CADTH Reimbursement Review
Tremelimumab (Imjudo) in Combination With Durvalumab (Imfinzi)
Sponsor: AstraZeneca Canada Inc.
Therapeutic area: Unresectable hepatocellular carcinoma
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Stakeholder Input
Clinical Review
Abbreviations
AASLD
American Association for the Study of Liver Diseases
AE
adverse event
ALBI
albumin-bilirubin
BCLC
Barcelona Clinic Liver Cancer
BICR
blinded independent central review
BOR
best objective response
CCRAN
Colorectal Cancer Resource & Action Network
CCSN
Canadian Cancer Survivor Network
CGOEN
Canadian Gastrointestinal Oncology Evidence Network
CLF
Canadian Liver Foundation
CI
confidence interval
CTCAE
Common Terminology Criteria for Adverse Events
CTLA-4
cytotoxic T-lymphocyte-associated protein 4
DCR
disease control rate
DCR-16w
disease control rate at 16 weeks
DCR-24w
disease control rate at 24 weeks
DoR
duration of response
ECOG PS
Eastern Cooperative Oncology Group Performance Status
EHS
extrahepatic spread
EORTC QLQ-C30
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30
EORTC QLQ-HCC18
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Hepatocellular Carcinoma 18
ESS
effective sample size
FAS
full analysis set
GHS
global health status
GI
gastrointestinal
HBV
hepatitis B virus
HCC
hepatocellular carcinoma
HCV
hepatitis C virus
HR
hazard ratio
HRQoL
health-related quality of life
IQR
interquartile range
irRECIST
immune-related Response Evaluation Criteria in Solid Tumors
ITC
indirect treatment comparison
ITT
intention-to-treat
MAIC
matching adjusted indirect comparison
MID
minimal important difference
mRECIST
modified Response Evaluation Criteria in Solid Tumors
MVI
macrovascular invasion
NAFLD
nonalcoholic fatty liver disease
NICE
National Institute for Health and Care Excellence
NMA
network meta-analysis
OR
odds ratio
ORR
objective response rate
OS
overall survival
PD-1
programmed cell death 1 protein 1
PD-L1
programmed cell death 1 ligand 1
PD-L2
programmed cell death 1 ligand 2
PFS
progression-free survival
PGIC
Patient’s Global Impression of Change
QoL
quality of life
RCT
randomized controlled trial
RECIST 1.1
Response Evaluation Criteria in Solid Tumors Version 1.1
SAE
serious adverse event
SMQ
Standardized Medical Dictionary for Regulatory Activities Query
TACE
transarterial chemoembolization
TKI
tyrosine kinase inhibitor
TTP
time to progression
Executive Summary
An overview of the submission details for the drug under review is provided Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Information on drug submitted for review | |
Drug product | Tremelimumab (Imjudo), 20 mg/mL, concentrate IV infusion in combination with durvalumab (Imfinzi), 50 mg/mL, concentrate for IV infusiona |
Sponsor | AstraZeneca Canada Inc. |
Indication | Proposed: Imjudo (tremelimumab for injection) in combination with durvalumab is indicated for the first-line treatment of adult patients with unresectable hepatocellular carcinoma who require systemic therapy |
Reimbursement request | Imjudo (tremelimumab for injection) in combination with Imfinzi (durvalumab) for the first-line treatment of patients with unresectable hepatocellular carcinoma |
Health Canada approval status | Under review (pre-NOC) |
Health Canada review pathway | Standard review |
NOC date | To be determined |
Recommended dose | Tremelimumab 300 mg as a single priming dose in combination with 1,500 mg at day 1 of cycle 1, followed by durvalumab 1,500 mg as a single drug every 4 weeks;1 this combination is referred to as the single tremelimumab regular-interval durvalumab (STRIDE) regimen Patients with a body weight of 30 kg or less must receive weight-based dosing, equivalent to tremelimumab 4 mg/kg as a single priming dose in combination with durvalumab 20 mg/kg followed by durvalumab 20 mg/kg as a single drug every 4 weeks until weight exceeds 30 kg |
NOC = Notice of Compliance.
aThis submission to CADTH will be a new drug submission for tremelimumab (in combination with durvalumab), which is also under review with Health Canada as a new drug submission. Tremelimumab is not intended for monotherapy use. The durvalumab product monograph will be updated to reflect the unresectable hepatocellular carcinoma indication via a supplement to a new drug submission following approval of the tremelimumab new drug submission.
Introduction
Primary liver cancer is among the fastest rising cancers in Canada, and it is estimated that 3,500 patients will be diagnosed with primary liver cancer and 1,650 patients in Canada will die from this disease in 2022.2-4 According to Statistics Canada’s Short-term cancer prevalence in Canada, 2018 report, the estimated 5-year prevalence of liver cancer is approximately 11.3 cases per 100,000 for both sexes.5 Hepatocellular carcinoma (HCC) is a severe form of liver cancer that represents about 90% of primary liver cancers globally6 and approximately 72% of liver cancers in Canada.2 HCC is the third leading cause of cancer deaths worldwide,7 with a 5-year survival rate of only 20% in Canada.2 It is most commonly diagnosed in people aged 70 years or older and it is 3 times more common in men than in women.4 Due to the insidious nature of the disease, the majority of patients are diagnosed with advanced disease, with a median survival following diagnosis of approximately 6 to 8 months, or 25% at 1 year.6,7 The predominant risk factors for HCC include chronic infections with the hepatitis B virus (HBV) or hepatitis C virus (HCV), alcohol abuse or alcoholic steatohepatitis, and nonalcoholic fatty liver disease (NAFLD) or nonalcoholic steatohepatitis.7-9 HCC is often diagnosed using noninvasive imaging, tissue biopsies, physical examinations, or blood tests.6,7
For advanced, unresectable HCC, the goal of treatment is to extend long-term survival, delay progression, and maintain and improve the patient’s quality of life (QoL),10 and guidelines recommend the use of systemic targeted therapies.11,12 According to the clinical experts consulted by CADTH, systemic treatment options have improved over the past several years with the introduction of lenvatinib, sorafenib, and the combination of atezolizumab and bevacizumab as first-line systemic treatment options in Canada.11,13-15 The clinical experts consulted by CADTH for this review identified a key limitation of the current first-line therapy of atezolizumab in combination with bevacizumab: patients with untreated or incompletely treated esophageal and/or gastric varices with bleeding or those at high risk for bleeding are not candidates for this therapy.13 Upper endoscopy is indicated for patients with cirrhosis or at high risk of bleeding.
The dossiers for tremelimumab in combination with durvalumab were submitted to CADTH as a pre–Notice of Compliance submission. The proposed Health Canada indication for tremelimumab in combination with durvalumab, for the first-line treatment of adult patients with unresectable HCC who require systemic therapy, generally aligns with the sponsor’s requested reimbursement criteria. Tremelimumab in combination with durvalumab received approval from the FDA in October 2022 for treatment of adult patients with unresectable HCC.16
The objective of this report Is to”revi’w and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of tremelimumab (Imjudo), 20 mg/mL, concentrate IV infusion in combination with durvalumab (Imfinzi), 50 mg/mL, concentrate for IV infusion for the treatment of unresectable HCC in adult patients.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient and clinician groups who responded to CADTH’s call for input and from clinical experts consulted by CADTH for the purpose of this review.
Patient Input
The Colorectal Cancer Resource & Action Network (CCRAN) in collaboration with the Canadian Cancer Survivor Network (CCSN), Canadian Liver Foundation (CLF), and Gastrointestinal (GI) Society provided a collective patient input for this review. The CCRAN is a national not-for-profit patient advocacy group championing the health and well-being of patients in Canada affected by colorectal cancer and those at risk of developing the disease. The CCSN, CLF, and GI Society thoughtfully collaborated with CCRAN to ensure that the perspectives of patients with advanced HCC and their caregivers were captured, represented, and well weaved into this submission. The CCRAN gathered information for this review from in-depth interviews with 2 patients with HCC (both had experience with the currently available treatment of HCC and 1 patient had experience with the drug under review), a literature review, and online public forums for patient-reported outcomes.
According to the patient input received from CCRAN, HCC is the most common primary liver cancer. The CCRAN noted that risk factors associated with HCC include cirrhosis, HBV and HCV infections, and alcohol intake. Both patient interviews indicated that they had not been experiencing any symptoms at the time of HCC diagnosis. The CCRAN indicated that a diagnosis and symptoms of HCC represent a substantial physiological and psychological burden for patients and their caregivers and can significantly affect their health-related quality of life (HRQoL). The CCRAN pointed to various symptoms of HCC that affected patient QoL and daily activities, including sleep disorders, sexual dysfunction, ascites, gynecomastia, pruritis, fatigue, muscle cramps, and lack of appetite, even after treatment. Both patient respondents emphasized that the daily activities that were most commonly affected included the ability to work, participate in activities they enjoy, and spend time with family and friends. One of the interviewed patients (a female aged 92 years who was diagnosed with HCC diagnosis at 71 years) cycled through the same stages of cancer grief — anger, depression, guilt, anxiety, hopelessness, and fear — which hit the patient hard at the time of the initial diagnosis and subsequent relapse.
The CCRAN Indicated that patients with HCC expect any new drug or treatment to come with improvements in the key outcomes of QoL, survival time, manageable side effects, maintained functionality, and the ability to engage in society and contribute to the workforce. According to the patient input received from the CCRAN, HCC is a unique carcinoma because the majority of cases will develop in patients with cirrhosis, and therapeutic options will therefore be limited due to the patient’s overall health status. The CCRAN indicated that patients with early-stage HCC are preferred candidates for resection, transplant, and local ablation, while patients at intermediate stages may be candidates for transarterial chemoembolization (TACE) and those with advanced disease will receive systemic therapies. The CCRAN noted that the current systemic treatments for HCC include lenvatinib, sorafenib, regorafenib, cabozantinib, and atezolizumab in combination with bevacizumab. The limited treatment tolerability, in part due to the side effects, was highlighted by the CCRAN as a major challenge to available systemic therapy for advanced HCC.
One of the interviewed patients (a male 74 years of age diagnosed with HCC at 68 years) had experience with treatment with the drug under review after TACE that negatively affected his QoL. The patient respondent, who had access to tremelimumab in combination with durvalumab through a clinical trial and resided in Cranbrook, British Columbia, indicated that the drug under review has had promising and durable treatment results, with no side effects other than an occasional skin rash. The patient also mentioned that tremelimumab in combination with durvalumab helped him regain functionality and pursue a livelihood, which reduced the burden on his caregivers and loved ones. The CCRAN advocated that tremelimumab in combination with durvalumab be approved for the indication under review and suggested that it will help alleviate the gaps in current HCC therapy.
Clinician Input
Input From Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of unresectable HCC.
The clinical experts consulted by CADTH for this review stated that the treatment goals for patients with unresectable HCC Include prolonging life and delaying progression. They mentioned that treatments have improved in the past several years with the introduction of lenvatinib, which has superior efficacy and lower toxicity compared with sorafenib, and atezolizumab in combination with bevacizumab. However, the benefits of current treatments have been incremental. Moreover, the use of atezolizumab in combination with bevacizumab is limited to patients who have had a recent upper endoscopy and were found not to have symptomatic varices. The clinical experts noted that tremelimumab in combination with durvalumab would be indicated in the first line for patients who would currently be indicated for atezolizumab and bevacizumab, and that the choice of therapy would depend on clinician and patient preferences. They added that it may be indicated for patients who had started tyrosine kinase inhibitor (TKI) therapy and progressed or experienced severe toxicity.
The clinical experts agreed that tremelimumab in combination with durvalumab would be recommended for patients with unresectable HCC and preserved liver function (Childs-Pugh class A) and a good performance status (potentially up to an Eastern Cooperative Oncology Group Performance Status [ECOG PS] of 2), and who are not indicated for local therapy such as TACE. They mentioned that patients who were on TKIs and/or other therapies but had severe side effects that led to permanent discontinuation would also be eligible for tremelimumab in combination with durvalumab. Patients who are not candidates for other immune checkpoint inhibitors would not be candidates for tremelimumab in combination with durvalumab.
The clinical experts mentioned that, in clinical practice, imaging would be obtained every 3 months to assess response to treatment. The most important outcomes are prolonged survival, delayed progression, disease control, and maintained QoL with a low toxicity profile. The clinical experts note that tremelimumab in combination with durvalumab should be discontinued in the event of disease progression or intractable severe immune-related adverse effects. According to the clinical experts consulted for this review, tremelimumab in combination with durvalumab can be administered in most systemic therapy suites in which cancer patients receive chemotherapy and immunotherapy. Administration of this therapy can be supervised by most medical oncologists experienced in treating HCC.
Clinician Group Input
The clinician group input was obtained from 2 clinician groups, including the Canadian Gastrointestinal Oncology Evidence Network (CGOEN) represented by 6 clinicians; the Ontario Health (Cancer Care Ontario) Gastrointestinal Cancer Drug Advisory Committee, represented by 5 clinicians; and a clinician from the Alberta Health Services Cancer Care at the University of Alberta.
The CGOEN indicated that, with modern systemic therapy, downsizing of disease has led to newer options for local regional treatments of the liver (i.e., stereotactic radiation, embolization, ablation, resection, or transplant). The CGOEN emphasized that patients with HCC may be at increased risk of bleeding due to the underlying liver disease and the vascular nature of the disease itself, and therapy that does not increase this risk will be key in this area. The clinician groups agreed that, given an acceptable safety profile, tremelimumab in combination with durvalumab will be another first-line HCC treatment option, particularly for patients with hypertension or varices, or when upper GI endoscopy is not available. The clinician from the University of Alberta indicated that tremelimumab in combination with durvalumab may become the preferred first-line immunotherapy option for treatment of patients with unresectable HCC. The CGOEN and the clinician from the University of Alberta noted that patients receiving tremelimumab in combination with durvalumab would have fewer clinic visits and less time in the clinic as they would be treated every 4 weeks and essentially with 1 drug except for the first cycle, while for atezolizumab in combination with bevacizumab, patients are treated every 3 weeks and with 2 drugs. The clinician groups identified several reasons for discontinuing tremelimumab in combination with durvalumab, including disease progression, unacceptable drug-related toxicities, or patient preference. The clinician groups emphasized that treatment with tremelimumab in combination with durvalumab should be provided by clinicians with expertise and experience in treating HCC. The GI Drug Advisory Committee noted that treatment with tremelimumab in combination with durvalumab should be performed in outpatient infusion clinics, including satellite clinics.
Drug Program Input
The Provincial Advisory Group identified the following jurisdictional implementation issues: relevant comparators and considerations for initiation of therapy, continuation or renewal of therapy, discontinuation of therapy, and prescribing of therapy. The clinical experts consulted by CADTH weighed evidence from the HIMALAYA study and other clinical considerations to provide responses to the Provincial Advisory Group’s drug program implementation questions (Table 4).
Clinical Evidence
Pivotal Studies and Randomized Controlled Trial Evidence
Description of Studies
The HIMALAYA study was a randomized, open-label, sponsor-blind, multicentre, global, phase III study to assess the efficacy and safety of tremelimumab in combination with durvalumab versus sorafenib in the treatment of patients with unresectable HCC who are not eligible for locoregional therapy and have not received prior systemic therapy for HCC in the first-line setting. The primary objective was to compare the overall survival (OS) in all randomized patients receiving tremelimumab in combination with durvalumab versus thos receiving sorafenib. Secondary objectives included comparing OS rates (at 18, 24, and 36 months), progression-free survival (PFS), time to progression (TTP), objective response rate (ORR), disease control rate (DCR), and duration of response (DoR) as assessed by investigators, patient-reported outcomes, and safety between both treatment groups. The study was funded by AstraZeneca Canada and included 9 study centres in Canada.
Patients were randomly assigned in a 1:1:1:1 ratio using an interactive web response system into 1 of 4 treatment groups: tremelimumab in combination with durvalumab (300 mg × 1 dose plus durvalumab 1,500 mg every 4 weeks; n = 393), sorafenib (400 mg twice daily; n = 389), durvalumab monotherapy (not included in this review; n = 389), and a different dosing regimen of tremelimumab in combination with durvalumab (n = 153, recruitment to group closed due to preliminary efficacy findings). Randomization was stratified according to macrovascular invasion (MVI; yes or no), etiology of liver disease (confirmed HBV versus confirmed HCV versus others), and ECOG PS (0 versus 1). Tumour imaging assessments were to be performed at randomization and then every 8 weeks (± 1 week) for the first 48 weeks following randomization, and every 12 weeks (± 1 week) thereafter until confirmed disease progression.
Patient demographic characteristics and key disease characteristics were balanced between both treatment groups. ||| |||| ||| || |||| |||||| ||| ||||||||||||| || ||||| ||| and up to 15% of the patients in both groups were aged 75 years or older. |||| |||||||| |||| |||| |||||| ||| ||| || |||||||| |||| ||||||| |||||| |||| |||| ||||| |||||| ||||| ||||| || |||||||| |||| |||||| |||||| ||| |||||||| ||| |||||||||| ||||| ||||||||. Approximately 80% of patients had a Barcelona Clinic Liver Cancer (BCLC) stage of C, and 20% had a BCLC stage of B. Half of the patients had extrahepatic spread (EHS), and a quarter of patients had MVI. |||| ||||||| || |||||||| ||||||| ||||||| |||||||||| |||| |||||| || |||||||| ||| ||||||||| ||||||||||| |||||||||||||||||||||||||| ||| ||| ||||||||| ||| ||||||| |||||||, and 12% in the tremelimumab in combination with durvalumab group and 10% in the sorafenib group had received prior radiotherapy.
Efficacy Results
Key efficacy results of the HIMALAYA trial for all randomized patients are summarized in Table 2. As of the final primary analysis data cut-off date of August 27, 2021, ||||||| ||||||||| |||| |||||| ||||||, and the median follow-up times were 33.2 months (95% confidence interval [CI], 31.7 to 34.5) in the tremelimumab in combination with durvalumab group and 32.2 months (95% CI, 30.4 to 33.7) in the sorafenib group. The median total treatment durations were 5.5 months (range = 0.4 to 42.7) in the tremelimumab in combination with durvalumab group and 4.1 months (range = 0.1 to 38.6) in the sorafenib group.
Table 2: Summary of Key Results From the HIMALAYA Study (FAS With Final Data Cut-Off of August 27, 2021)
Detail | Tremelimumab in combination with durvalumab N = 393 | Sorafenib N = 389 |
|---|---|---|
Overall survival | ||
Median follow-up duration in all patients, months (95% CI) | 33.2 (31.7 to 34.5) | 32.2 (30.4 to 33.7) |
Median OSa (95% CI), months | 16.4 (14.2 to 19.6) | 13.8 (12.3 to 16.1) |
Hazard ratio (96.02% CI)b | 0.78 (0.65 to 0.93) | |
P value (2-sided)b | 0.0035 | |
Progression-free survival | ||
Median follow-up duration in all patients, months (range) | 3.8 (0.0 to 41.5) | 3.8 (0.0 to 33.4) |
Median PFSa (95% CI), months | 3.78 (3.68 to 5.32) | 4.07 (3.75 to 5.49) |
Hazard ratio (95% CI) | 0.90 (0.77 to 1.05) | |
P valuec | 0.1625 | |
Progression-free at data cut-off, n (%) | 49 (12.5) | 19 (4.9) |
Objective response rate in patients with confirmed responsesd | ||
Objective response, n (%) | 79 (20.1) | 20 (5.1) |
Complete response | 12 (3.1) | 0 |
Partial response | 67 (17.0) | 20 (5.1) |
|||| |||||| ||||||||| |||||||| |||| ||| | |||| ||||| || ||||| | |
P valuee | < 0.0001 | |
Duration of response in patients with confirmed responsesd | ||
n | 79 | 20 |
Median (interquartile range) | 22.34 (8.54 to NR) | 18.43 (6.51 to 25.99) |
Best objective response in patients with unconfirmed responsesd | ||
|||||| |||| | || |||||| | || ||||| |
Complete response | 13 (3.3) | 0 |
Partial response | 81 (20.6) | 26 (6.7) |
|||||||||||| |||||| |||| | ||| |||||| | ||| |||||| |
|||||| ||||||| | ||| |||||| | ||| |||||| |
||||||||||| | ||| |||||| | ||| |||||| |
Time to progression | ||
|||||| |||| |||| |||| |||||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
||||||||| |||||||| || ||| |||||||| ||||||||| |||||| ||||||| | |||| ||||| || |||||| | |||| ||||| || |||||| |
|| |||| ||| | |||| ||||| || ||||| | |
||||||| |||||| | |||||| | |
CI = confidence interval; CR = complete response; ECOG PS = Eastern Cooperative Oncology Group Performance Status; FAS = final analysis set; HBV = hepatitis B virus; HBC = hepatitis C virus; MVI = macrovascular invasion; NR = not reached; OS = overall survival; PFS = progression-free survival; PR = partial response; RECIST = Response Evaluation Criteria in Solid Tumors; vs.= versus.
aCalculated using the Kaplan-Meier technique.
bThe adjusted alpha levels for the 2-sided superiority test of tremelimumab in combination with durvalumab vs. sorafenib and CI were derived from the exact number of OS events for each comparison using the Lan and DeMets approach that approximates the O’Brien-Fleming spending function. Analysis performed using a stratified log-rank test adjusting for treatment, etiology of liver disease (HBV vs. HCV vs. others), ECOG PS (0 vs. 1), and MVI (yes vs. no). P value has been adjusted for multiple testing.
cAnalysis performed using stratified log-rank test adjusting for treatment, etiology of liver disease (HBV vs. HCV vs. others), ECOG PS (0 vs. 1), and MVI (yes vs. no). P value has not been adjusted for multiple testing.
dA confirmed response of CR/PR means that a response of CR/PR was recorded at 1 visit and confirmed by repeat imaging not less than 4 weeks after the visit where response was first observed with no evidence of progression between the initial and confirmation visit. Unconfirmed responses were not confirmed by repeat imaging.
eAnalysis was performed using a logistic regression model adjusted for treatment with factors for etiology of liver disease, ECOG PS, and MVI. P value has not been adjusted for multiple testing.
Source: HIMALAYA Clinical Study Report17 (details from the table have been taken from the sponsor’s Summary of Clinical Evidence).
The efficacy analyses of OS in all randomized patients showed that patients in the tremelimumab in combination with durvalumab group had a longer OS than those in the sorafenib group. The median OS was 16.4 months (95% CI, 14.2 to 19.6) in the tremelimumab in combination with durvalumab group compared to 13.8 months (95% CI, 12.3 to 16.1) in the sorafenib group, with a hazard ratio (HR) of 0.78 (96.02% CI, 0.65 to 0.93; P = 0.0035). The OS rates at 36 months were 30.7% (95% CI, 25.8 to 35.7) in the tremelimumab in combination with durvalumab group and 20.2% (95% CI,15.8 to 25.1) in the sorafenib group. Effect estimates for all predefined subgroups were consistent with the overall OS analysis.
All secondary outcomes were based on investigator assessment according to Response Evaluation Criteria in Solid Tumors Version 1.1 (RECIST 1.1) and were not adjusted for multiplicity. Median PFS in the full analysis set (FAS) was 3.8 months in the tremelimumab in combination with durvalumab group and 4.1 months in the sorafenib group, with an HR of 0.90 (95% CI, 0.77 to 1.05). The ORRs were 20.1% (79 patients) in the tremelimumab in combination with durvalumab group and 5.1% (20 patients) in the sorafenib group. When comparing tremelimumab in combination with durvalumab against sorafenib, the || ||| ||| ||| |||| |||| ||| |||| || ||||| || |||||| || |||||||||||| || ||||||||||| |||| |||||||||||||||| |||| || |||||||| ||||||| || ||| |||||||||||| || ||||||||||| |||| |||||||||| ||||| ||| || |||||||| |||||| || ||| ||||||||| ||||| ||| |||||||| ||||| ||||||||| |||||||| ||||| || |||||||| |||||||| |||| || ||||||| |||||||| |||| ||||| || ||||||||||| ||||||||| |||||| ||||||||| ||| ||||||||| || |||||| ||||||| || ||||| |||||| ||||| ||| |||||| || |||||||||||| ||||||||||. Of the patients in the tremelimumab in combination with durvalumab group, 13 (3.3%) achieved a complete response, while none in the sorafenib group achieved a similar outcome. Among the 79 responders in the tremelimumab in combination with durvalumab group and 20 responders in the sorafenib group, the median DoRs based on investigator assessment according to RECIST 1.1 were 22.3 months (interquartile range [IQR] = 8.5 to not reached) and 18.4 months (IQR = 6.5 to 26), respectively. ||| |||||||||| || |||||||| ||||||||| || |||||||| || || |||||| ||||| || ||| |||||||||||| ||||||||| ||| ||||| || ||| |||||||||||| || ||||||||||| |||| |||||||||| ||||| ||| ||||| || ||| ||||||||| |||||| The median times to onset of response from randomization were 2.2 months (IQR = 1.8 to 4) in the tremelimumab in combination with durvalumab group and 3.8 months (IQR = 1.9 to 8.4) in the sorafenib group. The overall DCR (complete response, partial response, or stable disease) was similar for the 2 groups, with 236 patients (60.1%) in the tremelimumab in combination with durvalumab group and 236 patients (60.7%) in the sorafenib group achieving control of the disease. ||| |||||| |||| || ||||||||||| ||| ||| |||||| |||| ||| ||| || |||| || ||| |||||||||||| || ||||||||||| |||| |||||||||| ||||| ||| ||| |||||| |||| ||| ||| || |||| || ||| ||||||||| |||||| Results from the assessment of exploratory outcomes (based on blinded independent central review [BICR] assessments using modified RECIST 1.1 for HCC and immune-related Response Evaluation Criteria in Solid Tumors [irRECIST]) were not provided by the sponsor.
Results for patient-reported outcomes as assessed by the European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 (EORTC QLQ-C30) and European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Hepatocellular Carcinoma 18 (EORTC QLQ-HCC18) suggested a similar overall health status in both study groups at baseline, with no mean change scores from baseline reaching the minimal important difference (MID) of a mean change of 10 points or greater at any time point. However, |||||||||| ||||| ||||||||||| || ||| ||| ||| || |||| || || ||| |||||||||||| || ||||||||||| |||| |||||||||| ||| ||||||||| |||||| ||||||||||||. Median time to deterioration of scores for patients favoured tremelimumab in combination with durvalumab over sorafenib in the EORTC QLQ-C30 and Global Health Status (GHS)/QoL (7.5 versus 5.7 months; HR = 0.76; 95% CI, 0.61 to 0.96); physical functioning (12.9 versus 7.4 months; HR = 0.68; 95% CI, 0.53 to 0.87), ||||||| |||| || ||| ||||||| || |||| ||||| || ||||||| |||||| ||||| || |||| ||||||| || |||| ||||| || ||||||| |||||||| |||| ||||| || ||| ||||||| || |||| ||||| || ||||||| ||| ||||| ||||||||| ||||||||| |||| ||||| || ||| ||||||| || |||| ||||| ||||||| ||| ||||||||| |||||||| ||||| || |||| ||||||| || |||| ||||| |||||||| The improvement rate in ||||||||||||||||||||| ||| |||||| || ||| |||||||||||| || ||||||||||| |||| |||||||||| ||||| |||||||| || ||| ||||||||| |||||| |||||||| ||| ||||| |||||| || ||||||| |||| ||| |||||| ||| |||||||||| ||||||||||
Harms Results
A summary of harms in the HIMALAYA trial is presented in Table 3.
A total of 378 patients (97.4%) in the tremelimumab in combination with durvalumab group and 357 patients (95.5%) in the sorafenib group experienced at least 1 adverse event (AE). The most frequently reported treatment-emergent AEs in the tremelimumab-plus-durvalumab and sorafenib groups were diarrhea (26.5% versus 44.7%, respectively), pruritis (22.9% versus 6.4%, respectively), rash (22.4% versus 13.6%, respectively), fatigue (17% versus 19%, respectively), decreased appetite (17% versus 17.9%, respectively), and palmar-plantar erythrodysesthesia syndrome (0.8% versus 46.5%, respectively). ||||| |||| ||| |||||||| ||||||| || ||| |||||||||||| || ||||||||||| |||| |||||||||| |||||| ||| ||| |||||||| ||||||| || ||| ||||||||| ||||| ||| ||||||||||| || ||||| ||| |||| ||||| . A total of 157 patients (40.5%) in the tremelimumab in combination with durvalumab group and 111 patients (29.7%) in the sorafenib group experienced at least 1 serious adverse event (SAE). ||| |||| |||||||||| |||||||| ||||||| ||||||| |||||| |||||| || ||| |||||||||||| || ||||||||||| |||| |||||||||| ||| ||||||||| |||||| |||| |||||||| ||||| || ||||| |||||||||||||| |||||| ||||| || || |||||||||||||| ||| ||||||||| ||||| || ||||| |||||||||||||| Fifty-three patients (13.7%) in the tremelimumab in combination with durvalumab group and 63 patients (16.8%) in the sorafenib group stopped treatment due to AEs.
At the final data cut-off date of August 21, 2021, in the FAS, ||||| |||| ||| |||||| ||||||| || ||| |||||||||||| || ||||||||||| |||| |||||||||| ||||| ||| ||| |||||| ||||||| || ||| ||||||||| |||||| |||| |||||||| |||||||||| || ||| ||||||| |||||||||||. In the safety analysis set, || |||||||| |||||| ||| || |||||||| |||||| |||| || ||| |||||||||||| || ||||||||||| |||| |||||||||| ||||| ||| ||||||||| |||||| |||||||||||||
||||||| |||||| || ||||||| |||||||| ||||||| |||||||| |||| |||||||||| || |||||||| || ||| |||||||||||| || ||||||||||| |||| |||||||||| ||||| |||| ||| ||||||||| ||||| ||||||| |||| || |||| ||| |||||||| || ||| ||||||||||||||||||||||| ||||| |||| |||| |||||| || |||||||| |||||||| ||||||||||||||| || ||||| ||||| |||| || ||| |||||||||||||| ||| ||||| |||| |||| |||||||||| |||||||| || ||| |||||||||||| || ||||||||||| |||| |||||||||| ||||| |||||| ||| |||||||| || ||||||| ||||| ||| |||| |||||||||| |||||||| || ||| ||||||||| ||||| |||| |||||| |||| |||||||||||||| Immune-mediated AEs were also more frequently reported in patients in the tremelimumab in combination with durvalumab group than in the sorafenib group (36% versus 8%, respectively). Six patients in the tremelimumab in combination with durvalumab group died due to immune-mediated AEs (|||||||||||| |||||||| ||||||| |||||||||||| ||| |||||||||| ||||||| ||| || |||||| || ||| ||||||||| |||||| |||||||| |||||||| |||||| |||||||| || || |||||||| |||||| || ||| |||||||||||| || ||||||||||| |||| |||||||||| ||||| ||| ||||||||| |||||| || ||| ||||||||| ||||||
There were 144 patients (37.1%) in the tremelimumab in combination with durvalumab group with any hepatic Standardized Medical Dictionary for Regulatory Activities Query (SMQ) AE compared to 121 patients (32.4%) in the sorafenib group. ||| || ||| ||||||| |||||||| ||| ||| || ||||||| || ||| |||||||||||| || ||||||||||| |||| |||||||||| ||||| ||| ||||||| || ||| ||||||||| |||||. There were 44 patients (11.3%) in the tremelimumab in combination with durvalumab group with any hemorrhage SMQ AE compared to 56 patients (15%) in the sorafenib group. ||| || ||| |||||||||| ||| ||| || ||| || ||| |||||||||||| || ||||||||||| |||| |||||||||| ||||| ||| ||||||| || ||| ||||||||| |||||| In the HIMALAYA trial, tremelimumab in combination with durvalumab showed no increase in liver toxicity or risk of bleeding.
Table 3: Summary of Key Harms Results From the HIMALAYA Study (SAS With Final Data Cut-Off of August 27, 2021)
Harms, n (%) | Tremelimumab in combination with durvalumab (N = 388) | Sorafenib (N = 374) |
|---|---|---|
Patients with ≥ 1 adverse event | 378 (97.4) | 357 (95.5) |
|||||||| |||| || ||||||| ||||| ||||| | ||| |||||| | ||| |||||| |
Patients with ≥ 1 SAE | 157 (40.5) | 111 (29.7) |
Patients who stopped treatment due to AEs | 53 (13.7) | 63 (16.8) |
|||||| | || ||||| | || ||||| |
||||||| ||||||| | | ||||| | | ||||| |
|||| | ||| |||||| | ||| |||||| |
Immune-mediated AE | 139 (35.8) | 30 (8.0) |
|||| ||||| ||| | | || |||||| | || ||||| |
Immune-mediated AE grade 3 or 4 | 49 (12.6) | 9 (2.4) |
||||||||||||||| | ||| |||||| | ||| |||||| |
|||||||||||||||| | ||| |||||| | ||| |||||| |
||||||| |||||| | || |||||| | || |||||| |
||||||||||| |||||| | || |||||| | || ||||| |
|||||||||| |||||| | || |||||| | || ||||| |
||||| |||||||||||||||||| |||||| | || |||||| | || ||||| |
|||||||| |||||||| |||||| | || ||||| | | ||||| |
|||||||||||| |||||| | || ||||| | | ||||| |
Hepatic SMQc | 144 (37.1) | 121 (32.4) |
Hemorrhage SMQd | 44 (11.3) | 56 (15) |
AE = adverse event; AESI = AE of special interest; SAE = serious adverse event; SAS = safety analysis set; SMQ = Standardized Medical Dictionary for Regulatory Activities Query.
aAE with outcome of death.
bAESIs for tremelimumab in combination with durvalumab include, but are not limited to, events with a potential inflammatory or immune-mediated mechanism and which may require more frequent monitoring and/or interventions such as steroids, immunosuppressants, and/or hormone-replacement therapy.
cThe following hepatic SMQs were considered of relevance to the hepatocellular carcinoma patient population: cholestasis and jaundice of hepatic origin, hepatic failure, fibrosis and cirrhosis, and other liver damage-related conditions, hepatitis, noninfectious liver infections, liver malignant tumours, liver-related investigations, signs and symptoms, and liver-related coagulation and bleeding disturbances.
dHemorrhage SMQs included hemorrhage terms and hemorrhage laboratory terms.
Source: HIMALAYA Clinical Study Report17 (details from the table have been taken from the sponsor’s Summary of Clinical Evidence).
Critical Appraisal
Internal Validity
HIMALAYA was an open-label, sponsor-blind, randomized phase III study comparing the effect of tremelimumab in combination with durvalumab and sorafenib in adult patients with unresectable HCC who require systemic therapy. The sponsor stated that an open-label, sponsor-blind design was used due to the nature of the treatment administration (IV versus oral) and the different administration schedules (every 4 weeks versus twice daily). The study used an appropriate central randomization method sufficient for concealing allocation until assignment to the intervention. Randomization appeared to adequately balance baseline demographic and disease characteristics between the tremelimumab in combination with durvalumab and sorafenib groups. The open-label design can result in a risk of bias in the measurement of the outcomes, particularly for subjective outcomes, whether by unblinded assessors, such as PFS and ORR, or self-reported, such as HRQoL and subjective harms. With the exception of subjective harms, the bias will likely favour the experimental intervention, although the extent and direction of bias are uncertain. This bias would not be introduced into the measurement of objective outcomes such as OS, which is the primary outcome of the trial. At the first interim analysis after at least 32 weeks of follow-up, tumour response assessments were performed by BICR (which would minimize bias in the measurement of these outcomes) but in the final analysis, tumour response assessments were performed only by investigators. Results from the interim analysis were similar to those from the final analysis. In the final analysis, exploratory end points included assessment of the PFS, TTP, ORR, DCR, and DoR by BICR to mitigate this bias. However, the results of these assessments were not available. The study was powered to detect a treatment difference in the primary end point of OS between treatment group, and the enrolled sample size was adequate. However, the study was not powered for individual subgroup comparisons, and no multiplicity adjustments were made, rendering any conclusion uncertain. Multiplicity was not controlled for other outcomes, which may have increased the risk of false-positive conclusions. Patients rated maintaining and improving QoL as an important outcome, yet interpretation of results for the HRQoL instruments (i.e., the ability to assess trends over time and to make comparisons across treatment groups) is limited by the substantial decline in the number of patients available to provide assessments over time.
External Validity
According to the clinical experts consulted by CADTH for this review, the demographic and disease characteristics of the HIMALAYA study population were reflective of the Canadian population with unresectable HCC. There was a large number of screening failures in the study, as almost a third of screened patients were not randomized, most commonly due to eligibility criteria not being fulfilled. However, the eligibility criteria that were most commonly not fulfilled were clear contraindications to treatment with tremelimumab in combination with durvalumab, such as a lack of adequate organ and marrow function. The clinical experts noted that, while only including patients with a Child-Pugh class of A is reasonable in clinical trials, it may also be reasonable to include other patients (e.g., those with a Child-Pugh class of B7) in clinical practice. They also noted that, although the trial excluded patients who had received prior systemic therapy, in clinical practice a large number of patients would have already received prior systemic therapy. It is unclear if findings from this study can be generalized to patients beyond the first line of therapy. All patients in the trial had an ECOG PS of 0 or 1 due to the eligibility criteria, but the experts indicated this would not be reflective of clinical practice and that clinicians would require some flexibility in restricting treatment by performance status. The clinical experts consulted by CADTH indicated that, at the time of the HIMALAYA study design, sorafenib was the only approved treatment for unresectable HCC patients who were ineligible for locoregional therapy and who had not undergone prior systemic therapy. Sorafenib was therefore considered standard-of-care treatment for these patients and was selected as the active comparator in this study. According to the clinical experts and recent clinical guidelines, sorafenib is no longer the most common standard-of-care therapy and has been replaced by therapies that include atezolizumab in combination with bevacizumab, as well as lenvatinib.
Long-Term Extension Studies
No long-term extension studies were identified by the sponsor.
Indirect Comparisons
Description of Studies
Two matching adjusted indirect comparisons (MAICs) and a published indirect treatment comparison (ITC) submitted by the sponsor were summarized and appraised for this CADTH review.
In the absence of direct comparative evidence from trials, the aim of the MAICs conducted by the sponsor was to compare the efficacy and safety of tremelimumab in combination with durvalumab against atezolizumab in combination with bevacizumab (from the IMbrave150 trial), and lenvatinib (from the REFLECT trial) in patients with unresectable HCC. A MAIC was identified as the preferred option to adjust for suspected heterogeneity between trials with individual patient-level data for the HIMALAYA trial and aggregate data available from the comparator trials. Individual patient data from the HIMALAYA trial were used to match and adjust patients to those included in the IMbrave150 and REFLECT comparator trials). All 3 trials (HIMALAYA, IMbrave150, and REFLECT) were phase III, open-label, multicentre studies. The mean durations of follow-ups were 33.2 months in the HIMALAYA trial, 27.5 months in the REFLECT trial, and 8.5 months in the IMbrave150 trial. The efficacy end points included OS and PFS in both MAICs, and ORR and DoR were only assessed in the MAIC comparing tremelimumab in combination with durvalumab versus atezolizumab in combination with bevacizumab. For parameters related to disease progression, the HIMALAYA and IMbrave150 trials employed RECIST 1.1, while REFLECT used the modified Response Evaluation Criteria in Solid Tumors (mRECIST). Harms related to the use of tremelimumab in combination with durvalumab were evaluated in both MAICs, including AEs, SAEs, and AEs leading to treatment discontinuation. Patient-reported outcomes were only reported in the MAIC comparing tremelimumab in combination with durvalumab versus atezolizumab in combination with bevacizumab.
Efficacy Results
This section focuses on the findings of the sponsor-submitted MAICs.
Tremelimumab in Combination With Durvalumab Versus Atezolizumab in Combination With Bevacizumab
After restriction and reweighting, the HR was 1.09 (95% CI, 0.80 to 1.48) for OS, and |||| |||| ||| ||| || ||||| ||| |||| The odds ratio (OR) was 1.18 (95% CI, 0.44 to 3.21) for ORR, and |||| |||| ||| |||| || ||||| ||| |||| ||| || ||| |||| || ||||||||||||| ||||| || |||||||||||||| ||||||| || |||| ||||||| ||| |||| |||| ||| |||| || |||||| while the HR for ||| || ||||||||| |||||||| ||| |||| |||| ||| |||| || ||||||
|||||||||||| || ||||||||||| |||| |||||||||| |||||| |||||||||
||||| ||||||||||| ||| |||||||||||| ||| || ||| |||| |||| ||| |||| || ||||| ||| ||| ||| |||| |||| ||| |||| || ||||| ||| ||||
Harms Results
Tremelimumab in Combination With Durvalumab Versus Atezolizumab in Combination With Bevacizumab
After restriction and reweighting, the OR was 0.73 (95% CI, 0.44 to 1.19) for AEs with a grade a 3 or 4 severity as defined by the Common Terminology Criteria for Adverse Events (CTCAE) | |||| |||| ||| |||| || ||||| ||| ||||||| |||| ||| |||| |||| ||| |||| || ||||| ||| ||| ||||||| || ||||||||||||||||||||||||.
|||||||||||| || ||||||||||| |||| |||||||||| |||||| ||||||||||
||||| ||||||||||| ||| |||||||||||| ||| || ||| |||| |||| ||| |||| || ||||| ||| ||| || ||||| ||||| ||| || |||| |||| ||| |||| || ||||| ||| ||||||| |||| ||| |||| |||| ||| |||| || ||||| ||| ||| ||||||| || ||||||||| ||||||||||||||||
Critical Appraisal
Although the methodology for matching and adjustment was in line with the technical guidance, the sponsor-submitted MAICs had a number of limitations that challenge the interpretation of the internal and external validities of the findings. Overall, based on the methods detailed in the report, the systematic literature review involved a comprehensive search, and the screening strategies were sufficient to minimize error and selection bias. The risk of bias of the included studies was assessed for each study; however, it may have differed depending on the study outcomes (i.e., OS versus patient-reported outcomes). The clinical experts consulted by CADTH for this review noted that several studies published over the past year that provide updated efficacy and safety data from the IMbrave150 and REFLECT trials were not identified in this search and therefore were not included in the ITC.18-20 As a result, MAIC analyses did not select some efficacy outcomes (i.e., PFS, ORR, DoR, and patient-reported outcomes) based on the longer follow-up data on efficacy outcomes, particularly for the IMbrave150 trial, and this may have influenced the results. While the sponsor inadvertently omitted the reference to longer follow-up data for the IMbrave150 trial in the MAIC report and the clinical evidence document, OS results from the IMbrave150 trial used in the MAIC analysis (HR = 0.66; 95% CI, 0.52 to 0.85) were reported in the Cheng et al. (2022) publication. As the matching criteria were based on the inclusion and exclusion criteria for the IMbrave150 and REFLECT trials and the availability of comparable data from the HIMALAYA trial, matching was not possible for all criteria that may remove an important portion of the patient population from the HIMALAYA trial. The effective sample size was reduced after matching and adjustment in both MAICs (65.7% to 78% of the original sample size in the HIMALAYA trial), which implies that the weighted estimates are being influenced by a subset of the patients from the HIMALAYA trial that may not be representative of the entire study population and may limit the generalizability of the results. In addition, the MAIC analysis could not account for some sources of heterogeneity in trials, such as differences in observation times or definition of end points. The clinical experts noted that, given the time gap, there is a possibility of systemic differences between patients in the HIMALAYA trial (from 2017 to 2019) and the REFLECT trial (from 2013 to 2016), such as treatments received before systemic therapy (i.e., loco-regional treatment). Furthermore, as not all trials included the same subjective and objective measurements, the comparative efficacy and safety of relevant treatments remain unknown. While OS and PFS data were available in all 3 trials, the ORR and DoR were not assessed in the REFLECT trial. In addition, DCR, considered by the clinical experts consulted for this review to be an important outcome, was assessed only in the HIMALAYA trial. Results on patient-reported outcomes (QoL and abdominal swelling), which were considered by patients to be important for this review, were only reported in the MAIC comparing tremelimumab in combination with durvalumab versus atezolizumab in combination with bevacizumab in patients with unresectable HCC. In both MAICs, results for some efficacy and harm estimates were imprecise (i.e., accompanied by wide CIs favouring either tremelimumab in combination with durvalumab or the comparators), which precluded drawing superiority conclusions.
Fulgenzi et al. (2022)
In addition to the MAICs conducted by the sponsor, a published network meta-analysis (NMA) conducted by Fulgenzi et al. (2022) was also identified. A frequentist NMA using fixed-effects models was used to compare the efficacy and safety of first-line treatments for unresectable HCC from 2007 to 2022. Two analyses were performed: the first compared the efficacy of atezolizumab in combination with bevacizumab versus all other first-line treatments, and the second compared all first-line treatments with placebo. As tremelimumab in combination with durvalumab is of interest to this report, only a comparison of atezolizumab in combination with bevacizumab versus tremelimumab in combination with durvalumab is presented here. The results of the NMA showed that the HR for OS for atezolizumab in combination with bevacizumab compared with tremelimumab in combination with durvalumab was 0.74 (95% CI, 0.52 to 1.06). The HR for PFS for atezolizumab in combination with bevacizumab compared with tremelimumab in combination with durvalumab was 0.66 (95% CI, 0.49 to 0.87). The OR for ORR for atezolizumab in combination with bevacizumab compared with tremelimumab in combination with durvalumab was 0.60 (95% CI, 0.28 to 1.25).
The results of the published NMA are highly uncertain given the heterogeneity in the baseline characteristics of patients within the included trials, data sparseness, network structure, and differences in the duration of follow for efficacy outcomes. While the use of fixed-effect models appears to be appropriate given the sparsity of data, no rationale was provided for the selection of the model in the published NMA. Furthermore, the effect estimates from the NMA are imprecise due to the sparseness of data and wide CIs, which for many outcomes included the possibility of benefit, lack of benefit, or harm for atezolizumab in combination with bevacizumab compared with tremelimumab in combination with durvalumab. Because model fit was not evaluated, it is unclear how well the model estimated treatment differences. No results on patient-reported QoL were evaluated, which was considered by patients to be an important end point. In addition, there were no comparative effect estimates for the harms. These limitations must be considered when drawing conclusions on the results of the published NMA.
Conclusions
One randomized, open-label, sponsor-blind, multicentre phase III trial provided evidence regarding the efficacy and safety of tremelimumab in combination with durvalumab compared with sorafenib in patients with unresectable HCC. Compared to sorafenib, treatment with tremelimumab in combination with durvalumab (tremelimumab 300 mg as a single priming dose in combination with 1,500 mg at day 1 of cycle 1, followed by durvalumab 1,500 mg as a single drug every 4 weeks) showed a statistically significant OS benefit. The absolute difference in median OS in patients with unresectable HCC between treatment groups (approximately 3 months) was considered clinically meaningful by the clinical experts consulted by CADTH. Because HRQoL analyses were limited by high rates of missing data, changes over time could not be interpreted. The clinical experts noted that, although sorafenib was the standard of care at the time the trial was conducted, it is now considered outdated. No definitive conclusions could be drawn from the ITCs submitted by the sponsor comparing the efficacy and safety of tremelimumab in combination with durvalumab to contemporary first-line therapies (i.e., atezolizumab in combination with bevacizumab and lenvatinib) due to methodological limitations and imprecision in the effect estimates for some outcomes. Given the lack of robust comparative data between tremelimumab in combination with durvalumab and other first-line therapies in the first-line setting (atezolizumab in combination with bevacizumab or lenvatinib), the clinical experts consulted could not draw firm conclusions about place in therapy. They noted that tremelimumab in combination with durvalumab would be suitable in patients with a higher risk of bleeding who would not be eligible for atezolizumab in combination with bevacizumab as tremelimumab in combination with durvalumab showed no increase in liver toxicity or the risk of bleeding in the HIMALAYA trial. The clinical experts would recommend funding this combination in the first-line treatment of appropriate patients with unresectable HCC as an alternative to the current options, which include lenvatinib, sorafenib, and atezolizumab in combination with bevacizumab.
The safety profile of tremelimumab in combination with durvalumab in this study was consistent with the known safety profile of other immuno-oncology checkpoint inhibitors, and no additional safety signals were identified with tremelimumab in combination with durvalumab therapy.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of tremelimumab, 20 mg/mL, concentrate for IV infusion in combination with durvalumab, 50 mg/mL, concentrate for IV infusion in the treatment of unresectable HCC in adult patients.
Disease Background
Contents within this section were informed by materials submitted by the sponsor and clinical expert input, and then summarized and validated by the CADTH review team.
Primary liver cancer is 1 of the fastest rising cancers in Canada.2,3 In 2022, it was estimated that 3,500 patients would be diagnosed with primary liver cancer and 1,650 patients in Canada would die from this disease, with an age-standardized incidence rate of 7.1 cases per 100,000.2,4 According to Statistics Canada’s Short-term cancer prevalence in Canada, 2018 report, the estimated 5-year prevalence of liver cancer is approximately 11.3 cases per 100,000 for both sexes.5 HCC, which originates from hepatocytes as a result of a complex process, is the most common type of liver cancer.2,21 It is a severe form of liver cancer that represents about 90% of primary liver cancers globally6 and approximately 72% of liver cancers in Canada.2 HCC is the third leading cause of cancer deaths worldwide,7 with a 5-year survival rate of only 20% in Canada.2 It is most commonly diagnosed in people older than 70 years and it is 3 times more common in men than women.4
HCC classically develops and grows in a silent fashion, which makes it difficult to detect before the development of the later stages of the disease.22 In its early stages, HCC is generally asymptomatic or presents with nonspecific symptoms, including right upper abdominal or epigastric pain, weight loss, early satiety, and malaise.10 Due to the insidious nature of the disease, the majority of patients are diagnosed with advanced disease, with a median survival following diagnosis of approximately 6 to 8 months, or 25% at 1 year.6,7 The extrahepatic manifestations of HCC may be associated with both distant metastases (most commonly to the bone, lung, and abdominal viscera) and paraneoplastic phenomena (i.e., hypoglycemia, hypocalcemia, and polycythemia).22
The predominant risk factors for HCC include chronic infections with HBV or HCV, alcohol abuse or alcoholic steatohepatitis, and NAFLD or nonalcoholic steatohepatitis.7-9 HBV or HCV infections in Canada are associated with increasing immigration from regions of the world where these infections are endemic, which is reflected in part by higher rates of viral hepatitis-related HCC in provinces where most immigrants settle.2 Other risk factors for HCC include obesity, diabetes, and nicotine use, as well as rare conditions such as hemochromatosis or hereditary tyrosinemia type 1.7-9 The prevalence of HCC risk factors varies geographically, with HBV predominating in Asia, HCV in Japan, and NAFLD, nonalcoholic steatohepatitis, or alcohol abuse in North America and Europe.7
HCC is often diagnosed using noninvasive imaging (multiphasic CT and MRI), tissue biopsies, physical examinations, or blood tests.6,7 The Child-Pugh class, based on clinical and laboratory parameters (i.e., serum bilirubin, serum albumin, ascites, neurologic disorder, and clinical nutrition status), is now widely used to assess liver function in clinical practice.7 The BCLC algorithm is a well-respected staging system that subdivides patients with HCC into 5 clinical stages: very early stage (BCLC 0), early stage (BCLC A), intermediate stage (BCLC B), advanced stage (BCLC C), and terminal stage (BCLC D).13,23,24 It is defined by the variables related to tumour burden (number and size of tumours), physical status, liver functional status, and cancer-related symptoms.13,25 Stage B is defined as multifocal HCC with relatively preserved liver function, no cancer-related symptoms (ECOG PS 0), and no vascular invasion or extrahepatic spread.6,13,25 Stage C comprises patients with cancer-related symptoms (symptomatic tumours, ECOG PS 1 or 2), vascular invasion (either segmental or portal invasion), or EHS (lymph node involvement or metastases), and those who have preserved liver function.6,13,25
Standards of Therapy
Contents within this section were informed by materials submitted by the sponsor and clinical expert input and then summarized and validated by the CADTH review team.
In early stages of HCC, the primary goal for treatment is a cure, and guidelines recommend potentially curative approaches that include surgical resection, liver transplant, and/or local regional therapies such as radiofrequency ablation. The choice of therapy depends on several factors, including resectability, liver function, and patient performance status.12,13,26 For advanced, unresectable HCC, the goal of treatment is to extend long-term survival, delay progression, and maintain and improve the patient’s QoL.10 HCC is considered to be chemotherapy-refractory; cytotoxic chemotherapy is of limited value to patients due to low tolerability and has been removed from guidelines due to modest efficacy.27 Guidelines therefore recommend the use of systemic targeted therapies for patients with unresectable HCC at BCLC stage B and C.11,12 According to the clinical experts consulted by CADTH, systemic treatment options have improved over the past several years with the introduction of lenvatinib, sorafenib, and the combination of atezolizumab and bevacizumab as first-line systemic treatment options in Canada.11,13-15
Atezolizumab is a programmed cell death 1 ligand 1 (PD-L1) inhibitor used in combination with bevacizumab, which is an angiogenesis inhibitor targeting vascular endothelial growth factor A, and is indicated as first-line treatment for advanced HCC. Clinical evidence to support the efficacy of atezolizumab in combination with bevacizumab was demonstrated in the pivotal IMbrave150 study, in which the combination showed a superior OS benefit compared to sorafenib in unresectable HCC patients. An analysis conducted at 56% OS maturity provided an OS HR of 0.66 (95% CI, 0.52 to 0.85) with a median OS of 19.2 months (95% CI, 17.0 to 23.7) for atezolizumab in combination with bevacizumab versus 13.4 months (95% CI, 11.4 to 16.9) in the sorafenib group, with OS rates at 18 months of 52% for atezolizumab in combination with bevacizumab versus 40% for sorafenib (CIs were not reported in the published article).28 Atezolizumab in combination with bevacizumab is funded widely across provincial drug programs.
Lenvatinib is also a multiple kinase inhibitor that targets vascular endothelial growth factor receptors 1, 2, and 3 and fibroblast growth factor receptors 1 through 4. Lenvatinib is indicated as first-line treatment for advanced HCC in patients without main portal vein invasion and an ECOG PS of 0 to 1. Clinical evidence to support the efficacy of lenvatinib in HCC was based on the phase III REFLECT study, in which lenvatinib demonstrated noninferiority to sorafenib. The median OS for lenvatinib was 13.6 months (95% CI 12.1 to 14.9) and the median OS for sorafenib was 12.3 months (95% CI, 10.4 to 13.9), with an HR of 0.92 (95% CI, 0.79 to 1.06; noninferiority margin = 1.08).29 Lenvatinib is widely funded across Canada and may be considered for first-line treatment of patients who decline or are not appropriate candidates for atezolizumab in combination with bevacizumab or who do not have access to this combination, and second-line treatment of patients who experience disease progression following first-line treatment with atezolizumab in combination with bevacizumab.15
Sorafenib is an oral TKI that targets multiple kinases, including the vascular endothelial growth factor receptors 1, 2, and 3, and the BRAF protein. Sorafenib is indicated for the treatment of patients with advanced HCC and was the first systemic treatment available for HCC. Clinical evidence to support the efficacy of sorafenib in HCC was based on the phase III SHARP study, in which sorafenib demonstrated an improvement in median OS of 2.8 months compared to placebo (median OS of 10.7 months for sorafenib [95% CI, 9.4 to 13.3] versus 7.9 months for placebo [95% CI, 6.8 to 9.1]; HR = 0.69; 95% CI, 0.55 to 0.87; P < 0.001).30 According to the clinical experts consulted by CADTH, sorafenib is now considered outdated and is no longer used by the majority of Canadian physicians, and it is recommended in the first and second lines only for patients intolerant of lenvatinib.
For patients who progress after treatment with lenvatinib or sorafenib, other multitargeted TKIs such as regorafenib and cabozantinib are funded widely in Canada as second-line therapies.
The clinical experts consulted by CADTH identified a key limitation of the current first-line therapy with atezolizumab in combination with bevacizumab: patients with untreated or incompletely treated esophageal and/or gastric varices with bleeding or who are at a high risk for bleeding are not candidates for this combination therapy.13 Upper endoscopy is therefore indicated for patients with cirrhosis or at high risk of bleeding.
Drug Under Review
Key characteristics of durvalumab (for injection) in combination with tremelimumab (for injection) are summarized in Table 4, along with other first-line treatments available for treatment of unresectable HCC.
Tremelimumab (for injection) in combination with durvalumab (for injection) is indicated for the first-line treatment of adult patients with unresectable HCC who require systemic therapy.1 Durvalumab is an engineered monoclonal antibody that blocks the interaction of PD-L1 with its receptors, programmed cell death 1 protein 1 (PD-1) and CD80.31 Selective blockade of PD-L1/PD-1 and PD-L1/CD80 interactions leads to prolonged T-cell activation and enhanced antitumour activity.31 Durvalumab does not bind to programmed cell death 1 ligand 2 (PD-L2). Leaving the interaction between PD‐L2 and PD-1 may reduce the potential for relevant immune-related toxicities such as pneumonitis, given the role of PD-L2 in ameliorating airway inflammation.32 Tremelimumab is a selective, fully human immunoglobulin G2 antibody that blocks cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) interactions with CD80 and CD86, enhancing T-cell activation and proliferation and resulting in increased T-cell diversity and enhanced antitumour immune activity.1
Targeting both the PD-1 and CTLA-4 pathways using a dual checkpoint blockade could potentially improve clinical outcomes due to this additive antitumour effect, resulting in a stronger immune response because the mechanisms of action of these pathways are nonredundant and utilized at different times of immune activation and at different locations in the body.33 This mechanism of action could be beneficial in targeting HCC tumour cells in which a state of immune tolerance to the pathogen or tumour may exist.
The recommended dose of tremelimumab is 300 mg as a single priming dose in combination with durvalumab 1,500 mg at day 1 of cycle 1, followed by durvalumab 1,500 mg as a single drug every 4 weeks.1 Patients with a body weight of 30 kg or less must receive weight-based dosing, equivalent to tremelimumab 4 mg/kg as a single priming dose in combination with durvalumab 20 mg/kg followed by durvalumab 20 mg/kg as a single drug every 4 weeks until weight is greater than 30 kg.1 Treatment with tremelimumab in combination with durvalumab should continue as long as a clinical benefit is observed or until unacceptable toxicity. Dose reduction or escalation is not recommended during treatment with tremelimumab in combination with durvalumab. Treatment withholding or discontinuation may be required based on individual safety and tolerability.1 Both durvalumab and tremelimumab should be administered under the supervision of health care practitioners experienced in the treatment of cancer.1,31
Dossiers for tremelimumab in combination with durvalumab were submitted to CADTH as a pre–Notice of Compliance submission. The proposed Health Canada indication for tremelimumab in combination with durvalumab is for the first-line treatment of adult patients with unresectable HCC who require systemic therapy, which generally aligns with the sponsor’s requested reimbursement criteria. Tremelimumab in combination with durvalumab received approval from the FDA in October 2022 for treatment of adult patients with unresectable HCC.16
Table 4: Key Characteristics of First-line Treatments for Unresectable HCC
Detail | Tremelimumab in combination with durvalumab | Sorafenib | Lenvatinib | Atezolizumab in combination with bevacizumab |
|---|---|---|---|---|
Mechanism of action | Tremelimumab blocks cytotoxic T-lymphocyte-associated protein 4, interactions with CD80 and CD86, enhancing T-cell activation and proliferation, resulting in increased T-cell diversity and enhanced antitumour immune activity Durvalumab blocks the interaction of PD-L1 with its receptors PD-1 and CD80, which leads to prolonged T-cell activation and enhanced antitumour activity | An oral tyrosine kinase inhibitor that targets multiple kinases, including VEGFR 1 to 3, and BRAF | A multiple kinase inhibitor that targets VEGFR 1 to 3 and fibroblast growth factor receptor 1 to 4 | Atezolizumab is a PD-L1 inhibitor, used in combination with bevacizumab, which is an angiogenesis inhibitor targeting VEGF-A |
Indicationa | For the first-line treatment of patients with unresectable HCCb | For treatment of patients with unresectable HCC | For the first-line treatment of adult patients with unresectable HCC | For the first-line treatment of adult patients with unresectable or metastatic HCC who require systemic therapy |
Route of administration | IV | Oral | Oral | IV |
Recommended dose | Tremelimumab 300 mg as a single priming dose in combination with durvalumab 1,500 mg at day 1 of cycle 1, followed by durvalumab 1,500 mg as a single drug every 4 weeks | 400 mg (2 × 200 mg tablets) taken twice a day, without food or with a low-fat or moderate-fat meal | 8 mg (2 × 4 mg capsules) once daily for patients with a body weight < 60 kg, and 12 mg (3 × 4 mg capsules) once daily for patients with a body weight ≥ 60 kg | Atezolizumab 1,200 mg over 60 minutes, followed by 15 mg/kg of bevacizumab administered as IV infusions, every 3 weeks |
Serious adverse effects or safety issues | Immune-mediated endocrinopathies (i.e., hypothyroidism, hyperthyroidism, thyroiditis, type 1 diabetes mellitus), immune-mediated rash or dermatitis, nephritis, or myocarditis Immune-mediated adverse reactions (i.e., myasthenia gravis, myositis, polymyositis, immune thrombocytopenia, pancreatitis, encephalitis, and retinal detachment) Infusion-related reactions (i.e., pruritus, diarrhea, rash, fatigue, decreased appetite) Tremelimumab in combination with durvalumab can cause severe and fatal immune-mediated adverse reactions, including enterocolitis, intestinal perforation, hepatitis, dermatitis, Stevens-Johnson syndrome, endocrinopathy, pneumonitis, interstitial lung disease, myocarditis, neuropathy, encephalitis, and myasthenia gravis, as well as toxicities in other organ systems | Hypertension, hemorrhage and cardiac ischemia or infarction, rash, hand-foot skin reactions, diarrhea, and fatigue. | Hypertension, cardiac failure, arterial thromboembolism, gastrointestinal perforation and fistula formation, hepatotoxicity or hepatic failure, renal failure and impairment, hemorrhage, and posterior reversible encephalopathy syndrome | Bleeding, immune-mediated pneumonitis, hepatitis, colitis, immune-mediated endocrinopathies (i.e., hypothyroidism, hyperthyroidism, adrenal insufficiency, and type 1 diabetes mellitus), immune-mediated meningo-encephalitis, neuropathies, myocarditis, nephritis, skin reactions, infections, and infusion-related reactions |
HCC = hepatocellular carcinoma; PD-1 = programmed cell death 1 protein 1; PD-L1 = programmed cell death 1 ligand 1; VEGF-A = vascular endothelial growth factor A; VEGFR = vascular endothelial growth factor receptor.
aHealth Canada–approved indication.
bHealth Canada–proposed indication.
Source: Product monographs for Imjudo,1 Imfinzi,31 Nexavar,34 Tecentriq,35 and Lenvima.36
Stakeholder Perspectives
Patient Group Input
This section was prepared by CADTH staff based on input provided by patient groups. The full original patient inputs received by CADTH are included in the stakeholder section at the end of this report.
The CCRAN in collaboration with the CCSN, CLF, and GI Society provided a collective patient input for this review. The CCRAN is a national not-for-profit patient advocacy group championing the health and well-being of Canadians affected by colorectal cancer and those at risk of developing the disease. The CCSN, CLF, and GI Society collaborated with CCRAN to ensure that the perspectives of patients with advanced HCC and their caregivers were captured, represented, and well incorporated into this submission. The CCRAN gathered information for this review from in-depth interviews with 2 patients with HCC (both patients had experience with the currently available treatment of HCC, and only 1 patient had experience with drug under review), a literature review, and online public forums for patient-reported outcomes.
According to the patient input received from the CCRAN, HCC is the most common primary liver cancer. The CCRAN noted that risk factors associated with HCC include cirrhosis, HBV and HCV infections, and alcohol intake. Both patient interviews indicated that they had not been experiencing any symptoms at the time of HCC diagnosis. The CCRAN indicated that a diagnosis and symptoms of HCC represent a substantial physiological and psychological burden for patients and their caregivers and can significantly affect their HRQoL. The CCRAN pointed to various symptoms of HCC that affected QoL and daily activities, including sleep disorders, sexual dysfunction, ascites, gynecomastia, pruritis, fatigue, muscle cramps, and lack of appetite, even after treatment. Both patient respondents emphasized that the daily activities that were most commonly affected included the ability to work, participate in enjoyable activities, and spend time with family and friends. One of the interviewed patients (a female aged 92 years who was diagnosed with HCC diagnosis at 71 years) found herself cycling through the same stages of cancer grief — anger, depression, guilt, anxiety, hopelessness, and fear — which hit her hard at the time of the initial diagnosis and subsequent relapse.
The CCRAN indicated that patients with HCC expect the following key outcomes to be improved from any new drug or treatment: improved QoL, prolonged survival, manageable side effects, maintenance of functionality, and ability to engage in society and contribute to the workforce. According to the patient input received from the CCRAN, HCC is a unique carcinoma because the majority of cases will develop in patients with cirrhosis and, therapeutic options will therefore be limited due to the patient’s overall health status. The CCRAN indicted that patients with early-stage HCC are preferred candidates for resection, transplant, and local ablation, patients at intermediate stages may be candidates for TACE, and those with advanced disease will receive systemic therapies. The CCRAN noted that the current systemic treatments for HCC include atezolizumab in combination with bevacizumab, lenvatinib, sorafenib, regorafenib, and cabozantinib. Limited treatment tolerability, in part due to side effects, was identified by the CCRAN as a major challenge to available systemic therapy for advanced HCC.
One of the interviewed patients (a male aged 74 years diagnosed with HCC at 68 years) had experience with treatment with the drug under review after transarterial chemoembolization, which negatively affected his QoL. The patient had access to tremelimumab in combination with durvalumab through a clinical trial, and resided in Cranbrook, British Columbia. That patient indicated that the drug under review has had promising and durable treatment results, with no side effects other than an occasional skin rash. He also mentioned that tremelimumab in combination with durvalumab helped him regain functionality and the ability to pursue a livelihood, which reduced the burden on his caregivers and loved ones. The CCRAN advocated that use of tremelimumab in combination with durvalumab be approved for the indication under review and suggested that it will help alleviate gaps in current HCC therapy.
Clinician Input
Input From Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of unresectable HCC.
Unmet Needs
The clinical experts consulted by CADTH for this review stated that the treatment goals for patients with unresectable HCC include prolonging life and delaying progression. They mentioned that treatments have improved in the past several years with the introduction of lenvatinib, which has superior efficacy and lower toxicity compared with sorafenib, and atezolizumab in combination with bevacizumab. However, the benefits of current treatments have been incremental. Moreover, the use of atezolizumab in combination with bevacizumab is limited to patients who have had a recent upper endoscopy and no symptomatic varices.
Place in Therapy
The clinical experts noted that tremelimumab in combination with durvalumab would be indicated in the first line for patients who would currently be indicated for atezolizumab and bevacizumab, and that the choice of therapy would depend on clinician and patient preference. They added that it may be indicated for patients who had started TKI therapy and progressed or experienced severe toxicity.
Patient Population
The clinical experts agreed that tremelimumab in combination with durvalumab would be recommended for patients with unresectable HCC with preserved liver function (as indicated by a Childs-Pugh class of A) and good performance status (potentially up to an ECOG PS of 2), and those who are not indicated for local therapy such as TACE. They mentioned that patients who were on TKIs and/or other therapies but had severe side effects that led to permanent discontinuation would also be eligible for tremelimumab in combination with durvalumab. Patients who are not candidates for other immune checkpoint inhibitors would not be candidates for tremelimumab in combination with durvalumab.
Assessing the Response Treatment
The clinical experts noted that, in clinical practice, imaging would be obtained every 3 months to assess response to treatment. The most important outcomes are prolonged survival, delayed progression, disease control, and maintained QoL with a low toxicity profile.
Discontinuing Treatment
The clinical experts advised that tremelimumab in combination with durvalumab should be discontinued if there is disease progression or intractable severe immune-related adverse effects.
Prescribing Considerations
According to the clinical experts, tremelimumab in combination with durvalumab can be administered in most systemic therapy suites in which cancer patients receive chemotherapy and immunotherapy. Administration of this therapy can be supervised by most medical oncologists experienced in treating HCC.
Clinician Group Input
This section was prepared by CADTH staff based on input provided by clinician groups. The full original clinician group inputs received by CADTH are included in the stakeholder section at the end of this report.
The clinician group input was obtained from 2 clinician groups, including the CGOEN, represented by 6 clinicians; the Ontario Health (Cancer Care Ontario) GI Drug Advisory Committee, represented by 5 clinicians; and a clinician from the Alberta Health Services Cancer Care at the University of Alberta.
The clinician groups noted that the current HCC treatment paradigm includes atezolizumab in combination with bevacizumab followed by lenvatinib, or sorafenib in patients without contraindication to immunotherapy, and lenvatinib followed by cabozantinib and regorafenib in patients with autoimmune disorders or contraindications to bevacizumab. The CGOEN indicated that, with modern systemic therapy, downsizing of disease has led to newer options for locoregional treatments of the liver (i.e., stereotactic radiation, embolization, ablation, resection, or transplant). The clinical groups identified the following as key goals of new therapies in HCC: prolonging life, delaying disease progression, improving response rate, managing side effects, reducing the severity of symptoms, maintaining HRQoL, and delaying deterioration. The CGOEN emphasized that a patient with HCC may be at an increased risk of bleeding due to the underlying liver disease and the vascular nature of the disease itself, and a therapy that does not increase this risk will be key in this area.
The clinician groups agreed that, given a good safety profile, tremelimumab in combination with durvalumab will be another first-line HCC treatment option, particularly for patients with hypertension or varices, or when upper GI endoscopy is not available. The clinician from the University of Alberta indicated that tremelimumab in combination with durvalumab may become the preferred first-line immunotherapy option for treatment of patients with unresectable HCC. The CGOEN and the clinician form the University of Alberta noted that patients receiving tremelimumab in combination with durvalumab would make fewer clinic visits and spend less time in the clinic because they would be treated every 4 weeks and essentially with 1 drug except for the first cycle, while for atezolizumab in combination with bevacizumab, patients are treated every 3 weeks and with 2 drugs.
According to the input from clinician groups received for this review, treatment response should be assessed by diagnostic imaging, such as MRI or CT scans, and blood tests every 3 to 4 months or as clinically indicated. The clinician groups indicated that the outcomes used to evaluate response to treatment in patients with HCC include improved OS, improved or maintained QoL, and improved response rate, leading to a reduction of symptoms and the possibility of other modalities, such as locoregional therapies, to control the disease process. The clinician groups identified several factors that may warrant discontinuation of tremelimumab in combination with durvalumab, including disease progression, unacceptable drug-related toxicities, or patient preference. The clinician groups emphasized that treatment with tremelimumab in combination with durvalumab should be provided by clinicians with expertise and experience in treating HCC. The GI Drug Advisory Committee noted that treatment with tremelimumab in combination with durvalumab should be performed in outpatient infusion clinics, including satellite clinics.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may affect their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 5.
Table 5: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
What is the relative efficacy or safety of tremelimumab in combination with durvalumab vs. atezolizumab in combination with bevacizumab or lenvatinib? | According to the clinical experts, there are several differences between the 3 trials that assess these therapies (HIMALAYA, IMBRAVE-150, and LEAP-002) limit cross-trial comparisons, such as time trial conducted, patient characteristics, and therapies before receiving systemic therapy. However, both tremelimumab in combination with durvalumab and atezolizumab in combination with bevacizumab were superior to sorafenib. Safety did not appear to be significantly different to atezolizumab in combination with bevacizumab. |
Considerations for initiation of therapy | |
Is histologic confirmation of HCC required to be eligible for tremelimumab in combination with durvalumab? | The standard of care would not require histologic tissue diagnosis, except when imaging is not diagnostic. |
If patients discontinue therapy for reasons other than toxicity or progressive disease and/or loss of clinical benefit, should patients be eligible for re-treatment? If yes, what re-treatment protocol and duration would be appropriate? | In the HIMALAYA trial, re-treatment was not specified unless patients have progression. At that point, they can be re-treated with the dual therapy for 1 cycle. If treatment is stopped for longer than 6 months (other than toxicity), re-treatment would be reasonable. |
Should we allow time-limited switching from atezolizumab in combination with bevacizumab to tremelimumab in combination with durvalumab? | Switching should be event-driven for patients experiencing any serious side effects, such as severe proteinuria or gastrointestinal perforation, only in the absence of disease progression. |
Considerations for continuation or renewal of therapy | |
What are appropriate criteria for re-treating with tremelimumab in clinical practice? Should re-treatment with tremelimumab be limited, in the setting of progression, to after cycle 5 of durvalumab? (Median duration of exposure was 20 weeks, range = 2 to 185.) | It would be reasonable to re-treat patients with progression with tremelimumab after cycle 4 of durvalumab. |
Considerations for discontinuation of therapy | |
What are appropriate discontinuation criteria for tremelimumab in combination with durvalumab in clinical practice? Are the discontinuation criteria different if a patient has already received a tremelimumab re-treatment? | Discontinuation criteria include clinical deterioration or treatment-related toxicity. In practice, patients may be receiving several scans before progression is confirmed (and several treatments during that time), similar to confirmed progression in clinical trials. The discontinuation criteria for patients who received re-treatment with tremelimumab should not be different. |
Considerations for prescribing of therapy | |
Is there any evidence for weight-based dosing of tremelimumab? | Most therapies have weight-based dosing for patients < 30 kg, but this weight range is not common in clinical practice. |
Administration of tremelimumab requires a 0.2 or 0.22 µm inline filter. | Comment to inform pERC deliberations. |
Funding algorithm (oncology only) | |
Tremelimumab in combination with durvalumab may change the place in therapy of comparator drugs. PAG considered unresectable hepatocellular carcinoma to be a complex therapeutic space with multiple lines of therapy, subpopulations, or competing products. | Comment to inform pERC deliberations. |
Care provision issues | |
Tremelimumab will be available in a 25 mg and 300 mg single-use vial. Infusion will take 1 hour. Durvalumab is available as a 120 mg and 500 mg vial; infusions take 1 hour per dose. | Comment to inform pERC deliberations. |
Preparation of durvalumab is familiar to many jurisdictions as it has funding for other indications for use. Preparation for tremelimumab would be new for many jurisdictions and is similar in preparation complexity to many other immunotherapy already in use. | Comment to inform pERC deliberations. |
Stability of prepared tremelimumab is up to 28 days under refrigerated conditions (would be limited by NAPRA sterility maximums, thus would likely not be longer than 9 days). This extended stability is very helpful operationally to support pharmacy workflow and reduce risk of drug wastage. | Comment to inform pERC deliberations. |
Vial sharing with tremelimumab would not be likely given the single-dose/flat-dose/single-use vial corresponding to a full dose. Vial sharing with durvalumab would be more likely given the q.4.w. interval, and other indications already funded at weight-based dosing. | Comment to inform pERC deliberations. |
System and economic issues | |
Atezolizumab has confidential negotiation and bevacizumab biosimilars also have confidential prices. | Comment to inform pERC deliberations. |
HCC = hepatocellular carcinoma; NAPRA = National Association of Pharmacy Regulatory Authorities; PAG = Provincial Advisory Group; pERC = CADTH pan-Canadian Oncology Drug Review Expert Review Committee; q.4.w. = every 4 weeks; vs. = versus.
Clinical Evidence
The objective of CADTH’s Clinical Review is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of tremelimumab (20 mg/mL) in combination with durvalumab (50 mg/mL) for the first-line treatment of adult patients with unresectable HCC. The focus will be placed on comparing tremelimumab in combination with durvalumab to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of tremelimumab in combination with durvalumab is presented in 2 sections, and CADTH’s critical appraisal of the evidence is included after each section. The first section, the systematic review, includes pivotal studies and randomized controlled trials (RCTs) that were selected according to the sponsor’s systematic review protocol. The second section includes indirect evidence from the sponsor. No long-term extension studies or studies addressing gaps in the pivotal and RCT evidence were submitted by the sponsor.
Included Studies
Clinical evidence from the following is included in the CADTH review and appraised in this document:
1 pivotal phase III trial
1 sponsor-conducted ITC and 1 published NMA.
Pivotal Studies and Randomized Controlled Trial Evidence
Contents within this section have been informed by materials submitted by the sponsor. The following were summarized and validated by the CADTH review team.
Description of Studies
Characteristics of the included studies are summarized in Table 6.
Table 6: Details of Pivotal Study and Randomized Controlled Trial Evidence
Detail | HIMALAYA |
|---|---|
Designs and populations | |
Study design | Phase III, multicentre, randomized, open-label, sponsor-blind, global trial |
Locations | Patients were enrolled at 181 sites and randomized at 170 study sites in 16 countries: Brazil (13 centres), Canada (9), France (14), Germany (10), Hong Kong (5), India (10), Italy (8), Japan (27), South Korea (8), Russian Federation (10), Spain (6), Taiwan (9), Thailand (9), Ukraine (8), US (21) and Vietnam (3) |
Patient enrolment dates | Start date: October 11, 2017 End date: June 19, 2019 |
Data cut-off dates | Interim analysis 1: September 2, 2019 Interim analysis 2: May 22, 2020 Final analysis: August 27, 2021 |
Randomized (N) | Total = 1,324 Tremelimumab in combination with durvalumab, n = 393 patients Sorafenib, n = 389 patients Durvalumab monotherapy, n = 389 patients (not included in this review) Closed treatment group: tremelimumab 75 mg plus durvalumab, n = 153 patients (not included in this review) |
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Intervention | Single tremelimumab regular interval durvalumab: tremelimumab 300 mg IV infusion × 1 dose plus durvalumab 1,500 mg IV infusion, followed by durvalumab 1,500 mg monotherapy q.4.w. until confirmed PD at the investigator’s discretion, unacceptable toxicity, or any discontinuation criteria were met Durvalumab: 1,500 mg IV infusion q.4.w. until confirmed PD, unacceptable toxicity, or any discontinuation criteria were met Closed treatment group: tremelimumab 75 mg IV infusion × 4 doses plus durvalumab 1,500 mg IV infusion q.4.w. followed by durvalumab monotherapy 1,500 mg q.4.w. until confirmed PD, unacceptable toxicity, or any discontinuation criteria were met |
Comparator(s) | Sorafenib: 400 mg orally twice daily, until confirmed PD at the investigator’s discretion, unacceptable toxicity, or any discontinuation criteria were met |
Study duration | |
Screening phase | 4 weeks |
Treatment phase | A 1-time combination dose of tremelimumab 300 mg and durvalumab 1,500 mg on day 1 of cycle 1 followed by 28-day cycles of durvalumab 1,500 mg monotherapy until confirmed PD (investigator assessment according to RECIST 1.1), unacceptable toxicity, or any treatment discontinuation criteria were met |
Follow-up phase | Until confirmed PD (investigator assessment according to RECIST 1.1), unacceptable toxicity, or any treatment discontinuation criteria were met; long-term follow-up data (survival and SAEs) will be collected for 3 years after final data cut-off for patients continuing treatment |
Outcomes | |
Primary end point | OS comparing tremelimumab in combination with durvalumab with sorafenib for superiority |
Secondary and exploratory end points | Key secondary
Additional secondary
Exploratory
Safety
|
Publication status | |
Publications | Abou-Alfa et al. (2022)37 |
BCLC = Barcelona Clinic Liver Cancer; BICR = blinded independent central review; BOR = best objective response; CTCAE = Common Terminology Criteria for Adverse Events; CTLA-4 = cytotoxic T-lymphocyte-associated antigen 4; DBP = diastolic blood pressure; DCR = disease control rate; DCR-16w = disease control rate at 16 weeks; DCR-24w = disease control rate at 24 weeks; DoR = duration of response; ECOG PS = Eastern Cooperative Oncology Group Performance Status; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-HCC18 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Hepatocellular Carcinoma 18; GI = gastrointestinal; HBV = hepatitis B virus; HCC = hepatocellular carcinoma; HCV = hepatitis C virus; HDV = hepatitis D virus; HRQoL = health-related quality of life; INR = international normalized ratio; mRECIST = modified Response Evaluation Criteria in Solid Tumors; NCI = National Cancer Institute; ORR = objective response rate; OS = overall survival; PD = progressive disease; PD-1 = programmed cell death 1 protein 1; PD-L1 – programmed cell death 1 ligand 1; PFS = progression-free survival; PFSFR = progression-free survival from rechallenge; PFSNT = progression-free survival from first postdiscontinuation therapy; q.4.w. = every 4 weeks; RECIST 1.1. = Response Evaluation Criteria in Solid Tumors Version 1.1; SBP = systolic blood pressure; TTP = time to progression; ULN = upper limit or normal.
Source: HIMALAYA Clinical Study Report.17
Key characteristics of the HIMALAYA trial are summarized in Table 5. HIMALAYA is a randomized, open-label, sponsor-blind, multicentre, global, phase III study to assess the efficacy and safety of tremelimumab in combination with durvalumab versus sorafenib in the treatment of patients with unresectable HCC who are not eligible for locoregional therapy and have not received prior systemic therapy for HCC in the first-line setting. The sponsor was blinded to treatment assignment and did not have access to any aggregate summaries by treatment group during the study. Patients were enrolled up to June 19, 2019, at 181 sites and randomized at 170 study centres in 16 countries (9 study centres in Canada).17
The primary outcome was OS for tremelimumab in combination with durvalumab versus sorafenib. Noninferiority and superiority for OS for durvalumab monotherapy versus sorafenib were key secondary outcomes; however, these are not relevant to this review. Additional secondary end points included OS rates (at 18, 24, and 36 months), PFS, TTP, ORR, DCR, and DoR assessed by the investigator, patient-reported outcomes, and safety. The study was funded by AstraZeneca Canada Inc.
Randomization was stratified according to macrovascular invasion (yes or no), etiology of liver disease (confirmed HBV versus confirmed HCV versus others), and ECOG PS (0 versus 1).
Tumour assessments were performed at randomization and then every 8 weeks (± 1 week) for the first 48 weeks following randomization, and every 12 weeks (± 1 week) thereafter until confirmed disease progression. Tumors were evaluated according to RECIST 1.1. All patients were followed up for survival until the end of the study unless they withdrew consent to survival follow-up.
Patients were randomly assigned in a 1:1:1:1 ratio using an interactive web response system into 1 of 4 treatment groups:
Tremelimumab 300 mg × 1 dose plus durvalumab 1,500 mg every 4 weeks (n = 393)
Sorafenib 400 mg twice daily (n = 389)
Durvalumab 1,500 mg every 4 weeks (n = 389)
Closed treatment group: tremelimumab 75 mg every 4 weeks × 4 doses plus durvalumab 1,500 mg every 4 weeks (n = 153).
The sponsor noted that at the time of the HIMALAYA study design, sorafenib was the only approved treatment for unresectable HCC patients who were ineligible for locoregional therapy and who had not undergone prior systemic therapy. Sorafenib was therefore considered standard-of-care treatment for these patients and was selected as the active comparator.
According to the sponsor, the results from the preplanned analysis of Study 22 (a phase I and II trial conducted in patients with unresectable HCC where the primary objectives were dose-finding and safety) demonstrated that tremelimumab 75 mg plus durvalumab did not provide meaningful efficacy improvements over durvalumab monotherapy. This treatment group was therefore closed in the HIMALAYA trial following protocol amendment 3, on November 29, 2018. The remaining patients were randomly assigned 1:1:1 to receive durvalumab, sorafenib, or tremelimumab in combination with durvalumab. Patients already randomized to the tremelimumab 75 mg plus durvalumab group could continue assigned study treatment, provided the investigator and patient agreed it was in the best interest of the patient. The tremelimumab 75 mg plus durvalumab regimen is not an approved dose.
Tremelimumab in combination with durvalumab is the drug under review; results pertaining to the durvalumab monotherapy versus sorafenib are not reported as durvalumab monotherapy was out of scope of this review.
The study design is depicted in Figure 1. The final data cut-off date was August 27, 2021.
Figure 1: HIMALAYA Study Design
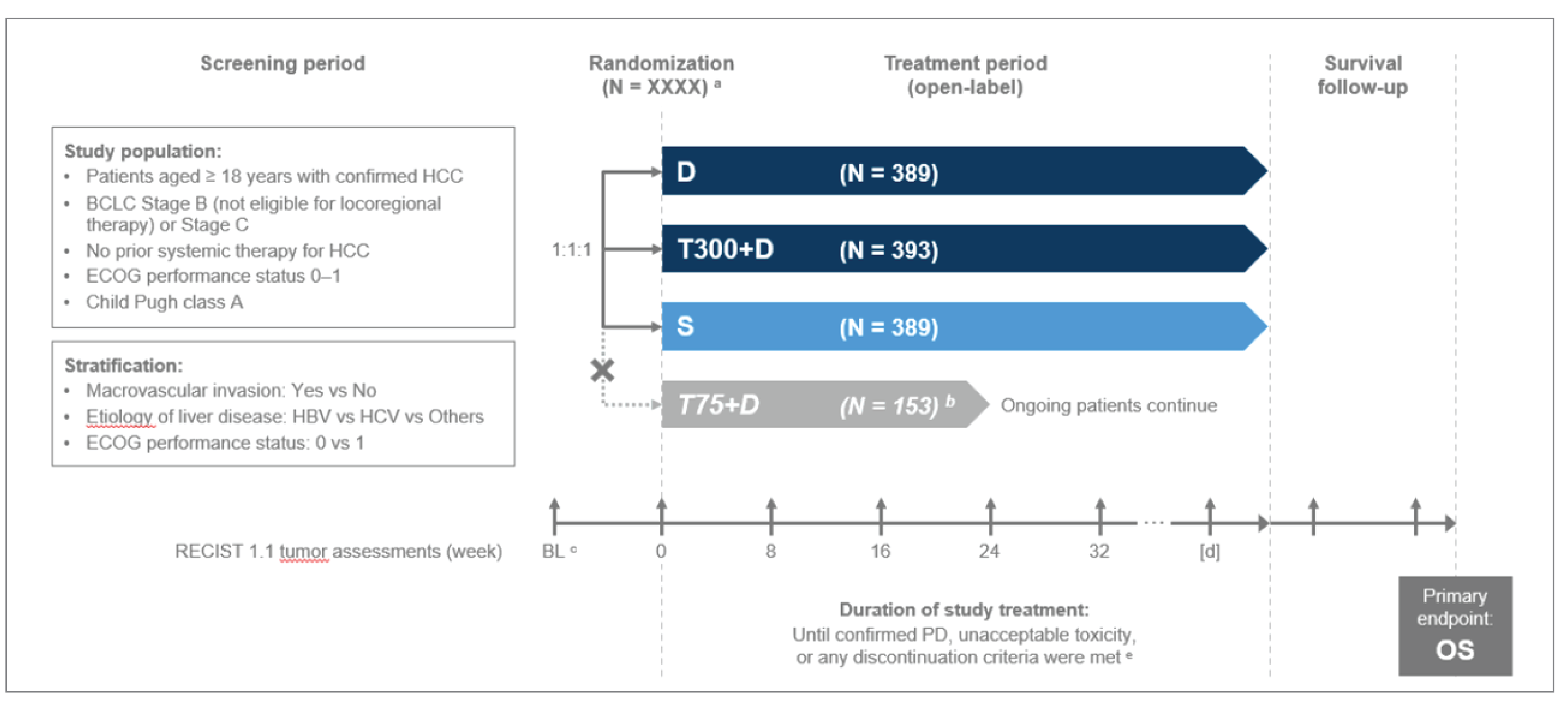
BCLC = Barcelona Clinic Liver Cancer; BL = baseline; D = durvalumab; ECOG = Eastern Cooperative Oncology Group; HBV = hepatitis B virus; HCC = hepatocellular carcinoma; HCV = hepatitis C virus; OS = overall survival; PD = progressive disease; S = sorafenib; T75+D = tremelimumab 75 mg plus durvalumab; T300+D = tremelimumab 300 mg plus durvalumab.
a Patient numbers shown are approximate and correspond to the planned enrolment.
b Enrolment into the T75+D group was closed following protocol amendment 3 (November 29, 2018) due to preliminary efficacy findings. Patients randomized to T75+D before protocol amendment 3 could continue on their assigned study treatment provided the investigator and patient agreed this was in the patient’s best interest. Patients randomized to T75+D who had not completed or started all 4 doses of tremelimumab could either complete the full schedule or continue with durvalumab monotherapy only.
c Baseline radiological tumour assessments were performed within 28 days before the date of randomization.
d Radiological tumour assessments were performed at randomization then every 8 weeks (± 1 week) for the first 48 weeks after randomization, and then every 12 weeks (± 1 week) until confirmed PD. The imaging schedule was followed regardless of any delays in dosing. Patients who permanently discontinued study treatment for reasons other than confirmed PD continued to have radiological scans performed following the same postrandomization schedule until confirmed PD.
e Patients with confirmed PD who, in the investigator’s opinion, continued to receive benefit from their assigned treatment and met the criteria for treatment in the setting of PD could continue to receive their assigned treatment regimen.
Source: HIMALAYA Clinical Study Report.17
Populations
Inclusion and Exclusion Criteria
Key inclusion and exclusion criteria for the HIMALAYA study are shown in Table 5. The study population included adult patients (18 years and older) with confirmed HCC, based on histopathological findings, and with preserved liver function (Child-Pugh class A). Patients must have been BCLC stage B or C, with an ECOG PS of 0 or 1 and a life expectancy of more than 12 weeks. They were included only if they were ineligible for locoregional therapy for unresectable HCC or progressed after locoregional therapy for HCC. Locoregional therapy must have been completed 28 days or more before the baseline scan. Patients must not have received any prior systemic therapy for unresectable HCC and must not have had clinically meaningful ascites within 6 months before the first scheduled dose.
Interventions
The HIMALAYA trial was an open-label, sponsor-blinded trial. Sponsor personnel refrained from accessing treatment records whenever possible and did not view data aggregated by treatment group during the course of the study, and interim analyses were performed by an independent data monitoring committee. Patients were randomized in a 1:1:1:1 ratio to receive 300 mg of tremelimumab in combination with durvalumab (N = 393), sorafenib (N = 389), durvalumab monotherapy (N = 389), or a different 75 mg of tremelimumab in combination with durvalumab (N = 153, closed during randomization due to preliminary efficacy findings). The latter 2 groups are not included in the CADTH systematic review as the drug under review is 300 mg of tremelimumab in combination with durvalumab N = 393).
In the tremelimumab in combination with durvalumab group, patients received 1 dose of tremelimumab (300 mg) by IV infusion over 1 hour (± 5 minutes) co-administered with 1 dose of durvalumab (1,500 mg) by IV infusion on day 1, followed by durvalumab 1,500 mg every 4 weeks until confirmed progressive disease, unacceptable toxicity, or any discontinuation criteria were met. Durvalumab was available as a 500 mg vial concentrate solution for infusion after dilution (50 mg/mL) and tremelimumab as a 400 mg vial concentrate solution for infusion after dilution (20 mg/mL). The dose schedule is presented in Figure 2. In the sorafenib group, patients received 400 mg (2 × 200 mg tablets) orally twice daily until confirmed progressive disease at the investigator’s discretion, unacceptable toxicity, or any discontinuation criteria were met.
Discontinuation criteria included withdrawal of consent, AEs that contraindicated further dosing, pregnancy or intent to become pregnant, noncompliance with study protocol, initiation of alternative anticancer therapy, and clinical progression. Patients could continue treatment after progression if the investigator determined that they were benefiting from treatment and they met the criteria for continuation after progressive disease. These criteria included that progression should not have occurred after confirmed response in the target lesions (regardless of the appearance of new lesions), and that there were no significant, unacceptable, or irreversible toxicities that indicate continuing treatment would not further benefit the patient.
Patients who did not continue treatment after disease progression were followed up for survival. Patients who discontinued treatment due to toxicity or symptomatic deterioration, or who commenced subsequent anticancer therapy, were followed up until confirmed disease progression and for survival. Crossover within the study was not permitted.
Figure 2: Tremelimumab in Combination With Durvalumab Dosing Schedule in HIMALAYA
D/C = discontinuation; Durva = durvalumab; PD = progressive disease; Treme = tremelimumab.
Source: HIMALAYA Clinical Study Report.17
Dose Modifications
Weight-based dosing modifications for durvalumab (20 mg/kg every 4 weeks) and tremelimumab (4 mg/kg) were permitted if a patient’s weight decreased to 30 kg or less. If the patient regained weight to more than 30 kg, they would receive the original assigned fixed dose of durvalumab 1,500 mg every 4 weeks.
Suspected sorafenib-related toxicities were managed based on the local approved product label. In countries where sorafenib was not approved, the sorafenib dose may be reduced to 400 mg (2 × 200 mg tablets) orally once daily. If an additional dose reduction was required, the sorafenib dose could be reduced to a single 400 mg dose (2 × 200-mg tablets) orally every other day.
Rechallenge With Tremelimumab
Patients receiving tremelimumab in combination with durvalumab who had evidence of disease progression (with or without confirmation according to RECIST 1.1) after their first 4 dosing cycles, but who were benefiting from treatment according to investigator opinion, were eligible for 1 round of re-treatment with tremelimumab (300 mg) combined with durvalumab. Patients had to meet rechallenge criteria, which were identical to the criteria for treatment through progression and included that no progression should have occurred after a confirmed response in the target lesions (regardless of the appearance of new lesions), and that there were no significant, unacceptable, or irreversible toxicities that indicated continuing treatment would not further benefit the patient.
Concomitant Medication
Treatments that were prohibited for both treatment groups included any other investigational anticancer therapy or monoclonal antibodies against CTLA-4, PD-1, or PD-L1, and any concurrent chemotherapy, radiotherapy, immunotherapy, or biologic or hormonal therapy for cancer treatment. Treatments prohibited for patients in the tremelimumab in combination with durvalumab group included immunosuppressive medications (e.g., systemic corticosteroids at doses exceeding 10 mg/day of prednisone or equivalent, methotrexate, azathioprine, and tumour necrosis factor-alpha blockers) unless clinically indicated. Sunitinib and drugs with laxative properties and herbal or natural remedies for constipation were prohibited within 90 days after the last dose of tremelimumab. Epidermal growth factor receptor TKIs were prohibited within 90 days after the last dose of durvalumab. Best supportive and prophylactic care (including antibiotics, nutritional support, correction of metabolic disorders, optimal symptom control, and pain management) was provided as necessary for all patients.
Outcomes
A list of efficacy end points assessed in this clinical review report is provided in Table 7 and further summarized in the following section. Summarized end points are based on those included in the sponsor’s Summary of Clinical Evidence as well as any identified by stakeholders (e.g., clinical experts, clinician groups, or patient groups) as important to this review. Only outcomes for the comparison relevant to this review (i.e., tremelimumab in combination with durvalumab versus sorafenib) are included.
Primary End Point
The primary outcome of the HIMALAYA trial was OS, defined as the time from the date of randomization until death due to any cause regardless of whether the patient withdrew from randomized therapy or received another anticancer therapy. Survival status was assessed at 2, 3, and 4 months (± 1 week), and then every 2 months (± 1 week) until the end of the study. The survival time for patients who were not known to have died at the data cut-off date was censored at the date of the last recorded date on which the patient was known to be alive. If that date or the date of death was after the data cut-off date, patients were censored at the data cut-off date.
Table 7: Outcomes Summarized From Pivotal Studies and RCT Evidence Identified by the Sponsor
Outcome measure | Place in HIMALAYA |
|---|---|
OS | Primary efficacy end pointa |
OS18, OS24, and OS36 | Other secondary efficacy end point |
ORR according to RECIST 1.1 and mRECIST by BICR | Other secondary efficacy end point |
BOR according to RECIST 1.1 and mRECIST by BICR | Other secondary efficacy end point |
DoR according to RECIST 1.1 and mRECIST by BICR | Other secondary efficacy end point |
PFS according to RECIST 1.1 using investigator assessment | Other secondary efficacy end point |
TTP according to RECIST 1.1 using investigator assessment | Other secondary efficacy end point |
ORR according to RECIST 1.1 using investigator assessment | Other secondary efficacy end point |
DCR according to RECIST 1.1 using investigator assessment | Other secondary efficacy end point |
DCR-16w and DCR-24w according to RECIST 1.1 using investigator assessment | Other secondary efficacy end point |
DoR according to RECIST 1.1 using investigator assessment | Other secondary efficacy end point |
EORTC QLQ-C30 | Other: Health economic outcomes research end point |
EORTC QLQ-HCC18 | Other: Health economic outcomes research end point |
BICR = blinded independent central review; BOR = best objective response; DCR = disease control rate; DCR-16w = disease control rate at 16 weeks; DCR-24w = disease control rate at 24 weeks; DoR = duration of response; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-HCC18 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Hepatocellular Carcinoma 18; mRECIST = modified Response Evaluation Criteria in Solid Tumors; ORR = objective response rate; OS = overall survival; OS18 = OS at 18 months; OS24 = OS at 24 months; OS36 = OS at 36 months; PFS = progression-free survival; RECIST 1.1 = Response Evaluation Criteria in Solid Tumors Version 1.1; TTP = time to progression.
aOS defined as time from date of randomization to date of death due to any cause. Statistical testing for these end points was adjusted for multiple comparisons. To control the familywise error rate at 5% (2-sided), an alpha of 0.1% was spent on the interim ORR and DoR analysis (first interim analysis). The remaining 4.9% alpha level was spent on all OS analyses.
Source: HIMALAYA Clinical Study Report17 (details from the table were taken from the sponsor’s Summary of Clinical Evidence).
Secondary and Exploratory End-Point Variables
Secondary end points included PFS, TTP, ORR, best objective response (BOR), DCR, disease control rate at 16 weeks (DCR-16w), disease control rate at 24 weeks (DCR-24w), and DoR based on investigator assessments using RECIST 1.1. Disease assessment by CT or MRI was conducted at baseline within 28 days of randomization, then every 8 weeks (± 1 week) for the first 48 weeks after randomization, and then every 12 weeks (± 1 week) until radiological progression as defined by RECIST 1.1 (followed by a subsequent scan if feasible). Guidelines for evaluation of objective tumour response using RECIST 1.1 included the recommendation that the same modality (and ideally the same unit) be used for each patient across all imaging time points.
In the first interim analysis, PFS, TTP, ORR, DCR, DoR, and time to response were evaluated based on BICR assessments according to RECIST 1.1, mRECIST, and irRECIST, and ORR was also evaluated based on investigator assessments according to RECIST 1.1. Secondary end points in this analysis were ORR, BOR, and DoR based on BICRs according to mRECIST. A BICR of radiological scans was performed on patients whose scans had been reviewed by at least 2 primary radiologists.
Progression-free survival was defined as the time from randomization until date of objective disease progression or death from any cause in the absence of progression, regardless of whether or not the patient withdrew from therapy or received another anticancer therapy before progression. Patients who were alive at the data cut-off date with no documented progression were censored at the date of their last evaluable RECIST 1.1 assessment. If a patient died after 2 or more consecutive missed visits, they were censored at the date of the last evaluable RECIST 1.1 assessment before the 2 missed visits. Patients with no baseline data were censored at the randomization date.
The TTP was defined as the time from randomization until objective tumour progression in the absence of death. Patients who died without progression were censored at the date of death.
The ORR was defined as the proportion of patients with at least 1 confirmed visit response of a complete response or partial response. A confirmed response refers to a response recorded at 1 visit and confirmed by repeat imaging after 4 weeks with no evidence of progression between the 2 visits. Unconfirmed responses were not confirmed by repeat imaging. Patients who had no disease progression between 2 nonconsecutive visit responses of a partial response were defined as responders. ORR assessments included data obtained until disease progression or the last evaluable assessment in the absence of progression.
A BOR was defined as the best response a patient had following randomization but before starting any subsequent cancer therapy and up to and including RECIST 1.1 progression or the last evaluable assessment in the absence of RECIST 1.1 progression. BORs were categorized to 1 of the following response categories: complete response, partial response, stable disease, no evidence of disease (applies only to patients entering the study with no disease at baseline), progressive disease, and not evaluable.
The DCR was defined as the proportion of patients with a BOR of complete response, partial response, or stable disease. The DCR-16w and DCR-24w were defined as the proportions of patients with a BOR of CR, PR, or stable disease for at least 16 weeks (± 7 days) and at least 24 weeks (± 7 days) following the start of treatment, respectively.
The DoR was defined as the time from the date of first documented response until the first date of documented progression or death in the absence of disease progression.
Exploratory end points included PFS, TTP, ORR, DCR, DCR-16w, DCR-24w and DoR based on BICR assessments using mRECIST for HCC and irRECIST. While RECIST 1.1 measures lesions in their longest diameter, mRECIST only measures the arterially enhanced portions of the HCC target lesions. Lesions categorized as progressive disease in RECIST 1.1 require verification in subsequent examinations to confirm progressive disease in irRECIST (to distinguish progressive disease from pseudoprogression).38
Progression-free survival from rechallenge in combination therapy groups was defined as the time from the date of rechallenge (first dose date in the rechallenge period) until the date of objective disease progression or death (by any cause in the absence of progression) regardless of whether the patient withdrew from therapy or received another anticancer therapy before progression. PFS from first postdiscontinuation therapy on next treatment was defined as the time from the date of first postdiscontinuation therapy (cancer therapy drug start date on the postinvestigational-product discontinuation systemic cancer therapy form) until the date of objective disease progression or death (by any cause in the absence of progression) regardless of whether the patient receives another anticancer therapy before progression. PFS from rechallenge (in combination therapy arms) and PFS on next treatment were determined by investigator assessments using RECIST 1.1.
Health-Related Quality of Life and Patient-Reported End Points
Health-related QoL was assessed as a secondary outcome using the EORTC QLQ-C30 and EORTC QLQ-HCC18. Exploratory outcomes included the EQ-5D 5-Level questionnaire (EQ-5D-5L), Patient’s Global Impression of Change (PGIC), and patient-reported outcome version of the CTCAE. Results of the EQ-5D 5-Level, PGIC, and patient-reported outcome of the CTCAE were not included in our review as they were not considered critical or important outcome measures for decision-making by the CADTH review team. Baseline assessments were completed at day 1 (except for PGIC), then assessments were completed every 8 weeks (± 7 days) for the first 48 weeks and every 12 weeks (± 7 days) thereafter until disease progression and up to 3 months after disease progression. The sponsor stated that approved translations of all tools underwent cultural and linguistic validation for the countries involved in the study before use.
The EORTC QLQ-C30 is a 30-item self-administered questionnaire comprising 5 functional scales (physical, role, cognitive, emotional, and social), 3 symptom scales (fatigue, pain, and nausea and/or vomiting), a GHS/QoL scale, and 6 single items (dyspnea, insomnia, appetite loss, constipation, diarrhea, and financial difficulties).39 Most questions have 4 response options (“not at all,” “a little,” “quite a bit,” and “very much”), with scores on these items ranging from 1 to 4. For the 2 items that form the global QoL scale, the response format is a 7-point Likert-type scale with anchors at 1 (“very poor”) and 7 (“excellent”). All scale and item scores were linearly transformed to a scale from 0 to 100. For the functional scales and the global QoL scale, a high score represents a good level of functioning. Conversely, high scores on the symptom scales and single items of the EORTC QLQ-C30 correspond to more severe symptoms.40 The sponsor defined an MID for clinically meaningful change as an absolute change in the score from baseline of 10 or more points. The main measures reported were GHS/QoL, physical function, and fatigue scales, along with the single items of appetite loss and nausea.
The EORTC QLQ-HCC18 is an 18-item self-administered additional questionnaire to the EORTC QLQ-30, which was developed and validated specifically for HCC.41,42 It consists of 6 multi-item symptom scales (fatigue, jaundice, nutrition, pain, fever, and body image), 2 single-item symptom scales (abdominal swelling and sexual interest); and 1 multi-item functional scale (body image). All scale and item scores were linearly transformed to a scale from 0 to 100. For all scales, a higher score indicates worse symptoms or poorer HRQoL. The sponsor defined an MID for clinically meaningful change as an absolute change in the score from baseline of 10 or more points on both scales based on a patient population with breast cancer and small-cell lung cancer.43 The CADTH review team did not identify any evidence for an MID among patients with unresectable HCC and the sponsor did not provide any evidence for this threshold for the EORTC QLQ-HCC18. The main measures reported were shoulder pain, abdominal pain, and abdominal swelling symptom scales.
Time to HRQoL or function deterioration was defined as the time from the date of randomization until the date of the first clinically meaningful deterioration (confirmed at a subsequent visit) or death in the absence of clinically meaningful deterioration, regardless of whether the patient discontinued study drug(s) or received another anticancer therapy before deterioration. The population for the analysis of time to GHS/QoL or function deterioration included a subset of the FAS with baseline scores of 10 or higher. Patients whose GHS/QoL or function did not show a clinically meaningful deterioration and who were alive at the time of the analysis were censored at the time of their last evaluable assessment of patient-reported outcomes. Patients whose GHS/QoL or function deteriorated or who died after 2 or more missed patient-reported outcome assessment visits were censored at the time of their last evaluable assessment. GHS or QoL and symptom improvement rates were defined as the number (%) of patients with a best overall score response of “improved” in GHS/QoL, function, or symptoms. The denominator consisted of a subset of the FAS with a baseline GHS/QoL or function score of 90 or lower, or a symptom score of 10 or higher.
Table 8: Summary of Patient-Reported Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
EORTC QLQ-C30 | The core questionnaire consists of 30 questions that are scored to create 5 multi-item functional scales (physical, role, emotional, cognitive, social), 3 multi-item symptom scales (fatigue, nausea and vomiting, pain), 6 single-item symptom scales (dyspnea, insomnia, appetite loss, constipation, diarrhea, financial difficulties), and a GHS/QoL scale.44,45 Presently, version 3.0 of the questionnaire is the most current version and was used in the HIMALAYA study.39 | The validity and reliability were assessed in advanced cancer patients with a short life expectancy and treated with palliative radiotherapy. The questionnaire was completed by 247 patients before palliative radiotherapy and 181 after palliative radiotherapy. In terms of responsiveness, the questionnaire was able to detect the effect of palliative radiotherapy over time. Scale reliability was excellent for all scales except the role functioning scale. Excellent criterion validity was found for the emotional functioning scale where it was correlated with the 20-item General Health Questionnaire (GHQ-20).46 | No MID identified in patients with hepatocellular carcinoma. The sponsor referred to a study in patients with breast cancer that reported between-group MID values ranging from 4 to 11.47 Patients with breast and small-cell lung cancer;43 > 10 points was considered more than a small change. Sponsor defined a difference of 10 points between study groups as clinically significant based on Osoba et al. (2018).43 |
EORTC QLQ-HCC18 | The questionnaire consists of 18 items comprising 6 multi-item symptom scales (fatigue, jaundice, nutrition, pain, fever, and body image), 2 single-item symptom scales (abdominal swelling and sexual interest), and 1 multi-item functional scale (body image).41 | The psychometric properties and clinical validity were assessed in an international field validation study.42 The EORTC QLQ-HCC18 was administered with the EORTC QLQ-C30, to 272 patients from 7 centres in 6 countries. Results of this assessment confirmed the scale structure and demonstrated that it is psychometrically and clinically valid, that it was able to detect differences between known subgroups, measure different constructs to the EORTC QLQ-C30, and that it is sensitive to changes over time in hypothetically relevant domains. | Not available. |
EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-HCC18 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Hepatocellular Carcinoma 18; GHS = global health status; HCC = hepatocellular carcinoma; MID = minimal important difference; QoL = quality of life.
Harms End Points
Safety and tolerability were assessed at all visits using the incidence of AEs, serious AEs, deaths, AEs leading to discontinuation, other AEs, AEs of special interest, and immune-mediated AEs. Treatment-emergent AEs were defined as any AEs with an onset on or after the date of the first dose, or pretreatment AEs that increased in severity on or after the date of the first dose, within 90 days following the date of last dose of study drug(s) or up to the date of initiation of the first subsequent therapy (whichever occurs first).
The AEs of special interest for tremelimumab in combination with durvalumab include, but are not limited to, events with a potential inflammatory or immune-mediated mechanism and that may require more frequent monitoring and/or interventions, such as steroids, immunosuppressants, and/or hormone-replacement therapy. AEs of special interests that were managed using immunosuppressants, and/or endocrine therapy were adjudicated by as immune-mediated AEs.
The sponsor noted that, due to the nature of HCC, AEs reported in the system organ class of hepatobiliary disorders and AEs of bleeding were of particular interest. The sponsor therefore presented a detailed analysis for the hepatic SMQ and hemorrhage SMQ. The following hepatic SMQs were considered relevant to the HCC patient population: cholestasis and jaundice of hepatic origin, hepatic failure, fibrosis and cirrhosis, and other liver-damage–related conditions, hepatitis, noninfectious liver infections, liver malignant tumours, liver-related investigations, signs and symptoms, and liver-related coagulation and bleeding disturbances. Hemorrhage SMQs included hemorrhage terms and hemorrhage laboratory terms.
Statistical Analysis
A summary of statistical analyses for trial end points is presented in Table 9.
Table 9: Statistical Analysis of Efficacy End points
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
HIMALAYA | ||||
Overall survival | Stratified log-rank test (for P value), HR from Cox model (with 95% CI) |
| If a patient is known to have died and only a partial death date is available, then the date of death will be imputed as the latest of the last date known to be alive + 1 from the database and the death date using the available information provided:
If there is evidence of death but the date is entirely missing, it will be treated as missing, i.e., censored at the last known alive date |
|
Progression-free survival | Stratified log-rank test using Investigator assessments per RECIST 1.1 (for P value), HR from Cox model (with 95% CI) |
| If the patient has no evaluable visits or does not have baseline data, they will be censored at randomization date unless they die within 2 visits of baseline (16 weeks plus 1 week allowing for a late assessment within the visit window), then they will be treated as an event with date of death as the event date | NA |
Time to progression | Stratified log-rank test using Investigator assessments per RECIST 1.1 (for P value), HR from Cox model (with 95% CI) |
| If patients died without tumour progression, they will be censored at the time of death | NA |
Objective response rate | First interim analysis: exact confidence intervals. Second and final analyses: logistic regression using investigator assessments per RECIST 1.1 (odds ratio with 95% CI and P value) |
| Patients who go off treatment without progression, receive a subsequent therapy, and then respond will not be included as responders in the ORR | NA |
Best objective response | Descriptive statistics | NA | For patients who die with no evaluable RECIST 1.1 assessments, if the death occurs ≤ 9 weeks (i.e., 8 weeks + 1 week to allow for a late assessment within the assessment window) after randomization, then BOR will be assigned to the progression category; for patients who die with no evaluable RECIST assessments, if the death occurs > 9 weeks after randomization the BOR will be assigned to the NE category | NA |
Duration of response | Descriptive statistics including Kaplan-Meier plot | NA | If a patient does not progress following a response, their DoR will use the PFS censoring time | NA |
Disease control rate (DCR, DCR-16w, DCR-24w) | Descriptive statistics | NA | NA | NA |
Proportion of patients alive at 18 months | Kaplan-Meier estimates of OS at 18 months | NA | NA | NA |
Proportion of patients alive at 24 months | Kaplan-Meier estimates of OS at 24 months | NA | NA | NA |
Proportion of patients alive at 36 months | Kaplan-Meier estimates of OS at 36 months Stratified chi-square test of difference in Kaplan-Meier estimators at a fixed time point (36 months) (for P value) | For chi-square test: adjustment for the stratification factors (macrovascular invasion, etiology of liver disease, and ECOG PS) will be applied only if there are sufficient number of events and patients at risk available in each stratum at 36 months | NA | NA |
PFS from rechallenge | Summarized by treatment group using investigator assessments following RECIST 1.1 | NA | If the patient has no evaluable visits or does not have baseline data, they will be censored at day 1 of rechallenge period unless they die within 2 visits of rechallenge period, then they will be treated as an event with date of death as the event date | NA |
PFS on next treatment | Summarized by treatment group | NA | If the patient has no evaluable visits or does not have first postdiscontinuation data, they will be censored at day 1 of first postdiscontinuation period unless they die within 2 visits of first postdiscontinuation period, then they will be treated as an event with date of death as the event date | NA |
Time to deterioration (EORTC QLQ-C30 and EORTC QLQ-HCC18) | Stratified log-rank test (for P value), HR from Cox model (with 95% CI), Kaplan-Meier plot | NA | NA | NA |
EORTC QLQ-C30, EORTC QLQ-HCC18 | Average change from baseline using a mixed model for repeated measures analysis and summary statistics | NA | NA | NA |
Improvement based best overall response (EORTC QLQ-C30, EORTC QLQ-HCC18) | Logistic regression with odds ratio, 95% CI and P value | NA | NA | NA |
PRO-CTCAE, PGIC, EQ-5D-5L | Descriptive statistics | NA | NA | NA |
AEs, SAEs, WDAEs, AESI | Descriptive statistics | NA | NA | NA |
AE = adverse event; AESI = adverse event of special interest; BOR = best objective response; CI = confidence interval; DCR = disease control rate; DCR-16w = disease control rate at 16 weeks; DCR-24w = disease control rate at 24 weeks; ECOG PS = Eastern Cooperative Oncology Group Performance Status; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-HCC18 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Hepatocellular Carcinoma 18; EQ-5D-5L = EQ-5D 5-Level questionnaire; HBV = hepatitis B virus; HCV = hepatitis C virus; HR = hazard ratio; NA = not applicable; NE = not evaluable; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; PGIC = Patient’s Global Impression of Change; PRO-CTCAE = patient-reported outcome version of the Common Terminology Criteria for Adverse Events; RECIST = Response Evaluation Criteria in Solid Tumors Version 1.1; SAE = serious adverse event; vs. = versus; WDAE = withdrawal due to adverse event.
Source: HIMALAYA Clinical Study Report17 (details from the table were taken from the sponsor’s Summary of Clinical Evidence).
Sample Size and Power Calculation
The sample size determination considered the comparison of the primary outcome of OS for tremelimumab in combination with durvalumab versus sorafenib for superiority. Because this determination assumed an exponential distribution of OS and a 2-month delay in the separation of the OS curves, an average HR of 0.70 was used. When estimating the analysis times, a nonuniform accrual of patients with a duration of 22 months was assumed with a follow-up duration of 15.5 months and a total duration of 37.5 months. No adjustment was made for dropouts. For the efficacy comparisons, the median OS for sorafenib was assumed to be 11.5 months, with an 18-month OS rate of 33.8%. The assumed average HR of 0.70 for tremelimumab in combination with durvalumab versus sorafenib translated to an increase in median OS from 11.5 months to 16.5 months and an increase in the 18-month OS rate from 33.8% to 46.8%. The final analysis was planned for when 515 OS events occurred in both treatment groups combined (67% maturity) which was approximately 37.5 months after the first patient was randomized, providing at least 97% power to demonstrate a statistically significant difference In OS at a 2-sided 4.25% significance level. No formal sample-size calculations were associated with planned first interim analysis.
Statistical Testing and Multiple-Testing Procedure
The end points for OS were compared between treatment groups using a stratified log-rank test, stratifying by etiology of liver disease (confirmed HBV versus confirmed HCV versus others), ECOG PS (0 versus 1), and MVI (yes versus no). The P value was generated using rank tests for association as the testing approach, which corresponds to a Cox regression with the Breslow approach for handling ties. The HR was estimated using a stratified Cox proportional hazards model and the Efron method to control for ties and stratification variables, and the 95% CI was calculated using a profile-likelihood approach. The stratification variable used the values recorded in the randomization system (an interactive web response system). Kaplan-Meier plots of OS were also estimated by treatment group.
This first interim analysis was performed after 100 patients in each treatment group were followed for at least 32 weeks after randomization and after all patients had been enrolled. A second interim analysis was performed when 415 OS events had occurred in the 2 treatment groups combined (52% maturity). The OS comparison was not statistically significant at a 2-sided alpha level of 0.0244 and the study continued to the final analysis. The final analysis was performed when 555 OS events had occurred in the 2 treatment groups combined (71% maturity), which was 46 months after the first patient was randomized. The smallest treatment difference that was statistically significant at the final analysis was an average HR of 0.84 (an increase in median OS from 11.5 months to approximately 13.7 months in tremelimumab in combination with durvalumab versus sorafenib).
To control for the familywise error rate at the 5% level (2-sided), an alpha level of 0.1% was applied to the interim ORR and DoR analysis (at the first interim analysis), while the remaining 4.9% alpha level was spent on all OS analyses. If all the OS analyses for all the alternative hypotheses were considered successful, as shown in Figure 3, the 4.9% alpha level was to be passed to test the difference in the 3-year survival rates between tremelimumab in combination with durvalumab and sorafenib; however, this was not achieved. Multiplicity for other outcomes was not controlled.
The key secondary analyses for the alternative hypotheses were to compare OS for durvalumab monotherapy versus sorafenib group (first for noninferiority then superiority). Exploratory analyses compared OS for the durvalumab group versus tremelimumab in combination with durvalumab group. These analyses were not relevant and are not included in this review.
Secondary Outcomes
The ORR was calculated using exact confidence intervals at the first interim analysis, and then logistic regression was used to calculate the odds ratio (with 95% CI and P value) at the final analysis. PFS and time to response were calculated using a stratified log-rank test for P value, and HR from the Cox model (with 95% CI). BOR, DoR and DCR were reported using descriptive statistics.
For the EORTC QLQ-C30 and EORTC QLQ-HCC18, average changes from baseline were reported using a mixed model for repeated measures analysis as well as summary statistics. Time to deterioration was assessed using a stratified log-rank test for the P value, HR from the Cox model, and a Kaplan-Meier plot. The improvement rate was assessed using logistic regression with an OR, 95% CI, and P value.
Subgroup Analyses
Subgroup analysis comparing OS between the tremelimumab in combination with durvalumab and sorafenib treatment groups was conducted for the following prespecified subgroups: PD-L1 expression level, etiology of liver disease, serum alpha-fetoprotein level, MVI, EHS, MVI and EHS, ECOG PS at screening, BCLC stage at study entry, sex, age at randomization, and region. For each subgroup level, the HR and 95% CI were calculated from a Cox proportional hazards model that only contained a term for treatment.
The following subgroups, planned a priori in the statistical analyses plan, were deemed clinically meaningful by the clinical experts consulted by CADTH for this review: etiology of liver disease, MVI, ECOG PS at screening, BCLC stage at study entry, and serum alpha-fetoprotein level.
Subgroup analyses were also performed for secondary end points PFS, TTP, ORR, BOR, DoR, DCR, DCR-16w, and DCR-24w in the PD-L1 expression, etiology of liver disease, and MVI subgroups.
Figure 3: Multiple-Testing Strategy in the HIMALAYA Trial
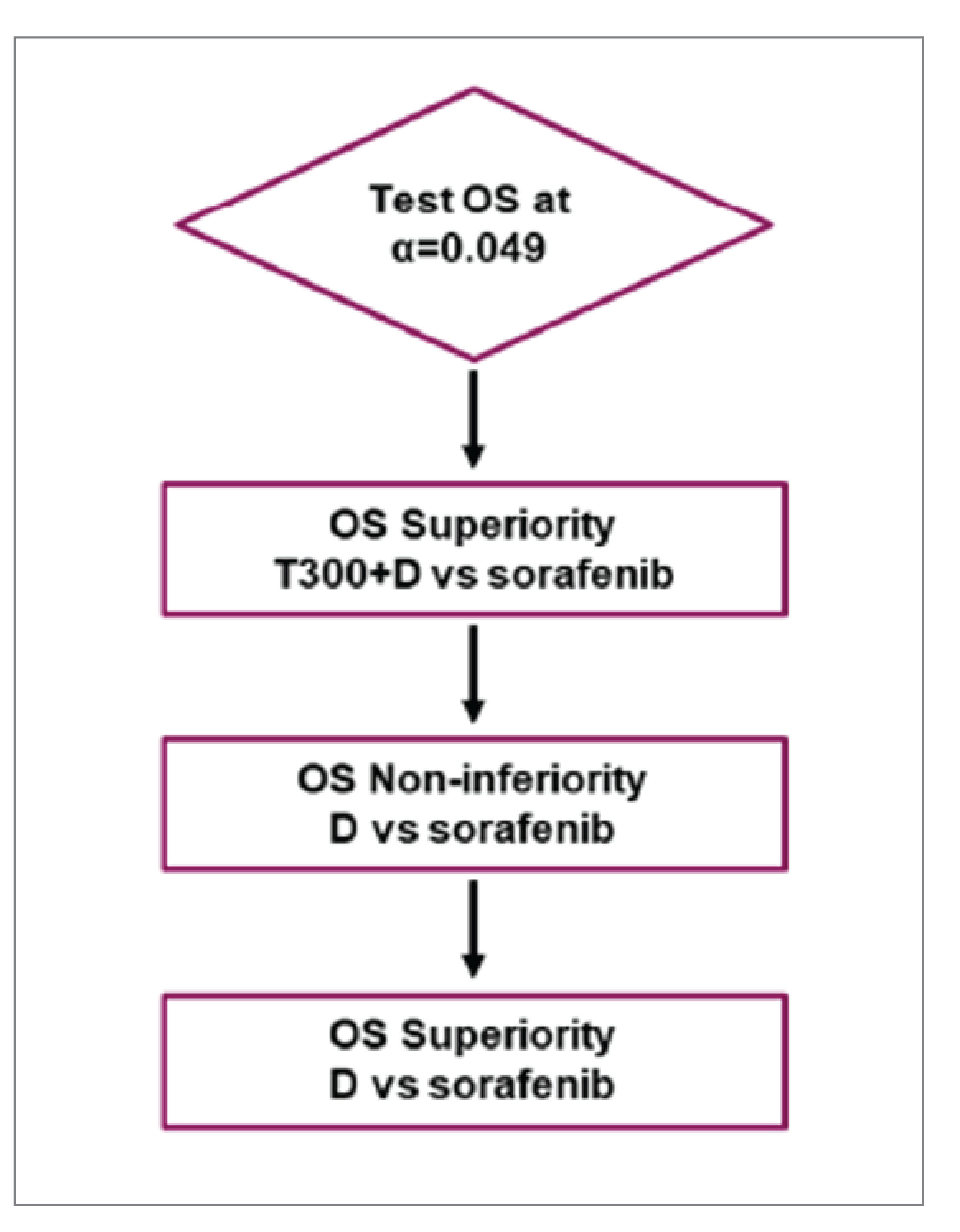
D = durvalumab monotherapy 1,500 mg every 4 weeks; OS = overall survival; sorafenib = sorafenib 400 mg twice daily; T300+D = tremelimumab 300 mg × 1 dose + durvalumab 1,500 mg every 4 weeks.
Source: HIMALAYA Clinical Study Report17 (details from the table were taken from the sponsor’s Summary of Clinical Evidence).
Sensitivity Analyses
Sensitivity analyses for OS were conducted to rule out attrition bias using a Kaplan-Meier plot of time to censoring, where the censoring indicator for OS was reversed (0 for patients who died and 1 for censored patients). An exploratory analysis was also conducted to assess the assumption of proportionality using a stratified MaxCombo test with the same stratification factors as the primary analysis. Nonproportionality was expected due to the delayed effects of immuno-oncology drugs. An additional sensitivity analysis was planned to assess the potential impact of the COVID-19 pandemic, with patients who died from COVID-19 to be censored at their infection death date. However, the sponsor noted that the pandemic did not meaningfully affect the study and only 7 cases of COVID-19 infection were reported during the study.
Analysis Populations
Key analysis populations of interest are presented in Table 10. The FAS was used in all efficacy analyses and included all randomized patients, including those randomized in error. Patients were analyzed in the group to which they were randomized. The safety analysis set included all patients who received any number of investigational products, including those who were randomized in error or not randomized and still started on study treatment. Patients were analyzed based on the treatment received.
Table 10: Analysis Populations in the HIMALAYA Trial
Population | Definition | Application |
|---|---|---|
FAS (intention-to-treat population) | All randomized patients, including those randomized in error | All efficacy analyses were performed on the FAS; patients were analyzed based on the randomized treatment received; those who were randomized but did not receive treatment were analyzed in the treatment group to which they were randomized |
Safety analysis set | All patients who received any amount of investigational products (durvalumab, tremelimumab, or sorafenib), including those who were randomized in error or not randomized and still started on study treatment | Safety data were not formally analyzed but summarized using the safety analysis set according to the investigational products received, i.e., erroneously treated patients were summarized according to the products they actually received |
FAS = full analysis set.
Source: HIMALAYA Clinical Study Report17 (details from the table have been taken from the sponsor’s Summary of Clinical Evidence).
Protocol Changes
The major changes to the study protocol were:
In protocol amendment 1, Protocol version 2.0 (December 20, 2017), the exploratory objective was included to assess PFS from rechallenge in the tremelimumab in combination with durvalumab group.
In protocol amendment 3, Protocol version 4.0 (November 29, 2018), enrolment into the tremelimumab 75 mg plus durvalumab group was closed due to preliminary efficacy findings in Study 22, the primary and secondary objectives were realigned (the original primary objective of OS for tremelimumab 75 mg plus durvalumab versus sorafenib was replaced with durvalumab versus sorafenib and tremelimumab 300 mg plus durvalumab versus sorafenib for OS), the multiple-testing strategy was updated to reflect the procedure for controlling the type I error as a result of the changes to the primary and secondary objectives, patient-reported outcome end points were added to the multiple-testing procedure and updated such that the first interim analysis was performed after approximately 100 patients per treatment group had the opportunity for 32 weeks rather than 24 weeks of follow-up.
In protocol amendment 5, Protocol version 6.0 (August 20, 2019), statistical analysis methods were revised to change dual primary objectives to a hierarchical approach with a single primary objective and 2 key secondary objectives, including ORR, following interim analysis of the ongoing Study 22 (the multiple-testing strategy was updated, ORR and patient-reported outcome end points were removed from the multiple-testing procedure, and the number of events, maturity, power and 2-sided significance levels for these analyses were updated). Efficacy assessments in the first interim analysis of patients with an opportunity for 32 weeks of follow-up were added as a secondary objective. This amendment was made before data cut-off for the first interim analysis on September 2, 2019.
Results
Patient Disposition
Patient disposition in the HIMALAYA study in the tremelimumab in combination with durvalumab and sorafenib groups is summarized in Table 11. Of ||||| |||||||| who were screened, ||||| |||||||| were randomized into 1 of the 4 original treatment groups. Of the 687 screening failures (34%), the majority (654 patients) were due to eligibility criteria not being fulfilled ||||||||| |||| || |||||||| ||||| ||| |||||| |||||||| |||| |||||||||| || ||||||||| ||| ||||| || ||||||||||||||||| |||||||| |||| ||||| ||||||| ||| ||||||||| ||| |||||||||| ||||| ||||| |||| || ||| |||| |||||||||| ||| ||||||| ||||| |||| ||| ||||| |||||||| |||||||| |||||||| || |||||||||| |||||||| ||| |||| |||||||| ||| |||||||||||||| |||| |||||| ||| ||||||||||| ||||| ||| ||||||||| |||||||||| |||| ||| |||||| |||||| || |||||||| |||||||| ||| ||||||
At the time of the data cut-off on August 27, 2021, 345 patients (88.7%) in the tremelimumab in combination with durvalumab group and 353 (94.4%) in the sorafenib group had discontinued study treatment. The most common reasons for discontinuing tremelimumab in combination with durvalumab and sorafenib were objective progressive disease (183 patients [47.0%] and 170 patients [45.5%], respectively), subjective progressive disease (61 patients [15.7%] and 66 patients [17.6%], respectively), and AEs (52 patients [13.4%] and 63 patients [16.8%], respectively).
Protocol Violations
| ||||| || ||||||||| |||||| || ||| |||||||||||| || ||||||||||| |||| |||||||||| ||||| ||| || |||||||| |||||| || ||| ||||||||| ||||| ||| || ||||| |||||||||| |||||||| |||||||||| ||| |||| |||||||| |||||||| |||| || ||||||||| ||| |||||||| || |||||| |||||| ||| |||| |||||||| ||| |||||||||| ||| ||| ||| ||||||| ||||| ||||||||| || |||||||| |||| || ||| |||||||||||| || ||||||||||| |||| |||||||||| ||||| ||| || |||||||| |||| || ||| ||||||||| |||||||
Table 11: Summary of Patient Disposition in the HIMALAYA Study
Patient disposition | Tremelimumab in combination with durvalumab | Sorafenib |
|---|---|---|
Randomized, N | 393 | 389 |
Received treatment, N (%) | 389 (99.0) | 374 (96.1) |
Discontinued from treatment, N (%) | 345 (88.7) | 353 (94.4) |
Reason for discontinuation, N (%) | ||
Objective progressive diseasea | 183 (47.0) | 170 (45.5) |
Subjective progressive disease | 61 (15.7) | 66 (17.6) |
Adverse event | 52 (13.4) | 63 (16.8) |
Patient decision | 19 (4.9) | 34 (9.1) |
Specific discontinuation criteria | 5 (1.3) | 2 (0.5) |
Lost to follow-up | 1 (0.3) | 0 |
Severe noncompliance | 0 | 1 (0.3) |
Other | 24 (6.2) | 17 (4.5) |
|||||||||| |||||| ||| | ||| ||||| | ||| ||||| |
Death | 260 (66.2) | 280 (72.0) |
||||| | ||| |||||| | || |||||| |
||||||||| ||||| ||||||| | ||| |||||| | || |||||| |
|||||||||| || ||||||| | | ||||| | || ||||| |
Lost to follow-up | 1 (0.3) | 7 (1.8) |
Status at final data cut-off (August 27, 2021) | ||
Ongoing studyc | 125 (31.8) | 80 (20.6) |
Ongoing study treatmentd | 44 (11.3) | 21 (5.6) |
aObjective progressive disease as confirmed by Response Evaluation Criteria in Solid Tumors Version 1.1.
bPatients confirmed alive in follow-up or on active study treatment at the time of final analysis reported “study completion” on the disposition electronic case report form.
cPatients ongoing in study are the same as patients who completed the final analysis.
dPercentages are calculated from the number of patients who received treatment in the global study. For combination therapy patients, durvalumab reason is reported.
Source: HIMALAYA Clinical Study Report17 (details from the table have been taken from the sponsor’s Summary of Clinical Evidence).
Table 12: Summary of Baseline Characteristics in HIMALAYA (Full Analysis Set)
Characteristic | Tremelimumab in combination with durvalumab (N = 393) | Sorafenib (N = 389) |
|---|---|---|
Median age, years (range) | 65.0 (22 to 86) | 64.0 (18 to 88) |
|||| ||| ||||| ||||| | |||| ||||||| | |||| ||||||| |
Age group by years, n (%) | ||
< 65 | 195 (49.6) | 195 (50.1) |
≥ 65 to < 75 | 145 (36.9) | 137 (35.2) |
≥ 75 | 53 (13.5) | 57 (14.7) |
Male sex, n (%) | 327 (83.2) | 337 (86.6) |
Race, n (%) | ||
||||| | ||| |||||| | ||| |||||| |
||||| | ||| |||||| | ||| |||||| |
Black | 7 (1.8) | 10 (2.6) |
Other | | ||||| | 5 (1.3) |
Missing | 1 (0.3) | 6 (1.5) |
Region, n (%) | ||
Asia (excluding Japan) | 156 (39.7) | 156 (40.1) |
Rest of world (including Japan)a | 237 (60.3) | 233 (59.9) |
|||||| | || ||||| | || ||||| |
||| | || ||||| | || |||||| |
ECOG PS score, n (%)b | ||
0 | 244 (62.1) | 241 (62.0) |
1 | 148 (37.7) | 147 (37.8) |
2 | 1 (0.3) | 1 (0.3)c |
Missing | | ||||| | | ||||| |
Child-Pugh class, n (%)e | ||
A | 392 (99.7) | 386 (99.2) |
B | 0 | 3 (0.8) |
C | 0 | 0 |
Missing | 1 (0.3) | 0 |
BCLC stage, n (%)f | ||
B | 77 (19.6) | 66 (17.0) |
C | 316 (80.4) | 323 (83.0) |
Virology status at baseline, n (%) | ||
HBV | 122 (31.0) | 119 (30.6) |
HCV | 110 (28.0) | 104 (26.7) |
Nonviralg | 161 (41.0) | 166 (42.7) |
Macrovascular invasion, n (%) | ||
Yes | 103 (26.2) | 100 (25.7) |
No | 290 (73.8) | 289 (74.3) |
Extrahepatic spread, n (%) | ||
Yes | 209 (53.2) | 203 (52.2) |
No | 182 (46.3) | 185 (47.6) |
Missing | 2 (0.5) | 1 (0.3) |
Alpha-fetoprotein, n (%) | ||
< 400 ng/mL | 243 (61.8) | 256 (65.8) |
≥ 400 ng/mL | 145 (36.9) | 124 (31.9) |
Missing | 5 (1.3) | 9 (2.3) |
PD-L1 status, n (%)h | ||
Positive | 148 (37.7) | 148 (38.0) |
Negative | 189 (48.1) | 181 (46.5) |
Missing | 52 (13.2) | 45 (11.6) |
|||||||||| ||| ||| ||||||| | | ||||| | || ||||| |
Prior disease-related radiotherapy, n (%) | 48 (12.2) | 37 (9.5) |
Previous treatment modalities, n (%) | ||
||||||||||| |||||||||||| | ||| |||||| | ||| |||||| |
||| ||||||| ||||||| | ||| |||||| | ||| |||||| |
|||||||| ||||||| | || |||||| | || |||||| |
|||||||| |||||||||||| | | ||||| | | ||||| |
||||||| |||||||| |||| | ||
|||||||||||| | ||| |||||| | ||| |||||| |
|||| |||||||| |||||||| | ||| |||||| | || |||||| |
||||||||| | | || |||||| | | |||||| |
||||||| ||||||||| | || |||||| | || |||||| |
||||||||| | | || |||||| | || |||||| |
BCLC = Barcelona Clinic Liver Cancer, ECOG = Eastern Cooperative Oncology Group Performance Status, HBV = hepatitis B virus; HCC = hepatocellular carcinoma; HCV = hepatitis C virus, PD-L1 = programmed cell death 1 ligand 1; SD = standard deviation.
aIncludes Brazil, Canada, France, Germany, Italy, Japan, Russia, Spain, Ukraine, and the US.
bThe ECOG PS scale ranges from 0 to 5, with higher numbers corresponding to greater disability.
cPatient’s ECOG PS was normal (0) at screening but deteriorated to 2 at randomization, while the Child-Pugh class was A at screening but B at randomization. The patient did not receive study treatment.
dECOG PS was not assessed at screening. At cycle 1, the ECOG PS was normal (0).
eThe Child-Pugh classification of liver disease severity is determined by the degree of ascites, serum concentrations of bilirubin and albumin, prothrombin time, and degree of encephalopathy and classified as follows: class A (well-compensated disease), score of 5 to 6; class B (significant functional compromise), score of 7 to 9; and class C (decompensated disease), score of 10 to 15.
fThe BCLC staging classification system includes stages 0 (very early), A (early), B (intermediate), C (advanced), and D (end stage).
gNo active viral hepatitis identified.
hBaseline PD-L1 results were not available for patients who were randomly assigned but not treated.
Source: HIMALAYA Clinical Study Report17 (details from the table have been taken from the sponsor’s Summary of Clinical Evidence).
Baseline Characteristics
The baseline demographic and disease characteristics of the tremelimumab in combination with durvalumab and sorafenib groups are summarized in Table 12. Baseline demographic and disease characteristics were generally well balanced between study groups. ||| |||| |||| || |||||||| || ||| |||||||||||| || ||||||||||| |||| |||||||||| ||| ||||||||| |||||| |||| || ||||| ||| ||||| |||||| ||| |||| ||||| ||| ||||| ||||||| ||||||||||||| ||||||||||||| ||| || |||||||| || |||| ||||||||| |||||| |||| ||||| ||| ||| |||| ||||||| |||||| |||| || ||| |||||||| |||| ||||| |||||||| ||||| |||| |||||| ||| |||| |||| || |||| |||||||| || |||||||| |||||| Almost all patients had a Child-Pugh class A score (| |||||||| |||||||||||| || ||||| ||||| |||||||||). Approximately 80% had BCLC stage C, and 20% had BCLC stage B. Approximately half had EHS, and a quarter of patients had MVI. With regards to previous disease-related treatment, more than a third of patients had undergone therapeutic embolization, ||||||||||||| ||| ||| ||||||||| ||| ||||||| |||||||, and 48 patients (12.2%) in the tremelimumab in combination with durvalumab group and 37 patients (9.5%) in the sorafenib group had received prior radiotherapy.
Exposure to Study Treatments
Patient exposure to study treatments is presented in Table 13. For all treated patients (in the safety analysis set) the median total treatment duration was 5.5 months (range = 0.4 to 42.7) in the tremelimumab in combination with durvalumab group and 4.1 months (range = 0.1 to 38.6) in the sorafenib group. ||||| |||| || |||||||||| || |||||||||||| |||| |||||||||| |||||||| ||| |||||||||||| ||||||| |||||||||| |||||||| ||||||||||||| |||||||| || ||||||||| |||||| ||| |||||||||| |||||| |||||||| || ||| ||||| |||||||| ||||| |||||||| ||| || ||| || ||| |||||||| ||||||||| ||||| |||| || |||||||||||| |||||||| ||||||||||||| || |||||||||| ||||||| ||||||||| |||| |||||||||| |||||||| || ||||||||||||| |||| || |||||||| |||| |||||||| ||||||||| |||| |||||||| ||| || ||| |||| |||||||| |||||||||
|||||||| | |||||||||||| || ||||||||||| |||| |||||||||| ||| |||| | |||||||||||| ||||| | ||
|---|---|---|---|---|
|||||||||||| | |||||||||| | |||
||||||||||||||||| | | ||||||||||| | ||||| ||||| | | ||||| ||||| | | |
||||| ||||||||| ||||||||| ||||||||| | | ||| | || | ||| | ||| |
|||| |||| | ||| |||||| | ||| |||||| | |||| ||||||| | ||| |||||| |
|||||| ||||||| | ||| |||| || |||| | ||| |||| || |||| | ||| |||| || ||||| | ||| |||| || ||||| |
||||| ||||||||| ||||| | |||| | ||| | ||||| | ||||| |
|||||| ||||||||| ||||||||| ||||||||| | | ||| | || | ||| | ||| |
|||| |||| | ||| |||||| | ||| |||||| | |||| ||||||| | ||| |||||| |
|||||| ||||||| | ||| |||| || |||| | ||| |||| || |||| | ||| |||| || ||||| | ||| |||| || ||||| |
||||| ||||||||| ||||| | |||| | ||| | ||||| | ||||| |
||||| |||||||| || ||||| |||||| |||||| ||||| ||||||||| | | ||| | ||| | ||| | ||| |
|||| |||| | || | ||| |||||| | ||| |||||| | || |
|||||| ||||||| | || | ||| |||| || ||| | ||| |||| || |||| | || |
NA = not applicable; SD = standard deviation.
Note: This table has been redacted at the request of the sponsor.
aInitial treatment phase includes the start of study treatment to last treatment or last treatment before rechallenge, where rechallenge occurred.
bTotal study exposure includes initial treatment and rechallenge phase, where rechallenge occurred.
cTotal treatment duration for immunotherapies = (last dose date + 27 days or date of death or data cut-off, whichever occurred earlier − first dose date + 1)/(365.25/12). Total treatment duration for sorafenib = (last dose date or date of death or data cut-off, whichever occurred earlier − first dose date + 1)/(365.25/12).
dActual treatment duration = (intended exposure – total duration of dose delays)/(365.25/12). Patients who took infusion earlier than planned were set to 0 for calculation.
Source: HIMALAYA Clinical Study Report17 (details from the table have been taken from the sponsor’s Summary of Clinical Evidence).
Concomitant Medications
Concomitant medications used by || ||||| || || |||||||| || |||||| ||||||||| ||||| ||| |||||||||| || ||||| ||| ||| || ||||||||||| ||||||||||| ||| ||||||||| |||| |||||||| ||||||| ||||||| ||| |||| |||||||||| |||||||| ||||||||||| ||||||||||| || |||| |||| ||| || |||||||| || |||| ||||| |||||| |||| |||||||||| ||| |||||||||| ||||||| ||||||||||||| |||||||||| ||||||| ||| |||||| |||| |||||||||| ||||||||
||| |||||||||||||| | | |||||||||||| || ||||||||||| |||| ||||||||||| || ||||| | |||||||||||| ||||| |
|---|---|---|
|||||||||| ||| |||||||||| ||||||| ||||||||||||| |||||||||| | || |||||| | || |||||| |
|||||| |||| |||||||||| | || |||||| | || |||||| |
||||||||||||||| ||||||||||| | || ||||| | || ||||| |
|||||||||| | || ||||| | || ||||| |
|||||||| | || ||||| | || ||||| |
||||| ||||||| | || ||||| | || ||||| |
||||| ||||||| | || ||||| | || ||||| |
||| ||| ||||||||| |||||||||| | || ||||| | || ||||| |
|||||||| ||||||||||| |||||||||| ||||||||| ||||||| | || ||||| | || ||||| |
||||||||||| ||||||||||| | || ||||| | || ||||| |
||||||||||| || |||||||| ||||||||| ||||| | || ||||| | || ||||| |
||||||||||||| ||||||||||||| ||| ||||||| ||||| |||||||| ||||| | || ||||| | || ||||| |
|||| ||||| ||| ||||||||||| | || ||||| | || ||||| |
||||||| ||||| ||||||||| | || ||||| | || ||||| |
||| ||||||||||| ||||| | || ||||| | || ||||| |
||||||||||||| | || ||||| | || ||||| |
||||||||||| ||||||||||| | || ||||| | | ||||| |
||||||||| |||||||| | | ||||| | || ||||| |
ACE = angiotensin-converting-enzyme; ATC = anatomic therapeutic chemical; carbamide products = hydrogen peroxide; FAS = full analysis set; H2 = histamine type-2; HMG CoA = hydroxymethylglutaryl coenzyme A.
Note: This table has been redacted at the request of the sponsor.
aA patient can have 1 or more generic term reported under a given ATC text. Patients with multiple concomitant medications with the same generic term under a given ATC text are counted once for that generic term. Therapy classification shown according to the WHO drug ATC classification system and mechanism of action.
Source: HIMALAYA Clinical Study Report.17
Subsequent Treatment
The details on subsequent anticancer therapy (defined as therapy started on or after the first dosing date) are presented in Table 15. Subsequent anticancer therapy was received by 40.7% of patients in the tremelimumab in combination with durvalumab group and 45% of patients in the sorafenib group. The most common therapy ||| |||||||| |||||||| ||||| ||||| || |||||||| || ||| |||||||||||| || ||||||||||| |||| |||||||||| ||||| ||| |||| || |||||||| || ||| ||||||||| ||||| |||||||| |||||||||| |||| |||||||| |||||||| |||||||||| || ||| |||||||||||| || ||||||||||| |||| |||||||||| ||||| |||| || ||||| ||| |||| |||||||| || ||| ||||||||| ||||| ||||||||
Efficacy
A summary of key efficacy outcomes from the HIMALAYA trial is presented in Table 16. Detailed discussions follow.
Overall Survival
At the final OS data cut-off on August 27, 2021, ||||||| ||||||||| |||| ||| ||| ||||||, and median follow-up time in the tremelimumab in combination with durvalumab group was 33.2 months (95% CI, 31.7 to 34.5) while in the sorafenib group it was 32.2 months (95% CI, 30.4 to 33.7). OS results are presented in Table 17. In the FAS, ||||| |||| ||| |||||| ||||||| || ||| |||||||||||| || ||||||||||| |||| |||||||||| ||||| ||| ||| |||||| ||||||| || ||| ||||||||| |||||. The HR, adjusted for stratification factors (determined by an interactive web response system), was 0.78 (96.02% CI, 0.65 to 0.93; stratified log-rank 2-sided P = 0.0035). The Kaplan-Meier estimates for median OS were 16.4 months (95% CI, 14.2 to 19.6) in the tremelimumab in combination with durvalumab group and 13.8 months (95% CI 12.3 to 16.1) in the sorafenib group. The OS rates at 36 months were 30.7% (95% CI, 25.8 to 35.7) in the tremelimumab in combination with durvalumab group and 20.2% (95% CI,15.8 to 25.1) in the sorafenib group. In the Kaplan-Meier plot of OS for tremelimumab in combination with durvalumab versus sorafenib presented in Figure 4 the curves appear to separate after 4 months, favouring tremelimumab in combination with durvalumab.
Subgroup Analysis of Overall Survival
The estimated effects in all predefined subgroups were consistent with the overall OS analysis as shown in Table 18.
||||||||||| |||||| || |||||| |||| |||||||| |||||||| |||||||||| ||||||||||||| || ||| ||||||||| ||||| ||||||| |||| ||| |||||||||||| || ||||||||||| |||| |||||||||| ||||| ||||||| |||||| |||| || ||| |||||||| ||| |||||||| |||||||||| ||||||| |||||||| |||| ||||||||||| ||||||| |||||| || ||| |||||||| || ||| |||||||||||| || ||||||||||| |||| ||||||||| ||||| ||| ||||| || ||| ||||||||| |||||||
||| || || |||||||| |||||||||||| |||| ||||||||||||| |||||| |||| ||| |||||||| |||||||| |||||||| |||||||||| |||||||||| |||||||| || |||||||| |||||||||| || |||||||| ||| |||| ||| |||||||||||||
Table 15: Summary of Subsequent Treatment in HIMALAYA (Full Analysis Set)
|||||||| | |||||||||||| || ||||||||||| |||| |||||||||| |||| | | |||||||||||| ||||| |
|---|---|---|
||||| |||||| || |||||||| | ||| |||||| | ||| |||||| |
||||||||||||| | || ||||| | || |||||| |
|||||||||||| | | ||||| | || ||||| |
||||||||| | | ||||| | || |||||| |
||||||||||||| | | ||||| | || ||||| |
||||||||| |||||||||||| | || ||||| | || ||||| |
|||||||| | | ||||| | | ||||| |
||||||||||| | | ||||| | | ||||| |
|||||||||||| | | ||||| | | ||||| |
|||||||||| | | ||||| | | ||||| |
|||||||| ||||||| | ||| |||||| | ||| |||||| |
||||||||| | ||| |||||| | || ||||| |
|||||||||| | || |||||| | || ||||| |
||||||||||| | || ||||| | || |||||| |
|||||||||||| | || ||||| | || ||||| |
|||||||||||||| ||||||| | || ||||| | || ||||| |
||||||||||| | | ||||| | || ||||| |
||||||||||| | | ||||| | | ||||| |
Notes: This table has been redacted at the request of the sponsor. Patients may have received more than 1 postinvestigational-product discontinuation therapy.
aTherapies taken following discontinuation of investigational product. Only therapies used by at least 1% of patients in either group are reported.
bIncludes intra-arterial administrations.
Source: HIMALAYA Clinical Study Report17 (details from the table have been taken from the sponsor’s Summary of Clinical Evidence).
Table 16: HIMALAYA Key Efficacy Outcomes (FAS With Final Data Cut-Off on August 27, 2021)
Outcomes | Tremelimumab in combination with durvalumab N = 393 | Sorafenib N = 389 |
|---|---|---|
Overall survival | ||
Median follow-up duration in all patients, months (95% CI) | 33.2 (31.7 to 34.5) | 32.2 (30.4 to 33.7) |
Median overall survival,a months (95% CI) | 16.4 (14.2 to 19.6) | 13.8 (12.3 to 16.1) |
Hazard ratio (96.02% CI) | 0.78 (0.65 to 0.93) | |
P value (2-sided)b | 0.0035 | |
Progression-free survival | ||
||||||||| |||||||| || ||| |||||||| ||||||||| |||||| ||||||| | ||| |||| |||||| | ||| |||| |||||| |
Median progression-free survival,a months (95% CI) | 3.78 (3.68 to 5.32) | 4.07 (3.75 to 5.49) |
Hazard ratio (95% CI) | 0.90 (0.77 to 1.05) | |
||||||| | |||||| | |
Progression-free at data cut-off, n (%) | 49 (12.5) | 19 (4.9) |
Objective response rate in patients with confirmed responsesd | ||
||||||||| ||||||||| |||| | || |||||| | || ||||| |
|||||||| |||||||| | || ||||| | || ||||| |
||||||| |||||||| | || |||||| | || ||||| |
|||| |||||| ||||||||| |||||||| |||| ||| | |||| |||||| ||||| | |
||||||| | ||||||| | |
Duration of response | ||
Median, months (IQR) | 22.34 (8.54 to NR) | 18.43 (6.51 to 25.99) |
Best objective response in patients with unconfirmed responses | ||
|||||| ||| | || |||||| | || ||||| |
|||||||| |||||||| | || ||||| | || ||||| |
||||||| |||||||| | || |||||| | || ||||| |
Time to progression | ||
Median TTPa (95% CI), months | |||| ||||| || ||||| | 5.55 (5.13 to 5.75) |
Treated ≥ 1 cycle beyond progression, n (%) | 182 (46.9) | 134 (34.4) |
||||||||| |||||||| || ||| |||||||| ||||||||| |||||| ||||||| | |||| ||||| ||||||| | |||| ||||| ||||||| |
|| |||| ||| | |||| ||||| || ||||| | |
||||||| |||||| | |||||| | |
CI = confidence interval; CR = complete response; FAS = final analysis set; IQR = interquartile range; OS = overall survival; PFS = progression-free survival; PR = partial response; RECIST = Response Evaluation Criteria in Solid Tumors; TTP = time to progression.
aCalculated using the Kaplan-Meier technique.
bThe adjusted alpha levels for the 2-sided superiority test of tremelimumab in combination with durvalumab vs. sorafenib and CI were derived from the exact number of OS events for each comparison using the Lan and DeMets approach that approximates the O’Brien-Fleming spending function. Analysis performed using a stratified log-rank test adjusting for treatment, etiology of liver disease (hepatitis B virus vs. hepatitis C virus vs. others), Eastern Cooperative Oncology Group Performance Status (0 vs. 1), and macrovascular invasion (yes vs. no). P value has been adjusted for multiple testing.
cAnalysis was performed using a logistic regression model adjusted for treatment with factors for etiology of liver disease, Eastern Cooperative Oncology Group Performance Status, and macrovascular invasion. P value has not been adjusted for multiple testing.
dA confirmed response of CR/PR means that a response of CR/PR was recorded at 1 visit and confirmed by repeat imaging not less than 4 weeks after the visit at which the response was first observed with no evidence of progression between the initial and confirmation visit. Unconfirmed responses were not confirmed by repeat imaging.
Source: HIMALAYA Clinical Study Report17 (details from the table have been taken from the sponsor’s Summary of Clinical Evidence).
Table 17: Overall Survival (Final Analysis Set With Final Data Cut-Off on August 27, 2021)
Overall survival | Tremelimumab in combination with durvalumab N = 393 | Sorafenib N = 389 |
|---|---|---|
||||||| |||| | ||| |||||| | ||| |||||| |
|||||||| ||||||||| |||| | ||| |||||| | || |||||| |
Lost to follow-up | 1 (0.3) | 7 (1.8) |
||||||||| ||||||| | | ||||| | || ||||| |
||||| |||||||||| | | ||| |||||| | || |||||| |
||||||||| |||||||| || |||||||| |||||||| ||||||||| |||||| ||||||| | |||| |||| |||||| | |||| |||| |||||| |
Median overall survival,b months (95% CI) | 16.4 (14.2 to 19.6) | 13.8 (12.3 to 16.1) |
Hazard ratio (96.02% CI)c | 0.78 (0.65 to 0.93) | |
P value (2-sided)c | 0.0035 | |
|| |||| || || ||||||| ||||| ||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
OS rate at 18 months, % (95% CI) | 48.7 (43.6 to 53.5) | 41.5 (36.5 to 46.4) |
OS rate at 24 months, % (95% CI) | 40.5 (35.6 to 45.3) | 32.6 (27.9 to 37.4) |
OS rate at 36 months, % (95% CI) | 30.7 (25.8 to 35.7) | 20.2 (15.8 to 25.1) |
Median (95% CI) follow-up in all patients (months) | 33.2 (31.7 to 34.5) | 32.2 (30.4 to 33.7) |
CI = confidence interval; IQR = interquartile range; OS = overall survival; PFS = progression-free survival; RECIST = Response Evaluation Criteria in Solid Tumors; TTP = time to progression; vs. = versus.
aPatients confirmed alive at follow-up or on active study treatment at the time of final analysis reported “study completion” on the disposition electronic case report form. Includes patients known to be alive at data cut-off.
bCalculated using the Kaplan-Meier technique.
cThe adjusted alpha levels for the 2-sided superiority test of tremelimumab in combination with durvalumab vs. sorafenib and CI were derived from the exact number of OS events for each comparison using a Lan and DeMets approach that approximates the O’Brien-Fleming spending function. Analysis performed using stratified log-rank test adjusting for treatment, etiology of liver disease (hepatitis B virus vs. hepatitis C virus vs. others), Eastern Cooperative Oncology Group Performance Status (0 vs. 1), and macrovascular invasion (yes vs. no). The values of the stratification factors were obtained from an interactive web response system.
Source: HIMALAYA Clinical Study Report17 (details from the table have been taken from the sponsor’s Summary of Clinical Evidence).
Table 18: OS by Subgroup of Interest (FAS With Final Data Cut-Off on August 27, 2021)
Subgroupa | Tremelimumab in combination with durvalumab | Sorafenib | HR | 95% CI | ||
|---|---|---|---|---|---|---|
N | Number of events (%) | N | Number of events (%) | |||
Viral etiology | ||||||
HBV | 122 | 82 (67.2) | 119 | 98 (82.4) | 0.64 | 0.48 to 0.86 |
HCV | 110 | 73 (66.4) | 104 | 64 (61.5) | 1.06 | 0.76 to 1.49 |
Nonviral | 161 | 107 (66.5) | 166 | 131 (78.9) | 0.74 | 0.57 to 0.95 |
Eastern Cooperative Oncology Group Performance Status | ||||||
0 | 244 | 147 (60.2) | 241 | 168 (69.7) | 0.79 | 0.63 to 0.98 |
1 | 148 | 114 (77.0) | 147 | 124 (84.4) | 0.74 | 0.57 to 0.95 |
Barcelona Clinic Liver Cancer stage | ||||||
B | 77 | 44 (57.1) | 66 | 44 (66.7) | 0.87 | 0.57 to 1.33 |
C | 316 | 218 (69.0) | 323 | 249 (77.1) | 0.76 | 0.63 to 0.91 |
Macrovascular invasion | ||||||
Yes | 103 | 78 (75.7) | 100 | 83 (83.0) | 0.78 | 0.57 to 1.07 |
No | 290 | 184 (63.4) | 289 | 210 (72.7) | 0.77 | 0.63 to 0.93 |
Extrahepatic spread | ||||||
Yes | 209 | 146 (69.9) | 203 | 160 (78.8) | 0.67 | 0.53 to 0.84 |
No | 182 | 114 (62.6) | 185 | 133 (71.9) | 0.87 | 0.67 to 1.11 |
Alpha-fetoprotein at baseline | ||||||
< 400 ng/mL | 167 | 109 (65.3) | 182 | 130 (71.4) | 0.82 | 0.63 to 1.05 |
≥ 400 ng/mL | 98 | 70 (71.4) | 71 | 60 (84.5) | 0.64 | 0.45 to 0.91 |
CI = confidence interval; FAS = full analysis set; HBV = hepatitis B virus; HCV = hepatitis C virus; HR = hazard ratio.
aHR and 95% CI were estimated from an unstratified Cox proportional hazards model with treatment as the only covariate and using the Efron method to control for ties.
Source: HIMALAYA Clinical Study Report.17
Sensitivity Analysis of Overall Survival
As there was a 4-month delay in the separation of Kaplan-Meier curves, an assessment of the assumption of nonproportionality was conducted. The linear interaction between treatment and time was tested, and no significant interaction was found |||||||| |||||| This was supported by a post hoc analysis that was conducted to calculate piecewise constant treatment effects for the comparison of tremelimumab in combination with durvalumab versus sorafenib. Beyond 9 months, the HR was 0.70 (95% CI, 0.56 to 0.89).
| ||||||| || |||||||| ||||| ||| |||| ||||||||||| || ||||||||| ||| |||||||| ||| ||||||| || ||| |||||||| || |||||| ||| |||||||||||| || ||||||||||| |||| |||||||||| ||| ||||||||| |||||| |||||||| ||||| || |||| ||| ||||| || ||||||| |||||||||| |||||| |||||||| ||||||||| ||| |||| |||||| || ||||| ||||| ||| || |||||||||| |||||||||| || ||| |||||| || ||||||||| ||||| ||||||| ||||||| || ||| |||||||| |||||||| |||| ||||||||| |||||||||| |||| ||||| || ||| ||||||| |||||||||
The results of another sensitivity analysis for OS based on the stratified Cox proportional hazard model, adjusted for EHS, albumin-bilirubin (ALBI) score, alpha-fetoprotein levels, and BCLC stage, were consistent with the primary OS analysis results.
Overall Survival for Patients Rechallenged With Tremelimumab
|| ||| || |||||||| ||| |||| |||||||||||| |||| |||||||||||| || ||| |||||||||||| || ||||||||||| |||| |||||||||| |||||| ||| |||||| || ||| ||||| |||||| |||| ||| ||||| || |||| ||| || ||||| |||| ||| |||| ||||| ||||| || ||||| || || ||||||| ||||| ||||| || ||||| || || ||||||| ||||| ||||| || ||||| || || ||||||| ||| ||||| ||||| || ||||| || || |||||||
Overall Survival for Patients Treated After Progression
A total of 182 patients (46.3%) in the tremelimumab in combination with durvalumab group and 192 (49.4%) in the sorafenib group received at least 1 dose of the study treatment after progressive disease as defined by RECIST 1.1. ||| |||||| || ||| |||| |||||| |||| ||| |||| || ||||| || ||| |||||||||||| || ||||||||||| |||| |||||||||| ||||| ||| |||| |||||| |||| ||| |||| || ||||| || ||| ||||||||| |||||| || ||||| |||| ||| || ||| |||||||||||| || ||||||||||| |||| |||||||||| |||||| ||||||||| |||||| |||| ||||| ||||| || ||||| || ||||| ||||| || ||||| || || ||||||| ||||| ||||| || ||||| || ||||| ||||| || ||||| || || ||||||| ||||| ||||| || ||||| || ||||| ||||| || ||||| || || |||||| ||| ||||| ||||| || ||||| || ||||| ||||| || ||||| || || |||||||
Progression-Free Survival
At the final data cut-off on August 27, 2021, the Kaplan-Meier estimates for median PFS in the FAS were 3.8 months (95% CI, 3.7 to 5.3) in the tremelimumab in combination with durvalumab group and 4.1 months (95% CI, 3.8 to 5.5) in the sorafenib group, with an HR of 0.90 (95% CI, 0.77 to 1.05) |||||||| |||||| |||||||| A Kaplan-Meier plot for PFS is presented in Figure 5. There were 49 patients (12.5%) in the tremelimumab in combination with durvalumab group and 19 patients (4.9%) in the sorafenib group who were progression-free. PFS results are presented in Table 19.
Table 19: PFS by Investigator Assessment According to RECIST 1.1 (FAS With Final Data Cut-Off on August 27, 2021)
Progression-free survival | Tremelimumab in combination with durvalumab N = 393 | Sorafenib N = 389 |
|---|---|---|
||||||||| |||||||| || ||| |||||||| ||||||||| |||||| ||||||| | ||| |||| || ||||| | ||| |||| || ||||| |
Median PFS,a months (95% CI) | 3.78 (3.68 to 5.32) | 4.07 (3.75 to 5.49) |
Hazard ratio (95% CI) | 0.90 (0.77 to 1.05) | |
||||||| |||| | |||||| | |
||| |||||| ||| | ||| |||||| | ||| |||||| |
|||||| ||||||||||| | ||| |||||| | ||| |||||| |
||||| || ||| ||||||| || ||||||||||| | || |||||| | || |||||| |
|||||||| ||||||||| |||| | || |||||| | || |||||| |
||||||||| ||||| | | || |||||| | || ||||| |
|||||||| ||||| | | | ||||| | || ||||| |
||||||||| ||||||| | | ||||| | | ||||| |
|||| || ||||||||| | | ||||| | | ||||| |
|||||||| |||||| ||||||||||| | | | ||||| | | ||||| |
||||||||| |||||||| || |||||||| |||||||| ||||||||| |||||| ||||||| | |||| |||| || ||||| | ||| |||| || ||||| |
Progression-free at data cut-off, n (%) | 49 (12.5) | 19 (4.9) |
CI = confidence interval; FAS = full analysis set; HBV = hepatitis B virus; HCV = hepatitis C virus; PFS = progression-free survival; q.4.w. = every 4 weeks; RECIST 1.1 = Response Evaluation Criteria in Solid Tumors Version 1.1.
aCalculated using the Kaplan-Meier technique.
bPatients who had not progressed or died, or who progressed or died after 2 or more missed visits, were censored at the latest evaluable RECIST 1.1 assessment, or day 1 if there were no evaluable visits. Patients who had no evaluable visits or baseline data were censored at day 1 unless they died within 2 visits of baseline.
cStudy completion refers to patients who were progression-free and ongoing in the study at the data cut-off.
dDeath occurred after 2 or more missed visits after last evaluable RECIST 1.1 assessment (or randomization).
eRECIST 1.1 progression event occurred after 2 or more missed visits after last evaluable RECIST 1.1 assessment (or randomization).
Source: HIMALAYA Clinical Study Report17 (details from the table have been taken from the sponsor’s Summary of Clinical Evidence).
Objective Response Rate
At the final data cut-off on August 27, 2021, the ORRs (in patients with confirmed responses) according to RECIST 1.1 and based on investigator assessment were 20.1% (79 patients) in the tremelimumab in combination with durvalumab group and 5.1% (20 patients) in the sorafenib group. |||| ||||||||| |||||||||||| || ||||||||||| |||| |||||||||| ||| |||||||||| ||| || ||| |||| |||| ||| |||| || ||||| ||||||| |||||||| || |||||| || |||||||||||| || ||||||||||| |||| |||||||||||
Overall, the results of the additional analyses of ORR, according to RECIST 1.1 and based on investigator assessments, were consistent with the results of the main analysis. In the first additional analysis, a logistic regression model adjusted for treatment with factors for etiology of liver disease, ECOG PS, and MVI was used. The confirmed ORR was 23.9% for the tremelimumab in combination with durvalumab group and 6.7% for the sorafenib group, and ||| || ||| |||| |||| ||| |||| || |||||| || ||| |||||| |||||||||| ||||||||| |||||||||| ||||||||||||||||||||||| |||| |||||||| ||| ||| |||| ||||||| ||| |||| ||| ||| || ||| |||| |||| ||| |||| || ||||||
|||||||| || ||| || |||||||||| ||||||||| || |||||||| ||||||||| || ||||| ||||||| ||| |||| |||||||||||| |||| ||| ||||||||| ||||||| || ||| ||||||||| |||| |||||||||| |||| ||||||| ||| |||||||||
Figure 4: Kaplan-Meier Plot of Overall Survival (FAS With Final Data Cut-Off on August 27, 2021)
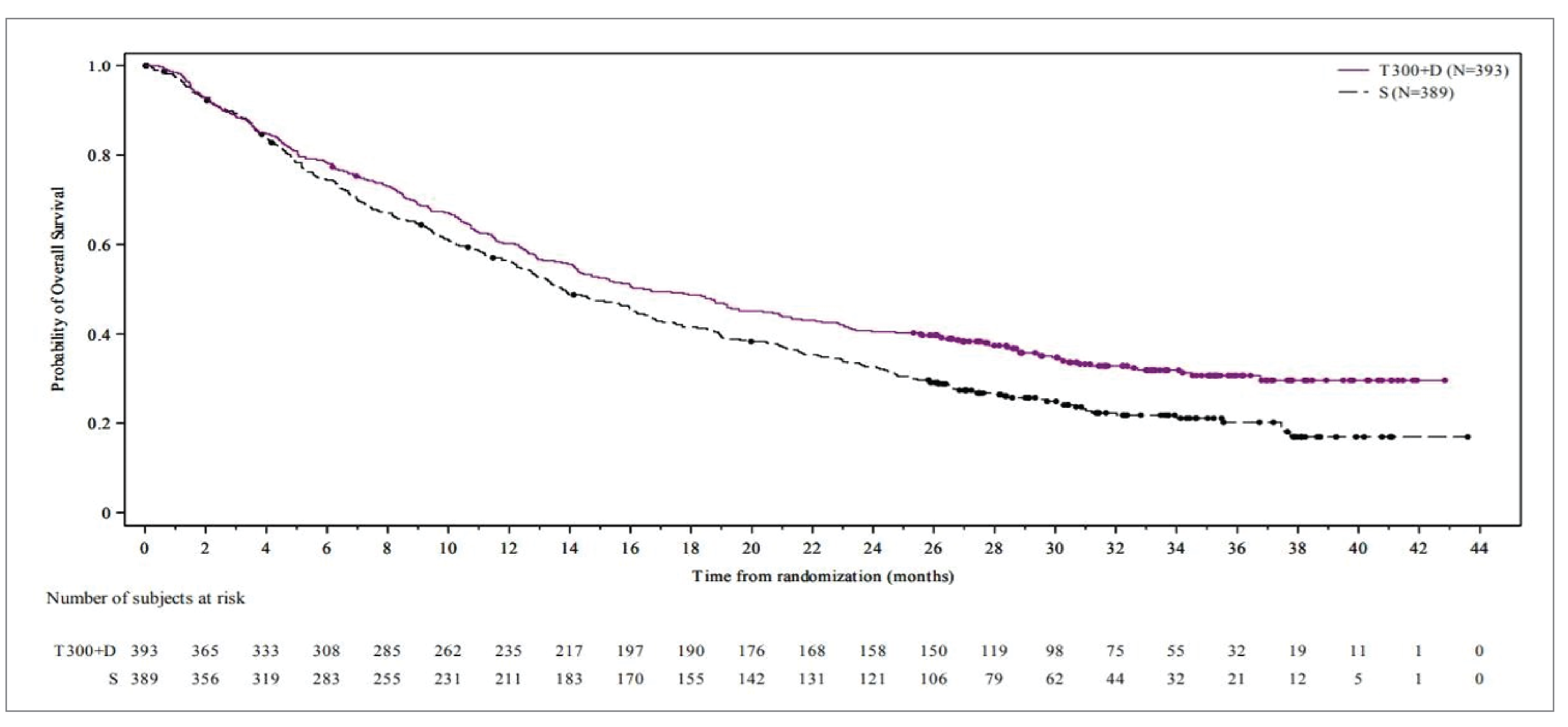
FAS = full analysis set; S = sorafenib; T300+D = tremelimumab 300 mg plus durvalumab.
Source: HIMALAYA Clinical Study Report.17
The descriptive results for ORR for patients at the final data cut-off and in the 32-week follow-up set (first interim analysis) are presented in Table 20. Overall, similar results were observed in terms of ORR in patients’ confirmed responses based on BICR according to RECIST 1.1 as well as in patients with unconfirmed responses (i.e., independent of imaging RECIST 1.1 methodology) based on BICR according to mRECIST, irRECIST, and RECIST 1.1.
Figure 5: Kaplan-Meier Plot for PFS (FAS With Final Data Cut-Off on August 27, 2021)
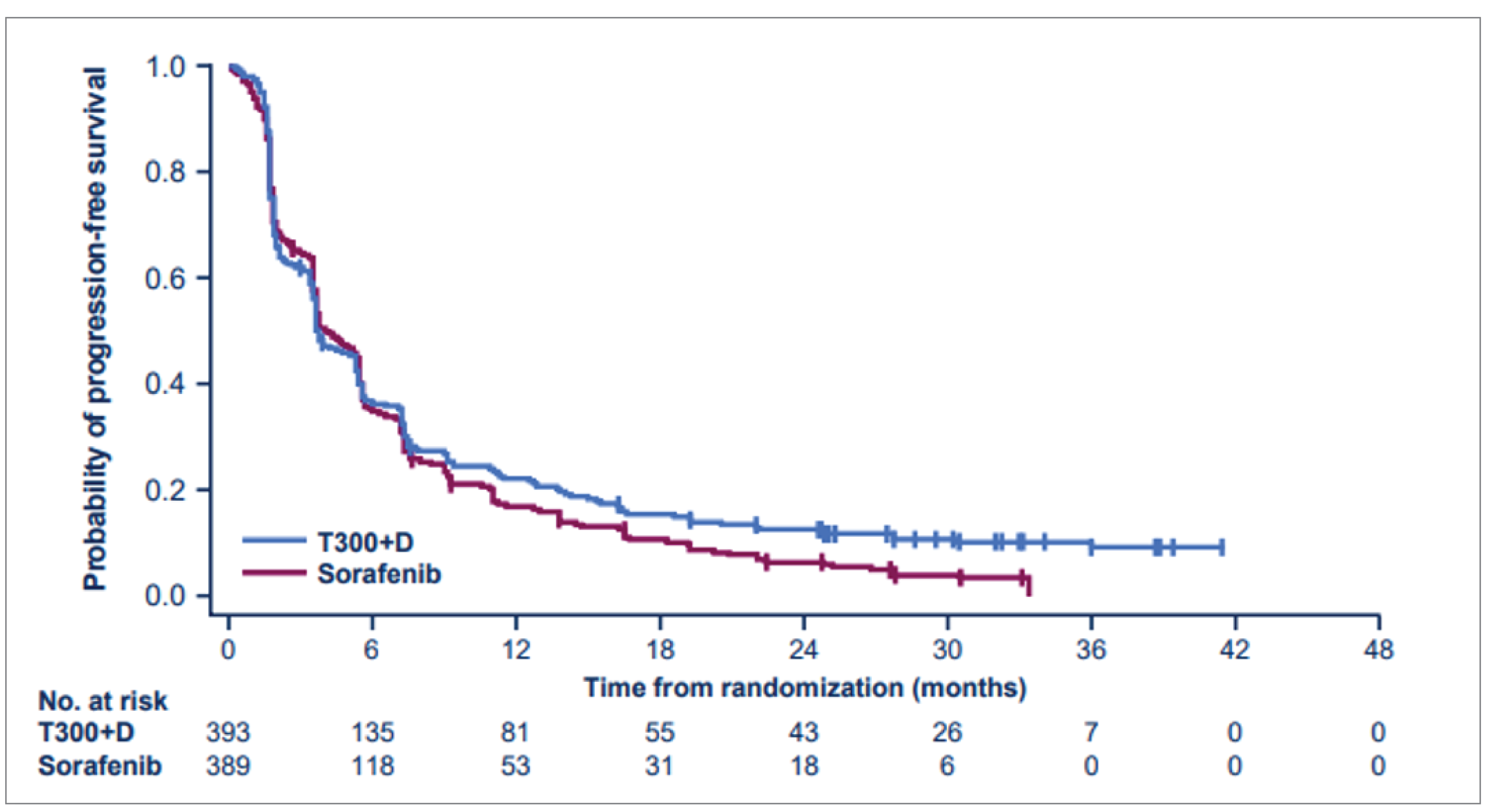
FAS = full analysis set; S = sorafenib 400 mg twice daily; T300+D = tremelimumab 300 mg × 1 dose + durvalumab 1,500 mg every 4 weeks.
Source: HIMALAYA Clinical Study Report.17
Table 20: ORR for Patients in the FAS Based on Investigator Assessment Using RECIST 1.1. (With Final Data Cut-off on August 27, 2021) and First Interim Assessment (FAS-32w With Data Cut-Off on September 2, 2019)
Objective response rate | Tremelimumab in combination with durvalumab | Sorafenib |
|---|---|---|
FAS — ORR by investigator assessment according to RECIST 1.1 | ||
N | 393 | 389 |
ORR (confirmed responses),a n (%) | 79 (20.1) | 20 (5.1) |
Complete response | 12 (3.1) | 0 |
Partial response | 67 (17.0) | 20 (5.1) |
|||| ||||| ||||||||| |||||||| |||| ||| | |||| ||||| || ||||| | |
||||||| | | |||||| | |
||| |||| ||||| || |||| | ||
|| ||||| | ||| | ||| |
||| |||||||||| |||||||||| ||||||||| || |||||| ||| |||| ||| | |||| |||||| || |||||| | ||| ||||| || ||||| |
||| |||||||||||| |||||||||| ||||||||| || |||||||| |||| ||| | |||| |||||| || |||||| | |||| ||||| || |||||| |
||| |||||||||||| |||||||||| ||||||||| || |||||||| ||| |||| ||| | |||| |||||| || |||||| | ||| ||||| || |||||| |
||| |||||||||||| |||||||||| ||||||||| || |||||| ||| |||| ||| | |||| |||||| || |||||| | ||| ||||| || |||||| |
BICR = blinded independent central review; CI = confidence interval; CR = complete response; FAS = full analysis set; FAS-32w = full analysis set at 32-week follow-up; ORR = objective response rate; PR = partial response; RECIST 1.1 = Response Evaluation Criteria in Solid Tumors Version 1.1.
aA confirmed response of CR/PR means that a response of CR/PR was recorded at 1 visit and confirmed by repeat imaging not less than 4 weeks after the visit where response was first observed with no evidence of progression between the initial and confirmation visit. Unconfirmed responses were not confirmed by repeat imaging.
bAnalysis was performed using a logistic regression model adjusted for treatment with factors for etiology of liver disease, Eastern Cooperative Oncology Group Performance Status, and macrovascular invasion. P value has not been adjusted for multiple testing.
Source: HIMALAYA Clinical Study Report.17
Best Objective Response
|| ||| ||||| |||| ||||||| || |||||| ||| ||||| || |||||||| ||||||| || ||| |||||||||||| || ||||||||||| |||| |||||||||| ||||| ||| || |||||||| |||||| |||||||| ||| || || || || ||||| || ||||||||||| ||||||||| || |||||||||||| ||||||||||| ||||| |||| || |||||||| |||||| || ||| |||||||||||| || ||||||||||| |||| |||||||||| ||||| ||| |||||||| ||||||||| ||||||||| ||| |||| || ||| ||||||||| |||||| ||| ||||||| ||| |||||||| |||| ||||||||| ||||||||| || |||||||||||| |||||||||| |||| |||||||| Results for BOR at the first interim analysis for patients in the 32-week follow-up set based on BICR assessments were also consistent with those reported at the final analysis for patients in the FAS based on investigator assessments. Results from the final analysis and interim analysis are presented in Table 21.
Table 21: Best Objective Response Based on Investigator Assessment and BICR Assessment (Confirmed Response) According to RECIST 1.1 at the FAS (With Final Data Cut-Off on August 27, 2021) and IA1 (FAS-32w With Data Cut-Off on September 2, 2019)
Best objective response | Tremelimumab in combination with durvalumab | Sorafenib |
|---|---|---|
FAS – Best objective response based on investigator assessment | ||
N | 393 | 389 |
Response total | 79 (20.1) | 20 (5.1) |
Complete response | 12 (3.1) | 0 |
Partial response | 67 (17.0) | 20 (5.1) |
Nonresponse total | 314 (79.9) | 369 (94.9) |
Stable disease | 157 (39.9) | 216 (55.5) |
||||||||||| | ||| |||||| | ||| |||||| |
|||||| ||||||||||| | ||| |||||| | || |||||| |
||||| | || ||||| | || ||||| |
||| ||||||||| | || ||||| | || ||||| |
||| |||| ||||| || |||| | ||
|| ||||| | ||| | ||| |
|||||||| ||||| | || |||||| | || ||||| |
|||||||| |||||||| | | ||||| | || ||||| |
||||||| |||||||| | || |||||| | || ||||| |
|||||||||||| ||||| | ||| |||||| | ||| |||||| |
|||||| ||||||| | || |||||| | ||| |||||| |
||||||||||| | || |||||| | || |||||| |
|||||| ||||||||||| | || |||||| | || |||||| |
||||| | || ||||| | || ||||| |
||||||| |||||||| | | ||||| | | ||||| |
||| ||||||||| | | ||||| | || ||||| |
BICR = blinded independent central review; FAS = full analysis set; FAS-32w = full analysis set at 32-week follow-up; IA1 = first interim assessment; RECIST 1.1 = Response Evaluation Criteria In Solid Tumors Version 1.1.
Source: HIMALAYA Clinical Study Report.17
Duration of Response
At the final data cut-off on August 27, 2021, among 79 patients with a confirmed response in the tremelimumab in combination with durvalumab group and 20 patients with a confirmed response in the sorafenib group, the median DoRs based on investigator assessments according to RECIST 1.1 were 22.3 months (IQR = 8.5 to NR) and 18.4 months (IQR = 6.5 to 26) respectively. ||| ||||||||| || |||||||| ||||||||| || |||||||| || || |||||| ||||| || ||| |||||||||||| ||||||||| ||| ||||| || ||| |||||||||||| || ||||||||||| |||| |||||||||| ||||| ||| ||||| || ||| ||||||||| |||||. Median time to onset of response from randomization was 2.2 months (IQR = 1.8 to 4.0) and 3.8 months (IQR = 1.9 to 8.4), respectively.
Disease Control Rate
At the final data cut-off on August 27, 2021, the overall DCR was similar between both groups, with 236 patients (60.1%) in the tremelimumab in combination with durvalumab group and 236 patients (60.7%) in the sorafenib group achieving controlled disease based on investigator assessments according to RECIST 1.1. ||| |||||||||| || |||||||| ||| |||||||| ||||||| ||||||| || || ||| || ||||| ||| ||||| ||| |||||| ||||||||||||| || ||| ||||||||||| || ||||||||||| |||| |||||||||| ||||| |||||||| |||| ||||| ||| |||||| respectively, in the sorafenib group as presented in Table 22.
Table 22: Disease Control Rate by Investigator Assessment According to RECIST 1.1 (FAS With Final Data Cut-Off August 27, 2021)
Disease controla | Tremelimumab in combination with durvalumab (N = 393) | Sorafenib (N = 389) |
|---|---|---|
Overall disease control | ||
Yes | 236 (60.1) | 236 (60.7) |
No | 157 (39.9) | 153 (39.3) |
||||||| | ||
||| | ||| |||||| | ||| |||||| |
|| | ||| |||||| | ||| |||||| |
||||||| | ||
||| | ||| |||||| | ||| |||||| |
|| | ||| |||||| | ||| |||||| |
DCR-16w = disease control rate at 16 weeks; DCR-24w = disease control rate at 24 weeks; FAS = full analysis set; RECIST 1.1 = Response Evaluation Criteria in Solid Tumors Version 1.1.
aDisease control = complete response + partial response + stable disease. Responses do not require confirmation.
Source: HIMALAYA Clinical Study Report17 (details from the table have been taken from the sponsor’s Summary of Clinical Evidence).
Time to response | Tremelimumab in combination with durvalumab (N = 393) | Sorafenib (N = 389) |
|---|---|---|
|||| ||||||||| |||||| |||| ||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
||||| |||||| || |||||| |||||||||| | ||| |||||| | ||| |||||| |
|||||||| ||||||||| |||| | ||| |||||| | ||| |||||| |
||||| || ||| ||||||| || ||||||||||| | | || |||||| | || |||||| |
||||| |||||||||| | | || |||||| | || ||||| |
||||||||| ||||||| | | ||||| | | ||||| |
|||| || ||||||||| | | ||||| | | ||||| |
|||||||| |||||| ||||||||||| | | | ||||| | | ||||| |
|||||||||||||||| || |||| || |||||||| | | ||||| | | ||||| |
|| |||| ||| | |||| ||||| || ||||| | |
||||||| ||||||| | |||||| | |
||||||||| |||||||| || ||| |||||||| ||||||||| |||||| ||||||| | |||| ||||| || |||||| | |||| ||||| || |||||| |
||||||||| |||||||| || |||||||| |||||||| ||||||||| ||||| | |||| ||||| || |||||| | |||| ||||| || |||||| |
CI = confidence interval; eCRF = electronic case report form; FAS = full analysis set; RECIST 1.1 = Response Evaluation Criteria in Solid Tumors Version 1.1; TTP = time to progression.
Note: This table has been redacted at the request of the sponsor.
aCalculated using the Kaplan-Meier technique.
bPatients who had not progressed, or who progressed after 2 or more missed visits, were censored at the latest evaluable RECIST assessment, or day 1 if there were no evaluable visits. Patients who have no evaluable visits or baseline data were censored at day 1 unless they died within 2 visits of baseline. Patients who die without tumour progression will be censored at the time of death.
cDeath occurred in the absence of progression or death occurred after 2 or more missed visits after last evaluable RECIST 1.1 assessment (or randomization).
dOther recorded on disposition eCRF with specified status of “study terminated by sponsor.” These patients were ongoing in the study at the data cut-off.
eRECIST 1.1 progression event occurred after 2 or more missed visits after last evaluable RECIST 1.1 assessment (or randomization).
fNot adjusted for multiplicity.
Source: HIMALAYA Clinical Study Report17 (details from the table have been taken from the sponsor’s Summary of Clinical Evidence).
Time to Progression
At the final data cut-off on August 27, 2021, the Kaplan-Meier estimates of median TTP according to RECIST 1.1 based on investigator assessments were ||| |||||| |||| ||| ||| || |||| in the tremelimumab in combination with durvalumab group and 5.6 months (95% CI, 5.1 to 5.8) in the sorafenib group. Results of TTP assessment are presented in Table 23.
HRQoL Measures (EORTC QLQ-C30 and EORTC QLQ-HCC)
The EORTC QLQ-C30 compliance rate (defined as the proportion of evaluable forms out of all expected forms) at baseline was 80% in the tremelimumab in combination with durvalumab group and 88% in the sorafenib group. ||||| |||||||| || ||| ||| ||| || |||| ||| ||| ||| ||| ||| || |||||| || ||| |||||||||||| || ||||||||||| |||| |||||||||| ||| ||||||||| |||||| ||||||||||||| ||| ||||| ||||||||| |||||||||| |||| || |||||||| ||| ||| ||| |||| ||| |||||||| || ||| ||| ||| || |||| ||| ||| ||| ||| ||| || |||||| || ||| |||||||||||| || ||||||||||| |||| |||||||||| ||| ||||||||| |||||| |||||||||||||
Change From Baseline
At baseline, mean scores for EORTC QLQ-C30 and QLQ-HCC items were comparable between treatment groups. Throughout follow-up, HRQoL remained stable in both treatment groups, with no mean change in scores from baseline reaching the MID (i.e., mean change ≥ 10 points) at any time point for either treatment group or between groups. Absolute scores at baseline and mean change from baseline scores using a mixed model for repeated measures at each visit up to week 60 are presented in Table 24 and Table 25.
|||||||| ||||| ||||| | |||||||||||| || ||||||||||| |||| |||||||||| ||| |||| | |||||||||||| |||| |
|---|---|---|
||||||| ||||||| |||||| |||||| | ||
||||||||| | | ||| | ||| |
|||||||| ||||| |||| | |||| ||||||| | |||| ||||||| |
|||||| |||| ||||||||| |||| || | | ||| | ||| |
|||||||| |||| |||| ||| | |||| |||||| || |||||| | |||| ||||||| || |||||| |
||||||||| |||||||||| |||| ||| | ||| |||||| || ||||| | |
|||||||| |||| |||| ||| | |||| |||||| || |||||| | |||| ||||||| || |||||| |
||||||||| |||||||||| |||| ||| | ||| |||||| || ||||| | |
|||||| |||| ||||||||| |||| ||| | | ||| | ||| |
||||||| ||||||||| ||||||||||| | ||
||||||||| | | ||| | ||| |
|||||||| ||||| |||| | |||| ||||||| | |||| ||||||| |
|||||| |||| ||||||||| |||| || | | ||| | ||| |
|||||||| |||| |||| ||| | |||| |||||| || |||||| | |||| |||||| || |||||| |
|||||| |||| ||||||||| |||| ||| | | ||| | ||| |
|||||||| |||| |||| ||| | |||| |||||| || |||||| | |||| ||||||| || |||||| |
||||||| |||||||| | ||
||||||||| | | ||| | ||| |
|||||||| ||||| |||| | |||| ||||||| | |||| ||||||| |
|||||| |||| ||||||||| |||| || | | ||| | ||| |
||||||||| |||||||||| |||| ||| | |||| |||||| || |||||| | |
|||||| |||| ||||||||| |||| ||| | | ||| | ||| |
|||||||| |||| |||| ||| | ||| ||||| || ||||| | ||| ||||| || |||||| |
||||||||| |||||||||| |||| ||| | |||| |||||| || |||||| | |
|||||| |||| ||||||||| |||| ||| | | ||| | ||| |
||||||| ||||||| | ||
||||||||| | | ||| | ||| |
|||||||| ||||| |||| | ||| ||||||| | ||| ||||||| |
|||||| |||| ||||||||| |||| || | | ||| | ||| |
|||||||| |||| |||| ||| | ||| ||||| || ||||| | ||| ||||| || ||||| |
||||||||| |||||||||| |||| ||| | |||| |||||| || ||||| | |
|||||| |||| ||||||||| |||| ||| | | ||| | ||| |
|||||||| |||| |||| ||| | ||| ||||| || ||||| | ||| ||||| || ||||| |
||||||| ||||||||| |||| | ||
||||||||| | | ||| | ||| |
|||||||| ||||| |||| | |||| ||||||| | |||| ||||||| |
|||||| |||| ||||||||| |||| || | | ||| | ||| |
|||||||| |||| |||| ||| | ||| ||||| || ||||| | |||| |||||| || |||||| |
||||||||| |||||||||| |||| ||| | |||| ||||||| || |||||| | |
|||||| |||| ||||||||| |||| ||| | | ||| | ||| |
CI = confidence interval; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; FAS = full analysis set; MMRM = mixed model for repeated measures; NE = not estimable.
Notes: This table has been redacted at the request of the sponsor. This table has been redacted at the request of the sponsor. The analysis set includes a subset of FAS with an evaluable baseline assessment and at least 1 evaluable postbaseline assessment. Change from baseline is derived using a MMRM analysis of all the postbaseline scores for each visit. The model includes treatment, visit, and treatment by visit interaction as explanatory variables and the baseline score as a covariate.
Some redacted rows have been deleted.
Source: HIMALAYA Clinical Study Report.17
|||||||| ||||| ||||| | |||||||||||| || ||||||||||| |||| |||||||||| ||| |||| | |||||||||||| |||| |
|---|---|---|
||||| ||||||||| ||||||||| |||| | ||
||||||||| | | ||| | ||| |
|||||||| ||||| |||| | |||| ||||||| | |||| ||||||| |
|||||| |||| ||||||||| |||| || | | ||| | ||| |
|||||||| |||| |||| ||| | ||| ||||||| ||||| | |||| ||||||| ||||| |
||||||||| |||||||||| |||| ||| | ||| ||||||| ||||| | |
|||||| |||| ||||||||| |||| ||| | | ||| | ||| |
|||||||| |||| |||| ||| | |||| ||||||| ||||| | |||| ||||||| ||||| |
||||| ||||||||| |||||||||| |||| | ||
||||||||| | | ||| | ||| |
|||||||| ||||| |||| | |||| ||||||| | |||| ||||||| |
|||||| |||| ||||||||| |||| || | | ||| | ||| |
|||||||| |||| |||| ||| | ||| ||||||| ||||| | ||| |||||| ||||| |
||||||||| |||||||||| |||| ||| | |||| |||||||| |||||| | |
|||||| |||| ||||||||| |||| ||| | | ||| | ||| |
|||||||| |||| |||| ||| | |||| ||||||| ||||| | ||| |||||| |||||| |
||||| ||||||||| |||||||||| |||||||| | ||
||||||||| | | ||| | ||| |
|||||||| ||||| |||| | |||| ||||||| | |||| ||||||| |
|||||| |||| ||||||||| |||| || | | ||| | ||| |
|||||||| |||| |||| ||| | ||| ||||||| ||||| | ||| |||||| ||||| |
|||||| |||| ||||||||| |||| ||| | | ||| | ||| |
|||||||| |||| |||| ||| | ||| ||||||| ||||| | ||| |||||| ||||| |
||||||||| |||||||||| |||| ||| | |||| ||||||| ||||| | |
CI = confidence interval; EORTC QLQ-HCC18 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Hepatocellular Carcinoma 18; FAS = full analysis set; MMRM = mixed model for repeated measures; NE = not estimable.
Note: This table has been redacted at the request of the sponsor.
Some redacted rows have been deleted.
Source: HIMALAYA Clinical Study Report.17
Time to Deterioration of HRQoL and HCC Symptoms
Time to deterioration was estimated among patients with symptom scores of 10 or lower, and/or GHS and/or physical function scores of 90 or higher at baseline. || ||| ||||| |||| ||||||| || |||||| ||| ||||| |||||| ||| || |||||| ||| |||||||| |||||||| |||||||||||| || ||||||||||| |||| |||||||||| |||| ||||||||| || ||||| ||||||| ||||||| |||| |||| ||| ||| || ||||| || ||| |||| ||| ||| || |||| ||||||| || ||||| |||| ||| |||| || ||||||| |||||||| ||||||||||| ||||| |||| ||| ||| || ||||| || ||| |||| ||| ||| || ||||| ||||||| || ||||| |||| ||| |||| || ||||||| ||||||| |||| |||| || |||| || ||| |||| || |||| ||||||| || ||||| ||||| || ||||||| |||||| ||||| |||| ||| |||| || ||| || |||| |||| ||| ||| || ||||| ||||||| || ||||| ||||| || ||||||| |||||||| |||| ||||| |||| ||| ||| || ||||| || ||| |||| ||| ||| || |||| ||||||| || ||||| ||||| || ||||||| ||| ||||| ||||||||| ||||||||| |||| ||||| || ||| ||||||| || |||| ||||| ||||||| ||| ||||||||| |||||||| ||||| || |||| ||||||| || ||||| ||||| || ||||||| ||| |||||||| ||| |||||||||||| || |||| ||||||||||| ||||||| |||||| || ||| ||| |||||||| |||| || |||||||| ||| ||| ||||| ||||||||| ||||| |||| ||| ||| || ||||| || ||| |||| ||| ||| || |||||| || ||||| |||| ||| |||| || ||||||| Time-to-deterioration results are presented in Table 26 and Table 27.
Table 26: Analysis of Time to Deterioration of EORTC QLQ-30 (FAS With Final Data Cut-Off on August 27, 2021)
Time to deterioration | Tremelimumab in combination with durvalumab (N = 393) | Sorafenib (N = 389) |
|---|---|---|
Global health status | ||
Total events, n (%) | 142 (47.0) | 162 (50.2) |
|||||||| ||||||||| |||| | | ||| |||||| | ||| |||||| |
Median time to deterioration, months (95% CI)b | 7.5 (5.82 to 10.84) | 5.7 (4.80 to 7.39) |
Hazard ratio (95% CI)c | 0.76 (0.61 to 0.96) | |
||||||| ||||||| | | |||||| | |
Physical function | ||
Total events, n (%) | 122 (40.1) | 148 (45.8) |
|||||||| ||||||||| |||| | | ||| |||||| | ||| |||||| |
Median time to deterioration, months (95% CI)b | 12.9 (9.23 to 16.82) | 7.4 (5.68 to 10.15) |
Hazard ratio (95% CI)c | 0.68 (0.53 to 0.87) | |
||||||| ||||||| | | |||||| | |
Fatigue | ||
Total events, n (%) | 150 (49.7) | 173 (55.3) |
Censored patients, n (%)a | 152 (50.3) | 140 (44.7) |
Median time to deterioration, months (95% CI)b | ||||||||| || ||||| | ||||||||| || ||||| |
Hazard ratio (95% CI)c | 0.71 (0.57 to 0.89) | |
||||||| ||||||| | | |||||| | |
Nausea | ||
Total events, n (%) | 90 (29.9) | 114 (36.0) |
Censored patients, n (%)a | 211 (70.1) | 203 (64.0) |
Median time to deterioration, months (95% CI)b | ||||||||||| || ||| | |||||||||| || |||||| |
Hazard ratio (95% CI)c | 0.65 (0.49 to 0.87) | |
||||||| ||||||| | | |||||| | |
Appetite loss | ||
Total events, n (%) | 114 (38.0) | 154 (48.7) |
Censored patients, n (%)a | 186 (62.0) | 162 (51.3) |
Median time to deterioration, months (95% CI)b | |||| ||||| || |||||| | ||| ||||| || ||||| |
Hazard ratio (95% CI)c | 0.59 (0.46 to 0.75) | |
||||||| ||||||| | | ||||||| | |
CI = confidence interval; ECOG PS = Eastern Cooperative Oncology Group Performance Status; EORTC QLQ-30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-HCC18 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Hepatocellular Carcinoma 18; FAS = full analysis set; GHS = global health status; HBV = hepatitis B virus; HCV = hepatitis C virus; MVI = macrovascular invasion; NR = not reached; QoL = quality of life; vs. = versus.
Note: The analysis includes a subset of the FAS with baseline scores of 10 or higher for EORTC QLQ-C30 GHS/QoL and functioning domains while baseline scores of up to 90 for EORTC QLQ-C30 and EORTC QLQ-HCC18 symptom domains or items.
aPatients who have not shown a clinically meaningful deterioration or died, or who shows a clinically meaningful deterioration or die after 2 or more missed visits, are censored at the latest evaluable patient-reported outcome assessment, or day 1 if there are no evaluable visits. Patients with a clinically meaningful deterioration within 2 visits of baseline who do not have any evaluable visits or do not have a baseline assessment are censored at day 1.
bCalculated using the Kaplan-Meier technique.
cAnalysis performed using a Cox proportional hazards model adjusting for treatment, etiology of liver disease (HBV vs. HCV vs. others), ECOG PS (0 vs. 1), and MVI (yes vs. no). A hazard ratio less than 1 favours immune-oncology treatment groups to be associated with a longer time to QoL deterioration than sorafenib.
dAnalysis was performed using a stratified log-rank test adjusting for treatment, etiology of liver disease (HBV vs. HCV vs. others), ECOG PS (0 vs. 1), and MVI (yes vs. no). P values were not adjusted for multiplicity.
Source: HIMALAYA Clinical Study Report.17
Table 27: Analysis of Time to Deterioration of EORTC QLQ-HCC18 (FAS With Final Data Cut-Off on August 27, 2021)
Time to deterioration | Tremelimumab in combination with durvalumab (N = 393) | Sorafenib (N = 389) |
|---|---|---|
Shoulder pain | ||
Total events, n (%) | 105 (36.5) | 121 (39.2) |
|||||||| ||||||||| |||| | | ||| |||||| | ||| |||||| |
|||||| |||| || ||||||||||||| |||||||| |||| ||| | | |||||||||| || |||||| | ||||||||| || |||||| |
Hazard ratio (95% CI)c | 0.82 (0.63 to 1.06) | |
||||||| ||||||| | | |||||| | |
Abdominal pain | ||
Total events, n (%) | 93 (32.0) | 132 (42.0) |
Censored patients, n (%)a | 198 (68.0) | 182 (58.0) |
Median time to deterioration, months (95% CI)b | ||||||||||| || ||| | ||||||||| ||||||| |
Hazard ratio (95% CI)c | 0.61 (0.47 to 0.80) | |
||||||| ||||||| | | |||||| | |
Abdominal swelling | ||
Total events, n (%) | 96 (33.1) | 115 (36.1) |
|||||||| ||||||||| |||| | | ||| |||||| | ||| |||||| |
Median time to deterioration, months (95% CI)b | ||||||||||| || |||||| | |||||||||| || |||||| |
Hazard ratio (95% CI)c | 0.74 (0.56 to 0.97) | |
||||||| ||||||| | | |||||| | |
CI = confidence interval; ECOG PS = Eastern Cooperative Oncology Group Performance Status; EORTC QLQ-HCC18 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Hepatocellular Carcinoma 18; GHS = global health status; HBV = hepatitis B virus; HCV = hepatitis C virus; FAS = full analysis set; MVI = macrovascular invasion; NR = not reached; QoL = quality of life; vs. = versus.
Notes: Others (for etiology of liver disease) is defined as no active viral hepatitis identified. The analysis includes a subset of the FAS who have baseline scores of 10 or higher for EORTC QLQ-C30 GHS/QoL and functioning domains while baseline scores of 90 or lower for EORTC QLQ-C30 and QLQ-HCC18 symptom domains/items.
aPatients who have not shown a clinically meaningful deterioration or died, or who show a clinically meaningful deterioration or die after 2 or more missed visits, are censored at the latest evaluable patient-reported outcome assessment, or day 1 if there are no evaluable visits.
bCalculated using the Kaplan-Meier technique.
cThe analysis was performed using a Cox proportional hazards model adjusting for treatment, etiology of liver disease (HBV vs. HCV vs. others), ECOG PS (0 vs. 1), and MVI (yes vs. no). A hazard ratio of less than 1 favours immune-oncology treatment groups to be associated with a longer QoL deterioration than sorafenib.
dThe analysis was performed using stratified log-rank test adjusting for treatment, etiology of liver disease (HBV vs. HCV vs. others), ECOG PS (0 vs. 1), and MVI (yes vs. no). P values were not adjusted for multiplicity.
Source: HIMALAYA Clinical Study Report.17
Improvement Rate
Improvement rate was estimated in the trial among patients with symptom scores of 10 or greater, and/or GHS and/or physical function scores less than or equal to 90 at baseline. At the final data cut-off on August 27, 2021, the odds of improvement (defined as the number of patients with a best overall score response of “improved”) were higher in the tremelimumab-plus-durvalumab group compared to the sorafenib group for fatigue according to the EORTC QLQ-C30 results (OR = 1.67; 95% CI, 1.13 to 2.47) and abdominal swelling according to the EORTC QLQ-HCC18 (OR = 2.28; 95% CI, 1.19 to 4.44) as shown in Table 28 and Table 29. For the improvement rate for all other HRQoL domains, the evidence was insufficient to show a difference between groups. The differences between groups were not tested statistically.
Table 28: EORTC QLQ-C30 Improvement Rate (FAS With Final Data Cut-Off on August 27, 2021)
||||| |||||| | |||||||||||| || ||||||||||| |||| |||||||||| ||| ||||| | |||||||||||| ||||| |
|---|---|---|
|||||| |||||| ||||||| | ||| | ||| |
|||||||| |||| |||||||||||| |||| | || |||||| | || |||||| |
|||| ||||| |||| ||| | |||| ||||| || ||||| | |
|||||||| ||||||||| | ||| | ||| |
|||||||| |||| |||||||||||| |||| | || |||||| | || |||||| |
|||| ||||| |||| ||| | |||| ||||| || ||||| | |
|||||||| | ||| | ||| |
|||||||| |||| |||||||||||| |||| | || |||||| | || |||||| |
|||| ||||| |||| ||| | |||| ||||| || ||||| | |
|||||| ||| ||||||||| | || | || |
|||||||| |||| |||||||||||| ||| | || |||||| | || |||||| |
|||| ||||| |||| ||| | |||| ||||| || ||||| | |
|||||||| ||||| | || | ||| |
|||||||| |||| |||||||||||| |||| | || |||||| | || |||||| |
|||| ||||| |||| ||| | |||| ||||| || ||||| | |
CI = confidence interval; EORTC = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; FAS = full analysis set.
Notes: This table has been redacted at the request of the sponsor. The analysis was performed using logistic regression model adjusted for treatment with factors for etiology of liver disease, Eastern Cooperative Oncology Group Performance Status, and macrovascular invasion.
aSubset of the FAS who have baseline scores of 90 or lower.
bSubset of the FAS who have baseline scores of 10 or higher.
Source: HIMALAYA Clinical Study Report.17
Table 29: EORTC QLQ-HCC18 Improvement Rate (FAS With Final Data Cut-Off on August 27, 2021)
||||| |||||| | |||||||||||| || ||||||||||| |||| |||||||||| ||| ||||| | |||||||||||| ||||| |
|---|---|---|
|||||||| ||||| | || | ||| |
|||||||| |||| |||||||||||| ||| | || |||||| | || |||||| |
|||| ||||| |||| ||| | |||| ||||| || ||||| | |
||||||||| ||||| | ||| | ||| |
|||||||| |||| |||||||||||| |||| | || |||||| | || |||||| |
|||| ||||| |||| ||| | |||| ||||| || ||||| | |
||||||||| |||||||| | || | || |
|||||||| |||| |||||||||||| ||| | || |||||| | || |||||| |
|||| ||||| |||| ||| | |||| ||||| || ||||| | |
CI = confidence interval; ECOG PS = Eastern Cooperative Oncology Group Performance Status; EORTC QLQ-HCC18 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Hepatocellular Carcinoma 18; FAS = full analysis set.
Notes: This table has been redacted at the request of the sponsor. The analysis was performed using logistic regression model adjusted for treatment with factors for etiology of liver disease, ECOG PS, and macrovascular invasion.
aSubset of the FAS who have baseline scores of 10 or greater.
Source: HIMALAYA Clinical Study Report.17
Harms
Table 30 provides detailed harms data from the August 27, 2021, data cut-off. Safety was analyzed in all treated patients in the group corresponding to the treatment received.
Adverse Events
Totals of 378 patients (97.4%) in the tremelimumab in combination with durvalumab group and 357 patients (95.5%) in the sorafenib group experienced at least 1 AE. The most frequently reported treatment-emergent AEs in the tremelimumab in combination with durvalumab and sorafenib groups, respectively, were diarrhea (26.5% versus 44.7%), pruritis (22.9% versus 6.4%), rash (22.4% versus 13.6%), fatigue (17% versus 19%), decreased appetite (17% versus 17.9%), and palmar-plantar erythrodysesthesia syndrome (0.8% versus 46.5%).
||||||||||| ||| |||||| |||||||| ||||||| || ||| |||||||||||| || ||||||||||| |||| |||||||||| |||||| ||| ||| |||||||| ||||||| || ||| ||||||||| ||||| ||||||||||| || ||||| ||| |||| ||||| ||| ||| |||| |||||||||| |||||||| ||||||||| || ||| ||||||| || ||| |||||||||||| || ||||||||||| |||| |||||||||| ||| ||||||||| ||||||| |||||||||||| |||| ||||||||| ||||||||| |||||||||||||||| ||||| ||| |||||| ||||||||| |||||| ||||| ||| |||||| |||||||||||| ||||| ||| |||||| ||| |||||||||||||| |||||||||||||||||| |||||||| || |||||||| || ||||||
Serious Adverse Events
A total of 157 patients (40.5%) in the tremelimumab in combination with durvalumab group and 111 patients (29.7%) in the sorafenib group experienced at least 1 SAE. ||| |||| |||||||||| |||||||| |||| || ||| |||||||||||| || ||||||||||| |||| |||||||||| ||| ||||||||| |||||| |||| |||||||| ||||| || |||||| |||||| ||||| || ||| ||| ||||||||| ||||| || |||||| ||| ||||| |||| |||||||| || |||| |||| || || |||||||| || |||| |||||
Withdrawals Due to Adverse Events
There were 53 patients (13.7%) in the tremelimumab in combination with durvalumab group and 63 patients (16.8%) in the sorafenib group stopped treatment due to AEs. No AEs led to discontinuation in more than 2% of patients in either study group.
Mortality
At the final data cut-off date of August 21, 2021, in the FAS, there was a total of ||| |||||| ||||||| || ||| |||||||||||| || ||||||||||| |||| |||||||||| ||||| ||| ||| |||||| ||||||| || ||| ||||||||| |||||| |||| |||||||| |||||||||| || ||| ||||||| ||||||||||| || |||||||||||| ||||||||||| ||| |||||| |||||||| |||| ||||| |||| || |||||||| |||||| ||| || |||||||| |||||| ||| ||||||||||| || ||||||| |||| ||| ||||||| ||||| || ||| |||||||||||| || ||||||||||| |||| |||||||||| ||||| ||| ||||||||| |||||| ||||||||||||| ||| |||| |||||| ||||| || ||||| ||| ||||||| ||||||| ||||| || |||||| || ||||| ||||| ||| |||||||| || |||| |||| || || |||||||| || |||||| ||||||||| |||||.
Adverse Events of Special Interest
||||| |||||||| |||| |||||||||| || |||||||| || ||| |||||||||||| || ||||||||||| |||| |||||||||| ||||| |||| ||| ||||||||| ||||| ||||||| |||| || ||||| |||||||| || ||| |||||||||||| || ||||||||||| |||| |||||||||| ||||| |||| |||| |||| |||||| || ||||||| |||||||| ||||||||||||||| ||| ||||| |||| || |||| ||| ||||||||||||| ||||| |||| |||| |||||||||| |||||||| || ||| |||||||||||| || ||||||||||| |||| |||||||||| ||||| |||||| ||| |||||||||||||||| |||| ||| |||| |||||||||| |||||||| || ||| ||||||||| ||||| |||| |||||| |||||
Immune-mediated AEs were reported in 36% of patients in the tremelimumab in combination with durvalumab group and 8% of patients in the sorafenib group. Six patients in the tremelimumab in combination with durvalumab group died due to immune-mediated AEs (|||||||||||| |||||||| ||||||| |||||||||||| ||| |||||||||| ||||||) and no deaths were reported in the sorafenib group.
|||||||| |||||||| |||||| |||||||| || || |||||||| |||||| || ||| |||||||||||| || ||||||||||| |||| |||||||||| ||||| ||| ||||||| |||||| || ||| ||||||||| ||||||
Other Significant Adverse Events
There were 144 patients (37.1%) in the tremelimumab in combination with durvalumab group with any hepatic SMQ AE compared to 121 patients (32.4%) in the sorafenib group. ||| || ||| ||||||| |||||||| ||| ||| || ||||||| || ||| |||||||||||| || ||||||||||| |||| |||||||||| ||||| ||| |||||| || ||| ||||||||| ||||||
There were 44 patients (11.3%) in the tremelimumab in combination with durvalumab group with any hemorrhage SMQ AE compared to 56 patients (15%) in the sorafenib group. ||| || ||| |||||||||| ||| ||| || ||||||| || ||| |||||||||||| || ||||||||||| |||| |||||||||| ||||| ||| |||||| || ||| |||||||||||||||
In the HIMALAYA trial, tremelimumab in combination with durvalumab showed no increase in liver toxicity or risk of bleeding.
Table 30: Summary of Harms in the HIMALAYA Trial (Safety Analysis Set)
Adverse events | Tremelimumab in combination with durvalumab (N = 388) | Sorafenib (N = 374) |
|---|---|---|
Most common adverse events,a n (%) | ||
≥ 1 adverse event | 378 (97.4) | 357 (95.5) |
Diarrhea | 103 (26.5) | 167 (44.7) |
Pruritus | 89 (22.9) | 24 (6.4) |
Rash | 87 (22.4) | 51 (13.6) |
Fatigue | 66 (17.0) | 71 (19.0) |
Decreased appetite | 66 (17.0) | 67 (17.9) |
Palmar-plantar erythrodysesthesia syndrome | 3 (0.8) | 174 (46.5) |
Most common adverse events grade ≥ 3,b n (%) | ||
≥ 1 adverse event grade ≥ 3 | ||| |||||| | ||| |||||| |
Increased aspartate transaminase | 20 (5.2) | 12 (3.2) |
Lipase increased | 24 (6.2) | 11 (2.9) |
Hypertension | 7 (1.8) | 23 (6.1) |
Palmar-plantar erythrodysesthesia syndrome | 0 | 34 (9.1) |
Serious adverse events,c n (%) | ||
|||||||| |||| | ||| | ||| |||||| | ||| |||||| |
|||||||| | | ||||| | | ||||| |
|||||| | | ||||| | || ||||| |
||||||||| | | ||||| | | ||||| |
Patients who stopped treatment due to adverse events,d n (%) | ||
Patients who stopped | 53 (13.7) | 63 (16.8) |
||||||||| |||||||||||||||| ||||||||| | | ||||| | | ||||| |
||||||||| | || ||||| | || ||||| |
|||||||| | || ||||| | || ||||| |
|||||||||||||| |||||||||||||||||| |||||||| | || ||||| | || ||||| |
||||||||| |||| | || ||||| | || ||||| |
Deaths,d n (%) | ||
Patients who died e | 30 (7.7) | 27 (7.2) |
||||||| ||||||| | | ||||| | | ||||| |
Adverse events of special interest, n (%) | ||
|||| | ||| |||||| | ||| |||||| |
Immune-mediated adverse events | 139 (35.8) | 30 (8.0) |
|||| ||||| || | | || |||||| | || ||||| |
Immune-mediated adverse events of grade 3 or 4 | 49 (12.6) | 9 (2.4) |
|||| |||||||| |||||||| ||||||||||||||| | || |||||| | || ||||| |
Immune-mediated adverse events, received systemic corticosteroids | 96 (24.7) | 16 (4.3) |
|||| |||||| | ||
||||||||||||||| | ||| |||||| | ||| |||||| |
|||||||||||||||| | ||| |||||| | ||| |||||| |
||||||| |||||| | || |||||| | || |||||| |
||||||||||| |||||| | || |||||| | || ||||| |
|||||||||| |||||| | || |||||| | || ||||| |
||||| |||||||||||||||||| |||||| | || |||||| | || ||||| |
|||||||| |||||||| |||||| | || ||||| | | ||||| |
|||||||||||| |||||| | || ||||| | | ||||| |
||||| |||||| | || ||||| | | ||||| |
SAE = serious adverse event.
Some redacted rows have been deleted.
a15% or greater of patients in either group.
b5% or greater of patients in either group.
c2% or greater of patients in either group.
d1% or greater of patients in either group.
eAdverse event with outcome of death.
Source: HIMALAYA Clinical Study Report17 (details from the table have been taken from the sponsor’s Summary of Clinical Evidence).
Pooled Safety Analysis
The sponsor also provided a pooled safety analysis of tremelimumab in combination with durvalumab in patients from the |||||||| ||||||||| ||| |||| ||||| || || ||||| ||||| ||||||||| || |||||||| |||| |||| ||| ||| |||||| |||||||||| ||| |||| |||||||||| ||| || ||| ||||||| ||||||||| |||| |||||||||| || ||| |||||| || ||| |||||||| ||||||| ||||||||||| || |||||||||||||| ||||||| ||||||||||| || || ||||| ||| || ||| |||||||| ||||||| || |||||||| ||||||||||| || |||| ||| ||| ||||||| || ||||||||||||||| || ||| ||||| ||||||||| |||| |||||||| || || |||||||| ||||||||
Critical Appraisal
Internal Validity
HIMALAYA was an open-label, sponsor-blind, randomized phase III study comparing tremelimumab in combination with durvalumab and sorafenib in adult patients with unresectable HCC. The study also included 2 additional treatment groups who received durvalumab monotherapy and a lower dose of tremelimumab in combination with durvalumab. These were not relevant to this review and closed before the end of randomization, respectively, and were not included in this report. The sponsor stated that an open-label design was used due to the nature of the treatment administration (IV versus oral) and the different administration schedules (every 4 weeks versus twice daily), which made blinding infeasible. Protocol amendments occurred after recruitment had started but before the first interim analysis, and do not appear to have influenced results. The study used an appropriate central randomization method sufficient for concealing allocation until assignment to the intervention (1:1:1:1 using an interactive web response system until the fourth treatment group was closed, after which patients were randomized at 1:1:1). Randomization appeared adequate in balancing baseline demographic and disease characteristics between the tremelimumab in combination with durvalumab and the sorafenib groups. Concomitant therapy use was also similar across the treatment groups.
||||| |||| |||||| ||||| || |||| ||||||||| |||||||| |||||||||| || ||| ||||||||| ||||| |||||| || || ||| |||||||||||| || ||||||||||| |||| |||||||||| ||||| ||||||| |||| |||||||| ||||||| |||||||| ||| |||||||||| ||| ||| |||||||||| ||||| |||||||||. Due to the limited number of patients who were affected by protocol deviations, no substantial impact on the comparative clinical efficacy of tremelimumab in combination with durvalumab versus sorafenib would be expected.
An open-label design can result in a risk of bias in the study, including the measurement of the outcomes, whether by unblinded assessor, such as PFS and ORR, or self-reported, such as HRQoL or harms. With the exception of harms, the bias will likely favour the experimental intervention, although the extent of bias is uncertain. This bias would not be introduced into the measurement of objective outcomes such as OS, which is the primary outcome of the trial. At the first interim analysis after at least 32 weeks of follow-up, tumour response was assessed by BICR to minimize bias in their measurement due to the investigators’ knowledge of the assigned interventions. Results from the interim analysis were similar to results from the final analysis. In the final analysis, tumour response assessments were performed only by investigators. In the final analysis, exploratory end points included assessment of PFS, TTP, ORR, DCR, and DoR by BICR to mitigate this bias; however, as the results of these assessments were not available it is not possible to determine the extent of any bias in the measurement of these outcomes. The trial used RECIST 1.1 primarily to assess tumour response, while some guidelines recommend the use of mRECIST, which more accurately measures tumour viability to targeted therapies (which are cytostatic rather than cytotoxic).27 However, the clinical experts consulted for this review noted that radiological reporting differs between the academic and clinical settings, and neither RECIST 1.1 nor mRECIST are used in clinical practice.
Statistical analyses were in general appropriate for the outcomes evaluated. Intention-to-treat (ITT) analyses (i.e., using the FAS) were used for efficacy outcomes, which is appropriate for estimating the effect of assignment to the intervention. At the time of the final analysis, 555 OS events had occurred across the groups, which, according to the prespecified analysis plan, provided greater than 97% power to demonstrate a statistically significant difference in OS (assuming a HR of 0.70). However, the study was not sized for individual subgroup comparisons and no multiplicity adjustments were made, rendering any conclusion uncertain. Moreover, although OS was assessed in the ITT population, it would still be influenced by treatments received after progression. Because of the 4-month delay in the separation of the Kaplan-Meier curves, the sponsor conducted additional analysis to assess the assumption of nonproportionality, finding no significant interactions. The study was not sized for secondary end points. ||| ||||||| ||||| |||||| ||||| ||||| || |||| ||| ||||| || ||| ||||||| |||||||| || ||| ||| ||| || ||||||| ||| |||||||||| ||||| |||| || ||| || ||||| ||||||||| ||||| ||| |||| ||| ||||| || ||| || ||||||||. However, only descriptive statistics were presented for ORR and DoR. The lack of multiplicity control for other outcomes may have increased the risk of false-positive conclusions.
Maintaining and improving QoL overall was rated as an important outcome by patients, yet the interpretation of results for the HRQoL instruments (i.e., the ability to assess trends over time and to make comparisons across treatment groups) is limited by the missing data at baseline and significant decline in the number of patients available to provide assessment over time. There was no evidence of validity or MID of the EORTC QLQ-C30 and EORTC QLQ-HCC18 in patients with HCC. However, the sponsor provided literature on patients with other cancers that supported the choice of MID. The clinical experts consulted by CADTH pointed out that HRQoL instruments are not used routinely in clinical practice and more weight is placed on clinical outcomes.
External Validity
According to the clinical experts CADTH consulted for this review, the HIMALAYA study population is considered reflective of the requested reimbursement population. The following considerations are of importance regarding the external validity of the study.
Population: According to the clinical experts consulted by CADTH for this review, the demographic and disease characteristics of the HIMALAYA study population were reflective of the Canadian population with unresectable HCC. There was a large number of screening failures in the study, with almost a third of screened patients not randomized, most commonly due to eligibility criteria not being fulfilled. However, the eligibility criteria that were most commonly not fulfilled |||| ||||| ||||||||||||||||| || ||||||||| |||| |||||||||||| || ||||||||||| |||||||||||||| |||| || |||| || |||||||| ||||| ||| |||||| |||||||| ||| || ||||||||| ||| ||||| || ||||||||||||||||| |||||||| |||| ||||| |||||||. It was required that patients have a Child-Pugh class of A, which excluded 87 patients after screening. The clinical experts noted that, while only including patients with a Child-Pugh class of A is reasonable in clinical trials, it may also be reasonable to include other patients (e.g., those with a Child-Pugh class of B7) in clinical practice. It is unclear if findings from this study can be generalized to patients beyond the first line of therapy. All patients in the trial had an ECOG PS of 0 or 1 as specified by the eligibility criteria, but the experts indicated this would not be reflective of clinical practice and that clinicians would require some flexibility in restricting treatment by performance status. The clinical experts noted that, while almost ||| || |||||||| || |||| ||||||||| |||||| ||| |||||||| |||||||||||| ||||||| ||| |||| this proportion would be lower for patients in Canada. Although most participants were Asian, the clinical experts noted that this is consistent with other HCC trials, and they did not expect this would limit generalizability to patients in Canadian clinical practice.
Appropriateness of comparator: The clinical experts consulted by CADTH indicated that at the time of the HIMALAYA study design, sorafenib was the only approved treatment for unresectable HCC patients who were ineligible for locoregional therapy or who had progressed after locoregional therapy and who had not undergone prior systemic therapy. In this study, sorafenib was considered standard-of-care treatment for these patients and was selected as the active comparator. According to the clinical experts and recent clinical guidelines, sorafenib is no longer the most common standard-of-care therapy and has been replaced by more effective therapies, including atezolizumab in combination with bevacizumab and lenvatinib. As such, the results of the trial may not be directly generalizable to current standard of care.
Relevance of end points: The clinical experts consulted by CADTH and clinician groups providing input agreed that, ideally, prolonging survival and delaying progression are the most important end points, followed by ORR, DCR, and toxicity profile with maintained HRQoL. Although information about HRQoL was collected, interpretation is limited due to missing data and increased risk of type I error. The clinical experts noted that, in clinical practice, imaging would be obtained every 3 to 4 months to assess response to treatment.
Setting: This study was a multinational, multicentre trial. The study population was drawn from a wide variety of sites across the globe, with 9 study centres (out of 170) and ||||||||||||| |||| || |||||||| || ||||||. The clinical experts indicated that there is no concern regarding generalizing the findings from the pivotal study to the Canadian clinical setting.
Long-Term Extension Studies
No long-term extension studies were identified by the sponsor.
Indirect Evidence
The contents within this section were informed by materials submitted by the sponsor and summarized and validated by the CADTH review team.
Objectives and Methods for the Summary of Indirect Evidence of the Sponsor-Submitted Indirect Treatment Comparison
The objective of this section is to summarize and critically appraise available indirect evidence comparing tremelimumab in combination with durvalumab to other relevant first-line treatments for unresectable HCC currently used in Canadian settings.
Description of the Sponsor-Submitted Indirect Treatment Comparison
The efficacy and safety of tremelimumab in combination with durvalumab against sorafenib have been previously assessed in the HIMALAYA trial.17 However, no head-to-head comparison of tremelimumab in combination with durvalumab against other first-line treatments for unresectable HCC was available for this review. Due to this gap in evidence, the sponsor submitted an ITC that included a systematic literature review49 with an ITC in the form of 2 MAICs50,51 submitted in separate reports that provide comparative evidence of the efficacy and safety of tremelimumab in combination with durvalumab relative to atezolizumab in combination with bevacizumab, and tremelimumab in combination with durvalumab relative to lenvatinib. Data from this ITC were used to inform the pharmacoeconomic model.
Study Selection Method
Table 31 shows the study selection criteria and key aspects of the methods for the systematic review.
Based on the prespecified eligibility criteria outlined in Table 31, the sponsor conducted a systematic literature search to assess first-line systemic treatments in patients diagnosed with unresectable HCC and to evaluate the comparative efficacy and safety of tremelimumab in combination with durvalumab and alternative treatments in this patient setting. The systematic literature review was conducted on |||||| ||| ||||| ||| ||||||| || |||||||| || ||||| Systematic literature searches were conducted in electronic databases (i.e., Ovid Embase, Ovid MEDLINE, Cochrane Central Register of Controlled Trials, and Cochrane Database of Systematic Reviews). In addition, hand searches were conducted, including a reference list of eligible studies, conference proceedings (i.e., American Society of Clinical Oncology, American Association for the Study of Liver Diseases [AASLD]), clinical trial registries, and global health technology assessment bodies (i.e., National Institute for Health and Care Excellence [NICE]).
The study screening, selection, and data-extraction processes were conducted by 2 independent reviewers. Studies identified by the systematic literature search were first screened based on the title and abstract. Full-text screening was then carried out for studies selected from the title and abstract screening stage. A third reviewer was involved to resolve any discrepancies during the title, abstract, and full-text review. An independent reviewer performed a data-extraction quality check by reviewing 20% of the extracted articles. Two independent reviewers assessed the risk of bias of the included studies using the checklist of the NICE single-technology appraisal user guide.52 A list of excluded studies was reported with reasons for exclusion.
| ||||| || ||| ||||||| |||| |||||||| || ||||| ||| ||||||||| || |||||| || |||||||||||| |||| |||||||| || |||| ||||| ||||| |||| ||||||||| ||| ||||||||||| |||||||| ||| |||||| || |||||||||| |||||||| || ||| |||||| |||||||||||||||||||| ||||| |||||||| ||| ||||||||||| || ||| |||||||||| ||| ||| |||||||||| ||||||||| || |||||||| |||| |||||||| || |||||||||| |||| || |||||| || |||||||||||| |||| |||||||||| || || |||||||| |||||||| || ||| |||||| |||| ||||||| || |||||| ||||||||| || |||||||| |||| |||||||| || |||||||||| ||| ||||||||| ||||||||| |||| ||||||||||| |||||| ||||||||| ||| || || ||| |||||| || |||||||| || ||| |||||||| |||||| ||| ||||||||| |||||||||||||||||||| || |||||||| |||||||||| ||||| |||| ||| ||| ||||| || ||| ||| |||||| |||||||| ||||| |||| || |||||| |||||||| |||| ||||||||||| ||| || |||| ||||||| |||||||||| || ||||| || |||||||| ||| ||| |||||||| |||||||||||||||||||| |||||||| || |||||||||||| ||||||||
To be eligible to be included in the ITC, the population, the control treatment, and the study design of studies identified from the systematic literature review had to be aligned with those from the HIMALAYA trial conducted by the sponsor. The studies that were potentially eligible for the ITC were assessed for feasibility:
Decision set trials: trials assessing only treatments included in the decision set of comparators, including both an intervention and a comparator of interest. |||| ||| |||||||| |||| |||| |||| || |||||||||||| || |||| || ||||||||||| || |||||||| ||| ||| |||| |||||||| ||||||||| |||| the IMbrave150 trial, and the HIMALAYA trial.
Analysis set trials: trials included in the decision set trials as well as additional trials that could be of interest to connect the network. |||| ||| |||||||| ||||| || ||||| |||| |||| |||| ||| |||||||| |||| ||| ||||||||||| ||||||| ||||| || || ||||||||| |||||||| || ||||||| ||| ||||||| || |||| ||||||| ||||||||| || |||||||||||| ||| ||||||| |||||| |||||||||| |||| || |||||||||| ||||||| ||||||||| ||| ||| || || ||| |||||||
After assessing the publications identified in the systematic literature review and based on the decision set, || |||||||||||| ||||||| || ||||| |||||||||||| || || |||||||| || ||| ||| ||||||||| ||||||||| |||| and the IMbrave150 trial) with the sponsor-conducted HIMALAYA trial (Table 32).
Table 31: Study Selection Criteria and Methods for ITC Submitted by Sponsor
Characteristics | Indirect comparison |
|---|---|
Population | Patients aged 18 years and older with unresectable, advanced, or metastatic HCC receiving systemic first-line treatment |
Intervention |
|
Comparator |
|
Outcome | Efficacy:
Safety and tolerability:
|
Study designs | Randomized controlled trials |
Publication characteristics | Journal articles and abstracts (English-language only) |
Exclusion criteria | The following were excluded:
|
Databases searched |
|
Language | English language only |
Country | No restriction |
Selection process | Trials were identified through an SLR, in which 2 reviewers independently screened titles and abstracts to identify studies included. Any discrepancy was resolved by discussion, and if there were any references that remain unresolved after this discussion a third reviewer adjudicated the final response. |
Data-extraction process | An independent reviewer undertook the quality check of the data extraction by reviewing 20% of the extracted articles. |
Quality assessment | To assess the quality of clinical trials, the reviewers used the list of assessment questions provided in the NICE single-technology appraisal template and in the guidance by the Centre for Reviews and Dissemination at the University of York:
The risk of bias for each trial was assessed by 2 independent reviewers. Any discrepancy was be resolved by discussion and if there were any references that remained unresolved after this discussion, a third reviewer adjudicated final responses. |
AE = adverse event; HCC = hepatocellular carcinoma; HTA = health technology assessment; ITC = indirect treatment comparison; NICE = National Institute for Health and Care Excellence; PFS = progressive-free survival; SLR = systematic literature review.
Note: Details from the table were taken from the sponsor’s Summary of Clinical Evidence.
Source: Sponsor systematic literature review49 and ITC reports.50,51
Table 32: Studies Included in the Sponsor-Conducted Indirect Treatment Comparison
Trial | Publication | Treatment groups |
|---|---|---|
HIMALAYA | Trial data17 (no publication was available) |
|
IMbrave150 | Finn et al. (2020)53 |||||||||| || || |||| |||||||||||||| || || |||| |||||||||||||| || || |||| ||||||||| |
|
||||||| | |||||||||| || || |||| |||| | |||||||||| || || |||| |||| |||||| || ||| || || |||| |||| |||||| || |||| |||||| |||| |
|||||||| ||| | |||||||||| || || |||| |||||||||||||| || || |||| |||| | ||||||||| ||| || || |||||||| ||||| ||||||||||||||||| ||| ||| |||||| ||||| ||||| |
| ||||||||| || ||| ||||||||||| ||| ||| |||||||||| ||||||||| ||| ||| || ||||||| ||| || ||| ||||||||| ||||||| || |||| ||| |||||
Source: Sponsor indirect treatment comparison reports.50,51
Design of Indirect Treatment Comparison Conducted by Sponsor
Evidence Network
The overall evidence network was constructed as part of a feasibility assessment, which was built for each outcome of interest based on data availability. The availability of hazard ratios or Kaplan-Meier curves was considered for time-to-event outcomes including OS, |||| |||| ||| |||| For binary outcomes, such as ORR, AEs of grade 3 || ||||||| ||||||| |||| and AEs leading to discontinuation, the availability of rates and/or number of patients with response or experiencing the AEs were considered to build the networks. Figure 6 presents the overall network of evidence for efficacy, patient-reported, and safety outcomes. However, MAIC analyses were performed by the sponsor to compare tremelimumab in combination with durvalumab relative to atezolizumab in combination with bevacizumab, ||| |||||||||||| || ||||||||||| |||| |||||||||| |||||||| || ||||||||||| ||||| ||||||||| || ||| ||||||||||| ||| ||| |||||||||| ||||||||| ||| ||| || ||||||| || || ||| ||||||||| ||||||| || |||| |||||||| ||| |||||||| ||||| |||||||| |||||||| |||||||||||| || ||||||||||| |||| |||||||||| || ||| |||| ||||| ||||||| ||||||||| ||||||| |||||||||| || ||| |||||||||| ||||||||||| |||||| ||||||||| ||| ||| ||||||||||||||||||||||| ||||||||||| ||| ||| || ||||| || |||| ||||||| ||| |||| |||||||| ||| |||||| |||||||||| || |||||||| |||| ||||||||| || |||||||||| ||||| |||| || |||||| |||||| OS and ||| were reported in all trials, while 2 additional efficacy outcomes, ORR and DoR, were only included in the MAIC comparing tremelimumab in combination with durvalumab versus atezolizumab in combination with bevacizumab, || ||||| |||||||| |||| ||| |||||||| || ||| ||||||| ||||||

Note: This figure has been redacted at the request of the sponsor.
Analysis Methods
Matching Adjusted Indirect Comparison Rationale
A feasibility assessment was performed to determine the method of the ITC. The comparability of the HIMALAYA trial with trials involving comparators was examined by comparing eligibility criteria and patients’ characteristics. Observed differences across eligible trials included region (specifically the proportion of patients from China mainland), etiology, and macrovascular invasion. There was also a limited amount of evidence in the network in terms of number of trials per comparison, leading to insufficient data to consider meta-regression to adjust for treatment-effect modifiers. In addition, individual patient-level data were available only for the HIMALAYA trial, and the authors concluded that ITCs based on summary-level data were likely to provide misleading results due to the presence of heterogeneity between trials. A MAIC was therefore chosen to adjust for suspected heterogeneity with aggregate data available from the trials involving comparators and individual patient-level data for the HIMALAYA trial. A simulated treatment comparison could also be applied but, given the additional assumptions required by the simulated treatment comparison related to the parametric fit of the time-to-event curves and the associated risk of bias in some instances, the MAIC was preferred.
||| anchored MAICs were performed to compare tremelimumab in combination with durvalumab versus atezolizumab in combination with bevacizumab (using the IMbrave150 trial), ||| |||||||||||| || ||||||||||| |||| |||||||||| |||||| |||||||||| |||||| ||| ||||||| ||||||| The sources of heterogeneity are discussed in the following sections.
Treatment-Effect Modifiers
Potential treatment-effect modifiers in unresectable or advanced HCC were identified to assess sources of heterogeneity between studies. Table 33 presents a summary of the lists of potential treatment-effect modifiers identified through a targeted literature review and the final list of potential treatment-effect modifiers identified based on the sponsor’s clinical expert’s opinion, and the review of relative treatment effect by subgroup reported in the trials included in the network of evidence.
Overview of Matching Adjusted Indirect Comparison Methods
An overview of the MAIC methods is provided in Table 34.
Table 33: List of Treatment-Effect Modifiers
Potential treatment-effect modifiers identified through a TLR | Final list of potential treatment-effect modifiers |
|---|---|
Age | Age |
Gender | Gender |
Region:
| Region |
Microvascular invasion | Microvascular invasion |
Extrahepatic spread | Extrahepatic spread |
Alpha-fetoprotein level ≥ 200 ng/mL | Alpha-fetoprotein level ≥ 200 ng/mL |
Etiology:
| Etiology:
|
Performance status:
| Performance status:
|
Barcelona Clinic Liver Cancer stage:
| Barcelona Clinic Liver Cancer stage |
Child-Pugh status:
| Child-Pugh status |
Albumin-bilirubin score | Albumin-bilirubin score |
Race | — |
Tumour burden:
| — |
Subsequent therapy (use of post-treatment anticancer therapy) | — |
Programmed cell death ligand 1 status at baseline | — |
| — |
Baseline weight | — |
| — |
| — |
ECOG PS = Eastern Cooperative Oncology Group Performance Status; TLR = targeted literature review; vs. = versus.
Source: Sponsor indirect treatment comparison reports.50,51
Table 34: Matching Adjusted Indirect Comparison Analysis Methods
Methods | Description |
|---|---|
Analysis methods | Population-adjusted ITCs were needed to adjust for the suspected heterogeneity, and a MAIC was preferred given that individual patient data were only available for the HIMALAYA trial. |
Proportional hazards assumption | The assessment of the proportional hazards assumption was conducted for OS, |||| ||| || |||| ||| ||||||||||| |||||||| ||| ||||||| ||| the IMbrave150 trial based on the published Kaplan-Meier curves, and based on individual patient data analysis for the HIMALAYA trial. |
Preliminary steps |
|
Implementation steps | Weights associated with each patient in the HIMALAYA trial were estimated by generating a logistic regression model based on a similar approach to propensity score weighting. |
Validation of MAIC | The distribution of weights was analyzed to detect any overly influential individual and to study the populations’ overlap. The rescaled weight was also calculated to examine the distribution of the weights as the rescaled weights are relative to the original unit weights of each individual. The ESS was estimated to assess the quality of the matching as it can detect extreme situations where few individuals have important weights driving the results. Descriptive statistics were generated between the comparator trial baseline characteristics and the reweighted HIMALAYA characteristics to assess whether imbalances previously observed between populations have been reduced through the weighting process. |
Outcomes and analysis populations | Nine outcomes were considered as outcomes of interest. Two types of populations were used for MAICs (defined in the HIMALAYA trial):
|
||||||||||| |||||||| | ||||| ||| |||||||||| || ||||||| |||| ||| |||| |||| ||| ||||| ||| ||| ||| ||||||||| ||||||||||||||||||||| |||||||| |||| ||||||||| || |||| ||||||| || |||||||||||||| ||| |||||||||| || |||||||| |||| ||||||| ||||||||||| || ||| ||||| || ||| |||| |||||||| |||| |||| ||||||||| ||| ||| |||| ||||||||| |||||||||||| ||||||||||||| |||| |||||||||| |||| |||||||||||| || ||||||||||| |||| |||||||||||| || || ||| ||||||| |
AE = adverse event; AS = abdominal swelling; CTCAE = Common Terminology Criteria for Adverse Events; DoR = duration of response; ESS = effective sample size; IPD = individual patient data; ITC = indirect treatment comparison; MAIC = matching adjusted indirect comparison; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; PHA = proportional hazards assumption; TTD = time to deterioration.
Note: Details from the table have been taken from the sponsor’s Summary of Clinical Evidence.
Source: Indirect treatment comparison reports.50,51
Preliminary Steps
There was an imbalance between trials regarding some key treatment-effect modifiers, such as etiology (HBV and HCV), MVI, and region. Because all trials included sorafenib as a common comparator, an anchored MAIC was identified as the most relevant approach to adjust for imbalances in treatment-effect modifiers.
The following steps were therefore taken for each MAIC: the first step was to select patients from the HIMALAYA trial who would be included in the analysis on the basis of the eligibility criteria for each comparator trial. Next, the characteristics of these patients were compared with those of each comparator trial to assess for imbalances in treatment-effect modifiers.
Generation of baseline descriptive statistics on the restricted HIMALAYA trial (i.e., after application of the exclusion criteria from the comparator trial when required) and comparison with baseline characteristics in the ||||||| || IMbrave150 trials to assess imbalances between trials:
A specific focus was made on the characteristics known to be potential treatment-effect modifiers.
Implementation Steps
After restriction of the HIMALAYA population, anchored MAICs were implemented through the following steps:
Weights associated with each HIMALAYA patient were estimated through the generation of a logistic regression model based on a similar approach to propensity score weighting:
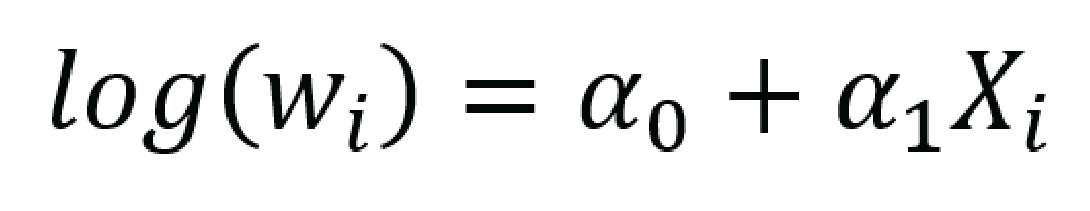
Where Xi is the covariate vector for the ith patient in the HIMALAYA trial and wi is the weight attributed to the ith patient treated with tremelimumab in combination with durvalumab or sorafenib.
All factors identified as being treatment-effect modifiers and available in HIMALAYA individual patient data and reported in ||||||| || IMbrave150 trial were included in the adjustment model, as recommended by the NICE Decision Support Unit.2,61
The method of moments2 was used to estimate these parameters so that the reweighted mean characteristics of the HIMALAYA trial matched the competitor’s trial. This meant minimizing
 when the vector of treatment-effect modifiers is null in the competitor’s trial, i.e.,
when the vector of treatment-effect modifiers is null in the competitor’s trial, i.e., 
An indirect comparison using the Bucher approach was then conducted on the weighted data from the HIMALAYA trial and published results from ||||||| ||| IMbrave150 trial. This method uses a common comparator group between 2 trials to estimate the relative treatment efficacy between 2 drugs that have previously not been investigated in a head-to-head trial, while maintaining randomization.
The Bucher formulas were applied to estimate the HR for time-to-event outcomes (e.g., PFS and OS) between the log HR obtained through the MAIC steps for the HIMALAYA trial and the log HR of the competitor’s trial (IMbrave150 ||| |||||||).
The Bucher formulas were applied to estimate the OR for binary outcomes (i.e., ORR, AEs of grade 3 || ||||||| ||||||| |||| and AEs leading to discontinuation) between the log OR obtained through the MAIC steps for the HIMALAYA trial and the log OR of the competitor’s trial.
Validation of Matching Adjusted Indirect Comparison
Finally, different steps were conducted for each MAIC to assess the validity of the analysis:
The distribution of weights was analyzed to detect any overly influential individual and to study the populations’ overlap. The rescaled weight was also calculated to examine the distribution of the weights as the rescaled weights are relative to the original unit weights of each individual. The rescaled weight was calculated as:
The effective sample size (ESS) was estimated to assess the quality of the matching as it can detect extreme situations where few individuals have important weights driving the results. The ESS was obtained by:
Descriptive statistics were generated between baseline characteristics in ||||||| ||| IMbrave150 trial and reweighted characteristics in the HIMALAYA trial to assess whether imbalances previously observed between populations were reduced through the weighting process.
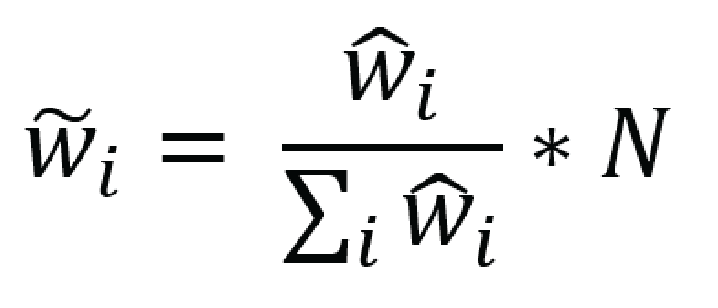
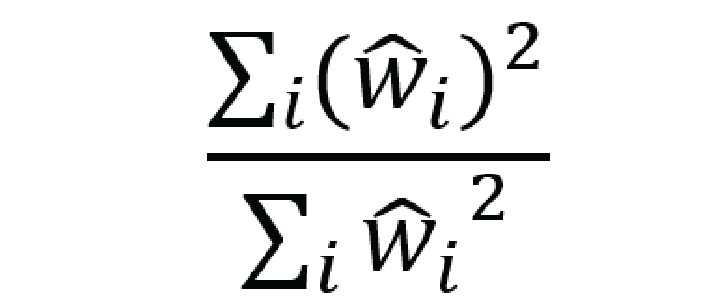
The proportional hazards assumption was examined for OS ||| ||| for all | trials, || |||| || ||| ||| || |||||| ||| ||| || ||||||||| ||||||||, for the IMbrave150 ||| ||||||| trials based on the published Kaplan-Meier curves, and for the HIMALAYA trial based on the analysis of individual patient data. The assessment was based on the visual inspection of the Kaplan-Meier curves, the log-log plot, the Schoenfeld residuals plot, and the Grambsch and Therneau test.62,63
Methods for the MAIC Comparing Tremelimumab in Combination With Durvalumab Versus Atezolizumab in Combination With Bevacizumab
The restriction and weighting process of the MAIC were applied to patients in the tremelimumab in combination with durvalumab and sorafenib groups in HIMALAYA.
Restricting HIMALAYA
Matching was not possible for all treatment-effect modifiers as, in some cases, individual patient-level data for some modifiers were not available, or eligibility criteria were more restrictive in the HIMALAYA trial. The comparison of the eligibility criteria of the HIMALAYA and IMbrave150 trials led to the identification of the following differences:
BCLC stage: no restriction in the IMbrave150 trial and few stage A included versus the HIMALAYA trial restricted to stages B and C.
Ascites: exclusion of moderate or severe ascites in the IMbrave150 trial versus exclusion of clinically meaningful ascites in the HIMALAYA trial.
Bleeding events: exclusion of prior bleeding event in prior 6 months in the IMbrave150 trial versus exclusion of active or prior gastrointestinal bleeding in prior 12 months in the HIMALAYA trial.
Countries: no patients coming from mainland China in the HIMALAYA trial, while 15.6% in the IMbrave150 trial originated in mainland China.
Some patients from the HIMALAYA trial presented a Child-Pugh class of B at baseline, while inclusion was restricted to a Child-Pugh class of A in both trials.
Some patients from the HIMALAYA trial presented an ECOG PS of 2 at baseline, while inclusion was restricted to an ECOG PS of 0 or 1 in both trials.
No matching was therefore possible for ascites, bleeding events, and region (China). In addition, the HIMALAYA trial was restricted to patients with a Child-Pugh class of A, BCLC stages B and C, and an ECOG PS of 0 or 1.
Adjustment
After restriction of HIMALAYA to patients eligible for IMbrave150, patients from the matched HIMALAYA population were adjusted by a number of (reweighted) treatment-effect modifiers to balance the baseline characteristics of the studies.
Table 35 presents the weighting model, including 9 treatment-effect modifiers that were considered as adjustment factors for the MAIC. The final list of modifiers was based on those from the initial list that were reported in both the index trial (HIMALAYA) and the comparator trial (IMbrave150). For the IMbrave150 trial, the distribution of the ALBI score was only reported in an abstract,29 and was related to a cohort that was not the ITT population. Adjustment was therefore not possible for the ALBI score; however, the distributions of the ALBI score were calculated for IMbrave150 and HIMALAYA populations for reference.50
Table 35: Factors Used for the MAIC Weighting Process Comparing Tremelimumab in Combination With Durvalumab Versus Atezolizumab in Combination With Bevacizumab
Variable identified as treatment-effect modifiers | Adjustment made on |
|---|---|
Age | Proportion of patients ≥ 65 years old |
Gender | Proportion of males |
Region | Proportion of patients from Asia, excluding Japan |
Macrovascular invasion | Proportion of MVI |
Extrahepatic spread | Proportion of EHS |
Alpha-fetoprotein | Proportion serum alpha-fetoprotein ≥ 400 ng/mL |
Etiology | Proportion of HBV Proportion of HCV |
ECOG PS | Proportion of ECOG PS 0 |
Barcelona Clinic Liver Cancer | Proportion of BCLC stage C |
ECOG PS = Eastern Cooperative Oncology Group Performance Status; HBV = hepatitis B virus; HCV = hepatitis C virus; MAIC = matching adjusted indirect comparison.
Note: Details from the table taken from the sponsor’s Summary of Clinical Evidence.
Source: Indirect treatment comparison report.50
||||||| ||| ||| |||| ||||||||| |||||||||||| || ||||||||||| |||| |||||||||| |||| |||||||||||||||||||||| ||||||||||||||||| ||| ||| |||||||| ||| ||| |||| ||| || |||| |||||| |||||||||| ||||||| |||| ||| |||| |||| |||| ||||||||||||| || ||||||||||| |||||||| |||| |||| ||||||||||| || ||||||||| ||| |||||||||| || ||| ||||||||||| |||||||| || |||||||| ||| ||||||| ||| || ||| |||||||||||||| || ||| ||||||||| |||||||||||| ||||||||| ||||||||| |||||||||||| |||||||||| ||||||| || ||||||||| ||||| ||| ||||||||| || |||||||||| || ||||||||||||||||| ||||||| ||||||||| || ||||| |||||||| ||||| || ||||| || |||| || ||||||| |||||| ||||||||| || |||||| || ||||| |||||||||||||||||||||||| || ||||| || |||||| || ||||||||||||||||| || |||||||| |||||| |||| ||||| |||||||| || |||||||| |||||| ||| |||||||| || ||||||||||||| |||||||| |||| |||||||| ||||||||| || |||| || || ||||||| || ||||||||||||| || || ||| |||| |||||||||||||||||| || |||||||| ||| |||||||| ||| |||||||| |||||||| ||||||| ||| |||||| |||||||| || ||||||||| ||| |||||||| ||||| ||| |||||||||| || |||||||| |||| |||| || || ||| || |||||||||||||||||||||||||||| || |||||||| || |||||||| |||||||| || |||||||| |||||||| |||| ||| ||||||| |||||||| |||||||||| |||| |||||||| || ||||||| || |||| |||||||||||| || ||||||| ||| |||||||| ||||||||||||||| || ||||||||||||||||| || |||||||| ||| ||||||||| ||||| ||| ||||| ||||||||| || |||| |||| |||| |||||||||| || |||||||||| |||||||| ||| ||||| |||| || |||| ||| ||||| || ||||| |||| ||| ||||||| |||| |||| |||| |||||||| || |||| ||| |||||||||| |||||||||| ||| ||| |||||||||| ||||| |||||||||| ||| |||||||| || ||||||||||| || ||| |||| ||||| || |||||||| ||| ||||||||| ||||||||| || ||| ||| |||| || ||| ||||||||| |||||||||
|||||||||| || ||||| | |||||||||| |||| || |
|---|---|
||| | |||||||||| || |||||||| ||| ||||| ||| ||| ||| ||||| |||||||||||||| || |||||||| || ||||| ||| |
|||||| | |||||||||| || ||||| |
|||||| | |||||||||| || |||||||| |||| |||| ||||||| |
||| | |||||||||| || ||| |
||| | |||||||||| || ||| |
||| | |||||||||| ||||| ||| ||||||||||||||| ||||| ||| |||| |
|||||||| | |||||||||| |||||||||||||| ||| |
|||| | |||||||||| |||| | |
|||||||||| | |||||||||| |||||||||| | |
|||| | |||||||||| |||| | |
||| ||||||||||||||||||| |||| ||||||||||||||||||| |||| ||||||||| |||||| ||||| ||||||| ||||| ||||||| ||||||||||| |||||||| |||||| ||| ||||||||||| ||||||| ||| ||||||||| |||||| ||| ||||||||| |||||| ||||| ||||||||||||||||| |||||||| ||||||||||| |||||||||||||||| ||||||||| ||| ||||||||| |||||| ||||||||||||||| ||||||| |||| ||| ||||| |||| |||| ||||| |||| ||| ||||||||| ||||||| || |||||||| ||||||||||||||||| ||| |||||
Note: This table has been redacted at the request of the sponsor.
Results of the Matching Adjusted Indirect Comparisons Conducted by Sponsor
Summary of Included Studies
The HIMALAYA, |||||||| and IMbrave150 trials used similar study designs and were phase III, randomized, open-label trials. All | trials were similar in terms of restriction on prior therapy, and HCC etiology. A small proportion of patients from the HIMALAYA tri had a Child-Pugh class of B and an ECOG PS of 2 at baseline, while for both IMbrave150 ||| ||||||| trials, inclusion was restricted to a Child-Pugh class of A and an ECOG PS of 0 or 1. Regarding HCC diagnosis, the HIMALAYA trial required a confirmed histological diagnosis, with the IMbrave150 trial ||| ||||||| also requiring the AASLD criteria for diagnosis. Unlike the IMbrave150 trial, the HIMALAYA trial ||| ||||||| included BCLC stage as an eligibility criterion, restricting patient enrolment to stages B or C. The time window between the latest bleeding event and study enrolment or randomization varied between studies. The HIMALAYA study excluded patients who had had bleeding events within the 12 months preceding enrolment, while the IMbrave150 trial excluded patients who had experienced bleeding in the 6 months preceding study entry ||| ||||||| |||||||| |||||||| ||| ||| |||||||| || |||||||||| ||||||||| |||||| || |||| ||||| || |||||||||||||| The median durations of study follow-ups were 33.18 months in the tremelimumab in combination with durvalumab group and 32.2 months in the sorafenib group in the HIMALAYA trial17 and 8.9 months in the atezolizumab in combination with bevacizumab group and 8.1 months in the sorafenib group in the IMbrave150 trial,53 ||| |||| |||||| || ||| |||||||||| ||||| ||| |||| |||||| || ||| ||||||||| ||||| || ||||||||
In terms of efficacy outcomes, OS and PFS were reported by all | trials in the ITT population. Additional efficacy outcomes for comparison of tremelimumab in combination with durvalumab with atezolizumab in combination with bevacizumab were ORR and DoR in the ITT population (Table 38). ||| ||||||| ||||| |||| |||||||| ||| |||||| |||| || || ||| ||| |||||||| || ||| ||| ||||||||| |||||||||||| || ||||||||||| |||| |||||||||| |||||| ||||||||||| Because PFS was available for all | trials, TTP was not considered an outcome of interest given the complementarity of these 2 outcomes. Overall, the definitions of OS, PFS, and ORR were similar across the trials. However, for parameters related to the disease progression, both the HIMALAYA and IMbrave150 trials employed RECIST v1.1, ||||| ||||||| |||| ||| |||||||| |||||||| |||||||||| |||||||| || ||||| |||||| ||||||||||
|| ||||| || |||||||||||||||| ||||||||| |||||||| |||| |||||||||| || || |||||||| ||| |||||||| || |||||| |||| || ||||||||||||| || ||||| ||||| || ||||| ||||||| ||| |||| || ||||||||||||| || ||||||||| |||||||| ||||| || ||||| ||||||||| |||||| |||| ||| |||| |||||||||||||||| ||||||| ||||||||||| ||| |||||||||| ||| |||||||||| ||||| ||| ||||||||| ||||||| || |||||||| |||| ||||||| |||||||||||| ||||| ||| |||||||||| || ||||||| |||| ||| |||||||||||||||| |||||||| |||| ||| ||||||| || ||||| ||| ||| ||| ||||||||| |||||||||| ||||||||||| |||||||| |||| ||||||||| || |||| ||||||| || ||||||||||| ||| ||| |||||||||| || |||||||| |||| ||||||| ||||||||||| || ||| ||||| || ||| ||||||||||||
In terms of safety outcomes, the most commonly reported outcomes across the 3 trials included AEs, AEs of CTCAE grade 3 || ||||||| ||||||| |||| and AEs leading to discontinuation, and these were considered of interest and analyzed in MAICs using the safety population (Table 37).
Table 37: List of Comparisons Performed in MAICs Conducted by Sponsor
Analysis | Comparator | OS | PFS | ORR | DoR | Grade 3 and higher TEAEs | ||||| | TEAEs leading to discontinuation | ||| || ||| | ||| || |
|---|---|---|---|---|---|---|---|---|---|---|
MAIC | Tremelimumab in combination with durvalumab vs. atezolizumab in combination with bevacizumab | Yes | Yes | Yes | Yes | Yes | ||||| | Yes | ||||| | ||||| |
|||||||||||| || ||||||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Yes = performed comparison; No = comparison data unavailable; AS = abdominal swelling; DoR = duration of response; MAIC = matching adjusted indirect comparison; OS = overall survival; ORR = objective response rate; PFS = progression-free survival; QoL = quality of life; TEAE = treatment-emergent adverse event; TTD = time to deterioration.
Notes: Because nivolumab is not recommended for the first-line treatment for hepatocellular carcinoma in Canada, it was not discussed further in this ITC report.15 Details from the table have been taken from the sponsor’s Summary of Clinical Evidence.
Source: Sponsor indirect treatment comparison reports.50,51
Table 38 is a summary of patient baseline characteristics for the HIMALAYA and IMbrave150 trials included in the MAIC comparing tremelimumab in combination with durvalumab versus atezolizumab in combination with bevacizumab, before and after reweighting. The largest baseline imbalances in terms of standardized mean difference were in MVI, EHS, etiology (HBV and HCV), and BCLC stage B, all of which were reduced by adjustment. After restriction, 766 of 782 patients in the tremelimumab in combination with durvalumab and sorafenib groups were kept from the HIMALAYA trial for the MAIC analysis based on the ITT population. Populations from the IMbrave150 trial and reweighted HIMALAYA trial were balanced with respect to known and measured treatment-effect modifiers. The reweighted HIMALAYA population differed from the original HIMALAYA population, as it had higher proportions of MVI, EHS, and HBV, as well as a lower proportion of HCV. Patients with grade 1 ALBI scores remained around 52% in HIMALAYA after restriction, and reweighting was not reported in the IMbrave150 trial.
||||| || ||||| |||||||| || ||||||| |||||||| ||||||||||||||| ||| ||| |||||||| ||| ||||||| |||||| |||||||| || ||| |||| ||||||||| |||||||||||| || ||||||||||| |||| |||||||||| |||||| ||||||||||| |||||| ||| ||||| ||||||||||||||||| |||||||||||| ||| ||| || ||| |||||||| || ||| |||||||||||| || ||||||||||| |||| |||||||||| ||| ||||||||| |||||| |||| |||| ||| |||||||| ||| ||| |||| ||||||||||||| ||||||| |||||||| |||||||||| || ||||| |||||||||||||| |||| |||||||||| |||| || |||| |||| ||| |||||||| |||| ||| ||||| ||| || ||||| |||| ||||||| || ||||||||||| ||||| |||||||||||| ||||||||||| |||| ||||||| ||| |||||||||| |||||||| |||| |||||||| |||| ||||||| || |||||||| |||||||| ||||||||| |||||| |||||||||| ||| |||||||||| |||||||| |||||||||| |||||||| |||| ||| |||||||| |||||||| ||||||||||| || || ||| |||||| |||||||||| || |||||||| |||| |||| |||||||| |||||| |||||||||| || |||| ||| ||||||| |||||| |||||||||| || ||| ||| ||||
The included studies were assessed for homogeneity. Important differences across trials for key characteristics are summarized in Table 42.
|||||||||||||| | |||||||||| | |||||||| ||||||||| | |||||||| |||||||||| | |||||||| ||||||||||| | |||
|---|---|---|---|---|---|---|---|
||| | ||||||||| | |||||||||||| || ||||| | ||||||||| | |||||||||||| || |||||| | ||||||||| | |||||||||||| || ||| | |
|||| | | ||| | ||| | ||| | ||| | ||| | ||||| | ||||| |
||| || |||||| ||| | |||| | |||| | |||| | |||| | |||| | |||| | |||| |
||||| ||| | |||| | |||| | |||| | |||| | |||| | |||| | |||| |
|||| ||||||||| | |||| | |||| | |||| | |||| | |||| | |||| | |||| |
|||| ||| | |||| | |||| | |||| | |||| | |||| | |||| | |||| |
|||| ||| | |||| | |||| | |||| | |||| | |||| | |||| | |||| |
|||| |||| ||| | |||| | |||| | |||| | |||| | |||| | |||| | |||| |
||||||||| || ||| | |||| | |||| | |||| | |||| | |||| | |||| | |||| |
||||||||| || ||| | |||| | |||| | |||| | |||| | |||| | |||| | |||| |
|||| || ||| | |||| | |||| | |||| | |||| | |||| | |||| | |||| |
|||| || ||| | |||| | |||| | |||| | |||| | |||| | |||| | |||| |
|||| || ||| | ||| | ||| | ||| | ||| | ||| | ||| | ||| |
||||| |||| || ||| | ||||| | |||| | |||| | ||||| | ||||| | ||||| | ||||| |
|||| || ||| | |||| | |||| | |||| | |||| | |||| | |||| | |||| |
|||| || ||| | |||| | |||| | |||| | |||| | |||| | |||| | |||| |
|||| ||||| || ||| | || | |||| | |||| | |||| | |||| | |||| | |||| |
|||| ||||| || ||| | || | |||| | |||| | |||| | |||| | |||| | |||| |
|||| ||||| || ||| | || | ||| | ||| | ||| | ||| | ||| | ||| |
|||||||||||||||||||| |||| ||||||||||||||||| |||||| |||| ||||||||| |||||| ||||| ||||||| |||| ||||||| ||||||||||| |||||||| |||||| |||||||||||||| ||||||| ||| |||||||| |||||| ||||| ||| |||||||||||||||||| ||| ||||||||||||| ||||||||| || ||||||||||||||||||| ||||||| |||| ||| ||||| |||| |||| ||||| |||| ||| ||||||||| ||||||| || |||||||| ||||||||||||||||| ||||||| ||| ||||
||| ||||||||| |||||| |||| || |||||||||| |||||||| ||| ||||| || ||| |||||||||||| || ||||||||||| |||| |||||||||| ||||| ||| ||||| || ||| ||||||||| |||||| |||||| |||||||| ||| |||||||| |||||| |||||| |||| || ||||| |||||||||||| |||||| ||| ||||| |||| ||| |||||| |||||| |||||
Note: This table has been redacted at the request of the sponsor.
Table 39: Distribution of Weights of MAIC Comparing Tremelimumab in Combination With Durvalumab Versus Atezolizumab in Combination With Bevacizumab: ITC
Detail | Rescaled weights | |||||||||||| ||||||| |
|---|---|---|
Minimum | 0.1437 | |||||| |
First quartile | 0.5702 | |||||| |
Median | 0.8309 | |||||| |
Mean | 1.0000 | |||||| |
Third quartile | 1.2076 | |||||| |
Maximum | 6.2042 | |||||| |
ITC = indirect treatment comparison; MAIC = matching adjusted indirect comparison.
Note: Details from the table were taken from the sponsor’s Summary of Clinical Evidence.
Source: Indirect treatment comparison report.50
|||||||||||||| | ||||||| | |||||||| |||||||| | |||||||| ||||||||||| | |||||||| |||||||||| | |||
|---|---|---|---|---|---|---|---|
||| | ||||||||| | |||||||||||| || |||||| | ||||||||| | |||||||||||| || |||||| | ||||||||| | |||||||||||| || ||||| | |
|||| | | ||| | ||| | ||| | ||| | ||| | ||||| | ||||| |
||| || || || |||||| ||| | |||| | |||| | |||| | |||| | |||| | |||| | |||| |
||| || |||||| ||| | |||| | |||| | |||| | |||| | |||| | |||| | |||| |
||||| ||| | |||| | |||| | |||| | |||| | |||| | |||| | |||| |
|||| |||||||| ||| | |||| | |||| | |||| | |||| | |||| | |||| | |||| |
|||| ||| | |||| | |||| | |||| | |||| | |||| | |||| | |||| |
|||| ||| | |||| | |||| | |||| | |||| | |||| | |||| | |||| |
||||| ||| | |||| | |||| | |||| | |||| | |||| | |||| | |||| |
||||||||| || ||| | |||| | |||| | |||| | |||| | |||| | |||| | |||| |
||||||||| || ||| | |||| | |||| | |||| | |||| | |||| | |||| | |||| |
|||| || ||| | |||| | |||| | |||| | |||| | |||| | |||| | |||| |
|||| || ||| | |||| | |||| | |||| | |||| | |||| | |||| | |||| |
|||| || ||| | ||| | ||| | ||| | ||| | ||| | ||| | ||| |
||| |||||||||||||||||||| |||| |||||||||||||||||| |||||| |||| |||||||||| |||||| ||||| ||||||| |||| |||||||| ||||||||||| |||||||| |||||| ||| |||||||||||| ||||||| ||| ||||||||| |||||| ||||| ||| ||||||||||||||||||| ||| |||||||||||| ||||||||| || ||||||||||||||||||| ||| ||||
||| ||| ||||| ||||||||| ||| ||||| || ||| |||||||||||| || ||||||||||| |||| |||||||||| |||||| ||| ||||| || ||| ||||||||| |||||| || ||||||| |||||||||| ||| |||||||||| ||||| || ||| ||||||| || ||| ||||||| |||||||| |||||| ||| |||| |||||| ||||
Note: This table has been redacted at the request of the sponsor. Some redacted rows have been deleted.
|||||||||||||| | |||||||| ||||||| | |||||||||||| ||||||| |
|---|---|---|
||| | |||||| | |||||| |
|| | |||||| | |||||| |
|||||| | |||||| | |||||| |
|||| | |||||| | |||||| |
|| | |||||| | |||||| |
||| | |||||| | |||||| |
|||| ||||||||||||||||| |||||||| ||||||||||| ||| |||||||| ||| |||||||| || ||||| ||||||||| || ||||| ||||||||||||||| ||||||| |||| ||| ||||| |||| |||| ||||| |||| ||| ||||||||| ||||||| || |||||||| ||||||||||||||||| ||| ||||||| ||||||| |||||||||| |||||| |||| |||||||| || ||| ||| ||| |||| ||||||| || ||||||||||| ||||||||||| || |||||||| ||| ||| |||||||| |||||| ||||||||| || ||| |||||||| || |||||||| |||||| || ||||||||||||| ||| |||||||||| ||||||||||| ||| |||||||| || ||| |||||||| |||| || |||| || ||| ||| |||||||| ||| ||||||| ||| |||||||| ||||||||| ||| |||| ||| |||||||| ||| ||||||| |||| |||| |||||| || ||| |||| || |||| ||| |||| ||| ||| ||||||||||| || |||||||||| ||||||||| |||||||||| ||| || |||| |||| || |||| ||| ||||||||| || |||||||||| |||||||||||| ||| ||||||| |||| || ||| |||| || ||||| ||| ||||||| |||| || ||||||| |||| || |||| ||| ||| ||||||||| || |||||||||||||||||| |||||||| ||| || ||| |||| || |||||| || ||||||| ||| |||||||| ||||||| |||||
Note: This table has been redacted at the request of the sponsor.
|||||||||||||| | ||||||||||| ||| |||||||| || ||||||||| |||||| ||||||||| |
|---|---|
||||||| |||||||| | ||| |||||| |||||||||| ||||||||||| ||| |||||||| |||||||| |||||||| |||| |||||||| || |||||||||| |||| |||||||| |||||||| |||||||| ||||||||| |||||||||||| ||||||||| || ||||||||| |||||||||| ||| ||||||| |||| |||||||| ||||| |||||||| ||| |||||||||| |
||||||||| ||||||| | ||| |||||| |||| ||||||| || ||||| || ||||||||||| || ||||| |||||||| |
||||| ||||||||||| |||||||| | || |||||| |||| ||||||| || ||||| || ||| ||||||||| |||| ||| ||| ||||| |||| ||||||||||||| ||||||||||| |||||||| ||| ||||||| |||||||| |||| ||||| || || ||||||||||||||||||||| ||||||||||| ||||||| |||||||||| || |||||| || |||||| |||| |||||| ||||||| ||| |||||| |||||||| ||||| ||| ||||| ||||||||| || ||||||||||||| |||||| ||||||| |||||||| |||||||||||| |||||||||| |||||||| |||||||| |||| |||||||||| |||||||||| |||||||| ||| ||||||| ||||||||||||||| |
|||||| || ||||||||||| | |||||| ||| ||||||| |||||||||| |||||| ||||||||||||||||||||| ||| || || |||||||| || |||||| |||| |||| |||| |||||||||| |||| || || ||||||||| |||||||| || |||||||||| |||| |||||||||||| ||||| |||||||||||||||||| |||| || |||| ||||||||||| || ||||| || |||||||| ||||| |||||||||||||||| || || |||| |||| |||||| || ||| || || |||| |||| |||||| ||||| |||||| |||| ||||| |||||||||| ||| ||| |||||| ||||| ||||| |
||||||||||| || ||||||||| | ||||||||| || |||| ||||| |||| |||||||| |||| ||||||| || || ||| |||| || ||||||||| ||| ||| ||| |||| |||||||| ||| |||||||||| || |||||||||||| || ||||||||||| |||| ||||||||||||| |||||||||||||| |||||||| ||| ||||||||||| || ||||||||| |||| ||||||| |||||| ||| ||||||| |||||||| ||| |||||||||| ||||||| || ||| ||||||| |||||||||||| ||||||||||| |||||||||| |||||||| ||| |||||| |||| ||||||||| ||||| ||||||| |||| ||| |||||||| |||||| ||||||||| ||||| |||||| |||||||||||| || ||||||| ||||||||||| ||||||||||||| ||||||||||| |
|||||| || |||||||| |||||||||| | |||||| |||||| || ||||| ||||||||| |||||| |||| ||| |||||| |||||||||||| || || |||||| ||||||||||| |
|||||||||| ||||||||| | ||| |||||||| || |||| ||||||||||| ||||||| ||||||||| ||||| |||| || |||| ||| || |||||||||| |||||||||| || ||||||||| ||||||| |||||| || |||| ||| |||||||||| ||| ||||||| |||||| ||| ||||||||| |
||||| |||||| | ||| |||||||||| |||||| |||| ||||| |||| ||||||||||| |||| ||||| ||||||| |
||||| ||||||||| ||||||||||| ||| ||| ||||| || ||||| ||||||||| |||| |||||||||||||||| ||||| ||||||| ||| ||||||||| || ||||||||| |||| || |||||||| ||||||||||| |||||||| ||||| ||||||||||| ||||||| ||| |||||||||||||| |||||||||| ||| |||||||| |||||||||||||||||||| |||| ||||||||||||||||| |||||||| ||||||||||| ||| |||||||| |||||||| ||||| || |||||| ||||||||| ||||||||||||||||||| ||||||||| |||||| ||||||| |||||||||| |||||||| || ||||| ||||||||||||||| ||||||| ||| |||||||
Note: This table has been redacted at the request of the sponsor.
Efficacy
This section summarizes the results of | MAICs comparing tremelimumab in combination with durvalumab versus atezolizumab in combination with bevacizumab| ||| |||||||||||| || ||||||||||| |||| |||||||||| |||||| ||||||||||| Details of efficacy results are presented in Table 43 and Table 44.
Overall Survival
Tremelimumab in Combination With Durvalumab Versus Atezolizumab in Combination With Bevacizumab
The proportional hazards assumption of the restricted and reweighted HIMALAYA data was examined and not rejected for OS. After restriction and reweighting, the HR for OS for tremelimumab in combination with durvalumab versus atezolizumab in combination with bevacizumab was 1.09 (95% CI, 0.80 to 1.48).
|||||||||||| || ||||||||||| |||| |||||||||| |||||| |||||||||||||| |||||||||||| ||||||| |||||||||| || ||| |||||||||| ||| |||||||||| |||||||| |||| ||| |||||||| ||| ||| ||| |||||||| ||| ||| ||||| ||||||||||| ||| |||||||||||||| || ||| || ||| |||||||||||| || ||||||||||| |||| |||||||||| |||||| |||||||||| ||| |||| |||| ||| |||| || ||||||
Progression-Free Survival
Tremelimumab in Combination With Durvalumab Versus Atezolizumab in Combination With Bevacizumab
Outcomes assessed by the investigator based on RECIST 1.1 were considered for PFS. The proportional hazards assumption of the restricted and reweighted HIMALAYA data was examined and not rejected for PFS. After restriction and reweighting, the HR for OS for tremelimumab in combination with durvalumab versus atezolizumab in combination with bevacizumab was |||| |||| ||| |||| || ||||||||||||||||| || ||||||||||| |||| |||||||||| |||||| |||||||||||||||||||||||| || ||| |||||||||| ||||||| |||||||| ||| ||||||| |||| ||||||||||| || ||||||||| ||| ||| |||||||||| ||| ||||| || ||| |||||| |||| ||||||||| |||||||| || |||||||||||||| || |||||||| |||||| || ||| |||| |||||||||| |||||| |||| ||||||| |||||||||| |||| ||| |||| || ||| |||| |||||||| |||| || ||||| || || ||| ||||||| |||||||| |||| || ||| ||| ||||||||||| ||||||| ||| ||||||| ||||||||| ||||| ||||||||||| || ||||| || |||||||||| ||||||| ||||||| ||| ||| |||||| ||| ||| ||||| || |||||||| |||||||| ||| |||||| ||| |||||||||| || ||||||||||| |||||||||| ||| ||| |||| ||| ||| |||||
|||||| |||||||| | |||| || |||||||||| | ||| || |||| |||||||||||||| || ||||||||| | |||||| ||| |||| ||| | |
|---|---|---|---|---|
|||||||||| | ||||||||| | |||
||||||| | |||||||||||| | |||| ||||| || ||||| | ||| |||| || |||| | ||| |||| || |||| |
||||||| | ||||||||||| | |||| ||||| || ||||| | ||| |||| || |||| | ||| |||| || |||| |
|||||| ||| | ||||||||||| | |||| ||||| || ||||| | ||| |||| || |||| | ||| |||| || |||| |
|||||||||||| ||||||||| || |||||| |||||| ||| ||||||||||||||||| ||||||||| |||||| ||||||||| |||||||||| |||||||| || ||||| |||||||| ||||||| ||||||||| ||||||| |||||| ||||||| |||| ||| ||||| |||| |||| ||||| |||| ||| ||||||||| ||||||| || ||||||||||||||||||||||||| ||||||| ||| ||||| |||| ||| ||||||| || ||| |||||||||| ||| |||||||||| |||||||| |||| ||| |||||||| ||| ||| ||| |||||||| ||| |||| ||||| ||||||||||| ||| |||||||||||| ||| || ||| ||| ||| |||||||||||| || ||||||||||| |||| ||||||||||||||| |||||||||| ||| |||| |||| ||| |||| || ||||||
Note: This table has been redacted at the request of the sponsor.
Objective Response Rate
Tremelimumab in Combination With Durvalumab Versus Atezolizumab in Combination With Bevacizumab
After restriction and reweighting, the OR for ORR of tremelimumab in combination with durvalumab versus atezolizumab in combination with bevacizumab was 1.18 (95% CI, 0.44 to 3.21).
Duration of Response
Tremelimumab in Combination With Durvalumab Versus Atezolizumab in Combination with Bevacizumab
The proportional hazards assumption of the restricted and reweighted HIMALAYA data was examined and not rejected for DoR. After restriction and reweighting, the HR for DoR of tremelimumab in combination with durvalumab versus atezolizumab in combination with bevacizumab was |||| |||| ||| |||| || ||||||
Table 44: Comparison of OS, PFS, ORR, and DOR for Tremelimumab in Combination With Durvalumab Versus Atezolizumab in Combination With Bevacizumab
Detail | HIMALAYA before weighting | HIMALAYA after weighting | IMbrave150 | ITC estimate |
|---|---|---|---|---|
Direct estimate: Tremelimumab in combination with durvalumab vs. sorafenib | Direct estimate: Tremelimumab in combination with durvalumab vs. sorafenib | Direct estimate: Atezolizumab in combination with bevacizumab vs. sorafenib | Tremelimumab in combination with durvalumab vs. atezolizumab in combination with bevacizumab | |
Overall survival | ||||
HR (95% CI) | 0.77 (0.66 to 0.92) | 0.72 (0.60 to 0.87) | 0.66 (0.52 to 0.85) | 1.09 (0.80 to 1.48) |
Progression-free survival | ||||
|| |||| ||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
Objective response rate | ||||
OR (95% CI) | |||| ||||| || ||||| | |||| ||||| || |||||| | |||| ||||| || |||||| | 1.18 (0.44 to 3.21) |
|||| | |||| | |||| | || | |
Duration of response | ||||
HR (95% CI) | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
CI = confidence interval; DoR = duration of response; HR = hazard ratio; ITC = indirect treatment comparison; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; vs. = versus.
Source: Sponsor indirect treatment comparison report.50
|||||| | |||||||| |||||| ||||||||| | |||||||| ||||| ||||||||| | ||||||| | ||| |||||||| |
|---|---|---|---|---|
|||||| ||||||||| |||||||| | |||||| ||||||||| |||||||||| | |||||||||| ||| |||| | |||||||||||| || ||||||||||| | |
||||||| |||||||| | ||||
|| |||| ||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
|||||||||||||||| |||||||| | ||||
|| |||| ||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
||||||||||| ||||||||| || |||||| |||||| ||| |||||||| ||||||||| ||||||||||| || ||||||| ||||||||| ||| ||||||||||||||||| ||||||||||||||||| ||||||| ||| ||||||
Note: This table has been redacted at the request of the sponsor.
Patient-Reported Outcomes
Tremelimumab in Combination With Durvalumab Versus Atezolizumab in Combination With Bevacizumab
Patient-reported outcomes included ||| || |||||| ||| ||| || ||||||||| ||||||||. The proportional hazards assumption of the restricted and reweighted HIMALAYA data was examined and not rejected for both patient-reported outcomes. For each patient-reported outcome, the population was restricted after the weighting process to patients with outcome information. ||| ||| ||| || ||||| ||||| ||||||||||| ||| ||||| || ||| |||||||||||| || ||||||||||| |||| |||||||||| |||||| ||| ||||| || ||| ||||||||| |||||| ||||| |||||||||||| ||| ||| ||| ||| || ||||||||| |||||||| ||| ||||| || ||| |||||||||||| || ||||||||||| |||| |||||||||| |||||| ||| ||||| || ||| ||||||||| |||||| ||||||||||| ||||| ||| |||||| || ||||||| |||| ||| ||| |||||||||
The HR for time to deterioration of QoL for tremelimumab in combination with durvalumab versus atezolizumab in combination with bevacizumab was |||| |||| ||| |||| || ||||||||| || ||| ||| || ||||||||| |||||||| ||| |||||||||||| || ||||||||||| |||| |||||||||| |||||| |||||||||||| || ||||||||||| |||| ||||||||||| ||| |||| |||| ||| |||| ||||||||
|||||||||||| || ||||||||||| |||| |||||||||| |||||| |||||||||||||| ||||||| || |||||||||||||||| |||||||| ||| |||||||||||| || ||||||||||| |||| |||||||||| |||||| |||||||||| |||| ||| |||||||||
Safety
Tremelimumab in Combination With Durvalumab Versus Atezolizumab in Combination With Bevacizumab
||||| ||||||||||| ||| ||| || |||||||| ||| ||| || ||| |||||||||||| || ||||||||||| |||| |||||||||| |||||| ||| ||| || ||| ||||||||| |||||| After reweighting, the safety populations from the IMbrave150 trial and reweighted HIMALAYA data were balanced for known and measured treatment-effect modifiers. ||| |||||||||| ||||||||||||||||| |||||||| |||| ||| |||||||| |||||||| |||||||||| || || ||| |||||| |||||||||| || |||| |||| ||| ||||||||| ||| |||| || |||||| |||||||||| || ||||||||| ||
The OR for AEs of CTCAE grade 3 or 4 for tremelimumab in combination with durvalumab versus atezolizumab in combination with bevacizumab was 0.73 (95% CI, 0.44 to 1.19). The OR for serious AEs for tremelimumab in combination with durvalumab versus atezolizumab in combination with bevacizumab was |||| |||| ||| |||| || |||||| The OR for AEs leading to discontinuation for tremelimumab in combination with durvalumab versus atezolizumab in combination with bevacizumab was 0.49 (95% CI, 0.23 to 1.04).
|||||||||||| || ||||||||||| |||| |||||||||| |||||| |||||||||||||||| |||||||||||| ||| ||| || |||||||| ||| || ||||| || ||| |||||||||||| || ||||||||||| |||| |||||||||| |||||| ||| ||||| || ||| |||||||||| ||| |||||||||||| || ||||||| ||||||||||||||| || ||| |||||||||| |||||| ||||||||||| |||| ||||||| ||| |||||||| ||| |||||||| ||| ||||| ||| |||||||| ||||| |||||| ||| |||||| || ||||||| |||||||| ||| |||||||| |||| |||||||| |||||| ||| |||||||||| ||||||| ||| |||||| |||||||||| |||||||| |||| ||| |||||||| |||||||| |||||||||| || || ||| |||||| |||||||||| || |||||||| |||| |||| |||||||| |||| ||||||||| || ||| ||||| |||||||||| || ||||||||| || |||| || ||| ||| || ||||| ||||| ||| |||||||||||||| || ||||||||||| |||| |||||||||| |||||| |||||||||| ||| |||| |||| ||| |||| || |||||| ||| || ||| ||||||| ||| ||| |||||||||||| || ||||||||||| |||| |||||||||| |||||| |||||||||| ||| |||| |||| ||| |||| || |||||| |||| ||| ||| ||| |||||||||||| || ||||||||||| |||| |||||||||| |||||| |||||||||| ||||||| || ||||||||||||||| ||| |||| |||| ||| |||| || ||||||
Critical Appraisal of MAICs Conducted by Sponsor
The sponsor-submitted MAICs had a number of limitations that challenge the interpretation of the internal and external validity of the findings. Studies included in the MAICs were selected from those identified by the systematic literature review. Overall, based on the methods detailed in the report, the systematic literature review involved a comprehensive search, and the screening strategies were sufficient to reduce the risk of error and selection bias. The criteria for the inclusion of studies for the ITC were provided and consistent with the objective. The literature search was conducted on |||||| ||| ||||| ||| ||||||| || |||||||| || ||||| The clinical experts consulted by CADTH for this review noted that several studies published over the past year that provide updated efficacy and safety data from the IMbrave150 trial |||||| |||| |||| |||| ||||||||| were not identified in this search and therefore were not included in the ITC.18-20 As a result, MAIC analyses did not select some efficacy outcomes |||||| |||| |||| |||| |||||||||||||||| ||||||||| based on the longer follow-up data, and this may have influenced the MAIC results. While the sponsor inadvertently omitted a reference to longer follow-up data for the IMbrave150 trial in the MAIC report and the clinical evidence document, OS results from the IMbrave150 trial used in the MAIC analysis (HR = 0.66; 95% CI, 0.52 to 0.85) were as reported in the Cheng et al. (2022) publication. The risk of bias of included studies was assessed for each study; however, it may differ depending on the study outcomes (i.e., OS versus patient-reported outcomes). || ||||||||| ||||| ||| |||| for the measurement of subjective outcomes due to the open-label study design. The eligible interventions for the ITC were restricted to those used in Canada for the treatment of patients with unresectable HCC to ensure that the comparators were relevant to the Canadian settings.
||| MAICs were conducted to compare efficacy and safety of tremelimumab in combination with durvalumab (from HIMALAYA) relative to atezolizumab in combination with bevacizumab (from the IMbrave150 trial), ||| |||||||||| ||||| ||||||||| The sponsor provided an adequate rationale for conducting the MAIC. Heterogeneity in the inclusion and exclusion criteria, patient baseline characteristics, and outcomes measured in the trials were reported and reviewed by the authors as part of the assessment of feasibility. The authors concluded that a population-adjusted indirect comparison was needed, and a MAIC was chosen to adjust for suspected heterogeneity with aggregate data available from the comparator trials and individual patient data for the HIMALAYA trial. In addition, the designs of all 3 trials included in MAICs were sufficiently similar and included a common comparison group (sorafenib).
The methodology for matching and adjustment was in line with NICE Decision Support Unit Technical Support Document 18.61 The list of potential treatment-effect modifiers was identified by a targeted literature review, a clinical expert’s opinion, and a review of relative treatment effects by subgroup reported in the trials included in the network of evidence. The matching criteria were based on the inclusion criteria for the IMbrave150 trial (for comparison of tremelimumab in combination with durvalumab versus atezolizumab in combination with bevacizumab) ||| ||||||| |||| |||||||||| || |||||||||||| || ||||||||||| |||| |||||||||| |||||| ||||||||||| and availability of comparable data from HIMALAYA. The final list of treatment-effect modifiers used in the adjustment included demographic variables (age, region, and gender), and a number of clinical variables, including MVI, EHS, alpha-fetoprotein level, etiology, performance status, BCLC stage, and Child-Pugh class. The clinical experts consulted by CADTH for this review agreed that the adjustment factors were found to be generally reasonable. Although the ALBI score, which was considered an important variable, was not included in the adjustment, the clinical experts consulted noted that omitting this factor could not result in potential bias. Another variable that was considered important but not included in the adjustment process was subsequent therapy; however, the clinical experts consulted mentioned that this variable is included in the BCLC staging to some extent.
The patient demographic characteristics before and after weighting were reported. After reweighting, populations from comparator trials (IMbrave150 ||| |||||||) and reweighted HIMALAYA data were balanced with respect to known, measured treatment-effect modifiers. However, it remains unclear how balanced the populations were for other variables that may be clinically relevant but could not be adjusted due to lack of data, or for those variables that were not part of the planned adjustment (unknown or unmeasured treatment-effect modifiers). In addition, the MAIC analysis could not account for some sources of trial heterogeneity, such as differences in observation times or definition of end points. The median duration of follow-up was 33.2 months in the HIMALAYA trial, versus 8.5 months in the IMbrave150 trial, ||| |||| |||||| || |||||||. For parameters related to disease progression, the HIMALAYA and IMbrave150 trials employed RECIST 1.1, while the REFLECT trial used mRECIST. While the clinical experts consulted for this review noted that mRECIST may be more accurate compared with RECIST 1.1 because it evaluates a viable tumour (enhancing area) using contrast-enhanced radiologic imaging,64,65 neither is typically used in clinical practice. The clinical experts indicated that, given the time gap, there is a possibility of systemic differences between patients in the HIMALAYA (from 2017 to 2019) and REFLECT (from 2013 to 2016) trials, such as treatments received before systemic therapy (i.e., local, or loco-regional treatment).
The ESS was reduced after adjustment in both MAICs. In the MAIC comparing tremelimumab in combination with durvalumab versus atezolizumab in combination with bevacizumab, the ESS was reduced to approximately 65.7% (513.9 of 782) of the original sample size in the HIMALAYA trial (69% in the tremelimumab in combination with durvalumab group and 63% in the sorafenib group). || ||| |||| ||||||||| |||||||||||| || ||||||||||| |||| |||||||||| |||||| ||||||||||| ||| ||||||||| |||||| |||| ||| ||||||| || ||||||||||||| ||| ||||||||||| || ||| |||||||| |||||| |||| || ||| |||||||| ||||||||||| || ||| |||||||||||| || ||||||||||| |||| |||||||||| ||||| ||| ||| || ||| ||||||||| ||||||| The reduction in the ESS reflects the heterogeneity between the trials among the variables, including in the weighting process. The matching of HIMALAYA patients based on the inclusion and exclusion criteria with comparator trials may remove an important portion of the patient population from the HIMALAYA trial. The small ESS therefore implies that the weighted estimates are being influenced by a subset of the patients from the HIMALAYA trial that may not be representative of the entire study population, which may limit the generalizability of the results.61 The distribution of weights was reported in both MAICs. || ||||||| |||||| |||||| |||| |||||||||| || ||| |||| ||||||||| |||||||||||| || ||||||||||| |||| |||||||||| |||||| ||||||||||| while a few extreme weight values were identified in the MAIC comparing tremelimumab in combination with durvalumab versus atezolizumab in combination with bevacizumab.
As not all trials included the same subjective and objective measurements, the comparative efficacy and safety of relevant treatments included remain uncertain. ||||| ||||||| |||||||| ||| |||||||||||||||| |||||||| |||| ||||||||| || ||| |||||||| ||||||||| |||||||| |||| ||| |||||||| || |||||||| |||| ||| |||||||| || |||||||||| |||||| In addition, DCR, which was considered an important outcome by the clinical experts consulted for this review, was assessed only in the HIMALAYA trial. Results on patient-reported outcomes (HRQoL and abdominal swelling), which were considered important outcome by patients, were reported only in the MAIC comparing tremelimumab in combination with durvalumab versus atezolizumab in combination with bevacizumab in patients with unresectable HCC. The results were reported as HRs, ORs, relative risks, and 95% CIs. In both MAICs, results in efficacy and harm estimates were imprecise (i.e., wide CIs) in the assessed end points, and the upper and lower boundaries of the CIs suggest the potential for different conclusions regarding the efficacy of tremelimumab in combination with durvalumab relative to the comparator drugs. No information was provided on how the missing data were handled for the outcomes in the included trials. The authors stated that 2 sensitivity analyses were performed on patient-reported outcomes (QoL and abdominal swelling) due to high rates of missing data; however, the results of the analyses were not reported. As a result, no superiority conclusions could be drawn from the MAICs submitted by the sponsor due to methodological limitations and imprecision in the effect estimates.
Fulgenzi et al. (2022) Network Meta-Analysis
In addition to the MAICs conducted by the sponsor, a published NMA conducted by Fulgenzi et al. (2022)66 was also identified. Details on how the NMA was identified by the sponsor, such as search strategy, screening, and selection procedure, were not reported.
Objectives
The NMA was conducted to compare first-line systemic therapy options for unresectable HCC based on studies published from 2007 to 2022.
Study Selection Methods
The research protocol was registered in PROSPERO, an international prospective register of systematic reviews (registration code CRD42022312489).
Based on the prespecified eligibility criteria outlined in Table 46, a systematic literature search was conducted for studies published from January 1, 2007, to February 28, 2022. The systematic literature searches were done in electronic databases (MEDLINE, the Cochrane Library, and Embase), and conference proceedings published in major scientific societies (American Society of Clinical Oncology, European Society for Medical Oncology, European Association for the Study of the Liver). Studies testing locoregional therapies alone or in combination with systemic treatments were excluded. Records retrieved by the searches were screened by 2 independent reviewers and disagreements were resolved by a third reviewer. The following data were extracted from the selected articles: study name, publication year, characteristics of experimental and control groups, age, region, MVI, EHS, etiology, Child-Pugh class, ECOG PS, BCLC stage, efficacy outcomes (OS, PFS, or ORR), and safety outcomes (the authors considered AEs of all types rather than focusing only on treatment-related AEs). Last, the authors collected HRs and corresponding 95% CIs for OS and PFS. Methods for data extraction were not reported. The risk of bias was evaluated according to the Cochrane risk-of-bias assessment tool.67 Methods for undertaking the risk of bias appraisals were not reported.
Analysis Methods of Published Network Meta-Analysis
A frequentist NMA was performed to compare the efficacy and safety of first-line treatments for unresectable HCC. Two analyses were performed: the first compared the efficacy of atezolizumab in combination with bevacizumab against all other first-line treatments, and the second compared all first-line treatments with placebo. The analyses were conducted using fixed-effect multivariable meta-regression models to estimate the indirect HR, with corresponding 95% CIs for OS and PFS, and ORs or relative risk, and corresponding 95% CIs for ORR and AEs, respectively. Forest plots were drawn to synthesize the results obtained for the comparisons of interest. Subgroup analyses were performed for atezolizumab in combination with bevacizumab against other first-line treatments by etiology: viral versus nonviral hepatitis. Homogeneity in the evidence network was assessed by comparing baseline descriptive statistics between the trials for variables considered potential effect modifiers. The assessment of the transitivity assumption was not reported in the NMA. The assessment of statistical consistency was not possible as no closed loops were included. The analysis was performed using the meta and netmeta packages in R, version 4.1.2.
Results of Published Network Meta-Analysis
A total of 13,709 records were screened by title and abstract. Of these, following removal of duplicates and exclusion of nonrelevant records, the full text of 70 publications were screened. Of these, 58 publications were removed, and another 7 publications were included after performing a hand search of relevant abstracts. Of the remaining 19 publications, 10 were removed because of irrelevant outcomes, design, or intervention. The 9 studies included in the NMA were phase III clinical trials, including SHARP (comparing sorafenib versus placebo), Asia Pacific (comparing sorafenib versus placebo), REFLECT (comparing lenvatinib versus sorafenib), Check-Mate 459 (comparing nivolumab versus sorafenib), IMbrave150 (comparing atezolizumab in combination with bevacizumab versus sorafenib), ORIENT-32 (comparing sintilimab in combination with IBI305 [a bevacizumab biosimilar] versus sorafenib), HIMALAYA (comparing tremelimumab in combination with durvalumab versus sorafenib), COSMIC-312 (comparing atezolizumab in combination with cabozantinib versus sorafenib) and the Qin et al. (2021) study (comparing donafenib versus sorafenib).
Table 46: Study Selection Criteria and Methods — Published Network Meta-Analysis
Characteristics | Indirect comparison |
|---|---|
Population | Patients with unresectable hepatocellular carcinoma receiving first-line systemic therapy |
Intervention | Immune checkpoint inhibitors or tyrosine kinase inhibitors:
|
Outcome | Efficacy:
Safety:
|
Study designs | Phase III randomized controlled trials |
Publication characteristics | Articles in English, published between January 2007 and February 2022 |
Databases searched |
|
Exclusion criteria | Locoregional therapies either alone or in combination with systemic treatments |
Selection process | The study screening and selection process was conducted by 2 independent reviewers based on the PICO framework and inclusion and exclusion criteria; any discrepancies were discussed and resolved with the contribution of a third independent author |
Data-extraction process | Methods for data extraction were not reported |
Quality assessment |
|
PICO = patient, intervention, comparison, outcome.
Source: Fulgenzi et al. (2022).66
Evidence Network
Figure 7 presents the network of evidence for the published NMA for efficacy and safety outcomes. In this network, all active treatments of interest, including tremelimumab in combination with durvalumab and atezolizumab in combination with bevacizumab, were connected to sorafenib. There were no closed loops. The HIMALAYA trial included 3 groups; as tremelimumab in combination with durvalumab is the drug under review and durvalumab monotherapy was out of scope of this review, results pertaining to the durvalumab monotherapy versus sorafenib are not reported.
Summary of Included Studies
This NMA compared atezolizumab in combination with bevacizumab with all other first-line treatment of HCC. Because tremelimumab in combination with durvalumab is of interest to this report, only a comparison of atezolizumab in combination with bevacizumab (from the IMbrave150 trial) versus tremelimumab in combination with durvalumab (from the HIMALAYA trial) was presented in this report. The HIMALAYA and IMbrave150 trials used similar study designs and were phase III, randomized, open-label trials. Both trials were similar in terms of restriction on prior therapy, HCC etiology, ECOG PS, and Child-Pugh class. Regarding HCC diagnosis, the HIMALAYA trial required a confirmed histological diagnosis, while the IMbrave150 trial also required AASLD criteria for diagnosis. Unlike the IMbrave150 trial, the HIMALAYA trial included the BCLC stage as an eligibility criterion, restricting patient enrolment to stages B or C. The HIMALAYA trial excluded patients who had had bleeding events within the 12 months preceding enrolment, while the IMbrave150 trial excluded patients who had experienced bleeding in the 6 months preceding study entry. The median durations of follow-up were 33.18 months in the tremelimumab in combination with durvalumab group and |||| months in the sorafenib group in the HIMALAYA trial, and 8.9 months in the atezolizumab in combination with bevacizumab group and 8.1 months in the sorafenib group in the IMbrave150 trial. In terms of efficacy outcomes, OS, and PFS were reported in both trials in the ITT population. The definitions of OS and PFS were similar across the trials, and parameters related to disease progression were based on RECIST 1.1.
Table 47 summarizes the baseline characteristics in the IMbrave150 and HIMALAYA trials. Only the groups of interest to this report are included. The most important sources of heterogeneity between the 2 studies were MVI, EHS, etiology, and BCLC B.
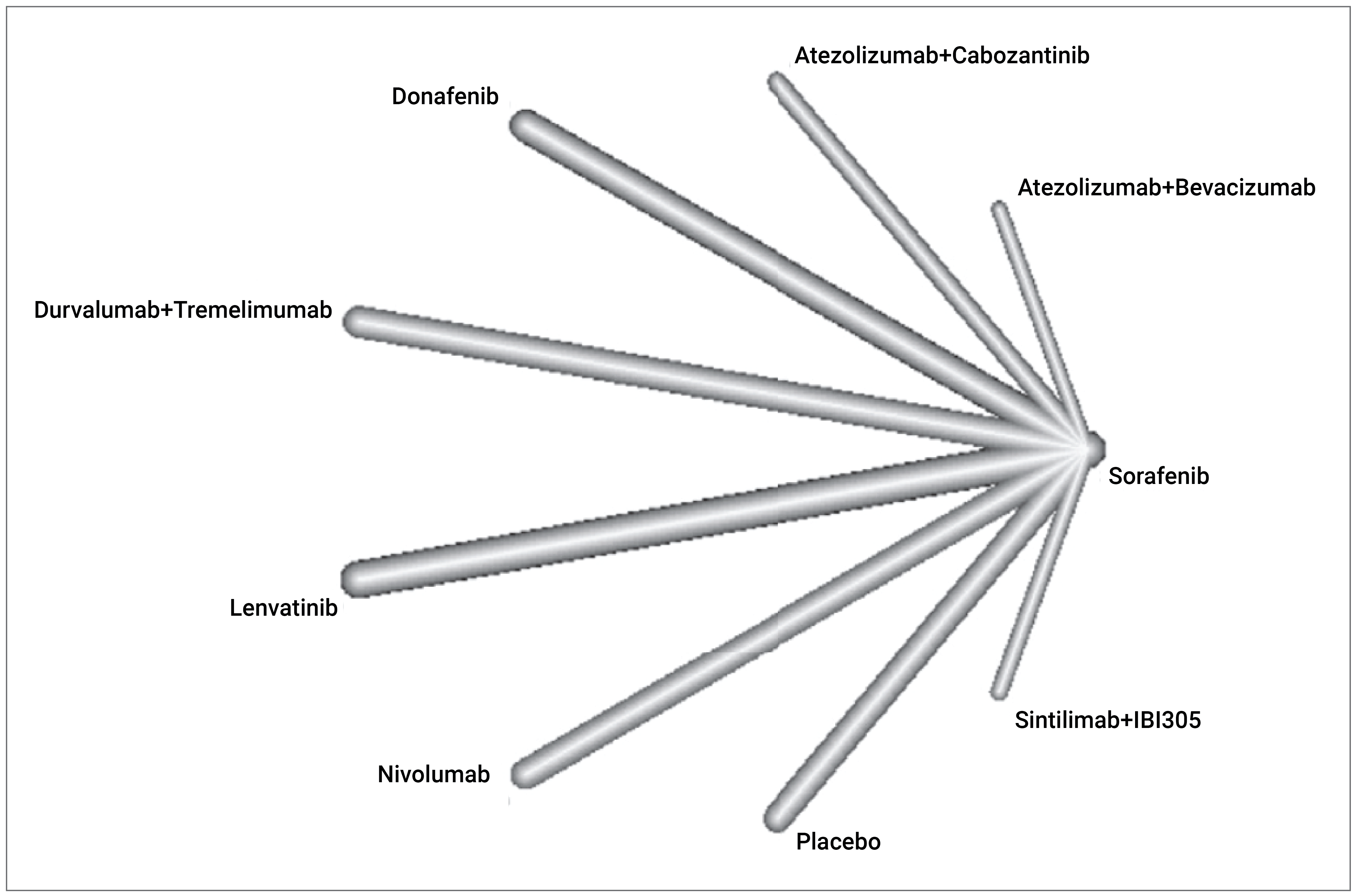
Source: Fulgenzi et al. (2022). Copyright 2022 Elsevier. Reprinted in accordance with Creative Commons Attribution Licence CC BY 4.0 DEED.66
Table 47: Baseline Characteristics of IMbrave150 and HIMALAYA Trials
Detail | IMbrave150 | HIMALAYA | ||
|---|---|---|---|---|
Atezolizumab in combination with bevacizumab N = 336 | Sorafenib N = 165 | Tremelimumab in combination with durvalumab N = 393 | Sorafenib N = 389 | |
Median age, in years | 64 | 66 | 65 | 64 |
Western region, n (%) | 60 | 59 | 60 | 57 |
Macrovascular invasion, % | 38 | 43 | 26 | 26 |
Extrahepatic spread, % | 63 | 56 | 53 | 52 |
Viral hepatitis, % | 70 | 68 | 59 | 57 |
Child-Pugh A, % | 100 | 100 | 100 | 100 |
ECOG PS 0, % | 62 | 62 | 62 | 61 |
BCLC C, % | 82 | 81 | 80 | 80 |
BCLC = Barcelona Clinic Liver Cancer; ECOG PS = Eastern Cooperative Oncology Group Performance Status.
Source: Fulgenzi et al. (2022).66
The authors found that the risk of bias was generally low across all the studies, with all the trials reporting a low risk in at least 5 out of 7 domains. The absence of blinding represented the major risk of bias for both the IMbrave150 and HIMALAYA trials. In addition, blinded independent radiologic review of imaging was performed in all the trials except for HIMALAYA.
Efficacy
The HR for OS for atezolizumab in combination with bevacizumab compared with tremelimumab in combination with durvalumab was 0.74 (95% CI, 0.52 to 1.06). Subgroup analyses were performed for OS by HCC etiology (nonviral versus viral hepatitis). The HRs for OS for atezolizumab in combination with bevacizumab compared with tremelimumab in combination with durvalumab in nonviral and viral patients were 1.23 (95% CI, 0.66 to 2.28) and 0.61 (95% CI, 0.39 to 0.94), respectively.
The HR for PFS for atezolizumab in combination with bevacizumab compared with tremelimumab in combination with durvalumab was 0.66 (95% CI, 0.49 to 0.87). The OR for ORR for atezolizumab in combination with bevacizumab compared with tremelimumab in combination with durvalumab was 0.60 (95% CI, 0.28 to 1.25).
Critical Appraisal of Published Network Meta-Analysis Submitted by Sponsor
The sponsor submitted a published NMA by Fulgenzi et al. (2022); however, because details on how it was identified were not reported, there is a risk of selection bias. The published NMA was conducted to compare atezolizumab in combination with bevacizumab with all other first-line treatment of unresectable HCC from 2007 to 2022.
The published NMA was based on a systematic literature review that identified studies according to prespecified inclusion criteria. Overall, based on the methods detailed in the paper, the systematic literature review involved a comprehensive search, and the screening strategies were sufficiently thorough to minimize the risk of error and selection bias. Although the risk of bias of individual studies were assessed in the systematic literature review, the methods for undertaking the risk-of-bias appraisals and the assessment results were not incorporated the paper. Other limitations of the NMA relate to data sparseness and network structure. The assessment of the transitivity assumption was not reported in the published NMA; however, important sources of heterogeneity across the included trials were noted. The networks for analyses were sparse (i.e., many comparisons but few trials), and an assessment of statistical consistency was not possible as no closed loops were included. A frequentist NMA was performed to compare the efficacy and safety of first-line treatments for unresectable HCC. Because tremelimumab in combination with durvalumab is of interest to this report, only a comparison of atezolizumab in combination with bevacizumab versus tremelimumab in combination with durvalumab is presented here. The use of fixed-effect models appears to be appropriate given the sparsity of data; however, no rationale was provided for the selection of the model in the published report. Furthermore, the effect estimates from the NMA are imprecise due to the sparseness of data and wide CIs, which for many outcomes included the possibility of benefit, lack of benefit, or harm for atezolizumab in combination with bevacizumab compared with tremelimumab in combination with durvalumab. As model fit was not evaluated, it is not clear how well the model estimated treatment differences.
Some important differences between the HIMALAYA and IMbrave150 trials increase the uncertainty of the NMA analyses. The HIMALAYA trial required a confirmed histological diagnosis of HCC, while the IMbrave150 trial also required AASLD criteria for diagnosis. Unlike the IMbrave150 trial, the HIMALAYA trial included BCLC stage as an eligibility criterion, restricting patient enrolment to stages B or C. The HIMALAYA trial excluded patients who experienced bleeding within the 12 months preceding enrolment, while the IMbrave150 trial excluded patients who had experienced bleeding in the 6 months preceding study entry. The definitions of end points were similar across the trials; however, the median duration of follow-up was 32.7 months in the HIMALAYA trial and 8.5 months in the IMbrave150 trial. Heterogeneity between the included studies would be expected to introduce bias into the study estimates observed between the comparators. Additional sensitivity analyses were not performed due to limited data. No results on patient-reported QoL, which was considered by patients to be an important end point for this review, were evaluated. In addition, there were no comparative effect estimates for the harms. These limitations must be considered when drawing conclusions on the results of the published NMA.
Studies Addressing Gaps in the Pivotal and Randomized Controlled Trial Evidence
No studies addressing gaps in the pivotal and RCT evidence were identified by the sponsor.
Discussion
Summary of Available Evidence
This systematic review included a single phase III, multicentre, randomized, open-label, sponsor-blind, global trial (HIMALAYA) comparing tremelimumab in combination with durvalumab to sorafenib in patients with unresectable HCC. A total of 1,324 patients were randomized in 1:1:1:1 ratio to receive tremelimumab in combination with durvalumab (N = 393), sorafenib (N = 389), durvalumab monotherapy (N = 389; not included in this review), and a lower dose of tremelimumab in combination with durvalumab (N = 153; closed during randomization due to preliminary efficacy findings). Randomization was stratified by macrovascular invasion, etiology of liver disease, and ECOG PS. Crossover within the study was not permitted.
The primary outcome was OS, which was defined as the time from the date of randomization until death due to any cause regardless of whether the patient withdrew from randomized therapy or received another anticancer therapy. PFS, TTP, ORR, BOR, DCR, DCR-16w, DCR-24w, and DoR based on investigator assessments using RECIST 1.1 were secondary outcomes. Exploratory outcomes included PFS, TTP, ORR, DCR, DCR-16w, DCR-24w, and DoR based on BICR assessments using mRECIST for HCC and irRECIST, as well as PFS from rechallenge and PFS on next treatment, and patient-reported HRQoL.
The HIMALAYA trial enrolled adult patients with confirmed HCC, based on histopathological findings rather than only clinical radiological findings, and with preserved liver function (a Child-Pugh class of A). Patients must have been assigned to BCLC stage B or C, exhibit an ECOG PS of 0 or 1, and have a life expectancy of more than 12 weeks. Patients had to be ineligible for locoregional therapy for unresectable HCC, and for patients who progressed after locoregional therapy for HCC, locoregional therapy must have been completed at least 28 days before the baseline scan. Patients must not have received any prior systemic therapy for unresectable HCC and must not have had clinically meaningful ascites within 6 months or active or prior documented GI bleeding within 12 months before the first scheduled dose.
||| MAICs and a published ITC submitted by the sponsor were summarized and appraised for this CADTH review. The sponsor-conducted systematic review and 2 MAICs evaluated the efficacy and safety of tremelimumab in combination with durvalumab against other first-line treatments (atezolizumab in combination with bevacizumab | ||| ||||||||||) in patients with unresectable HCC. The MAICs were considered necessary by the sponsor due to differences between trials in patient populations, with individual patient-level data available only for the HIMALAYA trial. The efficacy end points included OS and PFS in both MAICs, and ORR and DoR only in the MAIC comparing tremelimumab in combination with durvalumab versus atezolizumab in combination with bevacizumab. Harms related to the use of tremelimumab in combination with durvalumab, including AEs, SAEs, and AEs leading to treatment discontinuation, were also evaluated in both MAICs. Patient-reported outcomes (HRQoL and abdominal swelling) were only reported in the MAIC comparing tremelimumab in combination with durvalumab versus atezolizumab in combination with bevacizumab. The MAICs had a number of limitations that challenge the interpretation of the internal and external validity of the findings. Matching was not possible for all criteria due to substantial differences between the patient populations in the HIMALAYA and comparator trials. After reweighting, populations from comparator trials and reweighted HIMALAYA were balanced with respect to known, measured treatment-effect modifiers. However, it remains unclear how balanced the populations were for other variables that may be clinically relevant but could not be adjusted due to lack of data, or those variables that were not part of the planned adjustment (i.e., unknown or unmeasured treatment-effect modifiers). In addition, other sources of heterogeneity, such as differences in study follow-up duration or definition of the end points, could not be accounted for in the MAIC analysis. A small ESS implies that the weighted estimates are being influenced by a subset of the patients from the HIMALAYA trial who may not be representative of the entire study population, which may limit the generalizability of the results. In both MAICs, results in efficacy and harm estimates were imprecise (i.e., accompanied by wide CIs) in the assessed end points, precluding conclusions about comparative efficacy and harms. The sponsor submitted a published NMA, but it had important methodological limitations, and the results were highly uncertain.
Interpretation of Trial Results
Efficacy
Administration of the tremelimumab in combination with durvalumab combination therapy in the HIMALAYA study17 resulted in statistically significant prolongation of OS compared with sorafenib in all randomized patients (median OS of 16.4 months versus 13.8 months), a difference deemed clinically meaningful by the clinical experts. The OS rate at 36 months was also higher in the tremelimumab in combination with durvalumab group (30.7% versus 20.2%). The effect estimates in all predefined subgroups were consistent with the main effect; however, because the trial was not designed to detect differences in treatment effects across subgroups, no conclusions can be made based on subgroup results. There was no statistically significant difference in median PFS or median TTP between groups; the overall DCR between the 2 treatment groups was not tested statistically but appeared to be similar. The ORR in the final analysis was higher among patients in the tremelimumab in combination with durvalumab group compared to patients in the sorafenib group (20.1% versus 5.1%, respectively), which was consistent with results of the 32-week first interim analysis; however, because this end point was not adjusted for multiplicity there is an increased risk of false-positive conclusions.
At the time the HIMALAYA trial was conducted, sorafenib was the standard of care and was considered an appropriate comparator. However, based on feedback from clinical experts consulted for this review, as well as clinical guidelines, this is no longer the case. Current guidelines recommend the use of lenvatinib or atezolizumab in combination with bevacizumab instead. The clinical experts stated that, in the absence of comparative data for tremelimumab in combination with durvalumab versus atezolizumab-bevacizumab, it would be difficult to choose between the therapies in the first-line setting. They noted that tremelimumab in combination with durvalumab would be the only viable option for patients at high risk of bleeding and those who had started atezolizumab in combination with bevacizumab and experienced severe proteinuria or GI perforation. The clinician group input also noted that treatment with tremelimumab in combination with durvalumab would entail fewer clinic visits and less time in the clinic as patients would be treated every 4 weeks with 1 drug except for the first cycle, while for atezolizumab in combination with bevacizumab, patients are treated every 3 weeks and with 2 drugs.
HRQoL scores showed no mean change from baseline in reaching the estimated MID (a mean change ≥ 10 points, which was considered clinically relevant by sponsor and experts but not assessed in patients with HCC) at any time point for either treatment group. Improving QoL overall was described as an important outcome by patients, yet the interpretation of results for the HRQoL instruments is limited by missing data in both groups at baseline and a decline in the number of patients over time. Time to deterioration and improvement rates in QoL, function, and symptoms were longer and higher in most domains in the tremelimumab in combination with durvalumab group; however, because these results were also based on nonrandomized subgroups of the patient population and neither was adjusted for multiplicity, there is an increased risk of false-positive conclusions. The clinical experts consulted by CADTH noted that any worsening in HRQoL may be due to disease progression or adverse effects from treatment, which would occur early on in both groups, but also late in the tremelimumab in combination with durvalumab group due to immune-mediated AEs.
No superiority conclusions can be drawn from the MAICs due to imprecision of the effect estimates and other methodological limitations.
Harms
A total of 378 patients (97.4%) in the tremelimumab in combination with durvalumab group and 357 patients (95.5%) in the sorafenib group ||||||||||| || ||||| ||| ||||||| |||||| ||| ||||||| ||| |||||||| || ||||| || ||| |||||||| ||||||| || ||| |||||||||||| || ||||||||||| |||| |||||||||| ||||| ||| ||| ||||||| || ||| ||||||||| |||||. The proportion of patients discontinuing study treatment due to toxicity was slightly higher in the sorafenib group (63 patients [16.8%]) than the tremelimumab in combination with durvalumab group (53 patients [13.7%]). More patients experienced at least 1 SAE in the tremelimumab in combination with durvalumab group (157 patients [40.5%]) than the sorafenib group (111 patients [29.7%]). The number of deaths was similar in both groups ||| |||||||| |||||| |||||| || |||||||| |||||||| |||| |||||||| ||| || ||||||| ||||||| ||||| |||||| |||||| ||| ||||||||| || ||||| ||| |||||| || ||||||||||||||| || ||||||||||| |||| |||||||||| ||||| |||| |||||||| |||||||| |||||| ||| |||||||| ||||||||| ||| ||| |||||| || |||||||| ||||||||| |||||||| ||||||||||||||| ||| |||||||||| |||||| || ||| |||||||||||| ||||||||||||| ||| |||||||||| ||||| |||||| |||||| |||| Immune-mediated AEs, which are expected with immunotherapies, were reported 4 times more frequently in patients in the tremelimumab in combination with durvalumab group than in the sorafenib group (139 patients [35.8%] versus 30 patients [8%], respectively). ||||| |||| ||||||||| || ||| |||||||||||| || ||||||||||| |||| |||||||||| ||||| |||| |||| ||| || ||||| ||||||||||||| |||||||| ||||||| |||||||||||| ||| |||||||||| ||||||| ||| || ||||| || ||| ||||||||| |||||| Hemorrhage SMQ AEs occurred in 44 patients (11.3%) and ||| || ||||||| || ||| |||||||||||| || ||||||||||| |||| |||||||||| ||||| and 56 patients (15%) ||| ||| || ||||||| || ||| ||||||||| |||||| The clinical experts noted no new safety signals with tremelimumab in combination with durvalumab and added that the AEs appeared to be manageable in both groups.
Conclusion
One randomized, open-label, sponsor-blind, multicentre phase III trial provided evidence regarding the efficacy and safety of tremelimumab in combination with durvalumab compared with sorafenib in patients with unresectable HCC. Compared to sorafenib, treatment with tremelimumab in combination with durvalumab (tremelimumab 300 mg as a single priming dose in combination with 1,500 mg at day 1 of cycle 1, followed by durvalumab 1,500 mg as a single drug every 4 weeks) showed a statistically significant OS benefit. The absolute difference in median OS in patients with unresectable HCC between treatment groups (approximately 3 months) was considered clinically meaningful by the clinical experts consulted by CADTH. Because HRQoL analyses were limited by high rates of missing data, changes over time could not be interpreted. The clinical experts noted that, although sorafenib was the standard of care at the time the trial was conducted, it is now considered outdated. No definitive conclusions could be drawn from the ITCs submitted by the sponsor comparing the efficacy and safety of tremelimumab in combination with durvalumab to contemporary first-line therapies (i.e., atezolizumab in combination with bevacizumab and lenvatinib) due to methodological limitations and imprecision in the effect estimates for some outcomes. Given the lack of robust comparative data between tremelimumab in combination with durvalumab and other first-line therapies in the first-line setting (atezolizumab in combination with bevacizumab or lenvatinib), the clinical experts consulted could not draw firm conclusions about place in therapy. They noted that tremelimumab in combination with durvalumab would be suitable in patients with a higher risk of bleeding who would not be eligible for atezolizumab in combination with bevacizumab as tremelimumab in combination with durvalumab showed no increase in liver toxicity or the risk of bleeding in the HIMALAYA trial. The clinical experts would recommend funding this combination in the first-line treatment of appropriate patients with unresectable HCC as an alternative to the current options, which include lenvatinib, sorafenib, and atezolizumab in combination with bevacizumab. The safety profile of tremelimumab in combination with durvalumab in this study was consistent with the known safety profile of other immuno-oncology checkpoint inhibitors, and no additional safety signals were identified with tremelimumab in combination with durvalumab therapy.
References
1.PrImjudo® (tremelimumab): 20 mg/mL concentrate for solution for infusion [product monograph]. Mississauga (ON): AstraZeneca Canada Inc.; 2022 Dec 6.
2.Canadian Cancer Society’s Advisory Committee on Cancer Statistics. Canadian Cancer Statistics 2013. Toronto (ON): Canadian Cancer Society; 2013.
3.Hanumanthappa N, Cho BH, McKay A, et al. Epidemiology, clinical treatment patterns, and survival of hepatocellular carcinoma in Manitoba. Can Liver J. 2020;3(2):194-202. PubMed
4.Brenner DR, Poirier A, Woods RR, et al. Projected estimates of cancer in Canada in 2022. CMAJ. 2022;194(17):E601-E607. PubMed
5.Yao C, Billette JM. Short-term cancer prevalence in Canada, 2018. Health Rep. 2022;33(3):15-21. PubMed
6.EASL Clinical Practice Guidelines: management of hepatocellular carcinoma. J Hepatol. 2018;69(1):182-236. PubMed
7.Vogel A, Meyer T, Sapisochin G, Salem R, Saborowski A. Hepatocellular carcinoma. The Lancet. 2022;400(10360):1345-1362. PubMed
8.McGlynn KA, Petrick JL, El-Serag HB. Epidemiology of hepatocellular carcinoma. Hepatology. 2021;73(Suppl 1):4-13. PubMed
9.McGlynn KA, Petrick JL, London WT. Global epidemiology of hepatocellular carcinoma: an emphasis on demographic and regional variability. Clin Liver Dis. 2015;19(2):223-238. PubMed
10.Tinkle CL, Haas-Kogan D. Hepatocellular carcinoma: natural history, current management, and emerging tools. Biologics. 2012;6:207-219. PubMed
11.Hepatobiliary Cancers. Version 4.2022. NCCN Clinical Practice Guidelines in Oncology. Plymouth Meeting (PA): National Comprehensive Cancer Network; 2022 Dec 9.
12.Vogel A, Cervantes A, Chau I, et al. Hepatocellular carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2018;29(Suppl 4):iv238-iv255. PubMed
13.Reig M, Forner A, Rimola J, et al. BCLC strategy for prognosis prediction and treatment recommendation: The 2022 update. J Hepatol. 2022;76(3):681-693. PubMed
14.BC Cancer advanced hepatocellular carcinoma funding algorithm. Vancouver (BC): B.C. Cancer; [date unknown].
15.Provisional funding algorithm for unresectable hepatocellular carcinoma. CADTH drug implementation advice. Ottawa (ON): CADTH; 2021: https://www.cadth.ca/sites/default/files/pdf/ph0003-000_hcc-report-final.pdf. Accessed 2023 Jan 6.
16.FDA approves tremelimumab in combination with durvalumab for unresectable hepatocellular carcinoma. Silver Spring (MD): U.S. Food & Drug Administration; 2022: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-tremelimumab-combination-durvalumab-unresectable-hepatocellular-carcinoma. Accessed 2022 Jan 25.
17.Clinical Study Report: D419CC00002. A randomized, open-label, multi-center phase III study of durvalumab and tremelimumab as first-line treatment in patients with advanced hepatocellular carcinoma (HIMALAYA) [internal sponsor's report]. Gaithersburg (MD): AstraZeneca; 2022 Jan 19.
18.Vogel A, Qin S, Kudo M, et al. Lenvatinib versus sorafenib for first-line treatment of unresectable hepatocellular carcinoma: patient-reported outcomes from a randomised, open-label, non-inferiority, phase III trial. Lancet Gastroenterol Hepatol. 2021;6(8):649-658. PubMed
19.Li D, Toh HC, Merle P, et al. Atezolizumab plus bevacizumab versus sorafenib for unresectable hepatocellular carcinoma: results from older adults enrolled in the IMbrave150 randomized clinical trial. Liver Cancer. 2022;11(6):558-571. PubMed
20.Roy A. Updated efficacy and safety data from IMbrave150: atezolizumab plus bevacizumab vs. sorafenib for unresectable hepatocellular carcinoma. J Clin Exp Hepatol. 2022;12(6):1575-1576. PubMed
21.Mu X, Español-Suñer R, Mederacke I, et al. Hepatocellular carcinoma originates from hepatocytes and not from the progenitor/biliary compartment. J Clin Invest. 2015;125(10):3891-3903. PubMed
22.Bialecki ES, Di Bisceglie AM. Diagnosis of hepatocellular carcinoma. HPB (Oxford). 2005;7(1):26-34. PubMed
23.Bruix J, Sherman M. Management of hepatocellular carcinoma. Hepatology. 2005;42(5):1208-1236. PubMed
24.Sherman M, Burak K, Maroun J, et al. Multidisciplinary Canadian consensus recommendations for the management and treatment of hepatocellular carcinoma. Curr Oncol. 2011;18(5):228-240. PubMed
25.Pons F, Varela M, Llovet JM. Staging systems in hepatocellular carcinoma. HPB (Oxford). 2005;7(1):35-41. PubMed
26.Vogel A, Cervantes A, Chau I, et al. Hepatocellular carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2019;30(5):871-873. PubMed
27.Stuart K. Systemic treatment for advanced hepatocellular carcinoma. In: Post TW, ed. UpToDate. Waltham (MA): UpToDate; 2022: http://www.uptodate.com. Accessed 2023 Jan 04.
28.Cheng AL, Qin S, Ikeda M, et al. Updated efficacy and safety data from IMbrave150: Atezolizumab plus bevacizumab vs. sorafenib for unresectable hepatocellular carcinoma. J Hepatol. 2022;76(4):862-873. PubMed
29.Kudo M, Finn RS, Qin S, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase III non-inferiority trial. The Lancet. 2018;391(10126):1163-1173. PubMed
30.Llovet JM, Ricci S, Mazzaferro V, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359(4):378-390. PubMed
31.PrImfinzi® (durvalumab): 50 mg/mL concentrate for solution for infusion [product monograph]. Mississauga (ON): AstraZeneca Canada Inc.; 2022 Sep 28.
32.Akbari O, Stock P, Singh AK, et al. PD-L1 and PD-L2 modulate airway inflammation and iNKT-cell-dependent airway hyperreactivity in opposing directions. Mucosal Immunol. 2010;3(1):81-91. PubMed
33.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252-264. PubMed
34.PrNexavar®(sorafenib): 200 mg tablets. [product monograph]. Mississauga (ON): Bayer Inc.; 2022 Sep 27: https://www.bayer.com/sites/default/files/2020-11/nexavar-pm-en.pdf. Accessed 2023 Jan 25.
35.PrTecentriq® (atezolizumab): 60 mg/mL, 1200 mg/20 mL, and 840 mg/14 mL single use vials, concentrate for solution for infusion [product monograph]. Mississauga (ON): Hoffmann-La Roche Ltd.; 2022 Dec 21.
36.PrLenvima®(lenvatinib): capsules 4mg and 10mg capsules [product monograph]. Mississauga (ON): Eisai Ltd.; 2018 Dec 19.
37.Abou-Alfa GK, Lau G, Kudo M, et al. Tremelimumab plus durvalumab in unresectable hepatocellular carcinoma. NEJM Evidence. 2022;1(18).
38.Zhou M, Zhang C, Nie J, et al. Response evaluation and survival prediction following PD‐1 inhibitor in patients with advanced hepatocellular carcinoma: Comparison of the RECIST 1.1, iRECIST, and mRECIST criteria. Front Oncol. 2021:5198. PubMed
39.Fayers P, Bottomley A. Quality of life research within the EORTC-the EORTC QLQ-C30. European Organisation for Research and Treatment of Cancer. Eur J Cancer. 2002;38(Suppl 4):S125-133. PubMed
40.Li L, Mo FK, Chan SL, et al. Prognostic values of EORTC QLQ-C30 and QLQ-HCC18 index-scores in patients with hepatocellular carcinoma - clinical application of health-related quality-of-life data. BMC Cancer. 2017;17(1):8. PubMed
41.Blazeby JM, Currie E, Zee BCY, Chie W-C, Poon RT, Garden OJ. Development of a questionnaire module to supplement the EORTC QLQ-C30 to assess quality of life in patients with hepatocellular carcinoma, the EORTC QLQ-HCC18. Eur J Cancer. 2004;40(16):2439-2444. PubMed
42.Chie W-C, Blazeby JM, Hsiao C-F, et al. International cross-cultural field validation of an European Organisation for Research and Treatment of Cancer questionnaire module for patients with primary liver cancer, the European Organisation for Research and Treatment of Cancer quality-of-life questionnaire HCC18. Hepatology. 2012;55(4):1122-1129. PubMed
43.Osoba D, Rodrigues G, Myles J, Zee B, Pater J. Interpreting the significance of changes in health-related quality-of-life scores. J Clin Oncol. 1998;16(1):139-144. PubMed
44.Fayers PM, Aaronson NK, Bjordal K, et al. The EORTC QLQ-C30 Scoring Manual (3rd Edition). Brussels (BE): European Organisation for Research and Treatment of Cancer; 2001: https://www.eortc.org/app/uploads/sites/2/2018/02/SCmanual.pdf. Accessed 2022 Nov 28.
45.Aaronson NK, Ahmedzai S, Bergman B, et al. The European Organisation for Research and Treatment of Cancer QLQ-C30: a quality-of-life instrument for use in international clinical trials in oncology. JNCI. 1993;85(5):365-376. PubMed
46.Kaasa S, Bjordal K, Aaronson N, et al. The EORTC Core Quality of Life questionnaire (QLQ-C30): validity and reliability when analysed with patients treated with palliative radiotherapy. Eur J Cancer. 1995;31(13):2260-2263. PubMed
47.Musoro JZ, Coens C, Fiteni F, et al. Minimally important differences for interpreting EORTC QLQ-C30 scores in patients with advanced breast cancer. JNCI Cancer Spectr. 2019;3(3):pkz037. PubMed
48.Summary of Clinical Safety: durvalumab in combination with tremelimumab for the treatment of patients with unresectable hepatocellular carcinoma. Common technical document section 2.7.4. In: Drug Reimbursement Review sponsor submission: Imjudo (tremelimumab), 20 mg/mL, concentrate for solution for intravenous infusion in combination with Imfinzi (durvalumab), 50 mg/mL, concentrate for solution for intravenous infusion. Mississauga (ON): AstraZeneca Canada Inc.; 2022 Dec 15.
49.Systematic literature review in the first-line treatment of advanced unresectable hepatocellular carcinoma [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Imjudo (tremelimumab), 20 mg/mL, concentrate for solution for intravenous infusion in combination with Imfinzi (durvalumab), 50 mg/mL, concentrate for solution for intravenous infusion. Mississauga (ON): AstraZeneca Canada Inc.; 2022 Dec 15.
50.Indirect treatment comparison in the first-line treatment of advanced unresectable hepatocellular carcinoma: ITC report, version 2.0 [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Imjudo (tremelimumab), 20 mg/mL, concentrate for solution for intravenous infusion in combination with Imfinzi (durvalumab), 50 mg/mL, concentrate for solution for intravenous infusion. Mississauga (ON): AstraZeneca Canada Inc.; 2022 Dec 15.
51.Indirect treatment comparison in the first-line treatment of advanced unresectable hepatocellular carcinoma: ITC report - lenvatinib focus [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Imjudo (tremelimumab), 20 mg/mL, concentrate for solution for intravenous infusion in combination with Imfinzi (durvalumab), 50 mg/mL, concentrate for solution for intravenous infusion. Mississauga (ON): AstraZeneca Canada Inc.; 2022 Dec 15.
52.Systematic Reviews: CRD’s guidance for undertaking reviews in health care. York (UK): Centre for Reviews and Dissemination (CRD) University of York; 2009: https://www.york.ac.uk/media/crd/Systematic_Reviews.pdf. Accessed 2023 Jan 6.
53.Finn RS, Qin S, Ikeda M, et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med. 2020;382(20):1894-1905. PubMed
54.Finn RS, Qin S, Ikeda M, et al. IMbrave150: updated overall survival (OS) data from a global, randomized, open-label phase III study of atezolizumab (atezo) + bevacizumab (bev) versus sorafenib (sor) in patients (pts) with unresectable hepatocellular carcinoma (HCC) [abstract]. J Clin Oncol. 2021;39(3 Suppl):267-267.
55.Salem R, Li D, Sommer N, et al. Characterization of response to atezolizumab + bevacizumab versus sorafenib for hepatocellular carcinoma: results from the IMbrave150 trial. Cancer Med. 2021;10(16):5437-5447. PubMed
56.Qin S, Ren Z, Feng YH, et al. Atezolizumab plus bevacizumab versus sorafenib in the chinese subpopulation with unresectable hepatocellular carcinoma: phase III randomized, open-label IMbrave150 study. Liver Cancer. 2021;10(4):296-308. PubMed
57.Finn RS, Qin S, Ikeda M, et al. IMbrave150: Updated efficacy and safety by risk status in patients (pts) receiving atezolizumab (atezo) + bevacizumab (bev) vs sorafenib (sor) as first-line treatment for unresectable hepatocellular carcinoma (HCC) [abstract]. Cancer Res. 2021;81(13 Suppl):abstract CT009.
58.Yau T, Park JW, Finn RS, et al. LBA38_PR - CheckMate 459: a randomized, multi-center phase III study of nivolumab (NIVO) vs sorafenib (SOR) as first-line (1L) treatment in patients (pts) with advanced hepatocellular carcinoma (aHCC) [abstract]. Ann Oncol. 2019;30:v874-v875.
59.Bristol-Myers Squibb. NCT02576509: An investigational immuno-therapy study of nivolumab compared to sorafenib as a first treatment in patients with advanced hepatocellular carcinoma. ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine; 2022: https://clinicaltrials.gov/study/NCT02576509. Accessed 2022 Nov 1.
60.Yau T, Park J-W, Finn RS, et al. Nivolumab versus sorafenib in advanced hepatocellular carcinoma (CheckMate 459): a randomised, multicentre, open-label, phase III trial. Lancet Oncol. 2022;23(1):77-90. PubMed
61.Phillippo D, Ades AE, Dias S, Palmer S, Abrams KR, Welton NJ. NICE DSU Technical Support Document 18: methods for population-adjusted indirect comparisons in submissions to NICE. Sheffield (UK): University of Scheffield; 2016.
62.Kuitunen I, Ponkilainen VT, Uimonen MM, Eskelinen A, Reito A. Testing the proportional hazards assumption in cox regression and dealing with possible non-proportionality in total joint arthroplasty research: methodological perspectives and review. BMC Musculoskelet Disord. 2021;22(1):489. PubMed
63.Schoenfeld D. Partial residuals for the proportional hazards regression model. Biometrika. 1982;69(1):239-241.
64.Narang J, Bajpai S, Jarecha R, Sharma M, Bohnsack O. Comparison of response using mRECIST versus RECIST 1.1 criteria in advanced hepatocellular carcinoma: a retrospective analysis of multicenter clinical trials [abstract]. J Clin Oncol. 2020;38(15 Suppl):4595.
65.Yu H, Bai Y, Xie X, Feng Y, Yang Y, Zhu Q. RECIST 1.1 versus mRECIST for assessment of tumour response to molecular targeted therapies and disease outcomes in patients with hepatocellular carcinoma: a systematic review and meta-analysis. BMJ Open. 2022;12(6):e052294. PubMed
66.Fulgenzi CAM, D'Alessio A, Airoldi C, et al. Comparative efficacy of novel combination strategies for unresectable hepatocellular carcinoma: a network metanalysis of phase III trials. Eur J Cancer. 2022;174:57-67. PubMed
67.Higgins JPT, Altman DG, Gøtzsche PC, et al. The Cochrane Collaboration’s tool for assessing risk of bias in randomised trials. BMJ. 2011;343:d5928. PubMed
Pharmacoeconomic Review
Abbreviations
AE
adverse event
BCLC
Barcelona Clinic Liver Cancer
BIA
budget impact analysis
EQ-5D-5L
5-Level EQ-5D
HCC
hepatocellular carcinoma
HR
hazard ratio
ICER
incremental cost-effectiveness ratio
MAIC
matching adjusted indirect comparison
NICE
National Institute for Health and Care Excellence
OS
overall survival
PFS
progression-free survival
QALY
quality-adjusted life-year
STRIDE
single-dose tremelimumab in combination with regular-interval durvalumab
TTD
time to treatment discontinuation
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Tremelimumab (Imjudo) in combination with durvalumab (Imfinzi) for IV infusion |
Submitted price |
|
Indication | Proposed: for the first-line treatment of patients with unresectable HCC who require systemic therapy |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | August 31, 2023 |
Reimbursement request | As per indication |
Sponsor | AstraZeneca Canada Inc. |
Submission history | CADTH has previously reviewed durvalumab as monotherapy or in combination with other products but has never reviewed tremelimumab Indication: Durvalumab in combination with gemcitabine-based chemotherapy for the treatment of patients with locally advanced or metastatic biliary tract cancer Recommendation date: February 3, 2023 Recommendation: Reimburse with clinical criteria and/or conditions Indication: Durvalumab in combination with etoposide and either carboplatin or cisplatin for the first-line treatment of adult patients with extensive-stage small cell lung cancer Recommendation date: July 27, 2021 Recommendation: Reimburse with clinical criteria and/or conditions Indication: Durvalumab monotherapy for the treatment of patients with locally advanced, unresectable non–small cell lung cancer following curative-intent platinum-based chemoradiation therapy, for up to a maximum of 12 months Recommendation date: May 3, 2019 Recommendation: Reimburse with clinical criteria and/or conditions |
HCC = hepatocellular carcinoma; NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Partitioned survival model |
Target population | Adult patients with unresectable HCC who have not received prior systemic therapy (i.e., first-line treatment) |
Treatment | Single-dose tremelimumab in combination with regular-interval durvalumab |
Comparators |
|
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, life-years |
Time horizon | Lifetime (15 years) |
Key data source |
|
Submitted resultsa |
|
Key limitations |
|
CADTH reanalysis results |
|
HCC = hepatocellular carcinoma; ICER = incremental cost-effectiveness ratio; MAIC = matching adjusted indirect comparison; QALY = quality-adjusted life-year; STRIDE = single-dose tremelimumab in combination with regular-interval durvalumab; vs. = versus.
aThese results were estimated using rounded values from the sponsor’s technical report. Appendix 3 provides additional details of the unrounded values.
Conclusions
Based on the CADTH clinical review and HIMALAYA trial data, treatment with single-dose tremelimumab in combination with regular-interval durvalumab (STRIDE) in the first line for patients with unresectable hepatocellular carcinoma (HCC) resulted in a statistically significant improvement in overall survival (OS) compared to sorafenib. The difference in OS found in the trial (approximately 3 months) was considered clinically meaningful by the clinical experts consulted by CADTH. At the time of trial design, sorafenib was an appropriate comparator; however, sorafenib is no longer the most common standard of care therapy and has been replaced by therapies that include lenvatinib and atezolizumab plus bevacizumab. Because there is no direct comparative evidence for STRIDE versus atezolizumab plus bevacizumab or lenvatinib, the sponsor conducted matching adjusted indirect comparisons (MAICs) to estimate the comparative clinical efficacy for these comparators. However, the CADTH clinical review reported that no superiority conclusions could be drawn due to methodological limitations and imprecision in the effect estimates.
CADTH identified several limitations in the sponsor’s pharmacoeconomic analysis that have notable implications on the cost-effectiveness of STRIDE. For the CADTH base-case analysis, CADTH used the clinical efficacy data for sorafenib from the HIMALAYA trial, applied health-state utilities rather than treatment-specific utilities, and assumed that treatment was discontinued at the time of disease progression for all comparators. Results of the sponsor’s and CADTH’s base cases were similar: STRIDE is not cost-effective at a willingness-to-pay threshold of $50,000 per quality-adjusted life-year (QALY) gained. In the CADTH base case, the incremental cost-effectiveness ratio (ICER) of STRIDE versus sorafenib was $265,036 (incremental costs: $95,359; incremental QALYs: 0.36) per QALY gained, with a 0% probability of being cost-effective at a threshold of $50,000 per QALY gained. A price reduction of approximately 50% is required for STRIDE to be cost-effective at this threshold compared to sorafenib. Given the limitations in the comparative clinical efficacy data for the more commonly used comparators, a high degree of uncertainty remains in estimating the cost-effectiveness of STRIDE for the sponsor’s requested reimbursement population compared to lenvatinib and to atezolizumab plus bevacizumab; these comparisons were explored through scenario analyses only. In a scenario analysis assuming equivalent efficacy of atezolizumab and bevacizumab versus STRIDE, CADTH found that STRIDE was more costly and provided no additional QALYs compared to atezolizumab plus bevacizumab. CADTH also investigated the time at which the cumulative cost of STRIDE and atezolizumab plus bevacizumab would be equal (given the higher upfront cost but lower maintenance cost for STRIDE). After approximately 60 weeks (1.15 years) of continuous treatment, the cumulative costs of STRIDE and atezolizumab plus bevacizumab are equal. The scenario assuming the same efficacy for lenvatinib and sorafenib resulted in a higher ICER for STRIDE versus lenvatinib ($262,393 per QALY gained) compared to when the hazard ratios (HRs) from the MAICS were applied to lenvatinib.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process.
Patient input was jointly received from the Colorectal Cancer Resource & Action Network, Canadian Cancer Survivor Network, Canadian Liver Foundation, and Gastrointestinal Society. Patient input was gathered through interviews with 2 patients living in Canada who have advanced HCC, along with a review of qualitative scientific literature and patient forums on patient-reported outcomes. One patient was treated with STRIDE through a clinical trial program and a second patient was effectively treated with surgical resection and radiofrequency ablations. Systemic therapies such as atezolizumab plus bevacizumab, lenvatinib, sorafenib, regorafenib, and cabozantinib were the most commonly reported treatments for patients with advanced HCC. These treatments were associated with several adverse events (AEs) that significantly affect quality of life, including diarrhea, fatigue, hand-foot syndrome, hypertension, and other complications. Patient groups indicated that the clinical benefits of atezolizumab plus bevacizumab were superior to those of sorafenib; however, the need for a screening endoscopy 6 months before treatment initiation to mitigate the risk of bleeding associated with bevacizumab could delay treatment. Patient groups indicated a need for a treatment that would extend progression-free survival (PFS) and OS and improve quality of life. The patient with experience with STRIDE indicated that it provided another treatment option with manageable side effects and improved quality of life. Additionally, STRIDE reduces the frequency of administration visits and is not associated with the treatment-induced hypertension associated with bevacizumab.
Clinician group input was received from the Canadian Gastrointestinal Oncology Evidence Network and Gastrointestinal Cancer Drug Advisory Committee. Clinician groups noted that the current therapies for patients with unresectable HCC (i.e., lenvatinib, sorafenib, and atezolizumab plus bevacizumab) have shown a clinically meaningful improvement in OS but are associated with bleeding risks. Registered clinicians indicated that lenvatinib and sorafenib have been considered for patients for whom immunotherapy is contraindicated or who are at increased risk of bleeding. They noted that STRIDE is the first immunotherapy treatment targeted at nonvascular endothelial growth factor for patients with HCC, and is therefore associated with a lower risk of bleeding. There was a lack of head-to-head comparisons between STRIDE and atezolizumab plus bevacizumab, but clinician groups consulted for this review suggested a patient preference for STRIDE due to its advantages over the first-line therapy of atezolizumab plus bevacizumab in terms of dosage schedule and chair time. They mentioned that the improved response rates of STRIDE can lead to downsizing of disease and newer opportunities for local regional therapies to control the disease process (such as stereotactic radiation, embolization, ablation, resection, or transplant).
Drug plan input noted that sorafenib, along with alternative first-line therapies (atezolizumab plus bevacizumab and lenvatinib), are funded in most judications as first-line therapy for patients with HCC. The drug plans emphasized that patients enrolled in the HIMALAYA trial were required to have histologic confirmation, which may present practical challenge because HCC cannot always be confirmed histologically. Drug plans anticipated that durvalumab vials would sometimes be shared because other indications were already funded at weight-based dosing, but this may not be the case with tremelimumab given that it comes in a single full-dose vial. Questions were raised regarding STRIDE’s relative efficacy and safety compared to lenvatinib or atezolizumab plus bevacizumab, assessing discontinuation and re-treatment, potential implementation of weight-based dosing, and time-limited switching from atezolizumab-bevacizumab.
Several of these concerns were addressed in the sponsor’s model:
The sponsor’s model included relevant comparators (i.e., atezolizumab plus bevacizumab, lenvatinib, sorafenib).
PFS, OS, and quality of life were incorporated into the model.
Palliative treatment (i.e., terminal care) costs were included as a 1-time cost for patients who entered the death health state.
CADTH was unable to address the following concerns raised in stakeholder input:
Some important known AEs, including diarrhea and bleeding, were not included in the model.
Economic Review
The current review is for tremelimumab (Imjudo) in combination with durvalumab (Imfinzi) for the first-line treatment of adult patients with unresectable HCC who require systemic therapy.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis to assess the cost-effectiveness of STRIDE for patients with unresectable HCC compared to sorafenib, lenvatinib, and atezolizumab plus bevacizumab.1
Tremelimumab is available as a 20 mg/mL concentrated solution in 15 mL single-dose vials (total of 300 mg per vial) for IV infusion while durvalumab is available as a 50 mg/mL solution in single-dose vials of 2.4 mL (total of 120 mg per vial) and 10 mL (a total of 500 mg per vial) IV infusions. The recommended dosage of tremelimumab is a single infusion of 300 mg in combination with an initial infusion of 1,500 mg of durvalumab followed by 1,500 mg of durvalumab monotherapy every 4 weeks until disease progression or unacceptable toxicity.2,3 Patients with a body weight of 30 kg or less must receive weight-based dosing of tremelimumab 4 mg/kg as a single priming dose in combination with durvalumab 20 mg/kg followed by durvalumab 20 mg/kg monotherapy on the same schedule.1 The drug acquisition cost for a single dose of tremelimumab was $34,320. At the submitted prices of $939 per 120 mg and $3,911 per 500 mg vial, the cost of durvalumab for patients weighing more than 30 kg is $11,733 per 4-week cycle. The total cost of the proposed regimen (STRIDE) was $46,053 per 28 days in the initial cycle and $11,733 per 28 days thereafter. The sponsor calculated the cost of atezolizumab plus bevacizumab, lenvatinib, and sorafenib to be $14,124, $1,922 to 2,854, and $5,205 per 28 days, respectively.
Outcomes of the model included QALYs, and life-years over a 15-year lifetime time horizon (with a cycle length of 1 week) from the Canadian public health care payer perspective. Both costs and outcomes were discounted at 1.5% per annum.1
Model Structure
The sponsor submitted a partitioned survival model consisting of 3 health states: progression-free, progressed, and death (Figure 1).1 The proportions of patients who were progression-free or experienced disease progression or death at any time of the model’s time horizon were derived from survival curves using an area-under-the-curve approach. All patients entered the model in the progression-free state and patients were assigned to STRIDE or a comparator treatment (sorafenib, lenvatinib, or atezolizumab plus bevacizumab).1 Patients could then transition to the death state or postprogression state, where they would remain until they transitioned to the death state. The proportion of patients in the progression-free state was estimated based on PFS curves. The proportion of patients in the progressed disease state was calculated as the proportion of patients alive (based on the OS curve) minus the proportion of patients alive and progression-free. PFS curves were capped by the OS curve to ensure that PFS would not exceed OS.
Model Inputs
The model’s baseline population characteristics used to inform the model were characterized according to the phase III HIMALAYA trial (n = 782), with a mean age of |||||||| years and weight of |||||||| kg, and 83.7% were males.1
Key clinical efficacy and safety inputs for STRIDE were based on the results of the HIMALAYA trial. The data for OS, PFS, and time to treatment discontinuation (TTD) from the trial period were used to fit parametric survival curves and odds knot spline models to extrapolate observed trial data (maximum follow-up |||||| ||||||||||) over the 15-year modelled time horizon. OS and PFS for lenvatinib and atezolizumab plus bevacizumab were derived by applying HRs from MAICs relative to STRIDE, performed on the respective trials, to the PFS and OS curves for STRIDE. This method assumed no difference in the distribution of effect-modifying variables across trials. Similarly, sorafenib efficacy was derived from the reweighted HIMALAYA population analyzed in the lenvatinib MAIC. The sponsor also submitted a scenario analysis in which HIMALAYA trial data were used to inform sorafenib efficacy. The TTD curves for STRIDE and sorafenib were determined using independent extrapolation of Kaplan-Meier curves from the HIMALAYA trial. The TTD curves for atezolizumab plus bevacizumab and lenvatinib were assumed to be equal to their respective PFS curves. The survival extrapolation models selected in the sponsor’s base case were chosen based on the Akaike information criterion, Bayesian information criterion, visual inspection, and clinical plausibility. The Weibull model was chosen to extrapolate OS for STRIDE (Figure 2 and Figure 3), while 3 odds knot spline models were used to extrapolate PFS (Figure 4 and Figure 5). The OS and PFS curves for the comparators were extrapolated using HRs of each drug relative to STRIDE, based on their respective MAICs. The OS and PFS HR for atezolizumab plus bevacizumab versus STRIDE was |||||||||| and ||||||||||, respectively. The OS and PFS HRs for lenvatinib versus STRIDE were |||||||||| and ||||||||||, respectively. The OS and PFS HRs for sorafenib versus STRIDE were |||||||| and ||||||||||, respectively. In addition to the disease-specific mortality reported in the HIMALAYA trial, the model included age-specific and sex-weighted mortality consistent with that of the general population,4 and this was used to ensure the hazard of death predicted in the extrapolations was not lower than that of the general population.
The AE rates for both for STRIDE and sorafenib were informed by the HIMALAYA trial, while AE rates for other treatments were derived from the literature and National Institute for Health and Care Excellence (NICE) technology appraisals.5,6
In the sponsor’s base case, treatment-specific utility values for the progression-free and progressed disease states were estimated using the 5-Level EQ-5D (EQ-5D-5L) data from the HIMALAYA trial. Utilities corresponding to the EQ-5D-5L tool were calculated using Canadian-specific utility weights for EQ-5D-5L.7 Lenvatinib was assumed to have the same utility as sorafenib, and the utility value of atezolizumab plus bevacizumab was also assumed to equal that of STRIDE. The model accounted for the AE disutility value of −0.11 for all AEs (grade 3 or 4) based on the literature, and the value was converted to a weekly decrement applied while on treatment.8
The model included different types of costs from the public health care payer perspective, including drug acquisition and administration costs (both first-line and second-line therapies), health care resource use costs associated with disease management by treatment and/or health state, and the management of AEs. Drug acquisition costs of tremelimumab and durvalumab were based on the sponsor’s submitted prices, while the costs of comparators were obtained from the IQVIA DeltaPA database.9 Dosing of the STRIDE regimen was obtained from the HIMALAYA trial and the dosages for the comparators were based on their respective monographs.10-12 Lenvatinib and bevacizumab dosing is dependent on patient weight; doses of 12 mg lenvatinib and 1,050 mg bevacizumab were used in the model based on the mean weight in the HIMALAYA trial. The model also accounted for the bleeding risk of bevacizumab,13 and the sponsor assumed that 20% of patients on atezolizumab plus bevacizumab would delay bevacizumab treatment by 6 weeks to monitor bleeding. To model the efficacy of atezolizumab monotherapy during the delayed period, HRs of 1.28 and 1.82 for OS and PFS, respectively, were applied to the data for atezolizumab plus bevacizumab. The model considered a 100% relative dose intensity for all drugs and no vial sharing. Administration costs were obtained from the Ontario Schedule of Benefits.14 The proportion of patients receiving second-line treatment and the duration of treatment for each drug were informed by the HIMALAYA trial. Due to a lack of data, lenvatinib and sorafenib were assumed to have a similar proportion of patients requiring second-line treatment (||||%) and distribution of therapies (75% on cabozantinib and 25% on regorafenib). Likewise, equal proportions of patients receiving second-line treatment (||||||||%) and treatment distribution (95% on lenvatinib and 5% on sorafenib) were assumed between STRIDE and atezolizumab plus bevacizumab. Third-line therapies were not included. All patients accrued disease-management costs, including physician visits, hospitalization, and monitoring costs, in the model, and all patients were assigned a terminal care cost of $33,268 upon their deaths. The cost of serious or life-threatening (grade 3 or 4) AEs was obtained from the literature15 and applied weekly while on treatment. All costs were inflated to 2022 Canadian dollars.
Summary of Sponsor’s Economic Evaluation Results
The sponsor presented a probabilistic base-case analysis based on 1,500 iterations for the base-case and scenario analyses. Deterministic results were aligned with the probabilistic results. The probabilistic findings follow.
Base-Case Results
The sponsor’s probabilistic base-case analysis found that the ICER for STRIDE was $251,382 per QALY gained (incremental costs: $143,288; incremental QALYs: 0.57) and $219,363 per QALY gained (incremental costs: $129,424; incremental QALYs: 0.59), compared to lenvatinib and sorafenib, respectively. When compared with atezolizumab plus bevacizumab, STRIDE was associated with an incremental cost savings of $106,572 and 0.18 fewer incremental QALYs, resulting in an ICER of $592,067 per QALY gained (Table 3). First-line treatment costs accounted for 70%, 78%, 35%, and 30% in STRIDE, atezolizumab plus bevacizumab, sorafenib, and lenvatinib, respectively. Approximately 22% of the incremental QALYs accrued in the extrapolated period (from |||||| to 15 years).
Table 3: Summary of the Sponsor’s Pairwise Economic Evaluation Resultsa,b
Drug | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER vs. STRIDE ($ per QALY) |
|---|---|---|---|---|---|
STRIDE vs. sorafenib | |||||
Sorafenib | 146,253 | Reference | 1.36 | Reference | Reference |
STRIDE | 275,677 | 129,424 | 1.95 | 0.59 | 219,363 |
STRIDE vs. lenvatinib | |||||
Lenvatinib | 132,389 | Reference | 1.38 | Reference | Reference |
STRIDE | 275,677 | 143,288 | 1.95 | 0.57 | 251,382 |
STRIDE vs. atezolizumab plus bevacizumab | |||||
STRIDE | 275,677 | Reference | 1.95 | Reference | Reference |
Atezolizumab plus bevacizumab | 382,249 | 106,572 | 2.13 | 0.18 | 592,067 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; STRIDE = single-dose tremelimumab in combination with regular-interval durvalumab; vs. = versus.
Note: The submitted analyses are based on publicly available prices of comparators and may not reflect confidential, negotiated prices.
aGiven that the sponsor’s comparative efficacy data were derived from separate MAICs, a sequential analysis is inappropriate, and therefore only pairwise probabilistic results are presented.
bThese results were estimated using rounded values from the sponsor’s technical report. Appendix 3 provides additional details of the unrounded values.
Source: Sponsor’s pharmacoeconomic submission.1
Sensitivity and Scenario Analysis Results
The sponsor conducted several scenario analyses, including using sorafenib efficacy data from the HIMALYA trial, allowing vial sharing without wastage, alternative PFS assumptions for atezolizumab plus bevacizumab, subsequent therapy assumptions, patient weight, and AE costs. Generally, the conclusions of the sponsor’s base case were not largely impacted in these scenario analyses. In the scenario analysis using HRs derived directly from the HIMALAYA trial for sorafenib, STRIDE had an ICER of $259,714 per QALY gained, which was higher than the ICER from the base-case analysis. Results were also influenced by the choice of HR applied to the PFS of atezolizumab plus bevacizumab in that the lower the HR (i.e., the better atezolizumab plus bevacizumab performed relative to STRIDE) the more favourable STRIDE was due to lower treatment costs.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis:
Use of sorafenib efficacy data from the lenvatinib MAIC was inappropriate: In the sponsor’s base-case analysis, sorafenib efficacy was informed from the reweighted HIMALAYA population in the lenvatinib MAIC, despite the availability of direct comparative evidence for sorafenib and STRIDE from the HIMALAYA trial. The sponsor defended this assumption by suggesting that results of the economic model lacked face validity with respect to the comparison of sorafenib versus lenvatinib when using the HIMALAYA data for sorafenib. Specifically, the sponsor suggested that lenvatinib should result in superior survival outcomes compared to sorafenib based on prior economic evaluations and CADTH reports.15,16 However, CADTH notes that the incremental survival differences in the sponsor’s model are small (less than 0.05 incremental life-years) and this in itself is not sufficient justification to use indirect evidence (an anchored MAIC) rather than direct, head-to-head, phase III evidence. Moreover, head-to-head evidence from the REFLECT trial suggests lenvatinib is noninferior to sorafenib, which was also considered in the CADTH pan-Canadian Oncology Drug Review Expert Review Committee’s evaluation of atezolizumab plus bevacizumab for treatment of HCC.17,18 Given that the direct comparative evidence from the HIMALAYA trials offers the highest quality data to inform the relative efficacy of sorafenib versus STRIDE, this is more appropriate for the economic evaluation.
The CADTH reanalysis utilized sorafenib efficacy data from the HIMALAYA trial for OS and PFS.
A scenario analysis was conducted assuming equivalent efficacy of sorafenib and lenvatinib.
The comparative efficacy of lenvatinib and atezolizumab plus bevacizumab versus STRIDE is uncertain: In the absence of head-to-head evidence for lenvatinib and atezolizumab plus bevacizumab against STRIDE, the sponsor used indirect treatment comparisons to inform the efficacy of these comparators in the submitted model. CADTH’s clinical review team reported that no definitive conclusions could be drawn from the MAICs submitted by the sponsor due to methodological limitations. CADTH’s clinical review report emphasized several key limitations associated with the sponsor’s MAICs, including missing treatment-effect modifiers, reduced effective sample sizes from the HIMALAYA trial, the inability to account for some sources of heterogeneity between the trials, and potential systemic differences between patients, given the time periods over which the trials were conducted compared to the HIMALAYA trial. Furthermore, imprecision in the results (i.e., wide confidence intervals) led to uncertainty in the direction of clinical efficacy. Given the use of HRs derived from these MAICs to inform clinical efficacy relative to STRIDE, substantial uncertainty exists in the pairwise comparisons of STRIDE against lenvatinib and atezolizumab plus bevacizumab.
Additionally, the use of separate MAICs to derive comparative efficacy between treatments precluded CADTH from conducting a sequential analysis, which assumes that the populations informing each comparator are homogenous. CADTH therefore only presented pairwise results for each comparator.
Given the inability to draw conclusions from the MAIC, and the uncertainty in the long term effects of all comparator regimens, CADTH assumed equivalent efficacy of atezolizumab plus bevacizumab versus STRIDE in its scenario analysis.
The long-term extrapolation of clinical efficacy of STRIDE is uncertain: The sponsor fit several survival curves to extrapolate the OS and PFS observed in the HIMALAYA trial from the end of the trial period (maximum follow-up = |||||| years) to the end of the 15-year time horizon (Figure 2). With regard to OS, the clinical experts consulted by CADTH for this review reported that the proportion of patients predicted to still be alive at 5 years and beyond in the model was implausibly high for STRIDE and the comparators. Experts noted that OS would likely not exceed 5 years for the vast majority of patients, regardless of treatment. In the sponsor’s model, relative differences in survival between 5 and 15 years were considered uncertain by the clinical experts, potentially biasing the incremental survival results. The clinical experts consulted by CADTH also agreed that the sponsor’s PFS results were overestimated for all comparators. Because the sponsor used HRs to estimate the OS and PFS for comparators relative to STRIDE, the uncertainty in the extrapolated period for STRIDE translates to uncertainty for the comparators as well. For example, the sponsor’s submitted model predicted that approximately 10% of patients taking atezolizumab plus bevacizumab would be progression-free at 5 years, which was deemed implausible by clinical experts.
CADTH was unable to address this limitation in reanalysis, but notes that the sponsor-selected survival curves for STRIDE were among the more conservative options (i.e., the sponsor-selected curves predicted lower OS and PFS rates over time compared with other curves fit to the data, as Figure 2 and Figure 4 show). CADTH did not make changes to the selected OS and PFS curves.
Use of treatment-specific health-state utility values is inappropriate: The sponsor applied treatment-specific utilities for patients who were on or off treatment, whether or not they had experienced disease progression. The sponsor applied a lower utility value to patients who received lenvatinib and sorafenib (|||||||| for on treatment and |||||||||| for off treatment) and a higher utility value (|||||||| for on treatment and |||||||||| for off treatment) for patients who received STRIDE and atezolizumab plus bevacizumab. The use of treatment-specific utility values is contradictory to CADTH’s recommendation that utilities should reflect the health states in the economic model.19 The incorporation of AE-associated disutilities in the sponsor’s base case (which is the appropriate approach) further limits the justification for applying treatment-specific utilities.
Additionally, the clinical experts consulted by CADTH agreed that the utility values used in the sponsor’s submission did not meet face validity. Based on the Canadian utility norms from the 2013 to 2014 Canadian Community Health Survey, the reported utility for patients ages 60 to 70 years is 0.842.20 The sponsor applied a utility value of |||||||| to patients who were receiving STRIDE or atezolizumab plus bevacizumab, implying that people with unresectable HCC receiving these treatments have a higher level of well-being compared with the Canadian norm for people of an age similar to that of the modelled population. The NICE single-technology appraisal for lenvatinib for unresectable HCC applied health-state utilities for progression-free and progressed disease of 0.745 and 0.678, respectively, based on data from the REFLECT trial.6 The clinical experts consulted by CADTH agreed that the values used in the NICE review were more clinically plausible compared with the sponsor’s utility values.
The CADTH reanalysis applied utilities by health state rather than treatment type and being on or off treatment.
CADTH used utility values from the NICE lenvatinib review to better reflect the health-state utilities in this patient population. While the patient populations from the REFLECT trial may differ from those of the HIMALAYA trial, in the absence of alternative sources of utility values, these values were considered acceptable by clinical experts.
Time to treatment discontinuation was inconsistently modelled: The sponsor derived TTDs for STRIDE and sorafenib directly from the HIMALAYA trial for use in the economic model. According to the trial procedure, patients were allowed to continue taking these treatments after progression had occurred. Conversely, due to the absence of TTD curves from the IMbrave150 and REFLECT trials, TTD was assumed to equal the PFS curves for lenvatinib and atezolizumab plus bevacizumab, making it such that treatment was discontinued at the time of disease progression. The clinical experts consulted by CADTH suggested that, for all therapies, most patients would switch to a second-line therapy at the time of disease progression. They noted that, historically, a patient would be more likely to continue the same treatment after disease progression in the absence of viable, alternative second-line therapies, but given the second-line options currently available, most patients would move immediately to a second-line therapy.
In the CADTH reanalysis the TTD curve was assumed to be equal to the PFS curve for all comparators.
The model lacked transparency: The economic model submitted by the sponsor lacked transparency as it included numerous hidden sheets, columns, and rows, rendering it difficult to track inputs and outputs throughout. The model also included numerous IFERROR statements, which hinder the validation process.
CADTH was unable to address this limitation.
One additional limitation was identified but not considered to be a key limitation:
The model time horizon did not align with prior CADTH reviews of HCC: The sponsor assumed a 15-year time horizon in its submitted model. Prior CADTH reviews of treatments for unresectable HCC considered a 10-year time horizon which CADTH considered to be more appropriate.16,21 However, this was not considered a key limitation given the small proportion of the cohort still accruing costs and QALYs between 10 and 15 years.
The price of lenvatinib was out of date: The price of lenvatinib was updated in the Ontario Drug Benefit formulary between the time of the sponsor’s submission and CADTH reanalysis.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CADTH (Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CADTH comment |
|---|---|
Relative dose intensity for all treatments is 100%. | Appropriate. This aligns with prior CADTH reviews.22 |
Grade 1 and 2 AEs were excluded from the model. | Appropriate. Minor differences between groups are not expected to result in substantial effects on patient quality of life or health care resource costs. |
Although diarrhea and bleeding are important known AEs, they were not included in the model. | Not appropriate. Patient and clinical input noted the importance of these AEs for the available therapies. The risk of bleeding may differ across treatments. However, any harms or benefits associated with changes in bleeding risk between treatments cannot be captured in the submitted model. |
Subsequent therapy costs include only second-line therapies. | Appropriate. Only a small proportion of patients with unresectable HCC receive third-line therapies, and they are relatively inexpensive and unlikely to substantially differ between first-line therapy groups. |
UK-based resource use was adjusted to reflect Canadian practices (i.e., removal of clinical nurse specialist visits). | Uncertain. Ideally, Canadian resource use data would be used to inform the model. |
AE = adverse event; HCC = hepatocellular carcinoma.
CADTH Reanalyses of the Economic Evaluation
Base-Case Results
The CADTH base case was derived by making changes in model parameter values and assumptions, in consultation with clinical experts. These changes, summarized in Table 5, included using comparative efficacy data from the HIMALAYA trial for sorafenib, applying health-state utilities as opposed to treatment-specific utilities, and changes to TTD assumptions. The reanalysis is based on publicly available prices of the comparator treatments.
Table 5: CADTH Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
1. Lenvatinib price | 12 mg capsule: $101.92 | 12 mg capsule: $106.16 |
Changes to derive the CADTH base case | ||
1. Sorafenib comparative efficacy | Used sorafenib comparative efficacy derived from the lenvatinib MAIC | Used sorafenib comparative efficacy from the HIMALAYA trial |
2. Treatment-specific utilities | Applied treatment-specific utilities to patients who were on/off treatment | Applied health-state utilities for progression-free and progressed health states |
3. Time to treatment discontinuation | Assumed that STRIDE and sorafenib may be taken following progression, and that atezolizumab plus bevacizumab and lenvatinib would discontinue treatment at the time of disease progression | Assumed that treatment would be discontinued at the time of disease progression for all 4 comparators |
CADTH base case | — | Reanalyses 1 + 2 + 3 |
MAIC = matching adjusted indirect comparison; STRIDE = single-dose tremelimumab in combination with regular-interval durvalumab.
The CADTH base case focuses on the comparison of STRIDE versus sorafenib, the only comparison for which head-to-head trial data were available. The CADTH base case resulted in an ICER of $265,036 per QALY gained for STRIDE versus sorafenib (incremental cost = $95,359; incremental QALYs = 0.36) with a 0% probability of being cost-effective at a willingness-to-pay threshold of $50,000 per QALY gained. Cost-effectiveness was driven by the higher treatment cost of STRIDE compared to sorafenib, and influenced by both the higher acquisition cost and superior PFS of STRIDE, leading to patients remaining on therapy longer. The results of the stepped analysis are presented in Table 6.
Table 6: Summary of the Stepped Analysis of the CADTH Reanalysis Results (Deterministic)
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($ per QALY) |
|---|---|---|---|---|
Sponsor’s base casea | Sorafenib | 146,221 | 1.35 | Reference |
STRIDE | 274,912 | 1.94 | 219,960 | |
CADTH reanalysis 1 | Sorafenib | 148,984 | 1.44 | Reference |
STRIDE | 274,912 | 1.94 | 255,801 | |
CADTH reanalysis 2 | Sorafenib | 146,221 | 1.17 | Reference |
STRIDE | 274,912 | 1.61 | 289,694 | |
CADTH reanalysis 3 | Sorafenib | 133,788 | 1.34 | Reference |
STRIDE | 231,132 | 1.92 | 168,178 | |
CADTH base case (reanalyses 1, 2, and 3, deterministic) | Sorafenib | 139,377 | 1.25 | Reference |
STRIDE | 231,132 | 1.61 | 254,021 | |
CADTH base case (reanalyses 1, 2, and 3, probabilistic) | Sorafenib | 141,173 | 1.26 | Reference |
STRIDE | 236,532 | 1.62 | 265,036 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; STRIDE = single-dose tremelimumab in combination with regular-interval durvalumab.
aThese results were estimated using rounded values from the sponsor’s technical report. Appendix 3 provides additional details of the unrounded values.
Given the magnitude of uncertainty surrounding the sponsor’s MAIC and the long-term extrapolation of clinical benefits, CADTH was unable to derive a robust base-case estimate of the cost-effectiveness of STRIDE versus lenvatinib or atezolizumab plus bevacizumab. CADTH conducted additional reanalyses and scenario analyses to estimate the cost-effectiveness versus these comparators (Appendix 4).
Scenario Analysis Results
A price-reduction analysis based on the CADTH base case indicated that, at a willingness-to-pay threshold of $50,000 per QALY gained, a 50% price reduction would be required for the STRIDE regimen to be considered cost-effective compared to sorafenib (Table 7). Given that there is a confidential negotiated price for durvalumab for other indications, CADTH also conducted an analysis of the effect of a price reduction for tremelimumab only (Table 12). There is no price reduction for tremelimumab that would result in STRIDE being cost-effective compared to sorafenib at a threshold of $50,000 per QALY gained.
Table 7: CADTH Price-Reduction Analyses
Analysis | ICERs for STRIDE vs. sorafenib ($ per QALY) | |
|---|---|---|
Price reduction | Sponsor base casea,b | CADTH reanalysisa |
No price reduction | 219,363 | 265,036 |
10% | 186,402 | 221,773 |
20% | 153,441 | 178,510 |
30% | 120,480 | 135,246 |
40% | 87,519 | 91,982 |
50% | 54,558 | 48,719 |
60% | 21,597 | 5,455 |
70% | Dominant | Dominant |
80% | Dominant | Dominant |
90% | Dominant | Dominant |
100% | Dominant | Dominant |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; STRIDE = single-dose tremelimumab in combination with regular-interval durvalumab; vs. = versus.
aThe prices of both durvalumab and tremelimumab were reduced by the indicated percentage.
bThese results were estimated using rounded values from the sponsor’s technical report. Appendix 3 provides additional details of the unrounded values.
Additionally, CADTH conducted a series of scenario analyses to determine the impact of alternative assumptions on the cost-effectiveness of STRIDE:
In CADTH reanalysis A, the pairwise comparison for STRIDE versus atezolizumab plus bevacizumab is presented. Three scenario analyses (A1, A2, and A3) are also considered. Scenario analysis A1 assumes that the HR for atezolizumab plus bevacizumab compared to STRIDE is ||||||||||, reflecting the lower bound of the estimate from the MAIC. Scenario analysis A2 assumes equal efficacy of STRIDE and atezolizumab plus bevacizumab. Scenario analysis A3 assumes a 99% price reduction for atezolizumab.
CADTH reanalysis B presents the pairwise comparison of STRIDE versus lenvatinib. Scenario analysis B1 assumes equal efficacy (PFS and OS) for sorafenib and lenvatinib based on the CADTH pan-Canadian Oncology Drug Review Expert Review Committee recommendation for atezolizumab plus bevacizumab for unresected HCC.17
Results from these scenarios are presented in Table 13 and Table 14. When atezolizumab plus bevacizumab and STRIDE were assumed to have equal efficacy (scenario A2) STRIDE was more costly and provided no additional benefit.
Issues for Consideration
The pan-Canadian Pharmaceutical Alliance concluded negotiations with a letter of intent for atezolizumab plus bevacizumab for the treatment of adults with unresectable HCC,23 and the jurisdictional cancer formularies are now funding the regimen.24,25 As such, a confidential negotiated price exists for atezolizumab, while CADTH reanalyses are based on publicly available prices. Further price reductions for STRIDE may be warranted to ensure the cost neutrality of STRIDE compared to confidential prices for comparators.
The pan-Canadian Pharmaceutical Alliance concluded negotiations with a letter of intent for durvalumab for the treatment of adults with extensive-stage small cell lung cancer and non–small cell lung cancer and, as such, durvalumab has a confidential negotiated price and is currently funded by jurisdictional cancer formularies.24-27 The CADTH reanalyses are based on a sponsor-submitted price of durvalumab that may differ from the confidential price and may influence the results of the cost-effectiveness and budget impact analysis.
Overall Conclusions
Based on the CADTH clinical review and the HIMALAYA trial data, treatment with STRIDE in the first line for patients with unresectable HCC resulted in a statistically significant improvement in OS compared to sorafenib. The difference in OS of approximately 3 months found in the trial was considered clinically meaningful by the clinical experts consulted by CADTH. At the time of trial design, sorafenib was an appropriate comparator; however, sorafenib is no longer the most common standard of care therapy and has been replaced by therapies that include lenvatinib and atezolizumab plus bevacizumab. There is no direct comparative evidence for STRIDE versus lenvatinib or atezolizumab plus bevacizumab. The sponsor therefore conducted MAICs to estimate comparative clinical efficacy for these comparators. However, the CADTH clinical review reported that no superiority conclusions could be drawn due to methodological limitations and imprecision in the effect estimates.
CADTH identified several limitations in the sponsor’s pharmacoeconomic analysis that have notable implications on the cost-effectiveness of STRIDE. For the CADTH base-case analysis, CADTH used the clinical efficacy data for sorafenib from the HIMALAYA trial, applied health-state utilities rather than treatment-specific utilities, and assumed that treatment was discontinued at the time of disease progression for all comparators. Results of the sponsor’s and CADTH’s base cases were similar: STRIDE is not cost-effective at a willingness-to-pay threshold of $50,000 per QALY gained. In the CADTH base case, the ICER of STRIDE versus sorafenib was $265,036 (incremental costs: $95,359; incremental QALYs: 0.36) per QALY gained, with a 0% probability of being cost-effective at a threshold of $50,000 per QALY gained. A price reduction of approximately 50% is required for STRIDE to be cost-effective at this threshold compared to sorafenib. Given the limitations in the comparative clinical efficacy data for the more commonly used comparators, a high degree of uncertainty remains in estimating the cost-effectiveness of STRIDE for the sponsor’s requested reimbursement population compared to lenvatinib and atezolizumab plus bevacizumab; these comparisons were explored through scenario analyses only. In a scenario analysis that assumed equivalent efficacy of atezolizumab and bevacizumab versus STRIDE, CADTH found that STRIDE was more costly and provided no additional QALYs compared to atezolizumab plus bevacizumab. CADTH also investigated the time at which the cumulative cost of STRIDE and atezolizumab plus bevacizumab would be equal (given the higher upfront cost but lower maintenance cost for STRIDE). After approximately 60 weeks (1.15 years) of continuous treatment, the cumulative costs of STRIDE and atezolizumab plus bevacizumab are equal. The scenario assuming the same efficacy for lenvatinib and sorafenib resulted in a higher ICER for STRIDE versus lenvatinib ($262,393 per QALY gained) compared to when the HRs from the MAICS were applied to lenvatinib. In this scenario, STRIDE would require a price reduction of 63% to be cost-effective at a threshold of $50,000 per QALY gained compared to lenvatinib.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Imjudo (tremelimumab), 20 mg/mL, concentrate for solution for intravenous infusion in combination with Imfinzi (durvalumab), 50 mg/mL, concentrate for solution for intravenous infusion. Mississauga (ON): AstraZeneca Canada Inc.; 2022 Dec 15.
2.PrImjudo® (tremelimumab): 20 mg / mL concentrate for solution for infusion [product monograph]. Mississauga (ON): AstraZeneca Canada Inc.; 2023 Aug 31.
3.PrImfinzi® (durvalumab): 50 mg / mL concentrate for solution for infusion [product monograph]. Mississauga (ON): AstraZeneca Canada Inc.; 2022 Aug 18.
4.Table: 13-10-0114-01. Life expectancy and other elements of the life table, Canada, all provinces except Prince Edward Island. Ottawa (ON): Statistics Canada; 2022: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310011401. Accessed 2023 Mar 8.
5.Atezolizumab with bevacizumab for treating advanced or unresectable hepatocellular carcinoma. NICE technology appraisal guidance TA666: National Institute for Health and Care Excellence (NICE); 2020: https://www.nice.org.uk/guidance/ta666. Accessed 2023 Mar 8.
6.Lenvatinib for untreated advanced hepatocellular carcinoma. NICE technology appraisal guidance TA551: National Institute for Health and Care Excellence (NICE); 2018: https://www.nice.org.uk/guidance/ta551. Accessed 2023 Mar 8.
7.Xie F, Pullenayegum E, Gaebel K, et al. A Time Trade-off-derived Value Set of the EQ-5D-5L for Canada. Med Care. 2016;54(1):98-105. PubMed
8.Shabaruddin FH, Chen LC, Elliott RA, Payne K. A systematic review of utility values for chemotherapy-related adverse events. Pharmacoeconomics. 2013;31(4):277-288. PubMed
9.DeltaPA. [Ottawa (ON)]: IQVIA; 2023: https://www.iqvia.com/. Accessed 2023 Jun 2.
10.PrNexavar® (sorafenib): 200 mg tablets [product monograph]. Mississauga (ON): Bayer Inc.; 2014 Dec 17.
11.PrLenvima® (lenvatinib): 4 mg and 10 mg capsules [product monograph]. Mississauga (ON): Eisai Ltd.; 2018 Dec 19.
12.PrTecentriq® (atezolizumab): 60 mg/mL concentrate for solution for infusion as 1200 mg/20 mL and 840 mg/14 mL single use vials [product monograph]. Mississauga (ON): Hoffmann-La Roche Ltd; 2017 Apr 12.
13.Finn RS, Qin S, Ikeda M, et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med. 2020;382(20):1894-1905. PubMed
14.Schedule of benefits for physician services under the Health Insurance Act: effective October 1, 2021. Toronto (ON): Ontario Ministry of Health; 2022.
15.Meyers BM, Vogel A, Marotta P, et al. The cost-effectiveness of lenvatinib in the treatment of advanced or unresectable hepatocellular carcinoma from a Canadian perspective. Can J Gastroenterol Hepatol. 2021;2021:8811018. PubMed
16.Lenvatinib (lenvima) for hepatocellular carcinoma. pCODR final economic guidance report. Ottawa (ON): CADTH; 2019: https://www.cadth.ca/sites/default/files/pcodr/Reviews2019/10175LenvatinibHCC_inEGR_NOREDACT-ABBREV_EarlyCon_Post_24Jul2019_final.pdf. Accessed 2023 Mar 8.
17.pCODR Expert Review Committee (pERC) final recommendation: atezolizumab (Tecentriq) in combination with bevacizumab (Avastin). Ottawa (ON): CADTH; 2020: https://www.cadth.ca/sites/default/files/pcodr/Reviews2020/10217AtezolizumabBevacizumabHCC_fnRec_EC_Post17Nov2020_final.pdf. Accessed 2023 May 28.
18.Kudo M, Finn RS, Qin S, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet. 2018;391(10126):1163-1173. PubMed
19.Guidelines for the economic evaluation of health technologies: Canada. 4th ed. Ottawa (ON): CADTH; 2017: https://www.cadth.ca/guidelines-economic-evaluation-health-technologies-canada-4th-edition. Accessed 2023 Mar 8.
20.Guertin JR, Feeny D, Tarride JE. Age- and sex-specific Canadian utility norms, based on the 2013-2014 Canadian Community Health Survey. CMAJ. 2018;190(6):E155-E161. PubMed
21.Drug Reimbursement Review pharmacoeconomic report: atezolizumab (Tecentriq). Ottawa (ON): CADTH; 2020: https://www.cadth.ca/sites/default/files/pcodr/Reviews2020/10217AtezolizumabBevacizumabHCC_fnEGR_REDACTABBREV_EC_Post17Nov2020_final.pdf. Accessed 2023 Mar 8.
22.Pharmacoeconomic review. In: CADTH reimbursement review: enfortumab vedotin (Padcev). Can J Health Technol. 2022;2(1):99-127.
23.Tecentriq and bevacizumab (atezolizumab & bevacizumab). pCPA negotiation status report. Ottawa (ON): pan-Canadian Pharmaceutical Alliance; 2021: https://www.pcpacanada.ca/negotiation/21390. Accessed 2023 Mar 6.
24.Cancer Care Ontario: funded evidence-informed regimens. 2022; https://www.cancercareontario.ca/en/drugformulary/regimens. Accessed 2023 Mar 7.
25.Saskatchewan Cancer Agency. Drug Formulary. 2023; http://www.saskcancer.ca/health-professionals-article/drug-formulary. Accessed 2023 Mar 07.
26.Imfinzi (durvalumab). pCPA negotiation status report. Ottawa (ON): pan-Canadian Pharmaceutical Alliance; 2019: https://www.pcpacanada.ca/negotiation/21133. Accessed 2023 Jun 6.
27.Imfinzi (durvalumab). pCPA negotiation status report. Ottawa (ON): pan-Canadian Pharmaceutical Alliance; 2021: https://www.pcpacanada.ca/negotiation/21550. Accessed 2023 Jun 6.
28.Drug Reimbursement Review sponsor submission: Imjudo (tremelimumab), 20 mg/mL, concentrate for solution for intravenous infusion in combination with Imfinzi (durvalumab), 50 mg/mL, concentrate for solution for intravenous infusion [internal sponsor's package]. Mississauga (ON): AstraZeneca Canada Inc.; 2022 Dec 15.
29.Table: 17-10-0057-01 Projected population, by projection scenario, age and sex, as of July 1 (x 1,000). Ottawa (ON): Statistics Canada; 2022: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1710005701. Accessed 2023 Mar 8.
30.Exceptional Access Program (EAP). Toronto (ON): Ontario Ministry of Health; Ontario Ministry of Long-Term Care; 2023: http://www.health.gov.on.ca/en/pro/programs/drugs/odbf/odbf_except_access.aspx. Accessed 2023 Jun 2.
31.Abou-Alfa GK, Lau G, Kudo M, et al. Tremelimumab plus durvalumab in unresectable hepatocellular carcinoma. NEJM Evidence. 2022;1(8).
32.Clinical Study Report: D419CC00002. A randomized, open-label, multi-center phase III study of durvalumab and tremelimumab as first-line treatment in patients with advanced hepatocellular carcinoma (HIMALAYA) [internal sponsor's report]. Gaithersburg (MD): AstraZeneca; 2022 Jan 19.
33.Table: 17-10-0005-01. Population estimates on July 1st, by age and sex. Ottawa (ON: Statistics Canada; 2022: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1710000501. Accessed 2023 Jan 23.
34.Non-Insured Health Benefits program: First Nations and Inuit Health Branch: annual report 2020 to 2021. Ottawa (ON): Indigenous Services Canada; 2021: https://www.sac-isc.gc.ca/eng/1645718409378/1645718500555. Accessed 2023 Jan 23.
35.Canadian Cancer Society’s Advisory Committee. Canadian Cancer Statistics 2013: special topic: liver cancer. Toronto (ON): Canadian Cancer Society; 2013.
36.Canadian Cancer Statistics Advisory Committee in collaboration with the Canadian Cancer Society, Statistics Canada, and the Public Health Agency of Canada. Canadian Cancer Statistics 2021. Toronto (ON): Canadian Cancer Society; 2021: https://cdn.cancer.ca/-/media/files/research/cancer-statistics/2021-statistics/2021-pdf-en-final.pdf. Accessed 2023 Jun 2.
37.CancerMPact® Treatment Architecture: HCC U.S. [internal sponsor's report] In: Drug Reimbursement Review sponsor submission: Imjudo (tremelimumab), 20 mg/mL, concentrate for solution for intravenous infusion in combination with Imfinzi (durvalumab), 50 mg/mL, concentrate for solution for intravenous infusion. Mississauga (ON): AstraZeneca Canada Inc.; 2022 Dec 15.
38.Brenner DR, Poirier A, Woods RR, et al. Projected estimates of cancer in Canada in 2022. CMAJ. 2022;194(17):E601-E607. PubMed
39.Pires Amaro C, Allen MJ, Knox JJ, et al. Dosing, efficacy and safety of lenvatinb in the real-world treatment of hepatocellular carcinoma: results from a Canadian database. Liver Cancer Int. 2022;3(3):119-127.
40.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Imjudo (tremelimumab), 20 mg/mL, concentrate for solution for intravenous infusion in combination with Imfinzi (durvalumab), 50 mg/mL, concentrate for solution for intravenous infusion. Mississauga (ON): AstraZeneca Canada Inc.; 2022 Dec 15.
41.O’Sullivan DE, Boyne DJ, Syed IA, et al. Real-world treatment patterns, clinical outcomes, and health care resource utilization in advanced unresectable hepatocellular carcinoma. Can Liver J. 2022;5(4):476-492. PubMed
42.Avis transmis au ministre: LenvimaMC et CabometyxMC. Quebec (QC): Institut national d'excellence en santé et en services sociaux (INESSS); 2021: https://www.inesss.qc.ca/fileadmin/doc/INESSS/Inscription_medicaments/Avis_au_ministre/Mars_2021/Lenvima_Cabometyx_2021_02.pdf. Accessed 26 Jan 2023.
43.Alberta Health Services. Outpatient Cancer Drug Benefit Program. https://www.albertahealthservices.ca/services/page2328.aspx. Accessed 2023 Mar 6.
44.Saskatchewan Cancer Agency. Frequently Asked Questions - Health Professionals. http://www.saskcancer.ca/health-professionals-article/frequently-asked-questions-health-professionals. Accessed 2023 Mar 3.
45.Canadian Cancer Society’s Advisory Committee. Canadian Cancer Statistics: a 2018 special report on cancer incidence by stage. Toronto (ON): Canadian Cancer Society; 2018: https://cancer.ca/en/research/cancer-statistics/past-editions. Accessed 2023 Jun 2.
Appendix 1: Cost-Comparison Table
Note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical experts. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 8: CADTH Cost-Comparison Table for Treatment of Patients With Unresectable HCC
Treatment | Strength/ concentration | Form (vial size if single-use) | Price ($) | Recommended dosage | Daily cost ($) | Cost per 28 days ($) |
|---|---|---|---|---|---|---|
Tremelimumab (Imjudo) | 20 mg/mL | 15 mL (300 mg) Vial for IV infusion | 34,319.5779a | 300 mg single dose in combination with durvalumab on cycle 1, day 1b | 1,225.70 | 34,320 |
Durvalumab (Imfinzi) | 50 mg/mL | 2.4 mL (120 mg) 10 mL (500 mg) Vial for IV infusion | 938.6700 3,911.1100 | 1,500 mg every 4 weeksb for as long as clinical benefit is observed or until unacceptable toxicity | 419.05 | 11,733 |
Single tremelimumab regular-interval combined with durvalumab (STRIDE) regimen cost per 28 days | Initial cycle: 46,053 Thereafter: 11,733 | |||||
Atezolizumab plus bevacizumab | ||||||
Atezolizumab (Tecentriq) | 60 mg/mL | 14 mL (840 mg) 20 mL (1,200 mg) Vial for IV infusion | 4,743.2000 6,776.0000 | 1,200 mg in combination with bevacizumab every 3 weeks until loss of clinical benefit or unacceptable toxicity | 322.67 | 9,035 |
Bevacizumab (biosimilars) | 25 mg/mL | 4 mL (100 mg) 16 mL (400 mg) Solution for injection | 347.0000 1,388.0000 | 15 mg/kg every 3 weeks until loss of clinical benefit or unacceptable toxicity | 181.76c | 5,089c |
Atezolizumab plus bevacizumab (biosimilar) regimen cost per 28 days | 14,124 | |||||
Tyrosine kinase inhibitors | ||||||
Lenvatinib (Lenvima) | 8 mg 12 mg | Capsule | 70.7752d 106.1559d | 8 mg once daily for patients < 60 kg and 12 mg once daily for those ≥ 60 kg | 70.78 to 106.16 | 1,982 to 2,972 |
Sorafenib (Nexavar) | 200 mg | Tablet | 46.4689d | 400 mg twice daily until patient is no longer clinically benefiting or unacceptable toxicity | 185.88 | 5,205 |
Note: All prices are from the IQVIA Delta PA database (accessed June 2023)9 unless otherwise indicated. Daily and per-28-day costs assume wastage of excess medication.
aSponsor’s submitted prices.28
bPatients with a body weight of 30 kg or less must receive weight-based dosing of 4 mg/kg tremelimumab as a single dose on cycle 1, day 1, and 20 mg/kg durvalumab every 4 weeks until weight is greater than 30 kg.2
cBevacizumab costs assumed a mean patients weight of 70.9 kg based on the HIMALAYA clinical trial.29
dOntario Drug Benefit Exceptional Access Program list price (accessed June 2023).30
Appendix 2: Submission Quality
Note that this appendix has not been copy-edited.
Description | Yes/No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | Yes | No comment |
Model has been adequately programmed and has sufficient face validity | No | The clinical experts consulted by CADTH found that the proportion of patients still alive and/or progression-free in the extrapolated period beyond 5 years was clinically implausible. |
Model structure is adequate for decision problem | Yes | No comment |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | Yes | No comment |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | Yes | No comment |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | Yes | No comment |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note that this appendix has not been copy-edited.
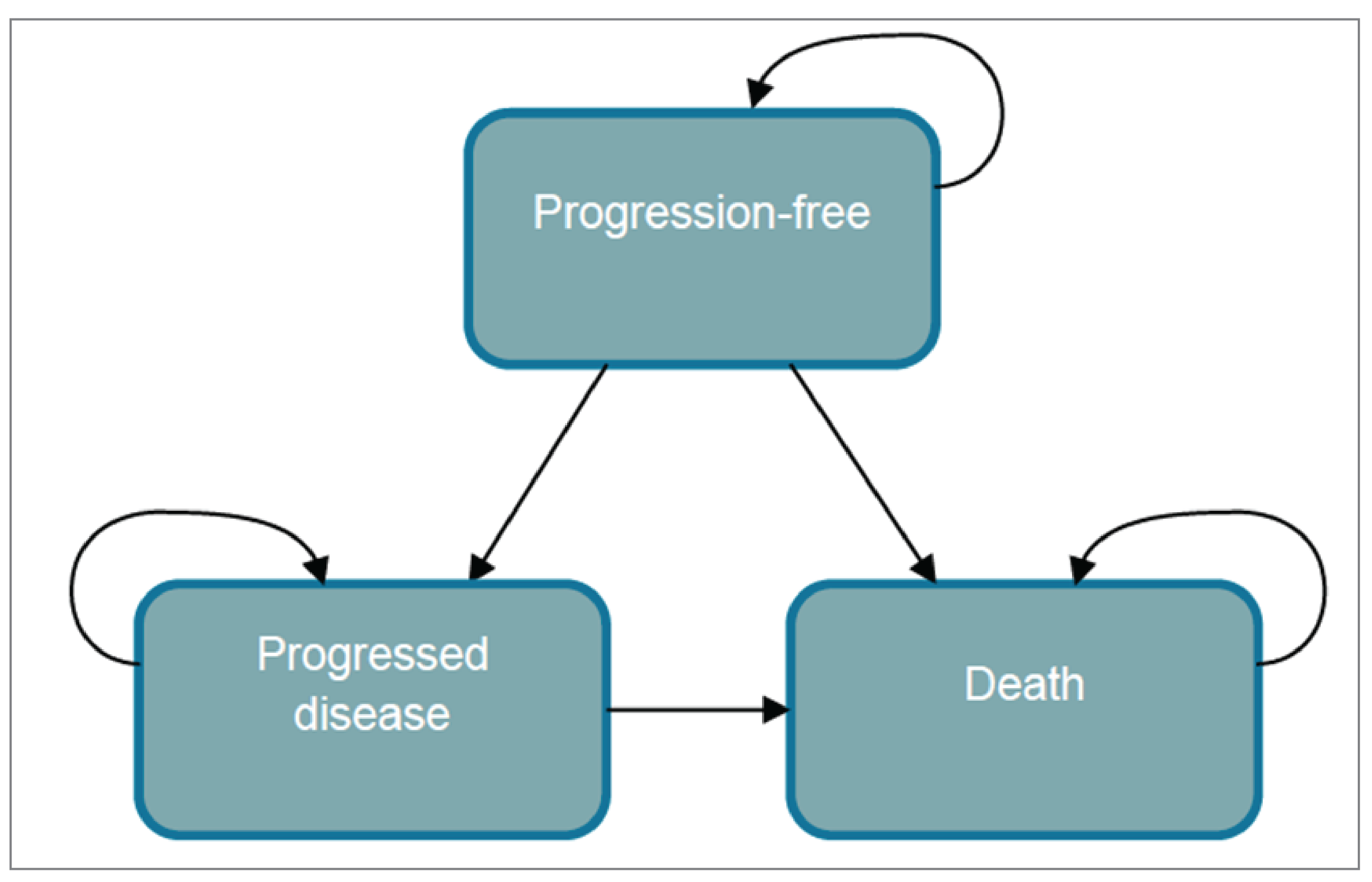
Figure 2: Kaplan-Meier and OS Extrapolation for STRIDE

KM = Kaplan-Meier; OS = overall survival; STRIDE = single-dose tremelimumab in combination with regular-interval durvalumab.
Notes: This figure has been redacted at the request of the sponsor. The Weibull distribution was selected in the sponsor’s base case.
Figure 3: Projected OS Curves for STRIDE, Sorafenib, Atezolizumab Plus Bevacizumab, and Lenvatinib

KM = Kaplan-Meier; OS = overall survival; STRIDE = single-dose tremelimumab in combination with regular-interval durvalumab.
Note: This figure has been redacted at the request of the sponsor.
Figure 4: Kaplan-Meier and PFS Spline Modelling Extrapolations for STRIDE

KM = Kaplan-Meier; PFS = progression-free survival; STRIDE = single-dose tremelimumab in combination with regular-interval durvalumab.
Notes: This figure has been redacted at the request of the sponsor. The hazard, 3 knots distribution was selected in the sponsor’s base case.
Figure 5: Projected PFS Curves for STRIDE, Sorafenib, Atezolizumab Plus Bevacizumab, and Lenvatinib

KM = Kaplan-Meier; PFS = progression-free survival; STRIDE = single-dose tremelimumab in combination with regular-interval durvalumab.
Note: This figure has been redacted at the request of the sponsor.
Detailed Results of the Sponsor’s Base Case
Table 10: Disaggregated Summary of the Sponsor’s Economic Evaluation Resultsa
Parameter | STRIDE | Sorafenib | Lenvatinib | Atezolizumab plus bevacizumab |
|---|---|---|---|---|
Discounted LYs | ||||
Total | 2.32 | 1.69 | 1.83 | 2.56 |
Progression-free | 0.76 | 0.59 | 1.08 | 1.61 |
Progressed | 1.56 | 1.10 | 0.76 | 0.95 |
Discounted QALYs | ||||
Total | 1.95 | 1.36 | 1.38 | 2.13 |
Progression-free | 0.65 | 0.49 | 0.79 | 1.36 |
Progressed | 1.29 | 0.88 | 0.60 | 0.78 |
Discounted costs ($) | ||||
Total | 275,677 | 146,253 | 132,389 | 382,249 |
Treatment costs | 193,471 | 51,167 | 40,114 | 299,983 |
Administration costs | 1,099 | 0 | 0 | 4,248 |
Subsequent treatment costs | 13,055 | 22,588 | 22,412 | 12,844 |
Subsequent administration costs | 0 | 0 | 0 | 0 |
Resource use costs | 34,909 | 39,917 | 36,308 | 33,021 |
End-of-life costs | 32,082 | 32,467 | 32,386 | 31,853 |
Adverse event costs | 73 | 115 | 1,169 | 299 |
ICER ($/QALY) | ||||
Sorafenib vs. STRIDE: 219,363 | — | |||
Lenvatinib vs. STRIDE: 251,381 | — | |||
STRIDE vs. Atezolizumab plus bevacizumab: 592,067 | — | |||
ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year; STRIDE = single-dose tremelimumab in combination with regular-interval durvalumab; vs. = versus.
aThese results were estimated using rounded values from the sponsor’s technical report. The unrounded probabilistic ICERs for each comparison were as follows: sorafenib vs. STRIDE: $221,503; lenvatinib vs. STRIDE: $255,469; and STRIDE vs. atezolizumab plus bevacizumab: $569,070.
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Note that this appendix has not been copy-edited.
Table 11: Disaggregated Summary of CADTH’s Economic Evaluation Results
Parameter | STRIDE | Sorafenib | Lenvatiniba | Atezolizumab plus bevacizumaba |
|---|---|---|---|---|
Discounted LYs | ||||
Total | 2.32 | 1.81 | 1.85 | 2.55 |
Progression-free | 0.76 | 0.64 | 1.08 | 1.60 |
Progressed | 1.56 | 1.17 | 0.77 | 0.94 |
Discounted QALYs | ||||
Total | 1.62 | 1.26 | 1.21 | 1.79 |
Progression-free | 0.56 | 0.47 | 0.69 | 1.15 |
Progressed | 1.06 | 0.80 | 0.52 | 0.64 |
Discounted costs ($) | ||||
Total | 236,532 | 141,173 | 135,853 | 381,353 |
Treatment costs | 155,660 | 43,353 | 43,152 | 299,235 |
Administration costs | 851 | 0 | 0 | 4,237 |
Subsequent treatment costs | 13,039 | 22,649 | 22,491 | 12,832 |
Subsequent administration costs | 0 | 0 | 0 | 0 |
Resource use costs | 34,856 | 42,678 | 36,662 | 32,886 |
End-of-life costs | 32,072 | 32,395 | 32,375 | 31,865 |
Adverse event costs | 54 | 97 | 1,173 | 298 |
ICER ($ per QALY) | ||||
Sorafenib vs. STRIDE: 265,036 | — | |||
Lenvatinib vs. STRIDE: 245,559 | — | |||
STRIDE vs. Atezolizumab plus bevacizumab: 863,185 | — | |||
ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year; STRIDE = single-dose tremelimumab in combination with regular-interval durvalumab; vs. = versus.
aThe results for these comparators are a part of CADTH’s scenario analyses (scenarios A and B) rather than the base-case analysis.
Scenario Analyses
Table 12: CADTH Price-Reduction Analysis — Tremelimumab Price Reduction
Analysis | ICERs for STRIDE vs. sorafenib ($ per QALY) | |
|---|---|---|
Price reduction | CADTH base case | Tremelimumab price reduction only |
No price reduction | 265,036 | 265,036 |
10% | 221,773 | 255,498 |
20% | 178,510 | 245,959 |
30% | 135,246 | 236,421 |
40% | 91,982 | 226,882 |
50% | 48,719 | 217,343 |
60% | 5,455 | 207,805 |
70% | Dominant | 198,266 |
80% | Dominant | 188,728 |
90% | Dominant | 179,189 |
100% | Dominant | 169,650 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; STRIDE = single-dose tremelimumab in combination with regular-interval durvalumab; vs. = versus.
Table 13 presents scenario analyses exploring the impacts of alternative assumptions for the comparison of STRIDE versus atezolizumab plus bevacizumab. CADTH reanalysis A represents the results from CADTH’s base case model, and scenarios A1 to A3 reflect changes made to reanalysis A.
Due to the uncertainty of the clinical benefit of STRIDE compared to atezolizumab plus bevacizumab, CADTH investigated the time at which the cumulative cost of STRIDE and atezolizumab plus bevacizumab would be equal. This investigation assumed that there would be no treatment discontinuation and no discounting of future costs. During the initial cycle of STRIDE, the single dose of tremelimumab results in a higher cycle cost compared to atezolizumab plus bevacizumab. However, in subsequent cycles the cost of durvalumab is lower than that of atezolizumab plus bevacizumab, when standardized to the same treatment cycle length. At approximately 60 weeks (1.15 years) of continuous treatment, the cumulative costs of STRIDE and atezolizumab plus bevacizumab are equal (Figure 6).
Table 14 presents scenario analyses exploring the impacts of alternative values for the comparison of STRIDE versus lenvatinib. CADTH reanalysis B represents the results from CADTH’s base case model, and scenario B1 reflects a change made to reanalysis B.
Table 13: Summary of CADTH’s Scenario Analysis Results — Reanalysis A (Atezolizumab Plus Bevacizumab)
Scenario analysis | Drug | Total costs ($) | Total QALYs | ICER ($ per QALY) |
|---|---|---|---|---|
CADTH reanalysis A (stepwise analysis 1 + 2 + 3 from base case)a | STRIDE | 236,532 | 1.62 | Reference |
Atezolizumab plus bevacizumab | 381,353 | 1.79 | 863,185 | |
CADTH scenario A1: alternative PFS for Atezolizumab plus bevacizumab = |||||||||| | STRIDE | 235,899 | 1.62 | Reference |
Atezolizumab plus bevacizumab | 296,755 | 1.77 | 394,213 | |
CADTH scenario A2:b equal efficacy of STRIDE and Atezolizumab plus bevacizumab (deterministic) | Atezolizumab plus bevacizumab | 221,460 | 1.62 | Reference |
STRIDE | 231,132 | 1.62 | More costly, equally effective | |
CADTH scenario A3: 99% price reduction atezolizumab | Atezolizumab plus bevacizumab | 194,665 | 1.77 | Reference |
STRIDE | 231,132 | 1.61 | Dominated |
ICER = incremental cost-effectiveness ratio; PFS = progression-free survival QALY = quality-adjusted life-year; STRIDE = single-dose tremelimumab in combination with regular-interval durvalumab.
Note: All results are presented probabilistically, unless otherwise stated.
aThis scenario analysis incorporates all of the changes made in CADTH’s base-case analysis (outlined in Table 5) except with atezolizumab plus bevacizumab as the comparator. Scenarios A1-A3 are conducted as changes made to Scenario A.
bIn this scenario, it was also assumed that there are no differences in AE disutilities and that no patients would delay bevacizumab treatment.
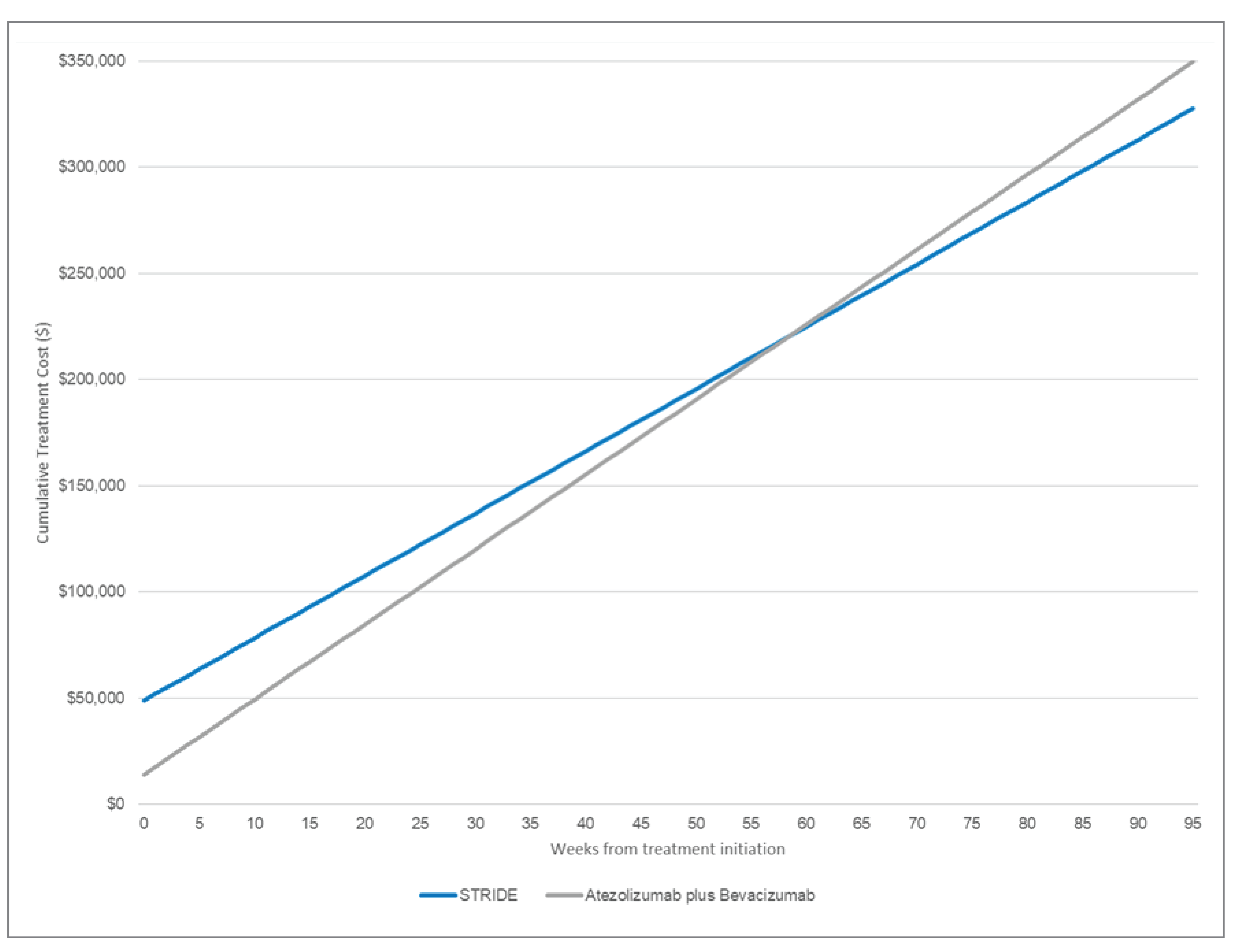
Table 14: Summary of CADTH’s Scenario Analysis Results – Reanalysis B (Lenvatinib)
Scenario analysis | Drug | Total costs ($) | Total QALYs | ICER ($ per QALY) |
|---|---|---|---|---|
CADTH reanalysis B (stepwise analysis 1 + 2 + 3 from base case) | Lenvatinib | 135,853 | 1.21 | Reference |
STRIDE | 236,532 | 1.62 | 245,559 | |
CADTH scenario B1: equal efficacy (OS and PFS) of lenvatinib and sorafenib | Lenvatinib | 123,338 | 1.27 | Reference |
STRIDE | 236,917 | 1.62 | 319,801a |
ICER = incremental cost-effectiveness ratio; OS = overall survival; PFS = progression-free survival; QALY = quality-adjusted life-year; STRIDE = single-dose tremelimumab in combination with regular-interval durvalumab.
aThis scenario analysis incorporates all of the changes made in CADTH’s base-case analysis (outlined in Table 5) except with lenvatinib as the comparator. Scenario B1 was conducted as a change made to Scenario B.
bSTRIDE would require a price reduction of 63% to be cost-effective at a $50,000 per QALY gained threshold compared to lenvatinib.
Appendix 5: Submitted Budget Impact Analysis and CADTH Appraisal
Note that this appendix has not been copy-edited.
Table 15: Summary of Key Take-Aways
Key take-aways of the Budget Impact Analysis |
|---|
|
Summary of Sponsor’s Budget Impact Analysis
The submitted budget impact analysis assessed the introduction of the STRIDE regimen for the first-line treatment of patients with unresectable HCC who require systemic therapy. The analysis was undertaken from the perspective of the Canadian health care payer using an epidemiological approach and included drug acquisition costs as well as the costs of administration, routine monitoring and disease management, subsequent therapy, and AE management. Durations of therapy, AE event rates, monitoring and disease management types and frequencies, proportions of patients receiving subsequent therapy, market shares of subsequent therapies, and all costs associated with AE management, monitoring and disease management, and subsequent therapies were as used in the pharmacoeconomic analysis presented in the main body of this report, with the exception of end-of-life costs which were not included in the budget impact analysis (BIA). A 3-year time horizon was considered, from 2024 through 2026, with 2023 as the base year. Data from the model were obtained from various sources including the HIMALAYA trial,31,32 the sponsor’s MAIC,28 Statistics Canada,33 the Non-Insured Health Benefits program,34 the Canadian Cancer Society,35,36 a CancerMPact report on HCC in the US,37 IQVIA data,9 the published literature,38,39 and expert opinion.40
The reference case scenario included atezolizumab-bevacizumab, sorafenib, and lenvatinib as potential treatments. The new drug scenario included the same comparators along with STRIDE. Key inputs to the BIA are documented in Table 15. Key assumptions included:
Excess medication in vials is wasted, and patients are dispensed full packages of tablets; thus, discontinuations midpack may also result in medication wastage.
Treatment patterns such as proportions of patients receiving liver transplants or resections and recurrence rates are similar between Canada and the US.
STRIDE will primarily displace sorafenib and lenvatinib, with some displacement of atezolizumab plus bevacizumab.
Figure 7: Derivation of the Eligible Population of Patients With Newly Diagnosed HCC
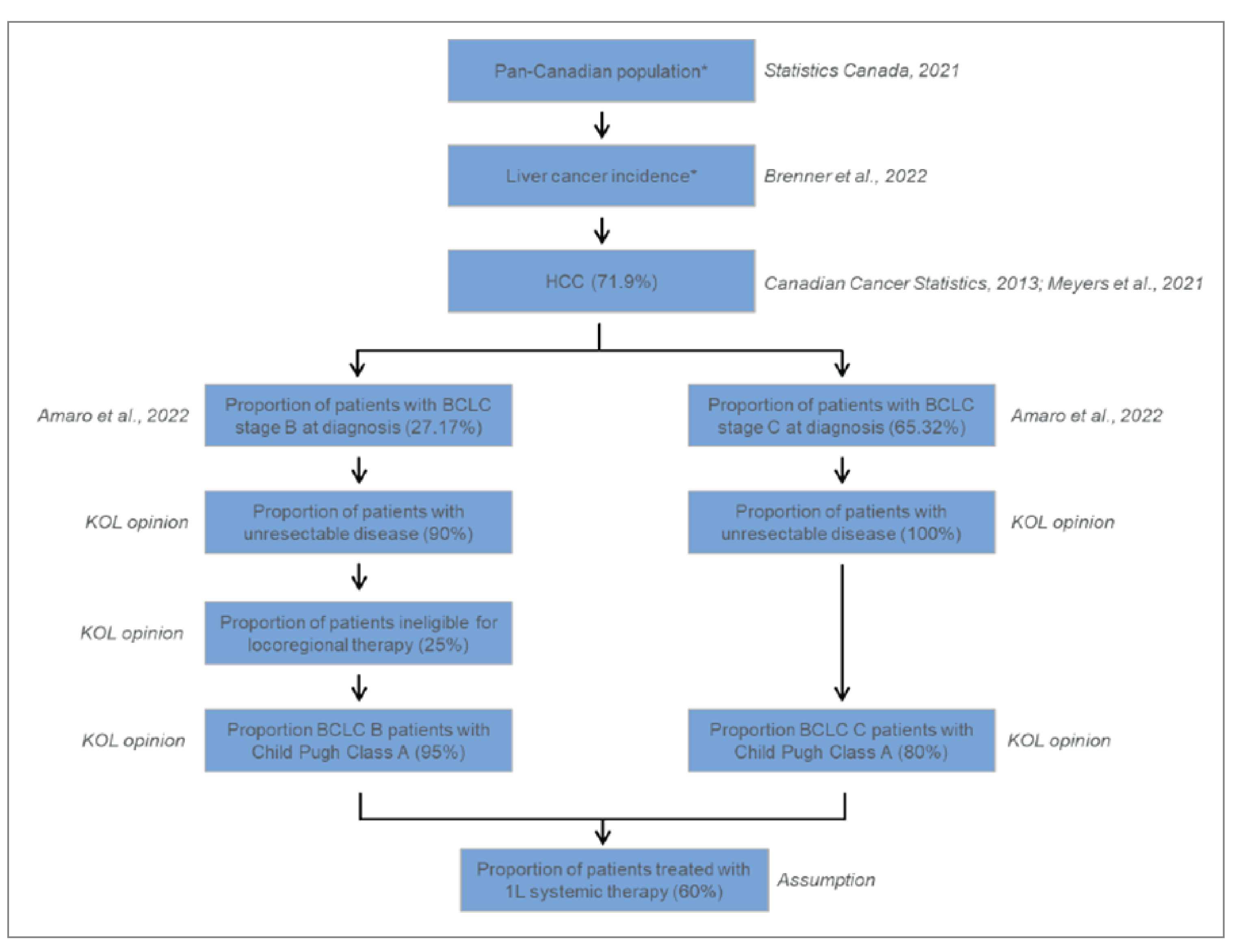
1L = first line; BCLC = Barcelona Clinic Liver Cancer; HCC = hepatocellular carcinoma.
Source: Sponsor’s submitted budget impact analysis.40
Figure 8: Derivation of the Eligible Population of Patients With Recurrent HCC

1L = first line; BCLC = Barcelona Clinic Liver Cancer; HCC = hepatocellular carcinoma.
Note: This figure has been redacted at the request of the sponsor.
Source: Sponsor’s submitted budget impact analysis.40
Table 16: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as Year 1 / Year 2 / Year 3 if appropriate) |
|---|---|
Target population — newly incident patients | |
Pan-Canadian population aged 18+ (excluding Québec) | |
Annual population growth rate | Varies by jurisdiction, range 0.11% to 1.74% per year33 |
Incidence of liver cancer | Varies by jurisdiction, range: 3.91 to 8.76 per 100,00038 |
Proportion of liver cancer patients with HCC | 71.9%35 |
Proportion of patients with HCC with BCLC stage B at diagnosis | 27.2%39 |
Proportion of BCLC-B patients with unresectable disease | 90%a |
Proportion of BCLC-B ineligible for locoregional therapy | 25%a |
Proportion of BCLC-B patients with Child Pugh Class A disease | 95%a |
Proportion of patients with HCC with BCLC stage C at diagnosis | 65.3%39 |
Proportion of BCLC-C patients with unresectable disease | 100%a |
Proportion of BCLC-C patients with Child Pugh Class A disease | 80%a |
Proportion of patients treated with first-line systemic therapy | 60%b |
Total newly incident patients eligible | 466 / 472 / 479 |
Target population — recurrent patients | |
Proportion of patients with HCC with BCLC stage A at diagnosis | 2.3%39 |
Proportion of BCLC-A patients receiving liver transplant | 8.0%37 |
Proportion of BCLC-A post–liver transplant recurrence | 10.0% at 2 years / 15.0% at 5 yearsa |
Proportion of BCLC-A patient eligible for liver resection | 52.0%37 |
Proportion of BCLC-A post–liver resection recurrence | ||||||||% at 2 years / ||||||||% at 5 yearsc |
Proportion of BCLC-A extrahepatic recurrence | ||||||||%a,37 |
Proportion of patients with HCC with BCLC stage B at diagnosis | 27.2%39 |
Proportion of BCLC-B patients with resectable disease | 10%a |
Proportion of BCLC-B patients eligible for liver resection | 100%a |
Proportion of BCLC-B post–liver resection recurrence (2 year/5 year) | ||||||||% at 2 years / ||||||||% at 5 yearsc |
Proportion of BCLC-B patients with unresectable disease | 90%a |
Proportion of BCLC-B patients eligible for locoregional therapy | 75%a |
Proportion of BCLC-B postlocoregional therapy recurrence | ||||||||% at 2 years / ||||||||% at 5 yearsc |
Proportion of BCLC-B extrahepatic recurrence | ||||||||%a,37 |
Proportion of recurrent patients treated with first-line systemic therapy | 60% |
Total recurrent patients eligible | 79 / 80 / 81 |
Total number of patients eligible for drug under review (incident + recurrent) | 545 / 552 / 560 |
Market uptake (reference scenario, 3 years)d | |
Sorafenib | |||| ||||| ||||| |
Lenvatinib | ||||| |||||| |||||| |
Atezolizumab + bevacizumab | ||||| |||||| |||||| |
Market uptake (new drug scenario, 3 years)d | |
STRIDE | |||| ||||| ||||| |
Sorafenib | ||||| |||||| |||||| |
Lenvatinib | ||||| |||||| |||||| |
Atezolizumab + bevacizumab | |||| ||||| ||||| |
Estimated cost of treatment (per patient per treatment course)e | |
STRIDE (median duration: |||||||| months) | $128,642 |
Sorafenib (median duration: |||||||| months) | Varies by jurisdiction, range: $38,564 to $38,950 |
Lenvatinib (median duration: |||||||| months) | Varies by jurisdiction, range: $5,942 to $19,342 |
Atezolizumab + bevacizumab (median duration: |||||||| months) | $140,313 |
BCLC = Barcelona Clinic Liver Cancer; HCC = hepatocellular carcinoma; STRIDE = single-dose tremelimumab in combination with regular-interval durvalumab.
aAssumption based on clinical expert opinion elicited by the sponsor.40
bAssumption based on a presumed increase in treatment rates from real world data from Alberta (2008 to 2018).41 This 60% estimate yields a patient count for Québec consistent with that reported by l’Institut national d'excellence en santé et services sociaux in its recent review of lenvatinib and cabozantinib for unresectable HCC.42
cAssumption based on clinical expert opinion elicited by the sponsor, consistent with similar recurrence data from the US CancerMPact HCC report with similar but not identical patient groupings and timelines.37
dMarket shares were based on the sponsor’s internal forecasting.1
eBased on the submitted prices for the STRIDE regimen,28 and unit costs reported by IQVIA’s Delta PA database for the comparator treatments.9
Summary of the Sponsor’s BIA Results
The sponsor’s estimated budget impact of funding STRIDE for the first-line treatment of adults with unresectable HCC was $10,256,695 in Year 1, $14,485,714 in Year 2, and $15,591,698 in year 3, for a 3-year total budget impact of $40,334,108.
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA.
Health care payer perspective inappropriate: CADTH submission guidelines stipulate that the BIA base case should represent the difference in costs that will be seen by a jurisdictional drug plan due to the introduction of the drug under review. As such, the perspective of such an analysis should primarily be that of a public drug plan. The sponsor’s analysis included drug acquisition costs of first and subsequent line therapies, but also included administration, monitoring and disease management, and AE management costs which are not consistent with the drug payer perspective.
In reanalyses, CADTH excluded costs associated with administration, patient monitoring and disease management, and AE management. Administration costs were included in a scenario analysis.
NIHB population was inappropriately calculated: While the sponsor appropriately removed NIHB clients from the provincial populations as reported by Statistics Canada to avoid double counting, 2 provincial jurisdictions (Alberta, Saskatchewan) fund oncology treatments for all residents within their borders,43,44 and 1 (Ontario) provides coverage for all residents aged 24 years and younger or 65 years and older. As such, the sponsor has overcounted patients who will be funded by NIHB, and undercounted those who will be funded by Alberta, Saskatchewan, and Ontario.
In reanalyses, CADTH included all NIHB clients aged 18+ years in Alberta and Saskatchewan, as well as those aged 18 to 24 years and 65+ years in Ontario, as part of their respective provincial populations for the purposes of this analysis, removing them from the NIHB population.
Cancer stage at diagnosis was inappropriately derived: The sponsor used a Canadian database study39 which reported the proportion of lenvatinib-treated patients who had been diagnosed with HCC at each BCLC stage as a proxy for the proportions of patients diagnosed with HCC in general. Patients within each BCLC stage at diagnosis were further filtered by the appropriate treatment regimens for that stage and by response and progression rates until an estimate of all patients who would be eligible for STRIDE and its comparators (sorafenib, lenvatinib, or atezolizumab plus bevacizumab) was reached. However, the study used by the sponsor to estimate staging at diagnosis reported the proportion of lenvatinib-treated patients who had been diagnosed at each BCLC stage, not the proportion of overall patients with unresectable who are diagnosed at each stage. As the sponsor’s eligibility derivation algorithm is intended to calculate the proportion of the overall unresectable HCC population by BCLC stage who will be eligible for STRIDE or its comparators, using staging-at-diagnosis proportions derived from patients who had already been deemed eligible for 1 of the comparators, lenvatinib, leads to an overestimation of the number of patients who will become eligible (e.g., the sponsor assumes no patients with HCC are diagnosed in BCLC stage D and therefore all patients with HCC are considered potentially eligible). In the absence of Canadian-specific unresectable HCC staging data, expert opinion solicited by CADTH indicated that the proportion of patients diagnosed with liver cancer in each BCLC stage would be a better proxy for the overall unresectable HCC population than proportions of those who had received lenvatinib. Alternately, the CancerMPact report37 cited elsewhere by the sponsor provides BCLC staging at HCC diagnosis estimates for the US population in 2021; these American HCC data are relatively consistent with Canadian staging at diagnosis data for liver cancer as a whole.
In the absence of Canadian data specific to HCC, CADTH used data for overall liver cancer from the 2018 Canadian Cancer Society report45 as a proxy to estimate that 31% of patients were diagnosed at BCLC stage A, 20% at stage B, and 31% at stage C, and 19% at stage D. Patients diagnosed in BCLC stage D were not considered eligible for treatment with STRIDE or its comparators and were therefore removed from the model in these reanalyses.
Some BCLC stage A patients inappropriately excluded: The sponsor’s analysis assumes that of patients who were diagnosed at BCLC stage A, 8% would receive a liver transplant and 52% would receive a liver resection, based on a 2021 HCC CancerMPact report from the US.37 Of these patients, many are assumed to later have an extrahepatic recurrence which would make them eligible for STRIDE and its comparators. However, the sponsor’s model excludes the remaining 40% of patients diagnosed at BCLC stage A who would initially receive an alternate therapy (e.g., transarterial chemoembolization, transarterial radioembolization, and radiofrequency ablation)37 and thereafter also have a risk of later extrahepatic recurrence leading to eligibility for STRIDE and its comparators. According to clinical expert opinion solicited by CADTH, recurrence rates for patients using these other treatments would be expected to be higher than 20%.
In reanalyses, CADTH assumed that of the remaining 40% of patients with HCC diagnosed at BCLC stage A, 20% would experience a recurrence by 2 years after therapy and 30% would experience a recurrence by 5 years. These patients were assumed to have the same risk of that recurrence being extrahepatic and of being treated with systemic therapy as other modelled patients who were stage A at diagnosis.
Time to treatment discontinuation was inappropriately modelled: As in the pharmacoeconomic evaluation described in the main body of this report, the sponsor assumed that a treatment’s TTD is relative to its respective PFS curve. In this approach, the TTD for STRIDE and sorafenib were based on data from the HIMALAYA trial and patients were assumed to continue taking these treatments after progression has taken place, while patients receiving lenvatinib and atezolizumab plus bevacizumab were assumed to discontinue at progression. Clinical experts consulted by CADTH agreed that most patients would switch therapies at the time of disease progression. The sponsor’s BIA model used median TTD derived from the PE model to calculate therapy costs.
In the CADTH PE reanalysis, the TTD curve was assumed to be equal to the PFS curve for all comparators, affecting median TTD for STRIDE and sorafenib relative to the sponsor’s submitted analysis. These new median TTDs were applied to CADTH’s budget impact reanalysis.
Proportion of patients receiving systemic therapy may be underestimated: The sponsor assumed that of patients who were otherwise eligible within the model, only 60% would be treated with a first-line systemic therapy (i.e., STRIDE or a comparator). This proportion was based on assumption due to an absence of current Canadian data, and derived by applying the model’s assumptions to the Québec population (which is excluded from the CADTH model) and comparing the resulting patient population estimate in Québec to that reported in the review of lenvatinib conducted by INESSS.42 This 60% estimate was then applied as a proxy to estimate the treated population in CADTH-participating jurisdictions. However, CADTH notes that once alternate BCLC staging proportions as described above are applied, the number of patients the model predicts would be treated in Québec is substantially lower than that predicted in the INESSS report. Additionally, CADTH notes that in the US-based CancerMPact HCC report cited by the sponsor for other inputs, 25% of patients diagnosed at BCLC stage A and 31% of patients diagnosed a BCLC stage B had disease progression after their first line of therapy and received a second line. In contrast, the sponsor’s model assumptions result in approximately 11% and 25% of patients diagnosed at stage A and B eventually receiving systemic therapy, respectively.
In a scenario analysis, CADTH assumed that 90% of patients who are otherwise eligible would receive STRIDE or 1 of its comparators. This 90% proportion was assumed because, when applied to the Québec population using CADTH base case inputs, it results in approximately the same number of eligible patients with unresectable HCC as was estimated in the INESSS review of lenvatinib.42 As alternate Canadian data were not found, CADTH considered this analysis to be exploratory.
Market displacement by STRIDE is uncertain: The sponsor assumed that of patients receiving STRIDE by Year 3 of the new drug scenario (||%), most would have otherwise received lenvatinib (|||||||%), while a smaller proportion would have otherwise received atezolizumab plus bevacizumab (|||||%) or sorafenib (||||||%). In contrast, clinical expert opinion solicited by CADTH expected that patients ultimately receiving STRIDE would be those for whom immunotherapy (STRIDE, atezolizumab plus bevacizumab) was deemed most appropriate or preferred, rather than those for whom a TKI (lenvatinib, sorafenib) was deemed the most appropriate or preferred therapy.
In a scenario analysis, CADTH assumed that atezolizumab plus bevacizumab, lenvatinib, and sorafenib would be displaced by STRIDE in each year of the new drug scenario proportionally to their assumed market shares in the reference scenario.
The price of drugs paid by public drug plans is uncertain: Both the sponsor’s and CADTH’s analyses are based on publicly available list prices for all comparators and may not reflect confidential, negotiated prices.
CADTH was unable to address this limitation.
CADTH Reanalyses of the BIA
CADTH revised the sponsor’s submitted analysis by incorporating a drug payer perspective, correcting NIHB reimbursement eligibility, revising the BCLC staging distribution of patients with HCC at diagnosis, incorporating all patients diagnosed at BCLC stage A into the model, and adjusting the median OS of sorafenib patients and the median TTD of STRIDE and sorafenib patients to match those of the CADTH pharmacoeconomic reanalysis. Additionally, CADTH updated the prices of some comparators to reflect changes in formulary list prices occurring after the sponsor’s submission.9,30 The changes applied to derive the CADTH base case are described in Table 17.
Table 17: CADTH Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
1. Updated lenvatinib 12 mg pricing | Nova Scotia: $33.9721 Ontario and NIHB: $101.9163 Newfoundland and Labrador: $110.5791 | Nova Scotia: $35.3853 Ontario and NIHB: $106.1559 Newfoundland and Labrador: $115.1792 |
Changes to derive the CADTH base case | ||
1. Drug payer perspective | Health care payer perspective: includes drug acquisition, administration, monitoring and maintenance, adverse event, and subsequent therapy costs. | Drug payer perspective: removal of all costs except drug acquisition and subsequent therapy |
2. NIHB reimbursed population | All adult NIHB clients subtracted from provincial jurisdiction adult populations. | The NIHB adult populations of Alberta and Saskatchewan, as well as those of Ontario aged 18 to 24 and 65+ were included in their respective provincial jurisdictions and subtracted from the NIHB population |
3. BCLC stage at diagnosis | Based on Canadian data for patients with HCC who had received lenvatinib39 Stage A: 2.31% Stage B: 27.17% Stage C: 65.32% Stage D: 0% | Based on Canadian data of liver cancer patients45 Stage A: 30.79% Stage B: 19.74% Stage C and unknown: 31.13% Stage D: 18.54% |
4. BCLC stage A receiving other treatments | Patients diagnosed at BCLC stage A who did not receive a liver transplant or resection (40%) were excluded from the model | The 40% of patients diagnosed at BCLC stage A were assumed to receive an alternate treatment and have 20% recurrence risk by 2 years, and a 30% recurrence risk by 3 years |
5. Median treatment discontinuation | STRIDE: |||||||| months Sorafenib: |||||||| months | STRIDE: 2.99 months Sorafenib: 2.53 months |
CADTH base case | 1 through 5 | |
BCLC = Barcelona Clinic Liver Cancer; HCC = hepatocellular carcinoma; MAIC = matching adjusted indirect comparison; NIHB = Non-Insured Health Benefit; STRIDE = single-dose tremelimumab in combination with regular-interval durvalumab.
The results of the CADTH stepwise reanalysis are presented in summary format in Table 18 and a more detailed breakdown is presented in Table 19.
CADTH reanalysis suggests that reimbursing the STRIDE regimen for the treatment of unresectable HCC would be associated with an incremental cost of $5,816,972 in Year 1, $6,532,047 in Year 2, and $6,053,880 in year 3, for a 3-year budgetary impact of $18,402,899.
CADTH conducted additional scenario analyses (Table 19) to highlight uncertainty in the model including: reintroducing administration costs, assuming 90% of otherwise eligible patients are treated with first-line systemic therapy, and assuming that the STRIDE regimen would displace comparators proportionally to their reference case market shares, and assuming a 50% price reduction for the STRIDE regimen resulting from the CADTH base case economic evaluation.
Table 18: Summary of the CADTH Reanalyses of the Budget Impact Analysis
Stepped analysis | Three-year total |
|---|---|
Sponsor’s submitted base case | $40,334,108 |
Corrected sponsor’s base case | $40,261,488 |
CADTH reanalysis 1: Drug payer perspective | $43,375,154 |
CADTH reanalysis 2: NIHB reimbursed population | $40,194,182 |
CADTH reanalysis 3: BCLC staging at diagnosis | $24,482,468 |
CADTH reanalysis 4: BCLC stage A receiving other treatments | $40,324,612 |
CADTH reanalysis 5: Median time to discontinuation | $27,943,208 |
CADTH base case (1 through 5) | $18,402,899 |
BCLC = Barcelona Clinic Liver Cancer; NIHB = Non-Insured Health Benefit.
Table 19: Detailed Breakdown of the CADTH Reanalyses of the Budget Impact Analysis
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | 3-year total |
|---|---|---|---|---|---|---|
Corrected sponsor’s base case | Reference | $36,743,840 | $64,226,645 | $72,822,745 | $77,221,906 | $214,271,295 |
New drug | $36,743,840 | $74,467,284 | $87,282,938 | $92,782,561 | $254,532,783 | |
Budget impact | $0 | $10,240,640 | $14,460,194 | $15,560,655 | $40,261,488 | |
CADTH base case | Reference | $17,131,124 | $30,230,252 | $34,718,047 | $37,182,793 | $102,131,092 |
New drug | $17,131,124 | $36,047,224 | $41,250,094 | $43,236,673 | $120,533,991 | |
Budget impact | $0 | $5,816,972 | $6,532,047 | $6,053,880 | $18,402,899 | |
CADTH scenario 1: Administration costs included | Reference | $17,323,580 | $30,583,639 | $35,134,124 | $37,636,629 | $103,354,392 |
New drug | $17,323,580 | $36,417,031 | $41,668,956 | $43,670,530 | $121,756,518 | |
Budget impact | $0 | $5,833,392 | $6,534,832 | $6,033,901 | $18,402,126 | |
CADTH scenario 2: 90% of eligible patients treated | Reference | $25,696,685 | $45,345,378 | $52,077,071 | $55,774,189 | $153,196,638 |
New drug | $25,696,685 | $54,070,836 | $61,875,141 | $64,855,009 | $180,800,986 | |
Budget impact | $0 | $8,725,458 | $9,798,070 | $9,080,820 | $27,604,348 | |
CADTH scenario 3: proportional comparator displacement | Reference | $17,131,124 | $30,230,252 | $34,718,047 | $37,182,793 | $102,131,092 |
New drug | $17,131,124 | $33,643,811 | $36,555,338 | $37,515,323 | $107,714,473 | |
Budget impact | $0 | $3,413,559 | $1,837,291 | $332,531 | $5,583,381 | |
CADTH scenario 4: 50% STRIDE price reduction | Reference | $17,131,124 | $30,230,252 | $34,718,047 | $37,182,793 | $102,131,092 |
New drug | $17,131,124 | $31,611,979 | $34,773,070 | $35,752,731 | $102,137,780 | |
Budget impact | $0 | $1,381,727 | $55,022 | −$1,430,061 | $6,688 |
BCLC = Barcelona Clinic Liver Cancer.
Stakeholder Input
Patient Input
Colorectal Cancer Resource & Action Network
About Colorectal Cancer Resource & Action Network
The Colorectal Cancer Resource & Action Network (CCRAN) is a national not for profit patient advocacy group championing the health and wellbeing of Canadians touched by colorectal cancer and those at risk of developing the disease. It has expanded its mandate to serve cancer patients outside the colorectal space by providing HTA patient input submissions within the oncology space for:
Patient groups who do not have the capacity to perform these submissions and
A therapeutic area wherein there currently exists no representative patient group to complete a submission (such as the therapy currently under review).
CCRAN led a collective patient input submission on tremelimumab for injection in combination with durvalumab for the first-line treatment of patients with unresectable hepatocellular carcinoma (HCC). The following patient advocacy groups thoughtfully collaborated with CCRAN to ensure the advanced hepatocellular cancer patient/caregiver perspective was captured, represented and well weaved throughout this submission:
Canadian Cancer Survivor Network (CCSN)
Canadian Liver Foundation
GI Society (https://badgut.org/)
All patient groups are registered with CADTH.
Information Gathering
Anticipating this could be a difficult file, in collaboration with the three patient advocacy groups, CCRAN employed a multi-faceted outreach strategy to help secure the advanced hepatocellular carcinoma (HCC) patient input.
On September 17, 2022, CCRAN reached out via email to 14 Canadian clinicians who treat HCC, some of whom served as investigators in the HIMALAYA trial. Having heard from very few of the clinicians, follow up emails were subsequently resent on October 1 and October 15, 2022, with accompanying phone calls. Email correspondences were also sent to 7 U.S. based clinicians including HIMALAYA investigators, as well as 3 European HIMALAYA investigators on the same three dates. All clinicians were supplied with a poster to share with any patients/caregivers having firsthand experience with the therapy under review, who would be willing to share their experience via a telephone interview with CCRAN.
On September 28, 2022, CCRAN contacted two U.S.-based patient advocacy groups to request assistance with patient recruitment: the American Liver Foundation and Liver Cancer Connect. Both organizations were keen to promote the poster through their social media channels (October 15 to November 30).
Additionally, CCSN designed and employed an online survey to help capture the advanced HCC patient’s experience with the disease, currently available treatments and the therapy under review. The online survey was administered between October 3, 2022 – January 3, 2023. The survey was sent to CCRAN, Canadian Liver Foundation, Blue Faery, GI Society, and the Coalition Priorité Cancer au Québec, all of whom circulated the survey to their memberships and through their social media channels except Blue Faery. While 14 users attempted to complete the survey, no survey respondents provided input and we are, therefore, unable to have captured any meaningful data from this online survey. This could be, in part, due to the absence of a clear and definitive statement having been included in the opening paragraph of the survey, inviting individuals with HCC who do not have experience with the treatment under review, but who might benefit from the treatment, to complete the survey, nonetheless.
The Canadian clinician outreach effort resulted in ONE patient telephone interview. The telephone interview was conducted by CCRAN on November 21, 2022. The advanced HCC patient provided firsthand compelling, and relevant qualitative input regarding their:
Experience with respect to the diagnosis of their cancer
Disease experience
Experience with respect to previous therapies
Experience with respect to the therapy under review
The qualitative data from the interview and will be referenced throughout this submission.
CCRAN, and the balance of the patient advocacy groups, anticipated this might be a difficult and challenging submission as it relates to the procurement of patient input due to the high mortality rates associated with this pathology. Couple that with a limited number of patients accessing the therapy under review (through a trial setting) who may not be well enough or available to respond to our plea for patient input. CCRAN diligently commenced the search for patient input months before the submission deadline, but our efforts were to no avail. Our efforts generated only two high quality patient interviews: both of whom had firsthand experience with the disease and currently available treatments (Patient A and Patient B) but only one had first had experience with the therapy under review (Patient A). And sadly, CCSN’s survey procured no survey respondents.
To complete this critically important patient evidence submission, CCRAN was required to pivot. We, therefore, decided to scour the literature and online public forums for patient reported outcomes (PROs) describing:
their advanced HCC journey,
the treatments’ (including the therapy under review) impact on their daily life, including physical, psychological functioning and well-being.
PROs in addition to patient stories and testimonials were sought and incorporated into this submission in the most comprehensive manner possible to ensure the advanced HCC patient voice is provided to help inform this committee’s deliberations.
Disease Experience
Hepatocellular carcinoma (HCC) is the most common primary liver cancer. It is the 6th most common cancer worldwide and 4th leading cause of cancer-related deaths. This cancer has been increasing due to the increased burden of Hepatitis C infection. Risk factors associated with HCC include cirrhosis, Hepatitis B and C infections and alcohol intake. Non-alcoholic steatohepatitis (NASH) has also emerged as an important cause of HCC. HCC symptoms represent a substantial physiological and psychological burden for patients and can significantly affect their health-related quality of life (HRQoL).
In HCC, both the cancer and its treatment can be severely debilitating. According to the health-related quality of life data captured by Gandhi et al, patients with HCC report several symptoms which are severe enough to affect their quality of life such as, sleep disorders, sexual dysfunction, ascites, gynecomastia, pruritis, fatigue, and muscle cramps. [J Gastrointest Oncol 2014: 5(4):296-317] and [Korean J Radiol 2020;21(6):633-646]. In patients who are symptomatic from HCC, authors describe the most common presenting clinical features as right upper quadrant pain, jaundice, weight loss, anemia or high concentration of red blood cells. Among late-stage HCC patients, the most severe symptoms at diagnosis were fatigue and distress and 90% of patients reported pain during and after treatment. Fatigue was the most serious symptom followed by sleep disturbance, distress, sadness, and lack of appetite even after treatment. These symptoms have a significant impact on the patient’s functioning and well-being. Emotional concerns associated with the disease give rise to anxiety in patients, which is understandably life altering. Emerging data suggest that HRQoL, particularly role functioning (involvement in life situations involving family, partner relationship, work or household chores), may be independently associated with survival in patients with advanced HCC [Hepatocellular carcinoma | Nature Reviews Disease Primers]. A study describing the presence, frequency, severity, and distress of symptoms in outpatients with advanced HCC toward the end of life reported lack of energy and pain as the most frequent and distressing symptoms for patients. Problems with sexual interest or activity was the fourth most present symptom after drowsiness. [Symptom Distress in Patients With Hepatocellular Carcinoma Toward the End of Life; November 2017: Oncology Nursing Forum 44(6):665-673]
Through a public forum, Their Stories. Our Purpose. | Susie & Robin MASTER.mp4 | Powered by Box , Robin described his HCC journey. He lived in the UK until 2020, at which point he then relocated permanently to France. He worked in the media industry for most of his life but is now semi-retired. His passions include flying and rebuilding cars. Robin was diagnosed with cirrhosis in 2010 after having felt quite unwell. His symptoms included vomiting and stomach bleed. He was advised at the time that his cirrhosis put him at higher risk of developing liver cancer but did not quite understand that he was almost certain to develop the pathology. When he was diagnosed with pneumonia in 2017, he insisted on an MRI which revealed a tumour on his liver. Diagnosed with HCC in December 2017, he started Transarterial Chemoembolization (TACE) in January 2018 for three months to reduce the size of the tumour but there had been little to no response. He notes there were no side effects from the TACE but was quite disappointed in its lack of efficacy. He was then advised to start the combination therapy Atezolizumab + Bevacizumab in 2020 but declined. Disease progression ensued so he started the combination therapy in June 2022.
Through another public forum as well Instagram account:
https://youtu.be/I6iDFx0unoE , https://youtu.be/ZBvwKfSprrw, https://youtu.be/LBcX88iavUI
https://www.instagram.com/p/Cmo3TX3p_PC/?igshid=MDJmNzVkMjY=
https://www.instagram.com/p/CmzScP8u4xd/?igshid=MDJmNzVkMjY=
https://www.instagram.com/p/CmEgnkUMgiw/?igshid=MDJmNzVkMjY=
Sharon describes her caregiver’s journey as well as her husband Jimmy’s HCC journey. Jimmy lives in Texas, U.S. and is also afflicted with COPD. He was diagnosed in the 1990’s with Hepatitis C and cirrhosis which worsened 2-3 years ago. He was admitted to hospital in December 2020 for pancreatitis due to severe pain. The initial scans revealed lesions on his liver. A diagnosis of HCC was confirmed 3 months later and went on to receive radiation therapy to shrink the largest tumour, but additional tumours developed in the interim. Jimmy was then prescribed Lenvima from which he experienced mouth/throat sores and subsequently whole-body sores. The treatment was stopped and then restarted. The Lenvima ceased working in December 2021, at which point he then started Atezolizumab + Bevacizumab therapy. Jimmy does note that he was certainly symptomatic from his cancer and did have a difficult time with the cancer itself.
With respect to our interviewed patients, Patient A is a 74-year-old male with four adult children. He was diagnosed with HCC in April 2016 at the age of 68. He had not been experiencing any symptoms whatsoever at the time of his HCC diagnosis but was diagnosed with “horrible cirrhosis from Hepatitis C” for which nothing could be done at the time. With a confirmed diagnosis of HCC, he was prescribed one treatment of TACE in 2017 to which there was no response.
Patient B is currently a 92-year-old female who was diagnosed with Hepatitis C in 1996 and widespread cirrhosis. She was subsequently diagnosed with HCC in April 2001 at the age of 71. Between 1996 and 2001, she relayed that her Hepatitis C and cirrhosis were quite inconsequential and did not impact her quality of life. She was not symptomatic, though she was under the regular care of a hepatologist who detected her HCC in its early stages. This qualified her for surgical resection of her HCC on May 3, 2001. In her words (Q7B):
“No, I wasn’t symptomatic, but I did have high blood levels of certain liver values. That is what I was told. Between my cirrhosis, Hepatitis C and the liver cancer, some of my levels were high. That was bad. And this worried me. I wasn’t young but I wasn’t old either in my opinion. I had a lot of living left to do. I wanted to enjoy my grandchildren who had just joined our family and I finally had retired from work, so I wanted to travel with my husband.”
Over the course of the next twenty-two years, Patient B has experienced 3 recurrences post-surgical resection, two of which were successfully treated with Radiofrequency Ablation (RFA) and the last of which is currently under a watch and wait surveillance approach.
When asked if there was any particular aspect of the disease that was difficult to control while on previous therapies, Patient B expressed how demoralizing and discouraging it has been to have continuously been assaulted emotionally with three recurrences. She found herself cycling through the same stages of cancer grief – anger, depression, guilt, anxiety, hopelessness, and fear – which hit her quite hard during her original diagnosis, but just as violently with each subsequent recurrence. She emotionally relays:
“I don’t know about the disease, but I guess it’s the cirrhosis that just kept giving me more and more tumours. It has been so demoralizing and discouraging. I even went on drugs to try to kill the Hepatitis C to help the cirrhosis, like Harvoni and sovaldi. But I just kept getting more tumours. My hepatitis got cured but my cirrhosis, there was little that could be done to get rid of that. I just couldn’t do anything about it. why wouldn’t it go away? And every time it came back, it was like the first time. I would get scared all over again, afraid for my future and family. So scared to the point of trembling. I did everything I could. But nothing helped. Why, why? My cancer just kept coming back in different spots of my liver. (cried)”
Experiences With Currently Available Treatments
HCC is a unique carcinoma because the majority of cases will develop in patients with cirrhosis and, therefore, therapeutic options will be limited due to the patient’s overall health status. Patients with early-stage HCC tumours are preferred candidates for resection, transplantation, and local ablation whereas patients at intermediate stages may be candidates for transarterial chemoembolization (TACE), while those with advanced disease will receive systemic therapies, which might include Atezolizumab + Bevacizumab, Lenvatinib, Sorafenib, Regorafenib, and Cabozantinib. Quality of life is especially important in this cancer population who have reported feelings of “fear, worry and anxiety” upon receiving a diagnosis of HCC due to the often-poor prognosis associated with advanced HCC (Cancers 2019, 11, 841; doi:10.3390/cancer11060841). The same paper reported on a global quality of life survey that captured the perspectives of 256 patients diagnosed with HCC. Several treatment-related symptoms such as fatigue, sexual dysfunction, abdominal pain, nausea, skin disorders, diarrhea and alopecia were reported. Of those patients who were working at the time they started HCC treatment, 60% stated the side effects they experienced caused them to stop working. Systemic treatment was reported to negatively affect patients’ relationships with family and caregivers, ability to perform daily activities, and their outlook for the future.
During the last decade, sorafenib, lenvatinib, atezolizumab/bevacizumab combination, cabozantinib, and regorafenib have proven efficacy with longer progression free survival (PFS) or overall survival (OS). These treatments have potential side effects, however, that can impact a patient’s quality of life – a clinical outcome just as important as PFS or OS in the management of patients diagnosed with advanced HCC.
Despite the improvement in overall survival with sorafenib alone, most patients potentially discontinue treatment due to poor tolerance of side effects and dose reductions are common. Drug resistance is commonly encountered for patients who adhere to the therapy. The most common adverse events associated with sorafenib include diarrhea, hand-foot skin reaction, weight loss and hypophosphatemia. Other agents have been approved for the management of the cancer (Cancers 2019, 11, 841) and (Curr Oncol 2022, 29, p. 5490) which include lenvatinib, regorafenib, cabozantinib and atezolizumab/bevacizumab combination, discussed below.
According to the same papers, the most frequent adverse events associated with lenvatinib were arterial hypertension, diarrhea and decreased appetite. Similar side effects to that of sorafenib were observed with regorafenib. The most common adverse events for patients treated with regorafenib include hypertension, hand-foot skin reaction, fatigue and diarrhea. The most frequent side effects associated with Cabozantinib were hand-foot skin reaction, hypertension, elevated transaminases, fatigue and diarrhea. Dose reductions were common as well. The most commonly reported side effects of the Atezolizumab + Bevacizumab therapy, were hypertension, proteinuria and low-grade diarrhea. Upper gastrointestinal endoscopies were and are still required within 6 months prior to receiving the therapy for the treatment of varices in all patients to mitigate the risk of bleeding associated with bevacizumab. This represents a change in practice, especially for the screening of patients in first line therapy, as upper gastrointestinal endoscopies will have to be performed prior to treatment initiation, which could potentially, delay treatment. They represent an additional burden on the patient – one that is necessary but nevertheless, a burden. (Nature Reviews, Disease Primers, 2021; 7:6)
Our online patient, Jimmy, accessed Lenvatinib for the treatment of his advanced HCC, after having tried to control the largest tumour in his liver through radiation therapy, which proved unsuccessful. The most notable side effect he reported from the lenvatinib therapy was the development of mouth and throat sores which then progressed into whole body sores. This necessitated treatment cessation, but he was able to eventually restart the therapy. The treatment was effective for approximately 9 months, at which point he then started Atezolizumab + bevacizumab therapy. Jimmy sadly succumbed to the disease on October 27, 2022.
Our second online patient, Robin, accessed TACE in January 2018 for 3 months to reduce the tumour burden in his liver. He reports no treatment induced side effects but was disappointed to learn of its complete lack of efficacy, for he learned of the disease progression which required accessing systemic therapy: Atezolizumab + Bevacizumab in June 2022.
Patient A was prescribed one treatment of TACE for the 3 tumours in his liver in 2017 to which there was no response whatsoever. Patient A was saddened to learn of the lack of response. In his words:
“I had no quality of life with this therapy. I just laid in bed, crippled, it was terrible, so much pain. And it didn’t even work for me. I would never undergo that again, no matter what. I was not able to do anything for 2-3 days. It was so nasty, just kept giving me pain meds to help with all the horrible pain I was having. I just laid there. That’s all that came of that therapy.”
And in Patient A’s words, TACE did not control his cancer, nor did it regress his cancer:
“Oh, God no. It did not. Based on the CT scan anyway. Actually, there has been progression, if anything. It has been slow growing, my disease, but progressive disease. The TACE therapy did not do anything at all. No regression at all.”
And,
“There was not regression at all. Yes, it was minimal, but my cancer was growing over the whole time. It never had a chance to stop or regress at any time!”
Patient B was diagnosed with early-stage HCC in April 2001. She underwent surgical resection for her disease on May 3, 2001, and two subsequent Radiofrequency Ablations (RFAs) in September 2008 and February 2019 respectively for two recurrences which were successful at eradicating the intended tumours. She is currently experiencing a third recurrence for which she has decided to forgo treatment. She describes her recovery from surgery at the age of 71 as difficult and challenging. Pain management was a daunting task, and she had a difficult time resuming her normal, everyday activities, such as bathing, dressing or walking. In her words (Q8A):
“That was not an easy surgery. I was in the hospital for two weeks! I had a hard time managing my pain and getting back to my normal life. I couldn’t do very much while I recovered. My husband, who was also in his late 70’s, had to take care of me. He did everything for me and so did my family. It took months before I was myself again, able to bathe myself, dress myself, be pain free, walk without hunching over, able to cook, or go for a walk. It was awful.”
She describes her experience with the RFA treatment as successful but not without some hardship. Post-RFA pain management was difficult to navigate because of limited pain-relieving options available to her. She shares:
“After surgery, I experienced three recurrences, two of which were treated really well with RFA. The first was in September 2008 and the second was in February 2019. The third, which I am experiencing now, I have chosen not to treat. RFA was a lot easier and there was no hospital stay but there was pain immediately after the procedure. I was limited on the pain medications that I could take because of my widespread cirrhosis so I would suffer for a couple of weeks after the procedure. But it was a lot better than the surgery, that’s for sure. And it treated the recurrence really well. It was a success. But the cancer just kept coming back. Oh boy.”
Surgical resection and RFA managed to keep Patient B’s HCC under control for well over twenty years. Her surgery kept her disease free for seven years and the RFAs achieved disease control with an excellent quality of life for eleven and three years.
Improved Outcomes
To help capture patients’ views on what outcomes should be considered when evaluating new therapies, CCRAN turned to the Lo et al study [Future Oncol. (2021)17(32);4275-4287] which aimed to understand patient preferences for characteristics of advanced HCC treatments. A stated preference survey was completed by 150 patients living in Europe, of whom 20% had a single tumour nodule, 55% had multifocal intrahepatic disease and 21% had extrahepatic disease.
The study examined what is important to people with HCC across four European countries when choosing a treatment for advanced HCC, given that OS was limited to approximately 1-2 years for these patients. The Discrete Choice Experiment (DCE) survey identified that patients placed most value on extending OS. However, the results also demonstrated that they were willing to forego several months of OS in order to maintain their quality of life, by avoiding the risk of treatment-related adverse events. The study results support clinician and patient discussion around shared decision making for treatment options in advanced HCC.
The study findings are consistent with the perspective provided by Patient A in Q25 of APPENDIX 2. When asked what improvements he would like to see in a drug therapy, Patient A thoughtfully provided the following:
“If I had my way, I would like to see a treatment that provides excellent quality of life, because that is most important to me. And of course, just as important is a treatment that provides survival. How the treatment is delivered is important too, like a capsule form via mouth. But you cannot have a better therapy than what I have had, except the infusional part.”
And when asked if he believed the therapy under review had those desired improvements, Patient A emphatically replied:
“Oh, yes, I sure do. It was the best, barring the capsule part.”
In light of the poor prognosis associated with advanced HCC, there is an urgent need to prioritize patient centered outcomes such as quality of life, together with overall survival. According to the study results and Patient A, the therapy under review addresses and provides these desired improvements.
Experience With Drug Under Review
There is no doubt that patients with unresectable HCC are in need of well-tolerated therapies that can meaningfully extend overall survival and provide that highly sought-after quality of life. Despite currently available treatments for the management of unresectable HCC, new options are required to improve long term disease control and patient survival because to date, there truly is an unmet need in this patient population as it relates to extending life with few to no side effects. The therapy under review [Tremelimumab (checkpoint blockade against CTLA-4) + Durvalumab (checkpoint blockade against PD-1)] may serve as that new, effective combination therapeutic that can significantly improve overall survival compared to what has been previously administered in the past.
After having accessed one treatment of TACE in 2017, Patient A articulately described the lack of response to that therapy, expressing his sheer disappointment to his progressing HCC. After failing TACE, Patient A was referred to a medical oncologist in Calgary, which he believed was the “best thing that could have ever happened to him”. He was referred to the Tremelimumab + Durvalumab study, in the second line setting, which he started in March 2018 and ended in May 2022. He received, he believes one or two doses of the Tremelimumab upfront, and then the balance of the therapy consisted of Durvalumab. In total, approximately 40 cycles were delivered via infusion. In terms of any adverse events or treatment induced side effects, he notes (Q15):
“No, not really, I would get the occasional rash on my skin, in particular on my lower legs, but nothing of consequence. I used to play golf twice a week while I was on the treatment the whole time. I would stay on this my whole life if I could if the study would let me and had not ended.”
“And I would definitely give it a 10 (rating)!”
The response to therapy was confirmed not only clinically but radiographically for Patient A, through MRI and CT every 3 and 6 months respectively. Patient A’s disease either disappeared entirely or has reduced in number and size (Q18A).
“4 or 5 spots shrank and now I am left with 2 or 3 very small spots and others have completely disappeared. It is so wonderful. What a great therapy.”
In describing the therapy’s ease of use (Q19B), Patient A relays:
“I would describe it or compare it to giving blood. It lasts only one hour so it is not time or effort intensive. It is really easy. Initially, I received Treme and then I started receiving the Durva. It is no big deal at all. It has saved my life, and it has been a really good quality of life.”
Quality of life is becoming a major endpoint in oncology research. Improvements in quality of life are captured by changes in patient reported outcomes, which result from clinical benefits from treatments and from the impact of adverse events and tolerability of potentially toxic drugs. Some of the challenges to systemic therapies in advanced HCC has been the limited tolerability of treatments, owing in part to adverse events from the treatments themselves. Patient A was happy to relay the deep and durable immune response experienced while on the therapy under review and the favourable adverse event profile in comparison to the previous therapy (TACE) he received, which, not only failed to provide benefit, but had also failed to provide any meaningful improvement in his quality of life. Quite the contrary: his quality of life had deteriorated quite drastically due to the TACE therapy. “I had no quality of life with this therapy. I just laid in bed, crippled, it was terrible, so much pain. And it didn’t even work for me….”
The Tremelimumab + Durvalumab therapy, on the other hand, provided an improved and sustained quality of life relative to the previous treatment, and a durable, robust, longstanding response that was highly welcomed and much appreciated by Patient A. When asked if it was worth having accessed the therapy (Q23), Patient A enthusiastically replied:
“Oh, my goodness…Absolutely 100% yes. Of course, it has been worth it. Where would I be today without it? There are so many wonderful people I met along the way in the centre who have enriched my life. The treatment itself has kept me alive, ALIVE!! And with no side effects!! Again, I am alive today because of this therapy. It was so very worth it. I am beyond grateful for this trial that my oncologist recommended!”
The therapy permitted Patient A the freedom and ability to live his life to the fullest. He was well enough to build his own home, play sports, travel, spend quality time with his family and friends. Every aspect of his life was attributed and indebted to this “wonderful therapy”, never having lost sight of how close he came to having gone down a different path and potentially not having survived long enough to be able to contribute to this submission. In his words (Q24):
“… gave me a chance to continue with my life. I actually got to build a home - my very own home with my own hands! I got to retire and play golf, travel lots, and be able to spend an abundant of time with my family all of whom I adore and cherish – all because of this therapy I got to access through a study. I wouldn’t have been able to do this on TACE! I certainly would not have been able to accomplish any of this, least of all build my own home on TACE. And now I just sold that home and got to reap the benefits! It was all because of this wonderful therapy that I was able to do all this and more! I have been able to lead a gratifying and fulfilling life on this therapy.”
When describing the toll his cancer journey has had on his wife, Patient A credits his wife’s emotional improvement to the study drugs (Q22):
“She worries more about me than I do. I travel to Calgary and come back a lot. She worries so much, which is understandable. It takes a toll. She travels with me too. The whole journey has impacted her emotionally but I have to say that this therapy which has been so therapeutic and without side effects moderated that emotional toll and ill being, so it got better in time. The caregiver always takes on the brunt of the journey for the patient. It is what they do. But this treatment has managed to mitigate some of that brunt. How wonderful…”
Patient B described, as she became emotional, the impact her cancer journey had on her family, in particular her husband (Q21):
“Initially, I lost sleep over this disease, I would stress, cried and worried a lot over this cancer, wondering if I would live to see more grandchildren, but as time went on, it got better. I even got to see great grandchildren be born which has brought me great joy. My husband did a lot of crying as well. He knew what a diagnosis of liver cancer meant but tried to hide it in front of me. But I caught him crying in the dark one night. I couldn’t bring myself to ask him why he was crying. But I knew why…………... I can tell that my family now is worried for me because I am frail and elderly. My husband is gone and my son and daughter in law are all I have. This cancer shows no mercy. It just keeps coming for you in a relentless and determined fashion. There has to be more to eventually put a final stop to this cancer.”
Companion Diagnostic Test
Reliable molecular markers to predict prognosis and treatment response to target therapy is lacking in the HCC space.
Anything Else?
HCC is one of the most malignant neoplasms in Canada, with a majority of the cases presenting at advanced stages. Regardless of the recent expansion in treatment options for patients diagnosed with advanced HCC, the prognosis of this patient population continues to be poor. The development of concrete sequencing strategies that include both a patient’s quality of life and survival as primary endpoints is an unmet need that this cancer patient population faces when undergoing therapeutic interventions.
In the last few years, there has been a dramatic change in the treatment algorithm, given new drug approvals in the advanced HCC space. Most importantly, the combination of atezolizumab and bevacizumab has demonstrated a clinical benefit in terms of response rate compared to sorafenib in the first line treatment of advanced HCC. However, given the bleeding risk associated with bevacizumab, all patients considered for the therapy are required to undergo endoscopic evaluation within 6 months prior to treatment initiation. Patients are carefully screened for bleeding risk, and varices have to be treated prior to the start of atezolizumab and bevacizumab therapy. This is an issue that does not impact patients who qualify for the therapy under review – a burden to which advanced HCC patients need not be subjected. Additionally, treatment-induced hypertension (a concern associated with bevacizumab) is not associated with the tremelimumab + durvalumab and while patients will start with a dual immunotherapy regimen, the tremelimumab will be eliminated after its first administration, leaving the durvalumab as a single infusion therapy to treat the patient’s advanced HCC. This clearly reduces the amount of time and effort spent at a clinic/cancer centre for the patient and their caregiver as well as, potentially, reducing the cost incurred to the health care system. The therapy under review is also administered once every four weeks (Q4W) versus once every three weeks for the atezolizumab + bevacizumab regimen, thereby, reducing the amount of visits/travel on behalf of the patient and their caregiver. Expenses on the whole are reduced. Time spent at the clinic/cancer centre is minimized. This is time that can be better spent by the patient. Lastly, there is another restriction regarding access to atezolizumab + bevacizumab therapy that does not apply to tremelimumab + durvalumab: low platelet count. This eligibility criteria will not impact the advanced HCC patient who qualifies for the therapy under review.
Therefore, tremelimumab + durvalumab combination is an excellent option for patients who are not suitable for atezolizumab + bevacizumab therapy, such as in the setting of elevated bleeding risk or low platelet count or any of the other aforementioned points.
Furthermore, based on the input captured herein, the side effect profile and tolerability to tremelimumab + durvalumab therapy was a major contributor towards improving and conserving quality of life in our advanced HCC patient (Patient A), which helped to achieve increased life expectancy. Patient A wished to express the following final thoughts to this kind committee (Q27):
“During my experience, I met many people who had liver cancer. They were on many different therapies, and I saw that their quality of life was nowhere near as good as mine and they also did not survive. I do not know what therapies they were on, but I do know they paled in comparison to me when it came to their quality of life and survival. I just think this therapy should be made available to those who qualify for the treatment because it is an easy treatment, and it extends life with excellent quality of life. I would recommend it to anyone who has liver cancer because it would be a blessing to them all.”
Patients value having access to new therapies that have few side effects, can improve their quality of life, allow them to be engaged in society, functioning and contributing members of the work force, and are able to be committed to their families and friends. This is a common theme expressed repeatedly by patients and their caregivers throughout various tumour types, but a critical unmet need exists for patients with advanced HCC who face limited treatment options that can extend progression free survival, overall survival and improve quality of life in a truly meaningful way. The therapy under review provides patients with another treatment option with manageable side effects, improved quality of life, durable and sustained response compared to previously accessed therapies, and increased longevity. To have observed the magnitude of the response in Patient A confirms that the therapy under review is effective and amenable for long term administration. If publicly funded, tremelimumab + durvalumab would be an extremely important therapeutic option for the HCC patient where surgical resection is unlikely or have progressed following treatment. Funding this therapeutic aligns well with the input captured within this submission.
We, therefore, strongly support and urge that a positive funding recommendation be issued for tremelimumab injection in combination with durvalumab for the treatment of patients with unresectable hepatocellular carcinoma. We believe a positive funding recommendation aligns well with the identified patient need for a new, effective, easily administered treatment option that is capable of maintaining a high quality of life, durable response and increased survival relative to previously administered therapies.
Conflict of Interest Declaration — Colorectal Cancer Resource & Action Network
To maintain the objectivity and credibility of the CADTH CDR and pCODR programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission?
No.
Did you receive help from outside your patient group to collect or analyze data used in this submission?
No.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review.
Table 1: Financial Disclosures for Colorectal Cancer Resource & Action Network
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Conflict of Interest Declaration — Canadian Liver Foundation
To maintain the objectivity and credibility of the CADTH CDR and pCODR programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission?
No.
Did you receive help from outside your patient group to collect or analyze data used in this submission?
No.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review.
Table 2: Financial Disclosures for Canadian Liver Foundation
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Astra Zeneca | — | — | — | X |
BMS | — | — | Xa | — |
Hoffmann-LaRoche Ltd | — | — | — | Xa |
Eisai Inc | — | — | — | X |
Ipsen Biopharmaceuticals | — | — | — | X |
aFunding received not exclusive to HCC. Funding supported other liver-related issues including Hepatitis C and liver transplantation.
Conflict of Interest Declaration — Gastrointestinal Society
To maintain the objectivity and credibility of the CADTH CDR and pCODR programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission? If yes, please detail the help and who provided it.
Only from others who are part of this submission.
Did you receive help from outside your patient group to collect or analyze data used in this submission? If yes, please detail the help and who provided it.
Only from others who are part of this submission.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review.
Table 3: Financial Disclosures for Gastrointestinal Society
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
AstraZeneca 2022 | — | — | X | — |
AstraZeneca 2021 | — | — | X | — |
Clinician Input
Ontario Health (Cancer Care Ontario) Gastrointestinal Cancer Drug Advisory Committee
About Ontario Health (Cancer Care Ontario) Gastrointestinal Cancer Drug Advisory Committee
OH-CCO’s Drug Advisory Committees provide timely evidence-based clinical and health system guidance on drug-related issues in support of CCO’s mandate, including the Provincial Drug Reimbursement Programs (PDRP) and the Systemic Treatment Program.
Information Gathering
The information was jointly discussed at a meeting.
Current Treatments and Treatment Goals
Atezolizumab-bevacizumab (atezo-bev) 1L followed by oral agents (e.g., sorafenib, lenvatinib)
In some patients, 1L lenvatinib may be preferred (per patient preference or atezo-bev contraindication), followed by regorafenib 2L and cabozantinib 3L.
The most important goals are prolonging life, delay disease progression, reduce the severity of symptoms, symptom management, delay deterioration, maintain health-related quality of life, minimize adverse effects.
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
Tremelimumab in combination with durvalumab (durva-treme) has a different side effect profile. For patients with hypertension and varices, durva-treme may be a better treatment option. None of the 1L treatment options have been compared in head-to-head comparison trials.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
Durva-treme can be used to treat a patient population for whom local therapies are no longer appropriate.
Durva-treme can be given to those who have contraindications to bevacizumab. Patients do not require pretreatment with upper endoscopy. Durva-treme would represent another 1L treatment option per treating clinician discretion and patient preference.
Which patients would be best suited for treatment with the drug under review? Which patients would be least suitable for treatment with the drug under review?
Best suited for durva-treme: per HAMALAYA eligibility criteria
No companion diagnostic test required.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice? How often should treatment response be assessed?
Disease response and patient tolerability.
Treatment response should be assessed every 3-4 months or as clinically indicated as assessed by cross-sectional imaging.
What factors should be considered when deciding to discontinue treatment with the drug under review?
Disease progression or unacceptable drug-related toxicities
What settings are appropriate for treatment with [drug under review]? Is a specialist required to diagnose, treat, and monitor patients who might receive [drug under review]?
Outpatient infusion clinics, including satellite clinics.
Conflict of Interest Declarations — Ontario Health (Cancer Care Ontario) Gastrointestinal Cancer Drug Advisory Committee
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please refer to the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
Yes. Ontario Health provided secretariat functions to the DAC.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission?
No.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician who contributed to the input.
Declaration for Clinician 1
Name: Dr. Erin Kennedy
Position: Lead, OH-CCO GI DAC
Date: 13-01-2023
Table 4: COI Declarations for OH-CCO Gastrointestinal Cancer Drug Advisory Committee — Clinician 1
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 2
Name: Dr. Jim Biagi
Position: Member, OH-CCO GI DAC
Date: 13-01-2023
Table 5: COI Declarations for OH-CCO Gastrointestinal Cancer Drug Advisory Committee — Clinician 2
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 3
Name: Dr. Tim Asmis
Position: Member, OH-CCO GI DAC
Date: 13-01-2023
Table 6: COI Declarations for OH-CCO Gastrointestinal Cancer Drug Advisory Committee — Clinician 3
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
AstraZeneca | X | — | — | — |
Declaration for Clinician 4
Name: Dr. Suneil Khanna
Position: Member, OH-CCO GI DAC
Date: 13-01-2023
Table 7: COI Declarations for OH-CCO Gastrointestinal Cancer Drug Advisory Committee — Clinician 4
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
AstraZeneca | X | — | — | — |
Declaration for Clinician 5
Name: Dr. Yoo-Joung Ko
Position: Member, OH-CCO GI DAC
Date: 13-01-2023
Table 8: COI Declarations for OH-CCO Gastrointestinal Cancer Drug Advisory Committee — Clinician 5
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
The Canadian Gastrointestinal Oncology Evidence Network
About The Canadian Gastrointestinal Oncology Evidence Network
The Canadian GI Oncology Evidence Network (CGOEN) is a virtual and inclusive network of Canadian GI Oncology clinicians who contribute to the knowledge of GI cancer and its treatments, including participating in clinical trials, conducting observational research, and involvement in local/provincial and national clinical guideline development and health technology assessment.
Information Gathering
Information gathered for this submission was based on relevant data from the HIMALAYA (Tremelimumab plus Durvalumab in Unresectable Hepatocellular Carcinoma) trial and expert evidence-based review by Canadian gastrointestinal cancer specialists.
Current Treatments and Treatment Goals
Hepatocellular carcinoma will affect 3500 Canadian a year and approximately 1650 will die from the disease.
Current systemic therapy options available for patients with unresectable HCC are Atezolizumab/Bevacizumab or Lenvatinib or Sorefenib. Lenvatinib or Sorafenib can be considered for patients who have a contraindication to immunotherapy or increased bleeding risk. Current treatments have demonstrated an increase in overall survival and a decrease in toxicity. The STRIDE regimen is a single dose of Tremelimumab and with Durvalumab. It has recently received Heath Canada approval and an access program is available. Patients have been receiving this combination through SAP.
The current treatments help improve overall survival. More modern treatments have demonstrated improved response rates which has translated into symptomatic benefit. In certain cases, the downsizing of disease has allowed for local regional therapies to be considered to debulk the liver disease or even provide the opportunity for transplantation. Transplantation in these cases would be curable – the majority of patients in this space would be considered palliative; however, with modern systemic therapy, downsizing of disease has led to newer opportunities for local regional treatments to the liver (such as stereotactic radiation, embolization, ablation, resection-or transplant).
The idea therapy would improve the overall survival for the patient while providing a reasonable side effect profile that does not overly impact on the patient’s quality of life. Response rates can also be a factor as some of the tumors can be large and either cause pain or biliary or vascular obstruction. Due to the underlying liver condition such as cirrhosis, the patient could be at an increased bleeding risk due to varices, or these tumors are vascular and larger tumors are prone to rupture. Therapies that do not increase this risk would also be key in this field.
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
All the current therapies have some bleeding risk associated with it as it targets the vascular endothelial growth factor pathway. Due to the underlying liver disease and the vascular nature of the disease itself – this disease is associated with increased bleeding risks either through varices due to underlying cirrhosis or bleeding from the tumor itself. The STRIDE regimen is the only pure immunotherapy treatment that does not target this pathway and therefore has a lower bleeding risk associated with it.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
The STRIDE regimen would be another first line therapy option. The population of HCC patients is heterogeneous. By having another option for physicians and patients – it allows a personalization of therapy depending on patient factors and preferences. It would be expected that patient could have a choice between STRIDE, Atezolizumab/Bevacizumab, or a TKI such as Lenvatinib or Sorafenib.
Additionally, in centers where urgent upper endoscopy is very difficult to obtain, the stride regimen allows patients to be treated with immunotherapy, something they would not be exposed to if they could not get an upper scope.
Which patients would be best suited for treatment with the drug under review? Which patients would be least suitable for treatment with the drug under review?
This therapy would be considered in patient with unresectable HCC. These patients would be identified by oncologists or reviewed in multiple disciplinary review. Adequate liver function is needed for therapy – patients should have a Child Pugh classification of A to be eligible. Although some patients with a B7 should also be considered. Patients who have an underlying auto-immune condition may not be eligible for treatment.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice? How often should treatment response be assessed?
Patients would undergo routine bloodwork and imaging to assess treatment response. A clinically meaningful response to treatment would be an improvement in overall survival. In addition, improvement or maintenance of quality of live is also important. Response rates can lead to downsizing of disease leading to a reduction of symptoms and the possibility of other modalities such as local regional therapies to control the disease process.
What factors should be considered when deciding to discontinue treatment with the drug under review?
Drug should be discontinued if there is disease progression, toxicity or patient preference. Most of the side effects are immune related. If these side effects cannot be effectively managed, then treatment should be discontinued.
What settings are appropriate for treatment with [drug under review]? Is a specialist required to diagnose, treat, and monitor patients who might receive [drug under review]?
Treatment should be provided by professional with knowledge treating HCC. These patients are often reviewed in a multi-disciplinary fashion by surgeon, radiologist, gastroenterologists, oncologists and the therapy are often delivered by an oncologist.
Additional Information
The STRIDE regimen provides the first non-VEGF targeted therapy that provides a survival benefit for patients. This allows for another therapy to be considered for these patients where a bleeding risk is considered to be an issue. It is important that these patients have the additional option of therapies so that treatment can truly be personalized for this heterogenous population of patients.
Additionally, there may be a patient preference for the Stride regimen due to
Dosing schedule advantage. The STRIDE regimen is composed of a single 300-mg dose of tremelimumab followed by durvalumab 1500 mg given every 4 weeks. Whereas atezolizumab (1200 mg intravenously) and bevacizumab (15 mg/kg intravenously) is administered every 3 weeks.
Chair time advantage. The STRIDE regimen requires a patient to receive intravenous treatment for only one treatment vs. two sequential treatments for (Atezo + Bev). Thus, the chair time required for the atezo + bev regimen is significantly longer.
Safety Profile: It should be not noted bevacizumab has been associated with an increased risk of bleeding of all grades. And while most bleeding events are mild, self-limited, and frequently mucosal without need for medical intervention or bevacizumab discontinuation, patients, when deciding on a treatment with their physician may consider this factor.
Treatment Algorithm for HCC
In April 2021 CADTH conducted a provisional funding algorithm project that appears to have been triggered by the CADTH recommendation with respect to atezolizumab in combination bevacizumab for first-line HCC.
There has been evolving new data so the algorithm should be reviewed in totality to address the place of STRIDE as well as other options within the HCC landscape.
Conflict of Interest Declarations — The Canadian Gastrointestinal Oncology Evidence Network
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please refer to the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission?
No.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission?
No.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician who contributed to the input.
Declaration for Clinician 1
Name: Sharlene Gill
Position: Medical Oncologist, BC Cancer - Vancouver
Date: 11-01-2023
Table 9: COI Declarations for The Canadian Gastrointestinal Oncology Evidence Network — Clinician 1
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Astra Zeneca Canada | X | — | — | — |
Roche Canada | X | — | — | — |
Eisai Canada | — | X | — | — |
Declaration for Clinician 2
Name: Jennifer Spratlin
Position: Medical Oncologist, Cross Cancer Institute; Associate Professor, University of Alberta
Date: 29-21-2022
Table 10: COI Declarations for The Canadian Gastrointestinal Oncology Evidence Network — Clinician 2
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Astra Zeneca | X | — | — | — |
Declaration for Clinician 3
Name: Ravi Ramjeesingh
Position: Medical Oncologist, Department of Medicine, Dalhousie University
Date: 21-12-2022
Table 11: COI Declarations for The Canadian Gastrointestinal Oncology Evidence Network — Clinician 3
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Amgen | X | — | — | — |
Astra-Zeneca | X | — | — | — |
Eisai | X | — | — | — |
Incyte | X | — | — | — |
Ipsen | X | — | — | — |
Declaration for Clinician 4
Name: Vincent Tam
Position: Medical Oncologist, Tom Baker Cancer Centre, University of Calgary
Date: 24-12-2022
Table 12: COI Declarations for The Canadian Gastrointestinal Oncology Evidence Network — Clinician 4
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
AstraZeneca | — | — | X | — |
Eisai | — | X | — | — |
Incyte | X | — | — | — |
Ipsen | — | X | — | — |
Roche | — | X | — | — |
Declaration for Clinician 5
Name: Petr Kavan
Position: Medical Oncologist, McGill University Health Centre
Date: 12-01-2023
Table 13: COI Declarations for The Canadian Gastrointestinal Oncology Evidence Network — Clinician 5
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Incyte | X | — | — | — |
Declaration for Clinician 6
Name: Howard Lim
Position: Medical Oncologist
Date: 12-01-2023
Table 14: COI Declarations for The Canadian Gastrointestinal Oncology Evidence Network — Clinician 6
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Roche | X | — | — | — |
Bayer | X | — | — | — |
Amgen | X | — | — | — |
AstraZeneca | — | X | — | — |
BMS | — | X | — | — |
Lilly | X | — | — | — |
Taiho | X | — | — | — |
Eisai | — | X | — | — |
Ipsen | X | — | — | — |
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
Redactions: Confidential information in this document may be redacted at the request of the sponsor in accordance with the CADTH Drug Reimbursement Review Confidentiality Guidelines
Stakeholder Input: The views expressed in each submission are those of the submitting organization or individual; not necessarily the views of CADTH or of other organizations. As such, they are independent of CADTH and do not necessarily represent or reflect the view of CADTH. No endorsement by CADTH is intended or should be inferred. By filing with CADTH, the submitting organization or individual agrees to the full disclosure of the information. CADTH does not edit the content of the submissions.
CADTH does use reasonable care to prevent disclosure of personal information in posted material; however, it is ultimately the submitter’s responsibility to ensure no identifying personal information or personal health information is included in the submission. The name of the submitting organization or individual and all conflict of interest information are included in the submission; however, the name of the author, including the name of an individual patient or caregiver submitting the patient input, are not posted.
Accessibility: CADTH is committed to treating people with disabilities in a way that respects their dignity and independence, supports them in accessing material in a timely manner, and provides a robust feedback process to support continuous improvement. All materials prepared by CADTH are available in an accessible format. Where materials provided to CADTH by a submitting organization or individual are not available in an accessible format, CADTH will provide a summary document upon request. More details on CADTH’s accessibility policies can be found here.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.