CADTH Reimbursement Review
Crisantaspase Recombinant (Rylaze)
Sponsor: Jazz Pharmaceuticals Canada Inc.
Therapeutic area: Acute lymphoblastic leukemia
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Stakeholder Input
Clinical Review
Abbreviations
AE
adverse event
ALL
acute lymphoblastic leukemia
ALT
alanine aminotransferase
AST
aspartate aminotransferase
CI
confidence interval
CTCAE
Common Terminology Criteria for Adverse Events
EDA
Erwinia-derived asparaginase
HRQoL
health-related quality of life
IM
intramuscular
ITC
indirect treatment comparison
LBL
lymphoblastic lymphoma
NMA
network meta-analysis
NSAA
nadir serum asparaginase activity
OR
odds ratio
SAA
serum asparaginase activity
SAE
serious adverse event
SD
standard deviation
TDM
therapeutic drug monitoring
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Item | Description |
|---|---|
Drug product | Crisantaspase recombinant (Rylaze), solution for intramuscular injection, 10 mg/0.5 mL (20 mg/mL) |
Indication | Indicated as a component of a multidrug chemotherapeutic regimen for the treatment of: acute lymphoblastic leukemia and lymphoblastic lymphoma in adult and pediatric patients 1 year or older who have developed hypersensitivity to Escherichia coli–derived asparaginase |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Project Orbis |
NOC date | September 2, 2022 |
Sponsor | Jazz Pharmaceuticals Canada Inc. |
NOC = Notice of Compliance.
Introduction
Acute lymphoblastic leukemia (ALL) and lymphoblastic lymphoma (LBL) are hematological malignancies characterized by a rapidly progressing transformation and proliferation of lymphoid blasts in the bone marrow, peripheral blood, and other organs. Both conditions are usually described as ALL/LBL because they are considered intersecting clinical presentations of the same disorder with likely multifactorial risks, such as endogenous and exogenous exposures and genetic vulnerability.1 ALL/LBL is classified according to the immunophenotype (i.e., if malignant cells originate from B cells or T cells).2 Approximately 80% to 85% of children with ALL have B-cell phenotypes and close to 75% of adults with ALL do.1,3,4 ALL/LBL has a bimodal age distribution, with a first peak in childhood (3 of 4 cases occur in children younger than 6 years) and a second peak in adults older than 60 years. In children, it is the most frequent malignancy, comprising 75% to 80% of acute leukemias, yet it only represents approximately 20% of all leukemias in adults.3,4
In Canada, the latest reported incidence of ALL/LBL, from 2018, is 1.3 cases per 100,000 persons of all ages.5 This incidence has been steady; in previous years, the incidence was approximately 1.4 cases per 100,000 persons. Worldwide, the estimated annual incidence is 1 to 5 cases per 100,000 population.6
Due to the complicated nature of the disease, clinical assessments for risk assignment, and effects of therapies, ALL/LBL has an important impact on the quality of life of patients and caregivers and there are also emotional, financial, and developmental effects.7,8 Prognosis is generally poor in adults (5-year survival of 40% to 70%), with older age associated with worse survival,9 while children and adolescents have better projections, including remission in 98% of patients, a 5-year survival greater than 90%, and long-term event-free survival of 85%.10
The objective of this report is to perform a systematic review of the beneficial and harmful effects of crisantaspase recombinant 10 mg/0.5 mL used as a component of a multidrug chemotherapeutic regimen for the treatment of ALL/LBL in adult and pediatric populations aged 1 year or older who have developed hypersensitivity to Escherichia coli–derived asparaginase.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient groups that responded to CADTH’s call for patient input and from clinical experts consulted by CADTH for the purpose of this review.
Patient Input
One patient group, the Leukemia & Lymphoma Society of Canada (LLSC), supplied patient group input for this review. The LLSC gathered input from 40 respondents (1 of whom had experience with crisantaspase recombinant) via an online survey distributed in English and French through social media networks and by email.
Interviewed patients often reported fatigue and/or weakness (68% of all respondents) and loss of appetite or weight loss (45% of respondents) as issues having a significant impact on quality of life. Disease symptoms had a significant impact on respondents’ ability to work, exercise, and continue daily activities (64% of respondents). Also, significant impacts on stress and/or anxiety and problems concentrating due to disease symptoms were reported by 68% and 64% of respondents, respectively.
In open-ended responses, side effects of treatments for their condition were highlighted as important, with some respondents providing comments such as, “The chemotherapy protocol is long and extremely tiring” or “Very difficult protocol of chemotherapy.” A total of 8 patients reported being in treatment for 4 or more years, and 6 mentioned having had more than 5 total lines of treatment. When starting new treatments, patients considered the least amount of travel needed, improved quality of life, and insurance coverage as crucial factors for decisions. Reduced side effects were also often mentioned when respondents were asked what improvements they would like to see for any new treatment for ALL.
The 1 respondent who reported experience with crisantaspase recombinant reported manageable side effects and indicated the disease responded completely to the treatment. The patient mentioned allergic reactions to other previously used chemotherapies and expressed a preference for the treatment to be in IV form rather than an intramuscular (IM) injection.
Clinician Input
Input From Clinical Experts Consulted by CADTH
The clinical experts consulted by CADTH agreed that the goals of asparaginase treatment as part of a multicomponent chemotherapeutic regimen in patients with ALL/LBL include maximizing cure rates while minimizing short-term and long-term side effects, improving health-related quality of life (HRQoL), and reducing caregiver burden. Pegylated E. coli–derived asparaginase (pegaspargase) is the mainstay asparaginase treatment option. Clinically evident hypersensitivity reactions to E. coli–derived asparaginase occur in 10% to 15% of patients. A smaller percentage of patients develop silent inactivation. The clinical experts noted that, given the established importance of asparaginase therapy for the treatment of ALL/LBL, the discontinuation of asparaginase would likely lead to inferior survival outcomes, and that a switch to Erwinia asparaginase would be the next best option. The global supply of Erwinia asparaginase has been limited due to manufacturing difficulties, highlighting the need for a more reliable supply of Erwinia-derived asparaginase (EDA).
The clinical experts indicated that the place in therapy of crisantaspase recombinant should be used as a component of a multidrug regimen for the treatment of ALL/LBL in patients who have developed a documented hypersensitivity reaction or silent inactivation to E. coli–derived asparaginase. The experts also noted that a potential advantage of crisantaspase recombinant is a better and more reliable supply chain.
The clinical experts noted that the patients best suited for treatment with crisantaspase recombinant are those who experience a true antibody-mediated hypersensitivity reaction or silent inactivation, documented by a decrease in nadir serum asparaginase activity (NSAA). Measurement of NSAA is required to detect patients with silent inactivation of asparaginase activity. Therefore, the clinical experts believed that measurement of NSAA should be considered a standard of care in patients receiving asparaginase products and it should be made widely available in Canada.
The outcomes used to determine whether a patient is responding to treatment of ALL/LBL include post hoc evaluations such as event-free survival, disease-free survival, and overall survival. For assessing response to treatment, therapeutic drug monitoring (TDM) should be performed if available. According to clinical experts, the potential reasons for discontinuing treatment with crisantaspase include development of hypersensitivity reaction or silent inactivation to the drug, development of other types of toxicities (e.g., pancreatitis, thrombosis, and hepatotoxicity), evidence of disease relapse, or change in treatment strategy that no longer requires asparaginase therapy (e.g., allogeneic bone marrow transplant).
The clinical experts indicated that any asparaginase preparation (including crisantaspase recombinant) can acutely result in anaphylaxis or other serious allergic reactions, hence these drugs should only be safely given in the inpatient or outpatient hospital setting with immediate availability of suitably trained personnel who can acutely assess the severity of adverse events (AEs) and provide emergency interventions, as required.
Clinician Group Input
Two clinician groups provided input for this CADTH submission: the Ontario Health-Cancer Care Ontario (OH-CCO), Hematology Cancer Drug Advisory Committee; and the Pediatric Oncology Group of Ontario (POGO). Both groups obtained advice by interviewing clinical experts in the field of cancer, with a focus for this CADTH submission on ALL/LBL in both adults and children.
The information provided by both groups was aligned with the input from the clinical experts consulted by CADTH regarding the importance of asparaginase treatment as part of a multidrug chemotherapy for patients with ALL/LBL and the need for a replacement in the case of overt allergy or silent inactivation, which occurs in up to 25% of patients (according to input from the pediatric group). The groups mentioned that currently Erwinia asparaginase is the only available choice for this purpose but there is a short supply, and it is unavailable for patients and clinicians in Canada. The clinician groups agreed that this situation has the risk of creating incomplete treatment schedules and poorer patient outcomes. The clinician groups and the clinician experts also agreed that the patients best suited for treatment with crisantaspase recombinant are those with overt allergy and silent inactivation. clinician groups also concurred that patients should be able to access the drug and that NSAA testing should be available to monitor response to treatment for anyone receiving asparaginase products.
Drug Program Input
The drug plans highlighted that patients were eligible for this trial if they experienced a grade 3 or greater allergic reaction to a pegylated E. coli–derived asparaginase, and they asked if results can be applied to patients with a lower-grade reaction. The clinical experts indicated that the treatment of patients with a lower-grade reaction depends on their asparaginase activity levels. To determine asparaginase activity levels, a test must be run. The clinical experts indicated that access to the test required to determine the levels is a challenge because it is not universally funded.
Regarding care provision issues, the drug plans asked that, as Erwinase is under Health Canada review, if supply becomes available, what circumstances would make Rylaze preferred over Erwinase (or vice versa). The clinical experts indicated that preference for Rylaze over Erwinase (or vice versa) is largely dependent on the availability of either product. if supply is not an issue, the clinical experts could not comment on which treatment is better or preferred based on the available evidence. The clinical experts also indicated that if supply is not an issue, it would be challenging to provide a rationale for the use Rylaze over Erwinase if Rylaze is more expensive. They indicated that this may result in off-label use of Rylaze (25 mg/m2 on Friday rather than 50 mg/m2).
Clinical Evidence
Pivotal Studies and Protocol-Selected Studies
Description of Studies
One study is included in this review,11 which is an ongoing, open-label, single-arm, multicentre, dose-confirmation pharmacokinetic trial of crisantaspase recombinant in pediatric and adult patients with ALL/LBL who have hypersensitivity (an allergic reaction and/or silent inactivation) to E. coli–derived asparaginases. The study was designed to assess the efficacy and harms of crisantaspase recombinant. The main efficacy end point was measured with asparaginase activity (with NSAA levels ≥ 0.1 international unit [IU]/mL as a meaningful threshold) assessed at 48 hours and 72 hours after dosing. The study was initiated on December 27, 2019, and the data cut-off for the available analysis was July 19, 2021. In this study, 6 doses of crisantaspase recombinant are substituted for each dose of a long-acting E. coli–derived asparaginase. Doses were administered on a Monday-Wednesday-Friday schedule for 2 consecutive weeks; 2 consecutive weeks of treatment with crisantaspase recombinant is defined as 1 course. The focus of this review was on cohort 1c of the study, which included 51 patients receiving a dosing schedule of 25 mg/m2 on Mondays and Wednesdays and 50 mg/m2 on Fridays, which is the approved Health Canada indication.
The population was young (age range, 3 to 25 years), and most (46 of 51; 90%) were diagnosed with ALL. Allergic reactions of grade 3 or higher to previous E. coli–derived asparaginase occurred in 44 of 51 patients, with 1 silent inactivation and 6 allergic reactions with inactivation.
Efficacy Results
The outcomes set in the protocol of this CADTH review were efficacy outcomes, such as overall survival, event-free survival, disease-free survival, complete clinical remission and/or minimal residual disease, HRQoL, and serum asparaginase activity (serum NSAA levels). Of these end points, only NSAA and harms were available in the pivotal trial. Serum asparaginase activity levels are a surrogate marker for asparagine depletion, and an NSAA level of 0.1 IU/mL or higher is the most widely accepted threshold to demonstrate adequate asparagine depletion in clinical practice.12-14
Overall, mean and median NSAA values showed that the dose of cohort 1c (25 mg/m2 on Monday and Wednesday, 50 mg/m2 on Friday) reached values above the 0.1 IU/mL threshold for both the last 48 hours (mean = 0.65 IU/mL; 95% confidence interval [CI], 0.53 to 0.77 IU/mL) and the last 72 hours (mean = 0.46 IU/mL; 95% CI, 0.34 to 0.58 IU/mL) assessments.
When measuring the number and proportion of patients with values at or above the 0.1 IU/mL threshold, 47 of 49 patients analyzed (95.9%; 95% CI, 90.4% to 100.0%) reached this at the last 48-hour time point (achieving the sponsor’s threshold for efficacy of 90%) and 44 of 49 patients (89.8%; 95% CI, 81.2% to 98.3%) at the last 72-hour assessment. Both proportions were considered clinically relevant by the clinical experts consulted by CADTH.
Harms Results
Harms outcomes included mortality, AEs, serious adverse events (SAEs), and withdrawal due to AEs. Notable harms comprised thrombosis, pancreatitis, hypersensitivity reaction, hepatotoxicity, and hypertriglyceridemia (in adults). The safety outcomes were assessed in the safety analysis set. As of the data cut-off date of July 19, 2021, a total of 167 patients (33 in cohort 1a [IM 25 mg/m2 Monday, Wednesday, and Friday], 83 in cohort 1b [IM 37.5 mg/m2 Monday, Wednesday, and Friday], and 51 in cohort 1c [25 mg/m2 Monday and Wednesday and 50 mg/m2 on Friday]) were included in the safety analysis set for part A (IM) of the study. The focus on this report is the cohort 1c since this group include the dose approved by Health Canada.
For cohort 1c, a total of 49 of 51 (96.1%) of patients reported at least 1 AE. In all patients in the IM cohort, 164 of 167 (98.2%) had at least 1 AE. The most frequent AEs in cohort 1c, up to the latest cut-off date, were anemia (52.9%; 27 of 51), platelet count decreased (45.1%; 23 of 51), neutrophil count decreased (47.1%; 24 of 51), and vomiting (33.3%; 17 of 51). In the whole IM cohort, these numbers were similar, with anemia (53.3% of patients [89 of 167]), platelet count decreased (43.7%; 73 of 167), neutrophil count decreased (43.1%; 72 of 167), and vomiting (42.5%; 71 of 167). These and other AEs, such as pyrexia, fatigue, febrile neutropenia, decreased white blood cell count, stomatitis, and increased alanine transaminase (ALT), were considered by the clinical experts consulted by CADTH as common occurrences in all patients with ALL/LBL treated with asparaginase as part of a multicomponent chemotherapy regimen. The clinical experts indicated that many of the AEs likely resulted from the chemotherapy regimen.
A total of 30 of 51 (58.8%) of those included in cohort 1c experienced at least 1 SAE. The most common SAEs (in at least 5% of patients) were febrile neutropenia (25.5%), pyrexia (11.8%), clinical investigations in the context of SAEs (11.8%), sepsis (9.8%), pancreatitis (any kind: 5.9%; acute: 2.0%), and renal and urinary disorders (5.9%). All other SAEs were reported in less than 5% of patients. In the total IM treated cohort (part A of the study), 107 of 167 patients (64.1%) experienced at least 1 SAE by the data cut-off of July 19, 2021. The most common AEs (in ≥ 5% of patients) were febrile neutropenia, anemia, gastrointestinal disorders, pyrexia, stomatitis, and sepsis. As with the AEs, these were considered by the clinical experts to be within the expected frequency and part of ALL/LBL therapy. No deaths occurred in cohort 1c, and 3 of 167 patients (1.8%) had a fatal event (1 in cohort 1a and 2 in cohort 1b).
Among the notable harms, allergic reactions (hypersensitivity and anaphylaxis) occurred in 18 (35.3%) patients in cohort 1c. The most frequently reported events related to allergic reactions in cohort 1c included rash (7.8%; 4 of 51]), dermatitis (7.8%; 4 of 51), and allergic transfusion reaction (7.8%; 4 of 51). As of the data cut-off date July 19, 2021, 6 of 51 patients in cohort 1c (11.8%) had pancreatitis. |||||||||| |||||||| |||| || || ||||||| || ||| || |||||||| || |||||| ||. Hepatic toxicity was observed in 13 of 51 patients (25.5%) in cohort 1c group consisting all of elevated measured levels of ALT, aspartate transaminase (AST), and bilirubin. Hypertriglyceridemia occurred in 6 of 51 patients (11.8%) in cohort 1c.
Table 2: Summary of Key Efficacy Results From the Pivotal Study
Key efficacy end point | Cohort 1c (IM) |
|---|---|
Serum asparaginase activity | |
NSAA (IU/mL) in last 48 hours | |
N | 49 |
Mean (95% CI) | 0.655 (0.537 to 0.773) |
Median (Q1 and Q3) | 0.599 (0.334 and 0.888) |
NSAA (IU/mL) in last 72 hours | |
N | 49 |
Mean (95% CI) | 0.468 (0.349 to 0.586) |
Median (Q1 and Q3) | 0.383 (0.168 and 0.587) |
Proportion of patients reaching NSAA levels ≥ 0.1 IU/mL or ≥ 0.4 IU/mL | |
Last 48 hours | |
N | 49 |
NSAA level ≥ 0.1 IU/mL, n (%) | 47 (95.9) |
95% CI | 90.4 to 100.0 |
NSAA level ≥ 0.4 IU/mL, n (%) | 32 (65.3) |
95% CI | 52.0 to 78.6 |
Last 72 hours | |
N | 49 |
NSAA level ≥ 0.1 IU/mL, n (%) | 44 (89.8) |
95% CI | 81.3 to 98.3 |
NSAA level ≥ 0.4 IU/mL, n (%) | 23 (46.9) |
95% CI | 33.0 to 60.9 |
CI = confidence interval; IM = intramuscular; Q1 = first quartile; Q3 = third quartile; NSAA = nadir serum asparaginase activity.
Source: Clinical Study Report of crisantaspase recombinant.11
Harm end pointsa | Cohort 1c (IM) | Group A (IM) total (N = 167) |
|---|---|---|
Patients with at least 1 AE, n (%) | 49 (96.1) | 164 (98.2) |
Patients with at least 1 SAE, n (%) | 30 (58.8) | 107 (64.1) |
Patients with at least 1 grade 3 or 4 AE, n (%) | 44 (86.3) | 140 (83.8) |
Patients with an AE leading to study drug discontinuation, n (%) | 5 (9.8) | 22 (13.2) |
Patients with an AE leading to death, n (%) | 0 | 3 (1.8) |
AEs, n (%) | ||
Most common AEsb | ||
Decreased platelet count | 23 (45.1) | 73 (43.7) |
Anemia | 27 (52.9) | 89 (53.3) |
Decreased neutrophil count | 24 (47.1) | 72 (43.1) |
Vomiting | 17 (33.3) | 71 (42.5) |
Nausea | 18 (35.3) | 58 (34.7) |
Pyrexia | 10 (19.6) | 56 (33.5) |
Fatigue | 11 (21.6) | 53 (31.7) |
SAEs, n (%) | ||
Most common SAEs, n (%) c | ||
Febrile neutropenia | 13 (25.5) | 45 (26.9) |
Pyrexia | 6 (11.8) | 17 (10.2) |
Investigations for SAE | 6 (11.8) | 9 (5.4) |
Sepsis | 5 (9.8) | 11 (6.6) |
Dehydration | 4 (7.8) | 14 (8.4) |
Renal and urinary disorders | 3 (5.9) | 8 (4.8) |
Pancreatitis | 3 (5.9) | 5 (3.0) |
AEs of special interest, n (%) | ||
Allergic reactions (including hypersensitivity and anaphylaxis) | 18 (35.3) | 69 (41.3) |
|||||||||| | | ||||| | | ||||| |
Pancreatitis | 6 (11.8) | 12 (7.2) |
Thrombocytopenia | 4 (7.8) | 7 (4.2) |
Hepatotoxicity | 13 (25.5) | 55 (32.9) |
Hypertriglyceridemia | 6 (11.8) | 16 (9.6) |
AE = adverse event; IM = intramuscular; SAE = serious adverse event.
aOnly cohort 1c and total of group A (which includes cohort 1c) considered the safety analysis set are presented. Percentages were calculated with the number of patients in the safety analysis set as a denominator. AEs were coded using Medical Dictionary for Regulatory Activities (MedDRA) version 22.1. The severity of AEs was recorded using Common Terminology Criteria for Adverse Events (CTCAE) version 5.0.
bFrequency > 30% in total group A.
cFrequency > 5% in cohort 1c.
Source: Clinical Study Report of crisantaspase recombinant.11
Critical Appraisal
The main limitation of the included study is the single-arm (noncomparative) design. Because of the lack of a comparator group and no control for confounding, the outcomes cannot be definitively attributed to the administration of crisantaspase recombinant. It is recognized that supply shortages of the main comparator (Erwinia-derived asparaginase) may have rendered a randomized comparison unfeasible. The selection criteria appear reasonable, and the risk of selection bias is probably low. No formal hypothesis testing was performed to assess the effect estimates in this study. Bias in the measurement of the outcome is not suspected because efficacy measurements, such as NSAA levels, were collected objectively. Overreporting of known harms is possible, but the extent to which this may have occurred cannot be quantified. An evaluation of the comparative efficacy of crisantaspase recombinant and Erwinia-derived asparaginase is limited by the shortcomings of this study. Even with these limitations and uncertainty about the information provided, crisantaspase recombinant may still behave similarly to the drug it aims to replace (E. coli–derived asparaginase) in the multicomponent chemotherapy regimen for patients with ALL/LBL; however, this was not tested. The efficacy analysis was performed on the efficacy analysis set, which included all patients who received at least 1 dose of the study drug and had at least one 48-hour or 72-hour NSAA assessment during course 1. Only 1 assigned patient is missing from this analysis set (compared to the enrolled set), therefore there are no concerns about bias related to this decision.
The pivotal study can only highlight that crisantaspase recombinant is associated with measurable asparaginase activity (an NSAA above a threshold of 0.1 IU/mL, which was previously set as an important limit of clinical importance).8,15,16 This end point, however, is used and should be interpreted as a surrogate for important clinical outcomes.
Indirect Comparisons
Description of Studies
One sponsor-submitted indirect treatment comparison (ITC)17 comparing crisantaspase recombinant and EDA was included. The body of evidence in this ITC consisted of 2 single-arm studies, 1 of crisantaspase recombinant and the other of EDA. Because a network meta-analysis (NMA) or other population-adjusted ITCs were unfeasible, the analysis was achieved through a naive comparison of the single-treatment arms in each study and the calculation of odds ratios (ORs) for the effect estimates. The populations and outcome definitions were, overall, similar between the studies for assessing a comparison. There were some differences in the inclusion and exclusion criteria, and patients in the crisantaspase recombinant trial were slightly older and were more recently diagnosed compared with those in the EDA trial. The EDA trial included only patients with ALL and no patients with LBL. The outcomes evaluated are the proportion of patients reaching an NSAA level of at least 0.1 IU/mL and harms. The ITC presented effect estimates with uncertainty due to risk of bias, confounding (single-arm studies without adjustments to balance unobserved prognostic factors or effect modifiers), and imprecision. Hence, any difference in effects between crisantaspase recombinant and EDA is difficult to ascertain.
Efficacy Results
Among the efficacy outcomes of interest in this review (overall survival, event-free survival, disease-free survival, complete clinical remission and/or minimal residual disease, and HRQoL), only the NSAA levels, measured as a proportion of patients reaching an NSAA level of at least 0.1 IU/mL, was evaluated. No long-term evaluation of any outcome is available at this point.
In the crisantaspase recombinant study, 47 of 49 (95.9%) patients reached an NSAA level of at least 0.1 IU/mL in the last 48-hour assessment time, whereas 38 of 41 (92.7%) of patients in the EDA study reached this threshold (OR = 1.86; 95% CI, 0.29 to 11.68). Similarly, 44 of 49 (89.8%) patients in the crisantaspase recombinant study reached an NSAA level of at least 0.1 IU/mL in the last 72-hour assessment time, whereas 38 of 43 (88.4%) of patients in the EDA study reached this level threshold (OR = 1.16; 95% CI, 0.31 to 4.31).
No other efficacy end points were evaluated in the study.
Harms Results
The sponsor-submitted ITC assessed specific outcomes of interest but no overall AEs. Also, it included patient discontinuations as an outcome, which was defined by the number of patients who had completed all remaining courses. Overall, no evidence of a difference was observed between the EDA and crisantaspase recombinant groups in the proportion of patients with an increased ALT level (grade 3 or 4), thrombosis, hyperglycemia, or trial completion.
An increased ALT level (grade 3 or 4) was observed in 7.8% of patients in the crisantaspase recombinant trial and in 5% of patients in the EDA trial. A total of 6 patients (11.8%) developed pancreatitis in the crisantaspase recombinant trial compared with 1 patient in the EDA trial. One patient developed drug hypersensitivity (2.0%) in the crisantaspase recombinant study, whereas 8 patients (14%) developed drug hypersensitivity in the EDA trial. The crisantaspase recombinant trial reported that all drug hypersensitivity events were of grade 3 or 4. Of the 8 patients who developed drug hypersensitivity in the EDA trial, it was reported that 5 (9% of total population) developed grade 3 or 4 hypersensitivity. || |||||||| || ||| ||| ||||| ||||||||| ||||||||||| ||||||| |||||||| || ||| ||||||||||||| ||||||||||| ||||| ||||||||| ||||||||||. A total of 7 patients (12%) developed hyperglycemia in the EDA trial, whereas 6 patients (11.8%) in the crisantaspase recombinant trial developed hyperglycemia. In the EDA trial, 55 (80%) patients completed all courses of planned asparaginase therapy, whereas in cohort 1c of the crisantaspase recombinant trial — still ongoing at the time of this analysis — it was assumed that a total of 42 patients (82.4%) had completed eligible courses to date.
Critical Appraisal
The main limitation of the ITC is mostly due to the characteristics of the individual studies contributing to the body of evidence. The ITC was achieved through a naive comparison of each treatment arm from each study because it was not feasible to perform an NMA or a population-adjusted ITC due to the small sample size of each single-arm trial. Overall, baseline demographics and study characteristics reported were similar, but due to the nature of the comparison, there is no certainty about the balance of unobserved prognostic factors or effect modifiers; hence, any difference in effects between crisantaspase recombinant and EDA is difficult to ascertain. There is large imprecision when estimating the OR for both comparisons due to large number of events in a small number of patients. The same limitations apply when evaluating harms. Overall, the crude numbers of AEs in both included studies were similar and within the expected incidence range, according to the clinical experts consulted by CADTH. The incidence of AEs is likely to be affected by the background chemotherapy regimens, which may have differed between trials as a result of the time period during which they were conducted. Additionally, there were some differences in definitions used for AEs between the trials, and differences in the inclusion criteria may have biased the naive comparison of AEs.
There are no major concerns in terms of external validity, aside from those previously noted for the trial of crisantaspase recombinant. Overall, the populations, interventions, and outcomes assessed in the ITC were considered by the clinical experts consulted by CADTH to be generalizable to the population and clinical practice in Canada for the specific question and indication assessed in this review. Some differences were noticed, such as in the condition included (for instance, there were no patients with LBL in the EDA study) and lack of adult patients (aged ≥ 25 years) representation in both studies.
Conclusions
Evidence from a single-arm, open-label study suggests that crisantaspase recombinant is associated with adequate asparaginase activity when used in patients with ALL/LBL who have developed hypersensitivity to E. coli–derived asparaginase, which was shown using NSAA serum levels, a surrogate measure of asparagine depletion. The NSAA serum levels reached above an a priori–determined threshold of 0.1 IU/mL, which was established by clinical consensus as an important threshold for providing clinical utility. The impact on clinically important outcomes, such as overall survival and disease remission, and patient-important outcomes such as HRQoL, is unknown. Clinical experts deemed the safety profile of crisantaspase recombinant and harms manageable and within the expected frequency of events observed in patients with ALL/LBL in Canadian practice. They believed that the toxicity of crisantaspase recombinant could be higher in older patients, who were not represented in the trial. The evidence has limitations due to the noncomparative, open-label design of the study, which precludes causal inferences. However, the mechanism of action of the drug suggests that a link to the surrogate end point is plausible.
Evidence from a naive indirect comparison between single-arm trials of crisantaspase recombinant and EDA showed a high proportion of patients reaching a threshold for adequate asparaginase activity and similar safety profiles in both arms. However, the body of evidence presents effect estimates with uncertainty due to risk of bias, confounding (single-arm studies without adjustments to balance unobserved prognostic factors or effect modifiers), and imprecision due to low number of patients and wide CIs. Any difference in effects between crisantaspase recombinant and EDA in terms of NSAA levels and harms is difficult to ascertain.
Overall, the evidence suggests that crisantaspase recombinant is associated with adequate asparaginase activity and a manageable safety profile, but uncertainty remains in terms of clinical end points relative to other relevant comparators.
Introduction
Disease Background
ALL and LBL are hematological malignancies characterized by a rapidly progressing transformation and proliferation of lymphoid blasts in the bone marrow, peripheral blood, lymph nodes (for lymphoma), and other organs. Both conditions are usually described as ALL/LBL because they are considered intersecting clinical presentations of the same disorder.1 For this reason, the systems used for diagnosis and clinical classification do not distinguish between leukemia and lymphoma. The cause of ALL/LBL is likely multifactorial, involving risk variables such as endogenous and exogenous exposures and genetic vulnerability.18
ALL/LBL is classified according to the immunophenotype (i.e., if malignant cells originate from B cells or T cells).2 In children, approximately 80% to 85% of the ALL cases are B-cell phenotypes, and close to 75% in adults.1,3,4 ALL/LBL has a bimodal age distribution, with a first peak in childhood (3 of 4 cases occur in children younger than 6 years) and a second peak in adults older than 60 years. In children, it is the most frequent malignancy, comprising 75% to 80% of acute leukemias, yet only represent about 20% of all leukemias in adults.3,4
In Canada, the most recent reported incidence of ALL/LBL (from 2018), is 1.3 cases per 100,000 persons of all ages.5 This incidence has been steady; in previous years it has been approximately 1.4 cases per 100,000 persons. Worldwide, the estimated annual incidence is 1 to 5 cases per 100,000 population.6
The clinical features of ALL/LBL include findings associated with bone marrow infiltration, such as anemia, neutropenia, and/or thrombocytopenia.9 Patients are affected by different clinical signs and symptoms, such as fatigue, infections, bleeding, bone pain, arthralgia, hepatomegaly, splenomegaly, and lymphadenopathy. Due to the complicated nature of the disease, clinical assessments for risk assignment, and the effect of therapies, ALL/LBL has an important impact on the quality of life of patients and caregivers and also has emotional, financial, and developmental impacts.7,8 The diagnosis requires clinical and laboratory tests (e.g., complete blood count, blood chemistry) and an oncologic evaluation with bone marrow aspirate and biopsy, lumbar puncture, flow cytometry, and cytogenetic testing, among others, to detect genetic alterations and factors of prognostic value.
Prognosis is generally poor in adults (5-year survival is 40% to 70%), with older age associated with worse survival,9 whereas children and adolescents have better projections, with 98% achieving remission, a 5-year survival above 90%, and long-term event-free survival of 85%.10 The overall prognosis is likely correlated with an increase in adverse cytogenetic and molecular risk factors in older age groups. Cytogenetics and the presence of minimal residual disease after induction and/or consolidation therapies are the most important factors for determining the prognosis.6
Standards of Therapy
Treatment of children and adults with ALL/LBL demands a complicated risk assessment, assignment of therapies, and the need for supportive care (e.g., transfusions; management of complications; and emotional, financial, and developmental support). The goal of therapy is to achieve initial remission or disease cure and to prolong life and improve HRQoL.1,6
The current treatment paradigm for ALL/LBL involves multidrug regimens divided into several phases (i.e., induction, consolidation, intensification, and maintenance), and includes therapy that targets the central nervous system.6,9,18 Allogeneic hematopoietic cell transplant is reserved for patients with high-risk disease or persistent minimal residual disease.6
Asparaginase (L-asparaginase) is an essential part of treatment of ALL/LBL in the induction, consolidation, and intensification phases. Leukemic cells are unable to synthesize asparagine due to lack of asparagine synthetase activity. Asparaginase catalyzes the conversion of L-asparagine into aspartic acid and ammonia, resulting in cytotoxicity to leukemic cells that depend on a steady source of asparagine.13
L-asparaginase consists of bacterial enzymes derived from either E. coli or Erwinia chrysanthemi (Erwinia). Preparations of E. coli–derived asparaginase include native E. coli asparaginase and pegylated asparaginase (pegaspargase, Oncaspar), in which the E. coli–derived enzyme is modified by the covalent attachment of polyethylene glycol.14
Before the development of crisantaspase recombinant (Rylaze), there were 3 sources of asparaginase: L-asparaginase (derived from native E. coli), pegaspargase, and Erwinia L-asparaginase.
Given the nonhuman origin of E. coli–derived asparaginases, patients can react by producing antibodies to the drug, which can substantially reduce the activity of asparaginase and affect the clinical evolution and outcomes of patients.14 Also of concern is the potential for silent inactivation, with the formation of neutralizing antibodies and reduction in asparaginase activity in the absence of clinical symptoms of hypersensitivity. Whether patients have a reaction or silent inactivation, the inadequate activity of asparaginase due to the presence of either is associated with poor clinical outcomes.19-21
EDA (Erwinase) is antigenically distinct from E. coli–derived asparaginase, and it is commonly used as an alternative for patients with hypersensitivity or allergic reactions to E. coli–derived asparaginase.13,22 However, it was cancelled postmarket in 2009 in Canada and can only be obtained via an exceptional importation Tier 3 designation. EDA continues to be used in clinical practice in patients with ALL/LBL who have developed hypersensitivity or silent inactivation; however, according to input from the clinical experts consulted by CADTH, supply shortages are common.
Drug
Crisantaspase recombinant is expressed in Pseudomonas fluorescens with an identical amino acid sequence to native Erwinia asparaginase.23 The solution for IM injection contains 10 mg/0.5 mL (20 mg/mL), and it is indicated as a component of a multidrug chemotherapeutic regimen for the treatment of ALL and LBL in adult and pediatric patients aged 1 year and older who have developed hypersensitivity to E. coli–derived asparaginase. The recommended dosage is 25 mg/m2 on Monday and Wednesday and 50 mg/m2 on Friday, for a total of 6 doses, to replace each planned dose of pegaspargase. This drug has not been previously reviewed by CADTH.
The mechanism of action is similar to other asparaginases, catalyzing the conversion of the amino acid L-asparagine into aspartic acid and ammonia, resulting in death of leukemic cells due to depletion of plasma asparagine. The sponsor’s reimbursement request is the same as the approved Health Canada indication. Crisantaspase recombinant obtained a Notice of Compliance from Health Canada on September 2, 2022. In Table 4, the key characteristics of this drug and its main comparator are presented.
Table 4: Key Characteristics of Crisantaspase Recombinant and Erwinia-Derived Asparaginase
Characteristic | Crisantaspase recombinant | Erwinia-derived asparaginase |
|---|---|---|
Mechanism of action | Conversion of the amino acid L-asparagine into aspartic acid and ammonia. The mechanism of action is based on the killing of leukemic cells due to depletion of plasma asparagine. Leukemic cells with low expression of asparagine synthetase have a reduced ability to synthesize asparagine, and therefore depend on an exogenous source of asparagine for survival. | The mechanism is based on a metabolic defect in asparagine synthesis of the malignant cells. Asparaginase hydrolyzes circulating asparagine, resulting in the starvation and death of the malignant cells. |
Indicationa | As a component of a multidrug chemotherapeutic regimen for the treatment of ALL and LBL in adult and pediatric patients 1 year and older who have developed hypersensitivity to E. coli–derived asparaginase. | Primarily in combination with other antineoplastic drugs to induce remission in children and adults with ALL. It may also be used to treat patients who have developed hypersensitivity (but not anaphylaxis) to L-asparaginase derived from E. coli. |
Route of administration | IM | IM, SC, or bolus IV |
Recommended dose | The recommended dosage is 25 mg/m2 on Monday and Wednesday and 50 mg/m2 on Friday, administered intramuscularly, for a total of 6 doses, to replace each planned dose of pegaspargase. | Depends on the regimen. From 6,000 IU/m2 of body surface IM 3 times weekly for 9 doses, to 10,000 U/m2 SC on days 1, 3, 5 of week 4 and day 1 of week 5. The lowest age range of children studied overall in trials was 2 to 6 months. |
Serious adverse effects or safety issues | Thrombosis, hemorrhage/bleeding, liver function abnormalities, pancreatitis, hypersensitivity reactions, increased triglycerides. | Pancreatitis, septicemia, bleeding, contact irritant, adverse reactions (hypersensitivity reactions), liver function abnormalities. Immunosuppressive activity reported. |
ALL = Acute lymphoblastic leukemia; IM = intramuscular; LBL = lymphoblastic lymphoma; SC = subcutaneous.
aHealth Canada–approved indication.
Sources: Product monographs for crisantaspase recombinant23 and Erwinia-derived asparaginase.24
Stakeholder Perspectives
Patient Group Input
This section was prepared by CADTH staff based on the input provided by patient groups. The full original patient inputs received by CADTH have been included in the stakeholder section at the end of this report.
One patient group, the Leukemia & Lymphoma Society of Canada (LLSC), supplied patient group input for this review. The LLSC gathered input from 40 respondents (1 of whom had experience with crisantaspase recombinant) via an online survey distributed in English and French through social media networks (e.g., Facebook, Twitter, Instagram) and by email. Respondents could skip questions not relevant to them.
Most respondents were between the ages of 35 and 44 years (10 respondents, 25%), followed by 25 to 34 years (8 respondents, 20%), 55 to 64 years (5 respondents, 13%), 65 to 74 years (4 respondents, 10%), and 45 to 54 years (3 respondents, 8%).
Fatigue or weakness (68% of the total respondents) and loss of appetite or weight loss (45% of respondents) were commonly reported to have a significant impact on quality of life (i.e., rated 4 or 5 on a scale from 1 [no impact] to 5 [extremely significant impact]). Disease symptoms also had a significant impact on respondents’ ability to work, exercise, and continue daily activities (64% of respondents); significant impacts on stress and/or anxiety and problems concentrating due to disease symptoms were reported by 68% and 64% of respondents, respectively.
In open-ended responses, side effects of treatments were highlighted by respondents as important; comments included “The chemotherapy protocol is long and extremely tiring” or “Very difficult protocol of chemotherapy.” A total of 8 patients reported being in treatment for 4 or more years, and 6 mentioned having had more than 5 total lines of treatment. When starting new treatments, patients considered the least amount of travel needed, improved quality of life, and insurance coverage as crucial factors for decisions. Reduced side effects were also often mentioned when respondents were asked what improvements they would like to see for any new treatment for ALL.
The 1 respondent who reported experience with crisantaspase recombinant reported manageable side effects and indicated the disease responded completely to the treatment. The patient mentioned allergic reactions to other previously used chemotherapies and expressed a preference for the treatment to be in IV form rather than IM injection.
Clinician Input
Input From Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of ALL/LBL.
Unmet Needs
The clinical experts consulted by CADTH indicated that treatment goals for patients with ALL/LBL include maximizing cure rates while minimizing short-term and long-term side effects, improving HRQoL, and reducing caregiver burden.
E. coli and Erwinia chrysanthemi are the primary sources for production of pharmaceutical asparaginase. The native form of E. coli–derived asparaginase is no longer available in Canada and has been replaced by the pegylated form of E. coli–derived asparaginase (pegaspargase). The clinical experts indicated that hypersensitivity reactions to pegaspargase treatment occur in about 10% to 15% of patients over the duration of therapy. A smaller percentage of patients develop silent inactivation. In these cases, 1 expert mentioned that options for modification of treatment include administering pegaspargase under desensitization protocols, discontinuing asparaginase therapy, or switching to Erwinia asparaginase. Although the use of desensitization protocols may allow patients with hypersensitivity reactions to be rechallenged with pegaspargase, the clinical experts noted that there are no data available on the potential impact of desensitization on survival outcomes. Given the established importance of asparaginase therapy in the treatment of ALL/LBL, the clinical experts stated that discontinuation of asparaginase therapy would likely lead to inferior survival outcomes. Therefore, based on the experience of the clinical experts, switching to Erwinia asparaginase is the best option and has demonstrated equivalent outcomes. However, in recent years, the global supply of Erwinia asparaginase has been limited due to manufacturing difficulties, highlighting the need for a more reliable supply of EDA.
Place in Therapy
According to the clinical experts, crisantaspase recombinant should be used as a component of a multidrug regimen for the treatment of ALL/LBL in patients who have developed a documented hypersensitivity reaction or silent inactivation to E. coli–derived asparaginase. The experts mentioned that it would be inappropriate to attempt a desensitization protocol before initiation of crisantaspase recombinant due to the lack of data supporting this approach, the risk of triggering another hypersensitivity reaction, and the substantial amount of health care resources required to perform the desensitization protocol safely. The clinical experts indicated that crisantaspase recombinant has the advantage of a more reliable supply chain.
Patient Population
The clinical experts indicated that patients who would be best suited for treatment with crisantaspase recombinant are those who experience a true antibody-mediated hypersensitivity reaction. Patients who experience infusion reactions due to non–antibody-mediated mechanisms do not require a change in asparaginase product. The clinical experts pointed out that distinguishing between the 2 types of reaction can be clinically challenging, and that although a companion diagnostic test is not required to diagnose clinically evident allergy to E. coli–derived asparaginase, the measurement of NSAA is required to detect patients with silent inactivation of asparaginase activity; therefore, the measurement of NSAA should be considered a standard of care in patients receiving asparaginase products and should be made widely available in Canada. Of note, the clinicians consulted by CADTH highlighted that patients with silent inactivation, which can only be detected through TDM, should be included in the indication for crisantaspase recombinant because these patients have no response to asparaginase, even if there are no obvious clinical signs.
Assessing Response to Treatment
The outcomes used to determine whether a patient was responding to treatment of ALL/LBL overall included post hoc evaluations, such as event-free survival, disease-free survival, and overall survival.
To assess response to treatment with crisantaspase recombinant, the clinical experts indicated that TDM should be performed, if available, and should be used to monitor levels of serum asparaginase activity (SAA) after drug dosing. It is important to determine if a therapeutic SAA level is achieved in a patient receiving crisantaspase recombinant. SAA levels can also be used to guide individualized dosing of crisantaspase recombinant. The optimal frequency of TDM is unknown. However, the clinical experts noted that TDM after each cycle of treatment for 3 or 4 cycles or until the stabilization of SAA levels is a reasonable approach.
Discontinuing Treatment
According to the clinical experts, potential reasons for discontinuing treatment with crisantaspase include development of hypersensitivity reaction or silent inactivation to the drug, development of other types of toxicities (e.g., pancreatitis, thrombosis, and hepatotoxicity), evidence of disease relapse, or change in treatment strategy that no longer requires asparaginase therapy (e.g., allogeneic bone marrow transplant).
Prescribing Conditions
The clinical experts mentioned that any asparaginase preparation (including crisantaspase recombinant) can acutely result in anaphylaxis or other serious allergic reactions, hence these drugs should only be safely given in the inpatient or outpatient hospital setting with immediate availability of suitably trained personnel who can acutely assess the severity of AEs and provide emergency interventions as required. This health care team would need to include physicians, nurses, nurse practitioners, and physician assistants who have undergone appropriate training to recognize and urgently manage complications in this oncology patient population.
Additional Considerations
The clinical experts emphasized the importance of readily available testing for NSAA levels across all Canadian practices for any patient receiving asparaginase therapy in regard to both optimizing outcomes and minimizing the cost of continuing to deliver what can be ineffective therapies.
Clinician Group Input
This section was prepared by CADTH staff based on the input provided by clinician groups. The full original clinician group inputs received by CADTH have been included in the stakeholder section at the end of this report.
Two clinician groups provided input for this CADTH submission: the Ontario Health-Cancer Care Ontario (OH-CCO), Hematology Cancer Drug Advisory Committee; and the Pediatric Oncology Group of Ontario. Both groups obtained advice by interviewing clinical experts in the field of cancer, with a focus for this CADTH submission on ALL/LBL in both adults and children.
The information provided by both groups was aligned with the input from the clinical experts consulted by CADTH regarding the importance of asparaginase treatment as part of a multidrug chemotherapy regimen for patients with ALL/LBL and on the need for a replacement in the presence of overt allergy or silent inactivation, which occurs in up to 25% of patients (according to the pediatric group input, while clinical experts consulted by CADTH mention approximately 15%). The groups mentioned that currently Erwinase is the only choice currently available for this purpose due to short supply and unavailability for patients and clinicians in Canada. The clinician groups agreed that this situation has the risk of creating incomplete treatment schedules and poorer patient outcomes.
The clinician groups and the clinician experts were concordant in their advice related to the characteristics of patients who would be best suited for treatment with crisantaspase recombinant: overt allergy and silent inactivation. The clinician groups also concurred with the clinical experts that patients should have readily available access to asparaginase treatment and asparaginase level (NSAA) testing to monitor response to treatment, as should anyone receiving asparaginase products.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that could have an impact on their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 5.
Table 5: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
Erwinase/Erwinia L-asparaginase is available in most jurisdictions for this indication. Erwinase is currently imported from the UK under Health Canada exceptional importation under Tier 3 drug shortage designation and is currently being re-evaluated by Health Canada after postmarket cancellation in 2021. | Comment from the drug programs to inform pERC deliberations. |
Considerations for initiation of therapy | |
Patients were eligible for this trial if they experienced a grade 3 or higher allergic reaction to a pegylated E. coli–derived asparaginase. Can the results be applied to patients with a lower-grade reaction? | The clinical experts indicated that the treatment of patients with a lower-grade reaction depends on their asparaginase activity levels. To determine asparaginase activity levels, a test must be run. The clinical experts indicated that access to the test required to determine the levels is an issue because it is not universally funded. |
Considerations for renewal of therapy | |
No questions included | — |
Considerations for discontinuation of therapy | |
No questions included | — |
Considerations for prescribing of therapy | |
Crisantaspase recombinant is administered 3 times per week at dosages of 25 mg/m2 IM on Mondays and Wednesdays and 50 mg/m2 IM on Fridays. | Comment from the drug programs to inform CDEC deliberations. |
Generalizability | |
No questions included | — |
Care provision issues | |
Erwinase is under Health Canada review. If supply becomes available, under what circumstances would Rylaze be preferred over Erwinase (and vice versa)? | The clinical experts indicated that preference for Rylaze over Erwinase (and vice versa) is largely dependent on the availability of either product. If supply is not an issue, the clinical experts could not comment on which treatment is better or preferred based on the available evidence. The clinical experts also indicated that if supply is not an issue, it would be challenging to provide a rationale for the use of Rylaze over Erwinase if Rylaze is more expensive. They indicated that this may result in off-label use of Rylaze (25 mg/m2 on Friday rather than 50 mg/m2). |
CDEC = CADTH Canadian Drug Expert Committee; IM = intramuscular; pERC = CADTH pan-Canadian Oncology Drug Review Expert Review Committee.
Clinical Evidence
The clinical evidence included in this review of crisantaspase recombinant is presented in 3 sections. The first section, the Systematic Review, includes pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as studies that were selected according to an a priori protocol. The second section includes indirect evidence from the sponsor and indirect evidence selected from the literature that met the selection criteria specified in the review. The third section includes sponsor-submitted long-term extension studies and additional relevant studies that were considered to address important gaps in the evidence included in the Systematic Review section.
Systematic Review (Pivotal and Protocol-Selected Studies)
Objectives
To perform a systematic review of the beneficial and harmful effects of crisantaspase recombinant 10 mg/0.5 mL for IM injection as a component of a multidrug chemotherapeutic regimen for the treatment of ALL and LBL in adult and pediatric populations aged 1 year and older who have developed hypersensitivity to E. coli–derived asparaginase.
Methods
Studies selected for inclusion in the systematic review included pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those meeting the selection criteria presented in Table 6. Outcomes included in the CADTH review protocol reflect outcomes considered to be important to patients, clinicians, and drug plans.
The literature search was performed by an information specialist using a peer-reviewed search strategy.
Two CADTH clinical reviewers independently selected studies for inclusion in the review based on titles and abstracts, according to the predetermined protocol. Full-text articles of all citations considered potentially relevant by at least 1 reviewer were acquired. Reviewers independently made the final selection of studies to be included in the review, and differences were resolved through discussion.
Table 6: Inclusion Criteria for the Systematic Review
Type | Criteria |
|---|---|
Patient population | Adult and pediatric populations aged 1 year and older receiving a multidrug chemotherapeutic regimen for the treatment of ALL and LBL who have developed hypersensitivity to E. coli–derived asparaginase Subgroups:
|
Intervention | Crisantaspase recombinant 25 mg/m2 on Monday and Wednesday and 50 mg/m2 on Friday, administered intramuscularly, for a total of 6 doses (to replace each planned dose of long-acting E. coli–derived asparaginase) |
Comparators | Erwinia-derived asparaginase |
Outcomes | Efficacy outcomes:
Harms outcomes:
|
Study design | Published and unpublished phase II and III RCTs |
AE = adverse event; ALL = acute lymphoblastic leukemia; CNS = central nervous system; HRQoL = health-related quality of life; LBL = lymphoblastic lymphoma; NSAA = nadir serum asparaginase activity; RCT = randomized controlled trial; SAE = serious adverse event; WDAE = withdrawal due to adverse event.
Findings From the Literature
A total of 737 studies were identified in the literature search and 2 reports of a single study were included in the systematic review (Figure 1). The included study is summarized in Table 7. A list of excluded studies is presented in Appendix 2.
Table 7: Details of Included Studies
Criteria | AALL1931 study |
|---|---|
Design and population | |
Study design | Phase II/III, open-label, dose-confirmation and pharmacokinetic, single-arm, multicentre study |
Locations | 67 sites across Canada (7 sites) and the US (60 sites) |
Patient enrolment dates | Initiated on December 27, 2019 Cut-off for this version of the Clinical Study Report is July 19, 2021 |
Rationale | The study was designed to assess the safety, tolerability, and efficacy of repeated doses of IM injection of crisantaspase recombinant (JZP-458) in patients with ALL/LBL who are hypersensitive to E. coli–derived asparaginases (part A) and to identify a dose level of IV crisantaspase recombinant for further evaluation (part B) |
Enrolled (N) | Part A Cohort 1a: 33 patients Cohort 1b: 83 patients Cohort 1c: 51 patients (the main cohort of interest for the CADTH review) |
Inclusion criteria | Pediatric and adult patients with a diagnosis of ALL or LBL and who had a ≥ grade 3 (parts A and B, cohort 1) or ≥ grade 2 (part A, cohort 2) allergic reaction to a long-acting E. coli–derived asparaginase or who had silent inactivation |
Exclusion criteria |
|
Drugs | |
Intervention | Crisantaspase recombinant (JZP-458) IM (in part A of the study) as single injection limited to 2 mL volume, as follows: Cohort 1a: 25 mg/m2 on a Monday-Wednesday-Friday schedule Cohort 1b: 37.5 mg/m2 on a Monday-Wednesday-Friday schedule Cohort 1c: 25 mg/m2 on Mondays and Wednesdays and 50 mg/m2 on Fridays |
Comparator(s) | None |
Duration | One course, which comprised 2 consecutive weeks; additional courses are administered based on each patient’s original treatment plan for as long as the patient derives clinical benefit |
Phase | |
Part A: dose confirmation | Cohort 1 (with subcohorts 1a, 1b, and 1c) is a repeat-dose, confirmatory cohort, with the initial dose based on data from the phase I healthy volunteer study; a final IM dose level is to be selected based on data from this cohort |
Part B: IV doses | In part B, crisantaspase recombinant is administered as an IV formulation (25 mg/m2 on Monday and Wednesday and 50 mg/m2 on Friday). Part B will not be discussed or included in this review because it is not part of the Health Canada indication under consideration in this submission |
Follow-up | Ongoing study: participants are followed up for at least 30 days after their last dose |
Outcomes | |
Primary end point |
|
Secondary and exploratory end points |
|
Notes | |
Publications | |
AE = adverse event; ALL = acute lymphoblastic leukemia; EDA = Erwinia-derived asparaginase; IM = intramuscular; LBL = lymphoblastic lymphoma; NSAA = nadir serum asparaginase activity.
Source: Clinical Study Report of crisantaspase recombinant.11
Description of Studies
The included study is an ongoing, phase II/III, open-label, single-arm, multicentre, dose-confirmation, pharmacokinetic study of crisantaspase recombinant (JZP-458) in pediatric and adult patients with ALL/LBL who are hypersensitive to E. coli–derived asparaginases (allergic reaction or silent inactivation). The study is designed to assess the tolerability and efficacy of crisantaspase recombinant, as measured by asparaginase activity (Table 7).
The study consists of 2 parts. Part A was devised to determine the dose and evaluate the safety of crisantaspase recombinant for IM administration in several cohorts (cohorts 1 and 2, with subcohorts included as needed). Part B was designed to define the optimal dose and schedule of IV doses. However, only part A (cohort 1c) is relevant to and will be evaluated in this CADTH review because it is the 1 that includes the drug dose and route of administration approved by Health Canada.
The primary objectives of the study included determining the efficacy of IM administration of crisantaspase recombinant, measured by the response in cohort 1 and cohort 2 and defined in the last 72-hour NSAA level of at least 0.1 IU/mL during the first course, and assessing the safety and tolerability of IM crisantaspase recombinant in patients with ALL/LBL who are hypersensitive to E. coli–derived asparaginases. The key secondary objective was to determine the efficacy of crisantaspase recombinant IM administration, measured by the response in cohort 1 and cohort 2 and defined as the last 48-hour NSAA level of at least 0.1 IU/mL during the first course. Pharmacokinetic and safety data are being assessed by a Study Data Review Committee at frequent intervals. As of July 19, 2021, the study had 7 active sites in Canada and 60 active sites in the US (including 6 satellite sites).
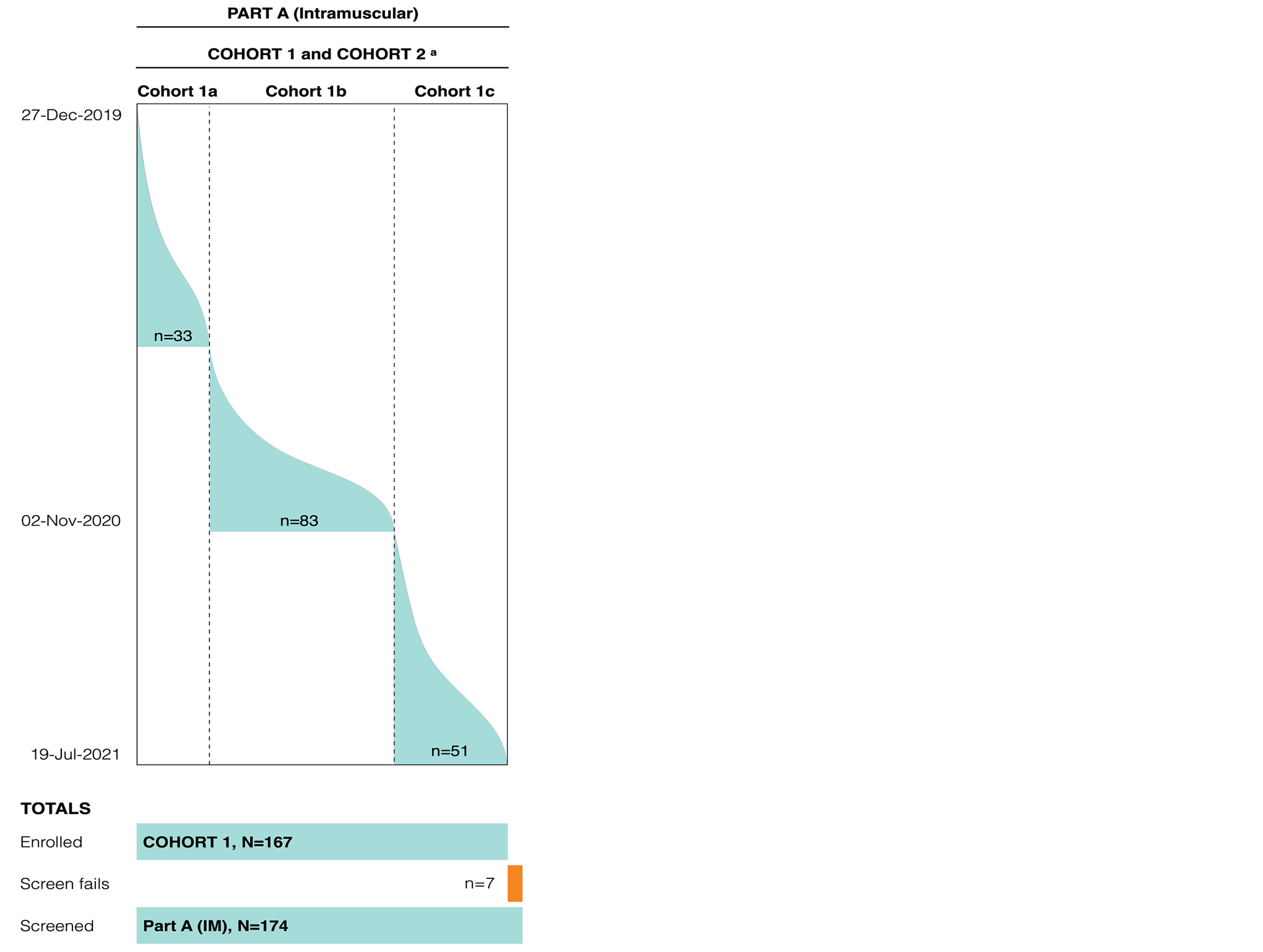
Figure 2 shows that 2 IM dose cohorts were planned for part A: Cohort 1 (with multiple subcohorts) is a crisantaspase recombinant repeat-dose, confirmatory cohort, and the initial dose was based on data from the phase I healthy volunteer study, with the objective of determining a final IM dose level; and cohort 2 is an expansion cohort to confirm the efficacy and safety of the final IM dose level and schedule. From the outset of the study, patients were integrated into a subcohort of cohort 1 (cohort 1a), to receive an initial 6-dose course of IM crisantaspase recombinant at a dosage of 25 mg/m2 on a Monday-Wednesday-Friday schedule over 2 weeks, based on available data from the phase I healthy volunteer study. Because the percentage of patients with postdose NSAA levels at the last 72 hours was below the protocol-defined threshold, and due to the acceptable safety profile at the IM 25 mg/m2 dose level, the sponsor decided to escalate to an IM dose of 37.5 mg/m2 on a Monday-Wednesday-Friday schedule over 2 weeks (cohort 1b).
On October 16, 2020, the Study Data Review Committee reviewed the data from cohort 1b (37 mg/m2 IM administered on a Monday-Wednesday-Friday schedule) and recommended that cohort 2, which was an expansion of cohort 1b, be opened at a dose of 37.5 mg/m2 administered on a Monday-Wednesday-Friday schedule. After reviewing preliminary data from the study that included modelled data from cohorts 1a and 1b, the protocol was amended (protocol amendment 2) and an additional subcohort of cohort 1 was initiated (cohort 1c). Patients included in cohort 1c received crisantaspase recombinant on a dosing schedule of 25 mg/m2 on Mondays and Wednesdays and 50 mg/m2 on Fridays. Parallel enrolment of patients in cohort 1 (1b and 1c) and cohort 2 could not occur because, per the protocol design, cohort 2 is defined as an expansion of cohort 1. Therefore, patients who were enrolled in cohort 2 were reassigned to cohort 1b.
Populations
Inclusion and Exclusion Criteria
In parts A and B, cohort 1 included pediatric and adult patients with a diagnosis of ALL or LBL who had an allergic reaction of at least grade 3 (using the Common Terminology Criteria for Adverse Events [CTCAE] version 5.0) to a long-acting E. coli–derived asparaginase or who had silent inactivation (Table 7). The study excluded patients who had relapsed ALL or LBL, a history of grade 3 or higher pancreatitis, an asparaginase-associated hemorrhagic event of grade 3 or higher, or an asparaginase-associated thrombus requiring anticoagulation therapy.
Figure 2: Study Design and Flow of Patients Included in Part A of the Study (IM) of Crisantaspase Recombinant
IM = intramuscular.
Note: The coloured areas represent the flow and number of patients and, at the bottom, the total number of patients included in the screened, screen fails, and enrolled groups is provided.
a Cohort 2 was created on October 16, 2020, as an expansion of cohort 1b. However, the sponsor determined to include it as part of cohort 1b (refer to text for details).
Source: Clinical Study Report of crisantaspase recombinant.11
Baseline Characteristics
The summary of baseline characteristics is presented in Table 8. For part A (IM) cohort 1c of the study, the majority of patients were identified or self-identified as white (33 of 51, or 64.7%); the median age at enrolment was 12 years (range, 3 to 25 years).
Of the 51 patients in cohort 1c, 11 (21.6%) were younger than 6 years, 14 (27.5%) were 6 to 12 years, 18 (35.3%) were 12 to 18 years, and 8 (15.7%) were 18 years or older. A higher percentage of male patients (31; 60.8%) than female patients (20; 39.2%) were included in the cohort. Most patients had ALL (46 patients; 37 [72%] with B-ALL and 9 [17.6%] with T-ALL), whereas LBL was the diagnosis in 5 patients (1 B-cell LBL, 4 T-cell LBL). The proportion of patients with central nervous system involvement was not reported. The majority of patients (39; 76.5%) had relatively recently been diagnosed within 0 to 3 months at study day 1. For all patients, Oncaspar was the asparaginase used as prior treatment before entering the study. Also, most patients (44; 86.3%) had an allergic reaction classified as grade 3 or higher, with 6 patients (11.8%) presenting with an allergic reaction with inactivation and 1 with purely silent inactivation.
Table 8: Summary of Baseline Characteristics — Safety Analysis Set, Cohort 1c (Cut-Off Date of July 19, 2021)
Variable | IM |
|---|---|
Sex, n (%) | |
Female, | 20 (39.2) |
Male | 31 (60.8) |
Ethnicity, n (%) | |
Hispanic or Latino | 17 (33.3) |
Not Hispanic or Latino | 32 (62.7) |
Declined to state | 2 (3.9) |
Race, n (%) | |
American Indian or Alaska Native | 3 (5.9) |
Asian | 1 (2.0) |
Black or African American | 8 (15.7) |
Native Hawaiian or other Pacific Islander | 0 |
White | 33 (64.7) |
Multiple | 0 |
Not reported | 6 (11.8) |
Age at enrolment (years) | |
Mean (SD) | 11.3 (5.4) |
Median (range) | 12 (3 to 25) |
Age subgrouping, n (%) | |
< 1 year | 0 |
1 year to < 6 years | 11 (21.6) |
6 years to < 12 years | 14 (27.5) |
12 years to < 18 years | 18 (35.3) |
≥ 18 years | 8 (15.7) |
Body surface area (m2) | |
n | 51 |
Mean (SD) | 1.35 (0.53) |
Median (range) | 1.29 (0.54 to 2.43) |
Body surface area, n (%) | |
0 < BSA ≤ 1 | 17 (33.3) |
1 < BSA ≤ 2 | 28 (54.9) |
2 < BSA | 6 (11.8) |
Primary disease, n (%) | |
ALL | 46 |
B-cell ALL | 37 (72.5) |
T-cell ALL | 9 (17.6) |
LBL | 5 |
B-cell LBL | 1 (2.0) |
T-cell LBL | 4 (7.8) |
Time since primary disease diagnosis to study day 1, n (%) | |
0 to 3 months | 39 (76.5) |
4 to 6 months | 11 (21.6) |
7 to 9 months | 1 (2.0) |
10 to 12 months | 0 |
> 12 months | 0 |
Prior asparaginase treatment, n (%) | |
Oncaspar | 51 (100) |
Calaspargase pegol-mknl (Asparlas) | 0 |
Erwinia chrysanthemi L-asparaginase | 0 |
Other | 0 |
Time since last asparaginase received to study day 1 (days) | |
n | 51 |
Mean (SD) | 23.8 (29.73) |
Median (range) | 10 (2 to 133) |
Eligibility criteria, n (%) | |
Grade ≥ 3 allergic reaction to an E. coli–derived asparaginase | 44 (86.3) |
Silent inactivation | 1 (2.0) |
Allergic reaction with inactivation | 6 (11.8) |
ALL = acute lymphoblastic leukemia; BSA = body surface area; IM = intramuscular; LBL = lymphoblastic lymphoma.
Note: Only the IM administration results are presented, as it is the approved dose formulation in Canada, with a focus on the group with a dosing schedule of 25 mg/m2 on Mondays and Wednesdays and 50 mg/m2 on Fridays. Percentages were calculated with the number of patients in the safety analysis set as the denominator.
Source: Clinical Study Report of crisantaspase recombinant.11
Interventions
In cohorts 1a, 1b, and 1c of part A of the study, crisantaspase recombinant was administered via the IM route as a single injection limited to 2 mL per injection site; multiple injection sites were used, as needed. A course was defined as 6 IM injections. Additional courses could be administered based on the patient’s original treatment plan and for as long as the patient derived clinical benefit. As previously noted, cohort 1c will be the focus of this review, as it is the only cohort that received crisantaspase recombinant in alignment with the indication approved by Health Canada. In cohort 1c, crisantaspase recombinant was administered IM at a dose of 25 mg/m2 on Mondays and Wednesdays and 50 mg/m2 on Fridays. Because this is an open-label, single-arm study, no blinding or comparators were used.
Patients continued with their original therapeutic regimen (chemotherapy) protocol for the treatment of ALL/LBL. Patients could not receive other investigational drugs and/or be treated with investigational devices at the same time of therapy with the first cycle of crisantaspase recombinant. All other treatments were allowed at the discretion of the treating physician.
Outcomes
Because crisantaspase recombinant is aimed at being used in lieu of an E. coli–derived asparaginase treatment in patients with ALL/LBL who have developed hypersensitivity to this asparaginase as part of a multidrug therapeutic regimen, the end points to consider and assess would be those of clinical relevance for efficacy and harms from crisantaspase recombinant compared with other possible options of treatment, if available, and to assess if the use of crisantaspase recombinant would provide similar benefits and a safety profile known to be obtained from E. coli–derived asparaginases. CADTH and the clinical experts consulted by CADTH identified several outcomes in the review protocol considered important for decision-makers, patients, and clinicians. These included overall survival, event-free survival, complete clinical remission and/or minimal residual disease, HRQoL, and NSAA levels. Of these end points, only NSAA levels and harms were evaluated in the included study.
The primary efficacy end point was response rate, defined as the proportion of patients with a last 72-hour NSAA level of 0.1 IU/mL or higher during the first course (last dose) of IM crisantaspase recombinant administration. NSAA is a valid and reliable method of measuring asparaginase efficacy.14 The analysis of NSAA was performed using a validated enzyme activity method to measure NSAA levels in human serum. Although no minimal critical value has been established for serum asparagine depletion, the literature has defined complete asparagine depletion as less than 0.1 μM to 0.2 μM, based on the limit of detection of the high-performance liquid chromatography assay used.12 Consensus expert opinion indicates that an NSAA level of at least 0.1 IU/mL appears to be a safe target level to ensure therapeutic benefit.14
Secondary efficacy end points included the proportion of patients with the last 48-hour NSAA level of 0.1 IU/mL or higher; the proportion of patients with the last 48-hour NSAA level of 0.4 IU/mL or higher; and the proportion of patients with the last 72-hour NSAA level of 0.4 IU/mL or during the first course of IM crisantaspase recombinant administration.
Harms or AEs were considered any untoward medical occurrence associated with the use of the study drug, whether or not deemed related to the study drug. All AEs, whether observed by the investigator, reported by the patient, determined from laboratory findings or other means were recorded. The severity of AEs was classified by the investigator using the National Cancer Institute CTCAE 5.0, from grade 1 (mild) to grade 5 (death related to AE). Treatment-emergent AEs leading to drug discontinuation and deaths were also recorded.
Statistical Analysis
The data presented in this report are from the second interim analysis. The primary efficacy end point was planned to be assessed using the efficacy analysis set for patients who were administered the final IM dose level with at least one 72-hour NSAA assessment collected within the protocol-defined sample collection window (± 2 hours) in course 1 of part A. The last observed 72-hour NSAA assessment collected within the protocol-defined sample collection window (± 2 hours) in course 1 was used in the calculation of the primary efficacy end point. Missing data were not imputed. The response rate, along with the 95% Wald CI, was assessed. The primary efficacy end point was considered to be met if the lower bound of the 95% CI of the response rate exceeded 90%.
SAA levels and descriptive summary statistics by time point, dose cohort, course, and schedule were analyzed in the same way as described for the primary end point and presented for the last 48-hour and last 72-hour SAA. Also, the proportion of patients with last 72-hour and last 48-hour NSAA levels of at least 0.1 IU/mL or at least 0.4 IU/mL during the first course of crisantaspase recombinant (efficacy analysis set; data cut-off of July 19, 2021) is presented, using the number of patients on each course and schedule as denominators.
The sample size for part A (IM administration, cohorts 1 and 2) of the study was initially planned to be 98 patients who had been administered the final IM dose level. For the final IM dose level, 13 evaluable patients were planned in part A, cohort 1 (IM dose confirmation) and 85 patients planned in cohort 2 (IM expansion) to obtain 98 patients in total at the final dose in part A for the primary efficacy analysis of the IM administration route. The sample size of 13 evaluable patients in part A, cohort 1 was planned to provide at least 80% posterior probability of the true response rate of 96% or higher given 100% response rate in cohort 1 and noninformative neutral beta prior.
Because the primary efficacy end point was considered to be met if the lower bound of the 95% Wald CI of the response rate exceeded 90%, a final sample size of 98 patients was planned, which would provide an 83% probability that the lower bound of the 95% Wald CI would exceed 90%, assuming a true response rate of 96% for the primary efficacy end point and a 5% dropout rate. With a sample size of 98 patients, the probability of observing at least 1 AE related to asparaginase with an incidence as low as 3% is 95%.
For the primary efficacy assessment at the primary analysis, a minimum of 93 patients in the efficacy analysis set was required. An interim analysis was planned, in which a sample size of 51 patients would provide a 70% probability that the lower bound of the 95% CI would exceed 90%, assuming a 96% true response rate and a 5% dropout rate. The probability of observing at least 1 AE related to asparaginase with an incidence as low as 3% is 79% with 51 patients.
No subgroup or sensitivity analysis was performed in the study.
Safety and tolerability were presented for the safety analysis set using descriptive statistics without any formal statistical testing.
Analysis Populations
The analysis sets included the enrolled analysis set contained all patients who signed the informed consent and met the inclusion and exclusion criteria, per investigator. The efficacy analysis set included all patients who received at least 1 dose of crisantaspase recombinant and had at least 1 48-hour or 72-hour NSAA assessment collected within the protocol-defined sample collection window (± 2 hours) in course 1. The safety analysis set included all patients who received at least 1 dose of crisantaspase recombinant. The pharmacokinetic analysis set included all patients who received at least 1 dose of crisantaspase recombinant and had at least 1 postdose evaluable SAA or pharmacokinetic concentration value.
Results
Patient Disposition
For part A (IM) of the study, 174 patients were screened as of the cut-off date of July 19, 2021; of these, 7 patients did not pass through screening (2 due to not meeting the inclusion or exclusion criteria, 4 withdrew consent, 1 “other”). Hence, a total of 167 patients were enrolled, which included 51 in cohort 1c (IM 25 mg/m2 [Monday and Wednesday]) and 50 mg/m2 [Friday]) (Table 9). As of the data cut-off date of July 19, 2021, 28 (16.8%) patients continued to receive treatment with crisantaspase recombinant in the study. A total of 9 of 51 patients (17.6%) discontinued treatment (5 of 51 [9.8%] due to AEs, 1 [2.0%] due to physician decision, 1 [2.0%] due to progressive disease, 1 [2.0%] due to patient withdrawal, and 1 [2.0%] was discontinued due to “other”). A total of 3 of 167 (1.8%) patients in part A had died as of the data cut-off of July 19, 2021; none of these patients were in cohort 1c (1 was in cohort 1a and 2 in cohort 1b).
Table 9: Disposition of All Screened Patients — Cut-Off Date July 19, 2021
Variable | Cohort 1c IM 25 mg/m2 (Monday, Wednesday) and 50 mg/m2 (Friday) |
|---|---|
All patients screened, N | 174 |
All patients enrolled, N | 167 |
All patients enrolled in cohort 1c, N | 51 |
Safety set: patients who received at least 1 dose of crisantaspase recombinant, n (% of screened patients) | 51 (100) |
Patients with ongoing crisantaspase recombinant treatment, n (%) | 24 (47.1) |
Patients who completed all planned crisantaspase recombinant treatment, n (%) | 18 (35.3) |
Patients who discontinued crisantaspase recombinant treatment, n (%) | 9 (17.6) |
Reasons for discontinuing crisantaspase recombinant treatmenta | |
Adverse event | 5 (9.8) |
Physician decision | 1 (2.0) |
Progressive disease | 1 (2.0) |
Recurrent disease | 0 |
Withdrawal by parent or guardian | 0 |
Withdrawal by patient | 1 (2.0) |
Other | 1 (2.0) |
Patients ongoing in the study | 35 (68.6) |
Patients who completed the study | 9 (17.6) |
Patients who discontinued the study | 7 (13.7) |
Reasons for discontinuing the study | |
Adverse event | 4 (7.8) |
Death | 0 |
Physician decision | 0 |
Progressive disease | 0 |
Protocol deviation | 1 (2.0) |
Recurrent disease | 0 |
Withdrawal by parent or guardian | 0 |
Withdrawal by patient | 1 (2.0) |
Other | 1 (2.0) |
IM = intramuscular.
aPatients who discontinued crisantaspase recombinant treatment may have been ongoing in the study for safety follow-up at the time of the data cut-off.
Source: Clinical Study Report of crisantaspase recombinant.11
Exposure to Study Treatments
In cohort 1c, the mean number of courses of treatment completed was 4.9 (standard deviation [SD] = 2.02). The mean percentage of planned doses administered among those in cohort 1c who had completed the study (n = 16) was 79.4%.
The most commonly reported concomitant medications included ondansetron (46 of 51 patients; 90.2%), vincristine (38; 74.5%), a combination of sulfamethoxazole and trimethoprim (40; 78.4%), methotrexate (39; 76.5%), cytarabine (34; 66.7%), paracetamol (33; 64.7%), mercaptopurine (34; 66.7%), famotidine (33; 64.7%), cyclophosphamide (30; 58.8%), and dexamethasone (30; 58.8%). Other drugs were also common (doxorubicin, lorazepam, diphenhydramine, lidocaine, and oxycodone were each used by more than 25% of the population). The clinical experts consulted by CADTH indicated these drugs are commonly used for patients with ALL/LBL who are receiving different chemotherapy regimens.
Efficacy
Only those efficacy outcomes identified in the review protocol are reported here.
The outcomes stated in this CADTH review protocol included efficacy outcomes such as overall survival, event-free survival, disease-free survival, complete clinical remission and/or minimal residual disease, NSAA levels, and HRQoL.
Of these end points, only NSAA levels and harms were evaluated in the included pivotal trial.
Serum Asparaginase Activity
A summary of the last 48-hour and last 72-hour NSAA results are presented in Table 10 from the efficacy analysis set, with a cut-off date of July 19, 2019. The mean (± SD) SAA versus time profiles for crisantaspase recombinant IM in course 1 in linear and semilogarithmic scales are presented in Figure 3. In Table 11, the proportion of patients with last 72-hour and last 48-hour NSAA levels of at least 0.1 IU/mL or at least 0.4 IU/mL during the first course of crisantaspase recombinant are presented.
SAA levels provide data as a surrogate marker for asparagine depletion, and an NSAA level of at least 0.1 IU/mL is the most widely accepted threshold to demonstrate adequate asparagine depletion in clinical practice.14
Overall, mean and median NSAA values showed the dose of cohort 1c (25 mg/m2 [Monday and Wednesday] and 50 mg/m2 [Friday]) reached values above the 0.1 IU/mL threshold for both the last 48-hour (mean = 0.655 IU/mL; 95% CI, 0.537 IU/mL to 0.773 IU/mL) and the last 72-hour (mean = 0.468 IU/mL; 95% CI, 0.349 IU/mL to 0.586 IU/mL) assessments.
Regarding the number and proportion of patients with values at or above the 0.1 IU/mL threshold, this was reached in 47 of 49 patients analyzed (95.9% [95% CI, 90.4% to 100.0%] above the sponsor’s predefined proportion efficacy threshold of 90%) at the last 48-hour time point, and 44 of 49 patients (89.8%; 95% CI, 81.2% to 98.3%) at the last 72-hour assessment (Table 11).
Table 10: SAA Results With IM Crisantaspase Recombinant — Efficacy Analysis Set, Cut-Off July 19, 2021
NSAA m(IU/mL) | Cohort 1c (IM) |
|---|---|
Last 48 hours | |
N | 49 |
Mean (95% CI) | 0.655 (0.537 to 0.773) |
Median (Q1 and Q3) | 0.599 (0.334 and 0.888) |
Last 72 hours | |
N | 49 |
Mean (95% CI) | 0.468 (0.349 to 0.586) |
Median (Q1 and Q3) | 0.383 (0.168 and 0.587) |
CI = confidence interval; IM = intramuscular; MW = Monday and Wednesday; NSAA = nadir serum asparaginase activity; Q1 = first quartile; Q3 = third quartile; SAA = serum asparaginase activity.
Note: The efficacy analysis set includes patients who received at least 1 dose of crisantaspase recombinant with at least one 48-hour or 72-hour NSAA assessment collected within the protocol-defined sample collection window (± 2 hours) in course 1. If the mean was less than the lower limit of quantitation, SD and CIs were not calculated.
Source: Clinical Study Report of crisantaspase recombinant.11
Figure 3: Mean (± SD) Serum Asparaginase Activity Versus Time Profiles for IM Crisantaspase Recombinant in Course 1 on Linear and Semilogarithmic Scales (Pharmacokinetic Analysis Set, Cut-Off July 19, 2021)
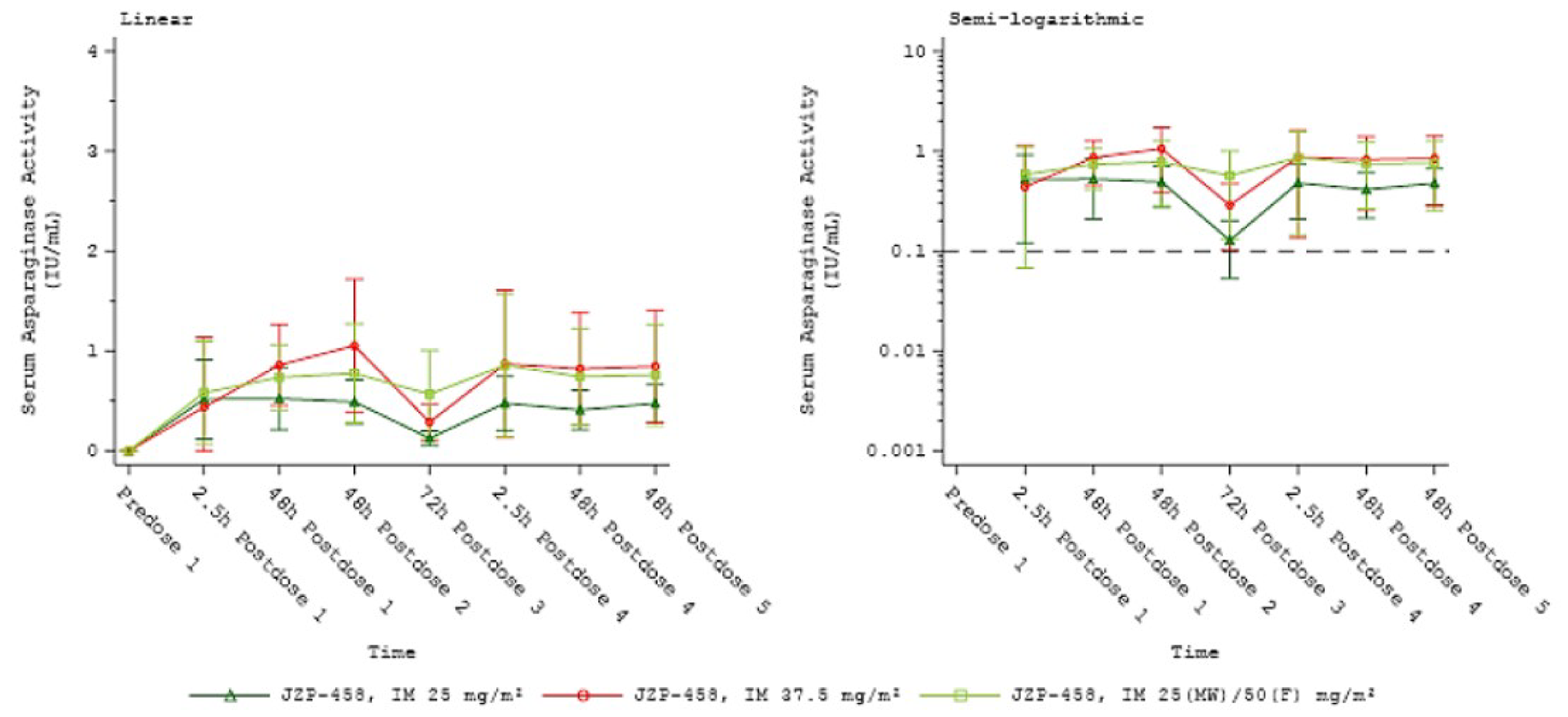
F = Friday; IM = intramuscular; JZP-485 = crisantaspase recombinant; MW = Monday and Wednesday; NSAA = nadir serum asparaginase activity; SD = standard deviation.
Note: Green values with triangle points are for cohort 1a, red values and circle points are for cohort 1b, and light green values with square points are for cohort 1c (the cohort of interest to this CADTH review).
Source: Clinical Study Report of crisantaspase recombinant.11
Table 11: Proportion of Patients With Last 72-Hour and Last 48-Hour NSAA Levels of 0.1 IU/mL or Higher or 0.4 IU/mL or Higher During the First Course of Crisantaspase Recombinant — Efficacy Analysis Set; Data Cut-Off July 19, 2021
NSAA (IU/mL) | Cohort 1c IM |
|---|---|
Last 48 hours | |
N | 49 |
NSAA level ≥ 0.1 IU/mL, n (%) | 47 (95.9) |
95% CI | 90.4 to 100.0 |
NSAA level ≥ 0.4 IU/mL, n (%) | 32 (65.3) |
95% CI | 52.0 to 78.6 |
Last 72 hours | |
N | 49 |
NSAA level ≥ 0.1 IU/mL, n (%) | 44 (89.8) |
95% CI | 81.3 to 98.3 |
NSAA level ≥ 0.4 IU/mL, n (%) | 23 (46.9) |
95% CI | 33.0 to 60.9 |
CI = confidence interval; IM = intramuscular; NSAA = nadir serum asparaginase activity.
Notes: Percentages were calculated with the number of patients for each course and schedule as a denominator.
The efficacy analysis set at 72 hours includes patients who received at least 1 dose of crisantaspase recombinant with at least one 72-hour NSAA assessment collected within the protocol-defined sample collection window (± 2 hours) in course 1. the efficacy analysis set at 48 hours includes patients who received at least 1 dose of crisantaspase recombinant with at least one 48-hour NSAA assessment collected within the protocol-defined sample collection window (± 2 hours) in course 1. The 95% CI was calculated using the Wald method.
Source: Clinical Study Report of crisantaspase recombinant.11
Harms
Only those harms identified in the review protocol are reported here. Refer to Table 12 for detailed harms data. Data for harms from all cohorts is included (including cohort 1c, which is the population and dose of interest) to evaluate any possible harm at any dose.
Harms outcomes included mortality, AEs, SAEs, and withdrawal due to adverse events. Harms of special interest identified by CADTH and reported in this study include thrombosis, pancreatitis, hemorrhage, hypersensitivity reaction, hepatotoxicity, and hypertriglyceridemia (in adults). The safety outcomes in this CADTH review were assessed in the safety analysis set, which included all patients who received at least 1 dose of crisantaspase recombinant. As of the data cut-off date of July 19, 2021, a total of 167 patients (33 in cohort 1a [IM 25 mg/m2 on Monday, Wednesday, and Friday], 83 in cohort 1b [IM 37.5 mg/m2 on Monday, Wednesday, and Friday], and 51 in cohort 1c [25 mg/m2 on Monday and Wednesday and 50 mg/m2 on Friday]) are included in the Safety Analysis Set for Part A (IM) of the study.
Adverse Events
In cohort 1c, 49 of 51 (96.1%) patients reported at least 1 AE. In all patients in the IM cohort, 164 of 167 (98.2%) had at least 1 AE.
The most frequent AEs in cohort 1c, up to the latest cut-off date, have been anemia (52.9%; 27 of 51), platelet count decreased (45.1%; 23 of 51), neutrophil count decreased (47.1%; 24 of 51), and vomiting (33.3%; 17 of 51). In the whole IM cohort, these numbers were similar, with anemia (53.3%; 89 of 167), platelet count decreased (43.7%; 73 of 167), neutrophil count decreased (43.1%; 72 of 167), and vomiting (42.5%; 71 of 167).
Serious Adverse Events
Of those included in cohort 1c, 30 of 51 (58.8%) experienced at least 1 SAE and 44 of 51 (86.3%) of patients experienced an AE of grade 3 or 4. The most common SAEs (in at least 5% of patients) were febrile neutropenia (25.5%), pyrexia (11.8%), investigations for SAE (11.8%), sepsis (9.8%), pancreatitis (any kind: 5.9%; acute: 2.0%), and renal and urinary disorders (5.9%). All other SAEs were reported in less than 5% of patients.
A total of 107 Of 167 patients (64.1%) in part A (IM) of the study experienced at least 1 SAE at the time of the data cut-off of July 19, 2021.
Withdrawal From Treatment Due to Adverse Events
Overall, in part A (IM), 22 of 167 patients (13.2%) experienced an AE that led to discontinuation of the study drug. Events that led to discontinuation of the study drug included drug hypersensitivity (3.6%; 6 of 167); pancreatitis and pancreatitis acute (each 3.0%; 5 of 167); anaphylactic reaction (1.8%; 3 of 167); and necrotizing fasciitis, ALT increased, and hyperammonemia (each 0.6%; 1 of 167).
Mortality
A total of 3 of 167 patients (1.8%) had a fatal event in the total of patients (group A), 1 occurring in cohort 1a, 2 in cohort 1b, and none in cohort 1c (Table 12). Grade 5 (fatal) SAEs included sepsis, aspiration pneumonia, and multiple organ dysfunction syndrome.
Table 12: Summary of Harms — Safety Analysis Set, Cut-Off Date July 19, 2021
Harms | IM dosage | IM total (N = 167) | ||
|---|---|---|---|---|
25 mg/m2 Monday, Wednesday, Friday | 37.5 mg/m2 Monday, Wednesday, Friday | 25 (Monday, Wednesday) and 50 mg/m2 (Friday) | ||
AEs, n (%) | ||||
At least 1 AE | 32 (97.0) | 83 (100.0) | 49 (96.1) | 164 (98.2) |
Most common eventsa | ||||
Anemia | 13 (39.4) | 49 (59.0) | 27 (52.9) | 89 (53.3) |
Decreased platelet count | 13 (39.4) | 37 (44.6) | 23 (45.1) | 73 (43.7) |
Decreased neutrophil count | 14 (42.4) | 34 (41.0) | 24 (47.1) | 72 (43.1) |
Vomiting | 12 (36.4) | 42 (50.6) | 17 (33.3) | 71 (42.5) |
Nausea | 9 (27.3) | 31 (37.3) | 18 (35.3) | 58 (34.7) |
Pyrexia | 10 (30.3) | 36 (43.4) | 10 (19.6) | 56 (33.5) |
Fatigue | 10 (30.3) | 32 (38.6) | 11 (21.6) | 53 (31.7) |
Febrile neutropenia | 10 (30.3) | 26 (31.3) | 14 (27.5) | 50 (29.9) |
Decreased appetite | 7 (21.2) | 28 (33.7) | 14 (27.5) | 49 (29.3) |
Decreased white blood cell count | 12 (36.4) | 26 (31.3) | 11 (21.6) | 49 (29.3) |
Headache | 12 (36.4) | 23 (27.7) | 11 (21.6) | 46 (27.5) |
Stomatitis | 8 (24.2) | 23 (27.7) | 14 (27.5) | 45 (26.9) |
Increased alanine aminotransferase | 5 (15.2) | 29 (34.9) | 10 (19.6) | 44 (26.3) |
Abdominal pain | 5 (15.2) | 26 (31.3) | 12 (23.5) | 43 (25.7) |
Diarrhea | 5 (15.2) | 23 (27.7) | 12 (23.5) | 40 (24.0) |
Back pain | 9 (27.3) | 22 (26.5) | 6 (11.8) | 37 (22.2) |
Decreased lymphocyte count | 8 (24.2) | 21 (25.3) | 8 (15.7) | 37 (22.2) |
Pain in extremity | 8 (24.2) | 18 (21.7) | 8 (15.7) | 34 (20.4) |
Increased aspartate aminotransferase | 4 (12.1) | 24 (28.9) | 5 (9.8) | 33 (19.8) |
Sinus tachycardia | 5 (15.2) | 19 (22.9) | 8 (15.7) | 32 (19.2) |
Hypokalemia | 3 (9.1) | 14 (16.9) | 11 (21.6) | 28 (16.8) |
Hyperglycemia | 7 (21.2) | 13 (15.7) | 6 (11.8) | 26 (15.6) |
Constipation | 4 (12.1) | 13 (15.7) | 7 (13.7) | 24 (14.4) |
Dehydration | 5 (15.2) | 13 (15.7) | 6 (11.8) | 24 (14.4) |
Cough | 5 (15.2) | 11 (13.3) | 7 (13.7) | 23 (13.8) |
Decreased weight | 1 (3.0) | 13 (15.7) | 7 (13.7) | 21 (12.6) |
Contusion | 4 (12.1) | 8 (9.6) | 7 (13.7) | 19 (11.4) |
Hypoalbuminemia | 4 (12.1) | 12 (14.5) | 3 (5.9) | 19 (11.4) |
Increased blood bilirubin | 2 (6.1) | 12 (14.5) | 4 (7.8) | 18 (10.8) |
Rhinorrhea | 4 (12.1) | 10 (12.0) | 4 (7.8) | 18 (10.8) |
Arthralgia | 5 (15.2) | 9 (10.8) | 3 (5.9) | 17 (10.2) |
Hypocalcemia | 4 (12.1) | 9 (10.8) | 4 (7.8) | 17 (10.2) |
Oropharyngeal pain | 2 (6.1) | 10 (12.0) | 5 (9.8) | 17 (10.2) |
Drug hypersensitivity | 2 (6.1) | 8 (9.6) | 1 (2.0) | 11 (6.6) |
SAEs,b n (%) | ||||
Number of patients with at least 1 SAE | 20 (60.6) | 57 (68.7) | 30 (58.8) | 107 (64.1) |
Febrile neutropenia | 9 (27.3) | 23 (27.7) | 13 (25.5) | 45 (26.9) |
Pyrexia | 3 (9.1) | 8 (9.6) | 6 (11.8) | 17 (10.2) |
Investigations for SAEs | 0 | 3 (3.6) | 6 (11.8) | 9 (5.4) |
Sepsis | 2 (6.1) | 4 (4.8) | 5 (9.8) | 11 (6.6) |
Dehydration | 3 (9.1) | 7 (8.4) | 4 (7.8) | 14 (8.4) |
Renal and urinary disorders | 1 (3.0) | 4 (4.8) | 3 (5.9) | 8 (4.8) |
Pancreatitis | 0 | 2 (2.4) | 3 (5.9) | 5 (3.0) |
Anemia | 0 | 3 (3.6) | 1 (2.0) | 4 (2.4) |
Vomiting | 1 (3.0) | 8 (9.6) | 2 (3.9) | 11 (6.6) |
Nausea | 2 (6.1) | 6 (7.2) | 1 (2.0) | 9 (5.4) |
Stomatitis | 3 (9.1) | 4 (4.8) | 2 (3.9) | 9 (5.4) |
Pancreatitis acute | 0 | 5 (6.0) | 1 (2.0) | 6 (3.6) |
Immune system disorders | 2 (6.1) | 8 (9.6) | 1 (2.0) | 11 (6.6) |
Drug hypersensitivity | 2 (6.1) | 4 (4.8) | 1 (2.0) | 7 (4.2) |
Toxicity from various drugs | 1 (3.0) | 2 (2.4) | 1 (2.0) | 4 (2.4) |
Decreased platelet count | 0 | 1 (1.2) | 1 (2.0) | 2 (1.2) |
Hyperglycemia | 0 | 1 (1.2) | 1 (2.0) | 2 (1.2) |
Acute kidney injury | 1 (3.0) | 4 (4.8) | 2 (3.9) | 7 (4.2) |
Patients who stopped treatment due to AEs | ||||
n (%) | 3 (9.1) | 14 (16.9) | 5 (9.8) | 22 (13.2) |
Most common events, n (%) | ||||
Pancreatitis | 0 (0) | 6 (7.2) | 4 (7.8) | 10 (6.0) |
Drug hypersensitivity | 2 (6.1) | 3 (3.6) | 1 (2.0) | 6 (3.6) |
Deaths (all cause) | ||||
n (%) | 1 (3.0) | 2 (2.4) | 0 | 3 (1.8) |
AE = adverse event; IM = intramuscular; SAE = serious adverse event.
Note: Percentages were calculated with the number of patients in the safety analysis set as a denominator. AEs were coded using Medical Dictionary for Regulatory Activities (MedDRA) 22.1. The severity of AEs was recorded using CTCAE 5.0.
aFrequency > 10%.
bAny frequency.
Source: Clinical Study Report of crisantaspase recombinant.11
Notable Harms
AEs of special interest identified by the CADTH team and the clinical experts consulted by CADTH included hypersensitivity reactions, pancreatitis, thrombosis, hepatotoxicity, hemorrhage, and hypertriglyceridemia (Table 13).
Allergic reactions regardless of causality, inclusive of hypersensitivity and anaphylaxis, occurred in 18 (35.3%) of patients in cohort 1c and in 69 of 167 patients (41.3%) in the total of part A group. The most frequently reported events related to allergic reactions in cohort 1c included rash (7.8% of patients; 4 of 51), dermatitis (7.8%; 4 of 51), and allergic transfusion reaction (7.8%; 4 of 51).
As of the data cut-off date (July 19, 2021), 6 of 51 patients in cohort 1c (11.8%) had experienced pancreatitis. One of these 6 cases was an acute pancreatitis. Meanwhile, 13 of 167 patients (7.2%) in part A (all IM) group had experienced an event of pancreatitis (either pancreatitis acute [6 patients] or pancreatitis [7 patients]).
|||||||||| |||||||| |||| || |||||||| || ||| || |||||||| || |||||| || |||||| || |||||||| |||||| ||||||||||| Meanwhile, thrombosis occurred in 5 of 167 patients in the part A group: 1 case of superior sagittal sinus thrombosis, 2 cases of pulmonary embolism, and 2 cases of jugular vein thrombosis.
Hepatic toxicity was observed in 13 patients (25.5%) in cohort 1c group, all including elevated measured levels of ALT, AST, and bilirubin. This was observed in 55 of 167 patients (32.9%) in the part A (IM) group.
Hypertriglyceridemia occurred in 6 of the 51 patients (11.8%) in cohort 1c and in 16 of the 167 (9.6%) patients in the total IM group.
Table 13: Harms of Special Interest — Safety Analysis Set, Cut-Off Date July 19, 2021
Patients with at least 1 AE of special interest, n (%)a | IM dosage | IM total | ||
|---|---|---|---|---|
25 mg/m2 Monday, Wednesday, Friday | 37.5 mg/m2 Monday, Wednesday, Friday | 25 (Monday, Wednesday) and 50 mg/m2 (Friday) | ||
Allergic reactions (including hypersensitivity and anaphylaxis) | 10 (30.3) | 41 (49.4) | 18 (35.3) | 69 (41.3) |
|||||||||| | ||||| | | ||||| | | ||||| | | ||||| |
Pancreatitis | 0 | 6 (7.2) | 6 (11.8) | 12 (7.2) |
Thrombocytopenia | 1 (3.0) | 2 (2.4) | 4 (7.8) | 7 (4.2) |
Hepatotoxicity | 8 (24.2) | 34 (41.0) | 13 (25.5) | 55 (32.9) |
Hypertriglyceridemia | 3 (9.1) | 7 (8.4) | 6 (11.8) | 16 (9.6) |
AE = adverse event; IM = intramuscular.
Note: Percentages were calculated with the number of patients in the safety analysis set as a denominator. AEs were coded using Medical Dictionary for Regulatory Activities (MedDRA) 22.1. The severity of AEs was recorded using CTCAE 5.0.
aDetermined as special interest for patients, clinicians, and stakeholders, as stated in the protocol of this review.
Source: Clinical Study Report of crisantaspase recombinant.11
Critical Appraisal
Internal Validity
The main limitation of the included study is the single-arm (noncomparative) design. As a result of the lack of comparator group and no control for confounding, the outcomes cannot be definitively attributed to the administration of crisantaspase recombinant. It is recognized that supply shortages of the main comparator (EDA) may have rendered a randomized comparison unfeasible. The selection criteria appear reasonable, and there were few (less than 5%) screen failures, indicating that the risk of selection bias is probably low. No formal hypothesis testing was performed to assess the effect estimates from this study.
The study was open label; however, bias in the measurement of the outcome is not suspected because efficacy measurements, such as NSAA levels, were collected objectively. Overreporting of known harms is possible, but the extent to which this may have occurred is not possible to quantify. An evaluation of the comparative efficacy of crisantaspase recombinant and EDA is limited by the shortcomings of this study. Noting these limitations and the uncertainty in the information provided, crisantaspase recombinant may still behave similarly to the drug it aims to replace (E. coli–derived asparaginase) in the multicomponent chemotherapy regimen for patients with ALL/LBL; however, this was not tested. The study can only highlight that crisantaspase recombinant is associated with measurable asparaginase activity (NSAA above a threshold of 0.1 IU/mL, previously set as an important limit of clinical importance).8,15,16 This end point, however, is used and should be interpreted as a surrogate for important clinical outcomes.
The efficacy analysis was performed on the efficacy analysis set, which included all patients who received at least 1 dose of the study drug and had at least one 48-hour or 72-hour NSAA assessment in course 1. Only 1 assigned patient is missing from this analysis set (compared to the enrolled set), therefore there are no concerns about bias related to this decision.
External Validity
The population included in the trial were from different centres in the US and Canada. Overall, the study population was deemed generalizable to the population in Canada, according to the clinical experts consulted by CADTH. The demographic, disease, and baseline characteristics, as well as the previous therapies received by patients, were in line with current clinical practice. The age of participants was representative of the most common age at presentation for ALL; however, no adults older than 25 years were included, which could affect the generalizability of results to older age groups. The clinical experts consulted by CADTH noted that the drug pharmacokinetics may differ in older populations, who may be prone to greater toxicity. Few LBL patients were included, however the clinical experts indicated that no difference in response would be expected for patients with ALL compared with those with LBL. Although all patients included in the trial had a previous grade 3 or higher hypersensitivity reaction to E. coli–derived asparaginase, the clinical experts consulted by CADTH noted that the findings would be applicable to those with lower-grade hypersensitivity reactions (e.g., grade 2 or higher). The trial did not include an assessment of patient-important outcomes such as HRQoL.
The study does not evaluate efficacy outcomes of interest such as overall survival, disease-free survival, or HRQoL; instead, the findings are limited to the surrogate marker of asparagine depletion, NSAA level. The NSAA threshold of 0.1 IU/mL or higher used in the trial is an accepted threshold based on reports from the literature and clinical expert consensus.12,14,16 The timing of the assessment of outcomes was deemed to be appropriate by the clinical experts and consistent with clinical practice, where usually the response to treatment is evaluated by measurements of NSAA levels, similar to the measurements performed in the study.
Indirect Evidence
Objectives and Methods for the Summary of Indirect Evidence
The objective of this section is to summarize and appraise the evidence from ITCs for the relative effectiveness and safety of crisantaspase recombinant for the treatment of adult and pediatric patients aged 1 year and older with ALL or LBL who have developed hypersensitivity to E. coli–derived asparaginases.
The aim of the ITC is to fill a gap created by the absence of trials directly comparing crisantaspase recombinant to relevant comparators (EDAs) in the Canadian landscape, as stated in the protocol of this CADTH review report.
A focused literature search for ITCs of Rylaze (crisantaspase recombinant) and ALL, LBL was run in MEDLINE All (1946–) on September 12, 2022. No limits were applied.
One ITC was submitted by the sponsor, and it is reviewed in this section. No other ITCs were identified in the literature search.
Description of Indirect Comparison
One naive ITC submitted by the sponsor17 aimed to compare crisantaspase recombinant with EDA for the treatment of patients with ALL or LBL. A feasibility assessment of the populations, treatments, and outcomes was conducted to determine whether the ITC methods were appropriate. The ITC evaluates 2 single-arm trials with no common comparator.
Methods of ITC
Objectives
The main objective of the ITC was to determine the relative efficacy and safety of crisantaspase recombinant compared with EDA for the treatment of patients with ALL/LBL, using indirect treatment comparisons of relevant clinical trials.
Study Selection Methods and Feasibility Assessment
Sources of data to support ITCs of crisantaspase recombinant and EDA were first determined. A targeted search of the literature (methods not further described) was conducted to identify any source of information for the crisantaspase recombinant and EDA treatment arms.
There is only 1 clinical trial of crisantaspase recombinant, which is a phase II/III, single-arm, open-label clinical trial conducted in the US and Canada and described in this CADTH review report.11 The trial enrolled 167 patients to 1 of 3 different IM dosing arms; patients in cohort 1a were initiated on an IM dose of 25 mg/m2 on a Monday-Wednesday-Friday schedule over 2 weeks, patients in cohort 1b were initiated on an IM dose of 37.5 mg/m2 on a Monday-Wednesday-Friday schedule over 2 weeks, and patients in cohort 1c were initiated on a dose of 25 mg/m2 on Mondays and Wednesdays and a dose of 50 mg/m2 on Fridays. For the purpose of the ITC, authors only included and evaluated cohort 1c as the comparator because the dosing regimen in this group represents the dosing schedule approved by Health Canada as a component of a multidrug chemotherapeutic regimen for the treatment of ALL/LBL in adult and pediatric patients 1 year and older who have developed hypersensitivity to E. coli–derived asparaginase.
For data relevant to EDA, the targeted search focused on identifying trials with the same efficacy outcome definitions as in the crisantaspase recombinant study. A single-arm, open-label clinical trial was found that enrolled 59 patients in the US to be treated with IM EDA.16 Another single-arm trial was identified but it used an IV formulation of EDA, so it was considered by the sponsor to not be relevant for the comparison.
Efficacy outcomes included in the ITC for comparative efficacy were defined as the proportion of patients with last 72-hour and last 48-hour NSAA levels of at least 0.1 IU/mL. No other outcomes of clinical interest to this CADTH report were included.
Harm outcomes (safety evaluations) included comparisons of AEs and patient discontinuations, which was defined by the number of patients who had completed all remaining courses of therapy.
A feasibility assessment was conducted to ensure that a naive ITC was an appropriate method for assessing the relative efficacy and safety of crisantaspase recombinant compared with EDA for the treatment of patients with ALL/LBL. The ability to conduct an ITC was dependent on the type of data available and whether the patient populations and outcome definitions in the trials were deemed comparable. Qualitative comparisons of the study design, eligibility criteria, patient baseline characteristics, and outcome definitions were thus made.
An ITC was deemed necessary to compare efficacy and safety data between the 2 therapies (crisantaspase recombinant and EDA), as there are no head-to-head clinical trials. The feasibility assessment concluded that only these 2 studies were available for comparison. The main characteristics of these studies is presented in Table 14, and the similarities between the 2 populations are described in the ITC results section.
Table 14: Comparison of Key Study Design Between the 2 Studies Included in the ITC
Variable | Crisantaspase recombinant study | Erwinia-derived asparaginase study |
|---|---|---|
Study completion date | 2021 | 2013 |
Design | Open-label, multicentre, clinical trial | Open-label clinical trial |
Phase | II/III | Not stated |
Blinding | No | No |
Country | US and Canada | US |
Population | Patients with ALL/LBL who developed hypersensitivity (grade ≥ 3a) to a long-acting E. coli–derived asparaginase | Patients with ALL with documented grade ≥ 2 allergy to pegaspargase and ≥ 1 remaining scheduled dose of pegaspargase |
Total patients | 51 | 58 |
Interventions | Six doses of IM Rylaze 25 mg/m2 on Monday and Wednesday and 50 mg/m2 on Friday | Erwinia asparaginase 6 doses of 25 000 IU/m2 on Monday, Wednesday, and Friday |
Supporting therapies | Chemotherapy (no additional details) | Chemotherapy (no additional details) |
Inclusion criteria | ||
Diagnosis | ALL or LBL | ALL |
Hypersensitivity | Grade ≥ 3 hypersensitivity to E. coli–derived asparaginase | Documented grade ≥ 2 allergy to pegaspargase with ≥ 1 remaining dose |
Age | Pediatric and Adult patients | > 1 year to < 30 years |
Exclusion criteria | ||
Prior therapy | Erwinia asparaginase or Rylaze | Erwinia asparaginase |
Adverse events | Grade ≥ 3 pancreatitis, asparaginase-associated grade ≥ 3 hemorrhagic event, or thrombus requiring anticoagulation therapy not catheter-related | Grade ≥ 2 pancreatitis |
Disease | Relapsed ALL or LBL | Not stated |
Concurrently receiving another investigational drug | Not at same time | Not stated |
Patient characteristics | ||
Age (years), mean (SD) [range] | 11.3 (5.41) [3 to 25] | 9.7 (5.2) [2 to 18] |
Sex (male), % | 60.8 | 58.6 |
Time since diagnosis (months), % | ||
0 to 3 months | 76.5 | 50 |
4 to 6 months | 21.6 | 44.8 |
7 to 9 months | 2 | 5.2 |
Number of courses of E. coli–derived asparaginase (median range) | NR | 3 (1 to 5) |
Number of courses of treatment (median range) | 4 (1 to 10) | 3 (1 to 9) |
Race or ethnicity, % | ||
Black or African American | 15.7 | 10.3 |
Hispanic or Latino | 33.3 | 34.5 |
White | 64.7 | 77.6 |
Precursor B-cell ALL, % | 72.5 | 88.0 |
T-cell ALL, % | 17.6 | 12.0 |
T-cell LBL, % | 7.8 | 0 |
ALL = acute lymphocytic leukemia; EDA = Erwinia asparaginase; IM = intramuscular; LBL = lymphoblastic lymphoma; NR = not reported; SD = standard deviation.
aPatient population for patients in part A and B, cohort 1, of the Rylaze trial, which encompassed the patients evaluated in this ITC.
Sources: Sponsor-submitted ITC,17 and based on Clinical Study Report of crisantaspase recombinant,11 and published study of Erwinia-derived asparaginase.16
Overall, the feasibility analysis concluded that the patient populations and efficacy outcome definitions between the trials had sufficient overlap to conduct an ITC.
ITC Analysis Methods
Because both trials only provide single-arm data with no common comparators, the authors did not consider a Bucher adjusted ITC method feasible. Given the small sample size in both trials, the large number of events for efficacy, and the low number of safety events, a population-adjusted ITC was not deemed appropriate to go through a population-adjusted ITC, such as an unanchored matching-adjusted indirect comparison or simulated treatment comparison, which adjust for imbalances between patient populations and mitigate bias to ensure the fairest comparison possible, given the data available.
Given the limitations mentioned, the ITC depended on a pairwise, naive, unanchored ITC method, comparing the EDA trial arm with cohort 1c of the crisantaspase recombinant trial. Outcomes assessed included the proportion of patients with last 48-hour and last 72-hour NSAA levels of at least 0.1 IU/mL during their first treatment course, as well as harm outcomes. To achieve this, comparisons were made to obtain ORs, with 95% CIs presented for each comparison. The 95% CIs for the ITC were calculated as per the National Institute for Health Care and Excellence (NICE) Technical Support Document 2.26 ORs and CIs were calculated using statistical algorithms described in the Cochrane handbook as implemented in RevMan 5.27 For comparisons of safety outcomes, when 1 trial reported no events, a continuity correction of 0.5 was added to each cell of the 2 × 2 table when calculating the OR.
Results of ITC
Summary of Included Studies
The 2 open-label, single-arm studies evaluating crisantaspase recombinant and EDA were included (Table 14).
As mentioned in the feasibility analysis, patient baseline characteristics were similar in the trials and treatment groups. Patients in the crisantaspase recombinant trial were slightly older and were more recently diagnosed. The EDA trial included only patients with ALL and a documented allergy to pegaspargase of grade 2 or higher, whereas patients in part A, cohort 1 of the crisantaspase recombinant trial could have had either ALL or LBL and were required to have had hypersensitivity to a long-acting E. coli–derived asparaginase of grade 3 or higher. Furthermore, the crisantaspase recombinant trial excluded patients with pancreatitis of grade 3 or higher, whereas the EDA trial excluded patients with pancreatitis of grade 2 or higher. Although the 2 trials had a similar proportion of patients with B-cell versus T-cell ALL, the crisantaspase recombinant trial also included patients with LBL (7.8% of patients), whereas the EDA trial did not include patients with LBL.
In terms of outcome definitions (Table 15), both trials assessed the proportion of patients achieving an NSAA level of at least 0.1 IU/mL at 72 hours and 48 hours during their first treatment course. Both trials evaluated the last 72-hour NSAA during course 1 before the following doses: dose 4 for patients receiving dose 1 on a Monday, dose 6 for patients receiving dose 1 on a Wednesday, and dose 5 for patients receiving dose 1 on a Friday. The timing of the evaluation of the 72-hour NSAA was aligned between the 2 trials. Similarly, both trials evaluated the 48-hour NSAA during course 1, before dose 6 for patients receiving dose 1 on a Monday or Friday, and before dose 5 for patients receiving dose 1 on a Wednesday.
Patients in the crisantaspase recombinant trial received a median of 4 courses (range, 1 to 10 courses), and patients in the EDA study received a median of 3 courses (range, 1 to 9 courses). Although the primary and secondary end points that assessed 48-hour and 72-hour NSAA levels occurred during course 1, AEs were assessed over the duration of all treatment courses.
For safety outcomes, the crisantaspase recombinant trial used the CTCAE 5.0 to identify and grade AEs, whereas CTCAE 3.0 was used for the EDA trial. AEs that were reported for both trials using the same definition included increased ALT (grade 3 or 4), pancreatitis (any grade), pancreatitis (grade 3 or 4), drug hypersensitivity, thrombosis, and hyperglycemia. Additionally, discontinuation was defined the same way in both trials. Both trials report the number of patients that completed all remaining courses of planned asparaginase therapy, which represents the number of patients who did not discontinue treatment. The trial of crisantaspase recombinant was still ongoing at the time of the analysis, so the sponsor assumed that all patients still on treatment at the time of the data cut-off would complete all remaining treatment courses.
Table 15: Assessment of Outcome Definitions for the Studies Included in the ITC
Outcome | Crisantaspase recombinant study | Erwinia-derived asparaginase study |
|---|---|---|
NSAA | Proportion of patients with last 72-hour and last 48-hour NSAA ≥ 0.1 IU/mL during the first treatment course. 48-hour: Based on samples before dose 6 for patients who received dose 1 on Monday or Friday and before dose 5 for patients receiving treatment on Wednesday. 72-hour: Based on samples collected before dose 4 for patients who began treatment on a Monday, before dose 6 for patients who started on a Wednesday, and before dose 5 for patients who started on a Friday. | Proportion of patients with 72-hour and 48-hour NSAA ≥ 0.1 IU/mL during the first treatment course. 48-hour: Based on samples before dose 6 for patients who received dose 1 on Monday or Friday and before dose 5 for patients who received treatment on Wednesday. 72-hour: Based on samples collected before dose 4 for patients who began treatment on a Monday, before dose 6 for patients who started on a Wednesday, and before dose 5 for patients who started on a Friday. |
Safety | AEs reported according to CTCAE 5.0. Discontinuation defined as number of patients who completed all remaining courses. | AEs reported according to CTCAE 3.0. Discontinuation defined as number of patients who completed all remaining courses. |
AE = adverse event; CTCAE = Common Terminology Criteria for Adverse Events; ITC = indirect treatment comparison; NSAA = nadir serum asparaginase activity.
Sources: Sponsor-submitted ITC,17 and based on Clinical Study Report of crisantaspase recombinant,11 and published study of Erwinia-derived asparaginase.16
Results
For efficacy end points, point estimates for the results of the comparison of the proportion of patients with last 48-hour and last 72-hour NSAA levels of at least 0.1 IU/mL are shown in Table 16. ORs with 95% CIs are presented for each comparison. In this comparison, 47 of 49 (95.9%) patients in the crisantaspase recombinant study reached an NSAA level of at least 0.1 IU/mL in the last 48-hour assessment time, as did 38 of 41 (92.7%) of patients in the EDA study (OR = 1.86; 95% CI, 0.29 to 11.68). Similarly, 44 of 49 (89.8%) patients in the crisantaspase recombinant study reached an NSAA level of at least 0.1 IU/mL in the last 72-hour assessment time, as did 38 of 43 (88.4%) of patients in the EDA study (OR = 1.16; 95% CI, 0.31 to 4.31).
Table 16: ITC of the Proportion of Patients With Last 48-Hour and Last 72-Hour NSAA Levels 0.1 IU/mL or Higher for Crisantaspase Recombinant Versus EDA
Efficacy assessment time | EDA n of N (%) | Crisantaspase recombinant n of N (%) | OR of ITC (95% CI), crisantaspase recombinant vs. EDA |
|---|---|---|---|
48 hours | 38 of 41 (92.7) | 47 of 49 (95.9) | 1.86 (0.29 to 11.68) |
72 hours | 38 of 43 (88.4) | 44 of 49 (89.8) | 1.16 (0.31 to 4.31) |
EDA = Erwinia-derived asparaginase; ITC = indirect treatment comparison; NSAA = nadir serum asparaginase activity; OR = odds ratio.
Sources: Sponsor-submitted ITC,17 and based on Clinical Study Report of crisantaspase recombinant,11 and published study of EDA.16
Comparisons of harm outcomes that were available in both trials are shown in Table 17. ORs with 95% CIs are presented for each comparison.
An increased ALT level (grade 3 or 4) was observed in 7.8% of patients in the crisantaspase recombinant trial and in 5% of patients in the EDA trial (OR = 1.56; 95% CI, 0.46 to 5.29). Six patients (11.8%) developed pancreatitis in the crisantaspase recombinant trial compared to 1 patient (1.8%) in the EDA trial (OR = 5.19; 95% CI, 0.96 to 28.05). Of the 6 patients who developed pancreatitis in the crisantaspase recombinant trial, 4 (7.8%) developed grade 3 or 4 pancreatitis, whereas no patients in the EDA trial had grade 3 or 4 pancreatitis (OR = 11.08; 95% CI, 0.13 to 930.6).
In the crisantaspase recombinant trial, 1 patient (2.0%) developed drug hypersensitivity, whereas 8 patients (14%) developed drug hypersensitivity in the EDA trial (OR = 0.13; 95% CI, 0.01 to 1.23). The crisantaspase recombinant trial reported that all drug hypersensitivity events were grade 3 or 4 events. Of the 8 patients who developed drug hypersensitivity in the EDA trial, it was reported that 5 of these patients (9% of total population) developed grade 3 or 4 hypersensitivity (OR = 0.21; 95% CI, 0.02 to 2.4).
|| |||||||| || ||| ||| ||||| ||||||||| |||||||||||| ||||||| |||||||| || ||| ||||||||||||| ||||||||||| ||||| ||||||||| |||||||||| ||| |||||| |||| ||||| ||| ||| |||| || |||||||| A total of 7 patients developed hyperglycemia in the EDA trial (12%), whereas 6 patients (11.8%) in the crisantaspase recombinant trial developed hyperglycemia (OR = 0.97; 95% CI, 0.49 to 1.93). Of the 7 patients who developed hyperglycemia in the EDA trial, 1 patient developed grade 3 or 4 hyperglycemia (2.0%). Of the 6 patients who developed hyperglycemia in the crisantaspase recombinant trial, 2 patients (3.9%) developed grade 3 or 4 hyperglycemia (OR = 2.33; 95% CI, 0.11 to 47.41).
In the EDA trial, 55 (80%) patients completed all remaining courses of planned asparaginase therapy. As cohort 1c of the crisantaspase recombinant trial was still ongoing at the time of this analysis, it was assumed that all patients on therapy at the time of the data cut-off would complete their remaining courses. At the July 19, 2021, data cut-off date, 9 patients (17.6%) had discontinued crisantaspase recombinant treatment; therefore, it was assumed that the remaining 24 patients (47.1%) would complete all remaining courses. These patients were added to the 18 patients who had already completed all courses, for a total of 42 patients (82.4%) who had completed eligible courses to date (OR versus EDA = 1.17; 95% CI, 0.72 to 1.90).
Table 17: ITC Comparison of Harm Outcomes for Crisantaspase Recombinant Versus EDA
AEs and discontinuations | EDA n of N (%) | Crisantaspase recombinant n of N (%) | OR of ITC (95% CI), crisantaspase recombinant vs. EDA |
|---|---|---|---|
ALT grade 3 or 4 | 3 of 58 (5) | 4 of 51 (7.8) | 1.56 (0.46 to 5.29) |
Pancreatitis (any grade, including acute) | 1 of 55 (1.8) | 6 of 51 (11.8) | 5.19 (0.96 to 28.05) |
Pancreatitis (grade 3 or 4) | 0 of 58 (0) | 4 of 51 (7.8) | 11.08 (0.13 to 930.61) |
Drug hypersensitivity (any grade) | 8 of 58 (14) | 1 of 51 (2.0) | 0.13 (0.01 to 1.23) |
Drug hypersensitivity (grade 3 or 4) | 5 of 58 (9) | 1 of 51 (2.0) | 0.21 (0.02 to 2.4) |
|||||||||| |||| |||||| | |||| |||| | |||| |||| | |||| |||||| || ||||||| |
Hyperglycemia (any grade) | 7 of 58 (12) | 6 of 51 (11.8) | 0.97 (0.49 to 1.93) |
Hyperglycemia (grade 3 or 4) | 1 of 58 (2) | 2 of 51 (3.9) | 2.33 (0.11 to 47.41) |
Patients completing all remaining treatment courses | 44 of 55 (80) | 42 of 51 (82.4)a | 1.17 (0.72 to 1.9) |
ALT = alanine aminotransferase; CI = confidence interval; EDA = Erwinia-derived asparaginase; ITC = indirect treatment comparison; OR = odds ratio.
aCohort 1c of the crisantaspase recombinant trial was still ongoing at the time of the analysis (data cut-off date July 19, 2021). It was therefore assumed that all patients still on treatment at the time of the data cut-off would complete their remaining treatment courses.
Sources: Sponsor-submitted ITC,17 Clinical Study Report of crisantaspase recombinant,11 and published study of EDA.16
Critical Appraisal of ITC
The main limitation of the ITC is mostly due to the characteristics of the individual studies conforming the body of evidence.
The literature search was aimed at identifying individual studies of EDA with a targeted search strategy. The evidence to inform the use of crisantaspase recombinant was known to be composed of the only single-arm clinical trial on this drug, which was obtained from the sponsor. Therefore, a description of the screening process, the selection of studies, data extraction, and assessment of risk of bias were not presented. Similarly, the risk of bias in the EDA trial was not described.
The ITC was achieved with a naive comparison of each treatment arm in each study because performing an NMA or a population-adjusted ITC was not feasible due to the small sample size of each single-arm trial. Although there is a description of the baseline, demographic, and study characteristics, where similarities between studies and populations are observed, there is no certainty about the balance of unobserved prognostic factors or effect modifiers, which increases the risk of residual confounding. Hence, any difference in effects between crisantaspase recombinant and EDA is difficult to ascertain.
There is large imprecision when estimating the OR for both comparisons due to high number of events in a small number of patients. The large uncertainty in estimating the OR for the proportion of patients with last 48-hour and last 72-hour NSAA levels of at least 0.1 IU/mL, combined with evidence that much lower thresholds are consistent with asparaginase depletion, suggests that there is limited evidence of a meaningful difference in effectiveness between therapies.
The same limitations apply when evaluating harms. Although ORs were calculated, overall, the crude numbers of AEs in both included studies were similar and within the expected incidence, according to the clinical experts consulted by CADTH. The incidence of AEs is likely to be affected by the background chemotherapy regimens, which may have differed across trials as a result of the time period during which they were conducted. Additionally, there were some differences in definitions used for AEs across the trials, and differences in inclusion criteria may have biased the naive comparison of AEs.
There were no major concerns in terms of the external validity, aside from those previously noted for the trial of crisantaspase recombinant. Overall, the populations, interventions, and outcomes assessed in the ITC were considered by the clinical experts consulted by CADTH to be generalizable to the population and clinical practice in Canada for the specific question and indication assessed in this review. Some differences were noticed, such as in the condition included (for instance, there were no patients with LBL in the EDA study) and in the lack of adult patients (≥ 25 years) represented in both studies.
Other Relevant Evidence
This section includes submitted long-term extension studies and additional relevant studies included in the sponsor’s submission to CADTH that were considered to address important gaps in the evidence included in the systematic review.
No long-term studies or other relevant studies addressing gaps in information were included in this review.
Discussion
Summary of Available Evidence
One study was included in this review, the pivotal trial of crisantaspase recombinant. The pivotal trial is an ongoing, open-label, single-arm, multicentre, dose-confirmation, pharmacokinetic trial of crisantaspase recombinant in pediatric and adult patients with ALL/LBL who are hypersensitive to E. coli–derived asparaginases (allergic reaction or silent inactivation). The study was designed to assess the efficacy and harms of crisantaspase recombinant. Efficacy was measured with asparaginase activity (an NSAA level ≥ 0.1 IU/mL was used as a meaningful threshold) assessed at 48 hours and 72 hours. The focus of this review was on cohort 1c of the study, which included 51 patients receiving a dosing schedule of 25 mg/m2 on Mondays and Wednesdays and 50 mg/m2 on Fridays, which is the approved Health Canada indication. The study was initiated on December 27, 2019, and the data cut-off date for the available analysis was July 19, 2021. In this study, 6 doses of crisantaspase recombinant were substituted for each dose of a long-acting E. coli–derived asparaginase. Doses were administered on a MWF schedule for 2 consecutive weeks, and 2 consecutive weeks of treatment with crisantaspase recombinant is defined as 1 course. The population was, overall, generalizable to the Canadian landscape, although no adults older than 25 years were included in the study.
Patient input obtained by CADTH suggests that, when starting new treatments, patients consider the least amount of travel needed, improved quality of life, fewer side effects, and insurance coverage as crucial factors for decisions. HRQoL end points were not available for this study.
One sponsor-submitted ITC comparing crisantaspase recombinant and EDA was included. The body of evidence in this ITC consisted of 2 single-arm studies: 1 of crisantaspase recombinant and the other of EDA. Because an NMA or other population-adjusted ITC was unfeasible, the analysis was achieved through a naive comparison of the single-treatment arm from each study and calculation of ORs for the effect estimates. The populations and outcome definitions were, overall, similar between the studies for assessing a comparison. The outcomes evaluated are the proportion of patients reaching an NSAA level of at least 0.1 IU/mL and harms. The ITC presented effect estimates with uncertainty due to risk of bias, confounding (single-arm studies without adjustments to balance unobserved prognostic factors or effect modifiers), and imprecision. Hence, any difference in effects between crisantaspase recombinant and EDA is difficult to ascertain.
Interpretation of Results
Efficacy
Among the outcomes of interest for this review (overall survival, event-free survival, disease-free survival, complete clinical remission and/or minimal residual disease, and HRQoL), only the surrogate outcome, asparaginase activity measured with NSAA levels, and harms were evaluated in the included study. Furthermore, no long-term evaluation of any outcome is available at this point.
At both time points evaluated (48 hours and 72 hours), the results showed that the dose of cohort 1c, the mean (95% CI) NSAA values were above the 0.1 IU/mL threshold set as important for establishing adequate asparaginase activity. These effects were also observed for the number and proportion of patients with values at or above the 0.1 IU/mL threshold at the 48-hour and 72-hour time points, where most patients exceeded the threshold. These findings were considered to be relevant by the clinical experts consulted by CADTH, but they noted that NSAA level is a surrogate marker of clinically important outcomes. The experts also noted that access to NSAA level assays is essential to manage these therapies, with at least 1 test per cycle (especially in children), or every 3 or 4 cycles, to identify patients with hypersensitivity. However, tests in clinical practice are rarely used because they are no longer funded.
Though the single-arm trial design precludes causal inferences, the mechanism of action of crisantaspase recombinant is equivalent to that of other asparaginases, supporting the plausibility of the link between the drug and the outcome. The clinical experts consulted by CADTH believed that the findings would be generalizable to patients seen in clinical practice in Canada.
Crisantaspase recombinant is indicated for patients with ALL/LBL who develop hypersensitivity to an E. coli–derived asparaginase as a replacement for asparaginase in a multicomponent chemotherapeutic regimen. Crisantaspase recombinant is anticipated to be used in a similar manner to EDA, which is currently the only option for use in patients with hypersensitivity (including silent inactivation). However, EDA has been cancelled postmarket by Health Canada, and it is currently being reviewed under the Priority Review policy, which adds to issues that had already been in place, such as short supply and scarcity for this option. The clinical experts consulted by CADTH considered that availability and safety profiles of each drug option are important factors they would consider when choosing either in clinical practice because direct comparative evidence on efficacy or patient-important outcomes is not available.
In the ITC, both included single-arm studies seemed similar enough in their population and outcome definitions to make an indirect comparison. Because both trials only provided single-arm data, with no common comparators, and the sample size was small in both trials, the number of events for efficacy was large, and the number of safety events was low, a population-adjusted ITC was not deemed appropriate. Overall, the proportion of patients who reached an NSAA level of at least 0.1 IU/mL was above 90% in both the crisantaspase recombinant and EDA arms, both at the 48-hour and 72-hour assessment time points. However, the estimated comparative effects calculated by the authors in the naive comparison were imprecise, with a risk of bias, plausible residual confounding, and some issues of generalizability (i.e., generally younger population included).
Harms
In the single-arm study, nearly all of the patients in the safety analysis set experienced at least 1 AE. These AEs and SAEs, such as pyrexia, fatigue, febrile neutropenia, decreased white blood cell count, stomatitis, and increased ALT, were considered by the clinical experts consulted by CADTH to be common occurrences and within the expected frequency in patients with ALL/LBL treated with asparaginase as part of a multicomponent chemotherapy regimen. The clinical experts consulted by CADTH noted that many harms were likely related to the background chemotherapy regimen.
No patients among the 51 included in cohort 1c and 3 of the 167 patients in all cohorts of the main study of crisantaspase recombinant died up until the latest cut-off of July 19, 2021.
The harms of special interest for this drug were also considered to be within the expected frequency by the clinical experts consulted by CADTH, including thrombosis, pancreatitis, hemorrhage, hypersensitivity reaction, and hepatotoxicity. The clinical experts consulted by CADTH noted that adults older than 25 years (not represented in the trial) may experience greater toxicity than what was observed in the trial.
In the ITC, the overall proportion of AEs is numerically similar in the crisantaspase recombinant and EDA arms, but with high imprecision, likely affected by a number of potential biases resulting from differences in inclusion criteria, potential background treatments, and AE definitions across the trials. According to the clinical experts consulted by CADTH, the proportion and type of AEs were within the range expected in current clinical context in Canada. There was no evidence of a meaningful difference between EDA and crisantaspase recombinant safety profiles, and the clinical experts considered this to be an important factor for decision-makers addressing patients with needs that either treatment may fill.
Conclusions
Evidence from a single-arm, open-label study suggests that crisantaspase recombinant is associated with adequate asparaginase activity when used in patients with ALL/LBL who have developed hypersensitivity to E. coli–derived asparaginase, which was shown using NSAA serum levels, a surrogate measure of asparagine depletion. The NSAA serum levels reached above an a priori–determined threshold of 0.1 IU/mL, which was established by clinical consensus as an important threshold for providing clinical utility. The impact on clinically important outcomes, such as overall survival and disease remission, and patient-important outcomes such as HRQoL, is unknown. Clinical experts deemed the safety profile of crisantaspase recombinant and harms manageable and within the expected frequency of events observed in patients with ALL/LBL in Canadian practice. They believed that the toxicity of crisantaspase recombinant could be higher in older patients, who were not represented in the trial. The evidence has limitations due to the noncomparative, open-label design of the study, which precludes causal inferences. However, the mechanism of action of the drug suggests that a link to the surrogate end point is plausible.
Evidence from a naive indirect comparison between single-arm trials of crisantaspase recombinant and EDA showed a high proportion of patients reaching a threshold for adequate asparaginase activity and similar safety profiles in both arms. However, the body of evidence presents effect estimates with uncertainty due to risk of bias, confounding (single-arm studies without adjustments to balance unobserved prognostic factors or effect modifiers), and imprecision due to low number of patients and wide CIs. Any difference in effects between crisantaspase recombinant and EDA in terms of NSAA levels and harms is difficult to ascertain.
Overall, the evidence suggests that crisantaspase recombinant is associated with adequate asparaginase activity and a manageable safety profile, but uncertainty remains in terms of clinical end points relative to other relevant comparators.
References
1.Hoelzer D, Bassan R, Dombret H, et al. Acute lymphoblastic leukaemia in adult patients: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2016;27(suppl 5):v69-v82. PubMed
2.Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391-2405. PubMed
3.Redaelli A, Laskin BL, Stephens JM, Botteman MF, Pashos CL. A systematic literature review of the clinical and epidemiological burden of acute lymphoblastic leukaemia (ALL). Eur J Cancer Care (Engl). 2005;14(1):53-62. PubMed
4.Dores GM, Devesa SS, Curtis RE, Linet MS, Morton LM. Acute leukemia incidence and patient survival among children and adults in the United States, 2001-2007. Blood. 2012;119(1):34-43. PubMed
5.Canada S. Table 13-10-0111-01 Number and rates of new cases of primary cancer, by cancer type, age group and sex. 2022; https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310011101.
6.Malard F, Mohty M. Acute lymphoblastic leukaemia. Lancet. 2020;395(10230):1146-1162. PubMed
7.Jankowska-Polanska B, Sliwinski M, Swiatoniowska N, Butrym A, Mazur G. Quality of life in children with acute lymphoblastic leukaemia. Scand J Caring Sci. 2020;34(2):380-389. PubMed
8.Lepretre S, Touboul C, Flinois A, et al. Quality of life in adults with acute lymphoblastic leukemia in France: results from a French cross-sectional study. Leuk Lymphoma. 2021;62(12):2957-2967. PubMed
9.Advani AS, Aster JC. Clinical manifestations, pathologic features, and diagnosis of B cell acute lymphoblasticleukemia/lymphoma. In: Larson RA, Rosmarin AG, eds. UpToDate: Wolters Kluwer; 2022.
10.Horton TM. Overview of the outcome of acute lymphoblastic leukemia/lymphoma in children and adolescents. In: Park JR, Rosmarin AG, eds. UpToDate: Wolters Kluwer; 2022.
11.Clinical Study Report: JZP458-201. An open-label, multicenter study of RC-P in patients with acute lymphoblastic leukemia (ALL)/lymphoblastic lymphoma (LBL) following hypersensitivity to E. coli-derived asparaginases [internal sponsor's report]. Palo Alto (CA): Jazz Pharmaceuticals Ireland Limited; 2022 Jan 24.
12.Asselin B, Rizzari C. Asparaginase pharmacokinetics and implications of therapeutic drug monitoring. Leuk Lymphoma. 2015;56(8):2273-2280. PubMed
13.Pieters R, Hunger SP, Boos J, et al. L-asparaginase treatment in acute lymphoblastic leukemia: a focus on Erwinia asparaginase. Cancer. 2011;117(2):238-249. PubMed
14.van der Sluis IM, Vrooman LM, Pieters R, et al. Consensus expert recommendations for identification and management of asparaginase hypersensitivity and silent inactivation. Haematologica. 2016;101(3):279-285. PubMed
15.Maese L, Loh ML, Lin T, et al. Initial Results from a Phase 2/3 Study of Recombinant Erwinia Asparaginase (JZP458) in Patients with Acute Lymphoblastic Leukemia (ALL)/Lymphoblastic Lymphoma (LBL) Who Are Allergic/Hypersensitive to E. coli-Derived Asparaginases. Blood. 2021;138(Supplement 1):2307-2307.
16.Salzer WL, Asselin B, Supko JG, et al. Erwinia asparaginase achieves therapeutic activity after pegaspargase allergy: a report from the Children's Oncology Group. Blood. 2013;122(4):507-514. PubMed
17.Recombinant Crisantaspase (Rylaze) for acute lymphoblastic leukemia (ALL) or lymphoblastic lymphoma (LBL): Indirect treatment comparison [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Crisantaspase recombinant. 2022.
18.Zhang Y, Le Beau MM, Aster JC. Classification, cytogenetics, and molecular genetics of acute lymphoblastic leukemia/lymphoma. In: Larson RA, Rosmarin AG, eds. UpToDate: Wolters Kluwer; 2022.
19.Silverman LB, Gelber RD, Dalton VK, et al. Improved outcome for children with acute lymphoblastic leukemia: results of Dana-Farber Consortium Protocol 91-01. Blood. 2001;97(5):1211-1218. PubMed
20.Vrooman LM, Stevenson KE, Supko JG, et al. Postinduction dexamethasone and individualized dosing of Escherichia Coli L-asparaginase each improve outcome of children and adolescents with newly diagnosed acute lymphoblastic leukemia: results from a randomized study--Dana-Farber Cancer Institute ALL Consortium Protocol 00-01. J Clin Oncol. 2013;31(9):1202-1210. PubMed
21.Panosyan EH, Seibel NL, Martin-Aragon S, et al. Asparaginase antibody and asparaginase activity in children with higher-risk acute lymphoblastic leukemia: Children's Cancer Group Study CCG-1961. J Pediatr Hematol Oncol. 2004;26(4):217-226. PubMed
22.Brown P, Inaba H, Annesley C, et al. Pediatric Acute Lymphoblastic Leukemia, Version 2.2020, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2020;18(1):81-112. PubMed
23.Rylaze (crisantaspase recombinant): 10 mg/0.5 mL (20 mg/mL), solution for intramuscular injection [product monograph]. Mississauga (ON): Jazz Pharmaceuticals Canada Inc; 2022 Sep 2.
24.Erwinase (Erwinia L-asparaginase): Freeze dried powder for injection (IM, SC, IV), 10,000 U/vial [product monograph]. Montreal (QC): CGF Pharmatec; 2016.
25.Maese LD, Loh ML, Choi LMR, et al. Efficacy and safety of intramuscular (IM) recombinant Erwinia asparaginase in acute lymphoblastic leukemia (ALL) or lymphoblastic lymphoma (LBL): The Children’s Oncology Group (COG) AALL1931 study. J Clin Oncol. 2022;40(16_suppl):7001-7001.
26.Dias S, Welton NJ, Sutton AJ, Ades AE. NICE DSU Technical Support Document 2: A Generalised Linear Modelling Framework for Pairwise and Network Meta-Analysis of Randomised Controlled Trials. London2014.
27.Cochrane Handbook for Systematic Reviews of Interventions version 6.3 (updated February 2022). Cochrane, 2022. In: Higgins JPT, Thomas J, Chandler J, et al., eds2022: http://www.training.cochrane.org/handbook
Appendix 1: Literature Search Strategy
Note this appendix has not been copy-edited.
Clinical Literature Search
Overview
Interface: Ovid
Databases:
MEDLINE All (1946 to present)
Embase (1974 to present)
Note: Subject headings and search fields have been customized for each database. Duplicates between databases were removed in Ovid.
Date of search: September 16, 2022
Alerts: Weekly search updates until project completion
Search filters applied: randomized controlled trials, controlled clinical trials
Limits:
Publication date limit: none
Humans
Language limit: none
Conference abstracts: excluded
Syntax | Description |
|---|---|
/ | At the end of a phrase, searches the phrase as a subject heading |
MeSH | Medical Subject Heading |
exp | Explode a subject heading |
* | Before a word, indicates that the marked subject heading is a primary topic; or, after a word, a truncation symbol (wildcard) to retrieve plurals or varying endings |
# | Truncation symbol for one character |
? | Truncation symbol for one or no characters only |
adj# | Requires terms to be adjacent to each other within # number of words (in any order) |
.ti | Title |
.ot | Original title |
.ab | Abstract |
.hw | Heading word; usually includes subject headings and controlled vocabulary |
.kf | Keyword heading word |
.dq | Candidate term word (Embase) |
.pt | Publication type |
.rn | Registry number |
.nm | Name of substance word (MEDLINE) |
medall | Ovid database code: MEDLINE All, 1946 to present, updated daily |
oemezd | Ovid database code; Embase, 1974 to present, updated daily |
Multidatabase Strategy
Asparaginase/ or (asparaginas* or rylaze* or crisantaspas* or erwinase* or erwinaze* or “jzp 458” or jzp458 or D733ET3F9O).ti,ab,kf,ot,hw,nm,rn.
1 use medall
*asparaginase/ or (asparaginas* or rylaze* or crisantaspas* or erwinase* or erwinaze* or “jzp 458” or jzp458).ti,ab,kf,dq.
3 use oemezd
4 not (conference review or conference abstract).pt.
2 or 5
(Randomized Controlled Trial or Controlled Clinical Trial or Pragmatic Clinical Trial or Equivalence Trial or Clinical Trial, Phase III).pt.
Randomized Controlled Trial/
exp Randomized Controlled Trials as Topic/
“Randomized Controlled Trial (topic)”/
Controlled Clinical Trial/
exp Controlled Clinical Trials as Topic/
“Controlled Clinical Trial (topic)”/
Randomization/
Random Allocation/
Double-Blind Method/
Double Blind Procedure/
Double-Blind Studies/
Single-Blind Method/
Single Blind Procedure/
Single-Blind Studies/
Placebos/
Placebo/
Control Groups/
Control Group/
(random* or sham or placebo*).ti,ab,hw,kf.
((singl* or doubl*) adj (blind* or dumm* or mask*)).ti,ab,hw,kf.
((tripl* or trebl*) adj (blind* or dumm* or mask*)).ti,ab,hw,kf.
(control* adj3 (study or studies or trial* or group*)).ti,ab,kf.
(Nonrandom* or non random* or non-random* or quasi-random* or quasirandom*).ti,ab,hw,kf.
allocated.ti,ab,hw.
((open label or open-label) adj5 (study or studies or trial*)).ti,ab,hw,kf.
((equivalence or superiority or non-inferiority or noninferiority) adj3 (study or studies or trial*)).ti,ab,hw,kf.
(pragmatic study or pragmatic studies).ti,ab,hw,kf.
((pragmatic or practical) adj3 trial*).ti,ab,hw,kf.
((quasiexperimental or quasi-experimental) adj3 (study or studies or trial*)).ti,ab,hw,kf.
(phase adj3 (III or “3”) adj3 (study or studies or trial*)).ti,hw,kf.
or/7-37
6 and 38
exp animals/
exp animal experimentation/ or exp animal experiment/
exp models animal/
nonhuman/
exp vertebrate/ or exp vertebrates/
or/40-44
exp humans/
exp human experimentation/ or exp human experiment/
or/46-47
45 not 48
39 not 49
remove duplicates from 50
Clinical Trials Registries
ClinicalTrials.gov
Produced by the US National Library of Medicine. Targeted search used to capture registered clinical trials.
[Search – Rylaze (crisantaspase recombinant), acute lymphoblastic leukemia (ALL), lymphoblastic lymphoma (LBL)]
WHO ICTRP
International Clinical Trials Registry Platform, produced by the WHO. Targeted search used to capture registered clinical trials.
[Search – Rylaze (crisantaspase recombinant), acute lymphoblastic leukemia (ALL), lymphoblastic lymphoma (LBL)]
Health Canada’s Clinical Trials Database
Produced by Health Canada. Targeted search used to capture registered clinical trials.
[Search – Rylaze (crisantaspase recombinant), acute lymphoblastic leukemia (ALL), lymphoblastic lymphoma (LBL)]
EU Clinical Trials Register
European Union Clinical Trials Register, produced by the European Union. Targeted search used to capture registered clinical trials.
[Search – Rylaze (crisantaspase recombinant), acute lymphoblastic leukemia (ALL), lymphoblastic lymphoma (LBL)]
Grey Literature
Search dates: September 6 to 13, 2022
Keywords: Rylaze (crisantaspase recombinant), acute lymphoblastic leukemia (ALL), lymphoblastic lymphoma (LBL)
Limits: none
Relevant websites from the following sections of the CADTH grey literature checklist Grey Matters: A Practical Tool for Searching Health-Related Grey Literature were searched:
Health Technology Assessment Agencies
Health Economics
Clinical Practice Guidelines
Drug and Device Regulatory Approvals
Advisories and Warnings
Drug Class Reviews
Clinical Trials Registries
Databases (free)
Internet Search
Open Access Journals
Appendix 2: Excluded Studies
Note this appendix has not been copy-edited.
No studies were excluded based on the screening of full texts.
Pharmacoeconomic Review
Abbreviations
ALL
acute lymphoblastic leukemia
BIA
budget impact analysis
BSA
body surface area
BSC
best supportive care
CI
confidence interval
EDA
Erwinia-derived asparaginase
EFS
event-free survival
EQ-5D-5L
5-Level EQ-5D
HUI
Health Utilities Index
ICER
incremental cost-effectiveness ratio
ITC
indirect treatment comparison
LBL
lymphoblastic lymphoma
OS
overall survival
POGO
Pediatric Oncology Group of Ontario
QALY
quality-adjusted life years
RST
relapse or secondary tumour
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Crisantaspase recombinant (Rylaze), solution for intramuscular injection |
Submitted price | 10 mg/0.5 mL vial: $1,091.91 |
Indication | As a component of a multidrug chemotherapeutic regimen for the treatment of acute lymphoblastic leukemia and lymphoblastic lymphoma in adult and pediatric patients 1 year or older who have developed hypersensitivity to Escherichia coli–derived asparaginase |
Health Canada approval status | NOC |
Health Canada review pathway | Project Orbis |
NOC date | September 2, 2022 |
Reimbursement request | As per indication |
Sponsor | Jazz Pharmaceuticals Canada Inc. |
Submission history | Previously reviewed: No |
ALL = acute lymphoblastic leukemia; LBL lymphoblastic lymphoma; NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis, Markov model |
Target populations | Adult and pediatric patients 1 year and older with ALL or LBL who have developed hypersensitivity (i.e., an allergic reaction) to an Escherichia coli–derived asparaginase. The population was split into 2 subgroups based on age: children and young adults (up to 25 years) and adult patients aged ≥ 25 years. |
Treatment | Crisantaspase recombinant |
Comparators | Base case: Erwinia L-asparaginase (Erwinia-derived asparaginase, EDA) Scenario: BSC, defined as an unspecified chemotherapy regimen without an asparaginase component |
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | Lifetime (100 years) |
Key data sources | The JZP458-201 trial, a sponsor-conducted ITC, and previous cost-effectiveness analyses |
Submitted results | Comparison to EDA Pediatric and young adult population: crisantaspase recombinant is dominated (incremental costs: $20,659; incremental QALYs: 0) Adult population (≥ 25 years): crisantaspase recombinant is dominated (incremental costs: $42,611; incremental QALYs: 0) |
Key limitations | The comparative safety and efficacy of crisantaspase recombinant and EDA are highly uncertain due to the reliance on a surrogate primary outcome and the absence of direct or non-naive indirect evidence. There is a lack of direct and indirect evidence comparing crisantaspase recombinant to BSC, forcing assumptions regarding relative clinical efficacy based on weighted mean EFS results from historical trials in which pegaspargase was compared with BSC regimens. Mortality is poorly modelled and highly uncertain, particularly in the adult population, where OS was assumed to equal EFS, and patients older than 25 years were thus not assumed to experience remission of ALL/LBL. The treatment duration of 4 asparaginase cycles assumed in the model may not be reflective of average use in Canada. Utility values are likely overestimated, particularly in the adult population who were assumed to have the same quality of life as the general population after year 2, despite very high mortality. The list price of EDA is uncertain, given pending changes to its regulatory status and availability. |
CADTH reanalysis results | CADTH was unable to derive a base-case reanalysis due to the lack of comparative clinical evidence and the structural limitations of the sponsor’s submitted pharmacoeconomic model. Instead, CADTH made minor corrections to the sponsor’s base case and then conducted a series of exploratory scenario analyses, establishing the sponsor’s results as sensitive to changes in the duration of asparaginase therapy, the mortality decrement applied to patients who could not access and/or tolerate a second-line asparaginase, the relative efficacy of EDA compared to crisantaspase recombinant, and the decreased utility values implemented for adult patients in year 3 and beyond. At the submitted price, a price reduction of 25% would be required for the cost of crisantaspase recombinant to be equivalent to the assumed price of EDA due to differences in their relative dosing schedules. Given the high degree of uncertainty around the effectiveness of crisantaspase recombinant compared to both EDA and BSC, a price reduction greater than 25% may be required. |
ALL = acute lymphoblastic leukemia; BSC = best supportive care; EDA = Erwinia-derived asparaginase; EFS = event-free survival; ITC = indirect treatment comparison; LBL = lymphoblastic lymphoma; LY = life-year; OS = overall survival; QALY = quality-adjusted life-year.
Conclusions
Based on the CADTH clinical review, crisantaspase recombinant has adequate asparaginase activity with what CADTH-obtained clinical expert input deemed a manageable safety profile. However, relative efficacy and safety of crisantaspase recombinant compared to Erwinia-derived asparaginase (EDA) was based on a naive comparison associated with uncertainty due to risk of bias, confounding (single-arm studies without adjustments to balance unobserved prognostic factors or effect modifiers), and imprecision due to small number of patients and wide confidence intervals (CIs) around effects. Any difference in effects between crisantaspase recombinant and EDA was difficult to ascertain.
CADTH was unable to derive a base-case reanalysis due to limitations in the model, including uncertainty in the relative efficacy of crisantaspase recombinant compared with either EDA or best supportive care (BSC), uncertainty in mortality, particularly in the adult population, uncertainty in utility values, and poor modelling practices limiting the ability to fully validate the model. None of these limitations could be addressed through reanalysis. The cost-effectiveness of crisantaspase recombinant compared with either EDA or BSC is, therefore, highly uncertain. All analyses assume a historical price of $1,091 per 10,000 IU vial for EDA; however, the actual costs paid by public plans are unknown. Also unknown is the cost of a new version of EDA that is currently under review by Health Canada.1
In its base case, the sponsor assumed equivalence between crisantaspase recombinant and EDA, effectively conducting a cost-minimization analysis. As such, crisantaspase recombinant was more costly but equally effective compared to EDA (i.e., crisantaspase recombinant is dominated). The unit price of crisantaspase recombinant would need to be reduced by 25% to reflect the same cost as EDA due to the increased dose per cycle (i.e., the double dose on Fridays). However, the assumption of clinical equivalence is highly uncertain, given the nature of the provided evidence and limitations in model structure.
Neither direct nor indirect evidence were available to estimate the comparative effectiveness of crisantaspase recombinant and BSC. An exploratory analysis conducted by CADTH found that the incremental cost-effectiveness ratio (ICER) of crisantaspase recombinant compared to BSC was highly sensitive to several key assumptions that could not be validated. Consequently, the cost-effectiveness of crisantaspase recombinant compared to BSC is unknown.
Given the degree of uncertainty in the comparative effectiveness evidence, there is insufficient evidence to justify a price premium for crisantaspase recombinant compared to EDA. Further, in the absence of clinical evidence for crisantaspase recombinant compared to BSC, the incremental cost-effectiveness is unknown. As EDA is currently available only through exceptional importation, it is reasonable to assume that, should another EDA product become available, the existence of 2 treatments that provide identical outcomes could justify a price reduction for both treatments to below the current price of EDA. The high degree of uncertainty around the assumption of equal effectiveness may justify an additional price reduction.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process.
CADTH received 1 patient input submission from the Leukemia & Lymphoma Society of Canada, collected through an online survey of 40 respondents from across Canada, 1 of whom had experience with crisantaspase recombinant. Most respondents were older than 35 years (22 of 40), 8 were aged 25 to 34 years, 2 were aged 18 to 24 years, 3 were younger than 18 years, and 5 did not specify their age. Patients responding to questions about disease symptoms that impact quality of life most often reported fatigue or weakness, loss of appetite or weight loss, headaches, blurred vision, nausea or vomiting, dizziness or light-headedness, and bone or joint pain, with their disease symptoms having an impact on the majority of respondents’ ability to work, travel, exercise, manage family responsibilities, and continue daily activities. Respondents also emphasized stress, anxiety, worry; problems concentrating; difficulty sleeping; feeling isolated; and financial impacts as psychological and social factors affecting quality of life after diagnosis. When patients start a new treatment, they consider the degree of certainty that their acute lymphoblastic leukemia (ALL) will respond to therapy, travel required, the effect on quality of life, and coverage by insurance or public plans as crucial factors. One respondent had experience with crisantaspase recombinant, having had previous allergic reactions to therapy, and reported their disease responded completely to treatment, with manageable side effects. This patient reported a preference to avoid intramuscular injections and would have preferred an IV form of crisantaspase recombinant.
Two clinician groups provided input for this submission: the Pediatric Oncology Group of Ontario (POGO) and the Ontario Health-Cancer Care Ontario Hematology Cancer Drug Advisory Committee. Both groups emphasized that asparaginase therapy is a crucial component of treatment for ALL and lymphoblastic lymphoma (LBL), and that the ability to deliver asparaginase therapy due to hypersensitivity reactions or silent inactivation (up to 25% of patients, according to the submission by POGO) impacts outcomes; POGO reported higher rates of relapse in patients who were unable to receive a complete substitution with EDA. Both clinician groups identified EDA as the current standard of care in cases of hypersensitivity or silent inactivation to pegaspargase; however, access to EDA has been limited, leading to many patients not receiving complete or timely substitution therapy. Both clinician groups suggested that all patients with ALL/LBL with allergy or silent inactivation should be eligible for crisantaspase recombinant in the absence of EDA availability, although POGO noted that if the supply of EDA is stabilized, there is no strong evidence to favour 1 product over the other.
Drug plan input noted that the crisantaspase recombinant pivotal trial included patients with an allergic reaction of grade 3 or higher to pegaspargase, leading to uncertainty about whether the results could be applied to patients with less severe allergic reactions. Additionally, the drug plans noted that while EDA has been subject to shortages and is only imported from the UK through Health Canada’s exceptional importation designation, an EDA product is currently under review by Health Canada, and thus determining under what circumstances crisantaspase recombinant or EDA would be preferred should both be available is of interest.
Several of these concerns were addressed in the sponsor’s model:
In light of EDA shortages, the sponsor included a scenario comparing crisantaspase recombinant to BSC.
Treatment for ALL/LBL was associated with decrements in quality of life.
CADTH was unable to address the following concerns raised from stakeholder input:
The impact on quality of life of specific forms of background chemotherapy.
The relative efficacy of crisantaspase recombinant compared with EDA, and thus the clinically preferable comparator.
Uncertainty about the efficacy of crisantaspase recombinant relative to BSC as used in current Canadian practice.
Economic Review
The current review is for crisantaspase recombinant (Rylaze) for the treatment of ALL or LBL in adult and pediatric patients 1 year and older who have developed hypersensitivity to Escherichia coli–derived asparaginase.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
Crisantaspase recombinant is indicated as a component of a multidrug chemotherapeutic regimen for the treatment of ALL and LBL in adult and pediatric patients aged 1 year and older who have developed hypersensitivity to E. coli–derived asparaginase.2 The sponsor submitted 2 cost-utility analyses: 1 to assess the cost-effectiveness of crisantaspase recombinant compared to EDA in children and young adults up to 25 years of age; and the other to assess its cost-effectiveness in adults aged 25 years and older.3 The reimbursement request is aligned with the Health Canada indication.2
Crisantaspase recombinant is available in 10 mg/0.5 mL vials for intramuscular injection, with a recommended dose of 25 mg/m2 of body surface area (BSA) on Monday and Wednesday and 50 mg/m2 on Friday for a total of 6 doses to replace each planned dose of pegaspargase (25 mg/m2 on Monday and Wednesday, 50 mg/m2 on Friday). The submitted price of crisantaspase recombinant is $1,091.91 per 0.5 mL vial, corresponding to a cost of $24,022 per 2-week course ($96,088 for 4 courses) in the pediatric and young adult population (mean BSA = 0.97 m2) and $43,676 per 2-week course ($174,706 for 4 courses) in adults older than 25 years (mean BSA = 1.95 m2). The cost of EDA therapy was based on a cost of $1,091.91 per 10,000 IU vial and reported as $19,654 per 2-week course ($78,618 for 4 courses) in pediatric and young adult patients (< 25 years) and $32,757 per 2-week course ($131,029 for 4 courses) in the adult (≥ 25 years) population. This difference in cost between crisantaspase recombinant and EDA is due to differences in the dosing regimens assumed. No drug costs for background chemotherapy regimens were included in the base case of the sponsor’s model.
The clinical outcome of interest was quality-adjusted life-years (QALYs). The sponsor adopted a lifetime (100-year) time horizon, with analyses conducted from the perspective of a publicly funded health care payer. Future costs and benefits were discounted at a rate of 1.5% per year.
Model Structure
The sponsor submitted 2 hybrid model structures consisting of decision trees with a 5-year duration, followed by a Markov model with a 1-year cycle length for the remainder of the patients’ lives.3 For both age-based populations, patients entered their respective decision trees when they experienced a first-line hypersensitivity to E. coli–derived asparaginase (Figure 1 and Figure 2). In the base-case analyses, they were assigned to receive either EDA or crisantaspase recombinant therapy. Patients in both age groups could have a second-line hypersensitivity reaction to their new asparaginase and receive no asparaginase therapy during the first year of treatment. The pediatric and young adult patients then remained in the event-free survival (EFS) health state, experienced a relapse or secondary tumour (RST), or died (Figure 1), whereas adult patients (≥ 25 years) were assumed to either remain in the EFS health state (assumed to be the same as overall survival) or to die (Figure 2). After 5 years, patients transitioned to the Markov model, which, for the pediatric and young adult population, consisted of 3 health states (EFS, RST, and dead; Figure 3), whereas for adults it consisted of 2 health states (EFS and dead; Figure 4), with the assumption that EFS was equivalent to overall survival (OS) in this population. For the remainder of the time horizon, patients had a transition probability to death that depended on their current health state (EFS or RST) and which age-based population they were in.
Model Inputs
The sponsor’s modelled mean age for the pediatric and young adult population was estimated from the median age and interquartile range reported in the UKALL2003 trial of treatment reduction for children with low-risk ALL,4 whereas the mean age for adult patients was derived from the UKALL14 trial of pegylated asparaginase during induction therapy for adult patients with ALL.5 The source of sex distribution assumptions (57% male for the pediatric and young adult population, 53% male for the adult population) were not specified, but are consistent with those reported in the UKALL2003 and UKALL14 trials, respectively.4,5 Mean BSA for both populations was derived from the crisantaspase recombinant pivotal trial, JZP458-201.6 All modelled patients were assumed to receive an unspecified background chemotherapy that was not modelled in the base case; it was assumed that it would be equal in the treatment arms.
Crisantaspase recombinant was assumed to have the same impact as EDA on efficacy, safety, quality of life, and long-term mortality, based on historical assumptions that all asparaginase products have identical efficacy.3 The sponsor conducted an unanchored indirect (naive) treatment comparison (ITC) between crisantaspase recombinant and EDA and concluded that the efficacy and safety of crisantaspase recombinant was comparable to that of EDA, despite considerable reported uncertainty.7
The primary measure of efficacy in the model were EFS and OS. Patients in the pediatric and young adult population were assumed to be standard risk (49.5%), intermediate risk (29.1%), or high risk (21.4%), based on the proportions reported in a 2019 cost-effectiveness analysis of pegaspargase8 and derived from the UKALL2003 trial of intensification with pegaspargase.4 Modelled EFS at 5 years for patients using either asparaginase product (EDA or crisantaspase recombinant) was assumed to be 90%, 85%, and 75% for the standard, intermediate, and high-risk groups,4,8 respectively, and 5-year OS was assumed to be 95%, 90%, and 80%, respectively, with the difference between EFS and OS proportions assigned to the RST health state. For patients in the adult population, OS was assumed to equal EFS, and the model assumed that 40% of patients younger than 40 years who received EDA or crisantaspase recombinant would still be alive at 5 years, as would 30% of patients older than 40 years;8 a weighted average of 33.2% was used for 5-year OS for the overall adult population. Modelled patients in both age-based populations had a 5% chance of having a hypersensitivity reaction and discontinuing EDA or crisantaspase recombinant, informed by the proportion of patients with allergic reactions in the JZP458-201 trial.6 Such patients were assigned an absolute 10.3% reduction to their probability of achieving EFS and/or OS at 5 years, as used in a 2019 cost-effectiveness analysis of EDA.9
Pediatric and young adult patients who were in the EFS health state at the end of the 5-year decision tree were assumed to be effectively cured and, therefore, mortality was assumed to be the same as the age-specific and sex-specific general population for the Markov model (i.e., the remaining time horizon).10 Pediatric and young adult patients in the RST state were assigned a mortality modifier of 1.9, which was also applied to that group in the general population, as assumed in the 2019 pegaspargase cost-effectiveness study.8 In contrast, for the adult population, mortality was predicted using a Weibull curve extrapolation between the proportion of patients in OS at 5 years and the assumption that OS at 40 years in the model would be 0%. Survival curves for the pediatric and young adult population and the adult population are reported in Figure 5 and Figure 6, respectively.
Health state utilities were informed by age-adjusted 5-Level EQ-5D (EQ-5D-5L) utility values from population norms in Alberta.11 Patients were assigned utility decrements in years 1 and 2 of the model based on the proportion of the year spent in each treatment phase (induction [4 weeks], central nervous system [3 weeks], intensification [30 weeks], and continuation [71 weeks]), using the mean of the Health Utility Index (HUI) 2 and HUI3 disutilities derived from Furlong et al. (2012).8,12 Additional disutilities were applied for patients in the RST health state (20%) who had a hypersensitivity reaction (0.014).8
Drug-acquisition costs for crisantaspase recombinant and EDA were based on the sponsor’s submitted price and the sponsor’s report of the historical cost of EDA per vial.3 The cost per vial of EDA could not be independently confirmed by CADTH due to its current lack of availability and its previous status as an exceptional importation.13 No costs were assumed for BSC under the assumption that background chemotherapy regimens would be identical between treatment groups.3 The model also applied a cost of $926 per hypersensitivity reaction14 and an end-of-life cost of $15,982, derived from costs accrued in the last 30 days of life in Canada, as reported in 2016.15
Summary of Sponsor’s Economic Evaluation Results
The sponsor presented a probabilistic base-case analysis based on 1,000 iterations, the results of which are presented here. Deterministic results were similar. More detailed results of the sponsor’s economic evaluations are presented in Appendix 3.
Base-Case Results
For the treatment of pediatric and young adult (< 25 years) patients with ALL or LBL, the use of crisantaspase recombinant was associated with an incremental cost of $20,659 compared to EDA and 0.0 incremental QALYs, resulting in crisantaspase recombinant being dominated (more costly, equally effective).
For the treatment of adult patients (≥ 25 years) with ALL or LBL, the use of crisantaspase recombinant was similarly associated with $42,611 in incremental costs compared to EDA and 0.0 incremental QALYs, resulting in crisantaspase recombinant being dominated (more costly, equally effective).
Table 3: Summary of the Sponsor’s Economic Evaluation Results Compared With EDA
Drug | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER vs. EDA ($/QALY) |
|---|---|---|---|---|---|
Pediatric and young adult patients (< 25 years) | |||||
EDA | 84,335 | Reference | 35.40 | Reference | Reference |
Crisantaspase recombinant | 104,994 | 20,659 | 35.40 | 0 | Dominateda |
Adult patients (≥ 25 years) | |||||
EDA | 155,562 | Reference | 4.09 | Reference | Reference |
Crisantaspase recombinant | 198,173 | 42,611 | 4.09 | 0 | Dominateda |
EDA = Erwinia-derived asparaginase; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: The sponsor’s cost-effectiveness results in their report did not match the individual cost, QALY tables, or the submitted model file. These results are consistent with those of the submitted model file and individual result tables.
aHigher costs, equal efficacy.
Source: Sponsor’s pharmacoeconomic submission.3
Sensitivity and Scenario Analyses Results
The sponsor also submitted scenario analyses in which crisantaspase recombinant was assumed to be the only asparaginase on the market, thus comparing it to BSC. The analysis for the pediatric and young adult population assumed no differences in background chemotherapy regimens, and thus only the crisantaspase recombinant group was associated with drug-acquisition costs. Crisantaspase recombinant was associated with $97,758 in incremental costs and 3.76 incremental QALYs, resulting in an ICER of $25,970 per QALY for the pediatric and young adult (< 25 years) population compared to BSC (Table 9).
In contrast, for the adult population (≥ 25 years), the sponsor included costs for a 3-week cycle of a regimen of doxorubicin, dexamethasone, cytarabine, and etoposide (total cost = $3,830 per patient) for the BSC group, but no chemotherapy regimen costs for the crisantaspase recombinant group. Crisantaspase recombinant was associated with $179,370 in incremental costs and 0.95 incremental QALYs, resulting in an ICER of $189,288 per QALY for the adult (≥ 25 years) population compared to BSC.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations to the sponsor’s analysis that have notable implications for the economic analysis:
Comparative efficacy and safety relative to EDA are highly uncertain: The sponsor has assumed clinical equivalence for crisantaspase recombinant and EDA on the basis of a naive ITC,16 which compared the proportion of patients with last 48-hour and last 72-hour nadir serum asparaginase activity (NSAA) levels of at least0.1 IU/mL in the treatment group that received 25 mg/m2 on Mondays and Wednesdays and 50 mg/m2 on Fridays in the JZP458-201 crisantaspase recombinant trial6 and in the ALL07P2 EDA trial17 (25,000 IU/m2 on Mondays, Wednesdays, and Fridays), as well as the proportion of patients experiencing a number of adverse events in these treatment groups. No direct evidence comparing these 2 products was provided or found. The indirect comparisons for each outcome were summarized as ORs, which all had CIs overlapping the null (i.e., statistically significant differences were not found). The ITC used data from a single arm from each trial, and no statistical adjustments were conducted to control for potential differences, such as study design, inclusion and exclusion criteria, baseline characteristics, outcome definitions, background therapies, and dosing differences. Additionally, the primary end point in both studies was the surrogate outcome NSAA; no comparison has been provided in terms of clinical outcomes such as EFS, OS, or remission. Thus, any difference of effect between crisantaspase recombinant and EDA was difficult to ascertain. Despite the uncertainty inherent in the quality of the available evidence, the sponsor assumed that the relative efficacy and safety of EDA and crisantaspase recombinant at these doses was identical and deterministic.
CADTH was unable to adjust for this limitation. The relative efficacy of crisantaspase and EDA is uncertain.
Lack of clinical evidence comparing crisantaspase recombinant with BSC: Feedback from participating drug plans indicated that EDA is not reimbursed for all patients in all jurisdictions, with some jurisdictions funding it only for pediatric patients, some for all patients, and some which do not yet fund it at all. Additionally, EDA shortages (refer to Issues for Consideration section) have led to limited access in some jurisdictions, even when funded. In jurisdictions or populations where EDA is not available and/or not funded, the comparator of primary interest for determining the cost-effectiveness of crisantaspase recombinant may be chemotherapy alone, called BSC by the sponsor. Although the sponsor provided a scenario analysis comparing crisantaspase recombinant to BSC, the sponsor did not include efficacy or cost information associated with a specific background chemotherapy regimen or regimens, but instead assumed a cost of $0, as the unspecified regimen would be identical between treatment groups, with the exception of a single course of doxorubicin, dexamethasone, cytarabine, and etoposide for adult patients who do not receive crisantaspase recombinant. In terms of efficacy, the sponsor assumed that for patients receiving BSC or, in the base case, for any patient who could not complete a course of EDA or crisantaspase recombinant due to another hypersensitivity reaction, there would be an absolute 10.3% reduction in EFS and OS, regardless of population or baseline risk.3 This 10.3% absolute reduction was based on a cost-effectiveness study by Kloos et al. (2019),9 using a weighted mean of the differences in 5-year EFS reported for pediatric patients with ALL in various studies, which individually had 5-year EFS differences ranging from 3.3% to 17%. Altering the EFS and OS decrement associated with BSC to reflect this range of EFS differences had a large impact on the ICER associated with crisantaspase recombinant in both the pediatric ($15,607 to $107,601 per QALY) and adult ($81,713 to $662,142 per QALY) populations.
CADTH was not able to address this limitation due to the simplicity of the sponsor’s model and the level of uncertainty inherent in almost all inputs.
Mortality is poorly modelled and highly uncertain: The sponsor assumed that pediatric patients who achieved EFS at 5 years were effectively cured and, thereafter, had the same mortality as the age- and sex-specific general population, whereas those in the RST state had a mortality multiplier of 1.9 times that of the general population, resulting in a plateau in mortality after 5 years and the majority of modelled pediatric and young adult patients living an additional 60 to 80 years (Figure 5). CADTH-obtained clinical expert feedback noted that a small number of pediatric patients would be expected to relapse even after 5 years of EFS, but overall considered these assumptions to be a reasonable simplification for the purposes of modelling. In contrast, adult patients were assumed to have a 5-year OS rate consistent with that used in the cost-effectiveness study by Hu et al. (2019)8 of 40% for patients aged 25 to 40 years and 30% for patients older than 40 years, leading to a weighted average of 33.2%. The study by Hu et al. (2019)8 cited the UKALL14 trial but did not provide a specific location for these data. The sponsor assumed that EFS was equal to OS despite other subpopulations in the UKALL14 trial showing distinct differences in the proportion of patients achieving OS and EFS.18,19 With no distinction between EFS and OS, modelled adult patients were not assumed to benefit from remission and, thus, did not experience a plateau in mortality after the initial 5-year period (Figure 6). CADTH-obtained clinical expert feedback, which emphasized that like pediatric patients, adult patients who were still alive at 5 years would have a substantially reduced risk of mortality thereafter, which is consistent with survival rates reported in both a rituximab UKALL14 publication19 and a Swedish registry study of adults with ALL.20 As such, the potential QALYs associated with asparaginase treatment, both crisantaspase recombinant and EDA, may have been underestimated in this population. Additionally, although the model assumes the lifetime time horizon ends at 100 years for the pediatric and young adult (< 25 years) population, modelled adult patients (≥ 25 years), despite their high mortality rate, had the potential to live to an implausible 146 years.
Due to inflexibility in the sponsor’s model, CADTH was unable to adjust mortality in the adult population to more appropriately reflect patterns seen in trial and real-world evidence, increasing uncertainty in the ICER associated with crisantaspase recombinant, particularly when compared to BSC. CADTH did apply a correction to the adult population analysis so that patients do not live past 100 years.
Treatment duration may not represent current clinical practice in Canada: The sponsor’s model assumes that all patients will receive 4 cycles (6 doses each) of EDA or crisantaspase recombinant, regardless of age group, consistent with the median number of cycles reported in the JZP458-201 trial.3,6 However, according to clinical expert input elicited by CADTH, standard-risk pediatric patients with ALL or LBL patients often receive just 2 cycles of asparaginase, whereas pediatric patients with high risk receive 4 cycles. In contrast, clinical expert input indicated that adult patients with ALL or LBL generally receive 10 cycles of asparaginase therapy over the course of 30 weeks,21 8 to 10 of which might be a second-line asparaginase, depending on the timing of the hypersensitivity reaction to E. coli–derived asparaginase.
CADTH conducted an exploratory reanalysis assuming pediatric and young adults would receive a weighted average of 2.43 cycles of crisantaspase recombinant or EDA therapy, based on the sponsor’s proportion of patients who are high risk versus lower risk, whereas adult patients were assumed to receive an average of 9 cycles. This scenario assumes patients receive the dosing recommended in the crisantaspase recombinant product monograph; should lower per-cycle dosing be used for adult patients in clinical practice, results may be more in line with those presented in the sponsor’s corrected analysis.
Utility values are uncertain: Utility values were based on age- and sex-specific population norms in Alberta,22 with utility decrements for each stage of treatment (induction, central nervous system, intensification, continuation) weighted in the applicable year (year 1 or year 2).8 In the pediatric population, after year 2, a utility decrement of 20% was applied to patients in the RST state; those in the EFS state were assumed to have the same utility as the general population,8 much like they are assumed to have the same mortality as the general population. Utility decrements based on treatment phase were also applied in the adult population in years 1 and 2, with patients assumed to also have the same utility as the general population thereafter. However, as the adult analysis does not distinguish between EFS and OS, there is no RST state with an associated utility decrement. As the model continues to assume a very high mortality rate for the adult population even after year 2 (refer to Figure 6), CADTH reviewers considered it implausible that such a population would have the same average quality of life as that of the general population, given the presumably continuing disease activity in most patients causing such mortality.
The sponsor’s model included health state utility values estimated using the EQ-5D-5L and estimated disutilities using the HUI. This is methodologically inappropriate, as the utility estimates produced from different multiattribute instruments are not synonymous. Differences in the size and direction of utility estimates generated by the EQ-5D and the HUI are specific to the instrument attribute affects and the disease area.23,24 This inappropriate pooling of utility measures adds an unknown amount of uncertainty to estimates of comparative effectiveness.
CADTH was unable to fully adjust for this limitation. In a scenario analysis, given the high mortality in the adult population, CADTH assumed that the mean utility in years 3 and beyond could be represented by applying the disutility modifier of 20%, as was used for the RST population in the sponsor’s pediatric analysis. CADTH did not propose alternative utility or disutility estimates to harmonize either the EQ-5D-5L or the HUI.
Poor modelling practices were employed: The sponsor’s submitted model included numerous IFERROR statements, which lead to situations in which the parameter value is overwritten with an alternative value without alerting the user to the automatized overwriting. The systematic use of IFERROR statements makes thorough auditing of the sponsor’s model impractical, as it remains unclear whether the model is running inappropriately by overriding errors. CADTH additionally notes that, for many parameters, a standard error of 10% from the mean was assumed. Given there are data from epidemiological studies and clinical trials, assuming an arbitrary 10% of the mean is unnecessary and does not reflect uncertainty in the available information. Finally, for all probabilistic parameters, the minimum and maximum acceptable values were set to equal each individual draw for that parameter, rather than confining the distribution to mathematically or logically possible limits. For example, the gamma distribution for mean age of the pediatric and young adult population should have been set so that the lower limit could not be below 1 year, as crisantaspase recombinant is not indicated for children younger than that, whereas the upper limit should have been set at just below 25, as that is how the sponsor defined this population. As submitted, the mean age of the pediatric and young adult population had a 9% probability of being under 1 year and a 0.8% probability of being above 25 years.
CADTH was unable to address most of these limitations and noted that a thorough validation of the sponsor’s model was not possible. CADTH incorporated a correction to the sponsor’s model in reanalyses by assigning appropriate minimum and maximum values to all probabilistic parameter distributions.
Additionally, the key assumptions made by the sponsor that have been appraised by CADTH are presented in Table 4.
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CADTH comment |
|---|---|
Mean starting age is atypically calculated and uncertain. | Uncertain. The sponsor estimated the mean age of modelled patients at baseline by incorporating the median, minimum, and maximum ages reported in the UKALL2003 and UKALL14 trials for the pediatric and young adult population and the adult population, respectively, using 2 different methods. For the younger population, the average of the median, minimum, and maximum ages was used ([5 + 3 + 10] / 3 years), whereas for the older population, the average of twice the median, minimum, and maximum was used ([46.5 + 46.5 + 25 + 65] / 4 years). Mean starting age impacts BSA, the choice of which impacts the relative number of vials chosen. |
Chosen BSA impacts relative cost of crisantaspase recombinant due to wastage assumption. | Uncertain. The relative cost of crisantaspase recombinant vs. EDA is sensitive to the choice of mean BSA for the modelled population due to the impact of rounding (due to the assumption of wastage) on the doubled Friday dose of crisantaspase recombinant compared to the single Friday dose of EDA. For example, when the assumed BSA for the pediatric population is 0.97 m2, 5 vials of crisantaspase recombinant are required for the 50 mg/m2 Friday dose, compared to 3 vials for the 25,000 IU/m2 dose of EDA. If the pediatric population instead had a mean BSA of 1.05 m2, the model would instead cost the Friday dose as requiring 6 vials, compared to 3 vials for EDA. Thus, the price reduction required for crisantaspase recombinant to be cost-neutral to EDA in the model depends on the mean BSA assumed for the population when wastage of excess medication is considered. When wastage is not included, the price reduction required to achieve cost-neutrality remains stable across BSA assumptions. |
The population cut-off at age 25 years is a meaningful distinction between clinical populations (age 20 in the budget impact analysis). | Uncertain. While studies of pediatric ALL may include patients up to the age of 25 or 30 years, the definition of pediatric patients in clinical practice goes up to age 18 years, with patients older than 18 years typically being seen by nonpediatric oncologists in nonpediatric settings. As such, patients older than 18 years may receive regimens according to hospital guidelines and jurisdictional reimbursement considerations for adult patients rather than pediatric patients. |
Hypersensitivity costs of $926 per event. | Uncertain. The sponsor assumed that the 5% of patients who suffered a hypersensitivity reaction to EDA or crisantaspase recombinant would incur a cost of $926 per event, based on the Ontario Case Costing Initiative data and unspecified allergy, and inflated to 2022 dollars.3 Removal of these costs did not have a substantial impact on the resulting ICERs. |
End-of-life costs of $15,982 per event. | Uncertain. The sponsor assumed end-of-life costs as reported by Bekelman et al. (2016)15 for the last 30 days of care for patients dying of cancer in Canada, converted and inflated to 2022 Canadian dollars. Removing these costs did not have a substantial impact on the resulting ICERs. |
ALL = acute lymphoblastic leukemia; BSA = body surface area, EDA = Erwinia-derived asparaginase; ICER = incremental cost-effectiveness ratio.
CADTH Reanalyses of the Economic Evaluation
Base-Case Results
Due to extensive limitations and the associated uncertainty in the clinical data, CADTH was unable to derive a base-case reanalysis.
CADTH made corrections to the sponsor’s base case, altering the range of all probabilistic comparators, removing randomization seeding, and limiting the mean patient age in the adult population to 100 years (outlined in Table 5). Results of this corrected analysis can be found in Table 6, and additional results, including corrections to the sponsor’s comparison of crisantaspase recombinant and BSC, can be found in Appendix 4.
Table 5: CADTH Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Correctionsa to sponsor’s base case | ||
1. Probabilistic corrections | Minimum and maximum of each parameter revised to equal each probabilistic draw result Randomization was seeded | Minimum and maximum of each parameter set to reasonable limitsb Randomization seeding removed |
2. Time horizon | 100 years from model start | Until mean patient age reaches 100 years |
aCorrections are minor errors (e.g., transcription errors between the report and model, misapplication of distributions or standard errors in probabilistic analyses.) that are not identified as limitations.
bFor example, pediatric age was limited to 1 to 24.99 years and adult age was limited to 25 to 80 years.
Table 6: Summary of the Sponsor’s Corrected Analyses
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER vs. EDA ($/QALYs) |
|---|---|---|---|---|
Pediatric and young adult population (< 25 years) | ||||
Sponsor’s base case | EDA | 84,507.24 | 35.40 | Reference |
Crisantaspase recombinant | 104,994.03 | 35.40 | Dominateda | |
Sponsor’s corrected base case | EDA | 84,709.08 | 35.38 | Reference |
Crisantaspase recombinant | 105,359.28 | 35.38 | Dominateda | |
Adult population (≥ 25 years) | ||||
Sponsor’s base case | EDA | 155,562.30 | 4.09 | Reference |
Crisantaspase recombinant | 198,173.00 | 4.09 | Dominateda | |
Sponsor’s corrected base case | EDA | 156,222.95 | 4.08 | Reference |
Crisantaspase recombinant | 198,964.68 | 4.08 | Dominateda | |
EDA = Erwinia-derived asparaginase; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: The sponsor did not conduct sequential analyses comparing all 3 comparators but instead conducted separate analyses comparing crisantaspase recombinant to EDA and crisantaspase recombinant to BSC. The efficacy of crisantaspase recombinant compared to BSC is mainly of interest in populations or jurisdictions that do not have access to or do not reimburse EDA and, thus, CADTH has not combined the results into a single sequential analysis.
aMore costly, equally effective.
Exploratory Scenario Analysis Results
To explore the impact of uncertainty in the sponsor’s model, CADTH also conducted several scenario analyses exploring uncertainty in the sponsor’s model (Table 13). The sponsor’s model was highly sensitive to the duration of treatment with second-line asparaginase, with incremental costs increasing as the number of cycles of EDA or crisantaspase recombinant increased (Table 14). Compared to EDA, the model is sensitive to small changes in relative efficacy in terms of EFS and OS between crisantaspase recombinant and EDA, and compared to BSC, the model is sensitive to changes in the mortality decrement associated with BSC when it is varied within the range reported in the literature. It was assumed that the adult population has a lower utility than the general population for years 3 and beyond.
Additionally, CADTH conducted price-reduction analyses (Table 15). Compared to EDA, and when wastage of excess medication in vials is assumed and the sponsor’s mean BSAs are considered, the cost of crisantaspase recombinant would need to be reduced by 21% for pediatric and young adult patients and 24% for adult patients to be equivalent to the cost of EDA at the dosing schedules described in the base case. When wastage is not assumed, a price reduction of 25% would be required for both populations. Compared with BSC, under the sponsor’s base-case assumptions, crisantaspase recombinant would be considered cost-effective in the pediatric and young adult population at a willingness-to-pay threshold of $50,000 per QALY, whereas a price reduction of 72% would be required for it to be cost-effective at the same threshold in the adult population.
Issues for Consideration
Erwinase has a history of repeated critical supply shortages, as worldwide demand has exceeded drug supply due to contamination issues in manufacturing.25,26 At the time of this review, Erwinase was unavailable through usual channels due to its status with Health Canada as Cancelled Post-Market.27 Instead, Erwinase was listed as a drug for exceptional importation and sale; however, this status expired on September 30, 2022.13 An EDA product from Porton Biopharma Ltd., presumably Erwinase, is currently under review at Health Canada.1 Porton Biopharma previously partnered with Jazz Pharmaceuticals, the current sponsor of Rylaze, to distribute Erwinase; however this partnership ended in 2021.28 It is unclear whether the potential approval of this product will lead to a more stabilized supply of EDA in Canada, and the price that will be paid by public plans for such a product, should it be approved, is unknown. For similar reasons, it is unknown what effect the availability of both products (Rylaze and the Porton EDA product) will have on the ability of drug plans to negotiate price reductions for either or both products, given the sponsor’s stated assumption of equal efficacy between them.
The submitted price of crisantaspase recombinant ($1,092 per 10 mg vial) is equivalent to that presented by the sponsor as the historical list price of EDA ($1,092 per 10,000 IU vial).3 However, the recommended dose of crisantaspase recombinant is 25 mg/m2 on Mondays and Wednesdays and 50 mg/m2 on Fridays,2 whereas the currently used regimen for EDA is 25,000 IU/m2 on Mondays, Wednesdays, and Fridays.29 All analyses in this review have been based on this difference in dosing, under the sponsor’s assumption that crisantaspase recombinant and EDA are clinically equivalent at these doses. It is unclear whether both comparators will continue to be used in clinical practice on these different schedules, or what the relative efficacy of crisantaspase recombinant and EDA would be if administered on the same schedule.
Overall Conclusions
Based on the CADTH clinical review, crisantaspase recombinant has adequate asparaginase activity and what CADTH-obtained clinical expert input deemed a manageable safety profile. However, relative efficacy and safety of crisantaspase recombinant compared to EDA was based on a naive comparison associated with uncertainty due to risk of bias, confounding (single-arm studies without adjustments to balance unobserved prognostic factors or effect modifiers), and imprecision due to the small number of patients and wide CIs around effects. Any difference in effects between crisantaspase recombinant and EDA was difficult to ascertain.
CADTH was unable to derive a base-case reanalysis due to limitations in the model, including uncertainty in the relative efficacy of crisantaspase recombinant compared with either EDA or BSC, uncertainty in mortality (particularly in the adult population), uncertainty in utility values, and poor modelling practices that limited the ability to fully validate the model. None of these limitations could be addressed through reanalysis. Therefore, the cost-effectiveness of crisantaspase recombinant compared to either EDA or BSC is highly uncertain. All analyses assume a historical price of $1,091 per 10,000 IU vial for EDA; however, actual costs paid by public plans are unknown. Also unknown is the cost of the new version of EDA that is currently under review by Health Canada.1
In their base case, the sponsor assumed equivalence between crisantaspase recombinant and EDA, effectively conducting a cost-minimization analysis. As such, crisantaspase recombinant was more costly than EDA, but equally effective (i.e., crisantaspase recombinant is dominated). The unit price of crisantaspase recombinant would need to be reduced by 25% to reflect the same cost as EDA due to the increased dose per cycle (i.e., the double dose on Fridays). However, the assumption of clinical equivalence is highly uncertain, given the nature of the provided evidence and the limitations in model structure.
Neither direct nor indirect evidence were available to estimate the comparative effectiveness of crisantaspase recombinant and BSC. Instead, the sponsor assumed a weighted mean decrement to EFS and OS, as reported in a previous cost-effectiveness analysis, which was informed by a wide range of EFS differences reported in the literature. This body of evidence primarily considered the efficacy of pegaspargase, rather than second-line asparaginase after a hypersensitivity reaction, compared to a number of different BSC regimens. CADTH conducted exploratory scenario analyses and found that the ICER of crisantaspase recombinant compared to BSC was highly sensitive to several key assumptions that could not be validated. Consequently, the cost-effectiveness of crisantaspase recombinant compared to BSC is unknown.
Given the degree of uncertainty in comparative effectiveness, there is insufficient evidence to justify a price premium for crisantaspase recombinant compared with EDA. Further, in the absence of clinical evidence for crisantaspase recombinant compared to BSC, the incremental cost-effectiveness is unknown. As EDA is currently available only through exceptional importation, it is reasonable to assume that, should another EDA product become available, the existence of 2 treatments that provide identical outcomes could justify a price reduction for both treatments below the current negotiated price of EDA. The high degree of uncertainty around the assumption of equal effectiveness may justify additional price reduction.
References
1.Drug and Health Product Submissions Under Review (SUR): New drug submissions under review. https://www.canada.ca/en/health-canada/services/drug-health-product-review-approval/submissions-under-review/new-drug-submissions-under-review.html. Accessed 2022 Oct 19.
2.Rylaze (crisantaspase recombinant): 10 mg/0.5 mL (20 mg/mL), solution for intramuscular injection [product monograph]. Mississauga (ON): Jazz Pharmaceuticals Canada Inc; 2022 Sep 2.
3.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Rylaze (crisantaspase recombinant), 25 mg/m2 on Monday and Wednesday and 50 mg/m2 on Friday, administrated intramuscularly. Mississauga (ON): Jazz Pharmaceuticals Canada Inc; 2022 Aug 18.
4.Vora A, Goulden N, Wade R, et al. Treatment reduction for children and young adults with low-risk acute lymphoblastic leukaemia defined by minimal residual disease (UKALL 2003): a randomised controlled trial. Lancet Oncol. 2013;14(3):199-209. PubMed
5.Patel B, Kirkwood AA, Dey A, et al. Pegylated-asparaginase during induction therapy for adult acute lymphoblastic leukaemia: toxicity data from the UKALL14 trial. Leukemia. 2017;31(1):58-64. PubMed
6.Clinical Study Report: JZP458-201. An open-label, multicenter study of RC-P in patients with acute lymphoblastic leukemia (ALL)/lymphoblastic lymphoma (LBL) following hypersensitivity to E. coli-derived asparaginases [internal sponsor's report]. Palo Alto (CA): Jazz Pharmaceuticals Ireland Limited; 2022 Jan 24.
7.Schedule of benefits for physician services under the Health Insurance Act: effective October 1, 2021. Toronto (ON): Ontario Ministry of Health; 2022: https://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master.pdf. Accessed 1800 Jan 1.
8.Hu X, Wildman KP, Basu S, Lin PL, Rowntree C, Saha V. The cost-effectiveness of pegaspargase versus native asparaginase for first-line treatment of acute lymphoblastic leukaemia: a UK-based cost-utility analysis. Health Econ Rev. 2019;9(1):40. PubMed
9.Kloos RQH, van Litsenburg RRL, Wolf S, et al. A cost-effectiveness analysis of Erwinia asparaginase therapy in children with acute lymphoblastic leukemia. Pediatr Blood Cancer. 2019;66(1):e27458. PubMed
10.Table: 13-10-0114-01. Life expectancy and other elements of the life table, Canada, all provinces except Prince Edward Island. Ottawa (ON): Statistics Canada; 2022: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310011401. Accessed 2022 Oct 06.
11.ASPERSU. Alberta Population Norms for EQ-5D-5L. 2018: https://apersu.ca/wp-content/uploads/2020/10/Alberta-Norms-Report_APERSU-1.pdf. Accessed 2022 Oct 06.
12.Furlong W, Rae C, Feeny D, et al. Health-related quality of life among children with acute lymphoblastic leukemia. Pediatr Blood Cancer. 2012;59(4):717-724. PubMed
13.Health Canada. List of drugs for exceptional importation and sale. 2022; https://www.canada.ca/en/health-canada/services/drugs-health-products/drug-products/drug-shortages/list.html. Accessed 2022 Oct 06.
14.Ontario Case Costing Initiative (OCCI). Toronto (ON): Ontario Health and Long-Term Care; 2017: https://data.ontario.ca/dataset/ontario-case-costing-initiative-occi. Accessed 2022 Jul 12.
15.Bekelman JE, Halpern SD, Blankart CR, et al. Comparison of Site of Death, Health Care Utilization, and Hospital Expenditures for Patients Dying With Cancer in 7 Developed Countries. JAMA. 2016;315(3):272-283. PubMed
16.Sponsor's ITC report title [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Drug name, doses and administration. Burlington (ON): Jazz Pharmaceuticals; 2022 Aug 18. 2022.
17.Salzer WL, Asselin B, Supko JG, et al. Erwinia asparaginase achieves therapeutic activity after pegaspargase allergy: a report from the Children's Oncology Group. Blood. 2013;122(4):507-514. PubMed
18.Mitchell RJ, Kirkwood AA, Barretta E, et al. IKZF1 alterations are not associated with outcome in 498 adults with B-precursor ALL enrolled in the UKALL14 trial. Blood Adv. 2021;5(17):3322-3332. PubMed
19.Marks DI, Kirkwood AA, Rowntree CJ, et al. Addition of four doses of rituximab to standard induction chemotherapy in adult patients with precursor B-cell acute lymphoblastic leukaemia (UKALL14): a phase 3, multicentre, randomised controlled trial. Lancet Haematol. 2022;9(4):e262-e275. PubMed
20.Lennmyr E, Karlsson K, Ahlberg L, et al. Survival in adult acute lymphoblastic leukaemia (ALL): A report from the Swedish ALL Registry. Eur J Haematol. 2019;103(2):88-98. PubMed
21.Cancer Care Ontario: funded evidence-informed regimens. 2022; https://www.cancercareontario.ca/en/drugformulary/regimens. Accessed 2022 Oct 26.
22.The APERSU Team. Alberta Population Norms for EQ-5D-5L. Edmonton AB: University of Alberta; 2018: https://apersu.ca/wp-content/uploads/2020/10/Alberta-Norms-Report_APERSU-1.pdf. Accessed 2022 Nov 03.
23.Lung T, Howard K, Etherton-Beer C, Sim M, Lewin G, Arendts G. Comparison of the HUI3 and the EQ-5D-3L in a nursing home setting. PLoS ONE. 2017;12(2):e0172796. PubMed
24.Polinder S, Haagsma JA, Bonsel G, Essink-Bot ML, Toet H, van Beeck EF. The measurement of long-term health-related quality of life after injury: comparison of EQ-5D and the health utilities index. Inj Prev. 2010;16(3):147-153. PubMed
25.Drug Shortages Canada. Drug Shortage Report for ERWINASE. 2021; https://www.drugshortagescanada.ca/shortage/133673. Accessed 2022 Oct 19.
26.Palmer E. FDA warning savages Porton for contamination that led to shortage of Jazz Pharma leukemia drug. 2017(2022 Oct 19). https://www.fiercepharma.com/manufacturing/fda-warning-savages-porton-for-contamination-led-to-shortage-jazz-pharma-leukemia.
27.Health Canada. Product Information: Erwinase. 2021; https://health-products.canada.ca/dpd-bdpp/info.do?lang=en&code=61174. Accessed 2022 Oct 19.
28.Jazz Pharmaceuticals. Jazz’s Commitment to ALL Patients: Next Steps in 2021. 2021; https://www.jazzpharma.com/eur-int-residents-only/. Accessed 2022 Oct 26.
29.Maese L, Rau RE. Current Use of Asparaginase in Acute Lymphoblastic Leukemia/Lymphoblastic Lymphoma. Front Pediatr. 2022;10:902117. PubMed
30.Table: 17-10-0057-01 Projected population, by projection scenario, age and sex, as of July 1 (x 1,000). Ottawa (ON: Statistics Canada; 2022: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1710005701. Accessed 2022 Oct 6.
31.Table: 13-10-0111-01. Number and rates of new cases of primary cancer, by cancer type, age group and sex: Acute lymphocytic leukemia. Ottawa (ON): Statistics Canada; 2022: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310011101&pickMembers%5B0%5D=2.1&pickMembers%5B1%5D=3.1&pickMembers%5B2%5D=4.50&cubeTimeFrame.startYear=2015&cubeTimeFrame.endYear=2019&referencePeriods=20150101%2C20190101. Accessed 2022 Oct 06.
32.Table: 13-10-0111-01. Number and rates of new cases of primary cancer, by cancer type, age group and sex: Non-Hodgkins lymphoma. Ottawa (ON): Statistics Canada; 2022: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310011101&pickMembers%5B0%5D=2.1&pickMembers%5B1%5D=3.1&pickMembers%5B2%5D=4.48&cubeTimeFrame.startYear=2015&cubeTimeFrame.endYear=2019&referencePeriods=20150101%2C20190101. Accessed 2022 Oct 06.
33.Indigenous Services Canada. Non-Insured Health Benefits program: First Nations and Inuit Health Branch: annual report 2019 to 2020. 2020: https://www.sac-isc.gc.ca/eng/1624462613080/1624462663098. Accessed 2022 Aug 22.
34.Portell CA, Sweetenham JW. Adult lymphoblastic lymphoma. Cancer J. 2012;18(5):432-438. PubMed
35.Halford ZP, M. Hypersensitivity Rates in Pediatric Patients Receiving Intravenous or Intramuscular Asparaginase Agents. Journal Hematol Oncol Pharm, 2020;10(1):7-12.
36.Lynggaard LS, Rank CU, Hansen SN, et al. Asparaginase enzyme activity levels and toxicity in childhood acute lymphoblastic leukemia: a NOPHO ALL2008 study. Blood Adv. 2022;6(1):138-147. PubMed
37.Stock W, Luger SM, Advani AS, et al. A pediatric regimen for older adolescents and young adults with acute lymphoblastic leukemia: results of CALGB 10403. Blood. 2019;133(14):1548-1559. PubMed
38.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Drug name, doses and administration. City (PROV): Sponsor name; 1800 Jan 1.
39.Dana-Farber Cancer Institute. About Childhood Lymphoblastic Lymphoma. https://www.dana-farber.org/childhood-lymphoblastic-lymphoma/.
40.Table: 17-10-0005-01 Population estimates on July 1st, by age and sex. Ottawa (ON: Statistics Canada; 2022: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1710000501. Accessed 2022 Nov 6.
41.Indigenous Services Canada. Non-Insured Health Benefits program: First Nations and Inuit Health Branch: annual report 2020 to 2021. 2021: https://www.sac-isc.gc.ca/eng/1645718409378/1645718500555. Accessed 2022 Aug 22.
42.Ravandi F, Kebriaei P. Philadelphia chromosome-positive acute lymphoblastic leukemia. Hematol Oncol Clin North Am. 2009;23(5):1043-1063, vi. PubMed
43.Alshomar A, El Fakih R. Philadelphia chromosome-positive lymphoblastic lymphoma-Is it rare or underdiagnosed? Hematol Oncol Stem Cell Ther. 2020;13(4):242-243. PubMed
Appendix 1: Cost Comparison Table
Note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical expert(s) and drug plans. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 7: CADTH Cost Comparison for Patients With ALL/LBL Who Are Hypersensitive to E. coli–Derived Asparaginase
Treatment | Strength / concentration | Form | Price | Recommended dosage | Cost per 14-day cycle | Cost per 4-cycle coursea |
|---|---|---|---|---|---|---|
Crisantaspase recombinant (Rylaze) | 10 mg/ 0.5 mL | 20 mg/mL solution for intramuscular injection in single dose vials | 1,091.9100 | 25 mg/m2 on Mondays and Wednesdays and 50 mg/m2 on Fridays for a total of 6 doses to replace each planned dose of pegaspargasea | Pediatric: $24,022 Adult: $43,676 | Pediatric: $96,088 Adult: $174,706 |
Erwinia-derived asparaginase (Erwinase) | 10,000 IU / vial | Freeze-dried powder for injection | 1,091.9100b | 25,000 IU on Mondays, Wednesdays, and Fridaysc | Pediatric: $19,654 Adult: $32,757 | Pediatric: $78,618 Adult: $131,029 |
aPatients in the JZP458-201 study received a median of 4 courses of crisantaspase recombinant (range, 1 to 15 courses).2,6
bAt the time of this review, Erwinase was only available in Canada through exceptional importation and sale (i.e., it is a designated drug).13,25 CADTH was unable to independently verify the list price of Erwinase, nor the costs paid by jurisdictional drug plans through exceptional importation.
cBased on currently used treatment protocols.29
Appendix 2: Submission Quality
Note that this appendix has not been copy-edited.
Description | Yes/No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | Clinical expert feedback elicited by CADTH did not consider the subgrouping of patients into those above and below 25 years of age to be clinically relevant. An analysis against appropriate comparators such as desensitization therapy or intensified chemotherapy were not conducted. |
Model has been adequately programmed and has sufficient face validity | No | The model included numerous IFERROR statements, making validation difficult (refer to Key Limitation section). Adult (25+ years) patients could reach the age of 146 years. |
Model structure is adequate for decision problem | Yes | — |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | No | Most parameters were assigned an arbitrary variation of 10% of the mean which did not adequately reflect uncertainty. Distributions were not constrained to plausible or logical minimum and maximum values. Efficacy and between treatment differences were not varied probabilistically despite high uncertainty, e.g., the clinical equivalence of EDA and crisantaspase, the reduction in EFS and OS associated with discontinuing asparaginase. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | No | — |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | No | The methodology described in the submitted pharmacoeconomic report was inconsistent with the methods used in the provided model (e.g., patients between 25 and 40 were not assumed to be cured after 5 years of EFS, nor were patients above 40 assumed to be dead at model cycle 40). |
EDA = Erwinia-derived asparaginase; EFS = event-free survival; OS = overall survival.
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note that this appendix has not been copy-edited.
Figure 1: Model Structure — Decision Tree for Pediatric and Young Adult Population (< 25 Years)
EFS = event-free survival; R/ST = relapse/secondary tumour.
Source: Sponsor’s pharmacoeconomic submission.3
Figure 2: Model Structure — Decision Tree for Adult Population (≥ 25 Years)
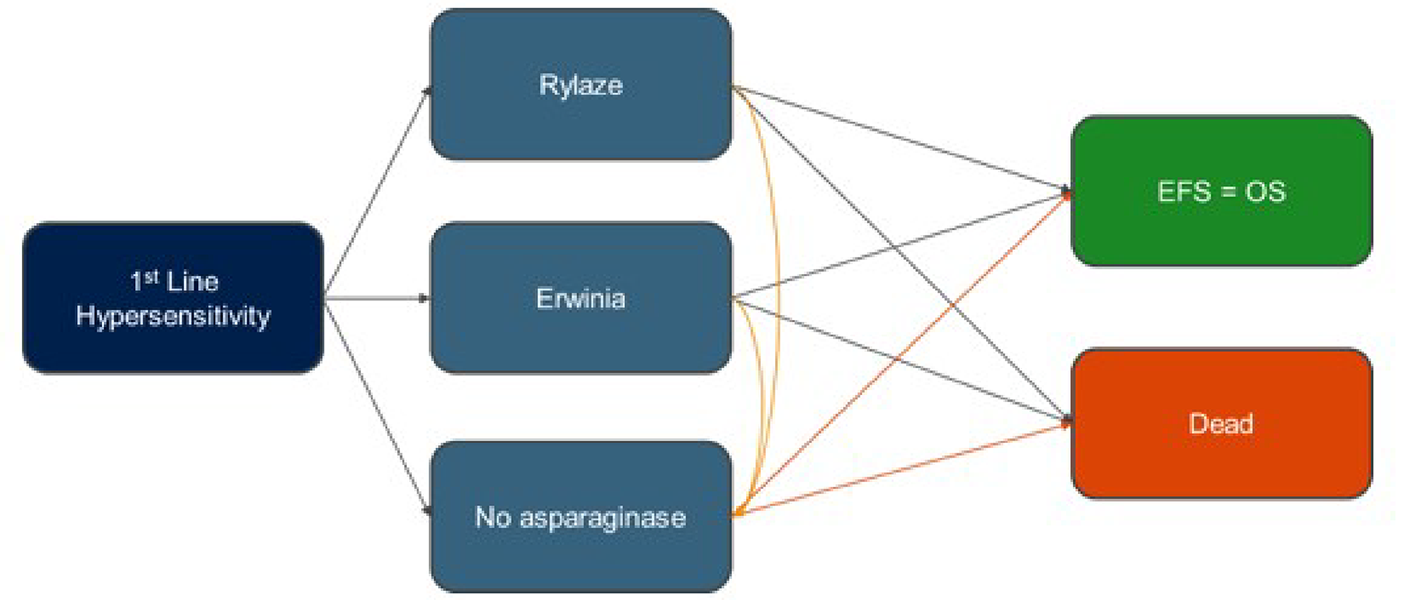
EFS = event-free survival; OS = overall survival.
Source: Sponsor’s pharmacoeconomic submission.3
Figure 3: Model Structure — Markov Model for Pediatric and Young Adult Patients (< 25 Years)
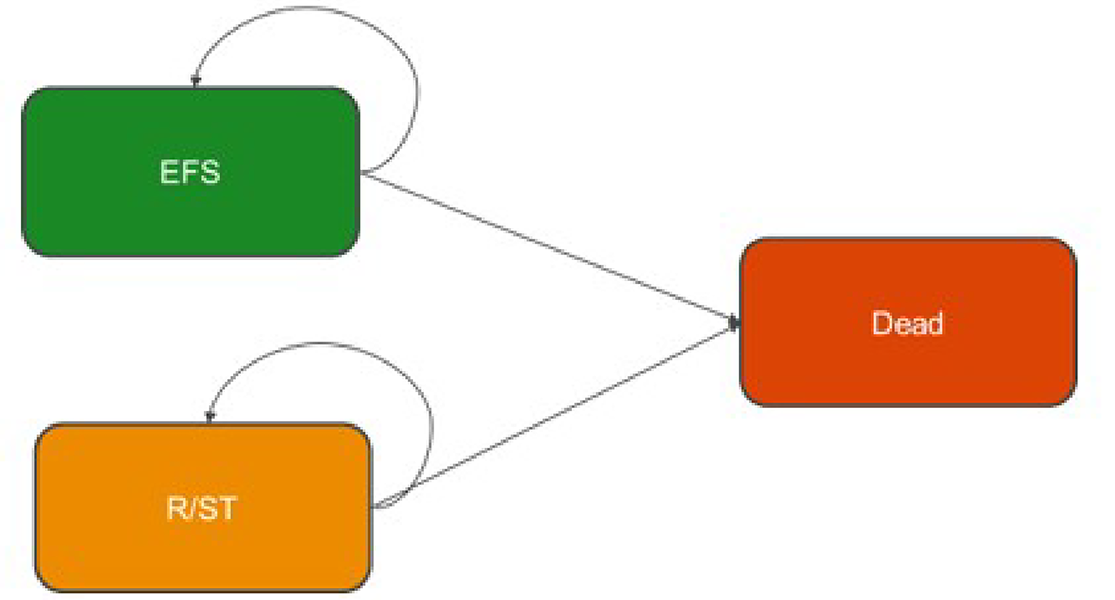
EFS = event-free survival; R/ST = relapse/secondary tumour.
Source: Sponsor’s pharmacoeconomic submission.3
Figure 4: Model Structure — Markov Model for Adult Patients (≥ 25 Years)

EFS = event-free survival.
Source: Sponsor’s pharmacoeconomic submission.3
Figure 5: Sponsor’s Predicted Survival — Pediatric and Young Adult Patients (< 25 Years)
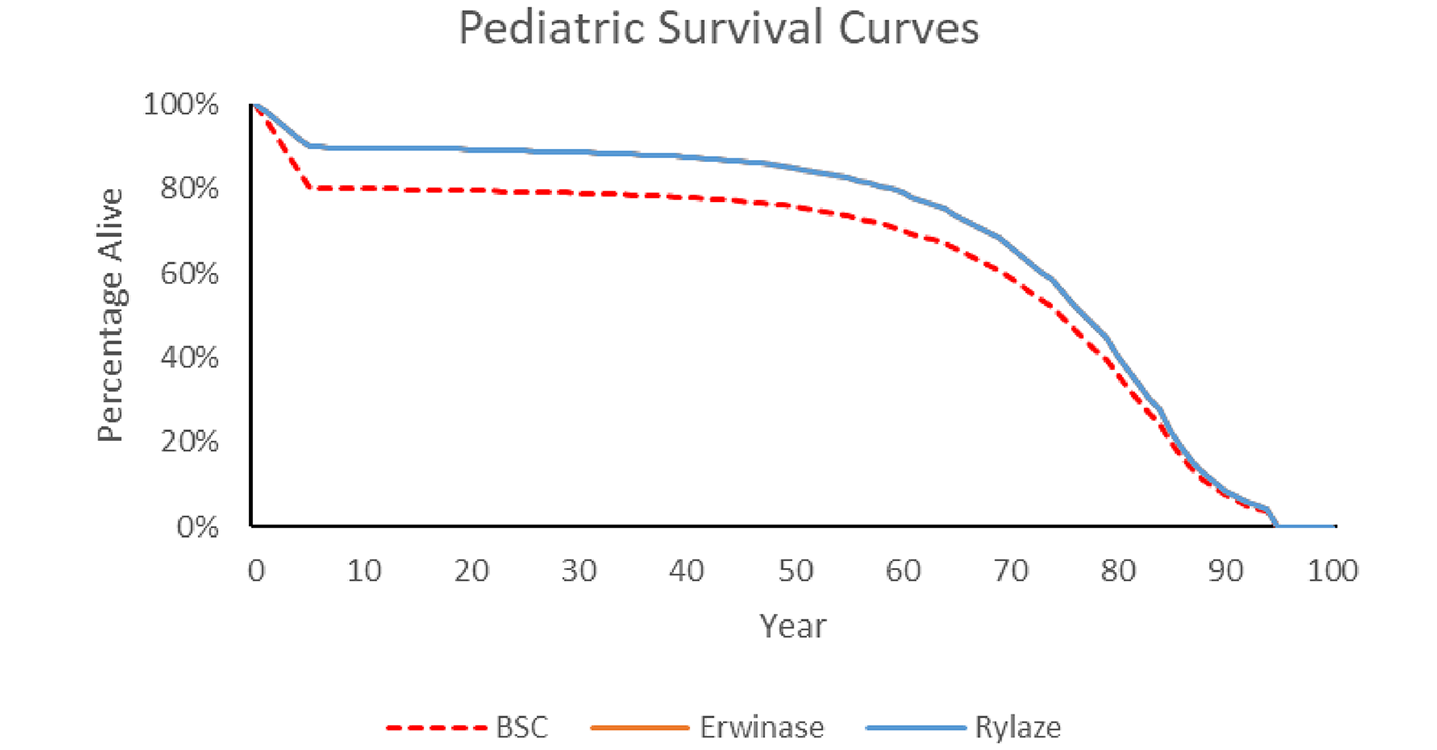
BSC = best supportive care.
Note: in this figure, the Erwinase (EDA) and Rylaze (crisantaspase recombinant) curves are identical. BSC represents patients who did not receive a second-line asparaginase (EDA or crisantaspase recombinant) due to having another hypersensitivity reaction (base case) or due to being assigned to BSC (scenario analysis).
Source: Sponsor’s pharmacoeconomic submission.3
Figure 6: Sponsor’s Predicted Survival — Adult Patients (≥ 25 Years)
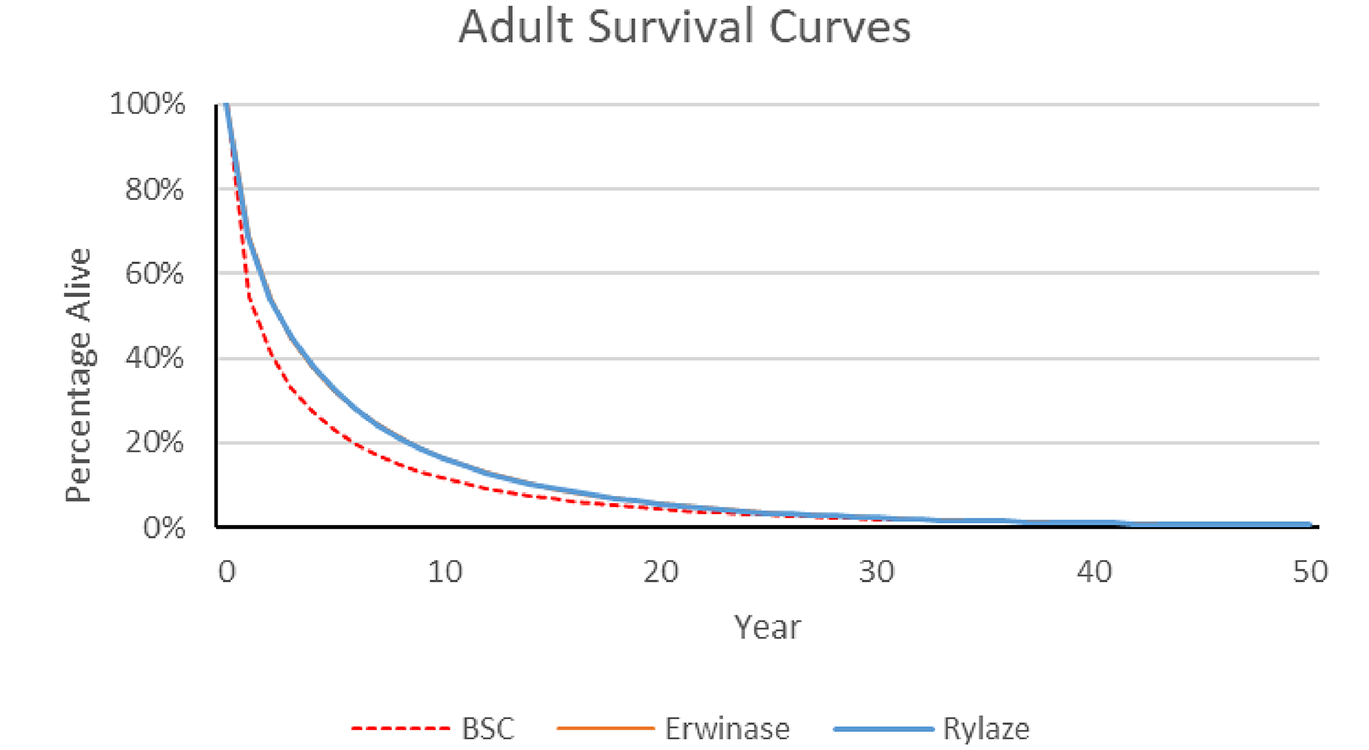
BSC = best supportive care.
Note: in this figure, the Erwinase (EDA) and Rylaze (crisantaspase recombinant) curves are identical. BSC represents patients who did not receive a second-line asparaginase (EDA or crisantaspase recombinant) due to having another hypersensitivity reaction (base case) or due to being assigned to BSC (scenario analysis).
Source: Sponsor’s pharmacoeconomic submission.3
Detailed Results of the Sponsor’s Base Case
Table 9: Summary of the Sponsor’s Economic Evaluation Results Compared to BSC
Drug | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER vs. EDA ($/QALY) |
|---|---|---|---|---|---|
Pediatric and young adult patients (< 25 years) | |||||
Best supportive care | 7,236 | Reference | 31.63 | Reference | Reference |
Crisantaspase recombinant | 104,994 | 97,758 | 35.40 | 3.76 | 25,970 |
Adult patients (25+ years) | |||||
Best supportive care | 18,803 | Reference | 3.15 | Reference | Reference |
Crisantaspase recombinant | 198,173 | 179,370 | 4.09 | 0.95 | 189,288 |
BSC = best supportive care; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Source: Sponsor’s pharmacoeconomic submission.3
Table 10: Disaggregated Summary of the Sponsor’s Economic Evaluation Results — Pediatric and Young Adult Population (< 25 years)
Parameter | Crisantaspase recombinant | Erwinia-derived asparaginase | Best supportive care | Incremental crisantaspase vs. EDA | Incremental crisantaspase vs. BSC |
|---|---|---|---|---|---|
Discounted LYs | |||||
Total | 40.98 | 40.98 | 36.67 | 0 | 4.30 |
Event-free survival | 38.95 | 38.95 | 34.64 | 0 | 4.31 |
Relapsed/secondary tumour | 2.03 | 2.03 | 2.03 | 0 | –0.01 |
Discounted QALYs | |||||
Total | 35.40 | 35.40 | 31.63 | 0 | 3.76 |
Event-free survival | 33.98 | 33.98 | 30.21 | 0 | 3.77 |
Relapsed/secondary tumour | 1.42 | 1.42 | 1.43 | 0 | –0.01 |
Hypersensitivity | –0.0007 | –0.0007 | 0 | 0 | 0.0007 |
Discounted costs ($) | |||||
Total | 104,994.03 | 78,067.20 | 7,235.92 | 20,658.94 | 97,758.11 |
Drug acquisition | 98,726.13 | 98,726.13 | 0 | 0 | 98,726.13 |
Hypersensitivity | 46.31 | 46.31 | 0 | 0 | 46.31 |
End of life | 6,221.59 | 6,221.59 | 7,235.92 | 0 | –1,014.33 |
Health state costs | 0 | 0 | 0 | 0 | 0 |
ICER, crisantaspase recombinant vs. EDA ($/QALY) | Dominated (more costs, equally effective) | ||||
ICER, crisantaspase recombinant vs. BSC ($/QALY) | $25,969.98 | ||||
BSC = best supportive care; EDA = Erwinia-derived asparaginase; ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year.
Table 11: Disaggregated Summary of the Sponsor’s Economic Evaluation Results — Adult Population (≥ 25 Years)
Parameter | Crisantaspase recombinant | Erwinia-derived asparaginase | Best supportive care | Incremental crisantaspase vs. EDA | Incremental crisantaspase vs. BSC |
|---|---|---|---|---|---|
Discounted LYs | |||||
Total | 4.91 | 4.91 | 3.79 | 0 | 1.12 |
Discounted QALYs | |||||
Total | 4.09 | 4.09 | 3.15 | 0 | 0.95 |
Event-free survival | 4.09 | 4.09 | 3.15 | 0 | 0.95 |
Hypersensitivity | –0.0007 | –0.0007 | 0 | 0 | –0.0007 |
Discounted costs ($) | |||||
Total | 198,173.00 | 155,562.30 | 18,802.74 | 42,610.70 | 179,370.26 |
Drug acquisition | 183,414.67 | 140,803.98 | 3,829.50 | 0 | 179,585.17 |
Hypersensitivity | 46.31 | 46.31 | 0 | 0 | 46.31 |
end of life | 14,712.01 | 14,712.01 | 14,973.23 | 0 | –261.22 |
Health state costs | 0 | 0 | 0 | 0 | 0 |
ICER, crisantaspase recombinant vs. EDA ($/QALY) | Dominated (more costs, equally effective) | ||||
ICER, crisantaspase recombinant vs. BSC ($/QALY) | $189,287.82 | ||||
BSC = best supportive care; EDA = Erwinia-derived asparaginase; ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year.
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Note that this appendix has not been copy-edited.
Detailed Results of CADTH Reanalysis
Table 12: Summary of the Sponsor’s Corrected Analyses, Including Best Supportive Care
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER vs. EDA ($/QALYs) | ICER vs. BSC ($/QALY) |
|---|---|---|---|---|---|
Pediatric and young adult population (< 25 years) | |||||
Sponsor’s base case | Erwinia-derived asparaginase | 84,507.24 | 35.40 | Reference | NA |
Crisantaspase recombinant | 104,994.03 | 35.40 | Dominateda | 25,969.98 | |
Best supportive care | 20,658.94 | 31.63 | NA | Reference | |
Sponsor’s corrected base case | Erwinia-derived asparaginase | 84,709.08 | 35.38 | Reference | NA |
Crisantaspase recombinant | 105,359.28 | 35.38 | Dominateda | 26,082.99 | |
Best supportive care | 20,650.20 | 31.62 | NA | Reference | |
Adult population (25+ years) | |||||
Sponsor’s base case | Erwinia-derived asparaginase | 155,562.30 | 4.09 | Reference | NA |
Crisantaspase recombinant | 198,173.00 | 4.09 | Dominateda | 189,287.82 | |
Best supportive care | 18,802.74 | 3.15 | NA | Reference | |
Sponsor’s corrected base case | Erwinia-derived asparaginase | 156,222.95 | 4.08 | Reference | NA |
Crisantaspase recombinant | 198,964.68 | 4.08 | Dominateda | 188,222.96 | |
Best supportive care | 18,812.17 | 3.12 | NA | Reference | |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; NA = not applicable.
The sponsor did not conduct sequential analyses comparing all 3 comparators, but instead conducted separate analyses comparing crisantaspase recombinant to EDA and crisantaspase recombinant to BSC. The efficacy of crisantaspase recombinant compared to BSC is of mainly of interest in populations or jurisdictions which do not have access to or do not reimburse EDA, and thus CADTH has not combined the results into a single sequential analysis.
aMore costly, equally effective.
A CADTH base-case reanalysis was not conducted due to extensive uncertainty associated with the sponsor’s model. A series of exploratory scenario analyses were instead conducted to explore some aspects of this uncertainty.
Exploratory Scenario Analyses
Table 13: CADTH’s Exploratory Scenario Analyses
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Changes to derive the CADTH exploratory scenario analyses | ||
1. Length of treatment with second-line asparaginase | 4 cycles | Pediatric: 2 (standard and intermediate risk patients) to 4 (high-risk patients) cycles; weighted average = 2.43 cycles Adult: 9 cycles |
2.a. BSC mortality decrement lower | 10.3% | 3.3% |
2.b. BSC mortality decrement higher | 10.3% | 17.0% |
3.a. EDA efficacy relative to crisantaspase recombinant is lower | EFS and OS RR: 1.0 | EFS and OS RR: 0.95 |
3.b. EDA efficacy relative to crisantaspase recombinant is higher | EFS and OS RR: 1.0 | EFS and OS RR: 1.05 |
4. RST utility decrement applied after year 2 (adult population only) | No utility decrement applied after year 2 | 20% utility decrement applied in year 3+ |
BSC = best supportive care; EDA = Erwinia-derived asparaginase; EFS = event-free survival; OS = overall survival; RR = relative risk; RST = relapsed/secondary tumour.
Table 14: Summary of CADTH’s Exploratory Scenario Analyses Results
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER vs. EDA | ICER vs. BSC |
|---|---|---|---|---|---|
Pediatric and young adult population (< 25 years) | |||||
Sponsor’s base case | Erwinia-derived asparaginase | 84,335.10 | 35.40 | Reference | NA |
Crisantaspase recombinant | 104,994.03 | 35.40 | Dominateda | 25,969.98 | |
Best supportive care | 7,235.92 | 31.63 | NA | Reference | |
Sponsor’s corrected base case | Erwinia-derived asparaginase | 84,709.08 | 35.38 | Reference | NA |
Crisantaspase recombinant | 105,359.28 | 35.38 | Dominateda | 26,082.99 | |
Best supportive care | 7,242.49 | 31.62 | NA | Reference | |
CADTH scenario 1 – average duration weighted by baseline risk of 2.43 cyclesb | Erwinia-derived asparaginase | 53,792.58 | 35.31 | Reference | NA |
Crisantaspase recombinant | 66,380.28 | 35.31 | Dominateda | 15,696.54 | |
Best supportive care | 7,263.04 | 31.54 | NA | Reference | |
CADTH scenario 2a – BSC mortality decrement 3.3% | Erwinia-derived asparaginase | 84,457.34 | 35.53 | Reference | NA |
Crisantaspase recombinant | 105,133.75 | 35.53 | Dominateda | 79,615.50 | |
Best supportive care | 6,521.50 | 34.29 | NA | Reference | |
CADTH scenario 2b – BSC mortality decrement 17.0% | Erwinia-derived asparaginase | 84,587.70 | 35.25 | Reference | NA |
Crisantaspase recombinant | 105,342.72 | 35.25 | Dominateda | 15,587.74 | |
Best supportive care | 7,947.33 | 29.00 | NA | NA | |
CADTH scenario 3a – EDA is less effective (EFS/OS RR: 0.95) | Erwinia-derived asparaginase | 84,722.65 | 33.81 | Reference | NA |
Crisantaspase recombinant | 104,892.07 | 35.33 | 13,251.75 | 25,844.27 | |
Best supportive care | 7,259.92 | 31.55 | NA | Reference | |
CADTH scenario 3b – EDA is more effective (EFS/OS RR: 1.05) | Erwinia-derived asparaginase | 84,017.28 | 36.84 | Reference | NA |
Crisantaspase recombinant | 104,798.72 | 35.32 | Dominated | 25,792.70 | |
Best supportive care | 7,263.55 | 31.54 | NA | Reference | |
Adult population (25+ years) | |||||
Sponsor’s base case | Erwinia-derived asparaginase | 155,562.30 | 4.09 | Reference | NA |
Crisantaspase recombinant | 198,173.00 | 4.09 | Dominateda | 189,287.82 | |
Best supportive care | 18,802.74 | 3.15 | NA | Reference | |
Sponsor’s corrected base case | Erwinia-derived asparaginase | 156,222.95 | 4.08 | Reference | NA |
Crisantaspase recombinant | 198,964.68 | 4.08 | Dominateda | 188,222.96 | |
Best supportive care | 18,812.17 | 3.12 | NA | Reference | |
CADTH scenario 1 – duration assumption of 9 cycles | Erwinia-derived asparaginase | 331,219.21 | 4.08 | Reference | NA |
Crisantaspase recombinant | 426,975.35 | 4.08 | Dominateda | 424,959.43 | |
Best supportive care | 18,813.37 | 3.12 | NA | Reference | |
CADTH scenario 2a – BSC mortality decrement 3.3% | Erwinia-derived asparaginase | 155,531.27 | 4.12 | Reference | NA |
Crisantaspase recombinant | 198,072.08 | 4.12 | Dominateda | 647,463.70 | |
Best supportive care | 18,613.58 | 3.84 | NA | Reference | |
CADTH scenario 2b – BSC mortality decrement 17.0% | Erwinia-derived asparaginase | 198,502.44 | 4.04 | Reference | NA |
Crisantaspase recombinant | 155,917.95 | 4.04 | Dominateda | 108,379.53 | |
Best supportive care | 19,015.89 | 2.39 | NA | Reference | |
CADTH scenario 3a – EDA is less effective (EFS/OS RR: 0.95) | Erwinia-derived asparaginase | 155,008.70 | 3.92 | Reference | NA |
Crisantaspase recombinant | 197,523.56 | 4.08 | 270,412.15 | 185,549.95 | |
Best supportive care | 18,814.11 | 3.11 | NA | Reference | |
CADTH scenario 3b – EDA is more effective (EFS/OS RR: 1.05) | Erwinia-derived asparaginase | 155,970.19 | 4.23 | Reference | NA |
Crisantaspase recombinant | 198,720.28 | 4.08 | Dominated | 186,651.54 | |
Best supportive care | 18,814.25 | 3.11 | NA | Reference | |
CADTH scenario 4 to 20% utility decrement applied to years 3+ | Erwinia-derived asparaginase | 156,039.74 | 3.48 | Reference | NA |
Crisantaspase recombinant | 198,475.73 | 3.48 | Dominateda | 224,165.00 | |
Best supportive care | 18,813.32 | 2.68 | NA | Reference | |
BSC = best supportive care; EDA = Erwinia-derived asparaginase; ICER = incremental cost-effectiveness ratio; NA = not applicable; QALY = quality-adjusted life-year; RR = relative risk.
The sponsor did not conduct sequential analyses comparing all 3 comparators, but instead conducted separate analyses comparing crisantaspase recombinant to EDA and crisantaspase recombinant to BSC. The efficacy of crisantaspase recombinant compared to BSC is of mainly of interest in populations or jurisdictions which do not have access to or do not reimburse EDA, and thus CADTH has not combined the results into a single sequential analysis.
aMore costly, equally effective.
bThis analysis assumes the 49.5% standard risk and 29.1% intermediate risk pediatric and young adult ALL or LBL patients assumed by the sponsor receive 2 courses each of second-line asparaginase, while the 21.4% high-risk patients each receive 4 courses.
Table 15: CADTH Price-Reduction Analyses
Analysis | ICERs for crisantaspase vs. Erwinia-derived asparaginase ($/QALY) | |||
|---|---|---|---|---|
Pediatric and young adult population (< 25 years) | Adult population (25+ years) | |||
Price reduction | Sponsor’s corrected base case vs. EDAa | Sponsor’s corrected base case vs. BSC | Sponsor’s corrected base case vs. EDAa | Sponsor’s corrected base case vs. BSC |
No price reduction | Dominated | 26,083 | Dominated | 188,223 |
10% | Dominated | 23,275 | Dominated | 167,053 |
20% | Dominated | 20,645 | Dominated | 147,887 |
21% | Dominant | 20,497 | Dominated | 146,879 |
24% | Dominant | 19,517 | Dominant | 140,793 |
30% | Dominant | 17,816 | Dominant | 128,188 |
40% | Dominant | 15,545 | Dominant | 112,167 |
50% | Dominant | 12,712 | Dominant | 89,860 |
60% | Dominant | 10,286 | Dominant | 72,384 |
70% | Dominant | 7,525 | Dominant | 52,575 |
72% | Dominant | 7,112 | Dominant | 49,601 |
BSC = best supportive care; EDA = Erwinia-derived asparaginase; ICER = incremental cost-effectiveness ratio.
aThe base-case analysis assumes wastage of excess medication in vials, with a mean body surface area of 0.97 m2 for the pediatric and young adult population, and 1.95 m2 for the adult population. If no wastage is assumed, then a price reduction of 25% is required for the cost of crisantaspase recombinant to equal that of Erwinia-derived asparaginase in both populations.
Appendix 5: Submitted BIA and CADTH Appraisal
Note that this appendix has not been copy-edited.
Table 16: Summary of Key Take-Aways
Key take-aways of the BIA |
|---|
|
Summary of Sponsor’s BIA
In the submitted budget impact analysis, the sponsor assessed the introduction of crisantaspase recombinant for the treatment of pediatric and adult patients with ALL and LBL who have developed hypersensitivity to E. coli–derived asparaginase. The BIA was undertaken from the perspective of a Canadian public drug plan payer over a 3-year time horizon (2024 to 2026) using an epidemiological approach. The sponsor’s base-case analysis included only drug-acquisition costs for crisantaspase recombinant and EDA, without markups or dispensing fees. Data from the model were obtained from various sources including Statistics Canada,30-32 the Non-Insured Health Benefits (NIHB) program,33 the published literature,34-37 the sponsor’s internal forecasts, and expert opinion.38 Key inputs to the BIA are documented in Table 18.
Key assumptions include:
ALL and LBL incidence rates are not changing over time.
Crisantaspase recombinant will not grow the market for second-line asparaginase.
Mean body surface area for adult and pediatric patients is the same as reported in the economic evaluation, despite a different age threshold separating the populations.
All patients receive a total of four 2-week courses of EDA or crisantaspase recombinant therapy.
Background chemotherapy regimens are identical between groups and do not need to be considered.
Second-line asparaginase therapy will only be given to patients with a grade 3+ hypersensitivity to E. coli–derived asparaginase.
Excess medication in vials is wasted.
Table 17: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (Year 1 / Year 2 / Year 3 if appropriate) |
|---|---|
Target population | |
Pan-Canadian populationa (projected) | 31,655,761 / 32,033,690 / 32,411,620 |
Incidence of ALL (all ages) | 0.001331 |
Proportion of ALL patients < 20 / 20+ years of age | 55% / 45%31 |
Incidence of NHL (all ages) | 0.0253%32 |
Proportion of NHL patients < 20 / 20+ years of age | 1% / 99%32 |
Proportion of pediatric NHL patients with LBL subtype | 35%39 |
Proportion of adult NHL patients with LBL subtype | 2%34 |
Proportion of ALL/LBL patients receiving E. coli–derived asparaginase | 100%b |
Proportion of patients with grade 3+ hypersensitivity to E. coli–derived asparaginase | 12%c |
Proportion of hypersensitive patients who switch to EDA | 90% |
Number of pediatric (< 20 years) patients eligible for drug under review | 28 / 28 / 28 |
Number of adult (20+ years) patients eligible for drug under review | 37 / 38 / 38 |
Market uptake (3 years, reference scenario), all populations | |
EDA | 100% / 100% / 100% |
Market uptake (3 years, new drug scenario), proportions switching to crisantaspase recombinant in each populationd | |
ALL pediatric population (< 20 years) | 30% / 40% / 50% |
ALL adult population (20+ years) | 40% / 45% / 50% |
LBL pediatric population (< 20 years) | 30% / 40% / 50% |
LBL adult population (20+ years) | 40% / 45% / 50% |
Cost of treatment (per patient, assumes four 2-week courses of therapy)e | |
Crisantaspase recombinant, pediatric (< 20 years) | $96,088 |
Crisantaspase recombinant, adults (20+ years) | $174,706 |
EDA, pediatric (< 20 years) | $78,618 |
EDA, adult (20+ years) | $131,029 |
Doxorubicin, dexamethasone, cytarabine, etoposide regiment (adults)f | $3,830 |
ALL = acute lymphoblastic leukemia; EDA = Erwinia-derived asparaginase; NHL = non-Hodgkin lymphoma; LBL = lymphoblastic lymphoma
aPan-Canadian population excludes Quebec and the 3 territories and adds NIHB clients. The population in future years is a linear projection of data from 2016 to 2021 (2016 to 2020 in the case of NIHB).40
bOpinion of 4 Canadian clinical experts interviewed by the sponsor.3
c12% estimated based on the ranges reported in 3 retrospective analyses of pediatric or young adult patients receiving asparaginase.35-37
dBased on sponsor’s internal forecasts. All patients who do not receive crisantaspase recombinant are assumed to receive EDA.
eBased on mean body surface areas reported for the pediatric and young adult populations (< 25 years) and adult populations (25+ years) as considered in the pharmacoeconomic evaluation.
fDefined by the sponsor as best supportive care, includes 3 doses of doxorubicin and a single course of dexamethasone, high dose cytarabine, and etoposide. Used in the BSC scenario for adult patients without access to an asparaginase.
Summary of the Sponsor’s BIA Results
Results of the sponsor’s analyses suggest that the reimbursement of crisantaspase recombinant, when considering only drug-acquisition costs and the displacement of EDA, will be associated with an incremental cost of $792,384 in year 1, $932,498 in year 2, and $1,075,694, in year 3, for a total budget impact of $2,800,576 over the first 3 years.
The sponsor also conducted a scenario which assumed EDA was not available, where all patients were assumed to use an unspecified BSC regimen and 50% of adult patients would also be receiving a cycle of doxorubicin, dexamethasone, cytarabine, and etoposide (i.e., on top of their underlying background chemotherapy regimen). In this scenario, the reimbursement of crisantaspase recombinant, which would be used by 100% of eligible patients, would be associated with an incremental cost of $9,055,367 in year 1, $9,163,477 in year 2, and $9,271,587 in year 3, for 3-year budget impact of $27,490,431.
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
The NIHB client population was doubled counted: The sponsor calculated the total population of CADTH-participating drug plans by adding the population of the provinces as reported by Statistics Canada, excluding Quebec, to the population of NIHB clients. However, NIHB clients living within the borders of the provincial jurisdictions are included in Statistics Canada population data, and thus the sponsor’s model double counts this population. Additionally, NIHB clients residing within Ontario are covered primarily by ODB if they are under 25 years or over 65 years of age, while the Alberta and Saskatchewan cancer agencies reimburse oncology products for all residents within their borders, including those otherwise reimbursed by NIHB.
CADTH corrected this by subtracting NIHB clients residing within the Atlantic provinces, Manitoba, British Columbia, and those in Ontario aged 25 to 64 from the applicable provincial population totals. NIHB clients residing within Alberta and Saskatchewan as well as those in Ontario under 25 or over 65 years of age were subtracted from the NIHB population total for the purposes of this analysis. CADTH also included updated 2020 to 2021 data available from NIHB to better inform population projections over the model’s time horizon.41
Treatment duration may not represent current Canadian clinical practice: As in the pharmacoeconomic analysis, the sponsor’s model assumes that all patients will receive 4 cycles (6 doses each) of EDA or crisantaspase recombinant, regardless of age group, consistent with the median number of cycles reported in the JZP458-201 trial.3,6 However, according to clinical expert input elicited by CADTH, standard-risk pediatric ALL/LBL patients often receive just 2 cycles of asparaginase, while high-risk pediatric patients receive 4 cycles. In contrast, clinical expert input indicated that adult ALL/LBL patients generally receive 10 cycles of asparaginase therapy over the course of 30 weeks,21 8 to 10 of which might be a second-line asparaginase depending on the timing of the hypersensitivity reaction to E. coli–derived asparaginase.
CADTH conducted a scenario analysis assuming pediatric and young adults would receive a weighted average of 2.43 cycles of crisantaspase recombinant or EDA therapy based on the sponsor’s proportion of high-risk versus lower risk patients, while adult patients were assumed to receive an average of 9 cycles. This scenario assumes patients receive the dosing recommended in the crisantaspase recombinant product monograph; should lower per-cycle dosing be used for adult patients in clinical practice, results may be more in line with those presented as the sponsor’s corrected analysis.
Asparaginase may not be given to all patients: According to CADTH-obtained clinical expert feedback, some treatment protocols may limit the use of asparaginase in adults to Philadelphia chromosome negative (Ph-negative) patients.21 The prevalence of Ph-positive ALL in adult patients has been reported to be 20 to 30%,42 while the prevalence of Ph-positive LBL thus far appears to be uncertain but more rare.43
CADTH conducted a scenario analysis reducing the eligible population of adult patients with ALL by 25% to explore the impact of assuming Ph-positive adult patients with ALL would not receive an asparaginase. The LBL and pediatric and young adult ALL populations were not altered in this scenario.
The availability of EDA is uncertain: The sponsor assumed that where EDA is available and reimbursed, 100% of eligible patients will receive EDA in the absence of crisantaspase recombinant, while 50% of eligible patients will instead receive crisantaspase recombinant should it be reimbursed by year 3. The sponsor also appropriately considered a scenario where EDA is not available. However, a third possibility exists where a supply of EDA may exist but be insufficient to treat all eligible patients in Canada. Therefore, a scenario where only some eligible patients are able to access EDA in the reference case, with the rest able to access crisantaspase recombinant if reimbursed may be relevant.
CADTH conducted a scenario assuming 50% of eligible patients would be able to access EDA in the reference case, with the remaining patients receiving BSC as assumed by the sponsor (i.e., pediatric patients receive no extra treatment, half of adult patients on BSC receive increased chemotherapy). Crisantaspase recombinant was then assumed to displace BSC in the new drug scenario, in proportions as assumed by the sponsor in years 1 and 2 (e.g., 30% in year 1, 40% in year 2, refer to Table 17), with 50% of patients still accessing EDA and 50% accessing crisantaspase recombinant by year 3.
CADTH Reanalyses of the BIA
CADTH corrected the sponsor’s analyses by removing NIHB clients who had been double counted as described in Table 18. As with the CADTH pharmacoeconomic review, CADTH also conducted a series of scenario analyses to explore areas of uncertainty in the sponsor’s BIA model.to highlight uncertainty in the potential budget impact, including reducing the duration of therapy in the pediatric population and increasing it in the adult, assuming adult patients with Ph-positive ALL would not receive an asparaginase, and assuming that the future supply of EDA will only be sufficient for half of eligible patients in the absence of crisantaspase recombinant. Finally, a scenario was also conducted assuming a price reduction of 25% as outlined in the CADTH pharmacoeconomic evaluation of the sponsor’s CUA.
Table 18: CADTH Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
1. NIHB population | NIHB population projected from 2019 to 2020 annual report33 NIHB added to pan-Canadian population (double counted). Total population 2021: 30,412,980 NIHB population 2021: 899,260 | Population projected from 2020 to 2021 annual report41 NIHB population removed from provincial populations where appropriatea Total population 2021: 29,629,921 NIHB population 2021: 513,108b |
Changes made for CADTH scenario analyses | ||
1. Duration of therapy | 4 cycles | Pediatric: 2 (standard and intermediate risk patients) to 4 (high-risk patients) cycles; weighted average = 2.43 cycles Adult: 9 cycles |
2. Ph-positive adult ALL patients removed | All adult patients with ALL are eligible for asparaginase therapy | Adult patients with Ph-positive ALL are assumed not to receive asparaginase |
3. Availability of EDA | Reference case market share: 100% EDA, 0% BSC | Reference case market share 50% EDA, 50% BSC |
4. Price reduction for crisantaspase recombinant | Submitted price | 25% price reduction |
ALL = acute lymphoblastic leukemia; EDA = Erwinia-derived asparaginase; NIHB = Non-Insured Health Benefits; Ph = Philadelphia chromosome.
aNIHB populations living within British Columbia, Manitoba, New Brunswick, Newfoundland and Labrador, Nova Scotia, and Prince Edward Island as well as those in Ontario aged 25 to 64 years were subtracted from the populations of those jurisdictions to avoid double counting. NIHB clients living within Alberta and Saskatchewan were subtracted from the NIHB population due to reimbursement by jurisdiction oncology plans. NIHB clients living within Ontario who are under age 25 and over age 65 years were subtracted from the NIHB population due to reimbursement by the province.
bThe number of NIHB clients reimbursed for oncology treatments by NIHB for the purposes of this analysis.
When the NIHB population was corrected, the 3-year budgetary incremental cost of reimbursing crisantaspase recombinant was $2,728,668 when EDA was assumed to also be available, and $26,784,584 when EDA was assumed to not be available.
The results of the sponsor’s corrected analyses as well as CADTH’s scenario analyses are presented in summary format in Table 19 and a more detailed breakdown is presented in Table 20.
Table 19: Summary of the CADTH Reanalyses of the BIA
Stepped analysis | Three-year total – EDA is available and reimbursed | Three-year total – EDA is unavailable or not reimbursed |
|---|---|---|
Submitted analyses | $2,800,576 | $27,490,431 |
Corrections to submitted analyses | $2,728,668 | $26,784,584 |
CADTH Scenario 1: Duration of therapy | $5,202,888 | $47,674,748 |
CADTH Scenario 2: Ph-positive adults with ALL removed | $2,438,160 | $24,232,822 |
CADTH Scenario 3: EDA available to 50% of patients in reference case | $11,983,084 | NA |
CADTH Scenario 4: 25% PR for crisantaspase recombinant | –$213,839a | $20,035,915a |
BIA = budget impact analysis; EDA = Erwinia-derived asparaginase; Ph = Philadelphia chromosome; PR = price reduction.
aAll analyses assume a body surface area of 0.97 m2 for pediatric and young adult patients, and 1.95 m2 for adult patients. When wastage of the excess medication in vials is considered, some body surface area assumptions lead to cost savings at equivalent pricing per mg of recommended dose due to rounding. For example, if pediatric patients are assumed to have a mean body surface area of 1.05 m2 rather than 0.97 m2, the 3-year budgetary impact of reimbursing crisantaspase recombinant at a 25% price reduction would be $0 when EDA is available, and $20,569,463 when EDA is not available or not reimbursed.
Table 20: Detailed Breakdown of the CADTH Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
EDA is available and reimbursed | ||||||
Submitted base case | Reference | 6,941,308 | 7,025,180 | 7,109,052 | 7,192,923 | 21,327,155 |
New drug | 6,941,308 | 7,817,564 | 8,041,549 | 8,268,617 | 24,127,731 | |
Budget impact | 0 | 792,384 | 932,498 | 1,075,694 | 2,800,576 | |
Corrected base case | Reference | 6,763,102 | 6,844,811 | 6,926,519 | 7,008,227 | 20,779,556 |
New drug | 6,763,102 | 7,616,851 | 7,835,074 | 8,056,300 | 23,508,224 | |
Budget impact | 0 | 772,040 | 908,555 | 1,048,073 | 2,728,668 | |
CADTH Scenario 1: Duration of therapy | Reference | 11,794,270 | 11,936,762 | 12,079,254 | 12,221,746 | 36,237,761 |
New drug | 11,794,270 | 13,442,915 | 13,811,910 | 14,185,825 | 41,440,649 | |
Budget impact | 0 | 1,506,153 | 1,732,656 | 1,964,079 | 5,202,888 | |
CADTH Scenario 2: Ph-positive adults with ALL removed | Reference | 6,133,310 | 6,207,410 | 6,281,509 | 6,355,608 | 18,844,527 |
New drug | 6,133,310 | 6,894,463 | 7,093,312 | 7,294,911 | 21,282,687 | |
Budget impact | 0 | 687,053 | 811,803 | 939,303 | 2,438,160 | |
CADTH Scenario 3: EDA only available to 50% of patients | Reference | 3,505,744 | 3,548,104 | 3,590,464 | 3,632,824 | 10,771,392 |
New drug | 3,505,744 | 6,905,690 | 7,580,169 | 8,268,617 | 22,754,476 | |
Budget impact | 0 | 3,357,586 | 3,989,705 | 4,635,793 | 11,983,084 | |
CADTH Scenario 4: 25% PR for crisantaspase recombinanta | Reference | 6,763,102 | 6,844,811 | 6,926,519 | 7,008,227 | 20,779,556 |
New drug | 6,763,102 | 6,792,085 | 6,855,379 | 6,918,253 | 20,565,717 | |
Budget impact | 0 | -52,725 | -71,140 | -89,974 | -213,839a | |
EDA is unavailable or not reimbursed | ||||||
Submitted base case | Reference | 70,181 | 71,029 | 71,877 | 72,725 | 215,630 |
New drug | 70,181 | 9,126,396 | 9,235,354 | 9,344,311 | 27,706,061 | |
Budget impact | 0 | 9,055,367 | 9,163,477 | 9,271,587 | 27,490,431 | |
Corrected base case | Reference | 68,379 | 69,205 | 70,031 | 70,857 | 210,093 |
New drug | 68,379 | 8,892,079 | 8,998,226 | 9,104,373 | 26,994,677 | |
Budget impact | 0 | 8,822,874 | 8,928,195 | 9,033,516 | 26,784,584 | |
CADTH Scenario 1: Duration of therapy | Reference | 68,379 | 69,205 | 70,031 | 70,857 | 210,093 |
New drug | 68,379 | 15,773,323 | 15,961,614 | 16,149,904 | 47,884,841 | |
Budget impact | 0 | 15,704,119 | 15,891,583 | 16,079,047 | 47,674,748 | |
CADTH Scenario 2: Ph-positive adults with ALL removed | Reference | 59,176 | 59,890 | 60,605 | 61,320 | 181,816 |
New drug | 59,176 | 8,042,211 | 8,138,213 | 8,234,215 | 24,414,638 | |
Budget impact | 0 | 7,982,320 | 8,077,607 | 8,172,894 | 24,232,822 | |
CADTH Scenario 4:b 25% PR for crisantaspase recombinanta | Reference | 68,379 | 69,205 | 70,031 | 70,857 | 210,093 |
New drug | 68,379 | 6,669,059 | 6,748,669 | 6,828,280 | 20,246,008 | |
Budget impact | 0 | 6,599,854 | 6,678,638 | 6,757,422 | 20,035,915a | |
BIA = budget impact analysis; EDA = Erwinia-derived asparaginase; Ph = Philadelphia chromosome; PR = price reduction.
aAll analyses assume a body surface area of 0.97 m2 for pediatric and young adult patients, and 1.95 m2 for adult patients. When wastage of the excess medication in vials is considered, some body surface area assumptions lead to cost savings at equivalent pricing per mg of recommended dose due to rounding. For example, if pediatric patients are assumed to have a mean body surface area of 1.05 m2 rather than 0.97 m2, the 3-year budgetary impact of reimbursing crisantaspase recombinant at a 25% price reduction would be $0 when EDA is available, and $20,569,463 when EDA is not available or not reimbursed.
bCADTH Scenario 3 is not relevant to analyses assuming EDA is not available and is therefore not reported.
Stakeholder Input
Patient Input
Leukemia & Lymphoma Society of Canada
About The Leukemia & Lymphoma Society of Canada
The Leukemia and Lymphoma Society of Canada is a national charitable status organization dedicated to finding a cure for blood cancers and its ability to improve the quality of life of people affected by blood cancers and their families by funding life-enhancing research and providing educational resources, services, and support. The Leukemia and Lymphoma Society of Canada is the largest charitable organization in Canada dedicated to blood cancer, our focus includes:
Funding research from bench to bedside
Rethinking how a person navigates their blood cancer experience
Providing targeted blood cancer information
Offering tools for psychological and emotional support
Empowering Canadians to take charge of their blood cancer experience through practical support and advocacy
To learn more, visit: bloodcancers.ca
Information Gathering
The LLSC created an online survey, that was distributed through social media networks [Facebook, Twitter, Instagram] and by email, between August 25 to September 8, 2022, in both English and French. The survey uses multiple choice, open-ended and rating questions, and uses skipping logic to allow respondents to pass on questions not relevant to them. Open-ended responses to surveys that reflected the sentiment of a majority are included verbatim to provide a deeper understanding of patient perspectives.
We received 40 respondents to the survey. Most respondents were from Quebec (18), followed by British Columbia (10), Ontario (4), Alberta (2) and Yukon (1). Five (5) respondents did not provide their province of residence. The majority of respondents were between the ages of 35-44 years (10 respondents), followed by 25-34 years (8 respondents), 55-64 years (5 respondents), 65-74 years (4 respondents), 45-54 years (3 respondents), 18-24 years (2 respondents) and finally 3 respondents were under 18 years of age. Five (5) respondents did not specify their age.
Disease Experience
Respondents were asked to rate on a scale of 1 (no impact) to 5 (extremely significant impact), the impact that the disease symptoms had affected their quality of life. Refer to Table 1.
68% of the respondents listed fatigue or weakness and having significant impact on their quality of life. This was followed by Loss of appetite or weight loss (45%) and Headaches, blurred vision, nausea, or vomiting (41%).
When asked how the disease impacted their day-to-day lives, ability to work (64%) and exercise (64%) were indicated as the most significantly impacted by the respondents. Refer to Table 2. The ability to travel (59%), manage family responsibilities (59%) and pursue activities or hobbies (59%) were also significantly affected by disease symptoms.
Most notably, respondents indicated that the psychological/social factors of the disease have significantly impacted their quality of life. Refer Table 3. Stress, anxiety, and worry had the most impact on the respondent’s quality of life after diagnosis (68%).
Table 1: Disease Symptoms Effects on Quality of Life
Side effect | Significant impact (4-5) |
|---|---|
Fatigue or weakness | 15 |
Fever | 5 |
Loss of appetite or weight loss | 10 |
Night sweats | 4 |
Spots under the skin (petechiae) | 1 |
Stomach pain | 6 |
Infections | 6 |
Dizziness or lightheadedness | 8 |
Feeling cold | 6 |
Shortness of breath | 7 |
Frequent or severe bleeding (nosebleeds, gums bleeding, unusual bleeding from minor cuts) or bruising | 3 |
Enlarged lymph nodes | 0 |
Bone or joint pain | 8 |
Headaches, blurred vision, nausea, or vomiting | 9 |
No response to this question | 18 |
Table 2: Disease Symptoms Effects on Day-to-Day Life Following Diagnosis
Effect | Significant impact (4-5) |
|---|---|
Ability to work | 14 |
Ability to travel | 13 |
Ability to exercise | 14 |
Personal image | 10 |
Ability to spend time with family and friends | 11 |
Intimate relationships | 12 |
Ability to continue daily activities | 14 |
Ability to concentrate | 11 |
Mental health | 8 |
Ability to manage family responsibilities | 13 |
Ability to pursue activities or hobbies | 13 |
No response to this question | 18 |
Table 3: Psychological/Social Factors Effects on Quality-of-Life Following Diagnosis
Psychological/social factors | Significant impact (4-5) |
|---|---|
Stress/anxiety/worry | 15 |
Depression | 10 |
Difficulty sleeping | 12 |
Feeling isolated | 12 |
Lack of support | 8 |
Difficulty with friend or family relationships | 8 |
Problems concentrating | 14 |
Loss of sexual desire | 9 |
Interruption of life goals/accomplishments (career, schooling, etc.) | 11 |
Financial impacts (cost of travel, inability to work, etc.) | 12 |
Loss of appetite | 10 |
No response to this question | 18 |
Experiences With Currently Available Treatments
We asked the respondents which previous treatments they had received following their diagnosis. 11 indicated chemotherapy, 8 chemotherapies with stem cell transplant, 7 radiation therapy and 1 person received targeted therapy. 18 provided no response to this question. Of those who responded, 6 mentioned having had more than 5 total lines of treatment.
We also inquired how many hospital visits were incurred (for Treatment, scans, follow ups, ER visits, etc.): 6 indicated 1-2 times per month, 2 indicated 3-4 times per month, 5 indicated 5 times per month and 10 respondents indicated more than 5 hospital visits per month. Table 4 shows the number of years the respondents have been in treatment and the number of kms travels to access care.
Table 4: Years in Treatment Versus Distance to Travel to Receive Care
Years in treatment | less than 100 kms | 100 to 199 kms | Over 400 kms |
|---|---|---|---|
1 year | 1 | 1 | 0 |
2 years | 5 | 1 | 0 |
3 years | 0 | 0 | 0 |
4 years | 3 | 0 | 0 |
5 years | 0 | 0 | 1 |
More than 5 years | 4 | 0 | 0 |
We asked the respondents what the impact of having to travel receive care and treatment were: extensive cost of travel and accommodations, emotional hardship, being away from support system for extended periods of time, and Impact to daily activities/routine were indicated as having significant impact. One respondent said, “Needed to fly to a major city, live with a family member and rent an apartment close to the hospital because treatment was longer than expected.”
When asked to rate the impact of treatment related side-effects, feelings of weakness (73%), fatigue (68%), lack of energy (59%), and inability to complete day-to-day activities (50%) were described as having the most significant impact on their lives. The side effects of the treatments had a significant impact on some of the respondents:
“The chemotherapy protocol is long and is extremely tiring.”
“Very difficult protocol of chemotherapy.”
“Intense and aggressive chemo protocol, difficult to tolerate, include intrathecal chemo, followed by allogenic stem cell transplant (2016). Very difficult, recovery over 2 years, major chicken pox affliction at 1.5 years from transplant, severely ill. Today I am doing well.”
“I developed Avascular necrosis as a result of the steroids taken during chemotherapy.”
“I had a hard time getting a diagnosis of ALL even though I had all the symptoms visible to the naked eye, an internist made a joke about pulses when my husband mentioned that my scalp was itchy, in addition to the other symptoms including my spleen hemorrhaging. Once I was diagnosed with ALL after 1 month, the Gatineau hospital transferred me to the Ottawa hospital where I was able to consider living again and received exceptional care. They were able to get me into pre-transplant remission despite the 95% leukemia cells in my marrow at the time of my transfer. The sad thing is that if my husband had not fought for further testing, I probably would have died before I could have been transferred to Ottawa. I am forever grateful to the Ottawa Hospital and its caregivers.”
Respondents also indicated that ease of access and that results were positive from the treatments received:
“I was lucky enough to live close to Princess Margaret and get treatment there.”
“I had quick access to health care professionals and appropriate treatment.”
Improved Outcomes
Respondents were asked to rate the most key factors or outcomes considered when deciding about taking a new treatment. Of the 21 responses received, the majority agreed that the degree of certainty that ALL will respond to treatment (100%) and least amount of travel required are significantly more important (95%). Improved quality of life and coverage by insurance/drug plan were the second most crucial factors when considering a new treatment. 19 people did not respond to this question.
Although, reduced side effects were note rated as highly as the above-mentioned factors, it was frequently mentioned in the open-ended question “What kinds of improvement would you like to see in any new treatment for ALL?”
"Fewer side effects during and after treatment.”
“Less side effects and adverse reactions.”
“Something more gentle than a BMT.”
“At home accessibility, remote nurses able to travel to homes or provide treatment in centers closer with less travel time.”
“Less side effects or if not, then to know what impact on our system.”
“Less time spent in the hospital and less harsh treatments, that don't take 2 and 1/2 years to complete. Also, would be nice to avoid having to require a stem cell transplant.”
“That has less side effects to the patient while doing the same amount of “damage” to the cancer.”
“Complications related to bone damage such as AVN are reduced or made more aware. More research on how different ethnicities would be affected and the level of risk.”
Experience With Drug Under Review
Of all 40 respondents to this survey, only one person had experience with Crisantaspase Recombinant (Rylaze) and had an overall positive experience with the drug. The respondent received Rylaze in a center close to home, so they did not incur any out-of-pocket expenses. The drug was also paid for by the cancer board/agency or government and follow-up care was received with their treating oncologist.
The potential side-effects were explained to the respondent; however, the potential side effects had no impact on their decision to receive treatment. Of the potential side effects, the respondent had no minor side-effects. Nausea, musculoskeletal pain, and fatigue were among the side-effects experienced but considered these manageable. The respondent also reported having experienced some allergic reactions to other previously used chemotherapies.
As compared to previous therapies, the respondent indicated Rylaze to be less difficult to other treatments and had positive impacts on their ability to perform daily activities and their mental health. The respondent provided one comment about the treatment under review:
“It was a good drug to use, but the problem is the injections. If they could make it in an IV form, it would much better for patients, in my opinion.”
The respondent also mentioned that their disease responded to Rylaze completely.
Companion Diagnostic Test
Not applicable.
Anything Else?
No.
Conflict of Interest Declaration — The Leukemia & Lymphoma Society of Canada
To maintain the objectivity and credibility of the CADTH reimbursement review process, all patients in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission? If yes, please detail the help and who provided it.
No.
Did you receive help from outside your patient group to collect or analyze data used in this submission? If yes, please detail the help and who provided it.
No.
List any companies or organizations that have provided your group with financial payment over the past 2 years AND who may have direct or indirect interest in the drug under review.
Jazz Pharmaceuticals Inc.– in excess of $50,000.
Clinician Input
Ontario Health Hematology Cancer Drug Advisory Committee
About the Ontario Health Hematology Cancer Drug Advisory Committee
OH-CCO’s Drug Advisory Committees provide timely evidence-based clinical and health system guidance on drug-related issues in support of CCO’s mandate, including the Provincial Drug Reimbursement Programs (PDRP) and the Systemic Treatment Program.
Information Gathering
The DAC gather the information jointly via email.
Current Treatments and Treatment Goals
Pegasparaginase is a critical part of ALL/LBL treatment and in ability to delivery this due to severe allergy would compromise outcomes. In addition, silent inactivation is a well recognized phenomenon that could impact outcomes. Thus, Rylaze would be necessary in these few cases, given lack of access to erwinase.
The treatment goal of Rylaze would be to continue to provide curative type therapy for ALL/LBL in a situation where pegasparaginase would carry significant risk or inactivity.
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
Access to erwinia is limited. Asparaginase level testing is not widely available or systematically funded within Ontario.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
Asparaginase products are standard of care in ALL/LBL patients.
Which patients would be best suited for treatment with the drug under review? Which patients would be least suitable for treatment with the drug under review?
All patients with ALL/LBL who are receiving curative style therapy and who cannot receive pegasparginase due to severe allergy or silent inactivation.
Patients who tolerate pegasparaginase should continue with that product.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice? How often should treatment response be assessed?
Standard response measures for ALL/LBL, including blood count and bone marrow testing for remission.
What factors should be considered when deciding to discontinue treatment with the drug under review?
Significant intolerance or treatment failure.
What settings are appropriate for treatment with [drug under review]? Is a specialist required to diagnose, treat, and monitor patients who might receive [drug under review]?
This drug would be given under the supervision of clinicians with expertise in ALL/LBL.
Additional Information
Not applicable.
Conflict of Interest Declarations — Ontario Health Hematology Cancer Drug Advisory Committee
To maintain the objectivity and credibility of the CADTH drug review programs, all patients in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please refer to the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
Yes, Ontario Health provided secretariat function to the DAC.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
No.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician who contributed to the input — please add more tables as needed (copy and paste). It is preferred for all declarations to be included in a single document.
Declaration for Clinician 1
Name: Dr. Tom Kouroukis
Position: Lead, Ontario Health CCO Hematology Cancer Drug Advisory Committee
Date: 14-08-2022
Table 5: COI Declaration for Ontario Health Hematology Cancer DAC — Clinician 1
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
Redactions: Confidential information in this document may be redacted at the request of the sponsor in accordance with the CADTH Drug Reimbursement Review Confidentiality Guidelines.
Stakeholder Input: The views expressed in each submission are those of the submitting organization or individual; not necessarily the views of CADTH or of other organizations. As such, they are independent of CADTH and do not necessarily represent or reflect the view of CADTH. No endorsement by CADTH is intended or should be inferred. By filing with CADTH, the submitting organization or individual agrees to the full disclosure of the information. CADTH does not edit the content of the submissions.
CADTH does use reasonable care to prevent disclosure of personal information in posted material; however, it is ultimately the submitter’s responsibility to ensure no identifying personal information or personal health information is included in the submission. The name of the submitting organization or individual and all conflict of interest information are included in the submission; however, the name of the author, including the name of an individual patient or caregiver submitting the patient input, are not posted.
Accessibility: CADTH is committed to treating people with disabilities in a way that respects their dignity and independence, supports them in accessing material in a timely manner, and provides a robust feedback process to support continuous improvement. All materials prepared by CADTH are available in an accessible format. Where materials provided to CADTH by a submitting organization or individual are not available in an accessible format, CADTH will provide a summary document upon request. More details on CADTH’s accessibility policies can be found here.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.