Drugs, Health Technologies, Health Systems
Reimbursement Review
Pembrolizumab (Keytruda)
Sponsor: Merck Canada Inc.
Therapeutic area: Biliary tract carcinoma
This multi-part report includes:
Clinical_Review
Pharmacoeconomic_Review
Clinical_Review
Abbreviations
AE
adverse event
BICR
blinded independent central review
BTC
biliary tract cancer
CCA
cholangiocarcinoma
CCRAN
Colorectal Cancer Resource & Action Network
CI
confidence interval
CPS
combined positive score
CrI
credible interval
DCO
data cut-off
GI
gastrointestinal
GRADE
Grading of Recommendations Assessment, Development and Evaluation
DOR
duration of response
ECOG
Eastern Cooperative Oncology Group
EORTC QLQ-BIL21
European Organization for the Research and Treatment of Cancer Quality of Life Questionnaire Cholangiocarcinoma and Gallbladder Cancer Module 21
EORTC QLQ-C30
European Organization for the Research and Treatment of Cancer Quality of Life Questionnaire Core 30
GBC
gallbladder cancer
GHS
Global Health Score
HR
hazard ratio
HRQoL
health-related quality of life
IA1
interim analysis 1
ITC
indirect treatment comparison
ITT
intention to treat
KM
Kaplan-Meier
LSM
least squares mean
MID
minimally important difference
NMA
network meta-analysis
OH-CCO
Ontario Health (Cancer Care Ontario)
ORR
objective response rate
OS
overall survival
PD-1
programmed cell death 1 protein
PD-L1
programmed cell death 1 ligand 1
PD-L2
programmed cell death 1 ligand 2
PFS
progression-free survival
PRO
patient-reported outcome
RCT
randomized controlled trial
RECIST
1.1 Response Evaluation Criteria in Solid Tumours Version 1.1
SAE
serious adverse event
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information for the Application Submitted for Review
Item | Description |
|---|---|
Drug product | Pembrolizumab (100 mg/4 mL vial solution for IV infusion). |
Sponsor | Merck Canada Inc. |
Indication | Pembrolizumab in combination with gemcitabine-based chemotherapy is indicated for the treatment of adult patients with locally advanced unresectable or metastatic biliary tract carcinoma (BTC). |
Reimbursement request | As per indication. |
Health Canada approval status | NOC |
Health Canada review pathway | Project Orbis |
NOC date | April 12, 2024 |
Recommended dose | Pembrolizumab is recommended to be administered before chemotherapy when given on the same day. The recommended dose in adults is either 200 mg every 3 weeks or 400 mg every 6 weeks until disease progression, unacceptable toxicity, or up to 24 months or 35 doses for 200 mg or 18 doses for 400 mg, whichever is longer, in patients without disease progression. The IV infusion is administered over 30 minutes. |
NOC = Notice of Compliance.
Introduction
Biliary tract cancer (BTC) is a heterogeneous group of tumours that originate in the biliary tree (cholangiocarcinoma [CCA]) or the gallbladder and cystic duct (gallbladder cancer [GBC]).1,2 Based on the location or origin of the tumour, CCA is generally classified into subtypes as intrahepatic and extrahepatic.1 Patients with early-stage BTC are usually asymptomatic; therefore, most patients (60% to 85%) are diagnosed with disease that is at the locally advanced unresectable or metastatic stage, at which time symptoms may still be nonspecific. Common symptoms associated with BTCs are jaundice, abdominal discomfort, malaise, hepatomegaly, weight loss, palpable abdominal mass, fever, night sweats, pruritis, dark urine, or clay-coloured or light-coloured greasy stools.3-8 Early diagnosis is challenging due to the asymptomatic nature of early BTCs, as patients do not experience symptoms until the disease becomes advanced.9
BTCs are rare and represent less than 1% of all cancers globally, comprising approximately 3% of all gastrointestinal cancers and 10% to 15% of primary liver cancers.1,2,9,10 In Western Europe and the US, the incidence of CCA and GBC ranged from 0.3 to 3.5 cases and 1.6 to 2.0 cases per 100,000 population, respectively.1 BTCs are aggressive and high mortality rates are reported in patients with BTCs.9,11 According to the American Cancer Society, between 2012 and 2018, the 5-year relative survival rates ranged from 3% to 9% for regional and distant intrahepatic CCA, and from 2% to 11% for regional and distant extrahepatic CCA, respectively.12 The 5-year relative survival rate ranged from 0% to 5% for unresectable GBC and extrahepatic BTC, respectively.13,14 The Canadian data support that BTCs comprise a rare group of malignancies with a poor prognosis. BTC comprises less than 0.5% of all cancer diagnoses each year in Canada.15,16 It is estimated that in 2025, there will be a total of 1,403 new cases of BTC and 1,263 new cases of locally advanced unresectable or metastatic BTC.15-19 The median survival for patients in Canada with unresectable BTC is approximately 6 to 12 months.17,18,20
According to the clinical experts Canada’s Drug Agency (CDA-AMC) consulted and the patient group input, the treatment goals for patients with locally advanced unresectable or metastatic BTC are to prolong life, delay disease progression, alleviate symptoms, and maintain patients’ quality of life. For patients with advanced BTC, high recurrence rates and the failure to meet eligibility criteria for surgery mean that systemic therapy plays a large role in the treatment.21 The clinical experts consulted by the review team identified the following unmet needs: a curative treatment regimen for patients with advanced BTC and a biomarker available for patient selection. Chemotherapy, mainly with gemcitabine plus cisplatin, was the first-line standard of care for patients with unresectable locally advanced or metastatic BTC more than a decade ago;10 however, the combination of a programmed cell death 1 ligand 1 (PD-L1) checkpoint inhibitor (durvalumab) plus chemotherapy has been introduced in recent years, and an improvement in overall survival (OS) with durvalumab plus chemotherapy compared with chemotherapy alone has been demonstrated in a phase III randomized controlled trial (RCT).22 Durvalumab in combination with gemcitabine-based chemotherapy is now the standard of care and is in widespread use throughout Canada for this particular patient population.23
Pembrolizumab is a high-affinity antibody against programmed cell death 1 protein (PD-1), which is an immune-checkpoint receptor that limits the activity of T lymphocytes in peripheral tissues. By inhibiting the PD-1 receptor from binding to its ligands, pembrolizumab reactivates tumour-specific cytotoxic T lymphocytes in the tumour microenvironment.24 In Canada, pembrolizumab has been issued market authorization for the treatment of various types of cancers.24 On April 12, 2024, pembrolizumab in combination with gemcitabine-based chemotherapy (herein referred to as pembrolizumab plus chemotherapy) was approved by Health Canada for the treatment of locally advanced unresectable or metastatic BTC. The sponsor’s reimbursement request is aligned with the Health Canada–approved indication. The recommended dosage of pembrolizumab for BTC treatment is either 200 mg every 3 weeks or 400 mg every 6 weeks (as IV infusion) until disease progression, unacceptable toxicity, or up to 24 months or 35 doses for 200 mg or 18 doses for 400 mg, whichever is longer, in patients without disease progression.
The objective of this Clinical Review Report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of pembrolizumab (100 mg/4 mL or 25 mg/mL) for IV infusion in the treatment of adult patients with locally advanced unresectable or metastatic BTC in combination with chemotherapy.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient and clinician groups that responded to our call for input and from the clinical experts we consulted for the purpose of this review.
Patient Input
One patient group, the Colorectal Cancer Resource and Action Network (CCRAN) provided input to the review of pembrolizumab used in combination with chemotherapy for BTC. CCRAN is a national not-for-profit patient advocacy group that collected inputs from patients and caregivers through a survey between October 20 and December 1, 2023, in collaboration with the Canadian Cancer Survivor Network (CCSN) and Gastrointestinal (GI) Society. CCRAN also reached out to the Cholangiocarcinoma Foundation, a US-based patient advocacy group dedicated to supporting patients with cholangiocarcinoma, to obtain additional patient input for this disease through telephone interviews, emails, and social media blasts. In total, 4 patients and 4 caregivers provided input on their experience with BTC and the treatments. Among them, 5 had experience with pembrolizumab, which was given without chemotherapy, following previous chemotherapy, or in combination with chemotherapy. The number of cycles of pembrolizumab that these patients received ranged from 2 to 40. Three patients received chemotherapy but did not report experience with pembrolizumab.
Based on the patient input, inoperable or metastatic BTC and the currently available treatments have significant negative impact on patient’s physical and psychosocial well-being, affecting their everyday life, work, and family. The patients often face great financial difficulties. The patients who had received treatment with pembrolizumab reported improved cancer-induced symptoms, fewer side effects, improved quality of life, and a shorter infusion time compared with other treatments.
The patient group input stated that the significant unmet need for patients with metastatic BTC is to have more treatment options than are currently available for this patient population. Outcomes important to patients included improved quality of life, delayed onset of symptoms, reduced side effects compared with the current treatments, and prolonged OS and progression-free survival (PFS). The respondents stated that the introduction of novel, more effective, better tolerated, and easily administered targeted therapies with equitable access is of paramount importance, particularly in the first-line setting.
Clinician Input
Input From the Clinical Experts CDA-AMC Consulted
The clinical experts indicated that for patients with locally advanced unresectable or metastatic BTC, the most important goals of treatment are to prolong life, delay disease progression, alleviate symptoms, and maintain patients’ quality of life. The experts identified 2 unmet needs associated with the current treatments for advanced BTC: to have curative therapies and biomarkers to help with patient selection.
The clinical experts indicated that, currently, durvalumab in combination with chemotherapy is widely used in the treatment of advanced BTC, and pembrolizumab will be the second immune-checkpoint inhibitor to be used along with chemotherapy for these patients. The experts also noted that pembrolizumab would be used as a first-line treatment, and it would be inappropriate to recommend that patients try other treatments before initiating pembrolizumab. Furthermore, the experts suggested that after a maximum of 8 cycles of combination therapy, treatment with pembrolizumab could be continued with or without gemcitabine.
The clinical experts noted that since no biomarkers have been identified in selecting patients who are suitable for the combination regimen, all patients with advanced BTC should be eligible for pembrolizumab plus chemotherapy, if this is not contraindicated in these patients. For example, patients with good performance status and with no comorbidities that might preclude them from receiving chemotherapy (e.g., cisplatin or carboplatin plus gemcitabine) would be eligible. The experts noted that in clinical practice, patients who are receiving treatment for advanced BTC would have regular imaging scans, such as CTs, to monitor their responses to treatment. Other assessments include patients’ functional status (e.g., Eastern Cooperative Oncology Group [ECOG] Performance Status) and disease status. Usually, these assessments are reviewed every 2 to 3 months for patients with advanced BTC. This practice is consistently adopted by treating physicians.
According to the clinical experts we consulted, treatment with a combination of pembrolizumab and chemotherapy would be discontinued if disease progression is detected by an imaging scan, or if the patients experience any intolerable adverse effects related to the treatment.
The clinical experts noted that, in general, patients should be treated by a medical oncologist who has knowledge of BTC management. They also noted that patients could receive the treatment in any setting, such as a community or academic centre.
Clinician Group Input
One clinician group provided input for the review of pembrolizumab in combination with chemotherapy: the Ontario Health (Cancer Care Ontario) (OH-CCO) Gastrointestinal Cancer Drug Advisory Committee.
In general, the clinician group input was consistent with the input provided by the clinical experts we consulted for this review. OH-CCO noted that the standard of care for patients with advanced BTC is gemcitabine plus cisplatin and gemcitabine plus carboplatin and the treatment goals would be prolonged life, delayed disease progression, and improved quality of life. OH-CCO added that the 1 available regimen has a poor duration of response; therefore, new regimens are required.
The clinician group stated that pembrolizumab can be safely added to first-line chemotherapy that is well tolerated, and all patients who align with the clinical trial criteria are best suited for the drug under review. The clinician group believes that clinical and/or radiologic progression, as assessed by the treating oncologist, determines whether a patient is responding to treatment in clinical practice, and treatment should be discontinued at the discretion of the treating oncologist if there is disease progression or toxicity. Additionally, the appropriate setting for treatment would be a hospital (outpatient clinic), and a specialist would be required.
Drug Program Input
Input was obtained from the drug programs that participate in our reimbursement review process. The following were identified as key factors that could potentially impact the implementation of a CDA-AMC recommendation for pembrolizumab in combination with chemotherapy:
relevant comparators
considerations for initiation of therapy
consideration for discontinuation of therapy
generalizability.
The clinical experts we consulted provided advice on the potential implementation issues raised by the drug programs.
Clinical Evidence
Systematic Review
Description of Studies
One phase III double-blind RCT (KEYNOTE-966; N = 1,069) met the inclusion criteria for the systematic review conducted by the sponsor.25 The purpose of this study was to evaluate the efficacy and safety of the combination of pembrolizumab plus chemotherapy versus placebo plus chemotherapy in patients with locally advanced unresectable or metastatic BTC. Patients were randomized at a 1:1 ratio to either pembrolizumab in combination with gemcitabine and cisplatin (chemotherapy) or placebo in combination with chemotherapy. More specifically, patients randomized to pembrolizumab received pembrolizumab 200 mg by IV infusion once every 3 weeks for a maximum of 35 cycles. Patients randomized to either pembrolizumab or placebo received treatment in combination with gemcitabine (1,000 mg/m2 on days 1 and 8 of each cycle every 3 weeks, with no maximum duration) and cisplatin (25 mg/m2 by IV infusion on days 1 and 8 of each cycle for a maximum duration of 8 cycles). The primary efficacy end point in the KEYNOTE-966 study was OS. Other relevant outcomes in this study included PFS, health-related quality of life (HRQoL) measured by the European Organization for the Research and Treatment of Cancer (EORTC) Quality of Life Questionnaire Core 30 (QLQ-C30) and the EORTC Quality of Life Questionnaire Cholangiocarcinoma and Gallbladder Module 21 (QLQ-BIL21), and safety. Overall, patients’ baseline characteristics were balanced between treatment groups. The trial randomized approximately equal proportions of females and males (51.6% male; n = 552). The median age was 64.0 years (range, 23 to 85 years). Most randomized patients were white (49.0%; n = 524), had an ECOG Performance Status score of 1 (54.4%; n = 582), and were from a region outside Asia (54.5%; n = 583). Most patients had metastatic disease (88.2%; n = 943) with an intrahepatic site of origin (59.2%; n = 633). Approximately 30% of these patients had received prior surgery (29.8%; n = 319).
Efficacy Results
The KEYNOTE-966 study met its primary end point at the final analysis (data cut-off [DCO] of December 15, 2022). The results suggested that treatment with pembrolizumab plus chemotherapy may be associated with prolonged OS compared with treatment with placebo plus chemotherapy, with a median OS of 12.7 months (95% confidence interval [CI], 11.5 to 13.6) versus 10.9 months (95% CI, 9.9 to 11.6), respectively. The hazard ratio (HR) for OS was 0.83 (95% CI, 0.72 to 0.95; P = 0.0034). Although the between-group difference in median OS was 1.8 months, given the poor prognosis in patients with advanced BTC (which has a median OS of less than 12 months), an improvement of 1.8 months in median survival is considered a clinically meaningful benefit according to the clinical experts we consulted. The between-group differences in the Kaplan-Meier (KM)–estimated OS rates at 6, 12, 18, and 24 months were 7.0% (95% CI, 2.0 to 12.0), 7.5% (95% CI, 1.6 to 13.4), 5.0% (95% CI, −0.5 to 10.5), and 6.8% (95% CI, 1.7 to 11.9), respectively. These estimates were affected by imprecision; the 95% CIs included the potential for trivial effects, based on a threshold for a clinically important between-group difference of 5%, as informed by the clinical experts we consulted. Results of prespecified subgroup analyses based on various patient baseline characteristics were consistent with those in the overall population.
PFS measured using Response Evaluation Criteria in Solid Tumours Version 1.1 (RECIST 1.1) was 1 of the key secondary end points in the KEYNOTE-966 study. At the December 15, 2021, DCO, the median PFS was 6.5 months (95% CI, 5.7 to 6.9) with pembrolizumab plus chemotherapy, and 5.6 months (95% CI, 5.1 to 6.6) with placebo plus chemotherapy. The corresponding HR was 0.86 (95% CI, 0.75 to 1.00; P = 0.0225), which did not meet the prespecified efficacy boundary for a statistically significant PFS benefit for pembrolizumab plus chemotherapy, according to the multiplicity scheme. Further, the HR cannot be interpreted reliably; based on visual inspection of the PFS curves, the proportional hazards assumption appears to have been violated. The between-group differences in the KM-estimated PFS rates were 6.2% (95% CI, 0 to 12.4), 5.7% (95% CI, −0.5 to 11.9), 5.6% (95% CI, −0.4 to 11.6), 6.3% (95% CI, 0.2 to 12.4), and 3.5% (95% CI, −2.8 to 9.8) for 6, 9, 12, 15, and 18 months, respectively. At 6 months of follow-up, the results showed that pembrolizumab plus chemotherapy may result in an increase in the KM-estimated probability of PFS when compared with placebo plus chemotherapy; however, the clinical importance of the increase (6.2%) is uncertain, and the 95% CI included the possibility of no difference between treatments. At 18 months, the evidence was very uncertain about the effect of pembrolizumab plus chemotherapy when compared with placebo plus chemotherapy on the KM-estimated probability of PFS (3.5%) owing to imprecision (the CI included the potential that either treatment could be favoured) and indirectness (due to uncertainty in the adequacy of RECIST 1.1 to measure PFS). Of note, although PFS is typically considered a surrogate for OS in oncology trials, assessing PFS in patients with BTC is complex and may not accurately reflect the PFS benefit gained in patients with BTC.26
HRQoL was measured by the EORTC QLQ-C30 and EORTC QLQ-BIL21. The latter is specific to patients with CCA and GBC. Both of these HRQoL measures were included as exploratory outcomes in the KEYNOTE-966 study. At week 18, approximately 60% of the patients completed the assessment and contributed to the analysis of the HRQoL data. As such, the results are at risk of bias due to missing outcomes data. At week 18, the between-group differences in the least squares mean (LSM) changes from baseline in the EORTC QLQ-C30 Global Health Status (GHS) (quality-of-life scales), physical functioning, and role functioning subscale scores were 0.04 (95% CI, −2.52 to 2.60; P = 0.9773), 1.24 (95% CI, −1.42 to 3.90; P = 0.3596), and 2.68 (95% CI, −0.76 to 6.11; P = 0.1264), respectively. The difference in the LSM changes from baseline in the EORTC QLQ-BIL21 jaundice and pain subscale scores were 0.26 (95% CI, −1.35 to 1.87; P = 0.7535) and −1.87 (95% CI, −4.26 to 0.53; P = 0.1265), respectively. For the EORTC QLQ-C30, the between-group minimally important differences (MIDs) ranged from 5 to 10 points for most scales. For the EORTC QLQ-BIL21, while no MID has been identified for patients with BTC, the MID can be extrapolated from other cancer types). Based on the MIDs for these 2 instruments, the HRQoL results suggested that compared with placebo plus chemotherapy, adding pembrolizumab to chemotherapy may not result in any clinically important difference in the subscale scores for GHS, physical functioning, and role functioning in the EORTC QLQ-C30, or in the in EORTC QLQ-BIL21 subscale scores for jaundice and pain.
Harms Results
The proportion of patients experiencing 1 or more adverse events (AEs) in the KEYNOTE-966 study was well balanced between the 2 treatment groups, which suggested that adding pembrolizumab to existing chemotherapy is not associated with an added risk of AEs; the comparisons between pembrolizumab plus chemotherapy and placebo plus chemotherapy were 99.1% versus 99.6% for any AEs, 52.2% versus 49.3% for serious adverse events (SAEs), and 26.1% versus 22.8% for treatment discontinuation due to AEs, respectively. For patients treated with pembrolizumab plus chemotherapy, commonly reported AEs were decreased neutrophil count (62.4%), anemia (61.1%), nausea (44.0%), decreased platelet count (39.9%), fatigue (35.3%), and constipation (35.2%). For patients who received placebo, commonly reported AEs included decreased neutrophil count (61.2%), anemia (58.6%), nausea (46.1%), decreased platelet count (39.7%), fatigue (32.2%), and constipation (35.6%). Commonly reported SAEs were cholangitis (5.9%), pyrexia (5.7%), decreased platelet count (3.6%), biliary tract infection (3.2%), anemia (2.5%), sepsis (2.5%), biliary obstruction (2.3%), decreased neutrophil count (2.1%), and pulmonary embolism (2.1%) in patients treated with pembrolizumab, and were cholangitis (4.5%), biliary tract infection (3.4%), sepsis (3.0%), biliary obstruction (3.0%), ascites (2.4%), pyrexia (2.2%), and liver abscess (2.1%) in patients treated with placebo plus chemotherapy. The most common reasons for treatment discontinuation due to AEs were decreased neutrophil count (3.6%) and decreased platelet count (3.6%) in patients treated with pembrolizumab plus chemotherapy, and decreased neutrophil count (3.0%) in patients treated with placebo plus chemotherapy.
The proportion of patients with AEs resulting in death was 5.9% (31 patients) in the pembrolizumab plus chemotherapy group and 9.2% (49 patients) in the placebo plus chemotherapy group. Patients in the pembrolizumab plus chemotherapy group reported more notable harms compared with those in the comparator group, 22.1% versus 12.9%, respectively. For the immune-mediated AEs (immune-mediated enterocolitis, hepatitis, or lung disease), it did not appear that pembrolizumab plus chemotherapy resulted in clinically important increases in these events (based on a threshold for a clinically important between-group difference of 5% to 10% as informed by the clinical experts we consulted); however, few events were reported in the KEYNOTE-966 study, which adds uncertainty to these results. There were no unusual safety signals observed for the treatment of pembrolizumab plus chemotherapy. The frequency, type, and severity of harms were consistent with pembrolizumab monotherapy, and the harms were not exacerbated by the combination of pembrolizumab plus chemotherapy. According to the clinical experts we consulted, the AEs observed in the KEYNOTE-966 study are manageable in clinical practice.
Critical Appraisal
In the KEYNOTE-966 study, patients’ baseline demographic and disease characteristics were balanced between the 2 treatment groups in general, although patients in the pembrolizumab plus chemotherapy group had a relatively better performance status compared with the placebo plus chemotherapy group. This imbalance is likely attributed to chance and, as such, does not introduce bias. The clinical experts we consulted noted that this imbalance would not significantly impact result interpretation.
A multiplicity testing procedure was applied to OS, PFS, and overall response rate to control for the type I error rate in the study and across interim analyses. However, other efficacy outcomes were analyzed without multiplicity adjustment, for example, HRQoL assessment using the EORTC QLQ-C30 and EORTC QLQ-BIL21. Nevertheless, there were no statistically significant results in any relevant domains.
HRQoL was assessed using disease-specific instruments, and 1 of these was specifically designed for patients with BTC. A specific MID for patients with BTC was not identified from the literature; however, a range of potential between-group MIDs was established based on clinical trials of 9 different cancer types and can be used to determine the clinical relevance of the study findings for HRQoL. Otherwise, the completion rate for the 2 EORTC questionnaires was approximately 60% in the 2 treatment groups. As such, the risk of bias due to missing outcomes data and its impact on the study findings is uncertain.
External Validity
Based on feedback from the clinical experts we consulted, the eligibility criteria and baseline characteristics of patients randomized in the KEYNOTE-966 study generally reflected a study population that is consistent with the patients in Canadian clinical practice who would receive the combination therapy of pembrolizumab plus chemotherapy, although the study population may be somewhat healthier. The clinical experts noted that the results from the KEYNOTE-966 study could be generalized to patients with advanced BTC in Canada who would be treated with pembrolizumab plus chemotherapy.
GRADE Summary of Findings and Certainty of the Evidence
For the pivotal studies and RCTs identified in the sponsor’s systematic review, the Grading of Recommendations Assessment, Development and Evaluation (GRADE) was used to assess the certainty of the evidence for outcomes considered most relevant to inform our expert committee deliberations. A final certainty rating was determined as outlined by the GRADE Working Group.27,28
Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty-of-evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null. The threshold for a clinically important effect was 5% to 10% for OS (as informed by the clinical experts we consulted) and null for PFS (due to uncertainties in the measurement and interpretation of the outcome). The threshold for a clinically important effect for the EORTC QLQ-C30 and EORTC QLQ-BIL21 scores was set according to the presence or absence of an important effect based on thresholds identified in the literature.29 For some harm events (e.g., immune-mediated AEs), due to the unavailability of the absolute difference in effects, the certainty of evidence was summarized narratively.
The selection of outcomes for GRADE assessment was based on the sponsor’s summary of clinical evidence, consultation with clinical experts, and input received from patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with members of the expert committee:
probability of OS at 12, 18, and 24 months
probability of PFS at 6 and 18 months
change from baseline in EORTC QLQ-C30 scores at 18 weeks
change from baseline in EORTC QLQ-BIL21 scores at 18 weeks
any AEs
any SAEs
AEs leading to treatment discontinuation
risk of immune-mediated AEs (enterocolitis, hepatitis, lung disease).
Long-Term Extension Studies
No relevant long-term extension studies were submitted by the sponsor.
Indirect Comparisons
Description of Studies
One network meta-analysis (NMA) was submitted by the sponsor to compare the efficacy and safety of pembrolizumab plus chemotherapy with durvalumab plus chemotherapy for the treatment of advanced BTC. A feasibility assessment was conducted to determine the feasibility of conducting an NMA in the study population for the outcome of interest and to assess the heterogeneities across the included studies. The NMAs of OS and PFS were conducted using reported HRs in a regression model with a contrast-based normal likelihood for the log HR. For the binary outcomes (e.g., harms), the NMAs were performed based on the proportion of patients experiencing the event of interest using a logistic regression model with a binomial likelihood and logit link.
The outcomes evaluated in this NMA included OS, PFS, and harms. In total, 2 phase III RCTs were included and contributed evidence in the NMA.
Table 2: Summary of Findings for Pembrolizumab in Combination With Chemotherapy Versus Placebo in Combination With Chemotherapy for Patients With Locally Advanced Unresectable or Metastatic BTC
Outcome and follow-up | Patients, N (studies) | Relative effect (95% CI) | Absolute effects (95% CI) | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
Placebo + chemotherapy | Pembrolizumab + chemotherapy | Difference | |||||
Efficacy (ITT population) | |||||||
OS | |||||||
Probability of OS at 12 months. Median follow-up: 25.6 months as of the DCO on December 15, 2022. | 1,069 (1 RCT) | NR | ███ ███ █████ | ██ █ ██████ | ██ ██████ █████ ███ ██ ██████ ████████ | Moderatea | Pembrolizumab plus chemotherapy likely results in a clinically important increase in the probability of OS at 12 months when compared with placebo plus chemotherapy. |
Probability of OS at 18 months. Median follow-up: 25.6 months as of the DCO on December 15, 2022. | 1,069 (1 RCT) | NR | ███ ███ █████ | ██ █ ██████ | ██ ██████ █████ ███ ██ ██████ ████████ | Moderateb | Pembrolizumab plus chemotherapy likely results in a clinically important increase in the probability of OS at 18 months when compared with placebo plus chemotherapy. |
Probability of OS at 24 months. Median follow-up: 25.6 months as of DCO on December 15, 2022. | 1,069 (1 RCT) | NR | ███ ███ █████ | ██ █ ██████ | ██ ██████ █████ ███ ██ ██████ ████████ | Moderatec | Pembrolizumab plus chemotherapy likely results in a clinically important increase in the probability of OS at 24 months when compared with placebo plus chemotherapy. |
PFS | |||||||
Probability of PFS at 6 months. Median follow-up: 13.6 months as of DCO on December 15, 2021. | 1,069 (1 RCT) | NR | ███ ███ █████ | ██ █ ██████ | ██ ██████ █████ ███ ██ ██████ ████████ | Lowd | Pembrolizumab plus chemotherapy may result in an increase in the probability of PFS at 6 months when compared with placebo plus chemotherapy. The clinical importance of the increase is uncertain. |
Probability of PFS at 18 months. Median follow-up: 13.6 months as of DCO on December 15, 2021. | 1,069 (1 RCT) | NR | ██ ███ ████████ | ██ █ ██████ | ██ ██████ █████ ███ ██ ██████ ████████ | Very lowe | The evidence is very uncertain about the effect of pembrolizumab plus chemotherapy on the probability of PFS at 18 months when compared with placebo plus chemotherapy. |
██████████████ ███████ ██ ████ ████ ████████ ████ | |||||||
EORTC QLQ-C30 (Global Health Status score) | |||||||
LS mean change from baseline could range from 0 (no or low symptom burden) to 100 (severe symptoms) points. Follow-up: 18 weeks. | 985 (1 RCT) | NA | −2.51 | −2.47 (−4.45 to −0.49) | 0.04 (−2.52 to 2.60) | Lowf | Pembrolizumab plus chemotherapy may result in little to no difference in LS mean change from baseline in the Global Health Status score when compared with placebo plus chemotherapy. |
EORTC QLQ-C30 (physical functioning score) | |||||||
LS mean change from baseline could range from 0 (no or low symptom burden) to 100 (severe symptoms) points. Follow-up: 18 weeks. | 985 (1 RCT) | NA | −7.66 | −6.42 (−8.34 to −4.49) | 1.24 (−1.42 to 3.90) | Lowg | Pembrolizumab plus chemotherapy may result in little to no difference in LS mean change from baseline in the physical functioning score when compared with placebo plus chemotherapy. |
EORTC QLQ-C30 (role functioning score) | |||||||
LS mean change from baseline could range from 0 (no or low symptom burden) to 100 (severe symptoms) points. Follow-up: 18 weeks. | 985 (1 RCT) | NA | −9.69 | −7.02 (−9.59 to −4.45) | 2.68 (−0.76 to 6.11) | Lowh | Pembrolizumab plus chemotherapy may result in little to no difference in LS mean change from baseline in the role functioning score when compared with placebo plus chemotherapy. |
EORTC QLQ-BIL21 (jaundice score) | |||||||
LS mean change from baseline could range from 0 (no or low symptom burden) to 100 (severe symptoms) points. Follow-up: 18 weeks. | 972 (1 RCT) | NA | −0.12 | 0.14 (−1.14 to 1.42) | 0.26 (−1.35 to 1.87) | Lowi | Pembrolizumab plus chemotherapy may result in little to no difference in LS mean change from baseline in the jaundice score when compared with placebo plus chemotherapy. |
EORTC QLQ-BIL21 (pain score) | |||||||
LS mean change from baseline could range from 0 (no or low symptom burden) to 100 (severe symptoms) points. Follow-up: 18 weeks. | 972 (1 RCT) | NA | −4.07 | −5.94 (−7.83 to −4.05) | −1.87 (−4.26 to 0.53) | Lowj | Pembrolizumab plus chemotherapy may result in little to no difference in LS mean change from baseline in the pain score when compared with placebo plus chemotherapy. |
Harms (safety analysis set) | |||||||
Any AEs | |||||||
Proportion of patients with any AEs. Median follow-up: 25.6 months as of DCO on December 15, 2022. | 1,063 (1 RCT) | NR | ███ ███ █████ | ██ █ ██████ | ████ ███ █████ ███ ████ ██ █ ████ ███ ██████ | Highk | Pembrolizumab plus chemotherapy results in little to no difference in the proportion of patients who experience any AEs when compared with placebo plus chemotherapy. |
Any SAEs | |||||||
Proportion of patients with any SAEs. Median follow-up: 25.6 months as of DCO on December 15, 2022. | 1,063 (1 RCT) | NR | ███ ███ █████ | ██ █ ██████ | ████ ███ █████ ███ ████ ██ █ ████ ███ ██████ | Moderatel | Pembrolizumab plus chemotherapy likely results in little to no clinically important difference in the proportion of patients who experience SAEs when compared with placebo plus chemotherapy. |
AEs leading to treatment discontinuation | |||||||
Proportion of patients with any AEs leading to treatment discontinuation. Median follow-up: 25.6 months as of the DCO on December 15, 2022. | 1,063 (1 RCT) | NR | ███ ███ █████ | ██ █ ██████ | ████ ███ █████ ███ ████ ██ █ ████ ███ ██████ | Moderatem | Pembrolizumab plus chemotherapy likely results in little to no clinically important difference in the proportion of patients who experience any AEs leading to treatment discontinuation when compared with placebo plus chemotherapy. |
Immune-mediated enterocolitis | |||||||
Proportion of patients with immune-mediated enterocolitis. Median follow-up: 25.6 months as of the DCO on December 15, 2022. | 1,063 (1 RCT) | NR | ███ █████ | ██ ███████ | ██ | Moderaten | Pembrolizumab plus chemotherapy likely results in little to no clinically important difference in the number of patients who experience immune-mediated enterocolitis when compared with placebo plus chemotherapy. |
Immune-mediated hepatitis | |||||||
Proportion of patients with immune-mediated hepatitis. Median follow-up: 25.6 months as of the DCO on December 15, 2022. | 1,063 (1 RCT) | NR | ██ ███ █████ | ██ █ ██████ | ██ | Moderateo | Pembrolizumab plus chemotherapy likely results in little to no clinically important difference in the number of patients who experience immune-mediated hepatitis when compared with placebo plus chemotherapy. |
Immune-mediated lung disease | |||||||
Proportion of patients with immune-mediated lung disease. Median follow-up: 25.6 months as of the DCO on December 15, 2022. | 1,063 (1 RCT) | NR | ██ ██████ | ██ █ ██████ | ██ | Moderatep | Pembrolizumab plus chemotherapy likely results in little to no clinically important difference in the number of patients who experience immune-mediated lung disease when compared with placebo plus chemotherapy. |
AE = adverse event; BTC = biliary tract cancer; CI = confidence interval; DCO = data cut-off; EORTC QLQ-BIL21 = European Organization for Research and Treatment of Cancer Quality of Life Questionnaire Cholangiocarcinoma and Gallbladder Cancer Module 21; EORTC QLQ-C30 = European Organization for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; FA = final analysis; IA1 = interim analysis 1; ITT = intention to treat; LS = least squares; MID = minimally important difference; NA = not applicable; NR = not reported; OS = overall survival; PFS = progression-free survival; RCT = randomized controlled trial; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1; RR = relative risk; SAE = serious adverse event.
Note: Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aRated down 1 level for serious imprecision. The clinical experts we consulted considered a 5% between-group difference to be clinically important. The 95% CI includes the potential for a trivial effect. The between-group differences were requested from the sponsor to aid in interpretation and were not part of the sponsor’s analysis plan.
bRated down 1 level for serious imprecision. The clinical experts we consulted considered a 5% between-group difference to be clinically important. The 95% CI includes the potential for a trivial effect. The between-group differences were requested from the sponsor to aid in interpretation and were not part of the sponsor’s analysis plan.
cRated down 1 level for serious imprecision. The clinical experts we consulted considered a 5% between-group difference to be clinically important. The 95% CI includes the potential for a trivial effect. The between-group differences were requested from the sponsor to aid in interpretation and were not part of the sponsor’s analysis plan.
dRated down 1 level for serious indirectness. Per the sponsor, RECIST 1.1 is not the best measure of PFS in this patient population. Rated down 1 level for serious imprecision. There is no known MID, so the null was used for the threshold. The 95% CI included the possibility of no difference. Between-group differences were requested from the sponsor to aid in interpretation and were not part of the sponsor’s analysis plan.
eRated down 1 level for serious indirectness. Per the sponsor, RECIST 1.1 is not the best measure of PFS in this patient population. Rated down 2 levels for very serious imprecision. There is no known MID, so the null was used for the threshold. The 95% CI included the possibility of no difference and harm. Between-group differences were requested from the sponsor to aid in interpretation and were not part of the sponsor’s analysis plan.
f,g,hRated down 2 levels for very serious study limitations due to risk of bias due to missing outcomes data (data were available for 635 patients at week 18). The between-group MID of EORTC QLQ-C30 subscales ranges from 5 to 10 points for most scales.29 Statistical testing for this outcome was not adjusted for multiplicity in the study and should be considered as supportive evidence.
I,jRated down 2 levels for very serious study limitations due to risk of bias due to missing outcomes data (data were available for 635 patients at week 18). There is no known MID; however, it was judged that the entire 95% CI likely included trivial effects. Statistical testing for this outcome was not adjusted for multiplicity in the study and should be considered as supportive evidence.
kEvidence was not rated down.
l,mRated down 1 level for serious imprecision. There is no established MID. The point estimate suggests little to no difference and the 95% CI included the possibility of important harm. Between-group differences were requested from the sponsor to aid in interpretation and were not part of the sponsor’s analysis plan.
n,o,pRated down 1 level for serious imprecision. There is no established MID; however, since the baseline risk for these immune-mediated AEs was very low in the KEYNOTE-966 study, rating down for imprecision for the certainty of evidence can be more conservative.
Sources: Clinical Study Report for the KEYNOTE-96625 study and additional information provided by the sponsor.30 Details included in the table are from the sponsor’s summary of clinical evidence.
Efficacy Results
The evidence from the NMA was insufficient to conclude whether pembrolizumab plus chemotherapy or durvalumab plus chemotherapy were favoured for OS or PFS. The credible intervals (CrIs) for the HRs were wide and spanned the null, such that either treatment could be favoured.
Harms Results
The evidence from the NMA was insufficient to conclude whether pembrolizumab plus chemotherapy or durvalumab plus chemotherapy was favoured for any AEs or AEs greater than grade 3. The CrIs for the odds ratios were wide and spanned the null, such that either treatment could be favoured.
Critical Appraisal
In this indirect treatment comparison (ITC), studies were identified by searching multiple databases based on prespecified inclusion and exclusion criteria. The reviewers of this ITC used methods for study selection and data extraction that were adequate to reduce the risk of error and bias. The risk of bias in the included studies was assessed using appropriate methods to reduce error and bias in the assessments. In this ITC, the analyses were based on the data from 2 RCTs (KEYNOTE-966 and TOPAZ-1). The 2 studies were comparable in study design, populations, and patients’ baseline characteristics. Note that the proportion of Asian patients was 46% in the KEYNOTE-966 trial and 56% in the TOPAZ-1 study. The impact of a 10% difference in the proportion of Asian patients between the 2 study populations on result interpretations was uncertain. Potential treatment-effect modifiers were identified by the sponsor, and clinical heterogeneity between studies was addressed. According to the clinical experts we consulted, the treatment-effect modifiers included in the feasibility appraisal are clinically relevant for the treatment of advanced BTC; however, it is not certain whether these represent all treatment-effect modifiers.
Efficacy and safety data were sparse (i.e., based on only 2 RCTs) in this NMA for the comparison of pembrolizumab plus chemotherapy versus durvalumab plus chemotherapy. Confidence in the effect estimates for efficacy and the harms of the study drugs was limited due to imprecision from the wide CrIs for these outcomes and precludes any conclusions as to which treatment may be favoured.
In this ITC, several efficacy and safety outcomes were analyzed, such as OS, PFS, and harms. However, other efficacy end points of interest to patients and clinicians, such as HRQoL, were not investigated. Therefore, the relative treatment effect of pembrolizumab plus chemotherapy versus other active treatments on patients’ HRQoL remains unknown.
Studies Addressing Gaps in the Evidence From the Systematic Review
No relevant studies addressing gaps in the evidence from the systematic review were submitted by the sponsor.
Conclusions
Locally advanced unresectable or metastatic BTCs are associated with a poor prognosis. The patient and clinician input highlighted the need for new treatments that prolong life, maintain HRQoL, and reduce side effects compared with the current treatments. Evidence from a randomized, double-blind, phase III RCT (KEYNOTE-966) showed that treatment with pembrolizumab plus chemotherapy likely results in modest increases in the probability of survival at 6, 12, 18, and 24 months compared with placebo plus chemotherapy for patients with locally advanced or metastatic BTC. Evidence from the trial also showed that pembrolizumab plus chemotherapy may result in an increase in the probability of PFS at 6 months, although the clinical importance of this increase is uncertain, and the evidence is very uncertain at longer follow-up (18 months). The evidence for PFS was additionally affected by uncertainty related to the ability to measure PFS appropriately using RECIST 1.1. Evidence on HRQoL suggested that adding pembrolizumab to chemotherapy may not result in any clinically important difference in patients’ HRQoL compared with chemotherapy alone; however, the evidence was rated as low certainty due to the limitations of the analyses, including the risk of bias due to missing data. In terms of safety, evidence from the KEYNOTE-966 study suggested that treatment with pembrolizumab plus chemotherapy did not result in an increased risk of any AEs, and likely did not result in an increased risk of SAEs, AEs leading to treatment discontinuation, or immune-mediated AEs, when compared with placebo plus chemotherapy.
There is a lack of direct comparative evidence between pembrolizumab plus chemotherapy and other active treatments for advanced BTC, namely, durvalumab plus chemotherapy. The indirect evidence from a sponsor-submitted NMA of 2 trials was insufficient to conclude whether treatment with pembrolizumab plus chemotherapy differs in terms of OS or PFS or the odds of AEs when compared with durvalumab plus chemotherapy. There was substantial uncertainty in the treatment-effect estimates (indicated by wide CrIs) from the NMA due to limited efficacy and safety data, and no comparisons of HRQoL outcomes that are important to patients and clinicians were conducted.
Introduction
The objective of this Clinical Review Report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of pembrolizumab (100 mg/4 mL or 25 mg/mL) for IV infusion in the treatment of adult patients with locally advanced unresectable or metastatic BTC, in combination with chemotherapy.
Disease Background
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following was summarized and validated by the CDA-AMC review team.
BTC is a heterogeneous group of tumours that originate in the biliary tree (CCA) or the gallbladder and cystic duct (GBC).1,2 Based on the location or origin of the tumour, CCA is generally classified into subtypes as intrahepatic CCA and extrahepatic CCA. Extrahepatic CCA can be further classified into perihilar CCA and distal CCA.1
Patients with early-stage BTC are usually asymptomatic; therefore, most patients (60% to 85%) are diagnosed with disease that is at the locally advanced unresectable or metastatic stage, at which time symptoms may still be nonspecific. Common symptoms associated with BTCs are jaundice, abdominal discomfort, malaise, hepatomegaly, weight loss, palpable abdominal mass, fever, night sweats, pruritis, dark urine, or clay-coloured or light-coloured greasy stools.3-8 Symptoms often reflect the location of the cancer. Patients with intrahepatic BTC often present with nonspecific symptoms including fever, weight loss, and/or abdominal pain, while patients with extrahepatic BTC present with jaundice due to biliary obstruction. Early diagnosis is challenging due to the asymptomatic nature of early BTCs, as patients do not experience symptoms until the disease becomes advanced.9 Diagnosis of BTC usually follows a stepwise process beginning with a medical history and physical exam to search for signs of jaundice and to palpate the abdomen for signs of lumps, hepatomegaly, tenderness, or ascites.31 There are no definitive laboratory tests that can reliably detect early BTC. Typical laboratory tests include blood chemistry to detect elevations in bilirubin or increased liver enzymes, which are indicative of liver dysfunction.31 Imaging scans routinely used in the diagnosis of BTC and cancer staging include abdominal ultrasound, CT, and MRI. Additional specialized testing may include endoscopic retrograde cholangiopancreatography (ERCP) when extrahepatic BTC is suspected. Other examination tools used for the diagnosis of BTC include percutaneous transhepatic cholangiography (PTC) and endoscopic ultrasound. A biopsy may be taken to confirm whether cancer cells are present, which can be done with ERCP, PTC, or fine needle aspiration. Lastly, a laparoscopy may be conducted to assist with staging the BTC, or a laparotomy may be performed to check the organs in the abdomen to determine whether the cancer is resectable.31
BTCs are rare and represent less than 1% of all cancers globally, comprising approximately 3% of all gastrointestinal cancers and 10% to 15% of primary liver cancers.1,2,9,10 In Western Europe and the US, the incidence of CCA and GBC ranged from 0.3 to 3.5 cases and 1.6 to 2.0 cases per 100,000 population, respectively.1 BTCs are aggressive and high mortality rates are reported in patients with BTCs.9,11 According to the American Cancer Society, the 5-year relative survival rate has ranged from 3% to 9% for regional and distant intrahepatic CCA, and from 2% to 11% for regional and distant extrahepatic CCA, respectively. These statistics are based on the patients diagnosed with bile duct cancer between 2012 and 2018.12 The 5-year relative survival rate ranged from 0% to 5% for unresectable GBC and extrahepatic BTC, respectively.13,14 The Canadian data support that BTCs comprise a rare group of malignancies with a poor prognosis. BTC comprises less than 0.5% of all cancer diagnoses each year in Canada.15,16 It was estimated that in 2025, there will be a total of 1,403 new cases of BTC and 1,263 new cases of locally advanced unresectable or metastatic BTC.15-19 The median survival for patients in Canada with unresectable BTC is approximately 6 to 12 months.17,18,20 Prognostic factors for BTC include the ability to resect the tumour, surgical margins, stage of BTC, tumour location, type of tumour, and tumour grade, and whether there is perineural invasion, vascular spread, or periductal invasion.32
Standards of Therapy
The content of this section was informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the CDA-AMC review team.
According to the clinical experts we consulted and the patient group input, the treatment goals for patients with locally advanced unresectable or metastatic BTC are to prolong life, delay disease progression, alleviate symptoms, and maintain patients’ quality of life. Despite therapeutic advances, high recurrence rates and the failure to meet eligibility criteria for surgery mean that systemic therapy plays a large role in the treatment of patients with locally advanced unresectable or metastatic BTC.21 The clinical experts consulted by the review team identified the following unmet needs: a curative treatment regimen for patients with advanced BTC and a biomarker available for patient selection.
Chemotherapy, mainly with gemcitabine plus cisplatin, was the first-line standard of care for patients with unresectable locally advanced or metastatic BTC more than a decade ago.10 According to the clinical experts we consulted, following the failure or intolerance of platinum-based chemotherapy, patients with IDH1 or FGFR2 gene mutations may be treated with ivosidenib (an IDH1 inhibitor) or pemigatinib (an inhibitor of FGFR1, FGFR2, FGFR3, and FGFR4), respectively. However, neither of these is publicly funded in Canada. Since then, the combination of a PD-L1 checkpoint inhibitor (durvalumab) and chemotherapy has been introduced. This combination was studied in a phase III, double-blind, randomized placebo-controlled trial (TOPAZ-1) that evaluated durvalumab plus chemotherapy versus placebo plus chemotherapy for up to 8 cycles in patients with previously untreated, unresectable locally advanced or metastatic BTC.22 A statistically significant and clinically meaningful improvement in OS with durvalumab plus chemotherapy was demonstrated in the TOPAZ-1 study. Durvalumab has been approved in Canada since September 28, 2022, for the treatment of patients with locally advanced or metastatic BTC in combination with gemcitabine-based chemotherapy and is currently the only approved therapy for this indication in Canada.33 Furthermore, durvalumab received a recommendation for reimbursement in combination with gemcitabine and platinum-based chemotherapy for the first-line treatment of patients with locally advanced unresectable or metastatic BTC from the pan-Canadian Oncology Drug Review Expert Review Committee.34 Currently, durvalumab in combination with chemotherapy is the standard of care and is in widespread use throughout Canada for this particular patient population.23
Drug Under Review
PD-1 is an immune-checkpoint receptor that limits the activity of T lymphocytes in peripheral tissues. The PD-1 pathway is an immune-control checkpoint that may be engaged by tumour cells to inhibit active T-cell immune surveillance. Pembrolizumab is a high-affinity antibody against PD-1, which exerts dual ligand blockade of the PD-1 pathway, including PD-L1 and PD-L2, on antigen-presenting and tumour cells. By inhibiting the PD-1 receptor from binding to its ligands, pembrolizumab reactivates tumour-specific cytotoxic T lymphocytes in the tumour microenvironment.24
In Canada, pembrolizumab has been issued market authorization without conditions for the treatment of patients with certain types of melanoma, non–small cell lung cancer, urothelial carcinoma, renal cell carcinoma, head and neck squamous cell carcinoma, colorectal cancer, carcinoma of the esophagus, cervical cancer and breast cancer, and with conditions (pending the results of trials to verify its clinical benefit) for the treatment of certain types of lymphoma, urothelial carcinoma (which is not eligible for any platinum-containing chemotherapy), bladder cancer, colorectal cancer, and endometrial cancer.24 On April 12, 2024, pembrolizumab in combination with gemcitabine-based chemotherapy (herein referred to as pembrolizumab plus chemotherapy) was approved by Health Canada for the treatment of locally advanced unresectable or metastatic BTC. The sponsor’s reimbursement request is aligned with that indication.
Pembrolizumab is provided as a single-use 100 mg/4 mL (25 mg/mL) vial and is administered as an IV infusion over 30 minutes in combination with chemotherapy. The recommended dose of pembrolizumab for BTC treatment is either 200 mg every 3 weeks or 400 mg every 6 weeks until disease progression, unacceptable toxicity, or up to 24 months or 35 doses for 200 mg or 18 doses for 400 mg, whichever is longer, in patients without disease progression. When given on the same day, pembrolizumab should be administered before chemotherapy.
In the most recent version of the National Comprehensive Cancer Network guidelines (Version 3.2023 dated November 8, 2023), pembrolizumab plus chemotherapy was added as a preferred regimen for the primary treatment of unresectable or metastatic BTC alongside durvalumab plus chemotherapy.35 In addition, consensus recommendations from a pan-Canadian panel of medical, surgical, and radiation oncologists positioned immunotherapy (durvalumab and pembrolizumab) plus chemotherapy as the preferred standard of care for patients with locally advanced unresectable or metastatic BTC.21
Key characteristics of pembrolizumab and another treatment (durvalumab) that is available, in combination with chemotherapy, for patients with locally advanced unresectable or metastatic BTC are summarized in Table 3.
Table 3: Key Characteristics of Pembrolizumab and Durvalumab
Characteristic | Pembrolizumab | Durvalumab |
|---|---|---|
Mechanism of action | Inhibiting the PD-1 receptor from binding to PD-L1 and PD-L2, and reactivating tumour-specific cytotoxic T lymphocytes. | Selective blockade of PD-L1 and PD-1 interactions and PD-L1 and CD80 interactions, which enhances antitumour immune responses. |
Indicationa | Treatment of adult patients with locally advanced unresectable or metastatic BTC in combination with chemotherapy. In addition, pembrolizumab is indicated for the treatment of other types of cancers. | Treatment of patients with locally advanced or metastatic BTC in combination with gemcitabine-based chemotherapy. In addition, durvalumab is indicated for the treatment of other types of cancers. |
Route of administration | IV | IV |
Recommended dose for BTC | Either 200 mg every 3 weeks or 400 mg every 6 weeks until disease progression, unacceptable toxicity, or up to 24 months or 35 doses for 200 mg or 18 doses for 400 mg, whichever is longer. | 1,500 mg in combination with gemcitabine-based chemotherapy every 3 weeks (21 days) followed by 1,500 mg every 4 weeks as monotherapy until disease progression or unacceptable toxicity. |
Serious adverse effects or safety issues | Immune-mediated adverse reactions (pneumonitis, colitis, hepatitis, nephritis and renal dysfunction, endocrinopathies, adrenal insufficiency, hypophysitis, type 1 diabetes mellitus, thyroid disorders, severe skin reactions), infusion-related reactions. | Immune-mediated adverse reactions (pneumonitis, hepatitis, colitis, endocrinopathies, adrenal insufficiency, hypophysitis or hypopituitarism, type 1 diabetes mellitus, nephritis, rash, myocarditis), infections, infusion-related reactions. |
BTC = biliary tract cancer; PD-1 = programmed cell death 1 protein; PD-L1 = programmed cell death 1 ligand 1; PD-L2 = programmed cell death 1 ligand 2.
aHealth Canada–approved indication.
Sources: Product monographs for pembrolizumab24 and durvalumab.33
Stakeholder Perspectives
Patient Group Input
This section was prepared by the CDA-AMC review team based on the input provided by patient groups. The full original patient input we received is included in the Stakeholder section of this report.
We received 1 patient input for this submission. CCRAN provided patient group input for this submission in collaboration with CCSN and the Gastrointestinal (GI) Society. CCRAN is a national not-for-profit patient advocacy group championing the health and well-being of Canadians touched by colorectal cancer and those at risk of developing the disease. It has expanded its mandate to serve patients with noncolorectal cancers by providing oncology-related submissions for patient groups that do not have the capacity to make these submissions or within therapeutic areas where there are currently no exclusively representative patient groups available to complete a submission (such as for the therapy currently under review).
In total, 4 patients and 4 caregivers provided input via an online survey or telephone interview. The CCSN conducted a survey that was promoted through its social media platforms and by the GI Society and CCRAN. This survey was administered between October 20 and December 1, 2023. In total, 2 female respondents completed the survey, 1 from Canada and 1 from the US, and only 1 of them had experience with pembrolizumab. CCRAN also reached out to the Cholangiocarcinoma Foundation, a US-based patient advocacy group dedicated to supporting patients with cholangiocarcinoma, which resulted in finding 4 interviewees, including 2 patients from the US and 2 caregivers (1 from the US and 1 from Canada). Two additional interviewees were secured through CCRAN’s email and social media blasts. Of the 6 interviewees, 4 (2 patients and 2 caregivers) had experience with pembrolizumab.
The signs and symptoms reported by the caregivers and patients who participated in the telephone interviews included jaundice, abdominal pain or discomfort, back pain, reflux, dark urine, fatigue, lack of energy, weight loss, elevated liver function results, sleep problems that included nights sweats, nausea and vomiting, dry mouth, GI symptoms, lack of appetite, neuropathy, and general feeling of unwellness. These were consistent with the signs and symptoms experienced by the 2 patients who participated in the CCSN survey. According to the patient group input, the impact of these symptoms on patients’ lives included anxiety, inability to do daily activities, depressive mood, trouble meeting the needs of the family, and financial difficulties. Three out of 6 interviewees described how they or their loved ones were required to undergo stenting for their obstructive disease or the insertion of biliary drains and described the complications that ensued, including life-threatening cholangitis.
The current therapies noted in the patient group input are gemcitabine plus cisplatin, pembrolizumab plus cyclophosphamide plus DPX-Survivac, gemcitabine plus cisplatin plus pembrolizumab, gemcitabine plus cisplatin followed by pembrolizumab, 5-fluorouracil plus oxaliplatin, FOLFOX (folinic acid, fluorouracil, and oxaliplatin), Yttrium-90, microwave ablation, stereotactic body radiation and conventional radiation therapy, and surgical therapy. Survey respondents mentioned that informed practitioners and mental health support are among the unmet needs of current treatment. CCRAN highlighted that there is a significant unmet need for more effective and tolerable treatment options for BTC, particularly in the first-line setting. Moreover, all interviewees stressed how important it was to undergo genomic profiling, a diagnostic tool that was not offered to them and for which they had to seek out on their own as an out-of-pocket expense. CCRAN clarified that the patients and caregivers interviewed stressed the importance of having access to a targeted therapy as a first-line treatment of metastatic BTC that has fewer side effects, allows for a cure of the disease, improves quality of life, offers equitable access and, if possible, oral administration.
According to the patient group input, among 4 interviewees who had experience with pembrolizumab, 1 patient was absolutely convinced that pembrolizumab had cured her, and another patient reported having hope and experiencing some signs of improvement; 1 caregiver reported that pembrolizumab had significant clinical benefits and that, if her late husband had had the chance to start pembrolizumab sooner, he could have avoided significant toxicity. Another caregiver mentioned that the trial (pembrolizumab plus cyclophosphamide plus the DPX-Survivac vaccine) had managed to stabilize his father and provided him with a satisfactory quality of life. CCRAN noted that all 3 interviewees from the US who had been treated with gemcitabine and cisplatin in combination with pembrolizumab, or gemcitabine and cisplatin followed by pembrolizumab, achieved a remarkable clinical and radiographic response, and had improvement in cancer-induced symptoms and quality of life. Based on the patient group input, the side effects related to pembrolizumab included compromised thyroid function and body aches. Two interviewees mentioned the shorter infusion time for pembrolizumab compared with gemcitabine plus cisplatin.
According to the patient group input, 1 respondent had received pembrolizumab in addition to gemcitabine and cisplatin, Taxotere, and therapies such as a liver resection, 3 video-assisted thoracoscopic lung surgeries, a thoracotomy, and adoptive T-cell therapy. This respondent noted fatigue and joint aches as side effects of pembrolizumab and reported that, compared with other therapies, pembrolizumab was “much better” in symptom and side effects management, ease of use, and controlling disease progression.
CCRAN stated that important patient outcomes include improved quality of life, delayed onset of symptoms, reduced side effects, prolonged OS and PFS, and providing a cure. CCRAN added that patients value having access to new therapies that have few side effects, can improve their quality of life, allow them to be engaged in society, and help them to be committed to their families and friends. CCRAN stated that 1 critical unmet need is to have more treatment options available to patients with metastatic BTC, as current options are limited. The introduction of novel, effective, easily administered, and less toxic targeted therapeutics is of paramount importance for this patient population.
Clinician Input
Input From the Clinical Experts Consulted by CDA-AMC
All our review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of BTC.
Unmet Needs
The clinical experts indicated that for patients with locally advanced unresectable or metastatic BTC, the most important goals of treatment are to prolong life, delay disease progression, alleviate symptoms, and maintain patients’ quality of life. The experts identified 2 unmet needs associated with the current treatments for advanced BTC: to have curative therapies and biomarkers that can help with patient selection.
Place in Therapy
The clinical experts indicated that currently, durvalumab in combination with chemotherapy is widely used in the treatment of advanced BTC, and pembrolizumab will be the second immune-checkpoint inhibitor to be used along with chemotherapy for these patients. The experts also noted that pembrolizumab would be used as a first-line treatment, and it is inappropriate to recommend that patients try other treatments before initiating pembrolizumab. Furthermore, the experts suggested that after a maximum of 8 cycles of combination therapy, treatment with pembrolizumab could be continued with or without gemcitabine.
Patient Population
Advanced BTCs comprise a rare group of malignancies with a poor prognosis. Overall, BTC comprises less than 0.5% of all cancer diagnoses in Canada each year. The clinical experts noted that since no biomarkers have been identified for selecting patients who are suitable for the combination regimen, all patients with advanced BTC should be eligible for pembrolizumab plus chemotherapy, if this is not contraindicated in these patients. For example, patients with good performance status and with no comorbidities that may preclude them from receiving chemotherapy (such as cisplatin or carboplatin plus gemcitabine) are eligible.
Assessing the Response Treatment
The experts noted that in clinical practice, patients who are receiving treatments for advanced BTC would have regular imaging scans, such as CTs, to monitor their responses to treatment. Other assessments include patients’ functional status (e.g., ECOG Performance Status) and disease status. Usually, these assessments are reviewed every 2 to 3 months for patients with advanced BTC. This practice is consistently adopted by the treating physicians.
Discontinuing Treatment
According to the clinical experts we consulted, treatment with a combination of pembrolizumab and chemotherapy will be discontinued if disease progression is detected based on the results of an imaging scan, or if the patients experience any intolerable adverse effects related to the treatment.
Prescribing Considerations
The clinical experts noted that, in general, patients should be treated by a medical oncologist who has knowledge of BTC management. The patients can receive the treatment in any setting, such as a community or academic centre.
Clinician Group Input
This section was prepared by the CDA-AMC review team based on the input provided by a clinician group. The full original clinician group input we received has been included in the Stakeholder section of this report.
The OH-CCO Gastrointestinal Cancer Drug Advisory Committee provided input for this submission. OH-CCO’s drug advisory committees provide timely evidence-based clinical and health system guidance on drug-related issues in support of OH-CCO’s mandate, provincial drug reimbursement programs, and OH-CCO’s Systemic Treatment Program. OH-CCO collected information by email from 2 clinicians.
OH-CCO noted that the standard of care is gemcitabine plus cisplatin and gemcitabine plus carboplatin, and the treatment goals would be prolonged life, delayed disease progression, and improved quality of life. OH-CCO added that the 1 available regimen has a poor duration of response; therefore, new regimens are required.
The clinician group stated that pembrolizumab can be safely added to first-line chemotherapy that is well tolerated, and all patients who align with the clinical trial criteria are best suited for the drug under review. The clinician group believes that clinical and/or radiologic progression, as assessed by the treating oncologist, determines whether a patient is responding to treatment in clinical practice, and treatment should be discontinued at the discretion of the treating oncologist if there is disease progression or toxicity. Additionally, the appropriate setting for treatment would be a hospital (outpatient clinic), and a specialist would be required.
Drug Program Input
The drug programs provide input on each drug reviewed through our reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts we consulted are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
In the KEYNOTE-966 study, patients received pembrolizumab in combination with chemotherapy (gemcitabine and cisplatin). The comparator was chemotherapy alone (gemcitabine and cisplatin). Durvalumab in combination with chemotherapy received a positive CADTH recommendation in April 2023 for the first-line treatment of patients with locally advanced (not amenable to surgery) or metastatic BTC. Other treatment options include chemotherapy alone, such as gemcitabine in combination with cisplatin.
|
|
Considerations for initiation of therapy | |
The KEYNOTE-966 study required histologic confirmation of unresectable locally advanced or metastatic extrahepatic cholangiocarcinoma (including mixed hepatocellular carcinoma and cholangiocarcinoma), gallbladder cancer, or intrahepatic cholangiocarcinoma.
| The clinical experts confirmed that a histologic diagnosis of unresectable locally advanced or metastatic BTC is required for the patients to be eligible for treatment with pembrolizumab. |
Should the criteria for pembrolizumab plus chemotherapy be similar to that of durvalumab plus chemotherapy for patients with locally advanced unresectable or metastatic BTC? | The clinical experts indicated that the criteria for pembrolizumab plus chemotherapy should be similar to that of durvalumab plus chemotherapy for patients with locally advanced unresectable or metastatic BTC. |
For patients who complete 2 years of treatment with pembrolizumab and experience disease progression or recurrence, are they eligible for re-treatment with pembrolizumab for up to 1 year (17 cycles)? If re-treatment is permitted, would this be as pembrolizumab monotherapy or in combination with chemotherapy? | The clinical experts noted that patients who have completed 2 years of treatment with pembrolizumab but experience disease progression or recurrence are eligible for re-treatment with pembrolizumab. Whether pembrolizumab is used as monotherapy or is combined with chemotherapy should be at the discretion of the treating oncologist. Patients’ response and possible intolerable toxicity from the previous chemotherapy should be evaluated. |
Considerations for discontinuation of therapy | |
If disease progression is identified during a drug holiday, can the treatment with pembrolizumab and chemotherapy be resumed? If a patient cannot tolerate the chemotherapy combination, are they able to continue with pembrolizumab alone? Is there a minimum number of chemotherapy cycles that must be given concurrently with pembrolizumab? | The clinical experts noted that if disease progression is identified during a drug holiday, treatment with pembrolizumab and chemotherapy can be resumed. The experts also noted that if the tumour grows within 2 to 3 months of re-treatment, this is counted as off-therapy growth, rather than disease progression. The experts indicated that if a patient cannot tolerate the chemotherapy combination, they are able to continue with pembrolizumab monotherapy. This is done in clinical practice and was the case in both clinical trials. The experts noted that a minimum of 1 cycle of chemotherapy would have to be given concurrently with pembrolizumab, as that is the recommended treatment regimen for this patient population. |
Considerations for the prescribing of therapy | |
For consistency, jurisdictions would plan on implementing pembrolizumab as weight-based dosing up to a cap (e.g., 2 mg/kg every 3 weeks to a maximum dose of 200 mg or 4 mg/kg every 6 weeks to a maximum of 400 mg), similar to other indications. | Comment from the drug programs to inform pERC deliberations. |
Generalizability | |
Should patients with an ECOG Performance Status of 2 or greater be eligible for treatment with pembrolizumab plus gemcitabine and cisplatin? | The clinical experts indicated that in the KEYNOTE-966 trial, only patients with an ECOG Performance Status 0 or 1 were included. In clinical practice, patients with an ECOG Performance Status of 2 may still be treated with pembrolizumab plus gemcitabine and cisplatin. |
Should patients with ampullary cancer be eligible for this treatment? | The clinical experts indicated that patients with ampullary cancer are not eligible for treatment with pembrolizumab plus gemcitabine and cisplatin. |
Should treatment with pembrolizumab be added to patients who are currently on, or who have just completed, a first-line chemotherapy regimen? | The experts indicated it is reasonable for treatment with pembrolizumab to be added to patients who are currently on or who have just completed a first-line chemotherapy, if disease progression is not observed. |
Funding algorithm (oncology only) | |
This drug may change the place in therapy of the comparator drugs. | Comment from the drug programs to inform pERC deliberations. |
Care provision issues | |
Pembrolizumab is already prepared and administered at facilities throughout Canada. Health care professionals have extensive experience with it. The preparation and administration times for pembrolizumab are relatively reasonable and would not be expected to significantly increase the use of health system resources. However, there is the additional cost related to drug wastage, since there is only 1 vial size available. | Comment from the drug programs to inform pERC deliberations. |
System and economic issues | |
At the time of this review, durvalumab is currently under pCPA negotiations. | Comment from the drug programs to inform pERC deliberations. |
BTC = biliary tract cancer; ECOG = Eastern Cooperative Oncology Group; pCPA = pan-Canadian Pharmaceutical Alliance; pERC = pan-Canadian Oncology Drug Review Expert Review Committee.
Clinical Evidence
The objective of this Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of pembrolizumab (100 mg/4 mL or 25 mg/mL) for IV infusion in the treatment of adult patients with locally advanced unresectable or metastatic BTC, in combination with chemotherapy. The focus will be placed on comparing pembrolizumab with relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence provided by the sponsor in the review of pembrolizumab is presented in 4 sections, with our critical appraisal of the evidence included at the end of each section. The first section, the systematic review, includes pivotal studies and RCTs that were selected according to the sponsor’s systematic review protocol. Our assessment of the certainty of the evidence in this first section using the GRADE approach follows the critical appraisal of the evidence. The second section is reserved for evidence from long-term extension studies; however, none were submitted by the sponsor. The third section includes indirect evidence from the sponsor. The fourth section is reserved for additional studies that were considered by the sponsor to address important gaps in the systematic review evidence; however, no studies addressing gaps were submitted.
Included Studies
Clinical evidence from the following is included in the our review and appraised in this document:
Systematic Review
Contents within this section have been informed by materials submitted by the sponsor. The following was summarized and validated by the CDA-AMC review team.
Description of Studies
The KEYNOTE-966 study is a global, multicentre, double-blind, randomized, placebo-controlled, phase III trial that compared pembrolizumab plus gemcitabine and cisplatin chemotherapy (hereafter referred to as pembrolizumab plus chemotherapy) versus placebo plus gemcitabine and cisplatin chemotherapy (hereafter referred to as placebo plus chemotherapy) in 1,069 adult patients with locally advanced unresectable or metastatic BTC (including intrahepatic BTC, extrahepatic BTC, and GBC). The primary objective of this trial was to compare the OS for pembrolizumab versus placebo (each combined with chemotherapy), as described in Table 5 and Figure 1.
The trial was conducted at 175 sites in 24 countries, including 5 sites in Canada. Following screening, patients were randomized 1:1 using a central interactive voice-response system to receive either pembrolizumab plus chemotherapy or placebo plus chemotherapy. Randomization was stratified by geographic region (Asia versus non-Asia), disease stage (locally advanced versus metastatic), and site of origin (gallbladder versus intrahepatic versus extrahepatic). Patients, investigators, and study personnel were blinded to treatment assignment.
The results reported in this submission are for the prespecified final analysis for OS corresponding with a DCO of December 15, 2022. The final analysis for the outcomes of PFS was prespecified to occur at interim analysis 1 (IA1) with a DCO of December 15, 2021. In addition, PFS data at the longest follow-up time points are included in this review. Study treatments were continued until disease progression, unacceptable toxicity, investigator decision, withdrawal of consent, or another reason, whichever occurred first. Crossover between treatment groups was not permitted.
The characteristics of the KEYNOTE-966 study are summarized in Table 5. The design of this study is illustrated in Figure 1.
Figure 1: Study Design of the KEYNOTE-966 Trial
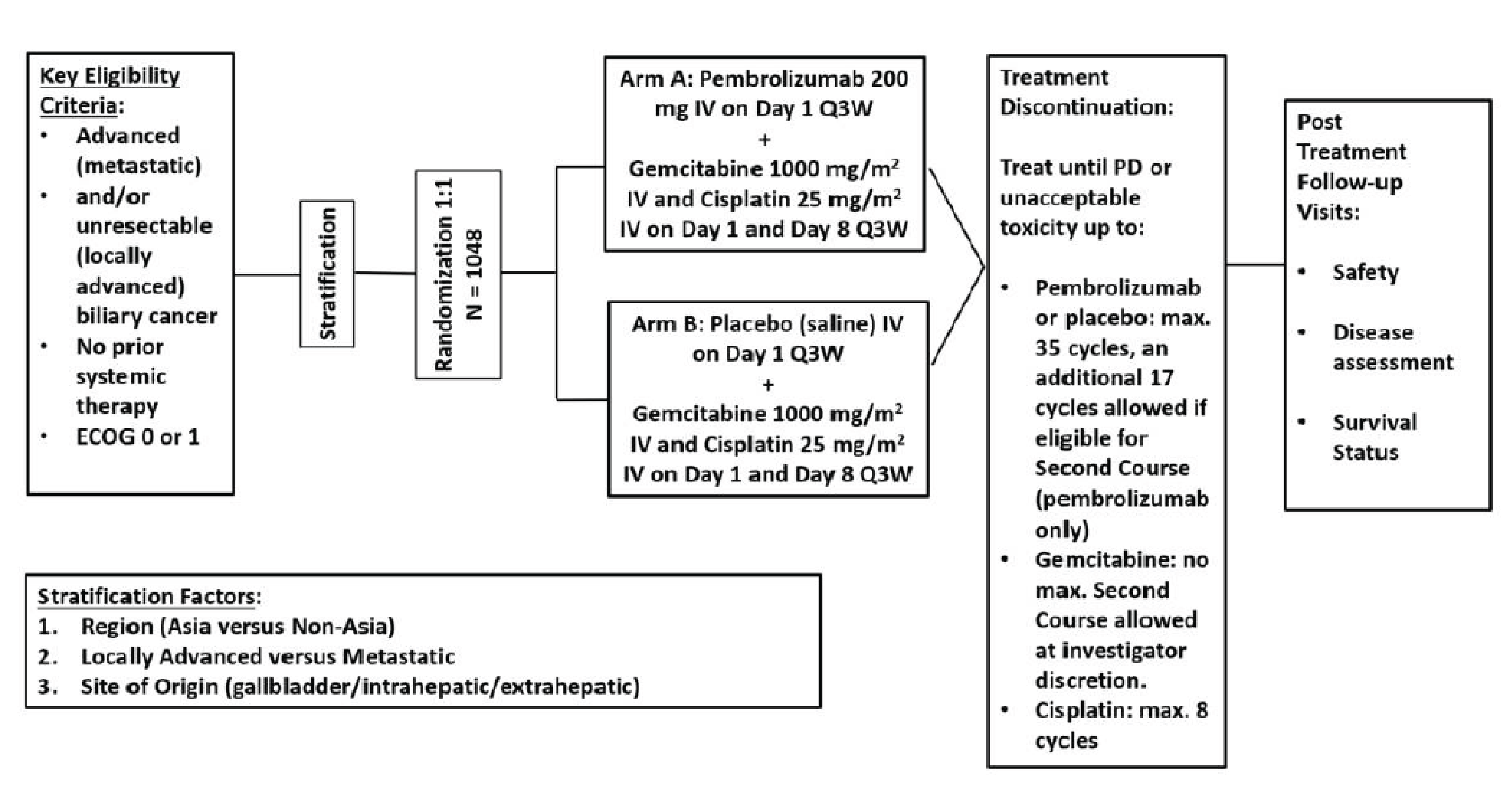
ECOG = European Cooperative Oncology Group; max. = maximum; PD = progressive disease; Q3W = every 3 weeks.
Note: The total projected enrolment in the global cohort was 1,048 patients; however, a total of 1,069 patients were enrolled. After enrolment was complete, the trial remained open to enrolment in China alone for the China extension cohort of 46 patients until a total of 158 Chinese patients had been enrolled across the global and extension parts to meet local regulatory requirements in China.
Source: Clinical Study Report for the KEYNOTE-966 study.25
Table 5: Details of Study Included in the Systematic Review
Details | KEYNOTE-966 Study |
|---|---|
Designs and populations | |
Study design | Phase III, DB, placebo-controlled RCT |
Locations | 175 centres in 24 countries or regions (Argentina, Australia, Belgium, Brazil, Canada, Chile, China, France, Germany, Hong Kong, Ireland, Israel, Italy, Japan, Malaysia, Netherlands, New Zealand, South Korea, Spain, Taiwan, Thailand, Turkey, UK, US) |
Patient enrolment dates |
|
Randomized (N) | A total of 1,069 (N) patients were randomized as follows:
|
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Intervention | Pembrolizumab, 200 mg on day 1 of each cycle q.3.w. by IV infusion (maximum 35 cycles) in combination with gemcitabine (1,000 mg/m2 on days 1 and 8 of each cycle q.3.w.; no maximum duration) and cisplatin (25 mg/m2 by IV infusion on days 1 and 8 of each cycle; maximum duration 8 cycles). |
Comparator | Placebo on day 1 of each cycle q.3.w. by IV infusion (maximum 35 cycles) in combination with gemcitabine (1,000 mg/m2 on days 1 and 8 of each cycle q.3.w.; no maximum duration) and cisplatin (25 mg/m2 by IV infusion on days 1 and 8 of each cycle; maximum duration of 8 cycles). |
Study duration | |
Screening phase | Up to 28 days before treatment randomization. |
Run-in phase | NA |
Treatment phase | Up to 35 cycles of pembrolizumab or placebo or until disease progression or unacceptable toxicity. |
Follow-up phase | Until PD, unacceptable toxicity, withdrawal of consent, or other discontinuation criteria are met, after which there was a 30-day safety follow-up after discontinuation (≤ 90 days for SAEs in the absence of new anticancer therapy). |
Outcomes | |
Primary end point | Overall survival |
Secondary and exploratory end points | Secondary:
Tertiary and exploratory:
|
Publication status | |
Publications | Kelley, RS et al. (2023);26 Finn, RS et al. (2020);37 Kelley, RS et al. (2020);38 Kelley RS et al. (2020);39 Valle, JW et al. (2020);40 Vogel, AF et al. (2020);41 Yoo et al. (2023);42 NCT04003636.43 |
anti-HBc = antihepatitis B core antibody; BTC = biliary tract cancer; CTLA-4 = cytotoxic T-lymphocyte antigen 4; DB = double blind; ECOG = Eastern Cooperative Oncology Group; EORTC QLQ-BIL21 = European Organization for Research and Treatment of Cancer Quality of Life Questionnaire Cholangiocarcinoma and Gallbladder Cancer Module 21; EORTC QLQ-C30 = European Organization for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; HbsAg = hepatitis B surface antigen; HBV = hepatitis B virus; HCV = hepatitis C virus; HRQoL = health-related quality of life; iRECIST = Response Evaluation Criteria in Solid Tumours Version 1.1 modified for immune-based therapies; LPLV = last patient last visit; NA = not applicable; PD = progressive disease; PD-1 = programmed cell death 1 protein; PD-L1 = programmed cell death 1 ligand 1; PD-L2 = programmed death 1 ligand 2; q.3.w. = every 3 weeks; q.6.w. = every 6 weeks; R2 = residual tumour classification 2; RCT = randomized controlled trial; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1; SAE = serious adverse event; SD = standard deviation; TACE = transarterial chemoembolization.
Source: Clinical Study Report for the KEYNOTE-966 study.25 Details included in the table are from the sponsor’s summary of clinical evidence.
Populations
Inclusion and Exclusion Criteria
Eligible patients included males and females aged 18 years or older who had a histologically confirmed diagnosis of locally advanced unresectable or metastatic BTC (i.e., intrahepatic or extrahepatic BTC, including GBC and mixed hepatocellular CCA), measurable disease as per RECIST 1.1 with no prior systemic therapy except for neoadjuvant and adjuvant therapy, an ECOG Performance Status of 0 or 1, adequate tumour tissue for biomarker assessment, adequate organ function, and a life expectancy of more than 3 months. Patients with controlled hepatitis B virus and past or ongoing hepatitis C virus infection were also permitted. Patients were excluded if they had ampullary cancer or had an active autoimmune disease that required systemic treatment in the previous 2 years. Full inclusion and exclusion criteria are provided in Table 5.
Interventions
The main study intervention was pembrolizumab at a fixed dose of 200 mg or placebo (i.e., normal saline) administered every 3 weeks by IV infusion at clinical sites on day 1 of each cycle, up to a maximum of 35 cycles. Both pembrolizumab or placebo were administered in combination with chemotherapy comprising gemcitabine 1,000 mg/m2 by IV infusion on days 1 and 8 of each cycle every 3 weeks until progressive disease or unacceptable toxicity, and cisplatin 25 mg/m2 by IV infusion on days 1 and 8 of each cycle every 3 weeks up to a maximum of 8 cycles. Dose reductions of pembrolizumab or placebo were not permitted. Crossover between treatment groups was not allowed.
Medications or vaccinations specifically prohibited in the exclusion criteria were not allowed during the study. If there was a clinical indication for any medication or vaccination specifically prohibited, discontinuation from study therapy or vaccination might be required.
Patients were to receive appropriate supportive care and rescue medications as deemed necessary by the treating investigator, such as oral or IV treatment with corticosteroids, or additional anti-inflammatory drugs if symptoms do not improve with the administration of corticosteroids, antihyperglycemic drugs, hormonal replacements, or antibiotics.
Outcomes
A list of efficacy end points assessed in this Clinical Review Report is provided in Table 6, followed by descriptions of the outcome measures. Summarized end points are based on outcomes included in the sponsor’s summary of clinical evidence as well as any outcomes identified as important to this review according to the clinical experts we consulted and in the stakeholder input from the patient and clinician groups and public drug plans. Using the same considerations, our review team selected end points that were considered to be most relevant to inform CDA-AMC’s expert committee deliberations and finalized this list of end points in consultation with members of the expert committee.
Efficacy assessments included contrast-enhanced CT (preferred) or MRI of the chest, abdomen, and pelvis within 4 weeks before randomization, 6 weeks after the first administration of the study treatment, and then every 6 weeks up to week 54 and every 12 weeks thereafter. Contrast-enhanced MRI (preferred) or CT of the brain and whole-body radionuclide bone scans were done as clinically indicated. Imaging continued until progressive disease (as measured using RECIST 1.1 and assessed by a blinded independent central review [BICR]), the start of a new anticancer therapy, death, or withdrawal of consent. Survival was assessed every 12 weeks until death, withdrawal of consent, or study end. HRQoL questionnaires were administered on day 1 of cycles 1 to 10, 12, 14, 16, and 18, at end of treatment, and at the 30-day safety follow-up.
All summarized efficacy end points were assessed using GRADE. Select notable harms outcomes considered important for informing our expert committee deliberations were also assessed using GRADE.
Table 6: Outcomes Summarized From the KEYNOTE-966 Study
Outcome measure | Time point | KEYNOTE-966 |
|---|---|---|
OS | Results at 6, 12, 18, and 24 months were reported | Primary outcome |
PFS | Results at 6, 9, 12, 15, and 18 months were reported | Secondary outcome |
HRQoL: EORTC QLQ-C30 and EORTC QLQ-BIL21 | Results at 18 weeks were reported | Exploratory outcome |
Any AEs | Results were based on a DCO of December 15, 2022, in the APaT population | Secondary outcome |
Any SAEs | ||
Any AEs leading to treatment discontinuation | ||
Immune-mediated AEs |
AE = adverse event; APaT = all participants as treated; DCO = data cut-off; EORTC QLQ-BIL21 = European Organization for Research and Treatment of Cancer Quality of Life Questionnaire Cholangiocarcinoma and Gallbladder Cancer Module 21; EORTC QLQ-C30 = European Organization for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; OS = overall survival; PFS = progression-free survival; SAE = serious adverse event.
Note: The outcomes of OS and PFS were adjusted for multiple comparisons. Other efficacy or safety outcomes or PROs in this table were not adjusted for multiple comparisons.
Source: Clinical Study Report for the KEYNOTE-966 study.25 Details included in the table are from the sponsor’s summary of clinical evidence.
The primary outcome of the KEYNOTE-966 study was OS, defined as the time from randomization to death due to any cause. Patients without documented death at the time of analysis were censored at the last known alive date or DCO date, whichever was the earliest.
The secondary outcomes and their definitions are as follows:
PFS: The time from randomization to the first documented instance of progressive disease per RECIST 1.1 assessed by BICR, or death due to any cause, whichever occurs first. For the primary analysis of PFS, patients were censored if they:
experienced an event (progressive disease or death) immediately after 2 or more missed disease assessments
initiated new anticancer therapy before documented progression
did not start new anticancer therapy and did not experience an event at the last disease assessment.
Safety and tolerability: Assessed by review of AEs, SAEs, study intervention discontinuations due to AEs, fatal AEs, laboratory parameters, and vital signs.
The patient-reported outcomes (PROs) measured in the KEYNOTE-966 study are described in Table 7.
EORTC Quality of Life Questionnaire Core 30
The EORTC QLQ-C30 is a widely used, cancer-specific, HRQoL instrument that contains 30 items. It measures 5 functional dimensions (physical, role, emotional, cognitive, and social), 3 symptom items (fatigue, nausea or vomiting, and pain), 6 single items (dyspnea, sleep disturbance, appetite loss, constipation, diarrhea, and financial impact), and a GHS quality-of-life scale.44 The validity, reliability, and responsiveness of the EORTC QLQ-C30 have been demonstrated in patients with BTC (further described in Table 7).44 For the GHS quality-of-life scales and the functional domains of the EORTC QLQ-C30, a higher score denotes improvement or better health. For the symptom domains, a higher score indicates a worsening of symptoms or more symptom burden. In general, the MIDs for between-group comparisons (within a range of 5 to 10 points) have been established based on data from clinical trials of different types of cancers.29
EORTC Quality of Life Questionnaire Cholangiocarcinoma and Gallbladder Cancer Module 21
The EORTC QLQ-BIL21 is a disease-specific questionnaire for the assessment of quality of life in patients with CCA and GBC. It was developed to supplement the EORTC QLQ-C30 questionnaire.45,46 Its validity and reliability have been evaluated in patients with BTC (Table 7). The EORTC QLQ-BIL21 consists of 21 questions where 3 single items assess treatment side effects, difficulties with drainage bags and tubes and concerns regarding weight loss. Eighteen items are grouped into 5 symptom scales related to: eating (4 items), jaundice (3 items), tiredness (3 items), pain (4 items) and anxiety (4 items). The response is provided on a 4-point Likert scale. For the symptom domains of the EORTC QLQ-BIL21, a higher score indicates a worsening of symptoms or more symptom burden. No MID for between-group comparisons was identified in patients with BTC.
Table 7: Summary of Outcome Measures and the Related Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
EORTC QLQ-BIL21 | A disease-specific module to be used in addition to the EORTC QLQ-C30 to assess HRQoL in patients with CCA and GBC.46 It consists of 21 questions, with 18 items grouped into 5 symptom scales: eating (4 items), jaundice (3 items), tiredness (3 items), pain (4 items), and anxiety (4 items). The remaining 3 items are single-item assessments of treatment side effects, difficulties with drainage bags or tubes, and concerns about weight loss.46 Patients complete the questionnaire based on a 1-week recall period by rating each item on a 4-point Likert scale (1 = not at all, 2 = a little, 3 = quite a bit, 4 = very much).46 The scores are then transformed linearly to a 0 to 100 scale to yield scale scores using EORTC guidelines, with higher scores indicating more severe symptoms.45,46 | An international study was conducted to validate the EORTC QLQ-BIL21 in patients with BTC.45 The study included 172 adult patients with CCA and 91 patients with GBC who had an expected minimum survival of 3 months and were undergoing treatment. Patients completed the EORTC QLQ-C30 and EORTC QLQ-BIL21, and KPS was recorded at ≤ 1 month before treatment and 2 months later. The analysis included 478 questionnaires. Patients were assigned to 1 of 3 groups based on the treatment received: surgical treatment; chemotherapy, radiotherapy, photodynamic, or laser therapy; or supportive care only. Validity: All items demonstrated item–scale convergence (construct) validity (Pearson r > 0.4, prespecified). For discriminant validity, no items had r > 0.70, indicating no items correlated with scales outside of the scale they were placed in. Known groups (construct) validity was demonstrated by the greater EORTC QLQ-BIL21 mean scores in patients with KPS < 70 at baseline compared with patients with KPS > 70 for all scales, with the exception of the jaundice and weight loss scales (N = 238 to 256). Reliability: Internal consistency was acceptable (alpha ≥ 0.70) for all multi-item scales at baseline (Cronbach alpha = 0.71 to 0.87) and when the assessment time points were pooled (Cronbach alpha = 0.71 to 0.89). Of note, the Cronbach alpha ranged from 0.61 to 0.93 at the 2-month assessment, with a coefficient of 0.61 and 0.68 for the jaundice and pain scales, respectively. Test–retest reliability was acceptable (ICC ≥ 0.70)47 for all scales, with the ICC ranging from 0.81 to 0.96 in 67 patients with clinically stable disease across all intervention groups in 2 weeks.a Responsiveness: Not identified for patients with BTC, including CCA and GBC. | Not identified for patients with BTC, including CCA and GBC. |
EORTC QLQ-C30 | A multidimensional, cancer-specific, patient-reported measure used to assess HRQoL in response to treatment in clinical trials.48 The core questionnaire consists of 30 items that make 5 multi-item functional scales: physical (5 items), role (2 items), emotional (4 items), cognitive (2 items), and social functioning (2 items). There are also 3 multi-item symptom scales: fatigue (3 items), nausea/vomiting (2 items), and pain (2 items). There are also 6 single-item symptom scales (dyspnea, insomnia, appetite loss, constipation, diarrhea, and financial impact), and a 2-item global QoL scale. Patients complete the questionnaire based on a 1-week recall period by rating most items on a 4-point Likert-type scale (1 = not at all; 2 = a little; 3 = quite a bit; 4 = very much). For the 2 items in the global QoL scale, the response format is a 7-point Likert-type scale (1 = very poor; 7 = excellent).49 Raw scores for each scale are computed as the average of the items that contribute to a particular scale. Each raw scale score is converted to a standardized score that ranges from 0 to 100 using a linear transformation. A decline in the symptom scale score reflects an improvement, whereas increases in the function and QoL scale scores reflect an improvement.49 According to the EORTC QLQ-C30 scoring algorithm, if there are missing items for a scale, the score for that scale can still be computed if there are responses for at least one-half of the items. In calculating the scale score, missing items are ignored.49 | The psychometric properties of the EORTC QLQ-C30 were evaluated in the validation study for the EORTC QLQ-BIL21 described previously.45 Validity: All items demonstrated item–scale convergence (construct) validity (Pearson r > 0.4, prespecified). Although the study stated that a known group comparison was performed for the EORTC QLQ-C30, the results were not reported. Reliability: Internal consistency was acceptable (alpha ≥ 0.70) for all scales, except for the physical function (alpha = 0.47), cognitive function (alpha = 0.65), and nausea/vomiting (alpha = 0.67) scales at baseline. Test–retest reliability was demonstrated by the ICCs that ranged from 0.52 to 0.92 in 67 patients with clinically stable disease across all intervention groups in 2 weeksa Responsiveness: Although the study stated that responsiveness to clinical change over time in the EORTC QLQ-C30 was measured, the results were not reported. | No MID was identified for patients with BTC, including CCA and GBC. For other types of cancers,29 the between-group differences in MIDs for improvement and deterioration ranged from 5 to 10 points across most scales:
|
BTC = biliary tract cancer; CCA = cholangiocarcinoma; EORTC QLQ-BIL21 = European Organization for the Research and Treatment of Cancer Quality of Life Questionnaire Cholangiocarcinoma and Gallbladder Cancer Module 21; EORTC QLQ-C30 = European Organization for the Research and Treatment of Cancer Quality of Life Questionnaire Core 30; GBC = gallbladder cancer; HRQoL = health-related quality of life; ICC = intraclass correlation coefficient; KPS = Karnofsky performance scale; MID = minimally important difference; QoL = quality of life.
aPatients who received IV chemotherapy at the time were excluded from the test–retest assessment.
The tertiary or exploratory outcomes that are included in this Clinical Review Report include efficacy outcomes (PFS, ORR, duration of response) per RECIST 1.1 modified for immune-based therapies as assessed by the investigator, time to deterioration (defined as the time to first onset of at least a 10-point decrease from baseline in the GHS quality-of-life scales of the EORTC QLQ-C30 and EORTC QLQ-BIL21), and change from baseline in the GHS quality-of-life scales of the EORTC QLQ-C30 and EORTC QLQ-BIL21. Note that if the outcomes were assessed by both BICR and the investigators, the Clinical Review Report focuses on the BICR assessments rather than the investigator’s assessments, due to the reduced risk of bias associated with BICR assessments.
Statistical Analysis
Sample Size and Power Calculation
The planned sample size was 1,048 patients; however, updated power calculations were based on the enrolment of 1,069 patients, which corresponds with the actual final number of randomized patients. Based on an estimated 818 deaths at the final OS analysis and 2 interim analyses, and assuming an exponential distribution of the HR (an HR of 1 for the first 2 months and an HR of 0.75 after 2 months), the trial would have approximately 93% power to identify a statistically significant OS benefit for pembrolizumab plus chemotherapy at a 1-sided alpha of 0.025. With an enrolment of 1,069 patients and assuming 786 events at the final PFS analysis and an exponential distribution of an HR of 1 for the first 2 months and an HR of 0.7 after 2 months, the trial would have approximately 92% power to identify a statistically significant PFS benefit for pembrolizumab plus chemotherapy at a 1-sided alpha of 0.0125.
Statistical Testing
Interim and Final Analyses
Two prespecified interim analyses and 1 final analysis were planned for the evaluation of the efficacy of pembrolizumab plus chemotherapy in patients with advanced BTC.
The first analysis (i.e., IA1) was performed when approximately 585 OS events had been observed and about 26 months had passed since the start of randomization (DCO of December 15, 2021) and the second interim analysis was performed when approximately 695 OS events had been observed and about 32 months had passed since the start of randomization (DCO of May 25, 2022). At IA1, an interim OS analysis was performed, as well as an analysis of the final PFS and ORR (not presented in this Clinical Review Report) if OS superiority was established. An interim OS analysis was performed at the second interim analysis.
A prespecified final analysis for OS was performed when approximately 818 OS events had been observed and about 38 months had passed from the start of randomization (DCO of December 15, 2022). Posthoc analyses of PFS were also performed at this time. The safety of the study drugs was assessed at a DCO of December 15, 2022. An independent data and safety monitoring committee provided study oversight and assessed efficacy and safety at each prespecified interim analysis.
A summary of the statistical analyses undertaken for each relevant clinical trial outcome is provided in Table 8. The nonparametric KM method was used to estimate OS at 12 and 24 months and PFS at 6 and 12 months at IA1, and 12 and 24 months at the final analysis. Between-group comparisons of OS and PFS were assessed using a stratified log-rank test; the magnitude of the treatment difference (i.e., HR) and the 95% CI were calculated using a stratified Cox regression model using the Efron method for tie handling and treatment as a covariate. Randomization stratification factors (geographic region, locally advanced versus metastatic, and site of origin) were applied to the stratified log-rank test and the stratified Cox regression model. The proportional hazards assumption on OS could be examined using both graphical and analytical methods, if warranted. The log (-log) of the survival function versus time for OS were plotted for the comparison between the pembrolizumab plus chemotherapy arm and placebo plus chemotherapy arm. If the curves are not parallel, indicating that hazards are not proportional, supportive analyses such as a sensitivity analysis based on the MaxCombo test with Fleming-Harrington (FH) weighted log-rank FH (0, 1) and FH (1, 1) tests at the final analysis of OS might be conducted to account for the possible non-proportional hazards effect associated with immunotherapies when the proportional hazard assumption was violated. The sponsor noted that the proportional hazards assumption has been examined through both a formal statistical test and visual examination. No violation of the assumption was found; therefore, no sensitivity analyses were performed.
The validity of the proportional hazards assumption on PFS was examined by formal tests and visual examinations.
For the analyses of HRQoL data, a constrained longitudinal data analysis model proposed by Liang and Zeger50 was applied, with the HRQoL score as the response variable, with treatment, time, the treatment by time interaction, strata of geographic region, disease status, and site of origin as covariates. The treatment difference in terms of the LSM change from baseline was estimated from this model together with the 95% CI. Model-based LSMs with 95% CIs were provided by treatment group for the HRQoL scores at baseline and postbaseline time points.
Table 8: Statistical Analysis of Efficacy End Points in the KEYNOTE-966 Study
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
Overall survival |
|
| Censored at the last known alive date | — |
Progression-free survival | Kaplan-Meier method
|
| Censoring rules for primary analysis:
| Censoring rules for sensitivity analyses:
|
PRO outcomes (i.e., EORTC QLQ-C30, EORTC QLQ-BIL21) | cLDA model | NA | NA | NA |
CI = confidence interval; cLDA = constrained longitudinal data analysis; CR = complete response; EORTC QLQ-BIL21 = European Organization for Research and Treatment of Cancer Quality of Life Questionnaire Cholangiocarcinoma and Gallbladder Cancer Module 21; EORTC QLQ-C30 = European Organization for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; HR = hazard ratio; NA = not applicable; PD = progressive disease; PRO = patient-reported outcome; SA = sensitivity analysis.
Source: Clinical Study Report for the KEYNOTE-966 study.25 Details included in the table are from the sponsor’s summary of clinical evidence.
Multiplicity Adjustment
The overall type I error rate over the primary and secondary hypotheses was controlled at 2.5% (1-sided alpha of 0.025) for all OS, PFS, and ORR analyses using the graphical method of Maurer and Bretz to control multiplicity for multiple hypotheses and the interim analyses.51 According to this approach, the initial alpha of 0.025 was assigned to test OS and, if the OS null hypothesis was rejected, the corresponding alpha was reallocated equally to the hypotheses of PFS and ORR (Figure 2). If the PFS hypothesis was rejected, the corresponding alpha was reallocated to ORR; if the ORR hypothesis was rejected, the corresponding alpha was reallocated to PFS.
Additionally, within each outcome, type I error control was maintained across the first and second interim analyses and the final analysis using the minimum alpha spending strategy with a Lan-DeMets spending function approximating O’Brien-Fleming boundaries. The 1-sided P value boundaries for declaring the superiority of pembrolizumab plus chemotherapy versus placebo plus chemotherapy were 0.0200 for OS, 0.0125 for PFS, and 0.0125 for ORR.
Figure 2: Multiplicity Diagram for Control of Type I Error From the KEYNOTE-966 Study
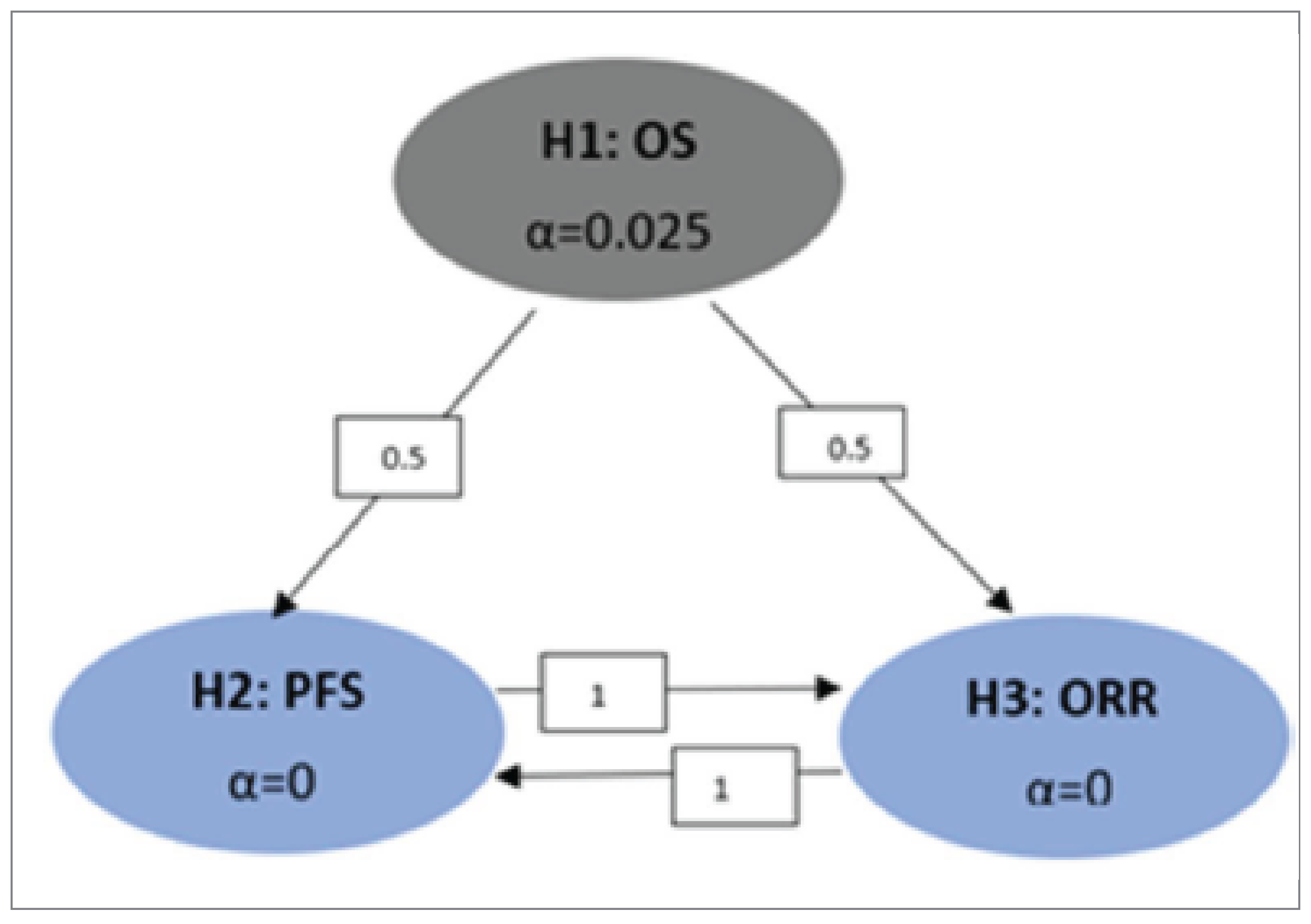
H1 = hypothesis 1; H2 = hypothesis 2; H3 = hypothesis 3; ORR = overall response rate; OS = overall survival; PFS = progression-free survival.
Source: Clinical Study Report for the KEYNOTE-966 study.25
Subgroup Analyses
Prespecified subgroup analyses were conducted to determine whether the treatment effect was consistent across various subgroups of interest. The between-group treatment effect for OS, PFS, and ORR (with nominal 95% CI) was estimated and plotted by treatment group within each category of the following classification variables:
geographic region (Asia versus Non-Asia)
disease stage (locally advanced versus metastatic)
site of origin (gallbladder versus intrahepatic versus extrahepatic)
age category (< 65 years versus ≥ 65 years)
gender (female versus male)
biliary stent and/or a biliary drain (yes, no)
antibiotics within 1 month of study start (yes, no)
prior radiation (yes, no)
prior chemotherapy (yes, no)
prior photodynamic therapy (yes, no)
smoking status (never, former, current)
microsatellite instable status (microsatellite stable, high microsatellite instability, indeterminate)
PD-L1 combined positive score 1 (CPS1) (CPS ≥ 1, CPS < 1, indeterminate)
PD-L1 CPS10 (CPS ≥ 10, CPS < 10, indeterminate)
ECOG Performance Status at randomization (0 versus 1).
An unstratified Cox model with treatment as a covariate was used to calculate the magnitude of the treatment difference in each subgroup category; the CIs for the subgroup analyses were at the nominal 95% CI level without adjustment for multiplicity. If the number of participants in a subgroup category comprised less than 5% of the intention-to-treat (ITT) population, a subgroup analysis was not performed for that category and the subgroup was not included in the forest plot.
Sensitivity Analyses
A sensitivity analysis could be performed based on the MaxCombo test with log rank FH (0, 1), FH (1, 1) at the final OS analysis to account for the potential loss of power with a log-rank test when the proportional hazard assumption was violated (results not reported in this Clinical Study Report).
To evaluate the robustness of the PFS end point per RECIST 1.1 assessed by BICR, 2 sensitivity analyses with different sets of censoring rules were performed. The first sensitivity analysis followed the ITT principle, that is, disease progressions or deaths were counted as events regardless of missed study visits or the initiation of a new anticancer therapy. The second sensitivity analysis considered discontinuation of study intervention due to reasons other than complete response or initiation of new anticancer treatment, whichever occurred later, to be a progressive disease event for patients without documented progressive disease or death. If a patient met multiple criteria for censoring, the censoring criterion that occurred earliest was applied.
Analysis Populations
The efficacy analysis population consisted of all randomized patients and the patients were analyzed using the ITT approach.
The safety analysis population included all randomized patients who received at least 1 dose of the study drug. This is also called the as-treated population.
The PRO analysis population included all randomized patients who received at least 1 dose of the study drug and completed at least 1 PRO assessment.
A summary of the analysis populations and how they are defined in the KEYNOTE-966 study is provided in Table 9.
Table 9: Analysis Populations in the KEYNOTE-966 Study
Population | Definition | Application |
|---|---|---|
Efficacy analysis population | All randomized patients. Patients were analyzed in the treatment group to which they were randomized (based on the ITT approach) | Analysis of all relevant efficacy outcomes |
Safety analysis population | All randomized patients who received at least 1 dose of the study intervention (i.e., based on the as-treated population) | Analysis of safety outcomes and treatment exposure |
PRO analysis population | All randomized patients who have received at least 1 dose of study intervention and completed at least 1 PRO assessment (i.e., based on the PRO FAS population) | Analysis of PRO outcomes |
FAS = full analysis set, ITT = intention to treat; PRO = patient-reported outcome.
Source: Clinical Study Report for the KEYNOTE-966 study.25 Details included in the table are from the sponsor’s summary of clinical evidence.
Results
Patient Disposition
Patient disposition as of the December 15, 2022, DCO for the final analysis is reported in Table 10. A total of 1,564 patients were screened; of these, 495 (31.6%) did not pass screening because they did not meet the inclusion or exclusion criteria. There were 1,069 patients randomized to the 2 treatment groups: 533 were randomized to the pembrolizumab plus chemotherapy group and 536 were randomized to the placebo plus chemotherapy group. Of these, 529 (99.2%) and 534 (99.6%) were treated, respectively; 4 patients in the pembrolizumab plus chemotherapy group and 2 patients in the placebo plus chemotherapy group were randomized but not treated. In the pembrolizumab plus chemotherapy group, 489 treated patients (92.4%) discontinued the study medication, mainly due to disease progression (n = 324; 61.2%). In the placebo plus chemotherapy group, 504 treated patients (94.4%) discontinued the study medication, also primarily due to disease progression (n = 354; 66.3%). As of the DCO for the final analysis of OS (December 15, 2022), 27 (5.1%) and 23 (4.3%) treated patients, respectively, were still ongoing in the trial.
Table 10: Patient Disposition in the KEYNOTE-966 Study (December 15, 2022, DCO)
Patient disposition | Pembrolizumab + chemotherapy (N = 533) | Placebo + chemotherapy (N = 536) |
|---|---|---|
Screened, N | 1,564 | |
Reason for screening failure, n (%) | ||
Did not meet inclusion criteria or did meet exclusion criteria | 495 (31.6) | |
Randomized, N (%) | 533 (100.0) | 536 (100.0) |
Discontinued from trial, n (%) | 414 (77.7) | 446 (83.2) |
Reason for discontinuation from trial, n (%) | ||
Death | 409 (76.7) | 443 (82.6) |
Lost to follow-up | 0 (0.0) | 1 (0.2) |
Withdrawal by patient | 5 (0.9) | 2 (0.4) |
Status for study medication of treatment phase, n (%) | ||
Treated | 529 (99.2) | 534 (99.6) |
Discontinued study medication, n (%)a | 489 (92.4) | 504 (94.4) |
Ongoinga | 27 (5.1) | 23 (4.3) |
Reason for discontinuation from study medication, n (%)a | ||
Adverse event | 67 (12.7) | 61 (11.4) |
Clinical progression | 35 (6.6) | 43 (8.1) |
Non-study anticancer therapy | 3 (0.6) | 6 (1.1) |
Physician decision | 32 (6.0) | 16 (3.0) |
Progressive disease | 324 (61.2) | 354 (66.3) |
Withdrawal by patient | 28 (5.3) | 24 (4.5) |
Efficacy analysis population, N (%) | 533 (100.0) | 536 (100.0) |
Safety analysis population or as-treated population, N (%) | 529 (99.2) | 534 (99.6) |
DCO = data cut-off.
Note: Data were obtained on December 15, 2022.
aThe denominator is the number of treated patients.
Source: Clinical Study Report for the KEYNOTE-966 study.25 Details included in the table are from the sponsor’s summary of clinical evidence.
Baseline Characteristics
The baseline characteristics were generally balanced between the 2 treatment groups. The trial enrolled approximately equal proportions of males and females (552 male patients, 51.6%; 517 female patients, 48.4%). The median age was 64.0 years (range, 23 to 85 years). Most enrolled patients were white (American Indian or Alaska Native: 3 patients, 0.3%; Asian: 495 patients, 46.3%; Black or African American: 14 patients, 1.3%; multiple ethnicities: 7 patients, 0.7%; Native Hawaiian or other Pacific Islander: 1 patient, 0.1%; white: 524 patients, 49.0%), an ECOG Performance Status score of 1 (ECOG 0: 486 patients, 45.5%; ECOG 1: 582 patients, 54.4%; ECOG ≥ 2: 1 patient, 0.1%), and were from a region outside Asia (non-Asia: 583 patients, 54.5%; Asia: 486 patients, 45.5%). Most patients had metastatic disease (locally advanced: 126 patients, 11.8%; metastatic: 943 patients, 88.2%) with an intrahepatic site of origin (intrahepatic: 633 patients, 59.2%; extrahepatic: 203 patients, 19.0%; gallbladder: 233 patients, 21.8%). Approximately 30% of these patients had received prior surgery (n = 319; 29.8%).
A summary of the baseline patient demographics and disease characteristics in the study population of the KEYNOTE-966 study is presented in Table 11.
Table 11: Summary of Baseline Characteristics in the KEYNOTE-966 Study (Efficacy Analysis Population)
Characteristic | Pembrolizumab + chemotherapy (N = 533) | Placebo + chemotherapy (N = 536) |
|---|---|---|
Sex, n (%) | ||
Male | 280 (52.5) | 272 (50.7) |
Female | 253 (47.5) | 264 (49.3) |
Age, years, n (%) unless otherwise specified | ||
< 65 | 269 (50.5) | 298 (55.6) |
≥ 65 | 264 (49.5) | 238 (44.4) |
Mean | 63.3 | 61.8 |
SD | 10.3 | 11.0 |
Median | 64.0 | 63.0 |
Range | 23 to 85 | 28 to 84 |
Race, n (%) | ||
American Indian or Alaska Native | 2 (0.4) | 1 (0.2) |
Asian | 245 (46.0) | 250 (46.6) |
Black or African American | 11 (2.1) | 3 (0.6) |
Multiple | 5 (0.9) | 2 (0.4) |
Native Hawaiian or other Pacific Islander | 1 (0.2) | 0 (0.0) |
White | 256 (48.0) | 268 (50.0) |
Missing | 13 (2.4) | 12 (2.2) |
Geographic region (by stratification factor), n (%) | ||
Asia | 242 (45.4) | 244 (45.5) |
Non-Asia | 291 (54.6) | 292 (54.5) |
Prior adjuvant therapy, n (%) | ||
Yes | 47 (8.8) | 48 (9.0) |
No | 486 (91.2) | 488 (91.0) |
Prior neoadjuvant therapy, n (%) | ||
Yes | 3 (0.6) | 1 (0.2) |
No | 530 (99.4) | 535 (99.8) |
Prior surgery, n (%) | ||
Yes | 157 (29.5) | 162 (30.2) |
No | 376 (70.5) | 374 (69.8) |
Prior radiation, n (%) | ||
Yes | 21 (3.9) | 28 (5.2) |
No | 512 (96.1) | 508 (94.8) |
Prior chemotherapy, n (%) | ||
Yes | 50 (9.4) | 48 (9.0) |
No | 483 (90.6) | 488 (91.0) |
Prior photodynamic therapy, n (%) | ||
No | 533 (100) | 536 (100) |
PD-L1 status (CPS cut-off 1), n (%) | ||
CPS < 1 | 113 (21.2) | 110 (20.5) |
CPS ≥ 1 | 363 (68.1) | 365 (68.1) |
Indeterminate | 57 (10.7) | 61 (11.4) |
PD-L1 status, (CPS cut-off 10), n (%) | ||
CPS < 10 | 273 (51.2) | 289 (53.9) |
CPS ≥ 10 | 203 (38.1) | 186 (34.7) |
Indeterminate | 57 (10.7) | 61 (11.4) |
ECOG Performance Status, n (%) | ||
0 | 258 (48.4) | 228 (42.5) |
1 | 274 (51.4) | 308 (57.5) |
≥ 2 | 1 (0.2) | 0 (0.0) |
Site of origin, n (%) | ||
Gallbladder | 115 (21.6) | 118 (22.0) |
Intrahepatic | 320 (60.0) | 313 (58.4) |
Extrahepatic | 98 (18.4) | 105 (19.6) |
Disease status, n (%) | ||
Locally advanced | 60 (11.3) | 66 (12.3) |
Metastatic | 473 (88.7) | 470 (87.7) |
HBV status, n (%) | ||
Chronic HBV infection | 14 (2.6) | 16 (3.0) |
Clinically resolved HBV infection | 150 (28.1) | 149 (27.8) |
Negative | 366 (68.7) | 366 (68.3) |
Missing | 3 (0.6) | 5 (0.9) |
HCV status, n (%) | ||
HCV infection | 1 (0.2) | 1 (0.2) |
Prior HCV infection | 18 (3.4) | 13 (2.4) |
Negative | 514 (96.4) | 520 (97.0) |
Missing | 0 (0.0) | 2 (0.4) |
Alcohol use status, n (%) | ||
Never used | 282 (52.9) | 316 (59.0) |
Used or using | 251 (47.1) | 219 (40.9) |
Missing | 0 (0.0) | 1 (0.2) |
Disease overall stage, n (%) | ||
I | 3 (0.6) | 2 (0.4) |
II | 19 (3.6) | 15 (2.8) |
III | 36 (6.8) | 47 (8.8) |
IV | 475 (89.1) | 472 (88.1) |
Biliary stent or drain, n (%) | ||
Yes | 33 (6.2) | 41 (7.6) |
No | 500 (93.8) | 495 (92.4) |
Antibiotics within 1 month of study start, n (%) | ||
Yes | 291 (54.6) | 273 (50.9) |
CPS = combined positive score; ECOG = Eastern Cooperative Oncology Group; HBV = hepatitis B virus; HCV = hepatitis C virus; PD-L1 = programmed cell death 1 ligand 1; SD = standard deviation.
Source: Clinical Study Report for the KEYNOTE-966 study.25 Details included in the table are from the sponsor’s summary of clinical evidence.
Exposure to Study Treatments
At the final analysis (December 15, 2022, DCO), the median duration of exposure to the study medications was 6.37 months (range, 0.03 to 36.40) in the pembrolizumab plus chemotherapy group and 5.54 months (range, 0.03 to 30.62) in the placebo plus chemotherapy group (Table 12).
Table 12: Patient Exposure in the KEYNOTE-966 Study (Safety Analysis Population)
Exposure | Pembrolizumab + chemotherapy (N = 529) | Placebo + chemotherapy (N = 534) |
|---|---|---|
Duration on therapy, months, median (range) | 6.37 (0.03 to 36.40) | 5.54 (0.03 to 30.62) |
Duration on therapy, months, mean (SD) | 8.04 (6.87) | 7.29 (6.32) |
Number of cycles, median (range) | 9.00 (1.00 to 51.00) | 8.00 (1.00 to 42.00) |
Number of cycles, mean (SD) | 11.46 (9.22) | 10.53 (8.55) |
Exposure duration, n (person-months) | ||
> 0 months | 529 (4,252.6) | 534 (3,895.0) |
≥ 1 months | 471 (4,226.9) | 480 (3,871.9) |
≥ 3 months | 388 (4,056.6) | 374 (3,659.9) |
≥ 6 months | 274 (3,549.7) | 249 (3,089.3) |
≥ 12 months | 117 (2,193.4) | 105 (1,896.9) |
≥ 18 months | 61 (1,389.3) | 46 (1,028.8) |
≥ 24 months | 24 (632.9) | 11 (285.9) |
n = number of patients; SD = standard deviation.
Note: Data were obtained on December 15, 2022.
Source: Clinical Study Report for the KEYNOTE-966 study.25 Details included in the table are from the sponsor’s summary of clinical evidence.
Concomitant Medications
The reported concomitant medications were generally balanced between the treatment groups (Table 13). The most frequently reported concomitant medications (used by more than 40% of patients overall) were dexamethasone (49.9%) under the category of “corticosteroids, dermatological preparations” and paracetamol (44.2%) under “analgesics.” The most frequently reported prior medications (more than 20% of patients overall) were also dexamethasone (21.9%) and paracetamol (20.3%) (data not shown).
Table 13: Concomitant Medications Use in the KEYNOTE-966 Study (Efficacy Analysis Population)
Medications | Pembrolizumab + chemotherapy (N = 533), n (%) | Placebo + chemotherapy (N = 536), n (%) |
|---|---|---|
Patients with at least 1 concomitant medications | 529 (99.2) | 533 (99.4) |
Patients with no concomitant medications | 4 (0.8) | 3 (0.6) |
Alimentary tract and metabolism | ||
Antidiarrheals, intestinal anti-inflammatory, or anti-infective drugs | 285 (53.5) | 253 (47.2) |
Antiemetics and antinauseants | 500 (93.8) | 505 (94.2) |
Bile and liver therapy | 194 (36.4) | 209 (39.0) |
Drugs for acid-related disorders | 404 (75.8) | 400 (74.6) |
Drugs for constipation | 406 (76.2) | 401 (74.8) |
Drugs for functional gastrointestinal disorders | 330 (61.9) | 338 (63.1) |
Drugs used in diabetes | 131 (24.6) | 130 (24.3) |
Mineral supplements | 372 (69.8) | 369 (68.8) |
Other alimentary tract and metabolism products | 108 (20.3) | 108 (20.1) |
Stomatological preparations | 483 (90.6) | 484 (90.3) |
Vitamins | 141 (26.5) | 131 (24.4) |
Anti-infectives for systemic use | ||
Antibacterials for systemic use | 317 (59.5) | 280 (52.2) |
Vaccines | 118 (22.1) | 107 (20.0) |
Antineoplastic and immunomodulating drugs | ||
Immunostimulants | 163 (30.6) | 188 (35.1) |
Blood and blood-forming organs | ||
Antianemic preparations | 153 (28.7) | 145 (27.1) |
Antithrombotic drugs | 236 (44.3) | 230 (42.9) |
Blood substitutes and perfusion solutions | 406 (76.2) | 413 (77.1) |
Cardiovascular system | ||
Drugs that act on the renin-angiotensin system | 160 (30.0) | 134 (25.0) |
Antihypertensives | 179 (33.6) | 158 (29.5) |
Calcium channel blockers | 124 (23.3) | 117 (21.8) |
Cardiac therapy | 116 (21.8) | 143 (26.7) |
Diuretics | 212 (39.8) | 222 (41.4) |
Lipid-modifying drugs | 131 (24.6) | 137 (25.6) |
Vasoprotectives | 423 (79.4) | 395 (73.7) |
Dermatologicals | ||
Antiacne preparations | 380 (71.3) | 363 (67.7) |
Antibiotics and chemotherapeutics for dermatological use | 205 (38.5) | 172 (32.1) |
Antipruritics (antihistamines, anesthetics, and so forth) | 214 (40.2) | 191 (35.6) |
Corticosteroids, dermatological preparations | 411 (77.1) | 384 (71.6) |
Medicated dressings | 231 (43.3) | 225 (42.0) |
Other dermatological preparations | 276 (51.8) | 272 (50.7) |
Preparations for treatment of wounds and ulcers | 235 (44.1) | 234 (43.7) |
Genitourinary system and sex hormones | ||
Gynecological anti-infectives and antiseptics | 190 (35.6) | 161 (30.0) |
Other gynecological drugs | 128 (24.0) | 149 (27.8) |
Urological drugs | 167 (31.3) | 173 (32.3) |
Musculo-skeletal system | ||
Anti-inflammatory and antirheumatic products | 166 (31.1) | 165 (30.8) |
Topical products for joint and muscular pain | 213 (40.0) | 217 (40.5) |
Nervous system | ||
Analgesics | 421 (79.0) | 415 (77.4) |
Anesthetics | 97 (18.2) | 115 (21.5) |
Antiepileptics | 254 (47.7) | 245 (45.7) |
Other nervous system drugs | 117 (22.0) | 122 (22.8) |
Psycholeptics | 301 (56.5) | 281 (52.4) |
Respiratory system | ||
Antihistamines for systemic use | 209 (39.2) | 191 (35.6) |
Cough and cold preparations | 304 (57.0) | 299 (55.8) |
Drugs for obstructive airway diseases | 364 (68.3) | 347 (64.7) |
Nasal preparations | 455 (85.4) | 450 (84.0) |
Throat preparations | 102 (19.1) | 123 (22.9) |
Sensory organs | ||
Ophthalmological and ontological preparations | 395 (74.1) | 368 (68.7) |
Ophthalmological drugs | 495 (92.9) | 500 (93.3) |
Otological drugs | 479 (89.9) | 463 (86.4) |
Systemic hormonal preparations, excluding sex hormones and insulins | ||
Corticosteroids for systemic use | 404 (75.8) | 377 (70.3) |
Various | ||
All other nontherapeutic products | 230 (43.2) | 222 (41.4) |
All other therapeutic products | 160 (30.0) | 160 (29.9) |
Diagnostic drugs | 222 (41.7) | 191 (35.6) |
General nutrients | 106 (19.9) | 117 (21.8) |
Homeopathic preparations | 294 (55.2) | 302 (56.3) |
Unspecified herbal and traditional medicine | 104 (19.5) | 125 (23.3) |
Note: Data were obtained on December 15, 2022. Only the concomitant medications received by at least 20% of the patients in either group are presented in this table.
Source: Clinical Study Report for the KEYNOTE-966 study.25 Details included in the table are from the sponsor’s summary of clinical evidence.
Subsequent Treatment
Overall, 47.5% of patients in the pembrolizumab plus chemotherapy group and 48.7% of patients in the placebo plus chemotherapy group received new oncology medications post treatment (Table 14). Forty-three percent of patients in each group received chemotherapy. Overall, 4.9% of patients in the pembrolizumab plus chemotherapy group and 7.1% in the placebo plus chemotherapy group received subsequent immune-checkpoint inhibitors, while 1.1% and 3.4% in each group, respectively, received targeted therapies.
Table 14: Subsequent Oncologic Therapies in the KEYNOTE-966 Study (Safety Analysis Population)
Therapy | Pembrolizumab + chemotherapy (N = 529), n (%) | Placebo + chemotherapy (N = 534), n (%) |
|---|---|---|
Chemotherapy | 230 (43.2) | 230 (42.9) |
Capecitabine | 49 (9.2) | 51 (9.5) |
Cisplatin | 50 (9.4) | 56 (10.4) |
Fluorouracil | 126 (23.6) | 127 (23.7) |
Gemcitabine | 41 (7.7) | 42 (7.8) |
Gimeracil, oteracil potassium, tegafur | 37 (6.9) | 33 (6.2) |
Irinotecan | 45 (8.4) | 42 (7.8) |
Oxaliplatin | 109 (20.5) | 111 (20.7) |
Immune-checkpoint inhibitors | 26 (4.9) | 38 (7.1) |
Other | 43 (8.1) | 50 (9.3) |
Note: Data were obtained on December 15, 2022. Only the subsequent therapies received by at least 5% of the patients in either group are presented in this table.
Source: Clinical Study Report for the KEYNOTE-966 study.25 Details included in the table are from the sponsor’s summary of clinical evidence.
Efficacy
Overall Survival
The primary efficacy end point in the KEYNOTE-966 study was OS at the final analysis (December 15, 2022, DCO). The median time from randomization to the DCO of December 15, 2022, in the ITT population was 25.6 months (range, 18.3 to 38.4 months).
At the final analysis, a total of 414 (77.7%) and 443 (82.6%) patients in the pembrolizumab plus chemotherapy and placebo plus chemotherapy groups, respectively, had died. The median OS was 12.7 months (95% CI, 11.5 to 13.6) in the pembrolizumab plus chemotherapy group and 10.9 months (95% CI, 9.9 to 11.6) in the placebo plus chemotherapy group. The corresponding HR was 0.83 (95% CI, 0.72 to 0.95; P = 0.0034), which met the prespecified efficacy boundary for a statistically significant OS benefit for pembrolizumab plus chemotherapy. The between-group differences in the KM-estimated OS rates at 6, 12, 18, and 24 months were 7.0% (95% CI, 2.0 to 12.0), 7.5% (95% CI, 1.6 to 13.4), 5.0% (95% CI, −0.5 to 10.5), and 6.8% (95% CI, 1.7 to 11.9), respectively (Table 15).
The KM OS curves are shown in Figure 3.
Descriptive subgroup analyses showed that the improvement in OS with pembrolizumab plus chemotherapy was generally consistent across the prespecified subgroups (Figure 4).
Table 15: Summary of OS in the KEYNOTE-966 Study (Efficacy Analysis Set, December 15, 2022, DCO)
Outcomes | Pembrolizumab + chemotherapy (N = 533) | Placebo + chemotherapy (N = 536) |
|---|---|---|
OS | ||
Number of patients randomized | 533 | 536 |
Number of events, n (%) | 414 (77.7) | 443 (82.6) |
Median (range) follow-up time, months | 25.6 (18.3 to 38.2) | 25.6 (18.3 to 38.4) |
Median OS, months (95% CI)a | 12.7 (11.5 to 13.6) | 10.9 (9.9 to 11.6) |
HR (95% CI)b | 0.83 (0.72 to 0.95) | Reference |
P valuec | 0.0034 | |
OS rate at various time points, % (95% CI)a | ||
6 months | 80.7 (77.1 to 83.8) | 73.7 (69.7 to 77.2) |
Between-group difference | 7.0 (2.0 to 12.0) | |
12 months | 51.6 (47.3 to 55.7) | 44.1 (39.9 to 48.3) |
Between-group difference | 7.5 (1.6 to 13.4) | |
18 months | 33.0 (29.1 to 37.0) | 28.0 (24.3 to 31.9) |
Between-group difference | 5.0 (−0.5 to 10.5) | |
24 months | 24.9 (21.2 to 28.8) | 18.1 (14.8 to 21.7) |
Between-group difference | 6.8 (1.7 to 11.9) | |
CI = confidence interval, DCO = data cut-off; HR = hazard ratio; OS = overall survival; sSAP = supplemental statistical analysis plan.
aFrom product-limit (Kaplan-Meier) method for censored data.
bBased on Cox regression model with Efron method of tie handling with treatment as a covariate stratified by geographic region (Asia vs. Non-Asia), disease status (locally advanced vs. metastatic), site of origin (gallbladder vs. intrahepatic vs. extrahepatic) with small strata collapsed as prespecified in the sSAP.
cOne-sided P value based on log-rank test stratified by geographic region (Asia vs. non-Asia), disease status (locally advanced vs. metastatic), site of origin (gallbladder vs. intrahepatic vs. extrahepatic) with small strata collapsed as prespecified in the sSAP.
Sources: Clinical Study Report for the KEYNOTE-966 study25 and additional information provided by the sponsor.30
Figure 3: Kaplan-Meier Estimates of Overall Survival in the KEYNOTE-966 Study (Efficacy Analysis Population)
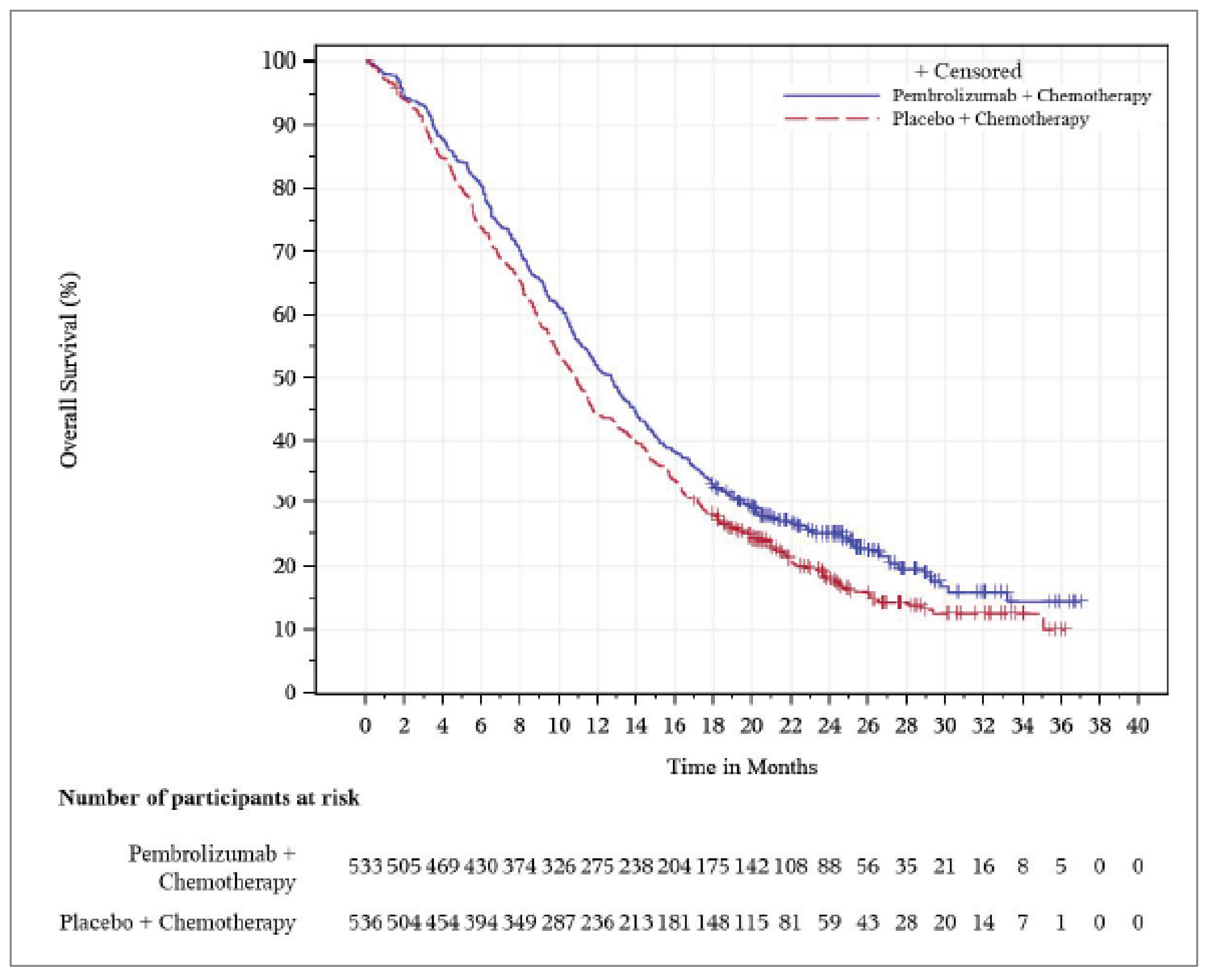
Source: Clinical Study Report for the KEYNOTE-966 study.25
Progression-Free Survival
The analysis of PFS was prespecified to occur at IA1 (December 15, 2021, DCO) using RECIST 1.1. The median time from randomization to the DCO for IA1 in the ITT population was 13.6 months (range, 6.3 to 26.4 months). Descriptive statistics for PFS at the final analysis were also reported.
At IA1, a total of 361 (67.7%) and 391 (72.9%) patients in the pembrolizumab plus chemotherapy and placebo plus chemotherapy groups, respectively, experienced a PFS event. The median PFS was 6.5 months (95% CI, 5.7 to 6.9) with pembrolizumab plus chemotherapy and 5.6 months (95% CI, 5.1 to 6.6) with placebo plus chemotherapy. The corresponding HR was 0.86 (95% CI, 0.75 to 1.00; P = 0.0225), which did not meet the prespecified efficacy boundary for a statistically significant PFS benefit for pembrolizumab plus chemotherapy, according to the multiplicity scheme. The between-group differences in KM-estimated PFS rates were 6.2% (95% CI, 0 to 12.4), 5.7% (95% CI, −0.5 to 11.9), 5.6% (95% CI, −0.4 to 11.6), 6.3% (95% CI, 0.2 to 12.4), and 3.5% (95% CI, −2.8 to 9.8) for 6, 9, 12, 15, and 18 months, respectively (Table 16).
The KM PFS curves are shown in Figure 5.
Figure 4: Forest Plot of OS by Prespecified Subgroups in the KEYNOTE-966 Study (Efficacy Analysis Population)
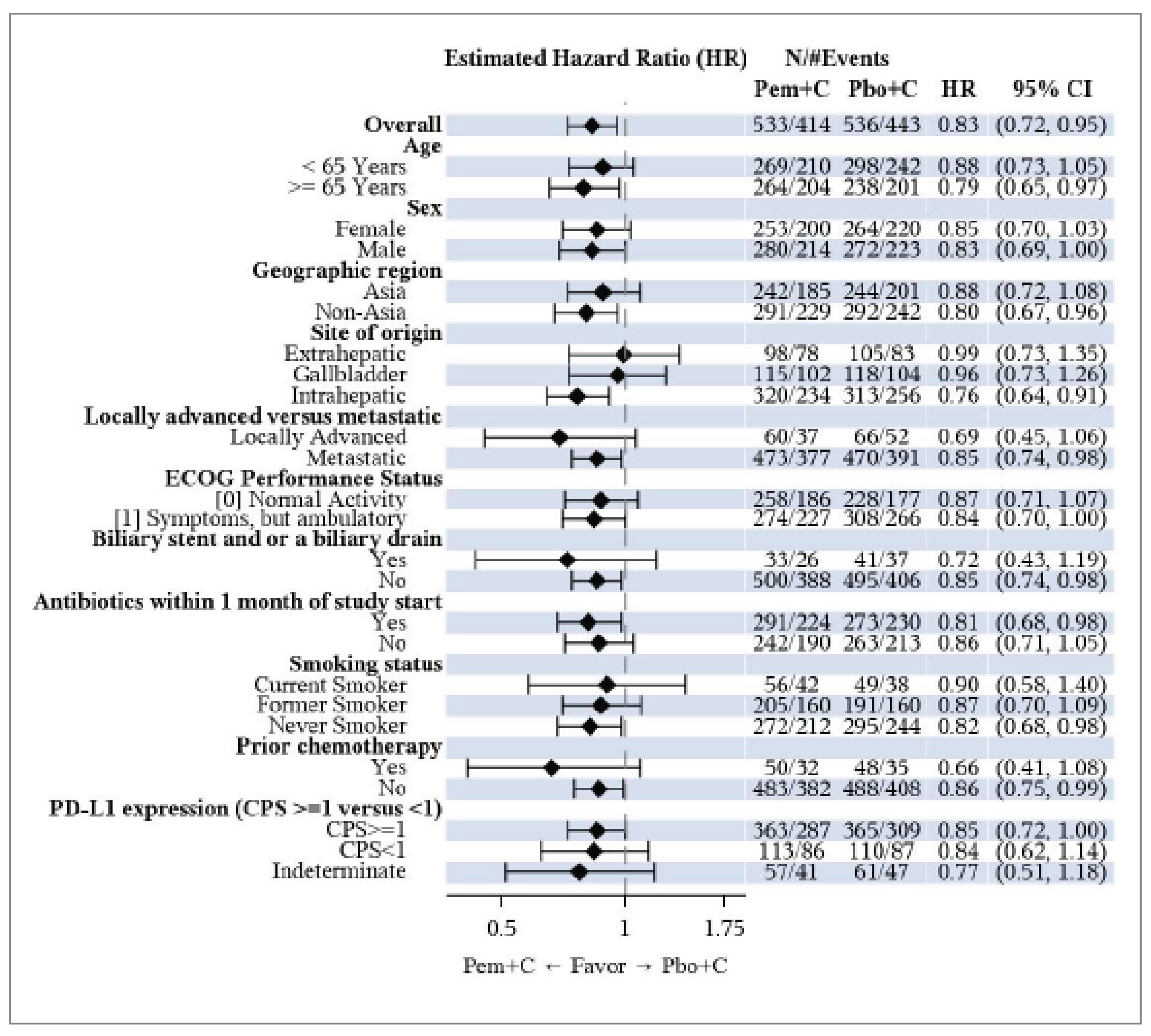
CI = confidence interval; CPS = combined positive score; ECOG = European Cooperative Oncology Group; HR = hazard ratio; ITT = intention to treat; PD-L1 = programmed cell death 1 ligand 1; Pem+C = pembrolizumab plus chemotherapy; Pbo+C = placebo plus chemotherapy.
Note: For the overall population, analysis is based on the same stratified Cox regression model as conducted for the primary analysis. Subgroup analyses were conducted using an unstratified Cox model with treatment as a covariate. If the number of participants in a category of a subgroup variable is less than 5% of the ITT population, the subgroup analysis will not be performed for this category of the subgroup variable. If there is only 1 category of a subgroup left (meaning other categories of a subgroup variable are all less than 5% of the ITT population), this subgroup variable was not displayed in the forest plot.
Source: Clinical Study Report for the KEYNOTE-966 study.25
Table 16: Summary of PFS in the KEYNOTE-966 Study (Efficacy Analysis Population, December 15, 2021, DCO)
Outcomes | Pembrolizumab + chemotherapy (N = 533) | Placebo + chemotherapy (N = 536) |
|---|---|---|
PFS | ||
Number of patients randomized | 533 | 536 |
Number of events, n (%) | 361 (67.7) | 391 (72.9) |
Death | 76 (14.3) | 84 (15.7) |
Documented progression | 285 (53.5) | 307 (57.3) |
Number of patients censored, n (%) | 172 (32.3) | 145 (27.1) |
New anticancer therapy | 49 (9.2) | 48 (9.0) |
PD or death immediately after ≥ 2 consecutive missed disease assessments, n (%) | 6 (1.1) | 3 (0.6) |
No PD or death as of the data cut-off date | 116 (21.8) | 94 (17.5) |
No adequate postbaseline disease assessment | 1 (0.2) | 0 (0.0) |
Median (range) follow-up time, months | 13.6 (6.3 to 26.3) | 13.6 (6.3 to 26.4) |
Median PFS, months (95% CI)a | 6.5 (5.7 to 6.9) | 5.6 (5.1 to 6.6) |
HR (95% CI)b | 0.86 (0.75 to 1.00) | Reference |
P valuec | 0.0225 | |
PFS rate at various time points, % (95% CI)a | ||
6 months | 52.3 (47.8 to 56.6) | 46.1 (41.7 to 50.4) |
Between-group difference | 6.2 (0.0 to 12.4) | |
9 months | 34.9 (30.5 to 39.4) | 29.2 (25.1 to 33.6) |
Between-group difference | 5.7 (−0.5 to 11.9) | |
12 months | 25.4 (21.1 to 29.9) | 19.8 (15.9 to 24.0) |
Between-group difference | 5.6 (−0.4 to 11.6) | |
15 months | 19.4 (15.0 to 24.2) | 13.1 (9.4 to 17.5) |
Between-group difference | 6.3 (0.2 to 12.4) | |
18 months | 11.9 (7.8 to 17.0) | 8.4 (4.8 to 13.4) |
Between-group difference | 3.5 (−2.8 to 9.8) | |
CI = confidence interval, HR = hazard ratio; PD = progressive disease; PFS = progression-free survival; sSAP = supplemental statistical analysis plan.
aFrom product-limit (Kaplan-Meier) method for censored data.
bBased on Cox regression model with Efron method of tie handling with treatment as a covariate stratified by geographic region (Asia vs. non-Asia), disease status (locally advanced vs. metastatic), site of origin (gallbladder vs. intrahepatic vs. extrahepatic) with small strata collapsed as prespecified in the sSAP.
cOne-sided P value based on log-rank test stratified by geographic region (Asia vs. Non-Asia), disease status (locally advanced vs. metastatic), site of origin (gallbladder vs. intrahepatic vs. extrahepatic) with small strata collapsed as prespecified in the sSAP.
Sources: Clinical Study Report for the KEYNOTE-96625 study and additional information provided by the sponsor.30
The descriptive subgroup analyses showed generally consistent results for PFS across the prespecified subgroups. The descriptive analyses of PFS at the final analysis (DCO of December 15, 2022, with a median follow-up of 25.6 months; range, 18.3 to 38.4 months) were consistent with the findings at IA1. Further, PFS sensitivity analyses also yielded results consistent with those of the primary analysis for the ITT population (data were available in the sponsor’s Clinical Study Report, but not shown in this Clinical Review Report).
Health-Related Quality of Life
In the KEYNOTE-966 study, the completion rate for treated patients at a specific time point was defined as the number of treated patients who completed at least 1 item (any 1 question) divided by the number of treated patients in the PRO analysis population. Compliance rate was defined as the number of treated patients who completed at least 1 item (any 1 question) divided by the number of eligible patients who are expected to complete the HRQoL assessment, not including the participants missing by design such as death, discontinuation from the trial, and/or translation not available (language barriers) for those who need it.
Figure 5: Kaplan-Meier Estimates of Progression-Free Survival at IA1 (Efficacy Analysis Population)
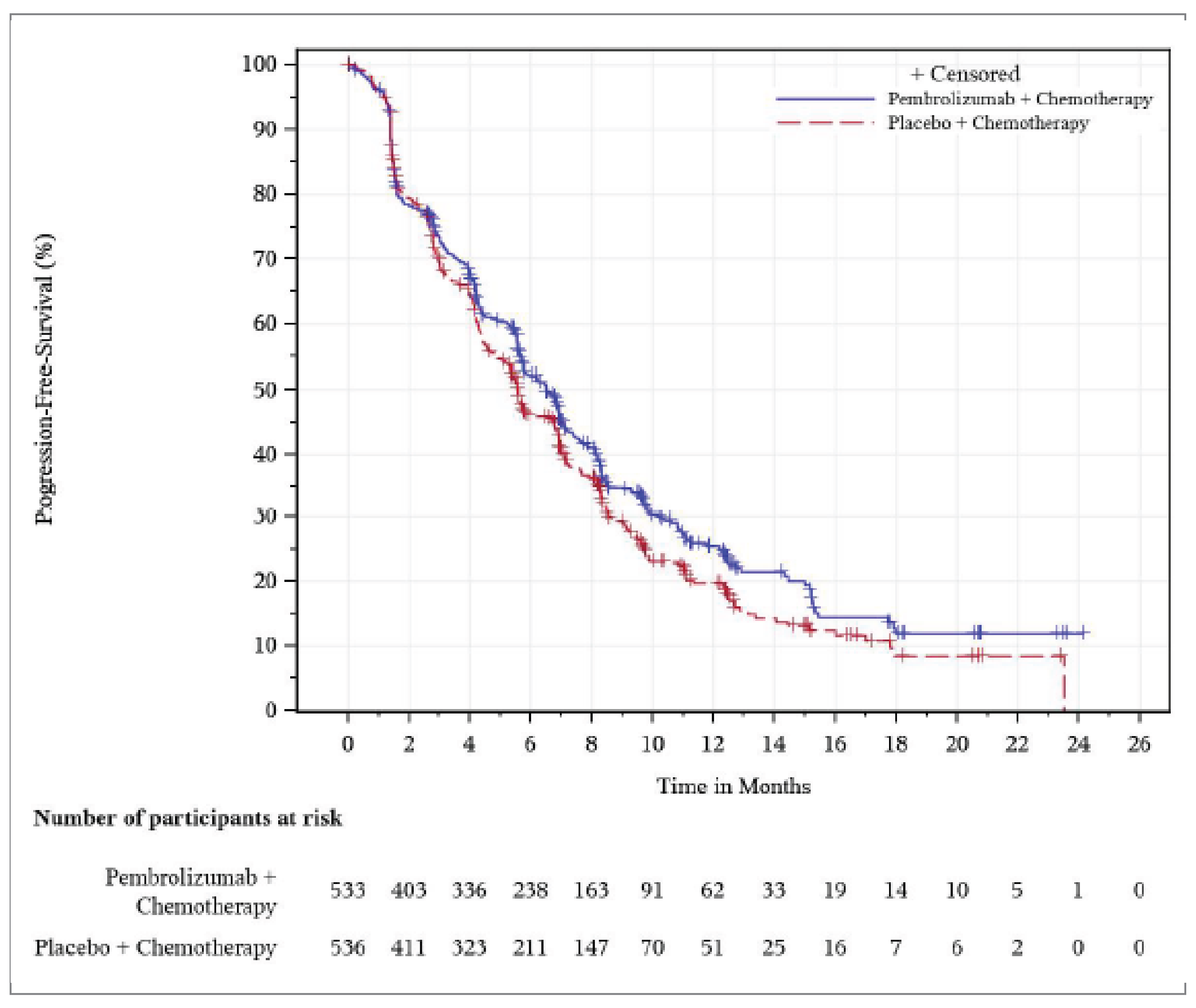
IA1 = interim analysis 1.
Source: Clinical Study Report for the KEYNOTE-966 study.25
EORTC Quality of Life Questionnaire Core 30
The compliance rate for the EORTC QLQ-C30 questionnaire at week 18 was 93.3% in the pembrolizumab plus chemotherapy group and 95.7% in the placebo plus chemotherapy group. The completion rate at week 18 was 61.5% and 60.9% in the 2 groups, respectively. At week 18, the between-group differences in LSM changes from baseline in the prespecified EORTC QLQ-C30 GHS (quality-of-life scales), physical functioning, and role functioning scores were 0.04 (95% CI, −2.52 to 2.60; P = 0.9773), 1.24 (95% CI, −1.42 to 3.90; P = 0.3596), and 2.68 (95% CI, −0.76 to 6.11; P = 0.1264), respectively (Table 17).
EORTC Quality of Life Questionnaire Cholangiocarcinoma and Gallbladder Cancer Module 21
Compliance with the EORTC QLQ-BIL21 at week 18 was 93.3% in the pembrolizumab plus chemotherapy group and 95.7% in the placebo plus chemotherapy group. The completion rate at week 18 was 61.5% in the pembrolizumab plus chemotherapy group and 61.0% in the placebo plus chemotherapy group. At week 18, the difference in the LSM changes from baseline in the EORTC QLQ-BIL21 jaundice and pain scores were 0.26 (95% CI, −1.35 to 1.87; P = 0.7535) and −1.87 (95% CI, −4.26 to 0.53; P = 0.1265), respectively (Table 17).
Table 17: Summary of PROs in the KEYNOTE-966 Study (PRO Analysis Population)
Instruments | Pembrolizumab + chemotherapy | Placebo + chemotherapy | |
|---|---|---|---|
EORTC QLQ-C30 | |||
Global Health Status (quality-of-life scales) | |||
Number of patients contributing to the analysis at baseline | 489 | 496 | |
Baseline, mean (SD) | 66.65 (19.91) | 66.99 (21.31) | |
Number of patients at week 18 | 320 | 315 | |
At week 18, mean (SD) | 66.85 (18.78) | 67.94 (17.09) | |
Change from baseline to week 18, LS mean (95% CI) | −2.47 (−4.45 to −0.49) | −2.51 (−4.49 to −0.53) | |
Treatment group difference vs. control, LS mean (95% CI) | 0.04 (−2.52 to 2.60) | ||
P valuea | 0.9773 | ||
Physical functioning | |||
Number of patients contributing to the analysis at baseline | 489 | 496 | |
Baseline, mean (SD) | 82.21 (18.16) | 81.21 (18.48) | |
Number of patients at week 18 | 320 | 315 | |
At week 18, mean (SD) | 79.17 (20.43) | 79.05 (18.43) | |
Change from baseline to week 18, LS mean (95% CI) | −6.42 (−8.34 to −4.49) | −7.66 (−9.58 to −5.74) | |
Treatment group difference vs. control, LS mean (95% CI) | 1.24 (−1.42 to 3.90) | ||
P valuea | 0.3596 | ||
Role functioning | |||
Number of patients contributing to the analysis at baseline | 489 | 496 | |
Baseline, mean (SD) | 81.53 (23.46) | 79.47 (25.54) | |
Number of patients at week 18 | 320 | 315 | |
At week 18, mean (SD) | 76.82 (25.11) | 75.71 (23.83) | |
Change from baseline to week 18, LS mean (95% CI) | −7.02 (−9.59 to −4.45) | −9.69 (−12.26 to −7.12) | |
Treatment group difference vs. control, LS mean (95% CI) | 2.68 (−0.76 to 6.11) | ||
P valuea | 0.1264 | ||
EORTC QLQ-BIL21 | |||
Jaundice | |||
Number of patients contributing to the analysis at baseline | 482 | 490 | |
Baseline, mean (SD) | 6.02 (12.36) | 6.35 (12.04) | |
Number of patients at week 18 | 320 | 315 | |
At week 18, mean (SD) | 5.87 (11.26) | 5.50 (10.54) | |
Change from baseline to week 18, LS mean (95% CI) | 0.14 (−1.14 to 1.42) | −0.12 (−1.40 to 1.17) | |
Treatment group difference vs. control, LS mean (95% CI) | 0.26 (−1.35 to 1.87) | ||
P valuea | 0.7535 | ||
Pain | |||
Number of patients contributing to the analysis at baseline | 482 | 490 | |
Baseline, mean (SD) | 23.17 (21.16) | 25.78 (22.63) | |
Number of patients at week 18 | 320 | 315 | |
At week 18, mean (SD) | 15.86 (17.02) | 17.30 (17.15) | |
Change from baseline to week 18, LS mean (95% CI) | −5.94 (−7.83 to −4.05) | −4.07 (−5.96 to −2.18) | |
Treatment group difference vs. control, LS mean (95% CI) | −1.87 (−4.26 to 0.53) | ||
P valuea | 0.1265 | ||
CI = confidence interval; cLDA = constrained longitudinal data analysis; EORTC QLQ-BIL21 = European Organization for Research and Treatment of Cancer Quality of Life Questionnaire Cholangiocarcinoma and Gallbladder Cancer Module 21; EORTC QLQ-C30 = European Organization for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; LS = least squares; PRO = patient-reported outcome; SD = standard deviation; sSAP = supplemental statistical analysis plan.
Note: The PRO analysis population includes all patients who received ≥ 1 dose of study treatment and completed ≥ 1 PRO assessment.
aThe P value was not adjusted for multiplicity. The P value was based on a cLDA model with the PRO scores as the response variable with covariates for the treatment by time interaction, stratification factors geographic region, disease status, site of origin with small strata collapsed as prespecified in the sSAP.
Source: Clinical Study Report for the KEYNOTE-966 study.25
Harms
All safety outcomes in the KEYNOTE-966 study were measured in the safety analysis population. Key harms at the December 15, 2022, DCO are summarized in Table 18.
Adverse Events
In the KEYNOTE-966 study, the proportion of patients who experienced at least 1 AE was 99.1% (524 patients) in the pembrolizumab plus chemotherapy group and 99.6% (532 patients) in the placebo plus chemotherapy group. The most frequently reported AEs (occurring 1 or more times in ≥ 20% of patients) in the pembrolizumab plus chemotherapy group were decreased neutrophil count (62.4%), anemia (61.1%), nausea (44.0%), decreased platelet count (39.9%), fatigue (35.3%), constipation (35.2%), decreased appetite (27.2%), decreased white blood cell count (26.7%), pyrexia (26.3%), and vomiting (23.1%). In the placebo plus chemotherapy group, the most frequently reported AEs were decreased neutrophil count (61.2%), anemia (58.6%), nausea (46.1%), decreased platelet count (39.7%), fatigue (32.2%), constipation (35.6%), decreased appetite (29.0%), decreased white blood cell count (23.8%), vomiting (24.0%), abdominal pain (22.8%), and increased alanine transaminase (21.2%).
Serious Adverse Events
The proportion of patients with SAEs was 52.2% (276 patients) in the pembrolizumab plus chemotherapy group and 49.3% (263 patients) in the placebo plus chemotherapy group. The most frequently reported SAEs (occurring 1 or more times in ≥ 2% of patients) in the pembrolizumab plus chemotherapy group were cholangitis (5.9%), pyrexia (5.7%), decreased platelet count (3.6%), biliary tract infection (3.2%), anemia (2.5%), sepsis (2.5%), biliary obstruction (2.3%), decreased neutrophil count (2.1%), and pulmonary embolism (2.1%). In the placebo plus chemotherapy group, the most frequent SAEs were cholangitis (4.5%), biliary tract infection (3.4%), sepsis (3.0%), biliary obstruction (3.0%), ascites (2.4%), pyrexia (2.2%), and liver abscess (2.1%).
Deaths
The proportion of patients with AEs resulting in death was 5.9% (31 patients) in the pembrolizumab plus chemotherapy and 9.2% (49 patients) in the placebo plus chemotherapy groups. A total of 8 (1.5%) and 3 (0.6%) patients in the 2 treatment groups, respectively, died of a drug-related AE, as assessed by the investigator. Of the 8 deaths in the pembrolizumab plus chemotherapy group considered by the investigator as related to the study intervention, 5 were considered related to chemotherapy. The AEs were abdominal abscess, cholangitis, lower respiratory tract infection, malignant neoplasm progression, myocardial infarction, pneumonia viral, pneumonitis, and septic shock.
Patients Who Stopped Treatment Due to Adverse Events
The proportion of patients with AEs resulting in treatment discontinuation of any drug was generally similar in the pembrolizumab plus chemotherapy group compared with the placebo plus chemotherapy group (26.1% versus 22.8%). The most frequently reported AEs resulting in treatment discontinuation (occurring in ≥ 1% of patients) in the pembrolizumab plus chemotherapy group were decreased neutrophil count (3.6%), decreased platelet count (3.6%), increased blood creatinine (1.5%), pneumonitis (1.3%), and fatigue (1.1%). In the placebo plus chemotherapy group, the most frequent AEs resulting in treatment discontinuation were decreased neutrophil count (3.0%), increased blood creatinine (1.5%), anemia (1.1%), and sepsis (1.1%).
Adverse Events of Special Interest
There were 117 patients (22.1%) in the pembrolizumab plus chemotherapy group and 69 patients (12.9%) in the placebo plus chemotherapy group who experienced an adverse event of special interest (AEOSI). The most frequent AEOSIs reported for pembrolizumab plus chemotherapy versus placebo plus chemotherapy were hypothyroidism, reported in 46 patients (8.7%) versus 14 patients (2.6%), respectively; pneumonitis, reported in 26 patients (4.9%) versus 10 patients (1.9%), respectively; and hyperthyroidism, reported in 19 patients (3.6%) versus 10 patients (1.9%), respectively (Table 18). Occurrences of immune-mediated AEs were low (< 2%) in both treatment groups: 0 versus 0.2% for enterocolitis, 0.9% versus 1.1% for hepatitis, and 0.4% versus 0 for lung disease for the pembrolizumab plus chemotherapy group and the placebo plus chemotherapy group, respectively.
Table 18: Summary of Harms Results From KEYNOTE-966 (Safety Analysis Population, December 15, 2022, DCO)
Adverse events | Pembrolizumab + chemotherapy (N = 529) | Placebo + chemotherapy (N = 534) |
|---|---|---|
Patients with ≥ 1 AE, n (%) | 524 (99.1) | 532 (99.6) |
AEs reported in ≥ 10% of patients in any treatment group, n (%) | ||
Neutrophil count decreased | 330 (62.4) | 327 (61.2) |
Anemia | 323 (61.1) | 313 (58.6) |
Nausea | 233 (44.0) | 246 (46.1) |
Platelet count decreased | 211 (39.9) | 212 (39.7) |
Fatigue | 187 (35.3) | 172 (32.2) |
Constipation | 186 (35.2) | 190 (35.6) |
Decreased appetite | 144 (27.2) | 155 (29.0) |
White blood cell count decreased | 141 (26.7) | 127 (23.8) |
Pyrexia | 139 (26.3) | 104 (19.5) |
Vomiting | 122 (23.1) | 128 (24.0) |
Diarrhea | 103 (19.5) | 98 (18.4) |
Abdominal pain | 92 (17.4) | 122 (22.8) |
Rash | 90 (17.0) | 49 (9.2) |
Aspartate aminotransferase increased | 88 (16.6) | 98 (18.4) |
Alanine aminotransferase increased | 87 (16.4) | 113 (21.2) |
Hypomagnesemia | 79 (14.9) | 79 (14.8) |
Pruritus | 77 (14.6) | 51 (9.6) |
Asthenia | 75 (14.2) | 95 (17.8) |
Edema peripheral | 73 (13.8) | 85 (15.9) |
Blood creatinine increased | 57 (10.8) | 58 (10.9) |
Alopecia | 55 (10.4) | 68 (12.7) |
Back pain | 54 (10.2) | 73 (13.7) |
Dyspnea | 53 (10.0) | 55 (10.3) |
Headache | 53 (10.0) | 46 (8.6) |
Weight decreased | 51 (9.6) | 63 (11.8) |
Blood bilirubin | 50 (9.5) | 65 (12.2) |
Hypokalemia | 48 (9.1) | 67 (12.5) |
Abdominal pain upper | 40 (7.6) | 57 (10.7) |
Dyspepsia | 33 (6.2) | 55 (10.3) |
SAEs, n (%) | ||
Patients with ≥ 1 SAE | 276 (52.2) | 263 (49.3) |
SAEs reported in ≥ 2% of patients in any treatment group | ||
Cholangitis | 31 (5.9) | 24 (4.5) |
Pyrexia | 30 (5.7) | 12 (2.2) |
Platelet count decreased | 19 (3.6) | 10 (1.9) |
Biliary tract infection | 17 (3.2) | 18 (3.4) |
Anemia | 13 (2.5) | 10 (1.9) |
Sepsis | 13 (2.5) | 16 (3.0) |
Biliary obstruction | 12 (2.3) | 16 (3.0) |
Neutrophil count decreased | 11 (2.1) | 1 (0.2) |
Pulmonary embolism | 11 (2.1) | 8 (1.5) |
Ascites | 6 (1.1) | 13 (2.4) |
Liver abscess | 4 (0.8) | 11 (2.1) |
Patients who stopped treatment due to AEs, n (%) | ||
Patients who stopped any drug | 138 (26.1) | 122 (22.8) |
Discontinued pembrolizumab or placebo | 77 (14.6) | 66 (12.4) |
Discontinued any chemotherapy | 124 (23.4) | 113 (21.2) |
Discontinued all drugs | 18 (3.4) | 14 (2.6) |
Deaths, n (%) | ||
Patients who died | 31 (5.9) | 49 (9.2) |
Death due to a drug-related AE (per investigator) | 8 (1.5) | 3 (0.6) |
AEOSIs, n (%) | ||
Patients with ≥ 1 AEOSI | 117 (22.1) | 69 (12.9) |
Adrenal insufficiency | 3 (0.6) | 2 (0.4) |
Colitis | 9 (1.7) | 6 (1.1) |
Immune-mediated enterocolitis | 0 (0.0) | 1 (0.2) |
Encephalitis | 2 (0.4) | 0 (0.0) |
Hepatitis | 9 (1.7) | 7 (1.3) |
Immune-mediated hepatitis | 5 (0.9) | 6 (1.1) |
Hyperthyroidism | 19 (3.6) | 10 (1.9) |
Hyperparathyroidism | 0 (0.0) | 1 (0.2) |
Hypophysitis | 2 (0.4) | 2 (0.4) |
Hypothyroidism | 46 (8.7) | 14 (2.6) |
Infusion reactions | 8 (1.5) | 6 (1.1) |
Myasthenic syndrome | 1 (0.2) | 1 (0.2) |
Myocarditis | 1 (0.2) | 1 (0.2) |
Myositis | 1 (0.2) | 0 (0.0) |
Nephritis | 2 (0.4) | 1 (0.2) |
Pancreatitis | 4 (0.8) | 6 (1.1) |
Pneumonitis | 26 (4.9) | 10 (1.9) |
Immune-mediated lung disease | 2 (0.4) | 0 (0.0) |
Severe skin reactions | 10 (1.9) | 3 (0.6) |
Thyroiditis | 3 (0.6) | 0 (0.0) |
Uveitis | 0 (0.0) | 2 (0.4) |
Vasculitis | 2 (0.4) | 1 (0.2) |
AE = adverse event; AEOSI = adverse event of special interest; DCO = data cut-off; SAE = serious adverse event.
Source: Clinical Study Report for the KEYNOTE-966 study.25 Details included in the table are from the sponsor’s summary of clinical evidence.
Critical Appraisal
Internal Validity
Appropriate methods of randomization, blinding, treatment allocation, and stratification were employed. There was a low risk of selective reporting bias, as the data were analyzed in accordance with the prespecified statistical plan. According to the study findings, the safety profile of pembrolizumab plus chemotherapy is similar to placebo plus chemotherapy; therefore, the chance of unblinding during the study due to the differences in harms would be small and thus less likely to affect the PROs. Although BTC is considered a rare disease, this multicentre study had a large sample size (N = 1,069) and would have sufficient statistical power to detect a statistically significant difference between treatment groups. Overall, patients’ baseline demographic and disease characteristics were balanced between the 2 treatment groups, for example, demographic characteristics, prior anticancer therapy, and cancer stage. The use of concomitant therapies and subsequent anticancer therapies was also generally balanced across the groups. Although patients in the pembrolizumab plus chemotherapy group had relatively better performance status at baseline (48.4% of the patients in the pembrolizumab plus chemotherapy group had an ECOG Performance Status score of 0 compared with 42.5% in the placebo plus chemotherapy group), this single imbalance is likely to have occurred by chance. The clinical experts we consulted noted that this would not significantly impact the interpretation of the results.
A multiplicity testing procedure was applied to OS, PFS, and overall response rate to control the type I error rate in the study, including across the interim analyses. However, other efficacy outcomes were analyzed without multiplicity adjustment, for example, HRQoL assessment using the EORTC QLQ-C30 and EORTC QLQ-BIL21. As such, there would be an increased risk of false-positive conclusions for statistically significant results; however, none of the reported results for these PROs were statistically significant.
In this study, some efficacy outcomes were measured using both RECIST 1.1 and RECIST 1.1 modified for immune-based therapies. The latter is specifically designed for tumour assessment in patients receiving immune-based therapies. The clinical experts we consulted confirmed the appropriateness of the use of both measures. In addition, some efficacy outcomes (e.g., PFS per RECIST 1.1) were assessed by both BICR (as secondary end points) and the investigators (as exploratory end points). The advantage of BICR-assessed outcomes is a reduced risk of bias in the measurement of outcomes. This Clinical Review Report focuses on the BICR assessments rather than the investigator’s assessments.
HRQoL was assessed using disease-specific instruments, and 1 of these was specifically designed for patients with BTC. The validity, reliability, and responsiveness of these questionnaires have been demonstrated in patients with BTC. A specific MID for patients with BTC was not identified from the literature; however, a range of potential between-group MIDs (within a 5- to 10-point change in general) was established based on clinical trials of 9 different cancer types and may be used to determine the clinical relevance of the study findings for HRQoL.29 On the other hand, the completion rates for these 2 questionnaires were comparable in the PRO FAS population (EORTC QLQ-C30, 61.5% versus 60.9%; EORTC QLQ-BIL21, 61.5% versus 61.0% in the pembrolizumab plus chemotherapy group and placebo plus chemotherapy group, respectively). A constrained longitudinal data analysis model with a longitudinal modelling approach was adopted to handle missing data; however, the benefits and limitations of this method in the study population were unclear. This method assumes that data are missing at random, but this assumption may not hold true in the KEYNOTE-966 study. As such, the risk of bias due to missing outcomes data and its impact on study findings is uncertain.
The use of PFS in assessing cancer drugs, especially for advanced cancer, is under debate in oncology drug research.52-54 In the current pembrolizumab submission, although the sponsor did not submit evidence for the validity of PFS as a surrogate for OS, moderate-certainty evidence was identified for modest improvements in OS associated with pembrolizumab in the KEYNOTE-966 study. In the analysis of PFS, the proportional hazards assumption for PFS was formally tested as well as visually examined to ensure its validity for PFS; no violation of this assumption was found. However, from visual inspection, the assumption appears to have been violated. As such, the HR for PFS cannot be reliably interpreted. In 1 published article,26 the sponsor indicated the following: “PFS assessed per RECIST 1.1 may not be the best measure of PFS in patients with BTC because the assessment of PFS is complex and often relies on nonradiographic factors such as biliary obstruction, liver function, and CA 19-9 [carbohydrate antigen 19-9] expression,” although no reference was provided to support this statement. The methods for testing the proportional hazards assumption for OS was described in the statistical analysis plan for the KEYNOTE-966 study; however, it is unclear whether this assumption has been formally tested, since no information was provided with respect to the results of these tests in the trial’s Clinical Study Report. Based on visual inspection, the assumption appeared reasonable. In the analyses of OS and PFS, the reasons for censoring were balanced between the 2 treatment groups.
Predefined sensitivity analyses were conducted to evaluate the robustness of the PFS end point per RECIST 1.1. Predefined subgroup analyses were conducted to examine the consistency of the primary and secondary outcomes across subgroups. Overall, the results of the sensitivity and prespecified subgroup analyses were generally aligned with the overall analysis for OS and PFS; however, the sensitivity and subgroup analyses were not adjusted for multiplicity and should be considered as supportive of the overall effect of pembrolizumab. The trial may not have been powered to detect subgroup differences and no tests for interaction effects were undertaken for differences in effects between subgroups.
External Validity
Based on feedback from the clinical experts we consulted, the eligibility criteria and baseline characteristics of patients randomized in the KEYNOTE-966 study generally reflected a study population that was consistent with the patients in Canadian clinical practice who would receive combination therapy of pembrolizumab plus chemotherapy, although the study population may be somewhat healthier; for example, patients in the KEYNOTE-966 study had better performance status and had fewer comorbidities. Patients with ampullary cancer were excluded from this study. According to the clinical experts, this is reasonable, since ampullary cancer is usually treated like pancreatic cancer or gastric cancer, for which different treatment regimens would be used. It was noted that this is a small patient group that is generally understudied. The clinical experts noted that the results from the KEYNOTE-966 study could be generalized to patients in Canada with advanced BTC who would be treated with pembrolizumab plus chemotherapy. The experts indicated that the outcome measures in the KEYNOTE-966 study are generally appropriate and clinically relevant in clinical trials of advanced BTC. The clinical experts noted that PFS is not as important to patients, given the poor prognosis of this disease.
In KEYNOTE-966, pembrolizumab in combination with chemotherapy was compared with chemotherapy alone. The clinical experts consulted for this review indicated that chemotherapy (gemcitabine plus cisplatin) alone is a relevant comparator for pembrolizumab plus chemotherapy in the study population. There is a lack of direct evidence to examine the relative efficacy and safety of the study drug versus other treatments, such as durvalumab plus chemotherapy.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For the pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered most relevant to inform our expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group:27,28
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate. The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited. The true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate. The true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as “very uncertain.”
Following the GRADE approach, the evidence from the RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty-of-evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null. The threshold for a clinically important effect was 5% to 10% for OS (as informed by the clinical experts we consulted) and null for PFS (due to uncertainties in the measurement and interpretation of the outcome). The threshold for a clinically important effect for EORTC QLQ-C30 and EORTC QLQ-BIL21 scores was set according to the presence or absence of an important effect based on thresholds identified in the literature.29 For some harm events (e.g., immune-mediated AEs), due to the unavailability of the absolute difference in effects, the certainty of evidence was summarized narratively.
Results of GRADE Assessments
Drug Versus Placebo
Table 2 presents the GRADE summary of findings for pembrolizumab plus chemotherapy versus placebo plus chemotherapy.
Long-Term Extension Studies
No long-term extension studies were submitted by the sponsor.
Indirect Evidence
Contents within this section have been informed by materials submitted by the sponsor. The following was summarized and validated by the CDA-AMC review team.
Objectives for the Summary of Indirect Evidence
As there was limited direct evidence comparing pembrolizumab plus chemotherapy with other relevant comparators for the treatment of adult patients with locally advanced unresectable or metastatic BTC, a review of indirect evidence was undertaken and submitted by the sponsor.36
The objective of this section is to summarize and critically appraise the sponsor-submitted ITC and to inform the pharmacoeconomic model.
Description of Indirect Comparisons
Objectives
The objective of the NMA described in this section is to compare the efficacy and safety of pembrolizumab plus chemotherapy, which was evaluated in the KEYNOTE-966 study, with durvalumab plus chemotherapy for patients with locally advanced unresectable or metastatic BTC.
Study Selection Methods
A summary of the study selection criteria and methodology used to conduct the NMA is described in Table 19. To meet requirements for health technology assessments that have been submitted in multiple countries, the search included other comparators in addition to durvalumab plus chemotherapy.
Table 19: Study Selection Criteria and Methods for ITC Submitted by the Sponsor
Characteristics | Indirect comparison |
|---|---|
Population | Adult (≥ 18 years old) patients with advanced (metastatic) and/or unresectable (locally advanced) BTC (intrahepatic or extrahepatic cholangiocarcinoma or gallbladder cancer):
|
Intervention | Pembrolizumab was administered as an IV infusion over 30 minutes. The recommended dosage of pembrolizumab in adults is either:
|
Comparator | Eligibility for inclusion Any of the following, alone or in combination with other drugs used as systemic therapies:
Eligibility for data extraction Any of the following guideline-recommended treatment regimens:
Note: Dosing information was not specified |
Outcome | Efficacy outcomes:
Safety outcomes:
Patient-reported outcomes:
|
Study designs | Eligibility for inclusion: Phase II and III clinical trials i.e., randomized controlled trials, nonrandomized controlled trials, and single-arm trials Eligibility for data extraction: Randomized controlled trials only |
Publication characteristics | Inclusion of published and/or unpublished studies, including conference proceedings and clinical trials registries |
Exclusion criteria |
|
Databases searched | Embase, MEDLINE, CENTRAL, Northern Light databases (ASCO, 2021 to 2022) and ESMO (2021 to 2022), manual searches of ASCO Gastrointestinal Cancers (2021 to 2022), Asian Pacific Association for the Study of the Liver (2021 to 2022), and ESMO Asia (2021 to 2022), US National Institutes of Health Clinical Trial Registry (http://www.clinicaltrials.gov), European Union Clinical Trial Registry (http://www.clinicaltrialsregister.eu), and Australian New Zealand Clinical Trials Registry. The databases were last searched on January 10, 2023. |
Selection process | Two reviewers, working independently, reviewed all abstracts and proceedings identified by the searches according to the selection criteria, except for outcome criteria, which were only applied during the screening of full-text publications. All studies identified as eligible during abstract screening were then screened at the full-text stage by the same 2 reviewers. Following reconciliation between the 2 reviewers, a third reviewer was included to reach a consensus for any remaining discrepancies at both stages. The full-text studies identified as meeting the eligibility criteria at this stage were included for data extraction. The process of study identification and selection was summarized with a Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) flow diagram. |
Data extraction process | Two reviewers, working independently, extracted data on study characteristics, treatment characteristics, patient characteristics, and outcomes for the final list of included studies. Following reconciliation between the 2 reviewers, a third reviewer was included to reach a consensus for any remaining discrepancies. |
Quality assessment | Two independent reviewers assessed the risk of bias in the included RCTs using the Cochrane Collaboration’s Risk of Bias tool, version 2. This instrument was used to assess the risk of bias in 5 domains: randomization process, deviations from the intended interventions, missing outcome data, measurement of the outcome, and selection of the reported result. The risk of bias instrument can be used to assign summary assessments of within-study bias: low risk of bias (low risk of bias for all key domains), some concerns (unclear risk of bias for 1 or more key domains), or high risk of bias (high risk of bias for 1 or more key domains). Following reconciliation between the 2 reviewers, a third reviewer was included to reach consensus for any remaining discrepancies. |
ASCO = American Society of Clinical Oncology; BPI = Brief Pain Inventory; BTC = biliary tract cancer; EORTC QLQ-C30 = European Organization for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-BIL2 = European Organization for Research and Treatment of Cancer Questionnaire Cholangiocarcinoma and Gallbladder Cancer Module 21; ESMO = European Society of Medical Oncology; FACT-G = Functional Assessment of Cancer Therapy–General; FACT-HEP = Functional Assessment of Cancer Therapy–Hepatic; ITC = indirect treatment comparison; S1 = tegafur-gimeracil-oteracil; SF-12 = Short Form (12) Health Survey; SF-36 = Short Form (36) Health Survey; RCT = randomized controlled trial.
Source: Sponsor-submitted ITC.36 Details included in the table are from the sponsor’s summary of clinical evidence.
ITC Analysis Methods
To gauge the appropriateness of proceeding with an NMA, a feasibility assessment was conducted that included:
a determination of whether the RCT evidence for the interventions of interest formed 1 evidence network for each population and outcome of interest
an assessment of the trial characteristics, treatment characteristics, distribution of baseline patient characteristics, and outcome definitions that may have affected treatment effects across direct comparisons in the evidence networks.
The baseline patient characteristics of age, sex, race or ethnicity, ECOG Performance Status, disease classification at baseline (e.g., locally advanced, metastatic, recurrent), cancer location (e.g., intrahepatic bile duct, extrahepatic bile duct, gallbladder, ampulla), and PD-L1 expression were explored as potential treatment-effect modifiers based on a review of subgroup results from the trials included in the systematic literature review and recently published narrative reviews and systematic reviews or meta-analyses.55-57
A summary of the indirect comparison analysis methods used for this NMA is presented in Table 20.
Table 20: ITC Analysis Methods
Methods | Description |
|---|---|
Analysis methods | Time-to-event outcomes using constant HRs: The proportional hazards assumption regarding time-to-event outcomes for each individual trial was assessed using the Grambsch and Therneau test and visual inspection Schoenfeld residual plots.58,59 Where no violations were observed, the NMAs of OS and PFS were conducted using reported HRs in a regression model with a contrast-based normal likelihood for the log HR (and corresponding standard error) of each trial (or comparison) in the network according to Dias et al.60 Time-to-event outcomes using Kaplan-Meier curves: Where there was evidence that the proportional hazards assumption was violated, NMA models allowing for time-varying HRs were utilized. In this analysis, the model introduced by Jansen was employed.61,62 For OS and PFS, the following competing survival distributions were considered using the multivariate NMA framework: Weibull, Gompertz, and second-order FPs including p1 = −1, −0.5, 0, 0.5, or 1, and p2 = −1, −0.5, 0, 0.5, or 1. In essence, the second-order FP models are extensions of the Weibull and Gompertz model and allow arc and bathtub-shaped hazard functions, which emulate parametric distributions such as log-normal and log-logistic. For the relative treatment effects in the second-order FP framework, we assessed models which assume that:
Binary outcomes: The NMAs were performed based on the proportion of patients experiencing the event of interest using a logistic regression model with a binomial likelihood and logit link. |
Priors | Time-to-event outcomes using constant HRs: Normal noninformative prior distributions for the parameters were estimated with a mean of 0 and a variance of 10,000.60 Time-to-event outcomes using Kaplan-Meier curves: Noninformative priors were used for both mean hazards and treatment effects. These are multivariate normal, with mean vectors centred at 0, and covariance matrices with diagonals of 10,000, and off-diagonal elements of 0. Binary outcomes: Normal noninformative prior distributions for the parameters were used with a mean of 0 and a variance of 10,000. Relative treatment effects were expressed as ORs. |
Assessment of model fit | Both fixed- and random-effects models were considered. In general, the assumptions of random-effects models are preferred, as they are expected to be more plausible than fixed-effect models; however, as there was insufficient evidence to estimate between-study heterogeneity, fixed-effect models were used. The DIC was used to compare the goodness of fit of competing survival models.63 |
Assessment of consistency | NA |
Assessment of convergence | The parameters of the different models were estimated using a Markov chain Monte Carlo method implemented in the JAGS software package. A first series of iterations (20,000) from the JAGS sampler was discarded as “burn-in” and the inferences were based on additional iterations (40,000) using 2 chains, with convergence assessed through a visual inspection of trace, density, and Gelman-Rubin plots. |
Outcomes | OS, PFS, ORR, and safety; PROs were not included due to lack of data availability. |
Model selection | The DIC was used to compare the goodness of fit of competing survival models.63 |
Subgroup analysis | As potential subgroup effects were noted in the TOPAZ-1 study based on geographic region (Asia vs. non-Asia), subgroup analyses were conducted to assess the impact of differences in the distribution of Asia-based patients in the TOPAZ-1 and KEYNOTE-966 studies on the estimated relative treatment effects. |
DIC = deviance information criterion; FP = fractional polynomial; HR = hazard ratio; ITC = indirect treatment comparison; NA = not applicable; NMA = network meta-analysis; OR = odd ratio; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; PRO = patient-reported outcome.
Source: Sponsor-submitted ITC.36 Details included in the table are from the sponsor’s summary of clinical evidence.
The results of the NMA are presented with estimates of treatment effects for each intervention relative to the reference treatment. The posterior distributions of relative treatment effects were summarized by the median and 95% CrIs, which were constructed from the 2.5th and 97.5th percentiles of the posterior distributions. NMA results are presented in cross-tables with relative treatment-effect estimates (i.e., HRs or odds ratios) between all interventions of interest, along with 95% CrIs for all outcomes.
Results
Summary of Included Studies
The searches yielded a total of 6,433 records, of which 5,240 were screened following the removal of duplicates. A total of 29 relevant reports describing 17 unique RCTs were initially included.
Sixteen of these trials connected in a network with the KEYNOTE-966 study; 8 of these evaluated treatment regimens recommended by the National Comprehensive Cancer Network or European Society of Medical Oncology (i.e., 5-fluorouracil plus leucovorin, 5-fluorouracil plus leucovorin plus oxaliplatin, capecitabine plus oxaliplatin, durvalumab plus gemcitabine plus cisplatin, gemcitabine alone, gemcitabine plus cisplatin, and gemcitabine plus oxaliplatin) and were included in the feasibility assessment [Figure 6]). This served to omit several trials evaluating tegafur-gimeracil-oteracil (S1), which is only approved for use in Japan, and other combinations of treatments that are unlikely to be relevant from a health technology assessment perspective outside of a select number of Asia-Pacific countries.
A summary of the assessment of homogeneity for the 9 trials (Schinzari [2017], Sharma [2010], Sharma [2019], Kim [2019], ABC-01, ABC-02, BT22, KEYNOTE-966, and TOPAZ-1) is provided in Table 21.
Based on the findings from the feasibility assessment, Schinzari (2017), Sharma (2010), and Sharma (2019) were excluded from the analyses due to substantial deviations in trial size, trial enrolment criteria, and baseline patient characteristics (i.e., age, sex, and performance status) compared with other trials in the restricted network. ABC-01, ABC-02, and BT22 were excluded since these trials enrolled patients with ampullary cancer. In addition, Kim (2019) was excluded from the analysis, as it was no longer connected to the other trials via Sharma (2019).
Results
The OS, PFS, and safety networks for the base case included 2 trials, KEYNOTE-966 and TOPAZ-1, that evaluated pembrolizumab plus chemotherapy and durvalumab plus chemotherapy, respectively, compared with placebo plus chemotherapy in patients with advanced BTC (Figure 7). A total of 1,069 patients were randomized in the KEYNOTE-966 study and 685 patients were randomized in the TOPAZ-1 study. Patients in the 2 trials were comparable in age (median age was 64 years in both trials), ECOG Performance Status (ECOG 0: 45% in the KEYNOTE-966 trial versus 49% in the TOPAZ-1 study), location of cancer (intrahepatic CCA: 59% versus 56%; extrahepatic CCA: 19% versus 19%; GBC: 22% versus 25%), and disease classification (metastatic BTC: 88% versus 86%). The proportion of Asian patients was 46% in the KEYNOTE-966 study and 56% in the TOPAZ-1 study.
Figure 6: Restricted Network of Evidence
5-FU/LV = 5-fluorouracil plus leucovorin; KN-966 = KEYNOTE-966 study.
Source: Sponsor-submitted indirect treatment comparison.36
Table 21: Assessment of Homogeneity for Sponsor-Submitted ITC
Characteristics | Description and handling of potential effect modifiers |
|---|---|
General trial characteristics | The KEYNOTE-966 and TOPAZ-1 trials were global, whereas the other trials were conducted in a single country. Schinzari (2017) and Sharma (2010) were relatively small trials, with fewer than 30 patients per treatment arm. |
Trial eligibility criteria | The ABC-01, ABC-02, BT22, and Schinzari (2017) studies enrolled patients with intrahepatic or extrahepatic bile duct, gallbladder, or ampullary cancer. The KEYNOTE-966, Kim (2019), and TOPAZ-1 studies enrolled only patients with intrahepatic or extrahepatic bile duct or gallbladder cancer. The Sharma (2010) and Sharma (2019) studies enrolled only patients with gallbladder cancer. |
Risk of bias | The ABC-02, KEYNOTE-966, Kim (2019), and Sharma (2019) studies had a low risk of bias, whereas ABC-01, BT22, Schinzari (2017), Sharma (2010), and TOPAZ-1 had some concerns of bias, mainly due to inadequate information reported on allocation sequence concealment. |
Treatment regimens | Regarding treatment regimens, there were slight between-trial differences in treatment regimens for gemcitabine + cisplatin and gemcitabine + oxaliplatin and marked between-trial differences in 5-FU/LV treatment regimens. For gemcitabine + cisplatin, both drugs were allowed until disease progression in ABC-01, ABC-02, and BT22; both drugs were restricted to a maximum of 8 cycles in the Sharma (2019) and TOPAZ-1 studies; and, in the KEYNOTE-966 study, cisplatin was restricted to a maximum of 8 cycles, but gemcitabine was allowed until disease progression. For gemcitabine + oxaliplatin, both drugs were allowed until disease progression in the Kim (2019) and Sharma (2010) trials, whereas the Sharma (2019) study restricted both drugs to a maximum of 6 cycles. For 5-FU/LV, there were multiple differences in the doses and schedules of both components of the treatment between the Sharma (2010) and Schinzari (2017) studies. |
Baseline patient characteristics | Age, sex, race or ethnicity, ECOG Performance Status, disease classification at baseline (e.g., locally advanced, metastatic, recurrent), cancer location (e.g., intrahepatic bile duct, extrahepatic bile duct, gallbladder, ampulla), and PD-L1 expression were explored as potential treatment-effect modifiers based on a review of subgroup results from trials included in the SLR and recently published narrative reviews and systematic reviews and meta-analyses.55-57 There was no notable between-trial heterogeneity in race or ethnicity, disease classification, or PD-L1 expression. However, there was notable between-study heterogeneity in age, sex, ECOG Performance Status, and cancer location. Regarding age, the Sharma (2010) and Sharma (2019) studies enrolled relatively young patients. Regarding sex, the Sharma (2010) and Sharma (2019) studies enrolled a relatively high proportion of female patients. Regarding ECOG Performance Status, the Sharma (2019) trial enrolled a relatively high proportion of patients with a score of 2, whereas the BT22 study enrolled a relatively high proportion of patients with a score of 0. |
Outcome definitions and availability | Regarding outcome definitions, the TOPAZ-1 study reported INV-assessed PFS, and the KEYNOTE-966 study reported both INV-assessed and BICR-assessed PFS. The remaining trials did not report the PFS assessment method. Regarding outcome availability, HRs and/or KM curves for PFS as well as response were available for all 9 trials, whereas HRs and/or KM curves for OS were available for only 8 trials. |
5-FU/LV = 5-fluorouracil plus leucovorin; BICR = blinded independent central review; ECOG = Eastern Cooperative Oncology Group; HR = hazard ratio; INV = investigator; ITC = independent treatment comparison; KM = Kaplan-Meier; NMA = network meta-analysis; OS = overall survival; PD-L1 = programmed cell death 1 ligand 1; PFS = progression-free survival; SLR = systematic literature review.
Source: Sponsor-submitted ITC.36 Details included in the table are from the sponsor’s summary of clinical evidence.
Figure 7: Network of Evidence for OS and PFS
KN-966 = KEYNOTE-966 study; OS = overall survival; PFS = progression-free survival.
The KEYNOTE-966 and TOPAZ-1 studies were included in the NMA, as these were the trials that were most similar in terms of study and patient characteristics. The relevant outcomes were OS, PFS, and all-cause AEs (any grade and grades 3 to 5). As potential subgroup effects were noted in the TOPAZ-1 study based on geographic region (Asia versus non-Asia), subgroup analyses were conducted to assess the impact of differences in the distribution of patients located in Asia in the TOPAZ-1 and KEYNOTE-966 studies on the estimated relative treatment effects for OS and PFS. Note that all analyses were based on the data cut with the longest available follow-up. According to the sponsor, fixed-effect models were used in the NMA because there was insufficient evidence to estimate between-study heterogeneity.
Detailed efficacy and safety results of the NMA are presented in Table 22.
Overall Survival
Considering the ITT population in both trials, results from fixed-effect models showed that the HR for OS was ████ ████ ███ ████ ██ █████ for the comparison of pembrolizumab plus chemotherapy versus durvalumab plus chemotherapy. Pembrolizumab plus chemotherapy was favoured over placebo plus chemotherapy.
In the subgroup analyses, the HR for OS was ████ ████ ███ ████ ██ █████ for patients in Asia and ████ ████ ███ ████ ██ █████ for patients outside Asia, for the comparison of pembrolizumab plus chemotherapy versus durvalumab plus chemotherapy.
Progression-Free Survival
Considering the ITT population in both trials, results from fixed-effect models showed that the HR for PFS was ████ ████ ███ ████ ██ █████ for the comparison of pembrolizumab plus chemotherapy versus durvalumab plus chemotherapy. Pembrolizumab plus chemotherapy was favoured over placebo plus chemotherapy.
Harms
In the as-treated population, results from the fixed-effect models showed that the odds ratio for any AEs was ████ ████ ███ ████ ██ █████, and the odds ratio for AEs of grade 3 or greater was ████ ████ ███ ████ ██ ██████ for the comparison of pembrolizumab plus chemotherapy versus durvalumab plus chemotherapy. The odds ratio for the comparison of pembrolizumab plus chemotherapy versus placebo plus chemotherapy for any AEs was ████ ████ ███ ████ ██ █████,and was ████ ████ ███ ████ ██ █████ for AEs of grade 3 or greater.
Table 22: Summary of Efficacy and Safety Outcome Measures in the Sponsor-Submitted ITC
Pembrolizumab + chemotherapy comparators | ITT population | Safety analysis population | ||
|---|---|---|---|---|
OS HR (95% CrI) | PFS HR (95% CrI) | AEs, any grade OR (95% CrI) | Grade 3 to 5 AEs OR (95% CrI) | |
Durvalumab + chemotherapy | █████████ | █████████ | █████████ | █████████ |
Placebo + chemotherapy | █████████ | █████████ | █████████ | █████████ |
AE = adverse event; CrI = credible interval; HR = hazard ratio; ITC = indirect treatment comparison; ITT = intention to treat; OR = odds ratio; OS = overall survival; PFS = progression-free survival.
Note: Bolded values indicate statistically meaningful difference between treatment groups at the 0.05 level.
Source: Sponsor-submitted ITC.36
Critical Appraisal of Sponsor-Submitted ITC
In the ITC submitted by the sponsor, the studies were identified by searching multiple databases based on prespecified inclusion and exclusion criteria. The studies were selected by 2 independent reviewers; thus, errors and bias in the study selection process were minimized. The reviewers of this ITC used appropriate methods to reduce the risk of bias and error in data extraction. The risk of bias in the included trials was assessed by 2 independent reviewers, which minimized the risk of error and bias in the assessments themselves. However, the risk of bias was assessed at the trial level rather than at the level of the reported results (i.e., per outcome), which ignores that the risk of bias can vary by reported result within a trial. There was no discussion on how any potential biases in the included trials could have an impact on the validity of the ITC, and no sensitivity analyses were conducted to assess the impact of trials with a risk of bias. In this ITC, the analyses were based on the data from 2 RCTs. The results were derived from fixed-effect models rather than random-effect models. According to the sponsor, this was justified because there was insufficient evidence in the network to estimate between-study heterogeneity.
One of the major concerns for the ITC and NMA is that the included trials could be highly heterogeneous in terms of study design and patient characteristics at baseline. Two studies (KEYNOTE-966 and TOPAZ-1) were included in the NMA. They were comparable in design (phase III, double-blind, multicentre, placebo-controlled RCT) and in patients’ baseline characteristics in general (demographic characteristics, cancer subtypes, performance status, site of origin, and so forth), although there was a 10% difference in race between the KEYNOTE-966 study (46% were Asian) and the TOPAZ-1 trial (56% were Asian). In this NMA, age, sex, race or ethnicity, ECOG Performance Status, disease classification at baseline (e.g., locally advanced, metastatic, recurrent), cancer location (e.g., intrahepatic bile duct, extrahepatic bile duct, gallbladder, ampulla), and PD-L1 expression were explored as potential treatment-effect modifiers. According to the sponsor, these effect modifiers were identified through subgroup analyses or systematic reviews. However, 1 key statistical limitation of using these subgroup analyses is that they are likely underpowered (the sample size of a clinical trial is not calculated based on a subset of patients but rather on all randomized patients to evaluate the primary objective of the study). In addition, it is unclear if these systematic reviews are sufficient to inform all relevant treatment-effect modifiers. According to the clinical experts we consulted, these are relevant effect modifiers for the treatment of advanced BTC. The impact of a 10% difference in the proportion of Asian patients between the 2 study populations on result interpretations was uncertain. Although subgroup analyses for OS were undertaken for patients in Asia and patients outside Asia, neither subgroup result was sufficiently precise to conclude which treatment was favoured.
The efficacy outcomes and safety outcomes were defined consistently between the 2 studies. The median length of follow-up was 16.8 months in the TOPAZ-1 study and 25.6 months in the KEYNOTE-966 trial. There were some discrepancies between the 2 studies in terms of the number of cycles of chemotherapy allowed; however, it is unlikely this would have an impact on the study results interpretation.
Given the lack of closed loops in the networks, the consistency in the ITC analyses could not be tested. All comparisons are therefore informed only by indirect evidence, which increases the level of uncertainty.
Efficacy and safety data were sparse in this NMA for the comparison of pembrolizumab plus chemotherapy versus durvalumab plus chemotherapy. The 95% CrIs for the point estimates were wide and spanned the null, therefore, confidence in the effect estimates for efficacy and harms of the study drugs was limited due to imprecision indicated by the wide CrIs for these outcomes and precludes any conclusions as to which treatment may be favoured.
In this ITC, several efficacy and safety outcomes were analyzed, such as OS, PFS, and harms. However, other efficacy end points of interest to patients and clinicians, such as HRQoL, were not investigated. Therefore, the relative treatment effect of pembrolizumab plus chemotherapy versus other active treatments on patients’ HRQoL remains unknown.
Studies Addressing Gaps in the Systematic Review Evidence
There were no relevant studies addressing the gaps in the systematic review evidence submitted for this review.
Discussion
Summary of Available Evidence
The evidence included in the systematic review consisted of 1 pivotal phase III double-blind RCT. The KEYNOTE-966 study (N = 1,069) met the inclusion criteria for the systematic review conducted by the sponsor. The purpose of this study was to evaluate the efficacy and safety of the combination of pembrolizumab plus chemotherapy versus placebo plus chemotherapy in patients with locally advanced unresectable or metastatic BTC. Patients were randomized to 1 of the following:
pembrolizumab 200 mg on day 1 of each cycle every 3 weeks by IV infusion (maximum 35 cycles) in combination with gemcitabine (1,000 mg/m2 on days 1 and 8 of each cycle every 3 weeks, with no maximum duration) and cisplatin (25 mg/m2 by IV infusion on days 1 and 8 of each cycle; maximum duration 8 cycles)
placebo in combination with gemcitabine and cisplatin.
The primary efficacy end point in the KEYNOTE-966 study was OS. Other relevant outcomes in this study included PFS, HRQoL measured by the EORTC QLQ-C30 and EORTC QLQ-BIL21, and safety. Overall, the patients’ baseline characteristics were balanced between treatment groups. The trial randomized approximately equal proportions of females and males (552 males; 51.6%). The median age was 64.0 years (range, 23 to 85 years). Most randomized patients were white (49.0%; n = 524), had an ECOG Performance Status score of 1 (54.4%; n = 582), and were from a region outside Asia (54.5%; n = 583). Most patients had metastatic disease (88.2%; n = 943) with an intrahepatic site of origin (59.2%; n = 633). Approximately 30% of these patients had received prior surgery (29.8%; n = 319).
The sponsor submitted 1 NMA to compare the treatment efficacy and safety of pembrolizumab plus chemotherapy with active therapies for the treatment of advanced BTC. The comparative efficacy and safety of pembrolizumab versus durvalumab were evaluated, in combination with chemotherapy with gemcitabine and cisplatin, based on evidence from 2 phase III RCTs.
Interpretation of Results
Efficacy
According to the patient group input, the clinical experts we consulted, and the clinician group that submitted input for this review, the important unmet needs that exist in current treatments for advanced BTC include having therapies that are curative and able to improve survival and patients’ HRQoL. The KEYNOTE-966 study met its primary end point at the final analysis (DCO of December 15, 2022). The results suggested that treatment with pembrolizumab plus chemotherapy may be associated with prolonged OS compared with treatment with placebo plus chemotherapy, with a median OS for the pembrolizumab plus chemotherapy group of 12.7 months (95% CI, 11.5 to 13.6) versus 10.9 months (95% CI, 9.9 to 11.6) for the placebo plus chemotherapy group. The HR for OS was 0.83 (95% CI, 0.72 to 0.95; P = 0.0034). Although the between-group difference in OS was 1.8 months, given the poor prognosis in patients with advanced BTC (with a median OS of less than 12 months), an improvement of 1.8 months in median survival is considered a clinically meaningful benefit, according to the clinical experts we consulted. In addition, the OS rates at various time points (6, 12, 18, and 24 months) showed that pembrolizumab plus chemotherapy likely results in a modest but clinically important increase in the probability of OS at all of the time points evaluated compared with placebo plus chemotherapy. The point estimates for the differences in KM-estimated survival probability between the 2 treatment groups ranged from 5.0% to 7.5%, depending on the follow-up time. These estimates were affected by imprecision; the 95% CIs included the potential for trivial effects, based on a threshold for a clinically important between-group difference of 5%, as informed by the clinical experts we consulted. The results of the prespecified subgroup analyses based on various patient baseline characteristics were consistent with those of the overall population. Although the survival benefit shown in the subgroups seems small, according to the clinical experts we consulted, they noted that this benefit may be considered meaningful to patients, given the poor prognosis of advanced BTC. However, the trial was unlikely to be sufficiently powered to detect subgroup differences, and tests for treatment by subgroup interactions were not undertaken.
PFS measured with RECIST 1.1 was 1 of the key secondary end points in the KEYNOTE-966 study. At the December 15, 2021, DCO, the median PFS was 6.5 months (95% CI, 5.7 to 6.9) with pembrolizumab plus chemotherapy and 5.6 months (95% CI, 5.1 to 6.6) with placebo plus chemotherapy. The corresponding HR was 0.86 (95% CI, 0.75 to 1.00; P = 0.0225), which did not meet the prespecified efficacy boundary for a statistically significant PFS benefit for pembrolizumab plus chemotherapy, according to the multiplicity scheme. Further, the HR cannot be interpreted reliably; based on visual inspection of the PFS curves, the proportional hazards assumption appears to have been violated. At 6 months of follow-up, the results showed that pembrolizumab plus chemotherapy may result in an increase in the KM-estimated probability of PFS when compared with placebo plus chemotherapy; however, the clinical importance of the increase (6%) is uncertain, and the 95% CI included the possibility of no difference between treatments. At 18 months, the evidence was very uncertain regarding the effect of pembrolizumab plus chemotherapy when compared with placebo plus chemotherapy on the KM-estimated probability of PFS, owing to imprecision (the CI included the potential that either treatment could be favoured) and indirectness (due to uncertainty in the adequacy of RECIST 1.1 to measure PFS). The sponsor noted that assessing PFS in patients with BTC is complex and often relies on nonradiographic factors, such as biliary obstruction, liver function, and serum carbohydrate antigen 19 to 9 expression.26 Thus, the sponsor indicated that PFS assessed based on RECIST 1.1 might not accurately reflect the PFS benefit gained in patients with BTC. PFS is typically considered a surrogate for OS in oncology trials. Although the sponsor provided no evidence for the surrogacy of PFS in this context, the KEYNOTE-966 study provided moderate-certainty evidence for a modest, albeit clinically important, OS benefit.
HRQoL measured by the EORTC QLQ-C30 and EORTC QLQ-BIL21 was an exploratory outcome in the KEYNOTE-966 study. At week 18, approximately 60% of the patients completed the assessments and contributed to the analysis of HRQoL data. As such, the results are at risk of bias due to missing outcomes data. Based on the between-group MIDs for these 2 instruments (the MID for the EORTC QLQ-C30 ranged from 5 to 10 points for most scales; while no MID for the EORTC QLQ-BIL21 was identified for patients with BTC, it can be extrapolated from other cancer types), the results suggested that treatment with pembrolizumab plus chemotherapy may result in little to no difference in HRQoL compared with placebo plus chemotherapy. A clinically important between-group difference in the change in the subscale scores in the EORTC QLQ-C30 for GHS, physical functioning and role functioning, and the subscale scores in the EORTC QLQ-BIL21 for jaundice and pain was not demonstrated.
For this submission, durvalumab plus chemotherapy was identified as the most relevant comparator for the indication under review. Comparative evidence of pembrolizumab plus chemotherapy versus durvalumab plus chemotherapy was only available through a sponsor-submitted NMA. Based on the results of this NMA, the evidence is insufficient to conclude whether pembrolizumab plus chemotherapy or durvalumab plus chemotherapy is favoured for prolonging OS or PFS in patients with advanced BTC. All estimates were affected by serious imprecision (i.e., wide 95% CrIs) that included the null, suggesting that either treatment could be favoured. Absolute effect estimates were not provided to infer the clinical importance of the HRs and their 95% CrIs. Other sources of uncertainty included a sparse network (included only 2 trials), reliance solely on indirect data, and a lack of information for other outcomes that are important to patients and clinicians (e.g., HRQoL).
Harms
The proportion of patients experiencing 1 or more AEs in the KEYNOTE-966 study was well balanced between the 2 treatment groups, which suggested that adding pembrolizumab to existing chemotherapy is not associated with an added risk of AEs; the comparisons between pembrolizumab plus chemotherapy and placebo plus chemotherapy were 99.1% versus 99.6% for any AEs, 52.2% versus 49.3% for SAEs, and 26.1% versus 22.8% for treatment discontinuation due to AEs, respectively. The proportion of patients with AEs resulting in death was 5.9% (31 patients) in the pembrolizumab plus chemotherapy group and 9.2% (49 patients) in the placebo plus chemotherapy group. Patients in the pembrolizumab plus chemotherapy group reported more notable harms compared with those in the comparator group (22.1% versus 12.9%, respectively). For the immune-mediated AEs (immune-mediated enterocolitis, hepatitis, or lung disease), few events were reported in both treatment groups in the KEYNOTE-966 study, and it did not appear that pembrolizumab plus chemotherapy resulted in clinically important increases in these events. When assessing the certainty of evidence, rating down for imprecision can be more conservative if the baseline risk is low in the treatment groups. There were no unusual safety signals observed for the treatment of pembrolizumab plus chemotherapy. The frequency, type, and severity of harms were consistent with pembrolizumab monotherapy, and the harms were not exacerbated by the combination of pembrolizumab with chemotherapy. According to the clinical experts we consulted, the AEs observed in the KEYNOTE-966 study are manageable in clinical practice.
In the sponsor-submitted ITC, the evidence was insufficient to conclude whether pembrolizumab plus chemotherapy or durvalumab plus chemotherapy were favoured for the odds of any AEs or AEs greater than grade 3. All estimates were affected by serious imprecision (i.e., wide 95% CrIs) that included the null, suggesting that either treatment could be favoured. Absolute effects were not provided to infer the clinical importance of the odds ratios and their 95% CrIs.
Conclusion
Locally advanced unresectable or metastatic BTCs are associated with a poor prognosis. Patients and clinicians highlighted the need for new treatments that prolong life, maintain HRQoL, and reduce side effects compared with the current treatments. Evidence from a randomized, double-blind, phase III RCT (KEYNOTE-966) showed that treatment with pembrolizumab plus chemotherapy likely results in modest increases in the probability of survival at 6, 12, 18, and 24 months compared with placebo plus chemotherapy for patients with locally advanced or metastatic BTC. Evidence from the trial also showed that pembrolizumab plus chemotherapy may result in an increase in the probability of PFS at 6 months, although the clinical importance of this increase is uncertain, and the evidence is very uncertain at longer follow-up (18 months). Evidence for PFS was additionally affected by uncertainty related to the ability of RECIST 1.1 to appropriately measure this outcome. Evidence on HRQoL suggested that adding pembrolizumab to chemotherapy may not result in any clinically important difference in patients’ HRQoL compared with chemotherapy alone; however, the evidence was rated as low certainty due to the limitations of the analyses, including the risk of bias due to missing data. In terms of safety, evidence from the KEYNOTE-966 study suggested that treatment with pembrolizumab plus chemotherapy did not result in an increased risk of any AEs, and likely did not result in an increased risk of SAEs, AEs leading to treatment discontinuation, or immune-mediated AEs, when compared with placebo plus chemotherapy.
There is a lack of direct comparative evidence between pembrolizumab plus chemotherapy and other active treatments for advanced BTC, namely, durvalumab plus chemotherapy. The indirect evidence from a sponsor-submitted NMA of 2 trials was insufficient to conclude whether the treatment of pembrolizumab plus chemotherapy differs in terms of OS or PFS or the odds of AEs when compared with durvalumab plus chemotherapy. There was substantial uncertainty in the treatment-effect estimates (indicated by wide CrIs) from the NMA due to limited efficacy and safety data, and no comparisons of HRQoL outcomes that are important to patients and clinicians were conducted.
References
1.Vogel A, Bridgewater J, Edeline J, et al. Biliary tract cancer: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann Oncol. 2023;34(2):127-140. PubMed
2.Tariq NU, McNamara MG, Valle JW. Biliary tract cancers: Current knowledge, clinical candidates and future challenges. Cancer Manag Res. 2019;11:2623-2642. PubMed
3.Forner A, Vidili G, Rengo M, Bujanda L, Ponz-Sarvise M, Lamarca A. Clinical presentation, diagnosis and staging of cholangiocarcinoma. Liver Int. 2019;39 Suppl 1(1):98-107.
4.Bridgewater J, Galle PR, Khan SA, et al. Guidelines for the diagnosis and management of intrahepatic cholangiocarcinoma. J Hepatol. 2014;60(6):1268-1289. PubMed
5.Lewis HL, Rahnemai-Azar AA, Dillhoff M, Schmidt CR, Pawlik TM. Current management of perihilar cholangiocarcinoma and future perspectives. Chirurgia (Bucur). 2017;112(3):193-207. PubMed
6.Mertens J. Clinical diagnosis and management of perihilar cholangiocarcinoma. Clin Liver Dis (Hoboken). 2014;3(3):60-64. PubMed
7.Dickson PV, Behrman SW. Distal cholangiocarcinoma. Surg Clin North Am. 2014;94(2):325-342. PubMed
8.Johns Hopkins Medicine. Ampullary Cancer [accessed by sponsor]. https://www.hopkinsmedicine.org/health/conditions-and-diseases/ampullary-cancer. Accessed 2023 Sep 12.
9.Banales JM, Marin JJG, Lamarca A, et al. Cholangiocarcinoma 2020: The next horizon in mechanisms and management. Nat Rev Gastroenterol Hepatol. 2020;17(9):557-588. PubMed
10.Valle J, Wasan H, Palmer DH, et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N Engl J Med. 2010;362(14):1273-1281. PubMed
11.Copur MS, Merani S, Vargas LM. Biliary cancer: Current multimodality treatment and future directions. Oncology (Williston Park). 2019;33(9). PubMed
12.American Cancer Society. Survival rates for bile duct cancer [accessed by sponsor]. https://www.cancer.org/cancer/types/bile-duct-cancer/detection-diagnosis-staging/survival-by-stage.html. Accessed 2024 Jan 28.
13.National Cancer Institute Surveillance, Epidemiology, and End Results (SEER) Program. Cancer Stat Facts: Liver and intrahepatic bile duct cancer [accessed by sponsor]. https://seer.cancer.gov/statfacts/html/livibd.html. Accessed 2023 Oct 11.
14.National Cancer Institute Surveillance, Epidemiology, and End Results (SEER) Training Modules. Pancreatic and biliary cancer. Five-year survival rates [accessed by sponsor]. https://training.seer.cancer.gov/biliary/intro/survival.html. Accessed 2023 Oct 11.
15.Xiao Y, Cattelan L, Lagacé F, et al. Epidemiologic trends and geographic distribution of patients with gallbladder and extrahepatic biliary tract cancers in Canada. HPB (Oxford). 2021;23(10):1541-1549. PubMed
16.Brenner DR, Poirier A, Woods RR, et al. Projected estimates of cancer in Canada in 2022. CMAJ. 2022;194(17):E601-E607. PubMed
17.Beaulieu C, Lui A, Yusuf D, et al. A population-based retrospective study of biliary tract cancers in Alberta, Canada. Curr Oncol. 2021;28(1):417-427. PubMed
18.Seung SJ, Saherawala H, Syed I, Clouthier DL, Shephard C, Chen E. Real-world treatment and outcomes for biliary tract cancer patients using administrative databases in Ontario, Canada. Paper presented at: European Society for Medical Oncology (ESMO) Gastrointestinal (GI) Congress; June 29-July 2, 2022, 2022.
19.Boutin M, Yang J, Safro M, Davies JM, Gill S. Trends in incidence, treatments, and clinical outcomes of cholangiocarcinoma diagnosed in British Columbia from 2005 through 2019 [accessed by sponsor]. ASCO Gastrointestinal Cancers; 2023.
20.Canadian Cancer Society. Cancer types: Bile duct. Prognosis and survival. Survival statistics for extrahepatic bile duct cancer [accessed by sponsor]. https://cancer.ca/en/cancer-information/cancer-types/bile-duct/prognosis-and-survival/survival-statistics. Accessed 2023 Sep 12.
21.Ramjeesingh R, Chaudhury P, Tam VC, et al. A practical guide for the systemic treatment of biliary tract cancer in Canada. Curr Oncol. 2023;30(8):7132-7150. PubMed
22.Oh DY, Ruth He A, Qin S, et al. Durvalumab plus gemcitabine and cisplatin in advanced biliary tract cancer. NEJM Evid. 2022;1(8):EVIDoa2200015.
23.Drug Intelligence. ONCO-CAPPS Patient Treatment Dynamics Advanced Biliary Tract Carcinoma, 2023 Q3 Update and Trends. Data on file. October 2023 [sponsor supplied reference].
24.Merck Canada Inc. KEYTRUDA (pembrolizumab) solution for infusion, 100 mg/4 mL vial. Product Monograph. Date of Initial Approval: May 19, 2015. Date of Revision: TBC (draft for proposed new indication) [sponsor supplied reference].
25.Merck. Merck Sharp & Dohme LLC, Rahway, NJ USA. [CONFIDENTIAL] Clinical Study Report P966V01MK3475. A phase 3 randomized, double-blind study of pembrolizumab plus gemcitabine/cisplatin versus placebo plus gemcitabine/cisplatin as first-line therapy in participants with advanced and/or unresectable biliary tract carcinoma. Study Completion Date: December 15, 2022 data cut-off. Date of Report: March 20, 2023 [sponsor supplied reference]. 2023.
26.Kelley RK, Ueno M, Yoo C, et al. Pembrolizumab in combination with gemcitabine and cisplatin compared with gemcitabine and cisplatin alone for patients with advanced biliary tract cancer (KEYNOTE-966): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2023;401(10391):1853-1865. PubMed
27.Balshem H, Helfand M, Schunemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-406. PubMed
28.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: Informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. PubMed
29.Musoro JZ, Coens C, Sprangers MAG, et al. Minimally important differences for interpreting EORTC QLQ-C30 change scores over time: A synthesis across 21 clinical trials involving nine different cancer types. Eur J Cancer. 2023;188:171-182. PubMed
30.Merck Canada Inc. Merck response to January 12, 2024 INITIAL CADTH request for additional information regarding KEYTRUDA CADTH review: between-group differences in efficacy outcomes [internal additional sponsor's information]. 2024.
31.Canadian Cancer Society. Cancer types: Bile duct. Diagnosis [accessed by sponsor]. https://cancer.ca/en/cancer-information/cancer-types/bile-duct/diagnosis. Accessed 2023 Sep 12.
32.Canadian Cancer Society. Cancer types: Bile duct. Prognosis and survival for bile duct cancer [accessed by sponsor]. https://cancer.ca/en/cancer-information/cancer-types/bile-duct/prognosis-and-survival. Accessed 2023 Sep 12.
33.AstraZeneca Canada Inc. Imfinzi (durvalumab for injection) 50 mg/mL concentrate for infusion. Product Monograph. Date of Initial Authorization: November 3, 2017. Date of Revision: April 21, 2023 [sponsor supplied reference].
34.CADTH Reimbursement Recommendation: Durvalumab (Imfinzi) in combination with gemcitabine-based chemotherapy, for the treatment of patients with locally advanced or metastatic biliary cancer. Can J Health Technol. 2023;3(2). https://www.cadth.ca/sites/default/files/DRR/2023/PC0296REC-Imfinzi_JH_GP1_KAS.pdf. Accessed 2023 Sep 12.
35.National Comprehensive Cancer Network (NCCN) Clinical Practice Guidelines in Oncology: Biliary tract cancers. Version 3.2023. November 8, 2023 [accessed by sponsor].
36.Systematic literature review and network meta-analysis of first-line treatments for patients with advanced and/or unresectable biliary tract cancer, Technical report [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Keytruda, 100 mg/4 mL vial solution for IV infusion. Kirkland (QC): Merck; 2023 Aug 26.
37.Finn RS, Kelley RK, Furuse J, et al. KEYNOTE-966: A randomized, double-blind, placebo-controlled, phase 3 study of pembrolizumab in combination with gemcitabine and cisplatin for the treatment of advanced biliary tract carcinoma. Cancer Res. 2020;80:CT283.
38.Kelley RK, Vogel A, Finn RS, et al. Pembrolizumab in combination with gemcitabine and cisplatin for the treatment of advanced biliary tract cancer: Phase 3 KEYNOTE-966 trial in progress. Paper presented at: International Liver Cancer Association (ILCA) 14th Annual Conference; September 11-13, 2020, 2020; ILCA Virtual Conference.
39.Kelley RK, Vogel A, Furuse J, et al. KEYNOTE-966: Trial in Progress: Pembrolizumab added to gemcitabine and cisplatin for advanced biliary tract cancer. Paper presented at: 71st Annual Meeting of the American Association for the Study of Liver Diseases (AASLD); November 13-17, 2020, 2020; Boston, MA.
40.Valle JW, Kelley RK, Furuse J, et al. KEYNOTE-966 Trial in Progress: Pembrolizumab plus gemcitabine and cisplatin for advanced biliary tract cancer. Paper presented at: European Society for Medical Oncology (ESMO) 45th Congress; September 14-October 18, 2020, 2020; Virtual Congress.
41.Vogel A, Finn RS, Kelley RK, et al. Pembrolizumab in combination with gemcitabine and cisplatin for the treatment of advanced biliary tract cancer: Phase 3 KEYNOTE-966 trial in progress. Ann Oncol. 2020;31:S122.
42.Yoo C, Finn RS, Klumpen HJ, et al. Health-related quality of life (HRQoL) in the phase 3 KEYNOTE-966 study of pembrolizumab (pembro) plus gemcitabine and cisplatin (gem/cis) versus placebo plus gem/cis for advanced biliary tract cancer (BTC). J Clin Oncol. 2023;41:4003.
43.Merck Sharp & Dohme LLC. NCT04003636: Pembrolizumab (MK-3475) plus gemcitabine/cisplatin versus placebo plus gemcitabine/cisplatin for first-line advanced and/or unresectable biliary tract carcinoma (BTC) (MK-3475-966/KEYNOTE-966). ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine; 2023: https://clinicaltrials.gov/study/NCT04003636. Accessed 2023 Sep 9.
44.Aaronson NK, Ahmedzai S, Bergman B, et al. The European Organization for Research and Treatment of Cancer QLQ-C30: A quality-of-life instrument for use in international clinical trials in oncology. J Natl Cancer Inst. 1993;85(5):365-376. PubMed
45.Kaupp-Roberts SD, Yadegarfar G, Friend E, et al. Validation of the EORTC QLQ-BIL21 questionnaire for measuring quality of life in patients with cholangiocarcinoma and cancer of the gallbladder. Br J Cancer. 2016;115(9):1032-1038. PubMed
46.Friend E, Yadegarfar G, Byrne C, et al. Development of a questionnaire (EORTC module) to measure quality of life in patients with cholangiocarcinoma and gallbladder cancer, the EORTC QLQ-BIL21. Br J Cancer. 2011;104(4):587-592. PubMed
47.Reeve BB, Wyrwich KW, Wu AW, et al. ISOQOL recommends minimum standards for patient-reported outcome measures used in patient-centered outcomes and comparative effectiveness research. Qual Life Res. 2013;22(8):1889-1905. PubMed
48.Giesinger JM, Kieffer JM, Fayers PM, et al. Replication and validation of higher order models demonstrated that a summary score for the EORTC QLQ-C30 is robust. J Clin Epidemiol. 2016;69:79-88. PubMed
49.Fayers PM, Aaronson NK, Bjordal K, et al. The EORTC QLC-C30 scoring manual (3rd Edition). Brussels (BE): European Organisation for Research and Treatment of Cancer (EORTC); 2001: https://www.eortc.org/app/uploads/sites/2/2018/02/SCmanual.pdf. Accessed 2022 Sep 14.
50.Liang K-YZSL. Longitudinal data analysis of continuous and discrete responses for pre-post designs. The Indian Journal of Statistics. 2000;62(1):134-148.
51.Maurer W, Bretz F. Multiple testing in group sequential trials using graphical approaches. Stat Biopharm Res. 2013;5(4):311-320.
52.Saad ED, Katz A, Hoff PM, Buyse M. Progression-free survival as surrogate and as true end point: Insights from the breast and colorectal cancer literature. Ann Oncol. 2010;21(1):7-12. PubMed
53.Fallowfield LJ, Fleissig A. The value of progression-free survival to patients with advanced-stage cancer. Nat Rev Clin Oncol. 2011;9(1):41-47. PubMed
54.Belin L, Tan A, De Rycke Y, Dechartres A. Progression-free survival as a surrogate for overall survival in oncology trials: A methodological systematic review. Br J Cancer. 2020;122(11):1707-1714. PubMed
55.Azizi AA, Lamarca A, McNamara MG, Valle JW. Chemotherapy for advanced gallbladder cancer (GBC): A systematic review and meta-analysis. Crit Rev Oncol Hematol. 2021;163:103328. PubMed
56.Rizzo A, Brandi G. First-line chemotherapy in advanced biliary tract cancer ten years after the ABC-02 trial: “And yet it moves!”. Cancer Treat Res Commun. 2021;27:100335. PubMed
57.Jansen H, Pape UF, Utku N. A review of systemic therapy in biliary tract carcinoma. J Gastrointest Oncol. 2020;11(4):770-789. PubMed
58.Grambsch PM, Terneau TM. Proportional hazards tests and diagnostics based on weighted residuals. Biometrika. 1994;81(3):515-526.
59.Schoenfeld D. Partial residuals for the proportional hazards regression model. Biometrika. 1982;69(1):239-241.
60.Dias S, Welton NJ, Sutton AJ, Ades A. NICE DSU Technical Support Document 2: A generalized linear modelling framework for pairwise and network meta-analysis of randomized controlled trials [accessed by sponsor]. 2011.
61.Jansen JP. Network meta-analysis of survival data with fractional polynomials. BMC Med Res Methodol. 2011;11:61. PubMed
62.Jansen JP, Cope S. Meta-regression models to address heterogeneity and inconsistency in network meta-analysis of survival outcomes. BMC Med Res Methodol. 2012;12(1):152. PubMed
63.Dempster AP. The direct use of likelihood for significance testing. Statistics and Computing. 1997;7(4):247-252.
Pharmacoeconomic_Review
Abbreviations
BIA
budget impact analysis
BTC
biliary tract cancer
CMA
cost-minimization analysis
KM
Kaplan-Meier
NMA
network meta-analysis
OS
overall survival
pCPA
pan-Canadian Pharmaceutical Alliance
PFS
progression-free survival
RDI
relative dose intensity
ToT
time on treatment
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Pembrolizumab (Keytruda), 100 mg/4 mL vial for IV infusion |
Indication | Pembrolizumab, in combination with gemcitabine-based chemotherapy, is indicated for the treatment of adult patients with locally advanced unresectable or metastatic biliary tract carcinoma (BTC). |
Health Canada approval status | NOC |
Health Canada review pathway | Project Orbis |
NOC date | April 12, 2024 |
Reimbursement request | As per indication |
Sponsor | Merck Canada Inc. |
Submission history | Previously reviewed: Yes. Pembrolizumab (Keytruda) has been reviewed for multiple indications. In 2023, CADTH reviewed it for the following indications:
|
BTC = biliary tract carcinoma; NOC = Notice of Compliance; TBD = to be determined.
Table 2: Summary of Economic Information
Component | Description |
|---|---|
Type of economic evaluation | Cost-minimization analysis. |
Target population | Patients with locally advanced unresectable or metastatic BTC who would be eligible to receive immunotherapy as a first-line treatment in combination with chemotherapy. |
Treatment | Pembrolizumab plus chemotherapy (gemcitabine and cisplatin). |
Dose regimen |
|
Submitted price | Pembrolizumab: $4,400.00 per 100 mg/4 mL vial for IV infusion. |
Submitted treatment cost | Pembrolizumab plus chemotherapy (as regimen): |
Comparator | Durvalumab plus chemotherapy (gemcitabine and cisplatin). |
Perspective | Canadian publicly funded health care payer. |
Time horizon | Lifetime (20 years). |
Key data source | Key assumption of equal treatment efficacy and safety between pembrolizumab plus chemotherapy and durvalumab plus chemotherapy based on an unpublished NMA conducted by the sponsor. |
Costs considered | Drug acquisition and treatment administration costs. |
Submitted results | Pembrolizumab plus chemotherapy was associated with cost savings of $48,118 relative to durvalumab plus chemotherapy. |
Key limitations |
|
CDA-AMC reanalysis results |
|
AE = adverse event; BTC = biliary tract cancer; CDA-AMC = Canada’s Drug Agency; CMA = cost-minimization analysis; HRQoL = health-related quality of life; ITC = indirect treatment comparison; NMA = network meta-analysis; OS = overall survival; pCPA = pan-Canadian Pharmaceutical Alliance; PFS = progression-free survival; q.3.w. = every 3 weeks; q.6.w. = every 6 weeks; RDI = relative dose intensity.
aCycle costs consider RDIs, vial sharing, 5% vial wastage, and assuming a body surface area of 1.8 m2.
Conclusions
Assuming similar clinical efficacy and safety between pembrolizumab plus chemotherapy and durvalumab plus chemotherapy, the sponsor submitted a cost-minimization analysis (CMA) comparing drug acquisition and treatment administration costs. Based on CDA-AMC’s clinical review of the sponsor-submitted network meta-analysis (NMA), the assumption of comparable clinical efficacy and safety between pembrolizumab and durvalumab is associated with substantial uncertainty in the treatment-effect estimates (indicated by wide credible intervals) from the NMA due to limited efficacy and safety data, and no comparisons of health-related quality of life outcomes that are important to patients and clinicians were conducted.
The CDA-AMC base case suggests that pembrolizumab plus chemotherapy is associated with cost savings of $58,930 over a lifetime horizon compared with durvalumab plus chemotherapy, driven by the assumption that pembrolizumab would be administered with weight-based dosing. The estimated incremental savings are based on publicly available list prices for durvalumab and may not reflect actual prices paid by Canadian public drug plans. If the pan-Canadian Pharmaceutical Alliance (pCPA) has concluded negotiations with large price reductions for durvalumab, pembrolizumab may no longer result in cost savings. Scenario analyses suggest that if pembrolizumab is administered with fixed dosing or if no vial sharing is allowed, the estimated cost savings are reduced to approximately half of the cost savings estimated by the CDA-AMC base case.
The CMA is based on the assumption of similar clinical efficacy and safety between pembrolizumab and durvalumab. In addition, the cost-effectiveness of pembrolizumab compared with chemotherapy alone in this population is unknown.
Economic Review
The current review is for pembrolizumab (Keytruda) for adult patients with locally advanced unresectable or metastatic biliary tract carcinoma (BTC), in combination with chemotherapy.
Economic Information
Summary of Sponsor’s Economic Information
The sponsor submitted a CMA for pembrolizumab in combination with gemcitabine and cisplatin (here forward referred to as pembrolizumab plus chemotherapy) compared with durvalumab in combination with gemcitabine and cisplatin (here forward referred to as durvalumab plus chemotherapy) for the treatment of patients with locally advanced unresectable or metastatic BTC who would be eligible to receive immunotherapy as a first-line treatment in combination with chemotherapy.1 The modelled population therefore did not align with the reimbursement request or the Health Canada indication, as neither specifies the treatment line for the indicated population.2 The sponsor assumed that the only relevant comparator for pembrolizumab plus chemotherapy is durvalumab plus chemotherapy. At the time of this review, durvalumab was under active negotiations with the pCPA for the indicated population, and any current access was provided through a patient support program.1,3
Pembrolizumab and durvalumab are assumed to be administered as a regimen with gemcitabine and cisplatin at a recommended dose of 1,000 mg/m2 and 25 mg/m2, respectively, on days 1 and 8 of a 21-day cycle.4,5 The sponsor assumed different treatment durations for gemcitabine, depending on the immunotherapy combination.1 The sponsor assumed that, when administered with pembrolizumab, gemcitabine would be continued until disease progression or unacceptable toxicity; whereas, when administered with durvalumab, the sponsor assumed that gemcitabine would be administered only during the first 8 cycles, which is aligned with the respective clinical trials, KEYNOTE-9664 and TOPAZ-1.5
Pembrolizumab is available as a 100 mg/4 mL vial for IV infusion at a submitted price of $4,400.00 per vial and the recommended dosage is 200 mg every 3 weeks, or 400 mg every 6 weeks, up to 24 months (35 cycles every 3 weeks or 18 cycles every 6 weeks) or until disease progression or unacceptable toxicity).2 The sponsor assumed that the prices of gemcitabine and cisplatin were $540.00 per 2,000 mg vial and $9.50 per 50 mg vial, respectively. In combination with pembrolizumab, cisplatin is assumed to be administered for the first 8 cycles only and gemcitabine is assumed to be continued until disease progression or unacceptable toxicity.1 The sponsor assumed that pembrolizumab, cisplatin, and gemcitabine had relative dose intensities (RDIs) of 93.0%, 90.7%, and 85.4%, respectively, based on the mean RDIs in the KEYNOTE-966 trial.1,6 Considering mean RDIs, vial sharing, 5% vial wastage, and assuming a body surface area of 1.8 m2, the sponsor estimated that pembrolizumab plus chemotherapy would cost $9,034.30 for the first 8 21-day cycles and $9,018.70 every 21 days thereafter. This results in treatment costs of $156,449 in the first year and $156,324 in the second year, as calculated by the sponsor.1
Durvalumab has a recommended dosage of 1,500 mg every 3 weeks for 8 cycles and 1,500 mg every 4 weeks as monotherapy thereafter.7,8 In combination with durvalumab, cisplatin and gemcitabine are assumed to be administered only during the first 8 cycles, after which durvalumab is continued as a monotherapy until disease progression or unacceptable toxicity.1 The sponsor assumed that durvalumab, cisplatin, and gemcitabine have RDIs of 100%, 93.8%, and 93.8% respectively, based on the median RDIs in the TOPAZ-1 trial.5 Similarly, considering median RDIs, vial sharing, and 5% vial wastage, the sponsor estimated that durvalumab plus chemotherapy would cost $12,666.26 for the first 8 21-day cycles and $11,733.33 every 28 days thereafter. This results in treatment costs of $183,463 in the first year and $152,533 in the subsequent years, as calculated by the sponsor.1
The sponsor assumed pembrolizumab plus chemotherapy was associated with similar efficacy and safety compared with durvalumab plus chemotherapy, based on an unpublished sponsor-submitted NMA.1,9 However, to estimate the time on treatment (ToT) for each regimen, the sponsor used 2 different approaches: for pembrolizumab plus chemotherapy, the sponsor fitted an exponential parametric function to the ToT Kaplan-Meier (KM) data from the KEYNOTE-966 trial (approximately 30 months of data); for durvalumab plus chemotherapy, the sponsor fitted a log-normal parametric function to the progression-free survival (PFS) KM data for pembrolizumab plus chemotherapy from the KEYNOTE-966 trial (approximately 29 months of data). The sponsor constrained ToT for both treatment regimens to be no greater than the overall survival (OS). To model OS, the sponsor fitted a log-logistic parametric function to the OS KM data for pembrolizumab plus chemotherapy from the KEYNOTE-966 trial (approximately 34 months of data).
The sponsor included treatment administration costs, specifically chair, pharmacy, and nursing time. The respective treatment administration durations were sourced from Ontario Health (Cancer Care Ontario),7,10-12 and unit costs were sourced from the literature and inflated to 2023 Canadian dollars.13
Summary of Sponsor’s Economic Evaluation Results
The analysis was conducted from the perspective of the publicly funded health care payer over a lifetime horizon (20 years). Costs were discounted at a rate of 1.5% per annum.1 All analyses were run probabilistically (5,000 iterations). The deterministic and probabilistic results were similar.
Base-Case Results
The sponsor’s base case estimated that, over a lifetime horizon, pembrolizumab plus chemotherapy was associated with cost savings of $48,118 per patient relative to durvalumab plus chemotherapy. Pembrolizumab plus chemotherapy was associated with a total cost of $103,898 per patient, while durvalumab plus chemotherapy was associated with a total cost of $153,009 per patient (Table 3). Approximately 39% of the cost savings were accrued during the extrapolated period of the model (i.e., after the first 29 months, as observed in the KEYNOTE-966 trial).
Table 3: Summary of the Sponsor’s Economic Evaluation Results, Probabilistic
Drug | Total drug costs ($) | Incremental drug costs ($) | Total costs ($) | Incremental costs ($) |
|---|---|---|---|---|
Durvalumab + chemotherapy | 153,009 | Reference | 158,402 | Reference |
Pembrolizumab + chemotherapy | 103,898 | −49,111 | 110,284 | −48,118 |
Note: Chemotherapy is assumed to comprise cisplatin and gemcitabine. The negative incremental costs represent cost savings.
Source: Sponsor’s economic submission.1
Sensitivity and Scenario Analysis Results
The sponsor conducted deterministic scenario analyses considering alternative discount rates, time horizons, rates of vial wastage, ToT parametric curve distributions, and assuming that the ToT for durvalumab plus chemotherapy was equal to the ToT for pembrolizumab plus chemotherapy. The latter scenario had the greatest impact, as cost savings were reduced to $25,905 per patient.
CDA-AMC Appraisal of the Sponsor’s Economic Information
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications for the economic analysis:
The assumption of comparable clinical efficacy and safety between pembrolizumab and durvalumab is uncertain: In the absence of a direct head-to-head comparison between pembrolizumab plus chemotherapy and durvalumab plus chemotherapy, the sponsor submitted an indirect treatment comparison assessing the comparative efficacy and safety of pembrolizumab plus chemotherapy (KEYNOTE-966 study) and durvalumab plus chemotherapy (TOPAZ-1 study).1,9 As noted in the CDA-AMC Clinical Review Report, CDA-AMC’s assessment of the sponsor-submitted NMA indicated there is not sufficient evidence to suggest that treatment with pembrolizumab plus chemotherapy is associated with improved survival benefits (OS and PFS) or an increased risk of adverse events when compared with durvalumab plus chemotherapy. Moreover, the assumption of comparable clinical efficacy and safety between pembrolizumab and durvalumab is associated with substantial uncertainty in the treatment-effect estimates (indicated by wide credible intervals) from the NMA due to limited efficacy and safety data, and no comparisons of health-related quality of life outcomes that are important to patients and clinicians were conducted. Should patients receiving pembrolizumab plus chemotherapy persist on treatment for longer and/or experience longer survival, greater health care costs could be accrued than for patients receiving durvalumab; therefore, a cost-utility analysis would be more appropriate than a CMA.
CDA-AMC was unable to address this limitation in reanalysis due to the structural limitations of the sponsor’s model.
Omission of relevant comparators: The sponsor considered durvalumab with chemotherapy to be the only relevant comparator. As noted in the market research provided by the sponsor,14 platinum-based regimens with gemcitabine (i.e., cisplatin plus gemcitabine, carboplatin plus gemcitabine, and oxaliplatin plus gemcitabine) were used to treat approximately ██% of prevalent cases of patients with BTC as of the third quarter of 2023; durvalumab plus chemotherapy was only used for ██% of prevalent cases of patients with BTC based on moving annual total data but increases to ██% based on patients who newly started treatment in the previous 6 months. Published guidelines, including those published by Alberta Health Services, list gemcitabine plus cisplatin as an alternative to durvalumab plus chemotherapy, the preferred treatment.15-17 The clinical expert input obtained by CDA-AMC noted that while durvalumab is available through compassionate access, as pCPA negotiations are ongoing,18 a proportion of patients continue to be treated with gemcitabine plus cisplatin without immunotherapy (i.e., chemotherapy alone). As chemotherapy alone is still used by a considerable number of patients (per the sponsor’s own estimates) and is still listed as an alternative therapy by numerous guidelines, the sponsor inappropriately excluded a relevant comparator. CDA-AMC’s Guidelines for the Economic Evaluation of Health Technologies: Canada states that all interventions currently used and potentially displaced should be considered in the analysis.19 Given the cost of adding pembrolizumab to the cost of chemotherapy alone, the exclusion of chemotherapy alone from the submitted CMA favours pembrolizumab. CDA-AMC notes that a cost-utility analysis comparing pembrolizumab plus chemotherapy versus chemotherapy alone would have been more appropriate, as the CDA-AMC Clinical Review Report notes that treatment with pembrolizumab plus chemotherapy likely results in modest increases in the probability of survival at 6, 12, 18, and 24 months compared with chemotherapy alone. However, as the sponsor did not submit a cost-utility analysis comparing pembrolizumab plus chemotherapy with chemotherapy alone, the cost-effectiveness of pembrolizumab plus chemotherapy relative to chemotherapy alone is unknown.
CDA-AMC was unable to address this limitation in reanalysis due to the structural limitations of the sponsor’s model.
Treatment costs are uncertain: First, ToT was constrained to be no greater than the OS for pembrolizumab for both immunotherapy options. However, the sponsor’s OS extrapolations beyond the trial period (34 months) using a log-normal parametric function predicted that at years 2, 3, and 5, ████%, ████%, and |% of patients were alive. The clinical expert input obtained by CDA-AMC noted that the sponsor’s extrapolations beyond the trial period seemed overly optimistic, as it is rare for the indicated population to survive long-term. Clinical experts consulted by CDA-AMC assessed the plausibility of the survival estimates at various time points generated by alternative extrapolation curves. Clinician input noted that although using a Gompertz parametric function to extrapolate OS produced estimates more reasonably aligned with clinical practice (OS at years 2, 3, and 5 was ████%, ███%, and ███%, respectively), there is still great uncertainty due to the lack of long-term evidence documenting survival after treatment with immunotherapy in the indicated population.
Second, the sponsor assumed ToT for durvalumab plus chemotherapy was equal to the PFS for pembrolizumab plus chemotherapy from the KEYNOTE-966 trial by fitting a log-normal parametric function to the KM data. The sponsor’s PFS curve predicted that at years 2, 3, and 5, ███%, ███%, and ███% of patients were progression-free and still receiving durvalumab maintenance therapy. Likewise, the clinical expert input noted that the sponsor’s estimates were overly optimistic. CDA-AMC also notes that the sponsor’s choice resulted in a higher proportion of patients receiving durvalumab plus chemotherapy relative to pembrolizumab plus chemotherapy (refer to Appendix 1, Figure 1) which favours the treatment costs of pembrolizumab. As the clinical expert input noted that PFS should be closely aligned with ToT, both immunotherapies should have a similar ToT as the sponsor has assumed similar efficacy and safety for durvalumab plus chemotherapy.1,9 Alternatively, the use of a gamma parametric function to estimate the PFS (and ToT for durvalumab) produced estimates more reasonably aligned with clinical practice (i.e., ███%, ███%, and ███% at years 2, 3, and 5, respectively) and more closely aligned to the ToT for both immunotherapies.
Third, per the KEYNOTE-966 and TOPAZ-1 trials,4,5 the sponsor assumed that gemcitabine would continue until disease progression or unacceptable toxicity when used in combination with pembrolizumab, but would only be administered for up to 8 cycles when used in combination with durvalumab. The clinical expert input received by CDA-AMC noted that the continuation of gemcitabine beyond 8 cycles will vary widely across Canada and be based largely on clinician judgment and discussions with the patient; the immunotherapy with which it is used is unlikely to have any impact on whether it is continued beyond 8 cycles or not. The clinician input further noted that after 8 cycles of chemotherapy, most patients suffer from cumulative toxicity and therefore need to consider stopping chemotherapy. However, if patients are doing relatively well, clinicians may be more inclined to continue gemcitabine. It was further noted in the clinician input that there is a lack of evidence to support the concept that the continuation of gemcitabine improves outcomes.
Fourth, the dose of pembrolizumab in the KEYNOTE-966 study was a fixed dose of 200 mg IV every 3 weeks or 400 mg every 6 weeks. Input from participating public drug plans indicated that jurisdictions would likely implement weight-based dosing for pembrolizumab of 2 mg/kg every 3 weeks to a maximum dose of 200 mg or 4 mg/kg every 6 weeks to a maximum of 400 mg, similar to other indications for which pembrolizumab is reimbursed, even though Health Canada has approved pembrolizumab for fixed dosing. CDA-AMC notes that weight-based dosing will increase the cost savings associated with pembrolizumab, as the sponsor’s base case assumes vial sharing and a mean body weight of 68.3 kg resulting in patients requiring a dosage of 136.6 mg every 3 weeks rather than 200 mg every 3 weeks when a fixed-dose approach is used. All patients are assumed in the submitted economic evaluation to receive pembrolizumab every 3 weeks rather than every 6 weeks.
Fifth, the sponsor incorporated RDIs for durvalumab plus chemotherapy based on the median RDI from the TOPAZ-1 trial and RDIs for pembrolizumab plus chemotherapy based on the mean RDI from the KEYNOTE-966 trial.1,5,6 Since the sponsor submitted a CMA instead of a cost-utility analysis, in which the underlying assumption is similar efficacy and safety for durvalumab plus chemotherapy, it is inappropriate to use different measures of central tendency (i.e., mean RDIs versus median RDI) between the 2 treatment options. Also, the consideration of RDI is complex, as this parameter can be influenced by several factors. The dose received by a patient may differ from the full planned dose of the drug due to dose delays, missed doses, dose reductions to manage toxicity, or subsequent dose re-escalation; each of these has differing impacts on drug costs. The clinical expert input received by CDA-AMC noted there is no reason for treatment compliance, dose delays, dose reductions to manage toxicity, or dose re-escalations to differ between the 2 regimens based on the evidence from the KEYNOTE-966 and TOPAZ-1 studies. Furthermore, it is unclear how these assumptions interact with drug wastage, as the sponsor applied a drug wastage setting that only accounted for partial drug wastage or vial sharing (i.e., 95% of the remaining vial quantity would be shared between patients and the remainder would result in product loss) for all treatments. The sponsor did not provide any supporting evidence to justify why 5% of the product would be wasted on a regular basis. Moreover, the clinical expert input indicated that the policies around vial sharing may vary across Canadian jurisdictions and that partial or full drug wastage might be an area of uncertainty.
Lastly, the sponsor assumed that the price of a 50 mg vial of cisplatin was $9.50, referencing a previous CDA-AMC report20 as its price source.1 Based on pricing information from IQVIA Delta PA,21 the price of a 50 mg vial of cisplatin is $135.00, as stated in the CDA-AMC report referenced by the sponsor.20
In all of the CDA-AMC reanalyses, the price of cisplatin was corrected to $135.00 per 50 mg vial.
CDA-AMC addressed the limitations concerning ToT for immunotherapies by using a Gompertz parametric function to extrapolate OS and a gamma parametric function to extrapolate PFS (and consequently ToT for durvalumab). CDA-AMC notes that changing the PFS distribution aligned the ToT for durvalumab plus chemotherapy with the ToT for pembrolizumab plus chemotherapy.
Additionally, CDA-AMC assumed that gemcitabine would be administered for a maximum of 8 cycles, aligning the stopping rule for gemcitabine when it is used in combination with either pembrolizumab or durvalumab. Also, CDA-AMC assumed pembrolizumab was administered with weight-based dosing, with the RDIs set to 100% for all treatments and with perfect vial sharing assumed.
CDA-AMC conducted scenario analyses assuming pembrolizumab was administered with fixed dosing (i.e., 200 mg every 3 weeks) and no vial sharing.
Lack of transparency and flexibility in the model: Several limitations were observed in the submitted model. CDA-AMC noted that the sponsor’s submission was unnecessarily complex for a CMA. This was commented on by the sponsor in the user guide accompanying its economic evaluation. Furthermore, table labels in its submitted economic evaluation were frequently incorrect, making the model validation process cumbersome. For example, the sponsor labelled 1 column “acquisition cost per dose,” but the column contained acquisition costs per treatment cycle, not the cost per dose. Additionally, probabilistic results were inconsistent with deterministic results when alternative OS parametric functions were fit to the KM data. The sponsor’s submitted model included numerous IFERROR statements, which lead to situations in which the parameter value is overwritten with an alternative value without alerting the user to the automatic overwriting. The systematic use of IFERROR statements makes thorough auditing of the sponsor’s model impractical, as it remains unclear whether the model is running inappropriately by overriding errors and whether this contributed to the discrepancy between the deterministic and probabilistic results.
CDA-AMC was unable to address this deficiency and found that the results from the submitted economic evaluation could not be fully validated. Due to large discrepancies between the probabilistic and deterministic results when selecting alternative OS parametric distribution, CDA-AMC’s base-case results were presented deterministically.
CDA-AMC Reanalyses of the Economic Information
The CDA-AMC base case was derived by making changes in model parameter values and assumptions, in consultation with the clinical experts. CDA-AMC undertook reanalyses that addressed key limitations within the submitted economic model, as summarized in Table 4. CDA-AMC was unable to address the omission of relevant comparators.
Table 4: CDA-AMC Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
Price of cisplatin | $9.50 per 50 mg vial | $135.00 per 50 mg vial |
Changes to derive the CDA-AMC base case | ||
1. Pembrolizumab dosage | 200 mg q.3.w. (i.e., fixed dosing) | 2 mg/kg q.3.w. up to a maximum of 200 mg q.3.w. (weight-based dosing) |
2. Stopping rule for gemcitabine | Gemcitabine is administered until disease progression or unacceptable toxicity when used in combination with pembrolizumab or up to 8 cycles when used in combination with durvalumab | Gemcitabine is administered up to a maximum of 8 cycles when used in combination with pembrolizumab or durvalumab |
3. Choice of parametric distribution for OS | Log-logistic | Gompertz |
4. Choice of parametric distribution for PFS | Log-normal | Gamma |
5. Vial wastage | 5% | 0% (i.e., perfect vial sharing) |
6. Relative dose intensity | ≤ 100% for all treatments | 100% for all treatments |
CDA-AMC base case | ― | Reanalysis 1 + 2 + 3 + 4 + 5 + 6 |
OS = overall survival; PFS = progression-free survival; q.3.w. = every 3 weeks.
CDA-AMC undertook a stepped analysis, incorporating the changes proposed in Table 4 into the sponsor’s base case to highlight the impact of each change (Table 5). All CDA-AMC reanalyses are presented deterministically.
Results from the CDA-AMC base case suggest that, over a lifetime horizon, pembrolizumab plus chemotherapy was associated with cost savings of $58,930 per patient relative to durvalumab plus chemotherapy. Pembrolizumab plus chemotherapy was associated with a total cost of $91,841 per patient, while durvalumab plus chemotherapy was associated with a total cost of $150,771 per patient. Only 5% of the cost savings were accrued during the extrapolated period of the model (i.e., after the first 29 months, as observed in the KEYNOTE-966 trial).
Table 5: Summary of the Stepped Analysis of the CDA-AMC Reanalysis Results, Deterministic
Stepped analysis | Drug | Total drug costs ($) | Incremental drug costs ($) | Total costs ($) | Incremental costs ($) |
|---|---|---|---|---|---|
Sponsor’s base case | Durvalumab + chemotherapy | 161,983 | Reference | 167,495 | Reference |
Pembrolizumab + chemotherapy | 108,736 | −53,246 | 115,312 | −52,183 | |
Sponsor’s corrected base case | Durvalumab + chemotherapy | 163,323 | Reference | 168,836 | Reference |
Pembrolizumab + chemotherapy | 109,995 | −53,328 | 116,571 | −52,265 | |
CDA-AMC reanalysis 1: Weight-based dosing for pembrolizumab | Durvalumab + chemotherapy | 163,323 | Reference | 168,836 | Reference |
Pembrolizumab + chemotherapy | 80,470 | −82,853 | 87,046 | −81,790 | |
CDA-AMC reanalysis 2: Gemcitabine continues for up to 8 cycles | Durvalumab + chemotherapy | 163,323 | Reference | 168,836 | Reference |
Pembrolizumab + chemotherapy | 109,596 | −53,727 | 116,071 | −52,764 | |
CDA-AMC reanalysis 3: Gompertz parametric distribution for OS | Durvalumab + chemotherapy | 155,823 | Reference | 161,228 | Reference |
Pembrolizumab + chemotherapy | 109,995 | −45,828 | 116,571 | −44,657 | |
CDA-AMC reanalysis 4: Gamma parametric distribution for PFS | Durvalumab + chemotherapy | 145,001 | Reference | 150,328 | Reference |
Pembrolizumab + chemotherapy | 109,995 | −35,006 | 116,571 | −33,757 | |
CDA-AMC reanalysis 5: No vial wastage | Durvalumab + chemotherapy | 163,283 | Reference | 168,796 | Reference |
Pembrolizumab + chemotherapy | 109,929 | −53,354 | 116,505 | −52,291 | |
CDA-AMC reanalysis 6: 100% RDI | Durvalumab + chemotherapy | 163,800 | Reference | 169,312 | Reference |
Pembrolizumab + chemotherapy | 119,326 | −44,474 | 125,901 | −43,411 | |
CDA-AMC base case (reanalysis 1 + 2 + 3 + 4 + 5 + 6) | Durvalumab + chemotherapy | 145,444 | Reference | 150,771 | Reference |
Pembrolizumab + chemotherapy | 85,365 | −60,079 | 91,841 | −58,930 |
OS = overall survival; PFS = progression-free survival; RDI = relative dose intensity.
Note: The CDA-AMC reanalysis is based on publicly available prices of the comparator treatments. Chemotherapy is assumed to be comprised of cisplatin and gemcitabine. The negative incremental costs represent cost savings.
Scenario Analysis Results
CDA-AMC conducted 2 scenario analyses: assuming fixed dosing for pembrolizumab and assuming no vial sharing (i.e., 100% of the unused portion of the vial is wasted). Additional details are available in Appendix 1, Table 8. In both of these scenarios, the estimated cost savings are reduced to approximately half of the cost savings estimated in the CDA-AMC base case.
As the price negotiations for durvalumab were still ongoing at the time of the submission,3 CDA-AMC conducted a threshold analysis (Table 6) using the CDA-AMC base case to examine the price reduction for durvalumab at which pembrolizumab would no longer be considered cost saving (i.e., cost-neutral). Assuming pembrolizumab is administered with weight-based dosing, a price reduction of at least 42.8% for durvalumab would have to be reached for pembrolizumab to be cost-neutral. Assuming pembrolizumab is administered with fixed dosing, a price reduction of at least 18.5% for durvalumab would have to be reached for pembrolizumab to be cost-neutral. CDA-AMC’s recommendation for durvalumab was for a price reduction of at least 93%.22 If the pCPA concluded durvalumab negotiations with a price reduction larger than 42.8%, pembrolizumab is no longer a cost saving for the indicated population at the submitted prices.
Table 6: CDA-AMC Price Reduction Analyses
Price reduction required for durvalumab to result in no cost savings compared with pembrolizumab | Durvalumab | Durvalumab price reduction neededa (%) | Reduced price of durvalumab ($) | Cost saving of pembrolizumab relative to a reduced price for durvalumabb ($) |
|---|---|---|---|---|
Scenario: Pembrolizumab weight-based dosing | 120 mg vial = 938.67 500 mg vial = 3,911.11 | 42.8 | 120 mg vial = 536.92 500 mg vial = 2,237.15 | 0.00 |
Scenario: Pembrolizumab fixed dosing | 18.5 | 120 mg vial = 765.02 500 mg vial = 3,187.55 |
aRelative to publicly available list prices of durvalumab.
bSavings based on the sponsor list price of pembrolizumab and the reduced price of durvalumab per patient per lifetime (20 years).
Issues for Consideration
pCPA negotiations for durvalumab were ongoing at the time of this review,18 and analyses are based on publicly available list prices. The sponsor’s submitted CMA was focused on a comparison of pembrolizumab versus durvalumab, with both drugs used in combination with gemcitabine and cisplatin, and assumes durvalumab is publicly available. Although the durvalumab negotiation process with the pCPA concluded with a letter of intent on February 28, 2024,23 during the review period, durvalumab had not yet been listed by participating drug plans, and the price of durvalumab had not been determined. The relevance of the sponsor’s submitted CMA depends on the availability and uptake of durvalumab as well as the negotiated price of durvalumab. This introduces notable uncertainty in the cost savings estimated for pembrolizumab plus chemotherapy. The cost-effectiveness of pembrolizumab compared with chemotherapy alone is unknown, as the sponsor did not submit a cost-utility analysis that included this comparator.
Pembrolizumab and durvalumab have different maintenance dosing schedules. The maintenance dose for pembrolizumab is once every 3 or 6 weeks, whereas durvalumab maintenance dosing is once every 4 weeks (until disease progression or unacceptable toxicity in the case of either drug). The clinical expert input noted that patients may find the maintenance dosing schedule for pembrolizumab more convenient than that for durvalumab, as it aligns with the chemotherapy portion of the regimen (for those patients who continue to receive chemotherapy beyond 8 cycles, at the clinician’s discretion).
The clinical evidence for the treatment efficacy of pembrolizumab plus chemotherapy is available only for first-line settings; the effectiveness and cost-effectiveness of pembrolizumab in subsequent lines of therapy are unknown. Thus, the budget impact of pembrolizumab being added to backbone chemotherapy in subsequent lines of therapy might have been underestimated.
Conclusions
Assuming that the clinical efficacy and safety for pembrolizumab plus chemotherapy versus durvalumab plus chemotherapy is similar, the sponsor submitted a CMA comparing the administration costs for drug acquisition and treatment. Based on CDA-AMC’s clinical review of the sponsor-submitted NMA, the assumption of comparable clinical efficacy and safety between pembrolizumab and durvalumab is associated with substantial uncertainty in the treatment-effect estimates (indicated by wide credible intervals) from the NMA due to limited efficacy and safety data, and no comparisons of health-related quality of life outcomes that are important to patients and clinicians were conducted.
The CDA-AMC base case suggests that pembrolizumab plus chemotherapy is associated with cost savings of $58,930 over a lifetime horizon compared with durvalumab plus chemotherapy, driven by the assumption that pembrolizumab would be administered with weight-based dosing. The estimated incremental savings are based on publicly available list prices for durvalumab and may not reflect the actual prices paid by Canadian public drug plans. If the pCPA has negotiated large price reductions for durvalumab, pembrolizumab may no longer result in cost savings. Scenario analyses suggest that if pembrolizumab is administered with fixed dosing or if no vial sharing is allowed, the estimated cost savings would be reduced to approximately half of the cost savings estimated by the CDA-AMC base case.
The CMA is based on the assumption of similar clinical efficacy and safety between pembrolizumab and durvalumab. In addition, the cost-effectiveness of pembrolizumab compared with chemotherapy alone in this population is unknown.
References
1.Pharmacoeconomic evaluation [internal sponsor’s report]. In: Drug Reimbursement Review sponsor submission: Keytruda (pembrolizumab), solution for infusion 100 mg/4 mL vial. Kirkland (QC): Merck Canada Inc; 2023 Dec 7.
2.Merck Canada Inc. KEYTRUDA (pembrolizumab) solution for infusion, 100 mg/4 mL vial. Product Monograph. Date of Initial Approval: May 19, 2015. Date of Revision: TBC (draft for proposed new indication) [sponsor provided reference].
3.pan-Canadian Pharmaceutical Alliance. Imfinzi (durvalumab). 2024; https://www.pcpacanada.ca/negotiation/22289. Accessed 2024 Jan 2.
4.Kelley RK, Ueno M, Yoo C, et al. Pembrolizumab in combination with gemcitabine and cisplatin compared with gemcitabine and cisplatin alone for patients with advanced biliary tract cancer (KEYNOTE-966): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2023;401(10391):1853-1865. PubMed
5.Oh DY, Ruth He A, Qin S, et al. Durvalumab plus gemcitabine and cisplatin in advanced biliary tract cancer. NEJM Evid. 2022;1(8):EVIDoa2200015.
6.Merck Canada response to February 2, 2024 CADTH request for additional information regarding Keytruda (pembrolizumab) CADTH review: Clinical evidence [internal sponsor’s report]. Kirkland (QC): Merck Canada Inc.; 2023 Feb 7.
7.Cancer Care Ontario. Product monograph: Durvalumab + gemcitabine + cisplatin [accessed by sponsor]. https://www.cancercareontario.ca/en/drugformulary/regimens/monograph/72866.
8.DURV(MNT): Regimen. Toronto (ON): Cancer Care Ontario; 2024: https://www.cancercareontario.ca/en/system/files_force/DURVMNT_LU_SC_P.pdf. Accessed 2024 Jan 2.
9.Systematic literature review and network meta-analysis of first-line treatments for patients with advanced and/or unresectable biliary tract cancer: Technical report [internal sponsor’s report]. In: Drug Reimbursement Review sponsor submission: Keytruda (pembrolizumab), solution for infusion 100 mg/4 mL vial. Kirkland (QC): Merck Canada Inc; 2023 Dec 7.
10.Cancer Care Ontario. Product monograph: Gemcitabine + cisplatin [accessed by sponsor]. https://www.cancercareontario.ca/en/drugformulary/regimens/monograph/46801.
11.Cancer Care Ontario. Product monograph: Pembrolizumab + gemcitabine + cisplatin [accessed by sponsor]. https://www.cancercareontario.ca/en/drugformulary/regimens/monograph/66861.
12.Cancer Care Ontario. Product monograph: Pembrolizumab monotherapy [accessed by sponsor]. https://www.cancercareontario.ca/en/drugformulary/regimens/monograph/66621.
13.Hannouf MB, Zaric GS, Blanchette P, et al. Cost-effectiveness analysis of multigene expression profiling assays to guide adjuvant therapy decisions in women with invasive early-stage breast cancer. Pharmacogenomics J. 2020;20(1):27-46. PubMed
14.Drug Intelligence. ONCO-CAPPS Patient Treatment Dynamics Advanced Biliary Tract Carcinoma, 2023 Q3 Update and Trends. Data on file. October 2023 [sponsor provided reference].
15.Alberta Health Services Guideline Resource Unit. Cholangiocarcinoma and gallbladder cancer. Clinical Practice Guideline GI-010 Version 6. Effective date: June 2023. 2023; https://www.albertahealthservices.ca/assets/info/hp/cancer/if-hp-cancer-guide-gi010-biliary.pdf. Accessed 2023 Sep 13.
16.Ramjeesingh R, Chaudhury P, Tam VC, et al. A practical guide for the systemic treatment of biliary tract cancer in canada. Curr Oncol. 2023;30(8):7132-7150. PubMed
17.National Comprehensive Cancer Network (NCCN) Clinical Practice Guidelines in Oncology: Biliary Tract Cancers. Version 3.2023. 2023; https://www.nccn.org/. Accessed 2023 Nov 8.
18.pan-Canadian Pharmaceutical Alliance. Imfinzi (durvalumab) in combination with gemcitabine-based chemotherapy for the treatment of patients with locally advanced or metastatic biliary tract cancer (BTC). 2023; https://www.pcpacanada.ca/negotiation/22289. Accessed 2023 Dec 22.
19.Guidelines for the economic evaluation of health technologies: Canada. 4th ed. Ottawa (ON): CADTH; 2017: https://www.cadth.ca/guidelines-economic-evaluation-health-technologies-canada-4th-edition. Accessed 2024 Feb 21.
20.CADTH Reimbursement Review: Pembrolizumab (KEYTRUDA) for persistent, recurrent, or metastatic cervical cancer [accessed by sponsor]. Can J Health Technol. 2023;3(3). https://www.cadth.ca/sites/default/files/DRR/2023/PC0292-Keytruda-Cervical.pdf.
21.DeltaPA. [Ottawa (ON)]: IQVIA; 2023: https://www.iqvia.com/. Accessed 2024 Jan 2.
22.CADTH Reimbursement Recommendation: Durvalumab (Imfinzi) in combination with gemcitabine-based chemotherapy, for the treatment of patients with locally advanced or metastatic biliary cancer [accessed by sponsor]. Can J Health Technol. 2023;3(2). https://www.cadth.ca/sites/default/files/DRR/2023/PC0296REC-Imfinzi_JH_GP1_KAS.pdf.
23.pan-Canadian Pharmaceutical Alliance. Imfinzi (durvalumab) in combination with gemcitabine-based chemotherapy for the treatment of patients with locally advanced or metastatic biliary tract cancer (BTC). 2023; https://www.pcpacanada.ca/negotiation/22289. Accessed 2024 Mar 12.
24.Merck Canada, Inc. Product Monograph for KEYTRUDA - Draft. May 05, 2023 [sponsor provided reference].
25.Budget Impact Analysis [internal sponsor’s report]. In: Drug Reimbursement Review sponsor submission: Keytruda (pembrolizumab), solution for infusion 100 mg/4 mL vial. Kirkland (QC): Merck Canada Inc; 2023 Dec 7.
26.Statistics Canada. Table 17-10-0005-01 Population estimates on July 1st, by age and sex (2022 data) [accessed by sponsor]. https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1710000501.
27.Xiao Y, Cattelan L, Lagacé F, et al. Epidemiologic trends and geographic distribution of patients with gallbladder and extrahepatic biliary tract cancers in Canada. HPB (Oxford). 2021;23(10):1541-1549. PubMed
28.Marcano-Bonilla L, Mohamed EA, Mounajjed T, Roberts LR. Biliary tract cancers: Epidemiology, molecular pathogenesis and genetic risk associations. Chin Clin Oncol. 2016;5(5):61. PubMed
29.CADTH Reimbursement Review: Durvalumab (Imfinzi) for biliary tract cancer [accessed by sponsor]. Can J Health Technol. 2023;3(4). https://www.cadth.ca/sites/default/files/DRR/2023/PC0296-Imfinzi_combined.pdf.
30.Beaulieu C, Lui A, Yusuf D, et al. A population-based retrospective study of biliary tract cancers in Alberta, Canada. Curr Oncol. 2021;28(1):417-427. PubMed
31.Boutin M, Yang J, Safro M, Davies JM, Gill S. Trends in incidence, treatments, and clinical outcomes of cholangiocarcinoma diagnosed in British Columbia from 2005 through 2019 [accessed by sponsor]. ASCO Gastrointestinal Cancers; 2023.
32.Seung SJ, Saherawala H, Syed I, Clouthier DL, Shephard C, Chen E. Real-world treatment and outcomes for biliary tract cancer patients using administrative databases in Ontario, Canada. Paper presented at: European Society for Medical Oncology (ESMO) Gastrointestinal (GI) Congress; June 29-July 2, 2022 [accessed by sponsor].
33.Ghidini M, Pizzo C, Botticelli A, et al. Biliary tract cancer: Current challenges and future prospects. Cancer Manag Res. 2019;11:379-388. PubMed
Appendix 1: Additional Economic Information
Table 7: CDA-AMC Cost Comparison Table for Locally Advanced Unresectable or Metastatic Biliary Tract Carcinoma
Treatment | Strength or concentration | Form (vial size if single-use) | Price ($) | Recommended dosage | Daily cost ($) | 21-day cost ($) |
|---|---|---|---|---|---|---|
CISPGEMC(W) + PEMB | ||||||
Pembrolizumab (Keytruda) | 25 mg/mL | 4 mL (100 mg) Vial for IV infusion | 4,400.0000a | 200 mg q.3.w. or 400 mg q.6.w. up to 24 months or disease progression or unacceptable toxicityb | 419.05 | 8,800 |
Cisplatin | 1 mg/mL | 50 mg 100 mg Vial for IV infusion | 135.0000 270.0000 | 25 mg/m2 on day 1 and 8 q.3.w. for up to 8 cyclesc | 12.86 | 270 |
Gemcitabine | 40 mg/mL | 1,000 mg 2,000 mg Vial for IV infusion | 270.0000 540.0000 | 1,000 mg/m2 on days 1 and 8 q.3.w. until disease progression or unacceptable toxicityc | 51.43 | 1,080 |
CISPGEMC + PEMB | Initial 8 cycles: 483.33 Maintenance: 470.48 | Initial 8 cycles: 10,150 Maintenance: 9,880 | ||||
CISPGEMC(W) + DURV | ||||||
Durvalumab (Imfinzi) | 50 mg/mL | 2.4 mL (120 mg) 10 mL (500 mg) Vial for IV infusion | 938.6700 3,911.1100 | 1,500 mg q.3.w. for 8 cycles; 1,500 mg q.4.w. as monotherapy thereafter until disease progression or unacceptable toxicity | Initial 8-cycles: 558.73 Maintenance: 419.05 | Initial 8-cycles: 11,733 Maintenance: 8,800 |
Cisplatin | 1 mg/mL | 50 mg 100 mg Vial for IV infusion | 135.0000 270.0000 | 25 mg/m2 on days 1 and 8 q.3.w. for up to 8 cycles | 12.86 | 270 |
Gemcitabine | 40 mg/mL | 1,000 mg 2,000 mg Vial for IV infusion | 270.0000 540.0000 | 1,000 mg/m2 on days 1 and 8 q.3.w. for up to 8 cycles | 51.43 | 1,080 |
CISPGEMC(W) + DURV | Initial 8 cycles: 623.02 Maintenance: 419.05 | Initial 8 cycles: 13,083 Maintenance: 8,800 | ||||
CISPGEMC | ||||||
Cisplatin | 1 mg/mL | 50 mg 100 mg Vial for IV infusion | 135.0000 270.0000 | 75 mg/m2 on day 1 q.3.w. or q.4.w. until disease progression or unacceptable toxicity | 14.46d | 304d |
Gemcitabine | 40 mg/mL | 1,000 mg 2,000 mg Vial for IV infusion | 270.0000 540.0000 | 1,000 mg to 1,250 mg/m2 on days 1 and 8 q.3.w. or 1,000 mg/m2 on days 1, 8, and 15 q.4.w. until disease progression or unacceptable toxicity | 57.86e | 1,215e |
CISPGEMC | 72.32 | 1,519 | ||||
CRBPGEMC | ||||||
Carboplatin | 10 mg/mL | 50 mg 150 mg 450 mg 600 mg Vial for IV infusion | 70.0000 210.0000 599.9985 775.0020 | AUC 5 to 6 on day 1 q.3.w. | 46.90 to 56.90 | 985 to 1,195 |
Gemcitabine | 40 mg/mL | 1,000 mg 2,000 mg Vial for IV infusion | 270.0000 540.0000 | 1,000 mg/m2 on days 1 and 8 q.3.w. for up to 8 cycles | 51.43 | 1,080 |
CRBPGEMC | 98.33 to 108.33 | 2,065 to 2,275 | ||||
AUC = area under the free carboplatin plasma concentration vs. time curve; CRBPGEMC = carboplatin plus gemcitabine; CISPGEMC(W) = cisplatin 25mg/m2 plus gemcitabine; CISPGEMC = cisplatin 75mg/m2 plus gemcitabine; DURV = durvalumab; PEMB = pembrolizumab; q.3.w. = every 3 weeks; q.4.w. = every 4 weeks; q.6.w. = every 6 weeks.
The comparators presented in the above table have been deemed to be appropriate based on feedback from clinical expert(s) and drug plans. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Note: Prices are wholesale prices from the IQVIA Delta PA database21 (accessed January 2024), unless otherwise indicated, and do not include dispensing fees. Cost calculations assume a body surface area of 1.80 m2 where applicable. Wastage of excess medication in vials is included in costs. Recommended dosage is based on Ontario Health (Cancer Care Ontario) monographs unless otherwise indicated.
aSponsor’s submitted price.
bRecommended dosage is based on the draft product monograph. Pembrolizumab treatment is capped at 24 months (35 doses of 200 mg or 18 doses of 400 mg).24 Drug plans noted that jurisdictions may administer pembrolizumab based with a capped weight-based dose approach (i.e., 2 mg/kg every 3 weeks to a maximum dose of 200 mg or 4 mg/kg every 6 weeks to a maximum of 400 mg), similar to other indications for pembrolizumab. Assuming a body weight of 70 kg and wastage of excess medication in vials, fixed dosing cost estimates remain unchanged.
cRecommended dosage is based on the KEYNOTE-966 trial.4 However, clinical input obtained by CDA-AMC indicated that clinical practice varies, and some clinicians may stop gemcitabine after 8 cycles, similar to cisplatin.
dCost assumes a recommended dosage of 75 mg/m2 q.4.w.
eCost assumes a recommended dosage of 1,000 mg/m2 on days 1, 8, and 15 q.4.w.
Note: This table has not been copy-edited.
Table 8: Scenario Analyses Conducted on CDA-AMC Base Case, Deterministic
Stepped analysis | Drug | Total drug costs ($) | Incremental drug costs ($) | Total costs ($) | Incremental costs ($) |
|---|---|---|---|---|---|
CDA-AMC base case | Durvalumab + chemotherapy | 145,444 | Reference | 150,771 | Reference |
Pembrolizumab + chemotherapy | 85,365 | −60,079 | 91,841 | −58,930 | |
Scenario 1: Fixed dosing for pembrolizumab | Durvalumab + chemotherapy | 145,444 | Reference | 150,771 | Reference |
Pembrolizumab + chemotherapy | 118,784 | −26,661 | 125,259 | −25,512 | |
Scenario 2: No vial sharing | Durvalumab + chemotherapy | 146,313 | Reference | 151,640 | Reference |
Pembrolizumab + chemotherapy | 120,268 | −26,045 | 126,744 | −24,896 |
Additional Details on the Sponsor’s Submission
Figure 1: Sponsor’s Extrapolations of Time on Treatment [Redacted]

Source: Sponsor’s economic submission.1
Additional Details on the CDA-AMC Reanalyses and Additional Analyses
Figure 2: CDA-AMC’s Extrapolations of Time on Treatment [Redacted]

Source: Sponsor’s economic submission.1
Appendix 2: Submitted Budget Impact Analysis and CDA-AMC Appraisal
Please note that this appendix has not been copy-edited.
Table 9: Summary of Key Take Aways
Key take aways of the budget impact analysis |
|---|
|
Summary of Sponsor’s Budget Impact Analysis
In the submitted budget impact analysis (BIA), the sponsor assessed the budget impact of reimbursing pembrolizumab for use by adult patients with locally advanced unresectable or metastatic BTC, in combination with gemcitabine and cisplatin.25 The BIA was undertaken from the perspective of a Canadian public payer over a 3-year time horizon (2025 to 2027) using an epidemiologic approach. The sponsor’s pan-Canadian estimates reflect the aggregated results from provincial budgets (excluding Quebec). The sponsor’s analysis only included drug acquisition costs for pembrolizumab plus chemotherapy and durvalumab plus chemotherapy. Data informing the model were obtained from various sources, including the published literature, a previous CDA-AMC report, and the sponsor’s internal data. Key inputs to the BIA are documented in Table 11.
Table 10: Summary of Key Model Parameters
Parameter | Sponsor’s estimate |
|---|---|
Target population | |
Population in Canada, excluding Quebec | 30,104,32326 |
Incident cases of extrahepatic and gallbladder cancer | 30.92 per million people per year27 |
Annual incident growth in BTC cases | |
Proportion of intrahepatic cancer | 28.9%30 |
Proportion of locally advanced unresectable or metastatic cancer | 90%29 |
Proportion of patients diagnosed earlier and progressed to advanced disease | 5.26%a |
First-line effective treatment rate | |
Proportion of patients eligible for immunotherapy | 78%29 |
Number of patients eligible for the drug under review | 534 / 536 / 539 |
Market uptake (3 years) | |
Uptake (reference scenario) Durvalumab with chemotherapy Clinical trials | 90% / 90% / 90% 10% / 10% / 10% |
Uptake (new-drug scenario) Pembrolizumab with chemotherapy Durvalumab with chemotherapy Clinical trials | 12% / 32% / 35% 78% / 58% / 55% 10% / 10% / 10% |
Cost of treatment (per patient, per year) | |
Pembrolizumab with chemotherapy Durvalumab with chemotherapy Clinical trials | Year 1 = $161,659; year 2 = $156,340; $14,484 annually thereafterb Year 2 = $185,176; year 2+ = $152,533 $0 |
BTC = biliary tract carcinoma.
Note: chemotherapy is assumed to be comprised of gemcitabine and cisplatin.
aAssumes that one-half of the 10% of patients who do not present with locally advanced unresectable or metastatic cancer at diagnosis and who subsequently undergo surgery because their condition is deemed operable will experience a relapse after the procedure.33
bCost includes the annual cost of gemcitabine as patients are assumed to remain on gemcitabine until disease progression or unacceptable toxicity.
The sponsor’s BIA also included the following key assumptions:
Chemotherapy alone is not a relevant comparator.
Vial sharing is allowed for IV treatments and 5% vial wastage was assumed.
Pembrolizumab plus chemotherapy is assumed to capture its maximum market share of 35% by week 78 (uptake is modelled linearly) for treatment in the first-line setting only.
ToT and PFS estimates from clinical trials were assumed to represent treatment duration patterns in Canada.
RDIs observed in the clinical trials were assumed to represent dose intensity in practice.
The reimbursement of pembrolizumab would not affect the market share attributed to clinical trials.
No patients received pembrolizumab every 6 weeks.
The total number of patients treated annually is the sum of weekly incident patients attributed to that year.
Summary of the Sponsor’s BIA Results
The sponsor estimated that the 3-year budget impact of reimbursing pembrolizumab plus chemotherapy for the treatment of adult patients with locally advanced unresectable or metastatic BTC is expected to be a savings of $4,306,929 (year 1 savings: $1,004,211; year 2 savings: $3,578,426; year 3 savings: $4,306,929).
CDA-AMC Appraisal of the Sponsor’s BIA
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Omission of relevant comparators: The sponsor considered durvalumab with chemotherapy to be the only relevant comparator, irrespective of durvalumab currently only being available through compassionate access as pCPA negotiations were ongoing.18 However, as estimated by the market research provided by the sponsor,14 platinum-based regimens with gemcitabine were used to treat approximately ██% of prevalent cases of patients with BTC as of the third quarter of 2023 based on moving annual total data and approximately ██% based on patients who newly started treatment in the previous 6 months. Chemotherapy alone is still listed as an alternative therapy by numerous guidelines.15-17 Given the added cost of pembrolizumab versus chemotherapy alone, the exclusion of chemotherapy alone favours pembrolizumab and can overestimate the cost savings.
CDA-AMC was unable to address this limitation due to the submitted model structure. Alternatively, CDA-AMC conducted an exploratory analysis to determine a proxy estimate of reimbursing pembrolizumab should durvalumab not be funded by public drug plans (i.e., all eligible patients receive pembrolizumab plus chemotherapy immediately; no patients receive durvalumab).
Treatment costs are uncertain: The sponsors assumed ToT differs between durvalumab and the chemotherapy components of the regimen. For durvalumab the sponsor assumed the same as pembrolizumab ToT, and for the chemotherapy components of the regimen the sponsor assumed ToT was equal to the PFS of pembrolizumab plus chemotherapy. These are different assumptions than those used to estimate the regimen costs in the submitted CMA (i.e., all regimen components of durvalumab plus chemotherapy were equal to PFS). These results in a higher proportion of patients receiving chemotherapy, when used in combination with durvalumab, relative to when used in combination with pembrolizumab. As clinical expert input noted that PFS should be closely aligned with ToT, both immunotherapies should have a similar ToT as the sponsor has assumed similar efficacy and safety for durvalumab plus chemotherapy.1,9
Additionally, several of the same limitations related to treatment costs addressed in the CMA were observed in the submitted BIA such as limitations about the dose assumption of pembrolizumab (fixed dosing), stopping rules for gemcitabine, vial wastage, and RDI.
In reanalysis, CDA-AMC updated ToT to reflect the same treatment duration in the pharmacoeconomic model. CDA-AMC obtained a modelled ToT from the CDA-AMC base case from the CMA. Additionally, in the CDA-AMC base case, CDA-AMC assumed that gemcitabine would be administered for a maximum of 8 cycles irrespective of the immunotherapy combination, assumed pembrolizumab was administered with weight-based dosing, set RDI for all treatments to 100%, and assumed perfect vial sharing.
CDA-AMC conducted a scenario analysis assuming pembrolizumab was administered with fixed dosing (i.e., 200 mg every 3 weeks).
Allocating market share to clinical trials is not appropriate: The sponsor assumed that 10% of patients were enrolled in clinical trials (in both the reference scenario and the new-drug scenario). As a result, patients are assumed to receive medications through the clinical trial and do not incur any treatment or drug costs. This artificially decreases the estimated market size and omits potential treatment-related costs that may be incurred by drug plans from patients participating in clinical trials. This assumption underestimates the budget impact, as patients currently enrolled in clinical trials could become eligible for pembrolizumab plus chemotherapy, if reimbursed.
In the CDA-AMC reanalysis, no market share was allocated to clinical trials in the new and reference drug scenario.
The market uptake of pembrolizumab plus chemotherapy is uncertain: The sponsor’s submitted base case assumed that 12%, 32%, and 35% of eligible patients would receive pembrolizumab plus chemotherapy in year 1, year 2, and year 3, respectively. Uptake was modelled using a linear curve in which the maximum market share (35%) was assumed to be reached in week 78. Clinician input received by CDA-AMC for this review noted that patients may find the maintenance dosing schedule for pembrolizumab more convenient than durvalumab as it aligns with the chemotherapy portion of the regimen for patients who continue to receive gemcitabine beyond 8 cycles, at the clinician discretion. Thus, uptake may be quicker and higher than the sponsor assumed.
The impact of assuming a faster uptake and increased market share was largely captured by the exploratory analysis conducted to address the omission of relevant comparators as it estimates the maximum cost to drug plans by assuming pembrolizumab plus chemotherapy captures 100% of the market.
Budget impact of patients diagnosed in years 1 to 3 not fully captured: the total number of patients eligible for treatment per year were assumed to be diagnosed and start treatment weekly (i.e., spread throughout the year). Although this approach potentially provides a more timely and accurate estimate of costs that are incurred in 3 years, the analysis omits a substantial impact on the budget that will be incurred in year 4. Those are the remaining costs related to the third year of treatment of those patients diagnosed toward the end of the budget year (i.e., full treatment costs will only be captured for those diagnosed in the first week as costs are incurred over a year). Likewise, this approach makes the BIA sufficiently more complex and difficult to validate.
CDA-AMC conducted a scenario analysis to estimate the budget impact of all incident patients starting treatment at the beginning of the calendar year in which they were diagnosed.
Poor modelling practices were employed: The sponsor’s model was poorly organized, with parameters repeated across multiple sheets. In many instances, the user-facing cells in the input sheets did not affect calculations in the model. For example, altering the user-facing value for the maximum number of cycles for gemcitabine when used in combination with pembrolizumab, did not have any effect as the “ToT KMs” sheet had been hard-coded to not permit a stopping rule. Additionally, CDA-AMC notes that incorrect formulas were used in calculating the total administration costs associated with durvalumab plus chemotherapy. This error is not expected to have any impact on the estimated budgetary impact of reimbursing pembrolizumab as administration costs have been excluded from base case analyses.
CDA-AMC Reanalyses of the BIA
CDA-AMC revised the sponsor’s base case by assuming that pembrolizumab is administered with weight-based dosing and that gemcitabine is administered up to a maximum of 8 cycles when used in combination with either pembrolizumab or durvalumab, and by aligning ToT with the CMA CDA-AMC base case, assuming no vial wastage, setting RDI to 100% for all treatments, and setting the clinical trial market share to 0%.
Table 11: CDA-AMC Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Changes to derive the CDA-AMC base case | ||
1. Pembrolizumab dosage | 200 mg q.3.w. (i.e., fixed dosing) | 2 mg/kg q.3.w. up to a maximum of 200 mg q.3.w. (weight-based dosing) |
2. Stopping rule for gemcitabine | Gemcitabine is administered until disease progression or unacceptable toxicity when used in combination with pembrolizumab or up to 8 cycles when used in combination with durvalumab | Gemcitabine is administered up to a maximum of 8 cycles when used in combination with pembrolizumab or durvalumab |
3. ToT |
| All components of the durvalumab plus chemotherapy regimen are equal to durvalumab ToT, sourced from the CMA CDA-AMC base case |
4. Vial wastage | 5% | 0% (i.e., perfect vial sharing) |
5. Relative dose intensity | ≤ 100% for all treatments | 100% for all treatments |
6. Clinical trial market share | 10% in years 1, 2, and 3 | No market share attributed to clinical trials |
CDA-AMC base case | ― | Reanalysis 1 + 2 + 3 + 4 + 5 + 6 |
KM = Kaplan-Meier; OS = overall survival; PFS = progression-free survival; q.3.w. = every 3 weeks; ToT = time on treatment.
The results of the CDA-AMC stepwise reanalysis are presented in summary format in Table 12 and a more detailed breakdown is presented in Table 13. In the CDA-AMC base case, the 3-year budget impact of reimbursing pembrolizumab for adult patients with locally advanced unresectable or metastatic BTC is expected to be a savings of $20,309,629 (year 1 savings: $1,797,999; year 2 savings: $7,680,385; year 3 savings: $10,831,246).
Table 12: Summary of the CDA-AMC Reanalyses of the BIA
Stepped analysis | Three-year total ($) |
|---|---|
Submitted base case | −8,889,566 |
CDA-AMC reanalysis 1: Weight-based pembrolizumab dosage | −18,417,178 |
CDA-AMC reanalysis 2: Gemcitabine administered for up to 8 cycles | −10,130,241 |
CDA-AMC reanalysis 3: Time on treatment | −10,873,062 |
CDA-AMC reanalysis 4: No vial wastage | −8,894,774 |
CDA-AMC reanalysis 5: 100% RDI for all treatments | −6,086,374 |
CDA-AMC reanalysis 6: No market share attributed to clinical trials | −8,889,566 |
CDA-AMC base case (reanalysis 1 + 2 + 3 + 4 + 5 + 6) | −20,309,629 |
BIA = budget impact analysis; RDI = relative dose intensity.
CDA-AMC conducted additional scenario analyses and an exploratory analysis to address remaining uncertainty, using the CDA-AMC base case. Results are provided in Table 13.
Assuming fixed dosing for pembrolizumab.
Assuming no vial sharing.
Assuming that the price of durvalumab is reduced by 42.8%, the price reduction at which pembrolizumab would be considered cost-neutral, over a lifetime horizon, compared with durvalumab in the CMA reanalysis (refer to Table 6).
Assuming that the price of durvalumab is reduced by 93%, CDA-AMC’s recommended price reduction for durvalumab.22
All incident patients start treatment at the beginning of each year.
Assuming all eligible patients receive pembrolizumab plus chemotherapy immediately (i.e., no patients receive durvalumab plus chemotherapy).
Results of CDA-AMC’s scenario analyses demonstrate that the budget impact is sensitive to assumptions regarding the price of durvalumab and the number of patients expected to receive pembrolizumab. CDA-AMC notes that if the price of durvalumab is reduced by 93% (CDA-AMC’s recommended price reduction for durvalumab),22 pembrolizumab is no longer considered cost saving and the 3-year budget impact increases to $20,047,388. If the price of durvalumab is reduced by 42.8% (the price reduction at which pembrolizumab would be considered cost-neutral over a lifetime), the small cost savings still estimated in the BIA are likely attributed to the gradual market share uptake of the model. The exploratory analysis estimates that the reimbursement of pembrolizumab could cost drug plans approximately $95,024,704 (Table 14) should durvalumab not be funded by public drug plans and replace chemotherapy alone instead. CDA-AMC scenario analyses suggest that the true budget impact of reimbursing pembrolizumab for adult patients with locally advanced unresectable or metastatic BTC is likely to lie in between cost savings of $23,715,235 (if all incident patients are assumed to start treatment in week 1 of each year) or an added cost of $95,024,704 (should durvalumab not be funded by public drug plans and pembrolizumab uptake the chemotherapy alone market instead).
Table 13: Detailed Breakdown of the CDA-AMC Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|---|
Submitted base case | Reference | 36,257,589 | 59,754,945 | 65,195,820 | 66,705,372 | 191,656,137 |
New drug | 36,257,589 | 58,750,734 | 61,617,394 | 62,398,443 | 182,766,571 | |
Budget impact | 0 | −1,004,211 | −3,578,426 | −4,306,929 | −8,889,566 | |
CDA-AMC base case | Reference | 42,077,440 | 69,807,839 | 75,939,724 | 77,519,025 | 223,266,588 |
New drug | 42,077,440 | 68,009,841 | 68,259,339 | 66,687,779 | 202,956,959 | |
Budget impact | 0 | −1,797,999 | −7,680,385 | −10,831,246 | −20,309,629 | |
CDA-AMC scenario analysis: fixed dosing for pembrolizumab | Reference | 42,077,440 | 69,807,839 | 75,939,724 | 77,519,025 | 223,266,588 |
New drug | 42,077,440 | 68,851,183 | 72,219,327 | 72,670,390 | 213,740,900 | |
Budget impact | 0 | −956,657 | −3,720,397 | −4,848,635 | −9,525,688 | |
CDA-AMC scenario analysis: no vial sharing | Reference | 42,456,496 | 70,272,744 | 76,406,953 | 77,988,590 | 224,668,287 |
New drug | 42,456,496 | 69,314,460 | 72,679,831 | 73,131,665 | 215,125,956 | |
Budget impact | 0 | −958,284 | −3,727,122 | −4,856,925 | −9,542,331 | |
CDA-AMC scenario analysis: incidence of cases to occur at the start of each year | Reference | 62,269,011 | 74,065,809 | 76,973,432 | 77,712,052 | 228,751,294 |
New drug | 62,269,011 | 70,825,893 | 67,762,778 | 66,447,387 | 205,036,059 | |
Budget impact | 0 | −3,239,916 | −9,210,654 | −11,264,665 | −23,715,235 | |
CDA-AMC scenario analysis: durvalumab price reduced by 42.8% | Reference | 25,528,419 | 41,720,897 | 45,237,289 | 46,149,648 | 133,107,833 |
New drug | 25,528,419 | 41,462,022 | 44,469,936 | 45,439,153 | 131,371,111 | |
Budget impact | 0 | −258,875 | −767,353 | −710,495 | −1,736,723 | |
CDA-AMC scenario analysis: durvalumab price reduced by 93% | Reference | 6,118,118 | 8,777,801 | 9,226,489 | 9,356,593 | 27,360,883 |
New drug | 6,118,118 | 10,324,160 | 16,567,411 | 20,516,699 | 47,408,270 | |
Budget impact | 0 | 1,546,359 | 7,340,922 | 11,160,106 | 20,047,388 |
BIA = budget impact analysis.
Table 14: Detailed Results of Pembrolizumab Plus Chemotherapy From CDA-AMC Exploratory Analysis New-Drug Scenario
Treatment | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|
Pembrolizumab plus chemotherapy | 0 | 23,211,598 | 40,045,499 | 43,042,901 | 106,299,997 |
Pembrolizumab | 0 | 19,942,173 | 36,052,546 | 39,029,984 | 95,024,704 |
Chemotherapy | 0 | 3,269,424 | 3,992,952 | 4,012,917 | 11,275,293 |
Note: In the exploratory analysis, 534, 536, and 539 patients are estimated to receive pembrolizumab plus chemotherapy in years 1, 2, and 3, respectively.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.