Drugs, Health Technologies, Health Systems
Reimbursement Review
Relugolix (Orgovyx)
Sponsor: Sumitomo Pharma Canada, Inc.
Therapeutic area: Advanced prostate cancer
This multi-part report includes:
Clinical_Review
Pharmacoeconomic_Review
Clinical_Review
Abbreviations
ADT
androgen deprivation therapy
AE
adverse event
ALT
alanine transaminase
ARAT
androgen-receptor axis therapy
AST
aspartate transaminase
CCS
Canadian Cancer Society
CI
confidence interval
CrI
credible interval
CRFS
castration resistance–free survival
DIC
deviance information criterion
EBRT
external beam radiation therapy
ECOG
Eastern Cooperative Oncology Group
EMA
European Medicines Agency
EORTC QLQ-C30
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30
EORTC QLQ-PR25
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Prostate-Specific 25 Items
FSH
follicle-stimulating hormone
GnRH
gonadotropin-releasing hormone
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HRQoL
health-related quality of life
ITC
indirect treatment comparison
LHRH
luteinizing hormone–releasing hormone
MACE
major adverse cardiovascular event
mCRPC
metastatic castration-resistant prostate cancer
mCSPC
metastatic castration-sensitive prostate cancer
mITT
modified intention to treat
NMA
network meta-analysis
PC
prostate cancer
PSA
prostate-specific antigen
RCT
randomized controlled trial
REIP
random effects with informed priors
REV
random effects with vague priors
RP
radical prostatectomy
RT
radiation therapy
SAE
serious adverse event
SMQ
standardized Medical Dictionary for Regulatory Activities query
TEAE
treatment-emergent adverse event
ULN
upper limit of normal
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Relugolix, (Orgovyx), 120 mg, oral tablets |
Sponsor | Sumitomo Pharma Canada, Inc. |
Indication | Orgovyx (relugolix) tablets, 120 mg, for the treatment of adult patients with advanced prostate cancer |
Reimbursement request | According to Health Canada–approved indication |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | October 10, 2023 |
Recommended dosage | A loading dose of 360 mg (3 tablets) on the first day and continue treatment with a 120 mg tablet taken orally once daily at approximately the same time each day |
NOC = Notice of Compliance.
Introduction
Prostate cancer (PC) is a malignancy in which prostate cells grow uncontrollably, often driven by testosterone-producing pathways. In its early stages, PC may be asymptomatic or present with nonspecific symptoms such as altered urination patterns, blood in the urine or semen, painful urination and/or ejaculation, pelvic-area pain, and erectile dysfunction. As the tumour grows or metastasizes, typically to bones in 90% of cases, symptoms such as bone pain or mobility issues can severely affect quality of life.
Prostate cancer spans various stages, from nonmetastatic, localized disease to metastatic castration-resistant prostate cancer (mCRPC). Advanced PC is a severe subset of PC with a high risk of progression or death, requiring androgen deprivation therapy (ADT). It includes a broad range of incurable disease states with diverse clinical options and survival times. High-risk locally advanced PC, as defined by the Canadian Urological Association 2022 guidelines and criteria such as those specified in the STAMPEDE trial (involving patients who have at least 2 of: a diagnosis of stage T3 or T4 cancer, a Gleason score of 8 to 10, and a prostate-specific antigen (PSA) level of 40 ng/mL or higher), falls under this category. Survival rates vary significantly, from nearly 100% over 5 years for localized and locally advanced PC to 34% for metastatic PC.
Detection of PC typically involves elevated PSA levels or digital rectal examinations, followed by diagnostic tests such as prostate biopsy, and imaging (transrectal ultrasound or MRI). The Gleason score (and more recently the Gleason grade group) system grades PC histologically, with higher scores indicating worse prognosis and higher metastatic potential. The tumour-node-metastasis system classifies PC into stages I through IV, based on primary tumour involvement, lymph node involvement, and metastasis.
In Canada, PC is the most common cancer among men, with about 24,600 diagnoses in 2022. An estimated 1 in 8 males in Canada will develop PC in their lifetime. The prevalence was 0.66% in 2018, calculated using prevalent cases and the adult male population at the time, and this rate is assumed to remain stable into 2024, balancing incidence and mortality rates.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient and clinician groups who responded to CADTH’s call for input and from 2 clinical experts consulted by CADTH for the purpose of this review.
Patient Input
Three patient groups submitted 3 inputs. The ADT Education Program supports patients living with PC undergoing hormone therapies. The Canadian Cancer Society (CCS) is the only national charity that supports patients living with all types of cancer across the country by conducting research, supplying a compassionate support system and establishing health policies. Both of these groups provided input based on 1 patient. The third patient group, PROCURE, is a charitable organization that educates, supports, and informs people affected by PC and promotes and contributes to financing research. PROCURE collected information from an online survey of 263 patients conducted in May 2022.
In ADT Education Program input, a patient living with advanced PC reported being on ADT almost continuously for more than 20 years and experienced many side effects. The patient, speaking on behalf of other patients, stated that the most disturbing side effects are hot flashes, fatigue, and loss of sexual interest. He added that they also regularly experience loss of muscle mass but gain weight as fat, making simple tasks like walking up stairs difficult. According to the patient, ADT affects memory, can lead to depression and insomnia, and makes patients feel weak, old, flabby, and demoralized. Last, he stated that the depot injection form of ADT drugs may cause inflammation at the injection site, producing discomfort for days after injections. According to the input, some patients may delay getting repeated injections or take risky drug holidays that can cause their cancer to fulminate, just to avoid injections. The PROCURE input noted that some patients opt for orchiectomy to avoid regular injections. Similarly, another patient consulted by the CCS described experiencing side effects such as weight gain, impacts on kidneys and liver, and reduced sexual desire, which was noted as a key side effect by the patient. In both the PROCURE and CCS inputs, patients said taking ADT can cause side effects that may require other medications such as antidepressants or kidney-protection drugs. The patient consulted by CCS reported feeling weak and tired, which reduced the motivation to exercise. The patient’s wife said she did not experience a significant impact on her life, besides the limited sexual desire the patient described as the side effect of treatment. According to PROCURE input, patients and partners often mourn the loss of a satisfying sexual relationship, and advanced cancer creates anxiety within couples. Also, according to the PROCURE input, children and family members may experience anxiety as their father passes away from the cancer, or they may be at risk of getting prostate, breast, or ovarian cancers if their father living with PC is a carrier of the BRCA mutation. The PROCURE input noted that injection hormone therapy and frequent travel to clinics or hospital for medical follow-up exams can be costly, and that it takes too long (from months to years) for testosterone levels to return to normal after the end of long-term ADT.
Based on these inputs, a key outcome important to patients was the safety of medication and minimizing side effects. Other key outcomes cited by patients as important included maintaining long-term survival (with ADT) and a good quality of life. The PROCURE input also stated that patients seek improved outcomes in treatment, such as slowing down the progression of cancer, extension of life expectancy, and decreased PSA levels. All inputs indicated that patients living with PC would appreciate a new, patient-friendly alternative form of ADT that is not a difficult to administer or invasive.
Clinician Input
Input From Clinical Experts Consulted by CADTH
Despite advancements in PC treatment leading to longer overall survival, resistance to therapies is inevitable, and most patients with PC will eventually succumb to the disease. All current ADT options effectively induce profound medical castration by suppressing testosterone.
One gap in current ADT options is the lack of oral administration, as the available injectable forms of therapy may not suit all patients. However, according to the clinical experts consulted for this review, no published evidence is available to inform the Canadian clinical practice regarding a preference for oral options or the position that injectables negatively affect compliance. Given that patients with advanced PC typically see their physicians twice a year, the current treatment regimen does not greatly burden the health care system. However, for patients in remote areas of Canada who find travel logistically or economically challenging, an oral ADT option could address this unmet need. Relugolix is positioned as a foundational ADT. It may be particularly beneficial for patients in remote locations, those who prefer oral medication, or those needing intermittent rather than continuous ADT (for example, where intermittent ADT is attempted to minimize the adverse effects of medical castration by withdrawing treatment in patients who have responded to continuous ADT), due to the rapid recovery in testosterone levels and quality of life associated with the drug.
The Canadian consensus recommends a castration-level threshold of up to 0.7 nmol/L for patients with metastatic castration-sensitive prostate cancer (mCSPC), along with androgen-receptor axis therapy (ARAT) intensification. Response measures include prolonging overall survival, progression-free survival, time to skeletal events, symptomatic deterioration, and castration resistance. For patients with clinical and/or biochemical relapse after curative local therapy, the goals include achieving castration levels of testosterone, extending overall and metastasis-free survival, and delaying castration resistance.
Discontinuation of ADT in the mCSPC setting is rare, except in cases of intolerable toxicities. In the high-risk curative setting, ADT may be stopped more frequently due to toxicities, and decisions are based on a risk-benefit analysis at that time. Most ADT toxicities are manageable. Relugolix is prescribed by specialist oncologists and self-administered orally by the patient.
Clinician Group Input
Two clinician groups, the ADT Education Program and the British Columbia Genitourinary Group with the Vancouver Prostate Centre, contributed insights on PC treatment, specifically focusing on the need for better-tolerated and more-convenient treatment options that enhance compliance. These groups support the development of an oral formulation of a luteinizing hormone–releasing hormone (LHRH) antagonist to overcome the disadvantages of injectable forms, including injection-site reactions, discomfort due to high dosage volume, and the need to travel to clinics.
The clinician groups highlighted the current unmet need in PC treatment: resistance to therapies due to androgen-independent mechanisms. They stated that an oral form of ADT would be particularly beneficial for patients living far from cancer centres. However, they cautioned that long-term ADT could lead to compliance issues or increased pill burden, particularly when combined with other therapies. The goal for an ideal PC treatment is a cure, but for advanced stages that have spread beyond the gland, the objectives shift to suppressing androgen with fewer side effects or less-invasive administration, prolonging survival, and improving quality of life. Other important goals of therapy include prolonging time to skeletal-related events, symptomatic deterioration, and castration resistance.
Patients best suited for relugolix include those with hormone-sensitive disease or newly diagnosed or substantial metastatic disease requiring prompt androgen suppression, patients needing short-term ADT, and those having difficulty accessing injection clinics. Additionally, relugolix would be beneficial for those preferring an oral medication or needing intermittent ADT.
Response to relugolix is measured via serum PSA concentrations or imaging, similar to current ADT drugs. For relugolix monotherapy, a “profound castration” level of testosterone (≤ 0.7 nm/L) is indicative of a response. Generally, ADT is continuous for patients with metastatic, locally advanced, or castration-resistant PC, unless contraindications or intolerable side effects arise. Relugolix can also be administered intermittently based on serum PSA levels or for a fixed duration in patients receiving ADT with curative-intent radiation.
Urologists, medical- or uro-oncologists, and radiation oncologists experienced in managing advanced PC should prescribe and monitor relugolix treatment. The medication can be dispensed in an outpatient setting, and patients take relugolix orally at home.
Drug Program Input
Drug plans submitted questions covering the appropriateness of comparison, study population, and response assessment.
Clinical Evidence
Systematic Review
Description of Studies
The HERO trial was a phase III, randomized, multicentre, open-label, parallel-group study conducted across 160 sites in 22 countries. The trial enrolled patients from April 2017 to October 2019 for the primary analysis and until August 2020 for the final analysis. A total of 934 patients were included in the primary analysis, with this number increasing to 1,078 in the final analysis. Patients were divided into 2 groups: 624 received relugolix and 310 received leuprolide in the primary analysis. Eligible participants were adult males with histologically or cytologically confirmed PC, candidates for at least 1 year of continuous ADT, and those who met specific criteria such as evidence of biochemical or clinical relapse, newly diagnosed androgen-sensitive metastatic disease, or advanced localized disease. Exclusions included a likely need for chemotherapy or surgery soon after ADT initiation, prior extensive ADT or systemic cytotoxic treatment, brain metastases, recent significant cardiac events, conduction-system abnormalities, and uncontrolled hypertension.
The intervention consisted of administering relugolix as a 120 mg tablet daily following a 360 mg oral loading dose on day 1, compared to leuprolide given as 22.5 mg depot injections every 12 weeks, both for a duration of 48 weeks. The study was structured into a 28-day screening phase, a 48-week treatment phase, and a follow-up phase of 30 days for safety and up to 90 days for assessing testosterone recovery. The primary end point was the sustained castration rate from week 5 to week 49. Secondary efficacy end points included sustained castration rate, profound castration rate, PSA response rate, follicle-stimulating hormone (FSH) level, castration resistance–free survival (CRFS) for patients with or without metastatic cancer (final analysis), and testosterone recovery rate. Other end points assessed changes in quality of life, serum concentrations of various hormones, and safety end points such as treatment-emergent adverse events (TEAEs), major adverse cardiovascular events (MACEs), clinical laboratory tests, vital signs, and electrocardiograms. Exploratory end points included overall survival and the presence of polymorphisms in germline genes.
The age distribution was similar between the 2 groups, with approximately 71% of patients in both groups aged 75 years or younger. The mean age was approximately 71 years, with a slightly higher median age in the relugolix group (72 years) compared to the leuprolide group (71 years). Ethnicity and race distributions were broadly comparable across both groups, with the majority being non-Hispanic or non-Latino and white. The study included participants from various geographic regions, with the largest proportion from Europe (close to 40% in both groups), followed by North America, Asia, and other regions.
Clinically, approximately half of the participants in both groups presented with evidence of biochemical or clinical relapse following local primary intervention with curative intent. Newly diagnosed androgen-sensitive metastatic disease and advanced localized disease not suitable for primary surgical intervention were other major disease presentations. The distribution of disease stages at study entry was similar across both groups, with approximately 32% having metastatic, 30% locally advanced, and 29% localized disease. Gleason scores were also similar, with the most common being 7 and 8 to 10. The majority of participants had an Eastern Cooperative Oncology Group (ECOG) Performance Status of 0. Prior ADT and radiation therapy (RT) histories were noted in both groups, with a slightly higher percentage of patients with a history of prior ADT in the relugolix group. Cardiovascular risk factors were prevalent in more than 90% of participants in both groups, with a notable proportion also having lifestyle risk factors and a history of MACEs.
Efficacy Results
The proportion of patients who achieved sustained testosterone suppression was 96.7% (95% confidence interval [CI], 94.9 to 97.9) in the relugolix treatment group compared with 88.8% (95% CI, 84.6 to 91.8) in the leuprolide group, with a mean difference between the relugolix and leuprolide treatment groups of 7.9% (95% CI, 4.1% to 11.8%). These results demonstrate the noninferiority of relugolix to leuprolide (the lower bound of the 95% CI for the difference between groups was greater than the prespecified noninferiority margin of −10%, with P < 0.0001), as well as the statistical superiority of relugolix compared with leuprolide (lower bound of the 95% CI > 0, with P < 0.0001).
Patients in the relugolix group had a shorter time to achieve castration compared to those in the leuprolide group at profound castration levels of testosterone (< 20 ng/dL). The median time to profound castration was 15 days in the relugolix group compared with 29 days in the leuprolide group. At day 15, the difference in the proportion of patients achieving profound castration was more pronounced in the relugolix group compared with the leuprolide group (78.38% versus 0.98%), with a statistically significant difference of 77.41% (95% CI, 73.98% to 80.83%; P < 0.0001).
Treatment with relugolix resulted in a higher proportion of patients achieving and maintaining profound castration (81.6%; 95% CI, 78.1% to 84.5%) compared with the leuprolide group (68.6%; 95% CI, 63.0% to 73.5%) from day 29 through 48 weeks, with a difference between groups of 13.0%.
Harms Results
Overall, the safety profile of relugolix appears to be consistent with that of the ADT therapeutic class. In the HERO trial, adverse events (AEs) were reported by a similar proportion of patients in both the relugolix (92.9%) and leuprolide (93.5%) groups. The most common AE for both groups was hot flushes, occurring in more than half of the patients. Gastrointestinal issues such as constipation and diarrhea were reported more frequently in the relugolix group. All cases of constipation and diarrhea were mild to moderate, with only 1 patient withdrawing from the study due to these AEs. Serious adverse events (SAEs) were slightly numerically less common in the relugolix group (12.2%) compared to the leuprolide group (15.3%). The SAEs in the relugolix group included myocardial infarction (0.8%), acute kidney injury (0.6%), and urinary tract infections (0.5%). Within the leuprolide group, SAEs included anemia (1.0%), cardio-respiratory arrest (1.0%), and urinary tract infection (0.6%). Grade 3 or 4 SAEs were slightly more common, numerically, in the leuprolide group.
Treatment discontinuation due to AEs was higher in the relugolix group (3.5%) compared to the leuprolide group (0.3%). The deaths reported were slightly numerically higher in the leuprolide group (2.9%) than in the relugolix group (1.1%), with cardiovascular-related deaths being more common in the leuprolide group. Vasomotor symptoms such as hot flushes and fatigue were common in both groups (56.1% for relugolix, 54.9% for leuprolide), but hepatic transaminase elevations were numerically higher in the relugolix group (7.6%) contrasted with the leuprolide group (5.5%). The incidence of MACEs was numerically higher in the leuprolide group. Loss of bone mineral density was reported in similar proportions in both groups, and there were no significant liver-related toxicities meeting Hy’s law criteria in either group.
Critical Appraisal
The HERO study, a phase III trial comparing relugolix with leuprolide in men with advanced PC, demonstrated a robust methodology in terms of randomization, stratification, and sample size. Its open-label design, while potentially introducing bias, is mitigated by the objective nature of the primary outcome. The sensitivity analyses for the primary outcome and the approach to handling missing data enhance the study’s robustness.
Externally, the HERO trial’s applicability to typical Canadian practice may be limited due to a number of factors, including its lack of a clear definition of what constitutes locally advanced disease in the inclusion criteria and patient population. The study’s focus on biomarkers such as testosterone and PSA, while relevant for advanced PC, does not fully capture the clinical outcomes of the disease. The study also does not address the combination of ADT and other systemic therapies, nor does it inform on the use of relugolix in patients undergoing RT. In addition, several additional standard-of-care medicines (available and reimbursed in Canada) that would ordinarily be combined with relugolix if approved in the mCSPC setting (abiraterone, enzalutamide, and apalutamide) were not permitted to be given concurrently in the HERO study. This raises concerns given the potential use of relugolix in the mCSPC setting.
GRADE Summary of Findings and Certainty of the Evidence
The selection of outcomes for the Grading of Recommendations Assessment, Development and Evaluation (GRADE) tool was based on the sponsor’s Summary of Clinical Evidence, consultation with clinical experts, and input received from patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members:
sustained castration rate
profound castration rate
MACE
loss of bone mineral density.
Table 2: Summary of Findings for Relugolix Versus Leuprolide for Patients With Advanced Prostate Cancer
Outcome and follow-up | Patients, N (studies) | Relative effect (95% CI) | Absolute effects (95% CI) | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
Leuprolide (N = 308) | Relugolix (N = 622) | Difference | |||||
Sustained castration rate | |||||||
Sustained castration rate (< 50 ng/dL) Follow-up: from day 29 to day 337 | 930 (1 RCT) | HR = 0.2621 (0.1489 to 0.4613) | 88.8 per 100 persons | 96.7 per 100 persons (94.9 to 97.9) | 7.9 more persons per 100 (95% CI, 4.1 to 11.8) | Higha | Relugolix likely results in an increase in the number of patients with sustained castration compared to leuprolide |
Profound castration rate | |||||||
Profound castration rate (< 20 ng/dL) Follow-up: day 15 | 930 (1 RCT) | NR | 0.98 per 100 persons | 78.38 per 100 persons (75.06 to 81.53) | 77.41 more persons per 100 (95% CI, 73.98 to 80.83) | Highb | Relugolix results in an increase in the number of patients with profound castration at day 15 compared to leuprolide |
Cumulative probability of profound castration rate (< 20 ng/dL) Follow-up: day 29 to day 337 | 930 (1 RCT) | NR | 68.6 per 100 persons | 81.6 per 100 persons (78.1 to 84.5) | 13.0 more persons per 100 (95% CI, 6.9 to 19.1) | Highb | Relugolix results in an increase in the number of patients with profound castration compared to leuprolide |
Harms | |||||||
MACE Follow-up: day 337 | 930 (1 RCT) | NR | 6.2 per 100 persons | 2.9 per 100 persons (NR) | NR | Very lowc | The evidence is very uncertain about the effects of relugolix compared to leuprolide on MACE |
Loss of bone mineral density Follow-up: day 337 | 930 (1 RCT) | NR | 3.9 per 100 persons | 3.2 per 100 persons (NR) | NR | Very lowc | The evidence is very uncertain about the effects of relugolix compared to leuprolide on loss of bone mineral density |
CI = confidence Interval; HR = hazard ratio; MACE = major adverse cardiovascular event; MID = minimal important difference; NR = not reported; RCT = randomized controlled trial.
Note: Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to rating down of the level of certainty are documented in the table footnotes.
aNo published between-group MID was identified, and the clinical experts consulted by CADTH were unable to estimate a threshold for clinically important effects, therefore the null was used. Did not rate down for imprecision; a between-group difference of larger than the null and a CI that excludes the null suggest a benefit compared to leuprolide as judged by the CADTH review team.
bNo published between-group MID was identified, and the clinical experts consulted by CADTH were unable to estimate a threshold for clinically important effects, therefore the null was used. Did not rate down for imprecision; a between-group difference of larger than the null and a CI that excludes the null suggest benefit compared to leuprolide as judged by the CADTH review team.
cRated down 2 levels for very serious concerns about imprecision due to very small number of events. Rated down 1 level for serious indirectness due to insufficient duration of follow-up for the outcome according to clinical expert input.
Sources: Clinical Study Report1 and the sponsor’s Summary of Clinical Evidence.77
Long-Term Extension Studies
No long-term extension studies were submitted.
Indirect Comparisons
Description of Studies
The sponsor submitted an indirect treatment comparison (ITC) designed to assess the efficacy and safety of relugolix compared to other medical ADTs available in Canada for adult male patients with advanced PC. The analysis included a network meta-analysis (NMA) of randomized controlled trials (RCTs) identified by a systematic literature search that reported on testosterone suppression to castration levels and MACE outcomes at a 12-month (± 3 months) time point. The quality assessment of these RCTs utilized the Cochrane Risk of Bias tool. The NMA used a Bayesian framework, employing various models to estimate treatment effects for each outcome. Model fit assessment relied on the deviance information criterion (DIC), resulting in the selection of the random effects with informed priors (REIP) model for testosterone castration and random effects with vague priors (REV) model for MACEs, as the primary analysis. An additional hierarchical approach was adopted, accounting for treatment class exchangeability and assuming normal distribution around class-specific means. Statistical heterogeneity was evaluated using the I2 statistic. Sensitivity analyses were conducted using different models and priors.
Efficacy Results
The NMA included 7 studies for testosterone suppression, defined as sustained chemical castration with testosterone levels lower than 50 ng/dL at 12 ± 3 months. ███ ████████ ███ ███ ████ ███████████ ████ ████████ ███ ████ ██ ███ ████████ █████████ ███████ █████████ ███ █████ ██████████ ████ ██████████ ██████████ ████████████ ███ ███████████ ████████ ███████████ ████████ █████████ ████████ ████████ ███ ████ ███████████ ███████████ ██████████
Harms Results
The NMA included 4 studies of MACEs, primarily comparing relugolix to degarelix and leuprolide. ███ ███████ ███ ███ ████ ███████████ ████ ████████ ███ ████ ██ ███ ████████ ████████ ███████ █████████ ███ █████ ███████████
Critical Appraisal
Various limitations of the ITC were noted, including the heterogeneity in study characteristics and patient populations. The exploration of between-study differences and potential biases was further limited by incomplete data in the published trials included in the networks. The clinical experts consulted for this CADTH review noted imbalances in certain prognostic factors and effect modifiers (baseline testosterone concentrations, metastatic status of participants, and previous hormonal treatment), which raises concerns about bias in the comparisons in the NMA. The clinical experts noted that MACE assessments in the Canadian clinical practice occur later than 12 ± 3 months and that profound castration levels (< 20 ng/dL) would have been a more appropriate outcome measure, and that this raises notable generalizability issues. Considering these limitations, there is a high risk of bias in the comparison in this NMA, the direction of that bias is unclear, and the findings of the sponsor-submitted ITC remain highly uncertain.
Studies Addressing Gaps in the Evidence From the Systematic Review
Description of Studies
The clinical evaluation of relugolix in treatment of advanced PC encompassed 3 key studies. The C27300 study, a phase II, open-label trial, focused on comparing relugolix with degarelix in patients with intermediate-risk localized PC, specifically assessing its role in neoadjuvant and adjuvant therapy alongside external beam radiation therapy (EBRT). The MVT-601 to 049 study, a phase I, open-label trial, investigated the combination of relugolix with abiraterone or apalutamide in men diagnosed with either mCSPC or castration-resistant PC. Last, the Apa-RP study, a phase II, open-label trial, evaluated the efficacy of ADT in combination with apalutamide in treatment-naive men after radical prostatectomy (RP), particularly those at high risk of metastases.
Efficacy Results
The C27300 study enrolled 103 patients, with 65 receiving relugolix and 38 degarelix. The study found that relugolix achieved a sustained castration rate of 95% and a profound castration rate of 82% by 24 weeks. In comparison, degarelix produced a sustained castration rate of 89% and a profound castration rate of 68% by 24 weeks. The MVT-601 to 049 study involved 25 patients and demonstrated consistent testosterone suppression in combinations of relugolix plus either abiraterone or apalutamide for 12 weeks. The Apa-RP study, with 108 patients in a 1-year main study and 12 patients in a 28-day substudy, resulted in a sustained castration rate of 100% in both the substudy and the main study.
Harms Results
In the C27300 study, the most common AEs were hot flushes (57%), fatigue (26%), and diarrhea (18%) in the relugolix cohort. Deterioration in quality of life during treatment followed by improving health-related quality of life (HRQoL) posttreatment was noted when assessed by the European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 (EORTC QLQ-C30) and European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Prostate-Specific 25 Items (EORTC QLQ-PR25). Part 1 of the MVT-601 to 049 study reported common AEs, including pain in the extremity (20%), increased alanine transaminase (ALT) levels (13.3%), and anemia (13.3%), with 1 incident (6.7%) of an SAE reported for a left femur fracture in the relugolix-plus-abiraterone cohort (n = 15). The Apa-RP study identified hot flushes (50%) as the most common AE in the relugolix cohort (n = 12), with no significant SAEs or treatment discontinuations due to AEs reported.
Critical Appraisal
The internal validity of these studies is limited due to their open-label nature and the absence of true comparators. This design potentially biases the reporting of AEs, which are typically reported by patients whose responses may be subjective. Furthermore, the objectives of phase I and phase II clinical trials are limited in terms of establishing causal inference. Additionally, the study durations may not be long enough to assess long-term outcomes, particularly MACEs, which the clinical experts consulted by CADTH described as notable AEs in patients with advanced PC. Externally, the studies’ applicability to the Canadian context is questionable, as none of the study sites was in Canada, and patients with cardiovascular diseases, a common comorbidity in the advanced PC patient population, were excluded.
Conclusions
The efficacy and safety of relugolix for patients with advanced PC was assessed in the HERO study, a phase III, open-label, controlled trial that compared relugolix against leuprolide. The HERO trial enrolled 934 patients for the primary analysis. Patients were randomized in a 2:1 ratio to receive either relugolix or leuprolide for 48 weeks. Key end points included sustained castration rate and profound castration rate. The study also evaluated testosterone recovery and quality-of-life indicators. The primary analysis was performed after 48 weeks of enrolment, with the final analysis including additional participants with metastatic disease. The HERO trial achieved its primary end point of showing noninferiority against leuprolide and a sustained castration rate of greater than 90% in patients taking relugolix. Furthermore, relugolix achieved a higher profound castration rate at day 15, and a higher cumulative profound castration rate from day 29 to day 337. Safety results suggest that relugolix has a safety profile similar to that of the ADT therapeutic class.
The GRADE assessment of relevant outcomes indicated that, compared to leuprolide, there is a high certainty that relugolix will likely lead to a sustained castration rate, and a high certainty that relugolix will likely lead to more patients experiencing profound castration. There is a very low certainty of any differences in MACEs or loss of bone density between relugolix and leuprolide.
Given the limited evidence available to inform the comparative efficacy and safety of relugolix versus other ADT treatments, an ITC was submitted by the sponsor. However, several limitations in the ITC suggest the resulting estimates may be biased, and firm conclusions about the comparative efficacy and safety of relugolix cannot be made.
The sponsor identified a number of evidence gaps, including a lack of evidence to inform on intensification therapy and adjuvant or neoadjuvant therapy. The sponsor submitted 3 phase I or phase II studies to address these gaps. However, due to the limitations in these studies, their results do not address these gaps with a high degree of certainty.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of 120 mg of relugolix administered as oral tablets in the treatment of advanced PC in adult patients.
Disease Background
Contents within this section were informed by materials submitted by the sponsor and clinical expert input. The following summary was validated by the CADTH review team.
Prostate cancer develops when healthy prostate cells undergo a malignant change and start growing uncontrollably.2,3 The testosterone-producing pathways are the main drivers of cancerous cell growth and disease progression in PC.
During its early stages, PC may go unnoticed or result in symptoms that are difficult to distinguish from other disease causes. Such symptoms include altered urination patterns; blood in the urine or semen; painful urination and/or ejaculation; pain in the pelvic area, hips, or lower back; and erectile dysfunction.4-6 Symptoms can intensify once the tumour grows locally or metastases begin to interfere with physiological functions. Bones are the most common sites for metastases; this occurs in 90% of patients with metastatic PC.7,8 These patients may experience bone pain, pathologic fractures, or mobility limitations that greatly affect their quality of life.9,10
The PC disease continuum consists of several disease states, from nonmetastatic castration-sensitive localized disease to mCRPC. The term “advanced PC” is often used to describe a more severe diagnosis and prognosis. The definition of advanced PC encompasses patients with a significant risk of disease progression and/or death.11,12 Treatment recommendations and guidelines for PC recognize localized both disease with high-risk features and metastatic disease as advanced PC, which includes all patients that require ADT.12-14 According to the clinical experts consulted by CADTH, advanced PC covers a heterogenous collection of disease presentations and clinical contexts. The clinical experts stated in broad terms that it represents PC that is incurable in nature but has a wide variety of clinical therapeutic options and survival times, ranging from weeks to many years. The clinical experts referred to the Canadian Urological Association 2022 guidelines update for a definition of locally advanced PC.15 Specifically, the guideline referred to the high-risk locally advanced inclusion criteria of the STAMPEDE trial, in which patients with at least 2 of a diagnosis of stage T3 or T4 cancer, a Gleason score of 8 to 10, and a PSA of 40 ng/mL or higher were considered in the trial.16 Survival is highly dependent on disease state. While patients with localized and locally advanced PC have a 5-year survival rate of almost 100%, patients with metastatic PC have a 5-year survival rate of only 34%.2,17 In 2022, an estimated 4,600 Canadians died due to PC, translating into 1 in 29 men.2,3
Prostate cancer is usually diagnosed upon the detection of elevated PSA or through a systematic digital rectal examination. Urologists are the primary contact for patients with suspected PC. Diagnostic tests include blood screening for PSA,4,13 physical digital rectal examination,13 biopsy of the prostate, and imaging through CT or MRI scans.4,8,18 Histologically, PC is graded using Gleason scores.8,18 Biopsy samples taken via invasive techniques are assigned a score between 6 and 10 to describe the tumour and the disease prognosis.8,18 Higher scores reflect a worse disease prognosis, indicating that the tumour is more likely to spread and become metastatic.4,8,18 Additionally, PC can be scored using the tumour-node-metastasis classification system,4,13,19 which involves 4 stages — from stage I (e.g., T1 N0 M0), representing the lowest chance that the cancer cells will spread, to stage IV (e.g., Tany N1 M1), representing the highest chance it will or has spread.4,13,19
In Canada, approximately 24,600 men received a PC diagnosis in 2022, and it is estimated that about 1 in 8 adult males in Canada will develop PC during their lifetime, making it the most common cancer among males in the country.2,3 The prevalence of PC was calculated using the most recent number of prevalent cases in 2018 (96,590), based on a special report on cancer prevalence published by the CCS in 2022, and the adult male Canadian population in 2018 (14,744,508), which led to an estimated prevalence of 0.66%.20,21 To estimate the prevalence of PC cases in 2024, the prevalence rate was assumed to be stable over time, implying that incidence and mortality rates balance each other out.22
Standards of Therapy
Contents within this section were informed by materials submitted by the sponsor and clinical expert input. The following summary was validated by the CADTH review team.
The Canadian Urology Association and Canadian Urologic Oncology Group have established guidelines for treating different stages of PC, as captured in the CADTH funding algorithm.23-25 For localized or locally advanced PC, patients can benefit from local treatments such as surgery (radical prostatectomy) or RT, with or without adjuvant (postsurgical) or neoadjuvant (presurgical) pharmaceutical interventions.25,26 These local treatments aim for a curative outcome.25,27
A crucial element of PC treatment, particularly after initial curative-intent procedures such as prostatectomy or RT, is ADT. Testosterone production is a major driver of PC progression, and ADT works by suppressing testosterone. Experts consulted by CADTH emphasized that ADT, which is recommended by national and international guidelines, controls advanced PC for some time and prolongs significant health outcomes, such as overall survival and time to various disease progressions, despite eventual development of resistance.25,27
Bilateral orchiectomy, a surgical method to halt testosterone production, is replicated pharmacologically by ADTs aiming to reduce testosterone to specific levels. These ADTs are reversible, unlike surgical castration, and can be administered through gonadotropin-releasing hormone (GnRH) and/or LHRH agonists or antagonists. They are recommended for patients without evidence of metastatic disease after curative intervention, particularly in high-risk cases. ADTs are administered continuously, with precautions such as concurrent antiandrogen treatment to prevent symptom flare-ups, such as pain from bone metastases or spinal-cord compression.28,29
In metastatic or castration-resistant cases of PC, guidelines recommend adding treatments to ADT for intensification. ADT remains foundational, and it is used continuously even in supportive-care settings to avoid pain flare-ups. As the disease progresses, additional therapies such as ARAT, chemotherapy, or radioligand therapy may be added, depending on the disease state.14,24,30
For various stages of PC, such as nonmetastatic castration-resistant PC and newly diagnosed androgen-sensitive metastatic disease, additional therapies such as darolutamide, apalutamide, and ARATs (e.g., abiraterone and enzalutamide) are combined with ADT. These combinations have shown improvements in critical end points, including metastasis-free survival and overall survival. In certain high-risk patients, triple therapy including cytotoxic chemotherapy is used. Additionally, for advanced localized disease, ADT is combined with RT, either as adjuvant or neoadjuvant therapy, to improve outcomes such as overall survival compared to local therapy alone.14,24,28-30
Drug Under Review
Key characteristics of relugolix along with other treatments available for PC are summarized in Table 3.
Relugolix (Orgovyx), 120 mg oral tablets, is indicated for the treatment of adult patients with advanced PC.31 The reimbursement request is aligned to the Health Canada indication (i.e., for the treatment of patients with advanced PC). CADTH has not reviewed relugolix in the past.
Relugolix is initiated with a loading dose of 360 mg (3 tablets) on the first day. Treatment is then continued with a single 120 mg tablet taken orally once daily at approximately the same time each day.31 While patients with metastatic PC often receive ADT until their death, patients with localized high-risk or locally advanced disease may need ADT (in addition to local treatment) only until they are cured.23
Relugolix is a nonpeptide GnRH or LHRH receptor antagonist that competitively binds to pituitary GnRH (or LHRH) receptors, thereby reducing the release of luteinizing hormone and FSH into the systemic circulation, and consequently reducing the production of testosterone in the testes. In humans, concentrations of FSH and luteinizing hormone rapidly decline after oral administration, and testosterone concentrations are suppressed to below physiologic concentrations.31
Table 3: Key Characteristics of Available ADT — Relugolix, Degarelix, and GnRH (or LHRH) Agonists
Characteristic | Mechanism of action | Indicationa | Route of administration and recommended dose | Serious adverse effects or safety issues | Other |
|---|---|---|---|---|---|
Drug under review | |||||
Relugolix (Orgovyx) | GnRH receptor antagonist at pituitary gland. Reduces release of LH and FSH, thereby, suppressing testosterone release by testes | For the treatment of adult patients with advanced PC31 | Oral tablets, loading dose of 360 mg, then 120 mg once daily | Decreased bone density, osteoporosis, increased risk of bone fracture (“Black box” warning) QTc prolongation | With an oral P-gp inhibitor or a combined P-gp and strong cytochrome P4503A inducer may increase or decrease the exposure to relugolix, respectively |
GnRH or LHRH antagonist | |||||
Degarelix (Firmagon) | Competitively and reversibly binds to GnRH receptors at pituitary gland, thereby reducing release of LH and FSH, which leads to reduced secretion of testosterone by testes | For testosterone suppression in patients with advanced hormone-dependent PC in whom androgen deprivation is warranted32 | SC, starting dose of 240 mg once followed by maintenance dose of 80 mg once monthly | QTc prolongation, osteoporosis, changes in glucose tolerance (development or aggravation of diabetes), anemia | Should be prescribed by a qualified health professional that is experienced in the use of hormonal therapy in PC; should be administered under the supervision of a physician |
GnRH or LHRH agonists | |||||
Leuprolide acetate (Lupron, Lupron Depot, Zeulide Depot, Eligard) | Nonapeptide analogue of naturally occurring GnRH or LHRH; inhibits pituitary gonadotropin secretion and suppresses testicular testosterone production | Lupron, Lupron Depot: For palliative treatment of sex hormone responsive advanced (stage D2) carcinoma of the prostate33 Zeulide Depot: For palliative treatment of advanced and/or metastatic PC34 Eligard: For palliative treatment of advanced PC (Stage D2)35 | Lupron Depot: IM, 7.5 mg once monthly, 22.5 mg every 3 months, and 30 mg every 4 months Zeulide Depot: IM, 3.75 mg once monthly and 22.5 mg every 3 months Eligard: SC, 7.5 mg once monthly, 22.5 mg every 3 months, 30 mg every 4 months, and 45 mg once every 6 months | Lupron, Lupron Depot: (“Black box” warning) Flare reaction, osteoporosis Zeulide Depot: (“Black box” warning) flare, osteoporosis, sudden cardiac deaths, drug-induced liver injury Eligard: Flare, changes in bone density, symptoms of hypogonadism | Should be prescribed by a qualified physician experienced in the use of hormonal therapy in prostate cancer; should be administered under the supervision of a health care professional |
Buserelin acetate (Suprefact Depot) | Peptide analogue of the natural GnRH or LHRH; inhibits gonadotropin release, and subsequent reduction of serum testosterone | For palliative treatment of patients with hormone-dependent advanced carcinoma of the prostate gland (Stage D)36 | SC implant, 6.3 mg once every 2 months and 9.45 mg once every 3 months About 7 days before the first injection, an antiandrogen should be administered and continued for 4 weeks after first injection of buserelin until testosterone level is within castration range | (“Black box” warning) Flare reaction, osteoporosis | Should be prescribed by a qualified physician experienced in the use of hormonal therapy in prostate cancer; should be administered by a health care professional |
Goserelin acetate (Zoladex, Zoladex Depot) | Decapeptide analogue of GnRH or LHRH. Inhibits gonadotropin (LH) production resulting in gonadal and consequently, accessory sex organ regression (suppressing testosterone) | For palliative treatment of patients with hormone-dependent advanced carcinoma of the prostate In combination with antiandrogen and RT for the management of locally advanced or bulky carcinoma of the prostate Adjuvant hormone therapy to external beam irradiation for patients with locally advanced PC37 | SC, 3.6 mg depot 8 weeks before RT, followed in 4 weeks by 10.8 mg depot, continue until completion of the RT Alternatively, 14.4 mg depot SC every 4 weeks, 2 depots preceding and 2 during RT until completion of RT | (“Black box” warning) tumour flare reaction, osteoporosis, injection-site injuries, and vascular injuries | Should be administered by a health care professional experienced in administering deep SC injections under the supervision of a physician |
Triptorelin (Trelstar) | Decapeptide analogue of naturally occurring GnRH or LHRH; inhibits gonadotropin secretion and suppresses testosterone production at testes | For palliative treatment of hormone-dependent advanced carcinoma of the prostate gland (stage D2)38 | IM, 3.75 mg once monthly, 11.25 mg every 3 months and 22.50 mg once every 6 months | (“Black box” warning) flare reaction, osteoporosis | Should be prescribed by a qualified health care professional experienced in the use of hormonal therapy in prostate cancer; should be administered by a health care professional |
ADT = androgen deprivation therapy; FSH = follicle-stimulating hormone; GnRH = gonadotropin-releasing hormone; IM = intramuscular injection; LH = luteinizing hormone; LHRH = luteinizing hormone–releasing hormone; PC = prostate cancer; RT = radiation therapy; SC = subcutaneous.
aHealth Canada–approved indication.
Sources: Sponsor’s Summary of Clinical Evidence77 and product monographs.31-38
Stakeholder Perspectives
Patient Group Input
This section was prepared by the CADTH review team based on the input provided by patient groups. The full original patient inputs received by CADTH are included in the Stakeholder section of this report.
Three patient groups each submitted an input. The ADT Education Program supports patients living with PC undergoing hormone therapies (ADTs). The CCS is the only national charity that supports patients living with all types of cancer across the country with research and a compassionate support system, and by establishing health policies. These patient groups were represented by 1 patient each in their submissions. The third patient group, PROCURE, is a charitable organization that educates, supports, and informs people affected by PC and promotes and contributes to financing research. PROCURE collected information from an online survey of 263 patients conducted in May 2022.
In ADT Education Program input, a patient living with advanced PC said he has been on ADT almost continuously for more than 20 years and experienced many side effects. The patient, speaking on behalf of other patients, stated that the most disturbing effects are hot flashes, fatigue, and loss of sexual interest. He added that they also regularly experience loss of muscle mass yet gain weight as fat, making simple tasks like walking upstairs difficult. Also, based on input, ADT affects memory, can lead to depression and insomnia, and makes patients feel weak, old, flabby, and demoralized. Last, he stated that the depot injection form of ADT drugs may cause inflammation at the injection site, producing discomfort for days after injections. According to the input, just to avoid injections, some patients may delay getting repeated injections or take risky drug holidays that can cause their cancer to fulminate. The PROCURE input noted that some patients decide to opt for orchiectomy to avoid regular injections. Similarly, another patient from CCS stated that he experienced side effects such as weight gain, impacts on kidneys and the liver, and reduced sexual desire, which was noted as a key side effect by the patient. In the PROCURE and CCS inputs, patients said taking ADT can cause side effects that may require other medications, such as antidepressants or a kidney-protective drug. The patient from the CCS said he felt weak and tired, which reduced his motivation to exercise. The patient’s wife did not report a significant impact on her life, other than the limited sexual desire the patient described as a side effect of treatment. According to the PROCURE input, patients and their partners often mourn the loss of a satisfying sexual relationship, and advanced cancer creates anxiety within the couples. In addition, PROCURE stated that children and family may experience anxiety as their father passes away from the cancer, or they may be at risk of getting prostate, breast, and ovarian cancers if their father is a carrier of the BRCA mutation. PROCURE added that frequent travel to clinics or hospital for medical follow-up exams associated with injection hormone therapy can be costly, and that it takes too long, i.e., from months to years, to see testosterone levels return to normal after long-term ADT ends.
Based on inputs, key outcomes important to patients were the safety of medication and minimizing side effects. Other key outcomes cited by patients as important included maintaining long-term survival (with ADT) and a good quality of life. The PROCURE input also stated that patients want improved outcomes in treatment such as slowing down the progression of cancer, extension of life expectancy, and decreased PSA levels. All inputs indicated that patients living with PC would appreciate a new patient-friendly alternative form of ADT treatment that is not difficult and invasive.
Clinician Input
Input From Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of PC.
Unmet Needs
Despite improvements in median overall survival that now lead to prognosis usually being measured in several years, the clinical experts noted that resistance to available therapies is inevitable and the majority of men will succumb to their disease. However, none of these treatment gaps in overall survival are related to ADT, but rather the androgen-independent mechanisms of resistance that occur once ADT combined with additional systemic therapy starts to fail. All available ADT options, in a suitable time frame, lead to profound levels of medical castration (testosterone suppression), and this is not a large unmet need from an “efficacy” point of view.
Based on input from the clinical experts, currently available ADT options lack oral administration, and injectable forms may not be convenient or preferred by all patients. The clinical experts stated that they are not aware of published evidence that has influenced Canadian clinical practice regarding patient preference for oral options, or that compliance is negatively affected by injectable options alone.
According to the clinical experts, most patients with advanced PC would visit their physicians at least twice a year to review disease control measures, symptoms, and toxicity management, and that such visits would be appropriate times for their next ADT dose. As such, the current treatment paradigm does not pose a large burden on the health care system. However, due to the geography of Canada, certain patients may not be able to travel and be present for ADT administration, creating an unmet need that can be addressed with an oral treatment option.
Place in Therapy
Relugolix is best placed as a foundational ADT, eligible for use as a single drug or in addition to ARAT intensification for men with advanced PC according to the patient populations specified in the HERO study. The available evidence does not support combining relugolix with other intensification measures that may be needed later, such as chemotherapy or polyADP ribose polymerase inhibitors or theragnostic. This is an issue that should be addressed.
Patient Population
The clinical experts consulted by CADTH described the potential population of patients who may choose relugolix as those living in remote locations far from cancer centres, those who have a strong preference for oral rather than injected ADT, and those for whom intermittent ADT is attempted to avoid the AEs of medical castration by withdrawing treatment in patients who have responded to continuous ADT (given that testosterone and HRQoL recovery is more rapid that conventional ADT). However, the clinical experts emphasized that treatment adherence becomes an important consideration for patients prescribed relugolix. The clinical experts were unaware of evidence that influenced Canadian clinical practices with respect to the long-term survival outcome of patients and the role of rapid testosterone recovery compared to lingering testosterone suppression after treatment discontinuation.
Assessing the Response Treatment
According to the clinical experts, in the MCSPC setting, and from an ADT-response point of view, the Canadian consensus statement recommends up to 0.7 nmol/L as the new castration threshold for patients with advanced PC.39 In this setting, patients should also be intensified with ARAT therapy, and in that context other measures of response include prolonging overall survival, prolonging progression-free survival (with progression defined either clinically by PSA levels or on a CT or bone scan), prolonging time to skeletal-related events, prolonging time to symptomatic deterioration, and prolonging time to castration resistance.
According to the clinical experts, for patients who experience clinical and/or biochemical relapse after curative-intent local therapy, the aim is to achieve castration levels of testosterone and prolong overall survival, metastasis-free survival, and time to castration resistance.
Discontinuing Treatment
According to the clinical experts, in the mCSPC setting, ADT is rarely ceased, unless toxicities are truly intolerable. In the high-risk curative setting, ADT may be ceased due to toxicities more often; however, such decisions must be based on balanced discussions of the risks and benefits at that time point. According to the clinical experts, most of the toxicities from ADT are easily managed.
Prescribing Considerations
Prescriptions should be made by a specialist oncologist (a medical, radiation, or urological surgeon). The drug is administered orally at home by the patient.
Clinician Group Input
This section was prepared by the CADTH review team based on the input provided by clinician groups. The full original clinician group inputs received by CADTH are included in the Stakeholder section of this report.
A total 2 inputs were submitted. The ADT Education Program, represented by 1 clinician and the British Columbia Genitourinary Group at the Vancouver Prostate Centre, represented by 6 clinicians, participated in submission. The ADT Education Program supports patients living with PC undergoing hormone therapies (ADTs) through activities such as educational courses. The BC Genitourinary Tumour Group advises stakeholders and leaders in health care on all matters related to the care of prostate, bladder, and kidney cancers, whereas the Vancouver Prostate Centre engages in basic, translational, and clinical research, including clinical trials, on genitourinary cancers. The latter 2 groups gathered information from peer-reviewed publications.
All clinician groups agreed that there is the need for a better-tolerated, more-convenient treatment that improves compliance. The clinical experts consulted by CADTH added that resistance to available therapies, which is not related to ADT but rather androgen-independent mechanisms of resistance, is the unmet need. The clinician groups stated that an oral formulation of an LHRH antagonist would be more advantageous than the injection forms that are associated with injection-site reactions, high dosages causing discomfort, travel to clinics, and high resource utilization. The clinical experts consulted by CADTH agreed that no oral options are available for androgen suppression for men with PC, and that an oral form would be beneficial for those who live far from cancer centres. However, the clinical experts noted that, because most regimens are available as 6 monthly doses, any claims of a large “burden” on the health care system due to the injectable ADT should be interpreted with caution. Additionally, the clinical experts cautioned that, because patients should take ADT for a long time, a pill form may lead to compliance issues and/or increased pill burden (with concurrent ARATs). The clinician groups stated that a cure is the goal of an ideal PC treatment. However, for noncurative PC that has spread past the gland, the goals are to suppress androgen with either reduced side effects or less-invasive administration, prolong survival, and increase quality of life.
The clinical experts consulted by CADTH agreed, and they added that prolonging time to skeletal-related events, symptomatic deterioration, and castration resistance are also goals of therapy. Both clinician groups stated that patients best suited for relugolix treatment include patients who have hormone-sensitive disease, those with newly diagnosed or substantial metastatic disease in need of prompt androgen suppression (e.g., widespread bony involvement causing pain or imminent risk of spinal-cord compression or a locally advanced tumour causing urinary retention and/or ureteral obstruction), those in need of short-term ADT (i.e., an adjuvant to RT or rapid recovery of testosterone is desirable after discontinuation), and patients having difficulty accessing injection clinics. The clinical experts consulted by CADTH agreed and added that those with a strong preference for an oral rather than injected ADT option (due to fear of local reactions) or those who need intermittent rather than continuous ADT (given the rapid recovery of testosterone and quality of life versus conventional ADT) are best suited for relugolix treatment.
According to the clinician groups, responses to relugolix treatment are measured in the same manner as currently available ADT drugs, either by serum PSA levels or imaging (CT, bone, or prostate-specific membrane antigen PET). The clinical experts added that the response to relugolix monotherapy would be reflected in the “profound castration” level of testosterone (≤ 0.7 nm/L). The clinician groups and clinical experts agreed that ADT is generally administered as a continuous treatment for patients with metastatic, locally advanced, or castration-resistant PC, unless contraindications or truly intolerable side effects (debilitating hot flushing, decreased libido, or anergia) exist. The British Columbia groups added that relugolix can be administered intermittently based on serum PSA Ievels in patients with biochemical recurrence or administered for a predetermined duration in patients receiving ADT with curative-intent radiation. All clinician groups and clinical experts agreed that a urologist, medical- or uro-oncologist, or radiation oncologist experienced in the management of patients with advanced PC should prescribe relugolix and monitor responses, while histopathology is involved in confirming diagnosis. The clinician groups noted that relugolix could be dispensed in outpatient settings and the clinical experts added that the patient takes relugolix by mouth at home.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s Reimbursement Review processes by identifying issues that may affect their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert responses |
|---|---|
Relevant comparators | |
Relevant comparators funded in most jurisdictions include leuprolide (comparator in the HERO trial), degarelix, buserelin, and goserelin, all of which are injectables. | This is a comment from drug plans for pERC. |
The primary efficacy outcome measure was medical castration rate, defined as achieving and maintaining serum testosterone suppression to castration levels (< 50 ng/dL) by day 29 through 48 weeks of treatment. Other key secondary end points included castration rates on day 4 and 15, castration rates with testosterone < 20 ng/dL at day 15, and PSA response rate at day 15, and FSH level at day 176 (week 25, day 1). In clinical practice what is the most appropriate frequency to determine treatment response? | Currently, most patients with advanced prostate cancer would visit their physicians at least twice a year for review of disease control and symptoms and toxicity management. In the management of mCSPC, the Canadian consensus statement recommends maintaining testosterone levels at or below 0.7 nmol/L, aligning with the “profound” castration level proposed by the drug sponsor. Additionally, patient treatment in this context should be intensified with ARAT therapy and PSA levels and clinical end points are primarily used to assess clinical response. |
Patients in the HERO trial with disease progression during the treatment period were encouraged to remain on study and, if indicated, may have received radiotherapy as prescribed by the investigator. If patients had PSA progression, they were allowed to receive enzalutamide or docetaxel during the study. What are the discontinuation criteria for relugolix? | In the mCSPC setting, ADT is rarely ceased, unless toxicities are truly intolerable. In the high-risk curative setting, ADT is more often ceased due to toxicities; however, it is a balanced discussion based on risks and benefits at that time point. According to the clinical experts, most of the toxicities from ADT are easily managed. |
Considerations for prescribing of therapy | |
Relugolix should be initiated at a loading dose of 360 mg (3 tablets) on the first day and continued with a 120 mg tablet taken once daily at approximately the same time each day. | This is a comment from drug plans for pERC. |
Generalizability | |
Can the trial results be generalized to patients with an ECOG Performance Status > 1? | The results are generalizable to patients with an ECOG Performance Status > 1. |
Funding algorithm | |
Under what clinical circumstances would relugolix be used over existing drugs? | Patient preference for oral treatment or preference for rapid return of testosterone to normal levels upon cessation of the drug may be factors where relugolix is used over existing drugs. |
Care provision | |
Relugolix has the potential for drug-drug and drug-laboratory interactions, requiring assessment and/or intervention. Would this limit its use in combination regimens (i.e., apalutamide is a strong CYP3A4/P-gp inducer, and abiraterone was a prohibited medication in the trial)? | The study did not include abiraterone and apalutamide, which are significant intensification options in this therapeutic area. The sponsor proposed a comprehensive listing for the use of relugolix in combination with all ARATs. However, it may be appropriate to consider restricting combination partners to those explicitly included in the study, such as the use of enzalutamide specifically in the context of metastatic castration-resistant prostate cancer. |
ADT = androgen deprivation therapy; ARAT = androgen-receptor axis therapy; CYP3A4/P-gp = cytochrome P450 3A4 and P-glycoprotein; ECOG = Eastern Cooperative Oncology Group; FSH = follicle-stimulating hormone; mCSPC = metastatic castration-sensitive prostate cancer; pERC = CADTH pan-Canadian Oncology Review Expert Review Committee; PSA = prostate-specific antigen.
Clinical Evidence
The objective of CADTH’s Clinical Review is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of relugolix, 120 mg, orally in the treatment of advanced PC in adult men. The focus will be placed on comparing relugolix to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of relugolix is presented in 4 sections, with CADTH’s critical appraisal of the evidence included at the end of each section. The first section, the systematic review, includes pivotal studies and RCTs that were selected according to the sponsor’s systematic review protocol. CADTH’s assessment of the certainty of the evidence in this first section using the GRADE approach follows the critical appraisal of the evidence. The second section includes any sponsor-submitted long-term extension studies (none were submitted). The third section includes indirect evidence from the sponsor. The fourth section includes additional studies that were considered by the sponsor to address important gaps in the systematic review evidence.
Included Studies
Clinical evidence from 1 RCT identified in the systematic review (an ITC), and 3 additional studies addressing gaps in evidence are included in the CADTH review and appraised in this document.
Systematic Review
Contents within this section were informed by materials submitted by the sponsor. The following summary was validated by the CADTH review team.
Description of Studies
Characteristics of the included studies are summarized in Table 5. The HERO study was a randomized, open-label, parallel, multicentre, phase III trial. The objective was to evaluate the efficacy and safety of relugolix 120 mg taken orally once daily after a single oral loading dose of relugolix 360 mg (three 120 mg tablets) on day 1 compared to leuprolide given as a 3-month, 22.5 mg depot suspension to males with advanced PC who required at least 1 year of continuous ADT. The trial consisted of 3 phases, including a screening period of up to 28 days, a 48-week treatment period, and 30-day follow-up safety period. A subset of patients was followed for up to 90 days to assess testosterone recovery.
At baseline, patients meeting all eligibility criteria at screening were enrolled in the study and randomized in a 2:1 ratio using an interactive voice-response system to receive either relugolix (120 mg once daily after a single oral loading dose of 360 mg) or leuprolide (22.5 mg [or 11.25 mg in Japan and Taiwan] by intramuscular or subcutaneous injection every 3 months) for 48 weeks. Randomization was stratified according to geographic region (North and South America, Europe, or Asia-Pacific region), metastatic disease (yes or no), and age (≤ 75 or > 75 years).
The study planned a primary analysis and a final analysis. For the primary analysis (cohort 1), 934 patients were randomized to the relugolix group (n = 624) or to the leuprolide group (n = 310,) in 155 study centres located in Australia, Austria, Belgium, Brazil, Canada (8 sites), Denmark, Finland, France, Germany, Italy, Japan, Netherlands, New Zealand, Poland, Republic of Korea, Slovakia, Spain, Sweden, Taiwan, the UK, and the US. For the final analysis, 144 additional participants (cohort 2) with metastatic disease had been enrolled and randomized to the relugolix group (n = 719, including participants from the primary analysis – cohort 1 and cohort 2) or to the leuprolide group (n = 359, including participants from the primary analysis – cohort 1 and cohort 2) from the 160 total locations, comprising sites from the primary analysis and other centres located in China. The database-lock dates were December 10, 2019, for the primary analysis and September 23, 2020, for the final analysis. In this section (section 2), results of the primary analysis are reported across all subsections, except for the castration resistance–free survival (CRFS) outcome, which is derived from the final analysis.
Table 5: Details of Studies Included in the Systematic Review
Detail | HERO trial |
|---|---|
Designs and populations | |
Study design | Phase III, randomized, multicentre, open-label, parallel-group study |
Locations | Patients enrolled across 160 sites in 22 countries in Europe, North and South America, and the Asia-Pacific region |
Patient enrolment dates | Start: April 2017 End: October 2019 (primary analysis) and August 2020 (final analysis) |
Randomized (N) | Primary analysisa: 934
Final analysis 1,078
|
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Intervention | Relugolix, 120 mg tablet, administered orally, once daily, following an oral loading dose of 360 mg (three 120 mg tablets) on day 1, for 48 weeks |
Comparator(s) | Leuprolide, 22.5 mg (or 11.25 mg in Japan, Taiwan, and China), 3-month depot injections, every 12 weeks for 48 weeks |
Study duration | |
Screening phase | In the 28 days before starting treatment |
Treatment phase | 48 weeks |
Follow-up phase | Safety: 30 days posttreatment Testosterone recoveryc: 30 days, 60 days and 90 days posttreatment |
Outcomes | |
Primary end point | Sustained castration rated (time frame: from week 5, day 1 [day 29] through week 49, day 1 [day 337]). |
Secondary efficacy end points | Key secondary end points
Other secondary end points
|
Safety end points |
|
Exploratory end points |
|
Records | Shore et al. (2020)40 Clinicaltrials.gov: NCT0308509541 |
ADT = androgen deprivation therapy; CFB = change from baseline; CRFS = castration resistance–free survival; ECG = electrocardiogram; ECOG = Eastern Cooperative Oncology Group; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-PR25 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Prostate-Specific 25 items; FSH = follicle-stimulating hormone; GnRH = gonadotropin-releasing hormone; LH = luteinizing hormone; PC = prostate cancer; PSA = prostate-specific antigen; MACE = major adverse cardiovascular event; QoL = quality of life; RP = radical prostatectomy; RT = radiation therapy; TEAE = treatment-emergent adverse event.
aPatients included in the primary analysis.
bPatients included in the final analysis.
cTestosterone recovery was only assessed in a subset of patients. The testosterone recovery rate was defined as the cumulative probability of testosterone recovery to greater than 280 ng/dL (lower limit of the normal range), to 50 ng/dL or greater (definition of castration), and to greater than 280 ng/dL or baseline level.
dSustained castration rate was defined as the cumulative probability of testosterone suppression to less than 50 ng/dL (1.7 nmol/L).
eCRFS was defined by disease progression despite achieving testosterone suppression to castrate levels of less than 50 ng/dL.
fProfound castration rate was defined as the cumulative probability of testosterone suppression to less than 20 ng/dL.
gPSA response defined as a reduction of greater than 50% in PSA from baseline and greater than 90% in PSA from baseline.
hUndetectable PSA rate was defined as the proportion of participants with PSA concentration less than 0.02 ng/mL.
iPSA progression was defined as the first increase in PSA of at least 25% and at least 2 ng/mL above the nadir with confirmation by a second consecutive PSA measurement at least 3 weeks later.
jMACE was defined as nonfatal myocardial infarction, nonfatal stroke, and death from any cause.
kOverall survival was defined as time from randomization to date of death before data cut-off date.
lPolymorphisms in germline genes related to the hypothalamic-pituitary androgen pathway, prostate cancer risk, or to drug metabolizing enzymes and transporter proteins that might be implicated in the drug disposition, safety, or efficacy of relugolix.
Sources: Clinical Study Reports,1,42 Shore et al. (2020),40 Clinicaltrials.gov: NCT03085095.41 and the sponsor’s Summary of Clinical Evidence.77
Populations
Inclusion and Exclusion Criteria
Patients eligible for enrolment in the HERO study were males aged 18 years and older diagnosed with androgen-sensitive advanced PC who required at least 1 year of continuous ADT. Patients with evidence of biochemical (PSA) or clinical relapse following local primary intervention with curative intent (e.g., surgery, RT, cryotherapy, or high-frequency ultrasound), newly diagnosed hormone-sensitive metastatic disease, and advanced localized disease unlikely to be cured by local primary treatment with either surgery or radiation were included. Patients who had a MACE within 6 months before trial initiation were excluded. Patients who previously received any form of ADT for a total duration of up to 18 months were allowed to participate if that therapy had been completed at least 3 months before baseline or as long as the dosing interval for leuprolide depot was longer than 3 months.
Interventions
During the randomized treatment period of the HERO study, patients received either a single oral loading dose of relugolix 360 mg (three 120 mg tablets) on day 1 followed by a 120 mg tablet orally once daily for 48 weeks, or leuprolide in a 3-month depot injection, 22.5 mg (or 11.25 mg in Japan, Taiwan, and China) every 12 weeks for 48 weeks (the last injection occurred 12 weeks before the end of the treatment period).
No dose adjustments were made in this study. The dosing schedule for relugolix required patients to take 1 tablet at a regular time each morning at least 1 hour before breakfast. If the dose was missed in the morning for any reason, the study drug could be taken later in the day, at least 1 hour before or 2 hours after eating a meal. For leuprolide depot, a first dose was administered subcutaneous or intramuscular injection on day 1 at the clinic, followed by subsequent doses at 12-week intervals. In Canada, Eligard 22.5 mg for subcutaneous use, and both Lupron Depot 11.25 mg and 22.5 mg for intramuscular use are approved by Health Canada and available on the market. However, in this study, Eligard was used exclusively in the trial across all sites for the 22.5 mg dose, encompassing locations in Canada. As for the 11.25 mg dose administered subcutaneously or intramuscularly, it was administered solely in Japan, Taiwan, and China, with Prostap being the brand used in these regions. Patients in the leuprolide group were allowed to receive an antiandrogen therapy for the initial 4 weeks, or even longer if deemed by the investigator as necessary for the management of disease flare-ups and/or based on disease status (e.g., in patients with extensive localized symptomatic disease or with metastatic disease).
Patients had to withhold or permanently discontinue treatment if any signs of hepatotoxicity were detected, following a decision made by the investigator and sponsor. This decision had to be based on the severity of the hepatotoxicity, or the occurrence of an event related to abnormal ALT, aspartate transaminase (AST), or total bilirubin levels on a liver test, or a liver disease (such as alcoholic hepatitis, autoimmune hepatitis, hepatobiliary tract disease, or drug-induced hepatotoxicity).
Outcomes
A list of efficacy end points assessed in this Clinical Review is provided in Table 6, followed by descriptions of the outcome measures. Summarized end points are based on outcomes included in the sponsor’s Summary of Clinical Evidence as well as any outcomes identified as important to this review by the clinical experts consulted by CADTH and stakeholder input from patient and clinician groups and public drug plans. Using the same considerations, the CADTH review team selected end points that were considered the most relevant to inform CADTH’s expert committee deliberations and finalized this list of end points in consultation with members of the expert committee. All summarized efficacy end points were assessed using the GRADE system. Select notable harms outcomes considered important for informing CADTH’s expert committee deliberations were also assessed using GRADE.
Table 6: Outcomes Summarized From Studies Included in the Systematic Review
Outcome measure | Threshold | Time point | HERO | Cohort (analysis) |
|---|---|---|---|---|
Efficacy | ||||
Sustained castration rate | Testosterone suppression to < 50 ng/dL | From week 5, day 1 (day 29) through week 49, day 1 (day 337) | Primarya | Primary |
Week 1, day 4 (day 4) | Key secondarya | |||
Week 3, day 1 (day 15) | ||||
Cumulative probability of profound castration rate | Testosterone suppression to < 20 ng/dL | Week 3, day 1 (day 15) | Key secondarya | Primary |
Week 5, day 1 (day 29) through week 49, day 1 (day 337) | Other secondary | |||
Week 25, day 1 (day 169) through week 49, day 1 (day 337) | ||||
Safety | ||||
Proportion of patients who experienced MACE | NA | Through 48 weeks | Key harms | Primary |
Loss of bone mineral density | NA | Through 48 weeks | Adverse events | Primary |
MACE = major adverse cardiovascular event; NA = not applicable.
aStatistical testing for these end points was adjusted for multiple comparisons (i.e., hierarchal testing). The order of the outcomes presented in this table does not represent the order in which the hierarchical testing was performed. The specific order of hierarchical testing is described further in this document.
Sources: Clinical Study Report,1,42 Shore et al. (2020),40 Clinicaltrials.gov: NCT03085095,41 and the sponsor’s Summary of Clinical Evidence.77
Castration Rate
The FDA and the European Medicines Agency (EMA) provide guidelines for evaluating anticancer medicinal products in males with castration-sensitive PC.43,44 For medicinal products aiming to achieve medical castration through GnRH analogues, the guidelines suggest a need to demonstrate maintenance of castration levels of testosterone (50 ng/dL and below) without breakthroughs and micro-surges. Additionally, the percentage of participants maintaining a testosterone level of less than 20 ng/dL (profound castration) should be considered an end point and included in labelling.
In the HERO trial, the primary end point was the sustained castration rate, defined as the cumulative probability of testosterone suppression to less than 50 ng/dL (1.7 nmol/L) while on study treatment from week 5, day 1 (study day 29) through week 49 (study day 337). The rate was estimated for each treatment group based on point estimates from the Kaplan-Meier curve and reported as the percentage of participants, with a 95% CI calculated by log-log transformation of the survival function in each treatment group. Serum testosterone concentration was obtained at baseline and at each study visit through the end of treatment visit, to assess outcomes related to testosterone castration levels. Serum testosterone was within a window of about 2 days from the baseline visit up to the week 3 visit, and within a window of about 7 days from week 5 onward until the 90-day follow-up visit. If a patient discontinued treatment for any reason, samples were collected following drug discontinuation at the week 49 visit. Censoring rules for this outcome are detailed in Table 7.
Table 7: Censoring Rules for Sustained Castration Rate
Scenario | Date of event | Censoring outcome |
|---|---|---|
Patients with testosterone escape, defined as any testosterone castration level ≥ 50 ng/dL from week 5, day 1 through week 49, day 1 | Time from the date of the first dose to date of the first testosterone escape | Time to event |
Patients who have not reached castration level at week 5, day 1 | Week 5, day 1 | Time to event |
Patients who discontinued from the study before week 5, day 1 | Week 5, day 1 | Censored |
Patients without a week 5, day 1 assessment | At week 5, day 1 | Time to event |
All patients including those who discontinue from the trial for reasons other than a noncastration testosterone level and without testosterone suppression to < 50 ng/dL | Last available testosterone assessment before follow-up visits | Censored |
Patients who had initiated therapies known to suppress testosterone | Last available testosterone assessment before the initiation of such therapies | Censored |
For patients reaching castration levels at week 5, day 1 | ||
Patients who had ≥ 1 consecutive missed visits (i.e., a visit gap of > 42 days) and had a noncastration assessment immediately after the missed visit(s) were considered as having an escape | At target day of the earliest missed visit before the noncastration assessment | Time to event |
Patients with 1 missed visit who has a castration assessment immediately before and after the missed visit were considered to be castrated | At missed visit | Time to event |
Patients who had ≥ 1 consecutive missed visits (i.e., a visit gap of > 70 days) and who had a castration assessment immediately before and after the missed visits | Last available testosterone assessment before the missed visits | Censored |
Source: HERO Statistical Analysis Plan.45
Key secondary end points included other time points for the sustained castration rate, assessed before dosing at week 1, day 4 (day 4) and before dosing at week 3, day 1 (day 15). Additionally, the cumulative probability of a profound castration rate of greater than 20 ng/dL before dosing at week 3, day 1 (day 15) was evaluated. Other secondary end points for the cumulative probability of a profound castration rate of greater than 20 ng/dL were recorded from week 5, day 1 (day 29) through week 49, day 1 (day 337) and week 25, day 1 (day 169) through week 49, day 1 (day 337). These outcomes were also summarized by treatment group using the Kaplan-Meier method and reported as percentage of participants.
Safety
Adverse events were collected from the first administration of study drugs through approximatively 30 days after the last dose of relugolix, 12 weeks and 30 days after the last injection of leuprolide, or the date of initiation of another therapy, whichever occurred first.
The main safety variables included the following:
TEAEs, in the form of AEs, SAEs, AEs leading to discontinuation, AEs leading to drug interruption, and AEs resulting in a fatal outcome. The severity grade of all AEs was evaluated by the investigator and was based on National Cancer Institute Common Terminology Criteria for Adverse Events, version 4.03.
The proportion of patients with a MACE, defined as nonfatal myocardial infarction, nonfatal stroke, and death from any cause. Specifically, a MACE was considered a composite outcome including myocardial infarction, central nervous system hemorrhages, and cerebrovascular conditions (as defined by a standardized Medical Dictionary for Regulatory Activities query), and deaths due to all causes (Table 8). These events were not adjudicated.
The proportion of patients with AEs of special interest for relugolix classified in the following categories: vasomotor symptoms, carbohydrate and lipid metabolic effects, hepatic transaminase elevations, AEs related to hypersensitivity, mood disorders, adverse cardiovascular events (including MACEs and ischemic heart disease), loss of bone mineral density, and corrected QT interval prolongation (Table 8). In the category of hepatic transaminase elevations, AEs of clinical interest, corresponding to patients with ALT and/or AST levels at least 3 times the upper limit of normal (ULN), were also recorded.
Table 8: Investigator’s Search Criteria Used for Safety Parameters of Interest
Category | Search criteria |
|---|---|
Loss of bone mineral density |
|
Corrected QT prolongation | Torsades de pointes/QT prolongation SMQ (broad) |
Hepatic transaminase elevations | Drug-related hepatic disorders SMQ (narrow) |
Carbohydrate and lipid metabolic effects |
|
Adverse cardiovascular events | Major adverse cardiovascular events:
Ischemic heart disease:
|
Vasomotor symptoms | The following 5 preferred terms were included:
|
Mood disorders | Depression and suicide/self-injury SMQ (broad) |
Hypersensitivity | Hypersensitivity SMQ (narrow) |
SMQ = standardized Medical Dictionary for Regulatory Activities Query.
Source: HERO Protocol.46
Statistical Analysis
Clinical Trial End Points
A summary of statistical analyses for all efficacy end points is presented in Table 9. In this table, all outcomes were reported in the primary analysis. Unless otherwise specified, efficacy analyses were conducted using the modified intention-to-treat (mITT) population (Table 12 provides a description of the population included in the analyses) and were stratified by the randomization stratification factors (geographic region, presence of metastatic disease, and baseline age). If the group of patients from any of the individual randomization stratification factors accounted for less than 10% of the entire mITT population, this stratification factor was collapsed for stratified analyses. In addition, if there were less than 15 patients in 1 of the 12 strata, stratification factors of presence of metastatic disease and baseline age were used in the stratified analysis for more robust strata-adjusted estimates of the treatment effect.
Kaplan-Meier estimates were calculated for sustained castration rate and cumulative probability of profound castration rate along with a 95% CI calculated by log-log transformation of survival function in each treatment group. For CRFS, the hazard ratio was used for treatment comparisons in the Cox proportional hazard model, and P values were obtained through an unstratified log-rank test for patients with metastatic disease and a stratified log-rank test for the mITT final analysis population. Proportionality in survival curves between the 2 treatment groups was assessed, and the CRFS rate at day 337 was estimated with a 95% CI for the treatment difference calculated by a linear transformation of the difference in survival function.
The HERO study used 2 separate evaluation criteria for the primary efficacy end point to support different regulatory requirements for assessing benefit. Evaluation criterion 1 was an FDA requirement and the trial success criterion for the primary efficacy end point. The lower bound of the 95% CI for the cumulative probability of sustained testosterone suppression in the relugolix treatment group had to be 90% or greater to meet this evaluation criterion. Evaluation criterion 2 was required by the EMA and used to establish the noninferiority of relugolix compared to leuprolide treatment. The lower bound of the 95% CI for the difference in the cumulative probability of sustained testosterone suppression between the 2 treatment groups had to be greater than or equal to a noninferiority margin of −10% to meet this evaluation criterion. If noninferiority was demonstrated, subsequent superiority testing was conducted. Superiority was established if the lower boundary of the 95% CI for the between-group difference was greater than zero.47 Both criteria were submitted in Canada, and the Health Canada approval was based on evaluation criterion 1.
To assess the robustness of the analyses of criteria 1 and 2 of the primary end point, 4 sensitivity analyses were performed. The first analysis repeated the analysis of the primary end point but in the per-protocol population. In the second sensitivity analysis, patients who had received concomitant medications and herbal supplements that could possibly affect testosterone levels during study treatment were excluded from the analysis. In the third analysis, patients who had missed 2 or more consecutive visits after week 5, day 1 or discontinued from the study early were considered to have an event at the target day of the earliest missed visit. To assess the impact of delayed testosterone suppression on castration level, patients who had not reached the castration level at week 5, day 1 were censored at week 5, day 1 for the fourth sensitivity analysis of the primary end point.
The handling of missing data for the primary end point of sustained castration rate was addressed using a set of predefined rules in the statistical analysis. First, patients who discontinued the study before week 5, day 1 were censored at the target day of week 5, day 1. This means their data were only considered up to that point and not beyond, even if they left the study earlier. For patients who did not have a week 5, day 1 assessment, the analysis treated them as if they had experienced the event (failure to maintain castration rate) at this target day. For patients with 1 or more consecutive missed visits, different rules were applied based on subsequent assessments. If a patient missed visits but then had a noncastration-level assessment immediately following the missed visits, they were considered to have had an escape (failure to maintain castration rate) at the earliest missed visit before the noncastration assessment. Conversely, if a patient had 1 missed visit with castration assessments both before and after the missed visit, it was assumed they remained castrated at the missed visit. However, if a patient missed 2 or more consecutive visits and had a castration assessment immediately before and after the missed visits, their data were censored at the last available testosterone assessment before the missed visits. Handling of missing data for each outcome is described in Table 9.
Table 9: Statistical Analysis of Efficacy End Points
End point (analysis) | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
HERO trial | ||||
Sustained castration rate ¾ primary end point (primary analysis) | Kaplan-Meier method | Stratified by the randomization stratification factors (geographic region, presence of metastatic disease, and baseline age) |
|
|
Cumulative probability of profound castration rate (primary analysis) | Kaplan-Meier method | Same as primary end point | Same as primary end point but changing the castration level (< 50 ng/dL) to profound castration level (< 20 ng/dL) | None |
Source: HERO Statistical Analysis Plan45 and the sponsor’s Summary of Clinical Evidence.
Sample Size and Power Calculation
The following assumptions were used to determine the sample size for this study:
true probabilities of sustained testosterone suppression are 94% and 96% for relugolix and leuprolide, respectively
2:1 randomization ratio (relugolix: leuprolide)
dropout rate: 15%.
For evaluation criterion 1, a total of 610 patients in the relugolix group was expected to be sufficient to provide approximately 90% power to rule out a fixed probability of sustained testosterone suppression of less than 90% at a 2-sided type I error rate of 0.05.
For evaluation criterion 2, with a noninferiority margin of −10% and an overall 2-sided type I error rate of 0.05, a total of approximately 915 patients (610 receiving relugolix, 305 receiving leuprolide) was expected to yield at least 99% power to declare the noninferiority of relugolix to leuprolide.
The 10% noninferiority margin for the comparison of relugolix versus leuprolide was based on the regulatory precedence of pivotal assessment of the GnRH antagonist degarelix versus leuprolide as well as studies of branded GnRH agonists.
The primary analysis was expected to be performed separately for each evaluation criterion using data collected through 48 weeks after enrolling approximately 915 patients.
Statistical Testing
The statistical model selected for each end point is described in the preceding section.
The primary and the key secondary efficacy analyses were performed at an overall 1-sided type I error of 0.05. A test was deemed statistically significant if the 2-sided P value rounded to 4 decimal places was less than 0.05. If the result of the primary end point analysis met the respective evaluation criterion, the key secondary end points were then tested with a fixed-sequence testing procedure (Table 10) to maintain the overall familywise error rate of 0.05 for the testing of primary and key secondary end points. None of the P values aside from the end points listed in the testing order were adjusted in multiplicity and were at a nominal level of 0.05.
The HERO study was composed of 2 analyses, a primary analysis and a final analysis. All but 1 of the end points were assessed at the primary analysis. The final analysis included an update of previously analyzed end points as well as assessments of castration resistance–free survival. No interim efficacy analysis was performed for this study.
Table 10: Testing Order and Timing of Analysis for Primary and Key Secondary End Points
End points | At primary analysis | At final analysis |
|---|---|---|
Sustained castration rate from evaluation criterion 1 (≥ 90% in relugolix) | 1 | Update |
Sustained castration rate from evaluation criterion 2 (noninferiority and superiority of relugolix compared with leuprolide) | 2 | Update |
Sustained castration rate on week 1, day 4 | 3 | Update |
Sustained castration rate on week 3, day 1 | 4 | Update |
Confirmed PSA response rate at week 3, day 1 | 5 | Update |
Cumulative probability of profound castration rate at week 3, day 1 | 6 | Update |
Follicle-stimulating hormone level at week 25, day 1 | 7 | Update |
Castration resistance–free survival during the 48-week treatment in patients with metastatic prostate cancera | NA | 8 |
Castration resistance–free survival during the 48-week treatment in patients with or without metastatic prostate cancera,b | NA | 9 |
Time to testosterone recovery back to > 280 ng/dL at the 90-day follow-up in patients participating in testosterone recovery follow-upc | 10c | NA |
NA = not applicable; PSA = prostate-specific antigen.
aCastration resistance–free survival and time to testosterone recovery back to greater than 280 ng/dL at the 90-day follow-up were tested at the final analysis only if all the end points reached statistical significance in the primary analysis. End points in the higher order were updated with descriptive statistics in the final analysis.
bAccording to the fixed-sequence testing procedure prespecified in the statistical analysis plan, because the CRFS end point did not achieve statistical superiority in the metastatic patients during the final analysis, the CRFS could not be tested formally as the next key end point on the modified intention-to-treat final analysis population.
cTesting the order of time to testosterone recovery was followed by CRFS in the final analysis.
Source: HERO Statistical Analysis Plan45 and the sponsor’s Summary of Clinical Evidence.77
Subgroup Analyses
Primary End Point
Subgroup analyses of the primary efficacy end point were performed to determine whether treatment effects were consistent across clinically important subgroups. Those analyses were prespecified in the statistical analysis plan. Testosterone castration rates at week 49, day 1 (day 337) for the relugolix arm and their 95% CIs were displayed in a forest plot. The difference in testosterone castration rates at week 49, day 1 between the relugolix and leuprolide arms and their 95% CIs were also displayed in a forest plot.
Subgroups reported in this statistical analysis plan included the following:
ethnicity (Hispanic or Latino, not Hispanic or Latino)
race (white, Black or African American, Asian, or others)
clinical disease-state presentations (biomedical or clinical relapse, newly diagnosed metastatic disease, advanced localized disease)
baseline PSA level (< 20 ng/mL, ≥ 20 ng/mL)
Gleason score at study entry (< 8, ≥ 8)
presence of metastatic disease (yes versus no).
Additional subgroup analyses of the HERO trial were reported in records identified by this systemic literature review from the primary Clinical Study Report:1
concomitant RT during the study (yes versus no)
concomitant enzalutamide or docetaxel during the study (yes versus no)
risk stratification of advanced localized PC (intermediate risk versus high risk).
Safety End Points
For the MACE end point, an additional analysis was performed after the database lock (i.e., MACE incidence by MACE medical history status). For AEs, SAEs, and all-cause mortality, additional subgroup analyses of the HERO trial were found in reports identified by this systemic literature review:
concomitant RT during the study (yes versus no)
concomitant enzalutamide or docetaxel during the study (yes versus no)
clinical disease-state presentations (biochemical or clinical relapse, newly diagnosed metastatic disease, advanced localized disease).
All these subgroup analyses, conducted in the primary analysis population (cohort 1) are presented in the Results section.
Analysis Populations
The analysis sets (i.e., population for analysis) for the primary analysis and the final analysis are described in Table 11 and Table 12, respectively. In the primary analysis, the safety set was equivalent to the mITT analysis set.
Table 11: Analysis Populations of the HERO Trial — Primary Analysis
Study | Population | Definition | Application |
|---|---|---|---|
HERO trial | mITT | All randomized patients who have taken at least 1 dose of any study treatment | Efficacy end points |
PP | Members of the mITT population who do not have important protocol deviations | Sensitivity analysis for the primary efficacy end point | |
Safety | All randomized patients who have taken at least 1 dose of any study treatment | Safety analyses |
mITT = modified intention to treat; PP = per-protocol.
Sources: HERO Statistical Analysis Plan45 and Sponsor’s Summary of Clinical Evidence.77
Table 12: Analysis Populations of the HERO Trial — Final Analysis
Study | Population | Definition | Application |
|---|---|---|---|
HERO trial | mITT | All randomized patients who have taken at least 1 dose of any study treatment. This population was separated in 2 subgroups:
|
|
CRFS = castration resistance–free survival; mITT = modified intention to treat.
Sources: HERO Statistical Analysis Plan45 and Sponsor’s Summary of Clinical Evidence.77
Results
Patient Disposition
Out of 1,327 patients initially screened, 393 (29.6%) did not qualify for the study, with exclusion criteria accounting for 61.8% of failures, followed by not meeting inclusion criteria (14.0%). Other reasons included patients’ refusal to participate due to various factors, such as visit complexity, travel burdens, or uncertainty about the clinical benefits of relugolix.
The relugolix group saw a 9.5% dropout rate, while the leuprolide group had a slightly higher dropout rate of 10.3%. The reasons for discontinuation in the relugolix group were primarily AEs (3.7%), patient withdrawal (2.7%), and physician decisions (1.4%). In contrast, the leuprolide group saw a higher proportion of discontinuations for “other” reasons (4.2%), in addition to AEs (2.6%) and patient withdrawal (1.9%). Completion rates for the 48-week treatment period were comparable in both groups, with 90.2% in relugolix and 89.0% in leuprolide completing the study. The number of follow-up participants was considerably higher in the relugolix group (137) compared to the leuprolide group (47).
Table 13: Summary of Patient Disposition From Studies Included in the Systematic Review
Patient disposition | HERO | |
|---|---|---|
Relugolix | Leuprolide | |
Screened, N | 1,327 | |
Reason for screening failure, N (%) | 393 | |
Patient refused due to visit complexity and/or frequency | 15 (3.8) | |
Patient refused due to burdensome travel to the site | 2 (0.5) | |
Patient refused due to uncertainty of clinical benefits of relugolix | 5 (1.3) | |
Patient refused due to deterioration of health/increase in clinical symptoms | 3 (0.8) | |
Patient no longer wants to participate in any clinical trial | 22 (5.6) | |
Inclusion criteria not met | 55 (14.0) | |
Exclusion criteria met | 243 (61.8) | |
Principal investigator anticipates patient will be noncompliant | 3 (0.8) | |
Other | 44 (11.2) | |
Not available | 1 (0.3) | |
Randomized, N | 624 | 310 |
Discontinued from study, N (%) | 59 (9.5) | 32 (10.3) |
Reason for discontinuation, N (%) | ||
Adverse event | 23 (3.7) | 8 (2.6) |
Protocol violation | 0 | 1 (0.3) |
Lost to follow-up | 2 (0.3) | 1 (0.3) |
Withdrawal by patient | 17 (2.7) | 6 (1.9) |
Physician decision | 9 (1.4) | 3 (1.0) |
Other | 8 (1.3) | 13 (4.2) |
Treated participants (mITT), N (%) | 622 (99.7) | 308 (99.4) |
Participants who completed 48 weeks of treatment, N (%) | 563 (90.2) | 276 (89.0) |
Safety, N (%) | 622 (99.7) | 308 (99.4) |
Follow-up participants,a N | 137 | 47 |
mITT = modified intention to treat.
aThis subset of patients corresponds with those who were monitored for testosterone recovery levels.
Sources: Clinical Study Report,1 Shore et al. (2020),40 and the sponsor’s Summary of Clinical Evidence.77
Baseline Characteristics
Baseline demographic characteristics of the participants were generally similar between the relugolix and the leuprolide groups. Both treatment groups exhibited a similar age distribution, with 71.4% of participants aged 75 years or younger and 28.6% older than 75 years in each group. The mean age was similar, with relugolix participants averaging 71.2 years (standard deviation [SD] = 7.75) and leuprolide participants 71.0 years (SD = 8.03). Ethnicity and race distributions showed slight numerical differences, with Hispanic or Latino participants comprising 8.4% of the relugolix group and 10.1% of the leuprolide group. The proportion of white participants was 69.8% in the relugolix group contrasted with 65.6% of the leuprolide group.
Clinical characteristics of the participants were similar across the 2 treatment arms. Regarding the clinical disease state presentation, 49.7% of patients in the relugolix group and 51.3% in the leuprolide group had evidence of biochemical or clinical relapse following local primary intervention with curative intent. The distribution of disease stages at study entry was comparable: metastatic cases constituted 31.8% of the relugolix group and 31.5% of the leuprolide group, while locally advanced cases accounted for 30.4% and 30.8%, respectively. The Gleason scores, which are critical in assessing the severity of PC, were similar across the groups, with slightly more patients in the relugolix group having higher scores (8 to 10).
A numerically higher number of patients had a history of prior ADT therapy in the relugolix group (13.0% in the relugolix group and 9.7% in the leuprolide group). Prior experience with RT was similar, with 30.5% of patients in the relugolix group and 29.9% in the leuprolide group having had prior RT. A history of MACEs was reported in 13.5% of patients in the relugolix group and 14.6% in the leuprolide group.
The mean PSA levels were higher in the relugolix group than in the leuprolide group, with large SDs reported. Specifically, PSA levels were 104.2 ng/mL (SD = 416.0) in the relugolix group and 68.6 ng/mL (SD = 244.0) in the leuprolide group. The median values were 11.7 ng/mL for relugolix and 9.4 ng/mL for leuprolide. Similarly, testosterone levels were numerically higher in the relugolix group compared with the leuprolide group. Specifically, the mean testosterone levels were 436.1 ng/dL (SD = 159.0) for relugolix and 410.0 ng/dL (SD = 149.1) for leuprolide. FSH levels were comparable between the 2 groups, with the mean FSH levels at 16.3 IU/L (SD = 12.8) in the relugolix group and 16.7 IU/L (SD = 14.5) in the leuprolide group.
The baseline characteristics outlined in Table 14 are limited to those that are most relevant to this review or were assumed to affect the outcomes or interpretation of the study results.
Table 14: Summary of Baseline Characteristics From Studies Included in the Systematic Review
Characteristic | HERO | |
|---|---|---|
Relugolix (N = 622) | Leuprolide (N = 308) | |
Age category (years) | ||
≤ 75 | 444 (71.4) | 220 (71.4) |
> 75 | 178 (28.6) | 88 (28.6) |
Age | ||
Mean (SD) | 71.2 (7.75) | 71.0 (8.03) |
Median | 72.0 | 71.0 |
Minimum to maximum | 48 to 91 | 47 to 97 |
Ethnicity, n (%) | ||
Hispanic or Latino | 52 (8.4) | 31 (10.1) |
Not Hispanic or Latino | 558 (89.7) | 269 (87.3) |
Unknown | 12 (1.9) | 8 (2.6) |
Race, n (%) | ||
Asian | 127 (20.4) | 71 (23.1) |
Black or African American | 30 (4.8) | 16 (5.2) |
White | 434 (69.8) | 202 (65.6) |
Other | 8 (1.3) | 7 (2.3) |
Multiple | 11 (1.8) | 4 (1.3) |
Not reported | 12 (1.9) | 8 (2.6) |
Geographic region, n (%) | ||
North America | 182 (29.3) | 87 (28.2) |
South America | 34 (5.5) | 19 (6.2) |
Europe | 247 (39.7) | 122 (39.6) |
Asia | 125 (20.1) | 70 (22.7) |
Rest of world | 34 (5.5) | 10 (3.2) |
Mean (SD) | 26.6 (3.8) | 26.2 (3.9) |
Clinical disease-state presentation, n (%) | ||
Evidence of biochemical (PSA) or clinical relapse following local primary intervention with curative intenta | 309 (49.7) | 158 (51.3) |
Newly diagnosed androgen-sensitive metastatic disease | 141 (22.7) | 70 (22.7) |
Advanced localized disease not suitable for primary surgical intervention with curative intent | 172 (27.7) | 80 (26.0) |
Intermediate risk | 114 (66.3) | 48 (60.0) |
High risk | 58 (33.7) | 32 (40.0) |
Disease stage at study entry,b n (%) | ||
Metastatic | 198 (31.8) | 97 (31.5) |
Locally advanced | 189 (30.4) | 95 (30.8) |
Localized | 178 (28.6) | 82 (26.6) |
Not classifiable | 57 (9.2) | 34 (11.0) |
Gleason score,c n (%) | ||
2 to 4 | 0 | 1 (0.3) |
5 to 6 | 98 (15.8) | 46 (14.9) |
7 | 237 (38.1) | 122 (39.6) |
8 to 10 | 267 (42.9) | 134 (43.5) |
Missing data | 20 (3.2) | 5 (1.6) |
ECOG Performance Status,d n (%) | ||
0 | 548 (88.1) | 271 (88.0) |
1 | 74 (11.9) | 36 (11.7) |
3e | 0 | 1 (0.3) |
History of prostate cancer therapies, n (%) | ||
Prior ADT | 81 (13.0) | 30 (9.7) |
Prior RT | 190 (30.5) | 92 (29.9) |
PSA (ng/mL) | ||
Mean (SD) | 104.2 (416.0) | 68.6 (244.0) |
Median | 11.7 | 9.4 |
Testosterone (ng/dL) | ||
Mean (SD) | 436.1 (159.0) | 410.0 (149.1) |
Median | 415.8 | 395.9 |
FSH (IU/L)e | ||
Mean (SD) | 16.3 (12.8) | 16.7 (14.5) |
Cardiovascular risk factors, n (%)f | 570 (91.6) | 290 (94.2) |
Lifestyle risk factorsg | 422 (67.8) | 202 (65.6) |
Cardiovascular or cerebrovascular risk factorsh | 488 (78.5) | 254 (82.5) |
History of MACEi | 84 (13.5) | 45 (14.6) |
ADT = androgen deprivation therapy; ECOG = Eastern Cooperative Oncology Group; FSH = follicle-stimulating hormone; MACE = major adverse cardiovascular event; MedDRA = Medical Dictionary for Regulatory Activities PSA = prostate-specific antigen; RT = radiation therapy; SD = standard deviation; TNM = tumour-node-metastasis.
aBiochemical relapse was defined by a rising PSA level.
bDisease stage at study entry is defined based on TNM stage at study entry, M1 as metastatic, T3/4 NX M0 or N1 M0 and any T N1 M0 as locally advanced, and T1 or T2 N0 M0 as localized.
cGleason score is determined by adding primary and secondary Gleason scores together. Gleason scores range from 2 to 10, with higher scores indicating a worse prognosis.
dECOG Performance Status ranges from 0 to 5, with higher scores reflecting greater disability. One patient in the leuprolide group had a surgical vascular procedure on his leg and was given an ECOG score of 3 at screening because of the use of crutches. By the baseline day 1 visit, the patient no longer used crutches, and his ECOG score had improved to 0.
eThe normal range of FSH values for adults is 1.5 to 12.4 IU/L.
fPatients with multiple risk factors were counted only once.
gLifestyle risk factors included tobacco smoking (current or past), heavy alcohol use, and a body mass index (the weight in kilograms divided by the square of the height in metres) of more than 30.
hCardiovascular or cerebrovascular risk factors included prespecified event terms in the MACE query and a manual search of known risk factors, including hypertension, dyslipidemia, diabetes, a history of myocardial infarction or cardiovascular disease, a history of stroke (transient ischemic attack, or cerebral hemorrhage), peripheral arterial disease, atrial fibrillation and other arrhythmias, heart-valve disease, chronic obstructive pulmonary disease, chronic kidney disease, chronic liver disease, carotid-artery stenosis or occlusion; venous thromboembolic events, and heart failure.
iSearch criteria included “myocardial infarction” (broad standardized MedDRA query) and “central nervous system hemorrhages and cerebrovascular conditions” (broad standardized MedDRA query).
Sources: Clinical Study Report1 and the sponsor’s Summary of Clinical Evidence.77
Exposure to Study Treatments
Study Treatments
Compliance was high across both groups, with more than 99% of participants being at least 80% compliant. One patient in the relugolix group and none in the leuprolide group had a compliance rate below 80%. Overall, 231 patients (24.8%) had a compliance rate of greater than 100% (refer to footnote of Table 15).
Treatment duration was comparable between the 2 groups, with the relugolix group having an average treatment duration of 45.9 weeks (SD = 8.3), while the leuprolide group was slightly numerically higher at 46.1 weeks (SD = 7.4).
Table 15: Summary of Patient Exposure From Studies Included in the Systematic Review
Exposure and compliance | HERO | |
|---|---|---|
Relugolix (N = 622) | Leuprolide (N = 308) | |
Treatment duration (weeks)a | ||
Mean (SD) | 45.9 (8.3) | 46.1 (7.4) |
Median (minimum to maximum) | 48.0 (0.4 to 51.4) | 48.1 (1.1 to 51.6) |
Compliance,b n (%) | ||
< 80% | 1 (0.2) | 0 |
≥ 80% to ≤ 100% | 449 (72.2) | 249 (80.8) |
> 100% | 172 (27.7) | 59 (19.2) |
SD = standard deviation.
aTreatment duration in weeks is calculated as (last dose date of any of the study drug – first dose date of study drug + 1)/7. For patients in leuprolide group, the treatment period ends 12 weeks after the last injection.
bCompliance rate is calculated for relugolix group as (number of tablets taken during the study / expected number of tablets during the study) × 100 and for leuprolide group with ([cumulative dose administrated]/[treatment duration in weeks]/12)/22.5 mg or 11.25 mg per treatment on day 1) × 100.
Sources: Clinical Study Report1 and the sponsor’s Summary of Clinical Evidence.77
Concomitant Medications and Co-interventions
Table 16 below summarizes relevant concomitant medications used during the treatment phase of the HERO study.
Patients were allowed to receive medications and therapeutic procedures, in addition to study interventions, unless they were listed as prohibited medications or alternative treatments. Medications taken by the patient and therapeutic procedures used by the patient from the first dose of the study drug until 30 days after the last dose of relugolix or 12 weeks plus 30 days after the last leuprolide injection were recorded as concomitant medications.
Overall, 97.3% of participants reported taking at least 1 concomitant medication, and the proportion of patients who reported taking at least 1 concomitant medication was comparable between treatment groups (96.6% in the relugolix group and 98.7% in the leuprolide group). The 3 medications most commonly reported were drugs acting on the renin-angiotensin system (50.0%), lipid-modifying drugs (42.9%), and antithrombotic drugs (39.2%).
Overall, 15 patients (1.6%) took at least 1 therapy known to suppress testosterone during treatment, and all of these patients were in the leuprolide group (4.9%). Of these, 12 patients received leuprorelin. All patients who started therapies known to suppress testosterone were censored at the time the therapies were initiated or at the time of last testosterone assessment before initiation of the therapies. The majority of these therapies (14 of the 15 patients) were ADT given as the subsequent therapy to continue their ADT regimen. However, given variations in week 49, day 1 visit scheduling, these therapies were given less than 12 weeks after the last leuprolide injection on study, and they were therefore captured as concomitant medications. One patient (in the leuprolide group) received leuprorelin before week 49, which was a protocol deviation (data on file).
Antiandrogen therapies were permitted in the first 4 weeks to manage clinical flare-ups, as GnRH agonists cause an initial testosterone surge that may result in a clinical flare-up of symptoms. As expected, the proportion of patients who received antiandrogen therapies (bicalutamide or flutamide) during the first 4 weeks of study treatment was higher in the leuprolide group (23.4%) compared with the relugolix group (0.2%). Some concomitant therapies, including bicalutamide, enzalutamide, docetaxel among others, were identified as possibly affecting PSA levels. More patients in the leuprolide group (29.9%) took at least 1 of these therapies compared with the relugolix group (5.3%).
Patients with disease progression (i.e., noncastration levels of testosterone or PSA failure) during the treatment period had the option to undergo RT, which was allowed to be directed at the prostate bed according to protocol after 2 months of study initiation. The majority of patients who received RT after study treatment initiation (10.4% overall) were in the primary treatment setting. Patients who required systemic antineoplastic therapies for malignancies other than PC were discontinued from the study.
Table 16: Concomitant Medications Used During the Study (mITT Population) — Primary Analysis
Concomitant medication and therapies | HERO | |
|---|---|---|
Relugolix (N = 622) | Leuprolide (N = 308) | |
Participants using concomitant medication during treatment period, % | ||
Any concomitant medications | 96.6 | 98.7 |
Renin-angiotensin system–acting drugs | 48.6 | 52.9 |
Lipid-modifying drugs | 43.1 | 42.5 |
Antithrombotic drugs | 37.0 | 43.8 |
Endocrine therapy | 4.5 | 35.1 |
Antineoplastic drugs | 2.3 | 1.9 |
Antiandrogen taken during the first 4 weeks, % | 0.2 | 23.4 |
Bicalutamide | 0.2 | 23.1 |
Flutamide | 0 | 0.3 |
Apalutamide | 0 | 0 |
Enzalutamide | 0 | 0 |
Nilutamide | 0 | 0 |
Testosterone blockers taken during treatment period, % | 0 | 4.9 |
Leuprolide | 0 | 3.9 |
Goserelin | 0 | 0.6 |
Degarelix | 0 | 0.3 |
Abarelix | 0 | 0 |
Histrelin | 0 | 0 |
Triptorelin | 0 | 0 |
Therapies that can affect or alter PSA during treatment period, % | 5.3 | 29.9 |
Bicalutamide | 0.8 | 26.6 |
Enzalutamide | 2.7 | 1.9 |
Docetaxel | 1.3 | 1.6 |
Sipuleucel-T | 0.2 | 0.6 |
Cisplatin | 0.3 | 0 |
Etoposide | 0.3 | 0 |
Fluorouracil | 0.3 | 0 |
Doxorubicin | 0.2 | 0 |
Flutamide | 0 | 0.3 |
Ifosfamide | 0.2 | 0 |
Paclitaxel | 0.2 | 0 |
Current radiotherapies during treatment period, % | 15.9 | 18.8 |
Setting in systemic therapy | ||
Primary treatment | 9.6 | 12.0 |
Salvage | 2.4 | 3.2 |
Palliative | 3.9 | 3.2 |
Unknown | 0.2 | 0.3 |
mITT = modified intent-to-treat; PSA = prostate-specific antigen.
Source: Clinical Study Report.1
Efficacy
Summary of Efficacy Outcomes
The summary of efficacy outcomes from HERO trial is presented in Table 17 for the primary analysis.
Table 17: Key Efficacy Outcomes During Treatment and Follow-up Phases (mITT) — Primary Analysis
Outcomes | HERO | |
|---|---|---|
Relugolix (N = 622) | Leuprolide (N = 308) | |
Primary end point | ||
Sustained castration rate (< 50 ng/dL) from day 29 to day 337 | ||
Response rate % (95% CI)a | 96.7 (94.9 to 97.9) | 88.8 (84.6 to 91.8) |
Difference from leuprolide (95% CI)b | 7.9 (4.1 to 11.8) | — |
P valuec | < 0.0001 | — |
HR to leuprolide (95% CI)d | 0.2621 (0.1489 to 0.4613) | — |
Other secondary end points | ||
Cumulative probability of testosterone suppression to < 20 ng/dL at day 15 (profound castration) | ||
Response rate % (95% CI)a | 78.4 (75.1 to 81.5) | 0.98 (0.3 to 3.0) |
Difference from leuprolide (95% CI)b | 77.4 (74.0 to 80.8) | — |
P valuec | < 0.0001 | — |
Cumulative probability of testosterone suppression to < 20 ng/dL from day 29 to day 337 (profound castration) | ||
Response rate % (95% CI)a | 81.6 (78.1 to 84.5) | 68.6 (63.0 to 73.5) |
Difference from leuprolide (95% CI)b | 13.0 (6.9 to 19.1) | — |
CI = confidence interval; HR = hazard ratio; mITT = modified intention to treat.
a95% CI in each treatment group was calculated by log-log transformation of survival function in each treatment group.
b95% CI for treatment difference was calculated by linear transformation of the difference in survival function.
cUnstratified test statistics via log-log transformation of the difference in survival function at a fixed time point was performed.
dHazard ratio in a comparison of relugolix to leuprolide was performed using a Cox proportional hazards model.
Sources: Clinical Study Report1 and Shore et al. (2020).40
Sustained Castration Rate (Testosterone Level Below 50 ng/dL) ¾ Primary End Point
The results for the sustained castration rate (testosterone level < 50 ng/dL) for the overall population and subgroups are provided in Table 18, with an overview of the Kaplan-Meier analysis presented in Figure 1.
Overall Population
The proportion of patients who achieved sustained testosterone suppression was 96.7% (95% CI, 94.9 to 97.9) in the relugolix treatment group compared with 88.8% (95% CI, 84.6 to 91.8) in the leuprolide group, with a mean difference between the relugolix and leuprolide treatment groups of 7.9% (95% CI, 4.1% to 11.8%). These results demonstrated the noninferiority of relugolix to leuprolide (the lower bound of the 95% CI for the difference between groups was greater than the prespecified noninferiority margin of −10%), with P < 0.0001), and the statistical superiority of relugolix compared with leuprolide (lower bound of the 95% CI > 0, with P < 0.0001).
Subgroup Analyses
Subgroups of interest included in the statistical analysis plan were the presence of metastatic disease (yes or no), race (white, Black or African American, and other), ethnicity (Hispanic or Latino, not Hispanic or Latino), clinical disease-state presentation (experiencing biochemical or clinical relapse, having newly diagnosed androgen-sensitive metastatic disease, or advanced localized disease unlikely to be cured), Gleason score at entry (< 8 or ≥ 8), and baseline PSA level (< 20 ng/mL or ≥ 20 ng/mL). Except for the subgroup of Black or African American (N = 46) and Hispanic or Latino patients (N = 83), response rates and differences in sustained castration rate between relugolix and leuprolide were consistent with the overall difference observed in the mITT population (Figure 2 and Figure 3). Furthermore, the noninferiority to leuprolide was also met except for the comparatively smaller subgroup of Black or African American patients (Figure 3).
Additional subgroups of interest were reported for the primary end point and included patients receiving or not receiving concomitant enzalutamide or docetaxel during the study, patients receiving or not receiving concomitant RT during the study, and, among patients with advanced localized PC, patients at intermediate or high risk. The cumulative sustained castration rates through 48 weeks of treatment were 95.8% for men receiving relugolix and concomitant enzalutamide or docetaxel,48 and 96.9% for those receiving relugolix and concomitant RT.49 As for leuprolide, the corresponding rates were 90.9% (with enzalutamide or docetaxel)48 and 96.4% (with RT).49 These results are in line with the rates observed in the mITT population. In the subgroups of patients with advanced localized PC who were at intermediate or high risk, the primary outcome results also aligned with those of the overall mITT population. The rates were 97.3% for patients at intermediate risk and 96.5% for those at high risk in the relugolix group. In the leuprolide group, the rates were 97.9%% for patients at intermediate risk and 90.5% for those at high risk.
Table 18: Sustained Castration Rate (Testosterone Level Below 50 ng/dL) During Treatment Phase (mITT Population and Subgroup Analyses) — Primary End Point — Primary Analysis
Primary end point | HERO | |
|---|---|---|
Relugolix | Leuprolide | |
Sustained castration rate from day 29 to day 337 (mITT population) — evaluation criterion 1 and 2 | ||
Number of patients contributing to the analysis | 622 | 308 |
Response rate, % (95% CI)a | 96.7 (94.9 to 97.9) | 88.8 (84.6 to 91.8) |
Difference from leuprolide (95% CI)b | 7.9 (4.1 to 11.8) | — |
P valuec | < 0.0001 | — |
HR to leuprolide (95% CI)d | 0.2621 (0.1489 to 0.4613) | — |
Sustained castration rate from day 29 to day 337 (intermediate-risk patients) — subgroup analysis | ||
Number of patients contributing to the analysis (%) | 114 | 48 |
Response rate, % (95% CI)a | 97.3 (91.8 to 99.1) | 97.9 (86.1, 99.7) |
Difference from leuprolide (95% CI)b | −0.6 (−5.7 to 4.4) | — |
P valuec | 0.8127 | — |
HR to leuprolide (95% CI)d | 1.2732 (0.1320 to 12.2787) | — |
Sustained castration rate from day 29 to day 337 (high-risk patients) — subgroup analysis | ||
Number of patients contributing to the analysis (%) | 58 | 32 |
Response rate, % (95% CI)a | 96.5 (86.7 to 99.1) | 90.5 (73.4, 96.8) |
Difference from leuprolide (95% CI)b | 6.0 (−5.3 to 17.3) | — |
P valuec | 0.2613 | — |
HR to leuprolide (95% CI)d | 0.3394 (0.0567 to 2.0327) | — |
Sustained castration rate from day 29 to day 337 (patients who received concomitant enzalutamide or docetaxel) — subgroup analysis | ||
Number of patients contributing to the analysis, % | 24 | 11 |
Response rate, % | 95.8 | 90.9 |
Difference from leuprolide | NR | — |
Sustained castration rate from day 29 to day 337 (patients who did not receive concomitant enzalutamide or docetaxel) — subgroup analysis | ||
Number of patients contributing to the analysis, % | 598 | 297 |
Response rate, % | 96.7 | 88.7 |
Difference from leuprolide | NR | — |
Sustained castration rate from day 29 to day 337 (patients who received concomitant radiation therapy) — subgroup analysis | ||
Number of patients contributing to the analysis, % | 99 | 58 |
Response rate, % | 96.9 | 96.4 |
Difference from leuprolide | NR | — |
Sustained castration rate from day 29 to day 337 (patients who did not receive concomitant radiation therapy) — subgroup analysis | ||
Number of patients contributing to the analysis, % | 523 | 250 |
Response rate, % | 96.7 | 87.0 |
Difference from leuprolide | NR | — |
CI = confidence interval; HR = hazard ratio; mITT = modified intention to treat; NR = not reported.
a95% CI in each treatment group was calculated by log-log transformation of survival function in each treatment group.
b95% CI for treatment difference was calculated by linear transformation of the difference in survival function.
cUnstratified test statistics via log-log transformation of the difference in survival function at a fixed time point was performed.
dHazard ratio in comparison of relugolix to leuprolide was performed using a Cox proportional hazards model.
Sources: Clinical Study Report,1 George et al. (2023),48 George et al. (2021) conference proceeding,49 and HERO Subgroup Analyses.1
Figure 1: Kaplan-Meier Survival Curve of Sustained Castration Rate (Below 50 ng/dL) — mITT Population
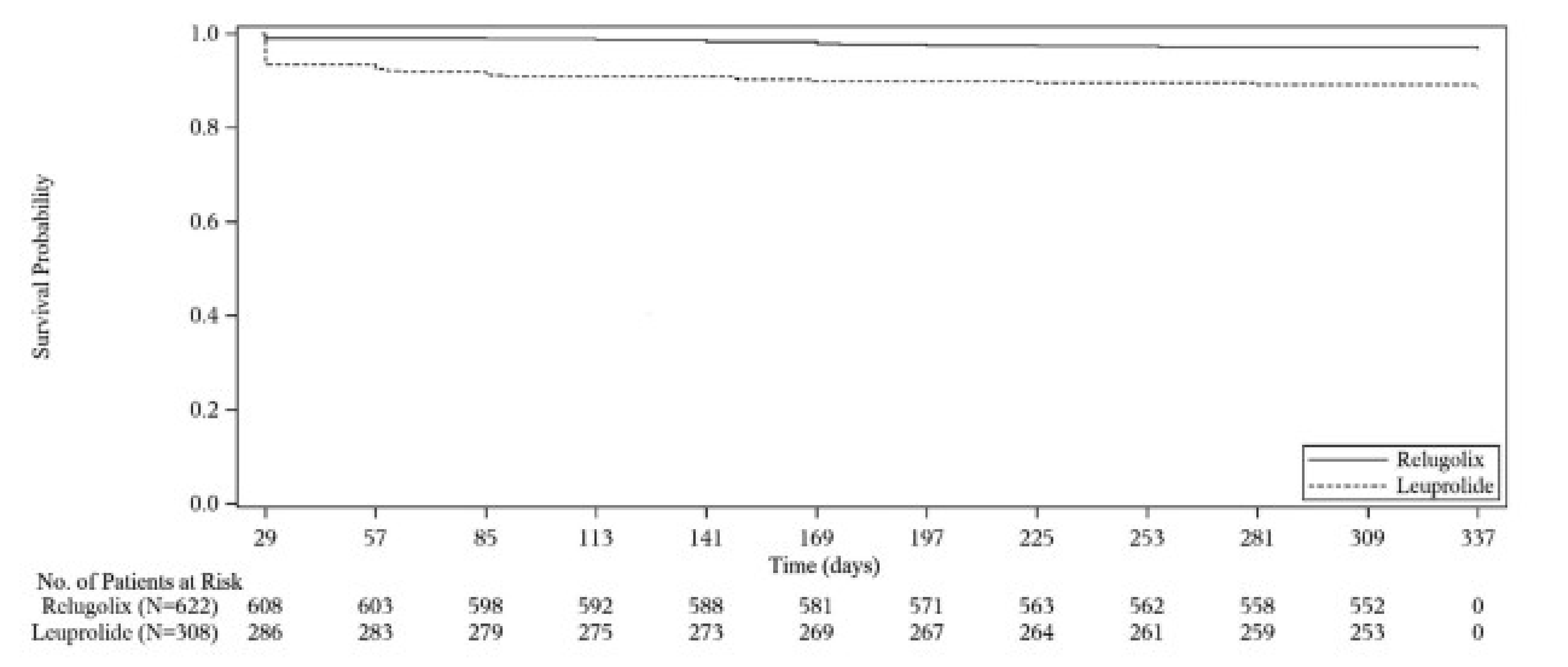
mITT = modified intention to treat.
Source: Clinical Study Report.1
Figure 2: Subgroup Analysis for Sustained Castration Rate (Below 50 ng/dL) — Evaluation Criterion 1 — mITT Population
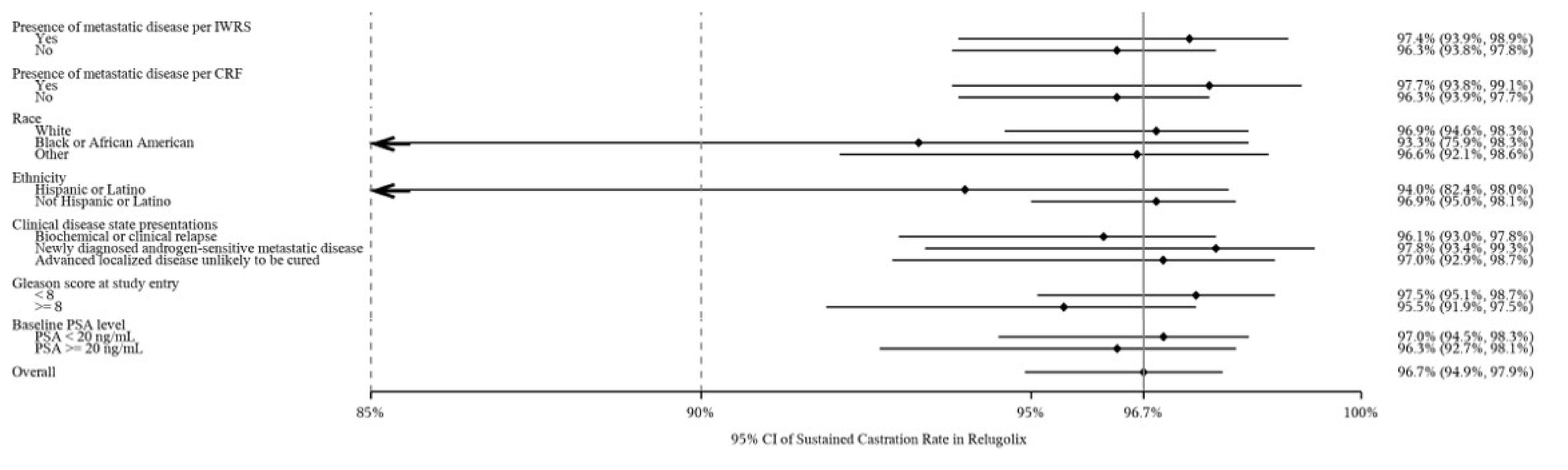
CI = confidence interval; CRF = case report form; IWRS = interactive web recognition system; mITT = modified intention to treat; PSA = prostate-specific antigen.
Notes: ← indicates 95% CI of sustained castration rate is extending to 75.9% in Black or African American and to 82.4% in Hispanic or Latino. Evaluation criterion 1 was used to determine whether the sustained castration rate (defined as the cumulative probability of testosterone suppression to < 50 ng/dL) while on study drug from week 5, day 1 through week 49, day 1 for relugolix is ≥ 90%. The lower bound of the 95% CI for the cumulative probability of sustained testosterone suppression in the relugolix treatment group was calculated and must have been at least 90% for this criterion to have been met.
Source: Clinical Study Report.1
Figure 3: Subgroup Analysis for Sustained Castration Rate (Below 50 ng/dL) — Evaluation Criterion 2) — mITT Population)
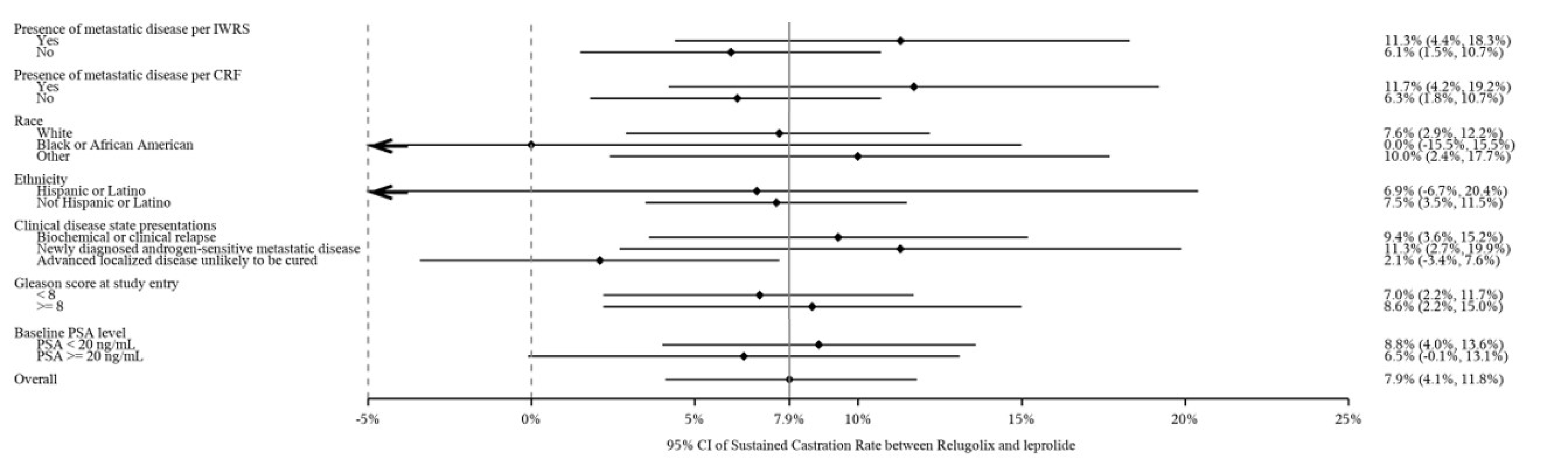
CI = confidence interval; CRF = case report form; IWRS = interactive web recognition system; mITT = modified intention to treat; PSA = prostate-specific antigen.
Notes: ← indicates 95% CI of sustained castration rate is extending to −15.5% in Black or African American and to −6.7% in Hispanic or Latino. Evaluation criterion 2 was used to establish the noninferiority of relugolix compared to leuprolide every 3-month depot injection as assessed by the cumulative probability of sustained testosterone suppression. The lower bound of the 95% CI for the difference in the cumulative probability of sustained testosterone suppression between the 2 treatment groups was calculated and must have been greater than or equal to the noninferiority margin of −10% for this criterion to have been met.
Source: Clinical Study Report.1
Sensitivity Analyses
Four sensitivity analyses were conducted on the primary end point. Results are presented in Table 19.
Sensitivity analysis 1: per-protocol population
Sensitivity analysis 2: excluding patients who had received concomitant medications or herbal supplements that could possibly affect testosterone levels during study treatment
Sensitivity analysis 3: excluding patients who had missed 2 or more consecutive visits after day 29 or discontinued early as having an event
Sensitivity analysis 4: censoring patients who had not reached castration levels at day 29.
Findings of all sensitivity analyses were consistent with those of the main analysis of the primary end point. The differences between relugolix and leuprolide in the sustained castration rate from day 29 through 48 weeks were 6.8 (95% CI, 2.9 to 10.7), 7.3% (95% CI, 2.9% to 11.7%), 5.0 (95% CI, 0.1 to 9.8), and 3.3% (95% CI, 0.2 to 6.4) for sensitivity analyses 1, 2, 3, and 4, respectively. All sensitivity analyses except analysis 3 successfully met both evaluation criteria 1 and 2 for the primary end point, demonstrating both noninferiority and superiority to leuprolide. In sensitivity analysis 3, although the lower bound of the 95% CI for the relugolix group was less than 90% (evaluation criteria 1), the between-group difference demonstrated not only inferiority but also statistical superiority to leuprolide (P value = 0.0368).
Table 19: Sustained Castration Rate (Testosterone Below 50 ng/dL) During Treatment Phase (mITT Population and Sensitivity Analyses) — Primary End Point — Primary Analysis
Primary end point | HERO | |
|---|---|---|
Relugolix | Leuprolide | |
Sustained castration rate from day 29 to day 337 (PP population) — sensitivity analysis 1 | ||
Number of patients in the PP population | 578 | 286 |
Cumulative number of patients with testosterone ≥ 50 ng/dL at day 337 | 19 | 29 |
Patients censored at day 337 | 559 | 257 |
Response rate, % (95% CI)a | 96.5 (94.5 to 97.7) | 89.7 (85.4 to 92.7) |
Difference from leuprolide (95% CI)b | 6.8 (2.9 to 10.7) | — |
P valuec | 0.0002 | — |
HR to leuprolide (95% CI)d | 0.3092 (0.1727 to 0.5535) | — |
Sustained castration rate from day 29 to day 337 (mITT population excluding patients who had received concomitant medications and herbal supplements that could possibly affect testosterone levels) — sensitivity analysis 2 | ||
Number of patients contributing to the analysis at day 1 | 591 | 214 |
Cumulative number of patients with testosterone ≥ 50 ng/dL at Day 337 | 17 | 22 |
Patients censored at day 337 | 574 | 192 |
Response rate, % (95% CI)a | 96.9 (95.0 to 98.1) | 89.6 (84.6 to 93.0) |
Difference from leuprolide (95% CI)b | 7.3 (2.9 to 11.7) | — |
P valuec | 0.0001 | — |
HR to leuprolide (95% CI)d | 0.2664 (0.1409 to 0.5035) | — |
Sustained castration rate from day 29 to day 337 (mITT population excluding patients who had missed 2 or more consecutive visits after day 29 or discontinued early as having an event) — sensitivity analysis 3 | ||
Cumulative number of patients with testosterone ≥ 50 ng/dL at day 337 | 69 | 50 |
Patients censored at day 337 | 553 | 258 |
Response rate, % (95% CI)a | 88.6 (85.8 to 90.9) | 83.7 (79.0 to 87.4) |
Difference from leuprolide (95% CI)b | 5.0 (0.1 to 9.8) | — |
P valuec | 0.0368 | — |
HR to leuprolide (95% CI)d | 0.6461 (0.4476 to 0.9326) | — |
Sustained castration rate from day 29 to day 337 (mITT population excluding patients who had not reached castrate levels of testosterone at day 29) — sensitivity analysis 4 | ||
Cumulative number of patients with testosterone ≥ 50 ng/dL at day 337 | 15 | 17 |
Patients censored at day 337 | 607 | 291 |
Response rate, % (95% CI)a | 97.3 (95.6 to 98.4) | 94.0 (90.5 to 96.2) |
Difference from leuprolide (95% CI)b | 3.3 (0.2, 6.4) | — |
P valuec | 0.0202 | — |
HR to leuprolide (95% CI)d | 0.4124 (0.2058 to 0.8263) | — |
CI = confidence interval; HR = hazard ratio; mITT = modified intention to treat; NR = not reported; PP = per-protocol.
a95% CI in each treatment group was calculated by log-log transformation of survival function in each treatment group.
b95% CI for treatment difference was calculated by linear transformation of the difference in survival function.
cUnstratified test statistics via log-log transformation of the difference in survival function at a fixed time point was performed.
dHazard ratio in comparison of relugolix to leuprolide was performed using a Cox proportional hazards model.
Source: Clinical Study Report.1
Cumulative Probability of Profound Castration rate (Testosterone Level Below 20 ng/dL) — Key Secondary End Point
The results for the cumulative incidence of time to profound castration (testosterone < 20 ng/dL) and Kaplan-Meier estimates are presented in Table 20 and Figure 4, respectively.
A shorter time to achieve castration in the relugolix group compared to the leuprolide group was also noticeable for profound castration levels of testosterone (< 20 ng/dL). The median time to profound castration was 15 days in the relugolix group compared with 29 days in the leuprolide group. At day 15, the difference in the number of subjects achieving profound castration was even more pronounced in the relugolix group compared with the leuprolide group (78.38% versus 0.98%), with a statistically significant difference of 77.41% (95% CI, 73.98% to 80.83%; P < 0.0001).
Table 20: Cumulative Probability of Profound Castration Rate (Testosterone Level Below 20 ng/dL) During Treatment Phase (mITT Population) — Key Secondary End Point — Primary Analysis
Profound castration rate and probability | HERO | |
|---|---|---|
Relugolix N = 622 | Leuprolide N = 308 | |
Time to initial profound castration in daysa | ||
Events, n (%) | 612 (98.4) | 302 (98.1) |
Censored events, n (%) | 10 (1.6) | 6 (1.9) |
Median (95% CI)b | 15.0 (NE to NE) | 29.0 (NE to NE) |
Profound castration rate (%) at day 15 (key secondary end point) | ||
Rate (95% CI)b | 78.38 (75.06 to 81.53) | 0.98 (0.32 to 3.00) |
Difference from leuprolide, % (95% CI)c | 77.41 (73.98 to 80.83) | — |
P valued | < 0.0001 | — |
CI = confidence interval; mITT = modified intention to treat; NE = not estimable.
aTime to initial profound castration was defined as the time from the date of first dose to the date of initial testosterone suppression to less than 20 ng/dL.
b95% CI in each treatment group is calculated by log-log transformation of survival function in each treatment group.
c95% CI of treatment difference is calculated by linear transformation of the difference in survival function.
dUnstratified test statistics via log-log transformation of the difference in survival function at a fixed time point was performed.
Source: Clinical Study Report.1
Figure 4: Cumulative Incidence of Time to Initial Profound Castration (Testosterone Level Below 20 ng/dL) — mITT Population
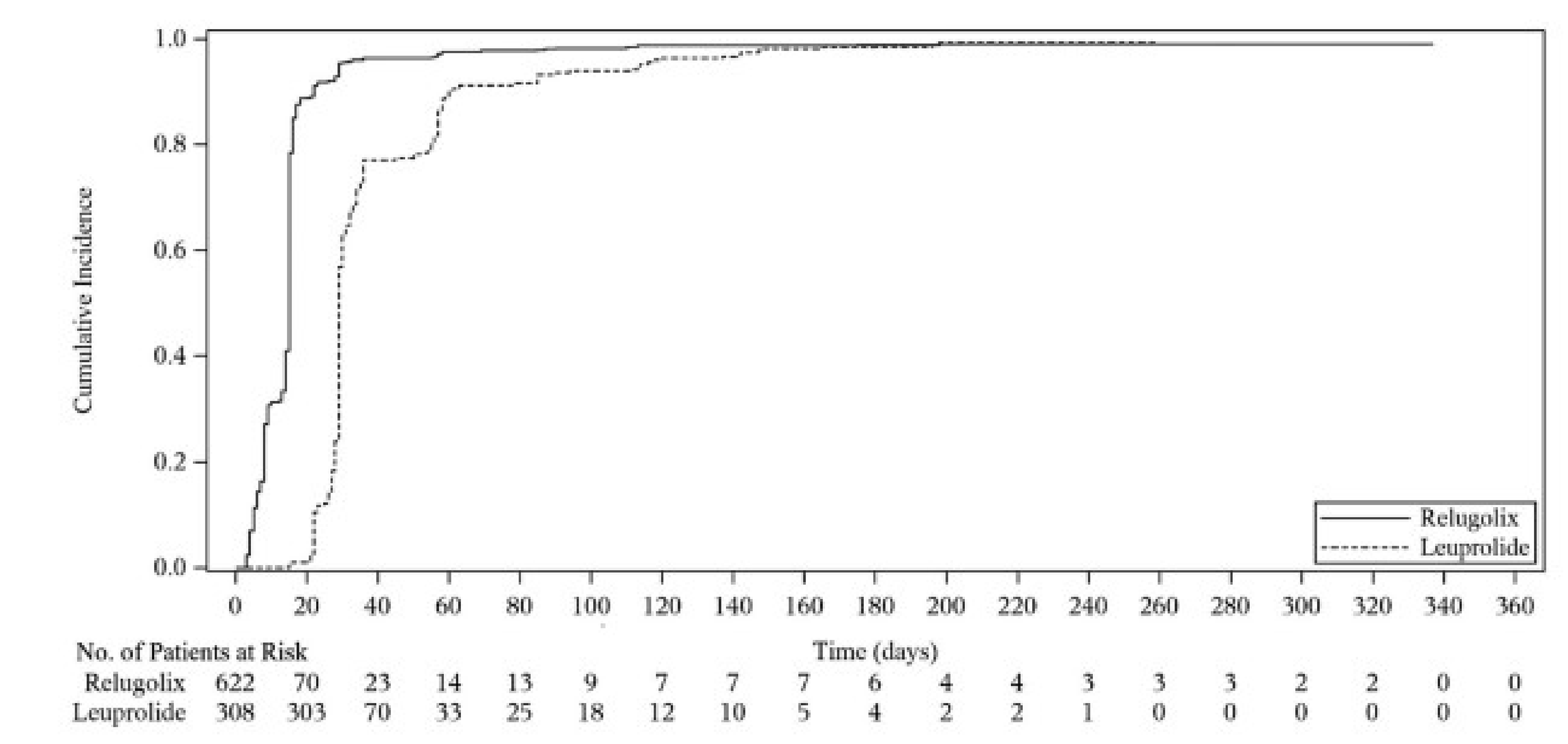
mITT = modified intent-to-treat.
Source: Clinical Study Report.1
Cumulative Probability of Profound Castration Rate (Testosterone Level Below 20 ng/dL) — Other Secondary End Points
An overview of the Kaplan-Meier Analysis for cumulative profound castration rate is presented in Figure 5, and the cumulative probability analyses are presented in Table 21.
Treatment with relugolix resulted in a higher rate of patients achieving and maintaining profound castration (81.6%; 95% CI, 78.1% to 84.5%) compared with the leuprolide group (68.6%; 95% CI, 63.0% to 73.5%) from day 29 through 48 weeks, with a difference between groups of 13.0%.
Table 21: Cumulative Probability of Profound Castration Rate (Testosterone Level Below 20 ng/dL) During Treatment Phase (mITT Population) — Other Secondary End Points — Primary Analysis
Profound castration rate and probability | HERO | |
|---|---|---|
Relugolix (N = 622) | Leuprolide (N = 308) | |
Profound castration rate (testosterone < 20 ng/dL) from day 29 to day 337 (other secondary end point) | ||
Response rate, % at day 337 (95% CI)a | 81.6 (78.1 to 84.5) | 68.6 (63.0 to73.5) |
Difference from leuprolide (95% CI)b | 13.0 (6.9 to 19.1) | — |
CI = confidence interval; mITT = modified intention to treat.
a95% CI in each treatment group was calculated by log-log transformation of survival function in each treatment group.
b95% CI for treatment difference was calculated by linear transformation of the difference in survival function.
Source: Clinical Study Report.1
Figure 5: Kaplan-Meier Survival Curve of Cumulative Profound Castration Rate (Testosterone Level Below 20 ng/dL) — mITT Population
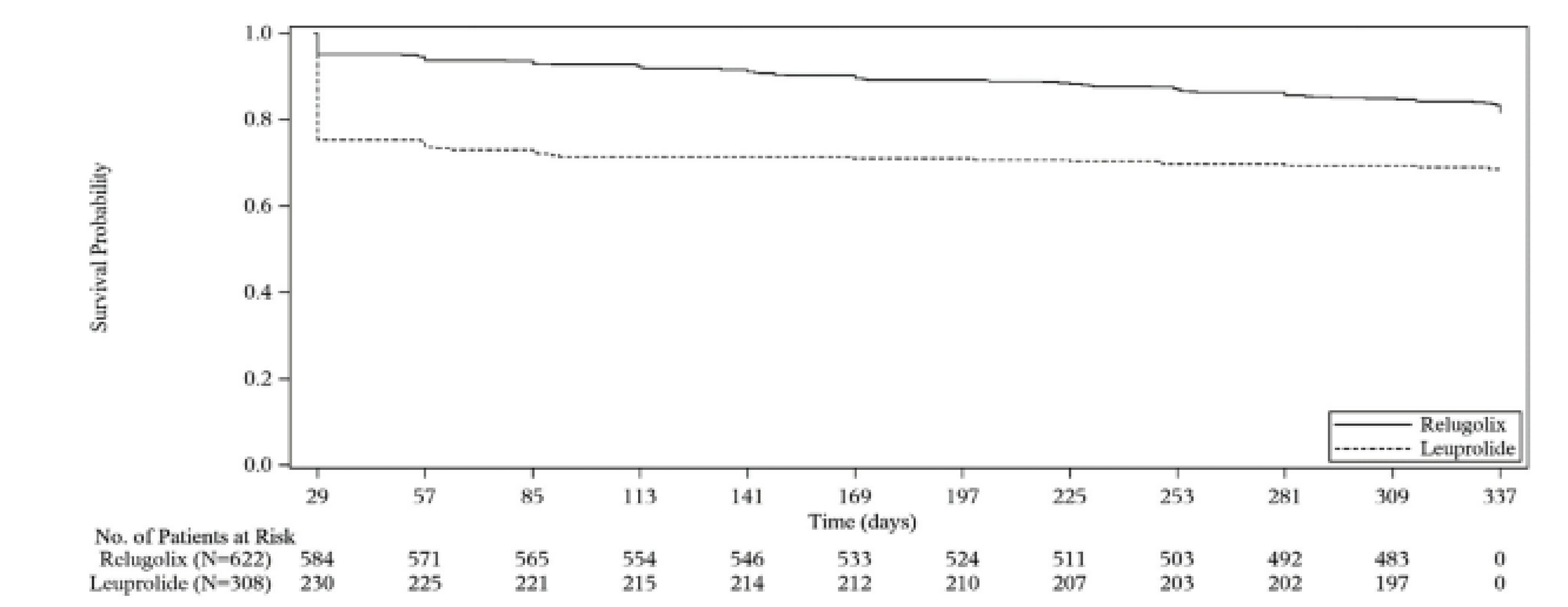
mITT = modified intention to treat.
Source: Clinical Study Report.1
Harms
Overview of Safety
Key harms data are presented in Adverse events classified as vasomotor symptoms occurred in 56.1% of patients in the relugolix group and 54.9% of patients in the leuprolide group. The most common AEs categorized as vasomotor symptoms were hot flashes (54.3% in the relugolix group and 51.6% in the leuprolide group) and fatigue (2.4% in the relugolix group and 5.2% in the leuprolide group). Hepatic transaminase elevations were reported for a higher proportion of patients in the relugolix group compared with the leuprolide group (7.6% versus 5.5%, respectively). The incidence of adverse cardiovascular events, and MACEs in particular, was higher in the leuprolide group (6.2%) compared with the relugolix group (2.9%). Loss of bone mineral density was reported in 3.2% of patients in the relugolix group and 3.9% in the leuprolide group.
Levels of ALT and/or AST at least 3 times the ULN were reported in a similar proportion of patients in both groups: 1.4% in the relugolix group and 1.3% in the leuprolide group. No patients had bilirubin levels more than twice the ULN in either group. In addition, no patients in either the relugolix or leuprolide group had ALT or AST levels 3 times the ULN or higher, with concurrent total bilirubin greater than twice the ULN with or without alkaline phosphatase less than twice the ULN (the definition of Hy’s law).
The overall incidence of AEs of any grade was comparable between the relugolix and leuprolide groups (92.9% versus 93.5%). The most common AE in both groups was hot flushes (54.3% in the relugolix group and 51.6% in the leuprolide group). Constipation and diarrhea were reported for a higher proportion of patients in the relugolix group (12.2% each) than in the leuprolide group (9.7% and 6.8%, respectively). All constipation and diarrhea AEs were mild or moderate (grade 1 or grade 2) in severity. There were no SAEs of constipation or diarrhea. One patient in the relugolix group (patient 206705) was withdrawn from the study due to a nonserious grade 2 AE of constipation; the patient also reported nonserious AEs associated with stomach cramps, taste disturbance, and smell disturbance that led to withdrawal of the study drug. No patients were withdrawn due to an AE of diarrhea. The median duration of constipation was shorter in the relugolix group compared with the leuprolide group (66.5 and 92.5 days, respectively). The median duration of diarrhea in both groups was short, but the median duration was longer in the relugolix group than in the leuprolide group (9.0 and 3.0 days, respectively).
Adverse Events
Treatment-emergent AEs were reported by 578 relugolix-treated subjects (92.9%) and 288 leuprolide-treated subjects (93.5%).
Serious Adverse Events
Reports of SAEs were made for 76 relugolix-treated patients (12.2%) and 47 leuprolide-treated patients (15.3%). SAEs in the relugolix group included acute myocardial infarction (0.8%), acute kidney injury (0.6%), urinary tract infection (0.5%), and PC metastatic (0.3%). Grade 3 or 4 SAEs occurred in 9.8% of patients treated with relugolix and 11.4% of patients treated with leuprolide in the mITT population. Grade 4 SAEs in the relugolix group included endocarditis, septic shock, acute myocardial infarction, myocardial infarction, ischemic stroke, suicidal ideation, and chronic kidney disease. In the leuprolide group, grade 4 SAEs included abnormal liver function, pulmonary edema, and rhabdomyolysis.
Withdrawal Due to Adverse Events
Permanent study discontinuation due to AEs was reported in 23 patients, including 22 (3.5%) in the relugolix group and 1 (0.3%) in the leuprolide group. AEs that led to study-drug interruption were reported for 17 patients (2.7%) in the relugolix group and no patients in the leuprolide group. The lower number of patients who withdrew due to AEs in the leuprolide group is likely attributable to the difference in the route of administration between the study drugs. Action can be more promptly and directly implemented for relugolix (which is administered orally on a daily basis) compared to leuprolide (administered every 3 months as a depot formulation). An atrioventricular block of the second degree (in 2 patients in the relugolix group and no patients in the leuprolide group) was the only AE resulting in study-drug withdrawal reported for more than 1 patient.
Mortality
Death occurred in 7 patients (1.1%) treated with relugolix and 9 patients (2.9%) treated with leuprolide. Cardiovascular-related fatal events in the relugolix group occurred in 2 patients, both due to myocardial infarction. In the leuprolide group, 6 patients experienced such events: 1 patient had congestive cardiac failure, 4 patients experienced cardio-respiratory arrest or cardiopulmonary failure, and 1 patient experienced cerebral hemorrhage.
Notable Harms
Adverse events classified as vasomotor symptoms occurred in 56.1% of patients in the relugolix group and 54.9% of patients in the leuprolide group. The most common AEs categorized as vasomotor symptoms were hot flashes (54.3% in the relugolix group and 51.6% in the leuprolide group) and fatigue (2.4% in the relugolix group and 5.2% in the leuprolide group). Hepatic transaminase elevations were reported for a higher proportion of patients in the relugolix group compared with the leuprolide group (7.6% versus 5.5%, respectively). The incidence of adverse cardiovascular events, and MACEs in particular, was higher in the leuprolide group (6.2%) compared with the relugolix group (2.9%). Loss of bone mineral density was reported in 3.2% of patients in the relugolix group and 3.9% in the leuprolide group.
Levels of ALT and/or AST at least 3 times the ULN were reported in a similar proportion of patients in both groups: 1.4% in the relugolix group and 1.3% in the leuprolide group. No patients had bilirubin levels more than twice the ULN in either group. In addition, no patients in either the relugolix or leuprolide group had ALT or AST levels 3 times the ULN or higher, with concurrent total bilirubin greater than twice the ULN with or without alkaline phosphatase less than twice the ULN (the definition of Hy’s law).
Table 22: Summary of Harms Results From Studies Included in the Systematic Review — Primary Analysis
Adverse events | HERO | |
|---|---|---|
Relugolix (N = 622) | Leuprolide (N = 308) | |
Most common adverse events (≥ 5%), n (%) | ||
Patients with ≥ 1 adverse event | 578 (92.9) | 288 (93.5) |
Patients with ≥ 1 adverse event with a severity of grade 3 or 4 | 112 (18.0) | 63 (20.5) |
Hot flushes | 338 (54.3) | 159 (51.6) |
Fatigue | 134 (21.5) | 57 (18.5) |
Constipation | 76 (12.2) | 30 (9.7) |
Diarrhea | 76 (12.2) | 21 (6.8) |
Arthralgia | 75 (12.1) | 28 (9.1) |
Nasopharyngitis | 59 (9.5) | 29 (9.4) |
Back pain | 50 (8.0) | 28 (9.1) |
Hypertension | 49 (7.9) | 36 (11.7) |
Increased weight | 49 (7.9) | 20 (6.5) |
Insomnia | 43 (6.9%) | 14 (4.5%) |
Pollakiuria | 37 (5.9%) | 20 (6.5%) |
Nausea | 36 (5.8) | 13 (4.2) |
Nocturia | 36 (5.8%) | 19 (6.2%) |
Dizziness | 35 (5.6%) | 17 (5.5%) |
Headache | 35 (5.6%) | 13 (4.2%) |
Pain in extremity | 33 (5.3%) | 19 (6.2%) |
Asthenia | 32 (5.1%) | 21 (6.8%) |
Urinary incontinence | 30 (4.8%) | 16 (5.2%) |
Hyperhidrosis | 15 (2.4%) | 16 (5.2%) |
Serious adverse events, n (%) | ||
Patients with ≥ 1 serious adverse event | 76 (12.2) | 47 (15.3) |
Patients with ≥ 1 serious adverse event with a severity of grade 3 or 4 | 61 (9.8) | 35 (11.4) |
Acute myocardial infarction | 5 (0.8) | 1 (0.3) |
Acute kidney injury | 4 (0.6) | 1 (0.3) |
Urinary tract infection | 3 (0.5) | 2 (0.6) |
Prostate cancer metastatic | 2 (0.3) | 2 (0.6) |
Anemia | 0 (0.0) | 3 (1.0) |
Cardio-respiratory arrest | 0 (0.0) | 3 (1.0) |
Cerebral hemorrhage | 0 (0.0) | 2 (0.6) |
Inguinal hernia | 0 (0.0) | 2 (0.6) |
Presyncope | 0 (0.0) | 2 (0.6) |
Syncope | 0 (0.0) | 2 (0.6) |
Transient ischemic attack | 0 (0.0) | 3 (1.0) |
Patients who were withdrawn from the study due to adverse events, n (%) | ||
Patients who discontinued permanently | 22 (3.5) | 1 (0.3) |
Patients who interrupted study drug due to adverse events, n (%) | ||
Total number of patients who interrupted study drug due to adverse events | 17 (2.7) | 0 |
Deaths, n (%) | ||
Total number of patients who died | 7 (1.1) | 9 (2.9) |
Adverse events of special interest, n (%) | ||
Adverse cardiovascular events | 24 (3.9) | 22 (7.1) |
Patients with ≥ 1 MACEa | 18 (2.9) | 19 (6.2) |
Patients with ≥ 1 MACE with a severity of grade 3 or 4 | 8 (1.3) | 4 (1.3) |
Without a history of MACEb | 15/538 (2.8) | 11/263 (4.2) |
With a history of MACEb | 3/84 (3.6) | 8/45 (17.8) |
Ischemic heart disease | 15 (2.4) | 5 (1.6) |
Vasomotor symptoms | 349 (56.1) | 169 (54.9) |
Carbohydrate and lipid metabolic effects | 53 (8.5) | 23 (7.5) |
Hypersensitivity | 44 (7.1) | 26 (8.4) |
Mood disorders | 32 (5.1) | 14 (4.5) |
Loss of bone mineral density | 20 (3.2) | 12 (3.9) |
QTc prolongation | 13 (2.1) | 6 (1.9) |
Hepatic transaminase elevations | 47 (7.6) | 17 (5.5) |
Liver function tests, n (%) | ||
Samples size, n | 619 | 306 |
AST and/or ALT > 3 × ULN | 9 (1.4) | 4 (1.3) |
AST or ALT > 3 × ULN concurrent with > 2 × ULN of total bilirubin | 0 (0.0) | 0 (0.0) |
AST or ALT > 3 × ULN concurrent with > 2 × ULN of bilirubin and ALP < 2 × ULN | 0 (0.0) | 0 (0.0) |
Total bilirubin > 2 × ULN | 0 (0.0) | 0 (0.0) |
ALP = alkaline phosphatase; ALT = alanine transaminase; AST = aspartate transaminase; MedDRA = Medical Dictionary for Regulatory Activities; QTc = corrected QT interval; ULN = upper limit normal.
aSearch criteria included “myocardial infarction” (broad standardized MedDRA query), “central nervous system hemorrhages and cerebrovascular conditions” (broad standardized MedDRA query), and deaths from any cause.
bFor the relugolix column: The percentage is calculated based on the subgroup of relugolix patients with a history of MACE (N = 84) and patients without a history of MACE (N = 538); For the leuprolide column: The percentage is calculated based on the subgroup of leuprolide patients with a history of MACE (N = 45) and patients without a history of MACE (N = 263)
Sources: Clinical Study Report1 and Shore et al. (2020).40
Critical Appraisal
Internal Validity
The HERO study, a randomized, multicentre, open-label, parallel-group, phase III trial, aimed to evaluate the efficacy and safety of relugolix compared to leuprolide in men with advanced PC requiring continuous ADT. A strength of the study is its robust randomization and stratification approach, using a 2:1 ratio and categorizing participants based on geographic region, metastatic disease, and age. Most baseline characteristics were similar between the groups, suggesting effective randomization. While the study was appropriately powered to meet study objectives, the noninferiority margin was based on previous regulatory precedence and lacked full reporting or clear evidence when estimating this margin. While the finding of superiority of relugolix over leuprolide negates concerns as to noninferiority, the EMA had concerns regarding type I error control in the superiority finding in the primary outcomes and did not endorse the view of superiority of relugolix over leuprolide for the sustained castration rate. The study gathered overall survival and patient-reported quality-of-life outcomes. However, due to the slow progressive nature of the disease and the relatively short duration of the study, these outcomes may not provide sufficient information for a reimbursement review of relugolix. The study followed a statistical analysis approach that is appropriate for the objectives outlined in the protocol. Type I error was controlled through hierarchical testing of the primary and key secondary outcomes. The handling of missing data used a conservative approach. A number of sensitivity analyses were conducted for the primary outcome that supported its robustness.
The study’s open-label design may have introduced bias in favour of the intervention. However, as most of the reported outcomes are objective in nature, this limitation is unlikely to exert a considerable effect, and the relatively large variability suggests that such a difference may not be a meaningful limitation. Also noted were the higher proportion of patients in the leuprolide group who received more therapies that can affect or alter PSA during treatment period, specifically bicalutamide. This may have biased the results in favour of the comparator.
External Validity
A number of limitations to the HERO trial may affect its external validity. In addition to patients with metastasis and patients with relapse, the study included patients with advanced PC (as determined by the investigator) if they had a locally advanced disease that is unlikely to be cured by local primary intervention with either surgery or radiation with curative intent. The last group of patients is not clearly defined, which makes it difficult to determine if the study population is representative of what is typically found in Canadian practice. Moreover, the study limited participation to patients with an ECOG Performance Status of 0 or 1, which may limit the generalizability to patients with an ECOG Performance Status higher than 1. However, the clinical experts emphasized that such patients would benefit and should receive the treatment if prescribed.
A relatively large proportion of patients failed screening, mostly due to meeting exclusion criteria. This could raise concerns as to the generalizability of the study. However, the clinical experts confirmed that the inclusion and exclusion criteria, as well as the resulting patients baseline characteristics, were sufficiently similar to the average patient they have in the clinic.
The primary and secondary outcomes reflect measurements of testosterone and PSA. These are biomarkers of disease activity and are considered surrogate in nature. PC can progress slowly, and capturing clinical outcomes may require a study with a longer duration. This is a major limitation of the study as it cannot inform on clinical outcomes related to PC due to its duration. Because ADT is an established treatment for patients with advanced PC, with a primary goal of suppressing testosterone to castration-like levels, testosterone-related outcomes are the most relevant. However, the lack of ability to inform on relevant clinical end points limits our understanding of the direct value that relugolix may provide patients. According to the clinical experts, the primary outcome may not be as useful to clinical practice as the secondary outcome of the profound castration rate because Canadian clinical practice emphasizes the need to reduce testosterone levels to less than 0.7 nmol/L (or 0.20 ng/mL).
An important limitation is the lack of intensification therapy. Current approaches to the treatment of PC emphasize the need for combining ADT with other systematic drugs, including docetaxel, darolutamide, and abiraterone, in patients with high-risk and/or high-volume disease, or abiraterone, apalutamide, or enzalutamide for other patients. The HERO trial cannot address the comparative or absolute efficacy of relugolix when combined with any other systemic therapy for advanced PC. Neither can it inform on the use of relugolix in patients undergoing RT. Finally, the HERO trial was not designed to measure the comparative efficacy and safety of relugolix versus any other comparator beyond leuprolide. These limitations to treatment-combination generalizability are due to the overall design of the trial, the limited number of patients who received ADT or RT beyond a certain progression threshold or who received antineoplastic drugs and were not discontinued, and the limitations on efficacy inferences from subgroup analyses.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered most relevant to inform CADTH’s expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group.50,51
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate — the true effect is likely close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited — the true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate — the true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as “very uncertain.”
Following the GRADE approach, evidence from RCTs were initially considered high-certainty evidence and could be rated down for concerns related to study limitations (which involve internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null.
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for relugolix versus leuprolide.
Long-Term Extension Studies
No long-term extension studies have been conducted.
Indirect Evidence
Contents within this section were informed by materials submitted by the sponsor. The following summary was validated by the CADTH review team.
Objectives for the Summary of Indirect Evidence
The HERO pivotal trial provided a head-to-head comparison between relugolix and leuprolide acetate in adult male patients with advanced PC. However, there is a lack of direct evidence comparing the effectiveness of relugolix with other available treatments in this population. An ITC is therefore warranted to address this evidence gap.
Description of Indirect Comparison
The sponsor submitted an NMA estimating the efficacy and safety of relugolix versus other medical ADTs available in Canada (Table 23).
Table 23: Study Selection Criteria and Methods for ITCs Submitted by the Sponsor
Characteristics | Indirect comparison |
|---|---|
Population | Adult men (aged ≥ 18 years) with advanced PC |
Intervention | ADTs and doses available in Canada:
|
Comparator |
|
Outcomes |
|
Study designs | RCTs |
Publication characteristics |
|
Exclusion criteria |
|
Databases searched |
Abstracts from the 2020 to 2022 meetings of the following congresses were reviewed, except for ASCO and ESMO, which were also searched in 2019: ASCO, ASCO GU Cancers Symposium, AUA, ESMO, ISPOR — International, European and Asia-Pacific annual congresses |
Selection process | Articles were screened independently by 2 reviewers, and any disagreements were resolved through consultation with a third reviewer |
Data extraction process | Data from the included studies were independently extracted into a data extraction table by 2 reviewers, and the entries were compared between the 2 reviewers; in case of disagreements, a third reviewer was consulted |
Quality assessment | Quality assessment of RCTs conducted using the Cochrane RoB version 1 |
ADT = androgen deprivation therapy; ASCO = American Society of Clinical Oncology; ASCO GU = American Society of Clinical Oncology –Genitourinary; AUA = American Urological Association; ESMO = European Society for Medical Oncology; ICTRP = International Clinical Trials Registry Platform; IM = intramuscular; ISPOR = International Society for Pharmacoeconomics and Outcomes Research; MACE = major cardiovascular event; PC prostate cancer; q.2.m. = every 2 months; q.3.m. = every 3 months; q.4.m. = every 4 months; q.4.w. = every 4 weeks; q.d. = once daily; RoB = Risk of Bias tool for randomized trials; RCT = randomized controlled trial; SC = subcutaneous; SLR = systematic literature review.
aRelevant SLRs and meta-analyses were excluded but flagged for handsearching of references against the list of included publications.
Source: Sponsor-submitted network meta-analyses.52
Indirect Treatment Comparison Design
Objectives
The sponsor-submitted ITC aimed to understand the relative efficacy and safety of relugolix in relation to other medical ADTs.
Study Selection Methods
The investigators conducted comprehensive systematic literature searches on March 30, 2020, and provided an update on February 1, 2022. The search was limited to articles published in the English language (Table 23). The ITC included RCTs investigating treatments in adults with advanced PC and reported on the following efficacy outcomes of interest to this CADTH review: testosterone suppression (castration level [< 50 ng/dL], time point: 12 ± 3 months) and MACE (defined as proportion of patients having an event, time point: 12 ± 3 months) (Table 23).
Two reviewers independently screened potentially eligible articles and extracted data. Discrepancies were resolved through discussion with a third reviewer until consensus was reached. Quality assessment of the included studies was conducted using version 1 of the Cochrane Collaboration Risk of Bias tool. The review protocol was not registered on any public platform.
Indirect Treatment Comparison Analysis Methods
Prior to conducting an NMA, a feasibility assessment was undertaken to determine the presence of clinical heterogeneity across the eligible studies. Outcome definitions and the timing of outcome assessment used across identified studies were explored, followed by an assessment of study designs and patients’ baseline characteristics. Details of the NMA methods are reported in Table 24.
The NMAs were conducted using a Bayesian framework. Each point estimate and 95% credible interval (CrI) was constructed using Markov chain Monte Carlo draws from the posterior distribution of the relevant effect estimand. The posterior distribution was estimated from 100,000 draws, with 10,000 burn-in runs across 3 Markov chains, using Gelman convergence assessment plots with a default REV model.
Various models were used to estimate individual treatment effects for each outcome of interest (fixed effect, REV, or REIP). Informed priors (mean = −3.23; SD = 1.88) were derived from the literature, using pharmacological versus pharmacological intervention comparison type and the semi-objective outcome type.53 Additional, a secondary analysis was conducted based on a hierarchical NMA described by Owen et al.,54 with both vague priors and informed prior values. This approach predicts individual treatment effects while accounting for exchangeability treatment classes and assumes a normal distribution of treatment effects around a class-specific mean and variance.
The assessment of model fit was based on the DIC and plausibility.
For the direct treatment comparisons, the assessment of statistical heterogeneity was based on the I2 statistic. The sponsor specified that comparisons showing substantial heterogeneity (i.e., with an I2 value > than 50%) were investigated.
To verify the consistency between direct and indirect comparisons for the castration rate outcome, a node-splitting approach with a standard P value threshold of 0.05 was considered.55 For MACE outcomes, a consistency assessment was not feasible due to the lack of closed loops.
The nodes in the network consisted of treatment arms, regardless of the administration schedules (e.g., every 4 weeks and every 12 weeks). Construction of nodes followed an equivalency assumption, assuming that the cumulative dosages are equivalent over a 12-week interval. For example, leuprolide acetate 22.5 mg administered every 12 weeks was considered equivalent to leuprolide acetate 7.5 mg every 4 weeks.
Three different approaches for sensitivity analysis were conducted and are reported in Table 29.
Table 24: Indirect Treatment Comparison Analysis Methods
Methods | Description | |
|---|---|---|
Testosterone suppression | MACE | |
Analysis methods | An NMA was performed using a Bayesian framework, with 5 models tested: FE, REV, REIP, REVH, and REIPH | |
Priors | Informed priorsa | Vague priors |
Assessment of model fit | Goodness of fit based on DIC | |
Assessment of consistency | Node-splitting used to assess consistency | Not available due to lack of a closed loops |
Assessment of convergence | Gelman convergence assessment plots | |
Outcomes | Castration rate defined as a cumulative and sustained incidence of testosterone suppression to castration level (< 50 ng/dL) | MACE and cardiovascular-related events, defined as a proportion of patients having an event (using study-specific definition) |
Follow-up time points | 12 months (± 3 months) | |
Construction of nodes |
|
|
Sensitivity analyses | Three sensitivity analyses: | Three sensitivity analyses: |
Subgroup analysis | Not reported | |
Methods for pairwise meta-analysis | Not reported | |
DIC = deviance information criterion; FE = fixed effects; HD = high-dose; MACE = major cardiovascular adverse event; NMA = network meta-analysis; REIP = random effects with informed priors; REIPH = random effects with informed priors – hierarchical; REV = random effects with vague priors.
aInformed priors (mean = −3.23 and standard deviation = 1.88) were derived from the literature (Turner et al., 2012), using pharmacological vs. pharmacological intervention comparison type and the semi-objective outcome type.53
bThe C35 study assessed the efficacy of goserelin compared to degarelix, and the efficacy outcomes was testosterone suppression from day 3 up to 12 months. As goserelin is an agonist, it might take longer to be effective compared with degarelix, which may explain why few patients receiving the former treatment achieved a response in this trial. To address this issue, the sponsor conducted a sensitivity analysis by excluding study CS35.
cMargel (2019) included a small sample of participants (< 100). The sponsor conducted a sensitivity analysis by excluding this trial due to its potential to cause publication bias.
Source: Sponsor-submitted NMA.25
Results of Indirect Treatment Comparison
Summary of Included Studies
Fifty-two publications, consisting of 35 unique trials, were assessed for eligibility for inclusion in the systematic review. Ultimately, 8 RCTs were retained in the quantitative synthesis (collectively including 3,830 randomized patients and 6 different treatment regimens), of which 7 studies included testosterone suppression and 4 included MACE as an outcome (Table 25). One trial was a phase II study, while the remaining trials were phase III studies. Treatment assignment was open-label for 6 trials and single-blind in 2 trials. The sponsors’ assessment of homogeneity for the included studies is reported in Table 26.
The sponsor assumed that the different treatment administration schedules (e.g., every 4 weeks and every 12 weeks) were equivalent if the cumulative dosages were equivalent over a specific period of time. All treatment arms included in the NMA are available in Canada, except for the high-dose degarelix arm, which was retained to increase the robustness of the network in the testosterone suppression analysis (Figure 6). For the MACE outcome analyses, degarelix at a regular dose and a high dose were collapsed because 1 of the included trials reported only combined estimates. Moreover, 1 of the included studies did not specify the comparator against which degarelix was being compared, and the sponsors’ analysis assumed the GnRH agonist against in question is leuprolide.
Regarding baseline characteristics, the proportion of patients with metastatic diseases varied greatly across the included studies (ranging from 18% to 64%) (Table 27). Baseline testosterone levels were reported in 6 trials, ranging from 326 ng/dL in the PRONOUNCE trial to 498 ng/dL in the Ozono (2018) study. All studies allowed the inclusion of patients on prior or ongoing hormone therapy in the local therapy setting as neoadjuvant or adjuvant treatment, but only 3 of them reported variable data on the proportion of patients receiving prior hormone therapy. Participants’ life expectancy was reported as eligibility criteria in 7 trials, of which 6 included individuals with expectancy greater than 1 year, whereas 1 required individuals to have an expectancy of 5 years or more. Seven studies reported participant level of independence as an inclusion criterion, requiring patients to fall within the range from 0 to 2 on the ECOG or WHO scale (0 being fully active and 2 being unable to work but capable of self-care), and higher than 40 on the Karnofsky core (indicating a need for special care and assistance). The exception was the HERO study, which required individuals to have an ECOG Performance Status of 0 or 1 (Table 27).
Results of the quality assessment of included studies was not provided in the sponsor’s submission.
Table 25: Description of Studies Included in the Sponsor-Submitted Network Meta-Analysis
Study name, author (year) | Experimental arm (N) | Control arm (N) | Outcome included in the NMA |
|---|---|---|---|
HERO, Shore et al. (2020) | Relugolix 360 mg/120 mg q.d. (622) | Leuprolide 22.5 mg q.3.m. (308) | TS, MACE |
Heyns et al. (2003) | Triptorelin 3.75 mg q.4.w. (137) | Leuprolide 7.5 mg q.4.w. (140) | TS |
Sun et al. (2020) | Goserelin 3.6 mg q.4.w. (142) | Degarelix 240 /80 mg q.4.w. (143) | TS |
Ozono et al. (2018) | Goserelin 3.6 mg q.4.w. (117) | Degarelix 240 mg/480 mg q.3.m. (117) | TS |
CS35, Shore et al. (2012) | Goserelin 3.6 mg q.4.w./10.8 mg q.3.m. (565) | Degarelix 240 mg/80 mg q.3.m. (282) | TS |
CS21, Tombal et al. (2010) | Degarelix 240 mg/80 mg q.4.w. (202) | Leuprolide 7.5 mg q.4.w. (201) | TS, MACE |
Degarelix 240 mg/160 mg q.4.w. (207) | |||
PRONOUNCE, Lopes et al. (2021) | Degarelix 240 mg/80 mg q.4.w. (276) | Leuprolide 22.5 mg q.3.m. (269) | TS, MACE |
Margel et al. (2019) | Degarelix 240 mg/80 mg q.4.w. (41) | GnRH agonista q.3.m. (39) | MACE |
MACE = major adverse cardiovascular event; q.d. = once daily; q.4.w. = every 4 weeks; q.3.m. = every 3 months; q.4.m. = every 4 months; TS = testosterone suppression.
aSponsor’s analysis assumes leuprolide acetate
Source: Sponsor-submitted network meta-analysis.25
Table 26: Assessment of Homogeneity for the Indirect Treatment Comparison
Characteristics | Description and handling of potential effect modifiers |
|---|---|
Disease severity — stage | Information on disease stage was specified in all but 1 clinical trial. When reported, all advanced PC patients from localized to metastatic disease were eligible to enter the trial. However, the proportion of patients with metastasis at baseline varied from 1 study to another, ranging from 18% to 64%. Outcomes data for subgroups stratified by presence of metastatic disease were not reported in any of the identified RCTs, except the HERO trial, and the degree to which differences in the prevalence of metastatic disease may modify treatment effects was not assessed. |
Treatment history | All studies excluded patients with HT, including ADT, for advanced disease but allowed prior or ongoing HT in the local therapy setting as neoadjuvant or adjuvant treatment. No exclusions were made for this characteristic. There was a wide range in proportions of patients with prior HT for local and/or localized disease or in a local setting. The Ozono (2018) study reported 0 to 0.9% prior HT, while Margel (2019) reported 99% for degarelix and 100% for GnRH agonist (leuprolide). In the HERO trial, only 12% of patients overall had received prior HT. It was not feasible to determine whether differences in the proportion of patients receiving prior HT was an effect modifier due to a lack of outcomes data for subgroups stratified by receipt of prior HT in the identified trials. |
Trial eligibility criteria | ECOG PS: Most trials required patients to be relatively independent, ranging from fully independent (ECOG PS = 0) to unable to work but capable of self-care (ECOG PS = 2). An exception was the HERO trial, which enrolled patients with an ECOG PS from 0 to 1. Heyns (2003) enrolled patients with a Karnofsky score ≥ 40, where 40 indicated a need for special care and assistance. Sun (2020) did not provide information on ECOG PS. Consequently, no studies were excluded based on this characteristic. Life expectancy: Eligibility criteria varied from ≥ 1 year to ≥ 5 years, with the PRONOUNCE trial not specifying life expectancy. Testosterone levels: All studies but Margel (2019) specified eligibility criteria for testosterone levels at baseline, which ranged from > 150 ng/dL to > 220 ng/dL. Differences in baseline testosterone levels may bias the analyses to the extent that treatment effects for testosterone suppression could be modified by higher or lower testosterone at baseline. However, when looking at testosterone levels at baseline, which was reported in 6 of the 8 trials, mean and median values remained in a similar range, from 326 ng/dL in the PRONOUNCE trial to 494 ng/dL in the Ozono (2018) study. As such, no studies were excluded for this characteristic. |
Dosing of comparators | When the cumulative dosages were equivalent over a specific interval, different administration schedules were considered equivalent. The following treatment arms were included in the NMA:
All treatments and dosages are available in Canada, except for the high-dose degarelix arm, which was retained to maintain a robust and comprehensive network. |
Placebo response | Not available |
Definitions of end points | Testosterone suppression (castration rate): Only studies reporting a castration rate, using the same testosterone threshold (i.e., < 50 ng/dL) were retained. Studies reporting a castration rate using other thresholds were excluded. MACE: MACE can be defined differently across trials. Definitions considered too dissimilar were excluded during the feasibility assessment process. Specifically, 1 study was excluded because only fatal cardiovascular-related events and/or all-cause deaths were reported. Another study was excluded because it reported both cardiac AEs and SAEs. Given that MACE is a category that may include both serious and nonserious AEs, it was not possible to determine which data to include in the analysis. In addition, a sensitivity analysis was conducted without the CS21 study, given that the definition of MACE in this study may have potentially introduced heterogeneity. |
Timing of end point evaluation | Testosterone suppression (castration rate): Only studies reporting a castration rate at 12 months (± 3 months) were eligible for inclusion in the NMA. Therefore, during the feasibility assessment process, 8 studies were excluded because the outcome was assessed at earlier than 9 months. These exclusions were implemented to reduce heterogeneity in timing of outcome assessment as a potential source of bias since a longer follow-up may be associated with a higher cumulative proportion of castration rate. MACE: The same approach was applied to MACEs, and only studies reporting MACEs at 12 months (± 3 months) were eligible for inclusion in the NMA. Therefore, during the feasibility assessment process, 8 studies were excluded because the time at which MACE was assessed was not reported, or it was assessed at earlier than 9 months. These exclusions were implemented to reduce heterogeneity in timing of outcome assessment as a potential source of bias since a longer follow-up may be associated with a higher probability of MACE. |
Withdrawal frequency | Not reported |
Clinical trial setting | Not reported |
Study design | Included studies were all RCTs. Only 1 trial was designed as a phase II study (with the remaining being phase III studies) and was not considered to be a potential source of material bias. No exclusions were made for this characteristic. |
Blinding | Blinding was open-label for 6 trials and single-blind in 2 trials. Open-label designs may be a source of bias to the extent that prior knowledge of the treatments may affect the way investigators collect outcomes data or the way clinical study personnel provide care to those enrolled in the trial. That most of the trials identified by the SLR were open-label likely reflects the difference in administration methods between treatments. Given that most of the identified trials, including the HERO trial, were open-label, no exclusions were made for differences in this characteristic. |
ADT = androgen deprivation therapy; AE = adverse event; ECOG PS = Eastern Cooperative Oncology Group Performance Status; GnRH = gonadotropin-releasing hormone; HT = hormone therapy; MACE = major adverse cardiovascular event; NMA = network meta-analysis; PC = prostate cancer; q.3.m = every 3 months; q.4.w = every 4 weeks; q.d. = once daily; RCT = randomized controlled trial; SAE = serious adverse event.
Source: Sponsor-submitted NMA.25
Table 27: Baseline Characteristics of Participants Across Trials Included in the NMAs
Study, author (year) | Treatment | Age, mean (SD or range) | Metastatic disease (%) | Mean or median testosterone, ng/dL (SD or IQR or rangea) | Prior HT (%) | Performance statusa |
|---|---|---|---|---|---|---|
HERO, Shore et al. (2020) | Leuprolide | 71b (47 to 97) | 32 | 436.1 (159.0) | 10 | ECOG PS 0 to 1 |
Relugolix | 72b (48 to 1) | 32 | 410.0 (149.1) | 13 | ||
CS21, Tombal et al. (2010) | Degarelix (240 mg/ 80 mg) | 72b (50 to 88) | 18 | 411 (305 to 532) | NR | ECOG PS 0 to 2 |
Degarelix HD (240 mg/160 mg) | 72b (51 to 89) | 20 | 378 (286 to 505) | NR | ||
Leuprolide | 74b (52 to 98) | 23 | 384 (291 to 501) | NR | ||
Sun et al. (2020) | Degarelix 240 mg/80 mg q.4.w. | 75b (52 to 86) | 63 | 460b (130 to 960) | NR | NR |
Goserelin 3.6 mg q.4.w. | 73b (47 to 91) | 64 | 460b (170 to 1,120) | NR | ||
Ozono et al. (2018) | Degarelix 240 mg/480 mg q.4.w. | 75.5 (6.1) | 20 | 498 (141) | 0.9% | ECOG PS 0 to 2 |
Goserelin 3.6 mg q.4.w. | 75.9 (5.9) | 18 | 494 (159) | NR | ||
CS35, Shore et al. (2012) | Goserelin 3.6 q.4.w./10.8 mg q.3.m. | 71.9 (8.3) | NR | NR | NR | ECOG PS 0 to 2 |
Degarelix 240 mg/80 mg q.4.w. | 71.1 (7.9) | NR | NR | NR | ||
Margel et al. (2019) | Degarelix 240 mg/80mg q.4.w. | 72b (66 to 77) | 27 | NR | 99 | WHO 0 to 2 |
GnRH agonist (leuprolide) | 71b (69 to 78) | 26 | NR | 100 | ||
Heyns et al. (2003) | Leuprolide | 71.6 (49 to 89) | NR | 348.12 | NR | Karnofsky > 40 |
Triptorelin | 70.5 (47 to 88) | NR | 346.97 | NR | ||
PRONOUNCE, Lopes et al. (2021) | Leuprolide 22.5 mg q.3.m. | 73.1 (7.2) | 17.8 | 338 (249 to 415) | NR | ECOG PS 0 to 2 |
Degarelix 240 mg/80 mg q.4.w. | 73.3 (7.3) | 22.9 | 326 (252 to 416) | NR |
ECOG PS = Eastern Cooperative Oncology Group Performance Status; HT = hormone therapy; IQR = interquartile range; NR = not reported; q.3.m. = every 3 months; q.4.w. = every 4 weeks; SD = standard deviation.
aStudy inclusion criteria.
bMedians reported instead of mean.
Source: Sponsor-submitted network meta-analysis.
Results
Seven studies were included in the NMA of testosterone suppression, defined as the cumulative probability of sustained chemical castration with levels of testosterone lower than 50 ng/dL (measured at 12 months ± 3 months) (Table 25 and Figure 6).
Direct evidence included 4 studies, of which 2 compared high-dose degarelix and goserelin57,59 and 2 compared degarelix to leuprolide.56,61 Statistical heterogeneity, assessed with the I2 statistic, was reported: C35 and Ozono trial: I2 = 99% (P < 0.01); and CS21 and PRONOUNCE: I2 = 0% (P = 0.35).
Sponsor reported that the best-fitting model for the NMA network of testosterone suppression was the REIP model (i.e., with the lowest DIC value of 86.8). Convergence of the model was achieved, with a shrink factor of 1.00 and 10,000 burn-in iterations. Although inconsistency testing reported nonsignificant findings (P values > 0.05), different and opposite directions of effects for direct and indirect estimators of comparative effect were observed (Appendix 1). Results for the comparison of relugolix to different treatments regarding castration levels are reported in Table 28. According to the estimated odds ratios and risk ratios in the REIP model, no significant differences were observed between treatments. Results of the 2 sensitivity analyses on castration levels, reported in Table 29, suggested opposite relative effects for certain comparisons. Nevertheless, CrIs included the value of 1 and were similar to the base-case analysis.
Four studies were included in the NMA of MACEs (Table 25 and Figure 7). The main analysis uses study-specific definitions of events (Table 30). Direct evidence included 3 studies, all of which compared degarelix to leuprolid.56,60,61 Statistical heterogeneity was observed (I2 = 54%; P = 0.12).
The best-fitting model for the NMA network of MACEs was a REV model, as suggested by the lowest DIC. Convergence of the model was achieved, with a shrink factor of 1.00 with a burn-in of 10,000 iterations. Consistency testing was not feasible due to the lack of closed loops. The MACE outcome estimates for the comparison of relugolix to different treatments suggested no significant differences between arms, as reported in Table 28. Two sensitivity analyses for MACEs using a REV model are summarized in Table 29. The outcomes from the sensitivity analysis, excluding a study with a different MACE definition, were consistent with the base case.
Figure 6: Network Meta-Analysis Evidence Network for Testosterone Suppression (7 Trials)
Source: Sponsor-submitted network meta-analysis.25
Figure 7: Network Meta-Analysis Evidence Network for MACE (4 Trials)
Source: Sponsor-submitted network meta-analysis.25
Table 28: ITC Relative Effects of Relugolix Compared to Different Treatments for Testosterone Suppression and MACE
Detail | Testosterone suppression at 12 ± 3 months | MACE at 12 months |
|---|---|---|
Number of studies, N (patients) | 7 studies (3,718 patients) | 4 studies (2,366 patients) |
Model | REIP | REV |
Relugolix vs. comparator, OR (95% CrI) and RR (95% CrI) | ||
Degarelix | ███ ████ █████ ██ █████████ ████ █████ ██ █████ | ████ ████ █████ ██ ███████████ ████ █████ ██ █████ |
Degarelix HD | ███ ████ █████ ██ █████████ ████ █████ ██ █████ | |
Goserelin | ███████ █████ ██ █████████████ █████ ██ █████ | NA |
Triptorelin | ███████ █████ ██ █████████████ █████ ██ █████ | NA |
Leuprolide | ███████ █████ ██ █████████████ █████ ██ █████ | ███ ████ █████ ██ █████████████ █████ ██ █████ |
CrI = credible interval; FE = fixed effects; HD = high-dose; IP = informed priors; ITC = indirect treatment comparison; MACE = major adverse cardiovascular event; OR = odds ratio; NA = not applicable; NMA = network meta-analysis; REV = random effects with vague priors; RR = risk ratio.
aAs the CS21 trial reported the total percentage of MACE for degarelix and degarelix HD combined, these rates were collapsed in the primary analysis.
Source: Sponsor-submitted NMA.25
Table 29: NMA Sensitivity Analyses for Testosterone Suppression and MACE
Drug | Sensitivity analysis for testosterone suppression (REIP model) | Sensitivity analysis for MACE (REV model) | ||
|---|---|---|---|---|
Non-zero correctiona | Nonzero correction, with CS35b removed | Without CS21c | Without Margel et al.d | |
Relugolix vs. comparator, OR (95% CrI) and RR (95% CrI) | ||||
Degarelix | ██ █ ████ █████ ██ ████████ █ ████ █████ ██ █████ | ██ █ ████ █████ ██ ███████ █ ████ █████ ██ █████ | ██ █ ████ █████ ██ ████████ █ ████ █████ ██ ██████ | ███ █ ████ █████ ██ █████████ █ ████ █████ ██ █████ |
Degarelix HD | ██ █ ████ █████ ██ █████ ███ █ ████ █████ ██ █████ | ██ █ ████ █████ ██ █████ ███ █ ████ █████ ██ █████ | ||
Goserelin | ██ █ ███ █████ ██ █████ ███ █ ████ █████ ██ █████ | ██ █ ████ █████ ██ █████ ███ █ ████ █████ ██ █████ | |||| | |||| |
Triptorelin | ██ █ ████ █████ ██ █████ ███ █ ████ █████ ██ █████ | ██ █ ████ █████ ██ █████ ███ █ ████ █████ ██ █████ | |||| | |||| |
Leuprolide | ██ █ ████ █████ ██ █████ ███ █ ████ █████ ██ ████ | ██ █ ████ █████ ██ █████ ███ █ ████ ██████ ██ █████ | ██ █ ████ █████ ██ ████████ █ ████ ████ ██ █████ | ██ █ ████ █████ ██ ████████ █ ████ █████ ██ █████ |
CrI = credible interval; FE = fixed effects; IP = informed priors; MACE = Major adverse cardiovascular event; NA = not applicable; NMA = network meta-analysis; OR = odds ratio; RE = random effects; REIP = random effects with informed priors; REV = random effects with vague priors.
Note: Because the CS21 trial reported the total percentage of MACE for degarelix and degarelix HD combined, these rates were collapsed in the primary analysis.
aSensitivity analysis for testosterone suppression were conducted for the arm containing a 100% response (goserelin in Ozono [2018]), reducing the number of responders by 0.5 to avoid possible calculation errors.
bThe C35 study assessed the efficacy of goserelin compared to degarelix, and the efficacy outcomes was testosterone suppression from day 3 up to 12 months. As goserelin is an agonist, it may take longer time to be effective than degarelix, which may explain why few patients receiving the former treatment achieved a response in this trial. To address this issue, the sponsor conducted a sensitivity analysis by excluding Study CS35.
cThe sponsor conducted a sensitivity analysis by excluding the CS21 study, which reported a different definition for MACE (which did not include all-cause mortality).
dThe sponsor conducted a sensitivity analysis by excluding the Margel et al. (2019) study due to its small sample size (n < 100) and potential to cause publication bias.
Source: Sponsors-submitted network meta-analysis.25
Table 30: Definition for MACE Outcomes Across Trials Included in the NMA
Author (year) | Study name | MACE definition |
|---|---|---|
Shore (2020) | HERO | Myocardial infarction (SMQ), central nervous system hemorrhages and cerebrovascular conditions (SMQ), and deaths due to all causes |
Lopes (2021) | PRONOUNCE | All causes of death, myocardial infarctions, or stroke (primary analysis), MACE-related AEs according to a broad SMQ |
Margel (2019) | Margel 2019 | Death, myocardial infarction, stroke, and heart catheterization with stent insertion |
Klotz (2008) | CS21 | Cardiovascular AEs (e.g., angina pectoris, atrial fibrillation, cardiac failure, and myocardial infarction) |
AE = adverse event; MACE = major adverse cardiovascular event; NMA = network meta-analysis; SMQ = standardized Medical Dictionary for Regulatory Activities query.
Source: Sponsor-submitted NMA.25
Critical Appraisal of Indirect Treatment Comparison
The sponsor’s ITC was conducted in line with well-known international guidelines, and adopted standard methods for conducting and reporting reviews, including defining the research question according to population, intervention, comparators, outcomes, and study design criteria, searching through multiple databases, comprehensive literature searches, adequate screening and data extraction processes (double reviewers), and quality control by an additional reviewer.62-64 No systematic review protocol was registered, which may raise concerns about the quality of the review methodology used to inform the NMA. CADTH reviewers could not evaluate whether changes were implemented in the review methodology over the 2-year course of the review. Moreover, because the results of the risk-of-bias assessment at the individual-study level were not reported, it was not possible to determine if quality issues affected the reported effect estimates of the NMA.
The sponsor conducted a feasibility assessment based on the outcome definition and time points of assessment, resulting in consistent outcome definitions across the included trials. Evaluation of clinical, methodological, and statistical heterogeneity was limited due to inadequate reporting of data in the published studies included in the NMA. Moreover, an investigation of the effects of possible effect-modifying factors on the NMA outcome estimates was not feasible due to the limited size of the network. Nevertheless, variability in patients’ baseline characteristics was observed, including metastatic status of the patients (range = 18% to 64%), baseline testosterone levels (> 150 ng/dL to > 220 ng/dL), prior hormone therapy (range = 0.9% to 100%), and patient performance status. According to the clinical experts consulted by CADTH, these differences could result in changing relative-treatment effects, and it is likely that these differences across studies in the network introduced bias to the comparisons in the NMA.
Results of the sensitivity analyses for both testosterone suppression and MACEs suggested inconsistent relative-treatment effects for certain comparisons within the network. However, the CrIs included the null value were wide and overlapped those from the base case. Regarding generalizability, the clinical experts consulted in the CADTH review noted that profound castration levels (< 20 ng/dL) would have been a more appropriate outcome in the Canadian clinical context. Moreover, the timeline for the outcome definition (i.e., 12 ± 3 months) is too short and inadequate for an assessment of MACEs, according to the experts. These issues pose serious generalizability concerns regarding the applicability of the NMA findings to the Canadian context.
Selection of the models for the base-case analyses was based on the lowest DIC, which resulted in the selection of the REIP model for testosterone castration and the REV model for MACE as the primary results. The selected model for testosterone suppression relies on informative priors derived from the literature as opposed to the alternative models that include a REV model and a fixed-effects model. Aside from the goodness-of-fit of these models suggested by the DIC, it is unclear which of these models would be most appropriate given both the heterogeneity in the network and the relatively sparse network. To create a connected network, the sponsor applied an equivalency assumption, which assumed the cumulative dosages of treatments are equivalent over a 12-week period. This assumption was considered appropriate by the clinical experts consulted by CADTH. The sponsor provided a series of exploratory analyses using different models with varying alternative prior distributions for both outcomes. The resulting point estimates were associated with wide CrIs, highlighting the uncertainty in the results. Moreover, these variabilities may suggest that the NMA findings are sensitive to prior selection, which raises additional uncertainty regarding the appropriateness of the selected model for the primary analyses.
Consistency between direct and indirect comparisons was assessed for castration outcome only. Statistical testing for inconsistency showed insignificant findings (P > 0.05), the point estimates were not entirely consistent, and the CrIs were wide and overlapped. Statistical heterogeneity assessment was based on I2 statistics (ranging from 0 to 99% for the 2 outcome comparisons), but the number of studies informing these analyses was low, limiting the interpretability of the assessment.
In reference to the comparators included in the sponsor’s NMA, the CADTH reviewers noted certain concerns. One study did not specify the GnRH agonist assessed in the trial, and the sponsor assumed the treatment in question was leuprolide.60 Moreover, the sponsor’s literature search yielded no studies on buserelin, even though this drug was specified as a comparator of interest. According to the data provided by the sponsor on market shares, buserelin only holds a small fraction of the Canadian market. Upon additional validation with the clinical experts regarding comparators, the CADTH reviewers concluded that there were no major generalizability issues with the representation of included treatments in the NMA.
Studies Addressing Gaps in the Systematic Review Evidence
Contents within this section were informed by materials submitted by the sponsor. The following summary was validated by the CADTH review team.
Table 31: Summary of Gaps in the Systematic Review Evidence
Evidence gap | Studies that address gaps | |
|---|---|---|
Study description | Summary of key results | |
The HERO study did not specifically investigate the efficacy and safety of relugolix as neoadjuvant and/or adjuvant therapy to RT. (Only a subgroup analysis was done on a limited number of patients who received RT with adjuvant relugolix in HERO trial.) | C27300 is a multicentre, phase II, open-label, randomized, active-control, parallel-group study of patients with intermediate-risk localized PC requiring neoadjuvant and/or adjuvant ADT with EBRT. | Treatment with either relugolix or degarelix in combination with EBRT was associated with sustained castration over 24 weeks, with castration rates (< 50 ng/dL) of 95% and 89%, respectively. Cumulative profound castration (20 ng/dL) over 24 weeks was achieved by 82% and 68% of patients receiving relugolix and degarelix, respectively. EORTC QLQ-C30 and EORTC QLQL-PR25 results demonstrated an increase in castration-related symptoms and a decline in QoL during treatment with either study drug. Upon discontinuation of treatment, QoL scores improved for both groups. |
The HERO trial included a limited number of patients undergoing treatment intensification with an oral androgen-receptor signaling inhibitor, i.e., enzalutamide and docetaxel. Use of ADT with other “add-on” therapies has not been studied in the HERO trial. | Multicentre, ongoing 52-week phase I, open-label, nonrandomized, parallel-group study of relugolix in combination with abiraterone in males with mCSPC or mCRPC (part 1) and apalutamide in those with mCSPC (part 2).a This study also provided data for participants who switched from injectable leuprolide acetate or degarelix to relugolix as the ADT component of their treatment. Study completion is planned for November 2023. | Based on an interim analysis of 15 participants in part 1 and 10 participants in part 2 who completed at least 12 weeks of treatment, combination therapy of relugolix with abiraterone or apalutamide demonstrated a consistent safety profile in the HERO trial, while maintaining effective suppression of testosterone to castration levels (< 50 ng/dL). |
The HERO study did not explore the combination regimen of ADT with antiandrogens other than enzalutamide (which was studied in a limited number of patients). | This was a substudy of a multicentre, phase II, open-label, single-arm study of ADT in combination with apalutamide in treatment-naive males who have undergone RP for nonmetastatic PC and who are at high risk for metastases. Study completion is planned for the end of October 2023. | Among the 11 patients who received relugolix (120 mg) plus apalutamide (240 mg) and were evaluated at day 28, all maintained castration levels (< 50 ng/dL) without requiring relugolix dose adjustment. Results from the main study indicated that, of the 10 patients who successfully completed a year of concurrent treatment with the standard 120 mg dose of relugolix plus apalutamide, 100% maintained castration (< 50 ng/dL) at 1 year. No patient required a dose modification of relugolix. |
ADT = androgen deprivation therapy; EBRT = external beam radiation therapy; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-PR25 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Prostate Cancer 25 Items; mCRPC = metastatic castration-resistant prostate cancer; mCSPC = metastatic castration-sensitive prostate cancer; PC = prostate cancer; QoL = quality of life.
aA part 3 was initially planned to study relugolix in combination with docetaxel with or without prednisone in patients with mCSPC but was discontinued due to a lack of enrolment and will not be reported in this report.
Source: Sponsor’s Summary of Clinical Evidence.
Description of Studies
Three clinical trials have been summarized to support relugolix in the treatment of advanced PC.
The HERO study did not specifically investigate the efficacy and safety of relugolix as neoadjuvant and/or adjuvant therapy to RT.65 Also, the evidence for combined use of relugolix with other antiandrogens (i.e., apalutamide and abiraterone) in patients with mCSPC or mCRPC is lacking.66 Last, the concurrent use of relugolix and an antiandrogen (apalutamide) as a adjuvant therapy in patients with high-risk localized PC after RP was not explored in the pivotal study.67 The following 3 trials have been summarized to address the gaps identified.
Table 32: Details of Studies Addressing Gaps in the Systematic Review Evidence
Detail | C27003 | MVT-601 to 049 | Apa-RP |
|---|---|---|---|
Designs and populations | |||
Study design | Phase II, randomized, open-label, parallel-group | Phase I, 3-part, open-label, parallel-cohort | Phase II, multicentre, open-label, single-arm |
Enrolled, N | 103 | 72 | 108 |
Key inclusion criteria |
|
|
|
Key exclusion criteria |
|
|
|
Drugs | |||
Intervention | Relugolix, oral tablets | Part 1 and part 3: relugolix 360 mg loading dose once orally, followed by 120 mg orally once daily Part 2: relugolix 360 mg loading dose once orally, followed by followed by 250 mg orally once daily Abiraterone: 500 mg or 1,000 mg orally once daily Prednisone: 5 mg orally once or twice daily Methylprednisolone: 4 mg orally twice daily Apalutamide: 240 mg orally once daily Docetaxel: 75 mg/m2 every 3 weeks as 1-hour IV infusion | Main study: apalutamide 240 mg once daily orally along with ADT for 12 cycles (each cycle is of 28 days) Substudy: apalutamide 240 mg once daily along with relugolix 120 mg once daily following a loading dose of 360 mg relugolix orally; relugolix is taken up to day 28, after which participants will be transitioned into the main study from cycle 2, day 1 and will continue to receive conventional (IM or SC) or oral ADT |
Comparator(s) | Degarelix (Firmagon), injection | NA | NA |
Outcomes | |||
Primary end point | Percentage of participants with effective castration rate over 25 weeksc | Incidence of AEs | Main study: time to BCRd Substudy: percentage of participants maintaining a testosterone level < 50 ng/dL through day 28 |
Secondary end points | TEAEs, SAEs, time to achieve effective or profound castration, number of participants with PSA response of ≥ 50% and ≥ 90% reduction, estimated TTR, AMS, EORTC QLQ-C30 and EORTC QLQ-PR25 | Mean testosterone serum concentrations, number and proportion of participants with testosterone concentrations ≥ 50 ng/dL at baseline (day 1), week 5, and week 13 | Main study: TTR Substudy: safety and tolerability (TEAEs) |
Notes | |||
Publications | Dearnaley (2020)65 | De La Cerda (2023)66 | Brown (2023)67 |
ADT = androgen deprivation therapy; AE = adverse event; AMS = Aging Male’s Symptoms; BCR = biochemical recurrence; EBRT = external beam radiation therapy; ECG = electrocardiogram; ECOG PS = Eastern Cooperative Oncology Group Performance Status; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-PR25 European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Prostate Cancer 25 Items; GnRH = gonadotropin-releasing hormone; IM = intramuscular; mCRPC = metastatic castration-resistant prostate cancer; mCSPC = metastatic castration-sensitive prostate cancer; MI = myocardial infarction; NCCN = National Comprehensive Cancer Network; nmCRPC = nonmetastatic castration-resistant prostate cancer; NYHA = New York Heart Association; PC = prostate cancer; PSA = prostate-specific antigen; RP = radical prostatectomy; SAE = serious adverse event; SC = subcutaneous; SoC = standard of care; TEAE = treatment-emergent adverse event; TTR = time to testosterone recovery; UA = unstable angina.
aDiagnostic definitions: mCSPC (parts 1, 2, and 3) defined as having at least 2 of 3 risk factors at the baseline (day 1) visit: total Gleason score of 6 or higher; and presence of at least 2 metastatic lesions on bone scan; or radiologic evidence of measurable visceral metastases with exception of hepatic metastases. Non-mCRPC (part 2 only) defined as disease progression despite maintaining castration levels of testosterone with ADT, as evidenced by an increase in consecutive PSA concentrations (2 measurements, at least 1 week apart). mCRPC (parts 1 and 3) defined as disease progression despite maintaining castration levels of testosterone with ADT: an increase in consecutive PSA (2 measurements at least 1 weeks apart), worsening clinical symptoms, radiologic evidence demonstrating enlarged metastatic lesions or the development of new metastases.
bAntiandrogens or docetaxel in combination with SoC: part 1: abiraterone acetate 1,000 mg or fine-particle abiraterone acetate 500 mg once daily plus prednisone 5 mg once daily for participants with mCSPC or twice daily for participants with mCRPC or methylprednisolone 4 mg once daily and in whom abiraterone has been well tolerated (without evidence of hepatotoxicity requiring dose adjustment for abiraterone). Part 2: apalutamide 240 mg once daily and in whom apalutamide has been well tolerated (without a fracture, fall, or seizure episode or need to dose adjust due to any AE). Part 3: docetaxel 75 mg/m2 and in whom docetaxel has been well tolerated (no evidence of hypersensitivity reaction, febrile neutropenia or neutrophils < 500 cells/mm3 for more than 1 week, severe or cumulative cutaneous reactions, or moderate neurosensory signs and/or symptoms despite dose reduction).
cCastration rate is defined as the observed percentage of participants who have testosterone concentrations below 50 ng/dL (1.73 nmol/L) at all scheduled visits.
dBCR is defined as a confirmed PSA above 0.2 ng/mL and time to BCR is analyzed using Kaplan-Meier method.
Sources: C27003 Clinical Study Report,68 Dearnaley et al. (2020),65 Clinicaltrials.gov: NCT02135445.69 De La Cerda et al. (2023),66 Clinicaltrials.gov: NCT04666129.70 Brown et al. (2023),67 Brown et al. (2023) conference proceeding,71 and Clinicaltrials.gov: NCT04523207.72
Description of Study C27003
The objective of the C27003 study was to evaluate the efficacy of neoadjuvant and adjuvant relugolix compared to degarelix for achieving and maintaining testosterone suppression in patients with localized intermediate-risk PC undergoing primary EBRT. Secondary objectives included the evaluation of safety and tolerability of relugolix in comparison to degarelix.
Study C27003 was a multicentre, phase II, open-label, randomized, active-controlled trial. A total of 103 patients were enrolled in 23 sites in the UK and the US, with no sites in Canada. Patients were included if they were diagnosed with localized PC of intermediate risk, for which a period of 6 months of neoadjuvant or adjuvant ADT to ERBT is indicated. Patients were randomized in a 3:2 ratio to oral relugolix (n = 65) or subcutaneous depot of degarelix (n = 38) for 24 weeks. The trial consisted of 3 phases, including a screening period of up to 28 days, a 24-week treatment period, and 12-week follow-up period.
Populations
Eligible patients were male, aged 18 years or older, with histologically confirmed, localized, intermediate-risk PC who required 6-month neoadjuvant or adjuvant ADT to EBRT. An intermediate risk was defined as the presence of 1 of the following, without any high-risk features: T2b-T2c disease, a Gleason score of 7, or a PSA concentration of 10 ng/L to 20 ng/L.73 Older patients with high-risk disease could be also considered for inclusion if they were deemed likely to benefit from 6 months of ADT based on the investigator discretion and a clinical assessment of the patient’s overall medical and disease status. EBRT was to begin at least 12 weeks after baseline visit.
Patients with metastatic disease or a history of significant cardiac conditions within 6 months before administration of the study drug were excluded. Patients who previously used or currently use a GnRH agonist or antagonist as a first-line hormone therapy were also excluded unless the total usage time was less than 6 months and was completed at least 1 year before the planned baseline visit.
Interventions
Patients were randomized in a 3:2 ratio to receive 1 of 2 treatments in the study:
relugolix 360 mg (3 × 120 mg tablets) single oral loading dose on day 1 followed by a single 120 mg tablet orally once daily for 24 weeks
degarelix 4-week depot injection, loading dose of 240 mg on day 1, then 80 mg every 4 weeks for 24 weeks (last injection occurred 4 weeks before the end of the treatment period).
Patients received their allocated neoadjuvant treatment 12 to 16 weeks before starting EBRT and their adjuvant therapy for the remainder of the 24-week period (8 to 12 weeks) during and following the completion of RT.
Outcomes
The primary efficacy end point was the rate of sustained castration between 4 and 24 weeks of treatment, defined as the estimated proportion of patients with a testosterone level below 50 ng/dL at all scheduled visits. Secondary efficacy end points included time to achieving castration (< 50 ng/dL) over 24 weeks, time to achieving profound castration (< 20 ng/dL) over 24 weeks, time to testosterone recovery (i.e., return of testosterone values to baseline or to > 280 ng/dL) at 12 weeks after treatment discontinuation, and PSA response rate (≥ 50% and ≥ 90% reduction from baseline) at 12 weeks.
Patient-reported HRQoL was assessed using 3 different instruments: the EORTC QLQ-C30,74 EORTC QLQ-PR25,75 and Aging Males’ Symptoms scale.76
The safety analysis included the incidence and severity of TEAEs recorded through 30 days after the last dose of relugolix or 4 weeks plus 30 days after degarelix injection.
Statistical Analysis
No stratification was implemented in the computer-generated randomization schedule.
The degarelix comparator group was included in the study to serve as a current ADT reference for qualitative comparisons. The study was not designed or powered to make a formal statistical inference of the difference between the effects of relugolix and those of degarelix. No subgroup was included in this study.
The primary and secondary end points were evaluated using the mITT population, which is defined as all randomized patients who have taken at least 1 dose of any study treatment. Time to castration (< 50 ng/dL) and time to profound castration (< 20 ng/dL) were summarized with Kaplan-Meier curves. Kaplan-Meier estimates were calculated for sustained castration rate, castration rate, profound castration rate, and testosterone recovery rate, along with a 95% CI calculated using the normal approximation method in each treatment group. Regarding time to castration and time to profound castration, if a patient’s testosterone measurements after the first dose remained consistently at or above a certain threshold (either ≥ 50 ng/dL or ≥ 20 ng/dL, depending on the type of castration), then the patient’s time to castration was censored at the last recorded testosterone measurement meeting or exceeding the specified threshold. For time to recovery, if a patient initiated ADT without experiencing testosterone recovery, their time to castration was censored at the last testosterone laboratory assessment conducted before starting the alternative ADT.
The change from baseline in the QoL total score, as assessed by the EORTC QLQ-C30, EORTC QLQ-PR25, and Aging Males’ Symptoms scale was summarized for each visit and treatment group, considering symptom, Global Health Status/quality-of-life and functional scale scores. A mixed model using repeated measures was conducted for the EORTC QLQ-PR25 and EORTC QLQ-C30, including baseline scores, treatment, visit, and the interaction between treatment groups and visit as covariates. The estimated means with 95% CIs from the mixed model using repeated measures was assessed for each treatment group at each visit.
A safety analyses set was defined as all randomized patients who received at least 1 dose of any study treatment.
Results
Patient Disposition
Table 33 presents detailed data on patient disposition.
Of 67 patients screened for the relugolix cohort, 65 were enrolled and 63 (97%) completed the 24 weeks of treatment followed by the follow-up period. Two patients (3%) discontinued the study in the relugolix cohort: 1 was lost to follow-up and 1 withdrew consent. In the degarelix cohort, of 39 patients screened, 38 were enrolled and 38 (100%) completed 24 weeks of treatment as well as the follow-up period.
Patient disposition | C27003 | |
|---|---|---|
Relugolix | Degarelix | |
Screened, N | 67 | 39 |
Randomized, N | 65 | 38 |
Discontinued from study, N (%) | 2 (3) | 0 |
Lost to follow-up | 1 (2) | 0 |
Withdrawal by patient | 1 (2) | 0 |
Participants who had completed 24 weeks of treatment, N (%) | 63 (97) | 38 (100) |
Follow-up participants, N | 63 (97) | 38 (100) |
Treated participants (mITT), N | 65 | 38 |
Safety, N | 65 | 38 |
mITT = modified intention to treat.
Sources: Sponsor’s summary of clinical evidence77 and C27003 Clinical Study Report.68
Baseline Characteristics
Table 34 provides detailed data on baseline characteristics.
In the mITT population, the mean and median ages of patients in both cohorts were 70 years and 71 years, respectively. In both cohorts, more than 80% of patients enrolled were white, while more Black or African American patients were enrolled in the degarelix (18%) cohort than in the relugolix cohort (11%). As for disease characteristics, the distribution of patients with different sites of primary tumour extension (T) at baseline was generally well balanced between the 2 cohorts. However, there were more patients with T2a/b in relugolix cohort (29%) than in the degarelix cohort (11%). More patients in the relugolix cohort had no node (N0) than in the degarelix cohort (60% versus 50%) at baseline. The majority of patients (> 70%) in both cohorts had had no distant metastasis (M0) at study entry, a Gleason score of 7 (62% to 68%), and an ECOG Performance Status of 0 (87% to 90%) at baseline. The median time since initial diagnosis for relugolix cohort was 0.2 years, whereas it was 0.1 years in the degarelix cohort. For both cohorts, the median PSA concentration at baseline was 7.3 ng/mL and median testosterone concentration was 12.4 nmol/L to 14 nmol/L.
Exposure to Study Treatments
The treatment duration was balanced between the relugolix and degarelix cohorts, with a median duration of exposure to treatment of 24.0 months. Compliance was high in both groups, with greater than 97% to 98% of participants being at least 80% compliant. None of the patients had a compliance rate below 80% in either of the 2 groups. Overall, 2 patients (2%) had a compliance rate above 120%, which could be attributed to the use of imputed data, which can lead to an overestimation of compliance (Table 35).
Overall, 97% of participants in both cohorts reported using at least 1 concomitant medication. The most commonly used concomitant medication was acetylsalicylic acid, which was taken by 46% in the relugolix group and 53% in the degarelix group. Other commonly used concomitant medications were ciprofloxacin, metformin, tamsulosin, tamsulosin hydrochloride, lisinopril, and simvastatin (Table 36).
Table 34: Summary of Baseline Characteristics of the C27300 Study — mITT Population
Characteristic | C27003 | |
|---|---|---|
Relugolix N = 65 | Degarelix N = 38 | |
Age | ||
Mean (SD) | 70.2 (5.65) | 70.3 (6.97) |
Median (minimum to maximum) | 71.0 (52 to 86) | 70.5 (52 to 81) |
Race, n (%) | ||
Black or African American | 7 (11) | 7 (18) |
White | 58 (89) | 31 (82) |
Primary tumour (T) at study entry, n (%) | ||
NA | 11 (17) | 8 (21) |
T1 | 21 (32) | 12 (32) |
T2 | 6 (9) | 5 (13) |
T2a | 12 (18) | 3 (8) |
T2b | 7 (11) | 1 (3) |
T2c | 7 (11) | 7 (18) |
T3 | 1 (2) | 1 (3) |
TX | 0 | 1 (3) |
Regional lymph nodes (N) at study entry, n (%) | ||
N0 | 39 (60) | 19 (50) |
NXa | 26 (40) | 19 (50) |
Distant metastasis (M) at study entry, n (%) | ||
M0 | 51 (78) | 28 (74) |
MXa | 14 (22) | 10 (26) |
Gleason score, n (%) | ||
6 | 5 (8) | 2 (5) |
7 | 40 (62) | 26 (68) |
8 | 5 (8) | 3 (8) |
9 | 2 (3) | 2 (5) |
Missing data | 13 | 5 |
ECOG Performance Status, n (%) | ||
0 | 60 (92) | 33 (87) |
1 | 4 (6) | 4 (11) |
Missing data | 1 | 1 |
Time since initial diagnosis, years | ||
Median (minimum to maximum) | 0.2 (0 to 6) | 0.1 (0 to 8) |
PSA concentration (ng/mL) | ||
Mean (SD) | 9.4 (6.0) | 14.6 (21.0) |
Median (interquartile range) | 7.3 (4.8 to 12.9) | 7.3 (5.5 to 11.2) |
Testosterone concentration (nmol/L) | ||
Mean (SD) | 14.1 (6.7) | 14.6 (5.6) |
Median (minimum to maximum) | 12.4 (5.2 to 44.8) | 14.0 (4.8 to 32.6) |
ECOG = Eastern Cooperative Oncology Group; mITT = modified intention to treat; PSA = prostate-specific antigen; SD = standard deviation.
aIncludes unknown, not available, and missing data.
Sources: Sponsor’s Summary of Clinical Evidence,77 C27003 Clinical Study Report,68 and Dearnaley et al. (2020).65
Table 35: Exposure and Compliance
Exposure and compliance | C27003 | |
|---|---|---|
Relugolix N = 65 | Degarelix N = 38 | |
Treatment duration (weeks)a | ||
Mean (SD) | 23.5 (2.7) | 24.1 (0.4) |
Median (minimum to maximum) | 24.0 (5.6 to 25.9) | 24.0 (23.6 to 25.1) |
Compliance,b n (%) | ||
≥ 80% to ≤ 120% | 64 (98) | 37 (97) |
> 120% | 1 (2) | 1 (3) |
SD = standard deviation.
aTreatment duration in weeks is calculated as (last dose date of study drug – first dose date of study drug + 1)/7 for relugolix. For patients in degarelix group, treatment duration = (last dose date of study drug – first dose date of study drug + 28) / 7.
bCompliance was assessed using 2 methods: first, by analyzing the data directly input by patients into a portable electronic device at home, and second, through onsite assessment involving the review of patient-reported compliance and an examination of pill counts. Compliance rate (%) = 100 × [total dose received (mg)/total dose expected (mg)]. If there were prescribed dose modifications, the expected dose was adjusted to account for prescription change and for missing dosing records from electronic diaries (relugolix only) without records confirming such doses missed by the patient, assuming the expected dose was taken when combined with a site assessment of the compliance (missing data imputed). The electronic diary was not used as a compliance measure for degarelix administration. For degarelix patients, a treatment duration greater than 165 days (3-day window) based on the site-entered date of last injection was considered as 24 weeks of treatment.
Sources: Sponsor’s Summary of Clinical Evidence77 and C27003 Clinical Study Report.68
Table 36: Concomitant Medications Used During the Study — mITT Population
Most common concomitant medications | C27003 | |
|---|---|---|
Relugolix (N = 65) | Degarelix (N = 38) | |
Participants using concomitant medication during treatment period, n (%)a | 63 (97) | 37 (97) |
Acetylsalicylic acid | 30 (46) | 20 (53) |
Ciprofloxacin | 6 (9) | 11 (29) |
Lisinopril | 12 (18) | 8 (21) |
Metformin | 8 (12) | 9 (24) |
Tamsulosin | 17 (26) | 7 (18) |
Tamsulosin hydrochloride | 12 (18) | 9 (24) |
Simvastatin | 12 (18) | 8 (21) |
Vitamins NOS | 13 (20) | 5 (13) |
mITT = modified intention to treat; NOS = not otherwise specified.
aFrequency of 20% or greater.
Source: Sponsor’s Summary of Clinical Evidence77 and C27003 Clinical Study Report.68
Efficacy
The results from the analysis of the sustained castration rate (testosterone level < 50 ng/dL) are provided in Table 37.
The proportions of patients who achieved sustained testosterone suppression from weeks 4 through 24 were 95% (95% CI, 87.1 to 99.0) in the relugolix treatment group, and 89% (95% CI, 75.2 to 97.1) in the degarelix group. Results of the sensitivity analysis (i.e., an analysis excluding patients who had high-risk localized PC) were consistent with the principal analyses of the primary end point. Profound castration was achieved by 82% and 68% of patients on relugolix and degarelix, respectively.
Responses to the EORTC QLQ-C30 Global Health Status domain, as well as the EORTC QLQ-PR25 sexual activity and hormone treatment–related symptom domains showed evidence of adverse effects during treatment in both the relugolix and degarelix groups. At week 24, The Global Health Status responses showed a deterioration of 10.1 points (SD = 18.9) in relugolix group and 7.5 points (SD = 13.7) in the degarelix group. Within 12 weeks after treatment discontinuation, the mean improvements in the sexual activity scores and hormonal treatment-related symptoms scores on the EORTC QLQ-PR25 were 12.1 (SD = 21.8) and −5.0 (SD = 10.3) in the relugolix group, and 6.6 (SD = 22.8) and −1.2 (SD = 9.1) in the degarelix group, respectively.
Table 37: Key Efficacy Outcomes During Treatment Phase — mITT Population
Key efficacy outcomes | C27003 | |
|---|---|---|
Relugolix N = 65 | Degarelix N = 38 | |
Primary end point | ||
Sustained castration (< 50 ng/dL) response rate from 4 to 24 weeks, % (95% CI)a | 95 (87.1 to 99.0) | 89 (75.2 to 97.1) |
Secondary end points | ||
Cumulative profound castration (< 20 ng/dL) response rate from 12 to 24 weeks, % (95% CI)a | 82 (70.0 to 90.1) | 68 (51.3 to 82.5) |
EORTC QLQ-C30 Global Health Status/quality-of-life score | ||
Baseline, n | 65 | 38 |
Mean (SD) | 82.6 (15.8) | 83.6 (13.6) |
Week 24, n | 60 | 38 |
Mean CFB (SD) | −10.1 (18.9) | −7.5 (13.7) |
Weeks 24 to 36 (12 weeks off-treatment), n | 58 | 38 |
Mean CFB (SD) | 2.3 (16.6) | 0.7 (15.5) |
Week 36 (at 12-week follow-up), n | 62 | 38 |
Mean CFB (SD) | −7.7 (17.8) | −6.8 (16.0) |
EORTC QLQ-PR25 sexual activity domain | ||
Baseline, n | 65 | 38 |
Mean (SD) | 35.1 (24.1) | 33.3 (24.5) |
Week 24, n | 60 | 38 |
Mean CFB (SD) | −19.7 (29.4) | −11.8 (36.3) |
Weeks 24 to 36 (12 weeks off-treatment), n | 58 | 38 |
Mean CFB (SD) | 12.1 (21.8) | 6.6 (22.8) |
Week 36 (at 12-week follow-up), n | 62 | 38 |
Mean CFB (SD) | −7.3 (30.0) | −5.3 (34.7) |
EORTC QLQ-PR25 hormone treatment–related symptoms domain | ||
Baseline, n | 65 | 38 |
Mean (SD) | 4.4 (6.7) | 2.5 (4.2) |
Week 24, n | 60 | 38 |
Mean CFB (SD) | 13.4 (12.1) | 12.9 (10.4) |
Weeks 24 to 36 (12 weeks off-treatment), n | 58 | 38 |
Mean CFB (SD) | −5.0 (10.3) | −1.2 (9.1) |
Week 36 (at 12-week follow-up), n | 62 | 38 |
Mean CFB (SD) | 8.5 (11.3) | 11.7 (10.2) |
CFB = change from baseline; CI = confidence interval; mITT = modified intent-to-treat; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-PR-25 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Prostate-Specific 25 Items; SD = standard deviation.
aThe 95% 2-sided CIs were calculated using the normal approximation method.
Sources: Sponsor’s summary of clinical evidence,77 C27003 Clinical Study Report,68 and Dearnaley et al. (2020).65
Harms
Key harms data are presented in Table 38.
The majority of patients in both cohorts reported at least 1 TEAE (86% for relugolix and 97% for degarelix). The most common AE in both treatment groups was hot flushes (57% in the relugolix cohort and 61% in the degarelix cohort). A higher percentage of individuals in the relugolix cohort reported fatigue (26% versus 16% in degarelix) and diarrhea (18% versus 13% in degarelix), whereas a higher percentage of individuals in degarelix reported dysuria (16% versus 8%), increased ALT (13% versus 0), increased blood testosterone (11% versus 3%), decreased urine flow (11% versus 2%), increased ALT (13% versus 0%), and injection-site erythema (11% versus 0%). The overall incidences of the AE of cataracts were 15% and 18% in the relugolix and degarelix groups, respectively.
One relugolix-treated patient (2%) and 3 degarelix-treated patients (8%) reported SAEs. In the relugolix cohort, SAEs included hypertension (2%) and headache (2%).
There were no AEs leading to study drug discontinuation or deaths in either treatment group.
Table 38: Key Harms Data During Treatment Phase — Safety Population
Adverse events | Relugolix N = 65 | Degarelix N = 38 |
|---|---|---|
Most common adverse events,a n (%) | ||
≥ 1 adverse eventa | 56 (86) | 37 (97) |
Hot flush b | 37 (57) | 23 (61) |
Fatigue | 17 (26) | 6 (16) |
Diarrhea | 12 (18) | 5 (13) |
Cataract | 10 (15) | 7 (18) |
Nocturia | 9 (14) | 5 (13) |
Pollakiuria | 8 (12) | 6 (16) |
Dysuria | 5 (8) | 6 (16) |
Increased blood testosterone | 2 (3) | 4 (11) |
Decreased urine flow | 1 (2) | 4 (11) |
Increased alanine transaminase | 0 (0) | 5 (13) |
Injection-site erythema | 0 (0) | 4 (11) |
Serious adverse events, n (%) | ||
Patients with ≥ 1 serious adverse event | 1 (2) | 3 (8) |
Ulcerative esophagitis | 0 (0) | 1 (3) |
Road traffic accident | 0 (0) | 1 (3) |
Malignant mesothelioma | 0 (0) | 1 (3) |
Headache | 1 (2) | 0 (0) |
Pleural effusion | 0 (0) | 1 (3) |
Hypertension | 1 (2) | 0 (0) |
Patients who stopped treatment due to adverse events, n (%) | 0 (0) | 0 (0) |
Deaths, n (%) | 0 (0) | 0 (0) |
aFrequency greater than 10%.
Sources: Sponsor’s Summary of Clinical Evidence,77 C27003 Clinical Study Report,68 and Dearnaley et al. (2020).65
Description of MVT-601 to 049
The MVT-601 to 049 study is a multicentre, phase I, open-label, 52-week, nonrandomized, parallel-group trial. During the 12-week primary study, 25 patients were enrolled at 17 sites in US, with no study site in Canada. Patients were enrolled in a 3-part parallel-cohort study to investigate the combination of relugolix with abiraterone plus a corticosteroid in men with mCSPC or mCRPC (part 1), and apalutamide in men with mCSPC (part 2). Part 3, which recruited patients treated with docetaxel, with or without prednisone, in the case of mCSPC or mCRPC (part 3) was discontinued due to a lack of enrolment and will not be discussed in this report. The ongoing MVT-601 to 049 study aims to assess the safety and tolerability of combining relugolix with abiraterone and apalutamide during the 12-week primary study treatment period. Additionally, the study aims to determine the proportion of patients who did not achieve castration levels of 50 ng/dL or higher in both part 1 and part 2 during the 12-week primary study treatment period.
The MVT-601 to 049 study consisted of a 45-day screening period followed by a 12-week treatment phase with 1 of the 2 combination treatments (parts 1 and 2) and a 40-week safety extension treatment period for a total of 52 weeks (ongoing). All participants must have undergone treatment with leuprolide or degarelix in conjunction with either abiraterone plus a corticosteroid (i.e., prednisone or methylprednisolone) or apalutamide before transitioning to relugolix. A total of 15 patients were enrolled in part 1, and an additional 10 patients were enrolled in part 2. Men diagnosed with advanced PC were to undergo a transition from leuprolide acetate or degarelix to relugolix (120 mg in part 1 or 240 mg in part 2, taken orally once daily following a single loading dose of 360 mg) as the ADT component of their treatment, initiated around the expected date of the next scheduled injection.
Populations
Patients eligible for inclusion in the MVT-601 to 049 study were required to have a documented medical history of either mCSPC for both part 1 and part 2, or mCRPC for part 1.
mCSPC (part 1 and part 2): at least 2 of the following factors:
a total Gleason score of 6 or higher and the presence of 2 or more metastatic lesions on bone scan
other radiologic evidence of measurable visceral metastases, excluding hepatic metastases
mCRPC (part 1): evidence of metastatic disease by imaging and demonstrated disease progression despite maintaining castration levels of testosterone with ADT, consistent with the Prostate Cancer Working Group 3 guidelines
Progression was evidenced by consecutive increases in PSA concentrations (2 measurements, at least 3 weeks apart) of 25% or higher and 2 ng/mL or greater above the nadir, along with the progression of pre-existing disease, including worsening symptoms and/or radiologic evidence of enlarged metastatic lesions, and/or the development of new metastases.
All patients were currently being treated with leuprolide acetate (3-, 4-, or 6-month injectable depot formulations delivered by intramuscular or subcutaneous injections) or degarelix in combination with either abiraterone plus a corticosteroid (part 1) or apalutamide (part 2). Prior treatment with abiraterone (part 1) or apalutamide (part 2) was required to have been well tolerated, defined as no evidence of hepatotoxicity requiring dose adjustment for a minimum of 12 weeks or no fracture, fall, or seizure episode, or the need for a dose adjustment due to an AE for a minimum of 6 weeks before the baseline visit, respectively.
Key exclusion criteria included having received combination therapy for a duration exceeding 24 months (for mCSPC) or 6 months (for mCRPC). Other exclusion criteria included a medical history of significant cardiac conditions within 6 months before screening.
Interventions
Patients were enrolled to receive 1 of 2 interventions in the study:
a single oral loading dose of relugolix 360 mg (three 120 mg tablets) administered on day 1, followed by a daily oral intake of a single 120 mg tablet; this was in combination with either abiraterone 1,000 mg once daily or fine-particle abiraterone 500 mg once daily, along with prednisone 5 mg once daily or twice daily (or methylprednisolone 4 mg twice daily) for a duration of 12 weeks
a single oral loading dose of relugolix 360 mg (three 120 mg tablets) administered on day 1, followed by a daily oral intake of 2 120 mg tablets; this was in combination with apalutamide 240 mg once daily for a duration of 12 weeks.
In part 2, the recommended daily dose of relugolix was adjusted to 240 mg. The decision to double the dose of relugolix considered that apalutamide is a weak-permeability glycoprotein inducer but a strong inducer of the CYP3A4 cytochrome. Moreover, doubling the dose of relugolix was a conservative approach based on the observation of reduced relugolix level with concomitant administration of rifampin (a strong permeability glycoprotein and CYP3A4 inducer that reduces exposure to relugolix by 55%).
Outcomes
The primary end point was the incidence and severity of TEAEs recorded from baseline to the end of the 12-week treatment period. A secondary end point included the percentage of patients who did not achieve castrate levels (≥ 50 ng/dL) throughout the 12-week period in parts 1 and 2 as measured by serum testosterone levels at baseline, as well as after 4 and 12 weeks of treatment.
Statistical Analysis
The primary and secondary end points were evaluated using the mITT population, which is defined as all randomized patients who have taken at least a single administration of relugolix in combination with abiraterone (part 1) or apalutamide (part 2). Analyses supporting the primary study objective involved a descriptive evaluation of safety and tolerability for each study part, based on safety assessments. Analyses supporting secondary study objectives included a descriptive summary of serum testosterone concentrations, along with the number and proportion of patients with testosterone concentrations ≥ 50 ng/dL at baseline, week 5, and week 13, categorized by study part (parts 1 and 2).
Results
Patient Disposition
Table 39 provides detailed data on patient disposition.
Of 15 patients enrolled in part 1 (relugolix plus abiraterone plus corticosteroids), 40% of patients completed 12 weeks of treatment followed by follow-up visits. The most common reason for discontinuing part 1 of the study was “not reported” (60%). Similarly, of 10 patients enrolled in part 2 (relugolix plus apalutamide), 30% completed the 12-week treatment period as well as follow-up visits. Most common reason reported for discontinuing part 2 of the study was “other reason” (60%), followed by “adverse event” (10%).
Table 39: Patient Disposition During Treatment Phase
Patient disposition | MVT-601 to 049 | |
|---|---|---|
Part 1 (relugolix + abiraterone + corticosteroids) | Part 2 (relugolix + apalutamide) | |
Screened, N | NR | |
Enrolled, N | 15 | 10 |
Discontinued from study, N (%) | 9 (60) | 7 (70) |
Adverse event | 0 (0) | 1 (10) |
Not reported | 9 (60) | 6 (60) |
Participants who had completed 12 weeks of treatment, N (%) | 6 (40) | 3 (30) |
Follow-up participants, N | 6 (40) | 3 (30) |
Treated participants (mITT), N | 15 | 10 |
mITT = modified intention to treat.
Sources: Sponsor’s Summary of Clinical Evidence77 and De La Cerda et al. (2023).66
Baseline Characteristics
Table 40 provides detailed data on baseline characteristics.
In mITT population, the mean age of patients in part 1 of the study was 72.3 years, whereas the mean age was 68.1 years for those in part 2 of the study. While 86.7% of patients had mCSPC and 13.3% had mCRPC at baseline in part 1 of the study, 100% of patients in part 2 had mCSPC. The majority of patients (90% to 100%) in both part 1 and 2 had an ECOG Performance Status of 0. In part 1 of the study, 53.3% of patients had a Gleason score of 8, and 20% had Gleason scores of 7 and 9 each. In part 2 of the study, 50% of patients had a Gleason score of 9, and 20% had Gleason scores of 8 and 1 each. Notable differences in PSA and testosterone concentrations were found between the cohorts. The median PSA concentration for the part 1 cohort was 0.05 ng/mL (range = 0.03 to 86.89) ng/mL, whereas that of part 2 was 0.08 ng/mL (range = 0.03 to 0.72). The median testosterone concentration for the part 1 cohort was 1 ng/mL (range = 1 to 8), whereas that of the part 2 cohort was 14.5 ng/mL (range = 10 to 26).
Table 40: Summary of Baseline Characteristics — mITT Population
Characteristic | MVT-601 to 049 | |
|---|---|---|
Part 1 (relugolix + abiraterone + corticosteroids) N = 15 | Part 2 (relugolix + apalutamide) N = 10 | |
Age | ||
Mean (SD) | 72.3 (7.87) | 68.1 (7.14) |
Median (minimum to maximum) | 71.0 (53 to 84) | 70.5 (56 to 76) |
Prostate cancer clinical state, n (%) | ||
mCSPC | 13 (86.7) | 10 (100.0) |
mCRPC | 2 (13.3) | 0 (0.0) |
ECOG Performance Status, n (%) | ||
0 | 15 (100.0) | 9 (90.0) |
1 | 0 (0.0) | 1 (10.0) |
Gleason score, n (%) | ||
7 | 3 (20.0) | 1 (10.0) |
8 | 8 (53.3) | 2 (20.0) |
9 | 3 (20.0) | 5 (50.0) |
10 | 1 (6.7) | 2 (20.0) |
PSA concentration (ng/mL) | ||
Mean (SD) | 8.64 (22.62) | 0.19 (0.23) |
Median (minimum to maximum) | 0.05 (0.03 to 86.89) | 0.08 (0.03 to 0.72) |
Testosterone concentration (ng/dL) | ||
Mean (SD) | 1.47 (1.81) | 15.50 (5.02) |
Median (minimum to maximum) | 1.00 (1.00 to 8.00) | 14.50 (10.00 to 26.00) |
ECOG = Eastern Cooperative Oncology Group; mCSPC = metastatic castration-sensitive prostate cancer; mCRPC = metastatic castration-resistant prostate cancer; mITT = modified intention-to-treat; SD = standard deviation.
Sources: Sponsor’s Summary of Clinical Evidence77 and De La Cerda et al. (2023).66
Exposure to Study Treatments
No information is available regarding exposure or adherence to study treatments.
Overall, all participants used at least 1 concomitant medication that was part of the treatment regimen. In part 1 of the study, 100% of patients received abiraterone and corticosteroid (either prednisone or methylprednisolone). In part 2 of the study, 100% of patients received apalutamide as part of their treatment regimen. Other concomitant medications potentially taken by patients were not reported.
Efficacy
All patients (100% in the mITT population, parts 1 and 2) achieved and maintained castration (testosterone < 50 ng/dL) after transitioning to relugolix at 4 and 12 weeks of treatment.
Harms
Key harms data are presented in Table 41.
The majority of patients in both groups reported at least 1 AE (86.7% for relugolix in combination with abiraterone with a corticosteroid; 80% for relugolix in combination with apalutamide). The most frequently reported AEs in both treatment groups (≥ 20%) included pain in an extremity, increased ALT, and anemia.
Serious adverse events were reported in 1 patient (6.7%) hospitalized due to a left femur fracture; the patient was receiving relugolix in combination with abiraterone with a corticosteroid (part 1), and in 1 patient with a neuroendocrine tumour (10%) who was treated with relugolix in combination with apalutamide (part 2).
One patient in the relugolix with apalutamide group discontinued from the study after approximately 4 weeks of treatment due to AEs, including fatigue, hot flushes, and body aches.
No deaths were reported in either treatment group.
Table 41: Key Harms Data During Treatment Phase — mITT Population
Adverse events | Relugolix + abiraterone + corticosteroidsa (N = 15) | Relugolix + apalutamideb (N = 10) |
|---|---|---|
Most common adverse events,a n (%) | ||
≥ 1 Adverse event | 13 (86.7) | 8 (80) |
Pain in extremity | 3 (20.0) | 1 (10) |
Increased alanine transaminase | 2 (13.3) | 2 (20) |
Anemia | 2 (13.3) | 2 (20) |
Serious adverse events, n (%) | ||
Patients with ≥ 1 serious adverse event | 1 (6.7) | 1 (10) |
Hospitalization due to a left femur fracture | 1 (6.7) | 0 (0) |
Neuroendocrine tumour | 0 (0) | 1 (10) |
Patients who stopped treatment due to adverse events, n (%) | 0 (0) | 1 (10) |
Deaths, n (%) | 0 (0) | 0 (0) |
mITT = modified intention to treat.
aFrequency of 20% or greater.
Sources: Sponsor’s Summary of Clinical Evidence77 and De La Cerda et al. (2023).66
Description of Apa-RP
The Apa-RP study was a multicentre phase II, open-label, single-arm trial. The trial was divided into a short-term substudy (N = 12) and a long-term main study. The Apa-RP substudy encompassed a peri-operative screening period covering RP conducted for all patients from day 29 to day 90. The pretreatment phase consisted of a 2-week period with relugolix monotherapy for substudy patients before enrolment, followed by a 12-month treatment phase and a subsequent 12-month follow-up period, resulting in a total duration of 24 months. Patients were enrolled if they were treatment-naive participants who had undergone RP for nonmetastatic PC and were at high risk for metastases.67,71 A total of 108 patients across 32 sites in the US were enrolled in the main study, with no study site in Canada.
The Apa-RP substudy aimed to assess whether the concurrent administration of apalutamide and relugolix could sustain castration levels of testosterone (< 50 ng/dL). Secondary objectives included evaluating the safety of relugolix monotherapy and the combination of relugolix with apalutamide. The Apa-RP main study investigated the rate of biochemical recurrence (a confirmed PSA > 0.2 ng/mL) in patients with high-risk localized PC after RP, following adjuvant therapy with apalutamide and relugolix. The main study also analyzed the time for testosterone levels to recover to at least 150 ng/dL and estimated the proportion of patients with testosterone recovery of 50 ng/dL or greater.
Populations
Eligible participants in the Apa-RP study were male, aged 18 years or older, and treatment-naive within 90 days post-RP. Participants were required to have PSA levels and histologically confirmed high-risk localized PC. A high risk for recurrent PC was defined as either a PSA of 20 ng/mL or higher or a Gleason score of 8 or higher (≥ 9 in any core on biopsy, or a Gleason score ≥ 8 [4 + 4 or 5 + 3] in greater than 80% of 2 cores, or a Gleason score of 8 [4 + 4 or 5 + 3] in 1 core with ≥ 5 other cores of a score ≥ 7).
Exclusion criteria encompassed patients with metastasis in soft tissue and/or bone or distant lymph nodes, a history of bilateral orchiectomy, a history of seizure, any condition or medication that may predispose to seizure, or MACE within 12 months before baseline.
Interventions
In the substudy, patients received a single oral loading dose of relugolix 360 mg (three 120 mg tablets) on day 1, followed by a daily oral intake of 120 mg as monotherapy for 2 weeks. Subsequently, their serum testosterone levels were confirmed to be less than 50 ng/dL. Following this confirmation, oral administration of apalutamide at a dosage of 240 mg once daily in combination with relugolix (120 mg once daily) was continued for 28 days. Substudy participants were then transitioned into the main study on day 28 and continued to receive a combination of relugolix plus apalutamide. In the main study, participants received oral apalutamide at a dosage of 240 mg once daily along with relugolix 120 mg once daily for 12 cycles, with each cycle lasting 28 days.
Outcomes
The primary efficacy end point in the substudy was the proportion of participants maintaining a castration testosterone level (< 50 ng/dL) through day 28, a parameter further evaluated throughout 1 year in the main study. This assessment was conducted using serum testosterone levels measured at baseline (day −14), day 1, and day 28, and subsequently every 3 months. Secondary objectives included evaluating the safety of relugolix monotherapy and relugolix plus apalutamide with TEAEs from days −14 to 28 (substudy) and up to 1 year (main study).
Statistical Analysis
The primary and secondary end points were assessed using the intention-to-treat population, encompassing all enrolled participants. Analyses supporting the primary study objective entailed a descriptive summary of the percentage of patients with testosterone levels below 50 ng/dL. According to the protocol, the substudy results would be deemed negative if 2 or more participants exhibited testosterone levels of 50 ng/dL or greater on day 28. Analyses supporting the secondary study objective in the substudy involved a descriptive evaluation of safety, with TEAEs recorded during both the relugolix monotherapy and relugolix-plus-apalutamide treatment periods. The proportion of patients with testosterone recovery of 50 ng/dL or greater was reported descriptively at 1 month.
No subgroup was included in this study.
Results
Patient Disposition
Of 19 patients screened, 12 patients were enrolled and all enrolled patients (100%) completed the substudy and rolled over to the main study. Two patients (16.7%) discontinued the main study by withdrawing consent.
Table 42: Patient Disposition — Treatment Phase
Patient disposition | Apa-RP study |
|---|---|
Screened, N | 19 |
Enrolled (relugolix plus apalutamide), N | 12 |
Discontinued from study, N (%) | 0 (0) |
Participants who had completed substudy and transitioned to main study, N (%) | 12 (100) |
Discontinuation from the main study (12 months), N (%) | 2 (16.7) |
Withdrawn consenta | 2 (16.7) |
Treated participants (ITT), N | 12 |
Safety, N | 12 |
ITT = intention to treat.
aOne participant was noncompliant with therapy, and the other withdrew for an unknown reason.
Sources: Sponsor’s Summary of Clinical Evidence77 and Brown et al. (2023).67
Baseline Characteristics
Table 43 provides detailed data on baseline characteristics.
In the ITT population, the median age of patients was 68 (range = 50 to 74) years. All but 1 patient (92%) had a metastasis staging of M0 at baseline, although information about metastasis staging for the 1 patient is missing. About two-thirds (67%) of patients had a nodal status of N0 at diagnosis, with 2 patients (17%) having N1 and NX each at initial diagnosis. A half (50%) of patients had tumour stage of T3 at diagnosis, whereas 3 patients (25%) had T1 and T2 each at initial diagnosis. Two-thirds (67%) of patients had Gleason score of 9 (4 + 5) at baseline with a median PSA of 7.4 ng/mL at initial diagnosis before RP.
Table 43: Summary of Baseline Characteristics, Apa-RP Substudy — ITT Population
Characteristic | Apa-RP |
|---|---|
Relugolix + apalutamide (N = 12) | |
Age, years | |
Median (minimum to maximum) | 68 (50 to 74) |
Metastasis stage at diagnosis, n (%) | |
M0 | 11 (92) |
Unknown | 1 (8.3) |
Nodal status at diagnosis, n (%) | |
N0 | 8 (67) |
N1 | 2 (17) |
NX | 2 (17) |
Tumour stage at diagnosis, n (%) | |
T1 | 3 (25) |
T2 | 3 (25) |
T3 | 6 (50) |
Gleason score, n (%) | |
7 (3 + 4) | 1 (8.3) |
8 (4 + 4) | 2 (17) |
9 (4 + 5) | 8 (67) |
10 (5 + 5) | 1 (8.3) |
PSA concentration at initial diagnosis before RP, ng/mL | |
Median (minimum to maximum) | 7.4 (4.2 to 26.2) |
ITT = intention to treat; PSA = prostate-specific antigen; RP = radical prostatectomy.
Sources: Sponsor’s Summary of Clinical Evidence77 and Brown et al. (2023).67
Exposure to Study Treatments
No information was provided by the sponsor regarding exposure to interventions. Without data from the sponsor, details on adherence to treatment cannot be described.
All 12 patients were prescribed concomitant medications, with lipid-modifying drugs (75%) and analgesics (67%) being the most frequently administered. Other commonly reported classes of medications included drugs acting on the renin-angiotensin system (50%) and vitamins (50%) (Table 44).
Table 44: Concomitant Medications Used During the Apa-RP Substudy — ITT Population
Concomitant medicationa | Apa-RP |
|---|---|
Relugolix + apalutamide (N = 12) | |
Participants using concomitant medication during treatment period, n (%) | 12 (100) |
Lipid-modifying drugs | 9 (75) |
Analgesics | 8 (67) |
Drugs acting on the renin-angiotensin system | 6 (50) |
Vitamins | 6 (50) |
ITT = intention-to-treat.
aConcomitant medications include those that are ongoing at the start of the first dose of relugolix treatment (day −14) or those taken during the treatment period, from the date of first relugolix dose through 28 days after the start of apalutamide plus relugolix treatment.
Sources: Sponsor’s Summary of Clinical Evidence77 and Brown et al. (2023).67
Efficacy
Key efficacy outcomes are presented in Table 45.
All 11 evaluable patients (i.e., with available data) achieved castration after 2 weeks of relugolix monotherapy (at day 1) and maintained castration testosterone levels by the end of combined apalutamide and relugolix treatment (day 28) without adjusting the relugolix dose. Specifically, the median baseline testosterone level was 348.5 ng/dL (range = 182 to 697), decreasing to 8.7 ng/dL (range = < 3 to 26) after 2 weeks of relugolix monotherapy. It then stabilized at 10.0 (range = < 3 to 35) ng/dL at day 28 of apalutamide and relugolix coadministration. These findings were confirmed at 1 year, with 100% of evaluable patients (10 of 12 initially) maintaining castration during concomitant therapy with relugolix plus apalutamide, and no patient requiring a dose modification of relugolix. The median testosterone level was 10.0 ng/dL after 1 year of treatment with relugolix (120 mg) combined with apalutamide, demonstrating no significant drug-drug interaction effects.
Table 45: Key Efficacy Outcomes During Treatment Phase, Apa-RT Substudy and Main Study — ITT Population
Key efficacy outcomes | Apa-RP |
|---|---|
Relugolix + apalutamide | |
Substudy | |
Primary end point: sustained castration rate (< 50 ng/dL) from day 1 through day 28, n | 11a |
Response rate, % | 100 |
Main Study | |
Sustained castration rate (< 50 ng/dL) from day 1 through 12 months), n | 10b |
Response rate, % | 100 |
ITT = intention to treat.
aOne patient inadvertently did not have testosterone measured at day 28.
bOne patient withdrew and another was noncompliant with therapy and subsequently withdrew. These 2 patients were not included in this analysis.
Sources: Sponsor’s Summary of Clinical Evidence,77 Brown et al. (2023) (peer-reviewed publication),67 and Brown et al. (2023) (conference proceeding).71
Harms
Key harms data are presented in Table 46.
A majority of patients (75%) reported at least 1 TEAE through 42 days of treatment, with 9 patients (75%) experiencing TEAEs during the 2 weeks of relugolix monotherapy and 8 (67%) during the 28-day relugolix-plus-apalutamide treatment. The most common was hot flushes (50% as monotherapy, 33% as combination therapy with apalutamide). No SAEs, AEs leading to drug discontinuation, or AEs leading to death occurred during the study period.
Table 46: Key Harms Data During the Treatment Phase, Apa-RT Substudy — ITT Population
Adverse events | Relugolix monotherapya N = 12 | Relugolix + apalutamideb N = 12 |
|---|---|---|
Adverse events, n (%) | ||
≥ 1 AE(s) | 9 (75) | 8 (67) |
Hot flush | 6 (50) | 4 (33) |
Hypertension | 0 (0) | 1 (8.3) |
Headache | 0 (0) | 1 (8.3) |
Hypoesthesias | 0 (0) | 1 (8.3) |
Restless leg syndrome | 0 (0) | 1 (8.3) |
Dry skin | 0 (0) | 1 (8.3) |
Rash | 1 (8.3) | 1 (8.3) |
Chest discomfort | 0 (0) | 1 (8.3) |
Fatigue | 4 (33) | 1 (8.3) |
Increased appetite | 0 (0) | 1 (8.3) |
Back pain | 0 (0) | 1 (8.3) |
Anxiety | 0 (0) | 1 (8.3) |
Diarrhea | 1 (8.3) | 0 (0) |
Urinary tract infection | 1 (8.3) | 0 (0) |
Serious adverse events, n (%) | 0 (0) | 0 (0) |
Patients who discontinued treatment due to AEs, n (%) | 0 (0) | 0 (0) |
AE leading to death, n (%) | 0 (0) | 0 (0) |
AE = adverse event; ITT = intention to treat; TEAE = treatment-emergent adverse event.
aTEAEs that occurred between day −14 and Day −1.
bTEAEs that occurred between day 1 and day 28.
Sources: Sponsor’s Summary of Clinical Evidence77 and Brown et al. (2023).67
Critical Appraisal
Internal Validity
The sponsor submitted additional studies that address some gaps in the evidence (i.e., the use of relugolix in neoadjuvant and/or adjuvant settings with RT and combination therapy in intensifying regimens other than enzalutamide). However, clinical experts consulted by CADTH noted that evidence supporting the use of relugolix in combination with other regimens, such as lutetium, is still missing, even though these patients have indications for ADT with add-on therapies. Moreover, major limitations with the submitted studies preclude drawing definitive conclusions. As all 3 studies were open-label and 2 did not include a comparator, the results from these studies cannot inform an analysis of the comparative effectiveness of relugolix. While the open-label design may not have influenced objective end points (i.e., testosterone levels, which are less likely to be influenced by performance and observation biases), reporting of AEs could still be influenced by these biases. Also, according to the clinical experts consulted by CADTH, patients with advanced PC typically require ADT indefinitely. In this regard, the durations of these studies may not be long enough to assess the efficacy and safety of relugolix, particularly with respect to MACEs that tend to occur over multiple years.
External Validity
None of the studies sites were located in Canada. Moreover, clinical experts consulted by CADTH noted that patients with cardiovascular diseases were excluded from these studies even though patients with advanced PC could have comorbidities such as cardiovascular diseases due to their older age and/or medications they usually take. Last, clinicians mentioned that inclusion of Black or African American patients up to a proportion of 18% in the C27300 study was noteworthy as this population is more likely to be affected by PC.
Discussion
Summary of Available Evidence
The HERO study was a phase II, multicentre, randomized, open-label evaluation of the efficacy and safety of relugolix, an oral medication, compared to injectable leuprolide, in men with advanced PC who required at least 1 year of continuous ADT. The study was conducted across 160 sites in 22 countries. Patients were stratified by geographic region, presence of metastatic disease, and age, and were randomized in a 2:1 ratio to receive either relugolix or leuprolide for 48 weeks. Key end points included sustained castration rate, profound castration rate, and PSA response rate. The study also evaluated testosterone recovery and quality-of-life indicators. The primary analysis was performed after 934 patients randomized to the study completed 48 weeks of treatment, and the final analysis occurred after an additional 144 metastatic patients completed treatment, bringing the total number of randomized patients to 1,078.
The sponsor provided an ITC that assessed the efficacy and safety of relugolix for patients with advanced PC compared to available treatments in Canada. Eight trials were eligible for inclusion in the NMAs, 7 of which were included in the network assessing testosterone suppression (< 50 ng/dL) as an efficacy outcome, and 4 were included in the network of MACEs as a safety outcome.
The sponsor submitted a number of phase I and II studies to address evidence gaps. The C27300 study is a multicentre, phase II, open-label, randomized, active-control, parallel-group study focusing on patients with intermediate-risk localized PC who require neoadjuvant and adjuvant ADT along with EBRT. Additionally, there is an ongoing 52-week, phase I, multicentre, open-label, nonrandomized, parallel-group study evaluating the combination of relugolix with abiraterone for adult males with mCSPC or mCRPC as the first part, and a combination of relugolix with apalutamide for males with mCSPC as a second part. This study also includes data from participants who transitioned from injectable leuprolide acetate or degarelix to relugolix as their ADT component, with an expected completion of November 2023. A substudy of a multicentre, phase II, open-label, single-arm study was also conducted. This substudy investigates ADT in combination with apalutamide in treatment-naive males who have undergone RP for nonmetastatic PC and are at high risk of metastasis, with the study anticipated to conclude by the end of October 2023.
Interpretation of Results
Efficacy
The HERO trial reported high efficacy in achieving and maintaining castration levels of testosterone with relugolix. The sustained castration rate was higher in the relugolix group (96.7%) compared to the leuprolide group (88.8%). Additionally, relugolix showed a faster and higher rate of profound castration. Subgroup analyses confirmed the consistency of treatment effects across various patient categories for the primary outcome. The safety profile of both treatments was similar, with the most common AEs being vasomotor symptoms. Relugolix demonstrated a lower numeric incidence of MACEs compared to leuprolide.
The lack of evidence for the efficacy of relugolix in combination with other treatments is a strong limiting factor. Contemporary treatment approaches emphasize combining ADT with systemic drugs such a docetaxel, darolutamide, abiraterone, or enzalutamide, particularly in patients with high-risk or high-volume disease.25,27 The HERO trial does not provide efficacy evidence for relugolix when used with these systemic therapies. Moreover, the study does not address the use of relugolix in patients undergoing RT The sponsor has tried to fill these evidence gaps with phase I and II studies. However, the nature and design of these studies poses significant limitations on the ability of the results to address the comparative efficacy and safety of relugolix and the evidence gaps.
Even though the ███ ████████ █████████ ██████████ ████████ ███ ██████ ██ █████████ ██████ █████ ████████ ████, various limitations of the ITC were noted, including the heterogeneity in study characteristics and patient populations. Limited data on between-study differences and potential biases in the trials included in the networks were available. Clinical experts consulted for this review noted imbalances in certain prognostic factors and effect modifiers (baseline testosterone concentrations, metastatic status of participants, and previous hormone therapy treatment), which raises concerns about possible bias in the NMA comparisons. Moreover, notable generalizability issues were brought up by the clinical experts, such as the short timeline of MACE assessment (i.e., 12 ± 3 months) and the fact that profound castration levels (< 20 ng/dL) would have been a more appropriate outcome in the clinical context in Canada. Considering these limitations, there is a high risk of bias in the comparison in this NMA, and the direction of that bias is unclear. The findings of the sponsor-submitted ITC therefore remain highly uncertain.
Harms
Overall, the safety profile of relugolix is consistent with that of the ADT therapeutic class. Relugolix exhibited a proportion of various AEs similar to that of leuprolide. The numerical incidence of MACEs in patients treated with relugolix was lower than in those treated with leuprolide.
Conclusion
The efficacy and safety of relugolix for patients with advanced PC was assessed in the HERO study, a phase III open-label, controlled trial that compared relugolix against leuprolide. The HERO trial enrolled 934 patients in its primary analysis. Patients were randomized in a 2:1 ratio to receive either relugolix or leuprolide for 48 weeks. Key end points included sustained castration rate and profound castration rate. The study also evaluated testosterone recovery and quality-of-life indicators. The primary analysis was performed after 48 weeks of enrolment, with the final analysis including additional participants with metastatic disease. The HERO trial achieved its primary end point of showing noninferiority against leuprolide and a sustained castration rate of greater than 90% in patients taking relugolix. Furthermore, relugolix achieved a higher profound castration rate at day 15, and a higher cumulative profound castration rate from day 29 through day 337. Safety results suggest that relugolix has a safety profile similar to that of the ADT therapeutic class.
A GRADE assessment of relevant outcomes indicated that, compared to leuprolide, there is a high certainty that relugolix will lead to a sustained castration rate, and a high certainty that relugolix will lead to more patients experiencing profound castration. There is a very low certainty of evidence to suggest any differences in MACEs or bone density loss between relugolix and leuprolide.
Given the limited evidence available to inform on relugolix suggest comparative efficacy and safety versus other ADT treatments, an ITC was submitted by the sponsor. However, limitations in the ITC the resulting estimates may be biased, and they do not support firm conclusions about the comparative efficacy and safety of relugolix.
A number of evidence gaps were identified by the sponsor, including the lack of evidence to inform on intensification therapy and adjuvant or neoadjuvant therapy. The sponsor submitted 3 phase I or phase II studies to address these gaps. However, due to the limitations in these studies, it is not possible to address these evidence gaps with a high degree of certainty.
References
1.Clinical Study Report (primary): MVT-601-3201. HERO: A multinational phase 3 randomized, open-label, parallel group study to evaluate the safety and efficacy of relugolix in men with advanced prostate cancer [internal sponsor’s report]. Basel (CH): Myovant Sciences GmbH; 2020 Mar 26.
2.Canadian Cancer Society. Prostate cancer statistics. 2022; https://cancer.ca/en/cancer-information/cancer-types/prostate/statistics. Accessed 2023 Feb 8.
3.Brenner DR, Poirier A, Woods RR, et al. Projected estimates of cancer in Canada in 2022. CMAJ. 2022;194(17):E601-E607. PubMed
4.Canadian Cancer Society. Prostate cancer. 2021; https://cancer.ca/en/cancer-information/cancer-types/prostate. Accessed 2023 Feb [sponsor supplied reference].
5.National Comprehensive Cancer Network. Guidelines for patients: Prostate cancer, early stage. 2022; https://www.nccn.org/patientresources/patient-resources/guidelines-for-patients/guidelines-for-patients-details?patientGuidelineId=49. Accessed 2023 Mar 15.
6.Bryant-Lukosius D, Browne G, DiCenso A, et al. Evaluating health-related quality of life and priority health problems in patients with prostate cancer: A strategy for defining the role of the advanced practice nurse. Can Oncol Nurs J. 2010;20(1):5-14. PubMed
7.Bubendorf L, Schopfer A, Wagner U, et al. Metastatic patterns of prostate cancer: An autopsy study of 1,589 patients. Hum Pathol. 2000;31(5):578-583. PubMed
8.Leslie SW, Soon-Sutton TL, R IA, Sajjad H, Skelton WP. Prostate cancer. StatPearls. Treasure Island (FL): StatPearls Publishing; 2024.
9.Saad F, Ivanescu C, Phung D, et al. Skeletal-related events significantly impact health-related quality of life in metastatic castration-resistant prostate cancer: Data from PREVAIL and AFFIRM trials. Prostate Cancer Prostatic Dis. 2017;20(1):110-116. PubMed
10.S AMEB, Salawu A, Brown JE. Bone health in men with prostate cancer: Review article. Curr Osteoporos Rep. 2019;17(6):527-537. PubMed
11.D’Amico AV, Moul J, Carroll PR, Sun L, Lubeck D, Chen MH. Cancer-specific mortality after surgery or radiation for patients with clinically localized prostate cancer managed during the prostate-specific antigen era. J Clin Oncol. 2003;21(11):2163-2172. PubMed
12.Moul JW. The evolving definition of advanced prostate cancer. Rev Urol. 2004;6 Suppl 8(Suppl 8):S10-17.
13.NCCN Clinical Practice Guidelines in Oncology (NCCN Guideline): Prostate Cancer. Version 1.2023. Plymouth Meeting (PA): National Comprehensive Cancer Network; 2022: https://nccn.org.
14.CADTH Reimbursement Review Provisional Funding Algorithm - Indication: Prostate cancer. Ottawa (ON): CADTH; 2023: https://www.cadth.ca/prostate-cancer-0. Accessed 2024 Mar 22.
15.So AI, Saad F. 2022 Update: Canadian Urological Association-Canadian Urologic Oncology Group guideline: Metastatic castration-naive and castration-sensitive prostate cancer summary of changes. Can Urol Assoc J. 2022;16(12):389-391. PubMed
16.Tannock IF, de Wit R, Berry WR, et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med. 2004;351(15):1502-1512. PubMed
17.Surveillance, Epidemiology, and End Results Program (SEER). SEER*Explorer: An interactive website for SEER cancer statistics. 2023; https://seer.cancer.gov/statistics-network/explorer/. Accessed 2023 Jun 15.
18.Gleason DF, Mellinger GT. Prediction of prognosis for prostatic adenocarcinoma by combined histological grading and clinical staging. J Urol. 1974;111(1):58-64. PubMed
19.Mary K. Gospodarowicz MK, Wittekind C, Brierley JD, eds. TNM Classification of Malignant Tumours, 8th edition. Hoboken (NJ): Wiley-Blackwell; 2016.
20.Canadian cancer statistics: A 2022 special report on cancer prevalence. TForonto (ON): Canadian Cancer Society; 2022: https://cdn.cancer.ca/-/media/files/research/cancer-statistics/2022-statistics/2022-special-report/2022_prevalence_report_final_en.pdf. Accessed 2024 Mar 22.
21.Statistics Canada. Table 17-10-0005-01 Population estimates on July 1st, by age and sex. 2023; https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1710000501. Accessed 2023 Sep 27.
22.Public Health Agency of Canada. Prostate cancer in Canada. 2020; https://www.canada.ca/en/public-health/services/publications/diseases-conditions/prostate-cancer.html. Accessed 2024 Mar 22.
23.So AI, Chi K, Danielson B, et al. 2022 Update: Canadian Urological Association-Canadian Urologic Oncology Group guideline: Metastatic castration-naive and castration-sensitive prostate cancer. Can Urol Assoc J. 2022;16(12):E581-E589. PubMed
24.Saad F, Aprikian A, Finelli A, et al. 2022 Canadian Urological Association (CUA)-Canadian Uro Oncology Group (CUOG) guideline: Management of castration-resistant prostate cancer (CRPC). Can Urol Assoc J. 2022;16(11):E506-e515. PubMed
25.Canadian Cancer Society. Treatments for locally advanced prostate cancer. 2021; https://cancer.ca/en/cancer-information/cancer-types/prostate/treatment/locally-advanced. Accessed 2023 Feb 8.
26.Cancer Care Alberta. Local Prostate Cancer - Clinical Practice Guideline GU-012 – Version 5. 2022; www.ahs.ca/guru. Accessed 2024 Mar 22.
27.National Cancer Institute. NCI Dictionary of cancer terms. https://www.cancer.gov/publications/dictionaries/cancer-terms. Accessed 2023 Mar 29.
28.Rosario DJ, Davey P, Green J, et al. The role of gonadotrophin-releasing hormone antagonists in the treatment of patients with advanced hormone-dependent prostate cancer in the UK. World J Urol. 2016;34(12):1601-1609. PubMed
29.Pokuri VK, Nourkeyhani H, Betsy B, et al. Strategies to circumvent testosterone surge and disease flare in advanced prostate cancer: Emerging treatment paradigms. J Natl Compr Canc Netw. 2015;13(7):e49-55. PubMed
30.Canadian Cancer Society. Treatments for metastatic castration-sensitive prostate cancer. 2021; https://cancer.ca/en/cancer-information/cancer-types/prostate/treatment/metastatic-castration-sensitive. Accessed 2023 Feb 8.
31.Orgovyx (relugolix): 120 mg oral tablets [product monograph]. Mississauga (ON): Sumitomo Pharma Switzerland GmbH; 2023 Oct 30.
32.Firmagon (Degarelix): 120 mg and 80 mg degarelix injection [product monograph]. North York (ON): Ferring Pharmaceuticals; 2016 Mar 18.
33.Lupron (leuprolide acetate): 3.75. 7.5, 11.25, 22.5, 30 mg pre-filled syringe for intramuscular injection [product monograph]. St-Laurent (QC): AbbVie Corporation; 2024 March 19.
34.Zeulide depot (leuprolide acetate): 3.75 mg and 22.5 mg powder for intramuscular injection [product monograph]. Mississauga (ON): Verity Pharmaceuticals Inc; 2024 Jan 08.
35.Eligard (leuprolide acetate): 7.5 mg, 22.5 mg, 30 mg and 45 mg powder for subcutaneous use [product monograph]. Oakville (ON): Tolmar International Ltd; 2024 Jan 26.
36.Suprefact (buserelin): 1 mg/mL injection and 1 mg/mL Nasal Solution [product monograph]. Mississauga (ON): Cheplapharm Arzneimittel GmbH; 2020 June 10.
37.Zoladex (goserelin): 13.6 mg depot injection [product monograph]. Mississauga (ON): TerSera Therapeutics LLC; 2017 Dec 17.
38.Trelstar (triptorelin): 3.75 mg, 11.25 mg and 22.5 mg powder for injectable suspension [product monograph]. Montreal (QC): Knight Therapeutics Inc; 2024 Mar 27.
39.Klotz L, Shayegan B, Guillemette C, et al. Testosterone suppression in the treatment of recurrent or metastatic prostate cancer: A Canadian consensus statement. Can Urol Assoc J. 2018;12(2):30-37. PubMed
40.Shore ND, Saad F, Cookson MS, et al. Oral relugolix for androgen-deprivation therapy in advanced prostate cancer. N Engl J Med. 2020;382(23):2187-2196. PubMed
41.Myovant Sciences GmbH. NCT03085095: A study to evaluate the safety and efficacy of relugolix in men with advanced prostate cancer (HERO). Bethesda (MD): U.S. National Library of Medicine; 2022: https://classic.clinicaltrials.gov/ct2/show/NCT03085095. Accessed 2023 Sept [sponsor supplied reference].
42.Clinical Study Report (final): MVT-601-3201. HERO: A multinational phase 3 randomized, open-label, parallel group study to evaluate the safety and efficacy of relugolix in men with advanced prostate cancer. Basel (CH): Myovant Sciences GmbH; 2021 Jan 14.
43.Appendix 4 to the guideline on the evaluation of anticancer medicinal products in man: Condition specific guidance (EMA/CHMP/703715/2012 Rev. 2). London (GB): European Medecines Agency; 2015: https://www.ema.europa.eu/en/documents/scientific-guideline/evaluation-anticancer-medicinal-products-man-appendix-4-condition-specific-guidance-revision-2_en.pdf. Accessed 2023 Oct 10.
44.Advanced prostate cancer: Developing gonadotropin releasing hormone analogues - guidance for industry. Silver Spring (MD): Food and Drug Administration; 2022: https://www.fda.gov/media/129027/download. Accessed 2023 Oct 10.
45.Clinical Study Statistical Analysis Plan: MVT-601-3201. HERO: A multinational phase 3 randomized, open-label, parallel group study to evaluate the safety and efficacy of relugolix in men with advanced prostate cancer. Basel (CH): Myovant Sciences GmbH; 2019 Nov 07.
46.Clinical Study Protocol: MVT-601-3201. HERO: A multinational phase 3 randomized, open-label, parallel group study to evaluate the safety and efficacy of relugolix in men with advanced prostate cancer. Basel (CH): Myovant Sciences GmbH; 2017 Jan 13.
47.Non-inferiority clinical trials to establish effectiveness: Guidance for industry. Silver Spring (MD): Food and Drug Administration; 2016: https://www.fda.gov/media/78504/download. Accessed 2024 Mar 22.
48.George DJ, Saad F, Cookson MS, et al. Impact of concomitant prostate cancer medications on efficacy and safety of relugolix versus leuprolide in men with advanced prostate cancer. Clin Genitourin Cancer. 2023;21(3):383-392.e382. PubMed
49.George DJ, Shore ND, Saad F, et al. Impact of concomitant prostate cancer therapy on efficacy and safety of relugolix versus leuprolide in men with advanced prostate cancer: Subgroup analysis from the phase III HERO study. J Clin Oncol. 2021;39(6_suppl):106-106. https://ascopubs.org/doi/abs/10.1200/JCO.2021.39.6_suppl.106. Accessed 2024 Mar 22.
50.Balshem H, Helfand M, Schunemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-406. PubMed
51.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: Informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. PubMed
52.Network Meta Analysis (NMA) of Therapies for Advanced Prostate Cancer [internal sponsor’s report]. In: Drug Reimbursement Review sponsor submission: Orgovyx (relugolix), 120 mg oral tablet. Mississauga (ON): Sumitomo Pharma Canada, Inc; 2023 Nov 13.
53.Turner RM, Davey J, Clarke MJ, Thompson SG, Higgins JP. Predicting the extent of heterogeneity in meta-analysis, using empirical data from the Cochrane Database of Systematic Reviews. Int J Epidemiol. 2012;41(3):818-827. PubMed
54.Owen RK, Tincello DG, Keith RA. Network meta-analysis: Development of a three-level hierarchical modeling approach incorporating dose-related constraints. Value Health. 2015;18(1):116-126. PubMed
55.Dias S, Welton NJ, Caldwell DM, Ades AE. Checking consistency in mixed treatment comparison meta-analysis. Stat Med. 2010;29(7-8):932-944. PubMed
56.Klotz L, Boccon-Gibod L, Shore ND, et al. The efficacy and safety of degarelix: A 12-month, comparative, randomized, open-label, parallel-group phase III study in patients with prostate cancer. BJU Int. 2008;102(11):1531-1538. PubMed
57.Ozono S, Tsukamoto T, Naito S, et al. Efficacy and safety of 3-month dosing regimen of degarelix in Japanese subjects with prostate cancer: A phase III study. Cancer Sci. 2018;109(6):1920-1929. PubMed
58.Weber F, Knapp G, Ickstadt K, Kundt G, Glass Ä. Zero-cell corrections in random-effects meta-analyses. Res Synth Methods. 2020;11(6):913-919. PubMed
59.Shore N, Crawford DE, Gittelman M, Tombal B, et al. Poster #84: Efficacy and safety of a 3-monthly depot formulation of degarelix compared with goserelin in prostate cancer. Conference proceedings of the 13th Annual Meeting of the Society of Urologic Oncology, November 28-30, 2012; Rockville, MD: https://suonet.org/docs/meetings/suo1211/2012_suo_program_book.aspx. Accessed 2023 Oct [sponsor supplied reference].
60.Margel D, Peer A, Ber Y, et al. Cardiovascular morbidity in a randomized trial comparing GnRH agonist and GnRH antagonist among patients with advanced prostate cancer and preexisting cardiovascular disease. J Urol. 2019;202(6):1199-1208. PubMed
61.Lopes RD, Higano CS, Slovin SF, et al. Cardiovascular safety of degarelix versus leuprolide in patients with prostate cancer: The primary results of the PRONOUNCE randomized trial. Circulation. 2021;144(16):1295-1307. PubMed
62.Systematic reviews: CRD’s guidance for undertaking reviews in health care. York (GB): Centre for Reviews and Dissemination, University of York; 2009: https://www.york.ac.uk/media/crd/Systematic_Reviews.pdf. Accessed 2022 Jun 15.
63.Higgins JPT, Thomas J, Chandler J, et al. Cochrane handbook for systematic reviews of interventions. 2022; www.training.cochrane.org/handbook. Accessed 2024 Mar 22.
64.National Institute for Health and Care Excellence. Single technology appraisal and highly specialised technologies evaluation: User guide for company evidence submission template. (NICE process and methods PMG24). 2000; https://www.nice.org.uk/process/pmg24/resources/single-technology-appraisal-and-highly-specialised-technologies-evaluation-user-guide-for-company-evidence-submission-appendices-10956190861/chapter/instructions-for-companies. Accessed 2022 Jun 15.
65.Dearnaley DP, Saltzstein DR, Sylvester JE, et al. The oral gonadotropin-releasing hormone receptor antagonist relugolix as neoadjuvant/adjuvant androgen deprivation therapy to external beam radiotherapy in patients with localised intermediate-risk prostate cancer: A randomised, open-label, parallel-group phase 2 trial. Eur Urol. 2020;78(2):184-192. PubMed
66.De La Cerda J, Dunshee C, Gervasi L, et al. A phase I clinical trial evaluating the safety and dosing of relugolix with novel hormonal therapy for the treatment of advanced prostate cancer. Target Oncol. 2023;18(3):383-390. PubMed
67.Brown G, Belkoff L, Hafron JM, et al. Coadministration of apalutamide and relugolix in patients with localized prostate cancer at high risk for metastases. Target Oncol. 2023;18(1):95-103. PubMed
68.Clinical Study Protocol: C27003. A Phase 2, randomized, open-label, parallel group study evaluating the safety and efficacy of TAK-385, an oral gonadotropin-releasing hormone (GNRH) antagonist, for patients with localized prostate cancer requiring neoadjuvant and adjuvant androgen deprivation therapy with external beam radiation therapy (EBRT) [internal sponsor’s report]. Cambridge (MA): Millennium Pharmaceuticals, Inc; 2016 Nov 21.
69.Millennium Pharmaceuticals Inc. NCT02135445: Safety and efficacy of TAK-385 for patients with localized prostate cancer. Bethesda (MD): U.S. National Library of Medicine; 2017: https://classic.clinicaltrials.gov/ct2/show/NCT02135445. Accessed 2023 Oct [sponsor supplied reference].
70.Sumitomo Pharma Switzerland GmbH. NCT04666129: Study of relugolix in men with metastatic castration-sensitive prostate cancer or non-metastatic or metastatic castration-resistant prostate cancer. Bethesda (MD): U.S. National Library of Medicine; 2022: https://classic.clinicaltrials.gov/ct2/show/NCT04666129. Accessed 2023 Oct [sponsor supplied reference].
71.Brown G, Belkoff L, Hafron J, et al. Maintenance of castration with concomitant relugolix and apalutamide (Apa) in patients with high-risk localized prostate cancer (HR-LPC): 1 year update. J Clin Oncol. 2023;41(16_suppl):e17094-e17094. https://ascopubs.org/doi/10.1200/JCO.2023.41.16_suppl.e17094. Accessed 2023 Oct [sponsor supplied reference].
72.Janssen Research & Development, LLC. NCT04523207: A study of apalutamide (adjuvant treatment) and androgen deprivation therapy (ADT) in participants who have undergone radical prostatectomy (RP) for non-metastatic prostate cancer and who are at high risk for metastases. Bethesda (MD): U.S. National Library of Medicine; 2023: https://classic.clinicaltrials.gov/ct2/show/NCT04523207. Accessed 2023 Oct [sponsor supplied reference].
73.NCCN Clinical Practice Guidelines in Oncology (NCCN Guideline): Prostate Cancer. Version 4.2013. Plymouth Meeting (PA): National Comprehensive Cancer Network; 2013: https://www.nccn.org/.
74.Aaronson NK, Ahmedzai S, Bergman B, et al. The European Organization for Research and Treatment of Cancer QLQ-C30: A quality-of-life instrument for use in international clinical trials in oncology. J Natl Cancer Inst. 1993;85(5):365-376. PubMed
75.O’Leary E, Drummond FJ, Gavin A, Kinnear H, Sharp L. Psychometric evaluation of the EORTC QLQ-PR25 questionnaire in assessing health-related quality of life in prostate cancer survivors: A curate’s egg. Qual Life Res. 2015;24(9):2219-2230. PubMed
76.Moore C, Huebler D, Zimmermann T, Heinemann LA, Saad F, Thai DM. The Aging Males’ Symptoms scale (AMS) as outcome measure for treatment of androgen deficiency. Eur Urol. 2004;46(1):80-87. PubMed
77.Orgovyx (relugolix) for the treatment of adult patients with advanced prostate cancer [internal sponsor’s report]. In: Drug Reimbursement Review sponsor submission: Orgovyx (relugolix), 120 mg oral tablet. Mississauga (ON): Sumitomo Pharma Canada, Inc; 2023 Nov 13.
Pharmacoeconomic_Review
Abbreviations
ADT
androgen deprivation therapy
BIA
budget impact analysis
CDH
Canadian Drug and Hospital
CMA
cost-minimization analysis
GnRH
gonadotropin-releasing hormone
ITC
indirect treatment comparison
MACE
major adverse cardiovascular event
NMA
network meta-analysis
PC
prostate cancer
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Relugolix (Orgovyx), 120 mg oral tablets |
Indication | For the treatment of adult patients with advanced prostate cancer |
Health Canada approval status | NOC |
Health Canada review pathway | Standard review |
NOC date | October 10, 2023 |
Reimbursement request | As per indication |
Sponsor | Sumitomo Pharma Canada, Inc. |
Submission history | Previously reviewed: No |
NOC = Notice of Compliance.
Table 2: Summary of Economic Information
Component | Description |
|---|---|
Type of economic evaluation | Cost-minimization analysis |
Target population | Adult patients with advanced prostate cancer |
Treatment | Relugolix |
Dosing regimen | 360 mg once (loading dose) then 120 mg once daily (maintenance dose) |
Submitted price | Relugolix: $9.00 per 120 mg oral tablet |
Submitted treatment cost | $3,303 in year 1; $3,285 in subsequent years |
Comparators | ADTs include:
|
Perspective | Canadian publicly funded health care payer |
Time horizon | Undefined (year 1 and subsequent year) |
Key data sources | Key assumption of equal treatment efficacy and safety of relugolix based on:
|
Costs considered | Drug-acquisition costs |
Submitted results | Compared to other ADTs, the cost savings associated with relugolix ranged from $185 to $1,864 in year 1 and from $5 to $1,872 in subsequent years |
Key limitations |
|
CADTH reanalysis results |
|
ADT = androgen deprivation therapy; ITC = indirect treatment comparison.
Conclusions
The CADTH Clinical Review of the 48-week HERO trial demonstrated the noninferiority of relugolix against leuprolide acetate and included the end points of sustained castration rate (defined as testosterone levels lower than 50 ng/dL at 12 ± 3 months), profound castration rates (< 20 ng/dL) and safety (e.g., major adverse cardiovascular events [MACEs]). The CADTH Clinical Review concluded that, compared to leuprolide, there is a high certainty that relugolix will likely lead to a sustained castration rate, and a high certainty that relugolix will likely lead to more patients experiencing profound castration. The evidence supporting any differences in MACEs or bone density loss between relugolix and leuprolide is of very low certainty. Although the sponsor submitted an indirect treatment comparison (ITC) comparing relugolix against other androgen deprivation therapies (ADTs), the CADTH Clinical Review identified various limitations surrounding the ITC, including heterogeneity in study characteristics and patient populations, as well as issues with generalizability. As such, the CADTH Clinical Review found that the ITC findings cannot support firm conclusions about the comparative efficacy and safety of relugolix versus other ADTs.
The existence and extent of cost savings with relugolix are highly dependent on the comparator ADT, the dosing form of choice (which varies by jurisdiction), and the participating drug plan prices of ADTs. CADTH accepted the sponsor’s cost-minimization analysis (CMA), which reports that relugolix is associated with cost savings ranging from $185 to $1,864 per patient in the first year of treatment and from $5 to $1,872 per patient in subsequent years of treatment when compared to weighted annual drug costs for each ADT. The largest estimated cost savings are relative to buserelin, which accounts for only 0.03% of ADTs used in clinical practice, as confirmed by clinical expert feedback. Conversely, the annual cost difference per patient of relugolix relative to the most commonly used forms of ADT (leuprolide acetate [Eligard], 45 mg and 22.5 mg) ranged from added costs of $13 per patient in year 1 to cost savings of $279 per patient annually in subsequent years, based on publicly available prices of comparators.
Economic Review
The current review is for relugolix (Orgovyx) for adult patients with advanced prostate cancer.
Economic Information
Summary of Sponsor’s Economic Information
The sponsor submitted a CMA comparing relugolix with other ADTs (buserelin, degarelix, goserelin acetate, leuprolide acetate, and triptorelin) for the treatment of adult patients with advanced prostate cancer from the perspective of a Canadian public health system.1 The analysis was conducted separately for the first year of treatment and subsequent years, with no discounting applied.
The sponsor assumed clinical equivalence (equal efficacy and safety) between relugolix and other ADTs based on the results of the phase III noninferiority HERO trial2 (compared to leuprolide acetate), a published network meta-analysis (NMA), and a sponsor-commissioned ITC (compared to other selected ADTs). The ITC included data for degarelix, leuprolide acetate, triptorelin, and goserelin acetate (the HERO, CS21, Sun [2020], Ozono [2018], CS35, Margel [2019], Heyns [2003], and PRONOUNCE studies) but not for buserelin.3 The ITC included outcomes of interest for efficacy (testosterone suppression defined as sustained chemical castration with testosterone levels lower than 50 ng/dL at 12 ± 3 months) and safety (MACEs). To further support the sponsor’s assumption of clinical equivalence, a published NMA comparing the safety and efficacy of gonadotropin-releasing hormone (GnRH) agonists to antagonists was also cited.3 However, the studies included in this published NMA (HERO, CS21, Sun [2020], and Ozono [2018]) were already identified in the sponsor’s ITC.3 Only drug-acquisition costs were included in the model (all other costs were assumed to be equal across treatments, and administration costs were included in a scenario analysis). All dosing information used to calculate drug costs was based on the respective product monograph information.1 Some comparators such as leuprolide acetate, buserelin, and triptorelin were available in multiple dosage forms and the sponsor therefore used claims data from the most recent Canadian Drug and Hospital (CDH) audit dated June 2022 to May 2023 to estimate the proportion of utilization per strength to calculate a weighted annual drug cost for each comparator.1,4 The sponsor estimated annual total drug costs for each treatment in year 1 and subsequent years and estimated incremental drug costs comparing relugolix to each comparator. Costs of concomitant antiandrogen therapy were also included as part of drug costs, assuming that 28.6% of patients treated with GnRH agonists (i.e., all ADTs included in the analysis except degarelix acetate and relugolix) would receive concomitant bicalutamide for 4 weeks ($35.53).1
At the submitted price of $9.00 per 120 mg oral tablet, the sponsor estimated that relugolix was associated with cost savings ranging from $185 to $1,864 per patient in the first year of treatment and $5 to $1,872 per patient in subsequent years of treatment when compared to other ADTs (Table 3).
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total treatment costs ($) | Incremental treatment costs ($) vs. relugolix | ||
|---|---|---|---|---|
Year 1 | Year 1+ | Year 1 | Year 1+ | |
Relugolix | 3,303 | 3,285 | Reference | Reference |
Degarelix | 3,758 | 3,290 | −455 | −5 |
Leuprolide acetate (Lupron Depot) | 4,326 | 4,316 | −1,023 | −1,031 |
Leuprolide acetate (Zeulide Depot) | 3,503 | 3,493 | −200 | −208 |
Leuprolide acetate (Eligard) | 3,488 | 3,478 | −185 | −193 |
Buserelin | 5,167 | 5,157 | −1,864 | −1,872 |
Triptorelin | 3,876 | 3,866 | −573 | −581 |
Goserelin acetate | 4,047 | 4,819 | −744 | −1,534 |
Note: The negative incremental costs represent cost savings. Results were presented deterministically, as only treatment-acquisition costs were included in the base case.
Source: Sponsor’s economic submission.1
The sponsor conducted sensitivity analyses that included a comparison of the cost of relugolix to the weighted average cost of all reimbursed ADTs (Table 5), the concomitant use of bicalutamide for all patients, and administration costs associated with intramuscular and subcutaneous treatments based on physician fees in Ontario. Relugolix remained cost-saving compared to other ADTs in all scenarios. In the scenario comparing the cost of relugolix to the weighted average cost of all reimbursed ADTs, relugolix was associated with cost savings of $601 (15.4%) and $835 (20.3%) per patient, in the first and subsequent years of treatment, respectively.
CADTH Appraisal of the Sponsor’s Economic Information
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis:
The sponsor’s assumption of clinical equivalence between relugolix and other ADTs is uncertain: The sponsor submitted a CMA assuming clinical similarity between relugolix and other ADTs used in clinical practice to treat adult patients with advanced prostate cancer. The pivotal trial2 (relugolix versus leuprolide acetate) and sponsor-submitted ITC3 (relugolix versus degarelix, leuprolide acetate, triptorelin, and goserelin acetate) examined sustained castration rate (defined as testosterone levels lower than 50 ng/dL at 12 ± 3 months) and safety (e.g., MACEs). According to the CADTH Clinical Review, the HERO trial demonstrated the noninferiority of relugolix compared with leuprolide acetate and suggested that relugolix has a safety profile similar to that of the ADT therapeutic class. Although ITC findings suggested |||||||||| |||||||| ||| |||||| || ||||||||| |||||| ||||| ||||, the CADTH Clinical Review identified various limitations surrounding the ITC, including heterogeneity in study characteristics and patient populations, as well as issues with generalizability. Limitations with generalizability were due to concerns with the short timeline of MACE assessment (e.g., 12 months) and the appropriateness of outcomes (e.g., profound castration levels would have been a more appropriate outcome in the Canadian context). As such, the CADTH Clinical Review found that the ITC findings are associated with high levels of uncertainty. Buserelin acetate was not included in the ITC, but its exclusion was not expected to have a large impact on the CMA estimates due to the limited use of buserelin in clinical practice. Other factors that may introduce uncertainty into the potential cost savings associated with relugolix include concerns related to adherence, according to clinical experts consulted by CADTH. Because relugolix requires once-daily oral administration, it is uncertain whether having to take another daily pill (instead of 1 injection monthly or every other month), in the context of advanced prostate cancer may be aligned with patient preference or convenience. This could potentially affect adherence to treatment and therefore the assumption of clinical equivalence.
CADTH was unable to address this limitation in reanalysis.
Cost savings associated with relugolix compared to other ADTs are highly variable depending on the treatment and dosing received: While results of the sponsor’s CMA indicate that relugolix generally results in cost savings compared to other ADTs in Canada, the effects on costs range from small incremental costs to costs savings. The largest incremental cost savings associated with relugolix are relative to buserelin ($1,872 per patient per year), which is a comparator that is not generally used in current clinical practice in Canada, according to feedback from the clinical experts. The estimated proportion of buserelin use in Canada is 0.03% according to IQVIA CDH audit data obtained by the sponsor for June 2022 to May 2023 (Table 5). However, data from the same audit also indicate that leuprolide acetate (Eligard) is the most frequently used ADT in Canada (37.36%), specifically its 45 mg vial size (used by 46.51% of all patients receiving leuprolide acetate [Eligard]) and 22.5 mg vial size (used by 38.82% of all patients receiving leuprolide acetate [Eligard]). When comparing the annual drug costs of relugolix to the 45 mg vial size of leuprolide acetate (Eligard), the cost savings per patient in subsequent years of treatment are limited to $5 per patient and require an additional $13 in expenditures in the first year of treatment. Similarly, when comparing the costs of relugolix to degarelix, the only other publicly reimbursed GnRH antagonist, the cost savings are approximately $5 per patient in subsequent years of treatment. When comparing the annual drug costs of relugolix to the 22.5 mg vial size of leuprolide acetate (Eligard), the cost savings per patient range from $261 to $279 per year, in year 1 and subsequent years, respectively. The sponsor included a scenario analysis comparing the cost of relugolix to a weighted average cost of all reimbursed ADTs according to CDH data (Table 5). Relugolix was associated with drug cost savings of $601 per patient in the first year of treatment and $835 per patient in subsequent years of treatment. According to these estimates based on the proportion of ADT use in Canadian clinical practice, the cost savings of relugolix are likely lower than the upper range estimated by the sponsor.
CADTH conducted a scenario analysis examining the drug cost savings associated with relugolix compared to the most commonly used ADT and its most frequently used drug formulations in practice in Canada (leuprolide acetate [Eligard] 45 mg and 22.5 mg). The estimated cost differences of using relugolix ranged from an incremental cost of $13 to a cost saving of $279 per patient per year, in year 1 and subsequent years respectively (Table 6). CADTH notes that these savings are different from the sponsors’ cost-comparison estimates for Eligard (Table 3) because the sponsor assumed weighted costs of all dosing forms of leuprolide.
The analysis was based on publicly available list prices: Both CADTH and the sponsor’s analyses are based on publicly available list prices for comparator ADTs. However, the confidentially negotiated prices for comparator ADTs are unknown and the submitted price of relugolix may require a price reduction to avoid incurring additional costs relative to comparators such as degarelix (the only other GnRH antagonist) or leuprolide acetate (Eligard) 45 mg or 22 mg (the most commonly used forms among all comparators). Similarly, the cost savings associated with relugolix relative to other comparator ADTs, such as goserelin and leuprolide acetate (Lupron Depot), may be less than those estimated by the sponsor.
CADTH was unable to address this limitation in reanalysis, as the negotiated prices of comparator ADTs are unknown.
CADTH Reanalyses of the Economic Information
CADTH did not conduct a base-case reanalysis and accepted the sponsor’s submitted base-case and scenario analyses.
CADTH conducted an additional scenario analysis in which the drug costs of relugolix were compared to the most-used ADT and its most frequently used drug formulations (leuprolide acetate [Eligard], 45 mg and 22.5 mg). These scenarios indicated that the total cost savings associated with relugolix versus the most commonly used ADT forms in clinical practice ranged from added costs of $13 per patient in year 1 to $279 cost savings per patient annually in subsequent years of treatment (Table 6).
Issues for Consideration
Clinical expert feedback obtained by CADTH indicated that relugolix may be considered for use in patients expecting to receive treatment with ADTs. However, given the accelerated testosterone recovery associated with relugolix, this would likely have impacts on the duration of testosterone suppression upon treatment discontinuation and there is currently no evidence to support the assumption that it confers the same survival benefits as the other available ADTs (i.e., injectables or implants). This should be taken into consideration in shared decision-making processes, particularly in the context of intermittent use as an attempt to minimize the adverse effects of medical castration, or for those patients with a strong preference for oral treatment or difficult access to the other ADT forms.
Relugolix is administered orally once daily, while comparator ADTs require administration costs associated with subcutaneous and intramuscular injections or implants. Cost savings may be greater due to lack of administration costs associated with relugolix; however, patient preference for less-frequent treatment (e.g., convenience of semiannual injection versus a once-daily pill) may limit the extent of cost savings in practice.
Conclusions
The CADTH Clinical Review of the 48-week HERO trial demonstrated the noninferiority of relugolix against leuprolide acetate and included the end points of sustained castration rate (defined as testosterone levels lower than 50 ng/dL at 12 ± 3 months), profound castration rates (< 20 ng/dL), and safety (e.g., MACEs). The CADTH Clinical Review concluded that, compared to leuprolide, there is a high certainty that relugolix will likely lead to a sustained castration rate, and a high certainty that relugolix will likely lead to more patients experiencing profound castration. The evidence supporting differences in MACEs or bone density loss between relugolix and leuprolide is of very low certainty. Although the sponsor submitted an ITC comparing relugolix versus other ADTs, the CADTH Clinical Review identified various limitations surrounding the ITC, including heterogeneity in study characteristics and patient populations, as well as issues with generalizability. As such, the CADTH Clinical Review found that the ITC’s findings cannot support firm conclusions about the comparative efficacy and safety of relugolix versus other ADTs.
The existence and extent of cost savings with relugolix are highly dependent on the comparator ADT, the dosing form of choice (which varies by jurisdiction), and the participating drug plan prices of ADTs. CADTH accepted the sponsor’s CMA, which reports that relugolix is associated with cost savings ranging from $185 to $1,864 per patient in the first year of treatment and from $5 to $1,872 per patient in subsequent years of treatment when compared to weighted annual drug costs for each ADT. The largest estimated cost savings are relative to buserelin, which is not widely used in clinical practice (0.03%), as confirmed by clinical expert feedback. Conversely, the cost difference per patient annually of relugolix relative to the most commonly used forms of ADT (leuprolide acetate [Eligard], 45 mg and 22.5mg) ranged from added costs of $13 per patient in year 1 to cost savings of $279 per patient annually in subsequent years, based on publicly available prices of comparators.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Orgovyx (relugolix tablets, 120 mg), oral. Mississauga (ON): Sumitomo Pharma Canada, Inc; 2023 Nov 13.
2.Clinical Study Report: MVT-601-3201. HERO: A multinational phase 3 randomized, open-label, parallel group study to evaluate the safety and efficacy of relugolix in men with advanced prostate cancer. Basel (SE): Myovant Sciences GmbH; 2020 Mar 26.
3.Sponsor's NMA report title [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Orgovyx (relugolix tablets, 120 mg), oral. Mississauga (ON): Sumitomo Pharma Canada, Inc; 2023 Nov 13.
4.IQVIA. Canadian Drugstore and Hospital Purchases Audit (CDH): May 2022-June 2023. https://www.iqvia.com/. Accessed 2023 Nov 28.
5.Ontario Ministry of Health. Ontario Drug Benefit (ODB) formulary. 2023; https://www.formulary.health.gov.on.ca/formulary/. Accessed 2023 Dec 15.
6.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Orgovyx (relugolix tablets, 120 mg), oral. Mississauga (ON): Sumitomo Pharma Canada, Inc; 2023 Nov 13.
7.Canadian Cancer Society. Prostate cancer statistics. 2022; https://cancer.ca/en/cancer-information/cancer-types/prostate/statistics. Accessed 2023 Feb 8.
8.Bolla M, de Reijke TM, Van Tienhoven G, et al. Duration of androgen suppression in the treatment of prostate cancer. N Engl J Med. 2009;360(24):2516-2527. PubMed
9.Lawton CAF, Lin X, Hanks GE, et al. Duration of androgen deprivation in locally advanced prostate cancer: Long-term update of NRG oncology RTOG 9202. Int J Radiat Oncol Biol Phys. 2017;98(2):296-303. PubMed
10.Hussain M, Tangen C, Higano C, Vogelzang N, Thompson I. Evaluating intermittent androgen-deprivation therapy phase III clinical trials: The devil is in the details. J Clin Oncol. 2016;34(3):280-285. PubMed
11.Hussain M, Tangen CM, Berry DL, et al. Intermittent versus continuous androgen deprivation in prostate cancer. N Engl J Med. 2013;368(14):1314-1325. PubMed
Appendix 1: Additional Economic Information
Please note that this appendix has not been copy-edited.
Cost-Comparison Table
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical experts. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 4: CADTH Cost-Comparison Table of ADTs for Adult Patients with Advanced PC
Treatment | Strength | Dosage form | Price ($) | Recommended dosage | Daily cost ($) | 28-day cost ($) |
|---|---|---|---|---|---|---|
Relugolix (Orgovyx) | 120 mg | Oral tablet | 9.000 | Loading dose: 360 mg once Maintenance dose: 120 mg once daily | Cycle 1: 9.64 Subsequent: 9.00 | Cycle 1: 270 Subsequent: 252 |
Other GnRH receptor antagonists | ||||||
Degarelix acetate (Firmagon) | 80 mg 120 mg | Powder for SC injection | 274.1760 370.9440 | Loading dose: 240 mg once Maintenance dose: 80 mg monthly one month after starting dose | Cycle 1: 26.50 Subsequent: 9.79 | Cycle 1:742 Subsequent: 274 |
GnRH receptor agonists | ||||||
Buserelin acetate (Suprefact depot) | 6.30 mg 9.45 mg | SC implant | 870.0704 1,289.3248 | 6.30 mg every 2 months 9.45 mg every 3 months | 15.35 15.54 | 430 435 |
Goserelin acetate (Zoladex) | 3.6 mg 10.8 mg | Prefilled syringe for SC injection | 422.6778 1,204.7322 | 3.6 mg monthly 10.8 mg every 3 months | 15.10 13.24 | 423 371 |
Leuprolide acetate (Eligard) | 7.5 mg 22.5 mg 30 mg 45 mg | Lyophilized powder for injection, Prefilled syringe for SC injection | 310.7200 891.0000 1,285.2000 1,645.0000 | 7.5 mg monthly 22.5 mg every 3 months 30 mg every 4 months 45 mg every 6 months | 11.10 9.79 10.56 9.01 | 311 274 296 252 |
Leuprolide acetate (Lupron depot) | 7.5 mg 22.5 mg 30 mg | Prefilled Syringe for IM injection | 387.9700 1,071.0000 1,428.0000 | 7.5 mg monthly 22.5 mg every 3 months 30 mg every 4 months | 13.86 11.77 11.74 | 388 330 329 |
Leuprolide acetate (Zeulide depot) | 3.75 mg 22.5 mg | Lyophilized powder for IM injection | 304.0000 873.0000 | 3.75 mg monthly 22.5 mg every 3 months | 10.86 9.59 | 304 269 |
Triptorelin (Trelstar) | 3.75 mg 11.25 mg 22.5 mg | Sterile vial of powder for IM injectable suspension | 346.3100 1,038.9700 1,659.9000 | 3.75 mg monthly 11.25 mg every 3 months 22.5 mg every 6 months | 12.37 11.42 9.10 | 346 320 255 |
ADT = androgen deprivation therapy; LHRH = luteinizing hormone-releasing hormone; PC = prostate cancer.
Note: All prices are from the Ontario Drug Benefit Formulary (accessed December 2023), unless otherwise indicated, and do not include dispensing fees.5 All dosing is from each treatment’s respective product monograph unless otherwise indicated.
Additional Details on the Sponsor’s Submission
Table 5: Sponsor’s Estimated Percentage of Use of ADTs in Canada
Treatment | Percentage of use | Weighted average cost other ADTs year 1 | Weighted average cost other ADTs year 1+ |
|---|---|---|---|
Degarelix | 3.65% | 3,905 | 4,120 |
Leuprolide acetate (Lupron Depot) | 23.63% | ||
Leuprolide acetate (Zeulide Depot) | 0.17% | ||
Leuprolide acetate (Eligard) | 37.36% | ||
Buserelin | 0.03% | ||
Triptorelin | 2.25% | ||
Goserelin acetate | 32.91% |
ADT = androgen deprivation therapy
Note: Percentage of use of each ADT in Canada as estimated from the CDH audit excluding Québec (June 2022 to May 2023).4 Weighted average annual cost was calculated using the percentage of use of each ADT in Canada, estimated from the CDH Audit excluding Québec (June 2022 to May 2023).4
Additional Details on the CADTH Reanalyses and Additional Analyses
Table 6: Scenario Analyses Conducted by CADTH on the Sponsor’s Base Case
Scenario analysis | Drug | Total drug costs ($) | Incremental drug costs ($) | ||
|---|---|---|---|---|---|
Year 1 | Year 1+ | Year 1 | Year 1+ | ||
CADTH Scenario 1: Relugolix vs. leuprolide acetate (Eligard) 45 mg | Relugolix | 3,303 | 3,285 | Reference | Reference |
Leuprolide acetate (Eligard) 45 mg | 3,290 | 3,290 | 13 | −5 | |
CADTH Scenario 2: Relugolix vs. leuprolide acetate (Eligard) 22.5 mg | Relugolix | 3,303 | 3,285 | Reference | Reference |
Leuprolide acetate (Eligard) 22.5 mg | 3,564 | 3,564 | −261 | −279 | |
Appendix 2: Submitted Budget Impact Analysis and CADTH Appraisal
Please note that this appendix has not been copy-edited.
Table 7: Summary of Key Takeaways
Key takeaways of the budget impact analysis |
|---|
|
Summary of Sponsor’s Budget Impact Analysis
In the submitted budget impact analysis (BIA), the sponsor assessed the budget impact of reimbursing relugolix for the treatment of adult patients with advanced prostate cancer.6 The analysis took the perspective of CADTH-participating Canadian public drug plans using a top-down, prevalence-based epidemiological approach over a 3-year time horizon. Data to inform the model were obtained from various sources, including the published literature, the sponsor’s internal data, and input from clinical experts consulted by the sponsor, and key inputs to the BIA are documented in Table 8.
The sponsor compared a reference scenario in which patients received currently reimbursed ADTs (degarelix, leuprolide acetate, buserelin acetate, triptorelin pamoate, and goserelin acetate) to a new drug scenario in which relugolix was reimbursed. The sponsor’s analysis included drug-acquisition costs and dosing modelled for relugolix reflected in the product monograph.
Figure 1: Sponsor’s Estimation of the Size of the Eligible Population in Base Year
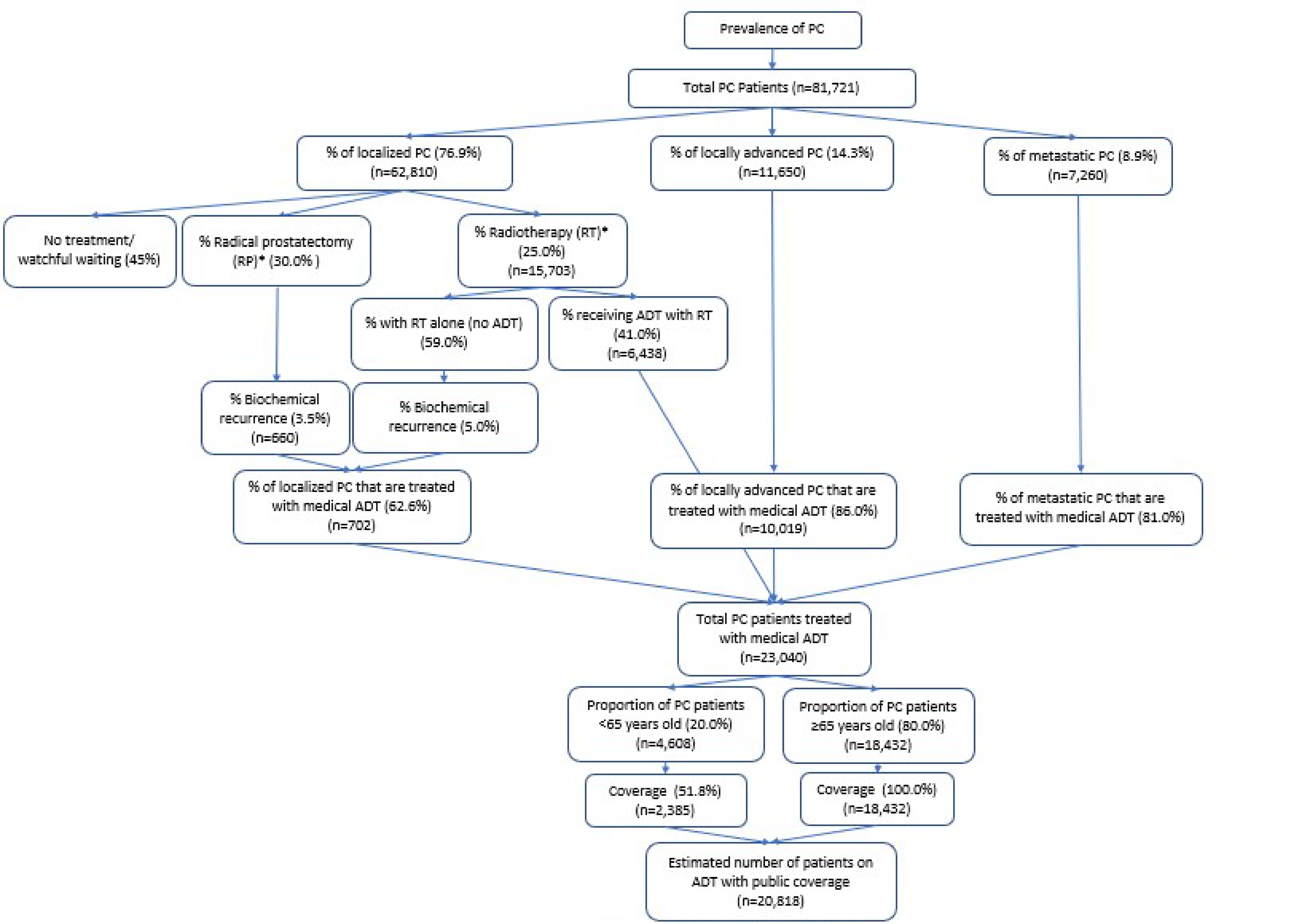
Source: Sponsor’s BIA submission.6
Table 8: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as Year 1 / Year 2 / Year 3 if appropriate) |
|---|---|
Target population | |
Number of patients eligible for drug under review | 21,155 / 21,490 / 21,816 |
Market uptake (3 years) | |
Uptake (reference scenario) Degarelix Leuprolide acetate (Lupron Depot) Leuprolide acetate (Zeulide Depot) Leuprolide acetate (Eligard) Buserelin Triptorelin Goserelin acetate | 3.76% / 3.76% / 3.76% 24.36% / 24.36% / 24.36% 0.18% / 0.18% / 0.18% 38.52% / 38.52% / 38.52% 0.03% / 0.03% / 0.03% 2.32% / 2.32% / 2.32% 30.83% / 30.83% / 30.83% |
Uptake (new drug scenario) Relugolix Degarelix Leuprolide acetate (Lupron Depot) Leuprolide acetate (Zeulide Depot) Leuprolide acetate (Eligard) Buserelin Triptorelin Goserelin acetate | 9.00% / 23.00% / 39.00% 3.43% / 2.90% / 2.30% 22.17% / 18.76% / 14.86% 0.16% / 0.14% / 0.11% 35.05% / 29.66% / 23.50% 0.03% / 0.02% / 0.02% 2.11% / 1.78% / 1.41% 28.05% / 23.74% / 18.80% |
Cost of treatment (per patient, per year) | |
Relugolix (year 1, year 1+) Degarelix (year 1, year 1+) Leuprolide acetate (Lupron Depot) Leuprolide acetate (Zeulide Depot) Leuprolide acetate (Eligard) Buserelin Triptorelin Goserelin acetate (year 1, year 1+) | $3,303, $3,285 $3,758, $3,290 $4,316 $3,493 $3,478 $5,157 $3,866 $4,037, $4,819 |
Note: Data from the most recent CDH audit dated June 2022 to May 2023 was used to estimate the proportion of utilization per strength to calculate a weighted annual drug cost for each comparator with multiple available dosages (e.g., leuprolide acetate, buserelin, and triptorelin).4 Costs of concomitant antiandrogen therapy were also included as part of drug costs assuming 28.6% of patients treated with gonadotropin-releasing hormone agonists would receive concomitant bicalutamide for 4 weeks (total costs of $35.53).6
Summary of the Sponsor’s BIA Results
The sponsor estimated that the budget impact of reimbursing relugolix for the treatment of adult patients with advanced prostate cancer would result in cost savings to the drug plans of $903,643 in Year 1, $2,806,850 in Year 2, and $5,228,552 in Year 3, for total savings of $8,939,046 across 3 years.
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
The sponsor’s approach to derive the target population was associated with uncertainty: First, the sponsor used a prevalence-based approach to calculate the number of patients with prostate cancer using estimates from the Canadian Cancer Society (2022).6 However, clinical experts consulted by CADTH noted that an incidence-based approach is likely more appropriate given that clinicians are unlikely to switch current patients with PC who already are receiving treatment with currently available ADTs to receiving relugolix. Clinical experts stated that relugolix would likely only be considered for incident patients with PC. CADTH notes that this results in notable impacts on the estimated target population eligible for treatment and therefore notable impacts to the cost savings associated with a prevalence-based versus incidence-based approach. CADTH re-estimated the BIA using an incidence-based approach based on data from the Canadian Cancer Society estimating 25,900 incident cases of prostate cancer in men living in Canada in 2023, resulting in an estimated incidence of 0.21%.7 Second, clinical experts consulted by CADTH indicated that the sponsor’s epidemiological breakdown to determine treatment eligibility did not meet face validity, particularly for patients with localized PC (Figure 1). Based on recent literature and current practice, clinical experts indicated that patients with localized PC should be divided into low, intermediate, and high-risk patients. Patients with low-risk localized PC are expected to remain under watchful waiting, and those who are intermediate risk are further broken down into favourable (receiving either radiotherapy or surgery without ADT; or remaining in active surveillance) and unfavourable (receiving radiotherapy alone, radiotherapy and ADT; or prostatectomy alone). Finally, high-risk patients are expected to receive radiotherapy and ADTs, or PT alone. Some structural and parameter changes to the epidemiological approach suggested by the clinical experts consulted by CADTH resulted in an increase in the number of eligible patients by approximately 50%. However, given the uncertainty in estimates and lack of available published literature to inform these exact estimates, CADTH was unable to address the limitations with the epidemiological top-down approach to derive the number of eligible patients. Instead, CADTH conducted a scenario analysis in which the number of eligible incident patients varies by ± 50% to assess the impact on the estimated cost savings of relugolix.
CADTH applied an incidence-based approach in reanalysis.
Two scenario analyses were conducted where the number of eligible incident patients was varied by ± 50% to reflect uncertainty in the approximate parameter estimates given by clinical experts and approximate a more clinically relevant approach to determining treatment eligibility based on the risk status of patients with localized PC.
The market uptake of relugolix may be overestimated: The sponsor’s submitted BIA assumed that relugolix would result in a market uptake of 9% in year 1, 23% in year 2, and 39% in year 3 based on the sponsor’s internal projections. However, CADTH obtained clinical expert feedback indicating that the projected market uptake does not align with clinical expectations and indicated the sponsor likely overestimated relugolix uptake. Clinical expert feedback also indicated that the uptake of relugolix would likely be similar to degarelix, the only other available GnRH antagonist in Canada. Clinical experts consulted by CADTH noted that relugolix results in faster recovery of testosterone levels after treatment discontinuation based on the HERO trial results, suggesting that ADTs currently used in practice confer a longer period of testosterone suppression (e.g., sustained treatment effect after discontinuation). This may impact the uptake of relugolix in practice, as the duration of testosterone suppression is known to have impacts on clinical outcomes in prostate cancer.8,9 If relugolix confers a shorter duration of testosterone suppression compared to ADTs after treatment is stopped, clinical experts consulted by CADTH noted that clinician preference may be to prescribe ADTs currently used in practice that have long-term data demonstrating biochemical control or survival benefit over relugolix, particularly for patients who are being treated intermittently or receiving short-term ADT, where their clinical outcomes for prostate cancer benefit from continuous suppression of testosterone on a long-term basis because intermittent ADT may not be associated with the same survival benefits compared to continuous treatment.10,11 Notably, there is no long-term clinical data examining the impact of treatment with relugolix on survival. Lastly, clinical expert feedback indicated that the sponsor’s predicted uptake may also be overestimated due to the administration method of relugolix, as daily oral administration may be less preferable to patients compared to bi-annual injections or subcutaneous implants. Clinical experts noted that the oral administration method of relugolix may only be preferable to needle-phobic patients.
To address this limitation, CADTH undertook a reanalysis by revising the market shares for relugolix in the new drug scenario to 2.90% in year 1, 3.43% in year 2, and 3.90% in year 3, aligned with degarelix. Equal displacement of market share among the comparators was assumed.
In a scenario analysis, CADTH explored the impact of assuming market uptake of relugolix was captured from the most commonly used ADT in Canadian clinical practice, leuprolide acetate.
CADTH included the sponsor’s forecasted market shares in a scenario analysis.
To reflect uncertainty in the predicted use of relugolix for patients receiving short-term or intermittent treatment, CADTH conducted a scenario analysis to estimate the budget impact of relugolix in patients who receive continuous treatment only (e.g., excluding patients who may be treated intermittently or in short-term).
The price of drugs paid by public drug plans is uncertain: Both the sponsor’s and CADTH’s analyses are based on publicly available list prices for all comparators. Actual costs paid by public drug plans are unknown. Confidential negotiated prices for ADT comparators may lead to budgetary savings being limited or eliminated.
CADTH could not address this limitation in reanalysis.
CADTH Reanalyses of the BIA
Table 9: CADTH Revisions to the Submitted BIA
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Changes to derive the CADTH base case | ||
1. Eligible patient population | Prevalence-based approach: 0.66% | Incidence-based approach: 0.21% |
2. Market uptake of relugolix | Relugolix: 9.00% / 23.00% / 39.00% | Relugolix: 2.90% / 3.43% / 3.90% |
CADTH base case | Reanalysis 1 + 2 | |
The results of the CADTH step-wise reanalysis are presented in summary format in Table 10 and a more detailed breakdown is presented in Table 11. The CADTH reanalysis of the BIA estimated that the budget impact of reimbursing relugolix for the treatment of adult patients with advanced prostate cancer would result in cost savings to the drug plans of $93,822 in year 1, $271,782 in year 2, and $498,778 in year 3, for a total savings of $864,382 across 3 years.
CADTH conducted additional scenario analyses to address remaining uncertainty using the CADTH base case. Results are provided in Table 11.
Assuming a 50% increase in the eligible incident patient population
Assuming a 50% reduction in eligible incident patient population
Applying the sponsor’s estimated market uptake of relugolix of 9% in year 1, 23% in year 2, and 39% in Year 3
Assuming market uptake of relugolix is derived from leuprolide acetate only (the only comparator with direct trial evidence)
Assuming only patients receiving continuous ADT would be eligible for treatment (i.e., metastatic PC only).
Table 10: Summary of the CADTH Reanalyses of the BIA
Stepped analysis | Three-year total |
|---|---|
Submitted base case | −$8,939,046 |
CADTH reanalysis 1 — incident approach | −$6,548,612 |
CADTH reanalysis 2 — market shares | −$1,372,302 |
CADTH base case | −$864,382 |
Table 11: Detailed Breakdown of the CADTH Reanalyses of the Budget Impact Analysis
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
Submitted base case (prevalence-based) | Reference | $86,891,961 | $92,995,658 | $94,465,020 | $95,896,323 | $283,357,001 |
New drug | $86,891,961 | $92,092,015 | $91,658,170 | $90,667,771 | $274,417,955 | |
Budget impact | $0 | −$903,643 | −$2,806,850 | −$5,228,552 | −$8,939,046 | |
CADTH base case (incidence-based) | Reference | $27,998,251 | $29,964,979 | $58,892,052 | $89,791,681 | $178,648,712 |
New drug | $27,998,251 | $29,871,157 | $58,620,270 | $89,292,903 | $177,784,330 | |
Budget impact | $0 | −$93,822 | −$271,782 | −$498,778 | −$864,382 | |
CADTH scenario analysis 1: increased eligible incident patients by 50% | Reference | $20,998,688 | $22,473,734 | $44,169,039 | $67,343,761 | $133,986,534 |
New drug | $20,998,688 | $22,403,368 | $43,965,203 | $66,969,677 | $133,338,248 | |
Budget impact | $0 | −$70,366 | −$203,837 | −$374,083 | −$648,286 | |
CADTH scenario analysis 2: reduced eligible incident patients by 50% | Reference | $13,999,125 | $14,982,489 | $29,446,026 | $44,895,840 | $89,324,356 |
New drug | $13,999,125 | $14,935,578 | $29,310,135 | $44,646,451 | $88,892,165 | |
Budget impact | $0 | −$46,911 | −$135,891 | −$249,389 | −$432,191 | |
CADTH scenario analysis 3: sponsor’s assumed market uptake | Reference | $27,998,251 | $29,964,979 | $58,892,052 | $89,791,681 | $178,648,712 |
New drug | $27,998,251 | $29,673,808 | $57,243,528 | $85,182,765 | $172,100,100 | |
Budget impact | $0 | −$291,171 | −$1,648,525 | −$4,608,916 | −$6,548,612 | |
CADTH scenario analysis 4: uptake captured from leuprolide | Reference | $27,998,251 | $29,964,979 | $58,892,052 | $89,791,681 | $178,648,712 |
New drug | $27,874,925 | $29,734,812 | $58,405,843 | $89,016,461 | $177,157,116 | |
Budget impact | −$123,325 | −$230,167 | −$486,209 | −$775,220 | −$1,491,596 | |
CADTH scenario analysis 5: eligible patients — continuous treatment only (i.e., metastatic patients) | Reference | $7,146,346 | $7,648,338 | $15,031,759 | $22,918,660 | $45,598,757 |
New drug | $7,146,346 | $7,624,391 | $14,962,389 | $22,791,350 | $45,378,130 | |
Budget impact | $0 | −$23,947 | −$69,370 | −$127,309 | −$220,627 |
Note: All analyses are based on publicly available prices of comparator treatments.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.