CADTH Reimbursement Review
Ibrutinib (Imbruvica)
Sponsor: Janssen Inc.
Therapeutic area: Chronic lymphocytic leukemia
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Stakeholder Input
Clinical Review
Abbreviations
AE
adverse event
ATC
average treatment effect among the control group
ATO
average treatment effect among the overlap
ATT
average treatment effect among the treated
beta-2M
beta-2 microglobulin
BM
bone marrow
BR
bendamustine and rituximab
BTK
Bruton tyrosine kinase
CI
confidence interval
CIRS
Cumulative Illness Rating Scale
CLL
chronic lymphocytic leukemia
CMH
Cochran-Mantel-Haenszel
CR
complete response
CrCl
creatinine clearance
CRi
complete response with incomplete bone marrow recovery
del(11q)
11q deletion
del(17p)
17p deletion
DOR
duration of response
ECOG PS
Eastern Cooperative Oncology Group Performance Status
EORTC QLQ-C30
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30
FACIT
Functional Assessment of Chronic Illness Therapy
FCR
fludarabine plus cyclophosphamide plus rituximab
FD
fixed duration
HR
hazard ratio
HRQoL
health-related quality of life
IA
investigator assessment
IGHV
immunoglobulin heavy chain variable region
IPD
individual patient data
IRC
independent review committee
ITC
indirect treatment comparison
ITT
intention to treat
iwCLL
International Workshop on Chronic Lymphocytic Leukemia
LC
Lymphoma Canada
LDH
lactate dehydrogenase
MAIC
matching-adjusted indirect treatment comparison
MID
minimally important difference
MRD
minimal residual disease
NE
not estimable
Neff
effective sample size
NGS
next-generation sequencing
nPR
nodular partial response
OH-CCO Hem DAC
Ontario Health (Cancer Care Ontario) Hematology Cancer Drug Advisory Committee
OR
odds ratio
ORR
overall response rate
OS
overall survival
PB
peripheral blood
PD
progressive disease
PFS
progression-free survival
PR
partial response
QoL
quality of life
RCT
randomized controlled trial
SAE
serious adverse event
SLL
small lymphocytic lymphoma
TEAE
treatment-emergent adverse event
TLS
tumour lysis syndrome
TTD
time to treatment discontinuation
TTNT
time to next treatment
VO
venetoclax plus obinutuzumab
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Ibrutinib (Imbruvica) 140 mg capsule, oral |
Sponsor | Janssen Inc. |
Indication | Ibrutinib in combination with venetoclax for the treatment of adult patients with previously untreated CLL, including those with 17p deletion |
Reimbursement request | Ibrutinib in combination with venetoclax for the treatment of adult patients with previously untreated CLL, including those with 17p deletion |
Health Canada approval status | Post-NOC |
Health Canada review pathway | Standard |
NOC date | March 20, 2023 |
Recommended dose | Ibrutinib should be administered as a single drug for 3 cycles (1 cycle is 28 days), followed by 12 cycles of ibrutinib plus venetoclax, starting at cycle 4. Venetoclax should be given as per the venetoclax product monograph |
CLL = chronic lymphocytic leukemia; NOC = Notice of Compliance.
Introduction
Chronic lymphocytic leukemia (CLL) is a lymphoproliferative B-cell malignancy characterized by the progressive expansion of monoclonal B lymphocytes in the blood, bone marrow (BM), lymph nodes, or other lymphoid tissue.1,2 CLL is a rare disease with low prevalence and incidence worldwide.3,4 However, it is the most common adult leukemia in Canada.5 In 2018, 1,725 patients were diagnosed with CLL (1,095 men and 630 women).5 Patients are usually diagnosed with CLL between the ages of 65 and 70 years;6 however, more than 10% of patients are diagnosed with CLL when they are younger than 55 years.7 Other than factors related to patient, disease, and treatment,8 several genetic alterations can influence prognosis, including the deletion of 17p (del[17p]) resulting in the loss of tumour protein 53 (TP53), 1 of the poorest prognostic factors for CLL. Other genetic alterations, including TP53 mutation without del(17p), unmutated immunoglobulin heavy chain variable region (IGHV) gene, deletion of 11q (del[11q]), and complex karyotype (i.e., more than 3 cytogenetic aberrations), are associated with a poor prognosis in CLL. For many patients with CLL, disease burden is increased by the presence of major comorbidities and frailty, as well as by toxicities associated with standard-of-care chemotherapy-based regimens. CLL is generally considered incurable. The 5-year net survival for CLL is 83%; in 2020, 554 patients in Canada died from CLL.5 Median life expectancy for patients with del(17p) or TP53 mutation is less than 2 to 3 years from the time of initial diagnosis.9
For patients deemed ineligible for fludarabine, cyclophosphamide, and rituximab (FCR), treatment with chemoimmunotherapy, such as chlorambucil plus obinutuzumab, venetoclax plus obinutuzumab (VO), and continuous Bruton tyrosine kinase (BTK) inhibitors (e.g., ibrutinib or acalabrutinib) may be used.10,11 For patients with high-risk features (i.e., del[17p] and/or TP53 mutation), regardless of age or fitness, BTK inhibitors are the preferred treatment option, even though VO is still available to them.10,11 For young patients (aged 18 to 64 years) without del(17p) or TP53 mutations, FCR is recommended as first-line treatment for those with mutated IGHV. For young and fit patients with unmutated IGHV, BTK inhibitors are preferred over FCR. VO is also a treatment option for these patients, albeit with less durable remission compared with BTK inhibitors, and is not reimbursed publicly.10,12 Based on clinician input collected by CADTH, continuous BTK inhibitors are most commonly used in Canada for younger patients with higher-risk mutations, such as TP53 mutations, 11q mutations, or unmutated IGHV.
Ibrutinib monotherapy is indicated for CLL and small lymphocytic lymphoma (SLL), mantle cell lymphoma, and Waldenström macroglobulinemia, all of which have been reviewed by CADTH.13-15 Venetoclax monotherapy is indicated for CLL and acute myeloid leukemia,16-18 both of which have been reviewed by CADTH. Venetoclax in combination with obinutuzumab is indicated for CLL, and venetoclax in combination with rituximab is indicated for CLL, both of which have been reviewed by CADTH.19,20 The requested indication for reimbursement is ibrutinib in combination with venetoclax for the treatment of adult patients with previously untreated CLL, including those with del(17p). The requested indication is the same as the Health Canada–approved indication (post–Notice of Compliance). Ibrutinib in combination with venetoclax is indicated for the treatment of CLL in Europe.21 The FDA has not approved the combination of ibrutinib and venetoclax as a treatment for any disease.22
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of ibrutinib (Imbruvica) (140 mg oral capsule) in combination with venetoclax (Venclexta) in the treatment of CLL in previously untreated adult patients, including those with del(17p).
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient and clinician groups who responded to CADTH’s call for input and from the clinical expert consulted by CADTH for the purpose of this review.
Patient Input
Lymphoma Canada (LC) is a national charity that engages in education, support, advocacy, and research activities for patients and the lymphoma community. LC conducted an online anonymous patient survey between March 22 and May 2, 2023. A total of 87 patients, including 49 patients from Canada, responded to the survey. As most patients with CLL experience no or minor symptoms, many respondents indicated that their daily activities were not strongly impacted by CLL at diagnosis. A total of 64 respondents rated fatigue (47%), high white blood cell counts (26%), and body aches and pains (25%) as having a highly negative impact (3 to 5 out of 5) at diagnosis. Among the 71 respondents who reported psychosocial impacts of their CLL diagnosis, anxiety or worry (61%), stress of diagnosis (59%), and difficulty sleeping (28%) were the most common. The most highly rated negative physical symptoms (3 to 5 out of 5) among 70 respondents included fatigue (44%), body aches and pains (27%), and indigestion, abdominal pain, or bloating (17%). The most negatively rated impacts on quality of life (QoL) among the 87 respondents included anxiety or worry (42%), difficulty sleeping (31%), and stress of diagnosis (28%). When considering a novel CLL treatment, respondents cited living longer (81%), controlling symptoms (75%), longer remission (71%), better QoL (66%), and fewer side effects (35%) as extremely important.
Of 10 patients with CLL who had specific experience with ibrutinib-venetoclax regimen, 5 patients were in remission for 2 to 5 years. In 10 patients treated with ibrutinib-venetoclax regimen, there was improvement in high white blood cell counts (80%), enlarged lymph nodes (70%), low platelet and red blood cell counts (60%), and weight loss (30%).
The input highlighted that a time-limited, oral ibrutinib-venetoclax therapy option would be especially beneficial for those living in rural areas and cost-saving for the health care system. Of note, 24% of respondents reported preference for the fixed duration (FD) of treatment. Also, 55% of patients reported having more treatment options is very important.
Clinician Input
Input From the Clinical Expert Consulted by CADTH
The clinical expert consulted by CADTH indicated that the most important goals of treatment for patients with CLL is to reverse symptoms and control the disease for as long as possible with treatments that have minimal toxicity and do not have a significant negative impact on QoL. The clinical expert stated that the biggest limitation of current treatments for patients with CLL is that tumour cell resistance usually occurs, and patients stop responding or relapse on therapy. Other limitations include toxicity and drug interactions, the requirement for continuous ongoing treatment, and that there are no curative treatments for patients with CLL. The clinical expert stated that other considerations might be addressed, including the opportunity for FD treatment versus continuous treatment and achieving undetectable minimal residual disease (MRD). The clinical expert stated that this combination therapy could be used in a first-line setting and possibly in recurrent disease and in patients with CLL who have some level of resistance or at least have been exposed to the drugs in the combination. In addition, the clinical expert speculated that it is possible that this combination therapy could be used in patients who are resistant to 1 of the drugs in the combination, although this would have to be shown in the evidence. Overall, the clinical expert stated that ibrutinib-venetoclax could be used in patients who have been exposed to, but are not necessarily resistant to, either of these drugs. The clinical expert stated that there is a possible shift in the current treatment paradigm with the combination therapy because the drugs are stopped after a fix period, which is different than the paradigm of continuous therapy that is used for the different BTK inhibitors. The clinical expert commented that older patients or younger patients with a high Cumulative Illness Rating Scale (CIRS) score or a reduced creatinine clearance (CrCl), regardless of del(17p) or TP53 mutations status, should be suited to ibrutinib-venetoclax. In addition, patients with bulky disease or high lymphocytes counts, or with impaired CrCl, who are considered at higher risk for tumour lysis syndrome (TLS), would also be suitable for ibrutinib-venetoclax because ibrutinib was given before venetoclax to reduce the risk of tumour lysis. The clinical expert mentioned that there were no factors identified in the subgroup analysis indicating any subset of patients who would be most likely to respond or, conversely, less likely to respond to the combination therapy. All symptomatic patients with CLL need treatment. The clinical expert indicated that response to treatment is assessed by changes in peripheral blood (PB) counts, which can easily be documented by clinicians looking after patients. In addition, per feedback from the clinical expert, MRD assessments were also used from PB and BM using 2 technologies: next-generation sequencing (NGS) and multicolour flow cytometry. The clinical expert stated that death and disease progression, measured by increasing lymphocyte count or enlarging lymph nodes or spleen, are major reasons for discontinuing the treatment of ibrutinib in combination with venetoclax. Similar to other BTK inhibitors, the clinical expert stated that ibrutinib in combination with venetoclax treatment should be managed by a specialist (i.e., hematologist or medical oncologist) who is familiar with this class of drug to optimally manage toxicities and dosing.
Clinician Group Input
Seven clinicians from the Ontario Health (Cancer Care Ontario) Hematology Cancer Drug Advisory Committee (OH-CCO Hem DAC) and 2 clinicians who treat patients with CLL and SLL in Canada submitted 2 separate clinician group inputs. The 2 clinician groups and a clinical expert consulted by CADTH agree that ibrutinib-venetoclax would be the first-line option for patients with CLL and also suitable for later settings in certain clinical situations. OH-CCO Hem DAC added that ibrutinib-venetoclax may be limited to a healthy (i.e., younger) population, due to safety concerns related to cardiovascular adverse effects. All clinicians agreed that having a time-limited treatment option is an unmet need, that discontinuation of therapy would be considered in the case of disease progression, and that hematologists and/or medical oncologists should be involved in the management of patients with CLL being treated with ibrutinib-venetoclax. Additionally, the clinical expert consulted by CADTH stated that the development of tumour cell resistance, drug interactions, and a lack of curative therapy are also unmet needs. Both clinician groups and the clinical expert consulted by CADTH agree that symptom control, disease control (undetectable MRD), and time off treatment are important goals of therapy with ibrutinib-venetoclax treatment. The 2 clinician groups added that improvement in progression-free survival (PFS) and QoL, along with minimal toxicity from ibrutinib-venetoclax treatment as added by the clinical expert, are the desirable goals of therapy. None of the clinicians had experience treating patients with CLL with the ibrutinib-venetoclax combination.
Drug Program Input
The drug programs that participate in the CADTH reimbursement review process identified potential implementation issues regarding the potential comparators for ibrutinib-venetoclax; the re-treatment of patients whose disease recurs after the completion of therapy; the dosing regimen for ibrutinib-venetoclax; the continuation of venetoclax monotherapy in patients who are intolerant to ibrutinib; access to ibrutinib-venetoclax for patients who are currently receiving ibrutinib monotherapy and have not experienced disease progression; the place in therapy for ibrutinib-venetoclax; care provision issues related to storage requirements; the potential of ibrutinib-venetoclax for drug-drug, drug-food, and drug-herb interactions, requiring assessment and/or intervention; and concerns regarding the feasibility of adoption.
Clinical Evidence
Pivotal Studies and Randomized Controlled Trial Evidence
Description of Studies
The GLOW trial is a multicentre, randomized, open-label phase III study that compared the efficacy and safety of the combination of ibrutinib-venetoclax to chlorambucil-obinutuzumab for the first-line treatment of patients with CLL. GLOW enrolled older patients (aged 65 years and older) with previously untreated CLL who were unfit for treatment with a fludarabine-based regimen. Participants with del(17p) or known TP53 mutation were excluded because these aberrations are associated with inferior outcomes with chemoimmunotherapy (i.e., chlorambucil-obinutuzumab). Participants (N = 211) were randomized in a 1:1 ratio to receive ibrutinib-venetoclax (n = 106) or chlorambucil-obinutuzumab (n = 105). The CAPTIVATE trial is a multicentre, phase II, single-arm study assessing time-limited treatment with the combination of ibrutinib-venetoclax in sequentially enrolled participants with treatment-naive CLL or SLL in either an MRD-guided discontinuation cohort (n = 164) or a FD cohort (n = 159). The MRD cohort will not be further discussed because the sponsor is not proceeding with this treatment regimen. This review focuses on the CAPTIVATE FD cohort, which enrolled 159 patients eligible for a fludarabine-based regimen. Eligible patients in the CAPTIVATE FD cohort were aged 18 to 70 years with previously untreated CLL or SLL requiring treatment, per International Workshop on Chronic Lymphocytic Leukemia (iwCLL) criteria, and with measurable nodal disease by CT, with an Eastern Cooperative Oncology Group Performance Status (ECOG PS) of 0 to 2, and with adequate hepatic, renal, and hematologic function. There were 2 study Canadian sites included in the GLOW trial and no sites in Canada for the CAPTIVATE FD cohort.
The primary end points were PFS per independent review committee (IRC) in the GLOW trial and complete response (CR) rate per investigator assessment (IA) in the CAPTIVATE FD cohort. Other secondary or exploratory outcomes of interest included PFS per IA, overall survival (OS), overall response rate (ORR) per IRC and IA, CR rate per IRC, improvement in hematological parameters (secondary outcome in the GLOW trial; exploratory outcome in the CAPTIVATE FD cohort), duration of response (DOR) per IRC and IA, time to next treatment (TTNT) (GLOW study only), MRD negativity rate, TLS risk reduction, and health-related quality of life (HRQoL) (GLOW study only).
For the fludarabine-ineligible patients included in the GLOW trial, the majority of patients enrolled were men (57.8%) and white (95.7%); 42.2% of patients were female and other races included Asian (0.5%), multiple (0.5%), and 3.3% of patients did not report their race. Median age was 71 years (range, 47 to 93 years), with 87.2% of patients 65 years and older and 34.1% of patients 75 years and older. Advanced stage disease at baseline was reported for 54.8% of patients based on Rai stage III or IV disease and 42.1% of patients based on Binet stage C disease. About half of patients had a baseline ECOG PS of 1 (53.1%). Overall, the proportion of patients with high-risk disease, defined by the presence of del(11q), unmutated IGHV, or TP53 mutation, was similar between treatment arms (59.4% versus 57.1% for ibrutinib-venetoclax versus chlorambucil-obinutuzumab). For fludarabine-eligible patients in the CAPTIVATE FD cohort, the median age at baseline was 60.0 years (range, 33 to 71 years), and 28.3% of patients were 65 years and older. More patients were male (66.7%), and the majority of patients were white (92.5%); 33.3% of patients were female and other races included Asian (1.9%), Black or African American (0.6%), and native Hawaiian or other Pacific Islander (0.6%), and 4.4% of patients did not report their race. At baseline, more patients (69.2%) had an ECOG PS score of 0. Cytogenetic characteristics indicative of poor prognosis (per hierarchical classification) were del(17p) (12.6%) and del(11q) (17.6%). Other poor prognostic characteristics included mutated TP53 (10.1%), mutated del(17p) or TP53 (17.0%), unmutated IGHV (56.0%), and complex karyotype (19.5%).
Efficacy Results
Unless otherwise specified, the key efficacy results of the GLOW and CAPTIVATE trials are summarized in Table 2. Refer to Appendix 1 for the hierarchical testing order of secondary end points for the GLOW study and detailed efficacy data for the GLOW study and the CAPTIVATE FD cohort.
Progression-Free Survival
In the GLOW study, PFS per IRC was the primary end point. At the time of primary analysis (data cut-off: February 26, 2021), the data recorded a median follow-up time of 27.7 months (95% confidence interval [CI], 27.50 to 27.83 months) in the ibrutinib-venetoclax arm and 27.89 months (95% CI, 27.53 to 28.58 months) in the chlorambucil-obinutuzumab arm. Median PFS per IRC was not reached in the ibrutinib-venetoclax arm and was 21.0 months in the chlorambucil-obinutuzumab arm. The hazard ratio (HR) for PFS events was 0.216 (95% CI, 0.131 to 0.357; P < 0.0001) based on IRC assessment. Events of disease progression or death were reported for 20.8% of patients in the ibrutinib-venetoclax arm (13 disease progression events and 9 deaths) and 63.8% of patients in the chlorambucil-obinutuzumab arm (65 disease progression events and 2 deaths). Subgroup analyses and prespecified sensitivity analyses of PFS per IA in the primary analysis were generally consistent with the primary analysis across all prespecified subgroups except for race and disease diagnosis at baseline. In general, results were similar at the time of the extended follow-up (data cut-off: August 25, 2022).
In the CAPTIVATE FD cohort, in the primary analysis (data cut-off: November 12, 2020), median PFS by IRC assessment was not reached in the all-treated FD cohort, based on an overall median follow-up of 27.9 months. The PFS event rate probabilities in the all-treated FD cohort were 14.5% (21 disease progression events and 2 deaths) at the time of primary analysis. Of note, results for median PFS per IRC for all patients and for patients with del(17p) were based on an immature time point after the median follow-up and were therefore considered to be unreliable by the sponsor. Generally, similar results were observed for PFS per IA and in the extended follow-up analysis (data cut-off: August 4, 2021).
Overall Survival
In the GLOW study, in the primary analysis (data cut-off: February 26, 2021), median OS was not reached in either arm. With a median follow-up of 27.7 months for the ibrutinib-venetoclax arm and 27.89 months for the chlorambucil-obinutuzumab arm, there were 11 (10.4%) deaths observed in the ibrutinib-venetoclax arm and 12 (11.4%) deaths observed in the chlorambucil-obinutuzumab arm (HR = 1.048; 95% CI, 0.454 to 2.419; nominal P = 0.9121). Similarly, median OS was not reached in either arm in the extended follow-up analysis (data cut-off: August 25, 2022).
In the CAPTIVATE FD cohort, in the primary analysis (data cut-off: November 12, 2020), median OS was not reached in the all-treated FD cohort based on an overall median follow-up of 27.9 months; there were 3 deaths (1.9%) reported in the all-treated FD cohort and no deaths reported in patients with del(17p). Most patients were alive and on study with OS probabilities of 98.1% at 24 months at the primary data cut-off date and at 36 months at the extended follow-up analysis data cut-off date (August 4, 2021) for the all-treated FD cohort. All patients with del(17p) were alive and on study at the primary data cut-off date and at the extended follow-up analysis data cut-off date.
Overall Response Rate
At the GLOW primary analysis (data cut-off: February 26, 2021), the IRC-assessed ORR (of partial response [PR] or better) was similar in the ibrutinib-venetoclax and chlorambucil-obinutuzumab arms (86.6% and 84.8%, respectively; relative response = 1.02; 95% CI, 0.92 to 1.14; P = 0.6991). Similar results were observed in the ORR based on IA. Similar results were observed in the extended follow-up analysis (data cut-off: August 25, 2022). Of note, as the difference in ORR based on IRC assessment between treatment arms was not statistically significant (P = 0.6991) in the primary analysis, the hierarchical statistical testing strategy ended at ORR per IRC. The remaining key secondary end points (i.e., OS, sustained hematological improvements, and time to improvement on the Functional Assessment of Chronic Illness Therapy [FACIT]-Fatigue Scale) and ORR per IA were considered not statistically significant.
In the CAPTIVATE FD cohort, in the primary analysis (data cut-off: November 12, 2020), the ORR per IRC assessment was 96.2% (95% CI, 93.3% to 99.2%) for all treated patients and 100.0% (95% CI, 100.0% to 100.0%) for patients with del(17p). Generally, similar results were observed with ORR per IA and in the extended follow-up analysis (data cut-off: August 4, 2021).
CR (CR and CR With Incomplete Bone Marrow Recovery) Rate
In the GLOW study, in the primary analysis (data cut-off: February 26, 2021), the IRC-assessed CR rate was higher in the ibrutinib-venetoclax arm compared with the chlorambucil-obinutuzumab arm (38.7% and 11.4%, respectively; relative response = 3.43; 95% CI, 1.91 to 6.15; P < 0.0001). Similar results were observed at the time of the 18-month extended follow-up (data cut-off: August 25, 2022) and with CR rate per IA.
In the CAPTIVATE FD cohort, CR rate by IA was assessed as the primary end point. In patients without del(17p) in the FD cohort, the CR rate per IA was 55.9% (95% CI, 47.5% to 64.2%), which exceeded the prespecified minimum CR rate of 37% (1-sided P < 0.0001) in the primary analysis (data cut-off: November 12, 2020). Similar results were observed in the extended follow-up analysis (data cut-off: August 4, 2021) with a CR rate of 58.1% (95% CI, 49.8% to 66.4%) based on IA. The investigator-assessed CR rate in the all-treated patients was 55.3% (95% CI, 47.6% to 63.1%) in the primary analysis and 57.2% (95% CI, 49.5% to 64.9%) in the extended follow-up analysis. For patients with del(17p), the CR rates per IA were identical, at 50.0% (95% CI, 28.1% to 71.9%), in the primary analysis and in the extended follow-up analysis.
Subgroup analyses of CR rate per IA, sensitivity analyses using CR rate per IRC and duration of CR, and supportive analysis of the duration of CR were generally consistent with the primary analysis. The subgroup analysis results were consistent across all prespecified subgroups except for bulky disease. Refer to Appendix 1 for detailed subgroup analyses data.
Sustained Hematologic Improvement
In the GLOW study, in the primary analysis (data cut-off: February 26, 2021), the proportion of patients with sustained improvement in hemoglobin was similar for the ibrutinib-venetoclax and chlorambucil-obinutuzumab arms (44.3% and 50.5%, respectively; nominal P = 0.3854). The proportion of patients with sustained improvement in platelets was similar for the ibrutinib-venetoclax and chlorambucil-obinutuzumab arms (24.5% and 29.5%, respectively; nominal P = 0.4346). Similar results were observed at the time of the 18-month extended follow-up (data cut-off: August 25, 2022).
In the CAPTIVATE FD cohort, in the primary analysis (data cut-off: November 12, 2020), the proportion of patients achieving a sustained improvement in hemoglobin was 41.5% (95% CI, 33.9% to 49.2%) in all treated patients and 60.0% (95% CI, 38.5% to 81.5%) in patients with del(17p). The proportion of patients with sustained improvement in platelets was 17.6% (95% CI, 11.7% to 23.5%) in all treated patients and 15.0% (95% CI, 0 to 30.6%) in patients with del(17p). Similar results were observed in the extended follow-up analysis (data cut-off: August 4, 2021).
Duration of Response
In the GLOW study, as of the data cut-off for the primary analysis (February 26, 2021), with an overall median follow-up of 27.7 months, the median DOR for patients who achieved an IRC-assessed PR or better was 28.9 months (95% CI, 28.7 to not estimable [NE]) in the ibrutinib-venetoclax arm and 21.1 months (95% CI,15.9 to 25.1) in the chlorambucil-obinutuzumab arm. Similar results were observed at the time of the 18-month extended follow-up (data cut-off: August 25, 2022) and for DOR per IA.
In the CAPTIVATE FD cohort, in the primary analysis (data cut-off: November 12, 2020), with a median follow-up of 27.9 months, the median DOR per IRC assessment for the FD cohort was not reached for all patients or for patients with del(17p) (with the lower end of the 95% CI of 18.9 months). The median DOR per IRC for patients with del(17p) in the primary analysis was considered not reliable due to the limited number (| | |) of patients at risk at 36 months after initial response. Similar results were observed for DOR per IA and for the extended follow-up analysis (data cut-off: August 4, 2021).
Time to Next Treatment
At the GLOW primary analysis (data cut-off: February 26, 2021), fewer patients in the ibrutinib-venetoclax arm received subsequent anticancer therapy compared to that in the chlorambucil-obinutuzumab arm (3.8% and 25.7%, respectively; HR = 0.143, 95% CI, 0.050 to 0.410; nominal P < 0.0001). Similar results were observed in the extended follow-up analysis (data cut-off: August 25, 2022).
TTNT was not reported in the CAPTIVATE FD cohort.
MRD Negativity
In the GLOW trial, a higher proportion of patients reported negative overall MRD by NGS in the ibrutinib-venetoclax arm than in the chlorambucil-obinutuzumab arm in BM (55.7% and 21.0%, respectively; relative response = 2.65; 95% CI, 1.75 to 3.99; P < 0.0001) and in PB (59.4% and 40.0%, respectively; nominal P = 0.0055) in the primary analysis (data cut-off: February 26, 2021). The MRD negativity rate was not assessed in the extended follow-up analysis (data cut-off: August 25, 2022).
In the all-treated FD cohort of the CAPTIVATE trial, overall MRD negativity rates by flow cytometry were as follows: 59.7% (95% CI, 52.1% to 67.4%) for all patients and 45.0% (95% CI, 23.2% to 66.8%) for patients with del(17p) in the BM; and 76.7% (95% CI, 70.2% to 83.3%) for all patients and 80.0% (95% CI, 62.5% to 97.5%) for patients with del(17p) in the PB. Identical results were reported for the overall MRD negativity rate in the extended follow-up analysis (data cut-off: August 4, 2021).
TLS Risk Reduction
In the ibrutinib-venetoclax arm of the GLOW study, 26 (24.5%) patients were at high risk for TLS based on tumour burden at baseline. After the ibrutinib lead-in, 22 (20.8%) patients shifted to medium or low risk in the primary analysis (data cut-off: February 26, 2021). TLS risk reduction was not assessed in the extended follow-up analysis (data cut-off: August 25, 2022).
In the FD cohort of the CAPTIVATE study, high tumour burden was observed in 34 of 159(21.4%) all-treated patients; among them, 1 (5%) patient had del(17p) at baseline. After 3 cycles of single-drug ibrutinib lead-in therapy, 33 (20.8%) patients shifted to medium or low risk in the primary analysis (data cut-off: November 12, 2020); among them, 1 (5%) patient had del(17p). TLS risk reduction was not assessed in the extended follow-up analysis (data cut-off: August 4, 2021).
Health-Related Quality of Life
In the GLOW trial, HRQoL was a secondary outcome. Generally, in the primary analysis of the GLOW trial (data cut-off: February 26, 2021), patients in the ibrutinib-venetoclax arm had early deteriorations and later improvements in HRQoL compared to patients in the chlorambucil-obinutuzumab arm, as measured by European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 (EORTC QLQ-C30) global health status, 5-Level EQ-5D questionnaire visual analogue scale (EQ-5D-5L VAS), EQ-5D-5L utility score, and FACIT-Fatigue Scale. All HRQoL results were considered not statistically significant according to the prespecified hierarchical statistical testing strategy because ORR per IRC failed to demonstrate statistical significance. The time to worsening or improvement in EORTC global health status, EQ-5D-5L VAS, EQ-5D-5L utility score, and FACIT-Fatigue Scale score were not assessed in the extended follow-up analysis (data cut-off: August 25, 2022).
HRQoL was not measured in the CAPTIVATE FD cohort.
Harms Results
Harms results for the GLOW trial and the CAPTIVATE FD cohort are summarized in Table 3. In fludarabine-ineligible patients reported in the GLOW trial, at least 1 adverse event (AE) was reported for a similar proportion of patients in the ibrutinib-venetoclax arm and the chlorambucil-obinutuzumab arm (99.1% versus 94.3% for ibrutinib-venetoclax versus chlorambucil-obinutuzumab) in GLOW. AEs of any grade were reported more frequently in the ibrutinib-venetoclax arm than in the chlorambucil-obinutuzumab arm, including diarrhea (50.9% versus 12.4%); nausea (26.4% versus 25.7%), infections and infestations (60.4% versus 48.6%); metabolism and nutrition disorders (42.5% versus 23.8%); respiratory, thoracic, and mediastinal disorders (35.8% versus 28.6%); musculoskeletal and connective tissue disorders (34.0% versus 25.7%); and nervous system disorders (30.2% versus 20.0%).
More patients in ibrutinib-venetoclax arm experienced at least 1 serious adverse event (SAE) of any grade than the chlorambucil-obinutuzumab arm (46.2% versus 27.6% for ibrutinib-venetoclax versus chlorambucil-obinutuzumab). More patients reported AEs leading to discontinuation in the ibrutinib-venetoclax arm (20.8% versus 7.6%). There were 11 (10.4%) patients in the ibrutinib-venetoclax arm and 12 (11.4%) patients in the chlorambucil-obinutuzumab arm who died during the study period and AE was the most frequent cause of death in the ibrutinib-venetoclax arm (6.6% versus 1.9%). In the GLOW study, more patients in the ibrutinib-venetoclax arm than in the chlorambucil-obinutuzumab arm reported atrial fibrillation (14.2% versus 1.9%). Grade 3 or 4 atrial fibrillation was reported in 2 (1.9%) patients in the ibrutinib-venetoclax arm while no patients in the chlorambucil-obinutuzumab arm. Similarly, a higher proportion of patients reported major hemorrhage in the ibrutinib-venetoclax arm than in the chlorambucil-obinutuzumab arm (3.8% versus 1.0%).
In fludarabine-eligible patients in the CAPTIVATE FD cohort, at least 1 AE was reported in 158 (99.5%) patients and AEs of grade 3 or 4 were reported in 98 (61.6%) patients. The most commonly reported AEs were diarrhea (62.3%), nausea (42.8%), neutropenia (41.5%), and arthralgia (33.3%). Grade 3 or 4 neutropenia was reported in 52 (32.7%) patients. There were 36 (22.6%) patients who experienced at least 1 SAE of any grade and 30 (18.9%) patients who experienced at least 1 SAE of grade 3 or 4. Eight (5.0%) patients had AEs leading to ibrutinib discontinuation, while 3 (1.9%) patients had AEs leading to venetoclax discontinuation. There were 3 (1.9%) patients who died during the study period and 7 (4.4%) patients who had atrial fibrillation, 2 (1.3%) of which were of grade 3 or 4. Major hemorrhage was reported in 3 (1.9%) patients.
Table 2: Summary of Key Results From Pivotal Studies and RCT Evidence (ITT Analysis Set for GLOW; All-Treated Analysis for CAPTIVATE)
Outcomes | GLOW primary analysis (data cut-off: February 26, 2021) | GLOW extended follow-up (data cut-off: August 25, 2022) | CAPTIVATE FD cohort | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
I+V (n = 106) | C+O (n = 105) | I+V (n = 106) | C+O (n = 105) | Primary analysis (N = 159) (data cut-off: November 12, 2020) | Extended follow-up (N = 159) (data cut-off: August 4, 2021) | |||||
All treated (N = 159) | Non-del(17p) (n = 136) | With del(17p) (n = 20) | All treated (N = 159) | Non-del(17p) (n = 136) | With del(17p) (n = 20) | |||||
PFS per IRC | ||||||||||
PFS events by IRC assessment, n (%) | 22 (20.8) | 67 (63.8) | 29 (27.4) | 78 (74.3) | 23 (14.5) | 18 (13.2) | 5 (25.0) | 40 (25.2) | 32 (23.5) | 7 (35.0) |
Progressive disease | 13 (12.3) | 65 (61.9) | 17 (16.0) | 71 (67.6) | 21 (13.2) | 16 (11.8) | NR | 38 (23.9) | 30 (22.1) | NR |
Death | 9 (8.5) | 2 (1.9) | 12 (11.3) | 7 (6.7) | 2 (1.3) | 2 (1.5) | NR | 2 (1.3) | 2 (1.5) | NR |
Censored | 84 (79.2) | 38 (36.2) | 77 (72.6) | 27 (25.7) | 136 (85.5) | 118 (86.8) | NR | 119 (74.8) | 104 (76.5) | NR |
PFS by IRC assessment, (months), median (95% CI) | NE (31.2 to NE) | 21.0 (16.6 to 24.7) | NE (NE to NE) | 21.7 (16.7 to 26.1) | NE (30.7 to NE) | NE (30.7 to NE) | NE (21.9 to NE) | 39.6 (39.0 to NE) | 39.6 (39.3 to NE) | 38.9a (38.6 to NE) |
HR by IRC assessment (95% CI; P value) | 0.216 (0.131 to 0.357; P < 0.0001) | 0.214 (0.138 to 0.334; P < 0.0001) | NA | |||||||
OS | ||||||||||
Deaths, n (%) | 11 (10.4) | 12 (11.4) | 15 (14.2) | 30 (28.6) | 3 (1.9) | 3 (2.2) | 0 | 3 (1.9) | 3 (2.2) | 0 |
Follow-up (months), median (95% CI) | 27.70 (27.50 to 27.83) | 27.89 (27.53 to 28.58) | 46.06 (45.83 to 46.29) | 46.06 (45.14 to 46.59) | 27.9 (27.7 to 28.1) | 27.8 (27.7 to 28.1) | 27.8 (27.7 to 28.1) | 38.7 (38.7 to 38.8) | 38.7(38.7 to 38.8) | 27.8 (27.7 to 28.1) |
OS, (months), median (95% CI) | NE (NE to NE) | 32.5 (32.5 to NE) | NE (NE to NE) | NE (NE to NE) | NE (NE to NE) | NE (NE to NE) | NE (NE to NE) | NE (NE to NE) | NE (NE to NE) | NE (NE to NE) |
HR (95% CI; P value) | 1.048 (0.454 to 2.419; nominal P = 0.9121) | 0.487 (0.262 to 0.907; nominal P = 0.0205) | NA | |||||||
ORR | ||||||||||
ORR by IRC assessment, % (95% CI) | 86.8 (80.3 to 93.2) | 84.8 (77.9 to 91.6) | 86.8 (80.3 to 93.2) | 84.8 (77.9 to 91.6) | 96.2 (93.3 to 99.2) | 95.6 (92.1 to 99.0) | 100.0 (100.0 to 100.0) | 96.2 (93.3 to 99.2) | 95.6 (92.1 to 99.0) | 100.0 (100.0 to 100.0) |
Rate ratio (95% CI; P value) | 1.02 (0.92 to 1.14; P = 0.6991) | 1.02 (0.92 to 1.14; nominal P = 0.6991) | NA | |||||||
CR (CR and CRi) rate | ||||||||||
CR rate by IRC assessment, % (95% CI) | 38.7 (29.4 to 48.0) | 11.4 (5.3 to 17.5) | 42.5 (33.0 to 51.9) | 12.4 (6.1 to 18.7) | 59.7 (52.1 to 67.4) | 61.0 (52.8 to 69.2) | 50.0 (28.1 to 71.9) | 62.3 (54.7 to 69.8) | 64.0 (55.9 to 72.0) | 50.0 (28.1 to 71.9) |
Rate ratio (95% CI; P value) | 3.43 (1.91 to 6.15; P < 0.0001) | 3.48 (2.00 to 6.05; nominal P < 0.0001) | NA | |||||||
CR rate by IA assessment, % (95% CI) | 45.3 (35.8 to 54.8) | 13.3 (6.8 to 19.8) | 51.9 (42.4 to 61.4) | 14.3 (7.6 to 21.0) | 55.3 (47.6 to 63.1) | 55.9 (47.5 to 64.2) | 50.0 (28.1 to 71.9) | 57.2 (49.5 to 64.9) | 58.1 (49.8 to 66.4) | 50.0 (28.1 to 71.9) |
Rate ratio (95% CI; P value) | 3.42 (2.01 to 5.82; P < 0.0001) | 3.65 (2.22 to 5.99; nominal P < 0.0001) | NA | |||||||
MRD negativity rate | ||||||||||
Overall MRD negativity rate,b % (95% CI) | ||||||||||
Bone marrow | 55.7 (46.2 to 65.1) | 21.0 (13.2 to 28.7) | NA | NA | 59.7 (52.1 to 67.4) | 61.8 (53.6 to 69.9) | 45.0 (23.2 to 66.8) | 59.7 (52.1 to 67.4) | 61.8 (53.6 to 69.9) | 45.0 (23.2 to 66.8) |
Rate ratio (95% CI; P value) | 2.65 (95% CI, 1.75 to 3.99; P value < 0.001) | NA | NA | NA | ||||||
Peripheral blood | 59.4 (50.1 to 68.8) | 40.0 (30.6 to 49.4) | NA | NA | 76.7 (70.2 to 83.3) | 76.5 (69.3 to 83.6) | 80.0 (62.5 to 97.5) | 76.7 (70.2 to 83.3) | 76.5 (69.3 to 83.6) | 80.0 (62.5 to 97.5) |
Rate ratio (95% CI; P value) | 1.48 (95% CI, 1.10 to 1.98; nominal P = 0.0055) | NA | NA | NA | ||||||
C+O = chlorambucil plus obinutuzumab; CI = confidence interval; CR = complete response; CRi = complete response with incomplete bone marrow recovery; del(17p) = deletion of 17p; FD = fixed duration; HR = hazard ratio; I+V = ibrutinib plus venetoclax; IA = investigator assessment; IRC = independent review committee; ITT = intention to treat; MRD = minimal residual disease; NA = not applicable; NE = not estimable; NR = not reported; ORR = overall response rate; OS = overall survival; PFS = progression-free survival; RCT = randomized controlled trial.
aEstimate is not reliable because only 3 patients were at risk at 39 months.
bNext-generation sequencing was used as the primary method of MRD analysis for the GLOW trial; flow cytometry method was used for the MRD analysis in the CAPTIVATE FD cohort.
Sources: GLOW primary analysis CSR;23 GLOW extended follow-up analysis CSR;24 GLOW PRO CSR;25 CAPTIVATE CSR.26 (Note: Details from the table have been taken from the sponsor’s Summary of Clinical Evidence.)
Table 3: Summary of Harms — Pivotal and RCT Evidence (Safety Analysis Set for GLOW; All-Treated Analysis for CAPTIVATE)
Adverse events | GLOW | CAPTIVATE | ||||
|---|---|---|---|---|---|---|
Ibrutinib-venetoclax (N = 106) | Chlorambucil-obinutuzumab (N = 105) | FD cohort (ibrutinib-venetoclax) (N = 159) | ||||
Any grade | Grade 3 or 4 | Any grade | Grade 3 or 4 | Any grade | Grade 3 or 4 | |
Harms, n (%) | ||||||
Patients with ≥ 1 adverse event | 105 (99.1) | 73 (68.9) | 99 (94.3) | 71 (67.6) | 158 (99.4) | 98 (61.6) |
Patients with ≥ 1 SAE | 49 (46.2) | 34 (32.1) | 29 (27.6) | 21 (20.0) | 36 (22.6) | 30 (18.9) |
TEAEs leading to discontinuation of 1 or more drugs | 22 (20.8) | NR | 8 (7.6) | NR | Ibrutinib: 8 (5.0) Venetoclax: 3 (1.9) | Ibrutinib: 7 (4.4) Venetoclax: 2 (1.3) |
Patients who died | 11 (10.4) | 12 (11.4) | 3 (1.9) | |||
Adverse events of special interest, n (%) | ||||||
Atrial fibrillation | 15 (14.2) | 7 (6.6) | 2 (1.9) | 0 (0.0) | 7 (4.4) | 2 (1.3) |
Major hemorrhage | 4 (3.8) | 4 (3.8) | 1 (1.0) | 1 (1.0) | 3 (1.9) | 2 (1.3) |
FD = fixed duration; NR = not reported (when incidence rates fall below the specified reporting threshold in either treatment arm within the specified adverse event severity); RCT = randomized controlled trial; SAE = serious adverse event; TEAE = treatment-emergent adverse event.
Note: At the GLOW extended 18-month follow-up from the primary analysis (data cut-off: August 25, 2022), no changes in safety data were noted, except for 1 additional patient in the chlorambucil-obinutuzumab treatment arm who was diagnosed with 2 new treatment-emergent SAEs, (myelodysplastic syndrome and myeloproliferative neoplasm) after primary analysis and 7 additional patients who were reported to have developed a nontreatment-emergent secondary malignancy (4 patients from the ibrutinib-venetoclax arm and 3 patients from the chlorambucil-obinutuzumab arm).
Sources: GLOW primary analysis CSR;23 CAPTIVATE CSR.26 (Note: Details from the table have been taken from the sponsor’s Summary of Clinical Evidence.)
Critical Appraisal
Fludarabine-Ineligible Population
The open-label design of the GLOW trial had the potential to introduce reporting bias into the assessment of subjective outcomes reported by patients, such as HRQoL and AEs. Disease response outcomes (PFS, ORR, DOR, CR rate) were assessed by investigator assessments and an IRC to help mitigate the biases associated with the open-label study design for the GLOW trial. In the GLOW trial, the overall median duration of exposure was substantially longer for the ibrutinib-venetoclax arm (13.8 months) than for the chlorambucil-obinutuzumab arm (5.1 months), which may bias the results in favour of ibrutinib-venetoclax. In addition, fewer patients in the chlorambucil-obinutuzumab arm discontinued treatment due to AEs compared to the ibrutinib-venetoclax arm, and a higher proportion of patients completed the study treatment in the chlorambucil-obinutuzumab arm than the ibrutinib-venetoclax arm, which indicates that patients in the chlorambucil-obinutuzumab arm had better treatment compliance than those in the ibrutinib-venetoclax arm in the GLOW trial, which may bias the results against the ibrutinib-venetoclax arm. A higher proportion of patients in the chlorambucil-obinutuzumab arm received subsequent anticancer therapy compared to the ibrutinib-venetoclax arm. The clinical expert consulted by CADTH indicated that the use of subsequent therapies would influence OS. The CADTH review team agreed with the clinical expert and noted that the use of subsequent anticancer therapy results in an indirectness of the estimated OS effect. It is difficult in this setting to isolate the direct effect of ibrutinib-venetoclax treatment on OS due to the intercurrent use of subsequent anticancer therapies. There were critical protocol amendments in the GLOW trial impacting the conduct of the trial after patients had first been randomized, which may have biased the results and increased uncertainty because of increased heterogeneity in the patient population for the ibrutinib-venetoclax arm. For example, the last 3 cycles (cycles 16, 17, and 18) of ibrutinib monotherapy were removed from the treatment in the ibrutinib-venetoclax arm. This may increase uncertainty in the estimate of treatment effect due to patient heterogeneity because there may be a mix of patients who received and did not receive 3 cycles of ibrutinib monotherapy. Due to limited data, the magnitude and direction of the bias is uncertain. In the GLOW trial, median OS was not reached in the ibrutinib-venetoclax arm in the primary analysis, so the OS data were considered to be immature.
Fludarabine-Eligible Population
The FD cohort in the CAPTIVATE trial was designed as a single-arm study and, given the lack of a comparator arm, the ability to make definitive conclusions about the comparative efficacy of ibrutinib-venetoclax in fludarabine-eligible patients with CLL is limited. In addition, the open-label design had the potential to introduce reporting bias in the assessment of subjective outcomes reported by patients (i.e., AEs). Disease response outcomes (PFS, ORR, DOR, CR rate) were assessed by investigators and by an IRC to help mitigate the biases associated with the open-label study design for the FD cohort in the CAPTIVATE trial. The Median of OS and PFS were not reached in the CAPTIVATE FD cohort in the primary analysis, so the OS data were considered immature. In addition, the CADTH review team noted that although the CAPTIVATE FD cohort included a subgroup of patients with del(17p), no formal statistical testing was performed between subgroups and the sample size for patients with del(17p) was small (n = 20); thus, no conclusions can be drawn from the study results for patients with del(17p). Although the subgroup analyses were prespecified, there is no evidence that the studies were powered to detect subgroup differences. HRQoL is considered a relevant outcome by patients with CLL and by clinicians. However, there was no assessment for HRQoL in the CAPTIVATE FD cohort; thus, it is uncertain whether treatment with ibrutinib-venetoclax would improve HRQoL in fludarabine-eligible patients with CLL.
External Validity
Both the GLOW trial and CAPTIVATE FD cohort required eligible patients to have measurable nodal disease. However, according to the clinical expert consulted by CADTH, there is a small proportion of patients who only have elevated white cell counts and cytopenia and may not have an enlarged lymph node in clinical practice; these patients are important and would fit in the patient population for the ibrutinib-venetoclax regimen. The clinical expert stated that there is a more diversified patient population that includes patients from Asia and other parts of the world in their clinical practice compared to the patient population in the GLOW trial and CAPTIVATE FD cohort. The baseline characteristics of the 2 studies may be indicative of the overrepresentation of white patients (≥ 92%) with CLL in both the fludarabine-eligible and fludarabine-ineligible populations and thus present an evidence gap in patients’ generalizability. Although the inclusion criteria mandated that eligible patients in the GLOW trial have a CrCl of less than 70 mL/min and/or a CRIS score higher than 6, the baseline CrCl for both treatment arms is higher than what the clinical expert would expect in high-risk patients with CLL who are ineligible to FCR in clinical practice, which may indicate that patients in the GLOW trial have better kidney functions than patients in clinical practice. This may compromise the study results for general fludarabine-ineligible patients with CLL. Generally, the risk profile of patients in the CAPTIVATE FD cohort is what the clinical expert would expect in fludarabine-eligible patients with CLL in clinical practice.
Long-Term Extension Studies
No long-term extension studies were identified for this review.
Indirect Comparisons and Observational Studies
Description of Studies
In patients who were ineligible to received fludarabine, the sponsor provided 2 indirect treatment comparisons (ITCs) and 2 individual patient data (IPD) observational studies that evaluated the efficacy and safety of ibrutinib-venetoclax versus bendamustine plus rituximab (BR),27 ibrutinib,28 VO,29 and acalabrutinib.30 In fludarabine-eligible patients, an IPD analysis was conducted comparing ibrutinib-venetoclax versus FCR.31 The ITC analyses were based on matching-adjusted indirect treatment comparison (MAIC) methods (comparison with acalabrutinib and VO) and the IPD analyses were based on propensity score methods (comparison with BR, ibrutinib, and FCR). All comparisons included patients with untreated CLL but varied in terms of age (all adults or ≥ 65 years only) and the presence of comorbidities and high-risk mutations, such as del(17p) and TP53. The median follow-up duration ranged from 38 months to 54.4 months, depending on the treatment group and the analysis. ||| |||| |||| |||| |||||| |||| |||||||| ||| | |||||||||| ||||| |||| ||||||||||| |||||| |||| ||||| ||| |||| |||||||| ||||||| ||| ||| |||||||| |||||||||| ||| || ||||||||| |||||||||||| ||||||||| |||| |||| |||||| ||||| ||||||||| ||| ||||||||| ||||||||| |||||| |||||||| ||| ||||||.
Efficacy Results
Fludarabine-Ineligible Population
For ibrutinib-venetoclax versus acalabrutinib, the time-varying analysis of PFS estimated an HR of |||| |||| ||| |||| || ||||| | | ||||| ||| | || |||||| ||| |||| |||| ||| |||| || ||||| | | ||||| for more than 12 months for the base-case model. The comparison with VO reported a base-case PFS HR of |||| |||| ||| |||| || ||||| | | ||||) for the first 12 months of follow-up; after 12 months, the estimated HR was |||| |||| ||| |||| || ||||| | | |||||).
The observational study comparing ibrutinib-venetoclax versus BR estimated a PFS HR of |||| |||| || |||| || ||||| | | ||||||) for the base-case analysis. For the comparison of ibrutinib-venetoclax versus ibrutinib, ||||| ||| ||||| of patients in the ibrutinib-venetoclax and ibrutinib groups, respectively (| | ||||), reported a progression event (|| ||| ||| || ||| ||||||||).
For all comparisons in the fludarabine-ineligible population, the comparative efficacy of ibrutinib-venetoclax on OS was unclear, as the results had high uncertainty due to the low number of events, the limited sample size, and, in many cases, ||| || |||| ||||||| ||| |||| ||| |||||| |||||||||.
Fludarabine-Eligible Population
In the base-case analysis for PFS, the model estimated an HR of |||| |||| ||| |||| || ||||| | | |||||), favouring ibrutinib-venetoclax over FCR. The analysis of OS also suggested a benefit favouring ibrutinib-venetoclax but was limited by small sample size and low event rates, and thus should be interpreted with caution.
Harms Results
No safety data were reported in the fludarabine-ineligible population.
Among fludarabine-eligible patients, grade 3 or 4 treatment-emergent adverse events (TEAEs) were reported by ||| || ||| of patients in the FCR group compared with ||| || ||| of those in the ibrutinib-venetoclax group, across the base-case and sensitivity analyses conducted. Comparative odds ratios (ORs) of grade 3 or 4 AEs were not reported. The incidence of treatment discontinuation ranged from || || || for ibrutinib-venetoclax to ||| || ||| for FCR across the analyses.
Critical Appraisal
Fludarabine-Ineligible Population
For the MAICs comparing ibrutinib-venetoclax with acalabrutinib and VO, there was poor overlap between trial populations in the GLOW study and comparator trials, which was evident, given the low effective sample size after weighting (||||| || || ||| ||||||||). The comparator trials included patients with high-risk mutations who were excluded from the GLOW study, and the presence of these mutations could not be controlled for in the adjusted analyses. The ability to achieve balance in effect modifiers was also limited due to missing covariate data, and imbalances between groups were noted for important patient characteristics in the base-case analysis. ||||||||| ||| |||||||||||| |||||| |||||||||| ||| ||| ||| ||| ||| ||| ||| ||| |||||||||||| ||| |||||||| |||||| ||||||||| ||||||| |||| |||||| |||||||||. Given these limitations, the findings of the MAIC were considered highly uncertain.
The selection of covariates for inclusion in the propensity score model is important for inference to be valid in observational studies. In these analyses, it is unclear if all known confounders and prognostic factors were included. Moreover, there were issues with missing covariate data which may have impacted the specification of the propensity scores. ||| |||||||||||| || |||||||||| |||||| |||||| | |||||||||||| |||||||||| || |||||||| ||| |||| ||| ||||||||||| || ||||||||| ||| ||||||||| |||||| |||||||||| ||||| |||| ||| ||||| |||||||| |||||| || |||| |||| |||||||| ||||||| |||||||| ||| |||| |||| | |||||||||||||||| ||||||||| || ||| ||||||||| ||||||| ||| |||||| ||||| |||| ||||||||| |||||| |||| |||| || |||||||| || ||| ||| |||||| ||| ||| ||| ||| |||||||| || ||| |||||||||| ||||||.
In the fludarabine-ineligible population, none of the pairwise comparisons reported safety outcomes, thus the comparative safety is unknown.
Regarding external validity, there were differences in the patients included in each pairwise comparison that should be considered when interpreting the results. The comparison with BR and ibrutinib included only patients 65 years and older (with or without comorbidities), whereas the comparison with VO included any adult (≥ 18 years) with comorbidities (CIRS score > 6 and CrCl ≤ 70 mL/min), and the comparison with acalabrutinib included all patients 65 years and older but only younger adults who had comorbidities. Thus, it appears that the patients may not be comparable across analyses and the external validity of each pairwise comparison should be assessed separately.
Fludarabine-Eligible Population
As there was no randomized control group in the CAPTIVATE study, the sponsor conducted an observational study based on propensity score methods. For the IPD analysis, it was unclear if all known confounders and prognostic factors were included in the model and, in addition, there were issues with missing covariate data. Therefore, there may be residual confounding from measured, as well as unmeasured, covariates that could bias the effect estimates. Also, the data were based on a small sample size (||| ||| ||||||||| ||| ||| |||||||| || ||| |||| |||| ||||||||), and for OS, there were low event rates. Limited safety data were reported; thus, the comparative safety is unclear. The external validity of the base-case findings may be limited by the inclusion of patients with comorbidities and high-risk mutations that would not be eligible to receive FCR in clinical practice.
Conclusions
Patients and clinicians highlighted the need for new effective treatments for CLL that prolong life, control disease and symptoms, maintain QoL, and reduce side effects compared to current treatments. According to 1 pivotal trial, ibrutinib-venetoclax demonstrated a clinically meaningful improvement in PFS compared with chlorambucil-obinutuzumab in fludarabine-ineligible patients with CLL without del(17p). In fludarabine-eligible patients with CLL without del(17p), ibrutinib-venetoclax demonstrated a meaningful improvement in the CR rate compared with the prespecified minimum CR rate of 37%. Analyses of secondary outcomes supported the efficacy of ibrutinib-venetoclax in prolonging PFS and delaying disease progression. Data on OS were considered immature and not interpretable at the time of the analysis. Due to limitations in the statistical analysis, a lack of identified minimally important difference (MID) estimates in fludarabine-ineligible patients with CLL, and the lack of assessment in fludarabine-eligible patients, no definitive conclusions can be drawn from the HRQoL analyses in the GLOW trial. The pivotal study results were subjected to key limitations, such as the exclusion of patients without measurable disease, a major amendment in protocol, and the use of a comparator treatment for fludarabine-ineligible patients that may not be currently relevant in Canada. No new safety signals were identified in either fludarabine-ineligible or fludarabine-eligible patients with CLL.
The supplementary comparative evidence from the ITCs and observational studies submitted by the sponsor had significant limitations that impact the internal validity of the findings. For each of these analyses, there was potential residual confounding from measured and unmeasured confounders, as the statistical methods were unable to control for all sources of heterogeneity in patient and study characteristics. In addition, most analyses were based on a small sample size, and there were low event rates for the OS analysis, with some results showing a wide 95% CI and poor precision. In the fludarabine-ineligible patient population, the comparative efficacy of ibrutinib-venetoclax versus ibrutinib, BR, VO, or acalabrutinib is unclear. The observational data comparing ibrutinib-venetoclax to FCR in fludarabine-eligible patients shared the same limitations, and thus definitive conclusions on comparative efficacy cannot be drawn from this study. In addition, the lack of comparative efficacy for patients with del(17p) were not sufficiently addressed by the supporting evidence.
No comparative safety data were available in the fludarabine-ineligible population and limited data in the fludarabine-eligible population, thus comparative safety of ibrutinib-venetoclax is unknown.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of ibrutinib (Imbruvica), (140 mg oral capsule), in combination with venetoclax (Venclexta) in the treatment of CLL in previously untreated adult patients, including those with del(17p).
Disease Background
The contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the CADTH review team.
CLL is a lymphoproliferative B-cell malignancy characterized by the progressive expansion of monoclonal B lymphocytes in the blood, BM, lymph nodes, or other lymphoid tissue.1,2 The accumulation of genetic lesions and the interactions of leukemic cells with antigen through the B-cell receptor, as well as the microenvironment, are believed to play a key role in the survival and proliferation of CLL cells.32
Patients are usually diagnosed with CLL between the ages of 65 and 70 years;6 however, more than 10% of patients are diagnosed with CLL when they are younger than 55 years.7 The prognosis of patients with CLL is dependent on a variety of patient-related (age, sex, comorbidities, performance status), disease-related (disease stage, cytogenetics, marrow failure, immunodeficiency, lymphomatous transformation, biomarkers), and treatment-related (type of treatment, response, toxicity, MRD status) factors.8 Several genetic alterations can influence prognosis, including del(17p), which can result in the loss of TP53 and 1 of the poorest prognostic factors for CLL. Other genetic alterations, including TP53 mutation without del(17p), unmutated IGHV, del(11q), and complex karyotype (i.e., more than 3 cytogenetic aberrations) are associated with a poor prognosis in CLL.
For many patients with CLL, the disease burden is increased by the presence of major comorbidities and frailty, as well as by toxicities associated with standard-of-care chemotherapy-based regimens. Patients with CLL rarely experience symptoms at the time of diagnosis, but symptoms that may arise include lymphadenopathy, weight loss, recurrent disease-related infections, anemia, night sweats, and bleeding.33-35 Patients with CLL experiencing symptoms can have a substantial impact on their QoL. Untreated patients with CLL have been shown to have a reduced QoL compared to healthy individuals.36 For patients receiving treatment, conventional therapy is thought to compound this impact on QoL, as these patients report poorer QoL compared with those receiving no therapy or healthy individuals.37-39 CLL is generally considered incurable. The 5-year net survival rate for CLL is 83% ; in 2020, 554 people died from CLL in Canada.5 Median life expectancy for patients with del(17p) or TP53 mutation is less than 2 to 3 years from the time of initial diagnosis.9
CLL is a rare disease with low prevalence and incidence worldwide.3,4 However, it is the most common adult leukemia in Canada.5 In 2018, 1,725 patients were diagnosed with CLL (1,095 men and 630 women).5 Disease prevalence by region is summarized in Table 4.
Table 4: Estimated Prevalence of CLL in Canada
Region | Estimated prevalence (per 100,000)a |
|---|---|
Pan-Canadian (excluding Quebec) | 10.8 |
Alberta | 10.2 |
British Columbia | 9.1 |
Manitoba | 12.6 |
New Brunswick | 20.2 |
Newfoundland and Labrador | 2.8 |
Northwest Territories | 11.1 |
Nova Scotia | 9.4 |
Nunavut | 0.0 |
Ontario | 11.0 |
Prince Edward Island | 6.6 |
Saskatchewan | 16.9 |
Yukon | 0.0 |
Non-Insured Health Benefits | 10.8b |
CLL = chronic lymphocytic leukemia; NR = not reported.
aLower and upper estimates have not been reported by the sponsor.
bPrevalence of CLL in the Non-Insured Health Benefits population is assumed to be the same as the pan-Canadian prevalence.
Source: Statistics Canada.40
To distinguish a diagnosis of CLL from other lymphoproliferative disease, a blood smear, immunophenotype, and sometimes the genetics of a patient’s circulating lymphoid cells must be evaluated.2 Diagnosis of CLL requires presence of 5 × 109/L or more B lymphocytes in PB for at least 3 months, with clonality confirmed with immunoglobulin light chain restriction using flow cytometry. Regarding immunophenotype, a panel of CD19, CD 5, CD 20, CD 23, kappa, and lambda is usually sufficient to establish the diagnosis. Molecular genetics may help predict prognosis.
Standards of Therapy
The content within this section has been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the CADTH review team.
Because CLL is incurable, the goal of treatment is to achieve effective and durable disease control, with minimal toxicity and an acceptable HRQoL. The clinical expert consulted by CADTH added that the most important goal is to reverse symptoms and control the disease for as long as possible with treatments that have minimal toxicity and no negative impact on QoL. Also, achieving undetectable MRD is also considered a goal, according to the clinician consulted by CADTH.
There are a number of treatment options in the first-line therapy setting based on individual patient and disease characteristics, such as fitness for fludarabine-based therapy and/or risk status defined by IGHV mutation, del(17p) or TP53 mutation, and age.10 Eligibility for fludarabine-based therapy is also based on patient fitness; patients who are fit have a CIRS score of less than 6, an ECOG PS of 0 to 2, an absence of cardiac and renal diseases (CrCl equal or greater than 60 mL/min), favourable cytogenic and/or mutational status, or have clinical rationale from their physician (Figure 1).
Among patients deemed ineligible for FCR due to age (adults older than 65 years), a lack of overall fitness, cytogenetic and/or mutational status, or other clinical rationale from their physicians, treatment options must account for toxicity and infectious complications. In these older patients or in patients with comorbidities without del(17p) and/or TP53 mutation, treatment with chemoimmunotherapy, such as chlorambucil-obinutuzumab, VO, or continuous BTK inhibitors (e.g., ibrutinib or acalabrutinib), may be used.10,11 Chemoimmunotherapy has been shown to be the least effective treatment and is only recommended in limited scenarios.10 VO is an option in patients with mutated and unmutated IGHV; however, it has shorter expected PFS compared to continuous BTK inhibitors in those with unmutated IGHV. According to the clinical expert consulted by CADTH, VO has good activity in CLL and could be used for those who prefer FD therapy or for patients with chronic comorbidities (e.g., hypertension, atrial fibrillation, or an indication for blood thinners) that may be exacerbated by BTK inhibitors. However, the clinical expert noted that VO could be less desirable if patients with CLL are at high risk of TLS (i.e., elevated creatinine, nodal bulk, or very high lymphocyte counts).
For patients with high-risk features (i.e., del[17p] and/or TP53 mutation), regardless of age or fitness, BTK inhibitors are the preferred treatment option even if VO is still available to them.10,11 The clinical expert mentioned that cross-trial comparisons of the SEQUOIA, ELEVATE-TN, and CLL 14 trials suggest longer PFS with BTK inhibitors for patients with mutated TP53. According to the clinical expert, ibrutinib is being used less because other BTK inhibitors may have less toxicity and FD treatments have become an option. For patients with unmutated IGHV regions, ibrutinib, acalabrutinib, or venetoclax and obinutuzumab are options. For patients who are much older and very frail, FD treatment with mild alkylating drugs, such as chlorambucil-obinutuzumab, are often offered to these patients (even though they would likely have better results with BTK inhibitors with access permitted).
Among young (ages 18 to 64) patients without del(17p) or TP53 mutations, FCR is recommended as a first-line treatment for those with mutated IGHV. According to the clinical expert, FCR with chemoimmunotherapy can induce very long remissions and perhaps even cures for younger patients with good CIRS scores without high-risk mutations. For young and fit patients with unmutated IGHV, BTK inhibitors are preferred over FCR. VO is also a treatment option for these patients albeit with less durable remission compared to BTK inhibitors and is not reimbursed publicly.10,12 Based on clinician input collected by CADTH, continuous BTK inhibitors are most commonly used in Canada for younger patients with higher-risk mutations, such as TP53 mutations, 11q mutations, or unmutated IGHV genes.
Figure 1: Current Treatment Paradigm of CLL
![Treatment algorithms for patients with chronic lymphocytic leukemia who are treatment naive. For young patients (aged 18 to 64 years) without del(17p) or TP53 mutations, FCR is recommended as first-line treatment. For patients deemed ineligible for fludarabine, VO, chemotherapy-induced thrombocytopenia, and continuous BTK inhibitors may be used. For patients with high-risk features (i.e., del[17p] and/or TP53 mutation), BTK inhibitors are the preferred treatment option, although VO is still available to them. For patients without del(17p) or TP53 mutations who received FCR, VO, BTK inhibitor, and chemotherapy-induced thrombocytopenia, acalabrutinib, idelalisib-rituximab, venetoclax-rituximab, and zanubrutinib are used as second-line treatments. For patients with del(17p) or TP53 mutations who received BTK inhibitor, venetoclax-rituximab is used as the second-line treatment.](https://canjhealthtechnol.ca/index.php/cjht/article/download/PC0317r/version/826/1780/6636/PC0317CL-fig01.png)
BR = bendamustine-rituximab; BTKI = Bruton’s tyrosine kinase inhibitor; CIT = chemoimmunotherapy; Chl-R = chlorambucil plus rituximab; CLL = chronic lymphocytic leukemia; del(17p) = deletion of 17p; FCR = fludarabine, cyclophosphamide, and rituximab; VO = venetoclax plus obinutuzumab.
a First-line treatment options are based on the 2021 CADTH provisional funding algorithm and publicly available provincial criteria, supplemented with the 2022 update to the Canadian evidence-based guideline for front-line treatment of CLL.
b Relapsed or refractory CLL treatment options are based on the 2021 CADTH provisional funding algorithm and publicly available provincial criteria. Canadian evidence-based guideline in the relapsed or refractory setting is currently in development.
c Idelalisib-rituximab is available only in cases of intolerance to a BTK inhibitor or for bridging to cellular therapy.
d Venetoclax monotherapy is only funded after failure of a BTK inhibitor.
e Zanubrutinib for the treatment of relapsed or refractory CLL is currently under CADTH review. Sequencing of zanubrutinib in the relapsed or refractory setting is not specified, as it is a pre–Notice of Compliance submission to CADTH.
Drug Under Review
Key characteristics of ibrutinib in combination with venetoclax are summarized in Table 5, along with other treatments available for CLL.
For treatment of untreated CLL with an ibrutinib-venetoclax combination, the recommended dosing schedule is ibrutinib 420 mg (three 140 mg capsules) administered orally once daily for 3 28-day cycles, followed by ibrutinib 420 mg plus venetoclax 400 mg administered orally daily for 12 28-day cycles. Venetoclax dosing should be initiated in cycle 4, with dose ramp-up over 5 weeks (20 mg, 50 mg, 100 mg, 200 mg, and 400 mg daily), and continued at 400 mg daily from cycle 5 onward.
Ibrutinib monotherapy is indicated for CLL and SLL, mantle cell lymphoma, and Waldenström macroglobulinemia, all of which have been reviewed by CADTH.13-15 Venetoclax monotherapy is indicated for CLL and acute myeloid leukemia, both of which have been reviewed by CADTH.16-18 Venetoclax in combination with obinutuzumab is indicated for CLL and venetoclax in combination with rituximab is indicated for CLL, both of which have been reviewed by CADTH.19,20
B-cell receptor signalling is a key mechanism of disease progression in B-cell malignancy, wherein BTK plays a role in the signalling cascade.41,42 Ibrutinib is an oral, first-in-class, targeted BTK inhibitor.43 Specifically, the target of ibrutinib and its active metabolite, PCI-45227, is a cysteine residue located on site 481 within the adenosine triphosphate binding domain of BTK. Both molecules bind covalently and irreversibly to this residue, providing potent and sustained inhibition of BTK enzymatic activity.41,42 Venetoclax is an oral inhibitor of B-cell lymphoma 2.44 Given their distinct and complementary mechanism of action, ibrutinib and venetoclax work synergistically to eradicate CLL by eliminating both dividing and resting leukemic subpopulations.44 Ibrutinib effectively inhibits tumour cell proliferation while mobilizing leukemic cells from protective lymphoid niches.44 Further, ibrutinib increases the sensitivity of CLL cells to B-cell lymphoma 2 inhibition, thereby accelerating apoptotic cell killing by venetoclax.45
The requested indication for reimbursement is ibrutinib in combination with venetoclax for the treatment of adult patients with previously untreated CLL, including those with del(17p). The requested indication is the same as the Health Canada–approved indication (post-NOC). Ibrutinib in combination with venetoclax is indicated for the treatment of CLL in Europe.21 The FDA has not approved the combination of ibrutinib and venetoclax as a treatment for any disease.22
Table 5: Key Characteristics of Comparator Regimens for Ibrutinib-Venetoclax in Previously Untreated Patients With CLL
Regimen | Mechanism of action | Indication(s) | Route and dose | Serious adverse effects or safety issues |
|---|---|---|---|---|
Ibrutinib-venetoclax | Ibrutinib: A small molecule that forms a covalent bond with a cysteine residue (Cys-481) in the BTK active site, thereby inhibiting BTK activity. BTK is implicated in pathogenesis of several B-cell malignancies, including CLL. Venetoclax: An oral, selective small-molecule BCL-2 inhibitor (a protein that inhibits apoptosis). BCL-2 overexpression has been associated with resistance to chemotherapies. | For treatment of adult patients with previously untreated CLL, including those with del(17p). | Cycles 1 to 3 (28 days each)
Cycles 4 to 15 (28 days each)
| Ibrutinib: Second primary malignancies, cardiac toxicity, cerebrovascular accidents, TLS, cytopenias, lymphocytosis, leukostasis, hemorrhage, hepatic impairment, infections, teratogenic risk. Venetoclax: TLS, myelosuppression ± infection, bleeding, dyspnea, edema. |
Chlorambucil-obinutuzumab | Chlorambucil: A cell-cycle phase-nonspecific bifunctional alkylating drug that forms a cross-linkage between 2 strands of DNA, interfering with DNA, RNA, and protein synthesis via radical formation. Obinutuzumab: A humanized recombinant type II anti-CD 20 monoclonal antibody that targets the CD 20 transmembrane antigen on B lymphocytes and induces cell death via antibody-dependent cellular cytotoxicity. | For the treatment of previously untreated CLL. | Cycle 1 (28 days)
Cycles 2 to 6 (28 days each)
| Chlorambucil: Bone marrow suppression, pulmonary toxicity, infertility, SJS or TEN. Obinutuzumab: Neutropenia, thrombocytopenia, infections, infusion-related reactions, PML. |
BR | Bendamustine: Exact mechanism unknown; may cause apoptotic and nonapoptotic death of malignant cells by damaging DNA, increasing proapoptotic gene expression and inhibiting mitotic control. Rituximab: A chimeric mouse-human monoclonal IgG1κ antibody. It binds to the CD 20 antigen expressed on B lymphocytes and depletes CD20-positive cells via antibody-dependent cell cytotoxicity and complement-mediated cell lysis. | For first-line treatment of CLL. | Cycle 1 (28 days)
Cycles 2 to 6 (28 days each)
| Bendamustine: Cardiac toxicity, infusion reactions, myelosuppression, SJS or TEN, TLS, carcinogenicity, mutagenicity. Rituximab: Neutropenia, infections, infusion-related symptoms, TLS, arterial thromboembolism, SJS or TEN, PRES, PML. |
Ibrutinib | A small molecule that forms a covalent bond with a cysteine residue (Cys-481) in the BTK active site, thereby inhibiting BTK activity. BTK is implicated in the pathogenesis of several B-cell malignancies, including CLL. | For the treatment of adult patients with previously untreated CLL, including those with del(17p). | Oral, 420 mg daily until disease progression or unacceptable toxicity | Second primary malignancies, cardiac arrhythmias and cardiac failure, PR interval prolongation, cerebrovascular accidents, TLS, cytopenias, lymphocytosis, leukostasis, hemorrhage, hepatic impairment, infections, teratogenic risk. |
VO | Venetoclax: An oral, selective small-molecule BCL-2 inhibitor (a protein that inhibits apoptosis). BCL-2 overexpression has been associated with resistance to chemotherapies. Obinutuzumab: A humanized recombinant type II anti-CD 20 monoclonal antibody that targets the CD 20 transmembrane antigen on B lymphocytes and induces cell death via antibody-dependent cellular cytotoxicity. | For previously untreated CLL in patients who are ineligible for fludarabine-based regimens, require treatment, and have good performance status. | Cycle 1 (28 days)
Cycle 2 (28 days)
Cycles 3 to 6 (28 days each)
| Venetoclax: TLS, myelosuppression ± infection, bleeding, dyspnea, edema. Obinutuzumab: Neutropenia, thrombocytopenia, infections, infusion-related reactions, PML. |
Acalabrutinib | Acalabrutinib (a small molecule) and its active metabolite, ACP-5862, form a covalent bond with a cysteine residue in the BTK active site, leading to irreversible inactivation of BTK. | In combination with obinutuzumab or as monotherapy in patients with previously untreated CLL. | Oral, 100 mg twice daily until disease progression or unacceptable toxicity | Atrial fibrillation, second primary malignancies, cytopenias, hemorrhage, (opportunistic) infections. |
FCR | Fludarabine: A synthetic fluorinated analogue of purine nucleoside antiviral that prevents elongation of DNA strands through direct incorporation into DNA as a false nucleotide; inhibits DNA polymerase, primase, ligase, ribonucleotide reductase; partially inhibits RNA polymerase II. Induces cytotoxicity and/or apoptosis. Cyclophosphamide: An alkylating drug that prevents cell division primarily by cross-linking DNA and RNA strands. Rituximab: A chimeric mouse-human monoclonal IgG1 kappa antibody that binds to the CD 20 antigen and depletes CD 20-positive cells via antibody-dependent cell cytotoxicity and complement-mediated cell lysis. | Treatment of anti-CD 20 antibody-naive, previously untreated or second-line RR CLL in patients for whom fludarabine-based therapy is considered appropriate. | Cycle 1 (28 days)
Cycles 2 to 6 (28 days each)
| Fludarabine: Neurotoxicity, immunosuppression and/or opportunistic infections, autoimmune hemolytic anemia, pulmonary toxicities. Cyclophosphamide: Immunosuppression, myelosuppression, infections, hemorrhagic cystitis, pulmonary toxicity, cardiac toxicity, secondary malignancies, liver disease. Rituximab: Neutropenia, infections, infusion-related symptoms, TLS, arterial thromboembolism, SJS or TEN, PRES, PML. |
BCL-2 = B-cell lymphoma; BR = bendamustine plus rituximab; BTK = Bruton tyrosine kinase; CLL = chronic lymphocytic leukemia; del(17p) = deletion of 17p; FCR = fludarabine, cyclophosphamide, and rituximab; IgG1 = immunoglobulin G, subclass 1; PML = progressive multifocal leukoencephalopathy; PRES = posterior reversible leukoencephalopathy syndrome; SJS = Stevens-Johnson syndrome; TEN = toxic epidermal necrolysis; TLS = tumour lysis syndrome; VO = venetoclax plus obinutuzumab.
Sources: Sponsor’s clinical summary,46 Cancer Care Ontario – Drug Formulary,47 BC Cancer.48
Stakeholder Perspectives
Patient Group Input
This section was prepared by CADTH staff based on the input provided by patient groups. The full original patient input(s) received by CADTH have been included in the stakeholder section at the end of this report.
LC is a national charity that engages in education, support, advocacy, and research activities for patients and the lymphoma community. LC conducted an online anonymous patient survey between March 22 and May 2, 2023. A total of 87 patients, including 49 patients from Canada, responded to the survey. As most patients with CLL experience no or minor symptoms, many respondents indicated that their daily activities were not strongly impacted by CLL at diagnosis. A total of 64 respondents rated fatigue (47%), high white blood cell counts (26%), and body aches and pains (25%) as having a highly negative impact (3 to 5 out of 5) at diagnosis. Among the 71 respondents who reported psychosocial impacts of their CLL diagnosis, anxiety or worry (61%), stress of diagnosis (59%), and difficulty sleeping (28%) were the most common. The most highly rated negative physical symptoms (3 to 5 out of 5) among 70 respondents included fatigue (44%), body aches and pains (27%), and indigestion, abdominal pain, or bloating (17%). The most negatively rated impacts on QoL among the 87 respondents included anxiety/worry (42%), difficulty sleeping (31%), and stress of diagnosis (28%). When considering a novel CLL treatment, respondents cited living longer (81%), controlling symptoms (75%), extending remission (71%), improving QoL (66%), and reducing side effects (35%) as extremely important.
Of the10 patients with CLL who had specific experience with ibrutinib-venetoclax regimen, 5 patients have been in remission for 2 to 5 years. In 10 patients treated with ibrutinib-venetoclax regimen, there was improvement in high white blood cell counts (80%), enlarged lymph nodes (70%), low platelet and red blood cell counts (60%), and weight loss (30%).
The input highlighted that a time-limited, oral ibrutinib-venetoclax therapy option would be especially beneficial for those living in rural areas and cost-saving for health care system savings. Of note, 24% of respondents reported preference for the FD of treatment. Also, 55% of patients reported that having more treatment options is very important.
Clinician Input
Input From the Clinical Expert Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 1 clinical specialist with expertise in the diagnosis and management of adult patients with previously untreated CLL.
Unmet Needs
According to the clinical expert consulted by CADTH, the first-line treatment options for adult patients with CLL depend upon the patient’s age, CIRS score, and the features of the CLL, including IGHV mutational status and cytogenetic profile. For younger patients with a good CIRS score and no high-risk mutations, chemoimmunotherapy with FCR can induce very long remissions, and perhaps even cure the disease. For younger patients with higher-risk mutations, such as TP53 mutations, 11q mutations, or unmutated IGHV, continuous BTK inhibitors are most commonly used in Canada. In addition, FD treatments with VO are now funded and being used for this patient population. For older patients, FD treatments with mild alkylating drugs, such as chlorambucil-obinutuzumab, are occasionally offered. The clinical expert also stated that many of these patients will have better results with less toxicity with BTK inhibitors, which are now accessible in the first-line setting.
The clinical expert consulted by CADTH indicated that the most important goals of treatment for patients with CLL is to reverse symptoms and control the disease for as long as possible with minimal toxicity and no significant negative impact on the QoL. The clinical expert stated that the biggest limitation to current treatments for patients with CLL is that tumour cell resistance usually occurs, and patients stop responding or relapse on therapy. Other limitations include toxicity and drug interactions, the requirement for continuous ongoing treatment, and the fact that there are no curative treatments for patients with CLL. The clinical expert stated that other considerations to be addressed include the opportunity for FD treatment versus continuous treatment and achieving undetectable MRD.
Place in Therapy
The clinical expert consulted by CADTH indicated that the addition of venetoclax to BTK inhibitor therapy is directed at the underlying disease process, as the mechanism of the combination therapy addresses a cell resistance mechanism that is biologically activated in many cancers. The clinical expert stated that this combination therapy could be used in the first-line setting where CLL cells have not acquired resistance to either of the drugs or mechanisms of cytotoxicity used by this combination. The clinical expert also indicated that the combination therapy could possibly be used in recurrent disease and in patients with CLL who have some level of resistance, or at least have been exposed to the drugs in the combination. In addition, the clinical expert speculated that it is possible that this combination therapy could be used in patients who are resistant to 1 of the drugs in the combination, although this would have to be shown in evidence. Overall, the clinical expert stated that ibrutinib-venetoclax could be used in patients who have been exposed but are not necessarily resistant to either of these drugs. This is relevant to venetoclax, which is used as FD therapy, and thus patients will eventually relapse not because of resistance but because the treatment has been stopped.
The clinical expert stated that there is a possible shift in the current treatment paradigm with the combination therapy because the drugs are stopped after a fix perioded, which is different than the paradigm of continuous therapy that is used for the different BTK inhibitors.
Patient Population
The clinical expert consulted by CADTH commented on the inclusion and exclusion criteria of the GLOW trial that older patients or younger patients with a high CIRS score or a reduced CrCl were all included. Although patients with del(17p) or TP53 mutations were excluded, the study did not exclude patients based on other disease characteristics, cytogenetics (except for del[17p] or TP53 mutation), or bulk of disease. The study also included patients with bulky disease, high lymphocyte counts, or impaired CrCl, which are considered at higher risk for TLS, according to the clinical expert. The clinical expert indicated that venetoclax therapy can result in TLS; therefore, ibrutinib was given before the venetoclax to reduce the risk of tumour lysis. The clinical expert mentioned that there were no factors identified in the subgroup analysis indicating that any subset of patients who would be most likely to respond or, conversely, less likely to respond to the combination therapy. All symptomatic patients with CLL need treatment. The clinical expert commented that patients with del(17p)/TP53 mutations were included in the CAPTIVATE trial and similar MRD negativity rates were observed between those patients with and without del(17p)/TP53 mutations; therefore, the presence of del(17p)/TP53 mutations should not be an exclusion criterion for patients with CLL.
According to the clinical expert consulted by CADTH, patients best suited for treatment with the drug under review can be identified by the treating physician, without the need for a companion diagnostic. In addition, the clinical expert stated that it is very unusual to misdiagnose patients with CLL.
Assessing the Response to Treatment
The clinical expert consulted by CADTH indicated that response to treatment is assessed by changes in PB counts, which can easily be documented by clinicians looking after patients. The clinical expert stated that repeat BM biopsies are not often performed in clinical practice but were required as part of the clinical trial formal response criteria. The clinical expert stated that DOR or response to next treatment are important end points used by clinicians to choose appropriate treatments and to inform prognosis. The clinical expert stated that objective responses often correlate with improvements in cytopenia, which may result in decreased transfusion requirements or decrease risk of infection. In addition, MRD assessments were also used from PB and BM using 2 technologies: NGS and multicolour flow cytometry.
Discontinuing Treatment
According to the clinical expert consulted by CADTH, death and disease progression (measured by increasing lymphocyte count or enlarging lymph nodes or spleen) are major reasons for discontinuing the treatment of ibrutinib in combination with venetoclax. The clinical expert indicated that increasing lymphocyte count is usually accompanied by anemia or thrombocytopenia.
Prescribing Considerations
The clinical expert stated that ibrutinib in combination with venetoclax treatment should be managed by a specialist (i.e., hematologist or medical oncologist) who is familiar with this class of drugs to optimally manage toxicities and dosing.
Additional Considerations
The clinical expert mentioned that, with FD treatment, if a patient has a good response (i.e., PFS of 3 years or more), they may be offered the same treatment again, as they are not necessarily resistant. This could be an advantage of the combination therapy over continuous BTK inhibitor treatment, as chronic BTK inhibitor therapy often results in accumulation of side effects over time. In addition, the undetectable MRD rates with the combination therapy are significantly higher than with BTK inhibitor monotherapy, which could indicate deeper responses.
Clinician Group Input
This section was prepared by CADTH staff based on the input provided by clinician groups. The full original clinician group input(s) received by CADTH have been included in the stakeholder section at the end of this report.
OH-CCO Hem DAC provides evidence-based clinical and health system guidance. Seven clinicians from OH-CCO Hem DAC submitted input. In addition, 2 clinicians who treat CLL and SLL in Canada submitted input based on literature and consultation with other clinicians, including those from LC.
Unmet Needs
Both clinician groups noted that time-limited, all-oral combination ibrutinib-venetoclax regimen is the important unmet need. The ibrutinib-venetoclax option would neither require IV access nor continuous treatment. This unmet need would reduce travel to clinics, caregiver cost, chronic side effects, and associated ongoing costs. The 2 clinicians said that ibrutinib-venetoclax can be used for any previously untreated patients with CLL and might be better tolerated than venetoclax-obinutuzumab (e.g., very low risks of TLS). Also, they added that patients should be able to be re-treated with ibrutinib or venetoclax in the second line if they are not refractory to either drug. In contrast, OH-CCO Hem DAC stated that ibrutinib-venetoclax replaces BTK inhibitor monotherapy and practical onboarding of venetoclax-obinutuzumab can be considered.
Place in Therapy
The 2 clinicians stated that ibrutinib-venetoclax will be an alternate first-line option among other funded therapies. In contrast, OH-CCO Hem DAC suggested that ibrutinib-venetoclax would be an option for time-limited therapy for the highest-risk groups (e.g., those with del[17p] or TP53 mutations) and/or would increase accessibility of a time-limited treatment option for those unable to easily access venetoclax-obinutuzumab.
Patient Population
The 2 clinicians mentioned that previously untreated patients are eligible for ibrutinib-venetoclax regimen, with access available for those who already take either drug as the first-line treatment. However, OH-CCO Hem DAC suggested that ibrutinib-venetoclax therapy be limited to patients without cardiovascular comorbidities and likely younger population due to safety concerns regarding sudden deaths reported in a study by Lampson et al. (2017).49 According to OH-CCO Hem DAC, molecular testing for genetic mutations is standard of care for CLL and SLL. If jurisdictions do not provide access to molecular testing, genetic testing should be required for TP53, IGVH, and del(17p) mutations to select the eligible population for ibrutinib-venetoclax therapy. OH-CCO Hem DAC also added that the population that may benefit the most are those taking continuous standard-of-care therapies or having access issues.
Assessing Response to Treatment
The 2 groups said that improved PFS and reduced symptoms would be the meaningful response and standard of outcomes for CLL studies. Additionally, the 2 clinicians and OH-CCO Hem DAC noted that time off treatment and QoL are measures to determine responses to treatment, respectively.
Discontinuing Treatment
Both clinician groups emphasized that ibrutinib-venetoclax regimen is time-limited therapy. The 2 groups mentioned that early disease progression while on therapy and significant intolerance are necessary factors to be considered for discontinuation of therapy.
Prescribing Conditions
Both groups agreed that clinicians familiar with the treatment of CLL (e.g., hematologists and medical oncologists) should be prescribers of the ibrutinib-venetoclax combination (as outpatient therapy). The 2 clinicians added that a cardiologist may also be required due to safety concern. Both groups emphasized that additional lab monitoring (e.g., weekly ramp-up intensive lab monitoring with venetoclax) is likely required.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical expert consulted by CADTH are summarized in Table 6.
Table 6: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
Relevant funded comparators include acalabrutinib, ibrutinib monotherapy, venetoclax plus obinutuzumab, obinutuzumab plus chlorambucil (comparator in GLOW trial), and other rituximab-based chemoimmunotherapy combinations (e.g., bendamustine-rituximab, chlorambucil-rituximab). Zanubrutinib is currently going through CADTH review for previously untreated and relapsed or refractory CLL or SLL in adult patients, but a recommendation has not yet been issued. | Comment from the drug programs to inform pERC deliberations. |
Considerations for initiation of therapy | |
Ibrutinib-venetoclax is given for a fixed duration. For patients whose disease recurs after the completion of therapy, is there evidence to support re-treatment? | The clinical expert consulted by CADTH stated that there is no evidence yet. However, if patients have a durable response to first treatment (i.e., at least 3 years DOR), then the clinical expert speculated that re-treatment with ibrutinib-venetoclax would be considered. |
Considerations for prescribing of therapy | |
Ibrutinib should be administered as a single drug at a dose of 420 mg, once daily, for three 28-day cycles, followed by ibrutinib 420 mg plus venetoclax 400 mg daily for 12 28-day cycles. Venetoclax should be initiated at cycle 4 with dose ramp-up over 5 weeks. | Comment from the drug programs to inform pERC deliberations. |
If a patient experiences intolerance to ibrutinib, can treatment with venetoclax monotherapy be continued? | The clinical expert consulted by CADTH confirmed that treatment with venetoclax monotherapy should be continued if a patient experiences intolerance to ibrutinib because venetoclax monotherapy is an active therapy in treating patients with CLL. The clinical expert noted that patients receiving venetoclax monotherapy may have an elevated risk of tumour lysis syndrome at the beginning of the treatment. According to the clinical expert consulted by CADTH, administrating ibrutinib in combination with venetoclax would reduce the risk of tumour lysis syndrome in patients with CLL; if patients have to stop ibrutinib due to intolerance, it is safe to continue venetoclax as monotherapy. |
Generalizability | |
Should patients who are currently receiving ibrutinib monotherapy and have not experienced disease progression be eligible for the addition of venetoclax? | According to the clinical expert consulted by CADTH, although there were no data available, the clinical expert speculated that it should be an acceptable treatment. The clinical expert indicated that adding fixed-duration venetoclax could produce undetectable MRD and provide a rationale for stopping the ibrutinib, although it is not a standard practice at present. For some patients with “low-grade” side effects related to ibrutinib, this could be desirable. |
Funding algorithm (oncology only) | |
Drug may change the place in therapy of comparator drugs. | Comment from the drug programs to inform pERC deliberations. |
Under what clinical circumstances would ibrutinib-venetoclax be used over existing first-line drugs?
| The clinical expert consulted by CADTH stated that there are several options for first-line treatment at present. There is a suggestion from a comparison of PFS results across trials that patients with TP53 mutations or 17p deletions may have longer PFS with BTK inhibitors rather than venetoclax and obinutuzumab. The clinical expert referred to the results from the CLL14 trial, in which PFS in patients with TP53 mutations was 60.4% at 36 months, compared to closer to 76% to 79% at 48 months for acalabrutinib (ELEVATE-TN trial) and ibrutinib. The clinical expert indicated that there is no direct comparison yet of BTK inhibitors to venetoclax-based combination therapies. BTK inhibitor treatment may be preferred over venetoclax-based therapy in these patients, assuming no other comorbid conditions that may make BTK inhibitors less preferred (i.e., atrial fibrillation, labile hypertension). The clinical expert stated that for patients without TP53 alterations, there is no strong rationale yet to choose BTK inhibitor therapy over venetoclax fixed-duration therapy. Therefore, according to the clinical expert, the preference for fixed-duration vs. indefinite therapy is a factor. Regarding the eligible patient population, the clinical expert mentioned that it is promising that the CAPTIVATE trial reported a 36-month PFS of about 80% for the subset of patients with TP53 mutations, which is similar to that seen in the GLOW trial, in which patients did not have del(17p) or TP53 mutations. However, the clinical expert pointed out that the CAPTIVATE trial was a nonrandomized trial and only included 27 patients with TP53 mutations. Thus, the clinical expert concluded that, at present, it is speculative and the data supporting the combination of venetoclax and ibrutinib as a preferred first-line option in patients with TP53 mutations is not solid. With respect to the impact on downstream sequencing, the clinical expert indicated that patients receiving fixed-duration therapy with ibrutinib-venetoclax could be eligible for re-treatment with the same treatment if they have a good response to the first-line course (i.e., greater than 36 months, although this number could be open to discussion). The clinical expert stated that the concept would be that these patients would not have acquired resistance mutations to ibrutinib, which could be tested. |
Care provision issues | |
Ibrutinib is supplied as a 140 mg capsule in a bottle of 90 capsules, The product monograph indicates to store at room temperature between 15°C and 30°C. | Comment from the drug programs to inform pERC deliberations. |
Ibrutinib has potential for drug-drug, drug-food, drug-herb, and drug-laboratory interactions, requiring assessment and/or intervention. | Comment from the drug programs to inform pERC deliberations. |
System and economic issues | |
Feasibility of adoption (budget impact) is a concern. | Comment from the drug programs to inform pERC deliberations. |
BTK = Bruton tyrosine kinase; CLL = chronic lymphocytic leukemia; DOR = duration of response; MRD = minimal residual disease; pERC = CADTH pan-Canadian Oncology Review Expert Review Committee; PFS = progression-free survival; SLL = small lymphocytic lymphoma.
Clinical Evidence
The objective of CADTH’s Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of ibrutinib (140 mg capsule, oral) in combination with venetoclax (10 mg, 50 mg, 100 mg tablet, oral) for the treatment of previously untreated adults with CLL, including those with del(17p). The focus was placed on comparing ibrutinib in combination with venetoclax to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of ibrutinib in combination with venetoclax is presented in 3 sections, and CADTH’s critical appraisal of the evidence is included after each section. The first section, the Systematic Review, includes pivotal studies and randomized controlled trials (RCTs) that were selected according to the sponsor’s systematic review protocol. The second section includes indirect evidence from the sponsor. The third section includes additional studies that were considered by the sponsor to address important gaps in the pivotal and RCT evidence.
Included Studies
Clinical evidence from the following is included in the CADTH review and appraised in this document:
2 pivotal studies: 1 phase III, open-label, multicentre RCT (GLOW) and a phase II, multicohort, international trial (CAPTIVATE)
5 nonrandomized comparisons: 2 ITCs using MAICs and 3 IPD observational studies.
Pivotal Studies and RCT Evidence
The contents of this section have been informed by materials submitted by the sponsor. The following information has been summarized and validated by the CADTH review team.
Description of Studies
Two pivotal trials (GLOW and CAPTIVATE) met in the inclusion criteria for the systematic review conducted by the sponsor. Their characteristics are summarized in Table 7.
GLOW Study (Fludarabine-Ineligible Population)
The objective of the GLOW study was to evaluate FD ibrutinib-venetoclax versus the standard chemoimmunotherapy combination of chlorambucil-obinutuzumab in older patients and/or those with comorbidities with previously untreated CLL. In this phase III, open-label trial, treatment was randomized and administered through a parallel assignment model. Patients were centrally randomized in a 1:1 ratio to either treatment arm, balanced by using permuted blocks and stratified by IGHV mutational status (mutated versus unmutated versus not available) and the presence of del(11q) (yes versus no). The trial enrolled patients aged 65 years or older and patients 18 to 64 years with a CIRS score greater than 6 and/or a CrCl less than 70 mL/min. All patients had active CLL or SLL requiring treatment, per iwCLL criteria. Patients were excluded if they had del(17p) or known TP53 mutations, bleeding disorders, central nervous system involvement, Richter syndrome, or uncontrolled autoimmune hemolytic anemia or thrombocytopenia. Between May 2018 and April 2019, 211 patients were enrolled and randomly assigned to treatment with either ibrutinib-venetoclax (n = 106) or chlorambucil-obinutuzumab (n = 105). The trial was conducted at 86 sites in 14 countries, including 2 sites in Canada. Patients in the ibrutinib-venetoclax arm received 3 cycles of ibrutinib lead-in at 420 mg once daily followed by 12 cycles of ibrutinib-venetoclax, with a venetoclax ramp-up from 20 to 400 mg/day over 5 weeks and 400 mg/day from cycle 5 onward. Patients in the chlorambucil-obinutuzumab arm received 6 cycles of obinutuzumab. In cycle 1, patients received 100 mg of obinutuzumab on day 1 and 900 mg on day 2, followed by 1,000 mg on days 8 and 15. In cycles 2 through 6, patients received 1,000 mg of obinutuzumab on days 1, 8, and 15. Patients received chlorambucil per the label (0.5 mg/kg body weight on days 1 and 15 of each cycle). A diagrammatic representation of the GLOW study design is presented in Figure 2.
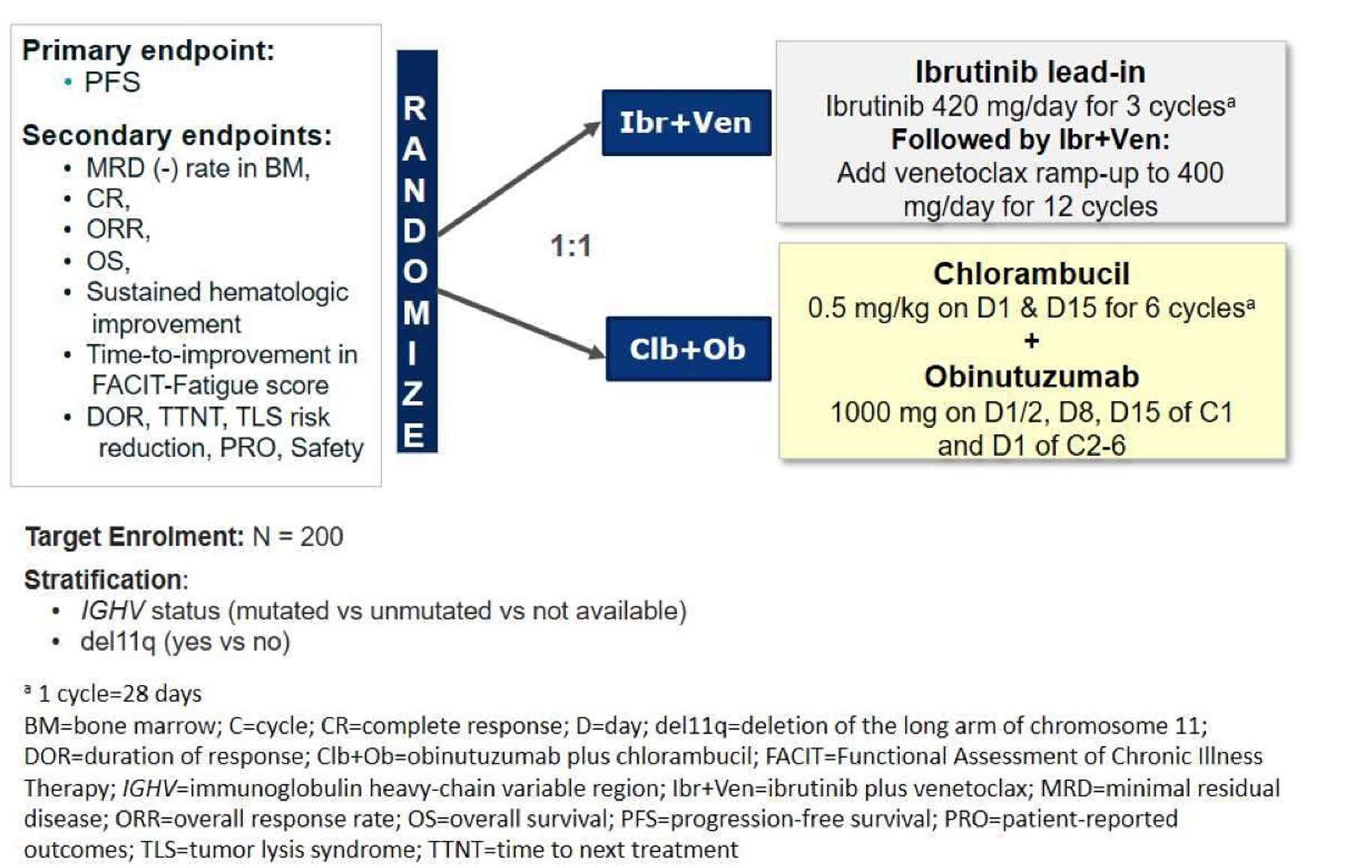
CAPTIVATE Study (Fludarabine-Eligible Population)
CAPTIVATE was a 2-cohort phase II study assessing both MRD-guided discontinuation (MRD cohort) and FD therapy (FD cohort) with the combination of ibrutinib-venetoclax in patients with treatment-naive CLL or SLL. The FD cohort was enrolled sequentially after the MRD cohort and is an open-label, single-arm cohort. For this review, only the FD cohort is discussed. The MRD cohort is not further discussed because the sponsor is not proceeding with this treatment regimen. The objective of the CAPTIVATE FD cohort was to evaluate the depth of response with the combination of ibrutinib-venetoclax administered for a FD of therapy by assessment of CR (CR and CR with incomplete BM recovery [CRi]) rate. Eligible patients for the CAPTIVATE FD cohort were aged 18 to 70 years with previously untreated CLL or SLL requiring treatment, per iwCLL criteria, and had measurable nodal disease by CT, an ECOG PS of 0 to 2, and adequate hepatic, renal, and hematologic function. Patients with known allergy to xanthine oxidase inhibitors and/or rasburicase were excluded because of the requirement for TLS prophylaxis per venetoclax prescribing information. At the start of the study, 159 patients enrolled in the FD cohort received 420 mg ibrutinib daily for 3 28-day cycles, followed by 12 cycles (28 days) of ibrutinib-venetoclax, with a target dose of 400 mg once daily after a 5-week ramp-up period, for a total of 15 cycles of open-label treatment.
Table 7: Details of Pivotal Studies and RCT Evidence Identified by the Sponsor
Details | GLOW | CAPTIVATE (FD cohort only) |
|---|---|---|
Designs and populations | ||
Study design | Phase III, open-label RCT | Phase II, multicohort, international trial FD cohort: single-arm, open-label |
Locations | 86 sites in 14 countries: Belgium, Canada, Czechia, Denmark, France, Israel, Netherlands, Poland, Russia, Spain, Sweden, Turkey, UK, US | 35 sites in 5 countries: Australia, Italy, New Zealand, Spain, US |
Patient enrolment dates | Start date: April 2018 End date: April 2019 | Start date: September 2016 End date: NA |
Randomized (N) | Total: 211 Ibrutinib-venetoclax: 106 Chlorambucil-obinutuzumab: 105 | 159 enrolled |
Inclusion criteria |
|
|
Exclusion criteria |
|
|
Drugs | ||
Intervention | Ibrutinib-venetoclax: 3 cycles of ibrutinib lead-in at 420 mg once daily, followed by 12 cycles of ibrutinib-venetoclax; venetoclax was initiated in cycle 4, with dose ramp-up per label over 5 weeks (20 mg/day, 50 mg/day, 100 mg/day, 200 mg/day, and 400 mg/day) and continued at 400 mg/day dose from cycle 5 onward; ibrutinib and venetoclax were administered on 28-day cycles. | Single-drug oral ibrutinib (420 mg once daily) lead-in for 3 cycles followed by 12 cycles of Ibrutinib-venetoclax (target dose 400 mg once daily after standard 5-week ramp-up). Treatment was administered in 28-day cycles. |
Comparator(s) | Chlorambucil-obinutuzumab: Received in six 28-day cycles of 1,000 mg/day of IV obinutuzumab (on day 1 [or 100 mg/day on day 1 and 900 mg/day on day 2], day 8, and day 15 of cycle 1 and day 1 of cycles 2 to 6) plus chlorambucil per label (0.5 mg/kg body weight on days 1 and 15 of each cycle) | NA |
Study duration | ||
Screening phase | Up to 30 days before randomization | Up to 30 days before initiation of study drug |
Run-in phase | NR | None in the FD cohort |
Treatment phase | Ibrutinib-venetoclax: Ibrutinib mean = 11.9 (SD = 3.84) months; venetoclax mean = 10.2 (SD = 2.32) months Chlorambucil-obinutuzumab: Chlorambucil mean = 5.0 (SD = 0.91) months; obinutuzumab mean = 4.7 (SD = 0.90) months | Ibrutinib mean = 13.3 (SD = 2.84) months; venetoclax mean = 11.1 (SD = 1.22) months |
Follow-up phase | Primary analysis: median = 27.7 months Extended follow-up: median = 46.1 months | Primary analysis: median = 27.9 months Extended follow-up: median = 38.7 months |
Outcomes | ||
Primary end point | PFS per IRC (up to 2 years and 10 months) | CR (CR and CRi) rate per investigator assessment |
Secondary and exploratory end points | Secondary:
| Secondary:
Exploratory:
|
Publication status | ||
Publications | Results of the GLOW trial were reported by Kater et al.44 NCT03462719 | Results of CAPTIVATE FD cohort were reported by Tam et al. (2022).50 NCT02910583 |
AE = adverse event; ANC = absolute neutrophil count; CIRS = Cumulative Illness Rating Scale; CLL = chronic lymphocytic leukemia; CNS = central nervous system; CR = complete response; CrCl = creatinine clearance; CRi = complete response with incomplete bone marrow recovery; del(17p) = deletion of 17p; DOR = duration of response; ECOG PS = Eastern Cooperativity Oncology Score Performance Status; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EQ-5D-5L = 5-Level EQ-5D; FACIT = Functional Assessment of Chronic Illness Therapy; FD = fixed duration; Hgb = hemoglobin; IRC = independent review committee; iwCLL = International Workshop on CLL; MRD = minimal residual disease; NA = not applicable; NR = not reported; ORR = overall response rate; OS = overall survival; PFS = progression-free survival; RCT = randomized controlled trial; SD = standard deviation; SLL = small lymphocytic leukemia; TLS = tumour lysis syndrome; TTNT = time to next treatment.
Note: One additional report was included: Health Canada report.51
Sources: GLOW primary analysis CSR;23 CAPTIVATE CSR.26 (Note: Details from the table have been taken from the sponsor’s Summary of Clinical Evidence.)
Populations
Inclusion and Exclusion Criteria
Both the GLOW and CAPTIVATE (FD cohort) studies required patients to be treatment naive and to be 18 years or older, with GLOW requiring patients younger than 65 years to have a CIRS score greater than 6 and/or a CrCl of less than 70 mL/min. Both studies had an inclusion criterion of an ECOG PS of 0 to 2. The GLOW study excluded patients with the presence of del(17p) and/or known TP53 mutations detected at a variable allele frequency of more than 10%, whereas the CAPTIVATE study allowed patients with del(17p) and/or TP53 mutations to enrol.
Interventions
Patients enrolled in the GLOW trial were randomized in a 1:1 ratio to treatment with either ibrutinib-venetoclax or chlorambucil-obinutuzumab, balanced by using permuted blocks and stratified by IGHV mutational status (mutated versus unmutated versus not available) and the presence or absence of del(11q). For patients in the ibrutinib-venetoclax treatment arm, ibrutinib was initiated with a lead-in phase consisting of 420 mg taken orally once daily for 3 cycles (28 days per cycle), then continued for 12 additional cycles in combination with venetoclax. Venetoclax was initiated in cycle 4 with a 5-week dose ramp-up phase in the sequence of 20 mg, 50 mg, 100 mg, 200 mg, then 400 mg daily administered orally, followed by continuous 400 mg per day treatment in combination with ibrutinib until the end of treatment. For the chlorambucil-obinutuzumab arm, patients received 6 cycles (28 days per cycle) of 1,000 mg daily of IV obinutuzumab on days 1, 8, and 15 of cycle 1, then day 1 for cycles 2 through 6. Chlorambucil 0.5 mg/kg on was given on day 1 and day 15 of each cycle. The first administration of obinutuzumab could be split into 100 mg on day 1 and 900 mg on day 2 for cycle 1 only. Patients assessed by IRC as having confirmed disease progression were eligible to receive subsequent therapy with single-drug ibrutinib, administered at 420 mg orally once daily until further disease progression or unacceptable toxicity.
Patients enrolled in the CAPTIVATE FD cohort received ibrutinib-venetoclax for 15 cycles of 28 days each, or until confirmed disease progression. Ibrutinib was initiated with a lead-in phase consisting of 420 mg taken orally once daily (three 140 mg capsules) for 3 cycles then continued for 12 cycles in combination with venetoclax. Venetoclax was given starting in cycle 4 with a target of 400 mg once daily taken orally after a standard 5-week ramp-up of 20 mg, 50 mg, 100 mg, 200 mg, and 400 mg doses.
Outcomes
A list of efficacy end points assessed in this report is provided in Table 8, and summarized subsequently. The summarized end points are based on those included in the sponsor’s Summary of Clinical Evidence as well as any identified as important to this review according to stakeholders, such as the clinical expert, clinician groups, or patient groups.
Table 8: Outcomes Summarized From Pivotal Studies Identified by the Sponsor
Outcome measure | Time point | GLOWa | CAPTIVATE (FD cohort)b |
|---|---|---|---|
PFS | GLOW: Up to 2 years and 10 months CAPTIVATE (FD cohort): At 24 months | Primaryc | Secondary |
OS | GLOW: Up to 3 years and 4 months CAPTIVATE (FD cohort): At 24 months | Secondaryc | Secondary |
ORR | GLOW: Up to 2 years and 10 months CAPTIVATE (FD cohort): Up to primary analysis data cut-off date (December 15, 2020) | Secondaryc | Secondary |
CR rate | Secondaryc | Primaryc | |
Improvement in hematologic parameters | GLOW: Up to 2 years and 10 months CAPTIVATE (FD cohort): From randomization date until before any reintroduced treatment | Secondaryc | Exploratory |
DOR | GLOW: Up to 2 years and 10 months CAPTIVATE (FD cohort): From initial documentation of a response until PD or death from any cause, whichever occurs first | Secondary | Secondary |
TTNT | GLOW: Up to 2 years and 10 months | Secondary | NR |
MRD negativity rate | GLOW: Up to 2 years 10 months CAPTIVATE (FD cohort): From randomization date until before any reintroduced treatment | Secondaryc | Secondary |
TLS risk reduction | GLOW: After ibrutinib 3-cycle lead-in CAPTIVATE (FD cohort): After ibrutinib 3-cycle lead-in before venetoclax | Secondary | Secondary |
Time to worsening (EQ-5D-5L, EORTC QLQ-C30, and FACIT-Fatiguec) | GLOW: Up to 2 years and 10 months | Secondary | NR |
Harms | GLOW: Up to 3 years and 4 months CAPTIVATE (FD cohort): From first dose until 30 days after last dose of study drug | Secondary | Secondary |
CR = complete response; DOR = duration of response; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EQ-5D-5L = 5-Level EQ-5D; FACIT = Functional Assessment of Chronic Illness Therapy; FD = fixed duration; MRD = minimal residual disease; NR = not reported; ORR = overall response rate; PD = progressive disease; PFS = progression-free survival; OS = overall survival; TLS = tumour lysis syndrome; TTNT = time to next treatment.
aFor the GLOW trial, the primary analysis data cut-off date was February 26, 2021, and the extended follow-up analysis data cut-off date was August 25, 2022.
bFor the CAPTIVATE FD cohort, the primary analysis data cut-off date was November 12, 2020, and the extended follow-up analysis data cut-off date was October 4, 2021.
cStatistical testing for these end points was adjusted for multiple comparisons (e.g., hierarchal testing).
Sources: GLOW primary analysis CSR;23 CAPTIVATE CSR.26 (Note: Details from the table have been taken from the sponsor’s Summary of Clinical Evidence.)
Progression-Free Survival
In the GLOW trial, PFS was the primary end point. PFS was defined as the time between the date of randomization and the date of progressive disease (PD) (as assessed by the IRC), or date of death due to any cause, whichever occurred first, regardless of the use of subsequent antileukemic therapy or treatment discontinuation before documented PD or death. Patients who were progression-free and alive were censored at the date of last disease assessment.
In the CAPTIVATE FD cohort, PFS was the primary end point. The definition of PFS was the time from the date of first study treatment to the date of disease progression, per IA, or date of death from any cause, whichever occurs first, regardless of the use of subsequent antineoplastic therapy before documented PD or death. For patients without documented PD or death, PFS was censored at the date of last adequate disease assessment.
Overall Survival
In both the GLOW and CAPTIVATE FD cohorts, OS was a secondary end point. In the GLOW study, OS was defined as the time from the date of randomization to the date of death from any cause, whereas in the CAPTIVATE FD cohort, OS was defined as the time from the date of first dose until the date of death due to any cause. If patients were alive or of unknown status by the end of study, their survival end point was censored at the date the patient was last known to be alive. In the GLOW trial, death due to COVID-19 was considered a potential competing risk to disease-related OS, and a composite variable strategy was adopted (i.e., considering death due to COVID-19 as an OS event) in the primary analysis of OS. If the number of patient deaths due to COVID-19 was more than 5% of total OS events, then a hypothetical outcomes strategy was adopted (censoring at the date of death due to COVID-19) as a supplementary analysis.
Overall Response Rate
ORR was a secondary end point in both the GLOW and CAPTIVATE trials. ORR was defined as the proportion of patients achieving a best overall response of either CR, CRi, nodular partial response (nPR), or PR, per iwCLL criteria, as evaluated by IRC or IA on or before initiation of subsequent antileukemic therapy (including subsequent single-drug ibrutinib). Disease response was assessed by investigators using CT or MRI as well as by IRC in the GLOW trial. Patients with missing postrandomization data were considered nonresponders.
CR Rate
In the GLOW study, CR rate was assessed as a secondary end point. CR rate was defined as the proportion of patients who achieved CR or CRi on or before initiation of subsequent antileukemic therapy (including subsequent single-drug ibrutinib).
In the CAPTIVATE FD cohort, CR rate for patients without del(17p), per IA, was the primary end point. CR rate was defined as the proportion of patients achieving a best overall response of CR or CRi on or before initiation of subsequent antineoplastic therapy or, if applicable, reintroduction of study treatment, whichever occurred first.
Improvement in Hematologic Parameters
Improvement in hematologic parameters was assessed as a secondary end point in the GLOW study and as an exploratory end point in the CAPTIVATE FD cohort. In both trials, sustained hemoglobin improvement was defined as the percentage of patients who achieved an increase in hemoglobin levels from baseline by 2 g/dL or more that lasted at least 56 days without blood transfusion or growth factors. In both trials, sustained platelet improvement was defined as the percentage of patients who achieved an increase in platelet levels from baseline by 50% or more that lasted for at least 56 days without blood transfusion or growth factors.
Duration of Response
In both trials, DOR was a secondary end point and was defined as the time elapsed between the date of initial documentation of a response, including PR with lymphocytosis, and the date of the first documented evidence of PD or death. Censoring of DOR data followed the same rule as for PFS. In the GLOW study, DOR was per IRC assessment, whereas in the CAPTIVATE FD cohort, it was per IA.
Time to Next Treatment
In the GLOW study, TTNT was assessed as a secondary end point. TTNT was measured from the date of randomization to the start date of any subsequent anticancer therapy. Patients without subsequent therapy were censored at the last known date to not have received subsequent therapy. TTNT was not assessed in the CAPTIVATE FD cohort.
MRD Negativity Rate
In both trials, the MRD negativity rate was assessed as a secondary end point and was defined as patients who achieved MRD negativity (< 1 CLL cell/104 leukocytes) in BM and PB. NGS was used as the primary method of MRD analysis, and flow cytometry was used for supplementary analyses in the GLOW study. In the CAPTIVATE FD cohort, only the flow cytometry method was used for the MRD analysis.
TLS Risk Reduction
TLS risk reduction was a secondary end point in both the GLOW and CAPTIVATE trials. In the GLOW trial, TLS risk reduction was defined as the proportion of patients whose TLS risk was reduced from high at baseline to medium or low after ibrutinib lead-in treatment; thus, it was only assessed for the ibrutinib-venetoclax arm. The CAPTIVATE FD cohort compared the proportion of patients who were at high risk of TLS after ibrutinib lead-in phase with the proportion at baseline. In both trials, TLS risk categories were defined per the venetoclax US package insert based on lymph node size and absolute lymphocyte count tumour burden measurements: Low risk was defined as all lymph nodes smaller than 5 cm and absolute lymphocyte counts less than 25 × 109/L; medium risk was defined as any lymph node 5 cm to less than 10 cm or absolute lymphocyte count 25 × 109/L or greater; and high risk was defined as any lymph node of at least 10 cm or absolute lymphocyte count 25 × 109/L or greater and any lymph node 5 cm or greater.
Health-Related Quality of Life
The GLOW trial also explored HRQoL by assessing time to worsening using the EQ-5D-5L, EORTC QLQ-C30, and FACIT-Fatigue instruments. The collection of patient-reported outcome assessments was to be stopped after the primary PFS analysis was completed.
EORTC QLQ-C30
EORTC QLQ-C30 includes 30 separate items resulting in 5 functional scales (physical functioning, role functioning, emotional functioning, cognitive functioning, and social functioning), 1 global health status scale, 3 symptom scales (fatigue, nausea and vomiting, and pain), and 6 single items (dyspnea, insomnia, appetite loss, constipation, diarrhea, and financial difficulties). Scores were derived using validated scoring algorithms, according to EORTC QLQ-C30 scoring manual. Scores range from 0 to 100; for functional and global QoL scales, higher scores indicate a better level of functioning. EORTC QLQ-C30 improvement or worsening is defined as a change of 10 points or more at each postbaseline assessment.52
EQ-5D-5L
The EQ-5D-5L consists of a 5-item descriptive system and the EQ-5D VAS of self-rated health, with scores ranging from 0 (worst imaginable health state) to 100 (best imaginable health state). Responses for the 5 dimensions are combined into a 5-digit number describing a respondents’ health state that can be converted into a single index value or utility score (using the UK weights), ranging from –1 to 1, where lower scores indicate a worse health status. A minimum difference of 0.07 points or more change in utility score is considered clinically important; for the VAS health rating, MID is a change of 7 points or more.53
FACIT-Fatigue
The FACIT-Fatigue Scale measures fatigue severity and its impact on daily activities. It includes 13 items that assess tiredness, weakness, and difficulty conducting usual activities due to fatigue. Scores range from 0 to 52, with high scores indicating less fatigue. FACIT improvement or worsening is defined as a change of 3 points or more at each postbaseline assessment.54
Harms
In the GLOW and CAPTIVATE trials, the proportion of patients who experienced AEs up until 30 days after the last dose of study treatment had their experiences recorded and reported using the National Cancer Institute Common Terminology Criteria for Adverse Events (version 4.03.26) coded with Medical Dictionary for Regulatory Activities (version 23.0). Specifically, hematological AEs were graded using iwCLL criteria, nonhematological AEs were graded using National Cancer Institute Common Terminology Criteria for Adverse Events (version 4.03.26), and TLS was assessed using Howard criteria.55 In both the GLOW and CAPTIVATE trials, AEs of special interest included atrial fibrillation and major hemorrhages.
Table 9: Summary of HRQoL Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
EORTC QLQ-C30 | EORTC QLQ-C30 includes 30 separate items, resulting in 5 functional scales (physical, role, emotional, cognitive, and social functioning), 1 global health status scale, 3 symptom scales (fatigue, nausea and vomiting, and pain), and 6 single items (dyspnea, insomnia, appetite loss, constipation, diarrhea, and financial difficulties). Scores are derived using scoring algorithms according to EORTC QLQ-C30 scoring manual. Scores range from 0 to 100 (for functional and global QoL scales; higher scores indicate a better level of functioning; for symptom scales or items, a higher score indicates a higher level of symptomatology).52 | Measurement properties of validity, reliability, and responsiveness have not been assessed in patients with CLL. A disease-specific module with 16 or 17 items for CLL (EORTC QLQ-CLL16 or EORTC QLQ-CLL17) has been developed and validated.56 | In patients with breast cancer and small cell lung cancer:57 EORTC QLQ-C30 improvement or worsening is defined as ≥ 10 points of change for each postbaseline assessment.52 |
EQ-5D-5L | The EQ-5D-5L consists of a 5-item descriptive system and the EQ VAS of self-rated health, with scores ranging from 0 (worst imaginable health state) to 100 (best imaginable health state). Responses to the 5 dimensions are combined into a 5-digit number describing a respondent's health state, which can be converted into a single index value or utility score (using the UK weights), ranging from –1 to 1, where lower scores indicate a worse health status.52 | Measurement properties of validity, reliability, and responsiveness have not been assessed in patients with CLL. | In cancer patients with 1 of 11 types of cancer, including lymphoma:53 Using an anchor-based approach, a minimum difference of ≥ 0.06 (US) or ≥ 0.09 (UK) points in utility score is considered clinically important; for the VAS health rating, MID is ≥ 7 points. Distribution-based methods (half SD, SEM) generated similar index scores (0.06 to 0.10), but higher VAS (9 to 11).53 |
FACIT-Fatigue | The FACIT-Fatigue Scale measures fatigue severity and its impact on daily activities. It includes 13 items that assess tiredness, weakness, and difficulty conducting usual activities due to fatigue. Scores range from 0 to 52 with high scores indicating less fatigue.52 | Validity Content validity in patients with CLL has been ensured with concept elicitation and cognitive debriefing interviews.58 Reliability When tested in patients with mixed-diagnosis cancers, internal consistency (Cronbach alpha = 0.93 to 0.95) was acceptable (> 0.7).54 In the same population, test-retest reliability was established using ICC of baseline and 3- to 7-day retest scores (r = 0.89).54 The responsiveness of measurement properties has not been assessed in patients with CLL. | In patients with mixed-diagnosis cancers:54
|
CLL = chronic lymphocytic leukemia; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EQ-5D-5L = 5-Level EQ-5D; FACIT = Functional Assessment of Chronic Illness Therapy; ICC = intraclass correlation coefficient; MID = minimally important difference; QoL = quality of life; SD = standard deviation; SEM = standard error of the mean; VAS = visual analogue scale.
Statistical Analysis
Sample Size and Power Calculation
A summary of statistical analysis of efficacy end points for the GLOW and CAPTIVATE trials is provided in Table 10.
With a 1:1 randomization, the GLOW study planned to enrol 200 patients (100 patients into ibrutinib-venetoclax and 100 patients into the chlorambucil-obinutuzumab treatment arm), expecting to observe 71 PFS events under an assumed alternative of HR = 0.5 and baseline exponential. Under the assumed alternative, the sample size would provide 80% power of rejecting a null of no difference in PFS using a log-rank test. Actual enrolment was 211 patients, with 106 and 105 randomly assigned to the ibrutinib-venetoclax and chlorambucil-obinutuzumab treatment arms, respectively.
The CAPTIVATE trial conducted separate sample size and power calculations for the FD cohorts. Sample sizes were estimated based on the primary end point of CR rate in patients without the del(17p) mutation (irrespective of TP53 mutation). A sample size of 125 patients was determined to be sufficient to exclude a minimum CR rate of 37%, with a 1-sided significance level of 0.025, with 83% power, assuming an alternative CR probability for ibrutinib-venetoclax of 50%.59
The choice of a minimum CR rate of 37% was based on the results of the CLL10 study, which evaluated first-line chemoimmunotherapy with BR versus FCR in patients with advanced CLL excluding patients with del(17p). In this study, a CR rate of 31% was observed in the BR arm, whereas treatment with FCR resulted in a CR rate of 40%.59 Based on that, a minimum CR rate of 37% was determined to ensure that the lower acceptable CR boundary for ibrutinib-venetoclax is comparable to the CR rate observed with FCR and BR in the CLL10 study, the standard of care for patients without del(17p) at the time the CAPTIVATE trial was designed.
Statistical Test or Model
In the GLOW study, all interval estimations were reported using 2-sided 95% CIs. No formal interim analysis was planned for efficacy outcomes due to the small sample size and short accrual period. Comparison tests across treatment groups were conducted using the Cochran-Mantel-Haenszel (CMH) chi-square test for discrete variables and a 2-sided log-rank test for time-to-event variables. The PFS analysis was performed on the intention-to-treat (ITT) population, defined as all randomized patients. The Kaplan-Meier method was used to estimate the distribution of PFS for each treatment arm. A difference in treatment effect of ibrutinib-venetoclax compared with chlorambucil-obinutuzumab on PFS was tested using a stratified log-rank test. The HR for ibrutinib-venetoclax relative to chlorambucil-obinutuzumab and its corresponding 95% CI were calculated using a stratified Cox regression model. In both cases, the stratification variables were those used for the block randomization: IGHV mutational status (mutated versus unmutated versus not available) and del(11q) (yes versus no). The GLOW study used a nonstratified Cox regression model to analyze the treatment effect on PFS as a sensitivity analysis. The model adjusted for select prespecified baseline factors as explanatory variables. The baseline factors are del(11q) (no, yes), IGHV (mutated, unmutated, not available), age group (< 65 years, > 65 years), sex (female, male), Rai stage (0 to II, III to IV), ECOG PS (0, 1 to 2), CIRS total score (< 6, > 6), elevated lactate dehydrogenase (LDH) (no, yes), bulky disease (< 5 cm, > 5 cm), cytopenia (no, yes), and serum beta-2 microglobulin (beta-2M) (< 3.5mg/L, > 3.5 mg/L).
In the CAPTIVATE trial, the estimate of CR rate per IA and the corresponding 95% CI based on normal approximation to the binomial distribution were provided. In the FD cohort non-del(17p) population, the P value for testing the CR rate (≤ 37% versus > 37%) was calculated using an asymptotic test for the binomial proportion. Planned analyses included a primary analysis and a final analysis. The primary analysis for the FD cohort was conducted after the last patient enrolled had the opportunity to be followed for at least 30 cycles (15 cycles of treatment plus 15 cycles of posttreatment follow-up), and all safety and efficacy outcomes were analyzed. The Kaplan-Meier method was used to estimate the median and the distribution of time-to-event variables (e.g., PFS, OS). The primary analysis was reported in the included publications for the FD cohort; as indicated in the statical analysis plan, a final analysis will be conducted when the study closes.
Multiple Testing Procedure
In the GLOW study, a serial gatekeeping procedure was used to account for multiplicity incurred from testing primary and secondary end points. Each hypothesis for secondary outcomes was only tested if the null hypotheses for the primary outcome and all preceding secondary outcomes were rejected. The secondary efficacy end points were tested in a hierarchal manner, at the nominal 0.05 significance level (2-sided), in the following order:
MRD negativity rate by NGS in BM
CR rate (per IRC)
ORR (ORR per IRC followed by ORR per IA)
OS
Rate of sustained platelet improvement
Rate of sustained hemoglobin improvement
Time to improvement in FACIT-Fatigue score.
In the CAPTIVATE FD cohort, hypothesis testing was performed independently (without multiplicity adjustment) for the primary end point of CR rate, which was tested in non-del(17) patients at a 1-sided alpha level of 0.025. Other end points were summarized descriptively, with 95% CIs whenever applicable.
Data Imputation Methods
There were no data imputation methods reported for the GLOW and CAPTIVATE trials.
Subgroup Analyses
The GLOW trial conducted predefined subgroup analyses for PFS per IRC for the following select demographic and baseline variables: age (< 65 years, ≥ 65 years), sex (male, female), race (white, nonwhite), diagnosis (CLL, SLL), Rai stage at screening for CLL patients only (0 to II, II to IV), Binet stage at screening for CLL patients only (A, B, C), ECOG PS (0, 1 to 2), CIRS total score (≤ 6, > 6), bulky disease (yes [longest diameter ≥ 5 cm], no [longest diameter < 5 cm]), IGHV mutation status (mutated, unmutated, not available), del(11q) mutation (yes, no), high-risk population (yes, no; defined as yes if patients presented with TP53 mutation or del[11q] or unmutated IGHV status at baseline), elevated LDH at baseline (yes [> upper limit of normal], no [≤ upper limit of normal]), cytopenia at baseline (yes, no; defined as yes if platelet count ≤ 100,000/μL, or Hgb ≤ 11 g/dL, or absolute neutrophil count ≤ 1,500/μL is observed), and serum beta-2M (≤ 3.5 mg/L, > 3.5 mg/L). The nonstratified log-rank test and Cox regression model were used for each of the predefined subgroup analyses to investigate whether the treatment effects on PFS persisted across populations. Estimated HRs across the 2 treatment arms within each subgroup and their 95% CIs were also provided.
The CAPTIVATE trial conducted predefined subgroup analyses for outcomes of CR rate, MRD negativity rate, and AEs for select variables. For CR rate and MRD negativity rate, select variables for subgroups included the following: age (< 65 years, ≥ 65 years), sex (male, female), race (white, nonwhite), ECOG PS (0, 1 to 2), Rai stage (0 to II, III to IV), bulky disease (< 5 cm, ≥ 5 cm), del(17p) mutated (yes, no), del(17p) or TP53 mutated (yes, no), fluorescence in situ hybridization (del[17p], del[11q], and others), and IGHV per central lab (unmutated, mutated). For AEs, select variables were age, sex, and race stratification, along with CrCl rate (< 60 mL/min, ≥ 60 mL/min) and National Cancer Institute Organ Dysfunction Working Group liver function classification (normal, abnormal).
Sensitivity Analyses
In the GLOW study, sensitivity and supplementary analyses were performed for PFS, as follows:
Nonstratified log-rank test and nonstratified Cox regression model were conducted for PFS based on IRC assessment in the ITT population.
Use of subsequent antileukemic therapy before documented PD or death: Patients were censored at the last disease assessment if there was no evidence of PD before the use of subsequent antileukemic therapy.
Disease assessment follow-up: Patients were censored at the last disease evaluation if they progressed or died after missing at least 2 consecutive planned disease evaluation visits.
PFS based on PD assessed by investigators: The same censoring rules and analysis methods used for primary analysis of IRC-reported PFS were used. The concordance rate between the IRC-assessed PD and investigator-assessed PD was evaluated. The number and percentage of PD events and non-PD cases assessed by investigator and by IRC were cross-tabulated.
The sponsor also planned to conduct a supplementary analysis censoring patients at the last disease assessment before pre-PD death due to COVID-19. However, the sponsor did not conduct this supplementary analysis, as patients with pre-PD death due to COVID-19 was less than the prespecified threshold of 5% of total PFS events.
In the CAPTIVATE trial, a sensitivity analysis for the primary end point, CR rate, was conducted, per IRC assessment. A supportive analysis included duration of complete response (duration of CR was defined as the interval between the date of initial CR or CRi until PD or death from any cause, whichever occurred first) for patients who achieved a CR or CRi, and durable CR rate (defined as the proportion of patients with a duration of CR of ≥ 336 days [12 cycles]). For discrete events (e.g., ORR), CIs were estimated using the normal approximation of the binomial distribution.
Secondary Outcomes of the Studies
Progression-Free Survival
In the CAPTIVATE trial, PFS was a secondary end point, and the PFS distribution was estimated using the Kaplan-Meier method; median PFS and 24-month and 36-month survival probability estimates with 2-sided 95% CIs were provided.
Overall Survival
In the GLOW study, OS was analyzed using similar analysis methods as used for the primary analysis of PFS. The sponsor also performed a supplementary analysis censoring patients at the date of death due to COVID-19.
In the CAPTIVATE trial, Kaplan-Meier estimates of median OS time and landmark time (i.e., 6-month, 12-month, 18-month, 24-month, 30-month, and 36-month) estimates with 2-sided 95% CIs were provided.
Overall Response Rate
ORR was estimated according to the crude proportion of confirmed responders (CR, CRi, nPR, or PR), based on the best overall response, and was summarized by treatment arm in the GLOW study. ORR was compared between treatment arms using the CMH chi-square test, adjusted for the randomization stratification factors. The relative response and its associated 95% CI were estimated according to CMH. Both ORR based on IRC and IA were analyzed based on the same analysis methods.
In the CAPTIVATE trial, the point estimate of ORR (per IRC and per IA) and the corresponding 95% CI based on normal approximation to the binomial distribution were provided.
Improvement in Hematologic Parameters
In both the GLOW and CAPTIVATE trials, the proportion of patients achieving sustained hemoglobin and platelet improvement were summarized by treatment arm, respectively. In the GLOW trial, these proportions were compared using the CMH chi-square test.
Duration of Response
DOR was analyzed using the same method as used for PFS in the GLOW study. Only patients who achieved a PR or better were included in the analysis of DOR. DOR was summarized descriptively using the Kaplan-Meier method, and no inferential comparison was made between treatment arms.
In the CAPTIVATE trial, Kaplan-Meier estimates of median DOR time (per IRC and per IA) and 6-month,12-month,18-month, 24-month, 30-month, and 36-month landmark estimates with 95% CIs were provided.
Time to Next Treatment
TTNT was analyzed using the same methods to those used for PFS outcome in the GLOW study. The number of patients who received subsequent antileukemic therapy were summarized by therapy type. TTNT was not assess in the CAPTIVATE trial.
MRD Negativity Rate
In the GLOW study, all randomized patients were included in this analysis; patients with missing MRD data were considered to be MRD positive. The overall MRD negativity rate in the BM, assessed by NGS, was the primary MRD analysis used for hierarchical testing. The MRD negativity rate by NGS in the PB was considered as the supportive analysis for this end point. The MRD negativity rate among patients who achieved a best overall response of CR or CRi per IRC assessment was conducted as a supplementary analysis. The CMH chi-square test adjusted for the randomization stratification factors was used to test for differences in MRD negativity rates across treatment groups.
In the CAPTIVATE trial, the point estimate of the MRD negativity rate and the corresponding 95% CI based on normal approximation to the binomial distribution were provided.
TLS Risk Reduction
In the GLOW study, descriptive statistics were provided. The proportion of patients with hospitalization indicated due to TLS risk (i.e., patients with TLS risk based on high tumour burden, patients with TLS risk based on medium tumour burden and CrCl < 80 mL/min) was also summarized at baseline and after the ibrutinib lead-in.
In the CAPTIVATE trial, the estimated difference in proportions of patients with high risk of TLS (at baseline after ibrutinib lead-in) and the corresponding 95% CI (adjusting correlation of paired data) were provided as primary analysis. Reduction of the proportion of patients with hospitalization indicated due to TLS risk (i.e., patients with high TLS risk and patients with medium risk of TLS and CrCl < 80 mL/min) were provided as a supportive analysis.
Health-Related Quality of Life
In the GLOW study, descriptive statistics were provided. Mixed-effects models for repeated measures analysis, time to deterioration (first clinically meaningful deterioration relative to baseline), and time to improvement (first clinically meaningful improvement relative to baseline) were provided for EORTC QLQ-C30 global health status, FACIT-Fatigue score, EQ-5D-5L utility score, and EQ-5D-5L VAS. HRQoL outcomes were not reported for the CAPTIVATE trial.
Table 10: Statistical Analysis of Efficacy End Points
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
GLOW | ||||
PFS |
| Stratification factors: IGHV mutation status; presence of del(11q) | Unless specified otherwise, missing values were not imputed |
|
MRD negativity rate | Cochran-Mantel-Haenszel chi-square test | Stratification factors: IGHV mutation status; presence of del(11q) | Patients with missing data were considered MRD positive |
|
CR rate | Cochran-Mantel-Haenszel chi-square test | Stratification factors: IGHV mutation status; presence of del(11q) | Unless specified otherwise, missing values were not imputed | NR |
ORR | Cochran-Mantel-Haenszel chi-square test | Stratification factors: IGHV mutation status; presence of del(11q) | Patients with missing postrandomization data were considered nonresponders | Based on investigator’s assessment |
DOR | Kaplan-Meier | NR | NR | NR |
OS |
| Stratification factors: IGHV mutation status; presence of del(11q) (unless number of events was < 10% of ITT set) | If the patient was alive or the vital status was unknown, the patient was censored at the date last known to be alive | Censoring patients at the date of death due to COVID-19 |
TTNT |
| NR | Patients without subsequent therapy were censored at the last known date to not have received subsequent therapy | NR |
Improvement in hematologic parameters | Cochran-Mantel-Haenszel chi-square test | NR | Patients without postbaseline assessment were considered as having no improvement | Patients with cytopenia at baseline |
AE | Descriptive statistics only | NA | NR | NR |
TLS risk reduction | Descriptive statistics only | NA | NR | NR |
Time to worsening (EQ-5D-5L) | MMRM analysis | NR | NR | NR |
Time to worsening (EORTC QLQ-C30) | MMRM analysis | NR | NR | NR |
Time to worsening (FACIT) | MMRM analysis | NR | NR | NR |
CAPTIVATE (FD cohort only) | ||||
CR rate | Based on normal approximation to the binomial distribution | NR | NR | Per IRC assessment |
ORR | Based on normal approximation to the binomial distribution | NR | NR | Per IRC assessment |
DOR | Kaplan-Meier | NR | NR | Per IRC assessment |
MRD negativity rate | Based on normal approximation to the binomial distribution | NR | NR |
|
PFS | Kaplan-Meier | NR | NR | NR |
OS | Kaplan-Meier | NR | NR | NR |
AE | Descriptive statistics only | NR | NR | NR |
TLS risk reduction | Difference in proportions from baseline to lead-in | Correlation of paired data | NR | Reduction in proportion of patients with high-risk or medium-risk disease and CrCl < 80 mL/min (supportive) |
Improvement in hematologic parameters | Based on normal approximation to the binomial distribution | NR | NR | NR |
AE = adverse event; BM = bone marrow; CLL = chronic lymphocytic leukemia; CR = complete response; CrCl = creatine clearance; CRi = complete response with incomplete bone marrow recovery; del(11q) = deletion of 11q; DOR = duration of response; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EQ-5D-5L = 5-Level EQ-5D; FACIT = Functional Assessment of Chronic Illness Therapy; FD = fixed duration; IRC = independent review committee; ITT = intention to treat; MMRM = mixed-effects models for repeated measures; MRD = minimal residual disease; NA = not applicable; NR = not reported; ORR = overall response rate; OS = overall survival; PB = peripheral blood; PD = progressed disease; PFS = progression-free survival; TLS = tumour lysis syndrome; TTNT = time to next treatment.
Sources: GLOW primary analysis CSR;23 CAPTIVATE CSR.26 (Note: Details from the table have been taken from the sponsor’s Summary of Clinical Evidence.)
Analysis Populations
A summary of analysis populations in the GLOW and CAPTIVATE trials is provided in Table 11.
The ITT analysis set was used for efficacy analyses in the GLOW study. The ITT population was defined as all patients randomized in the study, and it was analyzed according to assigned treatment group, regardless of the actual treatment received. The ITT population was used for all analyses of primary and secondary efficacy end points and for patient-reported outcomes, analyses of disposition, and demographic and baseline disease characteristics.
The safety population was defined as all randomized patients who receive at least 1 dose of any of the 4 study drugs (ibrutinib, venetoclax, chlorambucil, or obinutuzumab). The safety population is used for all safety analyses and analyses of exposure. All patients were analyzed according to the treatment they were assigned.
In the CAPTIVATE trial, the all-treated population was used in all analyses, including baseline, disposition, efficacy, and safety. The non-del(17p) population was used in the primary analysis of the primary and main secondary end points for the FD cohort.
Table 11: Analysis Populations of the GLOW and CAPTIVATE Studies
Study | Population | Definition | Application |
|---|---|---|---|
GLOW | Efficacy analysis set (ITT population) | All patients randomized to the study and analyzed according to assigned treatment group, regardless of the actual treatment received | Used for all analyses of primary and secondary efficacy end points and PROs, analyses of disposition, and demographic and baseline disease characteristics |
Safety analysis set | All randomized patients who receive at least 1 dose of any 1 of the 4 study drugs (ibrutinib, venetoclax, chlorambucil, or obinutuzumab) | Used for all safety analyses and analyses of exposure; all patients were analyzed according to the treatment they received | |
PFS subgroup analysis set | Subgroup analyses for age, ECOG PS, CIRS, Rai stage, bulky disease, elevated LDH, IGHV, and del(11q) | Hazard ratios of PFS for different baseline variables are reported | |
CAPTIVATE | All-treated population | Includes all enrolled patients who received at least 1 dose of study drug (ibrutinib or venetoclax or both) | All analysis including baseline, disposition, efficacy, and safety analysis will be based on this population |
Non-del(17p) population | Includes patients in the all-treated population without del(17p) abnormality according to nonmissing baseline FISH results | The primary analysis of the primary and main secondary end points for the FD cohort will be based on this population |
CIRS = Cumulative Illness Rating Scale; del(11q) = deletion of 11q; del(17p) = deletion of 17p; ECOG PS = Eastern Cooperativity Oncology Group Performance Status; FD = fixed duration; FISH = fluorescence in situ hybridization; ITT = intention to treat; LDH = lactate dehydrogenase; PFS = progression-free survival; PRO = patient-reported outcome.
Sources: GLOW primary analysis CSR;23 CAPTIVATE CSR.26 (Note: Details from the table have been taken from the sponsor’s Summary of Clinical Evidence.)
Results
Patient Disposition
Patient disposition for the GLOW trial and the CAPTIVATE FD cohort are summarized in Table 12.
GLOW
At the time of data cut-off for the primary analysis (February 26, 2021) in the GLOW trial, more patients in the ibrutinib-venetoclax arm had discontinued study treatment than in the chlorambucil-obinutuzumab arm (22.6% versus 4.8% for ibrutinib-venetoclax versus chlorambucil-obinutuzumab). The most common reason for treatment discontinuation was AE (10.4% versus 1.9%). Other reasons for study treatment discontinuation ibrutinib-venetoclax and chlorambucil-obinutuzumab arms included patient refusal of further study treatment (3.8% versus 1.0%), PD (2.8% versus 1.0%), and physician decision (1.9% versus 1.0%). A number of patients died during the study period (11 [10.4%] patients in the ibrutinib-venetoclax arm and 11 [10.5%] patients in the chlorambucil-obinutuzumab arm). Among them, 4 patients in the ibrutinib-venetoclax arm discontinued study treatment due to death, and the remaining 18 patients either completed the study treatment (2 patients in the ibrutinib-venetoclax arm and 9 patients in the chlorambucil-obinutuzumab arm) or discontinued study treatment due to AEs (4 patients in the ibrutinib-venetoclax arm), physician’s decision (1 patient in the ibrutinib-venetoclax arm), PD (1 patient in the chlorambucil-obinutuzumab arm), or patient refusal (1 patient in the chlorambucil-obinutuzumab arm) before they died. Five (2.4%) patients elected to terminate study participation prematurely (2 [1.9%] patients in the ibrutinib-venetoclax arm and 3 [2.9%] patients in the chlorambucil-obinutuzumab arm).
CAPTIVATE
In the FD cohort (n = 159), 147 patients (92.5%) completed the planned ibrutinib treatment and 149 patients (93.7%) completed the planned venetoclax treatment. The most common reason for treatment discontinuation was AEs (ibrutinib: 4.4%;venetoclax: 1.9%). The most common reason for study discontinuation was withdrawal by patient (3.8%).
Table 12: Summary of Patient Disposition From Pivotal Studies and RCT Evidence Submitted by the Sponsor (ITT Analysis Set for GLOW; All-Treated Analysis Set for CAPTIVATE)
Patient disposition | GLOW | CAPTIVATE | |
|---|---|---|---|
Ibrutinib-venetoclax (n = 106) | Chlorambucil-obinutuzumab (n = 105) | FD cohort (N = 159) | |
Screened, N | 263 | 159 | |
Reason for screening failure, N (%) | Not meeting criteria: 52 (19.8) | NR | |
Randomized, N | 106 | 105 | NA |
Completed study treatment, N (%) | 82 (77.4) | 100 (95.2) | Ibrutinib: 147 (92.5) Venetoclax:149 (93.7) |
Discontinued from study treatment, N (%) | 24 (22.6) | 5 (4.8) | Ibrutinib: 12 (7.5) Venetoclax: 4 (2.5) |
Reason for treatment discontinuation, N (%) | |||
AE | 11 (10.4) | 2 (1.9) | Ibrutinib: 7 (4.4) Venetoclax: 3 (1.9) |
Patient refusal | 4 (3.8) | 1 (1.0) | NR |
Death | 4 (3.8) | 0 | Ibrutinib: 1 (0.6) Venetoclax: 0 |
Progressive disease | 3 (2.8) | 1 (1.0) | Ibrutinib: 1 (0.6) Venetoclax: 1 (0.6) |
Investigator or physician decision | 2 (1.9) | 1 (1.0) | Ibrutinib: 1 (0.6) Venetoclax: 0 |
Consent withdrawal | NR | NR | Ibrutinib: 2 (1.3) Venetoclax: 0 |
Patient treated, N (%) | 106 (100) | 105 (100) | Ibrutinib: 147 (92.5) Venetoclax:149 (93.7) |
Discontinued from study from withdrawal by patient, N (%) | 2 (1.9) | 3 (2.9) | 6 (3.8) |
FAS, N | 106 | 105 | 159 |
Safety, N | 106 | 105 | 159 |
AE = adverse event; FAS = full analysis set; FD = fixed duration; ITT = intention to treat; NA = not applicable; NR = not reported; progressive disease; RCT = randomized controlled trial.
Sources: GLOW primary analysis CSR;23 CAPTIVATE CSR.26 (Note: Details from the table have been taken from the sponsor’s Summary of Clinical Evidence.)
Impact of COVID-19 on Patient Disposition
In the GLOW trial, in the primary analysis (data cut-off: February 26, 2021), no patient discontinued study treatment due to the COVID-19 pandemic. Six (2.8%) patients had not completed the FD treatment phase when the COVID-19 outbreak was declared a pandemic by WHO in March 2020. The sponsor considered the COVID-19 pandemic to have had a limited impact on patient disposition.
In the CAPTIVATE trial, in the primary analysis (data cut-off: November 12, 2020) and with extended follow-up (data cut-off: August 4, 2021), none of the patients in the FD cohort discontinued treatment or exited the study due to COVID-19.
Baseline Characteristics
The GLOW Study
The majority of patients in the trial were men (57.8%) and white (95.7%). Median age at enrolment was 71 years (range, 47 to 93 years), with 87.2% of patients aged 65 years or older and 34.1% of patients aged 75 years or older. Patient characteristics were generally balanced across treatment arms at baseline. However, a difference of more than 10% was observed for CIRS score greater than 6 (69.8% versus 58.1% for ibrutinib-venetoclax versus chlorambucil-obinutuzumab) and LDH elevation (33.0% versus 48.6%). CLL was the initial diagnosis in 93.4% of patients; 6.6% of patients had SLL. Advanced stage disease at baseline was reported for 54.8% of patients based on Rai stage III or IV disease and for 42.1% of patients based on Binet stage C disease. In total, 37.6% of patients had bulky disease based on the presence of a tumour lesion at least 5 cm in longest diameter and 58.3% of patients had cytopenia at baseline. About half the patients had a baseline ECOG PS of 1 (53.1%). Overall, the proportion of patients with high-risk disease, defined as the presence of del(11q), unmutated IGHV, or TP53 mutation, was similar in treatment arms (59.4% versus 57.1%). Although patients with del(17p) or known TP53 mutation at baseline were excluded from the study, patients with unknown TP53 mutation status were allowed to participate. After randomization, central laboratory testing identified 9 (4.3%) patients with a TP53 mutation, 7 (6.6%) in the ibrutinib-venetoclax arm and 2 (1.9%) in the chlorambucil-obinutuzumab arm.
The CAPTIVATE Study
In the FD cohort, the median age at baseline was 60.0 years (range, 33 to 71 years), with 28.3% of patients aged 65 years or older (Table 13). More patients were male (66.7%), and the majority of patients were white (92.5%). At baseline, more patients (69.2%) had an ECOG PS score of 0. Fewer patient had progressed to stage III or IV (27.7%), and 30.2% of patients had bulky disease of at least 5 cm. Cytogenetic characteristics indicative of poor prognosis (per hierarchical classification) in the study population were del(17p) (12.6%) and del 11q (17.6%). Other poor prognostic characteristics included mutated TP53 (10.1%), mutated del(17p) or TP53 (17.0%), unmutated IGHV (56.0%), and complex karyotype (19.5%).
The baseline characteristics outlined in Table 13 are limited to those that are most relevant to this review or were felt to affect outcomes or interpretation of the study results.
Table 13: Summary of Baseline Characteristics of Pivotal Studies and RCT Evidence Submitted by the Sponsor
Characteristic | GLOW | CAPTIVATE | |
|---|---|---|---|
Ibrutinib-venetoclax (n = 106) | Chlorambucil-obinutuzumab (n = 105) | FD cohort (N = 159) | |
Age (years) | |||
Mean (SD) | 71.0 (8.02) | 72.0 (6.16) | 58.0 (8.51) |
Median (range) | 71.0 (47 to 93) | 71.0 (57 to 88) | 60 (33 to 71) |
Gender, n (%) | |||
Male | 59 (55.7) | 63 (60.0) | 106 (66.7) |
Female | 47 (44.3) | 42 (40.0) | 53 (33.3) |
Race or ethnicity, n (%) | |||
Asian | 0 | 1 (1.0) | 3 (1.9) |
Black or African American | NR | NR | 1 (0.6) |
Native Hawaiian or other Pacific Islander | NR | NR | 1 (0.6) |
White | 101 (95.3) | 101 (96.2) | 147 (92.5) |
Multiple | 1 (0.9) | 0 | NR |
Not reported | 4 (3.8) | 3 (2.9) | 7 (4.4) |
ECOG PS score, n (%) | |||
0 | 35 (33.0) | 39 (37.1) | 110 (69.2) |
1 | 58 (54.7) | 54 (51.4) | 49 (30.8) |
2 | 13 (12.3) | 12 (11.4) | 0 (0) |
CIRS total score | |||
≤ 6 | 32 (30.2) | 44 (41.9) | NR |
> 6 | 74 (69.8) | 61 (58.1) | NR |
Histology, n (%) | |||
CLL | 96 (90.6) | 101 (96.2) | 146 (91.8) |
SLL | 10 (9.4) | 4 (3.8) | 13 (8.2) |
CrCl (mL/min), mean (range) | 69.2 (34.0 to 168.1) | 67.3 (32.3 to 180.9) | 95.8 (53 to 210) |
Cytopenia at baseline, n (%) | |||
Yes | 58 (54.7) | 65 (61.9) | 54 (34.0) |
No | 48 (45.3) | 40 (38.1) | 105 (66.0) |
Rai stage, n (%) | CLL only:96 | CLL only:101 | NR |
0 to II | 41 (42.7) | 48 (47.5) | 113 (71.1) |
III to IV | 55 (57.3) | 53 (52.5) | 44 (27.7) |
Missing | NR | NR | 2 (1.3) |
Binet stage, n (%) | CLL only: 96 | CLL only: 101 | NR |
A | 7 (7.3) | 8 (7.9) | NR |
B | 46 (47.9) | 53 (52.5) | NR |
C | 43 (44.8) | 40 (39.6) | NR |
Ann Arbor stage, n (%) | SLL only: 10 | SLL only: 4 | NR |
IV | 10 (100) | 4 (100) | NR |
Bulky disease, n (%) | |||
≥ 5 cm | 41 (39.0) | 38 (36.2) | 48 (30.2) |
≥ 10 cm | 0 (0) | 4 (3.8) | 5 (3.1) |
Elevated LDH, n (%) | 35 (33.0) | 51 (48.6) | NR |
IGHV status, n (%) | |||
Mutated | 27 (25.5) | 27 (25.7) | 66 (41.5) |
Unmutated | 55 (51.9) | 54 (51.4) | 89 (56.0) |
Unknown | 24 (22.6) | 24 (22.9) | 4 (2.5) |
Hierarchical cytogenetics classification, n (%) | |||
Del(17p) | NR | NR | 20 (12.6) |
Del(11q) | 20 (18.9) | 18 (17.1) | 28 (17.6) |
Trisomy 12 | NR | NR | 23 (14.5) |
Normal | NR | NR | 33 (20.8) |
Del(13q) | NR | NR | 54 (34.0) |
Unknown | NR | NR | 1 (0.6) |
TP53 mutation, n (%) | |||
Yes | 7 (6.6) | 2 (1.9) | 16 (10.1) |
No | NR | NR | 142 (89.3) |
Unknown | NR | NR | 1 (0.6) |
Del(17p) or TP53 mutation, n (%) | |||
Yes | NR | NR | 27 (17.0) |
No | NR | NR | 129 (81.1) |
Unknown | NR | NR | 3 (1.9) |
Complex karyotype, n (%) | |||
Yes | NR | NR | 31 (19.5) |
No | NR | NR | 102 (64.2) |
Unknown | NR | NR | 26 (16.4) |
CIRS = Cumulative Illness Rating Scale; CLL = chronic lymphocytic leukemia; CrCl = creatinine clearance; del(11q) = deletion of 11q; del(13q) = deletion of 13q; del(17p) = deletion of 17p; ECOG PS = Eastern Cooperativity Oncology Group Performance Status; FD = fixed duration; LDH = lactate dehydrogenase; NR = not reported; RCT = randomized controlled trial; SD = standard deviation; SLL = small lymphocytic leukemia.
Sources: GLOW primary analysis CSR;23 CAPTIVATE CSR.26 (Note: Details from the table have been taken from the sponsor’s Summary of Clinical Evidence.)
Exposure to Study Treatments
GLOW
A summary of exposure to study treatments is provided in Table 14.
At the time of the primary analysis (data cut-off: February 26, 2021), patients in the ibrutinib-venetoclax arm had a longer median duration of treatment than patients in the chlorambucil-obinutuzumab arm (13.8 months versus 5.1 months for ibrutinib-venetoclax versus chlorambucil-obinutuzumab). Ibrutinib and venetoclax dose reductions due to an AE were reported for 19.8% and 17% of patients, respectively. Dose interruptions (defined as doses missed for ≥ 7 consecutive days) for ibrutinib and venetoclax were noted in 52.8% and 36.8% of patients, respectively. For both study drugs, the majority of the dose interruptions were due to an AE. In the chlorambucil-obinutuzumab arm, the majority of patients received chlorambucil (79%) at the dose intended, per protocol, and without any prescribed dose reductions. Chlorambucil dosing was reduced or delayed because of an AE in 21.0% and 34.3% of patients, respectively. For obinutuzumab, which is administered intravenously, dose reductions were not permitted. Obinutuzumab dosing was skipped or delayed because of an AE in 2.9% and 39% of patients, respectively. Additionally, obinutuzumab infusion was temporarily interrupted, the infusion rate reduced, and/or the infusion aborted in 57.1% of patients due to an AE.
CAPTIVATE
In the FD cohort, the overall median treatment duration was 13.8 months at the primary analysis data cut-off date of November 12, 2020 (range, 0.5 to 24.9 months). The individual median treatment durations for ibrutinib and venetoclax were 13.8 months (range, 0.5 to 24.9 months) and 11.0 months (range, 1.3 to 22.1 months), respectively. One patient had a 2-day dose hold, but there were no other dose modifications.
Table 14: Summary of Patient Exposure From Pivotal Studies and RCT Evidence Submitted by the Sponsor (Safety Analysis Set for GLOW; All-Treated Analysis Set for CAPTIVATE)
Exposure | GLOW | CAPTIVATE | |||||
|---|---|---|---|---|---|---|---|
Ibrutinib-venetoclax (n = 106) | Chlorambucil-obinutuzumab (n = 105) | FD cohort (N = 159) | |||||
Ibrutinib | Venetoclax | Chlorambucil | Obinutuzumab | Ibrutinib | Venetoclax | ||
Treatment | 12 cycles of 28 days (plus 3 cycles of ibrutinib lead-in) | 6 cycles of 28 days | Single-drug oral ibrutinib for 3 cycles followed by 12 cycles of ibrutinib-venetoclax (cycles of 28 days) | ||||
Number of patients per protocol-specified treatment | 106 | 98 | 105 | 105 | NR | NR | |
Duration (months), mean (SD) | 11.89 (3.85) | 10.21 (2.32) | 5.05 (0.91) | 4.68 (0.90) | 13.3 (2.84) | 11.1 (1.22) | |
Duration (months), median (range) | 13.8 (0.7 to 14.7) | 11.0 (0.9 to 11.8) | 5.1 (0.0 to 7.9) | 4.7 (0.0 to 7.5) | 13.8 (0.5 to 24.9) | 11.0 (1.3 to 22.1) | |
Completed study treatment, n (%) | 82 (77.4) | 100 (95.2) | NR | ||||
Dose reduction or interruption,a or dose delay or dose skipped, n (%) | 56 (52.8) | 39 (36.8) | 45 (42.9)b | 3 (2.9) | NR | ||
Dose reduction or dose skipped due to adverse event, n (%) | 21 (19.8) | 18 (17.0) | 22 (21.0) | 3 (2.9) | NR | ||
Dose delay due to adverse event, n (%) | NR | NR | 36 (34.3) | 41 (39.0) | NR | ||
Reason for dose interruptiona or infusion-related action taken, n (%) | |||||||
Adverse event | 47 (44.3) | 31 (29.2) | NR | 60 (57.1) | NR | ||
Other reasons | 8 (7.5) | 4 (3.8) | NR | NR | NR | ||
FD = fixed duration; NR = not reported; SD = standard deviation.
aDose interruption is defined as missing a dose for ≥ 7 consecutive days.
bPatients with any prescribed dose reduction and/or dose delay due to adverse event.
Sources: GLOW primary analysis CSR;23 CAPTIVATE CSR.26 (Note: Details from the table have been taken from the sponsor’s Summary of Clinical Evidence.)
Concomitant Medications and Co-Interventions
A summary of concomitant medications and co-interventions in the GLOW and CAPTIVATE trials is presented in Table 15.
All patients in the primary analysis safety analysis set (data cut-off: February 26, 2021) received concomitant medication in both treatment arms in the GLOW trial. Concomitant medications reported with a frequency at least 15% higher in the ibrutinib-venetoclax arm compared to the chlorambucil-obinutuzumab arm include drugs acting on the renin-angiotensin system (61.3% versus 45.7% for ibrutinib-venetoclax versus chlorambucil-obinutuzumab); antithrombotic drugs (38.7% versus 13.3%); antidiarrheals, intestinal anti-inflammatory, and/or anti-infective drugs (34.9% versus 3.8%); and all other nontherapeutic products (27.4% versus 9.5%). Concomitant medications with a frequency at least 15% higher in the chlorambucil-obinutuzumab arm compared with the ibrutinib-venetoclax arm include analgesics (61.3% versus 100.0%), antihistamines for systemic use (21.7% versus 100.0%), corticosteroids for systemic use (17.0% versus 100.0%), and antiemetics and antinauseants (27.6% versus 9.4%). Similar results were observed in the extended follow-up analysis (data cut-off: August 25, 2022).
In CAPTIVATE FD cohort, in the primary analysis (data cut-off: November 12, 2020), the most common concomitant medications were antigout preparations (96.9%), antibacterials for systemic use (66.7%), and analgesics (54.7%). Similar results were observed in the extended follow-up analysis (data cut-off: August 4, 2021).
Table 15: Summary of Concomitant Treatmenta (Safety Analysis Set GLOW; All-Treated Analysis Set for CAPTIVATE)
Exposure | GLOW (data cut-off: February 26, 2021) | CAPTIVATE (data cut-off: November 12, 2020) | |
|---|---|---|---|
Ibrutinib-venetoclax (n = 106) | Chlorambucil-obinutuzumab (n = 105) | FD cohort (N = 159) | |
Patients who received any concomitant medication | 106 (100.0) | 105 (100.0) | 159 (100.0) |
Frequently reported concomitant medication (≥ 50% of either treatment group), n (%) | |||
Antigout preparations | 102 (96.2) | 88 (83.8) | 154 (96.9) |
Antibacterials for systemic use | 73 (68.9) | 60 (57.1) | 106 (66.7) |
Anticoagulants or antiplatelets | 66 (62.3) | 49 (46.7) | 61 (38.4) |
Analgesics | 65 (61.3) | 105 (100.0) | 87 (54.7) |
Paracetamol | 33 (31.1) | 105 (100.0) | 62 (39.0) |
Drugs acting on the renin-angiotensin system | 65 (61.3) | 48 (45.7) | 43 (27.0) |
Drugs for acid-related disorders | 54 (50.9) | 47 (44.8) | 70 (44.0) |
Antihistamines for systemic use | 23 (21.7) | 105 (100.0) | 29 (18.2) |
Corticosteroids for systemic use | 18 (17.0) | 105 (100.0) | 23 (14.5) |
FD = fixed duration.
aConcomitant treatment was defined as any medication administered while the study treatment was ongoing (i.e., during the treatment phase from first dose to last dose of the study treatment).
Sources: GLOW primary analysis CSR;23 CAPTIVATE CSR.26
Subsequent Treatment
Both the GLOW and CAPTIVATE trials reported on patients who received subsequent treatments. A summary of subsequent treatment is presented in Table 16.
In the GLOW study, at primary analysis (data cut-off: February 26, 2021), fewer patients received anticancer therapy in the ibrutinib-venetoclax arm than in the chlorambucil-obinutuzumab arm (3.8% versus 25.7% for ibrutinib-venetoclax versus chlorambucil-obinutuzumab). The commonly reported subsequent therapies include single-drug ibrutinib (0 versus 5.7% for ibrutinib-venetoclax versus chlorambucil-obinutuzumab), acalabrutinib (0 versus 1.0% for ibrutinib-venetoclax versus chlorambucil-obinutuzumab), and venetoclax (0 versus 1.0% for ibrutinib-venetoclax versus chlorambucil-obinutuzumab). Similar results were observed in the extended follow-up analysis (data cut-off: August 25, 2022).
In the CAPTIVATE FD cohort, at primary analysis (data cut-off: November 12, 2020), 9 (5.7%) patients of the FD cohort received subsequent therapy, 9 (5.7%) and 2 (1.3%) of whom received single-drug ibrutinib and venetoclax, respectively. Similar results were observed in the extended follow-up analysis (data cut-off: August 4, 2021).
Table 16: Summary of Subsequent Treatment From Pivotal Studies and RCT Evidence Submitted by the Sponsor (ITT Analysis Set for GLOW; All-Treated Analysis Set for CAPTIVATE)
Exposure | GLOW (data cut-off: February 26, 2021) | CAPTIVATE (data cut-off: November 12, 2020) | |
|---|---|---|---|
Ibrutinib-venetoclax (n = 106) | Chlorambucil-obinutuzumab (n = 105) | FD cohort (N = 159) | |
Received subsequent therapy, n (%) | 4 (3.8) | 27 (25.7) | 9 (5.7) |
Anticancer therapy,a n (%) | |||
Ibrutinib (single drug) | 0 | 6 (5.7) | 1 (0.6) |
Acalabrutinib | 0 | 1 (1.0) | NR |
Venetoclax | 0 | 1 (1.0) | 2 (1.3) |
FD = fixed duration; ITT = intention to treat; NR = not reported; RCT = randomized controlled trial.
aAny subsequent therapy other than reintroduced treatment, either ibrutinib or ibrutinib-venetoclax.
Sources: GLOW primary analysis CSR;23 CAPTIVATE CSR.26 (Note: Details from the table have been taken from the sponsor’s Summary of Clinical Evidence.)
Protocol Amendments
In the GLOW study, there were a total of 4 protocol amendments reported. Of these 4, the following was particularly of note and impactful. Protocol amendment 1, made June 6, 2018, removed eligibility for patients with galactose intolerance, Lapp lactase deficiency, or glucose-galactose malabsorption from GLOW study enrolment.
There were a total of 3 protocol amendments reported for the CATIVATE trial. No impactful amendment was identified by the CADTH review team.
Protocol Deviation
Both the GLOW and CAPTIVATE trials reported on patients who had major or important protocol deviations (Table 17).
In the GLOW study, at the time of primary analysis (data cut-off: February 26, 2021), a similar proportion of patients reported major protocol deviations in the ibrutinib-venetoclax and chlorambucil-obinutuzumab arms (5.7% versus 5.7% for ibrutinib-venetoclax versus chlorambucil-obinutuzumab). Five (4.7%) patients in the ibrutinib-venetoclax arm and 3 (2.9%) patients in the chlorambucil-obinutuzumab arm received the wrong treatment or an incorrect dose, including 1 patient in the ibrutinib-venetoclax arm who received expired venetoclax and 3 patients in the chlorambucil-obinutuzumab arm who received more than 175% of the prescribed chlorambucil daily dose. One (0.9%) patient in the ibrutinib-venetoclax arm and 3 (2.9%) patients in the chlorambucil-obinutuzumab arm missed a disease evaluation visit due to the COVID-19 pandemic; these were considered to be major protocol deviations because they potentially delayed the detection of PD. One (0.9%) patient in the ibrutinib-venetoclax arm did not meet an inclusion criterion (adequate organ function: absolute neutrophil count ≥ 750 cells/μL) for study eligibility.
In the CAPTIVATE trial, at the time of primary analysis (data cut-off: November 12, 2020), 2 patients had important protocol deviations related to not meeting all eligibility criteria, 1 patient was enrolled despite the need to be treated with 20 mg of prednisone during the screening period to control autoimmune hemolytic anemia, and 1 patient did not have an activated partial thromboplastin time coagulation test performed at screening. One patient refused to have CT scans performed at multiple time points, which was considered to have an impact on efficacy. On 3 separate occasions, 1 patient experienced grade 3 to 4 neutropenia related to venetoclax treatment, and the dose of venetoclax was not reduced or withheld per protocol requirements which was considered relating to safety.
Table 17: Summary of Patients With Major Protocol Deviations (ITT Analysis Set for GLOW; All-Treated Analysis Set for CAPTIVATE)
Protocol deviation | GLOW (data cut-off: February 26, 2021) | CAPTIVATE (data cut-off: November 12, 2020) | |
|---|---|---|---|
Ibrutinib-venetoclax (n = 106) | Chlorambucil-obinutuzumab (n = 105) | FD cohort (N = 159) | |
Patients with major protocol deviations | 6 (5.7) | 6 (5.7) | NR |
Received wrong treatment or incorrect dose | 5 (4.7) | 3 (2.9) | NR |
Other | 1 (0.9) | 3 (2.9) | NR |
COVID-19 related | 1 (0.9) | 3 (2.9) | NR |
Eligibility criteria not met | 1 (0.9) | 0 | 2 (1.3) |
Efficacy | NR | NR | 1 (0.6) |
Safety | NR | NR | 1 (0.6) |
FD = fixed duration; ITT = intention to treat; NR = not reported.
Sources: GLOW primary analysis CSR;23 CAPTIVATE CSR.26
Treatment Compliance
In the GLOW study, at the time of the primary analysis (data cut-off: February 26, 2021), for all 4 study drugs, around 95% of patients received the investigator-prescribed dose (96.2%, 96.9%, 94.3%, and 100% for ibrutinib, venetoclax, obinutuzumab, and chlorambucil, respectively). Three patients in the chlorambucil-obinutuzumab arm received a dose of chlorambucil that was more than 175% of the protocol-specified dose. These account for the chlorambucil mean compliance rate greater than 100%. All 3 overdose cases were reported as major protocol deviations.
In the CAPTIVATE trial, at the time of primary analysis (data cut-off: November 12, 2020), 147 patients (92.5%) completed the planned ibrutinib treatment and 149 patients (93.7%) completed the planned venetoclax treatment.
Efficacy
Unless otherwise specified, the key efficacy results of the GLOW and CAPTIVATE trials are summarized in Table 18. Refer to Appendix 1 for the hierarchical testing order of secondary end points for the GLOW trial and detailed efficacy data for the GLOW trial and CAPTIVATE FD cohort.
Progression-Free Survival
In the GLOW study, PFS per IRC was the primary end point. In the primary analysis (data cut-off: February 26, 2021), there were 89 IRC-assessed PFS events, 22 patients (20.8%) in the ibrutinib-venetoclax arm and 67 patients (63.8%) in the chlorambucil-obinutuzumab arm had experienced a PFS event. Of these, 13 (12.3%) had disease progression and 9 (8.5%) died in the ibrutinib-venetoclax arm, 65 (61.9%) had disease progression and 2 (1.9%) died in the chlorambucil-obinutuzumab arm. The median time on study was 27.7 months (95% CI, 27.50 to 27.83 months) in the ibrutinib-venetoclax arm and 27.89 months (95% CI, 27.53 to 28.58 months) in the chlorambucil-obinutuzumab arm. The primary analysis showed an improvement in PFS assessed by IRC for patients receiving FD ibrutinib-venetoclax compared with chlorambucil-obinutuzumab (HR = 0.216; 95% CI, 0.131 to 0.357; P < 0.0001). This represents a 78% reduction in the risk of PD or death for patients treated with ibrutinib-venetoclax compared with chlorambucil-obinutuzumab. Median PFS was not reached in the ibrutinib-venetoclax arm and was 21.0 months in the chlorambucil-obinutuzumab arm. The probability of not experiencing a PFS event by 24 months was 84.4% (95% CI, 75.8% to 90.1%) in the ibrutinib-venetoclax arm and 44.1% (95% CI, 34.2% to 53.6%) in the chlorambucil-obinutuzumab arm in the primary analysis. The Kaplan-Meier plot of PFS per IRC in the primary analysis is depicted in Figure 3.
Subgroup analyses of PFS per IRC in the primary analysis were general consistent with the primary analysis across all prespecified subgroups, except for race and disease diagnosis at baseline. Refer to Appendix 1 for detailed subgroup analyses data.
In addition, several prespecified sensitivity analyses based on the IRC assessment of PFS were included in the statistical analysis plan, including unstratified analysis, censoring patients who initiated subsequent anticancer therapy before PD, censoring patients at the last disease evaluation if they progressed or died after missing at least 2 consecutive planned disease evaluation visits, and using PFS per IA. The results were generally consistent with the results of the primary analysis and showed HR values ranging from 0.207 (95% CI, 0.120 to 0.357) to 0.233 (95% CI, 0.143 to 0.379). The overall concordance between IA and IRC assessments of PD and non-PD events was robust in both arms (93.4% versus 81.0% for ibrutinib-venetoclax versus chlorambucil-obinutuzumab). Refer to Appendix 1 for the Kaplan-Meier plot of PFS per IA. A planned supplementary PFS analysis censoring patients who died pre-PD due to COVID-19 was not conducted because only 1 pre-PD death due to COVID-19 was reported and the threshold specified in the statistical analysis plan (i.e., the number of pre-PD deaths > 5% of the total PFS events) was not met.
With a median time on study for all patients of 46.1 months (95% CI, 45.86 to 46.26 months) at the time of the extended follow-up (data cut-off: August 25, 2022), PFS analysis by IRC continued to show an improvement with FD ibrutinib-venetoclax compared to chlorambucil-obinutuzumab (HR = 0.214; 95% CI, 0.138 to 0.334; nominal P < 0.0001). This improvement represents a 79% reduction in the risk of PD or death with ibrutinib-venetoclax treatment compared with chlorambucil-obinutuzumab treatment. The Kaplan-Meier plot of PFS per IRC in the extended follow-up analysis is depicted in Figure 4.
In the CAPTIVATE FD cohort, in the primary analysis (data cut-off: November 12, 2020), median PFS by IRC assessment was not reached in the all-treated FD cohort based on an overall median follow-up of 27.9 months. PFS event probabilities in the all-treated FD cohort were 14.5% (21 disease progression events and 2 deaths) at the time of primary analysis and 25.2% (38 disease progression events and 2 deaths) in the follow-up analysis (data cut-off: August 4, 2021). The probability of not experiencing a PFS event based on IRC assessment was 88.9% (95% CI, 82.7% to 92.9%) at 24 months in the primary analysis and 85.5% (95% CI, 78.8% to 90.2%) at 36 months in the extended follow-up analysis for all treated patients. Among patients with del(17p), 5 (25.0%) reported a PFS event, the median PFS by IRC was not reached, and the PFS event-free probability was 75.0% (95% CI, 50.0% to 88.7%) at 24 months in the primary analysis and 80.0% (95% CI, 55.1% to 92.0%) at 36 months in the extended follow-up analysis. The Kaplan-Meier plot of PFS per IRC in the primary analysis is depicted in Figure 5; refer to Appendix 1 for the Kaplan-Meier plot of PFS per IRC in the extended follow-up analysis.
Generally, similar results were observed for PFS per IA, and median PFS by IA was not reached in the all-treated FD cohort. PFS event probabilities were 12.6% (18 disease progression events and 2 deaths) at the time of the primary analysis (data cut-off: November 12, 2020) and 17.6% (26 disease progression events and 2 deaths) in the extended follow-up analysis (data cut-off: August 4, 2021) for all treated patients. The probability of not experiencing a PFS event based on IA were 94.8% (95% CI, 89.8% to 97.3%) at 24 months in the primary analysis and 88.1% (95% CI, 81.7% to 92.3%) at 36 months in the extended follow-up analysis. In patients with del(17p), 4 (20.0%) reported a PFS event, median PFS by IA was not reached (the lower end of the 95% CI was 28.5 months), PFS event-free rates were 84.2% (95% CI, 58.7% to 94.6%) at 24 months in the primary analysis and 78.6% (95% CI, 52.5% to 91.4%) at 36 months in the extended follow-up analysis. The Kaplan-Meier plot of PFS per IA in the primary analysis was depicted in Figure 6; refer to Appendix 1 for the Kaplan-Meier plot of PFS per IA in the extended follow-up analysis.
Figure 3: Kaplan-Meier Plot of PFS per IRC at Primary Analysis for GLOW (ITT Analysis Set)
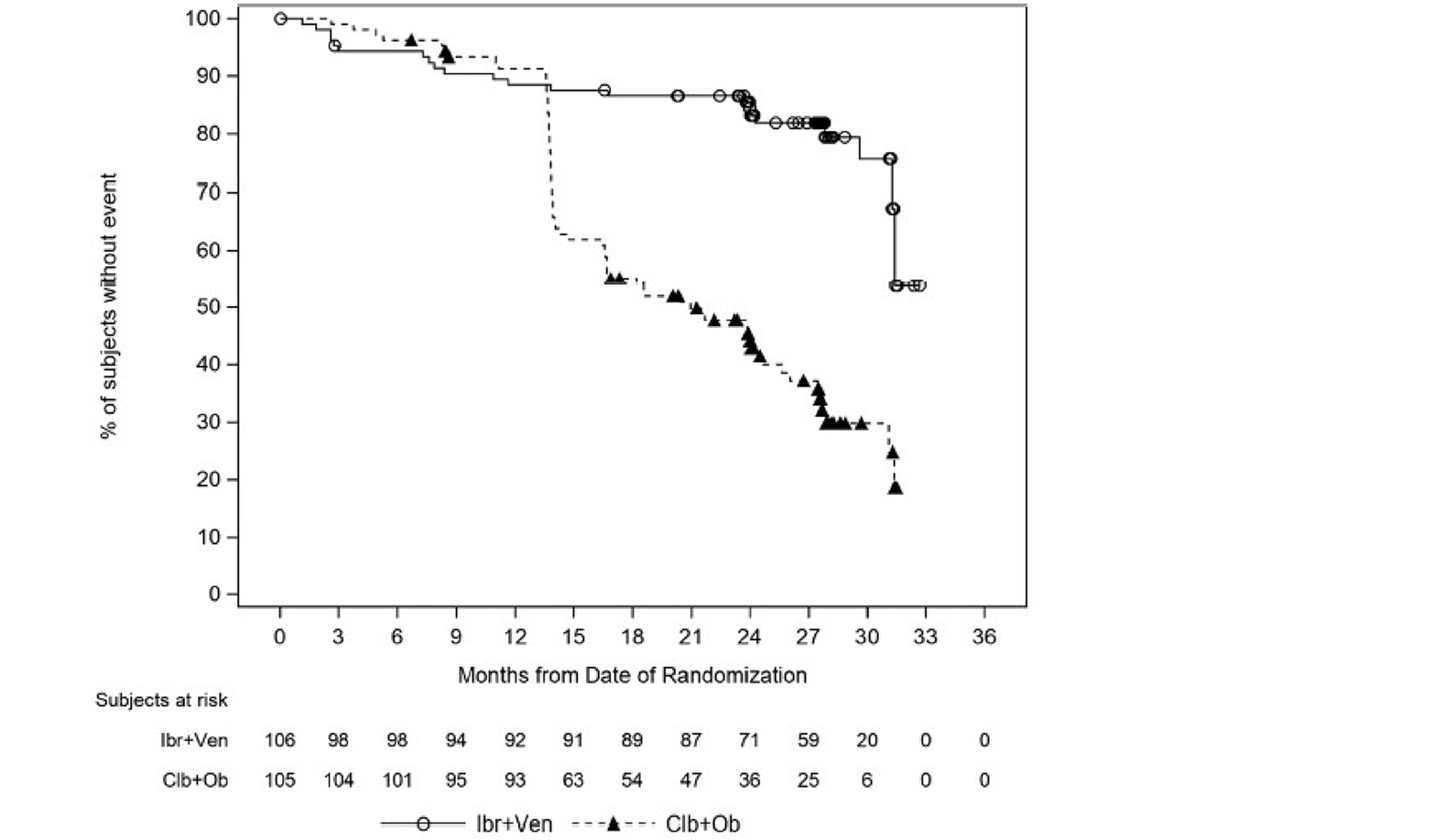
Clb+Ob = obinutuzumab plus chlorambucil; Ibr+Ven = ibrutinib plus venetoclax; IRC = independent review committee; ITT = intention to treat; PFS = progression-free survival.
Note: Data cut-off was February 26, 2021.
Source: GLOW Primary Analysis CSR.23
Figure 4: Kaplan-Meier Plot of PFS per IRC at Extended Follow-Up Analysis for GLOW (ITT Analysis Set)
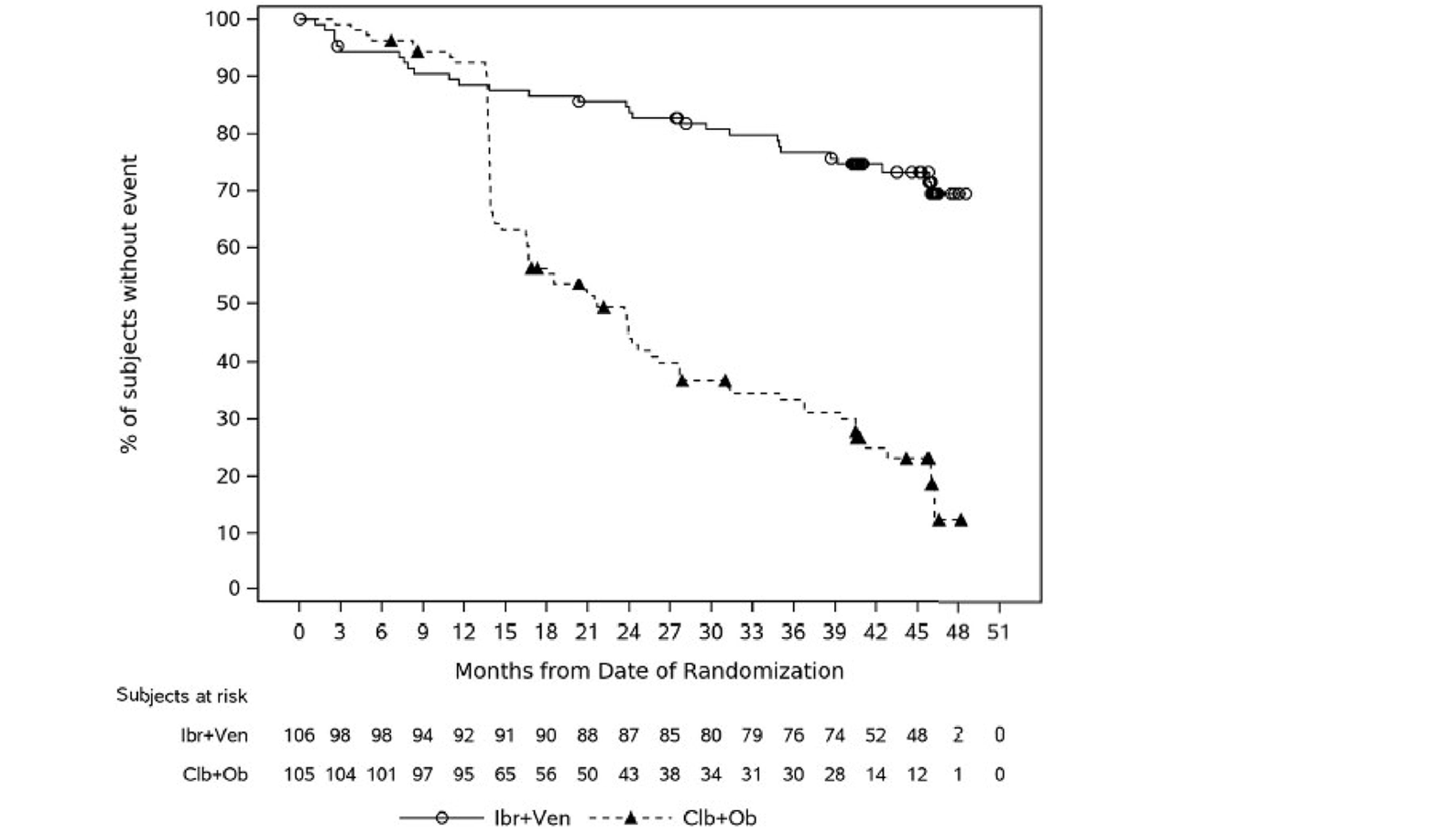
Clb+Ob = obinutuzumab plus chlorambucil; Ibr+Ven = ibrutinib plus venetoclax; IRC = independent review committee; ITT = intention to treat; PFS = progression-free survival.
Note: Data cut-off was August 25, 2022.
Source: GLOW extended follow-up analysis CSR.24
Figure 5: Kaplan-Meier Curves for PFS per IRC Assessment at Primary Analysis for CAPTIVATE (All-Treated Analysis Set)
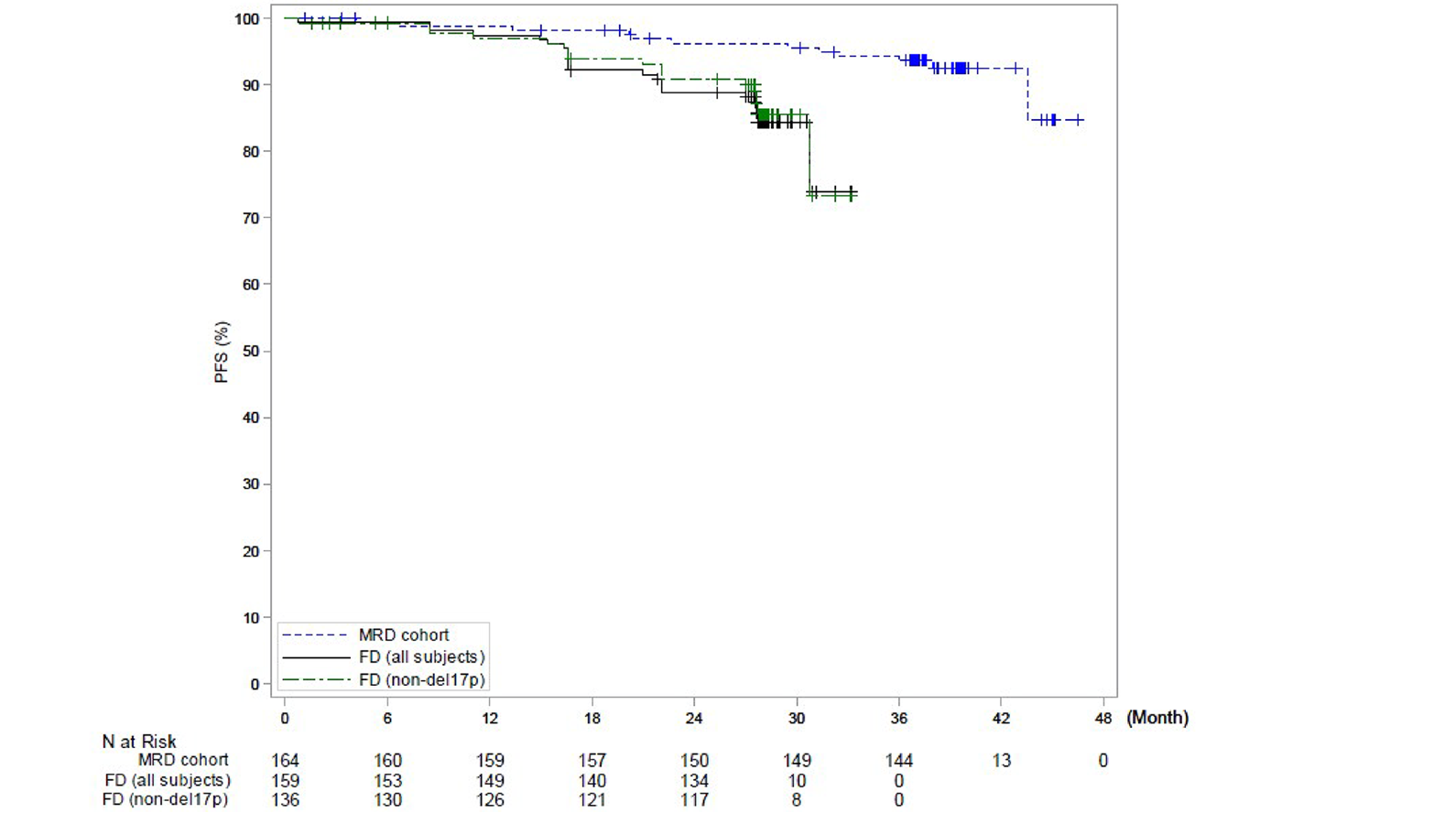
Del17P = deletion of 17p; FD = fixed duration; IRC = independent review committee; MRD = minimal residual disease; PFS = progression-free survival.
Note: Data cut-off was November 12, 2020.
Source: CAPTIVATE primary analysis CSR.
Figure 6: Kaplan-Meier Curves for PFS per IA at Primary Analysis for CAPTIVATE (All-Treated Analysis Set)
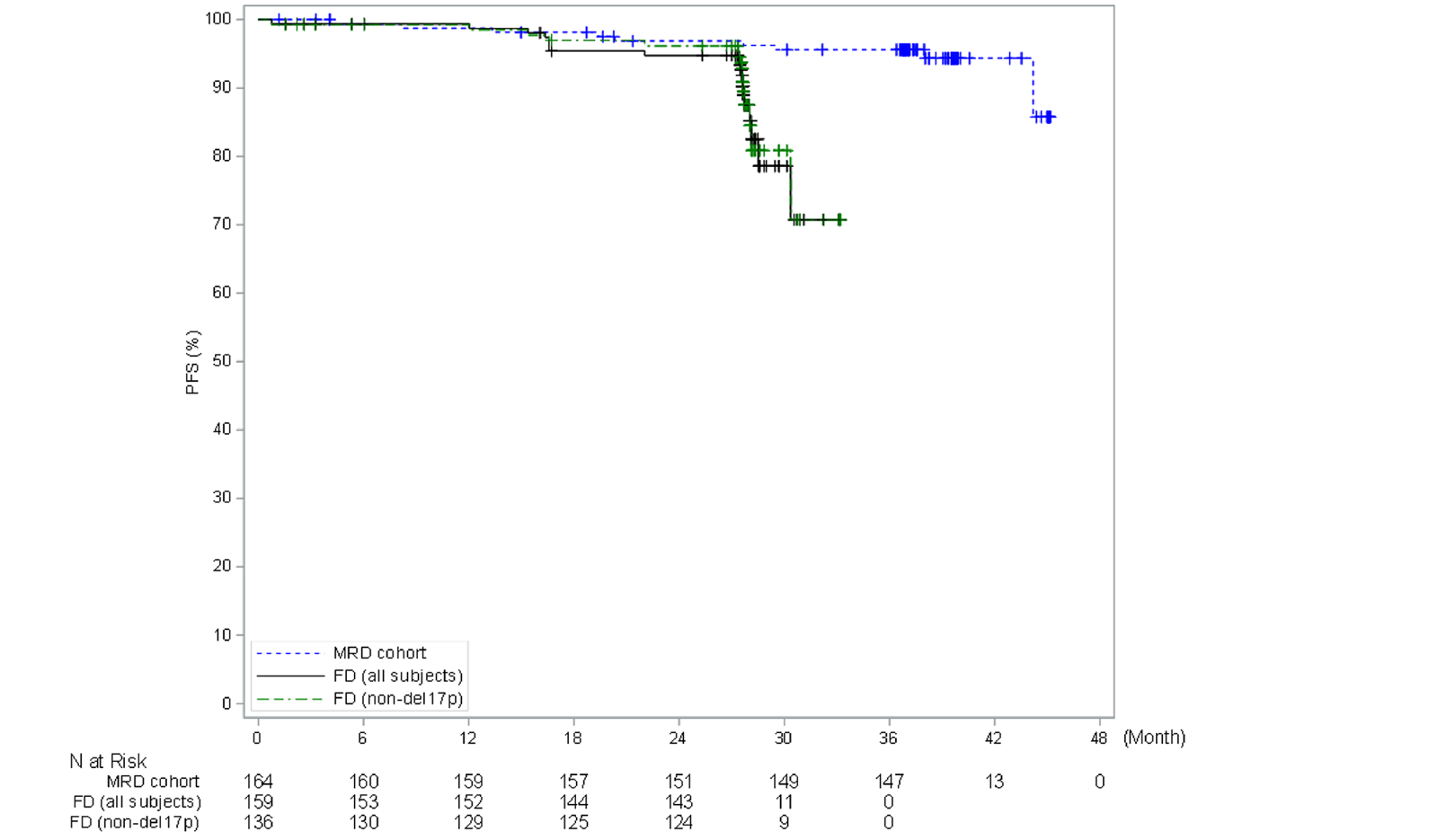
Del17P = deletion of 17p; FD = fixed duration; IA = investigator assessment; MRD = minimal residual disease; PFS = progression-free survival.
Note: Data cut-off was November 12, 2020.
Source: CAPTIVATE primary analysis CSR
Overall Survival
In the GLOW study, at the time of primary analysis (data cut-off: February 26, 2021), median OS was not reached in either arm. With a median follow-up of 27.7 months for the ibrutinib-venetoclax arm and 27.89 months for the chlorambucil-obinutuzumab arm, there were 11 (10.4%) deaths observed in the ibrutinib-venetoclax arm and 12 (11.4%) deaths observed in the chlorambucil-obinutuzumab arm (HR = 1.048; 95% CI, 0.454 to 2.419; nominal P = 0.9121). OS probabilities decreased from 91.4% at 12 months to 90.4% at 24 months in the ibrutinib-venetoclax arm and from 98.1% to 91.3%, respectively, in the chlorambucil-obinutuzumab arm. A supplementary OS analysis that censored patients who died due to COVID-19 showed results consistent with the primary OS analysis, with a HR of 1.298 (95% CI, 0.512 to 3.290). In the extended follow-up analysis (data cut-off: August 25, 2022), similar to the primary analysis, median OS was not reached in either treatment arm. With a median follow-up of 46.1 months, 15 (14.2%) death events were observed in the ibrutinib-venetoclax arm and 30 (28.6%) were observed in the chlorambucil-obinutuzumab arm. This corresponds to an HR of 0.487 (95% CI, 0.262 to 0.907; nominal P = 0.0205). OS survival probabilities at 42 months were 87.5% and 77.6% in the ibrutinib-venetoclax and chlorambucil-obinutuzumab arms, respectively. Results of a supplementary OS analysis that censored patients who died due to COVID-19 were consistent with the primary OS analysis, with an HR of 0.527 (95% CI, 0.268 to 1.035). The Kaplan-Meier plot of OS in the primary analysis is depicted in Figure 7.
In the all-treated CAPTIVATE FD cohort, at the time of primary analysis (data cut-off: November 12, 2020), median OS was not reached. Based on an overall median follow-up of 27.9 months, there were 3 deaths (1.9%) reported in the all-treated FD cohort, 3 deaths (2.2%) reported in patients without del(17p), and no deaths reported in patients with del(17p). Most patients were alive and on study, with OS probabilities of 98.1% at 24 months at the primary data cut-off date and 98.1% at 36 months at the extended follow-up analysis data cut-off date (August 4, 2021) for the all-treated FD cohort. All patients with del(17p) were alive and on study at the primary data cut-off date and at the extended follow-up analysis data cut-off date. The Kaplan-Meier plot of OS in the primary analysis is depicted in Figure 8.
Figure 7: Kaplan-Meier Plot of OS at Primary Analysis for GLOW (ITT Analysis Set)
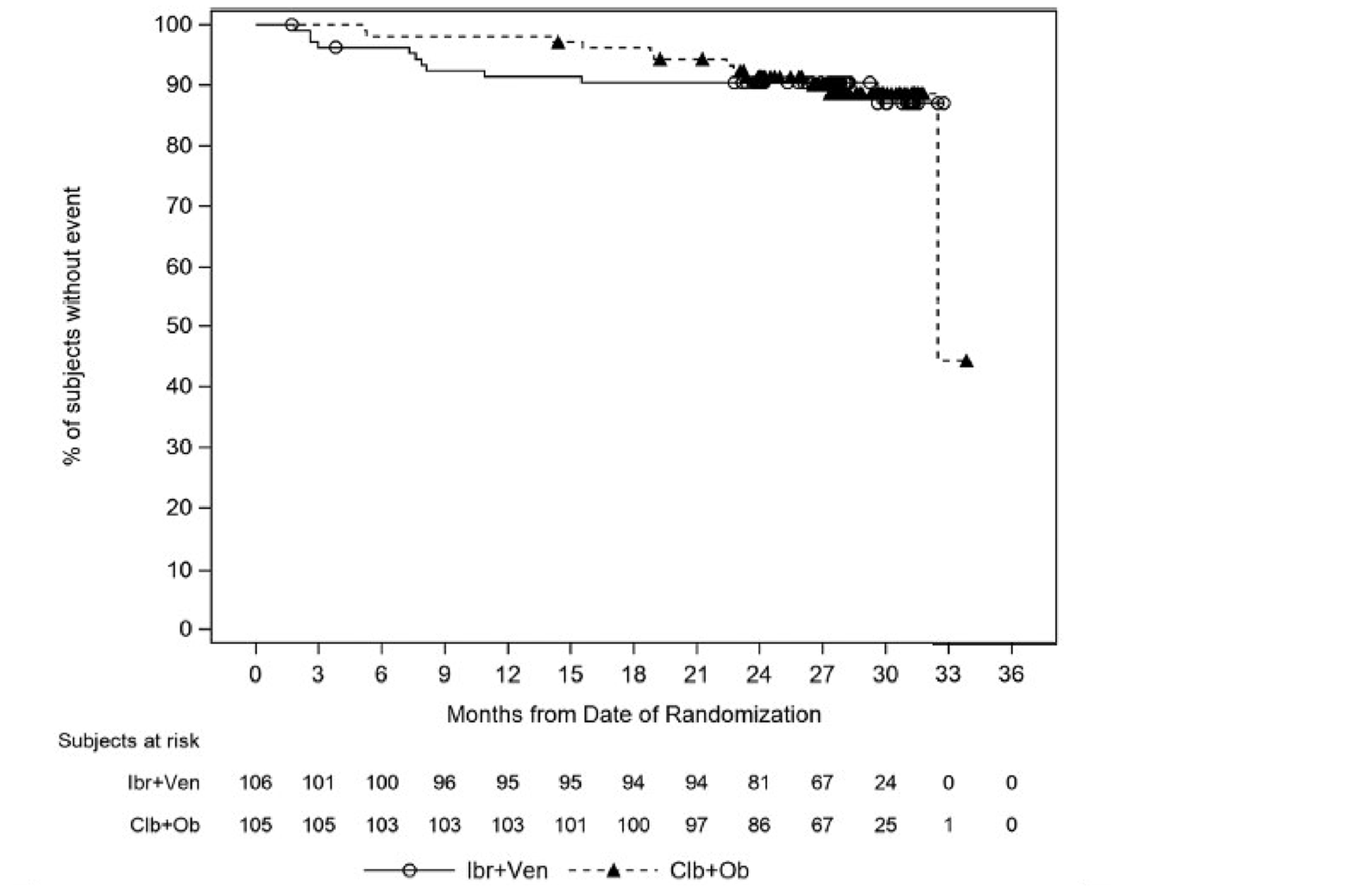
Clb+Ob = obinutuzumab plus chlorambucil; Ibr+Ven = ibrutinib plus venetoclax; ITT = intention to treat; OS = overall survival.
Note: Data cut-off was February 26, 2021.
Source: GLOW primary analysis CSR.23
Figure 8: Kaplan-Meier Curves for OS at Primary Analysis for CAPTIVATE (All-Treated Analysis Set)
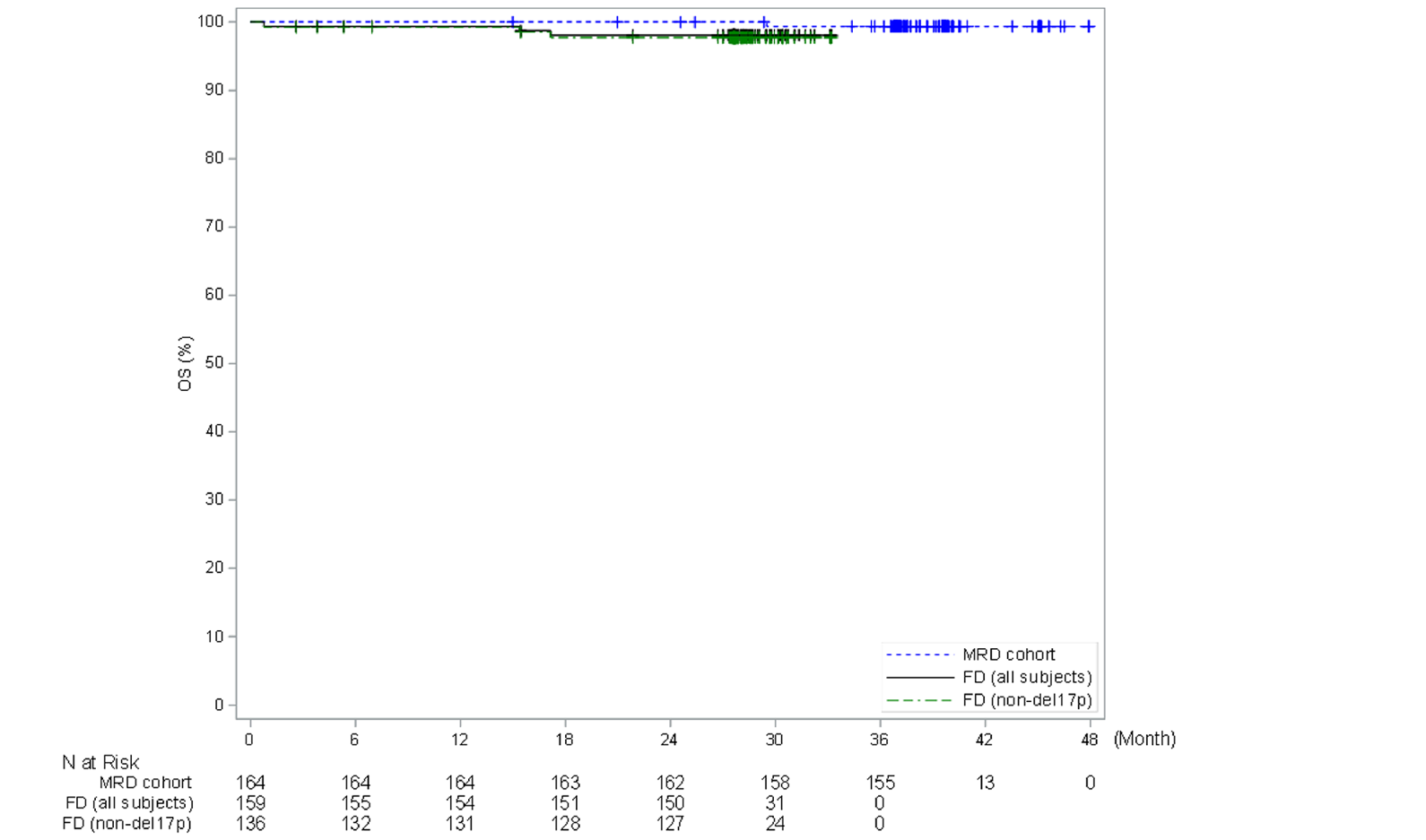
Del17P = deletion of 17p; FD = fixed duration; MRD = minimal residual disease; OS = overall survival.
Note: Data cut-off was November 12, 2020.
Source: CAPTIVATE primary analysis CSR.
Overall Response Rate
In the GLOW primary analysis (data cut-off: February 26, 2021), the IRC-assessed ORR (of PR or better) was similar in the ibrutinib-venetoclax and chlorambucil-obinutuzumab treatment arms (86.6% versus 84.8% for ibrutinib-venetoclax versus chlorambucil-obinutuzumab). The majority of patients had PR (44.3% versus 70.5), followed by CR (35.8% versus 11.4%), nPR (3.8% versus 2.9%), and CRi (2.8% versus 0%). In this analysis, the relative response of ORR for ibrutinib-venetoclax compared to chlorambucil-obinutuzumab was 1.02 (95% CI, 0.92 to 1.14; P = 0.6991). Similar results were observed in the ORR based on IA (92.5% versus 87.6%; relative response = 1.05; 95% CI, 0.96 to 1.15; nominal P = 0.2585). Generally, similar results were observed in the extended follow-up analysis (data cut-off: August 25, 2022).
Of note, as the difference in ORR based on IRC assessment between treatment arms was not statistically significant (P = 0.6991) in the primary analysis, the hierarchical statistical testing strategy ended at ORR per IRC. The remaining key secondary end points (i.e., OS, sustained hematological improvements, and time to improvement in the FACIT-Fatigue score) and ORR per IA were considered not statistically significant. Refer to Appendix 1 for the hierarchical testing order of secondary end points for the GLOW study.
In the CAPTIVATE FD cohort, in the primary analysis (data cut-off: November 12, 2020), the ORR per IRC assessment was 96.2% (95% CI, 93.3% to 99.2%) for all treated patients, 95.6% (95% CI, 92.1% to 99.0%) for patients without del(17p), and 100.0% (95% CI, NE) for patients with del(17p). The majority of all treated patients had CR (57.9%), followed by PR (35.2%), CRi (1.9%), and nPR (1.3%). Similar results were observed for patients with and without del(17p) and for ORR based on IA. Similar results were observed in the extended follow-up analysis (data cut-off: August 4, 2021).
CR (CR and CRi) Rate
In the GLOW study, in the primary analysis (data cut-off: February 26, 2021), the IRC-assessed CR rate was higher in the ibrutinib-venetoclax arm than in the chlorambucil-obinutuzumab arm (38.7% versus 11.4% for ibrutinib-venetoclax versus chlorambucil-obinutuzumab; relative response = 3.43; 95% CI, 1.91 to 6.15; P < 0.0001). Similar results were observed for the investigator-assessed CR probability in the primary analysis (45.3% versus 13.3%; relative response = 3.42; 95% CI, 2.01 to 5.82; nominal P < 0.0001). Generally, similar results were observed in the extended follow-up analysis (data cut-off: August 25, 2022).
In the CAPTIVATE FD cohort, investigator-assessed CR probability was assessed as the primary end point. In patients without del(17p) in the FD cohort, CR rate per IA was met, at 55.9% (95% CI, 47.5% to 64.2%), which exceeded the prespecified minimum CR probability of 37% (1-sided P < 0.0001) and the historical FCR comparator probability (40%) in the primary analysis (data cut-off: November 12, 2020). The investigator-assessed CR rate was 55.3% (95% CI, 47.6% to 63.1%) in all treated patients and 50.0% (95% CI, 28.1% to 71.9%) in patients with del(17p) in the primary analysis. Generally, similar results were observed in the extended follow-up analysis (data cut-off: August 4, 2021).
Subgroup analyses in the primary analysis of the CR probability based on IA for all patients in the FD cohort were, in general, consistent with the primary analysis across all prespecified subgroups except for bulky disease. Refer to Appendix 1 for detailed subgroup analyses data.
In sensitivity analysis, IRC-assessed results were similar to the results based on IA. The concordance of CR and CRi per investigator and IRC assessments was 78.0% at the primary analysis, and no appreciable change in concordance was identified with extended follow-up (76.1%). In the supportive analysis of durable CR in the primary analysis, around 50% of patients had a durable CR — 49.1% (95% CI, 41.3% to 56.8%) for all treated patients and 48.5% (95% CI, 40.1% to 56.9%) for patients without del(17p) — and similar results were seen in the extended follow-up analysis. The analysis of duration of CR in patients who achieved a CR was performed as a supportive analysis in the primary analysis, with a median follow-up of 27.9 months. The median duration of CR was not reached for all patients or for patients without del(17p), and similar results were observed in the extended follow-up analysis.
Sustained Hematologic Improvement
In the GLOW study, at the time of primary analysis (data cut-off: February 26, 2021), the proportion of patients with sustained improvement in hemoglobin was similar the ibrutinib-venetoclax arm compared with the chlorambucil-obinutuzumab arm (44.3% and 50.5%, respectively; nominal P = 0.3854). The proportion of patients with sustained improvement in platelets was also similar in the ibrutinib-venetoclax and chlorambucil-obinutuzumab arms (24.5% and 29.5%, respectively; nominal P = 0.4346). In the extended follow analysis (data cut-off: August 25, 2022), the proportions of patients with sustained improvement in hemoglobin and platelet counts in the ibrutinib-venetoclax and chlorambucil-obinutuzumab arms were similar to those in the primary analysis (hemoglobin: 52.8% versus 51.4% for ibrutinib-venetoclax versus chlorambucil-obinutuzumab; platelet count: 32.1% versus 32.4%).
In the CAPTIVATE FD cohort, in the primary analysis (data cut-off: November 12, 2020), the proportion of patients achieving a sustained improvement in hemoglobin was 41.5% (95% CI, 33.9% to 49.2%) in all treated patients and 60.0% (95% CI, 38.5% to 81.5%) in patients with del(17p). The proportion of patients with sustained improvement in platelets was 17.6% (95% CI, 11.7% to 23.5%) in all treated patients and 15.0% (95% CI, 0 to 30.6%) in patients with del(17p). Generally, similar results were observed in the extended follow-up analysis (data cut-off: August 4, 2021).
Duration of Response
In the GLOW study, as of the data cut-off for the primary analysis (February 26, 2021), with an overall median follow-up of 27.7 months, the median DOR for patients who achieved an IRC-assessed PR or better was 28.9 months (95% CI, 28.7 months to NE) in the ibrutinib-venetoclax arm and 21.1 months (95% CI,15.9 to 25.1 months) in the chlorambucil-obinutuzumab arm; based on investigator-assessed responses, median DOR was not reached in the ibrutinib-venetoclax arm and was 21.5 months (95% CI, 15.7 to 28.5 months) in the chlorambucil-obinutuzumab arm. In the extended follow-up analysis (data cut-off: August 25, 2022), with an overall median follow-up of 46.1 months, median DOR for patients who achieved an IRC-assessed PR or better was not reached in the ibrutinib-venetoclax arm and was 21.2 months (95% CI, 17.7, 28.6 months) in the chlorambucil-obinutuzumab arm; similarly, median DOR per IA was not reached in the ibrutinib-venetoclax arm and was 21.6 months (95% CI, 15.7 to 29.1 months) in the chlorambucil-obinutuzumab arm.
In the CAPTIVATE FD cohort, in the primary analysis (data cut-off: November 12, 2020), with a median follow-up of 27.9 months, the median DOR per IRC assessment in the FD cohort was not reached for all patients, patients without del(17p) (with the lower end of the 95% CI of 27.6 months), and patients with del(17p) (with the lower end of the 95% CI of 18.9 months). Similar results were observed for investigator-assessed DOR at the same analysis. In the extended follow-up analysis (data cut-off: August 4, 2021), with a median follow-up of 38.7 months, the median DOR per IRC was 36.4 months for all treated patients, not reached for patients without del(17p) (with the lower end of the 95% CI of 36.3 months), and 35.9 months (95% CI, 35.6 months to NE) for patients with del(17p). Of note, the median DOR for patients with del(17p) in the primary analysis was considered not reliable due to the limited number (n = 2) of patients at risk at 36 months after initial response. At the same analysis, similar results were observed for investigator-assessed DOR.
Time to Next Treatment
At the GLOW primary analysis (data cut-off: February 26, 2021), the proportion of patients who received subsequent anticancer therapy was lower in the ibrutinib-venetoclax arm (3.8%) compared with the chlorambucil-obinutuzumab arm (25.7%). The median time to treatment failure was not reached in either arm. The HR of subsequent anticancer therapy comparing the ibrutinib-venetoclax arm to the chlorambucil-obinutuzumab arm was 0.143 (95% CI, 0.050 to 0.410; nominal P < 0.0001). Similar results were observed in the extended follow-up analysis (data cut-off: August 25, 2022).
TTNT was not reported in the CAPTIVATE FD cohort.
MRD Negativity
In the GLOW trial, a higher proportion of patients reported negative overall MRD by NGS in the ibrutinib-venetoclax arm than in the chlorambucil-obinutuzumab arm in BM (55.7% versus 21.0% for ibrutinib-venetoclax versus chlorambucil-obinutuzumab; relative response = 2.65; 95% CI, 1.75 to 3.99; P < 0.0001) and in PB (59.4% versus 40.0% for ibrutinib-venetoclax versus chlorambucil-obinutuzumab; nominal P = 0.0055) in the primary analysis (data cut-off: February 26, 2021). The 12-month MRD negativity rate in PB was 49.1% in the ibrutinib-venetoclax arm and 12.4% in the chlorambucil-obinutuzumab arm. MRD negativity rate by NGS was not assessed in the extended follow-up analysis (data cut-off: August 25, 2022).
In the all-treated FD cohort of the CAPTIVATE trial, overall MRD negativity rates by flow cytometry were as follows: 59.7% (95% CI, 52.1% to 67.4%) for all patients, 61.8% (95% CI, 53.6% to 69.9%) for patients without del(17p), and 45.0% (95% CI, 23.2% to 66.8%) for patients with del(17p) in BM; and 76.7% (95% CI, 70.2% to 83.3%) for all patients, 76.5% (95% CI, 69.3% to 83.6%) for patients without del(17p), and 80.0% (95% CI, 62.5% to 97.5%) for patients with del(17p) in PB. Identical results were reported for overall MRD negativity rates in the extended follow-up analysis (data cut-off: August 4, 2021).
TLS Risk Reduction
In the ibrutinib-venetoclax arm of the GLOW study, 26 (24.5%) patients had a high TLS risk by tumour burden at baseline. After ibrutinib lead-in, 22 (20.8%) patients shifted to medium and/or low risk in the primary analysis (data cut-off: February 26, 2021). TLS risk reduction was not assessed in the extended follow-up analysis (data cut-off: August 25, 2022).
In the FD cohort of the CAPTIVATE study, a high tumour burden was observed for 34 (21.4%) all treated patients; among them, 1 (5%) patient had del(17p) at baseline. After 3 cycles of single-drug ibrutinib lead-in therapy, 33 (20.8%) patients shifted to medium or low risk in the primary analysis (data cut-off: November 12, 2020); among them, 1 (5%) patient had del(17p). TLS risk reduction was not assessed in the extended follow-up analysis (data cut-off: August 4, 2021).
EORTC QLQ-C30 Global Health Status
In the primary analysis of the GLOW trial (data cut-off: February 26, 2021), 47 (44.3%) patients in the ibrutinib-venetoclax arm and 43 (41.0%) patients in the chlorambucil-obinutuzumab arm reported a meaningful deterioration (decreased ≥ 10 points) in the EORTC QLQ-C30 global health status score. The median time to first meaningful deterioration was 14.95 months (95% CI, 8.38 months to NE) in the ibrutinib-venetoclax group and 24.18 months (95% CI, 13.86 months to NE) in the chlorambucil-obinutuzumab group, with an HR of 1.149 (95% CI, 0.756 to1.746; nominal P = 0.5130). At the same analysis, 65 (61.3%) patients in the ibrutinib-venetoclax arm and 58 (55.2%) patients in the chlorambucil-obinutuzumab arm reported a meaningful improvement (increased ≥ 10 points) in EORTC QLQ-C30 global health status. The median time to first meaningful improvement in EORTC QLQ-C30 global health status score was 5.72 months (95% CI, 3.88 to 13.86 months) in the ibrutinib-venetoclax group and 6.11 months (95% CI, 4.14 to 11.56 months) in the chlorambucil-obinutuzumab group, with an HR of 0.874 (95% CI, 0.610 to 1.253; nominal P = 0.4576). The time to worsening or improvement in EORTC QLQ-C30 global health status score was not assessed in the extended follow-up analysis (data cut-off: August 25, 2022).
EORTC QLQ-C30 global health status was not measured in the CAPTIVATE FD cohort.
EQ-5D-5L VAS
In the primary analysis of the GLOW trial (data cut-off: February 26, 2021), 52 (49.1%) patients in the ibrutinib-venetoclax arm and 43 (41.0%) patients in the chlorambucil-obinutuzumab arm reported a meaningful decrease (≥ 7 points) in the EQ-5D-5L VAS score. The median time to first meaningful deterioration was 8.34 months (95% CI, 5.65 months to NE) in the ibrutinib-venetoclax group and 24.18 months (95% CI, 11.27 months to NE) in the chlorambucil-obinutuzumab group, with an HR of 1.382 (95% CI, 0.916 to 2.084; nominal P = 0.1188). At the same analysis, 60 (56.6%) patients in the ibrutinib-venetoclax arm and 75 (71.4%) patients in the chlorambucil-obinutuzumab arm reported a meaningful improvement (an increase of ≥ 7 points) in the EQ-5D-5L VAS score. The median time to first meaningful improvement was 4.76 months (95% CI, 3.78 to 8.31 months) in the ibrutinib-venetoclax group and 3.42 months (95% CI, 2.14 to 3.81 months) in the chlorambucil-obinutuzumab group, with an HR of 1.436 (95% CI, 1.015 to 2.031; nominal P = 0.0359). The time to worsening or improvement in EQ-5D-5L VAS score was not assessed in the extended follow-up analysis (data cut-off: August 25, 2022).
EQ-5D-5L VAS was not measured in the CAPTIVATE FD cohort.
EQ-5D-5L Utility Score
In the primary analysis of the GLOW trial (data cut-off: February 26, 2021), 51 (48.1%) patients in the ibrutinib-venetoclax arm and 46 (43.8%) patients in the chlorambucil-obinutuzumab arm reported a meaningful decrease (≥ 0.07 points) in the EQ-5D-5L utility score. The median time to first meaningful deterioration was 14.29 months (95% CI, 8.15 months to NE) in the ibrutinib-venetoclax group and 24.11 months (95% CI, 8.34 months to NE) in the chlorambucil-obinutuzumab group, with an HR of 1.188 (95% CI, 0.793 to 1.779; nominal P = 0.3997). At the same analysis, 59 (55.7%) patients in the ibrutinib-venetoclax arm and 65 (61.9%) patients in the chlorambucil-obinutuzumab arm reported a meaningful improvement (increased ≥ 0.07 points) in the EQ-5D-5L utility score. The median time to first meaningful improvement was 5.59 months (95% CI, 3.81 to 11.30 months) in the ibrutinib-venetoclax group and 4.67 months (95% CI, 3.75 to 6.14 months) in the chlorambucil-obinutuzumab group, with an HR of 1.177 (95% CI, 0.817 to 1.695; nominal P = 0.3757). The time to worsening or improvement in the EQ-5D-5L utility score was not assessed in the extended follow-up analysis (data cut-off: August 25, 2022).
EQ-5D-5L utility score was not measured in the CAPTIVATE FD cohort.
FACIT-Fatigue Scale
In the primary analysis of the GLOW trial (data cut-off: February 26, 2021), 62 (58.5%) patients in the ibrutinib-venetoclax arm and 51 (48.6%) patients in the chlorambucil-obinutuzumab arm reported a meaningful decrease (≥ 3 points) in the FACIT-Fatigue Scale score. The median time to first clinically meaningful deterioration in the FACIT-Fatigue score was 8.15 months (95% CI, 3.98 to 10.94 months) in the ibrutinib-venetoclax group and 14.03 months (95% CI, 8.61 months to NE) in the chlorambucil-obinutuzumab group, with an HR of 1.5 (95% CI, 1.054 to 2.237; nominal P = 0.0235). At the same analysis, 61 (57.5%) patients in the ibrutinib-venetoclax arm and 73 (69.5%) patients in the chlorambucil-obinutuzumab arm reported a meaningful improvement (increased ≥ 3 points) in the FACIT-Fatigue Scale score. The median time to first meaningful improvement in FACIT-Fatigue score was 5.59 months (95% CI, 3.81 to 11.20 months) in the ibrutinib-venetoclax group and 3.75 months (95% CI, 2.20 to 5.75 months) in the chlorambucil-obinutuzumab group, with a HR of 1.369 (95% CI, 0.959 to 1.954; nominal P = 0.0776). The time to worsening or improvement in the FACIT-Fatigue score was not assessed in the extended follow-up analysis (data cut-off: August 25, 2022).
FACIT-Fatigue Scale was not measured in the CAPTIVATE FD cohort.
Table 18: Summary of Key Efficacy Results From Pivotal and RCT Evidence (ITT Analysis Set for the GLOW Study; All-Treated Analysis Set for the CAPTIVATE Study)
Outcomes | GLOW primary analysis (data cut-off: February 26, 2021) | GLOW extended follow-up (data cut-off: August 25, 2022) | CAPTIVATE FD cohort | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
I+V (n = 106) | C+O (n = 105) | I+V (n = 106) | C+O (n = 105) | Primary analysis (N = 159) (data cut-off: November 12, 2020) | Extended follow-up (N = 159) (data cut-off: August 4, 2021) | ||||||
All treated (N = 159) | Non-del(17p) (n = 136) | With del(17p) (n = 20) | All treated (N = 159) | Non-del(17p) (n = 136) | With del(17p) (n = 20) | ||||||
PFS per IRC | |||||||||||
Number of PFS events by IRC assessment, n (%) | 22 (20.8) | 67 (63.8) | 29 (27.4) | 78 (74.3) | 23 (14.5) | 18 (13.2) | 5 (25.0) | 40 (25.2) | 32 (23.5) | 7 (35.0) | |
Progressive disease | 13 (12.3) | 65 (61.9) | 17 (16.0) | 71 (67.6) | 21 (13.2) | 16 (11.8) | NR | 38 (23.9) | 30 (22.1) | NR | |
Death | 9 (8.5) | 2 (1.9) | 12 (11.3) | 7 (6.7) | 2 (1.3) | 2 (1.5) | NR | 2 (1.3) | 2 (1.5) | NR | |
Censored | 84 (79.2) | 38 (36.2) | 77 (72.6) | 27 (25.7) | 136 (85.5) | 118 (86.8) | NR | 119 (74.8) | 104 (76.5) | NR | |
PFS by IRC assessment (months), median (95% CI) | NE (31.2 to NE) | 21.0 (16.6 to 24.7) | NE (NE to NE) | 21.7 (16.7 to 26.1) | NE (30.7 to NE) | NE (30.7 to NE) | NE (21.9 to NE) | 39.6 (39.0 to NE) | 39.6 (39.3 to NE) | 38.9a (38.6 to NE) | |
PFS event-free rate by IRC assessment, % (95% CI) | |||||||||||
24 months | 84.4 (75.8 to 90.1) | 44.1 (34.2 to 53.6) | 84.6 (76.2 to 90.3) | 45.1 (35.2 to 54.4) | 88.9 (82.7 to 92.9) | 90.8 (84.3 to 94.7) | 75.0 (50.0 to 88.7) | 90.8 (85.0 to 94.5) | 92.3 (86.2 to 95.8) | 80.0 (55.1 to 92.0) | |
36 months | NA | NA | 76.7 (67.2 to 83.7) | 33.3 (24.2 to 42.7) | NA | 85.5 (78.8 to 90.2) | 86.0 (78.7to 91.0) | 80.0 (55.1 to 92.0) | |||
48 months | NA | NA | 69.4 (58.6 to 78.0) | 12.3 (3.70 to 26.3) | NA | NA | |||||
IRC assessment, HR (95% CI; P value) | 0.216 (0.131 to 0.357; P < 0.0001) | 0.214 (0.138 to 0.334; P < 0.0001) | NA | ||||||||
PFS per IA | |||||||||||
Number of PFS events by IA, n (%) | 17 (16.0) | 61 (58.1) | 27 (25.5) | 82 (78.1) | 20 (12.6) | 16 (11.8) | 4 (20.0) | 28 (17.6) | 23 (16.9) | 5 (25.0) | |
Progressive disease | 8 (7.5) | 59 (56.2) | 15 (14.2) | 74 (70.5) | 18 (11.3) | 14 (10.3) | NR | 26 (16.4) | 21 (15.4) | NR | |
Death | 9 (8.5) | 2 (1.9) | 12 (11.3) | 8 (7.6) | 2 (1.3) | 2 (1.5) | NR | 2 (1.3) | 2 (1.5) | NR | |
Censored | 89 (84.0) | 44 (41.9) | 79 (74.5) | 23 (21.9) | 139 (87.4) | 120 (88.2) | NR | 131 (82.4) | 113 (83.1) | NR | |
PFS by IA (months), median (95% CI) | NE (NE to NE) | 21.9 (16.9 to 31.1) | NE (NE to NE) | 23.0 (16.9 to 31.2) | NE (30.4 to NE) | NE (30.4 to NE) | NE (28.5 to NE) | NE (NE to NE) | NE (NE to NE) | NE (38.9 to NE) | |
Event-free rate by IA, % (95% CI) | |||||||||||
24 months | 85.6 (77.2 to 91.1) | 48.7 (38.8 to 57.9) | 85.6 (77.3 to 91.1) | 49.3 (39.5 to 58.5) | 94.8 (89.8 to 97.3) | 96.2 (91.1 to 98.4) | 84.2 (58.7 to 94.6) | 94.8 (89.8 to 97.3) | 96.2 (91.1 to 98.4) | 84.2 (58.7 to 94.6) | |
36 months | NA | NA | 78.7 (69.4 to 85.4) | 32.6 (23.8 to 41.7) | NA | 88.1 (81.7 to 92.3) | 89.1 (82.3 to 93.4) | 78.6 (52.5 to 91.4) | |||
48 months | NA | NA | 72.9 (62.9 to 80.6) | 18.5 (11.2 to 27.3) | NA | ||||||
HR per IA (95% CI; P value) | 0.207 (0.120 to 0.357; P < 0.0001) | 0.201 (0.129 to 0.315; P < 0.0001) | NA | ||||||||
OS | |||||||||||
Number of deaths, n (%) | 11 (10.4) | 12 (11.4) | 15 (14.2) | 30 (28.6) | 3 (1.9) | 3 (2.2) | 0 | 3 (1.9) | 3 (2.2) | 0 | |
Follow-up (months), median (95% CI) | 27.70 (27.50 to 27.83) | 27.89 (27.53 to 28.58) | 46.06 (45.83 to 46.29) | 46.06 (45.14 to 46.59) | 27.9 (27.7 to 28.1) | 27.8 (27.7 to 28.1) | 27.8 (27.7 to 28.1) | 38.7 (38.7 to 38.8) | 38.7(38.7 to 38.8) | 38.7(38.7 to 38.8) | |
OS (months), median (95% CI) | NE (NE to NE) | 32.5 (32.5 to NE) | NE (NE to NE) | NE (NE to NE) | NE (NE to NE) | NE (NE to NE) | NE (NE to NE) | NE (NE to NE) | NE (NE to NE) | NE (NE to NE) | |
OS rate, % (95% CI) | |||||||||||
24 months | 90.4 (82.9 to 94.7) | 91.3 (83.9 to 95.4) | 90.4 (82.9 to 94.7) | 91.3 (84.0 to 95.4) | 98.1 (94.2 to 99.4) | 97.7 (93.2 to 99.3) | 100.0 (100.0 to 100.0) | 98.1 (94.2 to 99.4) | 97.7 (93.2 to 99.3) | 100.0 (100.0 to 100.0) | |
36 months | NA | NA | 89.5 (81.8 to 94.0) | 84.5 (75.9 to 90.2) | NA | 98.1 (94.2 to 99.4) | 97.7 (93.2 to 99.3) | 100.0 (100.0 to 100.0) | |||
48 months | NA | NA | 85.1 (76.4 to 90.8) | 67.6 (56.4 to 76.5) | NA | ||||||
HR (95% CI; P value) | 1.048 (0.454 to 2.419; nominal P = 0.9121) | 0.487 (0.262 to 0.907; nominal P = 0.0205) | NA | ||||||||
ORR | |||||||||||
ORR by IRC assessment, % (95% CI) | 86.8 (80.3 to 93.2) | 84.8 (77.9 to 91.6) | 86.8 (80.3 to 93.2) | 84.8 (77.9 to 91.6) | 96.2 (93.3 to 99.2) | 95.6 (92.1 to 99.0) | 100.0 (100.0 to 100.0) | 96.2 (93.3 to 99.2) | 95.6 (92.1 to 99.0) | 100.0 (100.0 to 100.0) | |
Relative response (95% CI; P value) | 1.02 (0.92 to 1.14; P = 0.6991) | 1.02 (0.92 to 1.14; nominal P = 0.6991) | NA | ||||||||
Best overall response per IRC, n (%) | |||||||||||
CR | 38 (35.8) | 12 (11.4%) | 43 (40.6) | 13 (12.4) | 92 (57.9) | 81 (59.6) | NR | 97 (61.0) | 86 (63.2) | NR | |
CRi | 3 (2.8) | 0 | 2 (1.9) | 0 | 3 (1.9) | 2 (1.5) | NR | 2 (1.3) | 1 (0.7) | NR | |
nPR | 4 (3.8) | 3 (2.9) | 4 (3.8) | 3 (2.9) | 2 (1.3) | 2 (1.5) | NR | 2 (1.3) | 2 (1.5) | NR | |
PR | 47 (44.3) | 74 (70.5) | 43 (40.6) | 73 (69.5) | 56 (35.2) | 45 (33.1) | NR | 52 (32.7) | 41 (30.1) | NR | |
ORR by IA, % (95% CI) | 92.5 (87.4 to 97.5) | 87.6 (81.3 to 93.9) | 92.5 (87.4 to 97.5) | 87.6 (81.3 to 93.9) | 96.2 (93.3 to 99.2) | 95.6 (92.1 to 99.0) | 96.3 (89.2 to 100.0) | 96.2 (93.3 to 99.2) | 95.6 (92.1 to 99.0) | 96.3 (89.2 to 100.0) | |
Relative response (95% CI; P value) | 1.05 (0.96 to 1.15; nominal P = 0.2585) | 1.05 (0.96 to 1.15; nominal P = 0.2585) | NA | ||||||||
Best overall response per IA, n (%) | |||||||||||
CR | 38 (35.8) | 14 (13.3) | 45 (42.5) | 15 (14.3) | 83 (52.2) | 74 (54.4) | NR | 88 (55.3) | 79 (58.1) | NR | |
CRi | 10 (9.4) | 0 | 10 (9.4) | 0 | 5 (3.1) | 2 (1.5) | NR | 3 (1.9) | 0 | NR | |
nPR | 2 (1.9) | 4 (3.8) | 2 (1.9) | 4 (3.8) | 1 (0.6) | 1 (0.7) | NR | 1 (0.6) | 1 (0.7) | NR | |
PR | 48 (45.3) | 74 (70.5) | 41 (38.7) | 73 (69.5) | 64 (40.3) | 53 (39.0) | NR | 61 (38.4) | 50 (36.8) | NR | |
CR (CR and CRi) rate | |||||||||||
CR rate by IRC assessment, % (95% CI) | 38.7 (29.4 to 48.0) | 11.4 (5.3 to 17.5) | 42.5 (33.0 to 51.9) | 12.4 (6.1 to 18.7) | 59.7 (52.1 to 67.4) | 61.0 (52.8 to 69.2) | 50.0 (28.1 to 71.9) | 62.3 (54.7 to 69.8) | 64.0 (55.9 to 72.0) | 50.0 (28.1 to 71.9) | |
Relative response (95% CI; P value) | 3.43 (1.91 to 6.15; P < 0.0001) | 3.48 (2.00 to 6.05; nominal P < 0.0001) | NA | ||||||||
CR rate by IA assessment, % (95% CI) | 45.3 (35.8 to 54.8) | 13.3 (6.8 to 19.8) | 51.9 (42.4 to 61.4) | 14.3 (7.6 to 21.0) | 55.3 (47.6 to 63.1) | 55.9 (47.5 to 64.2) | 50.0 (28.1 to 71.9) | 57.2 (49.5 to 64.9) | 58.1 (49.8 to 66.4) | 50.0 (28.1 to 71.9) | |
Relative response (95% CI; P value) | 3.42 (2.01 to 5.82; P < 0.0001) | 3.65 (2.22 to 5.99; nominal P < 0.0001) | NA | ||||||||
Improvement in hematologic parameters | |||||||||||
Proportion of patients with sustained improvement in hemoglobin, % (95% CI) | 44.3 (NR) | 50.5 (NR) | 52.8 (NR) | 51.4 (NR) | 41.5 (33.9 to 49.2) | NR | 60.0 (38.5 to 81.5) | 45.9 (38.2 to 53.7) | NR | 60.0 (38.5 to 81.5) | |
Relative response (95% CI, P value) | 0.88 (0.66 to 1.17; nominal P = 0.3854) | 1.03 (0.80 to 1.33; nominal P = 0.8306) | NA | ||||||||
Proportion of patients with sustained improvement in platelets, % (95% CI) | 24.5 (NR) | 29.5 (NR) | 32.1 (NR) | 32.4 (NR) | 17.6 (11.7 to 23.5) | NR | 15.0 (0 to 30.6) | 19.5 (13.3 to 25.7) | NR | 15.0 (0.0 to 30.6) | |
Relative response (95% CI, P value) | 0.84 (0.54 to 1.31; nominal P = 0.4346) | 1.00 (0.68 to 1.48; nominal P = 0.9967) | NA | ||||||||
DOR | |||||||||||
Number of patients contributing to the analysis | 106 | 105 | 106 | 105 | 153 | 130 | 20 | 153 | 130 | 20 | |
DOR by IRC assessment (months), median (95% CI) | 28.9 (28.7 to NE) | 21.1 (15.9 to 25.1) | NE (NE to NE) | 21.2 (17.7 to 28.6) | NE (NE to NE) | NE (27.6 to NE) | NE (18.9 to NE) | 36.4 (36.0 to NE) | NE (36.3 to NE) | 35.9b (35.6 to NE) | |
DOR rate by IRC assessment, % (95% CI) | |||||||||||
24 months | 89.9 (79.6 to 95.1) | 41.2 (29.9 to 52.2) | 92.2 (84.4 to 96.2) | 44.5 (33.7 to 54.7) | 88.7 (82.5 to 92.8) | 90.6 (84.1 to 94.6) | 74.7 (49.4 to 88.6) | 90.7 (84.8 to 94.4) | 92.2 (86.0 to 95.7) | 80.0 (55.1 to 92.0) | |
36 months | NA | NA | 84.1 (74.5 to 90.2) | 34.0 (23.9 to 44.3) | NA | 68.7 (56.4 to 78.1) | 72.8 (60.5 to 81.8) | 40.0 (8.0 to 71.7) | |||
DOR by IA (months), median (95% CI) | NE (NE to NE) | 21.5 (15.7 to 28.5) | NE (NE to NE) | 21.6 (15.7 to 29.1) | NE (NE to NE) | NE (NE to NE) | NE (25.9 to NE) | NE (NE to NE) | NE (NE to NE) | NE (36.3 to NE) | |
DOR rate by IA, % (95% CI) | |||||||||||
24 months | 88.3 (79.7 to 93.4) | 44.6 (33.5 to 55.0) | 88.7 (80.6 to 93.6) | 47.4 (36.9 to 57.2) | 94.7 (89.6 to 97.3) | 96.1 (90.8 to 98.3) | 84.2 (58.7 to 94.6) | 94.7 (89.7 to 97.3) | 96.1 (90.9 to 98.4) | 84.2 (58.7 to 94.6) | |
36 months | NA | NA | 81.3 (72.0 to 87.8) | 32.5 (23.0 to 42.3) | NA | 83.0 (75.4 to 88.5) | 83.2 (74.8 to 89.1) | 78.6 (52.5 to 91.4) | |||
TTNT | |||||||||||
Number of events, n (%) | 4 (3.8) | 27 (25.7) | 8 (7.5) | 41 (39.0) | NR | NR | |||||
TTNT (months), median (95% CI) | NE (NE to NE) | NE (31.5 to NE) | NE (NE to NE) | NE (42.3 to NE) | NR | NR | |||||
TTNT rate, % (95% CI) | |||||||||||
24 months | 97.0 (90.9 to 99.0) | 81.3 (72.2 to 87.6) | 95.9 (89.5 to 98.5) | 81.3 (72.3 to 87.7) | NR | NR | |||||
36 months | NR | NR | 93.8 (86.7 to 97.2) | 63.0 (52.5 to 71.8) | NR | NR | |||||
48 months | NR | NR | 91.4 (83.5 to 95.6) | 50.2 (33.5 to 64.7) | NR | NR | |||||
HR (95% CI; P value) | 0.143 (0.050 to 0.410; nominal P < 0.0001) | 0.155 (0.072 to 0.333; nominal P < 0.0001) | NR | NR | |||||||
MRD negativity rate | |||||||||||
Overall MRD negativity rate,c | |||||||||||
Assessed by BM, % (95% CI) | 55.7 (46.2 to 65.1) | 21.0 (13.2 to 28.7) | NA | NA | 59.7 (52.1 to 67.4) | 61.8 (53.6 to 69.9) | 45.0 (23.2 to 66.8) | 59.7 (52.1 to 67.4) | 61.8 (53.6 to 69.9) | 45.0 (23.2 to 66.8) | |
Relative response, n (95% CI; P value) | 2.65 (1.75 to 3.99; P < 0.001) | NA | NA | NA | |||||||
Assessed by PB, % (95% CI) | 59.4 (50.1 to 68.8) | 40.0 (30.6 to 49.4) | NA | NA | 76.7 (70.2 to 83.3) | 76.5 (69.3 to 83.6) | 80.0 (62.5 to 97.5) | 76.7 (70.2 to 83.3) | 76.5 (69.3 to 83.6) | 80.0 (62.5 to 97.5) | |
Relative response, n (95% CI; P value) | 1.48 (1.10 to 1.98; nominal P = 0.0055) | NA | NA | NA | |||||||
TLS risk reduction | |||||||||||
Baseline high risk, n (%) | 26 (24.5) | NA | NA | NA | 34 (21.4) | NR | 1 (5.0) | NA | |||
Reduced to medium or low risk after ibrutinib lead-in phase, n (%) | 22 (20.8) | NA | NA | NA | 33 (20.8) | NR | 1 (5.0) | NA | |||
Time to worsening or improvement (EORTC QLQ-C30 global health status score)d | |||||||||||
Patients who showed deterioration, n (%) | 47 (44.3) | 43 (41.0) | NR | NR | NR | ||||||
Median time to worsening, months (95% CI) | 14.95 (8.38 to NE) | 24.18 (13.86 to NE) | NR | NR | NR | ||||||
HR (95% CI; P value) | 1.149 (0.756 to 1.746; nominal P = 0.5130) | NR | NR | NR | |||||||
Patients who showed improvement, n (%) | 65 (61.3) | 58 (55.2) | NR | NR | NR | ||||||
Time to improvement (months) median (95% CI) | 5.72 (3.88 to 13.86) | 6.11 (4.14 to 11.56) | NR | NR | NR | ||||||
HR (95% CI; P value) | 0.874 (0.610 to 1.253; nominal P = 0.4576) | NR | NR | NR | |||||||
Time to worsening or improvement (EQ-5D-5L VAS)e | |||||||||||
Patients who showed deterioration, n (%) | 52 (49.1) | 43 (41.0) | NR | NR | NR | ||||||
Time to worsening (months), median (95% CI) | 8.34 (5.65 to NE) | 24.18 (11.27 to NE) | NR | NR | NR | ||||||
HR (95% CI; P value) | 1.382 (0.916 to 2.084; nominal P = 0.1188) | NR | NR | ||||||||
Patients who showed improvement, n (%) | 60 (56.6) | 75 (71.4) | NR | NR | NR | ||||||
Time to improvement (months) median (95% CI) | 4.76 (3.78 to 8.31) | 3.42 (2.14 to 3.81) | NR | NR | NR | ||||||
HR (95% CI; P value) | 1.436 (1.015 to 2.031; nominal P = 0.0359) | NR | NR | NR | |||||||
Time to worsening or improvement (EQ-5D-5L utility score)f | |||||||||||
Patients who showed deterioration (%) | 51 (48.1) | 46 (43.8) | NR | NR | NR | ||||||
Time to worsening (months), median (95% CI) | 14.29 (8.15 to NE) | 24.11 (8.34 to NE) | NR | NR | NR | ||||||
HR (95% CI; P value) | 1.188 (0.793 to 1.779; nominal P = 0.3997) | NR | NR | NR | |||||||
Patients who showed improvement, n (%) | 59 (55.7) | 65 (61.9) | NR | NR | NR | ||||||
Time to improvement (months) median (95% CI) | 5.59 (3.81 to 11.30) | 4.67 (3.75 to 6.14) | NR | NR | NR | ||||||
HR (95% CI; P value) | 1.177 (0.817 to 1.695; nominal P = 0.3757) | NR | NR | NR | |||||||
Time to worsening or improvement (FACIT-Fatigue Scale)g | |||||||||||
Patients who showed deterioration (%) | 62 (58.5) | 51 (48.6) | NR | NR | NR | ||||||
Time to worsening months, median (95% CI) | 8.15 (3.98 to 10.94) | 14.03 (8.61 to NE) | NR | NR | NR | ||||||
HR (95% CI; P value) | 1.535 (1.054 to 2.237; nominal P = 0.0235) | NR | NR | NR | |||||||
Patients who showed improvement, n (%) | 61 (57.5) | 73 (69.5) | NR | NR | NR | ||||||
Time to improvement (months), median (95% CI) | 5.59 (3.81 to 11.20) | 3.75 (2.20 to 5.75) | NR | NR | NR | ||||||
HR (95% CI; P value) | 1.369 (0.959 to 1.954; nominal P = 0.0776) | NR | NR | NR | |||||||
BM = bone marrow; C+O = chlorambucil plus obinutuzumab; CI = confidence interval; CR = complete response; CRi = complete response with incomplete bone marrow recovery; del(17p) = deletion of 17p; DOR = duration of response; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EQ-5D-5L = 5-Level EQ-5D; FACIT = Functional Assessment of Chronic Illness Therapy; FD = fixed duration; HR = hazard ratio; I+V = ibrutinib plus venetoclax; IA = investigator assessment; IRC = independent review committee; ITT = intention to treat; MRD = minimal residual disease; NA = not applicable; NE = not evaluable; NR = not reported; nPR = nodular partial response; ORR = overall response rate; OS = overall survival; PB = peripheral blood; PFS = progression-free survival; PR = partial response; TLS = tumour lysis syndrome; TTNT = time to next treatment.
aEstimate is not reliable because only 3 patients were at risk at 39 months.
bEstimate is not reliable because only 2 patients were at risk at 36 months after initial response.
cNext-generation sequencing was used as the primary method of MRD analysis for the GLOW trial; the flow cytometry method was used for the MRD analysis in the CAPTIVATE FD cohort.
dDeterioration or improvement is defined as a change of ≥ 10 points in the EORTC QLQ-C30 global health status score for each assessment after baseline.
eMID of ≥ 7 points is considered clinically important for the EQ-5D-5L VAS health rating.
fA minimum difference of ≥ 0.07 points in EQ-5D-5L utility score is considered clinically important.
gFACIT-Fatigue improvement or worsening is defined as ≥ 3-point change for each assessment post baseline.
Sources: GLOW Primary Analysis CSR;23 GLOW Extended Follow-up Analysis CSR;24 GLOW PRO CSR;25 CAPTIVATE CSR.26 (Note: Details from the table have been taken from the sponsor’s Summary of Clinical Evidence.)
Harms
A summary of harms is provided in Table 19.
Adverse Events
In the GLOW study, at least 1 AE was reported in a similar proportion of patients in the ibrutinib-venetoclax arm compared with the chlorambucil-obinutuzumab arm (99.1% versus 94.3% for ibrutinib-venetoclax versus chlorambucil-obinutuzumab). AEs of any grade were reported more frequently in the ibrutinib-venetoclax arm, including diarrhea (50.9% versus 12.4%), nausea (26.4% versus 25.7%), infections and infestations (60.4% versus 48.6%), metabolism and nutrition disorders (42.5% versus 23.8%), respiratory, thoracic and mediastinal disorders (35.8% versus 28.6%), musculoskeletal and connective tissue disorders (34.0% versus 25.7% for), and nervous system disorders (30.2% versus 20.0% obinutuzumab). AEs of grade 3 or 4 were also reported in similar proportions in the ibrutinib-venetoclax arm and chlorambucil-obinutuzumab arm (68.9% versus 67.1%). The following AEs of grades 3 or 4 were reported more frequently (≥ 5% difference) in the ibrutinib-venetoclax arm than in the chlorambucil-obinutuzumab arm: diarrhea (10.4% versus 1.0% for obinutuzumab), vascular disorders (8.5% versus 1.9% for), and cardiac disorders (12.3% versus 2.9%).
In the CAPTIVATE FD cohort, at least 1 AE was reported in 158 (99.5%) patients, and AEs of grade 3 or 4 were reported in 98 (61.6%) patients. The most commonly reported AEs were diarrhea (62.3%), nausea (42.8%), neutropenia (41.5%), and arthralgia (33.3%). Grade 3 or 4 neutropenia was reported in 52 (32.7%) patients.
Serious Adverse Events
In the GLOW study, more patients in the ibrutinib-venetoclax arm experienced at least 1 SAE of any grade (46.2% versus 27.6% for ibrutinib-venetoclax versus chlorambucil-obinutuzumab). Of these SAEs, the most reported SAEs that were more frequently reported in the ibrutinib-venetoclax arm were cardiac disorders (13.2% versus 2.9%), infections (12.3% versus 8.6%), general disorders and administration-site conditions (5.7% versus 1.9%), skin and subcutaneous tissue disorders (4.7% versus 0%), and gastrointestinal disorders (3.8% versus 1.9%). Atrial fibrillation was reported more frequently in the ibrutinib-venetoclax arm (6.6% versus 0%) whereas TLS was more common in the chlorambucil-obinutuzumab arm (0% versus 2.9%).
In the CAPTIVATE FD cohort, 36 (22.6%) patients experienced at least 1 SAE of any grade and 30 (18.9%) patients experienced at least 1 SAE of grade 3 or 4.
Withdrawal Due to Adverse Events
In the GLOW trial, more patients reported AEs leading to discontinuation in the ibrutinib-venetoclax arm than in the chlorambucil-obinutuzumab arm (20.8% versus 7.6% for ibrutinib-venetoclax versus chlorambucil-obinutuzumab).
In the CAPTIVATE FD cohort, 8 (5.0%) patients had AEs leading to ibrutinib discontinuation and 3 (1.9%) had AEs leading to venetoclax discontinuation.
Mortality
In the GLOW trial, all patients had completed the study treatment at the time of the primary analysis (data cut-off: February 26, 2021). Overall, 11 (10.4%) patients in the ibrutinib-venetoclax arm and 12 (11.4%) patients in the chlorambucil-obinutuzumab arm died during the study period. AEs were the most frequent cause of death in the ibrutinib-venetoclax arm (6.6% versus 1.9% for ibrutinib-venetoclax versus chlorambucil-obinutuzumab), whereas other causes were the most frequent cause of death in the chlorambucil-obinutuzumab arm (2.8% versus 9.5%).
In the CAPTIVATE FD cohort, 3 (1.9%) patients died during the study period.
Notable Harms
AEs of special interest included atrial fibrillation and major hemorrhages. In the GLOW study, more patients reported atrial fibrillation (14.2% versus 1.9% for ibrutinib-venetoclax versus chlorambucil-obinutuzumab). Grade 3 or 4 atrial fibrillation was reported in 2 (1.9%) patients in the ibrutinib-venetoclax arm and in no patients in the chlorambucil-obinutuzumab arm. Similarly, a higher proportion of patients reported major hemorrhage in the ibrutinib-venetoclax arm than the chlorambucil-obinutuzumab arm (3.8% versus1.0% for ibrutinib-venetoclax versus chlorambucil-obinutuzumab).
In the CAPTIVATE FD cohort, 7 (4.4%) patients had atrial fibrillation, and 2 (1.3%) of these events were grade 3 or 4. Major hemorrhage was reported in 3 (1.9%) patients.
Table 19: Summary of Harms — Pivotal and RCT Evidence (Safety Analysis Set for GLOW; All-Treated Analysis Set for CAPTIVATE)
Adverse events | GLOW (data cut-off: February 26, 2021) | CAPTIVATE (data cut-off: November 12, 2020) | ||||
|---|---|---|---|---|---|---|
Ibrutinib-venetoclax (N = 106) | Chlorambucil-obinutuzumab (N = 105) | FD cohort (ibrutinib-venetoclax) (N = 159) | ||||
Any grade | Grade 3 or 4 | Any grade | Grade 3 or 4 | Any grade | Grade 3 or 4 | |
Most common TEAEs (≥ 20% in either treatment group), n (%)a,b | ||||||
Patients with ≥ 1 AE | 105 (99.1) | 73 (68.9) | 99 (94.3) | 71 (67.6) | 158 (99.4) | 98 (61.6) |
Gastrointestinal disorders | 71 (67.0) | 14 (13.2) | 43 (41.0) | 4 (3.8) | NR | NR |
Diarrhea | 54 (50.9) | 11 (10.4) | 13 (12.4) | 1 (1.0) | 99 (62.3) | 5 (3.1) |
Nausea | 28 (26.4) | 0 | 27 (25.7) | 0 | 68 (42.8) | 2 (1.3) |
Vomiting | 15 (14.2) | 1 (0.9) | 14 (13.3) | 0 | 35 (22.0) | 3 (1.9) |
Infections and infestations | 64 (60.4) | 16 (15.1) | 51 (48.6) | 11 (10.5) | NR | NR |
Upper respiratory tract infection | 13 (12.3) | NR | 14 (13.3) | 0 | 37 (23.3) | 0 |
Blood and lymphatic system disorders | 56 (52.8) | 36 (34.0) | 72 (68.6) | 58 (55.2) | NR | NR |
Neutropenia | 36 (34.0) | 30 (28.3) | 56 (53.3) | 47 (44.8) | 66 (41.5) | 52 (32.7) |
Thrombocytopenia | 12 (11.3) | 6 (5.7) | 28 (26.7) | 21 (20.0) | 21 (13.2) | NR |
Skin and subcutaneous tissue disorders | 52 (49.1) | 10 (9.4) | 27 (25.7) | 1 (1.0) | NR | NR |
Metabolism and nutrition disorders | 45 (42.5) | 16 (15.1) | 25 (23.8) | 11 (10.5) | NR | NR |
General disorders and administration-site conditions | 42 (39.6) | 5 (4.7) | 44 (41.9) | 3 (2.9) | NR | NR |
Fatigue | 16 (15.1) | 1 (0.9) | 10 (9.5) | 0 | 39 (24.5) | 1 (0.6) |
Respiratory, thoracic, and mediastinal disorders | 38 (35.8) | 3 (2.8) | 30 (28.6) | 2 (1.9) | NR | NR |
Musculoskeletal and connective tissue disorders | 36 (34.0) | 8 (7.5) | 27 (25.7) | 0 (0.0) | NR | NR |
Arthralgia | 12 (11.3) | 1 (0.9) | 7 (6.7) | 0 | 53 (33.3) | 2 (1.3) |
Nervous system disorders | 32 (30.2) | 4 (3.8) | 21 (20.0) | 2 (1.9) | NR | NR |
Vascular disorders | 27 (25.5) | 9 (8.5) | 24 (22.9) | 2 (1.9) | NR | NR |
Cardiac disorders | 26 (24.5) | 13 (12.3) | 14 (13.3) | 3 (2.9) | NR | NR |
Injury, poisoning, and procedural complications | 25 (23.6) | 6 (5.7) | 36 (34.3) | 6 (5.7) | NR | NR |
Muscle spasms | 9 (8.5) | 0 | 2 (1.9) | 0 | 47 (29.6) | 0 |
Headache | 7 (6.6) | 0 | 5 (4.8) | 1 (1.0) | 40 (25.2) | 1 (0.6) |
Increased tendency to bruise | NR | 0 | NR | NR | 35 (22.0) | 0 |
Patients with ≥ 1 SAE (≥ 2% of either treatment group), n (%)c,d | ||||||
Patients with ≥ 1 SAE | 49 (46.2) | 34 (32.1) | 29 (27.6) | 21 (20.0) | 36 (22.6) | 30 (18.9) |
Cardiac disorders | 14 (13.2) | 11 (10.4) | 3 (2.9) | 3 (2.9) | NR | NR |
Atrial fibrillation | 7 (6.6) | 5 (4.7) | 0 | 0 | NR | NR |
Cardiac failure | 3 (2.8) | 2 (1.9) | 0 | 0 | NR | NR |
Infections and infestations | 13 (12.3) | 9 (8.5) | 9 (8.6) | 6 (5.7) | NR | NR |
Pneumonia | 6 (5.7) | 4 (3.8) | 6 (5.7) | 5 (4.8) | NR | NR |
General disorders and administration-site conditions | 6 (5.7) | 1 (0.9) | 2 (1.9) | 1 (1.0) | NR | NR |
Nervous system disorders | 6 (5.7) | 2 (1.9) | 0 | 0 | NR | NR |
Blood and lymphatic system disorders | 5 (4.7) | 2 (1.9) | 5 (4.8) | 4 (3.8) | NR | NR |
Anemia | 3 (2.8) | 0 | 2 (1.9) | 0 | NR | NR |
Febrile neutropenia | 1 (0.9) | 1 (0.9) | 3 (2.9) | 3 (2.9) | NR | NR |
Skin and subcutaneous tissue disorders | 5 (4.7) | 5 (4.7) | 0 | 0 | NR | NR |
Gastrointestinal Disorders | 4 (3.8) | 4 (3.8) | 2 (1.9) | 2 (1.9) | NR | NR |
Diarrhea | 3 (2.8) | 2 (1.9) | 1 (1.0) | 1 (1.0) | NR | NR |
Injury, poisoning, and procedural complications | 4 (3.8) | 4 (3.8) | 7 (6.7) | 4 (3.8) | NR | NR |
Infusion-related reaction | 0 | 0 | 3 (2.9) | 1 (1.0) | NR | NR |
Neoplasms benign, malignant, and unspecified (including cysts and polyps) | 4 (3.8) | 2 (1.9) | 1 (1.0) | 0 | NR | NR |
Respiratory, thoracic, and mediastinal disorders | 4 (3.8) | 2 (1.9) | 1 (1.0) | 1 (1.0) | NR | NR |
Vascular disorders | 4 (3.8) | 2 (1.9) | 0 | 0 | NR | NR |
Renal and urinary disorders | 3 (2.8) | 3 (2.8) | 0 | 0 | NR | NR |
Metabolism and nutrition disorders | 2 (1.9) | 0 | 3 (2.9) | 3 (2.9) | NR | NR |
TLS | 0 | 0 | 3 (2.9) | 3 (2.9) | NR | NR |
Investigations | 1 (0.9) | 1 (0.9) | 3 (2.9) | 2 (1.9) | NR | NR |
Cellulitis | 1 (0.9) | 1 (0.9) | 0 | 0 | 4 (2.5) | 4 (2.5) |
Patients who stopped treatment due to AEs, n (%) | ||||||
TEAEs leading to discontinuation of 1 or more drugs | 22 (20.8) | NR | 8 (7.6) | NR | Ibrutinib: 8 (5.0) Venetoclax:3 (1.9) | Ibrutinib: 7 (4.4) Venetoclax: 2 (1.3) |
Deaths, n (%) | ||||||
Patients who died | 11 (10.4) | 12 (11.4) | 3 (1.9) | |||
AE | 7 (6.6) | 2 (1.9) | 1 (0.6) | |||
Progressive disease | 1 (0.9) | 0 | NR | |||
Other | 3 (2.8) | 10 (9.5) | 1 (0.6) | |||
COVID-19 related | 1 (0.9) | 4 (3.8) | NR | |||
Sudden death | NR | NR | 1 (0.6) | |||
AEs of special interest, n (%) | ||||||
Atrial fibrillation | 15 (14.2) | 7 (6.6) | 2 (1.9) | 0 (0.0) | 7 (4.4) | 2 (1.3) |
Major hemorrhage | 4 (3.8) | 4 (3.8) | 1 (1.0) | 1 (1.0) | 3 (1.9) | 2 (1.3) |
AE = adverse events; FD = fixed duration; NR = not reported (when incidence rates fall below the specified reporting threshold in either treatment arm within the specified AE severity); RCT = randomized controlled trial; SAE = serious adverse event; TEAE = treatment-emergent adverse event; TLS = tumour lysis syndrome.
Note: At the GLOW extended 18-month follow-up from primary analysis (data cut-off: August 25, 2022), no changes in safety data were noted, except for 1 additional patient in the chlorambucil-obinutuzumab treatment arm who was diagnosed with 2 new treatment-emergent SAEs (myelodysplastic syndrome and myeloproliferative neoplasm) after primary analysis and 7 additional patients who were reported to have developed a nontreatment-emergent secondary malignancy (4 patients from the ibrutinib-venetoclax arm and 3 patients from the chlorambucil-obinutuzumab arm).
aFor the GLOW study, any-grade TEAEs occurring in ≥ 20% of patients in any treatment arm by system organ class and preferred term, and grade 3 or 4 TEAEs occurring in ≥ 2% of patients in either arm by system organ class and preferred term are reported.
bFor the CAPTIVATE study, any-grade TEAEs occurring in ≥ 20% of patients by MedDRA preferred term and maximum severity, and grades 3 or 4 TEAEs occurring in ≥ 2% of patients by MedDRA preferred term and maximum severity are reported.
cFor the GLOW study, any-grade and grade 3 or 4 treatment-emergent SAEs occurring in ≥ 2% of patients in any treatment arm by system organ class and preferred term are reported.
dFor the CAPTIVATE study, any-grade and grade 3 or 4 serious TEAEs by MedDRA preferred term and maximum severity are reported if they occurred in ≥ 2% patients.
Sources: GLOW Primary Analysis CSR;23 CAPTIVATE CSR.26 (Note: details from the table have been taken from the sponsor’s Summary of Clinical Evidence.)
Critical Appraisal
Internal Validity
Fludarabine-Ineligible Population
The GLOW trial was a phase III, randomized study evaluating the efficacy and safety of the combination ibrutinib-venetoclax compared with chlorambucil-obinutuzumab in the first-line treatment of patients with CLL who are not eligible to fludarabine-based therapy. In the GLOW trial, patients were assigned to a treatment group via a computer-generated randomization schedule, which involved using IGHV mutation status and the presence of del(11q) as stratification factors were considered appropriate. There were imbalances reported in CIRS scores and LDH elevations, which are considered prognostic factors by the clinical expert consulted by CADTH. More patients in the ibrutinib-venetoclax arm than in the chlorambucil-obinutuzumab arm had a CIRS score higher than 6, and TP53 mutations were reported more frequently in the ibrutinib-venetoclax arm, which may bias the study against the ibrutinib-venetoclax arm, and more patients in the chlorambucil-obinutuzumab arm had LDH elevation, which may bias the study results in favour of the ibrutinib-venetoclax arm.
For the GLOW trial, the CADTH review team considered the open-label design to be reasonable, given the distinct dosing regimens of ibrutinib-venetoclax and chlorambucil-obinutuzumab, which would likely allow investigators and patients to make inferences about treatment assignment regardless of blinding. The CADTH review team noted that the open-label design of the GLOW trial had the potential to introduce reporting bias in the assessment of subjective outcomes reported by patients, such as HRQoL and AEs. Disease response outcomes (i.e., PFS, ORR, DOR, and CR rate) were assessed by IA and by IRC to help mitigate the biases associated with the open-label study design for the GLOW trial.
In the GLOW trial, the overall median duration of exposure was substantially longer in the ibrutinib-venetoclax arm (13.8 months) than for the chlorambucil-obinutuzumab arm (5.1 months). In addition, fewer patients in the chlorambucil-obinutuzumab arm discontinued treatment due to AEs compared to the ibrutinib-venetoclax arm, and a higher proportion of patients completed the study treatment in the chlorambucil-obinutuzumab arm than the ibrutinib-venetoclax arm, which indicates that patients in the chlorambucil-obinutuzumab arm had better treatment compliance than those in the ibrutinib-venetoclax arm in the GLOW trial, this may bias the results against the ibrutinib-venetoclax arm. A higher proportion of patients received concomitant analgesics, antihistamines, and corticosteroids in the chlorambucil-obinutuzumab arm than the ibrutinib-venetoclax arm in the GLOW trial. Although the clinical expert did not think the imbalances in concomitant treatments between treatment arms would bias the efficacy results, as these concomitant medications do not have any anti-CLL activity, except for corticosteroids, which were given at relatively low doses, the CADTH review team noted that these imbalances may bias the safety and HRQoL results against the ibrutinib-venetoclax arm. According to the clinical expert,, the use of concomitant medications is possibly a reflection of a patient population with comorbidities. Regarding subsequent anticancer therapies, in the GLOW trial, a higher proportion of patients in the chlorambucil-obinutuzumab arm received subsequent anticancer therapy compared to the ibrutinib-venetoclax arm. The clinical expert consulted by CADTH indicated that the use of subsequent therapies would influence OS. The CADTH review team agreed with the clinical expert and noted that the use of subsequent anticancer therapy results in an indirectness of the estimated OS effect. It is difficult in this setting to isolate the direct effect of ibrutinib-venetoclax treatment on OS due to the intercurrent use of subsequent anticancer therapies.
In the GLOW study, patients in both treatment arms had major protocol deviations. Most frequently, the wrong treatment or an incorrect dose was administered, which may increase uncertainty in the estimate of the treatment effect due to increased treatment heterogeneity and may be attributed to the difference between the treatment of interest and the treatment implemented.
Many of the outcomes used in the GLOW trial (PFS, OS, ORR, CR rate, DOR) are standard in oncology trials and in alignment with the end points used for similar indications for ibrutinib and other CLL therapies.51 In the GLOW trial (fludarabine-ineligible patients) the primary efficacy end point investigated was PFS as measured by IRC; PFS per IA was reported as a sensitivity analysis. Both IA and IRC used the iwCLL criteria for CLL, which were considered adequate for response assessment by CADTH and Health Canada.51 The primary end point was met at the February 26, 2021, cut-off for the GLOW trial as PFS per IRC demonstrated superiority in the ibrutinib-venetoclax arm compared to the chlorambucil-obinutuzumab arm.
The median of OS was not reached in the ibrutinib-venetoclax arm in the GLOW trial in the primary analysis, and OS data were considered immature. Although the sponsor conducted subsequent final PFS analyses in the extended follow-up analyses, there is uncertainty about the effect of ibrutinib-venetoclax on long-term OS for fludarabine-ineligible patients with CLL. In addition, in the GLOW trial, the sponsor conducted a sensitivity analysis that used death due to COVID as a censor in the analysis of OS; the CADTH review team considers censoring patients who died due to COVID is not sufficient to estimate the direct effect of treatment on OS in a COVID-free population, as the patients who died of COVID may have been different than the patients who stayed on trial in terms of other variables and baseline characteristics.
A serial gatekeeping procedure was employed to control the overall type I error for the primary end point and selected secondary end points in the GLOW trial for the primary analysis. Of note, the hierarchical statistical testing strategy ended at ORR per IRC. The remaining key secondary end points (i.e., OS, sustained hematological improvements, and time to improvement in FACIT-Fatigue score) and ORR per IA were considered not statistically significant due to the failure of ORR to demonstrate statistical significance, per statistical testing hierarchy. Therefore, any results with a P value less than the prespecified significance level should be interpreted with caution, considering the potentially inflated type I error rate. Although the subgroup analyses were prespecified, there is no evidence that the studies were powered to detect subgroup differences. In addition, no data imputation was performed in the GLOW trial.
HRQoL is considered a relevant outcome by patients with CLL and by clinicians. The EORTC QLQ-C30, EQ-5D-5L utility, EQ-5D VAS, and FACIT-Fatigue measurements used for HRQoL assessment in the GLOW study are commonly used in oncology trials; however, the validity, reliability, and responsiveness of these measurements have not been studied in patients with CLL. The minimal clinically important differences proposed by the sponsor were based on patients with breast cancer (EORTC QLQ-C30) or multiple types of cancer (EQ-5D-5L utility, and VAS, and FACIT-Fatigue). Given that AEs were the most common cause of discontinuation in the GLOW trial, there is a risk of reporting bias results from patients who remained in the trial, impacting the interpretability of HRQoL trends over time. Moreover, there was no multiplicity adjustment for the analysis of HRQoL, which may have further added uncertainty to the study results.
Fludarabine-Eligible Population
The CAPTIVATE trial was a phase II, multicohort, international trial with 2 cohorts: the MRD cohort and the FD cohort. This review focused on the CAPTIVATE FD cohort, which assessed the efficacy and safety of ibrutinib-venetoclax in fludarabine-eligible patients with treatment-naive CLL or SLL. Because the FD cohort of the CAPTIVATE trial was designed as a single-arm study, given the lack of a comparator arm, the ability to make definitive conclusions about the comparative efficacy of ibrutinib-venetoclax in fludarabine-eligible patients with CLL is limited. In addition, the open-label design had the potential to introduce reporting bias in the assessment of subjective outcomes reported by patients (i.e., AEs). Disease response outcomes (PFS, ORR, DOR, CR rate) were assessed by IA and by IRC to help mitigate the biases associated with the open-label study design for the FD cohort in the CAPTIVATE trial. Many of the outcomes used in the CAPTIVATE FD cohort (PFS, OS, ORR, CR rate, DOR) are standard in oncology trials and in alignment with the end points used for similar indications for ibrutinib and other CLL therapies. The primary efficacy end point was CR rate per IA; CR per IRC was also reported as a sensitivity analysis. Both IA and IRC used the iwCLL criteria for CLL, which were considered adequate for response assessment by CADTH and Health Canada.51
The primary end point for the CAPTIVATE FD cohort was met at the time of primary analysis (data cut-off: November 12, 2020) because the CR rate per IA in patients without del(17p) exceeded the prespecified minimum CR rate of 37%. The medians of OS and PFS were not reached in the CAPTIVATE FD cohort at the time of primary analysis, and OS data were considered immature. Therefore, there is uncertainty in the interpretation of long-term OS and PFS results for FCR-eligible patients with CLL in the CAPTIVATE FD cohort. In addition, the CADTH review team noted that the CAPTIVATE FD cohort included a subgroup of patients with del(17p); however, there was no formal statistical testing performed between subgroups and the sample size for patients with del(17p) is small (n = 20), thus no conclusions can be drawn from the study results for patients with del(17p). Although the subgroup analyses were prespecified, there is no evidence that the study were powered to detect subgroups differences. HRQoL is considered a relevant outcome by patients with CLL and by clinicians, but there was no assessment of HRQoL in the CAPTIVATE FD cohort, thus it is uncertain whether treatment with ibrutinib-venetoclax would improve HRQoL in fludarabine-eligible patients with CLL.
External Validity
Both the GLOW trial and CAPTIVATE FD cohort required eligible patients to have measurable nodal disease; however, according to the clinical expert consulted by CADTH, there is a small proportion of patients who only have elevated white blood cell counts and cytopenia and may not have an enlarged lymph node in clinical practice. These patients are important and would fit in the patient population for the ibrutinib-venetoclax regimen. In addition, the GLOW trial excluded patients with galactose intolerance, Lapp lactase deficiency, or glucose-galactose malabsorption from study enrolment at Protocol Amendment 1 made on June 6, 2018, which is not reflective of real-world clinical practice, as there are patients with lactose intolerance who take ibrutinib based on the feedback provided by the clinical expert consulted by CADTH. Furthermore, the exclusion of patients who are intolerant to galactose may underestimate the risk of abdominal pain and diarrhea in patients with CLL, as the formulation of ibrutinib contains lactose, and symptoms of lactose intolerance (i.e., abdominal pain and diarrhea) can occur in these patients, according to the clinical expert. The clinical expert stated that there is a more diversified patient population, including patients from Asia and other parts of the world, in heir clinical practice compared to the patient population in the GLOW trial and CAPTIVATE FD cohort. The baseline characteristics of the 2 studies may be indicative of the overrepresentation of white patients (≥ 92%) with CLL in both fludarabine-eligible and fludarabine-ineligible populations; this presents an evidence gap in the generalizability of results to all patients. Although the inclusion criteria mandated that eligible patients in the GLOW trial have a CrCl of less than 70 mL/min and/or a CRIS score greater than 6, the baseline CrCl for both treatment arms was higher than what the clinical expert would expect in high-risk patients with CLL who are ineligible to fludarabine-based therapy in clinical practice, which may indicate that patients in the GLOW trial have better kidney function than patients in clinical practice. This may compromise the generalizability of the study results to the general FCR-ineligible patients with CLL. Generally, the risk profile of patients in the CAPTIVATE FD cohort is what the clinical expert would expect in fludarabine-eligible patients with CLL in clinical practice.
The GLOW trial used chlorambucil-obinutuzumab as the treatment for the comparator arm. The clinical expert comment that chlorambucil-obinutuzumab may not be the best choice of comparator for ibrutinib-venetoclax, as it is an old historic treatment being used as a standard therapy in an older population of patients with CLL, which is about 5% of the patient population in the clinical expert’s clinical practice. Moreover, according to the clinical expert, the dosing regimen for the chlorambucil-obinutuzumab arm used in the GLOW trial (i.e., 6 cycles of 28 days) is a standard regimen commonly used in clinical practice; however, chlorambucil can be given in many different ways based on physician decision and patient preference in clinical practice in Canada (i.e., having a pause for 4 days every month or receiving daily low-dose chlorambucil), and there are variations in treatment length (i.e., 12-month duration). These factors may introduce uncertainty in the estimate of the treatment effect in practice in Canada due to limitations in the generalizability of the study results.
Long-Term Extension Studies
No long-term extension studies were identified for this review.
Indirect Evidence and Observational Evidence
The contents of this section have been informed by materials submitted by the sponsor. The following information has been summarized and validated by the CADTH review team.
Objectives for the Summary of Indirect Evidence
No comparative evidence was available in the pivotal trials for patients with CLL who were eligible for fludarabine nor direct comparative data to treatment regimens other than chlorambucil-obinutuzumab in the fludarabine-ineligible population. This section provides a summary and appraisal of the indirect and observational evidence submitted by the sponsor to address these knowledge gaps. These data were used to inform the pharmacoeconomic model.
Description of Indirect Comparisons and Observational Evidence
For patients who were ineligible to received fludarabine, the sponsor provided 2 ITCs and 2 IPD observational studies that evaluated the efficacy and safety of ibrutinib-venetoclax versus BR,27 ibrutinib,28 VO,29 and acalabrutinib.30 In fludarabine-eligible patients, an IPD analysis was conducted comparing ibrutinib-venetoclax versus fludarabine in combination with cyclophosphamide and rituximab.31 The ITC analyses were based on MAIC methods (comparison with acalabrutinib and VO) and the IPD analyses were based on propensity score methods (comparison with BR, ibrutinib, and FCR).
Indirect Treatment Comparison Design
Objectives
The aims of the sponsor-submitted analyses were to estimate the relative efficacy and safety of ibrutinib-venetoclax versus other treatment options in Canada for patients with previously untreated CLL.
Study Selection Methods
A systematic literature review was conducted to identify clinical trials evaluating first-line treatment of CLL in fludarabine-ineligible and fludarabine-eligible patients. The date of the last search update was October 27, 2022. The study selection criteria were broader than in the ITC criteria with respect to population, comparators, and outcomes. A summary of the selection criteria is presented in Table 20.
Although the sponsor reported having conducted a systematic review, no further information was provided on the number of trials that met the inclusion criteria, or their characteristics. According to the sponsor’s reports,27-31 the comparator studies used in the ITCs were selected based on the availability of IPD, had inclusion criteria that were similar to the ibrutinib-venetoclax trials (GLOW or CAPTIVATE FD cohort), or were used for regulatory approval of the drug.
Table 20: Study Selection Criteria and Methods for ITCs Submitted by the Sponsor
Characteristics | Indirect comparison | |
|---|---|---|
Population (study selection) | Adult patients receiving front-line therapy for CLL, small cell lymphoma, or small lymphocytic lymphoma | |
Intervention | Fixed-duration ibrutinib-venetoclax | |
Comparators (study selection) |
|
|
Outcomes |
| |
Study design (study selection) | RCT | |
Publication characteristics (ITC) | Phase III RCTs and 1 phase II trial (CAPTIVATE) | |
Exclusion criteria (study selection) |
| |
Databases searched | Embase, MEDLINE, Cochrane Collection Central Register of Clinical Trials (CENTRAL) | |
Selection process | Records were independently screened and assessed for eligibility by 2 reviewers | |
Data extraction process | Data were extracted by 1 reviewer and verified by a second reviewer | |
Quality assessment | Using a questionnaire tool based on National Institute for Health and Care Excellence–specified quality assessment summary tables | |
AE = adverse event, BR = bendamustine plus rituximab, CLL = chronic lymphocytic leukemia, CR = complete response, FCR = fludarabine plus cyclophosphamide plus rituximab, ITC = indirect treatment comparison, ORR = overall response rate, OS = overall survival, PFS = progression-free survival, RCT = randomized controlled trial, TTNT = time to next treatment, VO = venetoclax plus obinutuzumab.
Source: Sponsor’s Summary of Clinical Evidence.60
ITC Analysis Methods
In fludarabine-ineligible patients, 2 pairwise comparisons were conducted using MAIC methods to compare the relative efficacy and safety of ibrutinib-venetoclax with VO29 and acalabrutinib.30
Ibrutinib-Venetoclax Versus Acalabrutinib
The sponsor identified 2 RCTs that used a common comparator (chlorambucil-obinutuzumab) that could be used for the ITC comparing ibrutinib-venetoclax to acalabrutinib; however, there were differences in the recruited populations in these 2 trials and the sponsor concluded that a Bucher or network meta-analysis approach would yield biased results. IPD were available for ibrutinib-venetoclax from the GLOW trial (median follow-up 46 months) and aggregate data were available for acalabrutinib from the ELEVATE-TN study (median follow-up 46.9 months). Therefore, the sponsor opted to use anchored MAIC methods to compare the efficacy of ibrutinib-venetoclax versus acalabrutinib. Detailed methods, including outcomes, model estimation, covariates, and sensitivity analyses, are outlined in Table 21.
To conduct the MAIC, first the inclusion and exclusion criteria of the 2 trials were reviewed to determine the degree of overlap between the studies. The ELEVATE-TN study included patients who were not eligible to enrol in the GLOW study; specifically, patients with del(17p) or known TP53 mutation were included in the ELEVATE-TN study but not in the GLOW study. Given that the inclusion criteria for the ELEVATE-TN study were broader than the GLOW study, there were no criteria identified that could be used to exclude patients from the GLOW study that would otherwise have been excluded from the ELEVATE-TN study. In the next step of the analysis, patients from the GLOW study were weighted to match the mean values of relevant baseline characteristics for the comparator trial. Weighting was based on a propensity score model, in which patients in the GLOW study were weighted by the inverse odds of being in the GLOW trial rather than in the ELEVATE-TN study. The weighting used a generalized method of moments model to estimate the propensity scores, according to Signorovitch et al. (2012),51 and followed guidance from the NICE Decision Support Unit.61,62 Four variables were selected for the base-case propensity score model (age, ECOG PS, CIRS score, TP53 status) that were deemed most important by a UK clinical advisory board. The fully matched analysis included 17 variables that were reported for both studies. Region was not included in the fully matched model due to poor overlap between studies. Patients with missing covariate data were excluded from the propensity score model, under the assumption that they were missing completely at random.63 The outcomes planned for analysis were PFS, OS, and TTNT. No safety end points were analyzed. The Engauge Digitizer software was used to digitize Kaplan-Meier curves from the ELELVATE-TN study, which were then used to generate IPD (time and censoring status) for each curve based on the method by Guyot et al. (2012).64 Weighted patient data from the GLOW trial and simulated patient data from the ELEVATE-TN trial were analyzed using a weighted Cox proportional hazard model.
A time-varying Cox model was used for sensitivity analyses under a piecewise constant hazard assumption. The rationale for these analyses was that the proportional hazards assumption was violated for PFS and OS, and these violations persisted after matching. These analyses were run with a cut point of 12 months for PFS and 15.5 months for OS, based on the observed change in shape for the survival curves at these time points. A second sensitivity analysis was run that matched for all 17 variables plus region. In either case, the patient data from the GLOW trial were weighted using the propensity scores from the MAIC analysis.
Ibrutinib-Venetoclax Versus VO
The sponsor identified 2 RCTs that used a common comparator (chlorambucil-obinutuzumab) that could be used for the ITC comparing ibrutinib-venetoclax to VO. The sponsor concluded that a Bucher or network meta-analysis approach would yield biased results, as there were significant differences in the patient populations enrolled in the trials and the distribution of effect modifiers. Anchored MAIC methods were used to compare the efficacy of ibrutinib-venetoclax versus VO using IPD for ibrutinib-venetoclax from the GLOW trial (median follow-up 46 months) and aggregate data for VO from Study CLL14 (median follow-up 39.6 and 54.4 months). The methods used were largely the same as those used for the comparison with acalabrutinib; additional details are outlined in Table 21.
Based on the sponsor’s review of the inclusion and exclusion criteria of the 2 trials, patients who would not have qualified for entry in the CLL14 study were excluded from the GLOW dataset (CIRS score less than 6 and CrCl greater than 70 mL/min). Patients in the GLOW dataset were then weighted using the methods described for the acalabrutinib ITC. The selection and ranking of covariates used in the weighting were based on the availability of data and ranking by a UK clinical advisory board. The base-case analysis was matched for 4 variables (age, ECOG PS, CIRS score, TP53 status), and the fully weighted model included 5 additional variables (IGHV status, CrCl, sex, beta-2M level, and median time since initial diagnosis). The sponsor stated that the Binet stage was not included in the fully adjusted model due to poor overlap between trials on this variable. Patients with missing covariate data were excluded from the propensity score model under the assumption that they were missing completely at random.63
The outcomes planned for analysis were PFS, OS, and TTNT. No safety end points were analyzed. The primary analyses used 39.6-month median follow-up data from the CLL14 study, with a sensitivity analysis using 54.4-month data. Time-varying Cox models were run as sensitivity analyses with piecewise constant hazards for PFS (≤ 12 months, > 12 months) and OS (≤ 15.5 months, > 15.5 months), as noted for the comparison with acalabrutinib. A third sensitivity analysis was run that matched on 10 variables, including Binet stage.
Observational Studies Analysis Methods
In patients who were fludarabine-ineligible, 2 IPD analyses were conducted comparing ibrutinib-venetoclax to BR27 and to ibrutinib monotherapy.28 One IPD analysis was conducted in fludarabine-eligible patients to compare the relative efficacy and safety of ibrutinib-venetoclax with FCR.31
Ibrutinib-Venetoclax Versus BR
The sponsor had access to IPD from the ALLIANCE study, which enrolled a patient population similar to that in the GLOW study. However, the sponsor concluded that there were sufficient differences in the populations enrolled that a naive comparison, or a Bucher method analysis, would likely be biased. Instead, an analysis was conducted using IPD for ibrutinib-venetoclax from the GLOW study (median follow-up 46 months) and for BR from the ALLIANCE study (median follow-up 38 months). Common inclusion criteria from the trials were applied and patients were included in the analyses if they met the following criteria: did not have del(17p), were at least 65 years of age, had a Rai stage of I to IV, absolute neutrophil count of 1,000 cells/μL or greater, alanine aminotransferase and aspartate aminotransferase less than or equal to 3.0 times the upper limit of normal, and a platelet count of 50,000 cells/μL or greater.
The treatment propensity score weights were estimated using logistic regression for use in inverse probability treatment weighing estimation methods to compare the efficacy of ibrutinib-venetoclax versus BR. Average treatment among the treated (ATT) weighting was the primary analysis. Kaplan-Meier curves and weighted log-rank tests were also used to further present comparisons for PFS and OS. Analyses of CR and ORR were based on a weighted logistic regression model. Secondary weighting schemes included the average treatment among the controls (ATC) and the average treatment among the overlap (ATO). Patients with missing covariate data were assigned to a category “missing,” under the assumption that the reason of missingness is similar in both cohorts.63
Details of the analyses conducted, including methods, outcomes, covariates, and sensitivity analyses, are outlined in Table 21. The selection of variables for inclusion in the propensity model was based on a review of the variables adjusted for in prior reports of CLL, as well as the availability of data from both trials. Sensitivity analyses were conducted that excluded patients with missing covariates in the ALLIANCE study, and for the population that had no TP53 mutation as well as no del(17p). The sponsor reported balance diagnostic data in the form of standardized mean difference plots for the covariates included in the propensity model and in the distribution plots of propensity scores and weights.
Efficacy analyses included PFS and OS. Although safety outcomes were planned for analysis (TEAEs, grade 3 or 4 TEAEs, treatment discontinuation, and time to treatment discontinuation [TTD]), the sponsor stated that due to the limitations with the data available from the ALLIANCE trial, safety end points were not analyzed.63
Ibrutinib-Venetoclax Versus Ibrutinib
The sponsor stated that there were no RCTs comparing chlorambucil-obinutuzumab to ibrutinib, thus a network meta-analysis was not possible for this comparison. IPD were available for ibrutinib-venetoclax from the GLOW study and for ibrutinib from the ALLIANCE study. For this comparison, the same approach was taken as for the comparison of ibrutinib-venetoclax versus BR (Table 21). One additional sensitivity analysis was conducted based on pooled data from the ibrutinib and the ibrutinib plus rituximab groups in the ALLIANCE study.
Ibrutinib-Venetoclax Versus FCR
For patients who were eligible for fludarabine, IPD were available for ibrutinib-venetoclax from the FD cohort of the CAPTIVATE study (median follow-up 38.7 months and 49.7 months) and from the FCR group in the E1912 study (median follow-up of 48 months). For this comparison, the same approach was taken as for the ibrutinib-venetoclax comparisons versus BR and ibrutinib, with propensity score matching and inverse probability treatment weighting methods (ATT, ATC, and ATO weights). The outcomes analyzed included PFS, OS, ORR, CR, TEAEs, grade 3 or 4 TEAEs, treatment discontinuations, and TTD. Detailed methods, including outcomes, model estimation, covariates, and sensitivity analyses, are outlined in Table 21.
Based on the inclusion criteria that were common to both cohorts, the base-case population included adults aged 18 to 70 years with previously untreated CLL who were eligible to receive fludarabine, did not have the del(17p), and who had received at least 1 dose of the study drug. The selection of variables for inclusion in the propensity model was based on a review of the variables adjusted for in prior reports of CLL, as well as on the availability of data from both trials.
Three sensitivity analyses were planned that excluded patients with the TP53 mutation (3% and 5% in the CAPTIVATE and E1912 studies, respectively). The sponsor stated that TP53 mutation is of equal adverse prognostic value as the deletion of 17p, and therefore these patients are not suitable for FCR therapy. A second sensitivity analysis excluded patients with known TP53 mutation or missing TP53 status (18% in the E1912 study). A third analysis was limited to patients with a very good prognosis for treatment with FCR and included patients younger than 65 years with good renal function (CrCl ≥ 70 mL/min) and no TP53 mutation.
Table 21: Indirect Comparison and Observational Study Analysis Methods
Methods | Fludarabine-ineligible patients | Fludarabine-eligible patients | |||
|---|---|---|---|---|---|
Ibrutinib-venetoclax vs. acalabrutinib | Ibrutinib-venetoclax vs. VO | Ibrutinib-venetoclax vs. BR | Ibrutinib-venetoclax vs. ibrutinib | Ibrutinib-venetoclax vs. FCR | |
Analysis methods | Anchored MAIC | Anchored MAIC | Propensity score weighting, IPTW (ATT, ATC, ATO) | Propensity score weighting, IPTW (ATT, ATC, ATO) | Propensity score weighting, IPTW (ATT, ATC, ATO) |
Data sources | Ibrutinib-venetoclax: GLOW (n = 106) | Ibrutinib-venetoclax: GLOW (n = 106) VO: CLL1467 (n = 216) | Ibrutinib-venetoclax: GLOW (n = 106) BR: ALLIANCE68 (n = 183) | Ibrutinib-venetoclax: GLOW (n = 106) Ibrutinib: ALLIANCE68 (n = 182) | Ibrutinib-venetoclax: CAPTIVATE (FD cohort) (n = 136) FCR: E191269 (n = 158) |
Population (base case) | Patients with untreated CLL aged ≥ 65 years (or 18 to 64 years with a CIRS score > 6, CrCl < 70 mL/min, or CD20+ CLL)
Acalabrutinib population included patients with del(17p) or TP53 mutation, who were excluded from the Ibrutinib-venetoclax trial | Adults (≥ 18 years) with untreated CLL and
VO population included patients with del(17p) or TP53 mutation, and ECOG score of 3; these patients were excluded from the Ibrutinib-venetoclax trial | Patients with untreated CLL ≥ 65 years who received at least 1 dose of study drug and:
Platelet count ≥ 50,000 cells/μL | Same as for comparison with BR | Adults (18 to 70 years) with untreated CLL and an ECOG PS of 0 to 2 who were eligible for fludarabine treatment, had no del(17p) mutation, and received at least 1 dose of the study drug Both studies included patients with TP53 mutation and unmutated IGHV |
Outcomes |
|
| Primary:a
| Primary:
Secondary:b
| Primary:
Secondary:
|
Timing of end points (median follow-up) | GLOW: 46 months ELEVATE-TN: 46.9 months | GLOW: 46 months CLL14: 39.6 and 54.4 months | GLOW: 46 months ALLIANCE: 38 months | GLOW: 46 months ALLIANCE: 38 months | CAPTIVATE FD: 38.7 and 49.7 months (depending on outcome) E1912: 48 months |
Model estimation |
|
|
| ||
Covariates included | Base case:
Fully adjusted:
| Base case:
Fully adjusted:
| Base case:
| Same as for Ibrutinib-venetoclax vs. BR | Base case:
|
Criteria for selection of covariates | Ranking of covariates was based on feedback from a UK clinical advisory board Base case matched on 4 variables deemed to be most important to balance the trade-off between the number of characteristics matched and retaining the effective sample size | Based on covariates discussed in other ITCs and the availability of data within the studies Unable to incorporate the presence of bulky disease, complex karyotype, or CIRS score into model due to lack of data | Based on covariates discussed in other ITCs and the availability of data within the studies Unable to incorporate the presence of CIRS score, complex karyotype, and Binet stage due to lack of data | ||
Assessment of distribution or overlap in propensity score or patient weights | Effective sample size (Neff) was evaluated to assess the impact of matching and adjusting on the sample size | Distribution of weights and propensity score by treatment in unadjusted, ATT, ATC, and ATO weightings | Distribution of weights and propensity score by treatment in unadjusted, ATT, ATC, and ATO weightings | ||
Assessment of balance | Comparison of baseline characteristic summary statistics before and after matching and adjusting | SMDs were compared before and after weighing to assess degree of covariate balance | SMDs were compared before and after weighing to assess degree of covariate balance | ||
Sensitivity analyses | 1. Piecewise Cox model for time-varying analyses 2. Fully matched population, including region | 1. Extended VO arm follow-up (54.4 months) 2. Piecewise Cox model for time-varying analyses 3. Fully matched population, including Binet stage | 1. Excludes patients with missing covariate values in only 1 treatment group 2. Population of each study receiving at least 1 dose of study treatment, had no del(17p), and no TP53 mutation | 1. Excludes patients with missing covariate values in only 1 treatment 2. Population of each study receiving at least 1 dose of study treatment, had no del(17p) and no TP53 mutation 3. Pooled ibrutinib monotherapy and ibrutinib-rituximab vs. Ibrutinib-venetoclax | 1. Patients who received at least 1 dose of study treatment, had no del(17p) and no TP53 mutation, and were not missing TP53 mutation status 2. Received at least 1 dose of study treatment, had no del(17p) and no known TP53 mutation 3. Received at least 1 dose of study treatment, had no del(17p) and no TP53 mutation, aged < 65 years, and had CrCl ≥ 70 mL/min |
Subgroup analysis | NR | NR | NR | NR | NR |
ALT = alanine aminotransferase; ANC = absolute neutrophil count; AST = aspartate aminotransferase; ATC = average treatment effect in the control; ATO = average treatment effect in the overlap; ATT = average treatment effect in the treated; beta-2M = beta-2 microglobulin; BR = bendamustine plus rituximab; CI = confidence interval; CIRS = Cumulative Illness Rating Scale; CLL = chronic lymphocytic leukemia; CR = complete response; CrCl = creatinine clearance; CRR = complete response rate; Del(11q) = deletion of 11q; Del(17p) = deletion of 17p; ECOG PS = Eastern Cooperative Oncology Group Performance Status; FCR = fludarabine plus cyclophosphamide plus rituximab; FD = fixed duration; HR = hazard ratio; IPTW = inverse probability treatment weighting; ITC = indirect treatment comparison; LDH = lactate dehydrogenase; MAIC = matching-adjusted treatment comparison; NR = not reported; OR = odds ratio; ORR = overall response rate; OS = overall survival; PFS = progression-free survival; SMD = standardized mean difference; TEAE = treatment-emergent adverse event; TTD = time to treatment discontinuation; TTNT = time to next treatment; ULN = upper limit of normal; VO = venetoclax plus obinutuzumab.
aAnalysis of safety outcomes (incidence of TEAEs, grade 3 or 4 TEAEs, treatment discontinuation and TTD) were planned as secondary outcomes, but due to data limitations in the ALLIANCE study (no details provided), these outcomes could not be analyzed.
bAnalysis of safety outcomes (incidence of TEAEs, grade 3 or 4 TEAEs, and treatment discontinuation) were planned, but due to data limitations in the ALLIANCE study (no details provided), these outcomes could not be analyzed.
Sources: Sponsor’s Summary of Clinical Evidence,60 Sponsor’s ITC reports,27-31 and additional data supplied by the sponsor.63
Results for the Fludarabine-Ineligible Population
Summary of Included Studies
The sponsor did not report the results of the systematic review, thus the number of studies that met the inclusion criteria and their characteristics is unknown. Table 22 summarizes the characteristics of the trials used to inform the 4 pairwise comparisons of ibrutinib-venetoclax versus different standards of care. The 4 trials were multicentre, phase III RCTs, evaluating previously untreated patients with CLL ineligible for fludarabine-based therapy.
Differences in inclusion criteria and baseline characteristics between the ibrutinib-venetoclax study (GLOW) and the comparator studies (ALLIANCE, CLL14, ELEVATE-TN) were observed. A key difference was the inclusion of patients with del(17p) or TP53 mutations in the comparator trials (ALLIANCE, CLL14, ELEVATE-TN), whereas these patients were excluded from the ibrutinib-venetoclax study. Additionally, differences in patient age, CIRS score, and CrCl were observed, and are discussed further in the subsequent individual ITC sections. Dosing schedules, follow-up periods, and withdrawal frequencies are reported in Table 22. Of note, dosing of chlorambucil in the chlorambucil-obinutuzumab arm of the CLL14 study was given for 12 28-day cycles, in contrast to 6 cycles in both the GLOW and ELEVATE-TN studies. The sponsor stated that end point definitions were mostly similar, although some available reported time points varied in some instances.
The sponsor did not provide a quality assessment of the studies used to inform the analyses.
Table 22: Assessment of Homogeneity — Fludarabine-Ineligible Population
Characteristic | Assessment |
|---|---|
Disease severity | Ibrutinib-venetoclax vs. acalabrutinib: The ELEVATE-TN study included patients with del(17p) or known mutated TP53, whereas the GLOW study excluded these patients. The sponsor states that matching for mutated TP53 partially addressed this difference. The ELEVATE-TN study had fewer patients with advanced disease (Rai stage III to IV), more patients with an ECOG PS of 0, and a lower median CrCl than the GLOW study. Median CIRS score was substantially lower in the ELEVATE-TN study; however, the trial did not report CIRS scores for the entire study population but for only a subset of patients who were younger than 65 years at baseline. IGHV status was missing for 23% of patients in the GLOW study. According to the sponsor, when only patients with an available test result were considered, the GLOW study had a similar share of patients with unmutated IGHV status. Availability of characteristics not reported in the GLOW published source was checked and the following characteristics could also be used for matching: share of patients with CrCl < 60 mL/min, region, median time from CLL diagnosis, share of patients with ANC ≤ 1,500 μL, share of patients with hemoglobin ≤ 11 g/dL, share of patients with ≤ 100,000 μL, and share of patients with cytopenia at baseline. Ibrutinib-venetoclax vs. VO: Patients in the CLL14 study had a CIRS score > 6 or a CrCl < 70 mL/min regardless of age, whereas patients in the GLOW study who were aged ≥ 65 years were eligible regardless of CIRS score or CrCl. Differences in inclusion criteria of CIRS score and CrCl requirements were addressed by excluding patients in the GLOW study who did not meet these criteria. Inclusion of patients with del(17p) and an ECOG PS of 3 in the CLL14 study could not be directly addressed, as there were no patients with these characteristics in the GLOW study. The GLOW study excluded patients with del(17p) and known TP53 mutation, but the CLL14 study allowed inclusion of all. Mutated TP53 and del(17p) are considered equal in terms of treatment effect modification; thus, the sponsor stated that matching TP53 status partially addressed the exclusion of del(17p). The GLOW study had more patients with unmutated IGHV status when only patients with an available test result were considered (data missing for 23% of patients from the GLOW study). The CLL14 study had fewer patients with an ECOG PS of 0 and more patients with a CIRS score > 6, although median CIRS score was similar in the 2 studies. The following characteristics could not be assessed as they were not published for the CLL14 study: bulky disease, complex karyotype, Rai stage III or IV, and share of patients with a CrCl < 60 mL/min. Del(11q) was reported according to a hierarchical model and because del(11q) was not the first in the hierarchy, these data were considered unreliable for matching and not included. Availability of characteristics not reported in the GLOW published source was checked and the following additional characteristics could be used for matching: Binet stage, share of patients with CrCl < 70 mL/min, and median time from CLL diagnosis. Ibrutinib-venetoclax vs. BR and ibrutinib-venetoclax vs. ibrutinib: Compared with the ALLIANCE study, the GLOW study had a lower proportion of patients aged > 65 years, male, and with elevated LDH, but more patients with a worse ECOG PS and unmutated IGHV (although 20% of ibrutinib-venetoclax and 33% of BR patients were NR). The GLOW study excluded patients with del(17p) and known TP53 mutation, but the ALLIANCE study allowed inclusion of these patients. The ALLIANCE study excluded patients aged < 65 years, whereas in the GLOW study, patients had to be either > 65 years or meet comorbidity requirements for inclusion. The GLOW study included patients with any Rai stage, whereas the ALLIANCE study was limited to stages I to IV. |
Treatment history | All patients included in every analysis had previously untreated CLL. |
Trial eligibility criteria | Ibrutinib-venetoclax vs. acalabrutinib: Both the GLOW and ELEVATE-TN studies enrolled patients with previously untreated CLL who met the iwCLL criteria (2008 criteria for the ELEVATE-TN study) and had an ECOG PS of 0 to 2. Both trials included patients aged ≥ 65 years or 18 to 64 years with either a CIRS score > 6 or CrCl < 70 mL/min, whereas the ELEVATE-TN study included patients with neither of the former if they had a diagnosis of CD20+ CLL. The ELEVATE-TN study included patients with del(17p) and/or known TP53 mutation, whereas the GLOW study did not. Ibrutinib-venetoclax vs. VO: Both the GLOW and CLL14 studies included patients with previously untreated CLL who met iwCLL criteria (2008 criteria for the CLL14 study) and patients who were aged ≥ 18 years. The CLL14 study required patients to either have a total CIRS score > 6 or CrCl < 70 mL/min, whereas the GLOW study only required this of patients aged < 65 years. The CLL14 study included patients with an ECOG PS of 0 to 3, whereas the GLOW study included patients with scores of 0 to 2. The CLL14 study included patients with del(17p) and/or known TP53 mutation, whereas the GLOW study did not. Ibrutinib-venetoclax vs. BR and ibrutinib-venetoclax vs. ibrutinib: Both GLOW and ALLIANCE included patients with previously untreated CLL who met iwCLL criteria. The ALLIANCE study only included patients aged ≥ 65 years, whereas the GLOW study included these patients in addition to those aged 18 to 64 years. Both trials only enrolled patients with ECOG PS scores of 0 to 2. The ALLIANCE study included patients with del(17p) and/or known TP53 mutation, whereas the GLOW study did not, and the GLOW study included only patients with measurable nodal disease in at least 1 lymph node > 1.5 cm in longest diameter, whereas the ALLIANCE study did not have this criterion. |
Dosing of comparators | Ibrutinib-venetoclax vs. acalabrutinib: The comparator arm in common between the GLOW and ELEVATE-TN studies was chlorambucil-obinutuzumab. In both trials, chlorambucil-obinutuzumab dosing was administered as per outlined for the GLOW study, for six 28-day cycles. Ibrutinib-venetoclax vs. VO: The comparator arm in common for the GLOW and CLL14 studies was chlorambucil-obinutuzumab. In both trials, IV-administered obinutuzumab in 28-day cycles for 6 cycles starting with 100 mg on day 1 and 900 mg on day 2 (or 1,000 mg on day 1), 1,000 mg on day 8 and 1,000 mg on day 15 of cycle 1, and subsequently 1,000 mg on day 1 of cycles 2 through 6. In the GLOW study, chlorambucil was administered orally at 0.5 mg/kg of body weight on days 1 and 15 of each cycle until completion of 6 cycles, whereas in the CLL14 study, patients received the same dosing of chlorambucil, but for 12 cycles. Ibrutinib-venetoclax vs. BR: Rituximab 375 mg/m2 IV, then at 500 mg/m2 IV on day 1 of cycles 2 to 6. Bendamustine 90 mg/m2 IV on days 1 and 2 of each cycle (cycle 1 only, bendamustine may be given as 70 mg/m2 rather than 90 mg/m2 at investigator’s discretion). Ibrutinib-venetoclax vs. ibrutinib: Ibrutinib 420 mg oral daily until progression or unacceptable toxicity. |
Definitions of end points | PFS: All trials defined as time from randomization or study entry to the time of disease progression or death from any cause, whichever occurs first (regardless of subsequent therapy in the GLOW and ALLIANCE studies). There were differences in PFS assessment methods across studies. The GLOW study mandated MRI or CT scans at most study visits, whereas in the comparator studies, scans were conducted if there was a clinical suspicion of progression and there were fewer mandated scans at set time points. It was unclear if analyses of PFS were based on IRC-confirmed events or investigator-assessed events for each study. Some studies reported both IRC and investigator-assessed events (IRC PFS was the primary outcome in the GLOW and ELEVATE-TN studies, investigator-confirmed events were the primary outcome in the CLL14 study, and the PFS end point not stated for the ALLIANCE study) ORR: In all trials reporting this outcome, the definition was similar to the percentage of patients who achieved any response to treatment. CR: The GLOW study defined CR as the percentage of patients who achieved a CR or CRi. The ALLIANCE study defined it as the absence of lymphadenopathy > 1.5 cm, hepatomegaly, splenomegaly, clonal B-cells in blood, normal complete blood count, and normal bone marrow aspirate and biopsy. OS: All trials defined as time from randomization until death from any cause, except the ALLIANCE study, which counted the time from study registration. TTNT: Measured from the date of randomization to the start date of any subsequent antileukemic therapy in the GLOW and CLL14 studies. |
Timing of end point evaluation | Ibrutinib-venetoclax (GLOW study): 46 months Acalabrutinib (ELEVATE-TN study): 46.9 months VO (CLL14 study): 39.6 and 54.4 months BR and ibrutinib (ALLIANCE study): 38 months |
Withdrawal frequency | GLOW study: 22.6% of ibrutinib-venetoclax and 4.8% of chlorambucil-obinutuzumab group had discontinued study treatment at 27.7-month follow-up ALLIANCE study: 47% of both the BR and ibrutinib groups were off treatment at 55-month follow-up ELEVATE-TN study: 31% discontinued acalabrutinib treatment at 46.9-month follow-up CLL14 study: 25% of the chlorambucil-obinutuzumab group and 22% of the VO group stopped treatment at 39.6-month follow-up |
Clinical trial setting | All trials were multicentre and multinational; the ALLIANCE trial was the only one restricted to just 1 continent (North America). Patients were treated in clinical settings. |
Study design | GLOW, ALLIANCE, CLL14, ELEVATE-TN were phase III, randomized, controlled, double-blind studies. |
ANC = absolute neutrophil count; BR = bendamustine plus rituximab; CIRS = Cumulative Illness Rating Scale; CLL = chronic lymphocytic leukemia; CR = complete response; CrCl = creatinine clearance; CRi = complete response with incomplete bone marrow recovery; del(11q) = deletion of 11q; del(17p) = deletion of 17p; ECOG PS = Eastern Cooperative Oncology Group Performance Status; IRC = independent review committee; iwCLL = International Workshop on CLL, NR = not reported; ORR = overall response rate; OS = overall survival; PFS = progression-free survival; TTNT = time to next treatment; VO = venetoclax plus obinutuzumab.
Sources: GLOW trial: Clinicaltrials.gov (2018),70 Kater et al. (2022);45 ALLIANCE trial: Clinicaltrials.gov (2013),71 Woyach et al. (2018);68 ELEVATE-TN trial: Clinicaltrials.gov (2015),72 Sharman et al. (2019),65 Sharman et al. (2020);66 CLL17 trial: Fischer et al. (2019),67 sponsor’s ITC reports,27-31 and additional data supplied by the sponsor.63 (Note: Details included in the table are from the sponsor’s Summary of Clinical Evidence.)
Ibrutinib-Venetoclax Versus Acalabrutinib
There were differences in the inclusion and exclusion criteria of the 2 trials used in the ITC comparing ibrutinib-venetoclax to acalabrutinib, with the ELEVATE-TN study including patients who would have been ineligible to enter the GLOW study (Table 22). The key difference was that the ELEVATE-TN study included patients with del(17p) or TP53 mutation, whereas these patients were excluded from the GLOW study. In the GLOW study, TP53 testing was not mandatory, and it was later known that 9 patients enrolled had the TP53 mutation (7 and 2 patients in the ibrutinib-venetoclax and chlorambucil-obinutuzumab groups, respectively). In contrast, 41 patients (12%) in the ELEVATE-TN study had TP53 mutation and 32 patients (9%) had del(17p). Another key difference was in CIRS reporting. In the ELEVATE-TN study, this variable was missing for 35% of patients, as it was only collected for patients who were younger than 65 years. Dosing of chlorambucil-obinutuzumab was the same in both studies, and the follow-up time was similar. PFS and OS outcomes were defined similarly in both studies, although there were differences in the assessment of progression events. More rigorous detection methods were applied in the GLOW study, where PFS was assessed using MRI or CT imaging on most disease evaluation visits, regardless of suspicion of disease progression. In the ELEVATE-TN study, imaging was only performed if the investigator suspected progression. The sponsor stated that it was not possible to analyze TTNT because this end point was not reported for the 46.9-month follow-up period in the ELEVATE-TN study.
A summary of patient characteristics in the GLOW and ELEVATE-TN studies before and after weighting is shown in Table 23. In the base-case analysis, patients in the ibrutinib-venetoclax group were balanced to match the acalabrutinib group based on age, ECOG PS, CIRS score, and TP53 status, and the fully adjusted model added 12 more variables, including IGHV status, CrCl, del(11q) status, Rai stage, bulky disease, beta-2M level, and median time from diagnosis. The effective sample size was reduced from 211 to 127 patients in the base-case analysis, and from 154 to 69 patients in the full model, which represent 60% and 45% of the complete data cohort, respectively. The fully matched analysis had a reduced sample size, as any patients with missing covariate data were excluded, and also a low effective sample size, suggesting poor overlap in the populations. Region was not included in the fully matched model due to the poor overlap between the 2 studies on this variable (Neff [effective sample size] = 27). According to the sponsor, the selection of variables for the base case was a trade-off between the number of matching variables and retaining the effective sample size.
After weighting was applied to patients from the GLOW study for the base-case analysis, the proportion of patients in each age category, ECOG PS, mutated TP53 status, and median CIRS score was similar for those from the GLOW study compared with the ELEVATE-TN trial (Table 23). However, differences remained for other confounders, with the GLOW study showing more patients than the ELEVATE-TN study with CrCl less than 60 mL/min2 (33% versus 28%), hemoglobin 11 g/dL or less (48% versus 38%), and cytopenia at baseline (59% versus 48%), and fewer patients with Rai stage 0 to II disease (44% versus 53%) and bulky disease (59% versus 68%).
Table 23: Baseline Characteristics Before and After Matching — Ibrutinib-Venetoclax Versus Acalabrutinib MAIC
Characteristic | ELEVATE-TN N = 535 | GLOW N = 211 | GLOW (base case)a N = 211 Neff = 127 | GLOW (full matching)b N = 154 Neff = 69 |
|---|---|---|---|---|
Age (years), median | 70.3 | 71 | 70 | 70 |
Age category, % | ||||
< 65 years | 17 | 13 | 17 | 17 |
65 to < 75 years | 54 | 53 | 54 | 54 |
≥ 75 years | 29 | 34 | 29 | 29 |
ECOG PS score, % | ||||
0 or 1 | 93 | 88 | 93 | 93 |
2 | 7 | 12 | 7 | 7 |
CIRS-G score, median | 5.8 | 8 | 6 | 5 |
Mutated TP53, % | 12 | 4 | 12 | 12 |
Unmutated IGHV, % | 64 | 67 | 66 | 64 |
CrCl (mL/min), median | 73.8 | 64.8 | 66.5 | 72.3 |
CrCl < 60 mL/min, % | 28 | 38 | 33 | 28 |
Female, % | 39 | 42 | 41 | 39 |
del(11q), % | 18 | 18 | 19 | 18 |
Rai stage, % | ||||
0 to II | 53 | 45 | 44 | 53 |
III | 26 | 30 | 31 | 26 |
IV | 21 | 24 | 25 | 21 |
Bulky disease < 5 cm, % | 68 | 62 | 59 | 68 |
Time from initial diagnosis (months), median | 28.5 | 35.5 | 33.6 | 28.3 |
ANC ≤ 1,500 μL, % | 4 | 7 | 8 | 4 |
Hgb ≤ 11 g/dL, % | 38 | 46 | 48 | 38 |
Platelet count ≤ 100,000/μL, % | 21 | 27 | 27 | 21 |
Beta-2M > 3.5 mg/L, % | 76 | 72 | 71 | 76 |
Cytopenia at baseline, % | 48 | 58 | 59 | 48 |
Region: Europe, % | 52 | 90 | 90 | 91 |
Mutated del(17p), % | 9 | 0 | NA | NA |
ANC = absolute neutrophil count; beta-2M = beta-2 microglobulin; CIRS-G = Cumulative Illness Rating Scale-Geriatric; CrCl = creatinine clearance; del(11q) = deletion of 11q; del(17p) = deletion of 17p; ECOG PS = Eastern Cooperative Oncology Group Performance Status; Hgb = hemoglobin; MAIC = matching-adjusted treatment comparison; NA = not applicable; Neff = effective sample size.
aIn the base case, patients were matched on age, ECOG PS, CIRS score, and TP53 mutation status.
bIn the full analysis, patients were matched on all variables except region.
Source: Sponsor’s ITC report ibrutinib-venetoclax vs. acalabrutinib.30
Ibrutinib-Venetoclax Versus VO
For the analysis of ibrutinib-venetoclax versus VO, the CLL14 study enrolled patients with a higher comorbidity burden, who had either a CIRS score greater than 6 points or impaired renal function (CrCl < 70 mL/min). In contrast, only patients younger than 65 years were required to have a CIRS score greater than 6 or impaired renal function in the GLOW study. Patients 65 years and older were eligible regardless of CIRS score. The CLL14 study enrolled patients with an ECOG PS score of 0 to 3, whereas only patients with a score of 0 to 2 were eligible in the GLOW study. The CLL14 study included patients with any cytogenetic profile, including those with del(17p) or TP53 mutation; these patients were excluded from the GLOW trial.
Another notable difference between studies was the dosing of chlorambucil-obinutuzumab. In the GLOW study, chlorambucil-obinutuzumab was administered as a combination of the 2 drugs for a maximum of 6 cycles, whereas in the CLL14 study, chlorambucil-obinutuzumab combination therapy was administered for up to 6 cycles, then chlorambucil monotherapy was continued for another 6 cycles. There was also a discrepancy in the follow-up duration of the 2 trials. The most mature data for the GLOW study had a median follow-up of 46 months. For the primary analysis, 39.6-month data from the CLL14 study were used, and for the secondary analysis, 54.4-month follow-up data were used. Regarding outcome definitions, the issues present for PFS assessment were the same as for the comparison with acalabrutinib.
After patients who did not meet the inclusion criteria for the CLL14 study were excluded (CIRS score less than 6 and CrCl greater than 70 mL/min), 174 of 211 patients from the GLOW study were included in the analysis. In the base-case model, patients in the ibrutinib-venetoclax group were balanced to match the VO group based on age, ECOG PS, CIRS score, and TP53 status, and the fully adjusted model added IGHV status, CrCl, sex, beta-2M level, and median time from diagnosis. Differences in some study inclusion criteria could not be addressed in the analysis because patients with an ECOG PS score of 3 or del(17p) or known TP53 mutation were not enrolled in the GLOW study. It was also not possible to control for patients with bulky disease, complex karyotype, Rai stage III or IV, or share of patients with CrCl less than 60 mL/min, as published data were not available for these parameters for the CLL14 study. The sponsor reported that del11q data for the CLL14 study were unreliable for matching.
A summary of the baseline patient characteristics in the GLOW and CLL14 studies is shown before and after weighting in Table 24. The effective sample size was reduced from 174 to 118 patients in the base case, and from 136 to 70 patients in the full model, which represent 68% and 51% of the eligible sample, respectively. After weighting, the overall base-case population from the GLOW study was similar to the aggregate CLL14 population in terms of age, ECOG PS score, CIRS score, and TP53 status; however, differences were present in other variables. Specifically, the GLOW population had a higher proportion of patients with unmutated IGHV (72% versus 61%), CrCl < 70 mL/min2 (74% versus 58%), beta-2M > 3.5 mg/L (71% versus 61%), and median time from initial diagnosis (36.6 months versus 30.2 months).
Table 24: Baseline Characteristics Before and After Matching — Ibrutinib-Venetoclax Versus VO MAIC
Characteristic | CLL14 N = 432 | GLOWa N = 174 | GLOW (base case)b N = 174 Neff = 118 | GLOW (full matching)c N = 136 Neff = 70 |
|---|---|---|---|---|
Age (years), median | 71.5 | 72 | 72 | 72 |
Age ≥ 75 years, % | 35 | 38 | 35 | 35 |
ECOG PS score, % | ||||
0 | 45 | 32 | 45 | 45 |
1 | 43 | 56 | 43 | 43 |
2 or 3 | 12 | 13 | 12 | 12 |
CIRS score, median | 8 | 9 | 8 | 8 |
CIRS score > 6, % | 84 | 78 | 84 | 84 |
Mutated TP53, % | 10 | 5 | 10 | 10 |
Unmutated IGHV, % | 61 | 70 | 72 | 61 |
CrCl (mL/min), median | 66.4 | 61.1 | 61.5 | 66.5 |
CrCl < 70 mL/min, % | 58 | 73 | 74 | 58 |
Male, % | 67 | 59 | 58 | 67 |
Beta-2M > 3.5 mg/L, % | 61 | 74 | 71 | 61 |
Time from initial diagnosis (months), median | 30.2 | 35.3 | 36.6 | 30.5 |
Binet stage, % | ||||
A | 21 | 7 | 7 | 7 |
B | 36 | 49 | 49 | 50 |
C | 43 | 43 | 44 | 43 |
Mutated del(17p), % | 8 | 0 | NA | NA |
beta-2M = beta-2 microglobulin; CIRS = Cumulative Illness Rating Scale; CrCl = creatinine clearance; del(17p) = deletion of 17p; ECOG PS = Eastern Cooperative Oncology Group Performance Status; MAIC = matching-adjusted treatment comparison; NA = not applicable; Neff = effective sample size; VO = venetoclax plus obinutuzumab.
aAfter common study criteria were applied and patients with CIRS score less than 6 and CrCl greater than 70 mL/min were excluded from GLOW population.
bIn the base case, patients were matched on age, ECOG PS, CIRS and TP53 mutation status.
cIn the full analysis, patients were matched on all variable except Binet stage.
Source: Sponsor’s ITC report ibrutinib-venetoclax vs. VO.29
Ibrutinib-Venetoclax Versus BR
The sponsor identified differences in the patients enrolled in the GLOW and ALLIANCE trials (Table 22) and issues with missing covariate data that were required to calculate the propensity scores. After excluding patients with del(17p) mutation and patients younger than 65 years, and applying other common inclusion or exclusion criteria, 73 patients remained for GLOW and 153 for ALLIANCE. A summary of the unadjusted baseline patient characteristics is shown in Table 25. For the base-case analyses, patients from the ALLIANCE study were weighed on ECOG PS, del(11q) status, presence of cytopenia, sex, IGHV status, Rai stage, beta-2M, age, CrCl, LDH, and TP53 mutation status to balance these characteristics to patients in the GLOW study. The sponsor states that the ATT, ATC, and ATO weighting generally achieved balance in the covariates, except for some variables for which missing covariate values in 1 treatment group only were drivers of imbalance (i.e., beta-2M and TP53 mutation status). Beta-2M and TP53 mutation status were missing for 2% and 4% of patients from the BR group, respectively, and from no patients from the ibrutinib-venetoclax group. IGHV status was missing for 21% and 32% of patients in the ibrutinib-venetoclax and BR groups, respectively. The distribution of propensity scores showed some low values (i.e., near 0), and some extreme weights.
PFS, OS, and ORR outcomes were defined similarly in both studies, although there were differences in the assessment of progression events, but the GLOW trial used more rigorous detection methods for progression than the ALLIANCE study. CR was defined as the percentage of patients who achieved a CR or CRi in the GLOW study, and required the absence of lymphadenopathy greater than 1.5 cm, hepatomegaly, splenomegaly, clonal B-cells in blood, normal complete blood count, and normal BM aspirate and biopsy in the ALLIANCE study. There were differences in the follow-up durations of the 2 datasets, with patients who received ibrutinib-venetoclax followed for median of 46 months compared with 38 months for patients who received BR.
Ibrutinib-Venetoclax Versus Ibrutinib
The comparison between ibrutinib-venetoclax and ibrutinib monotherapy was also based on the GLOW and ALLIANCE studies and used the same approach as described for ibrutinib-venetoclax versus BR. The analysis included 73 patients in the GLOW study and 163 patients from the ibrutinib treatment group in the ALLIANCE study. Of the covariates used in the propensity score model, missing data were also an issue, with IGHV status missing for 21% and 32% of patients in the ibrutinib-venetoclax and BR groups, respectively. Beta-2M and TP53 mutation status were missing for 4% and 7% of patients in the ALLIANCE study and no patients in the GLOW study (Table 25). The sponsor states that the models generally achieved good balance in the covariates in the ATT and ATO weights, but with the ATC weights there were some imbalances related to beta-2M and TP53 mutation status due to missing data. The distribution of propensity scores showed some low values (i.e., near 0), and some extreme weights.
Table 25: Fludarabine-Ineligible Patients — Baseline Characteristics for ITC Ibrutinib-Venetoclax Versus BR and Ibrutinib (Base-Case Population)
Characteristic | GLOW All treated, non-del(17p) | ALLIANCE All treated, non-del(17p) | |
|---|---|---|---|
Ibrutinib-venetoclax (N = 73) | BR (N = 155) | Ibrutinib (N = 163) | |
Age category (years), % | |||
65 to < 70 | 26 | 44 | 41 |
70 to < 75 | 40 | 30 | 34 |
≥ 75 | 34 | 26 | 25 |
Male, % | 49 | 67 | 68 |
ECOG PS, % | |||
0 | 34 | 55.5 | 48.5 |
≥ 1 | 66 | 44.5 | 51.5 |
Del(11q), % | |||
Yes | 20.5 | 20 | 20.2 |
No | 79.5 | 79 | 79.1 |
Missing | 0 | 1 | 0.6 |
Cytopenia at baseline, % | 58 | 61 | 59 |
IGHV status, % | |||
Unmutated | 49 | 38 | 43 |
Mutated | 30 | 30 | 25 |
Missing | 21 | 32 | 32 |
TP53 mutation, % | |||
Yes | 5.5 | 5 | 7 |
No | 94.5 | 91 | 86 |
Missing | 0 | 4 | 7 |
Rai stage, % | |||
0 to II | 44 | 50 | 49 |
III to IV | 56 | 50 | 51 |
Beta-2M, % | |||
≤ 3.5 mg/L | 29 | 33 | 20 |
> 3.5 mg/L | 71 | 65 | 76 |
Missing | 0 | 2 | 4 |
Creatinine clearance, % | |||
< 60 mL/min | 37.0 | 32 | 33 |
60 to < 70 mL/min | 20.5 | 23 | 18 |
≥ 70 mL/min | 42.5 | 45 | 49 |
Lactate dehydrogenase, % | |||
Normal | 64 | 59 | 60 |
Elevated | 36 | 41 | 40 |
Beta-2M = beta-2 microglobulin; BR = bendamustine plus rituximab; del(11q) = deletion of 11q; del(17p) = deletion of 17p; ECOG PS = Eastern Cooperative Oncology Group Performance Status; ITC = indirect treatment comparison.
Sources: Sponsor’s ITC report ibrutinib-venetoclax vs. BR,27 Sponsor’s ITC report ibrutinib-venetoclax vs. ibrutinib.28
Efficacy
For fludarabine-ineligible patients, each of the 4 separate pairwise comparisons consisted of GLOW (ibrutinib-venetoclax versus chlorambucil-obinutuzumab) compared with ALLIANCE (BR versus ibrutinib versus ibrutinib-rituximab), CLL14 (VO versus chlorambucil-obinutuzumab), or ELEVATE-TN (acalabrutinib versus chlorambucil-obinutuzumab versus acalabrutinib plus obinutuzumab). The evidence networks are summarized in Figure 9. This diagram depicts comparisons in fludarabine-ineligible patients as part of a connected network, but this is not the case, as the analyses were conducted as independent pairwise comparisons.
Figure 9: ITC Networks for Fludarabine-Ineligible Patients
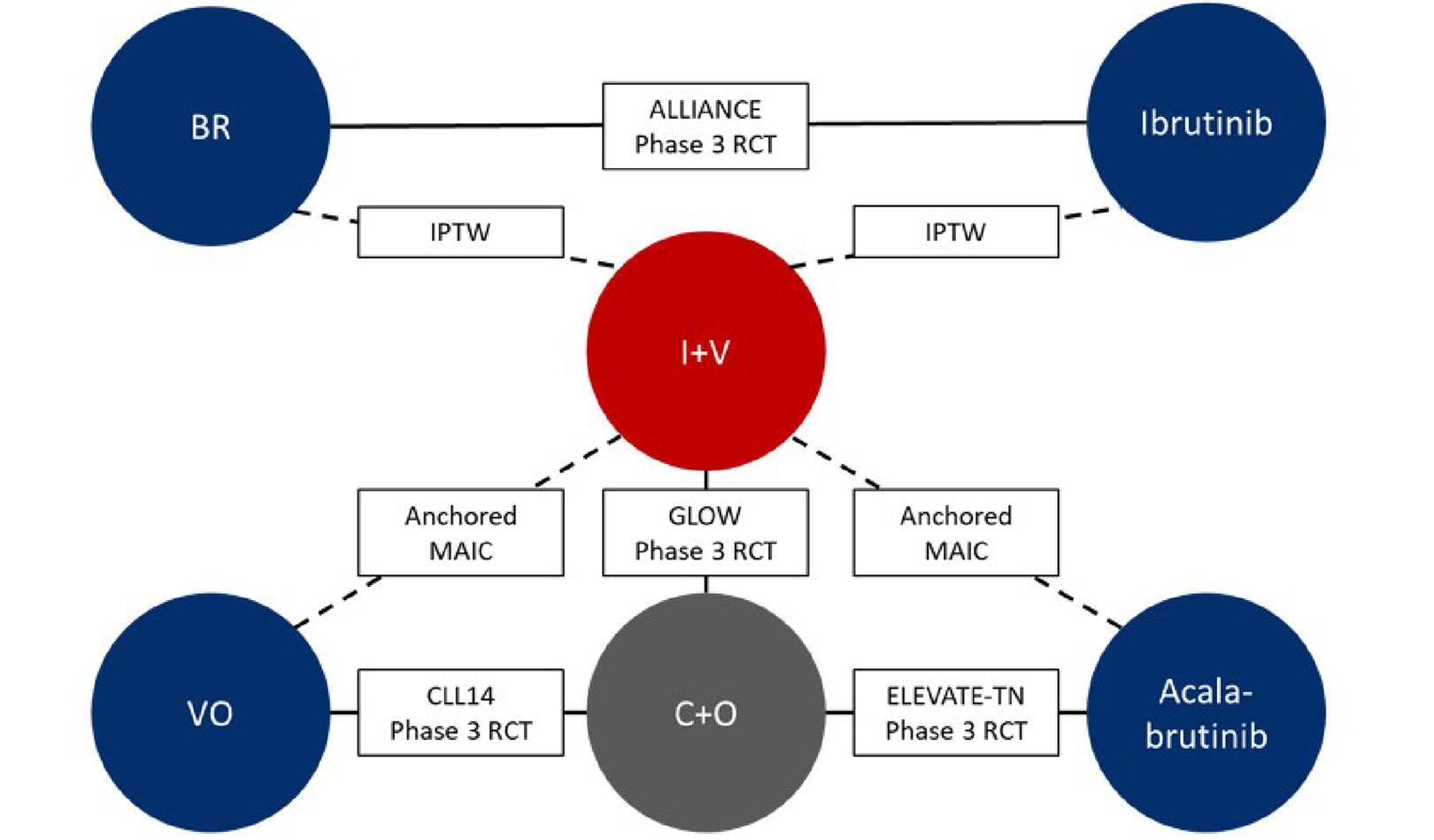
BR = bendamustine plus rituximab; C+O = chlorambucil plus obinutuzumab; IPD = individual patient data; IPTW = inverse probability treatment weighting; ITC = indirect treatment comparison; MAIC = matching-adjusted indirect comparison; RCT = randomized controlled trial; V+O = venetoclax plus obinutuzumab.
Note: Solid lines represent direct comparisons; dashed lines represent indirect comparisons.
Source: Sponsor’s Summary of Clinical Evidence.60
Ibrutinib-Venetoclax Versus Acalabrutinib
The estimated HR for PFS was |||| |||| ||| |||| || ||||| | | ||||| in the base-case model (Neff = 127 patients) and |||| |||| ||| |||| || ||||| | | ||||) in the fully adjusted model (Neff = 69) comparing ibrutinib-venetoclax to acalabrutinib (Table 26). ||| |||||||||||| ||||||| |||||||||| ||| |||||||| || |||| |||||||| ||||| |||| ||||||| ||| ||||| ||| |||||||||. For the base-case analysis up to 12 months, the HR for PFS was |||| |||| || |||| || ||||| | | ||||), and after 12 months was |||| |||| || |||| || ||||| | | |||||| ||| ||| || ||| ||| |||||||| ||||||| ||| ||||| The results for PFS and OS are shown in Table 26.
The sensitivity analysis that matched for ||| variables, including region, had an effective sample size of ||| patients and was deemed unreliable. Thus, these data have not been summarized in this report.
Table 26: Summary of PFS and OS in Fludarabine-Ineligible Patients for Ibrutinib-Venetoclax Versus Acalabrutinib
Outcome and analysis | Ibrutinib-venetoclax vs. acalabrutiniba HR (95% CI) | P value |
|---|---|---|
PFS | ||
Base caseb | |||| ||||||||||| | |||||| |
Fully matched populationc | |||| ||||||||||| | |||||| |
PFS time-varying sensitivity analysis | ||
Base case, ≤ 12 months | |||| ||||||||||| | |||||| |
Base case, > 12 months | |||| ||||||||||| | |||||| |
Fully matched population, ≤ 12 months | |||| |||||||||||| | |||||| |
Fully matched population, > 12 months | |||| ||||||||||| | |||||| |
OS base case | ||
Base caseb | |||| ||||||||||| | |||||| |
Fully matched populationc | |||| ||||||||||| | |||||| |
OS sensitivity analysesa | ||
Base case, ≤ 15.5 months | |||| |||||||||||| | |||||| |
Base case, > 15.5 months | |||| ||||||||||| | |||||| |
Fully matched population,b ≤ 15.5 months | |||| |||||||||||| | |||||| |
Fully matched population,b > 15.5 months | |||| ||||||||||| | |||||| |
CI = confidence interval; HR = hazard ratio; OS = overall survival; PFS = progression-free survival.
aWeighted Cox proportional hazards model for primary analyses; piecewise Cox model for time-varying analyses.
bMatched on age, ECOG PS, CIRS and TP53 status (N = 211, Neff = 127).
cMatched on 16 variables, excluding region (N = 154, Neff = 69).
Source: Sponsor’s ITC report for ibrutinib-venetoclax vs. acalabrutinib.30
Ibrutinib-Venetoclax Versus VO
For the comparison of ibrutinib-venetoclax versus VO, the MAIC for PFS reported a HR of |||| |||| ||| |||| || ||||| | | ||||) in the base-case model (N = 174, Neff = 118). The results were generally similar in the fully matched population (Table 27). |||||||||||| |||||||| |||| ||||||||| || ||| |||||||||||| ||||||| |||||||||| ||| ||| ||||. For the first 12 months of follow-up, the base-case PFS HR was |||| |||| ||| |||| || ||||| | | ||||), and after 12 months the HR was |||| |||| ||| |||| || ||||| | | |||||) for ibrutinib-venetoclax versus VO.
The base-case analysis of OS showed a 95% CI that overlapped the null, and time-varying analyses lacked precision, with wide CIs (Table 27). The TTNT favoured ibrutinib-venetoclax versus VO (base case || |||| |||| ||| |||| || ||||| | | |||||). Results were similar in the fully matched population.
Table 27: Summary of PFS, OS, and TNTT in Fludarabine-Ineligible Patients for Ibrutinib-Venetoclax Versus VO
Outcome and analysis | Ibrutinib-venetoclax vs. VOa HR (95% CI) | P value |
|---|---|---|
PFS (ibrutinib-venetoclax 46 months vs. VO 39.6 months follow-up) | ||
Base caseb | |||| ||||||||||| | |||||| |
Fully matched populationc | |||| ||||||||||| | |||||| |
PFS sensitivity analysis 1d (ibrutinib-venetoclax 46 months vs. VO 54.4 months follow-up) | ||
Base caseb | |||| ||||||||||| | |||||| |
Fully matched populationc | |||| ||||||||||| | |||||| |
PFS sensitivity analysis 2e (ibrutinib-venetoclax 46 months vs. VO 39.6 months follow-up) | ||
Base case,b ≤ 12 months | |||| ||||||||||| | |||||| |
Base case,b > 12 months | |||| ||||||||||| | |||||| |
Fully matched population,c ≤ 12 months | |||| ||||||||||| | |||||| |
Fully matched population,c > 12 months | |||| ||||||||||| | |||||| |
OS base case (ibrutinib-venetoclax 46 months vs. VO 39.6 months follow-up) | ||
Base caseb | |||| ||||||||||| | |||||| |
Fully matched populationc | |||| ||||||||||| | |||||| |
OS sensitivity analysis 1d (ibrutinib-venetoclax 46 months vs. VO 54.4 months follow-up) | ||
Base caseb | |||| ||||||||||| | |||||| |
Fully matched populationc | |||| ||||||||||| | |||||| |
OS sensitivity analysis 2e (ibrutinib-venetoclax 46 months vs. VO 39.6 months follow-up) | ||
Base case,b ≤ 15.5 months | |||| |||||||||||| | |||||| |
Base case,b > 15.5 months | |||| ||||||||||| | |||||| |
Fully matched population,c ≤ 15.5 months | |||| |||||||||||| | |||||| |
Fully matched population,c > 15.5 months | |||| ||||||||||| | |||||| |
TTNT (ibrutinib-venetoclax 46 months vs. VO 39.6 months follow-up) | ||
Base caseb | |||| ||||||||||| | |||||| |
Fully matched populationc | |||| ||||||||||| | |||||| |
TTNT sensitivity analysis 1d (ibrutinib-venetoclax 46 months vs. VO 54.4 months follow-up) | ||
Base caseb | |||| ||||||||||| | |||||| |
Fully matched populationc | |||| ||||||||||| | |||||| |
CI = confidence interval, HR = hazard ratio, OS = overall survival, PFS = progression-free survival, TTNT = time to next treatment, VO = venetoclax plus obinutuzumab.
aWeighted Cox proportional hazards model for primary analyses; piecewise Cox model for time-varying analyses.
bMatched on age, ECOG PS, CIRS score, and TP53 status (N = 174, Neff = 118).
cMatched on 9 variables, excluding Binet stage (N = 136, Neff = 70).
dExtended VO arm follow-up is a median of 54.4 months.
eTime-varying MAIC.
Source: Sponsor’s ITC report for ibrutinib-venetoclax vs. VO.29
Ibrutinib-Venetoclax Versus BR
For fludarabine-ineligible patients, PFS results were generally consistent across all propensity score weighting methods and sensitivity analyses. The base-case (ATT) analysis of PFS reported an HR of |||| |||| ||| |||| || ||||| | | ||||||) for ibrutinib-venetoclax versus BR (Table 28). For the analysis of OS, ||||| of patients died in the ibrutinib-venetoclax group compared with ||||| in the BR group (| | ||||) for the base-case (ATT) analysis. ||| || ||| ||| || ||| || ||| ||| |||||||| || ||| |||||||.
Regarding ORR, most patients responded to treatments. The percentage of patients who responded ranged from ||| || ||| (ibrutinib-venetoclax) compared to ||| || ||| (BR) across the base-case and sensitivity analyses. CR favoured ibrutinib-venetoclax versus BR (|| |||| ||| ||| |||| || ||||| | | |||||) in the base-case (ATT) and most sensitivity analyses (Table 28).
The analyses based on ATC and ATO weights generally showed results that were similar to the ATT analysis (Appendix 1, Table 36).
Table 28: Summary of PFS, OS, and CR in Fludarabine-Ineligible Patients for Ibrutinib-Venetoclax Versus BR
Outcome and analysisa | Ibrutinib-venetoclax vs. BR | P value | |
|---|---|---|---|
Ibrutinib-venetoclax | BR | ||
PFS base case,a HR (95% CI) ibrutinib-venetoclax vs. BRb | |||
Unadjusted | |||| ||||||||||| | |||||| | |
ATT | |||| ||||||||||| | |||||| | |
PFS sensitivity analysis (ATT), HR (95% CI) ibrutinib-venetoclax vs. BRb | |||
Sensitivity analysis 1c | |||| ||||||||||| | |||||| | |
Sensitivity analysis 2d | |||| ||||||||||| | |||||| | |
OS base case,a % patients with evente | |||
Unadjusted | ||||| | ||||| | |||||| |
ATT | ||||| | ||||| | |||||| |
OS sensitivity analysis (ATT), % patients with evente | |||
Sensitivity analysis 1c | ||||| | ||||| | |||||| |
Sensitivity analysis 2d | |||||| | ||||| | |||||| |
CR base case,a OR (95% CI) ibrutinib-venetoclax vs .BRf | |||
Unadjusted | |||| ||||||||||| | |||||| | |
ATT | |||| ||||||||||| | |||||| | |
CR sensitivity analysis (ATT), OR (95% CI) ibrutinib-venetoclax vs .BRf | |||
Sensitivity analysis 1c | |||| ||||||||||| | |||||| | |
Sensitivity analysis 2d | |||| ||||||||||| | |||||| | |
ATT = average treatment effect in the treated; BR = bendamustine plus rituximab; CI = confidence interval; CR = complete response; HR = hazard ratio; OR = odds ratio; OS = overall survival; PFS = progression-free survival.
aBase-case population (ibrutinib-venetoclax: N = 73; BR: N = 155).
bHR and 95% CI based on weighted Cox proportional hazard model; P value based on weighted log-rank test.
cPopulation that excludes patients with missing covariate values in only 1 treatment group (ibrutinib-venetoclax: N = 73; BR: N = 144).
dPopulation of each study receiving at least 1 dose of the study treatment, with no del(17p), and with no TP53 mutation (ibrutinib-venetoclax: N = 69; BR: N = 147) .
eP value based on weighted log-rank test. HR and 95% CI were not reported in the sponsor’s ITC report.
fOR and 95% CI based on weighted logistic regression model.
Source: Sponsor’s ITC report for ibrutinib-venetoclax vs. BR.27
Ibrutinib-Venetoclax Versus Ibrutinib
In the base-case (ATT) analysis, ||||| ||| ||||| of patients in the ibrutinib-venetoclax and ibrutinib groups, respectively, reported a progression event || | ||||) (Table 29). For OS, |||| |||||| |||| in the ibrutinib-venetoclax and ibrutinib groups reported an event (| | ||||, base-case ATT). The findings for PFS and OS were similar across all analyses, with the weighted log-rank test suggesting that no difference was detected, ||||||| ||| ||||||||||| || ||| ||| || |||| ||| ||||||||, thus the variance for the comparative effects is not known.
Overall response was reported by ||| || ||| of patients in the ibrutinib-venetoclax group and by ||| || ||| of patients in the ibrutinib group. In the ibrutinib-venetoclax group, ||| || ||| of patients achieved a CR, compared with || || ||| of patients in the ibrutinib group (with similar results across analyses). The estimated OR for CR was ||||| |||| ||| |||| || |||||| ||||||||) for the base-case ATT analysis (Table 30).
The analyses based on ATC and ATO weights generally showed results that were similar to the ATT analysis (Appendix 1, Table 36).
Table 29: Summary of PFS and OS in Fludarabine-Ineligible Patients for Ibrutinib-Venetoclax Versus Ibrutinib
Outcome and analysis | Patients with event, % | P valuea | |
|---|---|---|---|
Ibrutinib-venetoclax | Ibrutinib | ||
PFS base caseb | |||
Unadjusted | |||||| | |||||| | |||||| |
ATT | |||||| | |||||| | |||||| |
PFS sensitivity analysis (ATT) | |||
Sensitivity analysis 1c | |||||| | |||||| | |||||| |
Sensitivity analysis 2d | |||||| | |||||| | |||||| |
OS base caseb | |||
Unadjusted | ||||| | |||||| | |||||| |
ATT | ||||| | ||||| | |||||| |
OS sensitivity analysis (ATT)a | |||
Sensitivity analysis 1c | ||||| | |||||| | |||||| |
Sensitivity analysis 2d | |||||| | |||||| | |||||| |
ATT = average treatment effect in the treated; OS = overall survival; PFS = progression-free survival.
aWeighted log-rank test. HR and 95% CI were not reported in the sponsor’s ITC report.
bBase-case population (ibrutinib-venetoclax: N = 73; ibrutinib: N = 163).
cPopulation that excludes patients with missing covariate values in only 1 treatment group (ibrutinib-venetoclax: N = 73; ibrutinib: N = 146).
dPopulation of each study receiving at least 1 dose of study treatment, with no del(17p), and with no TP53 mutation (ibrutinib-venetoclax: N = 69; ibrutinib: N = 152).
Source: Sponsor’s ITC report for ibrutinib-venetoclax vs. Ibrutinib.28
Table 30: Summary of CR in Fludarabine-Ineligible Patients for Ibrutinib-Venetoclax Versus Ibrutinib
Outcome and analysis | Ibrutinib-venetoclax vs. ibrutiniba OR (95% CI) | P value |
|---|---|---|
CR base caseb | ||
Unadjusted | |||| |||||||||||| | ||||||| |
ATT | ||||| |||||||||||| | ||||||| |
CR sensitivity analysis (ATT) | ||
Sensitivity analysis 1c | ||||| |||||||||||| | ||||||| |
Sensitivity analysis 2d | ||||| |||||||||||| | ||||||| |
ATT = average treatment effect in the treated; CI = confidence interval; CR = complete response; OR = odds ratio.
aOR and 95% CI based on weighted logistic regression model.
bBase-case population (ibrutinib-venetoclax: N = 73; ibrutinib: N = 163).
cPopulation that excludes patients with missing covariate values in only 1 treatment group (ibrutinib-venetoclax: N = 73; ibrutinib: N = 146).
dPopulation of each study receiving at least 1 dose of study treatment, with no del(17p), and with no TP53 mutation (ibrutinib-venetoclax: N = 69; ibrutinib: N = 152).
Source: Sponsor’s ITC report for ibrutinib-venetoclax vs. Ibrutinib.28
Results for the Fludarabine-Eligible Population
Summary of Included Studies
For fludarabine-eligible patients, the CAPTIVATE study (ibrutinib-venetoclax FD cohort) was compared with the E1912 study (FCR versus ibrutinib-rituximab). The E1912 study was a multicentre, open-label, phase III RCT, and the CAPTIVATE study was an open-label, multicentre, multicohort, nonrandomized phase II trial. IPD from a single treatment group from each trial was used in the analysis. The sponsor did not provide a quality assessment of either study.
Differences in inclusion criteria and baseline characteristics between the ibrutinib-venetoclax study and the comparator study were observed in the unadjusted populations (Table 31). Both trials enrolled patients who were aged 18 to 70 years with previously untreated CLL, and who had an ECOG PS of 0 to 2. One key difference was that the E1912 study excluded patients with del(17p), whereas the CAPTIVATE cohort allowed these patients to enter the study.
For the pairwise comparison, patients with the del(17p) mutation were excluded. A total of 136 patients were included in the ibrutinib-venetoclax group and 158 were included in the FCR group; their baseline characteristics are shown in Table 32. After adjustment using the ATT inverse probability treatment weighting, the 11 covariates included in the model generally showed good balance between groups, except for beta-2M. The sponsor stated that missing data were a key driver of the imbalances. There was a substantial proportion of patients who had a propensity score near 0 in the FCR group and 1 in the ibrutinib-venetoclax group before weighting, which were eliminated when patients with missing covariate data were excluded from the analysis. In the base-case analysis, some extreme weights were noted, which could lead to unstable estimates.
Table 31: Assessment of Homogeneity — Fludarabine-Eligible Population
Characteristics | Ibrutinib-venetoclax vs. FCR |
|---|---|
Disease severity | Patients in the CAPTIVATE FD cohort were older and had a greater proportion of aged > 65 years than in E1912 (29% vs. 11%). Patients in the CAPTIVATE FD cohort also had better performance status and fewer instances of advanced disease (Rai stage III or IV). The CAPTIVATE FD cohort appeared to have fewer patients with mutated IGHV (40% verses 27%); however, it should be noted that 30% of patients in the E1912 study had no reported result. Most patients in the CAPTIVATE FD cohort had unmutated TP53 status (95%), compared with 79% in the E1912 study, with status missing for 0% and 18% of patients, respectively. The CAPTIVATE FD cohort did not collect or report CIRS score and the E1912 study did not report complex karyotype, so it is unclear if patients were similar across trials for these factors. Beta-2M levels were not reported in the CAPTIVATE FD cohort, but they were collected in the dataset. Other characteristics had similar distributions in both studies. |
Treatment history | All patients included in every analysis had previously untreated CLL. |
Trial eligibility criteria | Both the CAPTIVATE FD cohort and the E1912 study included patients with previously untreated CLL who met the iwCLL criteria, and both allowed the inclusion of patients with SLL. Both trials enrolled patients aged 18 to 70 years with ECOG PS scores of 0 to 2, and both included patients with TP53 mutation. The CAPTIVATE FD cohort included patients with del(17p), whereas the E1912 study excluded these patients. |
Dosing of comparators | Ibrutinib-venetoclax: Ibrutinib 420 mg once daily orally as monotherapy for the first 3 cycles, followed by ibrutinib plus venetoclax combination for a maximum of 12 cycles (28 days per cycle). Venetoclax was administered once daily orally at an increasing dose (20 mg week 1, 50 mg week 2, 100 mg week 3, and 200 mg week 4) for 4 weeks starting with day 1 of cycle 4 and the continued at 400 mg orally once daily until end of cycle 15. FCR: Fludarabine 25 mg/m2 IV on days 1, 2, and 3 of cycles 1 to 6, plus cyclophosphamide 25 mg/m2 IV on days 1, 2, and 3 of cycles 1 to 6, and rituximab 50 mg/m2 IV on day 1 of cycle 1, 325 mg/m2 on day 2 of cycle 1, then 500 mg/m2 on day 1 of cycles 2 to 6. Each cycle was 28 days. |
Placebo response | Not applicable. |
Definitions of end points | PFS: In both trials, defined as the time from randomization or study entry to the time of disease progression or death from any cause, whichever occurs first (regardless of subsequent therapy). ORR: In both trials, the definition was similar as the percentage of patients who achieved any response to treatment (CR, CRi, nPR or PR). Patients with missing postbaseline response data were considered nonresponders. CR: The CAPTIVATE trial defined CR as the percentage of patients who achieved a CR or CRi (patients with missing data were considered nonresponders). For the E1912 study, the definition was not readily available. OS: In both trials, OS is defined as the time from randomization until death from any cause. TTD: Both trials measured this outcome from the date of first study dose to the date of permanent discontinuation due to any cause during the follow-up of the original studies. |
Timing of end point evaluation | Ibrutinib-venetoclax (CAPTIVATE FD cohort): 38.7 and 49.7 months FCR (E1912 study): 48 months |
Withdrawal frequency | CAPTIVATE FD: 12 of 159 patients stopped treatment (7.5%) at the median 27.9-month follow-up. Discontinuation rate was not reported for E1912 study. |
Clinical trial setting | Both trials were multicentre and multinational. Patients were treated in clinical settings. |
Study design | E1912 was an open-label, phase III RCT, and CAPTIVATE was an open-label, nonrandomized, phase II study. Data from a single treatment group from each study were used in the analysis. |
Beta-2M = beta-2 microglobulin; CIRS = Cumulative Illness Rating Scale; CLL = chronic lymphocytic leukemia; CR = complete response; CRi = complete response with incomplete bone marrow recovery; del(17p) = deletion of 17p; ECOG PS = Eastern Cooperative Oncology Group Performance Status; FCR = fludarabine plus cyclophosphamide plus rituximab; FD = fixed duration; iwCLL = International Workshop on CLL; nPR = nodular partial response; ORR = overall response rate; OS = overall survival; PFS = progression-free survival; PR = partial response; RCT = randomized controlled trial; SLL = small lymphocytic lymphoma; TTD = time to treatment discontinuation.
Sources: Sponsor’s Summary of Clinical Evidence,60 sponsor’s ITC report for ibrutinib-venetoclax vs. FCR.31
Table 32: Fludarabine-Eligible Patients — Baseline Characteristics for ITC of Ibrutinib-Venetoclax Versus FCR (Base-Case Population)
Characteristic | CAPTIVATE FD All treated, non-del(17p) Ibrutinib-venetoclax (N = 136) | E1912 All treated, non-del(17p) FCR (N = 158) |
|---|---|---|
Age category, years (%) | ||
< 65 | 71 | 89 |
≥ 65 | 29 | 11 |
Male (%) | 65 | 70 |
ECOG PS (%) | ||
0 | 71 | 60 |
≥ 1 | 29 | 39 |
Del(11q) (%) | ||
Yes | 21 | 21 |
No | 79 | 79 |
Cytopenia at baseline (%) | 33 | 47 |
IGHV status (%) | ||
Unmutated | 57.4 | 42.4 |
Mutated | 40.4 | 27.2 |
Missing | 2.3 | 30.4 |
TP53 mutation (%) | ||
Yes | 5 | 3 |
No | 95 | 79 |
Missing | 0 | 18 |
Rai stage (%) | ||
0 to II | 73.5 | 58.9 |
III to IV | 25.0 | 41.1 |
Missing | 1.5 | 0 |
Beta-2M (%) | ||
≤ 3.5 mg/L | 55 | 52 |
> 3.5 mg/L | 34 | 48 |
Missing | 11 | 0 |
Creatinine clearance (%) | ||
< 60 mL/min | 4 | 8 |
60 to < 70 mL/min | 14 | 8 |
≥ 70 mL/min | 82 | 8 |
Bulky disease (%) | ||
Yes (≥ 5 cm) | 68 | 61 |
No | 32 | 35 |
Missing | 0 | 4 |
Beta-2M = beta-2 microglobulin; del(11q) = deletion of 11q; del(17p) = deletion of 17p; ECOG PS = Eastern Cooperative Oncology Group Performance Status; FCR = fludarabine plus cyclophosphamide plus rituximab; FD = fixed duration; ITC = indirect treatment comparison.
Source: Sponsor’s ITC report ibrutinib-venetoclax vs. FCR.31
Efficacy
The network diagram for the ITC in fludarabine-eligible patients is shown in Figure 10.
Figure 10: ITC Networks for Fludarabine-Eligible Patients

FCR = fludarabine plus cyclophosphamide plus rituximab; I+V = ibrutinib plus venetoclax; IPTW = inverse probability treatment weighting; ITC = indirect treatment comparison; RCT = randomized controlled trial.
Note: Solid lines represent direct comparisons; dashed lines represent indirect comparisons.
Source: Sponsor’s Summary of Clinical Evidence.60
The base-case analyses in the fludarabine-eligible population included 136 patients in the ibrutinib-venetoclax cohort and 158 patients in the FCR cohort (Table 33). In the base-case analysis for PFS, with ATT weighting, the model estimated an HR of |||| |||| ||| |||| || ||||| | | |||||) favouring ibrutinib-venetoclax versus FCR. The analyses that used ATC or ATO, as well as most sensitivity analyses, showed results that favoured ibrutinib-venetoclax (Appendix 1, Table 36).
The analyses of OS favoured ibrutinib-venetoclax versus FCR with an estimated HR of |||| |||| ||| |||| || ||||| | | |||||; base-case ATT), with sensitivity analyses showing consistent findings (Table 33). Of note, the sponsor stated that the sensitivity analyses with small sample sizes should be interpreted with caution, particularly for OS, which had low event rates.
For fludarabine-eligible patients, the ORR was ||| ||| ||| of patients for ibrutinib-venetoclax and FCR, respectively, in the base-case analysis. Across the sensitivity analyses, the results ranged from ||| || |||| (ibrutinib-venetoclax) compared to ||| || ||| (FCR). ||| ||||||| ||| ||| |||||| ||| || ||| ||| || ||| |||| |||||||, as they stated it was not considered informative, given the similar ORR between treatments.
The CR rate was an estimated ||| ||| of patients in the ibrutinib-venetoclax group compared to || ||| in the FCR group, with a || || |||| |||| ||| |||| || ||||| | | ||||| for the base-case ATT analysis. The sensitivity analyses generally showed a 95% CI that overlapped the null (Table 34).
Table 33: Estimated PFS and OS in Fludarabine-Eligible Patients for Ibrutinib-Venetoclax Versus FCR
Outcome and analysis | Ibrutinib-venetoclax vs. FCRa HR (95% CI) | P value |
|---|---|---|
PFS base caseb | ||
Unadjusted | |||| ||||||||||| | |||||| |
ATT | |||| ||||||||||| | |||||| |
PFS sensitivity analyses (ATT) | ||
Base case (only patients with no missing covariate data) | |||| ||||||||||| | |||||| |
Sensitivity analysis 1c | |||| ||||||||||| | |||||| |
Sensitivity analysis 2d | |||| ||||||||||| | |||||| |
Sensitivity analysis 3e | |||| ||||||||||| | |||||| |
OS base caseb | ||
Unadjusted | |||| ||||||||||| | |||||| |
ATT | |||| ||||||||||| | |||||| |
OS sensitivity analyses (ATT) | ||
Base case (only patients with no missing covariate data) | |||| ||||||||||| | |||||| |
Sensitivity analysis 1c | |||| ||||||||||| | |||||| |
Sensitivity analysis 2d | |||| ||||||||||| | |||||| |
Sensitivity analysis 3e | |||| ||||||||||| | |||||| |
ATT = average treatment effect in the treated; CI = confidence interval; FCR = fludarabine plus cyclophosphamide plus rituximab; HR = hazard ratio; OS = overall survival; PFS = progression-free survival.
aBased on weighted Cox proportional hazards model and weighted log-rank test.
bBase case: Population of each study receiving at least 1 dose of study treatment, with no del(17p) (number of patients: ibrutinib-venetoclax = 136, FCR = 158). The base-case analysis that was limited to patients with no missing covariate data included 117 patients in the ibrutinib-venetoclax group and 104 patients in the FCR group.
cPopulation of each study receiving at least 1 dose of study treatment, with no del(17p), with no TP53 mutation, and not missing TP53 mutation status .(number of patients ibrutinib-venetoclax = 129, FCR = 125).
dPopulation of each study receiving at least 1 dose of study treatment, with no del(17p), and with no known TP53 mutation (number of patients ibrutinib-venetoclax: = 129, FCR: = 154).
ePopulation of each study receiving at least 1 dose of study treatment, with no del(17p), with no TP53 mutation, aged < 65 years, and with CrCl ≥ 70 mL/min (number of patients ibrutinib-venetoclax: = 80, FCR: = 97).
Source: Sponsor’s ITC report for ibrutinib-venetoclax vs. FCR.31
Table 34: Summary of CR in Fludarabine-Eligible Patients for Ibrutinib-Venetoclax Versus FCR
Outcome and analysis | Ibrutinib-venetoclax vs. FCRa OR (95% CI) | P value |
|---|---|---|
CR base caseb | ||
Unadjusted | |||| ||||||||||| | |||||| |
ATT | |||| ||||||||||| | |||||| |
CR sensitivity analyses (ATT) | ||
Base case (only patients with no missing covariate data) | |||| ||||||||||| | |||||| |
Sensitivity analysis 1c | |||| ||||||||||| | |||||| |
Sensitivity analysis 2d | |||| ||||||||||| | |||||| |
Sensitivity analysis 3e | |||| ||||||||||| | |||||| |
ATT = average treatment effect in the treated; CI = confidence interval; CR = complete response; FCR = fludarabine plus cyclophosphamide plus rituximab; OR = odds ratio.
aBased on weighted logistic regression model.
bBase case: Population of each study receiving at least 1 dose of study treatment, with no del(17p) (number of patients: ibrutinib-venetoclax = 136, FCR = 158). The base-case analysis that was limited to patients with no missing covariate data included 117 patients in the ibrutinib-venetoclax group and 104 patients in the FCR group.
cPopulation of each study receiving at least 1 dose of study treatment, with no del(17p), with no TP53 mutation, and not missing TP53 mutation status .(number of patients ibrutinib-venetoclax = 129, FCR = 125).
dPopulation of each study receiving at least 1 dose of study treatment, with no del(17p) and with no known TP53 mutation (number of patients ibrutinib-venetoclax = 129, FCR = 154).
ePopulation of each study receiving at least 1 dose of study treatment, with no del(17p), with no TP53 mutation, aged < 65 years, and with CrCl ≥ 70 mL/min (number of patients ibrutinib-venetoclax = 80, FCR = 97).
Source: Sponsor’s ITC report for ibrutinib-venetoclax vs. FCR.31
Harms
No harms outcomes were analyzed for the ITCs of ibrutinib-venetoclax versus BR, VO, or acalabrutinib. The sponsor stated that due to availability and processing issues in the ALLIANCE study (BR and ibrutinib monotherapy), it was not possible to analyze the incidence rate of any-grade TEAEs or of grade 3 or 4 TEAEs for fludarabine-ineligible patients. Data for TTD were reported for the comparison with ibrutinib but not BR.
Incidence Rate of Any-Grade TEAEs
Regarding fludarabine-eligible patients, almost all patients in the ibrutinib-venetoclax and FCR groups experienced a TEAE; this was consistent across all analysis methods (ibrutinib-venetoclax range ||| || |||| and FCR range ||| || ||||).
Incidence Rate of Grade 3 or 4 TEAEs
Among fludarabine-eligible patients, grade 3 or 4 TEAEs were reported by ||| || ||| of patients in the FCR group compared with ||| || ||| of those in the ibrutinib-venetoclax group, depending on the analysis method. Comparative ORs of grade 3 or 4 AEs were not reported.
Incidence Rate of Treatment Discontinuation and TTD
Regarding fludarabine-ineligible patients, median TTD for ibrutinib-venetoclax was ||||| |||| ||| ||||| || |||||) months for the base-case ATT analysis. Ibrutinib monotherapy showed a median treatment exposure of 51.35 months (||| ||||||| || ||| ||||||||| ||||). The values remained similar for both drugs across all forms of analyses.
Regarding fludarabine-eligible patients, median treatment duration for ibrutinib-venetoclax was ||||| |||| ||| ||||| || |||||) months and ||| |||||| |||| || |||| || ||||| for FCR for the base-case ATT weighting, and was similar for the unadjusted, ATC, and ATO weighting methods. The incidence of treatment discontinuation ranged from 4% to 9% for ibrutinib-venetoclax versus 25% to 38% for FCR across the different statistical analyses.
Critical Appraisal of Indirect and Observational Evidence
Systematic Review
Although a systematic literature review was conducted to inform the indirect comparisons, the sponsor did not report the results of the literature review. Because there was no information available on the study characteristics or patient populations of the trials included in the systematic review, it is unclear if all potentially relevant comparator trials were considered for the ITCs. According to the sponsor, studies were selected as comparators for the ITCs or observational studies if they had similar study designs and patient populations with the GLOW and CAPTIVATE studies, had available IPD, or were trials used for regulatory approval of the comparator regimen. Also of note, no information was provided on the sponsor’s quality assessment of the comparator trials, thus the potential sources of bias for these studies is unknown.
Fludarabine-Ineligible Population
Each analysis used weighting methods to control for confounding; as with any nonrandomized study design, it is only possible to control for variables that are measured. There could be unreported or unobserved confounding factors that may bias the findings. In addition, there were limitations in each analysis in the ability to adjust for measured confounders, and as such, the potential for residual confounding should be considered when interpreting the findings.
Across the trials there were differences in how PFS was assessed, which could not be controlled for in the analyses. Although the trials all used the iwCLL criteria for progression, MRI or CT scans were mandated at most study visits in the GLOW study, whereas in other trials, scans were performed if there was clinical suspicion of progression, with fewer time points where scans were mandated. As such, there may be a difference in the detection of progression, with more events potentially identified among patients who received ibrutinib-venetoclax. This could bias the results in favour of BR and ibrutinib in the IPD analysis. The impact on the findings of the MAIC analysis is less clear, given that both the anchoring group (chlorambucil-obinutuzumab) and the ibrutinib-venetoclax group of the GLOW study would undergo the same assessment protocols. Furthermore, it is unclear if progression was consistently reported as either investigator-assessed events or IRC-confirmed events. The clinical expert consulted for this review stated that there is usually good concurrence between assessors.
In the fludarabine-ineligible population, there was no information on comparative safety. All pairwise comparisons analyzed PFS and OS end points, but TTNT was only reported for the comparison with VO. ORR and CR were reported for comparisons with BR and with ibrutinib. In some cases, the sponsor indicated that there were issues with data availability or data quality that prevented these other outcomes from being tested, but in many cases, the reasons for not conducting the analyses were not clear. The follow-up duration may be insufficient for the survival analyses, and the OS effect sizes may be biased due to intercurrent treatments.
Matching-Adjusted Indirect Comparisons
The MAICs comparing ibrutinib-venetoclax to acalabrutinib and to VO were conducted using similar methods, which appeared to be consistent with standard statistical approaches. Both analyses had the same key limitation: the comparator trials included a broader patient population than the GLOW study, thus there were important effect modifiers that could not be controlled for in the adjusted analyses. Specifically, patients with high-risk mutations, del(17p) and confirmed TP53, were allowed in the comparator trials (ELEVATE-TN and CLL14) but not in the GLOW trial. Some adjustment was attempted for TP53, as there was a small number of patients in the GLOW study in whom this mutation was detected after randomization (| || ||| ||| |||||| ||| | || ||| ||| |||||), but the adjustment may have limitations, given the small sample size. No adjustment was possible for del(17p), as there were no patients with this mutation in the GLOW study. In contrast, del(17p) was present in || || ||| and TP53 mutation was present in ||| || ||| of patients in the comparator trials. The lack of, or inadequate control for, these high-risk mutations could potentially bias the results in favour of ibrutinib-venetoclax.
The base-case models adjusted for 4 potential confounders (age, ECOG PS, CIRS score, TP53 mutation), which the clinical expert consulted for this review indicated were important confounders. Secondary analyses were conducted that included additional variables (a total of 9 for VO and 17 for acalabrutinib). Both fully adjusted models included other important effect modifiers (e.g., IGHV mutation, renal function, Rai or Binet stage); however, there were issues with missing covariate data. The sponsor stated that patients with missing covariate data were excluded from the analyses under the assumption that data were missing completely at random. This assumption may not hold true, as there were systemic issues with data availability. For example, in the comparison with acalabrutinib, CIRS data were only collected for patients younger than 65 years. IGHV status was missing for approximately a quarter of patients in the GLOW trial but were fully reported in the ELEVATE-TN and CLL14 studies. Other confounders, such as del(11q) or the presence of bulky disease, could not be controlled for in the comparison with VO due to data availability issues. There were variables excluded from the fully adjusted model because of poor overlap that resulted in a low Neff (i.e., region in the comparison with acalabrutinib; Neff = 27) and Binet stage in the comparison with VO (Neff = 48).
After weighting was applied for the base-case analyses, differences in patient characteristics remained for other clinically relevant factors for both MAICs. The sponsor noted that there was poor overlap between the populations in each comparison, as demonstrated with the substantial reductions in the effective sample size. The effective sample size in the base-case analyses was reduced to || ||| of the original sample for the comparison with acalabrutinib (N = 211; Neff = 127) and ||| for the comparison with VO (N = 174; Neff = 118), which suggests that there was poor overlap between the patients enrolled in the parent studies. The fully adjusted models had further reductions in effective sample size (acalabrutinib Neff = 69; VO Neff = 70), such that the treatment effect estimates may be considered unreliable, as the trial populations were too different to compare.
Another important limitation was the proportional hazard assumption was not met for PFS and OS in both comparisons, which persisted after weights were applied. This calls into question the interpretation of the effect estimates of the base-case results, as they may not be interpretable outside of the trials. Time-varying analyses were conducted, but the treatment effects lacked precision, showing wide 95% CIs.
Observational Studies
The comparisons between ibrutinib-venetoclax versus ibrutinib and BR were analyzed using propensity score weighting methods, based on IPD from the ALLIANCE and GLOW trials. Inverse probability treatment weighting methods use weights based on the propensity score to create a pseudosample in which the distribution of the measured baseline covariates is similar in the treatment and control groups. The selection of covariates for inclusion in the propensity score model is important, and in these analyses, it was unclear if all known confounders and prognostic factors were included. Eleven variables were used to generate the propensity scores (ECOG PS score, del(11q) status, presence of cytopenia, sex, IGHV status, Rai stage, beta-2M level, age, CrCl level, LDH level, and TP53 status). The model omitted bulky disease and CIRS score, which, according to the clinical expert consulted, are important confounders. It was not possible to adjust for race, as most patients in the GLOW study were white. In addition, there were issues with missing covariate data. The sponsor provided plots of the standardized mean differences for weighted and unweighted covariates, which generally showed better balance between groups after weighting. However, imbalances remained for some covariates with missing data (e.g., beta-2M level, TP53 mutation) in some analyses. Patients with missing data were assigned to a missing category, under the assumption that the reason for missingness was similar between groups. This assumption may not be met, and the extent of missing data for variables such as IGHV mutation status (missing for 20% to 33% of patients in the ibrutinib-venetoclax, BR, and ibrutinib groups) and TP53 status (missing for 4% to 7% of patients in the BR and ibrutinib groups), may have impacted the specification of the propensity scores. Patients with known TP53 mutation were excluded from the GLOW study, so only the 7 patients who were discovered to have this mutation after randomization were available for weighting in the ibrutinib-venetoclax group. Moreover, it is unclear if nonlinear relationships or interaction effects between covariates were explored to determine if the propensity score models were adequately specified. The distribution of propensity scores showed that a considerable proportion of patients had a very low probability of receiving the treatment (i.e., propensity score was near 0). These patients appear to have been assigned extreme weights, and thus have a disproportionate influence on the analysis. In addition, these unstable or inaccurate weights may inflate the CIs of the effect estimates. Although weight truncation or stabilization methods can be used to address extreme weights, these methods were not employed in the analyses and may not have been sufficient due to the heterogeneity across populations. It is unclear if an assessment was conducted to determine if the proportional hazards assumption was met. The Kaplan-Meier plots suggest this may be an issue for PFS for the comparison with BR.
Another limitation was the available sample size. The analyses excluded patients who did not meet the common inclusion criteria of the 2 trials and, as a result, the sample size of the ibrutinib-venetoclax group was low (number of patients ibrutinib-venetoclax = 73; BR = 155; ibrutinib = 163). The sensitivity analyses that excluded patients with missing data from the ALLIANCE study, and that excluded patients with the TP53 mutation, had further reductions in sample size. The sponsor noted that the number of survival events was low in all groups, and ATO weighting led to low sample sizes, thus these results should be interpreted with caution. The available data were less mature for the BR and ibrutinib groups (median 38 months follow-up) compared with the ibrutinib-venetoclax group (46 months follow-up).
The base-case analysis was based on the ATT weighting, which provides an estimate of the effect in treated patients (referring to the GLOW trial population). However, the preferred approach may be the ATO population estimates, as this describes a more general population, it has the most stable weights, and is the least sensitive to missing data and the lack of overlap between the trial populations. ATO weighting estimates were conducted as a secondary analysis and generally showed results that were consistent with the base-case findings.
The outcomes analyzed were PFS, OS, ORR, and CR. No safety data were analyzed for the comparison with BR, and time to discontinuation was the only safety end point reported for the comparison with ibrutinib. There was selective reporting of results. In some cases, the percentage of patients with events and P values were reported, but not the ORs or HRs and 95% CIs that were generated by the model.
For patients with the TP53 mutation, treatment with BR is not recommended. The base-case analysis included || || || of patients with this mutation. The sensitivity analysis that excluded these patients may be more applicable in the Canadian context.
Fludarabine-Eligible Population
As there was no randomized control group in the CAPTIVATE study, it was not possible to conduct a standard ITC analysis. The sponsor used IPD data from the FCR group of the E1912 study to conduct an inverse probability treatment weighting analysis versus ibrutinib-venetoclax. The methods used were consistent with those described for the comparisons with BR and ibrutinib and share similar limitations.
The base-case model adjusted for ECOG PS, del(11q) status, cytopenia presence, sex, IGHV status, Rai stage, bulky disease status, beta-2M level, age, TP53 status, and CrCl level, which included important confounders. There were, however, some issues with missing covariate data, namely for IGHV status, TP53 status, and bulky disease, which were missing for |||| |||| ||| || of patients from the E1912 study, respectively. In contrast, in the CAPTIVATE study, IGHV status was missing for ||| of patients, Rai status for ||||| of patients, and beta-2M for ||| of patients. Patients with missing covariate data were assigned to a missing category for the analysis, under the assumption that the reason for missing values is similar in both cohorts. It is unclear if this assumption is met, and it is possible that those variables with high rates of missing data may introduce bias in the effect estimates due to inappropriate methods used to account for confounding in the analysis. Sensitivity analysis that excluded patients with missing covariates generally showed results that were consistent with the main analyses; however, the sensitivity analyses rely on the same assumptions and therefore do not provide supportive evidence of the effect estimates.
The distribution of propensity scores showed that approximately ||| of patients in the FCR group had a propensity score of | ||| ||| of the ibrutinib-venetoclax group had a propensity score of 1 in the base-case population. As previously noted, propensity scores near 0 or 1 would generate extreme weights, which may disproportionately influence the results and inflate the 95% CIs. The overlap of propensity scores appeared to be poor, which adds to the uncertainty in the treatment effects.
The sample size was limited for the base-case analysis (ibrutinib-venetoclax: N = 136; FCR: N = 158), with further reductions in size for the sensitivity analyses that excluded patients with the TP53 mutation or missing TP53 status, and for the subgroup limited to patients with a good prognosis (< 65 years, CrCl ≥ 70 mL/min, and no TP53 mutation) (||| | | ||| ||| | | ||). The sponsor stated that considering the sample size of these sensitivity analyses and the low number of events, the results of these analyses should be interpreted with caution. It should be noted, however, that the sensitivity analyses most closely represent patients who may receive FCR in clinical practice, as typically older patients and those with high-risk mutations (e.g., TP53 or unmutated IGHV) would not be eligible for FCR.
Both safety and efficacy end points were reported for the comparison with FCR. There was selective reporting of results. In some cases, the percentage with events and P values were reported, but not the ORs and 95% CI that were generated by the model.
External Validity
In the fludarabine-ineligible population, the analyses comparing ibrutinib-venetoclax to acalabrutinib, VO, ibrutinib, and BR were presented as if they were part of a connected network (Figure 9); however, each analysis was conducted independently and represents a unique patient population. For the MAIC methods, the population from the GLOW trial was restricted in an attempt to match the population in the comparator trials (ELEVATE-TN, CLL14). Similarly for the comparison with BR and ibrutinib, common inclusion and exclusion criteria for the GLOW and ALLIANCE studies were applied and patients were eliminated who did not match these common characteristics. Although these are necessary steps to improve internal validity, they can impact external validity and may limit the comparability of patient populations across the analyses. As noted in Table 21, there were differences in the patients included in each analysis. The comparison with BR and ibrutinib included only patients 65 years and older (with or without comorbidities), whereas the comparison with VO included any adult (≥ 18 years) with comorbidities (CIRS score > 6 and CrCl ≤ 70 mL/min) and the comparison with acalabrutinib included all patients 65 years and older, but only younger adults who had comorbidities. Moreover, the comparator studies included patients with high-risk mutations (del[17p] and TP53) who were excluded from the GLOW trial. Thus, it appears that the patients may not be comparable across analyses and the external validity of each pairwise comparison should be assessed separately.
The base-case population for the comparison with BR and with FCR included patients who would not be eligible for the control group treatments, which may limit the generalizability of the findings. In these analyses, the populations in the sensitivity analyses may be more applicable to the Canadian context. According to the clinical expert consulted, BR is infrequently used in Canada, thus its relevance as a comparator may be limited.
Discussion
Summary of Available Evidence
The GLOW trial is a multicentre, randomized, open-label, phase III study comparing the efficacy and safety of the combination of ibrutinib-venetoclax to chlorambucil-obinutuzumab for the first-line treatment of patients with CLL. The GLOW trial was performed in the target patient population of older patients (aged 65 years and older) and patients with previously untreated CLL who were not suitable for treatment with a fludarabine-based regimen. Participants with del(17p) or known TP53 mutation were excluded because these aberrations are associated with inferior outcomes with chemoimmunotherapy (i.e., chlorambucil-obinutuzumab). Participants (N = 211) were randomized in a 1:1 ratio to receive ibrutinib-venetoclax (n = 106) or chlorambucil-obinutuzumab (n = 105). Dosing was in alignment with dosing instructions currently outlined in Canadian product monographs. The CAPTIVATE trial is a multicentre, phase II, single-arm study, also assessing time-limited treatment with the combination of ibrutinib-venetoclax for participants with treatment-naive CLL or SLL in either an MRD-guided discontinuation cohort (N = 164) or a FD cohort (N = 159), sequentially enrolled. This review focused on the CAPTIVATE FD cohort, which enrolled 159 patients who are eligible to fludarabine-based regimen. Eligible patients in the CAPTIVATE FD cohort were aged 18 to 70 years with previously untreated CLL or SLL requiring treatment, per iwCLL criteria, and had measurable nodal disease by CT, an ECOG PS of 0 to 2, and adequate hepatic, renal, and hematologic function. There were 2 study sites in Canada included in the GLOW trial and no sites in Canada in the CAPTIVATE FD cohort. The primary end point was PFS per IRC in the GLOW trial and CR rate per IA in the CAPTIVATE FD cohort. Other outcomes of interest included PFS per IA, OS, ORR (per IRC and IA), CR rate per IRC, improvement in hematological parameters, DOR (per IRC and IA), TTNT, MRD negativity rate, TLS risk reduction, and HRQoL (GLOW trial).
For the fludarabine-ineligible patients included in the GLOW trial, the majority of patients enrolled were men (57.8%) and white (95.7%), 42.2% of patients were female and other races included Asian (0.5%) and multiple (0.5%), 3.3% of patients did not report their race. Median age was 71 years (range, 47 to 93 years), with 87.2% of patients 65 years or older and 34.1% of patients 75 years or older. Advanced stage disease at baseline was reported for 54.8% of patients based on Rai stage III or IV disease and for 42.1% of patients based on Binet stage C disease. About half the patients had a baseline ECOG PS of 1 (53.1%). Overall, the proportion of patients with high-risk disease, defined by the presence of del(11q), unmutated IGHV, or TP53 mutation, was similar between treatment arms (59.4% versus 57.1% for ibrutinib-venetoclax versus chlorambucil-obinutuzumab). For fludarabine-eligible patients included the CAPTIVATE FD cohort, the median age at baseline was 60.0 years (range, 33 to 71 years), with 28.3% of patients 65 years or older. More patients were male (66.7%), and the majority of patients were white (92.5%), 33.3% of patients were female, and other races included Asian (1.9%), Black or African American (0.6%), and native Hawaiian or other Pacific Islander (0.6%), 4.4% of patients did not report their race. At baseline, more patients (69.2%) had an ECOG PS score of 0. Cytogenetic characteristics indicative of poor prognosis (per hierarchical classification) were del(17p) (12.6%) and del(11q) (17.6%). Other prognostic characteristics included TP53 mutated (10.1%), del(17p) or TP53 mutated (17.0%), unmutated IGHV (56.0%), and complex karyotype (19.5%).
For patient populations who were ineligible to received fludarabine, the sponsor provided 2 ITCs and 2 IPD observational studies that evaluated the efficacy and safety of ibrutinib-venetoclax versus BR, ibrutinib, VO, and acalabrutinib. In fludarabine-eligible patients, an IPD analysis was conducted comparing ibrutinib-venetoclax versus FCR. The ITC analyses were based on MAIC methods (comparison with acalabrutinib and VO) and the IPD analyses were based on propensity score methods (comparison with BR, ibrutinib, and FCR). All comparisons included patients with untreated CLL but varied in terms of age (all adults or ≥ 65 years only), presence of comorbidities, and high-risk mutations, such as del(17p) and TP53. The median follow-up duration ranged from 38 months to 54.4 months, depending on the treatment group and the analysis. The base-case MAIC models were adjusted for 4 covariates (age, ECOG PS score, CIRS score and TP53 status), whereas the IPD analyses controlled for 11 potential confounders, including age, ECOG score, renal function, and high-risk mutations (TP53, del[11q], and IGHV).
Interpretation of Trial Results
Efficacy
Ibrutinib in combination with venetoclax is indicated for the treatment of patients with CLL. The 2 pivotal trials, GLOW and the CAPTIVATE FD cohort, capture different patient populations; the GLOW trial included patients with CLL who were ineligible for fludarabine-based therapy, whereas the CAPTIVATE FD cohort included fludarabine-eligible patients with CLL. It was worthing noting that both studies included patients with SLL. The clinical expert consulted by CADTH confirmed that the inclusion of patients with SLL would not impact the interpretation of the results, as CLL and SLL are fundamentally the same disease, and it is appropriate to use the same treatments on them. In addition, the GLOW trial included patients with CrCl values higher than the clinical expert would expect in older patients with CLL, which indicates that the trial included patients with better kidney function than the general fludarabine-ineligible patient population in clinical practice. Moreover, the GLOW trial excluded patients with del(17p) or TP53 mutation; therefore, according to the clinical expert, the patient population in the GLOW study is a favourable patient population in general. The CADTH review team noted that there were 2 sites in Canada in the GLOW study and no study sites in Canada in the CAPTIVATE study, which may compromise the generalizability of the study results to clinical practice in Canada. In addition, in the GLOW trial, IGHV mutation status and the presence of del(11q) were used as stratification factors in randomization, which was considered appropriate; however, according to the clinical expert consulted by CADTH, there are other potential confounders, such as bulky disease, renal dysfunction, and disease stage, that need to be stratified or controlled for and could affect outcomes if unbalanced. The CADTH review team agreed with the clinical expert that not controlling for these confounders would not affect the interpretation of the study results, as baseline bulky disease, renal dysfunction, and disease stage were generally balanced between treatment groups.
According to the clinical expert consulted by CADTH, the chronic use of BTK inhibitor therapies often results in an accumulation of adverse effects (i.e., atrial fibrillation) with time. With FD ibrutinib-venetoclax treatment, if patients have a good response (i.e., PFS of 3 years or more), the clinical expert would likely offer patients the same treatment again, as those patients are not necessarily resistant. This could be an advantage over continuous BTK inhibitor treatments, according to the clinical expert consulted by CADTH. Overall, the clinical expert commented that a FD treatment (i.e., ibrutinib-venetoclax) might be attractive and good for older or higher-risk patients who have impaired renal function and/or atrial fibrillation, as those patients are more likely to experience AEs if they have been exposed to BTK inhibitors for a long time.
In the GLOW trial, the comparator was chlorambucil-obinutuzumab, which may not be a clinically relevant comparator, according to the clinical expert consulted by CADTH. The clinical expert commented that chlorambucil-obinutuzumab is an old historic treatment being used occasionally as a standard therapy in an older population of patients with CLL but not necessarily in a younger population of patients with CLL. The clinical expert confirmed that the use of chlorambucil-obinutuzumab for treatment might be generally reserved for an older patient population (i.e., older than 80 years) with comorbidities, which is about 5% of the patient population in the clinical expert’s practice. The clinical expert indicated that ibrutinib and venetoclax as monotherapies, chemoimmunotherapy such as BR, venetoclax in combination with obinutuzumab, and other BTK inhibitors (i.e., acalabrutinib and zanubrutinib) would be more appropriate comparators in the first-line setting for patients with CLL. Currently, according to the clinical expert, about 75% of older patients are eligible and funded for BTK inhibitors, and all those patients are getting BTK inhibitors at first-line treatment. However, the clinical expert acknowledged that, at the time of the study design (2017 to 2018), BTK inhibitors, venetoclax, and obinutuzumab were not being used as first-line treatment in clinical practice. Overall, there was no direct evidence available regarding the comparative efficacy and safety of ibrutinib-venetoclax to the previously mentioned comparators in the first-line-setting. Study CLL17 is an ongoing phase III, multicentre, randomized, prospective, open-label trial comparing the efficacy of continuous ibrutinib monotherapy with FD venetoclax-obinutuzumab and FD ibrutinib-venetoclax. This study may address the evidence gap of direct comparisons between ibrutinib-venetoclax and relevant comparators. There are no results published for Study CLL17, the details of which are summarized in Appendix 2.
Based on results from the GLOW trial reported in this review, ibrutinib-venetoclax demonstrated a superior and statistically significant improvement in the primary end point of PFS compared with chlorambucil-obinutuzumab for fludarabine-eligible patients with CLL who did not have del(17p) (HR = 0.216; 95% CI, 0.131 to 0.357; P < 0.0001). There was a notable drop in the Kaplan-Meier plot of PFS for the chlorambucil-obinutuzumab arm at approximately 15 months. Health Canada noted this and requested the sponsor for clarification. In the response to Health Canada’s request, the sponsor stated that the timing of protocol-specified imaging to assess treatment response may explain the marked drop in the Kaplan-Meier plot of PFS. The 6-month gap between the second (scheduled 9 months after randomization) and third imaging (scheduled 15 months after randomization) for patients randomized to the chlorambucil-obinutuzumab arm who were off treatment likely resulted in the pooled confirmation at the 15-month time point of PD events, which actually occurred between 9 and 15 months after randomization. Health Canada considered the sponsor’s response acceptable.51 The clinical expert consulted by CADTH is in alignment with Health Canada and confirmed that the Kaplan-Meier curve of PFS in the chlorambucil-obinutuzumab arm is consistent with what the clinical expert would expect in clinical practice. It is uncertain whether this benefit could be translated to an improvement in OS, as PFS is a surrogate outcome for OS and there is insufficient follow-up (after 46 months) available to accurately assess the treatment effect on OS. The CADTH review team noted that there was 1 major protocol amendment made after randomization that changed the treatment regimen in the ibrutinib-venetoclax by removing the last 3 cycles of ibrutinib monotherapy. This change in protocol may introduce uncertainty in the magnitude of the estimated treatment effect and compromise the generalizability of the study results to clinical practice, as the study results were based on a mixed patient population with and without the last 3 cycles of ibrutinib monotherapy. In addition, the benefit on PFS was observed primarily in white patients with better kidney function, and the majority of the study population was outside of Canada (only 2 study sites were in Canada). Moreover, the GLOW study excluded fludarabine-ineligible patients without measurable nodal disease, which are considered relevant and would be candidates for ibrutinib-venetoclax. Therefore, it remains unknown if this benefit could be generalizable to patients not well represented in the study. Overall, the benefit on PFS was generally consistent across various prespecified subgroup analyses (based on baseline demographic and disease characteristics), despite certain subgroups are too small to provide clear certainty on consistency (i.e., patients who were not white or had SLL). It is noteworthy that the observed benefit on PFS was compared to chlorambucil-obinutuzumab, which is not a clinically relevant therapy in Canada currently. In addition, ibrutinib-venetoclax also demonstrated benefit in achieving MRD negativity, CR or CRi, and overall best response compared to chlorambucil-obinutuzumab in fludarabine-ineligible patients.
The CAPTIVATE FD cohort met its primary end point of CR rate, indicating that treatment with ibrutinib-venetoclax is clinically meaningful in fludarabine-eligible non-del(17p) patients with CLL. The ORR results were consistent with the primary end point of CR rate, and the majority of all treated patients achieved a CR. According to the clinical expert consulted by CADTH, complete responders would do better than partial responders in terms of PFS and DOR. The clinical expert would consider the ORR results as supportive of the PFS results when making clinical decisions. There was a limited number of patients with del(17p) included in the CAPTIVATE FD cohort (n = 20), which may have contributed to inconsistent results in the subgroup analysis of patients with del(17p); therefore, it is uncertain whether the benefits of CR rate and ORR are consistent in patients with del(17p).
In fludarabine-eligible patients with CLL, due to the lack of a comparator and the absence of formal statistical testing in the CAPTIVATE study, the PFS results were difficult to interpret. The sponsor submitted an observational study that compared ibrutinib-venetoclax to FCR in adults (18 to 70 years) with previously untreated CLL and no del(17p) mutation. The results for PFS suggest that ibrutinib-venetoclax may be favoured over FCR (HR = 0.54; 95% CI, 0.31 to 0.95) in the base-case population, as well as in sensitivity analyses that excluded patients with the high-risk TP53 mutation, and in those with a poor prognosis (i.e., patients > 65 years or with impaired renal function). Most patients in each group achieved an ORR, but the analysis of the CR rate showed a 95% CI that included the null. The results of this study, however, should be interpreted with caution, given the potential for residual confounding, as it is unclear if all important confounders were adjusted for adequately. Further, the sample size was limited, and a substantial proportion of patients had propensity scores of 0 and 1, which would generate extreme weights that may disproportionately influence the results. Due to these limitations, there is substantial uncertainty in the findings of the IPD results.
Among patients who were ineligible for fludarabine, the sponsor provided 2 ITCs and 2 observational studies that estimated the comparative efficacy of ibrutinib-venetoclax versus standard of care. The analyses of PFS for ibrutinib-venetoclax versus acalabrutinib and VO showed wide 95% CIs that included the null for both the base-case and time-varying sensitivity analyses. The base-case analyses were adjusted for 4 confounders and could not control for the inclusion of patients with the high-risk del(17p) mutation in the control groups. The MAICs had poor overlap between the study populations and a low effective sample size after adjustment, which suggests that the patient populations were too different to compare. Two propensity score analyses compared ibrutinib-venetoclax to ibrutinib and BR. The PFS results suggested a possible difference favouring ibrutinib-venetoclax versus BR, whereas the comparison with ibrutinib did not estimate a significant difference based on the P value (HR and 95% CI were not reported). These IPD analyses adjusted for 11 covariates, but according to the clinical expert consulted by CADTH, other potential confounders, such as CIRS score, the presence of bulky disease, and race, were not adequately controlled for. Moreover, there were issues with missing covariate data that affected the ability of the model to balance covariates, and some patients had propensity scores near 0, which may result in extreme weights. The sample size was limited to 73 patients who received ibrutinib-venetoclax and 155 and 163 patients who received the comparator treatments. Overall, the limitations of the ITC and observational evidence were considered to be substantial by the CADTH review team, such that the results should be interpreted with caution. That said, the results do not support a conclusion that ibrutinib-venetoclax is more effective than ibrutinib, VO, or acalabrutinib, and nonstatistically significant findings cannot be interpreted as equivalence.
The OS data were considered immature and not interpretable at the time of the primary and extended follow-up analyses for the GLOW trial (median follow-up ranged from 27.70 months to 46.06 months) and the CAPTIVATE FD cohort (median follow-up ranged from 27.9 months to 38.7 months), based on a low number of events (event rate ranged from 1.9% to 28.6%); therefore, longer-term survival data (after 46 months for the GLOW trial and 39 months for the CAPTIVATE FD cohort) are required to assess the magnitude of treatment effect on an OS benefit. In addition, there were no comparative efficacy results in the CAPTIVATE FD cohort due to the lack of a comparator arm. Overall, these factors would introduce uncertainty and make the OS results difficult to interpret in both fludarabine-ineligible and fludarabine-eligible patients with CLL.
Generally, the benefits observed in improvement in hematological parameters, DOR, TTNT, MRD negativity rate, and TLS risk reduction in fludarabine-ineligible patients in the GLOW trial and in fludarabine-eligible patients in the CAPTIVATE FD cohort are clinical meaningful as per feedback from the clinical expert consulted by CADTH. The clinical expert indicated that the definitions of sustained improvement in hemoglobin and platelet levels are reasonable and relevant to patients and clinicians. The symptoms of CLL are related to anemia, thrombocytopenia, and neutropenia, according to the clinical expert, and improvements in hemoglobin and platelet levels for more than 2 months are indicative of an effective treatment in reducing the bulk of CLL in the BM, which would allow normal hematopoiesis to take place. The clinical expert also commented on MRD-negative response in BM and PB that the high undetectable MRD rates observed in both fludarabine-ineligible and fludarabine-eligible patients are significantly higher than BTK inhibitor monotherapy, which could indicate deeper responses in clinical practice. In addition, the clinical expert mentioned that some patients may have negative MRD in the PB but not in the BM, as MRD in BM is harder to clear; therefore, negative MRD in the BM would be a more significant result. The clinical expert confirmed that the TLS risk level definition is aligned with clinical practice, and stated that TLS may cause hypotension, renal dysfunction, confusion, and the need for hydration, which may require hospital admission. As a result, reducing TLS risk would translate into reducing the number of admissions to hospital to manage TLS, and even reducing the number of patients who need IV hydration, according to the clinical expert.
Patients and clinicians consider HRQoL a relevant outcome and expect the treatment of interest would be effective in improving QoL. In fludarabine-ineligible patients with CLL, HRQoL was measured using 4 measurements (EORTC QLQ-C30, EQ-5D-5L utility, EQ-5D VAS, and FACIT-Fatigue) in the GLOW trial. Generally, in the primary analysis of the GLOW trial, patients in the ibrutinib-venetoclax arm had early deteriorations and later improvements in HRQoL compared to patients in the chlorambucil-obinutuzumab arm as measured by the previously mentioned measurements. There were several limitations on the assessment of this outcome. The open-label design may impact the assessment of subjective HRQoL outcomes reported by patients; in addition, a higher proportion of patients received concomitant medications for the management of pain and AEs in the chlorambucil-obinutuzumab arm than the ibrutinib-venetoclax arm. No multiplicity adjustments were performed in the HRQoL analyses. Moreover, there are no published results regarding minimal clinically important difference estimates of EORTC QLQ-C30, EQ-5D-5L utility, EQ-5D VAS, and FACIT-Fatigue in patients with CLL, thus it is uncertain whether the results observed in fludarabine-ineligible patients with CLL in the GLOW trial are meaningful. It is also unknow if there is an HRQoL benefit associated with ibrutinib-venetoclax in fludarabine-eligible patients with CLL, as HRQoL was not measured in the CAPTIVATE FD cohort.
In the primary analysis, the type I error rate was adequately accounted for using a fixed sequence hierarchical testing in the GLOW study, whereas there was no multiplicity adjustment conducted for the CAPTIVATE FD cohort. Sensitivity analyses were conducted for PFS in the GLOW trial and for CR rate per IA in the CAPTIVATE FD cohort to assess the robustness of the findings, and overall, the results were consistent with the primary analyses.
Harms
Generally, no new safety signals were identified in the GLOW trial or the CAPTIVATE FD cohort in fludarabine-ineligible or fludarabine-eligible patients with CLL. More patients in the ibrutinib-venetoclax arm experienced at least 1 SAE, grade 3 to 4 AE, and withdrawal due to adverse events compared with the chlorambucil-obinutuzumab arm in the GLOW study. According to the clinical expert, the increase in AEs overall is aligned with what the clinical expert would expect in clinical practice in light of the dosing of the ibrutinib-venetoclax treatment. It is acknowledged that the high rate of withdrawal due to adverse events from the ibrutinib-venetoclax arm is consistent with the idea that there are more toxicities with ibrutinib-venetoclax compared to chlorambucil-obinutuzumab as per feedback from the clinical expert. Overall, similar rates of death were reported in the 2 treatment arms in the GLOW trial; however, death due to AEs was higher in the ibrutinib-venetoclax arm than in the chlorambucil-obinutuzumab arm. In addition, more patients died due to AEs n the ibrutinib-venetoclax arm compared to patients who died from PD. In addition, the higher frequency of atrial fibrillation reported in the ibrutinib-venetoclax arm in the GLOW trial was notable, and the clinical expert commented that the frequency of atrial fibrillation is higher compared to other BTK inhibitors.
No comparative safety data were available for the fludarabine-ineligible population, as the ITCs and observational studies did not address safety outcomes. Limited safety data were available from the observational study comparing ibrutinib-venetoclax to FCR, thus the comparative safety is unclear.
Conclusion
Patients and clinicians highlighted the need for new, effective treatments for CLL that prolong life, control disease and symptoms, maintain QoL, and reduce side effects compared with current treatments. According to 1 pivotal trial, ibrutinib-venetoclax demonstrated a clinically meaningful improvement of PFS compared with chlorambucil-obinutuzumab in fludarabine-ineligible patients with CLL without del(17p). In fludarabine-eligible patients with CLL without del(17p), ibrutinib-venetoclax demonstrated a meaningful improvement in CR rate compared with the prespecified minimum CR rate of 37%. Analyses of secondary outcomes supported the efficacy of ibrutinib-venetoclax in prolonging PFS and delaying disease progression. Data on OS were considered immature and not interpretable at the time of the analysis. Due to limitations in the statistical analysis and lack of identified MID estimates in fludarabine-ineligible patients with CLL, and the lack of assessment in fludarabine-eligible patients, no definitive conclusions can be drawn from the HRQoL analyses in the GLOW trial. The pivotal study results were subjected to key limitations, such as the exclusion of patients without measurable disease, a major amendment in protocol, and the use of a comparator treatment for fludarabine-ineligible patients that may not be currently relevant in Canada. No new safety signals were identified in either fludarabine-ineligible and fludarabine-eligible patients with CLL.
The supplementary comparative evidence from the ITCs and observational studies submitted by the sponsor had significant limitations that impacted the internal validity of the findings. For each of these analyses, there was potential residual confounding from measured and unmeasured confounders, as the statistical methods were unable to control for all sources of heterogeneity in the patient and study characteristics. In addition, most analyses were based on a small sample size and there were low event rates for OS analysis, with some results showing wide 95% CIs and poor precision. In the fludarabine-ineligible patient population, the comparative efficacy of ibrutinib-venetoclax versus ibrutinib, BR, VO, or acalabrutinib is unclear. The observational data comparing ibrutinib-venetoclax to FCR in fludarabine-eligible patients shared the same limitations, and thus definitive conclusions on the comparative efficacy cannot be drawn from this study. In addition, the lack of comparative efficacy for patients with del(17p) were not sufficiently addressed by the supporting evidence.
No comparative safety data were available in the fludarabine-ineligible population, and data were limited in the fludarabine-eligible population, thus comparative safety of ibrutinib-venetoclax is unknown.
References
1.Shanshal M, Haddad RY. Chronic lymphocytic leukemia. Disease-a-month: DM. 2012;58(4):153-167. PubMed
2.Hallek M, Cheson BD, Catovsky D, et al. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood. 2018;131(25):2745-2760. PubMed
3.Janssen Pharmaceuticals I. Epidemiology model: Assessment of Target Population - Frontline Chronic lymphocytic leukemia (CLL) [internal sponsor's report]. 2016.
4.Orphanet. B-cell chronic lymphocytic leukemia. 2023; https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=GB&Expert=67038. Accessed 2023 Jan 24.
5.Canadian Cancer Society. Chronic lymphocytic leukemia. https://www.cancer.ca/en/cancer-information/cancer-type/leukemia-chronic-lymphocytic-cll/statistics/?region=on. Accessed 2023 Apr 27.
6.Abrisqueta P, Pereira A, Rozman C, et al. Improving survival in patients with chronic lymphocytic leukemia (1980-2008): the Hospital Clinic of Barcelona experience. Blood. 2009;114(10):2044-2050. PubMed
7.Parikh SA, Rabe KG, Kay NE, et al. Chronic lymphocytic leukemia in young (≤ 55 years) patients: a comprehensive analysis of prognostic factors and outcomes. Haematologica. 2014;99(1):140-147. PubMed
8.Oscier D, Dearden C, Eren E, et al. Guidelines on the diagnosis, investigation and management of chronic lymphocytic leukaemia. Br J Haematol. 2012;159(5):541-564. PubMed
9.Schnaiter A, Stilgenbauer S. 17p deletion in chronic lymphocytic leukemia: risk stratification and therapeutic approach. Hematology/oncology clinics of North America. 2013;27(2):289-301. PubMed
10.Owen C, Banerji V, Johnson N, et al. Canadian evidence-based guideline for frontline treatment of chronic lymphocytic leukemia: 2022 update. Leuk Res. 2023;125:107016. PubMed
11.CADTH. PCODR Final clinical guidance report. Clinical Report. Venetoclax (Venclexta). 2020: https://www.cadth.ca/sites/default/files/pcodr/Reviews2020/10212VenetoclaxObinutuzumabCLL_fnCGR_REDACT_EC_Post17Nov2020_final.pdf. Accessed 27 Apr 2023.
12.CADTH. Provisional Funding Algorithm for CLL. Ottawa: CADTH; 2021: https://www.cadth.ca/sites/default/files/attachments/2021-06/PH0004-CLL-Provisional-Algorithm-final-may18-rev.pdf. Accessed 27 Apr 2023.
13.Drug Reimbursement Review clinical guidance report: ibrutinib (Imbruvica) for CLL/SLL. Ottawa (ON): CADTH; 2016: https://www.cadth.ca/sites/default/files/pcodr/pcodr_ibrutinib_imbruvica_cll-sll_fn_cgr.pdf. Accessed 2023 May 24.
14.Drug Reimbursement Review clinical guidance report: Ibrutinib (Imbruvica) for CLL/SLL (previously treated). Ottawa (ON): CADTH; 2015: https://www.cadth.ca/sites/default/files/pcodr/pcodr-ibrutinib-cll-sll-fn-cgr.pdf. Accessed 2023 May 24.
15.Drug Reimbursement Review clinical guidance report: Ibrutinib (Imbruvica) for WM. Ottawa (ON): CADTH; 2016: https://www.cadth.ca/sites/default/files/pcodr/pcodr_ibrutinib_imbruvica_wm_fn_cgr.pdf. Accessed 2023 May 24.
16.Drug Reimbursement Review clinical guidance report: venetoclax (Venclexta) for CLL. Ottawa (ON): CADTH; 2018: https://www.cadth.ca/sites/default/files/pcodr/pcodr_venetoclax_venclexta_cll_fn_cgr.pdf. Accessed 2023 May 24.
17.CADTH Drug Reimbursement Expert Review Committee final recommendation: venetoclax (Venclexta - AbbVie) Do not reimburse. Ottawa (ON): CADTH; 2021: https://www.cadth.ca/sites/default/files/DRR/2021/PC0239%20Venclexta%20%20-%20CADTH%20Final%20Rec.pdf. Accessed 2023 May 24.
18.CADTH Drug Reimbursement Expert Review Committee final recommendation: venetoclax (Venclexta - Abbvie) Reimburse with conditions. Ottawa (ON): CADTH; 2021: https://www.cadth.ca/sites/default/files/DRR/2021/PC0238%20Venclexta%20(AZA)%20-%20Final%20Rec.pdf. Accessed 2023 May 24.
19.Drug Reimbursement Review clinical guidance report: venetoclax (Venclexta) for CLL, in combinaiton with obinutuzumab. Ottawa (ON): CADTH; 2020: https://www.cadth.ca/sites/default/files/pcodr/Reviews2020/10212VenetoclaxObinutuzumabCLL_fnCGR_REDACT_EC_Post17Nov2020_final.pdf. Accessed 2023 May 24.
20.Drug Reimbursement Review clinical guidance report: venetoclax (Venclexta) for CLL, in combination with rituximab. Ottawa (ON): CADTH; 2019: https://www.cadth.ca/sites/default/files/pcodr/Reviews2019/10162VenetoclaxRituximabCLL_fnCGR_REDACT_Post_31May2019_final.pdf. Accessed 2023 May 24.
21.Product information: ibrutinib (Imbruvica). (European public assessment report). Amsterdam (Netherlands): European Medicines Agency; 2023: https://www.ema.europa.eu/en/medicines/human/EPAR/imbruvica. Accessed 2023 May 24.
22.Dana-Farber Cancer Institute, AbbVie, Pharmacyclics LLC. NCT04273139: Ibrutinib + Venetoclax in Untreated WM. ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine; 2022: https://clinicaltrials.gov/ct2/show/NCT04273139. Accessed 2023 May 24.
23.Clinical Study Report: 54179060CLL3011. GLOW: A Randomized, Open-label, Phase 3 Study of the Combination of Ibrutinib plus Venetoclax versus Chlorambucil plus Obinutuzumab for the First-line Treatment of Subjects with Chronic Lymphocytic Leukemia (CLL)/Small Lymphocytic Lymphoma (SLL) [internal sponsor's report]. Beerse (BE): Janssen Inc; 2021 Nov 12.
24.Clinical Study Report: 54179060CLL3011. GLOW: A Randomized, Open-label, Phase 3 Study of the Combination of Ibrutinib plus Venetoclax versus Chlorambucil plus Obinutuzumab for the First-line Treatment of Subjects with Chronic Lymphocytic Leukemia (CLL)/Small Lymphocytic Lymphoma (SLL): 46 month [internal sponsor's report] Beerse (BE): Janssen Inc; 2023 Jan 3.
25.Clinical Study Report: 54179060CLL3011. GLOW: Patient-Reported Outcomes Supplemental Analyses Report. A Randomized, Open-label, Phase 3 study of the Combination of Ibrutinib plus Venetoclax versus Chlorambucil plus Obinutuzumab for the First-line Treatment of Subjects with Chronic Lymphocytic Leukemia (CLL)/Small Lymphocytic Lymphoma (SLL)[internal sponsor's report]. Beerse (BE): Janssen Inc.; 2021.
26.Clinical Study Report: PCYC-1142-CA. CAPTIVATE: Phase 2 Study of the Combination of Ibrutinib Plus Venetoclax in Subjects with Treatment-Naive Chronic Lymphocytic Leukemia / Small Lymphocytic Lymphoma [internal sponsor's report]. Beerse (BE): Janssen Inc; 2021 Nov 11.
27.Cross-study comparisons of ibrutinib plus venetoclax versus bendamustine and rituximab combination and ibrutinib monotherapy in patients with previously untreated chronic lymphocytic leukemia (CLL): I+V vs BR ITC [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Imbruvica (ibrutinib), 140 mg, 280 mg, 420 mg, 560 mg tablets, 140 mg capsules, oral. Janssen Pharmaceutica NV.; 2022 Nov 1. Beerse (BE): Janssen Inc.
28.Cross-study comparisons of ibrutinib plus venetoclax versus bendamustine and rituximab combination and ibrutinib monotherapy in patients with previously untreated chronic lymphocytic leukemia (CLL): I+V vs Ibr ITC [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Imbruvica (ibrutinib), 140 mg, 280 mg, 420 mg, 560 mg tablets, 140 mg capsules, oral. Janssen Pharmaceutica NV; 2022 Nov 1. Beerse (BE): Janssen Inc.
29.Matching-adjusted indirect treatment comparison of ibrutinib in combination with venetoclax and venetoclax in combination with obinutuzumab [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Imbruvica (ibrutinib), 140 mg, 280 mg, 420 mg, 560 mg tablets, 140 mg capsules, oral. Janssen Pharmaceutica NV.; 2022 Nov 17. Beerse (BE): Janssen Inc.
30.Matching-adjusted indirect treatment comparison of ibrutinib in combination with venetoclax and acalabrutinib montherapy [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Imbruvica (ibrutinib), 140 mg, 280 mg, 420 mg, 560 mg tablets, 140 mg capsules, oral. Janssen Pharmaceutica NV.; 2022 Nov 9. Beerse (BE): Janssen Inc.
31.Cross-study comparisons of ibrutinib plus venetoclax versus fludarabine, cyclophosphamide and rituximab combination in patients with previously untreated chronic lymphocytic leukemia (CLL) [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Imbruvica (ibrutinib), 140 mg, 280 mg, 420 mg, 560 mg tablets, 140 mg capsules, oral. Janssen Pharmaceutica NV.; 2022 Nov 3. Beerse (BE): Janssen Inc.
32.Gaidano G, Foa R, Dalla-Favera R. Molecular pathogenesis of chronic lymphocytic leukemia. The Journal of clinical investigation. 2012;122(10):3432-3438. PubMed
33.Kipps TJ, Stevenson FK, Wu CJ, et al. Chronic lymphocytic leukaemia. Nature reviews Disease primers. 2017;3:17008. PubMed
34.D'Arena G, Guariglia R, La Rocca F, et al. Autoimmune cytopenias in chronic lymphocytic leukemia. Clinical & developmental immunology. 2013;2013:730131. PubMed
35.Hodgson K, Ferrer G, Montserrat E, Moreno C. Chronic lymphocytic leukemia and autoimmunity: a systematic review. Haematologica. 2011;96(5):752-761. PubMed
36.Kutsch N, Busch R, Bahlo J, et al. FCR front-line therapy and quality of life in patients with chronic lymphocytic leukemia. Leuk Lymphoma. 2017;58(2):399-407. PubMed
37.Holtzer-Goor KM, Schaafsma MR, Joosten P, et al. Quality of life of patients with chronic lymphocytic leukaemia in the Netherlands: results of a longitudinal multicentre study. Qual Life Res. 2015;24(12):2895-2906. PubMed
38.van den Broek EC, Oerlemans S, Nijziel MR, Posthuma EF, Coebergh JW, van de Poll-Franse LV. Impact of active surveillance, chlorambucil, and other therapy on health-related quality of life in patients with CLL/SLL in the Netherlands. Ann Hematol. 2015;94(1):45-56. PubMed
39.Kosmas CE, Shingler SL, Samanta K, et al. Health state utilities for chronic lymphocytic leukemia: importance of prolonging progression-free survival. Leuk Lymphoma. 2015;56(5):1320-1326. PubMed
40.Statistics Canada. Table 13-10-0751-01. Number of prevalent cases and prevalence proportions of primary cancer, by prevalence duration, cancer type, attained age group and sex. 2022; 10.25318/1310075101-eng.
41.Charalambous A, Schwarzbich MA, Witzens-Harig M. Ibrutinib. Recent Results Cancer Res. 2018;212:133-168. PubMed
42.Papanota AM, Ntanasis-Stathopoulos I, Kastritis E, Dimopoulos MA, Gavriatopoulou M. Evaluating ibrutinib in the treatment of symptomatic Waldenstrom's macroglobulinemia. J Blood Med. 2019;10:291-300. PubMed
43.Janssen Biotech Inc. Imbruvica® (ibrutinib) [drug label]. 2023 May; https://www.imbruvica.com/docs/librariesprovider7/default-document-library/prescribing-information.pdf. Accessed 2023 Apr 27.
44.Kater AP, Owen C, Moreno C, et al. Fixed-duration ibrutinib-venetoclax in patients with chronic lymphocytic leukemia and comorbidities. NEJM Evidence. 2022;1(7):1-13.
45.Deng J, Isik E, Fernandes SM, Brown JR, Letai A, Davids MS. Bruton's tyrosine kinase inhibition increases BCL-2 dependence and enhances sensitivity to venetoclax in chronic lymphocytic leukemia. Leukemia. 2017;31(10):2075-2084. PubMed
46.Drug Reimbursement Review sponsor submission: Imbruvica (ibrutinib), 140 mg, 280 mg, 420 mg, 560 mg tablets, 140 mg capsules, oral [internal sponsor's package]. Beerse (BE): Janssen Inc; 2023 May 4.
47.Cancer Care Ontario - Drug Formulary 2023; https://www.cancercareontario.ca/en/cancer-treatments/chemotherapy/drug-formulary. Accessed 2023 May 24.
48.BC Cancer. 2023; http://www.bccancer.bc.ca/. Accessed 2023 May 24.
49.Lampson BL, Yu L, Glynn RJ, et al. Ventricular arrhythmias and sudden death in patients taking ibrutinib. Blood. 2017;129(18):2581-2584. PubMed
50.Tam CS, Allan JN, Siddiqi T, et al. Fixed-duration ibrutinib plus venetoclax for first-line treatment of CLL: primary analysis of the CAPTIVATE FD cohort. Blood. 2022;139(22):3278-3289. PubMed
51.Health Canada reviewer's report: Imbruvica (Ibrutinib) [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Imbruvica (ibrutinib): 140 mg, 280 mg, 420 mg, 560 mg tablets, 140 mg capsules, oral. Beerse (BE): Janssen Inc.; 2023.
52.Clinical Study Report: CL3011. Title of report: Randomized, Open-label, Phase 3 study of the Combination of Ibrutinib plus Venetoclax versus Chlorambucil plus Obinutuzumab for the First-line Treatment of Subjects with Chronic Lymphocytic Leukemia (CLL)/Small Lymphocytic Lymphoma (SLL) - Patient-Reported Outcomes Supplemental Analyses Report. USA: Janssen Research & Development Inc.; 2021.
53.Pickard AS, Neary MP, Cella D. Estimation of minimally important differences in EQ-5D utility and VAS scores in cancer. Health Qual Life Outcomes. 2007;5:70. PubMed
54.Cella D, Eton DT, Lai JS, Peterman AH, Merkel DE. Combining anchor and distribution-based methods to derive minimal clinically important differences on the Functional Assessment of Cancer Therapy (FACT) anemia and fatigue scales. J Pain Symptom Manage. 2002;24(6):547-561. PubMed
55.Howard SC, Jones DP, Pui CH. The tumor lysis syndrome. N Engl J Med. 2011;364(19):1844-1854. PubMed
56.EORTC questionnaires - Chronic Lymphocytic Leukaemia. EORTC 2023; https://qol.eortc.org/questionnaire/qlq-cll17/. Accessed 2023 May 25.
57.Osoba D, Rodrigues G, Myles J, Zee B, Pater J. Interpreting the significance of changes in health-related quality-of-life scores. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 1998;16(1):139-144. PubMed
58.Chung H, Eek D, Krogh C, Blowfield M, Meyers O, Eyre TA. Functional Assessment of Chronic Illness Therapy-Fatigue Scale (FACIT-Fatigue): Cognitive Debriefing in Patients with Chronic Lymphocytic Leukemia (CLL). Blood. 2019;134(Supplement_1):4777-4777.
59.Eichhorst B, Fink AM, Bahlo J, et al. First-line chemoimmunotherapy with bendamustine and rituximab versus fludarabine, cyclophosphamide, and rituximab in patients with advanced chronic lymphocytic leukaemia (CLL10): an international, open-label, randomised, phase 3, non-inferiority trial. Lancet Oncol. 2016;17(7):928-942. PubMed
60.Imbruvica in combination with Venclexta for the treatment of patients with chronic lymphocytic leukemia [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Imbruvica (ibrutinib), 140 mg, 280 mg, 420 mg, 560 mg tablets, 140 mg capsules, oral. Beerse (BE): Janssen Inc.; 2023 Apr 17.
61.Signorovitch JE, Sikirica V, Erder MH, et al. Matching-adjusted indirect comparisons: a new tool for timely comparative effectiveness research. Value in health: the journal of the International Society for Pharmacoeconomics and Outcomes Research. 2012;15(6):940-947.
62.Phillippo DM, Ades AE, Dias S, Palmer S, Abrams KR, Welton NJ. Methods for Population-Adjusted Indirect Comparisons in Health Technology Appraisal. Medical decision making: an international journal of the Society for Medical Decision Making. 2018;38(2):200-211. PubMed
63.Janssen Inc. response to May 24, 2023 CADTH request for additional information regarding Imbruvica CADTH review: [internal additional sponsor's information]. Toronto (ON): Janssen Inc; 2023 Jun 6.
64.Guyot P, Ades AE, Ouwens MJ, Welton NJ. Enhanced secondary analysis of survival data: reconstructing the data from published Kaplan-Meier survival curves. BMC medical research methodology. 2012;12:9. PubMed
65.Sharman JP, Banerji V, Fogliatto LM, et al. ELEVATE TN: Phase 3 Study of Acalabrutinib Combined with Obinutuzumab (O) or Alone Vs O Plus Chlorambucil (Clb) in Patients (Pts) with Treatment-Naive Chronic Lymphocytic Leukemia (CLL). Blood. 2019;134:31.
66.Sharman JP, Egyed M, Jurczak W, et al. Acalabrutinib with or without obinutuzumab versus chlorambucil and obinutuzumab for treatment-naive chronic lymphocytic leukaemia (ELEVATE-TN): a randomised, controlled, phase 3 trial. The Lancet. 2020;395(10232):1278-1291. PubMed
67.Fischer K, Al-Sawaf O, Bahlo J, et al. Venetoclax and Obinutuzumab in Patients with CLL and Coexisting Conditions. New England Journal of Medicine. 2019;380(23):2225-2236. PubMed
68.Woyach JA, Ruppert AS, Heerema NA, et al. Ibrutinib Regimens versus Chemoimmunotherapy in Older Patients with Untreated CLL. New England Journal of Medicine. 2018;379(26):2517-2528. PubMed
69.Shanafelt TD, Wang XV, Kay NE, et al. Ibrutinib-Rituximab or Chemoimmunotherapy for Chronic Lymphocytic Leukemia. N Engl J Med. 2019;381(5):432-443. PubMed
70.Clinicaltrials.gov. A Study of the Combination of Ibrutinib Plus Venetoclax Versus Chlorambucil Plus Obinutuzumab for the First-line Treatment of Participants With Chronic Lymphocytic Leukemia (CLL)/Small Lymphocytic Lymphoma (SLL) (GLOW). 2018; https://clinicaltrials.gov/ct2/show/NCT03462719. Accessed 2023 Apr 27.
71.Clinicaltrials.gov. Rituximab and Bendamustine Hydrochloride, Rituximab and Ibrutinib, or Ibrutinib Alone in Treating Older Patients With Previously Untreated Chronic Lymphocytic Leukemia. 2013; https://clinicaltrials.gov/ct2/show/NCT01886872. Accessed 2023 Apr 27.
72.Clinicaltrials.gov. Elevate CLL TN: Study of Obinutuzumab + Chlorambucil, Acalabrutinib (ACP-196) + Obinutuzumab, and Acalabrutinib in Subjects With Previously Untreated CLL. 2015; https://clinicaltrials.gov/ct2/show/NCT02475681. Accessed 2023 Apr 27.
73.Clinicaltrials.gov. Ibrutinib Monotherapy Versus Fixed-duration Venetoclax Plus Obinutuzumab Versus Fixed-duration Ibrutinib Plus Venetoclax in Patients With Previously Untreated Chronic Lymphocytic Leukaemia (CLL) (CLL17). 2020; https://clinicaltrials.gov/ct2/show/NCT04608318. Accessed 2023 June 7.
Appendix 1: Detailed Outcome Data
Note that this appendix has not been copy-edited.
Table 35: Hierarchical Testing Order of Secondary End Points in GLOW
Secondary end points | P value | Significant |
|---|---|---|
MRD negativity rate in bone marrow | 0.0023 | Yes |
CR rate (per IRC) | 0.0001 | Yes |
ORR | ||
ORR per IRC | 0.6991 | No |
ORR per IA | 0.2585 | No |
OS | 0.9121 | No |
Rate of sustained platelet improvement | 0.4346 | No |
Rate of sustained hemoglobin improvement | 0.3854 | No |
Time to improvement in FACIT-Fatigue score | 0.0776 | No |
CR = complete response; FACIT = Functional Assessment of Chronic Illness Therapy; IA = investigator assessment; IRC = independent review committee; MRD = minimal residual disease; ORR = overall response rate; OS = overall survival.
Data cut-off: February 26, 2021
Source: GLOW Primary Analysis CSR23
Figure 11: Forest Plot of Progression-Free Survival per IRC for Subgroups Defined by Demographic and Baseline Clinical Disease Characteristics at Primary Analysis for GLOW (ITT Analysis Set; Part 1 of 3)
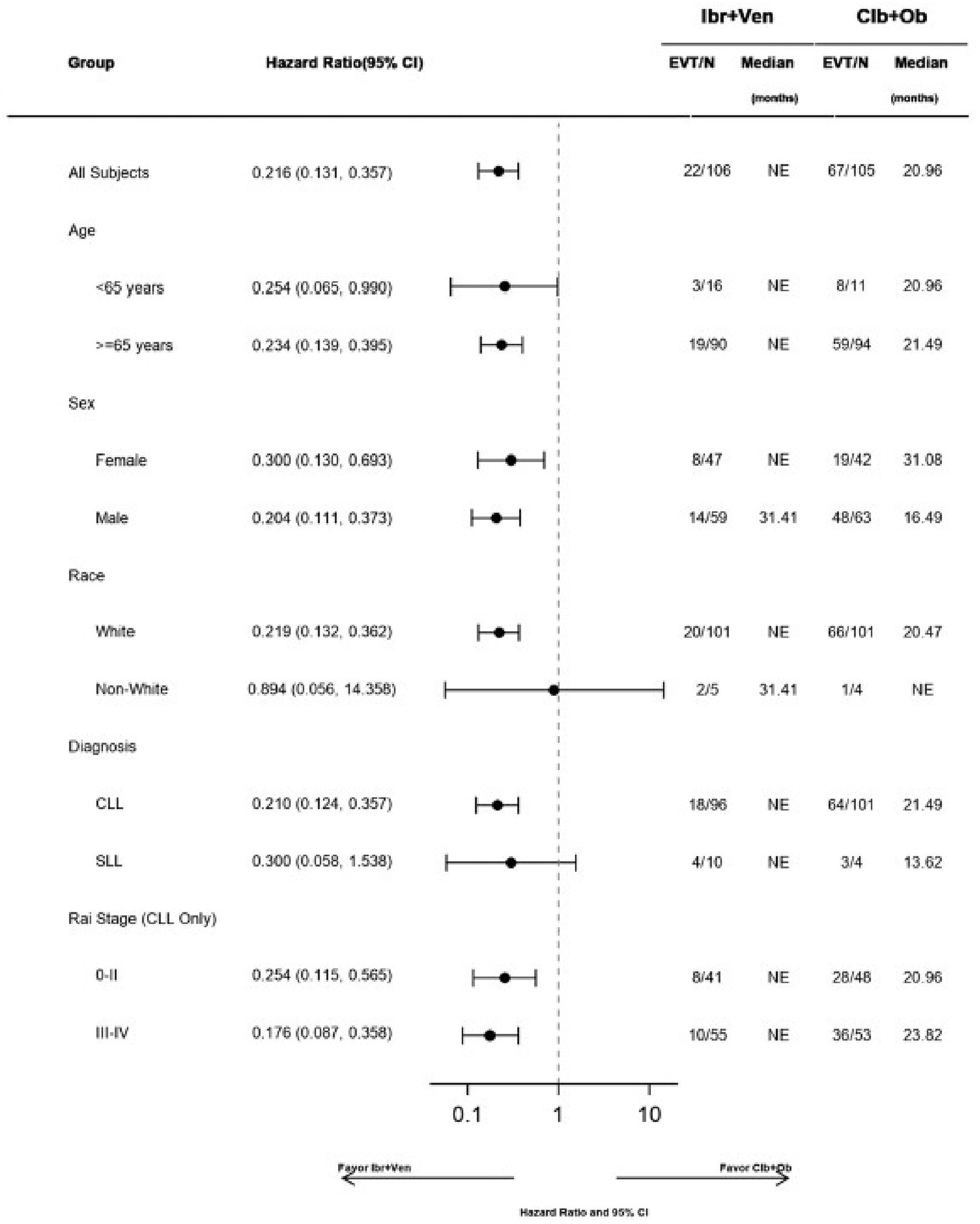
CI = confidence interval; CIRS = Cumulative Illness Rating Scale; CLL = chronic lymphocytic leukemia; ECOG = Eastern Cooperative Oncology Group; IGHV = immunoglobulin heavy chain variable; LDH = lactate dehydrogenase; NE = not estimable; SLL = small lymphocytic lymphoma.
Data cut-off: February 26, 2021.
Source: GLOW Primary Analysis CSR23
Figure 12: Forest Plot of Progression-Free Survival per IRC for Subgroups Defined by Demographic and Baseline Clinical Disease Characteristics at Primary Analysis for GLOW (ITT Analysis Set; Part 2 of 3)
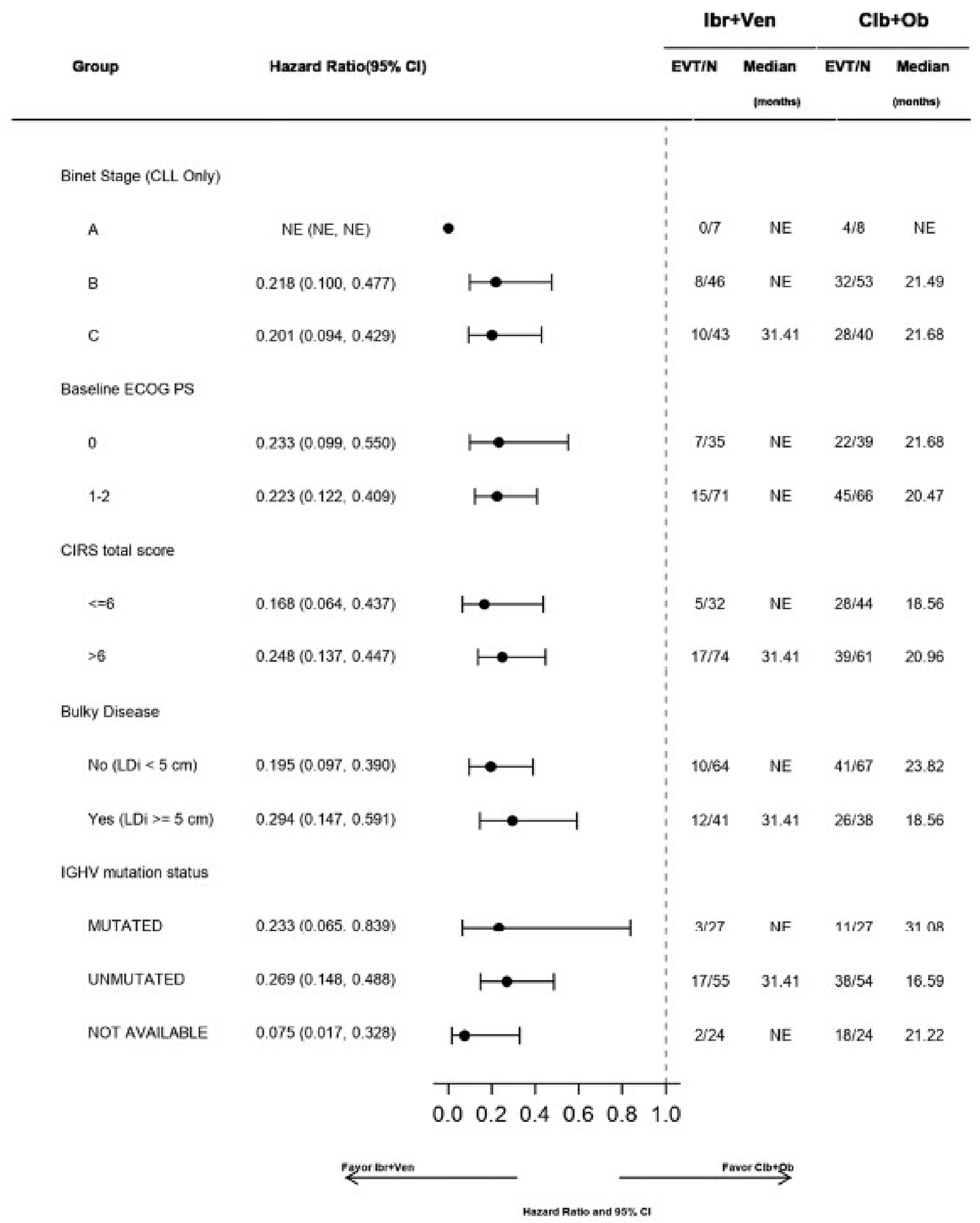
CI = confidence interval; CIRS = Cumulative Illness Rating Scale; CLL = chronic lymphocytic leukemia; ECOG = Eastern Cooperative Oncology Group; IGHV = immunoglobulin heavy chain variable; LDH = lactate dehydrogenase; NE = not estimable; SLL = small lymphocytic lymphoma.
Data cut-off: February 26, 2021.
Source: GLOW Primary Analysis CSR23
Figure 13: Forest Plot of Progression-Free Survival per IRC for Subgroups Defined by Demographic and Baseline Clinical Disease Characteristics at Primary Analysis for GLOW (ITT Analysis Set; Part 3 of 3)
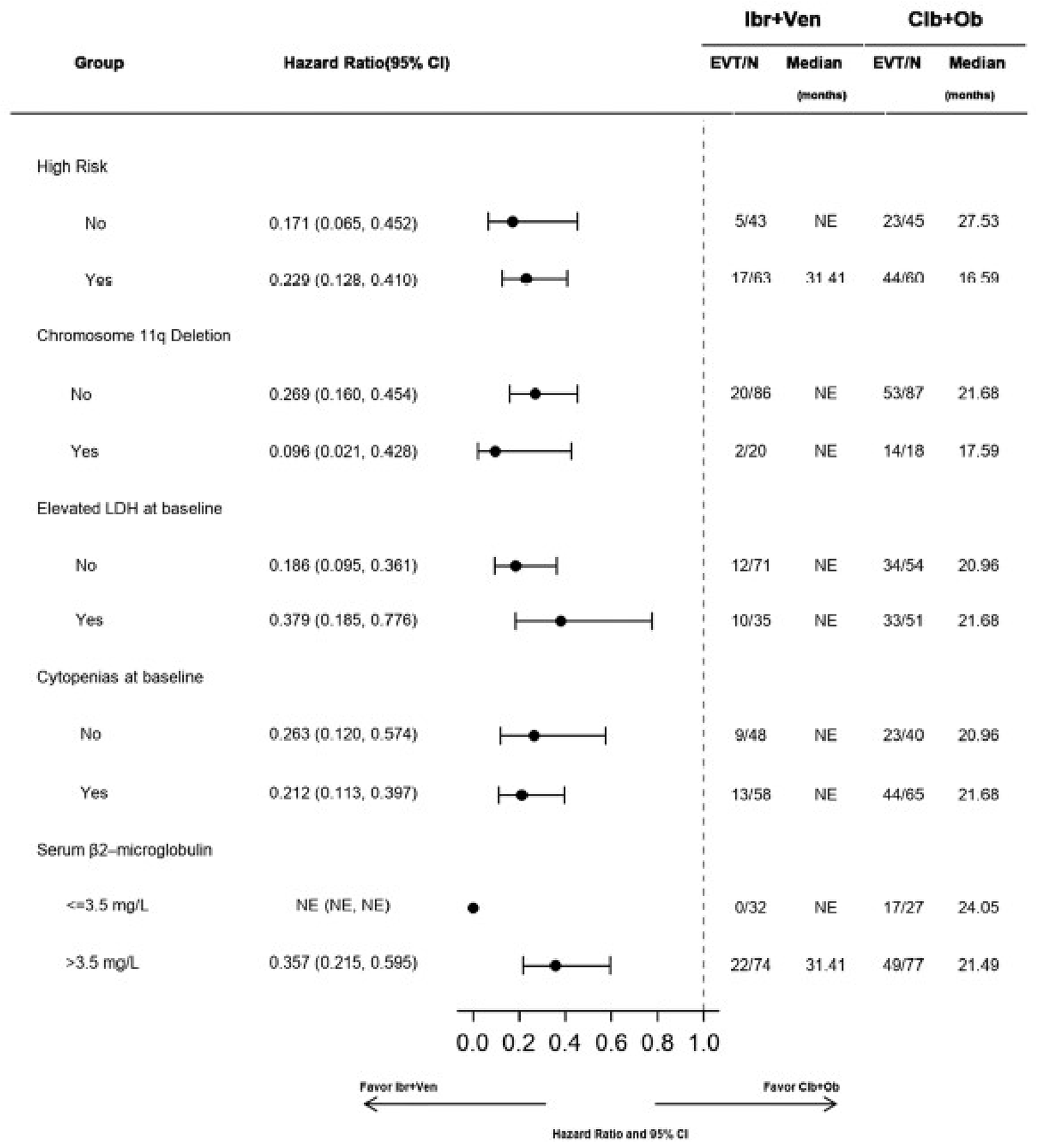
CI = confidence interval; CIRS = Cumulative Illness Rating Scale; CLL = chronic lymphocytic leukemia; ECOG = Eastern Cooperative Oncology Group; IGHV = immunoglobulin heavy chain variable; LDH = lactate dehydrogenase; NE = not estimable; SLL = small lymphocytic lymphoma.
Data cut-off: February 26, 2021.
Source: GLOW Primary Analysis CSR23
Figure 14: Kaplan-Meier Plot of Progression-Free Survival per IA at Primary Analysis for GLOW (ITT Analysis Set)
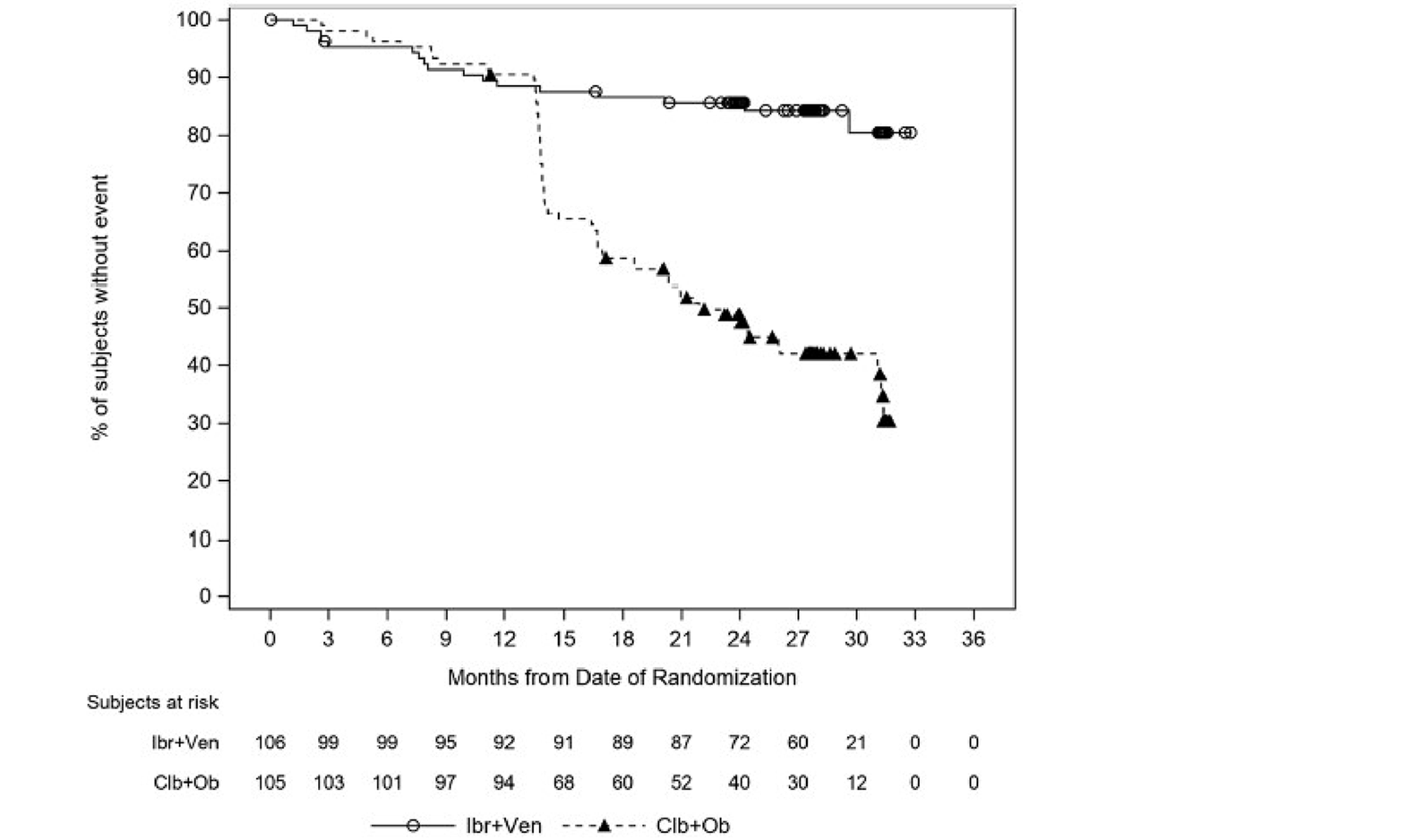
Data cut-off: February 26, 2021.
Source: GLOW Primary Analysis CSR23
Figure 15: Kaplan-Meier Plot of Progression-Free Survival per IA at Extended Follow-Up Analysis for GLOW (ITT Analysis Set)
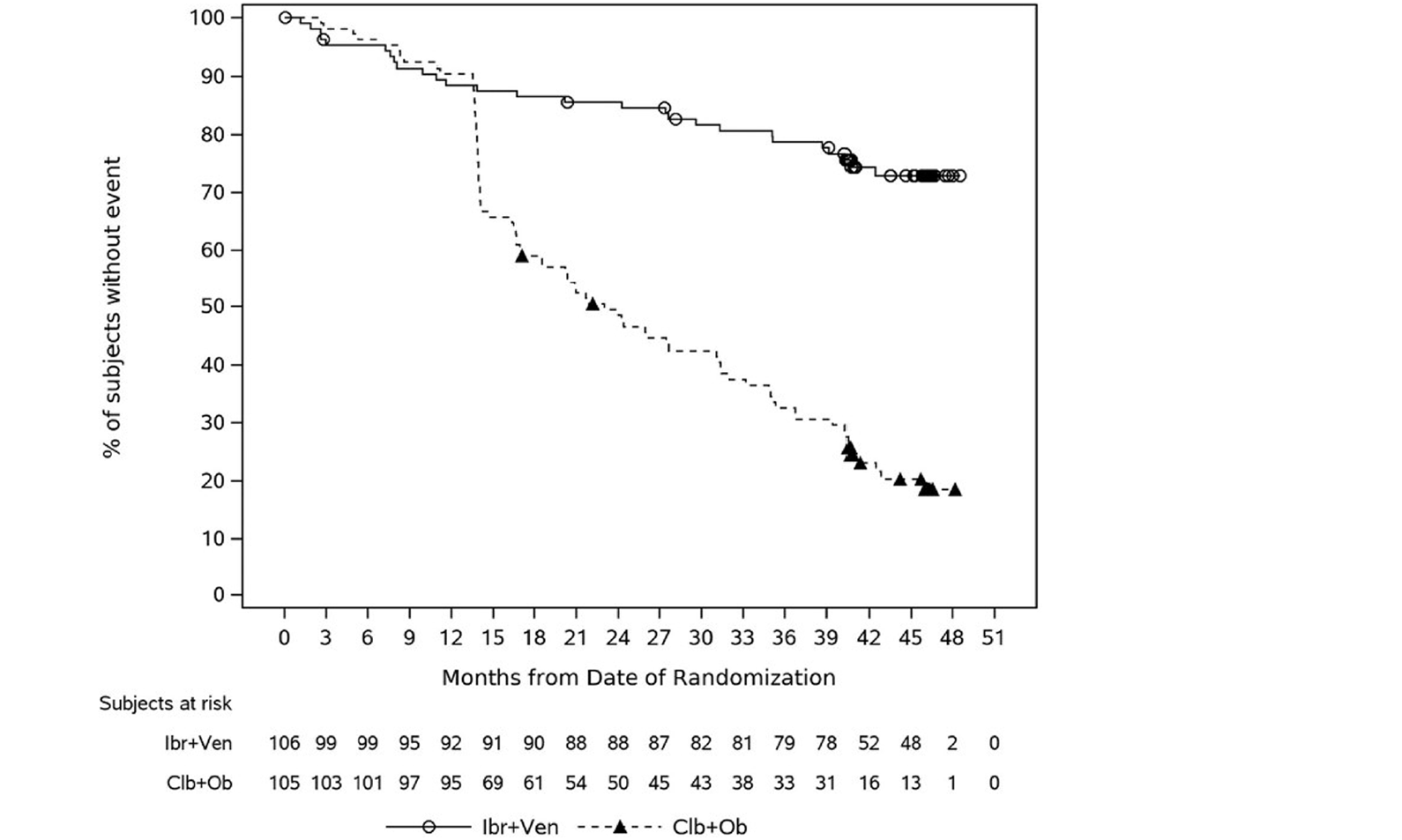
Data cut-off: August 25, 2022.
Source: GLOW Extended Follow-up Analysis CSR24
Figure 16: Kaplan-Meier Curves for Progression-Free Survival per IRC Assessment at Extended Follow-Up Analysis for CAPTIVATE FD Cohort (All-Treated Analysis Set)
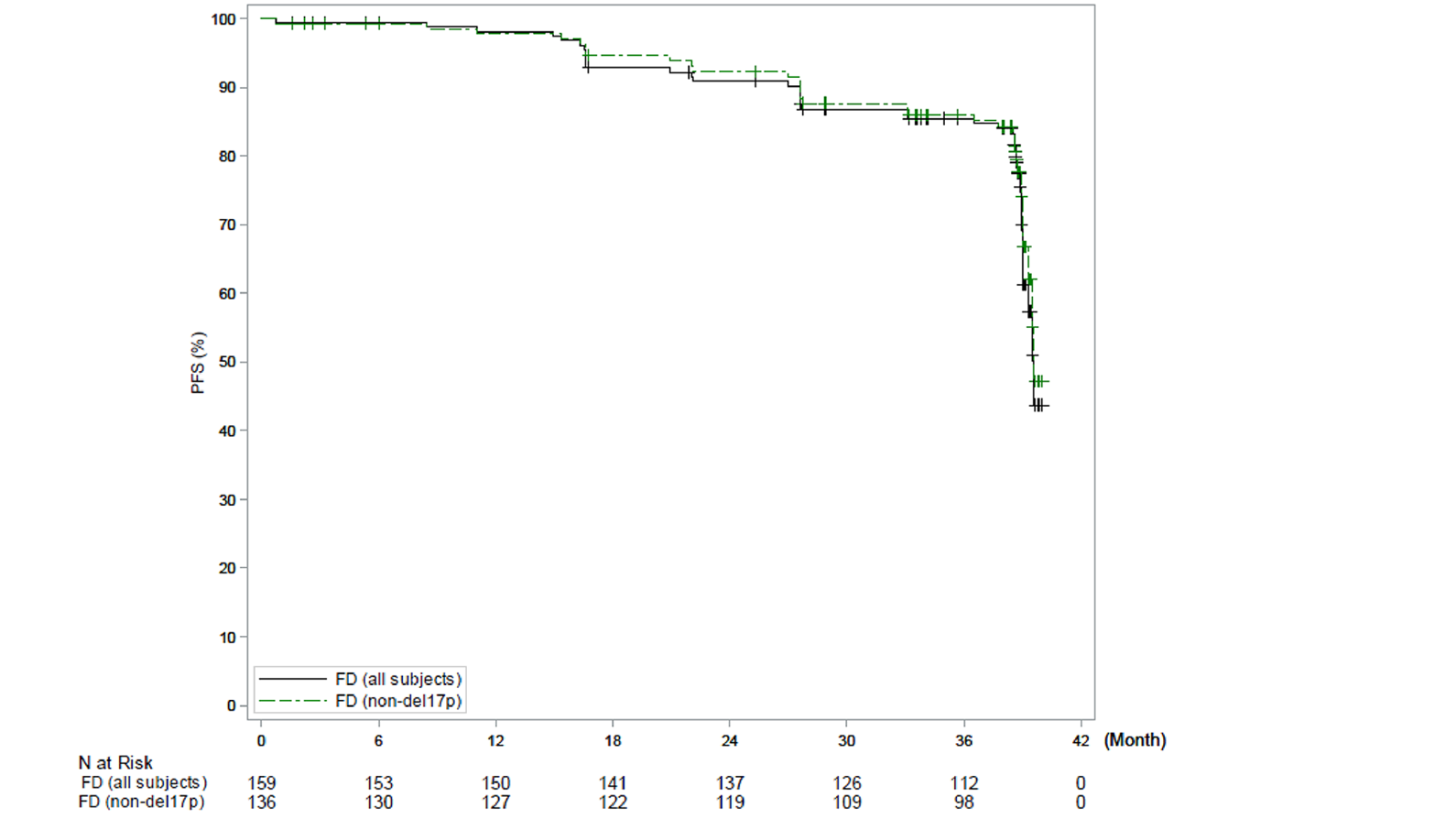
Data cut-off: August 4, 2021.
Source: CAPTIVATE Extended Follow-up Analysis CSR
Figure 17: Kaplan-Meier Curves for Progression-Free Survival per IA at Extended Follow-Up Analysis for CAPTIVATE FD Cohort (All-Treated Analysis Set)
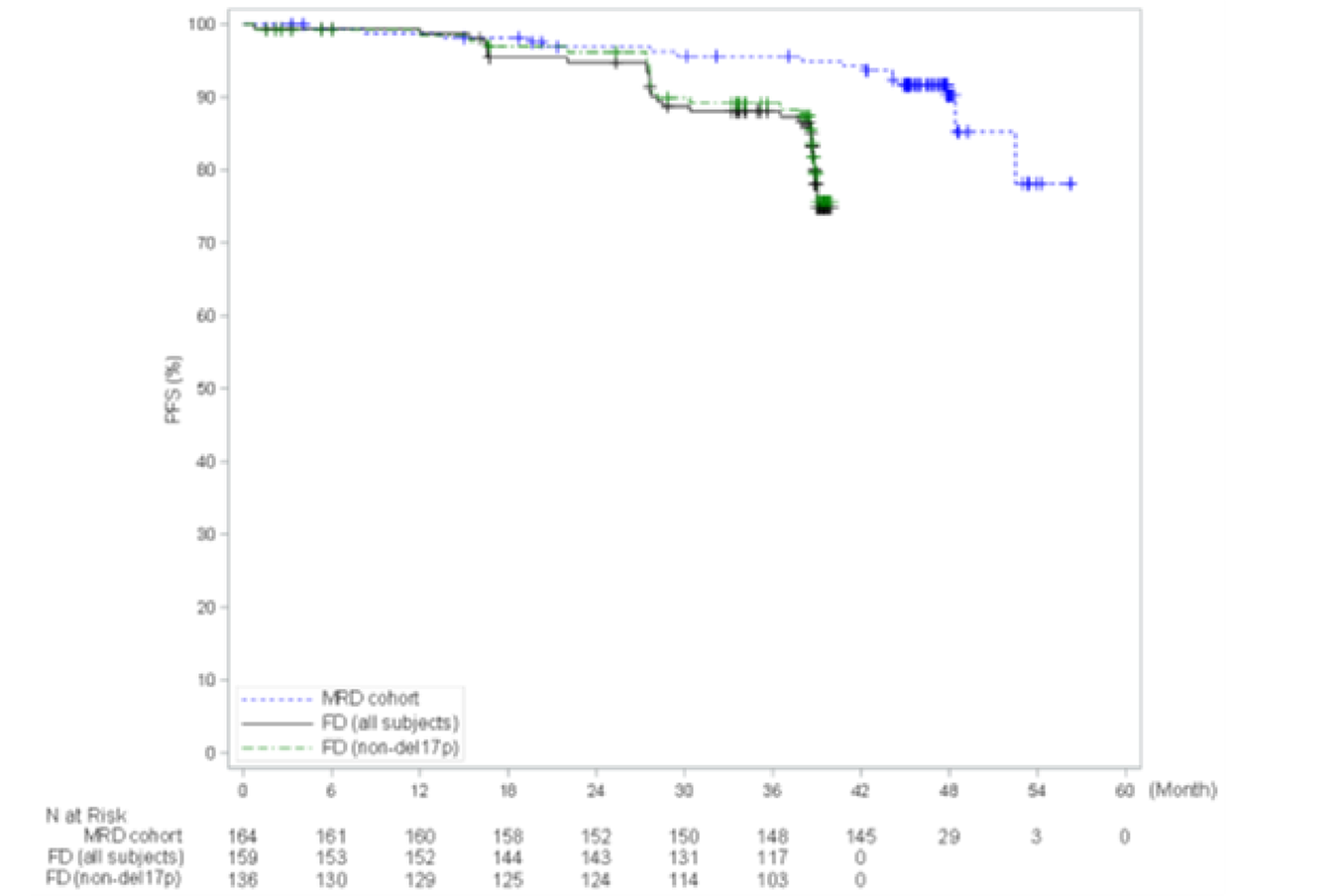
Data cut-off: August 4, 2021.
Source: CAPTIVATE Extended Follow-up Analysis CSR
Figure 18: Forest Plot of CR Rate Based on Investigator Assessment Subgroups at Primary Analysis for CAPTIVATE FD Cohort (All-Treated Analysis Set)
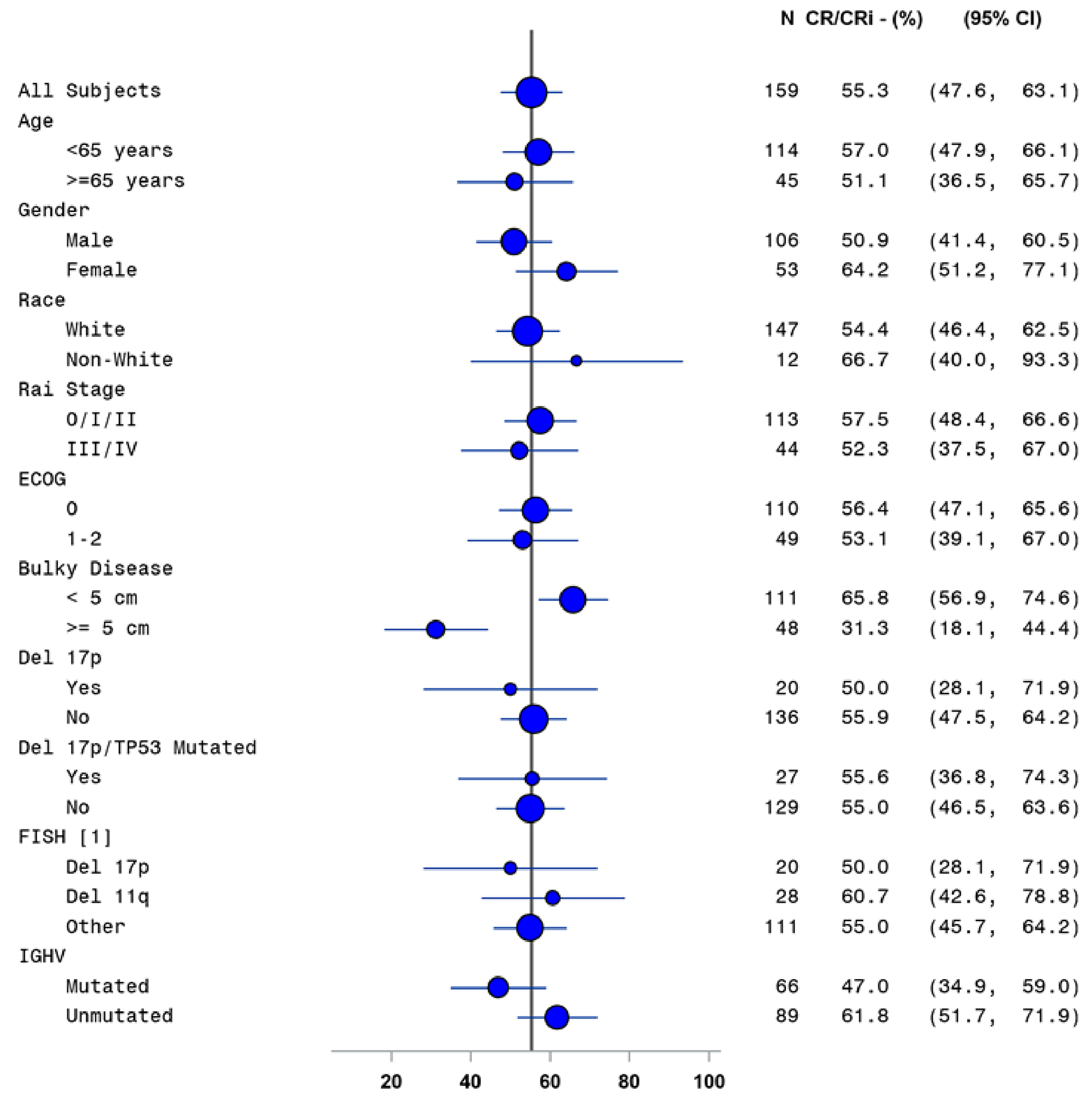
CI = confidence interval; CR = complete response; CRi = complete response with incomplete bone marrow recovery; del 11q = deletion of the long arm of chromosome 11; del(17p) = deletion of the short arm of chromosome 17; ECOG = Eastern Cooperative Oncology Group; FD = fixed duration; FISH = fluorescence in situ hybridization; IGHV = immunoglobulin heavy chain variable.
Data cut-off: November 12, 2020.
Source: CAPTIVATE Primary CSR.
Figure 19: Forest Plot of CR Rate Based on IRC Assessment Subgroups at Primary Analysis for CAPTIVATE FD Cohort (All-Treated Analysis Set)
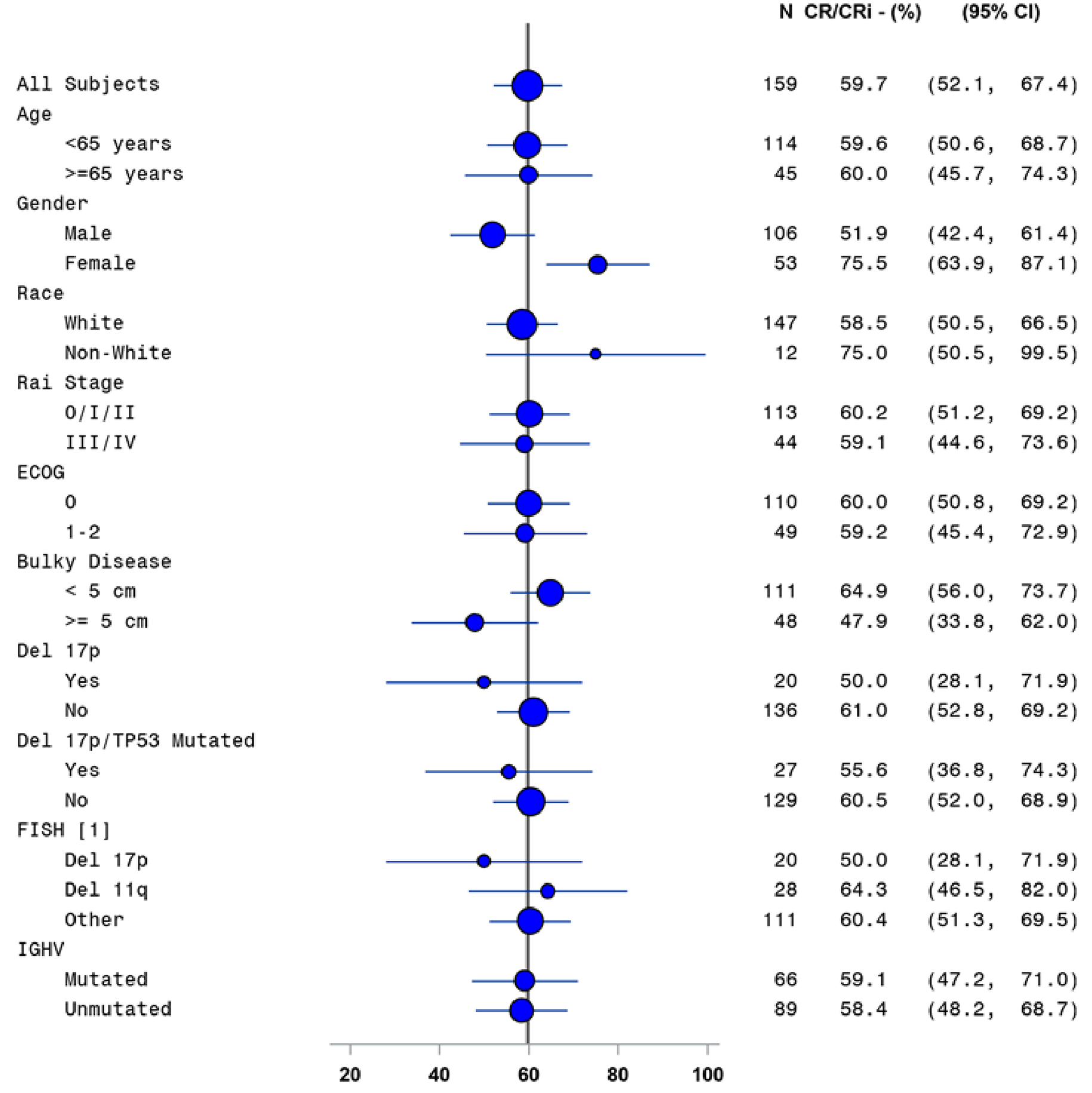
CI = confidence interval; CR = complete response; CRi = complete response with incomplete bone marrow recovery; del 11q = deletion of the long arm of chromosome 11; del(17p) = deletion of the short arm of chromosome 17; ECOG = Eastern Cooperative Oncology Group; FD = fixed duration; FISH = fluorescence in situ hybridization; IGHV = immunoglobulin heavy chain variable.
Data cut-off: November 12, 2020.
Source: CAPTIVATE Primary CSR.
Table 36: Secondary IPD Analyses for Ibrutinib-Venetoclax Versus BR, Ibrutinib, and FCR
Comparison, outcome, analysis | Results | P value | |
|---|---|---|---|
Fludarabine-ineligible: I+V vs. BR | |||
PFS | HR (95% CI) I+V vs. BRa | P valuea | |
ATC | |||| ||||||||||| | |||||| | |
ATO | |||| ||||||||||| | ||||||| | |
OS | I+V % patients with event | BR % patients with event | P valueb |
ATC | ||||| | ||||| | |||||| |
ATO | ||||| | ||||| | |||||| |
CR | OR (95% CI) I+V vs. BRc | P valuec | |
ATC | |||| ||||||||||| | |||||| | |
ATO | |||| ||||||||||| | |||||| | |
Fludarabine-ineligible: I+V vs. Ibrutinib | |||
PFS | I+V % patients with event | Ibrutinib % patients with event | P valueb |
ATC | |||||| | |||||| | |||||| |
ATO | |||||| | |||||| | |||||| |
OS | I+V % patients with event | Ibrutinib % patients with event | P valueb |
ATC | ||||| | |||||| | |||||| |
ATO | ||||| | |||||| | |||||| |
CR | OR (95% CI) I+V vs. Ibrutinibc | P valuec | |
ATC | |||| |||||||||||| | ||||||| | |
ATO | ||||| |||||||||||| | |||||| | |
Fludarabine-eligible: I+V vs. FCR | |||
PFS | HR (95% CI) I+V vs. FCRa | P valuea | |
ATC | |||| ||||||||||| | |||||| | |
ATO | |||| ||||||||||| | |||||| | |
OS | HR (95% CI) I+V vs. FCRa | P valuea | |
ATC | |||| ||||||||||| | |||||| | |
ATO | |||| ||||||||||| | |||||| | |
CR | OR (95% CI) I+V vs. FCRc | P valuec | |
ATC | |||| ||||||||||| | |||||| | |
ATO | |||| ||||||||||| | |||||| | |
ATC = average treatment effect in the control, ATO = average treatment effect in the overlap, BR = bendamustine plus rituximab, CI = confidence interval, CR = complete response; FCR = fludarabine plus cyclophosphamide plus rituximab; HR = hazard ratio, I+V = ibrutinib plus venetoclax, IPD = individual patient data; OR = odds ratio; OS = overall survival, PFS = progression-free survival.
aHR and 95% CI based on weighted Cox proportional hazard model; P value based on weighted log-rank test.
bP value based on weighted log-rank test.
cOR and 95% CI based on weighted logistic regression model.
Source: Sponsor’s ITC report I+V vs. BR27 Sponsor’s ITC report I+V vs. Ibrutinib.28 Sponsor’s ITC report I+V vs. FCR.31
Appendix 2: Ongoing Studies for the Indication
Note that this appendix has not been copy-edited.
Table 37: Details of Ongoing CLL17 Trial
Detail | CLL17 |
|---|---|
Designs and populations | |
Title | Ibrutinib Monotherapy vs. Fixed-duration Venetoclax Plus Obinutuzumab vs. Fixed-duration Ibrutinib Plus Venetoclax in Patients with Previously Untreated Chronic Lymphocytic Leukemia (CLL) |
Study design | phase III, multicentre, randomized, prospective, open-label trial |
Locations | Austria, Belgium, Denmark, Finland, Germany, Ireland, Israel, Italy, Netherlands, Norway, Spain, Sweden, Switzerland |
Populations | Adults (18 years and older) with untreated CLL |
Enrolment dates | |
Actual Study Start Date | March 1, 2021 |
Primary Completion Date: | March 2027 |
Study Completion Date: | March 2027 |
Participants | |
Estimated enrolment (N) | 897 participants |
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Intervention | Ibrutinib monotherapy
|
Comparators | FD I+V:
FD venetoclax plus obinutuzumab
|
Outcomes | |
Primary end points | Investigator-assessed PFS (up to 80 months): defined as time from randomization to the first occurrence of progression or relapse (determined using standard iwCLL guidelines), or death from any cause, whichever occurs first. |
Secondary end points |
|
Publications | |
Articles and presentations | N/A |
ALT = alanine transferase; ANC = absolute neutrophil count; AST = aspartate transferase; BM = bone marrow; CLL = chronic lymphocytic leukemia; CR = complete response; CRi = complete response with incomplete bone marrow recovery; ECOG PS = Eastern Cooperative Oncology Group Performance Status; FD = fixed duration; GFR = glomerular filtration rate; iwCLL = International Workshop on Chronic Lymphocytic Leukemia; I+V = ibrutinib plus venetoclax; MRD = minimal residual disease; N/A = not available; ORR = overall response rate; PB = peripheral blood; PFS = progression-free survival; PML = progressive multifocal leukoencephalopathy; PO = oral; uMRD = undetectable minimal residual disease.
Source: Clinicaltrials.gov - NCT04608318.73
Pharmacoeconomic Review
Abbreviations
AE
adverse event
AMR
annual mortality rate
BIA
budget impact analysis
BR
bendamustine plus rituximab
CIRS
Cumulative Illness Rating Scale
CLL
chronic lymphocytic leukemia
EQ-5D-3L
3-Level EQ-5D
EQ-5D-5L
5-Level EQ-5D
FCR
fludarabine plus cyclophosphamide and rituximab
HR
hazard ratio
ICER
incremental cost-effectiveness ratio
ITC
indirect treatment comparison
LY
life-year
MAIC
matching-adjusted indirect comparison
mIGHV
mutated immunoglobulin heavy chain variable region gene
NICE
National Institute for Health and Care Excellence
OS
overall survival
PF 1L
progression-free, first-line treatment
PF 2L
progression-free, second-line treatment
PFS
progression-free survival
PPS
postprogression survival
QALY
quality-adjusted life-year
uIGHV
unmutated immunoglobulin heavy chain variable region gene
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Ibrutinib (Imbruvica), capsule |
Submitted price | Ibrutinib: $99.84 per 140 mg capsule |
Indication | Ibrutinib with venetoclax for the treatment of adult patients with previously untreated CLL, including those with 17p deletion. |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | March 20, 2023 |
Reimbursement request | As per indication |
Sponsor | Janssen Inc. |
Submission history | Previously reviewed: Yes Indication: For the treatment of patients with CLL or small lymphocytic lymphoma with or without 17p deletion who have received at least 1 prior therapy and are not considered appropriate for treatment or re-treatment with a purine analogue (e.g., fludarabine)
Indication: For the treatment of patients with relapsed or refractory mantle cell lymphoma
Indication: For the treatment of patients with Waldenström’s macroglobulinemia who have received at least 1 prior therapy
Indication: Patients with previously untreated CLL
|
CLL = chronic lymphocytic leukemia; NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Semi-Markov model |
Target populations | Adults with previously untreated CLL. Two subpopulations were considered:
|
Treatments | Ibrutinib in combination with venetoclax |
Comparators |
|
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, life-years |
Time horizon |
|
Key data sources |
|
Submitted results |
|
Key limitations |
|
CADTH reanalysis results |
|
CIRS = Cumulative Illness Rating Scale; CLL = chronic lymphocytic leukemia; ECOG PS = Eastern Cooperative Oncology Group Performance Status; FCR = fludarabine plus cyclophosphamide and rituximab; ICER = incremental cost-effectiveness ratio; ITC = indirect treatment comparison; LY = life-year; QALY = quality-adjusted life-year.
Conclusions
The CADTH Clinical Review could not reach definitive conclusions regarding the relative efficacy of ibrutinib-venetoclax compared with the alternative treatments considered in the economic evaluation. CADTH raised concerns regarding the comparability of patient populations included in the submitted indirect treatment comparisons (ITCs). CADTH found high levels of uncertainty, particularly around relative overall survival (OS), between ibrutinib-venetoclax and all included comparators, including chlorambucil-obinutuzumab. CADTH was also not able to make definitive conclusions about the relative safety of ibrutinib-venetoclax compared to other available treatments in the fludarabine-eligible population. The uncertainty around the comparative efficacy of ibrutinib-venetoclax translates to uncertainty around the incremental cost-effectiveness of the therapy compared to other available treatments.
CADTH identified several additional limitations of the sponsor’s submitted economic evaluation that could not be addressed through reanalysis. Most prominently, the sponsor’s model did not adhere to accepted practices for the semi-Markov structure that was required to track time in state. In so doing, the model failed to correctly incorporate time dependency with respect to the initiation of second-line therapy. Given the limitations, CADTH was unable to assess the cost-effectiveness of ibrutinib-venetoclax at the submitted price.
The sponsor’s base-case results suggested that ibrutinib-venetoclax is associated with health care system cost savings, compared to some comparator therapies. This was due primarily to the fact that ibrutinib-venetoclax is prescribed as a fixed-duration dose (i.e., patients discontinue after 15 cycles), whereas ibrutinib monotherapy and acalabrutinib monotherapy are given until disease progression. Given the uncertainty associated with the economic model, CADTH conducted a comparison of drug costs between ibrutinib-venetoclax and the available alternatives. Compared to the other fixed-duration regimens, some comparator therapies (nongeneric bendamustine-rituximab [BR] and nongeneric fludarabine-cyclophosphamide-rituximab [FCR]) had higher daily acquisition costs than ibrutinib-venetoclax, whereas others (generic BR, generic FCR, and chlorambucil-obinutuzumab) were less costly. Ibrutinib-venetoclax had higher daily acquisition costs than the monotherapy regimen comparators. At publicly available list prices, cost savings could be realized from the fixed duration of ibrutinib-venetoclax if patients on ibrutinib or acalabrutinib monotherapy remain on treatment for at least 1.9 and 2.2 years, respectively. Conclusions related to total cost savings compared to other regimens are difficult to draw, as they depend on the relative effectiveness of each option and the risk of adverse events (AEs).
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process.
Patient input for this review was obtained from Lymphoma Canada, a national charity that aims to empower patients and the lymphoma community through education, support, advocacy, and research. Information for this submission was collected in an anonymous online patient survey conducted from March 22 to May 2, 2023. Of the 87 respondents to the survey, 100% were patients with chronic lymphocytic leukemia (CLL), 52% were female, and 49% were diagnosed 9 to 10 years earlier. Forty-nine (56%) respondents identified themselves as Canadian. Ten respondents had experience with the ibrutinib-venetoclax regimen. At the time of the survey, 2 were undergoing treatment, while the remaining patients were in remission. Nine of the 10 patients received access to the regimen through participation in a clinical trial. All 10 patients noted improvements in the management of white blood cell counts, lymph node size, platelet and red blood cell counts, and weight management. The fixed duration was an important attribute to respondents because it made the side effects of treatment more tolerable, compared with alternative strategies with indefinite treatment schedules. The respondents also acknowledged the benefit of an oral medication, which avoided the need to travel for treatment and receive IV administration.
Registered clinician input was received from the Ontario Health (Cancer Care Ontario) Hematology Cancer Drug Advisory Committee, as well as from a group of clinicians that treat CLL in Canada. The goal of CLL treatment is to achieve a reduction in symptoms and improvements in progression-free survival (PFS). Both submissions noted ibrutinib-venetoclax could be offered as a new first-line treatment option. However, the advisory committee’s submission suggested that this regimen would be an option for time-limited therapy for high-risk patients with specific genetic mutations or those who are unable to easily access treatments requiring IV administration. When compared with current treatment options, the submissions acknowledged that the time-limited, all-oral combination of this regimen addressed an important unmet need for the treatment of CLL. No impact on current clinical practice guidelines was noted in the clinical input.
Drug plan input sought clarification on the regimen’s place in therapy and whether patients would be eligible for re-treatment. Concerns were raised about the feasibility of adopting ibrutinib-venetoclax due to its budget impact.
Several of these concerns were addressed in the sponsor’s model:
relative cost-effectiveness of ibrutinib-venetoclax was considered against current treatment options.
CADTH was unable to address the following concerns raised from stakeholder input:
The submitted model failed to satisfy the requirements for the desired model structure and could not produce valid estimates of costs, life-years (LYs), or quality-adjusted life-years (QALYs). As a result, CADTH was unable to determine the value for money offered by the fixed-duration regimen of ibrutinib-venetoclax relative to other available treatment options.
Economic Review
The current review is for ibrutinib (Imbruvica) in combination with venetoclax for the treatment of adults with previously untreated CLL.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted an economic evaluation comparing ibrutinib-venetoclax with current first-line treatment strategies for CLL.1 As per the Health Canada indication, the population was restricted to adults with previously untreated CLL, including those with a 17p deletion. Two subpopulations were considered to reflect the increased use of novel treatment regimens that consider specific patient or disease characteristics in treatment selection. Each subpopulation was distinguished by the patient’s eligibility for treatment with fludarabine, which is part of an efficacious treatment regimen but cannot be considered for every patient with CLL. The fludarabine-ineligible subpopulation included patients older than 65 years without a 17p deletion as well as patients between 18 and 64 years with a Cumulative Illness Rating Scale (CIRS) score greater than 6 and the presence of cardiac or renal disease. The latter was defined using a creatinine clearance value less than 60 mL/min. Meanwhile, the fludarabine-eligible subpopulation included patients without a 17p deletion, a CIRS score less than or equal to 6, the absence of cardiac or renal disease, and an Eastern Cooperative Oncology Group Performance Status (ECOG PS) less than or equal to 2.1
Ibrutinib is available as a capsule for oral administration at a dose of 140 mg.2 The submitted price per capsule was $99.84. For the present indication, ibrutinib is considered in combination with venetoclax. In this regimen, the recommended dosage for ibrutinib is 420 mg once daily for fifteen 28-day cycles. Treatment with venetoclax is initiated in cycle 4, during which doses of 20 mg, 50 mg, 100 mg, and 200 mg are administered once daily for 7 days each, until a 400 mg daily dose is reached. Patients remain on the 400 mg daily dose of venetoclax in combination with ibrutinib until the conclusion of the 15th and final treatment cycle.2 At the submitted price for ibrutinib, this regimen will cost $512 per day and $214,851 per course.
Alternatives to ibrutinib-venetoclax considered in the submission were restricted to first-line treatment strategies specific to each subpopulation. In the fludarabine-ineligible subpopulation, these included chlorambucil-obinutuzumab, venetoclax-obinutuzumab, BR, as well as ibrutinib and acalabrutinib monotherapy. In the fludarabine-eligible subpopulation, ibrutinib-venetoclax was compared with a single regimen of FCR.1
Modelled outcomes included LYs and QALYs. Meanwhile, costs were estimated from the perspective of the Canadian public health care payer. Model outputs were generated using a 4-week cycle length over a 20-year time horizon in the fludarabine-ineligible subpopulation, and a 30-year time horizon in the fludarabine-eligible subpopulation. Costs and outcomes (LYs and QALYs) were discounted at 1.5%.1
Model Structure
The sponsor submitted a semi-Markov state transition model that tracked a cohort of patients from their first treatment strategy through best supportive care and death. As illustrated in Figure 1, the modelled health states were specific to a patient’s progression status and the line of therapy received. The submission considered 4 mutually exclusive health states: progression-free in first-line treatment (PF 1L), progression-free in second-line treatment (PF 2L), postprogression survival (PPS), and death. Movements between each health state were determined by time-dependent transition probabilities, with respect to time in the model and time on a given treatment.1
Upon initiation of the first-line treatment (ibrutinib-venetoclax or 1 of the subpopulation-specific alternatives) at model entry, patients were assumed to occupy the PF 1L state. After progression, patients were assumed to transition to the PF 2L state, during which second-line therapy was initiated. Eligible treatments in this state were restricted to a subpopulation-specific alternative not used as first-line therapy. Patients who experienced tumour progression in the PF 2L state transitioned to the PPS state, during which they were assumed to receive best supportive care until death. The progression risk in the PF 1L and PF 2L states was determined by 2 separate time-dependent probabilities of PFS specific to first-line therapy or second-line therapy. Meanwhile, transitions to the death state were informed by time-dependent inputs that characterized the risk of death specific to patients occupying the PF 1L, PF 2L, and PPS states.1
In addition to tracking the proportion of the cohort in each health state, the model also tracked the occurrence of AEs. Treatment-specific AE risks were assumed to be fixed throughout the model time horizon.
Model Inputs
Multiple inputs for the economic evaluation were obtained from the submitted pivotal trials and the systematic review of treatment strategies for adults with untreated CLL. The submission obtained data from 2 pivotal trials. For the fludarabine-ineligible subpopulation, data were obtained from the GLOW trial — a phase III, randomized, open-label study comparing ibrutinib-venetoclax with chlorambucil-obinutuzumab.3 Data specific to the fludarabine-eligible subpopulation were obtained from the CAPTIVATE trial, which was a phase II, triple-blind, randomized study assessing the time-limited treatment of ibrutinib-venetoclax in patients with treatment-naive CLL or small lymphocytic lymphoma.4 Subpopulation-specific data from the CAPTIVATE trial were restricted to results from the subgroup of trial patients without a 17p deletion in the fixed-duration arm.1,4 In addition to the 2 pivotal trials, the economic evaluation also included evidence from 4 additional trials identified from a systematic review conducted by the sponsor.1 The identified trials (and treatments) specific to the fludarabine-ineligible subpopulation included the CLL14 study (venetoclax-obinutuzumab, chlorambucil-obinutuzumab), the ELEVATE-TN study (acalabrutinib, chlorambucil-obinutuzumab), and the ALLIANCE study (ibrutinib, ibrutinib-rituximab, BR).5-7 Meanwhile, the review also identified the E1912 trial, which compared ibrutinib-rituximab with FCR and contained data relevant to the fludarabine-eligible subpopulation.8
Baseline demographic information, specific to each population, was obtained from 2 different trials. Data from the GLOW trial were used to determine the baseline age (median = 71 years), weight (mean = 77 kg), and sex (57.8% male) in the fludarabine-ineligible subpopulation.3 For the fludarabine-eligible subpopulation, baseline age (median = 58 years), weight (mean = 88.3 kg), and sex (67.3% male) were sourced from the E1912 trial.8
The economic model required evidence for the first-line PFS for each treatment. In the absence of direct evidence, hazard ratios (HRs) were obtained from 5 pairwise ITCs. The first ITC estimated the HR of venetoclax-obinutuzumab relative to ibrutinib-venetoclax using data from the GLOW and CLL14 trials in an anchored matching-adjusted indirect comparison (MAIC).1,3,5 The base case assumed a time-varying HR of 0.67 up to 12 months on treatment, and 2.23 thereafter.1 An anchored MAIC was used in the second ITC to estimate the HR (1.16) of acalabrutinib relative to ibrutinib-venetoclax.1,3,6 Because the treatments were not viewed to be statistically significantly different, the sponsor assumed no difference in treatment effectiveness (HR = 1) in the base case. This assumption was also made for the third ITC, which estimated the HR of ibrutinib relative to ibrutinib-venetoclax, using data from the ALLIANCE and GLOW trials. The fourth ITC used the propensity score method to estimate the HR (3.45) of BR relative to ibrutinib-venetoclax using data from the ALLIANCE and GLOW trials.1,3,7 Last, propensity score methods were also used to estimate the HR (0.44) of ibrutinib-venetoclax relative to FCR in the fludarabine-eligible subpopulation using data from the E1912 and GLOW trials.1,3,8
The model relied on 2 inputs to inform the time-dependent probability of remaining progression-free during the first or second line of therapy (PFS after first-line treatment and PFS after second-line treatment). Both inputs were estimated from extrapolations of treatment-specific survival functions. For a given cycle, the probability of remaining progression-free was calculated as the difference in survival probabilities between the current and preceding cycles, divided by the survival probability in the preceding cycle. Each treatment-specific survival function for first-line and second-line PFS were generated by combining the previously mentioned HRs with a reference parametric survival function.
For first-line PFS, parametric survival models were fit to data from the ibrutinib-venetoclax arm of the GLOW trial (fludarabine-ineligible subpopulation) and the FCR arm of the E1912 trial (fludarabine-eligible subpopulation).1,3,8 For both subpopulations, models were fit using the Gompertz, log-normal, exponential, Weibull, log-logistic, gamma, and generalized gamma distributions. Assessment of model fit statistics and consultation with clinical experts led to the base-case selection of the exponential distribution in the fludarabine-ineligible subpopulation and the Weibull distribution in the fludarabine-eligible subpopulation.1 The remaining survival curves (fludarabine-ineligible: acalabrutinib, BR, ibrutinib, venetoclax-obinutuzumab; fludarabine-eligible: ibrutinib-venetoclax) were generated by applying the treatment-specific HRs to the reference survival curve (fludarabine-ineligible: ibrutinib-venetoclax; fludarabine-eligible: FCR). For chlorambucil-obinutuzumab in the fludarabine-ineligible subpopulation, data from the chlorambucil-obinutuzumab arm of the GLOW trial were fitted to a flexible parametric survival model with 5 splines.1,3 This method was justified to capture the irregular hazard observed in the trial data.
For second-line PFS, parametric survival models were fit to individual patient data from the ibrutinib arm of the RESONATE trial.1,9 Data were restricted to patients with 1 or 2 prior lines of treatment and a median follow-up time of 65 months. Models were fit using the Gompertz, log-normal, exponential, Weibull, log-logistic, and generalized gamma distributions. Assessment of model fit statistics and consultation with clinical experts led to the base-case selection of the exponential distribution. It was assumed that second-line PFS would be equivalent for all treatments and subpopulations considered in the economic evaluation.1
Mortality risk was incorporated in the model as a time-dependent probability specific to each health state and treatment. Input values represented the maximum from a comparison of the general population mortality risk with state-specific estimates obtained from trial data. Statistics Canada Life Tables were used to estimate the general population mortality risk in each cycle.10 Values represented the sex-weighted mean for each age of the cohort over the time horizon of the model.1 State-specific mortality risks were calculated using the annual mortality rate (AMR), which was calculated as the number of deaths divided by the total number of patient years spent in that state (PF 1L, PF 2L, and PPS) for a specific trial.1 For the PF 1L state, the AMR was calculated using data obtained from the GLOW trial (fludarabine-ineligible subpopulation: ibrutinib-venetoclax and chlorambucil-obinutuzumab), CAPTIVATE trial (fludarabine-eligible subpopulation: ibrutinib-venetoclax), and E1912 trial (fludarabine-eligible subpopulation: FCR).1,3,4,8 It was assumed that the AMR for all treatments would be equal to the value specific to chlorambucil-obinutuzumab in the fludarabine-ineligible subpopulation and FCR in the fludarabine-eligible subpopulation.1 For the PF 2L and PPS states, the AMR for both subpopulations was calculated using data specific to the ibrutinib arm of the RESONATE trial.1,9 Each AMR was converted to a 28-day mortality risk, which was assumed to be a constant risk over the time horizon of the model.
AEs in the model were restricted to grade 3 or 4 events, which occurred in at least 5% of patients in at least 1 of the treatment arms from the available data sources. In the fludarabine-ineligible subpopulation, treatment-specific AE risks were sourced from GLOW (ibrutinib-venetoclax, chlorambucil-obinutuzumab), RESONATE (ibrutinib), CLL14 (venetoclax-obinutuzumab), ELEVATE-TN (acalabrutinib), and CLL10 (BR) trials.3,11-14 In the fludarabine-eligible subpopulation, treatment-specific AE risks were sourced from the CAPTIVATE trial and the E1912 trial (FCR).8,15 Specific AEs included allergy, anemia, arthritis, atrial fibrillation, cardiac-related AEs, cataract, gastrointestinal disorders, hyponatremia, infections, leukocyte- and lymphocyte-related AEs, musculoskeletal and connective tissue disorders, neutropenia, and thrombocytopenia. If a trial failed to report overall gastrointestinal disorders or infection rates, the rate of diarrhea and pneumonia were used instead.1
Health-related quality of life was captured in the model by combining health state utilities with disutilities associated with each AE. Health state utility values were obtained from 2 sources. For the PF 1L state, 5-Level EQ-5D (EQ-5D-5L) data from the GLOW trial were transformed to the 3-Level EQ-5D (EQ-5D-3L) scale to obtain age- and sex-adjusted utilities for each subpopulation. Preference weights were assumed to follow the general population of the UK. For the PF 2L and PPS states, EQ-5D-3L data were obtained from an economic evaluation published in 2016.16 It was assumed that there would be no quality-of-life difference between these 2 health states.
AE-specific disutilities were obtained from a patient preference study published in 2013 and a 2017 National Institute for Health and Care Excellence (NICE) technology appraisal of venetoclax for CLL.17,18 A specific AE disutility was recorded as the product of the disutility value, the assumed duration (in days) of the AE, and the proportion of patients experiencing the specific AE in a given cycle.1 It was assumed that AE disutilities would only apply to patients receiving their first-line of therapy for CLL.
The economic evaluation considered costs associated with treatment acquisition, administration, initiation, and monitoring. Treatment-acquisition costs were determined by applying treatment prices to the dosing schedule for each regimen considered in the model. The cost of each treatment was obtained from the sponsor’s submitted price for ibrutinib, as well as the Ontario Drug Formulary and the Ontario Exceptional Access Program.1,19,20 Prices for unlisted medications (obinutuzumab, bendamustine, fludarabine, cyclophosphamide) were obtained from a private distributor.1 Although the acquisition costs did not include dispensing fees or mark-ups, wastage based on treatment adherence (dose intensity) was included in the base case for first-line and second-line treatments. Treatment-administration costs were considered for any treatment that required IV administration. The model assumed an IV administration cost of $206.25, after inflating an estimate from 2010 to 2022.21 For second-line treatment, costs were calculated by combining the acquisition and administration costs weighted by the percent of patients assumed to be on each regimen. Furthermore, second-line treatment costs had to consider the delay between progression on first-line treatment and second-line initiation. As a result, the model assumed patients would only begin second-line treatment 14 cycles after entering the PF 2L state.
Initiation costs included tests and vaccination required before the initiation of first-line or second-line treatment. Physician costs to administer the influenza and pneumococcal vaccines were obtained the Ontario Schedule of Benefits.22 The Ontario Schedule of Benefits for Laboratory Services was used to obtain unit costs to test patients for immunoglobulin heavy chain variable region gene status, hepatitis B, hepatitis C, and cytomegalovirus.23 The cost of a HIV test was obtained from the British Columbia laboratory fee schedule.24
Monitoring costs included service use associated with routine follow-up and laboratory tests. After consultation with clinical experts, the sponsor assumed that routine follow-up would involve pharmacist consultation and regular visits with a hematologist. The cost of pharmacist time was obtained from data published by Statistics Canada, and the cost of a hematologist visit was obtained from the Ontario Schedule of Benefits and Fees for physician services.22,25 Monitoring tests were assumed to include a full blood count, chest X-ray, as well as a lactate dehydrogenase test, a urea and electrolytes test, a liver function test, and an immunoglobin blood test. Unit costs for laboratory tests were obtained for the Ontario Schedule of Benefits for Laboratory Services, and the cost of a chest X-ray was obtained from the Ontario Case Costing Initiative.23,26 The sponsor assumed that the frequency of follow-up for each service contact would align with the expectations of the consulted clinical experts.1 At treatment switch, patients are also assumed to undergo genetic testing for 17p deletion, as well as for tumour protein 53 mutation and 11q deletion, using Ontario prices.23 Monitoring costs also included immunoglobulin tests after an infection AE, as well as a bone marrow biopsy and hospitalization at progression.1 AE costs were obtained from the Ontario Case Costing Initiative, and values were inflated from 2017 to 2018 to 2022 Canadian dollars.26 Terminal care costs were included as a one-time cost at entry to the death state, based on a study of palliative care patients in Ontario in 2002 and 2003.27
Summary of Sponsor’s Economic Evaluation Results
For both subpopulations, the costs and QALYs of each alternative treatment strategy were generated using a Monte Carlo simulation of 500 iterations. Despite the nonlinear model structure, results from the deterministic and probabilistic simulations were aligned. The results from the probabilistic base case are presented here.
Base-Case Results
The submitted analysis was based on publicly available prices of the comparator treatments. Results from the base case of the submitted economic evaluation are presented in Table 3.
In the fludarabine-ineligible subpopulation, the expected costs and QALYs for ibrutinib-venetoclax combination therapy were $348,392 and 8.2, respectively. Only 1 other alternative, acalabrutinib, was identified to be on the cost-effectiveness frontier. This treatment was more expensive and more effective than ibrutinib-venetoclax, with an incremental cost-effectiveness ratio (ICER) of $2,386,278. At a willingness-to-pay threshold of $50,000 per QALY, ibrutinib-venetoclax had a 100% probability of being cost-effective compared to all other comparator treatments.
In the fludarabine-eligible subpopulation, the expected costs and QALYs for ibrutinib-venetoclax combination therapy were $496,651 and 12.3, respectively. Results indicated that ibrutinib-venetoclax was more costly and more effective than FCR, with an ICER of $37,698. At a threshold of $50,000 per QALY, ibrutinib-venetoclax had an 81% probability of being cost-effective.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
Fludarabine-ineligible subpopulation | |||
Ibrutinib-venetoclax | $348,392 | 8.168 | Reference |
BR | $387,101 | 6.717 | Dominated by ibrutinib-venetoclax |
Chlorambucil-obinutuzumab | $449,304 | 6.371 | Dominated by ibrutinib-venetoclax |
Venetoclax-obinutuzumab | $481,270 | 7.313 | Dominated by ibrutinib-venetoclax |
Acalabrutinib monotherapy | $988,749 | 8.436 | $2,386,278 |
Ibrutinib monotherapy | $1,106,920 | 8.436 | Dominated by acalabrutinib |
Fludarabine-eligible subpopulation | |||
FCR | $405,937 | 9.853 | Reference |
Ibrutinib-venetoclax | $496,651 | 12.259 | $37,698 |
BR = bendamustine plus rituximab; FCR = fludarabine plus cyclophosphamide plus rituximab; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Source: Sponsor’s pharmacoeconomic submission.1
Sensitivity and Scenario Analysis Results
In addition to the base case, several scenario analyses were considered. The sponsor examined the impact of alternative discount rates (undiscounted and 3%, as per CADTH guidelines), alternative time horizons (fludarabine-ineligible subpopulation: 10, 15, and 30 years; fludarabine-eligible subpopulation: 15, 25, and 40 years), and a broader societal perspective on costs. Scenario analyses also considered the impact of the exclusion of administration costs, drug wastage, second-line treatment, AEs, and terminal care costs. Finally, the sponsor considered alternative health state utility values for the PF 2L and PPS states, as well as different values for the HRs estimated via indirect comparison, and different distributions for the reference PFS survival curves.
None of the scenarios had a meaningful effect on the relative cost-effectiveness of ibrutinib-venetoclax.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis:
Comparative efficacy evidence is highly uncertain against relevant comparators: The submitted economic evaluation compared ibrutinib-venetoclax with relevant alternatives that would be considered for first-line treatment. In the fludarabine-ineligible subpopulation, these included chlorambucil-obinutuzumab, venetoclax-obinutuzumab, and BR, as well as acalabrutinib and ibrutinib monotherapy. In the fludarabine-eligible subpopulation, ibrutinib-venetoclax was compared with FCR. The CADTH Clinical Review of the direct evidence concluded that ibrutinib-venetoclax could achieve superior PFS compared with chlorambucil-obinutuzumab in the fludarabine-ineligible subpopulation, whereas conclusions could not be reached for OS due to a lack of mature data. The clinical experts consulted by CADTH suggested that chlorambucil-obinutuzumab is rarely used in clinical practice and that other comparators included in the economic evaluation were more reflective of current approaches to managing CLL. The comparative efficacy of ibrutinib-venetoclax with these other comparators was estimated through ITCs and observational studies. The CADTH Clinical Review could not reach definitive conclusions about the estimates of the relative efficacy of OS or PFS with these ITCs. The high degree of uncertainty surrounding estimates of relative OS and PFS translates to a high degree of uncertainty in estimates of incremental QALYs and costs.
CADTH was unable to address this limitation.
Methods used to calculate state membership were not aligned with best practices for the semi-Markov approach: The sponsor submitted a semi-Markov state transition model that sought to track health state membership (progression-free, progressed, and death) over 3 lines of therapy (first-line regimen, second-line regimen, and best supportive care). When using a Markov model, the cohort simulation involves a relatively straightforward process in which state membership in each model cycle is determined by partitioning the estimates from the preceding cycle according to a specified set of transition probabilities.28,29 The transition probability refers to the probability of making a transition from 1 state to another at a given point in time. In other words, membership in a specific health state must be calculated by multiplying the proportion of a cohort in each state by the probability of moving to the new state and adding the results together.28,29 This is not the approach that was adopted by the sponsor. As part of the review, CADTH contacted the sponsor to seek information (time in health states) and corrections to the calculation of state membership. The sponsor replied to CADTH’s request by noting that their approach was undertaken to address issues arising from the composite nature of the PFS end point (which encompasses both progression and death). However, although CADTH agrees that the composite nature of the PFS outcome is complex, these complexities relate to the misspecification of values specific to the transitions affected by that parameter. However, in a semi-Markov model, this would have no impact on how transition probabilities are used to generate a cohort trace. Alternate approaches to the modelling of survival data from a trial are needed to distinguish progression and death events in a single framework.30,31 As a result, CADTH is unable to validate estimates of costs, LYs, or QALYs from the model.
CADTH was unable to address this limitation. Correcting the calculation of state membership would have involved a complete redevelopment of the economic model. Such activity is beyond the scope of CADTH reviews.
Failure to incorporate time dependency for second-line treatment initiation: The model included a 14-cycle (56 week) delay between progression on first-line treatment and initiation of a second-line regimen. To implement this treatment-free interval, the sponsor assumed that initiation of a second-line regimen would be delayed until the 14th cycle in the PF 2L state. Although the 14th cycle in the PF 2L state represents a time-delay of 14 cycles following progression in the PF 1L state, it may not always be the case that a patient was on treatment when that progression occurred. This is attributable to the fact that the model included fixed (ibrutinib-venetoclax, BR, chlorambucil-obinutuzumab, venetoclax-obinutuzumab, and FCR) treatment schedules. In other words, the sponsor’s assumption failed to consider the fact that patients receiving a first-line regimen with a fixed duration will initiate second-line treatment at different times after entry into the PF 2L state, depending on their response to first-line treatment.
The sponsor’s assumption that the second-line regimen would be initiated in the 14th cycle in the PF 2L state would only apply to patients who progress before or upon completion of the first-line regimen. The correct implementation of this time dependency would also need to accommodate 2 additional scenarios. First, patients who progress 1 to 14 cycles after completing a fixed-duration regimen in the PF 1L state would need to initiate a second-line regimen in the second to 13th cycles in the PF 2L state. Second, patients who progress more than 14 cycles after completing a fixed-duration regimen in the PF 1L state would be eligible for second-line treatment in the first cycle of the PF 2L state. In response to CADTH’s request for clarification, the sponsor confirmed that these additional scenarios were not considered in the submitted economic evaluation.
The failure to incorporate time dependency related to the initiation of a second-line regimen has 2 important implications for the economic evaluation. First, it resulted in the incorrect calculation of treatment costs for every alternative with a fixed treatment duration (ibrutinib-venetoclax, BR, chlorambucil-obinutuzumab, venetoclax-obinutuzumab, and FCR). In addition to accruing treatment-acquisition costs at different points in time in the PF 2L state, different monitoring and treatment costs may be expected for patients who have completed first-line treatment but have yet to begin second-line therapy. The direction of this omission is unclear because the probability of experiencing progression in the PF 1L state was a time-dependent parameter specific to each regimen. Second, it revealed a logical inconsistency in the model in which patients receive the benefit of second-line treatment before its initiation. Patients transition from the PF 1L state to the PF 2L state because of progression yet are assumed to be progression-free in the first cycle of the PF 2L state before receiving any second-line treatment. In other words, the model was unable to distinguish between patients who progressed but have yet to initiate second-line therapy from patients who are progression-free after beginning second-line therapy. The model therefore did not accurately reflect the trajectory of patients moving through the different states of health, making its predictions of cost and QALYs invalid.
CADTH was unable to address this limitation.
Missing comparators: The sponsor acknowledged that fludarabine-rituximab, chlorambucil-rituximab, and rituximab-cyclophosphamide-vincristine-prednisone would all be appropriate treatments in the fludarabine-ineligible subpopulation. These potentially relevant alternatives were excluded from the economic evaluation because the clinical experts consulted by the sponsor suggested that they might not be commonly used.1 CADTH guidelines state that the identification of comparators should not be limited to a specific type or class of intervention. All interventions that may be used for treatment or those that may be displaced by a new technology should be considered in an economic evaluation.32 The cost-effectiveness of ibrutinib-venetoclax compared to these omitted comparators is unknown.
This limitation could not be addressed by CADTH.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CADTH (refer to Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CADTH comment |
|---|---|
Upon progression during first-line treatment, patients are assumed to be progression-free, even if second-line therapy has not been initiated. | Inappropriate. Clinical experts consulted by CADTH stated that progression status must be confirmed through follow-up assessment. The experts deemed it to be unlikely for patients to become progression-free following progression on first-line treatment without an active second-line therapy. |
There is no difference in HRQoL between patients who are progression-free on second-line treatment and those who are progressed. | Unclear. Clinical expert feedback solicited by CADTH confirmed that patients may experience worse quality of life due to the adverse events while on treatment. However, the extent to which the avoidance of adverse events is as preferable to the treatment benefits (i.e., delayed progression) is unclear. |
HRQoL = health-related quality of life.
CADTH Reanalyses of the Economic Evaluation
Base-Case Results
The inability to address several key limitations prevented CADTH from specifying a base case or conducting a reanalysis of the economic evaluation, as previously described. First, the specified model did not use a valid approach to determine state membership for a semi-Markov model. Verification of the equations to determine state membership revealed methods that were inconsistent with the established standards for the declared modelling approach.28,29 Second, the specified model failed to consider the potential time delays between entry into the PF 2L state and the initiation of second-line treatment. This was problematic because the treatment-acquisition costs would be inconsistent with the expected use of fixed-duration regimens. Third, the model could not distinguish progressed patients eligible for second-line therapy from those who would be progression-free after second-line initiation. This suggested that the model lacked face validity, as the distinction may affect the accumulation of costs and QALYs, and by extension the relative cost-effectiveness of each regimen. Most notably, the identified limitations in the clinical evidence meant that CADTH could not draw firm conclusions about the efficacy of ibrutinib-venetoclax compared to other available treatments, particularly those that are most frequently used in clinical practice. Consequently, CADTH could not derive valid estimates of costs, QALYs, or LYs for ibrutinib-venetoclax or any of its comparators in either the fludarabine-eligible subpopulation or the fludarabine-ineligible subpopulation. The cost-effectiveness of ibrutinib-venetoclax in adult patients with previously untreated CLL is unknown.
The sponsor’s base-case results suggested that ibrutinib-venetoclax is cost saving compared to some comparator therapies. This was due primarily to the fact that ibrutinib-venetoclax is prescribed as a fixed-duration dose (i.e., patients discontinue after 15 cycles), whereas other treatment regimens (ibrutinib monotherapy and acalabrutinib monotherapy) are given until disease progression. In the absence of a valid decision model to estimate cost-effectiveness, CADTH conducted cost comparison analyses based on the submitted publicly available list prices of all treatments, considering only the cost of the drugs themselves. As detailed in Table 5, the drug cost of the ibrutinib-venetoclax regimen is $512 per day and $214,851 for the complete fifteen 28-day cycle regimen. Compared to the other fixed-duration regimens, ibrutinib-venetoclax has higher acquisition costs than BR ($154,670) and FCR ($158,412) in the fludarabine-eligible subpopulation. Ibrutinib-venetoclax has higher acquisition costs than venetoclax-obinutuzumab ($126,935) and chlorambucil-obinutuzumab ($44,253) in the fludarabine-ineligible subpopulation. Meanwhile, the degree to which ibrutinib-venetoclax has lower acquisition costs than acalabrutinib ($7,615 per 28-day cycle) or ibrutinib ($8,386 per 28-day cycle) monotherapy will depend on the number of cycles that patients remain on either treatment. At 15 cycles (the fixed duration of ibrutinib-venetoclax), the acquisition cost of acalabrutinib and ibrutinib was calculated to be $114,219 and $125,790, respectively. To accumulate acquisition costs greater than ibrutinib-venetoclax, patients on acalabrutinib monotherapy would need to remain on treatment for twenty-nine 28-day cycles (2.22 years: $220,824). Meanwhile, patients on ibrutinib monotherapy would need to remain on treatment for twenty-six 28-day cycles (1.99 years: $218,040). These estimates are difficult to interpret for resource-allocation decisions, as they do not reflect the probability of remaining progression-free after 29 or 26 cycles on treatment. The extent to which 1 treatment is cost saving compared to another is a product not only of drug cost and duration of therapy, but also of survival and health care resource use as well. For the reasons outlined here, CADTH could not estimate these important outcomes and their associated costs.
Issues for Consideration
This section pertains to the full economic review and is based on issues identified by CADTH reviewers:
Publicly available list prices may not reflect actual acquisition costs incurred by public plans. The true acquisition costs paid by Canadian public drug plans may be lower than those listed on public formularies. CADTH’s cost comparison scenarios and budget impact analysis (BIA) are sensitive to this issue, as all drugs included in this review (either as monotherapies or in combination) have negotiated prices with various health care jurisdictions in Canada.
In May 2023, NICE published guidance from a single technology assessment of the ibrutinib-venetoclax regimen for untreated CLL in adults.33 The methodological summary suggested the economic evaluation was almost identical to the 1 considered for the present review. However, the NICE appraisal considered the model structure to be adequate for decision-making. Although concerns were identified with the estimation of transition probabilities, there was no mention of limitations regarding the calculation of state membership or the time dependency related to second-line treatment initiation.33 CADTH is unable to comment on the approaches that independent appraisal teams take to review and identify limitations in economic evaluations. In the context of the submission provided to CADTH, the methods used to determine state membership were inconsistent with the requirements of any Markov-type model structure. Additionally, the model failed to correctly incorporate the treatment-free assumption after first-line progression. As a result, the findings from the NICE appraisal have no bearing on CADTH’s assessment that the submitted model was inadequate for decision-making.
Overall Conclusions
The CADTH Clinical Review could not reach definitive conclusions regarding the relative efficacy of ibrutinib-venetoclax compared with the alternative treatments considered in the economic evaluation. CADTH raised concerns regarding the comparability of patient populations included in the submitted ITCs. CADTH found high levels of uncertainty, particularly around relative OS, between ibrutinib-venetoclax and all included comparators, including chlorambucil-obinutuzumab. CADTH was also not able to make definitive conclusions about the relative safety of ibrutinib-venetoclax compared to other available treatments in the fludarabine-eligible population. The uncertainty around the comparative efficacy of ibrutinib-venetoclax translates to uncertainty around the incremental cost-effectiveness of the therapy compared to other available treatments.
CADTH identified several additional limitations of the sponsor’s submitted economic evaluation that could not be addressed through reanalysis. Most prominently, the sponsor’s model did not adhere to accepted practices for the semi-Markov structure that was required to track time in state. In so doing, the model failed to correctly incorporate the time dependency with respect to the initiation of second-line therapy. CADTH identified these issues for the sponsor, who declined to address them. The consequence of these limitations is that the model lacked sufficient rigour to accurately reflect costs and outcomes for the modelled patient cohort. Therefore, CADTH was unable to assess the cost-effectiveness of ibrutinib-venetoclax at the submitted price.
The sponsor’s base-case results suggested that ibrutinib-venetoclax is cost saving compared to some comparator therapies. This was due primarily to the fact that ibrutinib-venetoclax is prescribed as a fixed-duration dose (i.e., patients discontinue after 15 cycles), whereas ibrutinib monotherapy and acalabrutinib monotherapy are given until disease progression. In the absence of a valid economic model, CADTH conducted a comparison of the acquisition costs between ibrutinib-venetoclax and the available alternatives. Compared to the other fixed-duration regimens, the acquisition cost for ibrutinib-venetoclax was in the middle of the alternatives included in the submission. Ibrutinib-venetoclax had higher daily acquisition costs than the included monotherapy regimens. At publicly available list prices, cost savings could be realized from the fixed duration of ibrutinib-venetoclax if patients on ibrutinib or acalabrutinib monotherapy remain on treatment for at least 1.9 and 2.2 years, respectively. Conclusions related to the total cost savings compared to other regimens are difficult to draw, as they depend on the relative effectiveness of each option and the risk of AEs.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Imbruvica (ibrutinib) 140 mg oral capsules [internal sponsor's package]. Toronto (ON): Janssen Inc.; 2023 Apr 21.
2.Imbruvica® (ibrutinib): tablets 140 mg, 280 mg, 420 mg, 560 mg, capsules 140 mg [product monograph]. Toronto (ON): Janssen Inc. ; 2023 Mar 15.
3.Clinical Study Report: A Randomized, Open-label, Phase 3 Study of the Combination of Ibrutinib plus Venetoclax versus Chlorambucil plus Obinutuzumab for the First-line Treatment of Subjects with Chronic Lymphocytic Leukemia (CLL)/Small Lymphocytic Lymphoma (SLL) GLOW [internal sponsor's report]. Janssen; 2022.
4.Clinical Study Report: Phase 2 Study of the Combination of Ibrutinib Plus Venetoclax in Subjects with Treatment-Naive Chronic Lymphocytic Leukemia / Small Lymphocytic Lymphoma. CAPTIVATE [internal sponsor's report]. Janssen; 2021.
5.Fischer K, Al-Sawaf O, Bahlo J, et al. Venetoclax and obinutuzumab in patients with CLL and coexisting conditions. New England Journal of Medicine. 2019;380(23):2225-2236. PubMed
6.Sharman JP, Egyed M, Jurczak W, et al. Acalabrutinib with or without obinutuzumab versus chlorambucil and obinutuzumab for treatment-naive chronic lymphocytic leukaemia (ELEVATE-TN): a randomised, controlled, phase 3 trial. The Lancet. 2020;395(10232):1278-1291. PubMed
7.Woyach JA, Ruppert AS, Heerema NA, et al. Ibrutinib regimens versus chemoimmunotherapy in older patients with untreated CLL. New England Journal of Medicine. 2018. PubMed
8.Shanafelt TD, Wang XV, Hanson CA, et al. Long-term outcomes for ibrutinib–rituximab and chemoimmunotherapy in CLL: updated results of the E1912 trial. Blood, The Journal of the American Society of Hematology. 2022;140(2):112-120. PubMed
9.Munir T, Brown JR, O'Brien S, et al. Final analysis from RESONATE: Up to six years of follow‐up on ibrutinib in patients with previously treated chronic lymphocytic leukemia or small lymphocytic lymphoma. American journal of hematology. 2019;94(12):1353-1363. PubMed
10.Table: 13-10-0114-01. Life expectancy and other elements of the life table, Canada, all provinces except Prince Edward Island. Ottawa (ON): Statistics Canada; 2022: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310011401. Accessed 2023 May 23.
11.Burger JA, Barr PM, Robak T, et al. Long-term efficacy and safety of first-line ibrutinib treatment for patients with CLL/SLL: 5 years of follow-up from the phase 3 RESONATE-2 study. Leukemia. 2020;34(3):787-798. PubMed
12.Al-Sawaf O, Zhang C, Tandon M, et al. Venetoclax plus obinutuzumab versus chlorambucil plus obinutuzumab for previously untreated chronic lymphocytic leukaemia (CLL14): follow-up results from a multicentre, open-label, randomised, phase 3 trial. The Lancet Oncology. 2020;21(9):1188-1200. PubMed
13.Sharman JP, Egyed M, Jurczak W, et al. Efficacy and safety in a 4-year follow-up of the ELEVATE-TN study comparing acalabrutinib with or without obinutuzumab versus obinutuzumab plus chlorambucil in treatment-naïve chronic lymphocytic leukemia. Leukemia. 2022;36(4):1171-1175. PubMed
14.Eichhorst B, Fink A-M, Bahlo J, et al. First-line chemoimmunotherapy with bendamustine and rituximab versus fludarabine, cyclophosphamide, and rituximab in patients with advanced chronic lymphocytic leukaemia (CLL10): an international, open-label, randomised, phase 3, non-inferiority trial. The lancet oncology. 2016;17(7):928-942. PubMed
15.Wierda WG, Siddiqi T, Flinn I, et al. Phase 2 CAPTIVATE results of ibrutinib (ibr) plus venetoclax (ven) in first-line chronic lymphocytic leukemia (CLL). American Society of Clinical Oncology; 2018.
16.Herring W, Pearson I, Purser M, et al. Cost effectiveness of ofatumumab plus chlorambucil in first-line chronic lymphocytic leukaemia in Canada. Pharmacoeconomics. 2016;34:77-90. PubMed
17.Tolley K, Goad C, Yi Y, Maroudas P, Haiderali A, Thompson G. Utility elicitation study in the UK general public for late-stage chronic lymphocytic leukaemia. The European Journal of Health Economics. 2013;14:749-759. PubMed
18.Venetoclax for treating chronic lymphocytic leukaemia. 2017 Nov 8; https://www.nice.org.uk/guidance/ta487. Accessed 2023 May 24.
19.Exceptional Access Program (EAP). Toronto (ON): Ontario Ministry of Health; Ontario Ministry of Long-Term Care; 2022: http://www.health.gov.on.ca/en/pro/programs/drugs/odbf/odbf_except_access.aspx. Accessed 2023 May 24.
20.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario drug benefit formulary/comparative drug index. 2022; https://www.formulary.health.gov.on.ca/formulary/. Accessed 2023 May 24.
21.Tam VC, Ko YJ, Mittmann N, et al. Cost-effectiveness of systemic therapies for metastatic pancreatic cancer. Current oncology. 2013;20(2):90-106. PubMed
22.Schedule of benefits for physician services under the Health Insurance Act: (January 25, 2022 (effective July 1, 2022)). Toronto (ON): Ontario Ministry of Health; 2022: https://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master.pdf. Accessed 2023 May 24.
23.Schedule of benefits for laboratory services: effective July 1, 2020. Toronto (ON): Ontario Ministry of Health; 2020: http://www.health.gov.on.ca/en/pro/programs/ohip/sob/lab/lab_mn2020.pdf. Accessed 2023 May 24.
24.Schedule of Fees for the Laboratory Services Outpatient Payment Schedule. Fee-for-Service Outpatient Laboratory Services in British Columbia. Victoria (BC): BC Ministry of Health; 2023: http://www.bccss.org/clinical-services/bcaplm/health-professionals/outpatient-payment-schedule. Accessed 2023 May 24.
25.Table 14-10-0064-01 Employee wages by industry, annual, median hourly wage, both sexes, full-time and part-time, 55 years and up, 2022 costs. 2022; https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1410006401. Accessed 2023 May 23.
26.Ontario Case Costing Initiative (OCCI). Toronto (ON): Ontario Health and Long-Term Care; 2017: https://data.ontario.ca/dataset/ontario-case-costing-initiative-occi. Accessed 2023 May 24.
27.Walker H, Anderson M, Farahati F, et al. Resource use and costs of end-of-life/palliative care: Ontario adult cancer patients dying during 2002 and 2003. Journal of palliative care. 2011;27(2):79-88. PubMed
28.Briggs A, Sculpher M, Claxton K. Decision modelling for health economic evaluation. Oup Oxford; 2006.
29.Sonnenberg FA, Beck JR. Markov models in medical decision making: a practical guide. Medical decision making. 1993;13(4):322-338. PubMed
30.Putter H, Fiocco M, Geskus RB. Tutorial in biostatistics: competing risks and multi‐state models. Statistics in medicine. 2007;26(11):2389-2430. PubMed
31.Williams C, Lewsey JD, Briggs AH, Mackay DF. Cost-effectiveness analysis in R using a multi-state modeling survival analysis framework: a tutorial. Medical Decision Making. 2017;37(4):340-352. PubMed
32.Guidelines for the economic evaluation of health technologies: Canada. 4th ed. Ottawa (ON): CADTH; 2017: https://www.cadth.ca/guidelines-economic-evaluation-health-technologies-canada-4th-edition. Accessed 2023 May 24.
33.Ibrutinib with venetoclax for untreated chronic lymphocytic leukaemia. London (UK): NICE; 2023: https://www.nice.org.uk/guidance/ta891. Accessed 2023 Jun 30.
34.DeltaPA. [Ottawa (ON)]: IQVIA; 2022: https://www.iqvia.com/. Accessed 2023 May 26.
35.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Imbruvica (ibrutinib) 140 mg oral capsules [internal sponsor's package]. Toronto (ON): Janssen Inc.; 2023 Apr 21.
36.Table 13-10-0751-01. Number of prevalent cases and prevalence proportions of primary cancer, by prevalence duration, cancer type, attained age group and sex. 2022; https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310075101.
37.Canadian Cancer Society. Incidence and mortality. 2023; https://cancer.ca/en/cancer-information/cancer-types/chronic-lymphocytic-leukemia-cll/statistics. Accessed 2023 Feb 1.
Appendix 1: Cost Comparison Table
Note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical expert(s). Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 5: CADTH Cost Comparison Table for Untreated CLL
Treatment | Strength / concentration | Form | Price | Recommended dosage | Daily cost | Course costa |
|---|---|---|---|---|---|---|
ibrutinib (Imbruvica) | 140 mg | Capsule | $99.8350a | 420 mg once daily for 15 cycles (28 days per cycle). | $299.50 | $125,793 |
venetoclax (Venclexta) | 10 mg 50 mg 100 mg | Tablet | $7.0800b $35.4000b $70.8000b | Initiated in cycle 4 on ibrutinib. Cycle 4: 20 mg once daily for 7 days. Repeat for 50 mg, 100 mg, and 200 mg. Cycle 5 to 15: 400 mg once daily. | $212.05 | $89,060 |
ibrutinib plus venetoclax | $511.55 | $214,852 | ||||
BR: bendamustine plus rituximab | ||||||
bendamustine (Treanda) | 25 mg | Injected Solution | $12.5000c | 90 mg/m2 days 1 and 2 for 6 cycles (28 days per cycle). | $5.79 | $972 |
rituximab (generic) | 10 mg/mL | Injected Solution | $29.7000 | Cycle 1: 375 mg/m2 day 1 Cycle 2 to 6: 500 mg/m2 day 1 | $91.49 | $15,370 |
rituximab (Truxima) | 10 mg/mL | Injected Solution | $297.0000 | $914.87 | $153,698 | |
BR: bendamustine plus rituximab (generic) | $97.27 | $16,342 | ||||
BR: bendamustine plus rituximab (Truxima) | $920.65 | $154,670 | ||||
C+O: chlorambucil plus obinutuzumab | ||||||
chlorambucil (Leukeran) | 2mg | Tablet | $1.9144 | 0.5 mg/kg on day 1 and 15 for 6 cycles (28 days per cycle) | $2.56 | $431 |
obinutuzumab (Gazyva) | 25 mg/mL | Injected Solution | $136.9460c | Cycle 1: 1,000 mg on days 1, 8, 15. Cycles 2 to 6: 1,000 mg on day 1. | $6.52 | $1,096 |
C+O | $9.09 | $1,527 | ||||
FCR: fludarabine plus cyclophosphamide and rituximab | ||||||
Fludarabine (Fludara) | 10 mg | Tablet | $41.8940 | 25 mg/m2 days 1 to 3 for 6 cycles (28 days per cycle) | $20.20 | $3,394 |
cyclophosphamide (Procytox) | 2000 mg | Injected Solution | $326c | 250 mg/m2 days 1 to 3 for 6 cycles (28 days per cycle) | $7.86 | $1,321 |
rituximab (generic) | 10 mg/mL | Injected Solution | $29.7000 | Cycle 1: 375 mg/m2 day 1 Cycle 2 to 6: 500 mg/m2 day 1 | $91.49 | $15,370 |
rituximab (Truxima) | 10 mg/mL | Injected Solution | $297.0000 | $914.87 | $153,698 | |
FCR: fludarabine plus cyclophosphamide and rituximab (generic) | $119.54 | $20,084 | ||||
FCR: fludarabine plus cyclophosphamide and rituximab (Truxima) | $942.92 | $158,412 | ||||
VO: venetoclax plus obinutuzumab | ||||||
venetoclax (Venclexta) | 10 mg 50 mg 100 mg | Tablet | $7.0800 $35.4000 $70.8000 | Cycle 1 (Day 22): 20 mg daily for 7 days Cycle 2: Once daily dose of 50 mg, 100 mg, 200 mg, and 400 mg for 7 days each. Cycle 3 to 12: 400 mg once daily. | $241.46 | $81,130 |
obinutuzumab (Gazyva) | 25 mg/mL | Injected Solution | $136.9460c | Cycle 1: 1,000 mg on days 1, 8, 15. Cycles 2 to 6: 1,000 mg on day 1. Cycles 7 to 12: None. | $3.26 | $1,096 |
VO: venetoclax plus Obinutuzumab | $244.72 | $82,226 | ||||
Monotherapies | ||||||
acalabrutinib (Calquence) | 100 mg | Capsule | $135.9750 | 100 mg twice daily until disease progression or intolerance | $271.95 | $7,615 every 28-days |
ibrutinib (Imbruvica) | 140 mg | Capsule | $99.8350 | 420 mg once daily until disease progression or intolerance | $299.50 | $8,387 every 28-days |
Note: All prices are from the Ontario Drug Benefit Formulary (accessed May 2023), unless otherwise indicated, and do not include dispensing fees. Costs assume a body weight of 75kg or a body surface area of 1.8m2 and include wastage of unused medication in vials.
aSponsor’s submitted price
bPrices obtained from the Ontario Drug Benefit Formulary Exceptional Access Program (Accessed May 2023).19
cIQVIA Delta PA wholesale price: $0.1630/mg. Accessed May 2023.34
Appendix 2: Submission Quality
Note that this appendix has not been copy-edited.
Description | Yes/No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | The criteria used to select treatment regimens for inclusion in the model were inconsistent with CADTH guidelines. See limitation: Missing comparators. |
Model has been adequately programmed and has sufficient face validity | No | The calculations used to determine state membership do not represent the requirements for a Semi-Markov model. Methods relied on basic arithmetic instead of applying rules of combining probabilities. See limitation: Incorrect Calculation of State Membership |
Model structure is adequate for decision problem | No | The model failed to incorporate the time dependency for second-line treatment initiation. Furthermore, it does not include a mechanism to distinguish patients who progressed on first-line treatment but had not yet initiated second-line treatment. See limitation: failure to incorporate the time dependency for second-line treatment initiation. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | No | Transition probabilities for PFS did not reflect the risk of progression at each cycle in the model. See limitation: Incorrect calculation of transition probabilities. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | No | The errors identified in the model meant that it could not be used to characterize the impact of parameter or structural uncertainty on a decision. See limitation: Incorrect calculation of State Membership and failure to incorporate the time dependency for second-line treatment initiation. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | No | The spreadsheet was poorly organized, and the programming logic was more complex than required for this model. |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note that this appendix has not been copy-edited.
PF 1L = progression-free in first-line treatment; PF 2L = progression-free in second-line treatment; PPS = postprogression survival; tx = treatment.
Detailed Results of the Sponsor’s Base Case
The sponsor’s base case results do not represent valid estimates of costs, LYs, or QALYs. As detailed in the key limitations section of the report, the submitted model relied on incorrect calculations to estimate state membership for a semi-Markov model and failed to incorporate the time dependency with respect to second-line treatment initiation. Given that this submission did not adhere to best practices and was not suitable for calculating costs and QALYs, CADTH did not report detailed results from the sponsor’s base case.
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Note that this appendix has not been copy-edited.
In the absence of a valid decision model, CADTH was unable to conduct any additional analysis to assess the cost-effectiveness of ibrutinib (in combination with venetoclax) for the treatment of adults with previously untreated CLL.
Appendix 5: Submitted BIA and CADTH Appraisal
Note that this appendix has not been copy-edited.
Table 7: Summary of Key Take-Aways
Key take-aways of the BIA |
|---|
|
Summary of Sponsor’s BIA
The sponsor submitted a budget impact analysis (BIA) for the introduction of ibrutinib-venetoclax for the first-line treatment of adult patients with previously untreated chronic lymphocytic leukemia (CLL). Estimates were generated from the perspective of CADTH participating drug plans (all but Quebec) and results were aggregated into pan-Canadian totals over a 3-year time horizon. An epidemiologic approach was used to estimate the eligible population for the analysis. New patients were added to the BIA based on jurisdictional specific population growth rates.35 Key inputs to the BIA are documented in Table 8.
To estimate the CLL population in each year of the model time horizon, the model multiplied the CLL incidence and prevalence rates to Canadian population estimates. A prevalence rate of 0.01071%, estimated from Statistics Canada data, was used to identify current untreated CLL patients at the start of the model.35,36 Incident CLL cases in each year of the model was calculated using an incidence rate of 0.004654%, based on data obtained from the Canadian Cancer Society.35,37
Annual treatment costs were estimated by multiplying the expected annual treatment dose by the corresponding unit price(s). Dosing schedules were used to determine the annual treatment consumption and were obtained from the respective pivotal trials for each treatment. Unit costs for each treatment were obtained from the price list of a medication distributor (McKesson Corporation). The annual acquisition costs were calculated without the use of mark-ups, dispensation fees, or confidential rebates. All treatment costs assumed 100% compliance and no wastage.35
In the reference scenario, it was assumed that patients would be eligible for 1 of the currently available first-line treatment regiments indicated for CLL. In the new drug scenario, it was assumed that the available first-line treatment regiments for CLL were expanded to include ibrutinib-venetoclax.
State the key assumptions:
50% of incident cases will initiate first-line treatment for CLL and 20% of prevalent (existing) cases will initiate first-line treatment for CLL following watch-and-wait.
Eligible treatments regimens included: ibrutinib plus venetoclax, ibrutinib monotherapy, acalabrutinib plus obinutuzumab, venetoclax plus obinutuzumab, chlorambucil plus obinutuzumab, bendamustine plus rituximab, and fludarabine plus cyclophosphamide and rituximab.
Age-specific criteria were used to define the fludarabine-ineligible subpopulation. For patients 65 years or older, it was assumed to include patients with unmutated immunoglobulin heavy chain variable (uIGHV) region genes, mutated immunoglobulin heavy chain variable (mIGHV) region genes, or deletion of the 17p gene. For patients younger than 65 years, the subpopulation was assumed to include patients with uIGHV or those with a 17p deletion. For the fludarabine-eligible subpopulation, it was assumed that fludarabine would be restricted to patients younger than 65 years with mIGHV.
Market share was estimated for 3 different subgroups within the fludarabine-ineligible subpopulation: patients with uIGHV, patients with mIGHV older than 65, and those with a 17p deletion.
Annual treatment costs assumed 100% compliance and no drug wastage.
Table 8: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3 if appropriate) | ||
|---|---|---|---|
Target population | |||
Pan-Canadian Population (Excluding Quebec) | 39,904,870 / 40,401,472 / 40,904,255 | ||
CLL Incidence Rate (New Cases) | 0.004654% | ||
CLL Prevalence Rate (Existing Cases) | 0.01071% | ||
New CLL Cases | 1,857 / 1,880 / 1,904 | ||
% Incident Patients that initiate therapy | 50% | ||
Existing Patients | 4,274 / 4,327 / 4,381 | ||
% Prevalent Patients that initiate therapy after watch-and-wait | 20% | ||
Initiate Therapy | 1,779 / 1,802 / 1,824 | ||
% ≥ 65 years old | 75% | ||
% ≥ 65 years old with public insurance | 100% | ||
% 18 to 64 years old | 25% | ||
% < 65 years old with public insurance | 15% | ||
% with ulGHV | 45% | ||
% with mlGHV | 40% | ||
% with del(17p) | 15% | ||
Fludarabine-Ineligible Subpopulation | 1,375 / 1,392 / 1,409 | ||
≥ 65 years old | 1,335 / 1,351 / 1,368 | ||
< 65 years old | 40 / 41 / 41 | ||
Fludarabine-Eligible Subpopulation | 27 / 27 / 27 | ||
≥ 65 years old | 0 / 0 / 0 | ||
< 65 years old | 27 / 27 / 27 | ||
Number of patients eligible for drug under review | 1,401 / 1,419 / 1,436 | ||
Market uptake (3 years) | |||
Fludarabine-ineligible subpopulation by subgroup | |||
Uptake (reference scenario) ibrutinib monotherapy acalabrutinib monotherapy acalabrutinib plus obinutuzumab venetoclax plus obinutuzumab chlorambucil plus obinutuzumab bendamustine plus rituximab | ||||||||| | ||| | ||||||| | ||| | |||||| | || | |||||| | ||| | |||||| | || | ||||| | || | || | ||||| | ||| ||||||| | ||| | |||||| | ||| | |||||| | || | |||||| | ||| | ||||||| | ||| | |||||| | || | || | ||| |||||||||||| | ||| | ||||||| | ||| | ||||||| | ||| | ||||||| | ||| | |||||| | || | ||||| | || | || |
Uptake (new drug scenario) ibrutinib plus venetoclax ibrutinib monotherapy acalabrutinib monotherapy acalabrutinib plus obinutuzumab venetoclax plus obinutuzumab chlorambucil plus obinutuzumab bendamustine plus rituximab | ||||||||| | |||| ||||||| | |||| |||||| | ||| | |||||| | || | |||||| | ||| | |||||| | || | ||||| | || | || | ||||| | ||| ||||||| | ||| | ||||||| | || | |||||| | ||| | |||||| | || | |||||| | ||| | ||||||| | ||| | |||||| | || | || | ||| ||||||||||| | || | |||||| | ||| | |||||| | ||| | ||||||| | ||| | ||||||| | ||| | |||||| | || | ||||| | || | || |
Fludarabine-eligible subpopulation | |||
Uptake (reference scenario) fludarabine plus cyclophosphamide and rituximab | ||||| | |||| | |||| | ||
Uptake (new drug scenario) Ibrutinib plus venetoclax fludarabine plus cyclophosphamide and rituximab | ||| | ||| | ||||||| | ||| | ||| | ||
Cost of treatment (per patient) | |||
Cost of treatment over 1-Year ibrutinib plus venetoclax ibrutinib monotherapy acalabrutinib monotherapy acalabrutinib plus obinutuzumab venetoclax plus obinutuzumab chlorambucil plus obinutuzumab bendamustine plus rituximab fludarabine plus cyclophosphamide and rituximab | $181,972 / $40,009 / $0 $109,319 / $109,319 / $109,319 $99,265 / $99,265 / $99,265 $141,469 / $99,265 / $99,265 $138,377 / $0 / $0 $43,704 / $0 / $0 $25,788 / $0 / $0 $27,604 / $0 / $0 | ||
uIGHV = unmutated immunoglobulin heavy chain variable region genes; mIGHV = mutated immunoglobulin heavy chain variable region genes.
Summary of the Sponsor’s BIA Results
In the sponsor’s base case, the net budget impact of ibrutinib-venetoclax for the total indicated population was estimated to be $10,007,998 in year 1, $5,957,940 in year 2, and –$6,977,824 in year 3. In the fludarabine-ineligible subpopulation, the net budget impact of ibrutinib-venetoclax was estimated to be $5,966,705 in year 1, $3,607,908 in year 2, and –$1,356,226 in year 3. In the fludarabine-eligible subpopulation, the net budget impact was of ibrutinib-venetoclax was estimated to be $4,011,293 in year 1, $2,350,033 in year 2, and –$5,621,598 in year 3. The 3-year net budget impact of ibrutinib-venetoclax was $8,988,115. By subpopulation, the 3-year budget impact was $8,248,387 and $739,728 in the fludarabine-ineligible and eligible subpopulations, respectively.
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Underestimation of the eligible population: To define the eligible population for treatment, the sponsor assumed that 20% of prevalent (existing) cases would initiate treatment after a watch-and-wait period. Clinical experts consulted by CADTH suggested this estimate was too low. In other words, a much higher proportion of prevalent cases may be expected to initiate treatment following a watch-and-wait period. This would increase the costs for all treatments in the budget impact model and may introduce some uncertainty toward the original estimate of the net budget impact of ibrutinib plus venetoclax.
CADTH addressed this limitation by assuming a higher estimate of treatment initiation following a watch-and-wait period. Clinical experts consulted by CADTH suggested a 50% estimate for the CADTH base.
CADTH Reanalyses of the BIA
Table 9: CADTH Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
None | — | — |
Changes to derive the CADTH base case | ||
1. Under-estimation of the eligible population | 20% of prevalent cases would initiate treatment following a watch-and-wait period. | 50% of prevalent cases will initiate treatment following a watch-and-wait period. |
CADTH base case | Reanalysis 1 | |
The results of the CADTH step-wise reanalysis are presented in summary format in Table 10 and a more detailed breakdown is presented in Table 11. All CADTH reanalysis were based on publicly available prices of the comparator treatments. In the CADTH base case, the net budget impact of ibrutinib plus venetoclax for the total indicated population was estimated to be $17,203,486 in year 1, $10,241,543 in year 2, and –$11,944,696 in year 3. When stratified by subpopulation, the net budget impact of ibrutinib plus venetoclax in years 1 to 3 was estimated to be $10,308,178, $6,201,899, and –$2,331,317 in the fludarabine-ineligible subpopulation and $6,895,307, $4,039,644, and –$9,663,379 in the fludarabine-eligible subpopulation. The 3-year net budget impact was $15,450,333 which could be stratified into $14,178,760 and $1,271,573 in the fludarabine-ineligible and eligible subpopulations, respectively.
The estimated BIA was highly sensitive to the assumed proportion of prevalent patients that would initiate treatment following a watch-and-wait period.
Table 10: Summary of the CADTH Reanalyses of the BIA
Stepped analysis | Three-year total |
|---|---|
Submitted base case | $8,988,115 |
CADTH reanalysis 1 (CADTH base case) | $15,450,333 |
BIA = budget impact analysis.
Table 11: Detailed Breakdown of the CADTH Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
Submitted base case | FI Subpopulation | |||||
Reference | $91,877,479 | $94,722,138 | $149,930,821 | $206,895,802 | $451,548,761 | |
New drug | $91,877,479 | $100,718,843 | $153,538,729 | $205,539,576 | $459,797,148 | |
Budget impact | $0 | $5,966,705 | $3,607,908 | -$1,356,226 | $8,248,387 | |
FE Subpopulation | ||||||
Reference | $54,576,278 | $56,819,454 | $81,642,309 | $119,094,090 | $257,555,853 | |
New Drug | $54,576,278 | $60,830,747 | $83,992,342 | $113,472,492 | $258,295,582 | |
Budget Impact | $0 | $4,011,293 | $2,350,033 | -$5,621,598 | $739,728 | |
Total Population | ||||||
Reference | $146,453,757 | $151,541,592 | $231,573,130 | $325,989,892 | $709,104,614 | |
New Drug | $146,453,757 | $161,549,590 | $237,531,071 | $319,012,069 | $718,092,729 | |
Budget Impact | $0 | $10,007,998 | $5,957,940 | -$6,977,824 | $8,988,115 | |
CADTH base case | FI Subpopulation | |||||
Reference | $157,934,972 | $162,824,866 | $257,727,141 | $355,648,447 | $776,200,454 | |
New drug | $157,934,972 | $173,133,044 | $263,929,040 | $353,317,130 | $790,379,214 | |
Budget impact | $0 | $10,308,178 | $6,201,899 | -$2,331,317 | $14,178,760 | |
FE Subpopulation | ||||||
Reference | $93,815,188 | $97,671,148 | $140,340,984 | $204,719,611 | $442,731,744 | |
New Drug | $93,815,188 | $104,566,456 | $144,380,629 | $195,056,233 | $444,003,317 | |
Budget Impact | $0 | $6,895,307 | $4,039,644 | -$9,663,379 | $1,271,573 | |
Total Population | ||||||
Reference | $251,750,160 | $260,496,014 | $398,068,126 | $560,368,058 | $1,218,932,197 | |
New Drug | $251,750,160 | $277,699,499 | $408,309,669 | $548,373,363 | $1,234,382,531 | |
Budget Impact | $0 | $17,203,486 | $10,241,543 | -$11,994,696 | $15,450,333 | |
BIA = Budget Impact Analysis; FI = fludarabine-ineligible; FE = fludarabine-eligible.
Stakeholder Input
Patient Input
Lymphoma Canada
About Lymphoma Canada
Lymphoma Canada is a national Canadian registered charity whose mission it is to empower patients and the lymphoma community through education, support, advocacy, and research. Based out of Mississauga (ON), we collaborate with patients, caregivers, healthcare professionals, and other organizations and stakeholders, to promote early detection, find new and better treatments for lymphoma patients, help patients access those treatments, learn about the causes of lymphoma, and work together to find a cure. Resources are provided in both English and French. www.lymphoma.ca
The mission of CLL Canada is to advocate and provide education to improve access to health care that will extend the lives of Canadians affected by Chronic Lymphocytic Leukemia (CLL) and Small Lymphocytic Lymphoma (SLL). CLL Canada is a volunteer driven organization. www.cllcanada.org
Information Gathering
Data presented in this submission was collected from an online anonymous patient survey, created by Lymphoma Canada. It was promoted by both Lymphoma Canada from March 22 to May 2, 2023. The link was promoted by via e-mail to patients registered in the LC national emailing list and made available via social media outlets, including Twitter, Instagram, and Facebook accounts. The survey was also promoted to CLL Canada members as well as on three international CLL patient forums: CLL Support on HealthUnlocked, CLL Archives on acor.org and CLLSLL@groups.io. The survey had a combination of multiple choice, rating, and open-ended questions. Skipping logic was built into the survey so that respondents were asked questions only relevant to them. Open-ended responses were noted in this report verbatim, to provide a deeper understanding of patient perspectives. 87 people responded to the survey, 49 identified as Canadians, 12 were from United States, 1 from Australia, and 25 others did not provide demographic information. The majority of patients were female (52%), diagnosed 9 – 10 years ago (49%), with a variety of chromosome or gene abnormalities, depicted in Table 1 to Table 4.
Information from a submission on Ibrutinib + Venetoclax (I+V) in untreated CLL to NICE, the UK health technology assessment agency, prepared by Leukaemia Care, Lymphoma Action and CLL Support
Association was used to highlight key takeaways of this indication. The submission was sent to NICE earlier in 2023 and was based on a survey conducted over the course of 2021, with 109 survey respondents.
Table 1: Age Range of Respondents From Lymphoma Canada Survey
Respondents | Age (years old) | |||||
|---|---|---|---|---|---|---|
45-54 | 55-64 | 65-74 | 75-89 | Skipped | Total | |
Patients with CLL | 2 | 12 | 26 | 21 | 25 | 62 |
Table 2: Gender of Respondents From Lymphoma Canada Survey
Respondents | Gender | |||
|---|---|---|---|---|
Female | Male | Skipped | Total | |
Patients with CLL | 32 | 30 | 25 | 62 |
Table 3: Number of Years Ago Respondents Were Diagnosed With CLL
Respondents | Years | ||||||
|---|---|---|---|---|---|---|---|
<1 | 1-2 | 3-5 | 5-8 | 9-10 | Skipped | Total | |
Patients with CLL | 4 | 10 | 15 | 8 | 36 | 14 | 74 |
Table 4: Chromosome or Gene Mutations of CLL Survey Respondents
Subtype of CLL | Number of respondents |
|---|---|
Deletion 17p | 8 |
Deletion 13q | 3 |
Deletion 11q | 1 |
TP53 mutation | 2 |
Trisomy 12 | 6 |
Unmutated IGHV | 10 |
I don’t know | 45 |
Skipped | 13 |
Total | 74 |
10 respondents had specific experience with Ibrutinib & Venetoclax (4 Canadians, 6 Americans, 1 skipped). All were diagnosed with Chronic Lymphocytic Leukemia.
Disease Experience
At Diagnosis
The development CLL is very different from other types of lymphomas, in that most patients are diagnosed through routine bloodwork and experience no or minor symptoms at the time of diagnosis. Respondents were asked to rate how much each symptom impacted their quality of life at diagnosis. The highest rated negative impacts (3, 4 or 5, out of 5) amongst 64 respondents were fatigue (47%,), high white blood cell counts (leukocytosis) (26%), body aches and pains (25%), enlarged lymph node(s) (23%), and night sweats (20%).
These results are consistent previous surveys LC and CLL Canada have undertaken for other HTA submissions, including the indication for Zanubrutinib which was submitted to CADTH earlier this year.
In terms of psychosocial impacts of CLL diagnosis, the most common factors of 71 respondents were anxiety/worry (61%), stress of diagnosis (59%), and difficulty sleeping (28%).
Current Quality of Life
Survey respondents were asked to rate physical symptoms and psychosocial factors which impacted their current quality of life (70 answered, 17 skipped). The most common negative physical symptoms whose impacts were rated 3, 4 or 5 out of 5, were fatigue (44%), body aches and pains (27%), and indigestion, abdominal pain, or bloating (17%).
CLL had a negative impact on the quality of life of 76% of 87 respondents, the most common impacts being anxiety/worry (42%), difficulty sleeping (31%) and stress of diagnosis (28%).
Daily Activities
Since many CLL & SLL patients do not experience physically debilitating symptoms during the “watch and wait” period before treatment, it is not surprising that many respondents indicated their daily activities were not strongly impacted by their diagnosis. Many respondents indicated their CLL symptoms did not limit their ability to contribute financially to household expenses (80%), ability to spend time with family & friends (55%) or ability to fulfill family obligations (49%).
While the impact of CLL is different from other types of lymphomas, many patients left comments that they still struggle with managing “overwhelming tiredness” and “fatigue which places limitations on daily activities”. One patient commented “the stress never really goes away, even after treatment because I am back to watch and wait”, while another wrote “the prompt diagnosis and treatment regime helped me to continue living quite normally except for some of the side effects”. This highlights the importance and need to continuously manage CLL treatment options in the frontline setting to allow Canadian lymphoma patients to have the best quality of life.
Summary of the Disease Experience
For many patients, to live with CLL means living with fatigue, anxiety and stress, all of which have a significant impact on a person’s quality of life.
Experiences With Currently Available Treatments
Due to the nature of CLL, many patients undergo a period of watchful waiting guided by their primary physician, before or ever needing to start treatment. Out of 68 patients which provided information on their CLL treatment, 21 indicated they have not received therapy, 26 respondents received one line of treatment, and 19 completed 2 or more treatments. Please refer to Table 5 to Table 7 for treatment options provided to CLL patients in first, second and third line of therapy.
Table 5: CLL Treatments in Firstline Therapy
CLL treatments | Number of respondents |
|---|---|
BTK inhibitor, Ibrutinib | 19 |
Chemoimmunotherapy (FCR and others) | 9 |
Ibrutinib + Venetoclax | 6 |
BTK inhibitor, Acalabrutinib | 3 |
Chemotherapy (unspecified) | 2 |
Chlorambucil + Obinutuzumab | 2 |
Venetoclax | 1 |
Rituximab + Bendamustine | 1 |
Acalabrutinib + Venetoclax | 1 |
Total | 21 |
Table 6: CLL Treatments in Second Line Therapy
CLL treatments | Number of respondents |
|---|---|
Ibrutinib | 6 |
Venetoclax | 3 |
Venetoclax + Ibrutinib | 2 |
Acalabrutinib | 2 |
Chemo FCR | 2 |
Rituximab | 2 |
Rituximab + methylprednisolone | 1 |
Zanubrutinib | 1 |
Venetoclax + Rituximab | 1 |
Lenalidomide + Rituximab | 1 |
Total | 21 |
Table 7: CLL Treatments in Third Line Therapy
CLL treatments | Number of respondents |
|---|---|
Venetoclax | 3 |
Ibrutinib | 3 |
Stem cell transplant | 1 |
Chemotherapy + ibrutinib | 1 |
Venetoclax + rituximab | 1 |
ABT-199 + Rituximab + Bendamustine | 1 |
Idelalisib + Rituximab | 1 |
Total | 11 |
All CLL patients were asked in the survey how strongly they agree with the following statement: “My treatment was able to manage my CLL symptoms.” 22% of patients strongly agreed with this sentence, providing a 10 out of 10 rating. The next highest rating was 9 of out 10, which 19% of patients selected. This indicates that the majority of CLL patients are content with the treatment options listed in Table 5 to Table 7 to manage their CLL symptoms. However, when asked which side effects were the most difficult to tolerate many patients, which indicated nausea, fatigue, joint pain, skin issues & bleeding, atrial fibrillation, diarrhea, inflammation, bodily aches and pain, headache, muscle weakness, heartburn, indigestion, night sweats, neuropathy, and frequent infections were very challenging during treatment. Furthermore, 26% of patients indicated their CLL treatment had a negative impact on their ability to travel, whereas 19% of patients had a negative impact on their ability to go to work, school, or volunteer. Here a few patient quotes collected from LC’s survey to highlight how CLL patients currently feel about their treatment options:
“I have liked the Venetoclax much more than the Ibrutinib. At the full dose It does increase my tiredness, but I am only going to have to take it for two years, so I like that.”
“I developed COVID with a blood clot in the lung. Was taken off ibrutinib and about a month later switched to Zanubrutinib. Less side effects.”
“I was extremely ill when I started treatment (fatigue, low blood counts, brain fog) so the treatments actually made me slowly feel better. The side effects were probably the symptoms of CLL. Currently the fatigue and brain fog are side effects of the Ibrutinib.”
“My third treatment (venetoclax) relieved my symptoms within a few weeks and there were no side effects. Treatment was finished after 22 months and offered 2 1/2 years of remission. My current treatment (Zanubrutinib) has been trouble free as well. It started in November 2022.”
Summary of the Current Available Therapies
Side effects of treatment and their impacts on the patient’s quality of life remain a significant issue for the majority of survey respondents, including those who believe their current therapy or therapies manage their CLL symptoms well. This indicates a need for alternative CLL treatments with an improved side effect profile.
Improved Outcomes
In the LC survey, patients were asked about factors important to them when considering a novel CLL treatment. The following factors were rated as extremely important: allow me to live longer (81%), control disease symptoms (75%), bring about a longer remission (71%), better quality of life (66%), and fewer side effects (35%). 56% of respondents indicated it is extremely important (10 out of 10) to have choice in their treatment decision, while 55% reported it is very important to have increased treatment options available to choose from.
This was echoed by several patients who indicated they would like choice when it comes to the therapy they receive:
“I would like to have a choice of medications rather than the 420 mg of daily Ibrutinib I have been on the past 3 years. It was the only option given to me.”
“More options for combination therapies with ibrutinib at start of treatment rather than waiting to see how it goes on its own. It has already been proven to be effective in combination.”
CLL patients were specifically asked in the LC survey “Would you prefer to take a novel therapy intravenously over a fixed duration vs. novel oral therapy required to take indefinitely?” 24% of patients reported preferring the fixed duration treatment versus 10% continuous oral therapy, and 66% reported they were not sure.
These results reflect the fact that the choice of treatment is rarely a simple, straightforward decision. Rather, multiple dimensions need to be considered, including the time and cost required to travel to a hospital as well as the constraints posed by family, caregiving and work responsibilities.
The answers to this question underline the importance of giving patients treatment options that they can discuss with their doctors and their loved ones. They can then make their treatment decision considering all the relevant factors, both medical (effectiveness, side effects, comorbidities, etc.) as well as the constraints and issues related to their life circumstances.
This is reflected by several patients which commented on what they want in novel CLL treatments:
“If available my preferred treatment would be a time-limited oral therapy without or minimal side effects. Ideally a treatment which is curative.”
“I am hopeful that a new drug therapy will be found to cure CLL so that patients once treated will be cured and no longer have to face repeated treatment therapies for the rest of their lives.”
“My expectations would be that a new drug therapy would offer symptom Relief, would increase the quality of life, a longer life expectancy with quality of life.”
Summary of Improved Outcomes
CLL patients identified factors important for novel treatments, which included longer life span, longer remission, better quality of life and fewer side effects.
A large majority of patients believe it is very important to have choice in their treatment decision and a variety of treatment options to choose from.
Experience With Drug Under Review
Of the 10 patients which confirmed they were treated with Ibrutinib + Venetoclax for their CLL treatment, 2 patients are currently undergoing treatment, 5 are in remission between 2 and 5 years, 2 patients are in remission longer than 2 years, and the final patient is in remission, but status is unknown. 9 of these patients accessed I+V through a clinical trial, and the other patient just started ibrutinib treatment and have not received Venetoclax yet. Table 8 summarizes how many years ago patients received I+V treatment for their CLL. Table 9 summarizes the genetic or molecular abnormalities of these 10 patients had.
Table 8: Number of Years Ago CLL Respondents Were Treated With I+V (Frontline Treatment)
Respondents | Years | ||||
|---|---|---|---|---|---|
1-2 | 3-5 | 6+ | Skipped | Total | |
Patients with CLL | 4 | 4 | 1 | 0 | 10 |
Table 9: Genetic/Molecular Abnormalities of CLL Patients Who Received I+V Treatment (Frontline)
CLL characteristics | Number of patients |
|---|---|
Deletion 17p | 1 |
Deletion 13q | 2 |
Trisomy 12 | 2 |
Unmutated IGHV | 2 |
I don’t know | 3 |
The most common CLL symptoms managed by I+V treatment in this patient cohort were high white blood cell counts (80%), enlarged lymph nodes (70%), low platelet counts (60%), low red blood cell count (60%), weight loss (30%). 10% of patients also experienced bodily aches and pains, neutropenia, indigestion, and night sweats.
Many positive comments were left by patients when asked to share about their I+V treatment experience:
“I am glad I was able to stop ibrutinib because of afib side effects.”
“I think it was a very good first line treatment. 4 years from start of trial 96% still in remission. Financially cheaper than doing 4 years of monotherapy (2-year trial).”
“I thought I would be hospitalized with Venetoclax but was not, just 8 hours of monitoring in clinic.”
“I have been MRD- for four years now after 13 months of treatment. I think I was MRD- after the first nine months. I believe that almost everyone did as well as me after 13 months. AMAZING!”
“I achieved MRD (measurable residual disease) undetectable status very quickly — within 10 month no evidence of disease. Side effects not terrible especially given what I got in return. Still MRD negative after 2 yrs past end of trial.”
“Highly recommend the I+V combo.”
“Seems to be a very good treatment with minimal side effects.”
“I have been in remission for 4 years following clinical trial treatment with ibrutinib and venetoclax at MD Anderson. Reached MRD negative after 9months treatment. Have been off all meds since 2019. My day-to-day life is not affected by CLL….”
Ibrutinib + Venetoclax (I+V) offers an appealing advantage to many CLL patients as it is an all-oral therapy and a fixed-duration schedule. In comparison to Obinutuzumab, which requires intravenous administration and frequent hospital visits, and BTK inhibitors which are taken indefinitely, I+V offers the benefits of oral and time-limited therapy. This treatment could be beneficial to the life and daily activities of many CLL patients, especially those that live in rural areas, work or are care takers themselves. These highlights were echoed in the UK health technology assessment of Ibrutinib + Venetoclax, which indicated there is a need for improved and long-lasting treatment benefits for those living with CLL.
Summary of Drug under Review
The patients who had undergone the I+V therapy highlighted its effectiveness in putting their CLL in remission as well as the low level of side effects they experienced.
The Ibrutinib + Venetoclax combination offers the appealing advantage of combining an all-oral therapy with a fixed-duration schedule.
Companion Diagnostic Test
The diagnosis of CLL or SLL needs to be confirmed through bloodwork (complete blood count) and the presence or absence of the following genetic/molecular markers: IGHV mutation status, deletion 17p, and TP53. These CLL markers are routinely identified through flow cytometry or fluorescence in situ hybridization. The current Canadian guidelines indicate IGHV testing needs to be conducted prior to first treatment only, and del(17p) and TP53 mutation testing should be done prior to each treatment.
Anything Else?
The possibility of taking an oral medication once daily allows for flexibility in administration according to a patient’s individual needs. It also makes it easier for caregivers to adjust their schedule to visit older patients who may not be diligent in taking their medication if left alone. Fixed duration therapy also another advantageous factor for patients who do not want to be on treatment for their entire life.
CLL is an incurable, chronic disease that must be managed through a series of treatments over many years, changing treatments as the disease reoccurs or the side effects become intolerable. Therefore, it is imperative to have many different treatments available that patients and their doctors can choose from as their disease evolves.
The data available on the I+V combination hold out the promise of long-lasting remissions for many patients, effectively getting them off the treatment treadmill. The long-lasting remission that appear to be possible with the I+V combination could make the constant worry of relapse and of unmanageable side effects a thing of the past for a large group of patients. We cannot overstate the importance and the need for a range of treatment options for patients with CLL given the heterogeneity of both the disease and patient population. Ibrutinib + Venetoclax would be a welcome and valuable addition for untreated CLL patients, especially for those with deletion 17p status. It would be greatly beneficial for patients to have an effective all oral, fixed-duration CLL therapy.
Conflict of Interest Declaration — Lymphoma Canada
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission? If yes, please detail the help and who provided it.
Lymphoma Canada & CLL Canada collaborated to complete this submission.
Did you receive help from outside your patient group to collect or analyze data used in this submission? If yes, please detail the help and who provided it.
Information from the National Institute for Health and Care Excellence (UK) was used for this submission.
List any companies or organizations that have provided your group with financial payment over the past 2 years AND who may have direct or indirect interest in the drug under review.
Table 10: Financial Disclosures for Lymphoma Canada
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Janssen | — | — | — | X |
AstraZeneca | — | — | — | X |
BeiGene | — | — | — | X |
Table 11: Financial Disclosures for CLL Canada
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Janssen | X | — | — | — |
AstraZeneca | X | — | — | — |
BeiGene | — | — | X | — |
Clinician Input
CLL Clinicians
About CLL Clinicians
We are a group of clinicians that treat CLL and SLL in Canada. Our practices are diverse and consist of academic and community practice members.
Information Gathering
The Literature was used to guide the efficacy and safety documents. Information was collated and circulated for input from the various clinicians.
Current Treatments and Treatment Goals
Currently there are many options for treatment for CLL. None of them are entirely oral combination options. The young fit, low risk (no molecular abnormalities) population has access to FCR, the fit but high risk patients have access to BTKI monotherapy or Ven 0. The older unfit patients have the access to BTKI monotherapy (in many provinces or restricted only to genetically high-risk patients in some provinces) or Ven 0 as they are both fludarabine ineligible or have high risk disease. The benefit of all therapies is increased disease control and progression free survival. Ven O also adds the treatment free benefit as well.
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
The unmet need comes down to practical considerations. A time limited treatment option to replace BTKi monotherapy (if that would otherwise have been used) is good for payers and for patients because it replaces continuous therapy reducing chronic side effects and ongoing costs. The other consideration is the practical onboarding of Ven 0. It is both resource intensive and can be considered inaccessible for those that may have to travel distances to receive care or be dependent on others and thus unable to maintain their independence. Unfortunately, the weekly ramp-up intensive lab monitoring with venetoclax is still required with the IV regimen but the risks of TLS are very low. It would be the first all-oral combination therapy with a time limited indication.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
Although the criteria in the studies would encompass all unfit patients regardless of risk, we already gave effective therapies in this space. The suggestion would be to have an option for time limited therapy in the highest risk patients, ex [ del)17p) or TP53 mutations] to enable time limited treatment, and/or increase the accessibility of a time limited treatment option for those unable to access Ven O easily.
Which patients would be best suited for treatment with the drug under review? Which patients would be least suitable for treatment with the drug under review?
The molecular testing for this indication is standard of care for CLL and SLL. But in jurisdictions that don’t have access, IGVH mutation, FISH for del 17p and TP53 mutation testing is required. The population that may benefit the most are those where continuous therapies are standard of care or access becomes an issue.
There is a safety concern with this treatment related early deaths in the IV (GLOW) study and increased deaths compared to the control arm. These are assumed to be related to the sudden death events which are the major concerns as clinicians (Blood 2017). The hesitancy in prescribing this combination is mainly from the safety concerns and not related to the efficacy. Thus, the patients will need to be free of cardiovascular comorbidities and likely younger as that population had lower cardiac events compared to the older population.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice? How often should treatment response be assessed?
Progression free survival and time off treatment is a meaningful response with this disease. There is also a reduction in disease related symptoms.
What factors should be considered when deciding to discontinue treatment with the drug under review?
Early disease progression while on therapy. This treatment is time limited otherwise.
What settings are appropriate for treatment with [drug under review]? Is a specialist required to diagnose, treat, and monitor patients who might receive [drug under review]?
A hematologist/oncologist should be the prescriber of this combination. Due to the safety concerns of Ibrutinib, a cardiologist may also be required.
Additional Information
Not applicable.
Conflict of Interest Declarations — CLL Clinicians
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please refer to the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
Lymphoma Canada.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission?
Not applicable.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician who contributed to the input.
Declaration for Clinician 1
Name: Versha Banerji
Position: Associate Professor and Clinician scientist, Manitoba
Date: 05-01-23
Table 12: COI Declaration for CLL Clinicians — Clinician 1
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Astra Zeneca | — | X | — | — |
Beigene | X | — | — | — |
Janssen | X | — | — | — |
Abbvie | — | X | — | — |
Merk | X | — | — | — |
Declaration for Clinician 2
Name: Carolyn Owen
Position: Associate Professor, Division of Hematology & Hematological Malignancies, Calgary
Date: 01-05-2023
Table 13: COI Declaration for CLL Clinicians — Clinician 2
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Janssen | X | — | — | — |
AbbVie | — | X | — | — |
Astrazeneca | — | X | — | — |
Beigene | — | X | — | — |
Ontario Health (Cancer Care Ontario) Hematology Cancer Drug Advisory Committee
About Ontario Health (Cancer Care Ontario) Hematology Cancer Drug Advisory Committee
OH-CCO’s Cancer Drug Advisory Committees provide timely evidence-based clinical and health system guidance on drug-related issues in support of CCO’s mandate, including the Provincial Drug Reimbursement Programs (PDRP) and the Systemic Treatment Program.
Information Gathering
Information was gathered via videoconferencing and email.
Current Treatments and Treatment Goals
This treatment would be considered for first line therapy for CLL. Currently, the standard of care would include venetoclax + obinutuzumab, Obinutuzumab + chlorambucil, bendamustine, and FCR. Also, a BTK inhibitor would be used for patients with high-risk genetics (i.e. del(17p), TP53, unmutated IGHV).
There is an ongoing review for zanubrutinib in the first line setting, not excluding the high-risk population.
The DAC notes that the previous CADTH provisional funding algorithm for CLL (https://www.cadth.ca/chronic-lymphocytic-leukemia) will be out of date.
If venetoclax +ibrutinib is administered for a fixed duration, there should be an opportunity to retreat with regimens including ibrutinib or venetoclax in the second line (these patients are not refractory to ibrutinib or venetoclax).
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
This combination can be used for any previously untreated CLL patients.
Both agents are administered orally. There is no need for IV access.
Venetoclax + ibrutinib might be better tolerated than venetoclax + obinutuzumab.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
This drug will be used as first-line therapy as an alternate choice to other funded therapies.
Which patients would be best suited for treatment with the drug under review? Which patients would be least suitable for treatment with the drug under review?
This treatment is for previously untreated patients. There will be a time-limited need to access this combination in patients already on either drug first-line.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice? How often should treatment response be assessed?
Standard CLL response outcomes, improvement in progression free survival, reduction in symptoms and improvement in quality of life.
What factors should be considered when deciding to discontinue treatment with the drug under review?
This is a fixed duration therapy. Earlier discontinuation will be based on significant intolerance.
What settings are appropriate for treatment with [drug under review]? Is a specialist required to diagnose, treat, and monitor patients who might receive [drug under review]?
Outpatient therapy by prescribers familiar with the treatment of CLL.
With the use of venetoclax + ibrutinib for CLL, additional lab monitoring may be needed.
Additional Information
Not applicable.
Conflict of Interest Declarations — Ontario Health (Cancer Care Ontario) Hematology Cancer Drug Advisory Committee
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please refer to the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
OH-CCO provided secretariat function to the group.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission?
No.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician who contributed to the input.
Declaration for Clinician 1
Name: Dr. Tom Kouroukis
Position: Lead, Ontario Health (Cancer Care Ontario) Hematology Cancer Drug Advisory Committee
Date: 23-03-2023
Table 14: COI Declaration for OH-CCO Hematology Cancer Drug Advisory Committee — Clinician 1
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 2
Name: Dr. Selay Lam
Position: Member, Ontario Health (Cancer Care Ontario) Hematology Cancer Drug Advisory Committee
Date: 06-04-2023
Table 15: COI Declaration for OH-CCO Hematology Cancer Drug Advisory Committee — Clinician 2
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 3
Name: Dr. Lee Mozessohn
Position: Member, Ontario Health (Cancer Care Ontario) Hematology Cancer Drug Advisory Committee
Date: 06-04-2023
Table 16: COI Declaration for OH-CCO Hematology Cancer Drug Advisory Committee — Clinician 3
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 4
Name: Mark Brown
Position: Member, Ontario Health (Cancer Care Ontario) Hematology Cancer Drug Advisory Committee
Date: 06-04-2023
Table 17: COI Declaration for OH-CCO Hematology Cancer Drug Advisory Committee — Clinician 4
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 5
Name: Dr Joanna Graczyk
Position: Member, Ontario Health (Cancer Care Ontario) Hematology Cancer Drug Advisory Committee
Date: 06-04-2023
Table 18: COI Declaration for OH-CCO Hematology Cancer Drug Advisory Committee — Clinician 5
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 6
Name: Dr Jordan Herst
Position: Member, Ontario Health (Cancer Care Ontario) Hematology Cancer Drug Advisory Committee
Date: 06-04-2023
Table 19: COI Declaration for OH-CCO Hematology Cancer Drug Advisory Committee — Clinician 6
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Janssen | X | — | — | — |
Declaration for Clinician 7
Name: Dr Pierre Villeneuve
Position: Member, Ontario Health (Cancer Care Ontario) Hematology Cancer Drug Advisory Committee
Date: 20-04-2023
Table 20: COI Declaration for OH-CCO Hematology Cancer Drug Advisory Committee — Clinician 7
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
Redactions: Confidential information in this document may be redacted at the request of the sponsor in accordance with the CADTH Drug Reimbursement Review Confidentiality Guidelines.
Stakeholder Input: The views expressed in each submission are those of the submitting organization or individual; not necessarily the views of CADTH or of other organizations. As such, they are independent of CADTH and do not necessarily represent or reflect the view of CADTH. No endorsement by CADTH is intended or should be inferred. By filing with CADTH, the submitting organization or individual agrees to the full disclosure of the information. CADTH does not edit the content of the submissions.
CADTH does use reasonable care to prevent disclosure of personal information in posted material; however, it is ultimately the submitter’s responsibility to ensure no identifying personal information or personal health information is included in the submission. The name of the submitting organization or individual and all conflict of interest information are included in the submission; however, the name of the author, including the name of an individual patient or caregiver submitting the patient input, are not posted.
Accessibility: CADTH is committed to treating people with disabilities in a way that respects their dignity and independence, supports them in accessing material in a timely manner, and provides a robust feedback process to support continuous improvement. All materials prepared by CADTH are available in an accessible format. Where materials provided to CADTH by a submitting organization or individual are not available in an accessible format, CADTH will provide a summary document upon request. More details on CADTH’s accessibility policies can be found here.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.