CADTH Reimbursement Review
Teclistamab (Tecvayli)
Sponsor: Janssen Inc.
Therapeutic area: Relapsed or refractory multiple myeloma
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
ADC
antibody-drug conjugate
ATO
average treatment effect in the overlap
ATT
average treatment effect in the treated
B2M
beta2-microglobulin
BCMA
B-cell maturation antigen
CAR
chimeric antigen receptor
CI
confidence interval
CMRG
Canadian Myeloma Research Group
CR
complete response
CRS
cytokine release syndrome
CTCAE
Common Terminology Criteria for Adverse Events
DSU
Decision Support Unit
ECOG PS
Eastern Cooperative Oncology Group Performance Status
EORTC QLQ-C30
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30
EQ VAS
EQ visual analogue scale
FAS
full analysis set
FISH
fluorescence in situ hybridization
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HR
hazard ratio
HRQoL
health-related quality of life
ICANS
immune effector cell-associated neurotoxicity syndrome
IMiD
immunomodulatory drug
IMWG
International Myeloma Working Group
IPTW
inverse probability of treatment weighting
IRC
independent review committee
ISS
International Staging System
ITC
indirect treatment comparison
ITT
intention-to-treat
LDH
lactate dehydrogenase
LSM
least squares mean
MAIC
matching-adjusted indirect comparison
MCT
meaningful change threshold
MGUS
monoclonal gammopathy of undetermined significance
MM
multiple myeloma
MRD
minimal residual disease
NE
not evaluable
NICE
National Institute for Health and Care Excellence
OH-CCO
Ontario Health (Cancer Care Ontario)
OR
odds ratio
ORR
overall response rate
OS
overall survival
PC
physician’s choice
pERC
pan-Canadian Oncology Drug Review Expert Review Committee
PFS
progression-free survival
PGI-S
Patient Global Impression–Severity
PI
proteasome inhibitor
PR
partial response
R-ISS
Revised International Staging System
RCT
randomized controlled trial
RRMM
relapsed or refractory multiple myeloma
RWPC
real-world physician’s choice
SC
subcutaneous
sCR
stringent complete response
SD
standard deviation
SMD
standardized mean difference
TEAE
treatment-emergent adverse event
TLS
tumour lysis syndrome
TTNT
time to next treatment
TTR
time to response
VGPR
very good partial response
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information on Application Submitted for Review
Item | Description |
|---|---|
Drug product | Teclistamab (Tecvayli), 10 mg/mL or 90 mg/mL single-dose vials containing 30 mg teclistamab in 3 mL solution or 153 mg teclistamab in 1.7 mL solution, subcutaneous injection |
Sponsor | Janssen Inc. |
Indication | Teclistamab injection is indicated for the treatment of adult patients with relapsed or refractory multiple myeloma who have received at least 3 prior lines of therapy, including a proteasome inhibitor, an immunomodulatory drug, and an anti-CD38 monoclonal antibody, and who have demonstrated disease progression on the last therapy. |
Reimbursement request | As per indication |
Health Canada approval status | NOC/c |
Health Canada review pathway | Advance consideration under NOC/c Project Orbisa |
NOC date | July 26, 2023 |
Recommended dosage | 1.5 mg/kg of actual body weight administered subcutaneously once a week, followed by step-up doses of 0.06 mg/kg and 0.3 mg/kg |
NOC/c = Notice of Compliance with Conditions.
aProject Orbis provides a framework for the collaborative review of new cancer treatments among international regulatory partners aiming to give patients access to promising cancer treatments across the globe. Health Canada is one of the Project Orbis partners.
Introduction
Multiple myeloma (MM) is a plasma cell cancer characterized by the clonal proliferation of malignant plasma cells (B-cells) and the overproduction of the abnormal immunoglobulin M protein.1 Older individuals and men of all ages (as opposed to women) are more likely to develop MM and it is twice as common in individuals who are Black compared to white or Asian individuals.2,3 In 2022, it was estimated that 4,000 people in Canada were diagnosed with MM and 1,650 people in Canada died from MM.4 The 5-year survival probability for patients with MM is estimated to be approximately 50%,5 and although survival rates have improved in recent years due to advances in therapeutic options, MM remains incurable.6,7 The majority of patients with MM will relapse and many patients will become refractory to commonly used therapies.8 The most common symptoms of MM are fatigue and bone pain,3 with other symptoms including kidney problems, recurrent infections, fever, and nervous system problems.9
Diagnosis of MM typically occurs during a visit to a primary care physician, occurring either incidentally when laboratory tests for other conditions are ordered, or if MM is suspected based on signs and symptoms.10 According to the International Myeloma Working Group (IMWG) criteria, a patient is considered to have relapsed and/or refractory MM if they have attained a minimal response or better at some point during previous treatments but the disease is currently nonresponsive on salvage therapy, or they experience disease progression within 60 days of their last therapy.11,12 Patients with relapsed or refractory multiple myeloma (RRMM) often undergo multiple rounds of treatment, with the duration of remission, the depth of response, progression-free survival (PFS), and overall survival (OS) decreasing with each subsequent line of therapy.3 According to the clinical experts consulted by CADTH for this review, the current approach to the treatment of MM at relapse depends on several factors, including patient factors (i.e., age, comorbidity, and beforexicity), line of therapy, and prior therapies received. There is no preferred standard of care for the treatment of RRMM in the fourth-line setting and beyond, and at this stage of the disease, patients may be exposed to a proteasome inhibitor (PI), an immunomodulatory drug (IMiD), and anti-CD38 monoclonal antibody,7,13 and in some cases receive more than 1 PI or iMiD, further limiting treatment options in later lines of therapy. The clinical experts consulted by CADTH noted that treatment options at relapse include a PI (bortezomib, carfilzomib) containing combinations such as cyclophosphamide in combination with bortezomib and dexamethasone, carfilzomib in combination with dexamethasone with or without cyclophosphamide, or selinexor in combination with bortezomib and dexamethasone. The clinical experts mentioned that later relapses can be treated through clinical trials or with regimens such as belantamab mafodotin, which is currently available through a special access program. The clinical experts and clinician groups consulted by CADTH for this review agreed that there is an unmet need for treatments beyond the third line that prolong survival, delay disease progression, prevent disease complications, improve quality of life, and minimize side effects.
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of teclistamab (1.5 mg/kg of body weight administered by subcutaneous [SC] injection) in the treatment of adult patients with RRMM who have received at least 3 prior lines of therapy, including a PI, an IMiD, and an anti-CD38 monoclonal antibody.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient and clinician groups that responded to CADTH’s call for input and from clinical experts consulted by CADTH for the purpose of this review.
Patient Input
CADTH received 1 patient group submission from Myeloma Canada, which has existed for more than 15 years to support the growing number of Canadians diagnosed with myeloma and those living longer than ever with the disease to access new and innovative therapies. Myeloma Canada gathered information for this review through a patient and caregiver survey (33 patients and 3 caregivers) that was conducted from August 28 to September 6, 2023.
Patient respondents indicated that their ability to travel was the factor most significantly impacted by symptoms associated with myeloma, followed by the ability to work and to exercise. Patient and caregiver respondents identified the following factors as those most important to myeloma treatment: improved quality of life, manageable side effects along with the effectiveness of treatment, especially in achieving remission and having a long and durable response, and treatment accessibility or portability (including fewer or minimal visits to the hospital or cancer centre). In terms of treatment outcomes, 13 of the 22 respondents rated improved quality of life as extremely important, 6 respondents rated it as very important, and 3 respondents rated it as somewhat important. A total of 17 of the 22 patients rated the estimated minimum of 1 year to 21 months of life extension as extremely desirable, and 5 patients rated it as very desirable. All caregiver respondents felt that caring for someone with myeloma had the most impact on “anxiety/worry,” followed by “interruption of life goals/accomplishments (e.g., career, retirement)”.
From August 28 to September 30, 2022, Myeloma Canada also conducted a different survey about a chimeric antigen receptor (CAR) T-cell therapy, which received more than 200 responses, only 2 of which had experience with the CAR T-cell therapy. While the teclistamab survey received far fewer total responses (33), there were 11 patients with teclistamab experience. Myeloma Canada emphasized that this is indicative of the comparative ease with which teclistamab can and has been made accessible to Canadians with triple-class exposed RRMM.
Clinician Input
Input From Clinical Experts Consulted by CADTH
The clinical experts consulted by CADTH for this review highlighted that the most important goals of treatment for patients with MM are to prolong survival, delay disease progression, prevent disease complications, improve quality of life, and minimize side effects. The clinical experts noted that clinicians try not to reuse the same drugs in subsequent lines of therapy, and after receiving 3 lines of therapy, the majority of patients will be triple-refractory and will need new families of drugs. The clinical experts also mentioned that beyond the third line of therapy, the treatment options get more restricted, and some patients do not respond to the current standard therapies. Thus, there is a need for treatments for fourth-line therapy and beyond that are tolerable for patients. The clinical experts consulted by CADTH indicated that, given that the prognosis of MM worsens as patients move on to subsequent lines of therapy, any patient with RRMM will require this intervention. The clinical experts noted that teclistamab is not the first approved treatment that targets underlying disease processes; however, this drug has a novel mechanism of action that is very different from any currently available therapies. The clinical experts agreed that there is no evidence that some patients are more likely to respond to treatment, and there are no disease-specific characteristics that would make a patient ineligible for treatment with teclistamab. The clinical experts noted that classic methodological assessments of interventions concern A versus B, comparing 1 versus the other without the consideration of sequencing beyond the initial drug failure; in this light, according to the clinical experts, the use of teclistamab represents a new class of drugs that can help sustain quality of life and extend the duration of life in the patient with RRMM.
The clinical experts identified OS, PFS, and clinical response outcomes as the most important outcomes for assessing the response to treatment. The clinical experts agreed that the best possible response to treatment would be complete remission that is minimal residual disease (MRD)–negative, and less deep responses that include complete response (CR), very good partial response (VGPR), partial response (PR), and stable disease. The clinical experts further noted that CR and VGPR are the most desirable outcomes in most situations; even achieving stable disease is acceptable. The clinical experts consulted by CADTH indicated that the main reason for discontinuing treatment with teclistamab would be the relapse of MM. The clinical experts further noted that as with any treatment, it can be expected that some patients will be forced to discontinue treatment due to intolerable side effects. The clinical experts consulted mentioned that currently, all patients receiving teclistamab are treated at tertiary care centres and are admitted to the hospital for the first few doses. The clinical experts also noted that depending on the situation, patients starting treatment with teclistamab will require treatment at a larger hospital capable of providing management and monitoring; however, after patients receive the first few doses of this treatment, they can receive ongoing therapy at community centres and smaller cancer centres.
Clinician Group Input
The clinician group input was obtained from 2 clinician groups: the Canadian Myeloma Research Group (CMRG) and the Ontario Health (Cancer Care Ontario) (OH-CCO) Hematology Cancer Drug Advisory Committee (OH-CCO’s Drug Advisory Committees). CMRG gathered information through teleconferences with physicians and OH-CCO’s Drug Advisory Committees gathered information through videoconferencing and email communications.
Both CMRG and OH-CCO’s Drug Advisory Committees mentioned that myeloma remains incurable, and patients eventually become refractory to all available funded drugs. One major unmet need mentioned by clinician groups is that patients with advanced disease who have received multiple lines of treatment and have already received the 3 major drugs (triple-class exposed or refractory) — including an IMiD, PI, and anti-CD38 monoclonal antibody — have no other substantial treatment options other than CAR T-cell therapy. CMRG also emphasized that the clinical features associated with advanced disease and short duration of responses lead to a poor quality of life, significant caregiver burden, and a shortened patient lifespan. Thus, this situation also represents 1 of the most pressing unmet needs in Canada for patients with MM. Another unmet need noted by OH-CCO’s Drug Advisory Committees is to achieve ease of administration (i.e., SC injection and no need for apheresis).
Both clinician groups agreed that teclistamab is another option for triple-class exposed patients. They believe that currently, it would be used in sequence after other lines of therapy for myeloma (i.e., after failure of multiple drugs); it is not expected to impact the sequencing of drugs earlier in the disease course or lead to a major change in treatment algorithms before patients becoming triple-class exposed or refractory.
Drug Program Input
The Provincial Advisory Group identified the following jurisdictional implementation issues: relevant comparators, considerations for the initiation of therapy, considerations for the discontinuation of therapy, considerations for the prescribing of therapy, generalizability, funding algorithm, care provision issues, and system and economic issues. The clinical experts consulted by CADTH weighed evidence from the MajesTEC-1 study and other clinical considerations to provide responses to the Provincial Advisory Group’s drug program implementation questions. Refer to Table 5 for more details.
Clinical Evidence
Systematic Review
Description of Studies
The MajesTEC-1 trial (n = 165) is a phase I and phase II, open-label, multicentre study assessing the efficacy and safety of teclistamab administered to adult patients with RRMM. The study is still ongoing and being conducted in 39 sites across 10 countries, including in Canada with patients who were enrolled at 4 Canadian sites. The MajesTEC-1 study was conducted in 3 parts: part 1 or dose escalation (phase I), part 2 or dose expansion (phase I) at a proposed recommended phase II dose (1.5 mg/kg subcutaneously weekly), and part 3 or dose expansion (phase II) in cohorts of patients with RRMM with unmet medical needs. The primary objectives reported in phase I of the MajesTEC-1 study were to identify the proposed recommended phase II dose and dose schedule assessed to be safe in part 1, and to characterize the safety and tolerability of teclistamab at the proposed recommended phase II dose in part 2. The primary objective of phase II of the MajesTEC-1 trial was to evaluate the efficacy and safety of teclistamab at the proposed recommended phase II dose. In phase II of the MajesTEC-1 study, cohort A enrolled patients with RRMM who had received at least 3 prior lines of therapy that included a PI, an IMiD, and an anti-CD38 monoclonal antibody, while cohort C enrolled patients who had received at least 3 prior lines of therapy that included a PI, an IMiD, an anti-CD38 monoclonal antibody, and an anti–B-cell maturation antigen treatment (CAR T-cells or an antibody-drug conjugate [ADC]). The primary efficacy outcome for the MajesTEC-1 trial was overall response rate (ORR), and the secondary efficacy outcomes included VGPR or better, CR or better, stringent complete response (sCR), time to response (TTR), duration of response, OS, PFS, MRD-negativity rate, and patient-reported outcomes. Time to next treatment (TTNT) was an exploratory outcome in phase II of the MajesTEC-1 study. Disease responses were evaluated by an independent review committee (IRC) using IMWG 2016 criteria in both phase I and phase II, cohort A.
In the MajesTEC-1 study, the median age of the patients was 64.0 years (range = 33.0 years to 84.0 years) |||| ||||| || ||| ||||| |||||||||| ||||| |||| |||| || ||||| || |||. Ninety-six (58.2%) patients were male and 69 (41.8%) patients were female. Most patients were white (81.2%), and 12.7% of patients identified as Black or African American. Most patients had an Eastern Cooperative Oncology Group Performance Status (ECOG PS) score of 1 (66.1%), while 33.3% of patients had an ECOG PS score of 0. The most common immunoglobulin isotypes were immunoglobulin G (55.2%) and immunoglobulin A (17.6%). The median time from diagnosis of MM to enrolment in the study was 6.0 years (range = 0.8 years to 22.7 years). Twenty-eight (17.0%) patients had 1 or more extramedullary plasmacytomas at baseline. Of the 147 patients with baseline cytogenetic data reported, 38 (25.9%) patients had at least 1 high-risk abnormality, including del(17p) (15.6%) and t(4;14) (10.9%) abnormality. Of the 162 patients with baseline International Staging System (ISS) data reported, 85 (52.5%) patients were ISS stage I while 20 (12.3%) patients were ISS stage III.
Efficacy Results
The primary analysis at the clinical cut-off date of September 2021 and the final analysis at the clinical cut-off date of August 2023 were prespecified analyses, and the Clinical Study Reports submitted by the sponsor with clinical cut-off dates of March 2022 and January 2023 were interim analyses. As the final report for the pivotal study is not yet available, information for the following sections was extracted from the more recent Clinical Study Report with a clinical cut-off date of January 4, 2023, submitted by the sponsor for this review. However, data from the clinical cut-off date of September 7, 2021, was also used to supplement the included data when necessary.
Overall Survival
At the time of analysis, using the January 4, 2023, data cut-off, the median duration of follow-up was 22.8 months (range = 0.3 months to 33.6 months). The estimated median OS was 21.9 months (95% confidence interval [CI], 15.1 months to not evaluable [NE] months). In the full analysis set (FAS), deaths were reported in || |||||||| ||||||| in phase I and || |||||||| ||||||| in phase II, cohort A, of the MajesTEC-1 trial. The 9-month OS probability was ||||| |||| ||| ||||| || ||||||, the 12-month OS probability was ||||| |||| ||| ||||| || ||||||, and the 24-month OS probability was ||||| |||| ||| ||||| || ||||||.
Progression-Free Survival
At the time of analysis, using the January 4, 2023, data cut-off, the estimated median PFS was 11.3 months (95% CI, 8.8 months to 16.4 months) in the MajesTEC-1 trial. In the FAS, the 9-month PFS probability was ||||| |||| ||| ||||| || ||||||, the 12-month PFS probability was ||||| |||| ||| ||||| || ||||||, and the 24-month PFS probability was ||||| |||| ||| ||||| || ||||||.
CR or Better
At the time of analysis, using the January 4, 2023, data cut-off, || |||||||| ||||||| |||| ||| ||||| || ||||||| in phase I and || |||||||| ||||||| |||| ||| ||||| || ||||||| attained CR or better (CR or sCR).
Stringent Complete Response
At the time of analysis, using the January 4, 2023, data cut-off, || |||||||| ||||||| |||| ||| ||||| || ||||||| in phase I and || |||||||| ||||||| |||| ||| ||||| || ||||||| in phase II, cohort A, attained sCR.
MRD-Negativity Status
Updated data regarding the MRD-negativity rate based on the January 4, 2023, clinical cut-off date are not available.
At the time of the data cut-off date of September 7, 2021, 37 (24.7%) patients (95% CI, 18.0% to 32.4%) attained MRD negativity at 10–5 bone marrow cells. Among 43 patients who attained CR or better, 18 (41.9%) patients (95% CI, 27.0% to 57.9%) attained MRD negativity at 10–5 bone marrow cells.
VGPR or Better
At the time of analysis, using the January 4, 2023, data cut-off, || |||||||| ||||||| |||| ||| ||||| || ||||||| in phase I and || |||||||| || ||||||||||||||||||||| ||| ||||| || ||||||| attained VGPR or better (VGPR, CR, or sCR).
Overall Response Rate
At the time of analysis, using the January 4, 2023, data cut-off, 104 (63.0%) patients (95% CI, 55.2% to 70.4%) had attained an overall response (PR or better), and ORR was similar across patients treated in phase I and phase II, cohort A, ||||||| |||| ||||||| || |||||| ||| |||||| |||| ||| ||||| || ||||||| |||||||||||||. Of the 104 responders (who attained PR or better), 51 (49.0%) patients maintained their response until the clinical cut-off date |||||| || |||||||| ||||| || ||||||||||| ||, including 46 (44.2%) patients who were still on treatment. Of the 104 responders, || |||||||| ||||||| had disease progression after initial response, of whom || ||||||| ||||||| died after disease progression, || |||||||| ||||||| discontinued the study treatment, and ||||||| |||||| remained on study treatment. A total of 19 (18.3%) patients died after achieving response and without experiencing disease progression, and |||||| |||||| had subsequent therapy after response and without progressive disease. Of the 63 respondents who changed their dosing schedule from weekly to biweekly (every 2 weeks) or monthly, 42 (66.7%) patients maintained their response until the clinical cut-off date of January 4, 2023, including 41 (65.1%) patients who remained on treatment.
Subgroup Analysis
Only results of the ORR subgroup analyses that were deemed clinically meaningful by the clinical experts consulted by CADTH for this review are reported. At the time of analysis, using the January 4, 2023, data cut-off, 32 of 43 (74.4%) patients who received 3 or fewer prior lines of therapy attained an overall response. Of the 122 patients who received more than 3 prior lines of therapy, 72 (59.0%) patients attained overall response, and 32 of 60 (53.3%) patients with high cytogenetic risk and/or extramedullary disease attained overall response.
Time to Response
At the time of analysis, using the January 4, 2023, data cut-off, in 104 responders, the median time to first response was 1.18 months (range = 0.2 months to 5.5 months) while the median time to best response was 3.96 months (range = 1.1 months to 18.7 months). Most patients demonstrated their first response rapidly, by the start of the second treatment cycle in the MajesTEC-1 study (depending on the dosing schedule, teclistamab was administered to patients in 21-day or 28-day cycles).
Duration of Response
At the time of analysis, using the January 4, 2023, data cut-off, the median duration of response was 21.6 months (95% CI, 16.2 months to NE months) in the MajesTEC-1 trial. Among 104 responders, || |||||||| ||||||| in phase I and || |||||||| ||||||| in phase II, cohort A, had disease progression or died due to any cause. The probability of patients remaining in response at 9 months was ||||| |||| ||| ||||| || ||||||. The probability of patients remaining in response at 18 months was ||||| |||| ||| ||||| || ||||||. The probability of patients remaining in response at 24 months was ||||| |||| ||| ||||| || ||||||.
Patient-Reported Outcomes
Patient-reported outcomes were assessed using the 30-item European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 (EORTC QLQ-C30) assessment, the EQ-5D-5L questionnaire, and the Patient Global Impression–Severity (PGI-S) scale. The health-related quality of life (HRQoL) results were reported only for phase II, cohort A, of the MajesTEC-1 study. Analyses were conducted in the HRQoL-evaluable population of patients who had evaluable assessment at baseline and follow-up time points for each domain of EORTC QLQ-C30 (i.e., cycle 2, day 1; cycle 3, day 1; and so forth).
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30
At the time of analysis, using the January 4, 2023, data cut-off, the results of a post hoc analysis of the EORTC QLQ-C30 showed |||||||||||| |||| |||| |||| |||||||| || |||| ||| ||||||| |||||| |||||| |||||||||||| |||| |||||||||||| || |||||| |||||| ||||||| ||| |||||||||||| |||| ||| |||||||| ||||||||| |||||||| || |||||| |||||||||||||||||||.
At the time of analysis, using the September 7, 2021, data cut-off, meaningful improvement from baseline (10 points using the literature-based meaningful change threshold [MCT]14) to cycle 2, cycle 4, and cycle 6 was reported by up to 35.8% of patients for global health status, up to 23.9% of patients for physical functioning, up to 68.7% of patients for fatigue system scale, and up to 78.8% of patients for pain.
EQ-5D-5L
At the time of analysis, using the January 4, 2023, data cut-off, the results of a post hoc analysis of the EQ-5D-5L showed |||||||||||| |||| |||| |||| |||||||| || ||||||| |||||| ||| ||| |||| |||||||||||.
At the time of analysis, using the September 7, 2021, data cut-off, meaningful improvement from baseline (7 points using the literature-based MCT15,16) in EQ VAS scores at cycle 2, cycle 4, and cycle 6 was reported by 23.8%, 28.6%, and 30.2% of patients, respectively. By cycle 8, 50% of patients had reported meaningful improvement in the EQ VAS score.
Time to Next Treatment
TTNT was an exploratory end point in phase II, cohort A, of the MajesTEC-1 study, and it was not reported in the Clinical Study Report at the clinical data cut-off date of January 4, 2023.
At the time of analysis, using the September 7, 2021, data cut-off, subsequent antimyeloma therapy and/or death due to progressive disease was reported for || |||||||| |||||||, with a median TTNT of |||| |||||| |||| ||| |||| || || |||||||.
Harms Results
At the time of analysis, using the January 4, 2023, data cut-off, patients in the MajesTEC-1 study had experienced at least 1 treatment-emergent adverse event (TEAE). The most common TEAEs occurring in at least 25% of patients in either phase of the study were cytokine release syndrome (CRS) (72.1%), neutropenia (71.5%), anemia (54.5%), thrombocytopenia (42.4%), lymphopenia (36.4%), diarrhea (33.9%), and pyrexia (31.5%). In the MajesTEC-1 study, || |||||||| ||||||| experienced TEAEs of Common Terminology Criteria for Adverse Events (CTCAE) grade 3, || |||||||| ||||||| experienced TEAEs of grade 4, and || |||||||| ||||||| experienced TEAEs of grade 5. The most common TEAEs of grade 3 or grade 4 were neutropenia (65.5%), anemia (37.6%), lymphopenia (34.5%), and thrombocytopenia (22.4%). The most common TEAEs of grade 5 were COVID-19 (10.8%) and general physical health deterioration (2.4%). At the time of analysis, using the January 4, 2023, data cut-off, 113 patients (68.5%) had experienced at least 1 serious TEAE in the MajesTEC-1 trial. The most common serious TEAEs occurring in at least 5% of patients in either phase of the study were COVID-19 (68.5%), pneumonia (20.6%), CRS (10.9%), pyrexia (8.5%), acute kidney injury (5.5%), and general physical health deterioration (5.5%). At the time of analysis, using the January 4, 2023, data cut-off, |||||||| |||||| stopped study treatment due to TEAEs in the MajesTEC-1 trial. The most common reasons for stopping study treatment included |||||||| ||||||||| ||||||| |||||||||||| ||||||||| ||||||||| ||||||| ||| |||||||||| ||||||||| ||||||. A total of || |||||||| ||||||| died during the study, including || |||||||| ||||||| who died due to TEAEs.
In the MajesTEC-1 study, several adverse events of clinical interest were identified, including CRS, neurologic adverse events and neurotoxicity, immune effector cell-associated neurotoxicity syndrome (ICANS), systemic administration-related reactions, injection-site reactions, hypogammaglobulinemia, cytopenia, and infections. At the time of analysis, using the January 4, 2023, data cut-off, 119 (72.1%) patients had experienced CRS events, of whom 83 (50.3%) patients experienced grade 1 events, and 35 (21.2%) patients experienced grade 2 events. One (0.6%) patient experienced CRS events of grade 3, and no patients experienced CRS events of grade 4 or grade 5. ||||| || || |||||||| ||||||| ||||||||||| || |||||||||||||||| ||||||| |||||| ||| || |||||||| ||||||| ||||||||||| ||||||||||||||||||||||| ||| |||| |||||| ||||||||||||| ||||| |||||||| |||||||| ||||||| |||||| |||||||| ||||||||||||||| ||||||||||||| |||||||| ||||||| ||||||||| ||||||| ||||||||||||| ||||||| ||| |||||||||| ||||||| |||||||||| ||||||| In the MajesTEC-1 study, a total of 132 (80.0%) patients had infections of any grade. The most common infections and infestations included |||||||| |||||||| ||||||||| |||||||| |||||||||| |||||||| ||||| ||||||||||| ||||||||| |||||||| ||||||||||||||| |||||||| ||||||||| |||||||| ||| ||||||| ||||| ||||||||| ||||||. In the MajesTEC-1 trial, 71 (43.0%) patients experienced at least 1 infection and infestation of grade 3 or grade 4, while 21 (12.7%) patients experienced at least 1 infection and infestation of grade 5. At the time of analysis, using the January 4, 2023, data cut-off, || |||||||| ||||||| had experienced at least 1 hypogammaglobulinemia TEAE, including || |||||||| ||||||| with a case of hypogammaglobulinemia, and ||||||| |||||| with a case of hypoglobulinemia. A total of 152 (92.1%) patients experienced at least 1 treatment-emergent cytopenic event, including neutropenia (71.5%), anemia (55.8%), thrombocytopenia (42.4%), and lymphopenia (36.4%). A total of 108 (65.5%) patients experienced treatment-emergent neutropenia of grade 3 or higher, 62 (37.6%) patients experienced anemia of grade 3 or higher, 37 (22.4%) patients experienced thrombocytopenia of grade 3 or higher, and 57 (34.5%) patients experienced lymphopenia of grade 3 or higher. A total of 61 (37.0%) patients experienced at least 1 case of injection-site reaction events, including 32.1% of cases of grade 1 and 4.8% of cases of grade 2.
Critical Appraisal
The MajesTEC-1 trial was a multicentre, single-arm, open-label, phase I and phase II study. Due to the lack of a comparator arm, the benefit of teclistamab compared to placebo or reference treatment was not documented. A single-arm study design is usually used when the purpose of the study is to provide preliminary evidence of the efficacy of a treatment and to collect additional safety data, and is not intended to be confirmatory for efficacy.17 Thus, a single-arm study design is a subject of several limitations that complicate the interpretation of the study results. The open-label design of the MajesTEC-1 study may increase uncertainty in subjective outcomes, including clinical response outcomes, PFS, HRQoL, and safety outcomes, introducing bias due to the inherent subjectivity of the outcome in an unblinded assessor. This bias would be less likely in more objective outcomes, such as OS, if assessed against a predetermined hypothesis. According to the FDA, the ORR can be evaluated in a single-arm study as a direct measure of a drug antitumour activity if it is defined as the sum of PRs plus CRs.18 In the MajesTEC-1 trial, the estimated ORR was tested against a predetermined hypothesis of an ORR greater than 45% (with a lower bound of the ORR 2-sided 95% CI above 30%). ORR achieved the predetermined threshold for a positive outcome in the MajesTEC-1 trial. However, for ORR, there was no adjustment for multiplicity across the various analyses of the outcome (i.e., the various data cut-offs), which may have increased the risk of false-positive conclusions. Additionally, this report presents interim analysis results because a prespecified final analysis was not available; therefore, there is the potential that the benefit of teclistamab is overestimated, but the presence and extent of any overestimation is uncertain.19-21
Disease responses were evaluated by the IRC using IMWG 2016 criteria in both phase I and phase II, cohort A. The time-to-event end points, including OS and PFS, were identified as important outcomes by clinical experts and patient and clinician groups consulted by CADTH for this review. However, OS and PFS were not adjusted for multiplicity in the MajesTEC-1 study, and the lack of a comparator group limited the estimation of relative effects of treatment with teclistamab. In addition, the longer-term efficacy of teclistamab for OS and PFS is unknown as the MajesTEC-1 study is ongoing. The clinical experts and patient and clinician groups consulted by CADTH for this review highlighted improvement in HRQoL as an important outcome and treatment goal for patients with RRMM. The analyses of HRQoL outcomes were undertaken post hoc, which introduced a risk of bias in the selection of the reported results. In addition, analyses for HRQoL were performed in HRQoL-evaluable patients and only for phase II, cohort A, rather than in the intention-to-treat (ITT) population of the MajesTEC-1 study, which may have biased the results; however, the extent of the bias with respect to the direction and magnitude of effect is uncertain. The size of the HRQoL-evaluable population in the MajesTEC-1 study gradually decreased over time and the rate of missing data was high among those who remained in the study at longer follow-up visits. Therefore, data from later time points should be interpreted with caution due to the possibility that HRQoL scores could have been overestimated if patients with better HRQoL were more likely to complete the questionnaires.22
According to the clinical experts, the patient population in the MajesTEC-1 study generally reflects patients in clinical practice in this setting. To be enrolled in the MajesTEC-1 study, patients with RRMM were required to have an ECOG PS score of 0 or 1 and have a measurable disease. The clinical experts consulted noted that this would not be reflective of clinical practice and that clinicians would prescribe teclistamab to patients with an ECOG PS score of 2 or 3 and to patients without biochemically measurable disease. Patients who had previously received antitumour therapy, such as a monoclonal antibody, or cytotoxic therapy within 21 days before the first dose of teclistamab, were excluded from the study; the clinical experts found this concerning as a washout period of 21 days is less relevant in this population. One of the exclusion criteria of the pivotal MajesTEC-1 study was any prior BCMA-targeted therapy. Additional supporting data were presented for phase II, cohort C, at the time of the clinical cut-off date of March 16, 2022, to address the use of teclistamab in patients previously treated with BCMA-targeted therapy in accordance with the Health Canada indication for teclistamab. Findings from phase II, cohort C, of the MajesTEC-1 study were consistent with the results from the pivotal cohort (phase I and phase II, cohort A); however, only 40 patients were included, which limits interpretation of the cohort findings. According to the clinical experts consulted by CADTH, the demographic and disease characteristics of the MajesTEC-1 study population were reflective of patients living in Canada with RRMM. The mean age of patients in the MajesTEC-1 trial was 64 years, with clinical experts noting that in the real-world setting, the mean age of patients with relapsed disease receiving fourth-line therapy and beyond would be around 70 years.23 About 26% of patients in the MajesTEC-1 trial had at least 1 high-risk abnormality, including del(17p) and t(4;14), although clinical experts noted that the proportion of patients with cytogenetic risk is slightly higher in clinical practice. In the MajesTEC-1 study, 63 (38.2%) patients switched from weekly to biweekly dosing of teclistamab, including 54 patients who met the response criteria and 9 patients who had switched from biweekly to monthly dosing. Clinical experts consulted by CADTH believed that there would be more patients in clinical practice switching to less frequent dosing of teclistamab. According to the clinical experts consulted by CADTH and patient and clinician group input, OS, PFS, clinical response outcomes, and HRQoL are the most important outcomes for assessing the response to treatment. However, due to its study design, the MajesTEC-1 trial provided no information about the efficacy and harms of teclistamab relative to treatments that would otherwise be used in this patient population in clinical practice. In the MajesTEC-1 trial, the study population was drawn from a number of sites around the globe, including in Canada. The clinical experts indicated no major concerns with generalizing the findings from the pivotal study to the Canadian clinical setting.
GRADE Summary of Findings and Certainty of the Evidence
The selection of outcomes for the Grading of Recommendations Assessment, Development and Evaluation (GRADE) assessment was based on the sponsor’s Summary of Clinical Evidence,83 consultation with clinical experts, and input received from patient and clinician groups and public drug plans. Table 2 presents the GRADE summary of findings for teclistamab in patients with RRMM in the MajesTEC-1 study.
Table 2: Summary of Findings for Teclistamab for Patients With Relapsed or Refractory Multiple Myeloma
Outcome and follow-up | Patients (study), N | Effect | Certainty | What happens |
|---|---|---|---|---|
OS | ||||
OS, months Median follow-up = 22.8 months | 165 (1 single-arm trial = phase I and phase II, cohort A) | Median (range) duration of OS of 21.9 (15.1 to NE) | Very lowa, b | The evidence is very uncertain about the effect of teclistamab on OS when compared with any comparator. |
PFS | ||||
PFS, months Median follow-up = 22.8 months | 165 (1 single-arm trial = phase I and phase II, cohort A) | Median (range) duration of PFS of 11.3 (8.8 to 16.4) | Very lowa, b | The evidence is very uncertain about the effect of teclistamab on PFS when compared with any comparator. |
CR or better (CR or sCR) | ||||
Proportion of patients who attained CR or better (95% CI) Median follow-up = 22.8 months | 165 (1 single-arm trial = phase I and phase II, cohort A) | 455 per 1,000 (377 to 534 per 1,000) | Very lowa, b | The evidence is very uncertain about the effect of teclistamab on CR or better when compared with any comparator. |
VGPR or better (VGPR, CR, or sCR) | ||||
Proportion of patients who attained VGPR or better (95% CI) Median follow-up = 22.8 months | 165 (1 single-arm trial = phase I and phase II, cohort A) | 594 per 1,000 (515 to 670 per 1,000) | Very lowa, b | The evidence is very uncertain about the effect of teclistamab on VGPR or better when compared with any comparator. |
ORR (PR, VGPR, CR, or sCR) | ||||
Proportion of patients who attained overall response (95% CI) Median follow-up = 22.8 months | 165 (1 single-arm trial = phase I and phase II, cohort A) | 630 per 1,000 (552 to 704 per 1,000) | Very lowa, b | The evidence is very uncertain about the effect of teclistamab on ORR when compared with any comparator. |
Duration of response | ||||
Duration of response (PR or better), months Median follow-up = 22.8 months | 165 (1 single-arm trial = phase I and phase II, cohort A) | Median (range) duration of response of 21.6 (16.2 to NE) | Very lowa, b | The evidence is very uncertain about the effect of teclistamab on duration of response when compared with any comparator. |
Harms | ||||
Proportion of patients with hypogammaglobulinemia Median follow-up = 22.8 months | 165 (1 single-arm trial = phase I and phase II, cohort A) | 212 per 1,000 | Very lowa, b | The evidence is very uncertain about the effect of teclistamab on hypogammaglobulinemia when compared with any comparator. |
Proportion of patients with infections Median follow-up = 22.8 months | 165 (1 single-arm trial = phase I and phase II, cohort A) | 800 per 1,000 | Very lowa, b | The evidence is very uncertain about the effect of teclistamab on infections when compared with any comparator. |
CI = confidence interval; CR = complete response; NE = not evaluable; ORR = overall response rate; OS = overall survival; PFS = progression-free survival; PR = partial response; sCR = stringent complete response; VGPR = very good partial response.
Note: Details included in Table 2 are from the sponsor’s Summary of Clinical Evidence.83
aIn the absence of a comparator arm, conclusions about efficacy relative to any comparator could not be drawn and certainty of evidence started at the level of very low.
b–1 level for serious study limitations. The reported result is from an interim analysis, and the effect may be overestimated.19-21 There was a risk of selection bias; it was not clear whether patients were enrolled consecutively.
Source: Clinical Study Report for the MajesTEC-1 study (2021).24
Additional Supporting Data
In the MajesTEC-1 study, efficacy and safety results for phase II, cohort C, were presented to support the results of the pivotal study in accordance with the Health Canada indication for teclistamab, and to address the question from the Provincial Advisory Group regarding the use of teclistamab in patients previously treated with BCMA-targeted therapy. Phase II, cohort C, enrolled patients with RRMM who had received 3 or more prior lines of therapy, including a PI, an IMiD, an anti-38 monoclonal antibody, and a BCMA-targeted treatment (e.g., CAR T-cell therapy, ADC). At the time of analysis, using the March 16, 2022, data cut-off, 40 patients had received at least 1 dose of teclistamab in phase II, cohort C, and were included in the FAS.
At the time of analysis, using the September 7, 2021, data cut-off, 38 patients were enrolled in phase II, cohort C, including 22 (57.9%) patients who were still on treatment. Baseline characteristics for these 38 patients are summarized as follows. More patients were enrolled in MajesTEC-1 study phase II, cohort C by the data cut-off date of March 16, 2022 (N = 40); however, the baseline data for the 40 patients were not reported in the Clinical Study Report.25 The median age of the patients was 63.5 years (range = 32 years to 82 years). A total of 24 (63.2%) patients were male and 14 (41.8%) were female. Most patients (89.5%) were white and 7.9% of patients identified as Black or African American. All patients were triple-class exposed, and a majority of patients were penta-exposed (78.9%). The most common immunoglobulin isotypes were immunoglobulin G, presenting in 18 (47.4%) patients. The median time from diagnosis of MM to enrolment in phase II, cohort C, was 6.5 years (range = 1.1 years to 24.1 years). Eleven (28.9%) patients had at least 1 extramedullary plasmacytoma at baseline. Of the 34 patients with baseline cytogenetic data reported, 11 (32.4%) patients had at least 1 high-risk abnormality, most commonly del(17p). A total of 20 (52.6%) patients were ISS stage I while 9 (23.7%) patients were ISS stage III. Prior anti-BCMA therapy included an ADC in 71.1% of patients and CAR T-cell therapy in 39.5% of patients.
Efficacy
At the time of analysis, using the March 16, 2022, data cut-off, the estimated median OS was 13.2 months (95% CI, 8.3 months to NE months). The median duration of follow-up was 12.5 months (range = 0.7 month to 14.4 months). In phase II, cohort C, deaths were reported in 17 (42.5%) patients in the FAS, and the proportion of patients who were censored (alive at the time of the data cut-off date) was 57.5% (23) of patients. The estimated 6-month OS probability among patients was ||||||||| ||| ||||| || ||||||, the 9-month OS probability was ||||| |||| ||| ||||| || ||||||, and the 24-month OS probability was ||||| |||| ||| ||||| || ||||||.
In phase II, cohort C, the estimated median PFS was ||| |||||| |||| ||| ||| || || |||||||. By the data cut-off date, a total of 24 (60.0%) patients had had a PFS event, and || |||||||| ||||||| were censored. The estimated 6-month PFS probability among patients in phase II, cohort C, was ||||| |||| ||| ||||| || ||||||. The 9-month PFS probability was ||||| |||| ||| ||||| || ||||||, and the 24-month PFS probability was ||||| |||| ||| ||||| || ||||||.
At the time of analysis, using the March 16, 2022, data cut-off, 11 (27.5%) patients (95% CI, 14.6% to 43.9%) in phase II, cohort C, attained CR or better (CR or sCR), 11 (27.5) patients (95% CI, 14.6% to 43.9%) attained sCR, 19 (47.5%) patients (95% CI, 31.5% to 63.9%) attained VGPR or better (VGPR, CR, or sCR), and 21 (52.5%) patients (95% CI, 36.1% to 68.5%) attained an overall response (PR or better). At the time of analysis, using the March 16, 2022, data cut-off, of the 21 patients who had attained PR or better, the median time to first response was 1.2 months (range = 0.2 month to 4.9 months), while the median time to best response was 2.9 months (range = 1.1 months to 9.5 months). At the time of analysis, using the March 16, 2022, data cut-off, the estimated median duration of response was not reached in phase II, cohort C. Among 21 responders (with a PR or better response), 15 (23.8%) patients had disease progression or died due to any cause. The estimated probability of patients remaining in response at 9 months was ||||| |||| ||| ||||| || ||||||, while the probability of patients remaining in response at 12 months was ||||| |||| ||| ||||| || ||||||.
Harms
At the time of analysis, using the March 16, 2022, data cut-off, all patients in phase II, cohort C, of the MajesTEC-1 study had experienced at least 1 TEAE. The most common TEAEs of any grade occurring in at least 20% of patients in phase II, cohort C, were CRS (67.5%), neutropenia (65.0%), anemia (50.0%), thrombocytopenia (45.0%), lymphopenia (45.0%), constipation (35.0), diarrhea (35.0%), pyrexia (32.5%), injection-site erythema (32.5%), and arthralgia (25.0%). In phase II, cohort C, 9 (22.5%) patients experienced TEAEs of grade 3, 20 (50.0%) patients experienced TEAEs of grade 4, and 8 (20.0%) patients experienced TEAEs of CTCAE grade 5. The most common TEAEs of grade 3 or grade 4 were neutropenia (62.5%), lymphopenia (42.5%), anemia (35.0%), and thrombocytopenia (30.0%). At the time of analysis, using the March 16, 2022, data cut-off, 24 (60.0%) patients had experienced at least 1 serious TEAE. The most common TEAEs were COVID-19 (10.0%), CRS and febrile neutropenia (7.5%), and anemia (5.0%). At the time of analysis, using the March 16, 2022, data cut-off, no patients had experienced a TEAE leading to treatment discontinuation. A total of 17 (42.5%) patients had died, of whom 8 (20.0%) patients had died within 30 days of the last dose of teclistamab. In phase II, cohort C, of the MajesTEC-1 study, several adverse events of clinical interest were identified, including CRS, neurologic adverse events and neurotoxicity, ICANS, injection-site reactions, hypogammaglobulinemia, cytopenia, infections, and tumour lysis syndrome (TLS). At the time of analysis, using the March 16, 2022, data cut-off, 26 (65.0%) patients in phase II, cohort C, had experienced 44 CRS events of any grade. A total of 21 (52.5%) patients in phase II, cohort C, had experienced at least 1 neurologic TEAE. The most common neurologic TEAEs included headache (22.5%), ICANS and insomnia (10.0%), encephalopathy (5.0%), peripheral sensory neuropathy (7.5%), dizziness (5.0%), and motor dysfunction (5.0%). At the time of analysis, using the March 16, 2022, data cut-off, a total of 10 (25.0%) patients had experienced at least 1 neurotoxicity event, including headache (12.5%) and ICANS (10.0%). A total of 26 (65.0%) patients in phase II, cohort C, had at least 1 treatment-emergent infection of any grade. The most common infections and infestations included COVID-19 (12.5%), bronchitis (10.0%), pneumonia (7.5%), cytomegalovirus infection reactivation (5.0%), implant site infection (5.0%), and laryngitis (5.0%). A total of 10 (25.0%) patients had experienced infections of grade 3 or grade 4, and 10 (25.0%) patients had experienced serious infections. At the time of analysis, using the March 16, 2022, data cut-off, proportions of patients with hypogammaglobulinemia were not reported. A total of 4 (10%) patients had experienced ICANS. All cases of ICANS were concurrent with CRS events, and no patients discontinued treatment due to ICANS. A total of 35 (87.5%) patients in phase II, cohort C, experienced at least 1 treatment-emergent cytopenic event, including neutropenia (67.5%), anemia (50.0%), thrombocytopenia (45.0%), and lymphopenia (45.0%). Hemorrhagic events were reported for 5 (12.5%) patients, 1 of which was of grade 2.
Findings from phase II, cohort C, of the MajesTEC-1 study were consistent with the results from the pivotal cohort (phase I and phase II, cohort A); however, only 40 patients were included, which limits interpretation of the cohort findings. Since the patients who took part in phase II, cohort C, were from the MajesTEC-1 study, it is reasonable to expect that the same limitations of the pivotal MajesTEC-1 study (phase I and phase II, cohort A) with respect to internal and external validity are relevant to phase II, cohort C, of the MajesTEC-1 study.
Long-Term Extension Studies
No long-term extension studies were submitted by the sponsor.
Indirect Comparisons
The efficacy and safety of teclistamab among adult patients with RRMM who had received at least 3 prior lines of therapy have been previously assessed in the MajesTEC-1 study. However, no head-to-head evidence of teclistamab compared against other treatments for RRMM was available for this review. Due to this gap in evidence, the sponsor submitted 6 indirect treatment comparisons (ITCs), of which 3 ITCs were used to inform the pharmacoeconomic model, including 2 ITCs comparing the relative efficacy of teclistamab with real-world physician’s choice (RWPC) therapy (from the LocoMMotion and daratumumab trials)26-29 and another ITC comparing the relative efficacy of teclistamab with ciltacabtagene autoleucel (from the CARTITUDE-1 trial).30 Of the 3 ITCs submitted by the sponsor that were not included in the pharmacoeconomic model, 1 published ITC compared the relative efficacy of teclistamab with RWPC therapy (from the Flatiron Health database),29,31 and 2 conference abstracts compared the relative efficacy of teclistamab with belantamab mafodotin (from the DREAMM-2 study),32 and selinexor in combination with dexamethasone (from the STORM study, part 2).33 No systematic review was reported by the sponsor.
Sponsor-Submitted ITCs Used to Inform Pharmacoeconomic Model
Three ITCs that were used to inform the pharmacoeconomic model were selected because they met the selection criteria. The sponsor stated that they included the most relevant comparators for the submission, including treatments that are reimbursed in Canada or have received a recommendation for reimbursement from CADTH for the indication under review. Given the absence of a comparator group in the MajesTEC-1 study, an external control group was used to establish the comparative efficacy of teclistamab versus treatments used in current clinical practice. To estimate the comparative efficacy, an inverse probability of treatment weighting (IPTW) estimator of the average treatment effect in the treated (ATT) was chosen for the main ITC analyses. This propensity score–based method allowed the RWPC cohorts from the LocoMMotion study and the daratumumab trials cohort, as well as the population in the CARTITUDE-1 study, to be reweighted to match the MajesTEC-1 trial’s population. There was sufficient overlap between patient characteristics between the MajesTEC-1 study and the LocoMMotion, CARTITUDE-1, and 4 daratumumab trials (the APOLLO, POLLUX, CASTOR, and EQUULEUS studies) to justify weighting techniques that do not depend on matching or excluding incompatible subpopulations. Propensity scores were estimated under an assumed logistic regression model using each cohort (the MajesTEC-1 study, the LocoMMotion study, the CARTITUDE-1 study, and the daratumumab trials cohort) as the dependent variable and selected baseline covariates as independent variables. The estimated propensity scores were then used to derive weights for each participant using the appropriate weighting formulas for the desired target population.
To ensure that the most important clinical factors were balanced between populations, an evidence-informed process (through a literature review of studies conducted to identify clinical outcomes in triple-class exposed patients with RRMM, and input from clinical experts) was used to select the prognostic factors for adjustment. In 2 ITCs comparing the relative efficacy of teclistamab with RWPC therapy from the LocoMMotion study and ciltacabtagene autoleucel from the CARTITUDE-1 study, treatment weights were rescaled to sum up to the original number of participants in the comparator studies. For the binary outcomes (e.g., ORR, CR or better, VGPR or better), a weighted logistic regression was used to derive an estimate of a conditional odds ratio (OR) and the corresponding 95% CI, transformed to response-rate ratio.34 For the time-to-event outcomes (i.e., PFS, duration of response, TTNT, and OS), a weighted Cox proportional hazards model was used to derive an estimate of the hazard ratio (HR) and the corresponding 95% CI. The appropriateness of the proportional hazards assumption used in the estimation of the HR of the survival outcomes was assessed based on visual inspection of the log-cumulative hazard plot, visual inspection of the Schoenfeld residuals plot, and the performance of the Grambsch-Therneau test (with a P value less than 0.05 considered to indicate a violation of the assumption).35
Teclistamab (MajesTEC-1 Study) Versus RWPC (LocoMMotion Study)
The MajesTEC-1 trial (n = 165) is an ongoing, phase I and phase II, multicentre, open-label, single-arm study. Eligible patients must have received a diagnosis of MM under IMWG diagnostic criteria, and have had prior exposure to at least 1 PI, 1 IMiD, and 1 anti-CD38 monoclonal antibody. In terms of efficacy outcomes, the primary outcome in the MajesTEC-1 trial is ORR; secondary outcomes include PR or better response, VGPR or better response, CR or better response, OS, PFS, MRD-negativity rate, duration of response, and TTR; and the exploratory outcome is TTNT. The LocoMMotion trial (n = 248) was a prospective, noninterventional study of real-life standard of care in patients with a documented diagnosis of MM according to IMWG diagnostic criteria who had received at least 3 prior lines of therapy including, at a minimum, PIs, IMiDs, and anti-CD38 monoclonal antibodies. The primary outcome in the LocoMMotion study was ORR; secondary outcomes included VGPR rate, CR rate, sCR rate, MRD-negativity rate, clinical benefit rate, duration of response, TTR, TTNT, PFS, and OS. Approximately 90 unique treatment regimens were used in the LocoMMotion study, including corticosteroids, PIs, IMiDs, alkylating drugs, and anti-CD38 monoclonal antibodies, and various combinations, reflecting the existing variety of real-life antimyeloma treatments in this population.28 A total of 17 prognostic factors that were identified a priori as important for population alignment were available from both studies. Before weighting, moderate (standardized mean difference [SMD] greater than 0.1 and less than or equal to 0.2) to substantial (SMD greater than 0.2) differences were observed for many of the variables included in the main IPTW analysis. After reweighting, observable differences remained in the ITT populations with regard to refractory status, and time to progression on last regimen. Cytogenetic risk was considered an important risk factor; however, it was not included in the main analyses due to a high level of missingness in the LocoMMotion study cohort (37.1%). As the LocoMMotion study population only included a low number of non-white patients, adding the race variable to the adjustment led to high weights for these patients and decreased balance for all the other variables.
Following adjustment, the estimated HRs of OS and PFS for teclistamab versus RWPC therapy were |||| |||| ||| |||| || ||||| ||| |||| |||||| |||| || |||||| ||||||||||||| || |||||| || |||||||||||. For duration of disease and TTNT, following adjustment, the estimated HR for teclistamab versus RWPC therapy was |||| |||| ||| |||| || ||||| ||| |||| |||| ||| |||| || |||||| ||||||||||||| || |||||| || |||||||||||. For ORR, following adjustment, the OR for teclistamab versus RWPC therapy was 4.89 (95% CI, 3.19 to 7.47), in favour of teclistamab. For CR or better, following adjustment, the OR for teclistamab versus RWPC therapy was |||||| |||| ||| ||||| || |||||||||| || |||||| || |||||||||||. For VGPR or better, following adjustment, the OR for teclistamab versus RWPC therapy was ||||| |||| ||| |||| || ||||||| || |||||| || |||||||||||. The results of the sensitivity analyses, including the analysis adjusted for all variables, were consistent with the main analysis results. No results for HRQoL and safety outcomes were reported in the ITC comparing the MajesTEC-1 and LocoMMotion trials.
Critical Appraisal
The sponsor-submitted ITC comparing the relative efficacy of teclistamab with RWPC therapy from the LocoMMotion trial had a number of limitations that challenged the internal and external validity of the findings. No systematic search was conducted to identify relevant studies; therefore, there is a risk of selection bias. There was variation in the design of the MajesTEC-1 and LocoMMotion studies, as the MajesTEC-1 study was a phase I and phase II trial while the LocoMMotion study was an observational, noninterventional study. Both studies were open label, so there is a risk of bias in the measurement of subjective outcomes, particularly PFS, and clinical response outcomes. Objective outcomes including OS should be unaffected by the open-label designs. The definitions of end points were similar across the studies; however, the median duration of follow-up was 14.1 months in the MajesTEC-1 study28 and 16.1 months in the LocoMMotion study.28 PFS and the clinical response outcomes were assessed based on IMWG criteria by an IRC in the MajesTEC-1 study and by an independent response review committee in the LocoMMotion study to reduce bias. In the MajesTEC-1 trial, there was a high degree of concordance between ORR assessments by the IRC and by the computerized algorithm used. The sensitivity analysis of ORR based upon investigator assessment was consistent with the primary analysis using IRC assessment based on IMWG response criteria, and similar comparisons were done with PFS in the MajesTEC-1 trial. The LocoMMotion study used a total of 90 different treatment regimens and given that not all treatment regimens are relevant to Canadian clinical practice in fourth-line settings and beyond (e.g., daratumumab, ixazomib, melphalan), the study results may not be generalizable to Canadian clinical practice. There was notable heterogeneity in the populations of the MajesTEC-1 and LocoMMotion studies. Of the 17 prognostic factors that were identified a priori, 15 variables were considered in the IPTW analyses for adjustment. Cytogenetic risk was considered an important risk factor by clinical experts; however, it was not included in the main analyses due to a high level of missingness in the LocoMMotion trial cohort (37.1%). The clinical experts consulted by CADTH noted that cytogenetic risk is an important prognostic factor, and omitting this factor could result in potential bias. After reweighting, populations from the MajesTEC-1 and LocoMMotion trials were more balanced, except for observed differences persisting in refractory status and time to progression on the final regimen. While the weighted populations were balanced with respect to known, measured prognostic factors, it remains unclear whether other unmeasured clinically relevant variables were unaccounted for. The variables not included in the planned adjustment set (unknown or unmeasured prognostic factors) can result in residual confounding and bias the estimates.36 Assessment of residual bias was not performed or reported. Therefore, the results of the IPTW analysis may be considered to have a high risk of residual bias;37 however, the magnitude and direction of any bias is unknown. In addition, the interpretation of the outcomes is challenging due to systematic differences in study design. The sponsor stated that due to a small sample size in the MajesTEC-1 and LocoMMotion trials, a scaled ATT weighting approach was used to scale treatment weightings so that they were summed to the original number of participants in the comparator studies. No information was reported in this IPTW analysis regarding the distribution of weights generated by the weighting process and the number of patients with extremely high and extremely low weights (including patients assigned 0 weight). Therefore, it remains unclear if patients with 0 weights (when there is no overlap with the target study) were excluded from the adjusted sample of the LocoMMotion study in accordance with the National Institute for Health and Care Excellence (NICE) Decision Support Unit (DSU) Technical Support Document,37 and what the effective sample size in the LocoMMotion study was after reweighting to estimate the number of nonweighted patients. Thus, due to the lack of clarity, the evidence obtained from this IPTW analysis remains uncertain, limiting the interpretation and generalizability of the results. Several sensitivity analyses were conducted, and results of the sensitivity analyses were consistent with the main analyses. For the OS, PFS, TTNT, and clinical response outcomes, the results of the adjusted treatment comparisons were consistent across end points, favouring teclistamab over RWPC therapy; this is consistent with the opinion of the clinical experts consulted for this review. However, these findings must be interpreted in the context of the methodological limitations of these studies. Safety outcomes were not analyzed in the ITC report and no justification was provided, which precludes a balanced judgment of comparative benefit relative to comparative harms. Outcomes that are important to patients, such as HRQoL, were also not analyzed in this ITC.
Teclistamab (MajesTEC-1 Study) Versus Ciltacabtagene Autoleucel (CARTITUDE-1 Study)
The MajesTEC-1 trial (n = 165) is an ongoing, phase I and phase II, multicentre, open-label, single-arm study. Patients in the MajesTEC-1 trial received teclistamab at a recommended dose of 1.5 mg/kg subcutaneously once a week, followed by step-up doses of 0.06 mg/kg and 0.3 mg/kg. The index date in the MajesTEC-1 study was defined as the date of the first dose for the MajesTEC-1 study. The CARTITUDE-1 study (n = 113) is an open-label, single-arm, phase Ib and phase II clinical trial evaluating the safety and efficacy of ciltacabtagene autoleucel in adult patients with RRMM. Eligible patients were diagnosed with MM according to IMWG diagnostic criteria and must have received at least 3 prior lines of therapy or must be double-refractory to an IMiD and a PI. In the CARTITUDE-1 study, following apheresis and premedication, ciltacabtagene autoleucel was administered as a single infusion dose of 0.75 × 106 CAR-positive viable T-cells per kg. The primary outcome in the CARTITUDE-1 study is ORR; secondary outcomes include VGPR rate, CR rate, sCR rate, MRD-negativity rate, clinical benefit rate, duration response, TTR, TTNT, PFS, and OS. The ITT population in the CARTITUDE-1 trial includes all patients who underwent apheresis with the index date defined as the date of apheresis.
Before weighting, moderate to substantial differences were observed for many of the main analysis variables. After weighting, populations in the MajesTEC-1 and CARTITUDE-1 studies were more balanced. After adjustment, differences remained in the ITT populations with regard to refractory status, age, hemoglobin level, and creatinine clearance. Following adjustment, the estimated HRs of OS and PFS for teclistamab versus ciltacabtagene autoleucel were |||| |||| ||| |||| || ||||| ||| |||| |||| ||| |||| || ||||||||||||||||||| || |||||| || |||||||||||||| ||||||||||. Following adjustment, the estimated HR of duration of response for teclistamab versus ciltacabtagene autoleucel was |||| |||| ||| |||| || |||||. Following adjustment, the estimated HR of TTNT for teclistamab versus ciltacabtagene autoleucel was |||| |||| ||| |||| || |||||| || |||||| || |||||||||||||| ||||||||||. No sensitivity analysis was performed in this ITC. No results for clinical response outcomes, including ORR, CR or better, and VGPR or better, were included in the ITC comparing the MajesTEC-1 and CARTITUDE-1 trials. No results for HRQoL and safety outcomes were included in the ITC comparing the MajesTEC-1 and CARTITUDE-1 studies.
Critical Appraisal
No systematic search was conducted to identify relevant studies; therefore, there is a risk of selection bias. Both the MajesTEC-1 and CARTITUDE-1 studies included in the ITC were presented with an unclear risk of bias for statistical analysis and a high risk for the measurement of subjective outcomes, such as PFS or clinical response outcomes, due to the open-label study design. Objective outcomes including OS should be unaffected by the open-label designs. The MajesTEC-1 trial cohort represented a broad population from Europe, Canada, and the US, whereas the results from the CARTITUDE-1 trial are drawn from US patients only. Therefore, it is unclear whether differences in clinical practice or treatment availability exist across regions, and the direction and magnitude of potential biases remain unclear. After weighting, populations in the MajesTEC-1 and CARTITUDE-1 trials were more balanced, although observable differences remained between the trials in refractory status, age, hemoglobin level, and creatinine clearance. While the weighted populations were balanced with respect to known, measured prognostic factors, it remains unclear whether other unmeasured clinically relevant variables were unaccounted for. The variables not included in the planned adjustment set (unknown or unmeasured prognostic factors) can result in residual confounding and bias the estimates.36 Assessment of residual bias was not performed or reported. Therefore, the results of the IPTW analysis may be considered to have a high risk of residual bias;37 however, the magnitude and direction of any bias is unknown. In addition, the interpretation of the outcomes is challenging due to systematic differences in study design and duration of follow-up. The sponsor stated that due to a small sample size in the MajesTEC-1 and CARTITUDE-1 studies, a scaled ATT weighting approach was used to scale treatment weightings so that they were summed to the original number of participants in the comparator studies. No information was reported in this IPTW analysis regarding the distribution of weights generated by the weighting process and the number of patients with extremely high and extremely low weights (including patients assigned 0 weight). Therefore, it remains unclear if patients with 0 weights were excluded from the adjusted sample of the CARTITUDE-1 study in accordance with the NICE DSU Technical Support Document,37 and what the effective sample size in the CARTITUDE-1 study was after reweighting to estimate the number of nonweighted patients. Thus, due to the lack of clarity, the evidence obtained from this IPTW analysis remains uncertain, limiting the interpretation and generalizability of the results. No methods for handling missing data were performed or reported in the ITC comparing teclistamab with ciltacabtagene autoleucel. For OS, PFS, and TTNT, the results of the adjusted treatment comparisons were consistent across end points, favouring ciltacabtagene autoleucel over teclistamab; however, these findings must be interpreted in the context of the methodological limitations of these studies. According to the clinical experts consulted by CADTH for this review, the population of the CARTITUDE-1 study was relatively healthy compared to the MajesTEC-1 trial’s population. Although all clinical response outcomes (ORR, CR or better, VGPR or better) were available in both studies, they were not assessed in this analysis. Safety outcomes were not analyzed in the ITC report, and no justification was provided, which precludes a balanced judgment of comparative benefit relative to comparative harms. Outcomes that are important to patients, such as HRQoL, were also not analyzed in this ITC.
Teclistamab (MajesTEC-1 Study) Versus Physician’s Choice Therapy (APOLLO, POLLUX, CASTOR, and EQUULEUS Studies)
The MajesTEC-1 trial (n = 165) is an ongoing, phase I and phase II, multicentre, open-label, single-arm study. Patients in the daratumumab trials were triple-class exposed and were treated with a physician’s choice (PC) of therapy after discontinuing the trial treatments. The daratumumab trials cohort consisted of patients from the long-term follow-up data from the POLLUX, CASTOR, EQUULEUS, and APOLLO studies. Because this ITC analysis retrospectively included patients participating in long-term follow-up clinical trials of daratumumab, it was possible to include patients in the earliest line of therapy initiated after all key selection criteria were met. However, this differed from the MajesTEC-1 study, in which patients may have received additional lines of therapy between the time at which they first met all eligibility criteria and the time at which they were enrolled in the clinical trial. To account for this difference, patients in the daratumumab trials became eligible for this analysis after having at least 3 prior lines of therapy, and patients who received multiple subsequent therapies after meeting eligibility criteria contributed multiple observations. Overall, 1,577 patients were initially included in the daratumumab trials cohort, of whom 642 patients were triple-class exposed and had received at least 1 treatment regimen. Of the 642 patients, 427 patients with 806 observations met the MajesTEC-1 trial’s key inclusion criteria.27 A total of 248 unique regimens were used in the RWPC from the daratumumab trials cohort. The primary outcome in the POLLUX study was PFS; secondary outcomes included time to progression, VGPR or better, MRD-negativity rate, ORR, OS, TTR, and duration of response. The primary outcome in the CASTOR study was PFS; secondary outcomes included time to progression, VGPR or better, MRD-negativity rate, ORR, OS, and TTR. The primary outcome in the EQUULEUS study was the proportion of adverse events and dose-limiting toxicities; secondary outcomes included ORR, OS, CR or better, and duration of response; and exploratory outcomes included PFS, MRD-negativity rate, and pharmacokinetics. The primary outcome in the APOLLO study was PFS; secondary outcomes included VGPR or better, MRD-negativity rate, ORR, OS, duration of response, TTNT, and TTR. In the daratumumab trials cohort, the index date was defined as the start of each eligible line of therapy.
Before weighting, moderate to substantial differences were observed for many variables. After weighting, substantial differences were observed with regard to prior stem cell transplant, ECOG PS, race, and type of MM. After adjustment, the resulting effective sample size in the daratumumab trials cohort was 264 patients compared to the original 806 patients. Following adjustment, the estimated HR for OS and PFS for teclistamab versus PC therapy was |||| |||| ||| |||| || ||||| ||| |||| |||| ||| |||||| |||||| ||||||||||||| || |||||| || |||||||||||. Following adjustment, the estimated HR of TTNT for teclistamab versus PC therapy was |||| ||||||| |||| || |||||| || |||||| || |||||||||||. For ORR, following adjustment, the OR for teclistamab versus PC therapy was |||| |||| ||| |||| || |||||| For CR or better, the OR for teclistamab versus PC therapy was ||||| |||| ||| |||| || ||||||. For VGPR or better, following adjustment, the OR for teclistamab versus PC therapy was ||||| |||| ||| |||| || ||||||. Results from the fully adjusted scenario analysis were consistent with the main analysis results. No results for HRQoL and safety outcomes were included in the ITC comparing the MajesTEC-1 study and the daratumumab trials cohort.
Critical Appraisal
There was variation in the design of the MajesTEC-1 study and the 4 daratumumab trials included in the IPTW analysis. The MajesTEC-1 study was a phase I and phase II trial while the POLLUX, CASTOR, and APOLLO studies were open-label, phase III, randomized controlled trials (RCTs), and the EQUULEUS trial was an open-label, nonrandomized, phase Ib study. Both the MajesTEC-1 study and the daratumumab trials were open label, so there was a risk of bias in the measurement of subjective outcomes, particularly PFS, and clinical response outcomes. Objective outcomes, including OS, should have been unaffected by the open-label designs. In addition, although 3 of the daratumumab trials included in the ITC were RCTs (the POLLUX, CASTOR, and APOLLO trials), and the EQUULEUS trial was an open-label, nonrandomized phase Ib study, patients selected from the daratumumab trials cohort were included in the analysis retrospectively. A total of 248 unique treatment regimens were used in the daratumumab trials cohort, many of which were not relevant to Canadian clinical practice; thus, the study results may not be generalizable to the Canadian setting. There was notable heterogeneity in the populations of the MajesTEC-1 study and the daratumumab trials cohort. Nine of the 17 prognostic factors identified a priori were used for ATT weighting in the main analysis. After weighting, populations from the studies were balanced with respect to known, measured prognostic factors. While the weighted populations were balanced with respect to known, measured prognostic factors, it remains unclear whether other unmeasured clinically relevant variables were unaccounted for. The variables not included in the planned adjustment set (unknown or unmeasured prognostic factors) can result in residual confounding and bias the estimates.36 Assessment of residual bias was not performed or reported. Therefore, the results of the IPTW analysis may be considered to have a high risk of residual bias;37 however, the magnitude and direction of any bias is unknown. In addition, IPTW cannot adjust for differences related to other sources of heterogeneity, such as differences in study design, or median duration of follow-up. After adjustment, the effective sample size was reduced to approximately 32.8% (264 of 804) of patients in the original sample size in the daratumumab trials cohort. A small effective sample size implies that the estimates are being influenced by a subset of the patients from the daratumumab trials and are caused by a violation of the transportability of the effects across cohorts.20 The proportional hazards assumption was tested for the time-to-event outcomes, and the Grambsch-Therneau test was significant for PFS and TTNT analyses, indicating potential violation of this assumption. The Cox proportional hazards model assumes that the HR across treatment groups does not change over time; therefore, violation of the proportional hazards assumption may lead to misleading and erroneous scientific conclusions.38,39 For the OS, PFS, TTNT, and clinical response outcomes, the results of the adjusted treatment comparisons were consistent across end points, favouring teclistamab over PC therapy; this is consistent with the opinion of the clinical experts consulted for this review. However, these findings must be interpreted in the context of the methodological limitations of these studies. Safety outcomes were not analyzed in the ITC report, and no justification was provided, which precluded a balanced judgment of comparative benefit relative to comparative harms. Outcomes that are important to patients, such as HRQoL, were also not analyzed in this ITC.
Other ITCs Not Included in the Pharmacoeconomic Model
The ITCs submitted by the sponsor had a number of limitations that challenged the internal and external validity of the findings. No systematic search was conducted to identify the comparator studies included in the 3 ITCs that were not used to inform the pharmacoeconomic model; therefore, there is a risk of selection bias. The selection criteria used to identify the comparator were consistent with the objective, and studies were included if they assessed treatment for RRMM, included patients with triple-class exposed RRMM who had received at least 3 prior lines of therapy, and had reported sufficient efficacy outcome data. However, no details were provided regarding the timing of the literature review, or the databases used. It is not possible to know whether the results may have differed if data from different RRMM studies or databases had been used. The list of excluded studies is not available and the risk of bias of the included studies was not assessed. Given the absence of a comparator group in the MajesTEC-1 study, an external control group was used to establish the relative efficacy of teclistamab versus treatments used in current clinical practice.
Teclistamab (MajesTEC-1 Study) Versus RWPC Therapy (Flatiron Health Database)
An ITC using the IPTW approach was conducted to assess the relative efficacy of teclistamab compared with RWPC therapy, using individual patient-level data from the MajesTEC-1 trial (for teclistamab) and the nationwide deidentified electronic health record–derived Flatiron Health database (for the RWPC cohort). Key eligibility criteria of the MajesTEC-1 study were applied to the RWPC therapy cohort, including a diagnosis of MM using IMWG criteria of prior exposure to 3 or more lines of therapy. Patients in the Flatiron Health database cohort who received multiple subsequent therapies after meeting eligibility criteria contributed multiple observations to the ITC analysis. The MajesTEC-1 trial cohort included data from 165 patients, while the unadjusted population of the RWPC Flatiron Health cohort included 420 unique patients, corresponding to 766 eligible lines of therapy. The propensity score–based method of IPTW with an ATT weighting was used to reweight the RWPC Flatiron Health cohort to align with the MajesTEC-1 trial’s population and adjust for imbalances between patient populations. For the MajesTEC-1 trial, a clinical cut-off of March 16, 2022, was used, with a median duration of follow-up of 14.1 months. For the RWPC Flatiron Health cohort, patients who had 2 or more documented clinical visits on or after January 2011 were included, with a median duration of follow-up of 18.2 months. After weighting, the effective sample size of the RWPC Flatiron Health cohort reduced to 42.6% of the original population.
Following adjustment, the estimated HR of OS for teclistamab versus RWPC therapy from the Flatiron Health cohort was 0.82 (95% CI, 0.59 to 1.14). For PFS, following adjustment, the estimated HR for teclistamab versus RWPC from the Flatiron Health cohort was 0.43 (95% CI, 0.33 to 0.56). For TTNT, following adjustment, the estimated HR for teclistamab versus RWPC from the Flatiron Health cohort was 0.36 (95% CI, 0.27 to 0.49). No results for safety or HRQoL were included in the ITC comparing the MajesTEC-1 study and the Flatiron Health cohort.
Critical Appraisal
As the Flatiron Health database was not selected using a systematic approach, there is a risk of selection bias. It is not possible to know whether the results may have differed if data from different RRMM studies or databases had been used. Numerous therapies were used in the RWPC groups from the Flatiron Health cohort, many of which were not relevant to Canadian clinical practice; thus, the ITC results may not be generalizable to the Canadian setting. Additionally, patients included in the present analysis initiated eligible lines of therapy between 2016 and 2021; however, clinical practice has changed since the enrolment of patients from these sources and may not be reflective of current treatment standards in Canada. Patients selected from the Flatiron Health cohort were included in the analysis retrospectively. Data analyzed retrospectively from databases are prone to unique biases (e.g., selection bias, confounding) compared with those collected from prospective interventional studies, which cannot be controlled using IPTW methods. Outcomes in the MajesTEC-1 study were assessed by the IRC while outcomes in the Flatiron Health cohort were assessed by the investigators; thus, the risk of bias in the outcome measurements is increased relative to the same outcomes as measured in the MajesTEC-1 study. The duration of follow-up in the MajesTEC-1 trial was 14.1 months versus 18.2 months in the Flatiron Health cohort. There were important differences in the design of the studies including in this ITC, as the MajesTEC-1 study was a phase I and phase II study while the Flatiron Health cohort was a real-world cohort from electronic health records in the US, which limits the ability to draw strong conclusions about the efficacy of teclistamab relative to RWPC therapy due to differences in clinician and patient behaviours, the heterogeneity of treatments for intercurrent events, and differences in data collection and intake. Such methodological differences could not be adjusted for in the IPTW analysis, and the magnitude and direction of any resulting bias is uncertain. There was evidence of heterogeneity between the MajesTEC-1 trial’s population and the Flatiron Health cohort. It remains unclear how balanced populations were for other variables that may be clinically relevant but could not be adjusted due to lack of data, or those variables that were not part of the planned adjustment (unknown or unmeasured prognostic factors), which leaves the potential for residual confounding.36 After weighting, the effective sample size of the Flatiron Health cohort was reduced by 57.4% from the included population. The reduction in the effective sample size reflects the heterogeneity between the trials among the variables included in the weighting process. A small effective sample size implies that the weighted estimates are being influenced by a subset of patients from the Flatiron Health cohort who may not be representative of the entire study population; this may limit the generalizability of the results. For OS and PFS, the results of the adjusted treatment comparisons were consistent across end points, favouring teclistamab over RWPC therapy from the Flatiron Health cohort. However, these findings must be interpreted in the context of the methodological limitations of these studies.
Teclistamab (MajesTEC-1 Study) Versus Belantamab Mafodotin (DREAMM-2 Study)
An unanchored matching-adjusted indirect comparison (MAIC) was conducted to compare the efficacy of teclistamab with belantamab mafodotin using individual patient data from the MajesTEC-1 trial (n = 150) and summary-level data from the DREAMM-2 trial (n = 97). The MajesTEC-1 trial was an open-label, single-arm, phase I and phase II study while the DREAMM-2 trial was an open-label, 2-arm, phase II study. The DREAMM-2 study’s eligibility criteria were applied to patients from the ITT population of the MajesTEC-1 study. Compared to patients in the DREAMM-2 study’s population, the MajesTEC-1 study’s population had a higher proportion of patients who had ISS stage I disease. All patients had triple-class exposed RRMM who had received at least 3 prior lines of therapy. Individual patient data from the MajesTEC-1 trial were weighted to match the aggregated DREAMM-2 trial baseline patient characteristics. The following factors were used to adjust for imbalances between patient populations: refractory status, cytogenetic profile, ISS staging, the presence of extramedullary disease, and the number of prior lines of therapy. The effective sample size of the MajesTEC-1 trial after propensity score matching was 33 patients. The comparative efficacy of teclistamab relative to belantamab mafodotin was estimated for ORR, CR or better, OS, PFS, and duration of response. For binary outcomes, the relative effects were quantified using an OR and 95% CI derived from a weighted logistic regression analysis, while time-to-event outcomes were estimated using a weighted Cox proportional hazards model.
Following adjustment, the estimated HR of OS for teclistamab versus belantamab mafodotin was 0.95 (95% CI, 0.47 to 1.92). For PFS, following adjustment, the estimated HR for teclistamab versus belantamab mafodotin was 0.63 (95% CI, 0.34 to 1.15), and the estimated HR of duration of response was 0.19 (95% CI, 0.05 to 0.73). Following adjustment, the OR of ORR for teclistamab versus belantamab mafodotin was 2.05 (95% CI, 0.92 to 4.57), while the OR of CR or better was 2.13 (95% CI, 0.80 to 5.65). No results for safety or HRQoL were included in the ITC comparing the MajesTEC-1 and DREAMM-2 trials.
Critical Appraisal
The open-label design of the studies can result in a risk of bias in the study conduct, including the measurement of the outcomes, and increase uncertainty in subjective outcomes such as PFS and ORR. The bias will likely favour the experimental intervention, although the extent of bias is uncertain. The effective sample size was reduced after adjustment in the MAIC analysis to approximately 22.0% (33 of 150) of the patients in the original population in the MajesTEC-1 study. The reduction in the effective sample size reflects the heterogeneity between the trials among the variables, including in the weighting process. Small effective sample size implies that the weighted estimates are being influenced by a subset of patients from the MajesTEC-1 study who may not be representative of the entire study population; this may limit the generalizability of the results. Populations from the studies were balanced with respect to known, measured prognostic factors. It remains unclear how balanced populations were for other variables that may have been clinically relevant but could not be adjusted due to lack of data, or those variables that were not part of the planned adjustment (unknown or unmeasured prognostic factors), which leaves the potential for residual confounding.36 In the MAIC analysis, the results in efficacy estimates were imprecise (i.e., wide CIs) in the end points assessed (including HR = 1), and the upper and lower boundaries of the CIs suggest the potential for different conclusions regarding the efficacy of teclistamab relative to the comparator drugs. Therefore, no superiority conclusions could be drawn from the MAIC submitted by the sponsor due to methodological limitations and imprecision in the effect estimates.
Teclistamab (MajesTEC-1 Study) Versus Selinexor in Combination With Dexamethasone (STORM Study, Part 2)
An unanchored MAIC was conducted to compare the efficacy of teclistamab with selinexor in combination with dexamethasone using individual patient data from the MajesTEC-1 trial (n = 150) and summary-level data from the STORM trial, part 2 (n = 122). The eligibility criteria of the STORM trial, part 2, were applied to patients from the ITT population of the MajesTEC-1 study. Compared to patients in the STORM trial, part 2, the MajesTEC-1 trial’s population had a higher proportion of patients with Revised International Staging System (R-ISS) stage II. The 2 populations were similar in age, ECOG PS, and cytogenetic status. All patients had triple-class exposed RRMM. After applying the eligibility criteria of the STORM trial, part 2, individual patient data from patients in the MajesTEC-1 study were weighted to match the aggregated baseline patient characteristics from the STORM trial, part 2. For binary outcomes, the relative effects were quantified using an OR and 95% CI derived from a weighted logistic regression analysis, while time-to-event outcomes were estimated using a weighted Cox proportional hazards model. The following factors were used to adjust for imbalances between patient populations: refractory status, cytogenetic profile, ISS staging, presence of extramedullary disease, and the number of prior lines of therapy. The effective sample size of the MajesTEC-1 study after matching was 37 patients.
Following adjustment, the estimated HR of OS for teclistamab versus selinexor in combination with dexamethasone was 0.52 (95% CI, 0.28 to 0.95). Following adjustment, the estimated HR of PFS for teclistamab versus selinexor in combination with dexamethasone was 0.58 (95% CI, 0.30 to 1.11), and the estimated HR of duration of response was 0.04 (95% CI, 0.01 to 0.10). Following adjustment, the OR of ORR for teclistamab versus selinexor in combination with dexamethasone was 3.14 (95% CI, 1.48 to 6.69), while the OR of CR or better was 16.3 (95% CI, 3.5 to 77.1). No results for safety or HRQoL were included in the ITC comparing the MajesTEC-1 trial and the STORM trial, part 2.
Critical Appraisal
The open-label design of the studies can result in a risk of bias in the study conduct, including the measurement of the outcomes, and increase uncertainty in subjective outcomes such as PFS and ORR. The bias will likely favour the experimental intervention, although the extent of bias is uncertain. The effective sample size was reduced after adjustment in the MAIC analysis to approximately 24.7% (37 of 150) of the patients in the original population in the MajesTEC-1 study. The reduction in the effective sample size reflects the heterogeneity between the trials among the variables, including in the weighting process. Small effective sample size implies that the weighted estimates are being influenced by a subset of patients from the MajesTEC-1 study who may not be representative of the entire study population; this may limit the generalizability of the results. It remains unclear how balanced populations were for other variables that may have been clinically relevant but could not be adjusted due to lack of data, or those variables that were not part of the planned adjustment (unknown or unmeasured prognostic factors), which leaves the potential for residual confounding.36 In the MAIC analysis, the results in efficacy estimates were imprecise (i.e., wide CIs) in the end points assessed, and the upper and lower boundaries of the CIs suggest the potential for different conclusions regarding the efficacy of teclistamab relative to the comparator drugs. Therefore, no superiority conclusions could be drawn from the MAIC submitted by the sponsor due to methodological limitations and imprecision in the effect estimates.
Studies Addressing Gaps in the Evidence From the Systematic Review
The sponsor submitted 2 ongoing head-to-head clinical studies comparing teclistamab to currently available fourth-line and beyond lines of therapy in patients with RRMM. The MajesTEC-3 trial is a multicentre, open-label, randomized, phase III study comparing the efficacy of teclistamab in combination with daratumumab versus investigator’s choice of daratumumab in combination with pomalidomide and dexamethasone, or daratumumab in combination with bortezomib plus dexamethasone in patients with RRMM. In the MajesTEC-3 trial, the primary outcome is PFS and the secondary outcomes include ORR, CR or better, MRD-negativity status, OS, and safety outcomes. The estimated completion date of the MajesTEC-3 study is October 2026. The MajesTEC-9 trial is a phase III, randomized, open-label, multicentre study comparing teclistamab with investigator’s choice of pomalidomide, bortezomib, and dexamethasone or carfilzomib and dexamethasone in patients with RRMM who have received 1 prior line to 3 prior lines of therapy, including an anti-CD38 monoclonal antibody, and lenalidomide. In the MajesTEC-9 trial, the primary outcome is PFS and the secondary outcomes include ORR, duration of response, and OS. The estimated completion date of the MajesTEC-9 study is May 2031.
Conclusions
One sponsor-submitted phase I and phase II, single-arm, open-label study (the MajesTEC-1 trial) provided evidence regarding the efficacy and safety of teclistamab for patients with RRMM who had received at least 3 prior lines of therapy. In general, the clinical experts consulted by CADTH for this review considered the OS, PFS, ORR, and CR results to be clinically meaningful, especially when indirectly compared to currently available therapies in this population. However, the evidence for the treatment effect of teclistamab is very uncertain due to the single-arm design of the study, which is not intended to be confirmatory for efficacy, and the lack of a comparator group, which limits the estimation of relative effects of treatment with teclistamab compared with other treatment options. Additionally, the estimates of the benefit of teclistamab may be overestimated because this report presents results of the interim analysis; however, the presence and extent of any overestimation is uncertain. The results for the HRQoL remained inconclusive due to a number of important limitations, including post hoc analysis, attrition bias, and increased risk of type I error. Notable harms that occurred more frequently in the MajesTEC-1 study included infections, CRS, and cytopenia. In general, TEAEs were stated by the clinical experts consulted by CADTH for this review to be manageable.
The evidence about the comparative efficacy of teclistamab relied on 1 single-arm study, and no direct evidence of teclistamab compared to other comparators was available for this review. Six ITCs were submitted by the sponsor comparing the efficacy of teclistamab with RWPC therapy, ciltacabtagene autoleucel, selinexor, and belantamab mafodotin in patients with RRMM. Although teclistamab was favoured over RWPC therapy for OS, PFS, and TTNT, the comparative efficacy estimates remain uncertain due to the methodological limitations, heterogeneity in the populations and studies, and potential for residual confounding. Although teclistamab was favoured over RWPC therapy for key response outcomes, no superiority conclusions could be drawn about the relative efficacy of teclistamab compared to RWPC for ORR, CR or better, or VGPR or better, due to methodological limitations and imprecision in the effect estimates (i.e., wide 95% CIs). Although the results suggest that ||||||||||| || |||||||| || |||||||||||||| |||||||||| ||| ||| |||| ||| ||||, these findings must be interpreted in the context of the methodological limitations of these studies. For OS and PFS, no superiority conclusions could be drawn about the relative efficacy of teclistamab compared to belantamab mafodotin and selinexor due to methodological limitations and imprecision in the effect estimates. No conclusions could be drawn from the 6 sponsor-submitted ITCs about the relative efficacy of teclistamab to the comparator drugs for the clinical response outcomes, including ORR, CR or better, or VGPR or better, due to methodological limitations and imprecision in the effect estimates (wide 95% CIs). No conclusions could be drawn on the relative safety of teclistamab to the comparative drugs because no safety analysis was performed. No HRQoL outcomes were evaluated in the sponsor-submitted ITCs.
There is an unmet need for fourth-line and beyond treatment options for RRMM, as patients with drug resistance after second-line or third-line therapy cannot be treated again with the same drug. In general, the indirect evidence was aligned with some outcomes identified as important to patients with RRMM who are seeking additional fourth-line and beyond treatment options that prolong survival and delay disease progression.
Introduction
Disease Background
Content in this section has been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the CADTH review team.
MM is a plasma cell cancer characterized by the clonal proliferation of malignant plasma cells (B-cells) and the overproduction of the abnormal M protein.1 Older individuals and men of all ages (as opposed to women) are more likely to develop MM and it is twice as common in individuals who are Black compared to white or Asian individuals.2,3 In 2022, it was estimated that 4,000 people in Canada were diagnosed with MM and 1,650 individuals living in Canada died from MM.4 The 5-year survival probability of patients with MM is estimated to be approximately 50%,5 and although survival rates have improved in recent years due to advances in therapeutic options, MM remains incurable.6,7 The majority of patients with MM will relapse and many patients will become refractory to commonly used therapies.8 Patients with RRMM often undergo multiple rounds of treatment, with the duration of remission, depth of response, PFS, and OS decreasing with each subsequent line of therapy.3
The majority of MM cases develop from an asymptomatic premalignant plasma cell condition called monoclonal gammopathy of undetermined significance (MGUS), which is characterized by the presence of abnormal M protein in the blood.3 MGUS can evolve into smouldering (asymptomatic) MM, which is a more advanced asymptomatic, premalignant stage that progresses into MM at a much faster rate compared to MGUS.3 The most common symptoms of MM are fatigue and bone pain,3 with other symptoms including kidney problems, recurrent infections, fever, and nervous system problems.9 Disease stage, along with other factors, can impact MM prognosis. Commonly recognized factors that impact the prognosis of MM include beta2-microglobulin (B2M) (high levels are associated with poor prognosis), serum albumin (low levels are associated with poor prognosis), lactate dehydrogenase (LDH) (high activity is predictive of poor prognosis), and chromosomal changes (shorter remission duration is associated with chromosome deletions or translocations).40 In addition to these factors, prognosis may be influenced by age, creatinine levels, and performance status. In general, older patients, those with high creatinine levels, and those with poor overall function tend to have worse outcomes compared to younger individuals, those with lower creatinine levels, and those with better overall function.41
The diagnosis of MM typically occurs during a visit to a primary care physician, occurring either incidentally when laboratory tests for other conditions are ordered, or if MM is suspected based on signs and symptoms.10 In 2016, the IMWG updated the diagnostic criteria for MM due to the emergence of novel treatment options, advances in laboratory and imaging techniques, and data indicating that early treatment in high-risk asymptomatic patients can improve survival.3,42 The updated criteria allows for earlier diagnosis and for treatment to be initiated before organ damage. The diagnosis of MM is based on the presence of 1 or more myeloma-defining events, along with either 10% or more clonal bone marrow plasma cells or biopsy-proven plasmacytoma.3 Myeloma-defining events include the presence of end-organ damage known as the CRAB criteria (hypercalcemia, renal insufficiency, anemia, and bone lesions) along with 3 specific biomarkers: a clonal bone marrow plasma cell percentage greater than or equal to 60%, a free light chain ratio greater than or equal to 100, and more than 1 focal lesion on MRI studies.3 It is recommended that the initial laboratory investigation for MM include a complete blood count to test, a peripheral blood smear, and a serum analysis, as well as baseline metabolic tests.43 To quantify the percentage of plasma cells relative to total nucleated cells, bone marrow aspiration and core biopsy are required. Finally, chromosomal aberrations should be assessed.43 The use of whole-body low-dose CT to detect osteolytic lesions in MM is becoming increasingly popular; however, if unavailable, a conventional radiography skeletal survey is a reasonable alternative.43
Several systems are used for staging MM, including ISS, the R-ISS, and the Durie-Salmon staging system.1,44,45 The Durie-Salmon staging system uses blood tests along with urine tests and X-rays to stage MM; however, unlike the ISS, the blood tests assess hemoglobin, calcium, and M-protein levels.44 The ISS is commonly used in Canada and uses blood tests that assess albumin levels and B2M levels to stage MM (advanced stage MM is associated with lower albumin and higher B2M levels):44
stage I — B2M is less than 3.5 mg/L; serum albumin is 3.5 g/dL or greater
stage II — B2M is less than 3.5 mg/L; serum albumin is less than 3.5 g/dL; or B2M is 3.5 mg/L to 5.5 mg/L, irrespective of serum albumin
stage III — B2M is greater than 5.5 mg/L.
The preferred staging system for MM is the R-ISS,45 which combines elements of tumour burden (ISS) and disease biology (the presence of high-risk cytogenetic abnormalities or an elevated LDH level) to create a unified prognostic index that helps in clinical care as well as in the comparison of clinical trial data. R-ISS uses serum B2M, serum albumin, serum LDH, and bone marrow fluorescence in situ hybridization (FISH) results to stratify patients into 3 risk groups:44,45
stage I — B2M is less than 3.5 mg/L, serum albumin is 3.5 g/dL or greater, normal LDH, and with no del(17p), t(4;14), t(14;16) by FISH
stage II — neither stage I nor stage III
stage III — B2M is 5.5 mg/L or greater, elevated LDH, and/or del(17p), t(4;14), t(14;16) by FISH.
According to the IMWG criteria, a patient is considered to have refractory MM when they are nonresponsive to therapy or experience disease progression within 60 days of their last line of therapy.11,12 A patient is considered to have relapsed MM if they experience disease progression after being previously treated and require a salvage therapy but do not meet the criteria for primary refractory MM or RRMM.11,12 Finally, a patient is considered to have relapsed and refractory MM if they have attained a minimal response or better at some point during previous treatments but the disease is currently nonresponsive on salvage therapy, or they experience disease progression within 60 days of their last therapy.11,12
Standards of Therapy
Content in this section has been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the CADTH review team.
According to the clinical experts and clinician groups consulted by CADTH for this review, initial therapy for patients with MM depends on whether patients are transplant eligible or ineligible at diagnosis, which is aligned with CADTH's Provisional Funding Algorithm for MM.13 According to the clinical experts and clinician groups, the majority of patients who are ineligible for transplant can be given daratumumab, lenalidomide, and dexamethasone as frontline therapy. A small number of patients will receive lenalidomide in combination with dexamethasone, and some other combinations of drugs that do not contain daratumumab, such as lenalidomide in combination with bortezomib and dexamethasone. The clinical experts consulted by CADTH mentioned that the current approach to the treatment of RRMM depends on several factors, including patient factors (i.e., age, comorbidity, and beforexicity), line of therapy, and prior therapies received. The clinical experts further noted that treatment options at relapse include a PI (bortezomib, carfilzomib) containing combinations such as cyclophosphamide in combination with bortezomib and dexamethasone, selinexor in combination with bortezomib and dexamethasone, or carfilzomib in combination with dexamethasone with or without cyclophosphamide. According to the clinical experts and clinician groups, patients who are eligible for transplant can receive induction therapy with either cyclophosphamide in combination with bortezomib and dexamethasone, or lenalidomide in combination with bortezomib and dexamethasone, and then lenalidomide maintenance until disease progression. The clinical experts consulted noted that the majority of patients who relapsed after a prior stem cell transplant will not have received daratumumab, and second-line therapy for these patients will likely include an anti-CD38 antibody. These combinations include isatuximab in combination with carfilzomib and dexamethasone, daratumumab in combination with lenalidomide and dexamethasone, isatuximab in combination with pomalidomide and dexamethasone, and selinexor in combination with bortezomib and dexamethasone. The clinical experts mentioned that later relapses can be treated through clinical trials or with regimens such as belantamab mafodotin, which is presently available through a special access program. Currently, access to bispecific antibodies for the treatment of relapsed myeloma is limited. Other treatments aim to control symptoms, such as those used to control pain, or bisphosphonates for bone diseases.
There is no preferred standard of care for the treatment of RRMM in the fourth-line setting and beyond, and at this stage of the disease, patients may be exposed to PIs, IMiDs, and anti-CD38s,7,13 and in some cases receiving more than 1 PI or iMiD, further limiting treatment options in later lines of therapy. Generally, the second-line treatment options available to a patient with RRMM depend on the patient’s response to the therapy received in first-line, and the choice of a third-line treatment option depends on their response to the therapy received in the second-line treatment. According to the Provisional Funding Algorithm for MM developed by CADTH,13 patients with drug resistance cannot be treated again with the same drug; however, in later lines, previously used treatments are often recycled when accessible due to a lack of novel options.46-48
The pan-Canadian Oncology Drug Review Expert Review Committee (pERC) recommends the reimbursement of carfilzomib in combination with dexamethasone for patients with RRMM with good performance status who have received 1 prior line to 3 prior lines of treatment, with reimbursement being conditional on cost-effectiveness being improved to an acceptable level.13,49 pERC recommends the reimbursement of pomalidomide combined with dexamethasone in patients with RRMM who displayed disease progression on their previous treatment regimen and who did not experience improvement with at least 2 prior lines of therapy, including both bortezomib and lenalidomide. Funding is conditional on cost-effectiveness being improved to an acceptable level.13,50,51 pERC recommends the reimbursement of selinexor in combination with bortezomib and dexamethasone for the treatment of adult patients with MM who have received at least 1 prior therapy.13,52 pERC recommends the reimbursement of ciltacabtagene autoleucel (Carvykti) for the treatment of adult patients with MM who have received at least 3 prior lines of therapy, including a PI, an IMiD, and an anti-CD38 antibody, and who are refractory to their last treatment.13,53 According to the Provisional Funding Algorithm for MM, selinexor in combination with bortezomib and dexamethasone is recommended for third-line therapy and beyond in patients who are sensitive to bortezomib but not to anti-CD38 monoclonal antibodies and lenalidomide.13
In Canada, coverage for pomalidomide in combination with dexamethasone with or without cyclophosphamide, and carfilzomib in combination with dexamethasone with or without cyclophosphamide, is determined on a case-by-case basis while ciltacabtagene autoleucel is under consideration for negotiation at the pan-Canadian Pharmaceutical Alliance.53 Coverage for selinexor varies across provinces in Canada. For example, selinexor is publicly funded in British Columbia54 but is only available through the Exceptional Access Program In Ontario.55
Drug Under Review
Teclistamab injection is indicated for treating adult patients with RRMM who have received at least 3 prior lines of therapy, including a PI, an IMiD, and an anti-CD38 monoclonal antibody, and who have demonstrated disease progression on their last therapy. The reimbursement request aligns with the Health Canada indication for teclistamab.
Teclistamab is a bispecific antibody that targets the CD3 receptor expressed on the surface of T-cells and BCMA expressed on the surface of malignant MM and healthy B-lineage cells and plasma cells.56 Teclistamab redirects CD3-positive T-cells to BCMA-expressing myeloma cells to induce the killing of tumour cells.57
The recommended dosage for teclistamab is 1.5 mg/kg of body weight after receiving step-up doses of 0.06 mg/kg and 0.3 mg/kg of body weight. One hour to 3 hours before each teclistamab step-up dose and the first full-strength treatment dose, all patients must receive a corticosteroid (16 mg oral or IV dexamethasone), an antihistamine (50 mg oral or IV diphenhydramine or equivalent), and an antipyretic (650 mg to 1,000 mg oral or IV acetaminophen or equivalent).58 Patients should be treated with teclistamab until disease progression or unacceptable toxicity. Dose delays may be required to manage toxicities related to teclistamab.58 The recommendations for restarting teclistamab after a dose is delayed is outlined in Table 3.
Table 3: Recommendations for Restarting Teclistamab After Dose Delay
Last dose administered | Duration of delay from the last dose administered | Action |
|---|---|---|
Step-up dose 1 | 7 days or less | Resume teclistamab step-up dosing schedule at step-up dose 2 (0.3 mg/kg).a |
More than 7 days | Restart teclistamab step-up dosing schedule at step-up dose 1 (0.06 mg/kg).a | |
Step-up dose 2 | 7 days or less | Resume teclistamab step-up dosing schedule at treatment dose (1.5 mg/kg).a |
8 days to 28 days | Resume teclistamab step-up dosing schedule at step-up dose 2 (0.3 mg/kg).a | |
More than 28 days | Restart teclistamab step-up dosing schedule at step-up dose 1 (0.06 mg/kg).a | |
Any treatment dose | 28 days or less | Resume teclistamab at treatment dose (1.5 mg/kg) once weekly. |
More than 28 days | Restart teclistamab step-up dosing schedule at step-up dose 1 (0.06 mg/kg).a |
aAdminister pretreatment medications before teclistamab dose and monitor patients accordingly.
Source: Product monograph for Tecvayli.58
Teclistamab is administered as an SC injection by a health care provider with appropriate medical equipment and medical personnel to manage administration-related reactions.58 Dose reductions are not recommended for teclistamab.58 Patients should remain in the proximity of a health care facility and be monitored daily for 48 hours for signs and symptoms of CRS after the administration of all step-up doses of teclistamab; alternatively, hospitalization for patients may be considered.58
The key characteristics of teclistamab and other therapies for the treatment of adult patients with RRMM in fourth-line therapy and beyond, as indicated in CADTH's Provisional Funding Algorithm for MM,13 are presented in Table 4.
Table 4: Key Characteristics of Pharmacotherapies for Multiple Myeloma
Characteristic | Teclistamab | Ciltacabtagene autoleucel | Selinexor | PIs (carfilzomib) | IMiDs (pomalidomide) |
|---|---|---|---|---|---|
Mechanism of action | A bispecific antibody targeting both BCMA (present on MM cells) and CD3 receptors (present on T-cells). Teclistamab redirects CD3-positive T-cells to BCMA-expressing myeloma cells to induce the killing of tumour cells. | BCMA-directed genetically modified autologous CAR T-cell immunotherapy. | Selinexor is a compound that specifically blocks XPO1, a nuclear export protein that transports cargo proteins within the cell. XPO1 inhibition by selinexor leads to the reduction of cancer cells. | Proteasome inhibition leads to the accumulation of misfolded protein in endoplasmic reticulum, resulting in apoptosis and the inhibition of cell proliferation. | Immunomodulatory and antineoplastic activity; inhibits proliferation and induces apoptosis of hematopoietic tumour cells. |
Indicationa | For the treatment of adult patients with RRMM who have received at least 3 prior lines of therapy, including a PI, an IMiD, and an anti-CD38 monoclonal antibody | For the treatment of adult patients with MM who have received at least 3 prior lines of therapy, including a PI, an IMiD, and an anti-CD38 antibody, and who are refractory to their last treatment | In combination with bortezomib and dexamethasone for the treatment of adult patients with MM who have received at least 1 prior therapy | In combination with dexamethasone alone, for patients with relapsed MM who have received 3 prior lines of therapy | In combination with dexamethasone for patients with MM for whom both bortezomib and lenalidomide had failed and who had received at least 2 prior regimens and demonstrated disease progression on the last regimen |
Route of administration | SC injection | IV infusion | Orally | IV infusion | Orally |
Recommended dosage | 1.5 mg/kg of body weight weekly after receiving step-up doses of 0.06 mg/kg and 0.3 mg/kg of body weight | Single infusion of 0.5 to 1.0 × 106 CAR-positive viable T-cells per kg body weight |
|
|
|
Serious adverse effects or safety issues | Cytokine release syndrome, neurologic toxicity (including immune effector cell-associated neurotoxicity syndrome), infections, hypogammaglobuli-nemia, cytopenias, injection-site infections | Cytokine release syndrome, neurologic toxicities (including ICANS), macrophage activation syndrome or hemophagocytic lymphohistiocytos-is | Fatigue, severe or life-threatening hyponatremia, nausea, vomiting, diarrhea, anorexia or weight loss, thrombocytopenia, neutropenia, infections, dizziness, cataracts | Infusion reactions, TLS, infections, cardiac disorders, venous thrombosis, hypertension, hemorrhage, thrombocytopenia, hepatoxicity, hepatitis B reactivation, posterior reversible encephalopathy syndrome, PML, acute renal failure, pulmonary toxicity | Neutropenia, thrombocytopenia, infections, DVT and pulmonary embolism, hepatoxicity, anaphylaxis, hepatitis B reactivation, severe rash (SJS, TEN, DRESS), TLS |
Other | None | None | Currently under negotiations | Premedication for carfilzomib recommended with dexamethasone (at least 30 minutes prior), to reduce incidence and severity of infusion reactions | Antithrombotic prophylaxis recommended |
BCMA = B-cell maturation antigen; CAR = chimeric antigen receptor; DRESS = Drug Rash with Eosinophilia and Systemic Symptoms; DVT = deep vein thrombosis; ICANS = immune effector cell-associated neurotoxicity syndrome; IMiD = immunomodulatory drug; MM = multiple myeloma; PI = proteasome inhibitor; PML = progressive multifocal leukoencephalopathy; RRMM = relapsed or refractory multiple myeloma; SC = subcutaneous; SJS = Stevens-Johnson syndrome; TEN = toxic epidermal necrolysis; TLS = tumour lysis syndrome.
aHealth Canada–approved indication.
Sources: Product monographs for Tecvayli,58 Carvykti,53,59 Xpovio,60 Pomalyst,61 and Kyprolis.62
Stakeholder Perspectives
Patient Group Input
This section was prepared by the CADTH review team based on the input provided by patient groups. The full original patient input received by CADTH has been included in the Stakeholder section of this report.
CADTH received 1 patient group submission from Myeloma Canada. Myeloma Canada has existed for more than 15 years to support the growing number of Canadians diagnosed with myeloma and those living longer than ever with the disease to access new and innovative therapies. Over the years, as a part of this mission, Myeloma Canada has collected data on the impact of myeloma and its treatments on patients and caregivers by conducting surveys. The results of a patient and caregiver survey conducted by Myeloma Canada from August 28 to September 6, 2023, were provided to the CADTH review. The results of the survey were shared across Canada and internationally via email and social media. A total of 33 patients and 3 caregivers were initially asked similar questions regarding disease experience. Upon verifying their eligibility for or experience with teclistamab, survey respondents were divided into 3 subsets as follows:
Subset 1: This subset consisted of 22 patient respondents (1 was from outside of Canada) who would currently be eligible for treatment with teclistamab from Alberta (n = 1), British Columbia (n = 5), New Brunswick (n = 1), Ontario (n = 8), Quebec (n = 5), Saskatchewan (n = 1), and France (n = 1). Twelve respondents identified themselves as male and 9 as female. One respondent was between 50 years and 59 years, 10 respondents were between 60 years and 69 years, 10 respondents were between 70 years and 79 years, and 1 respondent was between 80 years and 89 years. Eleven respondents lived in an urban area, 7 lived in a suburban area, and 4 lived in a rural area.
Subset 2: This subset consisted of 11 patient respondents who had received or were currently receiving treatment with teclistamab. Six respondents identified themselves as female, 5 respondents identified as male, 8 respondents lived in urban areas, 2 in a suburban area, and 1 in a rural area. One respondent was aged between 40 years and 49 years, 1 respondent was between 50 years and 59 years, 2 respondents were between 60 years and 69 years, and 7 respondents were between 70 years and 79 years.
Subset 3: This subset consisted of 3 caregivers of patients who would currently be eligible for treatment with teclistamab. All caregivers lived in an urban area and were female. One caregiver was between 40 years and 49 years, 1 caregiver was between 60 years and 69 years, and 1 caregiver was between 70 years and 79 years.
Patient respondents indicated that their ability to travel was the factor most significantly impacted by symptoms associated with myeloma, followed by the ability to work and exercise. Regarding the most significant financial implication of myeloma treatment on a household, 12 of 36 respondents identified the loss of income or pension funds due to absence from work, disability, or early retirement, and 12 of 36 respondents chose travel costs. Moreover, they felt that “loss of sexual desire” had the greatest impact on quality of life, and it was the option most frequently rated as severe impact. All caregivers felt that caring for someone with myeloma had the most impact on “anxiety/worry,” followed by “interruption of life goals/accomplishments (e.g., career, retirement)”. Eighteen of 33 patient respondents indicated that they needed a caregiver, 13 respondents answered that they did not need a caregiver, and 2 respondents indicated that they did not need a caregiver but would benefit from a caregiver’s help.
Patient and caregiver respondents identified the following factors as those most important to myeloma treatment: improved quality of life, manageable side effects along with the effectiveness of treatment, especially in achieving remission and having a long and durable response, and treatment accessibility or portability (including fewer or minimal visits to the hospital or cancer centre). Regarding the time it takes to make a round trip (to and from) the hospital or cancer centre to receive treatment, 20 of 36 respondents indicated “less than 1 hour 1 way,” 10 respondents selected “30 minutes to 1 hour 1 way,” 3 respondents selected “2.5 hours or more 1 way,” and 2 respondents selected “1 to 2 hours 1 way.” When all 33 patients and 3 caregivers were asked how often they visit the hospital or cancer centre for treatment, 19 respondents mentioned once a month, 7 respondents mentioned once a week, and 4 respondents mentioned every 2 weeks. In terms of the importance of controlling various aspects of myeloma, infections were the most important aspect to control, followed by kidney problems, mobility, gastrointestinal issues, relapse, and secondary cancers. Regarding receiving prior lines of therapy, 12 patient respondents indicated that they received 3 prior lines of therapy, 10 respondents indicated that they or the person they care for received 4 prior lines of therapy, and 3 respondents indicated that they or the person they care for had received 5 prior lines of therapy or more. Regarding receiving autologous stem cell transplant for myeloma treatment, 25 respondents indicated that they had received it, while 3 respondents indicated they or the person they care for were not eligible for an autologous stem cell transplant.
In terms of improving quality of life, 13 of the 22 respondents rated it as extremely important, 6 respondents as very important, and 3 respondents as somewhat important. A total of 17 of the 22 respondents rated the estimated minimum of 1 year to 21 months of life extension as extremely desirable, and 5 respondents as very desirable. A total of 11 respondents indicated having experience with teclistamab: 5 respondents received teclistamab as monotherapy, 1 patient indicated they were unsure, and 5 respondents received teclistamab in combination with another drug and provided the drug name(s). Of these 5 patient respondents, 2 patients are receiving daratumumab in combination with teclistamab, 1 respondent is receiving talquetamab in combination with teclistamab and the remaining 2 respondents described supportive care drugs, not a combination therapy (IV immunoglobulin, steroids, Benadryl). In terms of the impact of teclistamab on their quality of life compared to previous treatments, 5 respondents indicated no impact, 3 respondents mentioned some impact, 2 respondents stated significant impact, and 1 respondent indicated a little impact. In terms of side effects, 10 respondents rated respiratory infections as the least bearable side effect, followed by COVID-19, fungal infections, and ICANS.
Myeloma Canada also conducted another survey on CAR T-cell therapy from August 28 to September 30, 2022. It received more than 200 responses, only 2 of which had experience with the CAR T-cell therapy. While the teclistamab survey received far fewer total responses (33), there were 11 patients with teclistamab experience. Myeloma Canada emphasized that comments from patients currently receiving teclistamab were largely very positive, with multiple patients at different points in the survey indicating that it was the best treatment they had received for myeloma.
Clinician Input
Input From Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of MM.
Unmet Needs
The clinical experts consulted by CADTH for this review highlighted that the most important goals of treatment for patients with MM are prolonging survival, delaying disease progression, preventing disease complications, improving quality of life, and minimizing side effects. Clinical experts noted that although MM is traditionally considered an incurable malignancy, it is known that a small percentage of patients who undergo a stem cell transplant do not relapse. The clinical experts mentioned that the current treatment approach and choice of therapy for MM depends on several factors, including patient factors (i.e., age, comorbidity, and beforexicity), line of therapy, and prior treatments received. After diagnosis with MM, the lines of therapy that patients will undergo depend on whether or not they are transplant eligible. The clinical experts noted that clinicians try not to reuse the same drugs in subsequent lines of therapy. The common treatment options for MM relapse include a PI-containing combination, such as cyclophosphamide in combination with bortezomib and dexamethasone, selinexor in combination with bortezomib and dexamethasone, or carfilzomib in combination with dexamethasone with or without cyclophosphamide. The clinical experts also highlighted that after receiving 3 lines of therapy, the majority of patients will be triple-refractory and will need new families of drugs. The clinical experts also noted that beyond the third line of therapy, treatment options get more restricted, and some patients do not respond to the current standard therapies. The clinical experts indicated that, most importantly, very poor responses and very short remissions are observed in patients with high-risk cytogenetics. Thus, there is a need for treatments beyond the third line of therapy that are tolerable for patients.
Place in Therapy
The clinical experts consulted indicated that, given the level of evidence for the drug under review, it would be an appropriate treatment to use in the setting of RRMM. However, it is unknown whether teclistamab would be used as a complementary therapy, as the study assessing the efficacy of teclistamab when used in combination with other treatments (the MajesTEC-3 study) is ongoing. The clinical experts noted that teclistamab is not the first approved treatment that targets underlying disease processes; however, this drug has a novel mechanism of action that is very different from any currently available therapies. The clinical experts agreed that teclistamab would not be reserved for patients who are intolerant to other treatments or for whom other treatments are contraindicated, but instead would be available to any patient who meets eligibility requirements. The clinical experts agreed that teclistamab will not necessarily change the current treatment paradigm; however, this drug will be included in the treatment sequence, with the timing of treatment depending on the evidence for its use in different lines. The clinical experts also noted that as teclistamab is being introduced as a treatment for RRMM, it is expected that patients will have been treated with some other treatments before being eligible for this drug. The clinical experts noted that classic methodological assessments of interventions concern A versus B and implication of 1 or the other without the consideration of sequencing beyond the initial drug failure. In this light, the use of teclistamab represents a new class of drugs that can help sustain quality of life and extend the duration of life in the patient with RRMM.
Patient Population
The clinical experts consulted indicated that the MajesTEC-1 study enrolled a broad sample of patients with RRMM, including older patients who had received multiple prior lines of therapy and had measurable disease. Clinical experts agreed that there is no evidence that some patients are more likely to respond to treatment, and there are no disease-specific characteristics that would make a patient ineligible for treatment with teclistamab. Given that the prognosis of MM worsens as patients move on to subsequent lines of therapy, any patient with relapsed MM will require this intervention. This is especially true in cases where patients have exhausted other treatment options; however, it may also be appropriate to use teclistamab earlier in the relapse even if all other options have not been exhausted. The clinical experts highlighted that patients eligible for treatment will be identified at the time of MM relapse, usually based on biochemical evidence of relapse, and in some cases also based on other signs or symptoms of relapse. The clinical experts also noted that diagnostic tests for relapsed MM are well established and include serum protein electrophoresis, immunoglobulin free light chains, and other tests, and that a companion diagnostic test is not specifically required.
Assessing the Response Treatment
Clinical experts consulted by CADTH noted that response to treatment is usually assessed through regular monthly monitoring as part of the management of patients with RRMM. However, in many situations where the patient is stable on treatment that has predictable side effects, follow-up may be reduced to every 2 months, or sometimes less frequently. The clinical experts highlighted that if patients are followed relatively closely, difficulties or delays in diagnosing RRMM are unlikely. The clinical experts mentioned that the response to treatment for almost all patients is assessed by monitoring serum protein, electrophoresis, serum free light chains, or other tests. They further noted that these tests are objective and their interpretation does not really vary among physicians.
The clinical experts identified PFS, OS, and clinical response outcomes as the most important outcomes for assessing the response to treatment. The clinical experts agreed that the best possible response to treatment would be complete remission that is MRD-negative, and less deep responses include CR, VGPR, PR, and stable disease. Clinical experts further noted that CR and VGPR are the most desirable outcomes in most situations; even achieving stable disease is acceptable.
Discontinuing Treatment
The clinical experts consulted indicated that the main reason for discontinuing treatment with teclistamab would be relapse of MM. The clinical experts further noted that as with any treatment, it can be expected that some patients will be forced to discontinue treatment due to intolerable side effects.
Prescribing Considerations
The clinical experts consulted mentioned that currently, all patients receiving teclistamab are treated at tertiary care centres and are admitted to the hospital for the first few doses. The clinical experts indicated that depending on the situation, patients starting treatment with teclistamab will require treatment at a larger hospital capable of providing management and monitoring; however, after patients receive the first few doses of this treatment, they can receive ongoing therapy at community centres and smaller cancer centres. The clinical experts noted that the issue of intensive monitoring and hospitalization only affects the first 2 doses to 3 doses, after which treatment becomes more routine.
Clinician Group Input
This section was prepared by the CADTH review team based on the input provided by clinician groups. The full original clinician group input received by CADTH has been included in the Stakeholder section of this report.
CADTH received 2 clinician group submissions from the CMRG and OH-CCO’s Drug Advisory Committees. CMRG is a Canada-wide network of researchers aiming to develop better treatments for extending the life of patients with myeloma, enhancing quality of life for those living with myeloma and related diseases, and working to find a cure for these diseases and other plasma cell disorders. OH-CCO’s Drug Advisory Committees provide timely evidence-based clinical and health system guidance on drug-related issues in support of OH-CCO’s mandate, including the Provincial Drug Reimbursement Programs and the Systemic Treatment Program.
CMRG gathered information through teleconferences with physicians, and OH-CCO’s Drug Advisory Committees gathered information through videoconferencing and email communications. CMRG stated that newly diagnosed patients with myeloma living in Canada are classified into those who are transplant eligible or transplant ineligible, based on age and fitness. Transplant-eligible patients receive bortezomib-based induction with lenalidomide in combination with bortezomib and dexamethasone, or cyclophosphamide in combination with bortezomib and dexamethasone followed by high-dose melphalan and an autologous stem cell transplant, and then lenalidomide maintenance until disease progression. Transplant-ineligible patients were previously most often treated with lenalidomide in combination with dexamethasone or lenalidomide in combination with bortezomib and dexamethasone followed by single-drug lenalidomide (also given until disease progression). More recently, daratumumab-based combinations such as daratumumab in combination with lenalidomide and dexamethasone, or the combination of daratumumab, cyclophosphamide, bortezomib, and dexamethasone, and the combination of bortezomib, melphalan, and prednisone are preferred and include provisions for the long-term continuous administration of selected drugs. CMRG noted that second-line therapy depends on whether patients have progressed on lenalidomide (currently, this includes most patients who have undergone an autologous stem cell transplant or who are transplant-ineligible). Key in second-line therapy is the inclusion of an anti-CD38 antibody, such as daratumumab or isatuximab, which represents a high priority for virtually all patients. Other relevant anti-CD38 monoclonal antibody–containing regimens have been approved by Health Canada and could be used in second-line treatment and beyond. Treatment options for myeloma are extremely limited after the second line or third line of therapy. Both CMRG and OH-CCO’s Drug Advisory Committees emphasized that the overall treatment goals are to delay progression, improve OS, minimize adverse effects, control symptoms such as bone destruction and pain, renal failure, hypercalcemia, and low blood counts, and improve quality of life.
Both CMRG and OH-CCO’s Drug Advisory Committees mentioned that myeloma remains incurable, and patients eventually become refractory to all available funded drugs. One major unmet need is that patients with advanced disease who have received multiple lines of treatment and have already received the 3 lines of drugs (triple-class exposed and refractory), including an IMiD, PI, and anti-CD38 monoclonal antibody, have no other substantial treatment options other than CAR T-cell therapy. CMRG also emphasized that the clinical features associated with advanced disease and the short duration of responses lead to a poor quality of life, significant caregiver burden, and a shortened patient lifespan. Thus, this situation also represents 1 of the most pressing unmet needs in Canada for patients with MM. Another unmet need noted by OH-CCO’s Drug Advisory Committees is to achieve ease of administration (i.e., SC injection and no need for apheresis). Both clinician groups agreed that teclistamab is another option for triple-class exposed patients. They believe that it will currently be used sequentially after other lines of therapy for myeloma (i.e., after the failure of multiple drugs). Both clinician groups noted that teclistamab is not expected to impact the sequencing of drugs earlier in the disease course or lead to a major change in treatment algorithms before patients become triple-class exposed and refractory.
CMRG indicated that patients with a good performance status, minimal or no comorbidities, relatively low tumour burden, adequate organ function, and satisfactory blood counts are those most likely to have the best outcomes with teclistamab. CMRG further highlighted that the rates of immune-related complications are lower with bispecific antibodies such as teclistamab, in general, making them more broadly applicable to patients and more amenable to patients with more comorbidities (disease-related or otherwise). CMRG also mentioned that teclistamab represents an off-the-shelf treatment, which can be administered quickly even for rapidly proliferative myeloma. Chronological older age does not seem to be an exclusion factor for treatment with teclistamab. CMRG noted that overall, patients with poor disease-related prognostic factors, such as extramedullary myeloma and high-risk cytogenetics, should be eligible for the treatment under review. Both clinician groups expressed that treatment responses with teclistamab are based on standard myeloma response measures, CRS, and ICANS toxicity grading scales. CMRG further elaborated that responses to treatment are also based on the M protein markers in the serum and/or urine, bone marrow biopsy and, in some instances, on imaging studies (standardized IMWG criteria). These parameters are aligned with those used in the clinical trials, which also included the emerging parameter of marrow MRD. Clinically meaningful responses usually correlate with at least a partial remission by IMWG consensus criteria. These include improvement in symptoms (cessation of bone destruction with less pain, fractures, and need for radiotherapy), improvement in energy, and a greater ability to perform activities of daily living. In myeloma, responses are generally assessed every 1 month to 3 months, depending on clinical stability and the regimen used for therapy.
Both CMRG and OH-CCO’s Drug Advisory Committees agreed that treatment discontinuation is based on ongoing efficacy, disease progression, and long-term tolerability. Both clinician groups highlighted that teclistamab must be administered and monitored by hematologists or oncologists who have the knowledge and expertise to manage the potential short-term and long-term adverse events that can be associated with its use. Both clinician groups also recommended administration of the initial dosing in centres that have or are willing to develop the necessary infrastructure, experience, and support to safely administer T-cell redirecting therapies — for example, clinical assessment tools for CRS or ICANS grading or treatment, ICU support, and ready tocilizumab availability. CMRG recommended that, given that prior anti-BCMA exposure does not preclude responsiveness to subsequent anti-BCMA CAR T-cell therapy, patients previously treated with anti-BCMA therapy who did not progress during it (i.e., nonrefractory to anti-BCMA therapy other than anti-BCMA CAR T-cell therapy) should be given access to teclistamab.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 5.
Table 5: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
1. Issues with the choice of comparator in the submitted trial(s) The MajesTEC-1 study is a phase I and phase II open-label, single-arm trial. How does teclistamab compare to currently funded options in this therapeutic space (i.e., pomalidomide plus dexamethasone with or without cyclophosphamide, and carfilzomib plus dexamethasone with or without cyclophosphamide)? Selinexor with bortezomib and dexamethasone is funded in some jurisdictions as fourth-line therapy (and beyond) for patients sensitive to bortezomib but not anti-CD38 and lenalidomide. | The clinical experts mentioned that teclistamab in the MajesTEC-1 study has better progression-free survival when indirectly compared with that of pomalidomide plus dexamethasone with or without cyclophosphamide, or carfilzomib plus dexamethasone with or without cyclophosphamide. However, teclistamab is usually used when patients with RRMM have received pomalidomide or carfilzomib, or both. |
2. Other implementation issues regarding relevant comparators (e.g., access or funding, covered population) Cilta-cel is also used in this setting; however, it is under consideration for negotiation. The CADTH reimbursement conditions for cilta-cel specified that it should not be reimbursed in patients who have received prior treatment with therapy targeting BCMA. | No response required. For pERC consideration. |
Considerations for initiation of therapy | |
1. Other patient characteristics for eligibility (e.g., age restrictions, comorbidities) Should patients previously treated with BCMA-targeted therapy (e.g., cilta-cel) be eligible for teclistamab? Is there evidence to support this sequence? Should patients treated with teclistamab be eligible for CAR T-cell therapy (e.g., cilta-cel)? Is there evidence to support this sequence? | The clinical experts agreed that it is reasonable that patients previously treated with BCMA-targeted therapy would be eligible for teclistamab; however, there is little evidence to support this. There is no evidence to support the appropriateness of CAR T-cell therapy in patients previously treated with teclistamab since most studies of 1 drug exclude previous treatment with another, and vice versa. |
2. Prior therapies required for eligibility The Health Canada approval is for patients who have had at least 3 prior lines of therapy, including a PI, an IMiD, and an anti-CD38 antibody. Patients who could not tolerate a PI, IMiD, or anti-CD38 antibody were allowed per the MajesTEC-1 trial. Are 3 prior lines of therapy required if a patient is resistant to a PI, IMiD, and anti-CD38 antibody (e.g., received all 3 classes of these drugs, but across 2 lines of therapy)? | The clinical experts noted that patients who are resistant and intolerant to a PI, an IMiD, and an anti-CD38 antibody should be eligible to receive teclistamab, regardless of what line of therapy it is in, and teclistamab should not be limited to the fourth line of therapy in this situation. |
Considerations for discontinuation of therapy | |
1. Treatment interruptions Patients with prolonged treatment interruptions may require readministration of step-up dosing. | No response required. For pERC consideration. |
Considerations for prescribing of therapy | |
1. Dosing, schedule and/or frequency, dose intensity Teclistamab must be administered according to a step-up dosing schedule:
The 10 mg/mL vial is used for step-up dose 1 and step-up dose 2 while the 90 mg/mL vial is used for remaining doses. The trial protocol allowed patients to be switched to a biweekly dosing schedule (1.5 mg/kg SC every 2 weeks) if they attained a complete response or greater for a minimum of 6 months. Can pERC clarify the dosing schedule for teclistamab, including when biweekly dosing would be appropriate? | The clinical experts noted that switching from weekly to biweekly dosing should occur primarily due to side effects, toxicity, or patient choice. Clinical experts also noted that there is not enough evidence to say that teclistamab is as effective to switch to biweekly dosing without compromising efficacy. In clinical practice, physicians typically switch to less frequent dosing once the patient has responded to treatment. |
2. Drug administration CRS and ICANS can occur with teclistamab, although the severity and incidence appeared to be low in the trial. Tocilizumab may be needed to treat CRS. Can teclistamab be safely administered in the outpatient setting? | The clinical experts noted that patients starting treatment with teclistamab will receive the first 2 doses in the hospital, and after that they can safely receive ongoing therapy in an outpatient setting on a case-by-case basis. |
3. Concerns related to accessing clinical specialists and/or special settings The product monograph recommends that patients remain within proximity of a health care facility and monitor daily for 48 hours for signs and symptoms of CRS after the administration of all doses within the teclistamab step-up dosing schedule, or alternatively consider hospitalization for patients. Patients who experience higher than grade 1 CRS should be monitored daily for 48 hours following the next dose of teclistamab and remain within proximity of a health care facility. Jurisdictions may encounter capacity issues due to supportive care requirements. | No response required. For pERC consideration. |
Generalizability | |
1. Populations of interest matching the indication but with insufficient data Should teclistamab be used in:
| The clinical experts agreed that teclistamab can be used in patients with MM with an ECOG PS score of greater than 1, in patients with amyloidosis, and in patients with CNS disease that is under treatment or controlled, although this is rare. The clinical experts also noted that teclistamab can be used in patients with plasma cell leukemia at usual doses; however, these patients in general are excluded from the trials because their disease is more aggressive. |
2. Patients on active treatment with a time-limited opportunity to switch to the drug(s) under review At the time of funding, should patients receiving alternative therapies (i.e., pomalidomide in combination with dexamethasone, or carfilzomib in combination with dexamethasone with or without cyclophosphamide) be eligible to switch to teclistamab? | The clinical experts mentioned that physicians usually would not switch effective treatments until they no longer work; however, treatment can be switched to another drug if it stops working. |
Funding algorithm | |
Complex therapeutic space with multiple lines of therapy, subpopulations, or competing products | No response required. For pERC consideration. |
Another aspect is that there may be interest in sequencing teclistamab with other BCMA-targeted drugs. | No response required. For pERC consideration. |
Care provision issues | |
1. Drug preparation, storage, administration or dispensing Teclistamab is supplied as 153 mg/1.7mL (90 mg/mL) and 30 mg/3mL (10 mg/mL). However, drug wastage would be incurred due to the step-up and mg/kg dosing. There is a risk of medication error with 2 different concentrations. The drug may need to be initiated in the inpatient setting; in which case the drug cost would be outside of the drug program budget in some provinces. | No response required. For pERC consideration. |
System and economic issues | |
1. Concerns regarding the anticipated budget impact and sustainability Example: Provision of this drug in the first-line setting may translate into a substantial budget impact. A prioritization scheme may be required. There is concern about the feasibility of adoption (budget impact) in light of the cost of prior therapies and the potential for subsequent therapies. | No response required. For pERC consideration. |
2. Presence of confidential negotiated prices for comparators Example: Comparators A and B have successfully gone through price negotiations for the same indication. Generic pomalidomide is available, and confidential pricing exists for carfilzomib. | No response required. For pERC consideration. |
BCMA = B-cell maturation antigen; CAR = chimeric antigen receptor; cilta-cel = ciltacabtagene autoleucel; CNS = central nervous system; CRS = cytokine release syndrome; ECOG PS = Eastern Cooperative Oncology Group Performance Status; ICANS = immune effector cell-associated neurotoxicity syndrome, IMiD = immunomodulatory drug; MM = multiple myeloma; pERC = pan-Canadian Oncology Drug Review Expert Review Committee; PI = proteasome inhibitor; RRMM = relapsed or refractory multiple myeloma; SC = subcutaneous.
Clinical Evidence
The objective of CADTH’s Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of teclistamab (1.5 mg/kg body weight administered by SC injection) in the treatment of adult patients with RRMM who have received at least 3 prior lines of therapy, including a PI, an IMiD, and an anti-CD38 monoclonal antibody. The focus will be placed on comparing teclistamab to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of teclistamab is presented in 3 sections, with CADTH’s critical appraisal of the evidence included at the end of each section. The first section, the systematic review, includes pivotal studies that were selected according to the sponsor’s systematic review protocol. CADTH’s assessment of the certainty of the evidence in this first section using the GRADE approach follows the critical appraisal of the evidence. Another section includes indirect evidence from the sponsor. The last section includes a description of ongoing additional studies that were considered by the sponsor to address important gaps in the systematic review evidence.
Included Studies
Clinical evidence from the following is included in the CADTH review and appraised in this document:
One pivotal study identified in the systematic review
Six sponsor-submitted ITCs.
Systematic Review
Content in this section has been informed by materials submitted by the sponsor. The following has been summarized and validated by the CADTH review team.
Description of Studies
Characteristics of the included study are summarized in Table 6.
Table 6: Details of MajesTEC-1 Study
MajesTEC-1 study | |
|---|---|
Design and population | |
Study design | Phase I and phase II, open-label, multicentre, dose escalation study |
Locations | This study was conducted at 39 centres in 10 countries (Belgium, Canada, France, Germany, Italy, the Netherlands, Spain, Sweden, the UK, and the US). |
Patient enrolment dates | First patient enrolled: May 16, 2017 Cut-off for primary analysis: September 7, 2021 Cut-off for updated efficacy analysis: November 9, 2021 Cut-off for updated clinical data for pivotal data: March 16, 2022 Cut-off for updated clinical data for cohort C: March 16, 2022 Cut-off for the latest analysis for pivotal data: January 4, 2023 Last patient visit: Study is ongoing |
Randomized (N) | Randomization was not used in the study. Patients received the study treatment if they met eligibility criteria. |
Inclusion criteria | Patients with documented diagnosis of MMa Aged at least 18 years ECOG PS score of 0 or 1 Measurable disease at screening:
Prior treatments:
|
Exclusion criteria |
|
Drugs | |
Intervention | Phase I (part 1 and part 2) Dose escalation (part 1):
Dose expansion (part 2):
Phase II (part 3)
|
Comparator(s) | No comparator (single-arm study) |
Study duration | |
Screening phase | 28 days before the first dose of study drug |
Treatment phase | Began with the first administration of the study drug and continued until the completion of the end of treatment visit. An end of treatment visit was completed within 30 days (± 7 days) after the last dose of the study drug or before the start of a new anticancer therapy, whichever came first. |
Follow-up phase | The post-treatment follow-up phase started after the end of treatment visit (or treatment discontinuation if the end of treatment visit was not performed). If study treatment was discontinued before the onset of disease progression, as defined by IMWG, central laboratory disease evaluation was to continue to be performed every 3 weeks to 4 weeks (part 1 and part 2) or every 4 weeks (part 3) until confirmed disease progression, death, the start of a new anticancer treatment, the withdrawal of consent for study participation, or the end of the study, whichever occurred first. For patients in part 1 and part 2, post-treatment follow-up visits were to occur at 4 weeks and 8 weeks after the end of treatment visit (or treatment discontinuation if the end of treatment visit was not observed). Subsequent survival follow-up was to begin 8 weeks later and continue approximately every 16 weeks. |
Outcomes | |
Primary end point | Phase I (part 1 and 2 part)
Phase II (part 3) |
Secondary and exploratory end points | Secondary end points Phase I (part 1 and part 2):
Phase II (part 3):
Exploratory end points Phase I (part 1 and part 2):
Phase II (part 3):
|
Publication status | |
Publications | Moreau P et al. (2022)56 Usmani SZ et al. (2021)64 |
ADC = antibody-drug conjugate; AE = adverse event; BCMA = B-cell maturation antigen; CABG = coronary artery bypass graft; CAR = chimeric antigen receptor; CNS = central nervous system; CR = complete response; ECOG PS = Eastern Cooperative Oncology Group Performance Status; FLC = free light chain; HRQoL = health-related quality of life; IMiD = immunomodulatory drug; IMWG = International Myeloma Working Group; mAb = monoclonal antibody; MM = multiple myeloma; MRD = minimal residual disease; MRU = medical resource utilization; NK = natural killer; ORR = overall response rate; OS = overall survival; PFS = progression-free survival; PI = proteasome inhibitor; POEMS = polyneuropathy, organomegaly, endocrinopathy, M protein, and skin changes; PR = partial response; q.2.w. = every 2 weeks; RO = receptor occupancy; RP2D = recommended phase II dose; RRMM = relapsed or refractory multiple myeloma; SAE = serious adverse event; SC = subcutaneous; TTNT = time to next treatment; TTR = time to response; VGPR = very good partial response.
Note: Details included in Table 6 are from the sponsor’s Summary of Clinical Evidence.83
aAccording to IMWG diagnostic criteria, MM is defined as clonal bone marrow plasma cells of 10% or greater or biopsy-proven bony or extramedullary plasmacytoma and at least 1 of the following: i) evidence of end-organ damage, specifically: hypercalcemia: serum calcium is greater than 0.25 mmol/L (> 1 mg/dL) higher than the upper limit of normal or greater than 2.75 mmol/L (> 11 mg/dL); renal insufficiency: creatinine clearance is less than 40 mL per minute or serum creatinine is greater than 177 μmol/L (> 2 mg/dL); anemia: a hemoglobin value of more than 20 g/L below the lower limit of normal, or a hemoglobin value of less than 100 g/L; bone lesions: there are 1 or more osteolytic lesions on skeletal radiography, CT, or PET-CT; ii) any 1 or more of the following biomarkers of malignancy: clonal bone marrow plasma cell percentage of 60% or greater; involved to uninvolved serum free light chain ratio of 100 or more; more than 1 focal lesion on MRI studies.
bRelapsed or refractory myeloma is defined as disease that is nonresponsive while on salvage therapy, or progresses on or within 60 days of the last therapy in patients who have attained minimal response or better at some point previously before then progressing in their disease course.
cIf central laboratory assessment is not available, relevant local laboratory measurement must exceed the minimum required level by at least 25%.
dBased on the investigator’s determination of response by the IMWG 2016 criteria on or within 12 months of the last line of therapy.
Sources: Clinical Study Report for the MajesTEC-1 study65 and Clinical Study Protocol for the MajesTEC-1 study.66
Description of the MajesTEC-1 Study
The MajesTEC-1 trial is a phase I and phase II, open-label, multicentre study of teclistamab administered to adult patients with RRMM. The study is still ongoing and being conducted in 39 sites across 10 countries, including in Canada with patients who were enrolled at 4 Canadian sites; however, Canada, Germany, Italy, and the UK only participated in part 3 of the study. All patients received study treatment if all inclusion and exclusion criteria were met.
Description of Phases and Cohorts of the MajesTEC-1 Study
The MajesTEC-1 study was conducted in 3 parts, including part 1 or dose escalation (phase I), part 2 or dose expansion (phase I) at a proposed recommended phase II dose (1.5 mg/kg subcutaneously weekly), and part 3 or dose expansion (phase II) in cohorts of patients with RRMM with unmet medical needs (phase II). The main focus of the review is to assess the efficacy of receiving teclistamab at a dose of 1.5 mg/kg subcutaneously weekly within a population of patients with RRMM (40 patients in phase I and 125 patients in phase II, cohort A) who have received at least 3 prior lines of therapy, excluding anti-BCMA therapy. Additional supporting information was provided by the sponsor regarding the use of teclistamab in patients with RRMM previously treated with BCMA-targeted therapy in phase II, cohort C (n = 40).
Phase I of the MajesTEC-1 Study
The primary objectives reported in phase I of the MajesTEC-1 study were:
to identify the proposed recommended phase II dose and dose schedule assessed to be safe in part 1
to characterize the safety and tolerability of teclistamab at the proposed recommended phase II dose in part 2.
Treatment with teclistamab started with biweekly IV dosing and was then changed to weekly IV dosing after analysis of emerging safety and pharmacokinetic data. After a review of safety and efficacy data and considering the greater convenience of SC drug administration for patients and health care providers, the sponsor amended the protocol to evaluate SC administration. Dose escalation in phase I continued in parallel for both routes of administration. A wide range of escalating dose levels of teclistamab were evaluated. In part 1 of phase I, both accelerated and standard titration were used for dose escalation for IV administration of teclistamab, while for SC administration of teclistamab, the study drug was administered using standard titration only. Proposed recommended phase II doses for IV and SC administration of teclistamab were evaluated in part 2 of phase I, where up to 40 patients could receive teclistamab at the proposed recommended phase II doses identified in part 1 of phase I to further characterize antitumour activity and safety in additional patients at the relevant doses.
Phase II of the MajesTEC-1 Study
The primary objective of phase II was to evaluate the efficacy and safety of teclistamab at the proposed recommended phase II dose. The dose level and schedule selected as recommended phase II doses in phase I (1.5 mg/kg subcutaneously weekly) was evaluated in cohorts of patients with RRMM, including phase II, cohort A and cohort C. In the MajesTEC-1 study in phase II, cohort A and cohort C, enrolment began after at least 20 patients had received teclistamab 1.5 mg/kg subcutaneously weekly for at least 1 treatment cycle. Phase II, cohort A, enrolled patients with RRMM who had received at least 3 prior lines of therapy that included a PI, an IMiD, and an anti-CD38 monoclonal antibody. Phase II, cohort C, enrolled patients who had received at least 3 prior lines of therapy that included a PI, an IMiD, an anti-CD38 monoclonal antibody, and a BCMA-targeted treatment (CAR T-cells or an ADC). Enrolment in phase II, cohort B, was not planned because the intended patient population was enrolled in phase II, cohort A, including patients who were more heavily pretreated with at least 4 prior lines of therapy and considered penta-drug refractory.
In the MajesTEC-1 study, 340 patients were treated with teclistamab, with 165 patients in the pivotal study (patients in phase I and phase II, cohort A, who received SC teclistamab at a dose of 1.5 mg/kg weekly), 177 patients in the phase I group (dose escalation and dose expansion), and 38 patients in phase II, cohort C (patients with prior BCMA-targeted therapy who received 1.5 mg/kg SC teclistamab weekly) (Figure 1). At the time of the data cut-off date of March 16, 2022, 40 patients in phase II, cohort C, had received teclistamab at a dose of 1.5 mg/kg subcutaneously weekly.
Figure 1: Analysis Populations — MajesTEC-1 Study, Data Cut-Off Date of September 7, 2021
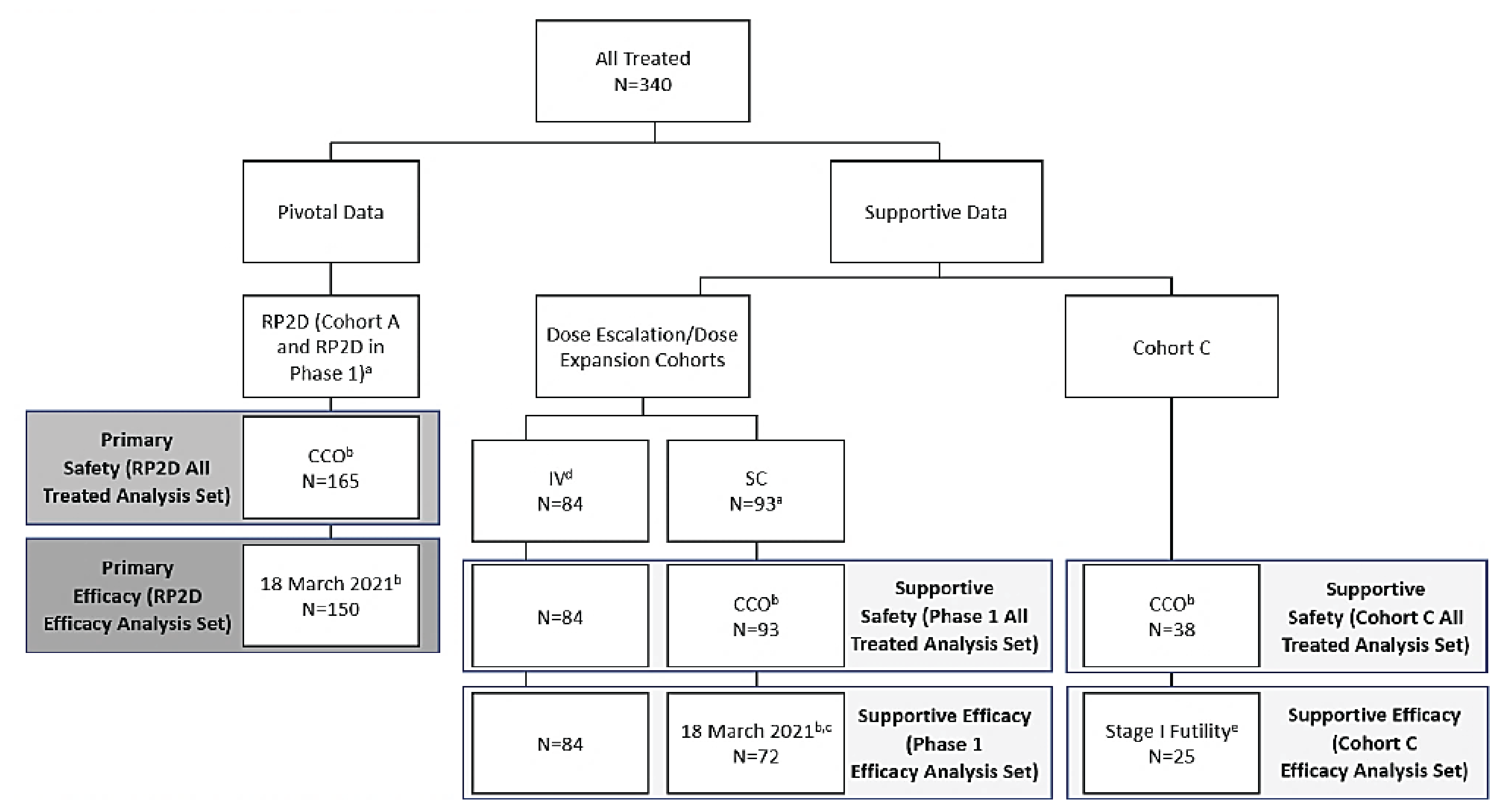
CCO = clinical cut-off; RP2D = recommended phase II dose; SC = subcutaneous.
Note: Details included in Figure 1 are from the sponsor’s Summary of Clinical Evidence.83
a Patients treated at RP2D (1.5 mg/kg teclistamab subcutaneously weekly) in phase I are presented in both pivotal RP2D data and phase I (dose escalation and dose expansion) data.
b A patient must have received the first dose of teclistamab on or before this date to be included in the indicated population. At the data cut-off date of January 4, 2023, both efficacy and safety analyses were performed in the all-treated analysis set (N = 165).
c One patient in the 6 mg/kg SC cohort who received their first step-up dose on March 17, 2021, was excluded from the efficacy analyses for phase I (dose escalation and dose expansion) because the cohort was incomplete at the time of the data cut-off for inclusion in the efficacy analysis and the only patient enrolled in it had not yet received a treatment dose.
d All patients treated with teclistamab intravenously received their first dose before March 18, 2021.
e Patients evaluated in stage I of the analysis for cohort C were included in the efficacy analysis, and all had received at least 1 dose of teclistamab on or before March 23, 2021. At the data cut-off date of March 16, 2022, both efficacy and safety analyses for cohort C were performed in the full analysis set (n = 40).
Source: Clinical Study Report for the MajesTEC-1 study (2021).65
Screening Phase
The screening phase of the MajesTEC-1 study began when the first screening assessment was performed. Screening procedures were performed 28 days before the first dose of the study drug. Eligibility criteria were reviewed and a full clinical evaluation was performed at screening, and before the first dose. Measurements collected at the time point closest to, but before, the first administration of the study drug were defined as baseline values for safety assessment and treatment decisions.
Treatment Phase
The treatment phase began with the first administration of the study drug and continued until the completion of the end of treatment visit. Patients received the study drug until disease progression, unacceptable toxicity, withdrawal of consent, death, or end of study defined as 2 years after the last patient’s first dose. An end of treatment visit was to be completed within 30 days with an additional week’s grace period after the last dose of the study drug or before the start of a new anticancer therapy, whichever comes first. An end of treatment visit was required for all patients, including those who discontinued treatment for any reason, except for lost to follow-up, death, or withdrawal of consent to participate in the study.
Follow-Up Phase
The follow-up phase started after the end of treatment visit or treatment discontinuation, if the end of treatment visit was not performed.
Populations
Inclusion and Exclusion Criteria
Eligible patients should have been aged at least 18 years and had a documented diagnosis of RRMM according to the IMWG criteria. Patients must have received at least 3 prior lines of antimyeloma therapy that included an IMiD, a PI, and an anti-CD38 monoclonal antibody, and have had progressive, measurable disease at screening informed by IMWG (except for in part 1, phase I). Patients must have had an ECOG PS score of 0 or 1, and laboratory values within the study specified the criteria for enrolment.
For patients in phase I and phase II, cohort A, previous treatment with a BCMA-targeted therapy was not allowed, while in phase II, cohort C, prior therapy must have included a BCMA-targeted therapy (ADC or CAR T-cell therapy) in addition to the requirement to have received at least 3 prior lines of therapy including a PI, an IMiD, and an anti-CD38 monoclonal antibody. Patients were excluded if they had prior antitumour therapy, such as targeted therapy within 21 days or at least 5 half-lives, or monoclonal antibody, cytotoxic, PI, or IMiD therapy within 21 days before the first dose of teclistamab. Patients were also excluded if they had received gene-modifying adoptive cell therapy (e.g., CAR T-cells, natural killer cells) within 3 months, or radiotherapy within 14 days or focal radiation within 7 days. Also, patients were excluded if they had received a cumulative dose of corticosteroids equivalent to 140 mg or more of prednisone within 14 days before receiving the first dose of the study drug, had an allogeneic stem cell transplant within 6 months, or had received an autologous stem cell transplant within at least 12 weeks before receiving the first dose of study drug.
Interventions
Phase I (Part 1 and Part 2) of the MajesTEC-1 Study
During dose escalation (part 1 of phase I), the following biweekly and weekly treatment dosages of teclistamab were examined up to the recommended phase II dosage:
teclistamab IV from 0.0003 mg/kg to 0.0192 mg/kg every 2 weeks
teclistamab IV from 0.0192 mg/kg to 0.72 mg/kg weekly
teclistamab SC from 0.08 mg/kg to 1.5 mg/kg weekly.
Patients treated with teclistamab intravenously were hospitalized for at least 36 hours from the start of any priming doses and the start of the first full dose of the study drug, and at least 24 hours from the start of the second full dose of the study drug. IV administration of teclistamab was administered over at least 4 hours in the controlled environment of a clinical research centre under the supervision of site staff. Patients treated in the initial cohort for SC administration were hospitalized for at least 48 hours from the start of any priming injection of the study drug and from the start of the first full injection of the study drug, and at least 24 hours from the start of the second full injection of the study drug. SC administration of teclistamab was administered by health professionals at treatment centres with adequate medical equipment and personnel to manage severe reactions. Additional cohorts received weekly weight-based treatment doses higher than the recommended phase II dose and other dosing schedules for the SC route of administration (with treatment doses up to 6 mg/kg and with flat dosing). Approximately half of the IV treatment doses and all SC treatment doses were preceded by step-up dosing.
Patients had the option to switch from weekly to biweekly dosing (day 1 and day 15) if they attained a PR or better and received at least 4 cycles of treatment (phase I) or attained a CR or better for at least 6 months (phase II). Patients could switch to monthly dosing with approval (phase I) or if they attained CR or better at cycle 12, day 1, or later, and had been receiving biweekly dosing for at least 6 months. Per investigator discretion, patients were permitted to switch to less frequent dosing to help manage toxicity.
During dose expansion (phase I, part 2), patients received treatment with teclistamab at dosages of 0.72 mg/kg IV weekly and 1.5 mg/kg teclistamab subcutaneously weekly (Figure 2). Depending on the dosing schedule, teclistamab was administered to patients participating in phase I in 21-day or 28-day cycles (Figure 3). Dosing schedules were either monthly, biweekly, or weekly. For monthly dosing, teclistamab was administered on day 1 in each 28-day cycle. For biweekly dosing, teclistamab was administered on day 1 and day 15 in each 28-day cycle and dose escalation was initiated at the minimum anticipated biological effect level–based starting dose of 0.0003 mg/kg. For weekly dosing, teclistamab was administered on day 1, day 8, and day 15 in each 21-day cycle. Dose escalation for IV dosing of teclistamab began at a dose level that was determined to be safe during the accelerated phase of dose escalation for biweekly dosing. Dose escalation for weekly SC dosing began at a cleared dose level approved by the safety evaluation team.
Phase II (Part 3) of the MajesTEC-1 Study
The step-up and treatment doses were selected for phase II (part 3) of the MajesTEC-1 study after review of safety, efficacy, pharmacokinetic, and pharmacodynamic data from patients treated during the dose escalation phase (part 1, phase I). During phase II, all patients received initial step-up doses of 0.06 mg/kg and 0.3 mg/kg administered on day 1 and day 3 (range = 2 to 7 days after step-up doses), respectively, after which teclistamab maintenance doses were administered weekly at a dose of 1.5 mg/kg via SC injection until disease progression, unacceptable toxicity, withdrawal of consent, death, or end of study.
Figure 2: Dose Escalation or Dose Expansion Cohorts Evaluated in Phase I of MajesTEC-1 Study
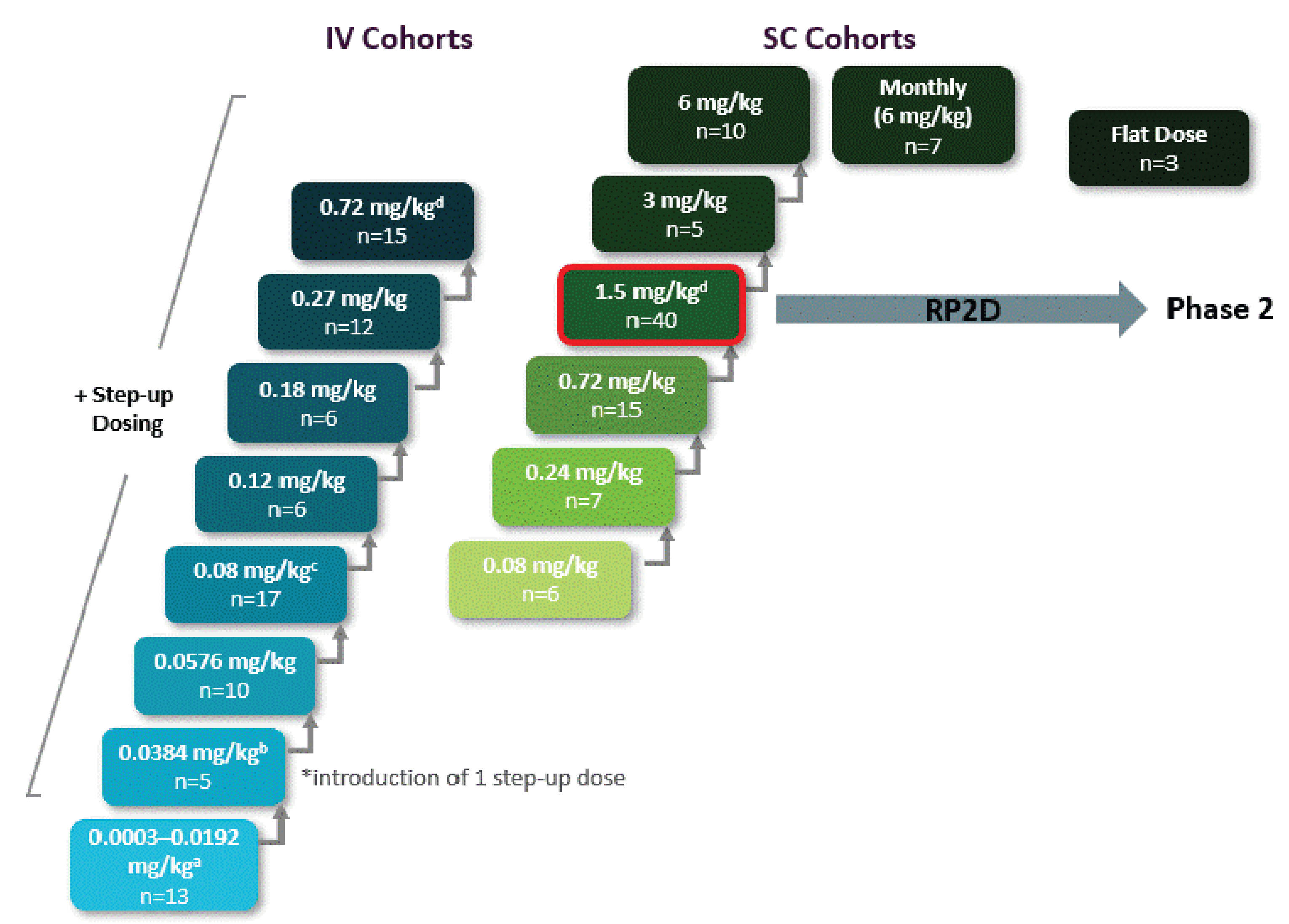
Q.2.w. = every 2 weeks; RP2D = recommended phase II dose; SC = subcutaneous.
Note: Details included in Figure 2 are from the sponsor’s Summary of Clinical Evidence.83
a Includes 8 cohorts (7 cohorts with q.2.w. dosing [N = 12] and 1 cohort with weekly dosing [N = 1]).
b Includes 1 cohort without step-up dosing (N = 1) and 1 cohort with a step-up dose (N = 4).
c Includes 1 cohort with 1 step-up dose (N = 12) and 1 cohort with 2 step-up doses (N = 5).
d Dose levels were expanded in part 2 (proposed RP2Ds).
Source: Clinical Study Report for the MajesTEC-1 study (2021).65
Dose Modifications
In phase I, patients were permitted to undergo dose reduction to the next lower dose level for the applicable route of administration. In phase II, dose reductions of teclistamab by 50% subcutaneously weekly could be considered in exceptional circumstances and after consultation with the sponsor.
In the MajesTEC-1 study, dose delay was the primary method for managing toxicities related to teclistamab. In the event of prolonged treatment interruption, disease evaluations were to continue every 4 weeks if possible. If a dose interruption was more than 28 days, treatment with teclistamab was to be permanently discontinued.
In phase I of the MajesTEC-1 study, patients who were enrolled in weekly cohorts could change from weekly dosing to biweekly or monthly dosing of teclistamab, if the patient attained a response of confirmed PR or better and received a minimum of 4 cycles of therapy. Patients in phase II could be treated biweekly if the patient attained a CR or better for a minimum of 6 months. The biweekly treatment dose was the same as the patient’s corresponding weekly dose.
Figure 3: Dosing Schedule for Cohorts Evaluated in Phase I — 21-Day and 28-Day Cycle Schedules
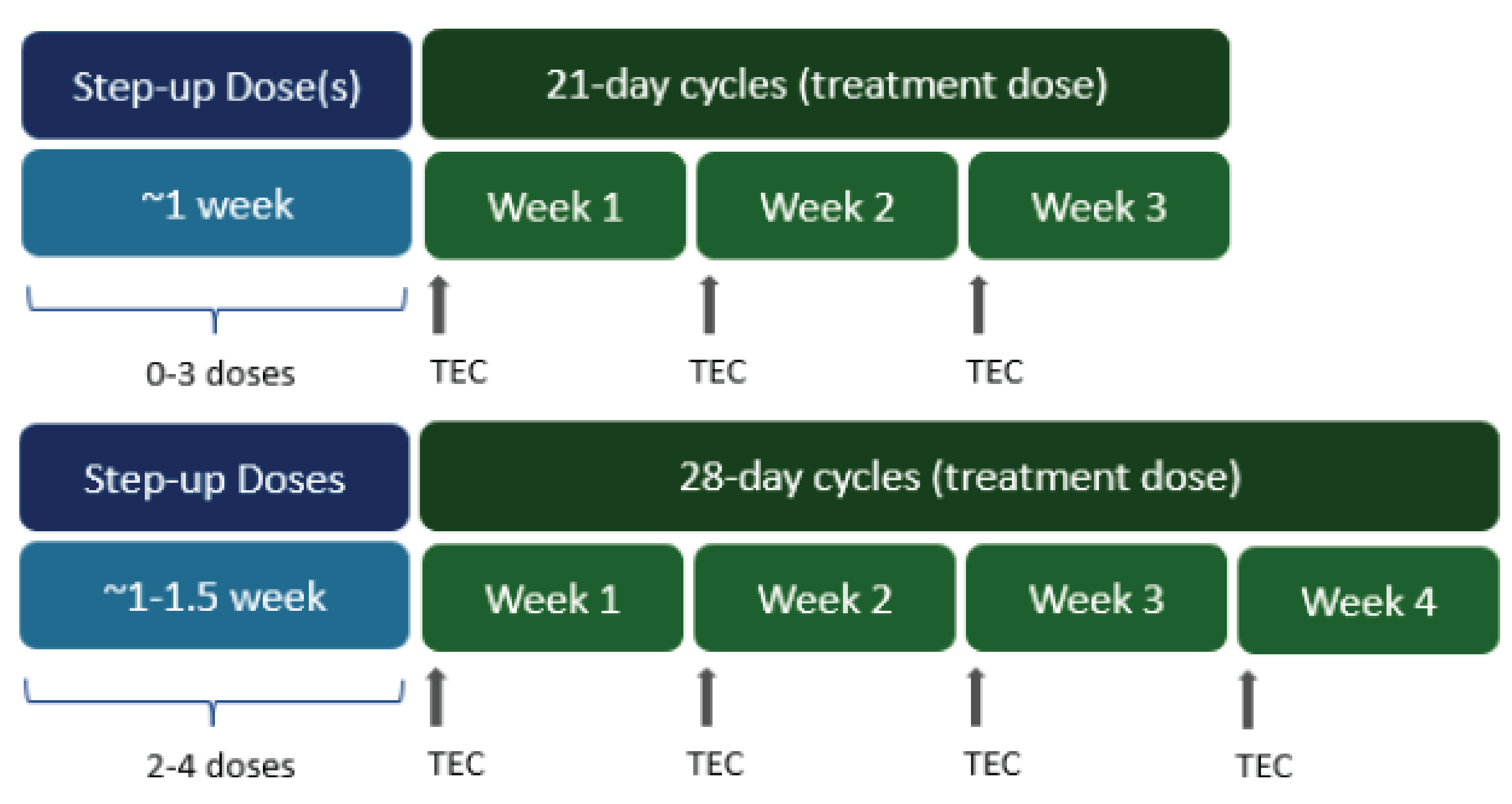
TEC = teclistamab.
Notes: Details included in Figure 3 are from the sponsor’s Summary of Clinical Evidence.83
One cohort exploring a treatment dose of 6 mg/kg administered monthly and preceded by 4 step-up doses (0.03 mg/kg, 0.09 mg/kg, 0.3 mg/kg, and 1.5 mg/kg) was evaluated in phase I.
Source: Clinical Study Report for the MajesTEC-1 study (2021).65
Disease Evaluations
Disease evaluations were performed at screening and within 3 days before day 1 of each cycle, before the study drug was administered. Disease evaluations were tested at a central laboratory until disease progression. In the event of prolonged treatment interruption, disease evaluations were to continue every 3 weeks to 4 weeks (phase I) or every 4 weeks (phase II), if possible. For patients who discontinued study treatment before disease progression, disease evaluations were to continue until confirmed disease progression, death, the start of a new treatment for MM, withdrawal of consent for study participation, lost to follow-up, or the end of the study, whichever occurred first. Responses for all patients treated with the recommended phase II dosage of teclistamab (phase I and phase II, cohort A) were assessed by an IRC, using the 2016 IMWG response criteria63 (Table 7).
In the MajesTEC-1 study, evaluations of quantitative immunoglobulins, serum M-protein quantitation by electrophoresis, serum immunofixation, urine immunofixation, and urine M-protein quantitation by electrophoresis were performed on day 1 of each cycle until disease progression. Serum free light chain evaluations were performed on day 1 of each cycle before dosing, until disease progression for patients with free light chain–only measurable disease, when CR was suspected and until sCR was confirmed, or before CR, if necessary. Bone marrow aspirate or biopsy was performed on day 1 of cycle 3, at the time of suspected CR or sCR, and at disease progression. For confirmed CR or sCR, an additional aspirate was collected 12 months after day 1 of cycle 1, and yearly thereafter. Skeletal surveys, including additional imaging (X-ray, CT scan, or MRI) were performed locally whenever clinically indicated based on symptoms. Evaluations of extramedullary plasmacytomas were assessed by clinical examination or radiologic imaging for all patients with a history of plasmacytomas, or if clinically indicated, at screening and then until the development of confirmed CR or confirmed disease progression — every 4 weeks for physical examination (if applicable) or every 12 weeks for radiologic assessment (for patients with a history of plasmacytomas or as clinically indicated for others).
Table 7: Criteria for Response to Multiple Myeloma Treatment
Response | Response criteria (IMWG 2016)63, a |
|---|---|
PR |
|
VGPR |
or
|
CRb | All of the following criteria:
|
sCR | All of the following criteria:
|
Minimal response |
|
Stable disease | Not meeting criteria for sCR, CR, VGPR, PR, MR, or progressive disease |
Progressive diseased | Any 1 or more of the following criteria:
|
CR = complete response; IgA = immunoglobulin A; IgG = immunoglobulin G; IgM = immunoglobulin M; IMWG = International Myeloma Working Group; MR = minimal response; PR = partial response; sCR = stringent complete response; VGPR = very good partial response.
aAll response categories (CR, sCR, VGPR, PR, MR, and progressive disease) required 2 consecutive assessments made at any time before the institution of any new therapy; CR, sCR, VGPR, PR, MR, and stable disease categories also required no known evidence of progressive or new bone lesions if radiographic studies were performed. VGPR and CR categories required serum and urine studies regardless of whether disease at baseline was measurable on serum, urine, both, or neither.
bClarifications were made to the criteria for coding CR and VGPR in patients in whom the only measurable disease was by serum free light chain levels. CR in such patients indicated a normal free light chain ratio of 0.26 to 1.65 (or reference range in testing laboratory) in addition to CR criteria listed earlier. VGPR in such patients required a 90% or greater decrease in the difference between involved and uninvolved free light chain levels.
cIn some cases, it was possible that the original M-protein light chain isotype was still detected on immunofixation but the accompanying heavy-chain component had disappeared; this would not be considered as a CR even though the heavy-chain component was not detectable, since it was possible that the clone evolved to one that secreted only light chains. Thus, if a patient had IgA lambda myeloma, then to qualify as CR, there should have been no IgA detectable on serum or urine immunofixation; if free lambda was detected without IgA, then it must have been accompanied by a different heavy-chain isotype (e.g., IgG, IgM).
dThese clarifications were made to the criteria for coding progressive disease: bone marrow criteria for progressive disease was to be used only in patients without measurable disease by M protein and by free light chain levels; “25% increase” referred to M protein and free light chains, and did not refer to bone lesions or soft tissue plasmacytomas; and the “lowest response value” did not need to be a confirmed value.
Source: Clinical Study Protocol for the MajesTEC-1 study.66
Safety Monitoring for Teclistamab Dosing
For phase II (part 3), all patients were hospitalized for safety monitoring for at least 48 hours after each step-up dose and the first treatment dose. Patients who experienced neurotoxicity of grade 2 or higher were to be hospitalized for at least 48 hours after the subsequent dose of teclistamab. Patients who experienced a systemic administration-related reaction or CRS of grade 2 or grade 3 were to be hospitalized for at least 48 hours after the subsequent dose of teclistamab.
Safety in the MajesTEC-1 trial was measured by the monitoring of adverse events, laboratory tests, vital sign measurements, physical triplicate electrocardiograms (phase I only), physical examination findings (including neurologic assessment and mini-mental state exam), the assessment of immune effector cell-associated encephalopathy tool score (phase II only), the assessment of ECOG PS grade, and concomitant medication usage. Adverse events were coded using the Medical Dictionary for Regulatory Activities Terminology (version 24.0). The severity of adverse events was assessed using the National Cancer Institute’s CTCAE (version 4.03), except for CRS, which was graded according to a CRS-revised grading system. Clinically significant abnormalities that persisted at the end of the study or at the time of early withdrawal were followed by the investigator until resolution or clinical stability. Safety monitoring may have been performed more frequently if clinically indicated, and adverse events were evaluated by the investigator according to standard practice.66
Pretreatment Medication
All patients in the MajesTEC-1 study were required to receive the following pretreatment medications before each step-up dose and the first treatment dose of teclistamab: steroids (dexamethasone at a dose of 16 mg), antihistamines (diphenhydramine at a dose of 50 mg or equivalent), and antipyretics (acetaminophen at a dose of 650 mg to 1,000 mg or equivalent). Additionally, patients who experienced events of CRS of grade 2 or higher or systemic administration-related reactions were required to receive dexamethasone before the next dose of teclistamab and patients who experienced CRS of any grade or systemic administration-related reactions were required to receive the antihistamine and antipyretic before at least the next dose of teclistamab. Histamine2 receptor antagonists (ranitidine at a dose of 50 mg or equivalent) and antiemetics (ondansetron at a dose of 16 mg or equivalent) could be administered as an optional pretreatment medication, per investigator discretion.
Concomitant Medications
During the MajesTEC-1 study, patients may be prescribed any concomitant medications or treatments deemed necessary for supportive care (treatments for adverse events or serious adverse events), including standard supportive care therapies (antiemetics, antidiarrheals, anticholinergics, antispasmodics, antipyretics, antihistamines, analgesics, antibiotics and other antimicrobials, histamine2 receptor antagonists or proton pump inhibitors, and other medications intended to treat symptoms or signs of disease), bisphosphonates, growth factor support, erythropoietin-stimulating drugs, transfusions, antibiotics or other anti-infective drugs, and corticosteroids used as pretreatment medications.
The following medications were prohibited during the study: any chemotherapy, anticancer immunotherapy (other than teclistamab), experimental therapy, radiotherapy, plasmapheresis (unless in exceptional circumstances unrelated to disease progression), corticosteroids in excess of 10 mg daily of prednisone or equivalent, other immunosuppressant drugs (unless used as protocol-specified pretreatment or post-treatment medications to treat an adverse event), vaccination with live, attenuated vaccine within 4 weeks before the first dose of teclistamab during treatment, IV contrast infusions, routine transfusions on study drug administration days, transdermal patches, and cytochrome P450 substrates with narrow therapeutic index. For patients receiving warfarin, investigators were to consider switching from warfarin to a different anticoagulant.
Subsequent Anticancer Therapy
Unless the patient was intolerant to the study drug, subsequent antimyeloma therapy could not be initiated until disease progression was established according to the IMWG criteria (in part 3 only). Once disease progression was confirmed, the choice of subsequent therapy was at the discretion of the investigator.
Outcomes
A list of efficacy end points assessed in this Clinical Review Report is provided in Table 8, followed by descriptions of the outcome measures. Summarized end points are based on outcomes included in the sponsor’s Summary of Clinical Evidence as well as any outcomes identified as important to this review according to the clinical experts consulted by CADTH, and stakeholder input from patient and clinician groups and public drug plans. Using the same considerations, the CADTH review team selected end points that were considered to be most relevant to informing CADTH’s expert committee deliberations and finalized this list of end points in consultation with members of the expert committee. Select efficacy and notable harms outcomes that were considered important for informing CADTH’s expert committee were assessed using GRADE.
The secondary outcomes for phase I used for the supportive efficacy analyses included ORR, VGPR or better, CR or better, sCR, OS, PFS, duration of response, and TTR.
For phase II of the MajesTEC-1 trial, the primary end point was ORR (PR or better), and secondary outcomes included VGPR or better, CR or better, sCR, TTR, duration of response, OS, PFS, MRD-negativity rate, and HRQoL outcomes. TTNT was an exploratory outcome in phase II of the MajesTEC-1 study.
Table 8: Outcomes Summarized From the MajesTEC-1 Study
Outcome measure | Time point | MajesTEC-1 study |
|---|---|---|
Efficacy outcomes | ||
OS | Time from the first dose of study drug to death | Secondary outcome for both phases |
PFS | The length of time between the date a patient first attained a PR or better to the first documented disease progression or death, whichever occurred first | Secondary outcome for both phases |
CR or better | At screening, and on day 1 of each cycle (or within 3 days before day 1) | Secondary outcome for both phases |
sCR | At screening, and on day 1 of each cycle (or within 3 days before day 1) | Secondary outcome for both phases |
HRQoL | Cycle 2, day 1; cycle 4, day 1; and cycle 6, day 1 | Secondary outcome for phase II |
MRD-negativity rate | At any time point after initial dose and before progressive disease or subsequent antimyeloma therapy | Secondary outcome for phase II |
VGPR or better | At screening, and on day 1 of each cycle (or within 3 days before day 1) | Secondary outcome for both phases |
ORR | At screening, and on day 1 of each cycle (or within 3 days before day 1) | Primary outcome for phase II Secondary outcome for phase I |
TTR | Time from the first dose of study drug to the first efficacy evaluation that the patient has met all criteria for PR or better | Secondary outcome for both phases |
Duration of response | Time from initial documentation of a response (PR or better) to the first documented evidence of progressive disease, or death due to disease progression, whichever occurred first | Secondary outcome for both phases |
TTNT | Time from the date of first dose of study drug to the start of the next line of treatment | Exploratory outcome for phase II |
Safety outcomes | ||
AEs, TEAEs | Continuous to 30 days after the last dose of study drug or until the start of subsequent anticancer therapy, if earlier | Exploratory |
Infections | Continuous to 30 days after the last dose of study drug or until the start of subsequent anticancer therapy, if earlier | Exploratory |
Hypogammaglobulinemia | Continuous to 30 days after the last dose of study drug or until the start of subsequent anticancer therapy, if earlier | Exploratory |
CRS | Continuous to 30 days after the last dose of study drug or until the start of subsequent anticancer therapy, if earlier | Exploratory |
Neurotoxicity | Continuous to 30 days after the last dose of study drug or until the start of subsequent anticancer therapy, if earlier | Exploratory |
ICANS | Continuous to 30 days after the last dose of study drug or until the start of subsequent anticancer therapy, if earlier | Exploratory |
Tumour lysis syndrome | Continuous to 30 days after the last dose of study drug or until the start of subsequent anticancer therapy, if earlier | Exploratory |
Cytopenias (neutropenia, thrombocytopenia and hemorrhagic events, lymphopenia, anemia) | Continuous to 30 days after the last dose of study drug or until the start of subsequent anticancer therapy, if earlier | Exploratory |
AE = adverse event; CR = complete response; CRS = cytokine release syndrome; HRQoL = health-related quality of life; ICANS = immune effector cell-associated neurotoxicity syndrome; MRD = minimal residual disease; ORR = overall response rate; OS = overall survival; PFS = progression-free survival; PR = partial response; sCR = stringent complete response; TEAE = treatment-emergent adverse event; TTNT = time to next treatment; TTR = time to response; VGPR = very good partial response.
Note: Details included in Table 8 are from the sponsor’s Summary of Clinical Evidence.83
Sources: Clinical Study Report for the MajesTEC-1 study (2021),24 Clinical Study Report for the MajesTEC-1 study (2021),65 and Clinical Study Protocol for the MajesTEC-1 study (2020).66
OS was defined as the time from the date of the first dose of the study intervention to the date of the patient’s death, due to any cause.
PFS was defined as the time from the date of the first dose of the study intervention to the date of the first documented disease progression, as defined in the IMWG criteria,12,63 or death due to any cause, whichever occurred first. Relapse from CR was not considered as disease progression.
CR or better was defined as the proportion of participants achieving CR or sCR according to the IMWG response criteria12,63 during or after the study intervention but before the start of subsequent antimyeloma therapy.
MRD-negativity status was defined as the proportion of patients who attained MRD-negative status (at 10–5) at any time point after the initial dose and before progressive disease or subsequent antimyeloma therapy. Participants with missing or unevaluable MRD status were grouped separately.
VGPR or better was defined as the proportion of patients achieving VGPR, CR, or sCR according to the IMWG response criteria12,63 during or after the study intervention but before the start of subsequent antimyeloma therapy.
sCR was defined as the proportion of patients achieving an sCR according to the IMWG criteria.12,63
ORR was defined as the proportion of participants who attained PR or better (i.e., PR, VGPR, CR, or sCR) according to the IMWG response criteria12,63 (Table 9) during or after study intervention but before the start of subsequent antimyeloma therapy, as assessed by the IRC.
TTR was defined as the time between the date of the first dose of study intervention and the first efficacy evaluation at which the patient had met all criteria for PR or better.
Duration of response was defined as the date of the initial documentation of a response (PR or better) to the date of the first documented evidence of progressive disease, as defined in the IMWG criteria,12,63 or death due to disease progression, whichever occurred first. Relapse from CR was not considered as disease progression.
TTNT was an exploratory outcome for phase II and was defined as the time from the date of the first dose of the study drug to the start of the next line of treatment.
Adverse events or serious adverse events were encoded using the Medical Dictionary for Regulatory Activities and graded according to the National Cancer Institute’s CTCAE version 4.0367 with the exception of CRS, which was evaluated according to the American Society for Transplantation and Cellular Therapy grading system.68 TEAEs are defined as adverse events with onset during the treatment phase or that are a consequence of a pre-existing condition that has worsened since baseline.66
Considerations that informed the selection of efficacy outcomes to be summarized and assessed using GRADE included the following:
survival outcomes were identified by the patient and clinician group input, and specified by the clinical experts consulted by CADTH to include OS and PFS (OS and PFS were also key inputs in the sponsor’s pharmacoeconomic model)
CR, sCR, VGPR, and ORR were identified as important by the clinical experts and by the patient and clinician group input (as response to treatment)
duration of response was identified as important by the clinical experts consulted by CADTH and patient and clinician group input
hypogammaglobulinemia and infections were identified as important safety outcomes by clinical experts consulted by CADTH for this review.
Table 9: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
EORTC QLQ-C30 | A cancer-specific, patient-reported outcomes standardized questionnaire that is commonly used in oncology clinical trials to evaluate HRQoL. The core questionnaire consists of 30 questions that are scored to create 5 multi-item functional scales (physical, role, emotional, cognitive, and social), 3 multi-item symptom scales (fatigue, nausea and vomiting, and pain), 6 single-item symptom scales (dyspnea, insomnia, appetite loss, constipation, diarrhea, and financial difficulties), and a global health status and QoL scale.69,70 Most items have 4 response options (“not at all,” “a little,” “quite a bit,” “very much”), with scores on these items ranging from 1 to 4, respectively. For the global health status and QoL scale, a 7-point Likert-type scale is used, with anchors between 1 (very poor) and 7 (excellent).70 The raw scores for each scale are computed as the average of the items that contribute to a particular scale. Each raw scale score is then converted to a standardized score that ranges from 0 to 100 using a linear transformation, with a higher score reflecting better function on the function scales, greater symptoms on the symptom scales, and better QoL.70 Thus, a decline in score on the symptom scale would reflect an improvement, whereas an increase in score on the function and QoL scales would reflect an improvement.70 | Osborne et al.71 conducted a systematic literature review of validated HRQoL tools in MM. For EORTC QLQ-C30, the sample included patients with newly diagnosed MM, relapsed MM, and mixed disease stages of MM and treatment experiences, and who were treated with HSCT.71 For the purposes of this review, data specific to only patients treated with HSCT were not reported in the following. Validity: For construct validity, the subscales for pain, fatigue, physical, and global QoL were considered able to discriminate between patients who improved (mixed disease stages and treatment experiences, including 69 [29%] patients with relapsed MM or disease progression) vs. patients who were stable or had deteriorated (N = 239).72 All subscales, with the exception of the single-item diarrhea scale, were considered able to discriminate between patients with newly diagnosed MM according to their performance and response status (N = 484).73 Reliability: For internal consistency, the Cronbach alpha ranged from 0.54 to 0.89 for all subscales in patients with newly diagnosed MM, and mixed disease stages and treatments, and who were treated with HSCT.73-76 Responsiveness: Responsiveness of the subscales to change over time varied depending on the sample population studied (listed previously).71 To assess responsiveness, Kvam et al.77 used the global rating of change to identify whether patients with mixed disease stages and treatments changed over time. Of note, 69 (29%) patients had relapsed or progressive disease.a, 77 For the global QoL scale, the SRMb in patients who reported improvement and deterioration over a period of 3 months was 0.32 and 0.57, respectively (N = 239). In patients rating themselves as unchanged, the SRM was negligible.77 Note that the results should be interpreted with caution as 86 (36%) patients did not receive treatment during the study77 (i.e., any change experienced was not due to treatment). | Cocks et al.78 examined 118 published studies on various types of cancer, such as breast, lung, or head and neck, and also used clinician expert input to determine meaningful differences and the magnitude of change in the EORTC QLQ-C30. According to the authors, a small change was considered a subtle, clinically relevant change.78 Note that patients with MM were not included in the meta-analysis.78 Based on the meta-analysis estimates, the thresholds for no difference (trivial change) in the subscales were as follows (any difference larger than this trivial change would be considered important):78
From the systemic review of validated HRQoL tools in MM conducted by Osborne et al.,71 the following estimated MIDs were reported for patients with mixed disease stages and treatments:
|
EQ-5D-5L | A generic measure of health status that comprises 2 parts. The descriptive system assesses health in 5 dimensions (mobility, self-care, usual activities, pain/discomfort and anxiety/depression).80 Each dimension has 5 increasing levels of severity or response (no problems, slight problems, moderate problems, severe problems, and unable to or extreme problems). A unique health state profile is generated as a 5-digit code. For example, 12345 indicates no problems with mobility, slight problems with self-care, moderate problems with usual activities, severe pain or discomfort, and extreme anxiety or depression.80 The health state can be converted to a summary index score based on societal (countries or regions) preference weights for the health state. Index scores range from less than 0 (negative values represent worse than dead, which is represented by 0) to 1 (full health), with higher scores representing higher health utility.80 A patient’s perceived health status on that day is also rated using the EQ VAS, ranging from 0 (worst imaginable health) to 100 (best imaginable health).80 | Responsiveness: Not assessed in patients with MM Validity: Not assessed in patients with MM Reliability: Not assessed in patients with MM. | Not estimated in patients with MM |
EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EQ VAS = EQ visual analogue scale; HRQoL = health-related quality of life; HSCT = hematopoietic stem cell transplant; MID = minimal important difference; MM = multiple myeloma; QoL = quality of life; SRM = standardized response mean; vs. = versus.
aThe European Group for Blood and Marrow Transplantation criteria for response were used to determine the patients’ disease phase.
bTo assess the magnitude of the difference in scores between patients who improved, deteriorated, and remained stable, SRMs were calculated and compared against Cohen’s rule of thumb for interpreting the magnitude of mean differences in HRQoL scores: 0.20 represents a small change, 0.50 represents a moderate change, and more than 0.80 represents a large change.
cThe MIDs were estimated using 2 approaches. The anchor-based approach was anchored to a structured QoL interview (response options were improved, deteriorated, or unchanged) while the distribution-based approach was based on the standard deviations of baseline scores as well as the receiver-operating characteristic curve method.
dThe MIDs were estimated using 2 approaches. The anchor-based approach was anchored to a structured QoL interview (response options were improved, deteriorated, or unchanged) while the distribution-based approach was based on the standard deviations of baseline scores.
Statistical Analysis
In the MajesTEC-1 trial, analysis of efficacy outcomes was based on the FAS for patients who received teclistamab at the recommended phase II dose (phase I and phase II, cohort A). The rates of ORR, VGPR or better, CR or better, and sCR with 2-sided 95% CIs were presented for each cohort (phase I and phase II, cohort A). VGPR, CR, or sCR after the start of subsequent anticancer therapy was not considered.
For the ORR calculation, patients who were NE for response were listed as such and were considered nonresponders. Patients with no postbaseline data were considered as nonresponders. Response after the start of subsequent therapy with teclistamab was not considered.
Time to first response was analyzed for patients in the FAS who attained a response (PR or better), using disease response as assessed by the IRC. Descriptive statistics (mean, standard deviation [SD], median, and range) were provided for each cohort. Time to first response, time to best response, time to CR or better response, and time to VGPR or better response were summarized similarly. Analysis was performed based on the IRC assessment.
The distribution of duration of response was estimated using the Kaplan-Meier method, and the corresponding median duration of response and its 95% CI were reported.
For OS, the Kaplan-Meier method was used to estimate the distribution of OS for each cohort, and the median OS with 95% CI was derived. Patients who were lost to follow-up were censored at the time of being lost to follow-up. Patients who died after consent withdrawal but with death data collected were considered as having an OS event. If the patient was alive or the vital status was unknown, then the patient’s survival time was censored at the date the participant was last known to be alive. The date of the patient being last known alive was determined by the maximum collection or assessment date from among selected data domains within the clinical database.
For PFS, the Kaplan-Meier method was used to estimate the distribution of PFS for each cohort and the median PFS with 95% CI was derived. For patients who had not progressed and were alive, their event times were censored at the date of the last disease evaluation before the start of any subsequent antimyeloma therapy. Patients without any postbaseline disease assessment were censored at the date of the first dose of the study intervention. The number and proportion of patients who had a PFS event or were censored were reported. The reasons for censoring were summarized accordingly. The Kaplan-Meier PFS curve was also plotted by cohort. Analysis was performed based on IRC assessment.
The MRD-negativity rate and its 2-sided 95% CI were estimated for each cohort (phase I and phase II, cohort A).
Safety Outcomes
Unless otherwise specified, all safety outcomes were based on the FAS. Descriptive statistics or frequency counts and percentages by cohort were derived for continuous and categorical safety variables, respectively.
Patient-Reported Outcomes
Three patient-reported outcome measures were evaluated using the EORTC QLQ-C30 and EQ-5D-5L questionnaires, and PGI-S. At the time of analysis, using the January 4, 2023, data cut-off, a post hoc analysis was performed in EORTC QLQ-C30 and EQ-5D-5L only in phase II, cohort A, in HRQoL-evaluable patients requiring a baseline and at least 1 postbaseline assessment. Within-group mean change in EORTC QLQ-C30 scales and the EQ-5D-5L tool was analyzed at the group level using mean change from baseline and at the individual level with the proportion of patients with a meaningful change from baseline. Change from baseline in the EORTC QLQ-C30 scales and the EQ visual analogue scale (EQ VAS) and utility were fitted to a mixed model of repeated measures including patients as a random effect, and baseline value and time as fixed effects. Compliance rates for completion of the 3 patient-reported outcome measures were assessed at each time point based on the number of participants in each cycle. Descriptive statistics by cohort were provided for EORTC QLQ-C30 scales and the EQ VAS and utility value. The frequency of responses for the PGI-S single item was summarized as number and percentage responding to each response category. Within-group changes were analyzed at the group level using mean change from baseline and at the individual level with the proportion of participants with a meaningful change from baseline.
At the time of analysis, using the September 7, 2021, data cut-off date, meaningful change at the individual level was assessed for the EORTC QLQ-C30 scales and EQ VAS using 2 methods. The first approach defined meaningful change based on a literature review and another relied on an MCT, defined as at least half of SD from baseline values. The achievement of a meaningful (10-point) improvement14 from baseline through cycle 6 using the literature-based MCT was summarized for global health status, physical functioning, fatigue, and pain of the EORTC QLQ-C30 tool. The achievement of a meaningful (7-point) improvement15,81 from baseline through cycle 6 using the literature-based MCT was summarized for the EQ VAS tool.
Sample Size and Power Calculation
For part 1 of phase I of the MajesTEC-1 study, at least 6 patients were enrolled at the safe and tolerable proposed recommended phase II dose(s). The total number of patients in part 1 depended on the frequency of the dose-limiting toxicities and when the recommended phase II dose(s) was determined. For part 2 of phase I of the MajesTEC-1 study, a sample size of up to 40 patients receiving the proposed recommended phase II dose was determined to provide a sufficient probability of observing at least 1 toxicity.
For part 3 (phase II) of the MajesTEC-1 study, the reported sample size requirements for hypothesis testing were as follows.
Cohort A: Approximately 100 patients treated with teclistamab would be sufficient to achieve at least 85% power such that the lower bound of the ORR 2-sided 95% CI would be above 30%, under an assumed ORR among those treated with teclistamab of 45%. Patients treated with teclistamab who had had a nonevaluable response were counted as nonresponders in the ORR assessment.
Cohort C: Simon’s 2-stage design was used to test the null hypothesis that the ORR was at most 15% against the alternative that the ORR was at least 35%. Cohort C required 34 response-evaluable patients; assuming a nonevaluable rate of 10%, a total of 38 patients were to be enrolled. After the first 21 patients were enrolled and had been followed for at least 2 cycles to be evaluable for response, the stage I analysis was to be performed. This design yields a type I error rate of 0.025 and a power of 80% under an assumed ORR of 35%.
Statistical Testing
General Statistical Testing Methods
No formal statistical hypothesis testing was conducted in part 1 or part 2 of the study. For each of the 3 parts of the study, data were summarized using descriptive statistics by dose level and by part. The number of observations, mean, SD, coefficient of variation, median, and range were used to summarize continuous variables, where appropriate. The number of observations and their percentages were used to summarize categorical variables.
Interim Analyses
In phase II, futility interim analyses occurred in cohort A. The ORR was analyzed after 30 patients in cohort A became the futility evaluable analysis set. The Lan-DeMets alpha spending function (O’Brien-Fleming type)82 was used as a beta-spending function to determine the futility boundary. With this beta-spending function, the stopping boundary in the cohort A interim analysis was identified as up to 6 responders of 30 patients.
In cohort C, the stage I analysis of futility was performed when the first 21 patients were enrolled and had been followed for at least 2 cycles to be evaluable for response. Further enrolment could be terminated if 3 or fewer responses were observed in the first stage. Otherwise, an additional 17 patients were to be enrolled to ensure there were a total of 34 response-evaluable patients with 2 stages combined. The null hypothesis was to be rejected if 10 or more responses were observed in 34 response-evaluable patients.
Subgroup Analyses
Subgroup analyses were performed during the primary efficacy analyses on the cut-off date of September 7, 2021, in the efficacy analysis set. ORR was estimated within the prespecified subgroups: sex, age, renal function, hepatic function, race, ECOG PS, number of lines of prior therapy, refractory status, prior autologous stem cell transplant, prior allogeneic stem cell transplant, type of myeloma, ISS, R-ISS, cytogenetic risk, bone marrow plasma cells, extramedullary plasmacytomas, prior anti-BCMA exposure, and BCMA tumour expression. Details pertaining to the subgroup analyses on the data cut-off date of January 4, 2023, were available only on select subgroups that were deemed clinically meaningful by the clinical experts consulted by CADTH for this review, including the number of lines of prior therapy, and cytogenetic risk.
Sensitivity Analyses
Sensitivity analyses were performed during the primary efficacy analyses on the cut-off date of September 7, 2021, using disease response based on computerized algorithm and investigator assessment according to IMWG in the efficacy analysis set.
There were no sensitivity analyses performed or reported at the time of the data cut-off date of January 4, 2023. No adjustment for multiple testing or type I error control was used.
Analysis Populations
Analysis sets for the MajesTEC-1 trial included in the systematic review are summarized in Table 11.
Table 10: Statistical Analysis of Efficacy End Points in MajesTEC-1 Study: Clinical Data Cut-Off of January 4, 2023
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
OS | Kaplan-Meier method | None | Patients who are lost to follow-up were censored at the time of loss to follow-up. | None |
PFS | Kaplan-Meier method | None | Patients without any postbaseline disease assessment were censored at the date of the first dose of study intervention. | None |
VGPR or better rate, CR or better rate, and sCR | 2-sided 95% exact confidence interval | None | None | None |
MRD-negativity rate | 2-sided 95% exact confidence interval | None | Patients with missing or unevaluable MRD status were considered as MRD-positive. | None |
ORR | 2-sided 95% exact confidence interval | None | Patients who had a nonevaluable response were counted as nonresponders in the ORR assessment. | None |
TTR | Descriptive statistics only (N, mean, SD, median, and range) | None | None | None |
Duration of response | Kaplan-Meier method | None | For patients who had not progressed or had died due to causes other than disease progression, data were censored at the last disease evaluation before the start of any subsequent antimyeloma therapy. | None |
Safety end points | Continuous variables were summarized by descriptive statistics by cohort (N, mean, SD, median, and range) Categorical variables were summarized by cohort using frequency counts and percentages | None | None | None |
EORTC QLQ-C30, EQ-5D-5L, and PGI-S | Descriptive statistics by cohort were provided for the EORTC QLQ-C30 scales and the EQ VAS and utility value. A line plot of mean with standard error over time was created. The frequency of responses for the PGI-S single item was summarized as number and percentage responding to each response category. Within-group mean change was assessed using a mixed model with repeated measures analysis to estimate the change from baseline at each time point for all participants with at least 1 postbaseline assessment. Change from baseline in the EORTC QLQ-C30 scales and the EQ VAS and utility value was fitted to a mixed model with repeated measures that included participants as a random effect, and baseline value and time as fixed effects. Meaningful change at the individual level was assessed for the EORTC QLQ-C30 scales and EQ VAS using 2 methods, both literature-based and distribution-based approaches. Distribution-based MCTs were defined as at least half of SD from baseline values. The number and percentage of participants meeting the MCT were summarized as the proportion of participants with meaningful change. | None | No imputation for missing data was performed. | None |
CR = complete response; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EQ VAS = EQ visual analogue scale; MCT = meaningful change threshold; MRD = minimal residual disease; ORR = overall response rate; OS = overall survival; PFS = progression-free survival; PGI-S = Patient Global Impression–Severity; sCR = stringent complete response; SD = standard deviation; TTR = time to response; VGPR = very good partial response.
Note: Details included in Table 10 are from the sponsor’s Summary of Clinical Evidence.83
Sources: Clinical Study Protocol for the MajesTEC-1 study66 and Clinical Study Reports for the MajesTEC-1 study.24,65
Table 11: Analysis Populations of MajesTEC-1 Study: Clinical Cut-Off Date of January 4, 2023
Study | Population | Definition | Application |
|---|---|---|---|
MajesTEC-1 study | All-treated analysis set, known as full analysis set | Patients who received at least 1 dose of teclistamab at RP2D on or before the clinical cut-off date of January 4, 2023 (last patient’s first dose was on August 13, 2021) | Efficacy analyses |
Safety analysis set | Patients who received at least 1 dose of teclistamab at RP2D on or before the clinical cut-off date of January 4, 2023 (last patient’s first dose was on August 13, 2021) | Safety analyses | |
Pharmacokinetics evaluable analysis set | Patients who received at least 1 dose of study intervention and had at least 1 evaluable concentration measurement; patients were excluded from the pharmacokinetic parameter analysis if individual data did not allow for adequate estimation of parameters | Pharmacokinetic evaluations | |
Immunogenicity analysis set | Patients who received at least 1 dose of study intervention and had at least 1 postdose immunogenicity sample | Immunogenicity evaluations |
RP2D = recommended phase II dose.
Sources: Clinical Study Reports for the MajesTEC-1 study.24,65
Protocol Amendments
Overall, there were 11 amendments (final amendment dated July 5, 2021) in the MajesTEC-1 trial. The amendment dated March 20, 2017, included the provision of additional clarity to safety measures, the revision of the definition of dose-limiting toxicity, the inclusion of patients with a disease that is relapsed or refractory to established therapies with known clinical benefit in RRMM or patients who are intolerant of those established MM therapies, and a candidate for treatment with teclistamab in the opinion of the treating physician; the amendment excluded allogeneic stem cell transplant up to 6 months before the first dose of the study drug, the modification of dose escalation rules, and the correction of a dose modification guideline. The amendment dated September 2, 2017, included revisions to the inclusion and exclusion criteria that were made for compliance with health agency regulations, the implementation of additional safety measures, and the addition of an explanation to the risks section to clarify that the risks with the study drug are unknown because the toxicological profiles in monkeys may not be relevant to humans. The amendment dated December 12, 2017, included the specification of the grade for TLS that is considered to be a dose-limiting toxicity, and the removal of the recommendation for anti–interleukin-6 therapy for the treatment of grade 2 or higher neurotoxicity. The amendment dated March 26, 2018, included increasing the dosing of teclistamab frequency from biweekly to weekly in new patients enrolled in the study. The dosing change decision was based on preliminary pharmacokinetic data from the first 7 patients who showed insufficient drug exposure beyond day 8 following the first dose of teclistamab. In addition, the patients remaining in the study who were originally dosed on a biweekly schedule would continue on the biweekly schedule.
The amendment dated October 3, 2018, included permission to modify the dosing schedule to align with pharmacokinetic data (weekly versus biweekly dosing schedule), the revision of dose escalation rules for part 1, and clarification of the administration of required preinfusion medication, the exclusion of specific postinfusion medication instructions, and the clarification of disease evaluation schedules and procedures. The amendment dated March 12, 2019, included the investigation of an alternative SC administration method of teclistamab, as it was hypothesized that SC administration should reduce the risk of CRS. The overall reason for this amendment was to add details to evaluate teclistamab administered subcutaneously in part 1 and part 2 of the MajesTEC-1 trial, and to update the required duration of hospitalization for dosing of the study drug at specific cycles and days based on recent pharmacokinetic and safety data. The amendment dated May 23, 2019, included adding time and events schedules to support twice-weekly dosing of the study drug, and updating the rules for dose escalation in the standard titration phase based on the safety and pharmacodynamic profile to date. The amendment dated May 23, 2019, included the addition of the option of a commercially available saline IV bag plus sponsor-provided diluent additive liquid for the dilution of the study drug before administration, an increase in the sample size to allow for continued enrolment in IV dose escalation, the allowance of a 2-hour infusion time for newly enrolled patients, and the updating of the intrasubject dose escalation criteria and discontinuation criteria. The amendment dated July 2, 2020, included the addition of phase II (or part 3) details to the MajesTEC-1 study, the addition of rules to stop enrolment during part 2 if the dose-limiting toxicity rate reaches a specified range, and the inclusion of the definition of women of childbearing potential. The amendment dated October 26, 2020, included providing updated data for the recommended phase II dose, providing a futility analysis for cohort A and cohort B in part 3, and clarifying the inclusion criteria for part 3 cohorts and the plan for enrolment of the cohorts.
Protocol Deviations
At the cut-off date of September 7, 2021, major protocol deviations were reported for 13 (7.9%) patients — none of which affected patient safety or data integrity. The most frequent major protocol deviations included not meeting eligibility criteria (4.3%) and receiving the wrong treatment or incorrect dose (3.0%). No major protocol deviations were related to COVID-19.
Protocol deviations based on the January 4, 2023; clinical cut-off date were not available at the time of review.
Results
Patient Disposition
Patient disposition in the MajesTEC-1 study is summarized in Table 12. At the time of analysis, using the January 4, 2023, data cut-off, patients were treated with teclistamab at the recommended phase II dose. In the MajesTEC-1 trial, 90 of 165 (54.5%) patients discontinued from the study. The reasons for study discontinuation included death (50.3%) and withdrawal by patient (4.2%).
At the time of analysis, using the January 4, 2023, data cut-off, similar proportions of patients discontinued the study treatment in phase I and phase II, cohort A (||||| ||| |||||| ||||||||||||). The reasons for study drug discontinuation included |||||||||||||||||| |||||||| ||||| |||||||| ||||||||| |||||||| ||||||| ||||||| ||||| ||||||| ||||||| || ||||||| ||||||||| ||||||| ||| ||||| |||||||||||| || || || ||| |||||||| ||||||| |||||||| || ||||||||| ||| ||| |||||||| || |||| ||||||| |||| ||||||||| |||| |||||||| |||||| |||||| |||||||| ||| |||||||||| ||||||| ||||||| || |||||||||||.
Table 12: Summary of Patient Disposition From MajesTEC-1 Study
Patient disposition | MajesTEC-1 study | ||
|---|---|---|---|
Phase I (N = 40) | Phase II, cohort A (N = 125) | Total (N = 165) | |
Treated, n | || | ||| | 165 |
Discontinued the study, n (%) | || |||||| | || |||||| | 90 (54.5) |
Reason for discontinuation, n (%) | |||
Death | || |||||| | || |||||| | 83 (50.3) |
Death due to COVID-19 | ||||| | || |||||| | 19 (11.5) |
Withdrawal by patient | || |||||| | || |||||| | || |||||| |
Discontinued study drug, n (%) | || |||||| | || |||||| | 118 (71.5) |
Reason for discontinuation, n (%) | |||
Progressive disease | || |||||| | || |||||| | || |||||| |
Death | || |||||| | || |||||| | || |||||| |
Death due to COVID-19 | || |||||| | || |||||| | || |||||| |
Physician decision | || |||||| | || |||||| | || |||||| |
Adverse event | || |||||| | || |||||| | || |||||| |
Adverse event due to COVID-19 | || |||||| | || |||||| | || |||||| |
Patient refused further treatmenta | || |||||| | || |||||| | || |||||| |
Other | || |||||| | || |||||| | || |||||| |
Remained on treatment, n (%) | || |||||| | || |||||| | 47 (28.5) |
FAS, N | || | ||| | 165 |
Safety, N | || | ||| | 165 |
FAS = full analysis set; RP2D = recommended phase II dose.
Note: Details included in Table 12 are from the sponsor’s Summary of Clinical Evidence.83
aPatients who refused further treatment included “(w)ithdrawal by patient” from phase I RP2D.
Source: Clinical Study Report for the MajesTEC-1 study (2021).24
Baseline Characteristics
The baseline characteristics outlined in Table 13 are limited to those that are most relevant to this review or were considered to affect the outcomes or interpretation of the study results. In the MajesTEC-1 trial, the median age of the patients was 64.0 years (range = 62.5 years to 64.0 years), with ||||| || ||| ||||| |||||||||| ||||| |||| |||| || ||||| || |||. Ninety-six (58.2%) patients were male and 69 (41.8%) patients were female. Most patients were white (81.2%), and 12.7% of patients identified as Black or African American. Most patients had an ECOG PS score of 1 (66.1%), and 33.3% of patients had an ECOG PS score of 0.
Table 13: Summary of Baseline Characteristics of MajesTEC-1 Study, Full Analysis Set
Characteristic | Phase I (N = 40) | Phase II, cohort A (N = 125) | Total (N = 165) |
|---|---|---|---|
Age, years | |||
Mean (SD) | 62.4 (10.0) | 64.4 (9.5) | 63.9 (9.6) |
Median (range) | 62.5 (39 to 84) | 64.0 (33 to 83) | 64.0 (33 to 84) |
< 65 years, n (%) | 23 (57.5) | 63 (50.4) | || |||||| |
65 to 74 years, n (%) | 12 (30.0) | 43 (34.4) | || |||||| |
≥ 75 years, n (%) | 5 (12.5) | 19 (15.2) | || |||||| |
Sex, n (%) | |||
Female | 14 (35.0) | 55 (44.0) | 69 (41.8) |
Male | 26 (65.0) | 70 (56.0) | 96 (58.2) |
Race, n (%) | |||
Asian | 0 (0.0) | 3 (2.4) | 3 (1.8) |
Black or African American | 1 (2.5) | 20 (16.0) | 21 (12.7) |
White | 34 (85.0) | 100 (80.0) | 134 (81.2) |
Multiple | 0 (0.0) | 1 (0.8) | 1 (0.6) |
Other | 1 (2.5) | 1 (0.8) | 2 (1.2) |
Not reported | 4 (10.0) | 0 (0.0) | 4 (2.4) |
Ethnicity, n (%) | |||
Hispanic or Latino | 2 (5.0) | 13 (10.4) | 15 (9.1) |
Not Hispanic or Latino | 33 (82.5) | 111 (88.8) | 144 (87.3) |
Not reported | 4 (10.0) | 1 (0.8) | 5 (3.0) |
Unknown | 1 (2.5) | 0 (0.0) | 1 (0.6) |
Weight, kg | |||
Mean (SD) | 77.8 (14.7) | 74.1 (17.3) | 75.0 (16.7) |
Median (range) | 76.1 (50.0 to 103.5) | 72.0 (41.0 to 138.9) | 73.0 (41.0 to 138.9) |
Baseline ECOG PS score, n (%) | |||
0 | 17 (42.5) | 38 (30.4) | 55 (33.3) |
1 | 23 (57.5) | 86 (68.8) | 109 (66.1) |
2 to 3 | 0 (0.0) | 1 (0.8) | 1 (0.6) |
Type of myeloma by immunofixation or serum FLC assay, n (%) | |||
IgG | 17 (42.5) | 74 (59.2) | || |||||| |
IgA | 8 (20.0) | 21 (16.8) | || |||||| |
IgM | 0 (0.0) | 2 (1.6) | || |||||| |
IgD | 2 (5.0) | 1 (0.8) | || |||||| |
IgE | 0 (0.0) | 0 (0.0) | || |||||| |
Light chain | 11 (27.5) | 25 (20.0) | || |||||| |
Kappa | 7 (17.5) | 8 (6.4) | || ||||| |
Lambda | 4 (10.0) | 16 (12.8) | || |||||| |
FLC-kappaa | 0 (0.0) | 1 (0.8) | || |||||| |
FLC-lambdab | 0 (0.0) | 0 (0.0) | || |||||| |
Biclonal | 2 (5.0) | 2 (1.6) | || |||||| |
Negative immunofixation | 0 (0.0) | 0 (0.0) | || |||||| |
Type of measurable disease per IMWG, n (%) | |||
Serum only | 15 (37.5) | 53 (42.4) | 68 (41.2) |
Serum and urine | 4 (10.0) | 24 (19.2) | 28 (17.0) |
Urine only | 4 (10.0) | 16 (12.8) | 20 (12.1) |
Serum FLC | 16 (40.0) | 31 (24.8) | 47 (28.5) |
NE | 1 (2.5) | 1 (0.8) | 2 (1.2) |
Time from MM diagnosis to first dose, years | |||
Mean (SD) | 5.9 (3.7) | 6.9 (3.8) | 6.6 (3.8) |
Median (range) | 5.6 (0.8 to 17.4) | 6.2 (0.9 to 22.7) | 6.0 (0.8 to 22.7) |
Number of lytic bone lesions, n (%) | |||
None | 5 (12.5) | 15 (12.0) | 20 (12.1) |
1 to 3 | 5 (12.5) | 15 (12.0) | 20 (12.1) |
4 to 10 | 11 (27.5) | 32 (25.6) | 43 (26.1) |
More than 10 | 19 (47.5) | 63 (50.4) | 82 (49.7) |
Number of extramedullary plasmacytomas, n (%) | |||
0 | 32 (80.0) | 105 (84.0) | 137 (83.0) |
≥ 1 | 8 (20.0) | 20 (16.0) | 28 (17.0) |
ISS staging, n (%)c | |||
Number of patients contributing to the analysis, n | 39 | 123 | 162 |
Phase I | 24 (61.5) | 61 (49.6) | 85 (52.5) |
Phase II | 11 (28.2) | 46 (37.4) | 57 (35.2) |
Phase III | 4 (10.3) | 16 (13.0) | 20 (12.3) |
R-ISS staging, n (%)d | |||
Number of patients contributing to the analysis, n | 37 | 119 | 156 |
Phase I | 15 (40.5%) | 28 (23.5%) | 43 (27.6%) |
Phase II | 19 (51.4%) | 81 (68.1%) | 100 (64.1%) |
Phase III | 3 (8.1%) | 10 (8.4%) | 13 (8.3%) |
Plasma cells, bone marrow biopsy or aspirate, n (%)e | |||
Number of patients contributing to the analysis, n | 38 | 122 | ||| |
< 5 | 16 (42.1) | 36 (29.5) | || |||||| |
5 to 30 | 14 (36.8) | 45 (36.9) | || |||||| |
31 to 59 | 5 (13.2) | 26 (21.3) | || |||||| |
60 or more | 3 (7.9) | 15 (12.3) | || |||||| |
Cytogenetic risk, n (%) | |||
Number of patients contributing to the analysis, n | 37 | 110 | 147 |
Standard risk | 25 (67.6) | 84 (76.4) | 109 (74.1) |
High risk | 12 (32.4) | 26 (23.6) | || |||||| |
del(17p) | 9 (24.3) | 14 (12.7) | || |||||| |
t(4;14) | 4 (10.8) | 12 (10.9) | || |||||| |
t(14;16) | 1 (2.7) | 3 (2.7) | || |||||| |
Bone marrow cellularity by biopsy, n (%) | |||
Number of patients contributing to the analysis, n | 23 | 45 | || |
Hypercellular | 4 (17.4) | 16 (35.6) | || |||||| |
Normocellular | 12 (52.2) | 20 (44.4) | || |||||| |
Hypocellular | 3 (13.0) | 6 (13.3) | || |||||| |
Indeterminate | 4 (17.4) | 3 (6.7) | || |||||| |
ECOG PS = Eastern Cooperative Oncology Group Performance Status; FLC = free light chain; IgA = immunoglobulin A; IgD = immunoglobulin D; IgE = immunoglobulin E; IgG = immunoglobulin G; IgM = immunoglobulin M; IMWG = International Myeloma Working Group; ISS = International Staging System; MM = multiple myeloma; NE = not evaluable; R-ISS = Revised International Staging System; RP2D = recommended phase II dose; SD = standard deviation.
Notes: Details included in Table 13 are from the sponsor’s Summary of Clinical Evidence.83
Percentages were calculated with the number of participants in the full analysis set with available data as the denominator.
aIncludes patients without a positive immunofixation but with evidence of FLC kappa by FLC testing.
bIncludes patients without a positive immunofixation but with evidence of FLC lambda by FLC testing.
cISS staging was derived based on serum beta2-microglobulin and albumin.
dR-ISS staging was derived based on the combination of serum beta2-microglobulin and albumin, genetic risk, and the level of lactate dehydrogenase.
eThe maximum value from either a bone marrow biopsy or bone marrow aspirate was selected if both results were available.
Source: Clinical Study Report for the MajesTEC-1 study (2021).65
The most common immunoglobulin isotypes were immunoglobulin G (55.2%) and immunoglobulin A (17.6%). The median time from diagnosis of MM to enrolment in the study was 6.0 years (range = 0.8 years to 22.7 years). Twenty-eight (17.0%) patients had 1 or more extramedullary plasmacytomas at baseline. Of the 147 patients with baseline cytogenetic data reported, 38 (25.9%) patients had at least 1 high-risk abnormality, including del(17p) (15.6%) and t(4;14) (10.9%) abnormality. Of the 162 patients with baseline ISS data reported, 85 (52.5%) patients were ISS stage I while 20 (12.3%) patients were ISS stage III.
In the MajesTEC-1 trial, 164 (99.4%) patients were refractory at any point to prior therapy and 148 (89.7%) patients were refractory to their last line of therapy. Of the 165 patients in the trial, 128 (77.6%) patients were triple-class refractory and 50 (30.3%) patients were penta-refractory (with at least 2 PIs, at least 2 IMiDs, and at least 1 anti-CD38 monoclonal antibody). The refractory status of patients in the MajesTEC-1 trial is summarized in Table 14.
Table 14: Summary of Refractory Status to Prior Multiple Myeloma Therapy for Patients in MajesTEC-1 Study, Full Analysis Set
Characteristic | Phase I (N = 40) | Phase II, cohort A (N = 125) | Total (N = 165) |
|---|---|---|---|
Refractory at any point to prior therapy, n (%) | 40 (100) | 124 (99.2) | 164 (99.4) |
Refractory status, n (%) | |||
Any PI | 34 (85.0) | 108 (86.4) | 142 (86.1) |
Any IMiD | 38 (95.0) | 114 (91.2) | 152 (92.1) |
Any anti-CD38 antibody | 39 (97.5) | 109 (87.2) | 148 (89.7) |
Double (PI and IMiD) | 33 (82.5) | 100 (80.0) | 133 (80.6) |
Triple (PI, IMiD, and anti-CD38 antibody) | 32 (80.0) | 96 (76.8) | 128 (77.6) |
Penta (2 PIs, 2 IMiDs, and anti-CD38 antibody) | 16 (40.0) | 34 (27.2) | 50 (30.3) |
Refractory to a drug(s), n (%) | |||
Bortezomib | 21 (52.5) | 62 (49.6) | 83 (50.3) |
Carfilzomib | 27 (67.5) | 68 (54.4) | 95 (57.6) |
Ixazomib | 8 (20.0) | 24 (19.2) | 32 (19.4) |
Lenalidomide | 34 (85.0) | 99 (79.2) | 133 (80.6) |
Pomalidomide | 29 (72.5) | 98 (78.4) | 127 (77.0) |
Thalidomide | 5 (12.5) | 11 (8.8) | 16 (9.7) |
Daratumumab | 39 (97.5) | 95 (76.0) | 134 (81.2) |
Isatuximab | 0 (0.0) | 21 (16.8) | 21 (12.7) |
Selinexor | 0 (0.0) | 4 (3.2) | 4 (2.4) |
Melphalan flufenamide | 1 (2.5) | 0 (0.0) | 1 (0.6) |
IMiD = immunomodulatory drug; PI = proteasome inhibitor.
Note: Details included in Table 14 are from the sponsor’s Summary of Clinical Evidence.83
Source: Clinical Study Report for the MajesTEC-1 study (2021).65
Exposure to Study Treatments
Patient exposure to study treatments in the MajesTEC-1 study is presented in Table 15. At the time of analysis, using the January 4, 2023, data cut-off, patients received a median of 9.3 months (range = 0.2 month to 33.6 months) of study treatment. The median duration of study treatment was |||| |||||| ||||||| ||| || |||| ||||||| in phase I, and ||| |||||| ||||||| ||| || |||| ||||||| in phase II, cohort A. Of the 165 patients, || |||||||| ||||||| |||||||| ||||||||||| ||| || |||||||||||| || |||||||| ||||||| |||||||| ||||||||||| ||| || ||||| || ||||||| ||| || |||||||| ||||||||||||||||||||||||| ||| || ||||| || ||||||. At the time of analysis, using the January 4, 2023, data cut-off, || |||||||| ||||||| |||| ||||| || ||||||||| |||| |||||||||||| ||| ||| |||||||| ||||||| |||||||||||| ||||| |||||||||| ||| |||||| |||| ||||||||| |||||| ||| ||||||||| |||||| ||| ||||||| |||||||||| ||||||| ||||| || ||||||| |||||||||||| ||| ||| |||||| |||||| || ||||| |||||||| ||| ||||||||||||| ||||| ||| |||||| |||||||| |||| ||||||||| ||| ||| |||||||||| |||||||||| || ||||||| || |||||| |||||| ||||||| |||||| ||| ||||| ||||||| ||||| || ||||||| (Table 15).
At the time of analysis, using the January 4, 2023, data cut-off, 63 (38.2%) patients had switched from weekly to biweekly dosing, including 54 patients who attained CR or better, and 9 patients who attained PR or VGPR or better. Of the 63 patients, 41 (65.1%) patients had maintained their response and were still receiving treatment at the time of analysis, using the January 4, 2023, data cut-off, with a range of 1 month to 25 months of follow-up after the initial dose schedule change. The median time to biweekly dosing was 11.3 months (range = 3.2 months to 29.5 months). The median duration of follow-up after a schedule change to biweekly dosing was 12.6 months (range = 1.0 month to 24.7 months). The reasons for the first schedule change (i.e., to biweekly dosing) among patients who did not meet protocol-defined criteria based on response included adverse events (4.8%), including neutropenia in 2 patients and injection-site rash in 1 patient and other (5%). Nine patients switched from biweekly to monthly dosing.
At the time of analysis, using the January 4, 2023, data cut-off, the cycle duration differed between phase I and phase II, cohort A, of the MajesTEC-1 trial. Among patients treated in phase I (21-day cycles), || |||||||| ||||||| |||| ||||||| ||| || ||||||||||| || |||||||||||||| |||| ||||||| ||| || |||||||||||| ||| || |||||||| ||||||| |||| ||||||| ||| || ||||| || ||||||| ||||| |||||||| ||||||| || ||||||||||||||||| |||||||| || |||||||| ||||||| |||| ||||||| ||| || |||||||||||| || ||||||||||||||| |||| ||||||| ||| || |||||||||||| ||| || |||||||| |||||| |||| ||||||| ||| || ||||| || ||||||.
Table 15: Summary of Treatment Exposure From MajesTEC-1 Study, Full Analysis Set
Characteristic | Phase I (N = 40) | Phase II, cohort A (N = 125) | Total (N = 165) |
|---|---|---|---|
Duration of study drug, months | |||
Mean (SD) | |||| |||||| | |||| ||||| | 11.4 (9.5) |
Median (range) | |||| |||| || ||||| | ||| |||| || ||||| | 9.3 (0.2 to 33.6) |
Dose intensity (all treatment cycles), mcg/kg per week)a | |||
|||| |||| | ||||||| ||||||| | ||||||| ||||||| | ||||||| ||||||| |
|||||| ||||||| | ||||||| |||||| || ||||||||| | ||||||| |||||| || |||||||| | ||||||| |||||| || |||||||| |
Number of doses | |||
Mean (SD) | |||| |||||| | |||| |||||| | 38.2 (28.4) |
Median (range) | |||| |||| || |||||| | |||| |||| || ||||| | 36.0 (3.0 to 112.0) |
Relative dose intensity, % | |||
|||| |||| | |||| ||||| | |||| |||||| | |||| |||||| |
|||||| ||||||| | |||| ||||| || |||||| | |||| ||||| || |||||| | |||| ||||| || |||||| |
Patients with a schedule change,b, c n (%) | || |||||| | || |||||| | 63 (38.2) |
Biweekly | || ||||||| | || ||||||| | 63 (100.0) |
Monthly | || ||||||| | || ||||||| | 9 (14.3) |
Weekly | || ||||||| | || ||||||| | 2 (3.2) |
Reason for switching to biweekly dose, n (%) | |||
||||||| ||| |||||||||||||||| |||||||| |||||||| | || ||||||| | || |||||| | || |||||| |
||| ||||||| ||| |||||||||||||||| |||||||| |||||||| | || ||||||| | || ||||||| | || ||||||| |
|| | || ||||||| | || ||||||| | || ||||||| |
||||| | || ||||||| | || ||||||| | || ||||||| |
Patients who remained on treatment, n (%) | || |||||| | || |||||| | 47 (28.5) |
Patients who had changed schedulee, f | || ||||||| | || |||||| | 42 (89.4) |
Patients who had not changed schedulee | || ||||||| | || ||||||| | 5 (10.6) |
Treatment cycles received | |||
Mean (SD) | |||| |||||| | |||| ||||| | 12.4 (10.2) |
Median (range) | |||| || || ||| | ||| || || ||| | 11.0 (1 to 40) |
Duration of exposure by cycles, n (%) | |||
|| |||||| || |||| | || ||||||| | ||| ||||||| | ||| ||||||| |
|| |||||| || |||| | || |||||| | ||| |||||| | ||| |||||| |
|| |||||| || |||| | || |||||| | || |||||| | ||| |||||| |
|| |||||| || |||| | || |||||| | || |||||| | ||| |||||| |
|| |||||| || |||| | || |||||| | || |||||| | ||| |||||| |
|| |||||| || |||| | || |||||| | || |||||| | ||| |||||| |
|| |||||| || |||| | || |||||| | || |||||| | ||| |||||| |
|| |||||| || |||| | || |||||| | || |||||| | || |||||| |
|| |||||| || |||| | || |||||| | || |||||| | || |||||| |
|| |||||| || |||| | || |||||| | || |||||| | || |||||| |
|| |||||| || |||| | || |||||| | || |||||| | || |||||| |
|| |||||| || |||| | || |||||| | || ||||| | || |||||| |
Duration of exposure by months, n (%) | |||
|| |||||| || |||| | || |||||| | ||| |||||| | ||| |||||| |
|| |||||| || |||| | || |||||| | || |||||| | ||| |||||| |
|| |||||| || |||| | || |||||| | || |||||| | ||| ||||||| |
|| |||||| || |||| | || |||||| | || |||||| | ||| |||||| |
|| |||||| || |||| | || |||||| | || |||||| | ||| |||||| |
|| |||||| || |||| | || |||||| | || |||||| | || |||||| |
|| |||||| || |||| | || |||||| | || |||||| | || |||||| |
|| |||||| || |||| | || |||||| | || |||||| | || |||||| |
|| |||||| || |||| | || |||||| | || |||||| | || |||||| |
|| |||||| || |||| | || |||||| | || |||||| | || |||||| |
|| |||||| || |||| | || |||||| | || |||||| | || |||||| |
|| |||||| || |||| | || |||||| | || |||||| | || |||||| |
SD = standard deviation.
Note: A summary of treatment exposure was presented at the time of analysis, using the January 4, 2023, data cut-off date.
aDose intensity (mcg/kg per week) was calculated as the sum of total treatment doses (mcg/kg) received (excluding step-up doses before cycle 1; any step-up doses that were received after cycle 1 were considered) divided by the protocol-specified cycle length in weeks on teclistamab after the step-up dosing period.
bFor all patients with a schedule change, the first schedule change was to biweekly dosing. A patient could later change to a monthly (or back to weekly) dosing schedule.
cOne patient may have had multiple schedule changes. (Consequently, numbers do not add up to the number of patients with a schedule change).
dThe protocol-defined criteria for a schedule change to biweekly dosing in phase I was if a patient had attained an investigator response of confirmed PR or better and received a minimum of 4 cycles of therapy. The protocol-defined criteria for a schedule change to biweekly dosing in phase II was if a patient had had an investigator response of CR or better for a minimum of 6 months. (Be advised that a phase II patient with an investigator response of CR or better for a minimum of 173 days was considered to have met the criteria to switch; this reflects the protocol-specified visit window of ± 7 days).
ePercentages were based on the number of patients who were still on treatment.
fFor all patients with a schedule change, the first schedule change was to biweekly dosing. A patient could later change to a monthly (or back to weekly) dosing schedule.
Source: Clinical Study Report for the MajesTEC-1 study (2021).24
Pretreatment Medications
All pretreatment medications (required and optional) administered at any time during the study are summarized in Table 16. In the MajesTEC-1 trial, all 165 patients (100.0%) received steroids, antipyretics (anilides), and antihistamines (histamine1 receptor antagonists) as a pretreatment medication. The median total dose of steroids received as a pretreatment medication was 64.0 mg (range = 48.0 mg to 340.0 mg).
Concomitant Medications
All 165 patients received concomitant medication during treatment with teclistamab (Table 17). The most frequently used medications were nucleosides and nucleotides excluding reverse transcriptase inhibitors (93.9%), followed by anilides (81.2%), natural opium alkaloids (80.6%), and combinations of sulfonamides and trimethoprim (52.7%).
Table 16: Pretreatment Medications, Full Analysis Set [Redacted]
Pretreatment medication | Phase I (N = 40) | Phase II, cohort A (N = 125) | Total (N = 165) |
|---|---|---|---|
|||||||||||| | || ||||| | ||| ||||| | ||| ||||| |
||||||||||||||||||| | || ||||| | ||| ||||| | ||| ||||| |
||||||||||| ||||||||||||||| | || ||||| | ||| ||||| | ||| ||||| |
||||||||||| ||||||||||||||| | ||| ||||| | ||| ||||| | ||| ||||| |
||||||||||| ||||||||||||||| | ||| ||||| | ||| ||||| | ||| ||||| |
||||||||||| ||||||||||||||| | ||| ||||| | ||| ||||| | ||| ||||| |
||||||||||| |||||||||||||||||||||||||| ||||||||||||||| | ||| ||||| | ||| ||||| | ||| ||||| |
|||||||||||||| ||||||||||||||| | ||| ||||| | ||| ||||| | ||| ||||| |
||||||| ||||| ||||||||||||| | ||| ||||| | ||| ||||| | ||| ||||| |
||||| ||||||||||||||| | ||| ||||| | ||| ||||| | ||| ||||| |
||||| |||||||||||||| ||| |||||||||||| | |||
||||||||||||||| | ||| ||||| | ||| ||||| | ||| ||||| |
|||| ||||||||||||| | ||| ||||| | ||| ||||| | ||| ||||| |
H1 = histamine1; H2 = histamine2.
Note: Details included in Table 16 are from the sponsor’s Summary of Clinical Evidence.83
Source: Clinical Study Report for the MajesTEC-1 study (2021).65
Table 17: Concomitant Medication Use, Full Analysis Set
Medication by class | Total (N = 165) |
|---|---|
Nucleosides and nucleotides, excluding reverse transcriptase inhibitors, n (%) | 155 (93.9) |
|||||||||||||||| | || |||||| |
|||||||||||||||| | || |||||| |
Anilides, n (%) | 134 (81.2) |
||||||||||| | ||| |||||| |
Natural opium alkaloids | 94 (57.0) |
||||||||| | || |||||| |
Combinations of sulfonamides and trimethoprim, including derivatives | 87 (52.7) |
Note: Details included in Table 17 are from the sponsor’s Summary of Clinical Evidence.83
Source: Clinical Study Report for the MajesTEC-1 study (2021).65
Subsequent Antimyeloma Treatment
At the time of analysis, using the September 7, 2021, data cut-off, of the 104 responders (with PR or more) in the MajesTEC-1 study, || |||||||| ||||||| received 1 or more subsequent antimyeloma therapies, with a median TTNT of 12.7 months. The most common antimyeloma treatments included glucocorticoids (17.0%), nitrogen mustard analogues (13.9%), PIs (12.1%), monoclonal antibodies (5.5%), and subsequent radiotherapy (5.5%).
At the time of analysis, using the January 4, 2023, data cut-off, subsequent antimyeloma therapy was reported for || |||||||| |||||||, including || |||||||| ||||||| in phase I and || |||||||| ||||||| in phase II, cohort A. The median time to subsequent antimyeloma treatment was 20.1 months (95% CI, 12.7 months to NE months); however, details pertaining to the specific subsequent therapies received by patients in this data cut-off are not reported.
Efficacy
The primary analysis at the data clinical cut-off of September 2021 and the final analysis at the data clinical cut-off of August 2023 were prespecified analyses, and the sponsor-submitted Clinical Study Reports with March 2022 and January 2023 clinical cut-offs present interim analyses. As the final clinical report for the pivotal study is not yet available, information for the following sections was extracted from the more recent Clinical Study Report with a clinical cut-off date of January 4, 2023, submitted by the sponsor for this review. However, data from the clinical cut-off date of September 7, 2021, was also used to supplement the included data when necessary.
Findings for key efficacy outcomes in the MajesTEC-1 trial are summarized in Table 20.
Overall Survival
At the time of analysis, using the January 4, 2023, data cut-off, the median duration of follow-up was |||| |||||| |||| ||| ||| || |||| ||||||||||| |||||| |||| ||| ||| || |||| ||||||||||| |||||| |||| ||| ||| || |||| ||||||| in phase I, and 22.3 months (range = 0.3 month to 26.5 months) in phase II, cohort A. The estimated median OS was |||| |||||| |||| ||| ||| || |||| ||||||||||| |||||| |||| ||| ||| || |||| ||||||| in phase I, and |||| |||||| |||| ||| ||| || |||| ||||||||||| |||||| |||| ||| ||| || |||| ||||||| in phase II, cohort A. In the FAS, deaths were reported in |||| |||||| |||| ||| ||| || |||| ||||||| in phase I and |||| |||||| |||| ||| ||| || |||| ||||||||||| |||||| |||| ||| ||| || |||| ||||||| in phase II, cohort A. The number of patients who were censored (alive at the time of data cut-off) was |||| |||||| |||| ||| ||| || |||| ||||||| and |||| |||||| |||| ||| ||| || |||| ||||||| in phase I and phase II, cohort A, respectively. In the FAS, the estimated 9-month OS probability |||| |||||| |||| ||| ||| || |||| ||||||||||| |||||| |, the 12-month OS probability was |||| |||||| |||| ||| ||| || |||| ||||||||||| |||||| |||| ||| ||| || |||| |||||||, and the 24-month OS probability was |||| |||||| |||| ||| ||| || |||| ||||||||||| |||||| |||| ||| ||| || |||| |||||||. The Kaplan-Meier curve of OS for patients in the FAS is presented in Figure 4.
Progression-Free Survival
At the time of analysis, using the January 4, 2023, data cut-off, the estimated median PFS was |||| |||||| |||| ||| ||| || |||| ||||||| in phase I, and |||| |||||| |||| ||| ||| || |||| ||||||| in phase II, cohort A. In the FAS, the estimated 9-month PFS probability was ||||| |||| ||| ||||||| ||||||, the 12-month PFS probability was ||||| |||| ||| ||||| || ||||||, and the 24-month PFS probability was ||||| |||| ||| ||||| || ||||||. The Kaplan-Meier curve for PFS for patients in the FAS is provided in Figure 5.
Figure 4: Kaplan-Meier Plot for Overall Survival, Full Analysis Set
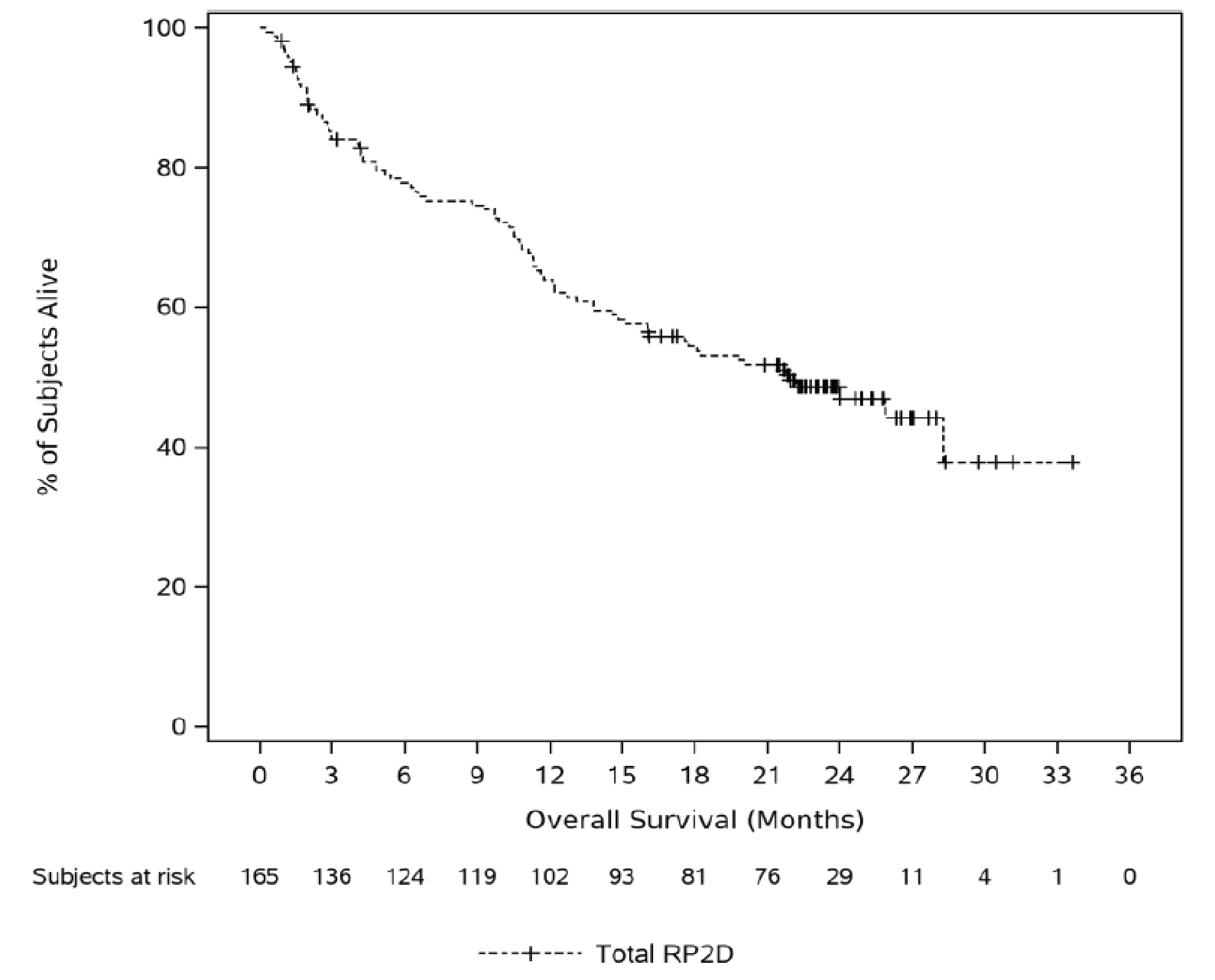
RP2D = recommended phase II dose.
Source: Clinical Study Report for the MajesTEC-1 study (2021).24
Figure 5: Kaplan-Meier Plot for Progression-Free Survival, Full Analysis Set
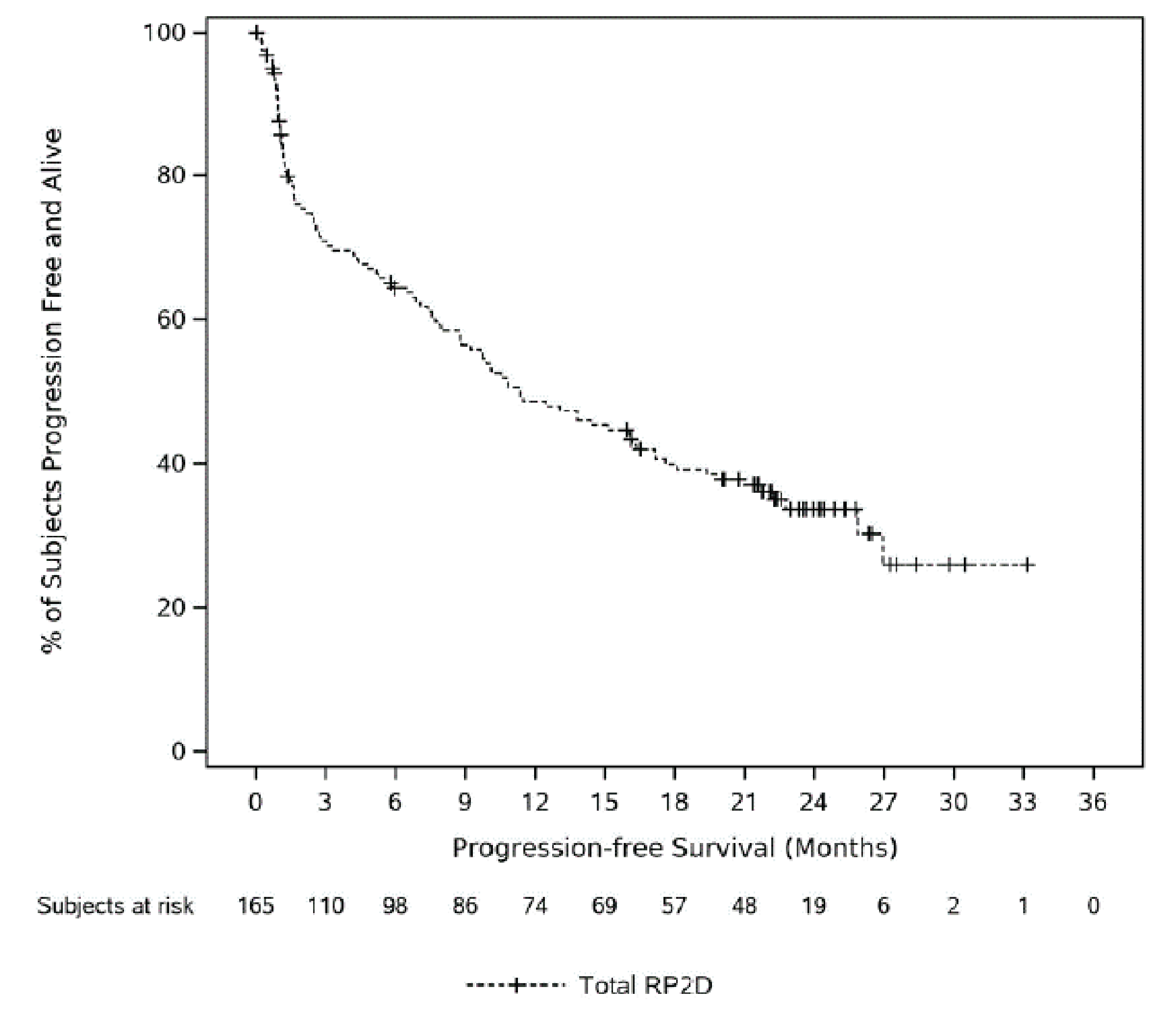
RP2D = recommended phase II dose.
Source: Clinical Study Report for the MajesTEC-1 study (2021).24
CR or Better
At the time of analysis, using the January 4, 2023, data cut-off, || |||||||| ||||||| |||| ||| ||||| || ||||||| in phase I and || |||||||| ||||||| |||| ||| ||||||| ||||||| in phase II, cohort A, attained CR or better (CR or sCR) (Figure 6).
sCR
At the time of analysis, using the January 4, 2023, data cut-off, || |||||||| ||||||| |||| ||| ||||| || ||||||| in phase I and || |||||||| ||||||| |||| ||| ||||||||||||| in phase II, cohort A, attained sCR (Figure 6).
Figure 6: Response Rate and Depth of Response, Full Analysis Set
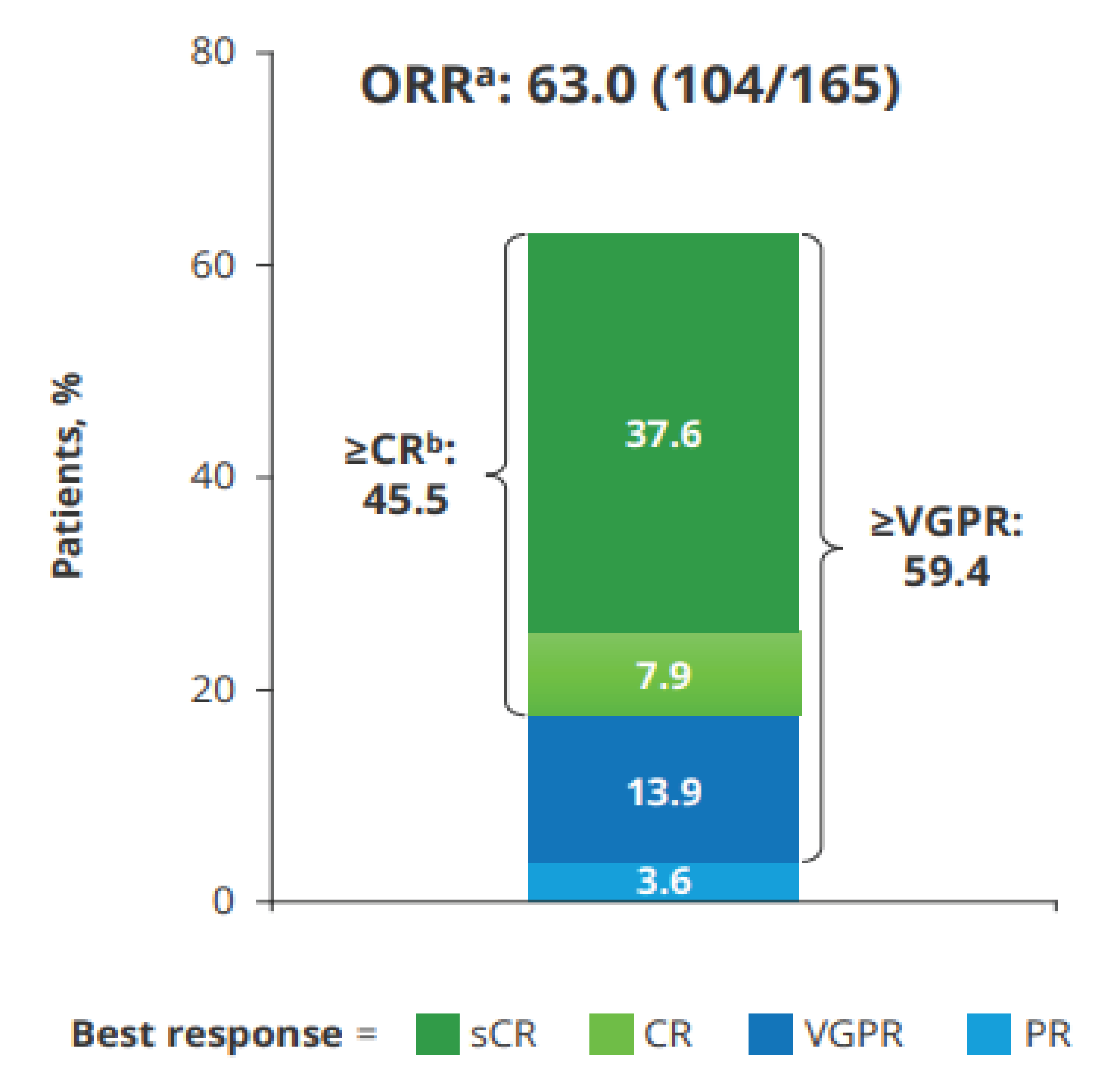
CR = complete response; IMWG = International Myeloma Working Group; IRC = independent review committee; PR = partial response; ORR = overall response rate; sCR = stringent complete response; VGPR = very good partial response.
a ORR is defined as the proportion of patients who attain PR or better (i.e., PR, VGPR, CR, or sCR) according to the IMWG response criteria,12,63 during or after study intervention but before the start of subsequent antimyeloma therapy, as assessed by the IRC.
b CR or better is defined as the proportion of patients achieving CR or sCR according to the IMWG response criteria,12,63 during or after the study intervention but before the start of subsequent antimyeloma therapy, as assessed by the IRC.
Source: Clinical Study Report for the MajesTEC-1 study (2021).24
MRD-Negativity Status
Updated data regarding the MRD-negativity status based on the January 4, 2023, clinical cut-off date are not available.
At the time of the data cut-off date of September 7, 2021, 37 (24.7%) patients (95% CI, 18.0% to 32.4%) had attained MRD negativity at 10–5 bone marrow cells. Among 43 patients who attained CR or better, 18 (41.9%) patients (95% CI, 27.0% to 57.9%) attained MRD negativity at 10–5 bone marrow cells.
VGPR or Better
At the time of analysis, using the January 4, 2023, data cut-off, || |||||||| ||||||| |||| ||| ||||| || ||||||| in phase I and || |||||||| ||||||| |||| ||| ||||||| ||||||| in phase II, cohort A, attained VGPR or better (VGPR, CR, or sCR) (Figure 6).
Overall Response Rate
In the MajesTEC-1 trial, ORR was the primary outcome for phase II, cohort A, and the secondary outcome for phase I.
At the time of analysis, using the January 4, 2023, data cut-off, 104 (63.0%) patients (95% CI, 55.2% to 70.4%) attained an overall response (PR or better), and ORR was similar across patients treated in phase I and phase II, cohort A ||||||| |||| ||| ||||| |||||||| ||| |||||| |||| ||| ||||| || ||||||, respectively) (Figure 6).
Of the 104 responders (who attained PR or better), 51 (49.0%) patients maintained their response until the clinical cut-off date |||||| || |||||||| ||||| || ||||||||||| ||, including 46 (44.2%) patients who were still on treatment (Table 18). Of 104 responders, || |||||||| ||||||| had disease progression after initial response, of whom || ||||||| ||||||| died after disease progression, || |||||||| ||||||| discontinued the study treatment, and ||||||| |||||| remained on study treatment. A total of 19 (18.3%) patients died after achieving response and without experiencing disease progression, and ||||||| |||||| had subsequent therapy after response and without progressive disease.
Table 18: Summary of Responders in MajesTEC-1 Study, Full Analysis Set [Redacted]
Detail | MajesTEC-1 study | ||
|---|---|---|---|
Phase I N = 26 | Phase II, cohort A N = 78 | Total N = 104 | |
|||||||| ||| |||||||||| |||||||| ||||| ||||| | || |||||| | || |||||| | || |||||| |
||||| |||||||||| |||| ||||| | || |||||| | || |||||| | || |||||| |
||||||| ||| ||| || ||||| ||||||||||||||| ||||| | || |||||| | || |||||| | || |||||| |
|||||||| ||||||| || ||| ||| |||| ||||| |||||||||||| ||||| | || |||||| | || |||||| | || |||||| |
|||||||| ||||||| || ||| ||| |||||||||| ||||||| ||||| |||||||||||| ||||| | || |||||| | || |||||| | || |||||| |
|||||||| ||||||| || ||| |||| |||||||||||| ||| |||||||||| |||||||| | || |||||| | || |||||| | || |||||| |
CCO = clinical cut-off; PD = progressive disease.
aIncludes patients who had not progressed, died, started subsequent therapy, or ended study until the CCO date of January 4, 2023.
bIncludes patients who died without PD being assessed (including deaths after subsequent therapy).
cIncludes patients who did not have PD assessed.
Source: Clinical Study Report for the MajesTEC-1 study (2021).24
Of the 63 responders who changed dosage schedule, || |||||||| ||||||| |||||||||| ||||| |||||||| ||||| ||| |||||||| ||||||| ||||| ||||||||| || |||||||| ||||||| ||| |||||| ||||||||||| (Table 19). |||| || || |||||||| ||||||| ||| ||||||| ||||||||||| ||||| ||||||| ||||||||| || ||||||||||| |||||| |||| ||||| ||||||| |||||||||||||||||||| ||||||| |||||||||||| ||| ||||| |||||||||| |||||||||| |||||| |||||| || ||||| ||||||||||||||| ||||||||| |||||| |||| ||||| ||||||||| |||||||| ||| ||||||| |||||||||||| ||||||| |||||||||||| ||||||||| |||||| ||| |||||||||| ||||||| ||||| |||||||| ||| ||||||||||||||||| ||||||||
Table 19: Summary of Responders With Schedule Change in MajesTEC-1 Study, Full Analysis Set [Redacted]
Detail | MajesTEC-1 study | ||
|---|---|---|---|
Phase I N = 22 | Phase II, cohort A N = 41 | Total N = 63 | |
|||||||| ||| |||||||||| |||||||| ||||| |||||| | || ||||||| | || ||||||| | || ||||||| |
||||| |||||||||| ||||||||||||||||||| | || ||||||| | || ||||||| | || ||||||| |
||||||| ||| ||| || ||||| |||||||||||||||||||| | || ||||||| | || ||||||| | || ||||||| |
|||||||| ||||||| || ||| ||| |||| ||||| |||||||||||||||| | || ||||||| | || ||||||| | || ||||||| |
|||||||| ||||||| || ||| ||| |||||||||| ||||||| ||||| |||||||||||||||| | || ||||||| | || ||||||| | || ||||||| |
|||||||| ||||||| || ||| |||| |||||||||||| ||| ||||||||| | || ||||||| | || ||||||| | || ||||||| |
CCO = clinical cut-off; IRC = independent review committee; PD = progressive disease.
aBased on time from first dosage schedule to last dose date, or disease progression as assessed by the IRC.
Source: Clinical Study Report for the MajesTEC-1 study (2021).24
Subgroup Analysis
Only results of the ORR subgroup analyses that were deemed clinically meaningful by the clinical experts consulted by CADTH for this review are reported.
At the time of analysis, using the January 4, 2023, data cut-off, 32 patients of 43 (74.4%) patients who received 3 or less prior lines of therapy attained an overall response. Of the 122 patients who received more than 3 prior lines of therapy, 72 (59.0%) patients attained overall response, and 32 of 60 (53.3%) patients with high cytogenetic risk and/or extramedullary disease attained overall response.
At the time of analysis, using the September 7, 2021, data cut-off, ORR was 60.8% (73 of 120) of patients, 62.1% (72 of 116) of patients, 61.4% (27 of 44) of patients, and 59.7% (80 of 134) of patients in those who were refractory to PI and IMiD, triple-refractory, penta-refractory, and refractory to last line of therapy, respectively.
Time to Response
At the time of analysis, using the January 4, 2023, data cut-off, of 104 patients who attained PR or better, the median time to first response was 0.9 months (range = 0.2 month to 2.3 months) while the median time to best response was 3.6 months (range = 1.7 months to 18.7 months). Most patients demonstrated their first response rapidly, by the start of cycle 2 (Figure 7).
Figure 7: Response and Follow-up in MajesTEC-1 Study per IRC Assessment, Full Analysis Set
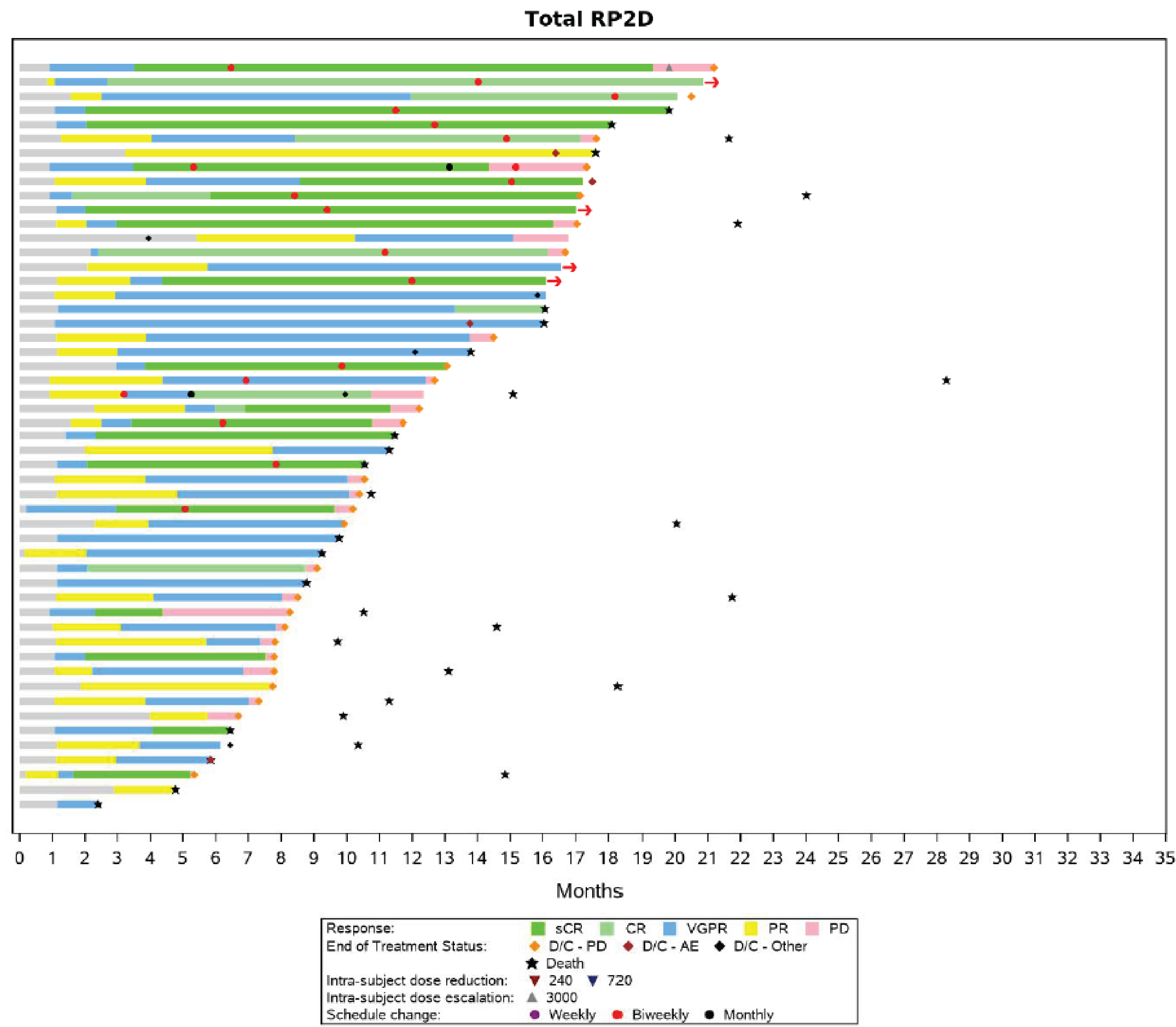
AE = adverse event; CR = complete response; D/C = discontinued; IRC = independent review committee; PD = progressive disease; PR = partial response; RP2D = recommended phase II dose; sCR = stringent complete response; VGPR = very good partial response.
Source: Clinical Study Report for the MajesTEC-1 study (2021).24
Duration of Response
At the time of analysis, using the January 4, 2023, data cut-off, with a median duration of follow-up of 22.8 months, the median duration of response was |||| |||||| |||| ||| |||| || || ||||||| in phase I and |||| |||||| |||| ||| |||| || || ||||||| in phase II, cohort A. Among 104 responders (with a PR or better response), || |||||||| ||||||| in phase I and || |||||||| ||||||| in phase II, cohort A, had disease progression or died due to any cause. The probability of patients remaining in response at 9 months was ||||| |||| ||| ||||| || ||||||| The probability of patients remaining in response at 18 months was ||||| |||| ||| ||||| || ||||||| The probability of patients remaining in response at 24 months was ||||| |||| ||| ||||| || |||||||
Table 20: Summary of Key Efficacy Results From MajesTEC-1 Study, Full Analysis Set
Variable | MajesTEC-1 study | ||
|---|---|---|---|
Phase I N = 40 | Phase II, cohort A N = 125 | Total N = 165 | |
Median follow-up time, months (range) | |||| ||||| || ||||| | |||| ||||| || ||||| | 22.8 (0.3 to 33.6) |
OS per IRC assessment | |||
OS events, n (%) | || |||||| | || |||||| | 84 (50.9) |
Primary cause of death, n (%) | |||
Adverse event | || ||||||| | || ||||||| | 25 (15.2) |
Disease progression | || ||||||| | || ||||||| | 48 (29.1) |
Other | || ||||||| | || ||||||| | 11 (6.7) |
Censored, n (%) | || ||||||| | || ||||||| | 81 (49.1) |
Mediana OS (95% CI), months | |||| ||||| || ||| | |||| ||||| || ||| | 21.9 (15.1 to NE) |
6-month OS probability,a % (95% CI) | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
9-month OS probability,a % (95% CI) | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
12-month OS probability,a % (95% CI) | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
18-month OS probability,a % (95% CI) | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
24-month OS probability,a % (95% CI) | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
PFS per IRC assessment | |||
PFS events, n (%) | || |||||| | || |||||| | ||| |||||| |
Progressive disease, n (%) | || |||||| | || |||||| | || |||||| |
Serum M protein | || ||||||| | || ||||||| | || ||||||| |
Urine M protein | || ||||||| | || ||||||| | || ||||||| |
Serum FLC | || ||||||| | || ||||||| | || ||||||| |
Bone lesion (increase in size) | || ||||||| | || ||||||| | || ||||||| |
Bone lesion (new) | || ||||||| | || ||||||| | || ||||||| |
Extramedullary plasmacytomas (increase in size) | || ||||||| | || ||||||| | || ||||||| |
Extramedullary plasmacytomas (new) | || ||||||| | || ||||||| | || ||||||| |
Circulating plasma cells (increase) | || ||||||| | || ||||||| | || ||||||| |
Without progressive disease, n (%) | || ||||||| | || ||||||| | || ||||||| |
Censored, n (%) | || ||||||| | || ||||||| | || ||||||| |
Reason for censoring, n (%) | |||
Data cut-off (ongoing) | || |||||| | || |||||| | || |||||| |
Subsequent therapy | || ||||||| | || ||||||| | || ||||||| |
Patient withdrawal | || ||||||| | || ||||||| | || ||||||| |
Mediana PFS (95% CI), months | |||| |||| || ||||| | |||| |||| || ||||| | 11.3 (8.8 to 16.4) |
6-month PFS probability,a % (95% CI) | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
9-month PFS probability,a % (95% CI) | |||| ||||| || ||||| | |||| |||||| ||||| | |||| ||||| || ||||| |
12-month PFS probability,a % (95% CI) | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
18-month PFS probability,a % (95% CI) | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
24-month PFS probability,a % (95% CI) | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
Patients who attained CR or better (CR or sCR) per IRC assessment | |||
n (%) | || |||||| | || |||||| | 75 (45.5) |
95% CI | |||| || |||| | |||| || |||| | 37.7 to 53.4 |
MRD-negativity statusb per IRC assessment | |||
Number of patients contributing to the analysis, n | redact | redact | 150 |
MRD-negativity rate (10–5), n (%) | || ||||||| | || ||||||| | 37 (24.7) |
95% CI | || ||||||| | || ||||||| | 18.0 to 32.4 |
Patients who attained VGPR or better (VGPR, CR, or sCR) per IRC assessment | |||
n (%) | || |||||| | || |||||| | 98 (59.4) |
95% CI | |||| || |||| | |||| || |||| | 51.5 to 67.0 |
Patients who attained overall response (PR, VGPR, CR, or sCR) per IRC assessment | |||
n (%) | || |||||| | || |||||| | 104 (63.0) |
95% CI | |||| || |||| | |||| || |||| | 55.2 to 70.4 |
Response category | |||
Patients who attained sCR | |||
n (%) | || |||||| | || |||||| | 62 (37.6) |
95% CI | |||| || |||| | |||| ||| |||| | 30.2 to 45.4 |
Patients who attained CR | |||
n (%) | || ||||||| | || ||||| | 13 (7.9) |
95% CI | ||| || |||| | ||| || |||| | 4.3 to 13.1 |
Patients who attained VPGR | |||
n (%) | || ||||||| | || |||||| | 23 (13.9) |
95% CI | || ||||||| | ||| || |||| | 9.0 to 20.2 |
Patients who attained PR | |||
n (%) | || ||||||| | || ||||||| | 6 (3.6) |
95% CI | || ||||||| | || ||||||| | 1.3 to 7.7 |
Patients who attained minimal response | |||
n (%) | || ||||||| | || ||||||| | 2 (1.2) |
95% CI | || ||||||| | ||| || ||| | 0.1 to 4.3 |
Stable disease | |||
n (%) | || ||||||| | || |||||| | 28 (17.0) |
95% CI | ||| || |||| | |||| || |||| | 11.6 to 23.6 |
Progressive disease | |||
n (%) | || ||||||| | || |||||| | 23 (13.9) |
95% CI | ||| ||| |||| | ||| || |||| | 9.0 to 20.2 |
NE | || ||||||| | || ||||||| | 8 (4.8) |
Time to response per IRC assessment | |||
Time to first response, monthsc | |||
Number of patients contributing to the analysis, n | || | || | 104 |
Mean (SD) | ||| ||||| | ||| ||||| | 1.5 (0.9) |
Median (range) | ||| |||| || |||| | ||| |||| || |||| | 1.2 (0.2 to 5.5) |
Time to best response, monthsc | |||
Number of patients contributing to the analysis, n | || | || | 104 |
Mean (SD) | ||| ||||| | ||| ||||| | 6.1 (4.7) |
Median (range) | ||| |||| || ||||| | ||| |||| || ||||| | 4.0 (1.1 to 18.7) |
Time to VGPR or better, months | |||
Number of patients contributing to the analysis, n | || | || | || |
Mean (SD) | ||| ||||| | ||| ||||| | ||| ||||| |
Median (range) | ||| |||| || |||| | ||| |||| || ||||| | ||| |||| || ||||| |
Time to CR or better, months | |||
Number of patients contributing to the analysis, n | || | || | || |
Mean (SD) | ||| ||||| | ||| ||||| | ||| ||||| |
Median (range) | ||| |||| || ||||| | ||| |||| || ||||| | ||| |||| || ||||| |
DoR per IRC assessment | |||
Number of patients contributing to the analysis, n | || | || | 104 |
Events,d n (%) | || |||||| | || |||||| | 50 (48.1) |
Censored, (n %) | || |||||| | || |||||| | 54 (51.9) |
Mediana DoR (95% CI), months | |||| ||||| || ||| | |||| ||||| || ||| | 21.6 (16.2 to NE) |
6-month event-free probability,a % (95% CI) | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
9-month event-free probability,a % (95% CI) | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
12-month event-free probability,a % (95% CI) | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
18-month event-free probability,a % (95% CI) | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
24-month event-free probability,a % (95% CI) | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
CI = confidence interval; CR = complete response; DoR = duration of response; IRC = independent review committee; MRD = minimal residual disease; NE = not evaluable; OS = overall survival; PFS = progression-free survival; PR = partial response; sCR = stringent complete response; SD = standard deviation; VGPR = very good partial response.
Note: Details included in Table 20 are from the sponsor’s Summary of Clinical Evidence.83
aKaplan-Meier estimate.
bThe MRD-negativity rate was reported as of the clinical data cut-off date of September 7, 2021.
cPR or better.
dThe number of events refers to the number of responders (PR or better) who developed disease progression or died due to any cause. DoR was calculated as the number of months from the first documented response to progression, death due to any cause, or the date of censoring.
Source: Clinical Study Report for the MajesTEC-1 study (2021).24
Patient-Reported Outcomes
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30: The results of a post hoc analysis for mean change from baseline in EORTC QLQ-C30 scales, including global health status, fatigue, pain score, and physical functioning, are presented in Table 21. Analyses were conducted in the HRQoL-evaluable population of patients who had evaluable assessment at baseline and follow-up time points for each domain of EORTC QLQ-C30 (i.e., cycle 2, day 1; cycle 3, day 1; and so forth). At the time of analysis, using the January 4, 2023, data cut-off, the EORTC QLQ-C30 compliance rate (defined as the proportion of evaluable forms of all expected forms) at baseline was ||||| |||||||| |||||||||| ||||| |||||||||||||| || ||||| || ||||| |||||| ||||||||| || ||||| ||| ||| ||||| |||||| ||||||||| || ||||| ||. The EORTC QLQ-C30 showed improvements over time from baseline on pain and fatigue system scale, improvements from cycle 4 onward on global health status, and improvements with the decrease initially observed in cycle 2 on physical functioning.
At the time of analysis, using the January 4, 2023, data cut-off, the least squares mean (LSM) of change from baseline in global health status was ||| |||| ||| ||| || ||||| || ||||| || ||| |||| ||| ||| || ||||| || ||||| ||| ||| |||| |||| ||| ||| || ||||| || ||||| ||. The LSM of change from baseline in fatigue symptom scale was ||| |||| ||| |||| || |||| || ||||| || |||| |||| ||| ||||| || ||||| || ||||| ||| ||| |||| |||||| || ||| || ||||| ||. The LSM of change from baseline in pain was ||||| |||| ||| ||| || ||||| || ||||| || ||| |||| ||| ||||| || |||||| || ||||| ||, and ||||| |||| ||| ||| || |||||| || ||||| ||. The LSM of change from baseline in physical functioning was |||| |||| ||| |||| || |||| || ||||| || ||| |||| ||| |||| || |||| || ||||| ||| ||| ||| |||| ||| |||| || ||||| || ||||| ||.
Table 21: Summary of Selected Scales for EORTC QLQ-C30 Assessment and Change From Baseline (Phase II, Cohort A) [Redacted]
Characteristic | Phase II, cohort A (N = 125) | |||||
|---|---|---|---|---|---|---|
Measured value | Change from baseline | |||||
N | Mean (SD) | Base mean | N | Mean (SD) | LSM (95% CI) | |
|||||| |||||| |||||| | ||||||
|||||||| | ||| | ||||| ||||||| | ||||| | || | |||| ||||||| | ||| ||||| || |||| |
|||||||||||| | || | ||||| ||||||| | ||||| | || | |||| ||||||| | ||| ||||| || |||| |
|||||||||||| | || | ||||| ||||||| | ||||| | || | |||| ||||||| | ||| |||| || ||||| |
|||||||||||| | || | ||||| ||||||| | ||||| | || | |||| ||||||| | ||| |||| || ||||| |
|||||||||||| | || | ||||| ||||||| | ||||| | || | |||| ||||||| | ||| |||| || ||||| |
|||||||||||| | || | ||||| ||||||| | ||||| | || | ||||| ||||||| | |||| |||| || ||||| |
|||||||||||| | || | ||||| ||||||| | ||||| | || | ||||| ||||||| | ||| |||| || ||||| |
|||||||||||| | || | ||||| ||||||| | ||||| | || | ||||| ||||||| | |||| |||| || ||||| |
|||||||||||| | || | |||| ||||||| | ||||| | || | ||||| ||||||| | |||| |||| || ||||| |
||| || ||||||||| ||||| | || | ||||| ||||||| | ||||| | || | ||||| ||||||| | |||| |||||| || ||||| |
||||||| ||||||| ||||| | ||||||
|||||||| | ||| | ||||| ||||||| | ||||| | || | |||| ||||||| | ||| ||||| || |||| |
|||||||||||| | || | ||||| ||||||| | ||||| | || | |||| ||||||| | ||| |||| || ||||| |
|||||||||||| | || | ||||| ||||||| | ||||| | || | ||||| ||||||| | ||| ||||| || || |
|||||||||||| | || | ||||| ||||||| | ||||| | || | ||||| ||||||| | ||| ||||| || |||| |
|||||||||||| | || | ||||| ||||||| | ||||| | || | ||||| ||||||| | |||| ||||| || |||| |
|||||||||||| | || | ||||| ||||||| | ||||| | || | ||||| ||||||| | |||| ||||| || |||| |
|||||||||||| | || | ||||| ||||||| | ||||| | || | |||||| ||||||| | |||| |||||| || ||||| |
|||||||||||| | || | ||||| ||||||| | ||||| | || | |||||| ||||||| | || |||||| || |||| |
|||||||||||| | || | ||||| ||||||| | ||||| | || | ||||| ||||||| | |||| |||||| || ||| |
||| || ||||||||| ||||| | || | ||||| ||||||| | ||||| | || | ||||| ||||||| | |||| |||| || ||||| |
|||| ||||| | ||||||
|||||||| | ||| | ||||| ||||||| | ||||| | || | ||||| ||||||| | |||| ||||| || |||| |
|||||||||||| | || | ||||| ||||||| | ||||| | || | ||||| ||||||| | |||| ||||| || |||| |
|||||||||||| | || | ||||| ||||||| | ||||| | || | |||||| ||||||| | |||| |||||| || ||||| |
|||||||||||| | || | ||||| ||||||| | ||||| | || | |||||| ||||||| | ||||| |||| || ||||| |
|||||||||||| | || | ||||| ||||||| | ||||| | || | |||||| ||||||| | ||||| |||||| || ||||| |
|||||||||||| | || | ||||| ||||||| | ||||| | || | |||||| ||||||| | ||||| |||||| || ||||| |
|||||||||||| | || | ||||| ||||||| | ||||| | || | |||||| ||||||| | ||| |||||| || |||||| |
|||||||||||| | || | ||||| ||||||| | ||||| | || | |||||| ||||||| | ||||| |||| || |||| |
|||||||||||| | || | ||||| ||||||| | ||||| | || | |||||| ||||||| | ||||| |||| || |||||| |
||| || ||||||||| ||||| | || | ||||| ||||||| | ||||| | || | |||| ||||||| | |||| |||| || ||||| |
|||||||| ||||||||||| | ||||||
|||||||| | ||| | ||||| ||||||| | ||||| | || | ||||| ||||||| | |||| ||||| || |||| |
|||||||||||| | || | ||||| ||||||| | ||||| | || | ||||| ||||||| | |||| |||||| || ||||| |
|||||||||||| | || | ||||| ||||||| | ||||| | || | |||| ||||||| | |||| ||||| || |||| |
|||||||||||| | || | ||||| ||||||| | ||||| | || | |||| ||||||| | |||| ||||| || |||| |
|||||||||||| | || | ||||| ||||||| | ||||| | || | |||| ||||||| | |||| ||||| || |||| |
|||||||||||| | || | ||||| ||||||| | ||||| | || | |||| ||||||| | |||| ||||| || |||| |
|||||||||||| | || | ||||| ||||||| | ||||| | || | |||| ||||||| | |||| ||||| || |||| |
|||||||||||| | || | ||||| ||||||| | ||||| | || | ||||| ||||||| | |||| ||||| || |||| |
|||||||||||| | || | ||||| ||||||| | ||||| | || | |||| ||||||| | |||| ||||| || |||| |
||| || ||||||||| ||||| | || | ||||| ||||||| | ||||| | || | ||||| ||||||| | |||| ||||| || |||| |
CI = confidence interval; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; LSM = least squares mean; SD = standard deviation.
Note: All the scores are presented in the range of 0 to 100 after linear transformation from raw scores (in the range of 1 to 4). A higher score indicates better health on the global health and functional scales (physical, role, emotional, cognitive, and social) and greater symptom severity on the symptom scales (fatigue, nausea and vomiting, pain, dyspnea, sleep disturbance, appetite loss, constipation, and diarrhea).
Source: Sponsor-submitted additional information.83
At the time of analysis, using the September 7, 2021, data cut-off, meaningful improvement from baseline (10 points using the literature-based MCT14) to cycle 2, cycle 4, and cycle 6 (i.e., the first 6 months) was reported by up to 35.8% of patients for global health status, up to 23.9% of patients for physical functioning, up to 68.7% of patients for fatigue system scale, and up to 78.8% of patients for pain.
EQ-5D-5L: The results of post hoc analysis for mean change from baseline in EQ-5D-5L scores, including utility score and EQ VAS, are presented in Table 22. At the time of analysis, using the January 4, 2023, data cut-off, the EQ-5D-5L compliance rate (defined as the proportion of evaluable forms of all expected forms) at baseline was ||||||||||| ||||||||| || ||||| || ||||| |||||| ||||||||| || |||||| ||||| |||||| ||||||||| || ||||| ||| ||| ||||| |||||| ||||||||| || ||||| ||. EQ-5D-5L showed improvements over time from baseline on utility scores and EQ VAS from cycle 4 onward.
Table 22: Summary of EQ-5D-5L Assessment and Change From Baseline (Phase II, Cohort A), Full Analysis Set [Redacted]
Characteristic | Phase II, cohort A (N = 125) | |||||
|---|---|---|---|---|---|---|
Measured value | Change from baseline | |||||
N | Mean (SD) | Base mean | N | Mean (SD) | LSM (95% CI) | |
|||||||| ||||||| ||||| | ||||||
||||| || ||| | || | |||| |||||| | |||| | || | |||| |||||| | ||| || || |||| |
||||| || ||| | || | |||| |||||| | |||| | || | |||| |||||| | ||| || || |||| |
||||| || ||| | || | |||| |||||| | |||| | || | |||| |||||| | ||| || || |||| |
||||| || ||| | || | |||| |||||| | |||| | || | |||| |||||| | ||| || || |||| |
||||| || ||| | || | |||| |||||| | |||| | || | |||| |||||| | ||| |||| || |||||| |
||||| || ||| | || | |||| |||||| | |||| | || | |||| |||||| | ||| |||| || |||| |
||||| || ||| | || | |||| |||||| | |||| | || | |||| |||||| | ||| || || |||| |
||||| || ||| | || | |||| |||||| | |||| | || | |||| |||||| | ||| |||| || |||| |
||||| || ||| | || | |||| |||||| | |||| | || | |||| |||||| | ||| |||| || |||| |
||| || ||||||||| ||||| | || | |||| |||||| | |||| | || | ||||| |||||| | |||| ||||| || ||||| |
|||||||| ||| | ||||||
||||| || ||| | || | ||||| ||||||| | ||||| | || | ||||| ||||||| | |||| ||||| || |||| |
||||| || ||| | || | ||||| ||||||| | ||||| | || | ||||| ||||||| | |||| ||||| || |||| |
||||| || ||| | || | ||||| ||||||| | ||||| | || | |||| ||||||| | ||| |||| || ||||| |
||||| || ||| | || | ||||| ||||||| | ||||| | || | |||| ||||||| | ||| |||| || ||||| |
||||| || ||| | || | ||||| ||||||| | ||||| | || | ||||| ||||||| | |||| |||| || ||||| |
||||| || ||| | || | ||||| ||||||| | ||||| | || | ||||| ||||||| | |||| |||| || ||||| |
||||| || ||| | || | ||||| ||||||| | ||||| | || | ||||| ||||||| | || |||| || ||||| |
||||| || ||| | || | ||||| ||||||| | ||||| | || | ||||| ||||||| | |||| |||| || ||||| |
||||| || ||| | || | ||||| ||||||| | ||||| | || | ||||| ||||||| | |||| |||| || ||||| |
||| || ||||||||| ||||| | || | ||||| ||||||| | ||||| | || | ||||| ||||||| | ||||| |||||| || ||||| |
CI = confidence interval; EQ VAS = EQ visual analogue scale; LSM = least squares mean; SD = standard deviation.
Notes: EQ VAS data (health today) are presented in the range of 0 to 100, with a high score indicating a high level of self-evaluated health status.
Utility score data are presented in the range of 0 to 1 (but this allows for values less than 0 by UK scoring algorithm), with a high score indicating a high level of utility.
Source: Sponsor-submitted additional information.83
At the time of analysis, using the September 7, 2021, data cut-off, meaningful improvement from baseline (7 points using the literature-based MCT15,16) in EQ VAS scores at cycle 2, cycle 4, and cycle 6 was reported by 23.8%, 28.6%, and 30.2% of patients, respectively. By cycle 8, 50% of patients had reported meaningful improvement in the EQ VAS score.
Patient Global Impression–Severity: At the time of analysis, using the September 7, 2021, data cut-off, patient compliance with the PGI-S assessment was 92.7% at baseline and ranged from 86.2% to 94.4% from cycle 2 to cycle 6. A total of 25.9%, 47.7%, and 55.4% of patients reported no or mild disease severity at cycle 2, cycle 4, and cycle 6, respectively.
Time to Next Treatment
TTNT was an exploratory end point in phase II, cohort A, of the MajesTEC-1 study, and it was not reported in the Clinical Study Report at the clinical data cut-off date of January 4, 2023.
At the time of analysis, using the September 7, 2021, data cut-off, subsequent antimyeloma therapy and/or death due to progressive disease was reported for || |||||||| |||||||, with a median TTNT of |||| |||||| |||| ||| |||| || || |||||||.
Harms
Harms data for the MajesTEC-1 trial are summarized in Table 23.
Adverse Events
At the time of analysis, using the January 4, 2023, data cut-off, all patients in the MajesTEC-1 study had experienced at least 1 TEAE. The most common TEAEs occurring in at least 25% of patients in either phase of the study were CRS (72.1%), neutropenia (71.5%), anemia (54.5%), thrombocytopenia (42.4%), lymphopenia (36.4%), diarrhea (33.9%), and pyrexia (31.5%). In the MajesTEC-1 study, || |||||||| ||||||| experienced TEAEs of grade 3, || |||||||| ||||||| experienced TEAEs of grade 4, and || |||||||| ||||||| experienced TEAEs of grade 5. The most common TEAEs of grade 3 or grade 4 were neutropenia (65.5%), anemia (37.6%), lymphopenia (34.5%), and thrombocytopenia (22.4%). The most common TEAEs of grade 5 were COVID-19 (10.8%) and general physical health deterioration (2.4%).
Serious Adverse Events
At the time of analysis, using the January 4, 2023, data cut-off, 113 (68.5%) patients had experienced at least 1 serious TEAE in the MajesTEC-1 trial. The most common TEAEs occurring in at least 5% of patients in either phase of the study were ||||||||||||||| ||||||||| |||||||| ||| |||||||| ||||||| ||||||| ||||| |||||| |||||| ||||||| ||| ||||||| |||||||| |||||| ||||||||||||| ||||||.
Withdrawals Due to Adverse Events
At the time of analysis, using the January 4, 2023, data cut-off, |||||||| |||||| had stopped study treatment due to TEAEs in the MajesTEC-1 trial. The most common reasons for stopping study treatment included |||||||| ||||||||| ||||||| |||||||||||| ||||||||| ||||||||| ||||||| ||||||||||| ||||||||| ||||||.
Mortality
At the time of analysis, using the January 4, 2023, data cut-off, || |||||||| ||||||| died during the study, including || |||||||| |||||| who died due to TEAEs. In the MajesTEC-1 study, || |||||||| ||||||| died within 30 days of the last study treatment dose, and || |||||||| ||||||| died within 60 days of the last study treatment dose.
Notable Harms
In the MajesTEC-1 trial, several adverse events of clinical interest were identified, including CRS, neurologic adverse events, neurotoxicity, ICANS, systemic administration-related reactions, injection-site reactions, hypogammaglobulinemia, cytopenia, and infections.
Cytokine Release Syndrome
At the time of analysis, using the January 4, 2023, data cut-off, 119 (72.1%) patients had experienced CRS events. At the time of the data cut-off date of September 7, 2021, 118 (71.5%) patients had experienced CSR events, of whom 83 (50.3%) patients had experienced grade 1 events and 35 (21.2%) patients had experienced grade 2 events. One (0.6%) patient had experienced CRS events of grade 3, and no patients had experienced CRS events of grade 4 or grade 5. There were 110 (66.7%) patients who received supportive measures to treat CRS events. The most common treatments for CRS were tocilizumab (60 [36.4%] patients), low-flow oxygen by nasal cannula (21 [12.7%] patients), and glucocorticoids (14 [8.5%] patients). A single vasopressor was administered in 1 (0.6%) patient.56 The median time from the last injection of teclistamab to new onset of CRS was 2 days (range = 1 day to 6 days), and the median duration of CRS was 2 days (range = 1 day to 9 days). All cases of CRS (100%) were effectively managed and resolved, and no patients discontinued teclistamab due to the development of a CRS event.
Neurologic Adverse Events
At the time of analysis, using the January 4, 2023, data cut-off, || |||||||| ||||||| had experienced at least 1 neurologic TEAE, and |||||||||| ||||||| had experienced at least 1 neurotoxicity TEAE. The most common neurologic TEAEs included headache (24.2%), dizziness (6.1%), and anxiety (4.8%).
Neurotoxicity
At the time of analysis, using the January 4, 2023, data cut-off, || |||||||| ||||||| had experienced at least 1 neurotoxicity TEAE in the FAS. The most common neurotoxicity TEAEs included |||||||| ||||||| |||||| |||||||| ||||||||||||||| ||||||||||||| |||||||| ||||||| ||||||||| ||||||| ||||||||||||||||||| |||||||||| ||||||| |||||||||| ||||||| ||| ||||||||| ||||||.
Infections
At the time of analysis, using the January 4, 2023, data cut-off, 132 (80.0%) patients had infections of any grade. The most common infections and infestations included |||||||| |||||||| ||||||||| |||||||| |||||||||| |||||||| ||||| ||||||||||| ||||||||| |||||||| ||||||||||||||| |||||||| ||||||||| |||||||| ||||||||| ||||| ||||||||| ||||||. In the MajesTEC-1 study, 71 (43.0%) patients had experienced at least 1 infection and infestation of grade 3 or grade 4, while 21 (12.7%) patients had experienced at least 1 infection and infestation of grade 5. A total of 18 (25.4%) patients had experienced an infection of grade 3 or higher within 12 months to 18 months after the start of treatment, 7 (13.0%) patients had experienced an infection of grade 3 or higher within 18 months to 24 months after the start of treatment, and 2 (10.5%) patients had experienced an infection of grade 3 or higher within 24 months or later after the start of treatment.
Hypogammaglobulinemia
At the time of analysis, using the January 4, 2023, data cut-off, || |||||||| ||||||| had experienced at least 1 hypogammaglobulinemia TEAE, including 34 (20.6%) patients with a case of hypogammaglobulinemia, and ||||||| |||||| with a case of hypoglobulinemia. A total of ||| |||||||| ||||||| had at least 1 episode of a postbaseline immunoglobulin G value of less than 500 mg/dL. In the MajesTEC-1 trial, 124 (75.2%) patients had a least 1 case of a hypogammaglobulinemia or postbaseline immunoglobulin G value of less than 500 mg/dL, of whom 79 (47.9%) patients received IV immunoglobulin.
Immune Effector Cell-Associated Neurotoxicity Syndrome
At the time of analysis, using the January 4, 2023, data cut-off, 5 (3.0%) patients had experienced ICANS, with 3 (1.8%) patients reporting cases of ICANS of a grade 1 event and 2 (1.2%) patients reporting cases of ICANS of a grade 3 event. No new events of ICANS were reported since the clinical cut-off date for the primary analysis for the MajesTEC-1 study (September 7, 2021). All cases of ICANS occurred relatively early in treatment, except for 1 case of a grade 1 event that occurred on cycle 3, day 8. There were 4 (2.4%) patients who received supportive treatments for ICANS, including tocilizumab (1.8%), corticosteroids (1.8%), levetiracetam (0.6%), and other (1.2%). The median time from last injection of teclistamab to new onset of ICANS was 4.0 days (range = 2 days to 5 days), and the median duration of ICANS was 3.0 days (range = 1 day to 20 days). All cases of ICANS (100%) were effectively managed and resolved without treatment discontinuation.
Cytopenia
At the time of analysis, using the January 4, 2023, data cut-off, 152 (92.1%) patients had experienced at least 1 treatment-emergent cytopenic event, including neutropenia (71.5%), anemia (55.8%), thrombocytopenia (42.4%), and lymphopenia (36.4%). Incidences of treatment-emergent cytopenic events of all grades were consistent with the primary analysis for the MajesTEC-1 study (clinical cut-off date of September 7, 2021). A total of 108 (65.5%) patients experienced treatment-emergent neutropenia of CTCAE grade 3 or higher, 62 (37.6%) patients experienced anemia of CTCAE grade 3 or higher, 37 (22.4%) patients experienced thrombocytopenia of CTCAE grade 3 or higher, and 57 (34.5%) patients experienced lymphopenia of CTCAE grade 3 or higher.
Injection-Site Reactions
At the time of analysis, using the January 4, 2023, data cut-off, 61 (37.0%) patients had experienced at least 1 case of injection-site reaction events, including 32.1% of cases of grade 1 and 4.8% of cases of CTCAE grade 2. A total of 19 (11.5%) patients received supportive measures to treat injection-site reactions, including ||||||| |||||||| ||||||| ||||||||||||| ||||||| ||| |||||||||||||| ||||| ||||||. The median time from last injection of teclistamab to new onset of injection-site reaction was 2 days (range = 1 day to 11 days), and the median duration of injection-site reaction was 5 days (range = 1 day to 328 days).
Tumour Lysis Syndrome
At the time of analysis, using the January 4, 2023, data cut-off, proportions of patients with TLS were not reported in the MajesTEC-1 study.
Table 23: Summary of Harms Results From Studies Included in the Systematic Review, Full Analysis Set
AE | MajesTEC-1 study | ||
|---|---|---|---|
Phase I (N = 40) | Phase II, cohort A (N = 125) | Total (N = 165) | |
Most common AEs | |||
Patients with ≥ 1 TEAE, n (%) | || ||||||| | ||| ||||||| | 165 (100.0) |
TEAEs in ≥ 20% in any group, n (%) | |||
CRS | || |||||| | || |||||| | 119 (72.1) |
Neutropenia | || |||||| | || |||||| | 118 (71.5) |
Anemia | || |||||| | || |||||| | 90 (54.5) |
Thrombocytopenia | || |||||| | || |||||| | 70 (42.4) |
Lymphopenia | || |||||| | || |||||| | 60 (36.4) |
Leukopenia | || |||||| | || |||||| | 33 (20.0) |
Hypogammaglobulinemia | || |||||| | || |||||| | 34 (20.6) |
Diarrhea | || |||||| | || |||||| | 56 (33.9) |
Pyrexia | || |||||| | || |||||| | 52 (31.5) |
Fatigue | || |||||| | || |||||| | 48 (29.1) |
Injection-site erythema | || |||||| | || |||||| | 43 (26.1) |
COVID-19 | || |||||| | || |||||| | 46 (27.9) |
Pneumonia | || |||||| | || |||||| | 31 (18.8) |
Nausea | || |||||| | || |||||| | 45 (27.3) |
Vomiting | || |||||| | || |||||| | 254 (15.2) |
Constipation | || |||||| | || |||||| | 36 (21.8) |
Cough | || |||||| | || |||||| | 44 (26.7) |
Arthralgia | || |||||| | || |||||| | 42 (25.5) |
Back pain | || |||||| | || |||||| | 30 (18.2) |
Headache | || |||||| | || |||||| | 40 (24.2) |
SAEs | |||
Patients with ≥ 1 SAE, n (%) | 21 (52.5) | 92 (73.6) | 113 (68.5) |
Serious TEAEs in ≥ 5% in any group, n (%) | |||
COVID-19 | || |||||| | || |||||| | || |||||| |
Pneumonia | || |||||| | || |||||| | || |||||| |
CRS | || |||||| | || |||||| | || |||||| |
Pyrexia | || |||||| | || |||||| | || |||||| |
Acute kidney injury | || |||||| | || |||||| | || |||||| |
General physical health deterioration | || |||||| | || |||||| | || |||||| |
Sepsis | || |||||| | || |||||| | || |||||| |
Maximum toxicity grade, n (%) | |||
Grade 1 | || |||||| | || |||||| | || |||||| |
Grade 2 | || |||||| | || |||||| | || |||||| |
Grade 3 | || |||||| | || |||||| | || |||||| |
Grade 4 | || |||||| | || |||||| | || |||||| |
Grade 5 | || |||||| | || |||||| | || |||||| |
TEAEs of grade 3 or grade 4 in ≥ 5% of patients in any group, n (%) | |||
Neutropenia | || |||||| | || |||||| | 108 (65.5) |
Anemia | || |||||| | || |||||| | 62 (37.6) |
Lymphopenia | || |||||| | || |||||| | 57 (34.5) |
Thrombocytopenia | || |||||| | || |||||| | 37 (22.4) |
Leukopenia | || |||||| | || |||||| | 15 (9.1) |
COVID-19 | || |||||| | || |||||| | 34 (20.6) |
Pneumonia | || |||||| | || |||||| | 22 (13.3) |
Hypophosphatemia | || |||||| | || |||||| | 11 (6.7) |
Hypertension | || |||||| | || |||||| | 10 (6.1) |
TEAEs of grade 5 in ≥ 1.5% of patients in any group, n (%) | |||
COVID-19 | || |||||| | || |||||| | 18 (10.8) |
General physical health deterioration | || |||||| | || |||||| | 4 (2.4) |
Respiratory distress | || |||||| | || |||||| | 2 (1.3) |
TEAEs leading to discontinuation of study drug | |||
Patients who stopped, n (%) | || |||||| | || |||||| | 8 (4.8) |
COVID-19 | || |||||| | || |||||| | 2 (1.2) |
Pneumocystis jirovecii pneumonia | || |||||| | || |||||| | 1 (0.6)a |
Pneumonia (adenoviral) | || |||||| | || |||||| | 1 (0.6)a |
Progressive multifocal leukoencephalopathy | || |||||| | || |||||| | 1 (0.6) |
Sepsis | || |||||| | || |||||| | 1 (0.6) |
Arthralgia | || |||||| | || |||||| | 1 (0.6) |
Arthritis | || |||||| | || |||||| | 1 (0.6) |
Brain neoplasm | || |||||| | || |||||| | 1 (0.6) |
Deaths | |||
Patients who died, n (%) | || |||||| | || |||||| | 84 (50.9) |
Patients who died due to TEAE, n (%) | || |||||| | || |||||| | 34 (20.6) |
Death due to COVID-19 | || |||||| | || |||||| | 18 (10.9) |
|||||||| ||| |||| |||||| || |||| || |||| ||||| ||||||||| |||||||| | || |||||| | || |||||| | || |||||| |
|||||||| ||| |||| |||||| || |||| || |||| ||||| ||||||||| |||||||| | || |||||| | || |||||| | || |||||| |
TEAEs leading to dose reduction | |||
≥ 1 TEAEs leading to dose reduction, n (%) | || |||||| | || |||||| | 1 (0.6) |
Neutropenia | || |||||| | || |||||| | 1 (0.6) |
AEs of special interest, n (%) | |||
CRS | || |||||| | || |||||| | 119 (72.1) |
Neurologic AEs | || |||||| | || |||||| | 94 (57.0) |
Neurotoxicity | || |||||| | || |||||| | 27 (16.4) |
Infections | || |||||| | || |||||| | 132 (80.0) |
Cytopenias | || |||||| | || |||||| | 152 (92.1) |
Hypogammaglobulinemia | || |||||| | || |||||| | 35 (21.2) |
ICANS | || |||||| | || |||||| | 5 (3.0) |
Injection-site reactions | || |||||| | || |||||| | 61 (37.0) |
AE = adverse event; CRS = cytokine release syndrome; ICANS = immune effector cell-associated neurotoxicity syndrome; SAE = serious adverse event; sARR = systemic administration-related reaction; TEAE = treatment-emergent adverse event.
Note: Details included in Table 23 are from the sponsor’s Summary of Clinical Evidence.83
aPneumocystis jirovecii pneumonia and pneumonia (adenoviral) occurred in the same patient.
Source: Clinical Study Report for the MajesTEC-1 study (2021).24
Additional Supporting Data
In this section, efficacy and safety results for phase II, cohort C, were presented to support the results of the pivotal study in accordance with the Health Canada indication for teclistamab and to address the question from the Provincial Advisory Group regarding the use of teclistamab in patients previously treated with BCMA-targeted therapy. Phase II, cohort C, enrolled patients with RRMM who had received 3 or more prior lines of therapy, including a PI, an IMiD, an anti-38 monoclonal antibody, and an anti-BCMA treatment (e.g., BCMA-targeted CAR T-cell therapy or ADC).
The Clinical Study Report with a data cut-off date of March 16, 2022,25 was the data source used for presenting all efficacy and safety end points for phase II, cohort C, of the MajesTEC-1 study.
At the time of analysis, using the March 16, 2022, data cut-off, 40 patients had received at least 1 dose of teclistamab in phase II, cohort C, and were included in the FAS.
Duration of Exposure
At the time of analysis, using the March 16, 2022, data cut-off, 40 patients were enrolled in phase II, cohort C, of the MjaesTEC-1 study. The median duration of study treatment was 5.2 months (range = 0.2 months to 13.6 months). A total of 19 (47.5%) patients received teclistamab for at least 6 months, 11 (27.5%) patients received teclistamab for at least 9 months, and 4 (10.0%) patients received teclistamab for at least 12 months. In phase II, cohort C, patients received a median of 5 treatment cycles (range = 1 cycle to 14 cycles). The median relative dose intensity for all study treatment, including step-up doses, was 95.9%. The median follow-up for the 40 enrolled patients in the FAS of phase II, cohort C, was 12.5 months (range = 0.7 month to 14.4 months). At the time of the data cut-off date of March 16, 2022, among the 21 responders (with a PR or better response), the median duration of follow-up was 11.8 months (range = 3.6 months to 13.8 months).
Treatment Disposition
At the time of analysis, using the March 16, 2022, data cut-off, 17 (42.5%) patients in phase II, cohort C, had discontinued the study due to death. The primary cause of death was disease progression for 10 patients, an adverse event in 6 patients, and subsequent therapy toxicity for 1 patient.
Baseline Characteristics
At the time of the data cut-off date of September 7, 2021, 38 patients were enrolled in phase II, cohort C, including 22 (57.9%) patients who were still on treatment. Baseline characteristics for these 38 patients are summarized as follows, since those of the 40 patients enrolled by March 16, 2022, were not reported in the Clinical Study Report. The median age of the patients was 63.5 years (range = 32 years to 82 years). A total of 24 (63.2%) patients were male and 14 (41.8%) patients were female. Most patients were white (89.5%), and 7.9% identified as Black or African American. All patients were triple-class exposed and a majority were penta-exposed (78.9%). Most patients had an ECOG PS score of 1 (73.7%) and 26.3% of patients had an ECOG PS score of 0. The most common immunoglobulin isotypes were immunoglobulin G, presenting in 18 (47.4%) patients. The median time from diagnosis of MM to enrolment in phase II, cohort C, was 6.5 years (range = 1.1 years to 24.1 years). Eleven (28.9%) patients had at least 1 extramedullary plasmacytoma at baseline. Of the 34 patients with baseline cytogenetic data reported, 11 (32.4%) patients had at least 1 high-risk abnormality, most commonly del(17p). A total of 20 (52.6%) patients were ISS stage I while 9 (23.7%) patients were ISS stage III.
In phase II, cohort C, all 38 patients enrolled by September 7, 2021, were triple-class exposed (PI, IMiD, and anti-CD38 monoclonal antibody) and 30 (78.9%) patients were penta-class exposed (at least 2 PIs, at least 2 IMiDs, and at least 1 anti-CD38 monoclonal antibody). Prior anti-BCMA therapy was required for patients enrolled in phase II, cohort C. A total of 27 (71.1%) patients received ADC (belantamab mafodotin or an investigational ADC), 15 (39.5%) patients received CAR T-cell therapy, and 4 patients received prior therapy with both ADC and CAR T-cells. In phase II, cohort C, 32 (84.2%) patients were refractory to their last line of therapy. A total of 32 (84.2%) patients were triple-class refractory (PI, IMiD, and anti-CD38 monoclonal antibody) and 12 patients (31.6%) were penta-refractory (at least 2 PIs, at least 2 IMiDs, and at least 1 anti-CD38 monoclonal antibody). A total of 14 patients who received a prior CAR T-cell therapy had documented, subsequently experienced disease progression, and 15 patients were considered refractory with a best response of stable disease. All patients in phase II, cohort C, received steroids, antipyretics (anilides), and antihistamines (histamine1 receptor antagonists) as a pretreatment medication.
Efficacy
Findings for key efficacy outcomes in phase II, cohort C, of the MajesTEC-1 study are summarized in Table 24.
Overall Survival
At the time of the data cut-off date of March 16, 2022, the median follow-up time was 12.5 months (range = 0.7 months to 14.4 months) in phase II, cohort C, of the MajesTEC-1 trial. The estimated median OS was 13.2 months (95% CI, 8.3 months to NE months). In cohort C, deaths were reported in 17 (42.5%) patients in FAS. The proportion of patients who were censored (alive at the time of data cut-off) was 57.5% (23) of patients. The estimated 6-month OS probability among patients was ||||| |||| ||| ||||| || ||||||. The 9-month OS probability was ||||| |||| ||| ||||| || ||||||, and the 24-month OS probability was ||||| |||| ||| ||||| |||||||. The Kaplan-Meier curve for OS for patients in the FAS is presented in Figure 8.
Progression-Free Survival
At the time of analysis, using the March 16, 2022, data cut-off, the median PFS was ||| |||||| |||| ||| ||| || || ||||||| in phase II, cohort C. By the data cut-off date, a total of 24 (60.0%) patients had had a PFS event, and || |||||||| ||||||| were censored. The estimated 6-month PFS probability among patients in phase II, cohort C, was ||||| |||| ||| ||||| || ||||||, the 9-month PFS probability was ||||| |||| ||| ||||| || ||||||, and the 24-month PFS probability was ||||| |||| ||| ||||| || ||||||. The Kaplan-Meier curve for PFS for patients in phase II, cohort C, is provided in Figure 9.
CR or Better
At the time of analysis, using the March 16, 2022, data cut-off, 11 (27.5%) patients (95% CI, 14.6% to 43.9%) in phase II, cohort C, had attained CR or better (CR or sCR).
Stringent Complete Response
At the time of analysis, using the March 16, 2022, data cut-off, 11 (27.5%) patients (95% CI, 14.6% to 43.9%) in phase II, cohort C, had attained sCR.
Figure 8: Kaplan-Meier Plot for OS in Phase II, Cohort C, of MajesTEC-1 Study, Full Analysis Set
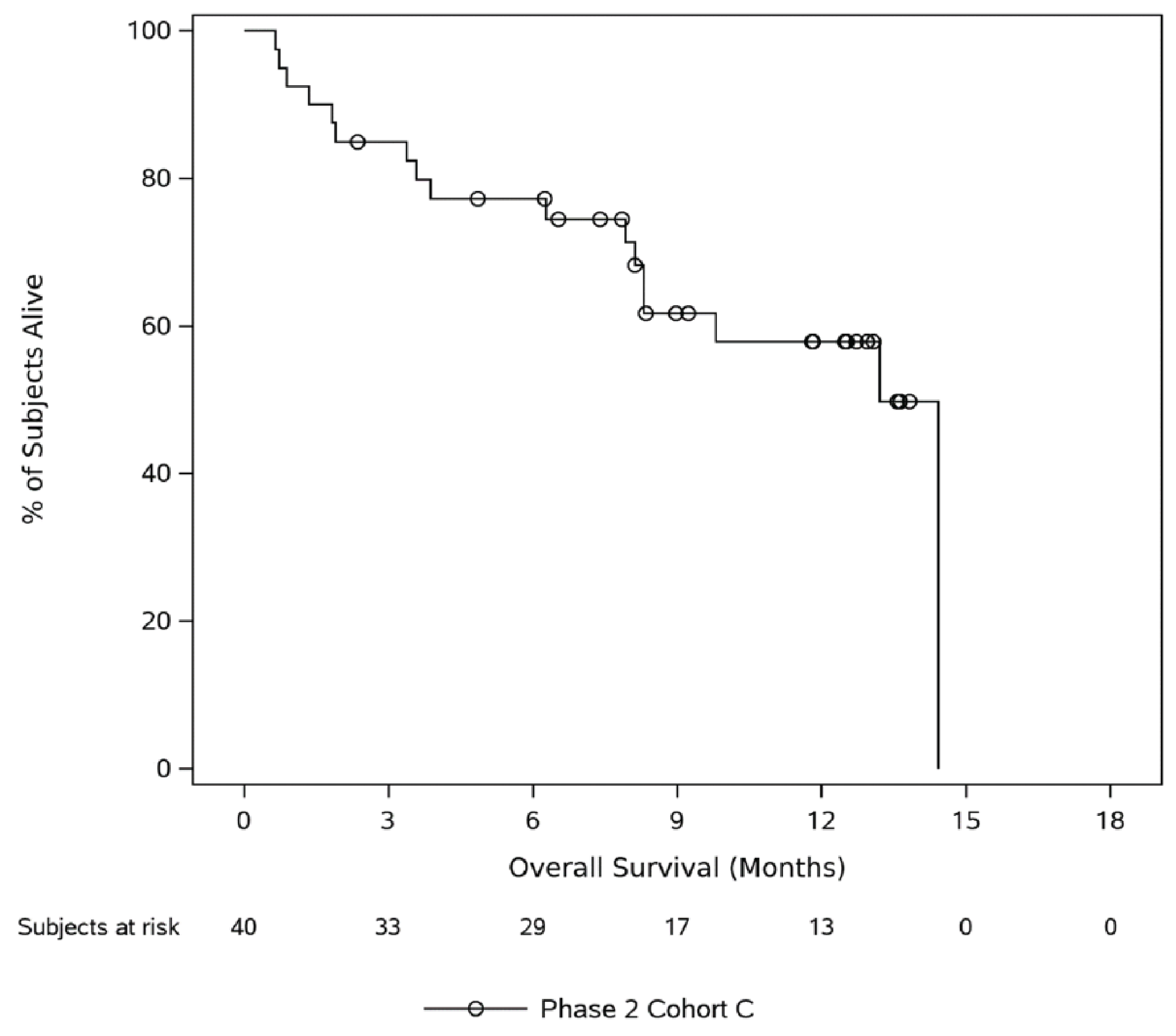
OS = overall survival.
Source: Clinical Study Report for cohort C, MajesTEC-1 study (2022).25
MRD-Negativity Status
At the time of analysis, using the March 16, 2022, data cut-off, 7 (17.5%) patients (95% CI, 7.3% to 32.8%) had attained MRD negativity at 10–5. Among 11 patients who attained CR or better per IRC assessment, 7 (63.6%) patients (95% CI, 30.8% to 89.1%) had attained MRD negativity at 10–5.
VGPR or Better
At the time of analysis, using the March 16, 2022, data cut-off, 19 (47.5%) patients (95% CI, 31.5% to 63.9%) in phase II, cohort C, had attained VGPR or better (VGPR, CR, or sCR).
Overall Response Rate
At the time of analysis, using the March 16, 2022, data cut-off, 21 (52.5%) patients (95% CI, 36.1% to 68.5%) had attained an overall response (PR or better) in cohort C, phase II, with similar ORR among patients with prior ADC exposure — a proportion of 55.2% (95% CI, 35.7% to 73.6%) — and patients with prior CAR T-cell therapy — a proportion of 53.3% (95% CI, 26.6% to 78.7%). Most responses occurred by the start of cycle 2. Responses were ongoing for 15 of 21 patients at the time of the cut-off date of March 16, 2022 (Figure 10).
Figure 9: Kaplan-Meier Plot for PFS in Phase II, Cohort C, of MajesTEC-1 Study, Full Analysis Set
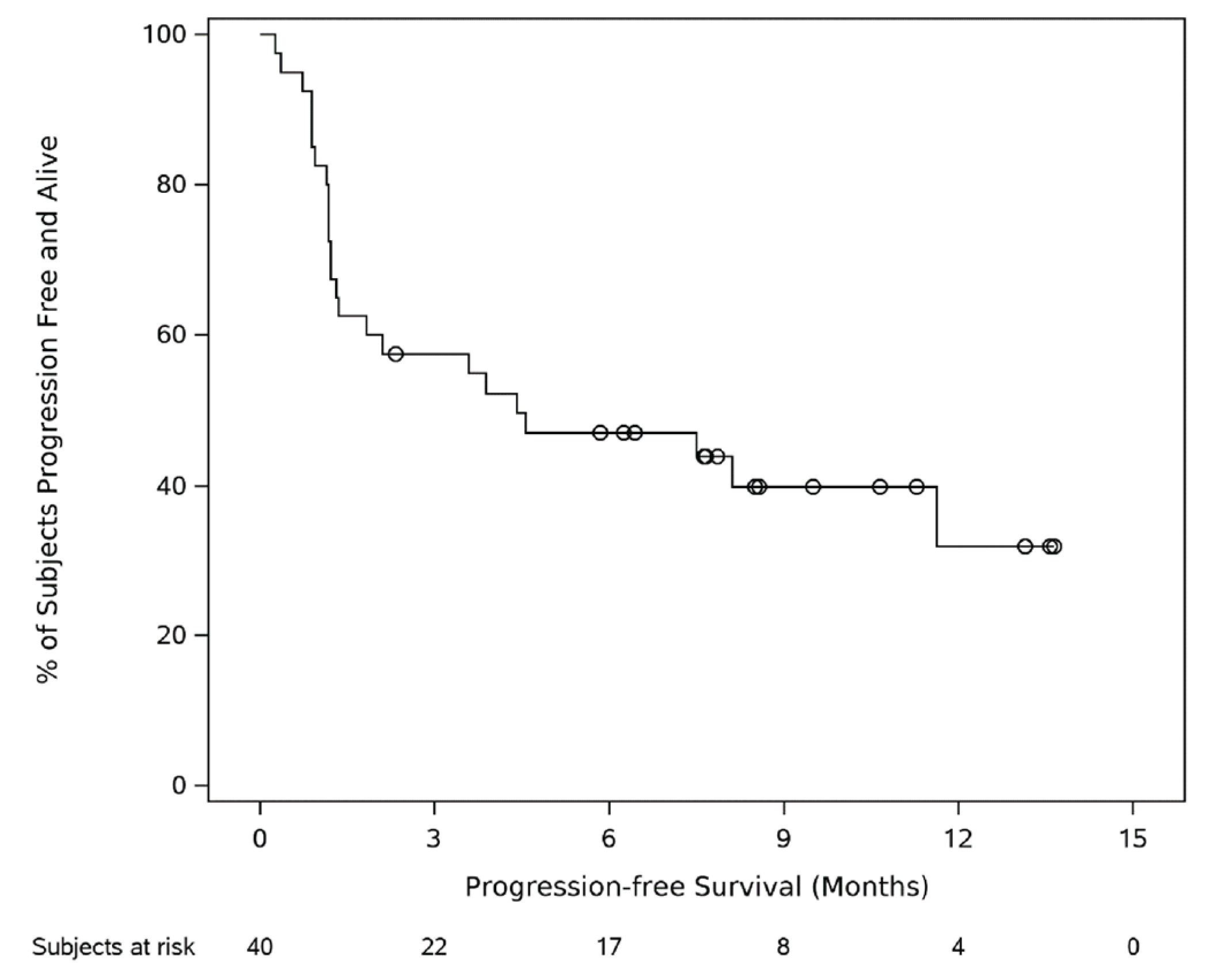
PFS = progression-free survival.
Source: Clinical Study Report for cohort C, MajesTEC-1 study (2022).25
Figure 10: Response and Follow-up in MajesTEC-1 Study per IRC Assessment, Phase 2 Cohort C
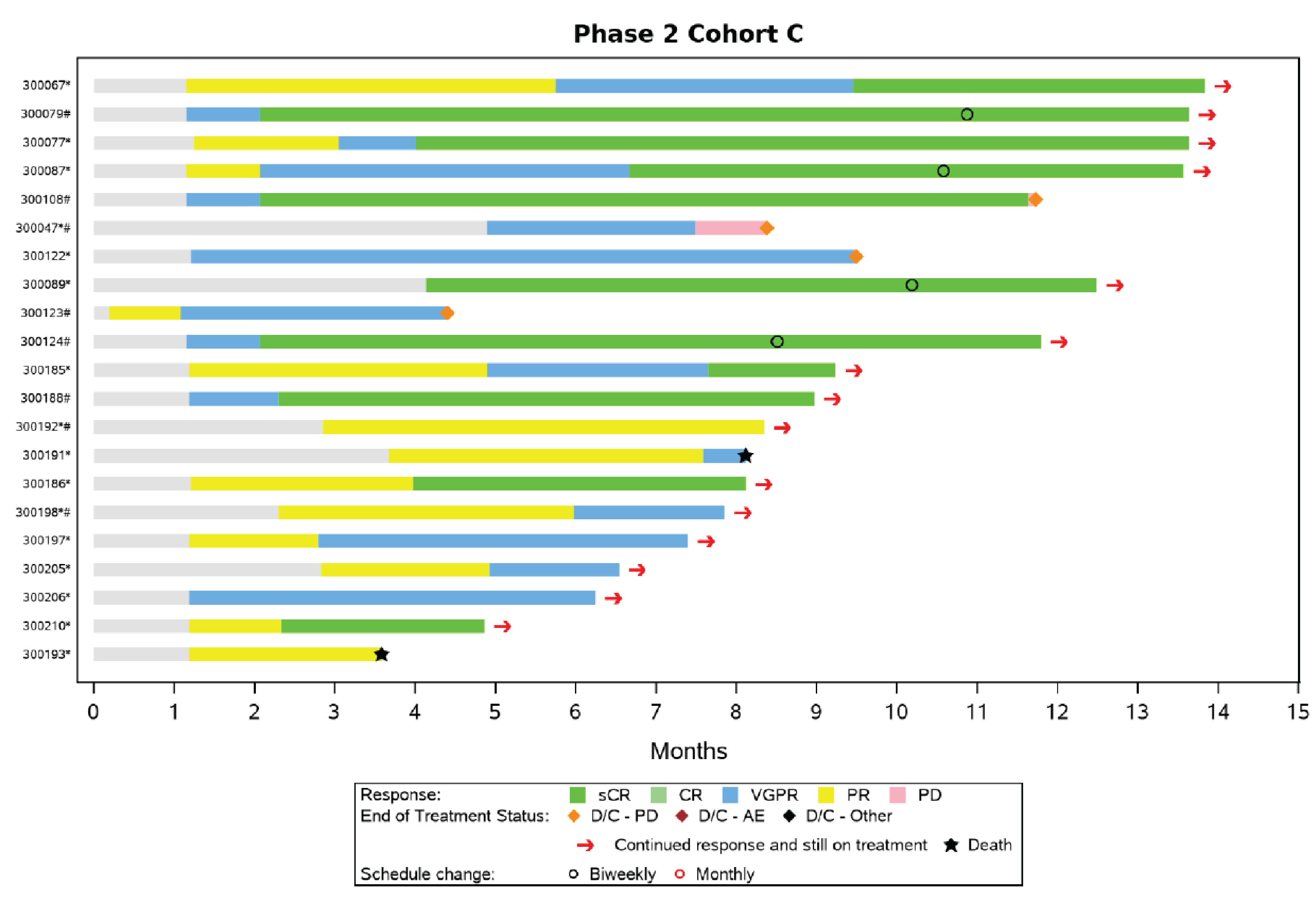
AE = adverse event; CR = complete response; D/C = discontinued; IRC = independent review committee; PD = progressive disease; PR = partial response; sCR = stringent complete response; VGPR = very good partial response.
Source: Clinical Study Report for cohort C, MajesTEC-1 study (2022).25
Time to Response
At the time of analysis, using the March 16, 2022, data cut-off, of the 21 patients who attained PR or better in phase II, cohort C, the median time to first response was 1.2 months (range = 0.2 months to 4.9 months) while the median time to best response was 2.9 months (range = 1.1 months to 9.5 months). Most patients demonstrated their first response rapidly, by the start of cycle 2.
Duration of Response
At the time of analysis, using the March 16, 2022, data cut-off, with a median follow-up time of 12.5 months, the median duration of response was not reached in phase II, cohort C. Among 21 responders (with a response of PR or better) in phase II, cohort C, 15 (23.8%) patients had disease progression or died due to any cause. The estimated probability of patients remaining in response at 9 months was ||||| |||| ||| ||||| || |||||| while the probability of patients remaining in response at 12 months was ||||| |||| ||| ||||| || ||||||. Kaplan-Meier curves for the duration of response for all responders and for responders with a schedule change are provided in Figure 11.
Figure 11: Kaplan-Meier Plot for Duration of Response per IRC Assessment, Full Analysis Set
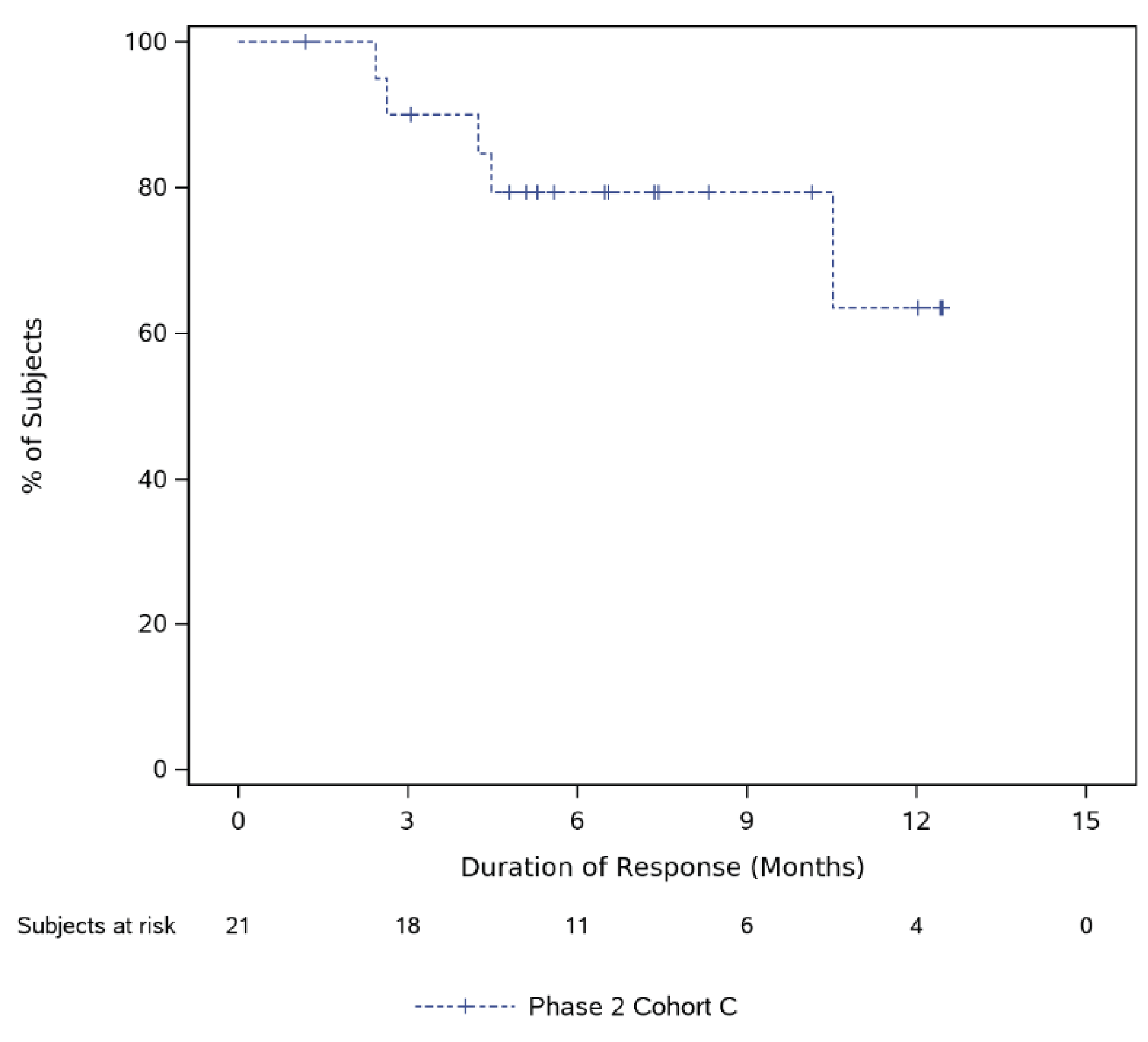
IRC = independent review committee.
Source: Clinical Study Report for cohort C, MajesTEC-1 study (2022).25
Table 24: Summary of Key Efficacy Results From Phase II, Cohort C, of MajesTEC-1 Study, Full Analysis Set
Variable | Phase II, cohort C (N = 40) |
|---|---|
Median follow-up time, months (range) | 12.5 (0.67 to 14.4) |
OS per IRC assessment | |
Events, n (%) | 17 (42.5) |
Censored, n (%) | 23 (57.5) |
Mediana OS (95% CI), months | 13.2 (8.3 to NE) |
6-month OS probability,a % (95% CI) | 77.3 (60.9 to 87.5) |
9-month OS probability,a % (95% CI) | 61.8 (13.8 to 75.5) |
12-month OS probability,a % (95% CI) | 57.9 (39.6 to 72.5) |
PFS per IRC assessment | |
PFS events, n (%) | 24 (60.0) |
Censored, n (%) | 16 (40.0) |
Mediana PFS (95% CI), months | 4.4 (1.3 to NE) |
6-month PFS probability,a % (95% CI) | 47.0 (31.0 to 61.5) |
9-month PFS probability,a % (95% CI) | 39.9 (24.0 to 55.3) |
12-month PFS probability,a % (95% CI) | 31.9 (14.6 to 50.8) |
Patients who attained CR or better per IRC assessment | |
n (%) | 11 (27.5) |
95% CI | 14.6 to 43.9 |
MRD-negativity status | |
MRD-negativity rate (10–5), n (%) | 7 (17.5) |
95% CI of MRD-negativity rate, % | 7.3 to 32.8 |
Patients who attained VGPR or better (VGPR, CR, sCR, or better) per IRC assessment | |
n (%) | 19 (47.5) |
95% CI | 31.5 to 63.9 |
Patients who attained overall response (PR, VGPR, CR, or sCR) per IRC assessment | |
n (%) | 21 (52.5%) |
95% CI | 36.1 to 68.5 |
Response category | |
Patients who attained sCR | |
n (%) | 11 (27.5) |
95% CI | 14.6 to 43.9 |
Patients who attained CR | |
n (%) | 0 (0.0) |
95% CI | NE |
Patients who attained VPGR | |
n (%) | 8 (20.0) |
95% CI | 9.1 to 35.6 |
Patients who attained PR | |
n (%) | 2 (5.0) |
95% CI | 0.6 to 16.9 |
Patients who attained minimal response | |
n (%) | 0 (0.0) |
95% CI | NE |
Stable disease | |
n (%) | 6 (15.0) |
95% CI | 5.7 to 29.8 |
Progressive disease | |
n (%) | 10 (25.0) |
95% CI | 12.7 to 41.2 |
Time to response per IRC assessment | |
Time to first response, monthsa | |
Number of patients contributing to the analysis, n | 21 |
Mean (SD) | 1.8 (1.2) |
Median (range) | 1.2 (0.2 to 4.9) |
Time to best response, monthsa | |
Number of patients contributing to the analysis, n | 21 |
Mean (SD) | 3.8 (2.5) |
Median (range) | 2.9 (1.1 to 9.5) |
Time to VGPR or better, monthsa | |
Number of patients contributing to the analysis, n | 19 |
Mean (SD) | 3.2 (2.0) |
Median (range) | 2.8 (1.1 to 7.6) |
Time to CR or better, monthsa | |
Number of patients contributing to the analysis, n | 11 |
Mean (SD) | 4.3 (2.6) |
Median (range) | 4.0 (2.1 to 9.5) |
DoR per IRC assessment | |
Number of patients contributing to the analysis, n | 21 |
Events,b n % | 5 (23.8) |
Censored, n % | 16 (76.2) |
Mediana duration of response (95% CI), months | NE (10.5 to NE) |
6-month event-free rate, % (95% CI) | 79.4 (54.0 to 91.7) |
9-month event-free rate, % (95% CI) | 79.4 (54.0 to 91.7) |
12-month event-free rate, % (95% CI) | 63.5 (26.0 to 85.8) |
CI = confidence interval; CR = complete response; DoR = duration of response; IRC = independent review committee; MRD = minimal residual disease; NE = not evaluable; OS = overall survival; PFS = progression-free survival; PR = partial response; sCR = stringent complete response; SD = standard deviation; VGPR = very good partial response.
aThe estimate was derived from the Kaplan-Meier estimator.
bThe number of events refers to the number of responders (PR or better) who developed disease progression or died due to any cause. DoR is calculated as the number of months from the first documented response to progression, death due to any cause, or the date of censoring.
Source: Clinical Study Report for cohort C, MajesTEC-1 study (2022).25
Harms
Harms data for phase II, cohort C, of the MajesTEC-1 study are summarized in Table 25. Safety data for the MajesTEC-1 study are presented from the most recent safety analyses (with a clinical cut-off date of March 16, 2022).
Adverse Events
At the time of analysis, using the March 16, 2022, data cut-off, all patients in phase II, cohort C, of the MajesTEC-1 study had experienced at least 1 TEAE. The most common TEAEs of any grade occurring in at least 20% of patients in phase II, cohort C, were neutropenia (67.5%), CRS (65.0%), anemia (50.0%), thrombocytopenia (45.0%), lymphopenia (45.0%), constipation (35.0), diarrhea (35.0%), and pyrexia (32.5%). In phase II, cohort C, 9 (22.5%) patients experienced TEAEs of grade 3, 20 (50.0%) patients experienced TEAEs of grade 4, and 8 (20.0%) patients experienced TEAEs of grade 5. The most common TEAEs of grade 3 or grade 4 were neutropenia (62.5%), lymphopenia (42.5%), anemia (35.0%), and thrombocytopenia (30.0%).
Serious Adverse Events
At the time of analysis, using the March 16, 2022, data cut-off, 24 (60.0%) patients in phase II, cohort C, had experienced at least 1 serious TEAE. The most common TEAEs were COVID-19 (10.0%), CRS (7.5%), febrile neutropenia (7.5%), and anemia (5.0%).
Withdrawals Due to Adverse Events
At the time of analysis, using the March 16, 2022, data cut-off, no patients had experienced a TEAE leading to treatment discontinuation. Treatment cycle delay was reported in 13 (32.5%) patients and dose delay in 6 (15.0%) patients in phase II, cohort C.
Mortality
At the time of analysis, using the March 16, 2022, data cut-off, 17 (42.5%) patients in phase II, cohort C, had died. A total of 8 (20.0%) patients died within 30 days of the last dose of teclistamab, of whom 3 (7.5%) patients died due to progressive disease and 5 (12.5%) patients died due to adverse events. A total of 6 (15.0%) patients died within 60 days of the first dose of teclistamab, of whom 3 (7.5%) patients died due to progressive disease, 1 (2.5%) patient died due to COVID-19, 1 (2.5%) patient died due to cardiac failure, and 1 (2.5%) patient died due to coronary artery dissection.
Table 25: Summary of Harms Results From Phase II, Cohort C, of MajesTEC-1 Study, Full Analysis Set
Adverse event | Phase II, cohort C (N = 40) |
|---|---|
Patients with ≥ 1 TEAE, n (%) | 40 (100.0) |
Maximum toxicity grade, n (%) | |
Grade 1 | 0 (0.0) |
Grade 2 | 3 (7.5) |
Grade 3 | 9 (22.5) |
Grade 4 | 20 (50.0) |
Grade 5 | 8 (20.0) |
TEAEs in ≥ 20% of patients of any grade, n (%) | |
Neutropenia | 27 (67.5) |
CRS | 26 (65.0) |
Anemia | 20 (50.0) |
Thrombocytopenia | 18 (45.0) |
Lymphopenia | 18 (45.0) |
Constipation | 14 (35.0) |
Diarrhea | 14 (35.0) |
Pyrexia | 13 (32.5) |
Injection-site erythema | 13 (32.5) |
Arthralgia | 10 (25.0) |
Headache | 9 (22.5) |
Dyspnea | 9 (22.5) |
Asthenia | 8 (20.0) |
Bone pain | 8 (20.0) |
TEAEs in ≥ 10% of patients of grade 3 or grade 4, n (%) | |
Neutropenia | 25 (62.5) |
Lymphopenia | 17 (42.5) |
Anemia | 14 (35.0) |
Thrombocytopenia | 12 (30.0) |
Patients with ≥ 1 serious TEAE, n (%) | 24 (60.0) |
TEAEs leading to discontinuation of study drug, n (%) | 0 (0.0) |
Deaths | |
Patients who died, n (%) | 17 (42.5) |
TEAEs leading to death, n (%) | 8 (20.0) |
Death due to COVID-19 | 2 (5.0) |
Primary cause of death, n (%) | |
Adverse event | 6 (15.0) |
Disease progression | 10 (25.0) |
Other | 1 (2.5) |
Patients who died within 30 days of last study treatment dose, n (%) | 8 (20.0) |
Patients who died within 60 days of last study treatment dose, n (%) | 6 (15.0) |
CRS = cytokine release syndrome; TEAE = treatment-emergent adverse event.
Source: Clinical Study Report for cohort C, MajesTEC-1 study (2022).25
Notable Harms
In phase II, cohort C, of the MajesTEC-1 study, several adverse events of clinical interest were identified, including CRS, neurologic adverse events and neurotoxicity, ICANS, injection-site reactions, hypogammaglobulinemia, cytopenias, infections, and TLS.
Cytokine Release Syndrome
At the time of analysis, using the March 16, 2022, data cut-off, 26 (65.0%) patients in phase II, cohort C, had experienced 44 CRS events of any grade. Most CRS events occurred during step-up or day 1 of cycle 1. The median time to the onset of CRS from the last injection of teclistamab was 2 days (range = 2 days to 6 days) and the median duration of CRS was 2 days (range = 1 day to 4 days). There were 23 (57.5%) patients who received supportive measures to treat CRS events, including tocilizumab in 12 (30.0%) patients.
Neurologic Adverse Events
At the time of analysis, using the March 16, 2022, data cut-off, 21 (52.5%) patients in phase II, cohort C, had experienced at least 1 neurologic TEAE. The most common neurologic TEAEs included headache (22.5%), ICANS (10.0%), insomnia (10.0%), encephalopathy (5.0%), peripheral sensory neuropathy (7.5%), dizziness (5.0%), and motor dysfunction (5.0%).
Neurotoxicity
At the time of analysis, using the March 16, 2022, data cut-off, 10 (25.0%) patients in phase II, cohort C, had experienced at least 1 neurotoxicity event, including headache (12.5%) and ICANS (10.0%).
Infections
At the time of analysis, using the March 16, 2022, data cut-off, 26 (65.0%) patients in phase II, cohort C, had had at least 1 treatment-emergent infection of any grade. The most common infections and infestations included COVID-19 (12.5%), bronchitis (10.0%), pneumonia (7.5%), cytomegalovirus infection reactivation (5.0%), implant site infection (5.0%), and laryngitis (5.0%). A total of 10 (25.0%) patients experienced infections of grade 3 or grade 4, and 10 (25.0%) patients experienced serious infections. At the time of the cut-off date of March 16, 2022, no patients in phase II, cohort C, had discontinued treatment due to infection. Opportunistic infections were reported in 7 (17.5%) patients, including cytomegalovirus infection reactivation (50.0%) and esophageal candidiasis (50.0%).
Hypogammaglobulinemia
At the time of analysis, using the March 16, 2022, data cut-off, the proportions of patients with hypogammaglobulinemia had not been reported in phase II, cohort C, of the MajesTEC-1 study.
Immune Effector Cell-Associated Neurotoxicity Syndrome
At the time of analysis, using the March 16, 2022, data cut-off, 4 (10%) patients in phase II, cohort C, had experienced ICANS. All cases of ICANS were concurrent with CRS events, and no patients discontinued treatment due to ICANS. In phase II, cohort C, the median time to onset of ICANS from the last injection of teclistamab was 2.5 days (range = 2 days to 4 days) and the median duration was 1.5 days (range = 1 day to 2 days). All cases of ICANS were reported as resolved. Supportive measures to treat ICANS were used in 2 patients, both of whom had received tocilizumab.
Cytopenia
At the time of analysis, using the March 16, 2022, data cut-off, 35 (87.5%) patients in phase II, cohort C, had experienced at least 1 treatment-emergent cytopenic event, including neutropenia (67.5%), anemia (50.0%), thrombocytopenia (45.0%), and lymphopenia (45.0%). Hemorrhagic events were reported for 5 (12.5%) patients, 1 of which was a grade 2 event.
Injection-Site Reactions
At the time of analysis, using the March 16, 2022, data cut-off, the proportions of patients with injection-site reactions had not been reported in phase II, cohort C, of the MajesTEC-1 study.
Tumour Lysis Syndrome
At the time of analysis, using the March 16, 2022, data cut-off, no events of TLS had been reported in phase II, cohort C, of the MajesTEC-1 study.
Since the patients who took part in phase II, cohort C, were from the MajesTEC-1 study, it is reasonable to expect that the same limitations of the pivotal MajesTEC-1 study (phase I and phase II, cohort A) with respect to internal and external validity are relevant to phase II, cohort C, of the MajesTEC-1 trial. Findings from phase II, cohort C, of the MajesTEC-1 study were consistent with the results from the pivotal cohort (phase I and phase II, cohort A); however, only 40 patients were included, which limits interpretation of the cohort findings.
Critical Appraisal
Internal Validity
The MajesTEC-1 trial was a multicentre, single-arm, open-label, phase I and phase II study. Due to the lack of a comparator arm, the benefit of teclistamab compared to placebo or reference treatment was not documented. A single-arm study design is usually used when the purpose of the study is to provide preliminary evidence of the efficacy of a treatment and to collect additional safety data, and is not intended to be confirmatory for efficacy.17 Thus, a single-arm study design is a subject of several limitations that complicates the interpretation of the study results. For example, it is impossible to distinguish whether the observed effect in the study is due to teclistamab, a placebo effect, or the effect of natural history of the disease in the absence of a frame of reference for comparison.17 The clinical experts consulted by CADTH for this review noted that conducting a phase III RCT in this population may be challenging because it would be difficult to recruit enough patients in this population. The open-label design of the MajesTEC-1 study may increase uncertainty in subjective outcomes, including clinical response outcomes, PFS, HRQoL, and safety outcomes, introducing bias due to inherent subjectivity of the outcome in an unblinded assessor. With the exception of harms, the bias will likely favour the experimental intervention, although the extent of bias is uncertain. This bias would be less likely in more objective outcomes, such as OS, if assessed against a predetermined hypothesis.
According to the FDA, the ORR can be evaluated in a single-arm study as a direct measure of a drug antitumour activity if it is defined as the sum of PRs plus CRs.18 ORR was the primary end point in phase II and the secondary end point in phase I of the MajesTEC-1 study. The estimated ORR was tested against a predetermined hypothesis of an ORR greater than 45% (with a lower bound of the 2-sided 95% CI above 30%). ORR achieved the predetermined threshold for a positive outcome in the MajesTEC-1 study. However, for ORR, there was no adjustment for multiplicity across the various analyses of the outcome (i.e., the various data cut-offs), which may have increased the risk of false-positive conclusions. Additionally, this report presents interim analysis results because a prespecified final analysis was not available. Therefore, there is the potential that the benefit of teclistamab is overestimated; however, the presence and extent of any overestimation is uncertain.19-21
The number of patients screened in this study, the proportion of patients who did not pass screening, and the patients who did not receive teclistamab were not reported. In MajesTEC-1 study, 54.5% of patients had discontinued study participation primarily due to death, and 71.5% of patients had discontinued study treatment primarily due to disease progression. The clinical experts consulted by CADTH for this review noted that the discontinuation rate in the MajesTEC-1 trial was high, although disease progression is a major issue in the population with RRMM who receive fourth-line and beyond therapy.
Disease responses were evaluated by the IRC using IMWG 2016 criteria in both phase I and phase II, cohort A, of the MajesTEC-1 study. The time-to-event end points, including OS and PFS, were identified as important outcomes by clinical experts and patient and clinician groups consulted by CADTH for this review. However, OS and PFS were not considered primary or key secondary outcomes in the MajesTEC-1 trial, and the lack of a comparator group limits the estimation of relative effects of treatment with teclistamab. In addition, the longer-term efficacy of teclistamab for OS and PFS is unknown as the MajesTEC-1 study is ongoing.
The clinical experts and patient and clinician groups consulted by CADTH highlighted improvement in HRQoL as an important outcome and treatment goal for patients with RRMM. HRQoL was measured using EORTC QLQ-C30 and EQ-5D-5L questionnaires. The analyses of HRQoL outcomes were undertaken post hoc, which introduces a risk of bias in the selection of the reported result. In addition, analyses for HRQoL were performed in HRQoL-evaluable patients at specific time points rather than in the ITT population. The results for HRQoL were reported only for phase II, cohort A, of the MajesTEC-1 study. The size of the HRQoL-evaluable population in the MajesTEC-1 study gradually decreased over time, and the rate of missing data was high among those who remained in the study at longer follow-up visits (e.g., data were missing for 49.6% [63 of 125] of all treated patients for the EORTC QLQ-C30 at cycle 6, day 1, and for 71.2% [36 of 125] of patients by cycle 16, day 1). Therefore, data from later time points should be interpreted with caution due to the possibility that HRQoL scores could be overestimated if patients with better HRQoL were more likely to complete the questionnaires.22 However, the extent of the bias with respect to the direction and magnitude of the effect is uncertain.
No sensitivity analyses were performed or reported for the study outcomes at the time of the data cut-off date of January 4, 2023. A number of subgroup analyses were prespecified a priori in the primary analysis of the MajesTEC-1 trial. However, at the time of the data cut-off date of January 4, 2023, data were available only on select subgroups that were deemed clinically meaningful by the clinical experts consulted by CADTH for this review, including the number of lines of prior therapy and cytogenetic risk. The results of subgroup analyses were consistent with the primary ORR analysis results across patient subgroups of interest; however, the interpretation of the analyses was limited by the small sample sizes of the groups.
External Validity
According to the clinical experts consulted by CADTH for this review, the patient population in the MajesTEC-1 study generally reflects patients in clinical practice in this setting. To be enrolled in the MajesTEC-1 study, patients with RRMM were required to have an ECOG PS score of 0 or 1 and have a measurable disease. The clinical experts consulted noted that this would not be reflective of clinical practice and that clinicians would prescribe teclistamab to patients with an ECOG PS score of 2 or 3 and to patients without biochemically measurable disease. Patients who had previously received antitumour therapy, such as a monoclonal antibody, or cytotoxic therapy within 21 days before the first dose of teclistamab, were excluded from the study; the clinical experts found this concerning as a washout period of 21 days is less relevant in this population. One of the exclusion criteria of the pivotal MajesTEC-1 study was any prior BCMA-targeted therapy. Additional supporting data were presented for phase II, cohort C, at the time of the clinical cut-off date of March 16, 2022, to address the use of teclistamab in patients previously treated with BCMA-targeted therapy in accordance with the Health Canada indication for teclistamab. Findings from phase II, cohort C, of the MajesTEC-1 study were consistent with the results from the pivotal cohort (phase I and phase II, cohort A); however, only 40 patients were included, which limits interpretation of the cohort findings.
According to the clinical experts consulted by CADTH for this review, the demographic and disease characteristics of the MajesTEC-1 study population were reflective of patients living in Canada with RRMM. The mean age of patients in the MajesTEC-1 study was 64 years, with clinical experts noting that in the real-world setting, the mean age of patients with relapsed disease receiving fourth-line therapy and beyond would be around 70 years.23 About 26% of patients in the MajesTEC-1 trial had at least 1 high-risk abnormality, including del(17p) and t(4;14), although clinical experts noted that the proportion of patients with cytogenetic risk is slightly higher in clinical practice. Most patients were white and non-Hispanic or non-Latino; however, the clinical experts noted that this would not limit generalizability to patients in Canadian clinical practice. In the MajesTEC-1 study, 63 (38.2%) patients switched from weekly to biweekly dosing of teclistamab, including 54 patients who met the response criteria, and 9 patients had switched from biweekly to monthly dosing. Clinical experts consulted by CADTH for this review believed that there would be more patients in clinical practice switching to less frequent dosing of teclistamab. They further noted that switching from weekly to biweekly dosing can occur primarily due to side effects, toxicity, or patient choice.
The clinical experts consulted by CADTH for this review did not have any major concerns with the end points used in the study. According to the clinical experts and patient and clinician group input, OS, PFS, clinical response outcomes, and HRQoL are the most important outcomes for assessing the response to treatment. However, due to its study design, the MajesTEC-1 trial provides no information about the efficacy and harms of teclistamab relative to treatments that would otherwise be used in this patient population in clinical practice.
In the MajesTEC-1 trial, the study population was drawn from a number of sites around the globe, including Canada. The clinical experts indicated that there were no major concerns with generalizing the findings from the pivotal study to the Canadian clinical setting.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For the pivotal study identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered most relevant to inform CADTH’s expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group.84,85
“High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate — The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word ‘likely’ for evidence of moderate certainty (e.g., ‘X intervention likely results in Y outcome’).
Low certainty: Our confidence in the effect estimate is limited — The true effect may be substantially different from the estimate of the effect. We use the word ‘may’ for evidence of low certainty (e.g., ‘X intervention may result in Y outcome’).
Very low certainty: We have very little confidence in the effect estimate — The true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as ‘very uncertain.’”
Although GRADE guidance is not available for noncomparative studies, the CADTH review team assessed pivotal single-arm trials for study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias to present these important considerations. Because the lack of a comparator arm does not allow for a conclusion to be drawn on the effect of the intervention versus any comparator, the certainty of evidence for single-arm trials started at very low certainty with no opportunity for rating up.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null.
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for teclistamab in patients with RRMM in the MajesTEC-1 trial.
Long-Term Extension Studies
No long-term extension studies were submitted by the sponsor.
Indirect Evidence
Content in this section has been informed by materials submitted by the sponsor. The following has been summarized and validated by the CADTH review team.
Objectives for the Summary of Indirect Evidence
The objective of this section is to summarize and critically appraise available evidence comparing teclistamab to other relevant treatments for patients with RRMM.
Description of Indirect Comparisons
The efficacy and safety of teclistamab among adult patients with RRMM who had received at least 3 prior lines of therapy have been previously assessed in the MajesTEC-1 study. However, no head-to-head evidence of teclistamab against other treatments for RRMM was available for this review. Due to this gap in evidence, the sponsor submitted 6 ITCs, of which 3 ITCs were used to inform the pharmacoeconomic model — including 2 ITCs comparing the relative efficacy of teclistamab with RWPC therapy (from the LocoMMotion and daratumumab trials)26-29 and another ITC comparing the relative efficacy of teclistamab with ciltacabtagene autoleucel (from the CARTITUDE-1 trial).30 Of the 3 ITCs submitted by the sponsor that were not included in the pharmacoeconomic model, 1 published ITC compared the relative efficacy of teclistamab with RWPC therapy (from the Flatiron Health database),29,31 and 2 conference abstracts compared the relative efficacy of teclistamab with belantamab mafodotin (from the DREAMM-2 study)32 and selinexor in combination with dexamethasone (from the STORM study, part 2).33
Study Selection Methods
No systematic review was reported by the sponsor. To estimate the efficacy of teclistamab relative to other therapies for RRMM, studies that assessed the efficacy of a treatment for RRMM were selected for inclusion in the ITCs. The studies included patients (or at minimum, a subset of patients) with triple-class exposed RRMM who had received at least 3 prior lines of therapy, and reported sufficient efficacy outcome data. A summary of the study selection criteria is presented in Table 26. Studies were required to be RCTs, nonrandomized clinical trials, observational studies, or database studies; the intervention and comparator could be any therapy used for the treatment of RRMM.
Table 26: Study Selection Criteria and Methods for ITCs Submitted by the Sponsor
Characteristics | Indirect comparison |
|---|---|
Population | Patients with MM diagnosis per IMWG Patients (or a subset of patients) with triple-class exposed RRMM (with prior exposure to PI, IMiD, and anti-CD38 monoclonal antibody) |
Intervention | Any therapy for RRMM |
Comparator | Any comparators |
Outcome | Efficacy:
|
Study designs | RCTs, nonrandomized clinical trials, observational studies, database studies |
Publication characteristics | Data on file and full-text publications |
Exclusion criteria | None |
Databases searched | NA |
Selection process | Records were independently screened and best available data for key RRMM comparators were selected. |
Data extraction process | Data were extracted by a single reviewer and confirmed by a second reviewer. |
Quality assessment | Quality assessment was not performed. |
CR = complete response; DoR = duration of response; IMiD = immunomodulatory drug; IMWG = International Myeloma Working Group; ITC = indirect treatment comparison; MM = multiple myeloma; NA = not applicable; ORR = overall response rate; OS = overall survival; PFS = progression-free survival; PI = proteasome inhibitor; RCT = randomized controlled trial; RRMM = relapsed or refractory multiple myeloma; TTNT = time to next treatment; VGPR = very good partial response.
Note: Details included in Table 26 are from the sponsor’s Summary of Clinical Evidence.83
No details were provided regarding the timing of the literature review or the databases used. The study screening was performed independently by 2 reviewers, extracted by 1 reviewer, and confirmed by a second reviewer. The risk of bias of the review process for including studies was not assessed. A list of excluded studies was not reported.
Three ITCs that were used to inform the pharmacoeconomic model were selected because they met the selection criteria. The sponsor stated that they included the most relevant comparators for the submission, including treatments that are reimbursed in Canada or have received a recommendation for reimbursement from CADTH for the indication under review.
Design of 3 ITCs Used to Inform Pharmacoeconomic Model
Analysis Methods
Given the absence of a comparator group in the MajesTEC-1 study, an external control group was used to establish the comparative efficacy of teclistamab versus treatments used in current clinical practice. The ITT populations in the MajesTEC-1, LocoMMotion, CARTITUDE-1, and daratumumab trials were considered comparable and therefore considered relevant for ITC analysis. In the MajesTEC-1 trial, individual patient data were included from all patients treated with teclistamab at a dose of 1.5 mg/kg weekly (clinical cut-off date of March 16, 2022)28 and compared with individual patient data from patients in the LocoMMotion study (clinical cut-off date of November 2021), the CARTITUDE-1 study (January 2022), and the daratumumab trials (September 2021 for the POLLUX trial, June 2021 for the CASTOR trial, July 2020 for the APOLLO trial, and 2017 to 2019 for the EQUULEUS trial). The treatment effect was estimated using IPTW methods. The index date was defined as the date of the first dose for the MajesTEC-1 trial; as day 1, cycle 1, of the real-life standard of care treatment for the LocoMMotion trial; as the date of apheresis in the CARTITUDE-1 trial; and as the start of each eligible line of therapy for the daratumumab clinical trials (the POLLUX, CASTOR, APOLLO, and EQUULEUS studies).
IPTW Rationale
A feasibility assessment was performed to determine the method of all 3 ITCs included in the pharmacoeconomic model. Poor overlap of prognostic factors between patient populations should be corrected, using the matching procedure employed to correct for observed imbalances in patient characteristics across trials. However, the LocoMMotion study was prospectively designed to recruit a patient population similar to the MajesTEC-1 study’s patient population, and there was sufficient overlap between patient characteristics to justify weighting techniques that do not depend on matching or excluding incompatible subpopulations. Similarly, there was sufficient overlap between patient characteristics between the MajesTEC-1 study and the CARTITUDE-1 and 4 daratumumab trials (the APOLLO, POLLUX, CASTOR, and EQUULEUS studies). To estimate the comparative efficacy, an IPTW estimator of ATT was chosen for the main ITC analyses. This propensity score–based method allowed the RWPC cohorts, as well as the population in the CARTITUDE-1 trial, to be reweighted to match the MajesTEC-1 study’s population. Also, IPTW is an efficient method when the sample size is small relative to the number of potential baseline confounding factors.86
Identification and Rank Ordering of Prognostic Factors
Teclistamab (MajesTEC-1 Study) Versus RWPC Therapy (LocoMMotion Study)
Imbalances in baseline patient characteristics between the MajesTEC-1 study and the RWPC cohort from the LocoMMotion study may lead to biased estimates of comparative efficacy if not adjusted for, due to confounding caused by factors that vary substantially across patient populations and are predictive of outcomes.
To ensure that the most important clinical factors were balanced between the 2 populations, an evidence-informed process (through studies from a literature review conducted to identify clinical outcomes in triple-class exposed patients with RRMM, as well as input from clinical experts) was used to select the prognostic factors for adjustment. This process considered the prognostic strength of potential covariates between the MajesTEC-1 study and the RWPC cohort from the LocoMMotion study. Prior to conducting the analyses, a pool of potential prognostic variables was identified through a targeted literature review. Clinical experts were consulted at multiple stages of the analysis to ensure clinical validity of the chosen covariates. All 17 factors that were identified a priori as important for population alignment were available from both populations. These factors were ranked according to availability and level of missingness within the included studies. Data for cytogenetic risk were missing for 37.1% of patients from the LocoMMotion trial and 10.3% of patients from the MajesTEC-1 trial, and this factor was not included in the analyses. In addition, race was not included in the analyses due to the extreme values of the estimated inverse probability weights. Variables with a high degree of missingness or, when included in the propensity score estimator, resulted in a lower overall balance were included for adjustment in the sensitivity analysis only.
The list of identified prognostic factors and those used for the adjustment process are presented in Table 27.
Teclistamab (MajesTEC-1 Study) Versus Ciltacabtagene Autoleucel (CARTITUDE-1 Study)
To identify prognostic factors requiring adjustment to balance the patient populations, a pool of potential prognostic variables was identified from the literature and validated; a rank ordering was conducted using input from clinical experts. Overall, 17 prognostic factors, selected at the index date, were included as adjustment variables in the propensity score methods (Table 27).
Teclistamab (MajesTEC-1 Study) Versus PC Therapy from Daratumumab Trials Cohort (APOLLO, POLLUX, CASTOR, and EQUULEUS Trials)
The following steps were undertaken for identifying and rank-ordering prognostic factors.
A pool of potential prognostic variables was identified by consulting studies from a literature review, originally conducted to identify clinical outcomes in triple-class exposed patients with RRMM.
Clinical experts were consulted to provide input on the most important factors that should be adjusted for in the analyses. The analyses including these top-ranked variables were considered as the base-case analyses.
The remaining factors were judged to be of lesser importance and adjusted for as a sensitivity analysis. These factors were ranked in order of importance considering both prognostic strength and the degree of imbalance between the populations, and were refined based on clinical input. Population differences between the MajesTEC-1 study and the PC cohort were assessed using SMDs (where an SMD between 0 and 0.1 was considered a small difference, an SMD of greater than 0.1 and less than or equal to 0.2 was a moderate difference, and an SMD greater than 0.2 was a substantial difference).87
The following prognostic factors (n = 9) were used for adjustment in the main analysis: refractory status, cytogenetic risk, ISS stage, the presence of extramedullary plasmacytomas, time to progression on the last line of therapy, the number of prior lines of therapy, years since MM diagnosis, age, and hemoglobin level. The fully adjusted sensitivity analysis included prior stem cell transplant, ECOG PS, race, sex, and type of MM in addition to the main analysis factors (n = 14) for adjustment in the propensity score model. The identified prognostic factors used for the adjustment process in these 3 ITCs are presented in Table 27.
Table 27: Availability of Prognostic Factors in MajesTEC-1 Study, RWPC From LocoMMotion Study, and Physician’s Choice from Daratumumab Trials Cohort (Main Analysis)
Prognostic factor | Teclistamab (MajesTEC-1 study) vs. RWPC therapy (LocoMMotion study) | Teclistamab (MajesTEC-1 study) vs. ciltacabtagene autoleucel (CARTITUDE-1 study) | Teclistamab (MajesTEC-1 study) vs. PC therapy (APOLLO, POLLUX, CASTOR, and EQUULEUS studies) |
|---|---|---|---|
Adjustment made on | Adjustment made on | Adjustment made on | |
Refractory statusa | Yes | Yes | Yes |
ISS stage | Yes | Yes | Yes |
Time to progression on last regimen | Yes | Yes | Yes |
Extramedullary plasmacytomab | Yes | Yes | Yes |
Number of prior lines of therapy | Yes | Yes | Yes |
Time since MM diagnosis | Yes | Yes | Yes |
Average duration of prior lines of therapy | Yes | Yes | No |
Age | Yes | Yes | Yes |
Hemoglobin level | Yes | Yes | Yes |
LDH level | Yes | Yes | No |
Creatinine clearance | Yes | Yes | No |
ECOG PS | Yes | Yes | No |
Sex | Yes | Yes | No |
Type of MM | Yes | Yes | No |
Prior stem cell transplant | Yes | Yes | No |
Race | No | Yes | No |
Cytogenetic risk profile | No | Yes | Yes |
ECOG PS = Eastern Cooperative Oncology Group Performance Status; ISS = International Staging System; LDH = lactate dehydrogenase; MM = multiple myeloma; PC = physician’s choice; RWPC = real-world physician’s choice; vs. = versus.
aRefractoriness was defined from the case report form as progressive disease or relapse (RWPC cohort) and by the International Myeloma Working Group consensus criteria (MajesTEC-1 study).64
bRefers to soft tissue mass that is not in contact with bone; does not include bone-based plasmacytomas.88
Source: Sponsor-submitted indirect treatment comparison reports.26-30
Model Specifications
In all 3 ITCs included in the pharmacoeconomic model, propensity scores were estimated under an assumed logistic regression model using each cohort (the MajesTEC-1, LocoMMotion, and CARTITUDE-1 studies and the daratumumab trials cohort [the APOLLO, POLLUX, CASTOR, and EQUULEUS trials]) as the dependent variable and selected baseline covariates as independent variables. The estimated propensity scores were then used to derive weights for each participant using the appropriate weighting formulas for the desired target population. Specifically, to target an ATT estimand, MajesTEC-1 study patients were assigned a weight of 1 and the population of the comparator studies reweighted to become similar to the MajesTEC-1 study’s population. Following weighting, balance between the MajesTEC-1 study’s ITT population and the population in the comparator studies was evaluated by comparing unweighted and weighted propensity score distributions. In 2 ITCs comparing the relative efficacy of teclistamab with RWPC therapy from the LocoMMotion trial and ciltacabtagene autoleucel from the CARTITUDE-1 trial, treatment weights were scaled to sum up to the original number of participants in the comparator studies.
The main analysis contained all covariates available for adjustment that did not have a high proportion of missing values or lead to a lower overall balance when included as adjustment variables. Estimates of comparative efficacy were derived for both the unadjusted comparison (before weighting) and the adjusted comparison (after weighting). For the binary outcomes (i.e., ORR, CR or better, VGPR or better), a weighted logistic regression was used to derive an estimate of the OR and the corresponding 95% CI, transformed to response-rate ratio.34 For the time-to-event outcomes (i.e., PFS, duration of response, TTNT, and OS), a weighted Cox proportional hazards model was used to derive an estimate of the HR and the corresponding 95% CI.
Handling of Missing Data
In the ITC comparing the relative efficacy of teclistamab with RWPC therapy from the LocoMMotion trial, low risk imputation was used to impute missing values of the independent variables in the MajesTEC-1 study cohort, and mode value was used to impute missing values for the RWPC cohort. Variables requiring imputation for the MajesTEC-1 study’s population were ISS stage, years since MM diagnosis, time to progression on last regimen, and average duration of prior lines of therapy. For the RWPC cohort from the LocoMMotion trial, several variables required imputation, including ECOG PS, ISS stage, hemoglobin levels, creatinine clearance, LDH levels, type of MM, time to progression on last regimen, and race.
In the ITC comparing the relative efficacy of teclistamab with PC therapy from the daratumumab trials cohort, mode value was used to impute missing values of the clinically important and prognostic covariates. In both the MajesTEC-1 study and the daratumumab trials cohort, the only variable requiring imputation was ISS stage. Sensitivity analyses were conducted by including missing values for covariates as a separate category to assess the impact of imputation.
No methods for handling missing data were performed or reported in the ITC comparing teclistamab with ciltacabtagene autoleucel.
Assessment of Proportional Hazards
The appropriateness of the proportional hazards assumption used in the estimation of the HR of the survival outcomes was assessed based on visual inspection of the log-cumulative hazard plot, visual inspection of the Schoenfeld residuals plot, and the performance of the Grambsch-Therneau test (with a P value of less than 0.05 considered to indicate a violation of the assumption).35
Subgroup Analysis
Subgroup analyses were not performed in any of the 3 ITCs.
Sensitivity Analysis
Teclistamab (MajesTEC-1 Study) Versus RWPC Therapy (LocoMMotion Study)
Multivariable regressions were conducted that included a binary treatment indicator (teclistamab or RWPC) and covariates for adjustment in the model. For the binary outcomes (e.g., ORR, CR or better, VGPR or better), an unweighted logistic regression model that included the selected baseline characteristics as covariates was used to generate a conditional ORs and the corresponding 95% CI. For the time-to-event outcomes (e.g., PFS, duration of response, TTNT, OS), an unweighted Cox proportional hazards model including the selected baseline characteristics as covariates was used to derive an estimate of the conditional HR and the corresponding 95% CI. The variance was estimated using a sandwich estimator. The fully adjusted sensitivity analysis included race and cytogenetic profile in addition to variables from the main analysis. IPTW using average treatment effect in the overlap (ATO) and average treatment effect weights was also used as sensitivity analyses.
Teclistamab (MajesTEC-1 Study) Versus Ciltacabtagene Autoleucel (CARTITUDE-1 Study)
No sensitivity analyses were performed in the ITC comparing the relative efficacy of teclistamab with ciltacabtagene autoleucel.
Teclistamab (MajesTEC-1 Study) Versus PC Therapy (APOLLO, POLLUX, CASTOR, and EQUULEUS Trials)
Several sensitivity analyses were conducted to complement the primary ITC of the relative efficacy of teclistamab with the daratumumab trials cohort using IPTW. Both a doubly robust estimator and a multivariable regression model (adjusted for all factors) were used to estimate the relative efficacy of the treatment options. A scenario analysis was conducted to investigate the impact on the treatment effect estimates, balance of populations, and effective sample size when adjusting for additional covariates in the analyses.
Detailed methods including outcomes, model specifications, covariates, and sensitivity analyses are presented in Table 28.
Outcomes
Outcomes evaluated in the ITC comparing the relative efficacy of teclistamab with RWPC therapy from the LocoMMotion study included ORR, CR or better, VGPR or better, PFS, OS, duration of response, and TTNT. Outcomes evaluated in the ITC comparing the relative efficacy of teclistamab versus ciltacabtagene autoleucel from the CARTITUDE-1 study included PFS, OS, duration of response, and TTNT. Outcomes evaluated in the ITC comparing the relative efficacy of teclistamab versus PC therapy from the daratumumab trials cohort included ORR, VGPR or better, PFS, and OS. Details of the outcomes included for each ITC are summarized in Table 29.
Table 28: Analysis Methods of ITCs Included in the Pharmacoeconomic Model
Methods | Teclistamab (MajesTEC-1 study) vs. RWPC (LocoMMotion study) | Teclistamab (MajesTEC-1 study) vs. cilta-cel (CARTITUDE-1 study) | Teclistamab (MajesTEC-1 study) vs. PC therapy (APOLLO, POLLUX, CASTOR, and EQUULEUS studies) |
|---|---|---|---|
Analysis methods | Unadjusted and IPTW (sATT) | Unadjusted and IPTW (sATT) | Unadjusted and IPTW (ATT) |
Model estimation |
|
|
|
Balance assessment | SMD was used to assess the degree of imbalance between the groups. Values of > 0.2 were considered to indicate important differences. Additionally, balance was assessed by comparing unadjusted and weighted propensity score distributions. | SMD was used to assess the degree of imbalance between the groups. Values of > 0.2 were considered to indicate important differences. Additionally, balance was assessed by comparing unweighted and weighted propensity score distributions. | SMD was used to assess the degree of imbalance between the groups. Values of > 0.2 were considered to indicate substantial differences. |
Sensitivity analyses |
| NA | A sensitivity analysis was conducted that adjusted for all factors |
Outcomes and analysis population | ITTa population for:
| ITT population for:
| ITT population for:
|
ATT = average treatment effect in the treated; CI = confidence interval; cilta-cel = ciltacabtagene autoleucel; CR = complete response; DoR = duration of response; HR = hazard ratio; IPTW = inverse probability of treatment weighting; ITC = indirect treatment comparison; ITT = intention-to-treat; NA = not applicable; OR = odds ratio; ORR = overall response rate; OS = overall survival; PC = physician’s choice; PFS = progression-free survival; RR = rate ratio; RWPC = real-world physician’s choice; sATT = scaled average treatment effect in the treated; SMD = standardized mean difference; TTNT = time to next treatment; VGPR = very good partial response; vs. = versus.
Note: Details included in Table 28 are from the sponsor’s Summary of Clinical Evidence.83
aITT population of the MajesTEC-1 trial.
Sources: Sponsor-submitted ITC reports.26-30
Outcomes in the MajesTEC-1 and CARTITUDE-1 studies were adjudicated by the IRC, in the LocoMMotion study by the response review committee, and in the daratumumab trials by the investigator. The outcomes were defined as follows:
OS was defined as the time from the index date to the date of a patient’s death due to any cause. Patients who died after consent withdrawal but with data collected as allowed by applicable regulations were considered as having an OS event. If a patient was alive or the vital status was unknown, then the patient’s data were censored at the date the patient was last known to be alive.
PFS was defined as the duration from the index date to the time of progressive disease or death from any cause, whichever occurred first. Patients who had not progressed and were alive at the data cut-off date were censored at the last disease evaluation before the start of any subsequent antimyeloma therapy. Patients who had not progressed and were alive at the data cut-off date were censored at the last disease evaluation before the start of any subsequent antimyeloma therapy.
TTNT was defined as the time from the index date to the initiation of the next therapy line or death, whichever occurred first. Patients who were still alive and did not initiate a next therapy line at the time of the data cut-off were censored at the last date known to be alive.
ORR was defined as the proportion of participants who attained a PR or better according to the IMWG criteria. Response after the start of subsequent therapy or re-treatment with teclistamab (in the MajesTEC-1 study) was not considered.
CR or better was defined as the percentage of participants achieving CR or sCR according to IMWG criteria.
VGPR was defined as the percentage of participants achieving VGPR or better response according to IMWG criteria.
Duration of response was defined as the time from the initial documentation of a PR or better to the date of disease progression, or death due to any cause, whichever occurred first (according to IMWG criteria). Patients who had not progressed and were alive at the data cut-off date were censored at the last disease evaluation before the start of any subsequent antimyeloma therapy or at the last follow-up date, whichever occurred first.
Table 29: List of Comparisons Performed in ITCs Submitted by Sponsor
Comparator | ORR | CR or better | VGPR or better | OS | PFS | DoR | TTNT |
|---|---|---|---|---|---|---|---|
Teclistamab vs. RWPC therapy (LocoMMotion study) | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
Teclistamab vs. ciltacabtagene autoleucel (CARTITUDE-1 study) | No | No | No | Yes | Yes | Yes | Yes |
Teclistamab vs. PC therapy (daratumumab trials cohort) | Yes | Yes | Yes | Yes | Yes | No | Yes |
CR = complete response; DoR = duration of response; ITC = indirect treatment comparison; ORR = overall response rate; OS = overall survival; PC = physician’s choice; RWPC = real-world physician’s choice; TTNT = time to next treatment; VGPR = very good partial response; vs. = versus.
Sources: Sponsor-submitted ITC reports.26-30
Results of ITCs Used to Inform Pharmacoeconomic Model
Summary of Included Studies
Of the 3 ITCs submitted by the sponsor, 2 ITCs compared the relative efficacy of teclistamab (from the MajesTEC-1 study) with PC therapy26-29 (using data from the LocoMMotion study and the daratumumab trials cohort [the APOLLO, POLLUX, CASTOR, and EQUULEUS trials]), and another ITC compared the relative efficacy of teclistamab with ciltacabtagene autoleucel using data from the CARTITUDE-1 trial.30
MajesTEC-1 Study
The MajesTEC-1 trial is an ongoing, phase I and phase II, open-label, multicentre, single-arm study. Eligible patients must have received a diagnosis of MM under IMWG diagnostic criteria, and have prior exposure to at least 1 PI, 1 IMiD, and 1 anti-CD38 monoclonal antibody. A total of 165 patients in the MajesTEC-1 trial (40 patients in phase I and 125 patients in phase II, cohort A) received teclistamab at a recommended dose of 1.5 mg/kg subcutaneously once a week, followed by step-up doses of 0.06 mg/kg and 0.3 mg/kg. In terms of efficacy outcomes, the primary outcome in the MajesTEC-1 study was ORR; secondary outcomes included PR or better, VGPR or better, CR or better, OS, PFS, MRD-negativity rate, duration of response, and TTR; and the exploratory outcome was TTNT. Patient-reported outcomes evaluated in the MajesTEC-1 study included change from baseline in HRQoL, symptoms, and functioning. The index date in the MajesTEC-1 trial was defined as the date of the first dose for the MajesTEC-1 trial.
LocoMMotion Study
The LocoMMotion trial was a prospective, noninterventional study of real-life standard of care in patients with a documented diagnosis of MM according to IMWG diagnostic criteria, who have received at least 3 prior lines of therapy; this included at minimum a PI, an IMiD, and an anti-CD38 monoclonal antibody. A total of 248 patients were enrolled in the LocoMMotion study. The primary outcome in the LocoMMotion trial was ORR; secondary outcomes included VGPR rate, CR rate, sCR rate, MRD-negativity rate, clinical benefit rate, duration response, TTR, TTNT, PFS, and OS. In the LocoMMotion trial, the index date was defined as day 1, cycle 1, of the real-life standard of care treatment.
CARTITUDE-1 Study
The CARTITUDE-1 study is a phase Ib and phase II, open-label, single-arm clinical trial evaluating the safety and efficacy of ciltacabtagene autoleucel in adult patients with RRMM. A total of 113 patients were enrolled in the CARTITUDE-1 study. Eligible patients were diagnosed with MM according to IMWG diagnostic criteria and must have received at least 3 prior lines of therapy or must be double-refractory to an IMiD and a PI. In the CARTITUDE-1 trial, following apheresis and premedication, ciltacabtagene autoleucel was administered as a single infusion dose of 0.75 × 106 CAR-positive viable T-cells per kg. The primary outcome in the CARTITUDE-1 trial was ORR; secondary outcomes included VGPR rate, CR rate, sCR rate, MRD-negativity rate, clinical benefit rate, duration response, TTR, TTNT, PFS, and OS. The ITT population in the CARTITUDE-1 trial included all patients who underwent apheresis, with the index date defined as the date of apheresis.
Daratumumab Clinical Trials
The choice of the 4 daratumumab trials was based on a consideration of existing daratumumab clinical trials for patients with RRMM and the selection of those trials that had completed their primary analysis, had follow-up data available at least 6 months after the primary database lock, and provided subsequent therapy information, including lines of therapy, best response, and progression status. Patients in the daratumumab trials were triple-class exposed and were treated with PC therapy after discontinuing the trial treatments. The daratumumab trials cohort consisted of patients from the long-term follow-up data from the POLLUX, CASTOR, EQUULEUS, and APOLLO trials. Since the patient population in the daratumumab trials cohort originated from various clinical trial populations, it was initially subjected to similar eligibility criteria as participants in the MajesTEC-1 study. Because this ITC analysis retrospectively included patients participating in long-term follow-up clinical trials of daratumumab, it was possible to include patients in the earliest line of therapy initiated after all key selection criteria were met. However, this differed from the MajesTEC-1 study, in which patients may have received additional lines of therapy between the time at which they first met all eligibility criteria and the time at which they were enrolled in the clinical trial. To account for this difference, patients in the daratumumab trials became eligible for this analysis after having at least 3 prior lines of therapy, and patients who received multiple subsequent therapies after meeting eligibility criteria contributed multiple observations (Figure 12). In the daratumumab trials cohort, the index date was defined as the start of each eligible line of therapy.
Overall, 1,577 patients were initially included in the daratumumab trials cohort, of whom 642 patients were triple-class exposed and had received at least 1 treatment regimen. Of the 642 patients, 427 patients with 806 eligible lines of therapy met the MajesTEC-1 study’s key inclusion criteria as they had received at least 3 prior lines of therapy, had an ECOG PS score of less than 2, had a creatinine clearance of less than or equal to 1.5 mg/dL, and had progressed on or within 12 months of the most recent line of therapy.27
Figure 12: Patient From the Physician’s Choice Cohort With Multiple Index Dates
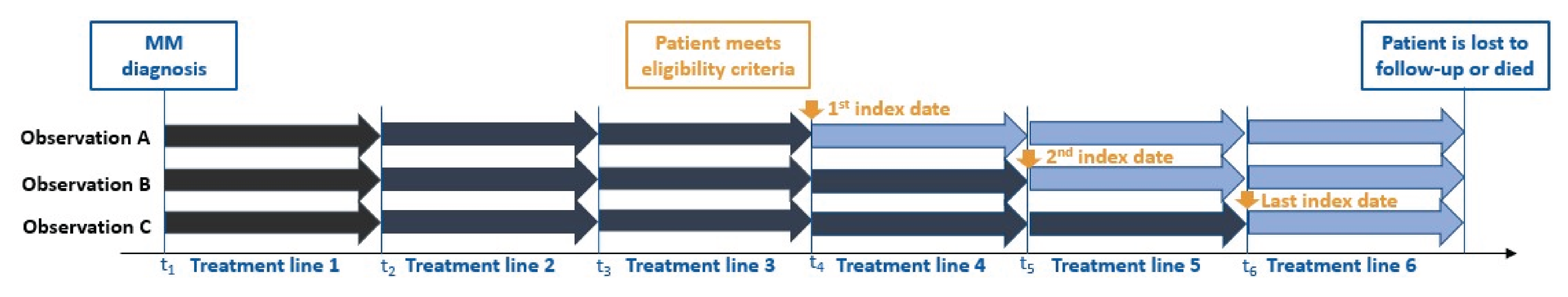
MM = multiple myeloma; t1 = treatment line 1; t2 = treatment line 2; t3 = treatment line 3; t4 = treatment line 4; t5 = treatment line 5; t6 = treatment line 6.
Source: Sponsor-submitted indirect treatment comparison.29
POLLUX Trial
The POLLUX study was an open-label, phase III RCT conducted to evaluate the effectiveness of daratumumab in combination with lenalidomide plus dexamethasone versus lenalidomide plus dexamethasone alone in adult patients with RRMM. A total of 569 patients with MM were randomized in the POLLUX study; they had documented MM, and had received 1 or more prior lines of therapy. The primary outcome in the POLLUX study was proportion; secondary outcomes included time to progression, VGPR or better response, MRD-negativity rate, ORR, OS, TTR, and duration of response.
CASTOR Trial
The CASTOR study was an open-label, phase III RCT conducted to evaluate the effectiveness of daratumumab in combination with bortezomib plus dexamethasone versus bortezomib plus dexamethasone alone in adult patients with RRMM. Patients were eligible for enrolment in the trial if they had documented MM, and had received 1 or more prior lines of therapy. A total of 498 patients were randomized to receive daratumumab in combination with bortezomib plus dexamethasone or bortezomib plus dexamethasone alone. The primary outcome in the CASTOR study was PFS; secondary outcomes included time to progression, VGPR or better response, MRD-negativity rate, ORR, OS, and TTR.
EQUULEUS Trial
The EQUULEUS study was an open-label, nonrandomized, phase Ib trial conducted to evaluate the safety, tolerability, and dose regimen of daratumumab when administered in combination with various treatment regimens for MM. A total of 103 patients with MM were enrolled in the EQUULEUS study who had documented MM and measurable disease. The primary outcome in the EQUULEUS study was the proportion of adverse events and dose-limiting toxicities; secondary outcomes included ORR, OS, CR or better, and duration of response; and exploratory outcomes included PFS, MRD-negativity rate, and pharmacokinetics.
APOLLO Trial
The APOLLO study was an open-label, phase III RCT conducted to evaluate the effectiveness of daratumumab in combination with pomalidomide and dexamethasone versus pomalidomide and dexamethasone alone in adult participants with RRMM. Patients were eligible for enrolment in the trial if they had measurable disease, had received 1 or more prior lines of therapy (including lenalidomide and a PI), and were refractory to lenalidomide if only 1 prior line of therapy had been received. In total, 304 participants were randomized in the APOLLO trial. The primary outcome in the APOLLO trial was PFS; secondary outcomes included VGPR or better, MRD-negativity rate, ORR, OS, duration of response, TTNT, and TTR.
The included studies were assessed for homogeneity. Important differences across trials for key characteristics are summarized in Table 30.
Table 30: Assessment of Homogeneity for ITCs Submitted by Sponsor
Characteristic | Description and handling of potential effect modifiers |
|---|---|
Disease severity | Teclistamab (MajesTEC-1 study) vs. RWPC therapy (LocoMMotion study) Compared to the LocoMMotion study, the MajesTEC-1 trial had a higher proportion of patients with ISS stage I disease, immunoglobulin G subtype, and extramedullary plasmacytomas, and who were aged younger than 65 years, were penta-refractory, had a prior stem cell transplant, and had creatinine clearance between 60 mL per minute and 90 mL per minute. Compared to the MajesTEC-1 trial, the LocoMMotion study had a higher proportion of patients with ISS stage III disease, and who were triple-refractory and had creatinine clearance of less than 60 mL per minute. Teclistamab (MajesTEC-1 study) vs. ciltacabtagene autoleucel (CARTITUDE-1 study) Compared to the CARTITUDE-1 trial, the MajesTEC-1 trial had a greater proportion of patients who were non–triple-refractory and white. The CARTITUDE-1 trial had a greater proportion of patients who were penta-refractory (to at least 2 IMiDs, 2 PIs, and an anti-CD38 mAB), had ISS stage III disease, and had more prior lines of therapy. Teclistamab (MajesTEC-1 study) vs. PC therapy (APOLLO, POLLUX, CASTOR, and EQUULEUS studies) Compared to the PC cohort, the MajesTEC-1 trial had a higher proportion of patients who had an ECOG PS score of 1, had prior stem cell transplant, and had hemoglobin levels < 12 g/dL. Compared to the MajesTEC-1 trial, the PC cohort had a greater proportion of patients who were triple- or quad-refractory and were aged 65 years to 74 years. |
Trial eligibility criteria | Teclistamab (MajesTEC-1 study) vs. RWPC therapy (LocoMMotion study) Both the MajesTEC-1 study and the LocoMMotion study included patients with a documented diagnosis of MM according to IMWG diagnostic criteria. The MajesTEC-1 trial required patients in part 1 and part 2 to have received prior lines of therapy that included a PI, an IMiD, and an anti-CD38 antibody in any order during the course of treatment, while the LocoMMotion trial required patients to have received at least 3 prior MM treatment lines or to be double-refractory to a PI and an IMiD. The MajesTEC-1 trial included patients who were aged at least 18 years whereas the LocoMMotion trial did not specify a lower age limit. Both studies enrolled patients with an ECOG PS score of 0 or 1. The MajesTEC-1 trial required patients in phase II to have documented disease progression (based on investigator assessment of response by IMWG criteria) on or within 12 months of the most recent line of therapy. Patients with documented evidence of disease progression within the previous 6 months and who were refractory or nonresponsive to their most recent line of therapy afterwards were also eligible. The LocoMMotion study required patients to have documented evidence of disease progression based on the study physician’s determination of response by the IMWG response criteria on or after the last treatment regimen. The MajesTEC-1 trial had specific exclusion criteria whereas the LocoMMotion trial had no exclusion criteria as the study was observational in nature. Teclistamab (MajesTEC-1 study) vs. ciltacabtagene autoleucel (CARTITUDE-1 study) Key eligibility criteria were generally aligned across the MajesTEC-1 and CARTITUDE-1 studies. Both trials included patients with a documented diagnosis of MM according to IMWG diagnostic criteria. Overall, both trials had similar inclusion and exclusion criteria. Specifically, the trials required patients to have received at least 3 prior MM treatment lines of therapy and included patients who were aged at least 18 years, with an ECOG PS score of 0 or 1. Both studies required patients to have documented disease progression (based on investigator assessment of response by IMWG criteria) on or within 12 months of the most recent line of therapy. Patients with documented evidence of progressive disease within the previous 6 months and who were refractory or nonresponsive to their most recent line of therapy afterwards were also eligible. Teclistamab (MajesTEC-1 study) vs. PC therapy from daratumumab trials cohort (APOLLO, POLLUX, CASTOR, and EQUULEUS studies) Key eligibility criteria were generally aligned across the MajesTEC-1 study and the daratumumab trials, including MM diagnosis per IMWG diagnostic criteria, evidence of disease progression during or after the patient’s last line of therapy, and prior exposure to at least 1 line of therapy. The EQUULEUS trial did not require evidence of disease progression on the last line of therapy; rather, patients were eligible if they progressed on a regimen that combined lenalidomide and bortezomib or consecutive regimens that contained lenalidomide and bortezomib separately. |
Timing of end point evaluation | Teclistamab (MajesTEC-1 study) vs. RWPC therapy (LocoMMotion study) In the MajesTEC-1 study, the median duration of follow-up was 14.1 months, with a clinical cut-off date of March 16, 2022.28 In the LocoMMotion study, the median duration of follow-up was 16.1 months, with a clinical cut-off date of November 2, 2021.28 End point evaluation in both cohorts was performed each cycle. Teclistamab (MajesTEC-1 study) vs. ciltacabtagene autoleucel (CARTITUDE-1 study) In the MajesTEC-1 study, the median duration of follow-up was 22.8 months, with a clinical cut-off date of January 4, 2023. The median duration of follow-up in the CARTITUDE-1 study was 27.7 months, with a clinical cut-off date of January 2022. End point evaluation in both cohorts was performed each cycle. Teclistamab (MajesTEC-1 study) vs. PC therapy (APOLLO, POLLUX, CASTOR, and EQUULEUS studies) In the MajesTEC-1 study, the median duration of follow-up was 14.1 months, with a clinical cut-off date of March 16, 2022.27 In the daratumumab trials cohort, the overall median duration of follow-up was 30.9 months.27 |
Definitions of end points | In general, the definitions of end points were similar across the included studies. However, outcomes were assessed by the IRC in the MajesTEC-1 trial, by the RRC in the CARTITUDE-1 trial, and by the investigator in the daratumumab trials cohort. |
Withdrawal frequency | Not reported in data extraction. |
Clinical trial setting | In the MajesTEC-1 trial, patients were recruited from study centres from Belgium, Canada, France, Germany, Italy, the Netherlands, Spain, Sweden, the UK, and the US. In the LocoMMotion trial, patients were enrolled at 76 sites across 9 European countries and the US. In the CARTITUDE-1 trial, patients were recruited from 16 study centres in the US. In the POLLUX trial, patients were enrolled at 135 sites that spanned 18 countries in Europe, North America, and the Asia-Pacific region. In the CASTOR trial, patients were enrolled at 115 centres in 16 countries across Europe, North America, South America, and the Asia-Pacific region. |
Study design | The MajesTEC-1 trial is an ongoing, phase I and phase II, multicentre, open-label, single-arm clinical trial. The LocoMMotion trial was a prospective, noninterventional study. The CARTITUDE-1 trial is a phase Ib and phase II, multicentre, open-label, single-arm clinical trial. The POLLUX, CASTOR, and APOLLO studies were open-label, phase III RCTs. The EQUULEUS study was an open-label, nonrandomized, phase Ib study. |
ECOG PS = Eastern Cooperative Oncology Group Performance Status; IMiD = immunomodulatory drug; IMWG = International Myeloma Working Group; IRC = independent review committee; ISS = International Staging System; ITC = indirect treatment comparison; mAb = monoclonal antibody; MM = multiple myeloma; PC = physician’s choice; PI = proteasome inhibitor; RWPC = real-world physician’s choice; RCT = randomized controlled trial; RRC = response review committee; vs. = versus.
Note: Details included in Table 30 are from the sponsor’s Summary of Clinical Evidence.83
Sources: Sponsor-submitted indirect treatment comparison reports.26-30
Evidence Network
Figure 13 presents the overall network of evidence for efficacy outcomes for the 6 ITCs submitted by the sponsor.
Teclistamab (MajesTEC-1 Study) Versus RWPC Therapy (LocoMMotion Study)
Table 31 shows a summary of the baseline characteristics of the patients in the MajesTEC-1 and LocoMMotion studies, both before and after weighting. Before weighting, moderate to substantial differences were observed for many of the variables included in the main IPTW analysis, including ISS stage, immunoglobulin subtypes, extramedullary plasmacytomas, refractory status, age, prior stem cell transplant, time to progression on last regimen, duration of prior lines of therapy, LDH levels, type of MM, creatinine clearance, race, and cytogenetic risk. After reweighting, populations in the MajesTEC-1 and LocoMMotion studies were more balanced. After adjustment, although there were no imbalances with a SMD greater than 0.2, observable differences remained in the ITT populations with regard to refractory status and time to progression on last regimen. Cytogenetic risk was considered an important risk factor; however, it was not included in the main analyses due to a high level of missingness in the LocoMMotion study cohort (37.1%). As the LocoMMotion study’s population only included a low number of non-white patients, including race as a variable in the adjustment set led to large weights for these patients and decreased balance for all other variables.
Table 31: Baseline Characteristics of MajesTEC-1 and LocoMMotion Studies Before and After Weighting, ITT Population [Redacted]
|||||||||||||| | ||||||||||||||| | ||||||||||||||| | |||
|---|---|---|---|---|---|
|||||||||||||||| | ||| | |||||||||| |||| |||| |||||||||| |||| |||||||| | ||| | ||
|||||||||| ||||||||| | |||||
|| ||||| |||||| |||||||||| | || |||||| | || |||||| | ||||| | || |||||| | ||||| |
|||||| |||||||||| | || |||||| | || |||||| | || |||||| | ||
|||| |||||||||| | || |||||| | || |||||| | || |||||| | ||
||||| |||||||||| | || |||||| | || |||||| | || |||||| | ||
||| |||||||| | |||||
||| | || |||||| | || |||||| | ||||| | ||| |||||| | ||||| |
|| | || |||||| | || |||||| | || |||||| | ||
||| | || |||||| | || |||||| | || |||||| | ||
|||| || ||||||||||| || |||| ||||||| || |||||||||| | |||||
||| | || |||||| | || |||||| | ||||| | || |||||| | ||||| |
||| | ||| |||||| | ||| |||||| | ||| |||||| | ||
|||||||||||||| |||||||||||||| | |||||
||| | || |||||| | || ||||| | ||||| | || |||||| | ||||| |
|| | ||| |||||| | ||| |||||| | ||| |||||| | ||
|||||| || ||||| ||||| || ||||||||||| | |||||
||| | || |||||| | ||| |||||| | ||||| | ||| |||||| | ||||| |
||| | || |||||| | ||| |||||| | ||| |||||| | ||
||||| ||||| || |||||||||| |||||||| | |||||
||| | || |||||| | ||| |||||| | ||||| | ||| |||||| | ||||| |
||| | || |||||| | ||| |||||| | ||| |||||| | ||
||||||| |||||||| || ||||| ||||| || |||||||| ||||||||| | |||||
||| | || |||||| | || |||||| | ||||| | || |||||| | ||||| |
||| | || |||||| | || |||||| | || |||||| | ||
||| | || |||||| | ||| |||||| | ||| |||||| | ||
||||||| | |||||
||| | || |||||| | || |||||| | ||||| | ||| |||||| | ||||| |
||| | || |||||| | ||| |||||| | ||| |||||| | ||
|||||||||| |||||||||| | |||||
||| | ||| |||||| | ||| |||||| | ||||| | ||| |||||| | ||||| |
||| | || |||||| | || |||||| | || |||||| | ||
||| |||||| ||||||||||||| | |||||
||| | ||| |||||| | ||| |||||| | ||||| | ||| |||||| | ||||| |
||| | || |||||| | || |||||| | || |||||| | ||
|||||||||| |||||||||||||| |||||| | |||||
||| | || |||||| | || |||||| | ||||| | || |||||| | |||||| |
||| | || |||||| | || |||||| | ||| |||||| | ||
||| | || |||||| | || |||||| | || |||||| | ||
|||| ||||||||| | |||||
||| | || |||||| | || |||||| | ||||| | || |||||| | ||||| |
||| | ||| |||||| | ||| |||||| | ||| |||||| | ||
|||||| | |||||
|||| | || |||||| | ||| |||||| | ||||| | ||| |||||| | ||||| |
|||||| | || |||||| | ||| |||||| | ||| |||||| | ||
|||| || ||||| | |||||
||| | || |||||| | ||| |||||| | ||||| | ||| |||||| | ||||| |
||||||| | || |||||| | ||| |||||| | || |||||| | ||
||||| |||| |||| ||||||||||||| | |||||
||| | ||| |||||| | ||| |||||| | ||||| | ||| |||||| | ||||| |
|| | || |||||| | || |||||| | || |||||| | ||
ECOG PS = Eastern Cooperative Oncology Group Performance Status; ISS = International Staging System; ITT = intention-to-treat; LDH = lactate dehydrogenase; LOT = line of therapy; mAb = monoclonal antibody; MM = multiple myeloma; sATT = scaled average treatment effect in the treated; SMD = standardized mean difference.
Source: Sponsor-submitted indirect treatment comparison report.9,38
Figure 13: Overall Evidence Network
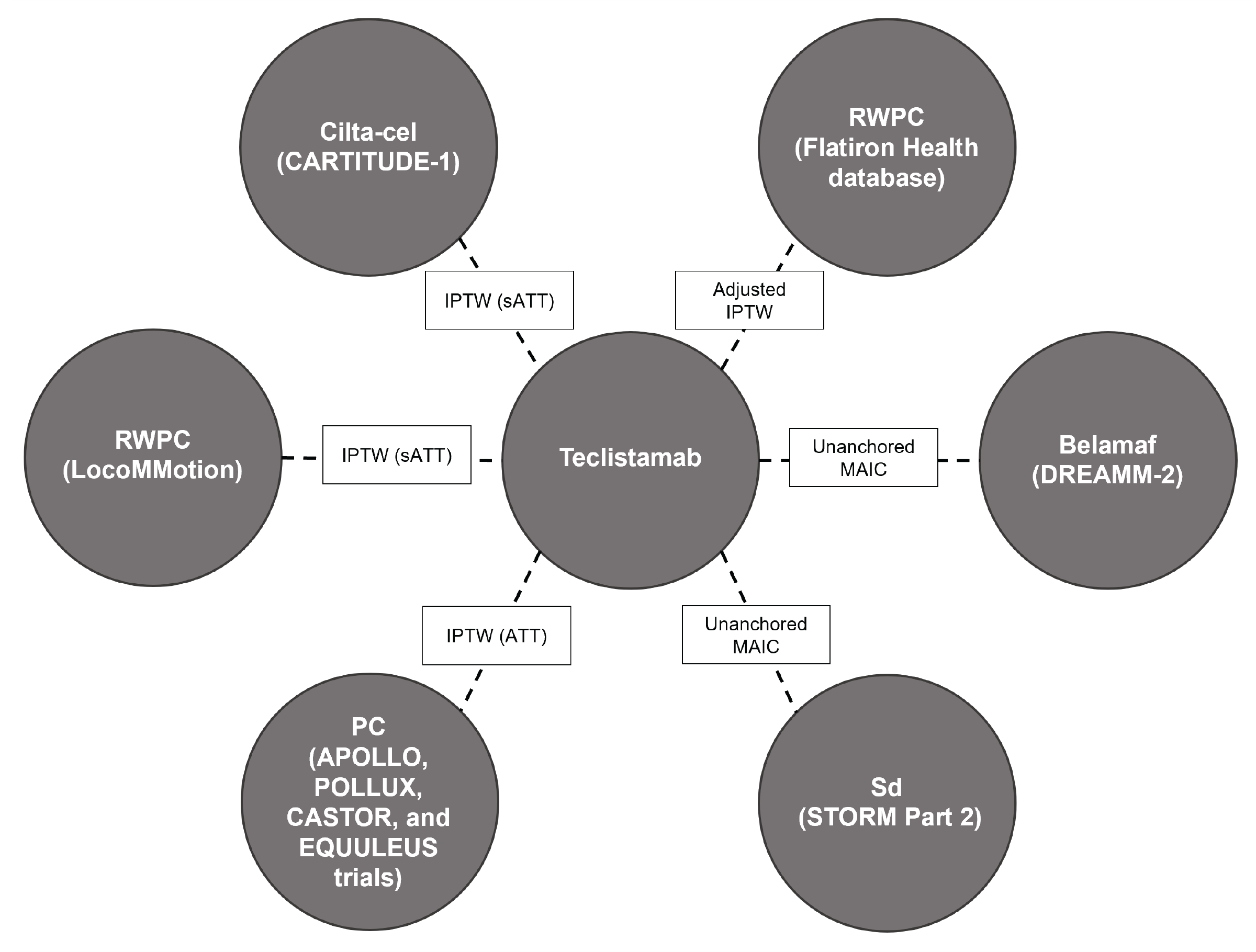
ATT = average treatment effect in the treated; belamaf = belantamab mafodotin; cilta-cel = ciltacabtagene autoleucel; IPTW = inverse probability of treatment weighting; MAIC = matching-adjusted indirect comparison; PC = physician’s choice; sATT = scaled average treatment effect in the treated; Sd = selinexor plus dexamethasone.
Source: Sponsor’s Summary of Clinical Evidence.83
Approximately 90 unique treatment regimens were used in the LocoMMotion study, including corticosteroids, PIs, IMiDs, alkylating drugs, and anti-CD38 monoclonal antibodies, and various combinations, reflecting the existing diversity of real-life antimyeloma treatments in this population.28 The most frequently used PI, IMiD, and anti-CD38 monoclonal antibody were carfilzomib (25.4%), pomalidomide (29.8%), and daratumumab (9.3%), respectively. Table 32 presents the real-world treatment combination received by more than 4 patients in the LocoMMotion study.
Table 32: Treatment Regimens in the LocoMMotion Study Cohort [Redacted]
Treatment regimen | Frequency, n (%) (N = 248) |
|---|---|
||||||||||| || ||||||||||| |||| ||||||||||||| | || |||||| |
|||||||||||||||| || ||||||||||| |||| ||||||||||||| |||| |||||||||||| | || |||||| |
||||||||||||| || ||||||||||| |||| |||||||||||| | || |||||| |
||||||||||||| || ||||||||||| |||| |||||||| |||| |||||||||||| | || ||||| |
|||||||||| || ||||||||||| |||| ||||||||||||| |||| |||||||||||| | || ||||| |
|||||||||||| || ||||||||||| |||| |||||||||| |||| ||||||||||||| | || ||||| |
||||||||||| || ||||||||||| |||| |||||||||||||||| |||| ||||||||||||| | || ||||| |
||||||||||||| || ||||||||||| |||| |||||||||| |||| |||||||||||| | || ||||| |
||||||||||||| || ||||||||||| |||| |||||||||||| | || ||||| |
|||||||||| || ||||||||||| |||| ||||||||||||| |||| ||||||||||| | || ||||| |
||||||||||| || ||||||||||| |||| ||||||||||||| |||| |||||||||||| | || ||||| |
||||||||||| || ||||||||||| |||| ||||||||||||| |||| |||||||||||| | || ||||| |
|||||||||| ||||||||| | || ||||| |
|||||||||||| || ||||||||||| |||| |||||||||| | || ||||| |
|||||||||||||||| || ||||||||||| |||| ||||||||||||| | || ||||| |
||||||||| | || ||||| |
Source: Sponsor-submitted indirect treatment comparison report.9,38
Efficacy Results
Overall Survival
The log-cumulative hazard plot and Schoenfeld residuals plots of OS were visually inspected; evidence of violation of the proportional hazards assumption was indicated before significant patient drop-off. However, the Grambsch-Therneau test was nonsignificant at the prespecified level (P value = 0.2646), indicating that there was insufficient evidence of a violation of the assumption.
Results comparing OS between teclistamab (MajesTEC-1 study) and RWPC therapy (LocoMMotion study) before and after adjustment are summarized in Table 33 and Table 34. In the MajesTEC-1 and LocoMMotion studies, the median OS were |||||||||| |||| ||| ||||| || || ||||||| ||| ||||| |||||| |||| ||| ||||| || ||||| |||||||| ||||||||||||| || |||||||||||| ||||||||| ||||||||||| ||| |||||| || ||| ||||| |||||| |||| ||| |||| || ||||| |||||||.
In the main analysis, following adjustment, the estimated HR of OS for teclistamab versus RWPC therapy was |||| |||| ||| |||| ||||||| || |||||| || |||||||||||. The results of the sensitivity analyses, including the analysis adjusted for all variables, were consistent with the main analysis results.
Progression-Free Survival
The log-cumulative hazard plot and Schoenfeld residuals plots of PFS were visually inspected; some evidence of violation of the proportional hazards assumption was indicated before significant patient drop-off. However, the Grambsch-Therneau test was nonsignificant at the prespecified level (P value = 0.1241), indicating that there was insufficient evidence of a violation of the assumption.
Results comparing PFS between teclistamab and RWPC therapy before and after adjustment are summarized in Table 33 and Table 34. In the MajesTEC-1 and LocoMMotion studies, the median PFS was ||||| |||||| |||| ||| |||| || ||||| ||||||| ||| |||| |||||| |||| ||| |||| || ||||||||||| ||||||||||||| || |||||||||||| ||||||||| ||||||||||| ||| |||||| ||| ||| |||| |||||| |||| ||| |||| || |||| |||||||| || |||||| || |||||||||||.
In the main analysis, following adjustment, the estimated HR of PFS for teclistamab versus RWPC therapy was |||| |||| ||| |||| ||||||| || |||||| || |||||||||||. The results of the sensitivity analyses, including the analysis adjusted for all variables, were consistent with the main analysis results.
Duration of Response
The log-cumulative hazard plot and Schoenfeld residuals plots of duration of response were visually inspected and some evidence of violation of the proportional hazards assumption was indicated before significant patient drop-off. However, the Grambsch-Therneau test was nonsignificant at the prespecified level (P value = 0.0831), indicating that there was insufficient evidence of a violation of the assumption.
Results comparing duration of response between teclistamab and RWPC therapy before and after adjustment are summarized in Table 33 and Table 34. Among responders (with PR or better) in the MajesTEC-1 trial (n = 104) and the LocoMMotion trial (n = 79), the median duration of response was ||||| |||||| |||| ||| ||||| || || ||||||| ||| |||| |||||| |||| ||| |||| || ||||| |||||||| ||||||||||||. Among responders in the LocoMMotion trial, following adjustment (n = 64), the median duration of response was |||| |||||| |||| |||||| || ||||| |||||||.
In the main analysis, following adjustment, the estimated HR for duration of response for teclistamab versus RWPC therapy was |||| |||| ||| |||| || |||||| || |||||| || |||||||||||. The results of the sensitivity analyses, including the analysis adjusted for all variables, were consistent with the main analysis results.
Time to Next Treatment
The log-cumulative hazard plot and Schoenfeld residuals plots of TTNT were visually inspected and some evidence of violation of the proportional hazards assumption was indicated before significant patient drop-off. However, the Grambsch-Therneau test was nonsignificant at the prespecified level (P value = 0.2656), indicating that there was insufficient evidence of a violation of the assumption.
Results comparing TTNT between teclistamab and RWPC therapy before and after adjustment are summarized in Table 33 and Table 34. In the MajesTEC-1 and LocoMMotion studies, the median TTNT was ||||| |||||| |||| ||| |||| || ||||| ||||||| ||| |||| |||||| |||| ||| ||||| |||| |||||||| ||||||||||||. In the LocoMMotion study, following adjustment, the median TTNT was |||| |||||| |||| ||| |||| || |||| |||||||.
In the main analysis, following adjustment, the estimated HR for TTNT for teclistamab versus RWPC therapy was 0.43 (95% CI, 0.33 to 0.56), in favour of teclistamab. The results of the sensitivity analyses, including the analysis adjusted for all variables, were consistent with the main analysis results.
Table 33: Unadjusted and Adjusted Kaplan-Meier Estimated OS, PFS, DoR, and TTNT [Redacted]
|||||| | |||||| |||||||| | ||
|---|---|---|---|
|||||||||||||||| | ||||||||||| | ||
|||||| |||||||||||||| | ||||| |||||||||||||| | ||
||||| |||||| || |||||| | |||
|||||| |||| |||| |||||| | ||||| |||||| || ||| | ||||| |||||| || |||||| | ||||| ||||| || |||||| |
||||| |||||| || |||||| | |||
|||||| |||| |||| |||||| | ||||| ||||| || |||||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
||||| |||||| || |||||| | |||
|||||| |||| |||| |||||| | ||||| |||||| || ||| | |||| ||||| || |||||| | |||| ||||| || |||||| |
||||| |||||| || |||||| | |||
|||||| |||| |||| |||||| | ||||| ||||| || |||||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
CI = confidence interval; DoR = duration of response; HR = hazard ratio; IPTW = inverse probability of treatment weighting; OS = overall survival; PFS = progression-free survival; RWPC = real-world physician’s choice; sATT = scaled average treatment effect in the treated; TTNT = time to next treatment.
Notes: Details included in Table 33 are from the sponsor’s Summary of Clinical Evidence.83
The main analysis was adjusted for refractory status, International Staging System stage, time to progression on last regimen, extramedullary plasmacytomas, number of prior lines of therapy, years since multiple myeloma diagnosis, average duration of prior lines of therapy, age, hemoglobin, lactate dehydrogenase level, creatinine clearance, ECOG PS score, sex, type of multiple myeloma, and prior stem cell transplant.
Source: Sponsor-submitted indirect treatment comparison report.9,38
Table 34: Comparison of OS, PFS, DoR, and TTNT for Teclistamab (MajesTEC-1 Study) With RWPC Therapy (LocoMMotion Study) [Redacted]
|||||||| | |||||||| |||||||||||| |||| ||| |
|---|---|
||||| |||||| || |||||| | |
|||||||||| | |||| ||||| || ||||| |
|||| ||||||||| |||| |||| |||| | |||| ||||| || ||||| |
||||||||||| |||||||| | |
|||||||||| ||||| |||||||| | |||| ||||| || ||||| |
|||||||| | |||| ||||| || ||||| |
|||||||| | |||| ||||| || ||||| |
||||||||||||| |||||||||| | |||| ||||| || ||||| |
||||| |||||| || |||||| | |
|||||||||| | |||| ||||| || ||||| |
|||| ||||||||| |||| |||| |||| | |||| ||||| || ||||| |
||||||||||| |||||||| | |
|||||||||| ||||| |||||||| | |||| ||||| || ||||| |
|||||||| | |||| ||||| || ||||| |
|||||||| | |||| ||||| || ||||| |
||||||||||||| |||||||||| | |||| ||||| || ||||| |
||||| |||||| || |||||| | |
|||||||||| | |||| ||||| || ||||| |
|||| ||||||||| |||| |||| |||| | |||| ||||| || ||||| |
||||||||||| |||||||| | |
|||||||||| ||||| |||||||| | |||| ||||| || ||||| |
|||||||| | |||| ||||| || ||||| |
|||||||| | |||| ||||| || ||||| |
||||||||||||| |||||||||| | |||| ||||| || ||||| |
||||| |||||| || |||||| | |
|||||||||| | |||| ||||| || ||||| |
|||| ||||||||| |||| |||| |||| | |||| ||||| || ||||| |
||||||||||| |||||||| | |
|||||||||| ||||| |||||||| | |||| ||||| || ||||| |
|||||||| | |||| ||||| || ||||| |
|||||||| | |||| ||||| || ||||| |
||||||||||||| |||||||||| | |||| ||||| || ||||| |
ATE = average treatment effect; ATO = average treatment effect in the overlap; CI = confidence interval; DoR = duration of response; HR = hazard ratio; IPTW = inverse probability of treatment weighting; OS = overall survival; PFS = progression-free survival; RWPC = real-world physician’s choice; sATT = scaled average treatment effect in the treated; TTNT = time to next treatment.
Notes: Details included in Table 34 are from the sponsor’s Summary of Clinical Evidence.83
The main analysis was adjusted for refractory status, International Staging System stage, time to progression on last regimen, extramedullary plasmacytomas, number of prior lines of therapy, years since multiple myeloma diagnosis, average duration of prior lines of therapy, age, hemoglobin, lactate dehydrogenase level, creatinine clearance, Eastern Cooperative Oncology Group Performance Status score, sex, type of multiple myeloma, and prior stem cell transplant. The sensitivity analysis including all variables was adjusted for all variables in the main analysis, plus race and cytogenetic profile.
Source: Sponsor-submitted indirect treatment comparison report.9,38
Clinical Response Outcomes
Results comparing the MajesTEC-1 and LocoMMotion studies for clinical response outcomes, including ORR, CR or better, and VGPR or better, are summarized in Table 35.
The proportion of patients with overall response was 63.0% for teclistamab and 31.9% for RWPC therapy. In the LocoMMotion study, following adjustment, the proportion of patients with overall response was 25.9%. In the main analysis, following adjustment, the OR of ORR for teclistamab versus RWPC therapy was |||| |||| ||| |||| || |||||| || |||||| || |||||||||||. The results of the sensitivity analyses, including the analysis adjusted for all variables, were consistent with the main analysis results.
For CR or better, the proportion was 45.5% for teclistamab and 0.4% for RWPC therapy. In the LocoMMotion study, following adjustment, the proportion of patients with CR or better was 0.4%. Following adjustment, the OR of CR or better for teclistamab versus RWPC therapy was |||||| |||||| || |||||||||| || |||||| || |||||||||||. The results of the sensitivity analyses, including the analysis adjusted for all variables, were consistent with the main analysis results.
For VGPR or better, the proportion was 59.4% for teclistamab and 13.3% for RWPC therapy. In the LocoMMotion study, following adjustment, the proportion of patients with VGPR or better was 10.3%. In the main analysis, following adjustment, the OR of CR or better for teclistamab versus RWPC therapy ||| ||||| |||| ||| |||| || ||||||| || |||||| || |||||||||||. The results of the sensitivity analyses, including the analysis adjusted for all variables, were consistent with the main analysis results.
Health-Related Quality of Life
No results for HRQoL were included in the study comparing the MajesTEC-1 and LocoMMotion trials.
Safety
No results for safety were included in the study comparing the MajesTEC-1 and LocoMMotion trials.
Teclistamab (MajesTEC-1 Study) Versus Ciltacabtagene Autoleucel (CARTITUDE-1 Study)
Table 36 presents a summary of baseline characteristics of the patients in the MajesTEC-1 and CARTITUDE-1 studies, both before and after sATT weighting. Before weighting, moderate to substantial differences were observed for many of the main analysis variables, including ISS stage, refractory status, age, prior stem cell transplant, time to progression on last regimen, duration of prior lines of therapy, creatinine clearance, ECOG PS score, years since MM diagnosis, race, and cytogenetic risk. After weighting, populations in the MajesTEC-1 and LocoMMotion trials were more balanced. After weighting, though there were no substantial imbalances, observable differences remained in the ITT populations with regard to refractory status, age, hemoglobin level, and creatinine clearance.
Table 35: Comparison of Clinical Response Results for Teclistamab (MajesTEC-1 Study) Versus RWPC Therapy (LocoMMotion Study) [Redacted]
|||||||| | |||||||||||||||||| | |||| ||||||||||||||| | || |||| ||| | || |||| ||| |
|---|---|---|---|---|
||| | ||||
|||||||||| | |||| | |||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
|||| ||||||||| |||| |||| |||| | |||| | |||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
||||||||||| |||||||| | ||||
|||||||||| ||||| |||||||| | |||| | |||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
|||||||| | |||| | |||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
|||||||| | |||| | |||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
||||||||||||| |||||||||| | |||| | |||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
|| || |||||| | ||||
|||||||||| | |||| | ||| | |||||| |||||| || ||||||||| | |||||| |||||| || ||||||| |
|||| ||||||||| |||| |||| |||| | |||| | ||| | |||||| |||||| || ||||||||| | |||||| |||||| || ||||||| |
||||||||||| |||||||| | ||||
|||||||||| ||||| |||||||| | |||| | ||| | |||||| |||||| || ||||||||| | ||||| |||||| || ||||||| |
|||||||| | |||| | ||| | |||||| ||||| || ||||||||| | ||||| ||||| || ||||||||| |
|||||||| | |||| | ||| | |||||| |||||| || ||||||||| | |||||| |||||| || ||||||| |
||||||||||||| |||||||||| | |||| | ||| | |||||| |||||| || ||||||||| | |||||| |||||| || ||||||| |
|||| || |||||| | ||||
|||||||||| | |||| | |||| | |||| ||||| || |||||| | |||| ||||| || ||||| |
|||| ||||||||| |||| |||| |||| | |||| | |||| | ||||| ||||| || |||||| | |||| ||||| || ||||| |
||||||||||| |||||||| | ||||
|||||||||| ||||| |||||||| | |||| | |||| | ||||| ||||| || |||||| | |||| ||||| || ||||| |
|||||||| | |||| | |||| | ||||| ||||| || |||||| | |||| ||||| || ||||| |
|||||||| | |||| | |||| | |||| ||||| || |||||| | |||| ||||| || ||||| |
||||||||||||| |||||||||| | |||| | ||| | ||||| ||||| || |||||| | |||| ||||| || ||||| |
ATE = average treatment effect; ATO = average treatment effect in the overlap; CI = confidence interval; CR = complete response; IPTW = inverse probability of treatment weighting; OR = odds ratio; ORR = overall response rate; RR = rate ratio; RWPC = real-world physician’s choice; sATT = scaled average treatment effect in the treated; VGPR = very good partial response.
Notes: Details included in Table 35 are from the sponsor’s Summary of Clinical Evidence.83
The main analysis was adjusted for refractory status, International Staging System stage, time to progression on last regimen, extramedullary plasmacytomas, number of prior lines of therapy, years since multiple myeloma diagnosis, average duration of prior lines of therapy, age, hemoglobin, lactate dehydrogenase level, creatinine clearance, Eastern Cooperative Oncology Group Performance Status score, sex, type of multiple myeloma, and prior stem cell transplant. The sensitivity analysis including all variables was adjusted for all variables in the main analysis, plus race and cytogenetic profile.
Source: Sponsor-submitted indirect treatment comparison report.9,38
Table 36: Baseline Characteristics of MajesTEC-1 and CARTITUDE-1 Studies Before and After Weighting, ITT Population [Redacted]
|||||||||||||| | ||||||||||||||| | ||||||||||||||| | |||
|---|---|---|---|---|---|
||||||||||||||| | ||| | |||||||||| |||| |||| | ||| | ||
|||||||||| |||||||||| | |||||
|| ||||| |||||| |||||||||| | || |||||| | || |||||| | ||||| | || |||||| | ||||| |
|||||| ||||||||| | || |||||| | ||| | || |||||| | ||
|||| ||||||||| | || |||||| | || |||||| | || |||||| | ||
||||| ||||||||| | || |||||| | || |||| | || |||||| | ||
||| |||||||||||||| | |||||
|||||| | || |||||| | || |||||| | ||||| | || |||||| | ||||| |
|||||| | || |||||| | || |||||| | || |||||| | ||
|||||| | || |||||| | || |||||| | || |||||| | ||
|||| || ||||||||||| || |||| ||||||| || |||||||||| | |||||
|||||| | || |||||| | || |||||| | |||||| | || |||||| | |||||| |
|||||| | ||| |||||| | || |||||| | || |||||| | ||
|||||||||||||| |||||||||||||| | |||||
||| | || |||| | || |||||| | |||||| | || |||||| | |||||| |
|| | ||| |||| | || |||||| | || |||||| | ||
|||||| || ||||| ||||| || |||||||||| | |||||
|||||| | || |||||| | || |||||| | |||||| | || |||||| | ||||| |
|||||| | || |||||| | || |||||| | || |||||| | ||
||||| ||||| || |||||||||| |||||||| | |||||
|||||| | || |||||| | || |||||| | ||||| | || |||||| | ||||| |
|||||| | || |||||| | || |||||| | || |||||| | ||
||||||| |||||||| || ||||| |||||| ||||||||| | |||||
|||| | || |||||| | || |||||| | ||||| | || |||||| | ||||| |
|||| || ||||| | || |||||| | || |||||| | || |||||| | ||
|||||| | ||| |||||| | || |||||| | || |||||| | ||
|||||| | |||||
|||||| | || |||||| | || |||||| | ||||| | || |||||| | |||||| |
|||||| | || |||||| | || |||||| | || |||||| | ||
|||||||||| |||||||||| | |||||
|||||| | ||| |||||| | || |||||| | |||||| | || |||||| | ||||| |
|||||| | || |||||| | || |||||| | || |||||| | ||
||| |||||| ||||||||||||| | |||||
|||||| | ||| |||||| | || |||||| | |||||| | || |||||| | ||||| |
|||||| | || |||||| | || |||||| | || |||||| | ||
|||||||||| ||||||||||||| | |||||
|||||| | || |||||| | || |||||| | ||||| | || |||||| | ||||| |
|||||| | || |||||| | || |||||| | || |||||| | ||
|||||| | || |||||| | || |||||| | || |||||| | ||
|||| |||||||||| | |||||
|||||| | || |||||| | || |||||| | ||||| | || |||||| | |||||| |
|||||| | ||| |||||| | || |||||| | || |||||| | ||
||||||| | |||||
|||| | || |||||| | || |||||| | |||||| | || |||||| | |||||| |
|||||| | || |||||| | || |||||| | || |||||| | ||
|||| || |||||| | |||||
||| | || |||||| | || |||||| | ||||| | || |||||| | |||||| |
||||||| | || |||||| | || |||||| | || |||||| | ||
||||| |||| |||| |||||||||||||| | |||||
||| | ||| |||||| | || |||||| | ||||| | || |||||| | |||||| |
|| | || |||||| | || |||||| | || |||||| | ||
|||||||| | |||||
||||| | ||| |||||| | || |||||| | ||||||| | || |||||| | |||||| |
||||||||| |||||||| | || |||||| | || |||||| | || |||||| | ||
||||||||||| ||||| | |||||
|||||||| |||| | ||| |||||| | || |||||| | ||||| | || |||||| | ||||| |
High risk | 38 (23) | 28 (24.8) | 26 (22.9) | ||
Missing | 17 (10.3) | 15 (13.3) | 12 (10.9) | ||
ECOG PS = Eastern Cooperative Oncology Group Performance Status; ISS = International Staging System; ITT = intention-to-treat; LDH = lactate dehydrogenase; LOT = line of therapy; mAb = monoclonal antibody; MM = multiple myeloma; sATT = scaled average treatment effect in the treated; SMD = standardized mean difference.
Source: Sponsor-submitted indirect treatment comparison.30
Efficacy Results
Overall Survival
Results comparing OS between teclistamab and ciltacabtagene autoleucel before and after adjustment are summarized in Table 37 and Table 38. In the MajesTEC-1 study, the median OS was 21.91 months (95% CI, 15.08 months to NE months). In the CARTITUDE-1 study, the median OS was not reached before and after weighting. Following adjustment, the estimated HR for OS for teclistamab versus ciltacabtagene autoleucel was |||| |||| ||| |||| || |||||| || |||||| || |||||||||||||| ||||||||||. No sensitivity analysis was performed for OS.
Progression-Free Survival
Results comparing PFS across teclistamab and ciltacabtagene autoleucel, both before and after adjustment, are summarized in Table 37 and Table 38. In the MajesTEC-1 and CARTITUDE-1 studies, the median PFS was 11.37 months (95% CI, 8.77 months to 16.36 months) and 31.47 months (95% CI, 22.44 months to NE months), respectively. In the CARTITUDE-1 study, following adjustment, the median PFS for ciltacabtagene autoleucel was ||||| |||||| |||| ||| ||||| || |||. Following adjustment, the estimated HR of PFS for teclistamab versus ciltacabtagene autoleucel was |||| |||| ||| |||| || |||||| || |||||| || |||||||||||||| ||||||||||. No sensitivity analysis was performed for PFS.
Duration of Response
Results comparing duration of response between teclistamab and ciltacabtagene autoleucel before and after adjustment are summarized in Table 37 and Table 38. In the MajesTEC-1 study, the median duration of response was 21.55 months (95% CI, 16.23 months to NE months). In the CARTITUDE-1 study, the median duration of response was not reached, while after adjustment, the median duration of response was ||||| |||||| |||| ||| ||||| || || |||||||. Following adjustment, the estimated HR of duration of response for teclistamab versus ciltacabtagene autoleucel ||| |||| |||| ||| |||| || ||||). No sensitivity analysis was performed for duration of response.
Time to Next Treatment
Results comparing TTNT between teclistamab and ciltacabtagene autoleucel before and after adjustment are summarized in Table 37 and Table 38. In the MajesTEC-1 study, the median TTNT was 12.68 months (95% CI, 8.71 months to 17.61 months). In the CARTITUDE-1 study, the median TTNT was not reached before and ||||| |||||||||. Following adjustment, the estimated HR of TTNT for teclistamab versus ciltacabtagene autoleucel was |||| |||| ||| |||| || |||||| || |||||| || |||||||||||||| ||||||||||. No sensitivity analysis was performed for TTNT.
Table 37: Unadjusted and Adjusted Kaplan-Meier Estimated OS, PFS, DoR, and TTNT [Redacted]
|||||| | |||||| |||||||| | ||
|---|---|---|---|
|||||||||| | ||||||||||| | ||
|||||||| | ||||| ||||||||| | ||
|||| |||||| || ||| | |||
|||||| |||| |||| |||||| | ||||| |||||| || ||| | || |||||| || ||| | || ||| || ||| |
||||| |||||| || ||| | |||
|||||| |||| |||| |||||| | ||||| ||||| || |||||| | ||||| |||||| || ||| | ||||| |||||| || ||| |
||||| |||||| || ||| | |||
|||||| |||| |||| |||||| | ||||| |||||| || ||| | || |||||| || ||| | ||||| |||||| || ||| |
|||||| |||||| || ||| | |||
|||||| |||| |||| |||||| | ||||| ||||| || |||||| | || |||||| || ||| | || |||||| || ||| |
CI = confidence interval; DoR = duration of response; HR = hazard ratio; IPTW = inverse probability of treatment weighting; NE = not evaluable; OS = overall survival; PFS = progression-free survival; sATT = scaled average treatment effect in the treated; TTNT = time to next treatment.
Notes: Details included in Table 37 are from the sponsor’s Summary of Clinical Evidence.83
The main analysis was adjusted for refractory status, International Staging System stage, time to progression on last regimen, extramedullary plasmacytomas, number of prior lines of therapy, years since multiple myeloma diagnosis, average duration of prior lines of therapy, age, hemoglobin, lactate dehydrogenase level, creatinine clearance, Eastern Cooperative Oncology Group Performance Status score, sex, type of multiple myeloma, prior stem cell transplant, race, and cytogenetic profile.
Source: Sponsor-submitted indirect treatment comparison.30
Clinical Response Outcomes
No results for clinical response outcomes, including ORR, CR or better, and VGPR or better, were included in the study comparing the MajesTEC-1 and CARTITUDE-1 trials.
Health-Related Quality of Life
No results for HRQoL were included in the study comparing the MajesTEC-1 and CARTITUDE-1 trials.
Safety
No results for safety were included in the study comparing the MajesTEC-1 and CARTITUDE-1 trials.
Teclistamab (MajesTEC-1 Study) Versus PC Therapy (APOLLO, POLLUX, CASTOR, and EQUULEUS Trials)
Overall, 1,577 patients were initially included in the daratumumab trials cohort, of whom 642 patients were triple-class exposed and had received at least 1 treatment regimen. Of the 642 patients, 427 patients with 806 observations met the MajesTEC-1 study’s key inclusion criteria, as they had received at least 3 prior lines of therapy, had an ECOG PS score of less than 2, had a creatinine clearance of less than or equal to 1.5 mg/dL, and had progressed on or within 12 months of the most recent line of therapy.27
Table 38: Comparison of OS, PFS, DoR, and TTNT for Teclistamab (MajesTEC-1 Study) With Ciltacabtagene Autoleucel (CARTITUDE-1 Study) [Redacted]
|||||||| | |||||||| |||||||||||| |||| ||| |
|---|---|
|||||| ||||| || ||||| | |
|||||||||| | |||| ||||| || ||||| |
|||| ||||||||| |||| |||| |||| | |||| ||||| || ||||| |
||||||| ||||| || ||||| | |
|||||||||| | |||| ||||| || ||||| |
|||| ||||||||| |||| |||| |||| | |||| ||||| || ||||| |
||||||| ||||| || ||||| | |
|||||||||| | |||| ||||| || ||||| |
|||| ||||||||| |||| |||| |||| | |||| ||||| || ||||| |
|||||||| ||||| || ||||| | |
|||||||||| | |||| ||||| || ||||| |
|||| ||||||||| |||| |||| |||| | |||| ||||| || ||||| |
CI = confidence interval; DoR = duration of response; HR = hazard ratio; IPTW = inverse probability of treatment weighting; OS = overall survival; PFS = progression-free survival; RWPC = real-world physician’s choice; sATT = scaled average treatment effect in the treated; TTNT = time to next treatment.
Notes: Details included in Table 38 are from the sponsor’s Summary of Clinical Evidence.83
The main analysis was adjusted for refractory status, International Staging System stage, time to progression on last regimen, extramedullary plasmacytomas, number of prior lines of therapy, years since multiple myeloma diagnosis, average duration of prior lines of therapy, age, hemoglobin, lactate dehydrogenase level, creatinine clearance, Eastern Cooperative Oncology Group Performance Status score, sex, type of multiple myeloma, prior stem cell transplant, race, and cytogenetic profile.
Source: Sponsor-submitted indirect treatment comparison.30
Table 39 shows a summary of baseline characteristics of patients in the MajesTEC-1 study and the daratumumab trials cohort before and after weighting. Before weighting, moderate to substantial differences were observed for many of the main analysis variables, including refractory status, the presence of extramedullary plasmacytomas, age, prior stem cell transplant, ECOG PS score, years since MM diagnosis, race, sex, and cytogenetic risk. After weighting, populations in the MajesTEC-1 and LocoMMotion trials were more balanced. After adjustment, the resulting effective sample size in the daratumumab trial cohort was 264 patients compared to the original 806 patients. After weighting, substantial differences were observed with regard to prior stem cell transplant, ECOG PS score, race, and type of MM.
A total of 248 unique regimens were used in the RWPC therapy from the daratumumab trials cohort (Table 40). The 2 regimens prescribed to the largest proportion of patients included ||||||||||| || ||||||||||| |||| ||||||||||||| ||||||| ||| |||||||||||| || ||||||||||| |||| ||||||||||||| ||||||.
Table 39: Baseline Characteristics of MajesTEC-1 and Daratumumab Trials Before and After Weighting, ITT Population [Redacted]
|||||||||||||| | ||||||||||||||| | ||||||||||| |||||| | |||
|---|---|---|---|---|---|
|||||| |||||||||||||| | ||| | ||||| ||||||||||||| | ||| | ||
|||||||||| ||||||||| | |||||
||||| |||||||||| | || |||||| | ||| |||||| | ||||| | || |||||| | ||||| |
|||||| || |||| |||||||||| | || |||||| | ||| |||||| | ||||| | ||| |||||| | ||||| |
||||| | || |||||| | || ||||| | ||||| | || |||||| | ||||| |
||| ||||||||| | |||||
||||| | || |||||| | ||| |||||| | ||||| | ||| |||||| | ||||| |
||||| | || |||||| | ||| |||||| | ||||| | || |||||| | ||||| |
||||| | || |||||| | || ||||| | ||||| | || |||||| | ||||| |
|||| || ||||||||||| || |||| ||||||| || |||||||||| | |||||
||||| | || |||||| | ||| |||||| | ||||| | ||| |||||| | ||||| |
||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||
|||||||||||||| |||||||||||||| | |||||
||| | ||| |||||| | ||| |||||| | ||||| | ||| |||||| | ||||| |
|| | || |||||| | || ||||| | || |||||| | ||
|||||| || ||||| ||||| || ||||||||||| | |||||
||||| | || |||||| | ||| |||||| | ||||| | ||| |||||| | ||||| |
||||| | || |||||| | ||| |||||| | ||| |||||| | ||
||||| ||||| || |||||||||| ||||||||| | |||||
||||| | || |||||| | ||| |||||| | ||||| | ||| |||||| | ||||| |
||||| | || |||||| | ||| |||||| | ||| |||||| | ||
||||||| | |||||
||||| | || |||||| | ||| |||||| | ||||| | ||| |||||| | ||||| |
||||| | || |||||| | ||| |||||| | ||||| | || |||||| | ||||| |
||||| | || |||||| | ||| |||||| | ||||| | || |||||| | ||||| |
|||||||||| |||||||||| | |||||
||||| | ||| |||||| | ||| |||||| | ||||| | ||| |||||| | ||||| |
||||| | || |||||| | ||| |||||| | || |||||| | ||
|||| |||||||||| | |||||
||||| | || |||||| | ||| |||||| | ||||| | ||| |||||| | ||||| |
||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||
|||||||||||| | |||||
|||| | || |||||| | ||| |||||| | ||||| | ||| |||||| | ||||| |
|||||| | || |||||| | ||| |||||| | ||| |||||| | ||
|||| || |||||||||| | |||||
||| | || |||||| | ||| |||||| | ||||| | ||| |||||| | ||||| |
||||| ||||| | || |||||| | ||| |||||| | ||||| | || |||||| | ||||| |
||||| | || |||||| | ||| |||||| | ||||| | || |||||| | ||||| |
||||| |||| |||| |||||||||||||| | |||||
||| | || |||||| | ||| |||||| | ||||| | ||| |||||| | ||||| |
|| | ||| |||||| | ||| |||||| | ||| |||||| | ||
||||||||||||| | |||||
||||| | ||| |||||| | ||| |||||| | ||||| | ||| |||||| | ||||| |
|||||| ||||||| |||||||| | || |||||| | || ||||| | ||||| | || ||||| | ||||| |
||| |||||||||||||| | || ||||| | ||| |||||| | ||||| | || |||||| | ||||| |
||||||||||| ||||||||||||| | |||||
|||| |||| | || |||||| | ||| |||||| | ||||| | || |||||| | ||||| |
|||||||| |||| | ||| |||||| | ||| |||||| | ||||| | ||| |||||| | ||||| |
||||||| | || |||||| | ||| |||||| | ||||| | || |||||| | ||||| |
ATT = average treatment effect in the treated; ECOG PS = Eastern Cooperative Oncology Group Performance Status; ISS = International Staging System; ITT = intention-to-treat; LDH = lactate dehydrogenase; LOT = line of therapy; mAb = monoclonal antibody; MM = multiple myeloma; SMD = standardized mean difference.
Source: Sponsor-submitted indirect treatment comparison.29
Table 40: Treatment Regimens in the Daratumumab Trials Cohort [Redacted]
Treatment | Frequency, n (%) (N = 248) |
|---|---|
||||||||||| || ||||||||||| |||| ||||||||||||| | || |||||| |
||||||||||||| || ||||||||||| |||| |||||||||||| | || |||||| |
||||||||||||||| |||| | || |||||| |
|||||||||||||||| || ||||||||||| |||| ||||||||||||| ||| |||||||||||| | || |||||| |
||||||||||||| || ||||||||||| |||| |||||||||||| | || |||||| |
||||||||||| || ||||||||||| |||| ||||||||||||| ||| |||||||||||| | || |||||| |
|||||||||||||||| || ||||||||||| |||| ||||||||||||| ||| ||||||||||| | || |||||| |
|||||||||||||||| || ||||||||||| |||| ||||||||||||| | || |||||| |
||||||||||||| || ||||||||||| |||| |||||||| ||| |||||||||||| | || |||||| |
||||||||| | || |||||| |
|||||||||| || ||||||||||| |||| ||||||||||||| | || |||||| |
||||||||||| || ||||||||||| |||| |||||||||||||||| ||| ||||||||||||| | || |||||| |
|||||||||||| | || |||||| |
|||||||||| || ||||||||||| |||| ||||||||||||| ||| |||||||||||| | || |||||| |
||||||||||| || ||||||||||| |||| ||||||||||||| ||| |||||||||||| | || |||||| |
|||||||||||||| || ||||||||||| |||| ||||||||| | || |||||| |
||||||||||||||| |||||||||||||| ||||| | || |||||| |
|||||||||||| || ||||||||||| |||| ||||||||||||| | || |||||| |
||||||||||| || ||||||||||| |||| |||||||||||||||| ||| ||||||||||||| | || |||||| |
Efficacy Results
Overall Survival
The log-cumulative hazard plot and Schoenfeld residuals plots of PFS were visually inspected; some evidence of violation of the proportional hazards assumption was indicated before significant patient drop-off. However, the Grambsch-Therneau test was nonsignificant at the prespecified level (P value = 0.9), indicating that there was insufficient evidence of a violation of the assumption.
Results comparing OS between teclistamab and PC therapy before and after adjustment are summarized in Table 41 and Table 42. In the MajesTEC-1 study and the daratumumab trials cohort, the median OS was 18.27 months (95% CI, 15.08 months to NE months) and 12.02 months (95% CI, 10.91 months to 14.09 months), respectively. Following adjustment, in the daratumumab trials cohort, the median OS was |||| |||| ||| |||| || |||||||
In the main analysis, following adjustment, the estimated HR of OS for teclistamab versus PC therapy was |||| |||| ||| |||| || |||||| || |||||| || |||||||||||. Results from the fully adjusted scenario analysis were consistent with the main analysis results.
Progression-Free Survival
The log-cumulative hazard plot and Schoenfeld residuals plots for PFS were visually inspected; some evidence of violation of the proportional hazards assumption was indicated before significant patient drop-off. The Grambsch-Therneau test was significant at the prespecified level (P value = 0.028), indicating a violation of this assumption.
Results comparing PFS between teclistamab and the daratumumab trials cohort before and after weighting are summarized in Table 41 and Table 42. In the MajesTEC-1 study and the daratumumab trials cohort, the median PFS was 11.30 months (95% CI, 8.77 months to 16.36 months) and 6.34 months (95% CI, 5.55 months to 7.03 months), respectively. Following adjustment, in the daratumumab trials cohort, the median ||| ||| |||| |||| ||| |||| || |||||| || |||||| || |||||||||||.
In the main analysis, after adjustment, the estimated HR of PFS for teclistamab versus PC therapy was |||| |||| ||| |||| || ||||). Results from the fully adjusted scenario analysis were consistent with the main analysis results.
Time to Next Treatment
The log-cumulative hazard plot and Schoenfeld residuals plots for TTNT were visually inspected; some evidence of violation of the proportional hazards assumption was indicated before significant patient drop-off. The Grambsch-Therneau test was significant at the prespecified level (P value = 0.041), indicating a violation of this assumption.
Results comparing TTNT between teclistamab and PC therapy before and after adjustment are summarized in Table 41 and Table 42. Before adjustment, the median TTNT was not reached in the MajesTEC-1 study while in the daratumumab trials cohort, the median TTNT was 4.83 months (95% CI, 4.50 months to 5.39 months). Following adjustment, in the daratumumab trials cohort, the median TTNT was |||| |||| ||| |||| || |||||| || |||||| || ||||||||||||
In the main analysis, following adjustment, the estimated HR for TTNT of teclistamab versus PC therapy was |||| |||| ||| |||| || |||||. Results from the fully adjusted scenario analysis were consistent with the main analysis results.
Table 41: Unadjusted and Adjusted Kaplan-Meier Estimated OS, PFS, DoR, and TTNT [Redacted]
Detail | MajesTEC-1 study N = 165 | Daratumumab trials cohort N = 806 | Daratumumab trials cohort N = 264 |
|---|---|---|---|
Before weighting | After weighting | ||
||||||| |||||| || |||||| | |||
|||||| |||| |||| |||||| | ||||| |||||| || ||| | ||||| |||||| || |||||| | |||| ||||| || |||||| |
|||||||| |||||| || |||||| | |||
|||||| |||| |||| |||||| | ||||| ||||| || |||||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
||||||||| |||||| || |||||| | |||
|||||| |||| |||| |||||| | || |||||| || ||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
CI = confidence interval; DoR = duration of response; HR = hazard ratio; IPTW = inverse probability of treatment weighting; NE = not evaluable; OS = overall survival; PFS = progression-free survival; sATT = scaled average treatment effect in the treated; TTNT = time to next treatment.
Notes: Details included in Table 41 are from the sponsor’s Summary of Clinical Evidence.83
The main analysis was adjusted for refractory status, cytogenetic profile, International Staging System stage, time to progression on last regimen, extramedullary plasmacytomas, the number of prior lines of therapy, years since multiple myeloma diagnosis, age, and hemoglobin.
Source: Sponsor-submitted indirect treatment comparison.29
Table 42: Comparison of OS, PFS, DoR, and TTNT for Teclistamab (MajesTEC-1 Study) With PC Therapy (Daratumumab Trials Cohort) [Redacted]
Analysis | HR (95% CI) |
|---|---|
|||||| ||||| || ||||| | |
|||||||||| | |||| ||||| || ||||| |
|||| ||||||||| |||| |||| ||| | |||| ||||| || ||||| |
|||||||| |||||||| | |
|||| |||| ||| ||||||| |||||| ||||||||| | |||| ||||| || ||||| |
||||||||||| |||||||| | |
|||| |||| ||| ||||||| ||||||| ||||||| | |||| ||||| || ||||| |
||||||||||||| |||||||||| | |||| ||||| || ||||| |
|| |||||||| | |||| ||||| || ||||| |
||||||| ||||| || ||||| | |
|||||||||| | |||| ||||| || ||||| |
|||| ||||||||| |||| |||| ||| | |||| ||||| || ||||| |
|||||||| |||||||| | |
|||| |||| ||| ||||||| |||||| ||||||||| | |||| ||||| || ||||| |
||||||||||| |||||||| | |
|||| |||| ||| ||||||| ||||||| ||||||| | |||| ||||| || ||||| |
||||||||||||| |||||||||| | |||| ||||| || ||||| |
|||| |||| ||| ||||||| |||||||||||||| | |||| ||||| || ||||| |
|| |||||||| | |||| ||||| || ||||| |
|||||||| ||||| || ||||| | |
|||||||||| | |||| ||||| || ||||| |
|||| ||||||||| |||| |||| ||| | |||| ||||| || ||||| |
|||||||| |||||||| | |
|||| |||| ||| ||||||| |||||| ||||||||| | |||| ||||| || ||||| |
||||||||||| |||||||| | |
|||| |||| ||| ||||||| ||||||| ||||||| | |||| ||||| || ||||| |
||||||||||||| |||||||||| | |||| ||||| || ||||| |
|| |||||||| | |||| ||||| || ||||| |
ATT = scaled average treatment effect in the treated; CI = confidence interval; DoR = duration of response; HR = hazard ratio; IPTW = inverse probability of treatment weighting; OS = overall survival; PC = physician’s choice; PFS = progression-free survival; RWPC = real-world physician’s choice; TTNT = time to next treatment.
Notes: Details included in Table 42 are from the sponsor’s Summary of Clinical Evidence.83
The main analysis was adjusted for refractory status, cytogenetic profile, International Staging System stage, time to progression on last regimen, extramedullary plasmacytomas, number of prior lines of therapy, years since multiple myeloma diagnosis, age, and hemoglobin. The fully adjusted analysis was adjusted for all variables in the base case, plus prior stem cell transplant, Eastern Cooperative Oncology Group Performance Status, race, sex, and type of multiple myeloma.
Source: Sponsor-submitted indirect treatment comparison.29
Clinical Response Outcomes
Results comparing the MajesTEC-1 study and the daratumumab trials cohort for clinical response outcomes including ORR, CR or better, and VGPR or better are summarized in Table 43.
For ORR, the observed proportion was 63.0% for teclistamab and 28.0% for PC therapy. Following adjustment, in the daratumumab trials cohort, the proportion of patients with overall response was |||||| In the main analysis, following adjustment, the OR of ORR for teclistamab versus PC therapy was |||| |||| ||| |||| || |||||| || |||||| || |||||||||||| Results from the fully adjusted analysis were consistent with the main analysis results.
For CR or better, the observed proportion was 39.4% for teclistamab and 2.0% for PC therapy from the daratumumab trials cohort. Following adjustment, in the daratumumab trials cohort, the proportion of patients with CR or better was ||||| In the main analysis, following adjustment, the OR of CR or better for teclistamab versus PC therapy was ||||| |||| ||| |||| || ||||||| || |||||| || |||||||||||| Results from the fully adjusted analysis were consistent with the main analysis results.
For VGPR, the observed proportion was 63.0% for teclistamab and 11.2% for PC therapy from the daratumumab trials cohort. Following adjustment, in the daratumumab trials cohort, the proportion of patients with VGPR or better was |||||| In the main analysis, following adjustment, the OR of VGPR for teclistamab versus PC therapy was ||||| |||| ||| |||| || ||||||| || |||||| || |||||||||||. Results from the fully adjusted analysis were consistent with the main analysis results.
Table 43: Comparison of Clinical Response Results for Teclistamab (MajesTEC-1 Study) Versus PC Therapy (Daratumumab Trials Cohort) [Redacted]
Detail | Teclistamab, response % | RWPC, response % | OR (95% CI) |
|---|---|---|---|
||| | |||
|||||||||| | |||| | |||| | |||| ||||| || ||||| |
|||| ||||||||| |||| |||| ||| | |||| | |||| | |||| ||||| || ||||| |
||||| |||||||| |||||||| |||||||| | |||| | ||||| | |||| ||||| || ||||| |
|| || |||||| | |||
|||||||||| | |||| | ||| | ||||| |||||| || |||||| |
|||| ||||||||| |||| |||| ||| | |||| | ||| | ||||| ||||| || |||||| |
||||| |||||||| |||||||| |||||||| | |||| | ||| | ||||| |||||| || ||||||| |
|||| || |||||| | |||
|||||||||| | |||| | |||| | ||||| ||||| || |||||| |
|||| ||||||||| |||| |||| ||| | |||| | |||| | ||||| ||||| || |||||| |
||||| |||||||| |||||||| |||||||| | |||| | ||| | ||||| ||||| || |||||| |
CI = confidence interval; CR = complete response; IPTW = inverse probability of treatment weighting; OR = odds ratio; ORR = overall response rate; RR = rate ratio; PC = physician’s choice; RWPC = real-world physician’s choice; sATT = scaled average treatment effect in the treated; VGPR = very good partial response.
Notes: Details included in Table 43 are from the sponsor’s Summary of Clinical Evidence.83
The main analysis was adjusted for refractory status, cytogenetic profile, International Staging System stage, time to progression on last regimen, extramedullary plasmacytomas, number of prior lines of therapy, years since multiple myeloma diagnosis, age, and hemoglobin. The fully adjusted analysis was adjusted for all variables in the base case, plus prior stem cell transplant, Eastern Cooperative Oncology Group Performance Status, race, sex, and type of multiple myeloma.
Source: Sponsor-submitted indirect treatment comparison.29
Health-Related Quality of Life
No results for HRQoL were included in the study comparing the MajesTEC-1 study and the daratumumab trials cohort.
Safety
No results for safety were included in the study comparing the MajesTEC-1 study and the daratumumab trials cohort.
Critical Appraisal of ITCs Used to Inform the Pharmacoeconomic Model
The ITCs submitted by the sponsor had a number of limitations that challenged the internal and external validity of the findings. No systematic search was conducted to identify the comparator studies included in the 3 ITCs that were used to inform the pharmacoeconomic model; therefore, there is a risk of selection bias. The selection criteria used to identify the comparators were consistent with the objective, and studies were included if they assessed treatment for RRMM, included patients with triple-class exposed RRMM who had received at least 3 prior lines of therapy, and reported sufficient efficacy outcome data. However, no details were provided regarding the timing of the literature review or the databases used. It is not possible to know whether the results may have differed if data from different RRMM studies or databases had been used. The list of excluded studies is not available, and the risk of bias of the included studies was not assessed.
Given the absence of a comparator group in the MajesTEC-1 study, an external control group was used to establish the relative efficacy of teclistamab versus treatments used in current clinical practice. Three IPTW analyses were submitted by the sponsor, including 2 IPTW analyses comparing teclistamab to RWPC therapy from the LocoMMotion study and the daratumumab trials cohort, and 1 IPTW analysis comparing teclistamab to ciltacabtagene autoleucel from the CARTITUDE-1 trial. The sponsor provided an adequate rationale for using IPTW analyses for these ITCs. The IPTW analyses with ATT weighting were chosen because there was sufficient overlap between patient characteristics between the MajesTEC-1 trial and comparator studies. The sponsor concluded that an IPTW estimator was identified as the preferred option to adjust for suspected heterogeneity within the studies. Also, IPTW is an efficient method when the sample size is small relative to the number of potential baseline confounding factors.86 In all 3 IPTW analyses, the MajesTEC-1 study was used as the reference study. The index date was defined as the date of the first dose for the MajesTEC-1 trial; as day 1, cycle 1, of the real-life standard of care treatment for the LocoMMotion trial; as the date of apheresis in the CARTITUDE-1 trial; and as the start of each eligible line of therapy for PC therapy.
Critical Appraisal of the ITC Comparing Teclistamab (MajesTEC-1 Study) Versus RWPC Therapy (LocoMMotion Study)
There was variation in the design of the MajesTEC-1 and LocoMMotion studies, as the MajesTEC-1 study was a phase I and phase II trial while the LocoMMotion trial was an observational and noninterventional study. Both studies were open label, so there was a risk of bias in the measurement of subjective outcomes, particularly PFS, and clinical response outcomes. Objective outcomes including OS should be unaffected by the open-label designs. The definitions of end points were similar across the studies; however, the median duration of follow-up was 14.1 months in the MajesTEC-1 study28 and 16.1 months in the LocoMMotion study.28 Both the MajesTEC-1 and LocoMMotion studies included patients with an ECOG PS score of 0 or 1. Thus, the ITC findings cannot be generalized to patients with an ECOG PS score of more than 1; however, clinical experts consulted by CADTH for this review noted that teclistamab might be prescribed to patients with RRMM with an ECOG PS score of 2 or 3. The MajesTEC-1 pivotal study, included in the IPTW analysis, excluded patients who had previously received BCMA-targeted therapy; thus, the study findings cannot be fully generalizable to the RRMM population
The top 5 real-world treatment combinations used in the LocoMMotion study were carfilzomib in combination with dexamethasone (14.1%), cyclophosphamide in combination with dexamethasone plus pomalidomide (14.1%), dexamethasone in combination with pomalidomide (11.7%), dexamethasone in combination with ixazomib plus lenalidomide (5.6%), and bortezomib in combination with dexamethasone plus panobinostat. The clinical experts consulted by CADTH for this review noted that some of these regimens are appropriate comparators for teclistamab in fourth-line and beyond settings for RRMM. However, the LocoMMotion study used a total of 90 different treatment regimens and given that not all treatment regimens are relevant to Canadian clinical practice in fourth-line and beyond settings (e.g., daratumumab, ixazomib, melphalan), the study results may not be generalizable to Canadian clinical practice.
There was notable heterogeneity in the populations of the MajesTEC-1 and LocoMMotion studies. Potential prognostic factors were identified via literature reviews and consultation with clinical experts, although few details of these methods were reported. Comparisons with externally generated cohorts were limited by the availability of information important to the analysis. Cytogenetic risk was considered an important risk factor by clinical experts; however, it was not included in the main analyses due to a high level of missingness in the LocoMMotion cohort (37.1%). The clinical experts consulted by CADTH noted that cytogenetic risk is an important prognostic factor, and omitting this factor could result in potential bias, while race is not considered important. As the LocoMMotion study’s population only included a low number of non-white patients, including race as a variable in the adjustment set led to large weights for those patients and decreased balance for all other variables.
The patient demographic characteristics before and after weighting were reported. Prior to adjustment, baseline covariates for prognostic factors demonstrated considerable heterogeneity in almost all variables. After reweighting, populations from the MajesTEC-1 and LocoMMotion trials were more balanced, except for observed differences persisting in refractory status and time to progression on the final regimen. While the weighted populations were balanced with respect to known, measured prognostic factors, it remains unclear whether other unmeasured clinically relevant variables were unaccounted for. The variables not included in the planned adjustment set (unknown or unmeasured prognostic factors) can result in residual confounding and bias the estimates.36 Assessment of residual bias was not performed or reported. Therefore, the results of the IPTW analysis may be considered to have a high risk of residual bias,37 as with any unanchored comparisons; however, the magnitude and direction of any bias is unknown. In addition, the interpretation of the outcomes is challenging due to systematic differences in study design.
The main IPTW analysis using an ATT weighting approach was performed adjusting for 15 prognostic factors. The sponsor stated that due to a small sample size in the MajesTEC-1 and LocoMMotion trials, a scaled ATT weighting approach was used to scale treatment weightings so that they were summed to the original number of participants in the comparator studies. However, this approach was used in the NICE working example only to present the distribution of the weights by scaling them, so that a rescaled weight of more than 1 means that an individual carries more weight in the reweighted comparator population than in the index study population, and a rescaled weight of less than 1 means that an individual carries less weight.37 No information was reported in this IPTW analysis regarding the distribution of weights generated by the weighting process and the number of patients with extremely high and extremely low weights (including patients assigned 0 weight). Therefore, it remains unclear if patients with 0 weights (when there is no overlap with the target study) were excluded from the adjusted sample of the LocoMMotion study in accordance with the NICE DSU Technical Support Document,37 and what the effective sample size was in the LocoMMotion study after reweighting to estimate the number of nonweighted patients. In the IPTW analysis submitted by the sponsor, the reported sample size of the LocoMMotion trial was 248 patients before and after weighting. Thus, due to the lack of clarity, the evidence obtained from this IPTW analysis remains uncertain, limiting the interpretation and generalizability of the results. Several sensitivity analyses were conducted, including fully adjusted analysis, as well as analyses balancing baseline covariates using the ATO and average treatment effect weighting approaches, and multivariate regression analysis. The results of safety analyses were consistent with the main analyses. No subgroup analyses were performed.
The definitions of the outcomes, including PFS and clinical response outcomes, assessed in both studies were similar and were assessed by the IRC in the MajesTEC-1 study and by the response review committee in the LocoMMotion study. In the MajesTEC-1 study, there was a high degree of concordance between ORR assessments by the IRC and by the computerized algorithm utilized. The sensitivity analysis of ORR based upon investigator assessment was consistent with the primary analysis using the IRC assessment based on IMWG response criteria, and similar comparisons were done with PFS in the MajesTEC-1 trial. Heterogeneity between populations after adjustment, systematic differences in study design, and the potential for residual confounding limited the ability to draw conclusions about the efficacy of teclistamab relative to RWPC therapy for the OS, PFS, TTNT, and clinical response outcomes (ORR, CR or better, or VGPR or better). The ITC estimates were too imprecise for clinical response outcomes as evidenced by the wide 95% CIs, and the upper and lower boundaries of the CIs suggest the potential for different conclusions regarding the efficacy of teclistamab relative to RWPC therapy. Safety outcomes were not analyzed in the ITC report, and no justification was provided, which precludes a balanced judgment of comparative benefit relative to comparative harms. Outcomes that are important to patients, such as HRQoL, were also not analyzed in this ITC.
Critical Appraisal of the ITC Comparing Teclistamab (MajesTEC-1 Study) Versus Ciltacabtagene Autoleucel (CARTITUDE-1 Study)
The MajesTEC-1 trial was a phase I and phase II study, and the CARTITUDE-1 trial was phase Ib and phase II study. Both studies included in the ITC were presented with an unclear risk of bias for statistical analysis and a high risk for the measurement of subjective outcomes, such as PFS or clinical response outcomes, due to the open-label study design. Objective outcomes, including OS, should be unaffected by the open-label designs. The median duration of follow-up was 22.8 months in the MajesTEC-1 trial and 27.7 months in the CARTITUDE-1 trial. The clinical experts consulted by CADTH indicated that ciltacabtagene autoleucel is an appropriate comparator for teclistamab for the treatment of RRMM in fourth-line and beyond settings. The MajesTEC-1 study cohort represented a broad population from Europe, Canada, and the US whereas the results from the CARTITUDE-1 study are drawn from US patients only. Therefore, it is unclear whether differences in clinical practice or treatment availability exist across regions, and the direction and magnitude of potential biases remain unclear. Both the MajesTEC-1 and CARTITUDE-1 studies included patients with an ECOG PS score of 0 or 1. Thus, the findings from the ITC cannot be generalized to patients with an ECOG PS score of 2 or 3, although clinical experts consulted by CADTH noted that teclistamab might be prescribed to patients with RRMM with an ECOG PS score of 2 or 3. The MajesTEC-1 pivotal study, included in the IPTW analysis, excluded patients who had previously received BCMA-targeted therapy; thus, the study findings cannot be fully generalizable to the RRMM population.
Heterogeneity in the eligibility criteria, patients’ baseline characteristics, and outcomes measured in the studies were reported and reviewed by the authors. Potential prognostic factors were identified via literature reviews and consultation with clinical experts, although few details of these methods were reported. The IPTW analysis using an ATT weighting approach was performed adjusting for 17 prognostic factors. Before weighting, moderate to substantial differences were observed for many of the main analysis variables. After weighting, populations in the MajesTEC-1 and CARTITUDE-1 trials were more balanced, although observable differences remained between trials in refractory status, age, hemoglobin level, and creatinine clearance. While the weighted populations were balanced with respect to known, measured prognostic factors, it remains unclear whether other unmeasured clinically relevant variables were unaccounted for. The variables not included in the planned adjustment set (unknown or unmeasured prognostic factors) can result in residual confounding and bias the estimates.36 Assessment of residual bias was not performed or reported. Therefore, the results of the IPTW analysis may be considered to have a high risk of residual bias;37 however, the magnitude and direction of any bias is unknown. According to the clinical experts consulted by CADTH for this review, the population of the CARTITUDE-1 study was relatively healthy compared to the MajesTEC-1 study’s population. In addition, the interpretation of the outcomes is challenging due to systematic differences in duration of follow-up.
The sponsor stated that due to a small sample size in the MajesTEC-1 and CARTITUDE-1 trials, a scaled ATT weighting approach was used to scale treatment weightings so that they were summed to the original number of participants in the comparator studies. However, the NICE working example cited by the sponsor used the scaling of weights for examining the distribution of the weights by scaling them, so that a rescaled weight of more than 1 means that an individual carries more weight in the reweighted comparator population than in the index study population, and a rescaled weight of less than 1 means that an individual carries less weight.37 No information was reported in this IPTW analysis regarding the distribution of weights generated by the weighting process and the number of patients with extremely high and extremely low weights (including patients assigned 0 weight). Therefore, it remains unclear if patients with 0 weights were excluded from the adjusted sample of the CARTITUDE-1 study in accordance with the NICE DSU Technical Support Document,37 and what the effective sample size was in the CARTITUDE-1 study after reweighting to estimate the number of nonweighted patients. In the IPTW analysis submitted by the sponsor, the reported sample size of the CARTITUDE-1 trial was 113 before and after weighting. Thus, due to the lack of clarity, the evidence obtained from this IPTW analysis remains uncertain, limiting the interpretation and generalizability of the results.
The definitions of the outcomes assessed in both studies were similar and were assessed by the IRC; thus, the risk of bias may have been reduced. Although all clinical response outcomes (ORR, CR or better, VGPR or better) were available in both studies, only selected outcomes were assessed in this analysis, including OS, PFS, duration of response, and TTNT. No methods for handling missing data were performed or reported in the ITC comparing teclistamab with ciltacabtagene autoleucel. For OS, PFS, and TTNT, the results of the adjusted treatment comparisons were consistent across end points, favouring ciltacabtagene autoleucel over teclistamab; however, heterogeneity between populations after adjustment, differences in the duration of follow-up, and the potential for residual confounding limit the ability to draw conclusions about the efficacy of teclistamab relative to ciltacabtagene autoleucel. Safety outcomes were not analyzed in the ITC report, and no justification was provided, which precludes a balanced judgment of comparative benefit relative to comparative harms. Outcomes that are important to patients, such as HRQoL, were also not analyzed in this ITC.
Critical Appraisal of the ITC Comparing Teclistamab (MajesTEC-1 Study) Versus PC Therapy (APOLLO, POLLUX, CASTOR, and EQUULEUS Trials)
There was variation in the design of the MajesTEC-1 study and the 4 daratumumab trials in the IPTW analysis. The MajesTEC-1 trial was a phase I and phase II trial while the POLLUX, CASTOR, and APOLLO trials were open-label, phase III RCTs, and the EQUULEUS trial was an open-label, nonrandomized, phase Ib study. Both the MajesTEC-1 study and the daratumumab trials were open label, so there is a risk of bias in the measurement of subjective outcomes, particularly PFS, and clinical response outcomes. Objective outcomes including OS should be unaffected by the open-label designs. In addition, although 3 of the daratumumab trials (the POLLUX, CASTOR, and APOLLO studies) included in the ITC were RCTs, and the EQUULEUS trial was an open-label, nonrandomized phase Ib study, patients selected from the daratumumab trials cohort were included in the analysis retrospectively. The index date in the MajesTEC-1 trial was defined as the date of the first dose while in the daratumumab trials cohort, the index date was defined as the start of each eligible line of therapy. A total of 248 unique treatment regimens were used in the daratumumab trials cohort, many of which were not relevant to Canadian clinical practice; thus, the study results may not be generalizable to the Canadian setting. Both the MajesTEC-1 study and the daratumumab trials cohort included patients with an ECOG PS score of 0 or 1. Thus, the findings from the ITC cannot be generalized to patients with an ECOG PS score of 2 or 3, although clinical experts consulted by CADTH noted that teclistamab might be prescribed to patients with RRMM with an ECOG PS score of 2 or 3. The MajesTEC-1 pivotal study included in the IPTW analysis excluded patients who had previously received BCMA-targeted therapy; thus, the study findings cannot be fully generalizable to the RRMM population.
Heterogeneity in the eligibility criteria, patients’ baseline characteristics, and outcomes measured in the MajesTEC-1 study and the daratumumab trials cohort were reported and reviewed by the authors. There was notable heterogeneity in the populations of the MajesTEC-1 study and the daratumumab trials cohort. The definitions of end points were similar across the studies; however, the median duration of follow-up was 14.1 months in the MajesTEC-1 trial and 30.9 months in the daratumumab trials cohort. Outcomes were evaluated by the IRC in the MajesTEC-1 trial and by the investigator using local laboratory values in the daratumumab trials cohort; thus, there is a risk of bias in the measurement of the outcomes due to the open-label design, likely in favour of teclistamab. Potential prognostic factors were identified via literature reviews and consultation with clinical experts, although few details of these methods were reported. Nine of the 17 prognostic factors identified a priori were used for ATT weighting in the main analysis, including refractory status, cytogenetic risk, ISS stage, the presence of extramedullary plasmacytomas, time to progression on last line of therapy, the number of prior lines of therapy, years since MM diagnosis, age, and hemoglobin level.
Baseline characteristics of patients in the MajesTEC-1 study and the daratumumab trials before and after ATT weighting were reported. Before weighting, moderate to substantial differences were observed for many of the main analysis variables. After weighting, populations in the MajesTEC-1 study and the daratumumab trials cohort were more balanced; however, substantial differences were observed with regard to prior stem cell transplant, ECOG PS score, race, and type of MM. Populations from the studies were balanced with respect to known, measured prognostic factors. While the weighted populations were balanced with respect to known, measured prognostic factors, it remains unclear whether other unmeasured clinically relevant variables were unaccounted for. The variables not included in the planned adjustment set (unknown or unmeasured prognostic factors) can result in residual confounding and bias the estimates.36 Assessment of residual bias was not performed or reported. Therefore, the results of the IPTW analysis may be considered to have a high risk of residual bias;37 however, the magnitude and direction of any bias is unknown. In addition, the interpretation of the outcomes is challenging due to systematic differences in study design and duration of follow-up.
After adjustment, the effective sample size was reduced to approximately 32.8% (264 of 804) of patients in the original sample size in the daratumumab trials cohort. The reduction in the effective sample size reflects the heterogeneity between the trials among the variables included in the weighting process. A small effective sample size implies that the estimates were being influenced by a subset of the patients from the daratumumab trials and are caused by a violation of the transportability of the effects across cohorts.20 Several sensitivity analyses were conducted, including a fully adjusted analysis, as well as supplementary analyses re-balancing baseline covariate distributions using the ATO and average treatment effect weighting approaches, and multivariate regression analysis. The fully adjusted sensitivity analysis weighted population on prior stem cell transplant, ECOG PS score, race, sex, and type of MM, in addition to the main analysis factors. The results of sensitivity analyses were consistent with the main analyses. No subgroup analyses were performed.
The proportional hazards assumption was tested for the time-to-event outcomes, and the Grambsch-Therneau test was significant for PFS and TTNT analyses, indicating potential violation of this assumption. The Cox proportional hazards model assumes that the HR across treatment groups does not change over time; therefore, violation of the proportional hazards assumption may lead to misleading and erroneous scientific conclusions.38,39 For OS, PFS, and TTNT, the results of the adjusted treatment comparisons were consistent across end points, favouring teclistamab over PC therapy from the daratumumab trials cohort; this is consistent with the opinion of the clinical experts consulted for this review. Heterogeneity between populations after adjustment, reduced effective sample size, differences in study design and duration of follow-up, and the potential for residual confounding limit the ability to draw conclusions about the efficacy of teclistamab relative to the comparator drug for the OS, PFS, TTNT, and clinical response outcomes. There was notable imprecision in the ITC estimates for clinical response outcomes as evidenced by the wide 95% CIs, and the upper and lower boundaries of the CIs suggest the potential for different conclusions regarding the efficacy of teclistamab relative to PC therapy. Safety outcomes were not analyzed in the ITC report, and no justification was provided, which precludes a balanced judgment of comparative benefit relative to comparative harms. Outcomes that are important to patients, such as HRQoL, were also not analyzed in this ITC.
Other ITCs Not Included in the Pharmacoeconomic Model
The ITCs submitted by the sponsor had a number of limitations that challenged the interpretation of the internal and external validity of the findings. No systematic search was conducted to identify the comparator studies included in the 3 ITCs that were not used to inform the pharmacoeconomic model; therefore, there is a risk of selection bias. The studies were included in these ITCs if they assessed treatment for RRMM, included patients with triple-class exposed RRMM who had received at least 3 prior lines of therapy, and reported sufficient efficacy outcome data. However, no details were provided regarding the timing of the literature review or the databases used. It is not possible to know whether the results may have differed if data from different RRMM studies or databases had been used. The list of excluded studies is not available, and the risk of bias of the included studies was not assessed. Given the absence of a comparator group in the MajesTEC-1 study, an external control group was used to establish the relative efficacy of teclistamab versus treatments used in current clinical practice.
Teclistamab (MajesTEC-1 Study) Versus RWPC Therapy (Flatiron Health Database)31
An ITC using the IPTW approach was conducted between teclistamab and RWPC using individual patient-level data from the MajesTEC-1 trial (for teclistamab) and the nationwide deidentified electronic health record–derived Flatiron Health database (for the RWPC cohort). Key eligibility criteria of the MajesTEC-1 study were applied to the RWPC cohort, including a diagnosis of MM using IMWG criteria, and prior exposure to 3 or more lines of therapy. Given that the Flatiron Health database is retrospective, it was possible to include patients in the ITC at the earliest line of therapy initiated after all key eligibility criteria were met. Patients in the Flatiron Health cohort who received multiple subsequent therapies after meeting eligibility criteria contributed multiple observations to the ITC analysis. The MajesTEC-1 study cohort included data from 165 patients while the unadjusted population of the RWPC Flatiron Health cohort included 420 unique patients, corresponding to 766 eligible lines of therapy. In the Flatiron Health cohort, the most common RWPC therapies, used as either monotherapy or in combination with other treatments, were dexamethasone (79.1%), daratumumab (30.6%), pomalidomide (24.4%), and carfilzomib (24.4%). Compared to patients in the Flatiron Health database, the MajesTEC-1 study’s population had a higher proportion of patients who had ISS stage I disease, had standard risk cytogenetics, and were aged younger than 65 years. The propensity score–based method of IPTW with an ATT weighting was used to reweight the RWPC Flatiron Health cohort to align with the MajesTEC-1 study’s population and adjust for imbalances between patient populations. Kaplan-Meier estimates were used to estimate survival curves and the median time to events. A pool of prognostic variables was identified by consulting studies from a review of the literature as well as input from clinical experts. The following prognostic factors were included in the main analyses for adjustment: refractory status, time to progression on the last line of therapy, cytogenetic risk status, ISS stage, the number of prior lines of therapy, years since MM diagnosis, age, and hemoglobin level. For the MajesTEC-1 trial, a clinical cut-off date of March 16, 2022, was used, with the median duration of follow-up being 14.1 months. For the RWPC Flatiron Health cohort, patients who had 2 or more documented clinical visits on or after January 2011 were included, with the median duration of follow-up being 18.2 months. After weighting, the effective sample size of the RWPC Flatiron Health cohort reduced to 42.6% of the original population.
Efficacy Results
The median OS for the MajesTEC-1 study and Flatiron Health cohort was 18.3 months (95% CI, 15.08 months to NE months) and 13.8 months (95% CI, 12.32 months to 15.67 months), respectively. Following adjustment, the median OS for the Flatiron Health cohort was 14.46 months (95% CI, 12.29 months to 18.56 months). Following adjustment, the estimated HR for OS of teclistamab versus RWPC therapy from the Flatiron Health cohort was 0.82 (95% CI, 0.59 to 1.14).
The median PFS for the MajesTEC-1 study and the Flatiron Health database was 11.30 months (95% CI, 8.77 months to 17.15 months) and 3.91 months (95% CI, 3.48 months to 4.30 months), respectively. Following adjustment, the estimated HR for PFS of teclistamab versus RWPC from the Flatiron Health cohort was 0.43 (95% CI, 0.33 to 0.56).
The median TTNT in the MajesTEC-1 study was not reached while in the Flatiron Health cohort, it was 5.19 months (95% CI, 4.63 months to 5.75 months). Following adjustment, the estimated HR for TTNT for teclistamab versus RWPC from the Flatiron Health cohort was 0.36 (95% CI, 0.27 to 0.49).
Safety
No results for safety were included in the study comparing the MajesTEC-1 study and the Flatiron Health cohort.
Critical Appraisal
As the Flatiron Health database was not selected using a systematic approach, there is a risk of selection bias. It is not possible to know whether the results may have differed if data from different RRMM studies or databases had been used. Numerous therapies were used in the RWPC groups from the Flatiron Health cohort, many of which were not relevant to Canadian clinical practice; thus, the ITC results may not be generalizable to the Canadian setting. Additionally, patients included in the present analysis initiated eligible lines of therapy between 2016 and 2021; however, clinical practice has changed since the enrolment of patients from these sources and may not be reflective of current treatment standards in Canada. Patients selected from the Flatiron Health cohort were included in the analysis retrospectively. Outcomes in the MajesTEC-1 study were assessed by the IRC while in the Flatiron Health cohort, outcomes were assessed by the investigators; thus, the risk of bias in the outcome measurements is increased relative to the same outcomes as measured in the MajesTEC-1 study. The duration of follow-up in the MajesTEC-1 trial was 14.1 months versus 18.2 months in the Flatiron Health cohort. There were important differences in the design of the studies included in this ITC, as the MajesTEC-1 study was a phase I and phase II study while the Flatiron Health cohort was a real-world cohort from electronic health records in the US, which limits the ability to draw strong conclusions about the efficacy of teclistamab relative to RWPC therapy due to differences in clinician and patient behaviours, the heterogeneity of treatments for intercurrent events, and differences in data collection and intake. Such methodological differences could not be adjusted for in the IPTW analysis and the magnitude and direction of any resulting bias is uncertain. There was evidence of heterogeneity between the population of the MajesTEC-1 study and the Flatiron Health cohort. It remains unclear how balanced populations were for other variables that may be clinically relevant but could not be adjusted due to lack of data, or those variables that were not part of the planned adjustment (unknown or unmeasured prognostic factors), which leaves the potential for residual confounding.36 After weighting, the effective sample size of the Flatiron Health cohort was reduced by 57.4% from the included population. The reduction in the effective sample size reflects the heterogeneity between the trials among the variables included in the weighting process. A small effective sample size implies that the weighted estimates are being influenced by a subset of patients from the Flatiron Health cohort who may not be representative of the entire study population, which may limit the generalizability of the results. For OS and PFS, the results of the adjusted treatment comparisons were consistent across end points, favouring teclistamab over RWPC therapy from the Flatiron Health cohort. However, heterogeneity between populations after adjustment, reduced effective sample size, a difference in study design, median duration of follow-up, retrospectively analyzed data, and the potential for residual confounding limit the ability to draw conclusions about the efficacy of teclistamab relative to the comparator drug.
Teclistamab (MajesTEC-1 Study) Versus Belantamab Mafodotin (DREAMM-2 Study)32
An unanchored MAIC was conducted to compare the efficacy of teclistamab with belantamab mafodotin using individual patient data from the MajesTEC-1 trial (n = 150) and summary-level data from the DREAMM-2 trial (n = 97). The MajesTEC-1 trial was an open-label, single-arm, phase I and phase II study while the DREAMM-2 trial was an open-label, 2-arm, phase II study. The DREAMM-2 trial’s eligibility criteria were applied to patients from the ITT population of the MajesTEC-1 study. Compared to patients in the DREAMM-2 study’s population, the MajesTEC-1 study’s population had a higher proportion of patients who had ISS stage I disease. All patients had triple-class exposed RRMM who had received at least 3 prior lines of therapy. Individual patient data from the MajesTEC-1 trial were weighted to match the aggregated DREAMM-2 trial’s baseline patient characteristics. The following factors were used to adjust for imbalances between patient populations: refractory status, cytogenetic profile, ISS staging, the presence of extramedullary disease, and the number of prior lines of therapy. The effective sample size of the MajesTEC-1 trial after propensity score matching was 33 patients. The comparative efficacy of teclistamab relative to belantamab mafodotin was estimated for ORR, CR or better, OS, PFS, and duration of response. For binary outcomes, the relative effects were quantified using an OR and 95% CI derived from a weighted logistic regression analysis, while time-to-event outcomes were estimated using a weighted Cox proportional hazards model.
Efficacy Results
Following adjustment, the HR for OS of teclistamab versus belantamab mafodotin was 0.95 (95% CI, 0.47 to 1.92). Following adjustment, the HR for PFS for teclistamab versus belantamab mafodotin was 0.63 (95% CI, 0.34 to 1.15) and the estimated HR of duration of response for teclistamab versus belantamab mafodotin was 0.19 (95% CI, 0.05 to 0.73). Following adjustment, the OR of ORR of teclistamab versus belantamab mafodotin was 2.05 (95% CI, 0.92 to 4.57) while the OR of CR or better for teclistamab versus belantamab mafodotin was 2.13 (95% CI, 0.80 to 5.65).
Safety
No results for safety were included in the study comparing the MajesTEC-1 and DREAMM-2 trials.
Critical Appraisal
The open-label design of the studies can result in a risk of bias in the study conduct, including the measurement of the outcomes, and increase uncertainty in subjective outcomes such as PFS and ORR. The bias will likely favour the experimental intervention, although the extent of bias is uncertain. The clinical experts consulted by CADTH noted that belantamab mafodotin is another option available to patients; however, this treatment is only available through special access and is not used frequently. The effective sample size was reduced after adjustment in the MAIC analysis to approximately 22.0% (33 of 150) of the patients in the original population in the MajesTEC-1 study. The reduction in the effective sample size reflects the heterogeneity between the trials among the variables included in the weighting process. A small effective sample size implies that the weighted estimates are being influenced by a subset of patients from the MajesTEC-1 study who may not be representative of the entire study population, which may limit the generalizability of the results. Populations from the studies were balanced with respect to known, measured prognostic factors. It remains unclear how balanced populations were for other variables that may be clinically relevant but could not be adjusted due to lack of data, or those variables that were not part of the planned adjustment (unknown or unmeasured prognostic factors), which leaves the potential for residual confounding.36 In the MAIC analysis, results in efficacy estimates were imprecise (i.e., wide CIs) in the end points assessed (including HR = 1), and the upper and lower boundaries of the CIs suggest the potential for different conclusions regarding the efficacy of teclistamab relative to the comparator drugs. Therefore, no superiority conclusions could be drawn from the MAIC submitted by the sponsor due to methodological limitations and imprecision in the effect estimates.
Teclistamab (MajesTEC-1 Study) Versus Selinexor in Combination with Dexamethasone (STORM Study, Part 2)33
An unanchored MAIC was conducted to compare the efficacy of teclistamab with selinexor in combination with dexamethasone using individual patient data from the MajesTEC-1 trial (n = 150) and summary-level data from the STORM trial, part 2 (n = 122). The STORM trial, part 2, eligibility criteria were applied to patients from the ITT population of the MajesTEC-1 study. Compared to patients in the STORM trial, part 2, the MajesTEC-1 study’s population had a higher proportion of patients with R-ISS stage II. The 2 populations were similar in age, ECOG PS, and cytogenetic status. All patients had triple-class exposed RRMM. After applying the STORM trial, part 2, eligibility criteria, individual patient data from patients in the MajesTEC-1 trial were weighted to match the aggregated baseline patient characteristics from the STORM trial, part 2. For binary outcomes, the relative effects were quantified using an OR and 95% CI derived from a weighted logistic regression analysis, while time-to-event outcomes were estimated using a weighted Cox proportional hazards model. The following factors were used to adjust for imbalances between patient populations: refractory status, cytogenetic profile, ISS staging, the presence of extramedullary disease, and the number of prior lines of therapy. The effective sample size of the MajesTEC-1 trial after matching was 37 patients.
Efficacy Results
Following adjustment, the estimated HR of OS for teclistamab versus selinexor in combination with dexamethasone was 0.52 (95% CI, 0.28 to 0.95). Following adjustment, the estimated HR of PFS for teclistamab versus selinexor in combination with dexamethasone was 0.58 (95% CI, 0.30 to 1.11), and the estimated HR of duration of response was 0.04 (95% CI, 0.01 to 0.10). Following adjustment, the OR of ORR for teclistamab versus selinexor in combination with dexamethasone was 3.14 (95% CI, 1.48 to 6.69) while the OR of CR or better was 16.3 (95% CI, 3.5 to 77.1).
Safety
No results for safety were included in the study comparing the MajesTEC-1 trial and the STORM trial, part 2.
Critical Appraisal
The open-label design of the studies can result in a risk of bias in the study conduct, including the measurement of the outcomes, and increase uncertainty in subjective outcomes such as PFS and ORR. The bias will likely favour the experimental intervention, although the extent of bias is uncertain. The effective sample size was reduced after adjustment in the MAIC analysis to approximately 24.7% (37 of 150) of the patients in the original population in the MajesTEC-1 study. The reduction in the effective sample size reflects the heterogeneity between the trials among the variables included in the weighting process. A small effective sample size implies that the weighted estimates are being influenced by a subset of patients from the MajesTEC-1 study who may not be representative of the entire study population, which may limit the generalizability of the results. It remains unclear how balanced populations were for other variables that may be clinically relevant but could not be adjusted due to lack of data, or those variables that were not part of the planned adjustment (unknown or unmeasured prognostic factors), which leaves the potential for residual confounding.36 In the MAIC analysis, results in efficacy estimates were imprecise (i.e., wide CIs) in the end points assessed, and the upper and lower boundaries of the CIs suggest the potential for different conclusions regarding the efficacy of teclistamab relative to the comparator drugs. Therefore, no superiority conclusions could be drawn from the MAIC submitted by the sponsor due to methodological limitations and imprecision in the effect estimates.
Studies Addressing Gaps in the Systematic Review Evidence
Content in this section has been informed by materials submitted by the sponsor. The following has been summarized and validated by the CADTH review team.
Table 44: Summary of Gaps in the Systematic Review Evidence
Evidence gap | Studies that address gaps | |
|---|---|---|
Study description | Summary of key results | |
No head-to-head studies of teclistamab compared to currently available fourth or higher lines of therapies in Canada | The MajesTEC-3 trial is a multicentre, open-label, randomized, phase III study that compares the efficacy of teclistamab in combination with daratumumab vs. daratumumab, pomalidomide, and dexamethasone, or daratumumab, bortezomib, and dexamethasone in patients with RRMM. | Ongoing |
The MajesTEC-9 trial is a randomized, open-label, multicentre, phase III study that compares teclistamab monotherapy vs. pomalidomide in combination with bortezomib and dexamethasone, or carfilzomib in combination with dexamethasone, in patients with RRMM who have received 1 to 3 prior lines of therapy, including an anti-CD38 monoclonal antibody, and lenalidomide. | Ongoing | |
RRMM = relapsed or refractory multiple myeloma; vs. = versus.
Note: Details included in Table 44 are from the sponsor’s Summary of Clinical Evidence.83
Description of Studies
The sponsor submitted 2 ongoing head-to-head clinical studies comparing teclistamab to currently available fourth-line and beyond lines of therapy in patients with RRMM. Details of the 2 studies are summarized in Table 45. The MajesTEC-3 trial is a multicentre, open-label, randomized, phase III study comparing the efficacy of teclistamab in combination with daratumumab versus investigator’s choice of daratumumab in combination with pomalidomide and dexamethasone, or daratumumab in combination with bortezomib plus dexamethasone in patients with RRMM. The MajesTEC-9 trial is a phase III, randomized, open-label, multicentre study comparing teclistamab with investigator’s choice of pomalidomide, bortezomib, and dexamethasone or carfilzomib and dexamethasone in patients with RRMM who have received 1 to 3 prior lines of therapy, including an anti-CD38 monoclonal antibody, and lenalidomide.
Table 45: Details of Studies Addressing Gaps in the Systematic Review Evidence
Detail | MajesTEC-3 Study | MajesTEC-9 Study |
|---|---|---|
Study design | Multicentre, open-label, randomized, phase III study | Multicentre, open-label, randomized, phase III study |
Enrolled, N | 560 | 590 |
Start date | October 2021 | January 2023 |
End date | October 2026 | May 2031 |
Key inclusion criteria |
|
|
Key exclusion criteria |
|
|
Intervention | Teclistamab in combination with daratumumab (28-day cycles) | Teclistamab (28-day cycles) |
Comparator(s) | Daratumumab in combination with pomalidomide and dexamethasone or Daratumumab in combination with bortezomib plus dexamethasone | Pomalidomide in combination with bortezomib and dexamethasone or Carfilzomib in combination with dexamethasone |
Outcomes | ||
Primary end point |
|
|
Secondary end points |
|
|
AE = adverse event; BCMA = B-cell maturation antigen; CNS = central nervous system; CR = complete response; CTCAE = Common Terminology Criteria for Adverse Events; ECOG PS = Eastern Cooperative Oncology Group Performance Status; IMWG = International Myeloma Working Group; MM = multiple myeloma; MRD = minimal residual disease; NCI = National Cancer Institute; ORR = overall response rate; OS = overall survival; PFS = progression-free survival; PI = proteasome inhibitor; POEMS = polyneuropathy, organomegaly, endocrinopathy, M protein, and skin changes.
Note: Details included in Table 45 are from the sponsor’s Summary of Clinical Evidence.83
Discussion
Summary of Available Evidence
The MajesTEC-1 trial is an ongoing, phase I and phase II, open-label study of teclistamab administered to adult patients with RRMM at a dose of 1.5 mg/kg subcutaneously weekly. The MajesTEC-1 trial enrolled 165 patients, including 40 patients in phase I and 125 patients in phase II, cohort A. Patients were enrolled in 39 sites across 10 countries, including Canada. The primary objectives of phase I in the MajesTEC-1 study were to determine the proposed recommended phase II dose of teclistamab and treatment schedule that was assessed to be safe, and to characterize the safety and tolerability of teclistamab at the recommended phase II dose. The primary objective of phase II was to evaluate the efficacy and safety of teclistamab at the proposed recommended phase II dose. In the MajesTEC-1 study (n = 165), enrolled patients had a documented diagnosis of RRMM, had received at least 3 lines of therapy (except BCMA-targeted therapy), and had progressive and measurable disease, with an ECOG PS score of 0 or 1. In the MajesTEC-1 trial, the median age of the patients was 64.0 years (range = 62.5 years to 64.0 years), with 52.1% of the study population being aged younger than 65 years. Ninety-six (58.2%) patients were male and 69 (41.8%) patients were female. Most patients were white (81.2%), and 12.7% of patients identified as Black or African American. Most patients had an ECOG PS score of 1 (66.1%) and 33.3% of patients had an ECOG PS score of 0. Phase II, cohort C (n = 40), enrolled patients who had received at least 3 prior lines of therapy that included a PI, an iMiD, an anti-CD38 monoclonal antibody, and an anti-BCMA treatment.
The sponsor submitted 6 ITCs, of which 3 ITCs were used to inform the pharmacoeconomic model — including 2 ITCs comparing the relative efficacy of teclistamab with RWPC therapy (from the LocoMMotion and daratumumab trials)26-29 and a third ITC comparing the relative efficacy of teclistamab with ciltacabtagene autoleucel (from the CARTITUDE-1 study).30 Of the 3 ITCs submitted by the sponsor that were not included in the pharmacoeconomic model, 1 published ITC compared the relative efficacy of teclistamab with RWPC therapy (from the Flatiron Health database),29,31 and 2 conference abstracts compared the relative efficacy of teclistamab with belantamab mafodotin (from the DREAMM-2 study)32 and selinexor in combination with dexamethasone (from the STORM study, part 2).33
The studies addressing gaps in the systematic review evidence included 2 ongoing studies. One study is the MajesTEC-3 study (n = 560), which is a multicentre, open-label, randomized, phase III study comparing the efficacy of teclistamab in combination with daratumumab versus daratumumab, pomalidomide, and dexamethasone, or daratumumab, bortezomib, and dexamethasone. The second study is the MajesTEC-9 study (n = 590), which is a randomized, open-label, multicentre phase III study comparing teclistamab monotherapy versus pomalidomide in combination with bortezomib and dexamethasone, or carfilzomib in combination with dexamethasone in patients with RRMM who have received 1 to 3 prior lines of therapy, including an anti-CD38 monoclonal antibody, and lenalidomide.
Interpretation of Results
Efficacy
A causal conclusion of the comparative effectiveness of teclistamab cannot be drawn from the MajesTEC-1 study, which is considered noninterpretable because of the lack of a comparator group and the relatively small number of patients. Other limitations of the MajesTEC-1 study included a high attrition rate for HRQoL and the risk of bias in the measurement of subjective outcomes, such as HRQoL and safety outcomes due to the open-label design. Additionally, this report presents interim analysis results because a prespecified final analysis was not available, and there is the potential that the benefit of teclistamab is overestimated; however, the extent of any overestimation is uncertain.19,20 Of note, there is an ongoing, randomized, phase III, open-label study (the MajesTEC-9 trial) comparing teclistamab monotherapy versus pomalidomide in combination with bortezomib and dexamethasone, or carfilzomib in combination with dexamethasone in patients with RRMM who have received 1 to 3 prior lines of therapy, including an anti-CD38 monoclonal antibody, and lenalidomide. However, the estimated completion date of the MajesTEC-9 study is May 2031.
In the MajesTEC-1 study (phase I and phase II, cohort A), ORR was the primary end point and achieved the predetermined threshold for a positive outcome based on the sponsor's assertion, an ORR greater than 45% (where the lower bound of the 95% CI of ORR is greater than 30%). However, the evidence for the treatment effect of teclistamab on ORR is very uncertain when compared with any comparator. According to the FDA, the ORR can be evaluated in a single-arm study as a direct measure of a drug antitumour activity if it is defined as the sum of PRs plus CRs.18 A total of 63.0% of patients (95% CI, 55.2% to 70.4%) attained an overall response (PR or better), which was considered clinically meaningful by the clinical experts consulted by CADTH for this review, especially when indirectly compared to currently available therapies in this population. However, there was no adjustment for multiplicity across the various analyses of the outcome (i.e., the various data cut-offs), which may have increased the risk of false-positive conclusions. Of the 104 responders (who attained PR or better), 51 (49.0%) patients maintained their response until the clinical cut-off date, including 46 (44.2%) patients who were still receiving treatment. The results from subgroup analyses based on the number of prior lines of therapy and cytogenetic risk that were deemed clinically meaningful by the clinical experts were consistent with the results of the primary ORR analysis; however, the analysis was limited by small sample sizes of the groups. A total of 75 (45.5%) patients (95% CI, 37.7% to 53.4%) attained CR or better, and 98 (59.4%) patients (95% CI, 51.5% to 67.0%) attained VGPR or better, which was considered clinically meaningful by the clinical experts consulted by CADTH. Clinical experts consulted by CADTH for this review identified clinical response outcomes as important outcomes and noted that CR and VGPR are the most desirable outcomes in most situations in the RRMM population. In the MajesTEC-1 trial, 63 (38.2%) patients switched from weekly to biweekly dosing, including 54 patients who attained CR or better and 9 patients who attained PR or VGPR or better. Of the 63 patients, 41 (65.1%) patients maintained their response and were still receiving treatment at the time of the data cut-off date. However, the number of patients with dose schedule change was very small, which limits interpretation of the findings.
In the MajesTEC-1 trial, which had a median duration of follow-up of 22.8 months, the median OS was 21.9 months (95% CI, 15.1 months to NE months) and reported an estimated 24-month OS probability of 48.7% (95% CI, 40.5% to 56.3%), which was considered clinically meaningful by the clinical experts consulted by CADTH. The median PFS was 11.3 months (95% CI, 8.8 months to 16.4 months) and the 24-month PFS probability was 33.7% (95% CI, 25.9% to 41.6%), which was also considered clinically meaningful by the clinical experts consulted by CADTH. The clinical experts consulted by CADTH mentioned that the median OS for current treatments for RRMM available in the fourth-line setting and beyond is approximately 24 months, and the median PFS is approximately 6 months to 12 months. However, the evidence for the treatment effect of teclistamab on OS and PFS is very uncertain when compared with any comparator. The median time to first response was 1.2 months (95% CI, 0.2 month to 5.5 months), and the median time to best response was 4.0 months (range = 0.2 month to 18.5 months). The median duration of response among 104 responders was 21.6 months (95% CI, 16.2 months to NE months) with a 24-months event-free probability of 49.9% (95% CI, 39.0% to 59.9%), which was considered clinically meaningful by the clinical experts consulted by CADTH. Updated data regarding the MRD-negativity status based on the January 4, 2023, clinical cut-off date were not available. At the time of the data cut-off date of September 7, 2021 (primary analysis), 37 (24.7%) patients (95% CI, 18.0% to 32.4%) had attained MRD negativity at 10–5 bone marrow cells. The clinical experts consulted by CADTH noted that MRD negativity correlates with the duration of response, and a single measurement of the MRD-negativity status is less meaningful than persistent MRD negativity over time.
The clinical experts and patient and clinician groups consulted by CADTH highlighted improvement in HRQoL as an important outcome and treatment goal for patients with RRMM. HRQoL was measured using the EORTC QLQ-C30 and EQ-5D-5L questionnaires. The EORTC QLQ-C30 questionnaire was generally validated in the RRMM, but the EQ-5D-5L questionnaire was not. The clinical experts consulted for this review noted that HRQoL measures are not used in routine clinical practice. The EORTC QLQ-C30 tool showed improvements over time from baseline on the pain and fatigue system scale, improvements in global health status from cycle 4 onward, and improvements in physical functioning from cycle 4 onward, with a decrease initially observed in cycle 2. The clinical experts noted that side effects of teclistamab that may affect HRQoL transiently, such as CRS and ICANS, are common in the first treatment cycle, and it will be some time before effective treatment reverses this trajectory. The clinical experts also noted that HRQoL measurements are not taken daily to better understand the inflection point. The analyses of HRQoL outcomes were undertaken post hoc, so there is a risk of bias in the selection of the reported result. The analyses were performed in the HRQoL-evaluable population of patients who had evaluable assessment at baseline and follow-up time points for each domain of EORTC QLQ-C30 or EQ-5D-5L, which limits the generalizability of the results. In addition, strong conclusions could not be drawn for HRQoL outcomes due to the high risk of attrition bias and increased risk of type I error.
The sponsor submitted 6 ITCs, which were summarized for this review to supplement the assessment of teclistamab compared to RWPC therapy, ciltacabtagene autoleucel, selinexor, and belantamab mafodotin for the treatment of patients with RRMM. The results of the adjusted treatment comparisons were consistent across end points, favouring teclistamab over RWPC therapy (from the LocoMMotion study, the daratumumab trials cohort, and the Flatiron Health database), including OS, PFS, TTNT, and key response outcomes. This is consistent with the opinion of the clinical experts consulted for this review; however, the comparative efficacy estimates remain uncertain due to the methodological limitations in these studies, the heterogeneity in the populations, and the potential for residual confounding. No conclusions could be drawn from the 6 sponsor-submitted ITCs about the relative efficacy of teclistamab to the comparator drugs on the clinical response outcomes (ORR, CR or better, or VGPR or better) due to methodological limitations and imprecision in the effect estimates (wide 95% CIs). Although the results suggest that teclistamab is inferior to ciltacabtagene autoleucel for OS, PFS, and TTNT, these findings must be interpreted in the context of the methodological limitations of these studies. For OS and PFS, no superiority conclusions could be drawn about the relative efficacy of teclistamab compared to belantamab mafodotin and selinexor due to methodological limitations and imprecision in the effect estimates (with wide 95% CIs). No safety or HRQoL outcomes were evaluated in any of these ITCs.
Additional supporting data were presented for phase II, cohort C, in the MajesTEC-1 trial to address the use of teclistamab in patients previously treated with BCMA-targeted therapy in accordance with the Health Canada indication for teclistamab. Findings from phase II, cohort C, were consistent with the results from the pivotal cohort; however, only 40 patients were included in phase II, cohort C, which limits interpretation of the findings.
Harms
All patients in the MajesTEC-1 study experienced at least 1 TEAE, and the most common TEAEs were CRS (72.1%), neutropenia (71.5%), anemia (54.5%), thrombocytopenia (42.4%), lymphopenia (36.4%), diarrhea (33.9%), and pyrexia (31.5%). In the MajesTEC-1 study, || |||||||| ||||||| experienced TEAEs of grade 3, || |||||||| ||||||| experienced TEAEs of grade 4, and || |||||||| ||||||| experienced TEAEs of grade 5. The clinical experts consulted for this review noted that the rate of TEAEs of grade 4 or grade 5 was relatively higher than would be expected in clinical practice. A total of 113 (68.5%) patients experienced at least 1 serious TEAE. A total of | |||||||| |||||| stopped study treatment due to TEAEs in the MajesTEC-1 trial, mostly due to COVID-19 infection, pneumocystis jirovecii pneumonia, and adenoviral pneumonia. Several adverse events of clinical interest were identified, including CRS, neurologic adverse events and neurotoxicity, ICANS, systemic administration-related reactions, injection-site reactions, hypogammaglobulinemia, cytopenia, and infections. The clinical experts consulted by CADTH noted that all adverse events of clinical interest were manageable. A total of 119 (72.1%) patients experienced CRS events, mostly at a level of grade 1 or grade 2. There were 110 (66.7%) patients who received supportive measures to treat CRS events. A total of 5 (3.0%) patients experienced ICANS, with 3 (1.8%) patients reporting cases of ICANS of a grade 1 event, and 2 (1.2%) patients reporting cases of ICANS of CTCAE grade 3. There were 4 (2.4%) patients who received supportive treatments for ICANS. Clinical experts noted that CRS and ICANS are common side effects in this population, but they represent short-term toxicities lasting the first few weeks. A total of 132 (80.0%) patients had infections of any CTCAE grade, including 21 (12.7%) patients with at least 1 infection and infestation of CTCAE grade 5. The clinical experts consulted for this review mentioned that the infection rate in the MajesTEC-1 trial was higher than would be expected in clinical practice; however, this study was conducted during the COVID-19 pandemic, and this high rate may be due to an overestimation of the number of infections associated with COVID-19. A total of 152 (92.1%) patients experienced at least 1 treatment-emergent cytopenic event, including neutropenia (71.5%), anemia (55.8%), thrombocytopenia (42.4%), and lymphopenia (36.4%). The clinical experts consulted by CADTH mentioned that cytopenia is very common in this population. A total of 35 (21.2%) patients experienced at least 1 hypogammaglobulinemia TEAE, including 34 (20.6%) patients with a case of hypogammaglobulinemia and 1 (0.6%) patient with a case of hypoglobulinemia. The clinical experts consulted by CADTH noted that the rate of hypogammaglobulinemia was low in the MajesTEC-1 study, as almost all patients treated with teclistamab in clinical practice experience hypogammaglobulinemia.
Safety outcomes were not analyzed in the sponsor-submitted ITC reports and no justification was provided, which precludes a balanced judgment of comparative benefit relative to comparative harms.
Conclusion
One sponsor-submitted phase I and phase II, single-arm, open-label, ongoing study (the MajesTEC-1 study) provided evidence regarding the efficacy and safety of teclistamab for patients with RRMM who had received at least 3 prior lines of therapy. In general, the clinical experts consulted by CADTH for this review considered the OS, PFS, ORR, and CR results to be clinically meaningful, especially when indirectly compared to currently available therapies in this population. However, the evidence for the treatment effect of teclistamab is very uncertain due to the single-arm design of the study, which is not intended to be confirmatory for efficacy, and the lack of a comparator group limits the estimation of relative effects of treatment with teclistamab compared with other treatment options. Additionally, the estimates of benefit of teclistamab may be overestimated because this report presents results of the interim analysis; however, the presence and extent of any overestimation is uncertain. The results for the HRQoL remained inconclusive due to a number of important limitations, including post hoc analysis, attrition bias, and increased risk of type I error. Notable harms that occurred more frequently in the MajesTEC-1 study included infections, CRS, and cytopenia. In general, TEAEs were stated by the clinical experts consulted by CADTH for this review to be manageable.
The evidence about the comparative efficacy of teclistamab relied on 1 single-arm study, and no direct evidence of teclistamab compared to other comparators was available for this review. Six ITCs were submitted by the sponsor comparing the efficacy of teclistamab with RWPC therapy, ciltacabtagene autoleucel, selinexor, and belantamab mafodotin in patients with RRMM. Although teclistamab was favoured over RWPC therapy for OS, PFS, and TTNT, the comparative efficacy estimates remain uncertain due to the methodological limitations, the heterogeneity in the populations and studies, and the potential for residual confounding. Although teclistamab was favoured over RWPC therapy for key response outcomes, no superiority conclusions could be drawn about the relative efficacy of teclistamab compared to RWPC for ORR, CR or better, or VGPR or better, due to methodological limitations and imprecision in the effect estimates (i.e., wide 95% CIs). Although the results suggest that ||||||||||| || |||||||| || |||||||||||||| |||||||||| for OS, PFS, and TTNT, these findings must be interpreted in the context of the methodological limitations of these studies. For OS and PFS, no superiority conclusions could be drawn about the relative efficacy of teclistamab compared to belantamab mafodotin and selinexor due to methodological limitations and imprecision in the effect estimates. No superiority conclusions could be drawn from the 6 sponsor-submitted ITCs about the efficacy of teclistamab relative to the comparator drugs for the clinical response outcomes, including ORR, CR or better, or VGPR or better, due to methodological limitations and imprecision in the effect estimates (wide 95% CIs). No conclusions could be drawn on the relative safety of teclistamab to the comparative drugs because no safety analysis was performed. No HRQoL outcomes were evaluated in the sponsor-submitted ITCs.
There is an unmet need for fourth-line and beyond treatment options for RRMM, as patients with drug resistance after second-line or third-line therapy cannot be treated again with the same drug. In general, the indirect evidence was aligned with some outcomes identified as important to patients with RRMM who are seeking additional fourth-line and beyond treatment options that prolong survival and delay disease progression.
References
1.Kyle RA, Rajkumar SV. Criteria for diagnosis, staging, risk stratification and response assessment of multiple myeloma. Leukemia. 2009;23(1):3-9. PubMed
2.Bird SA, Boyd K. Multiple myeloma: An overview of management. Palliat Care Soc Pract. 2019;13:1178224219868235. PubMed
3.Rajkumar SV. Multiple myeloma: 2022 update on diagnosis, risk stratification, and management. Am J Hematol. 2022;97(8):1086-1107. PubMed
4.Canadian Cancer Society. Multiple myeloma statistics [accessed by sponsor]. 2022; https://cancer.ca/en/cancer-information/cancer-types/multiple-myeloma/statistics. Accessed May 2023.
5.Canadian Cancer Society. Survival statistics for multiple myeloma [accessed by sponsor]. https://cancer.ca/en/cancer-information/cancer-types/multiple-myeloma/prognosis-and-survival/survival-statistics. Accessed May 2023.
6.Podar K, Leleu X. Relapsed/refractory multiple myeloma in 2020/2021 and beyond. Cancers (Basel). 2021;13(20):5154. PubMed
7.Mikhael J, Ismaila N, Cheung MC, et al. Treatment of multiple myeloma: ASCO and CCO joint clinical practice guideline. J Clin Oncol. 2019;37(14):1228-1263. PubMed
8.Kurtin SE. Relapsed or relapsed/refractory multiple myeloma [accessed by sponsor]. J Adv Pract Oncol. 2013;4(Suppl 1):5-14.
9.Canadian Cancer Society. Symptoms of multiple myeloma [accessed by sponsor]. https://cancer.ca/en/cancer-information/cancer-types/multiple-myeloma/signs-and-symptoms. Accessed May 2023.
10.Mayo Clinic. Multiple myeloma [accessed by sponsor]. https://www.mayoclinic.org/diseases-conditions/multiple-myeloma/diagnosis-treatment/drc-20353383. Accessed May 2023.
11.Bhatt P, Kloock C, Comenzo R. Relapsed/refractory multiple myeloma: A review of available therapies and clinical scenarios encountered in myeloma relapse. Curr Oncol. 2023;30(2):2322-2347. PubMed
12.Rajkumar SV, Harousseau JL, Durie B, et al. Consensus recommendations for the uniform reporting of clinical trials: Report of the International Myeloma Workshop Consensus Panel 1. Blood. 2011;117(18):4691-4695. PubMed
13.CADTH. Provisional funding algorithm for multiple myeloma [accessed by sponsor]. Ottawa (ON): CADTH; 2023: https://www.cadth.ca/sites/default/files/DRR/2023/PH0031%20Multiple%20Myeloma_Algorithm-Secured.pdf. Accessed August 2023.
14.Cocks K, King MT, Velikova G, Fayers PM, Brown JM. Quality, interpretation and presentation of European Organisation for Research and Treatment of Cancer quality of life questionnaire core 30 data in randomised controlled trials. Eur J Cancer. 2008;44(13):1793-1798. PubMed
15.Pickard AS, Neary MP, Cella D. Erratum to: Estimation of minimally important differences in EQ-5D utility and VAS scores in cancer [accessed by sponsor]. Health Qual Life Outcomes. 2010;8(1):4.
16.Pickard AS, Neary MP, Cella D. Estimation of minimally important differences in EQ-5D utility and VAS scores in cancer. Health Qual Life Outcomes. 2007;5(70):70. PubMed
17.Evans SR. Clinical trial structures. J Exp Stroke Transl Med. 2010;3(1):8-18. PubMed
18.U.S. Department of Health and Human Services, Food and Drug Administration, Oncology Center of Excellence, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER). Clinical trial endpoints for the approval of cancer drugs and biologics guidance for industry. 2018: https://www.fda.gov/media/71195/download Accessed 2023 Nov 10.
19.Bassler D, Briel M, Montori VM, et al. Stopping randomized trials early for benefit and estimation of treatment effects: Systematic review and meta-regression analysis. JAMA. 2010;303(12):1180-1187. PubMed
20.Briel M, Bassler D, Wang AT, Guyatt GH, Montori VM. The dangers of stopping a trial too early. J Bone Joint Surg Am. 2012;94 Suppl 1:56-60. PubMed
21.Wilcox RA, Djulbegovic B, Moffitt HL, Guyatt GH, Montori VM. Randomized trials in oncology stopped early for benefit. J Clin Oncol. 2008;26(1):18-19. PubMed
22.Fairclough DL, Peterson HF, Chang V. Why are missing quality of life data a problem in clinical trials of cancer therapy? Stat Med. 1998;17(5-7):667-677. PubMed
23.Tsang M, Le M, Ghazawi FM, et al. Multiple myeloma epidemiology and patient geographic distribution in Canada: A population study. Cancer. 2019;125(14):2435-2444. PubMed
24.Clinical Study Report: 64007957MMY1001. A Phase 1/2, First-in-Human, Open-Label, Dose Escalation Study of Teclistamab, a Humanized BCMA×CD3 Bispecific Antibody, in Subjects with Relapsed or Refractory Multiple Myeloma: MajesTEC-1 [internal sponsor's report]. Toronto (ON): Janssen Research & Development, LLC; 2021 Dec 6.
25.Updated Analysis Clinical Study Report: Updated Clinical Data for Cohort C for MajesTEC-1 (March 16, 2022) [internal sponsor’s report]. Toronto (ON): Janssen Inc.; 2022 Aug 25.
26.Janssen Research and Development. Adjusted comparison of teclistamab in MajesTEC-1 versus real-world physician’s choice in LocoMMotion for the treatment of patients with multiple myeloma with prior exposure to a proteasome inhibitor, an immunomodulatory agent and an anti-CD38 monoclonal antibody (04 January 2023 data cutoff) [accessed by sponsor]. 2023.
27.Mateos MV, Chari A, Usmani SZ, et al. Comparative efficacy of teclistamab versus physician's choice of therapy in the long-term follow-up of APOLLO, POLLUX, CASTOR, and EQUULEUS clinical trials in patients with triple-class exposed relapsed or refractory multiple myeloma. Clin Lymphoma Myeloma Leuk. 2023;23(5):385-393. PubMed
28.Moreau P, van de Donk N, Delforge M, et al. Comparative efficacy of teclistamab versus current treatments in real-world clinical practice in the prospective LocoMMotion Study in patients with triple-class-exposed relapsed and/or refractory multiple myeloma. Adv Ther. 2023;40(5):2412-2425. PubMed
29.Janssen Research and Development. Adjusted treatment comparison to evaluate the comparative efficacy of teclistamab in MajesTEC-1 versus physician’s choice of treatment in the long-term follow-up of APOLLO, POLLUX, CASTOR and EQUULEUS for the treatment of patients with multiple myeloma with prior exposure to a proteasome inhibitor, an immunomodulatory agent and an anti-CD38 monoclonal antibody [accessed by sponsor]. 2022.
30.Janssen Research and Development. Summary of indirect treatment comparison methods and results of teclistamab in MajesTEC-1 versus ciltacabtagene autoleucel in CARTITUDE-1 for the treatment of patients with multiple myeloma with prior exposure to a proteasome inhibitor, an immunomodulatory agent and an anti-CD38 monoclonal antibody [accessed by sponsor]. 2023.
31.Krishnan A, Nooka AK, Chari A, et al. Teclistamab versus real-world physician's choice of therapy in triple-class exposed relapsed/refractory multiple myeloma. J Comp Eff Res. 2023;12(6):e220186. PubMed
32.Moreau P, Usmani SZ, van de Donk NWCJ, et al. Matching-adjusted indirect treatment comparison (MAIC) of teclistamab (tec) versus belantamab mafodotin (belamaf) for the treatment of patients (pts) with triple-class exposed (TCE), relapsed/refractory multiple myeloma (RRMM) [accessed by sponsor]. J Clin Oncol. 2022;40(16_suppl):8035-8035.
33.Bahlis NJ, Usmani SZ, Rosiñol L, et al. Matching-adjusted indirect comparison (MAIC) of teclistamab (tec) versus selinexor-dexamethasone (sel-dex) for the treatment of patients (pts) with triple-class exposed (TCE) relapsed/refractory multiple myeloma (RRMM) [accessed by sponsor]. J Clin Oncol. 2022;40(16_suppl):e20028-e20028.
34.Lee J, Chia KS. Estimation of prevalence rate ratios for cross sectional data: An example in occupational epidemiology. Br J Ind Med. 1993;50(9):861-862. PubMed
35.Grambsch PM, Therneau TM. Proportional hazards tests and diagnostics based on weighted residuals [accessed by sponsor]. Biometrika. 1994;81(3):515-526.
36.Phillippo DM, Ades AE, Dias S, Palmer S, Abrams KR, Welton NJ. Methods for population-adjusted indirect comparisons in health technology appraisal. Med Decis Making. 2018;38(2):200-211. PubMed
37.Phillippo D, Ades T, Dias S, Palmer S, Abrams KR, Welton N. NICE DSU Technical Support Document 18: Methods for population-adjusted indirect comparisons in submissions to NICE. (Technical Support Documents). NICE Decision Support Unit. 2016; https://research-information.bris.ac.uk/ws/portalfiles/portal/94868463/Population_adjustment_TSD_FINAL.pdf. Accessed 2023 Nov 21.
38.Xue X, Xie X, Gunter M, et al. Testing the proportional hazards assumption in case-cohort analysis. BMC Med Res Methodol. 2013;13:88. PubMed
39.Mathoulin-Pelissier S, Gourgou-Bourgade S, Bonnetain F, Kramar A. Survival end point reporting in randomized cancer clinical trials: A review of major journals. J Clin Oncol. 2008;26(22):3721-3726. PubMed
40.Myeloma Canada. Multiple Myeloma Patient Handbook [accessed by sponsor]. 2014; https://myeloma.ca/pixms/uploads/serve/ckeditor/mmph_en-2.pdf. Accessed May 2023.
41.Canadian Cancer Society. Prognosis and survival for multiple myeloma [accessed by sponsor]. https://cancer.ca/en/cancer-information/cancer-types/multiple-myeloma/prognosis-and-survival. Accessed May 2023.
42.Rajkumar SV. Updated diagnostic criteria and staging system for multiple myeloma. Am Soc Clin Oncol Educ Book. 2016;35:e418-423. PubMed
43.Bergstrom DJ, Kotb R, Louzada ML, et al. Consensus guidelines on the diagnosis of multiple myeloma and related disorders: Recommendations of the Myeloma Canada Research Network Consensus Guideline Consortium. Clin Lymphoma Myeloma Leuk. 2020;20(7):e352-e367. PubMed
44.Canadian Cancer Society. Stages of multiple myeloma [accessed by sponsor]. https://cancer.ca/en/cancer-information/cancer-types/multiple-myeloma/staging. Accessed May 2023.
45.Greipp PR, San Miguel J, Durie BG, et al. International staging system for multiple myeloma. J Clin Oncol. 2005;23(15):3412-3420. PubMed
46.Bruno AS, Willson JL, Opalinska JM, et al. Recent real-world treatment patterns and outcomes in US patients with relapsed/refractory multiple myeloma. Expert Rev Hematol. 2020;13(9):1017-1025. PubMed
47.Mikhael J. Treatment options for triple-class refractory multiple myeloma. Clin Lymphoma Myeloma Leuk. 2020;20(1):1-7. PubMed
48.Weisel K, Martin T, Yong K, Qi K, Londhe A. Characteristics and treatment patterns of triple-class exposed patients with relapsed/refractory multiple myeloma who participated in clinical trials of daratumumab. (Presented at 47th annual EBMT meeting) [accessed by sponsor]. 2021.
49.CADTH pERC Reimbursement Recommendation: Carfilzomib (Kyprolis) in combination with dexamethasone (Dex) [accessed by sponsor]. Ottawa (ON): CADTH; 2017: https://www.cadth.ca/sites/default/files/pcodr/pcodr_carfilzomib_kyprolis_mm_rel_fn_rec.pdf. Accessed November 2023.
50.CADTH pERC Reimbursement Recommendation: Pomalidomide (Pomalyst) [accessed by sponsor]. Ottawa (ON): CADTH; 2014: https://www.cadth.ca/reimbursement-review-reports. Accessed November 2023.
51.CADTH Drug reimbursement review: Pomalidomide (Pomalyst) for multiple myeloma. Ottawa (ON): CADTH; 2019: https://www.cadth.ca/pomalyst-combination-dexamethasone-and-bortezomib-multiple-myeloma-second-line-or-beyond-details. Accessed 2023 Nov 1.
52.CADTH Drug reimbursement review: Selinexor (Xpovio) for multiple myeloma. Ottawa (ON): CADTH; 2022: https://www.cadth.ca/selinexor. Accessed 2023 Nov 1.
53.pan-Canadian Pharmaceutical Alliance (pCPA). Carvykti (ciltacabtagene autoleucel) [accessed by sponsor]. 2023; https://www.pcpacanada.ca/negotiation/22363. Accessed November 2023.
54.BC Cancer. Lymphoma and myeloma. 2023; http://www.bccancer.bc.ca/health-professionals/clinical-resources/chemotherapy-protocols/lymphoma-myeloma. Accessed 2023 Nov 6.
55.Cancer Care Ontario. Drug formulary. 2023; https://www.cancercareontario.ca/en/drugformulary/regimens/monograph/59751. Accessed 2023 Nov 6.
56.Moreau P, Garfall AL, van de Donk N, et al. Teclistamab in relapsed or refractory multiple myeloma. N Engl J Med. 2022;387(6):495-505. PubMed
57.Moreau P, van de Donk NW, Nahi H, et al. Plain language summary of the MajesTEC-1 study of teclistamab for the treatment of people with relapsed or refractory multiple myeloma. Future Oncol. 2023;19(12):811-818.
58.Janssen Pharmaceuticals. Tecvayli [accessed by sponsor]. 2023.
59.Carvykti (ciltacabtagene autoleucel): Cell suspensionin infusion bag, 0.5-1.0x106 CAR-positive viable T-cells per kg body weight with a maximum of 1x108 CAR-positive viable T-cells, for intravenous infusion [product monograph]. Toronto (ON): Janssen Inc; 2023 Feb 9.
60.Xpovio (selinexor tablets): Tablets, 20 mg, Oral, Antineoplastic Agent [product monograph]. Oakville (ON): FORUS Therapeutics Inc; 2024 Mar 22.
61.Pomalyst (Pomalidomide Capsules): 1 mg, 2 mg, 3 mg and 4 mg, for Oral Use [product monograph]. Saint-Laurent (QC): Bristol-Myers Squibb Canada; 2024 Jan 25.
62.Kyprolis (carfilzomib for injection): Powder for solution for intravenous infusion, 10, 30, 60 mg per vial [product monograph]. Mississauga (ON): Amgen Canada Inc; 2023 Jul 25.
63.Kumar S, Paiva B, Anderson KC, et al. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol. 2016;17(8):e328-e346. PubMed
64.Usmani SZ, Garfall AL, van de Donk N, et al. Teclistamab, a B-cell maturation antigen x CD3 bispecific antibody, in patients with relapsed or refractory multiple myeloma (MajesTEC-1): A multicentre, open-label, single-arm, phase 1 study. Lancet. 2021;398(10301):665-674. PubMed
65.Primary Analysis Clinical Study Report: A Phase 1/2, First-in-Human, Open-Label, Dose Escalation Study of Teclistamab, a Humanized BCMA×CD3 Bispecific Antibody, in Subjects with Relapsed or Refractory Multiple Myeloma [internal sponsor's report]. Toronto (ON): Janssen Research & Development, LLC; 2021 Dec 6.
66.Clinical Study Protocol: 64007957MMY1001. A Phase 1/2, First-in-Human, Open-Label, Dose Escalation Study of Teclistamab, a Humanized BCMA x CD3 Bispecific Antibody, in Subjects with Relapsed or Refractory Multiple Myeloma [internal sponsor's report]. Toronto (ON): Janssen Inc.; 2020 Oct 23.
67.National Cancer Institute. Common Terminology Criteria for Adverse Events (CTCAE) v4.03 [accessed by sponsor]. 2010; https://ctep.cancer.gov/protocoldevelopment/electronic_applications/ctc.htm#ctc_40.
68.Lee DW, Santomasso BD, Locke FL, et al. ASTCT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biol Blood Marrow Transplant. 2019;25(4):625-638. PubMed
69.Aaronson NK, Ahmedzai S, Bergman B, et al. The European Organization for Research and Treatment of Cancer QLQ-C30: A quality-of-life instrument for use in international clinical trials in oncology. J Natl Cancer Inst. 1993;85(5):365-376. PubMed
70.Fayers PM, Aaronson NK, Bjordal K, Groenvold M, Curran D, Bottomley A. The EORTC QLQ-C30 Scoring Manual (3rd Edition). Brussels (BE): European Organisation for Research and Treatment of Cancer; 2001: https://www.eortc.org/app/uploads/sites/2/2018/02/SCmanual.pdf. Accessed 2023 Nov 6.
71.Osborne TR, Ramsenthaler C, Siegert RJ, Edmonds PM, Schey SA, Higginson IJ. What issues matter most to people with multiple myeloma and how well are we measuring them? A systematic review of quality of life tools. Eur J Haematol. 2012;89(6):437-457. PubMed
72.Kvam AK, Wisloff F, Fayers PM. Minimal important differences and response shift in health-related quality of life: A longitudinal study in patients with multiple myeloma. Health Qual Life Outcomes. 2010;8(1):79. PubMed
73.Wisloff F, Eika S, Hippe E, et al. Measurement of health-related quality of life in multiple myeloma. Nordic Myeloma Study Group. Br J Haematol. 1996;92(3):604-613. PubMed
74.Danaher EH, Ferrans C, Verlen E, et al. Fatigue and physical activity in patients undergoing hematopoietic stem cell transplant. Oncol Nurs Forum. 2006;33(3):614-624. PubMed
75.Molassiotis A, Wilson B, Blair S, Howe T, Cavet J. Unmet supportive care needs, psychological well-being and quality of life in patients living with multiple myeloma and their partners. Psychooncology. 2011;20(1):88-97. PubMed
76.Ringdal K, Ringdal GI, Kaasa S, et al. Assessing the consistency of psychometric properties of the HRQoL scales within the EORTC QLQ-C30 across populations by means of the Mokken Scaling Model. Qual Life Res. 1999;8(1-2):25-43. PubMed
77.Kvam AK, Fayers PM, Wisloff F. Responsiveness and minimal important score differences in quality-of-life questionnaires: A comparison of the EORTC QLQ-C30 cancer-specific questionnaire to the generic utility questionnaires EQ-5D and 15D in patients with multiple myeloma. Eur J Haematol. 2011;87(4):330-337. PubMed
78.Cocks K, King MT, Velikova G, et al. Evidence-based guidelines for interpreting change scores for the European Organisation for the Research and Treatment of Cancer Quality of Life Questionnaire Core 30. Eur J Cancer. 2012;48(11):1713-1721. PubMed
79.Kvam AK, Fayers P, Wisloff F. What changes in health-related quality of life matter to multiple myeloma patients? A prospective study. Eur J Haematol. 2010;84(4):345-353. PubMed
80.EQ-5D-5L User Guide. Rotterdam (NL): EuroQol Research Foundation; 2019: https://euroqol.org/wp-content/uploads/2023/11/EQ-5D-5LUserguide-23-07.pdf. Accessed 2023 Nov 6.
81.Pickard AS, Neary MP, Cella D. Estimation of minimally important differences in EQ-5D utility and VAS scores in cancer. Health Qual Life Outcomes. 2007;5:70. PubMed
82.Gordon Lan KK, Demets DL. Discrete sequential boundaries for clinical trials [accessed by sponsor]. Biometrika. 1983;70(3):659-663.
83.Tecvayli (teclistamab) for the treatment of relapsed/refractory multiple myeloma: Clinical summary [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Tecvayli (teclistamab), 10 mg/mL or 90 mg/mL, single-dose vials containing 30 mg teclistamab in 3 mL solution or 153 mg teclistamab in 1.7 mL solution, SC injection. Toronto (ON): Janssen Inc.; 2023 Aug 31.
84.Balshem H, Helfand M, Schünemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. Journal of clinical epidemiology. 2011;64(4):401-406. PubMed
85.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. Journal of clinical epidemiology. 2020;119:126-135. PubMed
86.Hernan MA, Robins JM. Using big data to emulate a target trial when a randomized trial is not available. Am J Epidemiol. 2016;183(8):758-764. PubMed
87.Austin PC. Balance diagnostics for comparing the distribution of baseline covariates between treatment groups in propensity-score matched samples. Stat Med. 2009;28(25):3083-3107. PubMed
88.Caers J, Paiva B, Zamagni E, et al. Diagnosis, treatment, and response assessment in solitary plasmacytoma: Updated recommendations from a European Expert Panel. J Hematol Oncol. 2018;11(1):10. PubMed
Appendix 1: Detailed Outcome Data
This appendix has been copy-edited.
Figure 14: Kaplan-Meier Plot for Duration of Response per IRC Assessment, Full Analysis Set
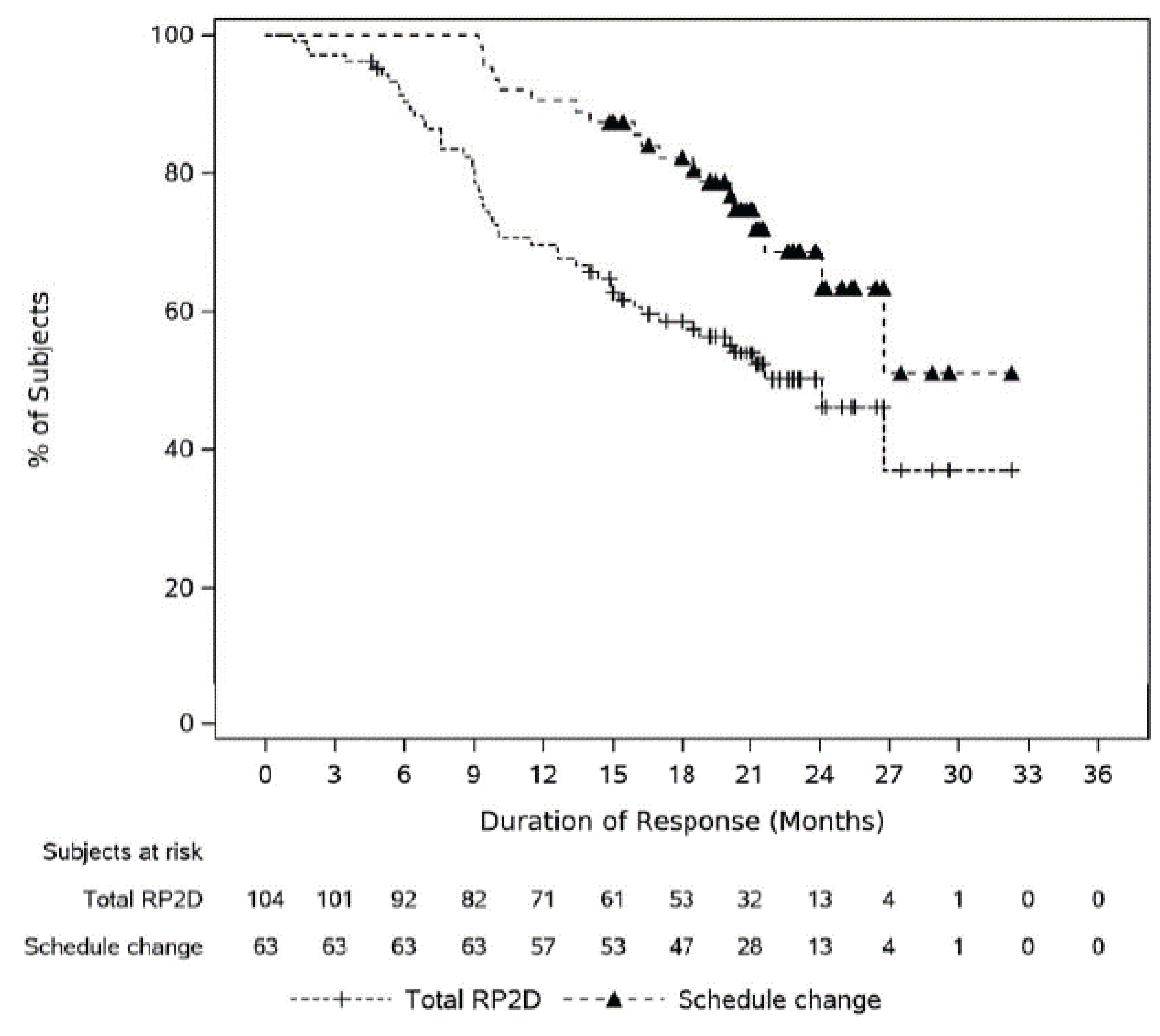
DoR = duration of response; IRC = independent review committee; RP2D = recommended phase II dose.
Source: Clinical Study Report for the MajesTEC-1 study (2021).24
Pharmacoeconomic Review
Abbreviations
AE
adverse event
BIA
budget impact analysis
CAR
chimeric antigen receptor
cilta-cel
ciltacabtagene autoleucel
CUA
cost-utility analysis
ICER
incremental cost-effectiveness ratio
IMiD
immunomodulatory drug
IPTW
inverse probability of treatment weighting
MM
multiple myeloma
NDMM
newly diagnosed multiple myeloma
OS
overall survival
PFS
progression-free survival
PI
proteasome inhibitor
PSM
partitioned survival model
QALY
quality-adjusted life-year
RRMM
relapsed or refractory multiple myeloma
TTTD
time to treatment discontinuation
WTP
willingness to pay
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Teclistamab (Tecvayli), 10 mg/mL (30 mg/3 mL) and 90 mg/mL (153 mg/1.7 mL), solution for subcutaneous injection |
Indication | Indicated for the treatment of adult patients with relapsed or refractory multiple myeloma who have received at least 3 prior lines of therapy, including a proteasome inhibitor, an immunomodulatory agent, and an anti-CD38 monoclonal antibody, and who have demonstrated disease progression on the last therapy |
Health Canada approval status | NOC/c |
Health Canada review pathway | Standard review |
NOC date | July 26, 2023 |
Reimbursement request | As per indication |
Sponsor | Janssen Inc. |
Submission history | Previously reviewed: No |
NOC/c = Notice of Compliance with Conditions.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis PSM |
Target population | Adult patients with RRMM with greater than or equal to 3 prior lines of therapy, including a PI, an IMiD, and an anti-CD38 monoclonal antibody, and who have demonstrated disease progression on the last therapy |
Treatment | Teclistamab |
Dose regimen | The recommended starting dose is 0.06 mg/kg on day 1, followed by 0.3 mg/kg on day 3 and 1.5 mg/kg on day 5; 1.5 mg/kg is then given once weekly thereafter. |
Submitted price | Teclistamab, 10 mg/mL (30 mg/3 mL) and 90 mg/mL (153 mg/1.7mL), solution for subcutaneous injection: $1,322 and $6,741 for 10 mg/mL and 90 mg/mL, respectively. |
Submitted treatment cost | The first 28-day costs of teclistamab are $29,608. Every 28-days after this the costs are $26,964. This assumes a weight of 75kg. |
Comparators |
|
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | Lifetime (30 years) |
Key data sources |
|
Submitted results |
|
Key limitations |
|
CADTH reanalysis results |
|
Cilta-cel = ciltacabtagene autoleucel; ICER = incremental cost-effectiveness ratio; IMiD = immunomodulatory drug; KCd = carfilzomib plus cyclophosphamide plus dexamethasone; Kd = carfilzomib plus dexamethasone; LY = life-year; OS = overall survival; PCd = pomalidomide plus cyclophosphamide plus dexamethasone; Pd = pomalidomide plus dexamethasone; PI = proteasome inhibitor; PSM = partitioned survival model; QALY = quality-adjusted life-year; RRMM = relapsed or refractory multiple myeloma; SVd = selinexor plus bortezomib plus dexamethasone; vs. = versus.
Conclusions
According to the clinical experts consulted by CADTH, evidence from the single-arm MajesTEC-1 trial suggests that treatment with teclistamab may be associated with clinically meaningful benefits, including improved progression-free survival (PFS) and overall survival (OS), in the Health Canada–approved indication. Based on the Grading of Recommendations Assessment, Development and Evaluation assessment of the MajesTEC-1 study, there is very low certainty associated with the potential benefit of teclistamab for PFS and OS due to a serious risk of selection bias, the interim nature of the analysis, and the noncomparative trial design. The CADTH clinical review identified limitations with the sponsor’s comparison of the MajesTEC-1 study to the LocoMMotion study, as well as the comparison of the MajesTEC-1 study to the CARTITUDE-1 study, which restricted the ability to interpret the relative treatment effects observed between teclistamab, physician’s choice (a mix of currently reimbursed combination therapies), and ciltacabtagene autoleucel (cilta-cel). This clinical uncertainty is reflected in the submitted economic analysis.
In the base case, CADTH updated the cost of pomalidomide and revised the dosing schedule of carfilzomib to be administered weekly rather than twice weekly. In the CADTH base case, teclistamab was more effective (incremental QALYs = 1.03) and associated with greater total costs (incremental costs = $522,024) than physician’s choice. This resulted in an incremental cost-effectiveness ratio (ICER) of $506,518 per QALY gained. Incremental QALYs were largely driven by the survival benefit associated with teclistamab relative to physician’s choice (incremental LYs = 1.37). The difference in cost was largely driven by higher drug costs associated with teclistamab, though an additional $32,173 is associated with adverse event (AE) management and administration costs associated with teclistamab. An 89% price reduction would be required for teclistamab to be considered cost-effective at a willingness-to-pay (WTP) threshold of $50,000 per QALY gained relative to physician’s choice. Relative to cilta-cel, teclistamab was found to be less costly and less effective (incremental costs = –$78,899; incremental QALYs = –2.46). Results from the CADTH base case closely resembled the economic analysis conducted by the sponsor, with the sole distinction being the reduced anticipated costs associated with physician's choice. This reduction is attributed to a lower list price for pomalidomide, and a decreased cost associated with carfilzomib regimens. The assumed incremental benefit remained consistent across both the CADTH and sponsor analysis.
CADTH notes that the base-case analysis assumes a large, sustained impact on OS favouring teclistamab relative to physician’s choice, assuming an additional 1.37 life-years. In the absence of robust head-to-head evidence, the extent of the survival benefit attributed to teclistamab compared to physician's choice remains uncertain. Consequently, the CADTH base case may overestimate the benefit associated with teclistamab. The estimates presented in the CADTH base case may represent the upper bound of the incremental efficacy gains that may be realized from teclistamab, and therefore higher price reductions may be required for teclistamab to be cost-effective. Finally, the sponsor did not consider selinexor as a relevant comparator in the economic analysis; as a result, the cost-effectiveness of teclistamab relative to selinexor is unknown.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process.
One patient group, Myeloma Canada, provided input through data collected from a patient (n = 32) and caregiver (n = 3) survey that was available from August 28 to September 6, 2023. The majority of respondents were living in Canada (n = 31) and 11 among them had received treatment with teclistamab. The most important outcomes for patients included delaying disease progression and achieving durable remission, with the ultimate objective of improving survival; reducing side effects from treatments; preserving independence to minimize the burden on caregivers; and maintaining quality of life. Overall, patients’ disease experience was influenced by the physical symptoms associated with multiple myeloma (MM) (e.g., fatigue, loss of sexual desire) and the psychosocial effects associated with the disease (i.e., anxiety associated with disease progression and the interruption of life goals due to absence from work and/or early retirement). Regarding prior treatment exposure, 12 respondents indicated that they had received 3 prior lines of therapy, 10 respondents indicated having received 4 prior lines of therapy, and 3 respondents indicated having received 5 or more prior lines of therapy. Additionally, the majority of patients surveyed (n = 25) had received autologous stem cell transplant for the treatment of MM, while 3 patients indicated that they were ineligible for the intervention. Patients emphasized the need for therapies to control various aspects of MM, including infections, kidney complications, decreased mobility, gastrointestinal issues, and secondary cancers. Among those who had experience with teclistamab, 10 patients indicated that they were hospitalized during the initial dosing titration period. Hospitalization time ranged between 4 nights and 4 weeks, with the most frequently reported length of stay falling between 9 and 10 nights. While on treatment with teclistamab, patients experienced a range of side effects, including respiratory infections, fungal infections, cytokine release syndrome, and immune effector cell-associated neurotoxicity syndrome. Input from patients currently receiving teclistamab was positive, with several patients indicating that teclistamab was effective in controlling the disease.
Registered clinician input was received from 2 groups: the Canadian Myeloma Research Group and the Ontario Health Hematology Cancer Drug Advisory Committee. The Canadian Myeloma Research Group stated that newly diagnosed myeloma patients in Canada are divided into 2 categories: those who are transplant eligible and those who are transplant ineligible, based on age and fitness. Transplant-eligible patients receive a bortezomib-based induction with bortezomib plus lenalidomide plus dexamethasone, or cyclophosphamide plus bortezomib plus dexamethasone, followed by high-dose melphalan plus autologous stem cell transplant and lenalidomide maintenance until disease progression. Transplant-ineligible patients would have previously received lenalidomide plus dexamethasone or bortezomib plus lenalidomide plus dexamethasone, followed by single-drug lenalidomide until disease progression. Recently, daratumumab-based combinations such as daratumumab in combination with lenalidomide plus dexamethasone, daratumumab in combination with cyclophosphamide plus bortezomib plus dexamethasone, and bortezomib in combination with melphalan plus prednisolone are preferred and include provisions for the long-term continuous administration of selected drugs. Second-line therapy depends on whether patients have progressed on lenalidomide. The inclusion of an anti-CD38 antibody (e.g., daratumumab or isatuximab) is key in second-line therapy, representing a high priority for patients. Other relevant anti-CD38 monoclonal antibody (mAb)–containing regimens have been approved by Health Canada and could be used in second-line therapy and beyond. Clinician groups also indicated that, since teclistamab is expected to be used after the failure of multiple drugs, it is not expected to impact the sequencing of drugs earlier in the disease course, or lead to a major change in treatment algorithms before patients becoming triple-class exposed or refractory. However, teclistamab is expected to shift the current treatment paradigm for patients with advanced disease given that it will provide an additional, more readily accessible T-cell redirecting therapy for patients refractory to the most commonly used drugs.
Participating drug plans were interested in understanding how teclistamab compared with currently funded options, including selinexor plus bortezomib plus dexamethasone. Drug plans questioned whether patients previously treated with B-cell maturation antigen–targeted therapy (e.g., cilta-cel) should be eligible for teclistamab, and vice versa. Drug plans were further interested in clarifying whether patients may be allowed to switch from a weekly to a biweekly (every 2 weeks) dosing schedule (as observed in the trial). Finally, drug plans indicated that jurisdictions may encounter capacity issues due to the supportive care recommended within the first 48 hours following teclistamab’s initial dose administration.
Several of these concerns were addressed in the sponsor’s model:
The impact of disease and treatment on patients’ quality of life was captured with utility values. AEs were incorporated as disutilities within the analysis.
For the 2 priming doses and the first treatment dose, hospitalization is required for at least 48 hours from the start of injection. Therefore, the model assumed a 6-day hospital stay in the first model cycle, the cost of which was reflected in the drug administration costs for teclistamab.
In addition, CADTH addressed some of the following concerns:
CADTH explored the impact of adopting a biweekly dosing schedule among patients treated with teclistamab who attain complete response for at least 6 months, as observed in the MajesTEC-1 trial. In this scenario analysis, CADTH assumed that 80% of patients receiving treatment at 11.3 months would switch to biweekly dosing.
Economic Review
The current review is for teclistamab (Tecvayli) for adult patients with relapsed or refractory multiple myeloma (RRMM) who have received at least 3 prior lines of therapy, including a proteasome inhibitor (PI), an immunomodulatory drug (IMiD), and an anti-CD38 mAb, and who have demonstrated disease progression on the last therapy.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis (CUA) of teclistamab compared with a basket of combination therapies (physician’s choice) and cilta-cel.1 Aligned with Health Canada’s indicated population, the modelled population comprised adult patients with RRMM who have received at least 3 prior lines of therapy, including a PI, an IMiD, and an anti-CD38 mAb, and who have demonstrated disease progression on the last therapy.1
Teclistamab is available as a solution for subcutaneous injection in 10 mg/mL and 90 mg/mL vials.2 The recommended starting dose is 0.06 mg/kg on day 1, followed by 0.3 mg/kg on day 3 and 1.5 mg/kg on day 5; 1.5 mg/kg is then given once weekly thereafter.2 Patients are treated with teclistamab until disease progression or unacceptable toxicity. The teclistamab treatment captured in the economic model reflected the Health Canada dosing regimen. The submitted price of teclistamab is $1,322 for a 10 mg/mL vial (30 mg/3 mL) and $6,741 for a 90 mg/mL vial (153 mg/1.7 mL),1 which at the recommended dose corresponds to a 28-day cost of $28,566 (first month on treatment) and $25,566 (month 2 onward) per patient, assuming a weight of 75 kg.
Physician’s choice encompassed a mix of therapies commonly used in Canadian clinical practice. Physician’s choice consisted of 33% carfilzomib plus dexamethasone, 7% carfilzomib plus cyclophosphamide plus dexamethasone, 28% pomalidomide plus dexamethasone, and 32% pomalidomide plus cyclophosphamide plus dexamethasone.1 The sponsor estimated the 28-day per patient drug acquisition cost of carfilzomib plus dexamethasone (cycle 1 = $12,324; cycle 2 onward = $15,290), carfilzomib plus cyclophosphamide plus dexamethasone (cycle 1 = $7,940; cycle 2 onward = $10,016), pomalidomide plus dexamethasone (cycle 1 = $8,523; cycle 2 onward = $8,310), and pomalidomide plus cyclophosphamide plus dexamethasone (cycle 1 = $8,534; cycle 2 onward = $8,320). Hence, the weighted 28-day per patient drug acquisition cost associated with physician’s choice was estimated to be $9,757 (cycle 1) and $10,736 (cycle 2 onward). Additionally, the sponsor applied a single cost at the time of cilta-cel infusion ($632,455) to the percentage of patients who ultimately received treatment in the weighted population of the CARTITUDE-1 trial (89%) to derive the 1-time per patient drug acquisition cost of $565,404.1 Vial-sharing was not incorporated by the sponsor.
The clinical outcomes modelled were OS, PFS, and time to treatment discontinuation (TTTD).1 The economic outcomes of interest were quality-adjusted life-years (QALYs) and life-years. The economic evaluation was conducted over a lifetime time horizon (30 years), from the perspective of the Canadian public health care payer. Costs and outcomes were discounted at 1.5% per annum.1
Model Structure
The sponsor submitted a partitioned survival model (PSM) to capture all costs and outcomes associated with teclistamab and comparators. The model included 3 health states: progression-free, progressed disease, and death, whereby transitions between health states occurred on a weekly cycle length (Figure 1).1 The proportion of patients in the progression-free, progressed disease, and death states was estimated over time based on the OS and PFS curves for each intervention, which were informed by the MajesTEC-1, LocoMMotion, and CARTITUDE-1 trials.1 The proportion of patients with progressed disease was estimated as the difference between the proportion of living patients (estimated from the OS curve) and the proportion of progression-free patients (estimated from the PFS curve). PFS was defined as the time from randomization to either progression or death due to any cause. Patients began in the progression-free health state, where they were assumed to initiate fourth-line treatment, and over time could progress to either the progressed disease health state or transition to the death state. Consistent with the natural history of RRMM, it was assumed that disease progression is irreversible; hence, patients in the progressed disease health state could either remain in this health state or transition to the death state (i.e., patients could not return to the progression-free health state).
Model Inputs
Baseline patient characteristics were derived from the MajesTEC-1 trial, a single-arm, phase I and phase II, open-label, dose escalation trial investigating the efficacy and safety of teclistamab among patients with RRMM who had received at least 3 prior lines of therapy, including a PI, an IMiD, and an anti-CD38 mAb, and who had demonstrated disease progression on the last therapy (n = 165).1 The average patient in the modelled cohort, which the sponsor assumed reflected the Canadian patient population, was aged 64 years, weighed 75 kg, and was more likely to be male (58%).1 These characteristics were derived from the enrolled patient population of the MajesTEC-1 trial and were used to inform the drug dosage regimens and the age- and gender-specific distribution of the general population mortality risk.
Clinical efficacy parameters used to characterize teclistamab, physician’s choice, and cilta-cel, including OS, PFS, and TTTD, were derived from various data sources. For teclistamab, inputs were based on the single-arm MajesTEC-1 trial (data cut-off = January 4, 2023; median follow-up = 22.8 months). For physician’s choice, inputs were derived from the prospective noninterventional LocoMMotion study (data cut-off = October 2022; median follow-up = 26.4 months). For cilta-cel, inputs were informed from the intention-to-treat population of the single-arm, phase Ib and phase II CARTITUDE-1 trial (data cut-off = January 2022; median follow-up = 27.7 months). The sponsor-submitted indirect treatment comparisons assessed the comparative efficacy of teclistamab relative to physician’s choice and cilta-cel in patients with RRMM who were triple-class exposed and who had demonstrated disease progression on the last therapy. The OS, PFS, and TTTD for physician’s choice were informed by the individual patient-level data from the LocoMMotion study. Patients from the LocoMMotion trial were weighted to be comparable with the MajesTEC-1 trial’s enrolled population using inverse probability of treatment weighting (IPTW). For cilta-cel, an IPTW comparison was conducted using individual patient-level data from the MajesTEC-1 trial such that they more resembled patients from the CARTITUDE-1 trial.
Parametric survival modelling was used to extrapolate OS, PFS, and TTTD beyond time points available in the studies. Distributions were selected based on clinical plausibility of long-term survival projections, the visual inspection of model fit, and the Akaike information criterion and Bayesian information criterion. The sponsor modelled the clinical efficacy parameters of the teclistamab population as the weighted sum of 2 subgroups, defined according to response status at the 8-week landmark (i.e., response-based approach). Hence, patients were stratified by their response into subgroups that were deemed to be homogenous in terms of progression or death hazard. All patients who either died or progressed at or before week 8 were categorized into the “progressed disease or death” subgroup, and the remaining patients (including those not evaluable) were placed in the “partial response or better, stable disease, or not evaluable” subgroup. Parametric distributions were fit to the OS, PFS, and TTTD outcomes of the partial response or better, stable disease, or not evaluable subgroup from the 8-week landmark onward, while distributions relevant to the progressed disease or death subgroup were fit from the start of the model. The sponsor selected distinct distributions to extrapolate OS in the partial response or better, stable disease, or not evaluable subgroup (exponential) and the progressed disease or death subgroup (log-normal). The sponsor also selected exponential distribution to extrapolate PFS and TTTD in the partial response or better, stable disease, or not evaluable patient subgroup, whereas the PFS and TTTD of patients in the progressed disease or death subgroup were modelled using the observed Kaplan-Meier data from the MajesTEC-1 study as no extrapolation was required. For the comparators, standard parametric models were independently fit to the LocoMMotion and CARTITUDE-1 trials’ individual patient-level data, which were weight-adjusted to the MajesTEC-1 study’s population. For physician’s choice, the sponsor selected the gamma distribution to model OS and the log-normal distribution to model PFS and TTTD, using data from the LocoMMotion study. For cilta-cel, the sponsor selected the exponential distribution to model OS and PFS beyond time points available in the CARTITUDE-1 trial.
Health state specific utility values were derived from an analysis of EQ-5D-5L index data collected from patients in the MajesTEC-1 trial, with UK-specific utility weights. Time-dependent utilities, which were estimated at each 28-day treatment cycle to reflect data from the MajesTEC-1 trial, were applied to all patients in the progression-free health state (irrespective of treatment) (Table 10). The time-dependent utility estimate at treatment cycle 24 (0.792) was carried forward throughout the rest of the model. A single postprogression utility value (0.670) was assigned to all patients in the progressed disease health state using data from the MajesTEC-1 trial. Although utility values were otherwise treatment-independent, the sponsor applied an off-treatment utility increment (0.060) to chimeric antigen receptor (CAR) T-cell–infused patients in the progression-free health state, which was estimated based on the CARTITUDE-1 study. The sponsor incorporated disutilities associated with AEs qualified as higher than or equal to grade 3, with a greater than or equal to 5% rate in any of the treatments considered. However, grade 1 and grade 2 cytokine release syndrome and neurotoxicity events were included. Treatment-related AE incidence for teclistamab, physician’s choice, and cilta-cel were based on data from the MajesTEC-1, LocoMMotion, and CARTITUDE-1 studies, respectively. AE-specific marginal disutilities were estimated from values reported in the literature while the duration of utility decrements was based on the MajesTEC-1 trial’s data. Disutilities were applied as a 1-time decrement in baseline utility during the first model cycle, assuming that serious AEs likely occurred at the time of treatment initiation.
Costs captured in the model included primary and subsequent treatment costs (i.e., drug acquisition and drug administration), follow-up medical costs, AE management costs, and terminal care costs. Drug acquisition costs for teclistamab were based on the sponsor’s submitted price. The dosing modelled for teclistamab is consistent with that described in the Overview section. Drug acquisition costs for physician’s choice therapies were sourced from the Ontario Drug Benefit Formulary, with dosing schedules based on the chemotherapy regimen monographs from OH-CCO. The sponsor assumed that 7% of all doses would be skipped from cycle 2 onward in alignment with the dose skipping proportion observed in the MajesTEC-1 trial. Additionally, the sponsor noted its intention to submit a biweekly dosing schedule to Health Canada and, thus, assumed that 80% to 95% of patients receiving teclistamab would switch to a biweekly dosing schedule at 11.3 months in scenario analyses. The sponsor estimated a weekly drug acquisition cost for teclistamab of $9,136 (week 1) and $6,391 (week 2+), which corresponded to a 28-day cycle cost of $28,566 (cycle 1) and $25,566 (cycle 2+). The sponsor estimated treatment cycle drug acquisition costs for each regimen encompassed within the physician’s choice therapy mix: carfilzomib plus dexamethasone (cycle 1 = $12,324; cycle 2+ = $15,290), carfilzomib plus cyclophosphamide plus dexamethasone (cycle 1 = $7,940; cycle 2+ = $10,016), pomalidomide plus dexamethasone (cycle 1 = $8,523; cycle 2+ = $8,310), and pomalidomide plus cyclophosphamide plus dexamethasone (cycle 1 = $8,534; cycle 2+ = $8,320). This resulted in weighted costs of $9,757 (cycle 1) and $10,760 (cycle 2+). For cilta-cel, the sponsor applied a 1-time per patient drug acquisition cost of $565,404, as well as costs associated with eligibility ($3,000), administration ($18,429), apheresis ($5,974), bridging therapy ($3,201), conditioning therapy ($1,993), and postinfusion monitoring ($2,514). Hence, the estimated per patient cost associated with the CAR T-cell procedure totalled $597,124. Treatment-specific AE costs were estimated based on data from the Ontario Case Costing Initiative and applied as 1-time costs in the first model cycle (teclistamab = $24,059; physician’s choice = $5,131; and cilta-cel = $42,252). Weighted 1-off subsequent therapy costs were applied when patients entered the progressed disease health state (teclistamab and cilta-cel = $128,822; physician’s choice = $125,277). All patients who transitioned to death were assumed to incur terminal care costs ($54,861) in the last cycle before death, based on de Oliveira et al. (2016).3
Summary of Sponsor’s Economic Evaluation Results
The sponsor conducted the base case via a probabilistic sensitivity analysis with 3,000 simulations.1 The deterministic and probabilistic results were similar. The probabilistic findings are presented as follows.
Base-Case Results
In the sponsor’s base case, teclistamab was associated with an incremental cost of $468,254 and an incremental QALY gain of 1.03 compared with physician’s choice, resulting in an ICER of $454,345 per QALY gained. Compared with cilta-cel, teclistamab was less costly and less effective (incremental costs = –$84,129; incremental QALYs = –2.46).
The sponsor’s analysis predicted that teclistamab was associated with a longer duration of life than physician’s choice, as well as a shorter duration of life than cilta-cel (i.e., incremental life-years of 1.37 and –3.13, respectively). Given the duration of the MajesTEC-1 trial (i.e., median follow-up = 23 months; maximum follow-up = 34 months) in contrast to the model’s time horizon (i.e., 30 years), it is important to note that the majority of the incremental QALYs realized by patients receiving teclistamab relative to physician’s choice (72%) was derived from the period beyond which there is observed trial data (i.e., extrapolated period). The key cost driver among patients receiving teclistamab was the drug acquisition cost, accounting for 80% of the total cost incurred. Likewise, the primary cost driver among patients receiving cilta-cel was the cost of drug acquisition, which accounted for 80% of the total estimated cost. The main cost drivers among patients receiving physician’s choice were drug acquisition costs (36%), followed by subsequent treatment costs (30%).
The probability that teclistamab was cost-effective at a $50,000 per QALY gained threshold was 0%. The sponsor’s submitted analysis is based on the publicly available prices for all drug treatments. Additional results from the sponsor’s submitted economic evaluation base case are presented in Appendix 3.
Table 3: Summary of the Sponsor’s Economic Evaluation Results, Pairwise
Drug | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER ($/QALY) |
|---|---|---|---|---|---|
Teclistamab vs. physician’s choice | |||||
Physician’s choice | 189,572 | Reference | 1.04 | Reference | Reference |
Teclistamab | 657,826 | 468,254 | 2.07 | 1.03 | 454,345 |
Teclistamab vs. cilta-cel | |||||
Cilta-cel | 741,955 | Reference | 4.53 | Reference | Reference |
Teclistamab | 657,826 | –84,129 | 2.07 | –2.46 | Less costly and less effective ICER for cilta-cel vs. teclistamab = $34,199a |
Cilta-cel = ciltacabtagene autoleucel; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
aThis represents the ICER if cilta-cel were funded in replacement of teclistamab. Cilta-cel costs an additional $84,129 but generates an additional 2.46 QALYs.
Source: Sponsor’s pharmacoeconomic submission.1
Sensitivity and Scenario Analysis Results
The sponsor assessed several model parameters and assumptions in probabilistic scenario analyses. These included applying different model time horizons; using alternative parametric distributions to extrapolate the OS, PFS, and TTTD of teclistamab and comparators; assessing the impact of biweekly dosing for teclistamab; using alternative utility value approaches; varying the source informing efficacy for physician’s choice; and including vial-sharing. The most influential parameters were alternative assumptions regarding biweekly dose switching among patients receiving teclistamab, as well as alternative parametric distributions to extrapolate OS for patients receiving physician’s choice. When selecting the log-normal parametric distribution to extrapolate OS for physician’s choice, the ICER associated with teclistamab increased to $601,437 per QALY gained (relative to physician’s choice). Moreover, when assuming that 95% of patients switch from a weekly to a biweekly dose at 11.3 months, the ICER decreased to $304,345 per QALY gained (relative to physician’s choice). All other scenarios resulted in ICERs ranging between $312,357 and $496,843 per QALY gained. All the scenario analyses comparing teclistamab with cilta-cel resulted in teclistamab being less costly and less effective than cilta-cel.
The sponsor conducted 2 pairwise scenario analyses from a societal perspective; these analyses included additional costs associated with losses in productivity and travel time due to treatment administration. Relative to physician’s choice, the ICER for teclistamab was $466,879 per QALY gained. When compared with cilta-cel, the scenario resulted in teclistamab being less costly and less effective than cilta-cel. Both were similar to the sponsor’s base-case analyses derived using a health care payer perspective.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations to the sponsor’s analysis that have notable implications for the economic analysis:
The impact of physician’s choice on long-term OS is uncertain. The sponsor derived clinical efficacy data for patients receiving physician’s choice from the LocoMMotion trial, a prospective noninterventional study of real-world treatments administered among patients with RRMM. Individual patient-level data from the LocoMMotion trial were weighted to be comparable with the enrolled population of the MajesTEC-1 trial across baseline covariates using IPTW. These data were then used to estimate OS for physician’s choice. However, in the absence of randomized evidence, uncertainty remains regarding the differences in OS between teclistamab and current treatment options. The sponsor extrapolated OS data for patients receiving physician’s choice from the LocoMMotion trial using a gamma distribution. Clinical experts consulted by CADTH noted that while it is reasonable to expect that fewer than 10% of patients may survive 5 years post-treatment initiation in the fourth-line setting, survival beyond the 5-year landmark remains uncertain due to the lack of robust data. Based on the selected OS curves for teclistamab and physician’s choice, the hazard ratio for OS is predicted to decrease over time. This means that the benefit of receiving teclistamab is expected to increase over time. There is insufficient evidence to note whether the hazard ratio decreases, remains the same, or increases over time, which would indicate a treatment waning effect.
CADTH explored the impact of long-term survival when using the Gompertz distribution, which generated higher survival estimates for patients treated with physician’s choice than predicted by the sponsor.
The generalizability of the modelled population to Canadian clinical practice is unclear. CADTH notes that there is uncertainty regarding the expected survival benefit in a Canadian setting following the implementation of teclistamab to a broader population that expands beyond the selective patient population recruited within the clinical trial. The MajesTEC-1 study consisted nearly exclusively of patients with an Eastern Cooperative Oncology Group Performance Status of 1 or less (99%) with a mean age of 64 years. Additionally, the majority of patients included in the trial were classified as International Staging System stage II or less (88%). A Canadian real-world population-based study of cancer registry databases estimated the mean age of patients with newly diagnosed multiple myeloma (NDMM) to be 70 years.4 As such, the MajesTEC-1 trial population may be younger than the expected patient population receiving fourth-line therapy in Canada. If teclistamab were to become available in clinical practice, where patients are likely to have more diverse clinical and demographic profiles, the magnitude of clinical benefit is uncertain.
CADTH was unable to address this limitation owing to the lack of clinical data.
The modelling approach may overestimate comparative efficacy. Results from the sponsor's model suggested that teclistamab was associated with longer survival after progression. The sponsor’s base case predicted that teclistamab extended life-years gained in a “progression-free” state (0.9 years), as well as in a “progressed disease” state (0.2 years), relative to physician’s choice, indicating that treatment with teclistamab is associated with reductions in the rate of postprogression mortality. Specifically, the sponsor’s modelling approach predicts that 17% of the incremental survival attributed to teclistamab relative to physician’s choice is due to a benefit that begins after the treatment has stopped controlling the disease (i.e., postprogression period). As the postprogression mortality transition is not modelled directly in the PSM approach, it is not possible to establish whether this effect was supported by the trial data or was an artifact of the modelling choice.
CADTH was unable to determine the extent to which the implied postprogression benefit was due to the effect of treatment versus due to structural bias within the PSM and could not address this in reanalysis.
Health state utility estimates are uncertain. The sponsor obtained health state utilities to derive patient quality of life from an analysis of EQ-5D-5L index data collected from patients in the MajesTEC-1 trial. In the submitted base case, all patients in the progression-free health state accrued time-dependent utilities, which were estimated at each 28-day treatment cycle. That is, the model assumed that treatment initiation would lead to an improvement in quality of life, and this improvement would persist as long as patients remained free from disease progression. Therefore, patients in the progression-free health state accrued utility values ranging from 0.639 (treatment initiation), to 0.757 (progression-free for 1 year) and 0.792 (progression-free for 2 years). Conversely, patients who experienced progressed disease were assigned a single postprogression utility value of 0.670. This resulted in a differential decline in quality of life experienced by patients with progressed disease, dependent on the length of time spent in the progression-free health state. For example, CADTH notes that the loss in quality of life was 4-fold higher among patients who were progression-free for 2 years compared with patients who had just started treatment. Clinical experts noted that it is uncertain whether patients with progressed disease who maintain freedom from progression for an extended period would experience a more significant decline in quality of life.
CADTH conducted a scenario analysis exploring the impact of uncertainty in the decline in quality of life experienced by patients with progressed disease by applying a single utility value in the progression-free health state.
The treatment schedule for carfilzomib plus dexamethasone is not reflective of Canadian practice. The sponsor assumed that patients receiving carfilzomib plus dexamethasone would receive the regimen on a twice weekly schedule. In consultation with clinical experts, it was noted that most centres in Canada use the once weekly schedule. This choice is attributed to greater convenience for patients and a potentially improved efficacy profile.5
CADTH updated the dosing schedule for carfilzomib and dexamethasone to align with the following once weekly dosing. Cycle 1 of the 28-day cycle: 20 mg/m2 on day 1, then 70 mg/m2 on day 8 and day 15; cycle 2 of the 28-day cycle: 70 mg/m2 on day 1, day 8, and day 15.
The cost of pomalidomide is not reflective of most jurisdictions. In the analysis, the sponsor estimated the cost of pomalidomide to be $425 per 4 mg capsule. Using the IQVIA DeltaPA database, $125 per 4 mg capsule was found to be the most frequently cited cost across Canada. The following jurisdictions use a price of $125 per 4 mg capsule of pomalidomide: New Brunswick, Nova Scotia, and Manitoba.
CADTH updated the cost of pomalidomide to $125 per 4 mg capsule.
The selection of all relevant comparators is not supported by current clinical practice. The sponsor omitted the selinexor plus bortezomib plus dexamethasone regimen from the base-case analysis. During the review process, both the participating drug plans and the clinical expert panel convened by CADTH indicated that the selinexor plus bortezomib plus dexamethasone regimen is prescribed in Canada for the treatment of adult patients with RRMM who are triple-class exposed and who have demonstrated disease progression on the last therapy. The sponsor indicated that it was infeasible to derive a reliable indirect treatment comparison between teclistamab and selinexor plus bortezomib plus dexamethasone, given that the breakdown of patient populations between the MajesTEC-1 and BOSTON trials by line of therapy is substantially different and the overlapping population by line of therapy between the trials is negligible. The cost-effectiveness of teclistamab versus selinexor plus bortezomib plus dexamethasone is, therefore, unknown.
CADTH could not address this limitation owing to the lack of comparative effectiveness data.
The proportion of patients who require subsequent therapy is uncertain. From the Majes TEC-1 trial, the sponsor estimated that of those who progress on teclistamab, 65.8% of patients go on to receive a subsequent line of therapy. Using the LocoMMotion study’s database, the sponsor estimated that of those who progress on physician’s choice, 73% of patients will go on to receive a subsequent line of therapy. This would indicate that patients who receive teclistamab and progress are less likely to receive a subsequent line of therapy. Data from Canada shows that most patients who progress will receive a subsequent treatment line.6 The main reason for attrition through treatment lines is due to death before progression, which the sponsor has already accounted for. However, this data only goes up to 3 lines of therapy. As teclistamab is a more intensive regimen than currently used options, this may influence whether a patient would receive a subsequent therapy should the treatment fail. In the absence of any robust head-to-head Canadian data, the impact that teclistamab has on subsequent therapy usage is uncertain.
If patients treated with teclistamab remain progression-free longer, relative to physician’s choice, fewer patients may require subsequent therapy as some patients are more likely to die before progressing. Therefore, there may be some cost savings associated with subsequent therapy, but the magnitude of savings is uncertain.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CADTH (refer to Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation
Sponsor’s key assumption | CADTH comment |
|---|---|
In the submitted model, the sponsor assumed that 7% of teclistamab doses would be skipped in alignment with the dose skipping proportion observed in the MajesTEC-1 trial (i.e., 461 doses skipped of a total of 6,449 patient visits). The sponsor expanded this assumption to the comparator interventions — that is, patients receiving physician’s choice and patients receiving bridging therapy before cilta-cel infusion. The dose skipping assumption was also applied to subsequent therapies across comparators. | Acceptable. CADTH notes that it is uncertain whether the trial-based proportion of doses skipped reflects real-world clinical practice as the doses received by patients may be different from the planned dosing schedule for several reasons (i.e., expected vs. observed doses). Moreover, the assumption that the dose skipping proportion observed for teclistamab could be applied to patients receiving treatments with varying administration routes is highly uncertain. CADTH notes that the majority of therapies (60%) for which a dose skipping assumption was made have an oral route of administration (e.g., dexamethasone, cyclophosphamide, pomalidomide). For oral therapies, Canadian pharmacies are likely to fill and dispense prescriptions in full. Therefore, it is unlikely that any unused tablets would result in lower prescription costs as it is improbable that they would be recuperated. CADTH notes that applying teclistamab’s dose skipping proportion across comparators resulted in conservative cost estimates for comparators. |
The sponsor modelled the clinical efficacy parameters of the teclistamab population as the weighted sum of 2 subgroups defined according to response status at the 8-week landmark (PR or better, SD, or NE vs. PD or death). | Acceptable. Patients were stratified by their response into subgroups that were deemed to be homogenous in terms of progression or death hazard. Conventional parametric distributions are inadequate for accurately representing abrupt shifts in Kaplan-Meier curves. Enhancing the fit of standard parametric distributions to observed survival data are achieved by categorizing patients into more homogeneous subgroups based on progression or death hazards. |
The utility value from treatment cycle 22 was carried over to treatment cycle 24 when deriving model cycle specific utility inputs. | Acceptable. The time-dependent preprogression utility value from treatment cycle 22 (0.792) was carried over to treatment cycle 24. The sponsor explained that this adjustment was made because the utility value in treatment cycle 24 was higher than the age- and gender-matched general Canadian population mortality, and hence did not meet face validity. Moreover, this utility value was based on low patient numbers (0.872 based on EQ-5D-5L data from 5 patients). |
In the submitted base case, teclistamab is administered on a weekly dosing schedule based on the dosage and administration considerations of the product monograph. The sponsor submitted a scenario analysis that explored the impact of adjusting teclistamab’s dosing schedule to biweekly. | Acceptable. CADTH notes that in the MajesTEC-1 trial, patients who attained a complete response or better for at least 6 months were eligible for dose switching. As a result, 63 patients switched to biweekly dosing after a median duration of 11.3 months. Since 79 patients were still on treatment, the dose switching rate at 11.3 months was 80%. Hence, exploring the impact of lower-frequency dosing in a probabilistic scenario analysis is warranted. |
The sponsor accounted for 100% drug wastage in drug costs (i.e., no vial-sharing). | Acceptable. While options to minimize wastage exist in real-world clinical settings, teclistamab is classified as a single-dose vial per label and is presented in the reference case as including wastage. |
NE = not evaluable; PD = progressed disease; PR = partial response; SD = stable disease; vs. = versus.
CADTH Reanalyses of the Economic Evaluation
Base-Case Results
CADTH identified the following limitations in the sponsor’s economic analysis: the generalizability of the modelled population to Canadian clinical practice is unclear, the modelling approach may overestimate comparative efficacy, the impact of physician’s choice on long-term OS is uncertain, health state utility estimates are uncertain, and the selection of comparators is not supported by current clinical practice. Although there is a large degree of uncertainty regarding the incremental benefit of teclistamab versus physician’s choice, due to the absence of nonrandomized evidence, the assumptions made by the sponsor fall within clinically plausible expectations based on feedback from clinical experts consulted by CADTH and the available clinical data. For the CADTH base case, the cost of pomalidomide was updated and carfilzomib was assumed to be administered weekly rather than twice weekly. Further uncertainties were explored using scenario analyses.
CADTH undertook the reanalyses outlined in Table 5 to address, where possible, the limitations within the sponsor’s submitted economic model. The CADTH base case was derived by making changes in model parameter values and assumptions, in consultation with clinical experts.
Table 5: CADTH Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
None | — | — |
Changes to derive the CADTH base case | ||
1. Cost of pomalidomide | $425 per 4 mg capsule | $125 per 4 mg capsule |
2. Dosing schedule for carfilzomib + dexamethasone | 28-day cycles:
| 28-day cycles:
|
CADTH base case | Reanalyses 1 + 2 | |
In the CADTH base case, teclistamab was associated with an ICER of $506,518 per QALY gained compared to physician’s choice (incremental costs = $522,024; incremental QALYs = 1.03) (Table 6). Compared with cilta-cel, teclistamab was less costly and less effective (incremental costs = –$78,899; incremental QALYs = –2.46). Hence, the ICER of cilta-cel vs. teclistamab was $32,068 per QALY gained. The results were primarily driven by the effect of each treatment on OS.
Table 6: Summary of the Stepped Analysis of the CADTH Reanalysis Results
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER vs. teclistimaba ($/QALY) |
|---|---|---|---|---|
Sponsor’s base case (deterministic) | Physician’s choice | 189,284 | 1.04 | 459,265 |
Teclistamab | 654,148 | 2.05 | Reference | |
Cilta-cel | 742,906 | 4.49 | Teclistamab is less costly and less effective. | |
CADTH reanalysis 1: Cost of pomalidomide | Physician’s choice | 148,151 | 1.04 | 489,828 |
Teclistamab | 643,950 | 2.05 | Reference | |
Cilta-cel | 729,228 | 4.49 | Teclistamab is less costly and less effective. | |
CADTH reanalysis 2: Dosing schedule for carfilzomib | Physician’s choice | 159,213 | 1.04 | 481,508 |
Teclistamab | 646,591 | 2.05 | Reference | |
Cilta-cel | 733,191 | 4.49 | Teclistamab is less costly and less effective. | |
CADTH base case (deterministic) | Physician’s choice | 118,079 | 1.04 | 512,071 |
Teclistamab | 636,393 | 2.05 | Reference | |
Cilta-cel | 719,513 | 4.49 | Teclistamab is less costly and less effective. | |
CADTH base case (probabilistic) | Physician’s choice | 118,216 | 1.04 | 506,518 |
Teclistamab | 640,240 | 2.07 | Reference | |
Cilta-cel | 719,140 | 4.53 | Teclistamab is less costly and less effective. The ICER for cilta-cel vs. teclistamab = $32,068.b |
Cilta-cel = ciltacabtagene autoleucel; ICER = incremental cost-effectiveness ratio; IPTW = inverse probability of treatment weighting; ITC = indirect treatment comparison; QALY = quality-adjusted life-year; vs. = versus.
aThis is not a sequential analysis given that the use of IPTW ITCs suggests that the populations enrolled in the relevant studies examining the efficacy of cilta-cel and physician’s choice are slightly different.
bThis represents the ICER if cilta-cel were funded in replacement of teclistamab. Cilta-cel costs an additional $78,899 but generates an additional 2.46 QALYs.
Scenario Analysis Results
CADTH undertook price reduction analyses based on the sponsor’s results and CADTH’s base case. The CADTH base case suggests that an 89% price reduction would be required for teclistamab to be considered cost-effective at a WTP threshold of $50,000 per QALY gained relative to physician’s choice. Price reduction analyses were also conducted for teclistamab versus cilta-cel (Table 14, Appendix 4).
Table 7: CADTH Price Reduction Analyses
Analysis | ICERs for teclistamab vs. physician’s choice ($/QALY gained) | |
|---|---|---|
Price reduction | Sponsor’s base case | CADTH’s base case |
No price reduction | 454,345 | 512,067 |
10% | 403,026 | 460,199 |
20% | 351,708 | 408,330 |
30% | 300,389 | 356,462 |
40% | 249,070 | 304,594 |
50% | 197,752 | 252,726 |
60% | 146,433 | 200,858 |
70% | 95,114 | 148,989 |
80% | 43,796 | 97,121 |
90% | Teclistamab is dominant. | 45,253 |
100% | Teclistamab is dominant. | Teclistamab is dominant. |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
CADTH conducted 3 scenario analyses to determine the impact of alternative assumptions on the cost-effectiveness of teclistamab relative to physician’s choice and cilta-cel, in line with clinical expert advice.
Selecting the Gompertz distribution to extrapolate OS for patients receiving physician’s choice.
Applying a single utility value (0.759) in the progression-free health state.
Adjusting teclistamab’s dosing schedule to biweekly at 11.3 months (dose switching rate = 80%).
When selecting the Gompertz distribution to extrapolate OS for patients receiving physician’s choice, the ICER of teclistamab relative to physician’s choice increased to $570,302 per QALY gained (incremental costs = $522,692; incremental QALYs = 0.92). When applying a single utility value in the progression-free health state, the ICER of teclistamab relative to physician’s choice increased to $519,590 per QALY gained (incremental costs = $522,024; incremental QALYs = 1.00). When assuming that 80% of patients receiving treatment at 11.3 months would switch to biweekly dosing, the ICER of teclistamab relative to physician’s choice decreased to $380,926 per QALY gained (incremental costs = $392,587; incremental QALYs = 1.03). In all scenarios, teclistamab was less costly and less effective than cilta-cel. The results of these analyses are presented in Table 13.
Issues for Consideration
CADTH notes that hospitalization is required for at least 48 hours from the start of injection for the 2 step-up doses (day 1 and day 3) and the first treatment dose (day 5) of teclistamab. To account for this, the model assumed a 6-day hospital stay in the first model cycle, the cost of which was reflected in the drug administration costs for teclistamab. Drug plans indicated that some jurisdictions may encounter capacity issues due to the supportive care recommended during teclistamab’s initial dose administration.
Although the product monograph currently describes a weekly dosing schedule for teclistamab, the sponsor stated that the adoption of biweekly dosing is planned for submission to Health Canada. In addition, drug plans were interested in clarifying whether patients may be allowed to switch from a weekly to a biweekly dosing schedule, as observed in the MajesTEC-1 trial. Clinical experts consulted by CADTH for this review noted that switching from weekly to biweekly dosing may occur primarily due to side effects, toxicity, or patient choice. Clinical experts further noted that there is insufficient evidence to conclude that transitioning to biweekly dosing would not undermine the effectiveness of teclistamab.
Overall Conclusions
According to the clinical experts consulted by CADTH, evidence from the single-arm MajesTEC-1 trial suggests that treatment with teclistamab may be associated with clinically meaningful benefits, including improved PFS and OS, in the Health Canada–approved indication. Based on the Grading of Recommendations Assessment, Development and Evaluation assessment of the MajesTEC-1 trial, there is very low certainty associated with the potential benefit of teclistamab for PFS and OS due to the serious risk of selection bias, the interim nature of the analysis, and the noncomparative trial design. The CADTH clinical review identified limitations with the sponsor’s comparison of the MajesTEC-1 study to the LocoMMotion study, as well as the comparison of the MajesTEC-1 study to the CARTITUDE-1 study, which restricted the ability to interpret the relative treatment effects observed between teclistamab, physician’s choice, and cilta-cel. This clinical uncertainty is reflected in the submitted economic analysis.
In the base case, CADTH updated the cost of pomalidomide and revised the dosing schedule of carfilzomib to be administered weekly rather than twice weekly. In the CADTH base case, teclistamab was more effective (incremental QALYs = 1.03) and associated with greater total costs (incremental costs = $522,024) than physician’s choice. This resulted in an ICER of $506,518 per QALY gained. Incremental QALYs were largely driven by the survival benefit associated with teclistamab relative to physician’s choice (incremental life-years = 1.37). The difference in cost was largely driven by higher drug costs associated with teclistamab, though an additional $32,173 is associated with AE management and administration costs associated with teclistamab. An 89% price reduction would be required for teclistamab to be considered cost-effective at a WTP threshold of $50,000 per QALY gained relative to physician’s choice. Relative to cilta-cel, teclistamab was found to be less costly and less effective (incremental costs = –$78,899; incremental QALYs = –2.46). Results from the CADTH base case closely resembled the economic analysis conducted by the sponsor, with the sole distinction being the reduced anticipated costs associated with physician's choice. This reduction is attributed to a lower list price for pomalidomide and a decreased cost associated with carfilzomib regimens. The assumed incremental benefit remained consistent across both the CADTH and sponsor analysis.
CADTH explored the impact of uncertainty associated with long-term OS and health-related quality of life. In these scenarios, the ICER of teclistamab relative to physician’s choice increased to $570,302 per QALY gained and $519,590 per QALY gained, respectively. CADTH further explored the impact of adopting a less frequent dosing schedule among patients treated with teclistamab who attain complete response for at least 6 months, as observed in the MajesTEC-1 trial. In this scenario, the ICER of teclistamab relative to physician’s choice decreased to $380,926 per QALY gained. CADTH notes that the base-case analysis assumes a large, sustained impact on OS favouring teclistamab relative to physician’s choice. In the absence of robust head-to-head evidence, the extent of the survival benefit attributed to teclistamab compared to physician's choice remains uncertain. Consequently, the CADTH base case may overestimate the benefit associated with teclistamab. The estimates presented in the CADTH base case may represent the upper bound of the incremental efficacy gains that may be realized from teclistamab, and therefore higher price reductions may be required for teclistamab to be cost-effective at the $50,000 per QALY gained WTP threshold. The sponsor did not consider selinexor as a relevant comparator in the economic analysis; therefore, the cost-effectiveness of teclistamab relative to selinexor is unknown.
References
1.Pharmacoeconomic Report Cost-effectiveness Analysis of Teclistamab in the Treatment of Patients with Relapsed or Refractory Multiple Myeloma Final. [internal sponsor's report]. Burlington (ON): Janssen Research & Development, LLC.; 2023 Aug 31.
2.Tecvayli (teclistamab): 153 mg/ 1.7 mL (90 mg/mL) and 30 mg / 3 mL (10 mg/mL) subcutaneous injection [product monograph]. Toronto (ON): Janssen, Inc; 2023 Jul 26.
3.de Oliveira C, Pataky R, Bremner KE, et al. Phase-specific and lifetime costs of cancer care in Ontario, Canada. BMC Cancer. 2016;16(1):809. PubMed
4.Tsang M, Le M, Ghazawi FM, et al. Multiple myeloma epidemiology and patient geographic distribution in Canada: A population study. Cancer. 2019;125(14):2435-2444. PubMed
5.Moreau P, Mateos MV, Berenson JR, et al. Once weekly versus twice weekly carfilzomib dosing in patients with relapsed and refractory multiple myeloma (A.R.R.O.W.): Interim analysis results of a randomised, phase 3 study. Lancet Oncol. 2018;19(7):953-964. PubMed
6.McCurdy A, Mian H, LeBlanc R, et al. Redefining attrition in multiple myeloma (MM): A Canadian Myeloma Research Group (CMRG) analysis. Blood Cancer J. 2023;13(1):111. PubMed
7.BC Cancer. BC Cancer protocol summary for therapy of multiple myeloma using carfilzomib and dexamethasone with or without cyclophosphamide. 2022; http://www.bccancer.bc.ca/chemotherapy-protocols-site/Documents/Lymphoma-Myeloma/UMYCARDEX_Protocol.pdf. Accessed 2023 Oct 10.
8.Cancer Care Ontario. Dexapoma regimen. 2020; https://www.cancercareontario.ca/en/drugformulary/regimens/monograph/46761. Accessed 2023 Oct 10.
9.Cancer Care Ontario. Cycldexapoma. 2019; https://www.cancercareontario.ca/en/drugformulary/regimens/monograph/47786. Accessed 2023 Oct 10.
10.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario drug benefit formulary/comparative drug index. 2022; https://www.formulary.health.gov.on.ca/formulary/. Accessed 2023 Oct 10.
11.DeltaPA. Ottawa (ON): IQVIA; 2022: https://www.iqvia.com/. Accessed 2023 Oct 10.
12.CADTH Reimbursement Review: Ciltacabtagene autoleucel (Carvykti) [accessed by sponsor]. Ottawa (ON): CADTH; 2023: https://www.cadth.ca/ciltacabtagene-autoleucel. Accessed 2023 Jun.
13.Abbreviated Clinical Study Report: EDMS-RIM-928241. A Phase 1/2, first-in-human, open-label, dose escalation study of teclistamab, a humanized BCMA x CD3 bispecific antibody, in subjects with relapsed or refractory multiple myeloma [internal sponsor's report]. Toronto (ON): Janssen Research & Development, LLC.; 2021 Mar 21.
14.Clinical Study Report A Phase 1b-2, Open-Label Study of JNJ-68284528, a Chimeric Antigen Receptor T cell (CAR-T) Therapy Directed Against BCMA in Subjects with Relapsed or Refractory Multiple Myeloma CARTITUDE-1. 2021. [internal sponsor's report]. Toronto (ON): Janssen Research & Development, LLC.; 2021 Mar 21.
15.Fonseca R, Usmani SZ, Mehra M, et al. Frontline treatment patterns and attrition rates by subsequent lines of therapy in patients with newly diagnosed multiple myeloma. BMC Cancer. 2020;20(1):1087. PubMed
16.Brenner DR, Poirier A, Woods RR, et al. Projected estimates of cancer in Canada in 2022. CMAJ. 2022;194(17):E601-E607. PubMed
17.Canadian Cancer Statistics Advisory Committee in collaboration with the Canadian Cancer Society, Statistics Canada, the Public Health Agency of Canada. Canadian cancer statistics 2021. Toronto (ON): Canadian Cancer Society; 2021: https://cdn.cancer.ca/-/media/files/research/cancer-statistics/2021-statistics/2021-pdf-en-final.pdf. Accessed 2023 Oct 10.
18.Canadian cancer statistics: A 2022 special report on cancer prevalence. Toronto (ON): Canadian Cancer Society; 2022: https://cdn.cancer.ca/-/media/files/research/cancer-statistics/2022-statistics/2022-special-report/2022_prevalence_report_final_en.pdf. Accessed 2022 Dec 1.
19.Bashir Q, Braunstein M, Buck T, et al. Overcoming barriers to autologous stem cell transplantation in multiple myeloma: Recommendations from a multidisciplinary roundtable discussion. Transplant Cell Ther. 2023;29(11):666-673. PubMed
Appendix 1: Cost Comparison Table
Table 8: CADTH Cost Comparison Table for Patients With Relapsed or Refractory Multiple Myeloma Who Have Received at Least 3 Prior Therapies
Treatment | Strength / concentration | Form | Price ($) | Recommended dosage | Daily cost ($) | 28-day cycle cost ($) |
|---|---|---|---|---|---|---|
Teclistamab (Tecvayli) | 30 mg 153 mg | Solution for subcutaneous injection | 1,322.0000 6,741.0000 | Step-up dosing schedule: 0.06 mg/kg on Day 1; 0.3 mg/kg on Day 3; 1.5 mg/kg on Day 5 Dosing schedule from week 2 onward: 1.5 mg/kg once weekly | Cycle 1: 973.31 Cycle 2 onward: 886.39 | Cycle 1: 29,608 Cycle 2 onward: 26,964 |
Carfilzomib + dexamethasone7 | ||||||
Carfilzomib | 10 mg 30 mg 60 mg | Powder in vial | 255.5500a 766.6590 1,533.3300 | 28-day cycles: Cycle 1: 20 mg/m2 on day 1; 70 mg/m2 on day 8 and day 15 Cycle 2+: 70 mg/m2 on day 1, day 8, and day 15 | Cycle 1: 273.81 Cycles 2+: 355.95 | Cycle 1: 7,667 Cycles 2+: 9,967 |
Dexamethasone | 0.5 mg 4 mg | Tablet | 0.1564 0.6112 | 28-day cycles: 40 mg on day 1, day 8, day 15, and day 22 | 0.87 | 24 |
Carfilzomib + dexamethasone regimen | Cycle 1: 307.64 Cycle 2+: 399.64 | Cycle 1: 7,691 Cycle 2+: 9,991 | ||||
Carfilzomib + cyclophosphamide + dexamethasone7 | ||||||
Carfilzomib | 10 mg 30 mg 60 mg | Powder in vial | 255.5500a 766.6590 1,533.3300 | 28-day cycles: Cycle 1: 20 mg/m2 on day 1; 70 mg/m2 on day 8 and day 15 Cycle 2+: 70 mg/m2 on day 1, day 8, and day 15 | Cycle 1: 273.81 Cycles 2+: 355.95 | Cycle 1: 7,667 Cycles 2+: 9,967 |
Cyclophosphamide | 25 mg 50 mg | Tablet | 0.3545 0.4773 | 28-day cycles: 300 mg/m2 on day 1, day 8, and day 15 | 0.60 | 17 |
Dexamethasone | 0.5 mg 4 mg | Tablet | 0.1564 0.6112 | 28-day cycles: 40 mg on day 1, day 8, day 15, and day 22 | 0.87 | 24 |
Carfilzomib + cyclophosphamide + dexamethasone regimen | Cycle 1: 275.24 Cycles 2+: 357.39 | Cycle 1: 7,707 Cycles 2+: 10,007 | ||||
Dexamethasone + pomalidomide8 | ||||||
Dexamethasone | 0.5 mg 4 mg | Tablet | 0.1564 0.6112 | 28-day cycles: 20 to 40 mg on day 1, day 8, day 15, and day 22 | 0.44 to 0.87 | 12 to 24 |
Pomalidomide | 1 mg 2 mg 3 mg 4 mg | Capsule | 125.0000 | 28-day cycles: 4 mg on day 1 to day 21 | 93.75 | 2,625 |
Dexamethasone + pomalidomide regimen | 94.19 to 94.62 | 2,637 to 2,649 | ||||
Cyclophosphamide + dexamethasone + pomalidomide9 | ||||||
Cyclophosphamide | 25 mg 50 mg | Tablet | 0.3545 0.4773 | 28-day cycles: 400 mg on day 1, day 8, and day 15 | 0.41 | 11 |
Dexamethasone | 0.5 mg 4 mg | Tablet | 0.1564 0.6112 | 28-day cycles: 20 to 40 mg on day 1, day 8, day 15, and day 22 | 0.44 to 0.87 | 12 to 24 |
Pomalidomide | 1 mg 2 mg 3 mg 4 mg | Capsule | 125.0000 | 28-day cycles: 4 mg on day 1 to day 21 | 93.75 | 2,625 |
Cyclophosphamide + dexamethasone + pomalidomide regimen | 94.60 to 95.03 | 2,649 to 2,661 | ||||
Bortezomib + dexamethasone + selinexor | ||||||
Bortezomib | 1 mg 2.5 mg 3.5 mg | Powder in vial | 400.6900a 1,001.7300 1,402.4200 | 35-day cycles: 1.3 mg/m2 on day 1, day 8, day 15, and day 22 | 114.48 | 3,206 |
Dexamethasone | 0.5 mg 4 mg | Tablet | 0.1564 0.6112 | 35-day cycles: 40 mg on day 1, day 8, day 15, day 22, and day 29 | 0.87 | 24 |
Selinexor | 20 mg | Tablet | 550.0000a | 35-day cycles: 100 mg on day 1, day 8, day 15, day 22, and day 29 | 392.86 | 11,000 |
Bortezomib + dexamethasone + selinexor regimen | 508.21 | 14,230 | ||||
CAR T-cell therapy | ||||||
Ciltacabtagene autoleucel | 0.5 to 1.0 × 106 CAR-positive viable T-cells per kg, with a maximum of 1 × 108 T-cells | Cell suspension in patient-specific single infusion bag | 632,455.0000b | 1-time dosec | NA | NA |
CAR = chimeric antigen receptor.
Notes The comparators presented in the above table have been deemed appropriate based on feedback from clinical expert(s) and participating drug plans. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table, and as such, the table may not represent the actual costs to public drug plans.
All prices are from the Ontario Drug Benefit Formulary (accessed October 2023),10 unless otherwise indicated, and do not include dispensing fees.
Calculations assume a patient body weight of 75 kg and a body surface area of 1.8 m2, based on the MajesTEC-1 trial.
aIQVIA DeltaPA database, accessed October 2023.11
bSponsor-submitted price reported in the CADTH pharmacoeconomic review of ciltacabtagene autoleucel.12
cCiltacabtagene autoleucel is delivered as a 1-time dose. Daily and annual costs were not calculated.
Note this appendix has not been copy-edited.
Appendix 2: Submission Quality
Note this appendix has not been copy-edited.
Description | Yes/no | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | Selinexor, which was identified as a relevant comparator by clinical experts, was not included in the sponsor’s economic analysis. |
Model has been adequately programmed and has sufficient face validity | No | The sponsor’s use of a PSM introduces structural assumptions about the relationship between PFS and OS that likely do not accurately reflect causal relationships within the disease pathway. These assumptions may produce a postprogression survival bias that favours teclistamab. Due to the assumed independence between OS and PFS end points in a PSM, extrapolations for each end point may reflect within-trial trends in the rates of relapse and death. However, as the postprogression mortality transition is not modelled directly in the PSM approach, it is not possible to establish from the model whether this effect was supported by the trial data or was generated entirely during extrapolation. |
Model structure is adequate for decision problem | Yes | No comment. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | Yes | No comment. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | Yes | No comment. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | Yes | No comment. |
OS = overall survival; PFS = progression-free survival; PSM = partitioned survival model.
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note this appendix has not been copy-edited.
Table 10: Health State Utility Values
Health state | Mean utility value |
|---|---|
Preprogression | |
Progression-free (28-day cycles) | |
0 | 0.63913 |
2 | 0.689 |
4 | 0.740 |
6 | 0.743 |
8 | 0.770 |
10 | 0.761 |
12 | 0.757 |
14 | 0.755 |
16 | 0.775 |
18 | 0.758 |
20 | 0.811 |
22 | 0.792 |
24 | 0.792 |
CAR T-cell off-treatment increment | 0.06014 |
Postprogression | |
Progressed disease | 0.67014 |
Source: Sponsor’s pharmacoeconomic submission.1
Detailed Results of the Sponsor’s Base Case
Table 11: Disaggregated Summary of the Sponsor’s Economic Evaluation Results
Parameter | Teclistamab | Physician’s choice | Cilta-cel |
|---|---|---|---|
Discounted LYs | |||
Total | 2.86 | 1.49 | 5.99 |
Preprogression | 1.75 | 0.64 | 3.31 |
Postprogression | 1.12 | 0.86 | 2.68 |
Discounted QALYs | |||
Total | 2.07 | 1.04 | 4.53 |
Preprogression | 1.34 | 0.47 | 2.76 |
Postprogression | 0.75 | 0.58 | 1.80 |
AE Disutility | –0.02 | 0.00 | –0.03 |
Discounted costs ($) | |||
Total | 657,826 | 189,572 | 741,955 |
Preprogression | |||
CAR T-cell costs | NA | NA | 597,142 |
Non–CAR T-cell therapy drug acquisition costs | 528,897 | 68,822 | NA |
Non–CAR T-cell therapy administration costs | 13,796 | 1,001 | NA |
Follow-up costs | 4,398 | 1,597 | 9,002 |
AE costs | 24,053 | 5,129 | 42,223 |
Postprogression | |||
Follow-up cost | 2,803 | 2,159 | 6,756 |
Subsequent treatment costs | 31,362 | 57,214 | 37,254 |
End of life costs | 52,518 | 53,651 | 49,578 |
AE = adverse event; CAR = chimeric antigen receptor; ICER = incremental cost-effectiveness ratio; NA = not applicable; QALY = quality-adjusted life-year.
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Note this appendix has not been copy-edited.
Table 12: Disaggregated Summary of CADTH’s Economic Evaluation Results
Parameter | Teclistamab | Physician’s choice | Cilta-cel |
|---|---|---|---|
Discounted LYs | |||
Total | 2.86 | 1.49 | 5.99 |
Preprogression | 1.75 | 0.64 | 3.31 |
Postprogression | 1.12 | 0.86 | 2.68 |
Discounted QALYs | |||
Total | 2.07 | 1.04 | 4.53 |
Preprogression | 1.34 | 0.47 | 2.76 |
Postprogression | 0.75 | 0.58 | 1.80 |
AE Disutility | –0.02 | 0.00 | –0.03 |
Discounted costs ($) | |||
Total | 640,240 | 118,216 | 719,140 |
Preprogression | |||
CAR T-cell costs | NA | NA | 595,225 |
Non–CAR T-cell therapy drug acquisition costs | 528,897 | 30,006 | NA |
Non–CAR T-cell therapy administration costs | 13,796 | 547 | NA |
Follow-up costs | 4,398 | 1,597 | 9,002 |
AE costs | 24,053 | 5,129 | 42,223 |
Postprogression | |||
Follow-up cost | 2,803 | 2,159 | 6,756 |
Subsequent treatment costs | 13,777 | 25,128 | 16,356 |
End of life costs | 52,518 | 53,651 | 49,578 |
AE = adverse event; CAR = chimeric antigen receptor; ICER = incremental cost-effectiveness ratio; NA = not applicable; QALY = quality-adjusted life-year.
Scenario Analyses
Table 13: Scenario Analyses of CADTH’s Economic Evaluation Results
Scenario analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
Sponsor's base case | Physician's choice | 118,216 | 1.04 | Reference |
Teclistamab | 640,240 | 2.07 | 506,518 | |
Scenario 1: Extrapolated OS for physician's choice (Gompertz) | Physician's choice | 117,548 | 1.15 | Reference |
Teclistamab | 640,240 | 2.07 | 570,302 | |
Scenario 2: Single health state utility in preprogression (0.759) | Physician's choice | 118,216 | 1.05 | Reference |
Teclistamab | 640,240 | 2.06 | 519,590 | |
Scenario 3: Biweekly dose switching at 11.3 months (80%) | Physician's choice | 118,216 | 1.04 | Reference |
Teclistamab | 510,803 | 2.07 | 380,926 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; OS = overall survival.
Table 14: CADTH Price Reduction Analyses
Analysis | ICERs for teclistamab vs. cilta-cel ($/QALY gained) | |
|---|---|---|
Price reduction | Sponsor’s base case | CADTH’s base case |
No price reduction | 34,199 | 33,996 |
10% | 55,508 | 55,469 |
20% | 76,956 | 76,942 |
30% | 98,404 | 98,414 |
40% | 119,853 | 119,887 |
50% | 141,301 | 141,360 |
60% | 162,749 | 162,833 |
70% | 184,197 | 184,306 |
80% | 205,645 | 205,778 |
90% | 227,093 | 227,251 |
100% | 251,106 | 248,724 |
Cilta-cel = ciltacabtagene autoleucel; ICER = incremental cost-effectiveness ratio; vs. = versus.
Note: This table presents the impact that price reductions for teclistamab may have on its ICER vs. cilta-cel. As teclistamab is less effective than cilta-cel, the ICER represents the additional cost per QALY gained associated with spending more on cilta-cel relative to teclistamab. As the cost of teclistamab decreases and the incremental costs increase, the ICER of cilta-cel relative to teclistamab increases. If the price of teclistamab was 7.5% lower, the ICER of cilta-cel relative to teclistamab would exceed $50,000 per QALY. That is, even though cilta-cel would be considered to be more effective than teclistamab, the additional costs would render cilta-cel not cost-effective at a willingness-to-pay threshold of $50,000 per QALY gained.
Appendix 5: Submitted Budget Impact Analysis and CADTH Appraisal
Note this appendix has not been copy-edited.
Table 15: Summary of Key Takeaways
Key takeaways of the BIA |
|---|
|
Summary of Sponsor’s Budget Impact Analysis
The sponsor submitted a budget impact analysis (BIA) to estimate the incremental 3-year budget impact of reimbursing teclistamab for the treatment of adult patients with RRMM who have received at least 3 prior lines of therapy, including a PI, an IMiD, and an anti-CD38 mAb, who have demonstrated disease progression on the last therapy, as per its reimbursement request. The analysis was performed from the perspective of the Canadian public drug plan formulary. The sponsor estimated the budget impact by comparing 2 scenarios: a reference scenario that estimated the total costs associated with the current standard of care (i.e., carfilzomib + dexamethasone, carfilzomib + cyclophosphamide + dexamethasone, pomalidomide + dexamethasone, pomalidomide + cyclophosphamide + dexamethasone, selinexor + bortezomib + dexamethasone, and cilta-cel) for the fourth-line treatment of adult patients with RRMM who have demonstrated disease progression on the last therapy; and a new drug scenario, where teclistamab is funded in the fourth-line setting. The sponsor estimated the eligible population using an epidemiological approach, leveraging data from multiple sources in the scientific literature and assumptions based on clinical expert input. The sponsor only considered drug acquisition costs. The dosing modelled for teclistamab reflected the product monograph. Key inputs to the BIA are documented in Table 16.
Key assumptions include:
Trial-based PFS data for teclistamab (median: 11.3 months), was used to standardize treatment duration to 1 year for all treatments. This assumes treatment duration on teclistamab is the same as all therapies for standard of care.
The sponsor applied weekly dosing for teclistamab in the base case, as per the product monograph.2 Biweekly dosing schedule was assumed in exploratory analyses where 38% of patients switched to biweekly dosing at 6 and 11.3 months.
The sponsor assumed that it requires 4 years for a patient with NDMM to receive fourth-line treatment.15
The uptake for teclistamab was assumed to be 20% in year 1, 22% in year 2, and 34% in year 3.
Table 16: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1/year 2/year 3 if appropriate) |
|---|---|
Target population | |
At-risk population | 30,788,795 |
Predicted prevalent cases of MMa | |
% of patients with NDMM reaching fourth-line | 18%15 |
Number of patients eligible for drug under review | 532 / 560 / 590 |
Market uptake (3 years) | |
Uptake (reference scenario) Kd KCd Pd PCd SVd Cilta-cel | 39% / 35% / 33% 10% / 9% / 8% 6% / 6% / 5% 32% / 29% / 27% 8% / 7% / 6% 5% / 14% / 20% |
Uptake (new drug scenario) Teclistamab Kd KCd Pd PCd SVd Cilta-cel | 20% / 22% / 34% 31% / 26% / 19% 8% / 7% / 5% 5% / 4% / 3% 26% / 22% / 16% 6% / 5% / 4% 5% / 14% / 20% |
Cost of treatment (per patient) | |
Annual cost of treatment Teclistamab Kd KCd Pd PCd SVd Cilta-cel | $353,176 $236,451 $147,891 $116,343 $116,617 $170,654 $632,455 |
KCd = carfilzomib plus cyclophosphamide plus dexamethasone; Kd = carfilzomib plus dexamethasone; MM = multiple myeloma; NDMM = newly diagnosed multiple myeloma; PCd = pomalidomide plus cyclophosphamide plus dexamethasone; Pd = pomalidomide plus dexamethasone; SVD = selinexor plus bortezomib plus dexamethasone.
aPredicted prevalence in each year based on incident (newly diagnosed) cases 4 years prior (assumes it takes 4 years for a newly diagnosed patient to receive fourth-line treatment).
Summary of the Sponsor’s BIA Results
Results of the sponsor’s base-case BIA suggest that the incremental expenditures associated with the reimbursement of teclistamab for the fourth-line treatment of adult patients with RRMM who have demonstrated disease progression on the last therapy, as per its reimbursement request, would be $19,170,558 in year 1, $22,218,458 in year 2, and $36,105,248 in year 3, for a 3-year cumulative total of $77,494,264.
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Uncertain calculation of the BIA. The sponsor uses an epidemiological approach to calculate prevalence of MM in the fourth-line setting. To do this, the sponsor examines incident cases of MM from 4 years prior and assumes 18% would receive fourth-line therapy in the current period. This assumes that individuals diagnosed before the 4-year time point will not receive a fourth-line therapy. While the sponsor acknowledges the analysis as a prevalence-based approach, technically, it would be classified as an incidence-based approach. This is because there are prevalent patients in Canada who are receiving a line 4 or later therapy and have had a MM diagnosis for more than 4 years who are excluded in this analysis. CADTH notes that there are notable cost implications associated with a prevalence-based versus an incidence-based approach. Using a prevalence-based approach, it is conventional to assign an annual cost of therapy to the size of the cohort every year. When using an incidence-based approach, the full costs of therapy incurred over the time horizon of the BIA should be applied to each incident case.
A prevalence-based approach is conventional when there is an expectation that the entry of a new drug will lead to patients switching to the new treatment from existing therapies. This means prevalent cases are relevant to the decision problem, not just new incident cases. Clinical expert feedback noted that there is no expectation that patients with RRMM would switch from current to new therapies unless they experienced unacceptable toxicity or progression at which point, they become an incident patient. Hence, only patients whose third line therapy or beyond fails (i.e., incident cases) are relevant to the decision problem. Finally, as time on therapy is different across the treatments considered in the analysis, a prevalence-based approach is also problematic as this is not reflected in the calculations. For example, CAR T-cell therapy is a 1-time cost; however, this is applied every year to the prevalent cohort in the sponsor’s model.
Given these reasons CADTH re-estimated the BIA using an incidence-based approach. CADTH extracted costs from the sponsor’s CUA model to estimate the teclistamab drug costs incurred for each incident case in the BIA. The CUA model was run with a 1, 2, and 3-year time horizon at a 0% discount rate.
When considering a 3-year time horizon, the CUA model indicates an average teclistamab drug acquisition cost of $417,063 for patients treated with teclistamab. Over the 3-year time horizon of the BIA, the average cost per patient for each incident case that receives teclistamab in year 1 is expected to be $417,063.
When considering a 2-year time horizon, the CUA model indicates an average teclistamab drug acquisition cost of $340,990. Over the time horizon of the BIA, the average cost per patient for each incident case that receives teclistamab in year 2 is expected to be $340,990.
Finally, when considering a 1-year time horizon, the CUA model indicates an average teclistamab drug acquisition cost of $218,128. Over the time horizon of the BIA, the average cost per patient for each incident case that receives teclistamab in year 3 is expected to be $218,128.
CADTH updated the costs associated with teclistamab: incident cases in year 1 are assumed to experience 3 years of costs equating to $417,063; incident cases in year 2 are assumed to experience 2 years of costs equating to $340,990; and incident cases in year 3 are assumed to experience 1 year of costs equating to $218,128.
For regimens included in the physician’s choice basket of therapies, it was assumed that all patients would receive 6 months of treatment. The disaggregated cost for each treatment for each regimen in physician’s choice was not considered in the sponsor’s CUA model. CADTH notes that since few patients remain on therapy with physician’s choice beyond 1 year, the absence of more precise cost information is unlikely to significantly impact the BIA.
Proportion of patients with NDMM receiving therapy in 4L is uncertain: As noted earlier, the sponsor excluded patients from the decision problem if they had been diagnosed more than 4 years prior. According to expert feedback elicited for this submission this assumption is inaccurate as daratumumab, for example, in the first-line setting has a median PFS of more than 5 years for transplant-ineligible patients. Second, the sponsor estimated the proportion of patients with NDMM who would become triple-refractory based on attrition rates from Fonseca et al. (2020). This publication leveraged data from US-based databases to examine treatment patterns and attrition rates by line of therapy in patients with NDMM.5 CADTH notes that Fonseca et al. likely overestimates attrition as it is challenging to capture reasons other than death for failing to receive a subsequent line of treatment. Fonseca et al. define attrition as the proportion of patients in a given line of therapy who receive only that line of therapy.15 However, it cannot be assumed that all patients who do not go on to receive subsequent therapy have either died or progressed without subsequent treatment. This is because there is a non-negligible proportion of patients treated with planned fixed-duration initial therapy who remain in remission, as well as a proportion of patients undergoing continuous first-line therapy who have maintained their response. These patients are incorrectly captured within the attrition rate estimated by Fonseca et al., thereby contributing to an overestimation of attrition. These patients may progress later, beyond the period of follow-up.
Given the aforementioned, CADTH attempted to estimate the eligible patient incident population using a different approach. CADTH examined data from the Canadian Cancer Society showing that the 25-year prevalence of MM in Canada is 1 in 2,505 (or 0.04%).18 Applying this estimate to jurisdictions included in the CADTH base case resulted in a total of 11,780 patients. This means there are 11,780 patients estimated to be alive with a MM diagnosis in 2023. To estimate how many patients will be eligible for teclistamab each year an assumption must be made as to what percentage of these patients will require an additional line of therapy after a third-line or later therapy each year. A Canadian-based study by McCurdy et al. (2023) analyzed retrospective cohort data from patients with NDMM who received at least 1 line of therapy from January 1, 2010 to December 31, 2020.6 McCurdy et al. categorized patients by transplant eligibility status and line of therapy. They further categorized patients receiving each therapy line based on whether they (1) were actively receiving treatment or were on remission off-treatment; (2) relapsed and went on to receive subsequent therapy; (3) progressed and opted to forego subsequent therapy; (4) died; or (5) were lost to follow-up.6 A summary of how these data were analyzed by CADTH is provided in Table 19. A total of 5,548 patients were identified. Of those, 1,409 patients died before reaching the fourth-line setting. Of the remaining 4,139 patients, 251 are alive and actively receiving third-line therapy, while 735 patients relapsed and went on to receive subsequent therapy in fourth-line setting and beyond. The study does not offer data regarding the distribution of patients in the fourth-line setting that are actively receiving therapy or have died. In the absence of data, CADTH assumed that 50% of patients who received therapy in fourth-line remained on active therapy. This assumption is based on the downward trend presented in the same study, which suggests that 10%, 17%, and 27% of patients treated in the first-line, second-line, and third-line settings, respectively, die before reaching the subsequent line.6 Assuming a 50% mortality risk in the fourth-line setting, the total number of patients who are alive in the cohort decreases to 3,771 (4,139 – (0.5 * 735)). Of those, 6.6% (251/3,771) are actively receiving third-line therapy, and 9.7% (0.5 * 735) / 3,771) are actively receiving treatment in the fourth-line setting and beyond. From this it is estimated that 1,885 patients (11,780 * (0.16)) are actively receiving a third-line or later therapy for MM in jurisdictions included in the CADTH base case.
The final assumption is what proportion of these patients move on to the next line of therapy each year. Of those actively receiving therapy in the third-line setting and beyond, some will die before failure or do not receive further treatment after treatment failure. In the McCurdy study, in the third-line setting, at the end of follow-up 40% of patients had gone on to receive a subsequent line and 40% did not. If we assume this same ratio applies to patients on active therapy this would mean 50% of patients who receive a third-line therapy will go on to receive a fourth-line and 50% will not. As the average time on third-line therapy and beyond is close to a year, on average it is likely that most people who progress will do so within a year. Based on this, CADTH assumed that 50% of patients actively receiving a third line or later therapy will receive a subsequent line every year. This equates to 966 incident patients each year requiring a fourth-line or later therapy (50% of 1,885 is 966). It was assumed this number would be stable over the 3-year time horizon (so every year we would expect an additional 966 patients to require a fourth-line or later therapy).
This approach is uncertain for several reasons. First, the data in McCurdy et al. is not broken down in the 4L or later setting so assumptions had to be made regarding the size of the surviving cohort. Second, these data are based on a time period (2010 to 2020) whereby some patients likely did not receive daratumumab. Daratumumab in the first-line setting in Canada has become more prominent in recent years and survival time on this regimen is much higher than alternative regimens. CADTH would therefore expect to encounter more patients diagnosed in recent years remain on active therapy in the first-line setting longer and therefore die on first-line treatment, meaning less patients in the fourth-line and latter settings. Finally, the prevalence estimate looks at MM cases over the past 25 years whereas the data from McCurdy looks at 10 years’ worth of diagnoses. Although the expected surviving cohort from 10 to 25 years plus is likely to be small in a MM population these patients may be more likely to be on later lines of therapy.
CADTH conducted a reanalysis that revised the eligible patient population assuming 966 patients will require a fourth-line or later therapy each year.
Noting considerable uncertainty owing to the lack of data informing relevant parameters in the fourth-line setting, it is plausible that the CADTH estimate may represent the upper bound of the eligible population given the changing treatment landscape with daratumumab becoming more prevalent in the first-line setting. CADTH conducted a scenario analysis that assumed a 50% reduction in the eligible patient population to offer a potential estimate of the lower bound.
The treatment schedule for carfilzomib plus dexamethasone is not reflective of Canadian practice. The sponsor assumed that patients receiving carfilzomib plus dexamethasone would receive the regimen on a twice weekly schedule. In consultation with clinical experts, it was noted that most centres in Canada use the once weekly schedule. This choice is attributed to the greater convenience for patients and a potentially improved efficacy profile.19
CADTH updated the dosing schedule for carfilzomib and dexamethasone to align with the following once weekly dosing: (28-day cycle) Cycle 1: 20 mg/m2 on day 1 then 70 mg/m2 on day 8 and day 15; Cycle 2: 70 mg/m2 on day 1, day 8, and day 15.
The cost of pomalidomide is not reflective of most jurisdictions. In the analysis, the sponsor estimates the cost of pomalidomide to be $425 per 4 mg capsule. Using the IQVIA DeltaPA database, $125 per 4 mg capsule was found to be the most cited cost across Canada.
CADTH updated the cost of pomalidomide to $125 per capsule.
Dosing schedule for teclistamab is uncertain. Although the product monograph currently describes a weekly dosing schedule for teclistamab, the sponsor stated that the adoption of biweekly dosing is planned for submission to Health Canada. In addition, drug plans were interested in clarifying whether patients may be allowed to switch from a weekly to a biweekly dosing schedule, as observed in the MajesTEC-1 trial. Clinical experts consulted by CADTH for this review noted that switching from weekly to biweekly dosing may occur primarily due to side effects, toxicity, or patient choice. Clinical experts further noted that there is insufficient evidence to conclude that transitioning to biweekly dosing would not undermine the effectiveness of teclistamab. CADTH notes that in the MajesTEC-1 trial, patients who attained a complete response or better for at least 6 months were eligible for dose switching. As a result, 63 of 104 responders (38%) switched to biweekly dosing after 23 months of follow-up.
CADTH explored a scenario analysis that assumed 38% of patients would switch to biweekly dosing at 11.3 months based on the median time to dose switching from the MajesTEC-1 trial.
Market share of teclistamab is likely underestimated. The sponsor assumed that teclistamab would have a market share of 20%, 22% and 34% in year 1, year 2, and year 3, respectively. Clinical expert feedback noted that the sponsor’s market share projections were potentially lower than they would anticipate in practice if a therapy like teclistamab were to be funded in fourth-line. This aligned with the feedback received from registered clinician groups in Canada who noted that teclistamab was expected to shift the current treatment paradigm given that it will provide a more readily accessible T-cell redirecting therapy for patients with advanced diseases who are refractory to the most used drugs. Clinical experts indicated that, if teclistamab were reimbursed, it would be reasonable to expect that two-thirds of patients would likely be treated with either teclistamab or cilta-cel in the fourth-line setting.
CADTH conducted a scenario reanalysis by adjusting the projected market share of teclistamab to 30%, 40%, and 47% in year 1, year 2, and year 3, respectively, based on feedback sought from clinical experts. In line with their input, teclistamab’s market uptake was assumed to increase gradually until the combined market share of teclistamab and cilta-cel constitutes 67% in year 3.
CADTH Reanalyses of the BIA
CADTH revised the sponsor’s submitted analysis by revising the calculation of the costs associated with teclistamab and physician’s choice therapies; revising the eligible patient population; adopting a once weekly dosing schedule for carfilzomib plus dexamethasone to once weekly; and adjusting the cost of pomalidomide. The changes applied to derive the CADTH base case are described in Table 17.
Table 17: CADTH Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
None | — | — |
Changes to derive the CADTH base case | ||
1. Calculation of the budget impact |
|
|
2. Eligible patient population | Year 1: 532 Year 2: 560 Year 3: 590 | Year 1: 966 Year 2: 966 Year 3: 966 |
3. Dosing schedule for carfilzomib + dexamethasone | 28-day cycles:
| 28-day cycles:
|
4. Cost of pomalidomide per 4 mg capsule | $425 | $125 |
CADTH base case | Reanalysis 1 + 2 + 3 + 4 | |
4L = fourth-line; NDMM = newly diagnosed multiple myeloma.
The results of the CADTH step-wise reanalysis are presented in summary format in Table 18 and a more detailed breakdown is presented in Table 20. The CADTH reanalysis of the BIA suggests that the incremental expenditures associated with the reimbursement of teclistamab for the fourth-line treatment of adult patients with RRMM who have demonstrated disease progression on the last therapy, as per its reimbursement request, would be $30,276,140 in year 1, $57,027,919 in year 2, and $92,228,347 in year 3, for a 3-year cumulative total of $179,532,406.
Table 18: Summary of the CADTH Reanalyses of the BIA
Stepped analysis | 3-year total ($) |
|---|---|
Submitted base case | 77,494,264 |
CADTH reanalysis 1: Calculation of the budget impact | 92,920,873 |
CADTH reanalysis 2: Revised eligible population | 132,250,764 |
CADTH reanalysis 3: Dosing schedule for carfilzomib + dexamethasone | 84,626,083 |
CADTH reanalysis 4: Cost of pomalidomide | 91,884,094 |
CADTH base case (1 + 2 + 3 + 4) | 179,532,406 |
BIA = budget impact analysis.
Table 19: Summary of Data Used From McCurdy et al.
Line of therapy | Transplant eligibility status | Total number of patientsa | Actively receiving treatment in the given lineb | Died on that line of therapyb |
|---|---|---|---|---|
1 | Transplant eligible | 3,111 | 1,151 (37%) | 156 (5%) |
Transplant ineligible | 2,437 | 463 (19%) | 414 (17%) | |
2 | Transplant eligible | 1,455 | 437 (30%) | 146 (10%) |
Transplant ineligible | 1,332 | 320 (24%) | 306 (23%) | |
3 | Transplant eligible | 787 | 126 (16%) | 157 (20%) |
Transplant ineligible | 657 | 125 (19%) | 230 (35%) | |
4 | Transplant eligible | 472 | NR | NR |
Transplant ineligible | 263 | NR | NR |
NR = not reported.
aValues in this column have been taken directly from McCurdy et al. (2023).
bValues in these columns have been calculated by applying the percentage from McCurdy et al. (2023) to the total number of patients for each line.
Source: McCurdy et al.6
CADTH conducted additional scenario analyses to address remaining uncertainty, using the CADTH base case. Results are provided in Table 20.
Assuming a 50% reduction in the eligible patient population to offer a probable estimate of the lower bound.
Assuming 38% of patients would switch to biweekly dosing at 11.3 months based on data from the MajesTEC-1 trial. This reduces 1, 2 and 3 year teclistamab costs to $213,318, $288,152, $333,891, respectively.
Assuming the projected market share of teclistamab is 30%, 40% and 47% in year 1, year 2, and year 3, respectively.
Table 20: Detailed Breakdown of the CADTH Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | 3-year total ($) |
|---|---|---|---|---|---|---|
Submitted base case | Reference | 82,425,894 | 104,321,635 | 132,940,874 | 156,356,734 | 393,619,243 |
New drug | 82,425,894 | 123,492,193 | 155,159,332 | 192,461,981 | 471,113,507 | |
Budget impact | 0 | 19,170,558 | 22,218,458 | 36,105,248 | 77,494,264 | |
CADTH base case | Reference | 59,426,870 | 86,998,590 | 136,608,194 | 169,731,005 | 393,337,789 |
New drug | 59,426,870 | 117,274,730 | 193,636,113 | 261,959,352 | 572,870,195 | |
Budget impact | 0 | 30,276,140 | 57,027,919 | 92,228,347 | 179,532,406 | |
CADTH scenario analysis 1: 50% reduction in the eligible patient population | Reference | 29,713,734 | 43,530,911 | 68,303,528 | 84,866,371 | 196,700,809 |
New drug | 29,713,734 | 58,661,261 | 96,824,689 | 130,994,983 | 286,480,934 | |
Budget impact | 0 | 15,130,350 | 28,521,162 | 46,128,612 | 89,780,124 | |
CADTH scenario analysis 2: Biweekly dose switching at 11.3 months | Reference | 59,426,870 | 86,998,590 | 136,608,194 | 169,731,005 | 393,337,789 |
New drug | 59,426,870 | 116,345,442 | 183,334,990 | 244,313,268 | 543,993,700 | |
Budget impact | 0 | 29,346,851 | 46,726,797 | 74,582,263 | 150,655,911 | |
CADTH scenario analysis 3: Increased market share for teclistamab | Reference | 59,426,870 | 86,998,590 | 136,608,194 | 169,731,005 | 393,337,789 |
New drug | 59,426,870 | 132,694,197 | 233,282,599 | 310,755,133 | 676,731,928 | |
Budget impact | 0 | 45,695,607 | 96,674,405 | 141,024,128 | 283,394,139 |
BIA = budget impact analysis; NDMM = newly diagnosed multiple myeloma.
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.