CADTH Reimbursement Review
Nivolumab and Relatlimab (Opdualag)
Sponsor: Bristol Myers Squibb Canada
Therapeutic area: Unresectable or metastatic melanoma
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Stakeholder Input
Clinical Review
Abbreviations
AE
adverse event
AJCC
American Joint Committee on Cancer
BICR
blinded independent central review
BMS
Bristol Myers Squibb
BOR
best overall response
CI
confidence interval
CNS
central nervous system
CR
complete response
CrI
credible interval
CTLA-4
cytotoxic T-lymphocyte–associated protein 4
DAC
drug advisory committee
DBL
database lock
DIC
deviance information criterion
DoR
duration of response
ECOG PS
Eastern Cooperative Oncology Group Performance Status
EWB
emotional well-being
EQ VAS
EQ visual analogue score
FACT-G
Functional Assessment of Cancer Therapy–General
FACT-M
Functional Assessment of Cancer Therapy–Melanoma
FDC
fixed-dose combination
FWB
functional well-being
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HR
hazard ratio
HRQoL
health-related quality of life
IA
interim analysis
ICI
immune checkpoint inhibitor
IgG4
immunoglobin G4
IMAE
immune-mediated adverse event
IO
immuno-oncology
IRT
Interactive response technology
ITC
indirect treatment comparison
ITC1
indirect treatment comparison 1
ITC2
indirect treatment comparison 2
ITT
intention-to-treat
LAG-3
lymphocyte activation gene 3
LDH
lactate dehydrogenase
MEK
mitogen-activated protein kinase enzyme
MID
minimally important difference
MS
melanoma scale
NMA
network meta-analysis
OESI
other event of special interest
OH-CCO
Ontario Health (Cancer Care Ontario)
OR
odds ratio
ORR
overall response rate
OS
overall survival
PD-1
programmed cell death protein 1
PD-L1
programmed cell death ligand 1
PFS
progression-free survival
PR
partial response
PWB
physical well-being
RCT
randomized controlled trial
RECIST 1.1
Response Evaluation Criteria in Solid Tumors Version 1.1
SAE
serious adverse event
SLR
systematic literature review
SWB
social well-being
SYSF
Save Your Skin Foundation
TT
BRAF-targeted therapy
TTR
time to response
VAS
visual analogue scale
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Nivolumab-relatlimab (Opdualag): nivolumab 12 mg/mL and relatlimab 4 mg/mL in a single-dose vial (FDC), administered as an IV infusion over 30 minutes The recommended dosage in adult patients is 480 mg nivolumab and 160 mg relatlimab, every 4 weeks The recommended dosage for pediatric patients who are aged at least 12 years and weigh at least 40 kg is the same as for adults A recommended dosage has not been established for pediatric patients who are aged 12 years or older and weigh less than 40 kg Nivolumab-relatlimab is supplied as a concentrate for solution for infusion: 240 mg of nivolumab per 20 mL (12 mg/mL) and 80 mg of relatlimab per 20 mL (4 mg/mL) in a single-dose vial (FDC) |
Sponsor | Bristol Myers Squibb Canada |
Indication | For the treatment of adult and pediatric patients aged 12 years or older with unresectable or metastatic melanoma who have not received prior systemic therapy for unresectable or metastatic melanoma |
Reimbursement request | For the treatment of adult and pediatric patients aged 12 years or older with unresectable or metastatic melanoma who have not received prior systemic therapy for unresectable or metastatic melanoma |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | September 13, 2023 |
FDC = fixed-dose combination; NOC = Notice of Compliance.
Introduction
Melanoma is a neoplasm originating from melanocytes or the pigment-producing cells of the skin. The clinical symptoms of advanced melanoma include swollen lymph nodes, a hard lump on the skin, unexplained pain, feeling very tired or unwell, and unexplained weight loss.1 The mean age at diagnosis of advanced metastatic melanoma is approximately 59 years in Canada.2,3 The diagnosis of melanoma is based on skin examination, physical examination, skin and/or lymph node biopsy, and diagnostic imaging (i.e., CT).4-7 According to the Canadian Cancer Society, 10.4% of all new melanomas are stage III at diagnosis and 3.9% are stage IV (i.e., metastatic disease). Poor prognostic factors include an Eastern Cooperative Oncology Group Performance Status (ECOG PS) of 2 or higher, elevated lactate dehydrogenase (LDH), nodal involvement and metastases, increased tumour thickness, ulceration, and mitoses of 1/mm2 or greater in thin T1 melanomas.3,5 Approximately 70% of metastatic melanomas have mutually exclusive mutations in the BRAF oncogene, NRAS homologue oncogene, and c-KIT, and GNAQ or GNA11 genes, which activate the mitogen-activated protein kinase pathway, leading to promotion of cell proliferation, prevention of apoptosis, and angiogenesis.8 About 38% to 51% of patients with stage III or IV melanoma had a mutation in the BRAF gene.9,10 An Australian study of patients with advanced melanoma reported a similar rate, with 48% of tumours testing positive for a V600 BRAF mutation.11 In Canada, melanoma accounted for 3.8% of new cancer cases and 1.5% of cancer deaths in 2021.12 An estimated 9,000 people in Canada were diagnosed with melanoma in 2022,13 with an age-standardized incidence rate of 23.5 per 100,000 in 2018 (excluding Quebec) based on data from Statistics Canada.14 The incidence is slightly higher in men than in women (25.9 versus 21.2 per 100,000, respectively).2,14 An estimated 1,200 persons died from melanoma in 2022 in Canada, with an age-standardized mortality rate of 2.7 per 100,000.13 In Canada, stage IV distant metastatic disease is associated with a 5-year survival rate of 18%.4 However, consistent with the observed decline in mortality rates, melanoma survival rates have improved in recent years with the introduction of novel immunotherapies and BRAF-targeted therapies (TT) based on inhibitors of the BRAF gene and mitogen-activated protein kinase enzyme (MEK) protein.
Important treatment goals of systemic therapy in metastatic advanced melanoma include prolonging survival, generating durable responses, providing symptom relief, minimizing treatment toxicities, and maintaining quality of life.15 According to the clinical experts consulted by CADTH for this review, immunotherapy is the first line of choice for melanoma regardless BRAF status.16,17 The immune checkpoint inhibitor (ICI) immunotherapies routinely used for the first-line treatment of metastatic melanoma in Canada include ipilimumab-nivolumab combination therapy,18-20 nivolumab (anti–programmed cell death protein 1 [PD-1]) monotherapy,19,21 ipilimumab (anti–cytotoxic T-lymphocyte–associated protein 4 [CTLA-4]) monotherapy,20,22 and pembrolizumab (anti–PD-1) monotherapy.23,24 However, the use of ipilimumab has been increasing.25 According to the clinical experts consulted for this review, ipilimumab-nivolumab is the first line of choice among the ICIs. After the first line, the treatment decisions are largely determined by BRAF mutation status.17 Patients with no BRAF mutation are treated with immunotherapies and patients with BRAF mutations are eligible for treatment with a TT.17 Among the TT regimens that have been approved by Health Canada and recommended for reimbursement by the CADTH pan-Canadian Oncology Drug Review Expert Review Committee are encorafenib (a BRAF inhibitor) combined with binimetinib (a mitogen-activated protein kinase enzyme [MEK] inhibitor),26-28 vemurafenib (a BRAF inhibitor) combined with cobimetinib (a MEK inhibitor),29-31 and dabrafenib (a BRAF inhibitor) combined with trametinib (a MEK inhibitor).32-34 The clinical experts CADTH consulted for this review indicated that TT use as a monotherapy is negligible and not reflective of clinical practice in Canada. It was reported that less than 5% of patients with advanced melanoma rarely receive a TT as a single drug.35 According to the clinical experts CADTH consulted for this review, there is an unmet medical need for an additional novel ICI combination therapy that can be used to treat metastatic melanoma regardless of BRAF mutation status. The novel therapy should offer increased efficacy relative to anti–PD-1 monotherapy and should have a favourable safety profile that does not result in the additive toxicities seen with conventional dual immunotherapy involving an ICI combined with a drug with a different mechanism of action.
Nivolumab is a humanized immunoglobin G4 (IgG4) monoclonal antibody ICI that binds to the PD-1 receptor and blocks its interaction with programmed cell death ligand 1 (PD-L1) and programmed cell death ligand 2, triggering PD-1 pathway–mediated inhibition of the immune response, including the antitumour immune response.36,37 Relatlimab is a novel, first-in-class ICI that targets the lymphocyte activation gene 3 (LAG-3) receptor.38,39 Relatlimab is a humanized IgG4 monoclonal antibody that binds to the LAG-3 receptor and prevents LAG-3–mediated inhibition of the immune response by blocking its interaction with ligands, ultimately leading to an antitumour response.37,40 LAG-3 and PD-1 are distinct ICI pathways, often co-expressed on tumour-infiltrating lymphocytes.40 They act synergistically on effector T-cells, leading to the development of T-cell exhaustion and impaired cytotoxic function. The recommended dosages of nivolumab-relatlimab for adult patients are 480 mg of nivolumab and 160 mg of relatlimab, every 4 weeks.
The recommended dosage of nivolumab-relatlimab for pediatric patients who are at least 12 years old and weigh at least 40 kg is the same as for adults. A recommended dosage has not been established for pediatric patients who are 12 years or older and weigh less than 40 kg. Nivolumab-relatlimab is supplied as fixed-dose combination (FDC) concentrate for solution for infusion: 240 mg of nivolumab per 20 mL (12 mg/mL) and 80 mg of relatlimab per 20 mL (4 mg/mL) in a single-dose vial.40
The Health Canada–approved indication of interest for this review is nivolumab-relatlimab FDC for the treatment of adult and pediatric patients aged 12 years or older with unresectable or metastatic melanoma who have not received prior systemic therapy for unresectable or metastatic melanoma.40 The CADTH reimbursement request aligns with this Health Canada indication. The nivolumab-relatlimab FDC was reviewed by Health Canada through the Standard Review Pathway. It has not been reviewed previously by CADTH.
The objective of this clinical review is to review the beneficial and harmful effects of nivolumab-relatlimab FDC for the treatment of adult and pediatric patients aged 12 years or older with unresectable or metastatic melanoma who have not received prior systemic therapy for unresectable or metastatic melanoma.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient and clinician groups who responded to CADTH’s call for input and from 2 clinical experts consulted by CADTH for the purpose of this review.
Patient Input
CADTH received patient-group submissions from Melanoma Canada and the Save Your Skin Foundation (SYSF). Data were gathered by Melanoma Canada via an online survey. A total of 119 individual patient responses combined with 84 caregiver responses were received. Among the patient respondents, 35 indicated they had no caregiver. Of the patient respondents, 81 were female and 38 were male. Regarding tumour staging, 26 patients had been diagnosed with stage 0 melanoma, 17 had stage I, 10 had stage II, 18 had stage III, 29 had stage IV, and 19 did not know their stage. Two patients in this survey were treated with nivolumab-relatlimab FDC.
Information was obtained by the SYSF through online surveys, virtual patient-roundtables and one-on-one conversations, which included 60 melanoma patients, of whom 12 had experience with the drug under review (nivolumab-relatlimab), that took place over the past 6 months. There were 37 females and 23 males aged between 18 and 89 years. A total of 18 (out of 60) respondents were from outside of Canada (US, Australia, and France).
Most patients reported that pain, scarring, lymphedema, fatigue, anxiety, fear, and depression are common impacts of the disease itself that affect the quality of life for patients and their families. Caregivers reported that the greatest impact of dealing with the diagnosis is the mental stress, followed by the negative financial impact on the family due to the loss of income from a working partner, and the additional responsibilities associated with taking care of the home, family, and loved one. Some of the respondents described the impact of melanoma using the terms scared, disbelief, unsettled, anxious, teary, disrupted life, and totally life changing.
In terms of current therapy options, based on input from 119 of the respondents who took part in Melanoma Canada’s survey, 55% had been treated with some form of drug therapy. Nine patients had been treated with multiple therapies. A total of 92% of the patients treated with available drug therapies indicated that they felt the side effects were worth tolerating for the anticipated results. Moreover, about 20% of patients experienced issues accessing treatment. The SYSF survey also found the same issues, as patients in remote areas of Canada have problems getting to treatment sites, paying for travel costs, taking time off from work, and dealing with the added concern of being treated far from home and their support system, all of which puts extra stress on patients, caregivers, and their families. Access to the drug under review is limited to Ontario and Quebec, and a number of patients in Canada could not obtain the drug under review, although it might have been their only option.
Both patient groups identified a vast opportunity for improvement if a wider variety of more-effective treatment options with minimal side effects and longer responses were made available.
Two respondents from Melanoma Canada’s survey and 12 respondents from the SYSF’s survey indicated that they had experience with the drug under review, the primary method of access to which was a clinical trial. Twelve respondents stated that the benefits outweighed the burdens of side effects, which were mainly rash and fatigue and were somewhat manageable.
Melanoma Canada reported that there is an ongoing need for superior options, and options when 1 therapy does not work or stops working. Melanoma Canada also noted that melanoma is difficult to treat once it has spread. Effective treatments, biomarkers, and earlier-stage treatments are needed to prevent some of the quality-of-life impacts from surgery, loss of income, duration of illness, and the impact on the mental health of both the patient and caregiver. According to Melanoma Canada, the drug under review is an improvement in treatment options for a cancer that continues to be on the rise and is complex to treat.
Clinician Input
The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of adult and pediatric patients (aged 12 years and older and weighing at least 40 kg) with unresectable or metastatic melanoma.
The clinical experts indicated that the goal is to increase overall rates, slow down progression, lessen symptoms, improve quality and quantity of life, and minimize toxicity, particularly long-term significant toxicities. The clinical experts indicated that formulations are needed to improve convenience. Currently the standard of care for metastatic melanoma in Ontario is ipilimumab-nivolumab if the patient is able to tolerate the potential toxicities of the drugs. Failure to respond to ipilimumab-nivolumab may prompt switching to a BRAF inhibitor plus a MEK inhibitor in patient who were BRAF positive. Pembrolizumab or nivolumab may be attempted as monotherapy if a patient experiences too many adverse events (AEs) due to ipilimumab-nivolumab combination treatment. The clinical experts indicated that immunotherapy is not 100% effective. The response rates of combination ipilimumab-nivolumab is approximately 56%. Patients may initially respond and eventually progress. After progression, new treatments are needed. Sometimes treatments are effective but accompanied by AEs that are not tolerable; treatment therefore has to be aborted despite efficacy, creating a need for less-toxic treatments that are more tolerable and less dangerous. This is an unmet need. According to the clinical experts, no beneficial second-line therapy superior to ipilimumab-nivolumab is currently available. New therapy is needed to increase response rates and reduce AEs. According to the clinical experts, the current standard practice is to discuss dual-drug versus single-drug immunotherapy if there are no contraindications. Factors that would be considered when determining the most suitable treatment include patient goals, age, comorbidities, bulk of disease, sites of disease, and pace of disease. Patients who choose a dual-drug therapy may de-escalate to a single drug to manage toxicity. If the patient progresses on dual therapy and has a BRAF mutation, a BRAF or MEK inhibitor would be offered. According to the clinical expert, dual immunotherapy has been recognized as a potentially curative regimen. In addition, many trials are based on fixed dosing and limited vial sizes. The clinical experts noted that many provinces reimburse these therapies based on weight, and that clinics are challenged to cohort patients to minimize drug wastage.
One clinical expert indicated that, given its equivalency to ipilimumab-nivolumab and fewer toxicities, the new treatment under review (nivolumab-relatlimab FDC) could be a first-line treatment for patients. The clinical experts emphasized that fewer AEs may mean improved patient compliance and superior outcomes overall, and that less toxicity may mean fewer hospital admissions, which is better for patients but also more cost-effective and would offset the extra cost of the drug. Nivolumab-relatlimab FDC could also be an alternative to ipilimumab-nivolumab, which is the current first-line treatment in Ontario. The other clinical expert indicated that, if this regimen is approved, then the options (ipilimumab-nivolumab versus nivolumab-relatlimab FDC), the outcomes of overall survival (OS), progression-free survival (PFS), and toxicities would be discussed with patients. The clinical expert also noted that nivolumab-relatlimab FDC is directly compared to nivolumab monotherapy in the RELATIVITY-047 trial; nivolumab-relatlimab FDC is the first-class drug; nivolumab-relatlimab FDC may be used as first or second line of ICIs; nivolumab-relatlimab FDC would not be reserved for those patients who are intolerant, but rather those who would benefit from an effective regimen with less toxicity; nivolumab-relatlimab FDC is expected to cause a shift in treatment paradigms; those candidates for single-drug immunotherapy would be offered nivolumab-relatlimab FDC; and those candidates considered for ipilimumab-nivolumab may be offered or choose nivolumab-relatlimab FDC. The clinical expert stated that this nivolumab-relatlimab FDC regimen may replace ipilimumab-nivolumab for less-robust patients.
The clinical experts indicated that all metastatic patients can be offered this treatment as it was beneficial regardless of BRAF status, PD-L1 and LAG-3 percentage, or stage. It is similar to other immunotherapy combinations and could be offered to all patients. The experts also stated that it will be important to follow OS data as they mature, to determine the efficacy in brain metastases, and whether the combination decreases or delays the occurrence of brain metastases.
The clinical experts indicated that an assessment of improvements in patient symptoms and the modified immunotherapy Response Evaluation Criteria in Solid Tumors Version 1.1 (RECIST 1.1) are needed as pseudoprogression can occur at the beginning of treatment. Usually, it can take up to 2 or 3 months to evaluate the true response. Initially, responses are assessed at 3-month intervals. As patients respond, the response assessment can be tailored and increased to every 6 months. Improved survival is the goal. The clinical experts noted that clinical outcomes assessments align with the clinical trial outcomes; physicians and patients review toxicities, symptom control, and objective evidence of disease response in an ongoing manner during active treatment.
Regarding discontinuation, the clinical experts indicated the nivolumab-relatlimab FDC should be discontinued when disease progression on imaging is obvious with no improvement in symptoms. According to the clinical experts, when harmful grade 3 or 4 AEs occur, patient should at the very least pause treatment of the disease in favour of treating the AE, then determine if disease treatment can be restarted at a lower dose.
The clinical experts noted that, ideally, treatments for metastatic melanoma should be provided by specialist oncologists and pharmacists in a Canadian cancer centre or, if at a community centre, treatment should be supervised or somehow connected to a cancer centre and experts who can be consulted for advice. According to the clinical experts, centres that administer and manage patients on ipilimumab-nivolumab are well equipped to manage this regimen.
Clinician Group Input
CADTH received 1 clinician-group submission from the Ontario Health (Cancer Care Ontario) (OH-CCO) Skin Cancer Drug Advisory Committee (DAC). At the time OH-CCO input, the proposed Health Canada indication was not line-specific (i.e., indicated for the treatment of adult and pediatric patients aged 12 years or older and weighing at least 40 kg with unresectable or metastatic melanoma). However, after the input was received, the indication was approved for the first line (i.e., for the treatment of adult and pediatric patients aged 12 years or older with unresectable or metastatic melanoma who have not received prior systemic therapy for unresectable or metastatic melanoma). In the metastatic or unresectable settings, first-line treatments can include single-drug nivolumab or pembrolizumab, ipilimumab-nivolumab, and TTs (for patients with BRAF mutations). The TT options are dabrafenib-trametinib, cobimetinib-vemurafenib, and binimetinib-encorafenib. If patients received pembrolizumab or nivolumab in the first line, the subsequent-line options are ipilimumab alone, or a TT (for patients with a BRAF mutation). If ipilimumab-nivolumab followed by nivolumab maintenance is used in the first line, only patients with a BRAF mutation have a second-line option to use a TT. Patients who received a first-line TT may be eligible for pembrolizumab, nivolumab, or ipilimumab-nivolumab in the second-line setting. If treated with pembrolizumab or nivolumab, the patient may be eligible to receive ipilimumab further downstream.
According to OH-CCO’s Skin Cancer DAC, the drug under review has a higher response rate compared with single-drug nivolumab in patients with unresectable or metastatic melanoma as shown by the RELATIVITY-047 trial. Although there was no head-to-head comparison trial, this combination also has less toxicity than ipilimumab-nivolumab, the treatment-related AEs for which are reported in the CheckMate 067 trial, and this combination may fill some of the unmet needs of the standard treatment.
The OH-CCO DAC reported that the algorithm in the first-line metastatic or unresectable setting should be: Patients who are not able to tolerate ipilimumab-nivolumab or who would be treated with a single-drug PD-1 inhibitor would be suitable for receiving the drug under review in the first-line metastatic or unresectable setting.
The DAC stated that a clinically meaningful response would be improved survival, reduction in the frequency and/or severity of symptoms, attainment of major motor milestones, ability to perform activities of daily living, improvement of symptoms, and stabilization (no deterioration) of symptoms. Treatment response will be routinely assessed clinically, and by CT and/or PET scans approximately every 3 months.
The DAC mentioned confirmed disease progression and/or unmanageable toxicities would be the most likely reasons to discontinue treatment. It also noted that the drug under review should be administered in an outpatient cancer clinic and prescribed by a oncologist.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s Reimbursement Review processes by identifying issues that may affect their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 4.
Clinical Evidence
Systematic Review
Description of Studies
One pivotal, phase II and III, double-blinded, randomized controlled and ongoing trial (RELATIVITY-047, N = 714)41 is included in the systematic review. The RELATIVITY-047 trial evaluated the comparative efficacy and safety of nivolumab-relatlimab FDC versus nivolumab monotherapy administered as a first-line therapy in the treatment of adult and pediatric patients aged 12 years or older with previously untreated, unresectable or metastatic melanoma. However, no adolescents (aged 12 to < 18 years) were enrolled. A total of 714 patients were randomized 1:1 to receive nivolumab-relatlimab FDC (N = 355) or nivolumab monotherapy (N = 359). The median age was 63 years (range = 20 to 94). The majority (N = 655, 91.7%) of patients had metastatic stage IV melanoma at study entry. The median duration from diagnosis to study treatment was 1.26 years. A total of 62 patients (8.7%) had received previous adjuvant or neoadjuvant treatment. A total of 275 patients (38.5%) were BRAF-positive. A total of 16 patients (2.2%) from Canada and 63 patients (8.8%) from the US were included. The primary outcome was PFS. The 2 secondary outcomes were OS and overall response rate (ORR). Tertiary and/or exploratory outcomes included duration of response (DoR), time to response (TTR) and health-related quality of life (HRQoL) measurements (Functional Assessment of Cancer Therapy–Melanoma [FACT-M] and EQ-5D-3L questionnaires). The sample size for the study was based on a primary end point of PFS using a blinded independent central review (BICR) for both the phase II and phase III studies. Results presented in this submission reflect the phase III component of the RELATIVITY-047 trial. The final analysis for PFS was conducted after a median follow-up of 13.2 months. The final analysis for OS and ORR were conducted after a median follow-up of 19.3 months. Results for median DoR and TTR were based on the updated descriptive analysis conducted after a median follow-up of 25.3 months. HRQoL measurements (FACT-M and, EQ-5D-3L) were recorded after a median follow-up of 19.3 months. The objective of the safety outcomes was to assess the overall safety and tolerability of nivolumab-relatlimab and nivolumab. Safety data reported in this review were based on a median follow-up of 25.3 months.
Efficacy Results
Based on the final analysis after a median follow-up of 13.2 months, the median PFS was 10.12 months (95% confidence interval [CI], 6.37 to 15.74) in the nivolumab-relatlimab FDC group, which was statistically significant and clinically meaningfully longer than the 4.63 months in the nivolumab monotherapy group (hazard ratio [HR] for nivolumab-relatlimab FDC versus nivolumab = 0.75; 95% CI, 0.62 to 0.92; P = 0.0055). The observed PFS benefit of nivolumab-relatlimab FDC compared with nivolumab monotherapy was shown in an updated descriptive analysis after a median follow-up of 25.3 months. Subgroup and sensitivity analyses of PFS were largely consistent with those of the primary analysis.
After a median follow-up of 19.3 months, the median OS was not reached in the nivolumab-relatlimab group compared to 34.10 months in the nivolumab group. The between-group difference (nivolumab-relatlimab FDC versus nivolumab) for median OS did not reach statistical significance at the final analysis of OS after a median follow-up of 19.3 months (HR = 0.80; 95% CI, 0.64 to 1.01; P = 0.0593). Similarly, after a median follow-up of 25.3 months, the median OS was not reached in the nivolumab-relatlimab group compared to 33.18 months in the nivolumab group in an updated descriptive analysis. The comparative OS of nivolumab-relatlimab FDC and nivolumab monotherapy was therefore uncertain.
Based on the descriptive final analyses, a total of 10.3% (95% CI, 3.4% to 17.3%) more patients in the nivolumab-relatlimab FDC group achieved an objective response compared with the nivolumab group after a median follow-up of 19.3 months. A consistent ORR benefit was also observed in the updated descriptive analysis after a median follow-up of 25.3 months. A total of 9.8% (95% CI, 2.8% to 16.8%) more patients in the nivolumab-relatlimab FDC group achieved an objective response compared with the nivolumab group.
In terms of complete response (CR) and progressive disease, no formal statistical or descriptive analysis was undertaken to report the between-group difference (or 95% CI). No HR (or 95% CI) was provided. The comparative rates of CR and progressive disease of nivolumab-relatlimab FDC compared with nivolumab monotherapy remain inconclusive.
After a median follow-up of 25.3 months, no statistical and clinical meaningful between-group difference was observed for DoR. The TTR appeared to be the same after a median follow-up of 25.3 months. However, no between-group difference and no HR were reported for TTR. The DoR and TTR of nivolumab-relatlimab FDC compared with nivolumab monotherapy remain uncertain.
After a median follow-up of 19.3 months, HRQoL (FACT-M and EQ-5D-3L utility index scores and EQ visual analogue scale [VAS] results) in the nivolumab-relatlimab FDC and nivolumab groups remained generally stable (no clinical meaningful improvement or deterioration) during the treatment period. There were little to no differences between nivolumab-relatlimab FDC and nivolumab monotherapy in FACT-M, EQ-5D-3L utility index scores, and EQ VAS.
No adolescents (aged ≥ 12 to 18 years) were enrolled in the pivotal study. However, in the Health Canada product monograph,40 the indication of nivolumab-relatlimab FDC includes pediatric patients aged 12 years or older and weighing at least 40 kg. The product monograph indicates that use of nivolumab-relatlimab FDC in pediatric patients is supported by predicted drug exposures at the recommended nivolumab-relatlimab FDC dose that is expected to result in safety and efficacy similar to those of adults. The safety and efficacy of nivolumab-relatlimab FDC have not been established in pediatric patients under the age of 12 years or in patients aged 12 years or older weighing less than 40 kg.40
Harms
The proportion of patients with at least 1 treatment-emergent AE appeared similar in the nivolumab-relatlimab FDC group compared with the nivolumab monotherapy group (99.2% in the nivolumab-relatlimab FDC group and 95.8% in the NIVO monotherapy group). However, the most common any-grade AEs (occurring in > 20% patients in either of the 2 groups) |||||||| || ||||| || |||| |||||||| || ||| ||||||||| |||| |||||||||| ||| ||||| |||| ||| ||||||||| ||||||||||| |||||| |||| || ||||||| |||||||||| ||| ||| ||||| ||||| ||| |||||| The frequency of serious adverse events (SAEs) appeared to be similar in both groups and individual SAEs were relatively rare. With the exception of malignant neoplasm progression, which occurred in 3.9% of patients receiving nivolumab-relatlimab FDC and in 5.6% of those receiving nivolumab monotherapy, no SAEs were reported in more than 2% of patients in either group. The frequency of withdrawal due to AEs also |||||||| ||||||||||| |||||| || ||| ||||||||| |||| |||||||||| ||| ||||| |||| || ||| ||||||||| ||||||||||| ||||| |||||||||| ||| ||| ||||| ||||| ||| ||||||| Discontinuation treatment due to specific AEs occurred in less than 2% patients in either of the groups, with the exception of malignant neoplasm progression, which occurred in 17% of patients receiving nivolumab-relatlimab FDC and 2.8% of those receiving nivolumab monotherapy. The frequency of death due to AEs (i.e., study drug toxicity) was rare in both groups (1.1% of the nivolumab-relatlimab FDC group and 0.6% of the nivolumab monotherapy group). ||||||| ||||||||||||| ||| |||||||||| || |||||||||| ||||||| ||||||||||||||| ||||||| ||||| ||||||| ||||| |||||||| ||||||||||| |||||| || ||| ||||||||| |||| |||||||||| ||| ||||| |||| || ||| ||||||||| ||||| ||||| ||||||||| The other particular notable harm, myocarditis, was rare, occurring in just 1.7% and 0.6% of patients, respectively). Grade 3 or 4 all-cause AEs were numerically more frequent with nivolumab-relatlimab FDC (44.8%) versus nivolumab (36.8%). Overall, the safety profile of nivolumab-relatlimab FDC was considered manageable and consistent with the known mechanisms of action of relatlimab or nivolumab. No new safety signal was identified.
Critical Appraisal
Appropriate methods of randomization, blinding and allocation concealment were reported. Objective outcomes and validated health-related outcomes were assessed. However, the minimally important between-group difference, which is the threshold used for all outcomes of the Grading of Recommendations Assessment, Development and Evaluation (GRADE), is not available. Clinical expert opinion therefore was used to inform the thresholds for determining whether the between-group difference observed for each outcome was clinically meaningful.
The proportion of patients with metastatic stage M1c was higher in the nivolumab-relatlimab FDC group (N = 151 [42.5%]) than in the nivolumab monotherapy group (N = 127 [35.4%]); however, the clinical experts consulted for this review stated that minor between-group imbalances of metastatic stage M1c would have been unlikely to affect the comparative results between the nivolumab-relatlimab FDC and nivolumab monotherapy groups.
Overall survival was assessed as a secondary outcome, and the study was not powered to assess the between-group difference in OS at the prespecified final analysis (after a median follow-up of 19.3 months) and updated analysis (after a follow-up of 25.3 months). The comparative efficacy on OS of nivolumab-relatlimab FDC compared with nivolumab therefore remains uncertain.
The statistical significance of ORR (according to BICR) could not be formally tested due to its position in the statistical hierarchy (the OS final analysis did not reach statistical significance). As a result, ORR, as well as CR and progressive disease (which were part of the overall response analysis), are based on only descriptive analyses after a median follow-up of 19.3 months. Only descriptive analyses without a between-group difference or HR were reported. Results for ORR, CR, and progressive disease should therefore be interpreted with caution.
The DoR and TTR were assessed as tertiary or exploratory outcomes but not subjected to the hierarchical testing procedure to control for type I error. Analyses of DoR and TTR were not statistically powered and were reported using descriptive statistics only. No between-group differences were reported for DoR or TTR, although an HR was reported for DoR. Overall, the findings of DoR and TTR should be viewed as supportive evidence only.
Similarly, FACT-M and EQ-5D-3L data were assessed as tertiary and/or exploratory outcomes but were not subjected to the hierarchical testing procedure to control for type I error. For these patients with reported HRQoL outcomes (FACT-M and EQ-5D-3L), there may have been differential recall bias. Overall, the magnitude and direction of the impact of recall bias on the patient-reported HRQoL outcomes are unknown. The HRQoL analyses were not statistically powered and were reported using descriptive statistics.42 Overall, the HRQoL findings should be viewed as supportive evidence only.
All subgroup analysis were excluded from the randomization scheme, and imbalances in characteristics may bias the results observed between the subgroups. In addition, the subgroup analysis may be not powered to detect the between-group difference in each subgroup. The findings of the subgroup analysis should therefore be viewed as supportive evidence only.
In addition, among the limitations of the RELATIVITY-047 trial is the lack of a comparison with current standard-of-care therapy, except for nivolumab monotherapy. The efficacy and safety of nivolumab-relatlimab FDC compared with ipilimumab-nivolumab, encorafenib-binimetinib, dabrafenib-trametinib, vemurafenib-cobimetinib, ipilimumab, pembrolizumab, dabrafenib, and trametinib are unknown.
It is uncertain whether the findings can be generalized to patients with central nervous system (CNS) metastases or those with an ECOG PS greater than 1 as no such patients were included in the study. Only 17 patients (2.4%) with brain metastasis were included (1.7% and 3.1% in the nivolumab-relatlimab FDC and nivolumab monotherapy groups, respectively). Patients with active CNS metastases were excluded. The clinical experts CADTH consulted for this review indicated that, while a higher ECOG PS (> 1) usually indicates more severe disease and is more likely to be accompanied by an unfavourable prognosis, the nivolumab-relatlimab FDC combination treatment could be extended to patients with an ECOG PS greater than 1. In terms of patients with CNS metastasis, the clinical experts CADTH consulted for this review indicated that additional studies are needed to understand the comparative efficacy and safety of nivolumab-relatlimab FDC versus nivolumab monotherapy in patients with CNS metastasis.
Finally, it should be noted that, although an age of 12 years or older was an inclusion criterion, no adolescents (aged ≥ 12 to < 18 years) were enrolled in the pivotal study. The comparative efficacy and safety profile of nivolumab-relatlimab FDC versus nivolumab monotherapy is therefore unknown, and whether the findings from the RELATIVITY-047 trial can be generalized to adolescent patients remains unknown. However, the Health Canada product monograph indicates that the use of nivolumab-relatlimab FDC in pediatric patients aged 12 years or older and weighing at least 40 kg is supported by predicted drug exposures at the recommended nivolumab-relatlimab FDC dosage, which is expected to result in safety and efficacy outcomes similar to those of adults. One clinical expert CADTH consulted for this review indicated that pediatric patients with unresectable or metastatic melanoma should be enrolled in clinical trials, if available, to assess the efficacy and safety profile of nivolumab-relatlimab FDC. The other clinical expert indicated that, because of the potential unfeasibility of the trials on pediatric patients, use of nivolumab-relatlimab FDC in adolescents should be considered on a case-by-case basis, particularly if body habitus is comparable or close to that of an adult. The clinical expert noted that immuno-oncology (IO) drugs are now given to the pediatric population, and they are well tolerated.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For pivotal studies and randomized controlled trials (RCTs) identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered most relevant to informing the deliberations of CADTH’s expert committee, and a final certainty rating was determined as outlined by the GRADE Working Group.43,44 Following the GRADE approach, evidence from RCTs was initially treated as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
The selection of outcomes for GRADE assessment was based on the sponsor’s Summary of Clinical Evidence, consultation with clinical experts, and input received from patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members: PFS, OS, ORR, DoR, HRQoL (i.e., FACT-M, EQ-5D-3L utility index, and EQ VAS), and change from cycle baseline after a median follow-up of 19.3 months and at a fixed-landmark time point of 24 months, as well as notable harms (i.e., myocarditis and adrenal insufficiency).
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or relative to the null. For this review, the target of the certainty of evidence assessment was based on the presence of absence of a clinically important effect, as informed by minimally important differences (MIDs) and thresholds suggested by the clinical experts (for all outcomes).
Results of GRADE Assessments
Table 2: Summary of Findings for Nivolumab-Relatlimab FDC Versus Nivolumab Monotherapy for the Treatment of Adult and Pediatric Patients (Aged 12 Years and Older and Weighing at Least 40 kg) With Unresectable or Metastatic Melanoma Who Have not Received Prior Systemic Therapy for Unresectable or Metastatic Melanoma
Outcome and follow-up | Patients (studies), N | Relative effect (95% CI) | Absolute effects (95% CI) | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
Nivolumab | Nivolumab-relatlimab | Difference | |||||
PFS | |||||||
PFS according to BICR using RECIST 1.1 Median follow-up: 13.2 months | 714 (1 RCT) | PFS events (i.e., disease progression or death) at data cut-off:
Median (95% CI) PFS at data cut-off, months:
| Higha | Nivolumab-relatlimab results in a clinically important increase in PFS when compared with nivolumab monotherapy | |||
OS | |||||||
OS according to DMC Median follow-up: 19.3 months | 714 (1 RCT) | OS events (i.e., deaths) at data cut-off:
Median (95% CI) OS at data cut-off, months:
| Lowb | Nivolumab-relatlimab may result in a clinically important increase in OS when compared with nivolumab monotherapy | |||
ORR | |||||||
ORR (CR plus PR) according to BICR using RECIST 1.1 Median follow-up: 19.3 months | 714 (1 RCT) | OR (95% CI): 1.58 (1.16 to 2.15) | 326 per 1,000 | 431 per 1,000 (379 to 484 per 1,000) | 103 more per 1,000 (34 to 173 more per 1,000) | Moderatec | Nivolumab-relatlimab likely results in a clinically important increase in ORR when compared with nivolumab monotherapy |
DoR | |||||||
DoR according to BICR using RECIST 1.1 Median follow-up: 25.3 months | 276 (1 RCT) | DoR events (i.e., progression or death, following first response) at data cut-off:
Median (95% CI) DoR at data cut-off
| Lowd | Nivolumab-relatlimab may result in little to no difference in DoR when compared with nivolumab monotherapy | |||
HRQoL (a median follow-up of 19.3 months and fixed-landmark time point of 24 months) | |||||||
FACT-M | |||||||
FACT-M total score Mean change from baseline (0 = worst HRQoL; 204 = best HRQoL) Median follow-up: 19.3 months | 151 (1 RCT) | NR | 3.563 | 1.756 (−1.763 to 5.275) | −1.807 (−6.561 to 2.947) | Lowe | Nivolumab-relatlimab may result in little to no difference in HRQoL as measured by FACT-M when compared with nivolumab monotherapy |
EQ-5D-3L utility index | |||||||
EQ-5D-3L utility score Mean change from baseline (0 = as bad as dead; 1 = perfect health) Median follow-up: 19.3 months | 150 (1 RCT) | NR | 0.002 | 0.009 (−0.036 to 0.053) | 0.007 (−0.052 to 0.066) | Lowe | Nivolumab-relatlimab may result in little to no difference in HRQoL as measured by EQ-5D-3L utility values when compared with nivolumab monotherapy |
EQ VAS | |||||||
EQ-5D-3L VAS Mean change from baseline (0 = worst health imaginable; 100 = best health imaginable) Median follow-up: 19.3 months | 150 (1 RCT) | NR | 2.084 | 2.840 (−0.454 to 6.135) | 0.757 (−3.651 to 5.164) | Lowe | Nivolumab-relatlimab may result in little to no difference in HRQoL as measured by the EQ VAS when compared with nivolumab monotherapy |
Notable harms (i.e., AEs of special interest) | |||||||
||||||||||||||||||| |||||||||| |||| |||||| | 714 (1 RCT) | || | ||| |||||| | || ||| ||||| |||| | || | ||| | |||||||||||||||||||| ||| |||||| || || |||||||| || ||| |||||||||| || |||||||| ||| |||||||||| ||||||||||| |||| |||||||| |||| ||||||||| |||||||||||| ||| |||||||| |||||||||| || ||| |||||||| || |||||||||| |
||||||| ||||||||||||||||||||| |||||||||| |||| |||||| | 714 (1 RCT) | || | || ||| ||||| | || ||| ||||| |||| | || | ||| | |||||||||||||||||||| ||| |||||| || || |||||||| || ||| |||||||||| || |||||||| ||| |||||||||| ||||||| ||||||||||||| |||| |||||||| |||| ||||||||| |||||||||||| ||| |||||||| |||||||||| || ||| |||||||| || |||||||||| |
AE = adverse event; BICR = blinded independent central review; CI = confidence interval; CR = complete response; DMC = data-monitoring committee; DoR = duration of response; EQ VAS = EQ visual analogue scale; FACT-M = Functional Assessment of Cancer Therapy–Melanoma; HR = hazard ratio; HRQoL = health-related quality of life; NA = not available (or not reached); NR = not reported; OR = odds ratio; ORR = overall response rate; OS = overall survival; PFS = progression-free survival; PR = partial response; RCT = randomized controlled trial; RECIST = Response Evaluation Criteria in Solid Tumors Version 1.1; vs. = versus.
Note: The analysis of ORR, DoR, and HRQoL (FACT-M total score and EQ-5D-3L) were not adjusted for multiple comparisons.
aIn the absence of available data for the between-group differences in event probabilities at clinically relevant time points, the judgment of imprecision was based on a 95% CI for the HR using the null as the threshold. The clinical importance of the between-group difference was judged based on the input of the clinical experts consulted by CADTH for the review.
bRated down 1 level for serious imprecision. In the absence of available data for the between-group difference in event probabilities at clinically relevant time points, the judgment of imprecision was based on the 95% CI for the HR using the null as the threshold. The 95% CI for the HR included the possibility of little to no difference (i.e., included the null). The clinical importance of the between-group difference was judged based on the input of the clinical experts consulted by CADTH for the review. Rated down 1 level for serious indirectness. The follow-up time was not sufficient to assess OS in this population.
cRated down 1 level for serious imprecision. Based on the threshold for a clinically important between-group difference suggested by the clinical experts of 50 to 100 events per 1,000 patients, the point estimate suggests a benefit; however, the lower bound of the 95% CI suggests little to no difference.
dRated down 2 levels for very serious imprecision. In the absence of available data for the between-group difference in event probabilities at clinically relevant time points, the judgment of imprecision was based on the 95% CI for the HR using the null as the threshold. The 95% CI for the HR included the possibility of both benefit and harm for nivolumab-relatlimab compared with nivolumab monotherapy.
eRated down 2 levels for a very serious risk of bias due to missing outcome data. Data were available for 21% of randomized patients. In the absence of a known threshold for a clinically important between-group difference, the null was used as the threshold.
fRated down 2 levels for very serious imprecision. The results are based on very few events in each group (6 of 355 vs. 2 of 359 for myocarditis and 20 of 355 vs. 4 of 359 for adrenal insufficiency in the nivolumab-relatlimab and nivolumab groups, respectively).
Long-Term Extension Studies
No long-term extensions studies are available.
Indirect Treatment Comparisons
Description of Studies
Two indirect treatment comparison (ITC) reports were submitted. Indirect treatment comparison 1 (ITC1), a Bayesian network meta-analysis (NMA), assessed the safety and efficacy of nivolumab-relatlimab relative to other IO drugs for adult patients in the first-line management of patients with advanced melanoma. Indirect treatment comparison 2 (ITC2), a patient-level propensity-weighted comparison, assessed nivolumab-relatlimab relative to ipilimumab-nivolumab among patients with advanced melanoma treated in the first line.
Efficacy Results
The first ITC, a Bayesian NMA, assessed nivolumab-relatlimab relative to nivolumab monotherapy, ipilimumab monotherapy, nivolumab (1 mg/kg) combined with ipilimumab (3 mg/kg), nivolumab (3 mg/kg) combined with ipilimumab (1 mg/kg), pembrolizumab, and cobimetinib-atezolizumab.
The results of ITC1 indicated that nivolumab-relatlimab is associated with improvements to OS relative || |||||||||| ||||||||||| ||||||||| ||| |||| ||||| |||||| For PFS, nivolumab-relatlimab is associated with improvements relative || |||||||||| |||| ||||| ||| |||| ||||| |||||| pembrolizumab (HR = 0.59; 95% credible interval [CrI], 0.35 to 0.97), and ||||||||||||||||||||||| |||| ||||| ||| |||| ||||| ||||||
For ITC2, the sponsor did not identify any association of the relative efficacy with respect to PFS or OS of nivolumab-relatlimab relative to ipilimumab (3 mg/kg) combined with nivolumab (1 mg/kg).
No data were available in either ITC with respect to ORR, time to progression, or any patient-reported outcome.
Harms Results
In ITC1, nivolumab-relatlimab was associated with higher proportions of patients having grade 3 or 4 treatment-related AEs when compared to nivolumab (odds ratio [OR] = 2.08; 95% CrI, 1.39 to 3.14 || |||||||||| |||| ||||| ||| ||||||||| ||||| and pembrolizumab (OR = 1.99; 95% CrI, 1.01 to 3.87), and was associated with lower proportions of patients experiencing these events relative to nivolumab (1 mg/kg) combined with ipilimumab (3 mg/kg) (OR = 0.43; 95% CrI, 0.25 to 0.73). For discontinuations due to AEs, nivolumab-relatlimab was associated with higher proportions of patients experiencing these events relative to nivolumab (OR = 1.59; 95% CrI, 1.10 to 2.32), and lower proportions of patients experiencing these events relative to nivolumab (1 mg/kg) combined with ipilimumab (3 mg/kg) (OR = 0.29; 95% CrI, 0.17 to 0.48). For discontinuations due to treatment-related AEs, nivolumab-relatlimab was associated with higher proportions of patients experiencing these events relative to nivolumab (OR = 2.21; 95% CrI, 1.41 to 3.56) and |||||||||| |||| ||||| ||| |||| ||||| |||||, and lower proportions of patients experiencing these events relative to nivolumab (1 mg/kg) combined with ipilimumab (3 mg/kg) (OR = 0.89; 95% CrI, 0.50 to 1.59).
No comparative data were presented from ITC2 with respect to safety outcomes, as no formal statistical comparison of the differences in safety events were conducted.
Critical Appraisal
Sponsor-submitted evidence from ITC1 was provided with comparisons to non-IO interventions of interest, such as BRAF and MEK inhibitors but, due to several challenges associated with mixed mutation status and the evidence from treatment nodes connecting to this network of evidence, no clear conclusions could be drawn about comparative efficacy and safety within this population. Several trials reporting on OS for the IO network of evidence still had ongoing observations for survival data at the time of analysis, and additional uncertainty may accompany these comparisons.
Studies Addressing Gaps in the Evidence from the Systematic Review
No studies addressing gaps in the evidence from the systematic review were available.
Conclusions
Evidence from the RELATIVITY-047 trial showed that nivolumab-relatlimab FDC therapy compared with nivolumab monotherapy results in a clinically meaningful benefit in terms of PFS (high certainty) in the treatment of adult patients with unresectable or metastatic melanoma who have not received prior systemic therapy for unresectable or metastatic melanoma. Nivolumab-relatlimab FDC may result in a clinically important increase in OS when compared with nivolumab monotherapy (low certainty). However, uncertainty remains in the OS results due to the inadequate length of follow-up for this outcome, and the results are imprecise (the CI included no difference between the nivolumab-relatlimab FDC and nivolumab monotherapy). Nivolumab-relatlimab FDC likely results in a clinically important increase in ORR when compared with nivolumab monotherapy (moderate certainty). However, the results were uncertain for the DoR of nivolumab-relatlimab FDC compared with nivolumab monotherapy after a median follow-up of 25.3 months (low certainty). Nivolumab-relatlimab FDC may result in little to no difference (either improvement or deterioration) when compared with nivolumab monotherapy in HRQoL as measured by FACT-M, EQ-5D-3L utility index, and EQ VAS (low certainty). Numerically more patients appeared to experience AEs (e.g., grade 3 or 4) in the nivolumab-relatlimab FDC group than in the nivolumab monotherapy group. However, the clinical experts CADTH consulted for this review indicated that the safety profile of nivolumab-relatlimab FDC appeared to be consistent with the known safety profile of each component drug (nivolumab and relatlimab) and was generally manageable. No additional safety signals were identified. It should be emphasized that the efficacy and safety profile of nivolumab-relatlimab FDC compared with nivolumab monotherapy is not available for pediatric patients (aged ≥ 12 to < 18 years). In addition, the efficacy and safety profile of nivolumab-relatlimab FDC compared with existing standard therapies except nivolumab monotherapy is not addressed in the RELATIVITY-047 trial. The sponsor-submitted ITCs were inconclusive with respect to nivolumab-relatlimab FDC relative to combination IO (ipilimumab [3 mg/kg] combined with nivolumab [1 mg/kg], ipilimumab [1 mg/kg] combined with nivolumab [3 mg/kg], and cobimetinib-atezolizumab) for PFS and OS but was associated with prolonged OS and PFS relative to ipilimumab monotherapy and prolonged PFS relative to pembrolizumab monotherapy. In the sponsor-submitted ITCs, nivolumab-relatlimab FDC demonstrated a favourable safety profile compared to a combination of nivolumab (1 mg/kg) and ipilimumab (3 mg/kg); however, compared to pembrolizumab, ipilimumab, and nivolumab monotherapy, the safety profile of nivolumab-relatlimab FDC was unfavourable.
Introduction
Disease Background
Contents within this section were informed by materials submitted by the sponsor and clinical expert input. The following summary was validated by the CADTH review team.
Melanoma is a neoplasm originating from melanocytes, the pigment-producing cells of the skin. It is 1 of the 3 main types of skin cancer, along with basal cell carcinoma and squamous cell carcinoma. Melanoma commonly arises in cutaneous primary locations (i.e., cutaneous melanoma).Other forms include mucosal, uveal, and acral melanoma.45,46 There are 4 main types of cutaneous melanoma: superficial spreading, nodular, lentigo maligna, and acral lentiginous.46,47 The majority of cases are superficial spreading or nodular, each accounting for approximately 37% of cases (73% combined) and each typically diagnosed at mean ages of 55 and 62 years, respectively.2,9 The clinical symptoms of advanced melanoma include hard or swollen lymph nodes, a hard lump on the skin, unexplained pain, feeling very tired or unwell, and unexplained weight loss.1 Melanoma is a debilitating disease that negatively affects patients’ physical, mental, and emotional well-being.48,49 The diagnosis tool of melanoma includes a skin examination, physical examination, skin and/or lymph node biopsy, and diagnostic imaging (i.e., CT).4-7 According the clinical experts CADTH consulted for this review, determining the molecular signature of a melanoma is critical. A blood test to assess serum LDH levels is an important prognostic marker of stage IV metastatic disease Most cases of melanoma are clinically identified early and can be cured with surgical excision alone.4,5,9
According to the Canadian Cancer Society, 10.4% of all new melanomas are stage III at diagnosis and 3.9% are stage IV (i.e., metastatic disease). Poor prognostic factors include an ECOG PS of 2 or higher, elevated LDH, nodal involvement and metastases, increased tumour thickness, ulceration, and mitoses measuring 1/mm2 or larger in thin T1 melanomas.3,5 Approximately 70% of metastatic melanomas have mutually exclusive mutations in BRAF, NRAS viral oncogene homologue, c-KIT, and GNAQ or GNA11 genes, which activate the MEK pathway, leading to promotion of cell proliferation, prevention of apoptosis, and angiogenesis.8 In a global systematic review, 38.5% of patients presented with genetic mutations in BRAF, 16.4% in NRAS, and 10% in c-KIT.10 However, a higher proportion of BRAF mutation-positive melanoma was reported in an analysis, in which 51% of patients with stage III or IV melanoma had a mutation in the BRAF gene.9 An Australian study of patients with advanced melanoma reported a similar rate, with 48% of tumours testing positive for a BRAF V600 mutation.11
In Canada, melanoma accounted for 3.8% of new cancer cases, approximately 4% of all diagnosed skin cancers in Canada,46,50 and 1.5% of cancer deaths in 2021.12 An estimated 9,000 people were diagnosed with melanoma in 2022 in Canada,13 with an age-standardized incidence rate of 23.5 per 100,000 in 2018 (excluding Quebec) based on data from Statistics Canada.14 The incidence was slightly higher in men than in women (25.9 versus 21.2 per 100,000).2,14 The mean age at diagnosis of advanced metastatic melanoma is approximately 59 years in Canada.2,3 The incidence of melanoma in Canada has continued to rise over the past 4 decades.12 Malignant cutaneous melanoma is responsible for 90% of all skin cancer–related deaths annually.4 An estimated 1,200 persons died from melanoma in 2022 in Canada, with an age-standardized mortality rate of 2.7 per 100,000.13 In contrast to incidence trends, mortality rates have declined over the past 4 decades.12 In Canada, stage IV distant metastatic disease is associated with a 5-year survival rate of 18%.4 However, consistent with the observed decline in mortality rates, melanoma survival rates have successively improved in recent years with the introduction of novel immunotherapies and TTs based on BRAF and MEK inhibitors. The advent of TTs and immunotherapies has resulted in a paradigm shift in outcomes for patients with advanced melanoma.
Standards of Therapy
Contents within this section were informed by materials submitted by the sponsor and clinical expert input. The following summary was validated by the CADTH review team.
Important treatment goals of systemic therapy in metastatic advanced melanoma include prolonging survival, generating durable responses, providing symptom relief, minimizing treatment toxicities, and maintaining quality of life.15 The current treatment paradigm in Canada for the first-line treatment of unresectable and/or metastatic advanced melanoma is based on the use of systemic therapies, which includes immunotherapy with ICIs as either monotherapy, dual therapy, or combination treatment with targeted BRAF or MEK inhibitors in BRAF mutation disease.16,51,52 These immunotherapies and TTs are funded with restrictions across the majority of Canadian provincial and territorial drug programs (excluding Quebec). According to the clinical experts CADTH consulted for this review, immunotherapy is the first line of choice for melanoma regardless BRAF status.16,17 The ICI immunotherapies that are approved by Health Canada and routinely used for the first-line treatment of metastatic melanoma in Canada include nivolumab (anti–PD-1) monotherapy,21,19 ipilimumab (anti–CTLA-4) monotherapy,20,22 pembrolizumab (anti–PD-1) monotherapy23,24 and ipilimumab-nivolumab combination therapy18-20 However, the use of nivolumab-ipilimumab has been increasing in patients with BRAF mutation melanoma,25 which is the treatment of choice among the ICIs. After the first line, the treatment decisions are largely determined by BRAF mutation status.17 Patients with no BRAF mutation are treated with immunotherapies and patients with BRAF mutations are eligible for treatment with a TT17 that has been approved by Health Canada and recommended for reimbursement by the CADTH pan-Canadian Oncology Drug Review Expert Review Committee, including encorafenib (a BRAF inhibitor) plus binimetinib (a MEK inhibitor),26-28 vemurafenib (a BRAF inhibitor) plus cobimetinib (MEK inhibitor),29-31 and dabrafenib (a BRAF inhibitor) combined with trametinib (a MEK inhibitor).32-34 TT use as a monotherapy is not reflective of typical clinical practice in Canada. Consultation with clinical experts in Canada revealed that patients with advanced melanoma rarely receive TT as a single drug (< 5%);35 standard chemotherapy (e.g., dacarbazine), surgery, and radiation therapy are not typically or commonly used in Canada for metastatic melanoma and have been displaced by either ICIs or TTs.4,52,53 There is an unmet medical need for an additional novel ICI combination therapy to treat metastatic melanoma, including a new therapy that can be used regardless of BRAF mutation status, offers increased efficacy in relation to anti–PD-1 monotherapy, and has a favourable safety profile that does not result in the additive toxicities seen with conventional dual immunotherapy combinations. A novel combination treatment regimen involving an ICI combined with a drug with a different mechanism of action would be preferred.
Drug Under Review
Key characteristics of nivolumab-relatlimab FDC and other relevant standard treatments available for patients with unresectable or metastatic melanoma who have not received prior systemic therapy for unresectable or metastatic melanoma are summarized in Table 3 and Table 4. Nivolumab is a humanized IgG4 monoclonal antibody ICI that binds to the PD-1 receptor and blocks its interaction with PD-L1 and programmed cell death ligand 2, triggering PD-1 pathway–mediated inhibition of the immune response, including the antitumour immune response.36,37 Relatlimab is a novel, first-in-class ICI that targets the LAG-3 receptor.38,39 Relatlimab is a humanized IgG4 monoclonal antibody that binds to the LAG-3 receptor and prevents LAG-3–mediated inhibition of the immune response by blocking its interaction with ligands, ultimately leading to an antitumour response.37,40 LAG-3 and PD-1 are distinct inhibitory immune checkpoint pathways, often co-expressed on tumour-infiltrating lymphocytes.40 They act synergistically on effector T-cells, leading to T-cell exhaustion and impaired cytotoxic function. Combined relatlimab (anti–LAG-3) and nivolumab (anti–PD-1)–mediated inhibition enables T-cell activation and restores effector function of exhausted T-cells, an effect that is greater than that of either antibody alone, leading to the initiation of an improved antitumour immune response.37,40
The recommended dosage of nivolumab-relatlimab FDC for adult patients is 480 mg of nivolumab and 160 mg of relatlimab, every 4 weeks. The recommended dosage for pediatric patients who are aged 12 years or older and weigh at least 40 kg is the same as for adults. A recommended dosage has not been established for pediatric patients aged 12 years or older and weighing less than 40 kg. Nivolumab-relatlimab FDC is supplied as a concentrate for solution for infusion: 240 mg of nivolumab per 20 mL (12 mg/mL) and 80 mg of relatlimab per 20 mL (4 mg/mL) in a single-dose vial (FDC).40 The Health Canada–approved indication of interest for this review is nivolumab-relatlimab FDC for the treatment of adult and pediatric patients aged 12 years or older with unresectable or metastatic melanoma who have not received prior systemic therapy for unresectable or metastatic melanoma.40 The CADTH reimbursement request aligns with the Health Canada indication.
Health Canada reviewed nivolumab-relatlimab FDC through the Standard Review Pathway. The treatment has not been reviewed previously by CADTH.
Table 3: Key Characteristics of Nivolumab-Relatlimab Encorafenib-Binimetinib, Dabrafenib-Trametinib, Vemurafenib-Cobimetinib, Dabrafenib, and Trametinib
Characteristics | PD-1 plus LAG-3 inhibitor | Target therapy (dual therapy) | Target therapy (monotherapy) | |||
|---|---|---|---|---|---|---|
Nivolumab-relatlimab | Encorafenib with binimetinib | Dabrafenib with trametinib | Vemurafenib with cobimetinib | Dabrafenib | Trametinib | |
Mechanism of action | Nivolumab is a PD-1 immune checkpoint inhibitor (mAb) Enhances antitumour responses by T-cells through blockade of PD-1 Relatlimab is a LAG-3 inhibitor and a human IgG4 monoclonal antibody that binds to the LAG-3 receptor and triggers the LAG-3–mediated inhibition of the immune response by blocking its interaction with ligands Combined relatlimab (anti–LAG-3) and nivolumab (anti–PD-1)–mediated inhibition enables T-cell activation and restores effector function of exhausted T-cells, which is greater than the effects of either antibody alone, leading to initiation of an improved antitumour immune response | Encorafenib (Braftovi) is a highly selective BRAF inhibitor that suppresses RAS/RAF/MEK/ERK pathways which inhibits BRAF V600 E, D, and K mutation-positive cell growth Binimetinib (Mektovi) is a MEK inhibitor that inhibits proliferation of human BRAF-mutant cell lines and tumour growth | Dabrafenib (Tafinlar) is a BRAF V600 inhibitor Trametinib (Mekinist) is a MEK inhibitor | Vemurafenib (Zelboraf) is a selective BRAF V600 inhibitor Cobimetinib (Cotellic) is a MEK inhibitor | Dabrafenib (Tafinlar) is a BRAF V600 inhibitor | Trametinib (Mekinist) is a MEK inhibitor |
Indicationa | Opdualag (nivolumab and relatlimab) is indicated for the treatment of adult and pediatric patients (12 years and older and weighing at least 40 kg) with unresectable or metastatic melanoma | Encorafenib in combination with binimetinib for the treatment of patients with unresectable or metastatic melanoma with a BRAF V600 mutation, as detected by a validated test | Dabrafenib in combination with trametinib for the treatment of patients with unresectable or metastatic melanoma with a BRAF V600 mutation, as detected by a validated test | Cobimetinib in combination with vemurafenib is indicated for treatment of patients with unresectable or metastatic melanoma with BRAF V600 mutation | Monotherapy for the treatment of patients with unresectable or metastatic melanoma with a BRAF V600 mutation | Monotherapy for the treatment of patients with unresectable or metastatic melanoma with a BRAF V600 mutation |
Route of administration | IV infusion | Oral | Oral | Oral | Oral | Oral |
Recommended dosage | For adult patients: 480 mg nivolumab and 160 mg relatlimab every 4 weeks (30-minute IV infusion) For pediatric patients aged 12 years and older and weighing at least 40 kg: The recommended dosage for pediatric patients who are aged 12 years or older and weigh at least 40 kg is the same as for adults. A recommended dosage has not been established for pediatric patients who are aged 12 years or older and weigh less than 40 kg | Encorafenib 450 mg (six 75 mg capsules) orally once daily and binimetinib 45 mg (three 15 mg tablets) orally taken twice daily, approximately 12 hours apart, until disease progression or unacceptable toxicity | Dabrafenib 150 mg orally twice daily (i.e., 300 mg daily) and trametinib 2 mg orally, once daily, until disease progression | Vemurafenib 960 mg twice daily and cobimetinib 60 mg daily for 21 days followed by a 7-day break | Dabrafenib 150 mg orally, twice daily, (total 300 mg daily) until disease progression | Trametinib 2 mg orally, once daily, until disease progression |
Serious adverse effects or safety issues (grade 3 and higher) | Immune-mediated AEs | New primary cutaneous malignancies, major hemorrhagic events, uveitis, venous thromboembolism, and QT prolongation | Hypertension, pyrexia, and elevated alanine aminotransferase, cutaneous squamous cell carcinoma, including keratoacanthoma | Increased ALA, increased AST, increased blood CPK, diarrhea, increased blood ALP, photosensitivity reaction, hyponatremia, and retinal detachment | Hypertension, pyrexia, and elevated ALA, cutaneous squamous cell carcinoma, including keratoacanthoma | Hypertension, pyrexia, and elevated ALA, cutaneous squamous cell carcinoma, including keratoacanthoma |
ALA = alanine transaminase; ALP = alkaline phosphatase; AST = aspartate transaminase; BRAF = B-Raf kinase protein; CPK = creative phosphokinase; IgG4 = immunoglobin G4; LAG-3 = lymphocyte activation gene 3; mAb = monoclonal antibody; MEK = mitogen-activated protein kinase enzyme; PD-1 = programmed cell death protein 1.
aHealth Canada–approved indication.
Sources: Nivolumab-relatlimab product monograph,40 Encorafenib in combination with binimetinib drug monographs,26,27 Clinical Guidance Report for dabrafenib and tramaetinib,54 and Clinical Guidance Report for vemurafenib and cobimetinib.55
Table 4: Key Characteristics of Ipilimumab-Nivolumab, Nivolumab, Ipilimumab, and Pembrolizumab
Characteristics | Immune checkpoint inhibitor (dual or monotherapy) | |||
|---|---|---|---|---|
Ipilimumab-nivolumab (CTLA-4 and PD-1 inhibitor) | Nivolumab | Ipilimumab | Pembrolizumaba | |
Mechanism of action | Ipilimumab:
Nivolumab:
| PD-1 immune checkpoint inhibitor (mAb) Enhances antitumour response by T-cell through blockade of PD-1 | CTLA-4 immune checkpoint inhibitor (mAb) Enhances antitumour response by T-cell through blockade of CTLA-4 | PD-1 immune checkpoint inhibitor (mAb) Enhances antitumour response by T-cells through blockade of PD-1 |
Indicationa | Nivolumab, in combination with ipilimumab, is indicated for the treatment of adult patients with unresectable or metastatic melanoma who have not received prior systemic therapy for unresectable or metastatic melanoma Nivolumab is indicated for the treatment of patients with unresectable or metastatic melanoma and disease progression following ipilimumab and, if BRAF V600 mutation-positive, a BRAF inhibitor PM 2015 version | Nivolumab as monotherapy is indicated for the treatment of adult patients with unresectable or metastatic melanoma who have not received prior systemic therapy for unresectable or metastatic melanoma. Opdivo is indicated for the treatment of patients with unresectable or metastatic melanoma and disease progression following ipilimumab and, if BRAF V600 mutation-positive, a BRAF inhibitor PM 2015 version, OPDIVO_EN_PM.pdf (bms.com) | Ipilimumab as a single drug is indicated for the treatment of unresectable or metastatic melanoma PM 2020 version, ipilimumab product monograph - Google Search | Pembrolizumab is indicated for the treatment of adult patients with unresectable or metastatic melanoma who have not received prior treatment with ipilimumab; subjects with BRAF V600-mutant melanoma may have received prior BRAF inhibitor therapy; pembrolizumab is indicated for the treatment of adult patients with unresectable or metastatic melanoma and disease progression following ipilimumab therapy and, if BRAF V600 mutation-positive, following a BRAF or MEK inhibitor PM 2023 version, KEYTRUDA-PM_E.pdf (merck.ca) |
Route of administration | IV infusion | IV infusion | IV infusion | IV infusion |
Recommended dosage | Ipilimumab in combination with nivolumab:
| Monotherapy:
| Monotherapy:
| 200 mg every 3 weeks, or 400 mg every 6 weeks |
Serious adverse effects or safety issues (grade 3 and higher) | Immune-mediated adverse events | Immune-mediated adverse events | Immune-mediated adverse events | Immune-mediated adverse events |
BRAF = B-Raf kinase protein; CTLA-4 = cytotoxic T-lymphocyte-associated antigen; mAb = monoclonal antibody; PD-1 = programmed cell death protein 1.
aHealth Canada–approved indication.
Sources: Keytruda product monograph,23 Opdivo product monograph,19 and Yervoy product monograph.20
Stakeholder Perspectives
Patient-Group Input
This section was prepared by the CADTH review team based on input provided by patient groups. The full original patient input received by CADTH is included in the Stakeholder section of this report.
CADTH received patient-group submissions from Melanoma Canada and the SYSF. Melanoma Canada is a national patient focused organization focused on the prevention and elimination of melanoma and skin cancers. Melanoma Canada provides patient support, advocacy, awareness, and education to the public and for health care professionals. The SYSF is a national patient-led not-for-profit group dedicated to the fight against nonmelanoma skin cancers, melanoma, and ocular melanoma through nationwide education, advocacy, and awareness initiatives. The SYSF provides a community of oncology-patient and caregiver support throughout the entire continuum of care, from prevention and diagnosis to survivorship.
Data were gathered by Melanoma Canada via an online survey. The survey link was emailed to a database of patients. Any patients and caregivers, regardless of stage or familiarity with the drug therapy in question, were asked to participate. Melanoma Canada also used its website and social media (e.g., Facebook) to promote the survey. The survey was made available June 15 to July 14, 2023. A total of 119 individual patient responses combined with 84 caregiver responses were received. Among the patients, 35 indicated they had no caregiver. Of the patient respondents, 81 were female and 38 were male. The survey was open to all patients, regardless of stage or whether they had been on the drug under review. There were 26 patients who had stage 0 melanoma, 17 were at stage I, 10 were at stage II, 18 were at stage III, 29 were at stage IV, and a further 19 did not know their stage. A total of 73 respondents were from Ontario, 15 were from Alberta, 11 from British Columbia, 8 from Quebec, 6 from Manitoba, and the remainder from other provinces. Two patients in this survey had been treated with the drug under review. Information was obtained by the SYSF through online surveys, virtual patient-roundtables, and one-on-one conversations with 60 melanoma patients, of whom 12 had experience with the drug under review, over the past 6 months. There were 37 females and 23 males aged between 18 and 89 years. There were 14 respondents from British Columbia, 6 from Alberta, 11 from Ontario, 1 from Nova Scotia, 7 from Quebec, 1 from Newfoundland and Labrador, and 2 from Prince Edward Island. A total of 18 respondents lived outside of Canada (in the US, Australia, and France).
Most patients reported that pain, scarring, lymphedema, fatigue, anxiety, fear, and depression are common impacts of the disease that affect the quality of life of patients and their families. Caregivers reported that the greatest impact associated with dealing with the diagnosis is the mental stress, followed by the negative financial impacts on the family due to the loss of income from a working partner, and the additional responsibilities of taking care of a home, family, and their loved one. Some of the respondents described the impact of melanoma using the following phrases: scared, disbelief, unsettled, anxious, teary, disrupted life, totally life changing, the diagnosis has taken a huge mental toll on me as I have always had a very large fear of cancer and I ended up living my worst nightmare, anxiety not knowing when disease may reoccur, had to stop working, disease affected my heart, general fatigue, less interest in activities, need to manage through therapy to address posttraumatic stress disorder, and fear.
In terms of current therapy options, based on 119 responses to the Melanoma Canada survey, 55% of patients had been treated with some form of drug therapy, and 9 had been treated with multiple therapies. A total of 92% of patients treated with available drug therapies indicated that they felt the side effects were worth tolerating, given the anticipated results. Moreover, about 20% of patients experienced trouble accessing treatment. The SYSF survey also mentioned that, for patients in remote areas of Canada, getting to treatment sites, paying for travel costs, and taking time off from work put extra stress on patients, caregivers, and their family, all of which was compounded by the added concern of being treated far from home and their support system. Access to the drug under review was limited to Ontario and Quebec, and a number of patients in Canada could not get access to the drug under review, even though it may have been their only option.
Regarding the improved outcomes, both patient groups identified a large opportunity for improvement if treatments with a wider variety of effective options with minimal side effects and longer responses were available.
Two respondents from Melanoma Canada’s survey and 12 respondents from SYSF’s survey indicated that they had experience with the drug under review, and the primary method of access to the drug was through a clinical trial. A total of 12 respondents explained that benefits of treatment outweighed the drawbacks of the side effects, which were mainly rash and fatigue and were somewhat manageable.
Melanoma Canada’s input did not mention a companion diagnostic test, while the SYSF asked all participants, not just those who received the drug under review, about their experiences with companion diagnostic testing. Respondents said they had received companion diagnostic testing, but 46 were unsure if they had received it. All patients were unsure how and when their companion diagnostic tests were conducted. For 1 patient, the testing process caused delays in starting treatment. All patients responded that there were no adverse effects associated with the testing procedure. Three patients had their testing expenses covered by private payer, 2 paid out of pocket, 1 received compassionate treatment, and 54 were not sure how treatment costs were covered, with most assuming health insurance was involved.
Melanoma Canada stated that there is an ongoing need for better options, and options for when 1 therapy does not work or stops working. Melanoma Canada also noted that melanoma is a complicated cancer with the highest level of mutations among cancers. It is difficult to treat once it has spread. Effective treatments, biomarkers, and earlier-stage treatments are needed to prevent some of the quality-of-life impacts from surgery, loss of income, duration of illness, and the impact on mental health for the patient and caregiver. According to Melanoma Canada, the drug under review represents an improvement and an option for the treatment of a cancer that continues to be increasingly prevalent and is complex to treat.
Clinician Input
Input From Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of adult and pediatric patients aged 12 years and older and weighing at least 40 kg with unresectable or metastatic melanoma.
Unmet Needs
The clinical experts indicated that the goal of the treatment is to increase OS rates, slow down progression, hopefully achieve a complete objective response, improve symptoms, and minimize toxicity, particularly long-term significant toxicities. They added that formulations are also needed to improve convenience. Currently the standard of care for metastatic melanoma in Ontario is ipilimumab-nivolumab (if the patient is able to tolerate potential toxicities).The general approach is that failure to respond to ipilimumab-nivolumab may prompt switching to a BRAF inhibitor plus a MEK inhibitor in BRAF-positive patients. Pembrolizumab or nivolumab may be attempted as monotherapy if too many AEs occur due to ipilimumab-nivolumab treatment. The clinical experts indicated that immunotherapy is not 100% effective, with response rates to ipilimumab-nivolumab of approximately 56%. Patients may initially achieve a response and then progress. After progression, new treatments are needed. Treatments may sometime be effective but accompanied by AEs that are not tolerable, necessitating discontinuing treatment despite efficacy. Less-toxic treatments that are more easily tolerable are therefore needed. According to the clinical experts, no currently available second-line therapy is more beneficial than ipilimumab-nivolumab combination therapy. A new therapy is needed to increase response rates with fewer AEs. According to the clinical expert, the current standard practice is to discuss dual-drug versus single-drug immunotherapy in the absence of contraindications. Factors that would be considered in determining the most suitable treatment include patient goals, age, comorbidities, bulk of disease, sites of disease, and pace of disease. A patient who chose a dual-drug therapy may de-escalate to a single drug to manage toxicity. If the patient progresses on dual therapy and has a BRAF mutation, BRAF and MEK inhibitors would be offered. According to the clinical experts, dual immunotherapy has been recognized as a potentially curative regimen. In addition, many trials are based on fixed dosing and limited vial sizes. The clinical experts noted that many provinces reimburse these therapies based on weight, and that clinics are challenged to cohort patients to minimize drug wastage.
Place in Therapy
One clinical expert indicated that, given its equivalency to ipilimumab-nivolumab and fewer toxicities, nivolumab-relatlimab FDC to be a first-line treatment for patients. The clinical experts emphasized that fewer AEs may mean more patient compliance and therefore superior outcomes overall, and that less toxicity may mean fewer hospital admissions, which would reduce burdens on patients and offset the extra cost of the drug. Nivolumab-relatlimab FDC could also be an alternative to ipilimumab-nivolumab, which is current first line of therapy in Ontario. The other clinical expert indicated that, if this regimen approved, then the choice between ipilimumab-nivolumab and nivolumab-relatlimab FDC with respect to OS, PFS, and toxicities would be discussed with patients. The clinical expert also noted that nivolumab-relatlimab FDC was directly compared with nivolumab monotherapy in the RELATIVITY-047 trial; nivolumab-relatlimab FDC is a first-class drug that may be used as a first- or second-line ICI. In addition, nivolumab-relatlimab FDC would not be reserved for those patients who are intolerant to ipilimumab-nivolumab, but made available to all those who would benefit from an effective regimen with reduced toxicity. Nivolumab-relatlimab FDC is expected to cause a shift in treatment paradigms; candidates for receiving a single-drug immunotherapy would be offered nivolumab-relatlimab FDC; and those considered for ipilimumab-nivolumab may also be offered nivolumab-relatlimab FDC. The clinical expert stated that the nivolumab-relatlimab FDC regimen may replace ipilimumab-nivolumab for less-robust patients.
Patient Population
The clinical experts indicated that all metastatic patients can be offered this treatment as it was beneficial regardless of BRAF status and PD-L1 and LAG-3 percentage or stage. It is similar to other immunotherapy combinations and could be offered to all patients. In addition, the experts stated that it will be important to follow OS data as they mature to determine the efficacy in brain metastases, and to know if the combination decreases or delays the occurrence of brain metastases.
Assessing the Response Treatment
The clinical experts indicated that it is necessary to assess responses in terms of improved patient symptoms and the modified immunotherapy RECIST criteria, as pseudoprogression can occur at the beginning of treatment. In most cases, it can take up to 2 or 3 months to evaluate a true response. Initially, responses should be assessed at 3-month intervals. As patients respond, the response assessment can be tailored and increased to every 6 months. Improved survival is the goal. The clinical experts noted that clinical-outcome assessments align with clinical trial outcomes; physicians and patients review toxicities, symptom control, and objective evidence of disease response in an ongoing fashion during active treatment.
Discontinuing Treatment
The clinical experts indicated that obvious disease progression on imaging with no improvement in symptoms will lead to discontinuation. When harmful grade 3 or 4 AEs occur, the patient should pause treatment in favour of treating the AE, after which the treatment can be restarted at a lower dose.
Prescribing Considerations
The clinical experts noted that these treatments for metastatic melanoma should be provided by specialist oncologists and pharmacists in a Canadian cancer centre or, if at a community centre, treatment should be supervised or connected to a cancer centre and experts who can dispense advice. According to the clinical experts, a centre administering or managing patients receiving ipilimumab-nivolumab are well equipped to manage this regimen.
Clinician-Group Input
This section was prepared by the CADTH review team based on the input provided by clinician groups. The full original clinician-group inputs received by CADTH are included in the Stakeholder section of this report. At the time of OH-CCO input, the proposed Health Canada indication was not line-specific (i.e., indicated for the treatment of adult and pediatric patients aged 12 years and older and weighing at least 40 kg with unresectable or metastatic melanoma). However, after the input was received, the indication was amended to first-line treatment of adult and pediatric patients aged 12 years or older with unresectable or metastatic melanoma who have not received prior systemic therapy for unresectable or metastatic melanoma.
CADTH received 1 clinician-group submission from the OH-CCO Skin Cancer DAC. The OH-CCO DACs provide timely evidence-based clinical and health system guidance on drug-related issues in support of OH-CCO mandate, which includes supporting the Provincial Drug Reimbursement Programs and the Systemic Treatment Program.
The OH-CCO DAC gathered data and information via videoconferencing and email. It stated that the indication for the drug under review is broad and can fit in all lines of therapy. In the first-line metastatic or unresectable setting, the current treatments can include single-drug nivolumab or pembrolizumab, combination ipilimumab-nivolumab and TTs (for patients with a BRAF mutation). The TT options are dabrafenib-trametinib, cobimetinib-vemurafenib, and binimetinib-encorafenib. For patients who received pembrolizumab or nivolumab in the first line, the subsequent-line options are ipilimumab alone, or a TT (for patients with a BRAF mutation). If ipilimumab-nivolumab followed by nivolumab maintenance is used in the first line, only patients with a BRAF mutation have a second-line option to use a TT. Patients who received a first-line TT may be eligible for pembrolizumab, nivolumab, or ipilimumab-nivolumab in the second-line setting. If treated with pembrolizumab or nivolumab, the patient may be eligible to use ipilimumab further downstream.
According to OH-CCO’s DAC, the drug under review has a higher response rate compared with single-drug nivolumab in patients with unresectable or metastatic melanoma, as demonstrated by the RELATIVITY-047 trial. Although the trial did not include a head-to-head comparison, this combination is also less toxic than ipilimumab-nivolumab, for which treatment-related AEs are reported in the CheckMate-067 trial, and this combination may fill some of the needs that are not met by the standard treatment.
OH-CCO’s DAC reported that following algorithm in the first-line metastatic and unresectable setting should be: Patients who are not able to tolerate ipilimumab-nivolumab or who would be treated with a single-drug PD-1 inhibitor would be suitable for receiving the drug under review in the first-line metastatic or unresectable setting.
For patients pretreated with ipilimumab-nivolumab, ipilimumab alone, or a PD-1 inhibitor alone: Patients who failed immunotherapy in the second line or beyond setting would also be suitable for this treatment.
In patients who are BRAF-negative:
If treatment with a PD-1 inhibitor failed, then ipilimumab would be next, followed by the drug under review.
If treatment with ipilimumab-nivolumab failed, then the patient can receive the drug under review.
In patients who are BRAF-positive:
If treated with PD-1 with or without cytotoxic CTLA-4 immunotherapy, the patient could then receive a BRAF and MEK inhibitor. The next line could be ipilimumab alone (if not previously treated with ipilimumab) in combination with PD-1 or the drug under review, which could also be used following all prior therapies (PD-1, CTLA-4, BRAF and MEK inhibitors).
According to the OH-CCO’s DAC, a clinically meaningful response would be improved survival and reduction in the frequency and/or severity of symptoms. Treatment response will be routinely assessed clinically, and by CT and/or PET scans approximately every 3 months. The DAC mentioned that the most likely reason to discontinue treatment would be confirmed disease progression and/or unmanageable toxicities. The DAC also noted that the drug under review should be administered in an outpatient cancer clinic and prescribed by an oncologist.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s Reimbursement Review processes by identifying issues that may affect their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 5.
Table 5: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert responses |
|---|---|
Relevant comparators | |
Issues with the choice of comparator in the submitted trial The comparator in the RELATIVITY-047 trial was single-drug nivolumab, which is publicly funded in Canada. Nivolumab-relatlimab is proposed as an alternative to currently available PD-1 inhibitors for unresectable or metastatic melanoma. Pembrolizumab and nivolumab are both publicly funded. | For pERC consideration. |
Other implementation issues regarding relevant comparators (e.g., access, funding, covered population) Several other therapies funded in Canada as potential comparators for unresectable or metastatic melanoma were not included as comparators in the RELATIVITY-047 trial:
| For pERC consideration. |
Considerations for initiation of therapy | |
Disease diagnosis, scoring or staging for eligibility The RELATIVITY-047 trial required PD-L1 and LAG-3 testing in all patients. Patients with expression or no expression were included. The study found that response was not predicted by expression of these markers.
| Routine testing is not required; however, it can be helpful for research purposes. LAG-3 testing is not routinely conducted. Most tests take 2 weeks; the turnaround time depends on whether it is next-generation sequencing or immunohistochemistry. |
Other patient characteristics for eligibility (e.g., age restrictions, comorbidities) The trial was in patients aged 12 years and older and weighing more than 40 kg. Should patients under the age of 12 years and weighing less than 40 kg be considered? Patients with active, known, or suspected autoimmune disease were excluded (exceptions: type 1 diabetes mellitus, hypothyroidism on hormone replacement, skin disorders). Should patients with autoimmune disorders be considered at the discretion of the treating physician? | One expert indicated that if the patient was aged less than 12 years, eligibility would depend on body habitus, and decisions would be made on a case-by-case basis. But in general, it is unlikely that such patients would be considered, as metastatic melanoma in this population would be extremely rare. The other expert indicated that patients with autoimmune disorders should be considered if their disease is not active. |
Prior therapies required for eligibility There are no other LAG-3 inhibitors currently available in Canada. Should the enrolment criteria regarding prior neoadjuvant and adjuvant treatment used in the RELATIVITY-047 trial be used as eligibility criteria? Should patients with potentially resectable disease be eligible? Patients enrolled had previously untreated unresectable or metastatic melanoma. Currently, CADTH’s provisional funding algorithm calls for single-drug PD-1 inhibitors to be funded in the first or second line after BRAF-targeted therapy. Are there data to support the use of nivolumab-relatlimab in the second line after BRAF-targeted therapy? | In general, the same patient population should be treated. In patients who received BRAF and MEK inhibitors (neoadjuvant or adjuvant) and are less than 6 months from completion, this combination could be considered. The RELATIVITY-020 trial provides support for considering this option in patients relapsing early. People with potentially resectable disease should be eligible. If the drugs make an unresectable patient resectable, they should have that option and this combination is more likely capable of making them resectable compared to immunomonotherapy alone. If patients have resectable disease with acceptable morbidity or mortality, the patient should have surgery. If patients have “borderline” resectable disease, or resectable disease with unacceptable morbidity or mortality risk or unresectable disease and obtain a response sufficient to have resection, they should not be denied surgery. The RELATIVITY-020 trial provides useful information. Nivolumab and Relatlimab in Patients With Advanced Melanoma That Had Progressed on Anti–Programmed Death-1/Programmed Death Ligand 1 Therapy: Results From the Phase I/IIa RELATIVITY-020 Trial (Nivolumab and Relatlimab in Patients With Advanced Melanoma That Had Progressed on Anti–Programmed Death-1/Programmed Death Ligand 1 Therapy: Results From the Phase I/IIa RELATIVITY-020 Trial | Journal of Clinical Oncology (ascopubs.org). The study conclusion states the following: “Nivolumab and relatlimab had a manageable safety profile and demonstrated durable clinical activity in a proportion of patients with heavily pretreated advanced melanoma with prior progression on anti–PD-(L)1-containing regimens.” One clinical expert indicated that if BRAF inhibitor therapy fails, the patient should be allowed to try this combination in the second line. |
Eligibility for re-treatment Should re-initiation of treatment be permitted for those who chose to take a treatment break but did not experience progression or unacceptable toxicity while on treatment? Should re-initiation be considered in the case of progression while off therapy? After a defined treatment break duration? | Case-by-case discussion with the treating oncologist is appropriate. Commonly, progression after a 6-month break is accepted as a guideline to reinstitute treatment; malignancies may still respond to treatment protocol. |
Special subtypes (not explicitly mentioned in the indication) to consider separately for eligibility Patients with active CNS disease were excluded. Should they be eligible? Patients with uveal melanoma were excluded. Should they be eligible? | No relevant data are available, but if nivolumab-relatlimab acts in the same manner as ipilimumab-nivolumab, then patients with active CNS disease may be excluded. A phase II or IV study should examine this issue and patient population (i.e., use knowledge of the pharmacokinetics to determine if it can pass the blood-brain barrier). One clinical expert stated that patients with uveal melanoma should not be eligible; a phase II and III study is required to address this issue. The other clinical expert indicated that making such a determination would be difficult, but in general, patients should have access to nivolumab-relatlimab. |
Considerations for discontinuation of therapy | |
Definition of loss of response, absence of clinical benefit, or disease progression In the RELATIVITY-047 trial, treatment was continued until progression or unacceptable toxicity. What is the most appropriate definition for progression? Patients could also continue treatment beyond progression if demonstrating a clinical benefit. Is this appropriate in any scenario other than pseudoprogression? | The definition is based on a subset of RECIST 1.1 for immunotherapy. One clinical expert indicated that, if patient symptoms are worsening or a tumour is progressing after 6 cycles of treatment, some pseudoprogression is occurring with immunotherapy and the treating physician would want to confirm it is true progression. If the patient symptomatically feels well, the drug may or may not be responsible and it may not be possible to determine if the disease is progressing. One clinical expert indicated that, pragmatically, a discussion with the patient should occur regarding options at the time of disease progression. First, treatment should continue if the patient is tolerating the drug well and progression is slowing down. Second, other options should be explored and/or accessed. Third, treatment could be continued to allow a patient to know of or reach an important life milestone. |
Treatment interruptions If treatment is interrupted, can it be resumed? Is there a specific time frame? Can treatment be resumed after holding for a toxicity that resolves to acceptable levels? | One expert indicated that the time frame includes the period after toxicity is resolved, as long as it is not life-threatening. The other expert indicated that treatment could be resumed on a case-by-case basis. |
Considerations for prescribing of therapy | |
Dosing, schedule/frequency, and dose intensity The FDC of 160 mg of relatlimab and 480 mg of nivolumab is given every 4 weeks. Is there potential for any other dosing options such as those based on weight? | Nivolumab-relatlimab FDC was manufactured as a fixed dose, and weight-based adjusted remuneration should not be based on weight dosing. |
Generalizability | |
Populations of interest matching the indication but with insufficient data Patients with an ECOG PS > 1 were excluded from the trial. Should they be eligible for treatment? | Patients with an ECOG PS > 1 should be eligible. The goal of treatment is to reduce an ECOG PS of 2 to an ECOG PS of 1. |
Populations outside the indication or reimbursement request but of interest to jurisdictions Should any patients considered appropriate for treatment with combination ipilimumab-nivolumab be considered for nivolumab-relatlimab? Is there any evidence or clinical rationale to choose nivolumab-relatlimab over nivolumab? | If approved and funded, both options would be presented to patients reviewing each of drugs. Nivolumab-relatlimab has fewer toxicities with similar efficacy compared to ipilimumab-nivolumab. If approved and funded, both options would be presented to patients reviewing each of drugs |
Patients on active treatment with a time-limited opportunity to switch to the drug(s) under review Should patients currently receiving first-line treatment for unresectable or metastatic melanoma with no disease progression be eligible to switch to nivolumab-relatlimab? Should patients who experience toxicity with ipilimumab and discontinue ipilimumab without progression be able to switch to nivolumab-relatlimab? Should patients being treated with second-line pembrolizumab or nivolumab (when BRAF-targeted therapy was used in the first line) be eligible to switch to nivolumab-relatlimab? | Patients currently receiving first-line treatment for unresectable or metastatic melanoma with no disease progression should not be eligible to switch to nivolumab-relatlimab. Patients who experience toxicity with ipilimumab and discontinue ipilimumab without progression should not be switched to nivolumab-relatlimab. Patients being treated with second-line pembrolizumab or nivolumab should not be eligible to switch to nivolumab-relatlimab if they response well to single-drug immunotherapy. However, in the event of failure of a second-line treatment, switching to nivolumab or ipilimumab-relatlimab can be considered. |
Funding algorithm | |
Drug may change place in therapy of comparator drugs. | For pERC consideration. |
Drug may change place in therapy of drugs reimbursed in previous lines. | For pERC consideration. |
Drug may change place in therapy of drugs reimbursed in subsequent lines Will patients be eligible for single-drug ipilimumab after progression? Will patients be eligible for any other ICI therapy after progression? Will patients with a BRAF mutation be eligible for BRAF-targeted therapy after progression? | Patients will be eligible for single-drug ipilimumab after progression. One clinical expert stated that this applies to a clinical trial context. Another clinical expert stated that, if new therapies become available, a patient could receive pembrolizumab. Patients with a BRAF mutation will be eligible for BRAF-targeted therapy after progression. |
Complex therapeutic space with multiple lines of therapy, subpopulations, or competing products. | For pERC consideration. |
Care provision issues | |
Companion diagnostics (e.g., access issues and timing of testing) Will LAG-3 testing be necessary? | LAG-3 testing will not be necessary. |
Other care provision issues In the event of toxicity to nivolumab-relatlimab, would switching to single-drug nivolumab be reasonable and permitted? | Switching to single-drug nivolumab would be reasonable and permitted if toxicity is related to relatlimab. |
System and economic issues | |
Additional costs to be considered (other than those related to care provision) Possible need for and cost of implementing LAG-3 testing in practice. | For pERC consideration. |
Presence of confidential negotiated prices for comparators Confidential prices for other first-line therapies (ICI and BRAF-targeted therapies). | For pERC consideration. |
ECOG PS = Eastern Cooperative Oncology Group Performance Status; ICI = Immune checkpoint inhibitor; pERC = CADTH pan-Canadian Oncology Drug Review Expert Review Committee; FDC = fixed-dose combination; LAG-3 = lymphocyte activation gene 3; MEK = mitogen-activated protein kinase enzyme; PD-1 = programmed cell death protein; PD-L1 = programmed cell death ligand 1.
Clinical Evidence
The objective of CADTH’s Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of nivolumab-relatlimab FDC (nivolumab 12 mg/mL and relatlimab 4 mg/mL) in a single-dose vial FDC), administered as an IV infusion over 30 minutes, in the treatment of adult and pediatric patients (aged 12 years and older and weighing at least 40 kg) with unresectable or metastatic melanoma who have not received prior systemic therapy for unresectable or metastatic melanoma. This review focuses on comparing nivolumab-relatlimab to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of nivolumab-relatlimab is presented in 4 sections, with CADTH’s critical appraisal of the evidence included at the end of each section. The first section, the systematic review, includes pivotal studies and RCTs that were selected according to the sponsor’s systematic review protocol. CADTH’s assessment of the certainty of the evidence in this first section using the GRADE approach follows the critical appraisal of the evidence. The third section includes indirect evidence from the sponsor. Of note, no evidence for long-term extension studies (second section) or studies addressing gaps in the systematic review evidence (fourth section) were submitted by the sponsor for this review.
Included Studies
Clinical evidence from the following is included in the CADTH review and appraised in this document:
1 pivotal and ongoing RCT (multinational, phase II and II RELATIVITY-047) identified in the systematic review
2 indirect treatment comparisons.
Systematic Review
Contents within this section were informed by materials submitted by the sponsor. The following summary was validated by the CADTH review team.
Description of Studies
Key characteristics of the included study are summarized in Table 6. A summary of its design is shown in Figure 1. The RELATIVITY-047 trial is a multinational, phase II and III, randomized, double-blind, and ongoing study of the efficacy and safety of nivolumab-relatlimab FDC relative to nivolumab monotherapy when administered as a first-line therapy in patients with previously untreated, unresectable, or metastatic melanoma. Interactive response technology (IRT) was used to randomize patients. The sponsor (Bristol Myers Squibb [BMS]), patients, investigators, and site staff were blinded to the assignment. The study was conducted in 114 sites across 25 countries, including 4 Canadian sites (Table 10).41,56 No adolescent patients (aged ≥ 12 to < 18 years) were enrolled. The median age was 63 years (range = 20 to 94). The majority (N = 655; 91.7%) of patients had metastatic stage IV melanoma at study entry. The median duration from diagnosis to study treatment was 1.26 years. A total of 62 patients (8.7%) had received previous adjuvant or neoadjuvant treatment. A total of 275 patients (38.5%) were BRAF-positive. A total of 16 patients (2.2%) from Canada and 63 patients (8.8%) from the US were included in the trial.
Table 6: Details of Study Included in the Systematic Review
Study detail | RELATIVITY-047 |
|---|---|
Designs and populations | |
Study design | Phase II and III, seamless, adaptive, double-blind, parallel, RCT of nivolumab-relatlimab vs. nivolumab monotherapy |
Locations | A total 114 sites in 25 countries (Argentina, Australia, Austria, Belgium, Brazil, Canada (N = 16; 2.2%), Chile, Columbia, Denmark, Finland, France, Germany, Greece, Israel, Italy, Mexico, New Zealand, Norway, Poland, Romania, Russia, Spain, Sweden, the UK, and the US (N = 63; 8.8%). |
Patient enrolment dates | Start date: April 11, 2018 Final PFS: median follow-up of 13.2 months Final OS analysis: median follow-up of 19.3 months Last updated data: median follow-up of 25.3 months Last subject randomized date: December 16, 2020 Clinical cut-off date (last patient’s last visit): January 25, 2021 End date: study is ongoing (estimated study completion is December 16, 2025) |
Randomized (N) | Total: N = 714; nivolumab-relatlimab: N = 355; nivolumab monotherapy: N = 359 |
Inclusion criteria |
|
Exclusion criteria | Active brain metastases or leptomeningeal metastases Patients with brain metastases were eligible if they had been treated with no MRI evidence of progression for at least 4 weeks after treatment and within 28 days before the first dose of study treatment; there must also have been no requirement for immunosuppressant therapy with systemic corticosteroids (> 10 mg/day prednisone equivalents) for at least 2 weeks before study treatment Uveal melanoma Active, known, or suspected autoimmune disease; patients with the following conditions were eligible:
Patients with a condition requiring systemic corticosteroids (> 10 mg daily prednisone equivalent) or other immunosuppressive therapies withing 14 days of initiating study treatment History of myocarditis Prior malignancy active within the previous 3 years, except for locally curable cancers that have been cured, such as basal cell carcinoma, squamous cell carcinoma, and superficial bladder cancer Prior treatment with an anti–PD-1, anti–PD-L1, anti–PD-L2, or anti–CTLA-4 antibody (except if given as adjuvant or neoadjuvant therapy for melanoma), or any other antibody or drug targeting T-cell co-stimulation or immune checkpoint pathways (except adjuvant or neoadjuvant interferon given for melanoma)
The following laboratory parameters must be met:
|
Drugs | |
Intervention | Nivolumab-relatlimab (480 mg nivolumab and 160 mg relatlimab FDC, IV, every 4 weeks) until disease progression, treatment discontinuation, unacceptable toxicity, withdrawal of consent, or end of study |
Comparator(s) | Nivolumab (480 mg, IV, every 4 weeks) until disease progression, treatment discontinuation, unacceptable toxicity, withdrawal of consent, or end of study |
Study duration | |
Screening phase | 14 days |
Run-in phase | A safety lead-in evaluation was performed on the first (up to) 18 patients; the lead-in followed a 6 + 6 + 6 design to monitor grade 3 and 4 infusion reactions; 6 patients were treated in each sequential set; there were no time-interval restrictions for patients to begin treatment within a set of 6, and the infusion reaction observation period was 48 hours |
Treatment phase | Treatment was given until disease progression, treatment discontinuation, unacceptable toxicity, withdrawal of consent, or end of study. The final analysis of the primary end point (PFS according to BICR) was conducted after a median follow-up of 13.2 months (March 9, 2021, DBL); the final analysis of the secondary end points (OS and ORR) was conducted after a median follow-up of 19.3 months (October 28, 2021, DBL); updated efficacy and safety analyses were conducted after a median follow-up of 25.3 months (October 27, 2022, DBL); all results presented at this DBL were descriptive |
Follow-up phase | Two follow-up visits following the end of treatment; the first follow-up visit was conducted 30 days after the last dose of study treatment, and the second follow-up visit was conducted 100 days from the last dose of treatment; survival visits are conducted 3 months after visit 2, with subsequent survival visits occurring every 3 months thereafter |
Outcomes | |
Primary end point | PFS according to BICRa,e using RECIST 1.1; final analysis conducted after a median follow-up of 13.2 months |
Secondary end point | OSb,e ORR according to BICR using RECIST 1.1 (including best overall response of partial or complete response)b,e Final analysis of OS and ORR conducted after a median follow-up of 19.3 months |
Safety | Rate of AEs, SAEs, select AEs, IMAEs, OESIs, AEs leading to treatment discontinuation, deaths, and laboratory abnormalitiesc; safety analysis reported based on a median follow-up of 25.3 months |
Tertiary and/or exploratory end pointsd | DoR (using RECIST 1.1) and TTR Subgroup analyses of PFS (according to BICR using RECIST 1.1), ORR (according to BICR), DoR, and OS based on LAG-3 expression (≥ 1% vs. < 1%) and PD-L1 expression (≥ 1% vs. < 1%) Pharmacokinetic parameters Incidence of antidrug antibody to relatlimab and nivolumab Integrated analysis of potential exposure-response relationship (pharmacodynamic effect, efficacy, and select safety) ORR, PFS, and OS by selected tumour or peripheral biomarkers Health-related quality of life Time to meaningful symptomatic deterioration as measured by FACT-M MS Summary measures of FACT-M total and subscale scores Item scores and postbaseline changes in FACIT GP5 item EQ visual analogue scale and 3-Level EQ-5D utility index scores WPAI:GH PFS, PFS2, ORR, DoR according to investigator using RECIST 1.1 Treatment-free interval, treatment-free survival Refer to the table footer for analysis time pointsd |
Publication status | |
Publications | Tawbi et al. (2022)57 Schadendorf et al. (2023)58 |
AJCC = American Joint Committee on Cancer; BICR = blinded independent central review; BMS = Bristol Myers Squibb; BRAF = B-Raf kinase protein; CTLA = cytotoxic T-lymphocyte-associated protein 4; DBL = database lock; DoR = duration of response; DoR = duration of response; ECOG PS = Eastern Cooperative Oncology Group Performance Status; FACIT-M GP5 = Functional Assessment of Chronic Illness Therapy Item GP5; FACT-M = Functional Assessment of Cancer Therapy–Melanoma; FDC = fixed-dose combination; IMAE = immune-mediated adverse event; MEK = mitogen-activated protein kinase enzyme; OESI = other event of special interest; ORR = overall response rate; OS = overall survival; PD-1 = programmed cell death 1 protein; PD-L1 = programmed cell death ligand 1; PD-L2 = programmed cell death ligand 2; PFS = progression-free survival; PFS2 = progression-free survival after the next line of subsequent therapy; RCT = randomized controlled trial; RECIST 1.1 = Response Evaluation Criteria in Solid Tumors Version 1.1; SAE = serious adverse event; TTR = time to response; ULN = upper limit of normal; vs. = versus; WPAI:GH = Work Productivity and Activity Impairment Questionnaire: General Health.
aThe final PFS analysis was conducted after a median follow-up of 13.2 months (March 9, 2021, DBL), with supportive data presented after a longer median follow-up of 25.3 months (October 27, 2022, DBL).
bThe final analysis of both secondary end points of OS and ORR was conducted after a median follow-up of 19.3 months (October 28, 2021, DBL), with supportive data presented after a longer median follow-up of 25.3 months (October 27, 2022, DBL).
cSafety data are based on the most recent data cut, representing a median follow-up of 25.3 months (October 27, 2022, DBL).
dThe list of tertiary and/or exploratory end points in Table 10 (T13) is comprehensive. Results are only reported in this submission for DoR and TTR; subgroup analyses of PFS, OS, and ORR; PFS2; FACT-M and the FACIT GP5 individual item; EQ-5D-3L visual analogue scale and utility index scores; and the WPAI:GH:
• DOR, TTR, and PFS2 results are based on a median follow-up of 25.3 months (October 27, 2022, DBL).
• Subgroup analyses of the primary and secondary end points are presented based on their respective final analysis time points, with supportive data presented after a longer median follow-up of 25.3 months (October 27, 2022, DBL).
• Results for patient-reported outcome measures (FACT-M, EQ-5D-3L, and WPAI:GH) are based on a median follow-up of 19.3 months (October 28, 2021, DBL), which reflects the most recent assessment time point.
ePrimary and secondary end points were tested using a statistical hierarchical procedure with group sequential testing, starting with PFS, followed by OS and ORR.
Sources: BMS (2021) Primary Clinical Study Report,41 BMS (2022), Addendum 01 – Primary Clinical Study Report,61 BMS (2023), Addendum 02 – Primary Clinical Study Report,62 BMS (2020), Clinical Protocol,63 BMS (2022), Statistical Analysis Plan,42 Clinicaltrials.gov (2022),56 Tawbi et al. (2022)57 Schadendorf et al. (2023).58
The primary outcome was PFS with OS and ORR as secondary outcomes. Tertiary and/or exploratory outcomes were DoR, TTR, and HRQoL measurements (i.e., FACT-M and EQ-5D-3L). Adults and adolescents aged 12 years and older were eligible for enrolment. A total of 714 patients were randomized 1:1 to receive nivolumab-relatlimab (N = 355) or nivolumab monotherapy (N = 359). All randomized patients received at least 1 dose of the study drug. Randomization was stratified according to the following factors:
LAG-3 expression status (≥ 1% versus < 1%)
Tumour cell PD-L1 expression status (≥ 1% versus < 1%)
BRAF V600 mutation status (mutation-positive versus wild-type)
American Joint Committee on Cancer (AJCC) 8th edition metastatic (M) stage.
The RELATIVITY-047 trial was designed as an adaptive and seamless phase II and III trial. A prespecified interim analysis (IA) of PFS was planned after a minimum of 400 patients were enrolled into phase II, or after at least 150 PFS events were observed by BICR, whichever came first. The IA of PFS at phase II was used to decide whether the trial should proceed to phase III or complete the study in phase II. Enrolment was paused until the final phase II analysis was conducted. On August 26, 2019, the IA of PFS in phase II met the prespecified threshold of an HR equal to or lower than 0.8, and therefore the study proceeded seamlessly to phase III enrolment in a double-blinded manner. Both the sponsor and site investigators were blinded to the results of the IA of PFS. In addition, an initial safety lead-in was performed on the first 18 patients randomized to assess potential grade 3 or 4 infusion reactions with nivolumab-relatlimab. This followed a 6 + 6 + 6 design, with 6 patients treated in each sequential set. As no safety risks were identified, enrolment in phase II continued as planned. Patients enrolled in phase II of the study continued on treatment and are part of the phase III analyses. The study design is presented in Figure 1. Results presented in this submission reflect phase III of the RELATIVITY-047 trial, which is still ongoing.
Figure 1: Study Design of RELATIVITY-047
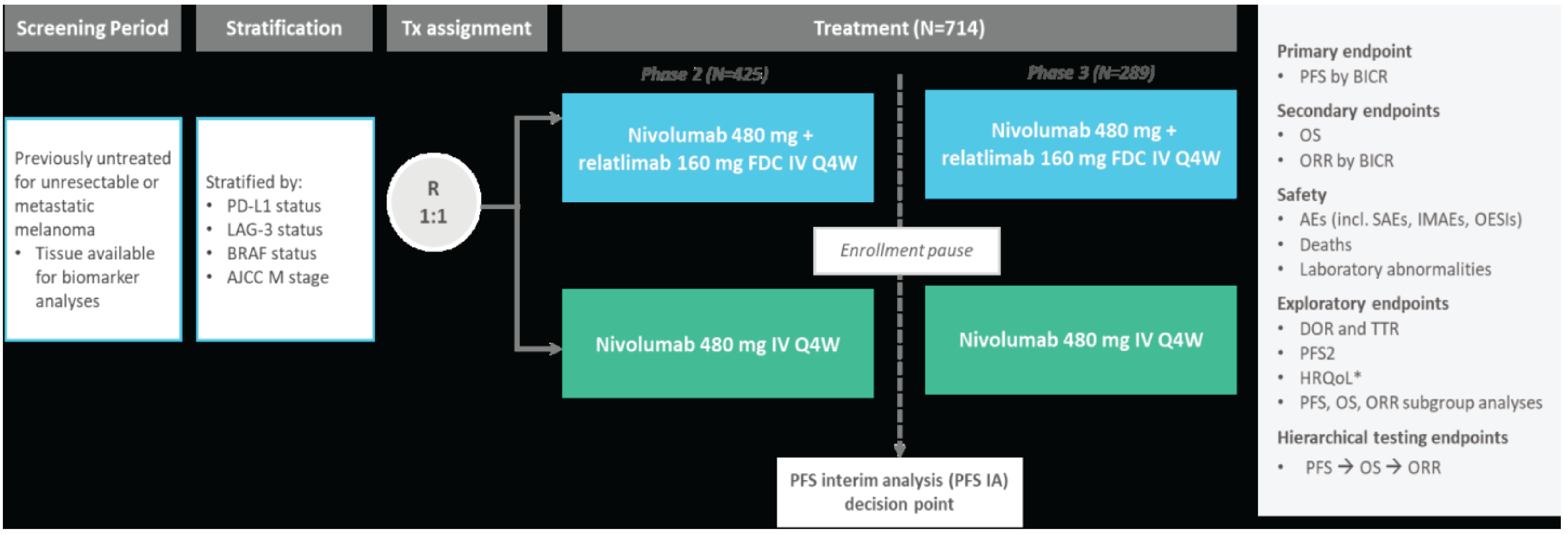
AE = adverse event; AJCC = American Joint Committee on Cancer; BICR = blinded independent central review; BMS = Bristol Myers Squibb; DoR = duration of response; BRAF = B-Raf kinase protein; EQ VAS = EQ visual analogue scale; FACIT-M GP5 = Functional Assessment of Chronic Illness Therapy Item GP5; FACT-M = Functional Assessment of Cancer Therapy–Melanoma; FDC = fixed-dose combination; HRQoL = health-related quality of life; IA = interim analysis; IMAE = immune-mediated adverse event; incl = including; LAG-3 = lymphocyte activation gene 3; M = presence of metastases; OESI = other event of special interest; ORR = overall response rate; OS = overall survival; PD-L1 = programmed cell death ligand 1; PFS = progression-free survival; PFS2 = progression-free survival after the next line of subsequent therapy; Q4W = every 4 weeks; RECIST 1.1 = Response Evaluation Criteria in Solid Tumors Version 1.1; SAE = serious adverse event; TTR = time to response; Tx = treatment; WPAI:GH = Work Productivity and Activity Impairment Questionnaire: General Health.
Notes: LAG-3 expression on immune cells (1%) determined by analytically validated immunohistochemistry assay (LabCorp Clinical Trials, Research Triangle Park, NC, US). PD-L1 expression on tumour cells (1%) determined by validated Agilent/Dako PD-L1 immunohistochemistry 28-8 pharmDx test (Agilent, Santa Clara, CA, US). The first tumour assessment (RECIST 1.1) was performed 12 weeks after randomization, every 8 weeks up to 52 weeks, and then every 12 weeks. HRQoL measures included FACT-M and the FACIT GP5 item, EQ VAS and utility index scores, and WPAI:GH.
Sources: BMS (2021), Primary Clinical Study Report41 and BMS (2020), Clinical Protocol.63
Populations
Inclusion and Exclusion Criteria
Patients were eligible for inclusion if they were aged 12 years or older with histologically confirmed, previously untreated, unresectable stage III or IV melanoma. Patients must have had measurable disease according to RECIST 1.1, expression of LAG-3 and PD-L1 that could be evaluated in tumour tissue, and an ECOG PS of 0 or 1. Patients must not have had prior systemic anticancer therapy for unresectable or metastatic melanoma, although they may have received prior neoadjuvant or adjuvant treatment if the final dose of therapy was administered at least 6 months before disease recurrence for anti–PD-1, anti–CTLA-4, or anti-BRAF or anti-MEK therapies, and at least 6 weeks before randomization for interferon. Patients were excluded if they had active brain metastases or leptomeningeal metastases, uveal melanoma, or an active, known, or suspected autoimmune disease (Table 6 provides more detail).
Interventions
Patients were randomized 1:1 to receive either nivolumab (480 mg) and relatlimab (160 mg) FDC or nivolumab monotherapy (480 mg). Both treatments were administered as an IV infusion every 4 weeks over 60 minutes in a clinic or hospital setting. Adolescents weighing less than 40 kg were to receive weight-based therapy (relatlimab 2 mg/kg and nivolumab 6 mg/kg), but no adolescents were enrolled in the study. Treatment continued until disease progression, treatment discontinuation, unacceptable toxicity, withdrawal of consent, or end of study.
Dose reductions were not permitted for either treatment group. Administration of study treatment could be delayed but not skipped.
Outcomes
A list of efficacy outcomes assessed in this Clinical Review Report is provided in Table 7, followed by descriptions of the outcome measures. Summarized outcomes are based on outcomes included in the sponsor’s Summary of Clinical Evidence as well as any outcomes identified as important to this review by the clinical experts consulted by CADTH and stakeholder input from patient and clinician groups and public drug plans. Using the same considerations, the CADTH review team selected outcomes that were considered to be most relevant to inform CADTH’s expert committee deliberations and finalized this list of outcomes in consultation with members of the expert committee. All summarized efficacy outcomes were assessed using GRADE. Select notable harms outcomes considered important for informing CADTH’s expert committee deliberations were also assessed using GRADE.
Table 7: Outcomes Summarized From the Study Included in the Systematic Review
Outcome measure | Time point | RELATIVITY-047 |
|---|---|---|
PFS | Final analysis conducted at the March 9, 2021, DBL (median follow-up of 13.2 months) | Primary end pointa |
OS | Final analysis conducted at the October 28, 2021, DBL (median follow-up of 19.3 months) | Secondary end pointa |
ORR | Final analysis conducted at the October 28, 2021, DBL (median follow-up of 19.3 months) | Secondary end pointa |
Safety | The most recent analysis of safety is available from the October 27, 2022, DBL, representing a longer length of exposure than previous data cuts (median follow-up of 25.3 months) | Safety assessmentb |
DoR and TTR | Data reported are based on the October 27, 2022, DBL (median follow-up of 25.3 months; reflects the latest analysis) | Tertiary and/or exploratory end pointb |
Subgroup analyses of PFS, OS, ORR | PFS: Final analysis conducted at the March 9, 2021, DBL (median follow-up of 13.2 months) OS and ORR: Final analysis conducted at the October 28, 2021 DBL (median follow-up of 19.3 months) Updated analyses for subgroups available from the October 27, 2022, DBL (median follow-up of 25.3 months; reflects the latest analysis) | Tertiary and/or exploratory end pointb |
EQ-5D-3L utility index scores and VAS | The most recent analyses for patient-reported measures are available from the October 28, 2021 DBL (median follow-up of 19.3 months) | Tertiary and/or exploratory end pointb,c |
FACT-M (and FACIT GP5 individual item) |
BICR = blinded independent central review; BMS = Bristol Myers Squibb; DBL = database lock; DoR = duration of response; FACIT GP5 = Functional Assessment of Chronic Illness Therapy Item GP5; FACT-M = Functional Assessment of Cancer Therapy–Melanoma; HRQoL = health-related quality of life; ORR = overall response rate; OS = overall survival; PFS = progression-free survival; TTR = time to response; VAS = visual analogue scale.
aA statistical hierarchical testing procedure was used to control for type I error in the primary and secondary end points. The hierarchy started with the primary analysis of PFS. If this was statistically significant at the applicable 2-sided alpha level (0.05), then the secondary end points would be tested in the order of OS followed by ORR. Other end points were not formally tested. Due to the position of ORR in the statistical testing hierarchy (after OS), ORR according to BICR was not formally tested for statistical significance as there was no significant difference in OS between the 2 treatment groups in the final OS analysis.
bSafety and tertiary/exploratory end points were assessed using descriptive analyses.
cThe most recent time point for analysis of HRQoL measures (exploratory) was the October 28, 2021, DBL. HRQoL was not assessed at the October 27, 2022, DBL.
Sources: BMS (2021), Primary Clinical Study Report;41 BMS (2022), Addendum 01 – Primary Clinical Study Report,61 BMS (2023), Addendum 02 – Primary Clinical Study Report,62 BMS (2022), Statistical Analysis Plan, and sponsor’s summary of clinical evidence.42
Progression-Free Survival
The primary end point was PFS as assessed by BICR using the RECIST 1.1 criteria with modifications by BMS, as presented in Table 9). Two definitions were used to assess PFS. The primary definition of PFS accounted for subsequent therapy (PFS truncated at subsequent therapy) by censoring at the last evaluable tumour assessment on or before the date of subsequent therapy. It was defined as the time between the date of randomization and the date of first documented tumour progression according to BICR, or death due to any cause, whichever occurred first. The secondary definition of PFS (intention-to-treat [ITT]) was irrespective of subsequent therapy and did not account for subsequent therapy. It was defined as the time between the date of randomization and the date of first documented tumour progression according to BICR, or death due to any cause, whichever occurred first.
Overall Survival
A secondary end point in the trial, OS was defined as the time between the date of randomization and the date of death due to any cause.
Overall Response Rate
The ORR was a secondary end point in the trial and was assessed by BICR and defined as the number of patients with a best overall response (BOR) of CR or partial response (PR) divided by the number of randomized patients in each treatment group (Table 9).61 As indicated previously, tumour assessments were conducted at 12 weeks, followed by every 8 weeks (through to week 52), and then every 12 weeks thereafter until disease progression or treatment discontinuation.
Tertiary and/or Exploratory End Points
While several tertiary and/or exploratory end points were assessed in the RELATIVITY-047 trial (Table 5), results presented in this submission focus on the key relevant exploratory end points of DoR, TTR, and HRQoL as assessed using the FACT-M and EQ-5D-3L.
Duration of Response
The DoR was defined as the time between the date of first response to the date of first documented tumour progression, as, assessed by BICR using RECIST 1.1 or death due to any cause, whichever occurred first.
The TTR was defined as the time from randomization to the date of the first documented response (CR or PR), assessed by BICR.
The DoR and TTR were only evaluated in responders (CR or PR).
Health-Related Quality of Life
The FACT-M and EQ-5D-3L (patient-reported outcomes) are validated instruments to assess the impact of treatment on HRQoL. Patient-reported outcome measures were assessed on day 1 of each cycle during the study, with 1 cycle lasting 4 weeks.
EQ-5D-3L
The EQ-5D-3L measure assessed patient-reported overall health status and consisted of a utility index score and the EQ VAS. The EQ-5D-3L descriptive system involves 5 domains (mobility, self-care, usual activities, pain or discomfort, and anxiety or depression), each with 3 levels (no problems, some problems, and severe problems). In the RELATIVITY-047 trial, utility index scores were calculated from self-reported health states based on a UK set of population-based preference weights; the scoring algorithm used was based on the UK MVH-A1 time trade-off value set.42
The EQ VAS assessed general health on a vertical scale, with scores ranging from 0 (worst health imaginable) to 100 (best health imaginable).42,64 The MID for the EQ-5D-3L was predefined as a change from baseline of 0.08 in health utility index scores, and a change of 7 for the EQ VAS.42,65
FACT-M
The FACT-M is a patient-reported measure used to evaluate the effects of disease symptoms on patient function and well-being.66 It includes 27 items from the Functional Assessment of Cancer Therapy–General (FACT-G) to assess physical well-being (PWB), social and/or family well-being (SWB), emotional well-being (EWB), and functional well-being (FWB). In addition, the FACT-M includes a 16-item disease-specific melanoma subscale (MS) and an 8-item melanoma surgery scale.42 Each item on the FACT-M is rated on a 5-point scale ranging from 0 (not at all) to 4 (very much).42 The FACT-G total score is generated by combining scores for the PWB, FWB, SWB, and EWB subscales, which reflects general quality of life. The FACT-M total score is generated by combining FACT-G and MS scores and provides a composite measure of general and targeted HRQoL. Scores are scaled so that higher scores indicate better functioning and lower symptom burden.42 The MID for the FACT-M 16-item MS has been estimated as a change of 2 to 4 points.67 In the RELATIVITY-047 trial, the midpoint of the range reported in the literature was used to define the MID (change ≥ 3 points). This has been applied in previous melanoma clinical trials.68 Using this approach, the MID for the FACT-M subscales was 5 for FACT-G and FACT-M total scores, 7 for trial outcome index, 2.5 for PWB and FWB, 2 for EWB, 3 for MS, and 1.5 for the melanoma surgery scale.41,67 The FACT scale is an acceptable indicator of patient HRQoL if the overall item response rate is greater than 80%.42
Safety
Safety assessments included the rate of AEs, SAEs, select AEs, AEs leading to study treatment discontinuation, immune-mediated adverse events (IMAEs), other events of special interest (OESIs), abnormalities in clinical laboratory assessments, and death.41 IMAEs were assessed to further evaluate AEs of clinical interest.42 They were defined as specific events or groups of preferred terms describing specific events and considered by the investigator as being potentially immune-mediated that met the following criteria:42 occurred within 100 days of the last dose; regardless of causality; treated with immune-modulating medication, and had no clear alternate etiology or an immune-mediated component. OESIs comprised a list of preferred terms grouped by specific category; for example, a myocarditis event and troponin elevation.42
Table 8: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MIDa |
|---|---|---|---|
Functional Assessment of Cancer Therapy–Melanoma (version 4)66,67 | Disease-specific questionnaire from the Functional Assessment of Chronic Illness Therapy catalogue of health-related quality-of-life questionnaires.66,67 The FACT-M is a patient-reported measure used to evaluate the effects of disease symptoms on patient function and well-being.66 It includes 27 items from the FACT-G to assess PWB, SWB, EWB, and functional well-being FWB. In addition, the FACT-M includes a 16-item disease-specific MS and an 8-item MSS.42 Each item on the FACT-M is rated on a 5-point scale ranging from 0 (not at all) to 4 (very much).42 Scores are scaled so that higher scores indicate better functioning and lower symptom burden.42 FACT-M total score: 51 items (0 to 204 points) FACT-G: 27 items (0 to 108 points) FACT-M subscale: 16 items (0 to 64 points) FACT MSS: 8 items (0 to 32 points)48 | Reliability: Excellent internal consistency and test-retest reliability observed for the FACT-M total score66,67 | The MID for the FACT-M MS has been estimated as a change of 2 to 4 points67 In the RELATIVITY-047 trial, the MID value of the range reported in the literature was used to define the MID (change ≥ 3 points). This has been applied in previous melanoma clinical trials.68 The MID for the FACT-M subscales was 5 for FACT-G and FACT-M total scores, 7 for trial outcome index, 2.5 for PWB and FWB, 2 for EWB, 3 for MS, and 1.5 for MSS41,66,67 The FACT scale is an acceptable indicator of patient HRQoL, as long as the overall item response rate is > 80%42 |
EQ-5D-3L | A generic, preference-based, self-reported HRQoL instrument that has been applied to a wide range of health conditions and treatments. The EQ-5D-3L assesses 5 domains: mobility, self-care, usual activities, pain/discomfort, and anxiety/depression Each domain has 3 levels: no problems, some problems, and severe problems The EQ-5D-3L also includes a VAS with anchors of 100 (best imaginable health) and 0 (worst imaginable health), on which patients provide a global assessment of their health42,64,69 | The validation of EQ-5D-3L is available across countries around the world and for various conditions64,69 No validity, reliability, and responsiveness were found for patients with melanoma | For cancers:65 MID (UK algorithm): 0.10 to 0.12 based on a PS range and 0.09 to 0.10 based on FACT-G score MID (US algorithm): 0.07 to 0.09 grouped by PS and 0.06 to 0.07 grouped by FACT-G score42,65 MIDs for VAS: 8 to 12 using PS and 7 to 10 using FACT-G quintiles42,65 No reported MID for patients with melanoma |
BMS = Bristol Myers Squibb; EWB = emotional well-being; FACT-G = Functional Assessment of Cancer Therapy–General; FACT-M = Functional Assessment of Cancer Therapy–Melanoma; FWB = functional well-being; HRQoL = health-related quality of life; MID = minimal important difference; MS = melanoma subscale; MSS = melanoma surgery scale; PS = performance status; PWB = physical well-being; SWB = social well-being; VAS = visual analogue scale.
Sources: Cormier et al. (2008);66 BMS (2022), Statistical Analysis Plan;42 and The EuroQol Group (1990).64
Table 9: Modified RECIST 1.1 for Evaluation of Target Lesions
Collection timing | Definition |
|---|---|
Complete response | Disappearance of all target lesions; any pathological lymph nodes (whether target or nontarget) must have a reduction in short axis to < 10 mm |
Partial response | At least a 30% decrease in the sum of diameters of target lesions, taking as reference the baseline sum diameters |
Progressive disease | At least a 20% increase in the sum of diameters of target lesions, taking as reference the smallest sum on study (this includes the baseline sum if that is the smallest on study); in addition to the relative increase of 20%, the sum must also demonstrate an absolute increase of at least 5 mm (the appearance of 1 or more new lesions was also considered progression) |
Stable disease | Neither sufficient shrinkage to qualify for partial response nor sufficient increase to qualify for progressive disease, taking as reference the smallest sum diameters while on study |
Not evaluable | If 1 or more target lesions cannot be measured or adequately assessed as either fully resolved or too small to measure (due to missing or poor-quality images), and the sum of diameters of the remaining measured target lesions (if any) has not increased sufficiently to meet progressive disease |
BMS = Bristol Myers Squibb; RECIST 1.1 = Response Evaluation Criteria in Solid Tumors Version 1.1.
Source: BMS (2020), Clinical Protocol.63
Statistical Analysis
A summary of the statistical analyses of the outcomes reported in this submission is provided in Table 13.
Primary Outcomes: Progression-Free Survival
Sample Size and Power Calculation
The study sample size for the RELATIVITY-047 trial was based on the primary outcome of PFS according to BICR for the phase II or III part of the study.42 As the IA of PFS at phase II met the prespecified HR threshold and the study transitioned to phase III, the sample-size justification was based on the phase III part of the study. The number of events required was simulated based on results from the CheckMate 067 (CA209067) study, with an estimated median PFS of 6.9 months for nivolumab monotherapy and 11.8 months for nivolumab-relatlimab, incorporating 35% of patients with durable response in the combined groups, and a piecewise HR, resulting in an effective HR of approximately 0.73.42 At least 365 PFS events were required to ensure approximately 85% power to detect an HR of 0.73, with an overall type I error of 0.05. The target HR was obtained via simulation modelling.42 The planned sample size for the phase III study was 700 patients, randomized to the 2 groups in a 1:1 ratio. This population comprised the phase II patient population plus enrolment of an additional 300 patients.42 The final analysis of PFS was planned to occur when 365 participants had a PFS event. Based on an anticipated screen failure rate of 30%, approximately 1,000 patients were required to be screened to randomize 700 patients meeting the eligibility criteria.42
Statistical Test or Model
The schedule of analyses of the primary end point and secondary outcomes are presented in Table 10.
The prespecified final analysis of the primary end point of PFS was conducted after a median follow-up of 13.2 months (March 9, 2021, database lock [DBL]). The prespecified final analysis of the secondary end points of OS and ORR were conducted after a median follow-up of 19.3 months (October 28, 2021, DBL).41,61 Updated efficacy and safety data are available after an additional 12 months of follow-up from the final OS and ORR analysis (October 27, 2022, DBL). Results from this most recent data cut are descriptive only and reflect a median follow-up of 25.3 months.62 The main results presented for the primary and secondary end points, as well as respective subgroup analyses, focus on the final analysis time points as prespecified in the statistical hierarchy (based on the March 9, 2021, DBL and October 28, 2021, DBL,61 respectively). Results for exploratory and/or descriptive end points of DoR, TTR, and safety are based on the most recent October 2022 DBL, reflecting the longest duration of follow-up.62 Results for exploratory HRQoL measures are based on the October 2021 DBL, which is the latest available data cut-off point (HRQoL was not assessed at the October 2022 DBL).61 The interim PFS analysis to determine whether the study continued onto phase III was performed once approximately 400 randomized patients had been followed up for a minimum of 12 weeks (or at least 150 PFS events according to BICR were observed).42 An administrative alpha penalty of 0.001 was used for the IA of PFS as the prespecified threshold was met and the study proceeded to phase III. The final PFS analysis was performed when at least 365 PFS events occurred to ensure approximately 85% power to detect an HR of 0.73 with an overall type I error of 0.05; all of the remaining unspent alpha was used (Table 10). At the March 9, 2021, DBL (median follow-up of 13.2 months) the results of the final analysis of PFS were statistically significant, which led to unblinding at the patient level, as prespecified in the statistical analysis plan.
Table 10: Schedule of Analyses of the Primary Outcome (PFS According to BICR)
Analysis | Interim PFS analysis | Final PFS analysis |
|---|---|---|
Population | All randomized patients | |
Conditions | When 150 PFS events are observed or all phase II randomized patients have been followed for at least 12 weeks (time of first scan) | When 365 events of PFS are observed |
Expected timings | Approximately 15 months | Approximately 34 months |
Nominal significance level | 0.001 | 0.049 |
Critical HR | HR ≤ 0.8a | HR ≤ 0.81 |
BMS = Bristol Myers Squibb; BICR = blinded independent central review; HR = hazard ratio; PFS = progression-free survival.
aNot formally tested (administrative alpha); had to pass this boundary for the study to continue to phase III.
Sources: BMS (2022), Statistical Analysis Plan and sponsor's summary of clinical evidence.42
When a stratified analysis is specified, the following stratifications factors are used: PD-L1 expression (≥ 1% versus < 1% or nonquantifiable immune cell surface expression); LAG-3 expression: (≥ 1% versus < 1% or nonquantifiable immune cell surface expression) (from IRT); BRAF mutation status: mutation-positive versus wild-type (from IRT); and AJCC (8th edition) M stage: M0/M1any[0] versus M1any[1]) (from lab value and electronic case report form). Each of the 4 stratification factors has 2 levels, with 16 strata in total. When there are fewer than 10 patients in a stratum, or in the case of unexpected model convergence issues due to small strata with too small event numbers, stratification factors are removed 1 at a time from the model until convergence is achieved. The CIs for the reported efficacy end points were not adjusted for multiplicity.
Subgroup Analyses
Exploratory subgroup analyses were conducted for PFS, OS, and ORR according to BICR among all randomized patients to assess the consistency of treatment effect across patient subgroups. Analyses of median PFS across subgroups were based on the Kaplan-Meier product-limit method with 2-sided 95% CIs. Unstratified HRs were produced for OS with 95% CIs, and unweighted differences in ORR and corresponding 95% 2-sided CIs used the Newcombe method. Subgroup analyses were not calculated for categories with fewer than 10 patients per treatment group (i.e., there must have been more than 10 patients in each subgroup category for the analysis to be conducted). Figure 4 and Figure 6 describe the subgroup analyses performed.
The RELATIVITY-047 trial was designed to assess the benefit of nivolumab-relatlimab FDC in all populations and was not powered to formally compare outcomes by any subgroups. Subgroup analyses were exploratory and may be limited by small sample sizes.
Sensitivity Analyses
In addition, prespecified sensitivity analyses to assess the robustness of the primary PFS analysis were conducted, as follows: censoring for subsequent therapy, constant hazards assumption, crossover of treatment effect across strata, adjustment for potentially important covariates, censoring for 2 missing images in a row, any differences between stratification values in IRT versus Medidata Rave RTSM Agile Randomization and Trial Supply Management, and an unstratified analysis.
A sensitivity analysis was planned of patients with no relevant protocol deviations greater than 10% of patients had relevant protocol deviations. A sensitivity analysis was planned if 10% of all PFS events were due to COVID-19; patients with a COVID-19 PFS event (i.e., death) would be censored on the previous evaluable image.
The potential effect of missing data (images) was assessed by performing a PFS analysis accounting for 2 or more consecutively missing disease assessments before the PFS event. Patients with 2 or more consecutive missing disease assessments were censored at the last disease assessment date before the 2 missing assessments.
Overall Survival
The analysis scheduled for OS is presented in Table 11. OS, the first key secondary end point in the hierarchical testing procedure, was formally assessed using a Lan-DeMets alpha-spending function with the O’Brien-Fleming type of boundary in East v6 for each end point. Initially, 2 interim analyses of OS were planned before the final analysis of OS. The first IA was performed by the data-monitoring committee at the time of the final PFS analysis, after 227 OS events (i.e., deaths) were observed (March 9, 2021, DBL). The first OS assessment was not statistically significant. However, it was decided that the second IA was no longer needed. The final analysis was conducted after approximately 300 OS events had occurred at the October 28, 2021, DBL (median follow-up of 19.3 months). The OS of nivolumab-relatlimab FDC versus nivolumab monotherapy at the first IA and final analysis was assessed using a 2-sided log-rank test, stratified by LAG-3 expression (≥ 1% versus < 1%), BRAF mutation status, and AJCC (8th edition) M stage. An O’Brien and Fleming alpha-spending function was used to determine the nominal significance levels for the interim and final OS analyses. In the final OS analysis, the O’Brien-Fleming boundary for significance was a P value less than 0.04302 (2-sided), with a cumulative design power of 69%, for a target HR of 0.75.
A sensitivity analysis was planned to assess the impact of COVID-19. This was to be performed if 10% of all OS events were due to COVID-19. Patients who died due to COVID-19 were censored on the start date of their COVID-19 AE.
The schedule of analyses for OS with the second IA removed is presented in Table 11.
Table 11: Schedule of Analyses of the Secondary End Point of Overall Survival (Without IA2)
Analysis | First interim OS analysis | Final OS analysis |
|---|---|---|
Population | All randomized patients | |
Assumed medians | Nivolumab-relatlimab FDC: 49.2 months Nivolumab monotherapy: 36.9 months | |
Conditions | 227 (75.7%) OS events observed at PFS final analysis | Approximately 300 (100%) OS events observed |
Nominal significance level (2-sided) OS efficacy boundary | 0.019 | 0.043 |
Cumulative alpha spent | 0.019 | 0.049 |
Critical HR | HR ≤ 0.733 | HR ≤ 0.792 |
Cumulative power | Approximately 43% | 69% |
BMS = Bristol Myers Squibb; HR = hazard ratio; IA2 = interim analysis 2; OS = overall survival; PFS = progression-free survival.
Sources: BMS (2022), Statistical Analysis Plan and sponsor's summary of clinical evidence.42
Overall Response Rate
ORR was assessed as the second key secondary outcome. As the final analysis of OS was not statistically significant, ORR was not formally tested for statistical significance given its position in the hierarchical testing procedure. ORR was only considered mature once all patients had had the potential for 7 months of follow-up, which was the time of the third on-study scan. The prespecified final analysis of ORR was carried out at the same time of the OS final analysis at the October 28, 2021, DBL (median follow-up of 19.3 months). However, because OS was not statistically significant, ORR could not be formally tested, and results are reported descriptively. The number and percentage of patients in each category of BOR according to BICR (CR, PR, stable disease, progressive disease, or unable to determine) were presented by treatment group. Estimates of response rate, with an exact 2-sided 95% CI based on the Clopper and Pearson method were presented by group, and a 2-sided 95% CI was calculated for the OR and the difference in response rates between the nivolumab-relatlimab FDC and nivolumab monotherapy treatment groups.
Subgroup analyses of ORR reported unweighted differences in ORR between groups based on the same variables as in the PFS analysis.
Duration of Response and Time to Response
Analyses of DoR and TTR were not statistically powered and were reported using descriptive statistics. The DoR for each treatment group was estimated using the Kaplan-Meier product-limit method for patients achieving a PR or CR. Median DoR and 2-sided 95% CIs were calculated based on a log-log transformation method. Patients censored in the Kaplan-Meier analysis of DoR were categorized as: ongoing follow-up (current [last scan within adequate window versus cut-off date], not current); off-study (lost to follow-up, withdraw consent, never treated); and received subsequent anticancer therapy. The TTR was summarized in all responders who achieved a confirmed PR or CR using descriptive statistics. No censoring was applied. DoR and TTR results presented in this submission are based on the most recent DBL, conducted on October 27, 2022, reflecting a longer duration of treatment and follow-up (25.3 months).
Health-Related Quality of Life (Patient-Reported Outcomes)
HRQoL was evaluated before dosing in each 4-week treatment cycle using the EQ-5D-3L and FACT-M. Analyses were conducted in all randomized patients with a baseline assessment and at least 1 postbaseline assessment. Analyses were not statistically powered and were reported using descriptive statistics. For the EQ-5D-3L utility index scores, EQ VAS, and FACT-M total, the mean score and mean change from baseline at each assessment time point was summarized by treatment group using descriptive statistics (N, mean, standard deviation, 95% CI, median, 25th and 75th percentiles, minimum, and maximum). The change from baseline in each HRQoL score was analyzed at time points with 10 or more patients using a longitudinal mixed model for repeated measures, with randomization strata, treatment, visit, interaction of treatment, and visit as fixed effects, and baseline HRQoL score as a covariate.
Multiple-Testing Procedure
A statistical hierarchical procedure was used to control for type I error of end points in the phase III part of the RELATIVITY-047 trial. The primary analysis was for PFS, and if this was significantly superior for the comparison of nivolumab-relatlimab FDC versus nivolumab monotherapy, then the secondary end points of OS followed by ORR could be tested. Other end points were not formally tested. A hierarchical testing procedure was employed for the phase III study to control for type I errors across end points (Figure 2). The overall alpha for the phase III study was 0.05 (2-sided). If the results of the PFS and OS analyses were statistically significant, then the ORR between treatment groups was to be tested when it had matured, which was after all randomized patients had the potential for 7 months of follow-up. Other end points were not formally tested.
Figure 2: Phase III Hierarchical Procedure With Group Sequential Testing in All Randomized Patients (Original Plan)
BMS = Bristol Myers Squibb; FA = final analysis; IA = interim analysis; IA2 = interim analysis 2; ORR = overall response rate; OS = overall survival; PFS = progression-free survival.
Notes: The second OS interim analysis (OS IA2) was removed, and the final OS analysis (OS FA) was directly performed.
Sources: BMS (2022), Statistical Analysis Plan and sponsor's summary of clinical evidence.42
Data Imputation Methods
The conventions used for imputing partial dates are presented in Table 12.
Table 12: Conventions Used for Imputing Partial Dates in Efficacy and Safety Analyses
Outcome measure | Convention |
|---|---|
Adverse events and safety | For missing and partial AE onset dates, imputation was planned to be performed using the Adverse Event Domain Requirements Specification For missing and partial AE resolution dates, imputation was planned to be performed as follows:
For death dates, the following conventions were planned for imputing partial dates:
|
Disease progression | Patients with 2 consecutive missing images were censored at the last assessment date before the 2 missing assessments for PFS For date of progression after the start of study therapy, the following conventions were planned for imputing partial dates:
For date of progression to prior therapies, the following conventions were planned for imputing partial dates:
For other partial or missing dates, the following conventions were used:
Last known alive date was based on all appropriate dates collected on the case report form |
HRQoL | FACT-M subscale scores could be prorated if there were missing items using the following formula:a prorated subscale score = (sum of item scores) × (N of items in subscale) / (N of items answered) |
Conversion factors | The following conversion factors were used to convert days to months or years: 1 month = 30.4375 days and 1 year = 365.25 days Duration (e.g., time to onset, time to resolution) was calculated as: duration = (last date − first date + 1) |
AE = adverse event; BMS = Bristol Myers Squibb; FACT-M = Functional Assessment of Cancer Therapy–Melanoma; PFS = progression-free survival.
aProrating by subscale when data are missing is acceptable providing more than 50% of the items are answered. The Functional Assessment of Cancer Therapy scale is considered an acceptable indicator of patient quality of life if overall item response rate is greater than 80%.
Note: Appendix 2 of the BMS (2022) Statistical Analysis plan provides imputation algorithms for missing and partial radiotherapy and surgery dates.
Source: BMS (2022), Statistical Analysis Plan.42
Table 13: Statistical Analysis of Efficacy Outcomes
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
PFS (primary) | 2-sided log-rank test stratified by LAG-3 expression (≥ 1% vs. < 1%), BRAF mutation status, and AJCC (8th edition) M stage.a HRs and 2-sided 95% CIs estimated using a Cox proportional hazards model with treatment group as a single covariate, stratified by the previously noted parameters Kaplan-Meier methodology used to estimate PFS curves, PFS medians, and 6- and 12-month PFS rates with 95% CIs Subgroup analyses of PFS reported median PFS based on the Kaplan-Meier product-limit method along with 2-sided 95% CIs | Adjustments made for subsequent therapies (primary PFS definition) Multivariate Cox regression model used to estimate the treatment effect after adjustment for imbalances potential prognostic factors in sensitivity analyses of PFS | Table 12 provides details | Censoring for subsequent therapy Constant hazards assumption Crossover of treatment effect across strata Adjustment for potentially important covariates Censoring for 2 missing images in a row Any differences between stratification values in IRT vs. RAVE An unstratified analysis PFS for patients with no relevant protocol deviations if > 10% of patients had relevant protocol deviations Impact of COVID-19 if 10% of all PFS events were due to COVID-19 |
OS (secondary) | Only interpreted if the primary end point of PFS is significantly superior according to the statistical hierarchical procedure: PFS at the March 9, 2021, DBL was statistically significant The resulting O’Brien-Fleming boundary for the OS FA was P < 0.04302 (2-sided) with a cumulative design power of 69% for a target HR of 0.75 OS FA assessed using a 2-sided log-rank test, stratified by same factors as PFS (LAG-3 status, BRAF mutation status, and AJCC M stage)a HR and 2-sided 95% CIs estimated using a Cox proportional hazards model with treatment group as a single covariate, stratified as previously noted. An O’Brien-Fleming alpha-spending function was employed to determine the nominal significance level for the FA OS estimated using Kaplan-Meier methodology 2-sided 95% CI for median OS computed via the log-log transformation method 6- and 12-month OS rates with 95% CIs were derived from Kaplan-Meier methodology and CIs derived based on the Greenwood formula for variance derivation and on log-log transformation applied on the survivor function | NA | Analysis performed of the impact of COVID-19 if 10% of all OS events were due to COVID-19 Patients with a death due to COVID-19 censored on the start date of their COVID-19 AE | |
ORR (secondary) | Only interpreted if the primary end point of PFS and secondary end point of OS were significantly superior, according to the statistical hierarchical procedure As OS was not significantly superior, ORR could not be formally tested and was only descriptively analyzed The number and percentage of patients in each category of BOR according to BICR (CR, PR, stable disease, progressive disease, or UTD) were assessed ORR estimates with exact 2-sided 95% CIs were reported and calculated using the Clopper Pearson method A 2-sided 95% CI was calculated for the OR of response between the treatment arms and for the difference in response rates between treatment arms | NA | NA (similar analyses were repeated based on the investigator’s assessment of ORR; not reported in this submission) | |
Safety (Safety) | Safety, including AEs, SAEs, AEs leading to discontinuation, select AEs, OESIs, IMAEs, deaths, and laboratory parameters, were assessed using descriptive statistics and graded for severity using the worst grade per the NCI-CTCAE v5.0) criteria and categorized by SOC and PT using the MedDRA v25.0 Frequency, management, and resolution of IMAEs and select AEs were analyzed | NA | NA | |
DoR and TTR (Tertiary and/or exploratory) | Evaluated in patients who achieved a CR or PR using descriptive statistics DoR estimated using Kaplan-Meier product-limit method and included median values, 2-sided 95% CIs, and range Median DoR, with 2-sided 95% CIs were computed based on a log-log transformation method TTR summarized in all responders (did not involve censoring) | NA | NA | |
Health-related quality of life | ||||
FACT-M (total score, subscales, and GP5 item); EQ-5D-3L (utility index and VAS) (tertiary and/or exploratory) | Assessed using descriptive analyses Mean score and CFB at each time point summarized using descriptive statistics (N, mean, standard deviation, 95% CI, median, 25th and 75th percentiles, minimum, and maximum) For GP5 only: the number and proportion of patients endorsing each response option at each assessment time point. Percentages based on the number of patients at each assessment time point EQ-5D utility index values were computed based using a scoring algorithm based on the UK MVH-A1 time trade-off value set CFB in each HRQoL score analyzed at time points with ≥ 10 patients using a mixed model for repeated measurements, with randomization strata, treatment, visit, and baseline HRQoL score considered Clinically meaningful CFB determined using prespecified MIDs: EQ-5D-3L utility index scores: CFB of 0.08b EQ VAS: CFB of 7b FACT-M: change of ≥ 3 pointsc | NA | NA | |
AE = adverse event; AJCC = American Joint Committee on Cancer; BICR = blinded independent central review; BMS = Bristol Myers Squibb; BOR = best overall response; CFB = change from baseline; CI = confidence interval; CR = complete response; DoR = duration of response; EQ VAS = visual analogue scale; FA = final analysis; FACT-M, = Functional Assessment of Cancer Therapy–Melanoma; HR = hazard ratio; HRQoL = health-related quality of life; IMAE = immune-mediated adverse event; IRT = interactive response technology; LAG-3 = lymphocyte activation gene 3; MedDRA v5.0 = Medical Dictionary for Regulatory Activities version 5.0; MID = minimal important difference; MS = melanoma scale; NA = not applicable; NCI-CTCAE v25.0 = National Cancer Institute Common Terminology Criteria for Adverse Events version 25.0; OESI = other event of special interest; ORR = overall response rate; OS = overall survival; PD-L1 = programmed cell death ligand 1; PFS = progression-free survival; PR = partial response; PT = preferred term; RAVE = Medidata Rave RTSM Agile Randomization and Trial Supply Management;70 SAE = serious adverse event; SOC = system organ class; TTR = time to response; UTD = unable to determine; vs. = versus.
aPD-L1 was removed from the models as a stratification factor because there were fewer than 10 subjects in at least 1 of the 16 strata.
bPrespecified in the statistical analysis plan based on Pickard et al. (2007).42,65
cPrespecified in the statistical analysis plan based on the midpoint of the range reported for MS in the literature.42,67,68
Sources: BMS (2021), Primary Clinical Study Report;41 BMS (2021), Addendum 01 – Primary Clinical Study Report;61 Addendum 02 – Primary Clinical Study Report;62 BMS (2022), Statistical Analysis Plan; and sponsor's summary of clinical evidence.42
Safety
Safety was assessed in all treated patients and reported by treatment group using descriptive statistics. Safety results presented in this submission are based on the most recent DBL, conducted on October 27, 2022, reflecting a longer duration of treatment and follow-up (25.3 months). Safety assessments included the frequency of deaths, AEs, SAEs, AEs leading to discontinuation of study treatment, select AEs, IMAEs, OESIs, and clinical laboratory abnormalities. Analyses were conducted using the 30-day and/or 100-day safety window from the day of the last dose received and were categorized by system organ class and preferred term using the Medical Dictionary for Regulatory Activities (version 25.0). The severity of AEs was graded using the worst grade according to the National Cancer Institute Common Terminology Criteria for Adverse Events (version 5.0). The frequency, management, and time to resolution of IMAEs and select AEs were analyzed. Laboratory parameters, which included hematology, chemistry, electrolytes, liver function, thyroid function, and renal function, were summarized using the worst grade per the Common Terminology Criteria for Adverse Events and conducted using the 30-day safety window.
The analysis population for each laboratory test was limited to patients who underwent that laboratory test. AEs were considered to be drug-related where the relationship to the study drug was missing.
Analysis Populations
The main analysis population sets in RELATIVITY-047 are presented in Table 14. The analysis of the primary end point was performed on the randomized analysis set, i.e., the ITT analysis set. Safety analyses were performed on the treated population, which included all patients who received at least 1 dose of the double-blind study drug. Of note, the randomized (ITT) analysis set is the same as the treated analysis set, as all randomized patients received at least 1 dose of the study drug.
Table 14: Analysis Populations of RELATIVITY-047
Population | Description | Application |
|---|---|---|
Enrolled participants | All patients who sign the informed consent and were registered into IRT | Patient disposition |
Randomized (ITT) | All patients who are randomized to any treatment group | Baseline demographic and disease characteristics, efficacy analyses, patient-reported outcomes analyses |
Treated and safety | All randomized patients who had at least 1 dose of the double-blind study treatment Data were analyzed based on randomized treatment, except in the following cases:
| Safety analyses |
Phase III | All patients randomized after August 26, 2019, date of data-monitoring committee recommendation to proceed to phase III | All phase III analyses, including final PFS, interim and final OS analysis, and ORR |
BMS = Bristol Myers Squibb; IRT = interactive response technology; ITT = intention-to-treat; ORR = overall response rate; OS = overall survival; PFS = progression-free survival.
Sources: BMS (2022), Statistical Analysis Plan and sponsor's summary of clinical evidence.42
Results
Patient Disposition
The disposition of patients in the RELATIVITY-047 trial is summarized in Table 15. Of the 1,281 patients enrolled (i.e., screened), a total of 714 patients (55.7%) were randomized to receive nivolumab-relatlimab FDC (N = 355) and monotherapy (N = 359). Although the reasons for discontinuation were not always similar between treatment groups, the overall proportion of patients discontinuing treatment was similar between the 2 treatment groups at all 3 DBLs for the final PFS analysis (median follow-up of 13.2 months), final OS and ORR analysis (median follow-up of 19.3 months), and at the updated descriptive analysis (median follow-up of 25.3 months). Disease progression was the most common reason for treatment discontinuation in both groups at all 3 DBL time points. A greater proportion of patients treated with nivolumab-relatlimab FDC discontinued treatment due to toxicity compared with those treated with nivolumab, although treatment discontinuation rates were largely similar between the 2 groups for each DBL.
Baseline Characteristics
Baseline characteristics of the all-randomized population of the RELATIVITY-047 trial are presented in Table 16. Overall, the enrolled patient population was generally representative of an untreated and unresectable or metastatic melanoma population, with a median age of 63 years and slightly more males than females (58.3% versus 41.7%, respectively). At baseline, the majority of patients had metastatic stage IV disease (AJCC, 8th edition) and 59% had disease with PD-L1 tumour cell expression levels of less than 1%. Although patients aged 12 years and older were eligible for enrolment, no adolescents (aged ≥ 12 to < 18 years) were enrolled.41 Baseline demographic and disease characteristics were well balanced between the nivolumab-relatlimab FDC and nivolumab monotherapy groups, including for key stratification factors of LAG-3 expression (< 1% versus ≥ 1%), PD-L1 expression (< 1% versus ≥ 1%), BRAF mutation status, and disease metastasis (M) stage based on the AJCC (8th edition).41 However, a higher proportion of patients in the nivolumab-relatlimab FDC group had M1c stage disease (42.5% versus 35.4%).41 A total of 60 patients had received adjuvant or neoadjuvant treatment before enrolment. Patients were not enrolled into the RELATIVITY-047 trial if they had received prior systemic anticancer therapy for unresectable or metastatic melanoma but could be enrolled if they had received prior adjuvant or neoadjuvant melanoma therapies, and all related AEs had either returned to baseline or stabilized.41,63
Table 15: Summary of Patient Disposition From Studies Included in the Systematic Review
Patient disposition | Final PFS analysis (March 9, 2021, DBL) | Final OS or ORR analysis (October 28, 2021, DBL) | Updated descriptive analysis (October 27, 2022, DBL) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
Nivo-Rela (N = 355) | Nivolumab (N = 359) | Total (N = 714) | Nivo-Rela (N = 355) | Nivolumab (N = 359) | Total (N = 714) | Nivo-Rela (N = 355) | Nivolumab (N = 359) | Total (N = 714) | ||
Screened (enrolled), n | 1281 (100.0) | |||||||||
Randomized, n (%) | 714 (55.7) | |||||||||
Not randomized, n (%) | 567 (44.3) | |||||||||
Reason for not randomized, n (%) | ||||||||||
Adverse event | 6 (0.5) | |||||||||
Subject withdrew consent | 41 (3.2) | |||||||||
Death | 12(0.9) | |||||||||
Poor/noncompliance | 2 (0.2) | |||||||||
Subject no longer meets study criteria | 465 (36.3) | |||||||||
Administrative reasons by sponsor | 6 (0.5) | |||||||||
Other | 35 (2.7) | |||||||||
Treated, n (%) | 355 (100.0) | 359 (100.0) | 714 (100.0) | — | — | — | — | — | — | |
Ongoing treatment, n (%) | 117 (33.0) | 126 (35.1) | 243 (34.0) | 86 (24.2) | 92 (25.6) | 178 (24.9) | 58 (16.3) | 58 (16.2) | 116 (16.2) | |
Completed treatment, n (%) | 1 (0.3) | 0 | 1 (0.1) | — | — | — | — | — | — | |
Discontinued treatment, n (%) | 237 (66.8) | 233 (64.9) | 470 (65.8) | 269 (75.8) | 267 (74.4) | 536 (75.1) | 297 (83.7) | 301 (83.8) | 598 (83.8) | |
Reason for treatment discontinuation, n (%) | ||||||||||
Disease progression | 129 (36.3) | 165 (46.0) | 294 (41.2) | 146 (41.1) | 183 (51.0) | 329 (46.1) | 156 (43.9) | 194 (54.0) | 350 (49.0) | |
Treatment toxicity | 63 (17.7) | 32 (8.9) | 95 (13.3) | 67 (18.9) | 36 (10.0) | 103 (14.4) | 76 (21.4) | 43 (12.0) | 119 (16.7) | |
Patient request | 19 (5.4) | 12 (3.3) | 31 (4.3) | 26 (7.3) | 17 (4.7) | 43 (6.0) | 32 (9.0) | 26 (7.2) | 58 (8.1) | |
AEs unrelated to study drug | 12 (3.4) | 14 (3.9) | 26 (3.6) | 15 (4.2) | 18 (5.0) | 33 (4.6) | 16 (4.5) | 20 (5.6) | 36 (5.0) | |
Death | 2 (0.6) | 3 (0.8) | 5 (0.7) | 1 (0.3) | 4 (1.1) | 5 (0.7) | 1 (0.3) | 3 (0.8) | 4 (0.6) | |
Maximum clinical benefit | 2 (0.6) | 1 (0.3) | 3 (0.4) | 3 (0.8) | 2 (0.6) | 5 (0.7) | 4 (1.1) | 3 (0.8) | 7 (1.0) | |
Withdrawal of consent | 1 (0.3) | 2 (0.6) | 3 (0.4) | 0 | 2 (0.6) | 2 (0.3) | 0 | 2 (0.6) | 2 (0.3) | |
Lack of efficacy | 1 (0.3) | 0 | 1 (0.1) | — | — | — | — | — | — | |
Poor or no compliance | 1 (0.3) | 0 | 1 (0.1) | 1 (0.3) | 0 | 1 (0.1) | 1 (0.3) | 0 | 1 (0.1) | |
Other | 7 (2.0) | 4 (1.1) | 11 (1.5) | 10 (2.8) | 5 (1.4) | 15 (2.1) | 11 (3.1) | 9 (2.5) | 20 (2.8) | |
Ongoing study, n (%) | 237 (66.8) | 227 (63.2) | 464 (65.0) | 210 (59.2) | 191 (53.2) | 401 (56.2) | 181 (51.0) | 163 (45.4) | 344 (48.2) | |
Discontinued study, n (%) | 118 (33.2) | 132 (36.8) | 250 (35.0) | 145 (40.8) | 168 (46.8) | 313 (43.8) | 174 (49.0) | 196 (54.6) | 370 (51.8) | |
Reasons for study discontinuation, n (%) | ||||||||||
Death | 107 (30.1) | 118 (32.9) | 225 (31.5) | 133 (37.5) | 153 (42.6) | 286 (40.1) | 157 (44.2) | 178 (49.6) | 335 (46.9) | |
Lost to follow-up | 5 (1.4) | 5 (1.4) | 10 (1.4) | 5 (1.4) | 6 (1.7) | 11 (1.5) | 5 (1.4) | 7 (1.9) | 12 (1.7) | |
Withdrew consent | 4 (1.1) | 9 (2.5) | 13 (1.8) | 6 (1.7) | 9 (2.5) | 15 (2.1) | 9 (2.5) | 9 (2.5) | 18 (2.5) | |
Other | 1 (0.3) | 0 | 1 (0.1) | 1 (0.3) | 0 | 1 (0.1) | 3 (0.8) | 2 (0.6) | 5 (0.7) | |
Not reported | 1 (0.3) | 0 | 1 (0.1) | — | — | — | — | — | — | |
Reason for discontinuation of study due to COVID-19 | — | — | — | 2 (0.6) | 4 (1.1) | 6 (0.8) | 2 (0.6) | 4 (1.1) | 6 (0.8) | |
Death | — | — | — | 2 (0.6) | 4 (1.1) | 6 (0.8) | 2 (0.6) | 4 (1.1) | 6 (0.8) | |
Intention-to-treat, N | 714 | — | — | — | — | — | — | — | — | |
Per-protocol, N | NR | — | — | — | — | — | — | — | — | |
Safety, N | 714 | — | — | — | — | — | — | — | — | |
AE = adverse event; BMS = Bristol Myers Squibb; DBL = database lock; ITT = intention-to-treat; Nivo = nivolumab; ORR = overall response rate; OS = overall survival; PFS = progression-free survival; Rela = relatlimab.
Sources: BMS (2021), Primary Clinical Study Report;41 BMS (2022), Addendum 01 – Primary Clinical Study Report;61 BMS (2023), Addendum 02 – Primary Clinical Study Report; and sponsor's summary of clinical evidence.62
Table 16: Summary of Baseline Characteristics From Studies Included in the Systematic Review (ITT Population)
Characteristic | Nivolumab-relatlimab (N = 355) | Nivolumab (N = 359) | Total (N = 714) |
|---|---|---|---|
Age category (%) | |||
≥ 12 and < 18 | 0 | 0 | 0 |
≥ 18 and < 65 | 187 (52.7) | 196 (54.6) | 383 (53.6) |
≥ 65 and < 75 | 102 (28.7) | 103 (28.7) | 205 (28.7) |
≥ 75 and < 85 | 60 (16.9) | 53 (14.8) | 113 (15.8) |
≥ 65 | 168 (47.3) | 163 (45.4) | 331 (46.4) |
≥ 75 | 66 (18.6) | 60 (16.7) | 126 (17.6) |
≥ 85 | 6 (1.7) | 7 (1.9) | 13 (1.8) |
Median age, years (range) | 63 (20 to 94) | 62 (21 to 90) | 63 (20 to 94) |
Sex | |||
Female, n (%) | 145 (40.8) | 153 (42.6) | 298 (41.7) |
Male, n (%) | 210 (59.2) | 206 (57.4) | 416 (58.3) |
Race, n (%) | |||
White | 342 (96.3) | 348 (96.9) | 690 (96.6) |
Black or African American | 0 | 5 (1.4) | 5 (0.7) |
American Indian or Alaska Native | 0 | 1 (0.3) | 1 (0.1) |
Other | 7 (2.0) | 4 (1.1) | 11 (1.5) |
Not reported | 6 (1.7) | 1 (0.3) | 7 (1.0) |
Initial stage of disease, n (%) | |||
III | 106 (29.9) | 90 (25.1) | 196 (27.5) |
IV | 111 (31.3) | 113 (31.5) | 224 (31.4) |
AJCC stage (8th edition) at study entry | |||
Unresectable stage III | 35 (9.9) | 23 (6.4) | 58 (8.1) |
Metastatic stage IV | 320 (90.1) | 335 (93.3) | 655 (91.7) |
Unknown/not reported | 0 | 1 (0.3) | 1 (0.3) |
Previous therapy, n (%) | 33 (9.3) | 29 (8.1) | 62 (8.7) |
Adjuvant | 31 (8.7) | 26 (7.2) | 57 (8.0) |
Neoadjuvant | 2 (0.6) | 1 (0.3) | 3 (0.4) |
Unknown or other | 0 | 2 (0.6) | 2 (0.3) |
Metastasis stage, n (%) | |||
M0 | 35 (9.9) | 23 (6.4) | 58 (8.1) |
M1 | 1 (0.3) | 3 (0.8) | 4 (0.6) |
M1a | 77 (21.7) | 107 (29.8) | 184 (25.8) |
M1b | 85 (23.9) | 88 (24.5) | 173 (24.2) |
M1c | 151 (42.5) | 127 (35.4) | 278 (38.9) |
M1d | 6 (1.7) | 11 (3.1) | 17 (2.4) |
Median time from initial diagnosis to randomization, years | 1.21 | 1.31 | 1.26 |
Melanoma subtype classification, n (%) | |||
Cutaneous acral | 41 (11.5) | 41 (11.4) | 82 (11.5) |
Cutaneous nonacral | 249 (70.1) | 254 (70.8) | 503 (70.4) |
Mucosal | 23 (6.5) | 28 (7.8) | 51 (7.1) |
Other | 42 (11.8) | 36 (10.0) | 78 (10.9) |
ECOG PS, n (%) (≥ 18 years) | |||
0 | 236 (66.5) | 242 (67.4) | 478 (66.9) |
1 | 119 (33.5) | 117 (32.6) | 236 (33.1) |
Tumour cell surface biomarker expression | |||
PD-L1 < 1% or nonquantifiable | 209 (58.9) | 212 (59.1) | 421 (59.0) |
PD-L1 ≥ 1% | 146 (41.1) | 147 (40.9) | 293 (41.0) |
LAG-3 < 1% or nonquantifiable | 87 (24.5) | 90 (25.1) | 177 (24.8) |
LAG-3 ≥ 1% | 268 (75.5) | 269 (74.9) | 537 (75.2) |
BRAF mutation status | |||
Mutation-positive | 136 (38.3) | 139 (38.7) | 275 (38.5) |
Wild-type | 219 (61.7) | 220 (61.3) | 439 (61.5) |
Lactase dehydrogenase | |||
≤ Upper limit of normal | 224 (63.1) | 231 (64.3) | 455 (63.7) |
> Upper limit of normal | 130 (36.6) | 128 (35.7) | 258 (36.1) |
Not reported | 1 (0.3) | 0 | 1 (0.3) |
AJCC = American Joint Committee on Cancer; BMS = Bristol Myers Squibb; ECOG PS = European Cooperative Oncology Group Performance Status; ITT = intention-to-treat; LAG-3 = lymphocyte activation gene 3; PD-L1 = programmed cell death ligand 1.
Sources: BMS (2021), Primary Clinical Study Report and sponsor's summary of clinical evidence.41
The baseline characteristics outlined in Table 16 are limited to those that are most relevant to this review or were likely to affect the outcomes or interpretation of the study results.
Exposure to Study Treatments
At the 3 DBLs for the final PFS analysis, final OS and ORR analysis, and updated descriptive analysis, the majority of patients in both treatment groups (ranging from 83.6% to 87.9%) had received 90% or more of the intended dose intensity.41,61,62 No meaningful difference was observed in the number of doses received between treatment groups at each of the 3 DBL time points (Table 17).41,61,62 At the time of the DBL for the updated descriptive analysis (October 27, 2022,), the median duration of treatment was approximately 2 months longer with nivolumab-relatlimab FDC than with nivolumab monotherapy (8.31 versus 6.47 months).62
Concomitant Medications and Co-Interventions
After a median follow-up of 25.3 months at the longer follow-up analysis, the majority of patients (97.9%) had received concomitant medication(s) during the treatment period.62 Concomitant medications used in 10% or greater of the total population are summarized in Table 18.
Patients were able to use topical, ocular, intra-articular, intranasal, and inhalational corticosteroids where there was minimal systemic absorption. Adrenal replacement steroid doses of greater than 10 mg of prednisone daily equivalent and a brief (< 3 week) course of corticosteroids to prevent or treat nonautoimmune conditions were also permitted.63 In addition, palliative radiotherapy and palliative surgery were allowed if patients met certain criteria and following discussion with the BMS medical monitor or designee.63 Immune-modulating concomitant medications recommended to treat certain AEs included corticosteroids, immune-modulating drugs, immunosuppressive drugs, and glucocorticoids.62 These were more frequently administered to manage AEs in patients receiving nivolumab-relatlimab FDC compared with nivolumab monotherapy (58.9% versus 44.0%).62 Surgical resection of solitary lesions was permitted only following consultation with the BMS medical monitor or designee and after the week 20 efficacy assessments.63 immunosuppressive drugs (except those used to treat a drug-related AE) and immunosuppressive doses of systemic corticosteroids were prohibited during the study63 (Table 18).
Subsequent Treatment
Approximately 46% of all randomized patients received subsequent anticancer therapy.61 The use of subsequent therapies was higher in the nivolumab group, including the use of PD-L1 or CTLA-4 inhibitors (15% and 18%, respectively), and targeted BRAF and MEK inhibitors (14% and 16%, respectively).61 The primary definition of PFS accounted for subsequent therapy by censoring at the last available tumour assessment on or before the date of subsequent therapy. Table 19 summarizes the subsequent anticancer therapies received by patients after a median follow-up of 25.3 months (on October 27, 2022, DBL).
Table 17: Summary of Patient Exposure From Study Included in the Systematic Review (All Treated Patients, ITT Population)
Exposure | PFS final analysis (March 9, 2021, DBL) | OS or ORR final analysis (October 28, 2021, DBL) | Updated descriptive analysis (October 27, 2022, DBL) | |||
|---|---|---|---|---|---|---|
Nivolumab-relatlimab (N = 355) | Nivolumab (N = 359) | Nivolumab-relatlimab (N = 355) | Nivolumab (N = 359) | Nivolumab-relatlimab (N = 355) | Nivolumab (N = 359) | |
Number of doses received, mean (SD) | 10.2 (8.67) | 10.5 (9.73) | 12.3 (10.71) | 12.8 (12.08) | 14.5 (13.81) | 15.1 (15.29) |
Mean cumulative dose, mg (SD) | 6,555.055 (5549.6498) | 5,043.354 (4674.8113) | 7,886.706 (6852.5687) | 6,143.209 (5800.9772) | 9,251.926 (8,833.4796) | 7,243.599 (7,340.6729) |
Relative dose intensity (%) | ||||||
≥ 110% | 0 | 1 (0.3) | 0 | 0 | 0 | 0 |
90% to < 110% | 309 (87.0) | 304 (84.7) | 309 (87.0) | 300 (83.6) | 312 (87.9) | 302 (84.1) |
70% to < 90% | 41 (11.5) | 45 (12.5) | 41 (11.5) | 53 (14.8) | 38 (10.7) | 51 (14.2) |
50% to < 70% | 5 (1.4) | 9 (2.5) | 5 (1.4) | 6 (1.7) | 5 (1.4) | 6 (1.7) |
< 50% | 0 | 0 | 0 | 0 | 0 | 0 |
Median duration of therapy, months (minimum to maximum) | 5.55 (0.0 to 31.5) | 4.86 (0.0 to 32.2) | 8.31 (0.03 to 38.83) | 6.47 (0.03 to 40.51) | 8.31 (0.03 to 49.87) | 6.47 (0.03 to 50.63) |
BMS = Bristol Myers Squibb; DBL = database lock; ITT = intention-to-treat; ORR = overall response rate; OS = overall survival; PFS = progression-free survival; SD = standard deviation
Sources: BMS (2021), Primary Clinical Study Report;41 BMS (2022), Addendum 01 – Primary Clinical Study Report;61 BMS (2023), Addendum 02 – Primary Clinical Study Report; and sponsor's summary of clinical evidence.62
Table 18: Concomitant Medication Use in 10% or More of the Total Population (All Treated Patients — Updated Descriptive Analysis)
Concomitant medication, n (%) | Nivolumab-relatlimab, n (%) (N = 355) | Nivolumab, n (%) (N = 359) | Total, n (%) (N = 714) |
|---|---|---|---|
Paracetamol | 149 (42.0) | 151 (42.1) | 300 (42.0) |
Prednisone | 126 (35.5) | 73 (20.3) | 199 (27.9) |
Omeprazole | 75 (21.1) | 55 (15.3) | 130 (18.2) |
Ibuprofen | 70 (19.7) | 43 (12.0) | 113 (15.8) |
Levothyroxine sodium | 63 (17.7) | 48 (13.4) | 111 (15.5) |
Acetylsalicylic acid | 59 (16.6) | 42 (11.7) | 101 (14.1) |
Pantoprazole | 54 (15.2) | 39 (10.9) | 93 (13.0) |
Tramadol | 49 (13.8) | 43 (12.0) | 92 (12.9) |
Levothyroxine | 46 (13.0) | 42 (11.7) | 88 (12.3) |
Hydrocortisone | 57 (16.1) | 24 (6.7) | 81 (11.3) |
Tozinameran | 39 (11.0) | 41 (11.4) | 80 (11.2) |
Metformin | 37 (10.4) | 42 (11.7) | 79 (11.1) |
Ondansetron | 41 (11.5) | 31 (8.6) | 72 (10.1) |
BMS = Bristol Myers Squibb.
Notes: October 27, 2022, DBL median follow-up of 25.3 months.
Sources: BMS (2023), Addendum 02 – Primary Clinical Study Report Supplementary Table S.6.30 and sponsor's summary of clinical evidence.62
Table 19: Subsequent Cancer Treatment (All Randomized Patients — Updated Descriptive Analysis)
Subsequent therapy category | Nivolumab-relatlimab (N = 355) | Nivolumab (N = 359) |
|---|---|---|
Patients with any subsequent therapy, n (%)a | 163 (45.9) | 166 (46.2) |
Patients who received subsequent radiotherapy, n (%) | 57 (16.1) | 51 (14.2) |
Patients who received allowed on-treatment radiotherapy, n (%)b | 48 (13.5) | 42 (11.7) |
Patients who received subsequent surgery, n (%) | 28 (7.9) | 34 (9.5) |
Patients who received allowed on-treatment surgery, n (%)b | 24 (6.8) | 28 (7.8) |
Patients who received subsequent systemic therapy, n (%) | 131 (36.9) | 136 (37.9) |
PD-1/CTLA-4 inhibitors | 53 (14.9) | 66 (18.4) |
Avelumab monotherapy | 0 | 1 (0.3) |
BMS 986218 | 1 (0.3) | 1 (0.3) |
Cemiplimab | 0 | 1 (0.3) |
Durvalumab | 0 | 1 (0.3) |
Ipilimumab monotherapy | 15 (4.2) | 23 (6.4) |
Nivolumab-ipilimumab | 18 (5.1) | 29 (8.1) |
Nivolumab monotherapy | 18 (5.1) | 23 (6.4) |
Pembrolizumab monotherapy | 7 (2.0) | 11 (3.1) |
Pembrolizumab-quavonlimab | 1 (0.3) | 0 |
Pembrolizumab–talimogene laherparepvec | 2 (0.6) | 1 (0.3) |
Quavonlimab monotherapy | 2 (0.6) | 0 |
Targeted BRAF and MEK monotherapy and combination therapy | 49 (13.8) | 57 (15.9) |
Binimetinib | 2 (0.6) | 0 |
Binimetinib-encorafenib | 16 (4.5) | 16 (4.5) |
Cobimetinib-vemurafenib | 4 (1.1) | 6 (1.7) |
Dabrafenib | 2 (0.6) | 1 (0.3) |
Dabrafenib mesilate | 1 (0.3) | 0 |
Dabrafenib-trametinib | 29 (8.2) | 42 (11.7) |
Encorafenib | 2 (0.6) | 0 |
Trametinib | 2 (0.6) | 1 (0.3) |
Trametinib dimethyl sulfoxide | 1 (0.3) | 1 (0.3) |
Vemurafenib | 0 | 1 (0.3) |
Otherc | 53 (14.9) | 60 (16.7) |
BMS = Bristol Myers Squibb; BMS 986218 = fixed-dose combination of nivolumab and relatlimab; CTLA-4 = cytotoxic T-lymphocyte-associated protein 4; PD-1 = programmed cell death protein 1.
Note: October 27, 2022, DBL median follow-up of 25.3 months.
aPatients may have received more than 1 type of subsequent therapy. Subsequent therapy was defined as therapy started on or after first dosing date (randomization date if the patient was never treated).
bOn-treatment radiotherapy and surgery were allowed if protocol-defined criteria were met.
cOther anticancer therapies included chemotherapies, cancer vaccines, imatinib, and investigational drugs.
Source: BMS (2023), Addendum 02 – Primary Clinical Study Report Supplementary Table.62
Efficacy
Only those efficacy outcomes presented in the sponsor’s evidence summary and included in CADTH GRADE Summary of Findings table and some supportive outcomes are reported here.
Progression-Free Survival According to Blinded Independent Review Committee (Primary End Point)
The primary final analysis of PFS was conducted after a median follow-up of 13.2 months (March 9, 2021, DBL). At the final analysis, a total of 391 events (180 in the nivolumab-relatlimab FDC group [50.7%] and 211 in the nivolumab group [58.8%]) had occurred. The median PFS was 10.12 months in the nivolumab-relatlimab FDC group versus 4.63 months in the nivolumab monotherapy group in all randomized patients. The HR for nivolumab-relatlimab FDC versus nivolumab) was 0.75 (95% CI, 0.62 to 0.92; P = 0.0055) (Table 20 and Figure 3). The PFS rates at 6 months were 57.2% (95% CI, 51.5% to 62.5%) in the nivolumab-relatlimab FDC group and 44.1% (95% CI, 38.5% to 49.5%) in the nivolumab monotherapy group. The PFS rates at 12 months were 47.7% (95% CI, 41.8% to 53.2%) in the nivolumab-relatlimab FDC group and 36.0% (95% CI, 30.5% to 41.6%) in the nivolumab group. The PFS rates over the fixed-landmark time points up to 48 months are presented in Table 20 and Figure 3. No between-group differences for PFS rates at the fixed time points were provided by the sponsor.
Table 20: Final Analysis of PFS According to BICR (Primary Definition) in RELATIVITY-047 (All Randomized Patients)
Variables | Primary analysis DBL on March 9, 2021, DBL (median follow-up 13.2 months) | |
|---|---|---|
Nivolumab-relatlimab (N = 355) | Nivolumab (N = 359) | |
Final PFS analysis, a median follow-up of 13.2 months (March 9, 2021, DBL) | ||
Events, n (%) | 180 (50.7) | 211 (58.8) |
Median PFS, months (95% CI) | 10.12 (6.37 to 15.74) | 4.63 (3.38 to 5.62) |
HR (95% CI) | 0.75 (0.62 to 0.92) | |
P value | 0.0055 | |
Updated PFS analysis, a median follow-up of 25.3 months (October 27, 2022, DBL) | ||
Events, n (%) | 219 (61.7) | 244 (68.0) |
Median PFS (95% CI), months | 10.18 (6.51 to 14.75) | 4.63 (3.48 to 6.47) |
HR (95% CI) | 0.81 (0.61 to 0.97) | |
P value | NRa | |
6-month PFS, % (95% CI)b on March 9, 2021, DBL (median follow-up of 13.2 months) | 57.2 (51.5 to 62.5) | 44.1 (38.5 to 49.5) |
Updated 6-month PFS, % (95% CI) on October 27, 2022, DBL (median follow-up of 25.3 months) | 57.9 (52.4 to 63.0) | 45.6 (40.2 to 50.8) |
12-month PFS, % (95% CI)a on March 9, 2021, DBL | 47.7 (41.8 to 53.2) | 36.0 (30.5 to 41.6) |
Updated 12-month PFS, % (95% CI) on October 27, 2022, DBL | 48.1 (42.6 to 53.3) | 37.2 (32.1 to 42.4) |
24-month PFS, % (95% CI) on March 9, 2021, DBL | 36.5 (30.0 to 43.1) | 28.7 (23.2 to 34.5) |
Updated 24-month PFS, % (95% CI) on October 27, 2022 DBL | 38.5 (33.1 to 43.9) | 30.6 (25.7 to 35.7) |
36-month PFS, % (95% CI) on October 27, 2022, DBL | 30.7 (25.3 to 36.3) | 27.2 (22.2 to 32.3) |
BMS = Bristol Myers Squibb; CI = confidence interval; DBL = database lock; HR = hazard ratio; PFS = progression-free survival.
Note: March 9, 2021, DBL median follow-up of 13.2 months.
aNo provision of P values for updated results on efficacy (PFS, OS, and ORR) for October 2022 DBL.71
bBased on Kaplan-Meier estimates
Sources: BMS (2021), Primary Clinical Study Report109;41 with additional data provided by the sponsor on September 11, 2023, and sponsor’s summary of clinical evidence.71
As shown in Figure 3, separation of the Kaplan-Meier curves occurred at approximately 3 months and remained separated over the course of follow-up.41 Similar PFS results were observed in the updated descriptive analysis conducted at the October 27, 2022, DBL after a longer median follow-up of 25.3 months. The median PFS with nivolumab-relatlimab FDC was 10.18 months versus 4.63 months with nivolumab monotherapy, representing a prolongation of PFS of 5.6 months (HR 0.81; 95% CI, 0.67 to 0.97) after a longer median follow-up of 25.3 months.62
Nivolumab-relatlimab FDC also resulted in a significant improvement in median PFS compared with nivolumab monotherapy based on the secondary definition of PFS (without censoring for subsequent therapy).41 At the final analysis (March 9, 2021, DBL), PFS results using the secondary definition were similar to those reported for the primary definition (HR = 0.76; 95% CI, 0.63 to 0.93; P = 0.0055).41
Progression-Free Subgroup Analysis
The PFS benefit of nivolumab-relatlimab FDC versus nivolumab monotherapy across key subgroups in exploratory prespecified subgroup analyses was largely consistent with that observed in the overall population (except at the ≥ 10% threshold) at the final PFS analysis (March 9, 2021, DBL median of 13.2 months follow-up) (Figure 4).41 In addition, PFS outcomes across subgroups remained consistent with those observed in the primary analysis after a longer median follow-up of 25.3 months at the October 27, 2022, DBL62 (data not presented in the sponsor’s evidence summary).
Progression-Free Sensitivity Analyses
Sensitivity analyses showed that the primary analysis of PFS according to BICR (median follow-up of 13.2 month) was also robust under the assumption that PFS according to BICR irrespective of subsequent therapy was similar to the primary analysis,41 indicating results were robust to the adjustment for subsequent therapy. The proportional hazards assumption was tested by the addition of a time-dependent covariate, defined by treatment-by-time interaction, to the stratified Cox regression model. A 2-sided Wald chi-square P value of less than 0.1 may indicate a potential nonconstant treatment effect. The P value in this analysis was 0.1497, indicating no evidence of a nonconstant hazard. There was no crossover of treatment effect between strata for PFS according to BICR (using the Gail and Simon test). Results were consistent with those of the primary analysis after adjusting for baseline covariates on multivariate analysis (LDH, age, sex, ECOG PS, and brain metastases). Missing data (2 consecutive missing disease assessment imaging sessions) did not affect the results; results from an unstratified analysis of PFS were consistent with the primary analysis. At the time of the final PFS analysis, the number of relevant protocol deviations was low (< 3% of the randomized patient population) and balanced between groups, with the most common deviations related to the timing of screening imaging and prohibited prior systemic therapy. As there were no on-treatment relevant protocol deviations, protocol deviations did not affect the interpretation of results, and COVID-19 did not affect the overall quality or outcome of study results.
Figure 3: Kaplan-Meier Analysis of PFS According to BICR (Primary Definition) — All Randomized Patients
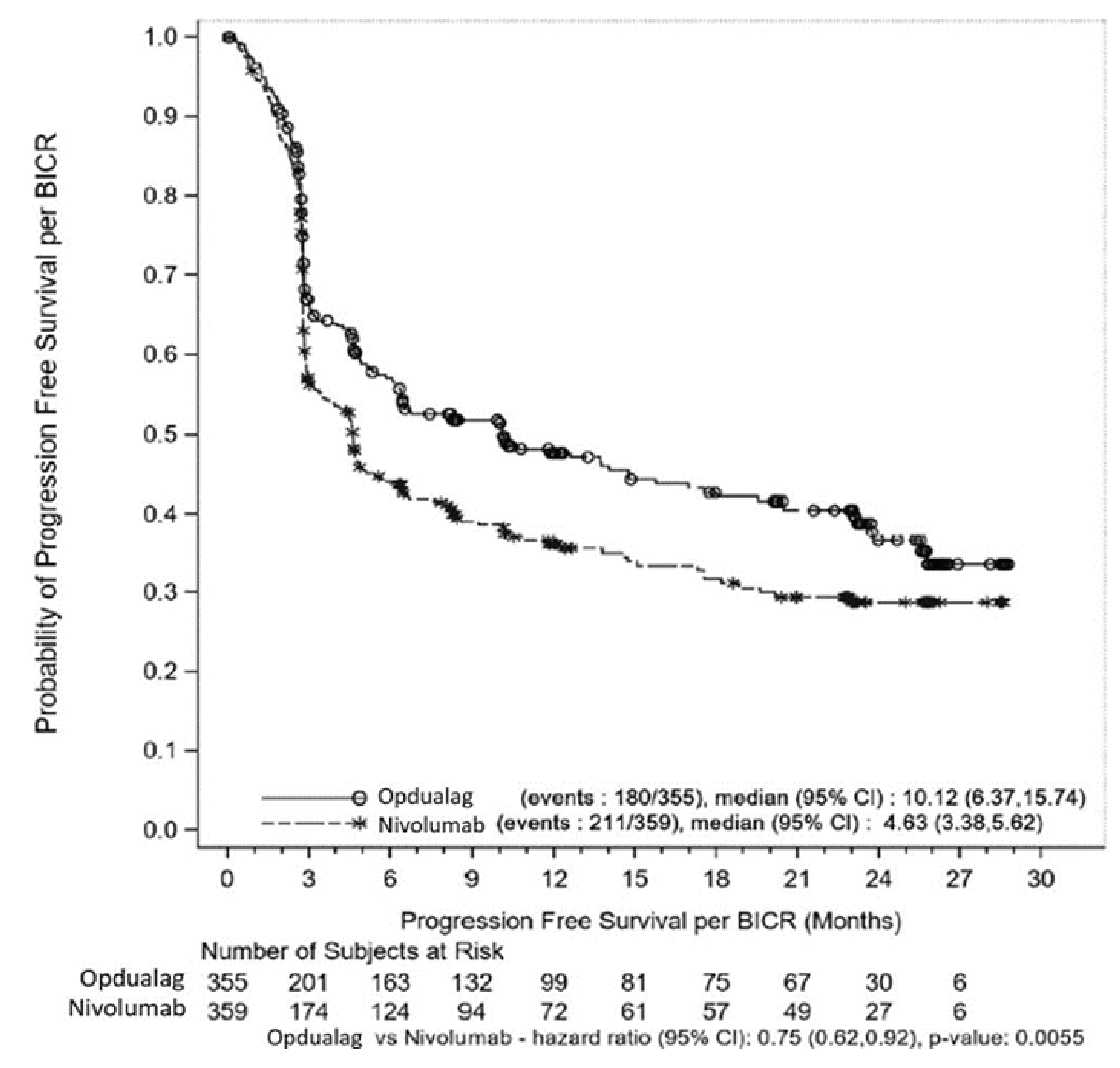
BMS = Bristol Myers Squibb; AJCC = American Joint Committee on Cancer; BICR = blinded independent central review; CI = confidence interval; HR = hazard ratio; PD-L1 = programmed cell death ligand 1; PFS = progression-free survival.
Notes: The statistical model to estimate HR and P values was a stratified Cox proportional hazard model and stratified log-rank test. Stratified by LAG-3 (≥ 1% versus < 1%), BRAF mutation status (mutation-positive versus wild-type), AJCC M stage (M0/M1any[0] versus M1any[1]). PD-L1 was removed from stratification because it led to subgroups with fewer than 10 patients. Symbols represent censored observations. The March 9, 2021, DBL median follow-up was 13.2 months.
Sources: BMS 2021, Primary Clinical Study Report and sponsor’s summary of clinical evidence.41
Figure 4: Forest Plot of Treatment Effect on PFS According to BICR (Primary Definition) in Predefined Subgroups (All Randomized Patients)
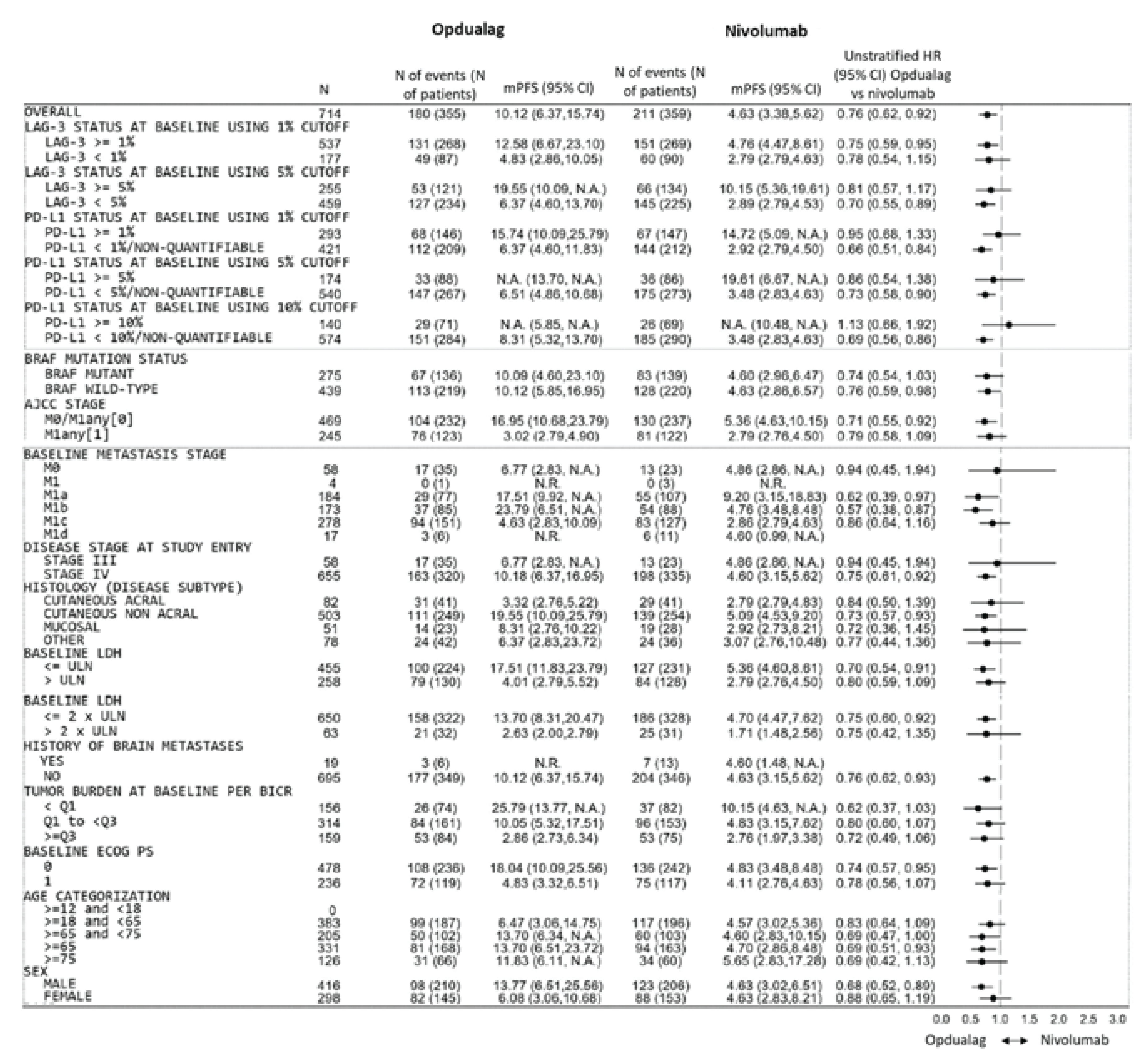
BMS = Bristol Myers Squibb; AJCC = American Joint Committee on Cancer; BICR = blinded independent central review; CI = confidence interval; DBL = database lock; ECOG PS = Eastern Cooperative Oncology Group Performance Status; HR = hazard ratio; IRT = interactive response technology; LAG-3 = lymphocyte activation gene 3; LDH = lactate dehydrogenase; M = metastasis stage; mPFS = median progression-free survival; N.A. = not available; N.R. = not reported; PD-L1 = programmed cell death ligand 1; PFS = progression-free survival; Q = quarter; ULN = upper limit of normal.
Notes: HR and median (displayed as N.R.) are not computed for subset category with less than 10 patients per treatment group. PD-L1 data were from LabCorp LA, LAG-3 and BRAF mutation status data were from IRT, and AJCC M-stage data were from Medidata Rave RTSM Agile Randomization and Trial Supply Management and laboratory values. N.A: not available (i.e., not reached), median or limit of CI not estimable. March 9, 2021, DBL median follow-up of 13.2 months.
Sources: BMS (2021), Primary Clinical Study Report and sponsor’s summary of clinical evidence.41
Overall Survival
The primary final analysis of OS was conducted after a median follow-up of 19.3 months (October 28, 2021, DBL). In the final analysis of OS, a total of 297 events (137 in the nivolumab-relatlimab FDC group [38.6%] and 160 in the nivolumab group [44.6%]) had occurred. The median OS was not reached (95% CI, 34.20 to not reached) in the nivolumab-relatlimab FDC group versus 34.10 months (95% CI, 25.23 to not reached) in the nivolumab monotherapy group in all randomized patients. The HR for nivolumab-relatlimab FDC versus nivolumab was 0.80 (95% CI, 0.64 to 1.01; P = 0.0593) (Table 21 and Figure 5). The OS rates at 6 months were 86.7% (95% CI, 82.75% to 89.8) in the nivolumab-relatlimab FDC group and 84.6% (95% CI, 84.4% to 88.0%) in the nivolumab monotherapy group. The OS rates at 12 months were 77.0% (95% CI, 72.2% to 81.1%) in the nivolumab-relatlimab FDC group and 71.6% (95% CI, 66.6% to 76.0%) in the nivolumab group. The OS rates at 24 months were 63.7% (95% CI, 58.1% to 68.7%) in the nivolumab-relatlimab FDC group and 58.3% (95% CI, 52.7% to 63.4%) in the nivolumab group. The OS rates at 36 months were 55.8% (95% CI, 49.8% to 61.4%) in the nivolumab-relatlimab FDC group and 48.8% (95% CI, 42.7% to 54.7%) in the nivolumab group. The OS rates at 48 months were 51.5% (95% CI, 45.9% to 56.9%) in the nivolumab-relatlimab FDC group and 42.5% (95% CI, 36.4% to 48.5%) in the nivolumab group. The OS rates over the fixed-landmark time points up to 48 months are presented in Table 21 and Figure 5. No provision was made by the sponsor for between-group differences for OS rates at landmark time points.71
Table 21: Final Analysis of Overall Survival in RELATIVITY-047 (All Randomized Patients)
Variables | Nivolumab-relatlimab (N = 355) | Nivolumab (N = 359) |
|---|---|---|
Final OS analysis on October 28, 2021, DBL (median follow-up of 19.3 months) | ||
Events, n (%) | 137 (38.6) | 160 (44.6) |
Median OS, months (95% CI) | NR (34.20 to NR) | 34.10 (25.23 to NR) |
HR (95% CI) | 0.80 (0.64 to 1.01) | |
P value | 0.0593 | |
Updated OS analysis on October 27, 2022, DBL (median follow-up of 25.3 months) | ||
Events, n (%) | 162 (45.6) | 185 (51.5) |
Median OS, months (95% CI) | NR | 33.18 (25.2 to 45.8) |
HR (95% CI) | 0.82 (0.67 to 1.02) | |
P value | NRa | |
6-month OS, % (95% CI)b on October 28, 2021, DBL | 86.7 (82.7 to 89.8) | 84.6 (80.4 to 88.0) |
Updated 6-month OS, % (95% CI) on October 27, 2022, DBL | 86.7 (82.7 to 89.8) | 84.6 (80.4 to 88.0) |
12-month OS, % (95% CI)b on October 28, 2021, DBL | 77.0 (72.2 to 81.1) | 71.6 (66.6 to 76.0) |
Updated 12-month OS, % (95% CI) on October 27, 2022, DBL | 76.7 (72.0 to 80.8) | 71.7 (66.7 to 76.0) |
24-month OS, % (95% CI)b on October 28, 2021, DBL | 63.7 (58.1 to 68.7) | 58.3(52.7 to 63.4) |
Updated 24-month OS, % (95% CI) (October 27, 2022, DBL | 61.8 (56.5 to 66.6) | 58.3 (52.9 to 63.2) |
36-month OS, % (95% CI)b on October 28, 2021, DBL | 55.8 (49.8 to 61.4) | 48.8 (42.7 to 54.7) |
Updated 36-month OS, % (95% CI) on October 27, 2022, DBL | 54.1 (48.6 to 59.3) | 48.4 (42.9 to 53.8) |
48-month OS, % (95% CI) on October 27, 2022, DBL | 51.5 (45.9 to 56.9) | 42.5 (36.4 to 48.5) |
BMS = Bristol Myers Squibb; CI = confidence interval; DBL = database lock; HR = hazard ratio; NR = not reached, median or limit of CI not estimable; OS = overall survival.
Notes: The statistical model to estimate HR and P values was stratified Cox proportional hazard model and stratified log-rank test. The O’Brien-Fleming boundary for statistical significance of the OS final analysis was P < 0.04302 (2-sided) analyzed at 69% power; target HR, 0.75. October 28, 2021, DBL median follow-up of 19.3 months.
aNo provision of P values for updated results on efficacy (progression-free survival, OS and overall response rate for October 2022 DBL.71
bBased on Kaplan-Meier estimates.
Sources: BMS (2022), Addendum 01 – Primary Clinical Study Report;61 additional data provided by the sponsor on September 11, 2023 and sponsor’s summary of clinical evidence.71
At the time of the final OS analysis, 61.4% of patients in the nivolumab-relatlimab FDC group and 55.4% in the nivolumab monotherapy group were censored for OS. The proportion of those censored for being off-study was equally low. The biphasic censoring pattern for the OS Kaplan-Meier analysis (Figure 5) is reflective of the adaptive design of the study with 2 enrolment periods (phases II and III) in addition to a slowdown in enrolment due to the COVID-19 pandemic.
In addition, OS continued to favour nivolumab-relatlimab FDC in the updated descriptive analysis conducted at the October 27, 2022, DBL (median follow-up of 25.3 months).62 After a longer median follow-up of 25.3 months, OS results were consistent with that of the final analysis; median OS was not reached with nivolumab-relatlimab FDC versus 33.18 months with nivolumab monotherapy (HR = 0.82; 95% CI, 0.67 to 1.02)62 (Table 21).
Overall Survival Subgroup Analysis
The trend toward an OS benefit with nivolumab-relatlimab FDC versus nivolumab monotherapy demonstrated in the overall population was consistent across key subgroups in prespecified subgroup analyses at the final analysis of OS (October 28, 2021, DBL; median follow-up of 19.3 months). As observed with the PFS subgroup analyses, there was an OS benefit with nivolumab-relatlimab FDC across the majority of subgroups, which was seen regardless of PD-L1 expression (except at the ≥ 5% and ≥ 10% thresholds), LAG-3 expression, LDH level, AJCC metastatic stage, and BRAF mutation status (HR < 1; Figure 6).61 In addition, OS outcomes across subgroups remained consistent with those observed in the primary analysis after a longer median follow-up of 25.3 months at the October 27, 2022, DBL.62 An OS benefit was seen with nivolumab-relatlimab FDC versus nivolumab monotherapy across the majority of key demographic and clinical subgroups in exploratory unstratified analyses, including PD-L1 less than 1%, LAG-3 less than 1%, elevated LDH, BRAF mutation-positive, and high burden of metastases (M1c) subgroups62 (data not provided in the sponsor’s evidence summary).
Figure 5: Kaplan-Meier Analysis of Overall Survival (All Randomized Patients)
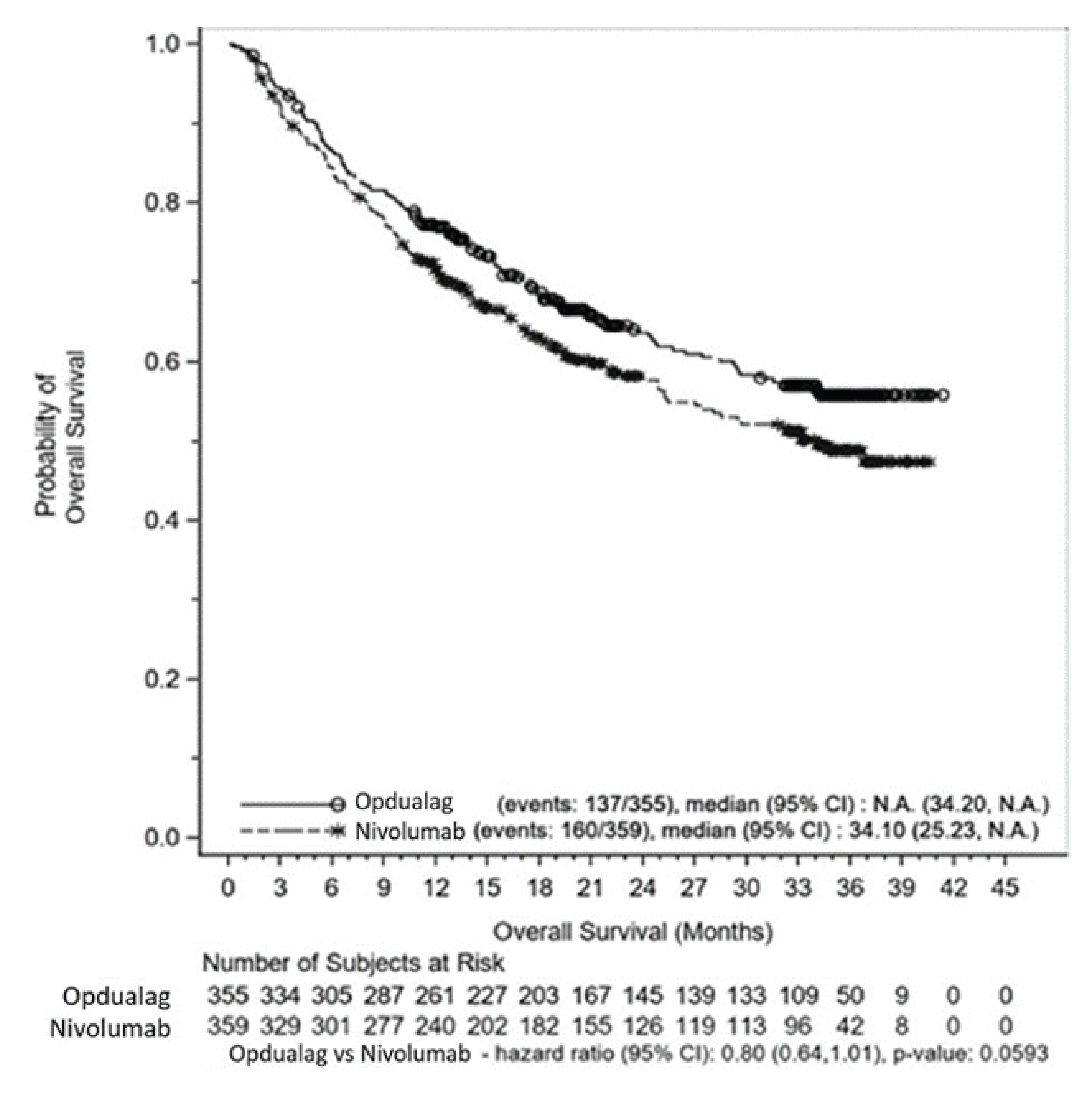
BMS = Bristol Myers Squibb; AJCC = American Joint Committee on Cancer; CI = confidence interval; DBL = database lock; HR = hazard ratio; LAG-3 = lymphocyte activation gene 3; NA = not available (not reached), median or limit of CI not estimable; PD-L1 = programmed cell death ligand 1.
Notes: The statistical model to estimate HR and P values was stratified by a Cox proportional hazards model and stratified log-rank test. Stratified by LAG-3 (≥ 1% versus < 1%), BRAF mutation status (mutation-positive versus wild-type), AJCC M stage (M0/M1any[0] versus M1any[1]). PD-L1 was removed from stratification because it led to subgroups with fewer than 10 patients. Symbols represent censored observations. The O’Brien-Fleming boundary for statistical significance of the OS final analysis was P < 0.04302 (2-sided) analyzed at 69% power; target HR = 0.75. October 28, 2021, DBL median follow-up of 19.3 months.
Sources: BMS (2022), Addendum 01 – Primary Clinical Study Report and sponsor’s summary of clinical evidence.61
Overall Survival Sensitivity Analysis
As the number of OS events due to COVID-19 or suspected COVID-19 infection was low in each group (3 patients in the nivolumab-relatlimab FDC group and 4 in the nivolumab monotherapy group), the prespecified sensitivity analysis that was planned if 10% of OS events were due to COVID-19 was not performed.61
Overall Response Rate
As the OS final analysis did not reach statistical significance, the significance of ORR (according to BICR) could not be formally tested due to its position in the statistical hierarchy (Figure 2).42,61 Based on descriptive analyses, after a median follow-up of 19.3 months (October 28, 2021, DBL). The OR was 1.58 (95% CI, 1.16 to 2.15; descriptive P value = 0.004). A greater proportion of patients in the nivolumab-relatlimab FDC than in the nivolumab monotherapy group achieved a CR and PR and BOR (i.e., CR plus PR) (Table 22).61 The improvement in ORR with nivolumab-relatlimab FDC was observed as early as the first 12-week scan (30.1% versus 21.7%).61
Across prespecified patient subgroups, ORR favoured nivolumab-relatlimab FDC over nivolumab in subgroup analyses (ORR difference > 0%), regardless of PD-L1 and LAG-3 expression, LDH level, AJCC metastatic stage except M0, and BRAF mutation status.61 As the ORR could not be formally tested for significance, a summary forest plot of ORR across all subgroups is not presented. Consistent findings with a between-groups difference of 9.8% (43.7% versus 33.7%) were observed in the updated descriptive analysis after a median follow-up of 25.3 months (Table 22). The RELATIVITY-047 trial was not powered to formally assess outcomes by subgroups. Subgroup analyses were exploratory, not statistically powered, and limited by small sample sizes.
Table 22: ORR According to BICR in RELATIVITY-047 (All Randomized Patients)
Variables | Nivolumab-relatlimab (N = 355) | Nivolumab (N = 359) |
|---|---|---|
Final analysis: a median follow-up of 19.3 months (October 28, 2021, DBL) | ||
ORR,a N of patients who were responders | 153 | 117 |
% of patients with responder, (95% CI) | 43.1 (37.9 to 48.4) | 32.6 (27.8 to 37.7) |
Between-group difference of ORRb,c (nivolumab-relatlimab – nivolumab), % (95% CI) | 10.3 (3.4 to 17.3) | |
P value | NRd | |
OR (95% CI)c,e | 1.58 (1.16 to 2.15) | |
P valuef (descriptive) | 0.004 | |
BOR, n (%) | ||
CR | 58 (16.3) | 51 (14.2) |
PR | 95 (26.8) | 66 (18.4) |
Stable disease | 61 (17.2) | 59 (16.4) |
Non-CR or non–progressive disease | 9 (2.5) | 6 (1.7) |
Progressive disease | ||
N, of patients with progressive disease | 105 | 149 |
% of patients with responder, (95% CI) | 29.6 (24.9 to 34.6) | 41.5 (36.4 to 46.8) |
Unable to determine | 27 (7.6) | 28 (7.8) |
Disease control rate (CR + PR + stable disease), n (%) | ||
N of patients with disease control (%) | 223 of 355 (62.8) | 182 of 359 (50.7) |
Disease control, % (95% CI) | 62.8 (57.6 to 67.9) | 50.7 (45.4 to 56.0) |
Updated ORR analysis (October 27, 2022, DBL) at a median follow-up of 25.3 months | ||
ORR,a N responders | 155 | 121 |
% of patients with events (95% CI) | 43.7 (38.4 to 49.0) | 33.7(28.8 to 39.9) |
Between-group difference (nivolumab-relatlimab – nivolumab) of ORRb,c, % (95% CI) | 9.8 (2.8, 16.8) | |
P value | Not provided | |
OR (95% CI)c,d | 1.54 (1.13 to 2.09) | |
BMS = Bristol Myers Squibb; AJCC = American Joint Committee on Cancer; BICR = blinded independent central review; BOR = best overall response; CI = confidence interval; CR = complete response; DBL = database lock; OR = odds ratio; ORR = overall response rate; PR = partial response; vs. = versus.
Note: October 28, 2021, DBL median follow-up of 19.3 months.
aCR + PR, CI based on the Clopper and Pearson method.
bStrata-adjusted difference in ORR (nivolumab-relatlimab over nivolumab) was based on the Cochran-Mantel-Haenszel method of weighting.
cStratified by LAG-3 (≥ 1% vs. < 1%), BRAF mutation status (mutation-positive vs. wild-type) AJCC M stage (M0/M1any[0] vs. M1any[1]).
dNo provision of P value for the between-group difference of objective response rates and no effect measures for comparison of progressive disease rates in October 2021, DBL.
eStrata-adjusted OR (nivolumab-relatlimab over nivolumab) conducted using the Mantel-Haenszel method.
fTwo-sided P value from stratified Cochran-Mantel-Haenszel test (descriptive P value).
Sources: BMS (2022), Addendum 01 – Primary Clinical Study Report61 with additional data provided by the sponsor on September 11, 2023.71
Complete Response
A total of 16.3% of patients in the nivolumab-relatlimab FDC group and 14.2% of patients in the nivolumab monotherapy group experienced a CR. No formal statistically analysis or any descriptive analysis was carried out to report the between-group difference and 95% CI. No HR (95% CI) was provided (Table 22).
Progressive Disease
A total of 29.6% (95% CI, 24.9% to 34.6%) of patients in the nivolumab-relatlimab FDC group and 41.5% (95% CI, 36.4% to 46.8%) of patients in the nivolumab monotherapy group experienced a CR. No formal statistically analysis or any descriptive analysis was carried out to report the between-group difference and 95% CI. No HR (95% CI) was provided (Table 22).
Figure 6: Forest Plot of Treatment Effect on Overall Survival in Predefined Subgroups (All Randomized Patients)

BMS = Bristol Myers Squibb; AJCC = American Joint Committee on Cancer; BICR = blinded independent central review; CI = confidence interval; DBL = database lock; ECOG PS = Eastern Cooperative Oncology Group Performance Status; HR = hazard ratio; IRT = interactive response technology; LAG-3 = lymphocyte activation gene 3; LDH = lactate dehydrogenase; mOS = median overall survival; N.A. = not available, median or limit of CI not estimable; N.R. = not reported; OS = overall survival; PD-L1 = programmed cell death ligand 1; ULN = upper limit of normal.
Notes: HR and median (displayed as N.R.) are not computed for subset category with fewer than 10 patients per treatment group. October 28, 2021, DBL median follow-up of 19.3 months.
Sources: BMS (2022), Addendum 01 – Primary Clinical Study Report and sponsor's summary of clinical evidence.61
Duration of Responses and Time to Response
The DoR and TTR were assessed as exploratory end points. Results for median DoR and TTR are based on the updated descriptive analysis conducted at the October 27, 2022, DBL (median follow-up of 25.3 months).62 For all confirmed responders, median DoR was not reached in either treatment group. Over the duration of follow-up (median of 25.3 months), the proportion of responders at fixed-landmark time points was comparable between the nivolumab-relatlimab FDC and nivolumab groups (Table 23).62 The HR (nivolumab-relatlimab FDC versus nivolumab) was 1.07 (95% CI, 0.71 to 1.63). The median TTR (according to BICR) was the same in both groups (2.79 months each). No provision of effect measures for comparison of treatments with respect to TTR, and no difference of DoR rates at specific time points were provided by the sponsor.
Table 23: TTR and DoR According to BICR in RELATIVITY-047 (All Randomized Patients)
Variables | Nivolumab-relatlimab (N = 155) | Nivolumab (N = 121) |
|---|---|---|
TRR | ||
TTR (October 28, 2021, DBL; median follow-up of 19.3 months) | ||
Median TTR, months (minimum to maximum) | 2.79 (1.2 to 12.2) | 2.79 (1.7 to 20.1) |
Mean TTR, months (SD)a | 3.52 (1.94) | 3.72 (2.2) |
TTR (October 27, 2022; DBL median follow-up of 25.3 months) | ||
Median TTR, months (minimum to maximum) | 2.79 (1.2 to 20.1) | 2.79 (1.7 to 42.3) |
Mean TTR, months (SD)a | 3.79 | 4.36 |
DOR (October 27, 2022; DBL median follow-up of 25.3 months) | ||
Median DoR, months | ||
N events of N responders (%) | 52 of 155 (33.5) | 38 of 121 (31.4) |
Median,b months (95% CI) | NA (39.36 to NA) | NA (39.82 to NA) |
Mininum to maximum, monthsc | 1.9+ to 47.9+ | 1.9+ to 45.5+ |
Hazard ratio (95% CI) | 1.07 (0.71 to 1.63) | |
Proportion of patients with DoR of at least 3 months (95% CI)c,a | ||
At 3 months | 0.99 (0.95 to 1.00) | 0.99 (0.94 to 1.00) |
At 1 year | 0.80 (0.73 to 0.86) | 0.83 (0.75 to 0.89) |
At 2 years | 0.65 (0.57 to 0.73) | 0.69 (0.59 to 0.77) |
At 3 years | 0.60 (0.51 to 0.69) | 0.63 (0.52 to 0.73 |
BMS = Bristol Myers Squibb; BICR = blinded independent central review; CI = confidence interval; DBL = database lock; DoR = duration of response; NA = not available (i.e., not reached; median or limit of CI not estimable); TTR = time to response.
Note: October 27, 2022, DBL median follow-up of 25.3 months.
aNo provision of effect measures for comparison of treatments with respect to TTR, and no difference of DoR rates at specific fixed-landmark time points.71
a“+” symbol indicates a censored value.
bMedian computed using the Kaplan-Meier method.
cBased on Kaplan-Meier estimates of the duration of response.
Sources: BMS (2023), Addendum 02 – Primary Clinical Study Report,62 with additional data provided by the sponsor on September 11, 2023.71
Health-Related Quality of Life
All HRQoL results, including FACT-M and EQ-5D-3L data from the RELATIVITY-047 trial were analyzed as exploratory end points and based on the October 28, 2021, DBL after a median follow-up of 19.3 months (Figure 8, Figure 9, and Table 24). HRQoL analyses were not updated at the subsequent DBL conducted on October 27, 2022 (median of 25.3 months follow-up). The findings of FACT-M, EQ-5D-3L utility index score and EQ VAS after a median follow-up of 19.3 months and their fixed-landmark time points (12 months and 24 months) are presented in Figure 8, Figure 9, and Table 24.
Functional Assessment of Cancer Therapy–Melanoma
In all randomized patients, HRQoL scores at baseline were similar to those of the US general population for the FACT-M (total and subscales) measures.41 Scores were also similar between treatment groups across patient-reported outcome measures.41 After a median follow-up of 19.3 months (October 28, 2021, DBL), the FACT-M total score in the nivolumab-relatlimab FDC group remained generally stable during the treatment period61 (Figure 8 and Table 24). HRQoL also remained stable in the nivolumab-relatlimab FDC group at time points beyond 128 weeks.61
Table 24: Summary of FACT-M, EQ-5D-3L, and EQ VAS Results From RELATIVITY-047
Outcomes | RELATIVITY −047 | |
|---|---|---|
Nivolumab-relatlimab (N = 355) | Nivolumab (N = 359) | |
Change from baseline in FACT-M | ||
Cut-off: October 28, 2021 (median follow-up of 19.3 months) | ||
Baseline | ||
Number of patients | 313 | 316 |
Baseline mean (95% CI) | 135.56 (132.89 to 138.23) | 136.40 (133.88 to 138.92) |
Median follow-up of 19.3 months | ||
Number of patients | 68 | 83 |
At median follow-up of 19.3 months, mean (95% CI) | 147.55 (142.59 to 152.51) | 148.12 (143.6 to 152.64) |
LSM change from baseline to median at 19.3 months (95% CI) | 1.756 (−1.763 to 5.275) | 3.563 (0.140 to 6.986) |
Between-group absolute mean difference in LSM change from baseline (95% CI) | −1.807 (−6.561 to 2.947) | |
P value | 0.456 | |
At 12 months | ||
Baseline | ||
Number of patients | 132 | 115 |
Baseline mean (95% CI) | 141.07 (137.19 to 144.95) | 138.74 (134.52 to 142.96) |
At 12 months | ||
Number patients) | 132 | 119 |
At 12 months, mean (95% CI) | 144.14 (140.52 to 147.76) | 144.20 (139.92 to 148.48) |
LSM change from baseline to 12 months, (95% CI) | 0.795 (−1.861 to 3.451) | 1.405 (−1.385 to 4.195) |
Between-group absolute mean difference in LSM change from baseline (95% CI) | −0.610 (−4.279 to 3.059) | |
P value | 0.744 | |
At 24 months | ||
Baseline | ||
Number of patients | 37 | 42 |
Baseline mean (95% CI) | 142.20 (135.94 to 148.46) | 145.87 (140.57 to 151.17) |
At 24 months | ||
Number of patients | 37 | 46 |
At 24 months, mean (95% CI) | 140.89 (133.33 to 148.45) | 152.97 (148.27 to 157.67) |
LSM change from baseline to 24 months (95% CI) | 0.604 (−3.889 to 5.098) | 4.242 (0.000 to 8.485) |
Between-group absolute mean difference in LSM change from baseline (95% CI) | −3.638 (−9.701 to 2.425) | |
P value | 0.239 | |
Change from baseline in EQ-5D-3L utility index scores | ||
Cut-off: October 21, 2021 (median follow-up of 19.3 months) | ||
Baseline | ||
Number of patients | 315 | 323 |
Baseline mean (95% CI) | 0.779 (0.752 to 0.806) | 0.779 (0.752 to 0.806) |
Median follow-up of 19.3 months (time points, months, or weeks) | ||
Number of patients | 67 | 83 |
At median 19.3 months, mean (95% CI) | 0.886 (0.842 to 0.93) | 0.898 (0.854 to 0.942) |
LSM change from baseline to median at 19.3 months (95% CI) | 0.009 (−0.036 to 0.053) | 0.002 (−0.040 to 0.043) |
Between-group absolute mean difference in LSM change from baseline (95% CI) | 0.007 (−0.052 to 0.066) | |
P value | 0.816 | |
At 12 months | ||
Baseline | ||
Number of patients | 136 | 121 |
Baseline mean (95% CI) | 0.830 (0.796 to 0.864) | 0.866 (0.835 to 0.897) |
At 12 months | ||
Number of patients | 136 | 121 |
At 12 months, mean (95% CI) | 0.847 (0.815 to 0.879) | 0.868 (0.833 to 0.903) |
LSM change from baseline to 12 months (95% CI) | −0.009 (−0.041 to 0.024) | −0.014 (−0.049 to 0.020) |
Between-group absolute mean difference in LSM change from baseline (95% CI) | 0.006 (−0.040 to 0.052) | |
P value | 0.798 | |
At 24 months | ||
Baseline | ||
Number of patients | 38 | 48 |
Baseline mean (95% CI) | 0.842 (0.787 to 0.897) | 0.906 (0.875 to 0.937) |
At 24 months | ||
Number of patients | 38 | 48 |
At 24 months, mean (95% CI) | 0.842 (0.776 to 0.908) | 0.944 (0.913 to 0.975) |
LSM change from baseline to 24 months (95% CI) | −0.012 (−0.070 to 0.046) | 0.030 (−0.022 to 0.082) |
Between-group absolute mean difference in LSM change from baseline (95% CI) | −0.042 (−0.119 to 0.035) | |
P value | 0.282 | |
Change from baseline in change from baseline in EQ VAS | ||
Cut-off: October 28, 2021 (median follow-up of 19.3 months) | ||
Baseline | ||
Number of patients | 315 | 323 |
Baseline mean (95% CI) | 77.67 (75.54 to 79.8) | 78.34 (76.22 to 80.46) |
Median follow-up of 19.3 months (time points, months, or weeks) | ||
Number of patients | 67 | 83 |
At median 19.3 months, mean (95% CI) | 85.94 (82.55 to 89.33) | 85.16 (81.35 to 88.97) |
LSM change from baseline to median 19. 3 months (95% CI) | 2.840 (−0.454 to 6.135) | 2.084 (−1.007 to 5.175) |
Between-group absolute mean difference in LSM change from baseline (95% CI) | 0.757 (−3.651 to 5.164) | |
P value | 0.736 | |
At 12 months | ||
Baseline | ||
Number of patients | 135 | 121 |
Baseline mean (95% CI) | 79.77 (76.6 to 82.94) | 81.86 (78.56 to 85.16) |
At 12 months | ||
Number of patients | 135 | 121 |
At 12 months, mean (95% CI) | 83.95 (81.3 to 86.6) | 84.00 (81.3 to 86.7) |
LSM change from baseline to 12 months (95% CI) | 1.958 (−0.477 to 4.393) | 1.383 (−1.154 to 3.919) |
Between-group absolute mean difference in LSM change from baseline (95% CI) | 0.575 (−2.813 to 3.963) | |
P value | 0.739 | |
At 24 months | ||
Baseline | ||
Number of patients | 38 | 48 |
Baseline mean (95% CI) | 76.11 (68.14 to 84.08) | 81.17 (75.06 to 87.28) |
At 24 months | ||
Number of patients | 38 | 48 |
At 24 months, mean (95% CI) | 81.87 (75.56 to 88.18) | 88.40 (85.31 to 91.49) |
LSM change from baseline to 24 months, (95% CI) | 3.388 (−0.876 to 7.652) | 5.482 (1.641 to 9.323) |
Between-group absolute mean difference in LSM change from baseline (95% CI) | −2.094 (−7.752 to 3.565) | |
P value | 0.468 | |
CI = confidence interval; EQ VAS = EQ visual analogue scale; FACT-M = Functional Assessment of Cancer Therapy–Melanoma; LSM = least squares mean.
Sources: Clinical Study Report41 and additional data provided by the sponsor on September 11, 2023.71
Figure 7: Kaplan-Meier Analysis of DoR According to BICR (Randomized Patients, All Responders)
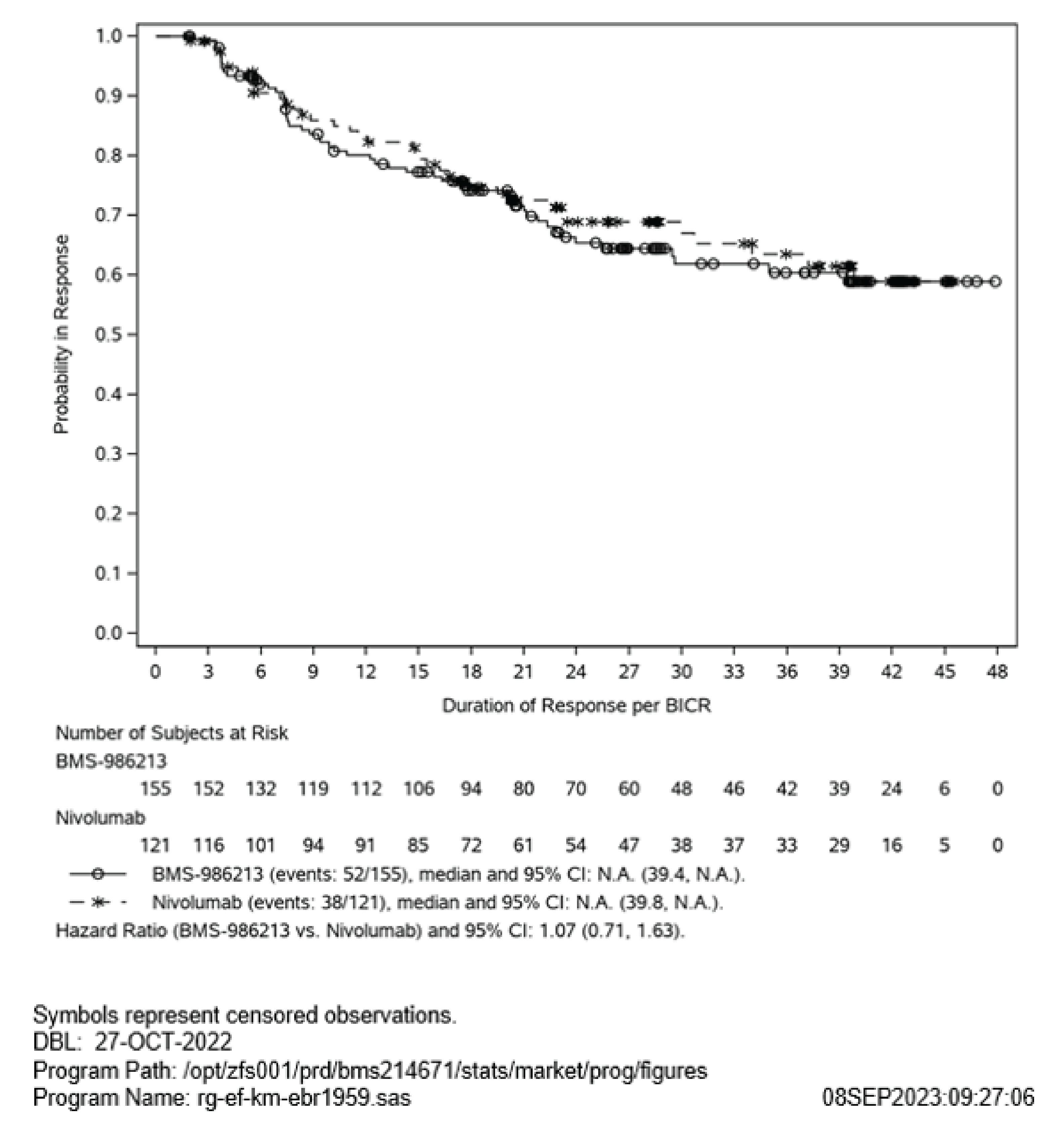
BICR = blinded independent review committee; BMS-72213 = fixed-dose combination of relatlimab and nivolumab; CI = confidence interval; N.A. = not available.
Figure 8: Change From Baseline in FACT-M Total Scores (All Randomized Patients)
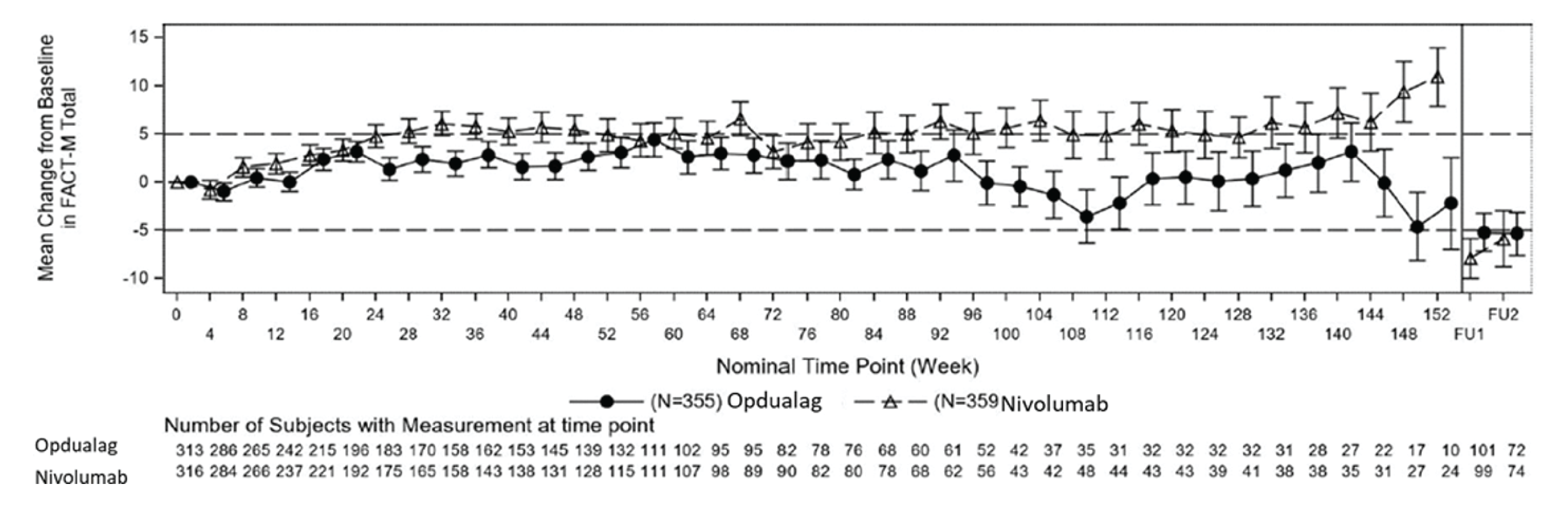
BMS = Bristol Myers Squibb; DBL = database lock; FACT-M = Functional Assessment of Cancer Therapy–Melanoma; FU1 = follow-up 1; FU2 = follow-up 2; HRQoL = health-related quality of life; MID = minimal important difference.
Notes: Error bars represent standard error for the mean. The horizontal reference line indicates MID. Only time points where data were available for 10 or more patients in each treatment group are plotted. All randomized patients must have had an assessment at baseline and at least 1 postbaseline. Higher scores indicate better HRQoL. October 28, 2021, DBL median follow-up of 19.3 months.
Source: BMS (2022), Addendum 01 – Primary Clinical Study Report Supplementary Figure S.10.5.3.61
EQ-5D-3L Utility Index Scores and VAS
In all randomized patients, HRQoL scores at baseline were similar to those of the US general population for the EQ-5D-3L (utility index and EQ VAS).41 Scores were also similar between treatment groups across patient-report outcome measures.41 EQ-5D-3L utility index scores were maintained from baseline in the nivolumab-relatlimab FDC group, as well as in the nivolumab group. EQ VAS scores were likewise maintained from baseline with nivolumab-relatlimab FDC and improved at later visits, after week 124 (Figure 9 and Table 24).
Even after completing treatment, health utility index and EQ VAS scores were maintained in the nivolumab-relatlimab FDC group during follow-up and survival visits.61 A similar trend was observed in the nivolumab monotherapy group.
Figure 9: EQ-5D-3L Utility Index Score and EQ VAS (All Randomized Patients)
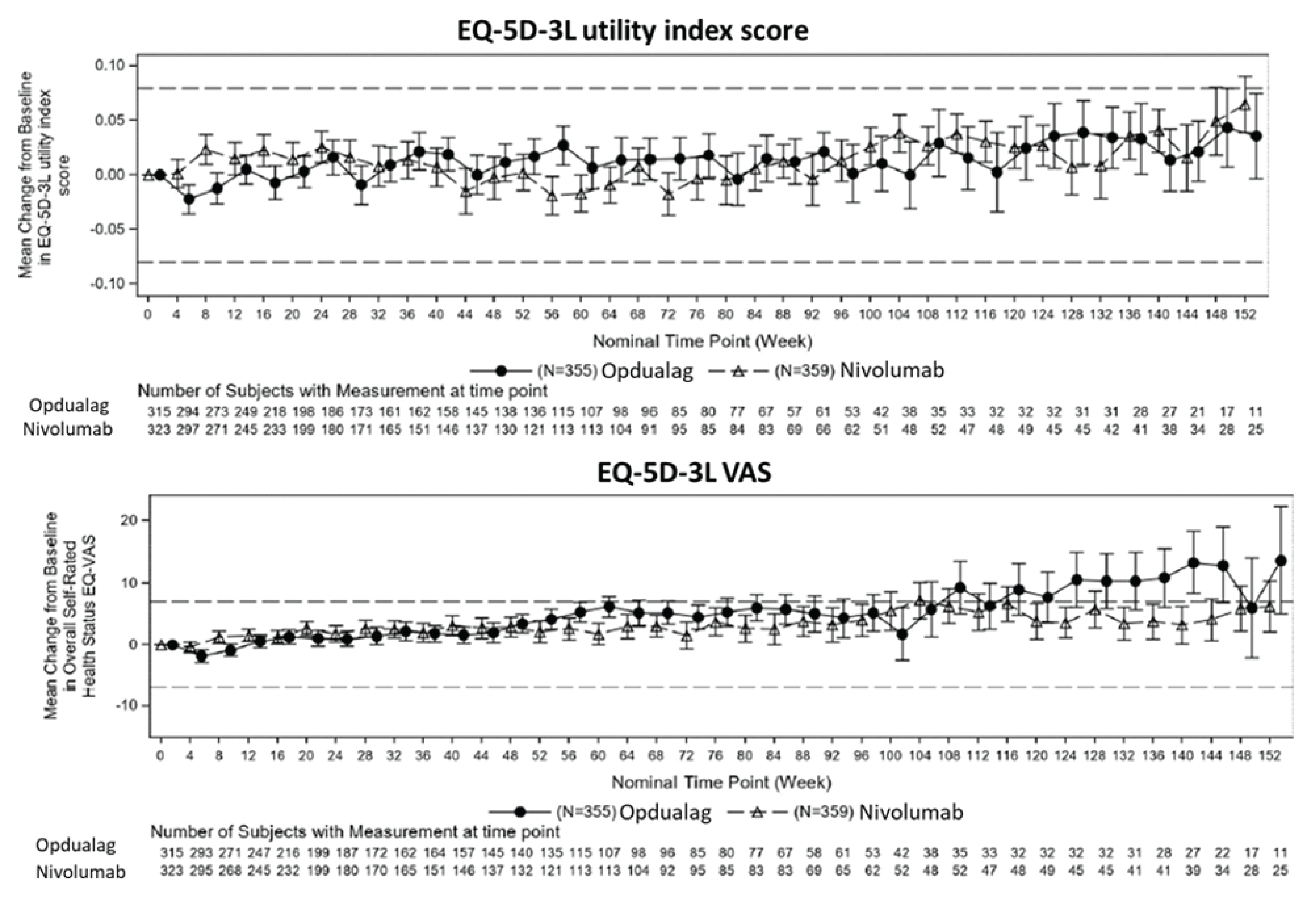
BMS = Bristol Myers Squibb; EQ VAS = EQ visual analogue scale; HRQoL = health-related quality of life; MID = minimal important difference.
Notes: Error bars represent standard error for the mean. The horizontal reference line indicates the MID. Only time points for which data are available for 10 or more patients in each treatment group are plotted. All randomized patients must have had an assessment at baseline and at least 1 postbaseline. Higher scores indicate better HRQoL for both measures. DBL: October 28, 2021 (median follow-up of 19.3 months).
Source: BMS (2022), Addendum 01 – Primary Clinical Study Report Supplementary Figures S.10.5.1 and S.10.5.2.61
Harms
Only harms identified in the sponsor’s evidence summary review protocol are reported here. Table 25 provides detailed harms data.
Table 25: Summary of Safety (All Treated Patients)
AEs | Nivolumab-relatlimab (N = 355) | Nivolumab (N = 359) |
|---|---|---|
Total all-cause AEs, n (%) | 352 (99.2) | 344 (95.8) |
Most common (> 20%) any-grade AEs, n (%) | ||
Fatigue | 109 (30.7) | 75 (20.9) |
Diarrhea | 99 (27.9) | 70 (19.5) |
Pruritus | 98 (27.6) | 65 (18.1) |
Arthralgia | 93 (26.2) | 63 (17.5) |
Nausea | 73 (20.6) | 55 (15.3) |
Headache | 71 (20.0) | 44 (12.3) |
Total all-cause SAEs | 138 (38.9) | 119 (33.1) |
Common (> 1%) SAE, n (%) | ||
Malignant neoplasm progression | 14 (3.9) | 20 (5.6) |
Diarrhea | 5 (1.4) | 2 (0.6) |
Anemia | 5 (1.4) | 1 (0.3) |
Adrenal insufficiency | 5 (1.4) | 0 |
Pneumonia | 4 (1.1) | 3 (0.8) |
Urinary tract infection | 4 (1.1) | 3 (0.8) |
Back pain | 4 (1.1) | 2 (0.6) |
Colitis | 4 (1.1) | 1 (0.3) |
Myocarditis | 4 (1.1) | 1 (0.3) |
Squamous cell carcinoma | 1 (0.3) | 4 (1.1) |
Pyrexia | 3 (0.8) | 4 (1.1) |
Total all-cause AEs leading to discontinuation | 82 (23.1) | 57 (15.9) |
Patients (> 1%) who discontinued treatment due to AEs, n (%) | ||
Malignant neoplasm progression | 6 (1.7) | 10 (2.8) |
Colitis | 4 (1.1) | 1 (0.3) |
Diarrhea | 4 (1.1) | 0 |
Pneumonitis | 5 (1.4) | 1 (0.3) |
Myocarditis | 5 (1.4) | 0 |
Deaths, n (%) | ||
Patients who died | 162 (45.6) | 185 (51.5) |
Primary reason for death | ||
Disease | 126 (35.5) | 154 (42.9) |
Study drug toxicity | 4 (1.1) | 2 (0.6) |
Unknown | 7 (2.0) | 10 (2.8) |
Othera | 25 (7.0) | 19 (5.3) |
AESIs, n (%) | ||
Total all-cause endocrine IMAEs within 100 days of last dose (> 5%) with or without immune-modulating medication | ||
Hypothyroidism and/or thyroiditis | 69 (19.4) | 54 (15.0) |
Hypothyroidism | 65 (18.3) | 51 (14.2) |
Adrenal insufficiency | 20 (5.6) | 4 (1.1) |
Hyperthyroidism | 24 (6.8) | 26 (7.2) |
Total all-cause OESIs within 100 days of last dose (> 1.5%) with or without immune-modulating medication | ||
Troponin event | 45 (12.7) | 36 (10.0) |
Uveitis | 6 (1.7) | 5 (1.4) |
Myocarditis | 6 (1.7) | 2 (0.6) |
Pancreatitis | 5 (1.4) | 6 (1.7) |
AE = adverse event; AESI = adverse event of special interest; BMS-986213 = fixed-dose combination of nivolumab and relatlimab; BMS = Bristol Myers Squibb; DBL = database lock; IMAE = immune-mediated adverse event; OESI = other event of special interest; SAE = serious adverse event.
Note: Safety data are based on the October 27, 2022, DBL after a median follow-up of 23.5 months, using the Medical Dictionary for Regulatory Activities version 25.0 and National Cancer Institute Common Terminology Criteria for Adverse Events version 5.0.
aThere were 3 additional deaths with a primary reason of “other” (i.e., deaths not due to study drug toxicity, disease progression, or unknown reasons) since the October 28, 2021, DBL (Addendum 01 Clinical Study Report 2): 2 in the BMS-986213 arm (verbatim terms: septic shock and lung infection, sepsis) and 1 in the nivolumab monotherapy group (verbatim terms: severe ketoacidosis and diabetes mellitus 2).
Sources: BMS (2023), Addendum 02 – Primary Clinical Study Report62 and BMS (2023), Addendum 02 – Primary Clinical Study Report supplementary tables.62
Adverse Events
After a median follow-up of 25.3 months, 99.2% of patients in the nivolumab-relatlimab FDC group and 95.8% in the nivolumab monotherapy group experienced at least 1 AE (Table 25). The most common AEs (≥ 20% in either of the groups) were fatigue (30.7% in the nivolumab-relatlimab FDC and 20.9% in the nivolumab group), diarrhea (27.9% and 19.5%, respectively), pruritus (27.6% and 18.1%, respectively), arthralgia (26.2% and 17.5%), nausea (20.6% and 15.3%), and headache (20.0% and 12.3%).
The overall rate of grade 3 and 4 AEs was higher in the nivolumab-relatlimab FDC than the nivolumab monotherapy group (44.8% versus 36.8%, respectively)62 (Table 26).
Table 26: Summary of All-Cause AEs by GRADE in RELATIVITY-047 (20% or Greater — All Treated Patients)
AE | Nivolumab-relatlimab (N = 355) | Nivolumab (N = 359) | ||
|---|---|---|---|---|
Any grade | Grade 3 or 4 | Any grade | Grade 3 or 4 | |
Total all-cause AEs, n (%) | 352 (99.2) | 159 (44.8) | 344 (95.8) | 132 (36.8) |
Fatigue | 109 (30.7) | 6 (1.7) | 75 (20.9) | 2 (0.6) |
Diarrhea | 99 (27.9) | 8 (2.3) | 70 (19.5) | 5 (1.4) |
Pruritus | 98 (27.6) | 0 | 65 (18.1) | 3 (0.8) |
Arthralgia | 93 (26.2) | 5 (1.4) | 63 (17.5) | 1 (0.3) |
Nausea | 73 (20.6) | 2 (0.6) | 55 (15.3) | 0 |
Headache | 71 (20.0) | 1 (0.3) | 44 (12.3) | 1 (0.3) |
AE = adverse event; BMS = Bristol Myers Squibb.
Notes: Safety data presented in the table are based on the Clinical Study Report 02 Addendum to the Primary Clinical Study Report at the October 27, 2022, DBL (follow-up of 25.3 months). The 10% or greater AE threshold used in the table is from that used in the product monograph (≥ 1% threshold), which also presents AE data for a slightly longer follow-up period compare with the Clinical Study Report. AE data at ≥ 1% or greater threshold is not reported in the Clinical Study Report; the higher 10% or greater threshold is presented based on data reported in the Clinical Study Report and for conciseness. Further details on the incidence of treatment-emergent AEs at a threshold of 5% or greater are provided in Table S.6.1.31.1 in the list of supplementary tables of the Clinical Study Report Addendum 02 of the Primary Clinical Study Report (October 27, 2022, DBL).62
Source: BMS (2023), Addendum 02 – Primary Clinical Study Report.62
Serious Adverse Events
After a longer median follow-up of 25.3 months, 38.9% of patients in the nivolumab-relatlimab FDC group and 33.1% in the nivolumab monotherapy group experienced at least 1 SAE (Table 25). The most common SAEs (≥ 1.1% in either of the groups) were malignant neoplasm progression (3.9% in the nivolumab-relatlimab FDC and 5.6% in the nivolumab group), diarrhea (1.4% and 0.6%, respectively), anemia (1.4% and 0.3%), and adrenal insufficiency (1.4% and 0%) (Table 25). The overall rate of grade 3 and 4 SAEs was higher in the nivolumab-relatlimab FDC group compared with the nivolumab monotherapy group (29.9% versus 21.7%)62 (Table 27).
Table 27: Summary of SAEs in RELATIVITY-047 (in More Than 1.1% of Patients — All Treated Patients)
SAE | Nivolumab-relatlimab (N = 355) | Nivolumab (N = 359) | ||
|---|---|---|---|---|
Any grade | Grade 3 or 4 | Any grade | Grade 3 or 4 | |
Total all-cause SAEs, n (%) | 138 (38.9) | 106 (29.9) | 119 (33.1) | 78 (21.7) |
Malignant neoplasm progression | 14 (3.9) | 13 (3.7) | 20 (5.6) | 13 (3.6) |
Adrenal insufficiency | 5 (1.4) | 5 (1.4) | 0 | 0 |
Anemia | 5 (1.4) | 3 (0.8) | 1 (0.3) | 1 (0.3) |
Diarrhea | 5 (1.4) | 3 (0.8) | 2 (0.6) | 2 (0.6) |
BMS = Bristol Myers Squibb; SAE = serious adverse event.
Note: October 27, 2022, median follow-up of 25.3 months.
Source: BMS (2023), Addendum 02 – Primary Clinical Study Report Supplementary Table S.6.3.1.2.1.62
Mortality
As of the October 27, 2022, DBL, after a median follow-up of 25.3 months, 45.6% of patients (N = 162) had died in the nivolumab-relatlimab FDC group and 51.5% (N = 185) had died in the nivolumab monotherapy group. Disease progression was the most common cause of death in both treatment groups62 (Table 28).
Deaths due to AEs occurred in 4 patients in the nivolumab-relatlimab group (1.1%) and 2 patients in the nivolumab monotherapy group (0.6%) (Table 28).
Table 28: Summary of Deaths in RELATIVITY-047 (All Treated Patients)
Adverse events | Nivolumab-relatlimab (N = 355) | Nivolumab (N = 359) |
|---|---|---|
As of the October 27, 2022, DBL (median follow-up of 25.3 months) | ||
Number of patients who died, n (%) | 162 (45.6) | 185 (51.5) |
Primary reason for death, n (%) | ||
Disease progression | 126 (35.5) | 154 (42.9) |
Study drug toxicity | 4 (1.1) | 2 (0.6) |
Unknown | 7 (2.0) | 10 (2.8) |
Other | 25 (7.0) | 19 (5.3) |
Number of patients who died within 30 days of last dose, n (%) | 11 (3.1) | 20 (5.6) |
Primary reason for death, n (%) | ||
Disease progression | 1 (0.3) | 9 (2.5) |
Study drug toxicity | 1 (0.3) | 1 (0.3) |
Unknown | 1 (0.3) | 2 (0.6) |
Other | 8 (2.3) | 8 (2.2) |
Number of patients who died within 100 days of last dose, n (%) | 55 (15.5) | 67 (18.7) |
Primary reason for death, n (%) | ||
Disease progression | 34 (9.6) | 47 (13.1) |
Study drug toxicity | 3 (0.8) | 1 (0.3) |
Unknown | 1 (0.3) | 3 (0.8) |
Other | 17 (4.8) | 16 (4.5) |
BMS = Bristol Myers Squibb; DBL = database lock.
Note: October 27, 2022, median follow-up of 25.3 months.
Source: BMS (2023), Addendum 02 – Primary Clinical Study Report.62
Withdrawals due to Adverse Events
After a median follow-up of 25.3 months, AEs of any grade leading to study drug discontinuation occurred in 23.1% of patients in the nivolumab-relatlimab FDC group and in 15.9% of the nivolumab monotherapy group. Grade 3 or 4 AEs leading to discontinuation occurred in 13.0% of patients in the nivolumab-relatlimab FDC group and 8.4% in the nivolumab monotherapy group (Table 29). Except for malignant neoplasm progression, grade 3 or 4 AEs resulting in treatment discontinuation occurred in less than 1% of patients in both groups.62
The most frequently reported (> 1% patients) AEs leading to treatment discontinuation in the nivolumab-relatlimab FDC group were malignant neoplasm progression (1.7%), pneumonitis (1.4%), myocarditis (1.4%), colitis (1.1%), and diarrhea (1.1%).62 In the nivolumab monotherapy group, the most common AE leading to discontinuation was malignant neoplasm progression (2.8%) (Table 29).62
Dose reductions were not permitted with either study treatment. In both treatment groups, the majority (> 93%) of treated patients received all doses of the study drug without an infusion interruption. Overall, treatment interruption occurred in a minority of patients (approximately 6% in both groups)62 (Table 29).
Table 29: Adverse Events Leading to Treatment Discontinuation or Interruption in RELATIVITY-047 (All Treated Patients)
Adverse events | Nivolumab-relatlimab (N = 355) | Nivolumab (N = 359) | ||
|---|---|---|---|---|
Any grade | Grade 3 or 4 | Any grade | Grade 3 or 4 | |
Adverse events leading to treatment discontinuation ≥ 1% of patients, n (%) | ||||
All-cause adverse events | 82 (23.1) | 46 (13.0) | 57 (15.9) | 30 (8.4) |
Malignant neoplasm progression | 6 (1.7) | 5 (1.4) | 10 (2.8) | 8 (2.2) |
Pneumonitis | 5 (1.4) | 2 (0.6) | 1 (0.3) | 1 (0.3) |
Myocarditis | 5 (1.4) | 2 (0.6) | 0 | 0 |
Colitis | 4 (1.1) | 2 (0.6) | 1 (0.3) | 0 |
Diarrhea | 4 (1.1) | 1 (0.3) | 0 | 0 |
Treatment interruption, n (%)a | ||||
Patients with ≥ 1 infusion interrupted | 24 (6.8) | 20 (5.6) | ||
Total number of infusions interrupted of total number of doses received | 32 of 5,135 (0.6) | 24 of 5,421 (0.4) | ||
Number of infusions interruption for each reason, n (%) | ||||
Hypersensitivity issues | 23 (71.9) | 15 (62.5) | ||
Infusion administration issues | 3 (9.4) | 5 (20.8) | ||
Other | 6 (18.8) | 4 (16.7) | ||
BMS = Bristol Myers Squibb.
Note: Database lock was on October 27, 2022; median follow-up of 25.3 months.
aPercentages are calculated based on the total number of doses interrupted by group.
Sources: BMS (2023), Addendum 02 – Primary Clinical Study Report62 and BMS (2023), Addendum 02 – Primary Clinical Study Report supplementary table.62
Notable Harms (Adverse Events of Special Interest)
The most common (> 5% in either of the treatment group) IMAEs reported in the nivolumab-relatlimab FDC group were hypothyroidism (18.3%), rash (12.7%), diarrhea and/or colitis (8.2%), hyperthyroidism (6.8%), hepatitis (6.2%), and adrenal insufficiency (5.6%). In the nivolumab monotherapy group, the previously noted most commonly reported IMAEs were hypothyroidism (14.2%), rash (8.6%), diarrhea and/or colitis (3.9%), hyperthyroidism (7.2%), hepatitis (3.3%) and adrenal insufficiency (1.1%).62 The majority of events in both groups were grade 1 or 2 in severity (Table 30).62 The sponsor indicated that no statistical comparisons between treatment groups were planned for any safety end point. Analyses are descriptive only and show the number of patients with events for both treatment groups. Due to different follow-up times for individual patients, the difference or OR based on incidences is not an appropriate effect measure to compare treatment groups for safety. For the notable harm (i.e., myocarditis), the number of patients with an event was low (< 10 patients per treatment group and in total), and even a descriptive analysis does not allow for any meaningful conclusions to be drawn.
Across OESIs, in the RELATIVITY-047 trial within 100 days of last dose, elevated troponin was the most commonly reported event in both treatment groups (12.7% in the nivolumab-relatlimab FDC group and 10.0% in the nivolumab group).62 The majority of events were grade 1 or 2 in severity and fewer than 1% of OESIs were grade 3 or 4 (Table 31).62 Among the notable harms, myocarditis and adrenal insufficiency are considered notable harms of particular interest. Myocarditis occurred infrequently and was reported in 1.7% of patients in the nivolumab-relatlimab FDC group and 0.6% in the nivolumab monotherapy group.61 Grade 3 or 4 myocarditis events occurred in 0.6% of patients in the nivolumab-relatlimab FDC group and no patients in the nivolumab monotherapy group (Table 31).62 Adrenal insufficiency occurred in 5.6% of patients the nivolumab-relatlimab FDC group and in 1.1% of the nivolumab monotherapy group (Table 30).
Critical Appraisal
Internal Validity
The included pivotal study (RELATIVITY-047) was a relatively well-designed, prospective, multicentre, double-blind, randomized, parallel, and active-controlled trial. The study used an appropriate randomization and allocation method (i.e., IRT). The primary analysis of PFS, OS, and ORR were based on an ITT analysis. No important protocol deviations were reported. Overall, most of the demographic and baseline characteristics were well balanced between groups (Table 25). The key effect or prognostic factors, such as an ECOG PS of 0 or 1, previous adjuvant and neoadjuvant therapy, and tumour cell surface biomarker expression (PD-L1 and LAG-3 inhibitors and BRAF mutation status) were well balanced between groups. The potential methodological limitations of the study are discussed in the following section.
Metastatic stage M1c accounted for a relatively high proportion of melanomas in the nivolumab-relatlimab FDC group (N = 151 [42.5%]) compared with the nivolumab monotherapy group (N = 127 [35.4%]); however, the clinical experts consulted for this review stated that minor between-group imbalances of metastatic stage M1c would have been unlikely to affect the comparative study results of the 2 groups.
Table 30: Summary of IMAEs in RELATIVITY-047 Within 100 Days of Last Dose (All Treated Patients)
IMAE | Nivolumab-relatlimab (N = 355) | Nivolumab (N = 359) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
% with IMAE | % with IMAEa receiving treatment | % with IMAE | % with IMAEa receiving treatment | |||||||
N | All grade | Grade 3 or 4 | IMM | High-dose CS | N | All grade | Grade 3 or 4 | IMM | High-dose CS | |
Hypothyroidism | 65 | 18.3% | 0% | 1.5% | 0% | 51 | 14.2% | 0% | 2.0% | 0% |
Rash | 45 | 12.7% | 0.8% | 100% | 13.3% | 31 | 8.6% | 1.7% | 100% | 25.8% |
Diarrhea/colitis | 29 | 8.2% | 1.7% | 100% | 82.8% | 14 | 3.9% | 1.4% | 100% | 78.6% |
Hyperthyroidism | 24 | 6.8% | 0% | 25.0% | 8.3% | 26 | 7.2% | 0% | 7.7% | 3.8% |
Hepatitis | 22 | 6.2% | 4.8% | 100% | 100% | 12 | 3.3% | 1.7% | 100% | 66.7% |
Adrenal insufficiency | 20 | 5.6% | 1.7% | 85.0% | 5.0% | 4 | 1.1% | 0% | 100% | 0% |
AE = adverse event; BMS = Bristol Myers Squibb; CS = corticosteroid; IMAE = immune-mediated adverse event; IMM = immune-modulating medication.
Notes: IMAEs include AEs of any grade occurring in 1% or more of patients considered by investigators to be potentially immune-mediated that met the following criteria: occurred within 100 days of the last dose, regardless of causality, treated with immune-modulating medication with no clear alternate etiology, or had an immune-mediated component. October 27, 2022, median follow-up of 25.3 months.
aBased on patients who experienced an IMAE.
Source: BMS (2023), Addendum 02 – Primary Clinical Study Report.62
Table 31: Summary of OESIs (Greater Than 1%) in RELATIVITY-047 Within 100 Days of Last Dose (All Treated Patients With or Without Immune-Modulating Medication)
OESI, n (%) | Nivolumab-relatlimab (N = 355) | Nivolumab (N = 359) | ||
|---|---|---|---|---|
All grade | Grade 3 or 4 | All grade | Grade 3 or 4 | |
Troponin event | 45 (12.7) | 1 (0.3) | 36 (10.0) | 3 (0.8) |
Uveitis | 6 (1.7) | 1 (0.3) | 5 (1.4) | 2 (0.6) |
Myocarditis | 6 (1.7) | 2 (0.6) | 2 (0.6) | 0 |
Pancreatitis | 5 (1.4) | 0 | 6 (1.7) | 1 (0.3) |
BMS = Bristol Myers Squibb; OESI = other event of special interest.
Note: October 27, 2022, median follow-up of 25.3 months.
Source: BMS (2023), Addendum 02 – Primary Clinical Study Report.41
Based on the study design, during the treatment phase, patients were prohibited from receiving additional concurrent anticancer therapies. Immune-modulating concomitant medications were recommended to treat certain AEs; these included corticosteroids, immune-modulating drugs, immunosuppressive drugs, and glucocorticoids. It was noted that they were more frequently administered to manage AEs in patients receiving nivolumab-relatlimab FDC compared with those receiving nivolumab monotherapy (58.9% versus 44.0%). In addition, palliative radiotherapy and palliative surgery were allowed if patients met certain criteria and following discussion with the BMS medical monitor or designee, but no detail information for palliative radiotherapy and palliative surgery use in each group was provided. The clinical experts CADTH consulted for this review indicated that the potential impact of minor imbalanced immune-modulating concomitant medications, palliative radiotherapy, and palliative surgery on the comparative efficacy between the treatment groups (nivolumab-relatlimab FDC versus nivolumab) would be negligible.
Subsequent systemic anticancer treatment (e.g., PD1 and/or PD-L1 inhibitors) may contribute to patients’ OS benefit in both groups. The impact of the subsequent systemic anticancer treatment on the comparative OS may introduce potential bias. However, the proportions of patients using the subsequent treatment in both groups were relatively comparable. It is therefore unlikely to have a substantial impact on the comparative OS findings observed in this study.
In terms of the OS assessment, OS was designed and assessed as a secondary outcome, and the study was not powered to assess OS between-group difference at the prespecified final analysis (after a median follow-up of 19. 3 months) and updated analysis (after a follow-up of 25.3 months). The efficacy on OS of nivolumab-relatlimab FDC compared with nivolumab therefore remains uncertain.
The statistical significance of ORR (according to BICR) could not be formally tested due to its position in the statistical hierarchy because the OS final analysis did not reach statistical significance. As a result, ORR, as well as CR and progressive disease (which were part of the overall response analysis), are based on only descriptive analyses after a median follow-up of 19.3 months. Only descriptive analyses without a between-group difference or HR were reported. Results for ORR, CR and progressive disease should be interpreted with caution.
The DoR and TTR were assessed as tertiary or exploratory outcomes but without a hierarchical testing procedure to control for type I error. Analyses of DoR and TTR were not statistically powered and were reported using descriptive statistics only. No between-group difference was reported for DoR or TTR, although an HR was reported for DoR; overall, the findings of DoR and TTR should be viewed as supportive evidence only.
Similarly, FACT-M and EQ-5D-3L were assessed as tertiary and/or exploratory outcomes but without hierarchical testing procedures to control for type I error. For these patient-reported HRQoL outcomes (FACT-M and EQ-5D-3L), a differential recall bias may have occurred, although the magnitude and direction of the impact of any recall bias on the patient-reported HRQoL outcomes is unknown. The HRQoL analyses were not statistically powered and were reported using descriptive statistics.42 Overall, the HRQoL findings should be viewed as supportive evidence only.
As the subgroup analysis were not part of the randomization scheme, imbalances in characteristics may bias the results observed between the subgroups. In addition, the subgroup analysis may be not powered to detect the between-group difference in each subgroup. The findings of the subgroup analysis should therefore be viewed as supportive evidence only.
Finally, 1 of the limitations of the RELATIVITY-047 trial is a lack of a comparison to current standard of care (i.e., dual-combination therapy and all available monotherapy except nivolumab). The efficacy and safety of nivolumab-relatlimab FDC compared with encorafenib-binimetinib, dabrafenib-trametinib, vemurafenib-cobimetinib, ipilimumab-nivolumab, ipilimumab, pembrolizumab, dabrafenib, and trametinib is unknown.
External Validity
Only 16 patients (2.2%) from Canada and 63 (8.8%) from the US were included in the trial. According to the clinical experts CADTH consulted for this review, the RELATIVITY-047 study population (i.e., adult patients with unresectable or metastatic melanoma who have not received prior systemic therapy for unresectable or metastatic melanoma) is considered reflective of patients in Canada. There is no concern regarding the generalizing findings from the pivotal study to Canadian clinical settings. The following considerations are of importance regarding the external validity of the RELATIVITY-047 study.
It is uncertain whether the finding can be generalized to patients with CNS metastases or patients with ECOG PS higher than 1 as no such patients were included in the study. Only 17 patients with brain metastasis (2.4%) were included (1.7% and 3.1% in the nivolumab-relatlimab FDC and nivolumab monotherapy groups, respectively). Patients with active CNS metastases were excluded. The clinical experts CADTH consulted for this review indicated that excluding patients with active CNS metastases and an ECOG PS higher than 1 in clinical trial is a common and reasonable practice for observing favourable benefits, risk ratios, and safety profiles. While a higher ECOG PS (> 1) usually indicates more severe disease and likely a more unfavourable prognosis, the nivolumab-relatlimab FDC combination treatments could be extended to patients with an ECOG PS higher than 1. In terms of patients with CNS metastasis, the clinical experts CADTH consulted for this review indicated that additional studies are needed to understand the comparative efficacy of relatlimab-nivolumab versus nivolumab monotherapy in patients with CNS metastasis.
Patients with active, known, or suspected autoimmune disease were excluded. The clinical experts CADTH consulted for this review indicated that patients with autoimmune disorders should be considered for nivolumab-relatlimab FDC treatment at the discretion of the treating physician.
Finally, although the Health Canada indication for nivolumab-relatlimab FDC is for the treatment of adult and pediatric patients 12 years of age or older with unresectable or metastatic melanoma who have not received prior systemic therapy for unresectable or metastatic melanoma, no children aged 12 years or older but younger than 18 years were enrolled in the pivotal study, Therefore, the comparative efficacy and safety profile of nivolumab-relatlimab FDC versus nivolumab monotherapy in children is not available. Whether the findings from the pivotal study (RELATIVITY-047) can be generalized to adolescent patients (≥ 12 to < 18 years old) remains unknown. However, the Health Canada product monograph indicates that the use of nivolumab-relatlimab FDC in pediatric patients 12 years of age or older and weighing at least 40 kg is supported by predicted drug exposures at the recommended nivolumab-relatlimab FDC dose that are expected to result in safety and efficacy similar to that of adults. One clinical expert CADTH consulted for this review indicated that pediatric patients with unresectable or metastatic melanoma should be enrolled in clinical trials if available to assess the efficacy and safety profile of the nivolumab-relatlimab FDC treatment. The other clinical expert indicated that, because of the potential unfeasibility of the trials on the pediatric patients, use of nivolumab-relatlimab FDC in adolescents should be considered on a case-by-case basis, particularly if the body habitus is similar to that of an adult. The clinical expert noted that currently IO is given to the pediatric population, and it is well tolerated.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered most relevant to inform CADTH’s expert committee deliberations. A final certainty rating was determined based on definitions established by the GRADE Working Group (Balshem et al. [2011] and Santesso et al. [2020]):43,44
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate — the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited — the true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate — the true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as “very uncertain.”
Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null. For this review, PFS, OS, CR, progressive disease, DoR, TTR, and notable harms use the null as a threshold.
The target of the certainty of evidence assessment was based on the presence or absence of a clinically important effect, as informed by MIDs suggested by the sponsor and agreed upon by the clinical experts consulted by CADTH for this review (for ORR and FACT-M and EQ-5D3L change from baseline).
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for nivolumab-relatlimab FDC versus nivolumab monotherapy in the treatment of adult patients with unresectable or metastatic melanoma who have not received prior systemic therapy for unresectable or metastatic melanoma.
Long-Term Extension Studies
No long-term extension studies were available.
Indirect Evidence
Contents within this section were informed by materials submitted by the sponsor. The following summary was validated by the CADTH review team.
Objectives for the Summary of Indirect Evidence
The objective of this section is to summarize and appraise the evidence from ITCs for the relative effects and safety of relatlimab plus nivolumab for the treatment of patients aged 12 years and over with unresectable or metastatic melanoma. While direct head-to-head evidence is available for the comparison of relatlimab plus nivolumab relative to nivolumab, there remains a lack of direct evidence for other relevant treatments of relevance to the Canadian population.
Description of Indirect Comparisons
Two ITCs were reviewed for this submission. ITC1 included a systematic literature review (SLR) and applied a Bayesian NMA to 2 major subpopulations: patients treated with immunotherapy only and a separate analysis for patients treated with either immunotherapy or BRAF and MEK inhibitors. ITC2 utilized patient-level data from the RELATIVITY-047 trial in conjunction with patient-level data from the CheckMate 067 trial to indirectly compare nivolumab-relatlimab FDC relative to nivolumab-ipilimumab by using an inverse probability of treatment-weighting approach to adjust for covariates between trials. As ITC2 did not include a literature review or study assessment process, a summary of the associated study inclusion criteria is only reported for ITC1, and its associated SLR is provided in Table 32.
Table 32: Study Selection Criteria and Methods for ITC1 Submitted by the Sponsor
Characteristics | Inclusion criteria |
|---|---|
Population | Adult patients with previously untreated unresectable or metastatic melanoma |
Intervention | Any of the following therapies either alone or in combination in patients previously untreated, with no restriction on dose or dose schedule: Immunotherapies:
Targeted therapies:
Chemotherapies:
|
Comparator | Placebo, or any therapy facilitating an indirect comparison from the interventions listed |
Outcome |
|
Study designs | Randomized controlled trials |
Publication characteristics | Published studies in the English language |
Exclusion criteria |
|
Databases searched |
|
Selection process | Abstracts screened against search criteria by 2 independent reviewers with adjudication by a third reviewer for discrepancies. Included abstracts were screened against search criteria by 2 independent reviewers with adjudication by a third reviewer for discrepancies for full-text article inclusion |
Data extraction process | Methods
|
Quality assessment | The Cochrane Collaboration Risk of Bias tool (version unspecified) was performed by 2 researchers independently with adjudication from a third reviewer for discrepancies. |
ITC1 = indirect treatment comparison 1.
Source: Sponsor-submitted ITC1.72
Indirect Treatment Comparison Design 1 — Bayesian Network Meta-Analysis
Objectives
The purpose of the first sponsor-submitted ITC and SLR was to assess the estimated comparative efficacy and safety of relatlimab and nivolumab relative to existing therapeutic regimens for the treatment of adult patients with previously untreated, unresectable, or metastatic melanoma.
Study Selection Methods
The SLR was conducted initially in 2017, followed by 7 updates, for a total of 8 searches, with the most recent search conducted in November 2022. The eligibility criteria for study inclusion are presented in Table 32. Using the Ovid platform, the following databases were reviewed for peer-reviewed publications: MEDLINE, Embase, and the Cochrane Central Register of Controlled Trials. Terms for RCTs were based on the study design filters recommended by the Scottish Intercollegiate Guidelines Network for MEDLINE and Embase. Non–peer-reviewed publications were considered as part of the study eligibility criteria, and the following scientific conferences were reviewed: annual meetings of the American Association for Cancer Research, American Society of Clinical Oncology, European Society of Medical Oncology, Society for Immunotherapy of Cancer, and Society of Melanoma Research. Publications were reviewed by 2 independent investigators with adjudication from a third investigator as required for disagreements on eligibility.
ITC1 Analysis Methods
Within this ITC, the sponsor provided a Bayesian NMA with varying outcome models conditional on the outcome being assessed. During dataset creation, the sponsor noted that several steps were taken to harmonize the associated data inputs. For time-to-event HR data (for OS and PFS), the sponsor utilized the HR with the shortest follow-up time in primary publications as the preferred parameter of interest. Where no HR was reported, Kaplan-Meier curves were presented, and the sponsor derived an HR using the Guyot algorithm on digitized and manually extracted curves using DigitizeIt. Imputation of standard errors used the average standard deviation among trials reporting dispersion of the outcome of interest. The mean standard deviation was then converted to a standard error using study-specific sample sizes. Both fixed- and random-effects models were considered.
For time-to-event outcomes (for OS and PFS) utilizing time-varying HRs, the sponsor used multidimensional treatment effects, which model hazard functions of interventions within a trial against known parametric survival functions or fractional polynomials, where differences in parameters are indirectly compared across studies. The sponsor assessed Weibull, Gompertz, and second-order fractional polynomials covering P1 = 0 or 1 and P2 = −1, 0.5, 0, 0.5, or 1. Goodness of fit for model selection was based on the deviance information criterion (DIC) utilizing the following steps:
Run full and less-complex fractional polynomial models for all combinations of P1 and P2.
Rank models according to DIC.
Review curve fits and extrapolation for top 4 best-fitting models according to DIC.
Compare full models with less-complex models based on best choice of P1 and P2; opt for the less-complex model if similar goodness of fit (most parsimonious model).
Noninformative priors were used for mean hazards and treatment effects. Specifically, they were reported as multivariate normals, with mean vectors centred at 0 and covariance matrices with diagonals of 10,000 and off-diagonal elements of 0.
For time-to-event outcomes (OS and PFS) utilizing reported constant HRs, the sponsor assessed the proportional hazard assumption with a Grambsch and Therneau test. Analyses conducted on constant HRs were performed using a regression model with a contrast-based normal likelihood for the log HR (and corresponding standard error) of each trial (or comparison) in the network. Normal noninformative priors were used, with a mean of 0 and a variance of 10,000.
For binary outcomes (used for safety outcomes), the sponsor reported on the proportion of patients experiencing the event of interest using a regression model with a binomial likelihood and logit link. Normal noninformative prior distributions for the parameters were used with a mean of 0 and a variance of 10,000. Relative treatment effects were expressed as ORs.
Consistency was evaluated between direct and indirect comparisons by edge-splitting.
Risk of bias was assessed using the Cochrane Collaboration’s risk-of-bias tool with 2 independent researchers reviewing and a third adjudicating in cases of divergent opinions.
Table 33: ITC1 Analysis Methods
Methods | Description |
|---|---|
Analysis methods | A Bayesian network meta-analysis, using fixed and time-varying hazards for survival outcomes, and a regression model with a binomial likelihood and logit link for binary outcomes |
Priors | Noninformative |
Assessment of model fit | For survival models, assessed via DIC and plausibility of HRs generated against the reported HR values where possible, prioritizing the most parsimonious model |
Assessment of consistency | Edge-splitting |
Assessment of convergence | Trace plots, density plots, Gelman-Rubin-Brooks plots, autocorrelation plots |
Outcomes | For survival outcomes:
For binary outcomes: a regression model with a binomial likelihood and logit link |
Follow-up time points | For survival outcomes, 48 months; for safety outcomes, not reported |
Construction of nodes | According to treatment and dose combination |
Sensitivity analyses | Constant HR analysis for survival outcomes, analyses without the CheckMate 066 trial, and analyses without the DreamSeq trial |
Subgroup analysis | BRAF-positive populations |
DIC = deviance information criterion; HR = hazard ratio; ITC1 = indirect treatment comparison 1.
Source: Sponsor-submitted ITC1.72
Results of ITC1
Of the 10,432 citations screened, the sponsor identified 16 unique trials (with an associated 121 citations) that met the study eligibility criteria. All trials were published between 2011 and 2021. The assessment of homogeneity of the included studies is summarized in Table 34.
One trial, DREAMseq, which compared nivolumab-ipilimumab against dabrafenib-trametinib, incorporated a crossover design element, with the sponsor reporting that approximately 20% to 30% of patients crossed over between groups following progression. The sponsor noted that OS data were not provided before crossover, and that PFS would not be affected by this design, as crossover occurred following a progression event. Accordingly, a sensitivity analysis was performed with the exclusion of the DREAMseq trial.
Another trial, CheckMate 066, was noted by the sponsor to have recruited an exclusively BRAF wild-type population. As this trial represents 1 of 2 trials that connect evidence between IO therapies and other therapeutic drugs, an assumption would have to be made that BRAF status does not act as a treatment-effect modifier for comparisons between nivolumab and dacarbazine. As this was not tested, sensitivity analyses were conducted with the exclusion of CheckMate 066.
With these exceptions, the sponsor did not consider any of the between-trial differences summarized in Table 34 to be sufficient to warrant excluding an individual trials from analysis.
Due to the differences among populations with respect to BRAF mutation status, the sponsor proposed separate analyses restricted to patients received IO therapies, in which 5 out of 6 trials reported a mixed BRAF mutation population, and 1 trial reported an exclusively BRAF wild-type population. Additionally, a broader network of evidence was provided to incorporate evidence on patients regardless of BRAF status. Additionally, sensitivity analyses were performed for outcomes in trials recruiting patients of mixed BRAF status, providing subgroup data for BRAF-positive patients.
Table 34: Assessment of Homogeneity for ITC1
Characteristics | Description and handling of potential effect modifiers |
|---|---|
Disease severity |
|
Treatment history |
|
Trial eligibility criteria |
|
Dosing of comparators |
|
Placebo response |
|
Definitions of outcomes |
|
Timing of end point evaluation |
|
Withdrawal frequency |
|
Clinical trial setting |
|
Study design |
|
AE = adverse event; AJCC = American Joint Committee on Cancer; ECOG PS = Eastern Cooperative Oncology Group Performance Status; ITC1 = indirect treatment comparison 1; OS = overall survival; PFS = progression-free survival; PD-L1 = programmed cell death ligand 1; RECIST = Response Evaluation Criteria in Solid Tumors Version; RECIST 1.1 = Response Evaluation Criteria in Solid Tumors Version 1.1.
Source: Sponsor-submitted ITC1.72
An overview of the efficacy analyses associated with ITC1 for the IO population is provided in Table 35. Overall, the sponsor noted improvements in OS at 48 months of nivolumab-relatlimab relative || |||||||||| |||| ||||| ||| |||| ||||| |||||| whereas the results relative to the other assessed therapies was not associated with improvements in OS. For PFS at 48 months, the sponsor noted improvements of nivolumab-relatlimab relative to |||||||||| |||| ||||| ||| |||| ||||| |||||| relative to ||||||||||||||||||||||| |||| ||||| ||| |||| ||||| ||||| and relative to pembrolizumab (HR = 0.59; 95% CrI, 0.35 to 0.97). Comparisons of nivolumab-relatlimab relative to other therapies were not associated with improvements in PFS. Comparisons relative to cobimetinib-atezolizumab are based on data extrapolated by the sponsors methodology, rather than from directly observed data.
An overview of the safety analysis of the IO population is presented in Table 36. When nivolumab-relatlimab was compared to nivolumab monotherapy, nivolumab-relatlimab was associated with an increased proportion of patients experiencing grade 3 or 4 treatment-related AEs (OR = 2.08; 95% CrI, 1.39 to 3.14), discontinuations due to AEs (OR = 1.59; 95% CrI, 1.10 to, 2.32), and discontinuations due to treatment-related AEs (OR = 2.21; 95% CrI, 1.41 to, 3.56), and no such association was noted compared to grade 3 or 4 AEs. When nivolumab-relatlimab was compared to ipilimumab monotherapy, nivolumab-relatlimab was |||||||||| |||| || ||||||||| |||||||||| || |||||||| |||||||||||| ||||| ||| ||||||||||||||||| ||||||| |||||| |||| ||||| ||| |||| ||||| |||||| ||| |||||||||||||||| ||| || ||||||||||||||||| ||||||| |||||| |||| ||||| ||| |||| ||||| |||||| ||| || |||| ||||||||||| ||| ||||| |||||||| || ||||| ||| ||||||| |||||| || |||||||||||||||| ||| || ||||||| ||||||. When nivolumab-relatlimab was compared to pembrolizumab monotherapy, nivolumab-relatlimab was associated with an increased proportion of patients experiencing grade 3 or 4 treatment-related AEs (OR = 1.99; 95% CrI, 1.01 to 3.87), and no such association was noted for discontinuations due to AEs or discontinuations due to treatment-related AEs. No data were presented for the effect of nivolumab-relatlimab relative to pembrolizumab monotherapy. When nivolumab-relatlimab was compared to nivolumab (1 mg/kg) combined with ipilimumab (3 mg/kg), nivolumab-relatlimab was associated with a decreased proportion of patients experiencing grade 3 or 4 adverse events (OR = 0.46; 95% CrI, 0.29 to 0.72), grade 3 or 4 treatment-related AEs (OR = 0.43; 95% CrI, 0.25 to 0.73), discontinuations due to AEs (OR = 0.29; 95% CrI, 0.17 to 0.48), and discontinuations due to treatment-related AEs (OR = 0.50; 95% CrI, 0.28 to 0.92).
Table 35: ITC1 Efficacy Analysis Data, Immuno-Oncology Population
Detail | Overall survival, 48 months | Progression-free survival, 48 months |
|---|---|---|
Number of studies, N | 6 | 6 |
Model | Time-varying Bayesian NMA, no restrictions on BRAF or PD-L1 mutation status, extrapolated data for cobimetinib-atezolizumab, P1 = 1, P2 = −1, scale and second shape, fixed effect | Time-varying Bayesian NMA, no restrictions on BRAF or PD-L1 mutation status, extrapolated data for cobimetinib-atezolizumab, P1 = 0, P2 = −1, scale and second shape, fixed effect |
Nivolumab-relatlimab comparator, HR (95% CrI) | ||
Nivolumab (3 mg/kg) | 0.85 (0.65 to 1.10) | 0.88 (0.67 to 1.16) |
|||||||||| | |||| |||||| ||||| | |||| |||||| ||||| |
||||||||||||||||||||||| | |||| |||||| |||||| | |||| |||||| |||||| |
Pembrolizumab | 0.66 (0.43 to 1.03) | 0.59 (0.35 to 0.97) |
Nivolumab (1 mg/kg) combined with ipilimumab (3 mg/kg) | 1.02 (0.72 to 1.47) | 1.12 (0.74 to 1.66) |
Nivolumab (3 mg/kg) combined with ipilimumab (1 mg/kg) | 0.89 (0.54 to 1.52) | 0.89 (0.50 to 1.59) |
CrI = credible interval; HR = hazard ratio; OR = odds ratio; ITC1 = indirect treatment comparison 1; PD-L1 = programmed cell death ligand 1; NA = not applicable; NMA = network meta-analysis.
aData extrapolated, with most recent time point available at 12 months.
Source: Sponsor-submitted ITC1, Table 7 (OS), Table 9 (PFS).72
Table 36: ITC1 Safety Analysis Data, Immuno-Oncology Population
Detail | Grade 3 to 4 adverse events | Grade 3 to 4 treatment-related adverse events | Discontinuations due to adverse events | Discontinuations due to treatment-related adverse events |
|---|---|---|---|---|
Number of IO studies, N | 2 | 4 | 3 | 4 |
Model | Regression model with a binomial likelihood and logit link, no restrictions on PD-L1 or BRAF mutation status, fixed effects | Regression model with a binomial likelihood and logit link, no restrictions on PD-L1 or BRAF mutation status, fixed effects | Regression model with a binomial likelihood and logit link, no restrictions on PD-L1 or BRAF mutation status, fixed effects | Regression model with a binomial likelihood and logit link, no restrictions on PD-L1 or BRAF mutation status, fixed effects |
Nivolumab-relatlimab comparator, OR (95% CrI) | ||||
Nivolumab (3 mg/kg) | 1.31 (0.98 to 1.77) | 2.08 (1.39 to 3.14) | 1.59 (1.10 to 2.32) | 2.21 (1.41 to 3.56) |
|||||||||| | |||| |||||| ||||| | |||| |||||| ||||| | |||| |||||| ||||| | |||| |||||| ||||| |
Pembrolizumab | NR | 1.99 (1.01 to 3.87) | 1.20 (0.60 to 2.40) | 1.77 (0.77 to 4.00) |
Nivolumab (1 mg/kg) and ipilimumab (3 mg/kg) | 0.46 (0.29 to 0.72) | 0.43 (0.25 to 0.73) | 0.29 (0.17 to 0.48) | 0.50 (0.28 to 0.92) |
CrI = credible interval; IO = immuno-oncology; ITC1 = indirect treatment comparison 1; NR = not reported; PD-L1 = programmed cell death ligand 1; OR = odds ratio.
Source: Sponsor-submitted ITC, Table 7 (OS) and Table 9 (PFS).72
Critical Appraisal of ITC1
A limitation associated with ITC1 is the assessment of evidence from the broader network (i.e., inclusive of BRAF and MEK inhibitors). Data on BRAF and MEK inhibitors are restricted to patient populations who are exclusively BRAF-positive. Conversely, in the IO network, 5 out of 6 trials reported a mixed BRAF mutation population, and 1 trial reported an exclusively BRAF wild-type population. Further, 1 of the 2 nodes that connect evidence from the IO network to the broader network is an exclusively BRAF wild-type population. No quantitative evidence was provided to determine whether the BRAF mutation status was an effect modifier for the included IO therapies. Nonquantitative summaries were provided on the CheckMate 067 and 069 trials, wherein the sponsor found “no difference” in PFS and OS for comparisons between nivolumab-ipilimumab and ipilimumab, but noted that comparisons of PFS between nivolumab and ipilimumab were closer to the null value among BRAF-positive patients. Accordingly, where comparisons were made between BRAF mixed populations within the IO network, and where comparisons were made between mixed BRAF and BRAF wild-type populations and the BRAF-positive populations, results are subject to potential bias from heterogeneity due to the differences in BRAF mutation status.
The sponsor did provide a BRAF mutation-positive scenario analysis, although this analysis itself is subject to additional limitations. First, the associated sample size among trials with mixed populations is substantially reduced, as the proportion of BRAF-positive patients within the available IO trials reporting subgroup data varied from 21% to 50.6%. Second, the provided network does not constitute an exclusively BRAF-positive population. Two trials consisted of exclusively BRAF wild-type patients, and 1 trial consisted of a mixed BRAF population. Finally, data were not provided on the balance of patient characteristics among the BRAF-positive population for trials with mixed populations. As such, it is possible that the distribution of characteristics may differ from the total trial population, representing a potential source of bias when interpreting this scenario analysis.
A second significant source of uncertainty with respect to the broader network encompassing BRAF and MEK inhibitors is the characteristics of the 2 trials that connect evidence from the IO network to the broader network. One trial, DREAMseq, which evaluated nivolumab-ipilimumab compared to dabrafenib-trametinib, incorporated a crossover design element in which the sponsor reported that approximately 20% to 30% of patients crossed over between arms following progression. While PFS outcomes would not be influenced by this crossover, OS and safety will be confounded by this mixing of treatment effects from the perspective of an NMA. The second trial connecting the IO network to the broader network consisted of an exclusively BRAF wild-type population. As noted earlier, the absence of BRAF-positive patients has an unknown effect on all of the outcomes presented by the sponsor. To mitigate the design characteristics of these 2 trials, the sponsor provided 2 scenario analyses that excluded 1 trial at a time from the network. The issue with interpreting results from this approach is that the source of bias is concentrated in a single trial. Given that the impact of an entirely BRAF wild-type population or the impact of a crossover design can have an influence on several outcomes, it is uncertain which trial is more influential on the associated indirect estimates obtained. Accordingly, such scenario analyses do not clearly mitigate the bias from the associated trials; rather, they amplify the influence of the remaining none-xcluded trial.
Taken in combination, the nonoverlapping BRAF-positive mutation status, and the trial characteristics of the 2 nodes that connect IO-evaluating results to BRAF- and MEK-inhibitor results, there are substantial limitations to any associated indirect estimate of relatlimab to the evaluated MEK- and BRAF-inhibitor products. These issues are in addition to the broader set of limitations indicated in the following section, which describe additional sources of uncertainty. Accordingly, efficacy and safety estimates of nivolumab-relatlimab relative to MEK and BRAF inhibitors should be evaluated with these substantial limitations in mind. The clinical experts consulted for this review indicted that, from their perspective, the comparison of IO therapies relative to one another was of greater importance than IO BRAF and MEK inhibitors due to their current place in clinical practice in Canada.
Evidence of nivolumab-relatlimab compared to the remaining network of evidence (whether IO-restricted or the broader analysis) is channelled through a single treatment node, nivolumab monotherapy. From the available data presented, efficacy outcomes (PFS and OS) demonstrated relatively consistent group averages with respect to median survival time. Conversely, a substantial difference was noted with respect to discontinuations due to overall AEs. Specifically, the proportion of patients discontinuing due to any AEs was reported as 7% in the CheckMate 066 trial, 50% in the CheckMate 067 trial, and 15.9% in the RELATIVITY-047 trial. Similar trends in the CheckMate 067 trial showed that higher proportions of patients experienced grade 3 or 4 AEs and overall discontinuations within the nivolumab monotherapy group relative to the CheckMate 066 and RELATIVITY-047 nivolumab monotherapy groups when compared to the other available studies assessing nivolumab monotherapy. As the CheckMate 067 trial represents an influential study in the network, and given its role as the single connection of evidence from nivolumab-relatlimab to the IO network and closed loop of evidence, this may have a significant influence on the safety estimates from this analysis.
With respect to OS and PFS, the sponsor noted that 1 trial, IMspire 170, which evaluated cobimetinib-atezolizumab versus pembrolizumab, used survival data extrapolated from 12 months through to 48 months. Accordingly, assessments of the relative efficacy of these 2 comparisons should be made with caution. The sponsor also provided evidence of goodness of fit for the OS network. Data from the CheckMate 069 trial, which evaluated nivolumab (1 mg/kg) combined with ipilimumab (3 mg/kg), appear to indicate observed data from this network until approximately 28 months only, although is not noted in the primary comparative analysis to be extrapolated data.
For OS, 5 trials had median data for which the upper bound of the CI reported within the trial could not be estimated. Accordingly, the estimates generated from these data may be subject to change following release of the associated trial data.
Overall, there was limited evidence with respect to the role of PD-L1 status in the included evidence. No information was available on PD-L1 status for 7 (44%) of the trials. All trials within the IO network had data presented on the evaluation of PD-L1 status. For trials reporting PD-L1 expression, the baseline proportions of patients considered to be PD-L1–positive (at a ≥ 1% tumour proportion score threshold) were only available in 2 trials (13%), and with proportions varying from 63% to 67.9%. The sponsor did not provide treatment-group-level proportions of patients with their associated PD-L1 positivity status. The sponsor noted that subgroup data were available for 7 trials (outcome unspecified) for patients with tumour proportion scores of less than 1% and 1% or greater, although no analyses of PD-L1 subpopulations were provided. Accordingly, the sponsor relied on 2 key assumptions: that the distribution of PD-L1 patient status is similar across all included trials where data are not available, and that, where this is not true, PD-L1 status does not constitute a meaningful effect modifier or prognostic factor. The sponsor did not provide direct within the report, but noted historical studies that include evidence indicating a role for PD-L1 as a prognostic and predictive factor for IO therapies, and mixed evidence of the effect that PD-L1 status has on BRAF and MEK inhibitors. Within the network, the sponsor noted that, in 1 included trial and 1 treatment group, PD-L1–positive patients had greater survival benefits over compared with PD-L1–negative patients. Overall, this constitutes a potential source of bias for both IO and BRAF- and MEK-inhibitor comparisons.
For safety outcomes, evidence was limited to 4 comparators, and only for grade 3 or 4 events (both treatment-related and overall) as well as for discontinuations due to AEs (both treatment-related and overall AEs). No data were presented on specific safety outcomes of potential interest, nor on overall discontinuations. The sponsor noted that discontinuations overall were not presented due to certain trials (data not presented) reporting 100% discontinuations by the end of study follow-up.
Several outcomes, specifically time to progression, ORR, CR, and PR, were noted to be extracted and relevant outcomes of interest during the SLR performed by the sponsor, but were not discussed in the sponsor’s feasibility assessment, nor were results presented in the associated report. As part of its SLR process, the sponsor also noted that DoR, stable disease rate, progressive disease rate, and discontinuations due to progressive disease or death were also captured but not analyzed, with no justification provided for their noninclusion.
No assessment of patient-reported outcomes was provided, and accordingly an indirect assessment of nivolumab-relatlimab relative to other interventions is not possible with the presented ITC report.
With respect to applicability to the Canadian patient population, data on the geographic distribution of included patients were not presented, making the influence of systematic differences in health care provision between the included geographies of patients among the trial populations and patients in Canada unclear.
Summary of ITC1
Overall, the evidence submitted by the sponsor for ITC1 indicates that, among trials evaluating IO therapies, nivolumab-relatlimab is associated with improvements in OS relative to ipilimumab monotherapy. For PFS, nivolumab-relatlimab is associated with improvements relative to ipilimumab, pembrolizumab, and cobimetinib-atezolizumab. For safety, nivolumab-relatlimab was associated with higher proportions of patients experiencing grade 3 or 4 treatment-related AEs when compared to nivolumab, ipilimumab, and pembrolizumab, and with lower proportions of patients experiencing these events relative to nivolumab (1 mg/kg) combined with ipilimumab (3 mg/kg). For discontinuations due to AEs, nivolumab-relatlimab was associated with higher proportions of patients experiencing these events relative to nivolumab, and lower proportions of patients experiencing these events relative to nivolumab (1 mg/kg) combined with ipilimumab (3 mg/kg). For discontinuations due to treatment-related AEs, nivolumab-relatlimab was associated with higher proportions of patients experiencing these events relative to nivolumab and ipilimumab, and lower proportions of patients experiencing these events relative to nivolumab (1 mg/kg) combined with ipilimumab (3 mg/kg).
The submitted evidence is subject to several important limitations with respect to potential effect modification due to between-trial differences in biomarkers, and in particular, evidence of the relative effect of nivolumab-relatlimab on MEK and BRAF inhibitors is substantially limited. Additionally, no patient-reported outcomes were provided, meaning that conclusions of the relative impact of nivolumab-relatlimab on patient quality of life cannot be assessed with the present network.
Indirect Treatment Comparison Design 2 — Patient-Level Propensity Model
Objectives
To estimate the indirect comparative efficacy and safety of first-line nivolumab-relatlimab relative to ipilimumab-nivolumab for the treatment of patients with advanced melanoma.
Study Selection Methods
This analysis consists of a posthoc analysis of 2 patient-level RCTs: RELATIVITY-047 and CheckMate 067. No details were provided on study selection.
ITC Analysis Methods
The sponsor used patient-level data from 2 trials. The RELATIVITY-047 trial evaluated nivolumab-relatlimab relative to nivolumab monotherapy whereas the CheckMate 067 trial evaluated 3 treatments: nivolumab-ipilimumab, ipilimumab monotherapy, and nivolumab monotherapy. For this analysis, the sponsor used the nivolumab-ipilimumab and the nivolumab monotherapy groups from the CheckMate 067 trial.
The sponsor used an inverse probability of treatment-weighting model, consisting of the following baseline covariates:
age (continuous)
sex (male or female)
geographic region (rest of world or US)
ECOG PS (≥ 1 or 0)
time from advanced melanoma diagnosis until randomization (continuous, years)
prior adjuvant therapy (yes or no)
AJCC M stage with LDH category 1 (M1any[1] or M0/M1any[0])
AJCC disease stage at study entry (stage III or stage IV)
melanoma subtype (cutaneous acral or cutaneous nonacral; mucosal or cutaneous nonacral; other or cutaneous nonacral)
BRAF mutation status (positive or wild-type)
baseline LDH category 1 (> upper limit of normal or ≤ upper limit of normal)
baseline LDH category 2 (> 2 × upper limit of normal or ≤ 2 × upper limit of normal)
PD-L1 expression category (≥ 1% or < 1%/nonquantifiable).
The propensity score model was employed at the treatment-group level through binary logistic regression for both nivolumab-relatlimab and ipilimumab-nivolumab in the all-comers population, using a subset of patients with nonmissing values. Patient weights were calculated as the inverse of the conditional probability of being exposed to a particular treatment given their included baseline characteristics. Stabilized weights were used in addition to truncation of weights at the fifth and 95th centiles. Assessment of balance was performed utilizing a threshold of less than 0.2 standardized mean differences.
For the efficacy outcomes assessed (OS and PFS), analyses utilized the Kaplan-Meier approach, with HRs calculated using a Cox proportional hazards model, Schoenfeld residuals were used to test for proportionality. For PFS, the sponsor used PFS data from investigator assessments. No definitions were reported for OS assessments. No details were provided by the sponsor on the use of statistical testing for safety outcomes.
For the included studies, truncation was applied during the evaluation of efficacy and safety outcomes. For efficacy outcomes (OS and PFS), data from the CheckMate 067 trial were truncated, with patients without events before August 1, 2016, artificially censored in an attempt to align the data with the median follow-up duration of the RELATIVITY-047 trial. The sponsor noted that this date is close to the September 13, 2016, DBL that constituted a prespecified per-protocol analysis of the CheckMate 067 population. For safety outcomes, only safety events that occurred within the first 28 months of follow-up were analyzed.
Results of ITC2
Two studies were included in ITC2. A summary of the key between-study differences is provided in Table 38.
Table 37: ITC2 Analysis Methods
Methods | Description |
|---|---|
Analysis methods | Inverse probability of treatment-weighting with a Cox proportional hazards model for efficacy (OS and PFS) |
Outcomes | OS PFS (investigator assessment) Safety (all-cause AEs, all-cause grade 3 or 4 AEs, treatment-related AEs (all grades), treatment-related AEs (grade 3 or 4), treatment-related AEs leading to discontinuation (all grades) |
Follow-up time points | 28 months, with truncation of data from CheckMate 067 (ipilimumab-nivolumab) |
Sensitivity analyses | Unweighted analysis, comparative analysis of shared comparator arm (nivolumab monotherapy) |
AE = adverse event; ITC2 = indirect treatment comparison 2; OS = overall survival; PFS = progression-free survival.
Source: Sponsor-submitted ITC2.72
Table 38: Assessment of Homogeneity for ITC2
Characteristics | Description and handling of potential effect modifiers |
|---|---|
Disease severity | Pre–weighting adjustment, several differences were noted between the 2 interventional arms of interest:
|
Treatment history |
|
Trial eligibility criteria |
|
Definitions of outcomes |
|
Timing of end point evaluation |
|
Withdrawal frequency |
|
Clinical trial setting |
|
Study design |
|
AE = adverse event; ITC2 = indirect treatment comparison 2; OS = overall survival; PFS = progression-free survival.
Source: Sponsor-submitted ITC2.72
Results
Prior to weighting, the evaluable sample size was 349 for patients receiving nivolumab-relatlimab, and 307 for patients receiving ipilimumab-nivolumab. Following weighting the effective sample size was 340 for patients receiving nivolumab-relatlimab and 298 for patients receiving ipilimumab-nivolumab.
For both studies, postweighting, the median OS was not reached for either treatment arm, with a lower OS CI of 37 months for nivolumab-relatlimab, and a lower OS CI of ipilimumab-nivolumab of 31.9 months. For OS, the effect of nivolumab-relatlimab relative to ipilimumab-nivolumab was presented (HR = 0.94; 95% CI, 0.74 to 1.19).
For investigator-assessed PFS, median survival data were not presented, although the effect of nivolumab-relatlimab relative to ipilimumab-nivolumab was presented (HR = 1.07; 95% CI, 0.87 to 1.31).
For safety, the sponsor did not provide formal between-group comparisons of the evaluated therapies but did provide summaries of event rates in the postweighting population. A summary of these data are presented in Table 39.
Table 39: ITC2 Safety Outcome Overview
Outcome | Nivolumab-relatlimab | Ipilimumab-nivolumab |
|---|---|---|
Effective sample size, n | 340 | 298 |
All-cause adverse events | 99.1% | 99.7% |
All-cause grade 3 or 4 adverse events | 45.7% | 78.5% |
Treatment-related adverse events | 84.8% | 95.6% |
Treatment-related grade 3 or 4 adverse events | 22.9% | 60.8% |
Treatment-related adverse events (any grade) leading to discontinuation | 17% | 40.1% |
ITC2 = indirect treatment comparison 2.
Sources: Sponsor-submitted ITC2 and sponsor's summary of clinical evidence.72
Critical Appraisal of ITC2
The sponsor-submitted ITC2 is subject to several limitations that should be considered when interpreting the comparative efficacy and safety.
A limitation inherent to all propensity-based analyses is the choice of covariates included within the model. No justification was included in the provided report on the choice of covariates with respect to their clinical significance as effect modifiers. For example, the geographic location was dichotomized between US and outside the US. While the overall balance (based on the sponsor’s proposed standardized mean difference threshold of 0.2) was met postweighting, any between-country differences may still introduce bias due to unmeasured covariates contained within the between-country differences. Further, the sponsor included only patients without missing values in its analysis population before weighting, although the evaluable sample size was only subject to a modest reduction when compared to the overall evaluable sample size.
For safety outcomes, data were not provided on the between-group differences of the shared nivolumab monotherapy group between trials. As no information was provided on AE definitions, and between-study AEs rates may vary due to local standard-of-practice differences in this nonrandomized context, comparing shared treatment groups is a critical step in contextualizing between-group differences. As noted in the sponsor’s ITC1, estimates of between-group differences when comparing all-cause and treatment-related AEs were highly variable. Given that patients treated in the CheckMate 067 trial had a longer minimum follow-up of 28 months, as opposed to 21 months for patients in the RELATIVITY-047 trial, this may disadvantage patients receiving ipilimumab-nivolumab relative to those receiving nivolumab-relatlimab.
With respect to OS data, the truncated and in-progress nature of the OS data (which made it impossible to a calculate median survival) means that long-term comparative OS cannot be interpreted with the information provided.
While the sponsor used a proportional hazards model, data were not provided on the approaches used to test the proportionality assumption. While the sponsor noted the use of Schoenfeld residuals to support the use of a proportional hazards model, the results of this test were not shown.
With respect to applicability to the patient population in Canada, data on the geographic distribution of included patients, making the influence of systematic differences in health care provision between the included geographies of patients among the trial populations and patients in Canada unclear.
Summary of ITC2
Overall, the sponsor-submitted ITC2 provides additional evidence beyond that of the primary ITC for the assessment of nivolumab-relatlimab relative to ipilimumab-nivolumab with respect to OS, PFS, and select safety outcomes. Within the submitted analyses, the sponsor did not detect differences in the relative effect of nivolumab-relatlimab relative to ipilimumab-nivolumab for either OS or PFS. Safety data of the 2 treatment groups were presented in a noncomparative manner. The submitted ITC was subject to some limitations with respect to details of the methods utilized and the use of in-progress OS data. The sponsor indicated that the findings from ITC2 were also supported by the results from an independently conducted NMA.72 However, the findings of the published NMA also should be interpreted with caution given its own methodological limitations.72
Studies Addressing Gaps in the Systematic Review Evidence
No studies addressing gaps in the systemic review evidence were provided.
Discussion
Summary of Available Evidence
The CADTH Clinical Review Report included input from patient groups, clinician groups, clinical experts, and drug programs; 1 ongoing, pivotal, phase II and III, double-blinded, randomized controlled trial (RELATIVITY-047); and 2 sponsor-provided ITCs. The RELATIVITY-047 trial is an ongoing, relatively well-designed multinational phase II and III, randomized, double-blind study of nivolumab-relatlimab FDC versus nivolumab monotherapy administered as a first-line therapy in patients with previously untreated unresectable or metastatic melanoma. The study involved a 14-day screening period. Adults and adolescents aged 12 years or older were eligible for enrolment.63 However, no adolescents aged 12 years or older were enrolled. A total of 714 patients were randomized 1:1 to receive nivolumab-relatlimab FDC (N = 355) or nivolumab monotherapy (N = 359). The median age was 63 years (range = 20 to 94). The majority of patients (N = 655, 91.7%) had metastatic stage IV melanoma at study entry. The median duration from diagnosis to study treatment was 1.26 years. A total of 62 patients (8.7%) had received previous adjuvant or neoadjuvant treatment. A total of 275 patients (38.5%) were BRAF-positive. A total of 16 patients (2.2%) from Canada and 63 patients (8.8%) from the US were included. The primary outcome was PFS. The 2 secondary outcomes were OS and ORR. Tertiary and/or exploratory outcomes included DoR, TTR, and HRQoL measurements (i.e., FACT-M and EQ-5D-3L). The sample size for the study was based on a primary end point of PFS using a BICR for either a phase II or a phase III study.
Results presented in this submission reflect the phase III component of the RELATIVITY-047 trial. The final analysis for PFS was conducted after a median follow-up of 13.2 months. The final analysis for OS and ORR was conducted after a median follow-up of 19.3 months. The median DoR and TTR were based on the updated descriptive analysis conducted after a median follow-up of 25.3 months. HRQoL measurements (FACT-M and EQ-5D-3L) were taken after a median follow-up of 19.3 months. Safety outcomes were assessed in terms of OS and tolerability of nivolumab-relatlimab FDC and nivolumab. Safety data reported in this review were based on a median follow-up of 25.3 months.
Interpretation of Results
Efficacy
Progression-Free Survival
Based on the prespecified final analysis of the nivolumab-relatlimab FDC group and after a median follow-up of 13.2 months, the median PFS was 10.12 months (95% CI, 6.37 to 15.74), which was statistically significant and clinically meaningfully longer compared to 4.63 months in the nivolumab monotherapy group (HR for nivolumab-relatlimab FDC versus nivolumab = 0.75; 95% CI, 0.62 to 0.92; P = 0.0055). The observed PFS benefit of nivolumab-relatlimab FDC compared with nivolumab monotherapy was extended in an updated analysis after a median follow-up of 25.3 months. Subgroup and sensitivity analyses of PFS were largely consistent with those of the primary analysis.
Overall Survival
Based on the stakeholder input from the patient group, clinical group, and the clinical experts CADTH consulted for this review, 1 of the treatment goals was to increase the OS rate. After a median follow-up of 19.3 months, the median OS was not reached in the nivolumab-relatlimab FDC group compared with an OS of 34.10 months in the nivolumab group. The between-group difference (nivolumab-relatlimab FDC versus nivolumab) for median OS did not reach statistical significance at the final analysis of OS after a median follow-up of 19.3 months (HR = 0.80; 95% CI, 0.64 to 1.01; P = 0.0593). The median OS was not reached in the nivolumab-relatlimab group compared with a median OS of 33.18 in the nivolumab monotherapy group in the updated analysis after a median follow-up of 25.3 months. The clinical experts CADTH consulted for this review indicated that the OS data observed in the pivotal study was early due to the limitation of the study design (i.e., it was not sufficiently powered and used a relatively short follow-up time) at the final and updated analyses. It will be important to follow OS data as they mature to determine the actual comparative efficacy on OS of nivolumab-relatlimab FDC over nivolumab monotherapy. Follow-up evaluations of OS in this trial are ongoing. The clinical experts CADTH consulted for this review suggested that a trial powered to assess OS as a primary end point comparing nivolumab-relatlimab FDC to either single-drug nivolumab or dual-drug nivolumab-ipilimumab would greatly help inform patient and care-provider decision-making.
Overall Response Rate
The ORR is an important outcome to patients because an improved ORR (based on radiographic evaluation) is usually correlated with improvement in important clinical outcomes, such as PFS and OS, or slower declines in ECOG PS, delays in clinical symptoms, and worsening of HRQoL, although they are not always absolutely proportionally correlated. As the final analysis of OS did not reach statistical significance, the significance of ORR (according to BICR) could not be formally tested. Based on descriptive final analyses, a total of 10.3% (95% CI, 3.4% to 17.3%) more patients in the nivolumab-relatlimab FDC group achieved the ORR compared with the nivolumab group after a median follow-up of 19.3 months. The results for ORR are in line with the survival benefit seen for PFS. A consistent ORR benefit was also observed in the updated descriptive analysis after a median follow-up of 25.3 months. A total of 9.8% (95% CI, 2.8% to 16.8%) more patients in the nivolumab-relatlimab FDC group achieved the ORR compared with the nivolumab group.
As part of ORR analysis (in terms of CR and progressive disease), no formal statistically analysis or any descriptive analysis was performed to report the between-group difference. No HR and 95% CI were provided. The comparative efficacy of nivolumab-relatlimab FDC versus nivolumab monotherapy in terms of CR and progressive disease is therefore uncertain based on the pivotal study.
Duration of Response and Time to Response
Patients expected a new treatment with a longer response (DoR). However, no statistical and clinical meaningful between-group difference were observed for DoR. The median DoR was not reached in either treatment group, indicating that a longer follow-up is needed to assess the between-group difference in DoR. In terms of TTR, no between-group difference and no HR were reported. Both DoR and TTR were assessed as exploratory end points. After a median follow-up of 25.3 months, the evidence is uncertain about the effects on DoR or TTR of nivolumab-relatlimab FDC compared with nivolumab monotherapy in the treatment of adult patients with unresectable or metastatic melanoma who have not received prior systemic therapy for unresectable or metastatic melanoma.
Health-Related Quality of Life
Outcomes regarding HRQoL based on FACT-M and EQ-5D-3L data were analyzed as exploratory end points. After a median follow-up of 19.3 months, FACT-M, EQ-5D-3L utility index scores, and EQ VAS responses in the nivolumab-relatlimab FDC group remained generally stable (no clinical meaningful improvement or deterioration) during the treatment period. There were no clinically meaningful differences between nivolumab-relatlimab FDC and nivolumab monotherapy in FACT-M, EQ-5D-3L utility index scores and EQ VAS responses. Several methodological study design limitations of HRQoL outcomes analysis are noted. For example, FACT-M, EQ-5D-3L utility index, and EQ VAS analyses were not statistically powered and were reported using descriptive statistics. In addition, there may have been differential recall bias for these patient-reported outcomes, although the magnitude and direction of the impact of the recall bias on the patient-reported HRQoL outcomes is unknown. In addition, the number of patients at the data cut-off included in the analysis was relatively small (not all patients were included). The HRQoL findings should be viewed as supportive evidence only and it is difficult to draw conclusions about the effect. The clinical experts CADTH consulted for this review indicated that the fixed-landmark follow-up time point of 2 years should be considered sufficient to assess the between-group difference in the HRQoL in this population due to the treatment. It is therefore reasonable to assume that no clinical meaningful between-group difference (nivolumab-relatlimab FDC versus nivolumab) was observed in terms of FACT-M, EQ-5D-3L utility index, and EQ VAS scores in the pivotal study.
Symptom reduction was identified an outcome by patient groups, clinical group, and clinical experts CADTH consulted for this review. Symptom reduction was not assessed as a separate outcome in the pivotal RELATIVITY-047 trial. However, the severity of symptoms, such as pain and discomfort, anxiety and depression, and overall well-being were assessed by the FACT-M and EQ-5D-3L instruments.
No adolescents (aged ≥ 12 to < 18 years) were enrolled in the pivotal study. However, the Health Canada product monograph40 indicates that the use of nivolumab-relatlimab FDC in pediatric patients aged 12 years or older and weighing at least 40 kg is supported by predicted drug exposures at the recommended nivolumab-relatlimab FDC dose, which is expected to result in safety and efficacy similar to that of adults. The safety and efficacy of nivolumab-relatlimab have not been established in pediatric patients under the age of 12 years or in patients aged 12 years or older and weighing less than 40 kg.40 One clinical expert CADTH consulted for this review indicated that pediatric patients with unresectable or metastatic melanoma should be enrolled in clinical trials if available to assess the efficacy and safety profile of nivolumab-relatlimab FDC. The other clinical expert advised that, because of the potential unfeasibility of trials involving pediatric patients, use of nivolumab-relatlimab FDC in adolescents should be considered on a case-by-case basis, particularly if body habitus resembles or is close to that of an adult. The clinical expert noted that IO is currently given to the pediatric population, and it is well tolerated.
One clinical expert indicated that patients with uveal melanoma also should be eligible for treatment.
Furthermore, the efficacy and safety of nivolumab-relatlimab FDC compared with existing standard therapies for the treatment of patients with unresectable or metastatic melanoma who have not received prior systemic therapy for unresectable or metastatic melanoma, such as encorafenib-binimetinib, dabrafenib-trametinib, vemurafenib-cobimetinib, ipilimumab-nivolumab, ipilimumab, pembrolizumab, dabrafenib, and trametinib, is lacking.
The sponsor-submitted ITCs were inconclusive when comparing nivolumab-relatlimab FDC against other combination IO therapies with respect to OS and PFS but did find prolonged OS and PFS relative to ipilimumab monotherapy and prolonged PFS relative to pembrolizumab monotherapy.
Harms
According to the clinical experts CADTH consulted for this review, the safety profile of nivolumab in the treatment of various cancers, including melanoma, has been well established by previous clinical trials. The proportions of patient with at least 1 treatment-emergent AE were similar in the 2 groups (99.2% in the nivolumab-relatlimab FDC group versus 95.8% in the nivolumab monotherapy group). However, the most common any-grade AEs, such as fatigue |||||||||| ||| ||| ||||| ||||| ||| ||||||| (occurring in > 20% patients in either of the 2 groups), appeared to occur in more patients in the nivolumab-relatlimab FDC group than in the nivolumab monotherapy group, The frequency of SAEs was similar in both groups (38.9% in the nivolumab-relatlimab FDC group and 33.1% in the nivolumab group). With the exception of malignant neoplasm progression (3.9% in the nivolumab-relatlimab FDC group and 5.6% in the nivolumab group), no other SAEs occurred in more than 2% of patients in either group. The frequency of withdrawal due to AEs was also numerically higher in the nivolumab-relatlimab FDC group compared with the nivolumab monotherapy group (23.1% versus 15.9%, respectively). Discontinuation treatment due to specific AEs occurred in less than 2% patients in each group with the exception of malignant neoplasm progression (1.7% in the nivolumab-relatlimab FDC group and 2.8% in the nivolumab group). The frequency of death due to AEs (i.e., study drug toxicity) was rare in both groups (1.1% in the nivolumab-relatlimab FDC group and 0.6% in the nivolumab group). The notable IMAEs (i.e., those of special interest for this review) were numerically higher in the nivolumab-relatlimab FDC group compared with the nivolumab group. Adrenal insufficiency, which was considered a particularly special IMAE in this review, occurred more often in the nivolumab-relatlimab FDC group than in the nivolumab group (5.6% versus 1.1%, respectively). Nivolumab-relatlimab FDC may result in an increase in the proportion of patients who experience adrenal insufficiency when compared with nivolumab monotherapy. Another particular notable harm, myocarditis, occurred rarely in both groups (1.7% and 0.6%, respectively). Nivolumab-relatlimab FDC may result in little to no clinically important difference in the proportion of patients who experience myocarditis when compared with nivolumab monotherapy. It was also noted that grade 3 and 4 treatment-related AEs appeared more frequently in patients treated with nivolumab-relatlimab FDC versus those treated with nivolumab monotherapy. The sponsor indicated that no statistical comparisons between treatment groups were planned for any safety end point. Analyses are descriptive only and show the number of patients with events for both treatment groups. Also, due to different follow-up times for individual patients, the difference or OR based on incidences is not an appropriate effect measure to compare treatment groups for safety. The clinical experts CADTH consulted for this review indicated that the overall safety profile of nivolumab-relatlimab FDC was largely similar to that of nivolumab monotherapy, although a numerically higher frequency of AEs and grade 3 or 4 AEs was observed in the nivolumab-relatlimab FDC group, and the safety profile of nivolumab-relatlimab FDC was manageable and consistent with the known mechanisms of action of relatlimab and nivolumab. No new safety signal was identified.
The sponsor-submitted ITCs showed that, for safety, when comparing nivolumab-relatlimab FDC relative to nivolumab (1 mg/kg) combined with ipilimumab (3 mg/kg), nivolumab-relatlimab FDC was associated with a lower proportion of patients experiencing grade 3 or 4 AEs (overall and treatment-related), as well as lower discontinuations due to AEs (overall and treatment-related). Conversely, nivolumab-relatlimab FDC was associated with higher proportions of patients experiencing grade 3 or 4 treatment-related AEs relative to nivolumab monotherapy, ipilimumab monotherapy, and pembrolizumab monotherapy. Further, nivolumab-relatlimab FDC was associated with increased proportions of patients discontinuing due to overall AEs relative to nivolumab monotherapy and discontinuations due to treatment-related AEs relative to nivolumab monotherapy and ipilimumab monotherapy. Safety data on other IO therapies were unavailable. No data on patient-reported outcomes were presented.
Conclusion
Evidence from the RELATIVITY-047 trial showed that nivolumab-relatlimab FDC therapy compared with nivolumab monotherapy resulted in clinically meaningful benefits in terms of PFS (high certainty) in the treatment of adult patients with unresectable or metastatic melanoma who have not received prior systemic therapy for unresectable or metastatic melanoma. Nivolumab-relatlimab FDC may result in a clinically important increase in OS when compared with nivolumab monotherapy (low certainty). However, uncertainty remains in the OS results due to both imprecision (the CI included no difference between the nivolumab-relatlimab FDC and nivolumab monotherapy) and the inadequate length of follow-up for this outcome. Nivolumab-relatlimab FDC likely results in a clinically important increase in ORR when compared with nivolumab monotherapy (moderate certainty). However, the results were uncertain for the DoR of nivolumab-relatlimab FDC when compared with nivolumab monotherapy after a median follow-up of 25.3 months (low certainty). Nivolumab-relatlimab FDC may result in little to no difference (either improvement or deterioration) when compared with nivolumab monotherapy in HRQoL (measured by FACT-M, EQ-5D-3L utility index and EQ VAS) (low certainty). Numerically more patients appeared to experience grade 3 or 4 AEs in the nivolumab-relatlimab FDC group than in the nivolumab monotherapy group. However, the clinical experts CADTH consulted for this review indicated that the safety profile of nivolumab-relatlimab FDC appeared to be consistent with the known safety profile of each individual drug (nivolumab and relatlimab) and was generally manageable. No additional safety signals were identified. It should be emphasized that the efficacy and safety profile of nivolumab-relatlimab FDC compared with nivolumab monotherapy are not available for pediatric patients (aged ≥ 12 to < 18 years). In addition, the comparative efficacy and safety profiles of nivolumab-relatlimab FDC compared with the existing standard therapies (except nivolumab monotherapy) are not provided in the RELATIVITY-047 trial. The sponsor-submitted ITCs were inconclusive with respect to nivolumab-relatlimab FDC relative to combination IO (ipilimumab [3 mg/kg] combined with nivolumab [1 mg/kg], ipilimumab [1 mg/kg] combined with nivolumab [3 mg/kg], and cobimetinib-atezolizumab) for PFS and OS but did show prolonged OS and PFS relative to ipilimumab monotherapy and prolonged PFS relative to pembrolizumab monotherapy. In the sponsor-submitted ITCs, compared to nivolumab (1 mg/kg) combined with ipilimumab (3 mg/kg), nivolumab-relatlimab FDC demonstrated a favourable safety profile; however, compared to pembrolizumab, ipilimumab, and nivolumab monotherapy, nivolumab-relatlimab FDC had an unfavourable safety profile.
References
1.Symptoms of advanced melanoma. Oxford (UK): Cancer Research UK; 2023: https://www.cancerresearchuk.org/about-cancer/melanoma/advanced-melanoma/symptoms-advanced-melanoma. Accessed 2023 Sep 18.
2.Ghazawi FM, Cyr J, Darwich R, et al. Cutaneous malignant melanoma incidence and mortality trends in Canada: a comprehensive population-based study. J Am Acad Dermatol. 2019;80(2):448-459. PubMed
3.Rigo R, Doherty J, Koczka K, et al. Real world outcomes in patients with advanced melanoma treated in Alberta, Canada: a time-era based analysis. Curr Oncol. 2021;28(5):3978-3986. PubMed
4.Burns D, George J, Aucoin D, et al. The pathogenesis and clinical management of cutaneous melanoma: an evidence-based review. J Med Imaging Radiat Sci. 2019;50(3):460-469.e461. PubMed
5.Nessim C, Rotstein L, Goldstein D, et al. Princess Margaret Cancer Centre clinical practice guidelines: melanoma. 2015: https://www.uhn.ca/PrincessMargaret/Health_Professionals/Programs_Departments/Documents/CPG_Melanoma.pdf. Accessed 2023 Nov 10.
6.Diagnosis of melanoma skin cancer. Toronto (ON): Canadian Cancer Society; 2023: https://cancer.ca/en/cancer-information/cancer-types/skin-melanoma/diagnosis. Accessed 2023 Nov 10.
7.Melanoma. Chapter 4. diagnosis. Vancouver (BC): BC Cancer,; 2014: http://www.bccancer.bc.ca/health-professionals/clinical-resources/cancer-management-manual/skin/melanoma#Diagnosis. Accessed 2023 Nov 10.
8.Sosman J. Translating BRAF mutations into effective therapy for patients with melanoma. Presented at 2011 ASCO Annual Meeting; Chicago, IL; Jun 3-7, 2011. https://meetinglibrary.asco.org/record/43645/edbook. Accessed 2023 Feb 24.
9.Gwadry-Sridhar F, McConkey H, Teng X, Ernst DS. The national melanoma research registry: a fundamental for disease characterization and epidemiology. Ann Oncol. 2018;29:viii460.
10.Gutierrez-Castaneda LD, Nova JA, Tovar-Parra JD. Frequency of mutations in BRAF, NRAS, and KIT in different populations and histological subtypes of melanoma: a systemic review. Melanoma Res. 2020;30(1):62-70. PubMed
11.Long GV, Menzies AM, Nagrial AM, et al. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol. 2011;29(10):1239-1246. PubMed
12.Canadian Cancer Statistics Advisory Committee, in collaboration with the Canadian Cancer Society, Statistics Canada, and the Public Health Agency of Canada. Canadian Cancer Statistics 2021. Toronto (ON): Canadian Cancer Society; 2021: https://cdn.cancer.ca/-/media/files/research/cancer-statistics/2021-statistics/2021-pdf-en-final.pdf. Accessed 2023 Nov 10.
13.Brenner DR, Poirier A, Woods RR, et al. Projected estimates of cancer in Canada in 2022. CMAJ. 2022;194(17):E601-E607. PubMed
14.Table 13-10-0111-01: number and rates of new cases of primary cancer, by cancer type, age group and sex. Ottawa (ON): Statistics Canada; 2022: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310011101&pickMembers%5B0%5D=2.1&pickMembers%5B1%5D=3.1&pickMembers%5B2%5D=4.26&cubeTimeFrame.startYear=2015&cubeTimeFrame.endYear=2019&referencePeriods=20150101%2C20190101. Accessed 2023 Mar 10.
15.Bhatia S, Tykodi SS, Lee SM, Thompson JA. Systemic therapy of metastatic melanoma: on the road to cure. Oncology (Williston Park). 2015;29(2):126-135. PubMed
16.Targeted therapy for melanoma skin cancer. Toronto (ON): Canadian Cancer Society; 2023: https://cancer.ca/en/cancer-information/cancer-types/skin-melanoma/treatment/targeted-therapy. Accessed 2023 Feb 24.
17.Provisional funding algorithm: melanoma. CADTH reimbursement review. Ottawa (ON): CADTH; 2023: https://www.cadth.ca/melanoma. Accessed 2023 Nov 10.
18.pCODR Expert Review Committee (pERC) final recommendation: nivolumab (Opdivo) in combination with ipilmumab (Yervoy) for metastatic melanoma. 2017: https://www.cadth.ca/sites/default/files/pcodr/pcodr_opdivo_yervoy_metmela_fn_rec.pdf. Accessed 2023 feb 24.
19.PrOPDIVO® (nivolumab): 10 mg nivolumab /mL in 40 mg and 100 mg single-use vials for intravenous infusion [product monograph]. Montreal (QC): Bristol-Myers Squibb Canada; 2022. Accessed 2023 Nov 10.
20.PrYERVOY® (ipilimumab): 5 mg/mL for intravenous infusion [product monograph]. Montreal (QC): Bristol-Myers Squibb Canada; 2022.
21.pCODR Expert Review Committee (pERC) final recommendation: nivolumab (Opdivo) for metastatic melanoma. Ottawa (ON): CADTH; 2016: https://www.cadth.ca/sites/default/files/pcodr/nivolumab_opdivo_mm_fn_rec.pdf. Accessed 2023 Feb 24.
22.pCODR Expert Review Committee (pERC) final recommendation: ipilimumab (Yervoy) for first line advanced melanoma. 2014: https://www.cadth.ca/sites/default/files/pcodr/pcodr-yervoy1st-fn-rec.pdf. Accessed 2023 Feb 24.
23.PrKEYTRUDA® (pembrolizumab): 100 mg/4 mL vial solution for infusion [product monograph]. Kirkland (QC): Merck Canada; 2023 Dec 12.
24.pCODR Expert Review Committee (pERC) final recommendation: pembrolizumab (Keytruda) for metastatic melanoma. 2015: https://www.cadth.ca/sites/default/files/pcodr/pcodr_pembrolizumab_keytruda_mm_fn_rec.pdf. Accessed 2023 Feb 24.
25.Atkins MB, Lee SJ, Chmielowski B, et al. Combination dabrafenib and trametinib versus combination nivolumab and ipilimumab for patients with advanced BRAF-mutant melanoma: the DREAMseq trial-ECOG-ACRIN EA6134. J Clin Oncol. 2023;41(2):186-197. PubMed
26.PrMEKTOVI® (binimetinib): 15 mg tablets [product monograph]. Kirkland (QC): Pfizer Canada; 2021 Mar 2.
27.PrBRAFTOVI® (encorafenib): 75 mg oral capsules [product monograph]. Kirkland (AC): Pfizer Canada ULC; 2022 Apr 7.
28.CADTH reimbursement recommendation: encorafenib (Braftovi) in combination with binimetinib (Mektovi). Can J Health Technol. 2021;1(7).
29.pCODR Expert Review Committee (pERC) final recommendation: cobimetinib (Cotellic) and vemurafenib (Zelboraf) for metastatic melanoma. Ottawa (ON): CADTH; 2016: https://www.cadth.ca/sites/default/files/pcodr/pcodr_cobimetinib_cotellic_metmela_fn_rec.pdf. Accessed 2023 Feb 24.
30.PrCOTELLIC® (cobimetinib): 20 mg tablets [product monograph]. Mississauga (ON): Hoffmann-La Roche; 2018 Jan 5.
31.PrZELBORAF® (vemurafenib): 240 mg film-coated tablet [product monograph]. Mississauga (ON): Hoffmann-La Roche; 2019 Dec 10.
32.PrTAFINLAR® (dabrafenib): 50 mg and 75 mg oral capsules, 10 mg tablets for suspension [product monograph]. Montreal (QC): Novartis Pharmaceuticals Canada; 2022.
33.PrMEKINIST® (trametinib): 0.5 mg and 2 mg tablets [product monograph]. Montreal (QC): Novartis Pharmaceuticals Canada; 2022.
34.pCODR Expert Review Committee (pERC) final recommendation: dabrafenib (Tafinlar) in combination with trametinib (Mekinist) for metastatic melanoma. 2015: https://www.cadth.ca/sites/default/files/pcodr/pcodr_tafinlar_mekinist_metmelanoma_fn_rec.pdf. Accessed 2023 Feb 24.
35.Melanoma specialist interviews for Canadian reimbursement submissions of Opdualag conducted March/April 2023 [internal sponsor's report]. Montreal (QC); Bristol-Myers Squibb Canada; 2023.
36.PrOPDUALAGTM (nivolumab and relatlimab): 240 mg nivolumab/20 mL (12 mg/mL) and 80 mg relatlimab/20mL (4 mg/mL) single-use vial for injection [draft product monograph]. Montreal (QC): Bristol-Myers Squibb; 2023.
37.Paik J. Nivolumab plus relatlimab: first approval. Drugs. 2022;82(8):925-931. PubMed
38.Raschi E, Comito F, Massari F, Gelsomino F. Relatlimab and nivolumab in untreated advanced melanoma: insight into RELATIVITY. Immunotherapy. 2023;15(2):85-91. PubMed
39.Lee A. Nivolumab/relatlimab in advanced melanoma: a profile of its use. Drugs Ther Perspect. 2023;39:89–95.
40.PrOPDUALAGTM (nivolumab and relatlimab): 240 mg nivolumab/20 mL (12 mg/mL) and 80 mg relatlimab/20mL (4 mg/mL) single-use vial solution for injection [product monograph] Montreal (QC): Bristol-Myers Squibb; 2023 Sep 13.
41.Primary Clinical Study Report: CA224047. A randomized, double-blind phase 2/3 study of relatlimab combined with nivolumab versus nivolumab in participants with previously untreated metastatic or unresectable melanoma [internal sponsor's report]. Montreal (QC): Bristol-Myers Squibb Canada; 2023 Jul 10.
42.Statistical analysis plan for CSR: a randomized, double-blind phase 2/3 study of relatlimab combined with nivolumab versus nivolumab in participants with previously untreated metastatic or unresectable melanoma V6.0. [internal sponsor's report]. Montreal (QC); Bristol-Myers Squibb Canada; 2023 Jul 10.
43.Balshem H, Helfand M, Schünemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-406. PubMed
44.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. PubMed
45.Kuk D, Shoushtari AN, Barker CA, et al. Prognosis of mucosal, uveal, acral, nonacral cutaneous, and unknown primary melanoma from the time of first metastasis. Oncologist. 2016;21(7):848-854. PubMed
46.Curti BD, Leachman S, Wj U. Chapter 72: Cancer of the skin. In: Jameson J, Fauci AS, Kasper DL, Hauser SL, Longo DL, Loscalzo J, eds. 20th ed. New York (NY): McGraw-Hill; 2019.
47.Types of melanoma skin cancer. Toronto (ON): Canadian Cancer Society; 2023: https://cancer.ca/en/cancer-information/cancer-types/skin-melanoma/what-is-melanoma/types-of-melanoma. Accessed 2023 Feb 22.
48.Lindqvist Bagge AS, Wesslau H, Cizek R, et al. Health-related quality of life using the FACT-M questionnaire in patients with malignant melanoma: A systematic review. Eur J Surg Oncol. 2022;48(2):312-319. PubMed
49.Cheung WY, Bayliss MS, White MK, et al. Humanistic burden of disease for patients with advanced melanoma in Canada. Support Care Cancer. 2018;26(6):1985-1991. PubMed
50.Joshua AM. Melanoma prevention: are we doing enough? A Canadian perspective. Curr Oncol. 2012;19(6):e462-467. PubMed
51.Immunotherapy for melanoma skin cancer. Toronto (ON): Canadian Cancer Society; 2023: https://cancer.ca/en/cancer-information/cancer-types/skin-melanoma/treatment/immunotherapy. Accessed 2023 Nov 10.
52.Treatments for metastatic melanoma skin cancer. Toronto (ON): Canadian Cancer Society; 2023: https://cancer.ca/en/cancer-information/cancer-types/skin-melanoma/treatment/metastatic. Accessed 2023 Feb 22.
53.Surgery for melanoma skin cancer. Toronto (ON): Canadian Cancer Society; 2023: https://cancer.ca/en/cancer-information/cancer-types/skin-melanoma/treatment/surgery. Accessed 2023 Feb 22.
54.Dabrafenib (Tafinlar) in combination with trametinib (Mekinist). (CADTH pan-Canadian Oncology Drug Review: final clinical guidance report). Ottawa (ON): CADTH; 2015: https://cadth.ca/sites/default/files/pcodr/pcodr_tafinlar_mekinist_metmelanoma_fn_cgr.pdf. Accessed 2021 Apr 23.
55.Cobimetinib (Cotellic) for metastatic melanoma. (CADTH pan-Canadian Oncology Drug Review: final clinical guidance report). Ottawa (ON): CADTH; 2016: https://cadth.ca/sites/default/files/pcodr/pcodr_cobimetinib_cotellic_metmela_fn_cgr.pdf. Accessed 2021 Apr 26.
56.Bristol-Myers Squibb. NCT03470922. A study of relatlimab plus nivolumab versus nivolumab alone in participants with advanced melanoma (RELATIVITY-047). Bethesda (MD): U.S. National Library of Medicine; 2022: https://clinicaltrials.gov/ct2/show/NCT03470922. Accessed 2023 Jun 26.
57.Tawbi HA, Schadendorf D, Lipson EJ, et al. Relatlimab and nivolumab versus nivolumab in untreated advanced melanoma. N Engl J Med. 2022;386(1):24-34. PubMed
58.Schadendorf D, Tawbi H, Lipson E, et al. Health-related quality of life with nivolumab plus relatlimab versus nivolumab monotherapy in patients with previously untreated unresectable or metastatic melanoma: RELATIVITY-047 trial. Eur J Cancer. 2023;187:164-173. PubMed
59.Long GV, Hodi FS, Lipson EJ, et al. Overall survival and response with nivolumab and relatlimab in advanced melanoma. NEJM Evidence. 2023;2(4):EVIDoa2200239.
60.Long GV, Hodi FS, Lipson EJ, et al. Overall survival and response with nivolumab and relatlimab in advanced melanoma [correction]. NEJM Evidence. 2023;2(4):EVIDoa2200239.
61.Addendum 01. In: Primary Clinical Study Report: CA224047. A randomized, double-blind phase 2/3 study of relatlimab combined with nivolumab versus nivolumab in participants with previously untreated metastatic or unresectable melanoma [internal sponsor's report]. Montreal (QC): Bristol-Myers Squibb Canada; 2023 Jul 10.
62.Addendum 02. In: Primary Clinical Study Report: CA224047. A randomized, double-blind phase 2/3 study of relatlimab combined with nivolumab versus nivolumab in participants with previously untreated metastatic or unresectable melanoma [internal sponsor's report]. Montreal (QC): Bristol-Myers Squibb Canada; 2023 Jul 10.
63.Clinical Protocol CA224047: A randomized, double-blind phase 2/3 study of relatlimab combined with nivolumab versus nivolumab in participants with previously untreated metastatic or unresectable melanoma [internal sponsor's report]. Montreal (QC): Bristol-Myers Squibb Canada; 2023 Jul 10.
64.The EuroQol Group. EuroQol--a new facility for the measurement of health-related quality of life. Health Policy. 1990;16(3):199-208. PubMed
65.Pickard AS, Neary MP, Cella D. Estimation of minimally important differences in EQ-5D utility and VAS scores in cancer. Health Qual Life Outcomes. 2007;5:70. PubMed
66.Cormier JN, Ross MI, Gershenwald JE, et al. Prospective assessment of the reliability, validity, and sensitivity to change of the Functional Assessment of Cancer Therapy-Melanoma questionnaire. Cancer. 2008;112(10):2249-2257. PubMed
67.Askew RL, Xing Y, Palmer JL, Cella D, Moye LA, Cormier JN. Evaluating minimal important differences for the FACT-Melanoma quality of life questionnaire. Value Health. 2009;12(8):1144-1150. PubMed
68.Robinson DW, Jr., Cormier JN, Zhao N, Uhlar CM, Revicki DA, Cella D. Health-related quality of life among patients with metastatic melanoma: results from an international phase 2 multicentre study. Melanoma Res. 2012;22(1):54-62. PubMed
69.Brooks R. EuroQol: the current state of play. Health Policy. 1996;37(1):53-72. PubMed
70.Medidata rave RTSM agile randomization and trial supply management [factsheet]. New York (NY): Medidata: https://www.medidata.com/wp-content/uploads/2019/05/Customer-Facing-Fact-Sheet-Rave-RTSM-May-19.pdf. Accessed 2023 Sep 19.
71.Additional data received from sponsor on Sept. 11, 2023. Montreal (QC); Bristol-Myers Squibb Canada.
72.Systematic literature review and network meta-analysis of relatlimab + nivolumab and first-line (1L) treatment options for advanced melanoma in a ITT population [internal sponsor's report]. Montreal (QC); Bristol-Myers Squibb Canada; 2023 Jul 10.
Pharmacoeconomic Review
Abbreviations
AE
adverse event
BIA
budget impact analysis
ICER
incremental cost-effectiveness ratio
MEK
mitogen-activated protein kinase enzyme
NMA
network meta-analysis
OS
overall survival
PFS
progression-free survival
PSM
partitioned survival model
QALY
quality-adjusted life-year
TTD
time to treatment discontinuation
WTP
willingness-to-pay
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Nivolumab plus relatlimab (Opdualag), single-use vial |
Submitted price | Nivolumab 240 mg plus relatlimab 80 mg, in a fixed-dose combination: $8,315 per 20 mL vial |
Indication | Adult and pediatric patients (aged 12 years and older and weighing at least 40 kg) with unresectable or metastatic melanoma who have not received prior systemic therapy for unresectable or metastatic melanoma |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | September 13, 2023 |
Reimbursement request | As per indication |
Sponsor | Bristol Myers Squibb Canada |
Submission history | Previously reviewed: No |
NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Partitioned survival model |
Target population | Adult and pediatric patients (aged 12 years and older and weighing at least 40 kg) with unresectable or metastatic melanoma |
Treatment | Nivolumab-relatlimab |
Comparators | Nivolumab monotherapy Ipilimumab monotherapy Pembrolizumab monotherapy Nivolumab-ipilimumab Vemurafenib-cobimetinib Dabrafenib-trametinib Encorafenib-binimetinib |
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, life-years |
Time horizon | Lifetime (25 years) |
Key data sources | RELATIVITY-047 Sponsor-submitted systematic review and network meta-analysis |
Submitted results | Nivolumab-relatlimab was more costly and more effective than the other treatments on the cost-effectiveness frontier (nivolumab monotherapy and nivolumab-ipilimumab); the ICER for nivolumab-relatlimab relative to nivolumab-ipilimumab was $610,904 per QALY gained (incremental cost = $124,726; incremental QALYs = 0.204) |
Key limitations |
|
CADTH reanalysis results |
|
ICER = incremental cost-effectiveness ratio; OS = overall survival; PSM = partitioned survival model; QALY = quality-adjusted life-year.
Conclusions
The CADTH Clinical Review found that treatment with nivolumab-relatlimab produces a clinically meaningful benefit over nivolumab monotherapy in terms of progression-free survival (PFS). but the evidence for an overall survival (OS) benefit was inconclusive. The Clinical Review also found that the submitted indirect treatment comparison of immunotherapies suggested that nivolumab-relatlimab offered a PFS benefit relative to ipilimumab and pembrolizumab as well as an OS benefit compared with ipilimumab. Meanwhile, estimates of relative efficacy between nivolumab-relatlimab and the BRAF and mitogen-activated protein kinase enzyme (MEK) inhibitors were subject to considerable uncertainty due to a failure to consider subpopulation-specific trial data. Both PFS and OS are key parameters that affect the estimates of state membership, and by extension expected costs and quality-adjusted life-years (QALYs), for each arm of the decision model.
The results of CADTH’s economic analysis concluded that nivolumab-relatlimab would not be cost-effective at a willingness-to-pay (WTP) threshold of $50,000 per QALY gained, which aligned with the sponsor’s base case. Relative to nivolumab-ipilimumab, nivolumab-relatlimab was more effective (0.178 incremental QALYs) and more costly ($72,548 in incremental costs), resulting in an incremental cost-effectiveness ratio (ICER) of $408,364 per QALY gained. CADTH’s base-case ICER was lower than that of the sponsor’s because of changes in survival assumptions, resulting in patients spending less time in the progression-free and progressed-disease health states. A price reduction would be required for nivolumab-relatlimab to be cost-effective at a threshold of $50,000 per QALY gained. The magnitude of the required price reduction is uncertain given the presence of an error in the model that was not corrected until after the review period (Appendix 4).
Clinical expert input suggested that treatment with BRAF and MEK inhibitors (dabrafenib-trametinib, vemurafenib-cobimetinib, and encorafenib-binimetinib) is commonly used in the first line for this patient population, while immunotherapy would be preserved as a second-line option. As a consequence of the uncertainty in the clinical evidence and the methodological limitations of the sponsor’s pharmacoeconomic model, CADTH was unable to estimate the cost-effectiveness of nivolumab-relatlimab in BRAF-positive patients. Given these uncertainties, and the inability to draw conclusions about whether nivolumab-relatlimab provides an OS benefit compared to other available treatments, a greater price reduction may be warranted.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process.
Patient input for this review was obtained from Melanoma Canada and the Save Your Skin Foundation. Information for both submissions was collected through an online survey, while the latter submission also included virtual patient-roundtables and one-on-one conversations. The Melanoma Canada submission included 203 responses, 119 of which were from patients (68% female) representing of each stage of metastatic disease. The submission from the Save Your Skin Foundation included 60 patients between the ages of 18 and 89 (62% female). Both submissions included responses from patients in Canada. Patients who had received current treatment options were willing to accept side effects in exchange for an effective treatment. The submissions noted that gaps in treatment included access for patients living in remote locations, as well as the availability of options with minimal side effects and longer response times. Two patients in the Melanoma Canada submission and 12 patients from the Save Your Skin Foundation submission had experience with combination nivolumab-relatlimab. The most frequent method of accessing the treatment was through participation in a clinical trial. While 2 patients had to discontinue treatment due to intolerance, the other respondents noted that they were willing to tolerate adverse events (AEs) in exchange for an effective treatment.
Registered clinician input was received from the provincial cutaneous tumour group of Alberta and the Ontario Health (Cancer Care Ontario) Drug Advisory Committee. In the first-line metastatic or unresectable setting, current treatments may include single-drug nivolumab and pembrolizumab and combination nivolumab-ipilimumab. Patients with a BRAF mutation are also eligible to receive BRAF-targeted agents (dabrafenib-trametinib, cobimetinib-vemurafinib, and binimetinib-encorafenib). Subsequent treatment options will depend on the first-line treatment selected. Following first-line use of pembrolizumab or nivolumab, patients may be eligible for ipilimumab alone or BRAF-targeted drugs. First-line use of combination nivolumab-ipilimumab will restrict second-line options to the BRAF-targeted drugs. Patients who receive first-line BRAF-targeted drugs may be eligible to receive second-line pembrolizumab, nivolumab, or combination ipilimumab-nivolumab. The submissions emphasized that nivolumab-relatlimab is expected to have a lower toxicity profile compared with combination ipilimumab-nivolumab.
Drug plans sought clarification on the appropriate place in therapy for nivolumab-relatlimab and the trade-offs of its use compared with currently available treatment options. Concerns were raised about the need for companion diagnostic testing, based on the eligibility criteria of the RELATIVITY-047 trial.
Two of these concerns were considered in the sponsor’s model:
the relative cost-effectiveness of nivolumab-relatlimab against current treatment options
the effect of AEs on patient health-related quality of life.
CADTH was unable to address the concern raised in stakeholder input regarding the need for companion diagnostic testing.
Economic Review
The current review is for nivolumab-relatlimab (Opdualag) for the treatment of adult and pediatric patients aged 12 years and older and weighing at least 40 kg with unresectable or metastatic melanoma.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted an economic evaluation comparing nivolumab-relatlimab with currently approved regimens for the treatment of adult or pediatric patients with unresectable or metastatic melanoma. This target population was aligned with the proposed Health Canada indication and the reimbursement request.
Nivolumab-relatlimab is available as a vial for IV administration at a dose of 240 mg of nivolumab and 80 mg of relatlimab. The submitted price was $8,315 per 20 mL vial. The recommended dosage is 480 mg of nivolumab and 160 mg of relatlimab every 4 weeks until disease progression or unacceptable toxicity. At the submitted price, this will cost $594 per day, or $16,630 every 28 days.
Following consultation with clinical experts, 7 alternatives were considered in the economic evaluation. These treatments were thought to reflect the current standard of care for unresectable or metastatic melanoma in Canada. The 4 alternative immunotherapies were: nivolumab monotherapy, ipilimumab monotherapy, pembrolizumab, and nivolumab-ipilimumab. In addition, the model considered 3 targeted therapies of BRAF and MEK inhibitors: dabrafenib-trametinib, encorafenib-binimetinib, and vemurafenib-cobimetinib.
Modelled outcomes included life-years and QALYs. Costs were estimated from the perspective of a public health care payer in Canada. Model outputs were generated over a lifetime horizon of 25 years, with a 7-day cycle length assumed for the first 28 cycles, after which a 28-day cycle length was used. Costs and outcomes were discounted at 1.5%.
Model Structure
The costs and effects of each treatment option were estimated using a partitioned survival model (PSM). This model structure relied on 2 independent survival curves (PFS and OS) to track patients across 3 distinct health states: progression-free, progressed disease, and death (Figure 1). State membership was determined using an “area under the curve” approach. A third survival curve, time to treatment discontinuation (TTD), was used to determine the proportion of patients in the PFS state no longer on treatment. This allowed the model to incorporate the possibility of treatment withdrawal before progression and capture the impact of medication use on costs.
Model Inputs
Costs and effects were estimated using a homogeneous baseline population. All data summarizing baseline characteristics of the cohort were obtained from the RELATIVITY-047 trial.1-3 This was a randomized phase II and III trial that directly compared nivolumab-relatlimab with nivolumab monotherapy in patients with previously untreated metastatic or unresectable melanoma.3 Data of interest included baseline age (mean = 62 years), sex (41.7% female), mean body weight (79.7 kg), and body surface area (1.82 m2).1-3
Estimates of the relative efficacy for PFS and OS were obtained from the submitted systematic review and network meta-analysis (NMA).1,4 This approach was justified by the absence of a direct comparison of nivolumab-relatlimab with every specified comparator in the economic evaluation. Studies of interest included first-line treatments of adult patients with previously untreated unresectable or metastatic melanoma.4 For both OS and PFS, a fractional polynomial NMA was used to estimate a time-varying hazard ratio for each alternative relative to nivolumab-relatlimab. Due to limitations in the network of evidence from the submitted trials, several simplifying assumptions would have been necessary to consider a single population using the estimates of relative efficacy. Instead, the sponsor used separate networks to establish the relative efficacy against the immunotherapies and BRAF- and MEK-targeted therapies. In the base case, the sponsor restricted the evidence network to the trials of the immunotherapy treatments. This meant that nivolumab-relatlimab was only compared against nivolumab-ipilimumab, nivolumab, ipilimumab, and pembrolizumab. Consideration of every specified alternative for the economic evaluation was explored in a separate scenario analysis, which considered the complete evidence network of immunotherapies and BRAF- and MEK-targeted therapies.1,4
The model relied on direct and indirect methods to generate the survival curves for PFS, OS, and TTD. For nivolumab-relatlimab and nivolumab, independent parametric survival curves for each event were fitted to the corresponding time-to-event data from the RELATIVITY-047 trial. These parametric models were used to predict survival probabilities for each cycle of the model.1 Parametric models that explored the relationship between the treatment arm and the survival event were considered in a separate scenario analysis. Models were fit using the exponential, log-logistic, log-normal, Weibull, Gompertz, gamma, and generalized gamma distributions.1,5 Based on assessment of model fit statistics, the submitted base case assumed a Gompertz distribution for PFS and OS and a Weibull distribution for TTD.1 Unlike the OS and TTD curves, the predicted values of the PFS curve were not generated from a single source. The sponsor assumed that the PFS values would follow the univariate Kaplan-Meier estimate in the first 3 months of the model’s time horizon and then switch to the predicted values from the parametric survival model.1 For the remaining treatments, the predicted values for OS and PFS were generated by applying an estimate of relative efficacy to a reference survival curve. This indirect prediction of the survival probability relied on 2 inputs: the time-varying hazard ratios obtained from the NMA and the use of the nivolumab-relatlimab reference curve for both PFS and OS.
Predicted values for the TTD survival probabilities were generated from a range of sources. Both nivolumab-ipilimumab and ipilimumab monotherapy were assumed to follow a univariate Kaplan-Meier estimate from the CheckMate-067 trial.1,6 At the end of the 7.5-year period covered by the data, it was assumed that all patients on these treatments would be off treatment.1 For pembrolizumab, it was assumed that TTD would follow the predicted values for nivolumab in the RELATIVITY-047 trial.1 In the model base case, a 2-year stopping rule was assumed for all immunotherapies. For the BRAF and MEK inhibitors, it was assumed that PFS would be an appropriate proxy for TTD.1
To ensure that the risk of death in the cohort would not fall below that expected for the general population, the predicted values for the OS curve were capped by the general population mortality risk. This was achieved using age- and gender-specific mortality risks estimated from Canadian Life Tables published by Statistics Canada.1,7
In addition to tracking the proportion of the cohort in each health state, the model also tracked the occurrence of AEs. The specific AEs included in the model were at least grade 3: nausea, diarrhea, vomiting, fatigue, arthralgia, colitis, pyrexia, abdominal pain, rash, hypertension, back pain, peripheral edema, pain in extremity, increased aspartate transaminase, and hepatitis.1 The treatment specific risk of each AE was obtained from the RELATIVITY-047 trial or 1 of the trials identified in the submitted systematic literature review.2,4,8-11
Health-related quality of life was captured in the model by combining health-state utilities with disutilities associated with each AE. The health-state utility values were obtained from the indirect measurement of patient preferences in the RELATIVITY-047 trial using the EQ-5D questionnaire. Mean utilities of 0.81 and 0.79 were estimated for the progression-free and progressed-disease health states, respectively, by applying tariffs that corresponded to preferences of the population in Canada.1,12 Values for AE-specific disutilities were sourced from a direct preference study of melanoma patients in Canada, a Cochrane review, and economic evaluation for chronic lymphocytic leukemia, and a technology appraisal from another jurisdiction.13-15 AE-specific disutilities were applied as a one-off in the first model cycle.1
The submission considered costs associated with the acquisition, administration, and monitoring of first-line therapy as well as those associated with the management of AEs, subsequent therapy, and end-of-life care. Treatment-acquisition costs were determined by applying the treatment prices to the dosing schedule for each alternative first-line therapy considered in the model. Dosing was established from the recommended dosage listed in the relevant product monographs. The price of nivolumab-relatlimab reflected the sponsor’s submitted price. The price of all other drugs was derived from the 3 previous CADTH reports.1,16-18 If different prices were identified for a single product, the sponsor selected the most recently published price.1 Treatment-administration costs, including infusion, pharmacy workload, and nursing workload, were considered for any treatment that required IV administration (nivolumab-relatlimab, nivolumab, ipilimumab, and pembrolizumab). For second-line treatment, costs were calculated by combining the acquisition and administration costs weighted by the percent of patients assumed to be on each alternative. The cost of such treatments was applied as a one-time cost upon treatment discontinuation.
Treatment-monitoring costs were determined using a unit-costing approach based on a resource-utilization study of patients with metastatic melanoma.1,19 In the base case, the frequency of each event was assumed to follow the estimates reported in the original resource-utilization study.1,19 A separate scenario was also considered in which the frequency of each event was assumed to follow estimates from a clinician survey conducted by the sponsor.1 Prices for each event were obtained from the Ontario Schedule of Benefits.20
Both AE costs and end-of-life costs were applied as one-off costs in the model. Costs related to AEs were applied in the first cycle of the model using data obtained from the Ontario Case Costing Initiative and Alberta’s interactive Health Data Application.1,21,22 However, end-of-life costs were applied upon entry to the death state using data obtained from a study by Yu et al. in the base case and the Ontario Case Costing Initiative in a separate scenario.1,21,23
Summary of Sponsor’s Economic Evaluation Results
The costs and QALYs of each alternative were generated using a Monte Carlo simulation. While results from the base case were generated from a simulation of 5,000 iterations, those for each scenario were limited to 350 iterations. Results from the probabilistic base case were aligned with those generated deterministically. Results from the probabilistic base case are summarized in the following section.
Base-Case Results
The submitted analysis was based on the publicly available prices of the comparator treatments. Results from the base case of the submitted economic evaluation are presented in Table 3.
The expected costs and QALYs for nivolumab-relatlimab were $369,420 and 6.891, respectively. Of the 5 immunotherapies included in the base case, 3 treatments were found to be on the cost-effectiveness frontier: nivolumab, nivolumab-ipilimumab, and nivolumab-relatlimab. Among these alternatives, nivolumab-relatlimab was the most expensive, with an ICER of $610,904 compared to nivolumab-ipilimumab. At a WTP threshold of $50,000 per QALY gained, nivolumab-relatlimab had a 0% probability of being cost-effective.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Total QALYs | ICER ($ per QALY) |
|---|---|---|---|
Base case: Immunotherapy network | |||
Nivolumab | 181,185 | 5.755 | Reference |
Nivolumab-ipilimumab | 244,694 | 6.687 | 68,106 |
Pembrolizumab | 268,928 | 5.122 | Dominated |
Ipilimumab | 271,336 | 3.533 | Dominated |
Nivolumab-relatlimab | 369,420 | 6.891 | 610,904 |
Scenario: Complete evidence network | |||
Nivolumab | 181,185 | 5.755 | Reference |
Nivolumab-ipilimumab | 244,694 | 6.687 | 68,106 |
Pembrolizumab | 266,928 | 5.122 | Dominated |
Ipilimumab | 271,336 | 3.533 | Dominated |
Nivolumab-relatlimab | 369,420 | 6.891 | 610,904 |
Dabrafenib-trametinib | 783,509 | 3.760 | Dominated |
Vemurafenib-cobimetinib | 820,130 | 3.192 | Dominated |
Encorafenib-binimetinib | 1,098,234 | 4.078 | Dominated |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Source: Sponsor’s pharmacoeconomic submission.1
Sensitivity and Scenario Analysis Results
Given that the submitted base case was restricted to the estimates of relative efficacy generated using the immunotherapy network, a separate analysis was conducted using all available evidence from the NMA. In this analysis, the expected costs and QALYs for nivolumab-relatlimab were $369,420 and 6.891, respectively. Of the 8 alternative treatments included in this scenario, only 3 were on the cost-effectiveness frontier: nivolumab, nivolumab-ipilimumab, and nivolumab-relatlimab. As with the submitted base case, nivolumab-relatlimab was the most expensive and the most effective, with an ICER of $610,904 relative to nivolumab-ipilimumab. At a WTP threshold of $50,000 per QALY gained, nivolumab-relatlimab had a 0% probability of being cost-effective.
In addition to the base case and NMA scenario, 13 different scenario analyses were considered. No scenario analysis was conducted using a perspective other than the health care payer. While each considered scenario had a slight impact on the estimated costs and effects, none had a meaningful effect on the conclusion for the cost-effectiveness of nivolumab-relatlimab.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis:
Estimates of relative treatment efficacy are highly uncertain: The submitted economic evaluation compared nivolumab-relatlimab with relevant alternatives that could be used as first-line treatment in the indicated population. These included alternative immunotherapies (nivolumab, ipilimumab, pembrolizumab, nivolumab-ipilimumab) and targeted BRAF and MEK inhibitors (dabrafenib-trametinib, encorafenib-binimetinib, vemurafenib-cobimetinib) for the BRAF-positive subpopulation. The CADTH Clinical Review of the direct evidence from the RELATIVITY-047 trial concluded that nivolumab-relatlimab could achieve a superior PFS benefit relative to nivolumab. Conclusions regarding an OS benefit in the direct comparison of nivolumab-relatlimab with nivolumab could not be drawn due to limitations in the trial design for this outcome. Importantly, the RELATIVITY-047 trial failed to enrol any adolescents aged between 12 and 18 years. As a result, the safety and efficacy of nivolumab-relatlimab have not been established in this subgroup of the indicated Health Canada population.
Relative treatment efficacy with the remaining comparators was also assessed in the sponsor-submitted NMA of PFS and OS. Among trials of immuno-oncology therapies, nivolumab-relatlimab was associated with improvements in OS relative to ipilimumab, but no other treatment. In addition, a PFS benefit was observed for nivolumab-relatlimab relative to ipilimumab and pembrolizumab. Despite these findings, the CADTH Clinical Review could not reach definitive conclusions regarding the relative efficacy for OS or PFS. Due to the nature of the compiled evidence network, evidence of the relative effect of nivolumab-relatlimab compared to BRAF and MEK inhibitors was substantially limited. Furthermore, the evidence used in the NMA for the immunotherapy network was subject to several important limitations with respect to potential effect modification from between-trial differences in BRAF mutation status and incomplete ascertainment of programmed cell death ligand 1 status between trials. The high degree of uncertainty in the estimates of relative treatment efficacy is a meaningful source of decision uncertainty (expected costs and QALYs) within the economic evaluation.
The sponsor’s analysis considers a homogeneous patient cohort: The economic evaluation assumed a homogeneous population of patients with unresectable or metastatic melanoma. In the submitted base case, concerns regarding the comparability of populations in the NMA led to the exclusion of the BRAF- and MEK-targeted therapies, despite their relevance to the decision problem. The sponsor attempted to address this limitation by considering the full network of evidence in each NMA (i.e., immunotherapies and BRAF- and MEK-targeted therapies). This was insufficient because it failed to consider differences in indication between the 2 different types of treatment. Clinical experts consulted by CADTH confirmed that BRAF and MEK treatments would be appropriate first-line alternatives, and that such treatments are indicated for patients with BRAF-positive tumours. However, the sponsor’s base case and scenario analysis relied on estimates of relative efficacy that did not distinguish patients by BRAF mutation status. Given the presence of such heterogeneity in the indicated population, the sponsor’s decision to conduct an analysis with a base case that excluded relevant comparators and an approach to evidence synthesis and economic evaluation that assumed a homogeneous population was not appropriate.
In the presence of such heterogeneity, CADTH guidelines recommend a stratified approach to economic evaluation in which the population is divided into homogeneous subgroups: BRAF-positive and BRAF-negative.5 This would allow for the estimation of expected costs and effects using subgroup-specific parameter estimates for the relative treatment efficacy (and potentially baseline characteristics).5 It appears such an approach may have been feasible, as the sponsor acknowledged the availability of subgroup-specific data for all trials included in the NMA.4 Rather than using a mixed trial population for both networks, the immunotherapy and complete evidence networks should have been restricted to the BRAF-negative and BRAF-positive subgroups. Given that the decision problem relates to the entire population, the expected costs and effects for each treatment would then need to be combined using a weighted average of the common treatments between each subgroup. Due to the differences in parameter uncertainty for each subpopulation, the use of subgroup-specific parameter values should return results that are considerably different from those of the sponsor’s base case or scenario analysis.
CADTH was unable to address this limitation. Relative efficacy parameters were only available for the mixed trial populations, and estimating subgroup-specific values was beyond the scope of CADTH review. Furthermore, the incorporation of the subgroup-specific estimates of relative efficacy would have necessitated a significant redevelopment of the submitted decision model. Such tasks are beyond the scope of this appraisal.
To ensure a homogeneous population, the CADTH base case considered the estimates of relative efficacy generated by the immunotherapy network in the submitted evidence synthesis.
An inappropriate method was used to calculate state membership in model: When estimating state membership with a PSM, the estimates for each mutually exclusive health state (progression-free, progressed disease, and death) must be established from a series of non–mutually exclusive survival functions.24 In the submitted economic evaluation, this requirement was not satisfied due to the decision to cap the predicted OS curves by the general population mortality risk. This is problematic because the 2 parameters are not interchangeable. The general population mortality risk refers to the probability of death at a specific age. In contrast, the survival probability refers to the probability that death will not occur after a specific point in time. Given that the general population mortality risk is not a survival probability, it cannot be used to determine state membership in a PSM.
CADTH modified the economic evaluation to remove the assumption capping the OS curve by the general population mortality risk.
Parameter uncertainty for PFS was improperly characterized: In predicting the PFS probabilities for nivolumab-relatlimab and nivolumab monotherapy, the sponsor assumed that the values would follow the univariate Kaplan-Meier estimates for 3 months and then switch to the predicted values from the selected parametric survival function. The sponsor justified this approach by noting that the parametric distributions did not represent a good fit to the observed short-term trial data. This approach was problematic because it did not allow the full range of predicted PFS values to be reflected in the characterization of parameter uncertainty. To avoid this situation, CADTH submission guidelines state that all parametric distributions must be implemented for the entire time horizon of the model.25
CADTH modified the economic evaluation to ensure a single source was used to estimate the predicted survival probabilities for PFS.
Extrapolations of long-term survival produced unrealistic results: In the submitted base case, the sponsor assumed that the predicted values for OS and PFS would both follow a Gompertz distribution for nivolumab-relatlimab and nivolumab. As illustrated in Figure 2, this represented the most optimistic long-term survival benefit for both outcomes and treatments. Given that the predicted values for the comparators were generated indirectly using the estimates of relative treatment effect, the sponsor’s base case may have overestimated the relevant improvement in OS and PFS for nivolumab-relatlimab. This can be attributed to the fact that the use of the Gompertz distribution implies that both nivolumab and nivolumab-relatlimab would be able to cure a meaningful number of patients. Clinical experts consulted by CADTH expressed serious concerns about whether this would be a likely outcome from long-term treatment. It was suggested that more appropriate estimates of PFS and OS would be generated from distributions that generate less-optimistic long-term survival estimates, such as exponential or gamma distributions.
CADTH modified the economic evaluation to generate an alternative estimate of long-term survival that was more aligned with the expectations of the clinical experts. The PFS and OS curves for nivolumab-relatlimab and nivolumab were assumed to follow an exponential distribution.
Decision uncertainty cannot be accurately characterized: Consistent with CADTH guidelines, the sponsor’s base case used a Monte Carlo simulation to characterize parameter uncertainty. However, the mechanism by which state membership is estimated in a PSM limits the usefulness of this approach.24 This can be attributed to the fact that the survival probabilities (PFS and OS) are predicted independently over the specified time horizon.24 As a result, there is a risk that some simulation trials represent scenarios in which PFS exceeds OS, which is clinically implausible. One way to avoid this problem would be to generate estimates of state occupancy using a Markov chain. In the context of a PSM, it would be necessary to resample the trial data using bootstrapping to produce a series of correlated survival curves for OS and PFS. While this may resolve concerns relating to the within-trial period, limitations for the extrapolated period will persist.24
CADTH was unable to address this limitation. The probabilistic results were preserved in the CADTH base case to capture the uncertainty in the remaining model parameters.
Administration costs for nivolumab are uncertain: Late in the review process, the sponsor notified CADTH of a calculation error in its submitted pharmacoeconomic model. This error concerned the dosing schedule for nivolumab. To address this error, the sponsor submitted an updated model file. The update to the model did not affect the model’s ability to estimate quality-adjusted survival but did have implications for cost. The costs of nivolumab monotherapy and nivolumab-ipilimumab are likely not accurately reflected in the sponsor’s original submission or in CADTH’s reanalysis.
CADTH was not able to validate the sponsor’s corrections to the model within the period of the review. CADTH performed a scenario analysis to explore the potential implications of the corrections. The results of this scenario analysis are presented in Appendix 4 (Table 11).
Additionally, the following key assumptions made by the sponsor were appraised by CADTH (Table 4).
CADTH Reanalyses of the Economic Evaluation
Base-Case Results
The CADTH base case was derived by making changes in model parameter values and assumptions, in consultation with clinical experts. A summary of the changes applied to the economic evaluation is presented in Table 4. To arrive at the CADTH base case, each independent change was applied in the sequence detailed in the table. In the analysis of the CADTH base case, the costs and effects for each alternative treatment strategy were generated from a Monte Carlo simulation of 4,000 iterations.
Table 4: CADTH Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
None | — | — |
Changes to derive the CADTH base case | ||
1. Predicted values for OS | Parametric OS curve capped by general population mortality risk | Removed; state membership was estimated using unmodified survival probabilities |
2. Predicted values for PFS | Predicted values for PFS were obtained from 2 sources:
| Predicted values for PFS were generated from a single source: parametric survival function |
3. Parametric distributions | Predicted values for OS and PFS were assumed to follow a Gompertz distribution | Predicted values for OS and PFS were assumed to follow an exponential distribution |
CADTH base case | Reanalysis 1 + 2 + 3 | |
OS = overall survival; PFS = progression-free survival.
Results from the CADTH base case are presented in Table 5. As with the sponsor’s base case, the results were generated using publicly available prices of comparator treatments. Results from the Monte Carlo simulation are summarized below.
The expected costs and QALYs for nivolumab-relatlimab were $354,869 and 3.713, respectively. Of the 5 immunotherapies included in the CADTH base case, 3 treatments were on the cost-effectiveness frontier: nivolumab, nivolumab-ipilimumab, and nivolumab-relatlimab. Among these alternatives, nivolumab-relatlimab was the most expensive and the most effective. Nivolumab-ipilimumab was associated with an ICER of $408,364 compared to nivolumab-relatlimab. At a WTP threshold of $50,000 per QALY gained, nivolumab-relatlimab had a 0% probability of being cost-effective compared to nivolumab-ipilimumab. Ipilimumab and pembrolizumab were more costly and less effective than (dominated by) nivolumab monotherapy. Additional details summarizing the CADTH base case are presented in Appendix 4.
Table 5: Summary of the CADTH Reanalysis Results
Drug | Total costs ($) | Total QALYs | ICER ($ per QALY) |
|---|---|---|---|
Nivolumab | 190,559 | 3.072 | Reference |
Nivolumab-ipilimumab | 282,285 | 3.535 | 198,150 |
Nivolumab-relatlimab | 354,869 | 3.713 | 408,364 |
Dominated treatments | |||
Ipilimumab | 212,078 | 1.925 | Dominated by nivolumab |
Pembrolizumab | 261,425 | 2.614 | Dominated by nivolumab |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Scenario Analysis Results
A series of scenario analyses were conducted to explore the impact of incremental reductions in the acquisition cost of nivolumab-relatlimab; the results are presented in Table 6. Using the deterministic results from the sponsor’s and CADTH’s base case, a price reduction of 65% would be necessary to achieve cost-effectiveness at a WTP threshold of $50,000 per QALY gained. Given the identified limitations of the underlying clinical evidence, and the fact that neither base case considered the impact of the parameter uncertainty on the decision, an even greater price reduction may be necessary. The magnitude of the price reduction is uncertain given the presence of an error in the model that was not corrected until after the review period (Appendix 4).
Issues for Consideration
All drugs on the cost-effectiveness frontier (nivolumab, relatlimab, and ipilimumab) are manufactured by the same sponsor. CADTH’s estimates of cost-effectiveness are based on publicly available list prices, which may not reflect actual acquisition costs incurred by public plans.
Table 6: CADTH Price-Reduction Analyses
Analysis | ICERs for nivolumab-relatlimab vs. alternatives ($ per QALY) | |
|---|---|---|
Price reduction | Sponsor base case | CADTH reanalysis |
No price reduction | 610,904 (vs. ipilimumab) | 408,364 (vs. ipilimumab) |
10% | 255,700 (vs. ipilimumab) | 299,357 (vs. nivolumab-ipilimumab) |
20% | 211,817 (vs. ipilimumab) | 192,599 (vs. nivolumab-ipilimumab) |
30% | 167,934 (vs. ipilimumab) | 158,406 (vs. nivolumab) |
40% | 124,051 (vs. ipilimumab) | 127,331 (vs. nivolumab) |
50% | 80,167 (vs. ipilimumab) | 96,225 (vs. nivolumab) |
60% | 58,078 (vs. nivolumab) | 65,180 (vs. nivolumab) |
65% | 49,357 (vs. nivolumab) | 49,642 (vs. nivolumab) |
70% | 40,637 (vs. nivolumab) | 34,104 (vs. nivolumab) |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Note: Data generated via deterministic simulation.
Overall Conclusions
The CADTH Clinical Review found that treatment with nivolumab-relatlimab produces a clinically meaningful benefit over nivolumab monotherapy in terms of PFS, but the evidence for an OS benefit is inconclusive. The Clinical Review also found that the submitted indirect treatment comparisons of immunotherapies suggested that nivolumab-relatlimab offers a PFS benefit relative to ipilimumab and pembrolizumab as well as an OS benefit relative to ipilimumab. Meanwhile, estimates of relative efficacy between nivolumab-relatlimab and the BRAF and MEK inhibitors were subject to considerable uncertainty due to a failure to consider subpopulation-specific trial data. Both PFS and OS are key parameters that affect the estimates of state membership, and by extension expected costs and QALYs, for each arm of the decision model.
CADTH identified additional limitations within the submitted economic evaluation. These limitations included inappropriate methods for calculating state membership, mixing of sources of efficacy data, and parametric extrapolations that lacked face validity. CADTH attempted to address some of these limitations through reanalysis. These changes involved elimination of heterogeneity from the exclusion of BRAF- and MEK-targeted therapies, corrections to the calculation of state membership, use of a single source for the prediction of PFS from the RELATIVITY-047 trial, and less-optimistic predictions of long-term PFS and OS.
In the CADTH base case, 3 alternatives were identified on the cost-effectiveness frontier: nivolumab, nivolumab-ipilimumab, and nivolumab-relatlimab. Relative to nivolumab-ipilimumab, nivolumab-relatlimab was more effective (0.178 incremental QALYs) and more costly ($72,584 in incremental costs). This resulted in an ICER of $408,364 compared to nivolumab-ipilimumab. There was a 0% probability of cost-effectiveness at a WTP threshold of $50,000 per QALY gained. The decrease in this ICER relative to the sponsor’s base case can be attributed to the changes in the distributions used to predict PFS and OS. As a result, patients spent less time in the progression-free and progressed-disease health states. A price reduction would be required for nivolumab-relatlimab to be considered cost-effective at that threshold. The magnitude of the price reduction is uncertain given the presence of an error in the model that was not corrected until after the review period (Appendix 4).
In both the sponsor’s base case and CADTH’s reanalysis, treatment with nivolumab-relatlimab was more costly and more effective compared with nivolumab monotherapy and nivolumab-ipilimumab in the full indicated population. Due to limitations within the sponsor’s submitted evidence, CADTH was not able to estimate the cost-effectiveness of nivolumab-relatlimab in BRAF-positive patients. Clinical expert input suggested that treatment with BRAF and MEK inhibitors (dabrafenib-trametinib, vemurafenib-cobimetinib, and encorafenib-binimetinib) is common among these patients as a first-line approach, while immunotherapy would be preserved as a second-line option. Consequently, CADTH’s estimate of the cost-effectiveness of nivolumab-relatlimab in BRAF-positive patients is subject to a high degree of uncertainty that is compounded by sources of uncertainty in the clinical evidence and the methodological limitations of the sponsor’s pharmacoeconomic model. Given these uncertainties, and the inability to draw conclusions about whether nivolumab-relatlimab provides an OS benefit compared to other available treatments, a greater price reduction may be warranted.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Opdualag (nivolumab plus relatlimab), 240 mg nivolumab/20 mL (12 mg/mL) and 80 mg relatlimab/20mL (4 mg/mL) single-use vial for injection. Montreal (QC); Bristol-Myers Squibb Canada; 2023 Jul 10.
2.Primary Clinical Study Report: CA224047. A randomized, double-blind phase 2/3 study of relatlimab combined with nivolumab versus nivolumab in participants with previously untreated metastatic or unresectable melanoma [internal sponsor's report]. Montreal (QC): Bristol-Myers Squibb Canada; 2023 Jul 10.
3.A randomized, double-blind phase 2/3 study of relatlimab combined with nivolumab versus nivolumab in participants with previously untreated metastatic or unresectable melanoma. In: Drug Reimbursement Review sponsor submission: Opdualag (nivolumab plus relatlimab). Montreal (QC); Bristol-Myers Squibb Canada; 2023 Jul 10.
4.Systematic literature review and network meta-analysis of relatlimab + nivolumab and first-line (1L) treatment options for advanced melanoma in a ITT population [internal sponsor's report]. Montreal (QC); Bristol-Myers Squibb Canada; 2023 Jul 10.
5.Guidelines for the economic evaluation of health technologies: Canada. 4th ed. Ottawa (ON): CADTH; 2017: https://www.cadth.ca/guidelines-economic-evaluation-health-technologies-canada-4th-edition. Accessed 2023 Aug 16.
6.Hodi FS, Sileni VC, Lewis KD, et al. Long-term survival in advanced melanoma for patients treated with nivolumab plus ipilimumab in CheckMate 067 [abstract]. J Clin Oncol. 2022;40(16_suppl):9522.
7.Table: 13-10-0114-01. Life expectancy and other elements of the life table, Canada, all provinces except Prince Edward Island. Ottawa (ON): Statistics Canada; 2022: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310011401. Accessed 2023 Jul 12.
8.Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Five-year survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. 2019;381(16):1535-1546. PubMed
9.Robert C, Ribas A, Schachter J, et al. Pembrolizumab versus ipilimumab in advanced melanoma (KEYNOTE-006): post-hoc 5-year results from an open-label, multicentre, randomised, controlled, phase 3 study. Lancet Oncol. 2019;20(9):1239-1251. PubMed
10.Dummer R, Flaherty KT, Robert C, et al. COLUMBUS 5-year update: a randomized, open-label, phase iii trial of encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF V600-mutant melanoma. J Clin Oncol. 2022;40(36):4178-4188. PubMed
11.Ascierto PA, Ferrucci PF, Fisher R, et al. Dabrafenib, trametinib and pembrolizumab or placebo in BRAF-mutant melanoma. Nat Med. 2019;25(6):941-946. PubMed
12.Bansback N, Tsuchiya A, Brazier J, Anis A. Canadian valuation of EQ-5D health states: preliminary value set and considerations for future valuation studies. PLoS One. 2012;7(2):e31115. PubMed
13.Hogg D, Osenenko K, Szabo SM, et al. Standard gamble utilities for advanced melanoma health states elicited from the Canadian general public [abstract]. Presented at: 7th International Melanoma Congress; Sydney, Australia; Nov 4-7, 2010.
14.Barbier M, Durno N, Bennison C, Ortli M, Knapp C, Schwenkglenks M. Cost-effectiveness and budget impact of venetoclax in combination with rituximab in relapsed/refractory chronic lymphocytic leukemia in Switzerland. Eur J Health Econ. 2022;23(5):837-846. PubMed
15.Heslop R, Roberts H, Flower D, Jordan V. Interventions for men and women with their first episode of genital herpes. Cochrane Database Syst Rev. 2016(8):CD010684. PubMed
16.CADTH reimbursement recommendation: encorafenib (Braftovi) in combination with cinimetinib (Mektovi). Can J Health Technol. 2021;1(7).
17.pCODR Expert Review Committee (pERC) final recommendation: cobimetinib (Cotellic) and vemurafenib (Zelboraf) for metastatic melanoma. Ottawa (ON): CADTH; 2016: https://www.cadth.ca/sites/default/files/pcodr/pcodr_cobimetinib_cotellic_metmela_fn_rec.pdf. Accessed 2023 Jul 12.
18.pCODR final economic guidance report: nivolumab (Opdivo) with ipilimumab (Yervoy) for metastatic melanoma. Ottawa (ON): CADTH; 2017: https://www.cadth.ca/sites/default/files/pcodr/pcodr_opdivo_yervoy_metmela_fn_egr.pdf. Accessed 2023 Jul 12.
19.McKendrick J, Gijsen M, Quinn C, Barber B, Zhao Z. Estimating healthcare resource use associated with the treatment of metastatic melanoma in eight countries. J Med Econ. 2016;19(6):587-595. PubMed
20.Schedule of benefits for physician services under the Health Insurance Act: (June 29, 2023 (effective July 24, 2023)). Toronto (ON): Ontario Ministry of Health; 2023: https://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master.pdf. Accessed 2023 Jul 12.
21.Ontario Case Costing Initiative (OCCI). Toronto (ON): Ontario Health and Long-Term Care; 2017: https://data.ontario.ca/dataset/ontario-case-costing-initiative-occi. Accessed 2023 Feb 28.
22.Interactive health data application. Edmonton (AB): Alberta Health; 2018: https://www.alberta.ca/interactive-health-data. Accessed 2023 May 31.
23.Yu M, Guerriere DN, Coyte PC. Societal costs of home and hospital end-of-life care for palliative care patients in Ontario, Canada. Health Soc Care Community. 2015;23(6):605-618. PubMed
24.Woods B, Sideris E, Palmer S, Latimer N, Soares M. Partitioned survival analysis for decision modelling in health care: a critical review. (NICE DSU Technical Support Document 19). Sheffield (UK): Decision Support Unit, ScHARR, University of Sheffield; 2017: http://nicedsu.org.uk/wp-content/uploads/2017/06/Partitioned-Survival-Analysis-final-report.pdf. Accessed 2023 Jul 12.
25.Procedures for CADTH Reimbursement Reviews. Ottawa (ON): CADTH; 2023: https://www.cadth.ca/cadth-procedures-reimbursement-reviews. Accessed 2023 Aug 16.
26.CADTH reimbursement review: nivolumab (Opdivo). Can J Health Technol. 2023;3(7).
27.CADTH reimbursement review: pembrolizumab (Keytruda). Can J Health Technol. 2021;1(12):134.
28.CADTH reimbursement review: nivolumab (Opdivo) in combination with ipilimumab (Yervoy). Can J Health Technol. 2021;1(9).
29.pan-Canadian Oncology Drug Review final economic guidance report: nivolumab (Opdivo) plus ipilimumab (Yervoy) for advanced renal cell carcinoma. Ottawa (ON): CADTH; 2018: https://www.cadth.ca/sites/default/files/pcodr/pcodr_opdivo_yervoy_rcc_fn_egr.pdf. Accessed 2023 Aug 31.
30.Ascierto PA, Dréno B, Larkin J, et al. 5-year outcomes with cobimetinib plus vemurafenib in BRAFV600 mutation-positive advanced melanoma: extended follow-up of the coBRIM study. Clin Cancer Res. 2021;27(19):5225-5235. PubMed
31.Tawbi HA, Schadendorf D, Lipson EJ, et al. Relatlimab and nivolumab versus nivolumab in untreated advanced melanoma. N Engl J Med. 2022;386(1):24-34. PubMed
32.Cowey CL, Liu FX, Black-Shinn J, et al. Pembrolizumab utilization and outcomes for advanced melanoma in US community oncology practices. J Immunother. 2018;41(2):86-95. PubMed
33.Long GV, Flaherty KT, Stroyakovskiy D, et al. Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K-mutant melanoma: long-term survival and safety analysis of a phase 3 study. Ann Oncol. 2017;28(7):1631-1639. PubMed
Appendix 1: Cost-Comparison Table
Note this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical expert(s). Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 7: CADTH Cost-Comparison Table for Unresectable or Metastatic Melanoma
Treatment | Strength / concentration | Form | Price ($) | Recommended dosage | Daily cost ($) | 28-Day cycle cost ($) |
|---|---|---|---|---|---|---|
Nivolumab and relatlimab (Opdualag) | Nivolumab: 240 mg (12 mg/mL) Relatlimab: 80 mg (4 mg/mL) | 20 mL vial for IV Infusion | 8,315.0000a | Adults and Pediatric (12 years old and at least 40kg): 480 mg nivolumab and 160 mg relatlimab every 4 weeks | 593.93 | 16,630 |
Immune checkpoint inhibitors | ||||||
Combination therapy | ||||||
Nivolumab (Opdivo) | 10 mg/mL | 4 mL or 10 mL Vial for IV Infusion | $782.2200b $1,955.0000b | 1 mg/kg on Day 1 for four 21-day cycles. | 74.50 | 2,086 |
Ipilimumab (Yervoy) | 5 mg/mL | 10 mL or 40 mL Vial for IV infusion | $5,800.0000b $23,200.0000b | 3 mg/kg on Day 1 for four 21-day cycles. | 1,380.95 | 38,667 |
nivolumab (Opdivo) + ipilimumab (Yervoy) | 1,455.45 | 40,753 | ||||
Monotherapies | ||||||
Ipilimumab (Yervoy) | 5 mg/mL | 10 mL or 40 mL Vial for IV infusion | 5,800.0000 23,200.0000 | 3 mg/kg on day 1, for four 21-day cycles. | 1,380 | 38,667 |
Nivolumab (Opdivo) | 10 mg/mL | 4 mL or 10 mL Vial for IV infusion | 782.2200b 1,955.0000 | 3 mg/kg (up to a maximum of 240 mg) every 2 weeks; or 6 mg/kg (up to a maximum of 480 mg) every 4 weeks | 347.10 359.95 | 9,719 10,079 |
Pembrolizumab (Keytruda) | 25 mg/mL | 4 mL Vial for IV infusion | 4,400.0000b | 2 mg/kg (maximum 200 mg) every 21 days; or 4 mg/kg (maximum 400 mg) every 42 days up to a maximum of two years. | 433.98 | 12,151 |
Targeted therapies: BRAF and MEK regimens | ||||||
Binimetinib (Mektovi) | 15 mg | Tablet | 37.7410 | 45 mg twice daily | 226.45 | 6,340 |
Encorafenib (Braftovi) | 75 mg | Capsule | 51.9585 | 450 mg daily | 311.75 | 8,729 |
Binimetinib (Mektovi) and encorafenib (Braftovi) | 538.20 | 15,070 | ||||
Cobimetinib (Cotellic) | 20 mg | Tablet | 125.1025 | 60 mg daily for 21 of 28 days in treatment cycle; repeat indefinitely. | 375.31 | 10,509 |
Vemurafenib (Zelboraf) | 240 mg | Tablet | 35.5539 | 960 mg twice daily | 284.43 | 7,964 |
Cobimetinib (Cotellic) and vemurafenib (Zelboraf) | 659.74 | 18,473 | ||||
Dabrafenib (Tafinlar) | 50 mg 75 mg | Capsule | 47.5667 71.2168 | 150 mg twice daily | 284.87 | 7,976 |
Trametinib (Mekinist) | 0.5 mg 2 mg | Tablet | 81.7520 325.6493 | 2 mg daily | 325.65 | 9,118 |
dabrafenib (Tafinlar) and trametinib (Mekinist) | 610.52 | 17,095 | ||||
Note: All prices are from the Ontario Exceptional Access Program e-formulary (accessed July 2023), unless otherwise indicated, and do not include dispensing fees. Costs assume a body weight of 75kg (adults), 40kg (pediatric), or a body surface area of 1.8m2 and include wastage of unused medication in vials.
aSponsor’s submitted price.1
bPrices obtained from CADTH reimbursement reviews for: ipilimumab (Yervoy); nivolumab (Opdivo; $19.55 per mg), pembrolizumab (Keytruda).26-29
Appendix 2: Submission Quality
Note this appendix has not been copy-edited.
Description | Yes/No | Commentsa |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | Sponsor assumed a homogeneous population. However, BRAF- and MEK-targeted therapies (an important class of comparators) are indicated for the subgroup of the indicated population with a BRAF-positive mutation. Total population estimates should have been generated from a weighted average approach. See limitation: Consideration of a homogeneous patient cohort. |
Model has been adequately programmed and has sufficient face validity | No | Errors identified in the calculation of state membership. See limitation: calculation of state membership |
Model structure is adequate for decision problem | Yes | No comment |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | No | Predicted values for Progression-Free Survival combined univariate Kaplan-Meier estimates and parametric survival model. See limitation: Use of 2 sources to generate survival probabilities is inconsistent with CADTH submission guidelines. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | No | Use of a partitioned survival model prevents the ability to characterize all relevant sources of parameter uncertainty. See limitation: failure to characterize decision uncertainty. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | Yes | No comment |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note this appendix has not been copy-edited.
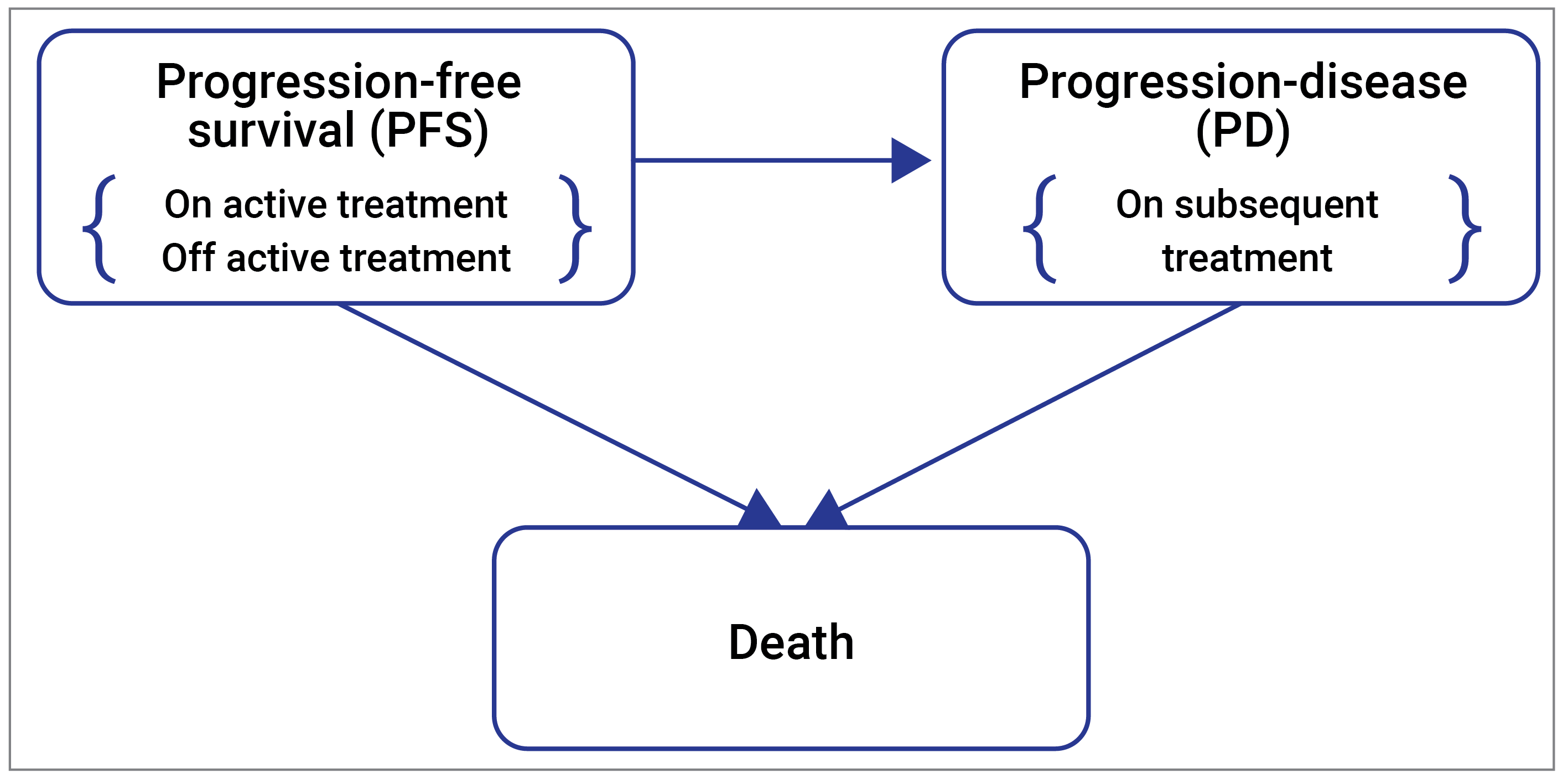
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Note this appendix has not been copy-edited.
Detailed Results of CADTH Base Case
To address some of the key limitations from the sponsor’s submission, a series of changes were implemented to arrive at the CADTH base case. Each revision listed in Table 4 was implemented independently and the results obtained from each revision are presented in Table 9, below. All estimates within Table 9 were obtained via Monte Carlo simulation of 4,000 iterations. Disaggregated results from the CADTH base case are presented in Table 10.
Table 9: Summary of the Stepped Analysis of the CADTH Reanalysis Results
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($ per QALY) |
|---|---|---|---|---|
Sponsor’s base case | Nivolumab | 181,185 | 5.755 | Reference |
Nivolumab-ipilimumab | 244,694 | 6.687 | 68,106 | |
Pembrolizumab | 268,928 | 5.122 | Dominated by nivolumab-ipilimumab | |
Ipilimumab | 271,336 | 3.533 | Dominated by nivolumab-ipilimumab | |
Nivolumab-relatlimab | 369,420 | 6.891 | 619,904 | |
CADTH reanalysis 1: Removal of OS Cap | Nivolumab | 199,624 | 6.151 | Reference |
Nivolumab-ipilimumab | 257,787 | 6.886 | 79,099 | |
Ipilimumab | 279,945 | 3.559 | Dominated by nivolumab, nivolumab-ipilimumab | |
Pembrolizumab | 281,649 | 5.244 | Dominated by nivolumab, nivolumab-ipilimumab | |
Nivolumab-relatlimab | 405,220 | 7.496 | 241,532 | |
CADTH reanalysis 2: Single Source (Parametric) PFS Predictions | Nivolumab | 208,218 | 5.743 | Reference |
Nivolumab-ipilimumab | 262,771 | 6.392 | 84,023 | |
Ipilimumab | 282,775 | 3.417 | Dominated by nivolumab, nivolumab-ipilimumab | |
Pembrolizumab | 294,054 | 4.948 | Dominated by nivolumab, nivolumab-ipilimumab | |
Nivolumab-relatlimab | 397,224 | 6.884 | 273,182 | |
CADTH reanalysis 3: Exponential distributions for OS and PFS | Nivolumab | 174,316 | 3.077 | Reference |
Ipilimumab | 213,523 | 1.923 | Dominated by nivolumab | |
Pembrolizumab | 249,131 | 2.609 | Dominated by nivolumab | |
Nivolumab-ipilimumab | 253,669 | 3.547 | 168,681 | |
Nivolumab-relatlimab | 337,091 | 3.729 | 459,416 | |
CADTH base case: 1 + 2 + 3 | Nivolumab | 190,559 | 3.072 | Reference |
Ipilimumab | 212,708 | 1.925 | Dominated by nivolumab | |
Pembrolizumab | 261,425 | 2.614 | Dominated by nivolumab | |
Nivolumab-ipilimumab | 282,285 | 3.535 | 198,150 | |
Nivolumab-relatlimab | 354,869 | 3.713 | 408,364 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Table 10: Disaggregated Summary of CADTH’s Economic Evaluation Results
Treatment | Component | Value | Incremental (vs. reference) | Incremental (sequential) |
|---|---|---|---|---|
Discounted LYs | ||||
Nivolumab | Progression-Free | 1.49 | NA | NA |
Progressed Disease | 2.37 | NA | NA | |
Total | 3.86 | NA | NA | |
Ipilimumab | Progression-Free | 0.75 | −0.74 | NA |
Progressed Disease | 1.69 | −0.68 | NA | |
Total | 2.44 | −1.42 | NA | |
Pembrolizumab | Progression-Free | 1.25 | −0.24 | 0.5 |
Progressed Disease | 2.04 | −0.33 | 0.35 | |
Total | 3.29 | −0.57 | 0.85 | |
Nivolumab-ipilimumab | Progression-Free | 1.89 | 0.4 | 0.64 |
Progressed Disease | 2.61 | 0.24 | 0.57 | |
Total | 4.49 | 0.63 | 1.2 | |
Nivolumab-relatlimab | Progression-Free | 1.94 | 0.45 | 0.05 |
Progressed Disease | 2.73 | 0.36 | 0.12 | |
Total | 4.67 | 0.81 | 0.18 | |
Discounted QALYs | ||||
Nivolumab | Progression-Free | 1.21 | NA | NA |
Progressed Disease | 1.86 | NA | NA | |
Total | 3.07 | NA | NA | |
Ipilimumab | Progression-Free | 0.61 | −0.6 | NA |
Progressed Disease | 1.34 | −0.52 | NA | |
Total | 1.92 | −1.15 | NA | |
Pembrolizumab | Progression-Free | 1.01 | −0.2 | 0.4 |
Progressed Disease | 1.61 | −0.25 | 0.27 | |
Total | 2.61 | −0.46 | 0.69 | |
Nivolumab-ipilimumab | Progression-Free | 1.53 | 0.32 | 0.52 |
Progressed Disease | 2.04 | 0.18 | 0.43 | |
Total | 3.54 | 0.47 | 0.93 | |
Nivolumab-relatlimab | Progression-Free | 1.57 | 0.36 | 0.04 |
Progressed Disease | 2.14 | 0.28 | 0.1 | |
Total | 3.71 | 0.64 | 0.17 | |
Discounted costs ($) | ||||
Nivolumab | Acquisition | 52,805 | NA | NA |
Administration | 1,144 | NA | NA | |
Disease Management | 122,391 | NA | NA | |
AEs | 162 | NA | NA | |
Subsequent Treatment | 14,057 | NA | NA | |
Total | 190,559 | NA | NA | |
Ipilimumab | Acquisition | 108,478 | 55,673 | NA |
Administration | 192 | −952 | NA | |
Disease Management | 90,447 | −31,944 | NA | |
AEs | 2,207 | 2,045 | NA | |
Subsequent Treatment | 10,753 | −3,304 | NA | |
Total | 212,078 | 21,519 | NA | |
Pembrolizumab | Acquisition | 137,173 | 28,695 | 28,695 |
Administration | 1,972 | 1,780 | 1,780 | |
Disease Management | 107,423 | 16,976 | 16,976 | |
AEs | 733 | −1,474 | −1,474 | |
Subsequent Treatment | 14,124 | 3,371 | 3,371 | |
Total | 261,425 | 49,347 | 49,347 | |
Nivolumab-ipilimumab | Acquisition | 135,088 | −2,085 | 82,283 |
Administration | 1,396 | −576 | 252 | |
Disease Management | 133,766 | 26,343 | 11,375 | |
AEs | 4,516 | 3,783 | 4,354 | |
Subsequent Treatment | 7,519 | −6,605 | −6,538 | |
Total | 282,285 | 20,860 | 91,726 | |
Mivolumab-relatlimab | Acquisition | 199,358 | 64,270 | 146,553 |
Administration | 648 | −748 | −496 | |
Disease Management | 139,788 | 6,022 | 17,397 | |
AEs | 447 | −4,069 | 285 | |
Subsequent Treatment | 14,628 | 7,109 | 571 | |
Total | 354,869 | 72,584 | 164,310 | |
ICER = incremental cost-effectiveness ratio; LY = life-year; NA = not applicable; QALY = quality-adjusted life-year; vs. = versus.
The sponsor reported a calculation error in their pharmacoeconomic model to CADTH. This error was detected and reported too late in the review process for their correction to be validated by CADTH. The sponsor claims, and CADTH agrees, that this error did not appear to contribute meaningfully to the estimated QALYs. In the interest of completeness and transparency, CADTH presents the sponsor’s revised estimates of cost-effectiveness in the following table. A scenario analysis was conducted in which CADTH’s changes were applied to the sponsor’s updated model. These results are also presented in the table.
Table 11: Scenario Analysis of the Economic Evaluation Results With Updated Model
Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|
Sponsor’s base case: Immunotherapy network (deterministic) | |||
Nivolumab | 235,576 | 5.745 | Reference |
Nivolumab-ipilimumab | 274,002 | 6.376 | 60,894 |
Nivolumab-relatlimab | 371,036 | 6.894 | 187,138 |
Dominated treatments | |||
Pembrolizumab | 268,007 | 4.934 | Dominated |
Ipilimumab | 268,964 | 3.405 | Dominated |
CADTH scenario analysis: Immunotherapy network (deterministic) | |||
Ipilimumab | 204,235 | 1.91 | Reference |
Nivolumab | 250,081 | 3.06 | 39,800 |
Nivolumab-ipilimumab | 312,989 | 3.52 | 138,196 |
Nivolumab-relatlimab | 355,258 | 3.71 | 226,144 |
Dominated treatments | |||
Pembrolizumab | 261,469 | 2.58 | Dominated |
Table 12: Price-Reduction Analysis of the Economic Evaluation Results With Updated Model
Analysis | ICERs for nivolumab-relatlimab vs. alternatives ($ per QALY) | |
|---|---|---|
Price reduction | Sponsor base case | CADTH reanalysis |
No price reduction | 187,138 (vs nivolumab + ipilimumab) | 226,144 (vs. nivolumab + ipilimumab) |
10% | 144,687 (vs nivolumab + ipilimumab) | 131,112 (vs. nivolumab) |
20% | 102,050 (vs nivolumab + ipilimumab) | 100,414 (vs. nivolumab) |
30% | 60,228 (vs. nivolumab) | 69,715 (vs. nivolumab) |
36% | 50,639 (vs. nivolumab) | 49,761 (vs. nivolumab) |
40% | 41,006 (vs. nivolumab) | 39,559 (vs. ipilimumab) |
50% | 21,784 (vs. nivolumab) | 28,474 (vs. ipilimumab) |
60% | 2,562 (vs. nivolumab) | 17,388 (vs. ipilimumab) |
70% | Dominant | 6,303 (vs. ipilimumab) |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Note: all price reductions are calculated using deterministic values and therefore do not properly reflect the joint effect of parameter uncertainty.
Appendix 5: Submitted Budget Impact Analysis and CADTH Appraisal
Note this appendix has not been copy-edited.
Table 13: Summary of Key Take-Aways
Key Take-aways of the Budget Impact Analysis |
|---|
|
Summary of Sponsor’s Budget Impact Analysis
The sponsor submitted a budget impact analysis (BIA) for the introduction nivolumab-relatlimab (nivolumab-relatlimab) for the treatment of adult and pediatric patients (12 years and older, weighing at least 40 kg) with unresectable or metastatic melanoma. Estimates were generated from the perspective of pCODR-participating drug plans (all but Quebec) and the results were aggregated into pan-Canadian totals over a three-year time horizon. Alternatives to nivolumab-relatlimab considered in the BIA included: nivolumab, nivolumab-ipilimumab, ipilimumab, pembrolizumab, dabrafenib-trametinib, encorafenib-binimetinib, vemurafenib-cobimetinib. An epidemiological approach was used to estimate the eligible population size for the analysis (Figure 3). Key inputs to the BIA are documented in Table 12.
In the reference scenario, it was assumed that patients eligible for treatment would receive 1 of the currently available immunotherapies or BRAF and MEK therapies. In the new drug scenario, it was assumed that nivolumab-relatlimab would displace market share from the treatments available in the reference scenario.
Key assumptions:
Patients were assumed to receive treatment until excess toxicity or disease progression. The duration of first-line treatment was assumed to follow the mean duration on treatment for reported in key-trials.8,10,30-33 To adhere to the requirements of the product monographs, the duration of treatment was capped for 2 alternatives. For pembrolizumab, the maximum treatment duration was assumed to be 2 years. For nivolumab-ipilimumab, the maximum treatment duration was assumed to be 4 21-day cycles, after which the patient was assumed to receive nivolumab monotherapy.
In the absence of data informing the duration of treatment for dabrafenib-trametinib, encorafenib-binimetinib, and vemurafenib-cobimetinib, the sponsor assumed the median value from 3 trials.10,30,33
The sponsor assumed that patients receive second-line treatment as a one-off cost upon discontinuation of first-line therapy. The proportion of patients on each of the possible second-line treatments followed estimates obtained from the RELATIVITY-047 and CheckMate-067 trials.
In the base case, it was assumed that half of the nivolumab-relatlimab market share would come from nivolumab-ipilimumab, with the remaining gains taken from nivolumab and pembrolizumab. The sponsor assumed that nivolumab-relatlimab would not take any market share from dabrafenib-trametinib, encorafenib-binimetinib, and vemurafenib-cobimetinib. Two additional market share scenarios were considered: i) postlaunch market share data in the US; and ii) clinical expert opinion.
Figure 3: Sponsor’s Estimation of the Size of the Eligible Population
Source: Sponsor’s pharmacoeconomic submission.1
Table 14: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as Year 1 / Year 2 / Year 3 if appropriate) |
|---|---|
Target population | |
Pan-Canadian Population (excluding Quebec) | 15,229,972 (Males); 15,470,294 (Females) |
Melanoma incidence per 100,000 | 29.1 (Males); 22.3 (Females) |
Average annual incidence percent change | 2.20% (Males); 1.40% (Females) |
% Stage I-III (resectable) diagnosis | 92.67% |
% that recur to stage III (unresectable) or IV | ||| |
% Stage III (unresectable/resectable) diagnosis | 12.75% |
% Stage III (unresectable) | ||| |
% Stage IV diagnosis | 4.78% |
Number of patients eligible for drug under review | 1,734 / 1,798 / 1,863 |
Market Uptake (3 years) | |
Uptake (reference scenario) | |
Nivolumab-relatlimab | |||||||||||||||| |
Nivolumab | |||||||||||||||| |
Nivolumab-ipilimumab | |||||||||||||||| |
Pembrolizumab | |||||||||||||||| |
Ipilimumab | |||||||||||||||| |
Dabrafenib-trametinib | |||||||||||||||| |
Encorafenib-binimetinib | |||||||||||||||| |
Vemurafenib-cobimetinib | |||||||||||||||| |
Uptake (new drug scenario) | |
nivolumab-relatlimab | |||||||||||||||| |
Nivolumab | |||||||||||||||| |
Nivolumab-ipilimumab | |||||||||||||||| |
Pembrolizumab | |||||||||||||||| |
Ipilimumab | |||||||||||||||| |
Dabrafenib-trametinib | |||||||||||||||| |
Encorafenib-binimetinib | |||||||||||||||| |
Vemurafenib-cobimetinib | |||||||||||||||| |
Cost of treatment (per patient) | |
Cost of treatment over: 1 month | |
Nivolumab-relatlimab | $18,077.70 |
Nivolumab | $10,203.78 |
Nivolumab-ipilimumab | $16,637.60 |
Pembrolizumab | $12,754.76 |
Ipilimumab | $42,032.74 |
Dabrafenib-trametinib | $16,802.11 |
Encorafenib-binimetinib | $15,816.06 |
Vemurafenib-cobimetinib | $16,531.22 |
Summary of the Sponsor’s Budget Impact Analysis Results
In the base case, the net budget impact of nivolumab-relatlimab was estimated to be $9,755,408 in Year 1, $26,558,529 in Year 2, and $34,363,795 in Year 3. The three-year net budget impact of nivolumab-relatlimab was $70,677,733.
CADTH Appraisal of the Sponsor’s Budget Impact Analysis
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Assumptions used to estimate market size: In the submitted BIA, an epidemiological approach was used to estimate the eligible population size. To identify the number of patients with unresectable or advanced melanoma, the sponsor included several assumptions relating to the diagnosis of patients at each stage of melanoma. Clinical experts consulted by CADTH suggested that 3 such estimates may be over-estimated: i) that 92.67% of patients would be diagnosed at Stage I-III (resectable); ii) 15% of patients initially diagnosed at Stage I-III resectable will recur to stage III (unresectable) or stage IV; and iii) 20% of patients will be diagnosed at stage III (unresectable).
In the CADTH base case, values used by the sponsor for all 3 parameters were replaced with the expectations of the clinical experts consulted by CADTH.
CADTH Reanalyses of the Budget Impact Analysis
Table 15: CADTH Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Changes to derive the CADTH base case | ||
1. Assumptions used to estimate market size. Percent diagnosed at Stage I-III (resectable) | 92.67% | 85% |
2. Assumptions used to estimate market size. Percent initially diagnosed at Stage I-III (resectable) that recur to stage III (unresectable) or Stage IV. | ||| | 5% |
3. Assumptions used to estimate market size. Percent of patients diagnosed at stage III (unresectable) | ||| | 10% |
CADTH base case | 1 + 2 + 3 | |
The results of the CADTH stepwise reanalysis summarized in Table 14 and a more detailed breakdown is presented in Table 15. All CADTH reanalyses were based on publicly available prices of the comparator treatments. In the CADTH base case, the three-year net budget impact of nivolumab-relatlimab was estimated to be $34,304,588. This decrease is attributable to the assumption that 5% (instead of 15%) of patients initially diagnosed at Stage I to III (resectable) will recur to stage III (unresectable) or Stage IV.
In addition to the CADTH base case, an additional scenario analysis was conducted to explore how the budget impact would be affected by the price reduction identified in the economic evaluation. Assuming a 70% price reduction, the three-year net budget impact of nivolumab-relatlimab was estimated to be savings of $53,061,958.
Table 16: Summary of the CADTH Reanalyses of the BIA
Stepped analysis | Three-year total |
|---|---|
Submitted base case | $70,677,733 |
CADTH reanalysis 1 | $66,846,618 |
CADTH reanalysis 2 | $39,824,819 |
CADTH reanalysis 3 | $67,071,018 |
CADTH base case | $34,304,588 |
BIA = budget impact analysis.
Table 17: Detailed Breakdown of the CADTH Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
Submitted base case | Reference | $304,539,498 | $407,512,369 | $422,606,287 | $438,011,334 | $1,268,129,990 |
New drug | $304,539,498 | $417,267,777 | $449,164,816 | $472,375,129 | $1,338,807,722 | |
Budget impact | $0 | $9,755,408 | $26,558,529 | $34,363,795 | $70,677,733 | |
CADTH base case | Reference | $147,813,202 | $197,792,761 | $205,118,840 | $212,595,930 | $615,507,531 |
New drug | $147,813,202 | $202,527,707 | $218,009,454 | $229,274,957 | $649,812,118 | |
Budget impact | $0 | $4,734,946 | $12,890,614 | $16,679,027 | $34,304,588 | |
CADTH scenario analysis: 65% price reduction | Reference | $147,813,202 | $197,792,761 | $205,118,840 | $212,595,930 | $615,507,531 |
New drug | $147,813,202 | $188,285,024 | $185,903,537 | $194,497,478 | $568,686,040 | |
Budget impact | $0 | -$9,507,737 | -$19,215,302 | -$18,098,452 | -$46,821,491 |
BIA = budget impact analysis.
Stakeholder Input
Patient Input
Melanoma Canada
About Melanoma Canada
Melanoma Canada has been registered with pCODR since 2012. We are a national patient focused organization focused on the prevention and elimination of melanoma and skin cancers. We provide patient support, advocacy, awareness and education for the public and for health care professionals.
Information Gathering
Data was gathered for this submission by way of an on-line survey. The survey link was emailed to our database of patients. Any patients and caregivers, regardless of stage or familiarity with the drug therapy in question, were asked to participate. We also used our website and social media via Facebook, etc. to promote the survey. The survey was made available June 15th to July 14th, 2023.
Demographics: We received a total of 119 individual patient responses combined with 84 caregiver responses. 35 patients indicated they had no caregiver. Of the total responses for patients, 81 were female and 38 were male. The survey was open to all patients, regardless of stage or whether or not they had been on the combination drug therapy. We had 26 patients that were stage 0; stage I – 17; stage II – 10; stage III – 18; stage IV – 29 and a further 19 did not know their stage. 73 respondents were from Ontario, 15 Alberta, 11 BC, 8 Quebec, 6 from Manitoba and the remainder from other provinces. 2 patients in our survey were treated with the combination therapy of nivolumab and relatlimab. The range of age of respondents were:
Table 1: Drug Therapy Survey Results by Age Group
Answer Choices | Responses | |
|---|---|---|
Percentage | Number of Participants | |
18 to 30 years | 0.84% | 1 |
31 to 40 years | 5.88% | 7 |
41 to 50 years | 5.04% | 6 |
51 to 60 years | 23.53% | 28 |
61 to 70 years | 42.02% | 50 |
71 years or greater | 22.69% | 27 |
Disease Experience
Pain, Scarring, lymphedema, fatigue, anxiety, fear and depression are common impacts of the disease itself that affect the quality of life for patients and their families. With these types of issues continuing to be reported year over year, and likely very common across cancer types, there is a need to address not only improved drug therapy, but improved early diagnosis, the impacts on mental health and the impact from surgery on patients’ ability to function. While melanoma has had several effective immune and targeted therapies developed in the last decade, there remains about 50% of patients that do not respond or have only partial response to available treatments. Both caregivers and patients agree that there is a continuing need to address this gap and find new therapies or combinations that have even better results and less side effects, albeit most are tolerable and short lived. There was significant commentary from patients which provides insight into the impact of the disease:
“Anxiety not knowing when disease may reoccur. Had to stop working. Disease affected my heart. General fatigue - less interest in activities.”
“Some physical limitations due to the lymphedema, weight gain due to depression.”
“Periods of depression and anxiety, especially awaiting pathology results.”
“My biopsy was done incorrectly. Walking post biopsy and post-surgery was limited and challenging as a result. I had to have a skin graft to cover the wide excision. The donor site was from my arm near my elbow. I now have a chunk out of my arm that I’m very self-conscious of. I no longer feel comfortable wearing t-shirts out in public. I also feel self-conscious of the disfigurement on my lower leg, and I try to keep that hidden as well. My initial dermatologist dismissed the mole with only a brief look. This made me second guess myself and not go back for quite some time. Then Covid hit and I was delayed further. By the time the mole was taken seriously, about 2 years had passed post discovery. This makes me feel anxious about recurrence.”
“In addition to scarring in my chest, abdomen, and leg I also have full lymphedema in one leg, I have difficulty getting up and walking, and serious stiffness in most joints. Financially, I am receiving reduced income with disability pay from my employer, but I am no longer able to contribute to a pension. I am no longer able to work and require assistance with outside yard week and inside cleaning. My social life is a fraction of what it was, I am tired a lot!”
“I worry that it will come back as I have many spots. I hide away from people as I do not want them to see my scars and feel sorry for me.”
“Leg and abdominal lymphedema limits activities. I now wear a compression garment every day. It is extremely unpleasant to wear in the hot weather. Sometimes the heat of the garment creates sores. The garment goes up to my waist and sometimes affects my breathing and definitely creates anxiety. It is difficult to find clothes to fit my now larger than normal legs. It is anxiety producing to explain to others the very obvious compression garment. I now need to plan time in the day to elevate my legs. Living with lymphedema adds extra stress and fatigue to my life. Purchasing garments every 3-4 months adds financial planning. I also feel unsupported by medical people with regards to lymphedema.”
“Difficult to continue with daily tasks due to dizziness, extreme fatigue and pain from gas buildup probably caused by cancer in the liver.”
“I avoided people, so I didn’t have to talk about my melanoma, thus making my anxiety worse.”
“I can't stand or walk for long. I take 2700 mg of Gabapentin a day to numb the pain. Can't do many activities I used to do. I am, however, grateful to be alive. Fear and sadness have also changed me.”
“Extreme stress for my daughter (an only child). Stress for me as the parent of an only child as I think about the what if...”
“Have to limit when I walk, cycle or golf outdoors to early morning or evenings. Friends and family annoyed with me because I try to avoid the sun. They think I’m over the top. As a result of melanoma my otherwise busy outdoor life has been curtailed so some have dropped including me in activities.”
“Need to manage through therapy to address PTSD and fear.”
“It’s frustrating going from great better than perfect vision to such a decrease. Struggling to see out of one eye is difficult to deal with sometimes.”
“Fatigue didn't allow me to work at full capacity and the headaches also would impact the work environment, meaning I wanted low lights. I also had a couple of situations where I fainted, probably due to the impact the cancer had on the brain.”
“I have missed outdoor activities due to high levels of anxiety and fear of being outside. I have been on a LTD leave from work for two years while I underwent surgery and treatments, resulting in a reduced income. I have been through therapy to try and co-exist with my fears, depression and anxiety and am now on daily medication in order to manage the depression and anxiety while still being a mom, wife and human. I have gained approximately 50lbs in the last two years as high calorie drinks and snacks were my "rewards" for doing treatments. I have lymphedema in my left leg and wear a full-length garment. My relationship with my husband was rocky while I was at my lowest as he tried to understand and help, but I was so deep I wouldn't let him in.”
“Lymphedema has been the worst side effect post op and post immunotherapy. Hate wearing compression stocking daily and for the rest of my life. Have to take it one day at a time.”
“Scarring from radical neck dissection (1987); mastectomy 2017, lung surgery 2020. Small day to day impact, but has made me feel more conspicuous in my clothing choices, in locker rooms, swimming areas etc. I have mild anxiety about the melanoma returning, but generally just before medical appointments when test results are going to be delivered.”
“I’ve had to adapt because of a partial amputation of my foot. My day to day is quite normal but it’s a new normal. I’m limited in how much I can do physically but I know my limit and try to not overdo it.”
“Since my surgery to remove a tumor from my spine I can no longer walk without canes, only walk short distances. Suffer now from Brown Squard Syndrome and I suffer from nerve pain everyday, all day. My life has totally changed. My husband has to do most of all of our day-to-day stuff on top of managing all my medical appointments and medicine. The constant fear of Melanoma growing and running out of treatment options cause my depression and anxiety to soar and I now take medication for that. The Melanoma and the nerve damage I suffer every day makes it hard to keep going.”
“It has affected my relationship with my wife, I feel I have let her down.”
“The multiple surgeries on dominant arm, resection of axilla, bicep muscle and other soft tissue in the arm removed. These large surgeries have been life changing and have left the arm unable to do many simple tasks such as prepare food, write, text, brush teeth. The list is huge, forced to learn to use non dominant arm for simple tasks. Some days the fatigue and frustrations are worse than other days.”
“Due to melanoma, my ability to dream or hope for the future is lost. I'll never become a mother. I've lost many friends as I am not the person I was before diagnosis. I can only work 5 hours a day as my cancer and targeted therapy leaves me with serious fatigue. I just can't do the things that I used to.”
Table 2: Health Issues Caused by Drug Therapy
Answer Choices | Responses | |
|---|---|---|
Percentage | Number of Participants | |
Pain | 25.21% | 30 |
Scarring or disfigurement | 57.98% | 69 |
Edema or fluid retention | 10.92% | 13 |
Lymphedema | 21.01% | 25 |
Mobility issues (unable to walk or impaired movement) | 11.76% | 14 |
Gastrointestinal issues | 9.24% | 11 |
Breathing problems | 4.20% | 5 |
Headaches | 11.76% | 14 |
Peripheral neuropathy (nerve pain or damage) | 10.92% | 13 |
Disrupted sleep | 30.25% | 36 |
Appetite loss or weight gain | 15.13% | 18 |
Fear or anxiety | 57.98% | 69 |
Fatigue | 36.13% | 43 |
Depression | 26.89% | 32 |
Post traumatic stress | 14.29% | 17 |
Cognitive impairment | 2.52% | 3 |
Nausea or vomiting | 2.52% | 3 |
Damage to organs, such a lung, liver, brain | 6.72% | 8 |
Negative impact to family or social life | 25.21% | 30 |
Financial loss or job loss | 11.76% | 14 |
Impact on sexuality | 9.24% | 11 |
None — there has been no impact | 8.40% | 10 |
Caregivers reported that the biggest impact on them of dealing with the diagnosis is the mental stress, followed by the negative financial impact to the family with the loss of income from a working partner, and as well, the additional responsibilities that they have to perform for the home and family and to care for their loved one.
Experiences With Currently Available Treatments
Of the 119 respondents, 55% had been treated with some form of drug therapy. 9 patients were treated with multiple therapies. 92% of patients treated with available drug therapies indicated that they felt the side effects were worth it for the anticipated results. Many comments mirrored the following:
“Small price to pay.”
“I'm still here!”
“I am here as opposed to dead!”
“I had a complete response and am currently cancer-free.”
“The side effects were worth it because at least it felt like I was doing something. Being diagnosed with cancer made me feel like there was a big part of my life I had no control over. Choosing surgery, the subsequent treatments of Interferon, and Nivolumab felt like I was doing everything I could to fight it. I've survived 3 diagnoses of Metastatic melanoma. So, it's worth it.”
“My Stage IIIC metastatic melanoma has not recurred since my lesion was removed in September 2021.”
“The skin rash was awful, thyroid issues and liver failure but since the treatment was successful for Stage IV melanoma it was definitely worth it.”
About 20% of patients experienced issues in accessing treatment which included:
Lost work opportunities, parking, the high price of gas, travel time.
Loss of income.
Difficulties in getting coverage for therapies – private insurance coverage limited or time consuming to arrange.
Table 3: Number of Participants Treated With Different Types of Drug Therapies
Answer Choices | Responses | |
|---|---|---|
Percentage | Number of Participants | |
Dabrafenib (Tafinlar) & trametinib (Mekinist) — combination therapy in the form of daily pills | 6.06% | 4 |
Vemurafenib (Zelboraf) & cobimetinib (Cotellic) — combination therapy in the form of daily pills | 1.52% | 1 |
Braftovi (Encorafenib) & Mektovi (Binimetinib) — combination therapy in the form of daily pills | 3.03% | 2 |
Trametinib (Mekinist) as a monotherapy | 0.00% | 0 |
Vemurafenib (Zelboraf) as a monotherapy | 0.00% | 0 |
Dabrafenib (Tafinlar) as a monotherapy | 0.00% | 0 |
Nivolumab (Opdivo) monotherapy administered in clinic by intravenous | 21.21% | 14 |
Nivolumab (Opdivo) in combination with ipilimumab (Yervoy) | 22.73% | 15 |
Ipilimumab (Yervoy) monotherapy administered in clinic by intravenous | 4.55% | 3 |
Pembrolizumab (Keytruda) monotherapy administered in clinic by intravenous | 18.18% | 12 |
Interleukin-2 (Aldesleukin, Proleukin) — injections into unresectable tumours | 1.52% | 1 |
Interferon alfa -2b (Intron A) | 4.55% | 3 |
Dacarbazine (DTIC) — chemotherapy | 0.00% | 0 |
None of the above | 3.03% | 2 |
Other (please specify) | 13.64% | 9 |
Table 4: Common Side Effects of Drug Therapies
Answer Choices | Responses | |
|---|---|---|
Percentage | Number of Participants | |
Skin rash | 39.39% | 26 |
Fatigue or weakness | 68.18% | 45 |
Diarrhea or Colitis | 36.36% | 24 |
Muscle or Joint pain | 31.82% | 21 |
Nausea | 21.21% | 14 |
Fever or flu like symptoms | 18.18% | 12 |
Headaches | 24.24% | 16 |
Hormone or thyroid problems | 21.21% | 14 |
Weight loss or Loss of appetite | 25.76% | 17 |
Several patients also indicated rarer side effects that included:
Vitiligo, alopecia (2)
Type 1 diabetes (2)
Developed myasthenia Gravis (1), uveitis (1), iritis (1)
Kidney failure (2)
Pancreatitis, atrial fibrillation (1)
Improved Outcomes
Both patients and caregivers would like to see a wider variety of options that prove more effective be made available. The trade-offs are not comparable as there are limited therapies available and with the once currently used there is a better quality of life and good rate of response. That being said, there is vast opportunity for improvement. Not everyone responds or can tolerate the therapies. It would be advantageous to have biomarker tests available that might predict response rates. We also need more effective options. When facing a dire outcome, patients and family members will put up with a lot of side effects, even if lasting, if it means the chance to live or live longer.
Experience With Drug Under Review
There were two respondents that had been treated with the new combination drug therapy under review. Both had access to the therapy through clinical trials. Both indicated they had skin rash and fatigue. One had pneumonitis, one had diarrhea and one had cognitive impairment. Both indicated the side effects were worth it. One said the side effects were worse than other therapies they had been given, the other said it was better. One indicated that it has eliminated the melanoma completely and the other said there has been a complete response so far, but they are still in treatment. Neither had any issue in accessing treatment. They and their families were thankful for the option. Waiting to see an hoping for a durable response.
Companion Diagnostic Test
There is no companion diagnostic test.
Anything Else?
There is an ongoing need for better options, and options when one therapy doesn’t work or stops working. Melanoma is a very complicated cancer with the highest level of mutations amongst cancers. It is very difficult to treat once it has spread. Effective treatments, biomarkers and earlier stage treatments are needed to prevent some of the quality-of-life impacts from surgery, loss of income, duration of illness and the impact on mental health for the patient and caregiver. This combination therapy is another improvement and option in a cancer that continues to be on the rise and is complex to treat.
Conflict of Interest Declaration — Melanoma Canada
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission?
No help received.
Did you receive help from outside your patient group to collect or analyze data used in this submission?
No help received.
List any companies or organizations that have provided your group with financial payment over the past 2 years AND who may have direct or indirect interest in the drug under review.
Table 5: Financial Disclosures for Melanoma Canada
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Bristol Myers Squibb | — | — | — | X |
Save Your Skin Foundation
About Save Your Skin Foundation
Save Your Skin Foundation (SYSF) is a national patient-led not-for-profit group dedicated to the fight against non-melanoma skin cancers, melanoma and ocular melanoma through nationwide education, advocacy, and awareness initiatives. SYSF provides a community of oncology patient and caregiver support throughout the entire continuum of care, from prevention and diagnosis to survivorship. www.saveyourskinca
Information Gathering
Information was obtained through online surveys, virtual patient roundtables and one-on-one conversations. Information collected for sections Disease Experience, Experiences With Currently Available Treatments, and Improved Outcomes included all Melanoma patients (60) inclusive of (12) on treatment under review and was gathered over the past 6 months.
Our surveys were offered in both English and French languages, and we have consolidated the data from both surveys here. There were (37) females and (23) males aged between 18 – 89 years old. (1) being between 18 – 29, (11) 30-49, (11) 50 – 59, (15) 60 – 69 and (17) 70-79 (4) 80 – 89. (1) did not specify.
(31) respondents were retired, (13) working full time, (7) PT, (2) on medical leave, (2) not employed and not looking for work and (5) not able to work due to health-related reasons.
There were (14) respondents from BC, (6) from Alberta, (11) from Ontario, (1) from NS. (7) from QC, (1) from NFLD and (2) from PEI. (18) from outside of Canada (USA, Australia, France)
Disease Experience
Fear and/or anxiety (all respondents)
Scarring and disfigurement (all respondents)
Fatigue (all respondents)
Fear of reoccurrence (all respondents)
Long, scary, roller coaster — surgeries, immunotherapy treatments, happy to be NED
Fearful of the future
Difficult and scary
Horrific. Horrific does not begin to describe the experience.
Physically exhausting, emotional experience and financially straining.
Patients quotes:
“Scared, disbelief, unsettled, anxious, teary, disrupted life & totally life changing.”
“No physical symptoms from the cancer itself. Mentally was experiencing some mild depression. Emotionally lots of feelings of anxiety and stress. Financially ok but it was stressful when taking time off work for surgery and treatment.”
“The diagnosis has taken a huge mental toll on me as I have always had a very large fear of cancer and I ended up living my worst nightmare.”
“Mentally and physically exhausting, time constraints, strains with a young family. Travelling to and from the hospital for treatments. Financially not being able to work full time.”
Experiences With Currently Available Treatments
Surgery
Immuno-oncology
Targeted therapy
Radiation
Patients in remote areas of Canada have problems getting to treatment if needed. Travel costs and time off from work puts extra stress on patients and caregivers. Fear and anxiety of reoccurrence. Some patients became isolated from friends and family. Huge expenses and increased stress to themselves and their family and the added concern of being treated far from home and their support system. There was very little access to the drug under review (ON and QC only) and there were a number of Canadian patients that could not get access to Opdualag, which might have been their only option.
Improved Outcomes
More new treatment options with minimal side effects
Less surgery
Less radiation
Treatments or procedures closer to home and to their support network
More innovative treatment options with fewer side effects
Treatment with longer response
Getting treatments to patients sooner and closer to where they live for the sake of both convenience and lessening financial burden.
Patients quotes:
“Luckily a trial was available when my last treatment stopped working and my tumours began to grow again.”
“Leaving our community and travelling repeatedly. Not being available to our kids and grandkids and my elderly mom.”
“We really only considered OS. I’m young and healthy enough that we knew tough treatment would likely be survivable. So, whatever it took to get the best OS.”
“There were no trade-offs. We did what we needed to do!”
Experience With Drug Under Review
(12) patients in total
Where survey participants are in the treatment process
(4) patients from a clinical trial
(8) not sure
How patients received treatment under review
(4) patients completed treatment
(6) patients still in treatment
(2) came off due to side effects
Side effects to treatment under review (survey participants could select multiple options)
(8) fatigue
(2) cognitive impairment
(2) fever
(1) nausea and/or vomiting
(5) skin rash
(1) damage to organs
(2) Gastrointestinal issues
(2) breathing problems
(3) weight loss or weight gain
(1) headaches
(1) loss of appetite
Rating of manageability of side effects of treatment under review
(1) not manageable
(5) somewhat manageable
(2) mostly manageable
(3) completely manageable
(1) not applicable
Responses when participants were asked if benefits outweighed the experience of side effects
(10) Yes
(2) No, had to come off due to side effects
Companion Diagnostic Test
Please note that in our surveys, all participants were asked about their experiences with companion diagnostic testing, not just those who received the treatment under review. (14) said they had received companion diagnostic testing, (46) were unsure if they had received it. All patients were unsure how their companion diagnostic tests were conducted and when. For (1) patient the testing process caused delays in starting their treatment. All patients responded that there were no adverse effects associated with the testing procedure. (3) patients had their testing expenses covered by private payer, (2) out of pocket, (1) compassionate, (54) not sure, most assumed by health insurance.
Patients quotes:
“I don't know if I had companion diagnostic testing.”
“Was not offered.”
“Do not know what this testing is.”
“Grateful that it was available to me.”
“Great. I knew they would do everything possible for me and I wanted holistic, comprehensive testing.”
“Fine, it was important.”
Anything Else?
Is there anything else specifically related to this drug review that CADTH reviewers or the expert committee should know?
As patient advocates, we are well aware that the proliferation of innovative treatments is allowing more people to survive melanoma than ever before. However, we also get to see the other side, where patients fall between the cracks of these offerings. If a treatment is not available in all urban centers, that means it is not accessible to all patients, or patients need to take on additional hardships to access it; if a treatment has a more aggressive side effect profile it is not appropriate for all patients, and therefore another option is needed to fill this gap. Patients have different lives that offer different access hardships, and different biology, meaning that different treatment options are better choices for some than for others. Patients are aware of this fact: access and the need for greater choice were issues consistently cited by participants in our survey, some of whom mentioned selecting care with challenging side effects because of a lack of choice and several citing the need for more options in case their cancer should return. The listing of the drug under review means that more patients would have access to melanoma care, patients who need a gentler side effect profile (the elderly, those with certain comorbidities) would have a better option, and there would be another recurrence.
As SYSF works with melanoma advocates globally, we have been able to get a sense of the lived experience of the treatment under review from our colleagues in Australia, where the combination has been available in trials for some time. Here, we want to highlight how the treatment under review has given Australian melanoma patients a great option when other treatments do not work for them or have aggressive side effects; these testimonials also demonstrate the high quality of life that the treatment under review has offered these patients. We hope that we can offer this additional choice to Canadians.
From a 70-year-old female patient, who started to receive the treatment under review 14 months ago:
“My tumours started reducing from the very first treatment. The largest tumour was 5½ cm and it reduced to 1½ cm very quickly. I did have some side effects as my thyroid function was affected. I am now on medication, and it is treated as a chronic condition. I also have dry eyes and a rash across my chest which has been controlled by cortisone cream. I do get fatigue and have a loss of appetite. RELA+NIVO saved my life and has given me back my life. I can now play with my grandchildren; they are no longer coming to see sick nanna. I am full of hope and now I have the freedom to drive - I can get out and enjoy life which is wonderful. Only 12 months ago I had the palliative care team working with me to work out how I would die. I feel so lucky that I still have a life to live and my beautiful family to encourage and the opportunity to see my grandchildren grow up.”
From a 39-year-old caregiver:
“It is three years since my husband had a headache one day – he was sent for a scan and he was immediately diagnosed with Stage 4 melanoma, it was in his brain. Our twin boys were only two years old. He had brain surgery to remove the melanoma. It was decided that he would be treated with IPI+NIVO due to him being an otherwise healthy 36-year-old and due to the brain metastasis. After just two treatments he developed hepatitis and had to withdraw from IPI+NIVO. Then two weeks later he had three new metastasis – they were treated by gamma knife radiation, and he had an incredible response. After a gap from the IPI+NIVO it was agreed to try one more IPI+NIVO infusion and he sadly got hepatitis immediately. He had a seizure and colitis. The risk of taking IPI again was too high he then went to a NIVO regime only. But the brain metastasis kept growing. We researched this new treatment and pushed for my husband to be granted RELA+NIVO on compassionate access grounds and we were successful. My husband started the RELA+NIVO treatment in June 2021 and since then he has been a complete responder – he is stable and there is no evidence of disease. Nothing new has grown in 12 months. It is amazing as he has had no side effects at all.
Previously he couldn’t work, couldn’t drive and was unable to work due to the side effects of other treatments – it was touch and go if he would survive.
Now he has been able to get back to a great level of physical health – he is running ultra- marathons, and no one would ever think that he had cancer. Everyone thinks he is totally fit and healthy.
Our twin boys are now age six – they have a dad who is now coaching their football team and able to enjoy them growing up. He has returned to work in his high-profile job and contributing to society.”
From a 39-year-old patient:
“My experience with RELA+NIVO treatment has been incredibly successful.
I had a mole removed in 2017 and I had a sentinel lymph node biopsy, but no lymph nodes were identified. I was diligent with my scans but in January last year I found a lump on my leg which I thought was a mosquito bite at first. I was subsequently diagnosed with Stage 4b melanoma.
I was offered the opportunity to join a clinical trial and I hoped that this would give me options as I have a nine-year-old daughter to care for. After my 5th dose out of a 24-dose trial. I started to experience some neurological side effects and after extensive testing it was decided that I could not receive any more infusions. The good news is that those 5 doses of treatment worked incredibly well. I heard the amazing words that I had a complete metabolic response to the treatment.
I was able to work right through the treatment and now look forward to a wonderful future with my daughter.”
Conflict of Interest Declaration — Save Your Skin Foundation
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission?
No.
Did you receive help from outside your patient group to collect or analyze data used in this submission?
No.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review.
Table 6: Financial Disclosures for Save Your Skin Foundation
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
BMS | — | — | — | X |
Clinician Input
Ontario Health (Cancer Care Ontario) Skin Cancer Drug Advisory Committee
About Ontario Health (Cancer Care Ontario) Skin Cancer Drug Advisory Committee
OH-CCO’s Drug Advisory Committees provide timely evidence-based clinical and health system guidance on drug-related issues in support of CCO’s mandate, including the Provincial Drug Reimbursement Programs (PDRP) and the Systemic Treatment Program.
Information Gathering
Information was gathered via videoconferencing and email.
Current Treatments and Treatment Goals
The indication for nivolumab and relatlimab is broad and can fit in all lines of therapy.
In the first line metastatic or unresectable setting, the current treatments can include single agent nivolumab, or pembrolizumab, combination IPI+NIVO and BRAF targeted agents (for patients with BRAF mutations). The BRAF targeted therapy options are dabrafenib-trametinib, cobimetinib-vemurafinib, and binimetinib-encorafenib.
If patients received pembrolizumab or nivolumab in first line, the subsequent line options are ipilimumab alone, or BRAF targeted therapy (for patients with BRAF mutation). If ipilimumab-nivolumab followed by nivolumab maintenance is used in first line, only patients with a BRAF mutation have a second line option to use BRAF targeted therapy.
Patients who received first line BRAF targeted therapy may be eligible for pembrolizumab, nivolumab, or IPI+NIVO in the second line setting. If treated with pembrolizumab or nivolumab, the patient may be eligible to use ipilimumab further downstream.
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
The combination of nivolumab and relatlimab has a higher response rate than single agent nivolumab in patients with unresectable or metastatic melanoma as per the Relativity 047 trial. Although there is no head-to-head comparison trial, this combination also has less toxicity than ipilimumab plus nivolumab whose treatment related adverse events are reported in checkmate 067 trial.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
First line metastatic or unresectable
Nivolumab and relatlimab can be an alternative first line option to the current treatments available for patients with unresectable or metastatic melanoma.
Patients with pre-treatment with ipi-nivo, ipi alone or pd-1 inhibitor alone
In subsequent line, using nivolumab-relatlimab as an option could be considered over single agent ipilimumab as it has fewer side effects than single agent ipilimumab.
Which patients would be best suited for treatment with the drug under review? Which patients would be least suitable for treatment with the drug under review?
First line metastatic / unresectable
Patients who are not able to tolerate ipilimumab-nivolumab or who would be treated with single agent PD-1 inhibitor would be suitable for receiving nivolumab-relatlimab in the first line metastatic or unresectable setting.
Patients with pre-treatment with ipi-nivo, ipi alone or pd-1 inhibitor alone
Patients who failed immunotherapy in the second line or beyond setting would also be suitable for this treatment.
BRAF Negative
If patient failed PD-1 inhibitor, then ipilimumab would be next, followed by nivolumab-relatlimab
If a patient failed Ipi-nivo, then the patient can receive rela-nivo.
BRAF Positive
If a patient failed PD-1 +/- CTLA4 immunotherapy, then they could receive BRAF/MEK inhibitor. The next line could be ipilimumab alone if not previously treated with this in combination with PD-1, or relatimab-nivolumab. Relatlimab-nivolumab could also be used following all prior therapies (PD-1, CTLA-4, BRAF/MEK).
What outcomes are used to determine whether a patient is responding to treatment in clinical practice? How often should treatment response be assessed?
Outcomes to determine response include improved survival, reduction in the frequency/severity of symptoms, attainment of major motor milestones, ability to perform activities of daily living, improvement of symptoms, and stabilization (no deterioration) of symptoms. Treatment response will be routinely assessed clinically, and by CT and/or PET approximately every 3 months.
What factors should be considered when deciding to discontinue treatment with the drug under review?
Confirmed disease progression and/or unmanageable toxicities.
What settings are appropriate for treatment with [drug under review]? Is a specialist required to diagnose, treat, and monitor patients who might receive [drug under review]?
This treatment should be administered in an outpatient cancer clinic, prescribed by a medical oncologist.
Additional Information
Not applicable.
Conflict of Interest Declarations — OH-CCO Skin Cancer Drug Advisory Committee
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please refer to the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
OH-CCO provided secretariat function to the group.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission?
No.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician who contributed to the input.
Declaration for Clinician 1
Name: Dr. Frances Wright
Position: Lead, Ontario Health (Cancer Care Ontario) Skin Cancer Drug Advisory Committee
Date: 03-07-2023
Table 7: COI Declaration for OH-CCO Skin Cancer Drug Advisory Committee — Clinician 1
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
BMS | X | — | — | — |
Declaration for Clinician 2
Name: Dr. Marcus Butler
Position: Member, Ontario Health (Cancer Care Ontario) Skin Cancer Drug Advisory Committee
Date: 09-06-2023
Table 8: COI Declaration for OH-CCO Skin Cancer Drug Advisory Committee — Clinician 2
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
BMS | X | — | — | — |
Declaration for Clinician 3
Name: Dr. Teresa Petrella
Position: Member, Ontario Health (Cancer Care Ontario) Skin Cancer Drug Advisory Committee
Date: 09-06-2023
Table 9: COI Declaration for OH-CCO Skin Cancer Drug Advisory Committee — Clinician 3
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
BMS | X | — | — | — |
Declaration for Clinician 4
Name: Dr. Xinni Song
Position: Member, Ontario Health (Cancer Care Ontario) Skin Cancer Drug Advisory Committee
Date: 09-06-2023
Table 10: COI Declaration for OH-CCO Skin Cancer Drug Advisory Committee — Clinician 4
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
BMS | X | — | — | — |
Declaration for Clinician 5
Name: Dr. Elaine McWhirter
Position: Member, Ontario Health (Cancer Care Ontario) Skin Cancer Drug Advisory Committee
Date: 09-06-2023
Table 11: COI Declaration for OH-CCO Skin Cancer Drug Advisory Committee — Clinician 5
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
BMS | X | — | — | — |
Declaration for Clinician 6
Name: Dr. Tara Baetz
Position: Member, Ontario Health (Cancer Care Ontario) Skin Cancer Drug Advisory Committee
Date: 09-06-2023
Table 12: COI Declaration for OH-CCO Skin Cancer Drug Advisory Committee — Clinician 6
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
BMS | X | — | — | — |
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
Redactions: Confidential information in this document may be redacted at the request of the sponsor in accordance with the CADTH Drug Reimbursement Review Confidentiality Guidelines
Stakeholder Input: The views expressed in each submission are those of the submitting organization or individual; not necessarily the views of CADTH or of other organizations. As such, they are independent of CADTH and do not necessarily represent or reflect the view of CADTH. No endorsement by CADTH is intended or should be inferred. By filing with CADTH, the submitting organization or individual agrees to the full disclosure of the information. CADTH does not edit the content of the submissions.
CADTH does use reasonable care to prevent disclosure of personal information in posted material; however, it is ultimately the submitter’s responsibility to ensure no identifying personal information or personal health information is included in the submission. The name of the submitting organization or individual and all conflict of interest information are included in the submission; however, the name of the author, including the name of an individual patient or caregiver submitting the patient input, are not posted.
Accessibility: CADTH is committed to treating people with disabilities in a way that respects their dignity and independence, supports them in accessing material in a timely manner, and provides a robust feedback process to support continuous improvement. All materials prepared by CADTH are available in an accessible format. Where materials provided to CADTH by a submitting organization or individual are not available in an accessible format, CADTH will provide a summary document upon request. More details on CADTH’s accessibility policies can be found here.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.