CADTH Reimbursement Review
Polatuzumab Vedotin (Polivy)
Sponsor: Hoffmann-La Roche Limited
Therapeutic area: Large B-cell lymphoma
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Stakeholder Input
Clinical Review
Abbreviations
ABC
activated B cell
AE
adverse event
BICR
blinded independent central review
CAR
chimeric antigen receptor
CCOD
clinical cut-off date
CI
confidence interval
CNS
central nervous system
COO
cell of origin
CR
complete response
DA-EPOCH-R
dose-adjusted etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin, and rituximab
DEL
double-expressor lymphoma
DFS
disease-free survival
DHL
double-hit lymphoma
DLBCL
diffuse large B-cell lymphoma
DOR
duration of response
EBV
Epstein-Barr virus
ECOG PS
Eastern Cooperative Oncology Group Performance Status
eCRF
electronic case report form
EFS
event-free survival
EFSall
event-free survival from all causes
EFSeff
event-free survival–efficacy
EORTC QLQ-C30
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30
EOT
end of treatment
FACT/GOG-NTX
Functional Assessment of Cancer Therapy/Gynecologic Oncology Group-Neurotoxicity
FACT-Lym
Functional Assessment of Cancer Therapy-Lymphoma
FISH
fluorescence in situ hybridization
GCB
germinal centre B cell
G-CSF
granulocyte colony-stimulating factor
HR
hazard ratio
HRQoL
health-related quality of life
iDMC
independent Data Monitoring Committee
IPI
International Prognostic Index
IRR
infusion-related reaction
ITT
intention to treat
LBCL
large B-cell lymphoma
LymS
lymphoma subscale
MDD
minimal detectable difference
MMAE
monomethyl auristatin E
NALT
nonprotocol or new antilymphoma treatment
NHL
non-Hodgkin lymphoma
NOS
not otherwise specified
ORR
objective response rate
OS
overall survival
PD
progressive disease
PFS
progression-free survival
PFS24
2-year progression-free survival
PML
progressive multifocal leukoencephalopathy
pola-R-CHP
polatuzumab vedotin, rituximab, cyclophosphamide, doxorubicin, and prednisone
PR
partial response
PRO
patient-reported outcome
R-CHP
rituximab, cyclophosphamide, doxorubicin, and prednisone
R-CHOP
rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone
RCT
randomized controlled trial
RMST
restricted mean survival time
SAE
serious adverse event
SD
standard deviation
SE
standard error
SOC
standard of care
TEAE
treatment-emergent adverse event
THL
triple-hit lymphoma
TLS
tumour lysis syndrome
TTD
time to deterioration
ULN
upper limit of normal
WDAE
withdrawal due to adverse event
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Information on drug submitted for review | |
Drug product | Polatuzumab vedotin (Polivy), 30 mg or 140 mg single-use vial, lyophilized powder for solution for IV infusion |
Sponsor | Hoffmann-La Roche Limited |
Indication | Polatuzumab vedotin in combination with R-CHP, indicated for the treatment of adult patients with previously untreated LBCL, including DLBCL NOS, high-grade B-cell lymphoma, EBV-positive DLBCL NOS, and T-cell/histiocyte-rich LBCL |
Reimbursement request | Polatuzumab vedotin in combination with R-CHP, indicated for the treatment of adult patients with previously untreated LBCL, including DLBCL NOS, high-grade B-cell lymphoma, EBV-positive DLBCL NOS, and T-cell/histiocyte-rich LBCL |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | November 14, 2022 |
Recommended dose | Patients with previously untreated LBCL: Polatuzumab vedotin 1.8 mg/kg given as IV infusion every 21 days for 6 cycles in combination with R-CHP. Polatuzumab vedotin, rituximab, cyclophosphamide, and doxorubicin can be administered in any order on day 1 after the administration of prednisone. Prednisone is administered on days 1 to 5 of each cycle. Cycles 7 and 8 consist of rituximab as monotherapy. |
DLBCL = diffuse large B-cell lymphoma; EBV = Epstein-Barr virus; LBCL = large B-cell lymphoma; NOC = Notice of Compliance; NOS = not otherwise specified; R-CHP = rituximab, cyclophosphamide, doxorubicin, and prednisone.
Source: Details included in the table are from the sponsor’s summary of clinical evidence1 and the product monograph for polatuzumab vedotin.2
Introduction
Non-Hodgkin lymphoma (NHL) is the fifth most common cancer, with an estimated 11,400 people diagnosed annually in Canada; approximately 3,000 will die from the disease.3,4 Diffuse large B-cell lymphoma (DLBCL) not otherwise specified (NOS) accounts for approximately 25% of NHL cases, and comprises a heterogeneous group of NHL histologic subtypes, including DLBCL transformed from indolent lymphoma or chronic lymphocytic leukemia, high-grade B-cell lymphoma, primary cutaneous DLBCL, Epstein-Barr virus (EBV)-positive DLBCL, and T-cell/histiocyte-rich large B-cell lymphoma (LBCL).5-9 The risk of DLBCL increases with age, with an average age at diagnosis of 65 years.10 DLBCL presents as a quickly growing, nonpainful enlarged lymph node in the neck, groin, or abdomen with high burden of symptoms including fever, weight loss, and night sweats, and poor health-related quality of life (HRQoL). According to the clinical experts consulted by CADTH for this review, nearly 50% to 60% of patients with advanced stage disease can be cured with first-line standard of care (SOC) treatment for LBCL in Canada using rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP). However, the clinical experts consulted reported that approximately 30% to 50% of patients will have disease progression or relapse (typically within the first 2 years), especially among high-risk subgroups (e.g., higher International Prognostic Index [IPI] score, activated B-cell [ABC] lymphoma, or double-hit lymphoma [DHL] or triple-hit lymphoma [THL]) with poor prognosis. According to the clinical experts, significant morbidity exists for patients who experience treatment failure in the first-line setting due to the need for salvage chemotherapy or other treatments that are associated with toxicities and lower cure rates. Overall survival (OS) for patients with primary refractory disease is estimated to be 15% to 20% at 5 years.11
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of polatuzumab vedotin 1.8 mg/kg administered as an IV infusion every 21 days for 6 cycles in combination with rituximab, cyclophosphamide, doxorubicin, and prednisone (R-CHP) in the treatment of previously untreated LBCL, including DLBCL NOS, high-grade B-cell lymphoma, EBV-positive DLBCL NOS, and T-cell/histiocyte-rich LBCL in adults.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient and clinician groups who responded to CADTH’s call for input and from clinical expert(s) consulted by CADTH for the purpose of this review.
Patient Input
One patient group, Lymphoma Canada, submitted input for this review. Lymphoma Canada is a national Canadian registered charity that empowers the lymphoma community through education, support, advocacy, and research. The input was based on an online anonymous patient survey among patients with a subtype of LBCL, created and promoted by Lymphoma Canada, available from February 2, 2023, to March 13, 2023. A total of 89 respondents were included in the patient input, with 4 confirmed responses for experience with polatuzumab vedotin. Most patients were living in Canada (94%), aged 55 to 74 years (64%), and diagnosed 1 year to 5 years ago (61%); more patients were female (58%) than male (42%).
The most frequently reported symptoms at diagnosis among respondents included fatigue, body aches and pains, night sweats, enlarged lymph nodes, and a reduced appetite. The psychosocial impacts of their diagnosis included stress, anxiety or worry, fear of progression, inability to continue daily activities, and difficulty sleeping. LBCL symptoms impacted respondents’ ability to exercise, travel, spend time with family, volunteer, and attend work or school. Most survey respondents received 1 line of treatment for their LBCL, with R-CHOP as the most common treatment regimen. Most patients were satisfied or very satisfied with their options for first-line treatment. When asked about accessing lymphoma therapy in Canada, many patients indicated they were required to travel long distances, which was challenging financially and required time off work. Among the 4 patients with experience with polatuzumab vedotin, rituximab, cyclophosphamide, doxorubicin, and prednisone (pola-R-CHP), 3 patients would recommend the treatment to other patients with LBCL and 2 patients indicated their overall experience with the treatment was very good. Side effects experienced by at least 2 patients on pola-R-CHP included fatigue, neutropenia, thrombocytopenia, decreased appetite, and diarrhea. According to the patient input received, expectations for new treatments include longer disease remission, control of disease symptoms, longer survival, normalized blood counts, and improved quality of life to be able to participate in daily activities.
Clinician Input
Input From Clinical Experts Consulted by CADTH
Two clinical experts provided input on the diagnosis and management of LBCL. The clinical experts identified that patients at high risk (IPI score 3 to 5), with advanced age, frailty, or other comorbidities experience poor outcomes due to a greater likelihood of refractory disease or relapse and would benefit from improved cure rates from first-line treatment. The experts reported using polatuzumab vedotin as a combined regimen with bendamustine and rituximab in the relapse or refractory setting. The clinical experts regarded pola-R-CHP to have a therapeutic role as first-line treatment in treating the underlying DLBCL disease, thereby reducing the need for salvage treatments (e.g., stem cell transplant and/or chimeric antigen receptor [CAR] T-cell therapy) among patients. Pola-R-CHP was anticipated by the experts to replace R-CHOP for DLBCL for patients with IPI score of 3 and greater. Its role in patients with an IPI score of 2 is less certain, but it was not considered to fill an unmet need for patients with limited-stage disease (IPI score 0 to 1) who typically experience high cure rates with current approaches including R-CHOP. The clinical experts expressed that these patients eligible for pola-R-CHP would also include those with an Eastern Cooperative Oncology Group Performance Status (ECOG PS) of 3 or 4 with pathological entities who were typically excluded from clinical trials (e.g., LBCL transformed indolent lymphoma, follicular grade 3B). The clinical experts consulted by CADTH reported the following outcomes to be important for patients with DLBCL: complete response (CR) at the end of treatment (EOT) as measured by PET and Lugano criteria, progression-free survival (PFS), especially at 2 years posttreatment (PFS24), and OS. According to the clinical experts, response to treatment is assessed using a CT scan after the first 3 or 4 cycles of therapy to identify responders, and PET at the EOT to determine remission or CR. CR maintained for 2 years was considered by the clinical experts to demonstrate cure. The experts indicated that treatment discontinuation should be considered when there is a lack of efficacy (i.e., no response or disease progression despite treatment) or unacceptable toxicity (e.g., serious adverse events [SAEs]), and emphasized regular monitoring of patients with supportive care in balancing the benefits versus harms of therapy. The clinical experts consulted by CADTH reported that specialists with experience treating patients with lymphoma could provide care and management of patients with DLBCL, including hematologists or oncologists.
Clinician Group Input
Clinician input was received from 2 groups: the Ontario Health (Cancer Care Ontario) Hematology Cancer Drug Advisory Committee, comprising 7 clinicians and a group of hematologists and oncologists practising in Canada and treating DLBCL, comprising 55 clinicians. Input from the clinician groups was generally aligned with the clinical experts consulted by CADTH. As highlighted by the clinical experts consulted by CADTH, the clinician groups noted that there remains a significant unmet need to improve the cure rate for patients with DLBCL with first-line therapy and to reduce the high rate of relapsed/refractory disease, thereby improving outcomes and reducing the need for patients to proceed to more toxic secondary options. The clinician groups stated that pola-R-CHP is an alternative to R-CHOP for patients with previously untreated DLBCL with an IPI score of 2 to 5, echoing the input of the clinical experts consulted by CADTH for the review. Outcomes used to assess patient response to treatment include PFS, which is a clinically meaningful end point that is used in clinical practice as well as PFS24, as most progressions or relapses will occur within this time frame. The input stated that the response during therapy is typically monitored by CT scan, and posttreatment patients are assessed by both CT scan and PET scan. This differed slightly according to the clinical experts consulted by CADTH who indicated posttreatment assessment to be conducted by PET scan. After therapy, clinician groups and the clinical experts consulted by CADTH alike reported that patients are typically monitored clinically every 3 months for 2 years, then every 6 to 12 months for evidence of progression. Disease progression or adverse events [AEs] were indicated as the primary reasons to discontinue treatment with the drug under review. The clinician groups also noted that treatment with pola-R-CHP has a similar safety profile to R-CHOP and it is anticipated that it can be safely administered in similar settings as R-CHOP. However, this opinion was not shared by the clinical experts consulted by CADTH, who highlighted concerns with greater toxicity with pola-R-CHP treatment. In general, pola-R-CHP is an outpatient systemic therapy that can be routinely administered by physicians with experience in oncology therapy (typically hematologists or oncologists).
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The drug plans identified implementation issues related to relevant comparators, considerations for continuation or renewal of therapy, discontinuation of therapy, and prescribing of therapy, as well as generalizability and system and economic issues. The clinical experts consulted by CADTH weighed evidence from the POLARIX trial and other clinical considerations to provide responses to the drug programs’ implementation questions. Refer to Table 4 for more details.
Clinical Evidence
Pivotal Studies and Randomized Controlled Trial Evidence
Description of Studies
One phase III, multicentre, randomized, double-blind, placebo-controlled trial (Study GO39942 or POLARIX; N = 879) assessed the efficacy and safety of pola-R-CHP compared with SOC in first-line treatment comprising R-CHOP in the treatment of adults with previously untreated LBCL, including DLBCL NOS, high-grade B-cell lymphoma, EBV-positive DLBCL NOS, and T-cell/histiocyte-rich LBCL. Outcomes identified to be important to patients and most relevant for clinicians included OS, PFS, CR, objective response rate (ORR), and patient-reported outcomes (PROs). PFS as assessed by the investigator was the primary outcome in the POLARIX trial, and OS and CR at EOT as assessed by blinded independent central review (BICR) were key secondary outcomes. Additional secondary efficacy outcomes included CR at EOT assessed by investigator and ORR assessed by BICR and by investigator. HRQoL was evaluated as a secondary outcome, assessed using time to deterioration (TTD) and responder analyses for the European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 (EORTC QLQ-C30) Physical Functioning and Fatigue scales and the Functional Assessment of Cancer Therapy-Lymphoma lymphoma subscale (FACT-Lym LymS). HRQoL was also assessed using rate of peripheral neuropathy on the Functional Assessment of Cancer Therapy/Gynecologic Oncology Group-Neurotoxicity (FACT/GOG-NTX). Treatment-emergent AEs (TEAEs), SAEs, withdrawal due to AEs (WDAEs), and deaths were reported in the POLARIX trial.
The POLARIX study included 7 sites in Canada. All patients enrolled had CD20-positive DLBCL, IPI score of 2 to 5, ECOG PS of 0 to 2 (84% with score of 0 to 1), and a life expectancy of 12 months or greater. Patients had a median study population age of 65 years. Most patients had advanced Ann Arbor stage III to IV (88.7%), and baseline lactate dehydrogenase greater than 1 time the upper limit of normal (ULN) (65.4%) at diagnosis. Patients were similar between treatment groups in stratification factors used for randomization (IPI score, bulky disease, and geographical region) and baseline characteristics. All patients in the safety population had at least 1 medical history condition with similar proportions between groups for the most common conditions.
Efficacy Results
Key efficacy results in the POLARIX study are summarized in Table 2. The analysis population for primary and secondary efficacy analyses consisted of the intention-to-treat (ITT) population (i.e., all randomized patients regardless of treatment received). The analysis population for HRQoL included the PRO-evaluable population (i.e., all randomized patients with a baseline and at least 1 postbaseline assessment). The primary analysis included patients followed up to the clinical cut-off date (CCOD) of June 28, 2021, for all efficacy and HRQoL outcomes. The updated analysis followed patients to the CCOD of June 15, 2022, for OS and PFS.
Overall Survival
OS, defined as time from randomization to date of death from any cause, was included in the hierarchical testing procedure as a key secondary end point. At the final analysis (CCOD of June 15, 2022), a total of 131 OS events were observed after a median survival follow-up of 39.7 months and 39.6 months for the pola-R-CHP and R-CHOP groups, respectively, (64 events [14.5%] and 67 events [15.3%], respectively). The stratified hazard ratio (HR) was 0.94 (95% confidence interval [CI], 0.67 to 1.33; P = 0.7326). OS rates for the pola-R-CHP and R-CHOP groups were 92.2% and 94.6%, respectively, at 12 months, and 88.7% and 88.6%, respectively, at 24 months.
Progression-Free Survival
The primary study end point in the POLARIX study was PFS, defined as the time from randomization to the first occurrence of disease progression or relapse as assessed by the investigator, using the Lugano response criteria for malignant lymphoma, or death from any cause, whichever occurred earlier. At the updated analysis with a CCOD of June 15, 2022, median follow-up time for PFS was 30.9 months (range, 0 to 46) and 30.8 months (range, 0 to 54) in the pola-R-CHP and R-CHOP groups, respectively. At this analysis, 118 (26.8%) patients had disease progression or had died in the pola-R-CHP group versus 143 (32.6%) patients in the R-CHOP group (stratified HR = 0.76; 95% CI, 0.60 to 0.97; P = 0.0298).
Subgroup Analyses
Subgroup analyses of PFS were exploratory in the POLARIX study, and the CADTH review focused on the subgroups of IPI score, bulky disease, and DLBCL subtype. Subgroup analysis suggested benefit with pola-R-CHP treatment compared with R-CHOP among patients with IPI score 3 to 5 (unstratified HR = 0.71; 95% CI, 0.53 to 0.95) and without bulky disease (unstratified HR = 0.59; 95% CI, 0.42 to 0.83). Unstratified investigator-assessed PFS subgroup analysis by baseline molecular DLBCL subtypes included centrally tested cell of origin (COO), centrally tested immunohistochemistry for BCL2 and MYC (double-expressor lymphoma [DEL]), and centrally tested fluorescence in situ hybridization (FISH) for rearrangements in MYC, BCL2, and BCL6 (DHL or THL), suggesting that treatment with pola-R-CHP compared with R-CHOP was associated with better PFS among patients in higher-risk subgroups: ABC-DLBCL subgroup (84.7% versus 56.1%; HR = 0.34; 95% CI, 0.21 to 0.56) and DEL subgroup (75.8% versus 63.1%; HR = 0.63; 95% CI, 0.42 to 0.94).
Based on the subgroup results for PFS among those with IPI score 3 to 5 and no bulky disease, the Health Canada reviewers12 requested the sponsor conduct additional subgroup analyses to examine the subgroups of patients with DLBCL who have an IPI score of 3 to 5 and no bulky disease. The results suggested that pola-R-CHP may have a greater PFS benefit compared to R-CHOP in this subgroup (unstratified HR = 0.40; 95% CI, 0.25 to 0.63). While concrete conclusions cannot be drawn on the results of these analyses, there is a signal that the benefit of treatment with pola-R-CHP may be most noticed in those with an IPI score of 3 to 5 and no bulky disease.
CR Rate at EOT (PET-CT, by BICR and by Investigator)
CR rate at EOT assessed using PET-CT by BICR was a key secondary end point included in the statistical testing hierarchy. At the EOT, BICR-assessed CR rate was 78.0% for pola-R-CHP (95% CI, 73.79 to 81.74) versus 74.0% for R-CHOP (95% CI, 69.66 to 78.07; difference = 3.9%; 95% CI, –1.9 to 9.7).
CR rate at EOT assessed using PET-CT by investigator assessment was a secondary efficacy end point that was not adjusted for multiplicity. Investigator-assessed CR rates at EOT were 75.0% for pola-R-CHP versus 72.2% for R-CHOP (difference = 2.79; 95% CI, –3.20 to 8.75; P = 0.3402).
ORR at EOT (PET-CT, by BICR and by Investigator)
ORR at EOT assessed using PET-CT by BICR and by investigator were secondary efficacy end points that were not adjusted for multiplicity. BICR-assessed ORR (i.e., partial response [PR] or CR) at EOT was 85.5% in the pola-R-CHP group versus 83.8% in the R-CHOP group (difference = 1.63%; 95% CI, –3.32 to 6.57; P = 0.4828). Investigator-assessed ORR at EOT was 84.5% in the pola-R-CHP group versus 80.9% in the R-CHOP group (difference = 3.68; 95% CI, –1.49 to 8.84; P = 0.1345).
Health-Related Quality of Life
HRQoL was assessed using the following secondary end points without adjustment for multiplicity: TTD in the EORTC QLQ-C30 Physical Functioning (≥ 10-point decrease) and Fatigue (≥ 10-point increase), FACT-Lym LymS (≥ 3-point decrease), and FACT-GOG-NTX; proportion of patients in each treatment group achieving clinically meaningful improvement in EORTC QLQ-C30 Physical Functioning (≥ 7-point increase) and Fatigue (≥ 9-point decrease), and FACT-Lym LymS (≥ 3-point increase); and a comparison of EORTC QLQ-C30 treatment-related symptoms and FACT/GOG-NTX peripheral neuropathy between the 2 treatment groups. There were no clear differences between the treatment groups for these outcomes.
Harms Results
Key safety results in the POLARIX study are summarized in Table 2. The analysis population for harms included all patients who received at least 1 dose of any study treatment component, with patients grouped according to the treatment received. Patients were followed for harms to the updated analysis (CCOD of June 15, 2022).
Table 2: Summary of Key Results From Pivotal Studies and RCT Evidence
Outcome | POLARIX pola-R-CHP n = 440 | POLARIX R-CHOP n = 439 |
|---|---|---|
OS (CCOD June 15, 2022) | ||
Patients with event, n (%) | 64 (14.5) | 67 (15.3) |
Median time to OS,a months (range) | NE (0 to 54) | NE (0 to 54) |
Stratifiedb HRc (95% Cl) | 0.94 (0.67 to 1.33) | Reference |
P value (log-rank)d | 0.7326 | Reference |
OS at 24 months | ||
Patients remaining at risk, n | 379 | 372 |
24-month OS rate (95% Cl) | 88.68 (85.70 to 91.67) | 88.69 (85.68 to 91.71) |
Difference in OS rate at 24 months (95% CI) | –0.01 (–4.25 to 4.23) | Reference |
Investigator-assessed PFS (CCOD June 15, 2022) | ||
Patients with events, n (%) | 118 (26.8) | 143 (32.6) |
Earliest contributing event, n | ||
Death | 22 | 21 |
Disease progression | 96 | 122 |
Median time to PFS,a months (range) | NE (0 to 46) | NE (0 to 54) |
Stratifiedb HRc (95% CI) | 0.76 (0.62 to 0.97) | Reference |
P value (log-rank)d | 0.0298 | Reference |
PFS at 24 months | ||
Patients remaining at risk, n | 313 | 284 |
24-month PFS ratec (95% CI) | 76.99 (72.99 to 80.99) | 70.35 (65.97 to 74.73) |
Difference in PFS rate at 24 months (95% CI) | 6.64 (0.70 to 12.58) | Reference |
EORTC QLQ-C30 (CCOD June 28, 2021) | ||
Physical Functioning | ||
Responderse, n (%) | 175 (42.4) | 163 (39.6) |
Difference in response rate, unstratified analysis (95% CI)f | 2.81 (–4.06 to 9.64) | Reference |
Time to deterioration,g patients with event, n (%) | 183 (41.6) | 187 (42.6) |
Stratifiedb HRc (95% CI) | 0.97 (0.79 to 1.19) | Reference |
Fatigue | ||
Responders,e n (%) | 309 (74.8) | 281 (68.2) |
Difference in response rate, unstratified analysis (95% CI)f | 6.61 (0.28 to 12.88) | Reference |
Time to deterioration,g patients with event, n (%) | 223 (50.7) | 230 (52.4) |
Stratifiedb HRc (95% CI) | 0.94 (0.78 to 1.13) | Reference |
FACT-Lym (CCOD June 28, 2021) | ||
LymS | ||
Responders,e n (%) | 340 (82.3) | 335 (81.3) |
Difference in response rate, unstratified analysis (95% CI)f | 1.01 (–4.43 to 6.45) | Reference |
Time to deterioration,g patients with event, n (%) | 148 (33.6) | 138 (31.4) |
Stratified HRd (95% CI) | 1.03 (0.81 to 1.30) | Reference |
B symptom, night sweats | ||
Time to deterioration,g patients with event, n (%) | 101 (23.0) | 119 (27.1) |
Stratifiedb HRc (95% CI) | 0.78 (0.60 to 1.02) | Reference |
FACT/GOG-NTX (CCOD June 28, 2021) | ||
Baseline, n | 407 | 406 |
Baseline, mean (SE) | 39.81 (0.22) | 39.49 (0.25) |
24 months, number of patients contributing to the analysis | 104 | 88 |
24 months, adjusted mean (SE) | –1.63 (0.46) | –1.60 (0.50) |
24 months, difference in adjusted means (95% CI) | –0.04 (–1.37 to 1.30) | Reference |
Harms, N (safety population) (CCOD June 15, 2022) | 435 | 438 |
AEs, n (%) | 426 (97.9) | 431 (98.4) |
SAEs, n (%) | 148 (34.0) | 134 (30.6) |
WDAE (from study treatment), n (%) | 26 (6.0) | 28 (6.4) |
Deaths, n (%) | 64 (14.7) | 69 (15.8) |
Notable harms, n (%) | ||
Peripheral neuropathy | 230 (52.9) | 236 (53.9) |
Infections | 216 (49.7) | 187 (42.7) |
Neutropenia including febrile neutropenia | 200 (46.0) | 188 (42.9) |
Anemia | 125 (28.7) | 119 (27.2) |
Thrombocytopenia | 58 (13.3) | 59 (13.5) |
Infusion-related reactions | 58 (13.3) | 70 (16.0) |
Hepatic toxicity | 46 (10.6) | 33 (7.5) |
Tumour lysis syndrome | 2 (0.5) | 4 (0.9) |
Progressive multifocal leukoencephalopathy | 0 | 0 |
AE = adverse event; CCOD = clinical cut-off date; CI = confidence interval; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; FACT-GOG/NTX = Functional Assessment of Cancer Therapy/Gynecologic Oncology Group-Neurotoxicity; FACT-Lym = Functional Assessment of Cancer Therapy-Lymphoma; HR = hazard ratio; IPI = International Prognostic Index; LymS = lymphoma subscale; NE = not estimable; OS = overall survival; PFS = progression-free survival; pola-R-CHP = polatuzumab vedotin, rituximab, cyclophosphamide, doxorubicin, and prednisone; R-CHOP = rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone; RCT = randomized controlled trial; SAE = serious adverse event; SE = standard error; vs. = versus; WDAE = withdrawal due to adverse event.
aSummaries of OS and PFS (median, percentiles) were Kaplan-Meier estimates. 95% CI for median was computed using the method of Brookmeyer and Crowley.
bStratified for IPI score (IPI 2 vs. IPI 3 to 5), bulky disease (1 lesion ≥ 7.5 cm; present vs. absent), and geographical region (Western Europe, US, Canada, and Australia vs. Asia vs. rest of world [remaining countries]).
cHRs were estimated by Cox regression.
dP value has been adjusted for multiple testing.
eResponder was defined as a patient with at least a 7-point scale score increase from baseline on the EORTC QLQ-C30 Physical Functioning, at least a 9-point scale score on the EORTC QLQ-C30 Fatigue, or at least a 3-point scale score increase on the FACT-Lym LymS.
f95% CI for difference in response rates are constructed using Wilson method.
gDeterioration was defined as a ≥ 10-point decrease in the EORTC QLQ-C30 Physical Functioning or Fatigue scale from baseline, a ≥ 3-point decrease in the FACT-Lym LymS, a ≥ 1-point increase in the B symptom raw score.
Source: POLARIX Clinical Study Reports.13,14
Most patients in the POLARIX study reported at least 1 AE (97.9% in pola-R-CHP group versus 98.4% in R-CHOP group). The most commonly reported AEs in the pola-R-CHP versus R-CHOP groups were nausea (41.6% versus 36.8%, respectively), constipation (28.7% versus 29.2%, respectively), fatigue (25.7% versus 26.5%, respectively), diarrhea (31% versus 20.1%, respectively), and alopecia (24.4% versus 24.0%, respectively).
The percentage of patient who experienced at least 1 SAE was 34.0% in the pola-R-CHP group and 30.6% in the R-CHOP group. The most common SAEs in the pola-R-CHP and R-CHOP groups were febrile neutropenia (9.9% versus 6.4%, respectively), pneumonia (4.1% versus 3.9%, respectively), diarrhea (2.3% versus 0.5%, respectively), and pyrexia (1.6% versus 1.8%, respectively).
The percentage of patients who experienced at least 1 AE that led to withdrawal of any study medication was 6.0% in the pola-R-CHP group and 6.4% in the R-CHOP group. The most common AEs that led to withdrawal of any study medication were infections (1.6% in pola-R-CHP group versus 2.3% in R-CHOP group) and nervous system disorders (0.7% in pola-R-CHP group versus 2.5% in R-CHOP group).
A total of 133 (15.2%) deaths occurred in the POLARIX study, with similar proportions between the pola-R-CHP and R-CHOP groups (14.7% and 15.8%, respectively). The primary cause of death among cases in the pola-R-CHP and R-CHOP groups were disease progression (7.8% and 8.0% of patients, respectively) and AEs (3.0% and 2.5% of patients, respectively).
Notable harms identified in the CADTH review included peripheral neuropathy, infections, neutropenia, anemia, thrombocytopenia, infusion-related reactions (IRRs), hepatic toxicities, tumour lysis syndrome (TLS), and progressive multifocal leukoencephalopathy (PML). The proportion of patients who experienced peripheral neuropathy was 52.9% and 53.9% in the pola-R-CHP and R-CHOP groups, respectively. A higher proportion of patients in the pola-R-CHP group compared with the R-CHOP group experienced infections (49.7% versus 42.7%), neutropenia including febrile neutropenia (46.0% versus 42.9%), and hepatic toxicity (10.6% versus 7.5%). Similar proportions of patients in the pola-R-CHP and R-CHOP groups experienced anemia (28.7% versus 27.2%) and thrombocytopenia (13.3% versus 13.5%). The proportion of patients who reported IRRs was 13.3% and 16.0% in the pola-R-CHP and R-CHOP groups, respectively. TLS was reported by 2 patients (0.5%) and 4 patients (0.9%) in the pola-R-CHP and R-CHOP groups, respectively. No patient reported experiencing PML in the POLARIX trial.
Critical Appraisal
The POLARIX study was a phase III, double-blind, placebo-controlled trial. There was low risk of bias for objective and subjective outcome assessments due to the blinded study design. Between-group proportions were similar in stratification factors for IPI score (2 versus 3 to 5), bulky disease, and geographical region, as well as other baseline demographics and disease characteristics, therefore the risk of selection bias from inappropriate randomization and allocation concealment was determined to be low. Few protocol deviations occurred to impact study conduct, assessments, or findings. There was a relatively high rate of discontinuations from the study (19.1%) with most losses due to deaths, which was similar between treatment groups. The large reduction in sample size makes it difficult to adequately assess the treatment effects on important outcomes such as PFS and HRQoL. A hierarchical gatekeeping approach was used to account for multiplicity for the primary efficacy outcome (PFS) and key secondary end points (OS and BICR-assessed CR rate). Analyses of additional secondary end points such as investigator-assessed CR rate, ORR, or HRQoL were not adjusted for multiplicity, therefore results for these end points were at increased risk of type I error. OS results were limited by the low number of events observed, relatively short duration of follow-up at the final analysis, and likely violation of the proportional hazards assumption. Most patients were censored for PFS because no progression event or death was recorded at the CCOD. Subgroup analyses were exploratory. HRQoL outcomes were not adjusted for multiplicity, and a high proportion of patients were lost to follow-up for HRQoL assessments at 24 months and later time points without adequate imputation of missing data.
The efficacy end points evaluated in the POLARIX trial were aligned with treatment outcomes important to patients and of relevance in clinical practice per the clinical experts consulted by CADTH, including PFS, OS, and CR rate. While the population enrolled in the POLARIX study were reported by the clinical experts to be representative of patients with DLBCL who they would consider eligible for pola-R-CHP treatment, there were limitations with the representativeness of the study population. Patients with ECOG PS 3 or 4, transformed indolent lymphoma, or with follicular lymphoma grade 3B, were excluded from the POLARIX study but considered to be eligible for treatment in current practice, as per the clinical experts. The clinical experts believed that higher-risk patients (IPI score 3 to 5) who typically experience poor outcomes with SOC R-CHOP are more likely to benefit from treatment with pola-R-CHP. There was uncertainty of benefit among patients with IPI score of 2 based on subgroup analyses, and those with IPI score of 0 to 1 were excluded from the POLARIX study. SOC R-CHOP is not routinely used in patients with specific molecular characteristics (e.g., DHL or THL) as other first-line approaches are preferred for these patients in Canada (e.g., dose-adjusted etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin, and rituximab [DA-EPOCH-R]). Moreover, patients with advanced age and/or frailty, or comorbidities are more likely to experience intolerability of R-CHOP requiring dose adjustments or alternative treatments, such that there is a lack of evidence from the POLARIX study for treatment with pola-R-CHP in these patients. PFS may be an acceptable surrogate for OS in DLBCL, though the strength of the correlation with OS beyond 5 years is uncertain. Nonetheless, the clinical experts considered PFS24 to be a reasonable outcome for assessing the effects of pola-R-CHP because most disease progression or relapses occur before this time point. However, there was uncertainty regarding whether the between-group difference in PFS observed in the POLARIX trial is clinically meaningful overall and at specific time points.
Long-Term Extension Studies
No long-term extension studies were submitted in the systematic review evidence.
Indirect Evidence
No indirect treatment comparisons were submitted in the systematic review evidence.
Studies Addressing Gaps in the Pivotal and Randomized Controlled Trial Evidence
No additional studies addressing important gaps in the systematic review evidence were identified.
Conclusions
In the POLARIX trial, the study population was limited in representativeness of patients with DLBCL, but likely representative of those considered to be eligible for treatment in clinical practice. Pola-R-CHP demonstrated a benefit for PFS compared to SOC R-CHOP in adult patients with previously untreated LBCL, including DLBCL NOS, high-grade B-cell lymphoma, EBV-positive DLBCL NOS, and T-cell/histiocyte-rich LBCL. However, there was uncertainty in whether the difference in PFS between groups was clinically meaningful. There were signals that the PFS benefit was primarily driven by treatment effects among the subgroup of patients with an IPI score of 3 to 5 and without bulky disease, but these findings were from exploratory subgroup analyses and may reflect differences in expected risk of progression among patients with an IPI score of 2 versus higher. It is uncertain whether pola-R-CHP is associated with an OS benefit because the data were immature, reflecting the relatively limited duration of follow-up, and the proportional hazards assumption was likely violated. Analyses of secondary outcomes showed numeric benefits with pola-R-CHP in achieving CR and ORR after treatment. There were no differences between the pola-R-CHP and R-CHOP groups for HRQoL, functioning, or key symptoms experienced by patients, including fatigue, diarrhea, and peripheral neuropathy. Patients treated with pola-R-CHP experienced similar frequencies of AEs, SAEs, WDAEs, and deaths as those treated with R-CHOP with no new safety signals identified.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of polatuzumab vedotin (Polivy) 1.8 mg/kg given as an IV infusion every 21 days for 6 cycles in combination with R-CHP in the treatment of previously untreated LBCL including DLBCL NOS, high-grade B-cell lymphoma, EBV-positive DLBCL NOS, and T-cell/histiocyte-rich LBCL in adults.
Disease Background
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the CADTH review team.
NHL comprises of a wide range of at least 60 closely related cancers of the lymphocytes.3 NHL is the fifth most common cancer diagnosed in Canada3 and in a given year it is estimated that 11,400 Canadians are diagnosed with NHL and 3,000 will die from it.4 DLBCL NOS is the most common histologic subtype of NHL accounting for approximately 25% of NHL cases.9 DLBCL cases remain heterogeneous regarding their morphology, genetics, and biological behaviour.9 Other subtypes include DLBCL transformed from follicular lymphoma or chronic lymphocytic leukemia; high-grade B-cell lymphoma; primary DLBCL of the central nervous system (CNS); primary cutaneous DLBCL, leg type; EBV-positive DLBCL; DLBCL associated with chronic inflammation; and T-cell/histiocyte-rich LBCL.5-8 Initial indicators of DLBCL include a quickly growing, nonpainful mass that is typically an enlarged lymph node in the neck, groin, or abdomen as well as fever, weight loss, and night sweats.9 Patients with DLBCL have demonstrated significantly worse HRQoL with respect to functional and symptom scores when compared with a general cancer reference population, highlighting the high symptom burden experienced by patients with DLBCL.15 Approximately 60% of patients present with advanced-stage disease16 and approximately half of patients with DLBCL have organ involvement at the time of diagnosis, commonly the digestive (gastrointestinal) tract and bone marrow.10 The risk of DLBCL increases with age and the average age at diagnoses is approximately 65 years.10
OS in patients with primary refractory disease is very poor, with only 15% to 20% surviving at 5 years.11 Patients with PR or CR to first-line treatment also have poor survival at relapse, with 38% and 42% surviving at 5 years, respectively.11 Patients with primary refractory disease or early relapsed disease (< 12 months) have worse outcomes compared to patients who relapse more than 12 months after first-line treatment.17,18
Standards of Therapy
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the CADTH review team.
According to the clinical experts consulted by CADTH, the goal of first-line treatment for DLBCL is curative intent.19-23 Approximately 50% to 60% of patients with previously untreated advanced DLBCL can be cured with R-CHOP.5,24 In Canada, the gold SOC in first-line treatment for DLBCL is R-CHOP. Based on clinician advisory boards convened by the sponsor across Canada consisting of 24 clinicians, approximately 85% to 90% of patients living in Canada are being treated with R-CHOP as first-line therapy. Optimization of first-line treatment for DLBCL requires consideration of several factors, including IPI risk factors (age of the patient, ECOG PS, Ann Arbor stage, number of extranodal sites, and lactate dehydrogenase level), presence of bulky disease, molecular features, CNS involvement, and patient factors such as comorbidities.19,25,26 For example, patients deemed to have worse health may receive a reduced dose of CHOP.26
While R-CHOP remains the SOC therapy in previously untreated DLBCL, approximately 40% of these patients will experience treatment failure or relapse with R-CHOP — with nearly 10% to 15% experiencing primary refractory disease (disease does not enter CR and/or progresses during or soon after treatment) — whereas an estimated 20% to 30% will experience relapse after complete remission.24,27,28 Most relapses will occur within the subsequent 2 years to 3 years after initial treatment29 and meta-analyses have found that patients who do not relapse within 2 years have similar survival outcomes as the general population.30,31 This remains a major cause of morbidity and mortality in DLBCL.32 In higher-risk subgroups (e.g., higher IPI score, certain molecular subtypes such as ABC), the likelihood for relapsed or refractory disease are even higher.33
In the setting of first-line DLBCL, most patients experience treatment failure within the first 2 years, with outcomes correlated with timing of progression or relapse. Recent analyses have demonstrated that patients with DLBCL who have remained in remission after this period have survival equivalent to that of the age-, sex-, and country-matched general population.34 However, approximately half of the patients will not respond to subsequent therapy because of refractory disease, and a significant number of patients are ineligible for these subsequent lines of therapy because of age, comorbidities, or chemotherapy-insensitive disease. The treatment approach for such patients in the second-line setting as well as for all patients beyond the second-line is a palliative approach.35
Drug Under Review
Polatuzumab vedotin is a CD79b-targeted antibody-drug conjugate that delivers an antimitotic agent, monomethyl auristatin E (MMAE), to B-cells, which results in the killing of malignant B-cells.2 The polatuzumab vedotin molecule consists of MMAE that is attached to a humanized immunoglobulin G1 monoclonal antibody. The monoclonal antibody binds to CD79b, which is a cell surface component of the B-cell receptor that is expressed in more than 95% of DLBCLs. Binding to CD79b enables delivery of MMAE, which binds to microtubules and kills dividing cells by inhibiting cell division and inducing apoptosis.2
The Health Canada indication under review is polatuzumab vedotin in combination with R-CHP for the treatment of adult patients with previously untreated LBCL, including DLBCL NOS, high-grade B-cell lymphoma, EBV-positive DLBCL NOS, and T-cell/histiocyte-rich LBCL. The sponsor’s reimbursement request is aligned with the approved Health Canada indication.
Polatuzumab vedotin is administered by IV infusion. The Health Canada recommended dose is 1.8 mg/kg every 21 days for 6 cycles in combination with R-CHP.2 Polatuzumab vedotin, rituximab, cyclophosphamide, and doxorubicin can be administered in any order on day 1 after the administration of prednisone. Prednisone is administered on days 1 to 5 of each cycle. Cycles 7 and 8 consist of rituximab as monotherapy.
Polatuzumab vedotin was previously reviewed by CADTH in combination with bendamustine and rituximab for the treatment of adult patients with relapsed or refractory DLBCL NOS, who are not eligible for autologous stem cell transplant and have received at least 1 prior therapy.36 Key characteristics of polatuzumab vedotin and R-CHOP are summarized in Table 3.
Table 3: Key Characteristics of Polatuzumab Vedotin and R-CHOP
Mechanism of action | Indicationa | Route of administration | Recommended doseb | Serious adverse effects or safety issues |
|---|---|---|---|---|
Polatuzumab vedotin | ||||
CD79b-targeted antibody-drug conjugate; delivers an antimitotic agent to B-cells | In combination with R-CHP, is indicated for the treatment of adult patients with previously untreated LBCL, including DLBCL NOS, high-grade B-cell lymphoma, EBV-positive DLBCL NOS, and T-cell/histiocyte-rich LBCL | IV infusion | 1.8 mg/kg every 21 days for 6 cycles in combination with R-CHP | Infusion-related reactions, peripheral neuropathy, tumour lysis syndrome, myelosuppression, hepatic toxicity, infections, progressive multifocal leukoencephalopathy |
Rituximab | ||||
Chimeric monoclonal antibody; binds to the transmembrane antigen CD20 | NHL | IV infusion, SC injection | 375 mg/m2 on day 1 of each cycle; second and following doses may be given as SC injection (fixed dose of 1,400 mg/11.7 mL) | Infections, neutropenia, hypotension, tumour lysis syndrome, infusion-related reactions |
Cyclophosphamide | ||||
Alkylating agent; cytotoxic action due to cross-linking of strands of DNA and RNA and inhibiting DNA synthesis | NHL | IV infusion | 750 mg/m2 on day 1 of each cycle | Secondary malignancy, cardiac toxicity, lung and liver injury, severe QT prolongation, serious allergic reaction |
Doxorubicin | ||||
Anthracycline antibiotic; binds to DNA and inhibits nucleic acid synthesis | Neoplastic diseases (adjunct treatment): NHL | IV infusion | 50 mg/m2 on day 1 of each cycle | Cardiomyopathy, secondary malignancies, extravasation and tissue necrosis, myelosuppression and sequelae, hepatic impairment |
Vincristine | ||||
Vinca alkaloid; binds with tubulin and disrupts progression of mitosis | Neoplastic diseases (adjunct treatment): NHL | IV infusion | 1.4 mg/m2 on day 1 of each cycle | Uric acid nephropathy, shortness of breath, severe bronchospasm, constipation, hair loss |
Prednisone | ||||
Synthetic corticosteroid; associated with anti-inflammatory and immune-modulating effects | Neoplastic diseases (adjunct treatment): NHL | Oral | 100 mg or 45 mg/m2 daily (round off dose to nearest 25 mg) on days 1 to 5 of each cycle | Osteoporosis, infections, cataracts, hypertension, Cushing syndrome, hyperglycemia, delayed wound healing |
DLBCL = diffuse large B-cell lymphoma; EBV = Epstein-Barr virus; LBCL = large B-cell lymphoma; NHL = non-Hodgkin lymphoma; NOS = not otherwise specified; R-CHOP = rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone; R-CHP = rituximab, cyclophosphamide, doxorubicin, and prednisone; SC = subcutaneous.
aHealth Canada–approved indication.
bR-CHOP recommended doses based on BC Cancer and Cancer Care Ontario treatment guidelines.23,37
Source: Product monographs for Polivy,2 Riabni,38 cyclophosphamide,39 doxorubicin,40 and prednisone.41
Stakeholder Perspectives
Patient Group Input
This section was prepared by CADTH staff based on the input provided by patient groups. The full original patient input received by CADTH has been included in the Stakeholder section at the end of this report.
One patient group, Lymphoma Canada, submitted input for this review. Lymphoma Canada is a national Canadian registered charity that empowers the lymphoma community through education, support, advocacy, and research. The input was based on an online anonymous patient survey among patients with a subtype of LBCL, created and promoted by Lymphoma Canada, available from February 2, 2023, to March 13, 2023. A total of 89 respondents were included in the patient input, with 4 confirmed responses for experience with polatuzumab vedotin. Most patients were living in Canada (94%), aged 55 to 74 years (64%), and diagnosed 1 year to 5 years ago (61%); more patients were female (58%) than male (42%).
The most common physical symptoms upon diagnosis that respondents found challenging included fatigue, body aches and pains, night sweats, enlarged lymph nodes, and a reduced appetite. The psychosocial impacts of their diagnosis included stress, anxiety or worry, fear of progression, inability to continue daily activities, and difficulty sleeping. LBCL symptoms impacted respondents’ ability to exercise, travel, spend time with family, volunteer, and attend work or school. Most survey respondents received 1 line of treatment for their LBCL, with R-CHOP as the most common treatment regimen. Most patients were satisfied or very satisfied with their options for first-line treatment. When asked about accessing lymphoma therapy in Canada, many patients indicated they were required to travel long distances, which was which challenging financially and required time off work. Among the 4 patients with experience with pola-R-CHP, 3 patients would recommend the treatment to other patients with LBCL and 2 patients indicated their overall experience with the treatment was very good. Side effects experienced by at least 2 patients on pola-R-CHP included fatigue, neutropenia, thrombocytopenia, decreased appetite, and diarrhea. According to the patient input received, expectations for new treatments include longer disease remission, control of disease symptoms, longer survival, normalized blood counts, and improved quality of life to be able to participate in daily activities.
Clinician Input
Input From Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of LBCL.
Unmet Needs
The clinical experts consulted by CADTH for polatuzumab vedotin identified that high-risk (IPI score 3 to 5) patients have lower cure rates and 40% to 50% experience primary refractory disease or early relapse (i.e., with < 1 year of SOC R-CHOP); thus, patients with higher-risk disease need improved cure rates from first-line treatment to avoid intensive salvage chemotherapy (e.g., autologous stem cell transplant, CAR T-cell therapy) that confers limited benefit with high toxicity risks. Additionally, the clinical experts reported that patients with advanced age or frailty consistently have lower cure rates due to a lack of standardized first-line treatment arising from intolerance to R-CHOP therapy.
Place in Therapy
The clinical experts consulted by CADTH regarded pola-R-CHP to treat underlying LBCL disease with curative intent, and therefore, to be offered as first-line treatment to patients who would be eligible for R-CHOP therapy. The clinical experts noted that polatuzumab vedotin has been successfully combined with other chemotherapy regimens (e.g., bendamustine and rituximab) available in Canada as a bridge to CAR T-cell therapy or for patients who are ineligible for CAR T-cell therapy with relapsed or refractory DLBCL. The clinical experts’ preference is to employ pola-R-CHP as a first-line therapy for increasing cure rates rather than as a later-line treatment to avoid salvage treatments. The clinical experts expressed that polatuzumab vedotin’s mechanism of action is independent of the other chemotherapy drugs in the pola-R-CHP combined regimen and works similarly to brentuximab vedotin in combination with CHP for T-cell lymphoma, supporting the use of the pola-R-CHP combination. The clinical experts anticipate pola-R-CHP could replace the current SOC first-line treatment of DLBCL for patients with an IPI score of 2 or greater, if the trial criteria are used; however, it was the clinical experts’ opinion that patients with an IPI score of 3 to 5 will benefit the most from pola-R-CHP treatment. The clinical experts indicated that patients with advanced age for whom dose reductions (e.g., > 75% of normal dosing of anthracycline or cyclophosphamide) may be necessary should also be included. The clinical experts consulted did not consider pola-R-CHP to have a therapeutic role for patients with limited-stage disease (i.e., IPI 0 to 1) as current approaches using R-CHOP (with or without radiotherapy) demonstrate high cure rates.
Patient Population
While the clinical experts supported the targeting of patients at high risk (IPI score 3 to 5) due to the potential to achieve the greatest benefit, there was uncertainty whether patients with intermediate risk (IPI score 2) would have a similar magnitude of benefit from treatment with pola-R-CHP. The clinical experts reiterated that patients with limited-stage disease experience highly successful outcomes with current R-CHOP approaches and they generally would not require treatment with pola-R-CHP.
The clinical experts noted that the IPI assessment is easy to conduct and used regularly in clinical practice to identify DLBCL patients at diagnosis and to guide treatment; however, the clinical experts expressed concerns regarding the diagnostic criteria for LBCL. Some pathological entities commonly excluded from clinical trials (e.g., LBCL transformed from indolent lymphoma, follicular grade 3B) often receive treatment identical to de novo DLBCL, and were considered by the clinical experts to be eligible for pola-R-CHP as a patient with LBCL who is deemed eligible for R-CHOP according to clinical practice guidelines.
Assessing the Response Treatment
The clinical experts consulted by CADTH considered outcomes such as CR at the EOT (as measured by PET and Lugano criteria), PFS, and OS to be relevant to clinical practice. According to the clinical experts, response to treatment is assessed using a CT scan after the first 3 or 4 cycles of therapy to determine partial remission at minimum, and to identify poor responders, and then CT or PET at the EOT to determine CR. While OS is the most important outcome, the clinical experts emphasized that PFS at 2 years is also an important outcome because the majority of patients relapse or experience disease progression in the first 1 year to 2 years posttreatment, and because relapse after 2 years of treatment is uncommon. The clinical experts considered patients who experienced CR maintained for 2 years as likely cured (late relapses can occur), and in this setting, follow-ups are conducted in the community setting with a physical exam, symptoms assessment, laboratory assessments, and additional imaging accordingly.
Discontinuing Treatment
The clinical experts consulted by CADTH for pola-R-CHP considered the same factors used for R-CHOP to discontinue treatment: lack of efficacy (no response or disease progression despite treatment) and unacceptable toxicity (e.g., severe toxicity such a neuropathy that is not amenable to a dose reduction, grade 3 anemia, grade 3 diarrhea). Lack of efficacy was reported by the clinical expert to be assessed clinically to inform subsequent imaging assessments whereas toxicity relies on clinical judgment for the balance of treatment benefits versus harms. The clinical experts emphasized the need to regularly monitor patients for tolerance to therapy and provide supportive care.
Prescribing Considerations
The clinical experts consulted by CADTH for the drug under review reported that hematologists or oncologists trained to treat patients with lymphoma could oversee the care of patients with DLBCL in diagnosis, treatment, and monitoring. The clinical experts indicated that the management of patients would be standard in a tertiary or community setting. Further, the clinical experts considered it to be reasonable for general physicians to oversee therapy in conjunction with a hematologist or oncologist. It was noted by the clinical experts that administration of pola-R-CHP for patients with LBCL would not necessitate special training above and beyond standard hematology or oncology training.
Clinician Group Input
This section was prepared by CADTH staff based on the input provided by clinician groups. The full original clinician group input received by CADTH has been included in the Stakeholder section at the end of this report.
Clinician input was received from 2 groups: the Ontario Health (Cancer Care Ontario) Hematology Cancer Drug Advisory Committee, comprising 7 clinicians, and a group of hematologists and oncologists practising in Canada and treating DLBCL, comprising 55 clinicians. Input from the clinician groups was generally aligned with the clinical experts consulted by CADTH. The clinician groups noted that there remains a significant unmet need to improve the cure rate for patients with DLBCL with first-line therapy, to reduce the high rate of relapsed/refractory disease, thereby improving outcomes and reducing the need for patients to proceed to more toxic secondary options. The clinician groups stated that pola-R-CHP is an alternative to R-CHOP for patients with previously untreated DLBCL with an IPI score of 2 to 5, echoing the input of the clinical experts consulted by CADTH for the review. Outcomes used to assess patient response to treatment include PFS, which is a clinically meaningful end point that is used in clinical practice as well as PFS24, as most progressions or relapses will occur within this time frame. The input stated that the response during therapy is typically monitored by CT scan, and posttreatment patients are assessed by both CT scan and PET scan. This differed slightly according to the clinical experts consulted by CADTH who indicated posttreatment assessment to be conducted by PET scan. After therapy, clinician groups and the clinical experts consulted by CADTH alike reported that patients are typically monitored clinically every 3 months for 2 years, then every 6 months to 12 months for evidence of progression. Disease progression or AEs were indicated as the primary reasons to discontinue treatment with the drug under review. The clinician groups also noted that treatment with pola-R-CHP has a similar safety profile to R-CHOP and it is anticipated that it can be safely administered in similar settings as R-CHOP. However, this opinion was not shared by the clinical experts consulted by CADTH, who highlighted concerns with greater toxicity with pola-R-CHP treatment. In general, pola-R-CHP is an outpatient systemic therapy that can be routinely administered by physicians with experience in oncology therapy (typically hematologists or oncologists).
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
The standard arm in the POLARIX study (GO39942) in previously untreated LBCL was R-CHOP, which is an appropriate comparator in Canada. In patients with “double-hit” or “double-expressor” LBLC (i.e., MYC translocation plus gene rearrangement of BCL2 or BCL6 [or both]), DA-EPOCH-R may be used in some jurisdictions as primary therapy in previously untreated patients. | This is a comment from the drug plans to inform pERC deliberations. The experts noted polatuzumab vedotin should not be added to DA-EPOCH-R until the combination has been demonstrated to be superior to standard DA-EPOCH-R. The experts also noted that polatuzumab vedotin plus DA-EPOCH-R is being actively studied in patients with previously untreated lymphoma.42 The clinical experts’ opinion was that patients with DLBCL and the listed genetic variants who are eligible for R-CHOP should also be eligible for pola-R-CHP. |
Considerations for initiation of therapy | |
Can patients who received pola-R-CHP as primary therapy be eligible for pola-BR at time of disease relapse if they are transplant-ineligible? If so, what is the appropriate progression-free interval from completion of pola-R-CHP to re-treat with polatuzumab vedotin as part of pola-BR? | For patients who relapse after 1 year, the experts would likely consider other available options (including clinical trials), and assess the patient’s experience with regard to adverse effects and their willingness to be retreated with polatuzumab vedotin. The experts would consider retreating with polatuzumab vedotin if no other options exist. For patients who experience durable remission (> 1 year) with first-line pola-R-CHP, the clinical experts indicated treatment with pola-BR would be reasonable provided other funding criteria are met. |
Would patients who are transplant-eligible benefit the same as patients who are transplant-ineligible? Is IPI scoring prognostic of treatment response to pola-R-CHP? | The clinical experts stated that pola-R-CHP should be offered to both transplant-eligible and transplant-ineligible patients, since data do not support whether transplant eligibility status influences outcomes, and there is no known impact on stem cell fitness or lymphocyte function with potential to impair subsequent collection, if needed. Therefore, IPI score is not applicable when pertaining to transplant eligibility. The experts indicated that first-line therapies are preferred over salvage therapy; therefore, treatment with pola-R-CHP may proceed without regard to transplant eligibility. |
Is IPI scoring prognostic of treatment response to pola-R-CHP? | The clinical experts noted that the IPI assessment is used regularly in clinical practice to identify patients with DLBCL at diagnosis and to guide treatment. The clinical experts anticipate pola-R-CHP could replace the current SOC first-line treatment of DLBCL for patients with an IPI score of ≥ 2, if the trial criteria are used; however, it was the clinical experts’ opinion that patients with an IPI score of 3 to 5 will benefit the most from pola-R-CHP treatment due to high risk of disease progression or relapse among these patients. Patients with IPI 0 to 1, who were excluded from the POLARIX trial, were reported by the clinical experts to experience high cure rates with SOC R-CHOP. |
Are patients with transformed follicular lymphoma, follicular lymphoma grade 3B, PMBCL, Burkitt lymphoma, CNS lymphoma (primary or secondary), or primary cutaneous DLBLC eligible? | The clinical experts reported that any lymphoma treated as de novo LBCL would be treated, including indolent lymphoma not previously systematically treated that has transformed to LBCL, follicular 3B, or primary cutaneous DLBCL. PMBCL would be excluded as this is a unique entity that responds well to R-CHOP with or without radiotherapy, or with DA-EPOCH-R strategies (better than de novo DLBCL). The experts noted that Burkitt lymphoma would not be treated with R-CHOP and therefore should not be considered for pola-R-CHP; these patients should be treated with other established protocols (e.g., Magrath, DA-EPOCH-R). The experts also did not consider primary CNS lymphoma to be eligible for pola-R-CHP as there are other available treatment options (e.g., MATRiX) and the benefit of this treatment has not been established for systemic lymphoma with CNS involvement. |
Are pediatric patients with previously untreated LBCL, including DLBCL NOS, high-grade B-cell lymphoma, Epstein-Barr virus-positive DLBCL NOS, and T-cell/histiocyte-rich LBCL eligible? | The clinical experts reported limited experience with pediatric (e.g., aged 16 years) patients with LBCL. However, given the experts’ experience with brentuximab in combination with chemotherapy in this setting, their opinion was that it would be reasonable to consider pola-R-CHP in a pediatric patient who would otherwise be treated with R-CHOP as polatuzumab vedotin theoretically would work similarly in this population. However, the experts expressed that while a 16-year-old patient may not be vastly different from an 18-year-old patient biologically, a reasonable cut-off age remains unclear given that the POLARIX trial only studied patients aged 18 years and older. The product monograph for polatuzumab vedotin outlines the following for pediatrics (aged younger than 18 years): Based on the data submitted and reviewed by Health Canada, the safety and efficacy of Polivy in pediatric patients has not been established; therefore, Health Canada has not authorized an indication for pediatric use. |
Considerations for prescribing of therapy | |
Polatuzumab vedotin is available in 30 mg and 140 mg vials that require reconstitution. The dose is 1.8 mg/kg IV every 21 days for 6 cycles. Polatuzumab vedotin, rituximab, cyclophosphamide, and doxorubicin can be administered in any order on day 1 after the administration of prednisone. Prednisone is administered on days 1 to 5 of each cycle. Cycles 7 and 8 consist of rituximab as monotherapy, which is an additional 2 cycles of therapy compared to the standard 6 cycles of R-CHOP that is usually administered, adding additional pharmacy workload and chair time visits. | This is a comment from the drug plans to inform pERC deliberations. |
The doses of cyclophosphamide, doxorubicin, vincristine, and prednisone in R-CHOP are sometimes reduced in older adults (mini R-CHOP). Is it appropriate to reduce the doses of cyclophosphamide, doxorubicin, and prednisone when used in the pola-R-CHP regimen in older or frail patients? | The clinical expert reported they would consider dose reduction of anthracycline/cyclophosphamide in the pola-R-CHP regimen to 75% to 80% of usual dosing in older or frail patients for whom a reasonably good performance status can be achieved and supported throughout treatment to limit toxicities (e.g., neuropathy, neutropenia). The experts indicated that they would assess tolerance based on the first cycle to make subsequent treatment decisions (i.e., continue vs. abandon), noting that patients themselves often declare adverse effects early on and those who tolerate treatment do well despite factors such as advanced age and/or poor risk disease. |
Can polatuzumab vedotin be used with rituximab SC or rituximab biosimilar as part of the pola-R-CHP regimen? | The clinical experts agreed there are no concerns with using either SC rituximab or biosimilars. |
It was noted that the initial dose of polatuzumab vedotin should be administered as a 90-minute IV infusion. Patients should be monitored for infusion-related reactions during the infusion and for at least 90 minutes following completion of the initial dose. If the prior infusion was well tolerated, the subsequent doses of polatuzumab vedotin may be administered as a 30-minute infusion and patients should be monitored during the infusion and for at least 30 minutes after completion of the infusion. This adds chair time compared to R-CHOP. | This is a comment from the drug plans to inform pERC deliberations. |
It was noted that for outpatient centres it may be difficult to administer cycle 1 within an 8-hour time frame. | This is a comment from the drug plans to inform pERC deliberations. |
In the monograph for polatuzumab vedotin, the following information complicates its preparation and may limit potential sites for administration in jurisdictions with centralized production:
| This is a comment from the drug plans to inform pERC deliberations. |
Generalizability | |
Patients with ECOG PS 0 to 2 were eligible in the POLARIX study. Are patients with ECOG PS > 2 eligible if performance status is related to their underlying lymphoma? | Since R-CHOP is standard first-line therapy for patients regardless of ECOG PS, the clinical experts considered patients with higher ECOG PS to be eligible for pola-R-CHP. The experts noted that ECOG PS is part of IPI risk scoring, and thus higher ECOG PS would increase the IPI risk score, thereby potentially conferring a greater benefit from pola-R-CHP among these patients. Therefore, the clinical experts stated that IPI score would be the preferred measurement for determining treatment vs. ECOG PS alone. |
Should patients currently receiving R-CHOP for previously untreated LBCL be allowed to switch to pola-R-CHP? | The clinical experts stated that there is no evidence to suggest changing regimens from R-CHOP to pola-R-CHP would benefit patients who are responding to SOC, and would continue patients on the current R-CHOP regimen. In patients who do not respond, the clinical experts would move to currently recommended salvage therapies, which may include polatuzumab vedotin, rather than use pola-R-CHP. Moreover, the clinical experts indicated that patients would still be considered eligible for polatuzumab vedotin in relapse if they demonstrated primary refractory disease with R-CHOP or if they experienced a short duration of remission. |
Funding algorithm (oncology only) | |
Drug may change place in therapy of drugs reimbursed in subsequent lines | This is a comment from the drug plans to inform pERC deliberations. |
Complex therapeutic space with multiple lines of therapy, subpopulations, or competing products | This is a comment from the drug plans to inform pERC deliberations. |
Care provision issues | |
It was noted the rate of febrile neutropenia is higher with pola-R-CHP compared to R-CHOP (9.9% vs. 6.4%). Are WBC growth factors (e.g., filgrastim, pegfilgrastim) required, either as primary or secondary prophylaxis with pola-R-CHP? (G-CSF was required in the POLARIX trial for 6 cycles of pola-R-CHP as primary prophylaxis. In many jurisdictions G-CSF is provided as primary prophylaxis for R-CHOP only in patients who are older than 65 years or younger than 65 years with higher risk of febrile neutropenia. Potential budgetary impact with larger population in first-line setting). | The clinical experts highlighted that given the higher rates of febrile neutropenia with pola-R-CHP, all patients would routinely receive G-CSF (as with R-CHOP); however, this may be a change in practice for centres that currently restrict G-CSF to patients at high risk for febrile neutropenia. |
When is it appropriate to consider dose reductions or delays? | The clinical experts indicated that dose reductions would be considered for patients who are older and/or frail, or who have poor tolerance to initial treatment. |
System and economic issues | |
The sponsor estimates that 396, 805, and 876 patients will initiate treatment with pola-R-CHP in years 1, 2, and 3 of funding, respectively, and the incremental budget impact in the base-case analysis of reimbursing pola-R-CHP for previously untreated adult patients with DLBCL will be $29.5 million for year 1, $59.4 million for year 2, and $64.2 million for year 3 with a total budget impact of $153 million over the 3-year period. The drug plans are concerned about the volume of patients, large budget impact analysis, and potential underestimation of the BIA. | This is a comment from the drug plans to inform pERC deliberations. |
CNS = central nervous system; DA-EPOCH-R = dose-adjusted etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin, and rituximab; DLBCL = diffuse large B-cell lymphoma; ECOG PS = Eastern Cooperative Oncology Group Performance Status; G-CSF = granulocyte colony-stimulating factor; IPI = International Prognostic Index; LBCL = large B-cell lymphoma; Magrath = cyclophosphamide, doxorubicin, vincristine, cytarabine, methotrexate, leucovorin, ifosfamide, mesna, and etoposide; MATRiX = methotrexate, cytarabine, thiotepa, and rituximab; NOS = not otherwise specified; pERC = CADTH pan-Canadian Oncology Review Expert Review Committee; PMBCL = primary mediastinal large B-cell lymphoma; pola-BR = polatuzumab vedotin, bendamustine, and rituximab; pola-R-CHP = polatuzumab vedotin, rituximab, cyclophosphamide, doxorubicin, and prednisone; R-CHOP = rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone; SC = subcutaneous; SOC = standard of care; vs. = versus; WBC = white blood cell.
Clinical Evidence
The objective of CADTH’s Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of polatuzumab vedotin 1.8 mg/kg IV in the pola-R-CHP regimen in the treatment of previously untreated LBCL, including DLBCL NOS, high-grade B-cell lymphoma, EBV-positive DLBCL NOS, and T-cell/histiocyte-rich LBCL in adults. The focus will be placed on comparing pola-R-CHP to relevant comparators, and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of pola-R-CHP is presented in 4 sections, and CADTH’s critical appraisal of the evidence is included after each section. The first section, the systematic review, includes pivotal studies and randomized controlled trials (RCTs) that were selected according to the sponsor’s systematic review protocol. The sponsor did not include long-term extension studies, indirect evidence, or additional studies addressing important gaps in the pivotal and RCT evidence.
Included Studies
Clinical evidence from 1 pivotal RCT is included in the CADTH review and appraised in this document.
Pivotal Studies and RCT Evidence
Contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the CADTH review team.
Description of Study
Characteristics of the included study are summarized in Table 5.
Table 5: Details of POLARIX Study
Characteristic | POLARIX |
|---|---|
Designs and populations | |
Study design | Phase III, multicentre, randomized, DB, placebo-controlled trial |
Locations | 879 patients (211 sites total) in 22 countries, in 3 regions
|
Patient enrolment dates | Start date: November 15, 2017 (first patient enrolled) End date: June 27th, 2019 (last patient randomized) |
Randomized (N) | N = 879 (440 in pola-R-CHP group; 439 in R-CHOP group) |
Inclusion criteria | Previously untreated patients with CD20-positive DLBCL who had 1 of the following diagnoses by 2016 WHO classification of lymphoid neoplasms:
For females and males: agreed to remain abstinent |
Exclusion criteria |
|
Drugs | |
Intervention | Polatuzumab vedotin 1.8 mg/kg IV, rituximab 375 mg/m2 IV (also given as monotherapy in cycles 7 and 8), cyclophosphamide 750 mg/m2 IV, doxorubicin 50 mg/m2 IV, placebo for vincristine, each given on day 1 and prednisone 100 mg/day orally given on day 15 of every 21-day cycle for 6 cycles |
Comparator(s) | Placebo for polatuzumab vedotin, rituximab 375 mg/m2 IV, cyclophosphamide 750 mg/m2 IV, doxorubicin 50 mg/m2 IV, and vincristine 1.4 mg/m2 IV (maximum 2 mg/dose) each given on day 1 and prednisone 100 mg/day orally given on day 15 of every 21-day cycle for 6 cycles. Rituximab 375 mg/m2 IV will be given as monotherapy in cycles 7 and 8. |
Study duration | |
Screening phase | 28 days before randomization to day 1 (randomization) |
Treatment phase | Polatuzumab vedotin in pola-R-CHP and vincristine in R-CHOP: 3.5 months Rituximab in both pola-R-CHP and R-CHOP: 4.9 months 90% of patients received treatment corresponding to a median of 3.5 to 3.6 months of treatment |
Follow-up phase | Median follow-up (primary analysis CCOD June 28, 2021): 28.2 months (range, 0 to 43 months; 28.1 months pola-R-CHP vs. 28.2 months R-CHOP) Median follow-up (updated CCOD June 15, 2022): 40.2 months Posttreatment follow-up visit: up to approximately 65 months Last patient last visit: study ongoing |
Outcomes | |
Primary end point | PFS (investigator) |
Secondary and exploratory end points | Key secondary:a
Secondary:b
Exploratory:
|
Publications | Tilly et al. (2022)43 ClinicalTrials.gov identifier: NCT03274492 |
ALT = alanine aminotransferase; aPTT = activated partial thromboplastin time; AST = aspartate aminotransferase; BICR = blinded independent central review; BOR = best overall response; CCOD = clinical cut-off date; CNS = central nervous system; CR = complete response; DB = double-blind; DFS = disease-free survival; DLBCL = diffuse large B-cell lymphoma; DOR = duration of response; EBV = Epstein-Barr virus; ECOG PS = Eastern Cooperative Oncology Group Performance Status; EFSall = event-free survival from all causes; EFSeff = event-free survival–efficacy; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EOT = end of treatment; FACT/GOG-NTX = Functional Assessment of Cancer Therapy/Gynecologic Oncology Group-Neurotoxicity; FACT-Lym LymS = Functional Assessment of Cancer Therapy-Lymphoma lymphoma subscale; FDG-PET = fluorodeoxyglucose PET; HTLV-1 = human T-lymphotrophic 1 virus; INR = international normalized ratio; IPI = International Prognostic Index; LBCL = large B-cell lymphoma; NOS = not otherwise specified; NYHA = New York Heart Association; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; PFS24 = 2-year progression-free survival; PML = progressive multifocal leukoencephalopathy; pola-R-CHP = polatuzumab vedotin, rituximab, cyclophosphamide, doxorubicin, and prednisone; PRO = patient-reported outcome; PT = prothrombin time; PTT = partial thromboplastin time; R-CHOP = rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone; TTD = time to deterioration; ULN = upper limit of normal.
Note: Details from the table have been taken from the sponsor’s summary of clinical evidence.
aKey secondary end points were included in the hierarchical testing procedure.
bSecondary end points were not adjusted for multiple testing (i.e., the type I error rate has not been controlled for).
Source: POLARIX Clinical Study Reports.13,14
The POLARIX study13,14 was a phase III, multicentre (7 sites in Canada), randomized, double-blind, placebo-controlled trial (N = 879) that examined the efficacy, safety, and PROs of polatuzumab vedotin plus chemoimmunotherapy (pola-R-CHP; n = 440) compared with SOC chemoimmunotherapy (R-CHOP; n = 439) in previously untreated patients with CD20-positive DLBCL with IPI score 2 to 5. At the CCOD for the primary analysis (June 28, 2021), patients had median duration of survival follow-up of 28.1 months in the pola-R-CHP group and 28.2 months in the R-CHOP group. The purpose of the updated Clinical Study Report (CCOD June 15, 2022) was to provide the results of the prespecified final OS analysis which was to be performed 36 months after the last patient was enrolled. The first patient was enrolled on November 15, 2017, and the study was ongoing at the time of the updated Clinical Study Report (September 2022).
Eligible patients were randomized in permuted blocks stratified by IPI score (2 versus 3 to 5), bulky disease defined as at least 1 lesion 7.5 cm or greater (presence versus absence), and geographical region (Western Europe, US, Canada, and Australia versus Asia versus remaining countries).
Patients were randomized in a 1:1 ratio to pola-R-CHP or R-CHOP. No crossover to the experimental arm was allowed. Patients were discontinued from treatment upon progressive disease (PD) including symptomatic deterioration attributable to PD, unacceptable toxicity, pregnancy, use of nonprotocol-specified anticancer therapy, or based on decision by patient, investigator, or sponsor.
Populations
Inclusion and Exclusion Criteria
The POLARIX trial included adults (aged 18 years to 80 years) with previously untreated CD20-positive DLBCL, including the following WHO 2016 classification of lymphoid neoplasms:
DLBCL NOS, including germinal centre B-cell (GCB)-type, and ABC
T-cell/histiocyte-rich LBCL
EBV-positive DLBCL, NOS
ALK-positive LBCL
HHV8-positive DLBCL, NOS
High-grade B-cell lymphoma with MYC and BCL2 and/or BCL6 rearrangements (DHL or THL)
High-grade B-cell lymphoma, NOS.
Of note, key eligibility criteria for enrolment in the POLARIX study included patients with IPI score of 2 to 5, ECOG PS of 0 to 2, and a life expectancy of 12 months or greater. Patients were excluded from the study if they had a diagnosis of follicular lymphoma grade 3B, B-cell lymphoma unclassifiable with features intermediate between DLBCL and classical Hodgkin lymphoma (grey-zone lymphoma), primary mediastinal (thymic) LBCL, Burkitt lymphoma, CNS lymphoma (primary or secondary involvement), primary effusion DLBCL, and primary cutaneous DLBCL.
Interventions
Patients in the POLARIX study received 6 cycles of either pola-R-CHP or standard R-CHOP chemoimmunotherapy regimens (Figure 1). In the pola-R-CHP intervention group, the components of chemoimmunotherapy comprised polatuzumab vedotin, rituximab, cyclophosphamide, doxorubicin, placebo for vincristine, and prednisone. Infusions of each component were administered on day 1 (except for prednisone on day 1 to day 5) of every 21-day cycle for 6 cycles in the following order: prednisone, rituximab, and polatuzumab vedotin. Infusions for placebo for vincristine, cyclophosphamide, and doxorubicin were administered thereafter according to institutional preference. In the R-CHOP comparator group, the chemoimmunotherapy regimen mirrored that of the intervention group, except for placebo for polatuzumab vedotin and treatment with vincristine. In both groups, rituximab was administered as monotherapy in cycle 7 and cycle 8.
Prior to study treatment initiation (i.e., prephase treatment), steroids were permitted to prevent TLS in patients with extensive disease and reduce toxicity (e.g., cytokine release syndrome) of the first cycle of study treatment. Premedication with antihistamines, antipyretics, and/or analgesics may have been administered at the discretion of the investigator. Other than the prednisone given as study treatment and prednisone that may be given as prephase treatment at the discretion of the treating investigator physician, corticosteroids may have been used only for the treatment of conditions other than lymphoma (e.g., asthma). In general, investigators should have managed a patient’s care with supportive therapies as clinically indicated, per local standard practice. Vitamin D substitution was encouraged for patients with vitamin D deficiency. All rituximab infusions were to be administered to patients after premedication.
Polatuzumab vedotin and placebo for polatuzumab vedotin was identical in dosage, preparation, and administration. Vincristine (1.4 mg/m2; maximum dose 2 mg) or placebo for vincristine (i.e., normal saline) were identical in dosage and administration. Vincristine was typically administered as an IV infusion via minibag over approximately 10 minutes to 30 minutes through a dedicated line.
Rituximab was administered as 375 mg/m2 IV infusion on day 1 of each cycle; no dose modifications were permitted. Rituximab was administered after the prednisone dosing, and before the cyclophosphamide, doxorubicin, polatuzumab vedotin (or placebo for polatuzumab vedotin), and vincristine (or placebo for vincristine) infusions.
Cyclophosphamide (750 mg/m2) and doxorubicin (50 mg/m2) were administered as IV infusions, administered after both rituximab and polatuzumab vedotin or its placebo. Oral prednisone (100 mg/day) was administered on day 1 to day 5 of each of 6 cycles, with prednisolone (100 mg/day) or IV methylprednisolone (80 mg/day) as substitutes; hydrocortisone was not permitted as a substitute.
Figure 1: Schematic of Pola-R-CHP and R-CHOP Regimens
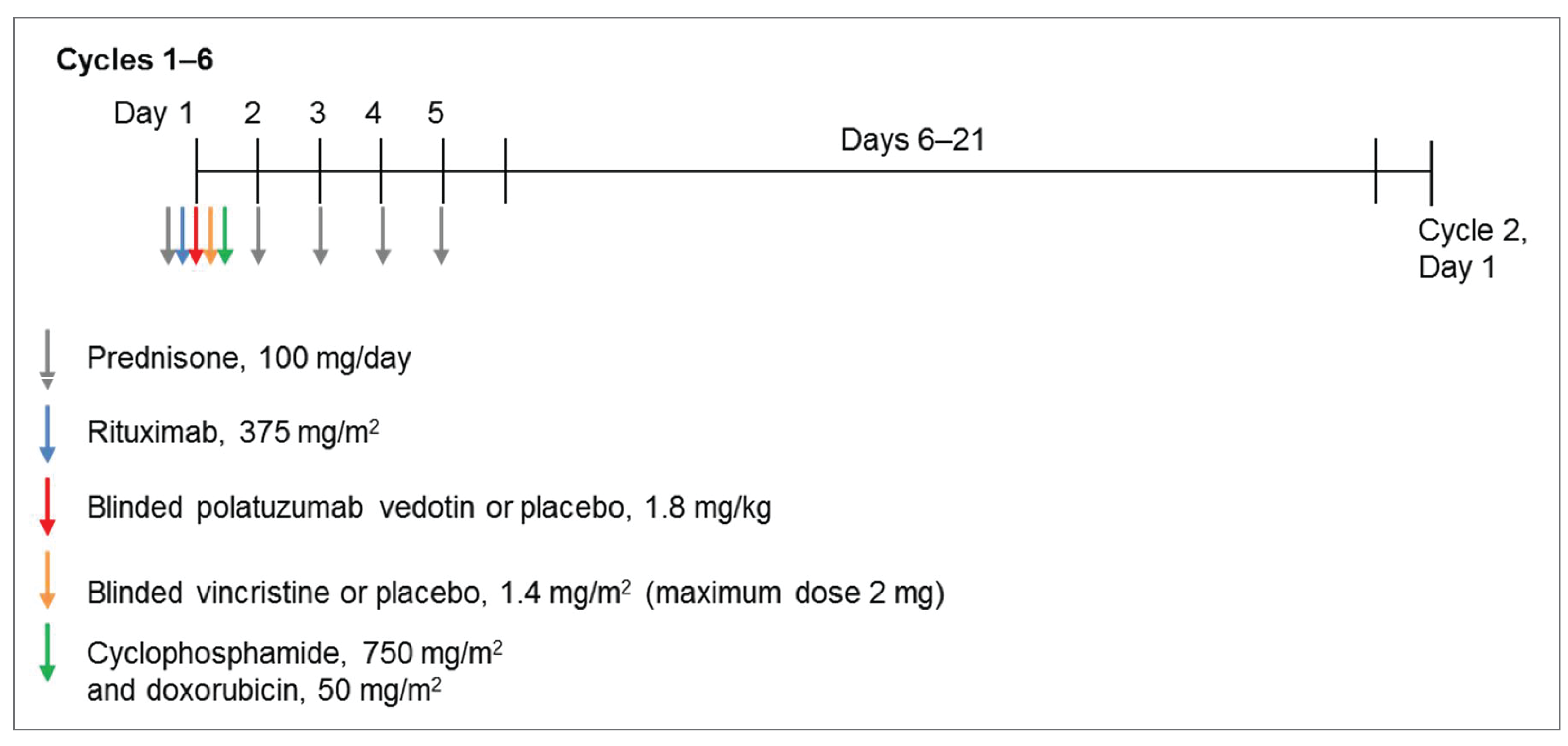
pola-R-CHP = polatuzumab vedotin, rituximab, cyclophosphamide, doxorubicin, and prednisone; R-CHOP = rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone.
Source: POLARIX Clinical Study Report.14
Patients who experience infusion-associated symptoms may be treated symptomatically with acetaminophen, ibuprofen, diphenhydramine, and/or H2-receptor antagonists (e.g., famotidine, cimetidine), or equivalent medications per local standard practice. Serious infusion-associated events manifested by dyspnea, hypotension, wheezing, bronchospasm, tachycardia, reduced oxygen saturation, or respiratory distress should be managed with supportive therapies as clinically indicated (e.g., supplemental oxygen and beta-2 adrenergic agonists).
Prophylaxis for CNS, hemorrhagic cystitis, neutropenia, and infection were recorded. CNS prophylaxis with intrathecal chemotherapy was only to be given according to institutional practice and its use documented in the electronic case report form (eCRF). CNS prophylaxis using high-dose IV methotrexate (e.g., 1 g/m2 per cycle) was not permitted and would be considered a nonprotocol or new antilymphoma treatment (NALT). Mesna was to be used as prophylaxis for hemorrhagic cystitis according to institutional practice. Granulocyte colony-stimulating factor (G-CSF) was required as primary prophylaxis for neutropenia in each cycle of therapy during cycles 1 through 6, typically starting 1 day to 3 days after administration of myelotoxic chemotherapy (doxorubicin, cyclophosphamide, and polatuzumab vedotin). Dosing of G-CSF was to follow each site’s institutional standards or may be at the investigator’s discretion. For patients who developed neutropenia despite prophylaxis, G-CSF is not routinely recommended for the treatment of uncomplicated neutropenia. However, G-CSF may be considered in patients with fever and neutropenia who are at high risk for infection-associated complications or who have prognostic factors predictive of poor clinical outcomes. Anti-infective prophylaxis for viral, fungal, bacterial, or Pneumocystis infections was permitted and instituted per institutional practice or investigator preference based on individual patient risk factors. Patients in countries where prophylactic antiviral medications for hepatitis B reactivation are the SOC may have been treated prophylactically.
Concomitant therapies that were permitted from 7 days before study drug initiation through to study completion or discontinuation included the following: prescription drugs, over-the-counter drugs, nonlive vaccines, herbal or homeopathic remedies (not intended for cancer treatment at discretion of investigator), nutritional supplements, and treatments for an IRR or an AE. Prednisone to a maximum dose of 30 mg/day or equivalent administered during the screening period was also considered concomitant therapy. Patients were permitted to use other therapies during the study including hormone replacement therapy, oral contraceptives, antihistamines, antipyretics, and/or analgesics.
Preplanned radiotherapy (i.e., radiation that was planned before randomization to be given at the end of study treatment) may have been administered to initial sites of bulky or extranodal disease according to institutional practice. If indicated, preplanned radiotherapy was initiated within 8 weeks after the last study drug treatment and started after all EOT assessments, including PET-CT scans for disease response assessment, were completed. Any radiotherapy should have been preplanned by the centre and documented before randomization and then entered in the eCRF once the patient is randomized. All unplanned radiotherapy administered to patients was considered to be a NALT.
NALT, including radiotherapy or systemically administered therapies, could be administered after the patient had completed study treatment. NALT was allowed to be administered with or without a disease progression documented in the patient.
Patients were withdrawn from study treatment if they were treated with nonprotocol antitumour drugs (e.g., cytotoxic therapies, immunotherapy or immunosuppressive therapy, unplanned radiotherapy, biologic therapies other than clinically indicated hematopoietic growth factors).
No crossover from the R-CHOP group to the pola-R-CHP group was permitted.
Outcomes
A list of efficacy end points assessed in this clinical review report are provided in Table 6. Summarized end points are based on those included in the sponsor’s summary of clinical evidence (refer to Table 6) as well as any identified as important to this review according to stakeholders, for example the clinical expert, clinician groups, or patient groups. Additional efficacy end points reported in the POLARIX trial and included in the sponsor’s summary of clinical evidence but not identified by stakeholders to be important for event-free survival (EFS), disease-free survival (DFS), and duration of response (DOR) are summarized in Appendix 1 (Table 20 and narratively).
The primary study end point in the POLARIX study was PFS as assessed by the investigator. PFS was defined as the time from randomization to the first occurrence of disease progression or relapse as assessed by the investigator, using the Lugano response criteria for malignant lymphoma, or death from any cause, whichever occurs earlier.
While the primary efficacy end point was investigator-assessed PFS, tumour assessments were collected by an independent review facility for the key secondary end point of PET-CT CR rate at the EOT by fluorodeoxyglucose-PET using the Lugano response criteria for malignant lymphoma. OS was included as a key secondary end point.
Table 6: Outcomes Summarized From POLARIX Study
Outcome measure | Time point | POLARIX |
|---|---|---|
OS Defined as the period from the date of randomization until the date of death from any cause | Date of death from any cause | Key secondarya |
PFS as determined by investigator Defined as time from randomization to the first occurrence of disease progression or relapse as assessed by the investigator using the Lugano response criteria for malignant lymphoma, or death from any cause, whichever occurs earlier | First occurrence of disease progression or relapse, or death from any cause, whichever occurs earlier; when there are approximately 228 PFS events, and after all patients in the study have been enrolled for at least 24 months, whichever occurs later. The number of PFS events was selected to achieve statistical power of 80% for the target hazard ratio at the primary analysis and 24 months follow-up, given that in patients with previously untreated DLBCL, most disease relapse occurs within this time frame. | Primarya |
CR rate at EOTa by FDG-PET as determined by BICR Defined as the percentage of patients with CR at the EOT by PET-CT as assessed by BICR | At EOT | Key secondarya |
CR rate at EOTb by FDG-PET as determined by investigator Defined as the percentage of patients with CR at the end of treatment by PET-CT as assessed by the investigator | At EOT | Secondary |
ORR at EOT by FDG-PET as determined by investigator Defined as the percentage of patients with CR or PR at EOT by PET-CT as determined by investigator | At EOT | Secondary |
ORR at EOT by FDG-PET as determined by BICR Defined as the percentage of patients with CR or PR at EOT by PET-CT as determined by BICR | At EOT | Secondary |
PFS24 Defined as PFS rate calculated through Kaplan-Meier method at 24 months after randomization. | At 24 months | Secondary |
PRO end points PRO end points include:
| Day 1 of cycles 1, 2, 3, and 5, at treatment completion visit, and posttreatment study visits every 6 months during the first 2 years, then every 12 months for the following 3 years | Secondary |
All remaining scales of the EORTC QLQ-C30, FACT-Lym LymS, and FACT/GOG-NTX | By visit and change from baseline | Exploratory |
CR rate at 24 months as determined by the investigator Defined as the percentage of patients with CR as determined by investigator at the visit within a 3-month window before or after 24 months from randomization | At 24 months (± 3 months) | Exploratory |
PFS end points by exploratory biomarkers and molecular DLBCL prognostic subtypes such as COO, DEL, and DHL or THL | On day 1 of cycle 1 and cycle 5, and at treatment completion | Exploratory |
Safety
All verbatim AE terms occurring on or after first study treatment were mapped to MedDRA | On day 1 of cycle 1 until 90 days after the last dose of study treatment; AESIs will be monitored from day 1 of cycle 1 until 12 months after the last dose of study treatment | Safety |
AE = adverse event; AESI = adverse event of special interest; BICR = blinded independent central review; COO = cell of origin; CR = complete response; DEL = double-expressor lymphoma; DLBCL = diffuse large B-cell lymphoma; DHL = double-hit lymphoma; EFS = event-free survival; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EOT = end of treatment; FACT/GOG-NTX = Functional Assessment of Cancer Therapy/Gynecologic Oncology Group-Neurotoxicity; FACT-Lym LymS = Functional Assessment of Cancer Therapy-Lymphoma lymphoma subscale; FDG-PET = fluorodeoxyglucose PET; MedDRA = Medical Dictionary for Regulatory Activities; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; PFS24 = progression-free survival at 24 months; PR = partial response; PRO = patient-reported outcome; SAE = serious adverse event; THL = triple-hit lymphoma; TTD = time to deterioration.
Note: Although EFS was measured as the key and other secondary outcomes in POLARIX, PFS assessments were determined to be more clinically relevant based on stakeholder input and more informative for the pharmacoeconomic evaluation.
Note: Details from the table have been taken from the sponsor’s summary of clinical evidence.
aStatistical testing for these end points was adjusted for multiple comparisons (e.g., hierarchical testing).
bEOT is defined as all planned chemoimmunotherapy treatment only; should any radiotherapy be administered, EOT tumour assessment shall occur before initiating radiotherapy.
Source: POLARIX Clinical Study Reports.13,14
Additional (non-key) secondary efficacy end points were CR rate as determined by investigator, ORR at EOT by BICR and by investigator, and PFS24.
Information on HRQoL and symptoms from self-administered paper questionnaires were assessed using the EORTC QLQ-C30, FACT-Lym LymS, FACT-GOG-NTX, and 5-Level EQ-5D on day 1 of cycles 1 (baseline), 2, 3, and 5. Questionnaires were translated into the local language as appropriate. PRO measures were completed at treatment discontinuation and posttreatment visits (every 6 months for the first 2 years after treatment completion visit or early termination visit, and every 12 months for the following 3 years). Secondary end points for HRQoL included: TTD in the EORTC QLQ-C30 Physical Functioning and Fatigue, FACT-Lym LymS, and FACT-GOG-NTX; proportion of patients in each treatment group achieving clinically meaningful improvement in EORTC QLQ-C30 Physical Functioning and Fatigue and FACT-Lym LymS; and a comparison of EORTC QLQ-C30 treatment-related symptoms and FACT/GOG-NTX peripheral neuropathy between the 2 treatment groups.
Exploratory end points included the following: subgroup analysis (demographics and baseline prognostic characteristics), descriptive summary statistics and change from baseline for EORTC QLQ-C30 treatment-related symptoms (constipation, diarrhea, physical functioning, fatigue, nausea, and vomiting), the FACT-Lym LymS, and the FACT/GOG-NTX. 5-Level EQ-5D health utilities was also an exploratory end point in the POLARIX study.
Safety was evaluated by monitoring all AEs; SAEs (≥ grade 3, including fatal AEs); AEs of special interest; AEs leading to discontinuation, dose reduction, or interruption of study drug; and abnormalities identified through physical examinations, vital signs, and laboratory assessments. Such events were graded using the National Cancer Institute Common Terminology Criteria for Adverse Events, version 4.0. Laboratory safety assessments included routine monitoring of hematology and blood chemistry, and tests of immunologic parameters. An independent Data Monitoring Committee (iDMC) monitored safety data. An iDMC was used to evaluate safety during the study every 6 months. All summaries and analyses by treatment group for the iDMC review were prepared by an external independent data coordinating centre. Members of the iDMC are external to the sponsor and follow a separate iDMC charter that outlines their roles and responsibilities, as well as a detailed monitoring plan.
Assessment
Patients were assessed for disease response by the investigator using regular clinical and laboratory examinations and fluorodeoxyglucose-PET (hereafter referred to as PET-CT) and dedicated CT scans (MRIs were performed if CT scans with contrast were contraindicated in the patient), according to the Lugano response criteria for malignant lymphoma. PET-CT and dedicated CT scans were obtained at screening and 6 weeks to 8 weeks after completion of study treatment. An interim assessment was obtained after cycle 4 and included PET-CT and dedicated CT. If local practice prohibited obtaining both assessments after cycle 4, PET-CT alone (preferred) or CT alone was obtained at this time point. During the follow-up period, CT scans (PET-CT also acceptable) were performed every 6 months (i.e., months 6, 12, 18, and 24) until the end of year 2 of follow-up (approximately 2.5 years after the first dose) in accordance with study (clinic) visits and included the neck (if involved at baseline), chest, abdomen, and pelvis. During years 3, 4, and 5 of follow-up, CT scans (PET-CT acceptable) of sites of prior involvement were obtained every 12 months (at months 36, 48, and 60). If disease in other areas was suspected, additional areas were imaged at all subsequent imaging assessments. During the study, diagnosis of disease progression based on clinical examination must have been confirmed by imaging (e.g., CT, PET-CT) within 30 days and before initiation of NALT.
Bone marrow assessments were required at screening and should have included biopsy for morphology. Repeat bone marrow examinations were required to confirm a radiological assessment of CR if bone marrow was involved with tumour at screening, or for confirmation of relapse in the bone marrow.
Samples for hematology, serum chemistry, pregnancy, hepatitis B and C serology, and quantitative immunoglobulin assessments were analyzed at each study site’s local laboratory. Laboratory, biomarker, and other biological samples were obtained up to 72 hours before start of study treatment administration on day 1 of the treatment cycle or scheduled visit.
Response was evaluated at the end of study treatment, or sooner in the event a patient discontinued early. After completion of therapy, all patients were followed at clinic visits conducted every 3 months for 2 years, and then every 6 months until month 60. Assessments at each visit up to the year 5, month 60 assessment (or until disease progression if it occurred before 5 years) included physical examination, standard hematologic and biochemistry assessments, vital signs, and B symptoms (i.e., weight loss, night sweats, or fever). After 5 years, patients were followed only for survival and initiation of a NALT by telephone contact approximately every 6 months until study termination, patient withdrawal of consent, or death. After disease progression, patients were followed by telephone contact for survival, applicable AE reporting, and initiation of a NALT. Patients who terminated study treatment early without PD completed their early study treatment termination visit and then were followed for progression, NALT, and OS. Patients who initiated NALT in the absence of PD also continued to be followed for progression, additional NALT, and OS. Patients who discontinued the protocol-defined treatment and needed to start a NALT in the absence of documented disease progression (e.g., if wrong diagnosis at screening and new diagnosis required a change of treatment) were followed for disease progression and survival. Patients who terminated study treatment early because of disease progression were followed for NALT and OS.
Statistical Analysis
Sample Size and Power Calculation
In total, enrolment of 875 patients was planned and expected to complete in approximately 23 months, leading to an average monthly recruitment of 38 patients per month. The sample size considerations for PFS were based on the following assumptions:
1:1 randomization ratio in pola-R-CHP versus R-CHOP
a 1-sided log-rank test
80% power at the 2.5% significance level
PFS in the control group was assumed to follow a piece-wise exponential distribution, and estimated using the historical data obtained from the GOYA study53 among patients with IPI score 2 to 5 who received R-CHOP; the hazard rate over time [h (t)] was estimated as follows: h (t = 1 month and 6 months) = 0.018217; h (6 months and 9 months) = 0.031606; h (9 months and 12 months) = 0.018519; h (12 months and 24 months) = 0.11737; h (24 months and 36 months) = 0.005636; and h (greater than 36 months) = 0.005958
using the hazard rate assumption for the control group and an HR of 0.69 for pola-R-CHP over R-CHOP (i.e., 31% reduction in the risk of disease progression, relapse, or death), the 3-year PFS rate is expected to improve from 62% to 72%
an assumed annual dropout rate of 5% for each treatment group.
Based on these assumptions, approximately 228 investigator-assessed PFS events were needed to detect an HR of 0.69 in PFS, with 80% power for the primary analysis of PFS. The minimal detectable difference (MDD) for PFS HR at the final PFS analysis was 0.771 (i.e., 22.9% reduction in the risk of disease progression, relapse, or death). Three-year PFS was expected to improve from 62% to 70% under the MDD. No interim analyses were planned for the primary end point of PFS. The PFS primary analysis was conducted approximately after 228 PFS events occurred in the ITT population and at least 24 months after enrolling the last patient (CCOD June 2021).
Table 7: Summary of Patient-Reported Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
EORTC QLQ-C30 | A 30-item patient self-administered questionnaire for evaluating the HRQoL of adult patients with cancer.44 Consists of 5 functional scales (physical, role, cognitive, emotional, and social), 3 symptom scales (fatigue, pain, and nausea and vomiting), 6 single-item scales (dyspnea, insomnia, appetite loss, constipation, diarrhea, and financial difficulties), and 1 global quality of life scale. Items are rated on a 4-point or 7-point Likert scale.44,45 Raw scores for each scale are converted to scores from 0 to 100 with higher scores reflecting better function, more symptoms, or better quality of life.44 | Measurement properties of validity, reliability, and responsiveness have not been assessed in patients with NHL. | In patients with various types of cancers: |
FACT-Lym LymS | A self-report measure of HRQoL relevant to patients with lymphoma. The full measure consists of the FACT-G physical, social/family, emotional, and functional well-being scales (27 items), as well as the LymS, a 15-item lymphoma specific subscale (i.e., pain, fever, swelling, and night sweats).48 In the POLARIX trial, only the LymS was administered to patients. Each item is rated on a 5-point response scale that ranges from 0 (not at all) to 4 (very much) with higher scores indicative of better HRQoL.14 | In a study48 of 84 patients with NHL with measurements taken at baseline, 3 to 7 days, and 8 to 12 weeks: Validity: LymS moderately correlated with POMS TMD, and the MCS and PCS of the SF-36 (r = 0.48 to 0.62). LymS differentiated between ECOG PS (0 to 2) and between patients on or off active treatment (i.e., radiation, chemotherapy).48 Reliability: Good test-retest reliability (ICC = 0.84) and internal consistency at measured time points (alpha = 0.79 to 0.85) for the LymS.48 Responsiveness: FACT-Lym subscale scores were sensitive to change in patient’s performance status over a 3- month period; effect sizes > 0.5.48 | Approximately 3 to 5 points for the LymS in a study of patients with NHL.48 |
FACT/GOG-NTX | A self-reported measure used to assess HRQoL concerns associated with chemotherapy-induced neuropathy. In the POLARIX trial, the instrument was used to assess vincristine-induced and polatuzumab vedotin-induced neuropathy.14 The full measure consists of the FACT-G (27 items) and a peripheral neuropathy symptoms scale (11 items).49 In the POLARIX trial, only the items that comprise the peripheral neuropathy scale were administered to patients. This scale contains 4 subscales that assess sensory, hearing, and motor neuropathy as well as dysfunction associated with neuropathy, which can be summed to create a total score. Each item is rated on a 5-point response scale that ranges from 0 (not at all) to 4 (very much), with higher scores indicative of more extreme neuropathy. Total subscale score ranges from 0 (no neuropathy) to 44 (most extreme neuropathy).49 | Measurement properties of validity, reliability, and responsiveness have not been assessed in patients with NHL.48 | Not assessed in NHL patients. General MID guideline for FACT cancer-specific subscales is 0.30 to 0.40 points per item.50 |
EQ-5D-5L | Generic, preference-based HRQoL instrument, consisting of an index score and VAS scale score. A higher score represents better HRQoL.51 The index score is based on 5 dimensions: mobility, self-care, usual activities, pain/discomfort, and anxiety/depression. Each dimension is rated on a scale of 1 (“no problems”) to 5 (“extreme problems”). The VAS scale ranges from 0 (worst health imaginable) to 100 (best health imaginable).51 | Measurement properties of validity, reliability, and responsiveness have not been assessed in patients with NHL. | Not assessed in patients with NHL. General population of people living in Canada (summarized mean of 0.056; SD = 0.011).52 |
ECOG PS = Eastern Cooperative Oncology Group Performance Status; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EQ-5D-5L = 5-Level EQ-5D; FACT = Functional Assessment of Cancer Therapy; FACT-G = Functional Assessment of Cancer Therapy-General; FACT/GOG-NTX = Functional Assessment of Cancer Therapy/Gynecologic Oncology Group-Neurotoxicity; FACT-Lym = Functional Assessment of Cancer Therapy-Lymphoma; HRQoL = health-related quality of life; ICC = intraclass correlation coefficient; LymS = lymphoma subscale; MCS = mental component summary; MID = minimal important difference; NHL = non-Hodgkin lymphoma; PCS = physical component summary; POMS = Profile of Mood States; SD = standard deviation; SF-36 = Short Form (36) Health Survey; TMD = total mood disturbance; VAS = visual analogue scale.
Sample size considerations for OS were based on the following assumptions:
1:1 randomization ratio in pola-R-CHP versus R-CHOP
a 1-sided log-rank test
a 27% reduction in the risk of death (i.e., OS HR = 0.73 for pola-R-CHP over R-CHOP)
OS in the control group was assumed to follow an exponential distribution with a hazard rate of 0.006923
an assumed annual dropout rate of 5% for each treatment group.
Based on these assumptions, approximately 134, 164, and 178 OS events were expected to be observed at the 2 interim analyses and the final analysis, respectively. The MDD for OS HR at the final OS analysis was 0.75. Overall power for detecting an OS HR of 0.73 was 52%, tested at a 1-sided alpha of 0.02.
Efficacy Analyses
The primary efficacy end point was PFS. To control the overall type I error rate at a 1-sided 0.025 level of significance, a hierarchical testing procedure, including possible alpha recycling, was used to adjust for multiple statistical testing of the primary and key secondary efficacy end points, in the following order.
First, the primary efficacy end point, PFS by investigator in the ITT population, was tested at an alpha of 0.025. If the 1-sided P value corresponding to the stratified log-rank test was less than 0.025, the null hypothesis was rejected, and it was concluded that pola-R-CHP prolongs the duration of PFS relative to the R-CHOP group. If the PFS hypothesis (1) was not rejected, the key secondary end points (EFSeff [event-free survival – efficacy], EOT CR rate by BICR, and OS) were not formally tested in the ITT population.
If the primary PFS end point was statistically positive, a formal statistical test of EFSeff by investigator in the ITT population between the 2 groups was performed at a 1-sided alpha of 0.025 using a stratified log-rank test with the same stratification factors as the PFS analysis. If the corresponding 1-sided P value was less than 0.025, then the null hypothesis in (2) was rejected and it was concluded that pola-R-CHP prolongs the duration of EFSeff relative to the control group. If the EFSeff hypothesis in (2) was not rejected, EOT CR rate by BICR and OS was not formally tested in the ITT population.
If the PFS hypothesis in (1) and EFSeff hypothesis in (2) were both rejected, then the EOT CR rate by BICR in the ITT population was tested using a stratified Cochran-Mantel-Haenszel test at a 1-sided alpha of 0.005. If the EOT CR rate by BICR result was statistically significant, then it was concluded that pola- R-CHP increases the chances of achieving CR at EOT relative to the control group.
If PFS hypothesis in (1) and EFSeff hypothesis in (2) were both rejected, then OS was tested using a log-rank test with the same stratification factors as the PFS analysis. If the EOT CR rate by BICR in (3) was statistically significant, then OS was tested at a 1-sided alpha of 0.02. If the OS result was statistically significant, then it was concluded that pola-R-CHP improves the duration of OS relative to the control group.
If the EOT CR rate in (3) was not rejected at a 1-sided alpha of 0.005 but OS in (4) was statistically significant in the ITT population at a 1-sided alpha of 0.02, then EOT CR rate by BICR in ITT was to be tested again at a 1-sided alpha of 0.025.
Key secondary end points included EFSeff, CR rate at EOT assessed by BICR, and OS. OS was defined as the period from date of randomization to date of death from any cause. Patients who had not died at the CCOD were censored on the last date known to be alive, as documented by the investigator. Patients without postbaseline information were censored at date of randomization.
Two formal OS analyses were conducted: at the time of the primary PFS analysis and approximately 36 months after enrolling the last patient (June 2022). OS analysis was only performed if the PFS efficacy boundary was crossed and the other secondary end points higher than OS in the hierarchical order had passed the corresponding significance levels. Due to the low likelihood of OS crossing the boundary at the interim OS analysis, a Haybittle-Peto boundary was chosen using a nominal alpha of 0.001 to control the type I error in the group sequential analysis of OS.
The POLARIX study statistical analysis plan is summarized in Figure 2.
Figure 2: Statistical Analysis Plan in POLARIX Study
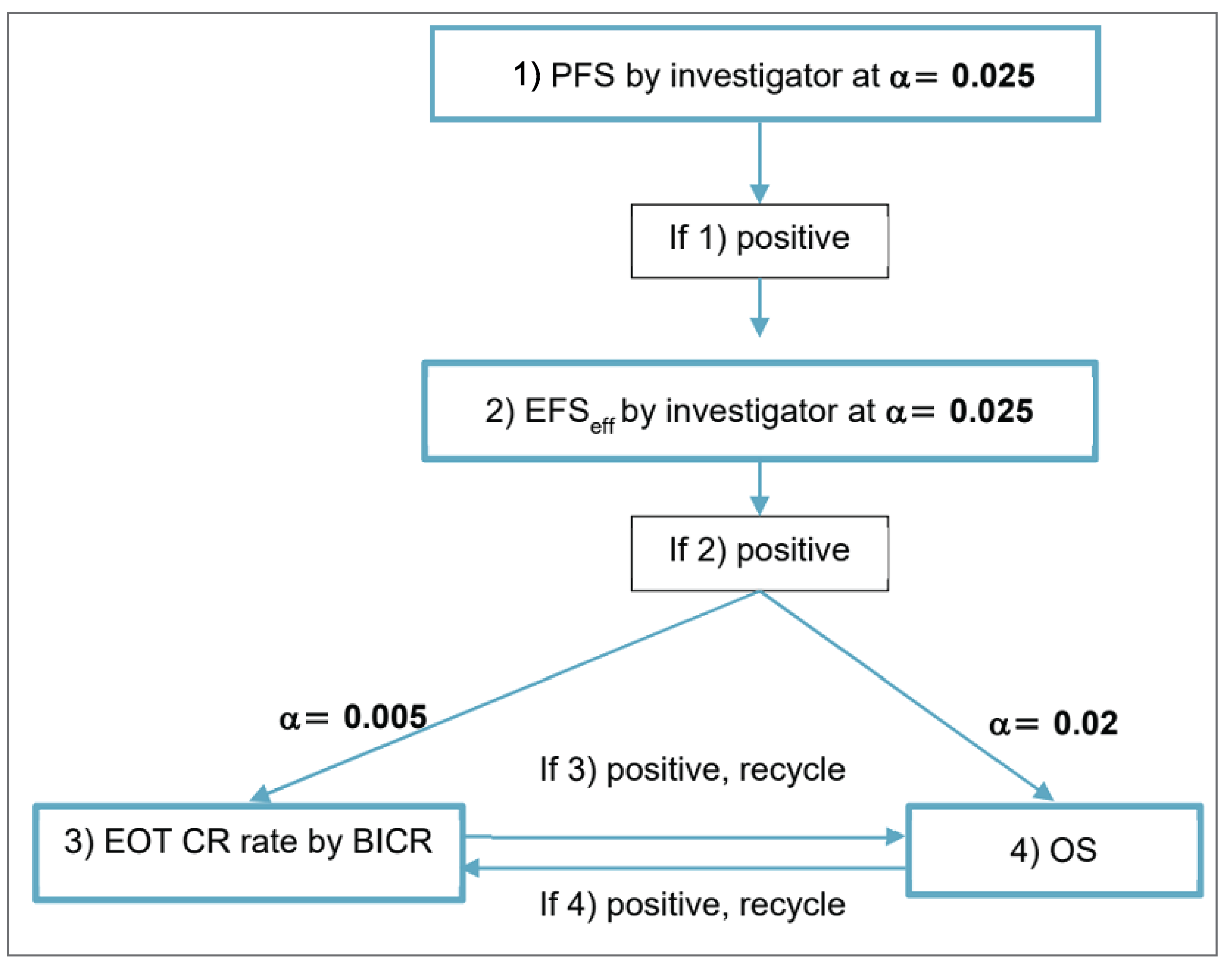
BICR = blinded independent central review; CR = complete response; EFSeff = event-free survival–efficacy; EOT = end of treatment; OS = overall survival; PFS = progression-free survival.
Source: POLARIX Clinical Study Report.14
Censoring
For primary efficacy end point PFS, patients who did not experience disease progression, relapse, or death at the data cut-off for analysis were censored on the date of last disease assessment when the patient was known to be progression-free. If no tumour assessments were performed after the baseline visit or postbaseline tumour assessment results were not evaluable, PFS was censored on the date of randomization. Censoring rules for PFS are summarized in Table 19.
For key secondary end points, CR rate at EOT by BICR for patients without response assessments were considered nonresponders. For OS, patients who had not died at the CCOD for analysis were censored on the last date when the patients were known to be alive, as documented by investigator.
For non-key secondary end points (CR rate at EOT by investigator and ORR), patients without valid or evaluable tumour assessments within the time window were considered patients who did not experience a CR.
For PRO end points, patients who did not have an observed deterioration at the time of clinical data cut-off were censored at the last nonmissing assessment date. Patients without a postbaseline assessment were censored at randomization. For EORTC QLQ-C30 subscales with 50% or more of constituent items completed, a prorated score was computed according to the scoring manuals and validation papers. Subscales of the EORTC QLQ-C30, FACT-Lym LymS, and FACT/GOG-NTX with fewer than 50% of items completed were considered as missing.
For handling incomplete deaths dates in safety analyses all deaths were included, from all sources, regardless of completeness of death date; participants who died with only a partial death date available were included. In efficacy analyses, a death was considered an event if and only if a complete death date was available; participants who died with only a partial death date available were censored.
Subgroup Analyses
To assess the consistency of the study results in prespecified subgroups defined by demographics (e.g., age) and baseline prognostic characteristics (e.g., IPI score, age-adjusted IPI, co-expression of BCL2 and MYC by immunohistochemistry [DEL], and MYC and BCL2 and/or BCL6 translocations by FISH [high-grade B-cell lymphoma]), PFS was examined in these subgroups. Summaries of PFS, including unstratified HRs estimated from Cox proportional hazards models and Kaplan-Meier estimates of 1-year and 2-year PFS rates, and median PFS (if reached), were produced separately for each level of the categorical variables for the comparisons between treatment groups and displayed in a forest plot. The analyses were exploratory and based on the investigator-assessed PFS. No statistical tests were performed to evaluate differences in effects between subgroups (i.e., tests for interaction) and analyses were not adjusted for multiplicity. The clinical experts consulted by CADTH identified age, IPI score, presence of bulky disease at baseline, and NHL histology and subtypes as the key clinical subgroups.
Biomarker Analyses
The following planned exploratory biomarker analyses included selected efficacy end points (e.g., PFS, PFS24, CR rate, ORR, OS, and PRO) by exploratory biomarkers and molecular DLBCL prognostic subtypes such as COO, circulating tumour DNA detectability in response subgroups based on fluorodeoxyglucose-PET, circulating tumour DNA as a method of molecular disease detection, and association of biomarkers (including molecular and proteomic subtypes and genomic profiles at baseline) with efficacy and/or AEs associated with pola-R-CHP and R-CHOP treatment.
Sensitivity Analyses
A sensitivity analysis based on interval censoring analysis methods was performed to account for the impact of missing scheduled tumour assessments on PFS events and because the actual timing of PFS events usually cannot be observed exactly. The PFS survival curves were estimated using the nonparametric maximum likelihood estimate for each treatment group; 1-year and 2-year rates of each treatment group were reported, and their 95% CIs were constructed based on the Greenwood method. For descriptive purposes, hypothesis testing was performed based on the log-rank test by Sun (1996)54 to compare PFS between treatment groups. The treatment effect was estimated using a stratified proportional hazard regression model (Finkelstein 1986)55 with a parametric assumption of piece-wise exponential distribution for the baseline hazard function (Friedman 1982; Royston and Parmar 2002).56,57
The impact of NALT before or in the absence of disease progression on PFS were assessed using the discount method by investigating what the PFS results would have been if NALT was not available (i.e., the time interval during which patients received NALT until the event or censoring time were discounted at 10%, 30%, and 50% for both groups). The primary analysis of PFS corresponded to a discount analysis with a discount rate of 0% on PFS time after NALT. Additional sensitivity analyses were also performed to assess the overall impact of NALT such that for patients who undertook NALT before or in the absence of subsequent death or disease progression, PFS was censored at the time of their last adequate tumour assessment before the first NALT.
Restricted mean survival time (RMST) method for PFS and OS was used to measure the difference in the average EFS time between the treatment and control group from the randomization through a prespecified time point. An unstratified nonparametric Kaplan-Meier estimate of RMST at months 12, 24, and 36 by group and the difference between groups was evaluated, with 95% CIs via the Greenwood method and P values by z test.
Secondary End Points
All non-key secondary end points were tested without adjusting for multiplicity and were based on the investigator’s assessment unless noted otherwise. For CR rate at EOT by investigator, estimates were calculated using the Clopper-Pearson method and 95% CIs using the Wilson method. CR rates comparing the treatment groups were calculated using the Cochran-Mantel-Haenszel test stratified by the same factors used in the PFS primary analysis. ORR at EOT by investigator and ORR at EOT by BICR were analyzed using the same methodologies as the CR rate. PFS24 was calculated using the Kaplan-Meier method and 95% CIs were calculated based on the normal approximation with standard errors (SEs) via the Greenwood method. The difference of PFS24 between treatment groups was informally tested using z test with SEs for the Kaplan-Meier estimates computed via the Greenwood method.
Patient-Reported Outcomes
For the EORTC QLQ-C30 Physical Functioning and Fatigue scales and the FACT-Lym LymS, visit summary and change from baseline analyses for each time point were performed and presented as summary statistics (number of patients, mean, standard deviation [SD], median, range) by treatment group. A repeated measures mixed effects model was used to compare scores between treatment groups for the subscale scores.
TTD analysis for HRs were estimated using a stratified Cox proportional hazards model with 95% CIs. Kaplan-Meier methodology was used to estimate 1-year and 2-year rates, including median TTD (if reached) per treatment group, with Kaplan-Meier curves produced. For EORTC QLQ-C30 Physical Functioning and Fatigue, TTD was defined as time from randomization to the first documented 10-point or greater decrease and increase, respectively, from baseline. For FACT-Lym LymS, TTD was defined as time from randomization to the first documented 3-point or greater decrease from baseline.
Responder analyses calculated the proportion of patients with clinically meaningful improvement on the EORTC QLQ-C30 Physical Functioning and Fatigue scales, and the FACT-Lym LymS using the Clopper-Pearson method for each treatment group, with 95% CIs. For the EORTC QLQ-C30 Physical Functioning and Fatigue scales, a clinically meaningful improvement was defined as a 7-point or greater increase and a 9-point or greater decrease, respectively. For the FACT-Lym LymS, a clinically meaningful improvement was defined as a 3-point or greater increase. For the FACT-Lym B symptoms (fever, weight loss, night sweats), a crude 1-point change was used. Differences in proportions between treatment groups were presented with 95% CIs based on a normal approximation to the binomial distribution.
For the EORTC QLQ-C30 treatment-related symptoms and the FACT-GOG-NTX peripheral neuropathy, a mixed effects model for repeated measures using an unstructured covariance matrix was used to compare scores between treatment groups for each time point with 95% CIs.
Safety Analyses
Descriptive statistics (i.e., frequencies) were summarized for each treatment group for AEs, SAEs, AEs of special interest, AEs leading to study drug discontinuation, and deaths.
Analysis Populations
The analysis population for the primary and secondary efficacy analyses consisted of all randomized patients, with patients grouped according to their assigned treatment. The ITT population included all patients randomized during the global enrolment phase (including patients enrolled in China during that phase), regardless of the treatment received. The global study included 879 patients randomized on or before June 27, 2019; this is the population for which the primary analysis has been performed. The China extension cohort included patients from mainland China who were randomized after June 27, 2019; this cohort was excluded from the ITT and the safety population. The safety population included all patients in the global study who received at least 1 dose of study treatment (any of the treatment components), with patients grouped according to the treatment regimen they actually received. The PRO-evaluable population included all randomized patients in the global study who had a baseline and at least 1 postbaseline assessment.
Patients were randomly assigned by an interactive voice or web response system.
Table 8: Statistical Analysis of Efficacy End Points in POLARIX Study
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
Investigator-assessed PFS | KM method with treatment comparison using stratified log-rank test. Estimates of treatment effect: HRs using a Cox PH analysis with 95% CIs. The PH assumption was first assessed via the cox.zph test in R and visual inspection of the cumulative log-hazards curve for PFS. | IPI score (IPI 2 vs. IPI 3 to 5), bulky disease (present vs. absent), and geographical region (Western Europe, US, Canada, and Australia vs. Asia vs. rest of world [remaining countries]). | PFS was censored on the date of last disease assessment when the patient is known to be progression-free. If no tumour assessments were performed after the baseline visit or postbaseline tumour assessment results were not evaluable, PFS was censored on the date or randomization. | Interval censoring analysis
RMST at prespecified time points (months 12, 24, and 36) |
OS | KM method with treatment comparison using stratified log-rank test. Estimates of treatment effect: HRs using a Cox PH analysis with 95% CIs. The PH assumption was assessed in the same manner as the primary efficacy analysis. | Stratified with the same stratification factors as the primary efficacy analysis. | For patients who had not died at the clinical cut-off date for analysis, OS was censored on the last date when the patients were known to be alive, as documented by investigator. | RMST at prespecified time points (months 12, 24, and 36) |
CR rate at EOT by BICR | Stratified CMH test | Stratified with the same stratification factors as the primary efficacy analysis | Non-CR imputations | None |
CR rate at EOT by investigator | Stratified CMH test | Stratified with the same stratification factors as the primary efficacy analysis | Non-CR imputations | None |
ORR at EOT by BICR or by investigator | Stratified CMH test | Stratified with the same stratification factors as the primary efficacy analysis | Non-CR imputations | None |
PFS24 | KM method with comparisons between treatment groups tested using the z test | Stratified with the same stratification factors as the primary efficacy analysis | PFS was censored on the date of last disease assessment when the patient is known to be progression-free. If no tumour assessments were performed after the baseline visit or postbaseline tumour assessment results were not evaluable, PFS was censored on the date or randomization. | None |
TTD in EORTC QLQ-C30 Physical Functioning and Fatigue | KM method with treatment comparison using stratified log-rank test. Estimates of treatment effect: HRs using a Cox PH analysis with 95% CIs. | Stratified with the same stratification factors as the primary efficacy analysis | Patients who did not have an observed deterioration at the time of clinical data cut-off were censored at the last nonmissing assessment date. Patients without a postbaseline assessment were censored at randomization. For subscales with ≥ 50% of the constituent items completed, a prorated score will be computed consistent with the scoring manuals and validation papers. For subscales with < 50% of the items completed, the subscale will be considered as missing. This approach will also be used for any missing data with the FACT-Lym LymS and FACT/GOG-NTX as per the scoring manual. | None |
TTD in FACT-Lym LymS | KM method with treatment comparison using stratified log-rank test. Estimates of treatment effect: HRs using a Cox PH analysis with 95% CIs. | Stratified with the same stratification factors as the primary efficacy analysis | Censoring and scoring were conducted in the same manner as EORTC QLQ-C30. | None |
Proportion of patients achieving meaningful improvement in EORTC QLQ-C30 Physical Functioning and Fatigue | MMRM | Stratified with the same stratification factors as the primary efficacy analysis | Scoring was done in the same manner as TTD analyses. | None |
Proportion of patients achieving meaningful improvement in FACT-Lym LymS | MMRM | Stratified with the same stratification factors as the primary efficacy analysis | Scoring was done in the same manner as TTD analyses. | None |
EORTC QLQ-C30 rate of treatment-related symptoms | MMRM | Stratified with the same stratification factors as the primary efficacy analysis | Scoring was done in the same manner as TTD analyses. | None |
FACT/GOG-NTX rate of peripheral neuropathy | MMRM | Stratified with the same stratification factors as the primary efficacy analysis | Scoring was done in the same manner as TTD analyses. | None |
BICR = blinded independent central review; CI = confidence interval; CMH = Cochran-Mantel-Haenszel; CR = complete response; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EOT = end of treatment; FACT/GOG-NTX = Functional Assessment of Cancer Therapy/Gynecologic Oncology Group-Neurotoxicity; FACT-Lym LymS = Functional Assessment of Cancer Therapy-Lymphoma lymphoma subscale; HR = hazard ratio; IPI = International Prognostic Index; KM = Kaplan-Meier; MMRM = mixed effect model with repeated measures; NALT = nonprotocol or new antilymphoma treatment; NPMLE = nonparametric maximum likelihood estimate; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; PFS24 = progression-free survival at 24 months; PH = proportional hazards; RMST = restricted mean survival time; TTD = time to deterioration; vs. = versus.
Note: Details from the table have been taken from the sponsor’s summary of clinical evidence.
Table 9: Analysis Populations of POLARIX Study
Population | Definition | Application |
|---|---|---|
ITT population | The ITT population is defined as all patients randomized during the global enrolment phase (i.e., the global study, whether or not the patients received the assigned treatment). | All efficacy analyses |
Safety population | The safety analysis population consists of all patients in the global study who received at least 1 dose of study treatment (polatuzumab vedotin, rituximab, cyclophosphamide, doxorubicin, vincristine, or prednisone), with patients grouped according to the treatment regimen actually received. | All safety analyses |
PRO-evaluable population | The PRO-evaluable population included all randomized patients in the global study who had a baseline and at least 1 postbaseline assessment. | All PRO analyses |
ITT = intention to treat; PRO = patient-reported outcome.
Note: Details from the table have been taken from the sponsor’s summary of clinical evidence.
Source: POLARIX Clinical Study Reports.13,14
Table 10: Summary of Patient Disposition From POLARIX Study, Data Cut-Off Date June 15, 2022
Patient disposition | POLARIX | |
|---|---|---|
pola-R-CHP n = 440 | R-CHOP n = 439 | |
Screened, N | 1,063 | |
Screening failure | 184 (17.3) | |
Randomized | 440 (100) | 439 (100) |
Discontinued from treatment | 49 (11.1) | 61 (13.9) |
Reason for treatment discontinuation | ||
Adverse events | 9 (2.0) | 17 (3.9) |
Death | 11 (2.5) | 4 (0.9) |
Physician decision | 10 (2.3) | 12 (2.7) |
Withdrawal by patient | 6 (1.4) | 9 (2.1) |
Progressive disease | 12 (2.7) | 16 (3.6) |
Other | 1 (0.2) | 3 (0.7) |
Discontinued from study | 81 (18.4) | 87 (19.8) |
Reason for study discontinuation | ||
Death | 63 (14.3) | 67 (15.3) |
Physician decision | 3 (0.7) | 4 (0.9) |
Withdrawal by patient | 10 (2.3) | 13 (3.0) |
Lost to follow-up | 4 (0.9) | 3 (0.7) |
Other | 1 (0.2) | 0 |
ITT, N | 440 | 439 |
Safety, N | 435 | 438 |
ITT = intention to treat; pola-R-CHP = polatuzumab vedotin, rituximab, cyclophosphamide, doxorubicin, and prednisone; R-CHOP = rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone.
Note: Values are n (%) unless otherwise indicated.
Note: Details from the table have been taken from the sponsor’s summary of clinical evidence.
Source: POLARIX Clinical Study Reports.13,14
Results
Patient Disposition
Of the 1,063 patients screened, 879 patients were randomized into the study and 184 patients did not meet eligibility criteria based on information collected in the interactive voice or web response system. The first patient was randomized on November 15, 2017. The last patient was randomized on June 27, 2019.
The main reasons for screen failure were patients not meeting the following inclusion criteria: IPI score of 2 to 5 (29 patients), availability of archival or freshly collected tumour tissue before study enrolment (28 patients), and provision of signed written consent (21 patients). A total of 873 and 825 patients were included in the safety and PRO-evaluable population, respectively.
As of the CCOD for the updated analysis, 763 patients (86.8%) had completed treatment and 142 patients (16.2%) had discontinued the study. A total of 387 patients (88.0%) in the pola-R-CHP group and 376 patients (85.6%) in the R-CHOP group completed treatment. The most frequent reason for patients discontinuing the study was due to death (14.8%). Fewer than 1% of patients were never treated in the POLARIX study. As of the updated analysis, 711 patients (80.9%) were still on study with 359 patients (81.6%) and 352 patients (80.2%) in the pola-R-CHP and R-CHOP groups, respectively.
Baseline Characteristics
The demographic and clinical characteristics of patients in the POLARIX study were similar between treatment groups, and there were no between-group imbalances in stratification factors (IPI score, bulky disease, and geographical region). Patients had a median study population age of 65 years. All patients in the safety population had at least 1 medical history condition with similar proportions between groups for the most common conditions. Most patients had ECOG PS of 0 to 1 (83.9%), IPI score 3 to 5 (62.0%), advanced Ann Arbor stage III to IV (88.7%), and baseline lactate dehydrogenase greater than 1 time the ULN (65.4%) at diagnosis. Treatment groups were balanced for baseline biomarker assessments performed centrally (i.e., COO, DEL, and DHL or THL). Most patients in the trial received at least 1 prior concomitant medication (84.1% in the pola-R-CHP versus 86.5% in the R-CHOP group). Approximately 38% of patients had prephase (within 7 days before day 1 of cycle 1) steroid treatment.
Patient demographics were unchanged between the CCOD of the primary analysis (June 28, 2021) and the update analysis (June 15, 2022). Updated biomarker data for 21 additional patients (7 and 21 in the pola-R-CHP and R-CHOP groups, respectively) for COO were obtained for the update analysis.
The baseline characteristics outlined Table 11 are limited to those which are most relevant to this review, or were felt to impact the outcomes or interpretation of the study results.
Table 11: Summary of Baseline Characteristics of POLARIX Study, Data Cut-Off Date June 28, 2021 — ITT Population
Baseline characteristics | POLARIX | |
|---|---|---|
pola-R-CHP n = 440 | R-CHOP n = 439 | |
Age (years) | ||
18 to 64 | 209 (47.5) | 203 (46.2) |
≥ 65 | 231 (52.5) | 236 (53.8) |
Mean (SD) | 63.1 (11.4) | 63.0 (11.9) |
Median (range) | 65.0 (19 to 80) | 66.0 (19 to 80) |
Sex | ||
Female | 201 (45.7) | 205 (46.7) |
Male | 239 (54.3) | 234 (53.3) |
Race | ||
White | 235 (53.4) | 236 (53.8) |
Asian | 85 (19.3) | 84 (19.1) |
Black or African American | 8 (1.8) | 8 (1.8) |
American Indian or Alaska Native [wording from original source] | 1 (0.2) | 2 (0.5) |
Native Hawaiian or other Pacific Islander | 0 | 3 (0.7) |
Other | 6 (1.4) | 6 (1.4) |
Unknown | 105 (23.9) | 100 (22.8) |
ECOG performance status | ||
Number of patients with data | 440 | 438 |
0 | 175 (39.8) | 173 (39.4) |
1 | 199 (45.2) | 190 (43.3) |
2 | 66 (15.0) | 75 (17.1) |
Stratification: IPI score (lxRS) | ||
2 | 167 (38.0) | 167 (38.0) |
3 to 5 | 273 (62.0) | 272 (62.0) |
Stratification: bulky disease (lxRS) | ||
Absent | 247 (56.1) | 247 (56.3) |
Present | 193 (43.9) | 192 (43.7) |
Stratification: geographic region (lxRS) | ||
Asia | 81 (18.4) | 79 (18.0) |
Rest of world | 57 (13.0) | 59 (13.4) |
Western Europe, US, Canada, and Australia | 302 (68.6) | 301 (68.6) |
Baseline LDH | ||
≤ 1 × ULN | 146 (33.2) | 154 (35.1) |
> 1 × ULN | 291 (66.1) | 284 (64.7) |
Bone marrow involvement at diagnosis | ||
Number of patients with data | 429 | 432 |
Negative | 342 (77.7) | 349 (79.5) |
Positive | 76 (17.3) | 72 (16.4) |
Indeterminate | 11 (2.5) | 11 (2.5) |
Ann Arbor stage | ||
I | 2 (0.5) | 9 (2.1) |
II | 45 (10.2) | 43 (9.8) |
III | 124 (28.2) | 108 (24.6) |
IV | 269 (61.1) | 279 (63.6) |
Number of extranodal sites | ||
0 to 1 | 227 (51.6) | 226 (51.5) |
≥ 2 | 213 (48.4) | 213 (48.5) |
NHL histologic diagnosis (eCRFa) | ||
DLBCL NOS, ABC, GCB | 373 (84.8) | 367 (83.6) |
HGBL NOS, DHL or THL | 43 (9.8) | 50 (11.4) |
Other large B-cellb | 24 (5.5) | 22 (5.0) |
COOc | ||
ABC | 102 (23.2) | 119 (27.1) |
GCB | 184 (41.8) | 168 (38.3) |
Unclassified | 44 (10.0) | 51 (11.6) |
Unknown | 110 (25.0) | 101 (23.0) |
DELd | ||
DEL | 139 (31.6) | 151 (34.4) |
Non-DEL | 223 (50.7) | 215 (49.0) |
Unknown | 78 (17.7) | 73 (16.6) |
DHL or THLd | ||
Double-hit/triple-hit-positive | 26 (5.9) | 19 (4.3) |
Double-hit/triple-hit-negative | 305 (69.3) | 315 (71.8) |
Unknown | 109 (24.8) | 105 (23.9) |
Patients with at least 1 prior concomitant medicatione | 366 (84.1) | 379 (86.5) |
Prior concomitant medication by ATC level 1a | ||
Alimentary tract and metabolism | 299 (68.7) | 302 (68.9) |
Nervous system | 227 (52.2) | 237 (54.1) |
Musculoskeletal system | 214 (49.2) | 216 (49.3) |
Cardiovascular system | 191 (43.9) | 203 (46.3) |
Dermatologicals | 187 (43.0) | 182 (41.6) |
Blood and blood-forming organs | 184 (42.3) | 164 (37.4) |
Sensory organs | 166 (38.2) | 175 (40.0) |
Patients with at least 1 prephased steroid treatment | 164 (37.7) | 169 (38.6) |
Time from diagnosis to study dose, days, n | 436 | 437 |
Mean (SD) | 29.99 (21.46) | 33.02 (35.74) |
Median (range) | 26.0 (1.0 to 195.0) | 27.0 (1.0 to 621.0) |
ABC = activated B-cell; ATC = Anatomical Therapeutic Classification; COO = cell of origin; DEL = double-expressor lymphoma; DHL = double-hit lymphoma; DLBCL = diffuse large B-cell lymphoma; ECOG = Eastern Cooperative Oncology Group; eCRF = electronic case report form; GCB = germinal centre B-cell; HGBL = high-grade B-cell lymphoma; IPI = International Prognostic Index; ITT = intention to treat; IxRS = interactive voice/web response system; LDH = lactate dehydrogenase; NHL = non-Hodgkin lymphoma; NOS = not otherwise specified; pola-R-CHP = polatuzumab vedotin, rituximab, cyclophosphamide, doxorubicin, and prednisone; R-CHOP = rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone; SD = standard deviation; THL = triple-hit lymphoma; ULN = upper limit of normal.
Note: Values are n (%) unless otherwise indicated.
Note: Details from the table have been taken from the sponsor’s summary of clinical evidence.
aBased on local diagnosis.
bEpstein-Barr virus-positive DLBCL and T-cell/histiocyte-rich large B-cell lymphoma.
cBased on central review.
dPrephase steroid treatment was within 7 days before day 1 of cycle 1.
eBased on safety population (435 patients in pola-R-CHP and 438 patients in R-CHOP).
Source: POLARIX Clinical Study Report.14
Exposure to Study Treatments
A total of 873 patients received treatment (435 patients in the pola-R-CHP group and 438 patients in the R-CHOP group by safety-evaluable definition) and were included in the safety-evaluable population. Approximately 94% and 91% of patients received at least 6 cycles of any study drug in the pola-R-CHP group and the R-CHOP group, respectively; 89.2% and 86.3% of patients received 8 cycles of rituximab in the pola-R-CHP group and the R-CHOP group, respectively. For the investigational drugs that were administered in a blinded fashion, a higher number of patients received all 6 planned doses of polatuzumab vedotin in the pola-R-CHP group (91.7%, among patients who received any dose of polatuzumab vedotin [n = 435]) compared to the number of patients who received all 6 planned doses of vincristine in the R-CHOP group (88.5%, among patients who received any dose of vincristine [n = 436]). A total of 44 (10.1%) and 37 (8.4%) patients had a treatment delay of more than 7 days in at least 1 treatment cycle in the pola-R-CHP group and R-CHOP group, respectively. Four (0.9%) patients in each group had more than 1 treatment cycle delayed by more than 7 days.
The median treatment duration with pola-R-CHP (polatuzumab vedotin = 3.5 months, rituximab = 4.9 months, cyclophosphamide = 3.5 months, doxorubicin = 3.5 months, prednisone = 3.6 months) was similar with R-CHOP (rituximab = 4.9 months, cyclophosphamide = 3.5 months, doxorubicin = 3.5 months, vincristine = 3.5 months, prednisone = 3.6 months). Median relative dose intensity, adjusted to account for dose modifications and delays, was greater than 99.8% for all components of treatment in each group.
Concomitant Therapies
Most patients in the safety-evaluable population received at least 1 prior concomitant medication (84.1% for the pola-R-CHP group and 86.5% for the R-CHOP group). All patients received at least 1 concomitant medication. Prior and concomitant treatments were unchanged at the update analysis (CCOD June 15, 2022).
Nonprotocol or New Antilymphoma Treatment
The total number of patients with at least 1 NALT was higher in the R-CHOP group (32.8%) compared to the pola-R-CHP group (24.3%) at the time of the updated analysis (Table 14). Similarly, the total number of NALT treatments administered was higher in the R-CHOP group (315 treatments) compared to the pola-R-CHP group (196 treatments). The number of patients each receiving radiotherapy, systemic therapy, stem cell transplants, and CAR T-cell therapy was also higher in the R-CHOP group compared to the pola-R-CHP group. The majority received NALT after a PFS event.
Table 12: Summary of Patient Exposure to Treatment in POLARIX Study, Data Cut-Off Date June 15, 2022 — Safety Population
Treatment exposure | pola-R-CHP n = 435 | R-CHOP n = 438 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
Pola n = 435 | RTX n = 435 | CYC n = 435 | DOX n = 435 | PRED n = 435 | RTX n = 438 | CYC n = 436 | DOX n = 436 | VIN n = 436 | PRED n = 438 | |||
Treatment duration, months | ||||||||||||
Mean (SD) | 3.4 (0.6) | 4.7 (0.9) | 3.5 (0.6) | 3.5 (0.6) | 3.6 (0.6) | 4.6 (1.2) | 3.4 (0.7) | 3.4 (0.7) | 3.4 (0.7) | 3.5 (0.7) | ||
Median (range) | 3.5 (0 to 5) | 4.9 (0 to 8) | 3.5 (0 to 5) | 3.5 (0 to 5) | 3.6 (0 to 5) | 4.9 (0 to 11) | 3.5 (0 to 8) | 3.5 (0 to 8) | 3.5 (0 to 8) | 3.6 (0 to 6) | ||
Number of cycles | ||||||||||||
Mean (SD) | 5.8 (0.8) | 7.6 (1.3) | 5.8 (0.8) | 5.8 (0.8) | 5.8 (0.8) | 7.4 (1.6) | 5.7 (1.0) | 5.7 (1.0) | 5.7 (1.0) | 5.7 (1.0) | ||
Median (range) | 6.0 (1 to 6) | 8.0 (1 to 8) | 6.0 (1 to 6) | 6.0 (1 to 6) | 6.0 (1 to 6) | 8.0 (1 to 8) | 6.0 (1 to 6) | 6.0 (1 to 6) | 6.0 (1 to 6) | 6.0 (1 to 6) | ||
Range, n (%) | ||||||||||||
1 to 5 | 36 (8.3) | 31 (7.1) | 29 (6.7) | 29 (6.7) | 29 (6.7) | 42 (9.6) | 39 (8.9) | 39 (8.9) | 50 (11.5) | 45 (10.3) | ||
6 | 399 (91.7) | 7 (1.6) | 406 (93.3) | 406 (93.3) | 406 (93.3) | 14 (3.2) | 397 (91.1) | 397 (91.1) | 386 (88.5) | 393 (89.7) | ||
7 | NA | 9 (2.1) | NA | NA | NA | 4 (0.9) | NA | NA | NA | NA | ||
8 | NA | 388 (89.2) | NA | NA | NA | 378 (86.3) | NA | NA | NA | NA | ||
Relative dose intensity, % | ||||||||||||
n | 432 | 431 | 431 | 431 | 435 | 435 | 433 | 433 | 436 | 438 | ||
Mean (SD) | 98.1 (5.2) | 99.0 (3.3) | 98.5 (3.9) | 98.5 (4.0) | 98.4 (7.7) | 99.1 (2.6) | 98.6 (3.9) | 98.7 (4.1) | 98.5 (5.0) | 98.4 (8.2) | ||
Median | 99.8 | 100.0 | 100.0 | 100.0 | 100.0 | 100.0 | 100.0 | 100.0 | 100.0 | 100.0 | ||
Range | 64 to 111 | 64 to 116 | 64 to 106 | 65 to 106 | 26 to 127 | 84 to 108 | 65 to 109 | 64 to 109 | 63 to 103 | 20 to 103 | ||
Total cumulative dose, mg | ||||||||||||
Mean (SD) | 774.5 (228.9) | 5,247.1 (1,141.2) | 7,983.6 (1,544.1) | 532.4 (103.0) | 2,863.6 (447.5) | 5,128.1 (1,284.7) | 7,864.6 (1,717.9) | 524.7 (115.2) | 11.2 (2.1) | 2,817.0 (539.0) | ||
Median | 762.0 | 5,380.0 | 8,150.0 | 540.0 | 3,000.0 | 5,329.0 | 8,042.1 | 540.0 | 12.0 | 3,000.0 | ||
Range | 102 to 2,125 | 600 to 9,318 | 1,200 to 14,198 | 80 to 947 | 500 to 3,800 | 570 to 9,450 | 750 to 14,185 | 66 to 948 | 2 to 12 | 100 to 3,700 | ||
CYC = cyclophosphamide; DOX = doxorubicin; NA = not applicable; pola = polatuzumab vedotin; pola-R-CHP = polatuzumab vedotin, rituximab, cyclophosphamide, doxorubicin, and prednisone; PRED = prednisone; R-CHOP = rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone; RTX = rituximab; SD = standard deviation; VIN = vincristine.
Note: Details from the table have been taken from the sponsor’s summary of clinical evidence.
Source: POLARIX Clinical Study Report.13
Table 13: Summary of Concomitant Therapies in POLARIX Study, Data Cut-Off Date June 15, 2022 — Safety Population
Medication | POLARIX | |
|---|---|---|
pola-R-CHP n = 435 | R-CHOP n = 438 | |
Patients with at least 1 prior concomitant medication | 366 (84.1) | 379 (86.5) |
Patients with at least 1 concomitant medication | 435 (100.0) | 438 (100.0) |
Anti-infective prophylaxis | 268 (61.6) | 250 (57.1) |
Related to adverse events | 398 (91.5) | 384 (87.7) |
Patients with at least 1 prephase steroid treatment | 164 (37.7) | 171 (39.0) |
Patients with concomitant G-CSF | 405 (93.1) | 417 (95.2) |
Patients with concomitant G-CSF for prophylaxis | 392 (90.1) | 408 (93.2) |
Patients with concomitant G-CSF for nonprophylaxis | 99 (22.8) | 83 (18.9) |
G-CSF = granulocyte colony-stimulating factor; pola-R-CHP = polatuzumab vedotin, rituximab, cyclophosphamide, doxorubicin, and prednisone; R-CHOP = rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone.
Note: Values are n (%) unless otherwise indicated.
Note: Details from the table have been taken from the sponsor’s summary of clinical evidence.
Source: POLARIX Clinical Study Report.13
Table 14: Follow-Up Nonprotocol or New Antilymphoma Treatments in POLARIX Study, Data Cut-Off Date June 15, 2022 — ITT Population
Follow-up treatments | POLARIX | |
|---|---|---|
pola-R-CHP n = 440 | R-CHOP n = 439 | |
Total number of patients with at least 1 subsequent antilymphoma treatmenta | 107 (24.3) | 144 (32.8) |
Total number of NALT treatments, n | 196 | 315 |
Number of patients with ≥ 1 NALT before PFS event | 9 (2.0) | 16 (3.6) |
Number of patients with ≥ 1 NALT after PFS event | 72 (16.4) | 104 (23.7) |
Number of patients with ≥ 1 NALT and without PFS event | 29 (6.6) | 31 (7.1) |
Patients with at least 1 radiotherapy treatment | 42 (9.5) | 61 (13.9) |
Preplanned radiotherapy | 11 (2.5) | 18 (4.1) |
Unplanned radiotherapy | 31 (7.0) | 43 (9.8) |
Patients who received at least 1 systemic therapyb | 83 (18.9) | 114 (26.0) |
Patients who received stem cell transplant | 19 (4.3) | 34 (7.7) |
Autologous transplant | 19 (4.3) | 31 (7.1) |
Allogeneic transplant | 0 | 3 (0.7) |
Patients who received CAR T-cell therapy | 9 (2.0) | 16 (3.6) |
Total number of patients who received platinum-based therapyc | 41 (9.3) | 65 (14.8) |
CAR = chimeric antigen receptor; EFS = event-free survival; ITT = intention to treat; NALT = nonprotocol or new antilymphoma treatment; PFS = progression-free survival; pola-R-CHP = polatuzumab vedotin, rituximab, cyclophosphamide, doxorubicin, and prednisone; R-CHOP = rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone.
Note: Values are n (%) unless otherwise indicated.
Note: Details from the table have been taken from the sponsor’s summary of clinical evidence.
aSubsequent NALT is defined as nonprotocol antilymphoma therapy, and does not include intrathecal central nervous system disease prophylaxis as part of treatment; preplanned radiotherapy is included within radiotherapy here, but is not included as an event in EFS analyses.
bIncludes any monotherapy, multidrug, or cell-based regimen.
cPlatinum-based therapy are regimens typically intended to proceed to consolidative transplant or cellular therapies (e.g., R-GDP [rituximab, gemcitabine, dexamethasone, and cisplatin], R-ICE [rituximab, ifosfamide, carboplatin, and etoposide], R-DHAP [rituximab, dexamethasone, cytarabine, and cisplatin], R-ESHAP [rituximab, etoposide, methylprednisolone, cytarabine, and cisplatin]).
Source: POLARIX Clinical Study Report.13
Protocol Deviations
Nearly 6% of patients in the POLARIX study had at least 1 major protocol deviation (Table 15), including exclusion criteria not met (1.9%), noncompliance with study drug treatment modification or stopping rule (0.9%), accidental unblinding (0.8%), and incorrect kit given or administered (0.7%).
Table 15: Summary of Major Protocol Deviations of Interest in POLARIX Study, Data Cut-Off Date June 28, 2021 — ITT Population
Description | POLARIX | |
|---|---|---|
pola-R-CHP n = 440 | R-CHOP n = 439 | |
Number of patients with at least 1 major protocol deviation | 27 (6.1) | 23 (5.2) |
Number of protocol deviations | 29 | 26 |
Exclusion criteria not met | 12 (2.7) | 5 (1.1) |
Inclusion criteria not met | 4 (0.9) | 1 (0.2) |
Incorrect medication kit given or administered | 2 (0.5) | 4 (0.9) |
Noncompliance with study drug treatment modification or stoppage rules (either temporary or permanent) | 3 (0.7) | 5 (1.1) |
> 2 tumour assessments not performed during posttreatment phase | 3 (0.7) | 2 (0.5) |
Accidental unblinding of site staff team member(s) | 1 (0.2) | 2 (0.5) |
Accidental unblinding of patient(s) | 3 (0.7) | 4 (0.9) |
Any tumour assessments not performed during treatment phase | 0 | 1 (0.2) |
ITT = intention to treat; pola-R-CHP = polatuzumab vedotin, rituximab, cyclophosphamide, doxorubicin, and prednisone; R-CHOP = rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone.
Note: Values are n (%).
Note: Details from the table have been taken from the sponsor’s summary of clinical evidence.
Source: POLARIX Clinical Study Report.14
Efficacy
Findings for key efficacy outcomes in POLARIX are summarized in Table 16.
Key Efficacy End Points
Overall Survival
A total of 53 deaths (12.0%) and 57 deaths (13.0%) were reported in the pola-R-CHP group and R-CHOP group, respectively. The stratified HR for OS was 0.94 (95% CI, 0.65 to 1.37; P = 0.7524). OS results for the pola-R-CHP and R-CHOP groups were 92.2% and 94.6%, respectively, at 12 months, and 88.7% and 88.6%, respectively, at 24 months.
At the time of the CCOD for the updated (final) analysis of OS (June 15, 2022), the median duration of survival follow-up was 39.7 months (range, 0 to 54) in the pola-R-CHP group and 39.6 months (range, 0 to 54) in the R-CHOP group. A total of 131 events had been reported, 64 (14.5%) in the pola-R-CHP group and 67 (15.3%) in the R-CHOP group. This represented 21 additional events since the time of the OS analysis for the primary analysis (11 in the pola-R-CHP group versus 10 in the R-CHOP group). The stratified HR was 0.94 (95% CI, 0.67 to 1.33; P = 0.7326). The Kaplan-Meier curve is presented in Figure 3.
Progression-Free Survival
In the primary analysis CCOD (June 28, 2021), fewer patients in the pola-R-CHP group had progressed or died compared to the R-CHOP group (107 [24.3%] versus 134 [30.5%]). Treatment of patients with previously untreated DLBCL with the pola-R-CHP regimen resulted in a statistically significant reduction in progression, relapse, or death compared with patients treated with the R-CHOP regimen (stratified HR = 0.73; 95% CI, 0.57 to 0.95; P = 0.0177).
At the time of the updated CCOD (June 15, 2022), the median duration of PFS follow-up was 30.9 months (range, 0 to 46) in the pola-R-CHP group and 30.8 months (range, 0 to 54) in the R-CHOP group, representing approximately an additional 12 months of follow-up compared with the primary CCOD.
Results of the updated time-to-efficacy analysis of PFS were consistent with the primary analysis, where fewer patients in the pola-R-CHP group had progressed or died compared to the R-CHOP group (118 [26.8%] versus 143 [32.6%], respectively; stratified HR = 0.76; 95% CI, 0.60 to 0.97).
Median PFS was not reached for either group at the time of the updated CCOD. Kaplan-Meier curves are shown in Figure 4. The majority of PFS events occurred within 2 years of randomization in both groups, with a higher proportion of patients remaining alive and progression-free in the pola-R-CHP group compared to the R-CHOP group at 1 year, 2 years, and 3 years, although results at later time points were represented by low numbers of patients.
Figure 3: Kaplan-Meier Plot of Time to OS in POLARIX Study, Data Cut-Off Date June 15, 2022 — ITT Population
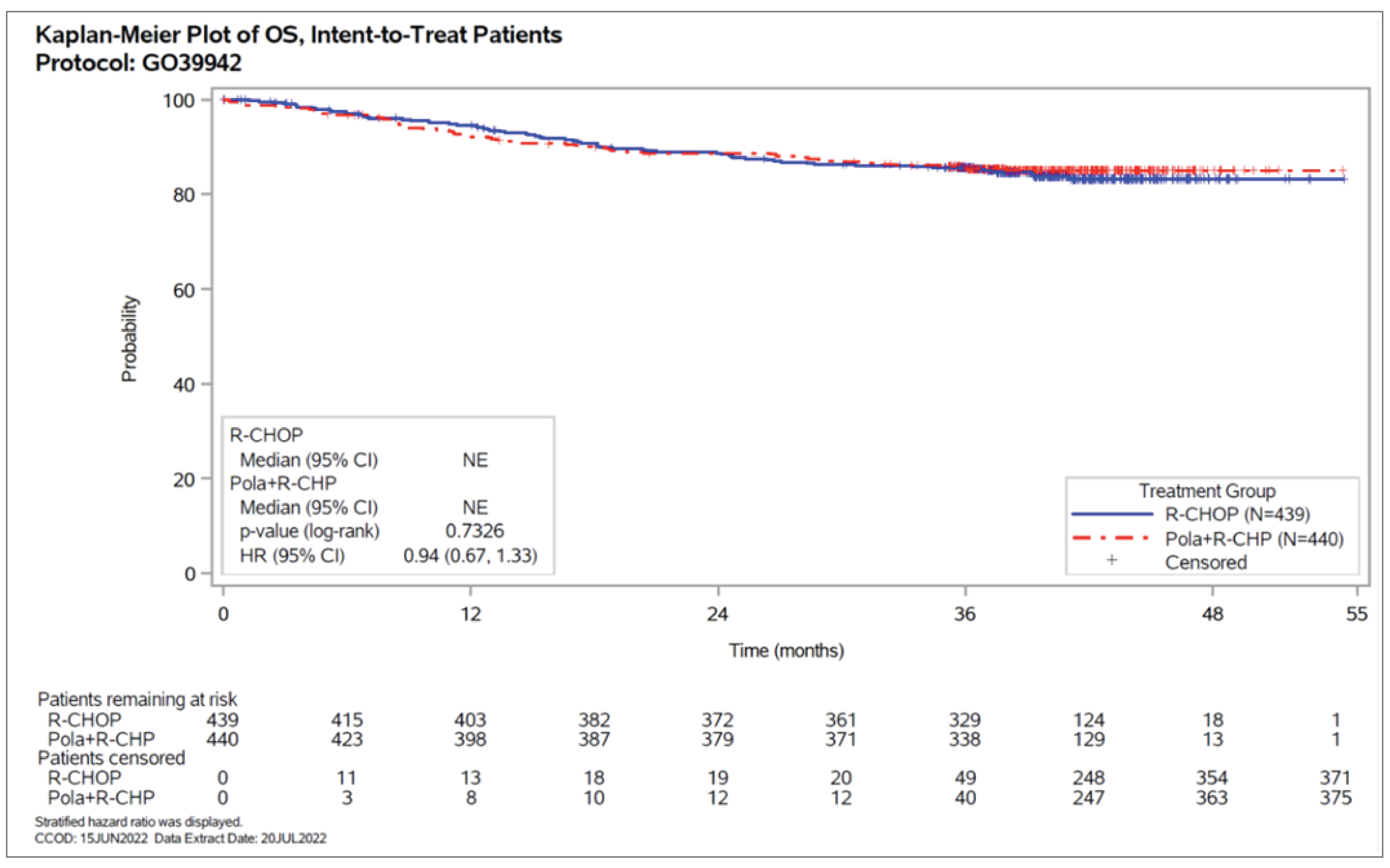
CI = confidence interval; HR = hazard ratio; ITT = intention to treat; NE = not estimable; OS = overall survival; pola-R-CHP = polatuzumab vedotin, rituximab, cyclophosphamide, doxorubicin, and prednisone; R-CHOP = rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone.
Source: POLARIX Clinical Study Report.13
Subgroup Analysis
The subgroup analyses of investigator-assessed PFS are presented in Figure 5, Figure 6, and Figure 7. The CADTH review focused on the subgroups of IPI score, bulky disease, and DLBCL subtype.
The HRs for PFS favoured treatment with pola-R-CHP compared with R-CHOP among patients with IPI score 3 to 5 (unstratified HR = 0.71; 95% CI, 0.53 to 0.95) and without bulky disease (unstratified HR = 0.59; 95% CI, 0.42 to 0.83). Unstratified investigator-assessed PFS subgroup analysis by baseline molecular DLBCL subtypes (centrally tested COO, centrally tested immunohistochemistry for BCL2 and MYC [DEL], and centrally tested FISH for rearrangements in MYC, BCL2, and BCL6 [DHL or THL]) suggested that treatment with pola-R-CHP compared with R-CHOP was associated with better PFS among patients in higher-risk subgroups: ABC-DLBCL subgroup (84.7% versus 56.1%; HR = 0.34; 95% CI, 0.21 to 0.56) and DEL subgroup (75.8% versus 63.1%; HR = 0.63; 95% CI, 0.42 to 0.94).
Figure 4: Kaplan-Meier Plot of Time to Investigator-Assessed PFS in Updated Analysis in POLARIX Study, Data Cut-Off Date June 15, 2022 — ITT Population
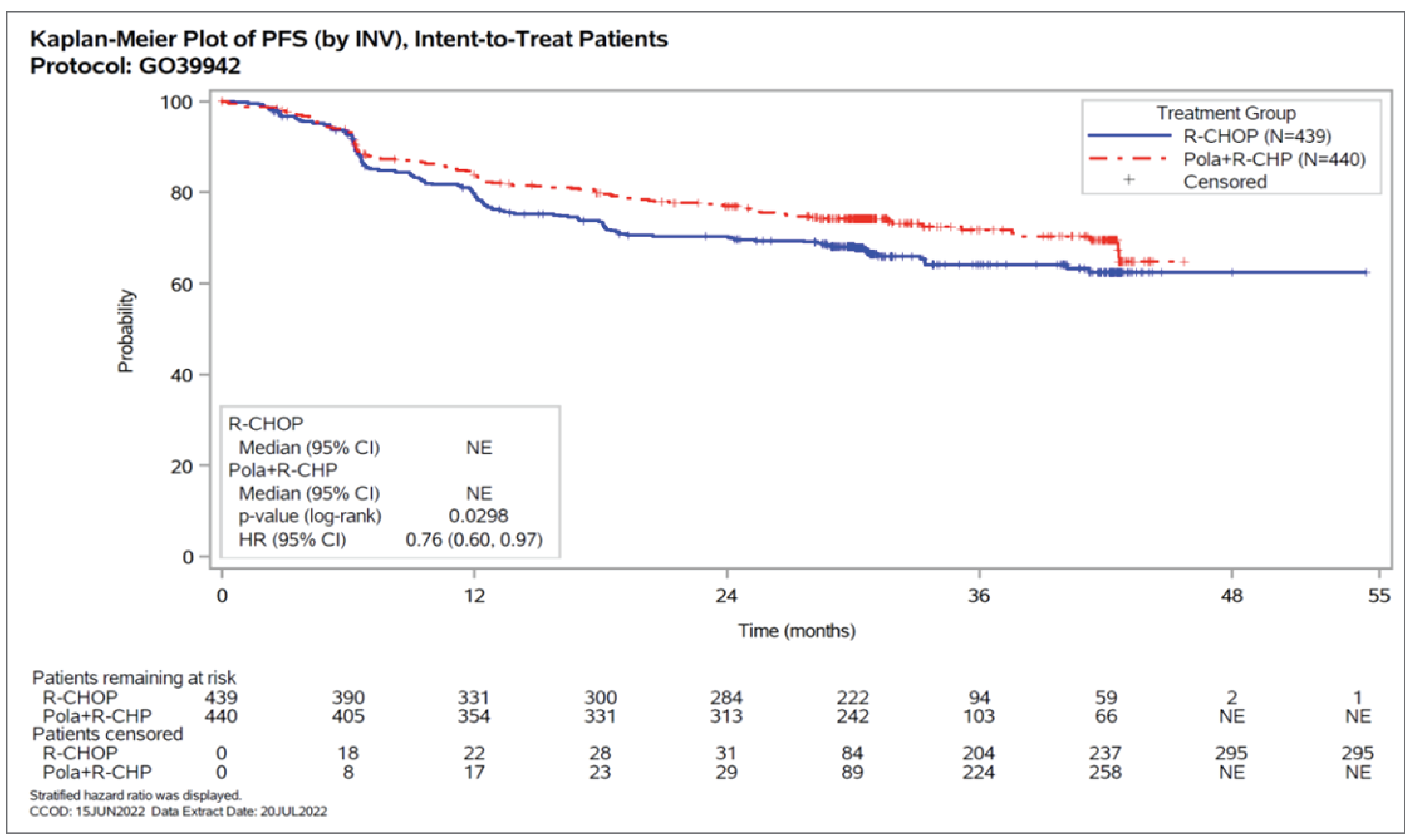
CI = confidence interval; HR = hazard ratio; INV = investigator; ITT = intention to treat; NE = not estimable; PFS = progression-free survival; pola-R-CHP = polatuzumab vedotin, rituximab, cyclophosphamide, doxorubicin, and prednisone; R-CHOP = rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone.
Source: POLARIX Clinical Study Report.13
Based on the subgroup results for PFS among those with IPI score 3 to 5 and no bulky disease, the Health Canada reviewers12 requested the sponsor conduct additional subgroup analyses combining these subgroups. The results of the post hoc subgroup analyses suggested the effects of pola-R-CHP versus R-CHOP on PFS were greatest among patients with IPI score of 3 to 5 and no bulky disease (26 of 139 patients versus 57 of 138 patients; unstratified HR = 0.40; 95% CI, 0.25 to 0.63).
Sensitivity Analyses
Interval censoring analysis of PFS appeared to be consistent with the primary PFS analysis, favouring pola-R-CHP (stratified HR = 0.77; 95% CI, 0.60 to 0.98). Similarly, the impact of NALT before or in the absence of disease progression was consistent with the primary PFS analysis after discounting time after the initiation of NALT by 10% (HR = 0.76; 95% CI, 0.60 to 0.97), 30% (HR = 0.76; 95% CI, 0.59 to 0.97), and 50% (HR = 0.76; 95% CI, 0.59 to 0.97) and favoured pola-R-CHP. When using the last adequate tumour assessment before initiating NALT as the method of censoring, the HR was 0.78 (95% CI, 0.60 to 1.01). Sensitivity analyses for RMST on PFS suggested that patients treated with pola-R-CHP had longer mean PFS duration compared to patients treated with R-CHOP at 12 months (treatment difference = 0.2; 95% CI, –0.1 to 0.5), 24 months (treatment difference = 1.0; 95% CI, 0.1 to 2.0), 36 months (treatment difference = 1.8; 95% CI, 0.2 to 3.5), and 42 months (treatment difference = 2.2; 95% CI, 0.3 to 4.2).
Figure 5: Forest Plot of Hazard Ratio of Investigator-Assessed PFS by Baseline Risk Factors (Part 1) in POLARIX Study — ITT Population
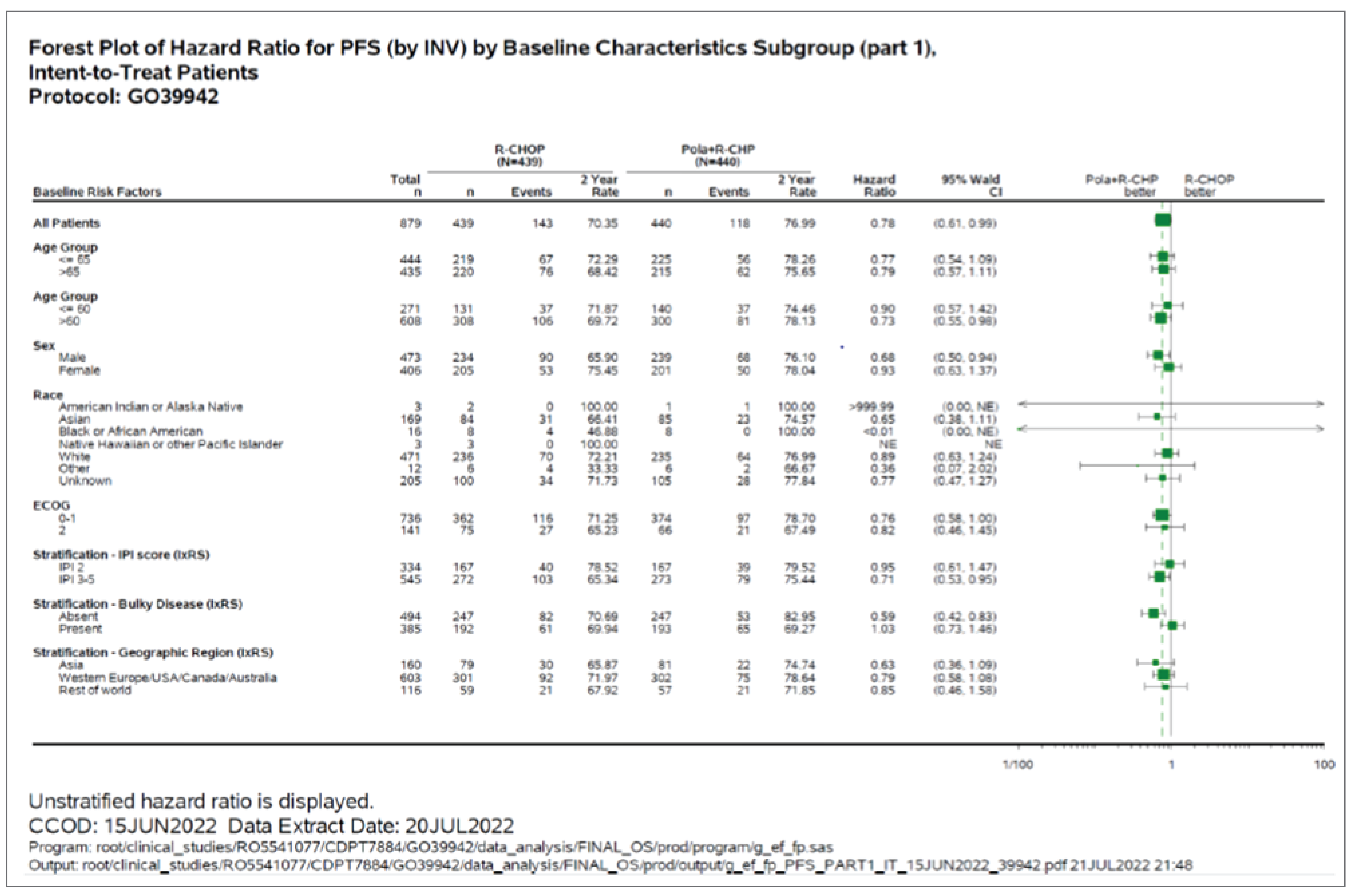
CI = confidence interval; ECOG = Eastern Cooperative Oncology Group; HR = hazard ratio; IPI = International Prognostic Index; ITT = intention to treat; INV = investigator; IxRS = interactive voice or web response system; NE = not estimable; PFS = progression-free survival; pola-R-CHP = polatuzumab vedotin, rituximab, cyclophosphamide, doxorubicin, and prednisone; R-CHOP = rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone.
Source: POLARIX Clinical Study Report.13
Figure 6: Forest Plot of Hazard Ratio of Investigator-Assessed PFS by Baseline Risk Factors (Part 2) in POLARIX Study — ITT Population
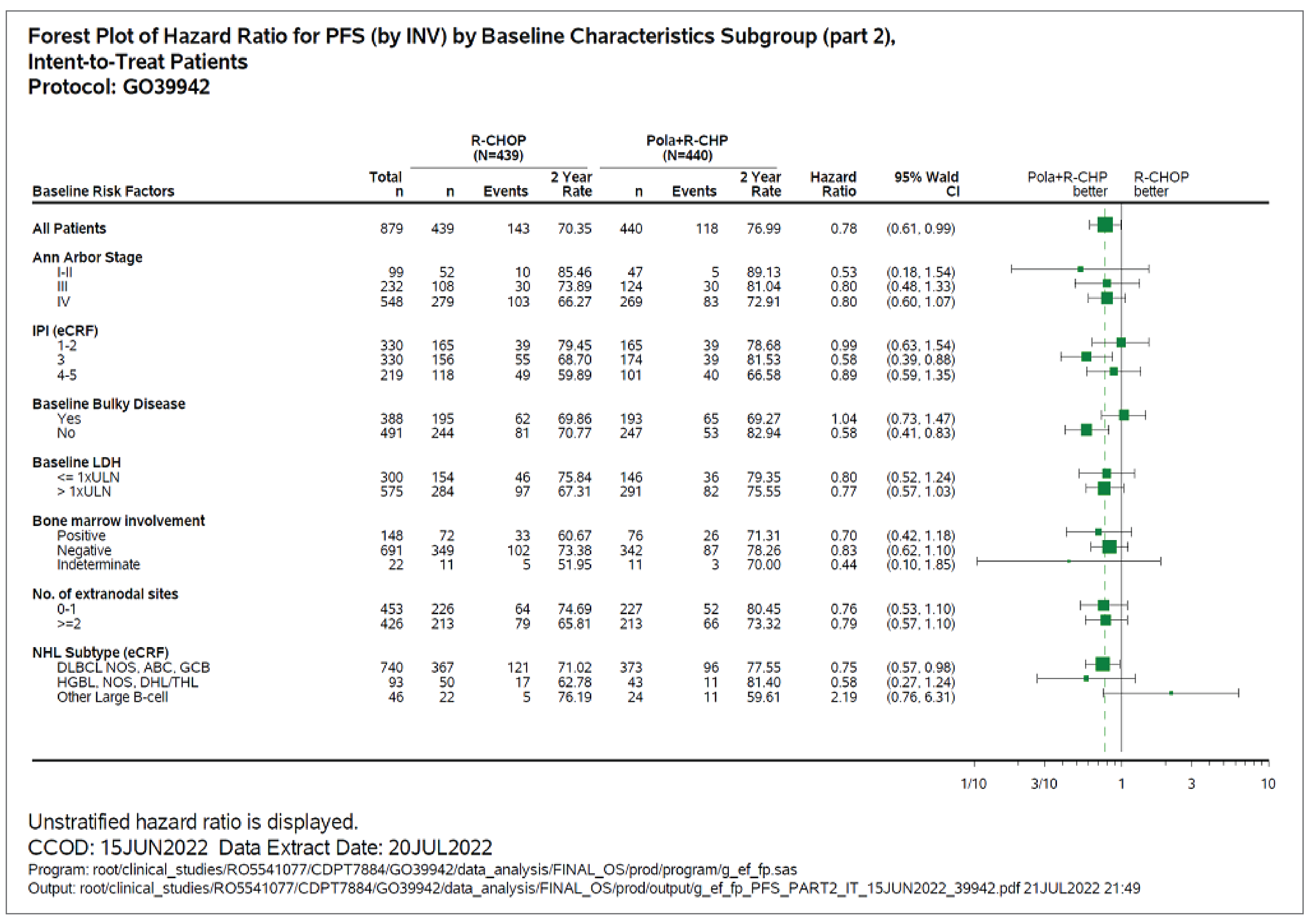
ABC = activated B-cell; CI = confidence interval; DHL = double-hit lymphoma; DLBCL = diffuse large B-cell lymphoma; eCRF = electronic case report form; GCB = germinal centre B-cell; HGBL = high-grade B-cell lymphoma; INV = investigator; IPI = International Prognostic Index; ITT = intention to treat; LDH = lactate dehydrogenase; NHL = non-Hodgkin lymphoma; NOS = not otherwise specified; PFS = progression-free survival; pola-R-CHP = polatuzumab vedotin, rituximab, cyclophosphamide, doxorubicin, and prednisone; R-CHOP = rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone; THL = triple-hit lymphoma.
Source: POLARIX Clinical Study Report.13
Figure 7: Forest Plot of Hazard Ratio of Investigator-Assessed PFS by Molecular DLBCL Subtypes in POLARIX Study — ITT Population
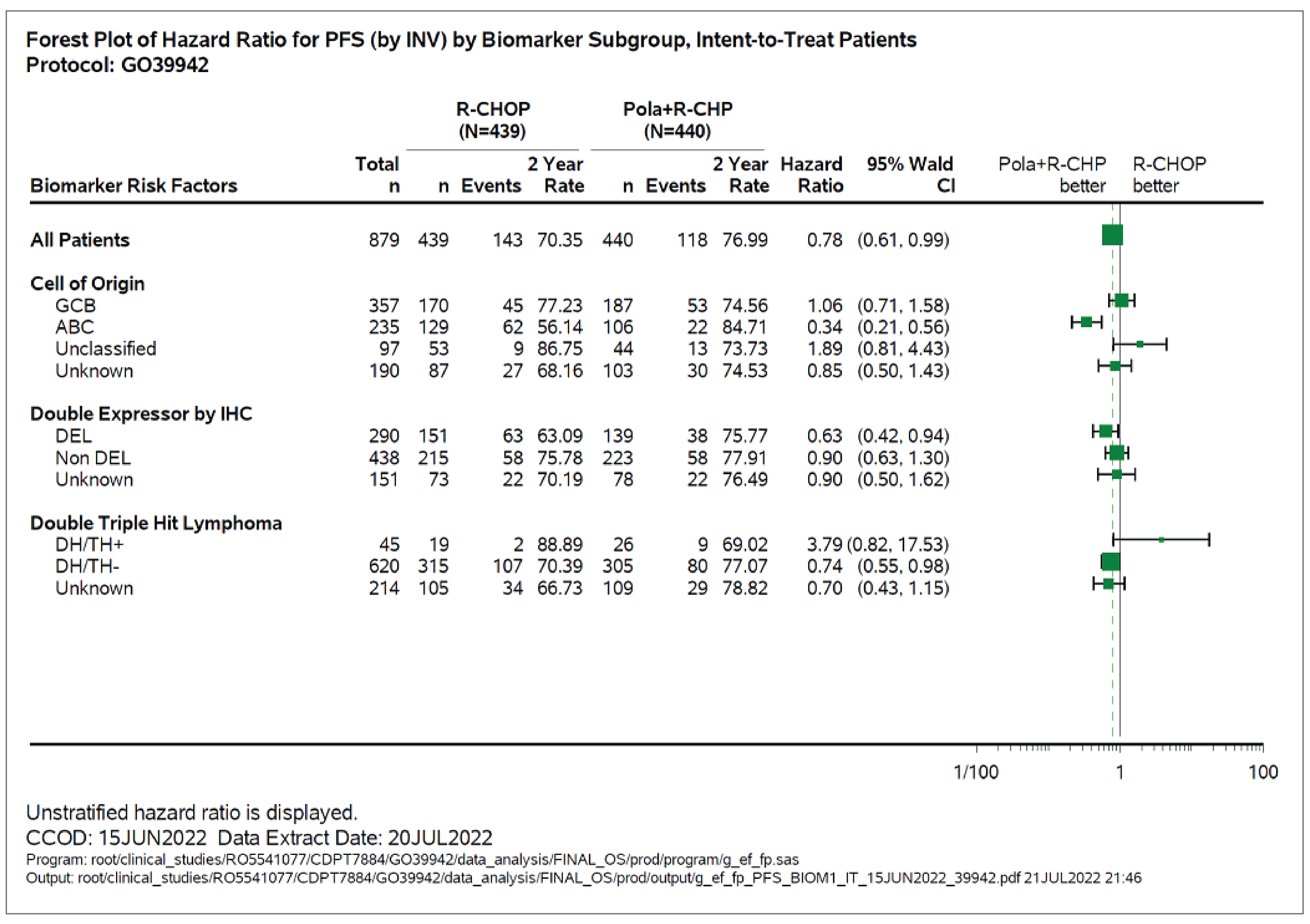
ABC = activated B-cell; CI = confidence interval; DEL = double-expressor lymphoma; DH/TH = double-hit lymphoma/triple-hit lymphoma; DLBCL = diffuse large B-cell lymphoma; GCB = germinal centre B-cell; IHC = immunohistochemistry; INV = investigator; ITT = intention to treat; PFS = progression-free survival; pola-R-CHP = polatuzumab vedotin, rituximab, cyclophosphamide, doxorubicin, and prednisone; R-CHOP = rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone.
Source: POLARIX Clinical Study Report.13
BICR-Assessed CR Rate at EOT by PET-CT
At the EOT, the BICR-assessed CR rate was greater than 70% in both treatment groups: 78.0% for pola-R-CHP (95% CI, 73.79 to 81.74) versus 74.0% for R-CHOP (95% CI, 69.66 to 78.07). The treatment difference was 3.9% (95% CI, –1.9 to 9.7).
Additional Secondary Efficacy End Points
Findings for investigator-assessed CR rate at EOT, ORR at EOT, and PFS24 are included in Table 16.
Investigator-Assessed CR Rate at EOT
Investigator-assessed CR rate at EOT was 75.0% for pola-R-CHP versus 72.2% for R-CHOP (difference = 2.79; 95% CI, –3.20 to 8.75; P = 0.3402). Findings for BICR-assessed (key secondary efficacy end point) and investigator-assessed CR rates demonstrated concordance that was similar between pola-R-CHP (88.9%) and R-CHOP (88.6%).
BICR-Assessed ORR at EOT
BICR-assessed ORR at EOT was 85.5% for pola-R-CHP versus 83.8% for R-CHOP (difference = 1.63%; 95% CI, –3.32 to 6.57; P = 0.4828).
Investigator-Assessed ORR at EOT
Investigator-assessed ORR at EOT was 84.5% for pola-R-CHP versus 80.9% for R-CHOP (difference = 3.68; 95% CI, –1.49 to 8.84; P = 0.1345).
Investigator-Assessed PFS24
The proportion of patients who had PFS24 after randomization was 77% in the pola-R-CHP group compared with 70% in the R-CHOP group (difference = 6.64; 95% CI, 0.70 to 12.58; P = 0.0284).
Patient-Reported Outcomes
There were no clear differences between the treatment groups for PROs (Table 17).
Table 16: Summary of Efficacy Results From POLARIX Study
Efficacy outcomes | POLARIX | |
|---|---|---|
pola-R-CHP n = 440 | R-CHOP n = 439 | |
Investigator-assessed PFS (June 15, 2022) | ||
Number of events, n (%) | 118 (26.8) | 143 (32.6) |
Earliest contributing event, n (%) | ||
Death | 22 (NR) | 21 (NR) |
Disease progression or relapse | 96 (NR) | 122 (NR) |
Median time to PFS,a months (range) | NE (0 to 46) | NE (0 to 54) |
Stratifiedb HRc (95% CI) | 0.76 (0.60 to 0.97) | Reference |
P value (log-rank)d | 0.0298 | Reference |
12 months, patients remaining at risk, n | 354 | 331 |
12-month PFS ratec (95% CI) | 83.94 (80.47 to 87.42) | 79.58 (75.73 to 83.44) |
Difference in PFS rate at 12 months (95% CI) | 4.36 (–0.82 to 9.55) | Reference |
24 months, patients remaining at risk, n | 313 | 284 |
24-month PFS ratec (95% CI) | 76.99 (72.99 to 80.99) | 70.35 (65.97 to 74.73) |
Difference in PFS rate at 24 months (95% CI) | 6.64 (0.70 to 12.58) | Reference |
P value (z test) | 0.0284 | Reference |
36 months, patients remaining at risk, n | 103 | 94 |
36-months PFS ratec (95% CI) | 71.78 (67.10 to 76.46) | 64.11 (59.07 to 69.14) |
Difference in PFS rate at 36 months (95% CI) | 7.67 (0.80 to 14.55) | Reference |
42 months, patients remaining at risk, n | 66 | 59 |
42-month PFS ratec (95% CI) | 69.54 (64.36 to 74.72) | 62.41 (56.98 to 67.83) |
Difference in PFS rate at 42 months (95% CI) | 7.13 (–0.37 to 14.63) | Reference |
OS (June 15, 2022) | ||
Patients with event, n (%) | 64 (14.5) | 67 (15.3) |
Median time to OS,a months (range) | NE (0 to 54) | NE (0 to 54) |
Stratifiedb HRc (95% Cl) | 0.94 (0.67 to 1.33) | Reference |
P value (log-rank)d | 0.7326 | Reference |
12 months, patients remaining at risk, n | 398 | 403 |
12-month OS rate (95% Cl) | 92.17 (89.65 to 94.70) | 94.63 (92.50 to 96.77) |
Difference in OS rate at 12 months (95% CI) | –2.46 (–5.77 to 0.85) | Reference |
24 months, patients remaining at risk, n | 379 | 372 |
24-month OS rate (95% Cl) | 88.68 (85.70 to 91.67) | 88.69 (85.68 to 91.71) |
Difference in OS rate at 24 months (95% CI) | –0.01 (–4.25 to 4.23) | Reference |
36 months, patients remaining at risk, n | 338 | 329 |
36-months OS rate (95% CI) | 85.62 (82.30 to 88.93) | 85.58 (82.23 to 88.93) |
Difference in OS rate at 36 months (95% CI) | 0.03 (–4.68 to 4.75) | Reference |
42 months, patients remaining at risk, n | 129 | 124 |
42-month OS rate (95% CI) | 85.04 (81.65 to 88.43) | 83.25 (79.47 to 87.02) |
Difference in OS rate at 42 months (95% CI) | 1.79 (–3.29 to 6.87) | Reference |
BICR-assessed CR rate at end of treatment (by PET-CT) (June 28, 2021) | ||
Complete responders, n (%) | 343 (78.0) | 325 (74.0) |
Difference in response rate (95% Cl) | 3.9 (–1.89 to 9.70) | Reference |
Stratified P valued (CMH) | 0.1557 | Reference |
Investigator-assessed CR rate at end of treatment (by PET-CT)2 (June 28, 2021) | ||
Complete responders, n (%) | 330 (75.0) | 317 (72.2) |
Difference in response rate (95% Cl) | 2.79 (–3.20 to 8.75) | Reference |
Stratified P value (CMH) | 0.3402 | Reference |
BICR-assessed ORR at EOT (June 28, 2021) | ||
Responders, n (%) | 376 (85.5) | 368 (83.8) |
Difference in response rate (95% CI) | 1.63 (–3.32 to 6.57) | Reference |
Stratified P value | 0.4828 | Reference |
CR, n (%) | 343 (78.0) | 325 (74.0) |
Partial response, n (%) | 33 (7.5) | 43 (9.8) |
Progressive disease, n (%) | 22 (5.0) | 28 (6.4) |
Investigator-assessed ORR at EOT (June 28, 2021) | ||
Responders, n (%) | 372 (84.5) | 355 (80.9) |
Difference in response rate (95% CI) | 3.68 (–1.49 to 8.84) | Reference |
Stratified P value | 0.1345 | Reference |
CR, n (%) | 330 (75.0) | 317 (72.2) |
Partial response, n (%) | 42 (9.5) | 38 (8.7) |
Progressive disease, n (%) | 34 (7.7) | 44 (10.0) |
BICR = blinded independent central review; CI = confidence interval; CMH = Cochran-Mantel-Haenszel; CR = complete response; EOT = end of treatment; HR = hazard ratio; IPI = International Prognostic Index; NE = not estimable; NR = not reported; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; pola-R-CHP = polatuzumab vedotin, rituximab, cyclophosphamide, doxorubicin, and prednisone; R-CHOP = rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone.
Note: Details from the table have been taken from the sponsor’s summary of clinical evidence.
aSummaries of PFS and OS (median, percentiles) were Kaplan-Meier estimates. 95% CI for median was computed using the method of Brookmeyer and Crowley.
bStratified for IPI score (IPI 2 vs. IPI 3 to 5), bulky disease (1 lesion ≥ 7.5cm; present vs. absent), and geographical region (Western Europe, US, Canada, and Australia vs. Asia vs. rest of world [remaining countries]).
cHRs were estimated by Cox regression.
dP value has been adjusted for multiple testing.
Source: POLARIX Clinical Study Reports.13,14
Table 17: Summary of PROs in POLARIX Study, Data Cut-Off Date June 28, 2021 — PRO-Evaluable Population
Outcomes | POLARIX | |
|---|---|---|
pola-R-CHP n = 413 | R-CHOP n = 412 | |
EORTC QLQ-C30 | ||
Physical Functioning | ||
Baseline, n | 411 | 409 |
Baseline, mean (SE) | 80.04 (22.01) | 80.55 (22.35) |
24 months, number of patients contributing to the analysis | 102 | 87 |
24 months, change from baseline, mean (SD) | 9.02 (23.69) | 4.52 (19.15) |
Responders,a n (%) | 175 (42.4) | 163 (39.6) |
Difference in response rate, unstratified analysis (95% CI)b | 2.81 (–4.06 to 9.64) | Reference |
Time to deterioration,c patients with event, n (%) | 183 (41.6) | 187 (42.6) |
Time to event, months, median (95% CI) | NE | 25.5 (17.6 to NE) |
Time to event, months, range | 0 to 34 | 0 to 32 |
Stratified HRd (95% CI) | 0.97 (0.79 to 1.19) | Reference |
24 months, patients remaining at risk, n | 125 | 107 |
24 months, event-free rate, % (95% CI) | 54.85 (49.93 to 59.78) | 53.31 (48.18 to 58.43) |
24 months, difference in event-free rate (95% CI) | 1.55 (–5.56 to 8.65) | Reference |
Fatigue | ||
Baseline, n | 411 | 409 |
Baseline, mean (SE) | 37.32 (1.34) | 35.11 (1.33) |
24 months, number of patients contributing to the analysis | 103 | 87 |
24 months, adjusted mean (SE) | –15.78 (1.60) | –16.44 (1.72) |
24 months, difference in adjusted means (95% CI) | 0.66 (–3.95 to 5.27) | Reference |
Responders,a n (%) | 309 (74.8) | 281 (68.2) |
Difference in response rate, unstratified analysis (95% CI)b | 6.61 (0.28 to 12.88) | Reference |
Time to deterioration,c patients with event, n (%) | 223 (50.7) | 230 (52.4) |
Time to event, months, median (95% CI) | 6.7 (2.9 to 24.3) | 3.0 (2.8 to 6.8) |
Time to event, months, range | 0 to 31 | 0 to 32 |
Stratified HRd (95% CI) | 0.94 (0.78 to 1.13) | Reference |
24 months, patients remaining at risk, n | 99 | 87 |
24 months, event-free rate, % (95% CI) | 45.15 (40.18 to 50.13) | 41.77 (36.75 to 46.79) |
24 months, difference in event-free rate (95% CI) | 3.38 (–3.68 to 10.45) | Reference |
Diarrhea | ||
Baseline, n | 407 | 405 |
Baseline, mean (SE) | 10.40 (1.07) | 9.22 (0.98) |
24 months, number of patients contributing to the analysis | 104 | 88 |
24 months, adjusted mean (SE) | –5.22 (1.18) | –4.68 (1.28) |
24 months, difference in adjusted means (95% CI) | –0.54 (–3.97 to 2.89) | Reference |
FACT-Lym | ||
LymS | ||
Baseline, n | 407 | 406 |
Baseline, mean (SE) | 39.81 (0.22) | 39.49 (0.25) |
24 months, number of patients contributing to the analysis | 102 | 88 |
24 months, change from baseline, mean (SD) | 7.21 (8.76) | 7.91 (9.52) |
Responders,a n (%) | 340 (82.3) | 335 (81.3) |
Difference in response rate, unstratified analysis (95% CI)b | 1.01 (–4.43 to 6.45) | Reference |
Time to deterioration,c patients with event, n (%) | 148 (33.6) | 138 (31.4) |
Time to event, months, median (95% CI) | NE | NE (30.9 to NE) |
Time to event, months, range | 0 to 34 | 0 to 32 |
Stratified HRd (95% CI) | 1.03 (0.81 to 1.30) | Reference |
24 months, patients remaining at risk, n | 145 | 134 |
24 months, event-free rate, % (95% CI) | 63.46 (58.63 to 68.29) | 63.99 (59.02 to 68.96) |
24 months, difference in event-free rate (95% CI) | –0.53 (–7.46 to 6.40) | Reference |
B symptom, night sweats | ||
Time to deterioration,c patients with event, n (%) | 101 (23.0) | 119 (27.1) |
Time to event, months, median (95% CI) | NE | NE |
Time to event, months, range | 0 to 32 | 0 to 33 |
Stratified HRd (95% CI) | 0.78 (0.60 to 1.02) | Reference |
24 months, patients remaining at risk, n | 166 | 137 |
24 months, event-free rate, % (95% CI) | 74.78 (70.39 to 79.18) | 68.88 (64.01 to 73.75) |
24 months, difference in event-free rate (95% CI) | 5.90 (–0.66 to 12.46) | Reference |
FACT/GOG-NTX | ||
Baseline, n | 407 | 406 |
Baseline, mean (SE) | 39.81 (0.22) | 39.49 (0.25) |
24 months, number of patients contributing to the analysis | 104 | 88 |
24 months, adjusted mean (SE) | –1.63 (0.46) | –1.60 (0.50) |
24 months, difference in adjusted means (95% CI) | –0.04 (–1.37 to 1.30) | Reference |
EQ-5D-5L VAS | 440 | 439 |
Baseline, n | 405 | 406 |
Baseline, mean (SD) | 68.74 (21.65) | 69.97 (19.84) |
24 months, n | 262 | 240 |
24 months, mean (SD) | 82.16 (15.03) | 82.43 (15.46) |
36 months, n | 104 | 77 |
36 months, mean (SD) | 78.97 (17.10) | 81.29 (14.93) |
CI = confidence interval; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EQ-5D-5L = 5-Level EQ-5D; FACT/GOG-NTX = Functional Assessment of Cancer Therapy/Gynecologic Oncology Group-Neurotoxicity; FACT-Lym = Functional Assessment of Cancer Therapy-Lymphoma; HR = hazard ratio; IPI = International Prognostic Index; LymS = lymphoma subscale; NE = not estimable; pola-R-CHP = polatuzumab vedotin, rituximab, cyclophosphamide, doxorubicin, and prednisone; PRO = patient-reported outcome; R-CHOP = rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone; SD = standard deviation; SE = standard error; VAS = visual analogue scale.
Note: The PRO-evaluable population (413 patients in pola-R-CHP and 412 patients in R-CHOP) included all randomized patients in the global study who had a baseline and at least 1 postbaseline assessment.
aResponder was defined as a patient with at least a 7-point scale score increase from baseline on the EORTC QLQ-C30 Physical Functioning, at least a 9-point scale score on the EORTC QLQ-C30 Fatigue, or at least a 3-point scale score increase on the FACT-Lym LymS.
b95% CI for difference in response rates are constructed using Wilson method.
cDeterioration was defined as a ≥ 10-point decrease in the EORTC QLQ-C30 Physical Functioning or Fatigue scale from baseline, a ≥ 3-point decrease in the FACT-Lym LymS, a ≥ 1-point increase in the B symptom raw score.
dHRs were estimated by Cox regression. Stratification factors (as applicable) included IPI score, bulky disease, and geographical region.
Source: POLARIX Clinical Study Report.14
Harms
Data for harms in the POLARIX trial are summarized for the safety population (435 patients in pola-R-CHP versus 438 patients in R-CHOP) at the updated analysis (CCOD June 15, 2022) in Table 18.
Table 18: Summary of Harms From Pivotal and RCT Evidence, Data Cut-Off Date June 15, 2022 — Safety Population
Adverse events | POLARIX | |
|---|---|---|
pola-R-CHP n = 435 | R-CHOP n = 438 | |
Patients with at least 1 adverse event | 426 (97.9) | 431 (98.4) |
Most commona adverse events | ||
Nausea | 181 (41.6) | 161 (36.8) |
Constipation | 125 (28.7) | 128 (29.2) |
Fatigue | 112 (25.7) | 116 (26.5) |
Diarrhea | 135 (31.0) | 88 (20.1) |
Alopecia | 106 (24.4) | 105 (24.0) |
Grade 3 to 4 adverse events | 251 (57.7) | 252 (57.5) |
Grade 5 adverse events | 13 (3.0) | 10 (2.3) |
Serious adverse events in ≥ 1% of patients | ||
Patients with ≥ 1 serious adverse event | 148 (34.0) | 134 (30.6) |
Pneumonia | 18 (4.1) | 17 (3.9) |
Febrile neutropenia | 43 (9.9) | 28 (6.4) |
Diarrhea | 10 (2.3) | 2 (0.5) |
Pyrexia | 7 (1.6) | 8 (1.8) |
Sepsis | 5 (1.1) | 7 (1.6) |
Urinary tract infection | 8 (1.8) | 3 (0.7) |
Anemia | 4 (0.9) | 6 (1.4) |
Neutropenia | 4 (0.9) | 6 (1.4) |
Vomiting | 5 (1.1) | 2 (0.5) |
Small intestinal obstruction | 0 | 5 (1.1) |
Adverse events leading to treatment discontinuation | ||
Patients who discontinued any study treatment due to adverse events | 26 (6.0) | 28 (6.4) |
Patients who discontinued polatuzumab vedotin/vincristine due to adverse events | 19 (4.4) | 22 (5.0) |
Pneumonia | 4 (0.9) | 4 (0.9) |
Peripheral neuropathy | 1 (0.2) | 4 (0.9) |
Peripheral motor neuropathy | 0 | 2 (0.5) |
Death | 2 (0.5) | 1 (0.2) |
Patients who discontinued any component of CHP due to adverse events | 15 (3.4) | 16 (3.7) |
Deaths | ||
Patients who died during the study from any cause | 64 (14.7) | 69 (15.8) |
Disease progression | 34 (7.8) | 35 (8.0) |
Adverse event | 13 (3.0) | 11 (2.5) |
Patients who died during adverse event reporting periodb | 13 (3.0) | 11 (2.5) |
Disease progression | 0 | 1 (0.2) |
Adverse event | 13 (3.0) | 10 (2.3) |
Notable harms | ||
Peripheral neuropathy | 230 (52.9) | 236 (53.9) |
Grade 3 | 7 (1.6) | 5 (1.1) |
Infections | 216 (49.7) | 187 (42.7) |
Grade 3 | 55 (12.6) | 43 (9.8) |
Grade 4 | 6 (1.4) | 6 (1.4) |
Grade 5 | 5 (1.1) | 6 (1.4) |
Neutropenia including febrile neutropenia | 200 (46.0) | 188 (42.9) |
Grade 3 | 67 (15.4) | 59 (13.5) |
Grade 4 | 115 (26.4) | 117 (26.7) |
Anemia | 125 (28.7) | 119 (27.2) |
Grade 3 | 49 (11.3) | 37 (8.4) |
Grade 4 | 3 (0.7) | 2 (0.5) |
Thrombocytopenia | 58 (13.3) | 59 (13.5) |
Grade 3 | 12 (2.8) | 11 (2.5) |
Grade 4 | 11 (2.5) | 10 (2.3) |
Infusion-related reactions | 58 (13.3) | 70 (16.0) |
Grade 3 | 5 (1.1) | 6 (1.4) |
Grade 4 | 0 | 1 (0.2) |
Hepatic toxicity | 46 (10.6) | 33 (7.5) |
Grade 3 | 8 (1.8) | 4 (0.9) |
Tumour lysis syndrome | 2 (0.5) | 4 (0.9) |
Grade 3 | 2 (0.5) | 3 (0.7) |
Progressive multifocal leukoencephalopathy | 0 | 0 |
CHP = cyclophosphamide, doxorubicin, and prednisone; pola-R-CHP = polatuzumab vedotin, rituximab, cyclophosphamide, doxorubicin, and prednisone; R-CHOP = rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone; RCT = randomized controlled trial.
Note: Values are n (%).
Note: Details from the table have been taken from the sponsor’s summary of clinical evidence.
aCommon adverse events included those reported by at least 20% of patients in either treatment group.
bAdverse event reporting period is defined as time from the first dose of any study drug through to 90 days after the last dose of any study drug or before nonprotocol or new antilymphoma treatment, whichever is earlier.
Source: POLARIX Clinical Study Report.13
Adverse Events
Almost all patients in each treatment group reported an AE (97.9% for pola-R-CHP and 98.4% for R-CHOP). The percentages between groups were similar for the most commonly reported AEs of nausea, constipation, fatigue, and alopecia, although the percentage of patients with diarrhea was larger with pola-R-CHP (31%) than with R-CHOP (20%). The frequency of grade 3 to grade 4 AEs was similar between pola-R-CHP (57.7%) and R-CHOP (57.5%) and the majority were associated with myelosuppression. The frequency of grade 5 AEs was similar between pola-R-CHP (3.0%) and R-CHOP (2.3%). Most of the grade 5 AEs in both groups were due to infections or complications of infection.
Serious Adverse Events
The percentage of patients who experienced at least 1 SAE was similar between the pola-R-CHP (34.0%) and R-CHOP (30.6%) groups. The most common SAEs in the pola-R-CHP and R-CHOP groups were febrile neutropenia (9.9% versus 6.4%), pneumonia (4.1% versus 3.9%), diarrhea (2.3% versus 0.5%), and pyrexia (1.6% versus 1.8%).
Withdrawals Due to Adverse Events
The percentage of patients who experienced at least 1 AE that led to withdrawal of any study medication was 6.0% in the pola-R-CHP group and 6.4% in the R-CHOP group. Most common AEs that led to withdrawal of any study medication were infections (1.6% pola-R-CHP versus 2.3% R-CHOP) and nervous system disorders (0.7% pola-R-CHP versus 2.5% R-CHOP).
Mortality
A total of 133 (15.2%) deaths occurred in the POLARIX study, with similar proportions between groups. Deaths reported during the AE reporting period (defined as time from first dose of any study drug to 90 days after the last dose of any study drug or before NALT, whichever is earlier) occurred in 13 (3.0%) and 11 (2.5%) patients in the pola-R-CHP and R-CHOP groups, respectively. The primary cause of death among cases were disease progression in 7.8% and 8.0% and AEs in 3.0% and 2.3% in the pola-R-CHP group and R-CHOP group, respectively.
Notable Harms
Neuropathy
The percentage of patients who experienced peripheral neuropathy was 52.9% and 53.9% in the pola-R-CHP and R-CHOP groups, respectively. Among patients who developed peripheral neuropathy, 1.7% (7 patients) and 1.0% (4 patients) were grade 3 to 5 events in the pola-R-CHP and R-CHOP groups, respectively. The percentage of patients with grade 3 peripheral neuropathy was 1.6% (7 patients) in the pola-R-CHP group and 1.1% (5 patients) in the R-CHOP group. No patient experienced grade 5 neutropenia. Peripheral neuropathy was reported as resolved at time of CCOD in 61.7% and 71.6% of patients in the pola-R-CHP and R-CHOP groups, respectively.
Neutropenia Including Febrile Neutropenia
The percentage of patients who experienced neutropenia was 46.0% in the pola-R-CHP group and 42.9% in the R-CHOP group. The percentage of patients who experienced a serious neutropenic event was 11.5% and 8.4% in the pola-R-CHP and R-CHOP groups, respectively; the incidence of serious febrile neutropenia was numerically higher in the pola-R-CHP group (9.9%) compared to the R-CHOP group (6.4%). Two patients (0.5%) and 0 patients in the pola-R-CHP and R-CHOP groups, respectively, experienced neutropenia that led to any study treatment discontinuation.
Anemia
The percentage of patients who experienced anemia was 28.7% in the pola-R-CHP group and 27.2% in the R-CHOP group. The percentage of patients with grade 3 to 4 anemia was 12.0% and 8.9% in the pola-R-CHP group and R-CHOP group, respectively. No patients experienced grade 5 anemia. Four patients (0.9%) in the pola-R-CHP group compared to 6 patients (1.4%) in the R-CHOP group experienced a serious anemic event. Anemia was reported to be resolved in 84.8% and 87.4% of patients in the pola-R-CHP and R-CHOP groups, respectively. No patients in either treatment group experienced anemia that led to study treatment discontinuation.
Thrombocytopenia
The percentage of patients who experienced thrombocytopenia was 13.3% in the pola-R-CHP and 13.5% in the R-CHOP group. The number of patients with grade 3 to 4 thrombocytopenia was numerically higher in the pola-R-CHP group (23 patients [5.3%]) compared with the R-CHOP group (21 patients [4.8%]). Two patients (0.5%) and 1 patient (0.2%) experienced a serious thrombocytopenia event in the pola-R-CHP and R-CHOP groups, respectively. Thrombocytopenia was reported as resolved in 94.8% of patients in the pola-R-CHP group and 88.1% in the R-CHOP group.
Infections
The percentage of patients who experienced infections was 49.7% and 42.7% in the pola-R-CHP and R-CHOP groups, respectively. The percentage of patients with grade 3 to 4 infections and serious infections was higher in the pola-R-CHP group (14.0% and 14.3%) compared to the R-CHOP group (11.2% and 10.3%). Five patients (1.1%) and 6 patients (1.4%) had grade 5 infections in the pola-R-CHP and R-CHOP groups, respectively. Infections were reported as resolved in 87.0% of patients in the pola-R-CHP group and 86.1% of patients in the R-CHOP group.
Hepatic Toxicity
The percentage of patients who experienced hepatic toxicity was 10.6% in the pola-R-CHP group and 7.5% in the R-CHOP group. Eight patients (1.8%) and 4 patients (0.9%) experienced grade 3 hepatotoxic events. No patient had hepatotoxic events rated as grade 4 or 5. One patient (0.2%) and 0 patients had a serious hepatotoxic event in the pola-R-CHP and R-CHOP groups, respectively. Most hepatotoxic events were low-grade liver enzyme elevations in both treatment groups. Hepatotoxic events were reported as resolved in 89.1% of patients in the pola-R-CHP group and 84.8% of patients in the R-CHOP group.
Tumour Lysis Syndrome
Two patients (0.5%) in the pola-R-CHP group and 4 patients (0.9%) in the R-CHOP group experienced TLS. Two patients (0.5%) and 3 patients (0.7%) reported grade 3 TLS in the pola-R-CHP and R-CHOP groups, respectively. No patient reported a grade 4 or 5 TLS. One patient (0.2%) in each treatment group experienced a serious TLS event. TLS was reported as resolved in all patients in the POLARIX study.
Progressive Multifocal Leukoencephalopathy
No patient in the POLARIX trial reported PML.
Infusion-Related Reactions
The percentage of patients with IRRs was 13.3% and 16.0% in the pola-R-CHP group and R-CHOP group, respectively. Five patients (1.1%) in the pola-R-CHP group and 7 patients (1.6%) in the R-CHOP group experienced grade 3 to 4 IRRs. No patient had a grade 5 IRR. Two patients (0.5%) and 3 patients (0.7%) experienced a serious IRR in the pola-R-CHP and R-CHOP groups, respectively.
Critical Appraisal
Internal Validity
The POLARIX study was a phase III, double-blind, placebo-controlled trial that used a centralized method (interactive voice/web response system) of randomizing patients to treatment by stratification factors for prognostic factors and geographical location that were considered appropriate. The likelihood of appropriate randomization was supported by similar between-group proportions for medical history, baseline prognostic indicators, and prior concomitant medications. Therefore, the risk of selection bias from inappropriate randomization and allocation concealment was determined to be low. Similarly, the population sampling strategy did not clearly lead to differential treatment effects.
Approximately 6% of patients had a protocol deviation, including accidental unblinding of site staff (n = 3) and patients (n = 7). The small number of unblinded individuals was unlikely to have a major impact on study conduct, assessments, and results. The primary outcome of PFS was assessed by the investigators. Although both investigator and independent review committee assessments for PFS would be preferred, the double-blinded study design would have helped limit potential bias from the investigator assessment method. Moreover, CR was assessed by BICR assessments as well as by investigators and showed reasonable concordance, thereby providing increased confidence in tumour assessments. Last, AEs were unlikely to have led to unblinding because pola-R-CHP generally had similar AEs at frequencies similar to R-CHOP, including neuropathies.
There was a relatively high rate of discontinuation from the study (168 patients [19.1%]) with most losses due to deaths. Given the aggressive nature of DLBCL, this was unsurprising, and there were few concerns with a differential risk of bias between groups due to dropouts since the proportion of study discontinuations (18.4% versus 19.8%) and deaths (14.3% versus 15.3%) were similar between the pola-R-CHP and R-CHOP groups, respectively. Nonetheless, the integrity of the data and validity of the results come into question when the total loss to follow-up is near or exceeds 20%.58 The potential impacts of the reduced sample size were notable in the later time points of the Kaplan-Meier curves for PFS and the imprecision in the RMST estimates for PFS. The sample sizes at the time of the final assessments for the PROs were less than one-third of what they were at the beginning of the study. While this is often observed in trials for interventions in oncology, the large reduction in sample size makes it difficult to adequately assess the treatment effects on these important outcomes. Therefore, the duration of the study combined with the discontinuations by the EOT assessments limits the interpretation of the longer-term effects of pola-R-CHP versus R-CHOP.
Appropriate hierarchical statistical testing methods were used to account for multiplicity for the primary end point and all key secondary end points, but were not employed for additional secondary end points such as investigator-assessed CR rate, ORR, or HRQoL. Due to the lack of adjustment for multiplicity, results for the additional secondary end points and exploratory end points are at increased risk of type I error.
PFS was the primary efficacy end point and a surrogate outcome for OS in the POLARIX study. The Kaplan-Meier method was used to estimate the PFS distribution and a visual inspection (of Kaplan-Meier plots, log (-log) plots, and Schoenfeld residuals with statistical testing) was used to assess the proportional hazards assumption. The PFS curves appeared to demonstrate separation at approximately 6 months, and from that point onward the proportional hazards assumption appeared to have been met. This was supported by the sensitivity analyses using RMST — which is independent of the proportional hazards assumption — showing that there was benefit for PFS favouring pola-R-CHP over R-CHOP at approximately 12 months, with greater differences between groups observed at 24 months and after. OS results were limited by the low number of events observed and relatively short duration of follow-up at the final analysis. Testing of the proportional hazards assumption for the analysis of OS was the same as for PFS. The Kaplan-Meier and cumulative log-hazards curves crossed at several time points suggesting that the proportional hazards assumption was violated. Additionally, RMST sensitivity analyses for OS did not show a difference between the curves at any time point. Therefore, CADTH clinical reviewers determined the validity of OS results from the POLARIX study was uncertain.
Most patients were censored for PFS because no progression event or death was recorded at the CCOD. The benefits observed in the R-CHOP treatment group were expected as per the clinical experts consulted by CADTH, as it is an established effective first-line therapy for DLBCL. Censoring rules were well reported and appeared reasonably conservative. Patients without a postbaseline tumour assessment or evaluable assessment were censored at randomization. While there was a similar distribution of censored patients in each group throughout the study, a total of 173 patients (19.7%) in the POLARIX study were censored by 30 months, and more patients were censored at later time points. The censoring rules related to NALT had an impact on the results as evidenced by sensitivity analyses. While the sensitivity analyses for NALT, including the various discounted percentages, showed results similar to the primary analysis, the more conservative censoring at the last adequate tumour assessment before the initiation of NALT demonstrated that the upper CI for the HR crossed unity (HR = 0.77; 95% CI, 0.59 to 1.01). Despite the apparent sensitivity of the results for PFS to these censoring rules, most analyses confirmed the primary PFS analysis favouring pola-R-CHP over R-CHOP. However, these analyses (and others) indicate there is uncertainty as to the clinical significance of the PFS results from the POLARIX study (refer to the Discussion).
The EORTC QLQ-C30, FACT/GOG-NTX, and 5-Level EQ-5D have not been validated in patients with LBCL; nevertheless, the EORTC QLQ-C30 and 5-Level EQ-5D are common HRQoL tools used in clinical trials, with the latter having been validated in the general population in Canada. These PROs were secondary and exploratory outcomes without adjustment for multiplicity in the POLARIX study. As mentioned, there were concerns for the validity of the EOT results due to a sizable proportion of losses to follow-up at 24 months in the EORTC QLQ-C30 (range, 72% to 77%), FACT-Lym LymS (77%), and FACT/GOG-NTX (77%) among randomized patients with baseline and at least 1 postbaseline assessment. Patients without a postbaseline assessment were censored at randomization, and questionnaires with fewer than 50% of constituent items completed were treated as missing, without data imputation, or employing appropriate methods of handling missing data. All PRO data are likely biased in favour of patients who respond to treatment and/or have few-to-no AEs, leading to an overestimate of the treatment effects. As such, the decreased number of patients contributing to the analysis of PROs over time creates uncertainty in the data.
External Validity
While the population enrolled in the POLARIX study were reported by the clinical experts to be representative of patients with LBCL who they would consider eligible for pola-R-CHP treatment, there were limitations with the representativeness of the study population. According to the clinical experts, patients initiating treatment with current standard first-line R-CHOP for DLBCL often present with ECOG PS of greater than 2 and therefore would be considered eligible for treatment with pola-R-CHP by clinicians. As well, those with transformed indolent lymphoma including follicular lymphoma grade 3B, and primary cutaneous DLBCL would be eligible for pola-R-CHP in practice yet were excluded from the POLARIX study. It was also noted that many patients have comorbidities that were exclusion criteria in the trial.
The clinical experts emphasized that the IPI score is widely used in clinical practice for prognostic assessment, and that patients with IPI score 3 to 5 (high risk) should be targeted for treatment with pola-R-CHP due to a reduced cure rate among this population, with an estimated 50% of patients with advanced stage disease experiencing relapse and requiring salvage chemotherapy. Furthermore, the experts expressed uncertainty regarding the magnitude of benefit for patients with IPI score 2 (intermediate risk) for whom treatment effectiveness has been established with R-CHOP. Patients with IPI score 0 or 1 (limited disease) were excluded from the POLARIX trial. The clinical experts reported that these patients typically experience high cure rates with R-CHOP (with or without radiotherapy) and therefore, in the absence of evidence, would not be targeted for treatment with pola-R-CHP in clinical practice. The clinical experts indicated that patients with DHL or THL, however, are not regularly treated with R-CHOP as DA-EPOCH-R tends to be the preferred first-line treatment in patients with these subtypes; therefore, the inclusion of these patients in the POLARIX study with R-CHOP as treatment comparator did not entirely reflect the SOC in Canada.
The clinical experts reported that while the intervention dose and regimen aligned with how it would be used in clinical practice, the additional 2 cycles of rituximab monotherapy following pola-R-CHP were not standard in current practice.
R-CHOP was an appropriate comparator since it is the SOC for most patients in the first-line setting. Pola-R-CHP is reported by the clinical experts to be an appropriate alternative to SOC R-CHOP in the first-line setting, and there is familiarity with using polatuzumab vedotin in the refractory or relapse setting including as a bridge therapy to CAR T-cell therapy or salvage chemotherapies. However, the clinical experts noted that patients with frailty or comorbidities are treated with dose-adjusted CHOP as first-line due to intolerability of AEs. The treatment groups were similar in terms of the relative percentage of treatment received and the percentages of patients who discontinued therapies for AEs. The percentage of patients with planned dose reductions because of AEs was generally similar in both treatment groups, except the planned dose reductions due to AEs in patients receiving polatuzumab vedotin was lower than the percentage of patients with planned vincristine dose reductions due to AEs in the R-CHOP group (6.9% pola-R-CHP versus 11.6% R-CHOP). Although it is reasonable for the trial to have planned to use standard doses of cyclophosphamide, doxorubicin, vincristine, and prednisone for all patients randomized to the R-CHOP group and dose adjust for AEs during treatment, it is unclear how similar the AE distribution and need for dose adjustments would have been in the R-CHOP group if eligible patients had initiated therapy on a reduced regimen. The clinical experts noted that standard R-mini-CHOP has lower cure rates than standard dose R-CHOP.
In the POLARIX study, patients were permitted to receive radiotherapy at the EOT if it was preplanned and documented before randomization. Preplanned radiotherapy was indicated if the patient had initial sites of bulky or extranodal disease. The clinical experts specified that this reflects the use of radiation in practice settings. Greater than 4% of patients randomized to R-CHOP had radiotherapy preplanned as part of their treatment compared with 2.5% of those randomized to pola-R-CHP. However, 9.8% and 7.0% of patients in the R-CHOP and pola-R-CHP groups, respectively, received radiotherapy that was unplanned. All unplanned radiotherapy was coded in the POLARIX study as a NALT (refer to internal validity for the impacts of NALT on the results). The clinical experts noted that unplanned radiotherapy is not consistent with practice and would not be considered to indicate PD. Nonetheless, in a trial setting, classifying unplanned radiotherapy as NALT likely improves internal validity (if rules are applied equally to treatment groups as they appeared to be in the POLARIX study) but reduces external validity.
In the POLARIX trial, G-CSF was required as primary prophylaxis for each of the 6 cycles of therapy, as well as for patients with prognostic factors or at high risk of infection or morbidity who exhibit fever and neutropenia during the study. The clinical experts noted that since the practice of G-CSF prophylaxis varied across clinical settings, mandating G-CSF as part of treatment with pola-R-CHP would necessitate a practice change for some sites that may have additional implications for resources and personnel. Moreover, the use of nonprophylaxis G-CSF in the POLARIX study primarily for AE management was high (22.1% in the pola-R-CHP group versus 18.9% in the R-CHOP group) and did not align with the clinical experts’ experience in practice.
The clinical experts consulted by CADTH agreed that the efficacy end points evaluated in the POLARIX trial were aligned with treatment outcomes of relevance in clinical practice, including PFS, OS, and CR rate (as measured by PET and Lugano criteria). The experts also agreed that assessing PFS24 is used to assess treatment response in clinical practice, especially for aggressive disease for which the experts noted leads to relapse or progression in most patients in the first year of treatment.
With respect to assessment and monitoring, the clinician groups and experts reported treatment response to be assessed every 3 months for 1 year followed by every 6 months in the second year (or slight variations such as every 3 months for 2 years) which appeared to be aligned with the frequency in the POLARIX study.
Long-Term Extension Studies
No long-term extension studies were submitted in the systematic review evidence.
Indirect Evidence
No indirect treatment comparisons were submitted in the systematic review evidence.
Studies Addressing Gaps in the Pivotal and RCT Evidence
No additional studies addressing important gaps in the systematic review evidence were identified.
Discussion
Summary of Available Evidence
One phase III, multicentre, randomized, double-blind, placebo-controlled trial (POLARIX; N = 849) assessed the efficacy and safety of polatuzumab vedotin 1.8 mg/kg in combination with R-CHP compared with R-CHOP, the SOC for first-line treatment, in the treatment of adults with previously untreated LBCL, including DLBCL NOS, high-grade B-cell lymphoma, EBV-positive DLBCL NOS, and T-cell/histiocyte-rich LBCL. The primary outcome was investigator-assessed PFS. Key secondary outcomes included OS and BICR-assessed CR at EOT. Additional secondary efficacy outcomes included investigator-assessed CR at EOT and investigator-assessed and BICR-assessed ORR. HRQoL was a secondary outcome, assessed using TTD and responder analyses for the EORTC QLQ-C30 Physical Functioning and Fatigue scales and the FACT-Lym LymS, and assessed using rate of peripheral neuropathy on the FACT/GOG-NTX. The remaining scales of these HRQoL tools were included as exploratory outcomes. TEAEs were reported in the POLARIX study.
The POLARIX study included 7 sites in Canada. All patients enrolled had CD20-positive DLBCL, IPI score of 2 to 5, ECOG PS of 0 to 2, and a life expectancy of 12 months or greater. Patients had a median study population age of 65 years. Most patients had advanced Ann Arbor stage III to IV (88.7%), and baseline lactate dehydrogenase greater than 1 time the ULN (65.4%) at diagnosis. Patients were similar between treatment groups in stratification factors used for randomization (IPI score, bulky disease, and geographical region) and baseline characteristics. All patients in the safety population had at least 1 medical history condition with similar proportions between groups for the most common conditions.
Interpretation of Trial Results
Efficacy
Patient input identified longer disease remission and survival, with controlled disease symptoms and improved quality of life to be important outcomes sought with new treatment for DLBCL. Similarly, clinicians identified PFS, OS, and CR as important outcomes when treating their patients, and these outcomes were assessed in the POLARIX trial. In the pivotal phase III POLARIX RCT, the primary end point of PFS was used to assess efficacy of pola-R-CHP compared to SOC R-CHOP. The clinical experts considered PFS to be an appropriate surrogate for OS given the limited duration of follow-up in the POLARIX study and the longer OS with R-CHOP, which in most cases is anticipated to be curative in the first-line setting for DLBCL. Studies reported moderate to high correlations between PFS and PFS24 with OS suggesting these outcomes are surrogates for OS in patients with DLBCL.30,31,59,60 However, these analyses used population bases largely drawn from RCTs with narrower enrolment criteria versus a real-world population, and the modelled associations appear to weaken at OS time points after 5 years.61
OS was a key secondary end point in the POLARIX trial, but the study was not adequately designed or statistically powered for OS. The HR for OS favoured pola-R-CHP (0.94; 95% CI, 0.67 to 1.33) but the upper CI crossed unity and an OS benefit cannot be concluded. A key limitation for the OS results was the insufficient number of events observed over the relatively short follow-up of nearly 40 months. Visual inspections of the curves for OS suggested nonproportionality such that the proportional hazards assumption was likely violated. Therefore, the treatment effects of pola-R-CHP for OS could not be determined based on the submitted evidence.
The primary findings from the POLARIX study indicate that pola-R-CHP provided greater efficacy for PFS compared with SOC R-CHOP at a median of nearly 31 months of follow-up. Sensitivity analyses generally supported the primary analysis results for PFS. The median PFS was not estimable for either treatment group, highlighting the overall benefits of both regimens in the first-line treatment of DLBCL. The clinical experts expressed uncertainty regarding whether the differences observed between groups in PFS at 12 months (4%), 24 months (6.5%), or in the incremental gain between these time points were clinically important. Given the aggressive nature of DLBCL and that relapsed disease is difficult to cure, the clinical experts remarked that preventing a single event of PD, relapse, or death compared with R-CHOP in first-line treatment may be clinically meaningful. The clinical experts and CADTH review team acknowledged that there is no empirically defined or consensus-based minimal important difference for PFS events with which to gauge between-group differences. RMST sensitivity analyses of PFS showed an overall trend of increasing magnitude of benefit with pola-R-CHP over SOC at months 12 (mean difference of 0.2 months), 24 (mean difference of 1 month), 36 (mean difference of 1.8 months), and 42 (mean difference of 2.2 months), but with increasingly wider 95% CIs observed with each subsequent time point, indicating reduced precision. As well, the sensitivity analyses conducted on the ITT population using time before NALT initiation was a conservative approach (i.e., PFS censored at the last adequate tumour assessment before the initiation) when compared to the discount method, suggesting imprecision in the estimated benefit with pola-R-CHP. While PFS assessment demonstrated some sensitivity to the censoring method used, this may be in part due to whether the true benefit of pola-R-CHP was realized in the ITT population or in a subgroup of patients. The subgroup results presented in Figure 4 identified potential signals of stronger benefit for PFS with pola-R-CHP versus R-CHOP in patients who have an IPI score of 3 to 5 and who do not have bulky disease. Although the subgroup analyses were exploratory, the IPI score and bulky disease were stratification factors and therefore randomization would have been preserved for the subgroup analyses. Health Canada clinical reviewers and biostatisticians likewise identified the notable differences in magnitude of effect in these subgroups.12 Health Canada requested additional subgroup analyses of PFS by IPI score and presence of bulky disease;12 refer to Figure 7 in this review report. Like CADTH, Health Canada concluded that much of the benefit in PFS with pola-R-CHP appears to be driven by those patients with an IPI score of 3 to 5 and who do not have bulky disease. The Health Canada review noted, “Interestingly, the 95% CIs for IPI score 3 to 5 without bulky disease and IPI score 3 to 5 with bulky disease do not overlap further strengthening that the difference is more than a chance event.”12 These subgroup data are included in the product monograph for polatuzumab vedotin.2 The clinical experts consulted by CADTH remarked that these results generally aligned with their opinion that pola-R-CHP may provide greater benefit in patients who have worse outcomes with current SOC (i.e., patients at high risk of disease progression or relapse) including patients with poor prognostic indicators (e.g., IPI score of 3 to 5). The experts noted that the uncertainty of the benefit in those with an IPI score of 2 likely reflects the benefit achieved by R-CHOP in these patients, which is consistent with their experience in practice. Despite the biological plausibility for the results and the strength of the association, the results were from exploratory subgroup analyses, therefore, firm conclusions may not be appropriate.
There were several distinguishable differences between the population included in the Health Canada indication, patients who were enrolled in the POLARIX trial, and patients deemed by the clinical experts to be eligible for treatment with pola-R-CHP in practice. While the Health Canada indication does not specify that patients have to have CD20-positive DLBCL, the trial used this as a criterion for enrolment. However, given that rituximab — an anti-CD20 monoclonal antibody — is part of the indicated regimen, the indication for polatuzumab vedotin in combination with R-CHP is implicit in its inclusion of patients with CD20-positive DLBCL. There was also uncertainty in the applicability of findings for children since patients younger than 18 years of age were excluded in the POLARIX trial. The approved indication does not specify the age of eligible patients, but the product monograph for polatuzumab vedotin notes that “the safety and efficacy of Polivy in pediatric patients has not been established; therefore, Health Canada has not authorized an indication for pediatric use.”2 The inclusion of patients with DHL and THL in the POLARIX study population was reported by the clinical experts to reflect a population whose preferred treatment in Canada is DA-EPOCH-R rather than R-CHOP. Therefore, there may be a small percentage of patients who could be treated with pola-R-CHP instead of DA-EPOCH-R; however, the experts agreed that a study comparing pola-R-CHP with DA-EPOCH-R in this subset of patients would be needed for a change in the preferred approach to treating these patients. The clinical experts noted that patients with ECOG PS of 3 or 4 were excluded from the trial. The clinical experts indicated that patients with worse performance status should be considered eligible for treatment with R-CHOP or pola-R-CHP, noting that many, if not most, of their patients present for initial treatment of DLBCL with ECOG PS of 3 or 4. The clinical experts stated that IPI score, which includes prognostic factors such as ECOG PS, is a better means of deciding eligibility for treatment. The clinical experts also noted that although the POLARIX trial excluded patients with transformed DLBCL, including those with follicular lymphoma grade 3B, these patients would otherwise be considered eligible for treatment in clinical practice.
HRQoL and symptoms that were identified as important by patient groups were assessed in the POLARIX study, including physical functioning, fatigue, peripheral neuropathy, diarrhea, and night sweats. There were no apparent differences between treatment groups for any of the HRQoL or symptom scales. While the sponsor suggested HRQoL was at least maintained with pola-R-CHP therapy, the results are difficult to interpret because the outcome measures were exploratory and there was a large decline in the sample size contributing to the analysis at later time points so that only approximately one-third of patients provided data at the final assessment time point. As well, it was unclear how missing data were handled and how disease progression may have affected patient responses to the various questionnaires. The Health Canada review stated, “Based on the way the information was presented in the CSR and the exploratory nature of the end point [QoL], it is not considered appropriate to highlight benefits of Polivy plus R-CHP vs. R-CHOP in the product monograph with respect to any quality of life measures, including improvements in peripheral neuropathy.”12
Harms
Overall, the safety profiles of pola-R-CHP and R-CHOP showed similar proportions of patients with TEAEs, SAEs, treatment and study discontinuations due to AEs, and deaths. Input from patients who have experience with polatuzumab vedotin reported experiencing AEs including fatigue, neutropenia, thrombocytopenia, and diarrhea, which were among the most commonly reported AEs in the POLARIX study. The clinical experts expressed concerns about neutropenia of any grade, grade 3 anemia, grade 3 diarrhea, and peripheral neuropathy among patients treated with polatuzumab vedotin. Rates of peripheral neuropathy were similar between groups in the POLARIX trial. Patients treated with pola-R-CHP had a higher rate of febrile neutropenia and infections than patients treated with R-CHOP, which was concerning to the clinical experts, and notable despite the trial having employed G-CSF prophylaxis for all patients. Furthermore, nearly 20% of patients had nonprophylaxis G-CSF to manage AEs. Additionally, a greater number of patients treated with pola-R-CHP compared with R-CHOP reported grade 3 anemia (49 patients [11.3%] versus 37 patients [8.4%]), hepatic toxicity (46 patients [10.6%] versus 33 patients [7.5%]), and serious diarrhea (10 patients [2.3%] versus 2 patients [0.5%]). The product monograph for polatuzumab vedotin included serious warnings and precautions for infections and myelosuppression (neutropenia, febrile neutropenia, thrombocytopenia, and anemia), which were included as AEs of special interest in the POLARIX trial. The Health Canada report outlined concerns regarding deaths that occurred earlier among patients treated with pola-R-CHP compared to R-CHOP, and while no OS differences between groups were detected, there was uncertainty regarding the potential increased toxicity with polatuzumab vedotin during treatment.12
The clinical experts reported that patients with advanced age and/or frailty and those with comorbidities may not be able to tolerate pola-R-CHP due to AEs, considering that some of these patients require a reduced dose regimen with SOC (e.g., R-mini-CHOP). As described previously, some patients in both treatment groups required pauses in treatment for AEs or had to permanently stop treatment for AEs. The study did not examine different doses of the components to assess potential effects on treatment tolerability, which is a gap in the evidence.
Conclusion
In the POLARIX trial, the study population was limited in representativeness of patients with DLBCL, but likely representative of those considered to be eligible for treatment in clinical practice. Pola-R-CHP demonstrated a benefit for PFS compared to SOC R-CHOP in adult patients with previously untreated LBCL, including DLBCL NOS, high-grade B-cell lymphoma, EBV-positive DLBCL NOS, and T-cell/histiocyte-rich LBCL. However, there was uncertainty in whether the difference in PFS between groups was clinically meaningful. There were signals that the PFS benefit was primarily driven by treatment effects among the subgroup of patients with an IPI score of 3 to 5 and without bulky disease, but these findings were from exploratory subgroup analyses and may reflect differences in expected risk of progression among patients with an IPI score of 2 versus higher. It is uncertain whether pola-R-CHP is associated with an OS benefit because the data were immature, reflecting the relatively limited duration of follow-up, and the proportional hazards assumption was likely violated. Analyses of secondary outcomes showed numeric benefits with pola-R-CHP in achieving CR and ORR after treatment. There were no differences between the pola-R-CHP and R-CHOP groups for HRQoL, functioning, or key symptoms experienced by patients, including fatigue, diarrhea, and peripheral neuropathy. Patients treated with pola-R-CHP experienced similar frequencies of AEs, SAEs, WDAEs, and deaths as R-CHOP with no new safety signals identified.
References
1.Clinical evidence template of Polivy (polatuzumab vedotin) plus rituximab, cyclophosphamide, doxorubicin, and prednisone (R-CHP) for the treatment of adult patients with previously untreated large B-cell lymphoma (LBCL), including diffuse large B-cell lymphoma (DLBCL) not otherwise specified (NOS), high grade B-cell lymphoma, Epstein-Barr virus-positive (EBV+) DLBCL NOS, and T-cell/histiocyte rich LBCL [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Polivy (polatuzumab vedotin), 30 mg or 140 mg single-use vial IV infusion. Mississauga (ON): Hoffman-La Roche Ltd.; 2022 Oct.
2.Polivy (polatuzumab vedotin): 30 mg or 140 mg single-use vial, lyophilized powder for solution for intravenous infusion only [product monograph]. Mississauga (ON): Hoffmann-La Roche Ltd.; 2023 Jan 20: https://pdf.hres.ca/dpd_pm/00069254.PDF. Accessed 2023 Apr 06.
3.Canadian Cancer Society. Non-Hodgkin Lymphoma. 2022; https://www.lymphoma.ca/lymphoma/non-hodgkin-lymphoma/. Accessed 2022 Aug 12.
4.Canadian Cancer Society. Non-Hodgkin Lymphoma statistics. 2022; https://cancer.ca/en/cancer-information/cancer-types/non-hodgkin-lymphoma/statistics. Accessed 2022 Sep 1.
5.Sehn LH, Salles G. Diffuse Large B-Cell Lymphoma. N Engl J Med. 2021;384(9):842-858. PubMed
6.Rosenwald A, Bens S, Advani R, et al. Prognostic Significance of MYC Rearrangement and Translocation Partner in Diffuse Large B-Cell Lymphoma: A Study by the Lunenburg Lymphoma Biomarker Consortium. J Clin Oncol. 2019;37(35):3359-3368. PubMed
7.Willenbacher E, Willenbacher W, Weger R, Dominik W, Manzl C, Brunner A. Patients with double/triple copy number gains on C-MYC, BCL2, and/or BCL6 treated with standard chemotherapy have a similarly poor prognosis than those with high-grade B cell lymphoma with C-MYC and BCL2 and/or BCL6 rearrangements: a single-center experience on a consecutive cohort of large B cell lymphomas. Ann Hematol. 2020;99(9):2125-2132. PubMed
8.Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127(20):2375-2390. PubMed
9.Freedman AS, Aster JC. Epidemiology, clinical manifestations, pathologic features, and diagnosis of diffuse large B cell lymphoma. In: Rosmarin AG, ed. UpToDate. Waltham (MA): UpToDate; 2020: http://www.uptodate.com. Accessed 2023 Mar 23.
10.Lymphoma Canada. Diffuse Large B cell Lymphoma (DLBCL). 2022. Accessed 2023 Mar 23.
11.Rovira J, Valera A, Colomo L, et al. Prognosis of patients with diffuse large B cell lymphoma not reaching complete response or relapsing after frontline chemotherapy or immunochemotherapy. Ann Hematol. 2015;94(5):803-812. PubMed
12.Health Canada Biologics Safety and Efficacy Assessment Report: Polivy (polatuzumab vedotin) [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Polivy (polatuzumab vedotin), 30 mg or 140 mg IV infusion. Mississauga (ON): Hoffman-La Roche Limited; 2022 Oct 31.
13.Clinical Study Report: Study GO39942 (POLARIX) Report No. 1116774. A phase III, multicenter, randomized, double-blind, placebo-controlled trial comparing the efficacy and safety of polatuzumab vedotin in combination with rituximab and CHP (R-CHP) versus rituximab and CHOP (R-CHOP) in previously untreated patients with diffuse large B-cell lymphoma. [internal sponsor's report]. Rouen (FR): Hoffman-La Roche Ltd.; 2022 Sept.
14.Clinical Study Report: Study GO39942 (POLARIX) Report No. 1106275. A phase III, multicenter, randomized, double-blind, placebo-controlled trial comparing the efficacy and safety of polatuzumab vedotin in combination with rituximab and CHP (R-CHP) versus rituximab and CHOP (R-CHOP) in previously untreated patients with diffuse large B-cell lymphoma [internal sponsor's report]. Rouen (FR): Hoffman-La Roche Ltd.; 2021 Oct 13.
15.Ma Q, Bailey A, Milloy N, Butcher J, Quek RG, Johnson PC. Real-World Health-Related Quality of Life in Patients with Diffuse Large B-Cell Lymphoma: Comparisons with Reference Populations and By Line of Therapy. Blood. 2021;138:4111.
16.Susanibar-Adaniya S, Barta SK. 2021 Update on Diffuse large B cell lymphoma: A review of current data and potential applications on risk stratification and management. Am J Hematol. 2021;96(5):617-629. PubMed
17.Gisselbrecht C, Glass B, Mounier N, et al. Salvage regimens with autologous transplantation for relapsed large B-cell lymphoma in the rituximab era. J Clin Oncol. 2010;28(27):4184-4190. PubMed
18.van Imhoff GW, McMillan A, Matasar MJ, et al. Ofatumumab Versus Rituximab Salvage Chemoimmunotherapy in Relapsed or Refractory Diffuse Large B-Cell Lymphoma: The ORCHARRD Study. J Clin Oncol. 2017;35(5):544-551. PubMed
19.Shafey M, Savage KJ, Skrabek P, et al. Canadian Evidence-Based Guideline for the Frontline Treatment of Diffuse Large B-Cell Lymphoma. Mississauga (ON): Lymphoma Canada; 2021: https://www.lymphoma.ca/wp-content/uploads/2021/09/LymphomaCanada_Guideline_Frontline_DLBCL_VF_Digital.pdf. Accessed 2023 Mar 23.
20.NCCN Guidelines for Patients: Diffuse Large B-cell Lymphoma [accessed by sponsor]. Plymouth Meeting (PA): National Comprehensive Cancer Network; 2020.
21.Lymphoma. (Clinical Practice Guideline LYHE-002 V19). Edmonton (AB): Alberta Health Services; 2021: https://www.albertahealthservices.ca/assets/info/hp/cancer/if-hp-cancer-guide-lyhe002-lymphoma.pdf. Accessed 2022 Aug 11.
22.Prica A, Baldassarre F, Hicks LK, et al. Rituximab in Lymphoma and Chronic Lymphocytic Leukemia: A Clinical Practice Guideline. (Guideline 6-8 Version 3). Toronto (ON): Cancer Care Ontario; 2015: https://www.cancercareontario.ca/en/guidelines-advice/types-of-cancer/721. Accessed 2023 Mar 23.
23.BC Cancer Protocol Summary for Treatment of Lymphoma with DOXOrubicin, Cyclophosphamide, vinCRIStine, predniSONE and riTUXimab (CHOP-R). [Victoria (BC)]: BC Cancer; 2022: http://www.bccancer.bc.ca/chemotherapy-protocols-site/Documents/Lymphoma-Myeloma/LYCHOPR_Protocol.pdf. Accessed 2023 Mar 23.
24.Crump M, Neelapu SS, Farooq U, et al. Outcomes in refractory diffuse large B-cell lymphoma: results from the international SCHOLAR-1 study. Blood. 2017;130(16):1800-1808. PubMed
25.Freedman AS, Friedberg JW. Initial treatment of limited stage diffuse large B cell lymphoma. In: Rosmarin A, ed. UpToDate. Waltham (MA): UpToDate; 2021: http://www.uptodate.com. Accessed 2023 Mar 23.
26.Freedman AS, Friedberg JW. Initial treatment of advanced stage diffuse large B cell lymphoma. In: Rosmarin A, ed. UpToDate. Waltham (MA): UpToDate; 2022: http://www.uptodate.com. Accessed 2023 Mar 23.
27.Sehn LH, Gascoyne RD. Diffuse large B-cell lymphoma: optimizing outcome in the context of clinical and biologic heterogeneity. Blood. 2015;125(1):22-32. PubMed
28.Zhou Z, Sehn LH, Rademaker AW, et al. An enhanced International Prognostic Index (NCCN-IPI) for patients with diffuse large B-cell lymphoma treated in the rituximab era. Blood. 2014;123(6):837-842. PubMed
29.Shafey M, Savage KJ, Skrabek P, Elsawy M, Bosch M, Kuruvilla J. Canadian Evidence-based Guideline for the Treatment of Relapsed/Refractory Diffuse Large B-Cell Lymphoma. Mississauga (ON): Lymphoma Canada; 2021: https://www.lymphoma.ca/resources/healthcare-professionals/dlbcl-diffuse-large-b-cell-lymphoma/. Accessed 2023 Mar 23.
30.Maurer MJ, Habermann TM, Shi Q, et al. Progression-free survival at 24 months (PFS24) and subsequent outcome for patients with diffuse large B-cell lymphoma (DLBCL) enrolled on randomized clinical trials. Ann Oncol. 2018;29(8):1822-1827. PubMed
31.Zhu J, Yang Y, Tao J, et al. Association of progression-free or event-free survival with overall survival in diffuse large B-cell lymphoma after immunochemotherapy: a systematic review. Leukemia. 2020;34(10):2576-2591. PubMed
32.Friedberg JW. Relapsed/refractory diffuse large B-cell lymphoma. Hematology Am Soc Hematol Educ Program. 2011;2011:498-505. PubMed
33.Salles G, Duell J, Gonzalez Barca E, et al. Tafasitamab plus lenalidomide in relapsed or refractory diffuse large B-cell lymphoma (L-MIND): a multicentre, prospective, single-arm, phase 2 study. Lancet Oncol. 2020;21(7):978-988. PubMed
34.Nastoupil LJ, Jain MD, Feng L, et al. Standard-of-Care Axicabtagene Ciloleucel for Relapsed or Refractory Large B-Cell Lymphoma: Results From the US Lymphoma CAR T Consortium. J Clin Oncol. 2020;38(27):3119-3128. PubMed
35.Gisselbrecht C, Glass B, Mounier N, et al. Salvage regimens with autologous transplantation for relapsed large B-cell lymphoma in the rituximab era. J Clin Oncol. 2010;28(27):4184-4190. PubMed
36.CADTH Drug Reimbursement Expert Review Committee final recommendaton: polatuzumab vedotin (Polivy - Hoffman-La Roche Limited). Ottawa (ON): CADTH; 2021 Apr 21: https://www.cadth.ca/sites/default/files/pcodr/Reviews2021/10227PolatuzumabDLBCL_fnRec_EC21Apr2021_final.pdf. Accessed 2023 May 26.
37.Cancer Care Ontario. Cancer Care Ontario drug formulary. Cyclophosphamide. 2019: https://www.cancercareontario.ca/en/drugformulary/drugs/monograph/44226. Accessed 2023 Apr 06.
38.Riabni (rituximab): 10 mg/mL Intravenous Infusion [product monograph]. Mississauga (ON): Amgen Canada Inc.; 2021 Mar 11: https://pdf.hres.ca/dpd_pm/00060341.PDF. Accessed 2023 Apr 06.
39.Cyclophosphamide: 500 mg, 1000 mg and 2000 mg per vial, lyophilized powder for injection [product monograph]. Kirkland (QC): Accord Healthcare Inc; 2022 Sep 14: https://pdf.hres.ca/dpd_pm/00067461.PDF. Accessed 2023 Apr 06.
40.Adriamycin (doxorubicin): solution, 2 mg/mL, for intravenous and intravesical use [product monograph]. Kirkland (QC): Pfizer Canada ULC; 2022 May 09: https://pdf.hres.ca/dpd_pm/00068415.PDF. Accessed 2023 Apr 06.
41.Teva-Prednisone (prednisone): Tablets 5 mg, and 50 mg [product monograph]. Toronto (ON): Teva Canada Limited; 2020 Dec 7: https://pdf.hres.ca/dpd_pm/00059252.PDF. Accessed 2023 Apr 06.
42.Lynch RC, Poh C, Ujjani CS, et al. Polatuzumab vedotin with infusional chemotherapy for untreated aggressive B-cell non-Hodgkin lymphomas. Blood Adv. 2023;7(11):2449-2458. PubMed
43.Tilly H, Morschhauser F, Sehn LH, et al. Polatuzumab Vedotin in Previously Untreated Diffuse Large B-Cell Lymphoma. N Engl J Med. 2022;386(4):351-363. PubMed
44.Aaronson NK, Ahmedzai S, Bergman B, et al. The European Organization for Research and Treatment of Cancer QLQ-C30: a quality-of-life instrument for use in international clinical trials in oncology. J Natl Cancer Inst. 1993;85(5):365-376. PubMed
45.Davda J, Kibet H, Achieng E, Atundo L, Komen T. Assessing the acceptability, reliability, and validity of the EORTC Quality of Life Questionnaire (QLQ-C30) in Kenyan cancer patients: a cross-sectional study. J Patient Rep Outcomes. 2021;5(1):4. PubMed
46.Cocks K, King MT, Velikova G, et al. Evidence-based guidelines for interpreting change scores for the European Organisation for the Research and Treatment of Cancer Quality of Life Questionnaire Core 30. Eur J Cancer. 2012;48(11):1713-1721. PubMed
47.Osoba D, Rodrigues G, Myles J, Zee B, Pater J. Interpreting the significance of changes in health-related quality-of-life scores. J Clin Oncol. 1998;16(1):139-144. PubMed
48.Hlubocky FJ, Webster K, Cashy J, Beaumont J, Cella D. The development and validation of a measure of health-related quality of life for non-Hodgkin’s lymphoma: the functional assessment of cancer therapy—lymphoma (FACT-Lym). Lymphoma. 2013;2013.
49.Huang HQ, Brady MF, Cella D, Fleming G. Validation and reduction of FACT/GOG-Ntx subscale for platinum/paclitaxel-induced neurologic symptoms: a gynecologic oncology group study. Int J Gynecol Cancer. 2007;17(2):387-393. PubMed
50.Yost K, Cella D, Chawla A, et al. Minimally important differences were estimated for the Functional Assessment of Cancer Therapy–Colorectal (FACT-C) instrument using a combination of distribution-and anchor-based approaches. J Clin Epidemiol. 2005;58(12):1241-1251. PubMed
51.van Reenen M, Janssen B, Oppe M, Kreimeier S, Greiner W, Stolk E. EQ-5D-Y-3L User Guide. Version 2.0. 2020; https://euroqol.org/publications/user-guides. Accessed 2023 Mar 24.
52.McClure NS, Sayah FA, Xie F, Luo N, Johnson JA. Instrument-Defined Estimates of the Minimally Important Difference for EQ-5D-5L Index Scores. Value Health. 2017;20(4):644-650. PubMed
53.Sehn LH, Martelli M, Trneny M, et al. A randomized, open-label, Phase III study of obinutuzumab or rituximab plus CHOP in patients with previously untreated diffuse large B-Cell lymphoma: final analysis of GOYA. J Hematol Oncol. 2020;13(1):71. PubMed
54.Sun J. A non-parametric test for interval-censored failure time data with application to AIDS studies. Stat Med. 1996;15(13):1387-1395. PubMed
55.Finkelstein DM. A proportional hazards model for interval-censored failure time data. Biometrics. 1986;42(4):845-854. PubMed
56.Friedman M. Piecewise Exponential Models for Survival Data with Covariates. The Annals of Statistics. 1982;10(1):101-113.
57.Royston P, Parmar MK. Flexible parametric proportional-hazards and proportional-odds models for censored survival data, with application to prognostic modelling and estimation of treatment effects. Stat Med. 2002;21(15):2175-2197. PubMed
58.Merrill RM, Timmreck TC. Introduction to Epidemiology. 4th ed. Burlington (MA): Jones & Bartlett Learning; 2006.
59.Shi Q, Schmitz N, Ou FS, et al. Progression-Free Survival as a Surrogate End Point for Overall Survival in First-Line Diffuse Large B-Cell Lymphoma: An Individual Patient-Level Analysis of Multiple Randomized Trials (SEAL). J Clin Oncol. 2018;36(25):2593-2602. PubMed
60.Maurer MJ, Habermann TM, Shi Q, et al. Utility of Progression-Free Survival at 24 Months (PFS24) to Predict Subsequent Outcome for Patients with Diffuse Large B-Cell Lymphoma (DLBCL) Enrolled on Randomized Clinical Trials: Findings from a Surrogate Endpoint in Aggressive Lymphoma (SEAL) Analysis of Individual Patient Data from 5853 Patients. Blood. 2016;128(22):3027-3027.
61.van der Galien HT, Hoogendoorn M, Kibbelaar RE, van Meerten T, van Rijn RS. Progression-free survival at 24 months (PFS24) and subsequent outcome for patients with diffuse large B-cell lymphoma (DLBCL) in the real-world setting. Ann Oncol. 2019;30(1):151-152. PubMed
Appendix 1: Detailed Outcome Data
Table 19: Censoring Rules for Primary Analysis of Investigator-Assessed PFS in POLARIX Study, Data Cut-Off Date June 15, 2022 — ITT Population
Scenarioa n (%) | Date of progression or censoring | Status | pola-R-CHP (n = 440) | R-CHOP (n = 439) |
|---|---|---|---|---|
No adequateb postbaseline assessment and no death | Randomization date | Censored | 7 (1.6) | 4 (0.9) |
No death and no disease progression before data cut-off | Date of last adequate assessment before data cut-off | Censored | 289 (65.8) | 318 (72.3) |
Withdrawal of treatment due to nonefficacy reason, no death, and no disease progression before data cut-off | Date of last adequate assessment before data cut-off | Censored | 10 (2.3) | 4 (0.9) |
Withdrawal of treatment due to nonefficacy reason, followed by disease progression or death | Date of earliest disease progression of death, before data cut-off | Event | 6 (1.4) | 5 (1.1) |
New anticancer treatmentc started due to efficacy reasons, followed by death or disease progression | Date of earliest disease progression of death, before data cut-off | Eventd,e | 6 (1.4) | 4 (0.9) |
New anticancer treatmentc started due to efficacy reasons, no death, or disease progression | Date of last adequate assessment before data cut-off | Censoredd,e | 2 (0.5) | 6 (1.4) |
New anticancer treatmentc started due to nonefficacy reasons, followed by death or disease progression | Date of earliest disease progression or death, before data cut-off | Evente | 13 (3.0) | 8 (1.8) |
New anticancer treatmentc started due to nonefficacy reasons, no death, or disease progression | Date of last adequate assessment before data cut-off | Censorede | 29 (6.6) | 19 (4.3) |
Death or disease progression following one or more consecutive missed assessmentsf | Date of earliest disease progression or death, before data cut-off | Event | 1 (0.2) | 5 (1.1) |
One or more missed assessments followed by no adequateb assessments or death | Date of last adequate assessment before data cut-off | Censored | 30 (6.8) | 27 (6.1) |
ITT = intention to treat; PFS = progression-free survival; pola-R-CHP = polatuzumab vedotin, rituximab, cyclophosphamide, doxorubicin, and prednisone; R-CHOP = rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone.
aSensitivity analyses may be performed for other situations if significant imbalances between groups are observed.
bTo be considered adequate, a tumour assessment not including PET should have CR, PR, SD and PD as outcome, and an assessment including PET-CT should have CMR, PMR, NMR, or PMD using Lugano criteria. Assessments that are “unevaluable” and “not done” were considered not adequate.
cNew anticancer treatment included all nonprotocol NALT for DLBCL. The protocol-permitted preplanned radiotherapy were not considered new anticancer treatment in any end point.
dIn sensitivity analyses, the impact of NALT before PD due to efficacy reason was assessed by discount method to investigate how the PFS results would have looked if the NALT was not available. More specifically, the time interval during which patients received NALT until the event or censoring time was discounted at 10%, 30%, and 50% for both groups. Note that the primary analysis of PFS corresponded to a discount analysis with a discount rate of 0% on PFS time after NALT.
eAs an additional sensitivity analysis to assess the overall impact of NALT, for patients who have taken NALT before or in the absence of subsequent death or disease progression, their PFS was censored at the time of their last adequate tumour assessment before the first NALT.
fThe impact of missing scheduled tumour assessments on PFS was assessed by performing a sensitivity analysis based on the interval censoring analysis methods.
Note that this table has not been copy-edited.
Source: POLARIX Clinical Study Report.13
Table 20: Additional Efficacy End Points in POLARIX Study, Data Cut-Off Date June 15, 2022 — ITT Population
Efficacy outcomes | Pola-R-CHP (N = 440) | R-CHOP (N = 439) |
|---|---|---|
Investigator-assessed EFSeffa | ||
Number of patients contributing to analysis | 440 | 439 |
Patients with event (%) | 124 (28.2) | 147 (33.5) |
Median time to EFSeff – Months (95% Cl) | NE (NE) | NE (NE) |
Stratified HR (95% Cl) | 0.79 (0.62 to 1.00) | Reference |
P value (log-rank)b | 0.0244 | Reference |
12-Month EFSeff rate (95% Cl) | 82.33 (78.72 to 85.94) | 78.48 (74.56 to 82.40) |
24-Month EFSeff rate (95% Cl) | 75.63 (71.55 to 79.71) | 69.54 (65.13 to 73.94) |
Investigator-assessed DFSa | ||
Number of patients contributing to analysis | 381 | 363 |
Patients with event, n (%) | 72 (18.9) | 89 (24.4) |
Median DFS, Months (95% Cl) | NE (NE) | NE (NE) |
Stratified HR (95% Cl) | 0.72 (0.53 to 0.99) | Reference |
12-Month DFS rate (95% Cl) | 90.08 (87.04 to 93.11) | 83.36 (79.45 to 87.27) |
24-Month DFS rate (95% Cl) | 81.79 (77.43 to 86.15) | 77.35 (72.73 to 81.96) |
Investigator-assessed DORa | ||
Number of patients contributing to analysis | 422 | 413 |
Patients with event, n (%) | 104 (24.6) | 125 (30.3) |
Median DOR, Months (95% CI) | NE (NE) | NE (NE) |
Stratified HR (95% Cl) | 0.77 (0.60 to 1.01) | Reference |
Unstratified HR (95% Cl) | 0.78 (0.60 to 1.02) | Reference |
12-Month DOR rate (95% Cl) | 83.81 (80.26 to 87.36) | 78.20 (74.15 to 82.25) |
24-Month DOR rate (95% Cl) | 75.67 (71.04 to 80.31) | 71.65 (67.10 to 76.20) |
EFSalla | ||
Patients with event, n (%) | 144 (32.7%) | 174 (39.6%) |
Median time to EFSall – Months (95% CI) | NE | NE |
Stratified HR (95% Cl) | 0.75 (0.60, 0.94) | Reference |
CI = confidence interval; DFS = disease-free survival; DOR = duration of response; EFSall = event-free survival from all causes; EFSeff = event-free survival–efficacy; HR = hazard ratio; NE = not estimable; pola-R-CHP = polatuzumab vedotin, rituximab, cyclophosphamide, doxorubicin, and prednisone; R-CHOP = rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone.
aSummaries of investigator-assessed EFSeff, DFS, DOR, and EFSall (median, percentiles) are Kaplan-Meier estimates. 95% Cl for median was computed using the method of Brookmeyer and Crowley. HRs were estimated by Cox regression.
bP value has been adjusted for multiple testing.
Note that this table has not been copy-edited.
Source: POLARIX Clinical Study Report.14
Additional Efficacy End Points
Investigator-Assessed Event-Free Survival (EFSeff)
EFSeff was a key secondary end point in POLARIX. EFSeff was defined as time from randomization to the earliest occurrence of disease progression or relapse, as assessed by the investigator, death, biopsy that is positive for residual disease after treatment completion, or start of a NALT due to efficacy reasons.
In the primary analysis (June 28, 2021), 112 patients (25.5%) in the pola-R-CHP group, and 138 patients (31.4%) in the R-CHOP group had an EFS event. Treatment of patients with previously untreated DLBCL with pola-R-CHP resulted in a statistically significant reduction in risk of disease progression or relapse, death, biopsy that is positive for residual disease after treatment completion, or start of a NALT due to efficacy reasons (stratified HR = 0.75; 95% CI, 0.58 to 0.96; P = 0.0244). At the time of the updated CCOD (June 15, 2022), 124 patients (28.2%) in the pola-R-CHP group, and 147 patients (33.5%) in the R-CHOP group had an EFS event. Results of the updated time-to-efficacy analysis of PFS were consistent with the primary analysis (stratified HR 0.79; 95% CI, 0.62 to 1.00).
Disease-Free Survival
DFS was defined as time from first occurrence of a documented CR to the date or relapse or death from any cause for the subgroup of patients with a BOR of CR, all assessed by the investigator. In the primary analysis (June 28, 2021), 62 patients (16.3%) in the pola-R-CHP group, and 79 patients (21.8%) in the R-CHOP group had progressed or died (stratified HR = 0.70; 95% CI, 0.50 to 0.98). At the time of the updated CCOD (June 15, 2022), treatment with pola-R-CHP among patients who experienced CR reduced the risk of progression or death (i.e., DFS) by 28% compared to treatment with R-CHOP (stratified HR: 0.72; 95% CI, 0.53 to 0.99).
Duration of Response
DOR was defined as time from first occurrence of a documented clinical response (CR or PR) to the date of progression, relapse, or death from any cause for the subgroup of patients with a BOR of CR or PR, all assessed by the investigator. In the primary analysis (June 28, 2021), 94 patients (22.3%) in the pola-R-CHP group, and 116 patients (28.1%) in the R-CHOP group had subsequent disease progression or death (stratified HR = 0.74; 95% CI, 0.56 to 0.98). At the time of the updated CCOD (June 15, 2022), treatment with pola-R-CHP among patients who experienced CR or PR reduced the risk of progression or death (i.e., DOR) by 23% compared to treatment with R-CHOP (stratified HR = 0.77; 95% CI, 0.60 to 1.01).
Event-Free Survival From All Causes
Event-free survival from all causes (EFSall) was defined as the time from randomization to disease progression or relapse, as determined by the investigator, death from any cause, or initiation of any NALT. In the primary analysis (June 28, 2021), 133 patients (30.2%) in the pola-R-CHP group, and 165 patients (37.6%) in the R-CHOP group had an EFSall event (stratified HR = 0.73; 95% CI, 0.58 to 0.92). At the time of the updated CCOD (June 15, 2022), the results for EFSall were consistent with the results for EFSeff (stratified HR = 0.75; 95% CI, 0.60 to 0.94).
DFS, DOR, and EFSall were (non-key) secondary end points that were not included in the statistical testing hierarchy.
Pharmacoeconomic Review
Abbreviations
AE
adverse event
BIA
budget impact analysis
CAR
chimeric antigen receptor
CUA
cost-utility analysis
DLBCL
diffuse large B-cell lymphoma
EBV
Epstein-Barr virus
GDP
gemcitabine, dexamethasone, and cisplatin
GemOx
gemcitabine and oxaliplatin
ICER
incremental cost-effectiveness ratio
IPI
International Prognostic Index
LBCL
large B-cell lymphoma
LY
life-year
NOS
not otherwise specified
OS
overall survival
PD
progressed disease
PFS
progression-free survival
pola-BR
polatuzumab, bendamustine, and rituximab
pola-R-CHP
polatuzumab vedotin, rituximab, cyclophosphamide, doxorubicin, and prednisone
PSM
partitioned survival model
QALY
quality-adjusted life-year
R-CHOP
rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone
R-CHP
rituximab, cyclophosphamide, doxorubicin, and prednisone
R-DHAP
rituximab, dexamethasone, cytarabine, and cisplatin
R-EPOCH
rituximab, etoposide, prednisone, vincristine, cyclophosphamide, and doxorubicin
R-GDP
rituximab, gemcitabine, dexamethasone, and cisplatin
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Polatuzumab vedotin (Polivy), 30 mg or 140 mg single-use vial, lyophilized powder for solution for IV infusion |
Submitted price | Polatuzumab vedotin: $3,160.71 per 30 mg vial or $14,750.00 per 140 mg vial |
Indication | In combination with rituximab, cyclophosphamide, doxorubicin, and prednisone (R-CHP) is indicated for the treatment of adult patients with previously untreated large B-cell lymphoma (LBCL), including diffuse large B-cell lymphoma (DLBCL) not otherwise specified (NOS), high grade B-cell lymphoma, Epstein-Barr virus-positive (EBV+) DLBCL NOS, and T-cell/histiocyte rich LBCL. |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | November 14, 2022 |
Reimbursement request | As per indication |
Sponsor | Hoffmann-La Roche Limited |
Submission history | Previously reviewed: Yes Indication: Relapsed or refractory DLBCL Recommendation date: April 21, 2021 Recommendation: Reimburse with clinical criteria and/or conditions |
DLBCL = diffuse large B-cell lymphoma; EBV = Epstein-Barr virus; LBCL = large B-cell lymphoma; NOC = Notice of Compliance; NOS = not otherwise specified; R-CHP = rituximab, cyclophosphamide, doxorubicin, and prednisone.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis PSM |
Target populations | Adult patients with previously untreated LBCL, including DLBCL NOS, high-grade B-cell lymphoma, Epstein-Barr virus-positive DLBCL NOS, and T-cell/histiocyte-rich LBCL |
Treatment | pola-R-CHP |
Comparator | R-CHOP |
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | Lifetime (60 years) |
Key data sources | POLARIX trial to inform PFS and OS for pola-R-CHP vs. R-CHOP GOYA trial extension used to support long-term extrapolation of PFS |
Submitted results | ICER = $86,627 per QALY gained (0.44 incremental QALYs; $37,868 incremental cost) |
Key limitations |
|
CADTH reanalysis results |
|
DLBCL = diffuse large B-cell lymphoma; ICER = incremental cost-effectiveness ratio; IPI = International Prognostic Index; KM = Kaplan-Meier; LBCL = large B-cell lymphoma; LY = life-year; NOS = not otherwise specified; OS = overall survival; PFS = progression-free survival; pola-R-CHP = polatuzumab vedotin, rituximab, cyclophosphamide, doxorubicin, and prednisone; PSM = partitioned survival model; QALY = quality-adjusted life-year; R-CHOP = rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone; vs. = versus.
Conclusions
Based on the CADTH clinical review of the POLARIX trial, polatuzumab vedotin, rituximab, cyclophosphamide, doxorubicin, and prednisone (pola-R-CHP) provided greater efficacy for progression-free survival (PFS) compared with the standard of care of rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP); however, whether the magnitude of benefit is clinically meaningful and whether such a benefit is maintained beyond the trial period is uncertain. The available overall survival (OS) data from the POLARIX trial did not demonstrate a difference during the observation period (median OS follow-up = 39.7 months), and CADTH’s clinical review reported that OS results from the POLARIX trial were uninterpretable. Overall, the safety profile of pola-R-CHP was comparable to R-CHOP. The CADTH clinical review also noted that the study findings were limited in generalizability.
CADTH identified several limitations in the sponsor’s pharmacoeconomic analysis that have notable implications on the cost-effectiveness of pola-R-CHP. For the CADTH base-case analysis, CADTH revised the assumptions regarding PFS extrapolation such that only data from the POLARIX study were used and no Kaplan-Meier data for PFS were applied directly in the model, which led to more plausible estimates of PFS; incorporated a waning of effect of pola-R-CHP on OS; changed the time point at which the model transitioned from Kaplan-Meier data to parametric survival estimates for OS to better reflect the available OS evidence, as well as the availability of second-line curative therapies; aligned the number and distribution of subsequent therapies for both pola-R-CHP and R-CHOP to be nearly equivalent and aligned with expectations of clinical practice in Canada; accounted for drug wastage; and incorporated additional chair time required for pola-R-CHP. The CADTH base-case analysis resulted in an incremental cost-effectiveness ratio (ICER) of $394,163 per quality-adjusted life-year (QALY) gained (incremental costs = $76,379; incremental QALYs = 0.19) for pola-R-CHP versus R-CHOP. The probability of pola-R-CHP being cost-effective at a $50,000 per QALY gained threshold was 6%. Based on CADTH’s reanalysis, in order for pola-R-CHP to be considered cost-effective compared to R-CHOP at a $50,000 per QALY gained threshold, the price of polatuzumab vedotin would need to be less than $5,015 per 21-day cycle, reflecting a price reduction of 66%.
Although the CADTH reanalysis attempted to address the identified limitations of the sponsor’s economic submission, some uncertainty remains. First, the misalignment of the trial population with the indicated population could not be addressed, and the cost-effectiveness of pola-R-CHP in some patient populations remains unknown. Specifically, uncertainty remains for patients with an International Prognostic Index (IPI) of 0 to 1 who were not included in the trial, and in situations where there is a standard of care that is different from R-CHOP. Second, due to structural limitations of the partitioned survival model (PSM) approach, only the costs of subsequent therapies are modelled and not changes in OS resulting from curative subsequent therapies. This structural assumption favours pola-R-CHP, given that QALY gains in the progressed disease (PD) health state would be underestimated for all patients with PD, of which there were more for R-CHOP. As this limitation could not be addressed, any analyses performed by CADTH or the sponsor are likely to underestimate the true ICER. Further price reductions (i.e., > 66%) may therefore be required.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process.
Patient input was received from Lymphoma Canada, which collected input from 89 patients diagnosed with large B-cell lymphoma (LBCL), the majority of whom were living in Canada, via an anonymous online survey. Patients reported that at the time of diagnosis they experienced several physical and psychosocial struggles that impacted their quality of life. Patients also reported that they experience mental health challenges associated with their cancer diagnosis including fear of progression or relapse. With regard to first-line treatment, almost all patients had some form of chemotherapy or chemoimmunotherapy and reported that they were overall satisfied with first-line treatment options. However, some patients did report they faced challenges and delays accessing first-line treatment due to living far from a hospital, being unsatisfied with the number of options available to them, and treatment waitlists. Input was gathered from 4 patients who had experience with polatuzumab vedotin for the first-line treatment of LBCL. Three of these patients would recommend pola-R-CHP to other patients with LBCL, and 2 patients reported that their overall experience was very good. The most common side effects reported by those treated with pola-R-CHP included fatigue, neutropenia, thrombocytopenia, and diarrhea.
Clinician input was received from the Canadian Hematologists/Oncologists Treating DLBCL (diffuse large B-cell lymphoma) and Ontario Health (Cancer Care Ontario) Hematology Cancer Drug Advisory Committee. The clinician input highlighted the first-line treatment goal is to cure LBCL and prevent the need for subsequent therapies. However, clinician input indicated that approximately 40% of patients will relapse or are refractory to the current first-line treatment and that the best chance of cure for patients with DLBCL is with first-line treatment, as opposed to subsequent therapies. The clinician feedback also highlighted the benefit of first-line cure due to concerns with worse quality of life due to higher toxicity associated with subsequent therapies. The clinician input indicated that pola-R-CHP’s place in therapy would be as an additional first-line option and that it would be appropriate to treat people who are eligible for the current first-line treatment with R-CHOP. Regarding safety, the clinician input noted that pola-R-CHP has a similar safety profile to R-CHOP.
Drug plan input raised questions regarding the eligibility and dosing of pola-R-CHP with regard to pediatric populations, older adults (who sometimes receive a reduced R-CHOP dose, known as mini-R-CHOP), transplant eligibility, and other types of patients with lymphoma. Additionally, the drug plan input noted the additional pharmacy workload and chair time compared to the current standard of care that would be required if the additional 2 cycles (7 and 8) with rituximab monotherapy as implemented in the POLARIX trial would be applied in Canada. Additional chair time would also be required for pola-R-CHP compared to the current standard of care due to the length of infusion and monitoring requirements of polatuzumab vedotin, and that some outpatient centres might have difficulty administering the first cycle within an 8-hour time frame. With regards to drug administration, drug plan input noted concerns about jurisdictions with centralized production due to the product monograph instructions for transportation of the prepared solution. Finally, the drug plans also noted that a substantial budget impact is anticipated due to the introduction of polatuzumab vedotin in a first-line regimen.
Several of these concerns were addressed in the sponsor’s model:
The sponsor’s submitted model accounted for quality of life and length of life.
The additional 2 cycles of rituximab monotherapy could be included or excluded in the submitted model.
In addition, CADTH addressed some of these concerns as follows:
the inclusion of additional chair time associated with pola-R-CHP.
CADTH was unable to address the following concern raised from stakeholder input:
the populations of interest that were not modelled and/or there was no clinical evidence for, including older adults receiving mini-R-CHOP, pediatric populations, and other types of patients with lymphoma.
Economic Review
The current review is for polatuzumab vedotin (Polivy), as used in the pola-R-CHP regimen, for the treatment of adult patients with previously untreated LBCL, including DLBCL not otherwise specified (NOS), high-grade B-cell lymphoma, Epstein-Barr virus (EBV)-positive DLBCL NOS, and T-cell/histiocyte-rich LBCL.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis (CUA) comparing costs and outcomes for pola-R-CHP with the current standard of care, R-CHOP. The model population comprised adults with previously untreated LBCL, including DLBCL NOS, high-grade B-cell lymphoma, EBV-positive DLBCL NOS, and T-cell/histiocyte-rich LBCL. The modelled population was aligned with the Health Canada–approved indication and the sponsor’s reimbursement request. The POLARIX trial population informing the sponsor’s model was narrower than the Health Canada–approved indication and modelled the population by restricting eligibility to patients with Eastern Cooperative Oncology Group Performance Status 0 to 2 and IPI 2 to 5.
Polatuzumab vedotin is administered intravenously, with the initial dose administered as a 90-minute infusion followed by 90 minutes of observation. Subsequent doses may be administered as 30-minute infusions with a 30-minute observation. The recommended dose of polatuzumab vedotin is 1.80 mg/kg every 21 days for a maximum of 6 cycles, in combination with rituximab, cyclophosphamide, doxorubicin, and prednisone (R-CHP).1 Polatuzumab vedotin is provided in either 30 mg or 140 mg vials, at a submitted price of $3,160.71 and $14,750.00 per vial, respectively. The cost per 21-day cycle of polatuzumab vedotin, based on an average patient weight of 75.92 kg was estimated to be $14,397.76; however, this included an assumption of perfect vial sharing. The treatment cost for six 21-day cycles of polatuzumab vedotin is $86,386.56. When used in combination with R-CHP, the cost per 21-day cycle was $17,243.84, for a total 6-cycle cost of $103,463.04. In addition to drug costs, there are associated administration and monitoring costs.
For the base-case analysis, the sponsor considered R-CHOP the only relevant comparator. The model used a 1-week cycle length and simulated costs, life-years (LYs), and QALYs for each treatment regimen, over a lifetime time horizon (60 years) from the perspective of Canada’s publicly funded health care system. Costs and QALYs were discounted at a rate of 1.5% per annum, and a half-cycle correction was applied.
Model Structure
The sponsor submitted a PSM with the following health states: PFS, PD, and death (Figure 1). All patients entered the model in the PFS state and could remain in the PFS state or transition to the PD or death health states each cycle. Patients in the PD health state could remain in the PD state or transition to the death state. The proportion of patients who were progression-free, had PD, or were dead at any time was derived from Kaplan-Meier data and survival extrapolations informed by trial data. Using a mixture cure model, a long-term remission fraction was derived from trial data and applied to the model to inform the proportion of patients experiencing long-term remission and then subsequently experiencing mortality aligned with the age-matched and sex-matched general population.
Model Inputs
The baseline population characteristics used to inform the model were based on the POLARIX trial. The age distribution applied in the model used a mean age of 63 years (range, 18 to 80). The mean height and weight were 167.97 cm (standard deviation = 10.23 cm) and 75.92 kg (standard deviation = 19.35 kg), respectively.
Clinical efficacy for both pola-R-CHP and R-CHOP was informed by the POLARIX trial (data cut-off: June 2022) and data from the GOYA trial was used to support PFS extrapolation. The POLARIX trial had a median duration of PFS follow-up of 30.9 months (range, 0 months to 46 months) in the pola-R-CHP arm and 30.8 months in the R-CHOP arm (range, 0 months to 54 months).2 The GOYA trial, a phase III study that compared the efficacy of obinutuzumab with cyclophosphamide, doxorubicin, vincristine, and prednisone against R-CHOP in patients with previously untreated DLBCL, had a median observation time of 47.4 months.3 The GOYA trial data were aggregated with the POLARIX study data using propensity score weighting to control for clinical baseline characteristics or geographical regions across trials.2 The GOYA-extended POLARIX trial data were used to estimate the long-term remission fractions for each treatment, which assumed that patients who would not progress could be considered long-term remission patients (i.e., cured). In this approach, the PFS is calculated as a product of background survival (for the proportion of patients estimated to be in long-term remission) and the cancer-specific survival (for those who do not experience long-term remission).4
In the submitted model, Kaplan-Meier data were used directly to inform PFS until 42 months. Following 42 months, the sponsor applied parametric survival curves as tails to the Kaplan-Meier data, while accounting for the long-term remission fraction. The base-case parametric functions were selected based on visual inspection of fit, statistical goodness of fit, and clinical plausibility. The sponsor chose the generalized gamma function to extrapolate PFS beginning at 42 months and assumed an equal treatment effect for both treatments from this time onward. The sponsor also included the option to model PFS using POLARIX trial data alone.
To model OS, the sponsor used a PFS-informed OS approach. Due to the immaturity of OS data in the trial, the long-term remission fraction that was estimated from PFS was used to extrapolate OS curves from 42 months, before which Kaplan-Meier data were directly applied. The generalized gamma curve was selected for the sponsor’s base case for both arms. Similar to PFS, equal treatment effects were assumed from 42 months onward.
In the submitted model, the sponsor assumed that all patients would receive 6 cycles of pola-R-CHP. The sponsor’s base case assumed that 8% of patients would receive 3 cycles of R-CHOP and the remainder would receive 6 cycles, resulting in an average of 5.76 cycles. Drug acquisition costs for comparators were obtained from the IQVIA Delta PA database.5 Perfect vial sharing was assumed (i.e., no drug wastage was accounted for). The sponsor’s base case assumed that all IV drugs would be administered at the same time, and the cost per hour of chair time was obtained from the literature.6 Routine monitoring costs were informed by a literature review and costs were obtained from the Ontario Schedule of Benefits for Laboratory Services,7 the Ontario Case Costing Initiative database,8 and the literature.
Health state utility values for the PFS and PD states were obtained from the GOYA trial. The sponsor used propensity score weighting to adjust the original GOYA study sample size to 1 that has a population more similar to the POLARIX trial. After weighting, the utility values were 0.82 and 0.73 for the PFS and PD health states, respectively. Utility values derived from alternative sources, including those collected from the POLARIX trial, were assessed by the sponsor, and tested in scenario analyses.9 In the model, everyone who reaches 2 years after treatment completion in the PFS health state switches to age-specific and sex-specific utility norms for the population of people living in Canada.10,11
The model included grade 3 to 5 adverse events (AEs) with an incidence of greater than 2% in either POLARIX trial arm. The value of the utility decrements associated with each AE was informed from the National Institute for Health and Care Excellence technology appraisal of pixantrone monotherapy for treating relapsed or refractory aggressive non-Hodgkin B-cell lymphoma, and the decrement took into account the AE duration from the same appraisal.12 Inpatient and outpatient AE costs were obtained from the Ontario Case Costing Initiative database.8 The proportion of patients treated as inpatients and outpatients for each AE was estimated from clinician input. Resource use costs and disutilities for AEs were applied as a 1-time event during the first treatment cycle in the model.
The model included subsequent therapies for patients who entered the PD health state. Subsequent treatment options included autologous stem cell transplant, chimeric antigen receptor (CAR) T-cell therapy, rituximab, gemcitabine, dexamethasone, and cisplatin (R-GDP), gemcitabine, dexamethasone, and cisplatin (GDP), gemcitabine and oxaliplatin (GemOx), polatuzumab vedotin, bendamustine, and rituximab (pola-BR), and allogenic hematopoietic cell transplant. Distributions and number of subsequent therapies specific to the first-line treatment option received were informed from the POLARIX trial. Subsequent therapy costs were calculated as a weighted average based on the proportion of patients expected to be treated in each arm, and then multiplied by the number of subsequent treatment cycles expected (2.03 in the pola-R-CHP arm and 2.26 in the R-CHOP arm). The cost was applied as a 1-time cost at the time of progression, applying a cost of $118,460.64 for pola-R-CHP and $172,821.25 for R-CHOP. A 1-time end-of-life cost of $39,367 was applied to both arms for those who died of DLBCL.13,14
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically (500 iterations for the base-case and scenario analyses). The deterministic and probabilistic results were similar. The probabilistic findings are presented subsequently.
Base-Case Results
In the sponsor’s base-case analysis, treatment with pola-R-CHP was associated with incremental costs of $37,868 and a gain of 0.44 QALYs compared with R-CHOP over the lifetime time horizon, resulting in an ICER of $86,627 per QALY gained (Table 3). The probability of pola-R-CHP being cost-effective at a $50,000 per QALY threshold compared to R-CHOP was 25%. Approximately 77% of the incremental QALYs in the sponsor’s base case were accrued beyond 46 months, the maximum follow-up of pola-R-CHP in the POLARIX trial. The submitted analysis is based on the publicly available list prices of all treatments, other than polatuzumab vedotin, including subsequent therapies.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER vs. reference ($/QALY) |
|---|---|---|---|---|---|
R-CHOP | 150,896 | Reference | 11.91 | Reference | Reference |
pola-R-CHP | 188,764 | 37,868 | 12.34 | 0.44 | 86,627 |
ICER = incremental cost-effectiveness ratio; pola-R-CHP = polatuzumab vedotin, rituximab, cyclophosphamide, doxorubicin, and prednisone; QALY = quality-adjusted life-year; R-CHOP = rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone; vs. = versus.
Note: The submitted analysis is based on the publicly available prices of the comparator treatments.
Source: Sponsor’s pharmacoeconomic submission.2
Sensitivity and Scenario Analysis Results
The sponsor conducted several scenario and sensitivity analyses testing alternative parameter values and assumptions. These included: excluding the use of efficacy data from the GOYA trial to inform long-term PFS and OS extrapolation, waning the duration of treatment effect over time, reducing the amount of vial sharing to account for some drug wastage, and testing alternate subsequent therapy usage. The sponsor’s base case was most influenced by assuming that the treatment effect is null after 36 months, with an ICER of $135,183 per QALY. Additional influential analyses included excluding the GOYA trial results, which resulted in an ICER of $131,193 per QALY, and assuming equivalent subsequent therapy usage regardless of first-line therapy received, resulting in an ICER of $116,219 per QALY.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis.
There is substantial uncertainty regarding PFS as modelled. The accuracy of the submitted model’s estimates of PFS over the model’s lifetime time horizon was uncertain due to key assumptions and methodological choices. First, the model incorporated an external data source, the GOYA trial, to extend the follow-up time of PFS for R-CHOP and fit the parametric distributions for both treatment groups. The inclusion of the GOYA trial data is difficult to validate in the context of this model and was not included in the CADTH clinical review appraisal. Additionally, using external data in the model has introduced additional uncertainty into the clinical parameters and extrapolations. Given that the follow-up time for PFS in the POLARIX trial was long enough to capture the majority of cures (i.e., longer than 2 years), there was inadequate justification to introduce the GOYA data and the accompanying uncertainty, particularly as it influenced the efficacy of pola-R-CHP in the model, which was not evaluated in the GOYA trial. The second limitation in the way in which PFS was modelled in the submitted evaluation was the use of Kaplan-Meier data directly from the trial until 42 months, instead of using a more robust parametric distribution for the entirety of the model time horizon. It is generally more accepted to use the extrapolated parametric distributions over the entire model time horizon as it results in more consistent hazard estimates, and avoids limitations around a small number of events at later times points of observed data, the stepped-nature of the intervals for assessment in trials and restricted mean estimates using incomplete data.15 Additional uncertainty around the magnitude of benefit of pola-R-CHP on PFS benefits, as well as the clinical meaningfulness of these benefits, was noted in CADTH’s clinical review, highlighting specific issues around the magnitude of benefit with widening confidence intervals at later time points in the trial follow-up period.
The CADTH base-case reanalysis did not use the GOYA trial extension data and removed the direct application of Kaplan-Meier data in the model. CADTH maintained the sponsor’s choice of the generalized gamma distribution to model PFS for both treatments.
CADTH ran a scenario analysis using the GOYA trial data to extend PFS follow-up time for data informing extrapolation for R-CHOP and support the extrapolation of PFS for pola-R-CHP.
The extrapolation of OS is uncertain. The submitted pharmacoeconomic model predicted a gain in LYs, due in part to the PFS-informed OS approach in the sponsor’s model, as well as some of the sponsor’s assumptions and methodological choices. CADTH identified several sources of uncertainty with regards to OS in the submitted model that further propagated the uncertainty of the predicted OS benefit. First, there is general uncertainty of an OS benefit with pola-R-CHP given that no OS benefit was observed in the available follow-up period in the POLARIX trial, and that some relevant long-term outcomes of pola-R-CHP that may impact OS have not yet been able to be assessed due to the duration of the trial. Due to the immaturity of OS data in the trial, the long-term remission fraction that was estimated from PFS was used to extrapolate OS curves from 42 months, before which Kaplan-Meier data were directly applied. This approach makes it such that the limitations and uncertainties related to PFS described previously are propagated onto the OS estimates in the model, including the general uncertainty of the magnitude of PFS benefit for pola-R-CHP in comparison with R-CHOP. Furthermore, there is uncertainty that an OS benefit would be realized even if trial follow-up had been long enough. The clinical experts consulted by CADTH indicated that it remains unknown whether first-line treatment with pola-R-CHP will have an impact on subsequent therapy efficacy for any of the currently used treatments (e.g., that subsequent therapy becomes more or less effective in patients who have been treated with pola-R-CHP) and that patients with PD may receive subsequent therapies that are curative and improve their chances of OS.
Beyond the issues with the PFS-informed OS approach, there were other limitations with the implementation of OS in the sponsor’s model. While the use of Kaplan-Meier data was viewed as inappropriate for the PFS estimates, the parametric distributions for OS extrapolations inappropriately separated the OS estimates for pola-R-CHP and R-CHOP during the trial period despite observing no separation of OS in the trial. For this reason, using Kaplan-Meier data directly provides more accurate estimates of OS. However, the time at which the model switches from Kaplan-Meier data to the parametric survival curve is influential. The sponsor applied Kaplan-Meier data up to 42 months; however, at this time point there were few patients at risk and few events taking place and clinical experts consulted by CADTH indicated that the Kaplan-Meier results become less certain at the time when the number at risk in the trial drops off. In the trial, between 36 and 42 months, the number of patients at risk changes from 338 to 129 for the pola-R-CHP trial arm.2
Further uncertainty in the extrapolation of OS arises with the assumption made by the sponsor that there is an indefinite treatment effect of pola-R-CHP on OS. Beyond the general uncertainty of an OS benefit as observed in the POLARIX trial, the statistical properties inherent in extrapolating OS data within a PSM over a lifetime time horizon contributes to the uncertainty in this extrapolation.16 When basing the extrapolation of OS for patients who experienced disease progression on the within-trial time horizon where the trial data has demonstrated a treatment benefit on PFS but not OS (as is the case with pola-R-CHP), it may lead to an overestimation of the OS benefit for the treatment with fewer progressed patients (i.e., pola-R-CHP). Clinical experts consulted by CADTH agreed that the assumption that the OS benefit of pola-R-CHP is indefinite is unrealistic and that over time the hazard ratio for OS will converge to 1 (i.e., approach no difference in OS over time) due to uncertainty of the overall OS benefit of pola-R-CHP and the use of curative-intent subsequent therapies.
The CADTH reanalysis maintained the use of Kaplan-Meier data in the model; however, only up until 39.7 months. This time point was chosen based on the decline in number of patients at risk declining significantly between 36 months and 42 months and aligning with the median OS follow-up time for pola-R-CHP from the POLARIX trial.
Given the influence of the time point at which Kaplan-Meier data are applied directly in the model, and that OS Kaplan-Meier data have many time points that are overlapping or where the OS data crosses over between treatment arms, CADTH conducted a scenario analysis using Kaplan-Meier data in the model until 30 months. This represents another time point that the clinical experts consulted by CADTH suggested after which the trial data becomes more uncertain.
The CADTH reanalysis assumed that the treatment duration would begin to wane at 30 months and be null by 60 months. This change produced extrapolated OS distributions that were more closely aligned with the clinical expectations of the clinical experts consulted by CADTH as the OS hazards of the comparators moved closer together over time.
The model structure was inappropriate for the decision problem. The sponsor used a PSM to estimate costs and outcomes associated with first-line treatment for LBCL. Although PSMs are routinely used to model oncology treatments, this approach was not suitable for this decision problem where the primary goal of both first-line and subsequent treatments is to achieve a cure. The model failed to account for patients who experienced long-term remission (i.e., were cured) with subsequent lines of therapy, and did not capture changes in OS or quality of life for these patients. While cure from first-line treatment is an important treatment goal, clinical experts consulted by CADTH estimate that an additional 25% to 50% of patients could be cured with salvage therapy depending on the subsequent treatment received (e.g., approximately 40% may experience a cure with subsequent CAR T-cell therapy and 40% may be cured following transplant). Therefore, the magnitude of the benefit in the extrapolated period was inaccurately estimated, as the model did not allow for the possibility of future cures and the related change in OS and quality of life. A more appropriate model for this decision problem would have been a Markov model with a long-term remission state, which could incorporate different survival assumptions based on subsequent therapy.
CADTH was unable to address this limitation within the submitted model.
The generalizability of the modelled population to the Health Canada–indicated population is uncertain. The sponsor submitted a model based on the efficacy data collected in the POLARIX trial. The trial population differs from that of the sponsor’s reimbursement request and the Health Canada–indicated population. Key generalizability issues arise from the clinical eligibility criteria for participation in the POLARIX trial and have been discussed in CADTH’s clinical review. In general, the clinical experts consulted by CADTH noted that patients initiating treatment with current standard first-line R-CHOP for DLBCL often present with Eastern Cooperative Oncology Group Performance Status of greater than 2 and therefore would be considered eligible for treatment with pola-R-CHP by clinicians. As well, those with transformed indolent lymphoma, including follicular lymphoma grade 3B and primary cutaneous DLBCL would be eligible for pola-R-CHP in practice yet were excluded from the POLARIX study. With regards to IPI, the trial included those with an IPI of 2 to 5 while the indicated population includes IPI 0 to 5. The clinical experts consulted by CADTH expressed uncertainty regarding the magnitude of benefit of pola-R-CHP for patients with an IPI score of 2 and indicated that treatment with pola-R-CHP would be focused on patients with an IPI of 3 to 5 due to poor cure rates with current treatment options in comparison with patients with better IPI scores. This expectation may be supported by exploratory trial findings suggesting that the true benefit of pola-R-CHP is in patients with IPI 3 to 5 and no bulky disease, and not those with an IPI of 2, as reported in CADTH’s clinical review. The submitted model does not allow for the assessment of the cost-effectiveness of pola-R-CHP for IPI 3 to 5. The POLARIX trial also included patients with double-hit and triple-hit lymphoma for whom first-line treatment is often rituximab, etoposide, prednisone, vincristine, cyclophosphamide, and doxorubicin (R-EPOCH) rather than R-CHOP,17 and the model did not include this as a comparator in the model.
CADTH could not address this limitation given the lack of clinical data on the efficacy of pola-R-CHP in all indicated patient populations. As such, the cost-effectiveness of pola-R-CHP in these populations or in comparison with relevant comparators other than R-CHOP is unknown.
Subsequent therapy assumptions were not reflective of clinical practice in Canada. In the submitted model, at the time of disease progression, a 1-time cost of subsequent therapy was applied based on the weighted cost of each subsequent therapy and the proportion of patients that received it. The sponsor assumed that the number and distribution of subsequent therapies received postprogression matched that of the POLARIX trial. In the POLARIX trial, patients who received pola-R-CHP had an average of 2.03 subsequent therapies, and patients who received R-CHOP had an average of 2.26 subsequent therapies. The distribution of which subsequent therapies were received also differed by first-line treatment group. Clinical experts consulted by CADTH indicated that in clinical practice in Canada, the approach to subsequent therapy would not differ between first-line therapy (i.e., whether a patient receives pola-R-CHP or R-CHOP) and they expected the distribution and number of subsequent therapies would be nearly equivalent between groups. They did, however, acknowledge that patients who received pola-R-CHP would not likely be treated with pola-BR subsequently, but that it would be used for patients who receive R-CHOP first-line. Additionally, clinical experts agreed that rituximab in combination with dexamethasone, cytarabine, and cisplatin (R-DHAP) is used in 1% of patients as salvage therapy, and that allogenic hematopoietic cell transplant is no longer used in clinical practice in Canada.
To address this limitation, the CADTH base case assumed that the number of subsequent therapies postprogression was equivalent for each treatment group. Additionally, CADTH adjusted the proportions of patients receiving each subsequent therapy to align with clinical practice in Canada, assuming nearly equivalent proportions for each therapy with the exception of 0% and 3% of patients receiving pola-BR following treatment with pola-R-CHP and R-CHOP, respectively (Table 13). The CADTH base case also assumed that 0% of patients receive allogenic hematopoietic cell transplant, and that 1% of patients from each group receive R-DHAP as subsequent therapy.
The assumption that perfect vial sharing would take place was inappropriate. In the submitted pharmacoeconomic model, the sponsor assumed perfect vial sharing would occur. This assumption is inappropriate because of differences in vial sharing abilities by jurisdiction due to differing patient volumes. Further, the product monograph for polatuzumab vedotin indicates that the reconstituted product is intended for single use only and to discard any unused portion.1 The submitted model does not allow the user to specify vial sharing for individual drugs, and so the vial sharing assumption applied to every drug included in pola-R-CHP and R-CHOP regimens.
The CADTH reanalysis assumed 0% vial sharing. While some of the included drugs might be likely to be shared, the implications of vial sharing for polatuzumab vedotin are more significant given the higher relative cost of the drug and so adhering to the product monograph for polatuzumab vedotin was considered to be the most appropriate assumption in the base case.
A scenario analysis that allowed for 50% of vial sharing was also included to account for the possibility that some treatment centres will be able to cluster patients to allow for vial sharing.
Poor modelling practices were employed. The sponsor’s submitted model included numerous IFERROR statements, which lead to situations in which the parameter value is overwritten with an alternative value without alerting the user to the automated overwriting. The systematic use of IFERROR statements makes thorough auditing of the sponsor’s model impractical and it remains unclear whether the model is running inappropriately by overriding errors.
CADTH was unable to address this limitation and notes that a thorough validation of the sponsor’s model was not possible.
Additional limitations were identified but were not considered to be key limitations. These limitations are outlined subsequently.
Administration costs are not appropriately captured. The sponsor assumed that all IV medications would be delivered simultaneously for both pola-R-CHP and R-CHOP, and that the total chair time was equivalent to that of the longest infusion (i.e., rituximab). This resulted in the assumption that the required chair time for both regimens was 4.25 hours for the first infusion, and 1.5 hours for subsequent infusions. However, drug plan input and clinical expert feedback obtained by CADTH indicated that IV medications would be administered sequentially, and that the administration of polatuzumab vedotin with R-CHP will require more chair time than the R-CHOP regimen. The additional chair time is associated with the infusion time being longer for polatuzumab vedotin than the drug it replaced in R-CHOP (vincristine), and due to the required observation time of polatuzumab vedotin (1.5 hours and 0.5 hours observation for the first and subsequent infusions, respectively). In fact, drug plan input and the clinical experts consulted by CADTH estimated that the first cycle of pola-R-CHP could take up to 10 hours, and that some outpatient treatment facilities are unable to accommodate a treatment day of that length.
The CADTH reanalysis assumed that IV treatments would be administered sequentially for both pola-R-CHP and R-CHOP and accounted for the additional observation time required to administer polatuzumab vedotin. The incremental difference in chair time for pola-R-CHP compared to R-CHOP is 2.83 hours, which is the difference in administration time when replacing vincristine (0.17-hour infusion time) with polatuzumab vedotin (3 hours and 1 hour for first and subsequent infusions, respectively).
CADTH was unable to address the implications of the total administration time for the first cycle potentially being longer than some jurisdictional outpatient facilities are currently able to accommodate and the economic implications (e.g., additional staffing to keep facility open longer).
Additionally, the following key assumptions were made by the sponsor and have been appraised by CADTH (refer to Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CADTH comment |
|---|---|
Patients who survive and have not experienced disease progression at 2.4 years are assumed to be in long-term remission. | Acceptable. Clinical experts consulted by CADTH noted that while relapse can take place beyond 2 years after treatment completion, it is rare, and they agreed that this is an acceptable assumption. |
Health-related quality of life returned to that of the general population at 2.4 years after treatment initiation for those who remained progression-free. | Uncertain. Clinical experts consulted by CADTH indicated that it is not likely that utility values would return to that of the general population at this time point due to ongoing anxiety of potential relapse and potential health effects because of toxic treatments. However, the impact on the results of the model is small when the time point is adjusted to later dates. |
Mortality risk returned to that of the general population at 2.4 years after treatment initiation for those that remained progression-free. | Uncertain. There is some uncertainty around loss of life expectancy for patients who have had LBCL based on disease characteristics and age at diagnosis. However, adjusting the mortality hazard for the cured population relative to the general population mortality estimates has a minimal impact on the results of the model. |
Price of rituximab biosimilar is used, as opposed to the brand price. | Acceptable. The biosimilar rituximab product is available and widely used in Canadian jurisdictions. |
Rituximab assumed to be administered IV rather than subcutaneously in subsequent cycles. | Acceptable. This assumption was applied for both pola-R-CHP and R-CHOP. |
Health resource utilization for patients were derived from the NICE technology appraisal for pola-R-CHP. | Ideally, Canadian resource use estimates would be used; however, the impact of these estimates on the model results is small. |
LBCL = large B-cell lymphoma; NICE = National Institute for Health and Care Excellence; pola-R-CHP = polatuzumab vedotin, rituximab, cyclophosphamide, doxorubicin, and prednisone; R-CHOP = rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone.
CADTH Reanalyses of the Economic Evaluation
Base-Case Results
The CADTH base case was derived by making changes in model parameter values and assumptions, in consultation with clinical experts. These changes, summarized in Table 5, included removal of the GOYA trial extension data and Kaplan-Meier data from PFS, adjustments to OS Kaplan-Meier data and treatment effect duration, modifications to subsequent therapy use, and changes to assumptions about vial sharing and administration times.
Table 5: CADTH Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
None. | — | — |
Changes to derive the CADTH base case | ||
1. Use of the GOYA extension trial to extrapolate PFS | Used PFS extrapolations informed by GOYA trial. | Used PFS extrapolations based solely on the POLARIX trial which excluded data from the GOYA trial. |
2. Use of KM data when modelling PFS | Applied KM data until 42 months. | No KM data applied. |
3. OS treatment effect | Indefinite treatment effect. | Treatment waning beginning at 30 months, treatment effect null at 60 months. |
4. OS KM data | Applied KM data until 42 months. | Applied KM data for 39.7 months. |
5. Subsequent therapies | Average number of subsequent therapies following disease progression different for R-CHOP (2.26) and pola-R-CHP (2.03), and distribution of subsequent therapies aligned with POLARIX trial (refer to Table 11). | Average number of subsequent therapies following disease progression equivalent for R-CHOP (2.03) and pola-R-CHP (2.03), and nearly equivalent distribution of subsequent therapies aligned with clinical practice in Canada (refer to Table 13). |
6. Vial sharing | 100% | 0% |
7. Administration costs | Simultaneous administration of all drugs, total chair time equivalent to that of the longest infusion for both regimens (i.e., rituximab, 4.25 hours and 1.5 hours for first and subsequent infusions, respectively). | Sequential administration of all drugs for both regimens. Added observation time (1.5 hours and 0.5 hours for first and subsequent infusions, respectively) for polatuzumab vedotin. |
CADTH base case | 1 + 2 + 3 + 4 + 5 + 6 + 7 | |
KM = Kaplan-Meier; OS = overall survival; PFS = progression-free survival; pola-R-CHP = polatuzumab vedotin, rituximab, cyclophosphamide, doxorubicin, and prednisone; R-CHOP = rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone.
The CADTH base-case analysis found that pola-R-CHP was associated with 0.19 additional QALYs at an additional cost of $76,379 versus R-CHOP. Therefore, the ICER of pola-R-CHP was $394,163 per QALY gained compared to R-CHOP. The probability of cost-effectiveness at a $50,000 per QALY threshold was 6%. Based on the probabilistic results of the CADTH base case, approximately 77% of the incremental QALYs for pola-R-CHP were accrued during the extrapolated period (i.e., after 46 months, the maximum follow-up time from the POLARIX trial). A summary of the CADTH stepped analysis and base-case results can be found in Table 6.
Table 6: Summary of the Stepped Analysis of the CADTH Reanalysis Results (Deterministic)
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
Sponsor’s base case (deterministic) | R-CHOP | 150,553 | 11.88 | Reference |
pola-R-CHP | 187,982 | 12.33 | 82,956 | |
CADTH reanalysis 1 | R-CHOP | 145,931 | 11.90 | Reference |
pola-R-CHP | 196,025 | 12.30 | 124,922 | |
CADTH reanalysis 2 | R-CHOP | 142,056 | 11.93 | Reference |
pola-R-CHP | 181,925 | 12.36 | 90,976 | |
CADTH reanalysis 3 | R-CHOP | 150,553 | 11.88 | Reference |
pola-R-CHP | 184,840 | 12.19 | 108,883 | |
CADTH reanalysis 4 | R-CHOP | 150,662 | 11.88 | Reference |
pola-R-CHP | 186,912 | 12.28 | 91,130 | |
CADTH reanalysis 5 | R-CHOP | 133,216 | 11.88 | Reference |
pola-R-CHP | 187,929 | 12.33 | 121,267 | |
CADTH reanalysis 6 | R-CHOP | 152,664 | 11.88 | Reference |
pola-R-CHP | 196,016 | 12.33 | 96,087 | |
CADTH reanalysis 7 | R-CHOP | 151,593 | 11.88 | Reference |
pola-R-CHP | 190,480 | 12.33 | 86,189 | |
CADTH base case (1 + 2 + 3 + 4 + 5 + 6 + 7, deterministic) | R-CHOP | 122,941 | 11.95 | Reference |
pola-R-CHP | 198,551 | 12.15 | 375,724 | |
CADTH base case (1 + 2 + 3 + 4 + 5 + 6 + 7, probabilistic) | R-CHOP | 121,805 | 11.98 | Reference |
pola-R-CHP | 198,184 | 12.18 | 394,163 |
ICER = incremental cost-effectiveness ratio; pola-R-CHP = polatuzumab vedotin, rituximab, cyclophosphamide, doxorubicin, and prednisone; QALY = quality-adjusted life-year; R-CHOP = rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone.
Scenario Analysis Results
CADTH undertook price reduction analyses based on the sponsor base case and the CADTH base case (Table 7). This analysis demonstrated that a price reduction of 66% would be necessary to achieve cost-effectiveness at a $50,000 per QALY gained threshold when considering the CADTH base case.
Table 7: CADTH Price Reduction Analyses
Analysis | ICERs for pola-R-CHP vs. R-CHOP ($/QALY) | |
|---|---|---|
Price reduction (21-day cycle cost)a | Sponsor base case | CADTH reanalysis |
No price reduction ($14,750) | 86,627 | 394,163 |
10% ($13,275) | 69,046 | 342,233 |
20% ($11,800) | 51,457 | 290,271 |
30% ($10,325) | 33,868 | 238,310 |
40% ($8,850) | 16,279 | 186,348 |
50% ($7,735) | Dominant | 134,387 |
60% ($5,900) | Dominant | 82,425 |
70% ($4,425) | Dominant | 30,464 |
80% ($2,950) | Dominant | Dominant |
ICER = incremental cost-effectiveness ratio; pola-R-CHP = polatuzumab vedotin, rituximab, cyclophosphamide, doxorubicin, and prednisone; QALY = quality-adjusted life-year; R-CHOP = rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone; vs. = versus.
aThe 21-day cycle cost of polatuzumab vedotin assumes a mean patient weight of 75.92 kg as per the sponsor’s submission and assumes wastage of excess medication.
Additionally, CADTH conducted a series of scenario analyses to determine the impact of alternative assumptions on the cost-effectiveness of pola-R-CHP:
50% of vial sharing to account for the possibility that some treatment centres will be able to cluster patients to allow for vial sharing
inclusion of GOYA trial data to extend PFS follow-up time for R-CHOP and support the extrapolation of PFS for pola-R-CHP
applying Kaplan-Meier data for OS up to 30 months.
Results from these scenarios are presented in Table 14. The scenario analysis that used the matched data from the GOYA trial to support the PFS extrapolation reduced the ICER to $261,884 per QALY gained. The scenario analysis that assessed the uncertainty around the use of Kaplan-Meier to model OS resulted in a higher ICER of $465,175 per QALY gained compared to the CADTH base-case analysis.
Issues for Consideration
The pan-Canadian Pharmaceutical Alliance concluded negotiations with a letter of intent for polatuzumab vedotin in combination with bendamustine and rituximab for the treatment of relapsed or refractory DLBCL. As such, a confidential negotiated price exists for polatuzumab vedotin, while CADTH reanalyses are based on the price submitted by the sponsor.18
Stakeholder input received by CADTH indicated that there are implementation considerations that are likely to have an economic impact that was not able to be addressed in the pharmacoeconomic submission. Issues around the centralized locations in some jurisdictions where polatuzumab vedotin would be reconstituted and need to be transported to treatment facilities were noted. Economic impacts of this may include increased administrative costs associated with coordinating the timely and temperature-controlled transportation of the reconstituted product and potential for drug wastage due to product aggregation. Additionally, there are treatment centres that may not be able to accommodate the length of the regimen infusion for pola-R-CHP. Accommodating this treatment may have economic impacts on extending facility hours and requiring additional staff hours. However, polatuzumab vedotin is approved for use in combination with bendamustine and rituximab (as noted previously), and thus physicians and treatment centres have experience with regard to implementing a regimen that includes this treatment.
Overall Conclusions
Based on the CADTH clinical review of the POLARIX trial, pola-R-CHP provided greater efficacy for PFS compared with standard of care R-CHOP; however, whether the magnitude of benefit is clinically meaningful and whether such a benefit is maintained beyond the trial period is uncertain. The available OS data from the POLARIX trial did not demonstrate a difference during the observation period (median OS follow-up = 39.7 months), and CADTH’s clinical review reported that OS results from the POLARIX trial were uninterpretable. Overall, the safety profile of pola-R-CHP was comparable to R-CHOP. The CADTH clinical review also noted that the study findings were limited in generalizability.
CADTH identified several limitations in the sponsor’s pharmacoeconomic analysis that have notable implications on the cost-effectiveness of pola-R-CHP. For the CADTH base-case analysis, CADTH revised the assumptions regarding PFS extrapolation such that only the POLARIX study data were used and no Kaplan-Meier data for PFS were applied directly in the model, which led to more plausible estimates of PFS; incorporated a waning of effect of pola-R-CHP on OS; changed the time point at which the model transitioned from Kaplan-Meier data to parametric survival estimates for OS, to better reflect the available OS evidence, as well as the availability of curative therapies for second line use; aligned the number and distribution of subsequent therapies for both pola-R-CHP and R-CHOP to be nearly equivalent and aligned with expectations of clinical practice in Canada; accounted for drug wastage; and incorporated additional chair time required for pola-R-CHP. The CADTH base-case analysis resulted in an ICER of $394,163 per QALY gained (incremental costs = $76,379; incremental QALYs = 0.19) for pola-R-CHP versus R-CHOP. The probability of pola-R-CHP being cost-effective at a $50,000 per QALY gained threshold was 6%. Based on CADTH’s reanalysis, in order for pola-R-CHP to be considered cost-effective compared to R-CHOP at a $50,000 per QALY gained threshold, the price of polatuzumab vedotin would need to be less than $5,015 per 21-day cycle, reflecting a price reduction of 66%.
Although the CADTH reanalysis attempted to address the identified limitations of the sponsor’s economic submission, some uncertainty remains. First, the misalignment of the trial population with the indicated population could not be addressed, and the cost-effectiveness of pola-R-CHP in some patient populations remains unknown. Specifically, uncertainty remains for patients with IPI 0 to 1 who were not included in the trial, and in situations where there is a standard of care that is different from R-CHOP. Second, due to structural limitations of the PSM approach, only the costs of subsequent therapies are modelled and not changes in OS resulting from curative subsequent therapies. This structural assumption favours pola-R-CHP, given that QALY gains in the PD health state would be underestimated for all patients with PD, of which there were more for R-CHOP. As this limitation could not be addressed, any analyses performed by CADTH or the sponsor are likely to underestimate the true ICER. Further price reductions (i.e., greater than 66%) may therefore be required.
References
1.Polivy (polatuzumab vedotin): 30 mg or 140 mg single-use vial for infusion [product monograph]. Mississauga (ON): Hoffmann-La Roche Ltd.; 2023.
2.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: polatuzumab vedotin: 1.8mg/kg for infusion. Mississauga (ON): Hoffmann-La Roche Ltd; 2023 Mar 1.
3.Sehn LH, Martelli M, Trněný M, et al. A randomized, open-label, Phase III study of obinutuzumab or rituximab plus CHOP in patients with previously untreated diffuse large B-Cell lymphoma: final analysis of GOYA. J Hematol Oncol. 2020;13(1):71. PubMed
4.Felizzi F, Paracha N, Pohlmann J, Ray J. Mixture Cure Models in Oncology: A Tutorial and Practical Guidance. Pharmacoecon Open. 2021;5(2):143-155. PubMed
5.DeltaPA. [Ottawa (ON)]: IQVIA; 2022: https://www.iqvia.com/. Accessed 2023 Apr.
6.Tam VC, Ko YJ, Mittmann N, et al. Cost-effectiveness of systemic therapies for metastatic pancreatic cancer. Curr Oncol. 2013;20(2):e90-e106. PubMed
7.Schedule of benefits for laboratory services: effective July 1, 2020. Toronto (ON): Ontario Ministry of Health; 2020: http://www.health.gov.on.ca/en/pro/programs/ohip/sob/lab/lab_mn2020.pdf. Accessed 2023 Apr.
8.Ontario Case Costing Initiative (OCCI). Toronto (ON): Ontario Health and Long-Term Care; 2017: https://data.ontario.ca/dataset/ontario-case-costing-initiative-occi. Accessed 2022 Nov.
9.Wang H, Manca A, Crouch S, et al. Pcn351 - Health-State Utility Values in Diffuse Large B-Cell Lymphoma. Value Health. 2018;21.
10.Guertin JR, Feeny D, Tarride JE. Age- and sex-specific Canadian utility norms, based on the 2013-2014 Canadian Community Health Survey. CMAJ. 2018;190(6):E155-E161. PubMed
11.Shi Q, Schmitz N, Ou FS, et al. Progression-Free Survival as a Surrogate End Point for Overall Survival in First-Line Diffuse Large B-Cell Lymphoma: An Individual Patient-Level Analysis of Multiple Randomized Trials (SEAL). J Clin Oncol. 2018;36(25):2593-2602. PubMed
12.National Institute for Health and Care Excellence. Pixantrone monotherapy for treating multiply relapsed or refractory aggressive non-Hodgkin's B-cell lymphoma (Technology appraisal guidance TA306) 2014; https://www.nice.org.uk/guidance/ta306. Accessed 2023 Apr.
13.Walker H, Anderson M, Farahati F, et al. Resource Use and costs of End-Of-Life/Palliative Care: Ontario Adult Cancer Patients Dying during 2002 and 2003. J Palliat Care. 2018;27(2):79-88. PubMed
14.de Oliveira C, Pataky R, Bremner KE, et al. Phase-specific and lifetime costs of cancer care in Ontario, Canada. BMC Cancer. 2016;16(1):809. PubMed
15.Latimer N. Survival analysis for economic evaluations alongside clinical trials - extrapolation with patient-level data. (NICE DSU Technical Support Document 14). Sheffield (UK): Decision Support Unit, ScHARR, University of Sheffield; 2013: https://www.ncbi.nlm.nih.gov/books/NBK395885/pdf/Bookshelf_NBK395885.pdf. Accessed 2023 May 5.
16.Woods B, Sideris E, Palmer S, Latimer N, Soares M. Partitioned survival analysis for decision modelling in health care: a critical review. (NICE DSU Technical Support Document 19). Sheffield (UK): Decision Support Unit, ScHARR, University of Sheffield; 2017: http://nicedsu.org.uk/wp-content/uploads/2017/06/Partitioned-Survival-Analysis-final-report.pdf. Accessed 2023 May.
17.Shafey M, Savage K, Skrabek P, et al. Canadian Evidence-Based Guideline for the Frontline Treatment of Diffuse Large B-Cell Lymphoma. Mississauga (ON): Lymphoma Canada; 2021: https://z9a2f9c2.rocketcdn.me/wp-content/uploads/2021/09/LymphomaCanada_Guideline_Frontline_DLBCL_VF_Digital.pdf. Accessed 2023 May 8.
18.pan-Canadian Pharmaceutical Alliance. Brand name drug negotiation status: Polivy (polatuzumab vedotin). 2021; https://www.pcpacanada.ca/negotiation/21471. Accessed 2023 May 8.
19.Brenner DR, Poirier A, Woods RR, et al. Projected estimates of cancer in Canada in 2022. CMAJ. 2022;194(17):E601-E607. PubMed
20.Sehn LH, Berry B, Chhanabhai M, et al. The revised International Prognostic Index (R-IPI) is a better predictor of outcome than the standard IPI for patients with diffuse large B-cell lymphoma treated with R-CHOP. Blood. 2007;109(5):1857-1861. PubMed
21.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: polatuzumab vedotin: 1.8mg/kg for infusion. Mississauga (ON): Hoffmann-La Roche Ltd; 2023 Mar 1.
Appendix 1: Cost Comparison Table
Note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical experts and CADTH-participating public drug plans. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 8: CADTH Cost Comparison Table for First-Line Treatment of DLBCL
Treatment | Strength / concentration | Form | Price | Recommended dosage | Daily cost | Average 28-day cost |
|---|---|---|---|---|---|---|
pola-R-CHP | ||||||
Polatuzumab vedotin (Polivy) | 20 mg/mL | 30 mg vial 140 mg vial | 3,160.7100a 14,750.0000a | 1.8 mg/kg on day 1 every 21 days for up to 6 cycles | 702.38 | 19,666.67 |
Cyclophosphamide (Procytox) | 20 mg/mL | 500 mg vial 1,000 mg vial | 97.8000b 177.2700b | 750 mg/m2 on day 1 every 21 days for up to 6 cycles | 13.10 | 366.76 |
Doxorubicin (generic) | 2 mg/mL | 10 mg vial 50 mg vial | 50.4500b 252.2500b | 50 mg/m2 on day 1 every 21 days for up to 6 cycles | 24.02 | 672.67 |
Prednisone (generic) | 5 mg 50 mg | Tablet | 0.0220c 0.1735c | 100 mg on days 1 to 5 every 21 days for up to 6 cycles | 0.08 | 2.31 |
Rituximab (biosimilar) | 10 mg/mL | 100 mg vial | 297.0000c | 375 mg/m2 on day 1 every 21 days for up to 6 cycles, with possibility of 2 additional cycles | 99.00 | 2,772.00 |
pola-R-CHP | 838.59 | 23,480.41 | ||||
R-CHOP | ||||||
Cyclophosphamide (Procytox) | 20 mg/mL | 500 mg vial 1,000 mg vial | 97.8000b 177.2700b | 750 mg/m2 on day 1 every 21 days for up to 6 cycles | 13.10 | 366.76 |
Doxorubicin (generic) | 2 mg/mL | 10 mg vial 50 mg vial | 50.4500b 252.2500b | 50 mg/m2 on day 1 every 21 days for up to 6 cycles | 24.02 | 672.67 |
Prednisone (generic) | 5 mg 50 mg | Tablet | 0.0220c 0.1735c | 100 mg on days 1 to 5 every 21 days for up to 6 cycles | 0.08 | 2.31 |
Rituximab (biosimilar) | 10 mg/mL | 100 mg vial | 297.0000c | 375 mg/m2 on day 1 every 21 days for up to 6 cycles with possibility of 2 additional cycles | 99.00 | 2,772.00 |
Vincristine (generic) | 1 mg/mL | 1 mL vial | 30.6000c | 1.4 mg per m2 on day 1 every 21 days for up to 6 cycles | 4.37 | 122.40 |
R-CHOP | 140.58 | 3,936.14 | ||||
Note: Assumes mean patient weight of 75.92 kg and BSA of 1.86m2 as per sponsor’s submission. Daily and cycle costs assume wastage of excess medication.
aSponsor’s submitted price.2
bPrices are from DeltaPA database (accessed April 2023), and do not include dispensing fees.
cPrices are from the Ontario Drug Benefit Formulary (accessed April 2023), and do not include dispensing fees.
Appendix 2: Submission Quality
Note that this appendix has not been copy-edited.
Description | Yes or no | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | The model does not include R-EPOCH as a comparator, which is the standard of care for patients with double and triple-hit lymphoma. There is also uncertainty regarding patients with IPI 0 to 1 given that they were excluded from the POLARIX trial. |
Model has been adequately programmed and has sufficient face validity | Yes | No comment. |
Model structure is adequate for decision problem | No | Refer to the CADTH appraisal regarding the inappropriate model structure. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | Yes | No comment. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | Yes | No comment. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough detail) | Yes | No comment. |
IPI = International Prognostic Index; R-EPOCH = rituximab, etoposide, prednisone, vincristine, cyclophosphamide, and doxorubicin.
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note that this appendix has not been copy-edited.
PD = progressed disease; PFS = progression-free survival.
Source: Sponsor’s pharmacoeconomic submission.2
Detailed Results of the Sponsor’s Base Case
Table 10: Disaggregated Summary of the Sponsor's Economic Evaluation Results
Parameter | pola-R-CHP | R-CHOP | Incremental |
|---|---|---|---|
Discounted LYs | |||
Total | 15.98 | 15.52 | 0.46 |
By health state or data source | |||
PFS | 13.58 | 12.11 | 1.47 |
Progressed disease | 2.40 | 3.41 | –1.01 |
Discounted QALYs | |||
Total | 12.34 | 11.91 | 0.44 |
By health state or data source | |||
PFS | 10.65 | 9.50 | 1.14 |
Progressed disease | 1.70 | 2.41 | –0.71 |
Discounted costs ($) | |||
Total | 188,764 | 150,896 | 37,868 |
PFS | 111,107 | 26,271 | 84,835 |
Progressed disease | 38,070 | 54,032 | –15,962 |
Subsequent therapy | 31,506 | 59,896 | –28,390 |
End of life | 8,082 | 10,697 | –2,615 |
ICER ($/QALY) | 86,627 | ||
ICER = incremental cost-effectiveness ratio; LY = life-year; PFS = progression-free survival; pola-R-CHP = polatuzumab vedotin, rituximab, cyclophosphamide, doxorubicin, and prednisone; QALY = quality-adjusted life-year; R-CHOP = rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone.
Source: Sponsor’s pharmacoeconomic submission.2
Table 11: Distribution of Subsequent Therapies Used in the Sponsor’s Economic Evaluation
Subsequent therapy | pola-R-CHP | R-CHOP |
|---|---|---|
Number of therapies | 2.03 | 2.26 |
ASCT | 14% | 16% |
R-GDP | 31% | 30% |
GDP | 12% | 8% |
GemOx | 8% | 8% |
R-GemOx | 27% | 23% |
Pola-BR | 1% | 4% |
CAR T-cell therapy | 7% | 9% |
AHCT | 0% | 2% |
R-DHAP | 0% | 0% |
AHCT = allogenic hematopoietic cell transplant; ASCT = autologous stem cell transplant; CAR = chimeric antigen receptor; GDP = gemcitabine, dexamethasone, and cisplatin; GemOx = gemcitabine and oxaliplatin; pola-R-CHP = polatuzumab vedotin, rituximab, cyclophosphamide, doxorubicin, and prednisone; pola-BR = polatuzumab vedotin, bendamustine, and rituximab; R-CHOP = rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone; R-DHAP = rituximab, dexamethasone, cytarabine, and cisplatin; R-GDP = rituximab, gemcitabine, dexamethasone, and cisplatin; R-GemOx = rituximab, gemcitabine, and oxaliplatin.
Source: Sponsor’s pharmacoeconomic submission.2
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Note that this appendix has not been copy-edited.
Detailed Results of CADTH Base Case
Table 12: Disaggregated Summary of CADTH’s Economic Evaluation Results
Parameter | pola-R-CHP | R-CHOP | Incremental |
|---|---|---|---|
Discounted LYs | |||
Total | 15.73 | 15.53 | 0.20 |
By health state or data source | |||
PFS | 13.63 | 12.94 | 0.69 |
Progressed disease | 2.10 | 2.60 | –0.49 |
Discounted QALYs | |||
Total | 12.18 | 11.99 | 0.19 |
By health state or data source | |||
PFS | 10.69 | 10.15 | 0.55 |
Progressed disease | 1.49 | 1.85 | –0.35 |
Discounted costs ($) | |||
Total | 198,184 | 121,805 | 76,379 |
PFS | 121,000 | 28,780 | 92,220 |
Progressed disease | 33,277 | 41,240 | –7,963 |
Subsequent therapy | 34,971 | 41,632 | −6,662 |
End of life | 8,936 | 10,153 | –1,217 |
ICER ($/QALY) | 394,163 | ||
ICER = incremental cost-effectiveness ratio; LY = life-year; PFS = progression-free survival; pola-R-CHP = polatuzumab vedotin, rituximab, cyclophosphamide, doxorubicin, and prednisone; QALY = quality-adjusted life-year; R-CHOP = rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone.
Table 13: Distribution of Subsequent Therapies Used in CADTH’s Economic Evaluation
Subsequent therapy | pola-R-CHP | R-CHOP |
|---|---|---|
Number of therapies | 2.03 | 2.03 |
ASCT | 14% | 14% |
R-GDP | 31% | 30% |
GDP | 12% | 11% |
GemOx | 8% | 8% |
R-GemOx | 27% | 26% |
Pola-BR | 0% | 3% |
CAR T-cell therapy | 7% | 7% |
AHCT | 0% | 0% |
R-DHAP | 1% | 1% |
AHCT = allogenic hematopoietic cell transplant; ASCT = autologous stem cell transplant; CAR = chimeric antigen receptor; GDP = gemcitabine, dexamethasone, and cisplatin; GemOx = gemcitabine and oxaliplatin; pola-R-CHP = polatuzumab vedotin, rituximab, cyclophosphamide, doxorubicin, and prednisone ; pola-BR = polatuzumab vedotin, bendamustine, and rituximab; R-CHOP = rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone; R-DHAP = rituximab, dexamethasone, cytarabine, and cisplatin; R-GDP = rituximab, gemcitabine, dexamethasone, and cisplatin; R-GemOx = rituximab, gemcitabine, and oxaliplatin.
Scenario Analyses
Table 14: Summary of CADTH’s Economic Evaluation Results — Scenario Analyses
Scenario | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
CADTH base case | R-CHOP | 121,805 | 11.99 | Reference |
pola-R-CHP | 198,184 | 12.18 | 394,163 | |
CADTH scenario analysis: 50% vial sharing | R-CHOP | 121,518 | 11.99 | Reference |
pola-R-CHP | 192,958 | 12.19 | 357,211 | |
CADTH scenario analysis: GOYA data included | R-CHOP | 126,763 | 11.91 | Reference |
pola-R-CHP | 188,580 | 12.15 | 261,884 | |
CADTH scenario analysis: OS KM cut-off at 30 months | R-CHOP | 122,928 | 11.98 | Reference |
pola-R-CHP | 198,009 | 12.14 | 465,175 |
ICER = incremental cost-effectiveness ratio; KM = Kaplan-Meier; LY = life-year; OS = overall survival; pola-R-CHP = polatuzumab vedotin, rituximab, cyclophosphamide, doxorubicin, and prednisone; QALY = quality-adjusted life-year; R-CHOP = rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone.
Appendix 5: Submitted Budget Impact Analysis and CADTH Appraisal
Note that this appendix has not been copy-edited.
Table 15: Summary of Key Take-Aways
Key take-aways of the budget impact analysis |
|---|
|
Summary of Sponsor’s BIA
The sponsor submitted a budget impact analysis (BIA) estimating the incremental budget impact of reimbursing pola-R-CHP for the treatment of adult patients with previously untreated LBCL, including DLBCL NOS, high-grade B-cell lymphoma, EBV-positive DLBCL NOS, and T-cell/histiocyte-rich LBCL. The analysis was undertaken using an epidemiologic approach from the perspective of the CADTH-participating Canadian public drug plans over a three-year time horizon (2024 to 2026). Beginning with an estimate of Canadian incident non-Hodgkin lymphoma cases,19 the sponsor narrowed the population using estimates of those that are DLBCL,17 those eligible for public coverage, and those fit for first-line therapy. The model accounted for incident DLBCL cases only, as prevalent cases were assumed to have received prior treatment and thus would not be eligible for pola-R-CHP. The submitted model can also estimate the budgetary impact for the POLARIX trial population (IPI 2 to 5), which is narrower than the Health Canada–indicated population. The sponsor compared a reference scenario where pola-R-CHP was not reimbursed, with a new drug scenario where pola-R-CHP was reimbursed, as per its Health Canada indication. The reference scenario included R-CHOP as the only comparator. The submitted model incorporated the number and distributions of subsequent therapies, and calculated these costs based on the PFS and OS from the POLARIX trial (Figure 2). Key inputs to the BIA are documented in Table 16. Key assumptions included the following:
The number and distribution of subsequent therapies used in the BIA were derived from the POLARIX trial, and reflected what was used in the sponsor’s submitted CUA.
The number of cycles per regimen used in the sponsor’s base case was 6 cycles for pola-R-CHP and 5.76 cycles for R-CHOP, due to the fewer number of regimens used for R-CHOP in patients with IPI 0 to 1 (28% of patients were assumed to have an IPI of 0 to 1).20
DLBCL is assumed to reflect all untreated LBCL in the eligible population.
Market uptake is calculated as a weighted uptake of IPI 0 to 1 and IPI 2 to 5 estimates.
Figure 2: Sponsor’s Estimation of the Budgetary Impact
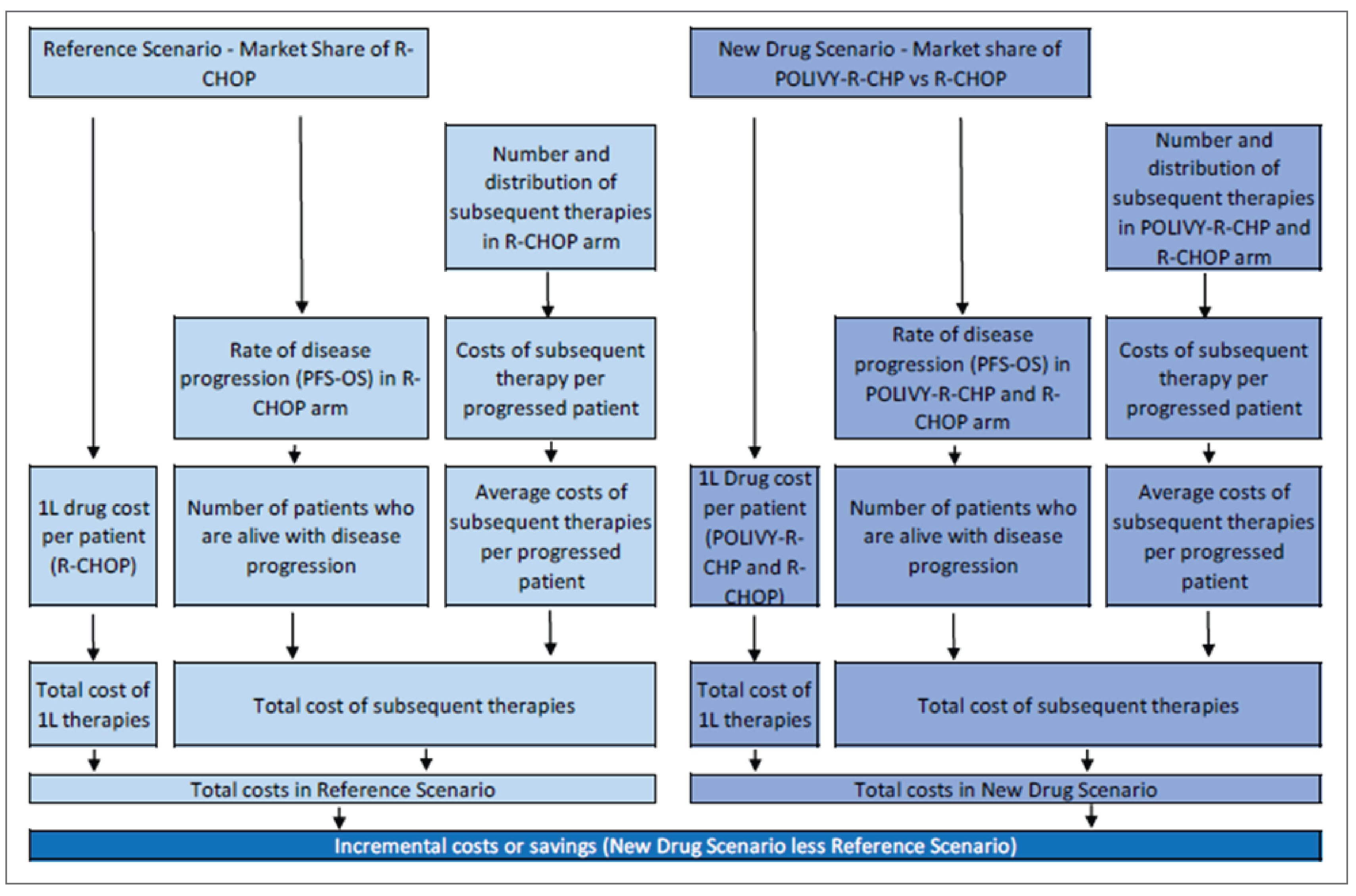
PFS = progression-free survival; OS = overall survival; POLIVY-R-CHP = polatuzumab vedotin, rituximab, cyclophosphamide, doxorubicin, and prednisone; R-CHOP = rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone.
Source: Sponsor’s BIA submission.21
Table 16: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3 if appropriate) |
|---|---|
Target population | |
Incident non-Hodgkin lymphoma cases in Canada (excluding Quebec) | 9,068 / 9,216 / 9,36619 |
Proportion that are DLBCL casesa | 35%17 |
Percent eligible for public coverage | 100% |
Proportion deemed fit to receive first-line chemotherapy treatment | 85% |
Number of patients eligible for drug under review | 2,698 / 2,742 / 2,786 |
Market uptake (3 years) | |
Uptake (reference scenario) pola-R-CHP R-CHOP | 0% / 0% / 0% 100% / 100% / 100% |
IPI 0 to 1 uptake (new drug scenario) pola-R-CHP R-CHOP | 1% / 2% / 3% 99% / 98% / 97% |
IPI 2 to 5 uptake (new drug scenario) pola-R-CHP R-CHOP | 20% / 40% / 42.5% 80% / 60% / 57.5% |
Weighted uptake (new drug scenario)b pola-R-CHP R-CHOP | 14.7% / 29.4% / 31.4% 85.3% / 70.6% / 68.6% |
Cost of treatment (per patient) | |
Cost of treatment over 21 days pola-R-CHP R-CHOP | $17,243.84 $2,907.27 |
DLBCL = diffuse large B-cell lymphoma; IPI = International Prognostic Index; pola-R-CHP = polatuzumab vedotin, rituximab, cyclophosphamide, doxorubicin, and prednisone; R-CHOP = rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone.
Note: Cost of treatment assumes perfect vial sharing (i.e., no wastage), and assumes a mean patient weight of 75.92 kg and BSA of 1.86m2 from the POLARIX trial.
aDLBCL is assumed to reflect all untreated LBCL in the eligible population.
bThe weighted uptake is based on 28% of eligible patients having IPI 0 to 1.20
Summary of the Sponsor’s BIA Results
The sponsor estimated the net budget impact of funding polatuzumab vedotin in combination with R-CHP for the treatment of adult patients with previously untreated LBCL, including DLBCL NOS, high-grade B-cell lymphoma, EBV-positive DLBCL NOS, and T-cell/histiocyte-rich LBCL to be $29,508,742 in year 1, $59,383,618 in year 2, and $64,154,138 in year 3, for a 3-year total budget impact of $153,046,498. A scenario analysis that assessed changes in the number of non-Hodgkin lymphoma cases that are DLBCL resulted in a change in the budgetary impact of plus or minus 14% when using the bottom and top of the estimated range (30% and 40%), suggesting that the budget impact model is sensitive to the number of patients treated. Scenarios that considered relevant costs from the health care payer perspective estimated the 3-year budget impact to be $153,208,267 when including administration costs, and $146,900,736 when transplant costs are included.
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
The market share of pola-R-CHP may be underestimated. The sponsor’s submitted BIA assumed that the pola-R-CHP regimen would have a market uptake of 1%, 2%, and 3% for patients with an IPI of 0 to 1 and 20%, 40%, 42.5% for patients with an IPI of 2 to 5 in years 1, 2, and 3. The market uptake used in the model was a weighted market share assuming that 28% of patients with DLBCL have an IPI of 0 to 1, resulting in the weighted market uptake of 14.7%, 29.4% and 31.4% in years 1, 2, and 3. CADTH obtained clinical expert feedback indicating that they agree that market uptake for patients with an IPI of 0 to 1 assumed by the sponsor to be reasonable. However, the market uptake in all 3 years for with the IPI 2 to 5 population does not align with clinical expectations, with clinical experts consulted by CADTH indicating the sponsor likely underestimated pola-R-CHP uptake, noting that in higher risk patients, 100% of patients would be treated with pola-R-CHP rather than R-CHOP, given that polatuzumab vedotin is a familiar drug to the prescribing clinicians and once available pola-R-CHP would likely become the new standard of care for the indication under review.
To address this limitation, CADTH undertook a reanalysis by revising the market shares for pola-R-CHP in the new drug scenario for patients with IPI 2 to 5 to 50% in year 1, and 100% in years 2 and 3. The resulting weighted market uptake in the CADTH reanalysis is 36%, 73% and 73% in years 1, 2, and 3.
Subsequent therapy assumptions were not reflective of clinical practice in Canada. The sponsor assumed that the number and distribution of subsequent therapies received postprogression in the BIA matched that of the POLARIX trial. In the POLARIX trial, patients who received pola-R-CHP had an average of 2.03 subsequent therapies, and patients who received R-CHOP had an average of 2.26 subsequent therapies. The distribution of which subsequent therapies were received also differed by first-line treatment group. Clinical experts consulted by CADTH indicated that in clinical practice in Canada, the approach to subsequent therapy would not differ between first-line therapy (i.e., whether a patient receives pola-R-CHP or R-CHOP) and that they expected the distribution and number of subsequent therapies would be nearly equivalent between groups.
The CADTH reanalysis applied the same number and distribution of subsequent therapies as the CADTH reanalysis of the CUA, as reported in Table 13.
The assumption that perfect vial sharing would take place was inappropriate. In the submitted BIA, the sponsor assumed perfect vial sharing would occur. This assumption is inappropriate because of differences in vial sharing abilities by jurisdiction due to differing patient volumes. Further, the product monograph for polatuzumab vedotin indicates that the reconstituted product is intended for single use only and to discard any unused portion.1 The submitted model does not allow the user to specify vial sharing for individual drugs, and so the vial sharing assumption was applied to every drug included in pola-R-CHP and R-CHOP regimens.
The CADTH reanalysis assumed 0% vial sharing, as aligned with the CADTH reanalysis of the CUA.
A scenario analysis that allowed for 50% of vial sharing was also included to account for the possibility that some treatment centres will be able to cluster patients to allow for vial sharing.
Additional limitations were identified but were not considered to be key limitations. These limitations include:
There are relevant cost considerations from the health care payer perspective that may not have been appropriately accounted for. When considering the health care payer perspective, administration costs are relevant, particularly for IV drugs. Drug plan input and clinical expert feedback solicited by CADTH indicated that the administration of polatuzumab vedotin with R-CHP will require more chair time than the R-CHOP regimen. The additional chair time is associated with the infusion time being longer for polatuzumab vedotin than the drug it replaced in R-CHOP (vincristine), and due to the required observation time of polatuzumab vedotin (1.5 hours and 0.5 hours observation for the first and subsequent infusion, respectively). In fact, drug plan input and the clinical experts consulted by CADTH estimated that the first cycle of pola-R-CHP could take up to 10 hours, and that some outpatient treatment facilities are unable to accommodate a treatment day of that length. Additionally, the introduction of pola-R-CHP is anticipated to change the use of subsequent therapies due to differences in PFS, and the health care payer perspective captures the change in costs associated with transplants, that are not captured in the public drug plan perspective.
CADTH conducted a scenario analysis from the health care payer perspective that included administration costs for both pola-R-CHP and R-CHOP and the inclusion of transplant costs as part of subsequent therapy. The infusion times used in the BIA were aligned with those used in the CADTH reanalysis of the CUA.
CADTH Reanalyses of the BIA
CADTH revised the sponsor’s submitted analysis by adjusting the market share for patients with IPI 2 to 5 to reflect expectations of clinical experts consulted by CADTH, revising the number and distribution of subsequent therapies, and accounting for the impact of drug wastage. The changes applied to derive the CADTH base case are described in Table 17.
Table 17: CADTH Revisions to the Submitted BIA
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
None. | — | — |
Changes to derive the CADTH base case | ||
1. Market share for IPI 2 to 5 | Year 1: 20% Year 2: 40% Year 3: 42.5% | Year 1: 50% Year 2: 100% Year 3: 100% |
2. Subsequent therapies | Number and distribution of subsequent therapies aligned with POLARIX trial (Table 11) | Number and distribution of subsequent therapies aligned with CUA (Table 13) |
3. Vial sharing | Perfect vial sharing | No vial sharing |
CADTH base case | 1 + 2 + 3 | |
BIA = budget impact analysis; CUA = cost-utility analysis; IPI = International Prognostic Index.
The results of the CADTH step-wise reanalysis are presented in summary format in Table 18 and a more detailed breakdown is presented in Table 19.
The CADTH reanalysis suggests that reimbursing pola-R-CHP for the first-line treatment of adults with DLBCL would be associated with an incremental cost of $80,865,544 in Year 1, $164,205,857 in Year 2, and $167,849,115 in Year 3, for a three-year budgetary impact of $412,920,515.
Table 18: Summary of the CADTH Reanalyses of the BIA
Stepped analysis | Three-year total |
|---|---|
Submitted base case | $153,046,498 |
CADTH reanalysis 1 | $368,167,639 |
CADTH reanalysis 2 | $167,274,752 |
CADTH reanalysis 3 | $157,338,292 |
CADTH base case | $412,920,515 |
BIA = budget impact analysis.
Table 19: Detailed Breakdown of the CADTH Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
Submitted base case | Reference | $95,805,338 | $109,846,312 | $111,636,322 | $113,431,036 | $334,913,670 |
New drug | $95,805,338 | $139,355,054 | $171,019,940 | $177,585,174 | $487,960,168 | |
Budget impact | $0 | $29,508,742 | $59,383,618 | $64,154,138 | $153,046,498 | |
CADTH reanalysis 1 | Reference | $95,805,338 | $109,846,312 | $111,636,322 | $113,431,036 | $334,913,670 |
New drug | $95,805,338 | $182,798,480 | $258,324,871 | $261,957,958 | $703,081,309 | |
Budget impact | $0 | $72,952,168 | $146,688,549 | $148,526,922 | $368,167,639 | |
CADTH reanalysis 2 | Reference | $79,992,639 | $89,931,181 | $91,396,840 | $92,865,754 | $274,193,775 |
New drug | $79,992,639 | $121,816,234 | $156,192,719 | $163,459,575 | $441,468,528 | |
Budget impact | $0 | $31,885,053.40 | $64,795,879 | $70,593,820 | $167,274,752 | |
CADTH reanalysis 3 | Reference | $97,162,867 | $111,293,535.57 | $113,107,139.54 | $114,925,473 | $339,326,148 |
New drug | $97,162,867 | $141,621,782.86 | $174,154,330.34 | $180,888,327 | $496,664,440 | |
Budget impact | $0 | $30,328,247.30 | $61,047,190.80 | $65,962,854 | $157,338,292 | |
CADTH base case | Reference | $81,306,945 | $91,323,967 | $92,812,333 | $94,303,977 | $278,440,277 |
New drug | $81,306,945 | $172,189,510 | $257,018,191 | $262,153,092 | $691,360,793 | |
Budget impact | $0 | $80,865,544 | $164,205,857 | $167,849,115 | $412,920,515 |
BIA = budget impact analysis.
CADTH conducted additional scenario analyses to address remaining uncertainty, using the CADTH base case. Results are provided in Table 20.
Price reduction of 66% to assess the budget impact if the price of the drug under review reflected the price in which the ICER would be at $50,000 per QALY in CADTH’s base-case CUA.
Assumed 50% vial sharing would take place, as aligned with the CUA scenario analysis.
Included administration costs to consider the budgetary impact from the health care payer perspective.
Assumed that pola-R-CHP is only funded for patients with IPI 2 to 5, aligned with the POLARIX trial population.
Assumed no difference in OS between treatments by applying the OS event estimates for pola-R-CHP from the POLARIX trial to R-CHOP.
Results of CADTH’s scenario analyses demonstrate that the estimated budget impact is highly sensitive to changes in drug cost (decrease of $291,933,180 form CADTH base case); however, the budget impact still exceeds 40 million in years 2 and 3 despite the price reduction. The remaining scenario analyses had limited impact on the estimated budgetary impact of polatuzumab vedotin.
Table 20: Detailed Breakdown of the CADTH Scenario Analyses of the BIA
Analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
CADTH base case | Reference | $81,306,945 | $91,323,967 | $92,812,333 | $94,303,977 | $278,440,277 |
New drug | $81,306,945 | $172,189,510 | $257,018,191 | $262,153,092 | $691,360,793 | |
Budget impact | $0 | $80,865,544 | $164,205,857 | $167,849,115 | $412,920,515 | |
CADTH scenario analysis: 66% price reduction | Reference | $81,306,945 | $91,323,967 | $92,812,333 | $94,303,977 | $278,440,277 |
New drug | $81,306,945 | $115,006,120 | $140,840,701 | $143,580,792 | $399,427,613 | |
Budget impact | $0 | $23,682,154 | $48,028,367 | $49,276,815 | $120,987,335 | |
CADTH scenario analysis: 50% vial sharing | Reference | $80,649,792 | $90,627,574 | $92,104,587 | $93,584,866 | $276,317,026 |
New drug | $80,649,792 | $170,473,814 | $254,241,506 | $259,322,904 | $684,038,225 | |
Budget impact | $0 | $79,846,241 | $162,136,919 | $165,738,038 | $407,721,198 | |
CADTH scenario analysis: health care payer perspective | Reference | $96,915,974 | $108,715,135 | $110,486,951 | $112,262,620 | $331,464,706 |
New drug | $96,915,974 | $190,159,843 | $275,879,262 | $281,396,780 | $747,435,884 | |
Budget impact | $0 | $81,444,708 | $165,392,310 | $169,134,160 | $415,971,178 | |
CADTH scenario analysis: Eligibility restricted to IPI 2 to 5 | Reference | $59,888,382 | $67,122,052 | $68,226,477 | $69,334,178 | $204,682,707 |
New drug | $59,888,382 | $146,632,839 | $229,813,625 | $233,738,648 | $610,185,112 | |
Budget impact | $0 | $79,510,786 | $161,587,149 | $164,404,470 | $405,502,405 | |
CADTH scenario analysis: Equivalent OS | Reference | $75,506,820 | $91,471,701 | $92,961,797 | $94,457,446 | $278,890,943 |
New drug | $75,506,820 | $174,475,972 | $259,287,175 | $262,210,620 | $695,973,767 | |
Budget impact | $0 | $83,004,271 | $166,325,378 | $167,753,174 | $417,082,824 |
BIA = budget impact analysis; IPI = International Prognostic Index; OS = overall survival.
Stakeholder Input
Patient Input
Lymphoma Canada
About Lymphoma Canada
Lymphoma Canada is a national Canadian registered charity whose mission it is to empower patients and the lymphoma community through education, support, advocacy, and research. Based out of Mississauga (ON), we collaborate with patients, caregivers, healthcare professionals, and other organizations and stakeholders, to promote early detection, find new and better treatments for lymphoma patients, help patients access those treatments, learn about the causes of lymphoma, and work together to find a cure. Resources are provided in both English and French. www.lymphoma.ca
Information Gathering
The data presented in this submission was collected from an online anonymous patient survey, created and promoted by Lymphoma Canada (LC) available from February 2 to March 13, 2023. The link was promoted via e-mail to patients registered in the LC national emailing list and made available via social media outlets, including Twitter, Instagram, and Facebook accounts. The survey had a combination of multiple choice, rating, and open-ended questions. Skipping logic was built into the survey so that respondents were asked questions only relevant to them. Open-ended responses were noted in this report verbatim, to provide a deeper understanding of patient perspectives. 89 responses were collected. Information from this survey was used to identify the main areas of concern for patients with Large B-cell lymphoma, with 4 confirmed responses for experience with Polatuzumab vedotin. Three of these patients indicated they live in Canada, and other in Italy. Please refer to Tables 1 to 4 below for demographic and relevant information of all survey respondents. The majority of patients lived in Canada (94%), between the age of 55 and 74 (64%), female (58%), and were diagnosed 1- 5 years ago (61%) with Diffuse Large B-cell lymphoma subtype (89%).
Table 1: Country of Respondents From Lymphoma Canada Survey
Respondents | CAN | USA | Italy | New Zealand | Skipped | Total |
|---|---|---|---|---|---|---|
Patients with Large B-cell lymphoma | 47 | 1 | 1 | 1 | 47 | 50 |
Table 2: Age Range of Respondents From Lymphoma Canada Survey
Respondents | Age (years old) | Total | |||||
|---|---|---|---|---|---|---|---|
25-34 | 35-54 | 55-64 | 65-74 | 75-89 | Skipped | ||
Patients with Large B-cell lymphoma | 3 | 6 | 12 | 20 | 9 | 47 | 50 |
Table 3: Gender of Respondents From Lymphoma Canada Survey
Respondents | Gender | Total | ||
|---|---|---|---|---|
Female | Male | Skipped | ||
Patients with Large B-cell lymphoma | 29 | 21 | 47 | 50 |
Table 4: Number of Years Ago Respondents Were Diagnosed With Large B-cell Lymphoma
Respondents | Years | Total | |||||
|---|---|---|---|---|---|---|---|
<1 | 1-2 | 3-5 | 5-8 | 9-10 | Skipped | ||
Patients with Large B-cell lymphoma | 9 | 21 | 20 | 9 | 10 | 20 | 69 |
Table 5: Subtype of Large B-cell Lymphoma of Survey Respondents
Subtype of Large B-cell Lymphoma | Number of respondents |
|---|---|
Diffuse Large B-cell Lymphoma (DLBCL) | 82 |
High grade B-cell Lymphoma | 3 |
EBV+ DLBCL | 1 |
T-cell/histiocyte-rich LBCL | 1 |
Transformed DLBCL | 2 |
Total | 89 |
Disease Experience
At Diagnosis
Through the online survey, patients were asked to rate a list of physical symptoms on a scale of 1 (no impact) to 5 (significant impact) in regard to their quality of life upon diagnosis. The most common reported symptoms rated as a three or higher were: fatigue (59%), bodily aches and pains (42%), night sweats (42%), enlarged lymph nodes (41%) and a reduced appetite (33%). Several patients left comments for this question indicating severe back pain and constipation were also negative symptoms experienced at the time of diagnosis.
Respondents of the survey were also asked to select from a list of psychosocial impacts they experience when diagnosed with LBCL. Of 69 patients, 81% were impacted by stress of diagnosis, 79% experienced anxiety/worry, and 60% were afraid of progression, 51% inability to continue daily activities, and 47% had difficulty sleeping. When asked to provide additional details about the challenges faced during diagnosis, several patients commented on how the COVID-19 pandemic felt them feeling isolated, with minimal support. Below is a selection of patient responses from the survey:
“I was diagnosed during Covid. Talked to my GP over phone where we discussed lab results, need for a biopsy, results, next steps etc. Covid made being given a cancer diagnosis very isolating.”
“While I had uncomfortable symptoms for a while, and was having lots of tests done, I was not at all expecting a cancer diagnosis until I did my own research and pretty much had it figured out. Receiving a diagnosis, over the phone, during the height of COVID was quite alarming.”
“I felt lonely, like nobody really cared. No nearby resources.”
“Due to Covid I was left to face this on my own since family was not allowed in hospital.”
“Challenged faced living in a rural community that I had to travel an hour and a half one way for chemo because my home hospital would not do this specific chemo. This had financial impacts as well as trying to find someone to drive me.”
“Putting your plans on hold (travel etc.) and not being able to participate safely in large group activities. Difficult telling family and friends about the diagnosis.”
Current Quality of Life
To understand the factors which currently impact patients with Large B-cell lymphoma, respondents were asked a similar style of questions from the diagnosis section of the survey. On a scale of 1 (no impact) to 5 (significant impact), majority of patients rated symptoms such as fatigue (42%), bodily aches (25%), and headaches (17%) as an impact of 3 or higher on their current quality of life. Patients also indicated they recently experienced mental health challenges such as fear of progression/relapse (66%), stress of having cancer (56%) and anxiety/worry (42%).
Daily Activities
Regarding day-to-day activities, patients with Large B-cell lymphoma rated several factors on a scale of 1 (no impact) to 5 (very significant impact) which impacted their daily life. Ability to exercise (41%), ability to travel (39%), spending time with family and friends (36%) and the ability to work, school and volunteer (34%) were rated as a 3 or higher by 59 patients. Many patients left comments in this section and a selection of quotes are included below:
“Afraid to do long walks/ hikes like I used too in case I run out of energy to return.”
“I still attend the Lymphoma Support Group of Ottawa to give hope to others Just as I was given hope by meeting people who had been cured or in remission for many years.”
“Some days I don’t have the energy to do things around the house. Other days I’m great and get a lot done. Energy levels are getting better now that treatment is complete. Working on getting more physical by walking. Still short of breath when walking. Working on improving that.”
“Being immunocompromised I mask indoors still so this limits restaurants or group eating events. I’m grateful for the Evusheld shots protection against covid BA-4 and BA-5. I’m less concerned about covid now than some of the other viruses circulating now. Ir RSV, etc.”
“Never felt as energetic after 3 years of treatment. Know this will probably come back.”
“I have an oncologist who I see every 3 months and she’s totally on top of my current health. I’m living a full life, making music, travelling, seeing friends, going to plays, concerts, walking, shopping, nothing holding me back. Being born in Canada is like winning the lottery!”
Summary of the Disease Experience
The most common physical symptoms LBCL patients found challenging at the time of diagnosis and on their current quality of life included fatigue, bodily aches and pains, night sweats, and headaches. Top-rated psychosocial factors included stress of diagnosis, anxiety/worry, and fear of progression. Many patients also left comments that the COVID-19 pandemic made them feel isolated during diagnosis.
There was a wide range of experiences in which LBCL symptoms impacted the daily lives of survey respondents. The ability to exercise and travel were factors which impacted most patients.
Experiences With Currently Available Treatments
Patients who completed the Lymphoma Canada survey were asked how many lines of treatment they received to treat their Large B-cell lymphoma. The majority of 56 patients indicated they received 1 (41%) or 3 (38%) lines of treatment, refer to Table 6. As the indication for this CADTH submission is for previously untreated Large B-cell lymphoma, the patient experience information in this section will focus on those treated with one line of therapy.
Table 6: Number of Lines of Therapy Survey Respondents Received
Respondents | Have not received therapy | 1 | 2 | 3 | Skipped | Total |
|---|---|---|---|---|---|---|
Patients with Large B-cell lymphoma | 3 | 23 | 9 | 21 | 33 | 56 |
In the front-line setting, almost half of patients (48%) received the chemoimmunotherapy (CIT) R-CHOP, almost all other patients had some form or chemotherapy or CIT such as R-CHP or EPOCH-R. These patients were asked: “How satisfied were you with the number of treatment options available to you for your lymphoma?” 28% of patients indicated they were satisfied with frontline treatment options and 57% indicated they were very satisfied.
67% of 52 survey declared they did not have any difficulty accessing treatment for their lymphoma. Although many comments were left from patients explaining challenges and delays in accessing treatment:
“I went to the U.S. for treatment because there were no other options other than RCHOP.”
“Standard treatment was available in my province. Could no longer work, spouse had to quit work. So moved in order to be closer to treatment.”
“I live 50 kilometres from the hospital.”
“Location was not very difficult to access but there was a waiting list, so treatment was delayed 1 month.”
The most common financial implications reported for treatment for LBCL was absence from work (46%), drug costs (32%), travelling costs (28%), and supplementary drug costs for side effects (22%). Survey respondents left several comments when asked about the difficulties of accessing treatment in Canada:
“It was impossible for me to get anything but RCHOP in Canada as a first line of treatment. My immune system was depleted and I did not think I would survive six rounds of RCHOP. After the first round I left Canada to look for other treatment options. It cost me everything financially, but I had a better quality of life during treatment and I’m alive today.”
“At the time CAR-T was new in the USA and not available in Canada. Now it's even available in my new hometown.”
“Could not be happier with the speed, efficiency, and results of my treatment. (All done at Sunnybrook, Toronto)”
“I was very fortunate not to have any difficulty accessing treatment. I know it is not the case for many people in Canada.
Summary of Currently Available Treatments
The majority of survey respondents received one line of treatment for their LBCL, with R-CHOP as the most common treatment regimen. Most patients were satisfied or very satisfied with their options for frontline treatment.
When asked about how to ask about accessing lymphoma therapy in Canada, many patients indicated they required to travel long distances, which was which challenging financially and required time off work.
Improved Outcomes
LBCL patients which completed the Lymphoma Canada survey were asked how important it was for a new drug to control/treat their Large B-cell lymphoma. 11 patients indicated factors such as longer disease remission (100%), control disease symptoms (91%), longer survival (100%), normalize blood counts (91%), and improved quality of life to perform daily activities (91%), were very important to them (10 out of 10). 8 of these patients indicated they would be willing to tolerate side effects to access new treatment and 7 patients indicated choice is important to them (8 or higher, of 10) in deciding to take a drug based on known side effects and expected outcomes of treatment.
A few patients left comments about managing expectations of novel lymphoma treatments:
“I would take the recommendation of my doctor.”
“I hope that they will have long lasting effects of remission.”
“I have had wonderful care in the treatment of my lymphoma, I have great trust in my doctors.”
9 out of 11 patients indicated they feel there is a need for more therapy options for patients with LBCL.
Summary of Currently Available Treatments
Factors important to LBCL patients when considering novel therapies include longer disease remission, controlled disease symptoms, longer survival, normalized blood counts and improved quality of life to include daily activities.
Majority of patients that completed this section of the survey feel there is a need for more therapeutic options for treatment of LBCL.
Experience With Drug Under Review
From survey responses, 4 patients indicated they were treated with Polatuzumab vedotin in combination with the chemotherapy R-CHP. Based on the information completed by LBCL patients in this section, the following information was gathered about the current drug under review:
3 patients would recommend Pola-R-CHP to other LBCL patients.
1 patient accessed this therapy through a clinical trial, 2 other patients accessed through Medicare or public care.
2 patients indicated their overall experience was very good with Pola-R-CHP and rated their experience as good.
Side effects patients experienced on Pola-R-CHP included: fatigue (3), neutropenia (2), thrombocytopenia (2), decreased appetite (2), diarrhea (2), cytokine release syndrome (1), fever (1), febrile neutropenia (1), low blood pressure (1), infections (1), nausea/vomiting (1), and joint or muscle pain (1).
Summary of Drug Under Review
Overall, the experience of Pola-R-CHP from LBCL patients was positive with minimal negative comments left about the ability to access treatment, financial implications, or challenges tolerating side effects.
Companion Diagnostic Test
Not applicable.
Anything Else?
Lymphoma Canada is a big advocate for lymphoma patients and their caregivers to have access to novel lymphoma therapies. An increased number of treatment options gives patients more choice to decide the therapy that is right for their personal goals, with their medical care team. A large majority of patients relapse from LBCL after first treatment, indicating there is a need for more and better therapeutic options for this subset of patients.
Conflict of Interest Declaration — Lymphoma Canada
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission?
No.
Did you receive help from outside your patient group to collect or analyze data used in this submission
No.
List any companies or organizations that have provided your group with financial payment over the past 2 years AND who may have direct or indirect interest in the drug under review.
Table 7: Financial Disclosures for Lymphoma Canada
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Roche | — | — | X | — |
Gilead | — | — | — | X |
Incyte | — | — | X | — |
Novartis | — | — | X | — |
BMS | — | — | — | X |
Clinician Input
Ontario Health (Cancer Care Ontario) Hematology Cancer Drug Advisory Committee
About Ontario Health (Cancer Care Ontario) Hematology Cancer Drug Advisory Committee
OH-CCO’s Cancer Drug Advisory Committees provide timely evidence-based clinical and health system guidance on drug-related issues in support of CCO’s mandate, including the Provincial Drug Reimbursement Programs (PDRP) and the Systemic Treatment Program.
Information Gathering
Information is gathered via video conferencing and emails.
Current Treatments and Treatment Goals
Current standard of care is R-CHOP.
Treatment goals: Cure and prevent need for salvage treatment (e.g., transplant)
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
A good proportion of patients still fails first-line therapy.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
Pola-R-CHP will be an alternative to 1L R-CHOP, as per POLARIX, for intermediate to high-risk population.
Which patients would be best suited for treatment with the drug under review? Which patients would be least suitable for treatment with the drug under review?
As per the study inclusion criteria, for intermediate to high-risk population.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice? How often should treatment response be assessed?
Per standard lymphoma response criteria and testing.
What factors should be considered when deciding to discontinue treatment with the drug under review?
Disease progression, adverse events
What settings are appropriate for treatment with [drug under review]? Is a specialist required to diagnose, treat, and monitor patients who might receive [drug under review]?
Outpatient administration
Additional Information
Not applicable.
Conflict of Interest Declarations — Ontario Health (Cancer Care Ontario) Hematology Cancer Drug Advisory Committee
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please refer to the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
OH-CCO provided secretariat function to the group.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission?
No.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician who contributed to the input.
Declaration for Clinician 1
Name: Dr. Tom Kouroukis
Position: Lead, OH-CCO Hematology Cancer Drug Advisory Committee (“Hem DAC”)
Date: 16-Feb-2023
Table 8: COI Declaration for OH-CCO Hematology Cancer Drug Advisory Committee — Clinician 1
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 2
Name: Dr. Jordan Herst
Position: Member, OH-CCO Hem DAC
Date: 16-Feb-2023
Table 9: COI Declaration for OH-CCO Hematology Cancer Drug Advisory Committee — Clinician 2
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 3
Name: Dr. Pierre Villeneuve
Position: Member, OH-CCO Hem DAC
Date: 16-Feb-2023
Table 10: COI Declaration for OH-CCO Hematology Cancer Drug Advisory Committee — Clinician 3
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 4
Name: Dr. Lee Mozessohn
Position: Member, OH-CCO Hem DAC
Date: 16-Feb-2023
Table 11: COI Declaration for OH-CCO Hematology Cancer Drug Advisory Committee — Clinician 4
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 5
Name: Dr. Selay Lam
Position: Member, OH-CCO Hem DAC
Date: 16-Feb-2023
Table 12: COI Declaration for OH-CCO Hematology Cancer Drug Advisory Committee — Clinician 5
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Roche | X | — | — | — |
Declaration for Clinician 6
Name: Dr. Guillaume Richard-Carpentier
Position: Member, OH-CCO Hem DAC
Date: 15-March-2023
Table 13: COI Declaration for OH-CCO Hematology Cancer Drug Advisory Committee — Clinician 6
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 7
Name: Dr. Joanna Graczyk
Position: Member, OH-CCO Hem DAC
Date: 19-March-2023
Table 14: COI Declaration for OH-CCO Hematology Cancer Drug Advisory Committee — Clinician 7
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Canadian Hematologists/Oncologists Treating DLBCL
About Canadian Hematologists/Oncologists Treating DLBCL
We are a group of Canadian hematologists/oncologists who treat patients with diffuse large B-cell lymphoma (DLBCL). We represent the specialty from across Canada in both academic and community settings and share the goals of improving the outcomes and quality of life of patients diagnosed with DLBCL.
Information Gathering
An initial draft was prepared by Dr. Laurie Sehn with the editorial assistance of a freelance medical writer (Cynthia N. Lank). The resulting final submission was distributed to a group of Canadian physicians who treat patients with DLBCL. Physicians who wished to support this Clinician Group Input submission are listed below.
Current Treatments and Treatment Goals
Diffuse large B-cell lymphoma (DLBCL) is the most common subtype of non-Hodgkin lymphoma (NHL), accounting for 30% to 40% of NHL cases. Using incidence data from the Canadian Cancer Statistics website and assuming 35% of NHL cases are DLBCL, the anticipated number of newly diagnosed DLBCL cases is estimated at 4048 in 2023 with a projected increase to 4249 in 2026. (Brenner D, et al. Projected estimates of cancer in Canada in 2022. Supplementary Table 3. CMAJ. 2022;194(17): E601-E607; DOI 10. 1503/cmaj.212097) Screening procedures are not available. The incidence of DLBCL increases with age (median age of 65 years at diagnosis). Without treatment, DLBCL is fatal. The primary goal of treatment is to cure DLBCL with first-line therapy, while minimizing treatment-related toxicities. However, with current therapy, approximately 40% of patients will relapse or are refractory (R/R) to first-line standard of care.
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
The first-line standard of care treatment for patients with DLBCL is rituximab with cyclophosphamide, doxorubicin, vincristine and prednisone (R-CHOP), which can cure approximately 60% of patients. Following front-line therapy, patients who remain progression-free at 24 months have a survival expectancy similar to the general population. The majority of patients who relapse will do so within the first 24 months after starting treatment and will require second and subsequent lines of therapy. The best chance of achieving cure is with first-line therapy. While secondary therapies may offer some patients a chance of cure, most are associated with a low chance of success and lowering of health-related quality of life (HRQoL) due to excessive risk of adverse events and side effects. Most patients who have R/R DLBCL will experience further relapse or die of their disease.
Since the introduction of R-CHOP over 20 years ago, there has been no advancement in treatment options for previously untreated DLBCL patients. Indeed, numerous phase III studies investigating intensive or novel therapies have failed to show additional benefit. There remains a significant unmet need to improve the cure rate for patients with DLBCL with first-line therapy, to reduce the high rate of relapsed/refractory disease, thereby improving outcomes and reducing the need for patients to proceed to more toxic secondary options. The greatest impact to patients with DLBCL, their families, and the healthcare system would be to prevent disease relapse or progression altogether.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
Polatuzumab vedotin is an antibody–drug conjugate developed to specifically target B-cell malignancies that express CD79b (> 95% of malignant B cells in DLBCL). Polatuzumab vedotin binds specifically to CD79b, which results in antibody–drug conjugate internalization and subsequent release of the potent microtubule inhibitor monomethyl auristatin E (MMAE) inside the targeted neoplastic B cells. This results in tumour apoptosis, while potentially minimizing the effects on normal healthy cells. Polatuzumab vedotin has been approved in combination with bendamustine and rituximab to treat patients with relapsed/refractory DLBCL. It has recently been evaluated in combination treatment in the first line setting and is the first novel combination to demonstrate a significant and clinically meaningful benefit over R-CHOP, with a well-tolerated and comparable safety and HRQoL profile.
The POLARIX trial investigated the use of polatuzumab vedotin in patients with previously untreated DLBCL. (Tilly H, et al. Polatuzumab vedotin in previously untreated diffuse large B-cell lymphoma. N Engl J Med. 2022;386:351-363. DOI: 10.1056/NEJMoa2115304) This was a phase III randomized, placebo-controlled, double-blind trial in which polatuzumab vedotin+R-CHP (pola-R-CHP) was compared to R-CHOP in patients with DLBCL and an International Prognostic Index (IPI) score of 2–5. The primary endpoint was progression-free survival (PFS). Patients treated with pola-R-CHP had a risk of disease progression, relapse or death that was 27% lower (stratified hazard ratio, 0.73; 95% confidence interval, 0.57 to 0.95; p = 0.02) than those treated with R-CHOP (i.e., ~1 out of 4 patients were spared from having a PFS event). At 2 years, PFS was 76.7% vs. 70.2% in the pola-R-CHP and R-CHOP groups, respectively. Patients receiving pola-R-CHP also had a statistically significant improvement in key secondary endpoints, including disease-free survival (DFS) and event-free survival (EFS). While an overall survival (OS) benefit has not yet been observed, longer follow-up may be required to assess potential impact. Importantly, more patients in the R-CHOP arm have required treatment with subsequent therapies (including stem-cell transplantation and chimeric antigen receptor T-cell therapy), which may have impacted OS. The course of DLBCL is characterized by an early risk of relapse followed by a relative plateau in the PFS curve. With a median follow-up at initial reporting of more than 2 years, the PFS results are felt to be mature and indicative of a higher cure rate in the first line setting with pola-R-CHP. Importantly, this was achieved with no differences in dose delivery and no new safety signals identified. The overall toxicity profile of pola-R-CHP was comparable to R-CHOP, with no difference in neuropathy observed.
Which patients would be best suited for treatment with the drug under review? Which patients would be least suitable for treatment with the drug under review?
The POLARIX trial included patients with previously untreated DLBCL with an International Prognostic Index (IPI) score of 2–5. These patients would be the recommended population for treatment with pola-R-CHP. The overall toxicity profile was comparable to R-CHOP. Thus, patients in clinical practice who would be suitable for R-CHOP would be appropriate to receive pola-R-CHP.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice? How often should treatment response be assessed?
The primary endpoint of the POLARIX study was progression-free survival (PFS), which is a clinically meaningful endpoint that is used in clinical practice. Importantly, PFS at 24 months has been shown to be a clinically meaningful endpoint, since most progressions/relapses will occur within 24 months. PFS is thus a surrogate for likely cure.
The response during therapy is typically monitored by CT scan, and post-treatment patients are assessed by both CT scan and PET scan. Post-therapy, patients are typically monitored clinically every 3 months for 2 years, then every 6 to 12 months for evidence of progression.
What factors should be considered when deciding to discontinue treatment with the drug under review?
The POLARIX trial demonstrated that pola-R-CHP had a similar safety profile to R-CHOP. As this is curative-intent therapy, the goal is to optimize treatment delivery by providing treatment-as-tolerated on the intended 3-weekly schedule. However, patients should be monitored for adverse events, including neuropathy, with dose reductions or interruptions if necessary. Patients should also be monitored for treatment response. If progression is observed, treatment discontinuation, and consideration for secondary therapies is most appropriate.
What settings are appropriate for treatment with polatuzumab vedotin? Is a specialist required to diagnose, treat, and monitor patients who might receive polatuzumab vedotin?
Treatment with pola-R-CHP has a similar safety profile to R-CHOP and it is anticipated that it can be safely administered in similar settings as R-CHOP. In general, it is out-patient systemic therapy that can be routinely administered by physicians with experience in oncology therapy (typically hematologists/oncologists). Requirements for the administration of pola-R-CHP are those typically required for other conventional cancer treatments. Pola-R-CHP is administered according to a similar schedule as R-CHOP, and it will not increase the number of infusions required or the expected clinic visits/hospitalizations for complications.
Additional Information
The POLARIX trial has demonstrated that pola-R-CHP is a more effective regimen than R-CHOP for the first-line therapy of patients with DLBCL, with comparable safety. The improvement in progression-free survival will result in a higher cure rate in the first line setting for Canadian patients with DLBCL. Fewer patients will develop relapsed/refractory disease and require secondary therapies that are generally associated with higher toxicity and cost. Sadly, most patients who require these subsequent lines of treatment for R/R DLBCL still die of their disease, so curing more patients in the first-line setting is an important goal.
Conflict of Interest Declarations — Canadian Hematologists/Oncologists Treating DLBCL
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict-of-interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please refer to the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
Medical writer, Cynthia Lank (Cynthia N. Lank Editorial Services, Halifax, NS), provided editorial and logistical support for the initial draft and incorporated feedback as directed by Dr. Sehn.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
Medical writer, Cynthia Lank (Cynthia N. Lank Editorial Services, Halifax, NS), provided editorial and logistical support for the initial draft and incorporated feedback as directed by Dr. Sehn.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician who contributed to the input.
Declaration for Clinician 1
Name: Dr. Laurie H. Sehn
Position: Chair, Lymphoma Tumour Group, BC Cancer Centre for Lymphoid Cancer, Clinical Professor of Medicine, Division of Medical Oncology, University of British Columbia, Vancouver, BC
Date: 27-03-2023
Table 15: COI Declaration for Canadian Hematologists/Oncologists Treating DLBCL — Clinician 1
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Roche | — | — | X | — |
Genentech | — | — | X | — |
Declaration for Clinician 2
Name: Dr. Sarah Anne Ingber
Position: General Internal Medicine Doctor, Hematologist, North York General Hospital, Toronto, ON
Date: 17-05-2023
Table 16: COI Declaration for Canadian Hematologists/Oncologists Treating DLBCL — Clinician 2
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 3
Name: Dr. Nicholas Allen Forward
Position: Assistant Professor, Division of Hematology and Hematologic Oncology, Department of Medicine, Dalhousie University/Nova Scotia Health, Halifax, NS
Date: 17-03-2023
Table 17: COI Declaration for Canadian Hematologists/Oncologists Treating DLBCL — Clinician 3
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Roche | — | — | X | — |
Declaration for Clinician 4
Name: Dr. Philip George Kuruvilla
Position: Oncologist, William Osler Health System, Brampton, ON
Date: 17-03-2023
Table 18: COI Declaration for Canadian Hematologists/Oncologists Treating DLBCL — Clinician 4
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Roche | — | — | X | — |
Declaration for Clinician 5
Name: Dr. Diego Villa
Position: Clinical Associate Professor, Division of Medical Oncology, University of British Columbia, Vancouver, BC
Table 19: COI Declaration for Canadian Hematologists/Oncologists Treating DLBCL — Clinician 5
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Roche | — | — | X | — |
Declaration for Clinician 6
Name: Dr. Joanne Hickey
Position: Associate Professor of Medicine, Division of Hematology, Memorial University, St. John's, NFL
Date: 17-03-2023
Table 20: COI Declaration for Canadian Hematologists/Oncologists Treating DLBCL — Clinician 6
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 7
Name: Dr. Alejandro Lazo-Langner
Position: Associate Professor of Medicine, Oncology, and Epidemiology and Biostatistics at Western University, London, ON
Date: 17-03-2023
Table 21: COI Declaration for Canadian Hematologists/Oncologists Treating DLBCL — Clinician 7
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 8
Name: Dr. Julie Stakiw
Position: Associate Professor, University of Saskatchewan; Medical Director, Saskatchewan Cancer Agency, Saskatoon, SK
Date: 17-03-2023
Table 22: COI Declaration for Canadian Hematologists/Oncologists Treating DLBCL — Clinician 8
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 9
Name: Dr. Carolyn Owen
Position: Associate Professor, Division of Hematology and Hematological Malignancies, U of Calgary
Date: 17-03-2023
Table 23: COI Declaration for Canadian Hematologists/Oncologists Treating DLBCL — Clinician 9
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Roche | — | — | X | — |
Declaration for Clinician 10
Name: Dr. Mohamed Elemary
Position: Professor, Division of Oncology, University of Saskatchewan, Saskatoon, SK
Date: 17-03-2023
Table 24: COI Declaration for Canadian Hematologists/Oncologists Treating DLBCL — Clinician 10
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 11
Name: Dr. Carolyn Ruth Faught
Position: Assistant Professor of Medicine, Hematology, University of Ottawa, Ottawa, ON
Date: 17-03-2023
Table 25: COI Declaration for Canadian Hematologists/Oncologists Treating DLBCL — Clinician 11
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 12
Name: Dr. Daniel Ontko
Position: Hematologist, Island Health, Nanaimo, BC
Date: 17-03-2023
Table 26: COI Declaration for Canadian Hematologists/Oncologists Treating DLBCL — Clinician 12
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 13
Name: Dr. Peter Duggan
Position: Associate Clinical professor of Medicine, University of Calgary, Calgary, AB
Date: 17-03-2023
Table 27: COI Declaration for Canadian Hematologists/Oncologists Treating DLBCL — Clinician 13
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Roche | — | — | X | — |
Declaration for Clinician 14
Name: Dr. Simon D. Baxter
Position: Clinical Assistant Professor of Medicine, Division of Medical Oncology, University of British Columbia, Medical Oncologist, BC Cancer, Kelowna, BC
Date: 17-03-2023
Table 28: COI Declaration for Canadian Hematologists/Oncologists Treating DLBCL — Clinician 14
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 15
Name: Dr. Nicol Alexander Macpherson
Position: Clinical Associate Professor, Department of Medicine, Faculty of Medicine, University of British Columbia, Department of Medical Oncology, BC Cancer Agency, Victoria, BC
Date: 17-03-2023
Table 29: COI Declaration for Canadian Hematologists/Oncologists Treating DLBCL — Clinician 15
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 16
Name: Dr. Hussein S Baden
Position: Assistant Professor, Department of Oncology - Hematology, University of Saskatchewan, Regina, SK
Date: 18-03-2023
Table 30: COI Declaration for Canadian Hematologists/Oncologists Treating DLBCL — Clinician 16
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 17
Name: Dr. Daryl Roitman
Position: Hematologist/Oncologist; Lecturer, Division of Medical Oncology, University of Toronto, Toronto, ON
Date: 18-03-2023
Table 31: COI Declaration for Canadian Hematologists/Oncologists Treating DLBCL — Clinician 17
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 18
Name: Dr. Eve St-Hilaire
Position: Hematologist, Dr Georges L Dumont University Hospital Centre, Moncton, NB
Date: 19-03-2023
Table 32: COI Declaration for Canadian Hematologists/Oncologists Treating DLBCL — Clinician 18
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 19
Name: Dr. Leslie N Zypchen
Position: Clinical Associate Professor, Division of Hematology, University of British Columbia
Date: 19-03-2023
Table 33: COI Declaration for Canadian Hematologists/Oncologists Treating DLBCL — Clinician 19
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 20
Name: Dr. Sathish Kumar Gopalakrishnan
Position: Director, Complex Malignant Haematology, Health Sciences North, Sudbury, ON
Date: 20-03-2023
Table 34: COI Declaration for Canadian Hematologists/Oncologists Treating DLBCL — Clinician 20
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 21
Name: Dr. Ardashes Avanessian
Position: Clinical Assistant Professor of Medicine, Division of Medical Oncology, University of British Columbia, Vancouver, BC
Date: 19-03-2023
Table 35: COI Declaration for Canadian Hematologists/Oncologists Treating DLBCL — Clinician 21
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
AstraZeneca | — | — | X | — |
Sanofi | — | — | X | — |
Roche | — | — | X | — |
Jazz Pharmaceuticals | — | — | X | — |
Declaration for Clinician 22
Name: Dr. Alina Gerrie
Position: Assistant Professor of Medicine, Division of Medical Oncology, University of British Columbia, Vancouver, BC
Date: 19-03-2023
Table 36: COI Declaration for Canadian Hematologists/Oncologists Treating DLBCL — Clinician 22
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
AstraZeneca | — | — | X | — |
AbbVie | — | — | X | — |
Janssen | — | — | X | — |
BeiGene | — | — | X | — |
Declaration for Clinician 23
Name: Dr. Mary-Margaret Keating
Position: Assistant Professor of Medicine, Division of Hematology and Hematologic Oncology, Dalhousie University, Halifax, NS
Date: 15-03-2023
Table 37: COI Declaration for Canadian Hematologists/Oncologists Treating DLBCL — Clinician 23
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Roche | — | — | X | — |
Declaration for Clinician 24
Name: Dr. Natasha Ann Pardy
Position: Adult Hematologist, Eastern Health; Clinical Assistant Professor, Discipline of Medicine, Memorial University
Date: 20-03-2023
Table 38: COI Declaration for Canadian Hematologists/Oncologists Treating DLBCL — Clinician 24
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Roche | — | — | X | — |
Declaration for Clinician 25
Name: Dr. Muhammad Saleem Raza
Position: Medical Oncologist, Dr. Everett Chalmers Hospital, Fredericton, NB
Date: 20-03-2023
Table 39: COI Declaration for Canadian Hematologists/Oncologists Treating DLBCL — Clinician 25
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 26
Name: Dr. Ariah Joshua Schattner
Position: Clinical Lead, Malignant Hematology, R S McLaughlin Durham Regional Cancer Centre, Oshawa ON
Date: 20-03-2023
Table 40: COI Declaration for Canadian Hematologists/Oncologists Treating DLBCL — Clinician 26
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 27
Name: Dr. Kuljit Grewal
Position: Associate Professor of Medicine (Hematology), Memorial University
Date: 20-03-2023
Table 41: COI Declaration for Canadian Hematologists/Oncologists Treating DLBCL — Clinician 27
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Roche | — | — | X | — |
AbbVie | — | — | X | — |
Janssen | — | — | X | — |
AstraZeneca | — | — | X | — |
Declaration for Clinician 28
Name: Randeep Sangha
Position: Associate Professor, Division of Medical Oncology, University of Alberta, Edmonton, AB
Date: 21-03-2023
Table 42: COI Declaration for Canadian Hematologists/Oncologists Treating DLBCL — Clinician 28
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Roche | — | — | X | — |
Declaration for Clinician 29
Name: Dr. Mark Bosch
Position: Associate Clinical Professor of Medicine, Hematological Oncology, Division of Oncology, University of Saskatchewan, Saskatoon, SK
Date: 21-03-2023
Table 43: COI Declaration for Canadian Hematologists/Oncologists Treating DLBCL — Clinician 29
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Roche | — | — | X | — |
Declaration for Clinician 30
Name: Dr. Sasha Smiljanic
Position: Medical Oncologist, Lions Gate Hospital, Vancouver, BC
Date: 22-03-2023
Table 44: COI Declaration for Canadian Hematologists/Oncologists Treating DLBCL — Clinician 30
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 31
Name: Dr. Nicole Marie Beatrice Laferriere
Position: Hematologist; Associate professor, Northern Ontario School of Medicine, Thunder Bay, ON
Date: 22-03-2023
Table 45: COI Declaration for Canadian Hematologists/Oncologists Treating DLBCL — Clinician 31
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 32
Name: Dr. Chantal Léger
Position: Clinical Associate professor, Dept of Medicine, Division of Hematology, University of British Columbia, St. Paul's hospital, Vancouver, BC
Date: 22-03-2023
Table 46: COI Declaration for Canadian Hematologists/Oncologists Treating DLBCL — Clinician 32
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 33
Name: Dr. Sindu Kanjeekal
Position: Assistant Professor, Department of Oncology, Windsor Regional Cancer Program, Schulich School of Medicine & Dentistry; Chief of Oncology, Windsor Regional Hospital, Windsor, ON
Date: 22-03-2023
Table 47: COI Declaration for Canadian Hematologists/Oncologists Treating DLBCL — Clinician 33
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 34
Name: Dr. Signy Chow
Position: Staff Hematologist and Assistant Professor, Odette Cancer Centre and Sunnybrook Health Sciences Centre, University of Toronto
Date: 22-03-2023
Table 48: COI Declaration for Canadian Hematologists/Oncologists Treating DLBCL — Clinician 34
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 35
Name: Dr. Yael Zaretsky
Position: Malignant Hematologist, Credit Valley Hospital, Toronto, ON
Date: 22-03-2023
Table 49: COI Declaration for Canadian Hematologists/Oncologists Treating DLBCL — Clinician 35
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 36
Name: Dr. Loree M Larratt
Position: Professor Emeritus, Clinical Hematology, University of Alberta, Edmonton, AB
Date: 22-03-2023
Table 50: COI Declaration for Canadian Hematologists/Oncologists Treating DLBCL — Clinician 36
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 37
Name: Dr. Waleed Sabry
Position: Clinical Associate Professor of Hematological Oncology, Division of Medical Oncology, University of Saskatchewan, Saskatoon, SK
Date: 22-03-2023
Table 51: COI Declaration for Canadian Hematologists/Oncologists Treating DLBCL — Clinician 37
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Roche | — | — | X | — |
AbbVie | — | — | X | — |
Novartis | — | — | X | — |
Bristol Myers Squibb | — | — | X | — |
Declaration for Clinician 38
Name: Dr. Neil Berinstein
Position: Professor, Departments of Medicine, Immunology and Medical Biophysics, University of Toronto; Medical Oncologist, Affiliate Scientist, Odette Cancer Research Program, Sunnybrook Research Institute, Toronto, ON
Date: 23-03-2023
Table 52: COI Declaration for Canadian Hematologists/Oncologists Treating DLBCL — Clinician 38
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 39
Name: Dr. Isabelle Andri Bence-Bruckler
Position: Associate Professor of Medicine, Division of Hematology, University of Ottawa, Ottawa, ON
Date: 23-03-2023
Table 53: COI Declaration for Canadian Hematologists/Oncologists Treating DLBCL — Clinician 39
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 40
Name: Dr. He Katharine Xing
Position: Medical Oncologist, BC Cancer Surrey, Surrey, BC
Date: 23-03-2023
Table 54: COI Declaration for Canadian Hematologists/Oncologists Treating DLBCL — Clinician 40
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 41
Name: Dr. Joy Mangel
Position: Associate Professor of Medicine, Division of Hematology, Western University, London, ON
Date: 23-03-2023
Table 55: COI Declaration for Canadian Hematologists/Oncologists Treating DLBCL — Clinician 41
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 42
Name: Dr. Pam Skrabek
Position: Associate Professor, Department of Medical Oncology and Hematology, Max Rady College of Medicine, University of Manitoba, Winnipeg, MB
Date: 23-03-2023
Table 56: COI Declaration for Canadian Hematologists/Oncologists Treating DLBCL — Clinician 42
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 43
Name: Dr. Edward Geoffrey Brooks
Position: Medical Oncologist, BC Cancer, Victoria, BC
Date: 23-03-2023
Table 57: COI Declaration for Canadian Hematologists/Oncologists Treating DLBCL — Clinician 43
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 44
Name: Dr. Kirsty Tompkins
Position: Associate Professor of Medicine (and Division Chair), Department of Hematology Memorial University
Date: 24-03-2023
Table 58: COI Declaration for Canadian Hematologists/Oncologists Treating DLBCL — Clinician 44
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 45
Name: Dr. Edward Leslie George Hardy
Position: Clinical Assistant Professor of Medicine, University of British Columbia, Vernon, BC
Date: 26-03-2023
Table 59: COI Declaration for Canadian Hematologists/Oncologists Treating DLBCL — Clinician 45
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 46
Name: Dr. Luke Chen
Position: Clinical Associate Professor of Medicine, Division of Hematology, University of British Columbia; Program Director, General Hematology Residency Program, Vancouver, BC
Vancouver General Hospital
Date: 27-03-2023
Table 60: COI Declaration for Canadian Hematologists/Oncologists Treating DLBCL — Clinician 46
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 47
Name: Dr. Sarah Elizabeth Zed
Position: Associate Professor, Division of Hematology, Department of Medicine, Saint John Regional Hospital (Dalhousie University), Saint John, NB
Date: 27-03-2023
Table 61: COI Declaration for Canadian Hematologists/Oncologists Treating DLBCL — Clinician 47
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 48
Name: Dr. Stephen Michael Reingold
Position: Clinical Investigator, Oncologist, William Osler Health System, Brampton, ON
Date: 27-03-2023
Table 62: COI Declaration for Canadian Hematologists/Oncologists Treating DLBCL — Clinician 48
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 49
Name: Dr. Andrew Daly
Position: Clinical Associate Professor, Department of Medicine (medical oncology), Cumming School of Medicine, University of Calgary, Calgary, AB
Date: 27-03-2023
Table 63: COI Declaration for Canadian Hematologists/Oncologists Treating DLBCL — Clinician 49
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 50
Name: Dr. Caroline Marie Hamm
Position: Medical Oncologist, Windsor Regional Hospital, Windsor, ON
Date: 27-03-2023
Table 64: COI Declaration for Canadian Hematologists/Oncologists Treating DLBCL — Clinician 50
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 51
Name: Dr. Graeme Fraser
Position: Associate Professor, Department of Oncology, McMaster University, Hamilton ON
Date: 27-03-2023
Table 65: COI Declaration for Canadian Hematologists/Oncologists Treating DLBCL — Clinician 51
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 52
Name: Dr. Martina Andrea Trinkaus
Position: Associate Professor, Hematologist, St. Michael's Hospital, University of Toronto, Toronto, ON
Date: 27-03-2023
Table 66: COI Declaration for Canadian Hematologists/Oncologists Treating DLBCL — Clinician 52
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Roche | — | — | X | — |
Declaration for Clinician 53
Name: Dr. Indermohan Singh Sandhu
Position: Medical Oncologist, Grey Bruce Health Services, Ontario
Date: 27-03-2023
Table 67: COI Declaration for Canadian Hematologists/Oncologists Treating DLBCL — Clinician 53
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 54
Name: Dr. Jesse Daniel Shustik
Position: Medical Oncologist, BC Cancer - Surrey Centre, Surrey, BC
Date: 27-03-2023
Table 68: COI Declaration for Canadian Hematologists/Oncologists Treating DLBCL — Clinician 54
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 55
Name: Dr. Kerry Savage
Position: Professor, Medical Oncology, University of British Columbia, Vancouver, BC
Date: 27-03-2023
Table 69: COI Declaration for Canadian Hematologists/Oncologists Treating DLBCL — Clinician 55
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
Redactions: Confidential information in this document may be redacted at the request of the sponsor in accordance with the CADTH Drug Reimbursement Review Confidentiality Guidelines
Stakeholder Input: The views expressed in each submission are those of the submitting organization or individual; not necessarily the views of CADTH or of other organizations. As such, they are independent of CADTH and do not necessarily represent or reflect the view of CADTH. No endorsement by CADTH is intended or should be inferred. By filing with CADTH, the submitting organization or individual agrees to the full disclosure of the information. CADTH does not edit the content of the submissions.
CADTH does use reasonable care to prevent disclosure of personal information in posted material; however, it is ultimately the submitter’s responsibility to ensure no identifying personal information or personal health information is included in the submission. The name of the submitting organization or individual and all conflict of interest information are included in the submission; however, the name of the author, including the name of an individual patient or caregiver submitting the patient input, are not posted.
Accessibility: CADTH is committed to treating people with disabilities in a way that respects their dignity and independence, supports them in accessing material in a timely manner, and provides a robust feedback process to support continuous improvement. All materials prepared by CADTH are available in an accessible format. Where materials provided to CADTH by a submitting organization or individual are not available in an accessible format, CADTH will provide a summary document upon request. More details on CADTH’s accessibility policies can be found here.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.