as outlined by the GRADE Working Group.35,36
Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refer to internal validity or risk of bias), indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null. The threshold for a clinically important effect for OS and EFS in the study population was not obtained. Therefore, the target of the certainty of evidence assessment was the presence or absence of any (non-null) effect for survival rates. The threshold for a clinically important effect for the EORTC QLQ-C30 score was set according to the presence or absence of an important effect based on thresholds identified in the literature.37 In addition, the target of the certainty of evidence assessment was the presence or absence of any non-null effect for CR, CR plus CR with incomplete hematologic recovery (CRi), and transfusion requirements. For some harm events (e.g., differentiation syndrome), because of the unavailability of the absolute difference in effects, the certainty of evidence was summarized narratively.
Table 2 presents the GRADE summary of findings for ivosidenib plus azacitidine versus placebo plus azacitidine.
The selection of outcomes for the GRADE assessment was based on the sponsor’s summary of clinical evidence, consultation with clinical experts, and input received from patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with members of the expert committee:
OS
EFS
CR
CR plus CRi
change from baseline in EORTC QLQ C-30 scores
transfusion requirements
any SAEs
risk of AEs of special interest (differentiation syndrome, infection).
Table 2: Summary of Findings for Ivosidenib Plus Azacitidine Versus Placebo Plus Azacitidine for Patients With IDH1-Mutated AML
Outcome and follow-up | Patients (studies), N | Relative effect (95% CI) | Absolute effects (95% CI) | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
Placebo + azacitidine | Ivosidenib + azacitidine | Difference | |||||
Efficacy (FAS) | |||||||
OSa | |||||||
Probability of OS at 12 months Median follow-up: ████ ██████ in the ivosidenib + azacitidine group and ████ ██████ in the placebo + azacitidine group as of DCO of June 30, 2022 | 148 (1 RCT) | NR | 383 per 1,000 | 629 per 1,000 (504 to 730 per 1,000) | 246 more per 1,000 (██ ██ ███ ████ per 1,000) | Moderateb | Ivosidenib + azacitidine likely results in a clinically important increase in the probability of OS at 12 months when compared with placebo + azacitidine. |
Probability of OS at 24 months Median follow-up: ████ ██████ in the ivosidenib + azacitidine group and ████ ██████ in the placebo + azacitidine group as of DCO of June 30, 2022 | 148 (1 RCT) | NR | 174 per 1,000 | 531 per 1,000 (404 to 642 per 1,000) | 357 more per 1,000 (███ ██ ███ ████ per 1,000) | Moderateb | Ivosidenib + azacitidine likely results in a clinically important increase in the probability of OS at 24 months when compared with placebo + azacitidine. |
EFS | |||||||
Probability of EFS at 6 months Median follow-up: approximately 15 months for both groups as of DCO of March 18, 2021 | 146 (1 RCT) | NR | 203 per 1,000 | 399 per 1,000 (286 to 510 per 1,000) | 197 more per 1,000 (██ ██ ███ ████ per 1,000) | Moderatec | Ivosidenib + azacitidine likely results in an increase in the probability of EFS at 6 months when compared with placebo + azacitidine. The clinical importance of the increase is uncertain. |
Probability of EFS at 12 months Median follow-up: approximately 15 months for both groups as of DCO of March 18, 2021 | 146 (1 RCT) | NR | 122 per 1,000 | 374 per 1,000 (259 to 489 per 1,000) | 253 more per 1,000 (██ ██ ███ ████ per 1,000) | Lowd | Ivosidenib + azacitidine may result in an increase in the probability of EFS at 12 months when compared with placebo + azacitidine. The clinical importance of the increase is uncertain. |
CR | |||||||
CR rate Median follow-up: approximately 15 months for both groups as of DCO of March 18, 2021 | 146 (1 RCT) | OR: 4.76 (2.15 to 10.50) | 149 per 1,000 | 472 per 1,000 (353 to 593 per 1,000) | 310 more per 1,000 (███ ██ ███ ████ per 1,000) | Lowe | Ivosidenib + azacitidine may result in an increase in the probability of CR when compared with placebo + azacitidine. |
CR + CRi rate Median follow-up: approximately 15 months for both groups as of DCO of March 18, 2021 | 146 (1 RCT) | OR: 5.90 (2.69 to 12.97) | 162 per 1,000 | 542 per 1,000 (420 to 660 per 1,000) | 370 more per 1,000 (███ ██ ███ ████ per 1,000) | Lowf | Ivosidenib + azacitidine may result in an increase in the probability of CR + CRi when compared with placebo + azacitidine. |
Transfusion requirement | |||||||
Rate of conversion to postbaseline transfusion independence (in a subset of patients who were transfusion dependent at baseline) | 80 (1 RCT) | OR: ███ ████ ██ ████ | ███ ███ █████ | ███ ███ █████ ████ ██ ███ ███ ██████ | ███ ████ ███ █████ ███ ████ ██ ███ ████ ███ ██████ | Lowg | Ivosidenib + azacitidine may result in an increase in the proportion of patients who became transfusion independent postbaseline when compared with placebo + azacitidine. |
Health-related quality of life | |||||||
EORTC QLQ C-30 (global health status score) | |||||||
LS mean change from baseline (0 [severe impairment] to 100 [good health]), points At 6 months | 146 (1 RCT) | NA | –2.0 | 10.6 (1.23 to 19.97) | 12.6 (1.51 to 23.65) | Very lowh | The effect of ivosidenib + azacitidine on the global health status score of EORTC QLQ C-30 from baseline to 6 months, when compared with placebo + azacitidine, is very uncertain. |
LS mean change from baseline (0 [severe impairment] to 100 [good health]), points At 12 months | 146 (1 RCT) | NA | 4.2 | 19.1 (8.51 to 29.72) | 14.9 (–2.09 to 31.97) | Very lowi | The effect of ivosidenib + azacitidine on the global health status score of EORTC QLQ C-30 from baseline to 12 months, when compared with placebo + azacitidine, is very uncertain. |
Harms (safety analysis set) | |||||||
Any SAEs | |||||||
Proportion of patients with any SAEs Median follow-up: ████ ██████ in the ivosidenib + azacitidine group and ████ ██████ in the placebo + azacitidine group as of DCO of June 30, 2022 | 148 (1 RCT) | NR | ███ ███ █████ | ███ ███ █████ █████ | ███ ████ ███ █████ ████ ████ ██ █ ████ ███ ██████ | Moderatej | Ivosidenib + azacitidine likely results in a reduction in the proportion of patients who experience SAEs when compared with placebo + azacitidine. |
Differentiation syndrome | |||||||
Proportion of patients with differentiation syndrome Median follow-up: ████ ██████ in the ivosidenib + azacitidine group and ████ ██████ in the placebo + azacitidine group as of DCO of June 30, 2022 | 148 (1 RCT) | NR | 81 per 1,000 | 139 per 1,000 (██) | 60 more per 1,000 (██ ████ ██ ███ ████ per 1,000) | Lowk | Ivosidenib + azacitidine may result in an increase in the proportion of patients who experience differentiation syndrome when compared with placebo + azacitidine. |
Infection | |||||||
Proportion of patients with infections Median follow-up: ████ ██████ in the ivosidenib + azacitidine group and ████ ██████ in the placebo + azacitidine group as of DCO of June 30, 2022 | 148 (1 RCT) | NR | 514 per 1,000 | 347 per 1,000 (██) | 170 less per 1,000 (███ ████ ██ ██ ████ per 1,000) | Moderatel | Ivosidenib + azacitidine likely results in fewer infections when compared with placebo + azacitidine. |
AML = acute myeloid leukemia; CI = confidence interval; CR = complete remission; CRi = complete remission with incomplete hematologic recovery; DCO = data cut-off; EFS = event-free survival; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; FAS = full analysis set; LS = least squares; NA = not applicable; NR = not reported; OR = odds ratio; OS = overall survival; RCT = randomized controlled trial; SAE = serious adverse event.
Notes: Study limitations (which refer to internal validity or risk of bias), indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes; the between-group differences of the efficacy and harm outcomes in this table were requested from the sponsor.
aThe outcome of OS at the DCO of June 30, 2022, was not multiplicity adjusted; however, significance was met at an earlier multiplicity-adjusted analysis at the DCO of March 18, 2021.
bRated down 1 level for serious imprecision. No threshold of clinical importance could be estimated, but it was considered that the effect estimate and entire CI were consistent with important benefit. The sample size and number of events are small, resulting in potential for overestimation of the true effect.
cRated down 1 level for serious imprecision. A threshold of clinical importance could not be estimated, but it was judged that the lower bound of the 95% CI includes the potential for little-to-no important difference.
dRated down 2 levels for very serious imprecision. The sample size is small for this composite end point; after the large majority of events assigned to the date of randomization due to treatment failure (which was the first component of the composite EFS end point), too few patients remained event-free to robustly assess the long-term effects on EFS.
e,fRated down 1 level for serious imprecision (results were from interim analysis of study with small sample size and low number of events). Rated down 1 level for risk of bias due to what appears to be a large amount of missing outcome data due to no postbaseline assessment.
gDid not rate down for risk of bias. Although only a subset of the population was represented, in which randomization may not be upheld, results appeared similar when compared to analysis of the full population. Rated down 2 levels for very serious imprecision. Using the null as the threshold, the point estimate suggests benefit while the lower bound of the CI suggests harm.
h,iRated down 2 levels for very serious study limitations because of risk of bias due to missing outcomes data (data were available for 9% to 33% of the study population). Rated down 1 level for serious imprecision. The between-group difference of EORTC QLQ-C30 subscales exceed the identified minimally important difference for the global health states subscale in this instrument. However, the 95% CI included the possibility of little-to-no difference. Statistical testing for this outcome was not adjusted for multiplicity in the study and should be considered as supportive evidence.
jRated down 1 level for serious imprecision. No threshold of clinical importance could be established; therefore, the null was used. The point estimate suggests benefit, but the 95% CI included the possibility of little-to-no difference.
kRated down 2 levels for very serious imprecision. No threshold of clinical importance could be established; therefore, the null was used. The point estimate suggests harm, but the 95% CI includes the possibility of little-to-no difference or benefit.
lRated down 1 level for serious imprecision. No threshold of clinical importance could be established; therefore, the null was used. The point estimate suggests benefit, but the 95% CI includes the potential for little-to-no difference.
Source: AGILE Clinical Study Report.38,39 Details included in the table are from the sponsor’s summary of clinical evidence.
Long-Term Extension Studies
No relevant long-term extension studies were submitted by the sponsor.
Indirect Comparisons
Description of Studies
One report of 4 indirect treatment comparisons (ITCs) — 1 network meta-analysis [NMA] and 3 matching-adjusted indirect comparisons [MAICs] — was submitted by the sponsor to compare the treatment benefits and harms of ivosidenib plus azacitidine with other active therapies for the treatment of IDH1-mutated AML. A feasibility assessment was conducted to determine the feasibility of conducting indirect comparisons in the study population for the outcome of interest and to assess the heterogeneities across the included studies. The efficacy of ivosidenib versus comparators (venetoclax plus azacitidine, azacitidine, LDAC, decitabine, venetoclax plus LDAC, and glasdegib plus LDAC) on OS, EFS, CR rates, and transfusion requirement were evaluated, based on evidence from 6 RCTs.
Efficacy Results
For this submission, venetoclax plus azacitidine was identified as the most relevant comparator. As per the clinical experts consulted for this review, it is currently the most commonly used therapy in the patient population of interest. Evidence comparing ivosidenib plus azacitidine to venetoclax plus azacitidine was only available through a sponsor-submitted ITC report. The rarity of the population of interest limits the size and number of clinical studies completed with potential comparators and adds to the practical challenges when indirectly comparing treatment options. Based on the results of the NMA and MAICs, the evidence is insufficient to conclude whether ivosidenib plus azacitidine differs from venetoclax plus azacitidine in terms of OS, EFS, CR rates, or transfusion requirement in patients with untreated AML. The limitations associated with the ITCs included limited evidence from 6 RCTs, heterogeneity in the included trials, and imprecision of study results from the wide credible intervals (CrIs) or CIs for these outcomes.
Harms Results
Harm outcomes were not assessed in the ITCs.
Critical Appraisal
There was no a priori protocol for the ITCs; therefore, it cannot be known whether the analyses presented were selected from multiple analyses of the data. Although appropriate methods were used to reduce the risk of bias and error in data extraction, it was unknown if the risk of bias in the included trials was assessed by 2 independent reviewers. In addition, risk of bias was assessed at the level of the trial, rather than at the level of the reported results (i.e., per outcome), which ignores that risk of bias can vary by reported result within a trial. Some of the studies included within the NMA had some potential for risk of bias.
Six RCTs were included in the NMA. Heterogeneities were identified in the analysis populations, which included IDH1 mutation status, gender, type of AML diagnosis, cytogenic risk, performance status, median bone marrow blast, differences in placebo effect across placebo-controlled studies, and differences in the definition of EFS. For the time-to-event comparisons (e.g., EFS), lengths of follow-up were different, and with longer follow-up it may be expected that the HR would be attenuated, even when the proportional hazards (PH) assumption is not formally violated. The bias would likely favour the study drug. These differences would undermine the validity of the NMA, which relies on the transitivity assumption being upheld. The use of fixed-effect models was chosen based on the deviance information criterion. However, the use of fixed rather than random effects models means that the CrIs are unlikely to adequately express the uncertainty arising from the heterogeneity. The limited number of included studies did not allow for meta-regression or other techniques to adjust for differences in effect modifiers across studies within the NMA. The rarity of the population of interest limits the size and number of clinical studies completed with potential comparators and adds to the practical challenges when indirectly comparing treatment options.
In the NMA, given the lack of closed loops in the networks, consistency in the ITC analyses could not be tested, which increases the level of uncertainty. When comparing ivosidenib plus azacitidine with other combination regimens, the 95% CrIs for the point estimates were wide for some efficacy outcomes and spanned the null; therefore, confidence in the relative effect estimates for efficacy was limited because of the imprecision indicated by the wide CrIs for these outcomes, which precludes any conclusions as to which treatment may be favoured.
In the MAICs, the following potential effect modifier or prognostic factors were identified through the literature and a deliberating process by the sponsor: age, gender, ECOG performance status, type of AML, cytogenetic risk of AML, bone marrow blasts, and IDH1 mutation. The clinical experts consulted for this review agreed that these are relevant effect modifiers and prognostic variables. However, it is unclear if the identification of potential effect modifiers through the literature would be sufficient to identify all relevant treatment effect modifiers. The populations in the AGILE study and the other comparator studies were weighted and matched. Within the unanchored MAIC there was no reported estimate of the potential residual bias due to unadjusted confounders; as a result, the magnitude of residual confounding remains uncertain.
Before adjustment, the median OS and EFS for the placebo plus azacitidine groups were substantially different, suggesting reduced comparability of the populations. The main differences for the 2 studies used (AGILE and VIALE-A) is that in the AGILE study, the patients were younger and had a better ECOG performance status and a lower proportion of the patients had high-risk cytogenic status. The effective sample size (ESS) for the anchored MAICs was reduced by approximately one-third, suggesting that the results are heavily influenced by a subset of the sample population in the trial who may not be representative of the full sample population. The reduction in the ESS and the sample size in general resulted in wide CIs. Furthermore, there is uncertainty about comparing the population with IDH1 mutation to the intention-to-treat (ITT) population in the VIALE-A study. It was not possible to adjust for this factor.
The study population for this review includes patients with AML with IDH1 mutation who are ineligible for intensive chemotherapy. However, most of the selected trials were not specifically for IDH1-mutated AML. No other studies included only patients with IDH1 mutation, and it is not clear in the other included trials whether there were separate results for this particular subgroup. The prognostic significance of IDH1 status in AML, or whether IDH1 status may be a treatment effect modifier, remains uncertain. According to the clinical experts consulted for this review, the effect modifiers identified in patients with AML by the sponsor are also considered effect modifiers in patients with IDH1-mutated AML.
In this ITC report, several efficacy outcomes were analyzed, such as OS, EFS, and CR rates (not evaluated in the MAICs). However, other efficacy end points of interest to patients and clinicians (e.g., HRQoL), as well as harms, were not investigated. Therefore, the relative treatment effect of ivosidenib plus azacitidine versus relevant comparators on patients’ HRQoL and on harms remains unknown.
Studies Addressing Gaps in the Evidence From the Systematic Review
No relevant studies addressing gaps in the evidence from the systematic review were submitted by the sponsor.
Conclusions
Adult patients with newly diagnosed AML with an IIDH1 R132 mutation who are not eligible to receive intensive induction chemotherapy have a poor prognosis. Patients and clinicians highlighted the need for new treatments that prolong life, improve remission, reduce transfusion requirements, and maintain HRQoL. Evidence from a randomized, double-blind, phase III RCT (the AGILE study) showed that treatment with ivosidenib plus azacitidine likely results in a clinically important increase in the probability of OS at 12 months and 24 months compared to placebo plus azacitidine in the target population. Evidence from the trial also showed that ivosidenib plus azacitidine likely results in a clinically important increase in the probability of EFS at 6 months. EFS was a composite end point driven by treatment failure events; postbaseline, too few patients remained event-free to robustly characterize other components of the end point (i.e., relapse and death). The rates of CR, as well as CR plus CRi, and the need for transfusions may be improved with treatment with ivosidenib plus azacitidine compared with placebo plus azacitidine. Evidence on HRQoL was very uncertain because of the limitations of the analyses, including risk of bias due to missing data and imprecision. In terms of harms, evidence from the AGILE study suggested that treatment with ivosidenib plus azacitidine may result in an increase in differentiation syndrome but likely results in fewer infections and SAEs than treatment with placebo plus azacitidine.
There is a lack of direct comparative evidence between ivosidenib plus azacitidine and other relevant treatments for patients with AML who are not eligible for intensive chemotherapy, such as venetoclax plus azacitidine, which is currently the most commonly used treatment in the target patient population. Indirect evidence from a sponsor-submitted NMA of 6 trials and 3 MAICs comparing patients from the AGILE study to patients treated with venetoclax plus azacitidine in the VIALE-A study was insufficient to conclude whether treatment with ivosidenib plus azacitidine differs from treatment with venetoclax plus azacitidine in terms of OS, EFS, CR rates, and transfusion requirements. There was substantial uncertainty in the treatment effect estimates (indicated by wide CrIs) from the ITCs because of limited efficacy data and important heterogeneity across studies. No comparisons of HRQoL or harms, which are important to patients and clinicians, were conducted.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of ivosidenib (tablets, 250 mg, oral use) in combination with azacitidine for the treatment of adult patients with newly diagnosed AML with an IIDH1 R132 mutation who are not eligible to receive intensive induction chemotherapy.
Disease Background
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by our review team.
AML is a heterogeneous hematologic malignancy characterized by the clonal expansion of myeloid blasts in the bone marrow, peripheral blood, and/or other tissues.1,2 Although the cause of AML is not known, several factors are associated with an increased risk of this disease, such as increasing age, male sex, genetic factors, environmental factors and lifestyle, drugs, chemical exposure, and antecedent blood disorders.40 Commonly reported signs of AML are anemia, leukopenia, neutropenia, and thrombocytopenia, which result from the dysfunctional clonal expansion of myeloid progenitor cells. Typical symptoms of AML include fatigue, pale skin, dyspnea, infection, dizziness, headache, and coldness in hands and feet.3-5 Furthermore, leukopenia and neutropenia increase the risk of infections and fever, while thrombocytopenia increases the likelihood of bruising, bleeding, frequent or severe nosebleeds, bleeding gums, and heavy menstrual bleeding. Other symptoms include weight loss, night sweats, and loss of appetite.6,7 Occasionally, patients experience hepatomegaly, splenomegaly, or a soft tissue mass due to myeloid sarcoma.5
AML is 1 of the most aggressive forms of leukemia.5 Poorer prognosis is associated with increased age,41,42 secondary AML (AML after prior diagnosis of myelodysplasia, myeloproliferative neoplasm, or aplastic anemia, as opposed to de novo AML, in which patients have no clinical history of prior myelodysplastic syndrome, myeloproliferative disorder, or exposure to potentially leukemogenic therapies or agents),43 and certain molecular subtypes.2 The Cancer Quality Council of Ontario has reported age-standardized 1-year (2017 to 2018) and 5-year survival rates (2014 to 2018) of 42.1% and 19.9%, respectively.8 Furthermore, AML mortality is strongly related to age, with the highest mortality rates in older people.41 Five-year net survival (based on the combined results from 2010 to 2012) reported by Statistics Canada was 62% for people aged 15 to 44 years, 44% for people aged 45 to 54 years, 24% for people aged 55 to 64 years, 10% for people aged 65 to 74 years, and 3% for people aged 75 years and older.42
The prevalence of AML ranges from 0.6 to 11.0 per 100,000 persons for all age categories, genders, and ethnicities globally.9,10 The national age-standardized incidence rate for AML was reported to be 3.8 per 100,000 persons by Statistics Canada in 2018.11 CCO and the Cancer Quality Council of Ontario have reported relatively higher age-standardized incidence rates of 4.4 and 4.6 per 100,000 persons in Ontario, in 2016 and 2018, respectively.44,45 Approximately 1,600 people in Canada were diagnosed with AML in 2022.12 It is estimated that 6% to 10% of all people with AML carry an IDH1 mutation (with an estimated incidence ranging from 0.24 to 0.40 per 100,000 persons).13-20 The incidence of IDH1-mutated AML is low, and it is considered to be a rare disease.21
Diagnosis of AML is based on morphology, immunophenotyping, cytogenetics and molecular cytogenetics, molecular testing, demographics and medical history, detailed family history, patient bleeding history, and performance status.1,46-48 Approximately 40% to 50% of people with newly diagnosed AML are ineligible for standard induction chemotherapy regimens because of older age, poor Karnofsky performance status or ECOG performance status, and/or comorbid conditions.12,22-25 Multiple international guidelines, such as those of the National Comprehensive Cancer Network, European LeukemiaNet, the American Society of Hematology and the College of American Pathologists, and the European Society for Medical Oncology, recommend testing for IDH1 mutations to identify patients who may benefit from IDH1-targeted treatments.1,47-49
Standards of Therapy
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by our review team.
The majority of patients with AML are aged 60 years or older. While results of treatment have improved steadily in younger adults over the past 20 years, there have been limited changes in outcomes among older adults. When treated with chemotherapy alone, this age group has an estimated 2-year survival probability of approximately 20% and 10% at 4 years and 5 years, respectively. The reasons for the unsatisfactory outcome in older adults likely relate to the increased frequency of unfavourable cytogenetics among older patients with AML, a greater frequency of antecedent myelodysplasia, as well as reduced ability to tolerate intensive chemotherapy. High-dose chemotherapy is not beneficial to older adults with AML. There has been an intense interest in the introduction of new treatment modalities.50 The patient and clinician groups that provided input for this submission and the clinical experts consulted for this review indicated that the unmet therapeutic need for patients with AML stems from the poor outcomes (e.g., disease progression or relapse after previous remission, transfusion dependency, intolerable side effects, short life expectancy) in this patient group despite the currently available treatments and from the limited treatment options available if the patients’ current therapies fail. According to the clinical experts, the treatment goals for patients with AML who are not eligible to receive intensive induction chemotherapy are to prolong life, extend time in remission, alleviate symptoms, reduce dependency on blood transfusion, reduce infections, and improve QoL.
Treatment options for patients with newly diagnosed AML who carry a mutation in the IDH1 enzyme and are ineligible for the standard intensive chemotherapy (because of poor performance status, a comorbid medical condition, or age) are limited. In Canada, treatments that are currently publicly funded for patients with AML who are ineligible for standard intensive chemotherapy, but not specific to those carrying an IDH1 mutation, include:1,26-29
venetoclax combined with azacitidine (currently the mainstay and most frequently used treatment in the target patient population)
monotherapy with azacitidine or LDAC if the patients are not considered candidates for combination or targeted therapy.
Before the introduction of venetoclax combination therapies, single-agent azacitidine or LDAC were recommended for patients with AML who were not eligible for intensive induction chemotherapy.51 Azacitidine and LDAC are widely available and reimbursed across Canada. Venetoclax (a small-molecule inhibitor of BLC-2, a protein that inhibits cells from programmed cell death26) plus azacitidine or LDAC, or glasdegib (an inhibitor of the Hedgehog signal transduction pathway27) plus LDAC, have been approved by Health Canada for the treatment of newly diagnosed AML in adult patients aged 75 years or older or who are otherwise not eligible to receive intensive induction chemotherapy. Glasdegib received a negative reimbursement recommendation in 2020 and, according to the clinical experts consulted for this review, is not routinely used in Canadian clinical practice. Venetoclax plus LDAC received a negative reimbursement recommendation in 2021, and according to the clinical experts consulted for this review, this regimen is not funded in jurisdictions in Canada, although some patients may have access to this treatment via a compassionate program. The clinical experts indicated that venetoclax plus azacitidine is currently the most commonly used treatment for the target patient population in Canada. Venetoclax plus azacitidine was recommended for reimbursement for patients with newly diagnosed AML aged 75 years or older or who have comorbidities that preclude the use of intensive induction chemotherapy. With the currently approved and reimbursed treatment options in Canada, the median OS in patients with AML, regardless of IDH mutation status, is approximately 5 months with LDAC,52-54 10 months with azacitidine,24,55 and 15 months with venetoclax plus azacitidine.55 In patients with AML with IDH1 mutation who are not eligible for intensive induction chemotherapy, the quality of evidence for treatment with venetoclax plus azacitidine is low, limited to post hoc subgroup analyses with a small number of patients.55-57
Drug Under Review
Ivosidenib has a non-cytotoxic mechanism of action. It is an inhibitor of the mutant IDH1 enzyme. Mutant IDH1 converts alpha-ketoglutarate to 2-hydroxyglutarate, which blocks cellular differentiation and promotes tumorigenesis in both hematologic and nonhematologic malignancies. The mechanism of action of ivosidenib beyond its ability to reduce 2-hydroxyglutarate and restore cellular differentiation is not fully understood.30
On July 19, 2024, ivosidenib in combination with azacitidine was approved by Health Canada for the treatment of adult patients with newly diagnosed AML with an IIDH1 R132 mutation who are not eligible to receive intensive induction chemotherapy. The sponsor’s reimbursement request is aligned with the Health Canada–approved indication. The IIDH1 R132 mutation must be confirmed before the combination regimen is initiated.30
Ivosidenib is provided as 250 mg film-coated tablets. The recommended dose is 500 mg ivosidenib (2 × 250 mg tablets) taken orally once daily. Ivosidenib should be started on cycle 1 day 1 and administered once daily during the 28-day cycle. It should be started in combination with azacitidine at 75 mg/m2 of body surface area, intravenously or subcutaneously, once daily on days 1 to 7 of each 28-day cycle. The first treatment cycle of azacitidine should be given at 100% of the dose. It is recommended that patients be treated for a minimum of 6 cycles. Treatment should be continued until disease progression or until treatment is no longer tolerated by the patient.30 Patients with AML and treated with ivosidenib have reported differentiation syndrome, which can be life-threatening or fatal if not treated.30
Key characteristics of ivosidenib plus azacitidine are summarized in Table 3, with other treatments available for untreated or newly diagnosed AML.
Table 3: Key Characteristics of Ivosidenib, Venetoclax, Azacitidine, and Cytarabine
Characteristic | Ivosidenib + azacitidine | Venetoclax + azacitidine | Azacitidine | LDAC |
|---|---|---|---|---|
Mechanism of action | Ivosidenib is an inhibitor of the mutant IDH1 enzyme | Venetoclax is a selective and orally bioavailable small-molecule inhibitor of BCL-2, a protein that inhibits cells from programmed cell death. | Multiple mechanisms, including inhibition of DNA, RNA, and protein synthesis; incorporation into RNA and DNA; and activation of DNA damage pathways. | Suppression of the development of cell-mediated immune responses, such as delayed hypersensitivity skin reaction to dinitrochlorobenzene. Suppression of antibody responses to E. coli VI antigen and tetanus toxoid in males. |
Indicationa | For the treatment of adult patients with newly diagnosed AML with an IIDH1 R132 mutation who are not eligible to receive intensive induction chemotherapy. | For the treatment of patients with newly diagnosed AML who are 75 years or older or who have comorbidities that preclude use of intensive induction chemotherapy. | For the treatment of adult patients who are not eligible for hematopoietic stem cell transplant with AML with 20% to 30% blasts and multi-lineage dysplasia, according to WHO classification. | Primarily for induction and maintenance of remission in acute leukemia in both adults and children. |
Route of administration | Ivosidenib: oral Azacitidine: SC or IV | Venetoclax: oral Azacitidine: SC | SC | IV |
Recommended dose | Ivosidenib: 500 mg orally once daily, 28-day cycles, until disease progression Azacitidine: 75 mg/m2 SC or IV for 7 days of 28-day cycles, until disease progression | Venetoclax: 100 mg orally on day 1, 200 mg orally on day 2, 400 mg orally on day 3, 400 mg orally on day 4 and onward, 28-day cycles, until disease progression Azacitidine: 75 mg/m2 SC for 7 days of 28-day cycles, until disease progression | 75 mg/m2 SC for 7 days of 28-day cycles, until disease progression It is recommended that patients be treated for a minimum of 6 cycles unless unacceptable toxicities occur, or standard supportive care has proved unsuccessful | Usually, cytarabine is used in combination with other cytotoxic drugs; dosing should be adapted based on the treatment effect and toxicities AML (induction remission) in adults: 200 mg/m2 daily by continuous infusion for 5 days, total dose 1,000 mg/m2 AML (maintenance) in adults: modifications of induction programs and, in general, similar schedules as were used during induction |
Serious adverse effects or safety issues | Differentiation syndrome | TLS; serious infections | Thrombocytopenia; renal failure including fatalities | Cardiomyopathy with subsequent death; GI toxicity, at times fatal; acute pancreatitis; CNS toxicity; severe neurologic adverse reactions, paraplegia, necrotizing leukoencephalopathy, and spinal cord toxicity; infection; pulmonary toxicity, ARDS, and pulmonary edema; myelosuppression |
AML = acute myeloid leukemia; ARDS = adult respiratory distress syndrome; CNS = central nervous system; GI = gastrointestinal; LDAC = low-dose cytarabine; RNA = ribonucleic acid; SC = subcutaneous; TLS = tumour lysis syndrome.
aHealth Canada–approved indication.
Sources: Product monographs for ivosidenib,30 venetoclax,26 and cytarabine.29
Perspectives of Patient, Clinical Input and Drug Program Input
Patient Group Input
This section was prepared by the review team based on the input provided by patient groups. The full original patient inputs received by us have been included in the Patient, Clinical Input, and Drug Program Input section of this report.
We received 2 patient group submissions, from the LLSC and from Heal Canada. The LLSC is a national organization with charitable status dedicated to finding a cure for blood cancers and improving the QoL of people affected by blood cancers and their families by funding life-enhancing research and providing educational resources, services, and support. Heal Canada is a registered not-for-profit organization that aims to empower patients, improve health care outcomes, and advocate for equitable access to quality health care across Canada.
Data for the LLSC input were gathered using 1 online survey, distributed through various social media channels and directly by email in March 2024. The survey was developed and distributed by the LLSC, in English only. Eighty-three respondents proceeded with the survey, of which 7 respondents identified as having the IDH1 mutation. The LLSC also conducted 2 1-on-1 interviews with patients currently living with AML.
Heal Canada launched an online survey to assess different characteristics of patients living with blood cancer on February 27, 2024. Of the 22 respondents, 5 had been diagnosed with AML. Information was also gathered from semistructured interviews with 2 patients and 2 caregivers.
Most respondents in both patient groups reported that the mental, physical, and financial effects of AML have significant impact on the lives of patients and caregivers. According to Heal Canada, the predominant symptoms of AML are extreme fatigue, weakness, and tiredness, which make it difficult to accomplish basic daily tasks such as showering, washing dishes, cleaning the house, and shopping. People with this condition tend to be heavily dependent on their caregivers. The LLSC revealed that both patients and caregivers are forced to change how, if, and when they can interact with the people close to them, which has both mental and physical impacts for those affected. The caregiver burden is significant, especially for older patients and those living alone before being diagnosed with AML.
In terms of the currently available treatments, the LLSC highlighted that doses of the prescribed medications have to be decreased, or treatment has to be discontinued, when there are intolerable side effects or no response to treatment. However, if the available treatments fail and stem cell or bone marrow transplant is not an option, the only alternative is often best supportive care until death. Heal Canada provided some details about patients who mentioned receiving azacitidine or best supportive care (e.g., blood transfusion) and noted that both treatments necessitate frequent blood transfusion, which remains the most critical burden for patients with AML. Heal Canada also indicated that current treatment options have limited efficacy and significant harms and that patients may not receive active treatment but rather best supportive care. Patients expressed that they often feel trapped, with no real options to treat their cancer and improve their QoL.
Both patient groups indicated that important patient outcomes included improved HRQoL (related to better control of anemia without transfusion or with fewer transfusions, as well as a lower infection rate), improved disease control, and prolonged survival.
No patients or caregivers from Heal Canada had experience with ivosidenib, while the LLSC interviewed 1 patient with previous experience with ivosidenib. The patient was initially diagnosed with IDH1-mutated AML in June 2021 and started induction chemotherapy treatments immediately. After relapse on induction chemotherapy, the patient started ivosidenib with great response and minimal side effects, and she had been in remission since then. She was aware of the option of getting a transplant, but she was also frustrated about not having a donor.
Heal Canada reported that the turnaround time of companion testing is different across the country, and the LLSC commented that treatment with ivosidenib may be delayed in some treatment facilities if laboratory results are not made available within a short window of time. The LLSC noted that testing for IDH1 mutation is part of the next-generation sequencing panel, which is conducted on all patients with AML, and does not require an additional blood test.
Clinician Input
Input From Clinical Experts Consulted for this Review
All our review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of AML.
Unmet Needs
The clinical experts identified the following unmet needs associated with the currently available treatments for patients with AML who are ineligible for intensive induction chemotherapy: first, not all patients respond to available therapies, and effective treatments for this patient population are lacking, making the outcomes for patients with AML (with or without IIDH1 R132 mutation) who are not eligible for intensive chemotherapy extremely poor; second, patients who respond to available therapy eventually relapse and succumb to their disease. Therefore, the clinical experts indicated that for patients in the target population, the most important treatment goals are to prolong remission and survival, reduce transfusion requirements, reduce the risk of infection and bleeding, and improve HRQoL.
Place in Therapy
The clinical experts indicated that based on its unique mechanism of action (inhibition of the mutated IIDH1 R132) and the available clinical evidence, ivosidenib would be reserved as first-line therapy for patients with AML who carry the IIDH1 R132 mutation and who are not eligible for intensive chemotherapy because of their age or comorbidities. Ivosidenib in combination with azacitidine could potentially replace the currently available combination therapy for these patients.
Patient Population
The clinical experts stated that only patients with a diagnosis of de novo AML with IIDH1 R132 mutation who are not eligible for induction chemotherapy would be eligible to receive treatment with ivosidenib. The experts also noted that testing for the IDH1 mutation is routinely performed in many specialized leukemia centres across Canada, although not in all jurisdictions (e.g., not in Manitoba). However, delays of days to weeks in receiving the test results have been reported, which makes it challenging for the clinician to select the appropriate treatment for patients with newly diagnosed AML; they can either initiate treatment with the currently used therapies before a patient’s IDH1 status is verified or wait until the patient’s IDH1 mutation status can be obtained. In addition, the clinical experts suggested that some flexibility should be applied in using ivosidenib plus azacitidine in patients with slightly lower ECOG performance status than in the trial.
Assessing the Response to Treatment
The experts noted that important outcomes for patients with AML are survival, HRQoL, response rates (in particular CR), and safety. Other outcomes of interest to the clinicians include transfusion requirements and infection rates. The experts also noted that in clinical practice, patients’ response to treatment are typically assessed every 28 days, corresponding to the length of treatment cycles for azacitidine.
Discontinuing Treatment
According to the clinical experts consulted for this review, treatment with a combination of ivosidenib and azacitidine will be discontinued if there is evidence of disease progression, as demonstrated by either an increased number of blasts in the bone marrow according to the standards of the International Working Group or, if a bone marrow aspiration is not performed, worsening of blood counts and/or an increased number of circulating blasts. Other reasons for treatment discontinuation include intolerable AEs related to the treatment and patient preference.
Prescribing Considerations
The clinical experts noted that patients should be treated by a hematologist and/or hematologist or oncologist with experience in AML management. Treatment with ivosidenib can be administered in both inpatient and outpatient settings.
Clinician Group Input
This section was prepared by the review team based on the input provided by clinician groups. The full original clinician group input(s) received by the team have been included in the Patient, Clinical Input and Drug Program Input section of this report.
Two clinician groups provided input for the review of ivosidenib in combination with azacitidine: the LLSC Clinician Network and the OH-CCO Hematology Cancer Drug Advisory Committee.
In general, the input from the 2 clinician groups was consistent with the input provided by the clinical experts consulted by the review team. The treatment goals for this patient population would be to prolong life, improve QoL, reduce transfusion requirements, and experience remission. The clinician groups noted that the current publicly funded treatment options for patients with AML who are not eligible for intensive chemotherapy include venetoclax plus azacitidine, single-agent azacitidine, LDAC, and best supportive care. The OH-CCO Drug Advisory Committee also mentioned venetoclax plus LDAC as an available therapy. However, not all patients respond to these therapies. In addition, both clinician groups suggested that treatment with azacitidine plus venetoclax is associated with increased risk of neutropenic fever and infections compared to azacitidine alone. According to the clinicians, infections may result in hospitalizations, which might last days to weeks depending on severity. The clinicians from LLSC Clinician Network added that no tumour lysis syndrome monitoring is required with ivosidenib plus azacitidine. The clinician groups noted that specific inhibitors may offer a chance for increased treatment response and suggested ivosidenib plus azacitidine be considered as first-line therapy and become the new standard of care for adult patients with newly diagnosed IDH1-mutated AML who are not eligible for intensive induction chemotherapy or stem cell or bone marrow transplant. Both clinician groups indicated that remission rate and stabilization and improvement in the frequency and severity of symptoms — such as improvement in blood counts, fewer transfusions, leukemia-free survival, and OS, using usual leukemia response timelines — are the outcomes used to determine whether a patient is responding to ivosidenib plus azacitidine. Reasons for treatment discontinuation identified by the clinician groups included disease progression, intolerable side effects, and patient preference. Both clinician groups noted that ivosidenib plus azacitidine can be given in the inpatient and outpatient settings, or even in community centres that have experience treating acute leukemias.
Both the LLSC Clinician Network and the OH-CCO Drug Advisory Committee noted that timely results of testing for IDH1 mutation are required to identify patients who would benefit from and be eligible for this treatment.
Drug Program Input
The drug programs provide input on each drug being reviewed through our reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by the review team are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Advice from the clinical experts |
|---|---|
Relevant comparators | |
In the AGILE study, ivosidenib + azacitidine was compared to placebo + azacitidine. Ivosidenib + azacitidine was not compared to other treatment options, such as azacitidine + venetoclax or LDAC in this study. | Comment from the drug programs to inform pERC deliberations. |
Considerations for initiation of therapy | |
Eligibility criteria in the AGILE study were age > 18 years, confirmed IDH1-mutated AML, ECOG performance status 0 to 2. Can patients with an ECOG performance status > 2 receive treatment with ivosidenib + azacitidine? | The clinical experts indicated that patients with an ECOG performance status score of 3 or 4 are usually excluded from the clinical trials. Even though a clinical benefit from treatment with ivosidenib may be derived for these patients, the extent of the benefit is unknown. The clinical experts noted that in clinical practice, some clinicians use a different scale to assess a patient’s performance status, such as KPS. This is a more detailed scale, with scores ranging from 0 (death) to 100 (normal), and provides more information than the ECOG performance status scale when quantifying a patient’s general well-being. The experts suggested that there may be patients whose ECOG performance status falls between the scores of 2 and 3 and who may benefit from treatment with ivosidenib. |
Why would ivosidenib + azacitidine be considered for treatment vs. venetoclax + azacitidine, and vice versa? Is 1 preferred over the other? | The clinical experts noted that in the AGILE study (pivotal study of this submission), all patients had an IDH1 mutation. In the VIALE-A study (venetoclax plus azacitidine vs. placebo plus azacitidine), eligible patients did not exclusively have an IDH1 mutation. Based on the mechanism of action of ivosidenib (inhibition of the mutant IDH1 enzyme), the clinical experts anticipated that ivosidenib plus azacitidine may be superior to venetoclax plus azacitidine in patients with AML with an IDH1 mutation. Therefore, patients without an IDH1 mutation would not be candidates for treatment with ivosidenib. |
Considerations for continuation or renewal of therapy | |
An ECG is required before treatment with ivosidenib + azacitidine, weekly for the first 3 weeks of therapy and monthly for the duration of therapy. | Comment from the drug programs to inform pERC deliberations. |
Considerations for discontinuation of therapy | |
In the AGILE study, treatment with the study drug was discontinued if disease progression or intolerable toxicities occurred. What is the definition of disease progression in patients with AML in clinical practice? | The clinical experts indicated that disease progression is observed if a patient obtained a response but thereafter lost the response or if the patient did not have a response after treatment initiation and the disease progressed. The clinical experts noted that disease progression is demonstrated if CR based on the bone marrow is lost and/or there is increased number of blasts in the bone marrow. |
Considerations for prescribing of therapy | |
In the AGILE study, ivosidenib was given as oral tablet of 500 mg (2 × 250 mg tablets) once daily until progression or until no longer tolerated. Should ivosidenib be given with alternative dosing schedules of azacitidine (6 day or 5 to 2-2)? | The clinical experts indicated that in the AGILE study, patients received ivosidenib once daily from day 1 to day 28. It is unclear whether changing the schedule of ivosidenib to 6 days or 5 to 2-2 would have an impact on the clinical effectiveness of the ivosidenib + azacitidine combination regimen. The experts also noted that in clinical practice, most clinicians would treat patients in line with the protocol of clinical trials. Therefore, ivosidenib may not be given with alternative dosing schedules of azacitidine. |
Ivosidenib is administered with SC azacitidine. On days 1 to 7 of each 28-day cycle, some jurisdictions will need to coordinate injectable (SC) and oral therapy (managed separately). | Comment from the drug programs to inform pERC deliberations. |
Generalizability | |
For patients who are currently on azacitidine therapy, can ivosidenib be added to azacitidine (time-limited need)? | The clinical experts indicated that for patients with an IDH1 mutation, it is reasonable to believe that patients who have received a limited number of cycles of azacitidine monotherapy could derive additional benefit if ivosidenib were to be added to azacitidine. The experts also suggested that the earlier the addition of ivosidenib (e.g., from cycle 1), the greater the benefit to patients. If patients are on venetoclax + azacitidine and respond well to the combination therapy, the treating clinician would usually continue the treatment and not switch the patients to ivosidenib. However, if the patients on venetoclax + azacitidine have a suboptimal response to this treatment (not obtaining a remission or remission with incomplete platelet recovery), they may be candidates to be switched to ivosidenib + azacitidine. It would be important for the patient and caregiver to have a detailed discussion with the treating clinician to guide this decision in the absence of robust direct evidence comparing venetoclax + azacitidine vs. ivosidenib + azacitidine. |
In the AGILE study, patients who had received previous treatment with an HMA (e.g., azacitidine or decitabine) for MDS or an IDH1 inhibitor were ineligible. In clinical practice, can patients who experience intolerance or toxicity with venetoclax + azacitidine be switched to ivosidenib + azacitidine? | The clinical experts indicated that some patients with an IDH1 mutation may be candidates to be switched to ivosidenib + azacitidine when experiencing intolerance or toxicity with venetoclax + azacitidine. However, the safety profile of venetoclax + azacitidine overlaps (except for differentiation syndrome) with that of ivosidenib + azacitidine, with the greatest toxicities for both combination regimens being related to cytopenia. Therefore, patients who do not tolerate treatment with venetoclax + azacitidine may not tolerate ivosidenib + azacitidine. |
Funding algorithm (oncology only) | |
The study drug may change the place in therapy of the comparator drugs. | Comment from the drug programs to inform pERC deliberations. |
Care provision issues | |
Drug preparation, storage, administration, or dispensing: During treatment with ivosidenib + azacitidine, there is a need to monitor the interactions between the study drug and CYP3A4. Potential dose reduction will be required if the drug is given in combination with CYP3A4 inhibitors. | Comment from the drug programs to inform pERC deliberations. |
Management of adverse effects:
| Comment from the drug programs to inform pERC deliberations. |
Companion diagnostics (e.g., access issues, timing of testing):
| The clinical experts noted that most, but not all, leukemia-treating centres have routine access to PCR testing for IDH1 mutation. The experts noted that the turnaround time varies across regions, ranging from a few days to up to 2 weeks. The experts indicated that the majority of the patients do not have an IDH1 mutation and will usually be treated with azacitidine or venetoclax + azacitidine initially. Approximately 5% of the older adult patients have an IDH1 mutation. It is reasonable to allow patients who are found to have an IDH1 mutation to be switched to ivosidenib + azacitidine once their IDH1 mutation status is confirmed. |
System and economic issues | |
Involvement of additional payers:
| Comment from the drug programs to inform pERC deliberations. |
Presence of confidential negotiated prices for comparators:
| Comment from the drug programs to inform pERC deliberations. |
5 to 2-2 = azacitidine was administered on days 1 through 5 and days 8 and 9 of each 28-day cycle; AML = acute myeloid leukemia; CR = complete remission; ECG = electrocardiogram; ECOG = Eastern Cooperative Oncology Group; HMA = hypomethylating agent; KPS = Karnofsky performance status; LDAC = low-dose cytarabine; MDS = myelodysplastic syndrome; PCR = polymerase chain reaction; pERC = CADTH pan-Canadian Oncology Review Expert Review Committee; SC = subcutaneous.
Clinical Evidence
The objective of this Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of ivosidenib (250 mg per tablet, oral use) in combination with azacitidine for the treatment of adult patients with newly diagnosed AML with an IIDH1 R132 mutation who are not eligible to receive intensive induction chemotherapy. The focus will be placed on comparing ivosidenib in combination with azacitidine to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of ivosidenib in combination with azacitidine is presented in 4 sections, with critical appraisal of the evidence included at the end of each section. The first section, the systematic review, includes the pivotal studies and RCTs that were selected according to the sponsor’s systematic review protocol. Our assessment of the certainty of the evidence in this first section using the GRADE approach follows the critical appraisal of the evidence. The second section would include sponsor-submitted long-term extension studies; however, none were submitted by the sponsor. The third section includes indirect comparisons from the sponsor. The fourth section would include additional studies that were considered by the sponsor to address important gaps in the systematic review evidence; however, no studies addressing gaps were submitted by the sponsor.
Included Studies
Clinical evidence from the following are included in the current review and appraised in this document:
Systematic Review
Contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
Description of Study
The AGILE study (also known as AG120-C-009) is an ongoing, phase III, multicentre, double-blind RCT assessing the efficacy and safety of ivosidenib in combination with azacitidine compared with placebo in combination with azacitidine in patients with newly diagnosed IDH1-mutated AML who were ineligible for intensive induction chemotherapy. The primary objective of the study was to compare EFS between ivosidenib and placebo (each combined with azacitidine), as described in Table 5. Key secondary objectives of this study included comparing the remission rates and OS in patients treated with ivosidenib plus azacitidine versus with placebo plus azacitidine. HRQoL, transfusion requirements, and harms were also assessed in the AGILE study.
All recruited patients underwent screening procedures within the 4 weeks before randomization to determine eligibility. A screening bone marrow aspirate (or peripheral blood sample if bone marrow aspirate was not available) was required for confirmation of IDH1 mutation at a central laboratory. Patients eligible for study treatment were randomized 1:1 to receive oral ivosidenib or placebo, both administered in combination with subcutaneous or IV azacitidine. The randomization schedule was generated by an independent statistical group, and the randomization assignment was implemented by interactive response technologies. The patients, investigators, sponsor, and clinical research unit staff who dealt directly with patients were blinded to the treatment assignment. Ivosidenib and matched placebo were packaged and labelled identically so that the study pharmacist remained blinded to treatment assignment. Randomization was stratified by de novo status (de novo AML and secondary AML) and geographic region (US and Canada; Western Europe, Israel, and Australia; Japan; and rest of the world). As of the DCO of March 18, 2021, 89 sites globally had enrolled patients, including 2 sites in Canada. A total of 295 patients were screened, of which 146 underwent randomization (72 to the ivosidenib plus azacitidine group and 74 to the placebo plus azacitidine group). As of the second DCO on June 30, 2022, 2 more patients were included in the AGILE study, 1 in each treatment group. Updated analyses based on the data available on the second DCO were performed for OS, transfusion requirements, and harms.
The IDMC reviewed the safety data as of March 18, 2021, based on the 146 patients enrolled in the AGILE study. A greater number of deaths were observed in the placebo plus azacitidine group than in the ivosidenib plus azacitidine group. This prompted another unblinded analysis for efficacy, which included OS, EFS, and clinical response, and led to the IDMC recommendation to halt recruitment to the study on May 12, 2021. Because of a notable difference in the number of deaths, which favoured ivosidenib, the IDMC recommended that trial recruitment should end early, treatment assignment should be unblinded, and crossover to ivosidenib should be allowed. Patients who were already receiving ivosidenib plus azacitidine could continue to receive treatment on the same assessment schedule. Before crossover to ivosidenib, the investigators evaluated the patients to determine their safety eligibility based on the inclusion and exclusion criteria of the study.
The characteristics of the AGILE study are summarized in Table 5.
Table 5: Details of Studies Included in the Systematic Review
Detail | AGILE |
|---|---|
Design and population | |
Study design | Phase III, multicentre, double-blind RCT |
Locations | 199 study sites participated in this study; 89 sites enrolled patients: Australia (3), Austria (2), Brazil (6), Canada (2), China (6), Czech Republic (1), France (14), Germany (7), Israel (3), Italy (6), Japan (4), Mexico (1), Netherlands (2), Poland (3), Russia (2), South Korea (5), Spain (13), Taiwan (5), UK (2), US (2) |
Patient enrolment dates | Start date: March 19, 2018 End date: May 27, 2021 (primary completion date was March 18, 2021) |
Randomized (N) | N = 146:
|
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Intervention | Ivosidenib 500 mg orally once daily + azacitidine 75 mg/m2/day, SC or IV, for 7 days, in 28-day cycles, until death, disease relapse, disease progression, development of unacceptable AE, confirmed pregnancy, withdrawal by patient, protocol violation, or end of study |
Comparator(s) | Placebo orally once daily + azacitidine 75 mg/m2/day, SC or IV, for 7 days, in 28-day cycles, until death, disease relapse, disease progression, development of unacceptable AE, confirmed pregnancy, withdrawal by patient, protocol violation, or end of study |
Study duration | |
Screening phase | 4 weeks |
Treatment phase | Treatment continued until death, disease relapse, disease progression, development of unacceptable AE, confirmed pregnancy, withdrawal by patient, protocol violation, or end of study |
Follow-up phase | All patients who discontinued study treatment without experiencing death, disease relapse, treatment failure, or withdrawal of consent were to be followed every day 1 (± 7 days) of weeks 9, 17, 25, 33, 41, and 53, and every 24 weeks thereafter for EFS until they experienced treatment failure, relapse, or death; until they withdrew from the study; until 173 EFS events had occurred or until deemed necessary by the IDMC Once the study was unblinded, survival follow-up continued; all patients who were alive after an EFS event were to be contacted every 8 weeks for survival follow-up until death, withdrawal by patient, loss to follow-up, or end of study |
Outcomes | |
Primary end point | EFS, defined as the time from randomization until treatment failure (i.e., patient does not experience CR by week 24), relapse from remission, or death from any cause, whichever occurred first |
Secondary and exploratory end points | Key secondary:
Additional secondary:
Exploratory: Evaluation of a variety of established and exploratory biomarkers for morphologic, functional, metabolic, and biologic changes over the course of treatment |
Publication status | |
Publications | Montesinos et al. (2022)21 Dohner et al. (2022)59 Institut de Recherches Internationales Servier (2024)60 |
AE = adverse event; AESI = adverse event of special interest; AML = acute myeloid leukemia; ANC = absolute neutrophil count; CNS = central nervous system; CR = complete remission; CRh = complete remission with partial hematologic recovery; CRi = complete remission with incomplete hematologic recovery; CRp = complete remission with incomplete platelet recovery; DOCR = duration of complete remission; DOCRh = duration of CR plus CRh; DOCRi = duration of CR plus CRi (including CRp); DOR = duration of response; ECG = electrocardiogram; ECHO = echocardiogram; ECOG PS = Eastern Cooperative Oncology Group performance status; EFS = event-free survival; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; HMA = hypomethylating agent; HRQoL = health-related quality of life; IDMC = Independent Data Monitoring Committee; LVEF = left ventricular ejection fraction; MDS = myelodysplastic syndrome; MLFS = morphologic leukemia-free state; MUGA = multigated acquisition; ORR = objective response rate; OS = overall survival; PR = partial remission; RBC = red blood cell; RCT = randomized controlled trial; SAE = serious adverse event; SC = subcutaneous; TTCR = time to complete remission; TTCRh = time to CR plus CRh; TTCRi = time to CR plus CRi (including CRp); TTR = time to first response; ULN = upper limit of normal.
Source: AGILE Clinical Study Report.38 Details in the table are from the sponsor’s summary of clinical evidence.
Populations
Inclusion and Exclusion Criteria
Eligible patients in the AGILE study were aged 18 years or older and had a centrally confirmed diagnosis of previously untreated AML with IIDH1 R132 mutation confirmed using an appropriate diagnostic test. Patients were required to be ineligible for intensive induction chemotherapy and to have an ECOG performance status score of 0 to 2 (on a 5-point scale, in which higher scores indicate greater disability). Additional eligibility criteria included no previous treatment with an IDH1 inhibitor or hypomethylating agent for myelodysplastic syndrome, as well as adequate hepatic and renal function. Patients were excluded if they were candidates for intensive induction chemotherapy for their AML, had received any prior treatment for AML (except for non-oncolytic treatments, such as hydroxyurea or leukapheresis) or prior hypomethylating agent for myelodysplastic syndrome, had received prior IDH1 inhibitor therapy, had severe cardiac disorder or pulmonary disorder, or had an active uncontrolled systemic fungal, bacterial, or viral infection without improvement despite appropriate antibiotics, antiviral therapy, and/or other treatment. Full inclusion and exclusion criteria, including the definition of ineligibility for intensive induction chemotherapy, in this study are provided in Table 5.
Interventions
Eligible patients in the AGILE study were randomly assigned to receive oral ivosidenib or placebo, both administered in combination with azacitidine. Ivosidenib 500 mg or matched placebo was administered orally once daily during weeks 1 to 4 in continuous 28-day cycles. Crossover between treatment arms was not permitted until the study was unblinded after the primary end point (EFS) reached statistical significance at an unplanned early interim analysis. Patients who were initially randomized to placebo plus azacitidine who met key safety eligibility criteria were given the opportunity to receive ivosidenib plus azacitidine following unblinding. Patients who were already receiving ivosidenib plus azacitidine could continue to receive this treatment on the same assessment schedule. Azacitidine at a dose of 75 mg/m2/day was administered subcutaneously or intravenously for 1 week every 4 weeks. A full 7 days of azacitidine was required, but as per institutional practice, a schedule of 5 days of daily dosing, followed by no dose received on the weekend, and 2 daily doses given again at the start of the next week, was allowed; the same schedule was to be used for each patient throughout the duration of treatment, when possible. Patients were to be treated for a minimum of 6 cycles of combination therapy.
Treatment was to be discontinued if any of the following occurred: death, disease relapse, disease progression, treatment failure (defined as patients with a response less than CR after receiving treatment for at least 24 weeks), clinical progression (within 24 weeks) not confirmed by International Working Group assessment, patient lost to follow-up, development of unacceptable AEs, confirmed pregnancy, withdrawal by patient, protocol violation, or end of study.
During the study, treatment with strong CYP3A4 inducers, sensitive CYP3A4 substrate medications with narrow therapeutic windows, and anticancer therapy (with the exception of hydroxyurea) were not allowed when patients were receiving the study treatment. Patients could receive analgesics, antiemetics, anti-infectives, antipyretics, antimicrobial prophylaxis, and blood products, as necessary. Information on subsequent therapies was collected during the EFS and OS follow-up periods.
Outcomes
A list of efficacy end points assessed in this Clinical Review Report is provided in Table 6, followed by descriptions of the outcome measures. Summarized end points are based on the outcomes included in the sponsor’s summary of clinical evidence as well as any outcomes identified as important to this review according to the clinical experts consulted for this review and input from patient and clinician groups and from public drug plans. Using the same considerations, the review team selected the end points considered most relevant to inform the expert committee deliberations and finalized this list of end points in consultation with members of the expert committee. OS, EFS, CR, HRQoL (measured with the EORTC QLQ C-30), and transfusion requirement were assessed using GRADE. SAEs and select notable harms outcomes considered important for informing the expert committee deliberations were also assessed using GRADE. Results for the ORR are presented in Appendix 1 but are not appraised for certainty using GRADE because the main treatment goals in the study population were to prolong survival and improve HRQoL, as indicated by the clinical experts consulted for this review and noted by the input, and because there is no evidence that ORR is a validated surrogate for OS.
Table 6: Outcomes Summarized From the AGILE Study
Outcome measure | Time point | Type of outcome |
|---|---|---|
EFSa | Reported for DCO of March 18, 2021 6-month and 12-month results are presented in SoF table (Table 2) | Primary outcome |
OSa | Reported for DCOs of March 18, 2021, and June 30, 2022 12-month and 24-month results are presented in SoF table (Table 2) | Secondary outcome |
Treatment response | ||
CRa | Reported for DCO of March 18, 2021 Assessed at screening; day 1 (± 7 days) of weeks 9, 17, 25, 33, 41, and 53; every 24 weeks thereafter; at end of treatment; as dictated by physical exam and/or blood counts; and/or any time that disease progression was suspected | Secondary outcome |
CR + CRi | Secondary outcome | |
EORTC QLQ C-30 | Reported for DCO of March 18, 2021 Assessed at screening; day 1 (± 7 days) of weeks 9, 17, 25, 33, 41, and 53; every 24 weeks thereafter; at end of treatment; as dictated by physical exam and/or blood counts; and/or any time that disease progression was suspected | Secondary outcome |
Transfusion requirement | Reported for DCOs of March 18, 2021, and June 30, 2022 (proportion of patients with postbaseline transfusion independence are presented in this report) Assessed at screening; day 1 (± 7 days) of weeks 9, 17, 25, 33, 41, and 53; every 24 weeks thereafter; at end of treatment; as dictated by physical exam and/or blood counts; and/or any time that disease progression was suspected | Secondary outcome |
Hospital stays | Reported for DCO of March 18, 2021 | Secondary outcome |
Safety | ||
AEs | Reported for DCOs of March 18, 2021, and June 30, 2022 Assessed at all time points throughout study SAEs and notable harms at DCO of June 30, 2022, were assessed using GRADE | Secondary outcome |
SAEs | Secondary outcome | |
WDAEs | Secondary outcome | |
Notable harm: Differentiation syndrome | Secondary outcome | |
AE = adverse event; CR = complete remission; CRi = complete remission with incomplete hematologic recovery; DCO = data cut-off; EFS = event-free survival; EORTC QLQ-C30 = European Organisation of Research and Treatment of Cancer Quality of Life Questionnaire Core 30; GRADE = Grading of Recommendations, Assessment, Development, and Evaluations; OS = overall survival; SAE = serious adverse event; SoF = Summary of Findings; WDAE = withdrawal due to adverse event.
aStatistical testing for these end points was adjusted for multiple comparisons (e.g., hierarchal testing). CR plus CR with partial hematologic recovery and objective response rate were included in the sponsor’s hierarchy tests; however, the certainty of the results of these outcomes was not assessed using GRADE in this Clinical Review Report.
Source: AGILE Clinical Study Report.38 Details included in the table are from the sponsor’s summary of clinical evidence.
Overall Survival
OS was defined as the time from date of randomization to the date of death due to any cause. Once the study was unblinded, survival follow-up continued. All patients who were alive after an EFS event were to be contacted every 8 weeks for survival follow-up until death, withdrawal by patient, loss to follow-up, or the sponsor ending the study. Results of OS on both DCO dates are available.
In the AGILE study, reasons for censoring for OS included withdrawal of consent, loss to follow-up, and being alive at the DCO.
Event-Free Survival
EFS was a composite end point, defined as the time from randomization until treatment failure, relapse from remission, or death from any cause, whichever occurred first. Treatment failure was defined as a patient not experiencing CR by week 24 (i.e., being on treatment > 24 weeks without CR or treatment discontinuation ≤ 24 weeks without CR); these patients were considered to have an event at day 1 of randomization.
All patients who discontinued study treatment without experiencing death, disease relapse, or treatment failure, or without withdrawing consent were followed every day 1 (± 7 days) of weeks 9, 17, 25, 33, 41, and 53, and every 24 weeks thereafter, for EFS until they experienced treatment failure, relapse, or death; until they withdrew from the study; until 173 EFS events had occurred; or until deemed necessary by the IDMC.
During the AGILE study, the initial primary end point was changed from OS to EFS. The sponsor’s justification was that the sample size estimation showed that this change allowed for a smaller sample (200 instead of 398), a more feasible trial size in this rare patient population. Furthermore, EFS was considered by the sponsor to more accurately describe the contribution of a novel therapy to clinical benefit by removing the potentially confounding effects of posttrial therapies and by capturing treatment failure as an event. A previous phase Ib/II study (AG-221-AML-005) provided encouraging preliminary safety and efficacy data (primary efficacy end point: overall response rate) comparing an IDH1 inhibitor plus azacitidine with azacitidine alone.61 These considerations supported the amendment of the protocol to include a primary end point of EFS as a measure of clinical benefit for the treatment of patients with AML who are ineligible for intensive induction chemotherapy. This change was recorded in protocol amendment 5, on January 9, 2020, which was before unblinding of the data. OS was kept as a key secondary end point in the AGILE study and included in a fixed-sequence testing procedure to control the overall type I error rate.21
Reasons for censoring for EFS included the following: CR by 24 weeks, starting subsequent anticancer therapy; CR by 24 weeks, relapse or death documented after 2 or more missing disease assessments; CR by 24 weeks, lost to follow-up; CR by 24 weeks, withdrawal by patient; and CR by 24 weeks, ongoing in study without relapse or death.
Treatment Response
Multiple outcome measures related to response and remission were included in the AGILE study:
Rate of CR, where CR is defined as bone marrow blasts less than 5% and no Auer rods, absence of extramedullary disease, absolute neutrophil count greater than or equal to 11,000/μL, platelet count greater than or equal to 100,000/μL, and independence from RBC transfusions. This outcome was assessed until the date of relapse. Only assessments performed on or before the start date of subsequent anticancer therapies were considered in the determination of this response end point. CR rate was 1 of the key secondary efficacy end points in the AGILE study.
Rate of CR plus CRi (including CR with incomplete platelet recovery), where CRi (including CR with incomplete platelet recovery) was defined as all CR criteria except for residual neutropenia where the absolute neutrophil count was less than 1,000/μL or thrombocytopenia where the platelet count was less than 100,000/μL without platelet transfusion for at least 1 week before disease assessment.
Health-Related Quality of Life
In the AGILE study, HRQoL was measured using the EORTC QLQ C-30 and the EQ-5D questionnaires. Results of the disease-specific instrument EORTC QLQ-C3062 are included in this Clinical Review Report. EORTC QLQ-C30 score is a self-reported measure of HRQoL for patients with cancer who are receiving cancer treatment. The EORTC QLQ-C30 contains 30 items in total, and each item is evaluated on a 4-point or 7-point Likert scale. These 30 items can be categorized into 1 global health status/QoL scale, 5 functional scales, 3 symptom scales, and 6 single-item scales. Each scale is scored from 0 to 100, with a higher score representing more of the concept (e.g., more functioning or more symptoms). Each of the multi-item scales includes a different set of items. No item occurs in more than 1 scale (Table 7):
one global health status/QoL scale (2 items)
five functional scales: physical functioning (5 items), role functioning (2 items), emotional functioning (4 items), cognitive functioning (2 items), social functioning (2 items)
three symptom scales: fatigue (3 items), nausea and vomiting (2 items), pain (2 items)
six single-item scales relating to dyspnea, insomnia, appetite loss, constipation, diarrhea, and financial difficulties.
Table 7: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
EORTC QLQ-C30 | A multidimensional, cancer-specific, patient-reported measure used to assess HRQoL in response to treatment in clinical trials.63 The core questionnaire consists of 30 items that make 5 multi-item functional scales (physical [5 items], role [2 items], emotional [4 items], cognitive [2 items], and social [2 items] functioning), 3 multi-item symptom scales (fatigue [3 items], nausea/vomiting [2 items], and pain [2 items]), 6 single-item scales (dyspnea, insomnia, appetite loss, constipation, diarrhea, and financial impact), and a 2-item global QoL scale. Patients complete the questionnaire based on a 1-week recall period by rating most items on a 4-point Likert-type scale (1 = not at all; 2 = a little; 3 = quite a bit; 4 = very much). For the 2 items in the global QoL scale, the response format is a 7-point Likert-type scale (1 = very poor; 7 = excellent).62 Raw scores for each scale are computed as the average of the items that contribute to a particular scale. Each raw scale score is converted to a standardized score that ranges from 0 to 100 using a linear transformation. A decline in the symptom scale score reflects an improvement, whereas an increase in the function and QoL scale scores reflects an improvement.62 According to the EORTC QLQ-C30 scoring algorithm, if there are missing items for a scale, the score for that scale can still be computed if there are responses for at least half the items. In calculating the scale score, missing items are ignored.62 | The psychometric properties of the EORTC QLQ-C30 were evaluated in the validation study, in which patients with cholangiocarcinoma and gallbladder cancer were enrolled.64 Validity: All items demonstrated item-scale convergence (construct) validity (Pearson r > 0.4, prespecified). Although the study authors stated that known-group comparison was performed for EORTC QLQ-C30 scores, the results were not reported. Reliability: Internal consistency was acceptable (alpha ≥ 0.70) for all scales, except for the physical functioning (alpha = 0.47), cognitive functioning (alpha = 0.65), and nausea/vomiting (alpha = 0.67) scales at baseline. Test-retest reliability was demonstrated by the ICCs, which ranged from 0.52 to 0.92 in 67 clinically stable patients across all intervention groups over 2 weeks.a Responsiveness: Although the study stated that responsiveness to clinical change over time was measured for the EORTC QLQ-30 scores, the results were not reported. | A MID was not identified for patients with AML. MIDs for other types of cancers:37
|
AML = acute myeloid leukemia; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; HRQoL = health-related quality of life; ICC = intraclass correlation coefficient; MID = minimally important difference; QoL = quality of life.
aPatients who were receiving IV chemotherapy at the time were excluded from the test-retest assessment.
Transfusion Requirements
The summary of on-treatment transfusion data included the number of patients with any on-treatment transfusions, the number of patients with each type of on-treatment transfusion (whole blood, packed RBCs, platelet, plasma, and other), the total number of units per patient with each type of on-treatment transfusion, and the reasons for which each type of transfusion was administered. Results of transfusion requirements on both DCO dates are available.
Hospitalization
Hospitalization due to AEs in the safety analysis population was evaluated in the AGILE study. No further information was available for the assessment on hospitalization. This outcome is presented in the Harms section in this report.
Harms
The harms of treatment with ivosidenib plus azacitidine were assessed by review of AEs, SAEs, discontinuations of the study intervention due to AEs, and AEs of special interest.
AE: any untoward medical occurrence associated with the use of a drug in humans, whether or not considered drug related.
SAE: an AE is considered serious if it results in death, a life-threatening condition, inpatient hospitalization or prolongation of existing hospitalization, a persistent or significant incapacity or substantial disruption of the ability to conduct normal life functions, congenital anomaly or congenital anomaly in a neonate or infant born to a parent exposed to study treatment, or an important medical event as assessed by the investigator or sponsor.
Differentiation syndrome and infections were identified by the clinical experts consulted for this review as important notable harms for treatment with ivosidenib plus azacitidine. Results of harms on both DCO dates are available.
Statistical Analysis
Sample Size and Power Calculation
Assumptions for the placebo plus azacitidine group in the AGILE study were based on results from study AZA-AML-001, a phase III RCT comparing azacitidine with conventional care regimens in patients with newly diagnosed AML. Based on results from previous clinical trials, the CR rate at 24 weeks was assumed to be 20% for the placebo plus azacitidine group. For patients who experienced CR by 24 weeks, the median EFS was assumed to be 14.6 months.
Assumptions for the ivosidenib plus azacitidine treatment group in the AGILE study were based on results from study AG-221-AML-005, a phase Ib/II study comparing enasidenib (an IDH2 inhibitor) plus azacitidine with azacitidine alone in patients with newly diagnosed AML and IDH2 mutation. The CR rate by 24 weeks was assumed to be 40%. For patients who experienced CR by 24 weeks, a target HR of 0.76 for EFS was assumed (equivalent to a median EFS among patients who experienced response to treatment of 14.6 months in the placebo plus azacitidine group versus 19.2 months in the ivosidenib plus azacitidine group, assuming an exponential distribution).
Based on simulation results, the average overall HR over 10,000 simulations for the entire population was 0.641. Given that the assumption of PH was not met based on the EFS definition, the overall HR was less meaningful in this context. Therefore, the overall HR for the entire population was not part of the study design assumptions. Under these assumptions, 173 EFS events were required to provide 80% power at a 1-sided alpha of 0.025 level of significance to reject the null hypothesis using a stratified log-rank test. Assuming a recruitment period of approximately 44 months, with an accrual rate of 3 patients per month during the first 10 months and 5 patients per month thereafter, along with an assumed 5% overall dropout rate, approximately 200 patients with previously untreated IDH1-mutated AML were planned to be randomized to the 2 treatment groups in a 1:1 ratio. Given these assumptions, it was estimated that the analysis of the primary end point for EFS would occur approximately 52 months after the first patient was randomized. However, the planned sample size was not achieved. Instead, study enrolment ended early on the advice of the IDMC.
Statistical Testing
There were no planned interim analyses for efficacy in this study. Following the recommendation of the IDMC, enrolment in the study was stopped before the planned number of patients had been enrolled. It was decided that the IDMC DCO of March 18, 2021, would be used for the study’s primary analysis, which was an unplanned analysis before reaching the protocol prespecified 173 EFS events for the primary end point of EFS.
In the AGILE study, EFS was not initially the primary efficacy end point. The sponsor amended the protocol, and EFS became the primary end point as of January 9, 2020 (refer to the Outcomes section for more details). EFS was tested using the log-rank test stratified by the randomization stratification factors. The basis for a claim of efficacy would be the statistical significance of EFS in favour of the ivosidenib plus azacitidine group when the 1-sided P value was less than 0.025. This was later adjusted to a P value of 0.0046 at the unplanned interim analysis. Kaplan-Meier estimates of EFS were presented by treatment group, together with a summary of associated statistics. In addition, the EFS rates at 1 day and at 3, 6, 9, 12, 18, 24, and 36 months were estimated with corresponding 2-sided 95% CIs. The HR was estimated using a Cox PH model stratified by the randomization strata. Given that the PH assumption was not met, based on the EFS definition, the overall HR may not be meaningful. In the analyses for EFS, if the PH assumption was violated when large departures from the PH assumption were observed, the log-rank test would be underpowered to detect differences in the survival distributions for the treatment groups, and a test of the difference in the restricted mean survival time between the treatment group and the control group may be more appropriate to determine the superiority of the treatment group compared to the control group with respect to the time-to-event end point. The associated 95% CI for the difference in restricted mean survival time and 1-sided P value were generated.
As EFS is a composite end point, the estimates for each component were summarized, including CR rate by 24 weeks, and EFS among patients who experienced CR by 24 weeks.
Key secondary end points in the AGILE study were CR, OS, CR plus CRh, and ORR. A description of the statistical analyses for each efficacy outcome reported in the AGILE study is provided in Table 8.
The HR of OS was estimated using a Cox PH model stratified by the randomization strata. Kaplan-Meier estimates (product-limit estimates) were presented by treatment group, together with a summary of associated statistics, including the median OS time with 2-sided 95% CIs. In particular, the OS rate at 3, 6, 9, 12, 18, 24, and 36 months was estimated, with corresponding 2-sided 95% CIs.
Multiplicity Control
To control the overall type I error rate, the fixed-sequence testing procedure was planned to adjust for multiple statistical testing of the primary and key secondary efficacy end points. These end points were intended to be tested in the following order:
EFS
CR rate
OS
CR plus CRh rate
ORR.
Table 8: Statistical Analysis of Efficacy End Points in the AGILE Study
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
OS | OS was tested using the log-rank test stratified by the randomization stratification factors. Kaplan-Meier estimates (product-limit estimates) were presented by treatment group, together with a summary of associated statistics, including the median OS time with 2-sided 95% CIs. In particular, the OS rate at 3, 6, 9, 12, 18, 24, and 36 months was estimated, with corresponding 2-sided 95% CIs. HR was estimated using a Cox PH model stratified by the randomization strata. | Randomization stratification factors:
| If a patient was not known to have died by the DCO date, then OS was censored at the date of last contact. | None. |
EFS | EFS was tested using the log-rank test stratified by the randomization stratification factors. Kaplan-Meier estimates (product-limit estimates) were presented by treatment group, together with a summary of associated statistics. In particular, the EFS rate at 1 day and at 3, 6, 9, 12, 18, 24, and 36 months was estimated, with corresponding 2-sided 95% CIs. HR was estimated using a Cox PH model stratified by the randomization strata. When the PH assumption was violated, RMST was used to measure the survival time distribution, as an alternative to the HR approach. The associated 95% CI for the difference in RMST and 1-sided P value was generated. | As for OS. | If a patient experienced CR by 24 weeks then started subsequent anticancer therapy (before relapse or no relapse), experienced CR by 24 weeks then relapsed or died after 2 or more missing or inadequate disease assessments, or experienced CR by 24 weeks and neither relapsed nor died, then the patient was censored at the last adequate disease assessment documenting no relapse before the start of subsequent anticancer therapy or missed response assessments. If a patient was on treatment ≤ 24 weeks, ongoing, and had not experienced CR yet, then the patient was censored at the date of randomization. | EFS was tested using the log-rank test stratified by the IRT randomization stratification factors and based on the FAS. The time of relapse or death was determined using the actual date of relapse or death, even in situations where relapse or death was observed after 2 or more missing disease assessments or the start of subsequent anticancer therapy. EFS was tested using the unstratified log-rank test and based on the FAS. EFS was tested using the log-rank test stratified by the IRT randomization stratification factors and based on the per-protocol set. EFS was tested using the log-rank test stratified by the randomization stratification factors derived based on data provided by the investigator on the eCRF and based on the FAS. EFS was tested using the log-rank test stratified by the IRT randomization stratification factors and based on the FAS. Patients who did not experience CR by week 24 were not considered to have had an EFS event at day 1 of randomization; the event time was either 24 weeks or EOT, whichever was earlier. |
Rate of CR and of CR + CRi | A CMH test stratified by the randomization stratification factors was used to compare the CR rate between the 2 treatment groups. The odds ratio and its associated 95% CI were presented. | As for OS. | Complete case analysis.a | None. |
Transfusion requirement | Summarized by treatment group using descriptive statistics. | None. | Best response was reported for all patients in line with the ITT analysis.a | None. |
EORTC QLQ-C30 | Transformed scores for each scale and the absolute and percent changes from baseline were summarized by treatment group at each visit. Mixed models were also applied in the analysis of the EORTC QLQ-C30. | Baseline score, treatment arm, time, randomization stratification factors, and interaction between treatment arm and time as fixed effects, and patient as random effect. | Handled implicitly in the model by assuming missing at random. | None. |
AML = acute myeloid leukemia; CI = confidence interval; CMH = Cochran-Mantel-Haenszel; CR = complete remission; CRi = complete remission with incomplete hematologic recovery; DCO = data cut-off; eCRF = electronic case report form; EFS = event-free survival; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EOT = end of treatment; FAS = full analysis set; HR = hazard ratio; IRT = interactive response technologies; ITT = intention to treat; OS = overall survival; PH = proportional hazards; RMST = restricted mean survival time.
aAccording to the sponsor-provided additional information, no imputation was performed to handle missing data in the analyses for CR, CR plus CRi, and transfusion requirement. Instead, best response was reported for all patients in line with the ITT analysis. A patient’s best response may have been reported as “not assessed” if they did not have any postbaseline disease assessment, or as “not evaluable.”
Source: AGILE Clinical Study Report.38 Details included in the table are from the sponsor’s summary of clinical evidence.
No control of the alpha level was made for the other analyses. To account for the unplanned interim analysis by the IDMC and early termination of the study, an individual set of group-sequential boundaries was applied separately to each of the efficacy end points. The stopping boundaries were calculated using the Lan and De Mets alpha spending function for 1-sided tests, accounting for the amount of information actually available (i.e., information fraction) at the time of the analysis.
For time-to-event end points, such as EFS and OS, the information fraction is the ratio of the number of actual events at the time of the analysis to the number of planned events as specified in the protocol. For binary end points, such as CR, the information fraction is the ratio of the number of patients who are randomized at the time of the analysis to the planned sample size as specified in the protocol. For EFS, CR, and OS, the 1-sided P value boundaries are 0.0046 (based on a 62.4% information fraction), 0.0087 (based on a 73.0% information fraction), and 0.0017 (based on a 51.0% information fraction), respectively.
The updated results of EFS, CR, and some other efficacy end points at the second DCO (June 30, 2022) were not available, because after unblinding the study and analyzing the primary efficacy end point, the invasive procedures and the necessary visits to the clinics for treatment response assessment (e.g., CR) were not warranted beyond those performed as per standard of care.
Sensitivity Analyses
Sensitivity analyses for EFS were performed to evaluate the robustness of the EFS end point. Details of these sensitivity analyses are presented in Table 8.
Subgroup Analyses
Prespecified subgroup analyses for EFS are presented in Table 9. Treatment groups were compared for EFS using a 2-sided unstratified log-rank test for each category, and the unstratified HR and its corresponding 95% CI was computed for each category and depicted in a Forest plot. If there was a small number of patients within a category (< 5% of the patients in the full analysis set [FAS]), the categories were pooled (if 3 or more categories are prespecified for the subgroup) or the subgroup would not be analyzed (if there are only 2 prespecified categories in the subgroup). Efficacy analyses in subgroups were purely exploratory and intended to evaluate the consistency of treatment effect.
Table 9: Subgroup Analyses Performed for EFS in the AGILE Study
Subgroup | Categories |
|---|---|
De novo status based on IRT | Yes; No |
De novo status based on investigator from eCRF | Yes; No |
Region | US and Canada; Western Europe, Israel, or Australia; Japan; rest of the world |
Age | < 75 years; ≥ 75 years |
Baseline ECOG PS | 0 or 1; ≥ 2 |
Sex | Female; Male |
Race | White; Asian; Black or African American; other |
Baseline cytogenetic risk status | American Indian or Alaska Native; Native Hawaiian or other Pacific Islander; not reported |
WHO classification of AML | Favourable risk; intermediate risk; poor risk |
Baseline WBC count | AML with genetic abnormalities; AML with myelodysplasia-related changes; therapy-related myeloid neoplasms; AML not otherwise specified |
Baseline percent bone marrow blastsa | ≤ 5 × 109/L; > 5 × 109/L |
AML = acute myeloid leukemia; ECOG PS = Eastern Cooperative Oncology Group performance status; eCRF = electronic case report form; EFS = event-free survival; IRT = interactive response technologies; WBC = white blood cell.
aFor bone marrow blasts, bone marrow aspirate was used as the primary source. If a bone marrow aspirate assessment was not available, a bone marrow biopsy assessment was used.
Source: AGILE Clinical Study Report.38 Details included in the table are from the sponsor’s summary of clinical evidence.
Analysis Populations
Analysis populations of the AGILE study are summarized in Table 10.
Table 10: Analysis Populations of the AGILE Study
Population | Definition | Application |
|---|---|---|
FAS | Included all patients who were randomized. Patients were classified according to the randomized treatment arm. This dataset was referred to as the ITT analysis set in the protocol. | Demographic and other baseline characteristics; disposition; major protocol deviations; subsequent therapies; efficacy |
SAS | Included all patients who received at least 1 dose of the study treatment. Patients were classified according to the treatment received, where treatment received was defined as:
| Demographic and other baseline characteristics; exposure and concomitant therapies; safety |
PPS | Subset of the FAS. Patients who met any of the following criteria were excluded from the PPS:
| Efficacy (primary and key secondarya), as supplemental analyses |
AML = acute myeloid leukemia; CR = complete remission; ECOG PS = Eastern Cooperative Oncology Group performance status; FAS = full analysis set; HMA = hypomethylating agent; ITT = intention to treat; PPS = per-protocol set; SAS = safety analysis set.
aKey secondary end points were CR rate, overall survival, CR plus CR with partial hematologic recovery rate, and objective response rate.
Source: AGILE Clinical Study Report.38 Details included in the table are from the sponsor’s summary of clinical evidence.
Results
Patient Disposition
A summary of patient disposition in the AGILE study is provided in Table 11.
As of the DCO of March 18, 2021, 144 patients (98.6%) were randomized and received at least 1 dose of the study treatment: 71 patients in the ivosidenib plus azacitidine group and 73 patients in the placebo plus azacitidine group. One patient in each treatment group died before receiving study treatment. Twenty-six patients (36.1%) were ongoing with ivosidenib plus azacitidine, and 12 patients (16.2%) were ongoing with placebo plus azacitidine as of this cut-off date.
A total of 106 patients discontinued ivosidenib or placebo: 45 (62.5%) in the ivosidenib plus azacitidine group and 61 (82.4%) in the placebo plus azacitidine group. The main reasons for treatment discontinuation were AEs (27.4%) and progressive disease (17.1%). Other reasons included patient withdrawal (10.3%), clinical progression (6.2%), lack of treatment benefit (6.2%), other (4.8%), and death (1 patient in the placebo plus azacitidine group), with similar proportions of patients in both groups within each category. For patients who discontinued azacitidine, the distribution of discontinuation rates due to these reasons were also similar across both groups.
Eighty-five patients (58.2%) discontinued the study: 34 in the ivosidenib plus azacitidine group and 51 in the placebo plus azacitidine group. Among them, 74 patients (50.7%) died (including 1 patient per group who died due to COVID-19), 10 patients (6.8%) withdrew, and 1 patient (0.7%) was lost to follow-up (placebo plus azacitidine).
Table 11: Summary of Patient Disposition From the AGILE Study (FAS, DCO March 18, 2021)
Patient disposition | Ivosidenib + azacitidine | Placebo + azacitidine |
|---|---|---|
Screened, N | 295 | |
Did not meet screening requirements, N | 149: ████ ███ ████ ████████ ██ ███████ ████████████ █████ ███████████ ████████ ███ █████ ███ | |
Randomized, N (%) | 72 (100.0) | 74 (100.0) |
Discontinued (not treated), n (%) | 1 (1.4) | 1 (1.4) |
Death, n (%) | 1 (1.4) | 1 (1.4) |
Completed (treated), n (%) | 71 (98.6) | 73 (98.6) |
Treatment status, n (%) | ||
Discontinued ivosidenib/placebo | 45 (62.5) | 61 (82.4) |
AE | 20 (27.8) | 20 (27.0) |
Progressive disease/relapse | 11 (15.3) | 14 (18.9) |
Withdrawal by patient | 5 (6.9) | 10 (13.5) |
Clinical progression (within 24 weeks) not confirmed by IWG assessment | 3 (4.2) | 6 (8.1) |
Lack of treatment benefit after > 24 weeks | 2 (2.8) | 7 (9.5) |
Death | 0 | 1 (1.4) |
Other | 4 (5.6) | 3 (4.1) |
Ongoing ivosidenib/placebo | 26 (36.1) | 12 (16.2) |
Discontinued azacitidine | 45 (62.5) | 61 (82.4) |
AE | 20 (27.8) | 20 (27.0) |
Progressive disease/relapse | 10 (13.9) | 14 (18.9) |
Withdrawal by patient | 5 (6.9) | 10 (13.5) |
Clinical progression (within 24 weeks) not confirmed by IWG assessment | 4 (5.6) | 6 (8.1) |
Lack of treatment benefit after 24 weeks | 3 (4.2) | 7 (9.5) |
Death | 0 | 1 (1.4) |
Other | 3 (4.2) | 3 (4.1) |
Ongoing azacitidine | 26 (36.1) | 12 (16.2) |
Study status, n (%) | 45 (62.5) | 61 (82.4) |
Discontinued study | 34 (47.2) | 51 (68.9) |
Death | 28 (38.9) | 46 (62.2) |
Lost to follow-up | 0 | 1 (1.4) |
Withdrawal by patient | 6 (8.3) | 4 (5.4) |
On study | 38 (52.8) | 23 (31.1) |
FAS with DCO of March 18, 2021, N (% of randomized) | 72 (100.0) | 74 (100.0) |
FAS with DCO of June 30, 2022, N (% of randomized)a | 73 (100.0) | 75 (100.0) |
SAS, N (% of randomized) | 71 (98.6) | 73 (98.6) |
PPS, N (% of randomized) | ██ ██████ | ██ ██████ |
AE = adverse event; DCO = data cut-off; FAS = full analysis set; IWG = International Working Group; PPS = per-protocol set; SAS = safety analysis set.
aAs of June 30, 2022, 2 more patients were enrolled in the AGILE study, 1 in each treatment group.
Source: AGILE Clinical Study Report.38 Details included in the table are from the sponsor’s summary of clinical evidence.
Baseline Characteristics
The baseline characteristics outlined in Table 12 are those that are most relevant to this review or were felt to affect the outcomes or interpretation of the study results.
The overall patient population was composed of a similar proportion of male and female patients (80 [54.8%] and 66 [45.2%], respectively; there were more male patients in the ivosidenib plus azacitidine group [58.3%] than in the placebo plus azacitidine group [51.4%]). The patients were primarily older than 75 years (82 patients [56.2%]). Race was unreported for most patients (84 [57.5%]).
The majority of patients (73.3% per investigator [76% per Interactive Web Response System]) had the subtype de novo (primary) AML at initial diagnosis, whereby AML arises as a new condition (the remaining patients in the trial had secondary AML, whereby the disease appears alongside a history of hematologic disorders).65 Based on the WHO classification of AML, fewer patients in the ivosidenib plus azacitidine group (22.2%) had AML with recurrent genetic abnormalities than in the placebo plus azacitidine group (32.4%); more patients in the ivosidenib plus azacitidine group (38.9%) had AML with myelodysplasia-related changes than in the placebo plus azacitidine group (35.1%). IIDH1 R132C was the most common polymorphism (65.8% of patients). In total, 63.9% of patients in the ivosidenib plus azacitidine group and 67.6% of patients in the placebo plus azacitidine group had an ECOG performance status score of 0 to 1. Cytogenetic risk status, as assessed by the investigators based on the 2017 National Comprehensive Cancer Network guidelines, was intermediate (63.0% of patients) or poor (24.7% of patients) for most patients at baseline. The baseline median bone marrow blast proportion was 52.5% (range, 17% to 100%).
A summary of baseline characteristics in the AGILE study is provided in Table 12.
Table 12: Summary of Baseline Characteristics From the AGILE Study (FAS)
Characteristics | Ivosidenib + azacitidine (N = 72) | Placebo + azacitidine (N = 74) |
|---|---|---|
Age | ||
Mean, years (SD) | 74.5 (6.18) | 75.2 (7.39) |
Median, years (range) | 76.0 (70.5 to 79.5) | 75.5 (70.0 to 80.0) |
Age category (years), range | 58 to 84 | 45 to 94 |
< 65, n (%) | 4 (5.6) | 4 (5.4) |
≥ 65, n (%) | 68 (94.4) | 70 (94.6) |
< 75, n (%) | 33 (45.8) | 31 (41.9) |
≥ 75, n (%) | 39 (54.2) | 43 (58.1) |
Sex, n (%) | ||
Male | 42 (58.3) | 38 (51.4) |
Female | 30 (41.7) | 36 (48.6) |
Race, n (%) | ||
Asian | 15 (20.8) | 19 (25.7) |
White | 12 (16.7) | 12 (16.2) |
Black or African American | 0 | 2 (2.7) |
Other | 1 (1.4) | 1 (1.4) |
Not reported | 44 (61.1) | 40 (54.1) |
Disease type, n (%) | ||
Nature of AML per investigator | ||
De novo | 54 (75.0) | 53 (71.6) |
Secondary | 18 (25.0) | 21 (28.4) |
Treatment-related AML | 2 (2.8) | 1 (1.4) |
History of MDS | 10 (13.9) | 12 (16.2) |
History of myeloproliferative neoplasms | 4 (5.6) | 8 (10.8) |
Other | 2 (2.8) | 0 |
Nature of AML per Interactive Web Response System | ||
De novo | 56 (77.8) | 55 (74.3) |
Secondary | 16 (22.2) | 19 (25.7) |
WHO classification of AML, n (%) | ||
AML with recurrent genetic abnormalities | 16 (22.2) | 24 (32.4) |
AML with myelodysplasia-related changes | 28 (38.9) | 26 (35.1) |
Therapy-related myeloid neoplasms | 1 (1.4) | 1 (1.4) |
AML not otherwise specified | 27 (37.5) | 23 (31.1) |
ECOG PS score, n (%) | ||
0 | 14 (19.4) | 10 (13.5) |
1 | 32 (44.4) | 40 (54.1) |
2 | 26 (36.1) | 24 (32.4) |
IDH1 mutation type based on central testing, n (%) | ||
R132C | 45 (62.5) | 51 (68.9) |
R132G | 6 (8.3) | 4 (5.4) |
R132H | 14 (19.4) | 12 (16.2) |
R132L | 3 (4.2) | 0 |
R132S | 2 (2.8) | 6 (8.1) |
Wild typea | 1 (1.4) | 0 |
Missinga | 1 (1.4) | 1 (1.4) |
Cytogenetic risk status by investigator, n (%) | ||
Favourable | 3 (4.2) | 7 (9.5) |
Intermediate | 48 (66.7) | 44 (59.5) |
Poor | 16 (22.2) | 20 (27.0) |
Other | 3 (4.2) | 1 (1.4) |
Missing | 2 (2.8) | 2 (2.7) |
Bone marrow blastsb | ||
n | 71 | 73 |
Mean, % (SD) | 55.2 (23.30) | 53.3 (23.45) |
Median, % (Q1, Q3) | 54.0 (32.0 to 75.0) | 48.0 (33.0 to 70.0) |
Range, % | 20 to 95 | 17 to 100 |
AML = acute myeloid leukemia; ECOG PS = Eastern Cooperative Oncology Group performance status; FAS = full analysis set; MDS = myelodysplastic syndrome; SD = standard deviation.
aIDH1 mutation for these patients was confirmed with local testing.
bFor bone marrow blasts, bone marrow aspirate was used as the primary source. If a bone marrow aspirate assessment was not available, a bone marrow biopsy assessment was used.
Source: AGILE Clinical Study Report.38 Details included in the table are from the sponsor’s summary of clinical evidence.
Exposure to Study Treatments
A summary of patient exposure in the AGILE study is provided in Table 13.
Overall, the median duration of exposure to ivosidenib plus azacitidine was 5.79 cycles (Q1: 1.25; Q3: 15.25), and the median duration of exposure to placebo plus azacitidine was 2.32 cycles (Q1: 1.25; Q3: 5.82).
Table 13: Patient Exposure in the AGILE Study (SAS, DCO March 18, 2021)
Exposure | Ivosidenib + azacitidine (N = 71) | Placebo + azacitidine (N = 73) |
|---|---|---|
Duration of exposure (4-week cycle) | ||
Total, patient-weeks or patient-years | NR | NR |
Mean (SD) | ████ ███████ | ████ ███████ |
Median (IQR or range) | ████ ██████ ██████ | ████ ██████ █████ |
Adherence, % | NR | NR |
DCO = data cut-off; IQR = interquartile range; NR = not reported; Q1 = first quartile; Q3 = third quartile; SAS = safety analysis set; SD = standard deviation.
Note: Duration of exposure (days) = (date of last dose – date of first dose + 1).
Source: AGILE Clinical Study Report.38 Details included in the table are from the sponsor’s summary of clinical evidence.
Concomitant Medications and Co-Interventions
███ ████████ ██ ████ █████████ ██████ ███████ received concomitant medications. Overall, the most frequently used medications in both groups were similar. In the ivosidenib plus azacitidine group (N = 71), the most frequently used medications (≥ 20 patients) included ███████████ ██ ██ ███████ █████████ ███████████ ██ ██ ███████ █████████ ████████████ ██████ ██ ██████████ ██████ ██ ██ ███████ █████████ ██████████ ██ ██ ███████ █████████ ███ ████████████████ ██ ██ ███████ ████████. In the placebo plus azacitidine group (N = 73), the most frequently used medications (≥ 20 patients) included ███████████ ██ ██ ███████ █████████ ██████████ ██ ██ ███████ █████████ ███████████ ██ ██ ███████ █████████ ████████████ ██ ██ ███████ █████████ ████████████ ██████ ██ ██████████ ██████ ██ ██ ███████ █████████ ██████████████ ██ ██ ███████ █████████ █████████ ██ ██ ███████ █████████ █████████ ██ ██ ███████ █████████ █████████ ████████ ██ ██ ███████ █████████ ███ ██████████████ █████████████ ██ ██ ███████ ████████. The most frequently used concomitant medications in the AGILE study are summarized in Table 14.
Table 14: Most Frequently Used Concomitant Medications in the AGILE Study (SAS, DCO March 18, 2021)
Preferred term | Ivosidenib + azacitidine (N = 71) | Placebo + azacitidine (N = 73) |
|---|---|---|
Received concomitant medications, n (%) | ██ ███████ | ██ ███████ |
Most frequently used medications (≥ 20 patients in either arm), n (%) | ||
Ondansetron | ██ ██████ | ██ ██████ |
Paracetamol | ██ ██████ | ██ ██████ |
Piperacillin sodium; tazobactam sodium | ██ ██████ | ██ ██████ |
Furosemide | ██ ██████ | ██ ██████ |
Hydroxycarbamide | ██ ██████ | ██ ██████ |
Levofloxacin | ██ ██████ | ██ ██████ |
Metoclopramide | ██ ██████ | ██ ██████ |
Meropenem | ██ ██████ | ██ ██████ |
Lactulose | ██ ██████ | ██ ██████ |
Potassium chloride | ██ ██████ | ██ ██████ |
Metoclopramide hydrochloride | ██████ | ██ ██████ |
DCO = data cut-off; SAS = safety analysis set.
Source: AGILE Clinical Study Report.38 Details included in the table are from the sponsor’s summary of clinical evidence.
Subsequent Treatments
A summary of subsequent anticancer therapies in the AGILE study is provided in Table 15. Fourteen patients (19.4%) in the ivosidenib plus azacitidine group (N for FAS = 72) and 16 patients (21.6%) in the placebo plus azacitidine group (N for FAS = 74) had at least 1 subsequent anticancer therapy. As of the DCO of March 18, 2021, 4 patients (5.6%) in the ivosidenib plus azacitidine group (N for safety analysis set [SAS] = 71) and 1 patient (1.4%) in the placebo plus azacitidine group (N for SAS = 73) had had an allogeneic hematopoietic stem cell transplant. As of the DCO of June 30, 2022, a total of 7 patients (4.7%) had had an allogeneic hematopoietic stem cell transplant: 5 (6.8%) in the ivosidenib plus azacitidine group and 2 (2.7%) in the placebo plus azacitidine group. Five patients initially treated with placebo plus azacitidine crossed over to the ivosidenib plus azacitidine group after March 18, 2021.39
Table 15: Subsequent Anticancer Treatments From the AGILE Study (FAS, DCO March 18, 2021)
Anatomic Therapeutic Chemical classification preferred term | Ivosidenib + azacitidine (N = 72) | Placebo + azacitidine (N = 74) |
|---|---|---|
Received subsequent therapy, n (%) | 14 (19.4) | 16 (21.6) |
Antimetabolites, n (%) | 11 (15.3) | 14 (18.9) |
Azacitidine | █████ | █████ |
Cytarabine | █████ | █████ |
Fludarabine | █████ | █████ |
Cladribine | █████ | █████ |
Decitabine | █████ | █████ |
Other antineoplastic agents, n (%) | █████ | █████ |
Venetoclax | 4 (5.6) | 7 (9.5) |
Amsacrine | █████ | █████ |
Bgb 324 | █████ | █████ |
Combinations of antineoplastic agents | █████ | █████ |
Flotetuzumab | █████ | █████ |
Ipilimumab | █████ | █████ |
Osimertinib | █████ | █████ |
Ivosidenib | 0 | 2 (2.7) |
Alkylating agents, n (%) | █████ | █████ |
Busulfan | █████ | █████ |
Cyclophosphamide | █████ | █████ |
Melphalan | █████ | █████ |
Immunosuppressants, n (%) | █████ | █████ |
Antilymphocyte serum | █████ | █████ |
Antithymocyte immunoglobulin (rabbit) | █████ | █████ |
Cytotoxic antibiotics and related substances, n (%) | █████ | █████ |
Daunorubicin hydrochloride | █████ | █████ |
Idarubicin | █████ | █████ |
Aclarubicin | █████ | █████ |
Aclarubicin hydrochloride | █████ | █████ |
Daunorubicin | █████ | █████ |
Investigational drug, n (%) | █████ | █████ |
DCO = data cut-off; FAS = full analysis set.
Notes: Patients with multiple medications within a preferred term are counted only once in that preferred term. Patients with multiple medications within an Anatomic Therapeutic Chemical classification are counted only once in that classification.
Source: AGILE Clinical Study Report.38 Details included in the table are from the sponsor’s summary of clinical evidence.
Efficacy
Event-Free Survival
EFS was the primary efficacy end point in the AGILE study. As of the DCO of March 18, 2021, the median EFS was 0.03 months (95% CI, 0.03 months to 11.01 months) in the ivosidenib plus azacitidine group and 0.03 months (95% CI, NE to NE) in the placebo plus azacitidine group. The median did not appear different between groups because of the majority of events being treatment failure, which were assigned the date of randomization. The corresponding HR was 0.33 (95% CI, 0.16 to 0.69; P = 0.0011). The primary end point met the defined boundary for statistical significance. Forty-two patients (58.3%) in the ivosidenib plus azacitidine group experienced treatment failure, as did 59 patients (79.7%) in the placebo plus azacitidine group, and were considered to have had an EFS event at day 1. Most of the treatment failure events were due to treatment discontinuation without CR: 36.1% in the ivosidenib plus azacitidine group and 64.9% in the placebo plus azacitidine group. Three patients (4.2%) in the ivosidenib plus azacitidine group and 2 patients (2.7%) in the placebo plus azacitidine group had relapsed AML. One patient (1.4%) in each group died. Data were censored for 26 patients (36.1%) in the ivosidenib plus azacitidine group and 12 patients (16.2%) in the placebo plus azacitidine group. The between-group difference in EFS rates for ivosidenib plus azacitidine versus placebo plus azacitidine were 19.7% (95% CI, ███ ██ █████) at 6 months and 25.3% (95% CI, ███ ██ █████) at 12 months (Table 16). The between-group differences in EFS rates at later time points were not reported in the study. In addition, updated EFS results at the second DCO (June 30, 2022) were not available, because after the study was unblinded and the primary efficacy end point analyzed, invasive procedures and the associated visits to the clinics were not warranted beyond those performed as part of standard of care.
Table 16: Summary of Event-Free Survival in the AGILE Study (FAS, DCO March 18, 2021)
EFS | Ivosidenib + azacitidine (N = 72) | Placebo + azacitidine (N = 74) |
|---|---|---|
Events, n (%) | 46 (63.9) | 62 (83.8) |
Treatment failure | 42 (58.3) | 59 (79.7) |
On treatment > 24 weeks without CR | 16 (22.2) | 11 (14.9) |
Treatment discontinuation ≤ 24 weeks without CR | 26 (36.1) | 48 (64.9) |
Relapse | 3 (4.2) | 2 (2.7) |
Death | 1 (1.4) | 1 (1.4) |
Patients censored, n (%) | 26 (36.1) | 12 (16.2) |
CR by 24 weeks, start subsequent anticancer therapy | 1 (1.4) | 0 |
CR by 24 weeks, relapse/death documented after 2 or more missing disease assessments | 0 | 0 |
CR by 24 weeks, lost to follow-up | 0 | 0 |
CR by 24 weeks, withdrawal by patient | 2 (2.8) | 0 |
CR by 24 weeks, ongoing without relapse or death | 20 (27.8) | 5 (6.8) |
On treatment ≤ 24 weeks, ongoing, not yet experienced CR | 3 (4.2) | 7 (9.5) |
EFS (months), median (95% CI)a | 0.03 (0.03 to 11.01) | 0.03 (NE to NE) |
HR (95% CI)b | 0.33 (0.16 to 0.69) | |
P valuec | 0.0011 | |
EFS rate, % (95% CI)d | ||
6 months | 39.9 (28.6 to 51.0) | 20.3 (12.0 to 30.0) |
Difference in EFS rate, % (95% CI) | 19.7 (███ ██ ████) | |
12 months | 37.4 (25.9 to 48.9) | 12.2 (4.3 to 24.4) |
Difference in EFS rate, % (95% CI) | 25.3 (███ ██ ████) | |
18 months | 33.3 (20.9 to 46.2) | 6.1 (0.7 to 20.9) |
Difference in EFS rate, % (95% CI) | NR | |
24 months | 22.2 (6.6 to 43.4) | NE |
Difference in EFS rate, % (95% CI) | NR | |
36 months | NE | NE |
Difference in EFS rate, % (95% CI) | NR | |
CI = confidence interval; CR = complete remission; DCO = data cut-off; EFS = event-free survival; FAS = full analysis set; HR = hazard ratio; NE = not estimable; NR = not reported.
aEstimated using the product-limit (Kaplan-Meier) method. The CIs are calculated using the Brookmeyer and Crowley method with log-log transformation.
bHR is estimated using a Cox proportional hazards model stratified by the randomization stratification factors (acute myeloid leukemia status and geographic region), with placebo plus azacitidine as the denominator.
cP value is calculated from the 1-sided log-rank test stratified by the randomization stratification factors (acute myeloid leukemia status and geographic region); the stopping boundary was 1-sided P = 0.0046.
dEFS rate is the estimated probability that a patient will remain event-free up to the specified time point. EFS rates are obtained from the Kaplan-Meier survival estimates. CIs are calculated using the Greenwood formula and log-log transformation.
Source: AGILE Clinical Study Report.38 Details included in the table are from the sponsor’s summary of clinical evidence.
The Kaplan-Meier EFS curves are shown in Figure 1.
The restricted mean survival time for EFS calculated up to 18.2 months was 7.1 months in the ivosidenib plus azacitidine group and 3.1 months in the placebo plus azacitidine group. The between-group difference was 4.0 months (95% CI, 1.5 to 6.5; P = 0.0009).
In general, the results of the sensitivity analyses were consistent with those of the primary analysis for EFS in terms of median EFS, HR, and proportion of censored patients, except for sensitivity analysis 5, where EFS was tested using the log-rank test stratified by randomization stratification factors and based on the FAS. In this sensitivity analysis, patients who did not experience CR by week 24 were not considered to have had an EFS event at day 1 of randomization; the event time was either 24 weeks or the end of treatment, whichever was earlier. In this analysis, the median EFS was ███ ██████ ████ ██ ███ ██ ████ in the ivosidenib plus azacitidine group versus ███ ██████ ████ ██ ███ ██ ████ in the placebo plus azacitidine group. The HR was similar to the main analysis ████ ████ ███ ████ ██ ██████ ████████████. In another sensitivity analysis, where EFS with treatment failure was defined as a lack of CR, CRi, or morphologic clearance of leukemic cells from the marrow after at least 24 weeks of treatment, the median EFS was 22.9 months (95% CI, 7.5 months to NE) with ivosidenib plus azacitidine and 4.1 months (95% CI, 2.7 months to 6.8 months) with placebo plus azacitidine (HR = 0.39; 95% CI, 0.24 to 0.64; P < 0.001).
Results of the subgroup analyses showed that the improvement in EFS with ivosidenib plus azacitidine was generally consistent across the prespecified subgroups (Figure 2).
Figure 1: Kaplan-Meier Plot of EFS in the AGILE Study (FAS, DCO March 18, 2021)
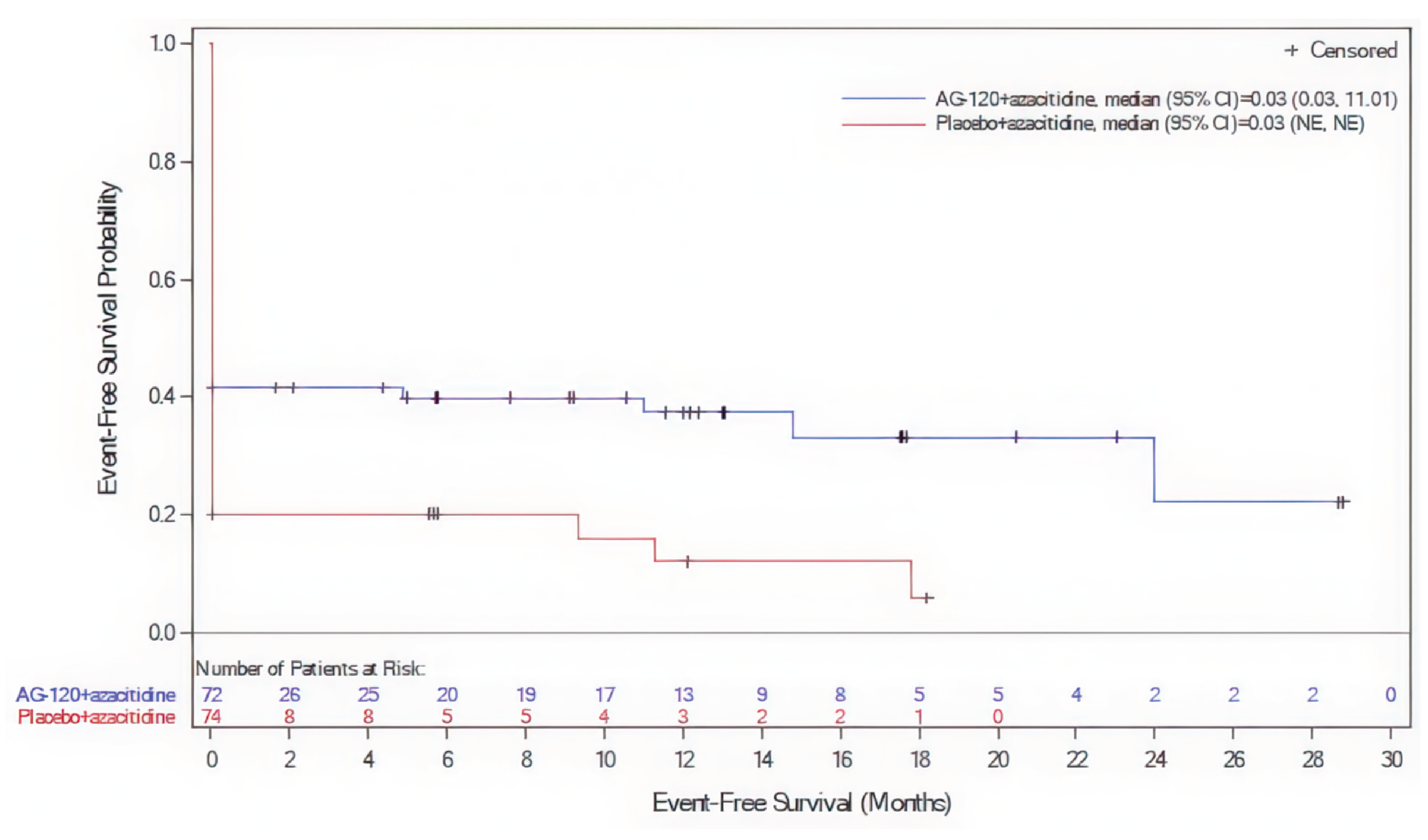
AG-120 = ivosidenib; CI = confidence interval; DCO = data cut-off; EFS = event-free survival; FAS = full analysis set; NE = not estimable.
Source: AGILE Clinical Study Report.38
Figure 2: Forest Plot of EFS by Subgroups in the AGILE Study (FAS, DCO March 18, 2021)
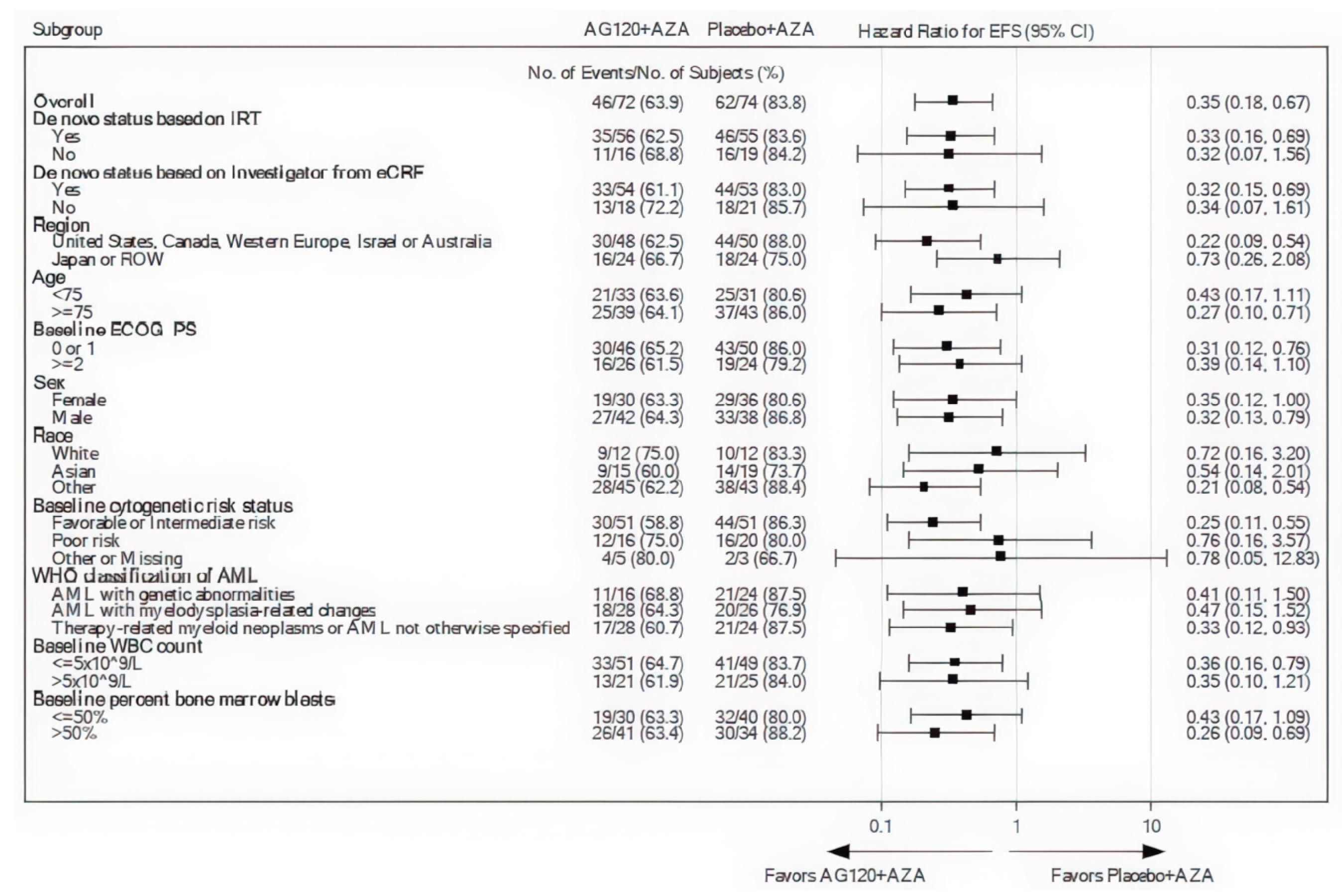
AG-120 = ivosidenib; AML = acute myeloid leukemia; AZA = azacitidine; CI = confidence interval; DCO = data cut-off; ECOG PS = Eastern Cooperative Oncology Group performance status; eCRF = electronic case report form; EFS = event-free survival; FAS = full analysis set; IRT = interactive response technologies; NE = not estimable; ROW = rest of world; WBC = white blood cell.
Notes: The hazard ratio is calculated from the unstratified Cox regression model, with placebo plus azacitidine as the denominator, with 2-sided 95% CI. “Greater than or equal to 20% of baseline blasts” was reported for 1 patient within the ivosidenib plus azacitidine arm. This patient is not included in the subgroup analyses for baseline percent bone marrow blasts.
Source: AGILE Clinical Study Report.38
Overall Survival
At the DCO of March 18, 2021, the median follow-up time was approximately 15 months for both treatment groups. Twenty-eight patients (38.9%) in the ivosidenib plus azacitidine group and 46 patients (62.2%) in the placebo plus azacitidine group had died (HR = 0.44; 95% CI, 0.27 to 0.73; P = 0.0005). OS met the predefined boundary for statistical significance. The median OS was 24.0 months (95% CI, 11.3 months to 34.1 months) in the ivosidenib plus azacitidine group and 7.9 months (95% CI, 4.1 months to 11.3 months) in the placebo plus azacitidine group (Figure 3). The OS rates were 45.4% (at 24 months) to 84.2% (at 3 months) in the ivosidenib plus azacitidine group and 20.5% (at 24 months) to 66.6% (at 3 months) in the placebo plus azacitidine group. Between-group differences in the OS rate at these time points were not available as of March 18, 2021. Treatment effect was maintained in the analysis of the per-protocol set (data not shown in this report).
After the DCO of June 30, 2022, the median follow-up time for OS was similar between the treatment groups in the FAS: ████ ██████ in the ivosidenib plus azacitidine group and ████ ██████ in the placebo plus azacitidine group. Two more patients were enrolled in the AGILE study. Five patients originally in the placebo group crossed over to the ivosidenib group. As of June 30, 2022, 95 OS events had occurred: 37 in the ivosidenib plus azacitidine group and 58 in the placebo plus azacitidine group (HR = 0.42; 95% CI, 0.27 to 0.65; P < 0.0001). The median OS was 29.3 months (95% CI, 13.2 months to NE) in the ivosidenib plus azacitidine group and 7.9 months (95% CI, 4.1 months to 11.3 months) in the placebo plus azacitidine group. The between-group difference in OS rate for ivosidenib plus azacitidine versus placebo plus azacitidine was 24.6% (95% CI, ███ ██ ████) at 12 months and 35.7% (95% CI, ████ ██ ████) at 24 months, respectively (Table 17). OS at the second DCO was not multiplicity adjusted (but significance was met at the earlier test).
Table 17: Summary of OS in the AGILE Study (FAS, DCO March 18, 2021, and June 30, 2022)
OS | Ivosidenib + azacitidine | Placebo + azacitidine |
|---|---|---|
DCO March 18, 2021 | N = 72 | N = 74 |
Median time of follow-up (months) | 15.2 | 15.3 |
Events, n (%) | 28 (38.9) | 46 (62.2) |
Patients censored, n (%) | 44 (61.1) | 28 (37.8) |
Alive | 38 (52.8) | 23 (31.1) |
Lost to follow-up | 0 | 1 (1.4) |
Withdrawal of consent | 6 (8.3) | 4 (5.4) |
OS (months), median (95% CI)a | 24.0 (11.3 to 34.1) | 7.9 (4.1 to 11.3) |
HR (95% CI)b | 0.44 (0.27 to 0.73) | |
P valuec | 0.0005 | |
OS rate, % (95% CI)d | ||
3 months | 84.2 (73.3 to 91.0) | 66.6 (54.4 to 76.2) |
6 months | 72.9 (60.4 to 82.0) | 56.3 (43.6 to 67.3) |
9 months | 67.5 (54.4 to 77.6) | 43.9 (30.9 to 56.1) |
12 months | 63.4 (49.8 to 74.2) | 36.9 (24.3 to 49.7) |
18 months | 60.9 (47.1 to 72.2) | 26.4 (14.7 to 39.6) |
24 months | 45.4 (26.8 to 62.2) | 20.5 (10.0 to 33.7) |
36 months | 0 | NE |
DCO June 30, 2022 | N = 73 | N = 75 |
Median time of follow-up (months) | ████ ██████ █████ | ████ ██████ █████ |
Events, n (%) | 37 (50.7) | 58 (77.3) |
Patients censored, n (%) | 36 (49.3) | 17 (22.7) |
Alive | 30 (41.1) | 9 (12.0) |
Lost to follow-up | 0 | 1 (1.3) |
Withdrawal of consent | 6 (8.2) | 7 (9.3) |
OS (months), median (95% CI) | 29.3 (13.2 to NE) | 7.9 (4.1 to 11.3) |
HR (95% CI) | 0.42 (0.27 to 0.65) | |
1-sided P valuee | < 0.0001 | |
OS rate, % (95% CI) | ||
3 months | 83.3 (72.4 to 90.1) | 67.8 (55.9 to 77.1) |
6 months | 73.1 (61.1 to 82.0) | 53.5 (41.3 to 64.1) |
9 months | 67.3 (55.0 to 76.9) | 44.5 (32.7 to 55.6) |
12 months | 62.9 (50.4 to 73.0) | 38.3 (27.0 to 49.5) |
Between-group difference | 24.6 (███ ██ ████) | |
18 months | 58.4 (45.9 to 69.0) | 29.1 (18.9 to 40.1) |
24 months | 53.1 (40.4 to 64.2) | 17.4 (8.9 to 28.2) |
Between-group difference | 35.7 (████ ██ ████) | |
36 months | 41.0 (26.7 to 54.7) | 11.9 (4.7 to 22.9) |
48 months | 35.8 (20.8 to 51.2) | NE |
CI = confidence interval; DCO = data cut-off; FAS = full analysis set; HR = hazard ratio; NE = not estimable; OS = overall survival.
aEstimated using the product-limit (Kaplan-Meier) method. The CIs are calculated using the Brookmeyer and Crowley method with log-log transformation.
bHR is estimated using a Cox proportional hazards model stratified by the randomization stratification factors (acute myeloid leukemia status and geographic region), with placebo plus azacitidine as the denominator.
cP value is calculated from the 1-sided log-rank test stratified by the randomization stratification factors (acute myeloid leukemia status and geographic region); the stopping boundary was 1-sided P = 0.0017.
dOS rate is the estimated probability that a patient will remain alive to the specified time point. OS rates are obtained from the Kaplan-Meier survival estimates. CIs are calculated using Greenwood’s formula and log-log transformation.
eOS at DCO of June 30, 2022, was not multiplicity adjusted.
Source: AGILE Clinical Study Report.38,39 Details included in the table are from the sponsor’s summary of clinical evidence.
Figure 3: Kaplan-Meier Plot of OS in the AGILE Study (FAS, DCO March 18, 2021)
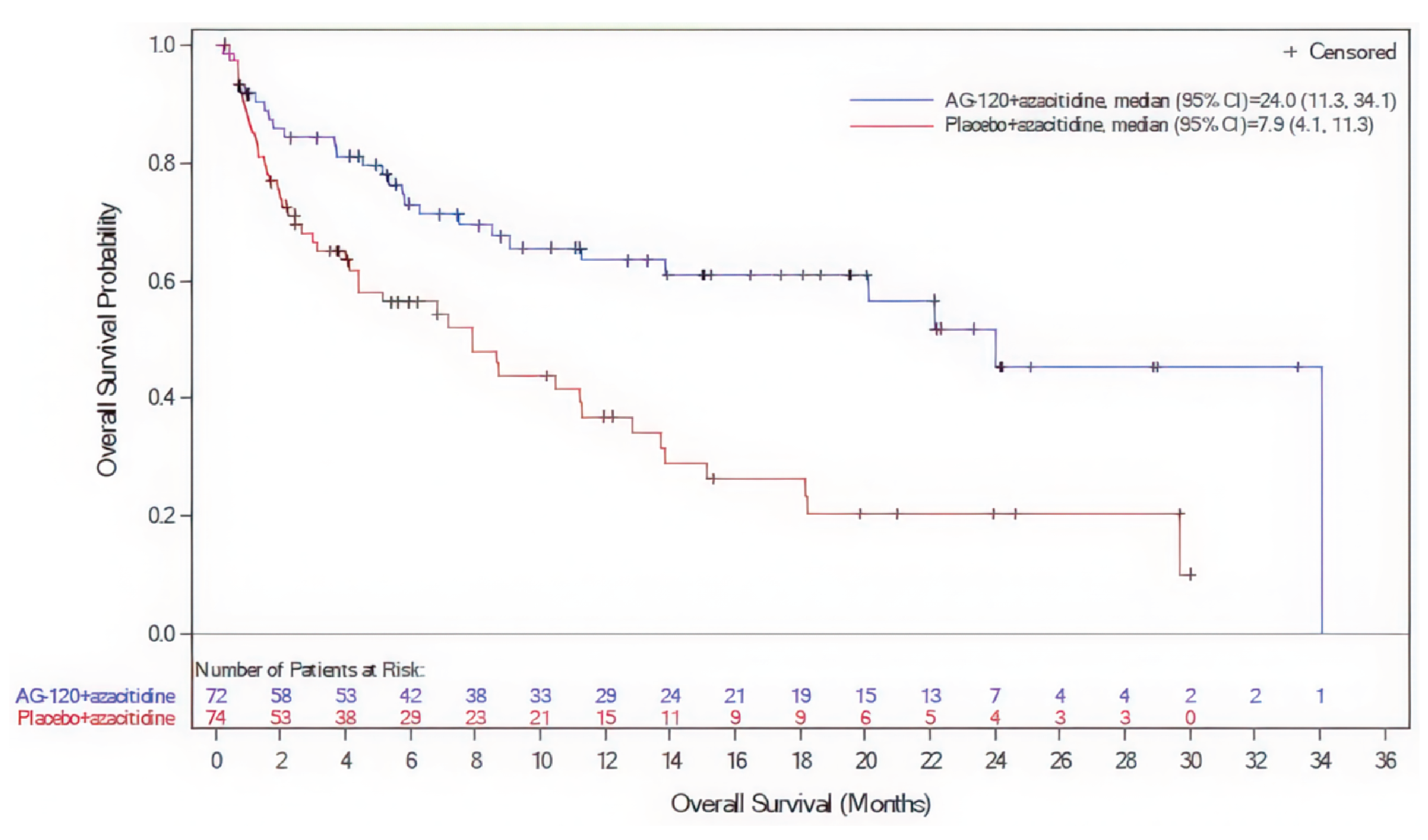
AG-120 = ivosidenib; CI = confidence interval; DCO = data cut-off; FAS = full analysis set; OS = overall survival.
Source: AGILE Clinical Study Report.38
Treatment Response
CR Rate
At the DCO of March 18, 2021, the CR rate was 47.2% in the ivosidenib plus azacitidine group and 14.9% in the placebo plus azacitidine group (odds ratio [OR] = 4.76; 95% CI, 2.15 to 10.50; 1-sided P < 0.0001). The between-group difference was 31% (95% CI, ██ ██ ███). For comparisons of the CR rate between ivosidenib plus azacitidine and placebo plus azacitidine, ████ ██████ ████ were NE and █████ ██████ █████ were not assessed because of a lack of postbaseline assessments.
CR Plus CRi Rate
At the DCO of March 18, 2021, the CR plus CRi rate was 54.2% in the ivosidenib plus azacitidine group and 16.2% in the placebo plus azacitidine group (OR = 5.90; 95% CI, 2.69 to 12.97; 1-sided P < 0.0001). The between-group difference in the CR plus CRi rate was 37% (95% CI, ██ ██ ███).
Detailed results of treatment response rates in the AGILE study are presented in Table 18.
HRQoL (Measured With EORTC QLQ C-30)
In the EORTC QLQ C-30, higher scores in the global health status subscale indicate better HRQoL.
At baseline, the mean scores for EORTC QLQ-C30 subscales were similar between the treatment groups (data not shown).
At 6, 12, and 18 months, the proportion of patients who were available to complete the HRQoL assessment was ██████ █████ ███ ████ of the FAS, respectively. The dropout rate was higher in the placebo plus azacitidine group than in the ivosidenib plus azacitidine group, which could be partially explained by there being more deaths in the former group.
Table 18: Summary of Treatment Response Rates in the AGILE Study (FAS, DCO March 18, 2021)
Response rates | Ivosidenib + azacitidine (N = 72) | Placebo + azacitidine (N = 74) |
|---|---|---|
CR rates, n (%) | 34 (47.2) | 11 (14.9) |
95% CIa | (35.3 to 59.3) | (7.7 to 25.0) |
Difference in CR rate, % (95% CI) | 31% (████ ██ ████) | |
OR (95% CI)b | 4.76 (2.15 to 10.50) | |
P valuec | < 0.0001 | |
CR + CRi rates, n (%) | 39 (54.2) | 12 (16.2) |
95% CIa | (42.0 to 66.0) | (8.7 to 26.6) |
Difference in CR + CRi (including CRp) rate, % (95% CI) | 37% (0.23 to 0.51) | |
OR (95% CI)b | 5.90 (█████ █████) | |
P valuec | < 0.0001 | |
CI = confidence interval; CMH = Cochran-Mantel-Haenszel; CR = complete remission; CRi = complete remission with incomplete hematologic recovery; CRp = complete remission with incomplete platelet recovery; DCO = data cut-off; FAS = full analysis set; OR = odds ratio.
aThe CI of the percentage is calculated with the Clopper and Pearson (exact binomial) method.
bThe CMH estimate for the OR is calculated with placebo plus azacitidine as the control (denominator).
cIf the primary analysis of EFS is significant, a stratified CMH test will be used to compare CR between the 2 treatment arms. The 1-sided P value is calculated from the CMH test stratified by the randomization stratification factors (acute myeloid leukemia status and geographic region); the stopping boundary was 1-sided P = 0.0087. CR plus CRi was not adjusted for multiplicity in the AGILE study.
Source: AGILE Clinical Study Report.38 Details included in the table are from the sponsor’s summary of clinical evidence.
Table 19: Summary of Change From Baseline in Global Health Status/QoL Subscale of EORTC QLQ-C30 (FAS, DCO March 18, 2021)
Subscales | Ivosidenib + azacitidine (N = 72) | Placebo + azacitidine (N = 74) |
|---|---|---|
Cycle 7, day 1: 6 months | ||
Patients contributing to the analysis, n | 31 | 17 |
Least squares mean change from baseline (95% CI) | 10.6 (1.23 to 19.97) | –2.0 (–12.80 to 8.84) |
Difference of least squares mean change from baseline (95% CI) | 12.6 (1.51 to 23.65) | |
P valuea | 0.0261 | |
Cycle 13, day 1: 12 months | ||
Patients contributing to the analysis, n | 18 | 5 |
Least squares mean change from baseline (95% CI) | 19.1 (8.51 to 29.72) | 4.2 (–11.94 to 20.28) |
Difference of least squares mean change from baseline (95% CI) | 14.9 (–2.09 to 31.97) | |
P value | 0.0854 | |
Cycle 19, day 1: 18 months | ||
Patients contributing to the analysis, n | 11 | 2 |
Least squares mean change from baseline (95% CI) | 18.5 (6.29 to 30.64) | –0.7 (–24.31 to 22.89) |
Difference of least squares mean change from baseline (95% CI) | 19.2 (–5.77 to 44.12) | |
P value | 0.1316 | |
CI = confidence interval; DCO = data cut-off; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; FAS = full analysis set; QoL = quality of life.
Notes: A 2-sided P value is reported. The least squares mean and 95% CI are estimated from the mixed effect model on the change from baseline across visits for all scales, with baseline score, treatment arm, time, randomization stratification factors (acute myeloid leukemia status and geographic region) and an interaction between treatment arm and time as fixed effects, and patient as a random effect. The unstructured covariance structure is used to define covariance between random effects. Unscheduled visits are excluded from the analysis.
aThis outcome was not multiplicity adjusted.
Source: AGILE Clinical Study Report.38 Details included in the table are from the sponsor’s summary of clinical evidence.
At 6, 12, and 18 months of treatment, patients in the ivosidenib plus azacitidine group reported increased global health status/QoL subscale scores from baseline; however, this trend was not observed in the placebo plus azacitidine group. The point estimates for the least squares mean between-group difference in this score exceeded the MIDs identified from the literature (a 3-point to 11-point increase indicates clinically meaningful improvement in this subscale). The MIDs identified from the literature were not specific to patients with AML.
Transfusion Requirement
At baseline, the proportion of patients who were transfusion dependent was 54.2% in the ivosidenib plus azacitidine group and 54.1% in the placebo plus azacitidine group. Among patients who were transfusion dependent at baseline, a higher proportion of patients receiving ivosidenib plus azacitidine (██ ███████ ████████) experienced RBC and/or platelet transfusion independence than those receiving placebo plus azacitidine (██ ███████ ████████) (OR ████ ███ ██ ███ ██ ████ ████████) at the DCO of March 18, 2021.
Similarly, at the DCO of June 30, 2022, a higher proportion of patients in the ivosidenib plus azacitidine group (██ ███████ ████████) experienced RBC and/or platelet transfusion independence than in the placebo plus azacitidine group (██ ███████ ████████) (OR ████ ███ ██ ███ ██ ██████████).
Table 20: Transfusion Requirements in the AGILE Study (FAS, DCO of March 18, 2021, and June 30, 2022)
Transfusion requirements | Ivosidenib + azacitidine | Placebo + azacitidine | |
|---|---|---|---|
DCO March 18, 2021 | N = 72 | N = 74 | |
Baseline RBC and/or platelet transfusion dependent, n (%) | 39 (54.2) | 40 (54.1) | |
Conversion from baseline transfusion dependent to postbaseline transfusion independent, n/Na (%) | ██████████ | ██████████ | |
95% CI | █████ ████ | █████ ████ | |
OR (95% CI) | ███ █████ ████ | ||
P value | ██████ | ||
DCO June 30, 2022 | N = 73 | N = 75 | |
Baseline RBC and/or platelet transfusion dependent, n (%) | ██ ██████ | ██ ██████ | |
Conversion from baseline transfusion dependent to postbaseline transfusion independent, n/Na (%) | ██████████ | ██████████ | |
95% CI | █████ ████ | █████ ████ | |
RD (95% CI) | ██ ██████ ██ █████ | ||
OR (95% CI) | ███ █████ ████ | ||
P value | ██████ | ||
CI = confidence interval; DCO = data cut-off; FAS = full analysis set; OR = odds ratio; RBC = red blood cell; RD = risk difference.
Note: The outcome of transfusion requirements was not adjusted for multiplicity.
aDenominators are the number of patients who required transfusion at baseline.
Source: AGILE Clinical Study Report.38,39 Details included in the table are from the sponsor’s summary of clinical evidence.
Harms
Summaries of safety data were presented by treatment group for the safety analysis set. In general, the safety results were consistent between the 2 DCO dates.
Adverse Events
As of the DCO of March 18, 2021, the proportion of patients who experienced at least 1 AE was 98.6% (70 patients) in the ivosidenib plus azacitidine group and 100% (73 patients) in the placebo plus azacitidine group. Patients treated with ivosidenib plus azacitidine were more likely (5% or more) to report the following TEAEs than patients treated with placebo plus azacitidine: vomiting (29 patients [40.8%] in the ivosidenib plus azacitidine group versus 19 patients [26.0%] in the placebo plus azacitidine group), neutropenia (20 [28.2%] versus 12 [16.4%]), thrombocytopenia (20 [28.2%] versus 15 [20.5%]), prolonged electrocardiogram QT interval (14 [19.7%] versus 5 [6.8%]), insomnia (13 [18.3%] versus 9 [12.3%]), differentiation syndrome (10 [14.1%] versus 6 [8.2%]), pain in extremity (10 [14.1%] versus 3 [4.1%]), hematoma (9 [12.7%] versus 1 [1.4%]), arthralgia (8 [11.3%] versus 3 [4.1%]), headache (8 [11.3%] versus 2 [2.7%]), leukocytosis (8 [11.3%] versus 1 [1.4%]), and leukopenia (6 [8.5%] versus 2 [2.7%]). In general, the severity of AEs was mild.
The following TEAEs were more likely (5% or more) to be reported in the placebo plus azacitidine group than in the ivosidenib plus azacitidine group: constipation (38 patients [52.1%] in the placebo plus azacitidine group versus 19 patients [26.8%] in the ivosidenib plus azacitidine group), pyrexia (29 [39.7%] versus 24 [33.8%]), febrile neutropenia (25 [34.2%] versus 20 [28.2%]), asthenia (24 [32.9%] versus 11 [15.5%]), pneumonia (23 [31.5%] versus 17 [23.9%]), hypokalemia (21 [28.8%] versus 11 [15.5%]), decreased appetite (19 [26.0%] versus 11 [15.5%]), edema peripheral (16 [21.9%] versus 8 [11.3%]), weight decrease (12 [16.4%] versus 4 [5.6%]), cough (11 [15.1%] versus 6 [8.5%]), and sepsis (6 [8.2%] versus 2 [2.8%]).
Grade 3 and higher AEs were reported in 66 patients (93.0%) in the ivosidenib plus azacitidine group and 69 patients (94.5%) in the placebo plus azacitidine group. In both groups, the commonly reported grade 3 and higher AEs were as follows (shown as ivosidenib plus azacitidine versus placebo plus azacitidine): anemia (25.4% versus 26.0%), febrile neutropenia (28.2% versus 34.2%), neutropenia (26.8% versus 16.4%), thrombocytopenia (23.9% versus 20.5%), and pneumonia (22.5% versus 28.8%).
As of the DCO of June 30, 2022, the number of patients who reported at least 1 AE was ██ ███████ in the ivosidenib plus azacitidine group and ██ ██████ in the placebo plus azacitidine group. Grade 3 and higher AEs were reported in ██ ███████ patients in the ivosidenib plus azacitidine group and ██ ███████ patients in the placebo plus azacitidine group. In both groups, the commonly reported grade 3 and higher AEs were as follows (shown as ivosidenib plus azacitidine versus placebo plus azacitidine): anemia (█████ ███ █████), febrile neutropenia (█████ ███ █████), neutropenia (█████ ███ █████), thrombocytopenia (█████ ███ █████), and pneumonia (█████ ███ █████).
Serious AEs
The proportion of patients who experienced SAEs was 69.0% (49 patients) in the ivosidenib plus azacitidine group and 82.2% (60 patients) in the placebo plus azacitidine group.
Commonly reported SAEs in the 2 treatment groups were febrile neutropenia (23.9% of patients in the ivosidenib plus azacitidine group versus 27.4% in the placebo plus azacitidine group) and pneumonia (19.7% versus 21.9%).
Results relating to SAEs at the DCO of June 30, 2022, were similar to those at the DCO of March 18, 2021.
Withdrawal Due to AEs
The overall incidences of TEAEs that led to combination treatment discontinuation were similar between the treatment groups: 19 patients (26.8%) in the ivosidenib plus azacitidine group and 19 patients (26.0%) in the placebo plus azacitidine group. Of the AEs leading to treatment discontinuation, only pulmonary embolism occurred in more than 1 patient (it occurred in 2 patients [2.8%] in the ivosidenib plus azacitidine group). The overall incidence of TEAEs that led to discontinuation of only ivosidenib or placebo was similar between the 2 groups (██████ patients in the ivosidenib plus azacitidine group versus ██████ in the placebo plus azacitidine group). TEAEs that led to discontinuation of only azacitidine occurred in █████ patients in the ivosidenib plus azacitidine group versus ██████ in the placebo plus azacitidine group and included febrile neutropenia (██████ patient per treatment group) and thrombocytopenia (██████ patient in the ivosidenib plus azacitidine group).
Mortality
Ten patients (14.1%) in the ivosidenib plus azacitidine group and 21 patients (28.8%) in the placebo plus azacitidine group died because of AEs during the study as of the DCO of March 18, 2021.
As of the DCO of June 30, 2022, ██ ████████ ███████ in the ivosidenib plus azacitidine group and ██ ████████ ███████ in the placebo plus azacitidine group died because of AEs during the study.
Notable Harms
Among the AEs of special interest reported by the sponsor, patients treated with ivosidenib plus azacitidine reported more cases of prolonged electrocardiogram QT intervals, leukocytosis, and differentiation syndrome (differentiation syndrome was identified as 1 of the most important AEs by the clinical experts consulted by the review team).
As of the DCO of June 30, 2022, differentiation syndrome was reported in 10 patients (13.9%) in the ivosidenib plus azacitidine group and 6 patients (8.1%) in the placebo plus azacitidine group. The majority of differentiation syndrome events occurring in patients in the ivosidenib plus azacitidine group were grade 2 (██████ █████████████), with only ██████ patients experiencing grade 3 differentiation syndrome. In the placebo plus azacitidine group, ██████ patients experienced grade 2 differentiation syndrome, ██████ patients experienced grade 3 differentiation syndrome, and ██████ patient experienced grade 4 differentiation syndrome.
Key harms at the DCOs of March 18, 2021, and June 30, 2022, are summarized in Table 21.
Table 21: Summary of Harms Results From the AGILE Study (SAS, DCO March 18, 2021, and June 30, 2022)
Harms | Ivosidenib + azacitidine March 18, 2021: N = 71 June 30, 2022: N = 72 | Placebo + azacitidine March 18, 2021: N = 73 June 30, 2022: N = 74 |
|---|---|---|
TEAEs, n (%) | ||
Patients with events, March 18, 2021 | 70 (98.6) | 73 (100.0) |
TEAEs occurring in ≥ 10% of patients in either treatment group | ||
Nausea | 30 (42.3) | 28 (38.4) |
Vomiting | 29 (40.8) | 19 (26.0) |
Diarrhea | 25 (35.2) | 26 (35.6) |
Pyrexia | 24 (33.8) | 29 (39.7) |
Anemia | 22 (31.0) | 21 (28.8) |
Febrile neutropenia | 20 (28.2) | 25 (34.2) |
Neutropenia | 20 (28.2) | 12 (16.4) |
Thrombocytopenia | 20 (28.2) | 15 (20.5) |
Constipation | 19 (26.8) | 38 (52.1) |
Pneumonia | 17 (23.9) | 23 (31.5) |
Prolonged electrocardiogram QT | 14 (19.7) | 5 (6.8) |
Insomnia | 13 (18.3) | 9 (12.3) |
Asthenia | 11 (15.5) | 24 (32.9) |
Decreased appetite | 11 (15.5) | 19 (26.0) |
Dyspnea | 11 (15.5) | 9 (12.3) |
Hypokalemia | 11 (15.5) | 21 (28.8) |
Differentiation syndrome | 10 (14.1) | 6 (8.2) |
Pain in extremity | 10 (14.1) | 3 (4.1) |
Fatigue | 9 (12.7) | 10 (13.7) |
Hematoma | 9 (12.7) | 1 (1.4) |
Arthralgia | 8 (11.3) | 3 (4.1) |
Headache | 8 (11.3) | 2 (2.7) |
Leukocytosis | 8 (11.3) | 1 (1.4) |
Edema peripheral | 8 (11.3) | 16 (21.9) |
Platelet count decreased | 8 (11.3) | 6 (8.2) |
Rash | 7 (9.9) | 9 (12.3) |
Cough | 6 (8.5) | 11 (15.1) |
Hemorrhoids | 5 (7.0) | 8 (11.0) |
Weight decrease | 4 (5.6) | 12 (16.4) |
Patients with events, June 30, 2022 | ██ ██████ | ██ █████ |
SAEs, n (%) | ||
Patients with events, March 18, 2021 | 49 (69.0) | 60 (82.2) |
Serious TEAEs occurring in ≥ 2% of patients in either treatment group | ||
Febrile neutropenia | 17 (23.9) | 20 (27.4) |
Pneumonia | 14 (19.7) | 16 (21.9) |
Differentiation syndrome | 6 (8.5) | 1 (1.4) |
Pyrexia | 4 (5.6) | 3 (4.1) |
Pulmonary embolism | 3 (4.2) | 1 (1.4) |
Bronchopulmonary aspergillosis | 2 (2.8) | 2 (2.7) |
COVID-19 | 2 (2.8) | 0 |
Hemorrhage intracranial | 2 (2.8) | 0 |
Pleural effusion | 2 (2.8) | 0 |
Renal failure | 2 (2.8) | 1 (1.4) |
Thrombocytopenia | 2 (2.8) | 1 (1.4) |
Sepsis | 1 (1.4) | 3 (4.1) |
Septic shock | 1 (1.4) | 2 (2.7) |
Anal abscess | 0 | 2 (2.7) |
Diarrhea | 0 | 2 (2.7) |
Diverticulitis | 0 | 2 (2.7) |
Epistaxis | 0 | 2 (2.7) |
General physical health deterioration | 0 | 2 (2.7) |
Parotitis | 0 | 2 (2.7) |
Patients with events, June 30, 2022 | ██ ██████ | ██ ██████ |
WDAEs, n (%) | ||
TEAE leading to discontinuation of study drug, March 18, 2021 | ||
Discontinuation of ivosidenib or placebo only | 3 (4.2) | 2 (2.7) |
Discontinuation of azacitidine only | 2 (2.8) | 1 (1.4) |
Discontinuation of both ivosidenib or placebo and azacitidine | 19 (26.8) | 19 (26.0) |
TEAE leading to discontinuation of study drug, June 30, 2022 | ||
Discontinuation of ivosidenib or placebo only | 3 (4.2) | 1 (1.4) |
Discontinuation of azacitidine only | 5 (6.9) | 3 (4.1) |
Discontinuation of both ivosidenib or placebo and azacitidine | 19 (26.4) | 19 (25.7) |
AEs leading to deaths, n (%) | ||
DCO: March 18, 2021 | 10 (14.1) | 21 (28.8) |
DCO: June 30, 2022 | ██ ██████ | ██ ██████ |
AESI, n (%) | ||
Differentiation syndrome | March 18, 2021: 10 (14.1) ≥ grade 3: 3 (4.2) June 30, 2022: ██ ██████ | March 18, 2021: 6 (8.2) ≥ grade 3: 3 (4.1) June 30, 2022: █████ |
Infection | March 18, 2021: 20 (28.8) ≥ grade 3: 15 (21.1) June 30, 2022: ██ ███████ ≥ grade 3: ██ ██████ | March 18, 2021: 36 (49.3) ≥ grade 3: 22 (30.1) June 30, 2022: ██ ███████ ≥ grade 3: ██ ██████ |
AE = adverse event; AESI = adverse event of special interest; DCO = data cut-off; SAS = safety analysis set; TEAE = treatment-emergent adverse event; WDAE = withdrawal due to adverse event.
Source: AGILE Clinical Study Report.38,39 Details included in the table are from the sponsor’s summary of clinical evidence.
Hospitalizations due to AEs
At the DCO of March 18, 2021, the rates of hospitalization for TEAEs were similar for both treatment groups. The days of hospitalization per person-year of drug exposure were ███ for the ivosidenib plus azacitidine group and ███ for the placebo plus azacitidine group (Table 22).
Hospitalization due to other reasons was not assessed in the AGILE study.
Table 22: Hospitalizations for Adverse Events in the AGILE Study (SAS, DCO March 18, 2021)
Hospitalizations | Ivosidenib + azacitidine (N = 71) | Placebo + azacitidine (N = 73) |
|---|---|---|
Events, n | ██ | ███ |
Days hospitalized, n | ████ | ████ |
Rate of event (unit not reported; variation not reported) | ███ | ███ |
Days hospitalized per person-year of drug exposure, n (variation not reported) | ███ | ███ |
DCO = data cut-off; SAS = safety analysis set.
Source: AGILE Clinical Study Report.38 Details included in the table are from the sponsor’s summary of clinical evidence.
Critical Appraisal
Internal Validity
In the AGILE study, appropriate methods of randomization and allocation concealment were employed. The randomization schedule was prepared by an independent statistical group and stratified by de novo status and geographic region. The allocation was implemented using interactive response technology. Several baseline patient characteristics were balanced between the 2 treatment groups, for example demographic characteristics, disease characteristics, and prior anticancer therapy. The use of concomitant therapies and subsequent anticancer therapies was also generally balanced across the groups and consistent with the clinical practice in Canada. There were some imbalances in baseline patient characteristics between the 2 treatment groups, for example gender, WHO classification of AML, and cytogenetic risk status as assessed by the investigator. These imbalances are likely to be the result of the small sample size, within which prognostic balance is not likely to be assured; as such, there is some risk that the observed effects are overestimated or underestimated. In addition, the postbaseline transfusion independence outcome was measured among approximately half the population who required transfusions at baseline. Randomization is not necessarily upheld in this population. However, the results of transfusion requirement in patients who were dependent on transfusion at baseline did not differ significantly from those in the overall population. Therefore, the potential for bias is unlikely to have an important impact on the study findings specific to this outcome.
The study originally had no planned interim analyses. Observations of a notable difference in the number of deaths (favouring ivosidenib) by the IDMC prompted an unplanned interim analysis before the protocol-defined number of events. To control for multiplicity, new stopping boundaries were calculated based on the observed information fraction that were not outlined in the original statistical analysis plan. Because the results are from an unplanned interim analysis (which became the final analysis), even though the new stopping boundaries are appropriate, there is a risk of overestimation of the true effects of the study drug. Some of the important clinical outcomes were analyzed without multiplicity adjustment, for example HRQoL assessment using the EORTC QLQ-C30. As such, there would be an increased risk of false-positive conclusions (i.e., erroneously rejecting the null hypothesis); however, the reported results for these patient-reported outcomes were not statistically significant at later time points.
The patients, investigators, sponsor, and clinical research unit staff who deal directly with patients were blinded to treatment allocation until the final analysis for the primary end point unless emergency unblinding was required. Following the early interim analysis, the AGILE study was unblinded, and patients who received placebo plus azacitidine could switch to ivosidenib plus azacitidine.
HRQoL was assessed using a cancer-specific instrument. Even though the EORTC QLQ C-30 is not an AML-specific instrument and an MID for patients with AML was not identified from the literature, a range of potential between-group MIDs (3 to 11 points for improvement and –5 to –13 points for deterioration on the global QoL scale) were established based on clinical trials of 9 cancer types and may provide some guidance when determining the clinical relevance of the findings for HRQoL in the AGILE study. Even though no threshold of clinical importance could be estimated in patients with AML, the review team leaned on these MID ranges identified in other cancer types when assessing the GRADE imprecision domain for the EORTC QLQ C-30 results in the AGILE study. The completion rate of the EORTC QLQ C-30 was low. The completion rates were ██████ █████ ███ ████ at 6 months, 12 months, and 18 months of the study. Missing data were implicitly imputed within the mixed model, with the assumption of “missing at random.” However, there were no sensitivity analyses, and it is unlikely that the missing at random assumption is plausible. As a result, there is a high risk of bias because of the large amount of missing outcome data.
In the analysis of EFS, patients were censored if CR was documented by 24 weeks and 1 of the following occurred: the patients started subsequent anticancer therapy, relapse or death was documented after 2 or more missing disease assessments, the patient was lost to follow-up, withdrawal by patient, or the CR was ongoing without patient relapse or death. For patients who experienced CR by 24 weeks, no 1 was lost to follow-up. In the analysis of OS, patients were censored if they were alive or lost to follow-up or if they withdrew consent. The proportion of patients who were lost to follow-up was very low. Therefore, the effect of missing data on survival outcomes was not considered significant. For other binary end points, such as CR and CR plus CRi, there appeared to be a large amount of data missing, labelled as “not assessed”: █████ in the ivosidenib plus azacitidine group versus █████ in the placebo plus azacitidine group were not assessed because of a lack of postbaseline assessments for CR assessment. Subsequently, a high risk of bias may be introduced with unclear direction; no reason for the missing data was reported.
EFS was the primary efficacy outcome in this study. This is a composite end point, which was defined as the time from randomization until treatment failure (i.e., patient does not experience CR by week 24), relapse from remission, or death from any cause, whichever occurred first. In the AGILE study, almost all events occurred at baseline (i.e., 1 component of the end point). As such, there were few patients left at risk postbaseline; as a result, the EFS could not robustly characterize the long-term efficacy of the study drug.31 The correlations between EFS and OS were modest in the published research that provided trial-level information. However, 1 major limitation of these studies was that they were not specific to the population nor the drug class of interest, and therefore the ability to generalize the study findings was not clear.32-34
Predefined sensitivity analyses were conducted to evaluate the robustness of the primary EFS results. Overall, the results of the sensitivity analyses were generally aligned with the primary analysis of EFS, which supported the robustness of the results. Prespecified subgroup analyses generally supported consistency in the overall direction of effect of ivosidenib across subgroups; some subgroups were small, resulting in wide CIs.
There was low risk of selective reporting bias; all presented end points had been specified in the statistical analysis plan; however, as mentioned previously, the interim analysis was unplanned.
As described in the Outcomes section, OS was the primary efficacy end point at the beginning of this trial, but it was replaced with EFS. The sponsor’s justification was that this change allowed for a smaller sample size and therefore a more feasible trial in this rare patient population. Furthermore, EFS was considered by the sponsor to more accurately describe the contribution of a novel therapy to clinical benefit by removing the potentially confounding effects of posttrial therapies and by capturing treatment failure as an event. Meanwhile, previous research provided encouraging preliminary safety and efficacy data when comparing an IDH1 inhibitor plus azacitidine with azacitidine alone. All these factors supported the amendment of the protocol of the AGILE study to use EFS as a measure of clinical benefit for the treatment of patients with AML who are ineligible for intensive induction chemotherapy. Moreover, this change was done before unblinding of the data and therefore was not likely to bias the study results.
External Validity
According to feedback from the clinical experts consulted for this review, the eligibility criteria and baseline characteristics of the patients randomized in the AGILE study generally reflected a patient population in Canadian clinical practice that would receive combination therapy of ivosidenib plus azacitidine. The clinical experts noted that the results from the AGILE study could be generalized to patients with IDH1-mutated AML in Canada who would be treated with ivosidenib plus azacitidine. The clinical experts also indicated that in clinical practice, ECOG performance status criteria would not always be used; in addition, some flexibility should be applied in terms of using ivosidenib plus azacitidine in patients with slightly worse ECOG performance status than in the trial. The potential benefits and risks of this treatment for individual patients need to be assessed. Patients’ IDH1 mutation status should be confirmed before the treatment. The experts indicated that the outcome measures in the AGILE study were generally appropriate and clinically relevant for clinical trials of AML.
In the AGILE study, ivosidenib in combination with azacitidine was compared with azacitidine monotherapy. The clinical experts consulted for this review indicated that azacitidine alone is not the most appropriate comparator for the study drug combination in the study population. Instead, venetoclax plus azacitidine is currently the most commonly used combination therapy in the target patient population.
In practice, monotherapy with azacitidine would typically be used for patients with increased frailty that would make treatment with the combination of venetoclax and azacitidine unreasonable. There is a lack of direct evidence within the AGILE study with which to examine the relative efficacy and safety of the study drug compared with other combination regimens.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for the outcomes considered most relevant to inform the expert committee deliberations, and a final certainty rating was determined, as outlined by the GRADE Working Group.35,36
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate — The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited — The true effect may be substantially different from the estimate of the effect. We use the word may for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate — The true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as “very uncertain.”
Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null. The threshold for a clinically important effect for OS and EFS in the study population was not obtained. Therefore, the target of the certainty of evidence assessment was the presence of absence of any (non-null) effect for survival rates. The threshold for a clinically important effect for the EORTC QLQ-C30 score was set according to the presence or absence of an important effect based on thresholds identified in the literature.37 In addition, the target of the certainty of evidence assessment was the presence or absence of any non-null effect for CR, CR plus CRi, and transfusion requirements. For some harm events (e.g., differentiation syndrome), because of the unavailability of the absolute difference in effects, the certainty of evidence was summarized narratively.
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for ivosidenib plus azacitidine versus placebo plus azacitidine.
Long-Term Extension Studies
There were no relevant long-term extension studies submitted for this review.
Indirect Evidence
Contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
Objectives for the Summary of Indirect Evidence
Aside from the comparison to placebo plus azacitidine, there was no direct evidence comparing ivosidenib plus azacitidine against other relevant comparators for the treatment of newly diagnosed or untreated patients with IDH1-mutated AML; therefore, a review of indirect evidence was undertaken and submitted by the sponsor.58 A research protocol is not available for this study; however, detailed selection criteria and methods of analyses were provided in the ITC report submitted by the sponsor.
Description of Indirect Comparisons
Objectives
The objective of the submitted ITC report was to derive estimates of the relative efficacy of ivosidenib plus azacitidine versus existing therapies for patients with treatment-naive or newly diagnosed AML with IDH1 mutation who are ineligible for intensive chemotherapy, by means of either an NMA or an MAIC.
Study Selection Methods
A summary of the study selection criteria and methodology used to conduct the systematic review contributing to the ITCs is given in Table 23. Overall, clinical trials of adult patients with newly diagnosed or untreated AML who were ineligible for intensive chemotherapy were included. Eligible patients could include those aged 75 years or older; had severe heart, pulmonary, liver, or renal disorders; or had a greater than 20% blast count. Treatments of interest included ivosidenib plus azacitidine, venetoclax plus azacitidine, azacitidine monotherapy, LDAC, venetoclax plus LDAC, and glasdegib plus LDAC. The outcomes considered in this ITC report included OS, EFS, treatment response rates, transfusion independence, and transfusion burden, which were deemed most important to capture and convey the treatment benefit to payers and clinicians and to provide outputs amenable for economic modelling. Multiple databases, conference abstracts, and trial registries were searched to identify relevant evidence. The search was last updated on January 31, 2023. Study selection was carried out by 2 reviewers independently. Data was extracted using a standardized data extraction form; however, it was unclear if this was completed by the 2 independent reviewers. Risk of bias in the included RCTs was assessed at the study level using the Cochrane Risk of Bias tool v2.0; the number of reviewers who contributed was not reported.
Table 23: Study Selection Criteria and Methods for Systematic Review Contributing to the ITCs Submitted by the Sponsor
Characteristics | Systematic review contributing to the ITCs |
|---|---|
Population | Adults (≥ 18 years old) with first-line/treatment-naive/newly diagnosed AML not eligible for intensive chemotherapy, which may include the following criteria:
|
Intervention | Ivosidenib 500 mg once daily + azacitidine 75mg/m2 for 7 days, every 28 days |
Comparator | Relevant comparators:
Additional comparators:a
|
Outcome | OS, EFS, DOR, CR, CR + CRi, CR + CRh, transfusion independence, transfusion burden. A broad list of outcomes in the SLR eligibility criteria is available in the sponsor-submitted ITC report. |
Study designs | Clinical trials (any phase) |
Publication characteristics | Published and unpublished studies |
Exclusion criteria | Patient population:
Interventions/comparators: Any treatments or therapies not listed in the inclusion criteria Outcomes: Studies not providing data on the specific outcomes of interest Study design:
Other restrictions: Non-English language studies |
Databases searched |
In addition, various oncology conferences were searched to identify abstracts presented between 2019 and 2021. To ensure all relevant trials were captured, searches of the Clinicaltrials.gov, ICTRP, and Clinicaltrialsregister.eu registries for completed trials were undertaken. |
Selection process | Abstract and full-text reviews were conducted independently by 2 reviewers based on the PICOS criteria; 10% of the abstracts were quality checked by a third independent reviewer. Any uncertainty, or any disagreements, about including certain publications were resolved either through “reconciliation” (discussion between the 2 reviewers) or through “arbitration” by a third independent reviewer, where the majority view determined inclusion or exclusion. |
Data extraction process | Extraction of data on the outcomes of interest from the full-text studies identified by the searches was conducted using a standardized data extraction template. |
Quality assessment | The quality of RCTs retained for data extraction was assessed using the revised Cochrane Risk of Bias tool (RoB 2.0), with assessment of 5 components: randomization process, deviations from intended interventions, missing outcome data, measurement of the outcome, and selection of the reported results. The overall risk of study bias was rated as low risk, some concerns, or high risk. |
AML = acute myeloid leukemia; CR = complete remission; CRh = complete remission with partial hematologic recovery; CRi = complete remission with incomplete hematologic recovery; DOR = duration of response; ECOG PS = Eastern Cooperative Oncology Group performance status; EFS = event-free survival; FEV1 = forced expiratory volume in 1 second; ICTRP = International Clinical Trials Registry Platform; ITC = indirect treatment comparison; LDAC = low-dose cytarabine; LVEF = left ventricular ejection fraction; OS = overall survival; PICOS = population, intervention, comparison, outcomes and study; RCT = randomized controlled trial; SLR = systematic literature review; ULN = upper limit of normal
aVenetoclax plus LDAC and glasdegib plus LDAC combinations are not recommended for reimbursement in Canada; however, they were included in the analyses for consistency with the SLR strategy and the ITC PICOS criteria.
Source: Sponsor-submitted ITC report.58 Details included in the table are from the sponsor’s summary of clinical evidence.
Feasibility Appraisal
Before the analysis, a comprehensive feasibility assessment was conducted to verify whether an ITC could be made. This assessment looked at the ability to pool across studies within each treatment group and the presence and extent of between-study heterogeneity. A rationale for conducting an NMA and various MAICs was not provided in this ITC analysis.
ITC Analysis Methods
Network Meta-Analyses
The AGILE study was the only study comparing treatments exclusively in patients with IDH1 mutation. The relative efficacy of ivosidenib plus azacitidine versus existing therapies was therefore estimated for adults with previously untreated AML ineligible for intensive chemotherapy irrespective of mutation status, using an NMA for the outcomes of OS, EFS, CR rates, CR plus CRi rates, CR plus CRh rates, and transfusion independence. In addition to the NMA, 3 MAICs were conducted in comparison with venetoclax plus azacitidine to account for population imbalances: an anchored MAIC of OS for ivosidenib plus azacitidine versus venetoclax plus azacitidine in the ITT population of the VIALE-A study; an unanchored MAIC of OS for ivosidenib plus azacitidine versus the venetoclax plus azacitidine group from the IDH1-mutated subgroup in the VIALE-A study; an anchored MAIC of EFS for ivosidenib plus azacitidine versus venetoclax plus azacitidine in the ITT population of the VIALE-A study.
A summary of the NMA methods is presented in Table 24.
Table 24: Network Meta-Analysis Methods
Methods | Description |
|---|---|
Analysis methods | Bayesian approach |
Priors | Vague (flat/uninformative) |
Assessment of model fit | Assessment of DIC and total residual deviance |
Assessment of consistency | Not applicable, no closed loops |
Assessment of convergence | Assessment of BGR diagnostic in OpenBUGS; assessment of Monte Carlo error; visual inspection of trace-density plots |
Outcomes |
|
Follow-up time points | Update data cut-off for OS:
For other outcomes:
|
Construction of nodes | Review of data availability for each outcome of interest, combined with the assessment of patient baseline characteristics and study design characteristics, for all included studies enabled the assessment of the feasibility of establishing networks of evidence and conducting analyses for each outcome of interest |
Sensitivity analyses | Not performed |
Subgroup analysis | Not performed |
Methods for pairwise meta-analysis | Not performed |
BGR = Brooks-Gelman-Rubin; CR = complete remission; CRi = complete remission with incomplete hematologic recovery; CrI = credible interval; DIC = deviance information criterion; EFS = event-free survival; HR = hazard ratio; OR = odds ratio; OS = overall survival.
Source: Sponsor-submitted indirect treatment comparison.58 Details included in the table are from the sponsor’s summary of clinical evidence.
Analysis Framework
Analyses were run in a Bayesian framework.66
Assessment of Convergence
Under a Bayesian approach, posterior densities for the unknown parameters are estimated using Markov chain Monte Carlo simulations for each model. The proposed analyses were based on a burn-in of (at least) 20,000 iterations and a further sample of (at least) 40,000 iterations or until convergence was achieved. Convergence was assessed by checking the Brooks-Gelman-Rubin diagnostic in OpenBUGS. In addition, the Monte Carlo error was captured, which reflects both the number of simulations and the degree of autocorrelation. This should be no more than 5% of the posterior standard deviation of the parameters of interest. Finally, visual inspection of trace-density plots was carried out.
As suggested by the National Institute for Health and Care Excellence (NICE) Decision Support Unit (DSU) report,66 a normal distribution with zero mean and variance equal to 104 was used for treatment effects and a uniform distribution with range zero to 2 was used for the between-trial standard deviation. Vague (flat/uninformative) priors were used for all calculations.
Inconsistency Assessment
As none of the evidence networks for OS, EFS, CR, CR + CRi, or transfusion requirement have closed loops, a consistency assessment as per NICE DSU Technical Support Document 4 was not possible.67
Model Selection
The conducted analyses consisted of binary outcomes (CR, CR plus CRi, transfusion independence) and time-to-event outcomes (hazard rates for OS and EFS). A binomial model with a logit link function was employed for binary outcomes and a normal model with an identity link function was employed for time-to-event outcomes, based on NICE guidance.66
Both fixed effects and random effects models were considered for each analysis, and results from each model were run. However, only 1 model was chosen from which to draw inferences. The deviance information criterion was used to choose the appropriate model for the data.66
Following the feasibility assessment, meta-regression was not carried out to adjust for differences in study-level effect modifiers because of a lack of data.
Presentation of Results
For categorical outcomes, ORs were used to reflect the relative treatment effects between interventions; for time-to-event outcomes, HRs were used. Forest plots are presented using the posterior median of OR or HR for each pairwise treatment comparison. The 2.5th and 97th percentiles to capture the 95% CrI of OR or HR are also provided. For time-to-event outcomes (OS and EFS), a median HR less than 1 indicates favourable results for ivosidenib plus azacitidine. For categorical outcomes, a median OR greater than 1 indicates favourable results for ivosidenib plus azacitidine.
Matching-Adjusted Indirect Comparisons: The VIALE-A study is a phase III RCT comparing venetoclax plus azacitidine with placebo plus azacitidine in patients with treatment-naive AML.55 A study by Pollyea et al.56 pooled data from the VIALE-A study and a prior phase Ib study (single-arm study of patients with treatment-naive AML, investigating the safety and pharmacokinetics of venetoclax combined with decitabine or azacitidine). None of these studies was specific to patients with IDH1-mutated AML. Since an anchored MAIC was not feasible to adjust for within-study imbalances in potential effect modifiers in these studies, an unanchored MAIC for OS was conducted for the comparison with the IDH1 mutation subgroup, where the baseline characteristics of patients in the ivosidenib plus azacitidine group in the AGILE study were matched to the baseline characteristics of patients in the venetoclax plus azacitidine group in Pollyea et al.56 The baseline characteristics in Pollyea et al. reflect the population with IDH1/2 mutations, as the baseline characteristics for patients with IDH1 mutations were not reported in the VIALE-A study or in Pollyea et al. For the comparisons of the OS and EFS outcomes with the ITT population from the VIALE-A study, an anchored MAIC was feasible and therefore conducted.
Identification of Effect Modifiers and Prognostic Variables: The final list of treatment effect modifiers to be adjusted for in the MAIC analyses was determined through a deliberative process that considered statistical analyses conducted using the AGILE study individual patient data, and a review of the effect modifiers identified in a previous published MAIC,68 and a simulated treatment comparison69 of therapies in the indication of interest. Input from the clinical experts consulted by the sponsor was also sought to validate the covariate selection process.
Quantitative Analysis
Analyses were performed using the AGILE study individual patient data for a wide set of variables identified through the literature as potential prognostic variables and/or effect modifiers for first-line treatment of AML.68,69 For the end points of interest (i.e., OS and EFS), the following variables that were commonly reported in both studies were considered and assessed:
Median age
Gender
ECOG PS
Type of AML
Intermediate cytogenetic risk
Poor cytogenetic risk
Bone marrow blasts
IDH1 mutation based on central testing
Multivariable regression models were fitted for the OS and EFS end points, including all potential prognostic variables as covariates. For OS and EFS, a Cox PH model was fitted. Variable selection was then performed based on the statistical significance of each prognostic variable in the model. Specifically, statistical testing to assess prognostic variable status consisted of likelihood ratio tests between a Cox model, with the variable of interest as a covariate, and a null intercept-only model. Given the relatively small sample size of the ivosidenib plus azacitidine group in the AGILE study (i.e., 72 patients), the classical significance threshold (i.e., 5%) was relaxed, and more conservative significance levels of 10%, 15%, and 20% were used. The most appropriate threshold was discussed and agreed with clinical experts consulted by the sponsor. This method violates the recommendations of the NICE DSU that the list of variables be identified before the analysis.
In addition to the likelihood ratio test, a stepwise selection process was used for the covariate selection, where the model with the lowest Akaike Information Criterion value was considered the best fitting. For the selection of the effect modifiers, the same process was followed as that described for prognostic variables, with the only difference being that the interaction between covariates and the treatment was explored.
Covariates Considered in Previous Studies
In previously published MAICs for AML,68,69 the following covariates were adjusted for: age, AML type, bone marrow blast count, cytogenetic risk, ECOG PS, gender, neutrophil count, platelet count, poor cytogenetic risk category, and response status.
Estimation of MAIC Weights: To enable an adjusted comparison between ivosidenib plus azacitidine and the available comparative evidence sources, individual patients in the AGILE study were assigned statistical weights that adjust for their overrepresentation or underrepresentation relative to the average prognostic variables observed in the comparative evidence source. As a result, after weighting, the average baseline characteristics (mean and variance or proportion of patients within a category) were balanced for the patients in the AGILE study and the comparator populations.
Weights were derived using a form of propensity score weighting. In the absence of patient-level data for the comparative evidence source, a method of moments and the quasi-Newton optimization Broyden-Fletcher-Goldfarb-Shanno algorithm was used to allow a propensity score logistic regression model to be estimated and ensure that the weights balanced the mean covariate values.
Following the estimation of the weights, the distribution of the rescaled weights was visually examined to determine whether specific patient(s) or groups of patients (based on covariate values) are overrepresented or underrepresented in the analysis.
The robustness of the analyses was also evaluated by approximating the ESS.
Missing Data: During the matching process, the estimation of patient-specific weights required that matching covariates were available for all patients enrolled in the AGILE study. Missing data (if any) for patients in the AGILE study were identified once the covariates to adjust for had been selected. Specifically, 1 patient in the ivosidenib plus azacitidine group and 1 patient in the azacitidine group did not report the percentage of bone marrow blasts; thus, in scenarios where this covariate was used in the matching process, these patients were removed from the initial sample size.
Statistical Analyses Incorporating MAIC Weights: After the matching procedure was conducted and the weights were derived, efficacy outcomes were compared between balanced treatment groups using analyses that incorporate the derived weights. For the OS and EFS end points, a reweighted relative treatment effect (and standard error) for ivosidenib plus azacitidine versus the relevant comparator treatments was estimated using the reweighted absolute effect of ivosidenib plus azacitidine and the reported absolute effect of the relevant comparator treatment. The same statistical approach was followed in the anchored and unanchored cases, with the only difference being that the relative treatment effect of ivosidenib plus azacitidine versus venetoclax plus azacitidine was established via the common comparison against azacitidine in the anchored case.
Model Fitting and Model Selection: For survival outcomes (OS and EFS), the assumption of PH was assessed by visually inspecting the log-cumulative hazard plots for nonlinearities and by inspecting the Schoenfeld residuals. HRs were obtained by fitting a weighted Cox PH model whenever the PH assumption was met. When the PH assumption did not hold, survival models were fitted to the original and weighted AGILE study data as well as to the digitized comparator data. Alternative survival parametric models, including exponential, Weibull, log-logistic, log-normal, Gompertz, and generalized gamma distributions, were fitted to the weighted AGILE study and the digitized comparator data. Model selection included visual comparison as well as calculation of the Akaike Information Criterion and the Bayesian Information Criterion, where lower values indicated better fit to the data.
Results of the Feasibility Assessment
Ten studies were identified through the systematic literature review for the ITCs and were included in the feasibility assessment.
Key considerations in the feasibility assessment included the availability of the outcomes of interest, study design, characteristics of patient populations, posology of evaluated interventions, definitions, and methods for ascertainment of outcomes. Several limitations for the ITCs were identified by the feasibility assessment:
None of the comparator studies were conducted in the target population (specifically, in relation to IDH1 mutation).
In studies reporting mutation subgroup data, IDH1 is based on post hoc analyses with small patient numbers, and IDH1 is not a stratification factor for randomization in those studies.
Population baseline characteristics for the IDH1 subgroup are not available for venetoclax plus azacitidine; the IDH1/2 baseline characteristics in Pollyea et al. are unbalanced between treatment arms.
Notable differences in placebo arm rates are observed across placebo-controlled studies (i.e., the AGILE study and the IDH1 mutation subgroup from the VIALE-A study as reported in Pollyea et al.), which suggest differences in populations across the studies and raise concerns about outcome homogeneity.
The feasibility assessment identified heterogeneity in the analysis populations arising from a lack of published subgroup data for patients with IDH1 mutation, heterogeneity in other patient demographic and disease characteristics (gender, type of AML, cytogenic risk, ECOG performance status, and median bone marrow blast), differences in placebo arm rates across placebo-controlled studies, and differences in the definition of EFS.
Table 25: Assessment of Homogeneity of Studies Included in the ITCs
Characteristics | Description and handling of potential effect modifiers |
|---|---|
Demographic characteristics |
|
Disease characteristics |
|
Trial eligibility criteria |
|
Definitions of end points |
|
Study design |
|
AML = acute myeloid leukemia; CR = complete remission; CRi = complete remission with incomplete hematologic recovery; ECOG PS = Eastern Cooperative Oncology Group performance status; EFS = event-free survival; ITC = indirect treatment comparison; LDAC = low-dose cytarabine; OS = overall survival; RBC = red blood cell; RCT = randomized controlled trial.
Source: Sponsor-submitted ITC report.58 Details included in the table are from the sponsor’s summary of clinical evidence.
Results of the NMA
Following the feasibility assessment, 6 studies were selected to contribute to the evidence networks for 6 outcomes of interest and the following treatments:
ivosidenib plus azacitidine
azacitidine
LDAC
decitabine
venetoclax plus azacitidine
venetoclax plus LDAC
glasdegib plus LDAC.
Results specific to patients with IDH1 mutation are reported only for venetoclax plus azacitidine in the VIALE-A study by DiNardo et al. and a pooled analysis by Pollyea et al. (pooled data from the VIALE-A study and a single-arm phase Ib study) but are based on post hoc subgroup analyses with small sample sizes (specifically, fewer than 20 patients with IDH1 mutation were enrolled in the azacitidine group in the VIALE-A study, which does not meet the sample size inclusion criterion in the feasibility assessment).
Studies were excluded from the NMA if there were serious quality concerns (results of risk of bias assessment are presented in Appendix 1), if outcomes of interest were not reported, or if there were highly uncertain study results. During the development of the NMA, new data cuts from the AGILE study (June 30, 2022; median follow-up: 28.6 months) and the VIALE-A study (December 1, 2021; median follow-up: 43.2 months) were available for the analysis of OS. The newest data cut from the VIALE-A study was ultimately published early in 2024,71 and data from the new data cut were used in updated data analyses of OS.
Of the 6 studies included in the NMA, 3 were considered to be at low risk of bias (DiNardo et al. [2020]; Dombret et al. [2015]; Wei et al. [2021]), concerns of risk of bias applied to 2 (Heuser et al. [2021]; Kantarjian et al. [2012]), and the AGILE study was not assessed.
The diagrams for the network of evidence for OS, EFS, CR, CR plus CRi, and transfusion independence are presented in Figures 4 to 8.
Figure 4: Network of Evidence for Overall Survival
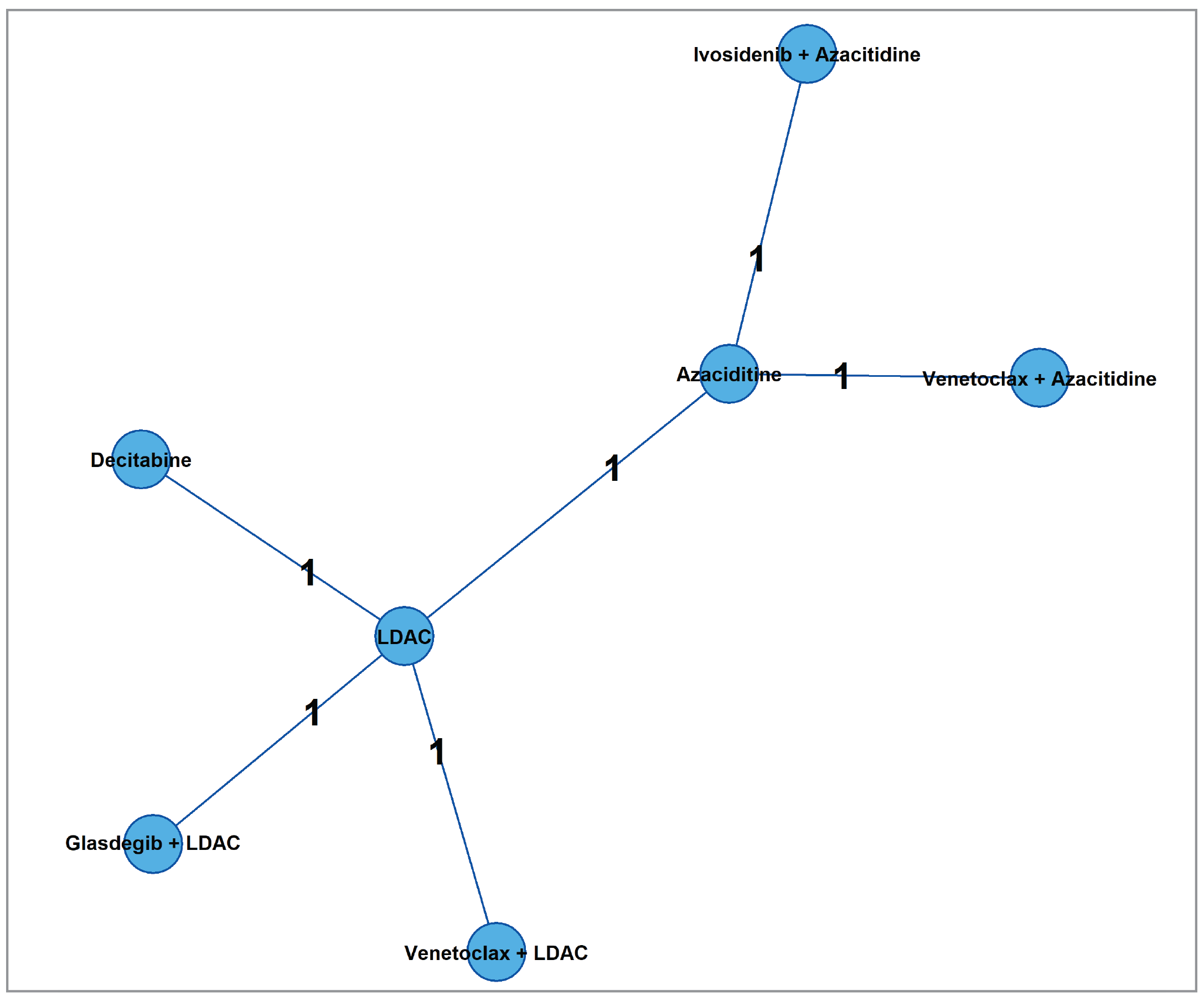
LDAC = low-dose cytarabine.
Source: Sponsor-submitted indirect treatment comparison.58
Results
Network Meta-Analyses
Both fixed-effect and random-effect models were fitted to the data, and the fixed-effect model was preferred across all analyses in this NMA.
For the comparison of ivosidenib plus azacitidine to venetoclax plus azacitidine and venetoclax plus LDAC, associated CrIs were wide, suggesting uncertainty about which regimen could be favoured. Results suggest that ivosidenib plus azacitidine was favoured over LDAC monotherapy for all outcomes and that azacitidine monotherapy was favoured for all outcomes except for transfusion independence (the CrI was wide).
Detailed results from the NMA are presented in Table 26.
Figure 5: Network of Evidence for Event-Free Survival
LDAC = low-dose cytarabine.
Source: Sponsor-submitted indirect treatment comparison.58
Figure 6: Network of Evidence for Complete Remission
LDAC = low-dose cytarabine.
Source: Sponsor-submitted indirect treatment comparison.58
Figure 7: Network of Evidence for CR Plus CR With Incomplete Hematologic Recovery
CR = complete remission; LDAC = low-dose cytarabine.
Source: Sponsor-submitted indirect treatment comparison.58
Figure 8: Network of Evidence for Transfusion Requirements
LDAC = low-dose cytarabine.
Source: Sponsor-submitted indirect treatment comparison.58
Table 26: Summary of Efficacy Outcome Measures in the Sponsor-Submitted ITCs, Results From NMA, Fixed-Effect Models
Outcome | Ivosidenib + azacitidine vs.: | |||
|---|---|---|---|---|
Venetoclax + azacitidine | Azacitidine | LDAC | Venetoclax + LDAC | |
OS HR (95% CrI) in ITT population | ████ ████ | ████ ████ | ████ ████ | ████ ████ |
OS with new data cut from the AGILE study and the VIALE-A study HR (95% CrI) in ITT population | ████ ████ | ████ ████ | ████ ████ | ████ ████ |
EFS HR (95% CrI) in ITT population | ████ ████ | ████ ████ | ████ ████ | ████ ████ |
CR OR (95% CrI) in safety analysis population | ████ ████ | ████ ████ | ████ ████ | ████ ████ |
CR + CRi OR (95% CrI) in safety analysis population | ████ ████ | ████ ████ | ████ ████ | ████ ████ |
Transfusion requirement OR (95% CrI) in safety analysis population | ████ ████ | ████ ████ | ████ ████ | ████ ████ |
CR = complete remission; CRi = complete remission with incomplete hematologic recovery; Crl = credible interval; EFS = event-free survival; HR = hazard ratio; ITC = indirect treatment comparison; ITT = intention to treat; LDAC = low-dose cytarabine; NMA = network meta-analysis; OR = odds ratio; OS = overall survival.
Notes: Venetoclax plus LDAC is not recommended for reimbursement in Canada. Bolded values indicate statistically significant differences between the 2 treatments. An HR less than 1 indicates “favours ivosidenib” for OS and EFS; an OR less than 1 indicates “favours comparator” for CR, CR plus CRi, and transfusion independence.
Source: Sponsor-submitted indirect treatment comparison.58
Matching-Adjusted Indirect Comparisons
During the MAIC analyses, each covariate selection approach identified different sets of covariates for inclusion in the MAIC analysis per outcome of interest. Three scenarios have been proposed to determine the covariates to adjust for in the matching process:
unanchored MAIC for OS: matching the AGILE study to the IDH1 subgroup in the VIALE-A study, with matching based on the IDH1/2 population described in the Pollyea et al. study
anchored MAIC for OS: matching the AGILE study to the ITT population from the VIALE-A study
anchored MAIC for EFS: matching the AGILE study to the ITT population from the VIALE-A study.
The rationale for matching against the IDH1 population in the VIALE-A study is that, despite the limitations of the data, this population reflects the target population for decision-making and is hence presented even if the results should be interpreted with great caution. The rationale for matching against the ITT population in the VIALE-A study, which includes a broader population of patients than in the AGILE study, is to explore whether IDH1 mutation status is an effect modifier for venetoclax plus azacitidine and to overcome the main limitation of the IDH1 mutation data as they are based on a post hoc subgroup with small sample size, where randomization is broken with explained and unexpected effect modification.
The baseline characteristics in the AGILE study before and after matching to the IDH1 population or ITT population are presented in Appendix 1, Tables 30 to 33. There may not be adequate information to determine if the matching is adequate when only the baseline characteristics on which the sponsor matched were provided, rather than all baseline characteristics (which could have become unbalanced during the matching of the other variables). In addition, for the ITT population, it was not possible to match on IDH1 status, because all patients in the AGILE study have an IDH1 mutation.
In the unanchored MAIC for OS in the IDH1 population, the following were matched for the base-case analysis: age, sex, ECOG performance status, AML type, cytogenetic risk, and bone marrow blasts; age and bone marrow blasts were matched for scenario analysis 1; and age, bone marrow blasts, and ECOG performance status were matched for scenario analysis 2.
In the anchored MAIC for OS in the ITT population, the following were matched for the base-case analysis: age, sex, ECOG performance status, AML type, cytogenetic risk, and bone marrow blasts; bone marrow blasts were matched for scenario analysis 1.
In the anchored MAIC for EFS in the ITT population, the following were matched for the base-case analysis: age, sex, ECOG performance status, AML type, cytogenetic risk, and bone marrow blasts; sex and ECOG performance status were matched for scenario analysis 1; and sex, cytogenetic risk, and ECOG performance status were matched for scenario analysis 2.
The results from the unanchored MAIC for OS in the IDH1 population showed that after matching (base case), the median OS was ████ ██████ ████ ██ ████ ██ █████ with ivosidenib plus azacitidine, compared to ████ ██████ ████ ██ ███ ██ ███ ████████ with venetoclax plus azacitidine.
The results from the anchored MAIC for OS in the ITT population showed that after matching, the median OS was ████ ██████ ████ ██ ███ ██ █████ with ivosidenib plus azacitidine, compared to ████ ██████ ████ ██ ███ ██ ███ ████████ with venetoclax plus azacitidine.
The results from the anchored MAIC for EFS in the ITT population showed that after matching, the median EFS was ████ ██████ ████ ██ ███ ██ ███ ████████ with ivosidenib plus azacitidine, compared to ███ ██████ ████ ██ ███ ██ █████ with venetoclax plus azacitidine.
Detailed results from the MAIC are presented in Table 27.
Table 27: Median Overall Survival and Event-Free Survival Before and After Matchings
Outcomes | AGILE | VIALE-A | Between-group difference HR (95% CI) | ||
|---|---|---|---|---|---|
Ivosidenib + azacitidine (N = 71) | Placebo + azacitidine (N = 73) | Venetoclax + azacitidine (N = 286) | Placebo + azacitidine (N = 145) | ||
Median OS in months (95% CI) Unanchored MAIC for IDH1 population | ████ ████ █████████ █████████ █████████ | ████ ████ ████████ | ████ ████ ████████ | ████ ████ ████████ | ████ ████ █████████ █████████ █████████ |
Median OS in months (95% CI) Anchored MAIC for ITT population | ████ ████ █████████ █████████ █████████ | ████ ████ ████████ | ████ ████ ████████ | ████ ████ ████████ | ████ ████ █████████ █████████ █████████ |
Median OS in months (95% CI) Anchored MAIC for ITT population (new data cut) | ████ ████ █████████ █████████ █████████ | ████ ████ ████████ | ████ ████ ████████ | ████ ████ ████████ | ████ ████ █████████ █████████ █████████ |
Median EFS in months (95% CI) Anchored MAIC for ITT population | ████ ████ █████████ █████████ █████████ | ████ ████ ████████ | ████ ████ ████████ | ████ ████ ████████ | ████ ████ █████████ █████████ █████████ |
BC = base case; CI = confidence interval; EFS = event-free survival; ESS = effective sample size; HR = hazard ratio; ITT = intention to treat; MAIC = matching-adjusted indirect comparison; NA = not applicable; NR = not reached; OS = overall survival; SA = scenario analysis.
Source: Sponsor-submitted indirect treatment comparison.58
Critical Appraisal of ITCs
There was no a priori protocol for the ITCs; therefore, it cannot be known whether the analyses presented were selected from multiple analyses of the data (e.g., based on the magnitude and direction of observed effects). In this ITC report, studies were identified by searching multiple databases based on prespecified inclusion and exclusion criteria. Studies were selected by 2 independent reviewers; thus, the error and bias in the study selection process were minimized. Appropriate methods were used to reduce the risk of bias and error in data extraction. It was unknown if the risk of bias in the included trials was assessed by the 2 independent reviewers. In addition, risk of bias was assessed at the level of the trial, rather than at the level of the reported results (i.e., per outcome), which ignores that risk of bias can vary by reported result within a trial. Some of the studies included within the NMA had some potential for risk of bias. Risk of bias in the AGILE study was not assessed.
One of the major concerns for the ITCs is that the included trials could have been highly heterogeneous in terms of study design and patient characteristics at baseline. Six RCTs were included in the NMA. Heterogeneities were identified in the analysis populations, which included IDH1 mutation status, gender, type of AML diagnosis, cytogenic risk, performance status, median bone marrow blast, differences in placebo arm rates across placebo-controlled studies, and differences in the definition of EFS. For the time-to-event comparisons (e.g., EFS), lengths of follow-up were different, and with longer follow-up it may be expected that the HR would be attenuated, even without formal violation of the PH assumption. The bias would likely favour the study drug. These differences would undermine the validity of the NMA, which relies on the transitivity assumption (i.e., that the trials are similar for all important effect modifiers) being upheld. The use of fixed-effect models was chosen based on the deviance information criterion. However, the use of fixed-effect models (assuming no between-study heterogeneity) rather than random effects models means that the CrIs are unlikely to adequately express the uncertainty arising from the heterogeneity. The limited number of included studies did not allow for meta-regression or other techniques to adjust for differences in effect modifiers across studies within the NMA. The rarity of the population of interest limits the size and number of clinical studies completed with potential comparators and adds to the practical challenges when indirectly comparing treatment options.
In the NMA, given the lack of closed loops in the networks, consistency in the ITC analyses could not be tested. All comparisons are therefore informed only by indirect evidence, which increases the level of uncertainty. Efficacy data were sparse in this NMA for the comparison of ivosidenib versus placebo in combination with azacitidine. The 95% CrIs for the point estimates were wide for some efficacy outcomes and spanned the null when comparing with other combination regimens; therefore, confidence in the effect estimates for efficacy of the study drugs was limited because of the imprecision indicated by the wide CrIs for these outcomes, which precludes any conclusions as to which treatment may be favoured.
In the MAICs, the following potential effect modifier or prognostic factors were identified through the literature and a deliberating process by the sponsor: age, gender, ECOG performance status, type of AML, cytogenetic risk of AML, bone marrow blasts, and IDH1 mutation. The clinical experts consulted for this review agreed that these are relevant effect modifiers and prognostic variables. However, it is unclear if the identification of potential effect modifiers through the literature would be sufficient to identify all relevant treatment effect modifiers. The populations in the AGILE study and the other comparator studies were weighted and matched. Within the unanchored MAIC there was no reported estimate of the potential residual bias due to unadjusted confounders; as a result, the magnitude of residual confounding remains uncertain.
Before adjustment, the median OS and EFS for the placebo plus azacitidine groups were substantially different, suggesting reduced comparability of the populations. The main differences for the 2 studies used (AGILE and VIALE-A) is that in the AGILE study, the patients were younger and had a better ECOG performance status and a lower proportion of the patients had high-risk cytogenic status. The unanchored MAIC matched the characteristics of the patients with IDH1/2 mutation from the VIALE-A study because the characteristics for IDH1 were unavailable. In the anchored MAICs, the ESS reduced by approximately one-third after the weighting process, suggesting that the results are heavily influenced by a subset of the sample population in the trial who may not be representative of the full sample population. The reduction in the ESS and the sample size in general resulted in wide CIs. Furthermore, there is uncertainty about comparing the population with IDH1 mutation to the ITT population in the VIALE-A study. It was not possible to adjust for this factor.
The study population for this review is patients with AML with IDH1 mutation who are ineligible for intensive chemotherapy. However, most of the selected trials were not specifically for IDH1-mutated AML. No other studies included only patients with IDH1 mutation, and it is not clear in the other included trials whether there were separate results for this particular subgroup. The prognostic significance of IDH1 status in AML, or whether this IDH1 status may be a treatment effect modifier, remains uncertain. According to the clinical experts consulted for this review, the aforementioned patient characteristics (e.g., de novo AML status, region, age, baseline ECOG performance status, sex, race, baseline cytogenetic risk status, WHO classification of AML, baseline white blood cell count, and baseline percent bone marrow blasts) were considered treatment effect modifiers in patients with AML and IDH1-mutated AML.
In this ITC report, several efficacy outcomes were analyzed, such as OS, EFS, and CR (not evaluated in the MAICs). However, other efficacy end points of interest to patients and clinicians (e.g., HRQoL), as well as harms, were not investigated. Therefore, the relative treatment effect of ivosidenib plus azacitidine versus relevant comparators on patients’ HRQoL and on harms remains unknown.
Studies Addressing Gaps in the Systematic Review Evidence
There were no relevant studies addressing the gaps in the systematic review evidence submitted for this review.
Discussion
Summary of Available Evidence
The evidence included in the systematic review consisted of 1 pivotal phase III, double-blind RCT, the AGILE study (N = 146). The purpose of this study was to evaluate the efficacy and safety of the combination of ivosidenib plus azacitidine versus placebo plus azacitidine in adult patients with newly diagnosed AML with an IIDH1 R132 mutation who are not eligible to receive intensive induction chemotherapy. Patients were randomized either to ivosidenib 500 mg orally once daily plus azacitidine 75 mg/m2/day, subcutaneous or IV, for 7 days, in 28-day cycles, or to placebo in combination with azacitidine. The primary efficacy end point in the AGILE study was EFS. Other relevant outcomes in this study included OS, remission rates, HRQoL measured by the EORTC QLQ-C30, transfusion requirement, and harms. The majority (73.3% per investigator [76% per Interactive Web Response System]) of patients had de novo AML at initial diagnosis. Based on the WHO classification of AML, fewer patients in the ivosidenib plus azacitidine group (22.2%) had AML with recurrent genetic abnormalities than in the placebo plus azacitidine group (32.4%), and more patients in the ivosidenib plus azacitidine group (38.9%) had AML with myelodysplasia-related changes than in the placebo plus azacitidine group (35.1%). IIDH1 R132C was the most common polymorphism (65.8% of patients). In total, 63.8% of patients in the ivosidenib plus azacitidine group and 67.6% of patients in the placebo plus azacitidine group had an ECOG performance status score of 0 to 1. Cytogenetic risk status, as assessed by the investigators based on the 2017 National Comprehensive Cancer Network guidelines, was intermediate (63.0%: 66.7% in ivosidenib plus azacitidine group versus 59.5% in placebo plus azacitidine group) or poor (24.7%: 22.2% in ivosidenib plus azacitidine group versus 27.0% in placebo plus azacitidine group) for most patients at baseline. The median bone marrow blast proportion at baseline was 52.5% (range, 17% to 100%).
Two DCOs were available for the AGILE study. The first DCO (March 18, 2021) represents an unplanned early interim analysis by the IDMC, which occurred before the protocol-specified number of events for the planned analysis. Because of a notable difference in the number of deaths, which favoured ivosidenib, the IDMC recommended that trial recruitment should end early, treatment assignment should be unblinded, and crossover to ivosidenib should be allowed. The stopping boundaries were therefore adjusted, and this became the final analysis. A later DCO (June 30, 2022) was available for OS, transfusion independence, and harms. The results of the interim analysis of efficacy end points are at risk of overestimating the true effects of ivosidenib plus azacitidine.
One ITC report (comprising 1 NMA and 3 MAICs) was submitted by the sponsor to compare the treatment efficacy and safety of ivosidenib plus azacitidine with other active therapies (e.g., venetoclax plus azacitidine, azacitidine monotherapy, LDAC monotherapy, and venetoclax plus LDAC) for the treatment of IDH1-mutated AML. The comparative efficacy of ivosidenib versus venetoclax, in combination with azacitidine, was evaluated based on evidence from 6 RCTs.
Interpretation of Results
Efficacy
According to the patient groups and the clinical experts consulted for this review and the clinician groups that submitted input for this review, important unmet needs for patients with IDH1-mutated AML who are not eligible for intensive chemotherapy, include therapies that offer durable remission, can prolong life, can reduce transfusion dependency, and would improve patients’ HRQoL. The AGILE study met its primary end point at an unplanned interim analysis by the IDMC that occurred at the DCO of March 18, 2021. The results suggested that treatment with ivosidenib plus azacitidine is likely to be associated with a clinically important improvement in EFS rates, compared with treatment with placebo plus azacitidine: the between-group difference in EFS rate was 19.7% (95% CI, ███ ██ ████) at 6 months, favouring ivosidenib plus azacitidine. The between-group differences were affected by imprecision, where the lower bound of the CI included effects that might not be considered clinically important. Improvement in EFS was largely driven by the proportion of patients who experienced treatment failure, assigned an event time of the date of randomization: 42 patients (58.3%) in the ivosidenib plus azacitidine group versus 59 patients (79.7%) in the placebo plus azacitidine group experienced treatment failure. EFS is a composite end point, and the sample size of the AGILE study was small; following the large number of events of treatment failure, too few patients remained event-free to robustly characterize the long-term treatment effect of ivosidenib plus azacitidine on EFS.31
Treatment with ivosidenib plus azacitidine was associated with prolonged OS. At the DCO of March 18, 2021, OS met the stopping boundary, leading to the unplanned interim analysis for a statistically significant OS benefit for ivosidenib plus azacitidine. At the updated DCO of June 30, 2022, 37 patients (50.7%) in the ivosidenib plus azacitidine group and 58 (77.3%) in the placebo plus azacitidine group had died. The median OS was 29.3 months (95% CI, 13.2 months to NE) in the ivosidenib plus azacitidine group and 7.9 months (95% CI, 4.1 to 11.3 months) in the placebo plus azacitidine group (P < 0.0001). The corresponding HR was 0.42 (95% CI, 0.27 to 0.65). In addition, the OS rates at various time points showed that ivosidenib plus azacitidine likely results in a clinically relevant increase in the probability of OS at 1 year and 2 years, compared with placebo plus azacitidine. The between-group differences in the Kaplan-Meier–estimated OS rate were 24.6% (95% CI, ███ ██ ████) at 12 months and 35.7% (95% CI, ████ ██ ████) at 24 months. There was some potential for overestimation of the true effect due to small sample size. The results of the prespecified subgroup analyses for OS and EFS based on various patient baseline characteristics were consistent with those in the overall population.
Treatment with ivosidenib plus azacitidine may be associated with higher CR rates than treatment with placebo plus azacitidine. As of the DCO of March 18, 2021, the CR rate was 47.2% (95% CI, 35.3% to 59.3%) in the ivosidenib plus azacitidine group and 14.9% (95% CI, 7.7% to 25.0%) in the placebo plus azacitidine group. However, these estimates were affected by high risk of bias due to missing data. As of the DCO of June 30, 2022, a higher proportion of patients in the ivosidenib plus azacitidine group (██ ███████ ████████) were RBC and/or platelet transfusion independent than in the placebo plus azacitidine group (██ ███████ ████████). This was measured in a nonrandomized subset of the population. According to the clinical experts, improved CR rates and reduced transfusion dependence are considered clinically meaningful changes, and better CR rates and, in their opinion, reduced transfusion dependence can subsequently be translated to improved HRQoL and potentially prolonged survival.
HRQoL measured by the EORTC QLQ-C30 was a secondary outcome in the AGILE study. The evidence for HRQoL was considered to be very uncertain because of large amounts of missing data and imprecision; the CIs included the potential for little-to-no clinically meaningful difference between groups.
For this submission, venetoclax plus azacitidine was identified as the most relevant comparator for the indication under review. Comparative evidence of ivosidenib plus azacitidine versus venetoclax plus azacitidine was available through a sponsor-submitted ITC analysis. The rarity of the population of interest limits the size and number of clinical studies completed with potential comparators and adds to the practical challenges when indirectly comparing treatment options. Based on the results of the NMA and the MAICs, the evidence is insufficient to conclude whether ivosidenib plus azacitidine differs from venetoclax plus azacitidine in terms of OS, EFS, CR rates, or transfusion requirement in patients with untreated AML because of the limitations associated with the ITC report, such as limited evidence from the 6 RCTs, heterogeneity existing in the included trials, and imprecision of study results from the wide CrIs or CIs for these outcomes. There was no evidence to compare impacts on HRQoL for ivosidenib plus azacitidine versus any comparators outside the AGILE trial. For the comparisons between ivosidenib plus azacitidine and azacitidine or LDAC monotherapies, the results were in the same direction as results observed in the AGILE study and were as expected by clinical experts based on their experience with combination and monotherapies in clinical practice.
Harms
Overall, the safety results from the 2 DCOs are consistent. As of the DCO of March 18, 2021, in the AGILE trial the proportion of patients who experienced at least 1 AE was 98.6% (70 patients) in the ivosidenib plus azacitidine group and 100% (73 patients) in the placebo plus azacitidine group. Patients treated with ivosidenib plus azacitidine were more likely (5% or more) to report the following AEs than patients treated with placebo plus azacitidine: vomiting (29 patients [40.8%] versus 19 patients [26.0%]), neutropenia (20 [28.2%] versus 12 [16.4%]), thrombocytopenia (20 [28.2%] versus 15 [20.5%]), prolonged electrocardiogram QT interval (14 [19.7%] versus 5 [6.8%]), insomnia (13 [18.3%] versus 9 [12.3%]), differentiation syndrome (10 [14.1%] versus 6 [8.2%]), pain in extremity (10 [14.1%] versus 3 [4.1%]), hematoma (9 [12.7%] versus 1 [1.4%]), arthralgia (8 [11.3%] versus 3 [4.1%]), headache (8 [11.3%] versus 2 [2.7%]), leukocytosis (8 [11.3%] versus 1 [1.4%]), and leukopenia (6 [8.5%] versus 2 [2.7%]).
Evidence from the AGILE study showed that ivosidenib plus azacitidine is likely to be associated with a reduction in SAEs compared to placebo plus azacitidine. The proportion of patients who experienced SAEs was 69.0% (46 patients) in the ivosidenib plus azacitidine group and 82.2% (60 patients) in the placebo plus azacitidine group. Commonly reported SAEs in the 2 treatment groups were febrile neutropenia (23.9% of patients in the ivosidenib plus azacitidine group versus 27.4% in the placebo plus azacitidine group) and pneumonia (19.7% versus 21.9%). The clinical experts noted that the increased incidence of febrile neutropenia and pneumonia in the placebo plus azacitidine group may be related to disease progression in this treatment group, rather than being an adverse effect from the study drug.
The overall incidences of TEAEs that led to combination treatment discontinuation were similar between the treatment groups: 19 patients (26.8%) in the ivosidenib plus azacitidine group and 19 patients (26.0%) in the placebo plus azacitidine group.
Ivosidenib plus azacitidine may increase the rate of differentiation syndrome compared to placebo plus azacitidine; however, this was informed by few events. As of June 30, 2022, differentiation syndrome was reported in 10 patients (13.9%) in the ivosidenib plus azacitidine group and 6 patients (8.1%) in the placebo plus azacitidine group. Infection was likely to be reduced with ivosidenib plus azacitidine: infection was reported in 25 patients (34.7%) in the ivosidenib plus azacitidine group and 38 patients (51.4%) in the placebo plus azacitidine group.
There was no direct or indirect evidence comparing the harms of ivosidenib plus azacitidine to any other relevant comparators, including venetoclax plus azacitidine.
Conclusion
Adult patients with newly diagnosed AML with an IIDH1 R132 mutation who are not eligible to receive intensive induction chemotherapy have a poor prognosis. Patients and clinicians highlighted the need for new treatments that prolong life, improve remission, reduce transfusion requirements, and maintain HRQoL, compared to the current treatments. Evidence from a double-blind, phase III RCT (the AGILE study) showed that treatment with ivosidenib plus azacitidine likely results in a clinically important increase in the probability of OS at 12 months and 24 months compared to placebo plus azacitidine in the target population. Evidence from the trial also showed that ivosidenib plus azacitidine likely results in a clinically important increase in the probability of EFS at 6 months. EFS was a composite end point driven by treatment failure events: postbaseline, too few patients remained at risk to robustly characterize other components of the end point (i.e., relapse and death). The rates of CR, as well as CR plus CRi, and the need for transfusions may be improved with treatment with ivosidenib plus azacitidine compared with placebo plus azacitidine. Evidence on HRQoL was very uncertain because of the limitations of the analyses, including risk of bias due to missing data and imprecision. In terms of harms, evidence from the AGILE study suggested that treatment with ivosidenib plus azacitidine may result in an increase in differentiation syndrome but likely results in a reduction in the proportion of patients who experience SAEs and infections compared with treatment with placebo plus azacitidine.
There is a lack of direct comparative evidence between ivosidenib plus azacitidine and other relevant active treatments for patients with AML who are not eligible for intensive chemotherapy, such as venetoclax plus azacitidine, which is currently the most commonly used treatment in the target patient population. Indirect evidence from a sponsor-submitted NMA of 6 trials and 3 MAICs comparing patients from the AGILE study to patients treated with venetoclax plus azacitidine in the VIALE-A study was insufficient to conclude whether treatment with ivosidenib plus azacitidine differs from treatment with venetoclax plus azacitidine in terms of OS, EFS, CR rates, and transfusion dependence. There was substantial uncertainty in the treatment effect estimates (indicated by wide CrIs) from the ITCs because of limited efficacy data and important heterogeneity across studies. No comparisons of HRQoL or harms were conducted.
References
1.National Comprehensive Cancer Network. Clinical Practice Guidelines in Oncology (NCCN Guidelines®). Acute Myeloid Leukemia Version 1.2024. 2024; https://www.nccn.org/guidelines/guidelines-detail?category=1&id=1411. Accessed 2024 Mar 7.
2.Chennamadhavuni A, Lyengar V, Mukkamalla SKR, Shimanovsky A. Leukemia [accessed by sponsor]. StatPearls. Treasure Island (FL): StatPearls Publishing LLC; 2024: https://www.ncbi.nlm.nih.gov/pubmed/32809325.
3.Vakiti A, Reynolds SB, Mewawalla P. Acute Myeloid Leukemia. StatPearls. Treasure Island (FL): StatPearls Publishing Copyright © 2023, StatPearls Publishing LLC.; 2024.
4.Pleyer C, Afzal A, Shomali W, et al. Impact of red blood cell and platelet transfusions in acute myeloid leukemia (AML) patients undergoing remission induction chemotherapy. Blood. 2015;126(23):2127-2127.
5.Shiffer CA, Wang ES. Overview of the complications of acute myeloid leukemia. In: Post TW, ed. UpToDate. Waltham (MA): UpToDate: https://www.uptodate.com. Accessed 2023 Oct 27.
6.National Organization for Rare Disorders. Acute Myeloid Leukemia. 2018; https://rarediseases.org/rare-diseases/acute-myeloid-leukemia/. Accessed 2023 Oct 27.
7.American Cancer Society. What Is Acute Myeloid Leukemia (AML)? [accessed by sponsor]. https://www.cancer.org/cancer/types/acute-myeloid-leukemia/about/what-is-aml.html. Accessed 2023 Oct 27.
8.Cancer Quality Council of Ontario. Cancer Survival. 2021; https://www.csqi.on.ca/en/2020/indicators/cancer-survival. Accessed 2023 Oct 27.
9.Lubeck DP, Danese M, Jennifer D, Miller K, Richhariya A, Garfin PM. Systematic literature review of the global incidence and prevalence of myelodysplastic syndrome and acute myeloid leukemia. Blood. 2016;128(22):5930-5930.
10.Turbeville S, Francis KM, Behm I, et al. Prevalence and incidence of acute myeloid leukemia may be higher than currently accepted estimates among the ≥65 year-old population in the United States. Blood. 2014;124(21):958-958.
11.Statistics Canada. Number of new cases and age-standardized rates of primary cancer, by cancer type and sex. 2023; https://www150.statcan.gc.ca/t1/tbl1/en/cv.action?pid=1310074701. Accessed 2023 Oct 2.
12.CADTH Reimbursement Review: Venetoclax (Venclexta). Can J Health Technol. 2023;1(10). https://www.cadth.ca/sites/default/files/DRR/2021/PC0238-combined-report-FINAL.pdf. Accessed 2023 Oct 26.
13.Patel JP, Gonen M, Figueroa ME, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366(12):1079-1089. PubMed
14.Abbas S, Lugthart S, Kavelaars FG, et al. Acquired mutations in the genes encoding IDH1 and IDH2 both are recurrent aberrations in acute myeloid leukemia: prevalence and prognostic value. Blood. 2010;116(12):2122-2126. PubMed
15.Chotirat S, Thongnoppakhun W, Promsuwicha O, Boonthimat C, Auewarakul CU. Molecular alterations of isocitrate dehydrogenase 1 and 2 (IDH1 and IDH2) metabolic genes and additional genetic mutations in newly diagnosed acute myeloid leukemia patients. J Hematol Oncol. 2012;5:5. PubMed
16.Chou WC, Hou HA, Chen CY, et al. Distinct clinical and biologic characteristics in adult acute myeloid leukemia bearing the isocitrate dehydrogenase 1 mutation. Blood. 2010;115(14):2749-2754. PubMed
17.Marcucci G, Maharry K, Wu YZ, et al. IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol. 2010;28(14):2348-2355. PubMed
18.Mardis ER, Ding L, Dooling DJ, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361(11):1058-1066. PubMed
19.Paschka P, Schlenk RF, Gaidzik VI, et al. IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. J Clin Oncol. 2010;28(22):3636-3643. PubMed
20.Bullinger L, Dohner K, Dohner H. Genomics of Acute Myeloid Leukemia Diagnosis and Pathways. J Clin Oncol. 2017;35(9):934-946. PubMed
21.Montesinos P, Recher C, Vives S, et al. Ivosidenib and Azacitidine in IDH1-Mutated Acute Myeloid Leukemia. N Engl J Med. 2022;386(16):1519-1531. PubMed
22.Sadashiv SK, Hilton C, Khan C, et al. Efficacy and tolerability of treatment with azacitidine for 5 days in elderly patients with acute myeloid leukemia. Cancer Med. 2014;3(6):1570-1578. PubMed
23.Cashen AF, Schiller GJ, O’Donnell MR, DiPersio JF. Multicenter, phase II study of decitabine for the first-line treatment of older patients with acute myeloid leukemia. J Clin Oncol. 2010;28(4):556-561. PubMed
24.Dombret H, Seymour JF, Butrym A, et al. International phase 3 study of azacitidine vs conventional care regimens in older patients with newly diagnosed AML with >30% blasts. Blood. 2015;126(3):291-299. PubMed
25.Estey EH. Acute myeloid leukemia: 2012 update on diagnosis, risk stratification, and management. Am J Hematol. 2012;87(1):89-99. PubMed
26.AbbVie Corporation. VENCLEXTA® (venetoclax) Product Monograph. April 5, 2022 [sponsor provided reference].
27.Pfizer Canada ULC. DAURISMO® (glasdegib) Product Monograph. October 31, 2022 [sponsor provided reference].
28.Celgene Inc, a Bristol Myers Squibb company. VIDAZA® (azacitidine) Product Monograph. February 13, 2024 [sponsor provided reference].
29.Pfizer Canada I. Cytarabine Injection (100 mg/mL) Product Monograph. November 16, 2017 [sponsor provided reference]. 2017.
30.Tibsovo (ivosidenib): 250 mg tablets, oral [product monograph]. Laval (QC): Servier Canada Inc.; 2024 Jul 19.
31.Woods A, Norsworthy KJ, Wang X, et al. FDA Approval Summary: Ivosidenib in Combination with Azacitidine for Treatment of Patients with Newly Diagnosed Acute Myeloid Leukemia with an IDH1 Mutation. Clin Cancer Res. 2024;30(7):1226-1231. PubMed
32.Othus M, van Putten W, Lowenberg B, et al. Relationship between event-free survival and overall survival in acute myeloid leukemia: a report from SWOG, HOVON/SAKK, and MRC/NCRI. Haematologica. 2016;101(7):e284-286. PubMed
33.Assouline S, Wiesinger A, Spooner C, Jovanovic J, Schlueter M. Validity of event-free survival as a surrogate endpoint in haematological malignancy: Review of the literature and health technology assessments. Crit Rev Oncol Hematol. 2022;175:103711. PubMed
34.Norsworthy KJ, Gao X, Ko CW, et al. Response Rate, Event-Free Survival, and Overall Survival in Newly Diagnosed Acute Myeloid Leukemia: US Food and Drug Administration Trial-Level and Patient-Level Analyses. J Clin Oncol. 2022;40(8):847-854. PubMed
35.Balshem H, Helfand M, Schunemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-406. PubMed
36.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. Journal of clinical epidemiology. 2020;119:126-135. PubMed
37.Musoro JZ, Coens C, Sprangers MAG, et al. Minimally important differences for interpreting EORTC QLQ-C30 change scores over time: A synthesis across 21 clinical trials involving nine different cancer types. Eur J Cancer. 2023;188:171-182. PubMed
38.Clinical Study Report AG120-C-009: A Phase 3, Multicenter, Double-Blind, Randomized, Placebo-Controlled Study of AG-120 in Combination with Azacitidine in Subjects ≥18 Years of Age with Previously Untreated Acute Myeloid Leukemia with an IDH1 Mutation [internal sponsor’s report]. Suresnes (FR): Institut de Recherches Internationales Servier; 2021 Dec 8.
39.Clinical Study Report AG120-C-009 (addendum): A Phase 3, Multicenter, Double-Blind, Randomized, Placebo-Controlled Study of AG-120 in Combination with Azacitidine in Subjects ≥18 Years of Age with Previously Untreated Acute Myeloid Leukemia with an IDH1 Mutation. Suresnes (FR): Institut de Recherches Internationales Servier; 2023 Aug 21.
40.American Cancer Society. Risk Factors for Acute Myeloid Leukemia (AML) [accessed by sponsor]. https://www.cancer.org/cancer/types/acute-myeloid-leukemia/causes-risks-prevention/risk-factors.html. Accessed 2023 Oct 27.
41.Klepin HD, Rao AV, Pardee TS. Acute myeloid leukemia and myelodysplastic syndromes in older adults. J Clin Oncol. 2014;32(24):2541-2552. PubMed
42.Statistics Canada. Age-specific five-year net survival estimates for primary sites of cancer, by sex, three years combined. 2020; https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310015801. Accessed 2023 Oct 2.
43.Cheson BD, Bennett JM, Kopecky KJ, et al. Revised recommendations of the International Working Group for Diagnosis, Standardization of Response Criteria, Treatment Outcomes, and Reporting Standards for Therapeutic Trials in Acute Myeloid Leukemia. J Clin Oncol. 2003;21(24):4642-4649. PubMed
44.Cancer Care Ontario. Ontario Cancer Statistics 2020 Ch 5: Cancer Incidence Rates and Trends. 2020; https://www.cancercareontario.ca/en/statistical-reports/ontario-cancer-statistics-2020/ch-5-cancer-incidence-rates-trends. Accessed 2023 Oct 27.
45.Cancer Quality Council of Ontario. Cancer Incidence. 2021; https://www.csqi.on.ca/en/2020/indicators/cancer-incidence. Accessed 2023 Oct 27.
46.Dohner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129(4):424-447. PubMed
47.Dohner H, Wei AH, Appelbaum FR, et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood. 2022;140(12):1345-1377. PubMed
48.Arber DA, Borowitz MJ, Cessna M, et al. Initial Diagnostic Workup of Acute Leukemia: Guideline From the College of American Pathologists and the American Society of Hematology. Arch Pathol Lab Med. 2017;141(10):1342-1393. PubMed
49.Heuser M, Ofran Y, Boissel N, et al. Acute myeloid leukaemia in adult patients: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2020;31(6):697-712. PubMed
50.Lowenberg B, Griffin JD, Tallman MS. Acute myeloid leukemia and acute promyelocytic leukemia. Hematology Am Soc Hematol Educ Program. 2003;2003(1):82-101. PubMed
51.Brandwein JM, Zhu N, Kumar R, et al. Treatment of older patients with acute myeloid leukemia (AML): revised Canadian consensus guidelines. Am J Blood Res. 2017;7(4):30-40. PubMed
52.Kantarjian HM, Thomas XG, Dmoszynska A, et al. Multicenter, randomized, open-label, phase III trial of decitabine versus patient choice, with physician advice, of either supportive care or low-dose cytarabine for the treatment of older patients with newly diagnosed acute myeloid leukemia. J Clin Oncol. 2012;30(21):2670-2677. PubMed
53.Cortes JE, Heidel FH, Hellmann A, et al. Randomized comparison of low dose cytarabine with or without glasdegib in patients with newly diagnosed acute myeloid leukemia or high-risk myelodysplastic syndrome. Leukemia. 2019;33(2):379-389. PubMed
54.Wei AH, Montesinos P, Ivanov V, et al. Venetoclax plus LDAC for newly diagnosed AML ineligible for intensive chemotherapy: a phase 3 randomized placebo-controlled trial. Blood. 2020;135(24):2137-2145. PubMed
55.DiNardo CD, Jonas BA, Pullarkat V, et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N Engl J Med. 2020;383(7):617-629. PubMed
56.Pollyea DA, DiNardo CD, Arellano ML, et al. Impact of Venetoclax and Azacitidine in Treatment-Naive Patients with Acute Myeloid Leukemia and IDH1/2 Mutations. Clin Cancer Res. 2022;28(13):2753-2761. PubMed
57.Committee for Medicinal Products for Human Use. Assessment report: Venclyxto (venetoclax). (European public assessment report). Amsterdam (NL): European Medicines Agency; 2021: https://www.ema.europa.eu/en/documents/variation-report/venclyxto-h-c-4106-ii-0030-epar-assessment-report-variation_en.pdf. Accessed 2023 Nov 8.
58.Tibsovo in first-line acute myeloid leukemia patients with IDH1 mutation ineligible for intensive chemotherapy - indirect treatment comparison report (version 8.0) [internal sponsor’s report]. Kirkland (QC): IQVIA Inc.; 2023 Apr 18.
59.Dohner H, Montesinos P, Polo SV, et al. AML-295 AGILE: A Global, Randomized, Double-Blind, Phase 3 Study of Ivosidenib + Azacitidine Versus Placebo + Azacitidine in Patients With Newly Diagnosed Acute Myeloid Leukemia With an IDH1 Mutation. Clin Lymphoma Myeloma Leuk. 2022;22:S234.
60.Institut de Recherches Internationales Servier. NCT03173248: Study of AG-120 (Ivosidenib) vs. Placebo in Combination With Azacitidine in Participants With Previously Untreated Acute Myeloid Leukemia With an IDH1 Mutation (AGILE). ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine; 2024: https://classic.clinicaltrials.gov/ct2/show/NCT03173248. Accessed 2024 May 25.
61.DiNardo CD, Schuh AC, Stein EM, et al. Enasidenib plus azacitidine versus azacitidine alone in patients with newly diagnosed, mutant-IDH2 acute myeloid leukaemia (AG221-AML-005): a single-arm, phase 1b and randomised, phase 2 trial. Lancet Oncol. 2021;22(11):1597-1608. PubMed
62.Fayers PM, Aaronson NK, Bjordal K. The EORTC QLC-C30 scoring manual (3rd Edition). Brussels (BE): EORTC; 2001: https://www.eortc.org/app/uploads/sites/2/2018/02/SCmanual.pdf. Accessed 2024 May 25.
63.Giesinger JM, Kieffer JM, Fayers PM, et al. Replication and validation of higher order models demonstrated that a summary score for the EORTC QLQ-C30 is robust. J Clin Epidemiol. 2016;69:79-88. PubMed
64.Kaupp-Roberts SD, Yadegarfar G, Friend E, et al. Validation of the EORTC QLQ-BIL21 questionnaire for measuring quality of life in patients with cholangiocarcinoma and cancer of the gallbladder. Br J Cancer. 2016;115(9):1032-1038. PubMed
65.Reid J. Secondary vs de novo acute myeloid leukemia. 2022; https://aml-hub.com/medical-information/secondary-vs-de-novo-acute-myeloid-leukemia. Accessed 2024 Aug 15.
66.Dias S, Welton NJ, Sutton AJ, Ades AE. NICE DSU Technical Support Document 2: A Generalised Linear Modelling Framework for Pairwise and Network Meta-Analysis of Randomised Controlled Trials. Report by the Decision Support Unit. August 2011 (last updated April 2014) [accessed by sponsor]. 2011; https://www.ncbi.nlm.nih.gov/books/NBK310366/pdf/Bookshelf_NBK310366.pdf.
67.Dias S, Welton NJ, Sutton AJ, Caldwell DM, Lu G, Ades AE. NICE DSU Technical Support Document 4: Inconsistency in Networks of Evidence Based on Randomised Controlled Trials [accessed by sponsor]. NICE Decision Support Unit Technical Support Documents. London (GB): National Institute for Health and Care Excellence (NICE); 2014: https://www.ncbi.nlm.nih.gov/pubmed/27466656.
68.Chen C, Huls GA, Cameron H, et al. Matching Adjusted Indirect Comparison (MAIC) of Oral Azacitidine (Oral-AZA) Versus Injectable AZA in Patients (pts) with Acute Myeloid Leukemia (AML) in First Remission after Intensive Induction Chemotherapy. Blood. 2021;138(Supplement 1):3015-3015.
69.Tremblay G, Daniele P, Bell T, Chan G, Brown A, Cappelleri JC. Comparative effectiveness of glasdegib versus venetoclax combined with low-dose cytarabine in acute myeloid leukemia. J Comp Eff Res. 2021;10(7):603-612. PubMed
70.Phillippo D, Ades T, Dias S, Palmer S, Abrams KR, Welton NJ. NICE DSU Technical Support Document 18: Methods for population-adjusted indirect comparisons in submissions to NICE [accessed by sponsor]. Vol 18: NICE Decision Support Unit; 2016.
71.Pratz KW, Jonas BA, Pullarkat V, et al. Long-term follow-up of VIALE-A: Venetoclax and azacitidine in chemotherapy-ineligible untreated acute myeloid leukemia. Am J Hematol. 2024;99(4):615-624. PubMed
Appendix 1: Detailed Outcome Data
Please note that this appendix has not been copy-edited.
Table 28: Summary of ORR in the AGILE Study (FAS, DCO March 18, 2021)
ORR | Ivosidenib + azacitidine (N = 72) | Placebo + azacitidine (N = 74) |
|---|---|---|
Rates, n (%) | 45 (62.5) | 14 (18.9) |
95% CIa | (50.3 to 73.6) | (10.7 to 29.7) |
Difference in CR rate, % (95% CI) | NR | |
OR (95% CI)b | 4.76 (2.15 to 10.50) | |
P valuec | < 0.0001 | |
CR + CRi rates, n (%) | 39 (54.2) | 12 (16.2) |
95% CIa | (42.0 to 66.0) | (8.7 to 26.6) |
Difference in CR + CRi (including CRp) rate, % (95% CI) | 37% (0.23 to 0.51) | |
OR (95% CI)b | 5.90 (2.69 to 12.97) | |
P valuec | < 0.0001 | |
CI = confidence interval; DCO = data cut-off; FAS = full analysis set; NR = not reported; OR = odds ratio; ORR = objective response rate.
Note: ORR was one of the key secondary outcomes in the AGILE study. It was defined as the rate of CR, CR with incomplete hematologic recovery (CRi) [including CR with incomplete platelet recovery (CRp)], partial remission (PR), and morphologic leukemia-free state (MLFS).
aCI of percentage is calculated with the Clopper and Pearson (exact Binomial) method.
bCochran-Mantel-Haenszel (CMH) estimate for OR is calculated with placebo plus azacitidine as the control (denominator).
cIf the primary analysis of EFS, CR, OS and CR plus CRh are significant, a stratified Cochran-Mantel-Haenszel (CMH) test will be used to compare ORR between the 2 treatment arms. 1-sided P value is calculated from CMH test stratified by the randomization stratification factors (AML status and geographic region).
Source: AGILE Clinical Study Report.38 Details included in the table are from the sponsor’s summary of clinical evidence.
Table 29: RoB-2 Tool for Assessing Risk of Bias in Randomized Trials
Study | Author | Randomization process | Deviations from intended interventions | Missing outcome data | Measurement of the outcomes | Selection of the reported result | Overall |
|---|---|---|---|---|---|---|---|
PETHEMA-FLUGAZA | Vives, 2021 | Low risk | Some concerns | Low risk | Some concerns | Low risk | Some concerns |
VIALE-A | DiNardo, 2020 | Low risk | Low risk | Low risk | Low risk | Low risk | Low risk |
BRIGHT AML 1003 | Cortes, 2019/ Heuser 2021 | Some concerns | Some concerns | Low risk | Some concerns | Low risk | Some concerns |
DACO-016 | Kantarjian, 2012 | Low risk | Some concerns | Low risk | Some concerns | Low risk | Some concerns |
NR | Mohammed, 2021 | Some concerns | Some concerns | Low risk | Some concerns | Some concerns | Some concerns |
PETHEMA-FLUGAZA | Ayala, 2021 | Low risk | Some concerns | Low risk | Some concerns | Low risk | Some concerns |
AZA-AML-001 | Dombret, 2015 | Low risk | Low risk | Low risk | Low risk | Low risk | Low risk |
VIALE-C | Wei, 2020 | Low risk | Low risk | Low risk | Low risk | Low risk | Low risk |
VIALE-A+ Phase Ib | Pollyea, 2022a | NA | NA | NA | NA | NA | NA |
AG120-C-009 | NA | NA | NA | NA | NA | NA | NA |
NA = not available.
aStudy was recommended by Servier.
Source: Sponsor-submitted indirect treatment comparison.58
Table 30: Baseline Characteristics in the AGILE Study Before and After Matching to IDH1 Population for OS (Unanchored MAIC)
Analysis | Baseline characteristic | AGILE IPD prematching | AGILE IPD postmatching | Pooled VIALE-A + phase Ib – (IDH1/2) |
|---|---|---|---|---|
BC | Age (≥ 75) (%) | ████ | ████ | ████ |
Sex, male (%) | ████ | ████ | ████ | |
ECOG (0 or 1) (%) | ████ | ████ | ████ | |
AML type (de novo / primary) (%) | ████ | ████ | ████ | |
AML type (secondary) (%) | ████ | ████ | ████ | |
Cytogenetic risk (intermediate) (%) | ████ | ████ | ████ | |
Cytogenetic risk (poor) (%) | ████ | ████ | ████ | |
Bone marrow blasts (< 30%) (%) | ████ | ████ | ████ | |
Bone marrow blasts (≥ 30% to 50%) (%) | ████ | ████ | ████ | |
SA1 | Age (≥ 75) (%) | ████ | ████ | ████ |
Bone marrow blasts (< 30%) (%) | ████ | ████ | ████ | |
Bone marrow blasts (≥ 30% to 50%) (%) | ████ | ████ | ████ | |
SA2 | Age (≥ 75) (%) | ████ | ████ | ████ |
Bone marrow blasts (< 30%) (%) | ████ | ████ | ████ | |
Bone marrow blasts (≥ 30% to 50%) (%) | ████ | ████ | ████ | |
ECOG (0 or 1) (%) | ████ | ████ | ████ |
AML = acute myeloid leukemia; BC = base case; ECOG = Eastern Cooperative Oncology Group; IPD = individual patient data; SA = scenario analysis.
Source: Sponsor-submitted indirect treatment comparison.58
Table 31: Baseline Characteristics in the AGILE Study Before and After Matching to ITT Population for OS (Anchored MAIC)
Analysis | Baseline characteristic | AGILE IPD prematching | AGILE IPD postmatching | VIALE-A (ITT) |
|---|---|---|---|---|
BC | Age (≥ 75) (%) | ████ | ████ | ████ |
Sex, male (%) | ████ | ████ | ████ | |
ECOG (0 or 1) (%) | ████ | ████ | ████ | |
AML type (de novo / primary) (%) | ████ | ████ | ████ | |
AML type (secondary) (%) | ████ | ████ | ████ | |
Cytogenetic risk (intermediate) (%) | ████ | ████ | ████ | |
Cytogenetic risk (poor) (%) | ████ | ████ | ████ | |
Bone marrow blasts (< 30%) (%) | ████ | ████ | ████ | |
Bone marrow blasts (≥ 30% to 50%) (%) | ████ | ████ | ████ | |
SA1 | Bone marrow blasts (< 30%) (%) | ████ | ████ | ████ |
Bone marrow blasts (≥ 30% to 50%) (%) | ████ | ████ | ████ |
AML = acute myeloid leukemia; BC = base case; ECOG = Eastern Cooperative Oncology Group; IPD = individual patient data; ITT = intention to treat; MAIC = matching-adjusted indirect comparison; OS = overall survival; SA = scenario analysis.
Source: Sponsor-submitted indirect treatment comparison.58
Table 32: Baseline Characteristics in the AGILE Study Before and After Matching to ITT Population for OS (Anchored MAIC) With New Data Cut for the AGILE Study and the VIALE-A Study
Analysis | Baseline characteristic | AGILE IPD prematching | AGILE IPD postmatching | VIALE-A (ITT) |
|---|---|---|---|---|
BC | Age (≥ 75) (%) | ████ | ████ | ████ |
Sex, male (%) | ████ | ████ | ████ | |
ECOG (0 or 1) (%) | ████ | ████ | ████ | |
AML type (de novo / primary) (%) | ████ | ████ | ████ | |
AML type (secondary) (%) | ████ | ████ | ████ | |
Cytogenetic risk (intermediate) (%) | ████ | ████ | ████ | |
Cytogenetic risk (poor) (%) | ████ | ████ | ████ | |
Bone marrow blasts (< 30%) (%) | ████ | ████ | ████ | |
Bone marrow blasts (≥ 30% to 50%) (%) | ████ | ████ | ████ | |
SA1 | Bone marrow blasts (< 30%) (%) | ████ | ████ | ████ |
Bone marrow blasts (≥ 30% to 50%) (%) | ████ | ████ | ████ |
AML = acute myeloid leukemia; BC = base case; ECOG = Eastern Cooperative Oncology Group; IPD = individual patient data; ITT = intention to treat; MAIC = matching-adjusted indirect comparison; OS = overall survival; SA = scenario analysis.
Source: Sponsor-submitted indirect treatment comparison.58
Table 33: Baseline Characteristics in the AGILE Study Before and After Matching to ITT Population for EFS (Anchored MAIC)
Analysis | Baseline characteristic | AGILE IPD prematching | AGILE IPD postmatching | Pooled VIALE-A + phase Ib – (IDH1/2) |
|---|---|---|---|---|
BC | Age (≥ 75) (%) | ████ | ████ | ████ |
Sex, male (%) | ████ | ████ | ████ | |
ECOG (0 or 1) (%) | ████ | ████ | ████ | |
AML type (de novo / primary) (%) | ████ | ████ | ████ | |
AML type (secondary) (%) | ████ | ████ | ████ | |
Cytogenetic risk (intermediate) (%) | ████ | ████ | ████ | |
Cytogenetic risk (poor) (%) | ████ | ████ | ████ | |
Bone marrow blasts (< 30%) (%) | ████ | ████ | ████ | |
Bone marrow blasts (≥ 30% to 50%) (%) | ████ | ████ | ████ | |
SA1 | Sex, male (%) | ████ | ████ | ████ |
ECOG (0 or 1) (%) | ████ | ████ | ████ | |
SA2 | Sex, male (%) | ████ | ████ | ████ |
Cytogenetic risk (intermediate) (%) | ████ | ████ | ████ | |
Cytogenetic risk (poor) (%) | ████ | ████ | ████ | |
ECOG (0 or 1) (%) | ████ | ████ | ████ |
AML = acute myeloid leukemia; BC = base case; ECOG = Eastern Cooperative Oncology Group; EFS = event-free survival; IPD = individual patient data; MAIC = matching-adjusted indirect comparison; SA = scenario analysis.
Source: Sponsor-submitted indirect treatment comparison.58
Pharmacoeconomic Review
Abbreviations
AE
adverse event
AML
acute myeloid leukemia
AZA
azacitidine
BIA
budget impact analysis
CDA-AMC
Canada’s Drug Agency
CR
complete remission with complete hematologic recovery
Cri
complete remission with incomplete hematologic recovery
CrI
credible interval
EFS
event-free survival
HR
hazard ratio
ICER
incremental cost-effectiveness ratio
LDAC
low-dose cytarabine
NMA
network meta-analysis
OS
overall survival
PSM
partitioned survival model
QALY
quality-adjusted life-year
RDI
relative dose intensity
WTP
willingness to pay
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Ivosidenib (Tibsovo), 250 mg oral tablets |
Indication | Ivosidenib in combination with azacitidine is indicated for the treatment of adult patients with newly diagnosed AML with an IDH1 R132 mutation who are not eligible to receive intensive induction chemotherapy |
Health Canada approval status | pre-NOC |
Health Canada review pathway | Standard |
NOC date | July 19, 2024 |
Reimbursement request | As per indication |
Sponsor | Servier Canada Inc. |
Submission history | Previously reviewed: No |
NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Partitioned survival model |
Target population | Adults with newly diagnosed AML with an IIDH1 R132 mutation who are not eligible to receive intensive induction chemotherapy |
Treatment | Ivosidenib in combination with AZA |
Dose regimen | 500 mg of ivosidenib taken orally once daily for a 28-day cycle in combination with AZA at 75 mg/m2, intravenously or subcutaneously, once daily on days 1 to 7 of each 28-day cycle; patients should receive ivosidenib for a minimum of 6 cycles |
Submitted price | Ivosidenib: $332.60 per tablet |
Submitted treatment cost | Ivosidenib: $16,616 per 28-day cyclea,b |
Comparators |
|
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, life-years |
Time horizon | Lifetime (25 years) |
Key data source | Efficacy of ivosidenib plus AZA and AZA alone informed by the AGILE study; efficacy of venetoclax plus AZA and LDAC informed by a sponsor-submitted network meta-analysis |
Submitted results | Ivosidenib plus AZA was associated with an ICER of $332,590 per QALY gained compared to venetoclax plus AZA (incremental costs: $319,036; incremental QALYs: 0.96) |
Key limitations |
|
CDA-AMC reanalysis results |
|
AML = acute myeloid leukemia; AZA = azacitidine; CDA-AMC = Canada’s Drug Agency; CR = complete remission with complete hematologic recovery; CRi = complete remission with incomplete hematologic recovery; EFS = event-free survival; ICER = incremental cost-effectiveness ratio; LDAC = low-dose cytarabine; OS = overall survival; QALY = quality-adjusted life-year; RDI = relative dose intensity.
aIvosidenib + AZA: $23,827 per 28-day cycle.
bAssuming ████% RDI.
Conclusions
Based on the clinical review by Canada’s Drug Agency (CDA-AMC), data from the AGILE trial suggests that ivosidenib plus azacitidine (AZA) likely improves OS at 12 months and 24 months compared to placebo plus AZA in the indicated population. Evidence from the AGILE trial also suggests that ivosidenib plus AZA likely improves event-free survival (EFS) versus placebo plus AZA at 6 months; however, the findings beyond 6 months were uncertain. There have been no direct head-to-head trials comparing ivosidenib plus AZA with venetoclax plus AZA or other currently available treatments other than AZA alone. Indirect evidence submitted by the sponsor was insufficient to conclude whether treatment outcomes with ivosidenib plus AZA differ from venetoclax plus AZA in terms of OS, EFS, and complete remission with complete hematologic recovery (CR) or complete remission with incomplete hematologic recovery (CRi) because of limited efficacy data and important heterogeneity across studies, and health-related quality of life and harms were not assessed in the sponsor’s network meta-analysis (NMA). Thus, there is insufficient clinical evidence to support a price premium for ivosidenib versus venetoclax when used in combination with AZA.
Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, clinician groups, and drug plans that participated in the CDA-AMC review process.
Patient group input was received from the Leukemia & Lymphoma Society of Canada (informed by an online survey conducted in March 2024) and from Heal Canada. In total, 92 respondents provided feedback, with 7 identified as having the IDH1 mutation. Patients with acute myeloid leukemia (AML) and their caregivers noted that the disease affects all aspect of their lives, resulting in an overall negative impact on their quality of life. The respondents noted that currently available treatments are associated with toxicities and unstable blood counts and that there remains an unmet need for new therapies for patients for whom current treatment options are not effective or cannot be tolerated or for patients who experience disease relapse. The respondents also expressed an interest in therapies that can improve overall outcomes, remain tolerable, lower the rate of infections, and reduce resource use burden, such as hospital visits. One interview was conducted with a patient who had experience with ivosidenib, who noted overall improvements in their quality of life with no adverse effects.
Clinician input was received from the Leukemia & Lymphoma Society of Canada Clinician Network and from the Ontario Health (Cancer Care Ontario) Hematology Cancer Drug Advisory Committee. The clinicians noted that the goal of treatment is to improve survival, improve quality of life, and attain remission. Currently available treatments include venetoclax plus AZA, low-dose cytarabine (LDAC), venetoclax plus LDAC, AZA alone, and supportive care. The clinician input noted that better tolerated treatments are needed as patients with AML frequently experience myelosuppression and hospitalizations, which impact quality of life. The clinicians noted that ivosidenib may become the standard of care for newly diagnosed adult patients with AML with the IIDH1 R132 mutation who are not eligible to receive intensive induction chemotherapy and are also not eligible for stem cell or bone marrow transplant.
CDA-AMC–participating drug plans noted concerns with the choice of comparator in the AGILE trial, given that currently available treatments for adults with newly diagnosed AML with an IIDH1 R132 mutation who are not eligible to receive intensive induction chemotherapy were not included. The drug plans inquired how clinicians would determine which patients would be eligible for ivosidenib plus AZA versus venetoclax plus AZA and if switching between the 2 treatments could occur. Lastly, the drug plans questioned the current states of diagnostic testing for patients with AML across Canada, as IDH1 testing is required for treatment with ivosidenib plus AZA.
Several of these concerns were addressed in the sponsor’s model:
Health-related quality of life was incorporated in the sponsor’s model by use of the EQ-5D-5L data captured in the AGILE trial.
Health care resources associated with AML and myelosuppression were considered.
CDA-AMC was unable to address the following concerns raised from input:
The omission of venetoclax plus LDAC as a comparator in the model; however, CADTH notes that venetoclax plus LDAC may not be a funded regimen in all participating jurisdictions.
Economic Review
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis to assess the cost-effectiveness of ivosidenib in combination with AZA for the treatment of newly diagnosed AML in adults with an IIDH1 R132 mutation who are not eligible to receive intensive induction chemotherapy.1 In the model, the sponsor compared ivosidenib plus AZA to venetoclax plus AZA, AZA alone, and LDAC. The modelled population is in line with the Health Canada indication and was based on patients enrolled in the AGILE trial.2
Ivosidenib is available as 250 mg oral tablets.1 The recommended dose of ivosidenib is 500 mg once daily. Ivosidenib should be started in combination with AZA (75 mg/m2 on days 1 to 7 of each 28-day cycle, IV or subcutaneous), and ivosidenib should be given for a minimum of 6 cycles.1 At the submitted price of $332.60 per ivosidenib tablet, the sponsor estimated the 28-day cost of ivosidenib plus AZA to be $23,827 per patient based on a relative dose intensity (RDI) of 89.2% and 90% for ivosidenib and AZA, respectively.2 The sponsor estimated the 28-day cost of venetoclax plus AZA, AZA alone, and LDAC to be $14,285, $7,560, and $151, respectively.
The analysis was conducted from the perspective of the Canadian public health care payer. Cost and outcomes (quality-adjusted life-years [QALYs] and life-years) were estimated over a lifetime horizon (25 years; 28-day cycle length). Discounting (1.5% per annum) was applied to both costs and outcomes.
Model Structure
The sponsor submitted a partitioned survival model (PSM) with 3 health states: EFS, progressed disease, and death (Figure 1).2 The proportion of patients who were event-free, who experienced progressed disease, or who were dead at any time over the model horizon was derived from non–mutually exclusive survival curves. Patients in the EFS health state could experience disease progression or death, with the proportion of patients in each state based on the area under the survival curves (OS and EFS). Specifically, OS was partitioned to estimate the proportion of patients in the dead state, while the EFS curve was used to estimate the proportion of patients in the progression-free health state. The difference between the OS curve and the EFS curve was partitioned at each time point to estimate the proportion of patients in the progressed disease health state. All patients entered the model in the EFS health state. EFS was further stratified into 2 groups of patients: patients who experienced CR or CRi (termed “CR/CRi” in Figure 1), and patients without CR or CRi (termed “no CR/CRi” in Figure 1).2 Patients who received ivosidenib plus AZA or venetoclax plus AZA were considered by the sponsor to be cured if they remained in the EFS health state (with CR or CRi) for more than 5 years; these patients were assumed to no longer be at risk of disease progression or disease-related mortality.2 Patients who received ivosidenib plus AZA or AZA alone could discontinue treatment based on the time-on-treatment curves from the AGILE trial, while patients who received venetoclax plus AZA or LDAC were assumed to discontinue treatment if they progressed to the progressed disease health state. After discontinuation, the cost of first-line treatment was no longer incurred, but patients in the progressed disease health state incurred the cost of subsequent therapy.
Model Inputs
The baseline characteristics used to inform the model were based on the AGILE trial (mean age ███ ██ years, ███ ██% female, mean weight ███ ██ kg, mean body surface area ████ m2).2 In the AGILE trial, patients with newly diagnosed IDH1-mutated AML who were ineligible for intensive induction chemotherapy were randomly assigned to receive ivosidenib plus AZA or placebo plus AZA as first-line treatment.
Key clinical efficacy inputs (EFS, OS, and CR or CRi) were derived from the AGILE trial for ivosidenib plus AZA and AZA alone and from a sponsor-submitted NMA for venetoclax plus AZA and LDAC. In the AGILE trial, EFS was defined as the time from randomization until progressed disease, relapse, treatment failure, or death. The sponsor fitted parametric survival curves to patient-level survival data from the AGILE trial to derive EFS and OS for ivosidenib plus AZA and AZA alone for the entire model time horizon. The sponsor chose the parametric survival distribution used in the base case based on fit statistics, visual inspection, and clinical and external validity. In the base case, the lognormal distribution was selected by the sponsor for both EFS and OS. The sponsor derived EFS and OS curves for relevant comparators via hazard ratio (HR) adjustment. The HRs were obtained from an NMA conducted by the sponsor for this review and were applied to the ivosidenib plus AZA curves. Time-on-treatment for ivosidenib plus AZA and AZA alone were informed by data extrapolated from the AGILE trial. The sponsor chose the lognormal distribution as the best-fitting parametric survival curve for ivosidenib plus AZA and the exponential distribution for AZA alone. In the absence of patient-level data for venetoclax plus AZA and LDAC, the sponsor modelled time-on-treatment for these comparators as time until progressed disease or relapse, which was assumed to align with the recommendation to “treat until progression,” as indicated in the product monographs.2
In the base-case analysis, the sponsor assumed that patients who received ivosidenib plus AZA or venetoclax plus AZA and remained in the EFS health state (with CR or CRi) after 60 months were cured and no longer at risk of disease progression or relapse.2 Cured patients were assumed to experience similar mortality to the general population of Canada.2
Health state utility values were derived from mean EQ-5D-5L data collected in the AGILE trial for EFS with CR or CRi (███ ██), cured patients (███ ██), EFS with no CR or CRi (███ ██), and progressed disease (███ ██).2 Utility values were adjusted for age and sex based on Canadian utility norms obtained from the literature.3,4
Grade 3 and 4 adverse events (AEs) occurring in at least 5% of patients, as well as differentiation syndrome, for ivosidenib plus AZA and AZA alone, were informed by data from the AGILE trial and were considered as a one-off cost in the first cycle of the model. AE rates for venetoclax plus AZA were informed by clinical trial data using a cut-off of greater than or equal to 10%; AE rates for LDAC were informed by a previous CADTH report using a cut-off of greater than or equal to 15%.5,6 Utility decrements for AEs were included in the first model cycle, with disutility values obtained from the literature.7-10
The model included costs related to drug acquisition and administration, disease management and monitoring, AEs, and end of life. Drug acquisition costs were calculated by the sponsor as a function of unit drug costs, dosing schedules, RDI, and the proportion of patients on treatment. The cost of ivosidenib was based on the sponsor’s submitted price, while all other drug acquisition costs were obtained from a prior CADTH report or from the Ontario Drug Benefit Formulary.6,11 Drug administration costs were informed by the Ontario Schedule of Benefits of Physician Services.12 The sponsor incorporated an administration cost of $54 for IV and subcutaneous drugs and a 1-time administration cost of $26 for oral drugs. Disease management and monitoring costs included costs associated with outpatient treatment, emergency department hospitalization, diagnostics, and blood transfusions. The frequency of disease management and monitoring use were informed by clinical expert feedback obtained by the sponsor, with unit costs informed by the Ontario Schedule of Benefits of Physician Services, the Canadian Institute for Health Information patient cost estimator, and published literature.12-15 Costs associated with AEs were informed by the Ontario Schedule of Benefits of Physician Services and the Canadian Institute for Health Information patient cost estimator. End-of-life costs were obtained from Hu et al. (2021), with the costs originally derived from a paper examining the cost of end-of-life care for patients in Ontario, Canada.13,16 All costs were reported in 2023 Canadian dollars.
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically (1,000 iterations). The deterministic and probabilistic results were similar. The probabilistic findings are presented in this section. The submitted analysis was based on the submitted price for ivosidenib and public list prices for comparators. Additional results from the sponsor’s submitted economic evaluation base case are presented in Appendix 3.
Base-Case Results
In the sponsor’s probabilistic base case, ivosidenib plus AZA was associated with an estimated cost of $969,460 and 3.17 QALYs over a lifetime horizon (Table 3). In sequential analysis, ivosidenib plus AZA was associated with an incremental cost-effectiveness ratio (ICER) of $332,590 versus venetoclax plus AZA (incremental cost: $319,036; incremental QALYs: 0.96). At a willingness-to-pay (WTP) threshold of $50,000 per QALY gained, ivosidenib plus AZA had a 0% probability of being considered the optimal treatment.
The main drivers were the predicted gain in life-years and the costs related to drug acquisition and health care resource use. The sponsor’s model predicted that ivosidenib plus AZA results in an additional 1.66 life-years relative to venetoclax plus AZA. Of the 3.17 QALYs predicted by the sponsor’s deterministic results to be gained with ivosidenib plus AZA, approximately 84% were accrued beyond the trial follow-up period (approximately 15 months). At the end of the model horizon (i.e., 25 years), approximately 1% of patients are predicted to remain alive in the ivosidenib plus AZA treatment group versus 2% in the venetoclax plus AZA group.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/ QALY) |
|---|---|---|---|
LDAC | 189,531 | 0.82 | Reference |
Venetoclax + AZA | 650,424 | 2.21 | 330,836 vs. LDAC |
Ivosidenib + AZA | 969,460 | 3.17 | 332,590 vs. venetoclax + AZA |
AZA = azacitidine; ICER = incremental cost-effectiveness ratio; LDAC = low-dose cytarabine; QALY = quality-adjusted life-year; vs. = versus.
Note: Only treatments that are on the efficiency frontier are reported.
Source: Sponsor’s pharmacoeconomic submission (probabilistic results). Deterministic results are provided in Appendix 4 (Table 11).2
Sensitivity and Scenario Analysis Results
The sponsor conducted several scenario analyses, including adopting alternative discount rates and time horizons and excluding IDH1 testing costs. Results of these analyses were largely aligned with the sponsor’s base-case analysis; however, only pairwise analyses were provided, limiting interpretation of the results. When compared with venetoclax plus AZA, the scenarios with the greatest impact on the ICERs were changes in time horizon (10-year horizon: $474,230 per QALY gained; 15-year horizon: $381,163 per QALY gained).
The sponsor additionally conducted a scenario analysis from a societal perspective, which included additional costs associated with productivity loss. In the pairwise analysis, the ICER was $373,656 per QALY gained compared to venetoclax plus AZA when productivity costs were included. This was higher than the sponsor’s base-case analysis using a health care payer perspective.
CDA-AMC Appraisal of the Sponsor’s Economic Evaluation
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis:
The comparative clinical efficacy of ivosidenib plus AZA is uncertain. There is a lack of direct head-to-head evidence comparing ivosidenib plus AZA to venetoclax plus AZA and to LDAC. To inform the economic model (i.e., OS, EFS, and CR rate), the sponsor conducted an NMA to estimate the relative efficacy of ivosidenib plus AZA to these regimens. Credible intervals from the sponsor’s NMA cross the null value, suggesting that there may be no statistically significant difference between ivosidenib plus AZA and venetoclax plus AZA for OS (HR ███ ██; 95% credible interval [CrI], ███ ██ to ███ ██), EFS (HR ███ ██; 95% CrI, ███ ██ to ███ ██), or CR or CRi (HR ███ ██; 95% CrI, ███ ██ to ███ ██); however, the CDA-AMC clinical report identified notable limitations with the sponsor’s NMA, including heterogeneity in study design and baseline patient characteristics, as well as substantial imprecision, all of which preclude meaningful conclusions from being made. As noted in the CDA-AMC clinical review, the indirect evidence submitted by the sponsor was insufficient to determine whether treatment outcomes differ between ivosidenib plus AZA versus venetoclax plus AZA or LDAC. Health-related quality of life and harms were not assessed in the sponsor’s NMA.
Given the lack of direct evidence comparing ivosidenib plus AZA to venetoclax plus AZA and limitations with the sponsor’s NMA, it remains uncertain whether ivosidenib plus AZA provides a net clinical benefit relative to venetoclax plus AZA. In scenario analyses, CDA-AMC assumed no difference in the efficacy of ivosidenib plus AZA and of venetoclax plus AZA.
The sponsor’s cure assumptions are highly uncertain. The sponsor assumed that patients who received ivosidenib plus AZA or venetoclax plus AZA and remained in the EFS health state (with CR or CRi) for more than 5 years were cured. After 5 years, such patients were assumed by the sponsor to discontinue treatment, no longer be at risk of progression, and have the same risk of death as the general population. Clinical expert input received by CDA-AMC for this review indicated that the inclusion of a cure assumption for this patient population is associated with considerable uncertainty, as patients in clinical practice typically experience relapse within 3 years and therefore are not likely to experience cure. Clinician input noted that although a small proportion of patients may stay in remission long-term, these patients remain at risk of disease progression and death due to long-term cancer complications.
The sponsor additionally assumed that AML could only be cured for patients who received ivosidenib plus AZA or venetoclax plus AZA; that is, patients who received either LDAC or AZA alone could not transition into the cured health state in the sponsor’s model. The clinical experts consulted by CDA-AMC for this review indicated that it is inappropriate to only apply a cure assumption to ivosidenib plus AZA and venetoclax plus AZA. Based on extrapolation of the sponsor’s data in the economic model, approximately 1.2% to 1.4% of patients who receive LDAC or AZA remained in the EFS health state (with CR or CRi) at 5 years.
In the CDA-AMC base case, a longer interval was adopted before assuming that AML is cured (i.e., 10 years). In this analysis, patients were considered cured if they remained in the EFS health state (with CR or CRi) for at least 10 years. explored uncertainty in the cure assumption in scenario analyses.
The long-term clinical benefits of ivosidenib plus AZA are uncertain. The sponsor submitted a PSM, in which treatment efficacy is represented by EFS and OS curves, informed by observations from the AGILE trial and extrapolated over the model’s lifetime horizon. In the pharmacoeconomic analysis, the long-term extrapolation of EFS and OS resulted in an incremental gain of approximately 1.89 life-years and 1.14 QALYs for ivosidenib plus AZA compared to treatment with venetoclax plus AZA. Of the QALYs predicted to be gained with ivosidenib plus AZA, approximately 84% were accrued after the AGILE trial on the basis of extrapolation. Based on the indirect evidence submitted by the sponsor, there may be no statistically significant difference in EFS or OS between ivosidenib plus AZA and venetoclax plus AZA. Owing to the absence of long-term data and limitations with the sponsor’s NMA, the extent of QALYs that will be gained and the magnitude of any incremental gain in EFS or OS with ivosidenib plus AZA compared with venetoclax plus AZA are highly uncertain.
In the economic model, the sponsor extrapolated EFS and OS data for ivosidenib plus AZA from the AGILE trial to estimate outcomes over the lifetime horizon (lognormal distribution for EFS and OS) and applied HRs from the sponsor-submitted NMA to the ivosidenib plus AZA OS and EFS curve to estimate outcomes for the comparators. Clinical expert feedback received by CDA-AMC suggests that these extrapolations resulted in OS and EFS rates that were likely overestimated. For example, the sponsor’s model estimated that approximately 27% of patients on venetoclax plus AZA would be alive at around 45 months, whereas data from the VIALE-A study suggests that the estimated OS at 45 months for patients with IDH1/2 mutations who received venetoclax plus AZA is approximately 20%.17
CDA-AMC raised further concerns regarding the predicted survival benefits of ivosidenib plus AZA, attributing this uncertainty to the sponsor’s choice of a PSM. While this modelling approach is suitable for the decision-making context, a PSM model relies on the structural assumption that the proportion of the population that is event-free is independent of the proportion of patients who remain on treatment and the assumption that the proportion of patients who remain alive is independent of the proportion of alive patients who are event-free. Such assumptions may suggest optimistic postprogression survival for patients treated with ivosidenib plus AZA and comparators. CADTH notes that the use of a PSM resulted in EFS exceeding OS in some iterations of the sponsor’s probabilistic analysis, which the sponsor attempted to address by capping EFS to OS. For these iterations, this resulted in zero patients experiencing progressed disease or relapse in each treatment group including AZA alone, which lacks face validity. As noted earlier, the clinical experts consulted by CDA-AMC indicated that patients who receive AZA alone would be expected to relapse within 3 years of treatment initiation.
In the CDA-AMC base case, CADTH adopted alternative extrapolation models for EFS and OS for ivosidenib plus AZA, which resulted in OS and EFS estimates more closely aligned with clinical expert input. CADTH was unable to fully address this limitation, however, because of the dependent nature of the efficacy of comparators on the ivosidenib plus AZA OS and EFS curves, the lack of alternate HRs, and the reliance on capping of EFS with OS. The CADTH reanalysis thus presents the results of the deterministic analyses.
Health state utility values lack face validity. In the sponsor’s base case, health state utility values were estimated based on EQ-5D-5L utility data collected in the AGILE trial. These utility values suggest that patients in the EFS health state (without CR or CRi) will have lower health-related quality of life than patients in the progressed disease health state (i.e., ███ ██ versus ███ ██, respectively). Clinical expert feedback received by CDA-AMC indicated that it is unlikely that patients will have better health-related quality of life after disease progression than before progression (with or without CR or CRi). Thus, the utilities adopted by the sponsor lack face validity and may underestimate health-related quality of life for patients in the EFS health state. Clinical expert feedback received by CDA-AMC noted that it is reasonable to assume that patients in the EFS health state have similar quality of life with or without CR or CRi.
In the CDA-AMC base case, CADTH assumed that all patients in the EFS health state (i.e., with or without CR or CRi) have the same utility value (i.e., ███ ██).
Health care resource use is highly uncertain. The sponsor incorporated costs related to disease management (i.e., health care resource use) in the economic model. Overall, the cost of health care resource use was assumed to vary both by health state (EFS with CR or CRi, EFS without CR or CRi, progressed disease) and by treatment received (Table 10). The sponsor used clinical expert opinion to estimate the monthly frequency of resource use for patients in each health state, “considering the overall care of a typical AML patient” and then adjusted these estimates for treatment-specific considerations. CDA-AMC identified several limitations with this approach.
First, the sponsor assumed that patients who received ivosidenib plus AZA would require 61% fewer resources than those who received venetoclax plus AZA, based on a retrospective chart review of “unscheduled acute care” usage in the first 12 weeks of treatment among patients in the US who received ivosidenib plus hypomethylating agents versus venetoclax plus hypomethylating agents. This study has been published only as an abstract, and full methodologic details are unavailable. CDA-AMC was unable to determine what resources were included by the study authors as part of “unscheduled acute care.” Further, because of the study methodology (retrospective chart review), it is highly uncertain whether differences in resource use are related to treatment received or are a result of confounding. It is also highly uncertain whether resource use in the US can be generalized to the Canadian context owing to differences in access to health care across jurisdictions. Finally, although the study assessed “acute care” resource use in the first 12 weeks of treatment, the sponsor assumed that these differences would persist for the model’s lifetime horizon.
Second, the sponsor assumed that patients who received venetoclax plus AZA would require 1.5 times more resources related to myelosuppression (blood transfusions, hospitalizations, hematologist visits, and nurse visits) than patients who received ivosidenib plus AZA, AZA alone, and LDAC. This assumption was based on clinical expert input obtained by the sponsor. Clinical expert feedback received by CDA-AMC was aligned with that received by the sponsor in that myelosuppression is associated with increased resource use. However, the clinical experts consulted by CDA-AMC noted that whether the rate of increased resource use in such a case is 1.5 times that of patients who received ivosidenib plus AZA is uncertain.
Overall, the clinical expert feedback received by CDA-AMC indicated that the majority of differences in health care resource use are expected to be correlated with a patient’s health state (i.e., EFS or progressed disease) and that health state–specific resource use between ivosidenib plus AZA versus venetoclax plus AZA are not expected. Instead, impacts on resource use should be captured in the model based on how long a patient stays in each health state versus treatment-specific adjustments.
In the CDA-AMC base case, patients were assumed to incur health care resource costs based on health state (i.e., not based on treatment received). CADTH explored the impact of assuming differences in myelosuppression-related health care resource use in scenario analyses.
Genetic testing cost assumptions are highly uncertain. The AGILE trial enrolled patients with newly diagnosed AML with IIDH1 R132 mutation. In the economic model, the sponsor assumed that all patients with AML will undergo genetic testing via next-generation sequencing as part of routine clinical practice for AML and thus assumed that there will be no additional change to the proportion of patients who undergo testing with the introduction of ivosidenib. The sponsor incorporated a cost of $1,277 per patient, obtained from the literature,18 and assumed that 10% of patients will test positive for the IIDH1 R132 mutation. Clinical expert feedback received by CDA-AMC noted that, at present, IIDH1 R132 mutation testing is jurisdiction dependent and therefore is not routinely performed across Canada; that is, not all patients currently undergo genetic testing. Expert input further noted that the reimbursement of ivosidenib may increase the proportion of patients who undergo genetic testing, as ivosidenib is the first mutation-specific treatment for AML. As such, the reimbursement of ivosidenib may result in additional costs related to IIDH1 R132 mutation testing in jurisdictions that do not currently perform genetic testing for all patients.
In the economic analysis, the sponsor assumed that 10% of tested patients would be positive for the IDH1 mutation, while in the submitted budget impact analysis (BIA), the sponsor assumed that 8% would have the mutation. Clinical experts consulted by CDA-AMC indicated that the proportion of patients positive for the IIDH1 R132 mutation is likely around 6% to 10%, in line with published literature. Therefore, use of a middle value (i.e., 8%) is reasonable. CADTH additionally notes that, of the patients screened for eligibility in the AGILE trial, approximately 61% were positive for the IIDH1 R132 mutation; however, this may not reflect the prevalence of the mutation in the general population owing to selection of the trial population.
CDA-AMC was unable to account for potentially higher rates of genetic testing in some jurisdictions because of the structure of the sponsor’s model. Given that the sponsor’s assumed that all patients with AML will undergo genetic testing as part of routine care in practice, adjustments to the proportion of patients who undergo genetic testing will be positive for the IIDH1 R132 mutation to align with the sponsor’s BIA do not impact the results.
AEs were not adequately considered in the model. In the sponsor’s base-case analysis, AEs for ivosidenib plus AZA and AZA alone were informed by grade 3 or 4 events that occurred in at least 5% of patients in the AGILE trial, while AEs occurring in more than 10% and more than 15% of patients were used to inform rates for venetoclax plus AZA and LDAC, respectively. CDA-AMC notes that the sponsor’s use of different thresholds to capture AE rates for comparators may not accurately represent the costs and disutilities associated with AEs. Furthermore, AEs were included via naive comparison (i.e., without adjustment or accounting for differences in patient characteristics) and included in the model as a one-off cost in the first cycle of the model. Owing to the direct use of clinical trial data, it is not possible to determine if any observed differences between the therapies are solely due to the treatment or, rather, due to bias or confounding factors.
CDA-AMC was unable to address this limitation.
Use of RDI may underestimate actual drug costs. In the sponsor’s base-case analysis, RDI observations from the AGILE trial (for ivosidenib plus AZA), from a prior CADTH review (for AZA alone and LDAC), or based on assumption (for venetoclax plus AZA) were used to derive the drug acquisition costs. The inclusion of RDI may underestimate the total drug costs in clinical practice as changes in RDI can result from numerous factors, including clinical judgment, dose delays, missed doses, or dose reductions, and such adjustments impact drug costs differently, especially when considering drug wastage.
In the CDA-AMC base case, an RDI of 100% was assumed for all treatments.
Model lacked transparency. The sponsor’s submitted model included numerous IFERROR statements, including on the model engine sheets. IFERROR statements may lead to situations in which the parameter value is overwritten with an alternative value without alerting the user to the automatic overwriting. The systematic use of IFERROR statements makes thorough auditing of the sponsor’s model impractical, as it remains unclear whether the model is running inappropriately by overriding errors.
CDA-AMC was unable to address this limitation and notes that a thorough validation of the sponsor’s model was not possible.
Additional limitations were identified but were not considered to be key limitations:
Inappropriate cost calculation. In the sponsor’s submission, the cost of cytarabine (used as part of LDAC) was based on the per millilitre cost. CDA-AMC noted that the analysis should be based on the cost per vial.
CDA-AMC corrected the cost of cytarabine within the sponsor’s submission, as well as the CADTH base-case and scenario analyses.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CDA-AMC (Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted As Limitations to the Submission)
Sponsor’s key assumption | CDA-AMC comment |
|---|---|
The sponsor’s analysis included only venetoclax plus AZA, AZA only, and LDAC as comparators. | Reasonable. Clinical expert feedback received by CDA-AMC noted that some patients may receive venetoclax plus LDAC or best supportive care. For example, venetoclax plus LDAC may be used for a subset of patients who have a history of hypomethylating agent exposure or who have severe liver or kidney dysfunction. Because of the lack of comparative evidence of ivosidenib plus AZA vs. venetoclax plus AZA or best supportive care, CADTH was unable to address this limitation. The cost-effectiveness of ivosidenib plus AZA vs. AZA vs. venetoclax plus AZA or best supportive care is unknown. Venetoclax plus LDAC may not be a funded regimen in some participating jurisdictions. |
The duration of treatment with ivosidenib plus AZA and AZA alone was informed by time-on-treatment data from the AGILE trial. For venetoclax plus AZA and for LDAC, the sponsor assumed that patients would remain on treatment until disease progression or relapse. | Uncertain. The use of different approaches to model time on treatment for different treatments may introduce additional uncertainty into the findings. CDA-AMC was unable to address this limitation owing to a lack of provided time-on-treatment data for venetoclax plus AZA and LDAC. |
A 1-time administration cost of $26 was included for drugs taken orally. | Inappropriate. The sponsor assumed that orally administered drugs were associated with a 1-time cost of $26. Given that oral drugs are self-administered by the patients, inclusion of administration costs for oral treatments would overestimate the cost of oral chemotherapies. However, as the cost of oral administration was assumed to be a 1-time cost, this assumption is unlikely to have a meaningful impact on the incremental cost-effectiveness ratio. |
AE = adverse event; AZA = azacitidine; CDA-AMC = Canada’s Drug Agency; LDAC = low-dose cytarabine.
CDA-AMC Reanalyses of the Economic Evaluation
Base-Case Results
The CDA-AMC base case was derived by making changes in model parameter values and assumptions, in consultation with clinical experts (Table 5). CADTH was unable to determine the probability that ivosidenib plus AZA is cost-effective at a WTP threshold (e.g., of $50,000 per QALY) owing to the structural limitations of the sponsor’s model (that is, all CADTH analyses are deterministic and do not reflect uncertainty).
Table 5: CADTH Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
1. LDAC cost calculation | Cytarabine: $15.37 per mL | Cytarabine: $76.85 per vial |
Changes to derive the CDA-AMC base case | ||
1. Cure assumption | Those who remain in the EFS CR/CRi state for more than 5 years were assumed to be cured | Those who remain in the EFS CR/CRi state for more than 10 years were assumed to be cured |
2. Extrapolation of EFS for ivosidenib plus AZA | Lognormal | Exponential |
3. Extrapolation of OS for ivosidenib plus AZA | Lognormal | Exponential |
4. Health state utility values | EFS CR/CRi = ███ ██ EFS no CR/CRi = ███ ██ Progressed disease = ███ ██ | EFS = ███ ██ Progressed disease = ███ ██ |
5. Health care resource use | Patients receiving ivosidenib plus AZA in the EFS health state were assumed to have 61% lower resource use (hematology visit, nurse visit, hospitalization, and transfusion) than patients in the same health state who received venetoclax plus AZA. Patients in the EFS health state who received venetoclax plus AZA were assumed to require 1.5 times more resource use than the typical patient with AML. | Health care resource use was assumed to be related to the health state (EFS, progressed disease) not treatment received. |
6. RDI | Ivosidenib = ███ ██ Venetoclax = 89.2% AZA (in combination with either ivosidenib or venetoclax) = ███ ██ AZA only = 90% LDAC = 98% | 100% for all |
CDA-AMC base case | ― | Reanalysis 1 + 2 + 3 + 4 + 5 + 6 |
AML = acute myeloid leukemia; AZA = azacitidine; CDA-AMC = Canada’s Drug Agency; CR = complete remission with complete hematologic recovery; CRi = complete remission with incomplete hematologic recovery; EFS = event-free survival; LDAC = low-dose cytarabine; OS = overall survival; RDI = relative dose intensity.
In the CDA-AMC base case, ivosidenib plus AZA was associated with the highest total costs ($985,719) and greatest QALYs (1.71) over the lifetime horizon. In sequential analysis, ivosidenib plus AZA was more expensive (incremental costs: $577,580) and produced more QALYs (incremental QALYs: 0.48) than venetoclax plus AZA, with an ICER of $1,206,919 per QALY gained (Table 6).
Table 6: Summary of the CDA-AMC Reanalysis Results
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
Sponsor base case (probabilistic) | |||
LDAC | 189,531 | 0.82 | Reference |
Venetoclax + AZA | 650,424 | 2.21 | 330,836 vs. LDAC |
Ivosidenib + AZA | 969,460 | 3.17 | 332,590 vs. venetoclax + AZA |
Sponsor’s corrected base case (probabilistic) | |||
LDAC | 189,248 | 0.86 | Reference |
Venetoclax + AZA | 633,776 | 2.26 | 318,377 vs. LDAC |
Ivosidenib + AZA | 961,722 | 3.19 | 351,063 vs. venetoclax + AZA |
CDA-AMC base case (deterministic) | |||
LDAC | 142,094 | 0.67 | Reference |
Venetoclax + AZA | 408,139 | 1.23 | 470,079 vs. LDAC |
Ivosidenib + AZA | 985,719 | 1.71 | 1,206,919 vs. venetoclax + AZA |
AZA = azacitidine; CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; LDAC = low-dose cytarabine; QALY = quality-adjusted life-year; vs. = versus.
Note: Only treatments that are on the efficiency frontier are reported.
Scenario Analysis Results
CDA-AMC conducted scenario analysis to explore the impact of assuming that AML is not cured (i.e., removal of the 10-year time point at which AML was considered cured in the CADTH base case), of including increased myelosuppression health care resource use for venetoclax plus AZA, and of assuming no difference in efficacy (i.e., equivalent OS, EFS, and CR or CRi) between ivosidenib plus AZA and venetoclax plus AZA. In the scenario where no cure was assumed, the results were similar to the base-case analysis in that ivosidenib plus AZA was associated with more costs and QALYs (ICER = $1,148,877; incremental costs = $549,803; incremental QALYs = 0.48) compared to venetoclax plus AZA. Results were again similar to the base-case analysis in the scenario that included the increased myelosuppression health care resource use for venetoclax plus AZA, where ivosidenib plus AZA was associated with more costs and QALYs (ICER = $1,148,877; incremental costs = $549,803; incremental QALYs = 0.48) relative to venetoclax plus AZA. Lastly, in the scenario analysis where efficacy was assumed equal between ivosidenib plus AZA and venetoclax plus AZA, ivosidenib plus AZA was associated with more costs ($985,719 versus $555,003) and similar QALYs (1.71 versus 1.70), resulting in an ICER of $27,891,608 per QALY gained.
Results of price reduction analyses undertaken using the CDA-AMC base case suggest that there is no price of ivosidenib that would result in ivosidenib plus AZA being considered cost-effective relative to venetoclax plus AZA at a WTP threshold of $50,000 per QALY (Table 7). At a 100% price reduction for ivosidenib, ivosidenib plus AZA was associated with incremental costs of $78,603 compared to venetoclax plus AZA, resulting in an ICER of $164,250 per QALY gained. This incremental cost difference is primarily due to health care resource use (incremental cost: $45,932) as patients who received ivosidenib plus AZA were estimated to spend more time in EFS and progressed disease health states than patients who received venetoclax plus AZA in the CADTH base case.
Additional price reduction analyses were conducted to explore the price reduction of AZA that would be required for ivosidenib plus AZA to be cost-effective at a WTP of $50,000 per QALY (Table 7). For ivosidenib plus AZA to be considered cost-effective at this threshold compared to venetoclax plus AZA, the cost of ivosidenib would need to be reduced by approximately 99.7% and the cost of AZA would need to be reduced by 25%.
Table 7: CDA-AMC Price Reduction Analyses Versus Venetoclax Plus AZA
Price reduction | Unit drug cost ($) | ICERs for ivosidenib + AZA vs. venetoclax + AZA ($/QALY) | ||
|---|---|---|---|---|
Sponsor’s corrected base case | CDA-AMC reanalysis (assuming list price for AZA) | CDA-AMC reanalysis (assuming a 25% price reduction for AZA) | ||
No price reduction | 333 | 335,853 | 1,206,919 | 1,089,362 |
10% | 299 | 299,300 | 1,102,652 | 985,096 |
20% | 266 | 262,746 | 998,386 | 880,829 |
30% | 233 | 226,193 | 894,119 | 776,562 |
40% | 200 | 189,639 | 789,852 | 672,295 |
50% | 166 | 153,086 | 685,585 | 568,028 |
60% | 133 | 116,532 | 581,318 | 463,761 |
70% | 100 | 79,979 | 477,051 | 359,494 |
80% | 67 | 43,425 | 372,784 | 255,227 |
90% | 33 | 6,872 | 268,517 | 150,960 |
100% | 0 | Dominated | 164,250 | 46,693 |
AZA = azacitidine; CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Issues for Consideration
Genetic testing for IIDH1 R132 mutations may not be routinely performed for all people with AML in all jurisdictions within Canada. Should ivosidenib plus AZA be reimbursed, testing frequency may increase in some jurisdictions, which would result in higher costs to the health care system.
The sponsor’s analyses rely on publicly accessible list prices and do not reflect existing confidential prices negotiated by public plans. Venetoclax has previously received a positive recommendation from the CADTH pan-Canadian Oncology Review Expert Review Committee and has successfully undergone price negotiations with the pan-Canadian Pharmaceutical Alliance for the treatment of patients newly diagnosed with AML who are aged 75 years or older or who have comorbidities that preclude use of intensive induction chemotherapy. It is likely that the price paid by public drug plans for venetoclax is lower than the value used in the sponsor’s analyses.
Overall Conclusions
Based on the CDA-AMC clinical review, evidence from the AGILE trial suggests that ivosidenib plus AZA likely improves OS at 12 months and 24 months compared to placebo plus AZA in the indicated population. Evidence from the AGILE trial also suggests that ivosidenib plus AZA likely improves EFS at 6 months; however, the findings beyond 6 months were uncertain, and no evidence beyond 24 months was submitted for CADTH’s review. CADTH additionally notes that the impact of ivosidenib plus AZA versus AZA alone on health-related quality of life is very uncertain, with 95% confidence intervals that include the potential for no differences compared to placebo plus AZA.
There have been no direct head-to-head trials comparing ivosidenib plus AZA to currently available treatments other than AZA alone. To inform the economic model, the sponsor submitted indirect evidence comparing ivosidenib plus AZA to venetoclax plus AZA and to LDAC. Indirect evidence submitted by the sponsor was deemed insufficient to conclude whether treatment outcomes with ivosidenib plus AZA differ from those with venetoclax plus AZA in terms of OS, EFS, and CR or CRi because of limited efficacy data and important heterogeneity across studies. Health-related quality of life and harms were not assessed in the sponsor’s NMA.
In addition to the uncertainty in the clinical evidence, CDA-AMC identified several additional sources of uncertainty in the sponsor’s economic model. CADTH undertook reanalyses to address some of the identified limitations, which included adjusting the cure assumption, adopting alternative survival curves for OS and EFS for ivosidenib plus AZA, adopting alternative health state utility values for patients in EFS without CR/CRi, adopting health state–specific resource use, and using 100% RDI for drug acquisition costs. Results of the CADTH base case are aligned with the sponsor’s results: ivosidenib plus AZA is not a cost-effective option at a WTP threshold of $50,000 per QALY gained. In the CADTH base case, ivosidenib plus AZA was associated with an ICER of $1,206,919 per QALY gained compared to venetoclax plus AZA. Price reduction analyses conducted by CADTH suggest that there is no price for ivosidenib at which ivosidenib plus AZA would be cost-effective at a WTP threshold of $50,000 per QALY gained, primarily owing to increased health care resource use with ivosidenib plus AZA as a result of the longer time spent in the EFS and progressed disease health states compared to with venetoclax plus AZA. For ivosidenib plus AZA to be considered cost-effective compared to venetoclax plus AZA at a WTP threshold of $50,000 per QALY gained, the cost of ivosidenib would need to be reduced by approximately 99.7% and the cost of AZA would need to be reduced by 25%.
There have been no direct head-to-head trials comparing ivosidenib plus AZA with venetoclax plus AZA, and indirect evidence was insufficient to conclude whether treatment outcomes differ between ivosidenib plus AZA and venetoclax plus AZA. The CDA-AMC base case predicts a smaller incremental gain in QALYs than is predicted in the sponsor’s base case; however, these results are still predicated on improved EFS and OS with ivosidenib plus AZA compared to venetoclax plus AZA. If these gains are not realized in practice, it is likely that the CADTH base case underestimates the true ICER.
References
1.Tibsovo (ivosidenib): 250 mg, film-coated tablets, oral [product monograph]. Laval (QC): Servier Canada Inc.; 2024 Jul 19.
2.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Tibsovo (ivosidenib), 250 mg, film-coated tablets, oral. Laval (QC): Servier Canada Inc.; 2024 Mar 19.
3.Guertin JR, Feeny D, Tarride JE. Age- and sex-specific Canadian utility norms, based on the 2013-2014 Canadian Community Health Survey. CMAJ. 2018;190(6):E155-E161. PubMed
4.Statistics Canada. Canadian Community Health Survey - Annual Component (CCHS) [accessed by sponsor]. 2014; https://www23.statcan.gc.ca/imdb/p2SV.pl?Function=getSurvey&Id=164081.
5.DiNardo CD, Jonas BA, Pullarkat V, et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N Engl J Med. 2020;383(7):617-629. PubMed
6.CADTH Reimbursement Recommendation: Venetoclax (Venclexta). Can J Health Technol. 2021;1(8). https://www.cadth.ca/sites/default/files/DRR/2021/PC0238%20Venclexta%20(AZA)%20-%20Final%20Rec.pdf. Accessed 2023 Nov 8.
7.Wehler E, Storm M, Kowal S, Campbell C, Boscoe A. A Health State Utility Model Estimating the Impact of Ivosidenib on Quality of Life in Patients with Relapsed/Refractory Acute Myeloid Leukemia [accessed by sponsor]. 2018.
8.Beusterien KM, Davies J, Leach M, et al. Population preference values for treatment outcomes in chronic lymphocytic leukaemia: a cross-sectional utility study. Health Qual Life Outcomes. 2010;8:50. PubMed
9.Lachaine J, Mathurin K, Barakat S, Couban S. Economic evaluation of arsenic trioxide compared to all-trans retinoic acid + conventional chemotherapy for treatment of relapsed acute promyelocytic leukemia in Canada. Eur J Haematol. 2015;95(3):218-229. PubMed
10.Neumann PJ, Cohen JT. ICER's Revised Value Assessment Framework for 2017-2019: A Critique. Pharmacoeconomics. 2017;35(10):977-980. PubMed
11.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario drug benefit formulary/comparative drug index. 2024; https://www.formulary.health.gov.on.ca/formulary/. Accessed 2024 Mar 19.
12.Schedule of benefits for physician services under the Health Insurance Act: (June 29, 2023 (effective July 24, 2023)). Toronto (ON): Ontario Ministry of Health; 2023: https://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master.pdf. Accessed 2024 Mar 19.
13.Hu Y, Charaan M, van Oostrum I, Heeg B, Bell T. The cost-effectiveness of glasdegib in combination with low-dose cytarabine, for the treatment of newly diagnosed acute myeloid leukemia in adult patients who are not eligible to receive intensive induction chemotherapy in Canada. J Med Econ. 2021;24(1):150-161. PubMed
14.Canadian Institute for Health Information. Patient Cost Estimator [accessed by sponsor]. 2019; https://www.cihi.ca/en/patient-cost-estimator.
15.Workplace Safety and Insurance Board Ontario. Fee schedule: Nurse Practitioner. https://www.wsib.ca/en/fee-schedule-nurse-practitioner. Accessed 2024 Apr 12.
16.Yu M, Guerriere DN, Coyte PC. Societal costs of home and hospital end-of-life care for palliative care patients in Ontario, Canada. Health Soc Care Community. 2015;23(6):605-618. PubMed
17.Pratz KW, Jonas BA, Pullarkat V, et al. Long-term follow-up of VIALE-A: Venetoclax and azacitidine in chemotherapy-ineligible untreated acute myeloid leukemia. Am J Hematol. 2024;99(4):615-624. PubMed
18.Kumar S, Bennett A, Campbell PA, et al. Costs of Next-Generation Sequencing Assays in Non-Small Cell Lung Cancer: A Micro-Costing Study. Curr Oncol. 2022;29(8):5238-5246. PubMed
19.BCCA Protocol Summary for Therapy of Acute Myeloid Leukemia Using Low Dose Cytarabine (BC Cancer Agency Protocol Summary LKAMLCYT, Revised: 1 June 2021). Vancouver (BC): BC Cancer; 2021: http://www.bccancer.bc.ca/chemotherapy-protocols-site/Documents/Leukemia-BMT/LKAMLCYT_Protocol.pdf. Accessed 2024 Mar 19.
20.Clinical Study Report: AG120-C-009. A Phase 3, Multicenter, Double-Blind, Randomized, Placebo-Controlled Study of AG-120 in Combination with Azacitidine in Subjects ≥18 Years of Age with Previously Untreated Acute Myeloid Leukemia with an IDH1 Mutation [internal sponsor’s report]. Laval (QC): Servier Canada, Inc; 2021 Dec 8.
21.BC Cancer. Drug Funding [accessed by sponsor]. http://www.bccancer.bc.ca/health-professionals/clinical-resources/pharmacy/drug-funding.
22.Alberta Health Services. Outpatient Cancer Drug Benefit Program [accessed by sponsor]. https://www.albertahealthservices.ca/findhealth/Service.aspx?id=1025651.
23.Saskatchewan Cancer Agency. Frequently Asked Questions [accessed by sponsor]. http://www.saskcancer.ca/health-professionals-article/frequently-asked-questions-health-professionals.
24.Cancer Care Manitoba. Manitoba Home Cancer Drug Program [accessed by sponsor]. https://www.cancercare.mb.ca/Treatments/pharmacy/home-cancer-drug-program.
25.Sutherland G, Dinh T. Understanding the Gap: A Pan-Canadian Analysis of Prescription Drug Insurance Coverage [accessed by sponsor]. Conference Board of Canada; 2017: https://books.google.ca/books?id=4qh10AEACAAJ.
26.Statistics Canada. Number of new cases and age-standardized rates of primary cancer, by cancer type and sex. 2023; https://www150.statcan.gc.ca/t1/tbl1/en/cv.action?pid=1310074701. Accessed 2023 Oct 2.
27.Statistics Canada. Number and rates of new cases of primary cancer, by cancer type, age group and sex. 2023; https://www150.statcan.gc.ca/t1/tbl1/en/cv.action?pid=1310011101. Accessed 2023 Oct 2.
28.National Institute for Health and Care Excellence. Venetoclax with azacitidine for untreated acute myeloid leukaemia when intensive chemotherapy is unsuitable (NICE Technology appraisal guidance TA765). 2022; https://www.nice.org.uk/guidance/ta765/evidence. Accessed 2024 Mar 19.
29.Abbas S, Lugthart S, Kavelaars FG, et al. Acquired mutations in the genes encoding IDH1 and IDH2 both are recurrent aberrations in acute myeloid leukemia: prevalence and prognostic value. Blood. 2010;116(12):2122-2126. PubMed
30.Patel JP, Gonen M, Figueroa ME, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366(12):1079-1089. PubMed
31.Chotirat S, Thongnoppakhun W, Promsuwicha O, Boonthimat C, Auewarakul CU. Molecular alterations of isocitrate dehydrogenase 1 and 2 (IDH1 and IDH2) metabolic genes and additional genetic mutations in newly diagnosed acute myeloid leukemia patients. J Hematol Oncol. 2012;5:5. PubMed
32.Chou WC, Hou HA, Chen CY, et al. Distinct clinical and biologic characteristics in adult acute myeloid leukemia bearing the isocitrate dehydrogenase 1 mutation. Blood. 2010;115(14):2749-2754. PubMed
33.Marcucci G, Maharry K, Wu YZ, et al. IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol. 2010;28(14):2348-2355. PubMed
34.Mardis ER, Ding L, Dooling DJ, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361(11):1058-1066. PubMed
35.Bullinger L, Dohner K, Dohner H. Genomics of Acute Myeloid Leukemia Diagnosis and Pathways. J Clin Oncol. 2017;35(9):934-946. PubMed
36.Paschka P, Schlenk RF, Gaidzik VI, et al. IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. J Clin Oncol. 2010;28(22):3636-3643. PubMed
Appendix 1: Cost Comparison Table
Please note that this appendix has not been copy-edited.
The comparators presented in Table 8 have been deemed to be appropriate based on feedback from clinical expert(s). Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 8: CDA-AMC Cost Comparison Table for Acute Myeloid Leukemia
Treatment | Strength / concentration | Form | Price | Recommended dosagea | Daily cost ($) | 28-day cycle cost ($) |
|---|---|---|---|---|---|---|
Ivosidenib (Tibsovo) | 250 mg | Tablet | 332.6000b | 500 mg orally once daily | 665.20 | 18,626 |
Azacitidine | 100 mg | Vial for powdered suspension | 599.9900 | 75 mg/m2 IV or SC once daily on days 1 to 7 of each 28-day cycle | 300.00 | 8,400 |
Ivosidenib + azacitidine | 965.20 | 27,025 | ||||
Venetoclax plus azacitidine | ||||||
Venetoclax (Venclexta) | 10 mg 50 mg 100 mg | Tablet | 7.0800 35.4000 70.8000 | 100 mg on day 1; 200 mg on day 2; 400 mg on day 3; 400 mg on day 4 and onwards | Cycle 1: 270.56 Cycle 2+: 282.20 | Cycle 1: 7,576 Cycle 2+: 7,930 |
Azacitidine | 100 mg | Vial for powdered suspension | 599.9900 | 75 mg/m2 IV or SC once daily on days 1 to 7 of each 28-day cycle | 300.00 | 8,400 |
Venetoclax plus azacitidine | Cycle 1: 571.55 Cycle 2+: 583.20 | Cycle 1: 15,975 Cycle 2+: 16,329 | ||||
Venetoclax plus LDAC | ||||||
Venetoclax (Venclexta) | 10 mg 50 mg 100 mg | Tablet | 7.0800 35.4000 70.8000 | 100 mg on day 1; 200 mg on day 2; 400 mg on day 3; 400 mg on day 4 and onwards | Cycle 1: 270.56 Cycle 2+: 282.20 | Cycle 1: 7,576 Cycle 2+: 7,930 |
Low-dose cytarabine | 100 mg/mL (5 mL vial) | Injectable solution | 76.8500 (15.3700 per mL) | 20 mg twice daily for 10 days every 4 to 6 weeksc | 27.45 | 769 |
100 mg/mL (20 mL vial) | Injectable solution | 306.5000 (15.3250 per mL) | ||||
Venetoclax plus low-dose cytarabine | Cycle 1: 298.01 Cycle 2+: 309.65 | Cycle 1: 8,345 Cycle 2+: 8,699 | ||||
Nonintensive chemotherapies | ||||||
Azacitidine | 100 mg | Vial for powdered suspension | 599.9900 | 75 mg/m2 IV or SC once daily on days 1 to 7 of each 28-day cycle | 300.00 | 8,400 |
Low-dose cytarabine | 100 mg/mL (5 mL vial) | Injectable solution | 76.8500 (15.3700 per mL) | 20 mg twice daily for 10 days every 4 to 6 weeksc | 27.45 | 769 |
100 mg/mL (20 mL vial) | Injectable solution | 306.5000 (15.3250 per mL) | ||||
CDA-AMC = Canada’s Drug Agency.
Note: All prices are from the IQVIA DeltaPA (accessed April 2024), unless otherwise indicated, and do not include dispensing fees. Calculations were informed assuming a patient body surface area of 1.8 m2.
aDosing information as informed by respective product monographs, unless otherwise stated.
bSponsor-submitted price.
cLow-dose cytarabine dosing as per British Columbia Cancer Agency protocol; note costing calculations assume doses every 4 weeks.19
Appendix 2: Submission Quality
Please note that this table has not been copy-edited.
Description | Yes or no | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | Refer to CDA-AMC critical appraisal. |
Model has been adequately programmed and has sufficient face validity | No | Refer to CDA-AMC critical appraisal. |
Model structure is adequate for decision problem | No | Refer to CDA-AMC critical appraisal. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | No | Refer to CDA-AMC critical appraisal. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | Yes | No comment. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | No | CDA-AMC identified several discrepancies between the sponsor-submitted model and report with regard to methodology used and inputs. For example, the reported utility values in the sponsor-submitted economic report did not align with those used within the economic model and until clarification made via an additional information request to the sponsor, it was not documented in their economic report that the cure assumption only applied to ivosidenib plus AZA and venetoclax plus AZA. |
CDA-AMC = Canada’s Drug Agency.
Appendix 3: Additional Information on the Submitted Economic Evaluation
Please note that this appendix has not been copy-edited.
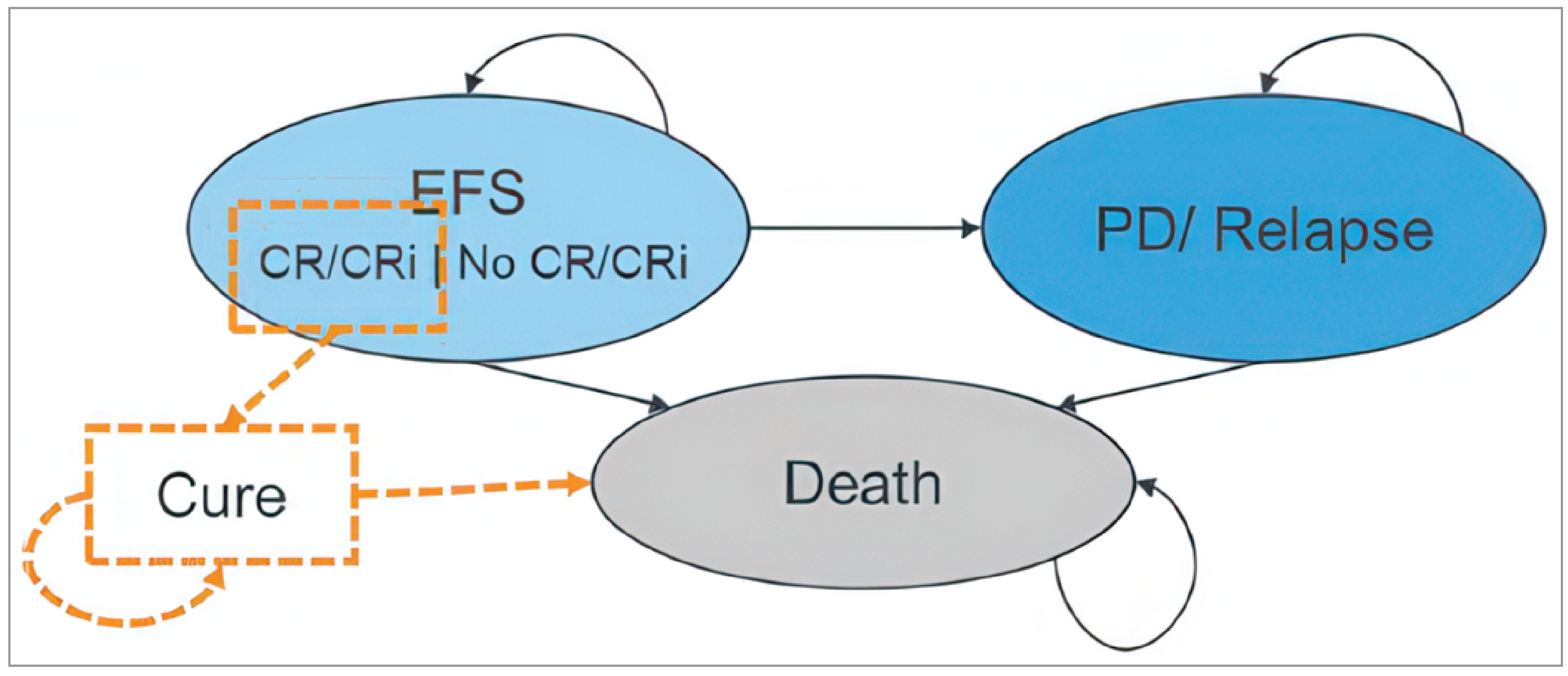
Detailed Results of the Sponsor’s Base Case
Table 10: Summary of the Sponsor’s Disease Management and Monitoring Costs per Cycle, by Health State and Treatment
Treatment | EFS, CR/CRi ($) | EFS, no CR/CRi ($) | Progressed disease ($) |
|---|---|---|---|
Ivosidenib plus AZA | 3,411 | 5,416 | 6,937 |
AZA | 3,998 | 6,406 | 6,937 |
Venetoclax plus AZA | 5,592 | 8,879 | 6,937 |
LDAC | 3,998 | 6,406 | 6,937 |
AZA = azacitidine; CR = complete remission; CRi = complete remission with incomplete hematologic recovery; EFS = event-free survival; IVO = ivosidenib; LDAC = low-dose cytarabine; VEN = venetoclax.
Note: One cycle = 28 days.
Source: Sponsor’s pharmacoeconomic submission.2
Table 11: Disaggregated Summary of the Sponsor’s Economic Evaluation Probabilistic Results
Parameter | IVO + AZA | AZA | VEN + AZA | LDAC |
|---|---|---|---|---|
Discounted LYs | ||||
Total LYs | 6.15 | 1.73 | 4.26 | 1.35 |
EFS, CR/CRi | 1.14 | 0.10 | 0.58 | 0.05 |
EFS, no CR/CRi | 1.97 | 0.53 | 1.09 | 0.61 |
Cured | 1.24 | 0.00 | 0.41 | 0.00 |
Progressed disease | 1.72 | 1.09 | 2.09 | 0.68 |
Cured (SCT)a | 0.08 | 0.01 | 0.09 | 0.02 |
Discounted QALYs | ||||
Total QALYs | 3.50 | 0.95 | 2.36 | 0.75 |
EFS, CR/CRi | 0.79 | 0.07 | 0.41 | 0.03 |
EFS, no CR/CRi | 1.04 | 0.30 | 0.59 | 0.34 |
Cured | 0.75 | 0.00 | 0.25 | 0.00 |
Progressed disease | 0.89 | 0.60 | 1.10 | 0.37 |
Cured (SCT) a | 0.05 | 0.01 | 0.06 | 0.01 |
Loss from AE | –0.02 | –0.02 | –0.04 | –0.01 |
Discounted costs ($) | ||||
Total cost | 1,131,486 | 281,131 | 651,723 | 165,088 |
Drug acquisition | 664,427 | 65,669 | 208,411 | 1,153 |
Drug administration | 406 | 3,299 | 406 | 953 |
Concomitant medication | 2,763 | 2,614 | 2,329 | 2,117 |
Subsequent treatment | 22,818 | 18,354 | 26,759 | 8,573 |
AE management | 10,327 | 12,387 | 14,506 | 6,713 |
Resource use | 430,745 | 178,808 | 399,313 | 145,580 |
mIDH1 testing cost | 12,770 | 12,770 | 12,770 | 12,770 |
Monitoring, EFS | 209,202 | 49,243 | 168,351 | 53,334 |
Monitoring, cure from remission | 30,031 | 0 | 9,854 | 0 |
Monitoring, on treatment | 3,320 | 0 | 0 | 0 |
Monitoring, progressed disease | 156,014 | 98,846 | 188,896 | 61,325 |
Monitoring, cure from SCT | 2,036 | 291 | 2,301 | 426 |
End of life | 17,372 | 17,658 | 17,141 | 17,725 |
AE = adverse event; AZA = azacitidine; CR = complete remission; CRi = complete remission with incomplete hematologic recovery; EFS = event-free survival; IVO = ivosidenib; LDAC = low-dose cytarabine; LY = life-year; mIDH1 = mutant isocitrate dehydrogenases 1; QALY = quality-adjusted life-year; SCT = stem cell transplant; VEN = venetoclax.
aIncludes patients who received SCT as subsequent treatment and deemed cured at the 5-year post-SCT time point.
Source: Sponsor’s pharmacoeconomic submission (probabilistic results).2
Appendix 4: Additional Details on the CDA-AMC Reanalyses and Sensitivity Analyses of the Economic Evaluation
Please note that this appendix has not been copy-edited.
Detailed Results of CDA-AMC Base Case
Table 12: Summary of the Stepped Analysis of the CDA-AMC Base-Case Results
Stepped analysis | Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|---|
Sponsor base case (deterministic) | LDAC | 159,317 | 0.72 | Reference |
AZA | 254,226 | 0.91 | Extendedly dominated | |
Venetoclax plus AZA | 611,860 | 2.15 | 315,970 | |
Ivosidenib plus AZA | 952,714 | 3.17 | 335,853 | |
Sponsor’s corrected base case (deterministic) | LDAC | 163,865 | 0.72 | Reference |
AZA | 254,226 | 0.91 | Extendedly dominated | |
Venetoclax plus AZA | 611,860 | 2.15 | 312,795 | |
Ivosidenib plus AZA | 952,714 | 3.17 | 335,853 | |
CDA-AMC reanalysis 1: Cure assumption | LDAC | 163,865 | 0.72 | Reference |
AZA | 254,226 | 0.91 | Extendedly dominated | |
Venetoclax plus AZA | 654,403 | 2.08 | 361,044 | |
Ivosidenib plus AZA | 1,151,180 | 3.05 | 511,301 | |
CDA-AMC reanalysis 2: Extrapolation of EFS | LDAC | 165,007 | 0.72 | Reference |
AZA | 256,144 | 0.90 | Extendedly dominated | |
Venetoclax plus AZA | 582,861 | 2.06 | 310,730 | |
Ivosidenib plus AZA | 1,064,553 | 3.13 | 453,911 | |
CDA-AMC reanalysis 3: Extrapolation of OS | LDAC | 141,288 | 0.60 | Reference |
AZA | 209,551 | 0.68 | Extendedly dominated | |
Venetoclax plus AZA | 451,706 | 1.32 | 430,412 | |
Ivosidenib plus AZA | 761,513 | 1.96 | 486,272 | |
CDA-AMC reanalysis 4: Health state utility values | LDAC | 163,865 | 0.80 | Reference |
AZA | 254,226 | 0.97 | Extendedly dominated | |
Venetoclax plus AZA | 611,860 | 2.28 | 301,748 | |
Ivosidenib plus AZA | 952,714 | 3.39 | 308,112 | |
CDA-AMC reanalysis 5: Health care resource use for myelosuppression | LDAC | 163,891 | 0.72 | Reference |
AZA | 254,244 | 0.91 | Extendedly dominated | |
Venetoclax plus AZA | 567,695 | 2.15 | 281,940 | |
Ivosidenib plus AZA | 968,414 | 3.17 | 394,841 | |
CDA-AMC reanalysis 6: RDI | LDAC | 163,981 | 0.72 | Reference |
AZA | 259,721 | 0.91 | Extendedly dominated | |
Venetoclax plus AZA | 641,234 | 2.15 | 333,223 | |
Ivosidenib plus AZA | 1,024,120 | 3.17 | 377,269 | |
CDA-AMC base case (reanalysis 1 + 2 + 3 + 4 + 5 + 6; deterministic) | LDAC | 142,094 | 0.67 | Reference |
AZA | 216,470 | 0.75 | Extendedly dominated | |
Venetoclax plus AZA | 408,139 | 1.23 | 470,079 | |
Ivosidenib plus AZA | 985,719 | 1.71 | 1,206,919 |
AZA = azacitidine; CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; LDAC = low-dose cytarabine; QALY = quality-adjusted life-year; RDI = relative dose intensity.
Note: The CDA-AMC reanalysis is based on publicly available prices of the comparator treatments. The results of all steps are presented deterministically unless otherwise indicated.
Table 13: Disaggregated Summary of CDA-AMC’s Economic Evaluation Deterministic Results
Parameter | IVO + AZA | AZA | VEN + AZA | LDAC |
|---|---|---|---|---|
Discounted LYs | ||||
Total LYs | 2.78 | 1.22 | 2.04 | 1.07 |
EFS, CR/CRi | 0.90 | 0.09 | 0.45 | 0.04 |
EFS, no CR/CRi | 0.76 | 0.46 | 0.58 | 0.52 |
Cured | 0.01 | 0.00 | 0.00 | 0.00 |
Progressed disease | 1.09 | 0.67 | 1.01 | 0.50 |
Cured (SCT)a | 0.02 | 0.00 | 0.01 | 0.00 |
Discounted QALYs | ||||
Total QALYs | 1.77 | 0.75 | 1.26 | 0.67 |
EFS, CR/CRi | 0.63 | 0.06 | 0.32 | 0.03 |
EFS, no CR/CRi | 0.53 | 0.33 | 0.41 | 0.37 |
Cured | 0.01 | 0.00 | 0.00 | 0.00 |
Progressed disease | 0.61 | 0.39 | 0.57 | 0.29 |
Cured (SCT) a | 0.01 | 0.00 | 0.01 | 0.00 |
Loss from AE | –0.02 | –0.02 | –0.04 | –0.01 |
Discounted costs ($) | ||||
Total cost | 1,018,914 | 218,243 | 414,459 | 143,282 |
Drug acquisition | 748,246 | 55,044 | 191,211 | 5,264 |
Drug administration | 406 | 2,488 | 406 | 987 |
Concomitant medication | 2,513 | 2,559 | 2,207 | 2,101 |
Subsequent treatment | 14,165 | 11,219 | 12,563 | 6,249 |
AE management | 10,327 | 12,387 | 14,506 | 6,713 |
Resource use | 243,256 | 134,546 | 193,566 | 121,968 |
mIDH1 testing cost | 12,770 | 12,770 | 12,770 | 12,770 |
Monitoring, EFS | 110,815 | 43,226 | 71,822 | 45,933 |
Monitoring, cure from remission | 239 | 0 | 5 | 0 |
Monitoring, on treatment | 2,275 | 0 | 0 | 0 |
Monitoring, progressed disease | 98,910 | 60,750 | 90,967 | 45,459 |
Monitoring, cure from SCT | 461 | 11 | 214 | 17 |
End of life | 17,785 | 17,789 | 17,789 | 17,789 |
AE = adverse event; AZA = azacitidine; CDA-AMC = Canada’s Drug Agency; CR = complete remission; CRi = complete remission with incomplete hematologic recovery; EFS = event-free survival; IVO = ivosidenib; LDAC = low-dose cytarabine; LY = life-year; mIDH1 = mutant isocitrate dehydrogenases 1; QALY = quality-adjusted life-year; RL = relapsed; SCT = stem cell transplant; VEN = venetoclax.
aIncludes patients who received SCT as subsequent treatment and deemed cured at the 5-year post-SCT time point.
Scenario Analyses
Table 14: Summary of CDA-AMC Scenario Analyses
Scenario | Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|---|
CDA-AMC base case | LDAC | 142,094 | 0.67 | Reference |
AZA | 216,470 | 0.75 | Extendedly dominated | |
Venetoclax plus AZA | 408,139 | 1.23 | 470,079 | |
Ivosidenib plus AZA | 985,719 | 1.71 | 1,206,919 | |
CDA-AMC scenario analysis: no cure | LDAC | 142,094 | 0.67 | Reference |
AZA | 216,470 | 0.75 | Extendedly dominated | |
Venetoclax plus AZA | 408,145 | 1.23 | 470,147 | |
Ivosidenib plus AZA | 1,007,855 | 1.71 | 1,261,300 | |
CDA-AMC scenario analysis: 1.5 × more myelosuppression resource use for venetoclax plus AZA | LDAC | 142,094 | 0.67 | Reference |
AZA | 216,470 | 0.75 | Extendedly dominated | |
Venetoclax plus AZA | 435,915 | 1.23 | 519,158 | |
Ivosidenib plus AZA | 985,719 | 1.71 | 1,148,877 | |
CDA-AMC scenario analysis: equal efficacy between Ivosidenib plus AZA and venetoclax plus AZA | LDAC | 142,094 | 0.67 | Reference |
AZA | 216,470 | 0.75 | Extendedly dominated | |
Venetoclax plus AZA | 555,003 | 1.70 | 401,244 | |
Ivosidenib plus AZA | 985,719 | 1.71 | 27,891,608 |
AZA = azacitidine; CDA-AMC = Canada’s Drug Agency; LDAC = low-dose cytarabine; QALY = quality-adjusted life-year.
Note: All analyses were conducted deterministically.
Appendix 5: Submitted Budget Impact Analysis and CDA-AMC Appraisal
Please note that this appendix has not been copy-edited.
Table 15: Summary of Key Take Aways
Key Take Aways of the Budget Impact Analysis |
|---|
|
Summary of Sponsor’s BIA
The sponsor submitted a BIA to estimate the three-year budget impact of reimbursing ivosidenib plus AZA for the treatment of adult patients with newly diagnosed AML with an IIDH1 R132 mutation who are not eligible to receive intensive induction chemotherapy. The analysis was taken from the perspective of the Canadian public drug plan over a three-year time horizon (2025 to 2027). The target population size was derived using an epidemiological approach and included drug acquisition costs. Key inputs to the BIA are documented in Table 16.
The sponsor compared a reference scenario in which patients received venetoclax plus AZA, AZA alone, or LDAC to a new drug scenario in which ivosidenib plus AZA was also available. In the new drug scenario, uptake of ivosidenib plus AZA was assumed to be 25%, 50%, and 75% in year 1, year 2, and year 3, respectively, based on internal estimates made by the sponsor. The sponsor assumed that ivosidenib plus AZA would capture market share proportionally from all comparators. Complete coverage was assumed for patients residing in British Columbia, Alberta, Saskatchewan, Manitoba. For the remaining jurisdiction, age distribution of AML patients not eligible to receive intensive induction chemotherapy was assumed to align with the AGILE study population (5.5% < 65 years old; 94.5% ≥ 65 years old) where patients aged ≥ 65 years old were assumed to be eligible for coverage and those that were < 65 years old were eligible based on rates published by the Conference Board of Canada.20-25 Wastage and administration costs were not included.
Table 16: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (year 1 / year 2 / year 3) |
|---|---|
Target population | |
Population of Canada AML Incidence % Adults % Eligible for Drug Plan Coverage % Ineligible for Intensive Induction Chemotherapy % with IDH1 mutation % Receiving Systemic Therapy | 31,498,616 / 31,924,211 / 32,355,777 0.0041% / 0.0042% / 0.0043%26 96.9%27 97.9% / 98.0% / 98.0%a 40%28 100% |
Number of patients eligible for drug under review | 40 / 41 / 42 |
Market Uptake (3 years) | |
Uptake (reference scenario) Ivosidenib plus AZA AZA Venetoclax plus AZA LDAC | 0.0% / 0.0% / 0.0% 95.0% / 95.0% / 95.0% 2.5% / 2.5% / 2.5% 2.5% / 2.5% / 2.5% |
Uptake (new drug scenario) Ivosidenib plus AZA AZA Venetoclax plus AZA LDAC | 25.0% / 50.0% / 75.0% 71.3% / 47.5% / 23.8% 1.9% / 1.3% / 0.6% 1.9% / 1.3% / 0.6% |
Cost of treatment (per patient, per 28-day cycle) | |
Ivosidenib plus AZA AZA Venetoclax plus AZA LDAC | $27,025 $16,329 $8,400 $769 |
AML = acute myeloid leukemia; AZA = azacitidine; LDAC = low-dose cytarabine.
aThe sponsor assumed 100% coverage in British Columbia, Alberta, Saskatchewan, and Manitoba. For jurisdictions without 100% coverage, the sponsor assumed that the age distribution of AML patients not eligible to receive intensive induction chemotherapy would be aligned with the AGILE study population (94.5% ≥ 65 years old) and that patients aged 65 years or older would be eligible for public drug plan coverage, while those aged less < 65 years old would have coverage rates based on rates published by the Conference Board of Canada.20-25
Summary of the Sponsor’s BIA Results
The sponsor estimated the 3-year budget impact of reimbursing ivosidenib plus AZA for the treatment of adult patients with newly diagnosed AML with an IIDH1 R132 mutation who are not eligible to receive intensive induction chemotherapy to be $16,245,416 (year 1: $1,399,495; year 2: $5,338,176; year 3: $9,507,745).
CDA-AMC Appraisal of the Sponsor’s BIA
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Exclusion of relevant comparators. As per the Health Canada indication and the sponsor’s submitted reimbursement request, the submitted budget impact model for ivosidenib plus AZA is indicated for the treatment of adults with newly diagnosed AML with an IIDH1 R132 mutation who are not eligible to receive intensive induction chemotherapy. Clinical expert feedback received by CDA-AMC noted that while venetoclax plus AZA is the most relevant comparator, other comparators for this patient population include venetoclax plus LDAC and BSC. Clinical expert feedback received by CDA-AMC indicated that the proportion of patients on these treatments may be low; however, they are still considered relevant for the budgetary analysis.
CDA-AMC was unable to address this limitation.
The proportion of patients with AML with an IIDH1 R132 mutation is uncertain. The sponsor estimated that 8% of patients had an IIDH1 R132 mutation based on using the midpoint of estimates from published literature,29-33,35,36 while in their CUA they adopted an estimate of 10%. Clinical expert feedback received by CDA-AMC acknowledged that there is some uncertainty in the estimate for the proportion of patients with the IIDH1 R132 mutation. CADTH notes that, of the patients screened for eligibility in the AGILE trial, approximately 61% were positive for the IIDH1 R132 mutation; however, this may not reflect the prevalence of the IDH1 mutation in Canadian practice owing to selection of the trial population.
To explore the impact of alternative inputs, CDA-AMC conducted a scenario analysis estimating that 10% of patients had an IIDH1 R132 mutation.
The uptake of ivosidenib plus AZA is likely underestimated. Ivosidenib is the first targeted treatment for the IIDH1 R132 mutation in this population. As such, clinical expert feedback obtained by CDA-AMC suggests that, should ivosidenib be reimbursed by the public drug plans, the uptake of ivosidenib plus AZA will likely be higher than expected by the sponsor. The sponsor’s estimated uptake of 75% by eligible patients by year 3 of the analysis was based on the sponsor’s internal estimates and was thought to be underestimated by clinical experts consulted by CDA-AMC. Expert input indicated that the uptake of ivosidenib plus AZA by year 3 likely to be in the range of 90% of eligible patients.
CDA-AMC addressed this limitation by setting the market share for ivosidenib plus AZA equal to 25%, 75% and 90% in years 1, 2, and 3, respectively.
CDA-AMC Reanalyses of the BIA
CDA-AMC revised the sponsor’s submitted analysis by modifying the expected market uptake of ivosidenib plus AZA. The changes made to derive the CADTH base case are described in Table 17.
The results of the CDA-AMC step-wise reanalysis are presented in summary format in Table 18 and a more detailed breakdown is presented in Table 19. In the CADTH base case, the 3-year budget impact of reimbursing ivosidenib plus AZA for the treatment of adult patients with newly diagnosed AML with an IIDH1 R132 mutation who are not eligible to receive intensive induction chemotherapy is expected to be $21,105,093 (year 1: $1,399,495; year 2: $6,778,829; year 3: $12,926,769).
CDA-AMC conducted a scenario analysis to explore uncertainty in the proportion of patients with an IIDH1 R132 mutation, using the CADTH base case (Table 19).
Table 17: CDA-AMC Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Changes to derive the CDA-AMC base case | ||
1. Ivosidenib plus AZA market share | Year 1: 25% Year 2: 50% Year 3: 75% | Year 1: 25% Year 2: 75% Year 3: 90% |
CDA-AMC base case | Reanalysis 1 | |
AZA = azacitidine; CDA-AMC = Canada’s Drug Agency.
Table 18: Summary of the CDA-AMC Reanalyses of the Budget Impact Analysis
Stepped analysis | 3-year total ($) |
|---|---|
Submitted base case | 16,245,416 |
CDA-AMC reanalysis 1 | 21,105,093 |
CDA-AMC base case | 21,105,093 |
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency.
Table 19: Detailed Breakdown of the CDA-AMC Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | 3-year total ($) |
|---|---|---|---|---|---|---|
Submitted base case | Reference | 8,115,438 | 8,971,165 | 9,234,933 | 9,507,386 | 27,713,484 |
New drug | 8,115,438 | 10,370,660 | 14,573,109 | 19,015,131 | 43,958,900 | |
Budget impact | 0 | 1,399,495 | 5,338,176 | 9,507,745 | 16,245,416 | |
CDA-AMC base case | Reference | 8,115,438 | 8,971,165 | 9,234,933 | 9,507,386 | 27,713,484 |
New drug | 8,115,438 | 10,370,660 | 16,013,762 | 22,434,155 | 48,818,577 | |
Budget impact | 0 | 1,399,495 | 6,778,829 | 12,926,769 | 21,105,093 | |
CDA-AMC scenario analysis: 10% of patients have an IIDH1 R132 mutation | Reference | 10,144,297 | 11,213,956 | 11,543,666 | 11,884,233 | 34,641,855 |
New drug | 10,144,297 | 12,963,325 | 20,017,202 | 28,042,694 | 61,023,221 | |
Budget impact | 0 | 1,749,369 | 8,473,536 | 16,158,461 | 26,381,366 |
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency.
Testing Procedure Assessment
Abbreviations
AML
acute myeloid leukemia
NGS
next-generation sequencing
PCR
polymerase chain reaction
Objective
The objective of this Testing Procedure Assessment is to identify and describe important health system implications of testing for IIDH1 R132 mutations in adult patients with newly diagnosed acute myeloid leukemia (AML) who are not eligible to receive intensive induction chemotherapy, which is the proposed indication for ivosidenib in combination with azacitidine.
Methods
Contents within this section have been informed by materials submitted by the sponsor, a literature search, and clinical expert input. Materials submitted by the sponsor related to companion diagnostic testing for IIDH1 R132 mutations were validated and summarized by the review team. The clinical expert input was provided by 2 clinical specialists with expertise in the diagnosis and management of AML.
An information specialist conducted a literature search on key resources including MEDLINE, the Cochrane Database of Systematic Reviews, the International HTA Database, and the websites of Canadian and major international health technology agencies, as well as a focused internet search. The search approach was customized to retrieve a limited set of results, balancing comprehensiveness with relevancy. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concepts were next generation sequence testing and AML. Secondary searches were conducted with search filters applied to limit retrieval to citations related to economics and equity considerations. The search was completed on April 17, 2024, and limited to English-language documents published since January 1, 2019. We conducted handsearching to identify additional information on genetic testing and AML more broadly.
Context
What Are IIDH1 R132 Mutations?
AML is a clonal disease caused by genetic mutation, leading to altered self-renewal, differentiation, and proliferation of myeloid hematopoietic progenitor cells.1 In AML, mutations in the IDH1 gene occur at conserved arginine residues within the enzymatic active site, specifically at the R132 codon.2 An IIDH1 R132 mutation in people with AML causes an overproduction of 2-hydoxyglutarate, which impairs the differentiation of immature hematopoietic cells into mature blood cells, contributing to oncogenesis.3 Multiple IDH1 amino acid changes related to the R132 codon have been identified, including R132C, R132G, R132H, R132L, and R132S.4 R132H and R132C are the most prevalent mutations in IDH1, occurring in more than 50% of people with IDH1 mutation–positive AML.4 According to the clinical experts consulted for this review, it is estimated that approximately 6% to 10% of people with AML have an IDH1 mutation.1,5 The age-adjusted incidence rate of IDH1 mutation–positive AML is less than 1 per 100,000 individuals per year.5 As per the intended indication of ivosidenib, adults with IIDH1 R132 mutation–positive AML who are not eligible to receive intensive induction chemotherapy represent less than 5% of the total population of people with AML.5
Why Test for IIDH1 R132 Mutations?
Ivosidenib is an IIDH1 R132–targeted therapy. It is a selective and orally active small molecule inhibitor of the IDH1 enzyme that supresses 2-hydoxyglutarate production and restores differentiation of the malignant cells.3 AML is considered the most aggressive form of leukemia, and according to the National Comprehensive Cancer Network guidelines and the Canadian Cancer Society, first-line treatment should be initiated promptly to improve patient outcomes.6-8 Thus, identifying people with AML who have an IIDH1 R132 mutation in an efficient and timely manner has potentially significant health impacts for individuals who may benefit from IIDH1 R132–targeted therapy.
How Are IIDH1 R132 Mutations Identified in People With AML?
There are 2 pathways for the identification of IIDH1 R132 mutation in people with AML. Next-generation sequencing (NGS) is currently the standard of care testing for the identification of all AML-associated oncogenic driver mutation, including IDH1 mutations,9 while polymerase chain reaction (PCR) testing can be used to identify specific single nucleotide variants of the IIDH1 R132 codon. For a patient to receive IIDH1 R132–targeted therapy, such as ivosidenib, an IIDH1 R132 mutation would need to be confirmed using NGS or a PCR test. Both methods of testing requires blood or bone marrow samples collected for IDH1 mutation diagnosis.
NGS testing in relation to AML is used for subclassification diagnostics of specific entities that have unique causality, prognostic, and therapeutic implications.9 NGS testing may be conducted during routine AML diagnosis to better prognosticate and risk-stratify the AML based on detected mutations.5 Additionally, NGS is used to assess baseline co-mutations. Identifying potential co-mutations can help determine a patient’s likely clinical response to treatment.1,3
PCR assay testing is also used to identify IDH1 mutations. The PCR test detects specific single nucleotide variants of the IIDH1 R132 codon among its 5 most common versions (C, G, H, L, and S) by using PCR technology with homogenous real-time fluorescence detection.3 Identification of the specific single nucleotide variant of IIDH1 R132 by PCR may confirm the IDH1 mutation, inform treatment pathways, and provide insight into a patient’s likely response to treatment.
What Are NGS and PCR Testing?
NGS is a massively parallel sequencing technology that allows for rapid, precise, and cost-effective sequencing of multiple genes, whole exomes, and genomes.1 NGS can provide for accuracy in variant classification, prognostic stratification, and treatment and response assessment in AML diagnosis.1 In recent years, NGS has become the gold standard for detecting the AML mutations that define AML subtypes.9 The relative disadvantages of NGS include technical complexity; relatively long turnaround times, which may range from hours to days and may complicate urgent, targeted therapeutic decision-making; the need for bioinformatics expertise and specialized variant expertise; upfront instrument costs; and the potential for identifying “unintended” variants of unknown significance, thus complicating therapeutic decision-making.1,9 The sample quality and the time needed to run the sequencing may also be impacted by the regions of interest sequenced, the sample number, the read size, the number of megabases in the panel, the quality control methods applied, and the sequencing platform used.1
PCR testing is a laboratory test used to amplify particular gene segments to determine specific gene variants.6 PCR testing is reported to be useful in accurately diagnosing and determining the prognosis and therapeutic disease management for single-gene mutations associated with leukemia.6,9 To optimize PCR testing performance, appropriate sample collection, handling, preparation, and storage is required, and any tampering with the sample may impact the accuracy of the results.4 Additionally, personnel who have appropriate training in molecular diagnostic assay procedures are required to analyze the testing sample.4 The relative disadvantage of PCR testing is that it cannot be used to analyze multiple genes; thus, PCR testing may be used to confirm suspected single-gene mutations or single-gene mutations identified using NGS.1
How Is AML Diagnosed in Canada?
A person with suspected AML typically requires multiple evaluations, including clinical and laboratory testing and pathology testing, to confirm an AML diagnosis. Testing for AML typically begins with an evaluation of the person’s history, a complete blood count with leukocyte differential count, a review of a blood smear, and comprehensive metabolic panelling.6,10 Pathology testing includes bone marrow or blood aspirate, which are evaluated by microscopy, flow cytometry, and cytogenetic or molecular evaluations.6,10 Additional imaging of the central nervous system and a lumbar puncture may be required for unexplained neurologic abnormalities.10 The diagnosis of AML requires demonstration of myeloid blasts by microscopy, immunophenotyping, and cytogenetic findings according to either the International Consensus Classification or the WHO Classification of Tumours, 5th edition, criteria.10 Testing for AML-associated genetic mutations may be used to risk-stratify patients and identify specific mutations, such as IIDH1 R132, to help inform patient prognosis and clinical decision-making.1,3
Findings
Health System Considerations
What Is the Availability of IIDH1 R132 Testing in Canada?
The availability of testing for IIDH1 R132 mutations in people with AML varies across jurisdictions within Canada. For example, according to the clinical experts consulted for this review, NGS testing that includes IDH1 is performed to risk-stratify patients as part of routine diagnostic practice in AML treatment centres in Ontario, but IIDH1 R132 testing is not currently undertaken at diagnosis for people with AML in Manitoba. Information related to the availability of testing for IIDH1 R132 mutations in people with AML in other provinces and territories is insufficient or uncertain, but the clinical experts’ input suggests that it might depend on whether IIDH1 R132–targeted therapies are funded within the jurisdiction of interest. It is possible that IDH1 mutation testing may be implemented more broadly if IIDH1 R132–targeted therapy, such as ivosidenib, were to be funded in Canada.
How Many Individuals in Canada Would Be Expected to Require the Testing Procedure?
AML is the most common form of acute leukemia among adults.11 The most recent Canadian estimates suggest that approximately 1,160 new cases of AML were diagnosed in 2019.11 According to the clinical experts and depending on jurisdictional availability, each person suspected to have AML would be tested for IDH1 mutations as part of routine AML stratification efforts. According to the clinical experts, approximately 6% to 10% of people with diagnosed AML have an IDH1 mutation and may be eligible for IIDH1 R132–targeted therapy.1,7
What Is the Expected Timing and Frequency of IIDH1 R132 Testing?
According to the clinical experts, testing for IDH1 mutations is part of routine diagnostic practice for people in jurisdictions with testing availability. In line with the National Comprehensive Cancer Network guideline,8 the clinical experts recommended that the turnaround time to confirm the status of an IIDH1 R132 mutation should be approximately 3 to 5 days. Rapid testing response time is important specifically for people with AML who are suspected to have an IIDH1 R132 mutation because identifying patients that are likely to benefit from first-line IIDH1 R132–targeted therapy could improve overall survival outcomes and avoid unnecessary exposure to treatment that is not specific to their mutation status. One clinical expert indicated that a testing sample is only needed to be collected once and, in most cases, any subsequent confirmatory IIDH1 R132 mutation testing can be done using the original blood or bone marrow sample. This distinction in sample collection can also expedite the process if additional confirmatory testing is needed. Additionally, 1 clinical expert indicated that in most cases there are no safety concerns associated with initiating IIDH1 R132–targeted therapy before receiving testing confirmation results; however, no literature was identified to support this statement. One clinical expert indicated that repeat testing is not necessary for people with confirmed or suspected IIDH1 R132 mutation–positive AML.
What Are the Expected Impacts of Incorporating IIDH1 R132 Testing?
For jurisdictions that do not currently test for IDH1 mutations, implementation of routine IIDH1 R132 testing for people with AML may have impacts on health system infrastructure and patient-related treatment decisions. Implementing routine genetic testing with NGS would potentially require upscaling current AML-related testing infrastructure, including personnel, laboratory equipment, and genetic counselling services for clinical decision-making.12 One clinical expert indicated that incorporating PCR testing for diagnosing IIDH1 R132 mutation may not have significant health system impacts if there is general established accessibility to PCR testing and if a relatively low number of PCR tests would be needed to identify and diagnose an IDH1 mutation in people with AML. Based on the information available, it is unclear how many jurisdictions currently do or do not have capacity for IIDH1 R132 mutation testing for people with AML.
How Is IIDH1 R132 Testing Funded?
Funding to support testing for IIDH1 R132 mutations varies across jurisdictions, and information to identify funding status is insufficient or uncertain. Based on clinical expert input, it appears that IIDH1 R132 testing is not currently in use or funded for routine AML diagnosis in Manitoba, while IDH1 testing is part of routine AML diagnosis testing and is currently reimbursed in Ontario.13 However, it is unclear how the test is funded in Ontario (e.g., through provincial, individual hospital, or laboratory funding budgets). British Columbia uses an NGS myeloid panel test as part of routine AML diagnostic testing, which can be used to identify IIDH1 R132 mutations and is funded through the provincial government.14 Testing information from the McGill University Health Centre, which provides care to populations in Quebec, has indicated that IDH1 gene testing is included within 2 approved NGS panel tests conducted out of the province; however, no funding information related to the use of these tests was provided.15 Currently, there are no publicly funded or private genetic testing facilities in the Northwest Territories, Nunavut, or Yukon.16 No additional information could be obtained regarding which other provinces conduct IIDH1 R132 testing as part of routine AML diagnosis or if IIDH1 R132 testing is funded.
Patient-Oriented Considerations
Patient-related considerations for IIDH1 R132–specific AML testing include informed decision-making regarding initial diagnostic testing and confirmation testing, possible psychological impacts of AML-related testing, adequate communication of testing procedures and possible outcomes, timing considerations of testing, and other barriers to testing such as access to testing.17,18 Because of the aggressive nature of AML, timely access to testing is often necessary to determine potential treatment options.5 The testing process may be emotionally burdensome for some patients where adequate time to emotionally process the testing procedures and testing implications may be limited.6 The indication for these testing procedures is to identify people with IIDH1 R132 mutation–positive AML who may benefit from targeted therapy, which is intended for patients ineligible for intensive induction chemotherapy, such as those older than 75 years. Generally, older patients with AML (i.e., patients older than 60 years) require more inpatient care and are likely to encounter longer hospital stays, impacting both the patient and their caregivers.5 One clinical expert indicated that people living in remote or rural areas may also encounter additional barriers, such as timely access to testing and the need to ensure appropriate collection, management, and possible shipment of patient testing samples to testing centres for accurate results. One clinical expert indicated that in jurisdictions that provide IDH1 mutation testing as part of routine AML diagnostics, no additional testing-related costs would be incurred by the patients.
Clinical Considerations
Clinical Utility
According to the clinical experts, identification of IIDH1 R132 mutations in people with AML using NGS would typically be carried out for AML risk stratification efforts, and testing for a specific IIDH1 R132 mutation using NGS or PCR testing would provide input for treatment decision-making. The intention for these testing procedures would be to identify and determine patients who are likely to be eligible for IIDH1 R132–targeted therapy, such as ivosidenib. NGS can also provide information related to potential co-mutations, which may impact a patient’s clinical response to treatment.3 In 1 exploratory study provided by the sponsor assessing translational biomarkers, an ACE Extended Cancer Panel NGS test was used to detect co-mutations among people with AML and IIDH1 R132 mutations.3 The results from this exploratory study indicated that having an IDH1 mutation and receptor tyrosine kinase pathway co-mutations was associated with experiencing primary resistance to ivosidenib monotherapy; however, patients with an IIDH1 R132 mutation and a receptor tyrosine kinase co-mutation who were treated with ivosidenib and azacitidine showed enriched clinical response rates compared to placebo and azacitidine.3
Diagnostic testing can also be carried out using a standard PCR assay test to determine the type of IIDH1 R132 mutation (i.e., C, G, H, L, or S). The exploratory study provided by the sponsor assessed the effect of the IIDH1 R132 mutation on treatment sensitivity. This analysis was carried out using the Abbott RealTime IDH1 in vitro PCR assay test and found that in patients who had an R132C mutation, treatment with ivosidenib and azacitidine was associated with more favourable clinical responses, event-free survival, and overall survival compared to treatment with placebo and azacitidine.3 Other R132 mutations (i.e., G, H, L, and S) were not associated with significant difference in clinical outcomes between treatments (i.e., ivosidenib and azacitidine or placebo and azacitidine).3 The study therefore suggests that the specific single nucleotide variant of the IDH1 mutation may provide insight into patient treatment responses; however, given the small proportion of patients analyzed in the subgroups, any slight or modest differences should be interpreted with caution.3
Diagnostic Test Accuracy
According to the clinical experts, the sensitivity and specificity of IDH1 mutation testing is high. Based on the testing used in the literature provided by the sponsor, the NGS test used a genetic panel (the ACE Extended Cancer Panel) that was described to have 500 times the average target cover for the full codon region (more than 1,400 genes) and a detection limit of 2%.3 The Abbott RealTime IDH1 in vitro PCR assay test used in the literature provided by the sponsor is reported to have a detection rate of 100% at mutation levels of 2% or higher for all IDH1 mutations combined and a detection rate of 98% at a mutation level of 1% or higher for all IDH1 mutations.4
Cost Considerations
The current cost of NGS panel testing for people with AML is not publicly available. Estimates based on a Canadian micro-costing study of NGS assays in non–small cell lung cancer used to inform the economic analysis for ivosidenib was $1,227. Additional estimates from a 2015 publication by the Institut national d’excellence en santé et services sociaux (INESSS) regarding the prognostic stratification of AML by NGS panel testing showed that the cost of analyzing 9 genes was estimated to be between $810 and $2,040, or approximately $1,000 to $2,525 adjusted to 2023 Canadian dollars.19 These estimates were based on 4 patient and control samples corresponding to the $810 estimate and 1 patient sample and 1 control sample corresponding to the $2,040 estimate.19
The current cost of PCR testing using the Abbott RealTime IDH1 in vitro PCR assay test in Canada is not publicly available. Based on 1 US Medicare reimbursement code for the Abbott Realtime IDH1 PCR test, the estimated cost of a PCR test is US$193.25, or CA$262.32.20 However, a confirmatory PCR test may only be necessary in people who have a suspected IIDH1 R132 mutation based on the NGS AML stratification analysis.
References
1.Leisch M, Jansko B, Zaborsky N, Greil R, Pleyer L. Next Generation Sequencing in AML-On the Way to Becoming a New Standard for Treatment Initiation and/or Modulation? Cancers (Basel). 2019;11(2). PubMed
2.Issa GC, DiNardo CD. Acute myeloid leukemia with IDH1 and IDH2 mutations: 2021 treatment algorithm. Blood Cancer J. 2021;11(6):107. PubMed
3.Clinical Study Report: AG120-C-009 – Translational-Biomarker. A Phase 3, Multicenter, Double-Blind, Randomized, Placebo-Controlled Study of AG-120 in Combination with Azacitidine in Subjects ≥18 Years of Age with Previously Untreated Acute Myeloid Leukemia with an IDH1 Mutation [internal sponsor’s report]. Suresnes (FR): Institut de Recherches Internationales Servier; 2021 Dec 7.
4.Abbott RealTime IDH1 (Reference 08N90-090 51-608283/R4). Abbott Park (IL): Abbott; 2023: https://www.molecular.abbott/content/dam/add/molecular/products/oncology/realtime-idh1/Abbott%20RealTime%20IDH1%20Package%20Insert.pdf. Accessed 2024 May 15.
5.Tibsovo (ivosidenib) in combination with azacitidine for the treatment of adult patients with newly diagnosed acute myeloid leukemia (AML) with an isocitrate dehydrogenase-1 (IDH1) R132 mutation who are not eligible to receive intensive induction chemotherapy: Clinical summary [internal sponsor’s report]. In: Drug Reimbursement Review sponsor submission: Tibsovo® (ivosidenib) tablets, 250 mg, Oral. Laval (QC): Servier Canada Inc.; 2024 Mar 19.
6.Canadian Cancer Society. Diagnosis of leukemia. 2024; https://cancer.ca/en/cancer-information/cancer-types/acute-myeloid-leukemia-aml/diagnosis. Accessed 2024 May 17.
7.Clinical Study Report: AG120-C-009. A Phase 3, Multicenter, Double-Blind, Randomized, Placebo-Controlled Study of AG-120 in Combination with Azacitidine in Subjects ≥18 Years of Age with Previously Untreated Acute Myeloid Leukemia with an IDH1 Mutation [internal sponsor’s report]. Suresnes (FR): Institut de Recherches Internationales Servier; 2021 Dec 8.
8.National Comprehensive Cancer Network. Acute Myeloid Leukemia: Version 1.2024. 2024; https://www.nccn.org/guidelines/guidelines-detail?category=1&id=1411. Accessed 2024 May 17.
9.Yang F, Anekpuritanang T, Press RD. Clinical Utility of Next-Generation Sequencing in Acute Myeloid Leukemia. Mol Diagn Ther. 2020;24(1):1-13. PubMed
10.Schiffer CA, Gurbuxani S. Acute myeloid leukemia: Clinical manifestations, pathologic features, and diagnosis. In: Post TW, ed. UpToDate. Waltham (MA): UpToDate; 2024: https://www.uptodate.com. Accessed 2024 May 17.
11.Canadian Cancer Society. Acute myeloid leukemia statistics. 2024; https://cancer.ca/en/cancer-information/cancer-types/acute-myeloid-leukemia-aml/statistics. Accessed 2024 May 17.
12.Borle K, Kopac N, Dragojlovic N, et al. Where is genetic medicine headed? Exploring the perspectives of Canadian genetic professionals on future trends using the Delphi method. Eur J Hum Genet. 2022;30(5):496-504. PubMed
13.Cancer Care Ontario. Consensus Pathology Recommendations for Complex Malignant Hematology. 2016; https://www.cancercareontario.ca/en/guidelines-advice/types-of-cancer/43061. Accessed 2024 Jun 19.
14.Cancer Genetics and Genomic Laboratory, BC Cancer. Myeloid Panel. 2024; https://cancergeneticslab.ca/genes/myeloid-panel/. Accessed 2024 May 23.
15.List of testing done out of Quebec. Montreal (QC): McGill University Health Centre; 2024: https://cusm.ca/sites/default/files/docs/m-Labs/list-testing-done-out-quebec.pdf. Accessed 2024 Jun 10.
16.Canadian Cancer Survivor Network. Genetic Testing. https://survivornet.ca/learn/health-concerns-for-cancer-patients/genetic-testing/. Accessed 2024 Jun 10.
17.Samuel GN, Dheensa S, Farsides B, Fenwick A, Lucassen A. Healthcare professionals' and patients' perspectives on consent to clinical genetic testing: moving towards a more relational approach. BMC Med Ethics. 2017;18(1):47. PubMed
18.Beachy SJ, SG. Olson, S. Berger, AC. Assessing Genomic Sequencing Information for Health Care Decision Making: Workshop Summary. Washington (DC): National Academies Press (US); 2014: https://www.ncbi.nlm.nih.gov/books/NBK241343/. Accessed 2024 May 24.
19.Stratification pronostique des leucémies myéloïdes aiguës par séquençage de nouvelle génération: Panel de gènes Quebec (QC): Institut national d’excellence en santé et services sociaux; 2016: https://www.inesss.qc.ca/fileadmin/doc/INESSS/Analyse_biomedicale/Juin_2016/INESSS-Avis_analyses_bm-juin16_8_Stratification_pronostique_leucemies_myeloides_aigues_sequencage_nouv_gen_panel_genes.pdf. Accessed 2024 May 23.
20.CodeMap, LLC. Molecular Diagnostics at Abbott. 2023: https://www.codemap.com/abbottmolecular/RealTimeIDH1.cfm. Accessed 2024 May 17.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.